


































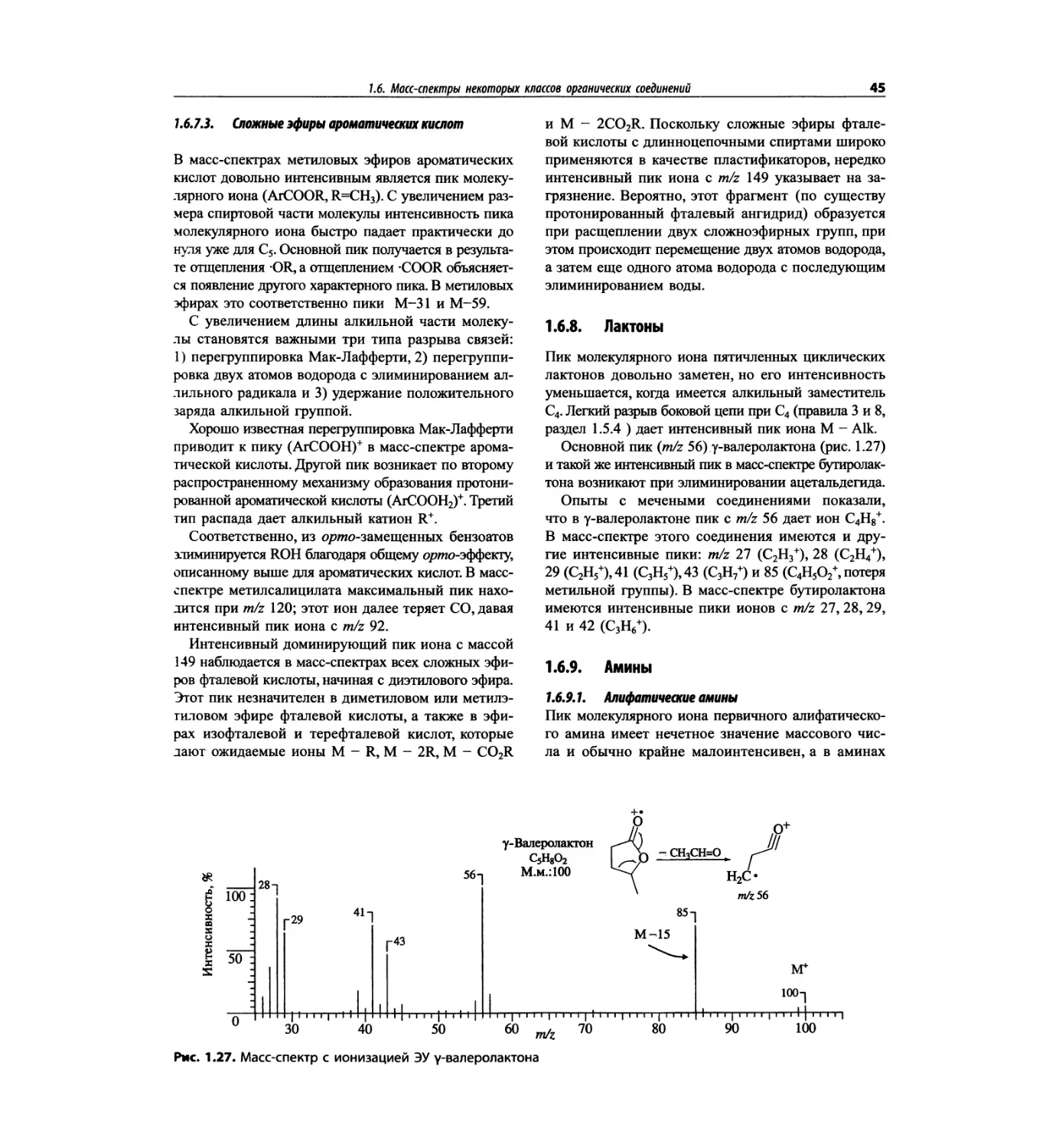





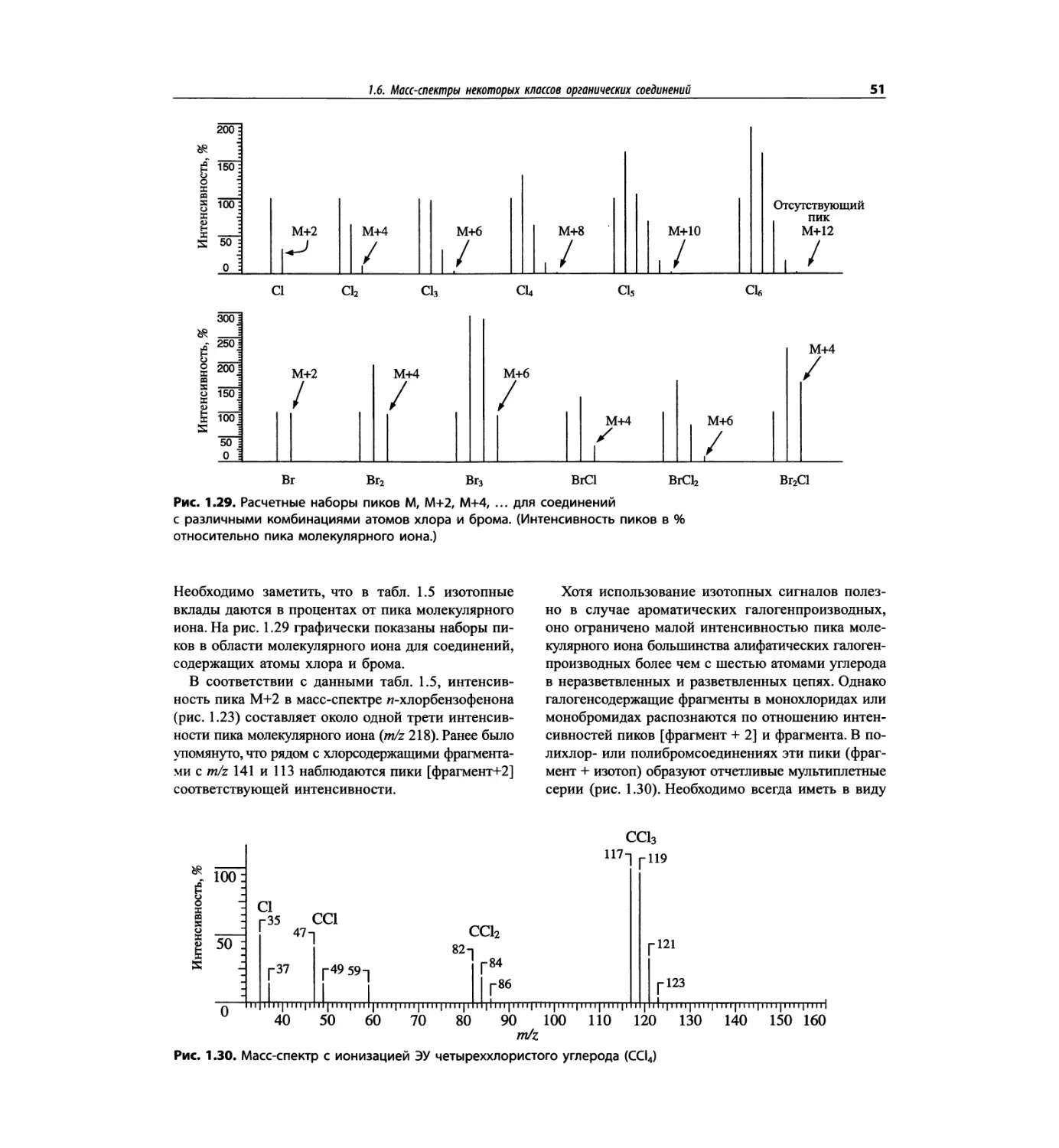



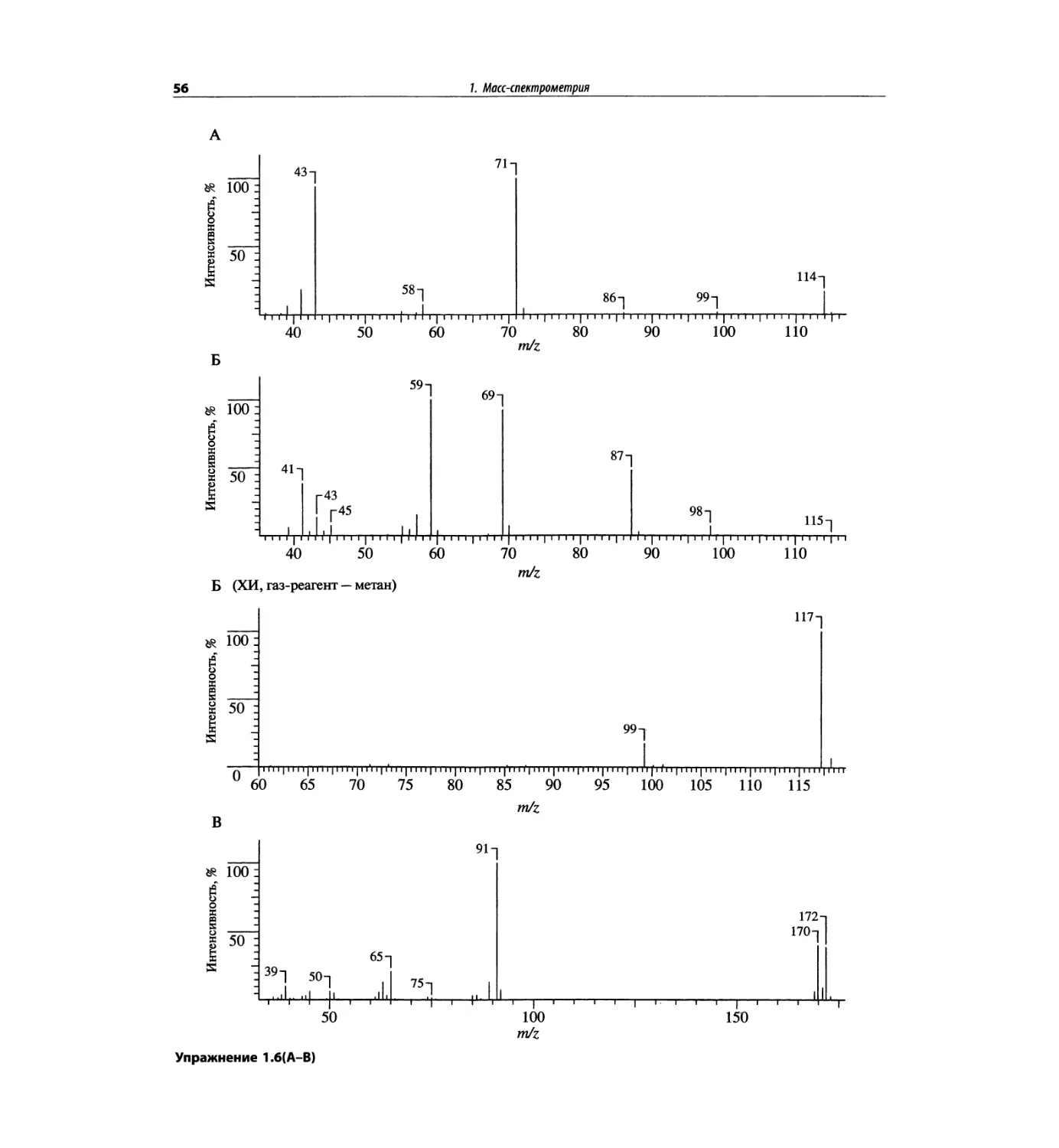








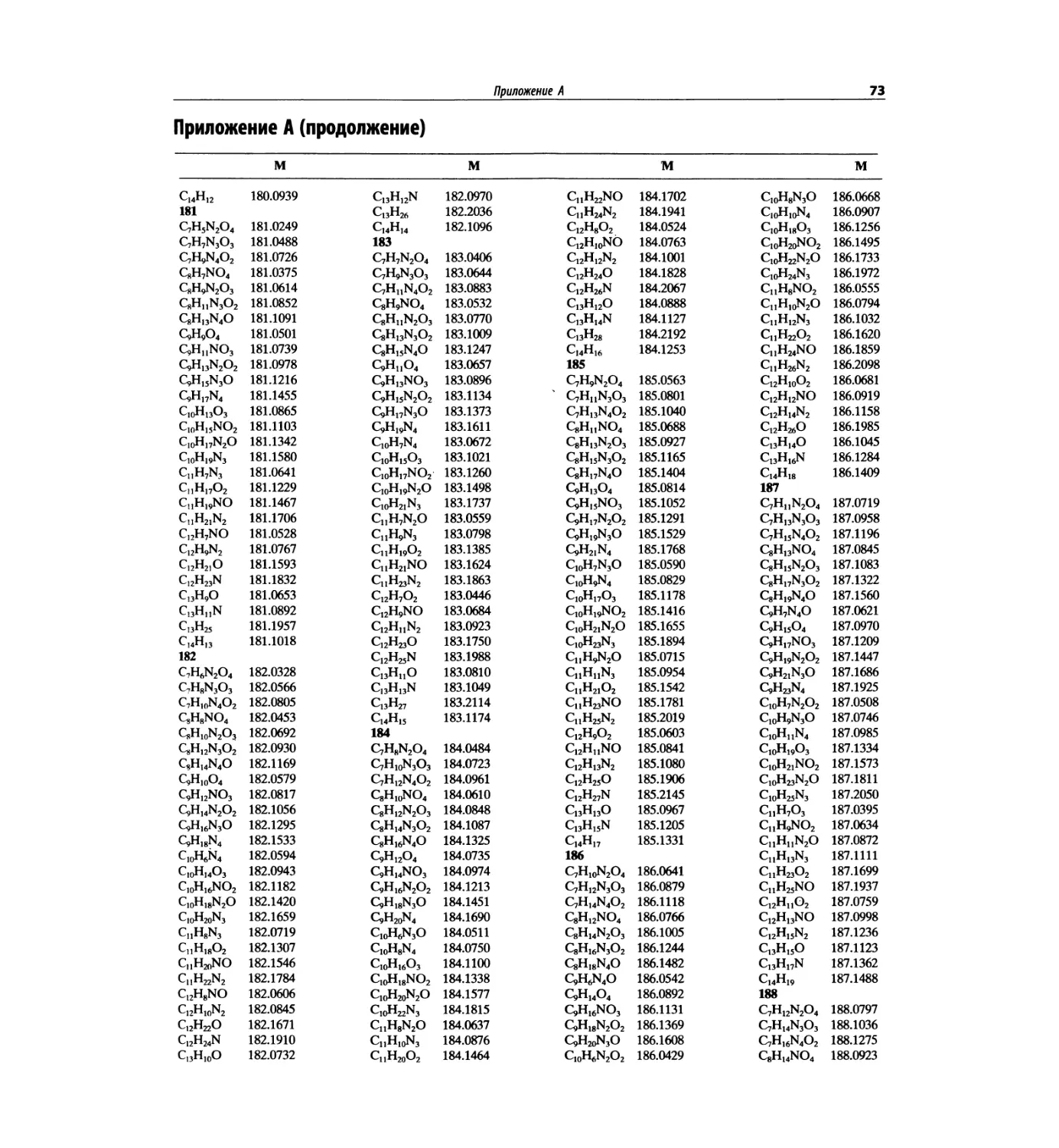
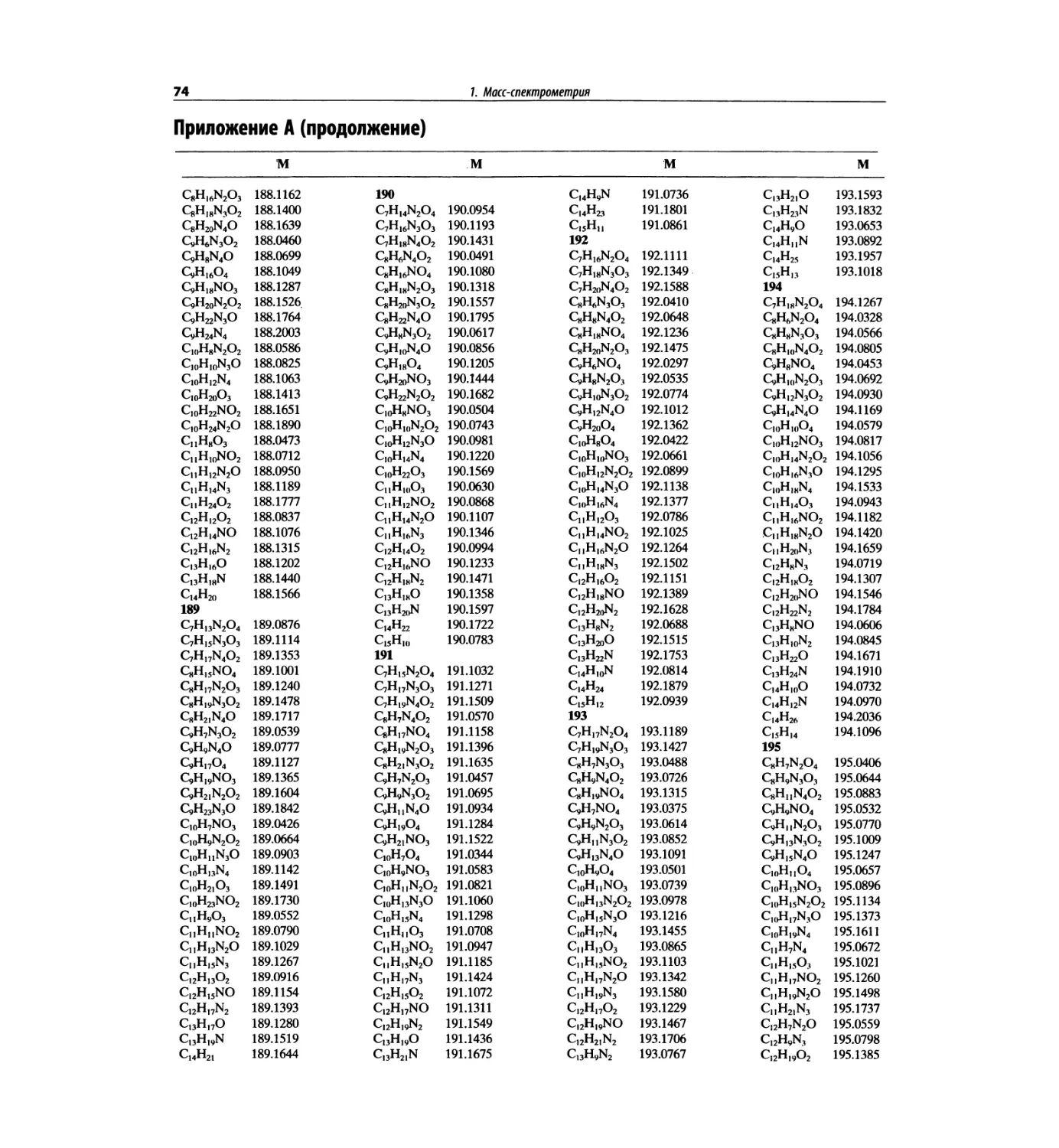
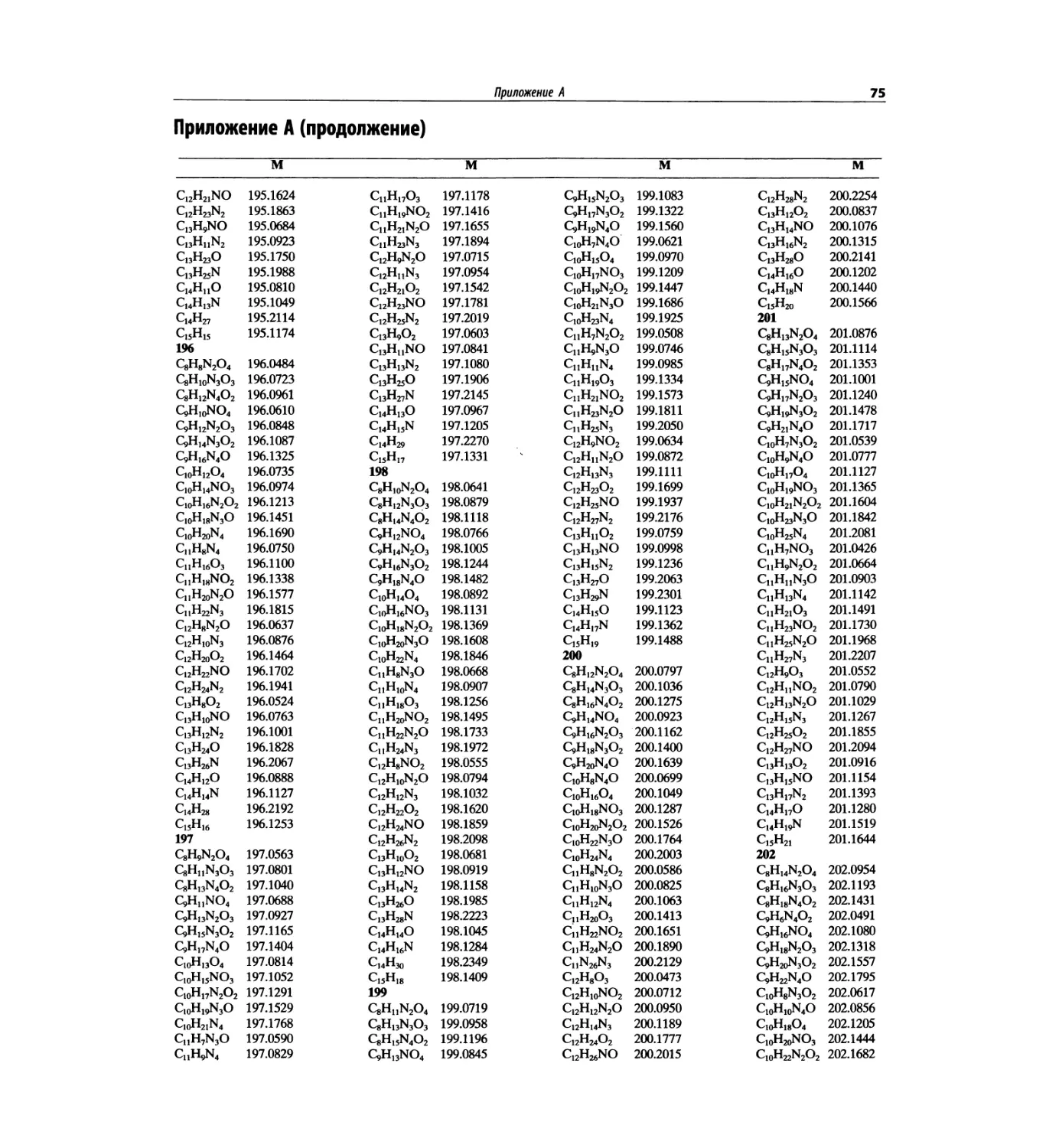



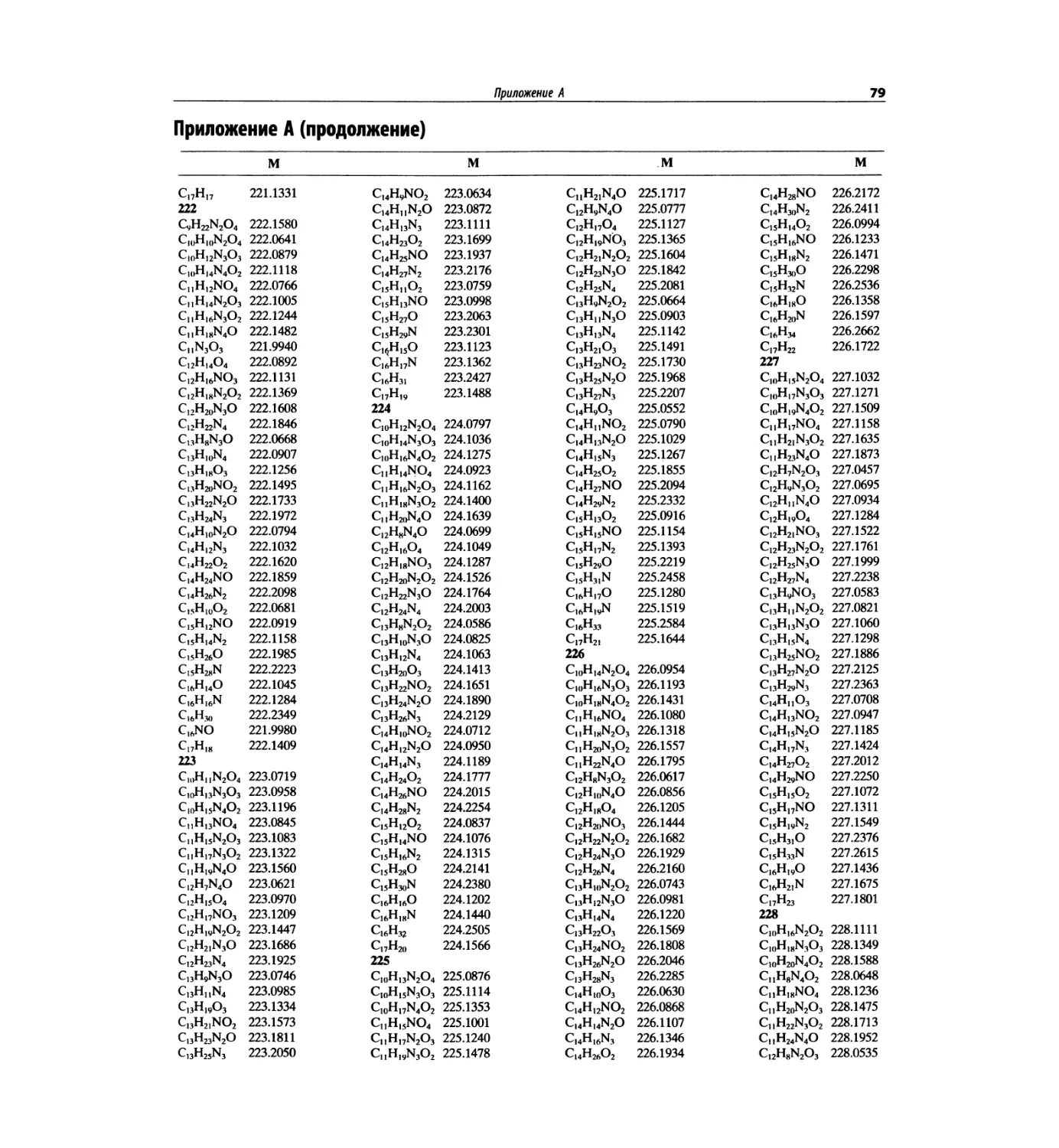




















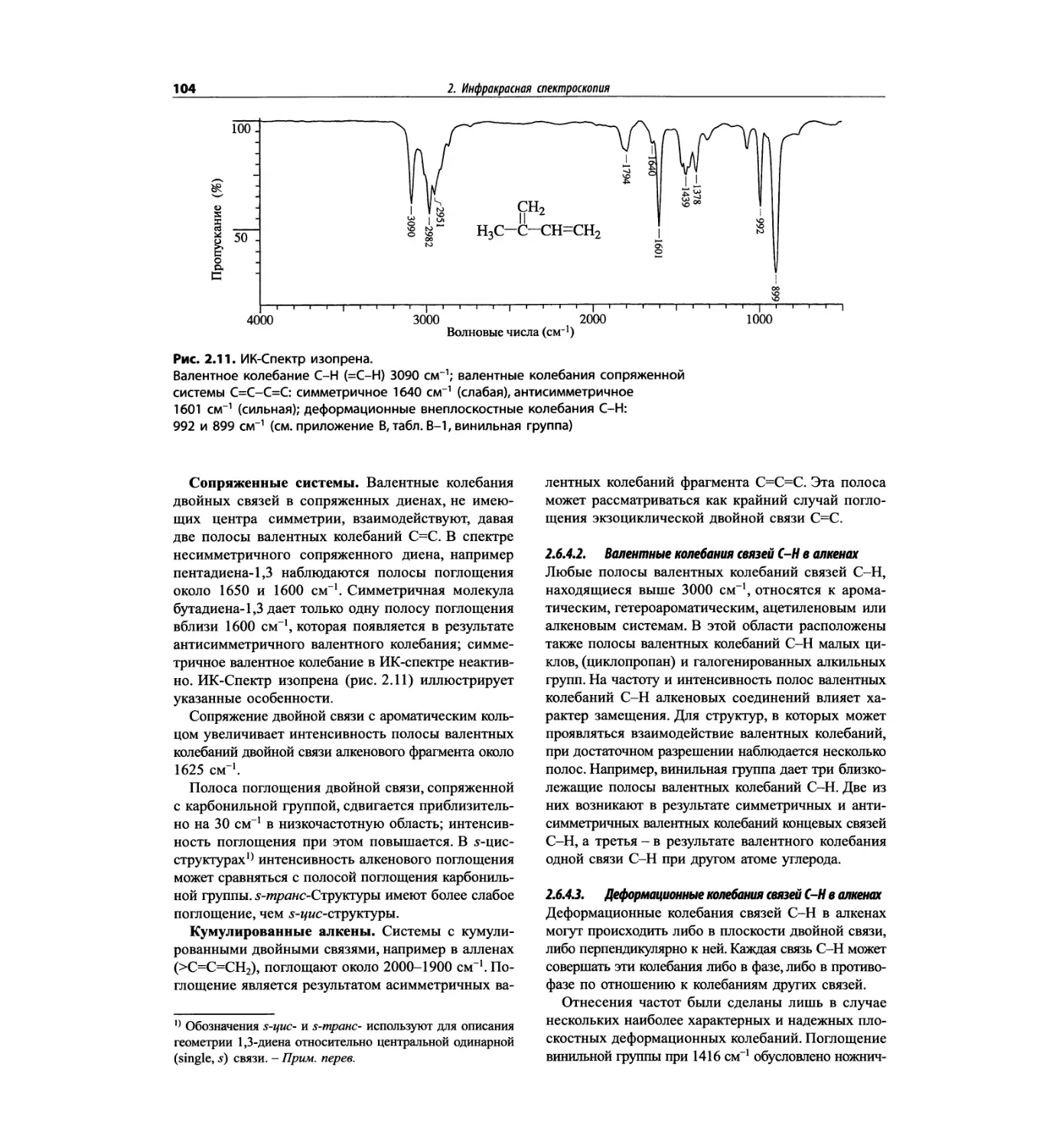



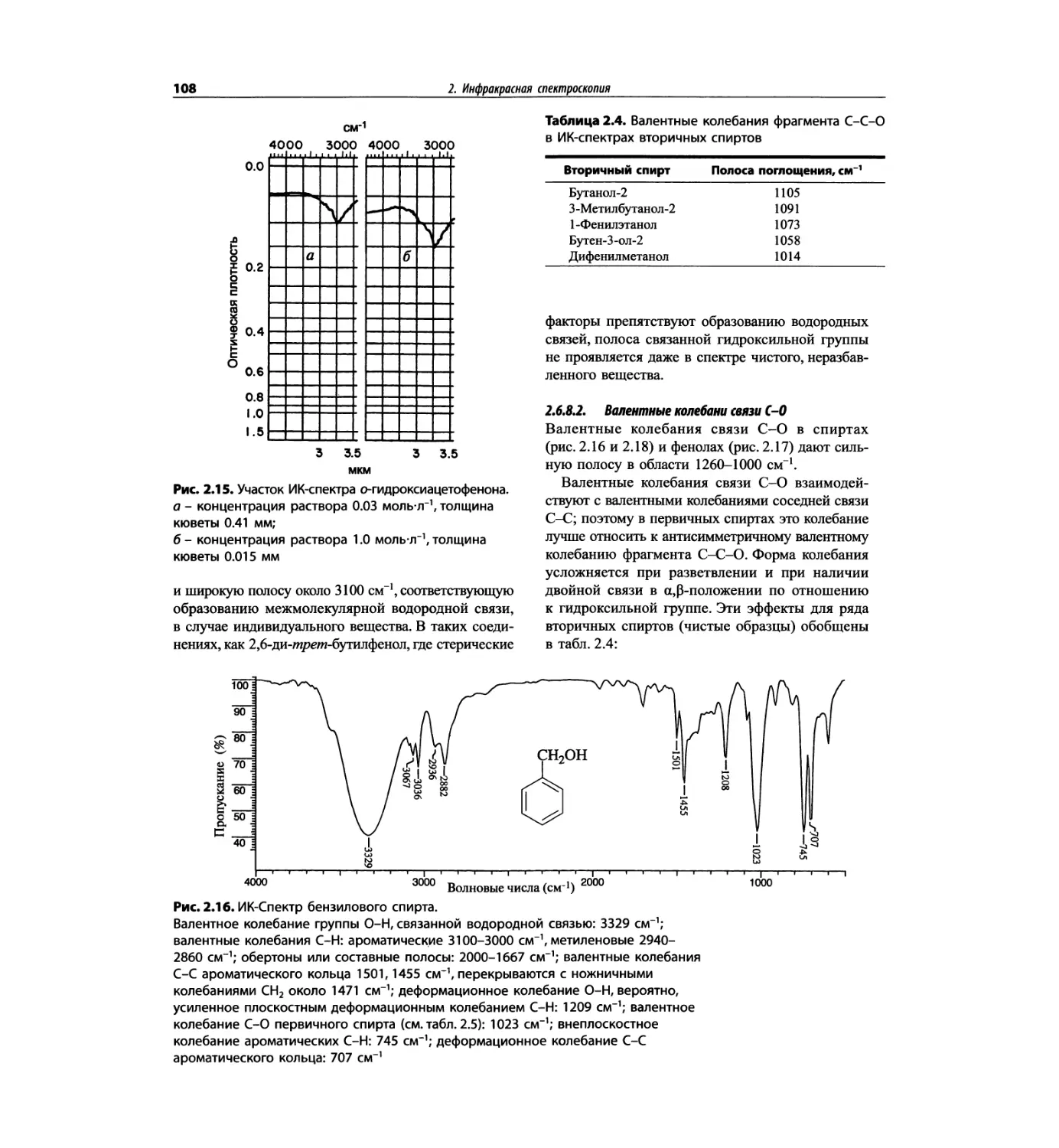

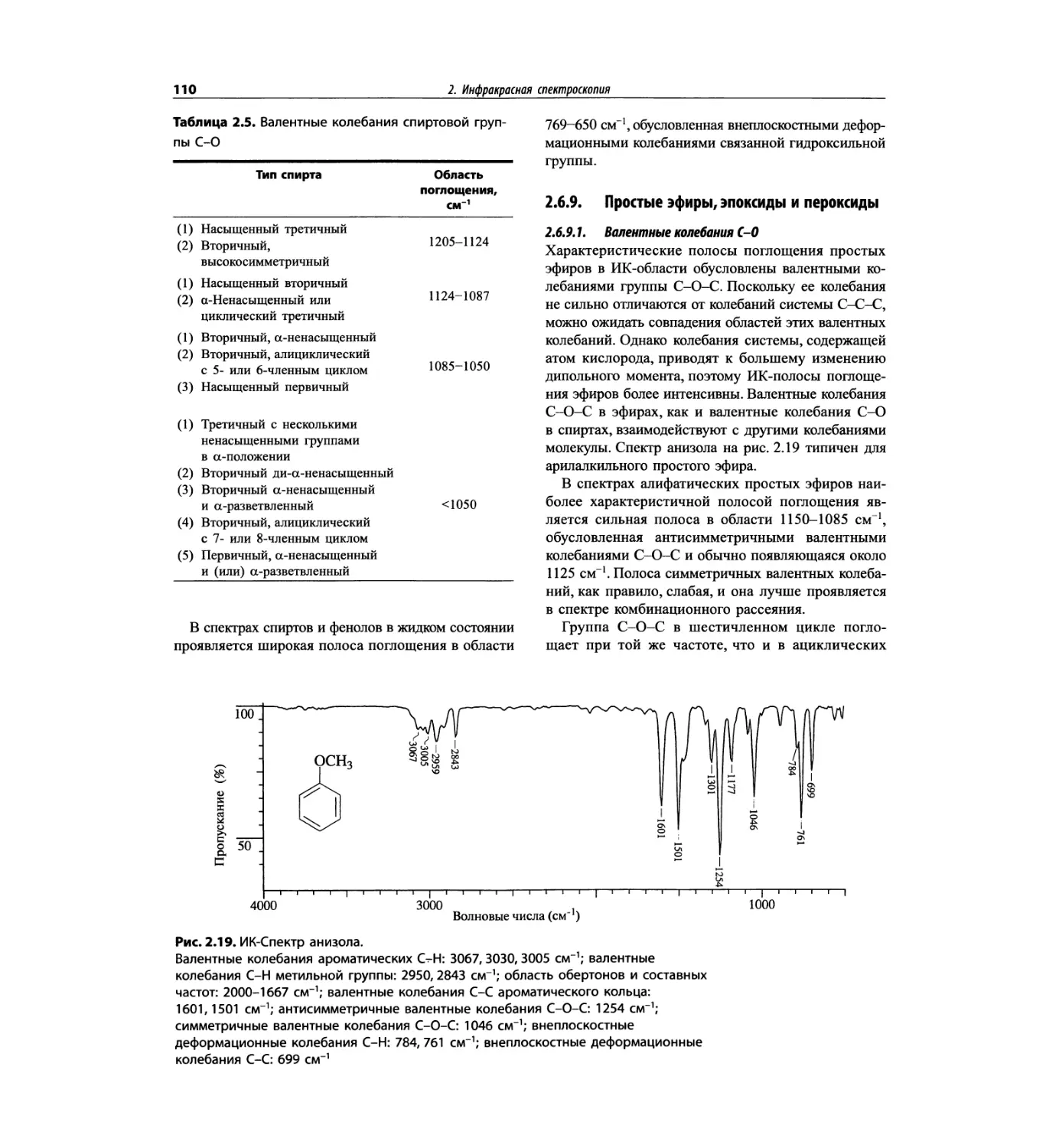

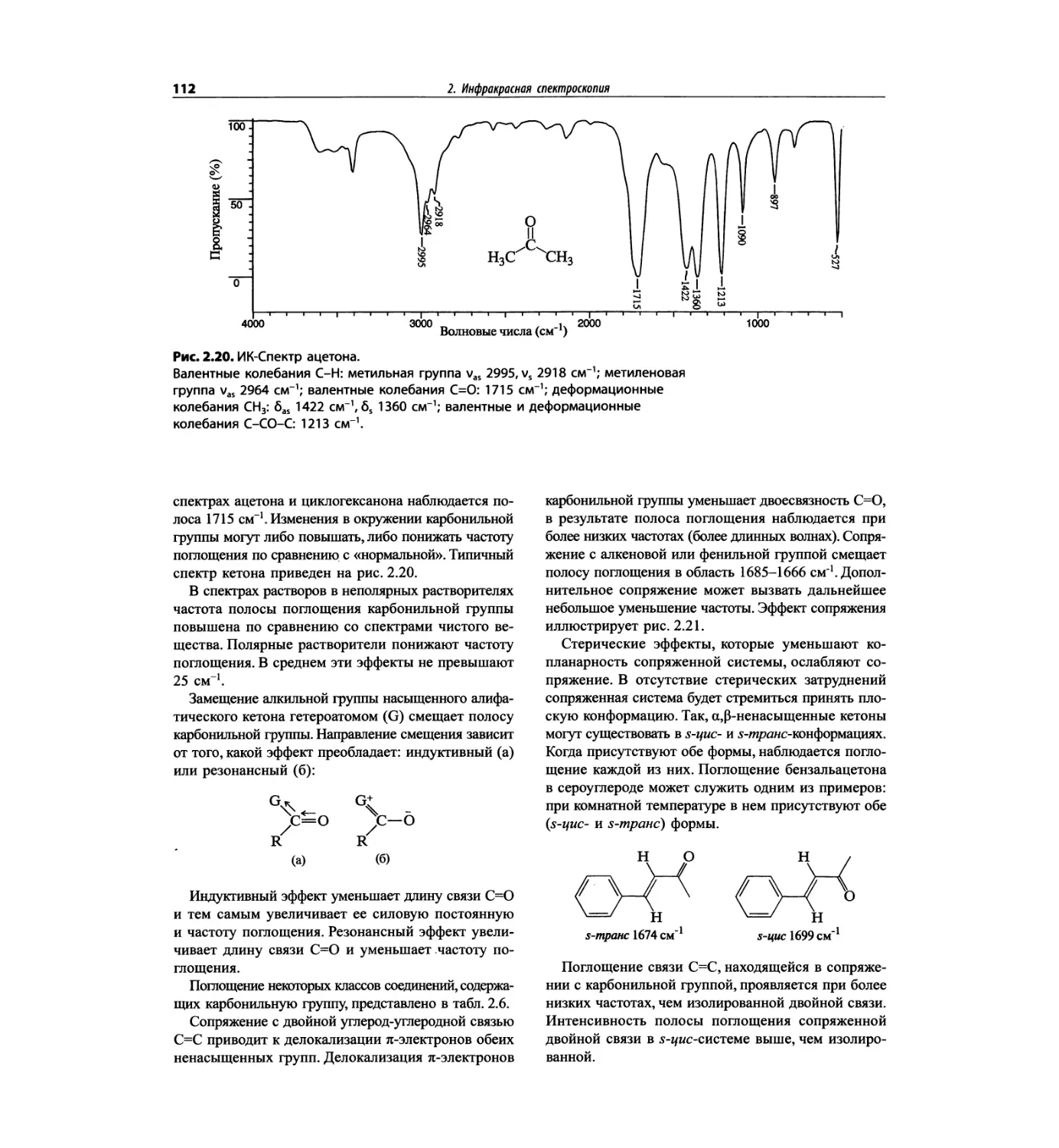

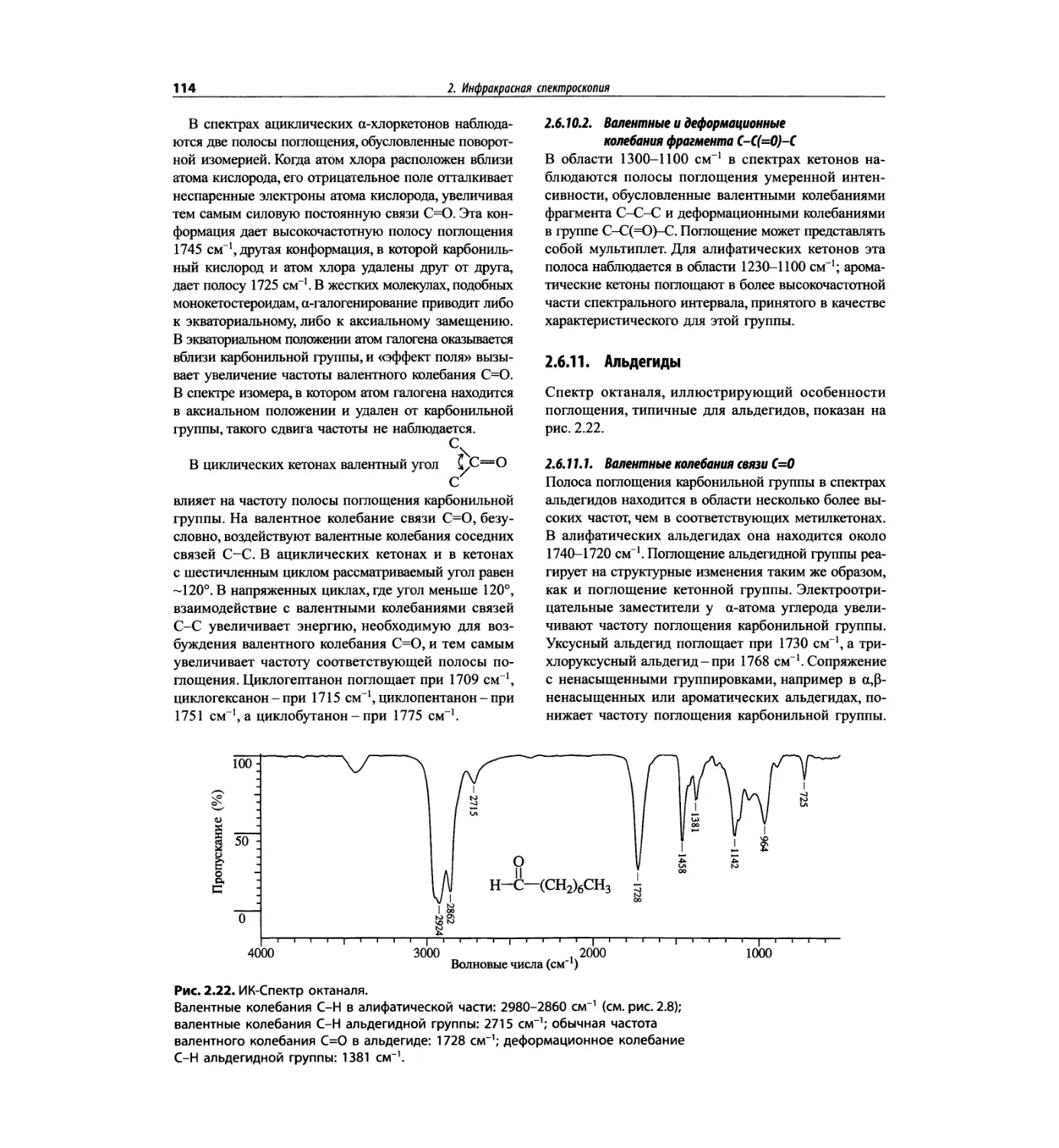
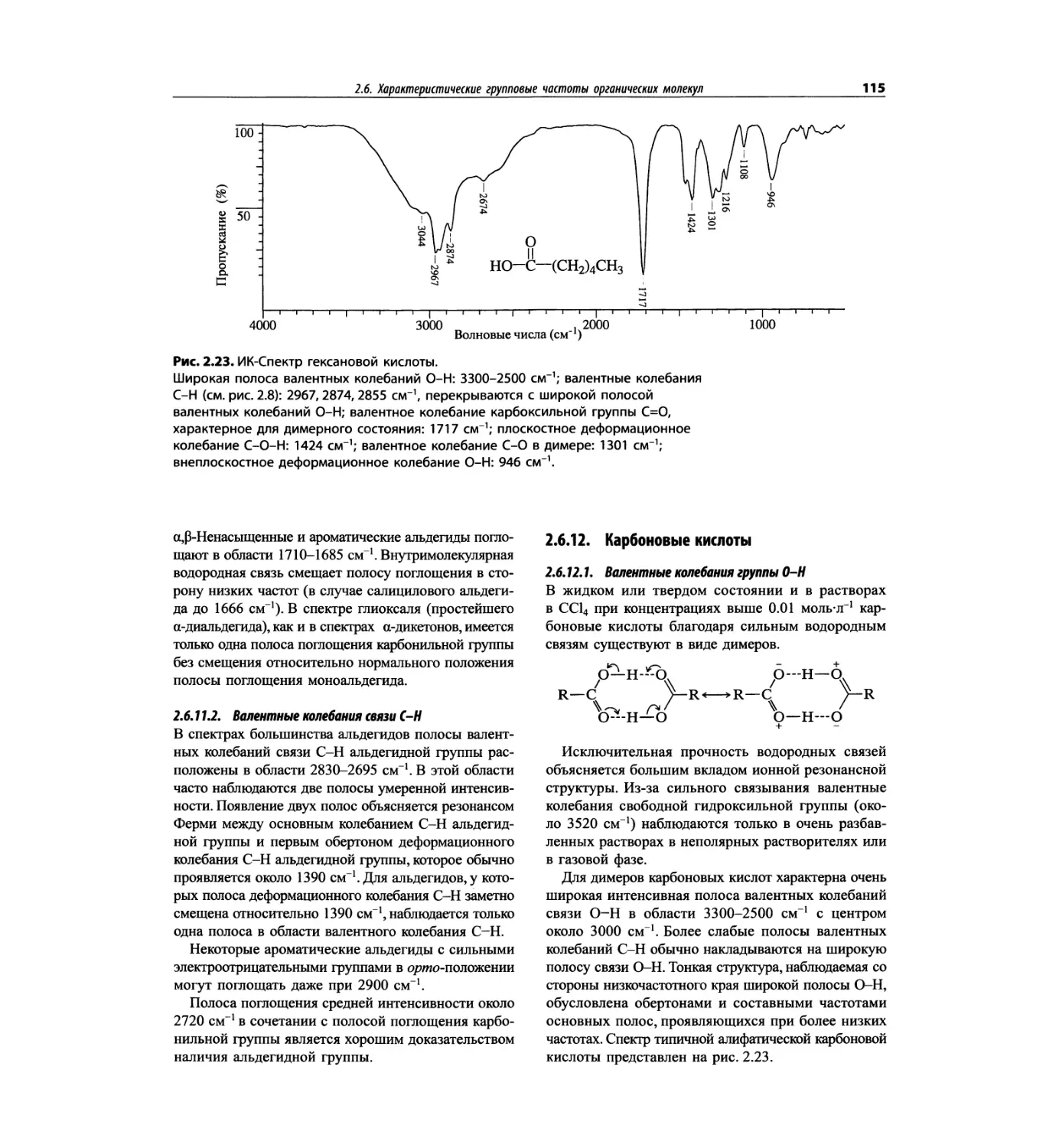

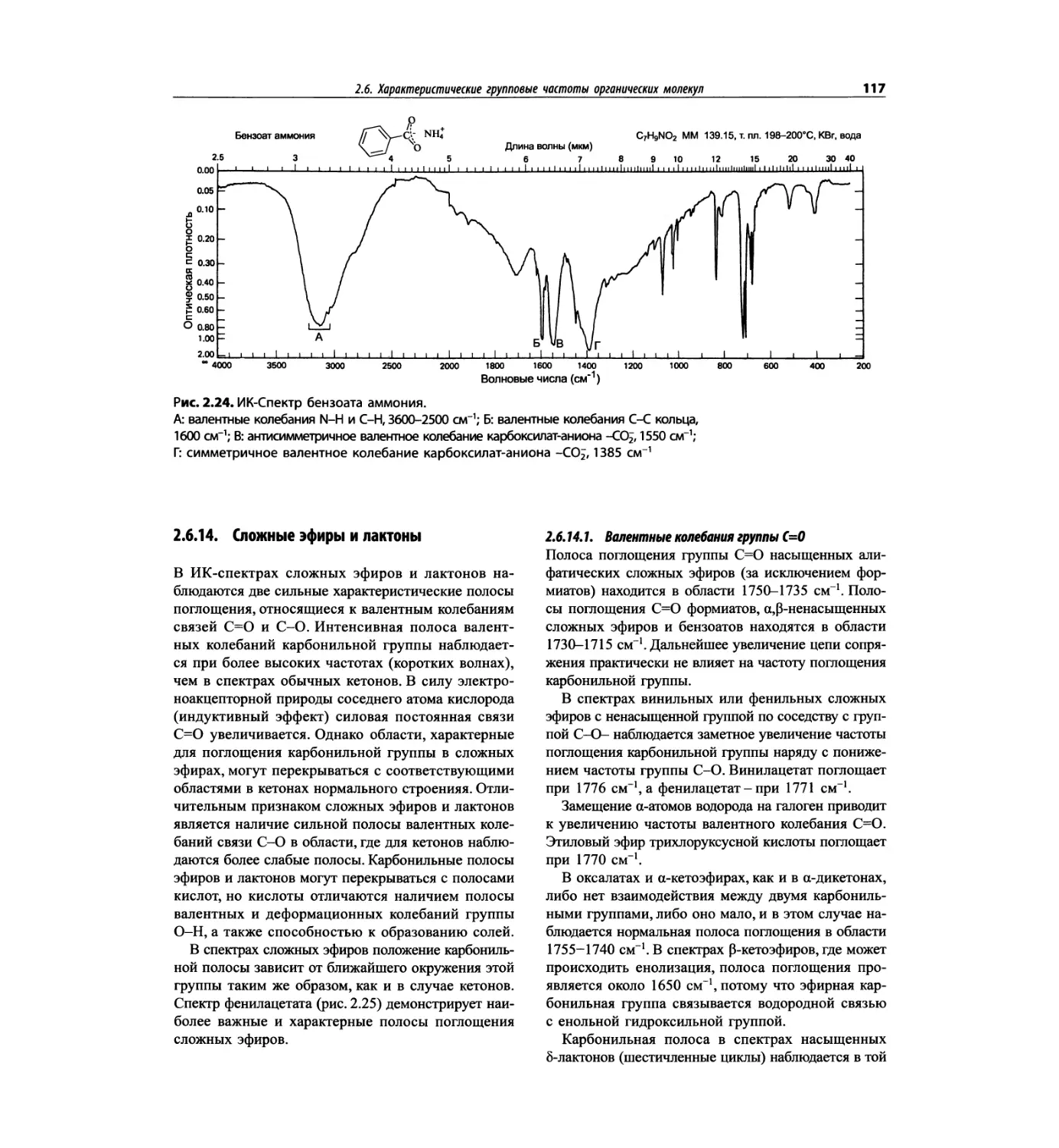
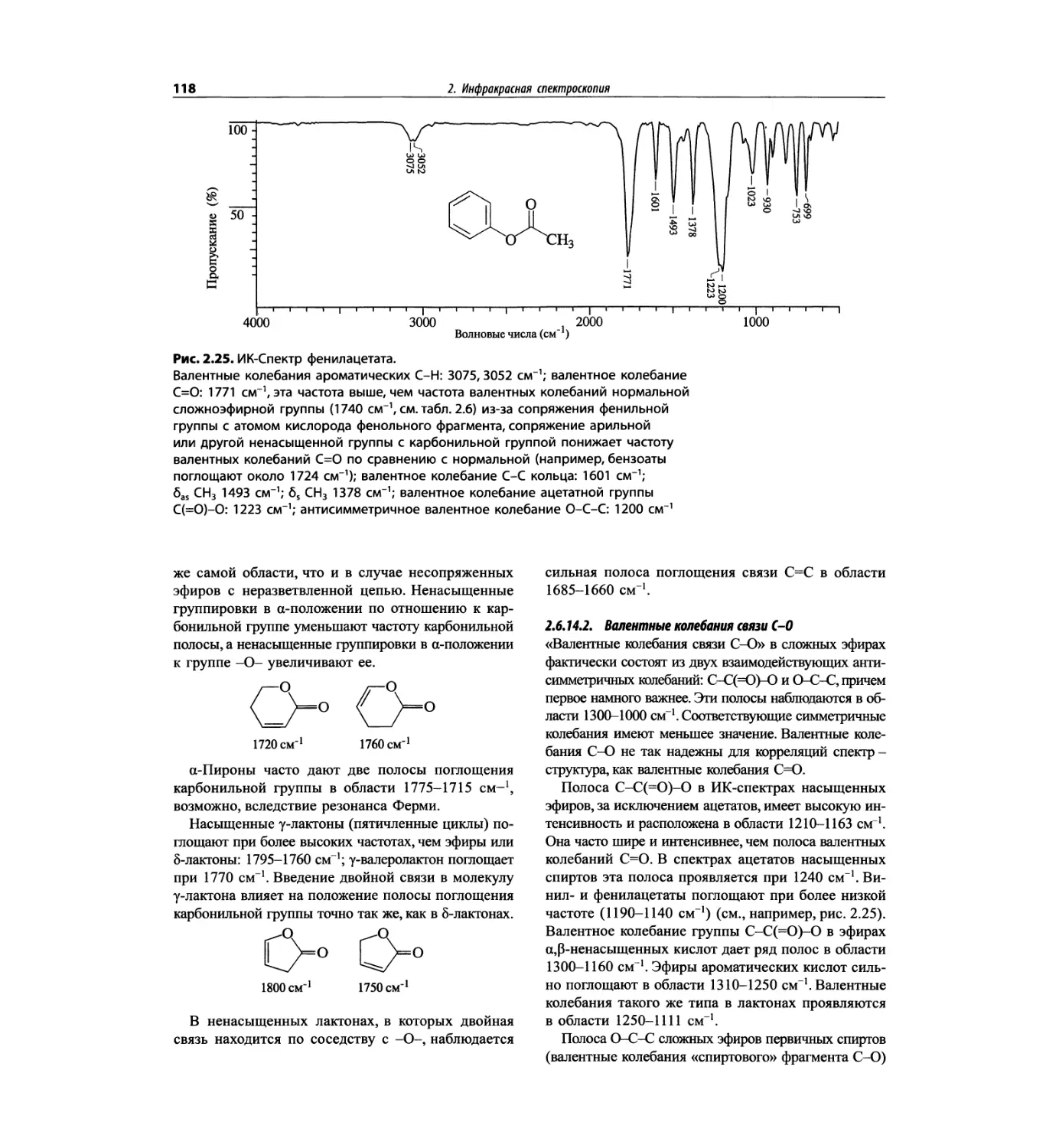


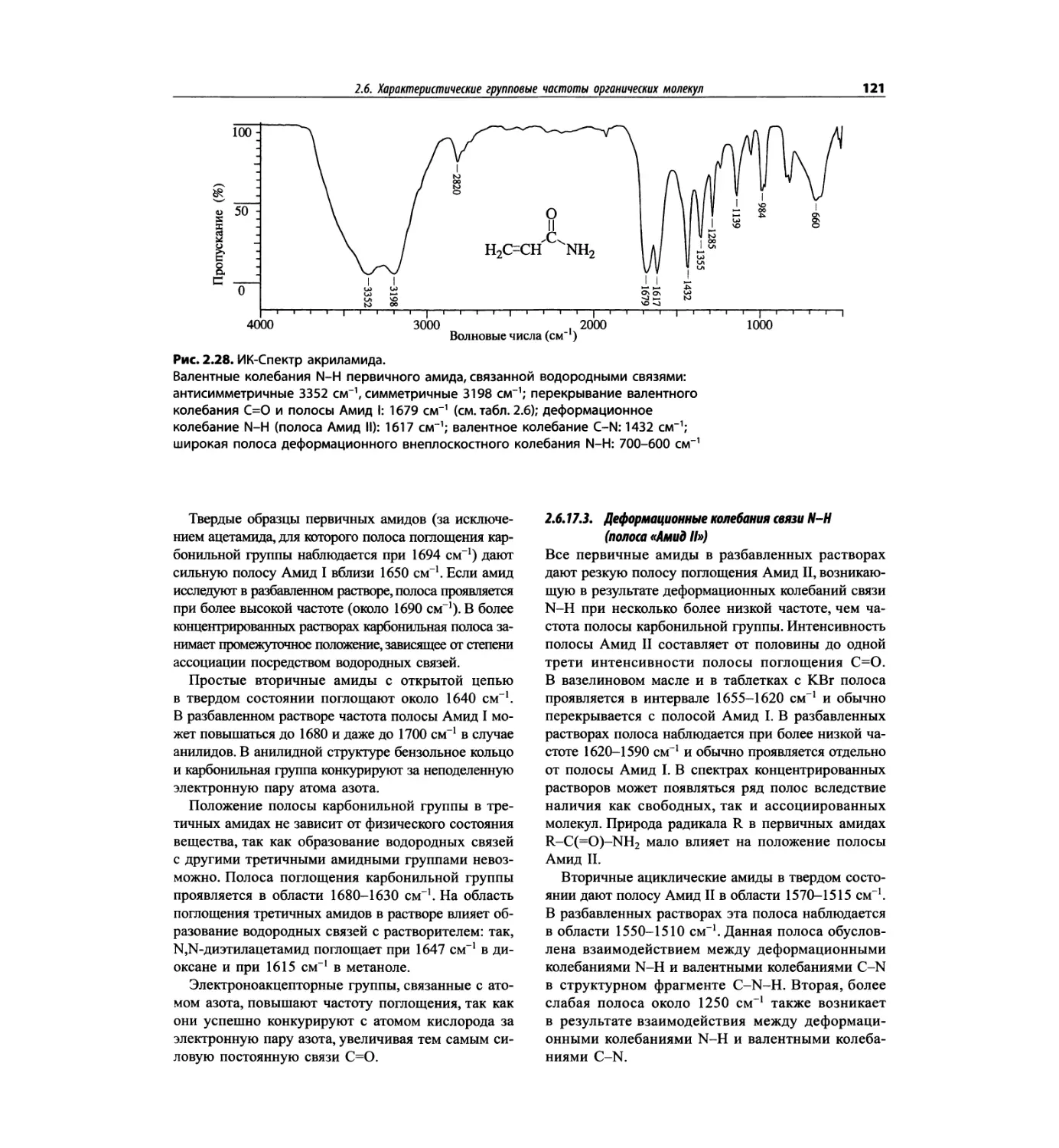
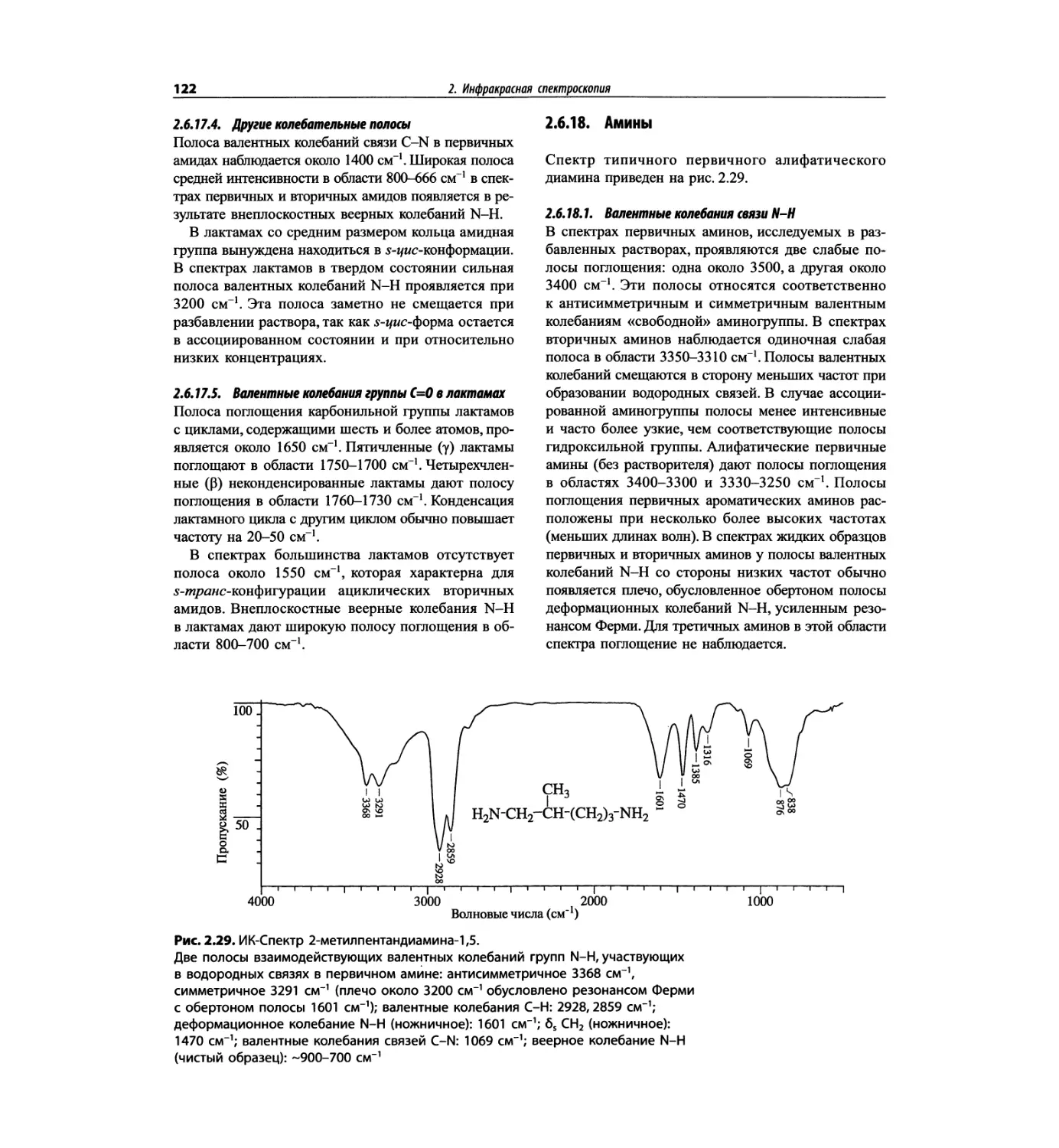

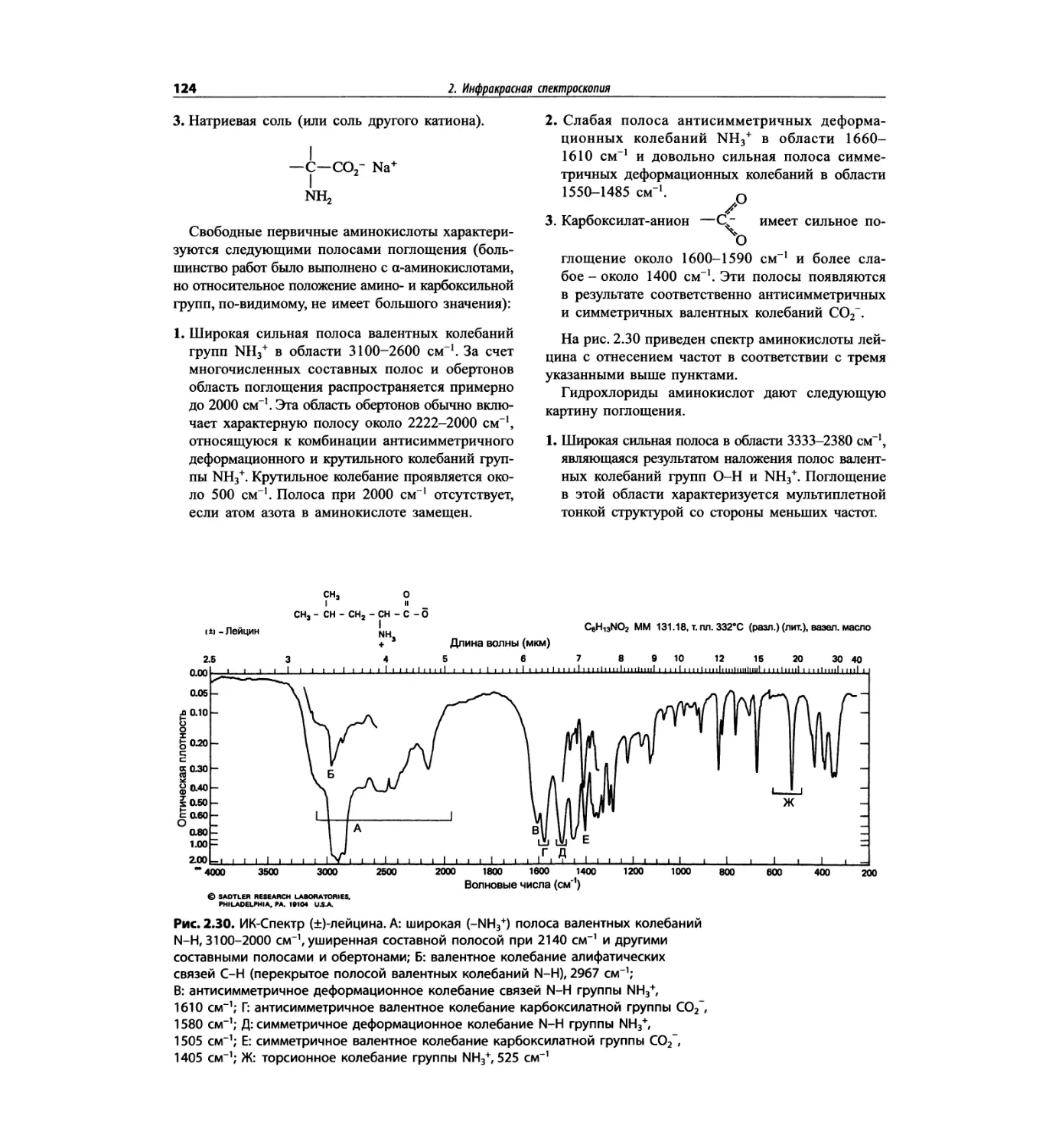




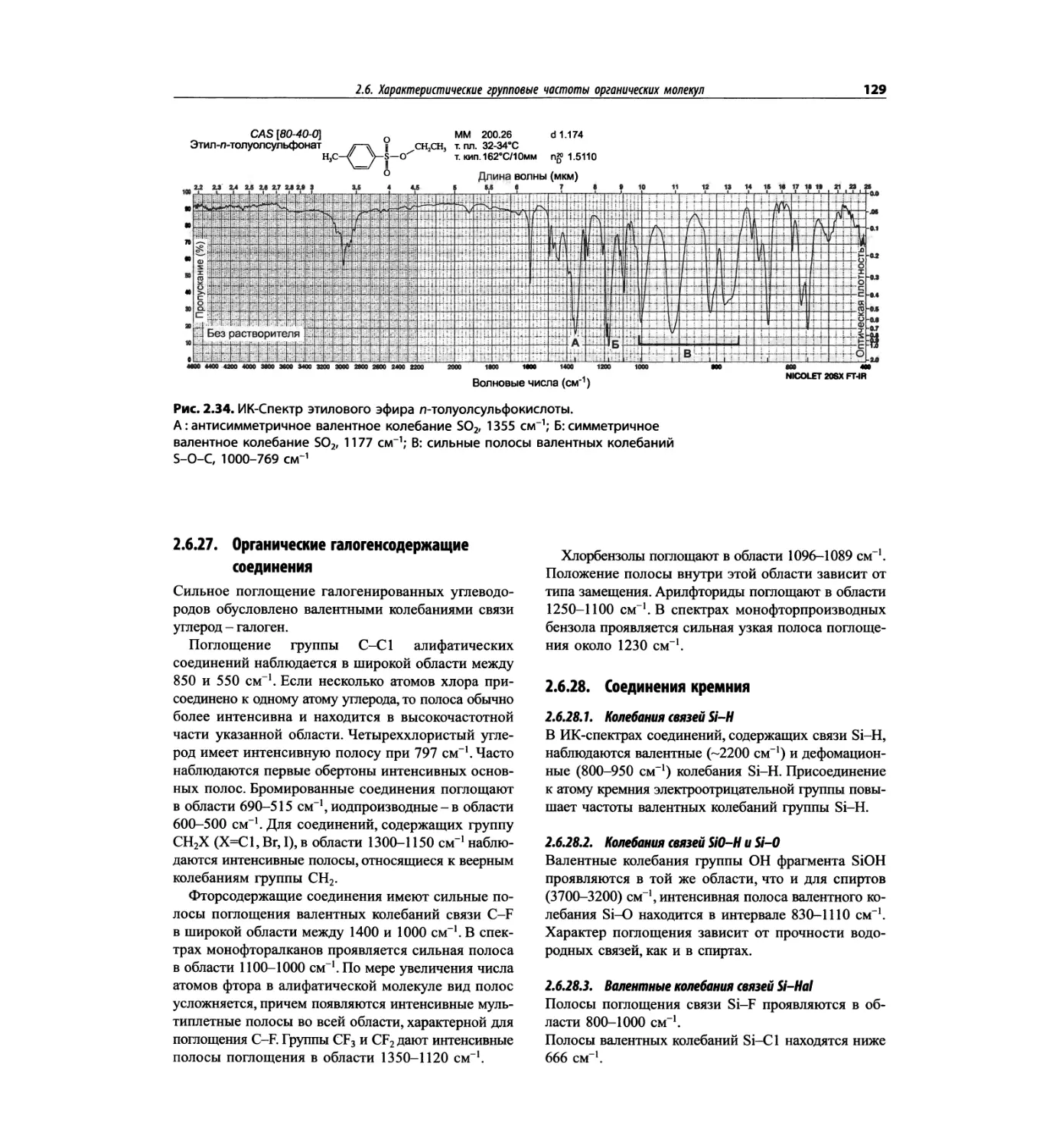



















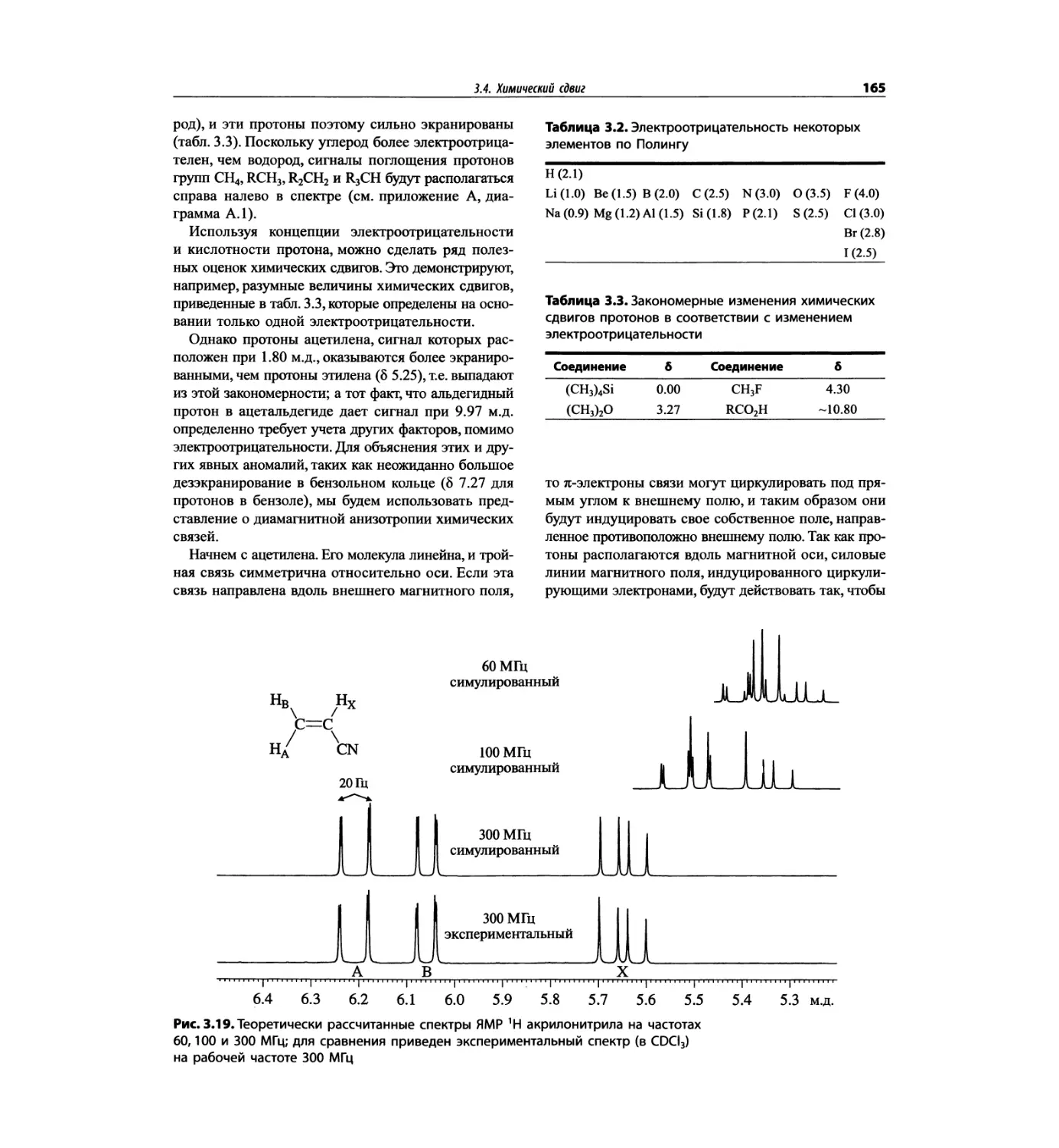

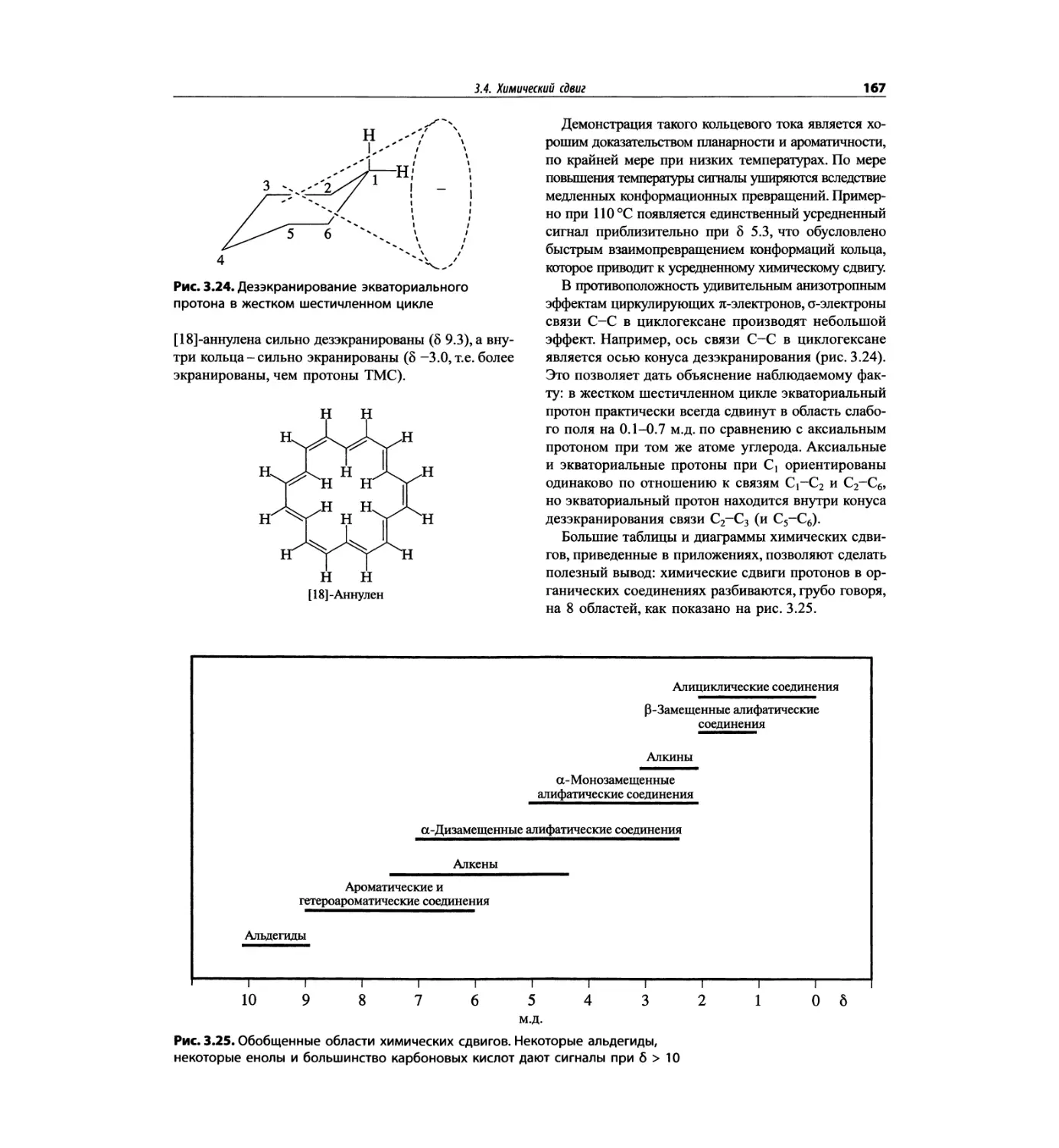

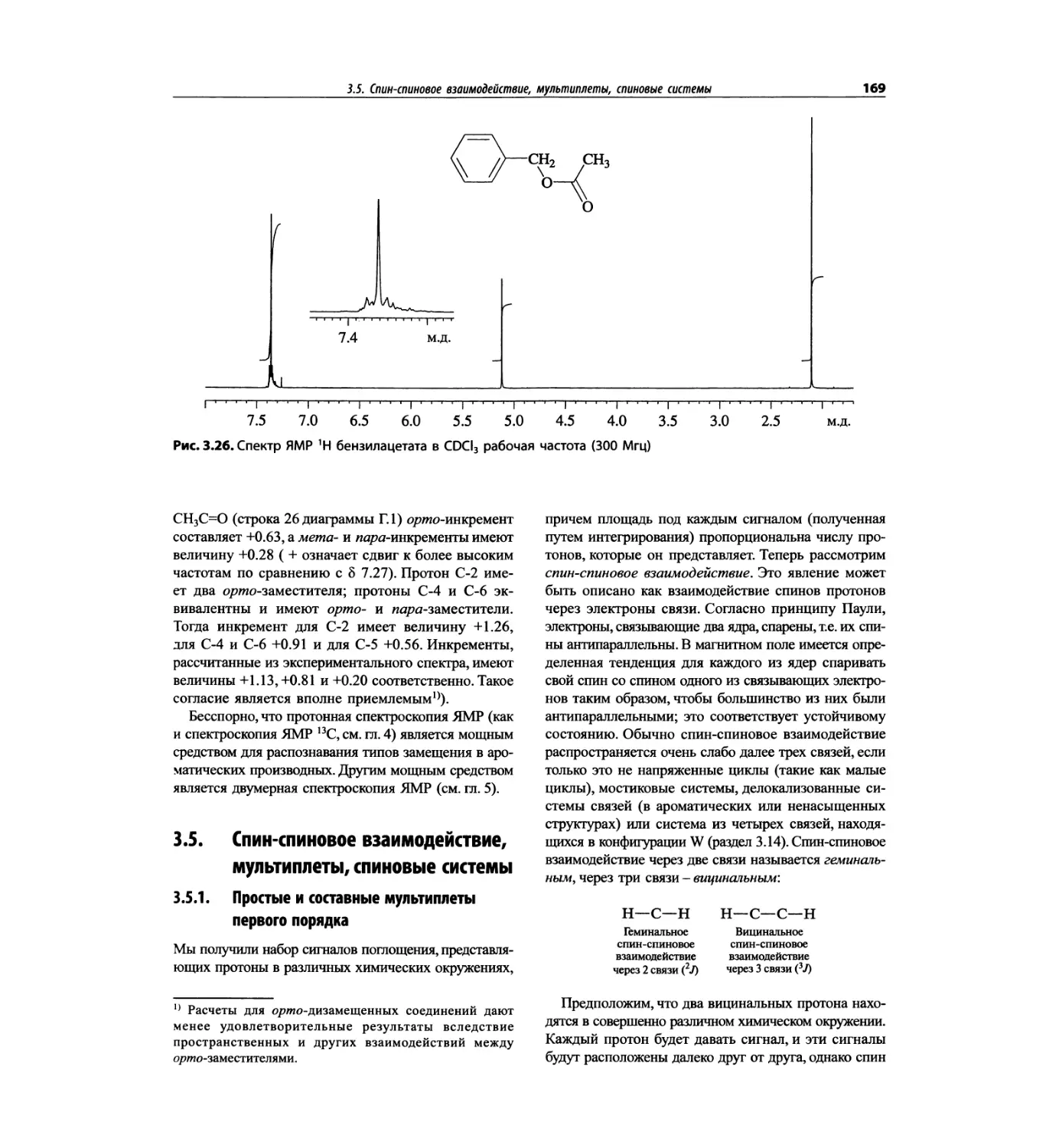



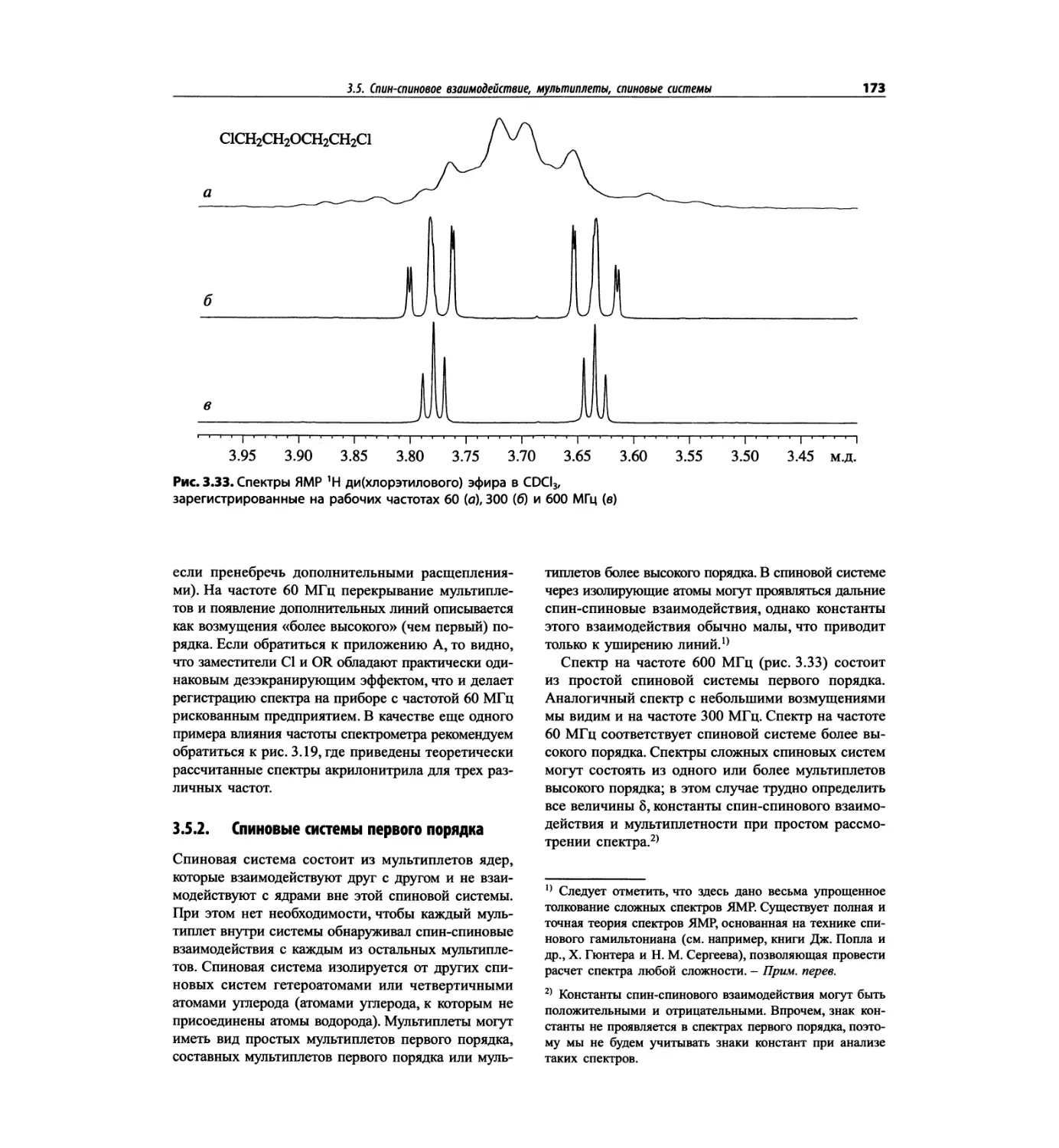













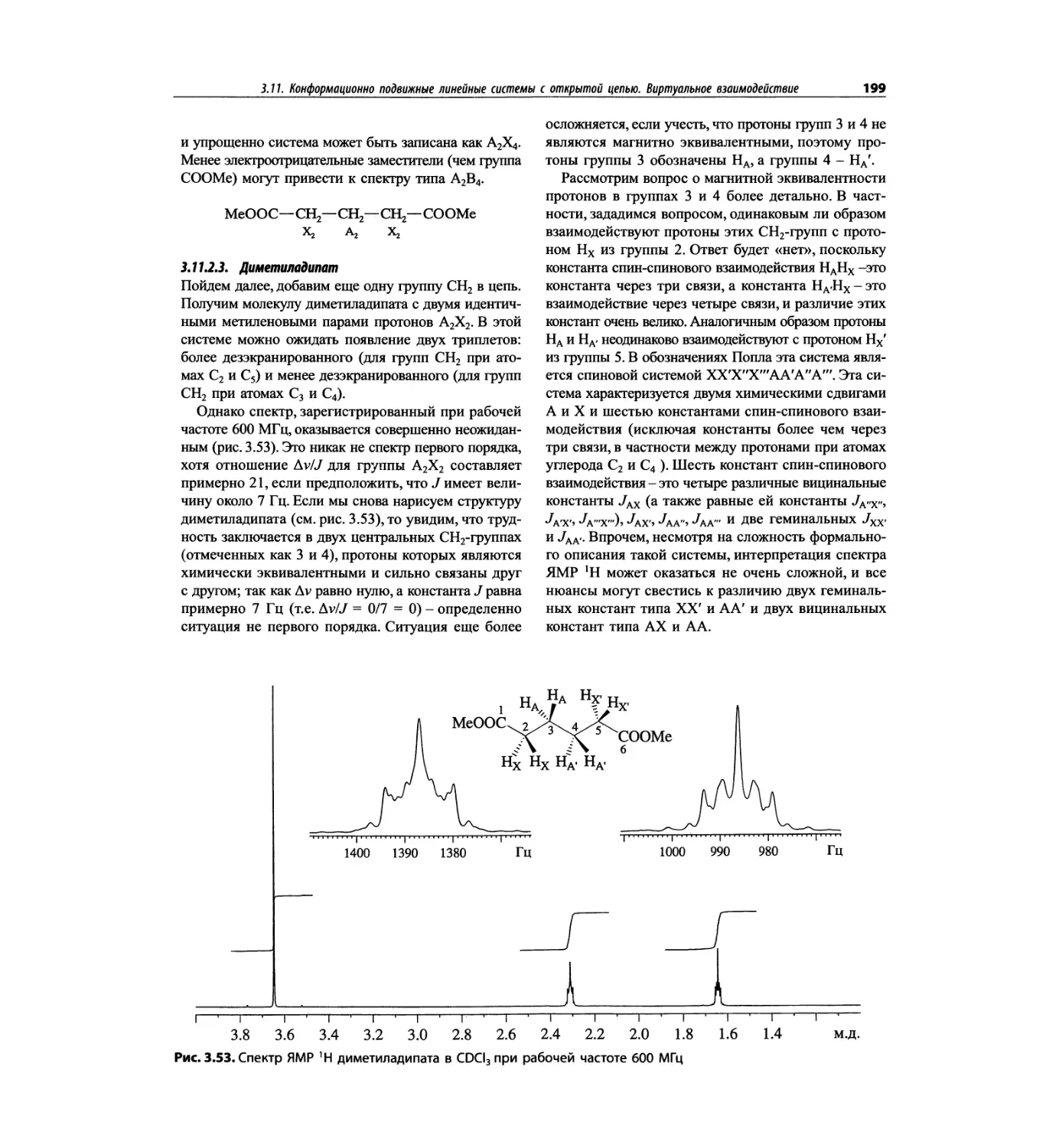
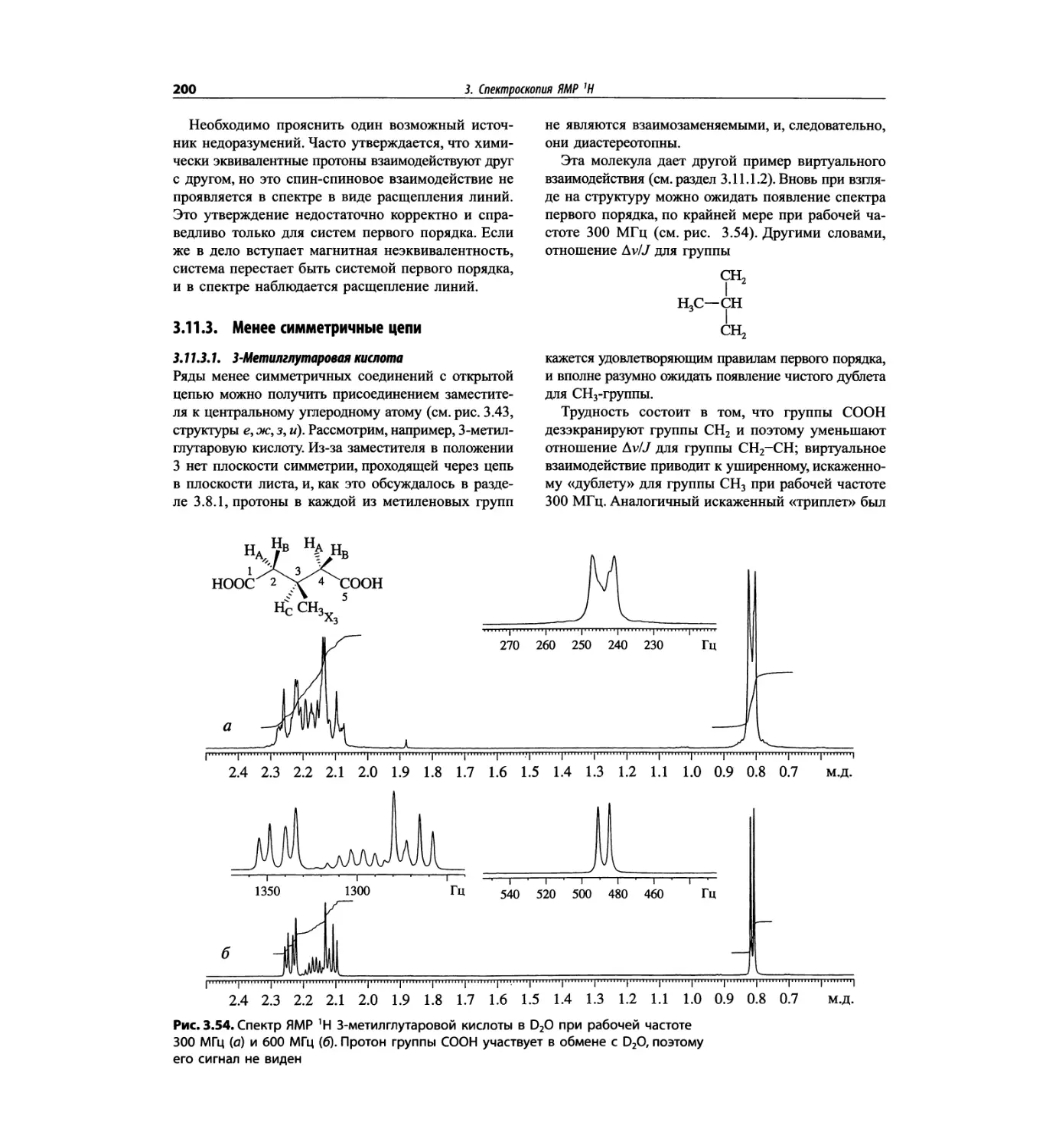

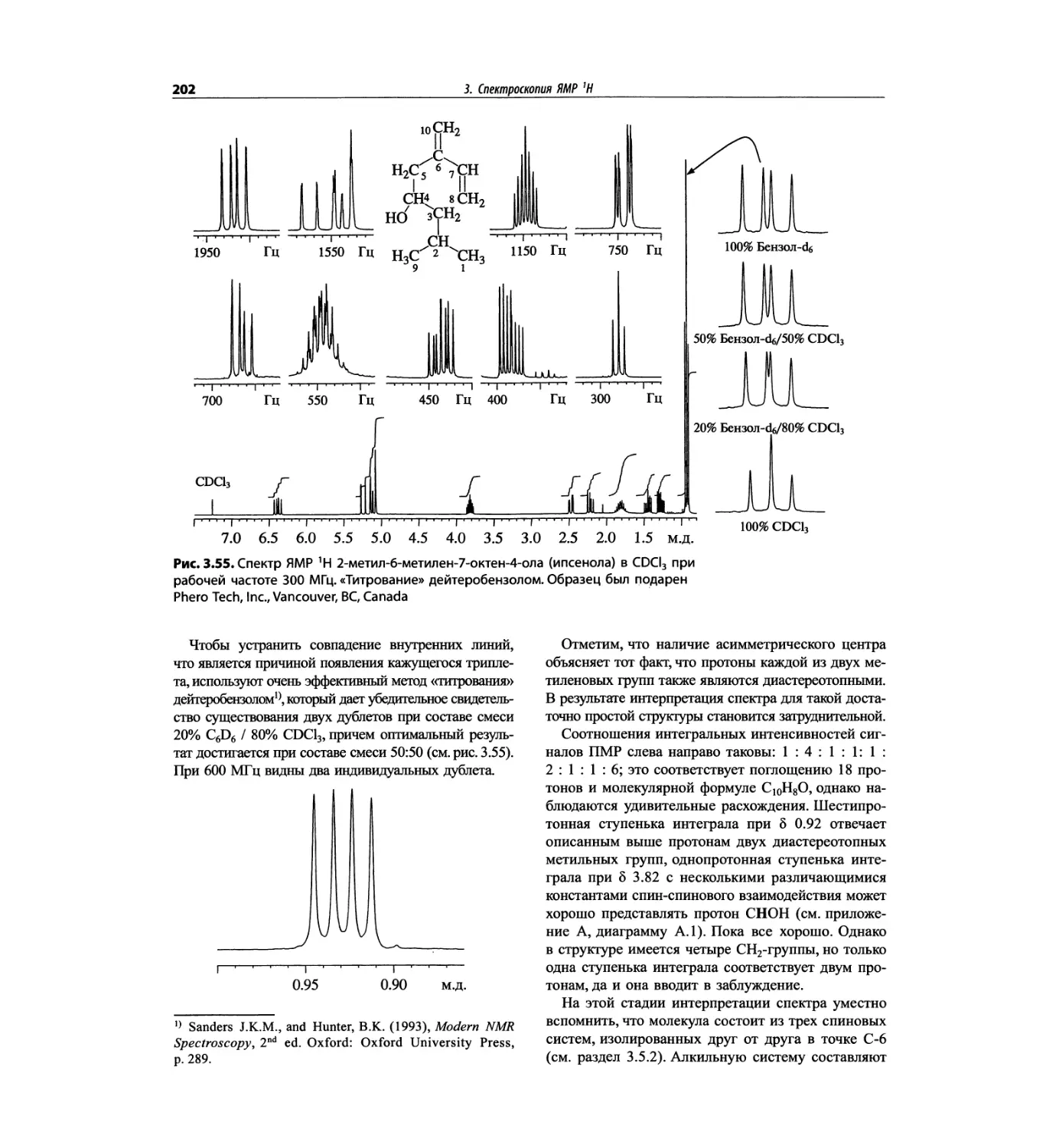




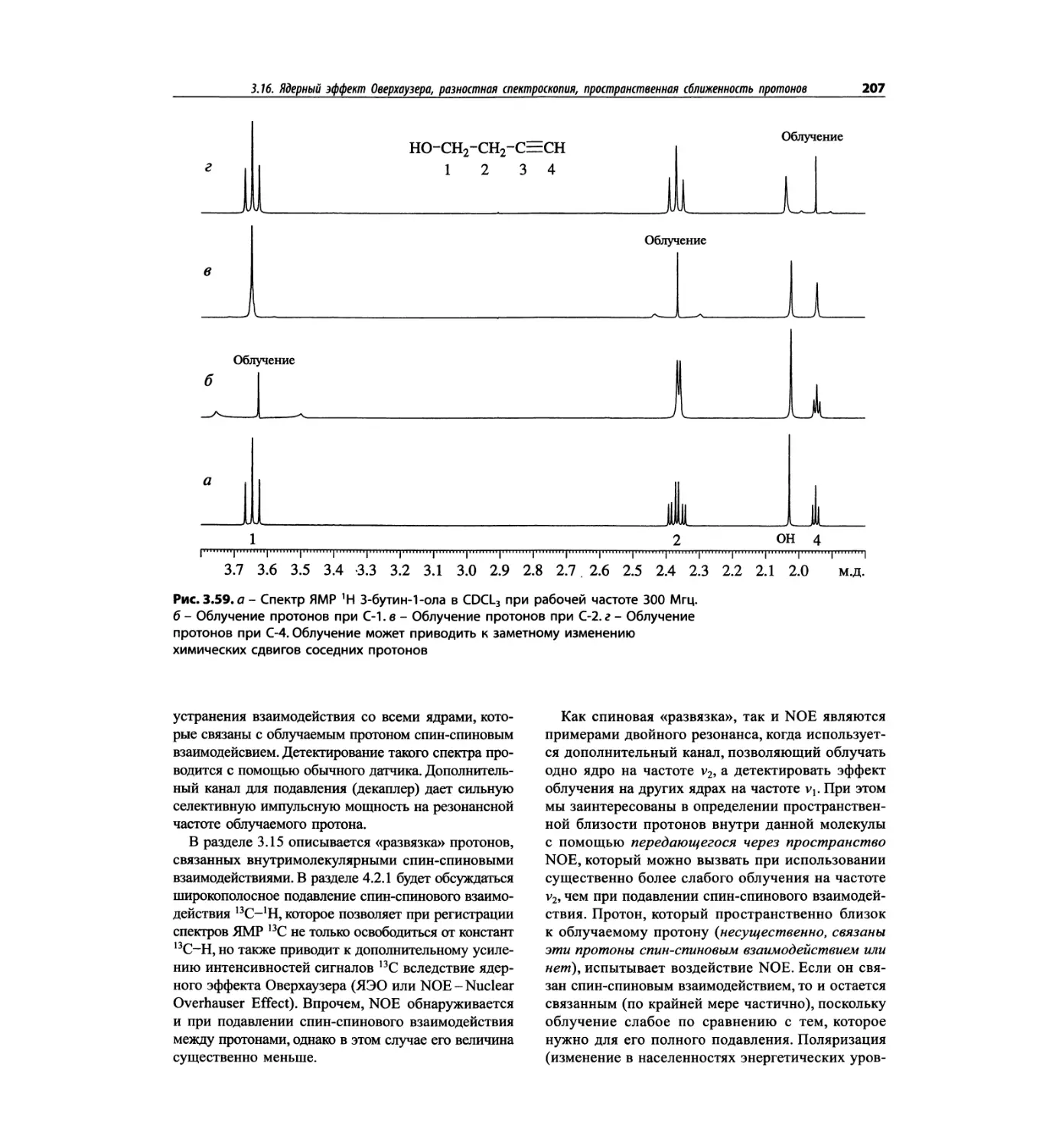




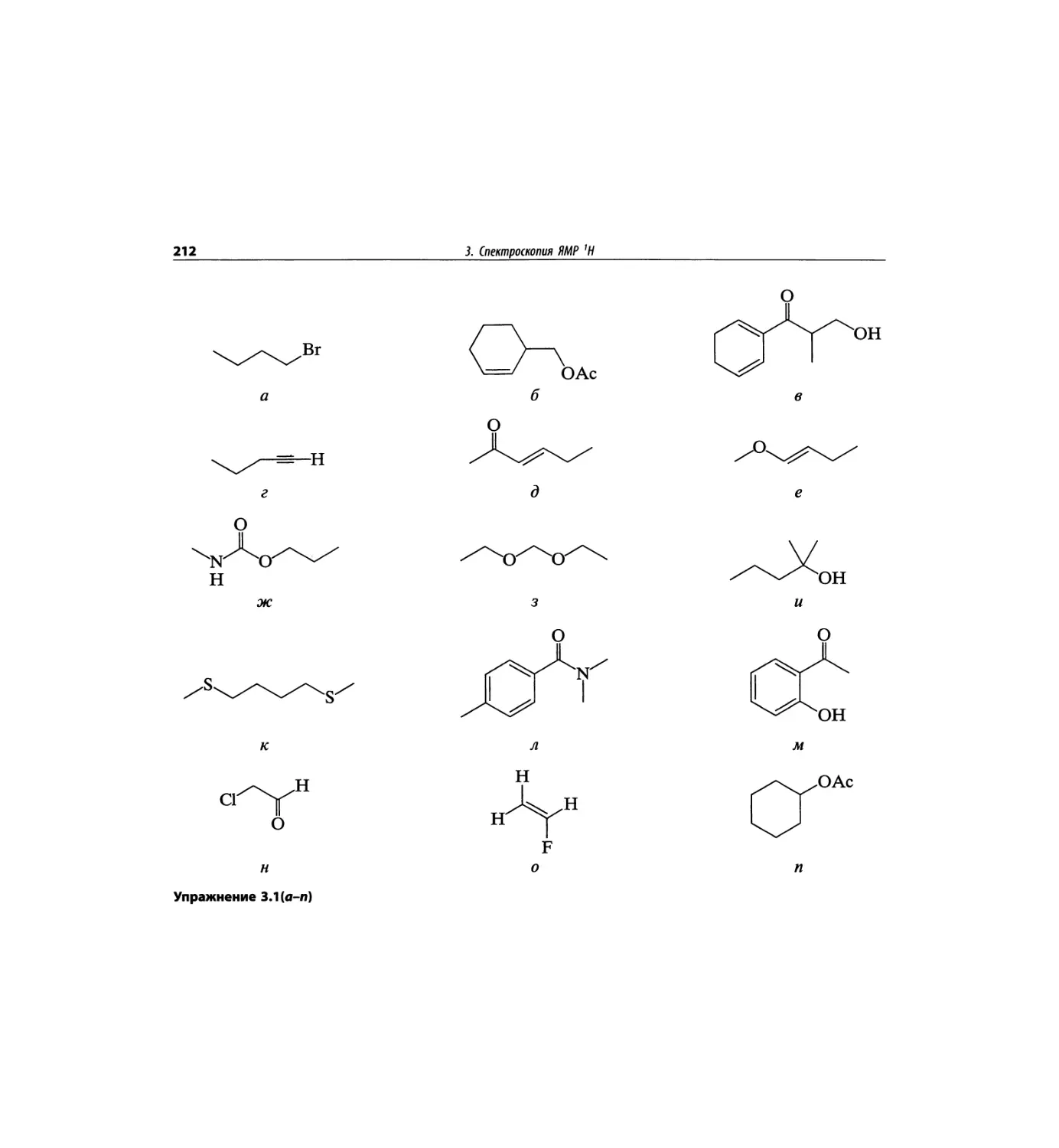
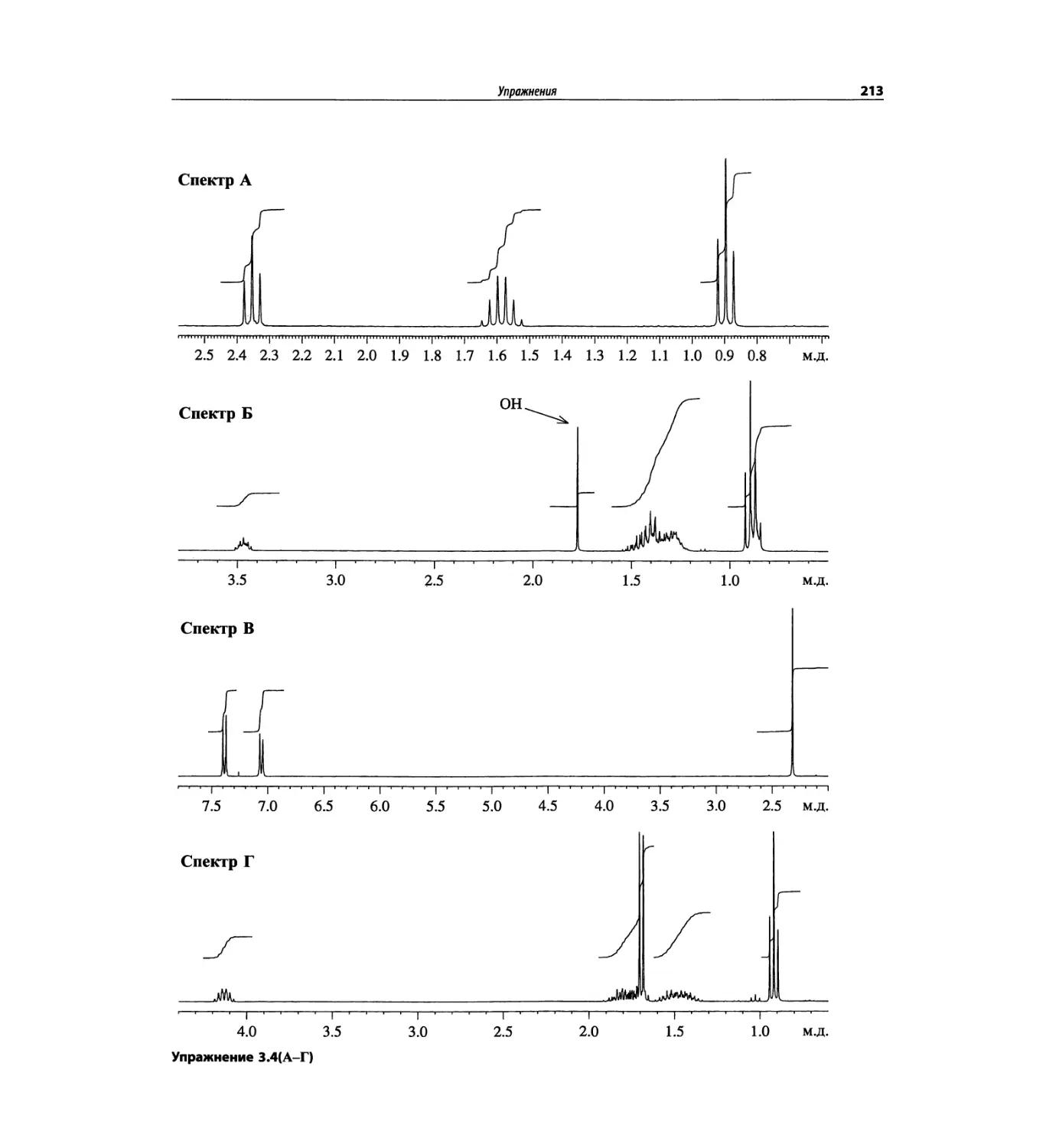
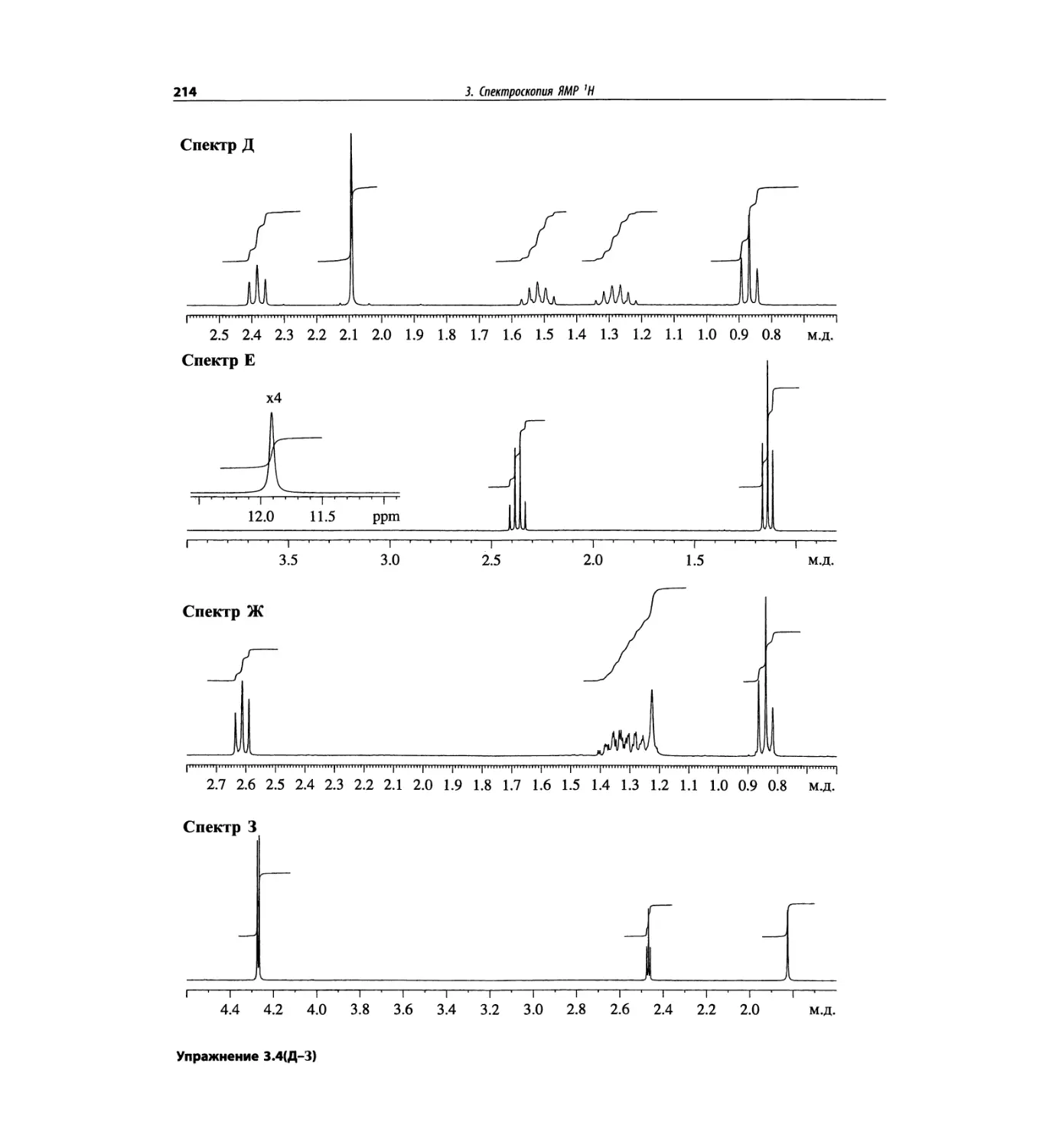
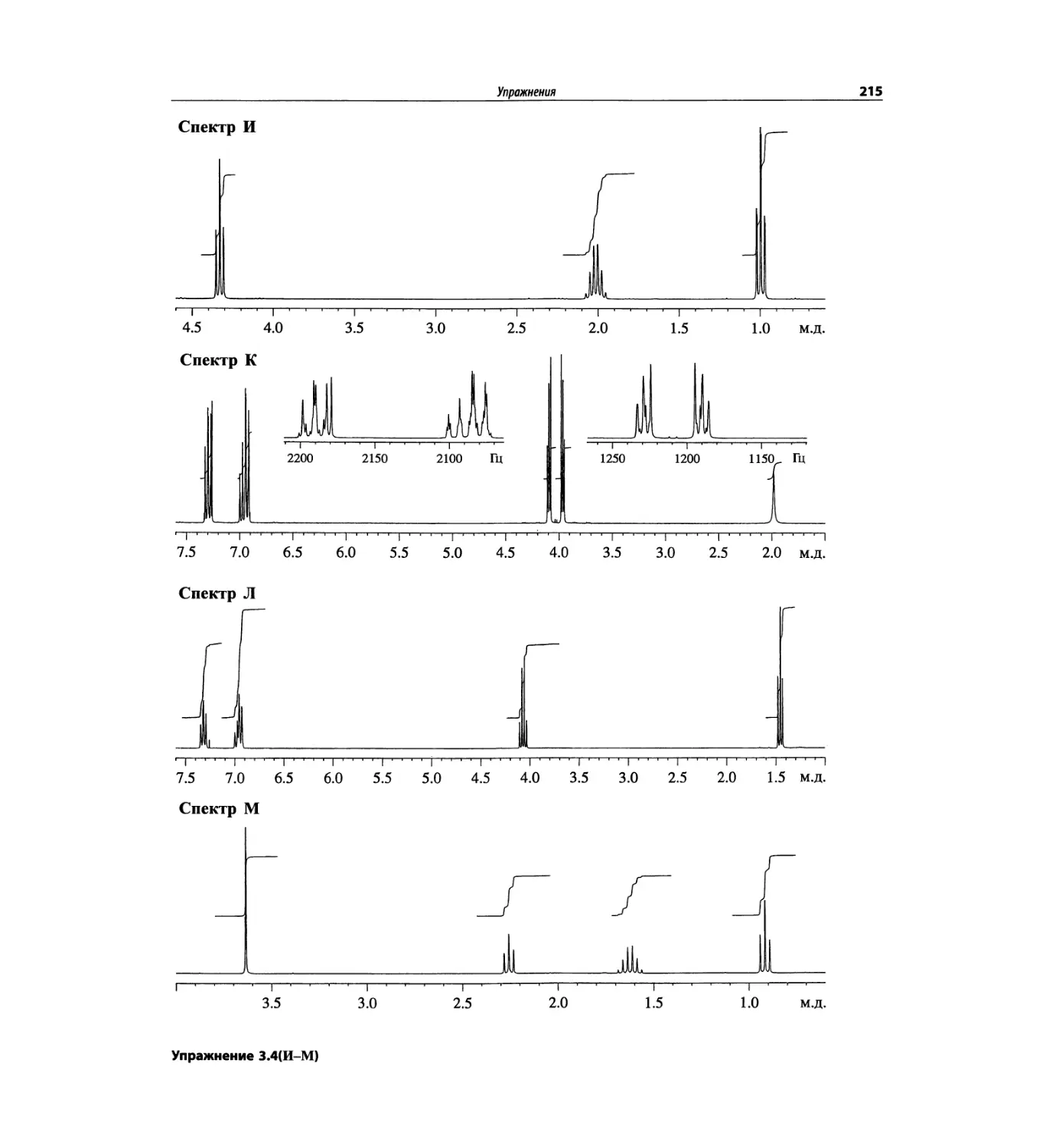
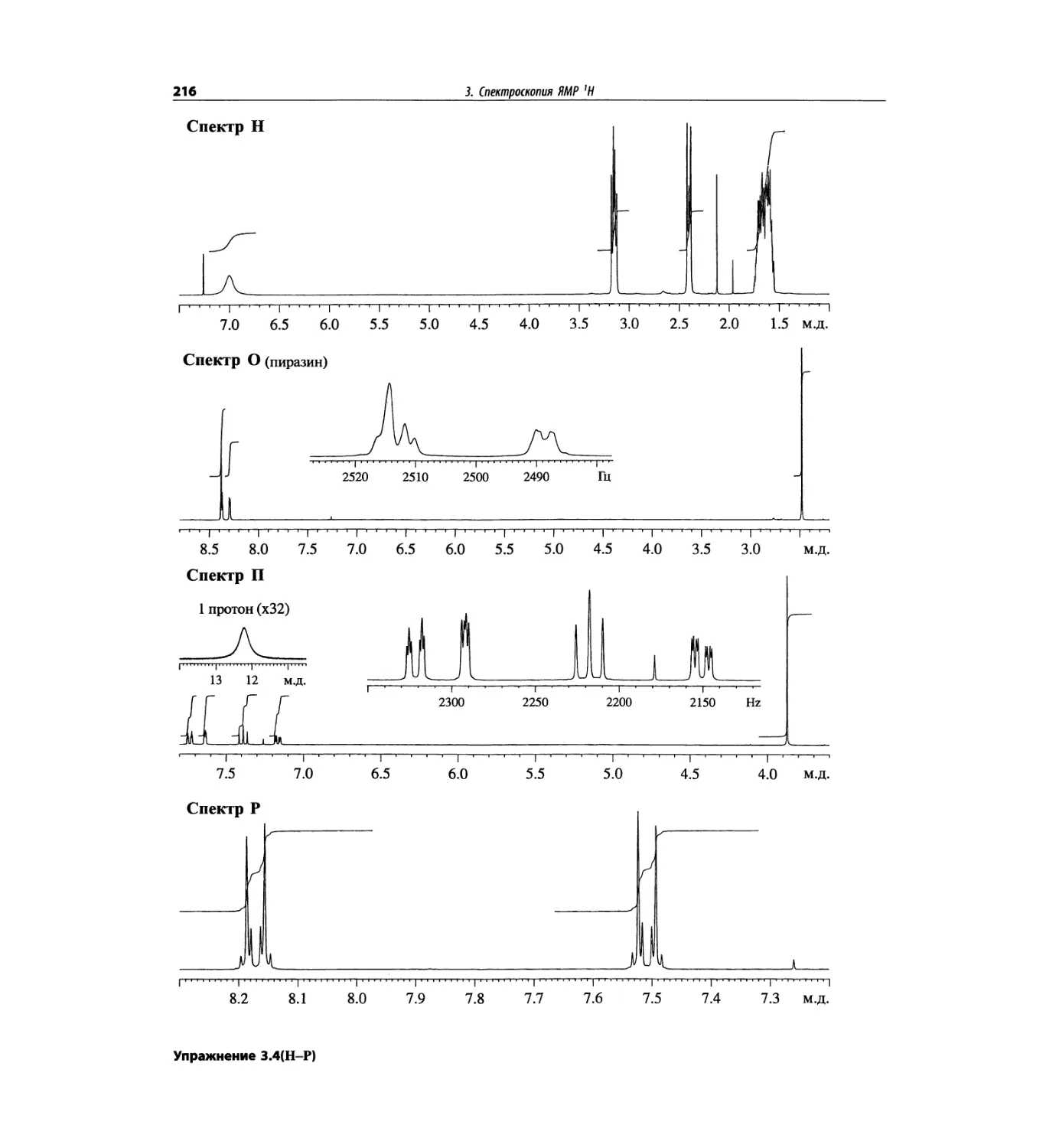
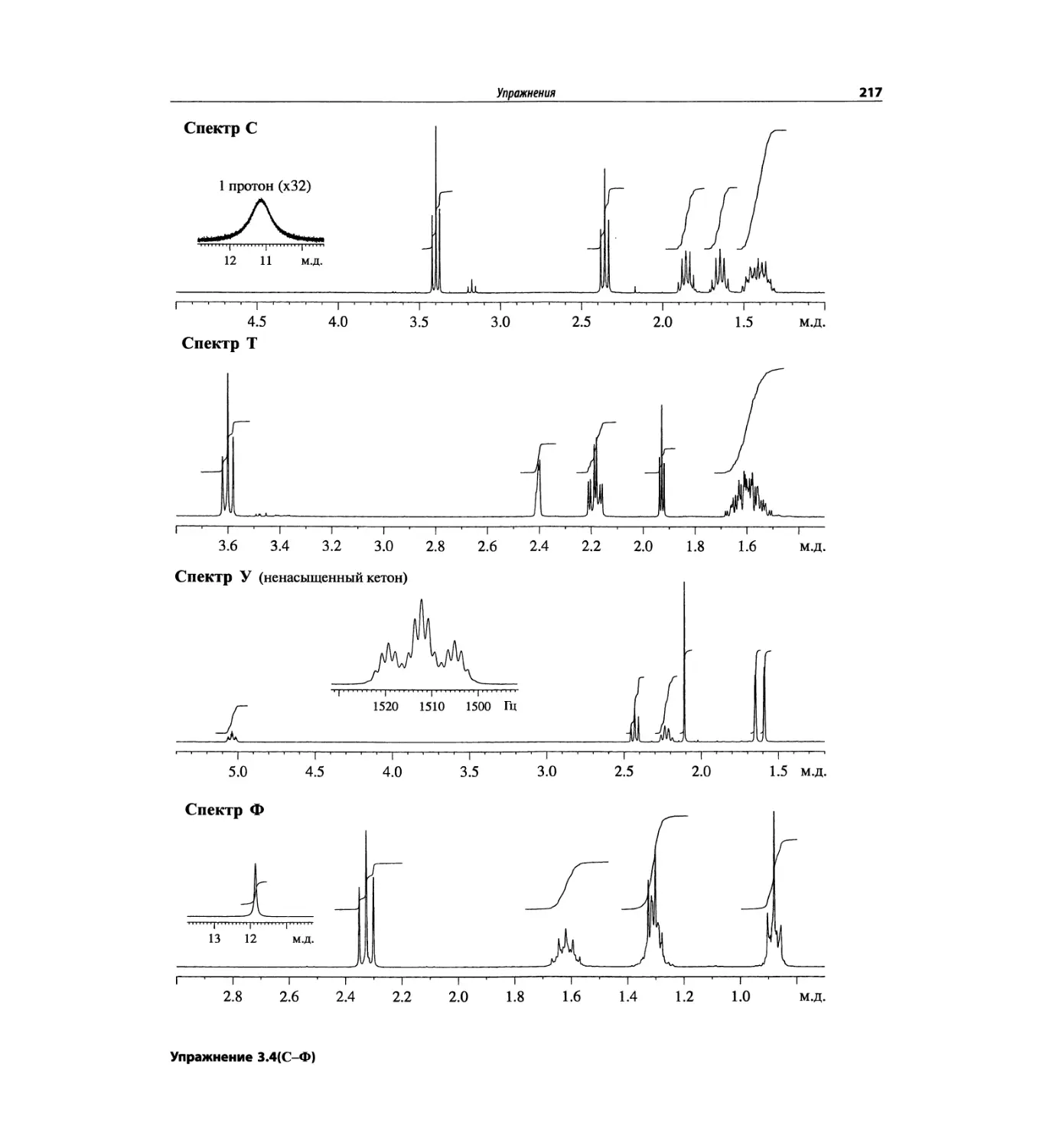
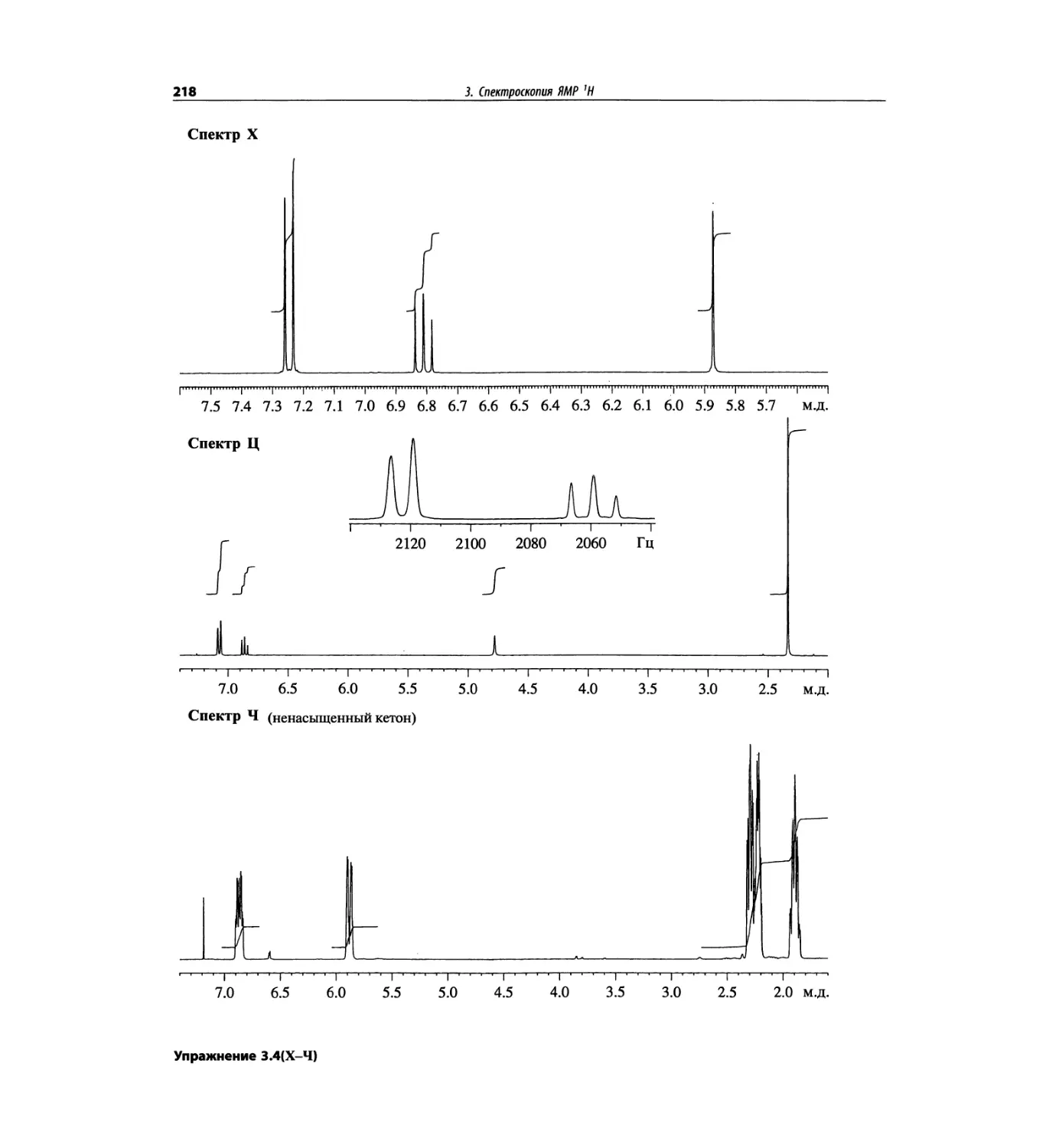
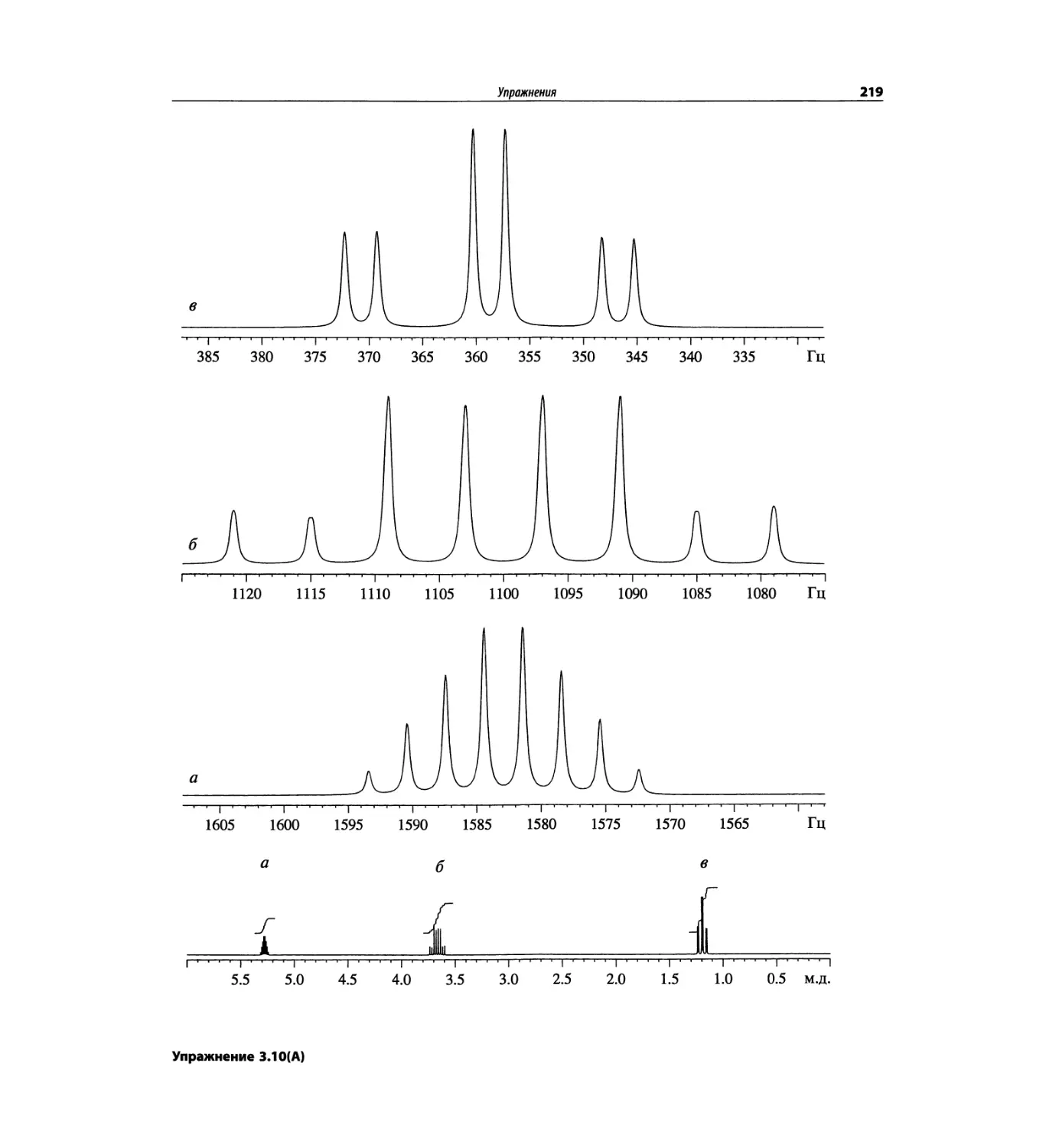


















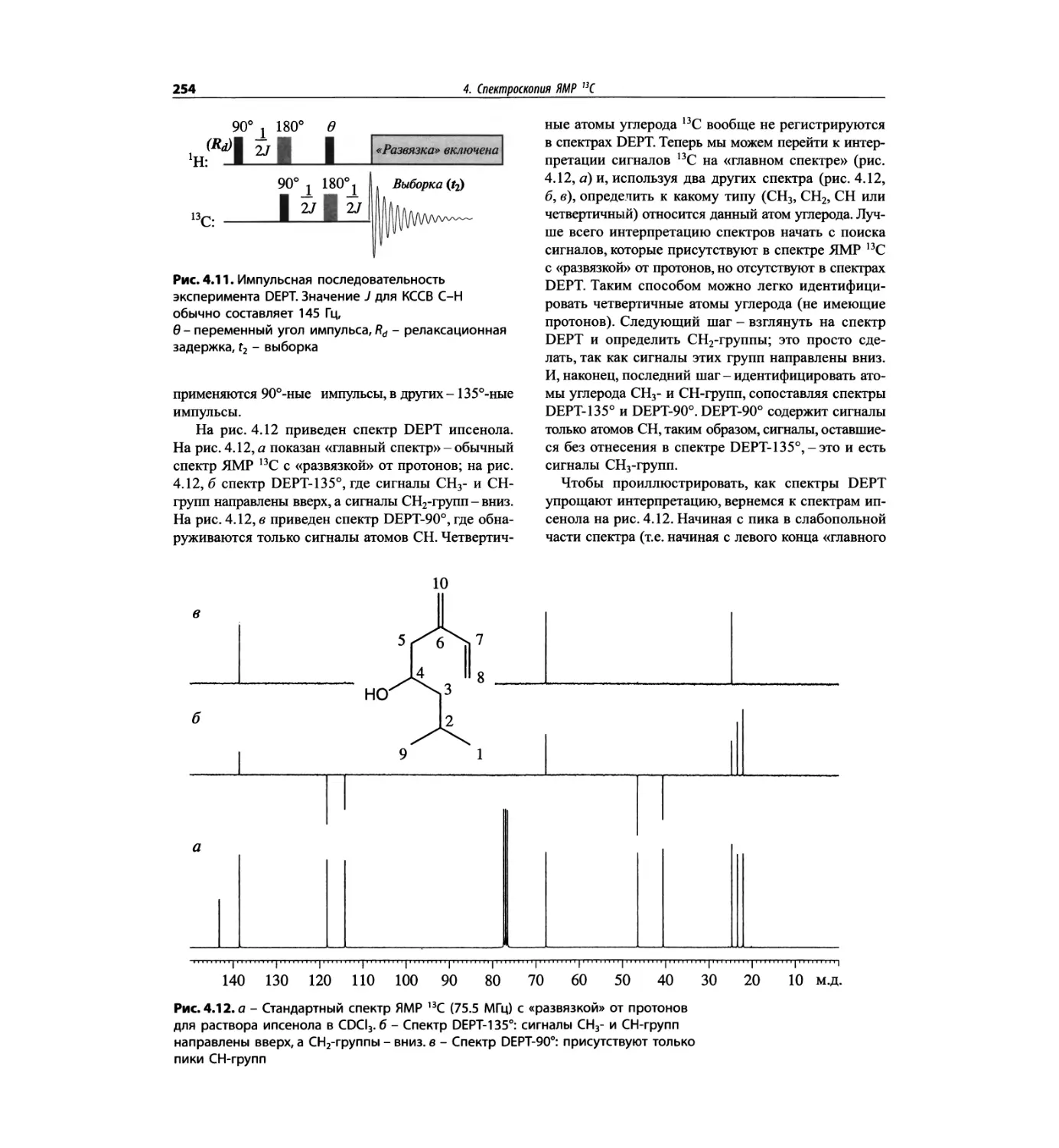



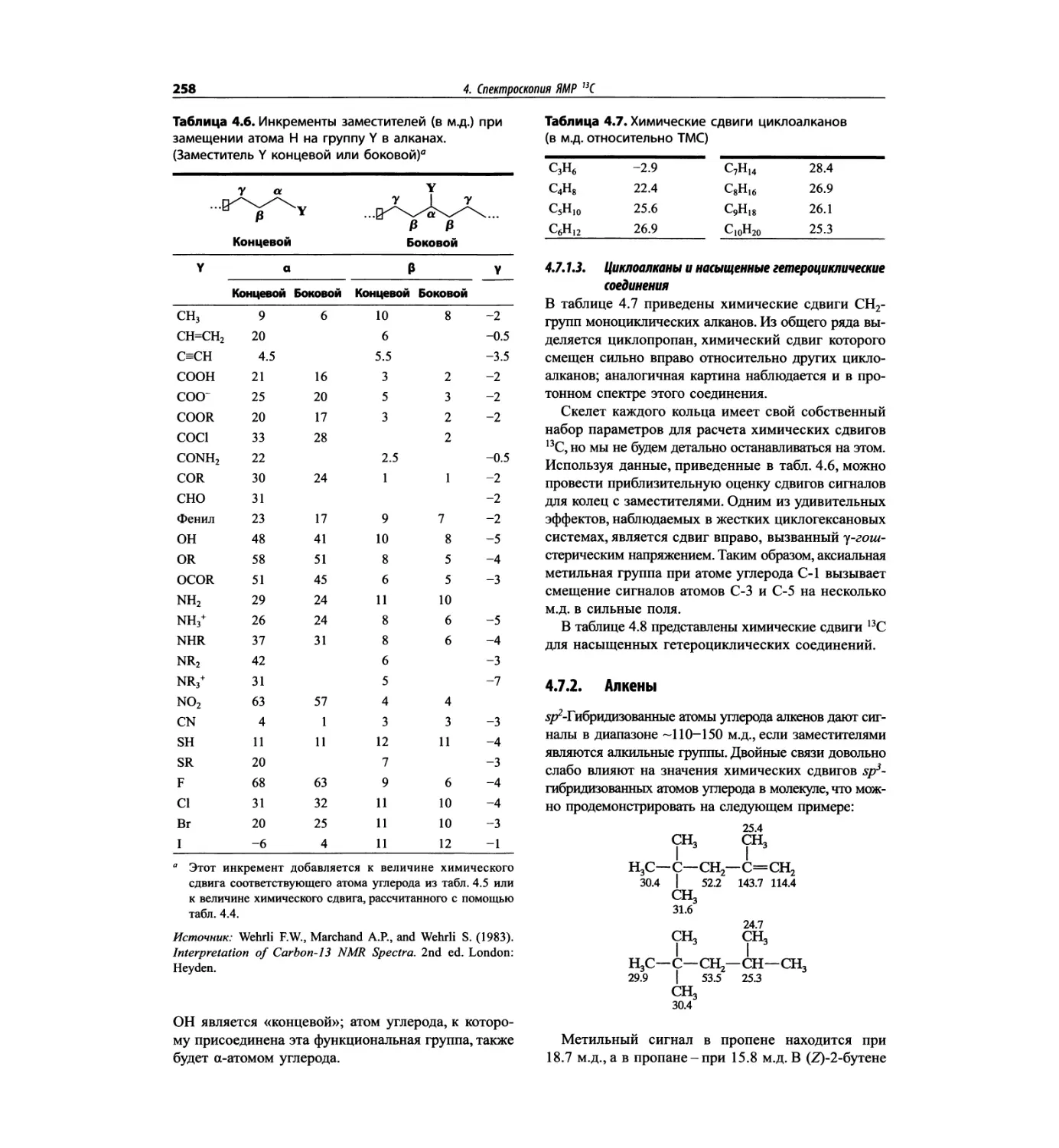
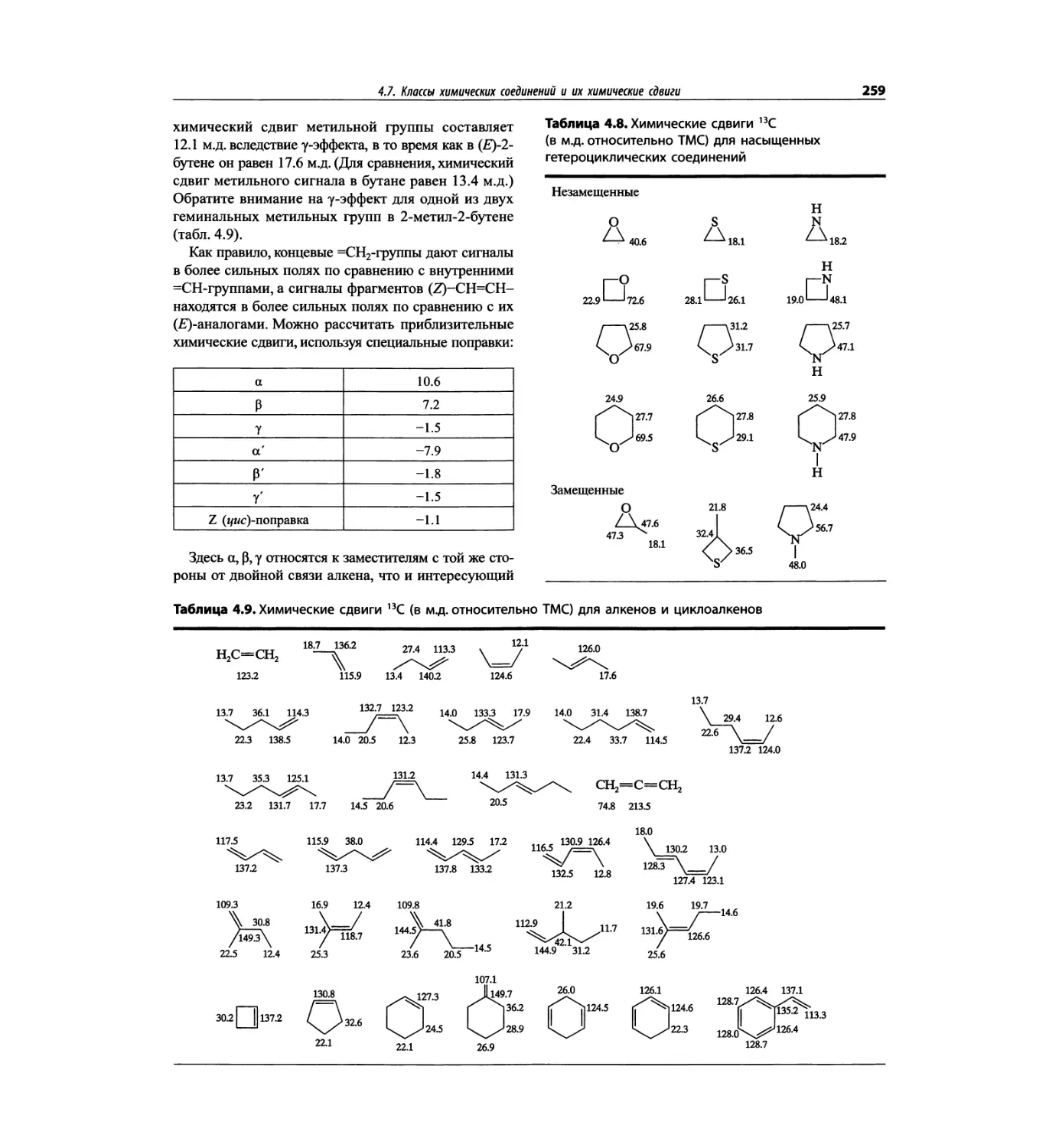


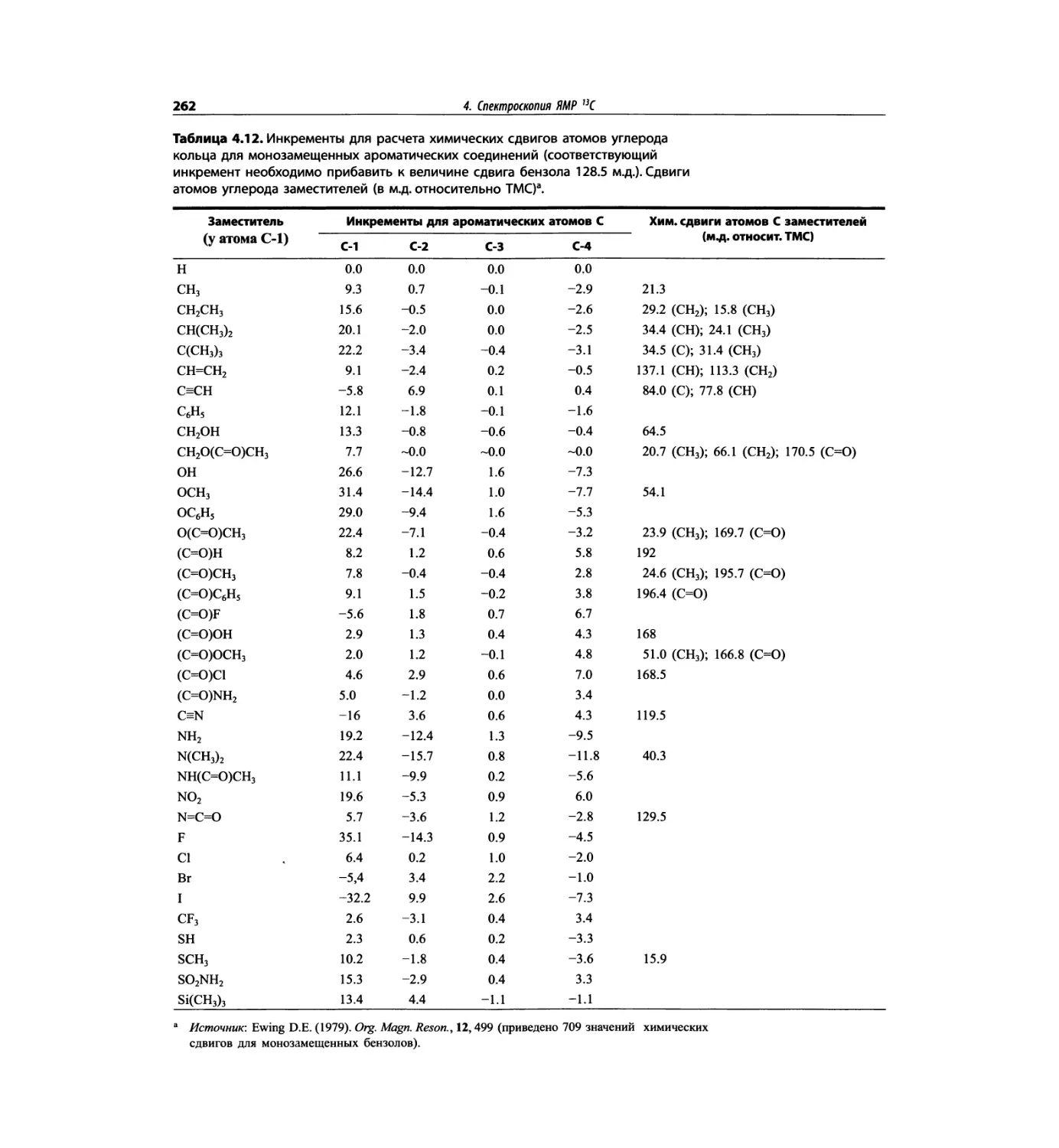




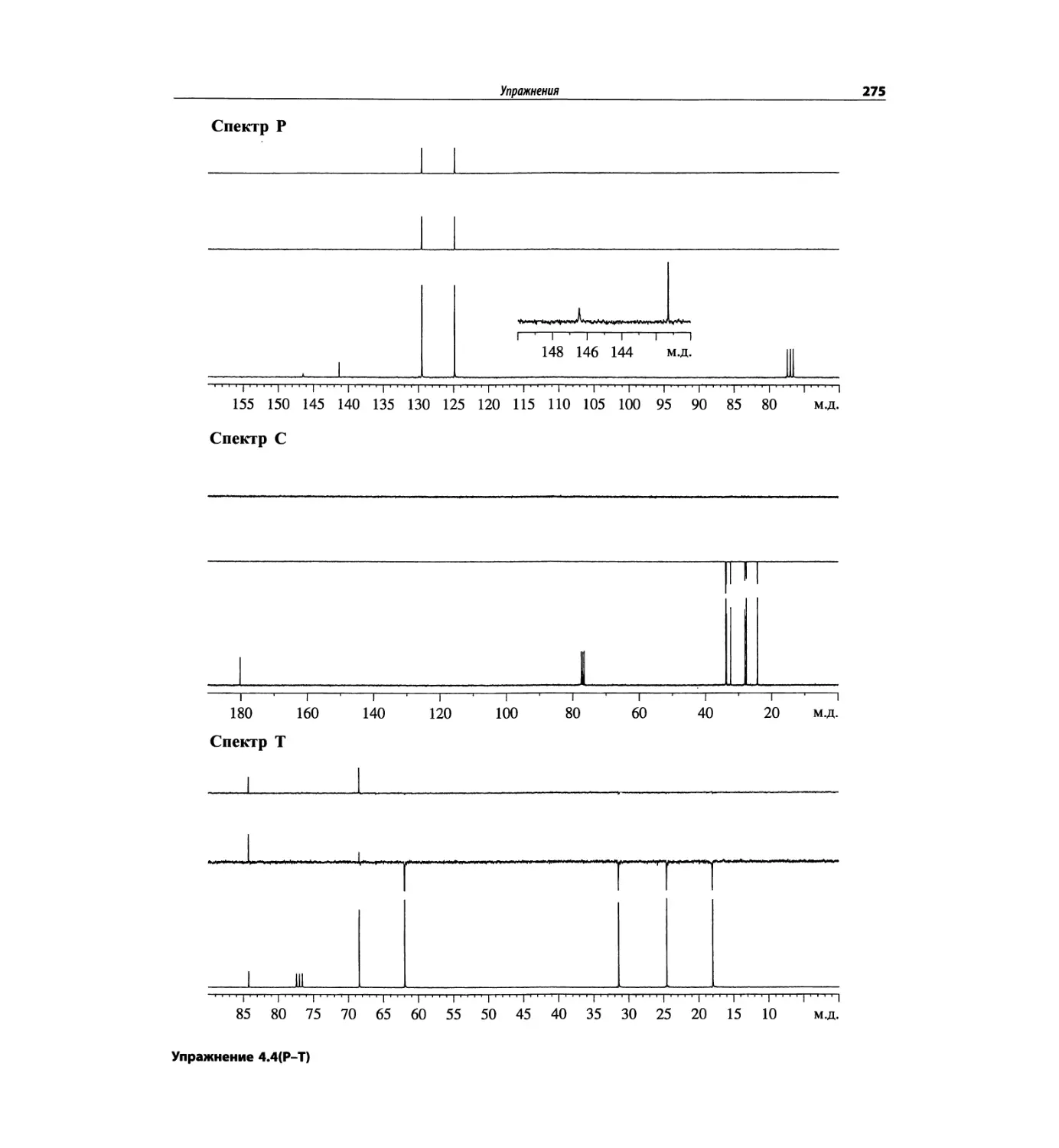
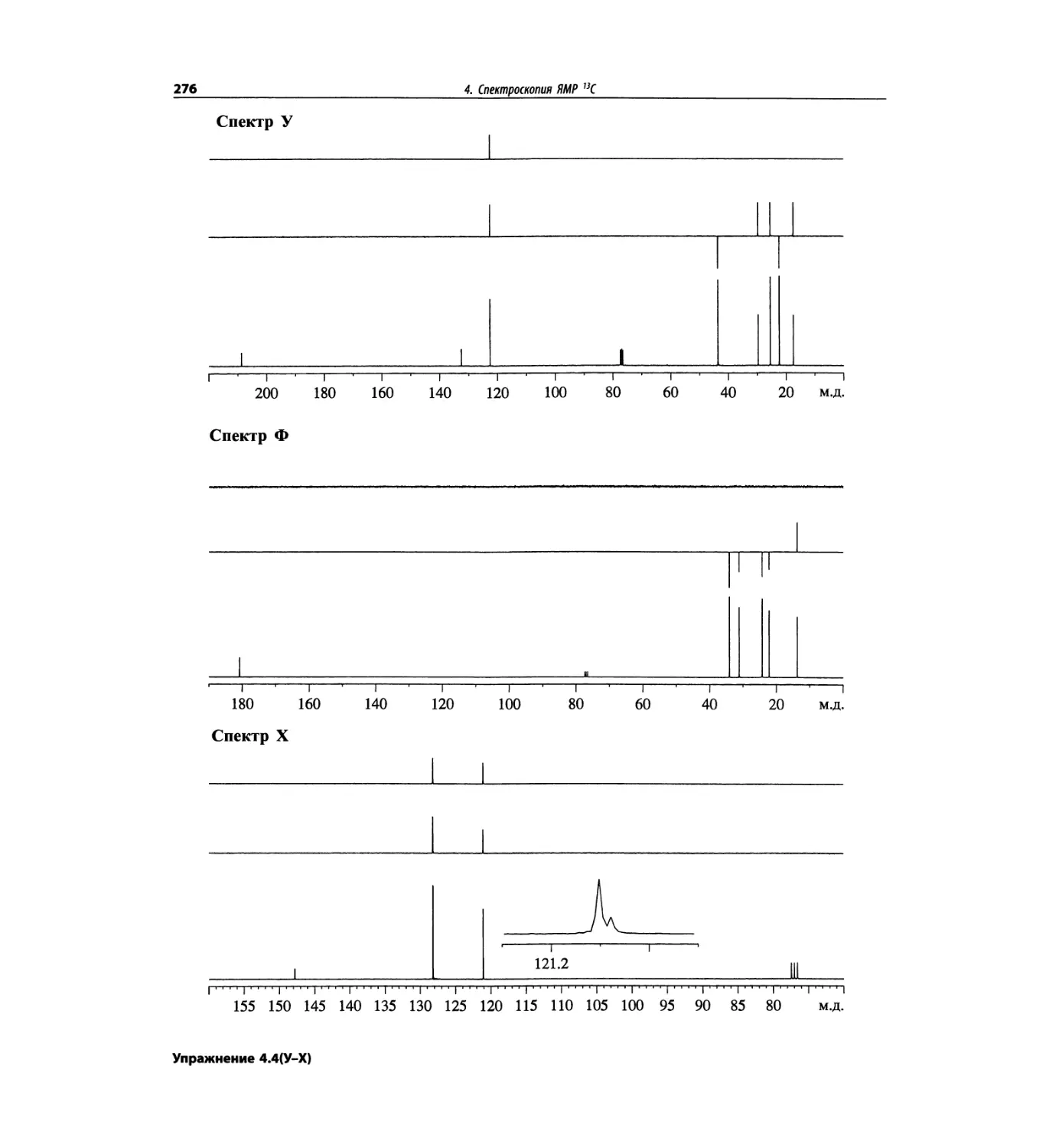
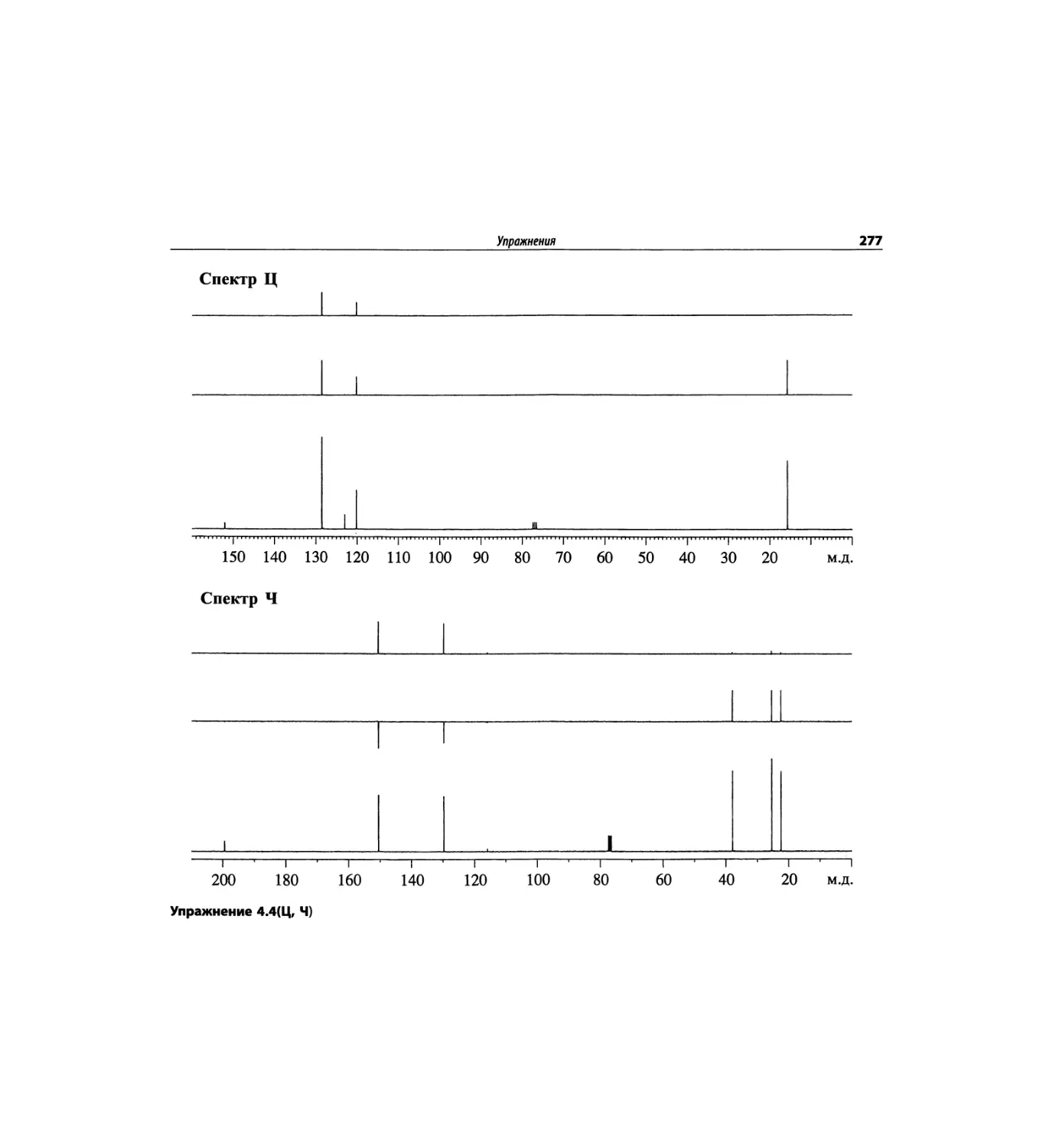
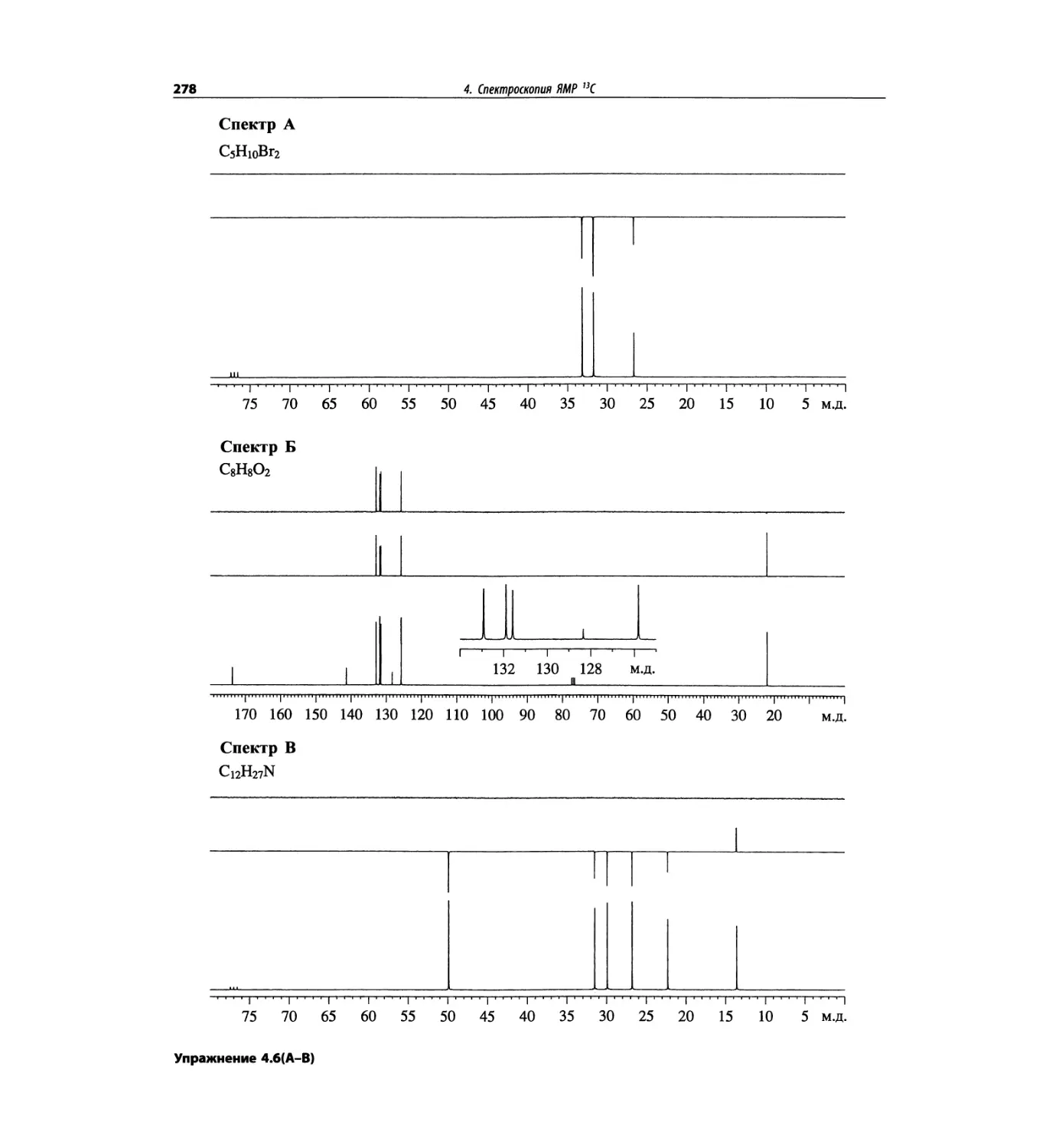
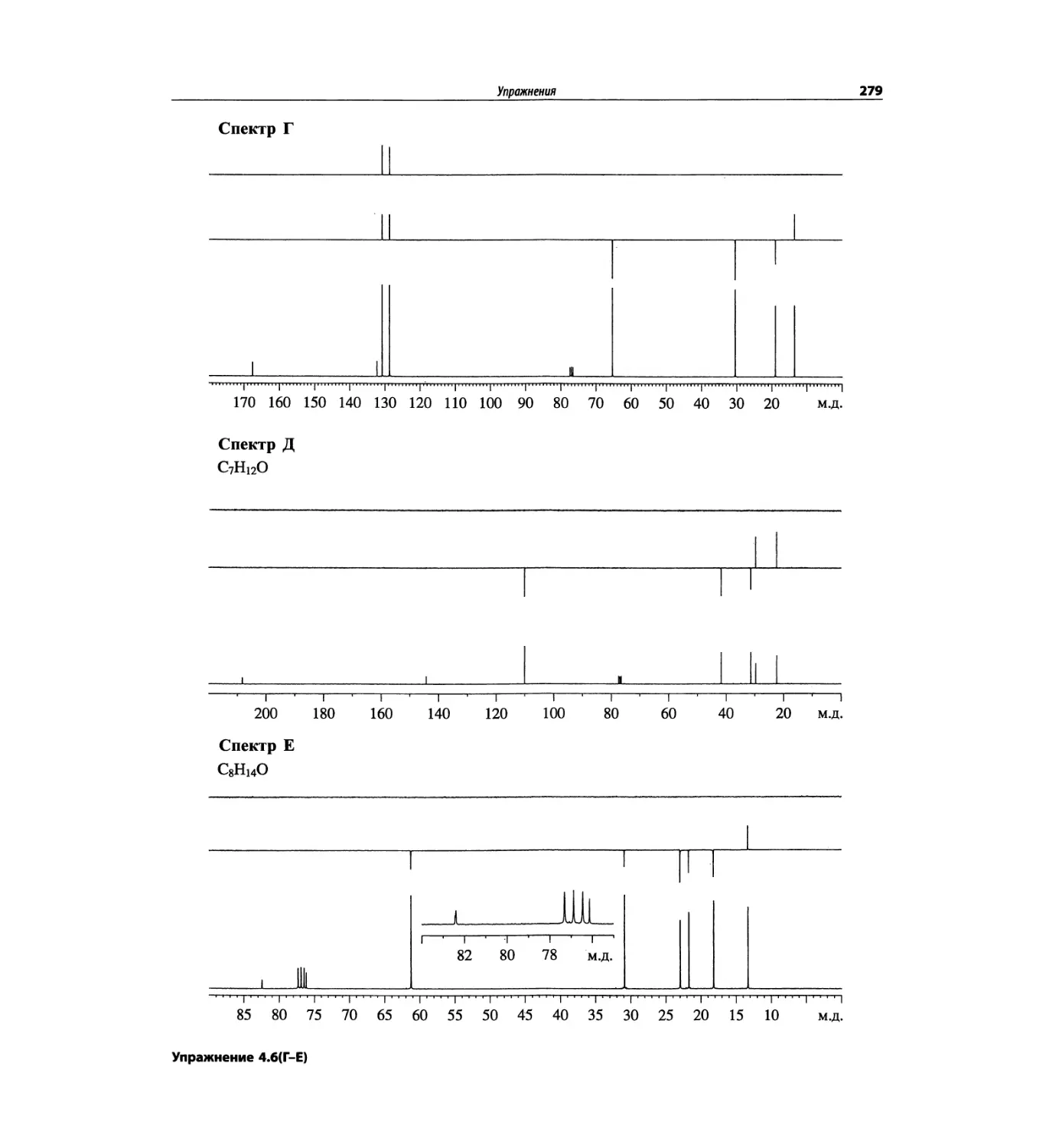
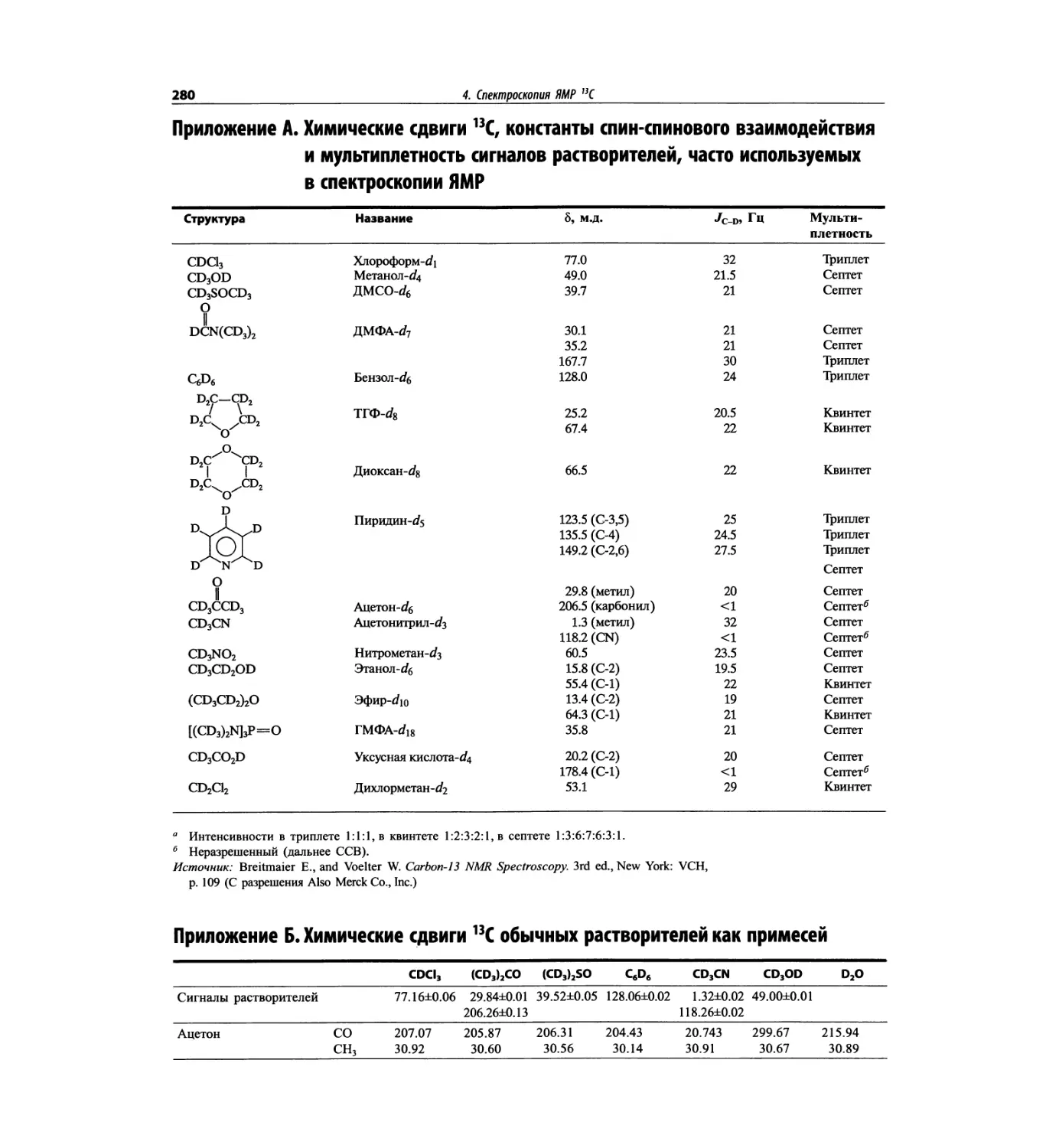
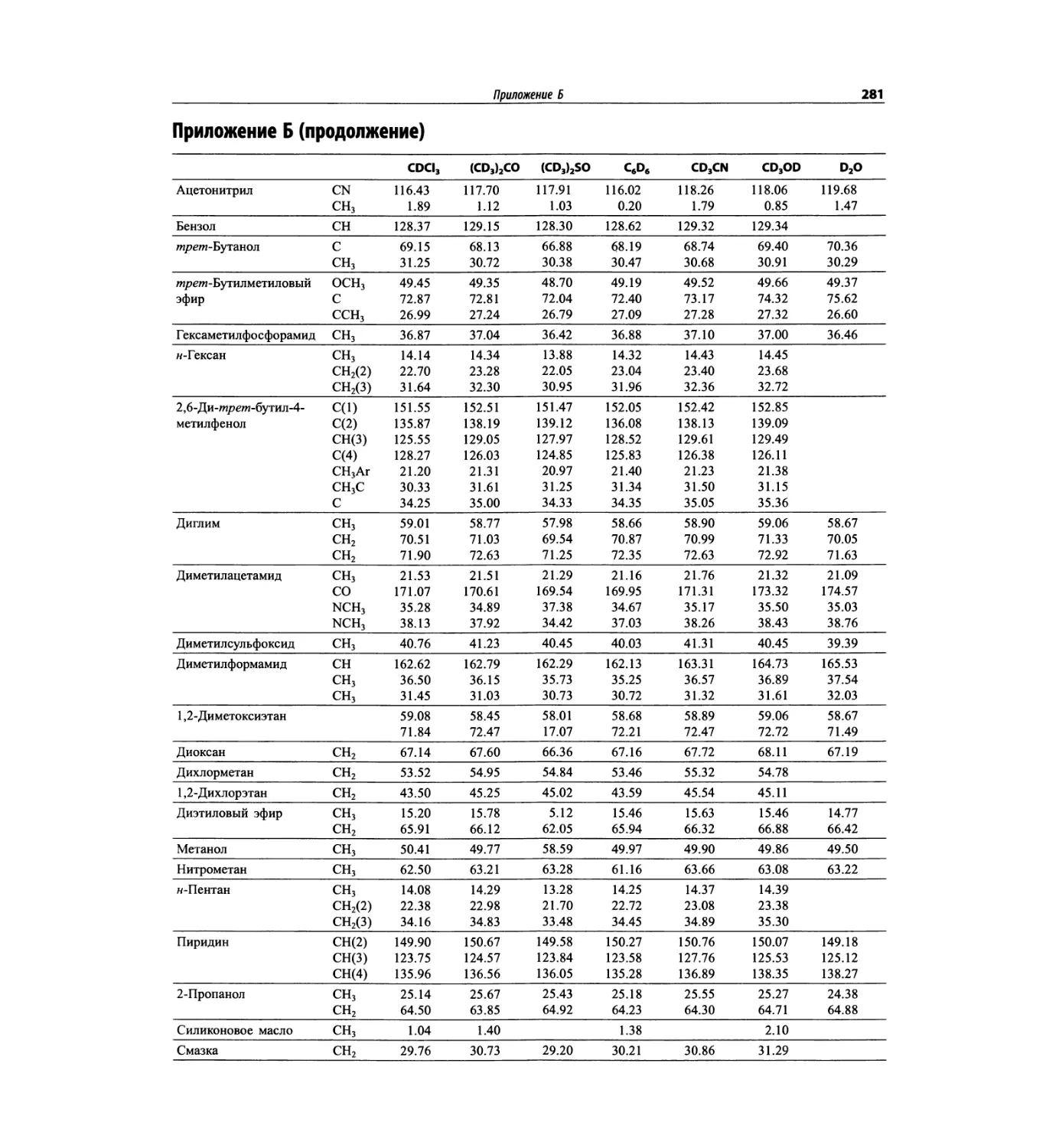
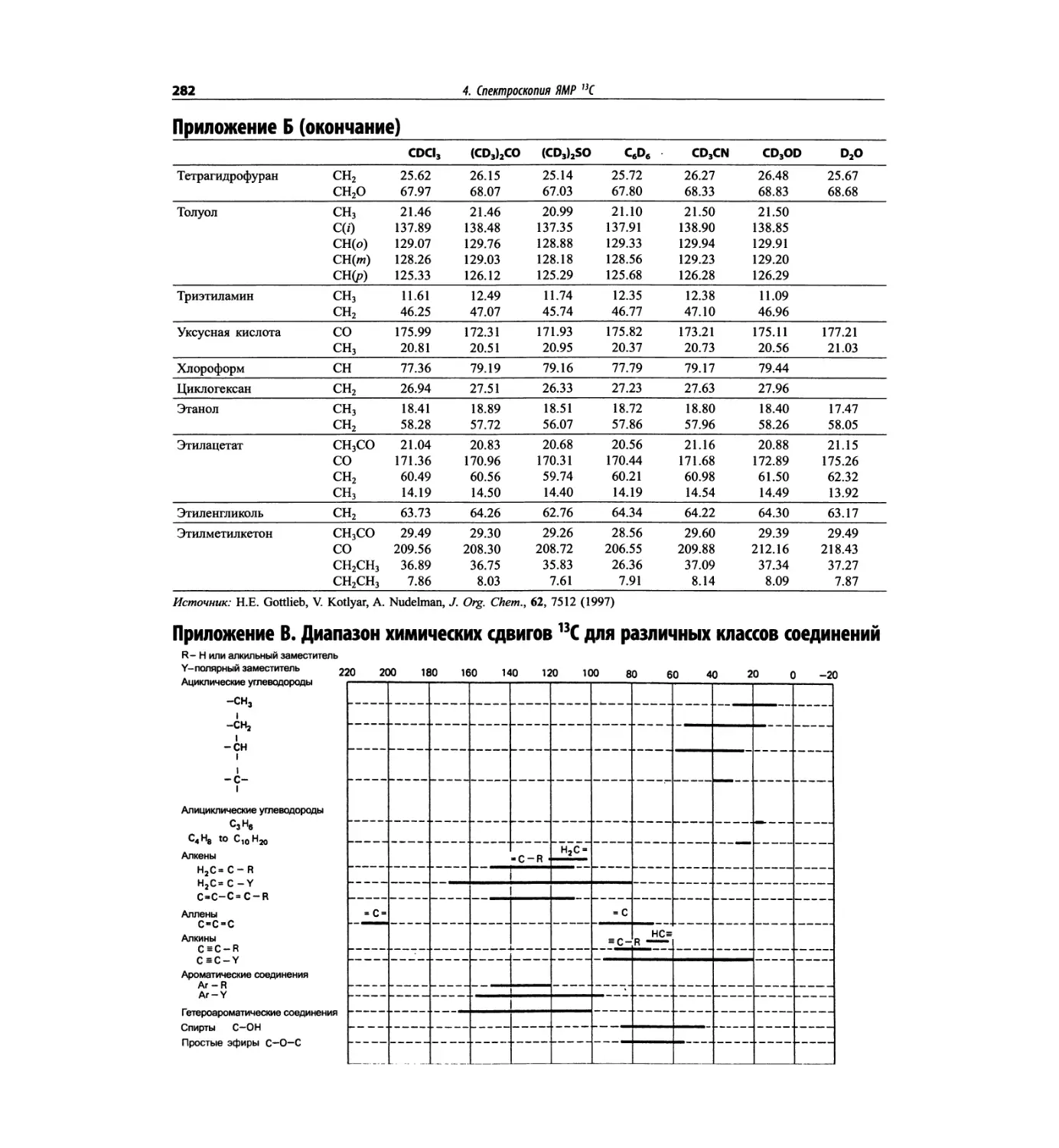


















































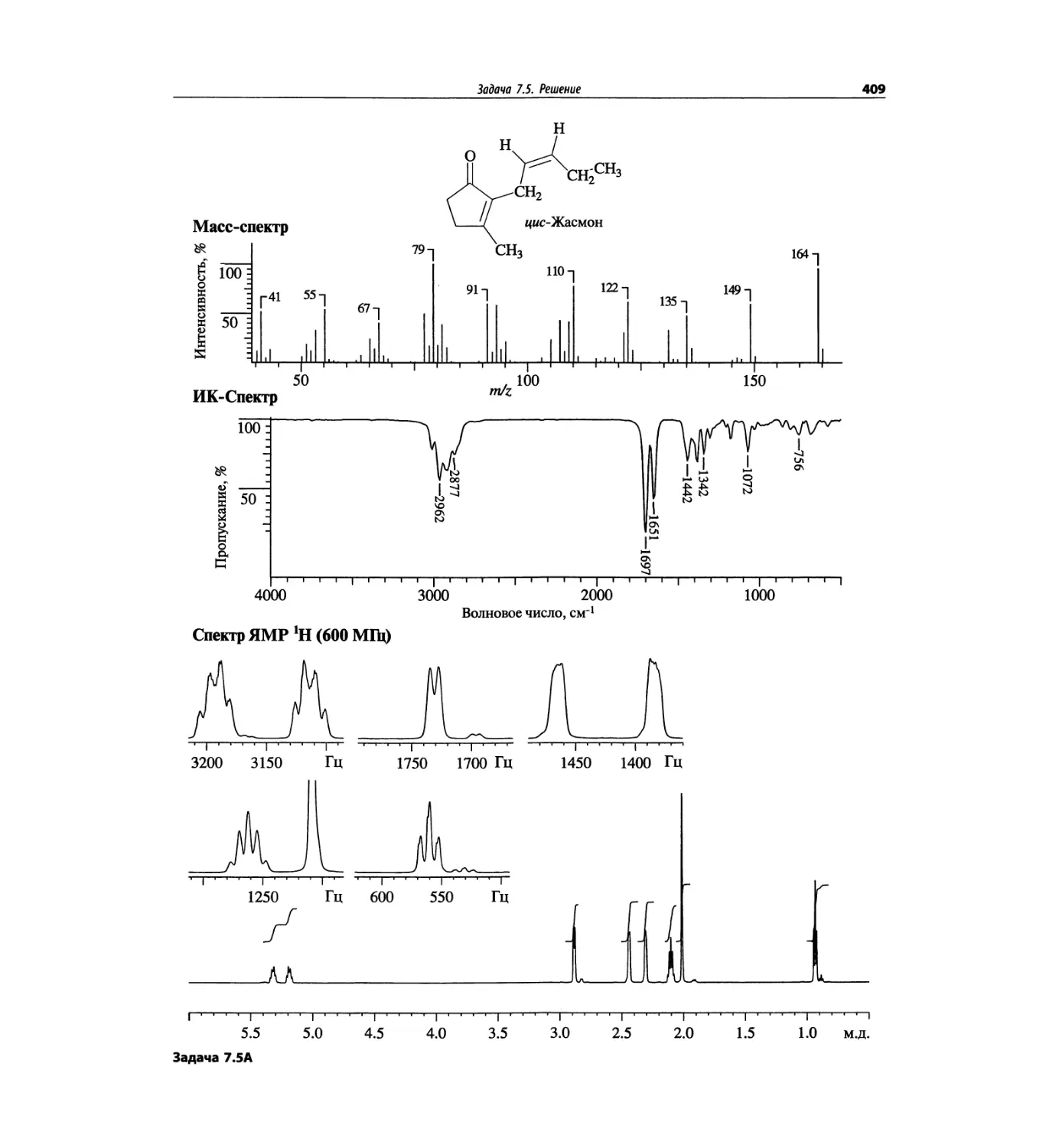
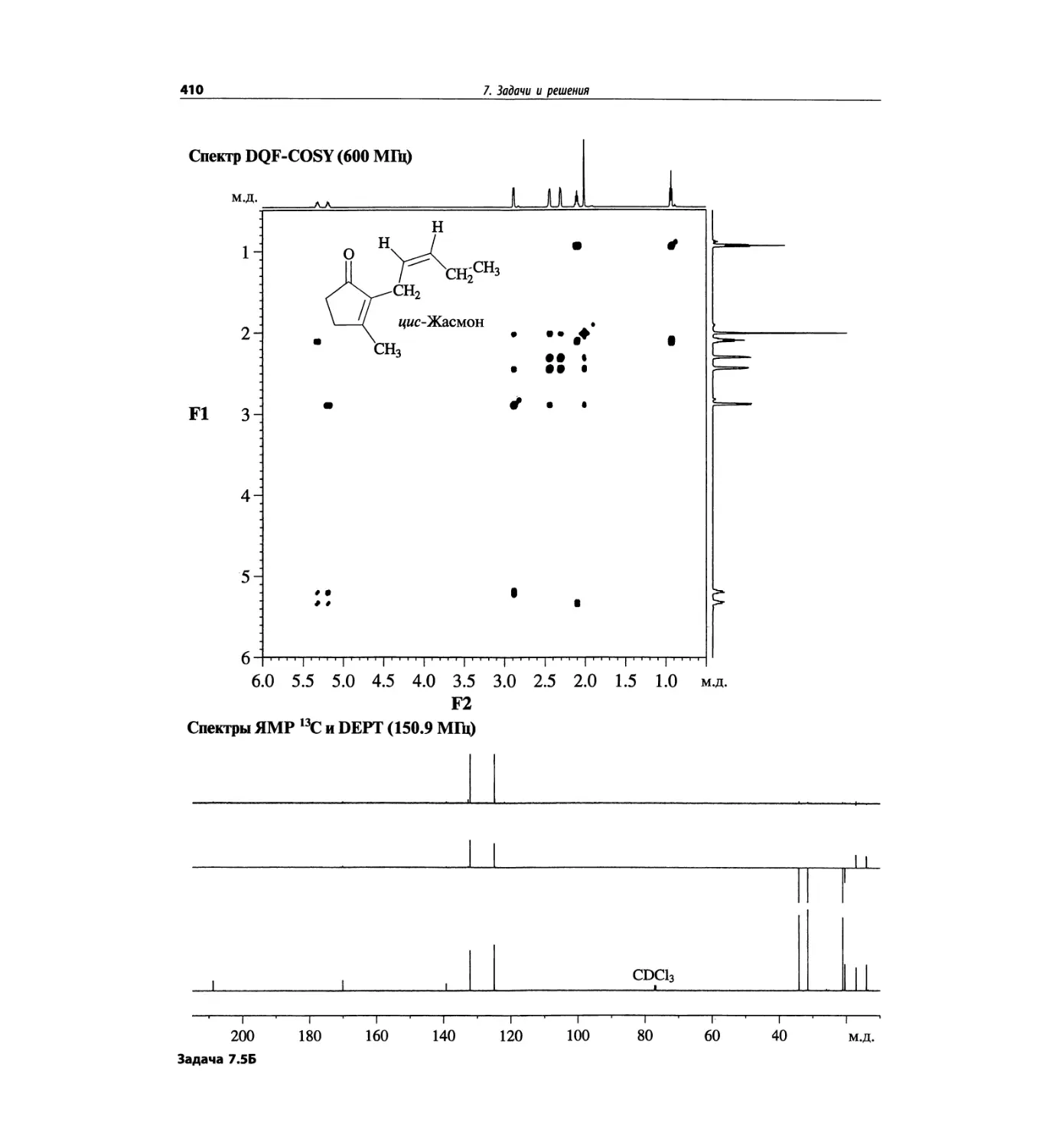
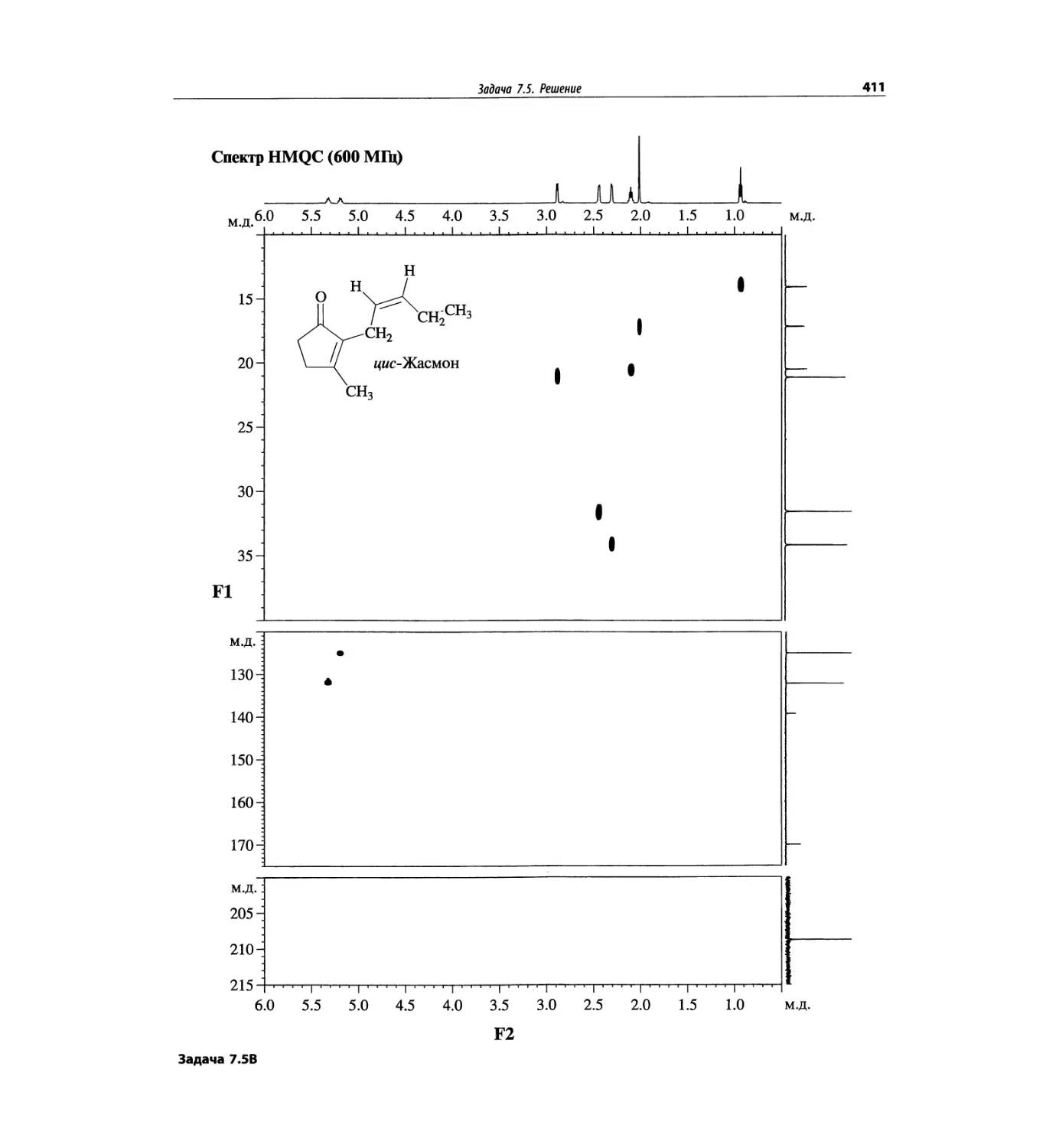
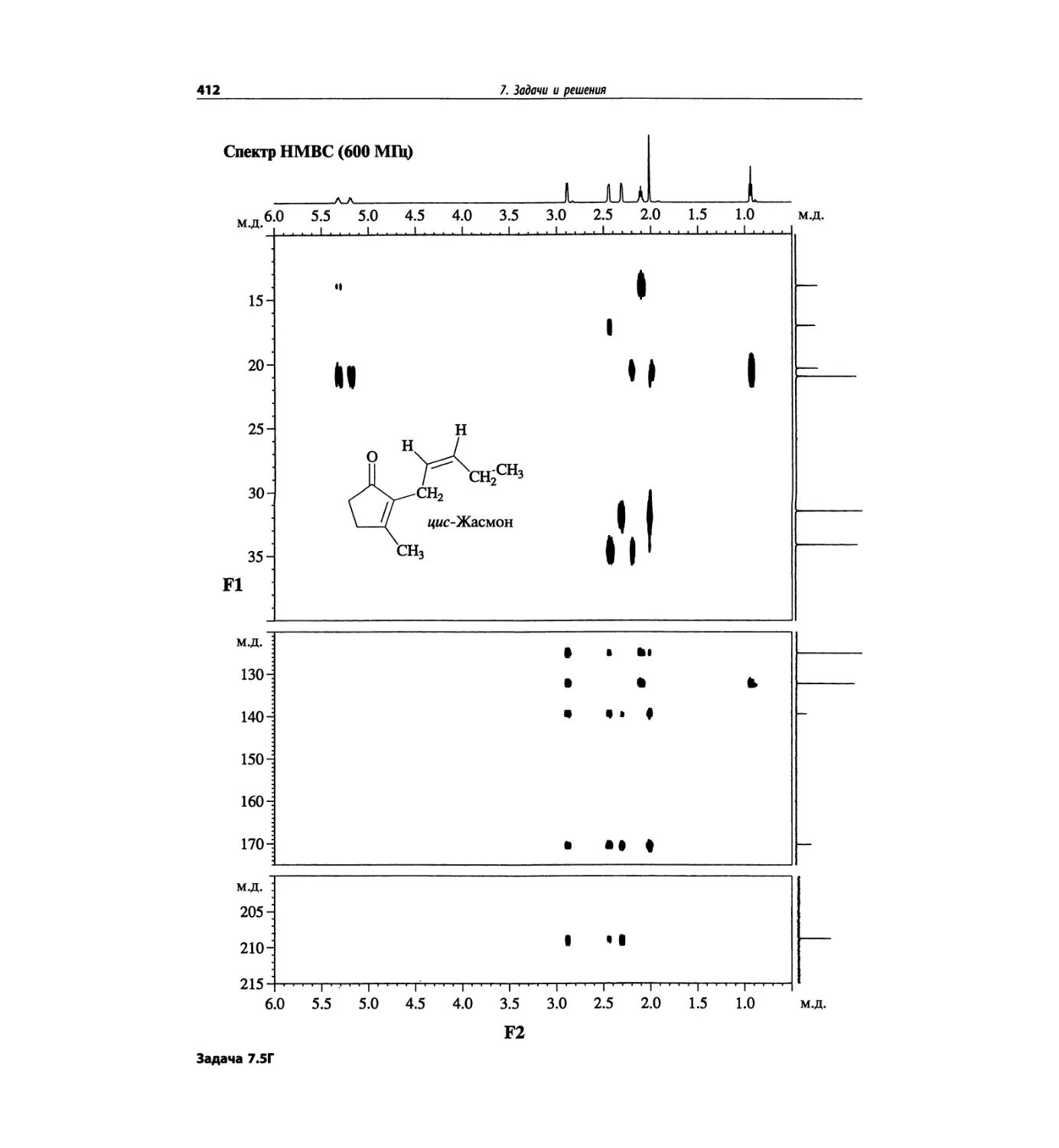





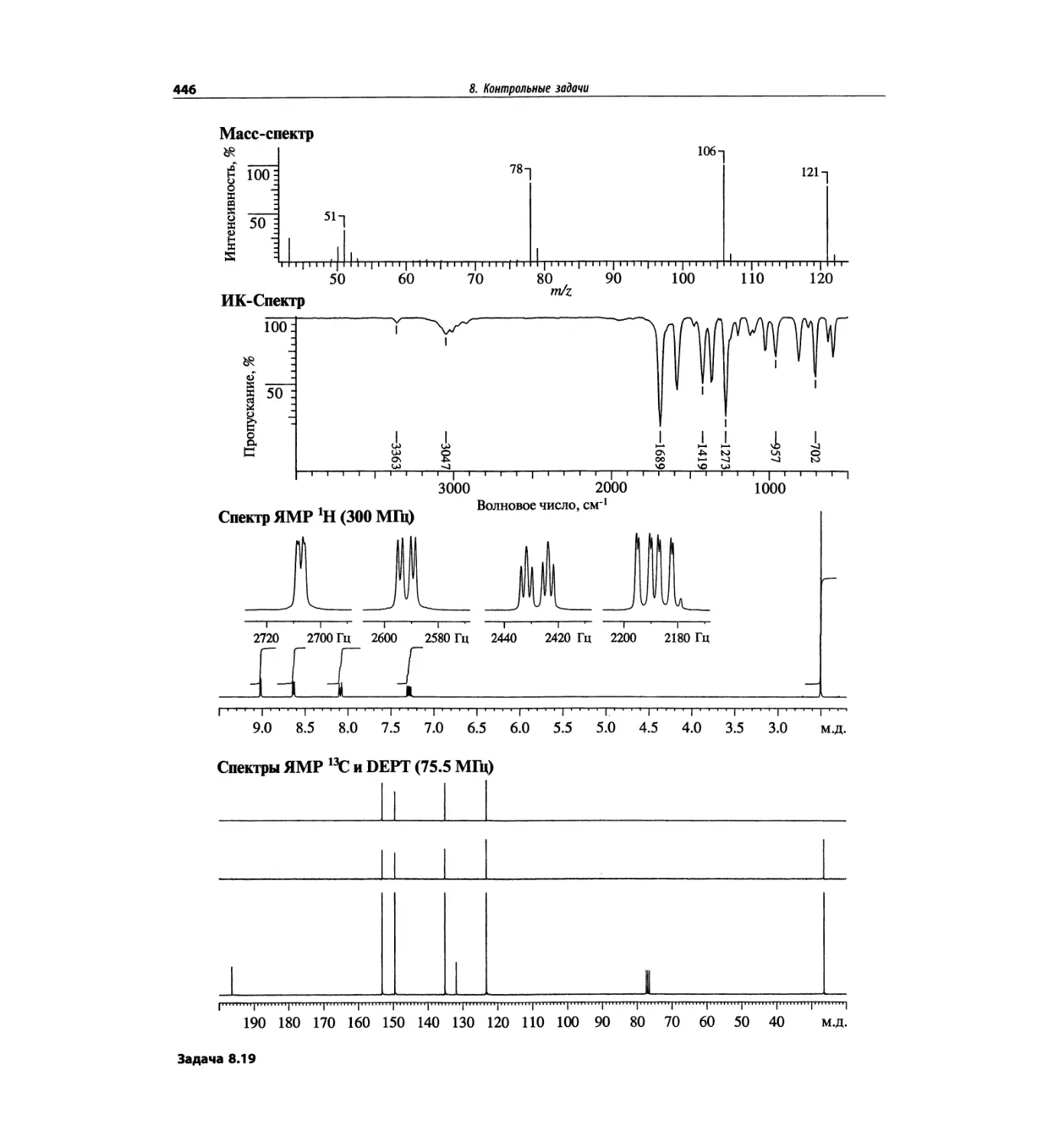
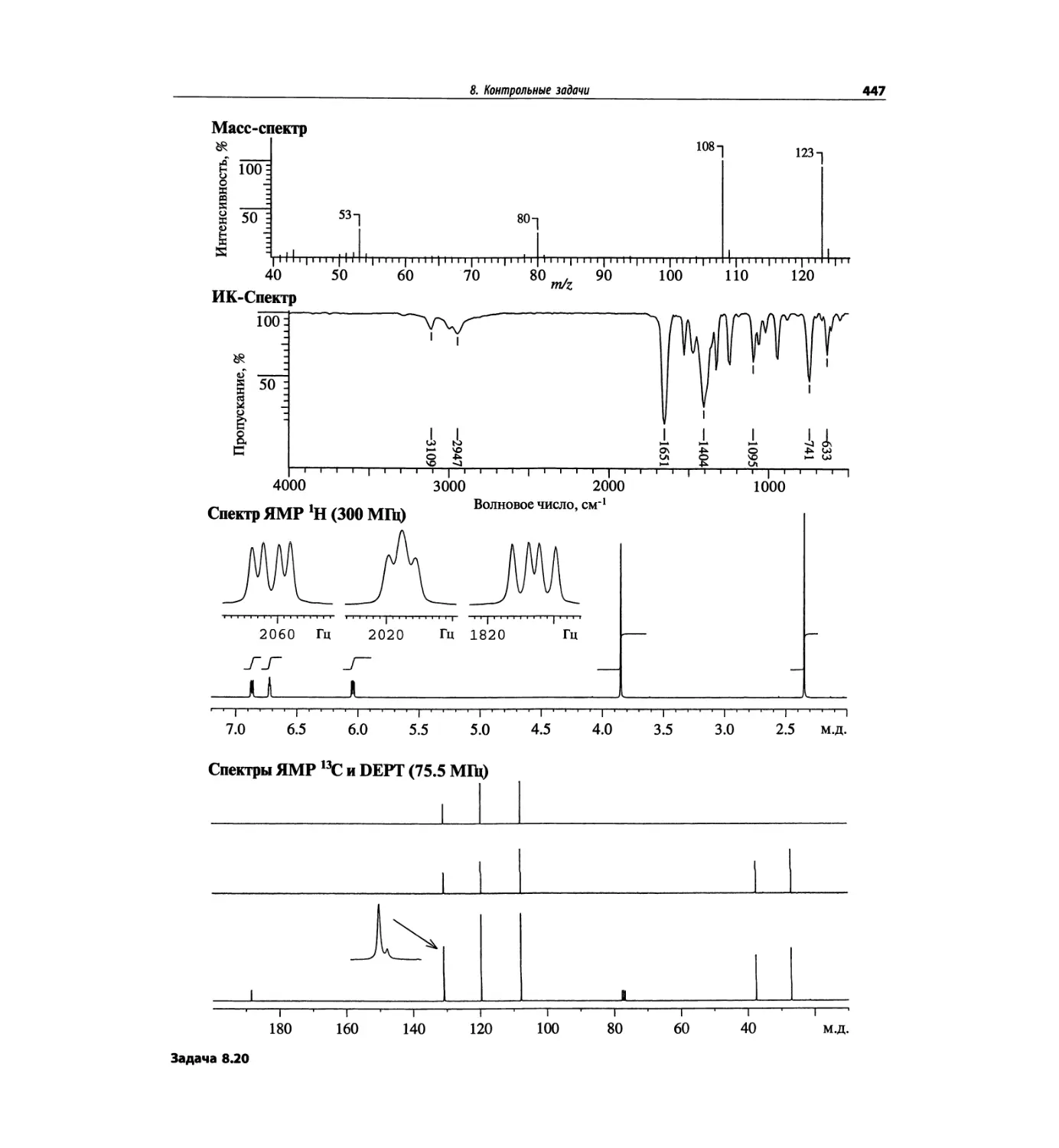
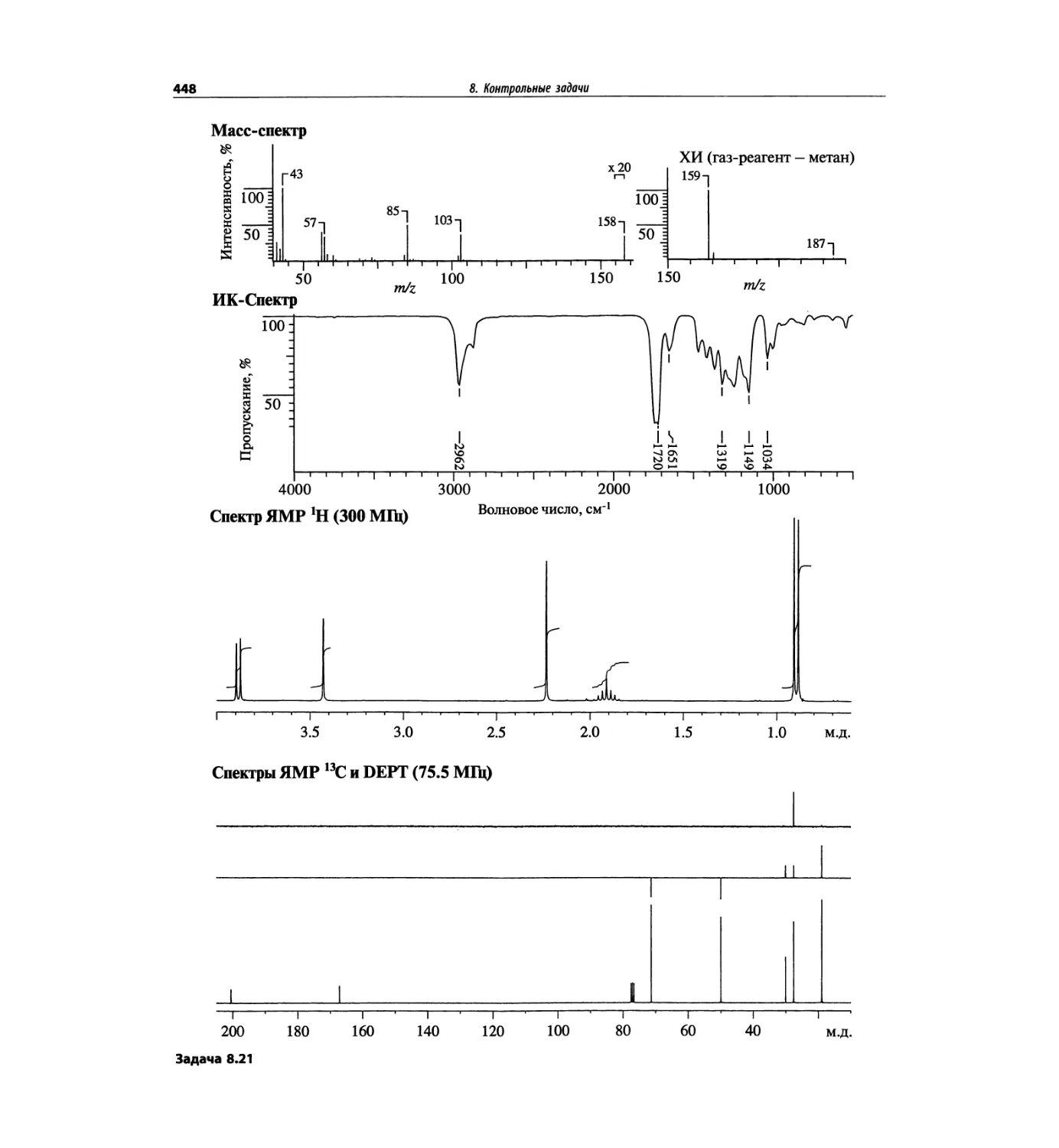
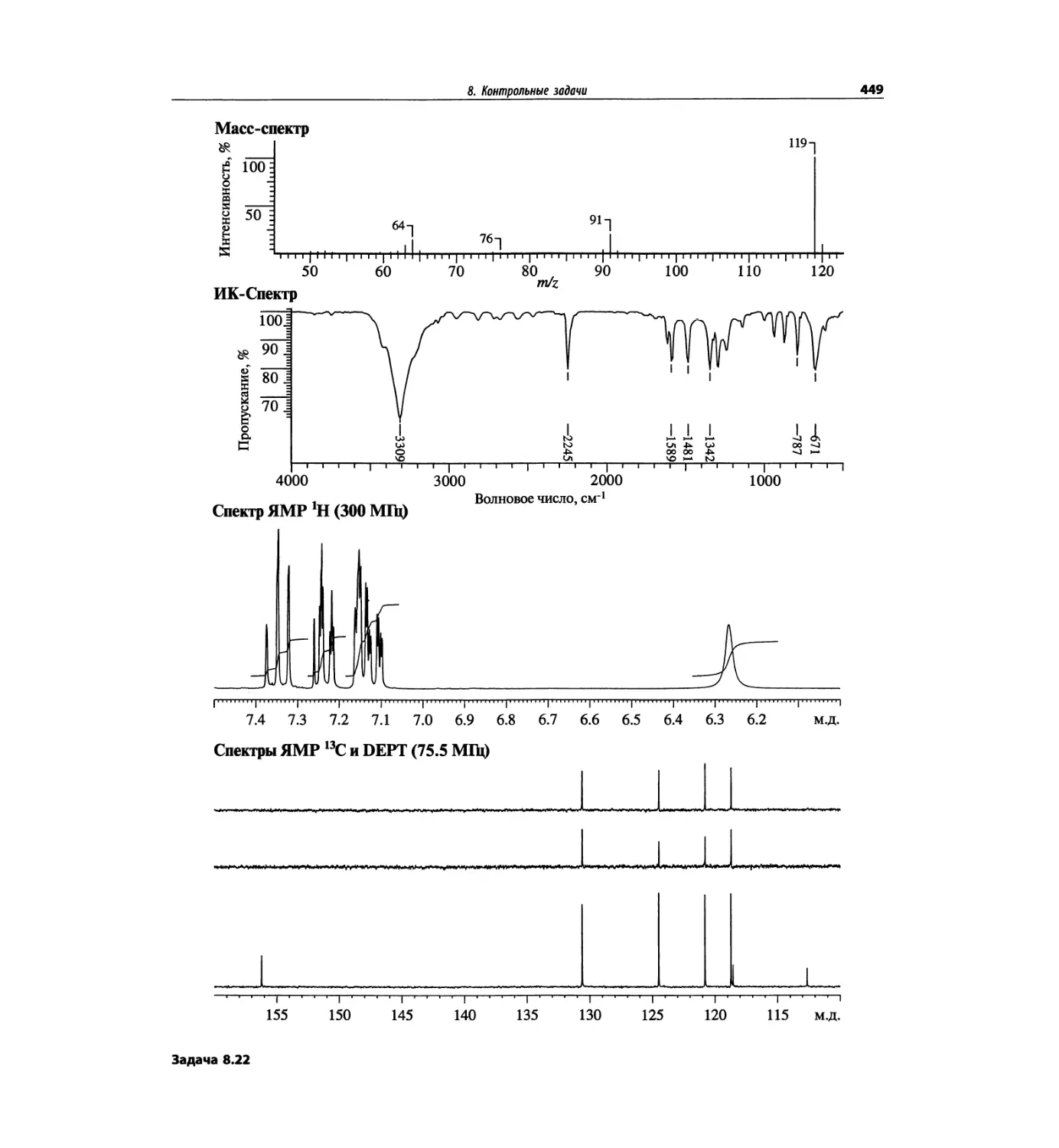


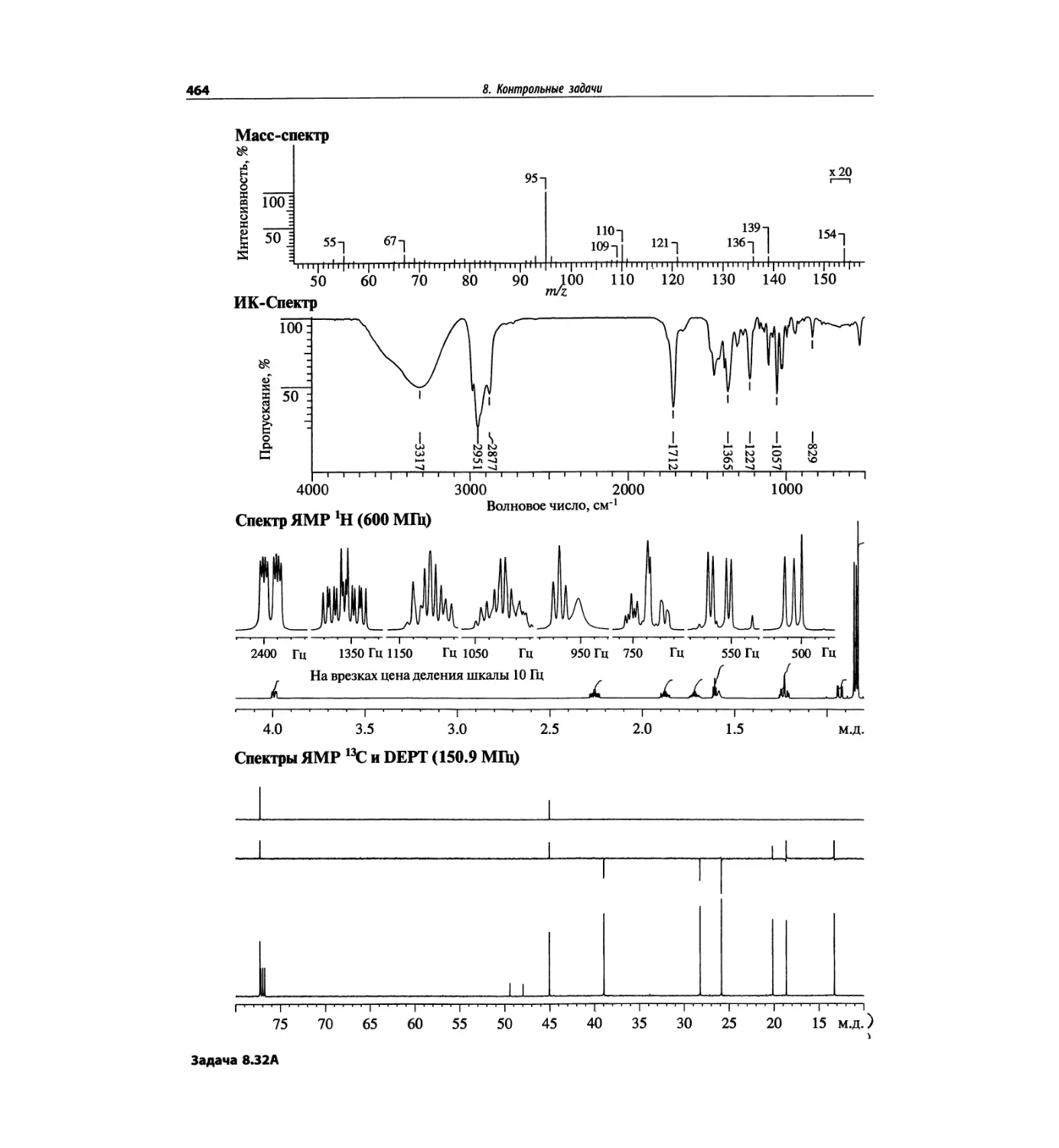
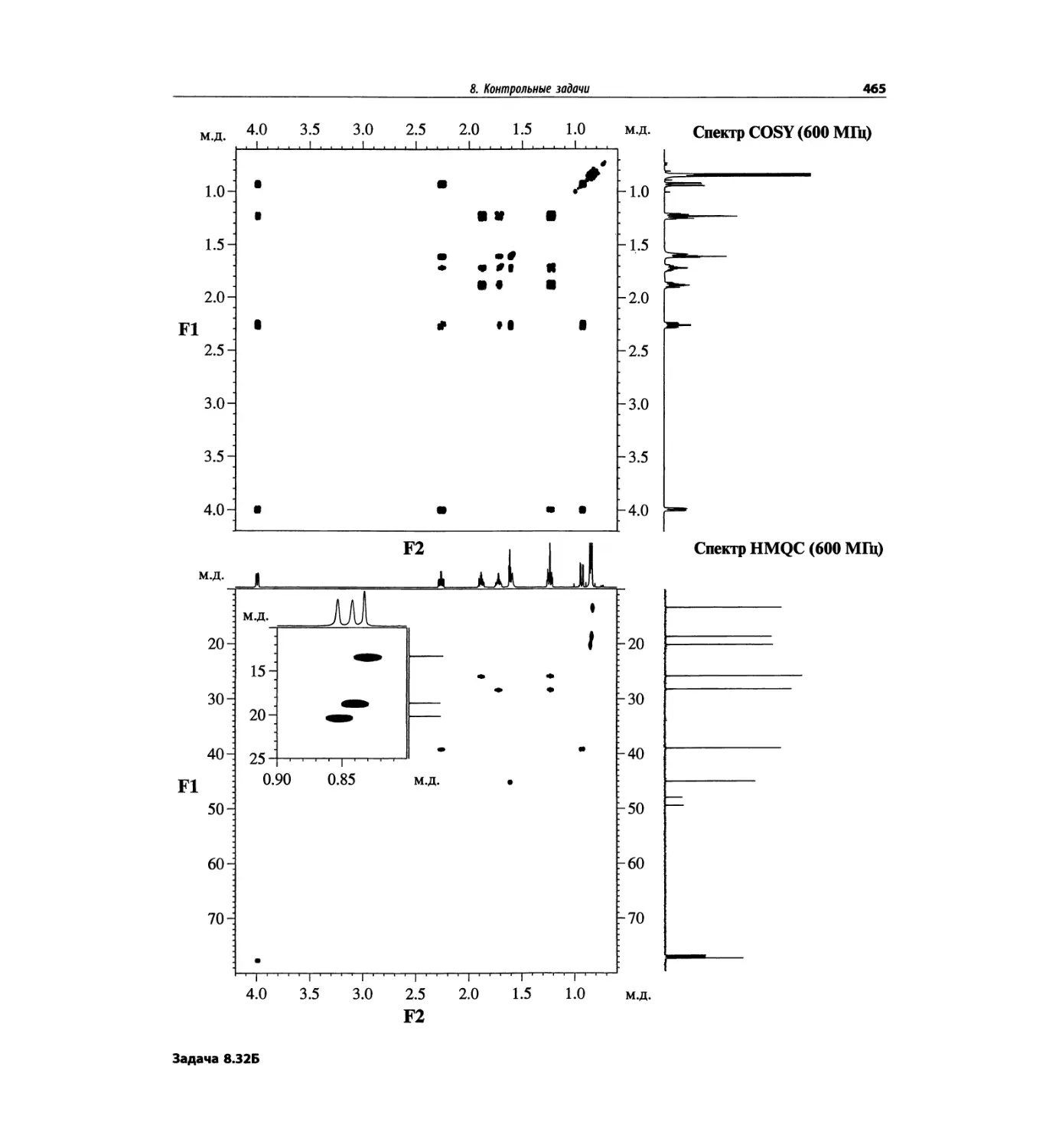
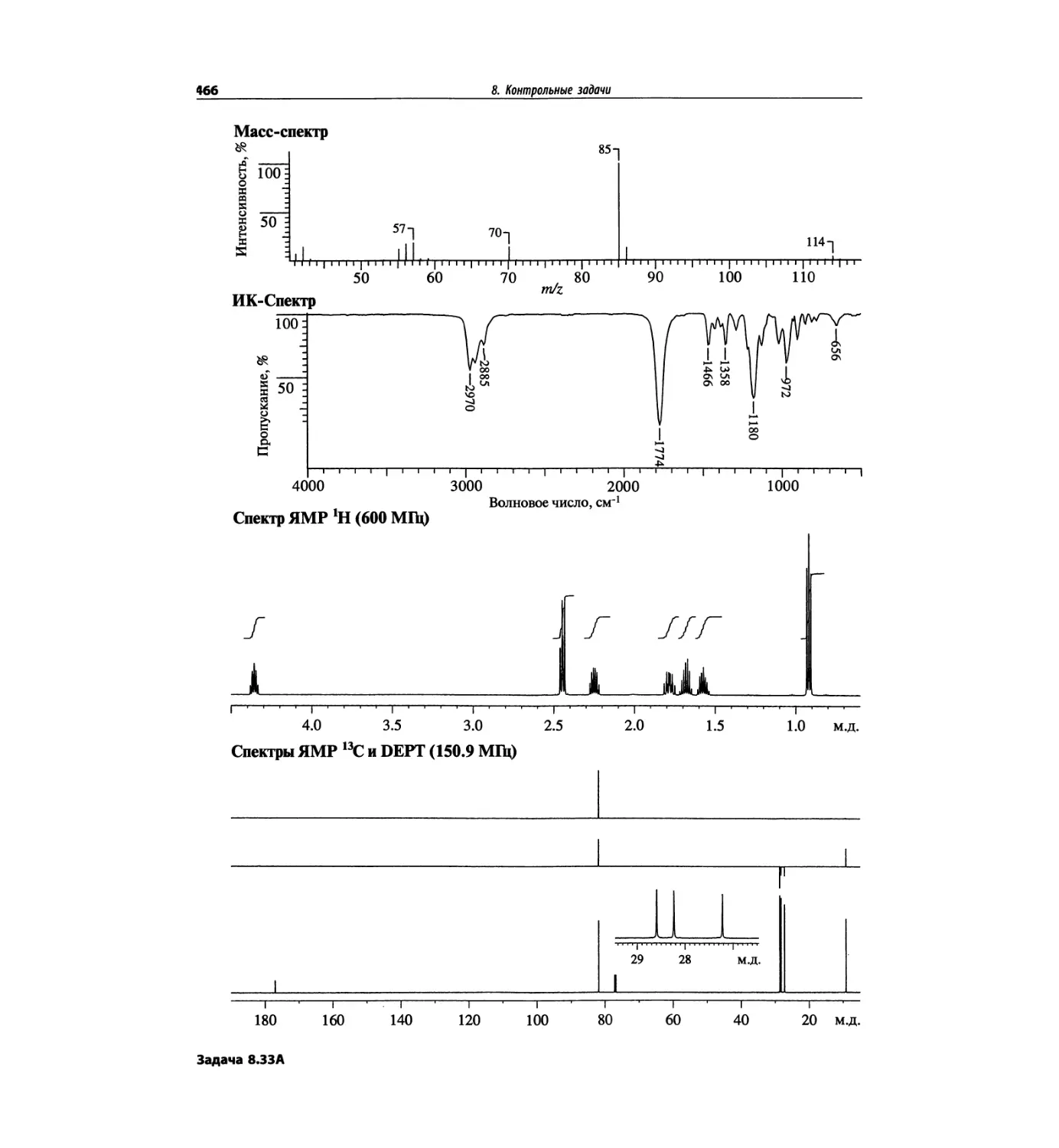
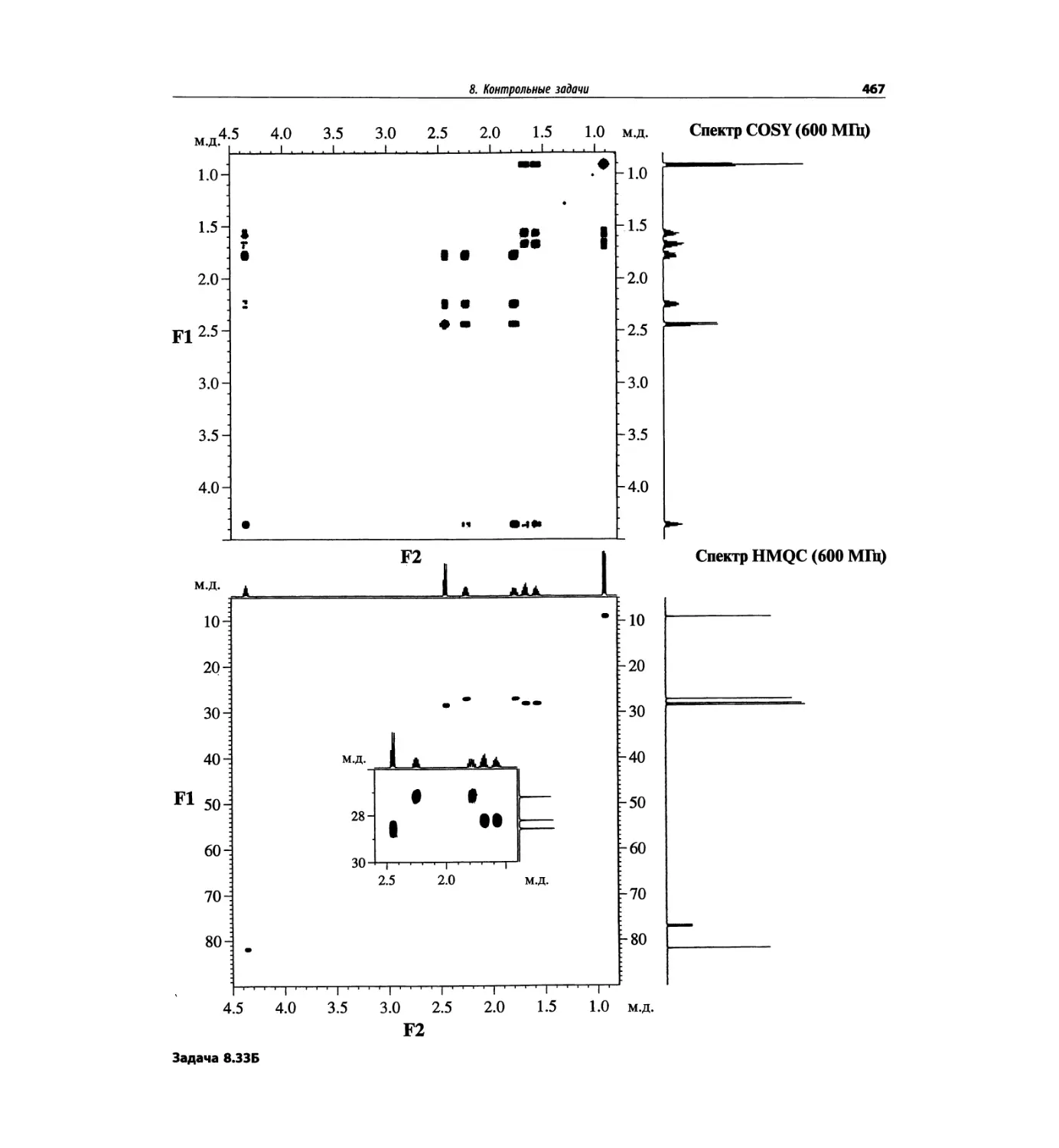





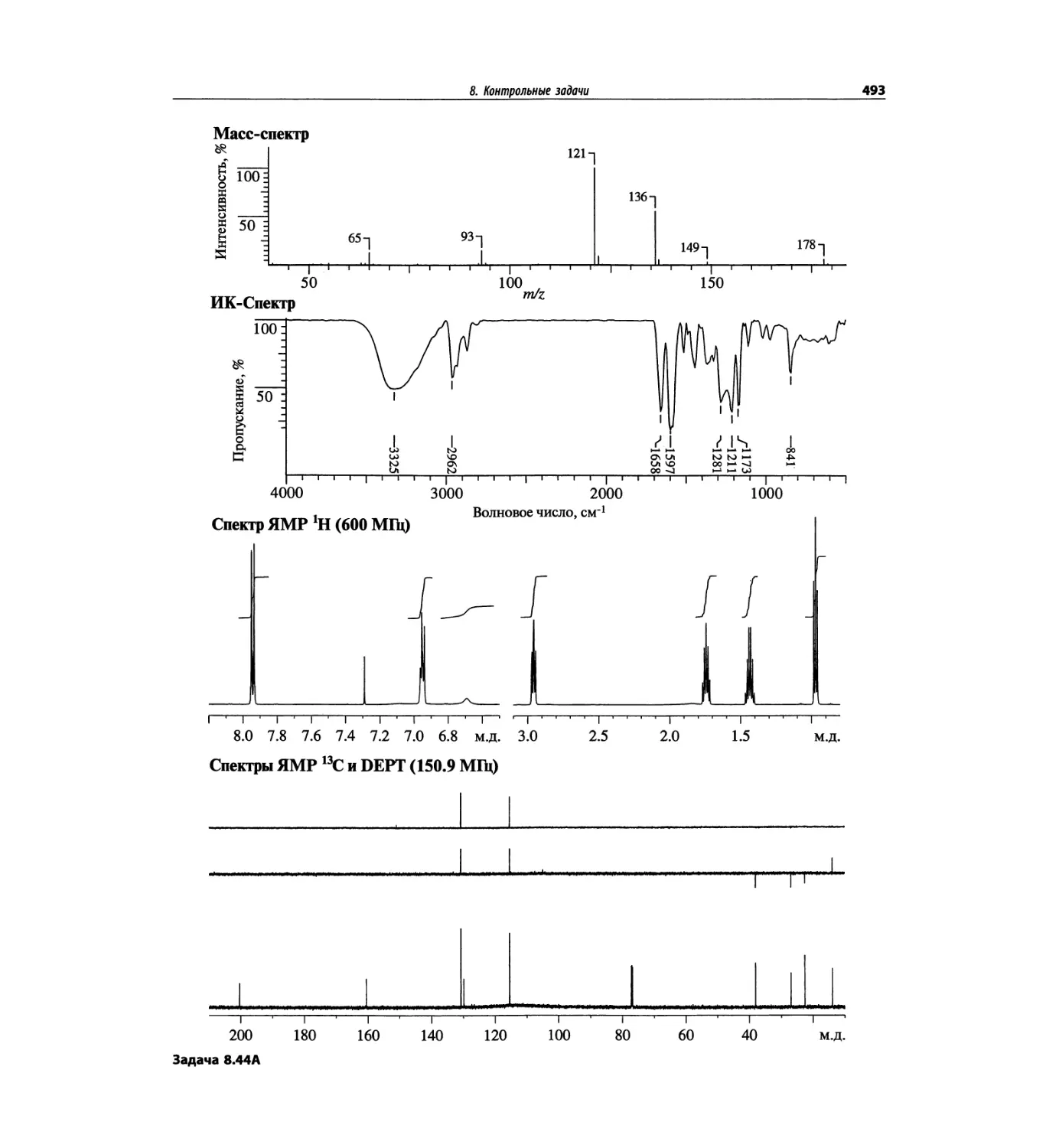
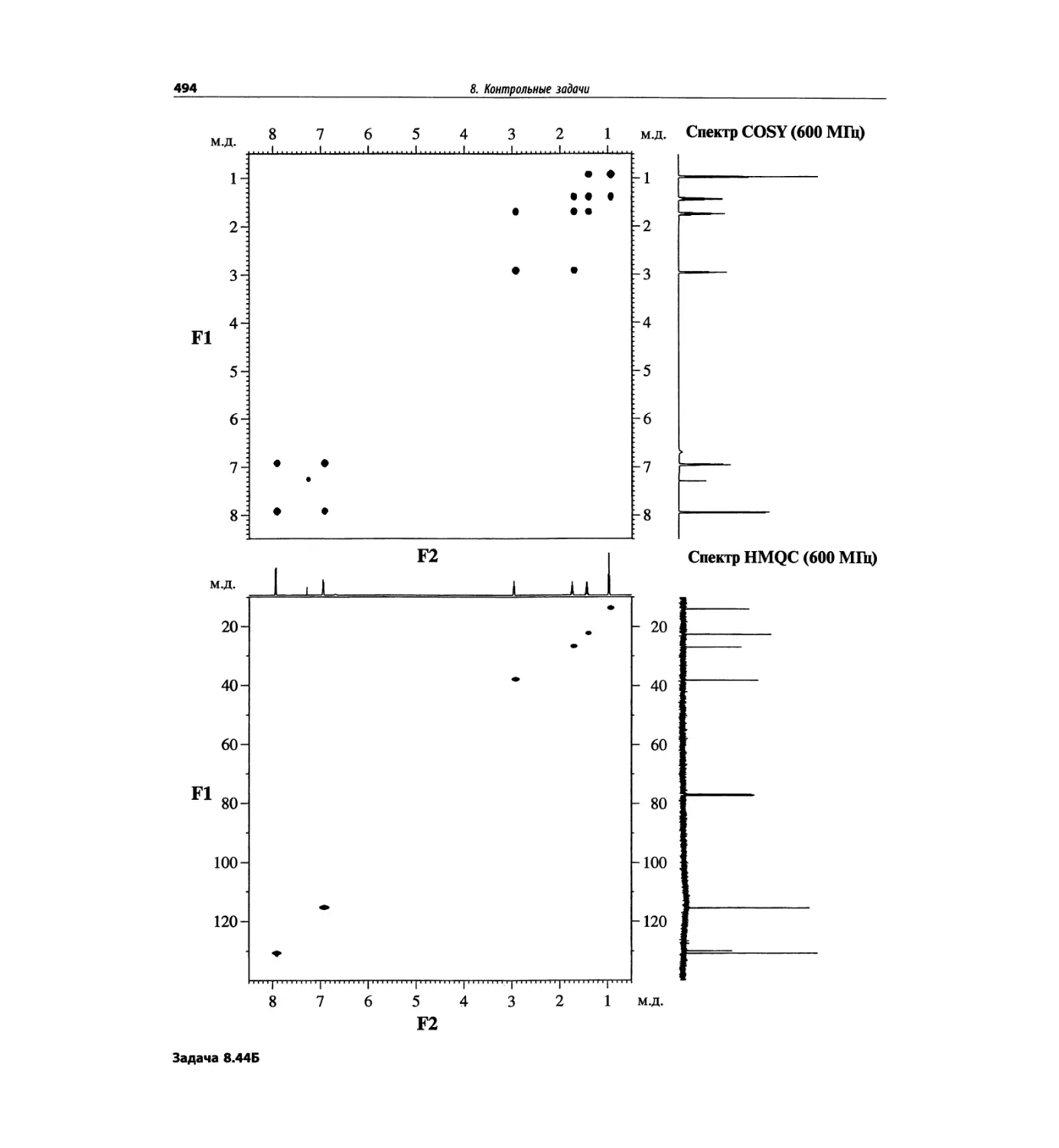
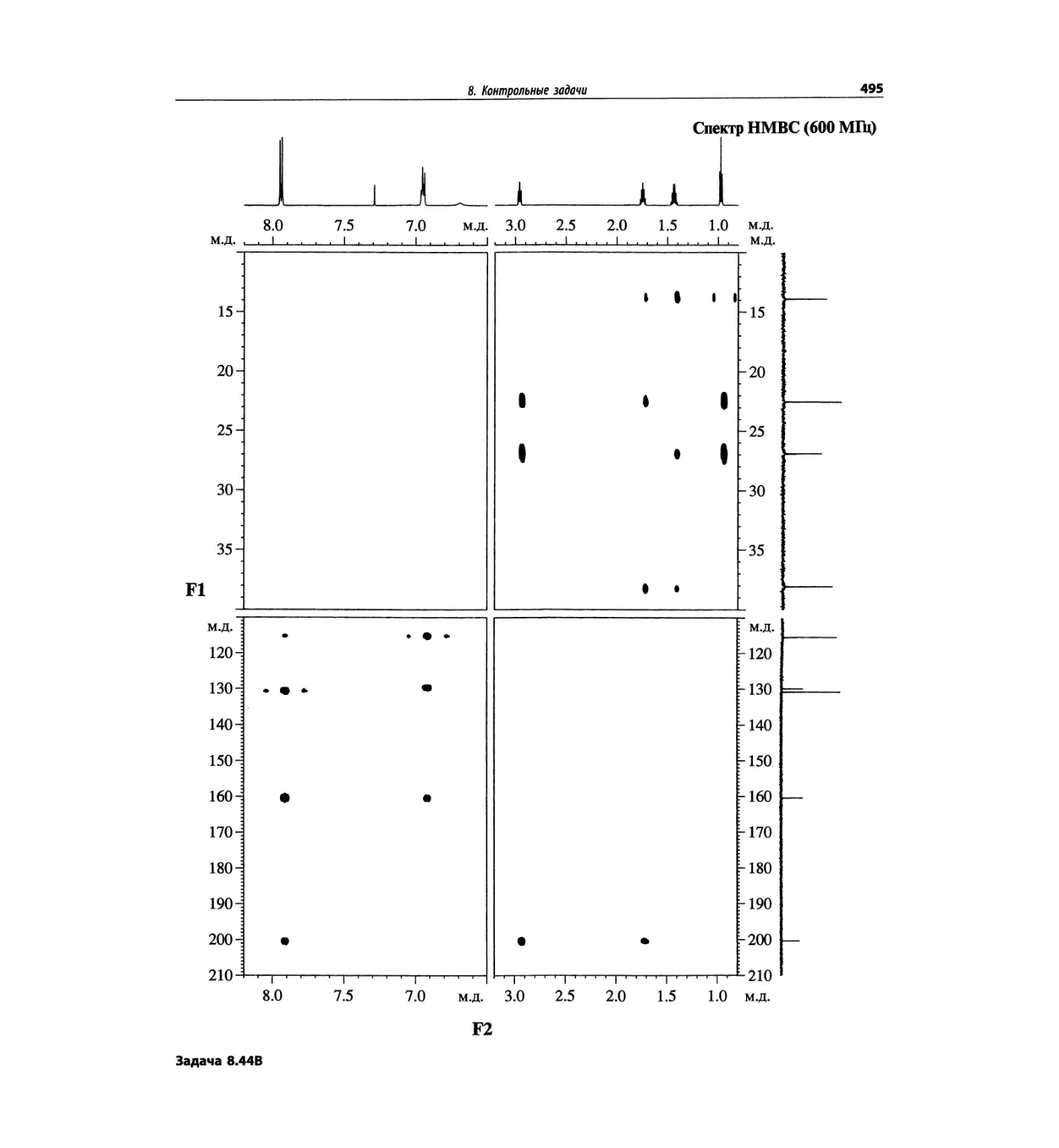
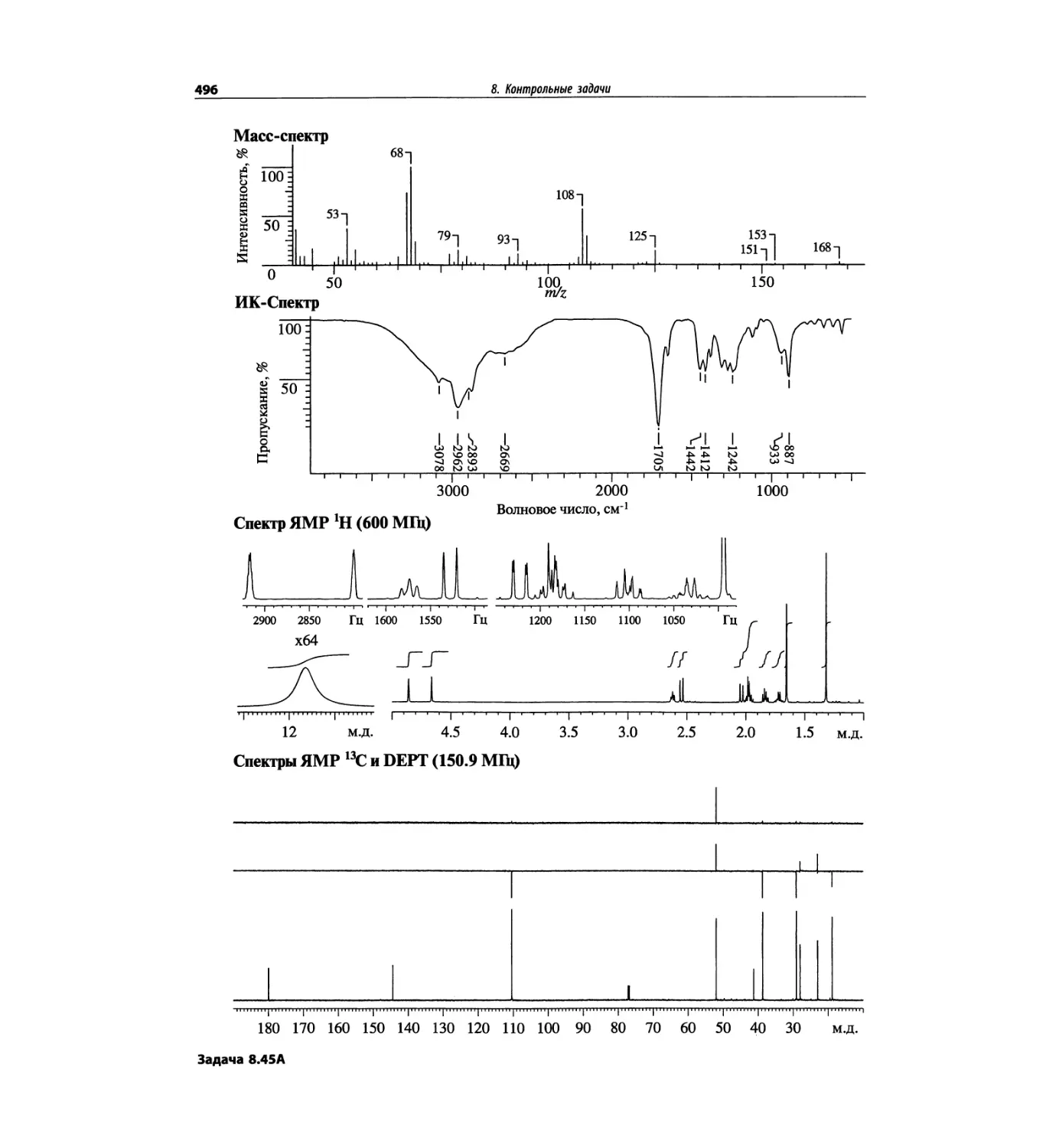







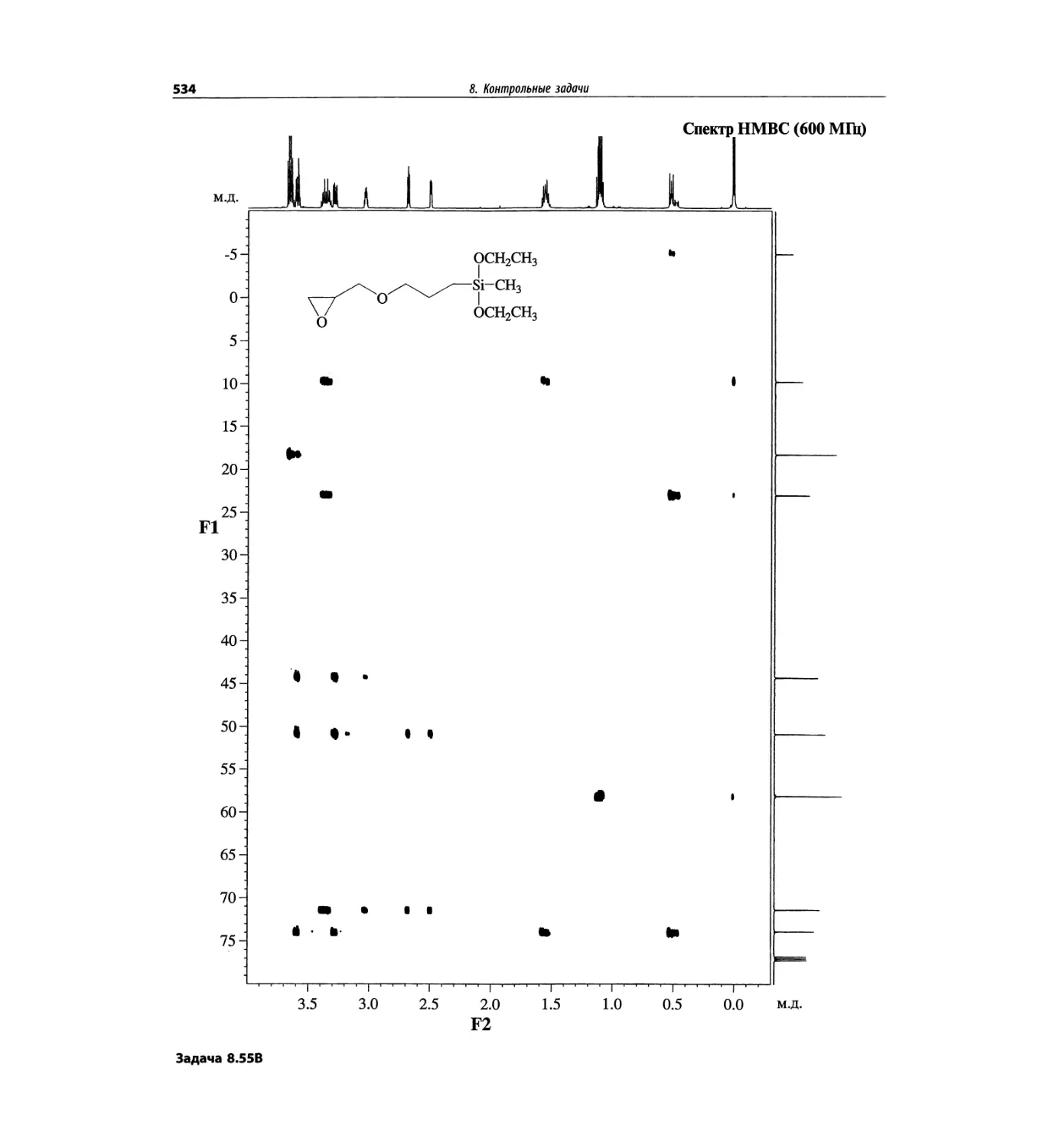
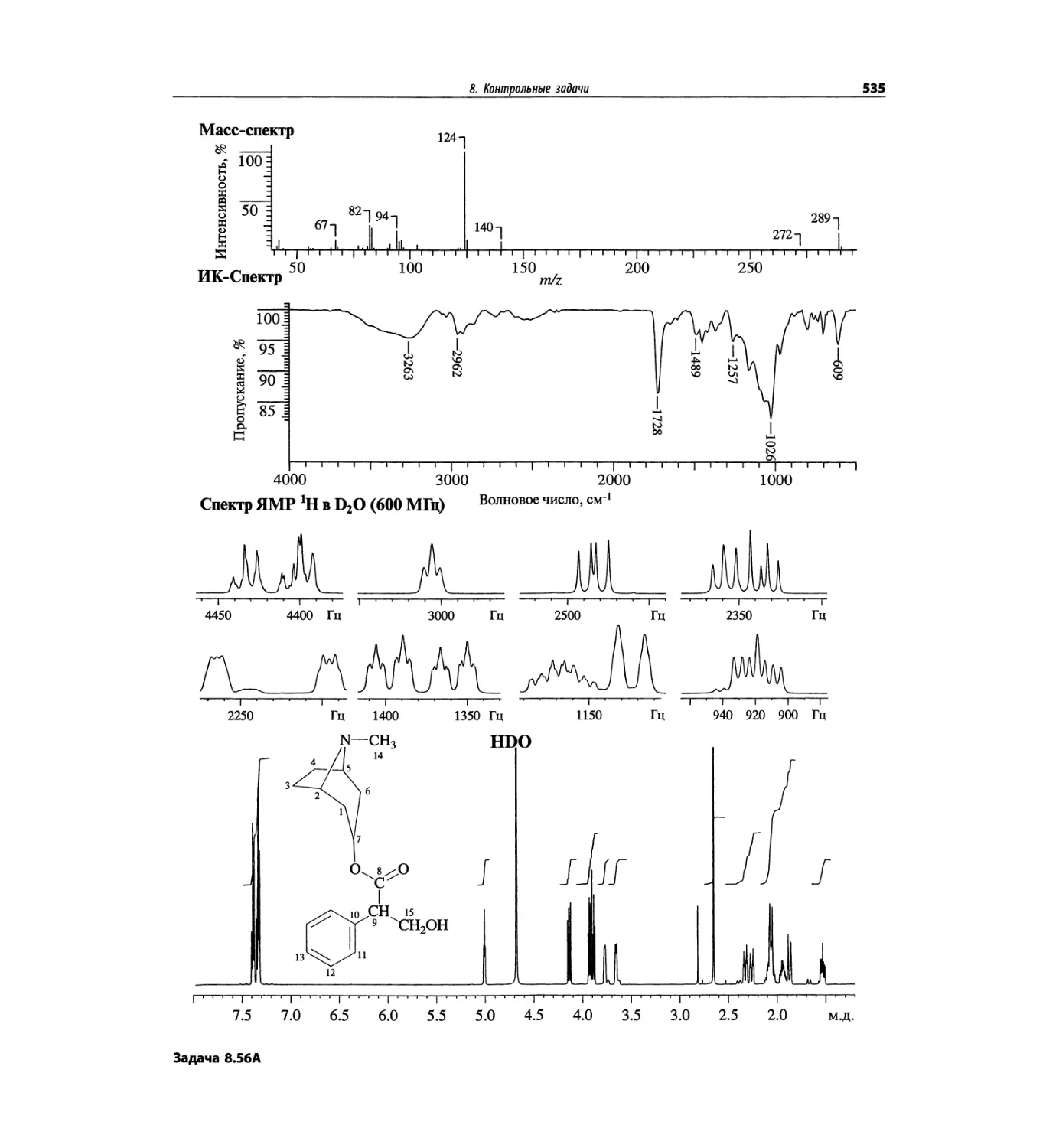
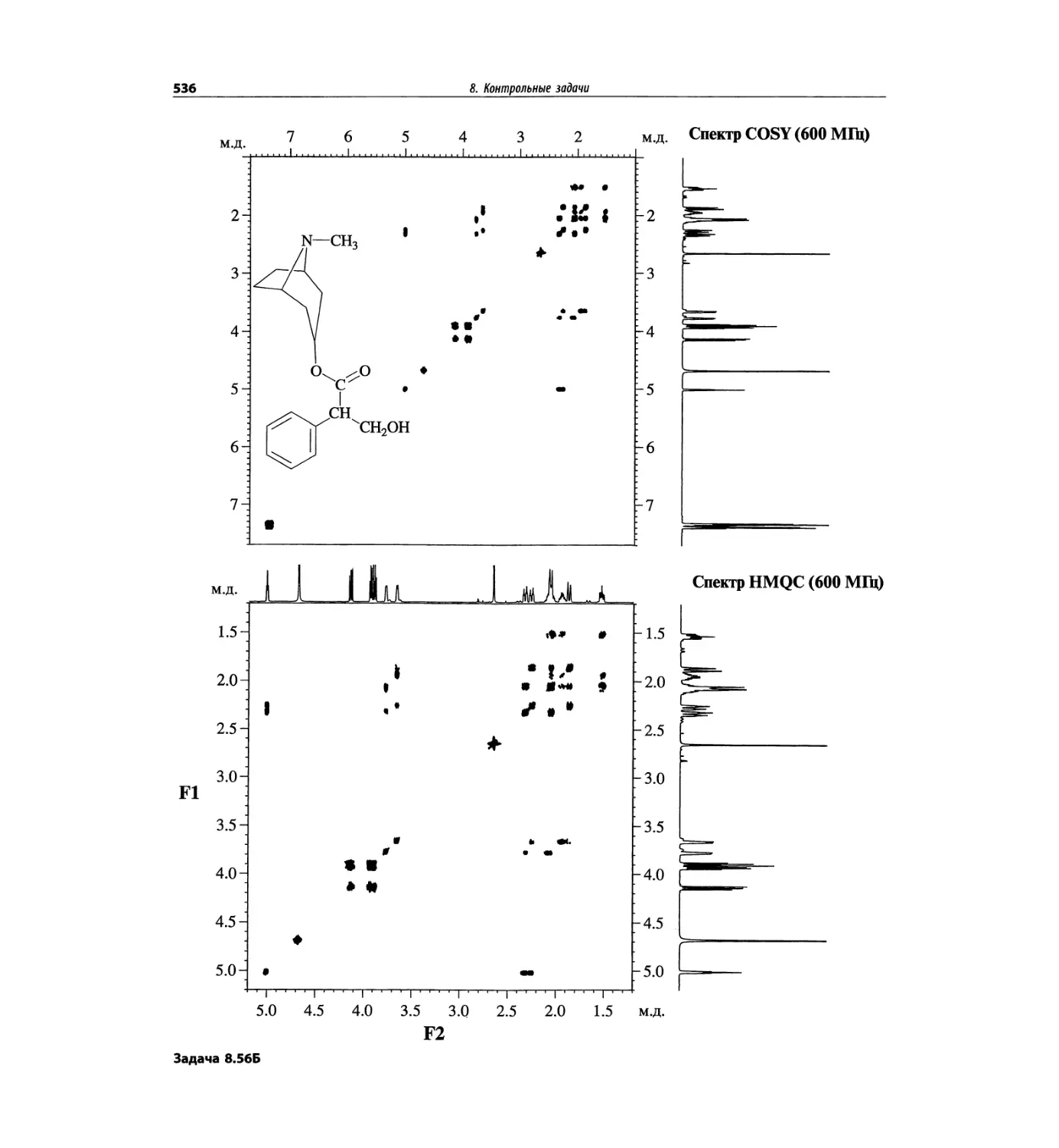
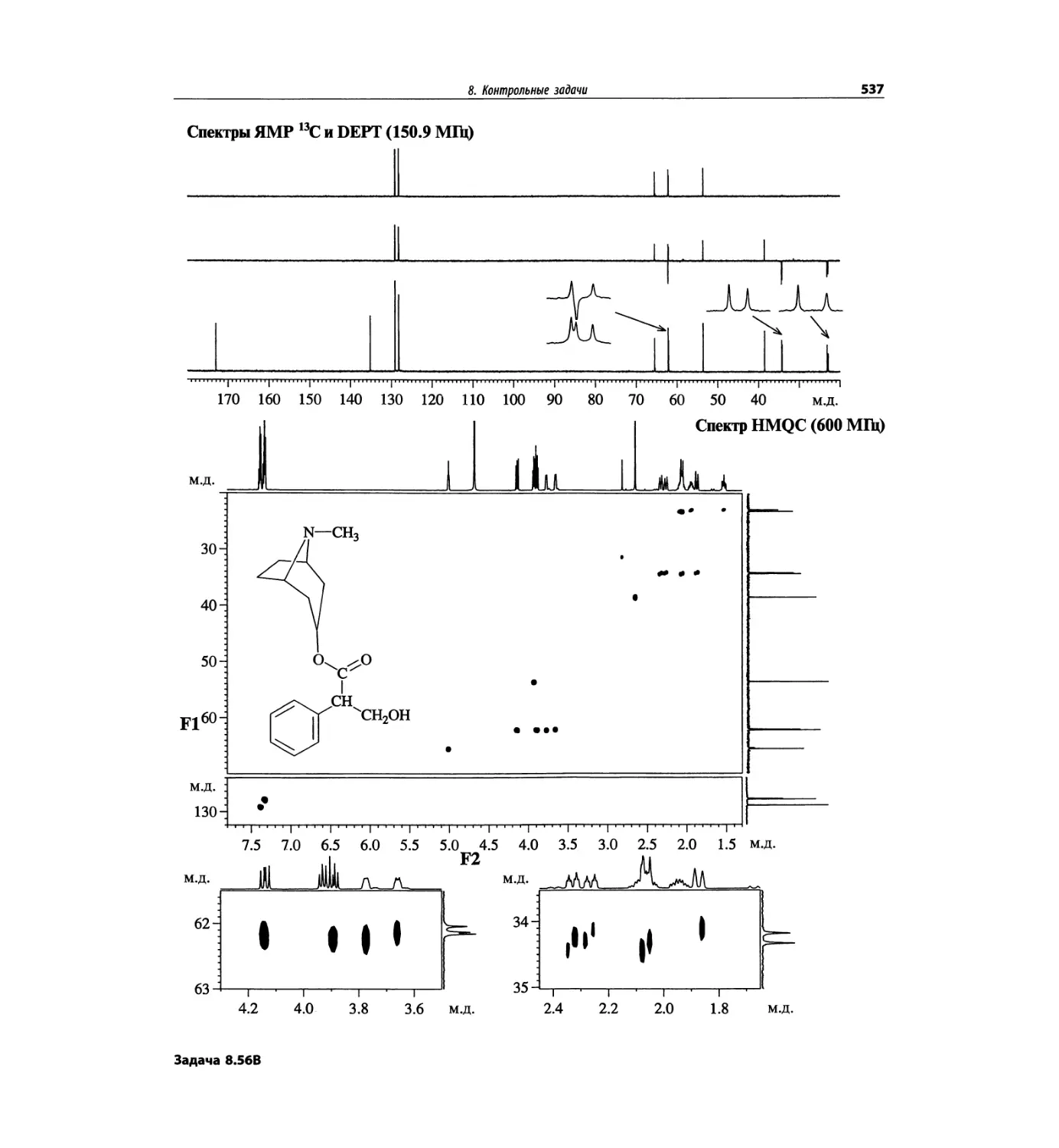

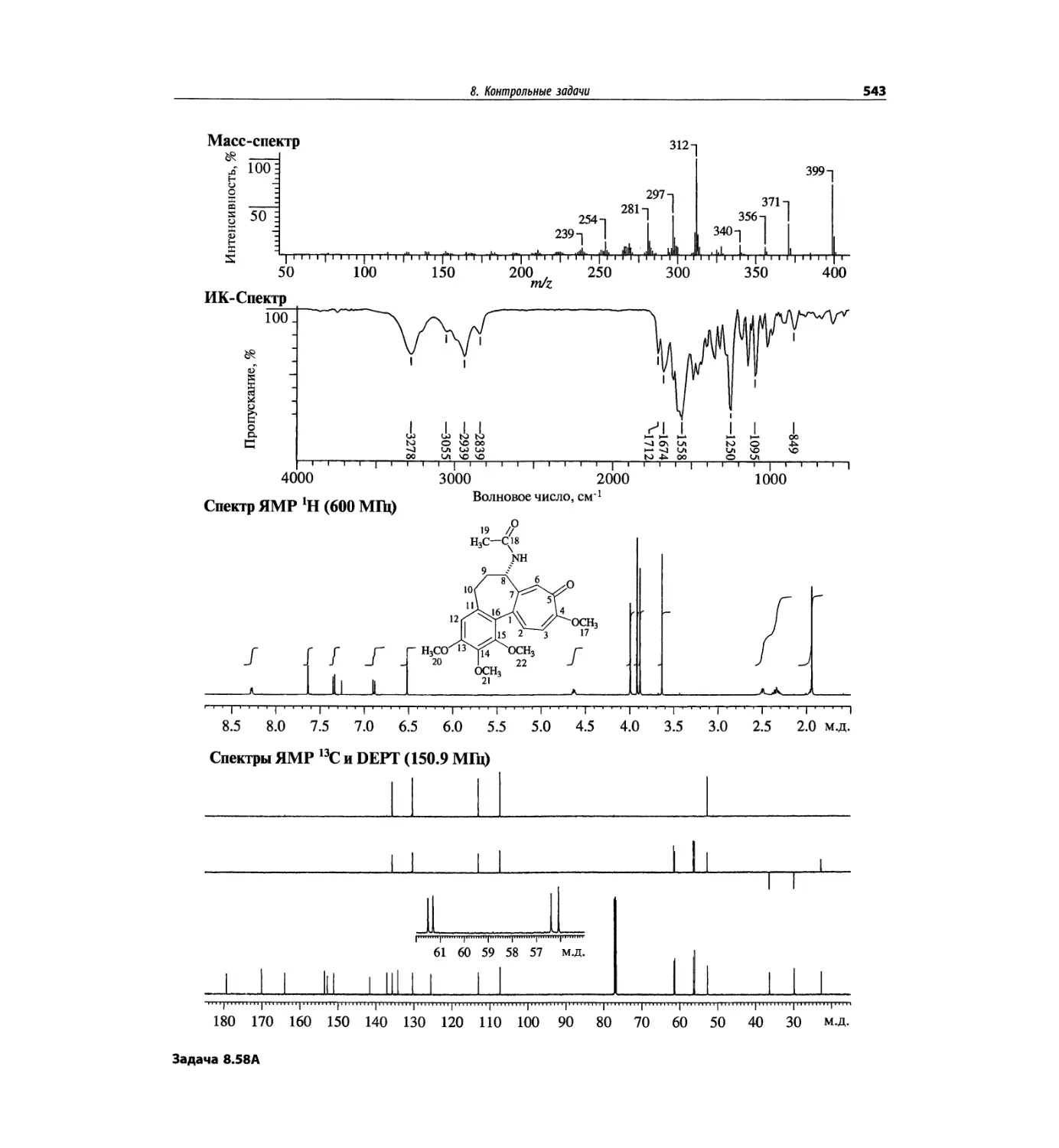

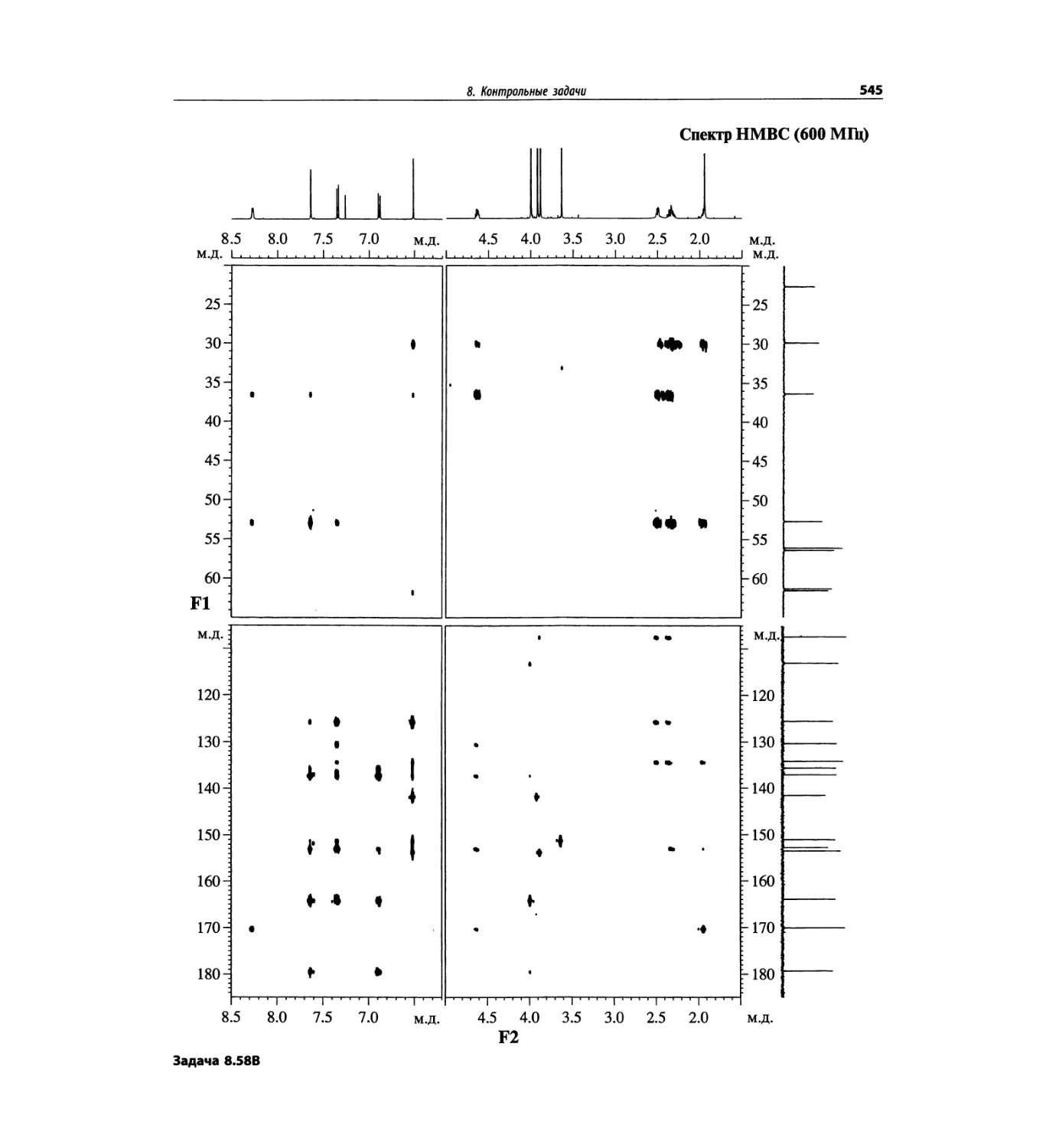















Автор: Сильверстейн Р. Вебстер Ф. Кимл Д.
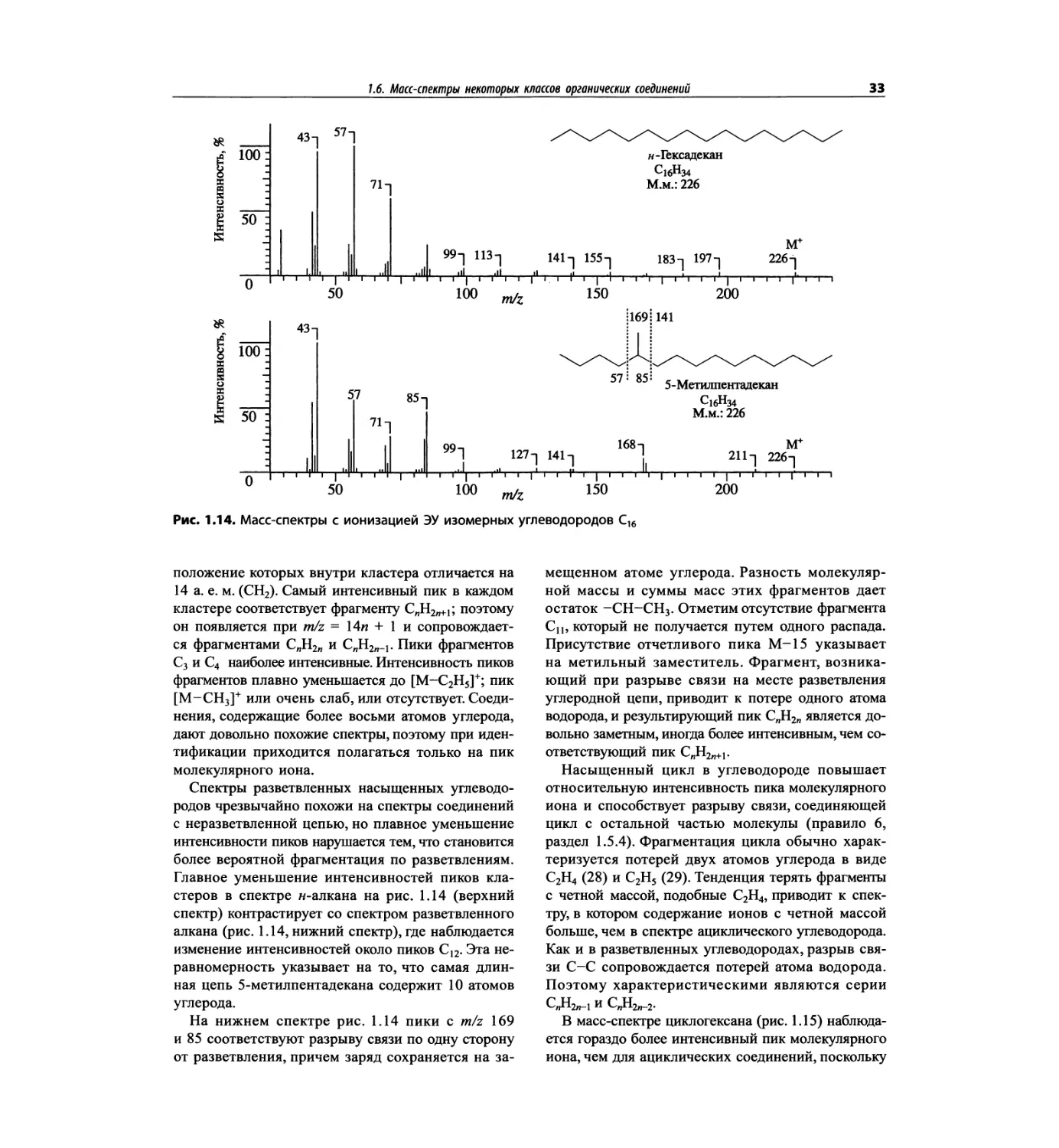
Теги: спектральные методы анализа оптические методы анализа органическая химия спектрометрия
ISBN: 978-5-94774-392-0
Год: 2014




Текст
методы в химии
Р. Сильверстейн, Ф. Вебстер, Д. Кимл
СПЕКТРОМЕТРИЧЕСКАЯ ИДЕНТИФИКАЦИЯ
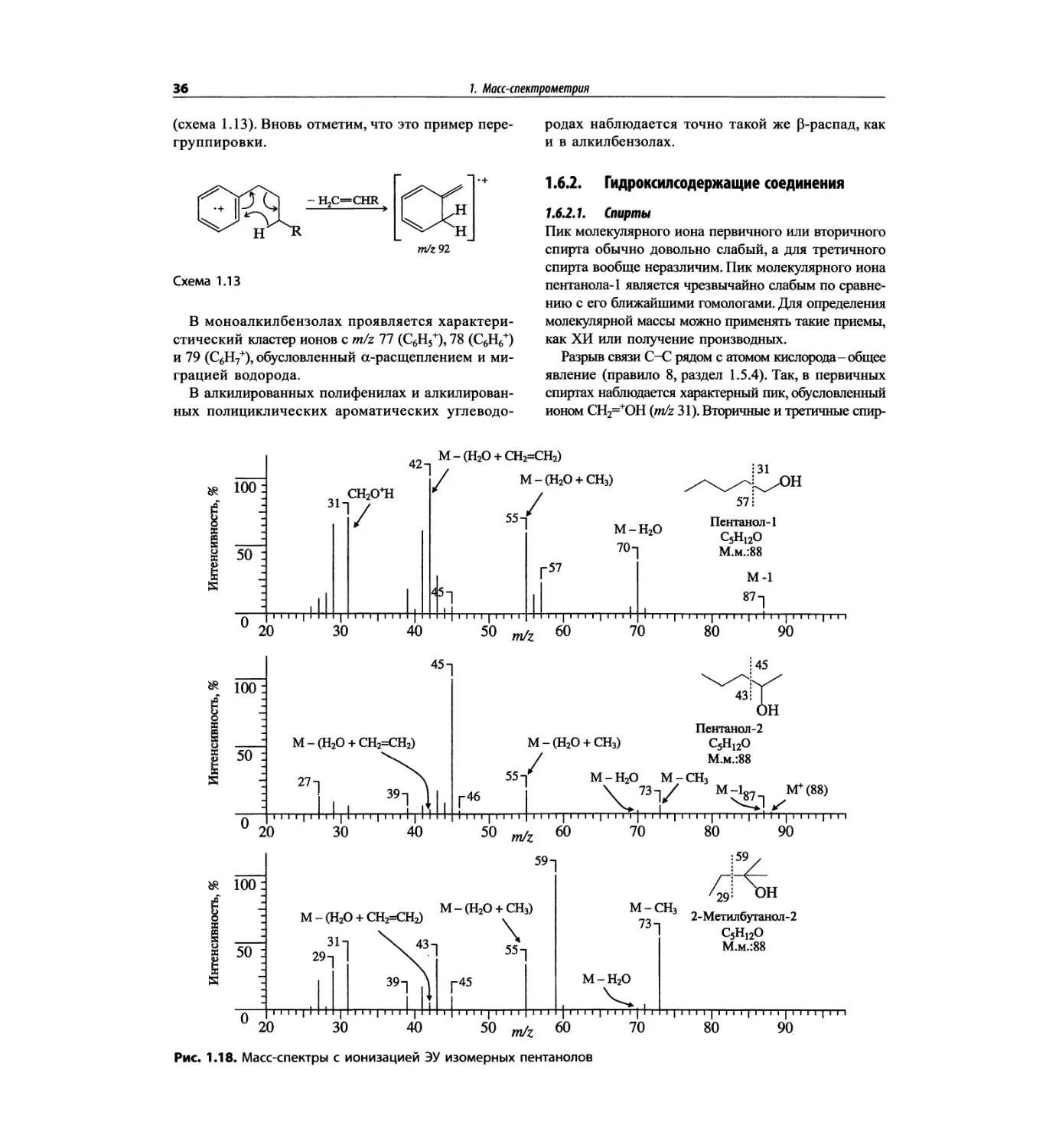
ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Перевод с английского
профессора, доктора хим. наук Н.М. Сергеева
и канд. хим. наук Б. Н. Тарасевича
Москва
БИНОМ. Лаборатория знаний
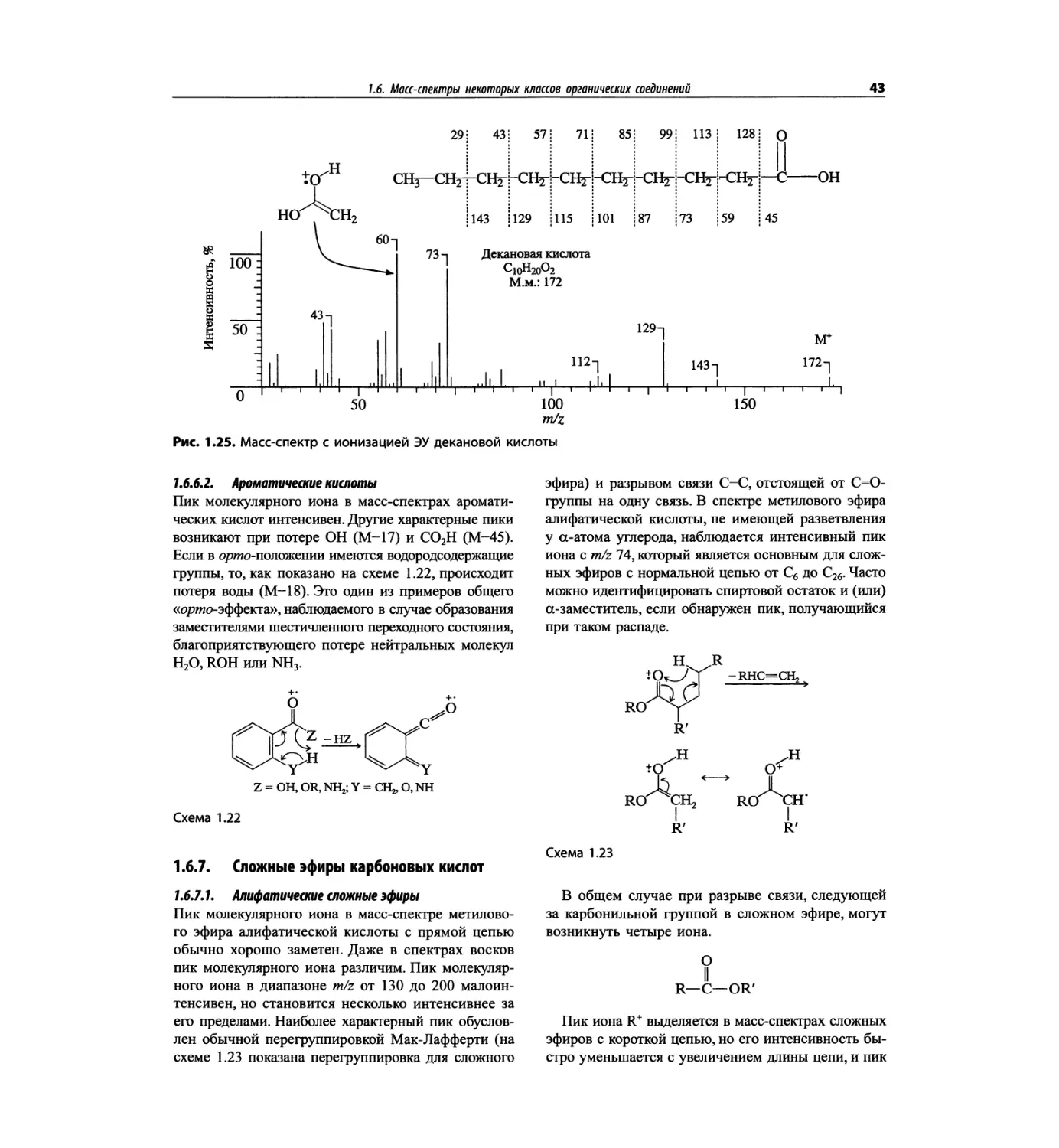
УДК 543.42
ББК 24.2я73
С36
Серия основана в 2003 г.
Сильверстейн Р.
С36 Спектрометрическая идентификация органических соедине-
ний / Р. Сильверстейн, Ф. Вебстер, Д. Кимл ; пер. с англ. —
М. : БИНОМ. Лаборатория знаний, 2014. —557 с. : ил. — (Методы
в химии).
ISBN 978-5-94774-392-0
Учебное издание, написанное американскими учеными с большим опытом
преподавательской деятельности, посвящено определению строения органиче-
ских соединений с использованием совокупности современных физико-химиче-
ских методов исследования (масс-спекгрометрии, ИК-спектроскопии, спектро-
скопии ЯМР на ядрах 1Н, 13С и др ). Изложены теоретические основы методов,
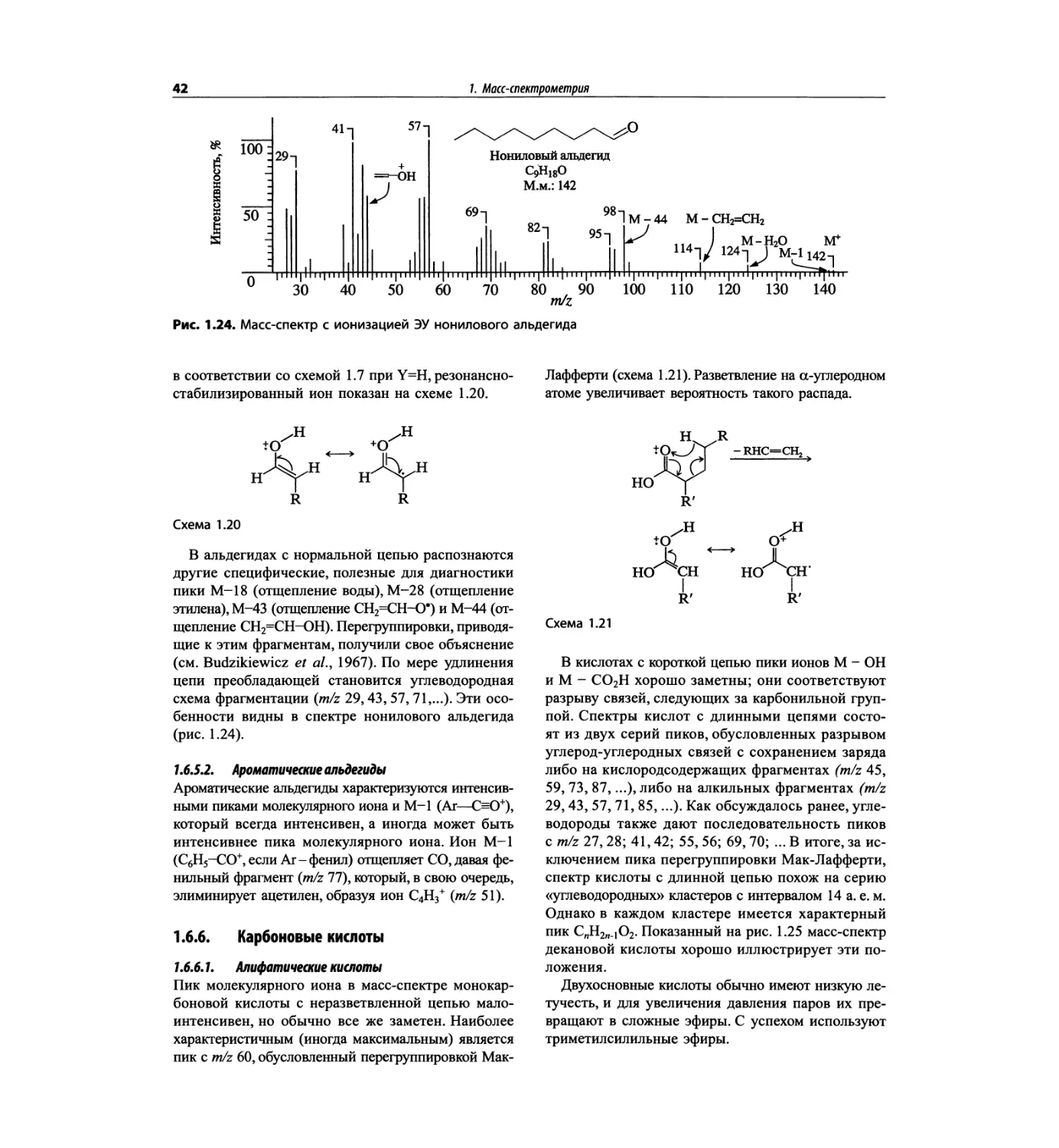
описаны особенности эксперимента и расшифровки спектров. Приведены кон-
кретные примеры установления структуры соединений по спектральным данным.
Книга великолепно иллюстрирована, содержит большое число упражнений для
студентов, подробную библиографию к каждой главе и обширный справочный
материал.
Для студентов старших курсов, аспирантов, преподавателей и научных
работников.
УДК 543.42
ББК 24.2я73
По вопросам приобретения обращаться:
«БИНОМ. Лаборатория знаний»
Телефон: (499)157-5272
e-mail: binom@Lbz.ru, http://www.Lbz.ru
ISBN 978-5-94774-392-0
Copyright © 2005 John Wiiey & Sons, Inc.
All rights reserved.
This translation published under license.
© Перевод на русский язык. БИНОМ.
Лаборатория знаний. 2011
SPECTROMETRIC IDENTIFICATION
OF ORGANIC COMPOUNDS
SEVENTH EDITION
Robert M. Silverstein
Francis X. Webster
David J. Kiemle
State University of New York
College of Environmental Science & Forestry
JOHN WILEY & SONS, INC.
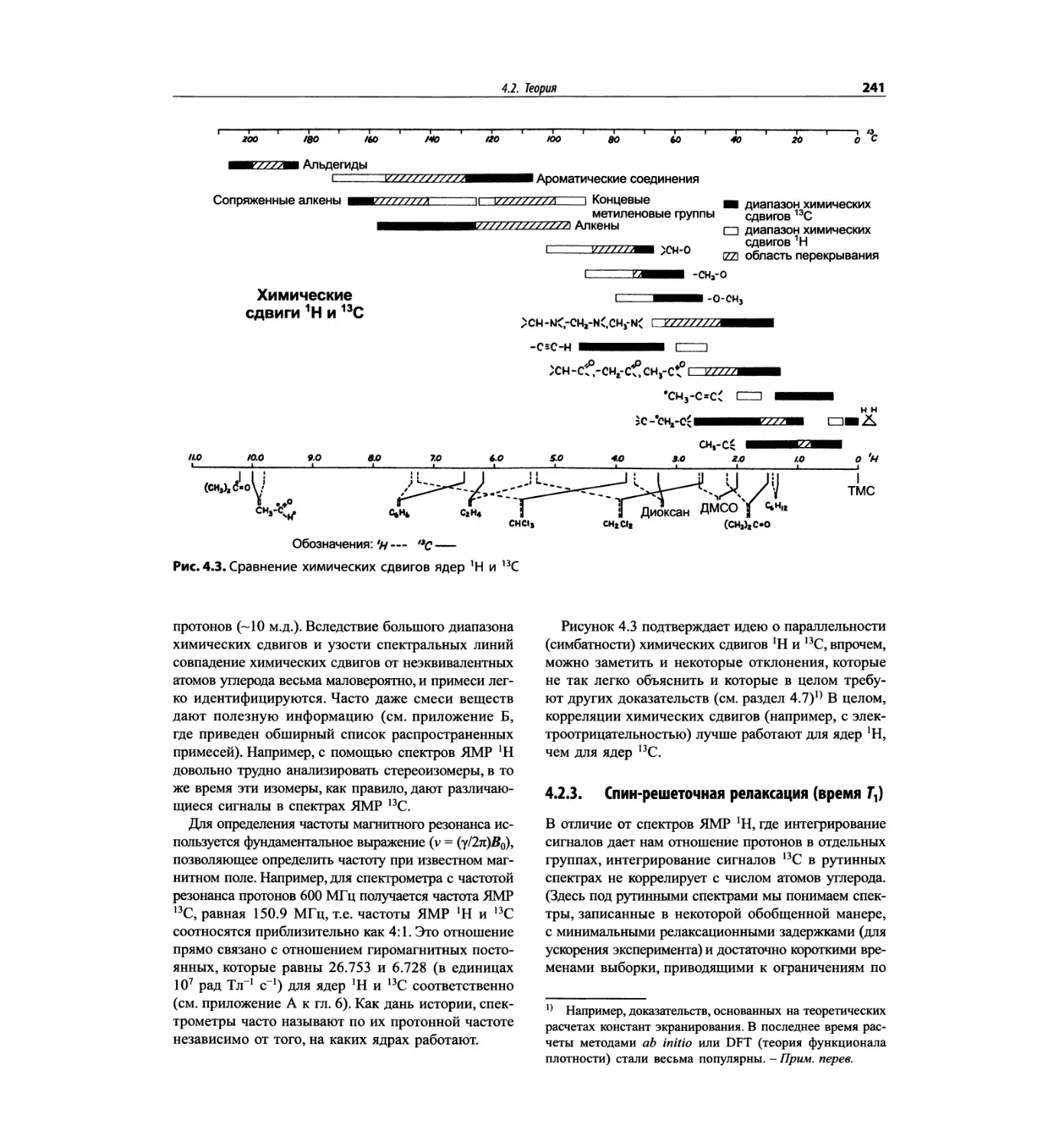
Предисловие к русскому изданию
Физические методы исследования, прежде всего спек-
трометрические методы, позволяют наиболее быстро
и эффективно решать задачи, связанные с идентифи-
кацией веществ и установлением строения химиче-
ских соединений. Спектрометрические методы при-
меняются уже около полувека, при этом постоянно
совершенствуются техника измерений, способы рас-
шифровки спектров, которые становятся более точ-
ными и однозначными, многие стадии эксперимента
и обработки результатов автоматизируются.
В настоящее время в указанной области исследова-
ний происходят серьезные качественные изменения,
начало которым было положено в конце прошлого
века. Во-первых, быстрыми темпами продолжается
смена поколений приборов и внедрение в практику
новейших приемов работы. В ИК-спектроскопии стали
широко применяться фурье-спектрометры; в спек-
троскопии ЯМР разрабатываются сложные много-
импульсные методики и двумерное представление
спектральных данных, что существенно раздвигает
границы возможностей метода. Усовершенствования
затронули и масс-спектрометрию. Во-вторых, в об-
ласти приборостроения завершился период компью-
теризации и началось активное развитие сетевых
технологий. Представление результатов спектральных
измерений в цифровом виде изменило сам характер
обработки данных, для этого стало применяться спе-
циализированное программное обеспечение с новы-
ми возможностями, в том числе с использованием
электронных библиотек и баз данных.
Совершенствование техники эксперимента и раз-
витие методологии привели к существенному рас-
ширению области применения упомянутых методов.
Не отказываясь от решения традиционных задач
классической органической химии, исследователи
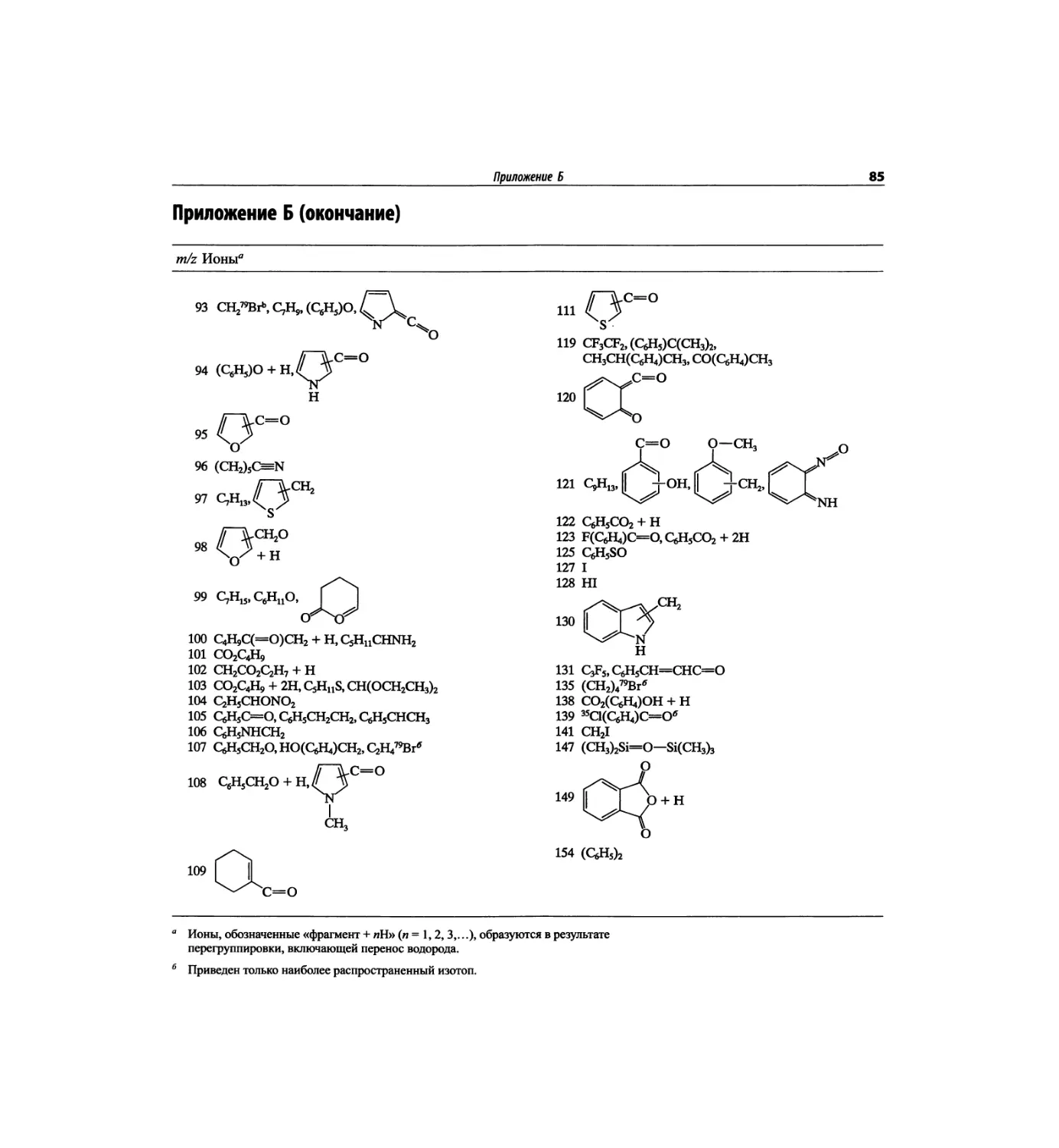
перешли к постановке и решению более сложных
проблем биохимии и биологии, химической техноло-
гии, химической защиты и экологии, фармацевтики,
метабономики, протеомики и пр.
Кардинальное обогащение методического арсенала
качественно изменило практику применения физиче-
ских, прежде всего спектроскопических, методов, что
потребовало обновления соответствующих учебных
пособий. Новым вызовам в значительной степени
отвечает предлагаемая читателям книга Р. Сильвер-
стейна, Ф. Вебстера и Д. Кимла «Спектрометрическая
идентификация органических соединений», в которой
развивается концепция комплексного использования
физико-химических методов для установления струк-
туры органических соединений.
За четыре десятилетия книга выдержала
шесть изданий (1-е издание - в 1963 г.). Тре-
тье издание (1974 г.) было переведено на рус-
ский язык в 1977 г. (см. Сильверстейн Р.,
Басслер Г., Моррил Т. Спектрометрическая иден-
тификация органических соединений / Пер. с англ,
под ред. А. А. Мальцева. - М.: Мир, 1977). Эта
книга в течение многих лет активно использовалась
в учебном процессе. Наконец, 7-е издание (перевод
которого мы с удовольствием предлагаем) подго-
товлено Р. Сильверстейном с новыми соавторами -
Ф. Вебстером и Д. Кимлом, что позволило суще-
ственно расширить изложение методов регистрации
спектров и приемов их расшифровки.
Книга полезна для широкого круга специалистов
любой квалификации. Она может использоваться при
самостоятельном освоении методов даже теми, кто
не имеет специальной подготовки в области спек-
троскопии. Для студентов это учебное пособие при
изучении обязательного для химиков-органиков курса
«Применение физических методов в органической
химии». Специалистам эта книга может пригодить-
ся как справочник в повседневной работе. Лектор
и преподаватель найдут здесь превосходные примеры,
задачи, иллюстративный материал.
Структура настоящего издания сложилась под
влиянием многолетнего использования его в учебном
процессе и существенно отличается от того, что
было в первых изданиях. Глава 1 содержит краткое,
но чрезвычайно полезное изложение физических
основ многочисленных методов масс-спектрометрии.
В главе 2 достаточно традиционно представлен ме-
тод ИК-спектроскопии: изложены основы теории
и техники эксперимента, принципы интерпретации
спектров и даны очень полезные комментарии, ка-
сающиеся характеристических областей поглоще-
б
Предисловие к русскому изданию
ния функциональных групп. Главы 3-6 посвящены
методам спектроскопии ЯМР: на протонах (гл. 3),
на ядрах 13С (гл. 4), на других ядрах со спином 1/2
(гл. 6). Следует также отметить доступное изложение
основ двумерной спектроскопии ЯМР (гл. 5), что
пока довольно слабо представлено в отечествен-
ной учебной литературе. В главах 1-6 приведены
контрольные задания для студентов. Подробная ли-
тература, сопровождающая каждую из глав (1-6),
включает как научные источники, так и фундамен-
тальные учебные издания, специальные моногра-
фии, справочники, атласы спектров. Кроме того,
главы 1-4 и 6 включают в качестве приложений
справочный материал, суммированный в виде та-
блиц и диаграмм.
В главе 7 подробно разобраны решения 6 струк-
турных задач с использованием совокупности физи-
ческих методов исследования. Изучение этой главы
чрезвычайно полезно для освоения общих подходов
к расшифровке сложных структур. В главе 8 приве-
дено около 60 задач разной степени сложности (без
решений) для самостоятельного разбора.
Особую ценность представляют многочисленные
задачи с решениями, приведенные в главах 4, 5 и 7.
Тщательный анализ этих решений позволит студенту
или начинающему исследователю приобрести прак-
тические навыки работы по интерпретации спектров
и спектрометрической идентификации органических
соединений.
К сожалению, несмотря на все достоинства ориги-
нального издания (на английском языке) там встре-
чаются некоторые досадные неточности, в частности
в задачах и их решениях. Этот недостаток Ф. Веб-
стеру и Д. Кимлу (проф. Р. Сильверстейн скончался
в 2008 г.) удалось частично исправить, опубликовав
в издательстве John Wiley & Sons специальный до-
кумент с поправками, что было учтено нами при
подготовке русского издания. Другие уточнения были
сделаны нами в виде примечаний.
Перевод выполнен Б. Н. Тарасевичем (главы 1, 2,
7, 8) и Н. М. Сергеевым (главы 3-6).
Надеемся, что книга будет высоко оценена спе-
циалистами и приобретет заслуженную популярность
среди студентов.
Н. М. Сергеев
Б. Н. Тарасевич
8
Предисловие
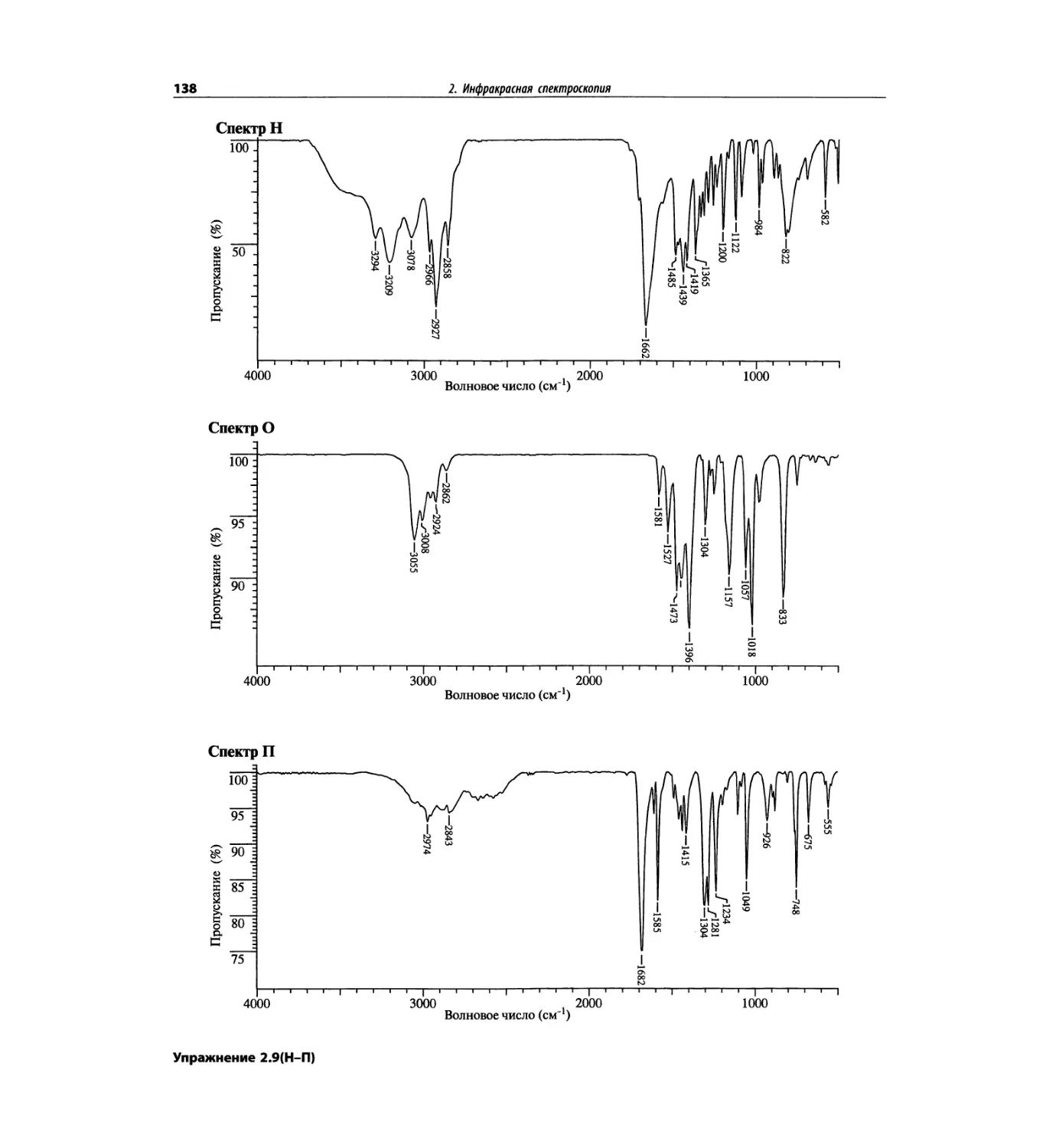
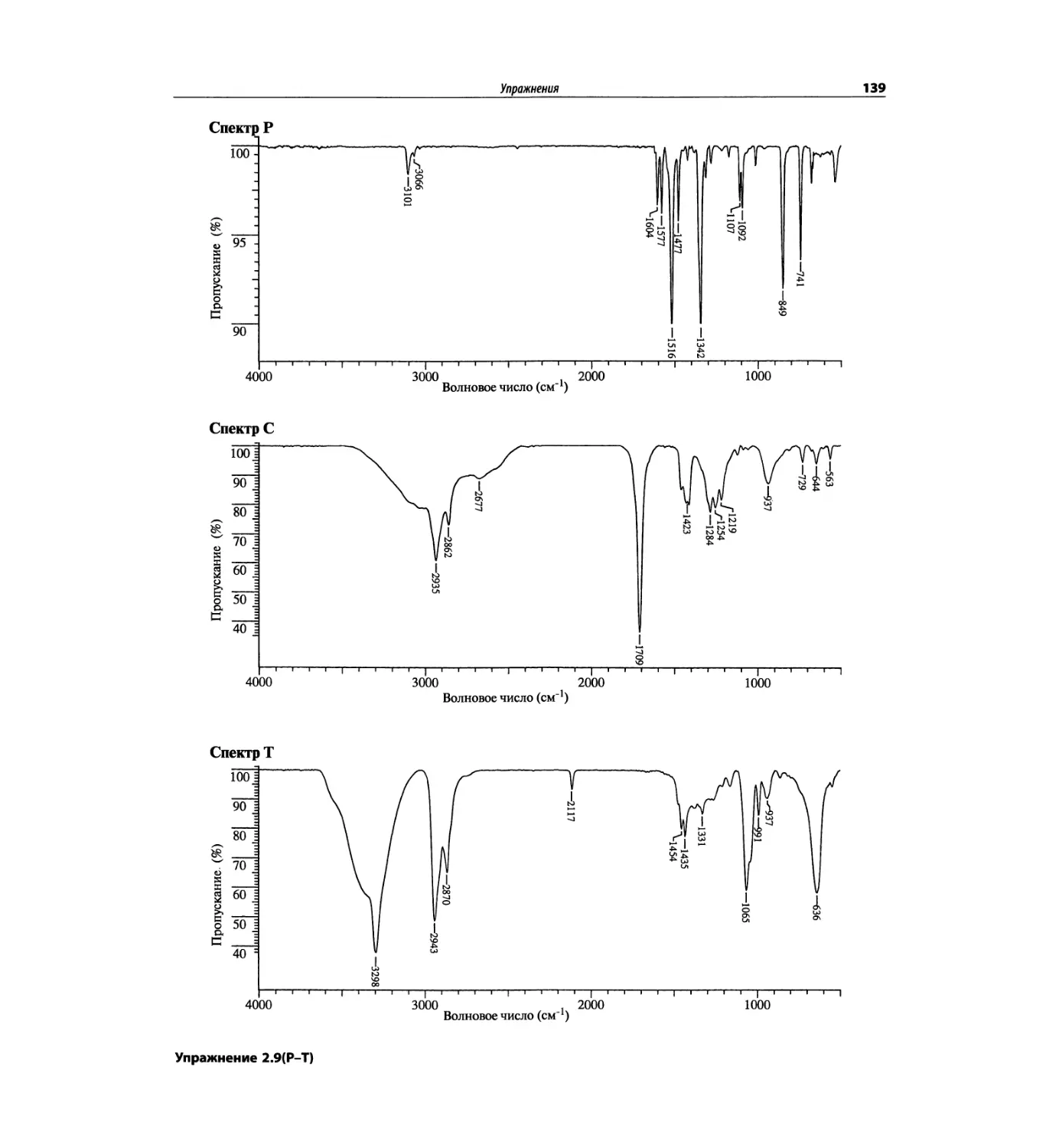
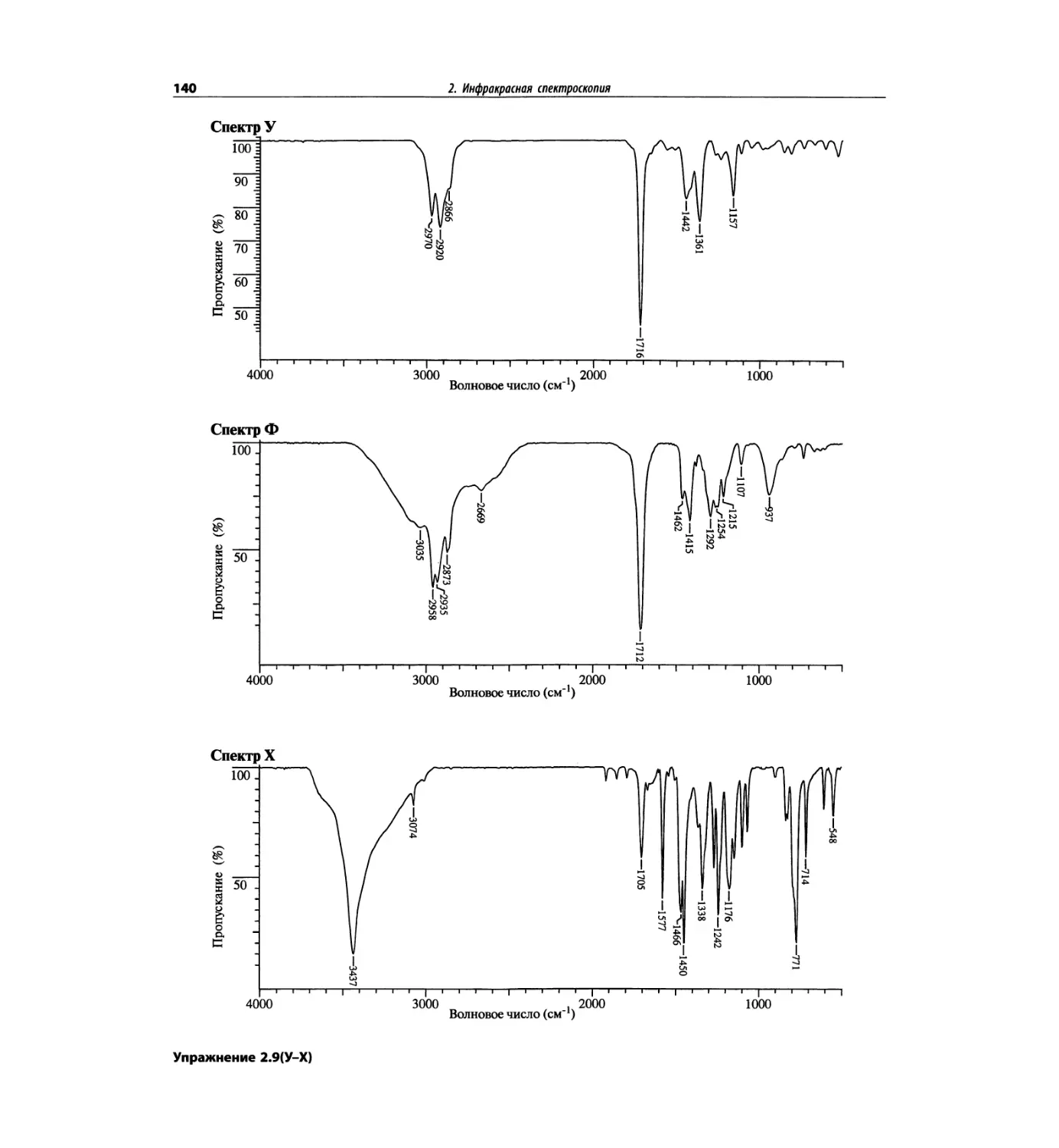
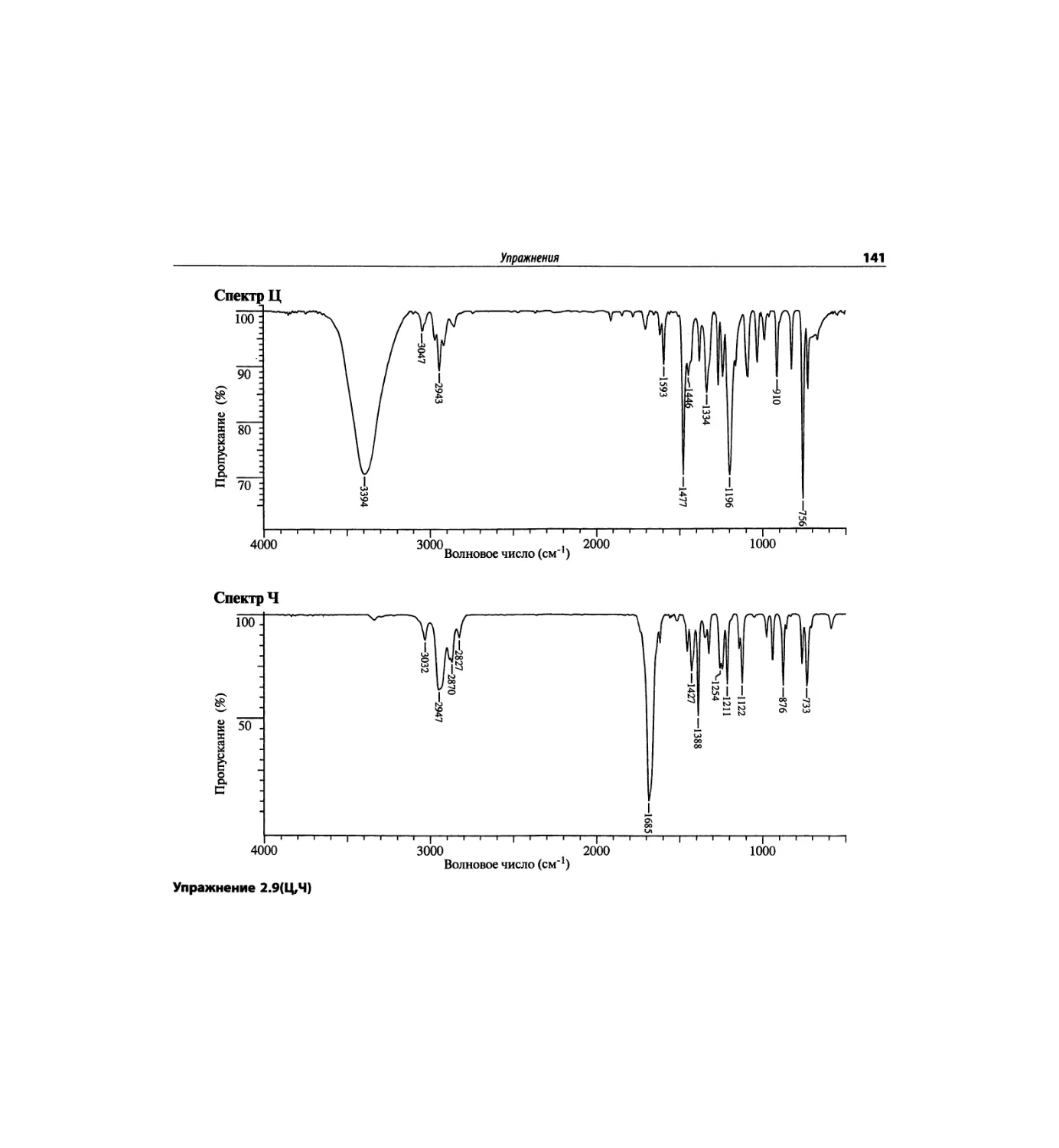
Инфракрасная спектроскопия (глава 2)
По-прежнему важно, чтобы химик-органик достаточ-
но свободно ориентировался в теоретических осно-
вах метода ИК-спектроскопии и был хорошо знаком
с современными спектрометрами. Мы уверены, что
изложение концепции характеристических групп, диа-
граммы, характеристические спектры, литературные
ссылки и упражнения полезны для студентов. Боль-
шинство ИК-спектров обновлены.
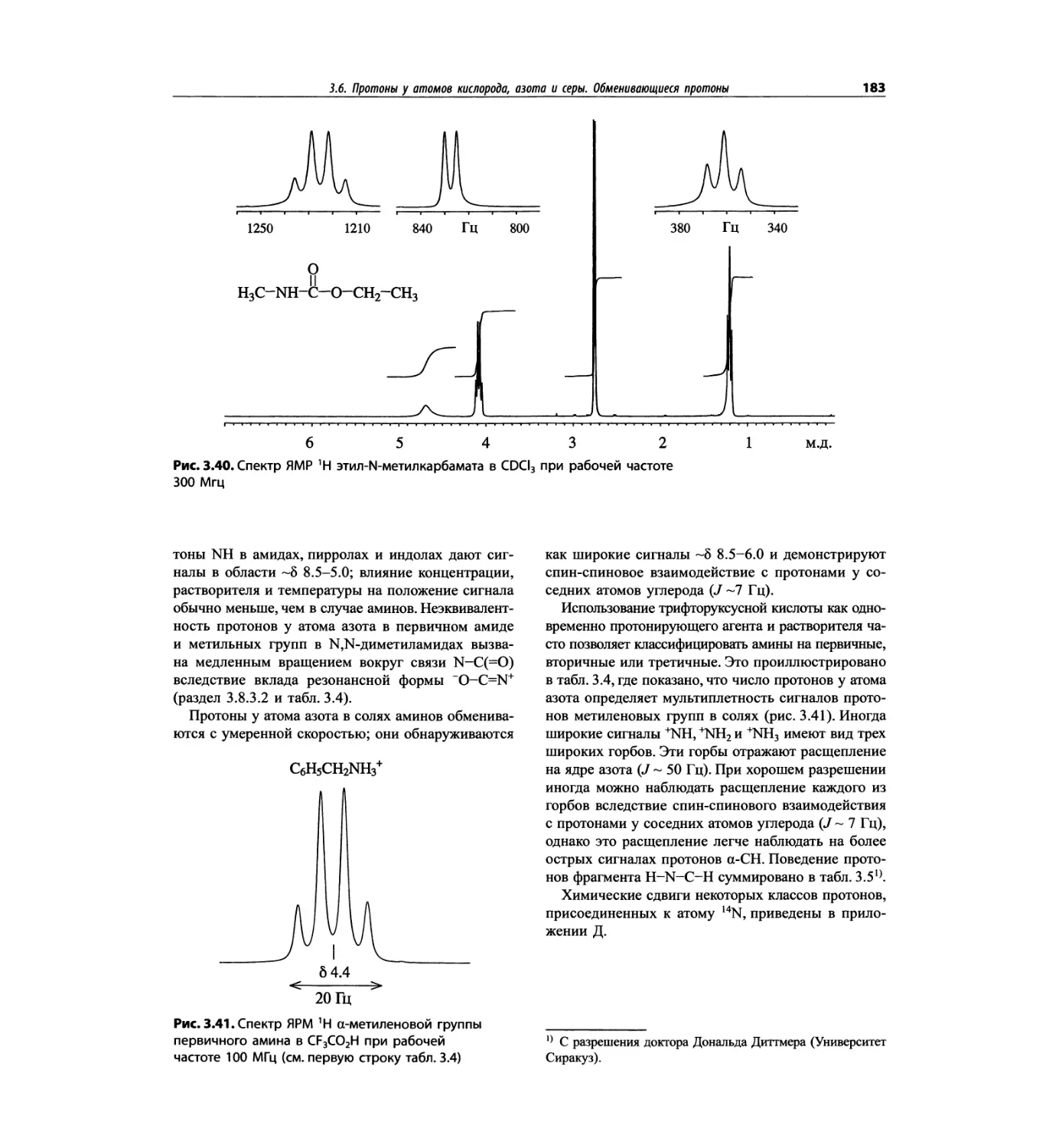
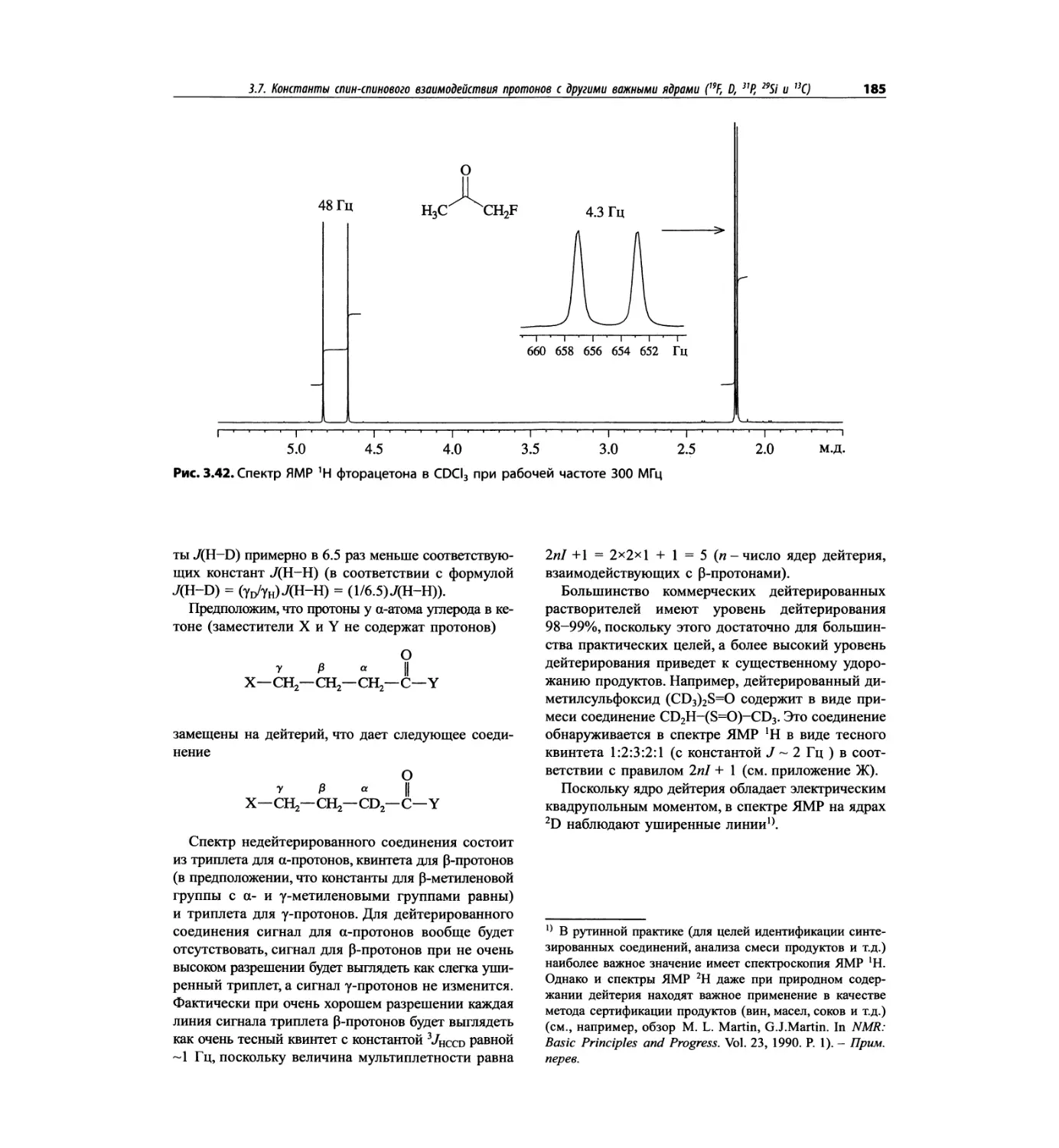
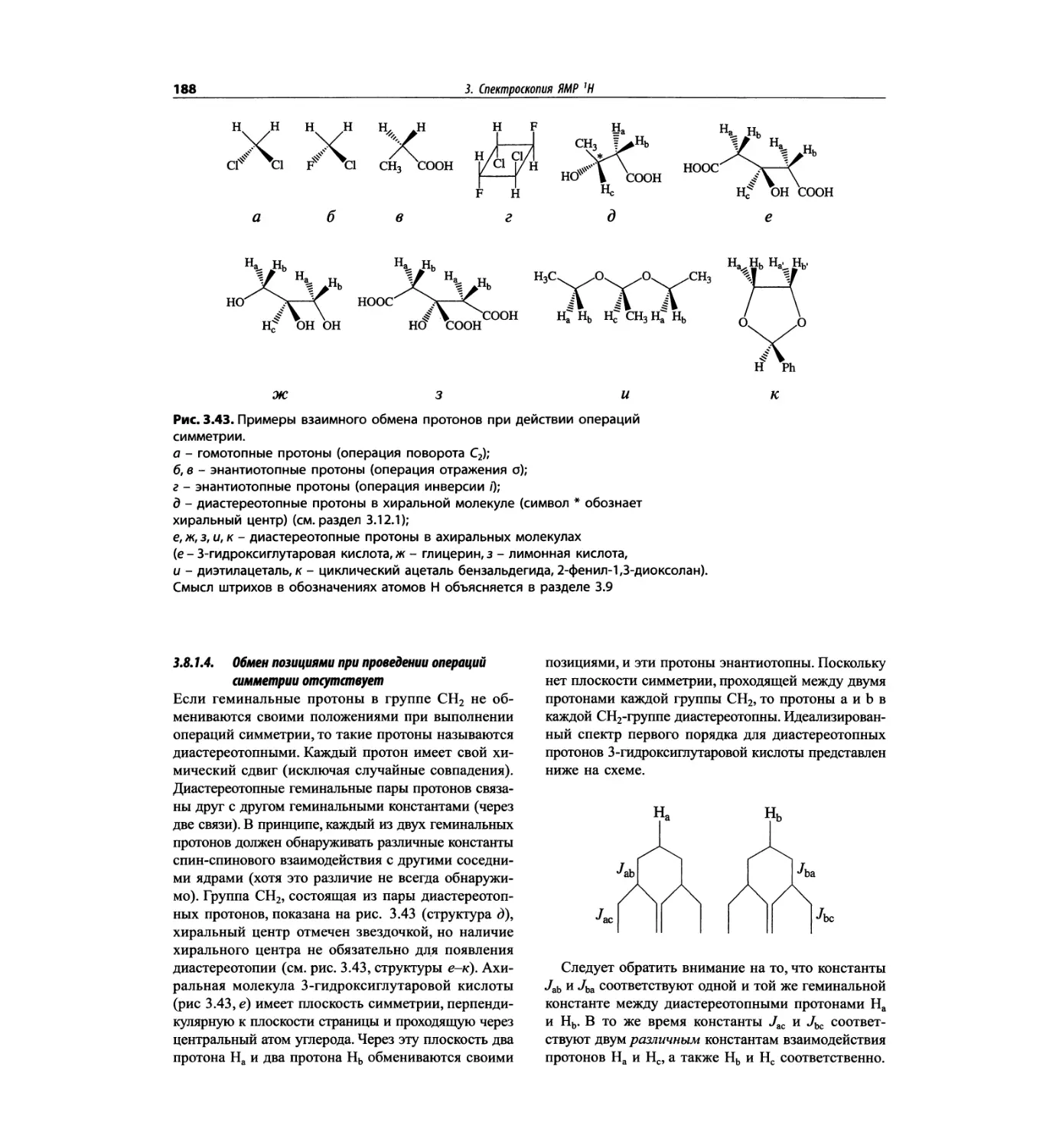
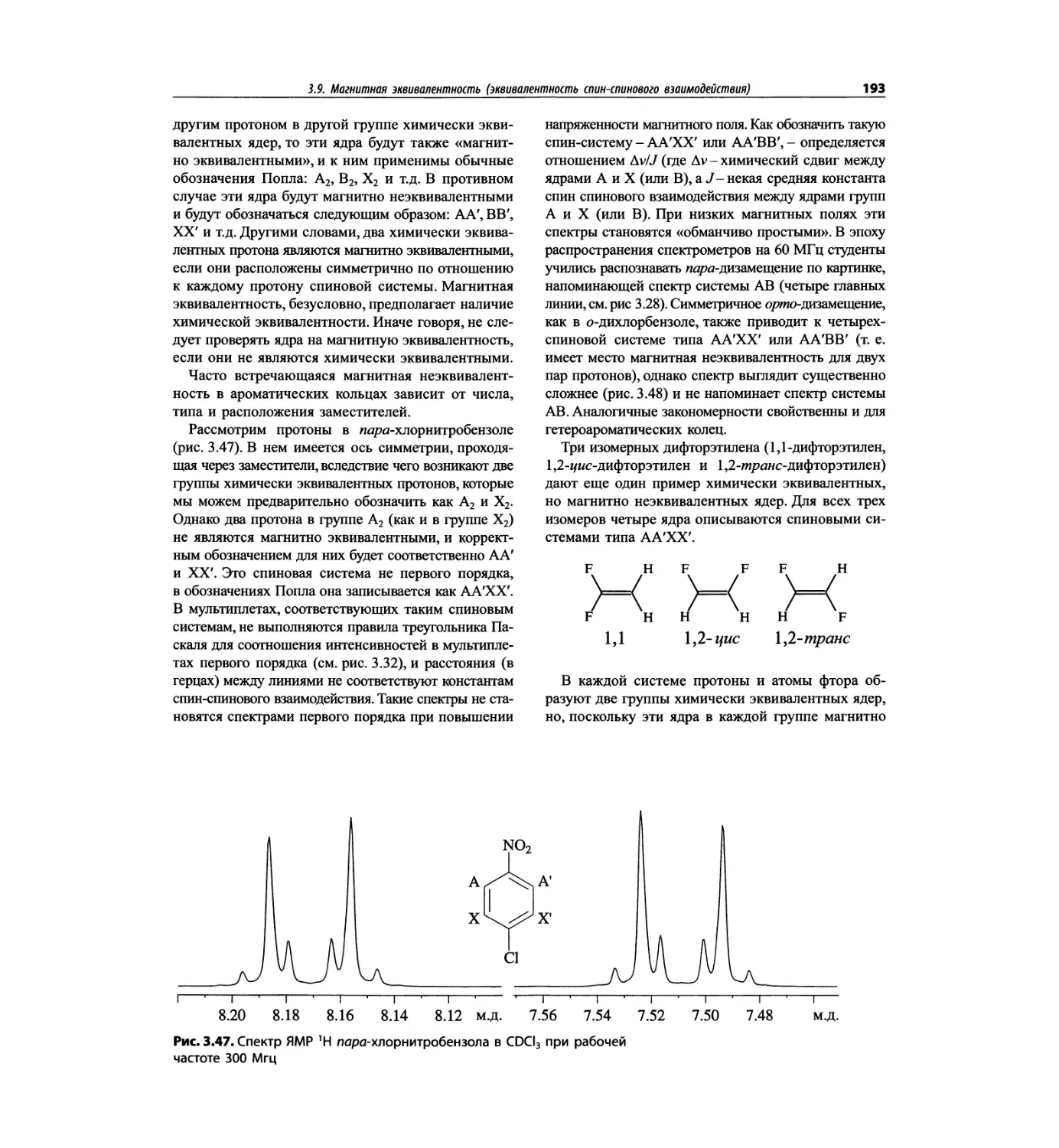
Спектроскопия ЯМР 'Н (глава 3)
В этой главе изложены общие основы спектроскопии
ядерного магнитного резонанса и более подробно - спек-
троскопия ЯМР на протонах. Главная цель - интерпре-
тация протонных спектров. Метод ЯМР с самого начала
развивался прежде всего как ЯМР *Н; и современные
достижения во многом касаются ЯМР протонов.
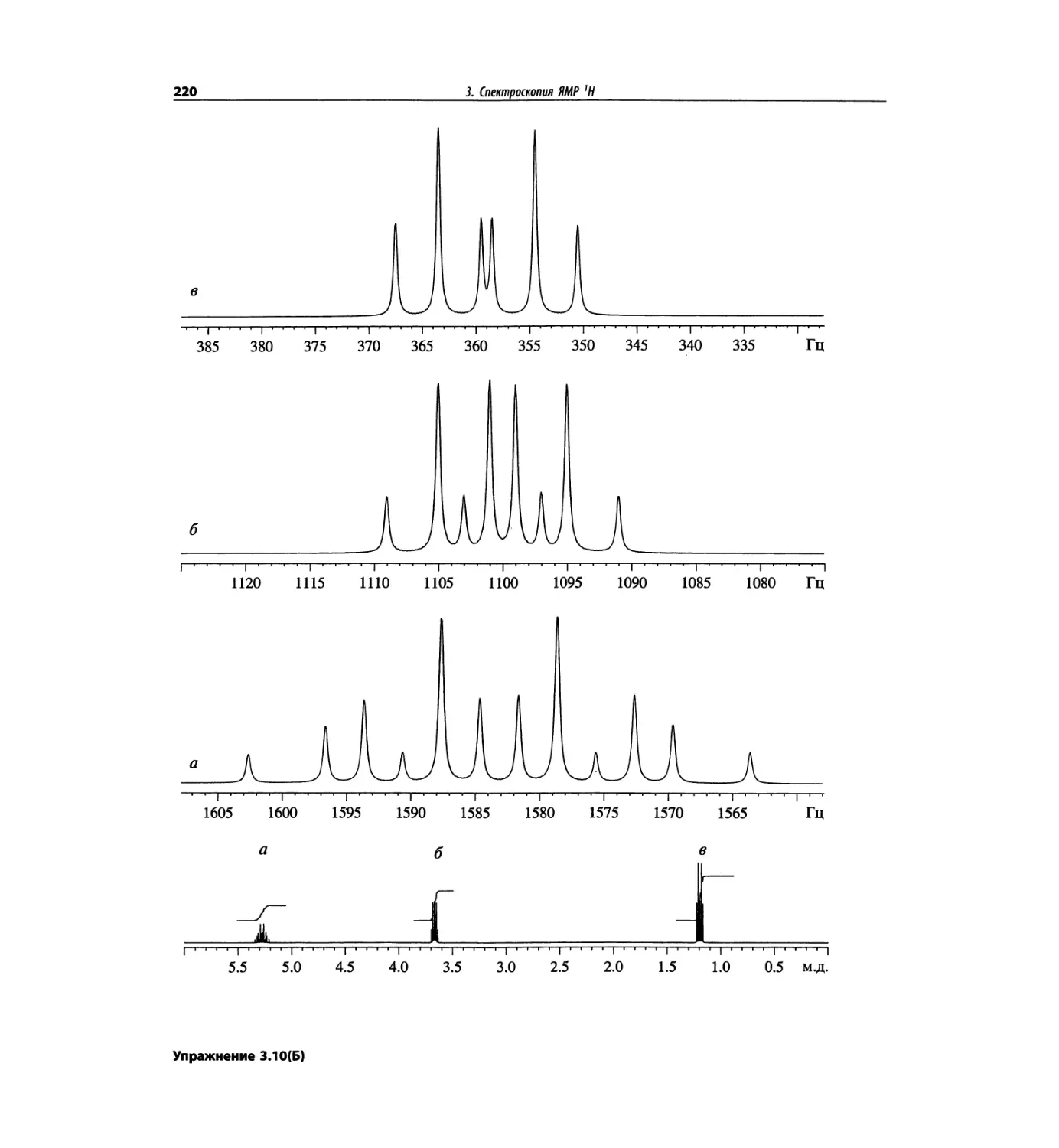
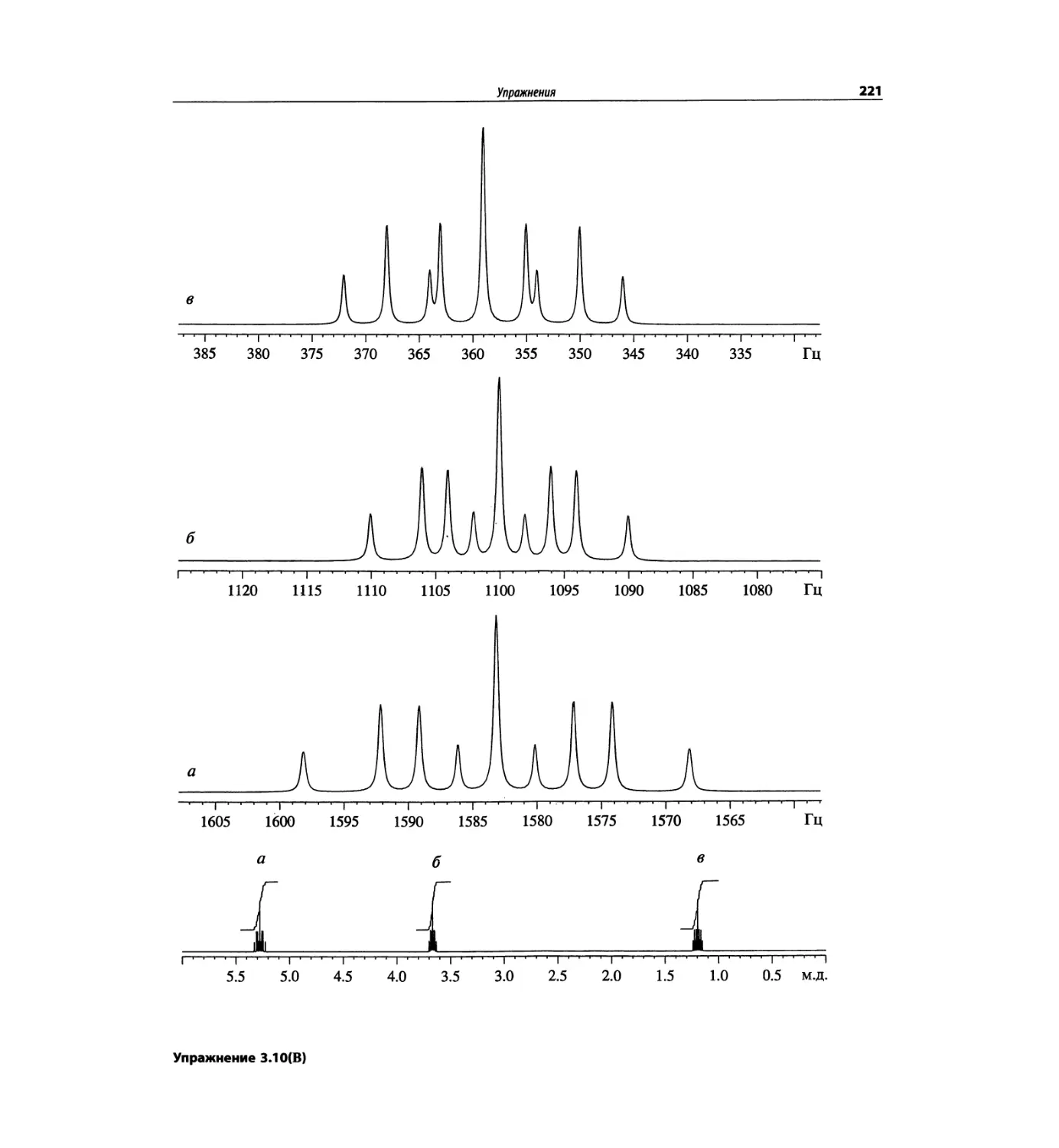
Чтобы не говорить о 17 разделах этой главы, просто
отметим, что она значительно расширена и совершенно
переделана. Акцент сделан на ЯМР с преобразовани-
ем Фурье, особенно, на теорию метода. Обновлена
большая часть рисунков и добавлены новые, включая
спектры на рабочей частоте 600 МГц. Для охвата
всего обсуждаемого материала увеличено число упраж-
нений. Растянутые по частотной шкале протонные
мультиплеты должны помочь студентам осваивать
концепцию спектров первого порядка. Это важное
понятие подробно обсуждается в данной главе.
Следующее замечание касается разделения спек-
трометрии !Н и 13С по главам 3 и 4. Мы считаем,
что этот принятый в предыдущих изданиях подход
является целесообразным; переходим к гл. 4.
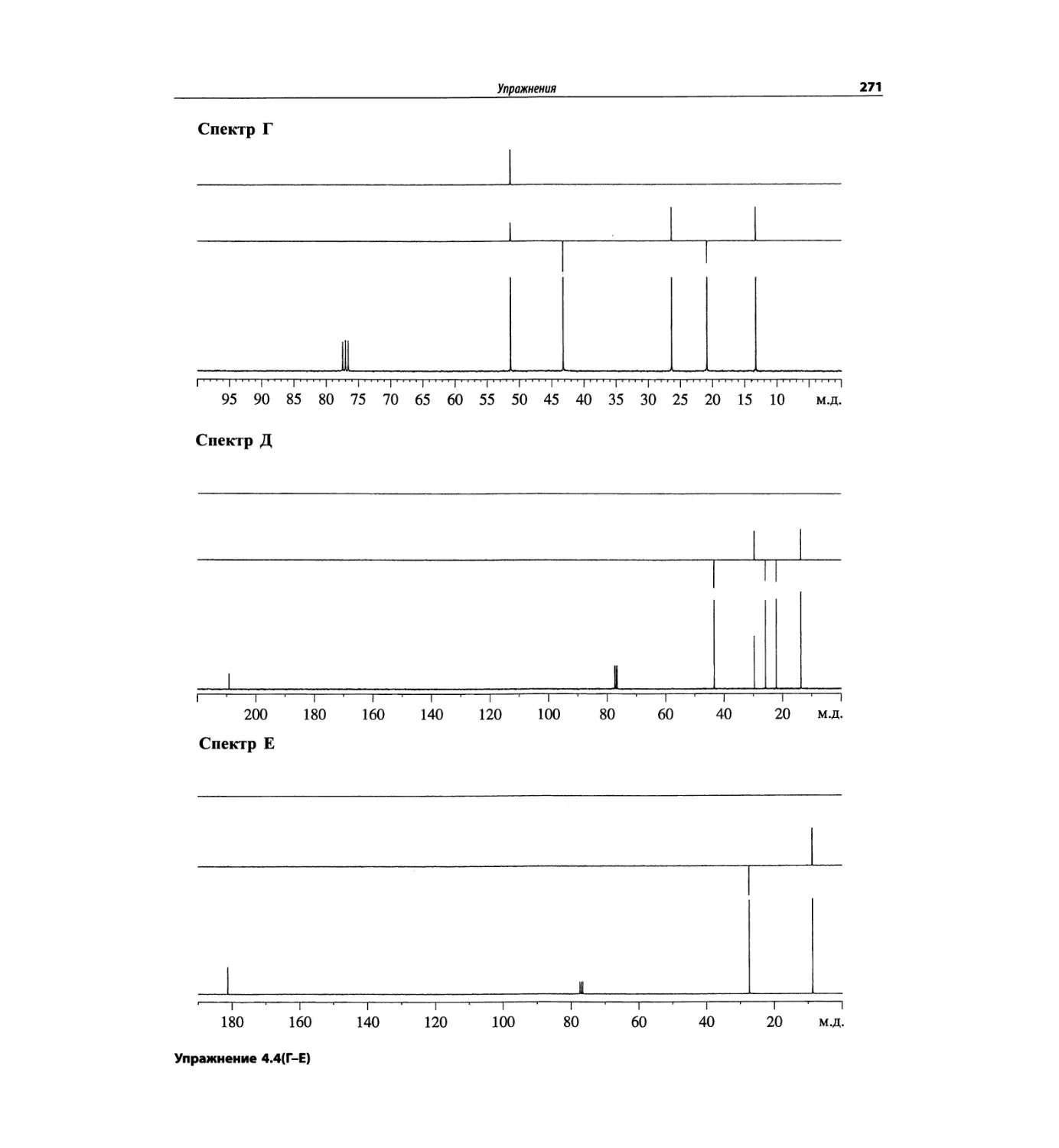
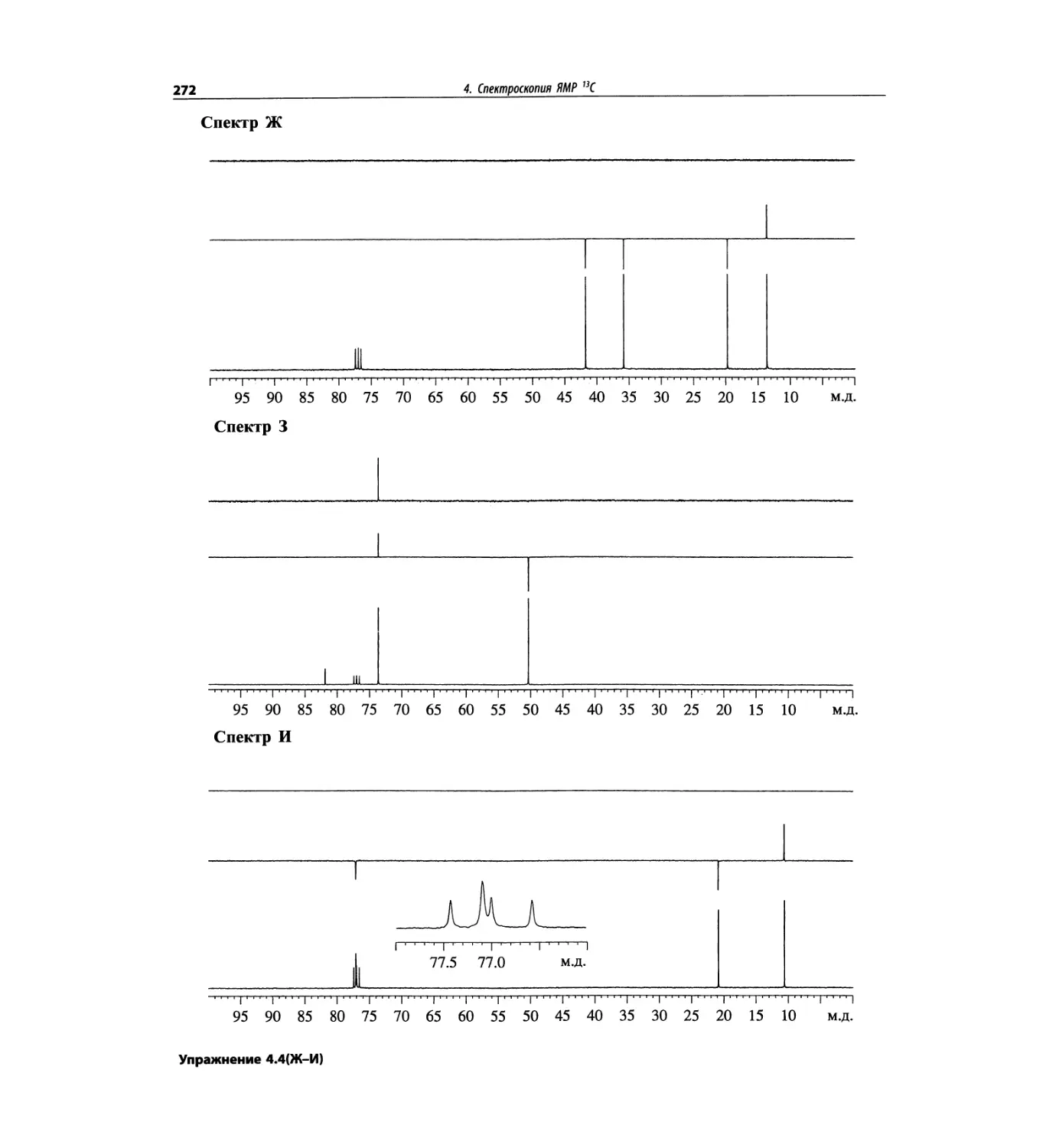
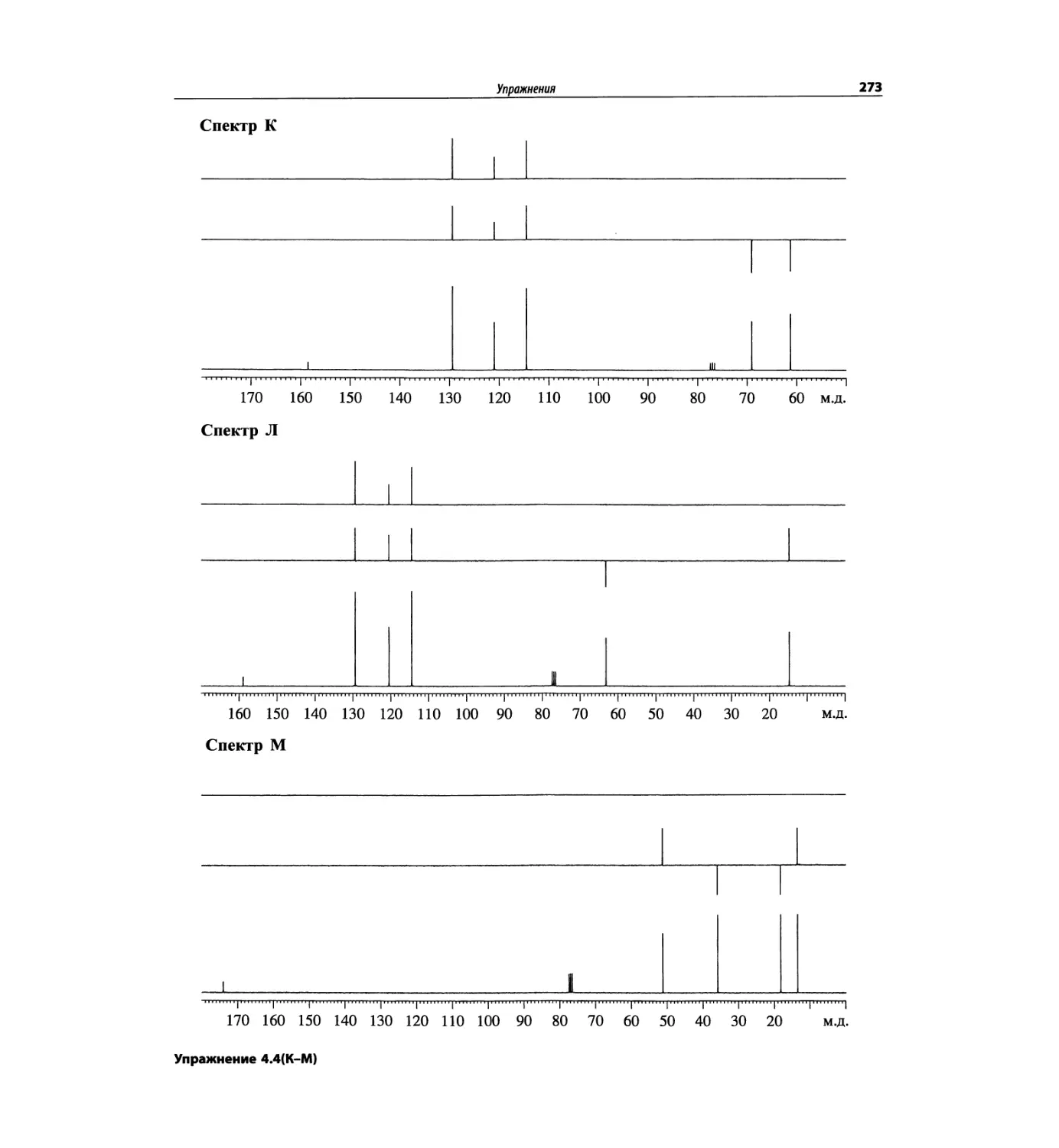
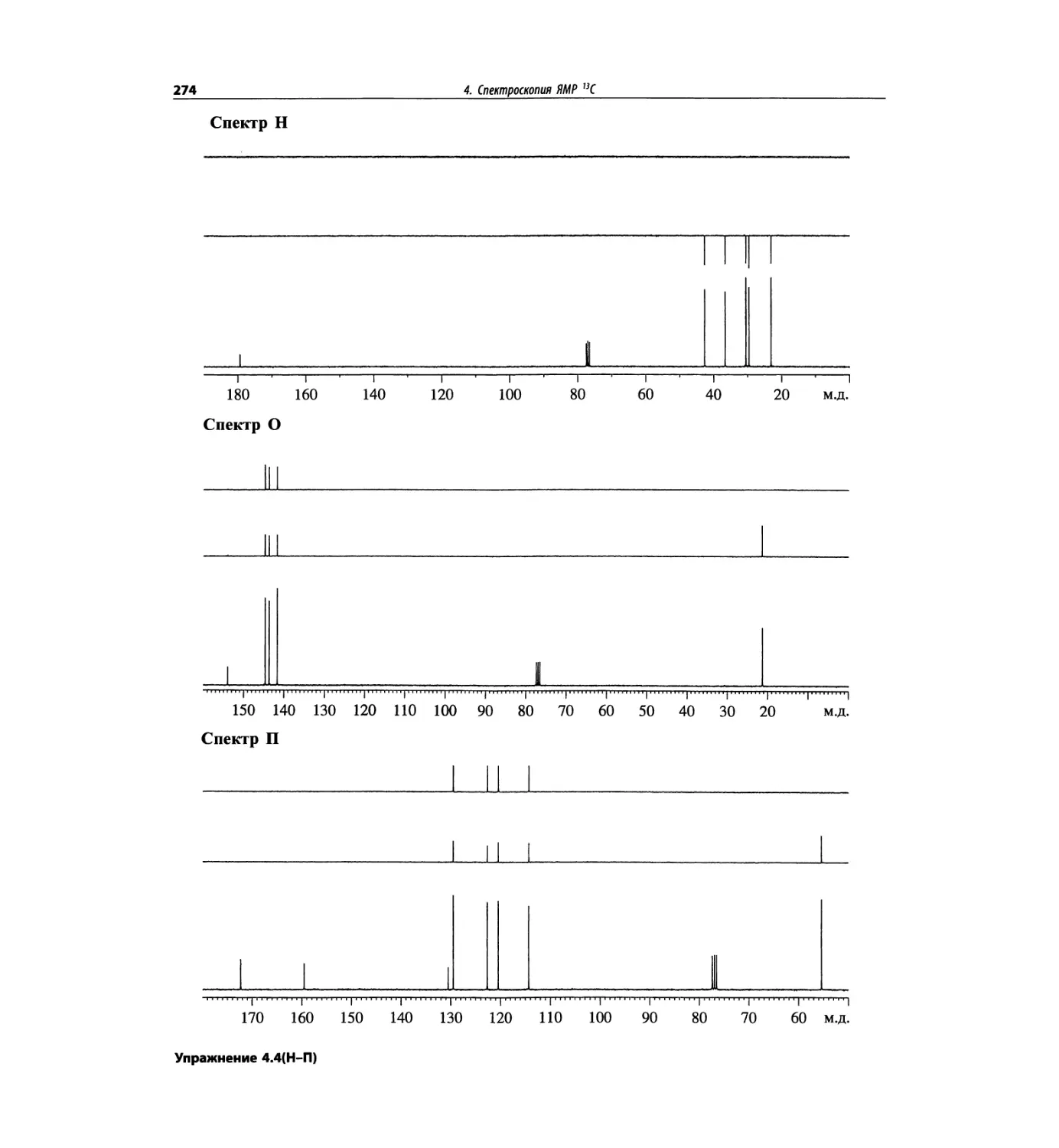
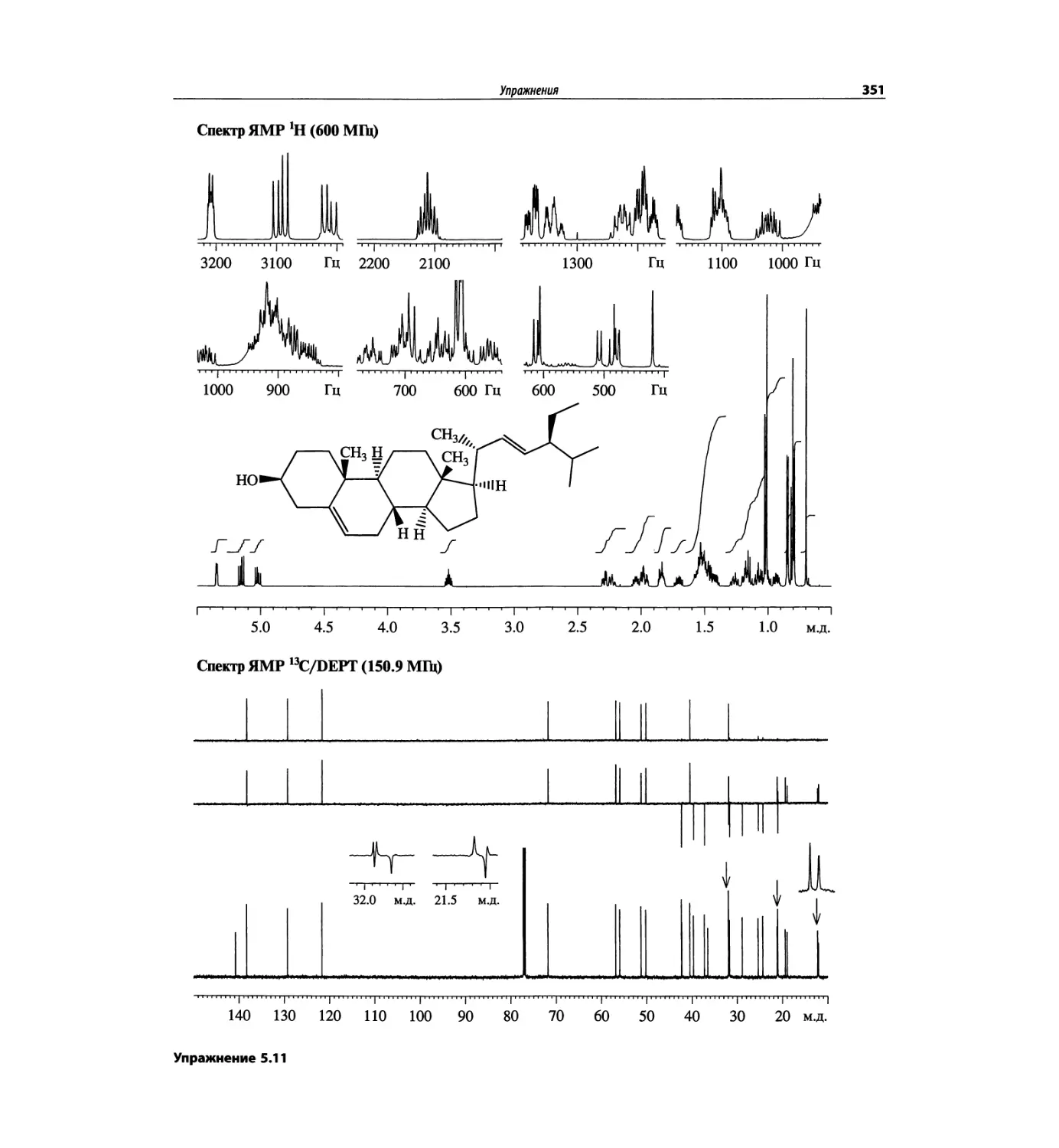
Спектроскопия ЯМР 13С (глава 4)
Эта глава также полностью пересмотрена. Все рисун-
ки обновлены, а спектры зарегистрированы на рабо-
чих частотах 75.5 МГц (что эквивалентно 300 МГц
для протонов) или 150.9 МГц (эквивалентно 600 МГц
для протонов). Расширены многие таблицы химиче-
ских сдвигов сигналов ядер 13С.
Много внимания уделено спектрам DEPT. По сути
они используются во всех упражнениях вместо вы-
шедших из употребления спектров 13С с подавле-
нием спин-спинового взаимодействия с протонами.
Спектры DEPT дают информацию о распределении
атомов углерода в соответствии с числом связанных
атомов водорода.
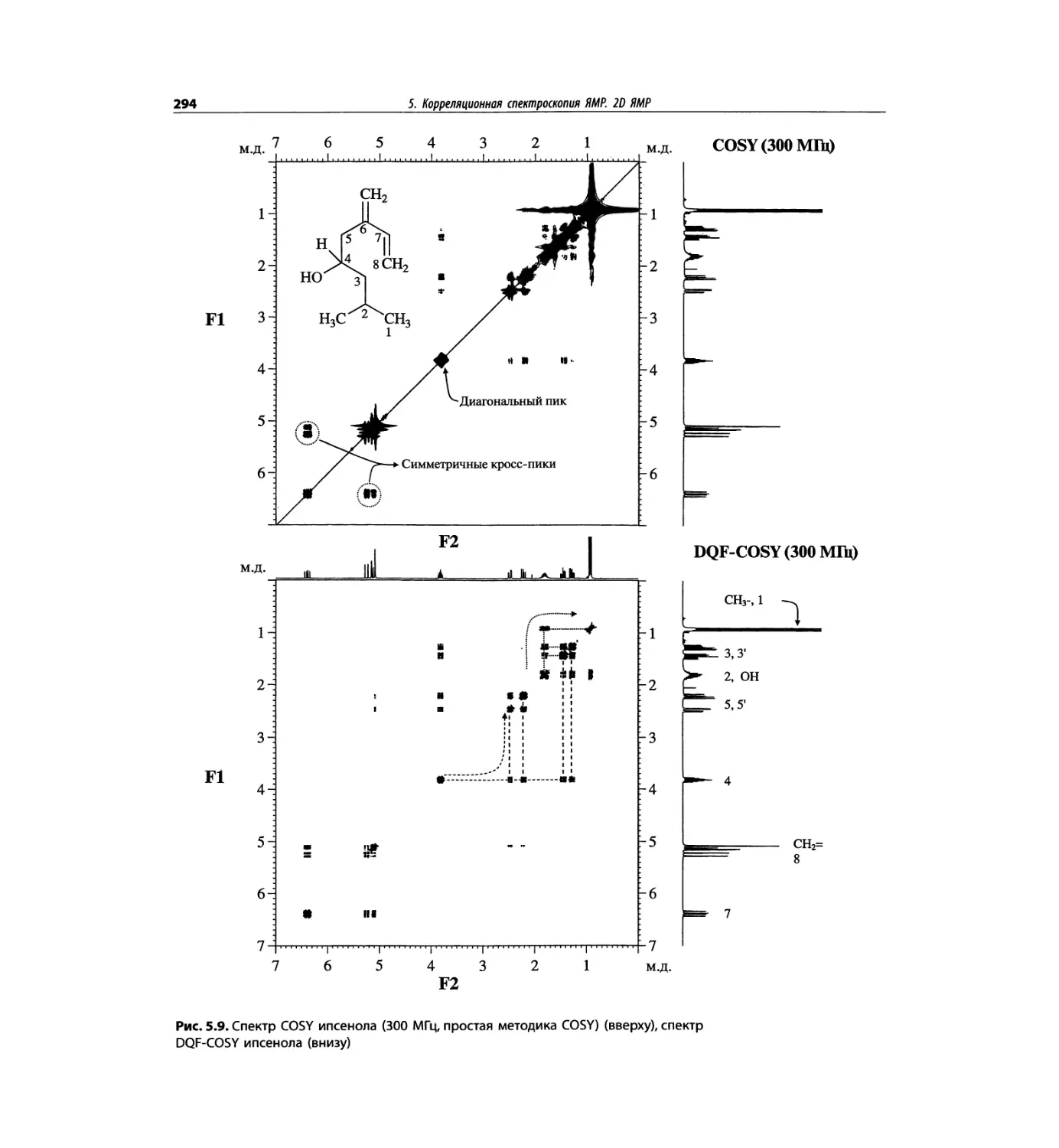
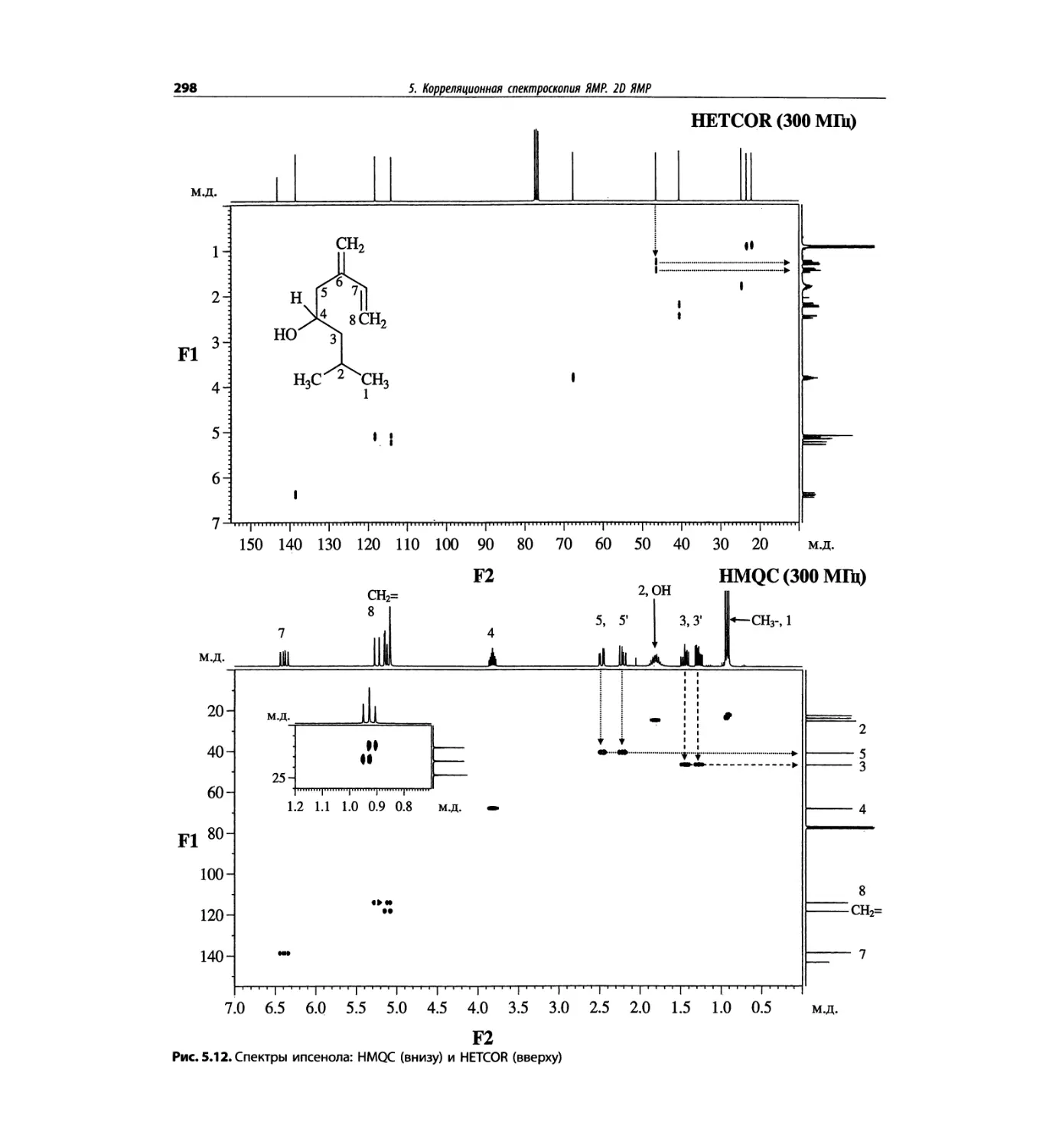
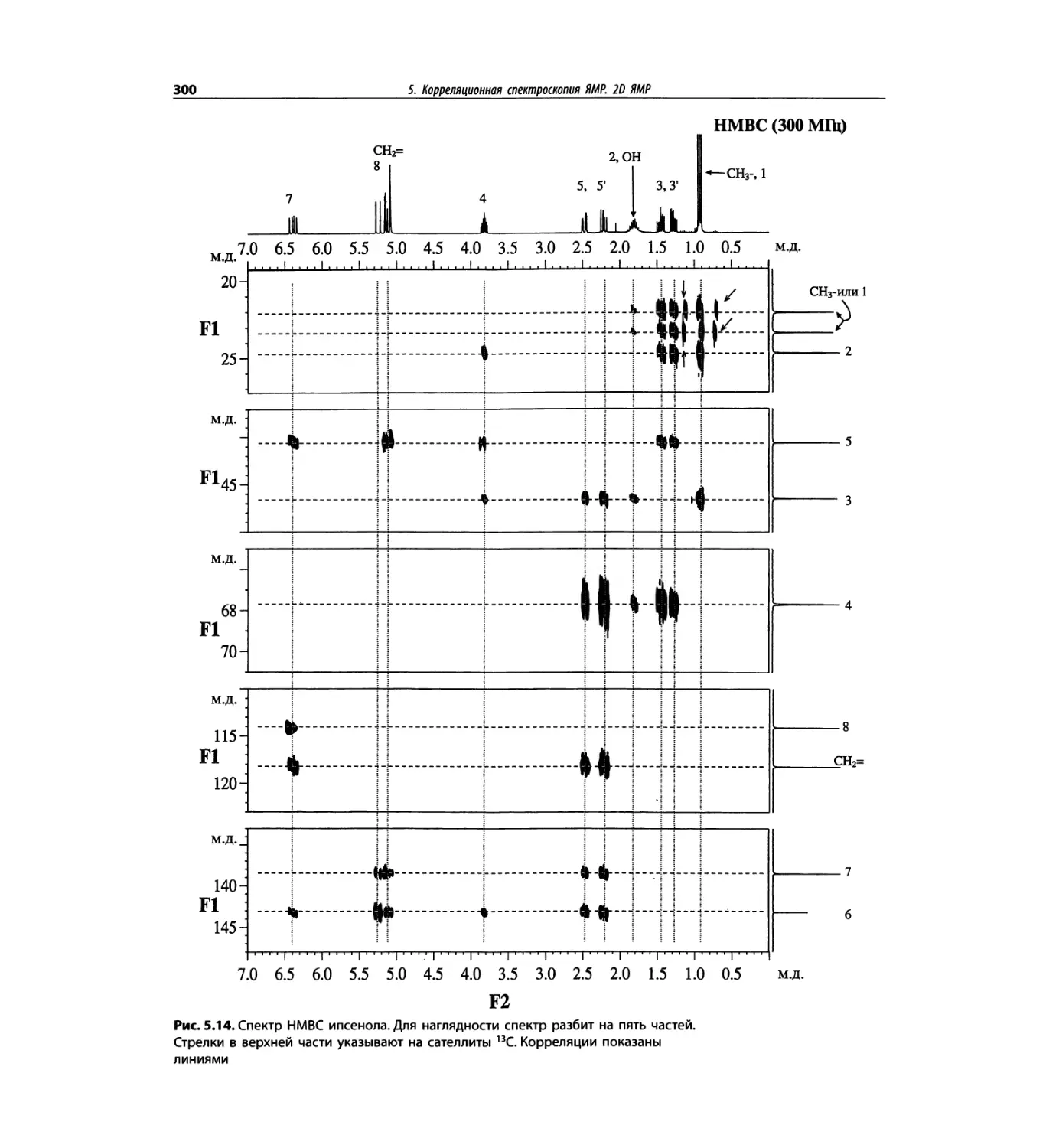
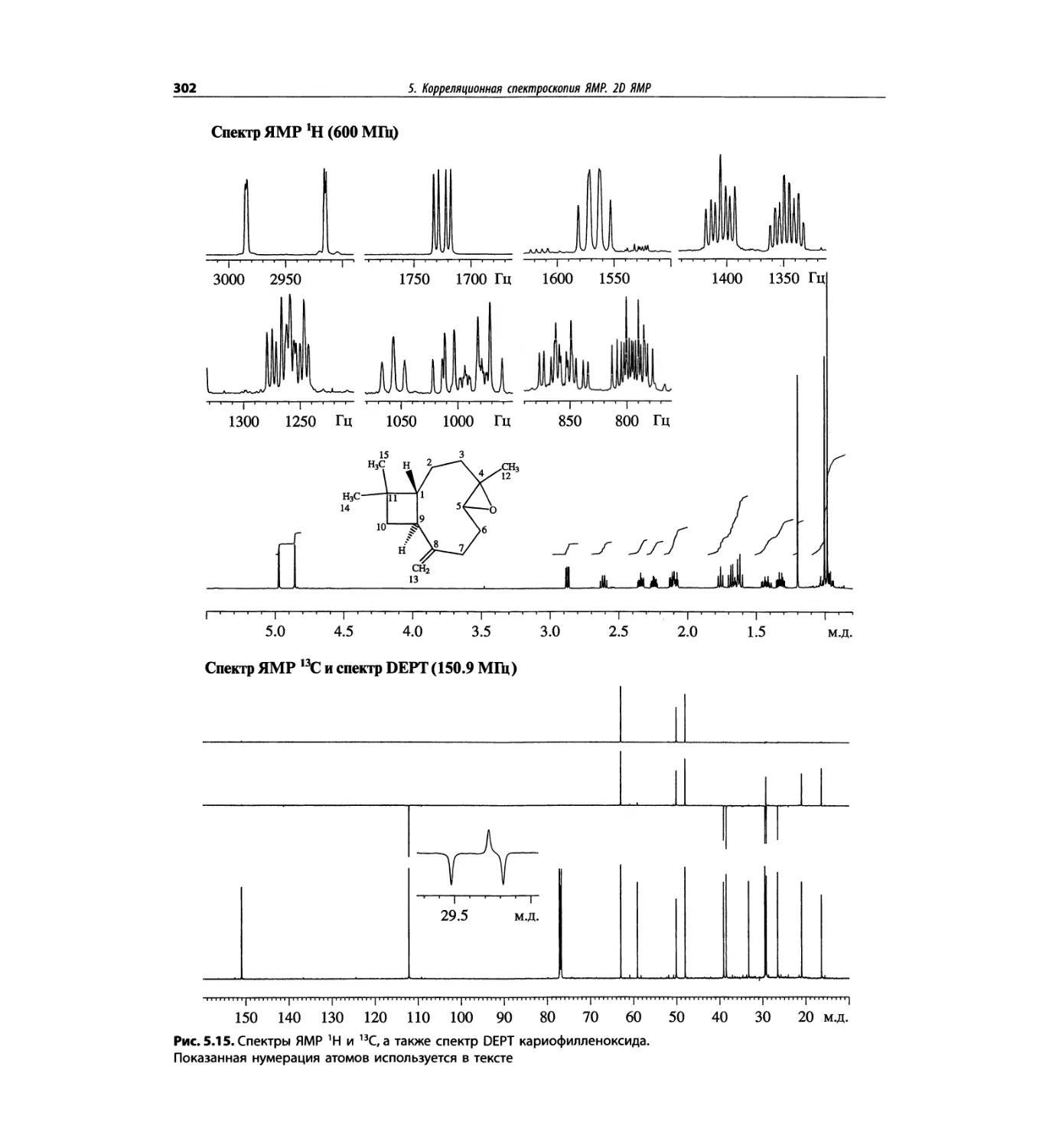
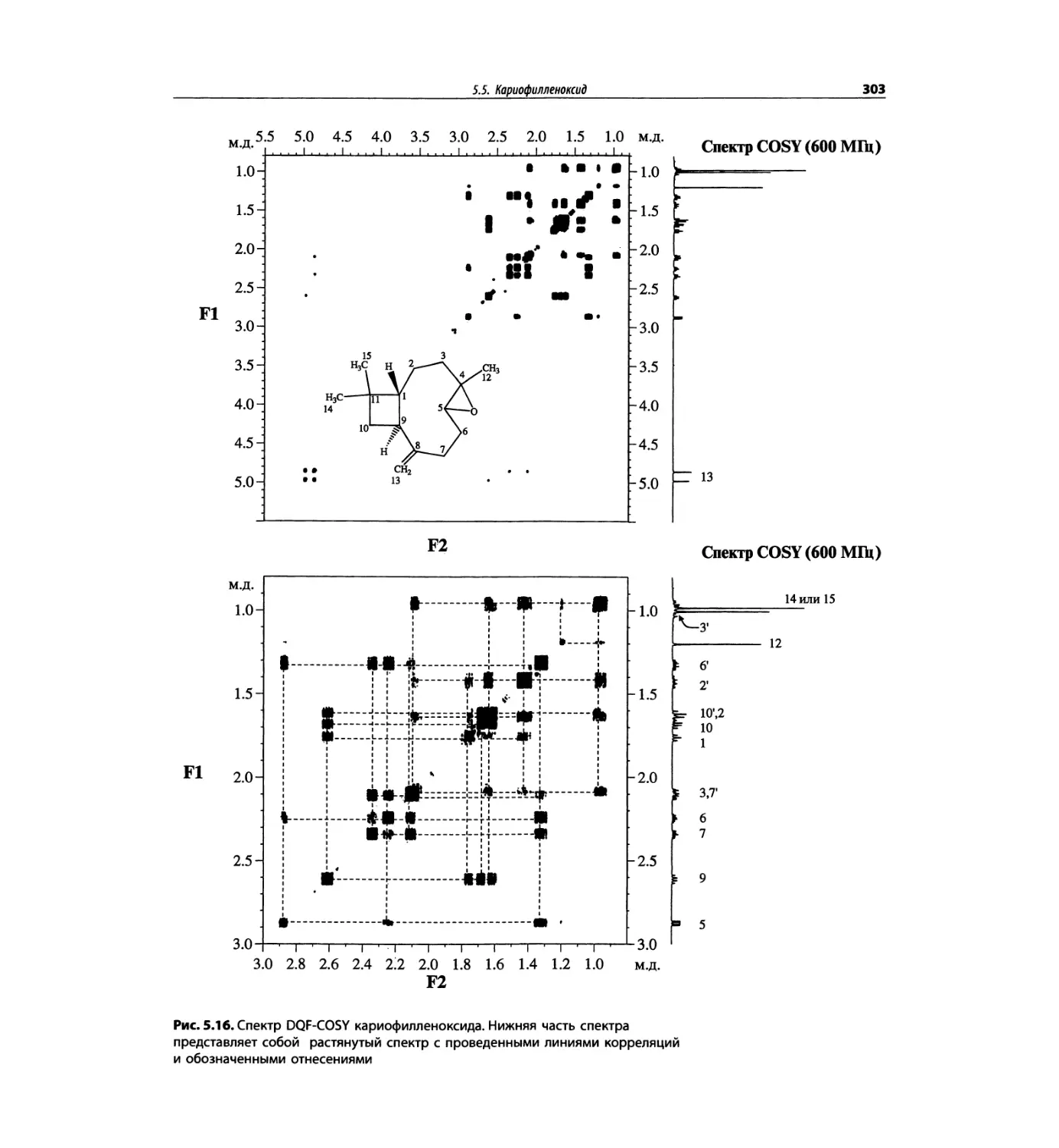
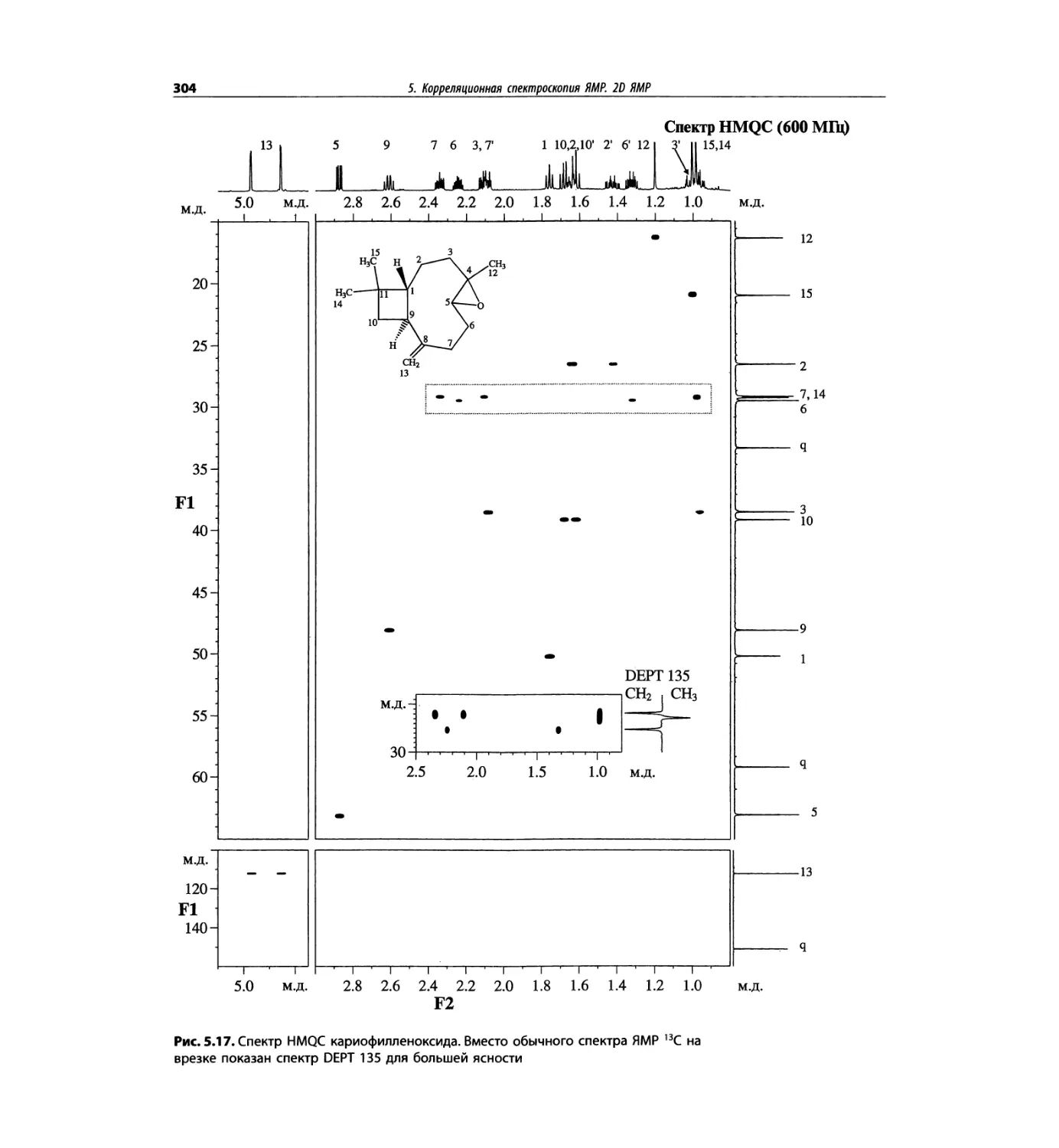
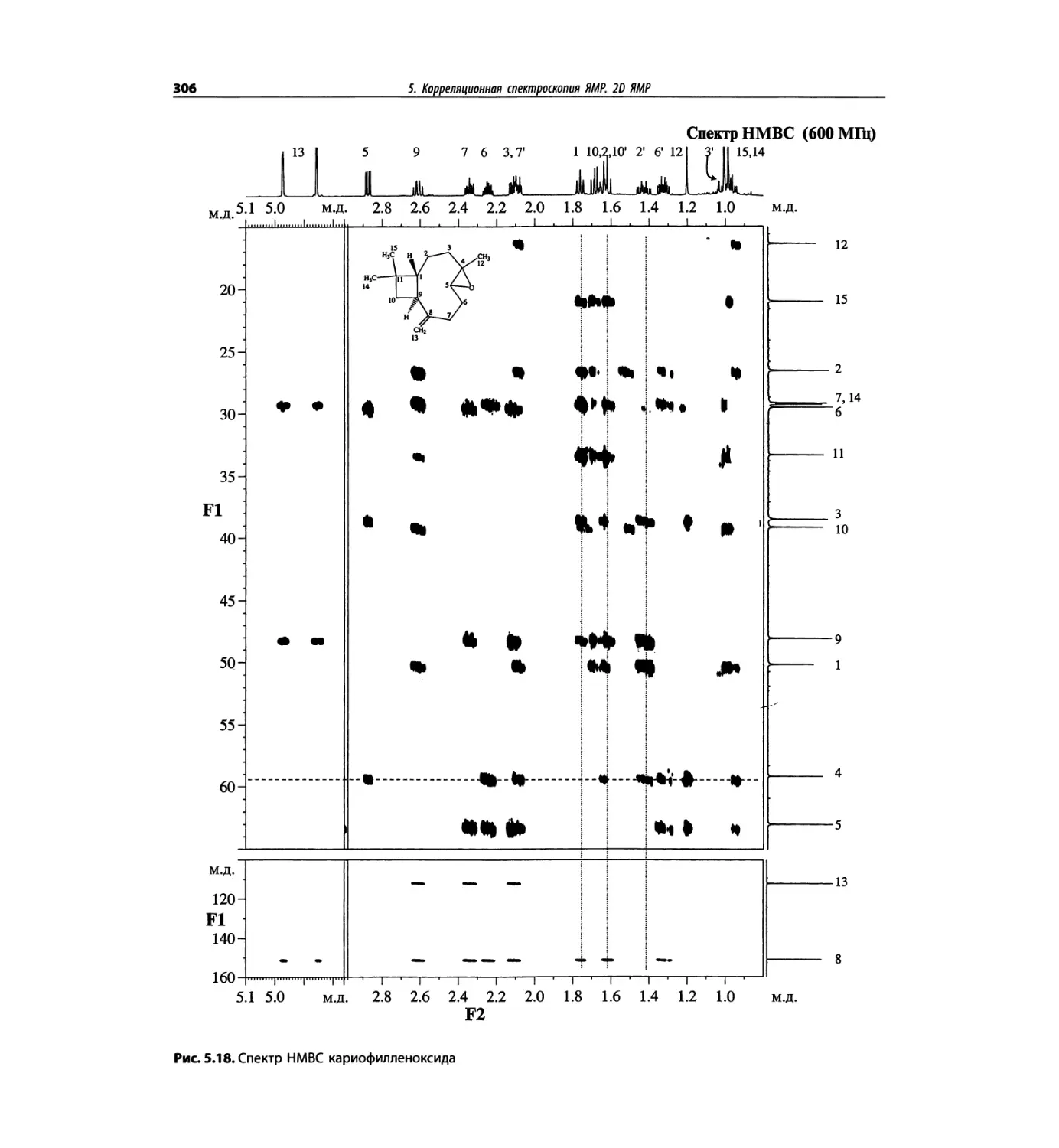
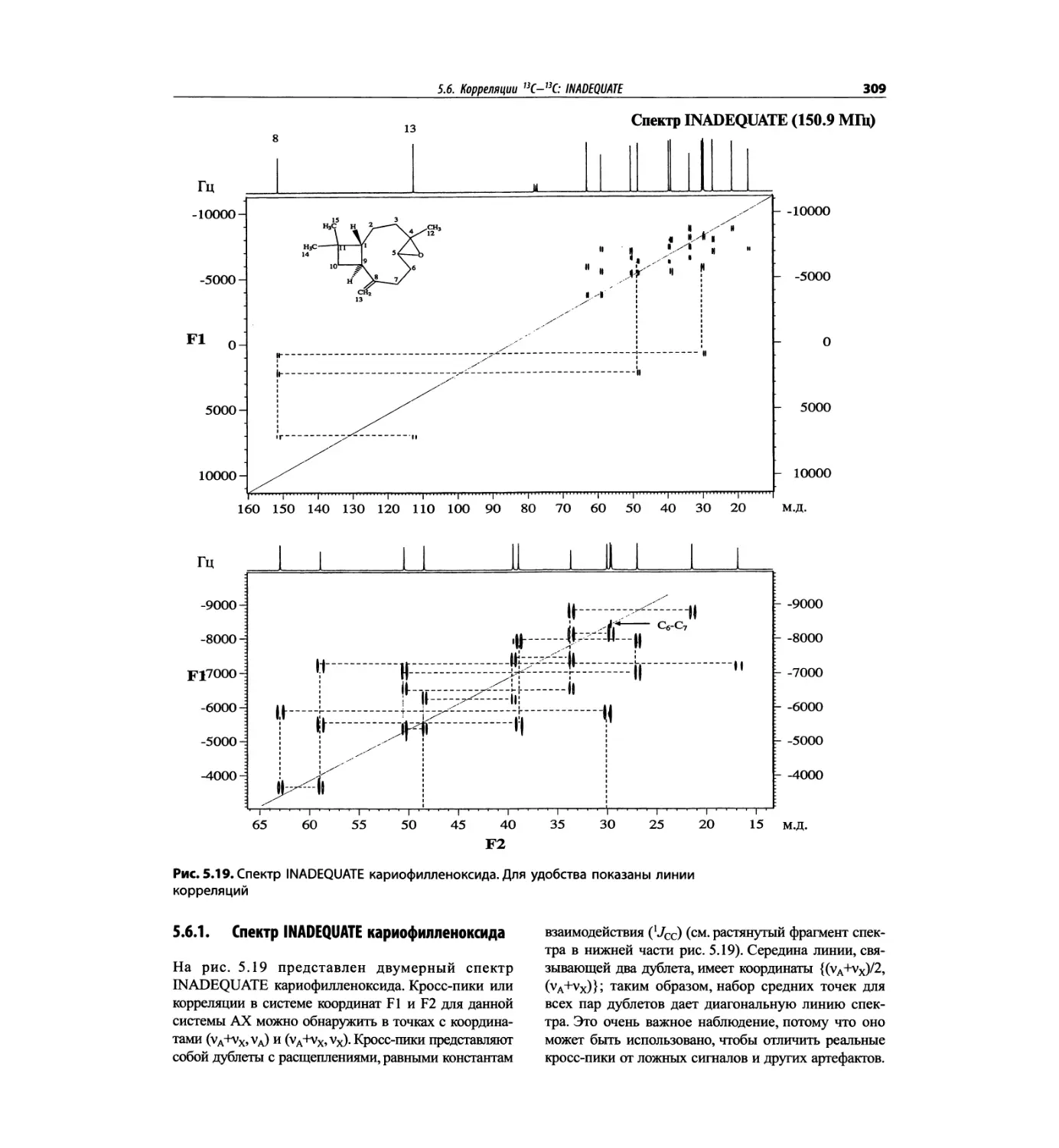
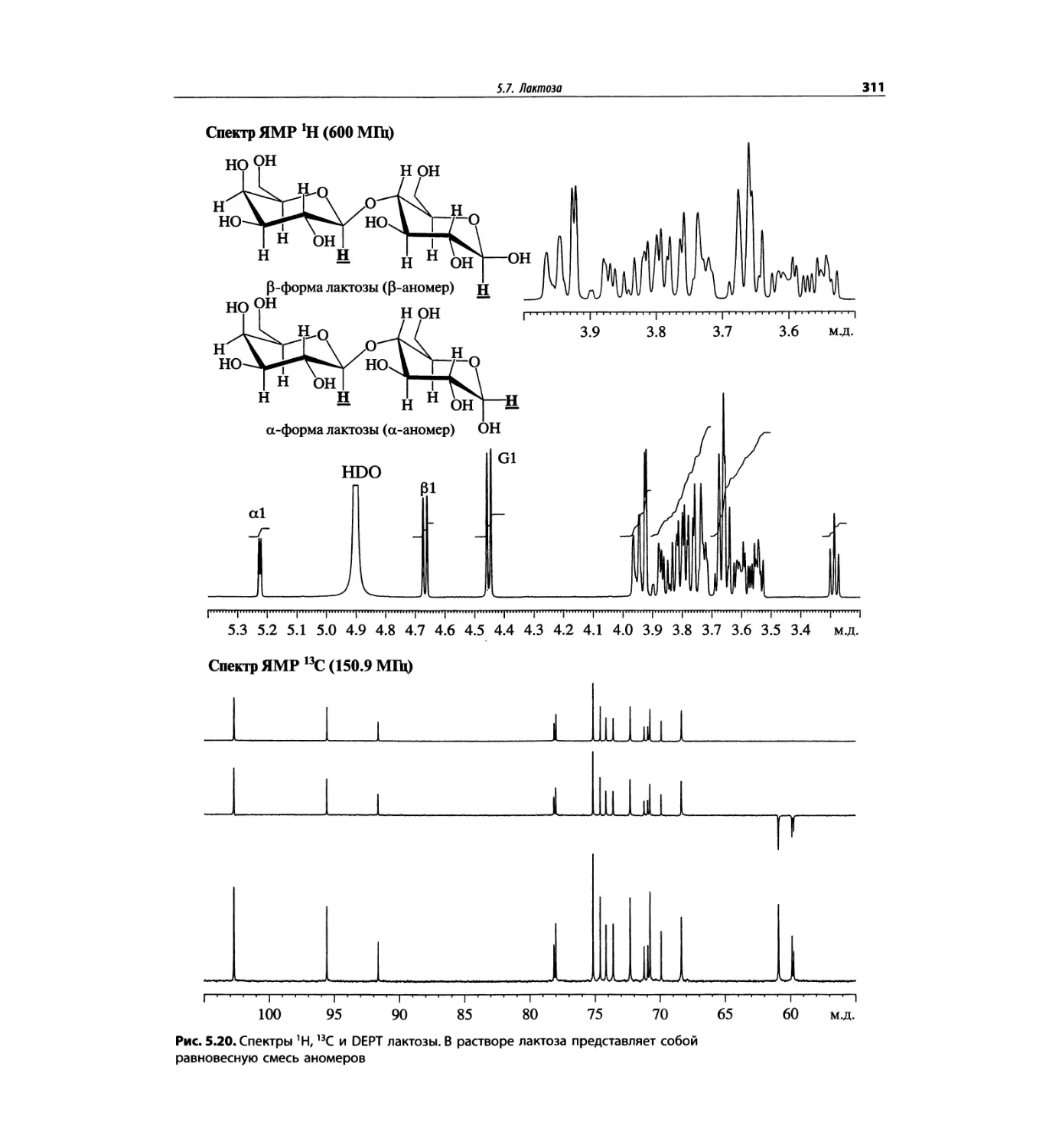
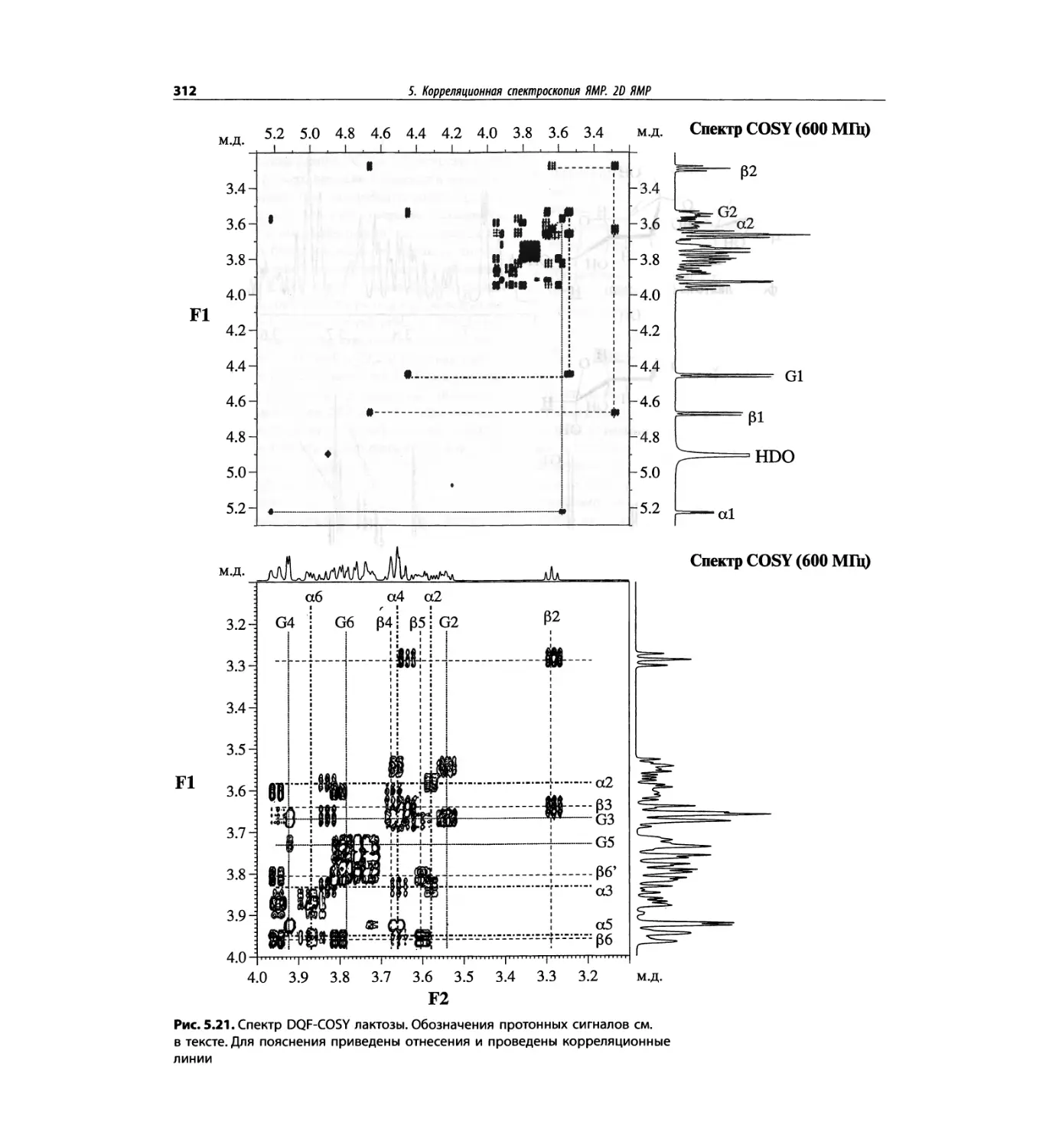
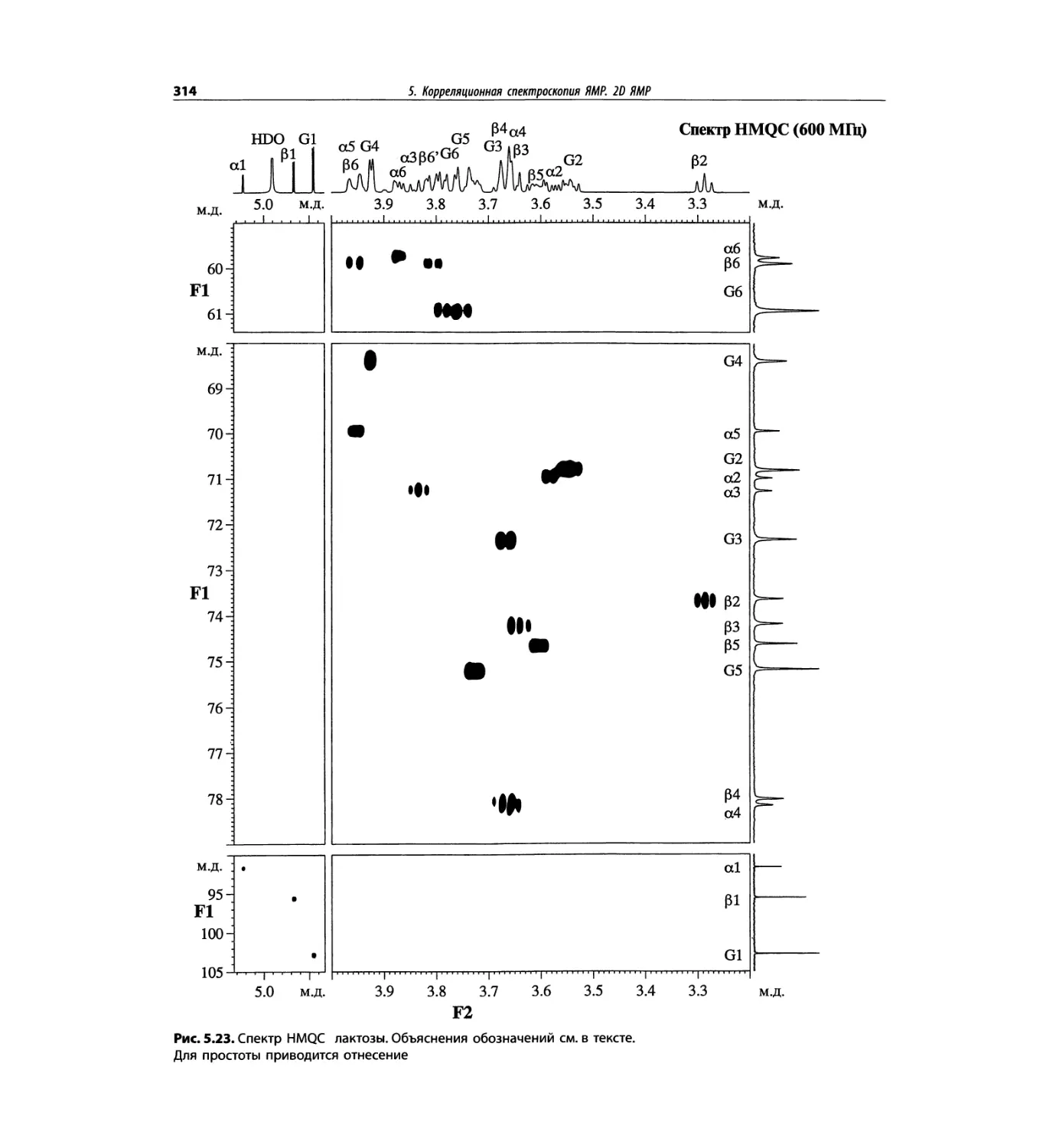
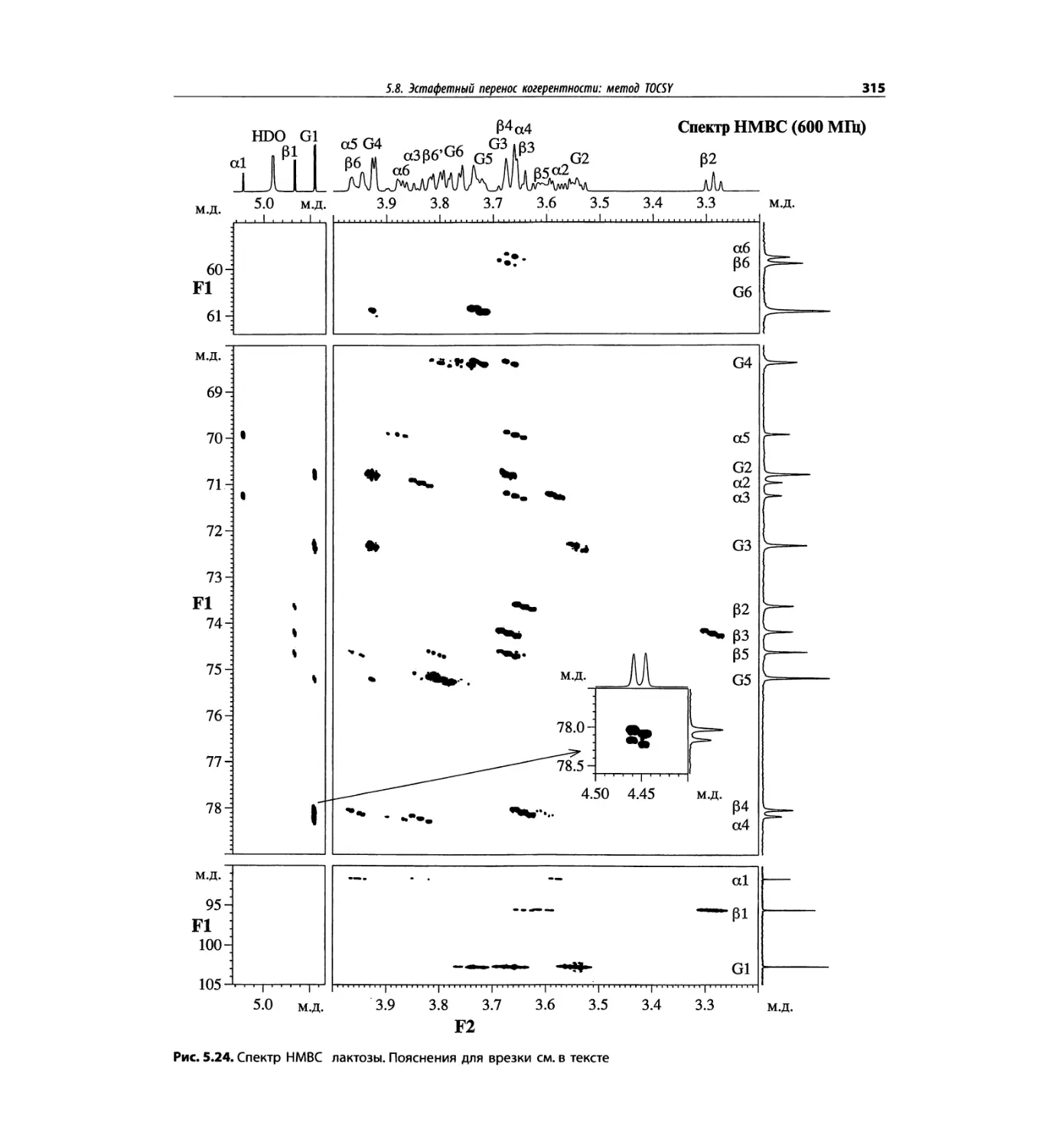
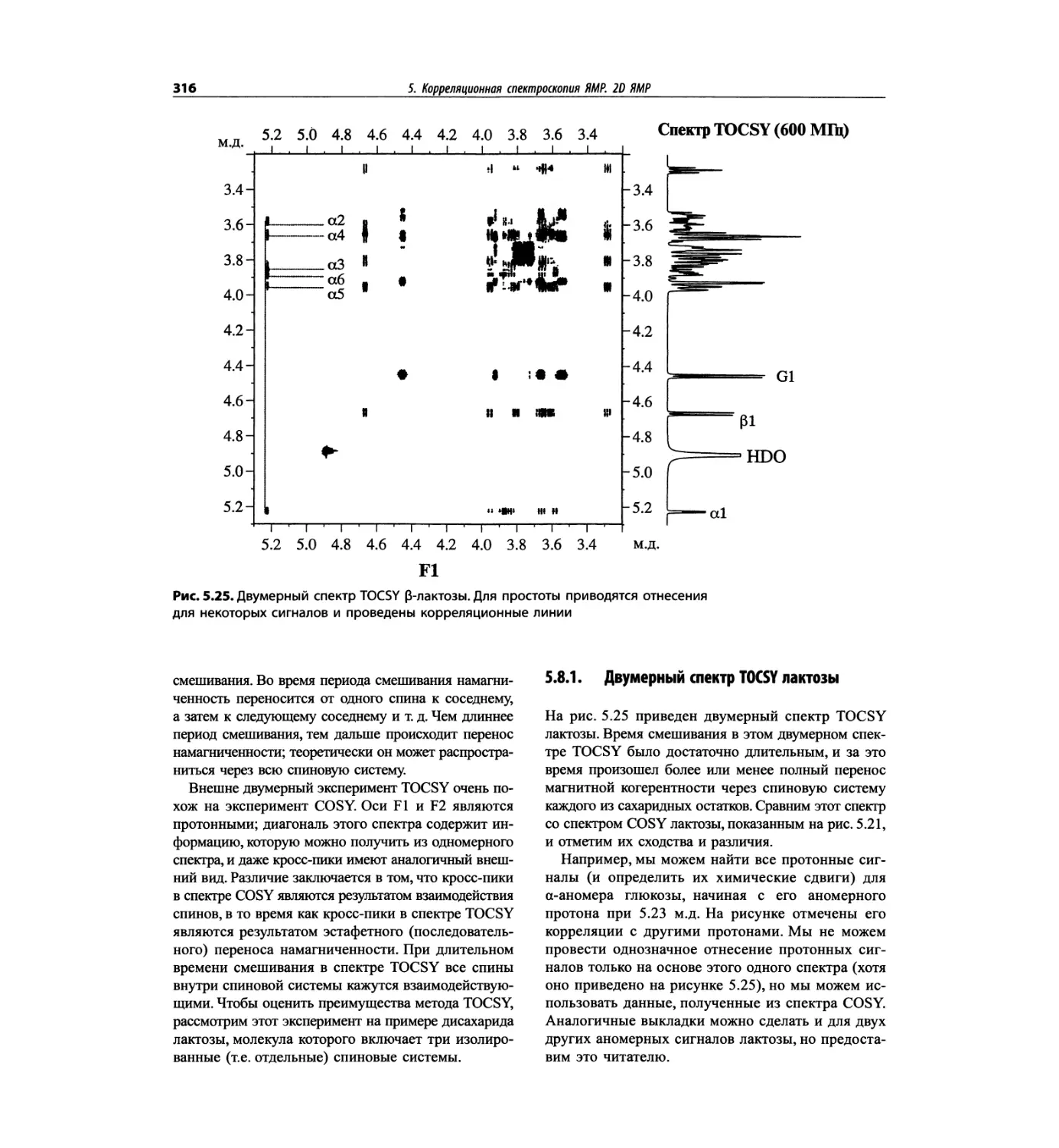
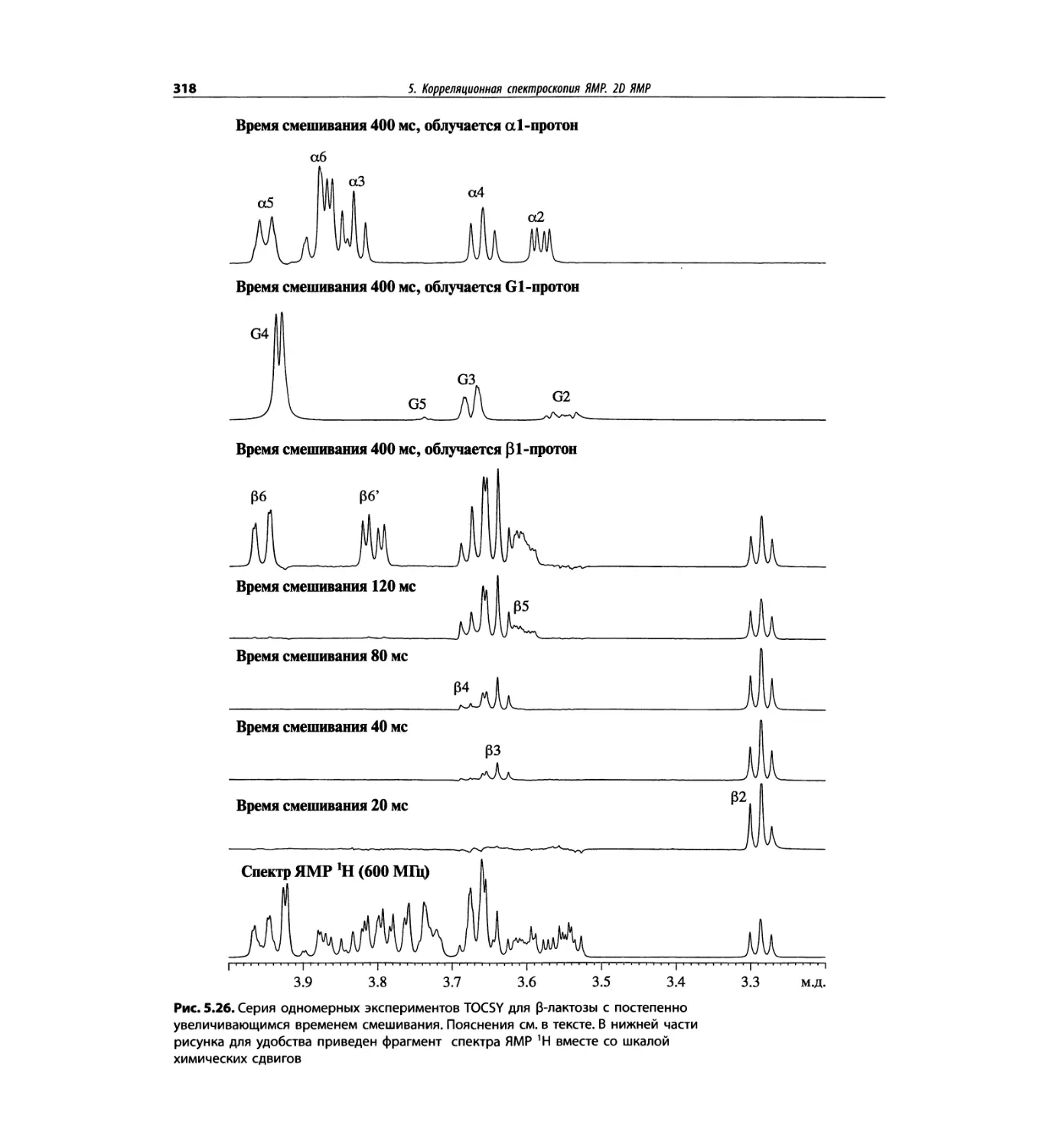
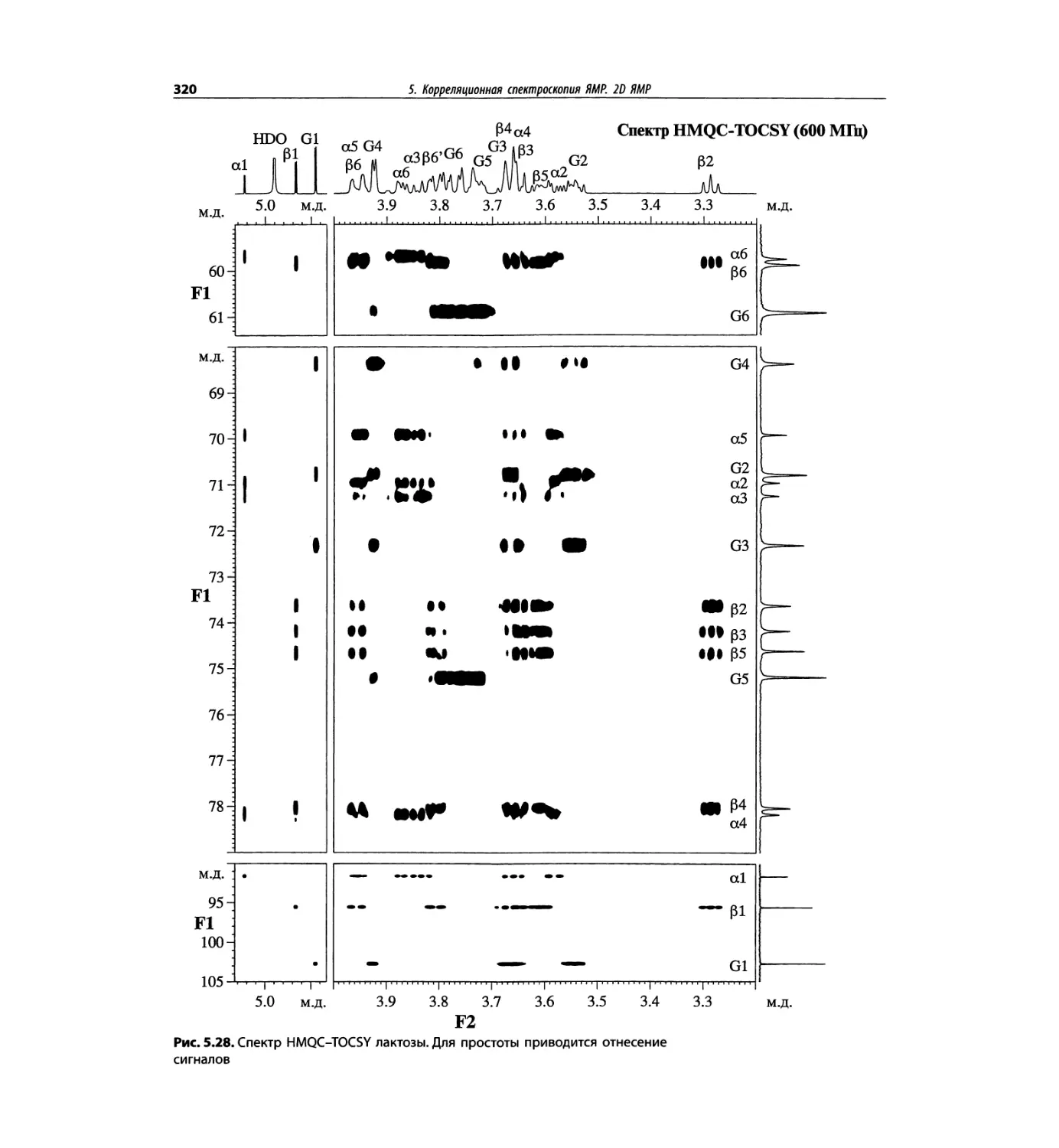
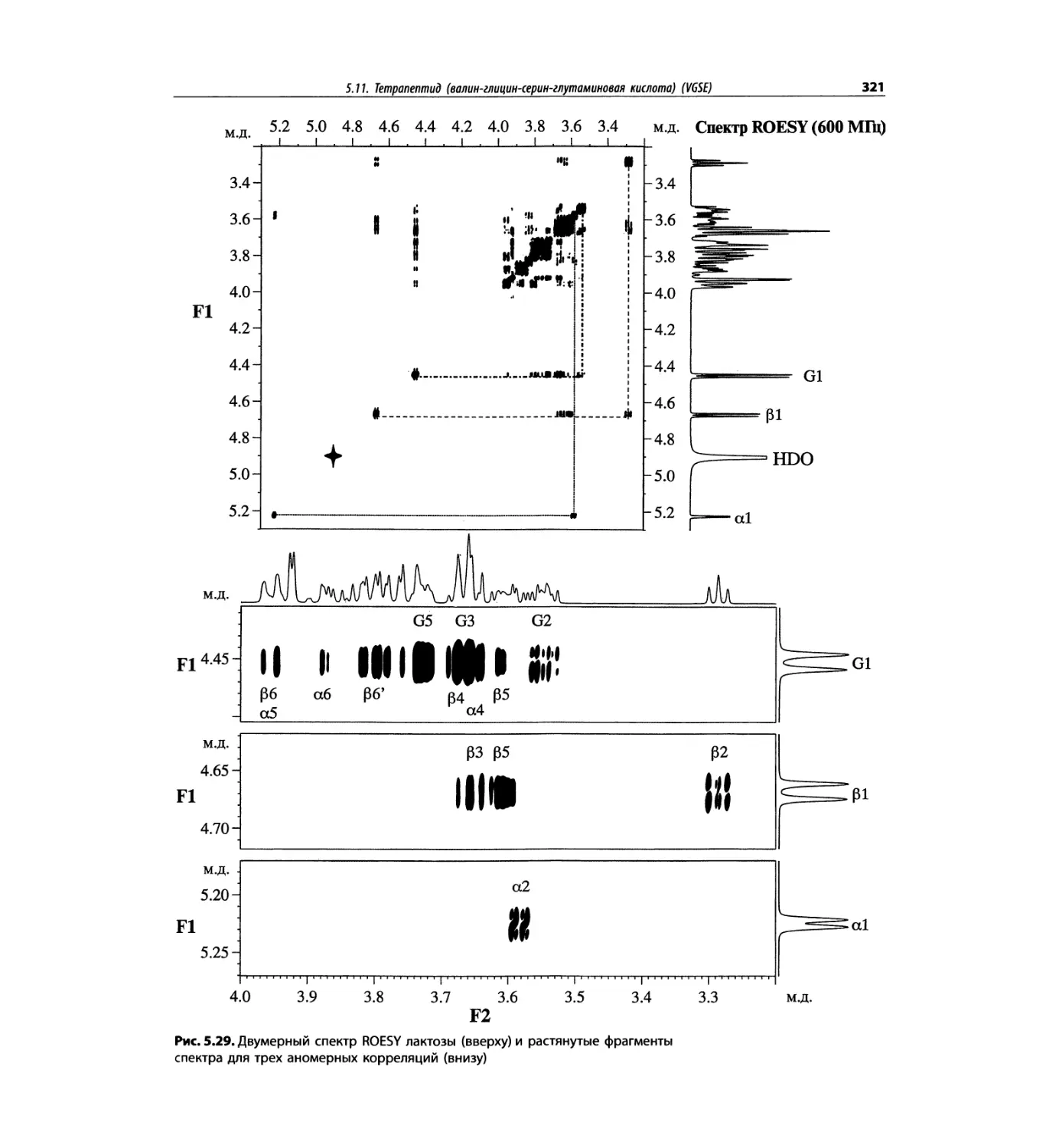
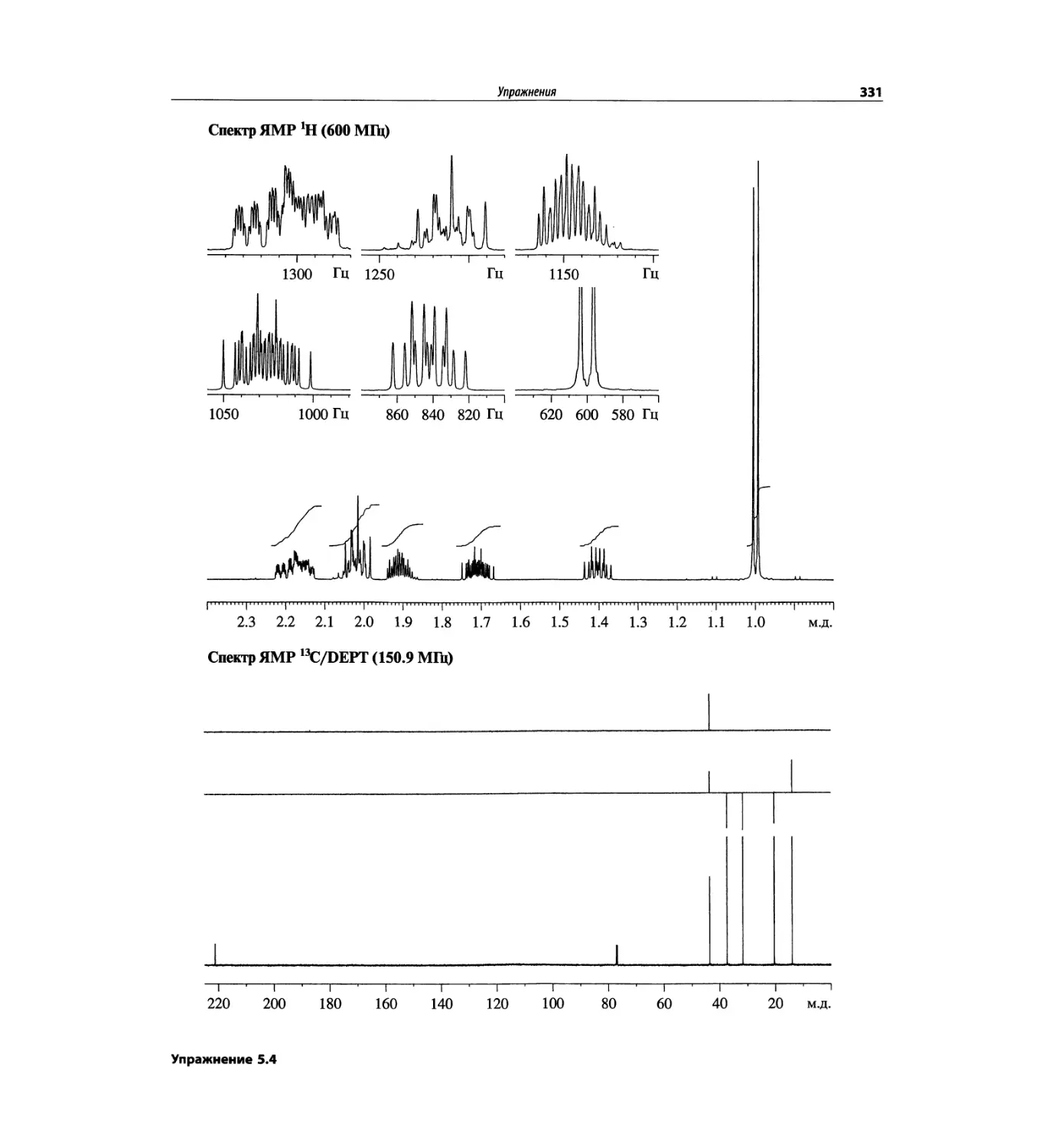
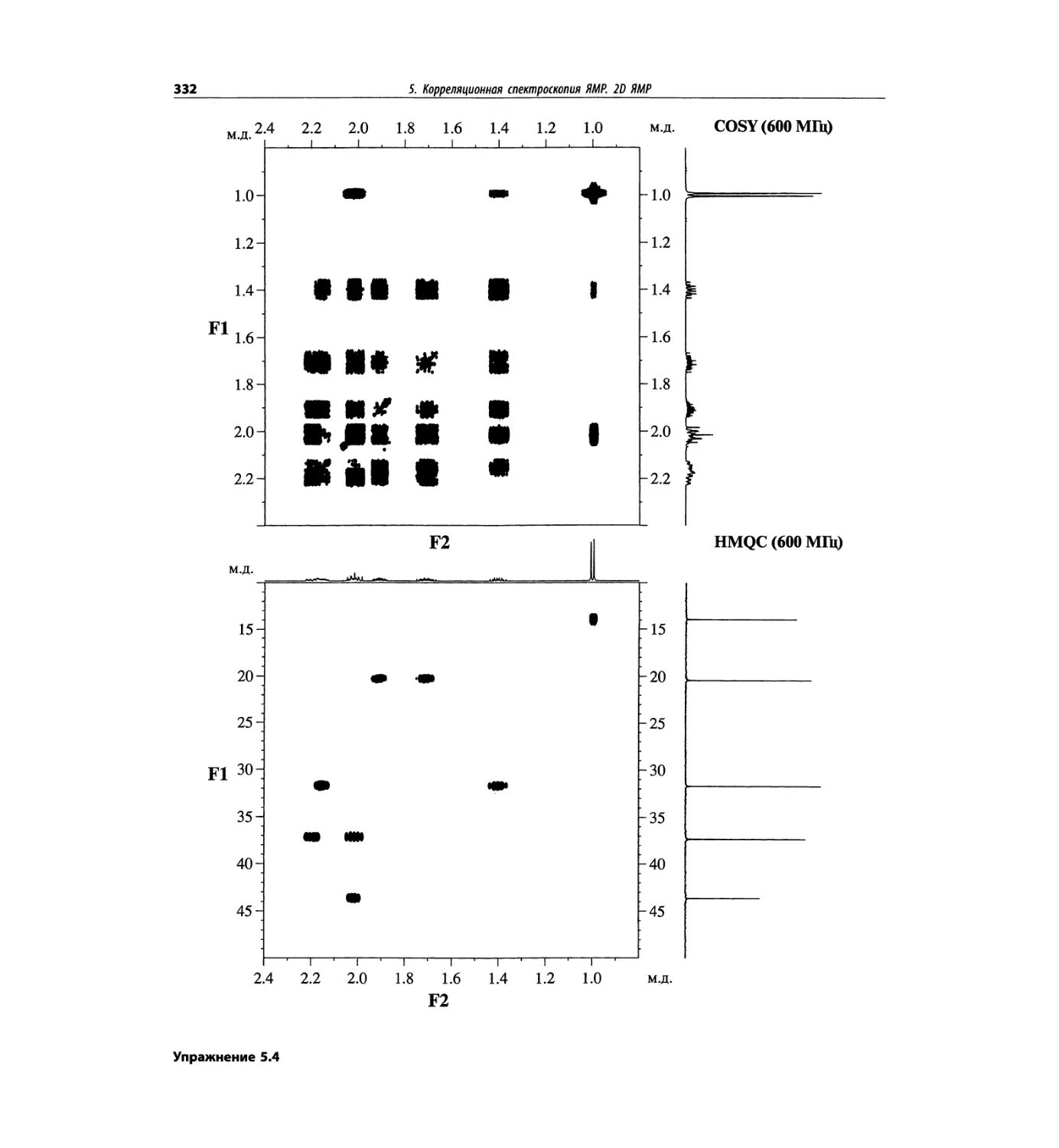
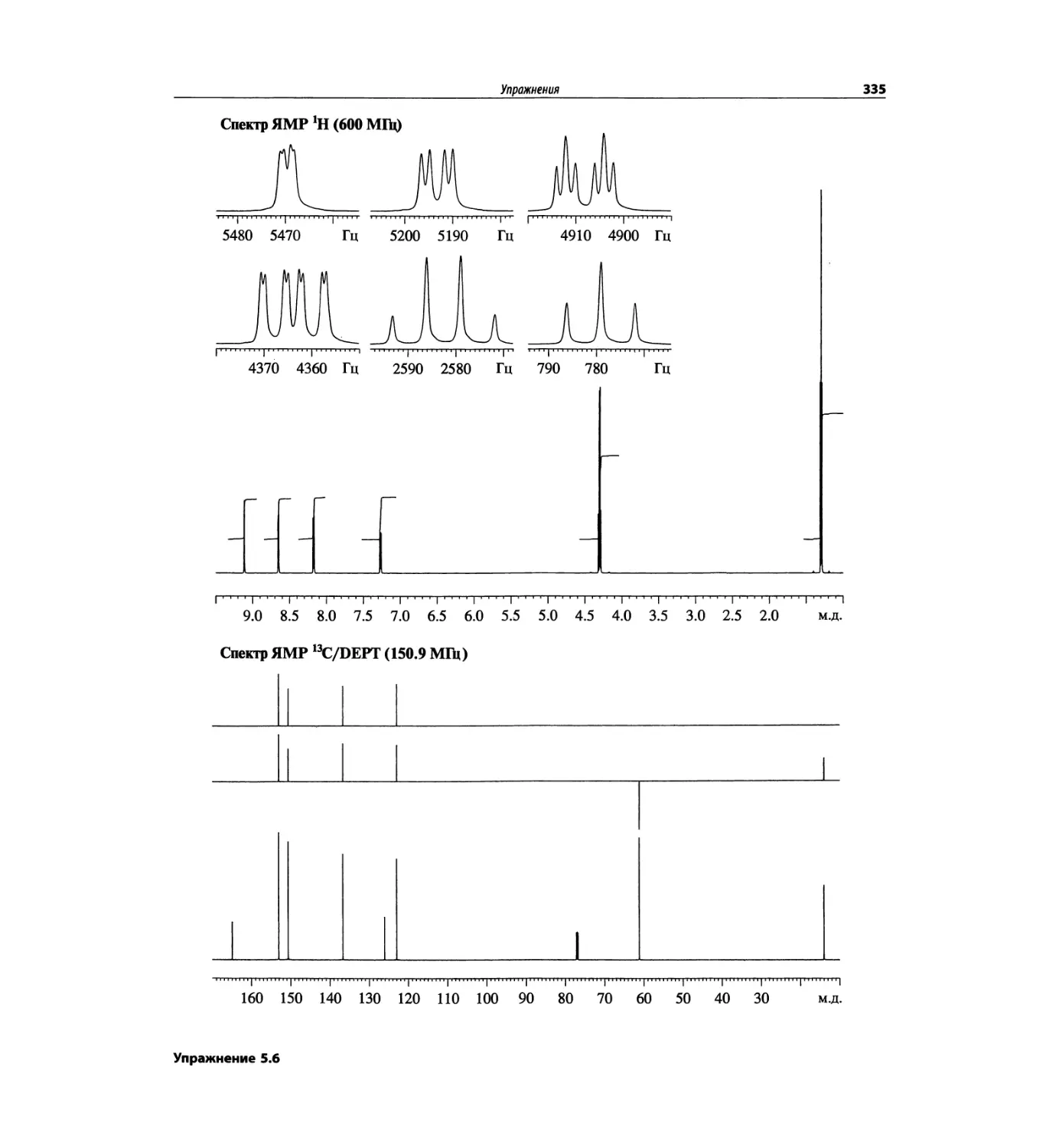
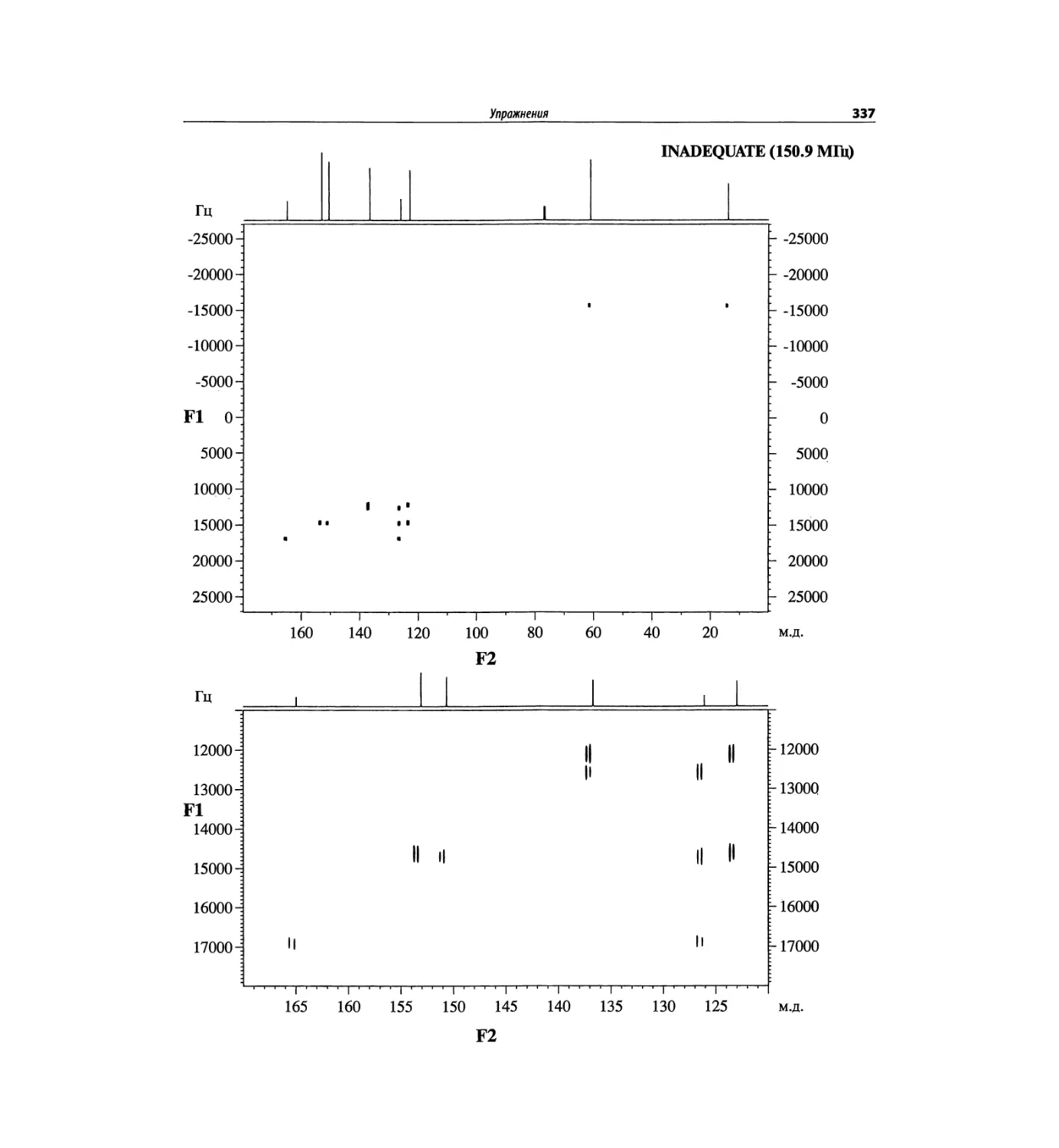
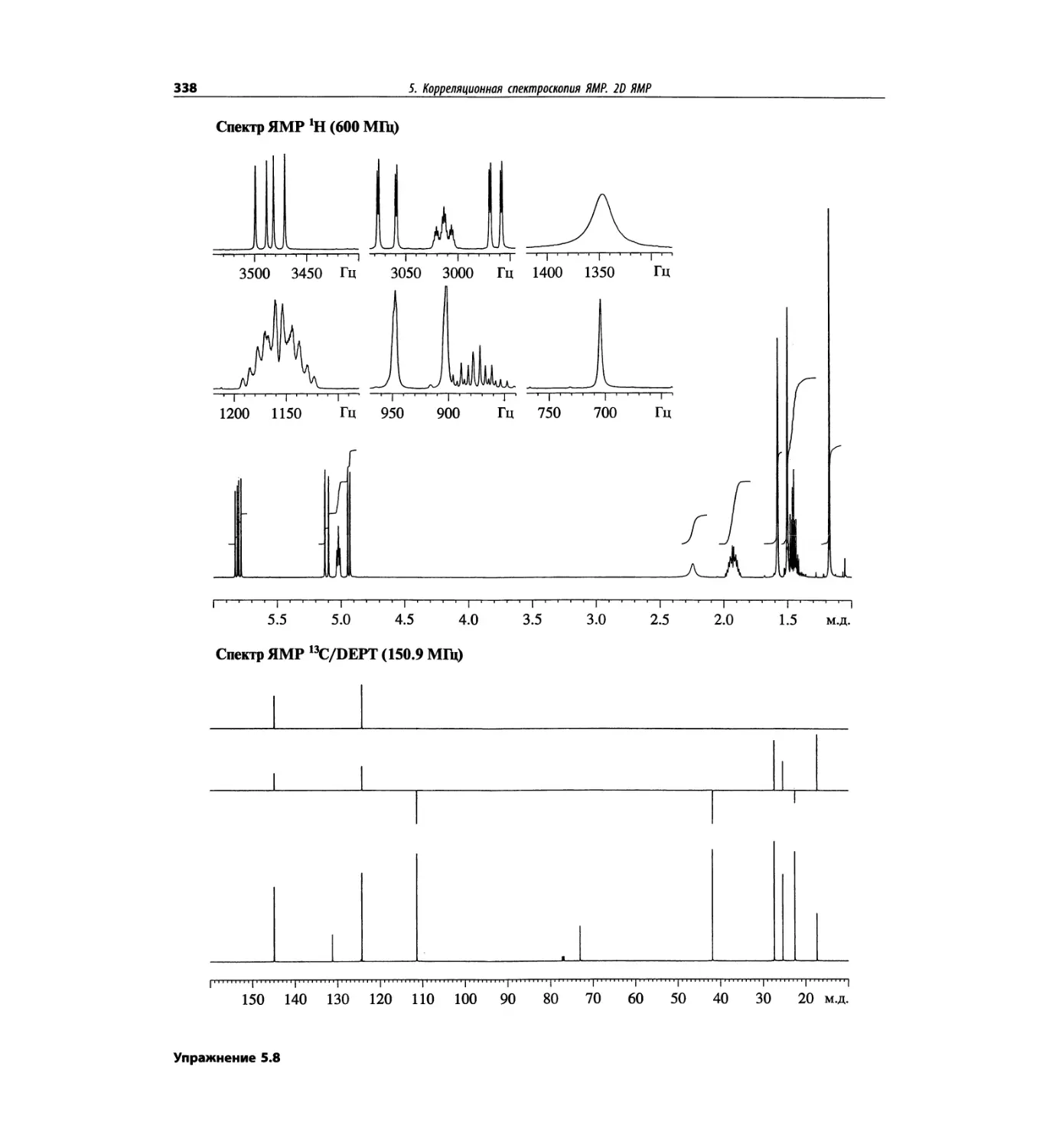
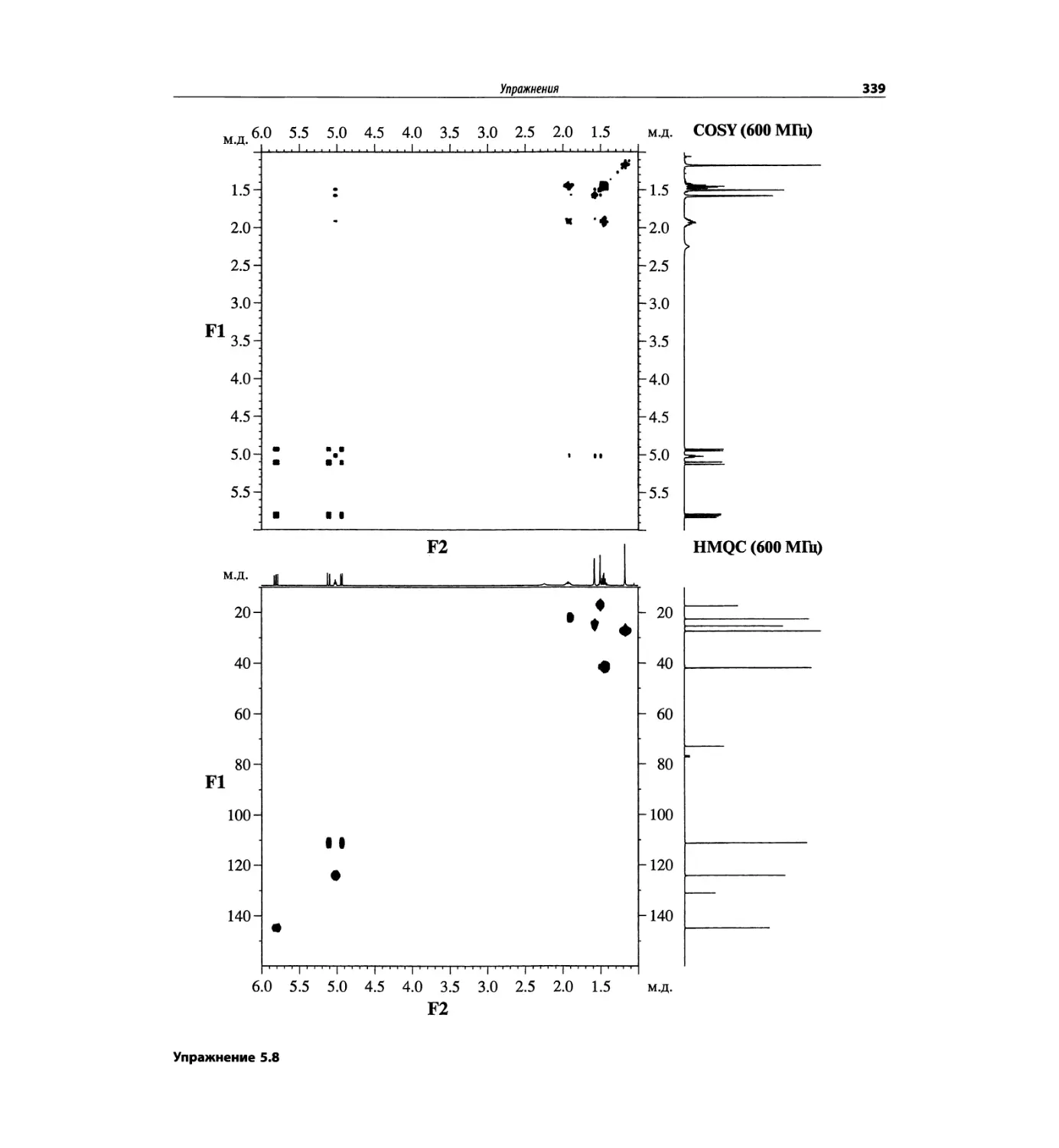
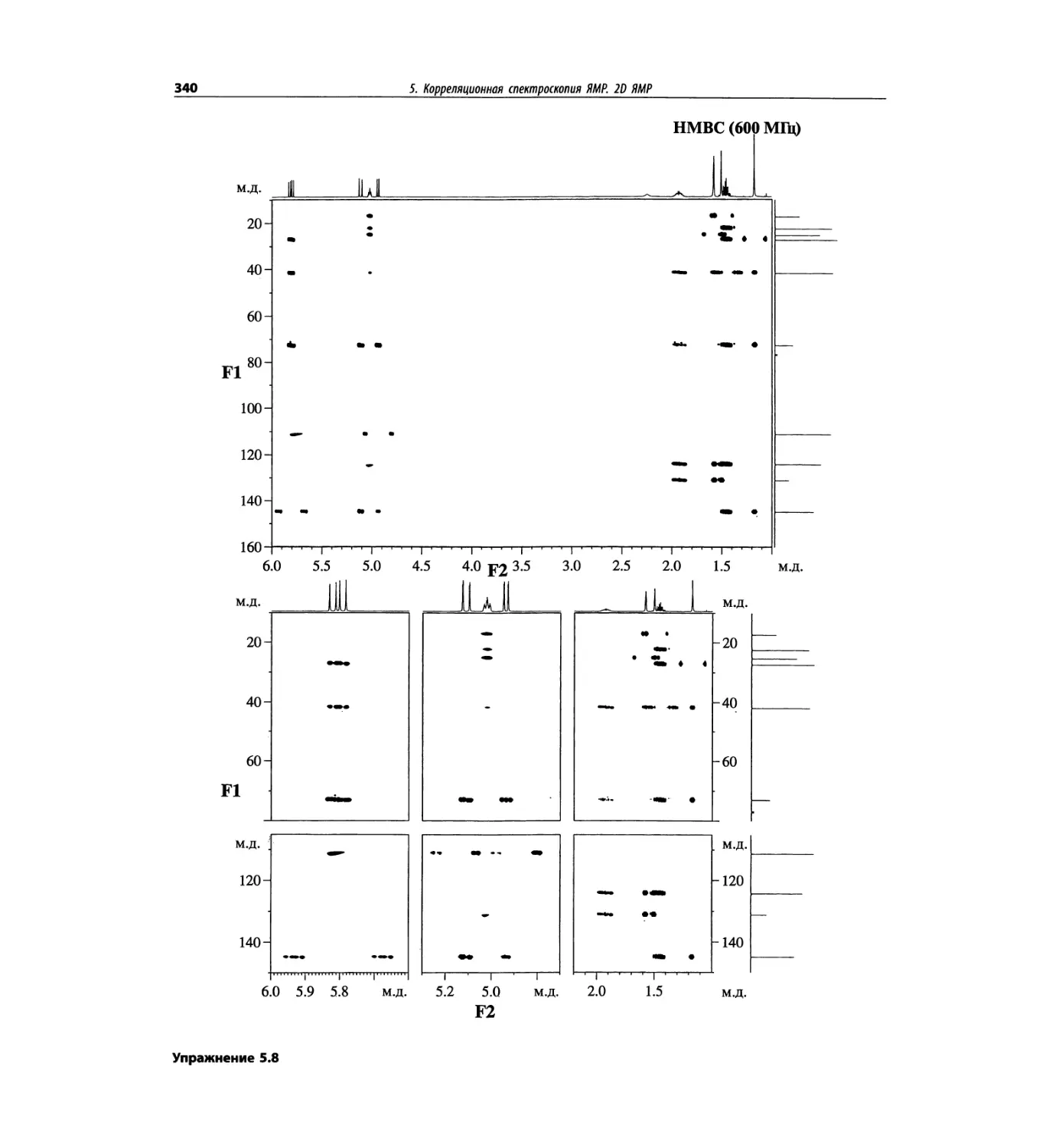
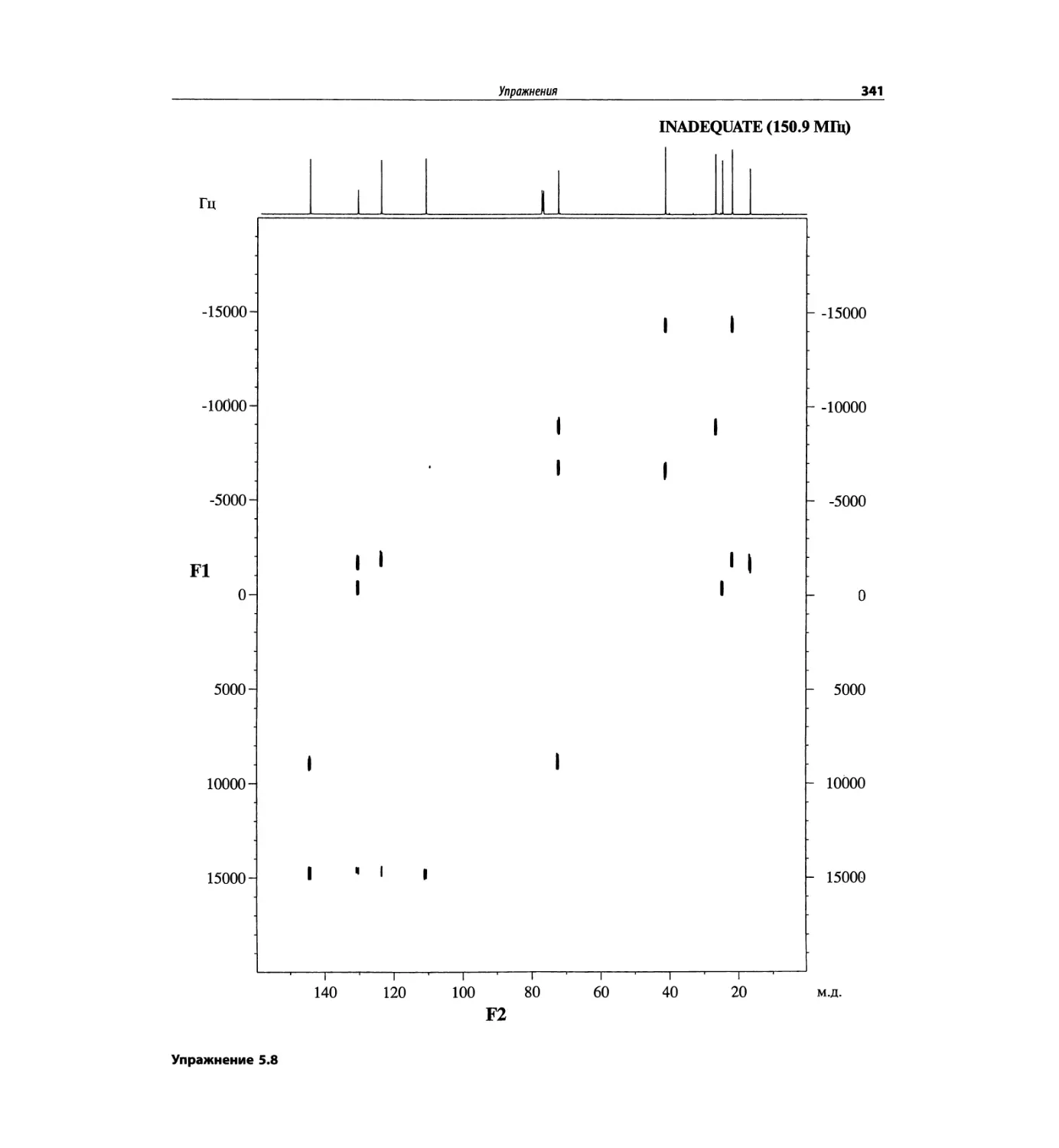
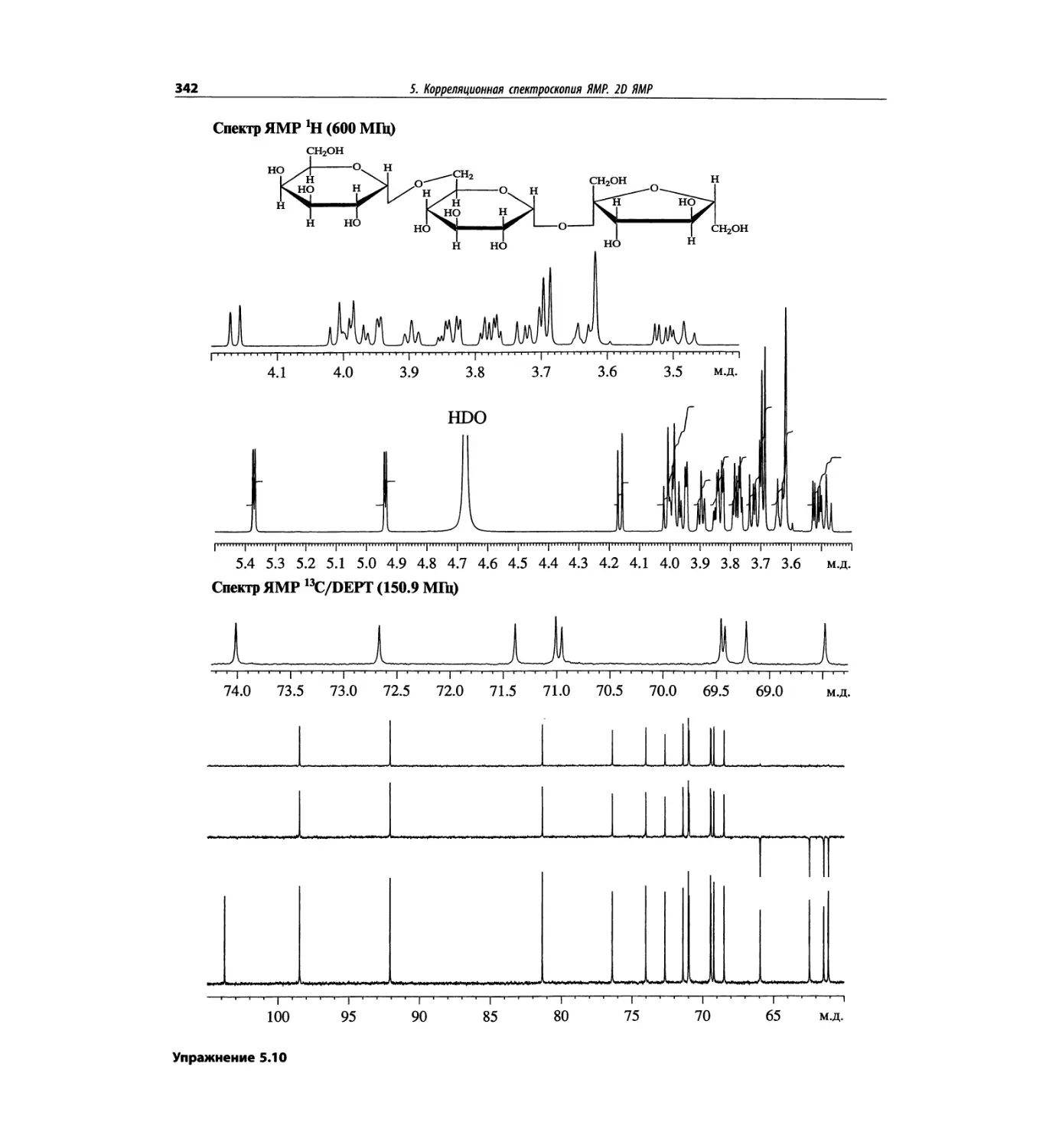
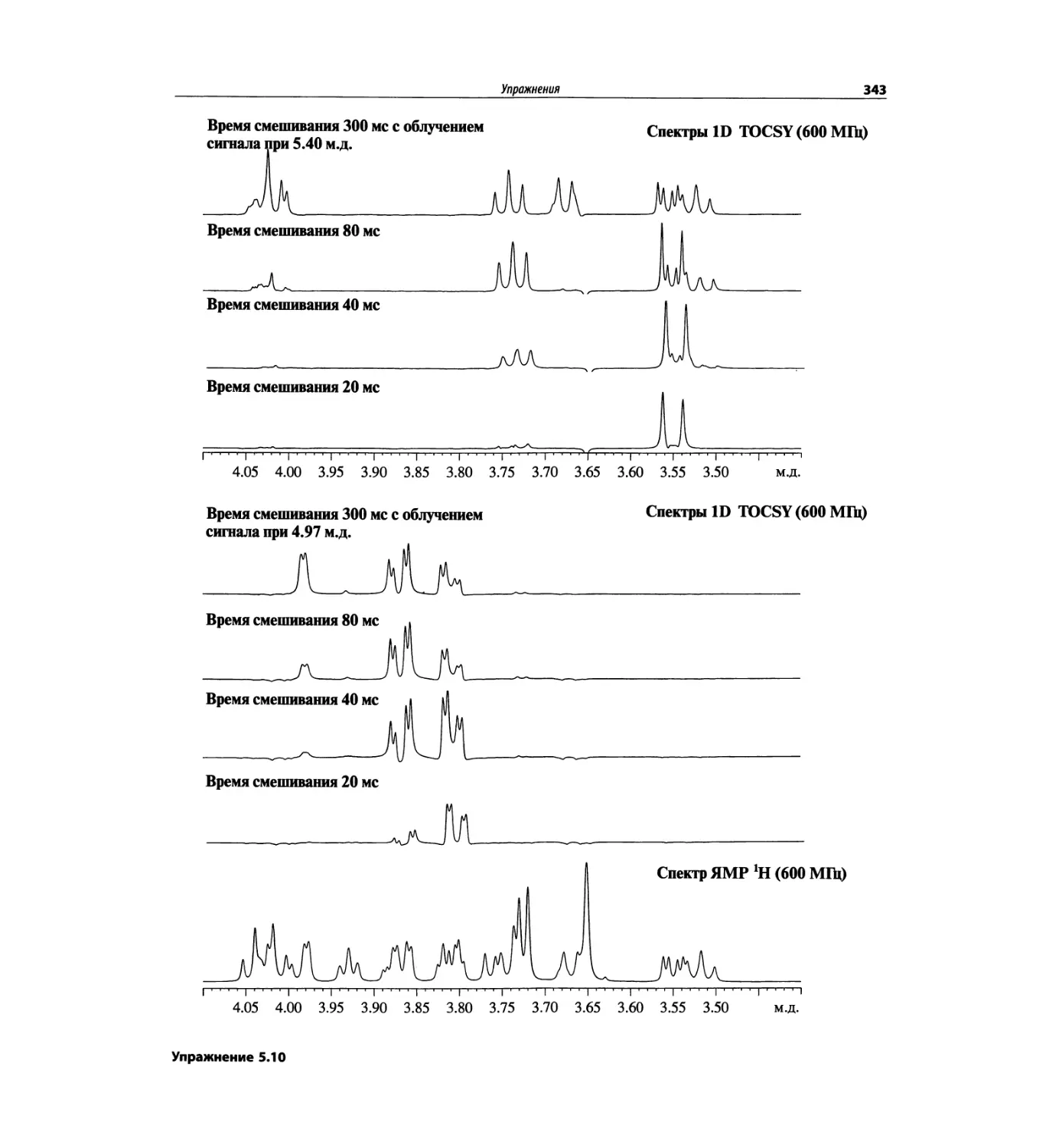
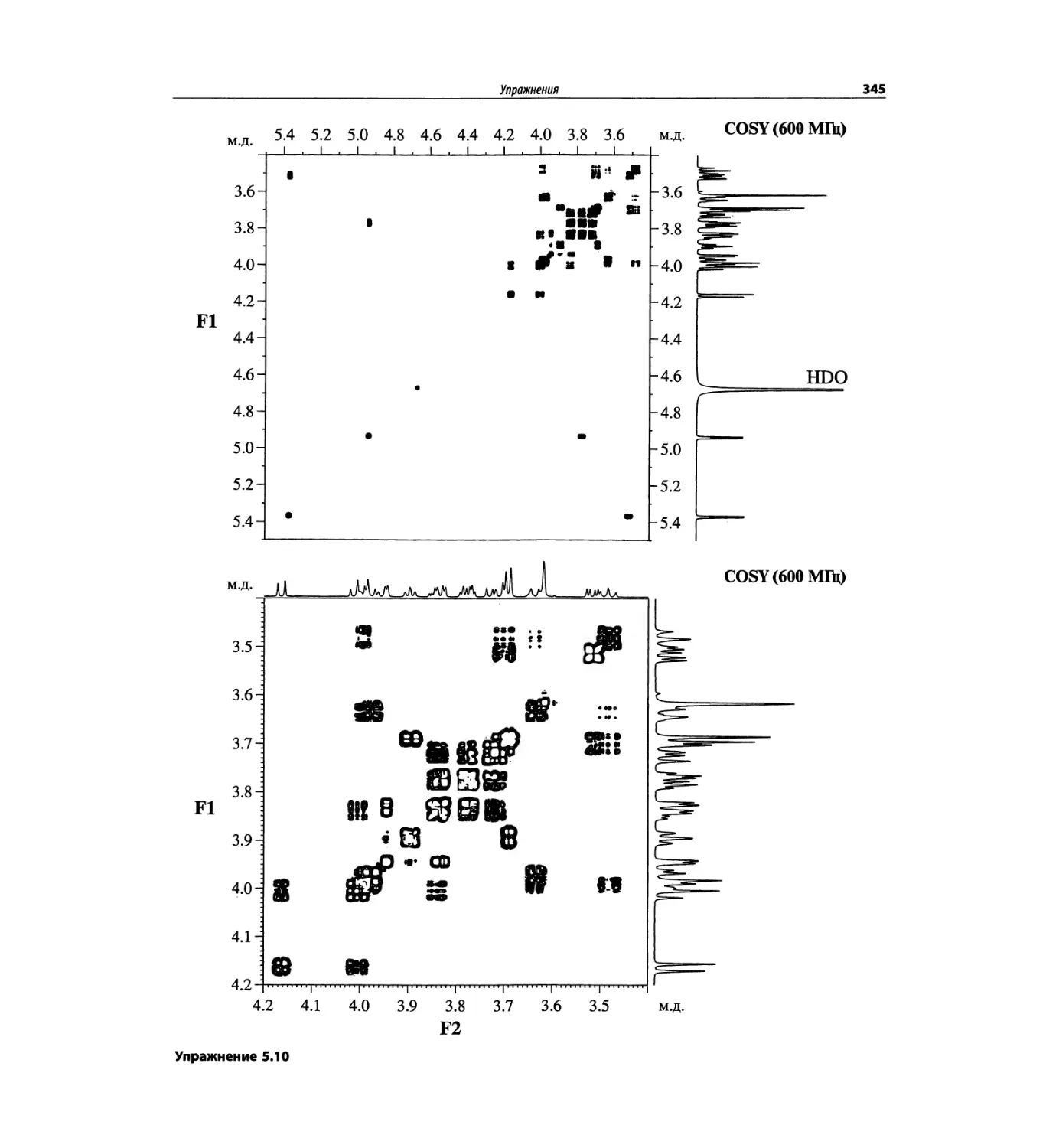
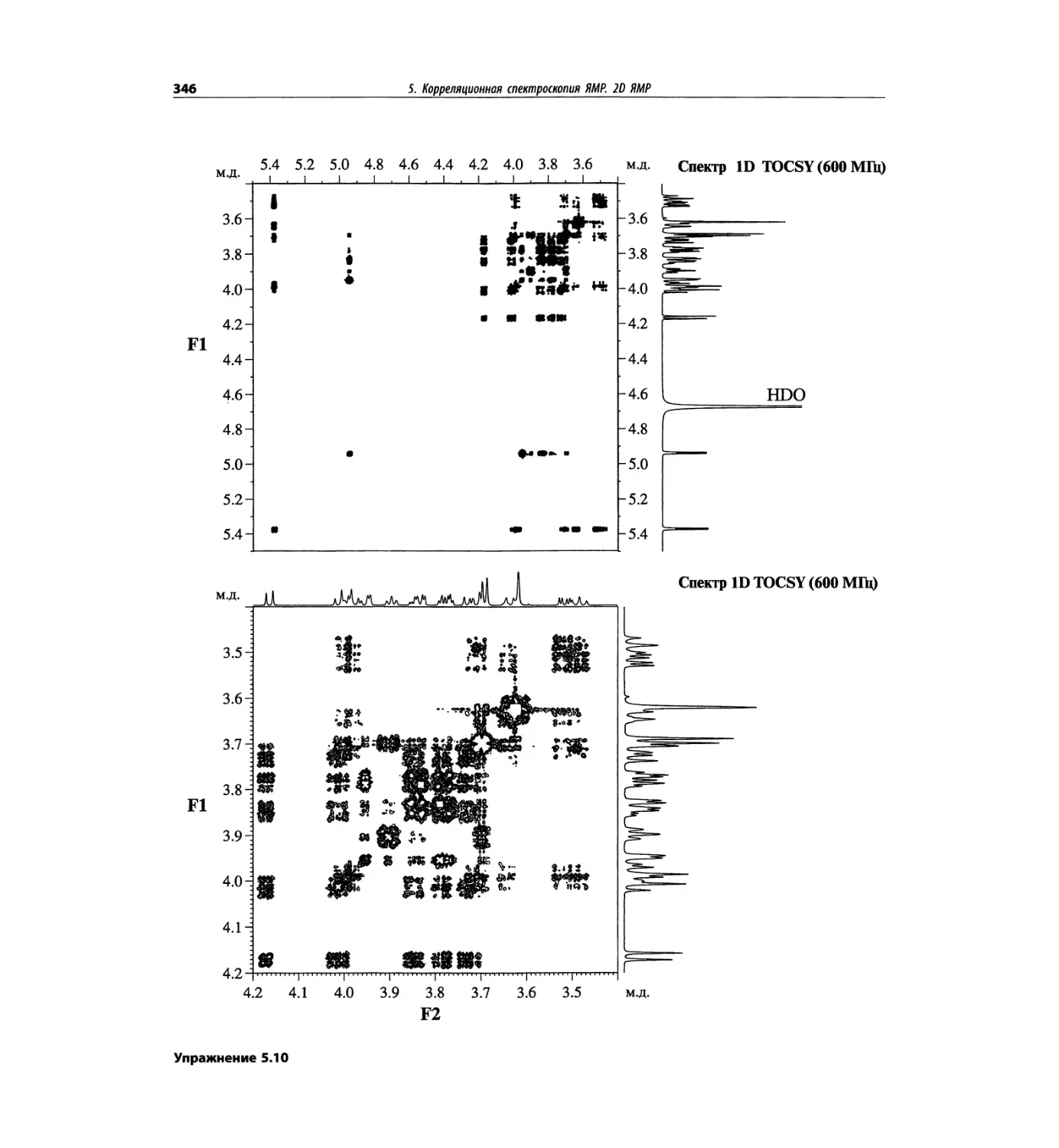
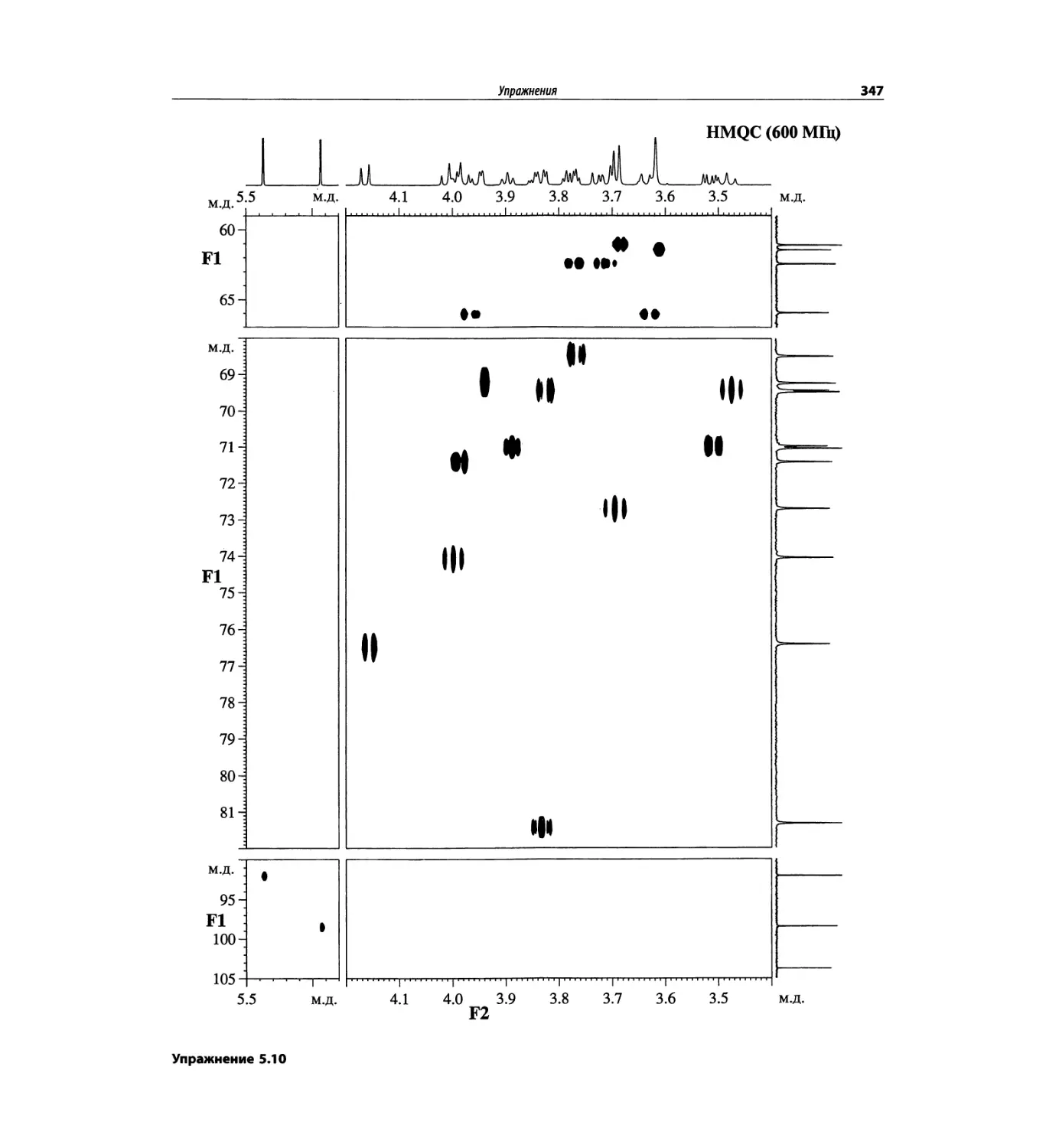
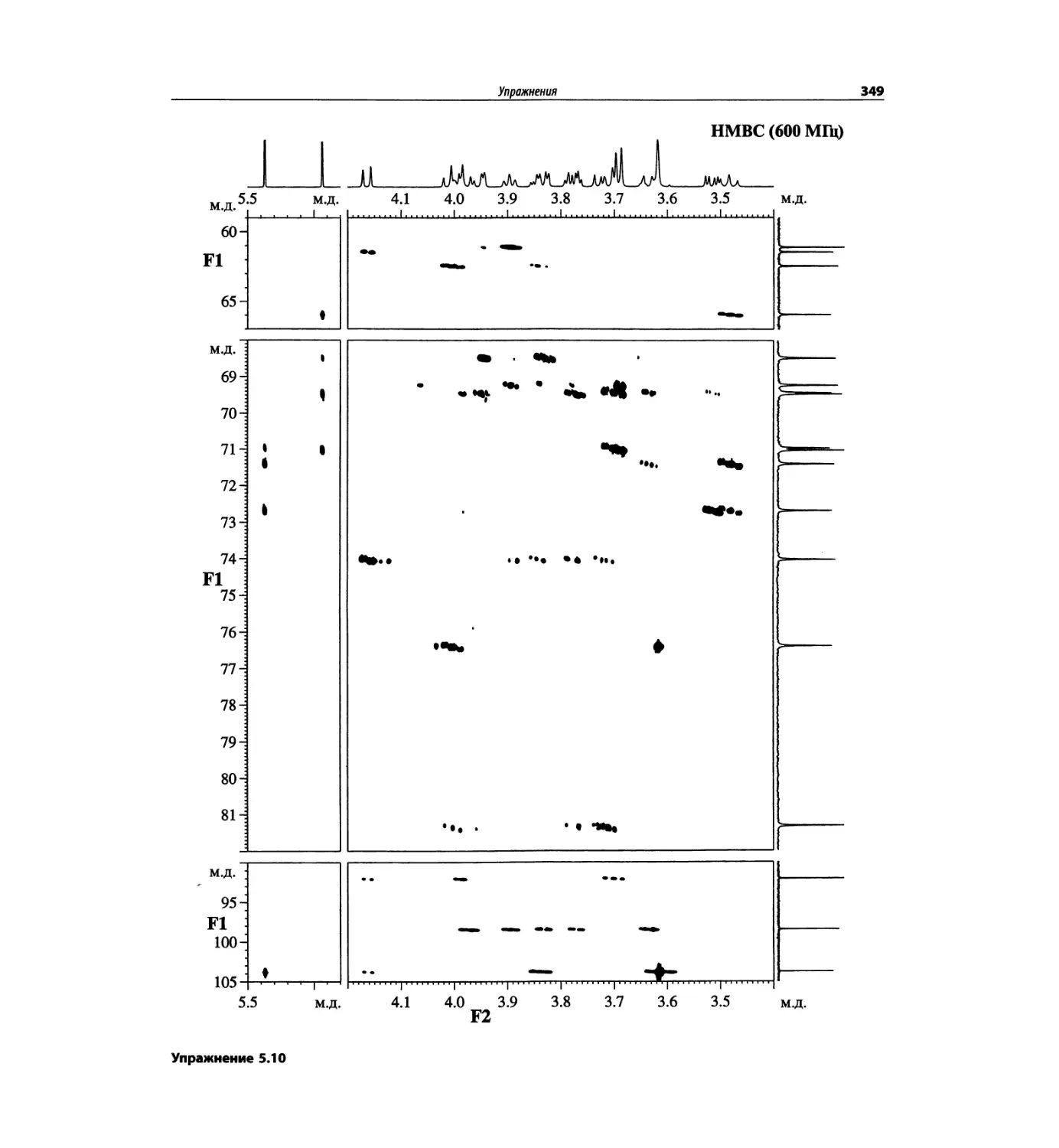
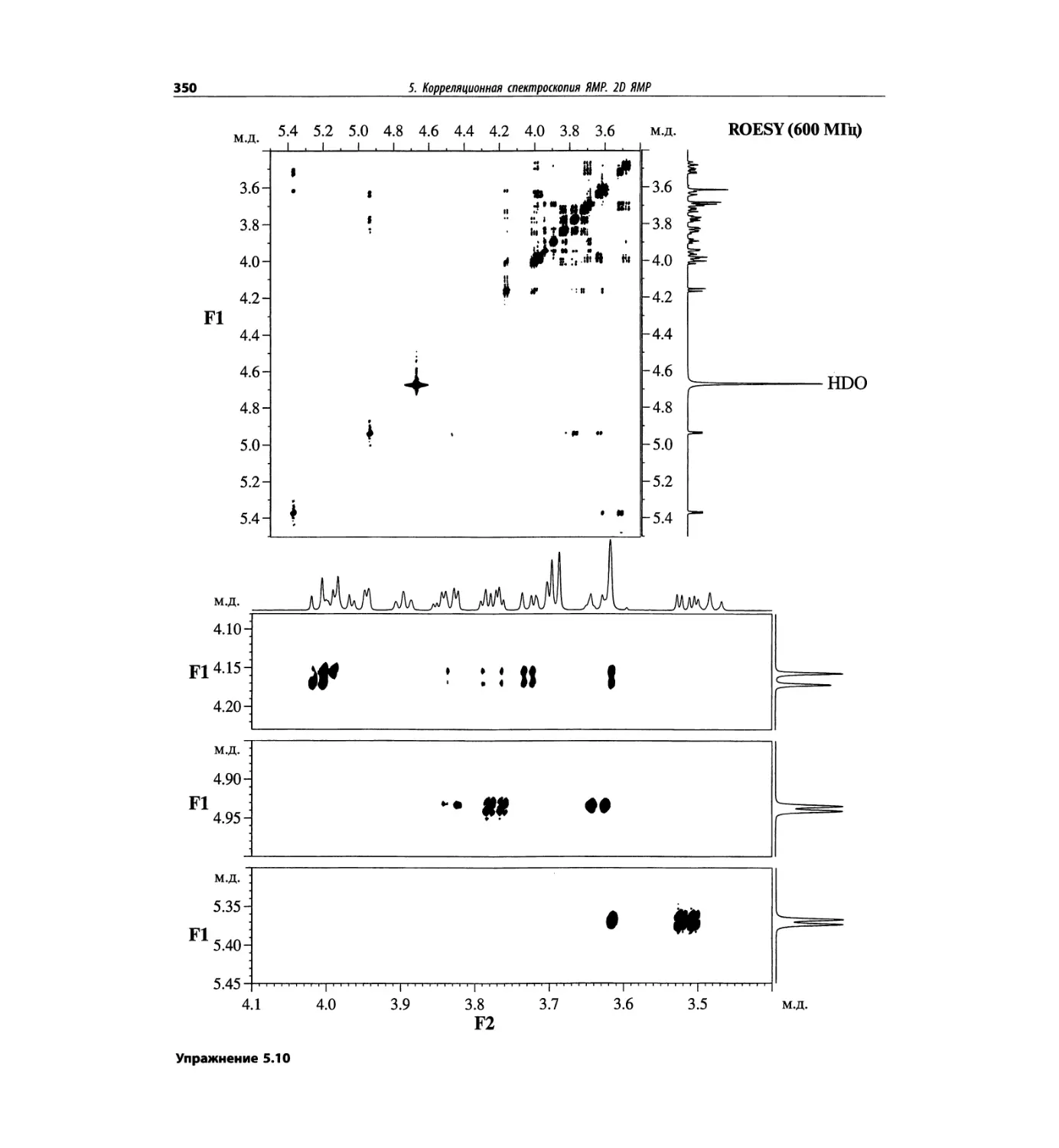
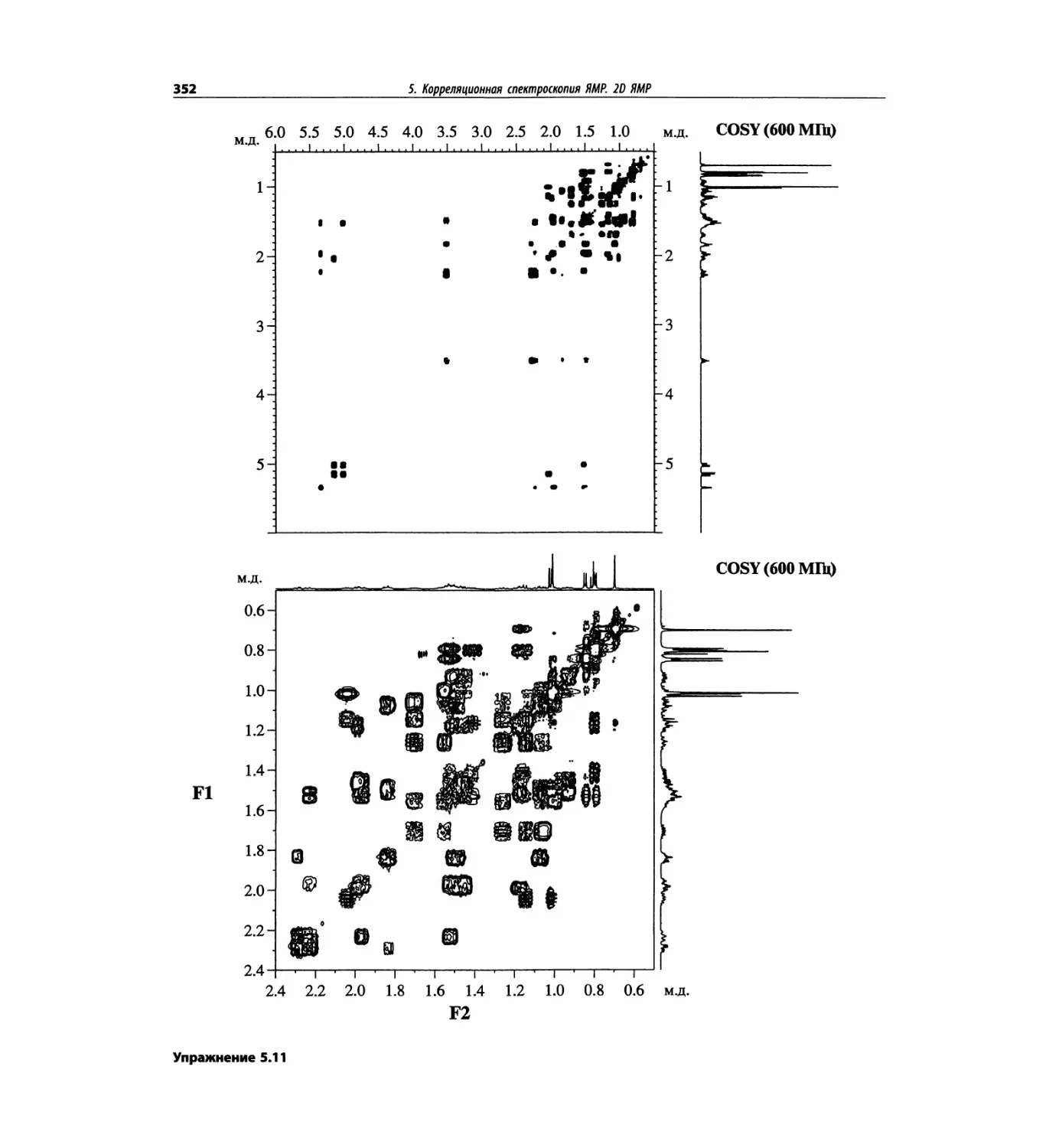
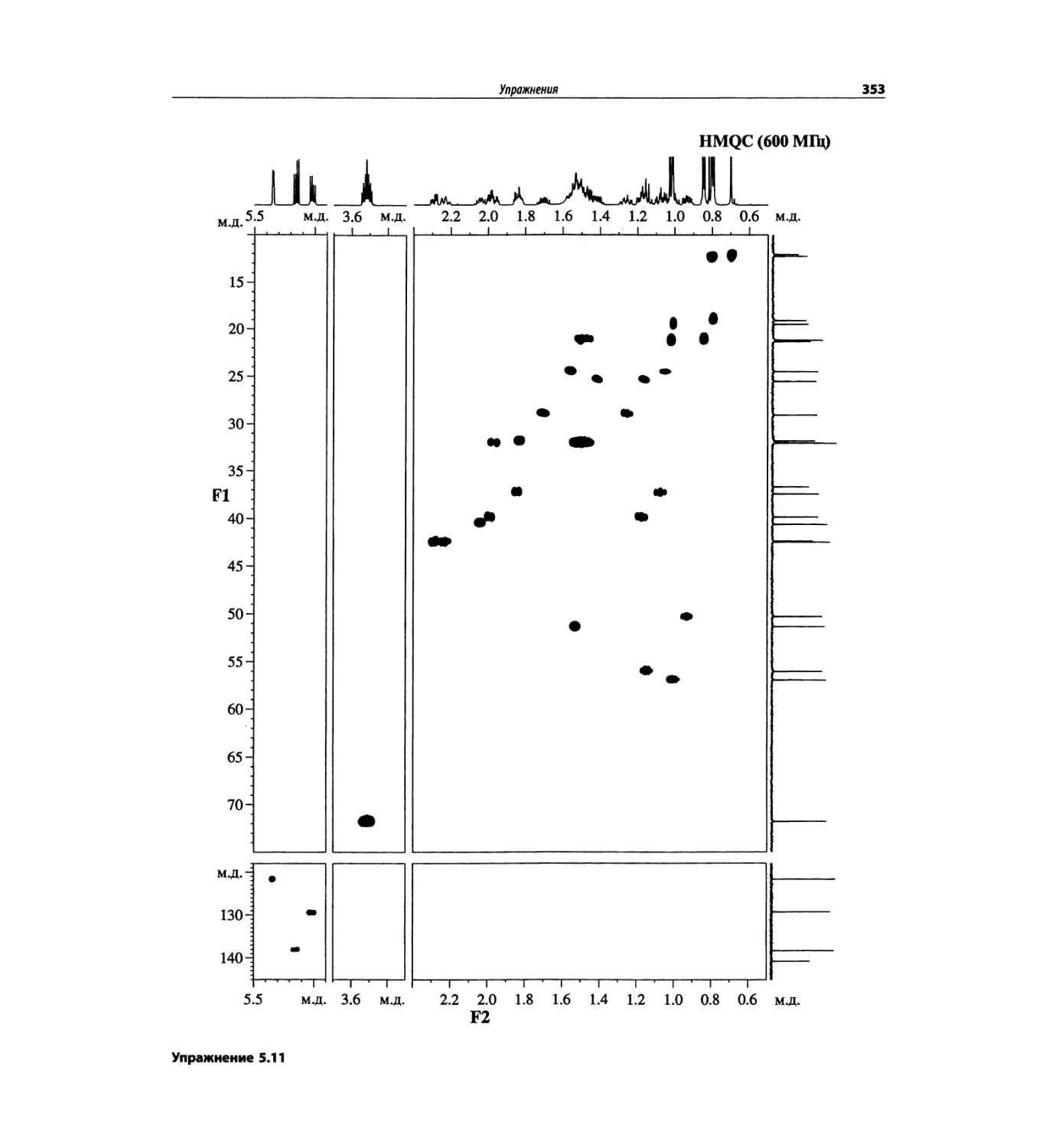
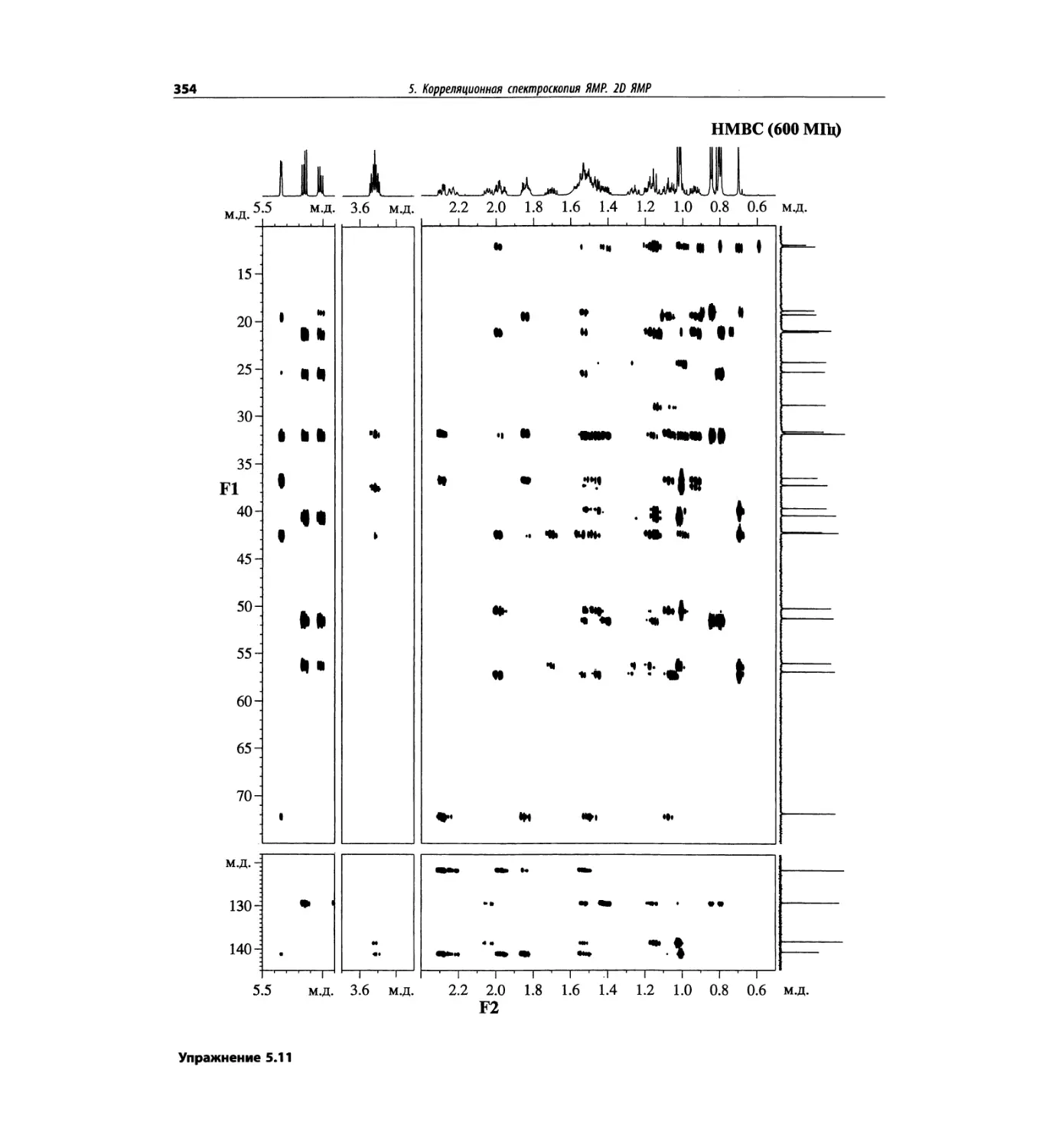
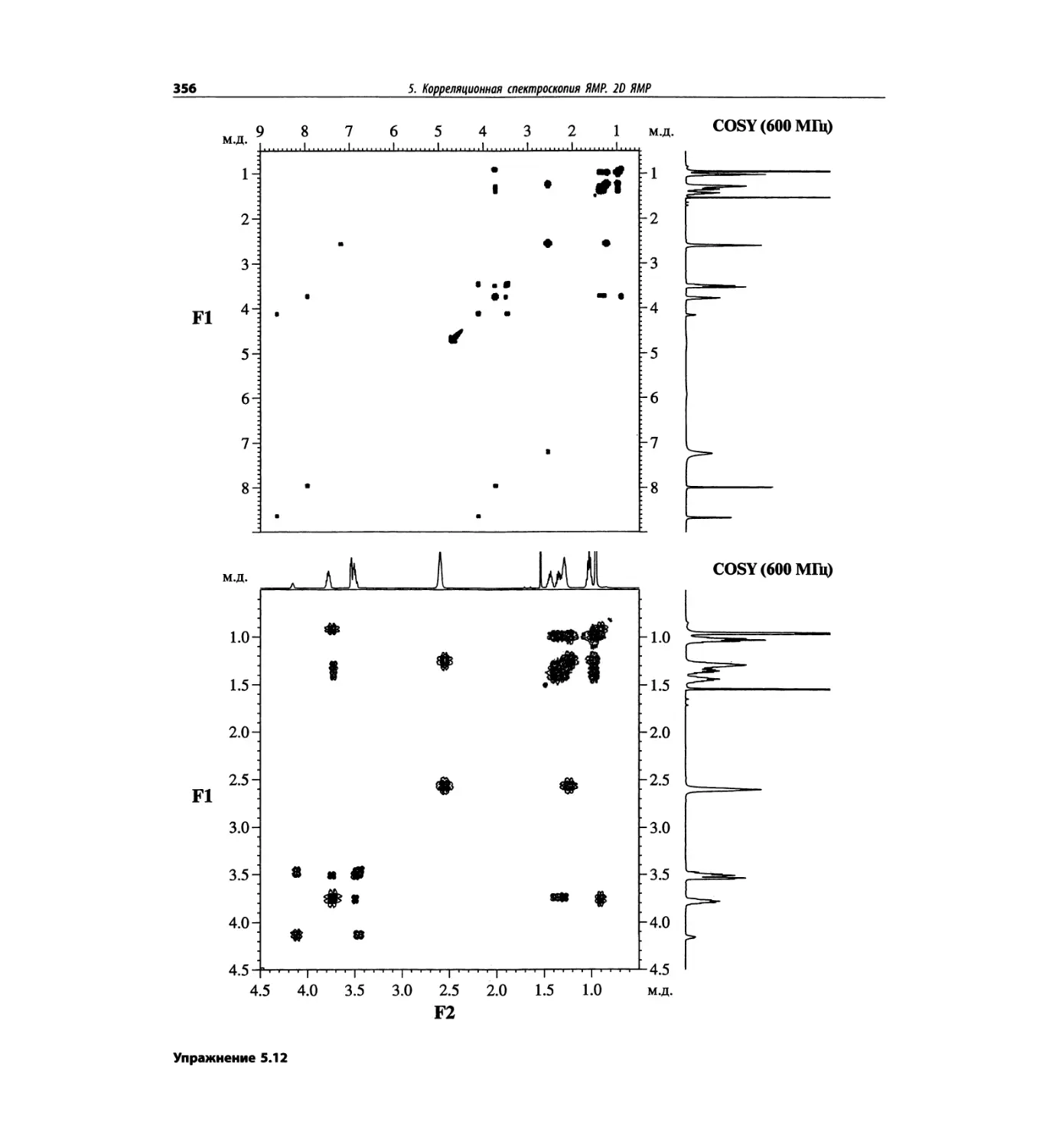
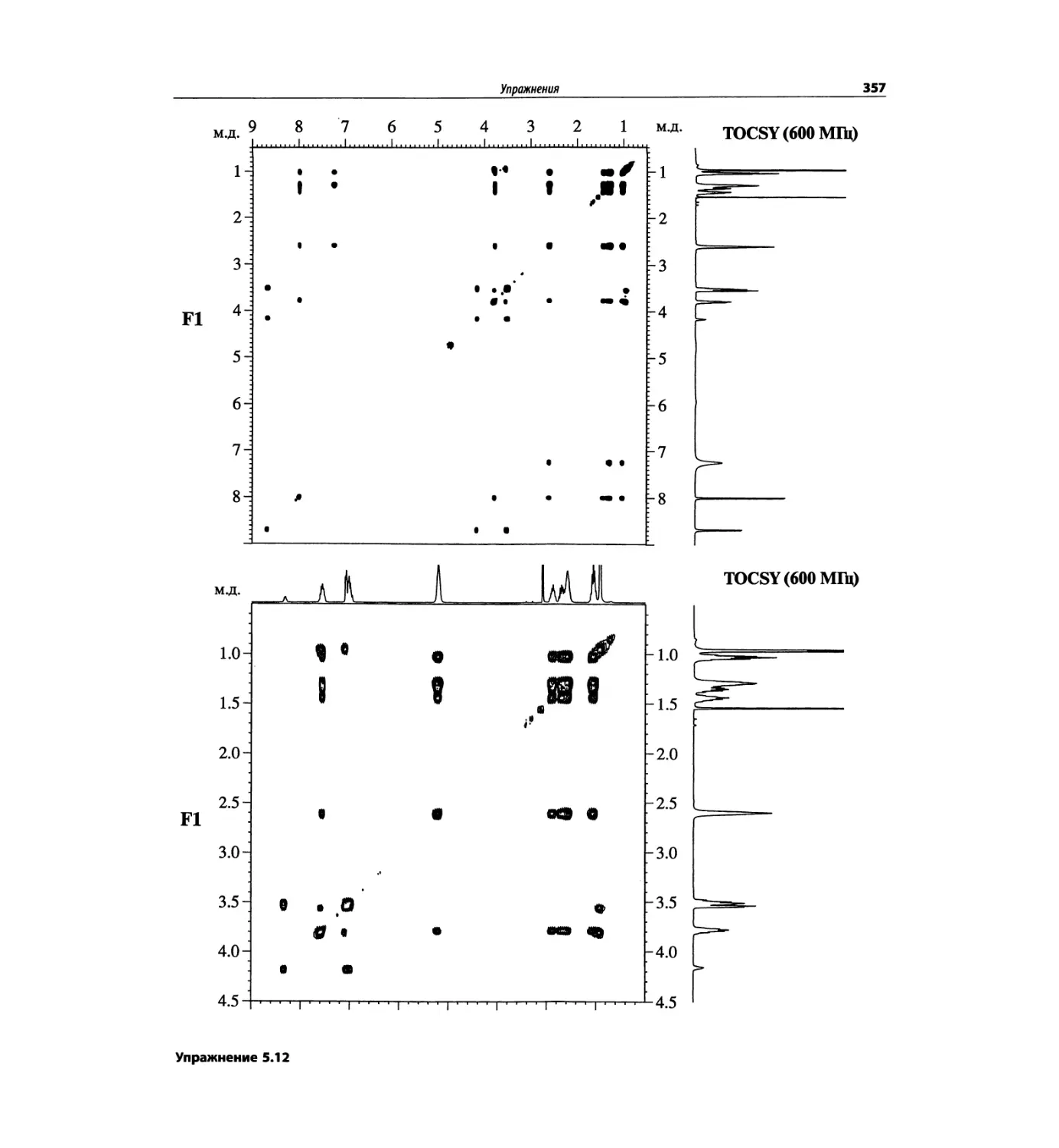
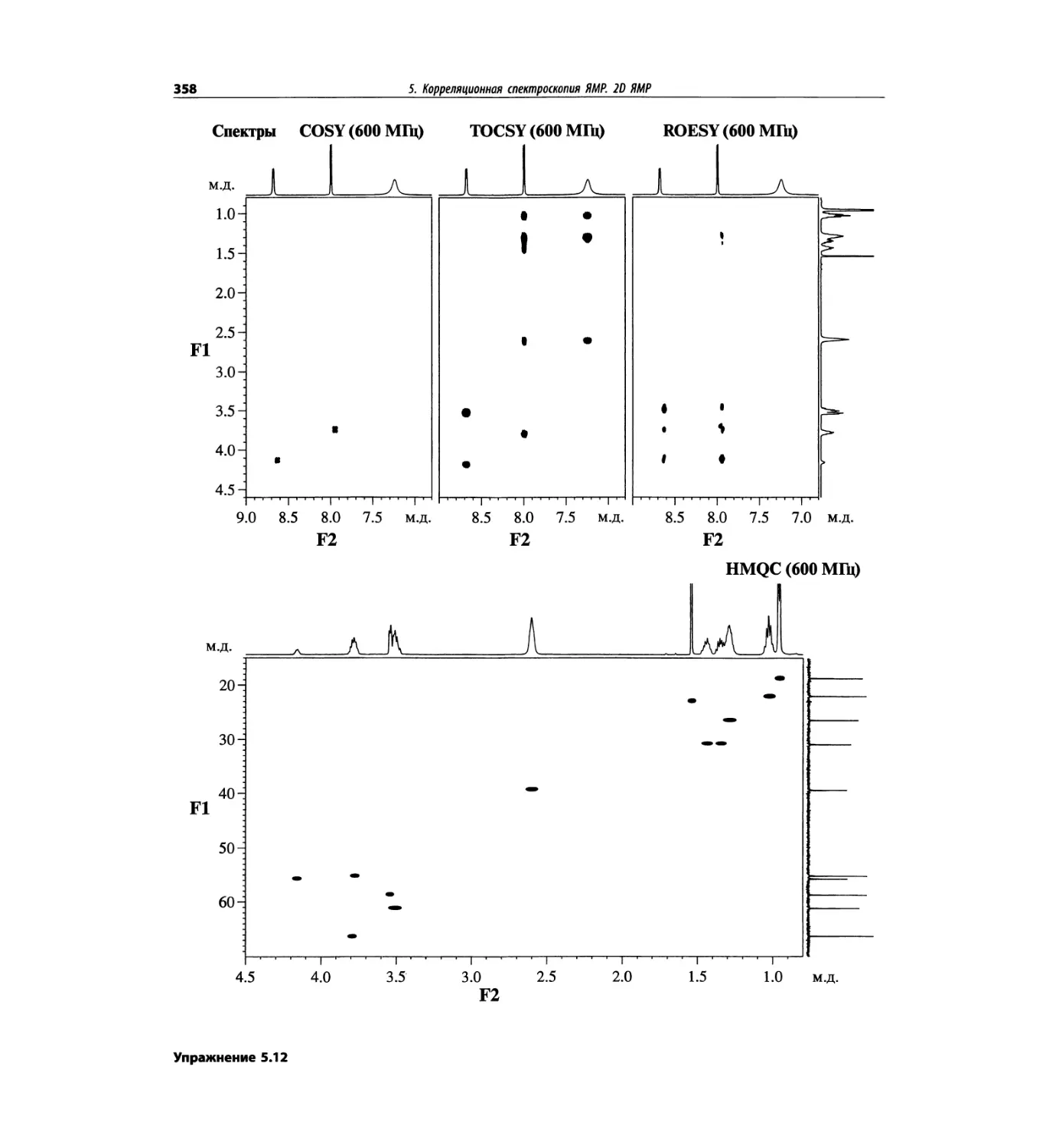
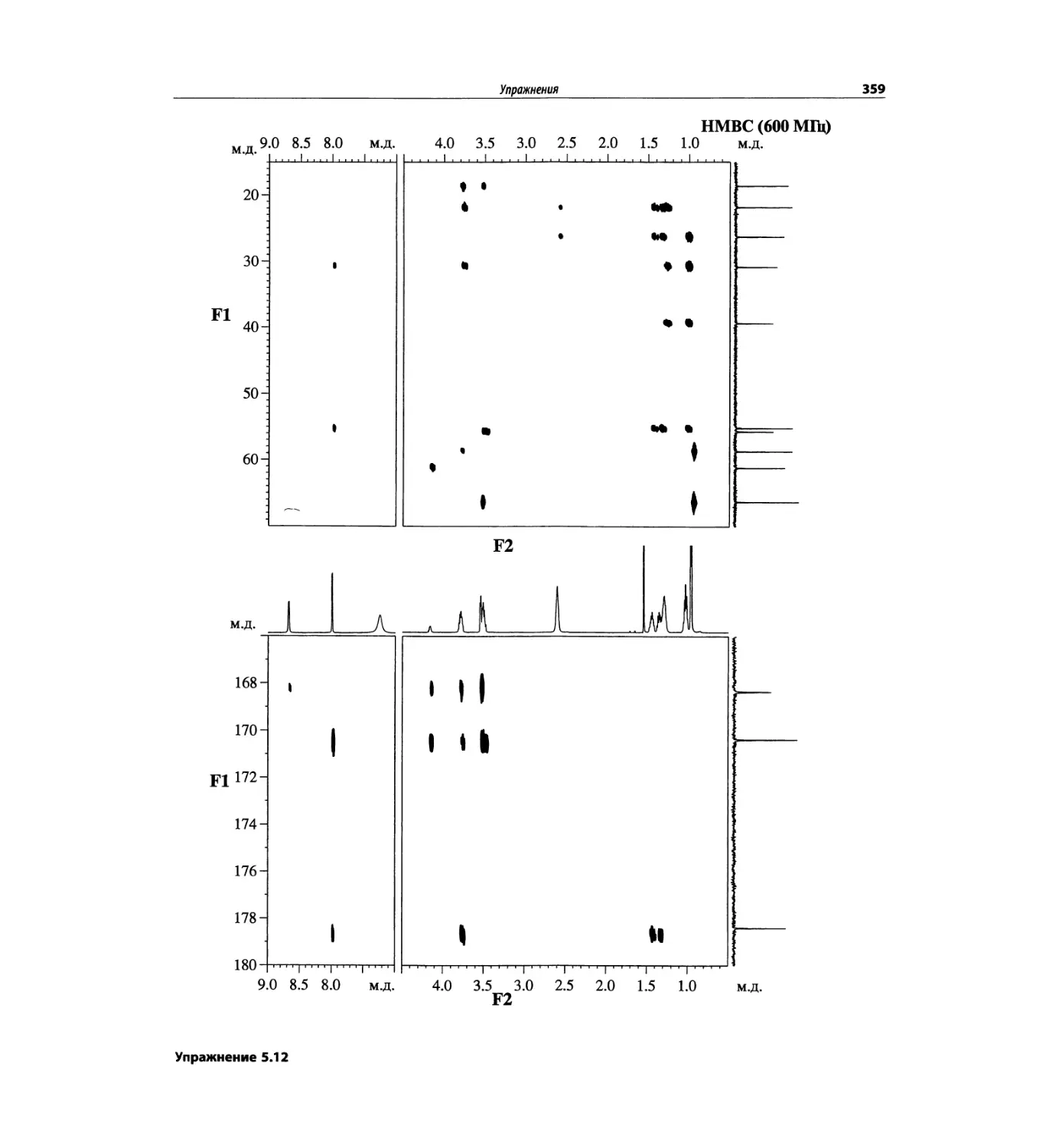
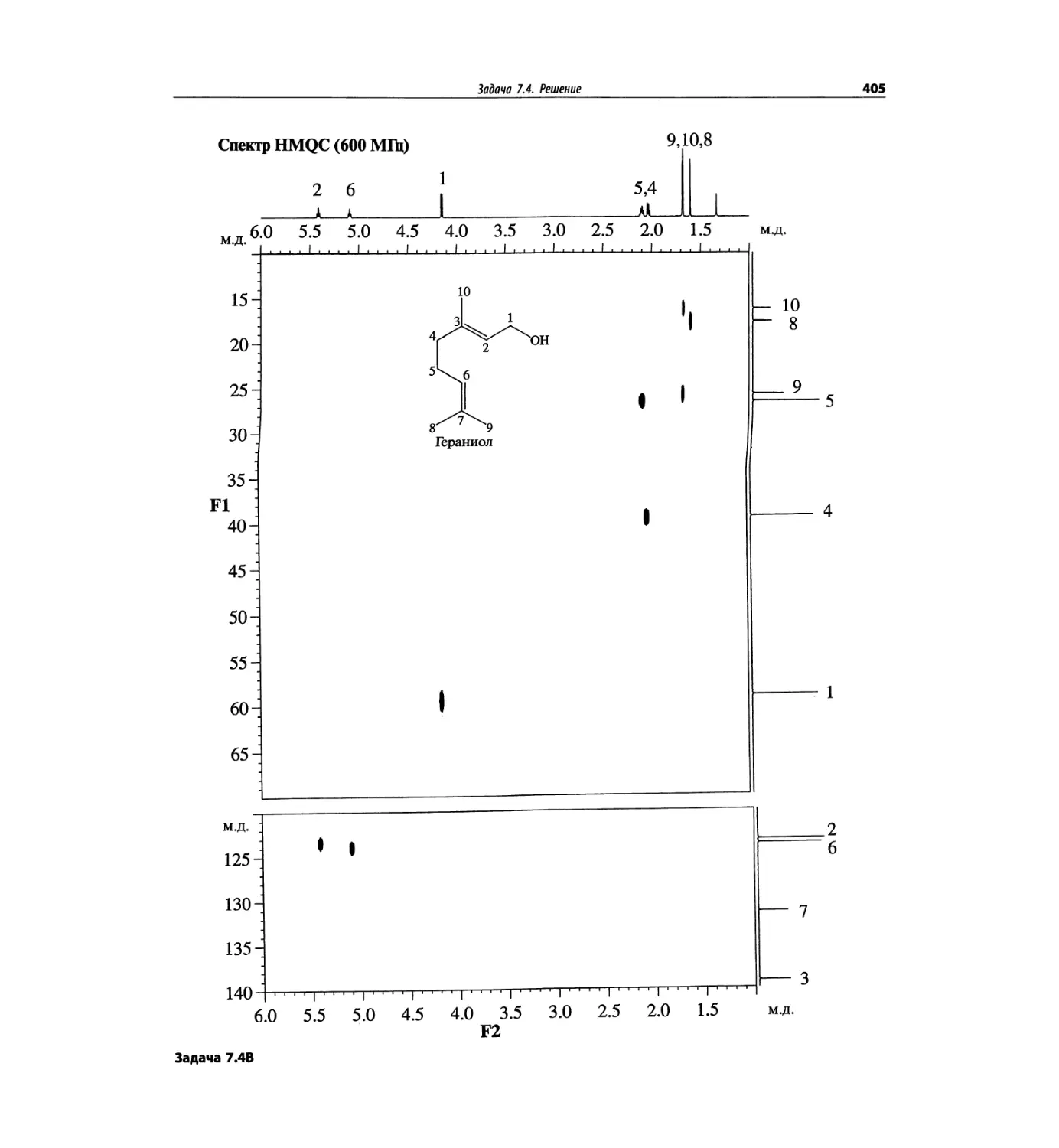
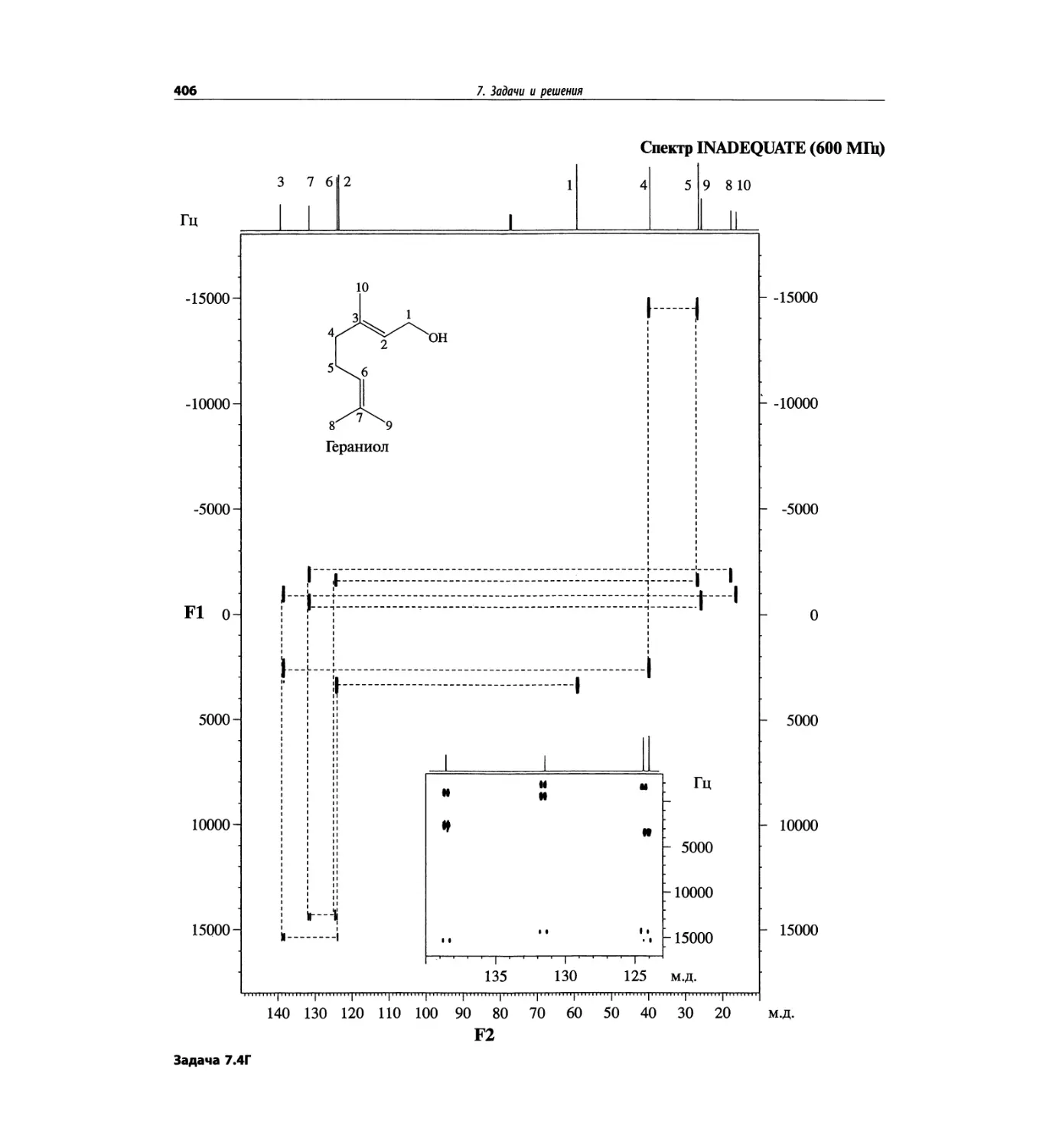
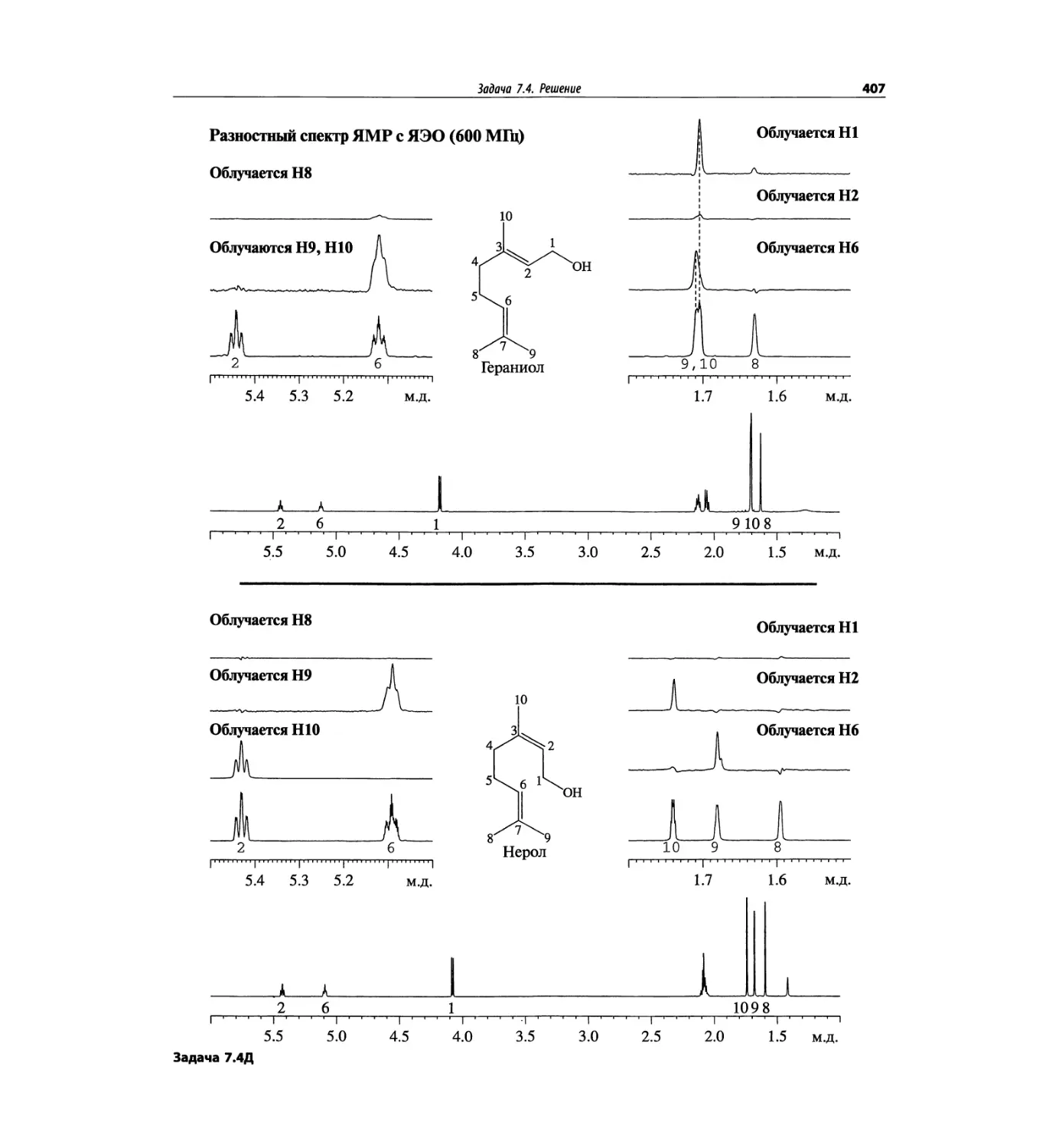
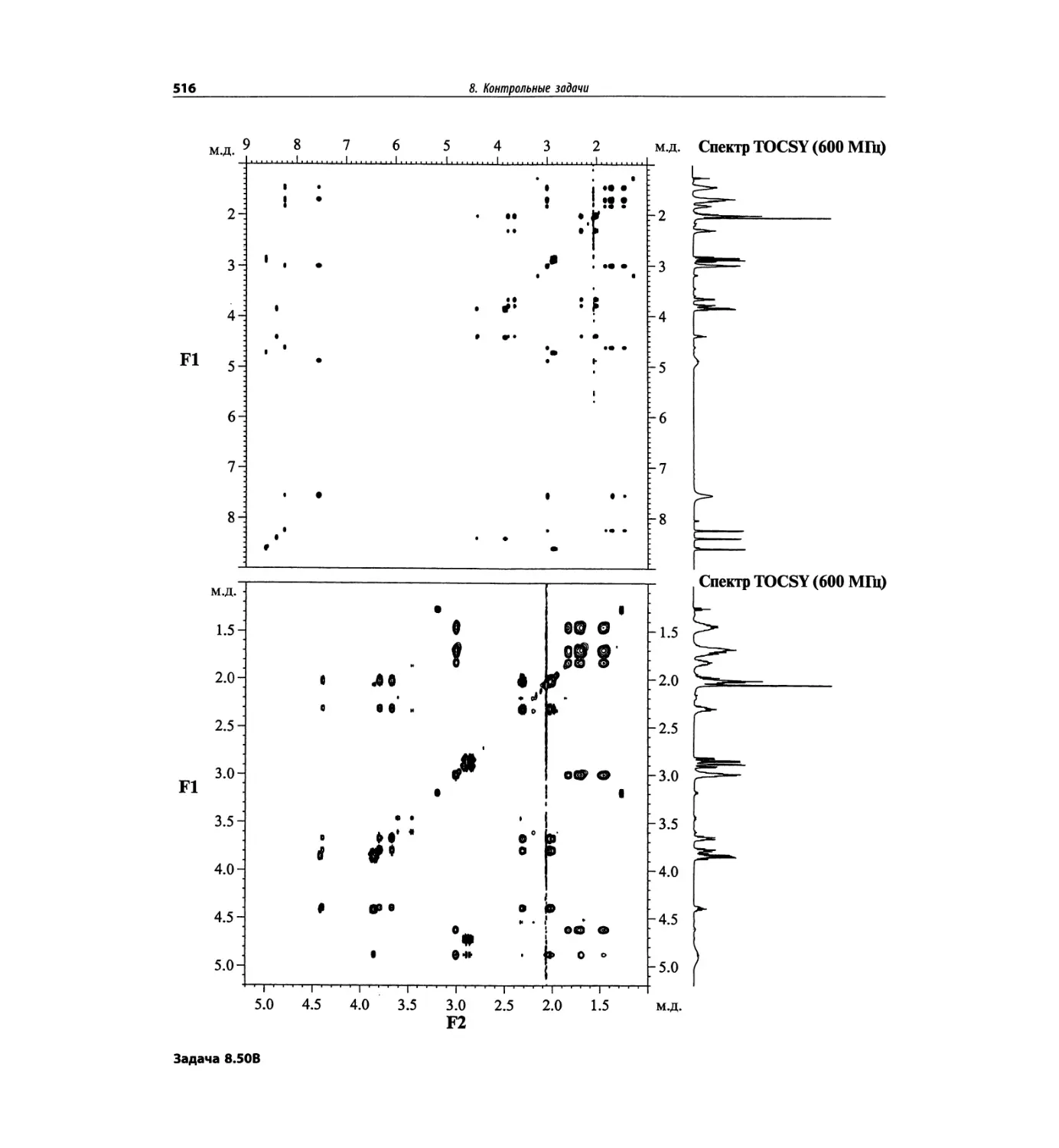
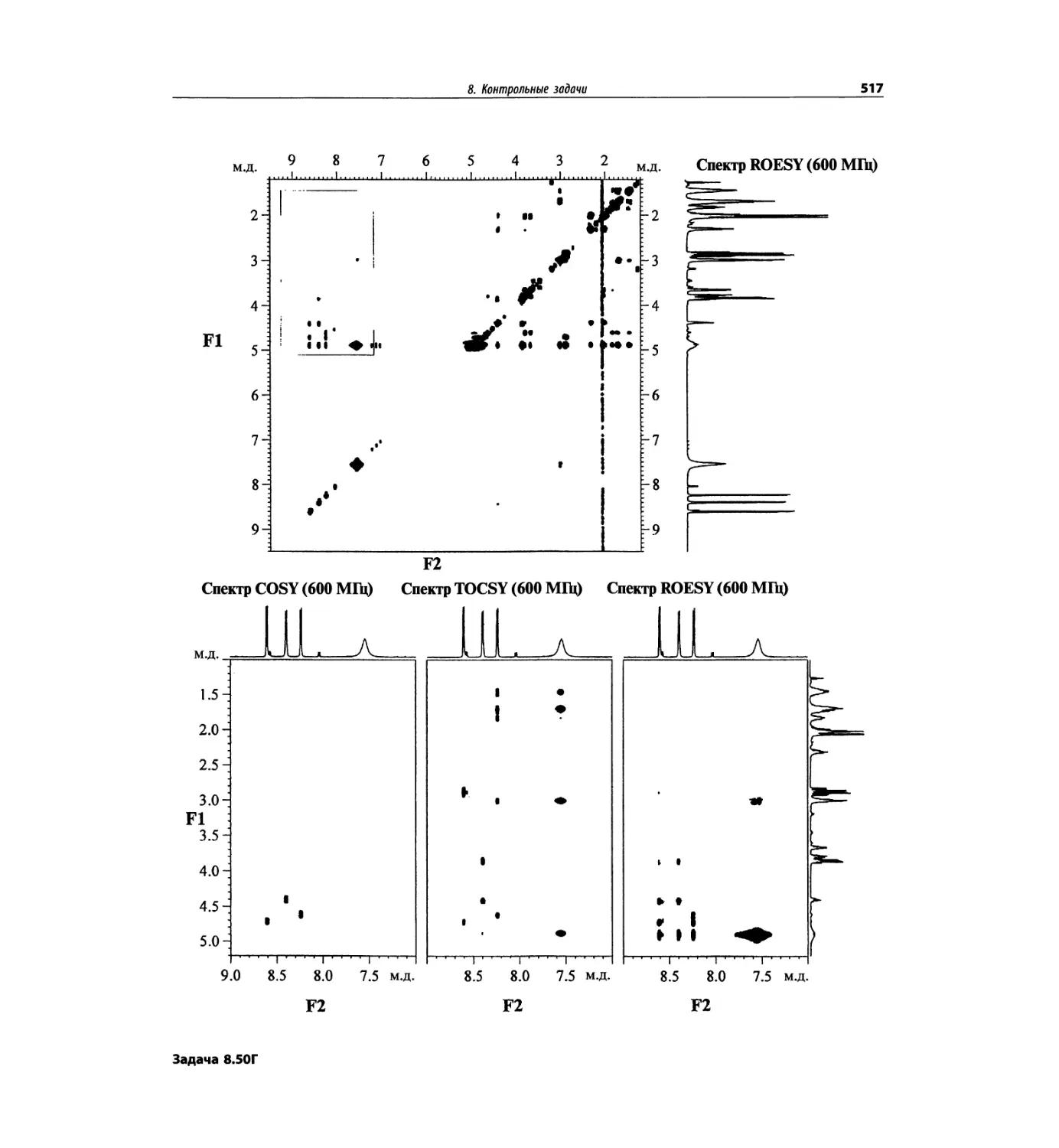
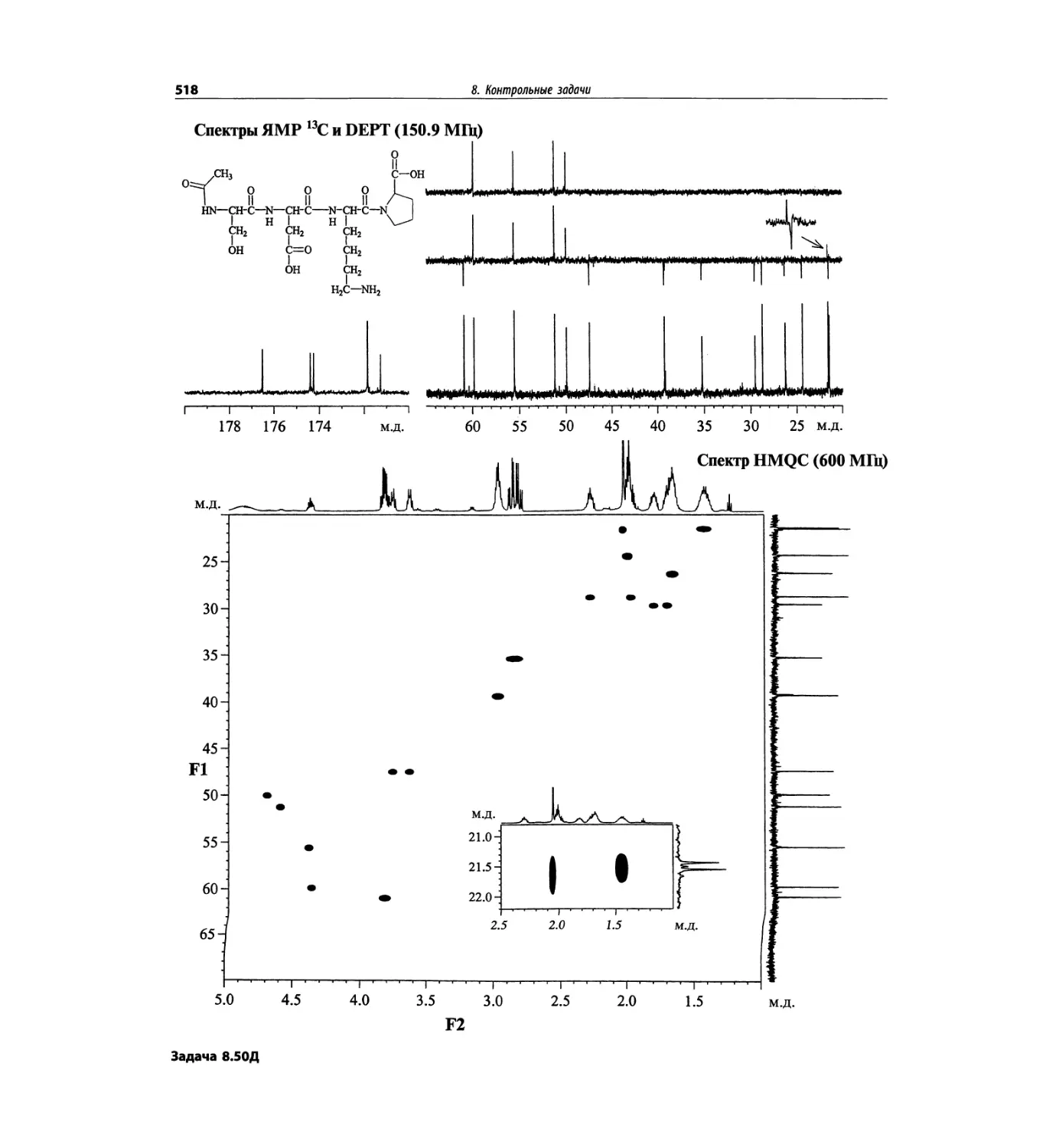
Корреляционная спектроскопия ЯМР. 2D-KMP (глава 5)
Глава 5 по-прежнему посвящена двумерной корреля-
ционной спектроскопии, но она существенно пересмо-
трена, расширена и обновлена, что отражает возросшее
значение этих спектров. В результате перегруппировки
материала все спектры конкретного соединения по-
мещены вместе и каждый пример рассматривается
отдельно: ипсенол, кариофилленоксид, лактоза и те-
трапептид. Для большей части экспериментов пред-
ставлены импульсные последовательности. Особое
внимание уделено новым двумерным экспериментам,
таким как ROESY, и гибридным методам, например
HMQC-TOCSY. Включено много новых упражнений.
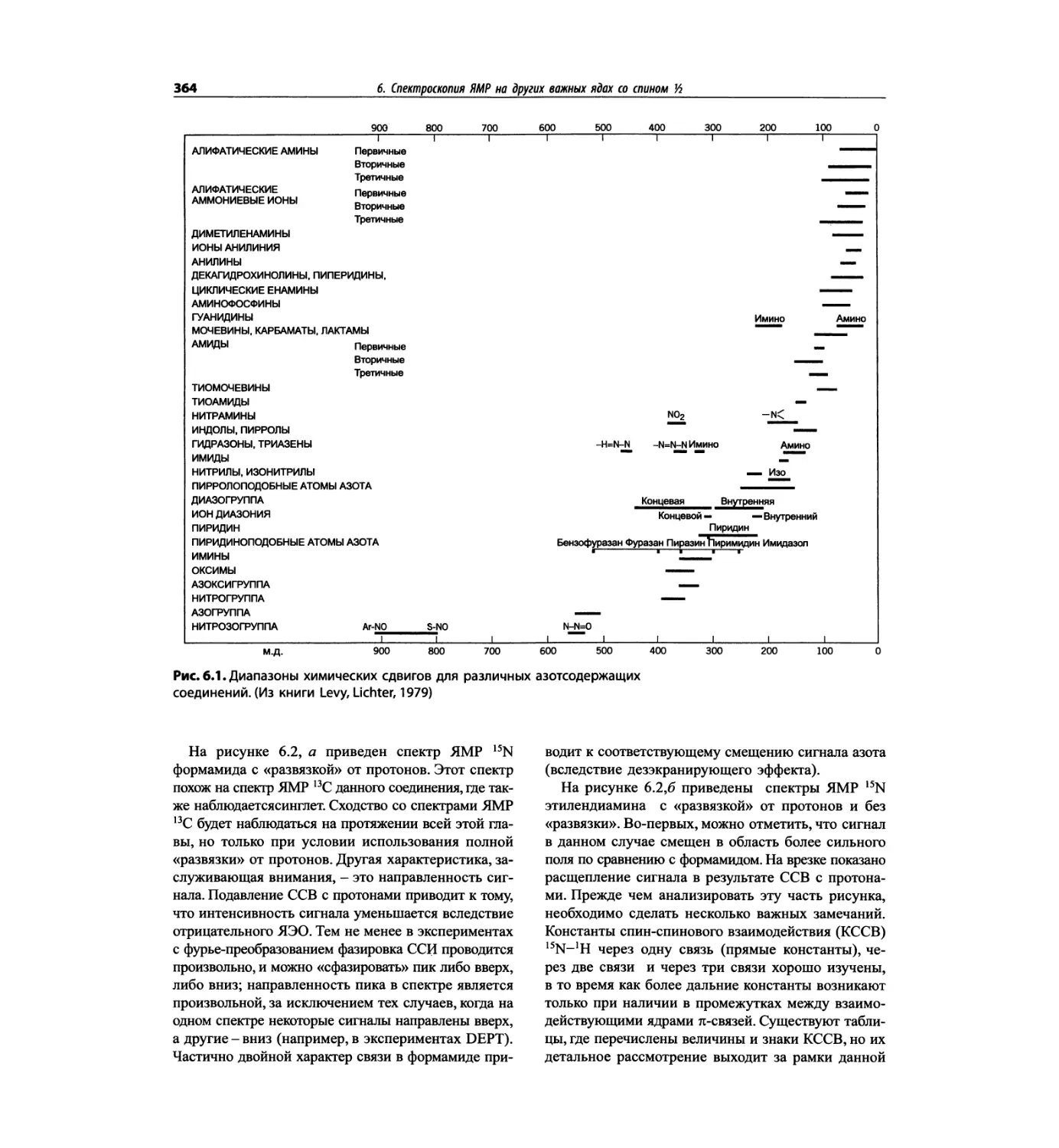
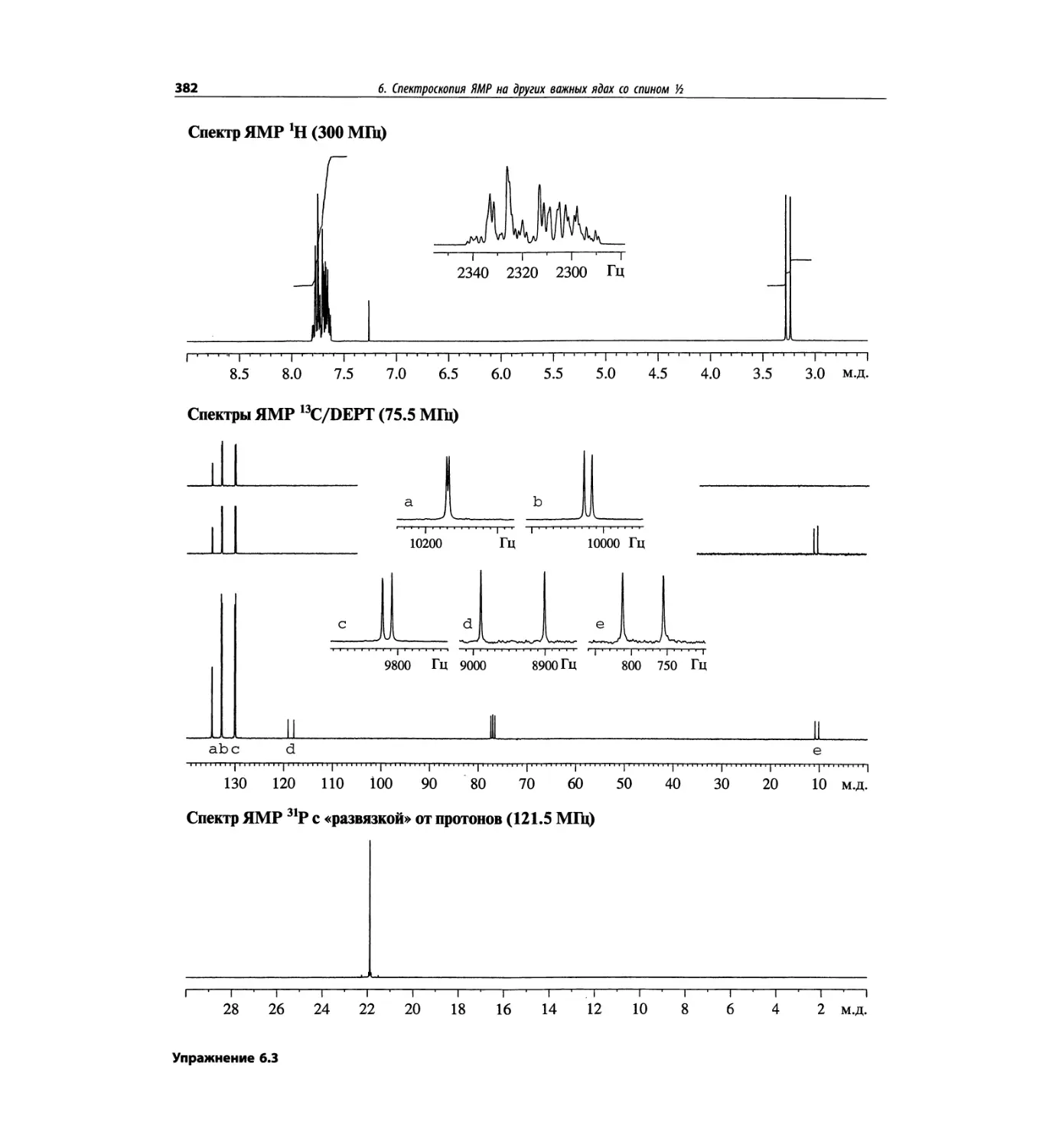
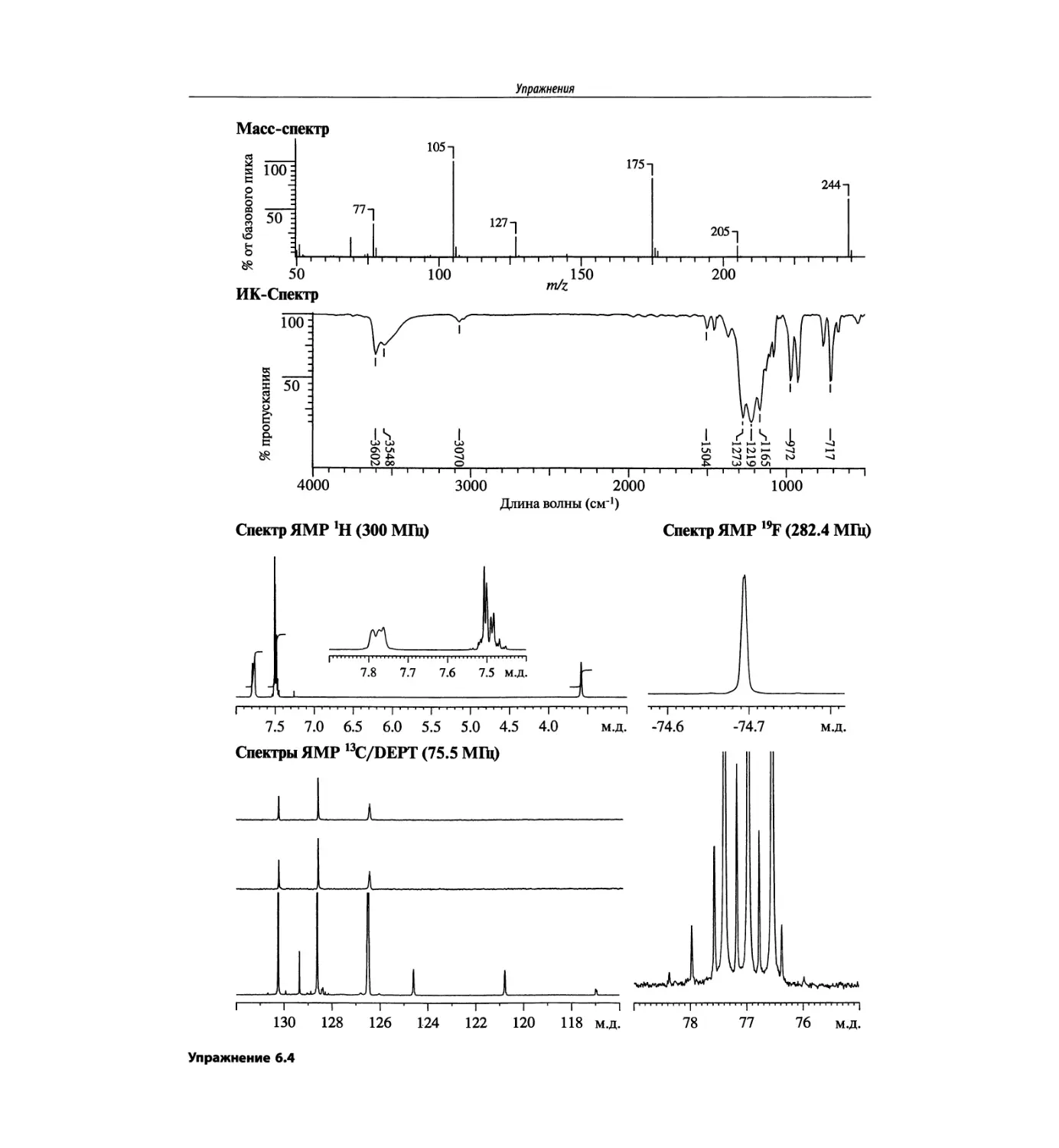
Спектроскопия ЯМР на других важных ядрах
со спином 1/2 (глава 6)
Глава 6 расширена за счет примеров, более подроб-
ных таблиц и улучшенного представления спектров.
Изложение материала служит выявлению химических
корреляций на ядрах 15N, 19F, 29Si, 31Р и включает не-
сколько 2О-спектров.
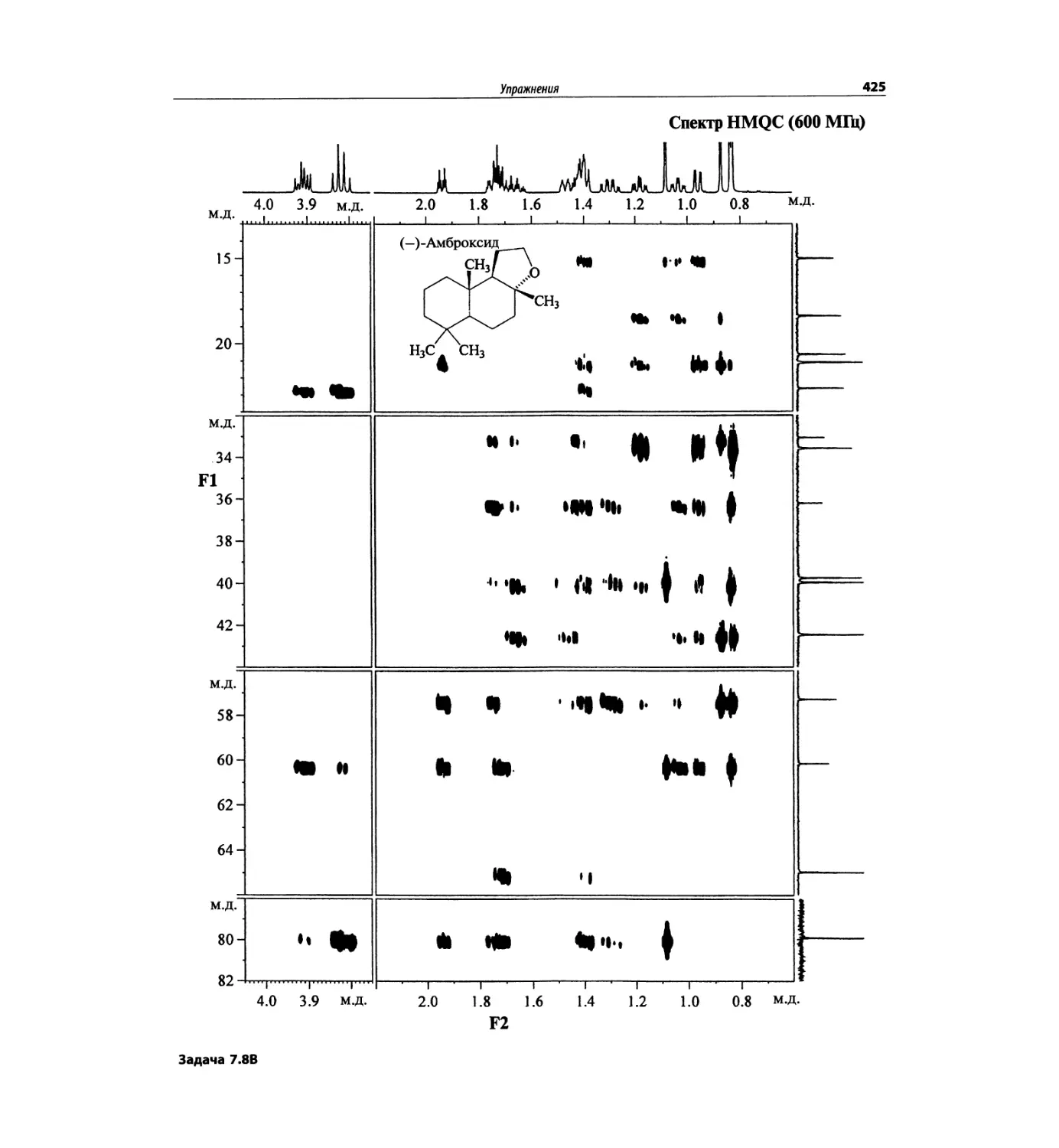
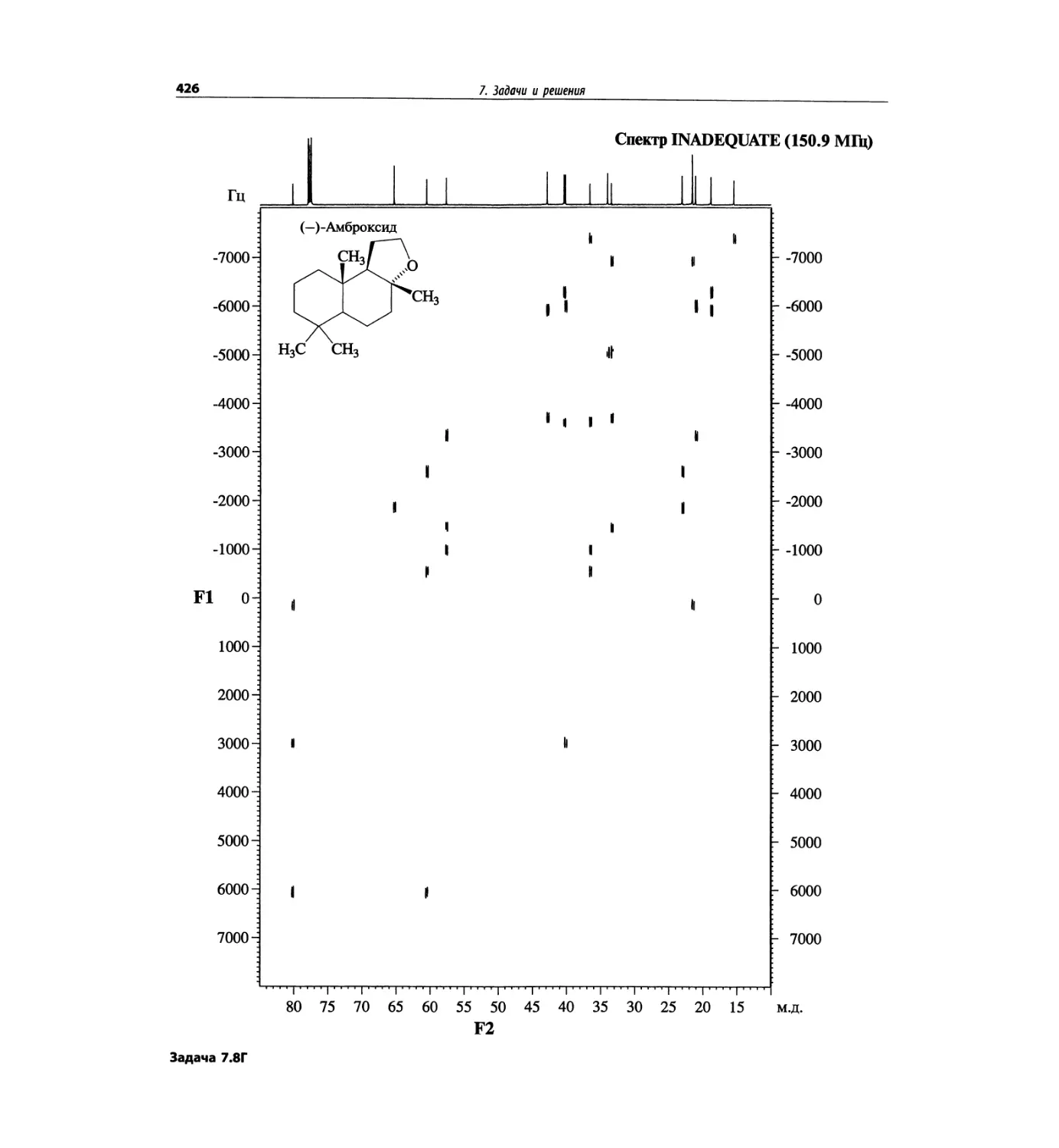
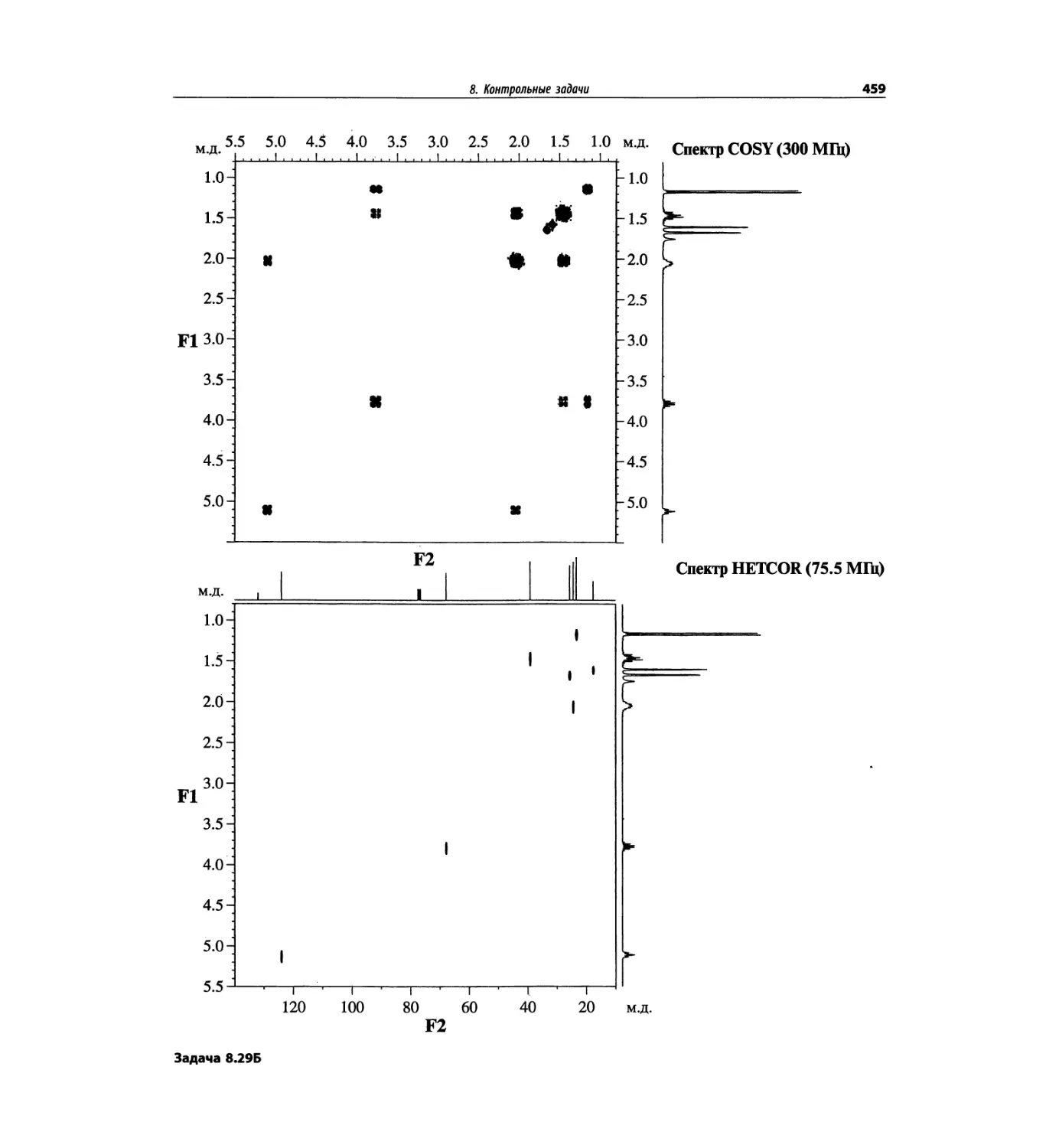
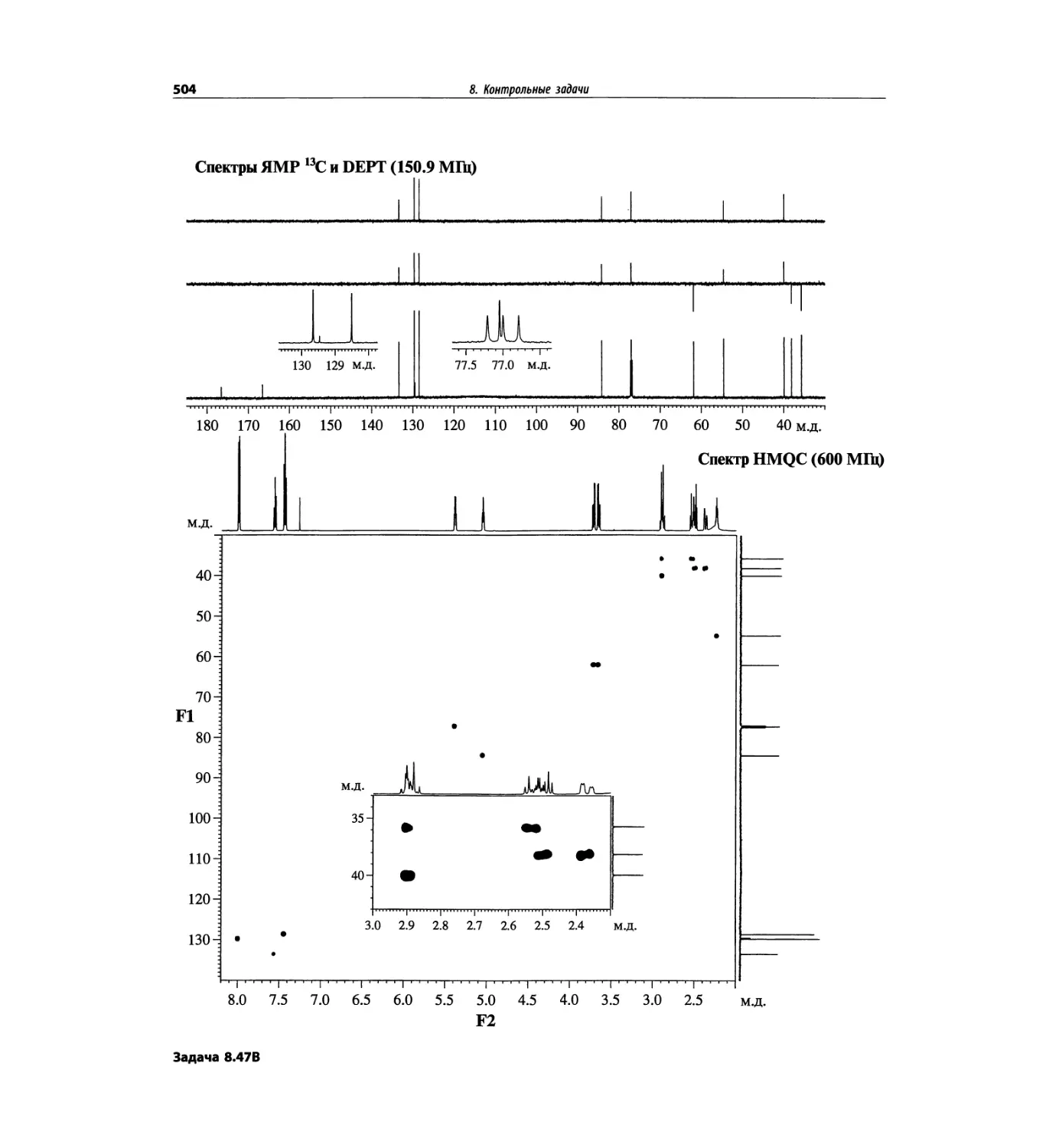
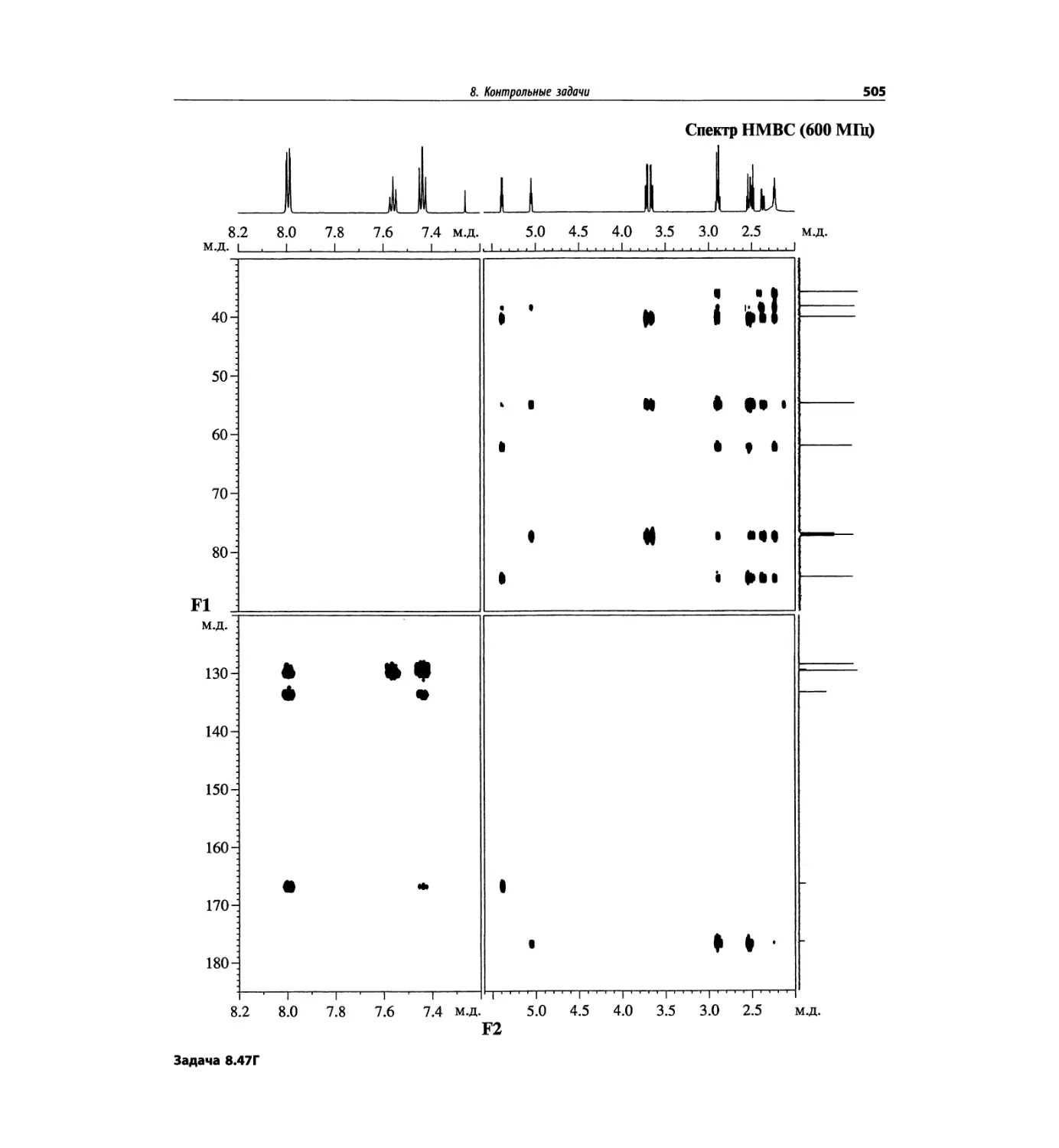
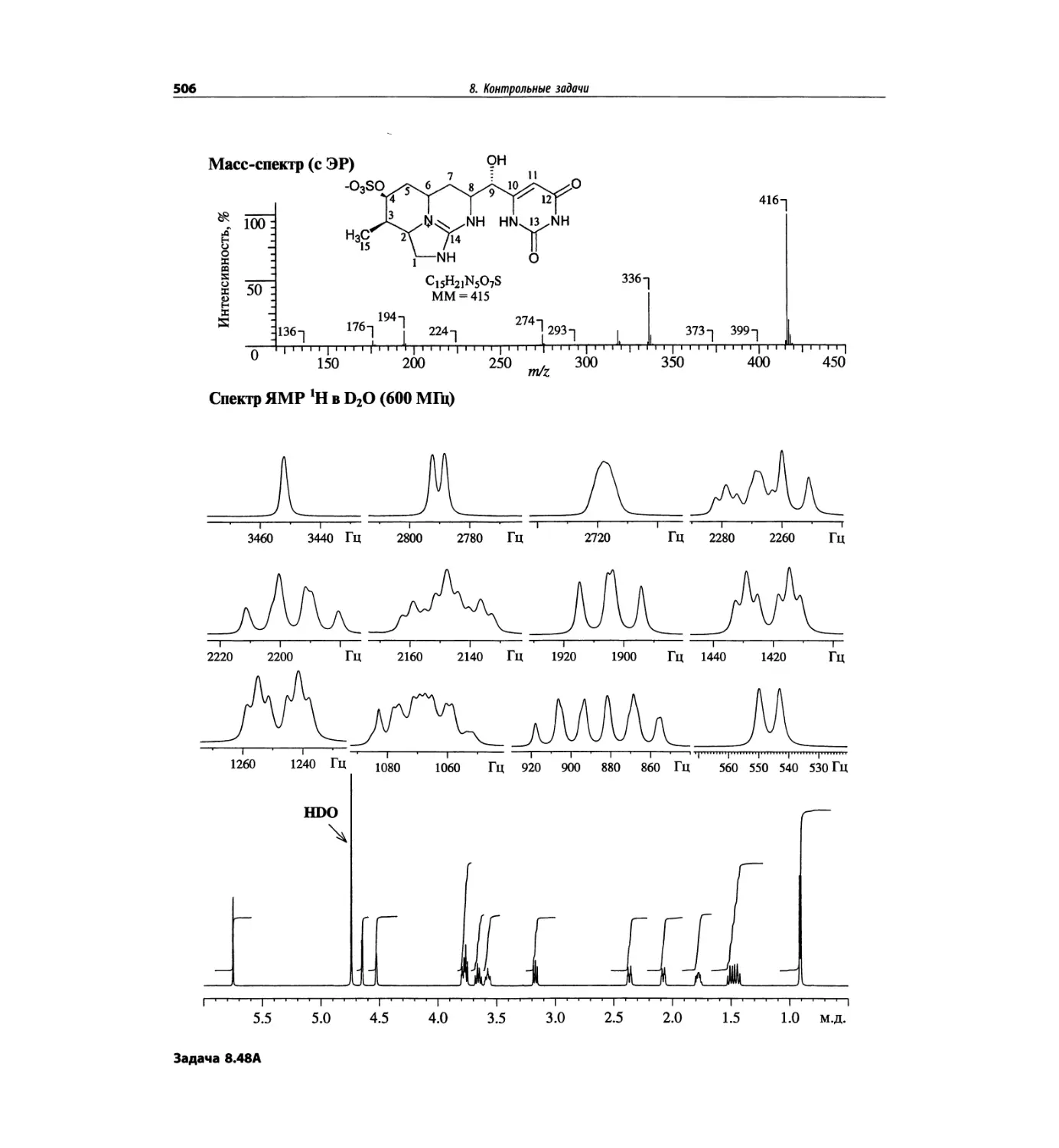
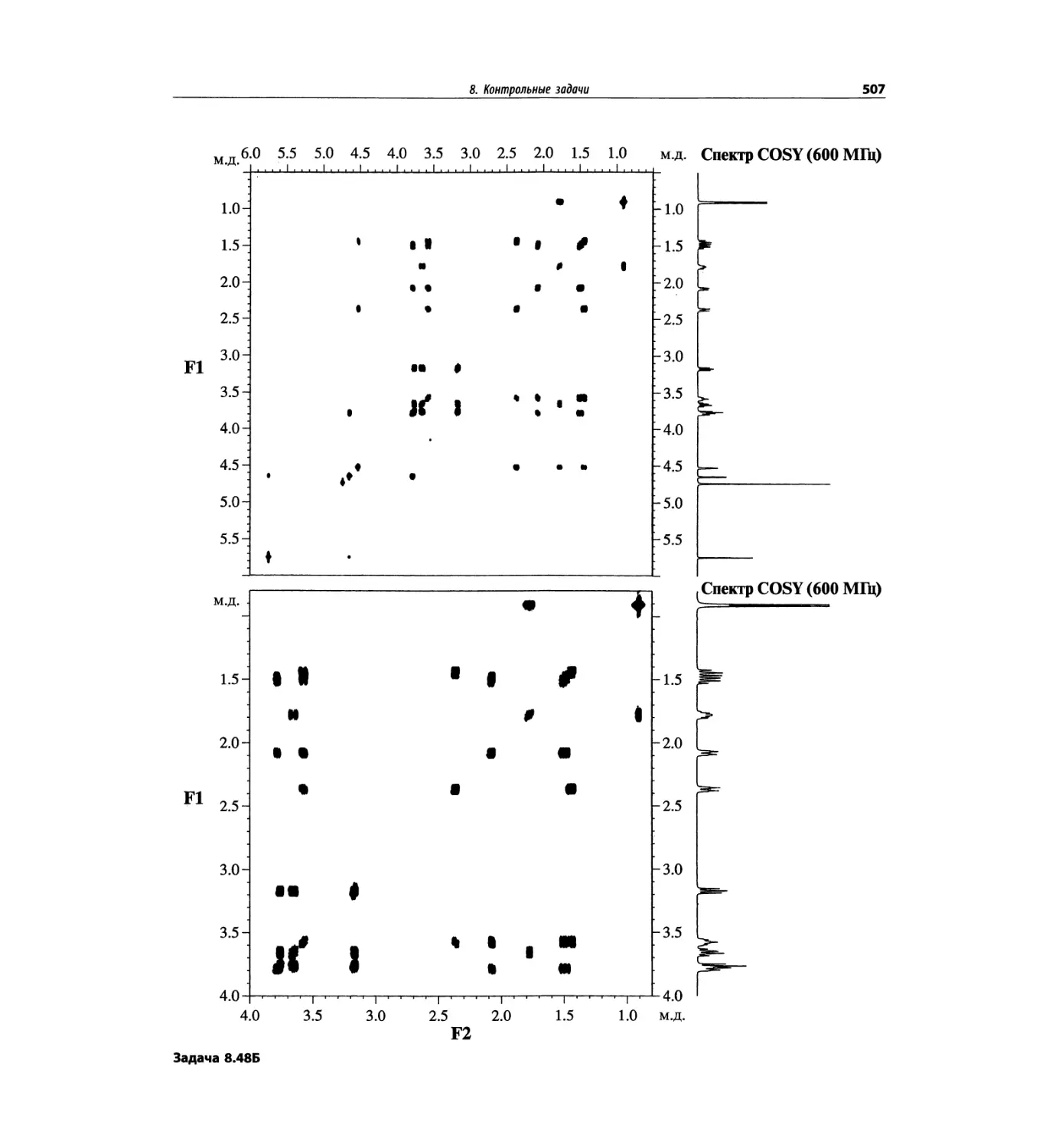
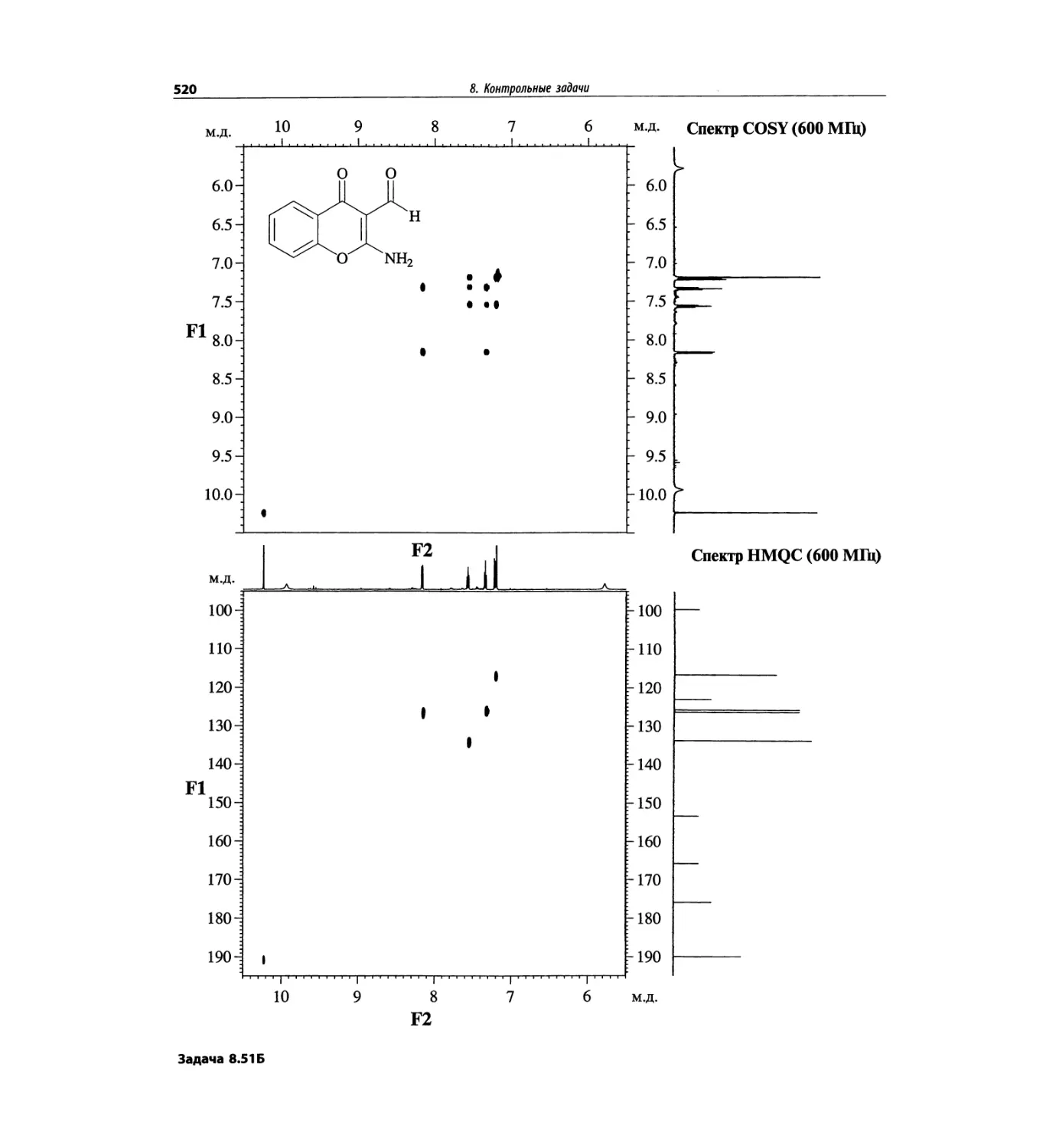
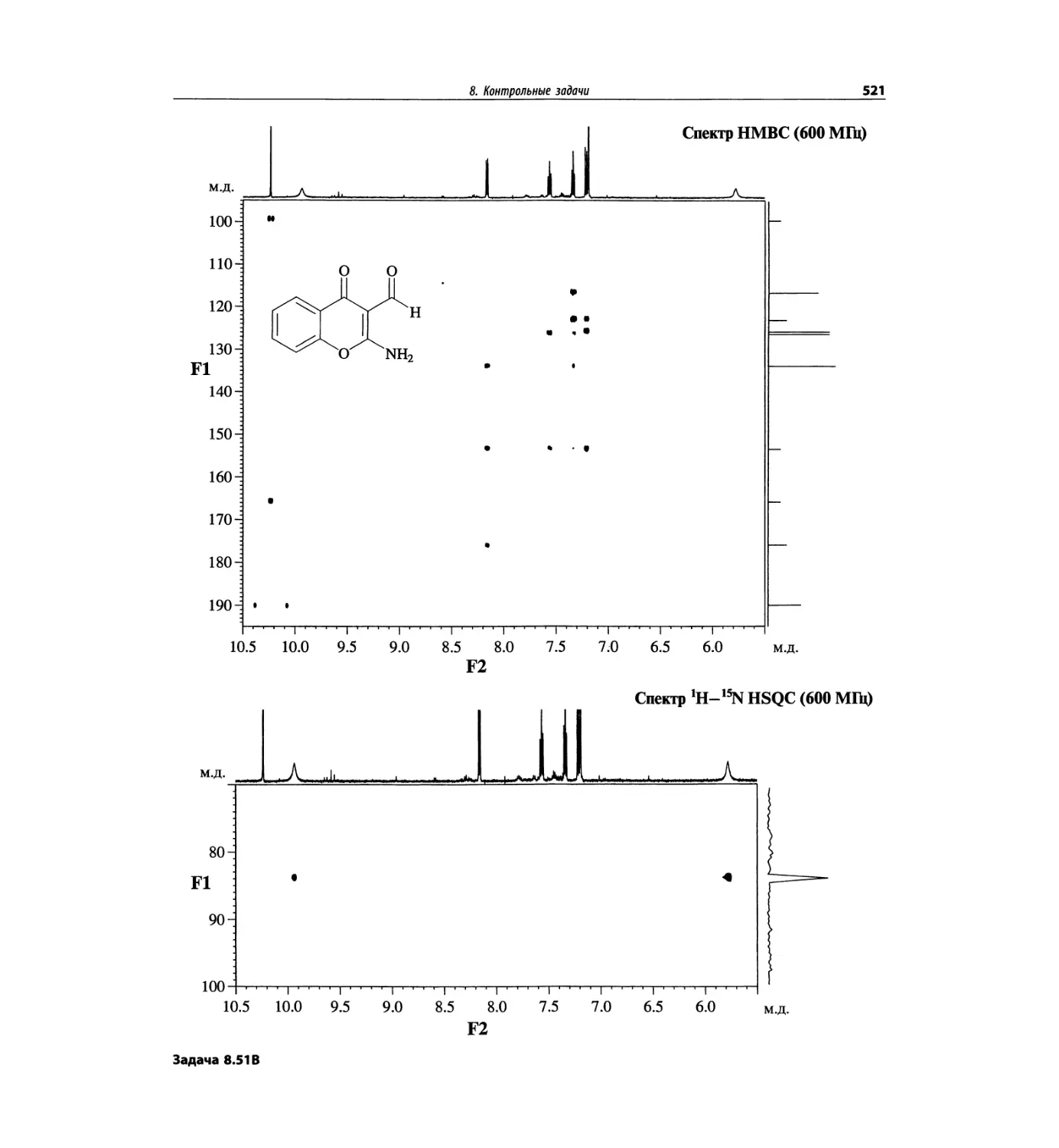
Задачи и решения (глава 7)
Глава 7 состоит из введения и шести разобранных за-
дач. Предложенные в этой главе подходы применимы
для решения других задач и полезны для студентов.
Мы воздержались от чрезмерно жестких предписаний.
Мы советуем студентам развивать собственные под-
ходы к решениям и предостерегаем от поспешности.
Эти шесть упражнений расположены в порядке возрас-
тания сложности. В главу добавлены два упражнения,
в которых предлагается провести отнесение и под-
твердить предложенные структуры. Дополнительные
упражнения такого типа помещены в конце гл. 8.
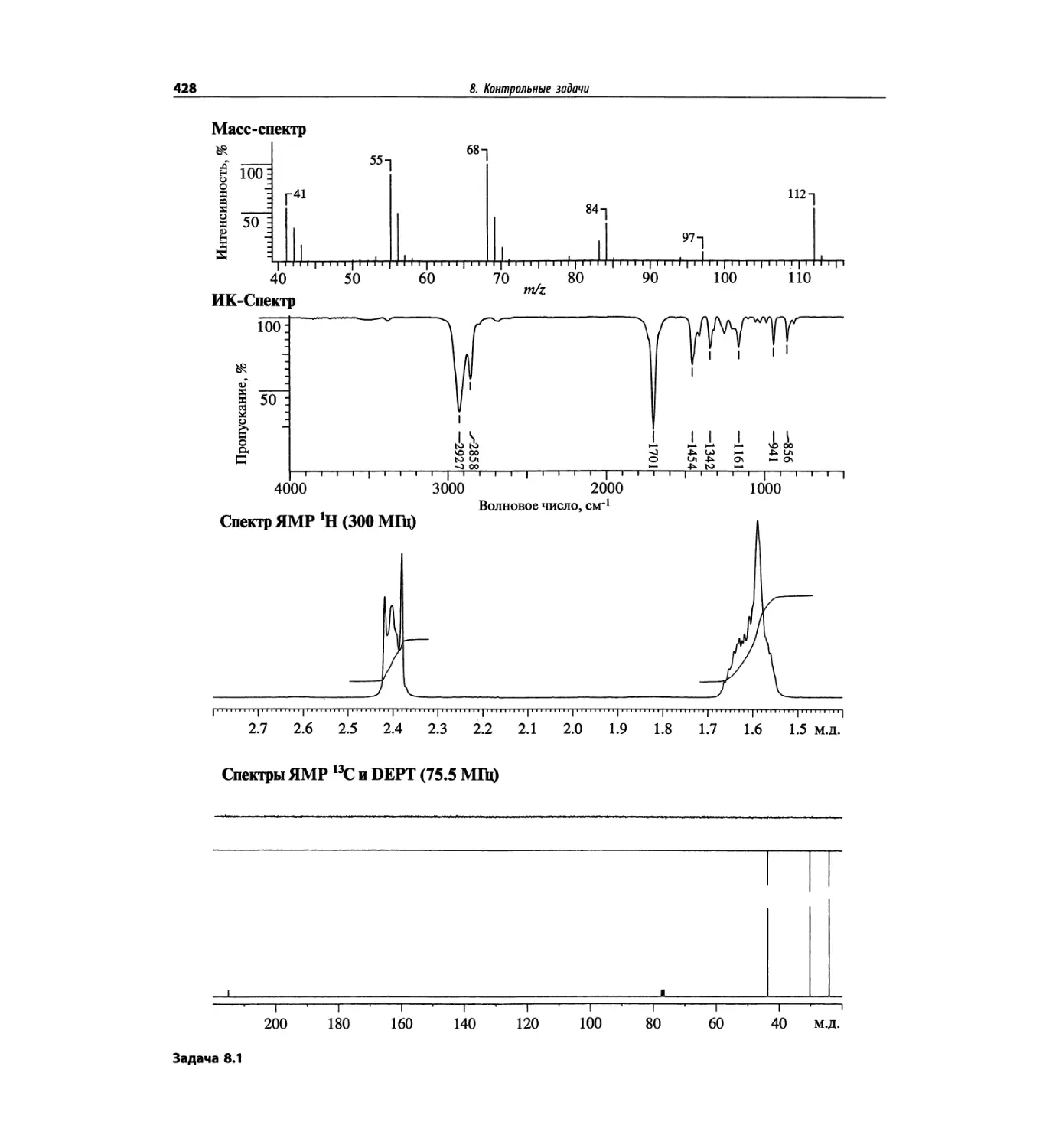
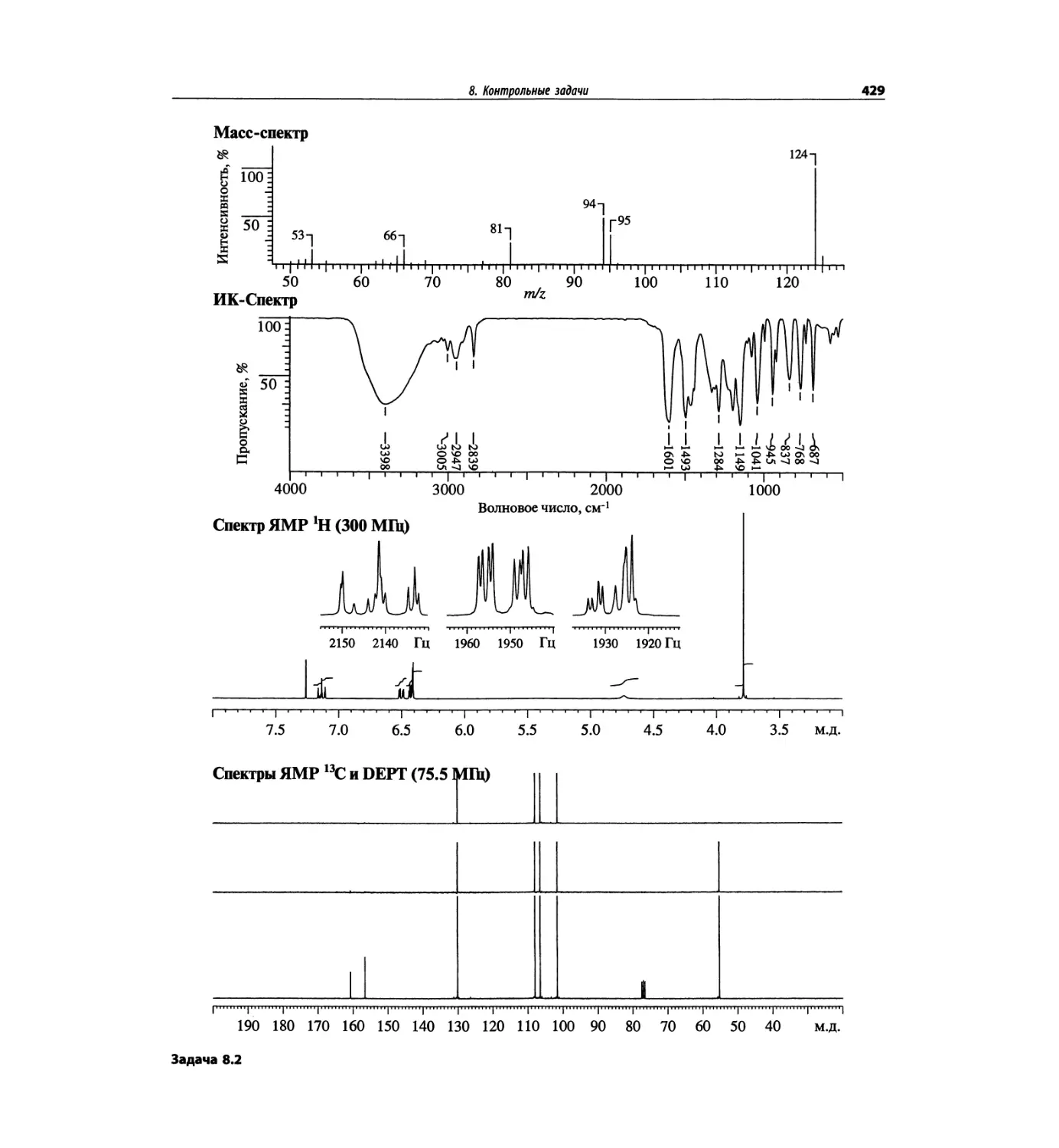
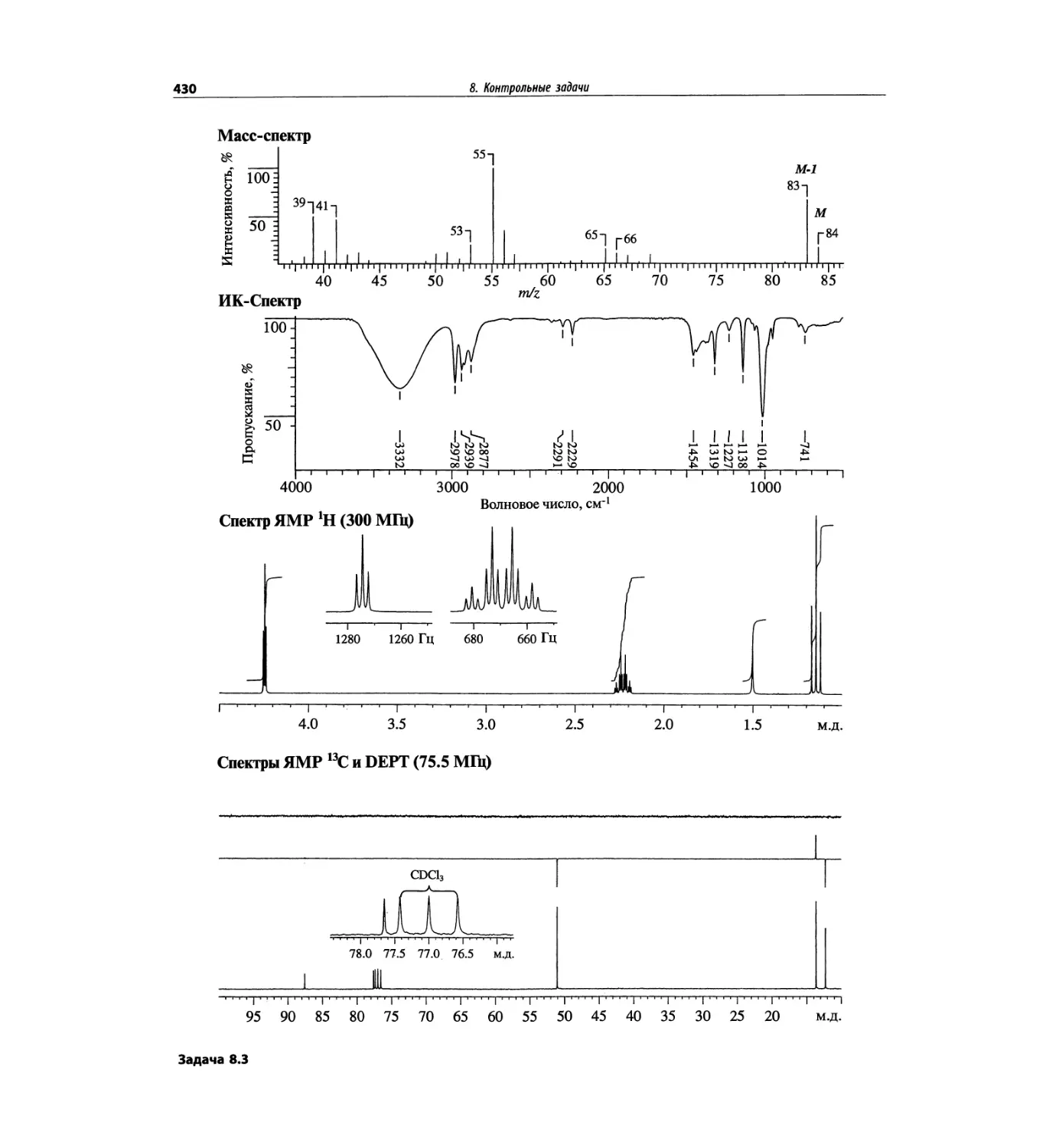
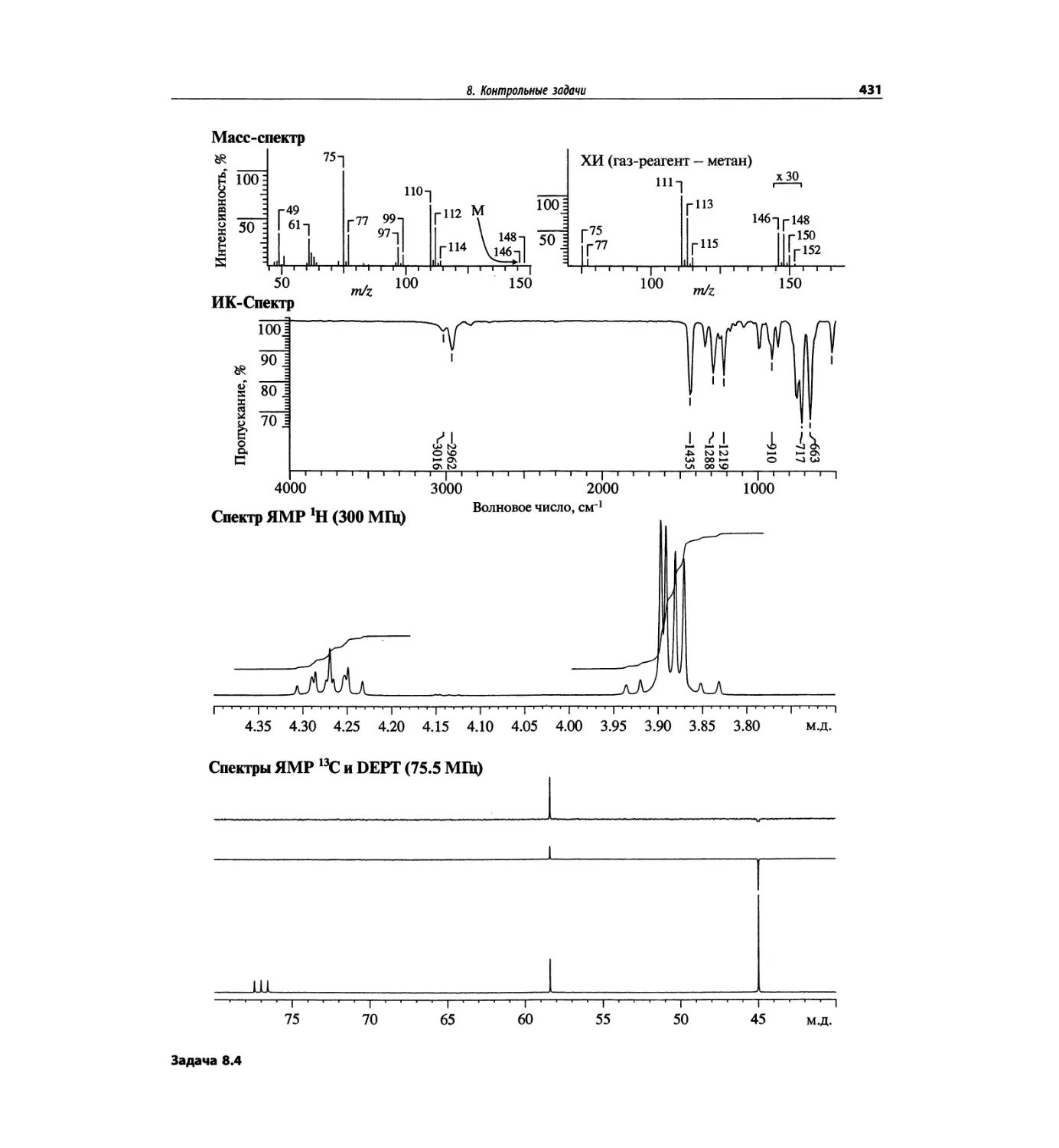
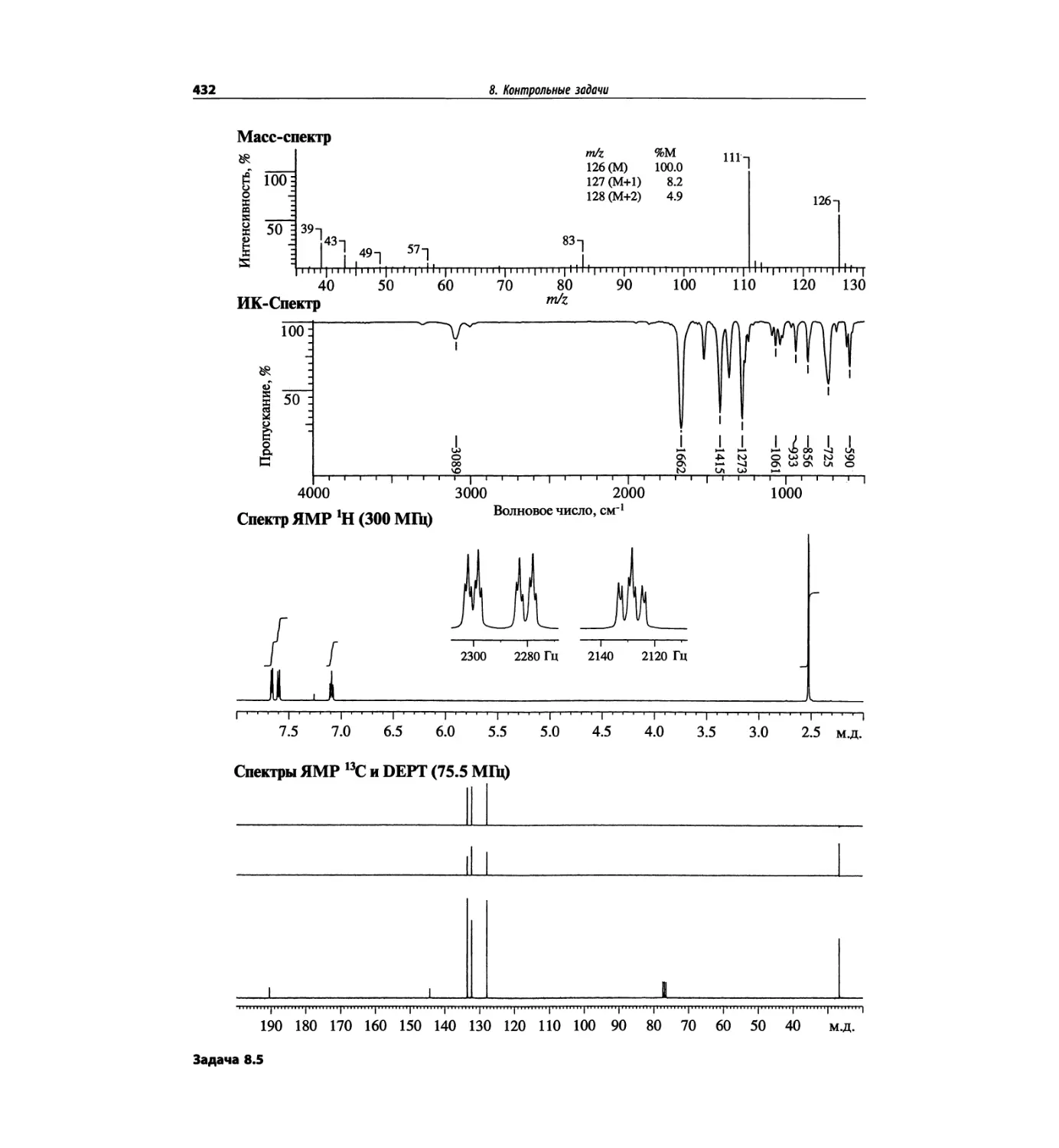
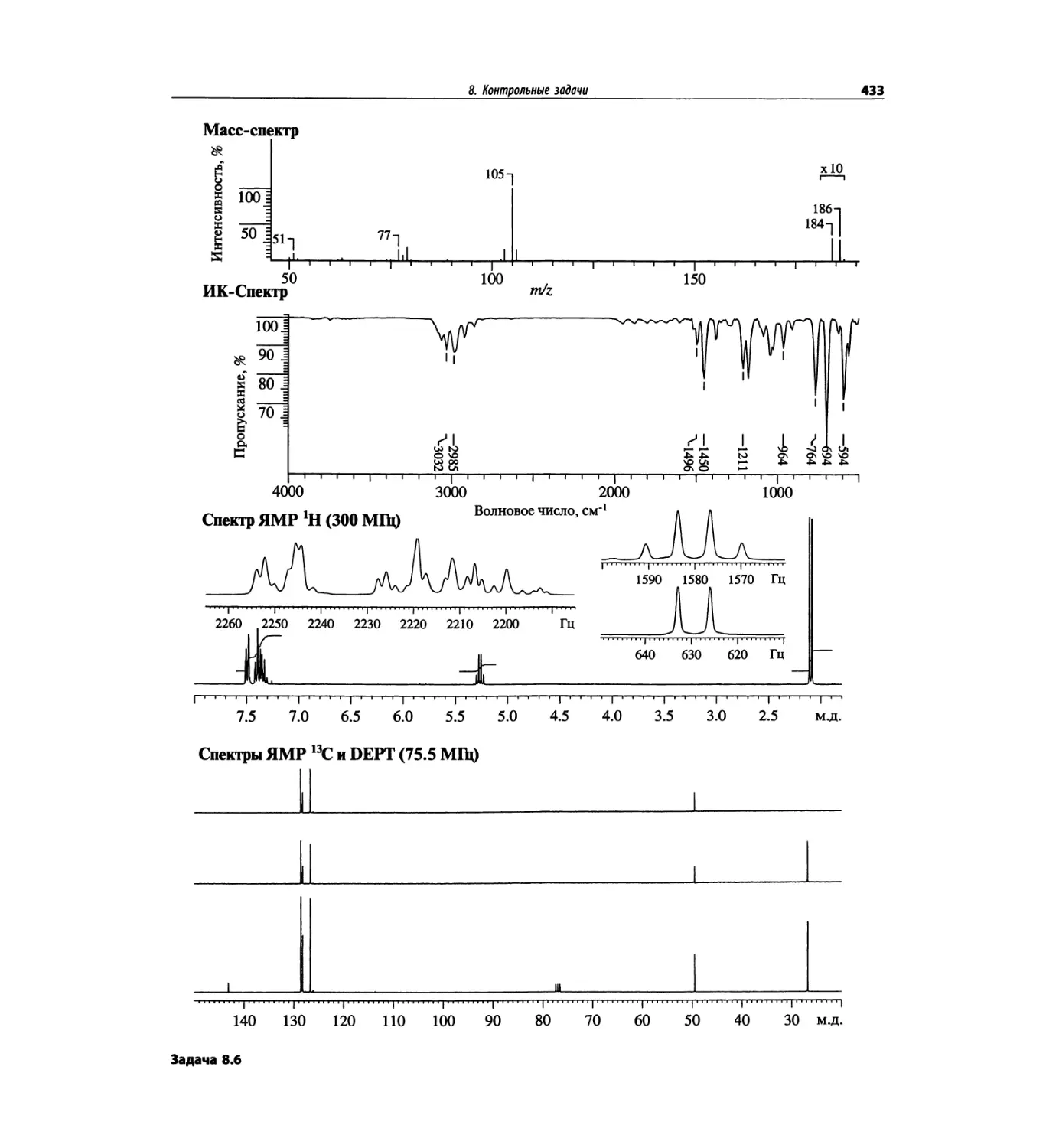
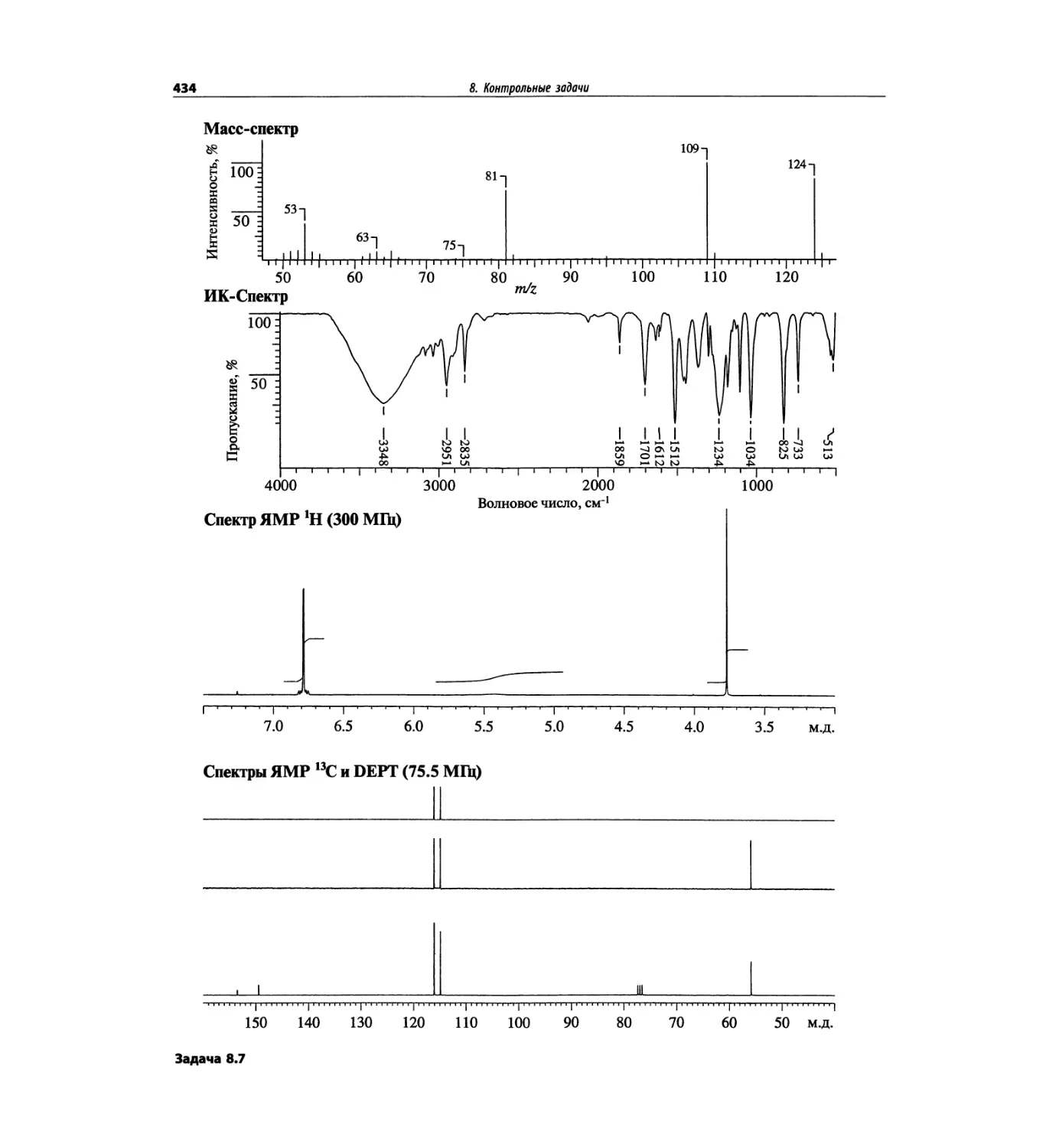
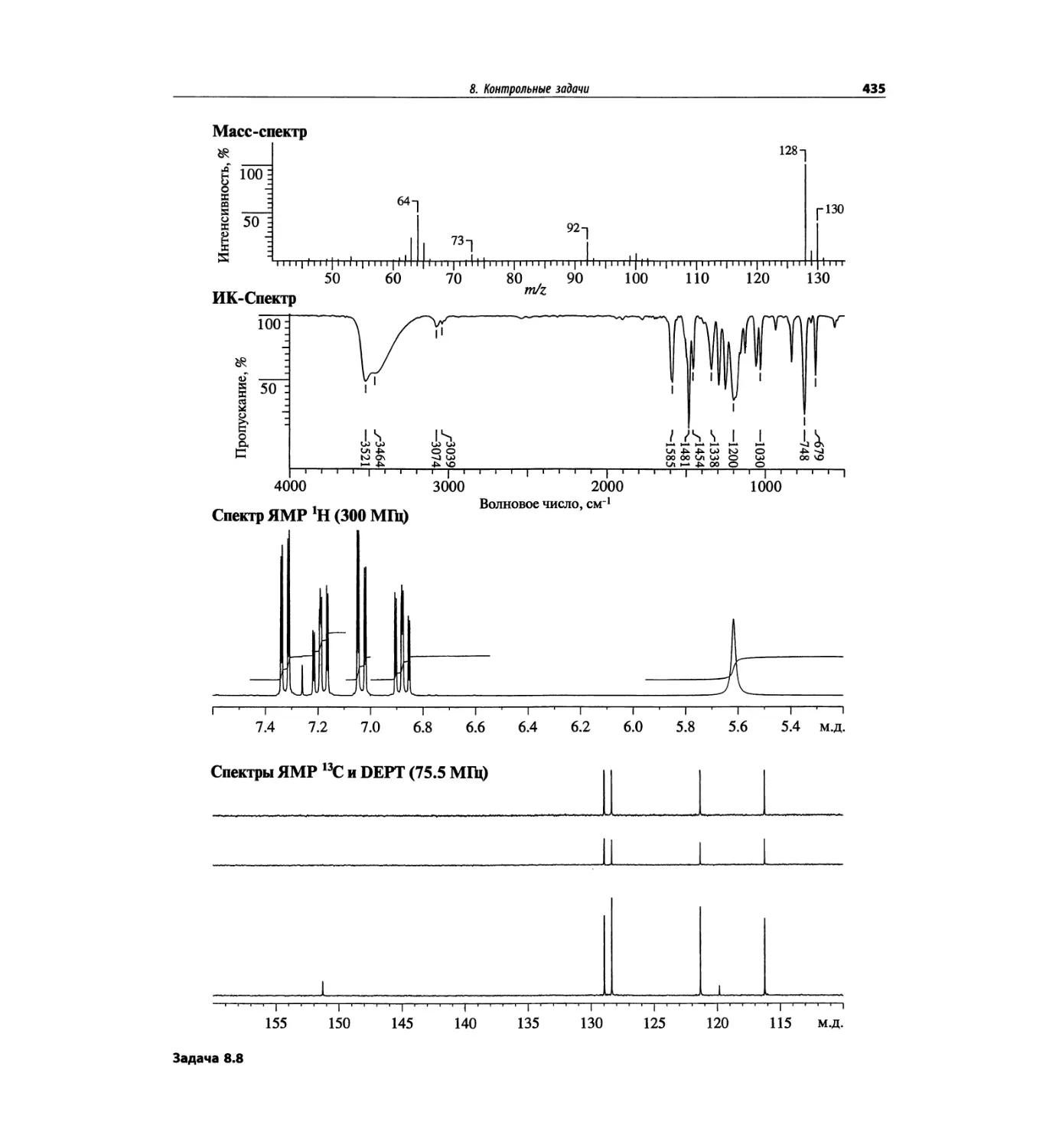
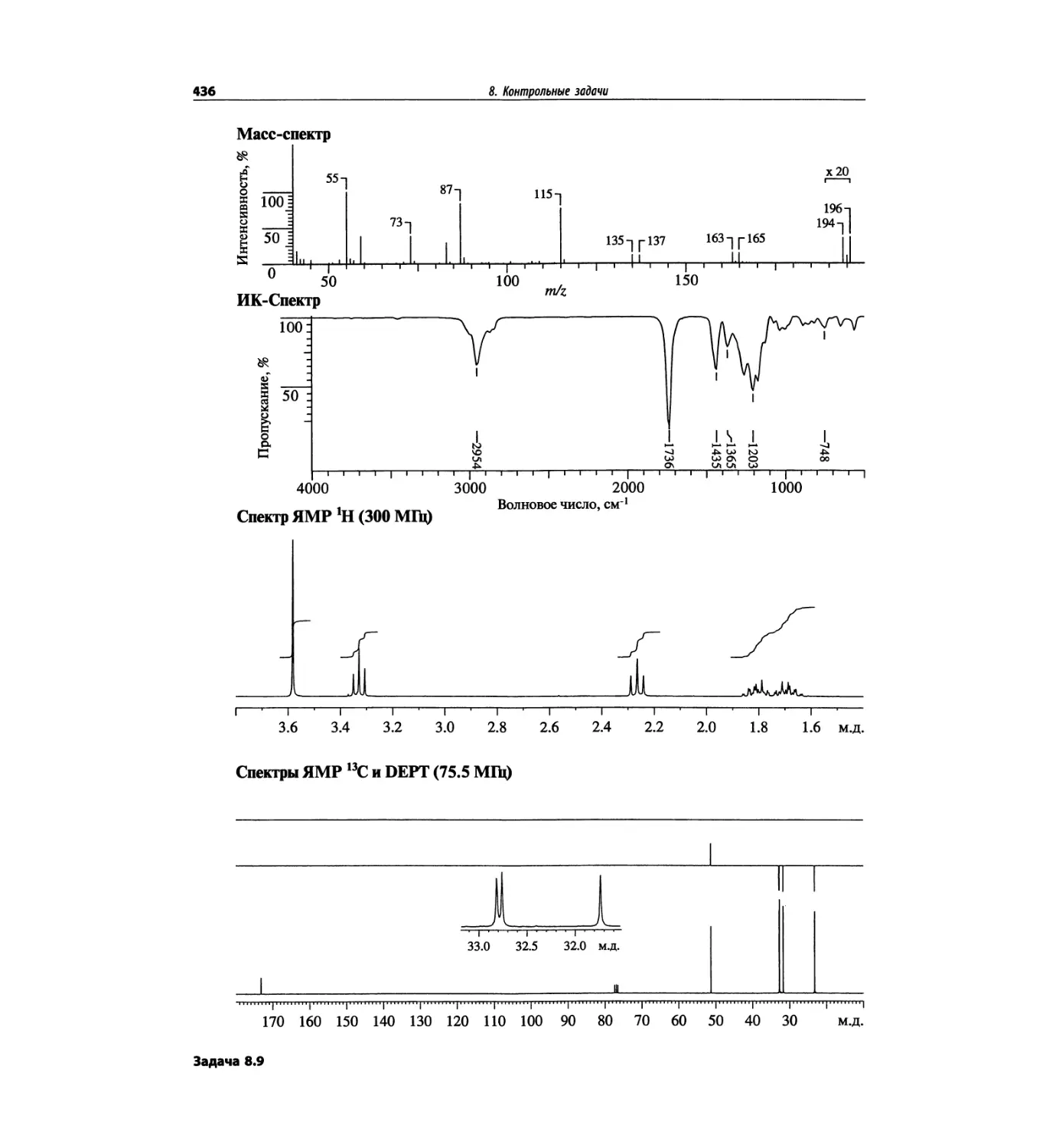
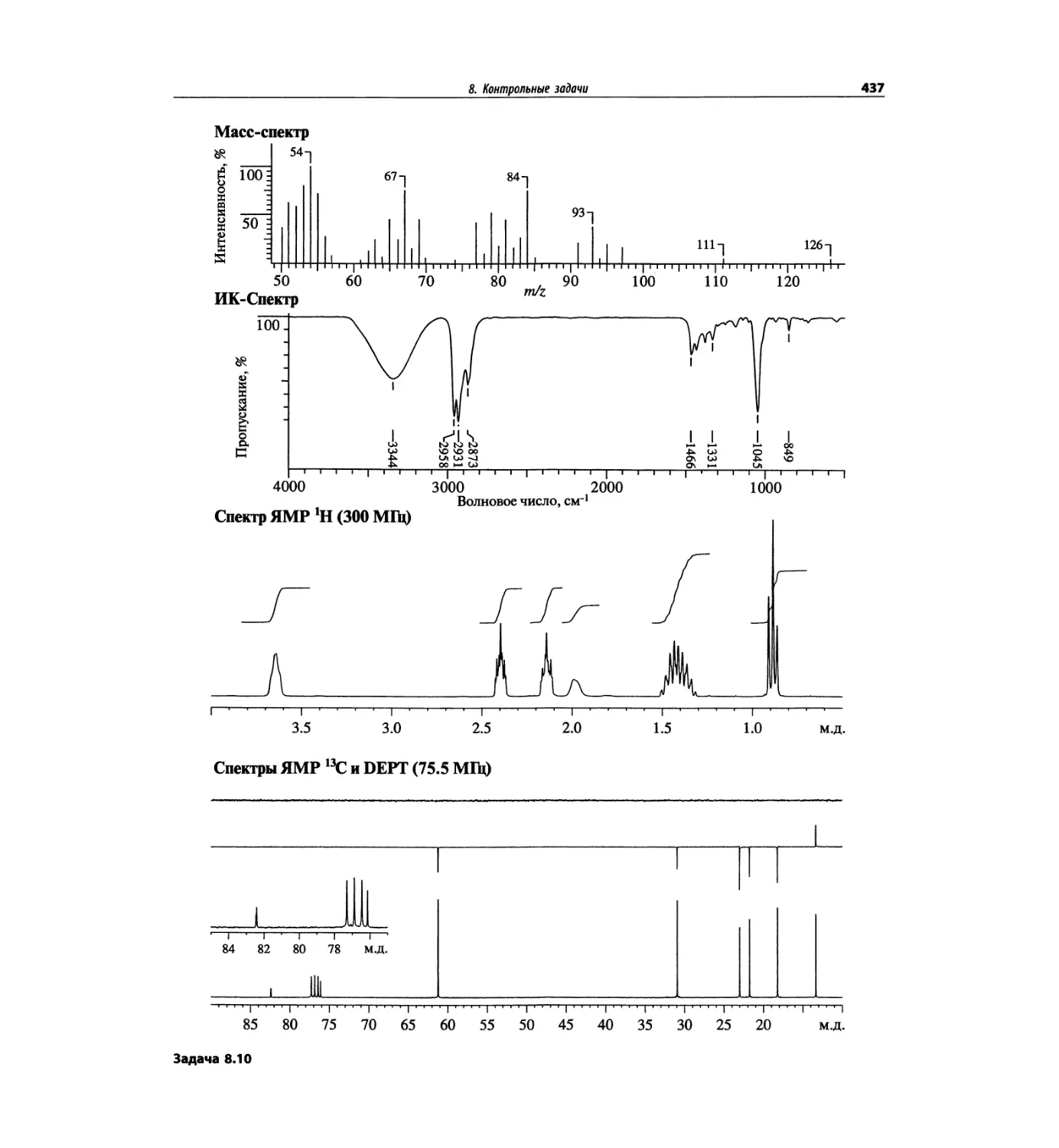
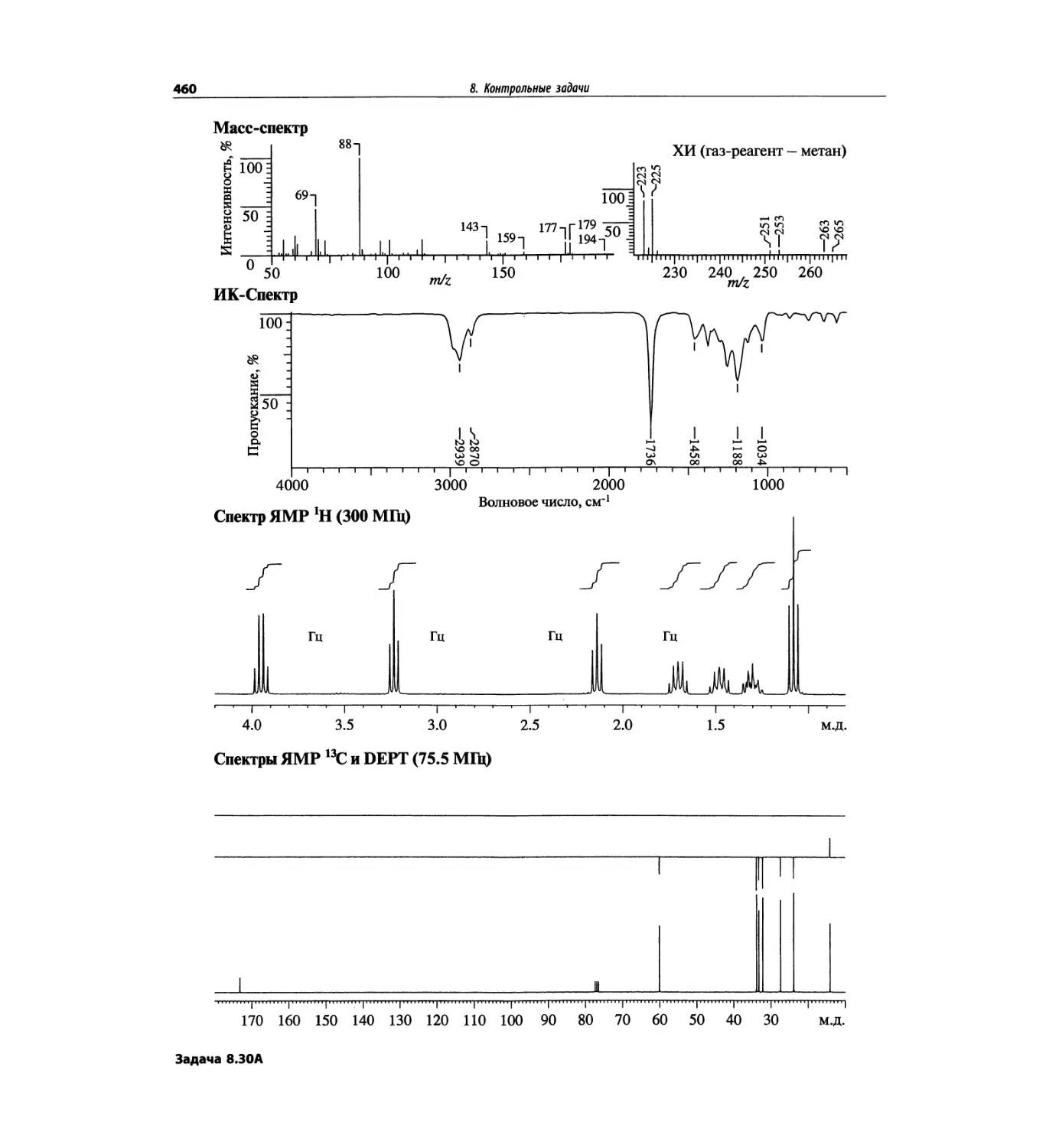
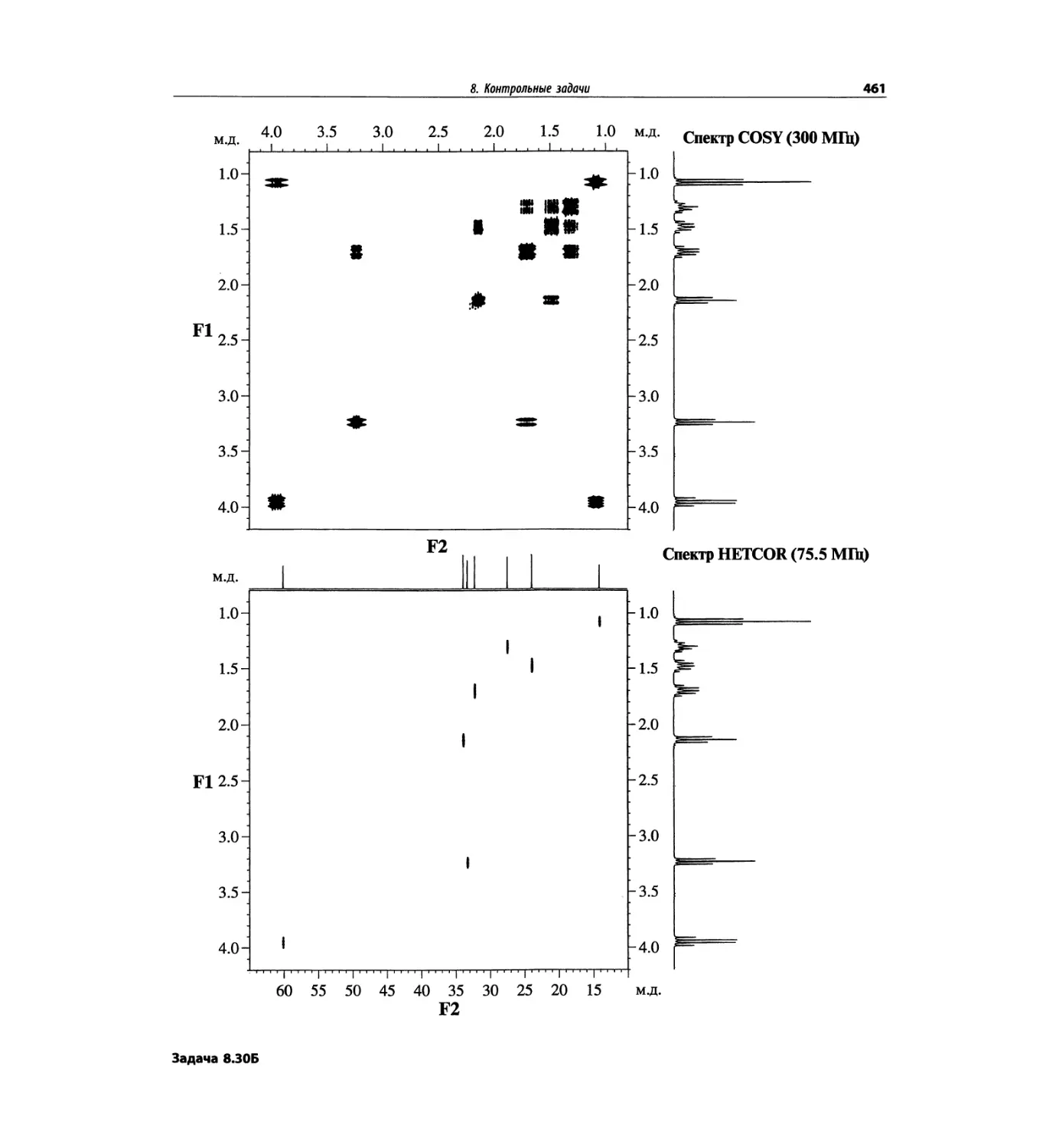
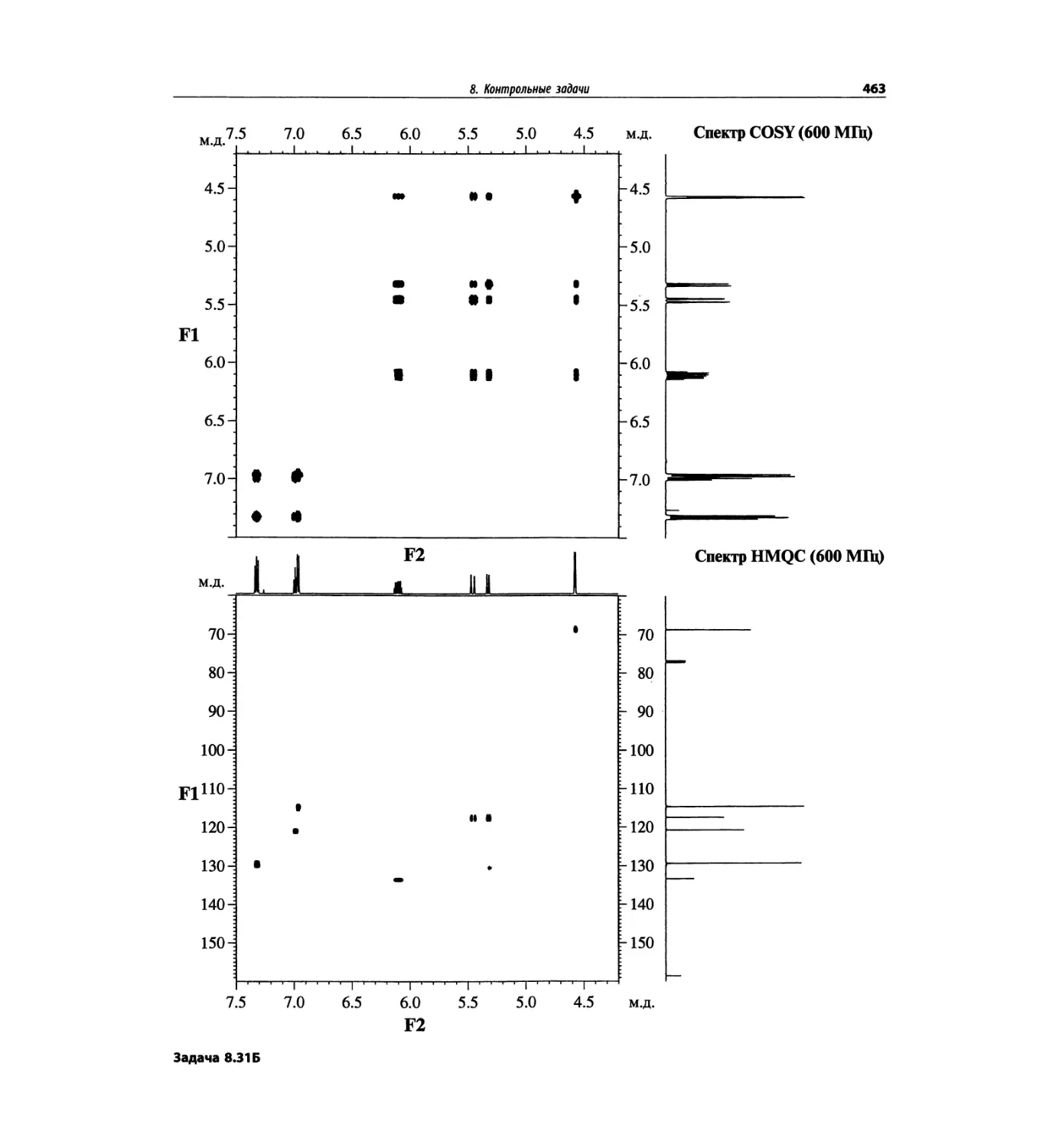
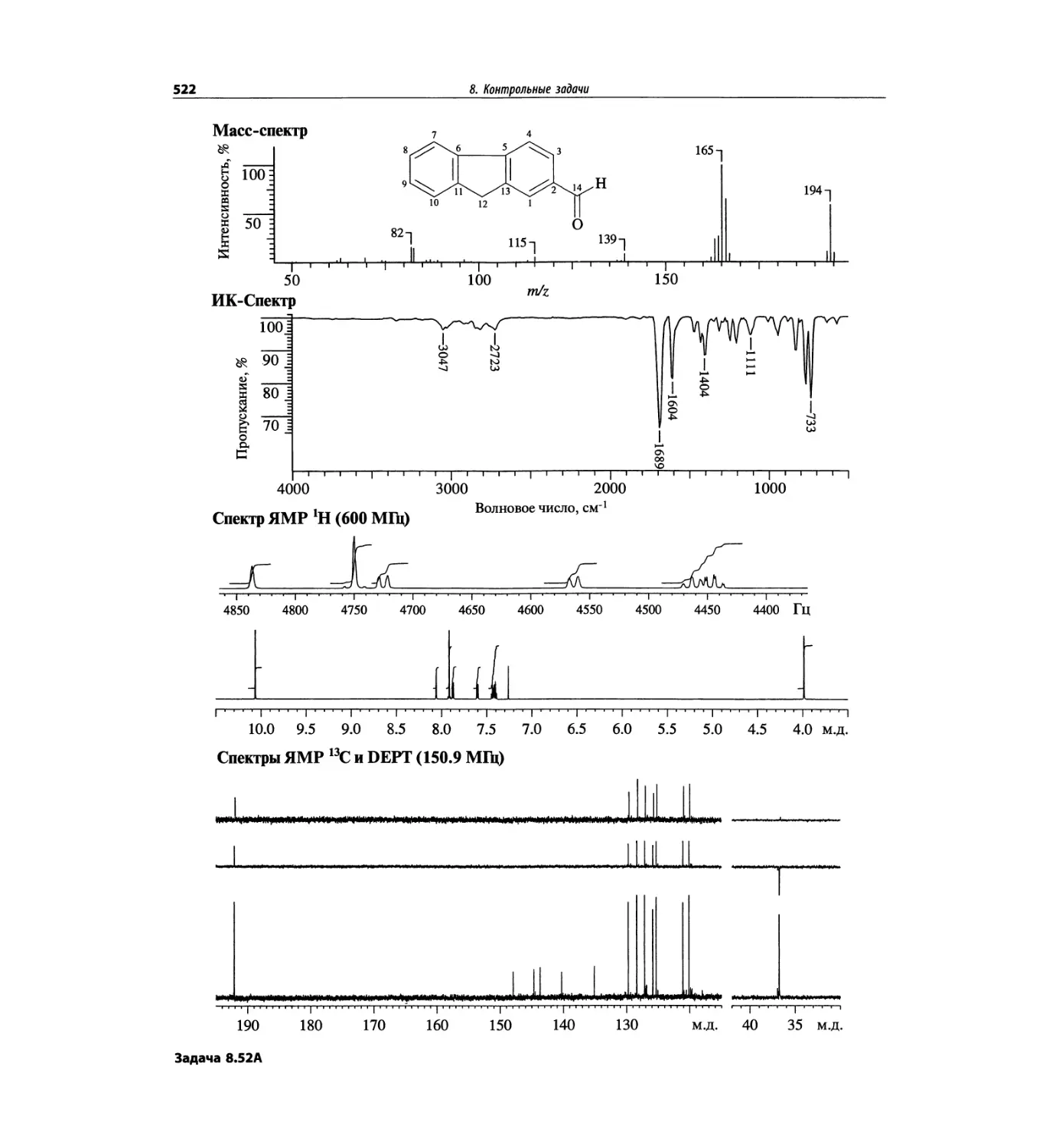
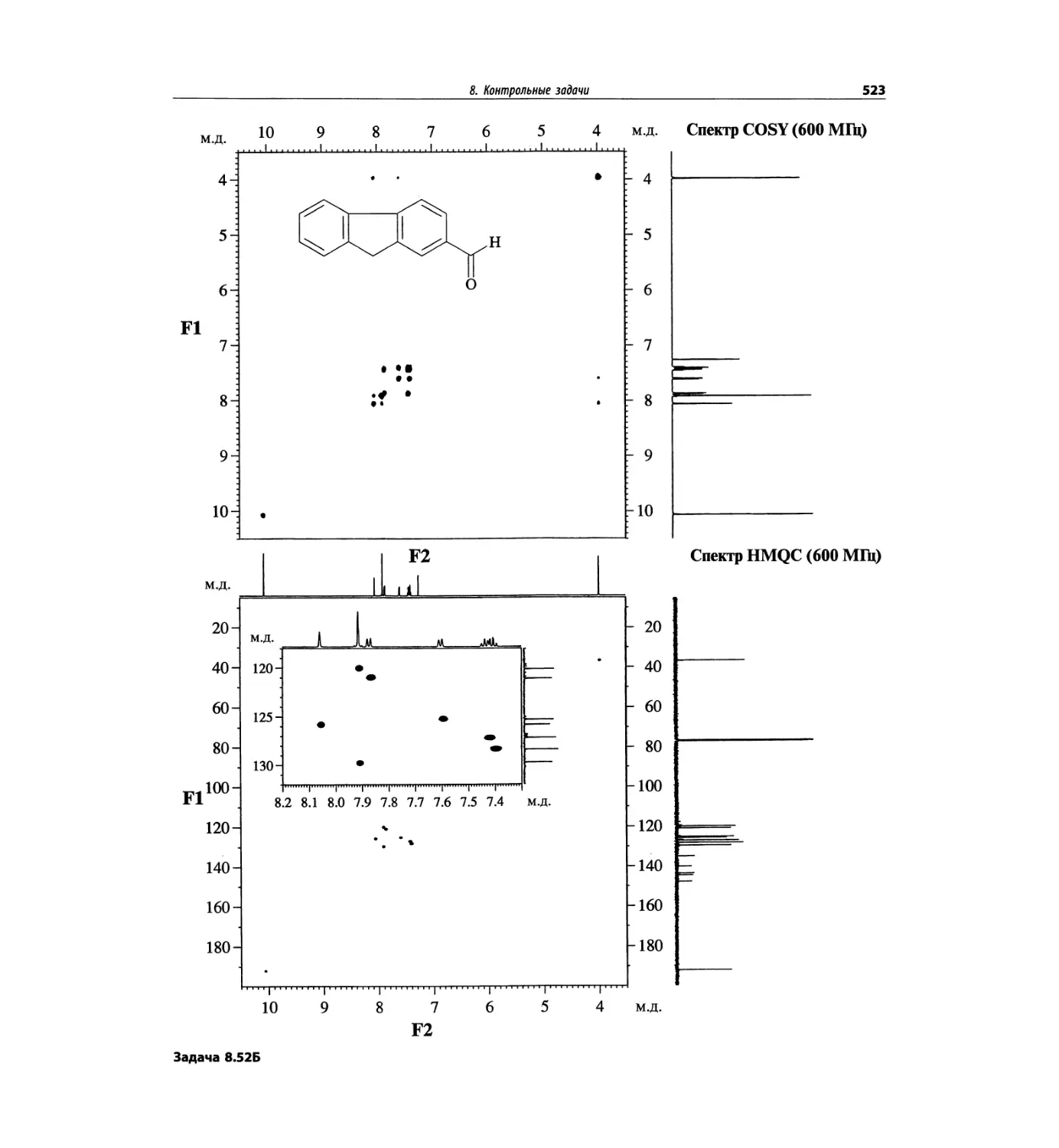
Контрольные задачи (глава 8)
Глава 8 полностью переделана. Задания расположены
в порядке усложнения структур, особое внимание уде-
лено двумерным спектрам. В некоторых более сложных
заданиях даны структуры, и от студента требуется её
проверка и проведение всех отнесений в спектрах.
Преподаватели и другие профессионалы мо-
гут получить ответы к упражнениям в форма-
те PDF, послав запрос к издателю по почте. До-
полнительные упражнения можно найти на сайте
http://www.wiley.com/college/silverstein.
Заключение
Большинство спектроскопических методов сейчас яв-
ляются рутинными для химиков-органиков в обычных
лабораториях. Получение высококачественных спек-
тров ЯМР, ПК- и масс-спектров больше не является
лимитирующей стадией в процессе идентификации
химической структуры. Напротив, анализ данных
стал для химика основным препятствием, как это
было для знающего спектроскописта в течение мно-
гих лет. Сейчас для оценки и предсказания спектров
ЯМР, масс- и ИК-спектров разработаны программные
Предисловие
9
средства, основанные на вводе данных о структу-
ре, в то же время все более доступными становятся
основанные на спектроскопических данных методы
автоматизированной оценки структуры. Такие мощ-
ные инструменты предполагают, что и квалифициро-
ванный, и неквалифицированный экспериментаторы
при интерпретации данных нуждаются в помощнике.
Существует ряд доступных методов предсказания
спектров (см., например, http://www.acdlabs.com для
более детальной информации), которые различаются
и по сложности, и по возможностям.
Этот учебник предназначен для студентов стар-
ших курсов и выпускников. Он может оказаться
полезным для практикующих химиков-органиков.
В очередной раз подчеркиваем, что цель состоит
в интерпретации спектров с использованием сово-
купной и взаимодополняющей спектрометрической
информации. Мы старались представлять спектры
в наиболее удобной для читателя форме. Это осо-
бенно важно для спектров ЯМР. Студенты быстро
осознают ценность мультиплетов первого порядка,
полученных на спектрометрах с рабочей частотой
300 и 600 МГц, и понимают пользу многочисленных
растянутых врезок. То же можно сказать и о пре-
подавателях.
Благодарности
Мы благодарим Э. Уильямса, вице-президента ком-
пании Advanced Chemistry Development (ACD), воз-
главляющего исследовательские работы, за предо-
ставление программного обеспечения для обработки
ИК- и масс-спектров, которое использовано в четырех
из восьми глав; оно позволило нам легко представить
высококачественные данные.
Мы также благодарим П. Коупа из корпорации
Bruker BioSpin за предоставление программ для об-
работки спектров ЯМР. Без этих программ данная
книга не состоялась бы.
Мы благодарим Дж. Ии, С. Вольфман-Робичоуд
и других сотрудников издательства John Wiley and
Sons за сотрудничество в преобразовании разных
частей этой сложной рукописи в прекрасное вось-
мое издание.
Наши рецензенты, Дж. Монтгомери из Универси-
тета в Уэйне, С. МакГован из Колледжа в Мерри-
маке, В. Фелд из Университета Райта, Дж.С. Новик
из Калифорнийского университета в Эрвине, Ирвин
и Мэри Чисхольм из Беренд-Колледжа Пенсильван-
ского университета в Эри, поддержали нас и внесли
много полезных предложений. Мы благодарны им
за их усилия и потраченное время.
Наконец, мы благодарим д-ра А. Стипановича,
директора аналитической и технической службы,
за разрешение использовать аналитическое обо-
рудование, принадлежащие SUNY ESF (Сиракузы,
Нью-Йорк).
Наши жены (Оливия, Кэтрин и Сандра) постоян-
но проявляли терпение и оказывали поддержку. Нет
слов, чтобы выразить нашу признательность.
Роберт М. Сильверстейн
Фрэнсис К. Вебстер
Дэвид Дж. Кимл
Слева направо: Роберт М. Сильверстейн, Фрэнсис К. Вебстер, Дэвид Дж. Кимл
Предисловие к первому изданию
В течение нескольких последних лет мы были заняты
выделением малых количеств органических веществ
из сложных смесей и их идентификацией спектроме-
трическими методами.
По предложению д-ра А. Дж. Кастро из Колледжа
в Сан-Хосе, мы расширили один из разделов курса,
назвали его «Спектрометрическая идентификация
органических соединений» и представили в весен-
нем семестре 1962 г. студентам старшего курса,
специализирующимся в области промышленной хи-
мии. Эта книга в значительной степени изменилась
по сравнению с материалом, собранным для курса
лекций с тем же названием. (Краткое изложение
методологии спектрометрической идентификации
было опубликовано: R.M. Silverstein and G.C. Bassler,
J. Chem. Educ, 39, 546 (1962).)
Мы хотели бы выразить признательность за фи-
нансовую поддержку, полученную нами из двух ис-
точников - корпорации Perkin Elmer и Стэнфордского
исследовательского института.
Мы чрезвычайно благодарны нашим коллегам из
Стэнфордского института. Мы воспользовались по-
мощью слишком многих, чтобы составить поименный
список, но особо мы хотели бы поблагодарить д-ра
С.А. Фукуа за полезные обсуждения по спектрометрии
ЯМР. Выражаем признательность за организационную
помощь д-ру С.М. Химелю, возглавляющему отдел
органических исследований, и д-ру Д.М. Коулсону,
возглавляющему отдел аналитических исследований.
Компания Varian внесла свой вклад благодаря
сотрудникам ее лаборатории прикладного ЯМР.
Мы находимся в долгу перед м-ром Н.С.Бхакка,
м-ром Л.Ф. Джонсоном и д-ром Дж.Н. Шулери за
представленные спектры ЯМР и огромную помощь
в интерпретации спектров.
Приглашение преподавать в Колледже Сан-Хосе
было сделано д-ром Б.М. Моррисом, деканом хи-
мического факультета, который доброжелательно
и оперативно решал административные проблемы.
Рукопись была прочитана д-ром Р.Г. Истманом
из Стэнфордского университета, чьи комментарии
наиболее полезны и точны.
Наконец, мы хотим поблагодарить наших жен.
Автор в муках творчества - это тяжелейшее испы-
тание для терпения окружающих. Наши жены не
только все выдержали, они поддерживали, помогали
в работе и вдохновляли.
Р.М. Силъверстейн
ГК. Басслер
Менлоу Парк
Калифорния
Апрель 1963
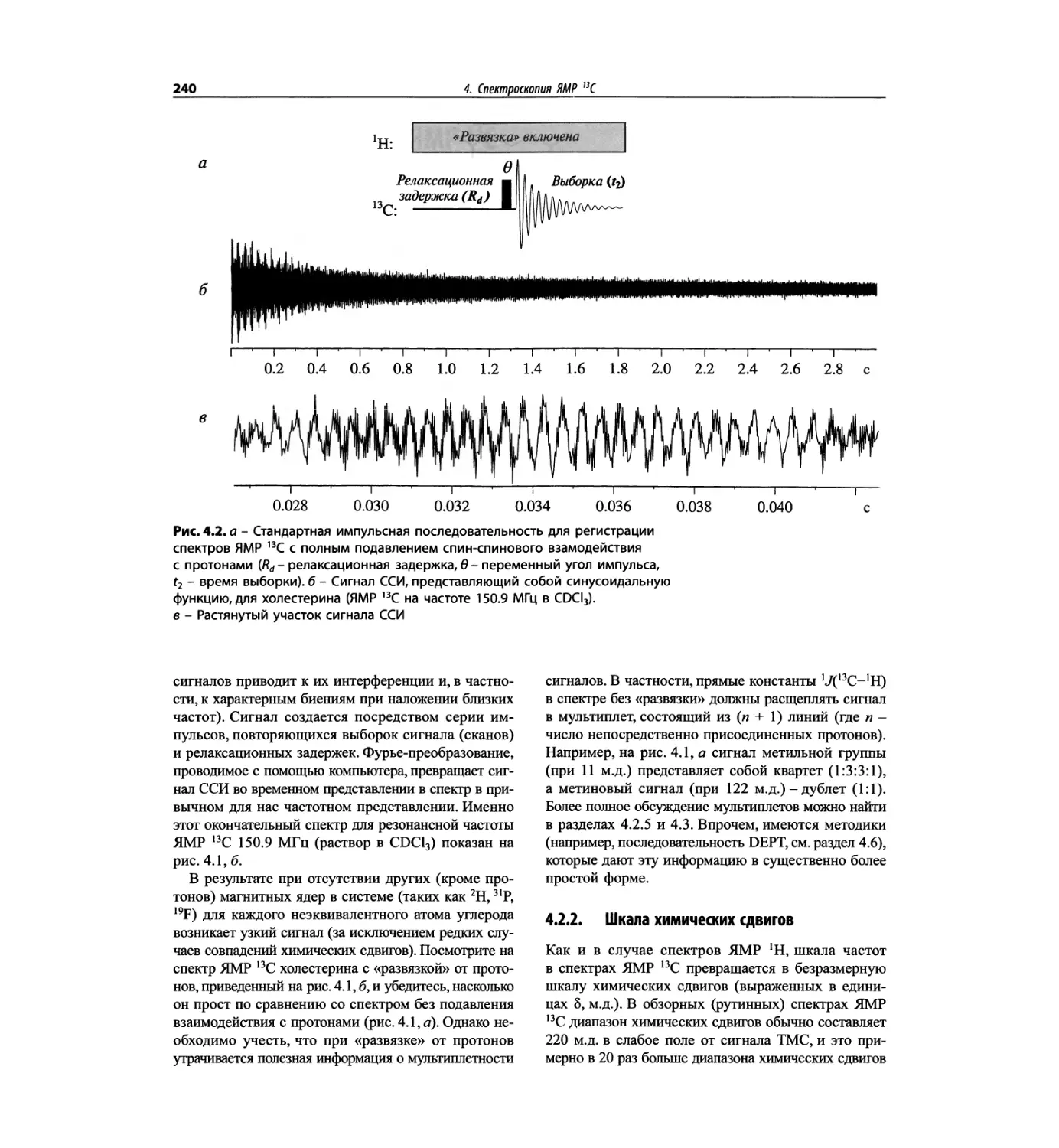
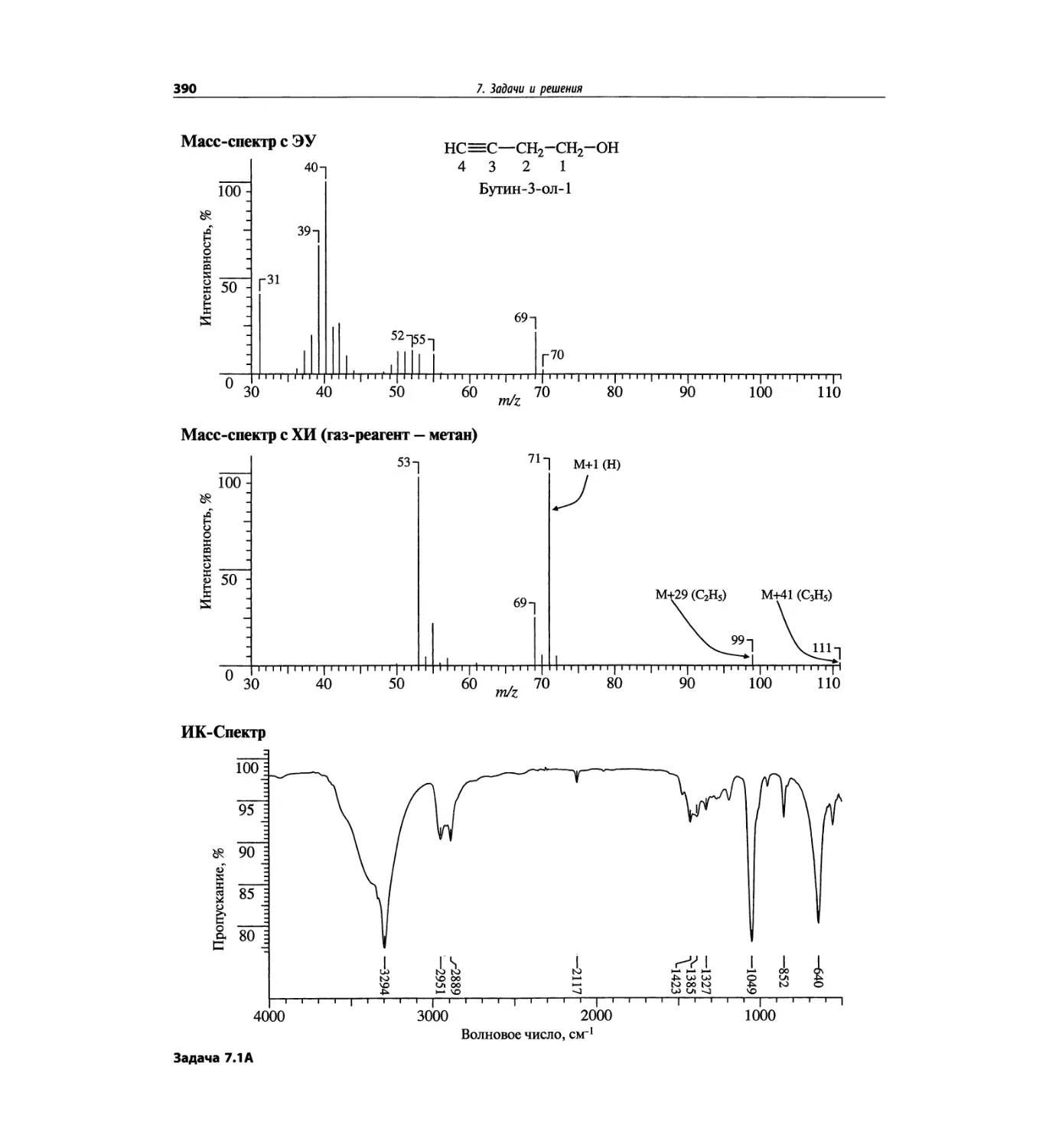
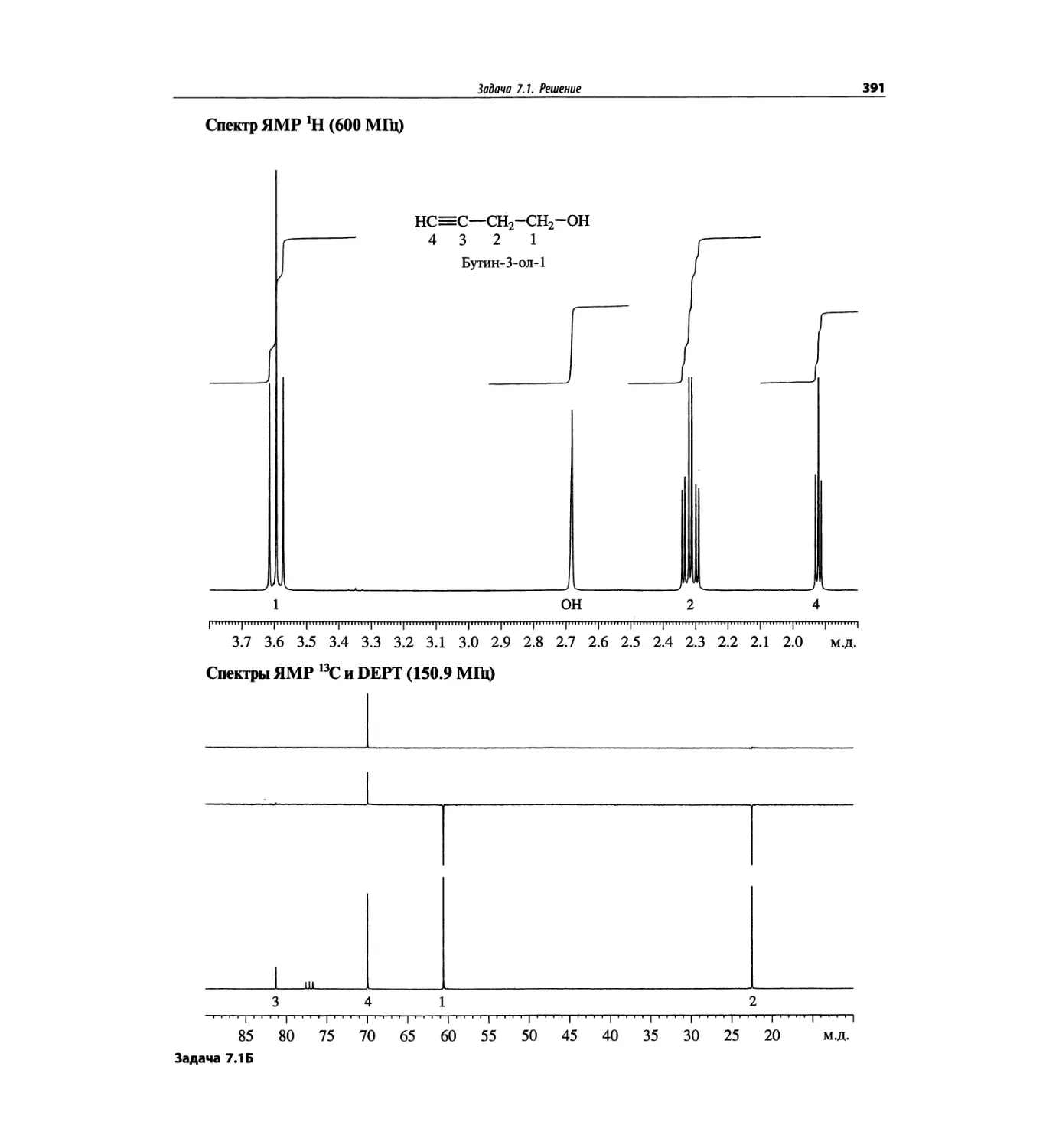
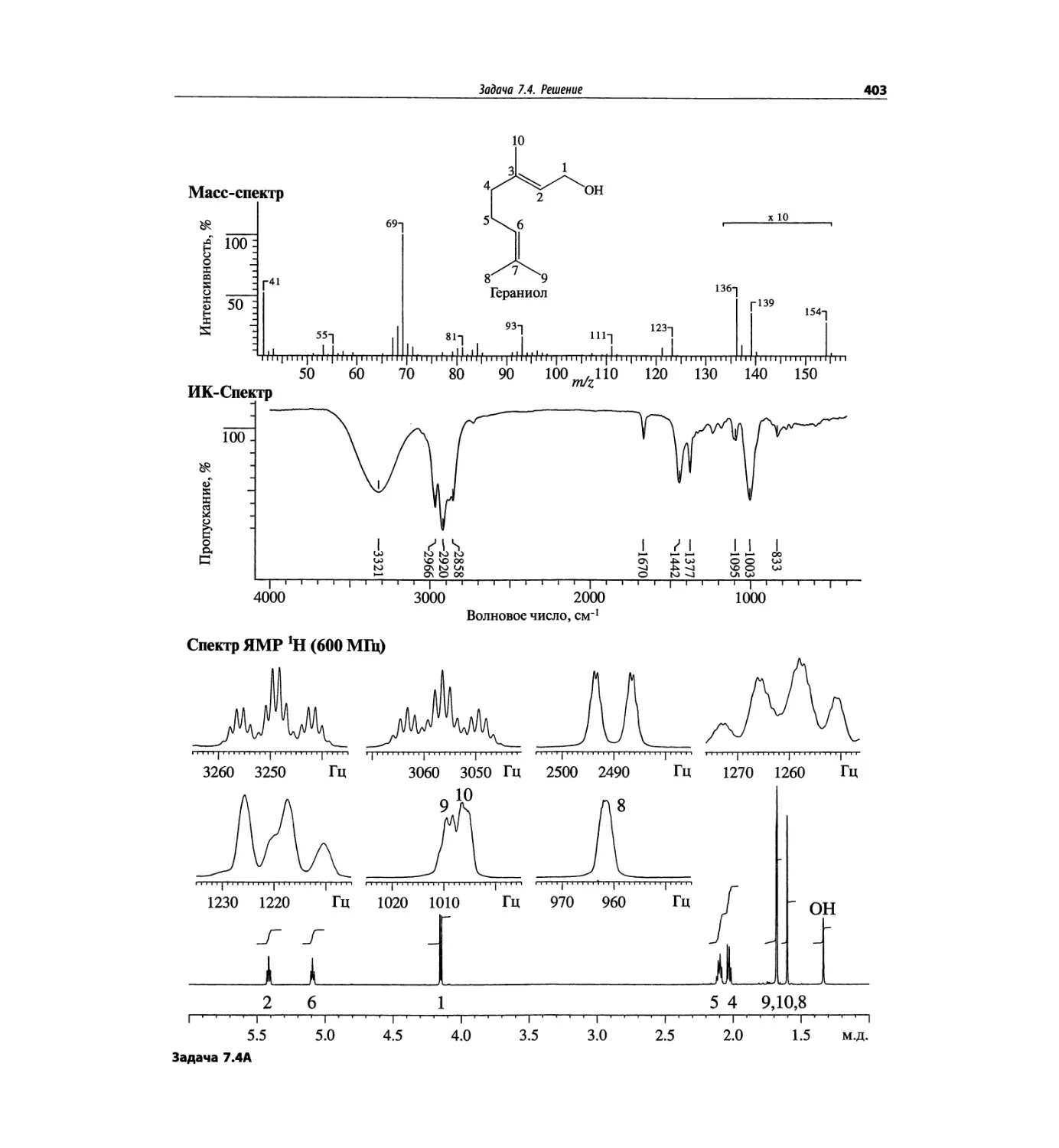
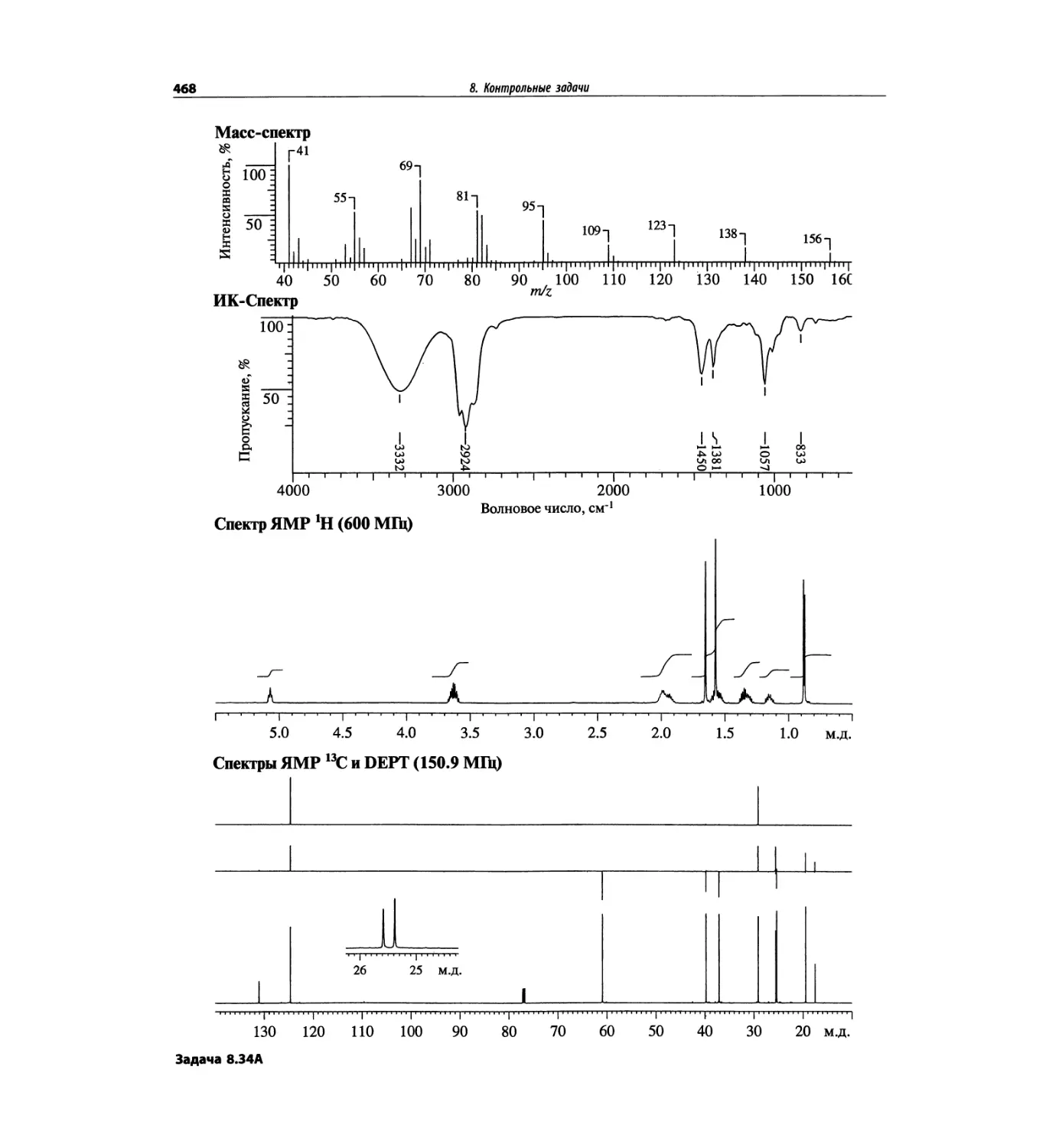
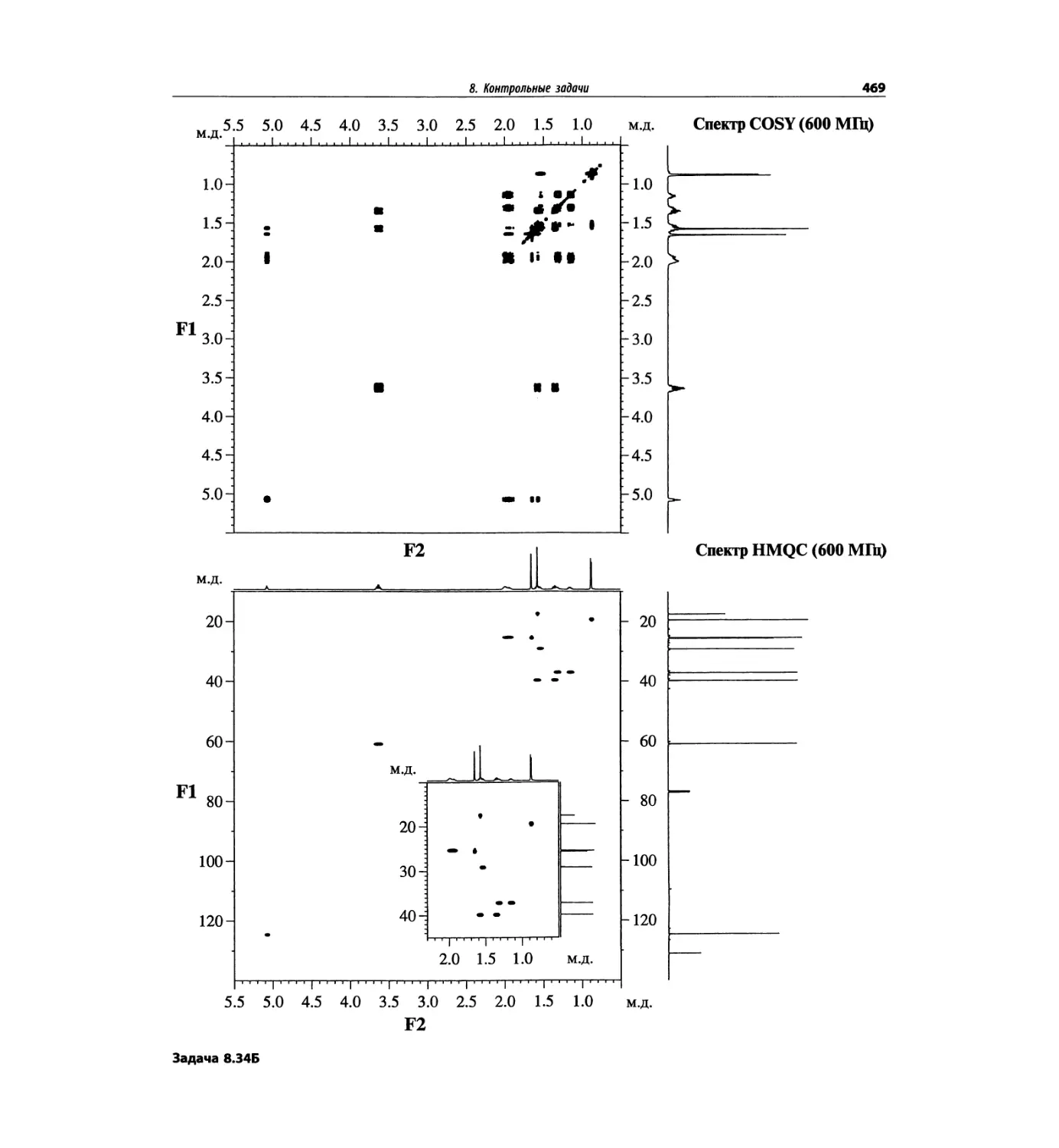
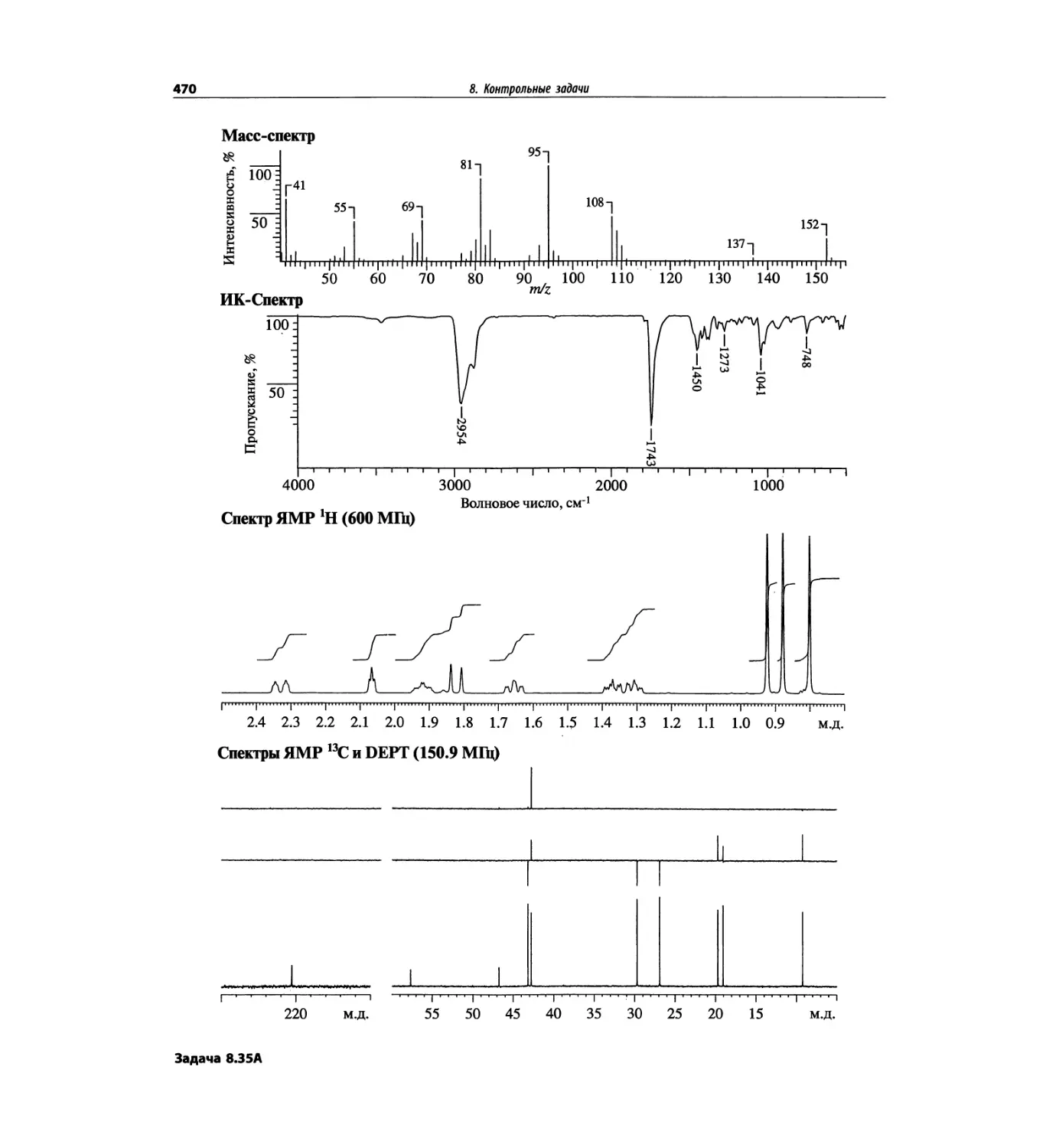
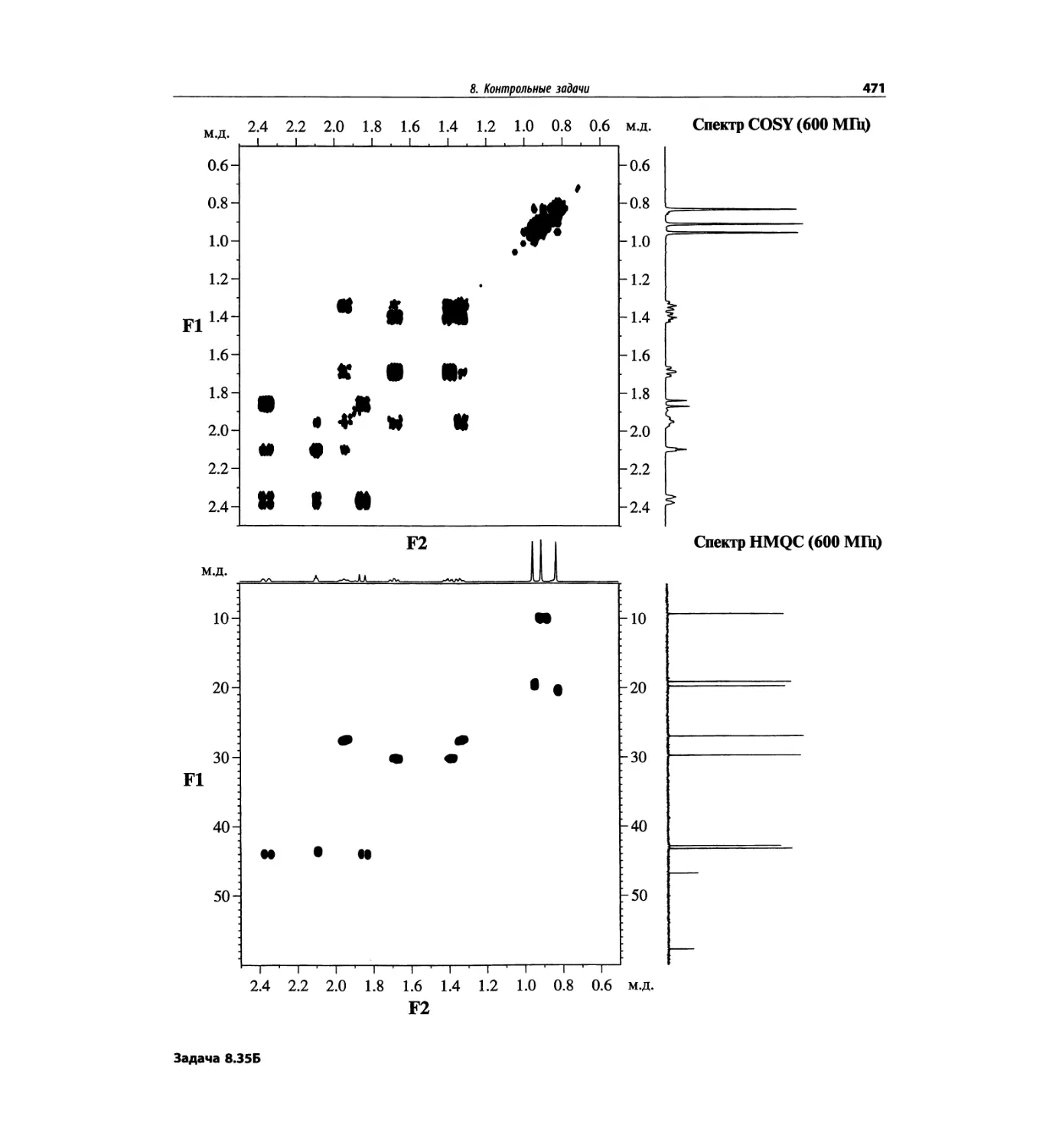
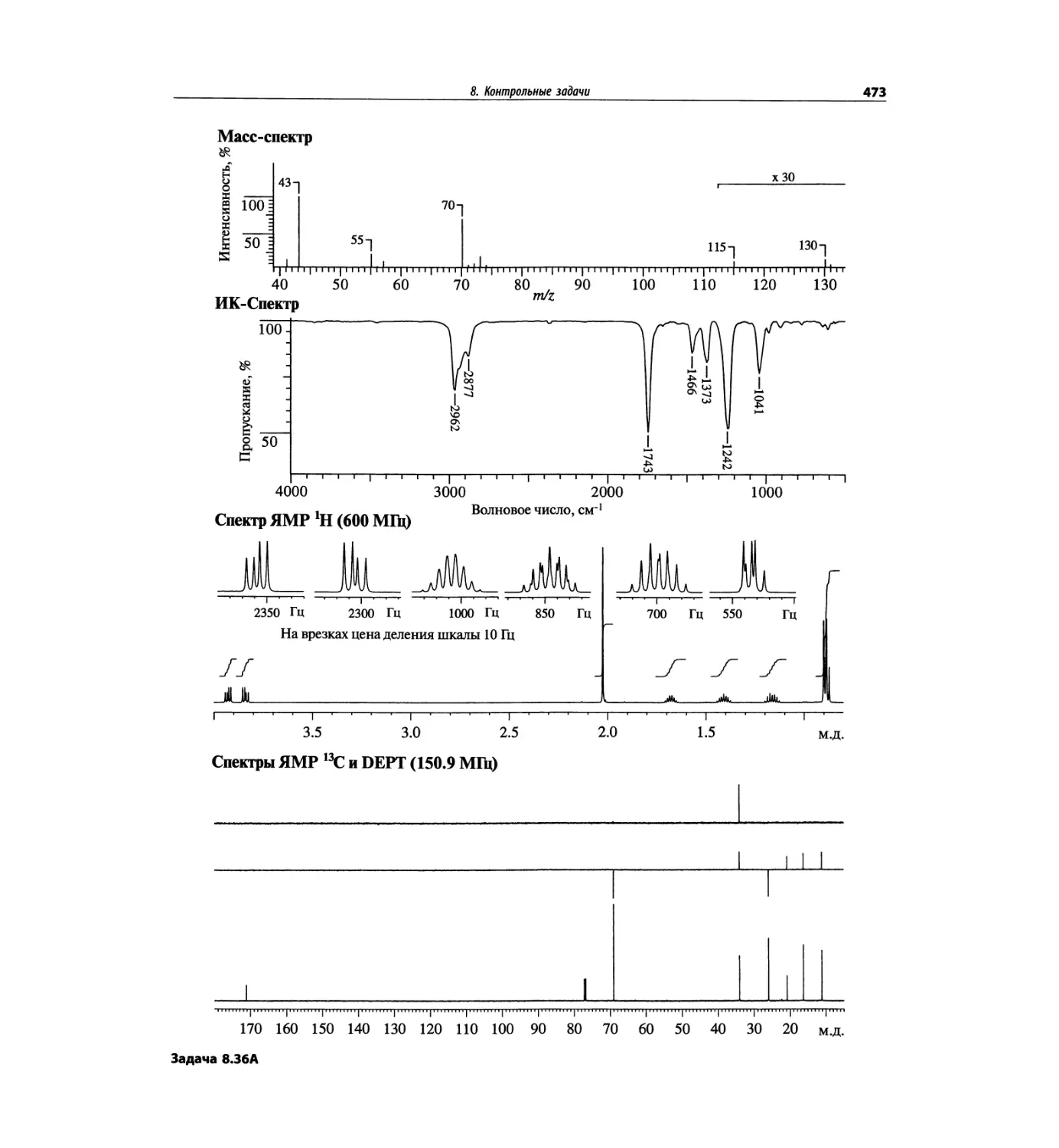
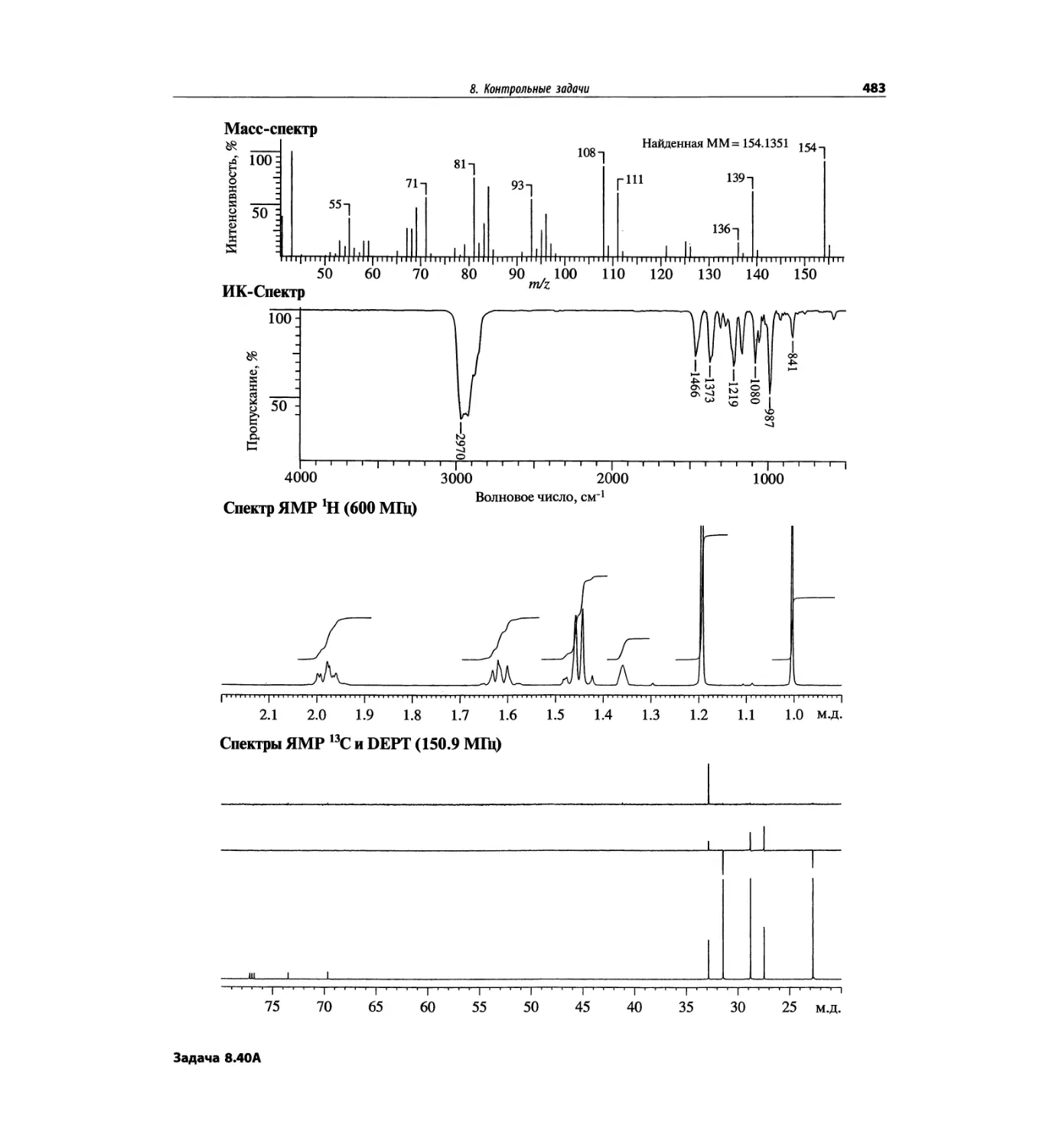
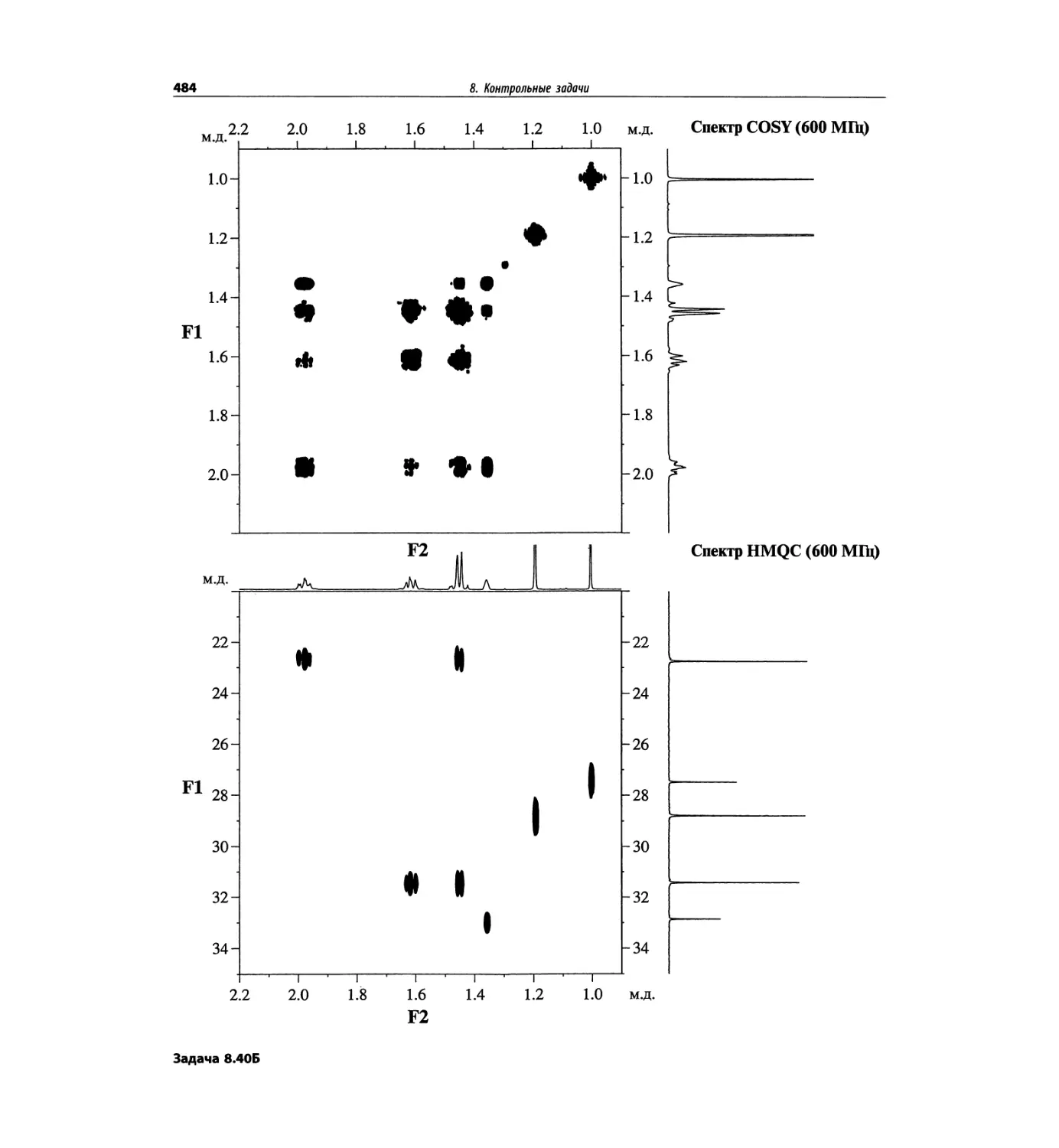
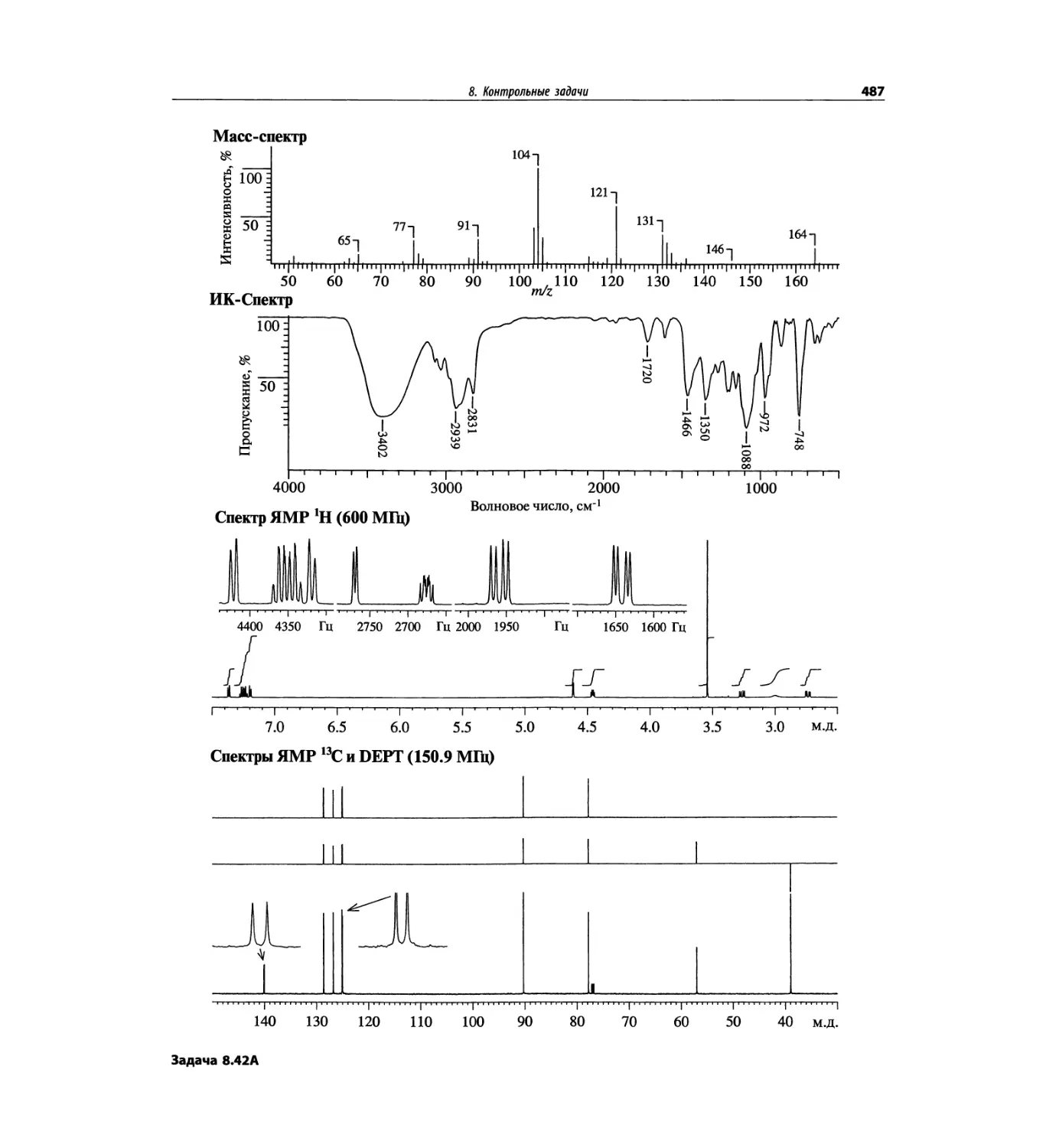
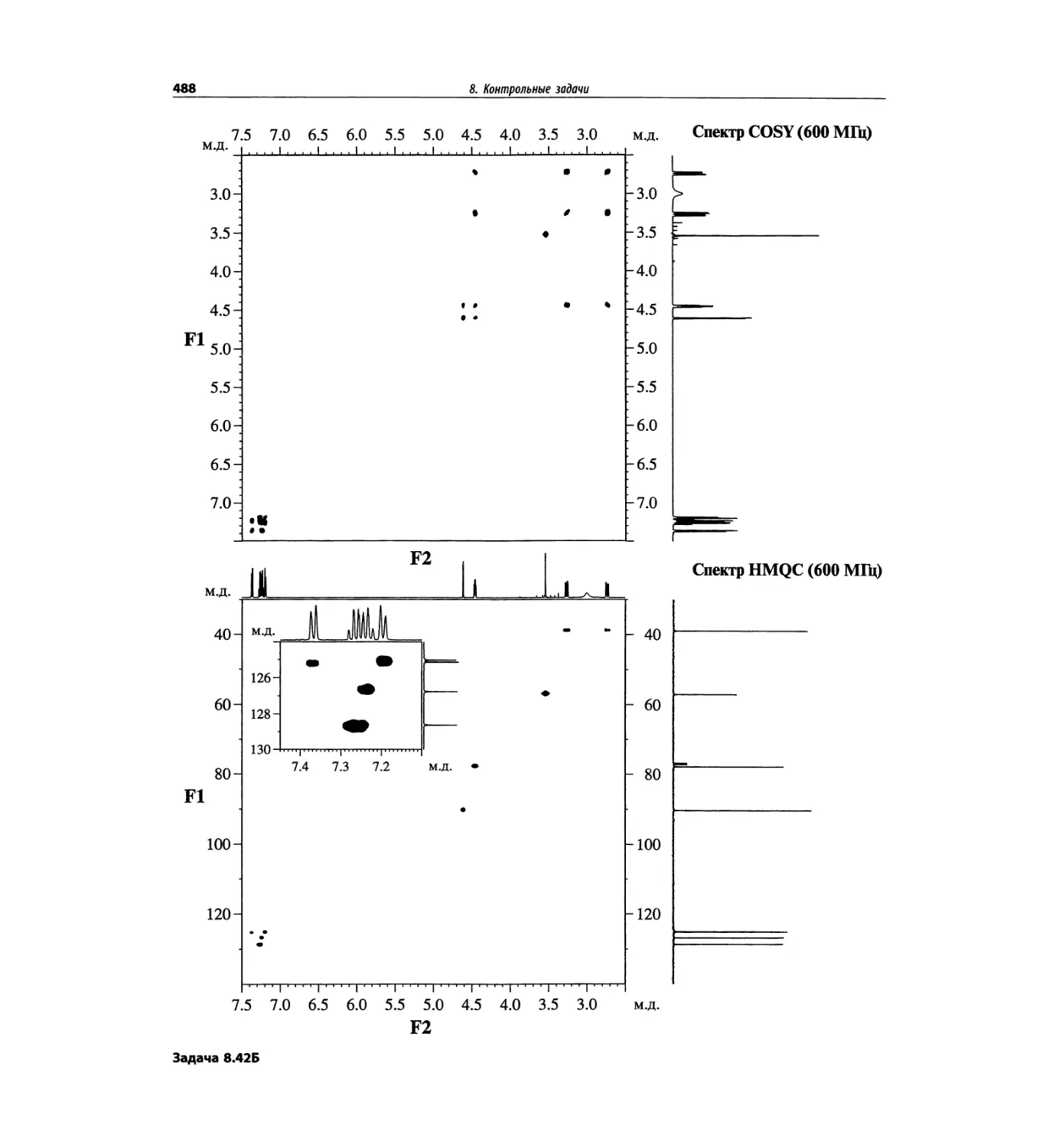
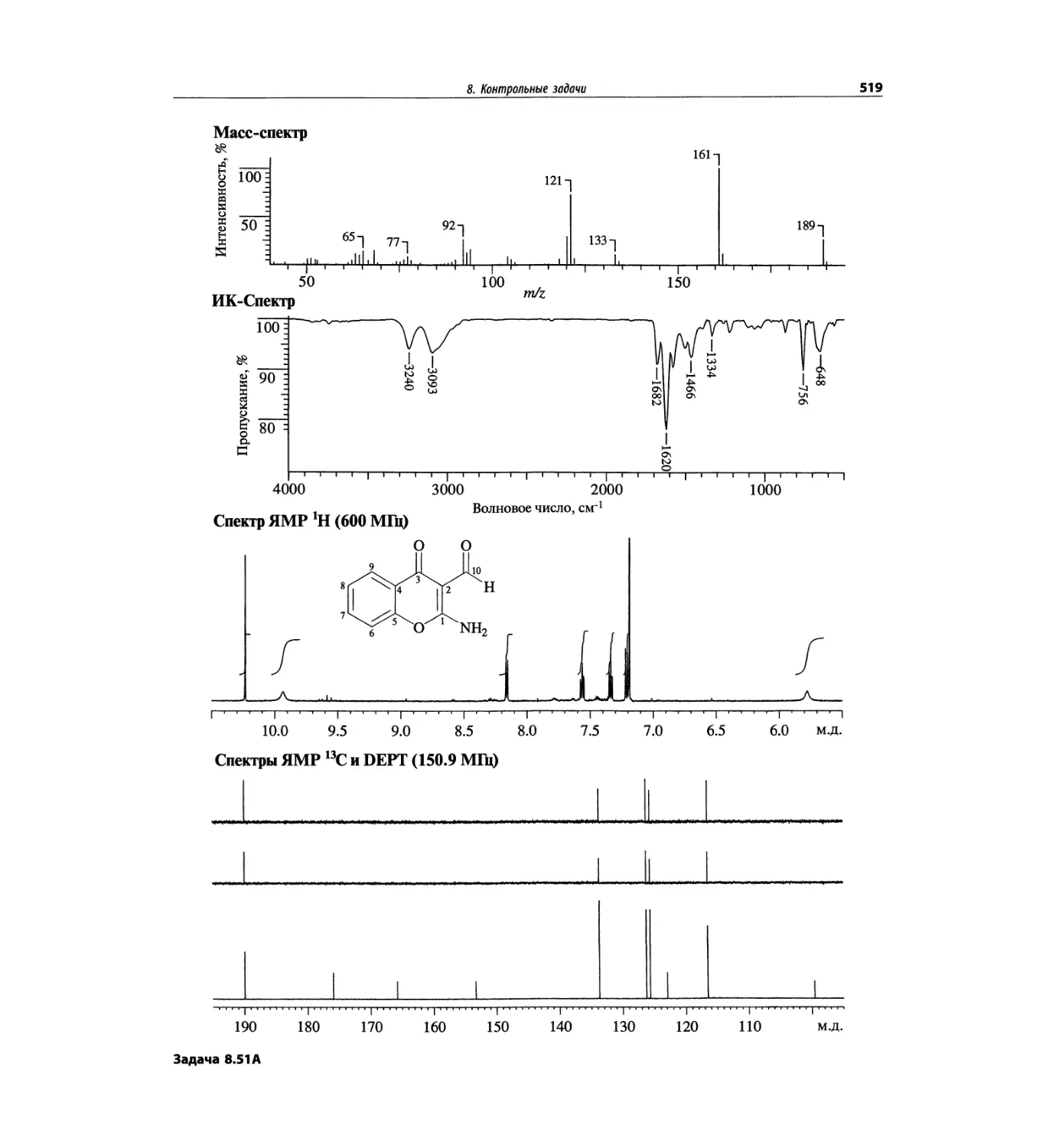
1. Масс-спектрометрия
1.1. Введение
В основе метода масс-спектрометрии лежит относи-
тельно простая идея: молекулы вещества подвергают-
ся ионизации тем или иным способом, образовавшиеся
ионы сортируются по величинам их отношения масса/
заряд, затем регистрируется число ионов для каждого
значения этого отношения в виде спектра. Например,
в широко используемом масс-спектрометре с иони-
зацией электронным ударом (ЭУ, electron-impact, EI)
молекулы в паровой фазе бомбардируются пучком
электронов с высокой энергией, а результат регистри-
руется в виде спектра положительных ионов, которые
разделены по величинам масса/заряд (m/z)V
Для иллюстрации рассмотрим масс-спектр с иони-
зацией электронным ударом бензамида, показанный
на рис. 1.1 и представляющий диаграмму относи-
тельного содержания (высота вертикальных линий)
в зависимости от m/z. Пик положительного иона с m/z
121 обусловлен целой молекулой (М), потерявшей
при ударной ионизации электронным пучком один
электрон. Он называется пиком молекулярного иона
и обозначается М>+. Из возбужденного молекулярного
нона возникает ряд фрагментных, или осколочных,
ионов, образование некоторых их них объяснено на
схеме на рис. 1.1.
Обычной практикой является объединение масс-
спектрометра с газовым (ГХ-МС) или жидкостным
ЖХ-МС) хроматографом. Масс-спектрометрия широко
используется как для анализа соединений с извест-
ными масс-спектрами, так и для анализа совершенно
неизвестных соединений. В случае известных соедине-
ний компьютерный поиск проводится сравнением их
экспериментальных масс-спектров с библиотечными.
Совпадение спектров является убедительным доказа-
тельством идентичности веществ и часто приемлемо
лаже для суда. В случае неизвестного соединения на
Единицей массы является дальтон (Да), по определению
тдзный 1/12 массы атома изотопа 12С, которой приписано
значение 12.0000.... массовых единиц. Как правило, интен-
сивность пиков выражается в % относительно главного
тика.
основе данных по молекулярному иону, характеру
фрагментации и других спектроскопических методов
(например, ИК и ЯМР) его можно идентифицировать.
Цель данной главы состоит в развитии у студентов
умения использовать метод масс-спектрометрии
в дальнейшей работе. В конце главы приведен спи-
сок книг с более подробным изложением метода
и атласов масс-спектров.
1.2. Техника эксперимента
В последнее десятилетие происходило быстрое раз-
витие и совершенствование масс-спектрометров.
Не обсуждая устройство отдельных моделей при-
боров, отметим, что они подразделяются по типам
в зависимости от 1) способа ионизации, 2) метода
разделения ионов. Вообще говоря, способ ионизации
не зависит от метода разделения ионов и наоборот,
хотя имеются исключения. Иногда способ ионизации
зависит от особенностей границы хроматографиче-
ского фронта (например, ЖХ-МС), в других методах
использование хроматографического введения пробы
затруднено (например, в методах FAB и MALDI, см.
далее). Прежде чем углубляться в технику экспери-
мента, отметим различие между двумя типами масс-
спектрометров, основанное на их разрешении.
Минимальное требование химика-органика к масс-
спектрометру состоит в способности прибора реги-
стрировать молекулярную массу анализируемого
соединения с точностью до ближайшего целого
числа. Предположим, если спектре появляется пик,
соответствующий массе 400, то он должен отличаться
от пиков, отвечающих массе 399 или 401. Для выбора
возможных молекулярных формул на основании из-
мерения интенсивностей изотопных пиков (см. раз-
дел 1.5.2.1) соседние пики должны четко различать-
ся. Высота провала (считая от уровня фона) между
двумя соседними пиками не должна превышать 10%
от высоты более интенсивного пика. Это разрешение
называется «единичным» и на доступных приборах
с «единичным разрешением» может быть достигнуто
приблизительно до 3000 Да.
12
1, Маа-спектрометрия
Бензамид
C7H7NO
М.м.: 121
Рис. 1.1. Масс-спектр с ионизацией ЭУ бензамида и схема, объясняющая
образование главных фрагментных ионов
I I I I I I I I I I I I I I I I I I I I I I I I I I I I I I I I I I I I Т| I I П-| I Н I | I I I I I I I I I | I ! I I | I I I I | I I н
60 m/z 70 80 90 100 110 120
Для определения разрешения прибора сравнивают
два соседних пика примерно равной интенсивности.
Эти пики нужно выбирать так, чтобы высота провала
между ними не превышала 10% от их интенсивности.
Разрешение (Л) равно
п Мп
R =----2—,
м-мт
п т
где Мп - большее массовое число пары соседних пи-
ков, а Мт - меньшее.
Масс-спектрометры подразделяют на два класса:
приборы низкого (единичного) и высокого разрешения.
Спектрометры низкого разрешения можно условно
определить как приборы, на которых можно разделить
целые массы до m/z 3000 (R = 3000/(3000-2999) = 3000).
На приборе с высоким разрешением (например,
R = 20000) можно различить С16Н26О2 и C15H24NO2
[R = 250.1933/(250.1933 - 250.1807) = 19857]. На
масс-спектрометрах этого важного класса с R более
100000 можно измерять массу иона с точностью,
достаточной для определения атомного состава (мо-
лекулярной формулы).
Все масс-спектрометры обладают общими особен-
ностями (см. рис. 1.2). Для введения пробы в масс-
спектрометр обычно применяют какой-либо вид
хроматографии, хотя во многих приборах имеется
возможность для прямого ввода образца в ионизаци-
онную камеру. Во всех масс-спектрометрах имеются
устройства для ионизации пробы и разделения ионов
по величине m/z. Они обсуждаются более детально
ниже. После разделения нужно детектировать ионы
и измерить их количество. Типичный коллектор
ионов состоит из коллимирующих щелей, которые
направляют в коллектор в данный момент только
ионы одного вида, где они детектируются, а сигнал
усиливается электронным умножителем. Метод де-
тектирования ионов в определенной степени зависит
от метода их разделения.
В настоящее время компьютеры контролируют
хроматографирование пробы, накопление и хранение
данных, обеспечивают графический вывод спектра
в виде линейчатой диаграммы или в табличном виде
почти во всех моделях масс-спектрометров.
1.3. Методы ионизации
13
1.3. Методы ионизации
Большое разнообразие существующих методов иони-
зации, некоторые из которых очень специфичны,
не позволяет рассмотреть их полностью. В целом
методы ионизации можно разделить на три большие
группы - ионизация в газовой фазе, ионизация при
десорбции и ионизация при испарении, - принципы
которых кратко изложены ниже.
1.3.1. Методы ионизации в газовой фазе
В масс-спектрометрии самыми старыми и наиболее
популярными являются методы ионизации в газо-
вой фазе. Они применимы к соединениям, имеющим
минимальное давление паров около 10"6 мм рт. ст.
при температуре, при которой соединение сохраняет
устойчивость; этому критерию отвечает большинство
неионных органических молекул с молекулярной мас-
сой менее 1000.
1.3.1.1. Ионизация электронным ударом
Электронный удар (ЭУ, electron impact, El) - наи-
более распространенный метод ионизации в масс-
спектрометрии. Молекула вещества пробы в газовой
фазе подвергается бомбардировке пучком электронов
с высокой энергией (обычно 70 эВ) и выбрасывает
электрон, образуя катион-радикал, называемый моле-
кулярным ионом. Потенциал ионизации молекулы ор-
ганического соединения обычно ниже 15 эВ, поэтому
бомбардировка электронами с энергией 50 эВ и выше
сообщает избыточную внутреннюю энергию возни-
кающему молекулярному иону. Эта энергия частично
рассеивается за счет разрыва ковалентных связей,
энергия которых находится в пределах от 3 до 10 эВ.
Обычно такой распад происходит избирательно,
захватывает широкий круг связей, является высоко-
воспроизводимым и характеристическим для данно-
го соединения. Более того, процессы фрагментации
предсказуемы, и именно они обусловливают широкие
возможности масс-спектрометрии для структурного
анализа. Часто избыточная энергия молекулярного
иона слишком велика, что приводит к исчезновению
его пика в масс-спектре (причина этого может за-
ключаться и в нестабильности молекулярного иона).
Понижение энергии электронного пучка является
общепринятым приемом получения молекулярного
иона, при этом степень фрагментации значитель-
но уменьшается. Недостаток этого приема состоит
в том, что спектр изменяется и его сравнение со
«стандартным» литературным спектром становится
невозможным.
Для многих масс-спектрометрия с ЭУ является
синонимом масс-спектрометрии вообще. Эта точ-
ка зрения понятна по двум причинам. Во-первых,
исторически первым, до того как были разработаны
другие методы ионизации, был ЭУ. Большинство ран-
них работ по масс-спектрометрии было выполнено
с ЭУ. Во-вторых, основные библиотеки и базы масс-
спектрометрических данных, на которые так часто
ссылаются, получены на приборах с ЭУ. Некоторые
из общедоступных баз данных содержат спектры
ЭУ более 390 000 соединений, которые легко най-
ти с помощью поисковых алгоритмов. Уникальность
масс-спектра для данного органического соединения,
даже для стереоизомеров, почти несомненна. Эта уни-
кальность соединяется с большой чувствительностью
метода, что делает ГХ-МС таким мощным и попу-
лярным аналитическим методом.
1.3.1.2. Химическая ионизация
Ионизация электронным ударом часто приводит к та-
кой глубокой фрагментации, что молекулярный ион
не наблюдается. Решить эту проблему можно, приме-
няя методы «мягкой ионизации», из которых важней-
шим является химическая ионизация (ХИ, chemical
ionization, CI). При использовании ХИ молекулы ис-
14
7. Маа-спектрометрия
следуемого вещества в паровой фазе не подвергаются
бомбардировке электронами с высокой энергией. Газ-
реагент (обычно используют метан, изобутан, аммиак
и др.) вводится в источник и ионизируется. Молекулы
исследуемого вещества сталкиваются с ионизирован-
ными молекулами газа-реагента (СН5, С4Н$ и т. д.) при
относительно высоком давлении в камере химической
ионизации и подвергаются вторичной ионизации пу-
тем переноса протона с образованием ионов [М+1]+
и электрофильного присоединения с образованием
[М+15]+, [М+29]+, [М+41]+, [М+18]+ (в случае NH4+).
Ион [М]+ редко образуется за счет переноса заряда.
Из-за гидридного переноса в спектрах с ХИ иногда
наблюдаются заметные сигналы квазимолекулярных
ионов [М-1]+. Так возникают ионы с четным числом
электронов. Избыточная энергия, которая переносит-
ся на молекулы образца во время ионизации, мала,
обычно менее 5 эВ, что гораздо ниже энергии фраг-
ментации. Как следствие, фрагментация оказывается
незначительной, а чувствительность повышенной,
потому что общий ионный ток определяется лишь
несколькими ионами. Однако информации о струк-
туре молекул из спектров ХИ получается меньше.
Обычно квазимолекулярные ионы довольно устойчи-
вы и легко детектируются. При ХИ часто образуются
только один или два фрагментных иона, а иногда
и ни одного.
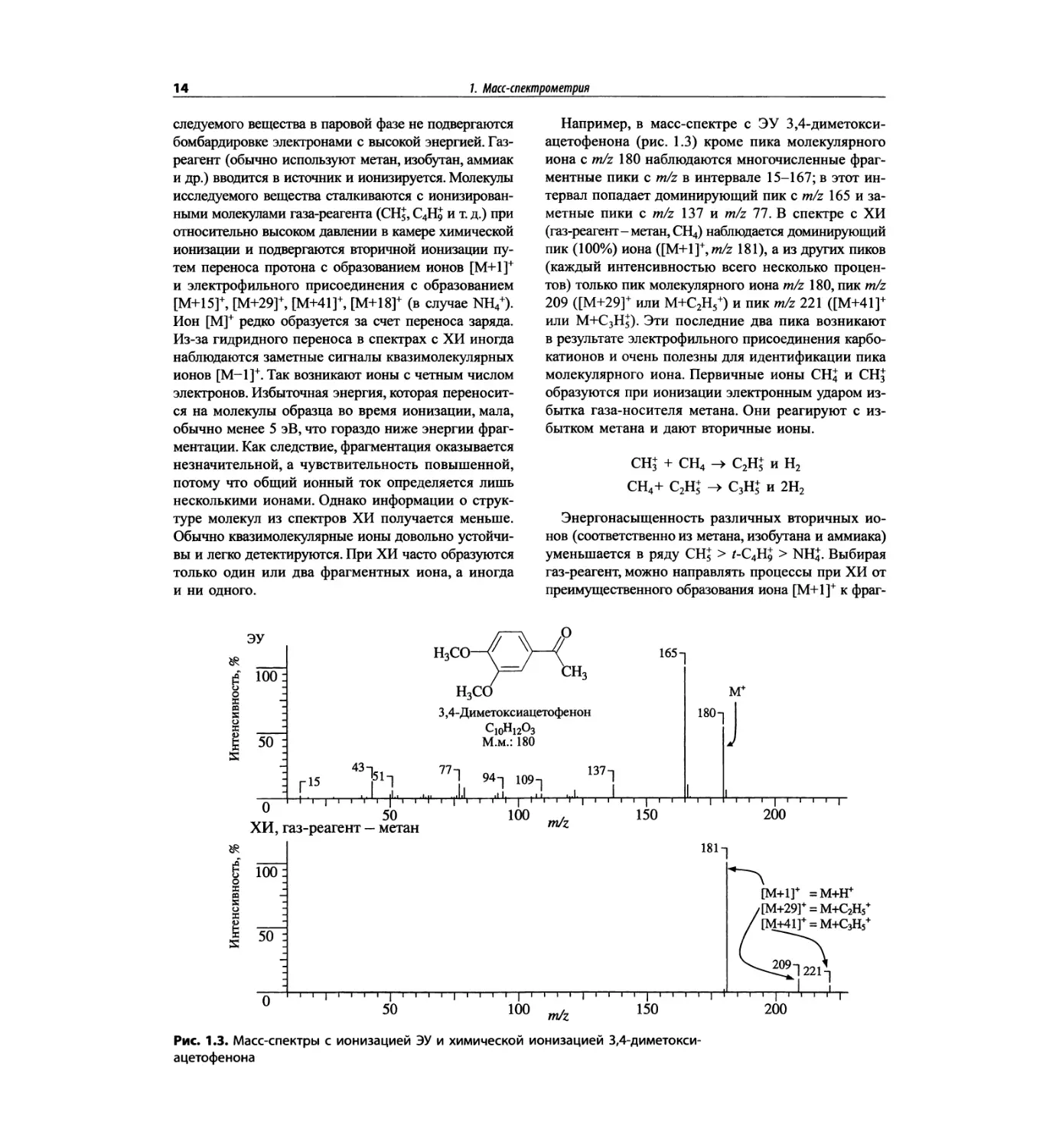
Например, в масс-спектре с ЭУ 3,4-диметокси-
ацетофенона (рис. 1.3) кроме пика молекулярного
иона с m/z 180 наблюдаются многочисленные фраг-
ментные пики с m/z в интервале 15-167; в этот ин-
тервал попадает доминирующий пик с m/z 165 и за-
метные пики с m/z 137 и m/z 77. В спектре с ХИ
(газ-реагент-метан, СН4) наблюдается доминирующий
пик (100%) иона ([M+l]+, m/z 181), а из других пиков
(каждый интенсивностью всего несколько процен-
тов) только пик молекулярного иона m/z 180, пик m/z
209 ([М+29]+ или М+С2Н5+) и пик m/z 221 ([М+41]+
или M+QH5). Эти последние два пика возникают
в результате электрофильного присоединения карбо-
катионов и очень полезны для идентификации пика
молекулярного иона. Первичные ионы CHJ и СН3
образуются при ионизации электронным ударом из-
бытка газа-носителя метана. Они реагируют с из-
бытком метана и дают вторичные ионы.
СЩ + СН4 -> С2Щ и Н2
СН4+ С2Н£ С3Н£ и 2Н2
Энергонасыщенность различных вторичных ио-
нов (соответственно из метана, изобутана и аммиака)
уменьшается в ряду СН5 > /-С4Н9 > NH4. Выбирая
газ-реагент, можно направлять процессы при ХИ от
преимущественного образования иона [М+1]+ к фраг-
ЭУ
100 z
3,4-Диметоксиацетофенон
С10Н12О3
М.м.: 180
165-1
М+
180-]
Г15
л . I . . . . I . .
и 50
ХИ, газ-реагент — метан
771
94-| 109-]
100 т/г
137 -|
“Т^
150
200
50 :
181-1
ё йхП
д -
и -
S
о
д
о
I 50 -
S
100 т/г
150
Рис. 1.3. Масс-спектры с ионизацией ЭУ и химической ионизацией 3,4-диметокси-
ацетофенона
Предисловие
Первое издание настоящего учебного пособия было
опубликовано в 1963 г. для обучения химиков-
органиков методам идентификации органических
соединений на основе совокупной информации, по-
лучаемой из масс-спектрометрии (МС), инфракрасной
(ИК) спектроскопии, спектроскопии ядерного маг-
нитного резонанса (ЯМР) и ультрафиолетовой (УФ)
спектроскопии. Эти энергетические зонды переводят
молекулу в возбужденное состояние, и ее отклики
регистрируются в виде спектров. УФ-Спектроскопия
применяется для других целей; в настоящие время
этот метод редко используется для идентификации
органических соединений, поэтому мы исключили
УФ-спектроскопию из шестого издания книги.
Для отражения замечательных достижений в раз-
витии ЯМР в данном издании потребовалось четыре
главы. Сейчас идентификация сложных соединений,
как показано в главах 5, 6, 7 и 8, практически не-
возможна без двумерных спектров ЯМР.
Непросто сохранить баланс между теоретическими
представлениями и практическими задачами. Мы ста-
рались избежать, с одной стороны, таинственных
областей электронных представлений и квантовой ме-
ханики, но, с другой стороны, неприемлемо и прибли-
жение черного ящика. Мы ушли от этих крайностей
с помощью наглядных иллюстраций и представления
некоторых вопросов в упрощенном нематематиче-
ском виде. Поскольку главная цель - интерпретация
спектров, то диаграммы и прекрасные спектры пред-
ставлены при каждом удобном случае.
Оценка даже на таком скромном уровне позволяет
решать немалое число задач по идентификации. На прак-
тике обычно доступна и дополнительная информация:
источник получения образца, особенности выделения,
метод синтеза, сведения об аналогичных веществах.
Сложные молекулы часто можно идентифицировать
по известным фрагментам их структуры и с помощью
точно сформулированных вопросов; процесс, таким
образом, является скорее подтверждением структуры,
а не идентификацией. В практической работе при под-
готовке малых проб вещества возникают следующие
трудности: улавливание, вымывание из адсорбентов,
удаление растворителя, предохранение от загрязнений
и продуктов разложения неустойчивых соединений.
Вода, воздух, смазка из кранов, примеси, содержащиеся
в растворителях, и пластификаторы затрудняют многие
исследования. В педагогических целях мы рассматри-
ваем только чистые органические соединения. «Чистое
вещество» в данном контексте является относительным
понятием; на самом деле лучше сказать так: чем чище,
тем лучше. Во многих случаях идентификация может
быть проведена на долях милиграмма или даже на
микрограммовых пробах вещества. Выполнение иден-
тификации для миллиграммов вещества относится
к рутинной практике. Конечно, не для всех молекул
определение получается легко. В некоторых случаях
может потребоваться химическая обработка, а получае-
мая из спектров информация позволит оптимизировать
эти операции.
Для достижения поставленных целей в данной книге
собран необходимый материал. Исчерпывающие схемы
и таблицы удобным образом расположены в тексте.
В конце глав имеются многочисленные подборки
упражнений. Глава 7 состоит из шести задач с пред-
ставительными подборками спектров, для которых
проведено детальное обсуждение. Глава 8 состоит из
упражнений для студентов, расположенных в порядке
(более или менее) возрастания сложности.
Авторы с удовольствием пользуются благоприят-
ной возможностью, чтобы включить новые данные,
исключить устаревшие и улучшить представление
материала. Ниже кратко указаны основные изменения
в содержании каждой главы.
Маа-спектрометрия (глава 1)
Основное содержание этой главы по-прежнему по-
священо фрагментации в спектрах с ионизацией элек-
тронным ударом. Описание приборов было написано
заново, расширено, основное внимание обращено на
методы ионизации молекул и разделения ионов. Все
спектры в этой главе были пересняты заново, помеще-
ны спектры новых соединений. Схемы фрагментации
были пересмотрены и исправлены. Отредактировано
обсуждение путей фрагментации в спектрах с иониза-
цией электронным ударом. Упражнения для студентов
в конце главы обновлены и существенно расширены.
В этом издании помещена таблица формуль-
ных масс (с точностью до четырех знаков после
запятой), чтобы на основании масс молекулярных
и фрагментных пиков при единичном разрешении
подбирать пробные молекулярные формулы. Отме-
тим, что в первом абзаце введения к главе 7 име-
ется рекомендация для студентов начинать именно
с определения молекулярной формулы.
1.3. Методы ионизации
15
ментации. Например, в масс-спектре диоктилфталата
при использовании метана в качестве газа-реагента
доминирует пик ([М+1]+с m/z 391); более важными
являются фрагментные пики (например, с m/z 113
и 140) с интенсивностями 30-60% от интенсивности
основного пика. При использовании изобутана в ка-
честве газа-реагента пик [М+1]+ остается основным,
а интенсивности фрагментных пиков составляют ~5%.
Масс-спектры, полученные с использованием ХИ,
не пригодны ни для поиска (ручного или компью-
терного) в спектроскопических базах данных, ни для
определения структуры. В основном они используются
для детектирования молекулярных ионов и опреде-
ления молекулярных масс.
1.3.2. Методы десорбционной ионизации
Методы десорбционной ионизации основаны на том,
что ионизированные молекулы пробы десорбируются
непосредственно из конденсированной фазы в газо-
вую. Первоначально метод применяли для больших
молекул, нелетучих или ионных соединений. У данного
приема имеются значительные неудобства. Для обыч-
ных образцов десорбционные методы в общем не
эффективны. Часто такие спектры несут ограничен-
ную информацию. В случае неизвестных соединений
методы десорбции используют в первую очередь для
определения молекулярной массы, иногда с высокой
точностью. Однако даже для этих целей метод нужно
использовать с осторожностью, так как идентификация
молекулярного или квазимолекулярного иона может
оказаться ненадежной. Получающийся спектр часто
бывает усложнен ионами вещества матрицы.
7.3.2.1. Полевая десорбция
В методе полевой десорбции (ПД, field desorbtion,
FD) проба помещается на металлический эмиттер,
на поверхность которого нанесены тонкие углерод-
ные микроиглы. Эти микроиглы активируют поверх-
ность, на которую подается ускоряющий потенциал
и которая функционирует как анод. Очень высокий
градиент потенциала на концах игл ионизирует моле-
кулу пробы, и возникший катион выталкивается с по-
верхности эмиттера. Такие ионы имеют небольшую
избыточную энергию, фрагментация происходит в ми-
нимальной степени, т. е. обычно наблюдается только
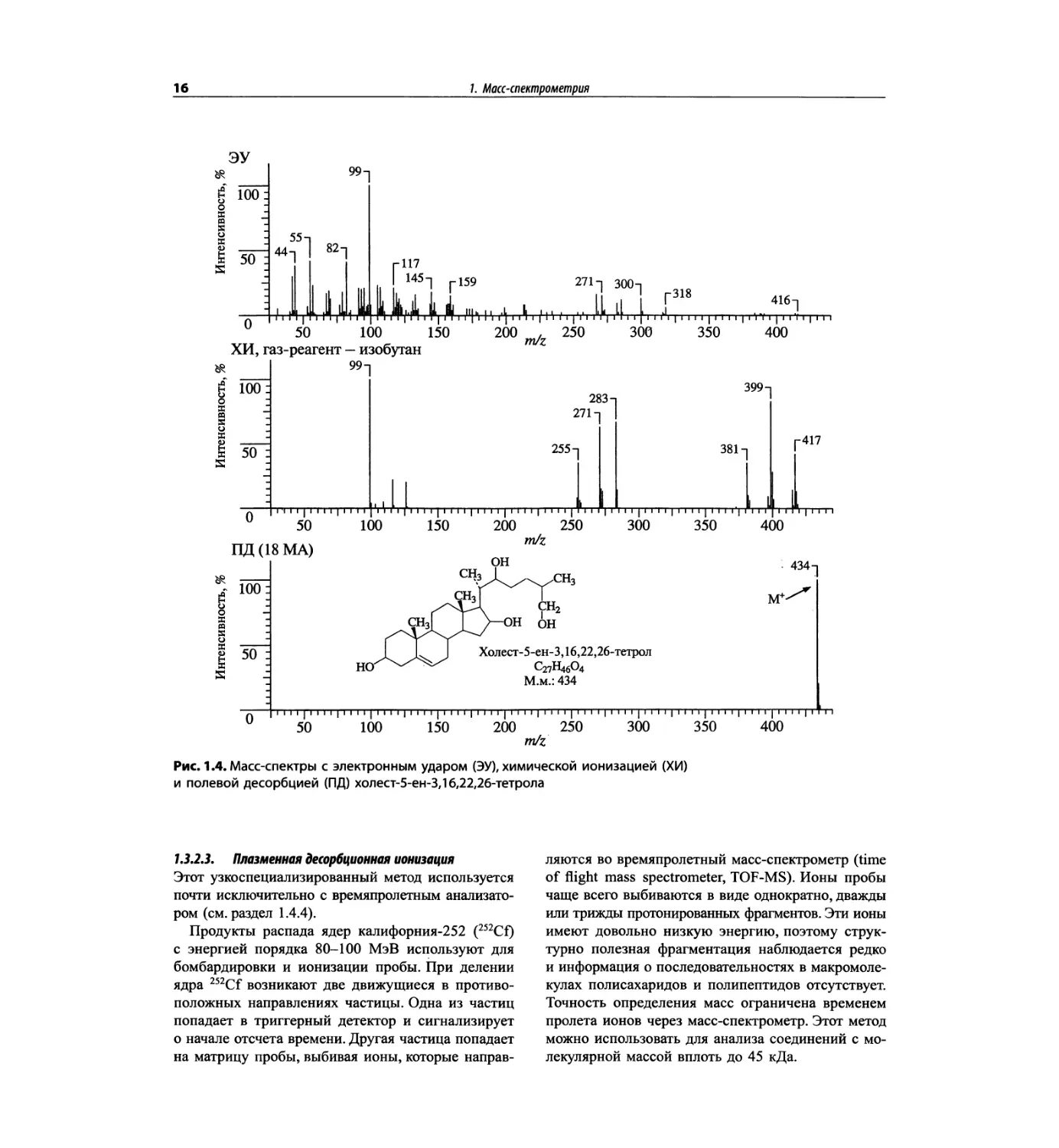
пик молекулярного иона. Например, в возбуждаемых
методами ЭУ и ХИ спектрах стероида холест-5-ен-
3,16,22,26-тетрола пик молекулярного иона невидим.
Однако в масс-спектре с ПД (рис. 1.4) доминирует
пик молекулярного иона и фактически не наблюдается
фрагментация.
Полевая десорбция отошла на второй план с по-
явлением метода ББА (бомбардировка быстрыми
атомами, см. следующий раздел). Несмотря на то
что метод ПД для неполярных соединений часто
полезнее, чем ББА, и в нем отсутствует влияние
уровня фона, который возникает при разложении
вещества матрицы, он не стал таким распростра-
ненным, как ББА; вероятно, из-за того, что произ-
водители оборудования активно внедряют именно
ББА.
1.3.2.2. Бомбардировка быстрыми атомами
При ионизации в процессе бомбардировки быстрыми
атомами (ББА, fast atom bombardment, FAB) ксенона
и аргона с высокой энергией (6-10 кэВ) облучают
пробу в виде раствора в жидкости с низким давлением
паров (например, в глицерине). Матрица защищает
пробу от избыточного радиационного разрушения.
Близкий метод ионизации - жидкостная вторичная
ионизация или вторично-ионная масс-спектрометрия
(ВИМС, liquid secondary ionization mass spectrometry,
LSIMS) - отличается использованием ионов цезия
с более высокой энергией (10—30 кэВ)
В обоих методах в результате присоединения ка-
тионов образуются положительные ионы [М+1]+ или
[М+23, Na]+ и отрицательные ионы, возникающие
при депротонировании [М-1]+; ионы обоих типов
обычно имеют единичный заряд. В зависимости от
прибора ББА можно использовать для регистрации
масс-спектров высокого разрешения; ББА главным
образом применяют при исследовании больших не-
летучих молекул, особенно для определения моле-
кулярной массы. Для большинства классов соеди-
нений часть спектра в области низких масс менее
информативна, из-за того что там могут проявиться
ионы вещества матрицы. Однако для соединений
некоторых классов с молекулами, составлеными из
«строительных блоков» (таких как полисахариды
и полипептиды), фрагментация обычно происходит
по гликозидным и пептидным связям соответствен-
но, что дает возможность определять структурные
последовательности в этих соединениях.
Верхний предел массы для ББА (и ВИМС) находит-
ся между 10 и 20 кДа, однако ББА наиболее полезен
до 6 кДа. Этот метод ионизации чаще используется
в приборах с двойной фокусировкой с разрешением
около 0.3 m/z во всем интервале масс. Метод сочета-
ется с большинством типов анализаторов масс. Самая
сложная проблема, возникающая при использовании
ББА, заключается в высоком уровне сигналов ионов
вещества матрицы, что ограничивает чувствительность
и скрывает важные фрагментные ионы.
16
7. Маа-спектрометрия
ЭУ
Рис. 1.4. Масс-спектры с электронным ударом (ЭУ), химической ионизацией (ХИ)
и полевой десорбцией (ПД) холест-5-ен-3,16,22,26-тетрола
1.3.2.3. Плазменная десорбционная ионизация
Этот узкоспециализированный метод используется
почти исключительно с времяпролетным анализато-
ром (см. раздел 1.4.4).
Продукты распада ядер калифорния-252 (252Cf)
с энергией порядка 80-100 МэВ используют для
бомбардировки и ионизации пробы. При делении
ядра 252Cf возникают две движущиеся в противо-
положных направлениях частицы. Одна из частиц
попадает в триггерный детектор и сигнализирует
о начале отсчета времени. Другая частица попадает
на матрицу пробы, выбивая ионы, которые направ-
ляются во времяпролетный масс-спектрометр (time
of flight mass spectrometer, TOF-MS). Ионы пробы
чаще всего выбиваются в виде однократно, дважды
или трижды протонированных фрагментов. Эти ионы
имеют довольно низкую энергию, поэтому струк-
турно полезная фрагментация наблюдается редко
и информация о последовательностях в макромоле-
кулах полисахаридов и полипептидов отсутствует.
Точность определения масс ограничена временем
пролета ионов через масс-спектрометр. Этот метод
можно использовать для анализа соединений с мо-
лекулярной массой вплоть до 45 кДа.
1.3. Методы ионизации
17
1.3.2.4. Лазерная десорбционная ионизация
Для ионизации проб в масс-спектрометрии можно
использовать пульсирующий лазерный луч. Так как
этот метод ионизации импульсный, его нужно исполь-
зовать либо с времяпролетным масс-спектрометром,
либо с масс-спектрометром с преобразованием Фурье
(см. раздел 1.4.5). Широкое распространение получили
два типа лазеров: на СО2, которые излучают в даль-
ней ИК-области, и твердотельные на основе иттрий-
алюминиевого граната с примесью неодима (Nd/YAG)
с учетверенной частотой, излучающие в УФ-области
при 266 нм. Без использования матрицы метод огра-
ничен молекулами с низкими массами (< 2 кДа).
Возможности метода значительно увеличиваются
при использовании твердого раствора исследуемого
вещества в органической матрице (матричная лазерная
десорбционная ионизация, matrix assisted laser desorp-
tion ionization, MALDI). Два вещества - никотиновая
и синапиновая кислоты (а также другие соедине-
ния), которые получили широкое распространение
в качестве матриц, имеют полосы поглощения, со-
впадающие со спектрами лазерного излучения. Этим
методом были успешно проанализированы вещества
с молекулярными массами до 200-300 кДа. Несколько
пикомолей пробы смешивают с матрицей, подвергают
импульсному лазерному облучению, приводящему
к образованию однозарядных ионов вещества про-
бы (но иногда наблюдаются многозарядные ионы
и димеры), которые выбрасываются из матрицы
в масс-спектрометр.
Ионы обладают малой избыточной энергией и име-
ют небольшую склонность к фрагментации. По этой
причине метод бывает полезен для анализа смесей.
Точность измерения масс невелика при использовании
с методомТОР-MS, но с методом FT-MS может быть
достигнуто очень высокое разрешение. Как и другие
матричные методы, MALDI испытывает мешающее
влияние фона матрицы, которое усиливается при об-
разовании аддуктов с матричным веществом. Таким
образом, отнесение молекулярного иона неизвестного
соединения может оказаться неопределенным.
1.3.3. Методы ионизации при испарении
Существуют два важных метода, которые основаны
на том, что ионы или (реже) нейтральные молекулы,
находящиеся в растворе (растворитель часто содержит
муравьиную кислоту), при испарении растворителя
одновременно подвергаются ионизации и последую-
щему анализу. В сочетании с жидкостной хромато-
графией эти методы стали чрезвычайно популярны.
1.3.3.1. Масс-спектрометрия с термораспылением
В масс-спектрометрии с термораспылением (ther-
mospray) раствор пробы вводится в спектрометр
посредством нагретого капилляра. При распылении
и частичном испарении растворителя образуется
поток мелких капель, которые попадают в ионный
источник. После полного испарения растворителя
ионы пробы попадают в анализатор. В этом методе
используют большие скорости потока исследуемого
раствора и буферного газа; на ранних этапах развития
такие масс-спектрометры объединяли с жидкостными
хроматографами. В дальнейшем метод в значительной
степени был вытеснен электрораспылением.
1.3.3.2. Масс-спектрометрия с электрораспылением
Источник ионов с электрораспылением (electrospray,
ES, ЭР), схема которого показана на рис. 1.5, работа-
ет при атмосферном давлении, а метод называется
ионизацией при атмосферном давлении (atmospheric
pressure ionization, API). Раствор пробы (обычно в по-
Частицы аэрозоля
с избыточным
Заряженные пластины
Рис. 1.5. Схема образования ионов при испарении растворителя в методе
электрораспыления
18
/. Маа-спектрометрия
лярном летучем растворителе) попадает в ионный
источник через капилляр из нержавеющей стали,
который окружен коаксиальным потоком распыляю-
щего газа-азота. На наконечник капилляра подается
высокий потенциал (относительно вспомогательного
электрода), создающий неоднородное электрическое
поле с градиентом до 5 кВ/см. При выходе из капил-
ляра раствор превращается в аэрозоль из заряженных
капелек. Поток распыляющего газа направляет аэро-
золь в масс-спектрометр.
По мере испарения растворителя капельки аэро-
золя сжимаются, а концентрация ионов пробы уве-
личивается. При достижении критического размера
капель силы поверхностного натяжения становятся
меньше сил кулоновского отталкивания. Происходит
так называемый «кулоновский взрыв», приводящий
к ионизации вещества пробы. После фокусировки
сепараторами ионы попадают в анализатор масс-
спектрометра.
В 90-е гг. XX в. началось быстрое развитие масс-
спектрометрии с электрораспылением главным обра-
зом в применении к соединениям, молекулы которых
могут нести несколько зарядов в разных местах. На-
пример, белки могут образовывать многозарядные
ионы. Так как в масс-спектрометрии измеряются не
массы, а отношения массы к заряду (m/z), эти много-
зарядные ионы регистрируются при кажущихся вели-
чинах масс 1/2, 1/3,...,1/и от их действительных значе-
ний, где п - число зарядов (z). На больших молекулах
белков может находиться до 40 или более зарядов,
в результате ион с массой до 100 кДа можно заре-
гистрировать на обычном квадрупольном с ионной
ловушкой или магнитном секторном спектрометре.
В этом случае в масс-спектре появляется ряд пиков
с увеличивающимися массами, которые отвечают псев-
домолекулярным ионам, имеющим меньшее число
протонов и, соответственно, меньшие заряды.
Для определения фактической массы иона нужно
знать его заряд. Если можно идентифицировать два
пика, которые различаются на один заряд, то вычисле-
ние сводится к простой алгебре. Напомним, что каж-
дый ион молекулы пробы (Ms) в общем виде можно
представить как (Ms + zH)z+, где Н- масса протона
(1.0079 Да). Для двух отличающихся на один заряд
ионов массы составляют тх = [Ms + (z + \)H]/(z + 1)
и т2 = [(Ms + z H)/z\. Совместное решение двух
уравнений дает заряд z = (тх - Н)/(т2 - тх). Про-
стая программа позволяет выполнять такой расчет
для каждого пика в спектре и непосредственно вы-
числять массы молекул.
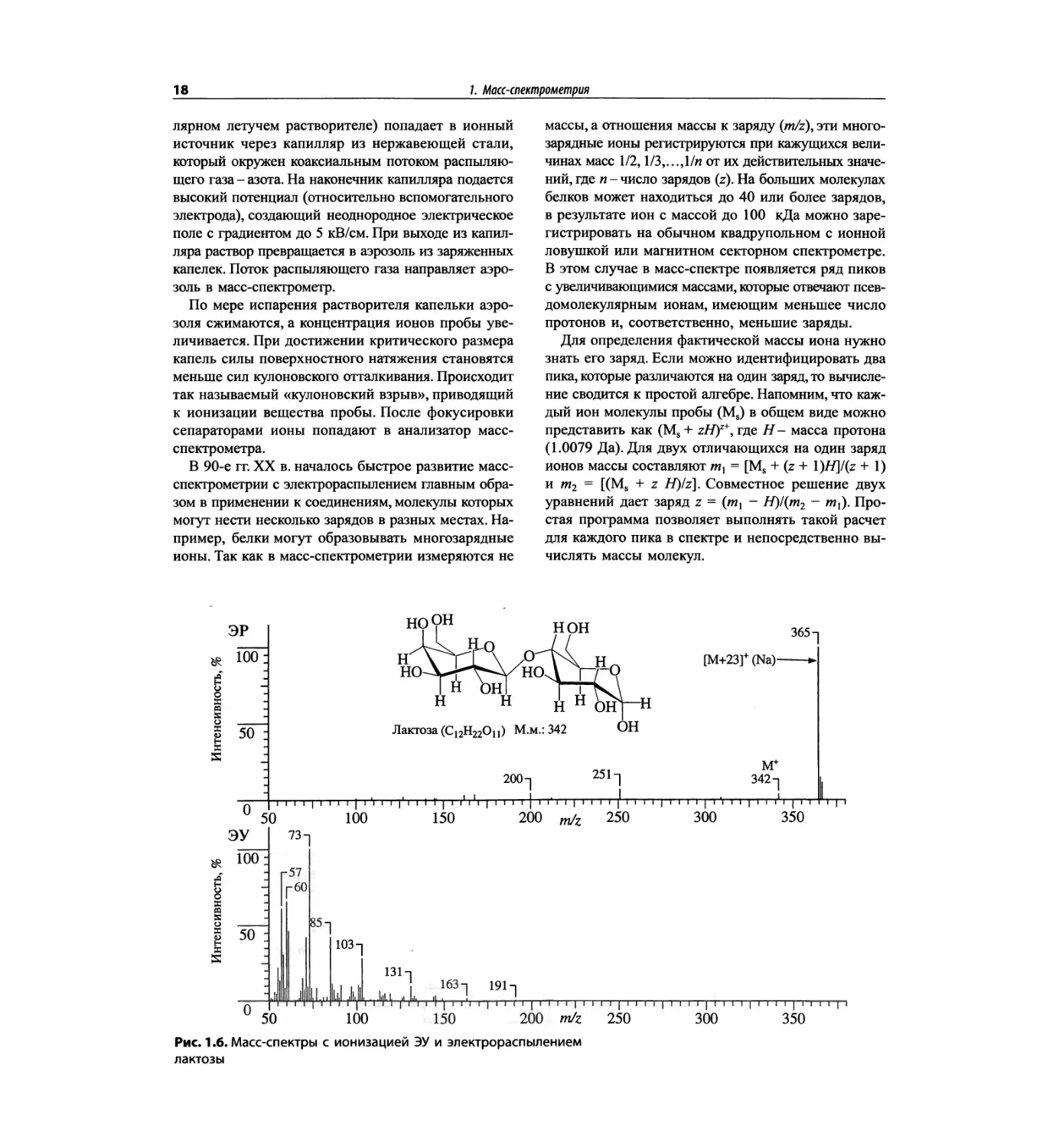
Рис. 1.6. Масс-спектры с ионизацией ЭУ и электрораспылением
лактозы
1.3. Методы ионизации
19
он
5C=O
ЭР
4СН3
I 5
он
4СН2
зсн2
зсн2
ЗСН-СНз ЗСН2 ЗСН2
I : 2 1 : I 1 : I
H2N—сн-с--N—CH2€-:-N—CH-C-i-N—СН-С—ОН
2 ||1;н II :Н 2 II ;н 2 ||1
о
О
Валин (V) Глицин (G)
C5H10ON QHjON
М.м.: 100 М.м.: 57
о
о
Серин (S) Глутаминовая
C3H5O2N
М.м.: 87
В
о
я
0
S
о
к
S
К
S
100:
50^ M-<E’S>
Z г 157
i *i । ।
кислота (Е)
C5H8O4N
М.м.: 146
M-(V,G)
235-1
M-(V)
150
200
М-(Е)
г 244
292-|
250
300
m/z
350
[М + If
[М + Н]
М-17
373-j
[М + 23Г
[M + Na]'
400
Рис. 1.7. Масс-спектр с электрораспылением тетрапептида, структура которого
показана на рисунке (пояснения см. в тексте)
Многие производители выпускают недорогие масс-
спектрометры с электрораспылением по двум при-
чинам. Во-первых, применение этого метода очень
успешно и в то же время он продолжает оставаться
достаточно простым в использовании. Во-вторых,
возросло значение анализа макромолекул белков и
пептидов, и лучшим методом для этого является
электрораспыление.
На рис. 1.6 сравнивается масс-спектр ЭУ лактозы
(в нижней части рисунка) со спектром, полученным
с использованием ЭР (верхняя часть рисунка). Более
подробно спектр лактозы рассматривается в гл. 5.
Таблица 1.1. Методы ионизации
Метод ионизации Образующиеся ионы Чувстви- тельность Преимущества Недостатки
Электронный удар М+ нг-пг Возможность использования поисковых систем и баз данных, структурная информация М+ иногда отсутствует
Химическая ионизация М+1, М+18, И т.д. нг-пг Обычно присутствует молекулярный ион М+ Мало информации о структуре молекулы.
Полевая десорбция М+ мкг-нг Нелетучие соединения Специальное оборудование
Бомбардировка быстрыми атомами М+1, М+катион М+матрица мкг-нг Нелетучие соединения, информация о порядке соединения фрагментов Мешающее влияние матрицы, трудности в интерпретации
Плазменная десорбция М+ мкг-нг Нелетучие соединения Мешающее влияние матрицы
Лазерная десорбция М+1, М+матрица мкг-нг Нелетучие соединения, выброс ионов Мешающее влияние матрицы
Термическое распыление М+ мкг-нг Нелетучие соединения Вышел из употребления
Электрораспыление м+, м++, м+++ и т. д. нг-пг Нелетучие соединения, сочетается с методом ЖХ (водной), образуются многозарядные ионы Ограниченные классы соединений, мало информации о структуре молекул
20
/. Масс-спектрометрия
Масс-спектр с ЭУ совершенно бесполезен, поскольку
у лактозы низкое давление паров, она термически
неустойчива, и в ее спектре нет характеристических
пиков. В масс-спектре с ЭР наблюдается слабый пик
молекулярного иона при m/z 342 и характерный пик
[М+23]+ (молекулярный пик плюс натрий). Появле-
ние повсеместно распространенных ионов натрия
в водном растворе носит общий характер.
На рис. 1.7 показан масс-спектр с ЭР тетрапептида,
состоящего из валина, глицина, серина и глутаминовой
кислоты (VGSE). Этот пример также рассмотрен в
гл. 5. Основной пик [М+1]+ проявляется при m/z 391,
90% от его интенсивности составляет пик [М+23]+,
обусловленный присутствием натрия. Кроме этого
полезную дополнительную информация дает харак-
терная для каждой из аминокислот фрагментация.
Для малых пептидов нет ничего необычного в такой
фрагментации, но для макромолекул белков этот про-
цесс менее вероятен.
Методы ионизации обобщены в табл. 1.1.
1.4. Анализаторы масс
Анализатор масс, который разделяет смесь возни-
кающих в процессе ионизации ионов по величинам
m/z, что приводит к образованию спектра, является
сердцем любого масс-спектрометра. Существует не-
сколько классов приборов с различными параметра-
ми. Характеристики важнейших типов анализаторов
масс представлены ниже. В заключение этого разде-
ла кратко обсудим тандемную масс-спектрометрию
и родственные процессы.
1.4.1. Магнитные секторные масс-спектрометры
Для детектирования движущихся по криволиней-
ной траектории ионов в магнитном секторном масс-
спектрометре (MS-MS) используется магнитное поле
(рис. 1.8). Магнитные секторные масс-спектрометры
были первыми доступными приборами и их важное зна-
чение сохраняется. Разделение ионов основано на том,
что при данной величине заряда в отношении масса/
заряд, более легкие ионы отклоняются сильнее, чем тя-
желые. Ионы с зарядом z, попавшие в магнитное поле
из источника, уже получили при ускорении потенциалом
V одинаковую кинетическую энергию Е = zV = mu2/2.
Когда ускоренный ион попадает в магнитное поле (В),
он испытывает отклоняющую силу (Bzu), направлен-
ную перпендикулярно первоначальному направлению
и искривляющую его путь. Теперь ион движется по
круговой траектории радиуса г, определяемого выра-
жением Bz\) = rm2/г. Объединение этих уравнений дает
хорошо известное соотношение для магнитного сектора
m/z = В2!2 12 V. Так как радиус прибора имеет постоянную
величину, при изменении величины магнитного поля
в область щели детектора попадают ионы с последова-
тельно изменяющимся соотношением m/z. Как показы-
вают эти уравнения, магнитный секторный инструмент
разделяет ионы по их моментам (т. е. произведениям
массы и скорости), а не только по массам, поэтому ионы
с одинаковой массой, но с разными кинетическими энер-
гиями будут фокусироваться в разных точках.
Электростатический анализатор (ЭСА) существенно
сужает энергетическое распределение в ионном пучке,
направляя ионы с одинаковыми зарядом (z) и кине-
тической энергией (независимо от массы) по одной
F J
Компьютер
Рис. 1.8. Схема простого масс-спектрометра с магнитным 180-градусным
секторным анализатором. Направление магнитного поля перпендикулярно
плоскости страницы. Радиус кривизны различается для разных приборов
1.4. Анализаторы маа
21
Ввод пробы
Источник
ионизации
Ускоряющие
электроды
Щель коллектора
г=35см
Фт = 65^
g
S
5
<т>
о
§
Sc
Фокусирующая щель
1 __Фокусирующее
устройство
Компьютер
Детектор
Рис. 1.9. Схема масс-спектрометра с двойной фокусировкой
траектории. Выходная щель ЭСА дополнительно фо-
кусирует ионный пучок перед его входом в детектор.
Сочетание ЭСА и магнитного сектора образует систему
с двойной фокусировкой, в которой нейтрализуются
эффекты рассеяния по направлению и скорости.
При использовании крайне узких щелей разре-
шение прибора с секторным магнитом и двойной
фокусировкой (рис. 1.9) может достигать 100 000.
Высокое разрешение позволяет измерять «точные
массы», однозначно дающие молекулярные форму-
лы, что чрезвычайно полезно. Такое высокое разре-
шение в значительной степени реализуется за счет
снижения чувствительности. Для сравнения скажем,
что в обычном инструменте с секторным магнитом
при разрешении около 5000 и точности 0.5 m/z по
всей области масс, т.е. при «единичном разрешении»,
верхний предел измерения m/z составляет около
15 000. Теоретически возможно повышение этого
предела, но практически недостижимо.
1.4.2. Квадрупольные масс-спектрометры
Квадрупольный анализатор масс гораздо меньше
и дешевле, чем магнитный секторный инструмент.
Квадруполь (схематически показанный на рис. 1.10)
состоит из четырех стержней круглого или гипер-
болического сечения длиной 100-200 мм, располо-
женных параллельно друг другу по углам квадрата.
Строгий математический анализ принципов действия
квадрупольного масс-анализатора сложен, здесь мы
потенциалы
Рис. 1.10. Схема квадрупольного «фильтра масс» или ионного сепаратора
22
/. Маа-спектрометрия
обсудим его работу в упрощенном виде. К стержням
приложено постоянное напряжение и дополнительно
радиочастотная компонента. Ионы вводятся в «тун-
нель», образованный четырьмя электродами квадру-
поля в центре квадрата, на одном конце электродов
и пролетают вдоль оси.
Для любой данной комбинации постоянного на-
пряжения и частоты переменного напряжения толь-
ко ионы с определенной величиной m/z обладают
устойчивой траекторией и способны пролетать через
квадруполь к детектору. Все ионы с другими величи-
нами m/z движутся по неустойчивыми траекториям
и сталкиваются с одним из электродов, рассеивают-
ся или проходят вне квадруполя. Совсем упрощая
картину, квадрупольный масс-анализатор можно рас-
сматривать как перестраиваемый фильтр масс. Если
с одной стороны попадают разные ионы, то через
анализатор проходит ион только с одним значением
m/z. Такая фильтрация в действительности происходит
очень быстро, и полный интервал масс сканируется
менее чем за одну секунду.
По разрешению и диапазону масс квадрупольный
масс-анализатор уступает магнитному сектору. Напри-
мер, верхний предел массы для квадруполя составляет
менее 5000 m/z. В то же время его чувствительность
обычно высока, так как не требуется щелей, которые
отсекают часть ионов. Важное преимущество ква-
друполей состоит в их высокой эффективности для
ионов с низкой скоростью; это означает, что ионные
источники могут работать при потенциале, близком
к потенциалу земли (т. е. при низком напряжении).
Так как входящие ионы обычно имеют энергию менее
100 эВ, квадрупольный масс-спектрометр идеален
для объединения с жидкостными хроматографами
и для методов ионизации при атмосферном давлении,
таких как электрораспыление (см. раздел 1.3.3.2).
Высокая эффективность для ионов с низкой энергией
объясняется тем, что лишь редкие столкновения с
высокой энергией происходят до того, как они по-
падут в квадруполь.
1.4.3. Масс-спектрометр с ионной ловушкой
Иногда ионная ловушка рассматривается как один
из вариантов квадруполя из-за их одновременного
появления и сходства принципа действия. Однако
ионная ловушка потенциально более универсальна
и имеет большие перспективы развития. Одно время
ионная ловушка имела плохую репутацию, потому
что ее ранние конструкции давали худшие резуль-
таты по сравнению с квадруполями. Эти результаты
часто зависели от концентрации; относительно боль-
шие пробы обычно давали интенсивные пики массы
[ион+1], что затрудняло поиск спектра в стандартных
библиотеках масс-спектров ЭУ. Со временем эти про-
блемы были преодолены, и спектры ЭУ, полученные
с ионной ловушкой, теперь полностью подходят для
поиска в обычных базах данных. Ионная ловушка чув-
ствительнее, чем квадруполь, и ее конструкция более
приемлема для проведения тандемных экспериментов
без чрезмерного усложнения оборудования.
Название ионной ловушки, возможно, возникло
из-за того, что в отличие от квадруполя, который
работает как фильтр масс, она может удерживать
ионы в течение относительно длительного времени.
Задержанные ионы последовательно выбрасываются
в детектор, что приводит к образованию обычного
масс-спектра. Прежде чем кратко рассмотреть дру-
гие пути использования пойманных ионов, полезно
ознакомиться с устройством ионной ловушки.
Фокусирующие
линзы
Рис. 1.11. Устройство ионной ловушки (вид в разрезе)
Электростатическая Центральный
1.4. Анализаторы маа
23
Обычная ионная ловушка состоит из трех электро-
дов: одного кольцевого с гиперболической внутренней
поверхностью и двух гиперболических полюсных
электродов с каждого конца (поперечный разрез
ионной ловушки показан на рис. 1.11). На кольцевой
электрод подается синусоидальное радиочастотное
поле, в то время как полюсные электроды работают
в одном из трех режимов: при потенциале земли,
при постоянном напряжении или при переменном.
Математический аппарат, который описывает дви-
жение ионов внутри ионной ловушки основан на
уравнении Матье. Детальное обсуждение трехмерных
диаграмм устойчивости ионов можно найти в книге
(March, Hughes, 1989). Достоинство ионной ловушки
заключается в возможности контроля трех параме-
тров-напряжений радиочастотного поля, переменного
тока и постоянного тока, что позволяет выполнять
разнообразные эксперименты (более подробно см.
March, Hughes, 1989).
Ионная ловушка может функционировать в трех
основных режимах. В первом при постоянном напря-
жении радиочастотного поля постоянный ток между
полюсными и кольцевым электродами отсутствует,
все ионы с отношением m/z, превышающим некоторую
предельную величину, будут удерживаться в ловушке.
По мере увеличения напряжения радиочастотного поля
граничное значение m/z контролируемо увеличивается,
ионы последовательно вылетают из ловушки и детекти-
руются. В результате получается обычный масс-спектр,
а эксперимент этого типа называется «нестабильным
масс-селективным сканированием». В этом эксперимен-
те верхние значения масс ограничены максимальный
величиной потенциала радиочастотного поля, которое
подается на электроды. Ионы с массами, превышающими
верхний предел, удаляются, после того как потенциал
радиочастотного поля снижается обратно до нуля.
В экспериментах во втором режиме используется
постоянный потенциал между полюсными электро-
дами. Результат состоит в том, что теперь имеются
низший и верхний пределы значения m/z ионов.
Возможности такой схемы эксперимента огромны,
и большинство приборов с ионными ловушками от-
носятся к этому типу. Можно выбрать даже один
ион. Мониторинг ионов конкретного типа является
важным приложением таких экспериментов. Массы
выбранных ионов практически не ограничены.
В экспериментах в третьем режиме, который по-
хож на второй, добавляется осциллирующее поле
между полюсными электродами, которое сообщает
добавочную кинетическую энергию выбранному иону.
Это поле малой амплитуды увеличивает кинетическую
энергию иона медленно, в течение времени, которо-
го достаточно для столкновительной фрагментации
с эффективностью близкой к 100%. Если присущая
ионной ловушке чувствительность сочетается с почти
100% тандемной эффективностью, то использование
ионной ловушки для тандемной масс-спектрометрии
значительно превосходит три последовательно свя-
занных квадруполя (см. ниже).
Другой путь реализации этой добавочной кинетиче-
ской энергии состоит в удалении из ионной ловушки
нежелательных ионов. Такие ионы могут возникать
из растворителя или матрицы в экспериментах ББА
или ВИМС. Поле с постоянной частотой при высоком
напряжении во время ионизации будет селективно
удалять ионы одного вида. В таком эксперименте
можно отделять ионы и нескольких видов.
1.4.4. Времяпролетный масс-спектрометр
Идея времяпролетного (time-of-flight, TOF) масс-
спектрометра проста. Ионы сначала ускоряются по-
тенциалом (V), а затем медленно «дрейфуют» к детек-
тору. Если предполагается, что все ионы, достигающие
начала области дрейфа, имеют одинаковую энергию
zeV= wru2/2, то ионы с разными массами будут иметь
разные скорости u = (2zeP7m)1/2. При длине области
дрейфа L время пролета для иона определяется вы-
ражением t = (L/mllzeV^12, из которого легко вычис-
ляется масса иона.
Решающим условием для работы этого в других
отношениях простого прибора является необходимость
получения ионов в точно известный начальный мо-
мент времени и с определенным положением в про-
странстве. Это требует использования во времяпро-
летных спектрометрах только методов импульсной
ионизации, которые включают плазменную и лазерную
десорбцию (например, MALDI).
Из-за неизбежного разброса энергий ионов раз-
решающая способность времяпролетных масс-
спектрометров обычно менее 20 000. Кроме того,
поскольку разница во времени достижения детектора
может быть менее 10-7 с, для достаточного разреше-
ния требуется применение электроники с высоким
быстродействием. Достоинством этого метода явля-
ется то, что область доступных масс не ограничена
и, как и у квадрупольных приборов, у них отличная
чувствительность из-за отсутствия ограничивающих
щелей. По этим причинам применение времяпролет-
ных масс-спектрометров оказалось наиболее полезным
для исследования больших биологических молекул.
1.45. Маа-спектрометр с преобразованием Фурье
Масс-спектрометры с преобразованием Фурье (FT)
из-за их высокой стоимости не получили широко-
24
1. Маа-спектрометрия
го распространения. Со временем, по мере прогресса
в области производства сверхпроводящих магнитов их
применение может расшириться. В масс-спектрометре
с преобразованием Фурье ионы удерживаются в ячейке
с улавливающим электрическим потенциалом в силь-
ном магнитном поле. В этой ячейке каждый из ионов
движется по орбите в плоскости, перпендикулярной на-
правлению магнитного поля, с частотой, пропорциональ-
ной m/z иона. Радиочастотный импульс, приложенный
к ячейке, содержит все круговые частоты одновременно,
давая интерферограмму, подобную спаду свободной маг-
нитной индукции (FID) в ЯМР или интерферограмме
в ИК-фурье-спекгроскопии. Интерферограмма, которая
является спектром во временном представлении, по-
средством преобразования Фурье превращается в спектр
в частотном представлении, который уже дает обычный
спектр m/z. Импульсная спектрометрия с преобразова-
нием Фурье в приложении к спектрометрии ядерного
магнитного резонанса обсуждается в гл. 3-5.
Так как прибор работает при постоянной напряжен-
ности магнитного поля, можно использовать мощные
сверхпроводящие магниты. Кроме того, прямая зави-
симость интервала масс от напряженности магнитного
поля позволяет детектировать очень большие массы.
Наконец, поскольку все ионы, возникающие при одно-
кратной ионизации, улавливаются и анализируются,
этот метод очень чувствителен и хорошо сочетается
с импульсными ионизационными методами. Преи-
муществом масс-спектрометрии с преобразованием
Фурье является ее высокое разрешение, что позволяет
сделать выбор в ее пользу по сравнению с другими
масс-анализаторами. Масс-спектрометр с преобразова-
нием Фурье можно объединить с хроматографом и с
разными методами ионизации, что даст возможность
исследовать и малые молекулы. Более подробную
информацию о масс-спектрометрах с преобразованием
Фурье можно найти в других изданиях.
1.4.6. Тандемная масс-спектрометрия
Тандемная масс-спектрометрия (МС-МС, «МС в ква-
драте») полезна для изучения известных и неизвест-
ных соединений; с ионными ловушками возможна
МС до п-й (МС(л)), где п от 2 до 9. На практике
и редко превышает 2 или 3. В случае МС-МС вы-
деляется «родительский» ион, возникающий при
начальной фрагментации (начальная фрагментация
приводит к обычному масс-спектру), который под-
вергается дальнейшей фрагментации с образованием
«дочерних» ионов. В случае сложных смесей такие
дочерние ионы указывают на присутствие известного
соединения. Для неизвестных или новых соединений
эти дочерние ионы дают информацию о структуре.
Одно из распространенных применений МС-МС
состоит в исследовании неочищенной пробы, избира-
тельном «вылавливании» характеристического для
изучаемого соединения иона и получении диагно-
стического спектра дочерних ионов, образующихся
из этого иона. Таким путем соединение может быть
однозначно идентифицировано в пробе сырья без
предшествующего хроматографического или иного
разделения. Таким образом, МС-МС может быть
очень полезным инструментом скрининга. Этот при-
ем уменьшает необходимость сложного разделения
смесей для выполнения многих рутинных анализов.
Например, анализ проб мочи людей (или живот-
ных, таких как скаковые лошади) на присутствие
допинга или метаболитов может быть выполнен
методом МС-МС на рутинном уровне без предва-
рительной очистки и разделения. В случае неиз-
вестных соединений дочерние ионы могут нести
структурную информацию.
Один из путей выполнения МС-МС-эксперимента
заключается в последовательном объединении двух
или более анализаторов масс, что дает возможность
выделять ион с данным значением m/z (либо ро-
дительский, либо дочерний) и регистрировать его
фрагментацию. Например, три последовательно
связанных квадруполя (так называемый «triple
quad») образуют тандемный масс-спектрометр.
В этой схеме первый квадруполь выделяет кон-
кретный ион для дальнейшего анализа, второй
квадруполь функционирует как столкновитель-
ная ячейка (распад, индуцированный столкнове-
ниями, CID) в радиочастотном режиме, третий
квадруполь разделяет образовавшиеся ионы и дает
спектр дочерних ионов. Область тандемной масс-
спектрометрии уже вполне сложилась, по этому
методу доступны неплохие книги.
Чтобы на приборе можно было проводить экспе-
рименты МС-МС, он должен быть способен выпол-
нять три указанные выше операции. Однако, как мы
видели, системы с ионной ловушкой, на которых воз-
можно проведение экспериментов МС-МС и МС(л),
совсем не используются в качестве тандемных масс-
анализаторов, для выполнения всех трех операций
одновременно предпочитают применять отдельную
ионную ловушку. Как уже отмечалось, эти экспери-
менты с тандемным масс-спектрометром с ионной
ловушкой очень чувствительны, а приборы удобны
для работы. Ионная ловушка дает возможность про-
водить МС-МС-эксперименты на компактном обо-
рудовании и относительно дешево.
В табл. 1.2 в кратком виде обобщены основные
характеристики масс-анализаторов и методов иони-
зации.
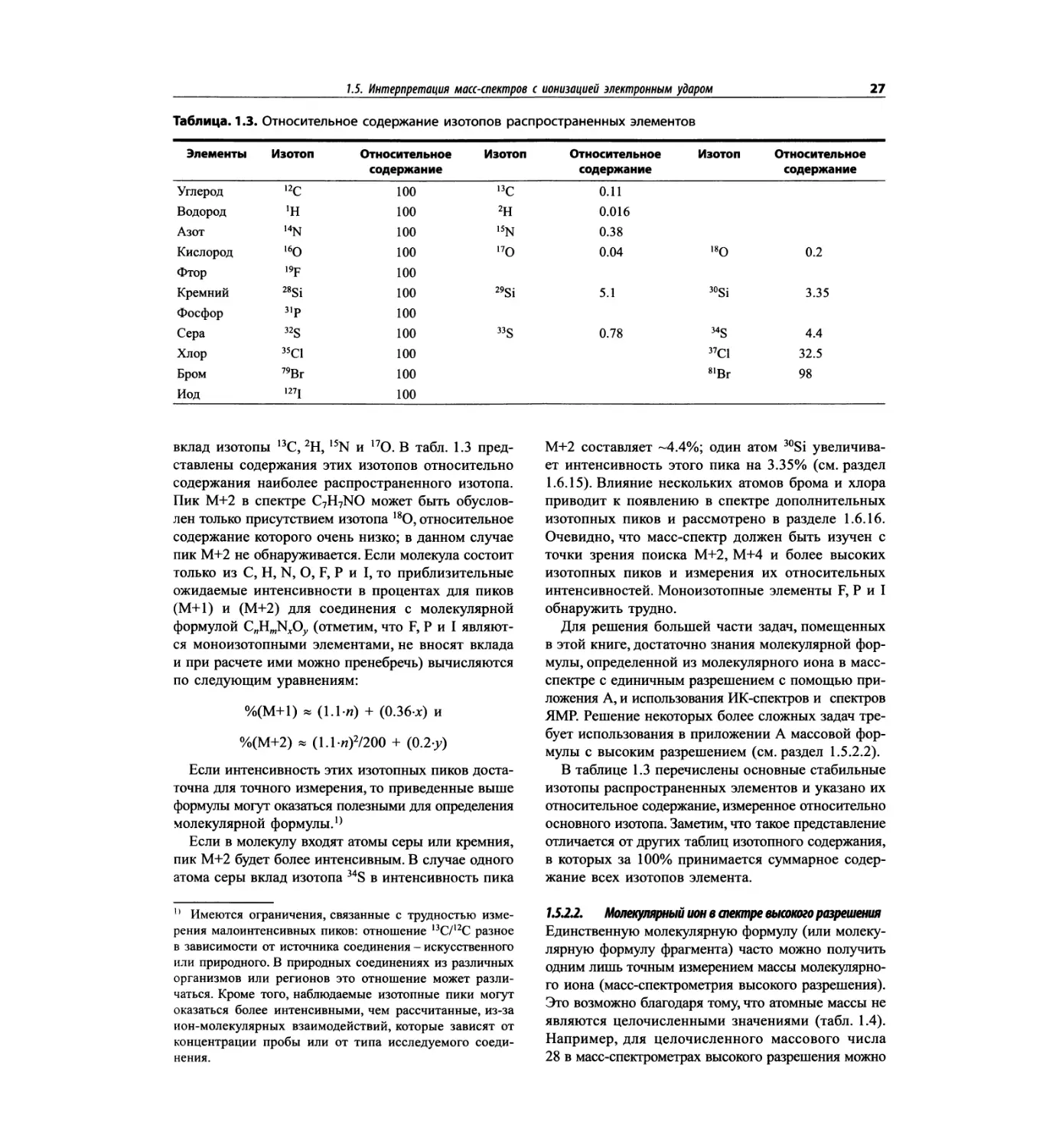
1.5. Интерпретация маа-спектров с ионизацией электронным ударом_____________________25
Таблица 1.2. Основные характеристики масс-анализаторов
Масс-анализатор Диапазон масс Разрешение Чувстви- тельность Преимущества Недостатки
Магнитный сектор mlz 1-15000 0.0001 Низкая Высокое разрешение Низкая чувствительность; высокая стоимость; сложен технически
Квадруполь mlz 1-5000 единица Высокая Прост в использовании, недорогой; высокая чувствительность Низкое разрешение; узкий диапазон масс
Ионная ловушка mlz 1-5000 единица Высокая Прост в использовании, недорогой; высокая чувствительность; тандемная МС (МСЛ) Низкое разрешение; узкий диапазон масс
Времяпролетный Не ограничен 0.0001 Высокая Широкий диапазон масс; простая конструкция Высокое разрешение
С преобразованием Фурье До 70 кДа 0.0001 Высокая Очень высокое разрешение; широкий диапазон масс Высокая стоимость; сложен технически
1.5. Интерпретация масс-спектров
с ионизацией электронным ударом
Дальнейшее обсуждение масс-спектров ограничивает-
ся масс-спектрометрией с ионизацией электронным
ударом. Фрагментация в этом случае несет богатую
информацию о структуре, владение данным методом
особенно полезно для химиков-органиков.
Масс-спектры обычно получают при энергии
электронного пучка 70 эВ. В простейшем случае мо-
лекула в газовой фазе под действием электронного
пучка теряет один электрон, что приводит к обра-
зованию молекулярного иона, который представляет
собой катион-радикал. Например, метанол образует
молекулярный ион, как показано на схеме 1.1. Одна
точка означает оставшийся нечетный электрон; когда
заряд локализуется на отдельном атоме, знак заряда
указывается на этом атоме.
СНзОН
СН3ОН + е"----> CH3OH+(m/z 32) + 2е“
Схема 1.1
Многие из этих молекулярных ионов распадаются
за время от 1О-10 до 10"3 с и дают в простейшем
случае положительно заряженный осколок и ради-
кал. Так образуется ряд осколочных ионов, каждый
из которых может распасться на еще более мелкие
фрагменты. Возможные пути распада для метанола
показаны на схеме 1.2.
СН3ОН +----> СИрЫ (m/z 31) + Н’
СН3ОН’+----> СН3+ (m/z 15) + ОН
СН2ОН+-----> СНО+ (m/z 29) +
Схема 1.2
Если некоторые из молекулярных ионов имеют
достаточно большое время жизни, то они достигают
детектора и регистрируются в виде пика молекулярного
иона. Распознать эти пики очень важно, так как они
дают молекулярную массу изучаемого соединения.
В случае единичного разрешения величина этой моле-
кулярной массы равна ближайшему целому числу.
Масс-спектр - это представление относительных
концентраций положительно заряженных осколков
(включая молекулярный ион) в зависимости от их
масс. Высота наиболее интенсивного в спектре пика,
называемого максимальным (основным), принимается
за 100%, а интенсивности (высота, умноженная на ко-
эффициент чувствительности) других пиков, включая
пик молекулярного иона, выражаются в процентах
от максимального пика. Конечно, иногда максималь-
ным пиком может быть и пик молекулярного иона.
На рис. 1.1 пик молекулярного иона имеет величину
m/z 121, а максимальный пик m/z 77.
Спектры можно представлять в табличном или
графическом виде. Преимущество графической за-
писи состоит в том, что опытный сотрудник может ее
быстро расшифровать. Однако график должен быть
построен так, чтобы без труда различать единицы
масс. Например, если ошибочно принять пик с m/z
26
1. Маа-спектрометрия
79 за пик с m/z 80, это может привести к полной
путанице. Пик молекулярного иона обычно является
пиком с наибольшим массовым числом, за исклю-
чением пиков изотопных ионов.
1.5.1. Идентификация пика
молекулярного иона
Распознавание пика молекулярного иона (М)+ в спек-
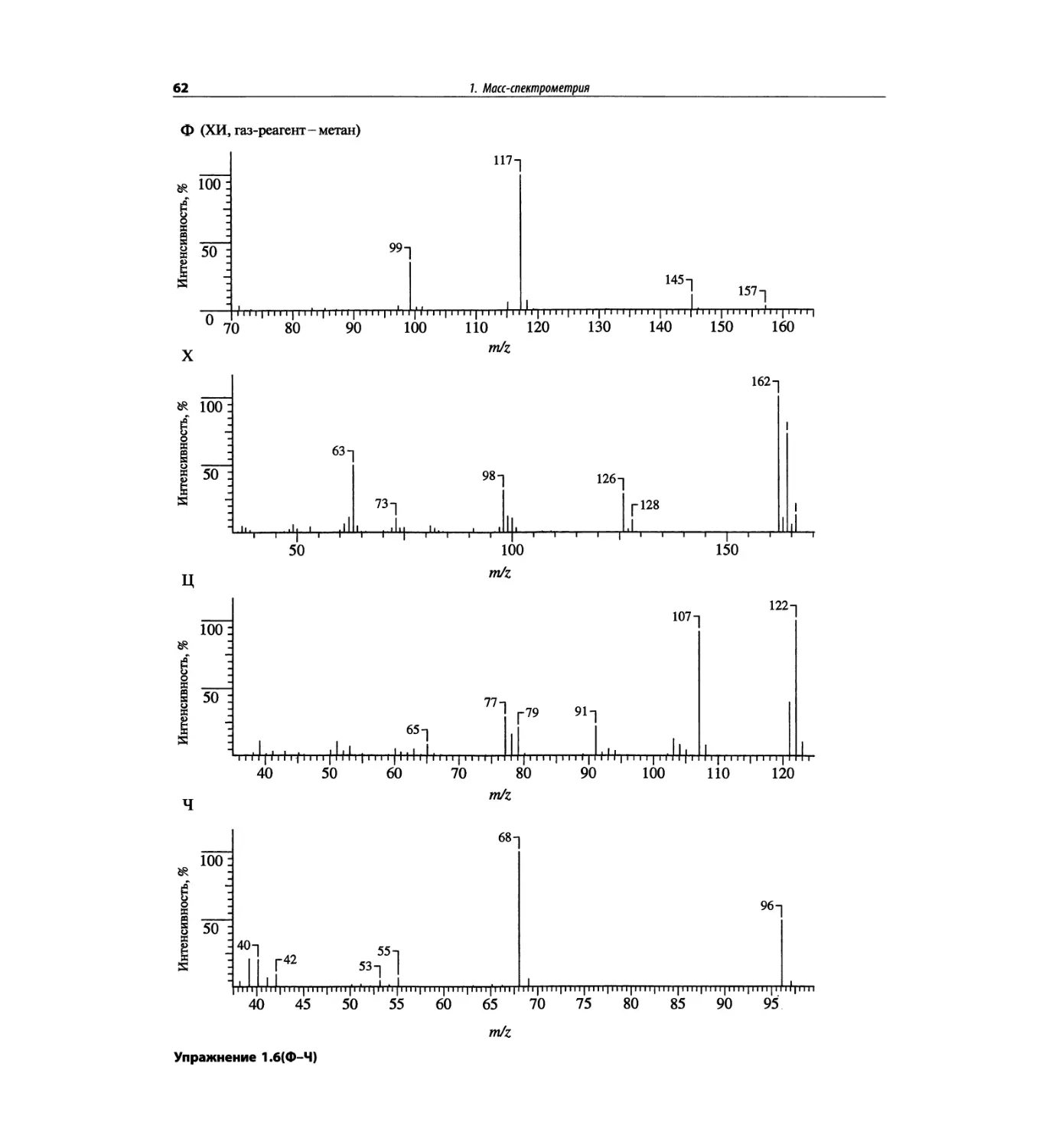
трах с ионизацией электронным ударом довольно ча-
сто представляется проблематичным. Этот пик может
оказаться очень слабым или не появиться совсем. Как
убедиться, что данный пик является молекулярным,
а не фрагментным или примесным? Часто лучшее
решение состоит в регистрации масс-спектра с хи-
мической ионизацией (см. раздел 1.3.1.2); при этом
получается спектр с интенсивным пиком [М+1]+ при
незначительной фрагментации.
Многие пики можно исключить из числа возмож-
ных пиков молекулярных ионов на основе разумных
структурных ограничений. Часто бывает полезным
«азотное правило». Оно устанавливает, что молеку-
ла с четной молекулярной массой либо не должна
содержать азот, либо число атомов азота должно
быть четным; нечетная молекулярная масса требует
нечетного числа атомов азота.Это правило спра-
ведливо для всех соединений, содержащих углерод,
водород, кислород, азот, серу и галогены, а также
многие другие реже встречающиеся атомы, такие
как фосфор, бор, кремний, мышьяк и щелочнозе-
мельные металлы.
Полезный вывод состоит в том, что простой раз-
рыв одинарной связи (без перегруппировки) дает
осколочный ион с нечетной массой из молекуляр-
ного иона с четной массой и, наоборот, осколочный
ион с четной массой образуется из молекулярного
иона с нечетной массой. Для выполнения этой за-
кономерности важно, чтобы такой осколочный ион
содержал все атомы азота (если они есть), имеющиеся
в молекулярном ионе.
Анализ фрагментации в сочетании с другой инфор-
мацией помогает идентифицировать пики молекуляр-
ных ионов. Следует иметь в виду, что в приложении
А содержатся формулы как фрагментов, так и молекул.
При решении конкретной задачи некоторые из этих
формул можно отбросить как тривиальные.
Интенсивность пика молекулярного иона зависит
от стабильности самого иона. Наиболее устойчивы-
ми молекулярными ионами являются ионы чисто
ароматических систем. Если имеются заместители,
° Для соблюдения азотного правила используются только
целочисленные молекулярные массы.
которые дают преимущественное направление рас-
пада, то пик молекулярного иона будет менее ин-
тенсивным, а относительные интенсивности пиков
осколков возрастут. В общем случае способность
органических соединений давать пик молекуляр-
ного иона уменьшается в ряду ароматические со-
единения > сопряженные алкены > насыщенные
циклические соединения > органические сульфиды
> неразветвленные алканы с короткими цепями >
тиолы. Обычно заметные пики молекулярных ио-
нов образуют следующие соединения (в порядке
убывания этой способности): кетоны > амины >
сложные эфиры > простые эфиры > карбоновые
кислоты ~ альдегиды ~ амиды ~ галогенпроизводные.
Пик молекулярного иона часто не обнаруживает-
ся для алифатических спиртов, нитритов, нитратов,
нитросоединений, нитрилов и сильноразветвленных
соединений.
Считается, что наличие в спектре пика М-15 (по-
теря СН3), пика М-18 (потеря Н2О), пика М-31 (по-
теря ОСН3 из метиловых сложных эфиров) и т. д.
является подтверждением соответствующего пика
молекулярного иона. Часто наблюдается пик М-1,
иногда возникает пик М-2 (потеря Н2 либо при фраг-
ментации, либо при термолизе), реже встречается пик
М-3 (в случае спиртов). Однако пики в области от
М-3 до М-14 указывают на возможность загряз-
нения или что предполагаемый пик молекулярного
иона является на самом деле пиком осколочного
иона. Маловероятны потери фрагментов с массой
от 19 до 25 (за исключением потери F = 19 или
HF = 20 фторсодержащими соединениями). Потеря
16(0), 17(ОН) или 18(Н2О) вероятна, если в молекуле
имеется атом кислорода.
1.5.2. Определение молекулярной формулы
7.5.2.7. Молекулярный ион и изотопные пики
в масс-спектрах низкого разрешения
До сих пор мы обсуждали масс-спектры низкого
(единичного) разрешения (с целочисленными значе-
ниями масс), т. е. масса молекулярного иона C7H7NO
(рис. 1.1) с m/z 121 складывается из целочисленных
масс наиболее распространенных изотопов (7x12
[для 12С]) + (7x1 [для !Н]) + (1x14 [для 14N]) + (1x16
[для 16О]) = 121.
Кроме того, существуют молекулярные частицы,
в состав которых входят изотопы с более низким
природным содержанием, что приводит к появлению
«изотопных пиков» с М+1, М+2 и т.д. На рис. 1.1 пик
М+1 имеет интенсивность около 8% относительно
интенсивности пика молекулярного иона, которая при-
нята за 100%. В интенсивность этого пика вносят
1.5. Интерпретация маа-спектров с ионизацией электронным ударом_____________________27
Таблица. 1.3. Относительное содержание изотопов распространенных элементов
Элементы Изотоп Относительное содержание Изотоп Относительное содержание Изотоп Относительное содержание
Углерод 12с 100 ,3С 0.11
Водород >н 100 2Н 0.016
Азот 14N 100 15N 0.38
Кислород 16о 100 ,7О 0.04 18О 0.2
Фтор 19р 100
Кремний 28Si 100 29Si 5.1 30Si 3.35
Фосфор 31Р 100
Сера 32s 100 33s 0.78 34S 4.4
Хлор 35С1 100 37С1 32.5
Бром 79Вг 100 8|Вг 98
Иод 1271 100
вклад изотопы 13С, 2Н, 15N и 17О. В табл. 1.3 пред-
ставлены содержания этих изотопов относительно
содержания наиболее распространенного изотопа.
Пик М+2 в спектре C7H7NO может быть обуслов-
лен только присутствием изотопа 18О, относительное
содержание которого очень низко; в данном случае
пик М+2 не обнаруживается. Если молекула состоит
только из С, Н, N, О, F, Р и I, то приблизительные
ожидаемые интенсивности в процентах для пиков
(М+1) и (М+2) для соединения с молекулярной
формулой C„HwNxOy (отметим, что F, Р и I являют-
ся моноизотопными элементами, не вносят вклада
и при расчете ими можно пренебречь) вычисляются
по следующим уравнениям:
%(М+1) « (1.1и) + (0.36-х) и
%(М+2) « (1.1и)2/200 + (0.2-у)
Если интенсивность этих изотопных пиков доста-
точна для точного измерения, то приведенные выше
формулы могут оказаться полезными для определения
молекулярной формулы.1)
Если в молекулу входят атомы серы или кремния,
пик М+2 будет более интенсивным. В случае одного
атома серы вклад изотопа 34S в интенсивность пика
!) Имеются ограничения, связанные с трудностью изме-
рения малоинтенсивных пиков: отношение 13С/12С разное
в зависимости от источника соединения - искусственного
или природного. В природных соединениях из различных
организмов или регионов это отношение может разли-
чаться. Кроме того, наблюдаемые изотопные пики могут
оказаться более интенсивными, чем рассчитанные, из-за
ион-молекулярных взаимодействий, которые зависят от
концентрации пробы или от типа исследуемого соеди-
нения.
М+2 составляет -4.4%; один атом 30Si увеличива-
ет интенсивность этого пика на 3.35% (см. раздел
1.6.15). Влияние нескольких атомов брома и хлора
приводит к появлению в спектре дополнительных
изотопных пиков и рассмотрено в разделе 1.6.16.
Очевидно, что масс-спектр должен быть изучен с
точки зрения поиска М+2, М+4 и более высоких
изотопных пиков и измерения их относительных
интенсивностей. Моноизотопные элементы F, Р и I
обнаружить трудно.
Для решения большей части задач, помещенных
в этой книге, достаточно знания молекулярной фор-
мулы, определенной из молекулярного иона в масс-
спектре с единичным разрешением с помощью при-
ложения А, и использования ИК-спектров и спектров
ЯМР. Решение некоторых более сложных задач тре-
бует использования в приложении А массовой фор-
мулы с высоким разрешением (см. раздел 1.5.2.2).
В таблице 1.3 перечислены основные стабильные
изотопы распространенных элементов и указано их
относительное содержание, измеренное относительно
основного изотопа. Заметим, что такое представление
отличается от других таблиц изотопного содержания,
в которых за 100% принимается суммарное содер-
жание всех изотопов элемента.
1.5.2.2. Молекулярный ион в спектре высокого разрешения
Единственную молекулярную формулу (или молеку-
лярную формулу фрагмента) часто можно получить
одним лишь точным измерением массы молекулярно-
го иона (масс-спектрометрия высокого разрешения).
Это возможно благодаря тому, что атомные массы не
являются целочисленными значениями (табл. 1.4).
Например, для целочисленного массового числа
28 в масс-спектрометрах высокого разрешения можно
28
7. Маа-спектрометрия
Таблица 1.4. Точные величины масс изотопов
Элемент Атомная масса Нуклид Масса
Водород 1.00794 >н 1.00783
D(2H) 2.01410
Углерод 12.0115 ,2С 12.00000
13с 13.00336
Азот 14.0067 ,4N 14.0031
15N 15.0001
Кислород 15.9994 16O 15.9949
17о 16.9991
18О 17.9992
Фтор 18.9984 19р 18.9984
Кремний 28.0855 28Si 27.9769
29Si 28.9765
30Si 29.9738
Фосфор 30.9738 3,р 30.9738
Сера 32.0660 32s 31.9721
33s 32.9715
34S 33.9679
Хлор 35.4527 35C1 34.9689
37C1 36.9659
Бром 79.9094 79Br 78.9183
81Br 80.9163
Иод 126.9045 127j 126.9045
увидеть различия между СО, N2, CH2N и С2Н4. Точное
значение массы СО 12.0000 (для 12С) + 15.9949 (для
16О) = 27,9949; точное значение массы N2 равно
2x14.60031 (для 14N) = 28.0062; Подобные вычисле-
ния дают точные значения масс 28.0187 для CH2N
и 28.0312 для С2Н4.
Таким образом, масса, наблюдаемая для молеку-
лярного иона СО, представляет собой сумму точных
масс наиболее распространенных изотопов углерода
и кислорода. Она отличается от молекулярной массы
СО, основанной на атомных массах, которые являются
средними значениями масс всех естественных изо-
топов элемента (например, С = 12.01, О = 15.999).
В табл. 1.4 представлены массы распространенных
ядер с точностью до четырех-пяти десятичных зна-
ков, здесь же показаны привычные значения атомных
масс (усредненные величины для элементов).
В приложении А приведены молекулярные фор-
мулы в порядке увеличения целочисленных масс.
Для целых масс формулы приведены b соответствии
с системой, принятой в Chemical Abstract. Формульная
масса (ФМ) приведена с точностью до четырех деся-
тичных знаков для каждой молекулярной формулы.
Приложение А предназначено для поиска в предпо-
ложении, что у студента имеется масса молекуляр-
ного иона с единичным разрешением масс-спектра
и дополнительная информация из других спектров.
Обратите внимание на то, что в таблицу включены
только ядра С, Н, N и О.
1.5.3. Использование молекулярной формулы.
Показатель водородной ненасыщенности
Если бы химик-органик должен был выбрать только
один вид информации среди всех других, которые
получают химическим путем или из спектров, то он,
конечно, выбрал бы молекулярную формулу.
Кроме типа и числа атомов молекулярная формула
дает показатель (индекс) ненасыщенности по водороду.
Показатель водородной ненасыщенности представляет
собой число пар атомов водорода, которые должны
быть удалены из соответствующей «насыщенной»
формулы, чтобы получить молекулярную формулу
исследуемого соединения. Показатель водородной
ненасыщенности также называется степенью нена-
сыщенности; это определение неполно, так как во-
дородная ненасыщенность может быть обусловлена
как циклической структурой, так и кратными связями.
Этот показатель является суммой числа циклов, числа
двойных связей и удвоенного числа тройных связей.
Индекс водородной ненасыщенности можно
вычислить для соединений, содержащих угле-
род, водород, азот, галоген, кислород и серу.
Для обобщенной молекулярной формулы
C„HmXxNyOz
показатель водородной ненасыщенности = (п) - (т/2)
- (х/2) + (у/2) + 1.
Для соединения с молекулярной формулой C7H7NO
показатель водородной ненасыщенности равен 7 -
3.5 + 0.5 +1 = 5. Отметим, что двухвалентные атомы
(кислород и сера) не учитываются.
В самом общем виде для формулы оцРпУцДу ин-
декс = (IV) - (1/2) + (Ш/2) + 1, где а - Н, D или
галоген (т. е. любой одновалентный атом), р - О,
S или любой двухвалентный атом, у - N, Р или другой
(С6Н5)3Р~ О
Диметил- Нитрометан Трифенил-
сульфоксид фосфиноксид
Рис. 1.12. «Полярные» структуры Льюиса
молекул диметилсульфоксида, нитрометана
и трифенилфосфиноксида, для которых получаются
правильные величины индексов водородной
ненасыщенности
1.5. Интерпретация маа-спектров с ионизацией электронным ударом
29
трехвалентный атом, 8 - С, Si или другой четырехва-
лентный атом. Римские цифры I - IV обозначают одно-,
двух-, трех- и четырехвалентные атомы соответственно.
Для простых молекулярных формул показатель
водородной ненасыщенности можно получить сравне-
нием рассматриваемой формулы с молекулярной фор-
мулой соответствующего насыщенного соединения.
Индексы ненасыщенности для С6Н6 и С6Н14 равны
4 и 0 соответственно.
Индекс ненасыщенности для C7H7NO равен 5,
и, возможно, этой брутто-формуле соответствует
структура бензамида (см. рис. 1.1). Естественно, воз-
можны и другие изомеры (т. е. соединения с такой
же молекулярной формулой), например
Отметим, что само бензольное кольцо при под-
счете дает четыре «ненасыщенных позиции» - три
для двойных связей и одну для цикла.
В случае соединений, содержащих атом в более
высоком валентном состоянии, такой как сера или
фосфор, нужно использовать полярные структуры.
Например, если мы рассматриваем серу в диметил-
сульфоксиде (ДМСО) формально в качестве двухва-
лентного атома, то индекс ненасыщенности получается
равным 0, что согласуется со структурой на рис. 1.12.
При определении индекса ненасыщенности нужно
использовать только формулы с заполненными ва-
лентными оболочками для всех атомов при условии
выполнения правила октетов Льюиса.
Аналогичным образом мы рассматриваем атом азота
в нитрометане как трехвалентный, что дает совмести-
мый с формулой на рис. 1.12 индекс ненасыщенно-
сти, равный 1. В трифенилфосфиноксиде валентность
фосфора приравнивается трем, что дает показатель
ненасыщенности 12 и согласуется с показанной на
рис 1.12 структурой Льюиса. В качестве примера
рассмотрим молекулярную формулу C13H9N2O4BrS.
Индекс ненасыщенности должен быть 13 - 10/2 +
2/2+1 = 10, что согласуется со структурой:
(Показатель ненасыщенности равен 4 для каждого
бензольного кольца и 1 для каждой группы NO2.)
Приведенную выше формулу для показателя не-
насыщенности можно применить к фрагментным
и молекулярным ионам. В случае ионов с четным
числом электронов (все электроны спарены) пока-
затель всегда получается равным нечетному числу,
умноженному на 0.5. В качестве примера рассмотрим
С7Н5О+ с индексом ненасыщенности 5.5. Удовлетво-
рительной структурой будет
так как потребовалось бы пять с половиной пар ато-
мов водорода, чтобы получить соответствующую на-
сыщенную формулу С7Н16О (C„H2w+2O). Осколочные
ионы с нечетным числом электронов всегда будут
давать целочисленное значение показателя водородной
ненасыщенности.
При исследовании терпенов часто приходится делать
выбор между двойной связью и циклической струк-
турой. Этот вопрос может быть легко разрешен с по-
мощью микровесов при каталитическом гидрировании
образца и повторном получении масс-спектра. Если
нет еще каких-либо легко восстанавливаемых групп,
то увеличение массового числа пика молекулярного
иона является мерой числа двойных связей; другими
«ненасыщенными позициями» должны быть циклы.
Такие простые соображения дают химику инфор-
мацию о структуре. Еще один пример: можно быстро
определить, является ли соединение, содержащее один
атом кислорода, простым эфиром или карбонильным
соединением, просто подсчитывая «ненасыщенные
позиции».
1.5.4. Фрагментация
На первый взгляд кажется, что из данных по фраг-
ментации молекул, происходящей при воздействии
колоссального избытка энергии, можно только гру-
бо оценивать их структуру. Разработанные логиче-
ские обоснования корреляций между масс-спектром
и структурой можно оценить как исключительно
изящные, хотя иногда и произвольные. Проница-
тельность первых исследователей, таких как Мак-
Лафферти, Бейнон, Стенхаген, Райхейдж и Майерсон,
привела к ряду рациональных механизмов фрагмен-
тации. Они были тщательно разработаны и мастерски
обобщены (см, например, Biemann (1962), Budzikiewicz
(1967) и др.).
Обычно стараются рассматривать молекулярный
ион с локализованным зарядом. В приближении Буд-
зикевича и соавторов (1967), положительный заряд
локализован либо на л-связи (за исключением со-
пряженных систем), либо на гетероатоме. Независимо
30
/. Маа-спектрометрия
от того, является ли эта концепция вполне строгой или
нет, в ней, по крайней мере, проявляется педагогическая
изобретательность. В этой книге мы будем использовать
такие молекулярные ионы с локализованными зарядами.
Например, на рис. 1.13 изображен молекулярный
ион циклогексадиена в виде структур А, В и С.
В структурной формуле А на один электрон меньше,
чем в исходном незаряженном диене, и она отражает
делокализацию неспаренного электрона и положи-
тельного заряда по л-системе. Так как молекулярный
ион образуется при удалении л-электрона, можно
использовать другие структуры (В или С). В таких
структурных формулах, как В и С, электрон и по-
ложительный заряд локализованы, поэтому они по-
лезны для описания процесса фрагментации.
Фрагментацию иницирует электронный удар. Толь-
ко малая часть переносимой электронным пучком
энергии расходуется на фрагментацию. Главная часть
этой энергии переносится на структуру частицы
и расходуется на возбуждение электронных и ко-
лебательных состояний катион-радикала.
Фрагментация молекулярного иона с нечетным чис-
лом электронов (катион-радикала М*+) по одинарной
связи может происходить по гомолитическому или
гетеролитическому пути. В случае гомолитического
распада (схема 1.3, I) каждый электрон «движется»
независимо, как показано стрелками типа рыболовного
крючка (с полуострием): фрагментами являются катион
с четным числом электронов и свободный радикал (с
нечетным числом электронов). Во избежание загро-
мождения формул достаточно изображать только одну
из пары изогнутых стрелок (схема 1.3, Др. При ге-
теролитическом распаде пара электронов «движется»
в одном направлении к заряженному месту, как по-
казано обычной изогнутой стрелкой; фрагментами
также являются катион с четным числом электронов
и радикал, но в данном случае положительный заряд
находится на алкильном продукте (схема 1.3,111).
I О^СнХо—R----------> СН3 +1^0=6—R
II СН3—/СН^— б—R----> СН3+Н2С=б—R
III СН3—СН2— СН2-^Вг--->СН3—СН2—СН++Вг’
IV сн3—сн2— сн2+ —> сн3++
Схема 1.3
В отсутствие колец (фрагментация которых требует
разрыва двух или большего числа связей), большая
часть узнаваемых фрагментов в масс-спектре являются В
В дальнейшем сохранен авторский вариант схем рас-
падов и перегруппировок. - Прим, перев.
Рис. 1.13. Разные способы изображения катион-
радикала циклогексадиена
катионами с четным числом электронов, образован-
ными, как было показано выше, при разрыве одинар-
ных связей. Дальнейшая фрагментация такого катиона
обычно приводит к другому катиону с четным числом
электронов и нейтральной молекуле или фрагменту
тоже с четным числом электронов (схема 1.3 IV).
Одновременный или последовательный разрыв
нескольких связей может происходить, когда воз-
никает выигрыш в энергии благодаря образованию
высокостабилизированного катиона и/или стабиль-
ного радикала или нейтральной молекулы, часто по
вполне определенному низкоэнергетическому пути.
Эти схемы рассмотрены в разделе 1.5.5 (перегруп-
пировки) и в разделе 1.6 при обсуждении отдельных
классов органических соединений.
Вероятность разрыва конкретной связи обусловлена ее
прочностью, вероятностью переходов с низкой энергией
и устойчивостью образовавшихся как заряженных, так
и нейтральных фрагментов. Для предсказания наиболее
вероятных направлений распада молекулярного иона мож-
но в определенной степени использовать представления
о пиролитическом распаде. Из-за крайне низкого давле-
ния паров в масс-спектрометре столкновения осколков
маловероятны и их распад является преимущественно
мономолекулярным. Это предположение, опирающееся
на большую базу стандартных спектров, лежит в основе
обширной информации, получаемой из картины фраг-
ментации молекулы. В то время как обычная органи-
ческая химия имеет дело с химическими реакциями,
вызванными теплом или светом, в масс-спектрометрии
исследуют результаты воздействия на органическую мо-
лекулу ионизирующего пучка электронов при давлении
паров около 10“6 мм рт. ст.
Используя общепринятые представления физиче-
ской органической химии, можно сформулировать
ряд общих правил для предсказания наиболее ин-
тенсивных пиков в спектре.
1. Относительная интенсивность пика молекуляр-
ного иона максимальна для неразветвленных
соединений и уменьшается по мере увеличения
разветвленности (см. правило 3).
2. С увеличением молекулярной массы в гомологи-
ческом ряду относительная интенсивность пика
1.5. Интерпретация маа-спектров с ионизацией электронным ударом
31
молекулярного иона обычно уменьшается. Ис-
ключением являются сложные эфиры жирных
кислот.
3. Разрыв связей происходит преимущественно по
алкилзамещенным атомам углерода. Чем боль-
ше степень замещения, тем вероятнее распад.
Это является следствием повышения устойчи-
вости третичного карбокатиона по сравнению с
вторичным, который, в свою очередь, устойчивее
первичного.
Порядок увеличения устойчивости катионов:
СН3+ < RCH2+ < R2CH+ < R3C+
Как правило, самый крупный заместитель
в узле разветвления легче отщепляется в виде
радикала; вероятно, потому, что радикал с длин-
ной цепью легче стабилизируется за счет дело-
кализации одного электрона.
4. Двойные связи, циклические структуры и, осо-
бенно, ароматические (или гетероароматические)
циклы стабилизируют молекулярный ион и, та-
ким образом, увеличивают вероятность его по-
явления.
5. Двойные связи способствуют аллильному распаду
с образованием резонансно-стабилизированного
аллильного карбокатиона. Это правило наруша-
ется для простых алкенов по причине легкой
миграции двойной связи, но выполняется для
циклоалкенов.
6. Насыщенные циклы склонны терять боковые
цепи при a-связи. Это просто особый случай
разветвления (правило 3). Положительный заряд
остается на циклическом фрагменте, (см. схе-
му 1.4.).
Ненасыщенные циклы могут подвергаться ре-
трореакции Дильса-Альдера (схема 1.5):
Схема 1.5
7. Распад ароматических алкилзамещенных соеди-
нений наиболее вероятен по P-связи относитель-
но цикла. В результате получается резонансно-
стабилизированный бензильный ион или,
что более вероятно, ион тропилия (см. схему 1.6).
Схема 1.6
8. Следующие за гетероатомом связи С-С часто
разрываются, оставляя заряд на содержащем гете-
роатом фрагменте, несвязывающие электроны ко-
торого обеспечивают резонансную стабилизацию.
9. Распад часто сопровождается элиминированием
небольших устойчивых нейтральных молекул,
таких как монооксид углерода, олефины, вода,
аммиак, сероводород, циановодород, меркаптаны,
кетены или спирты, часто с перегруппировкой
(раздел 1.5.5).
Нужно иметь в виду, что представленные выше
правила фрагментации применимы к масс-спектрам,
которые получены с использованием электронного
удара. Другие способы ионизации (например, хими-
ческая и др.) часто приводят к молекулярным ионам
с гораздо более низкой энергией или квазимолеку-
лярным ионам с сильно различающимися путями
фрагментации, распад этих молекулярных ионов
подчиняется другим правилам.
1.5.5. Перегруппировки
Перегруппировочные ионы представляют собой
осколки, происхождение которых нельзя описать
только простым разрывом связей в молекулярном
ионе. Они получаются в результате внутримолеку-
лярной перегруппировки атомов в процессе фрагмен-
тации. Особенно широко распространены перегруп-
пировки, включающие миграцию атомов водорода
в молекулах, содержащих гетероатом. Одним из
важных примеров является так называемая пере-
группировка Мак-Лафферти; которая общем виде
показана на схеме 1.7.
32
1. Маа-спектрометрия
Первые масс-спектрометристы, работавшие в не-
фтеперерабатывающей промышленности, отмечали
«случайные» перегруппировки углеводородов. На-
пример, перегруппировка неопентильного катион-
радикала в этильный катион, показанная на схеме 1.8,
простому объяснению не поддается.
Схема 1.7
Схема 1.8
Чтобы молекула могла претерпеть перегруппиров-
ку Мак-Лафферти, она должна иметь подходящим
образом расположенные гетероатом (например, О),
л-систему (обычно двойную связь) и способный
к отщеплению атом водорода в у-положении отно-
сительно группы С=О.
Такие перегруппировки часто приводят к появле-
нию интенсивных характеристических пиков, крайне
полезных для идентификации. Их можно объяснить
низкоэнергетическими структурными переходами
и повышенной устойчивостью продуктов. Широко
распространены перегруппировки, сопровождающиеся
элиминированием устойчивых нейтральных молекул
(например, отщепление олефина в процессе пере-
группировки Мак-Лафферти); они встретятся нам
при обсуждении масс-спектров отдельных типов
органических соединений.
Пики перегруппировочных ионов можно иден-
тифицировать, рассматривая массовые числа (m/z)
осколочных ионов и соответствующих им молекуляр-
ных ионов. Простой (без перегруппировки) распад
молекулярного иона с четным массовым числом
дает осколочный ион с нечетным массовым числом,
а простой распад молекулярного иона с нечетным
массовым числом дает осколок с четным массовым
числом. Если наблюдаемая масса осколочного иона
отличается на единицу от ожидаемой массы, по-
лучающейся при простом распаде (например,
осколок с четным массовым числом получается из
молекулярного иона с четным массовым числом),
то это указывает на миграцию атома водорода, со-
провождающую фрагментацию. Пики перегруппи-
ровочных ионов можно выявить при рассмотре-
нии следствия из «азотного правила» (разд. 1.5.1).
Пик с четным массовым числом, возникающий из
молекулярного иона с четным массовым числом,
получается в результате двух распадов, которые
могут сопровождаться перегруппировкой.
1.6. Масс-спектры некоторых классов
органических соединений
В этом разделе кратко охарактеризованы масс-спектры
некоторых классов органических соединений с точ-
ки зрения наиболее полезных для идентификации
общих правил. Для более подробного ознакомления
с этим вопросом можно обратиться к дополнитель-
ной литературе (в особенности к полному обобще-
нию спектров, сделанному в издании Budzikiewicz,
Djerassi, Williams, 1967). Базы данных доступны из
литературы и прилагаются к программному обеспе-
чению приборов. Приведенный список литературы
не носит исчерпывающего характера. Таблица наи-
более распространенных осколочных ионов дана
в приложении Б. Таблица чаще всего элиминируе-
мых незаряженных фрагментов и некоторые структу-
ры представлены в приложении В. Более полные
перечни обычных фрагментных ионов имеются
в литературе.
1.6.1. Углеводороды
1.6.1.1. Насыщенные углеводороды
В большинстве ранних работ по масс-спектрометрии
исследовались углеводороды, представляющие инте-
рес для нефтяной промышленности. В этих случаях
полностью применимы правила 1-3 (раздел 1.5.4);
перегруппировочные пики обычно не являются ин-
тенсивными (происходят случайные перегруппировки),
а в литературе имеется большое число примеров
для сравнения.
В масс-спектрах насыщенных углеводородов
с неразветвленной цепью всегда присутствует пик
молекулярного иона, хотя для соединений с длинной
цепью он имеет низкую интенсивность. Фрагмента-
ция характеризуется кластерами (группами) пиков,
1.6. Маа-спектры некоторых классов органических соединений
33
100:
43-, 571
н -Гексадекан
С16Н34
М.м.: 226
50 :
0
I 99-] 113-| 141-] 155-] 183^ 197-| 226-]
L]—j-uUi-j—! Ij|l | julll—( pldj J jjdLj J jlL-]—J p_J p_.и | , [ j j1 j , , , ,
50 100 150 200
43-i
100:
169:141
57: 85: c _
5-Метилпентадекан
С1бН34
М.м.: 226
57
85-1
ос _ 168-i M+
П 127-| 141-] 1 211-] 226-]
I I*1* J I Г1* I I i"1 Г 'I I Г ' | I I I" г J 1—I I г 1 "I ' I1' I Г I* I 1 I I
100 150 200
Рис. 1.14. Масс-спектры с ионизацией ЭУ изомерных углеводородов С16
положение которых внутри кластера отличается на
14 а. е. м. (СН2). Самый интенсивный пик в каждом
кластере соответствует фрагменту С„Н2л+1; поэтому
он появляется при m/z = 14и + 1 и сопровождает-
ся фрагментами С„Н2и и CJi^. Пики фрагментов
С3 и С4 наиболее интенсивные. Интенсивность пиков
фрагментов плавно уменьшается до [М-С2Н5]+; пик
[М-СН3]+ или очень слаб, или отсутствует. Соеди-
нения, содержащие более восьми атомов углерода,
дают довольно похожие спектры, поэтому при иден-
тификации приходится полагаться только на пик
молекулярного иона.
Спектры разветвленных насыщенных углеводо-
родов чрезвычайно похожи на спектры соединений
с неразветвленной цепью, но плавное уменьшение
интенсивности пиков нарушается тем, что становится
более вероятной фрагментация по разветвлениям.
Главное уменьшение интенсивностей пиков кла-
стеров в спектре н-алкана на рис. 1.14 (верхний
спектр) контрастирует со спектром разветвленного
алкана (рис. 1.14, нижний спектр), где наблюдается
изменение интенсивностей около пиков С12. Эта не-
равномерность указывает на то, что самая длин-
ная цепь 5-метилпентадекана содержит 10 атомов
углерода.
На нижнем спектре рис. 1.14 пики с m/z 169
и 85 соответствуют разрыву связи по одну сторону
от разветвления, причем заряд сохраняется на за-
мещенном атоме углерода. Разность молекуляр-
ной массы и суммы масс этих фрагментов дает
остаток -СН-СН3. Отметим отсутствие фрагмента
Сп, который не получается путем одного распада.
Присутствие отчетливого пика М-15 указывает
на метильный заместитель. Фрагмент, возника-
ющий при разрыве связи на месте разветвления
углеродной цепи, приводит к потере одного атома
водорода, и результирующий пик С„Н2„ является до-
вольно заметным, иногда более интенсивным, чем со-
ответствующий пик С„Н2л+1.
Насыщенный цикл в углеводороде повышает
относительную интенсивность пика молекулярного
иона и способствует разрыву связи, соединяющей
цикл с остальной частью молекулы (правило 6,
раздел 1.5.4). Фрагментация цикла обычно харак-
теризуется потерей двух атомов углерода в виде
С2Н4 (28) и С2Н5 (29). Тенденция терять фрагменты
с четной массой, подобные С2Н4, приводит к спек-
тру, в котором содержание ионов с четной массой
больше, чем в спектре ациклического углеводорода.
Как и в разветвленных углеводородах, разрыв свя-
зи С-С сопровождается потерей атома водорода.
Поэтому характеристическими являются серии
C„H2w-i и СлН2и_2.
В масс-спектре циклогексана (рис. 1.15) наблюда-
ется гораздо более интенсивный пик молекулярного
иона, чем для ациклических соединений, поскольку
34
/. Маа-спектрометрия
для фрагментации в этом случае требуется разрыв
двух связей углерод-углерод. В его спектре наблю-
дается основной пик при m/z 56 (из-за потери С2Н4)
и интенсивный пик при m/z 41, который является
фрагментом с п = 3 в серии C„H2w-i-
1.6.1.2. Алкены (олефины)
Пик молекулярного иона алкенов, особенно полиенов,
обычно проявляется отчетливо. Определение положе-
ния двойной связи в ациклических алкенах затрудне-
но вследствие легкости ее миграции во фрагментах.
В циклических (особенно полициклических) алкенах
положение двойной связи часто очевидно благодаря
выраженной склонности к аллильному расщеплению
почти без миграции двойной связи (правило 5, раз-
дел 1.5.4). Сопряжение с карбонильной группой также
фиксирует положение двойной связи. Подобно насы-
щенным углеводородам, ациклические алкены характе-
ризуются кластерами пиков с интервалами в 14 а. е. м.
В этих группах пики C„H2^i и С„Н2„ более интенсивны,
чем пики С„Н2п+1.
Масс-спектр терпена Р-мирцена показан на рис
1.16. Пики с m/z 41, 55 и 69 соответствуют форму-
ле C„H2„-i с п = 3, 4, 5 соответственно. Образование
фрагмента с m/z 41 должно включать изомеризацию.
Пики с m/z 67 и 69 обусловлены фрагментацией при
разрыве связи между двумя аллильными фрагментами
(схема 1.9).
Схема 1.9
Появление пика при m/z 93 объясняет схема 1.10.
Ион С7Н9+ образуется при изомеризации двойной
связи (приводящей к увеличению сопряжения) с по-
следующим аллильным распадом. Ион с m/z 93 имеет
по крайней мере две важные резонансные структуры,
Рис. 1.16. Масс-спектр с ионизацией ЭУ Р-мирцена
1.6. Маа-спектры некоторых классов органических соединений
35
которые его стабилизируют. В качестве упражнения
студентам предлагается их изобразить.
Схема 1.10
Циклические алкены обычно дают отчетливый пик
молекулярного иона. Единственным типом распада
является ретрореакция Дильса-Альдера. Эта реакция
иллюстрируется на примере лимонена (схема 1.11).
В этом примере в результате ретро-реакции Диль-
са-Альдера получаются две молекулы изопрена. По-
скольку реакция является примером перегруппировки,
один из изопреновых остатков представляет собой
нейтральную молекулу.
Схема 1.11
1.6.1.3. Ароматические и алкилароматические
углеводороды
Ароматическое кольцо в молекуле стабилизирует мо-
лекулярный ион (правило 4, раздел 1.5.4), пик кото-
рого является достаточно интенсивным для точных
измерений М+1 и М+2.
На рис. 1.17 показан масс-спектр нафталина.
Пик молекулярного иона является основным, а ин-
тенсивность самого большого пика фрагментного
иона с m/z 51 составляет только 12.5% от его ин-
тенсивности.
Алкилзамещенное бензольное кольцо дает характер-
ный пик (часто максимальный) с m/z 91 (С6Н5СН2+).
Разветвление у а-углеродного атома приводит к мас-
сам, большим чем 91, с инкрементами 14, причем
самый крупный заместитель элиминируется легче
(правило 3, раздел 1.5.4). Явное наличие пика иона
с массой 91 не исключает, однако, возможности раз-
ветвления у а-углеродного атома, так как этот очень
устойчивый фрагмент может получаться в результате
перегруппировок. При подобном бензильном раз-
рыве связи С-Н образуется заметный, а иногда
значительный пик М-1.
Предполагается, что в большинстве случаев ион
с массой 91 имеет строение тропилиевого, а не
бензильного катиона. Это объясняет легкую поте-
рю метильной группы ксилолами (схема 1.12), хотя
толуол ее так легко не теряет. Образовавшийся
в начальный момент молекулярный ион-радикал
ксилола перегруппировывается в метилциклогепта-
триеновый ион-радикал, который затем распадается
с образованием тропилиевого иона (С7Н7+). Часто
наблюдающийся пик при m/z 65 появляется в ре-
зультате элиминирования нейтральной молекулы
ацетилена из тропилиевого иона.
Схема 1.12
Если алкильная группа длиннее С2, то миграция
водорода с элиминированием нейтральной моле-
кулы алкена приводит к появлению пика с m/z 92
Рис. 1.17. Масс-спектр с ионизацией ЭУ нафталина
36
1. Маа-спектрометрия
(схема 1.13). Вновь отметим, что это пример пере-
группировки.
- h2c=chr
Схема 1.13
В моноалкилбензолах проявляется характери-
стический кластер ионов с m/z 77 (С6Н5+), 78 (С6Н6+)
и 79 (С6Н7+), обусловленный а-расщеплением и ми-
грацией водорода.
В алкилированных полифенилах и алкилирован-
ных полициклических ароматических углеводо-
родах наблюдается точно такой же Р-распад, как
и в алкилбензолах.
1.6.2. Гидроксилсодержащие соединения
1.6.2.1. Спирты
Пик молекулярного иона первичного или вторичного
спирта обычно довольно слабый, а для третичного
спирта вообще неразличим. Пик молекулярного иона
пентанола-1 является чрезвычайно слабым по сравне-
нию с его ближайшими гомологами. Для определения
молекулярной массы можно применять такие приемы,
как ХИ или получение производных.
Разрыв связи С—С рядом с атомом кислорода-общее
явление (правило 8, раздел 1.5.4). Так, в первичных
спиртах наблюдается характерный пик, обусловленный
ионом СН2=+ОН (m/z 31). Вторичные и третичные спир-
Рис. 1.18. Масс-спектры с ионизацией ЭУ изомерных пентанолов
1.6. Маа-спектры некоторых классов органических соединений
37
ты расщепляются аналогично, давая характерные пики,
обусловленные ионами CHR=+OH (m/z 45,59,73 и т.
д.) и CRR-+ОН (m/z 59,73,87 и т. д.) соответственно.
Самый большой заместитель отщепляется легче всего
(правило 3). Иногда разрывается следующая за атомом
кислорода связь С-Н, этот менее предпочтительный
процесс вносит вклад в пик М-1.
Первичные спирты, кроме основного разрыва связи
С-С около атома кислорода, дают гомологическую се-
рию пиков, интенсивность которых постепенно умень-
шается. Эти пики получаются в результате разрыва
связей С-С, последовательно все более удаленных от
атома кислорода. В спиртах с длинной цепью (боль-
ше С6) преобладающей становится фрагментация по
типу углеводородов; фактически спектр становится
похож на спектр соответствующего алкена. Спектр
первичного спирта иногда осложняется малоинтен-
сивными пиками М-2 и М-3 рядом с очень слабым
или неразличимым пиком молекулярного иона.
Заметный, а иногда и значительный пик обычно
находится при М-18, что соответствует отщеплению
молекулы воды. Этот пик наиболее заметен в спек-
трах первичных спиртов. Такое элиминирование при
электронном ударе можно объяснить механизмом,
в котором 5-атом водорода отрывается, как показано
на схеме 1.14, 7. Аналогичный механизм можно за-
писать для отрыва у-атома водорода. Интенсивность
пика М-18 часто увеличивается в результате тер-
мического разложения высших спиртов на горячей
поверхности системы ввода. Элиминирование воды
вместе с элиминированием алкена из первичных спир-
тов (см. схему 1.14, II) объясняет появление пика
М-(алкен + Н2О), т. е. пиков М-46, М-74, М-102...
В спектрах спиртов, имеющих разветвления и со-
держащих метильные группы (например, терпеновых
спиртов), часто наблюдается довольно интенсивный
пик иона М-33 в результате потери одновременно
СН3 и Н2О.
Фрагментация циклических спиртов происходит
сложными путями; например, циклогексанол (М =
m/z 100) дает ион С6НПО+ при отрыве а-водородного
атома, С6Н10+ (который, по-видимому, имеет более
чем одну возможную мостиковую бициклическую
структуру) при потере Н2О и образует С3Н5О+ (m/z 57)
в результате сложного расщепления цикла.
В случае первичного спирта пик иона с m/z 31 (см.
выше) хорошо идентифицируется и более интенсивен,
чем пики ионов с m/z 45, 59, 73... Однако первона-
чально образовавшийся ион вторичного спирта мо-
жет распадаться далее, давая пик с m/z 31 умеренной
интенсивности.
На рис. 1.18 представлены характерные масс-
спектры изомерных первичных, вторичных и тре-
тичных спиртов С5.
Бензиловые спирты и их замещенные гомологи
и аналоги составляют отдельный класс. Как пра-
вило, в их масс-спектрах пик молекулярного иона
интенсивен. Как и следовало ожидать, в результа-
те Р-расщепления относительно кольца образуется
бензильный пик (М - ОН) средней интенсивности.
Сложная последовательность превращений приводит
к характерным пикам М-1, М-2 и М-3. При фрагмен-
тации самого бензилового спирта последовательно
образуются следующие ионы: М-1, С6Н7+ при отрыве
СО и С6Н5+ при отрыве Н2 (см. схему 1.15).
Схема 1.15
Потеря Н2О приводит к заметному пику М-18,
что является общей закономерностью, особенно явно
проявляющейся для некоторых opwo-замещенных бен-
зиловых спиртов. Потеря воды, показанная на схеме
1.16, происходит одинаково хорошо и в случае ато-
ма кислорода в ортио-положении (фенол). При этом
также выделяется ароматический кластер с m/z 77,
78 и 79, возникающий при сложном распаде.
Схема 1.16
1.6.2.2. Фенолы
Заметный пик молекулярного иона облегчает иденти-
фикацию фенолов. В самом феноле пик молекулярного
38
1. Маа-спектрометрия
иона является основным, а пик М-1 имеет небольшую
интенсивность. В крезолах пик М-1 больше молеку-
лярного пика из-за легкого расщепления бензильной
связи С-Н. В масс-спектрах фенолов обычно наблю-
даются пик осколочного иона с m/z 77 и пики, воз-
никающие при потере СО (М-28) и СНО (М-29).
На рис. 1.19 показан масс-спектр типичного фено-
ла - о-этилфенола. Из этого спектра видно, что ме-
тильная группа отщепляется гораздо легче, чем а-атом
водорода.
1.6.3. Простые эфиры
1.6.3.1. Алифатические эфиры и ацетали
Пик молекулярного иона (он на две атомные единицы
массы больше, чем у соответствующего углеводоро-
да) невелик. При увеличении напуска, что обычно
делается, становятся заметными пик молекулярного
иона или пик М + 1 (возникающий при переносе Н*
в результате ион-молекулярных столкновений).
Наличие атома кислорода в молекуле можно
определить по интенсивным пикам ионов с m/z 31,
45, 59, 73... Они соответствуют фрагментам RO+
и ROCH2+. Фрагментация идет по двум основным
направлениям:
1. Разрыв связи С-С рядом с атомом кислорода
(а,Р-связь, правило 8, раздел 1.5.4). Основной
пик может соответствовать одному из этих кис-
лородсодержащих ионов. В случае, показанном
на рис. 1.20, разрыв первого типа является пред-
почтительным (т.е. около разветвления, с потерей
большего фрагмента). Первичный фрагмент рас-
падается далее с потерей этилена, давая основной
пик; это направление становится важным, когда
а-углеродный атом замещен (см. перегруппиров-
ку Мак-Лафферти, раздел 1.5.5).
Рис. 1.20. Масс-спектр с ионизацией ЭУ этил-втор-бутилового эфира
1.6. Маа-спектры некоторых классов органических соединений
39
2. Разрыв связи С-О, с сохранением заряда на алкиль-
ном фрагменте. В спектрах эфиров с длинной цепью
преобладает углеводородный тип фрагментации.
Ацетали - особый класс простых эфиров. Их масс-
спектры характеризуются крайне слабым пиком
молекулярного иона, характерными пиками М - R,
М - OR (и/или М - OR') и слабым пиком М - Н.
Распад по этим схемам легко происходит при уча-
стии атома кислорода. Как обычно, предпочтительно
элиминирование большей группы. Как и в алифа-
тических эфирах, первоначально образовавшиеся
кислородсодержащие фрагменты могут распадать-
ся далее с миграцией водорода и элиминированием
олефина. Кетали ведут себя аналогичным образом.
Н
R-t-C-{-OR
OR'
как было отмечено выше для алкилбензолов. Оче-
видно, что разрыв скорее происходит при участии
кольца, а не атома кислорода. Разрыв связи С-С,
следующей за атомом кислорода, значения не имеет:
Схема 1.17
Дифениловые эфиры в результате сложных пере-
группировок дают пики М - Н, М - СО и М - СНО.
1.6.4. Кетоны
1.63.2. Ароматические эфиры
В масс-спектрах ароматических эфиров пик молеку-
лярного иона интенсивный. Первичный акт фрагмен-
тации происходит по связи в 0-положении к коль-
цу, и образовавшийся ион может распадаться далее.
Так, в масс-спектре анизола (рис. 1.21, молекулярная
масса 108) наблюдаются ионы с m/z 93 и 65 и ха-
рактеристические ароматические пики с m/z 78 и 77.
Когда алкильная группа в арилалкиловом эфи-
ре содержит два или более атомов углерода, рас-
щепление в 0-положении по отношению к кольцу
сопровождается миграцией водорода (схема 1.17),
1.6.4.1. Алифатические кетоны
Пик молекулярного иона в масс-спектрах кетонов
обычно выражен довольно отчетливо. Интенсивные
пики фрагментов алифатических кетонов обуслов-
лены разрывом связи С-С, соседней с атомом кис-
лорода, причем заряд сохраняется на резонансно-
стабилизированном ацилий-катионе (схема 1.18).
Таким образом, как и в случае спиртов и простых
эфиров, разрыв происходит при содействии атома кис-
лорода. Это расщепление приводит к пикам ионов
с m/z 43, 57 или 71... Основной пик очень часто обу-
словлен отрывом большей алкильной группы.
Рис. 1.21. Масс-спектр с ионизацией ЭУ анизола
40
1. Маа-спектрометрия
•+ — R’ i + +
ZC=O -^R-C=o<—>R—С=О
R
Схема 1.18
Когда одна из алкильных цепей, связанных с груп-
пой С=О, содержит три или более атомов углерода,
происходит сопровождающийся миграцией водорода
разрыв связи С-С, которая находится через одну от
карбонильной группы (а,0-связь). Основной пик воз-
никает в результате перегруппировки Мак-Лафферти
(схема 1.19). Простой разрыв а,0-связи не наблю-
дается, так как при этом получался бы ион низкой
стабильности с двумя соседними положительными
центрами.
Схема 1.19
Заметим, что в кетонах с длинной цепью угле-
водородные пики не отличаются от ацильных (на
спектрометрах с низким разрешением), так как масса
фрагмента С=О (28) такая же, как у двух метилено-
вых групп. Сложные схемы расщепления в кетонах
иногда затрудняют определение конфигурации угле-
родной цепи. Восстановление карбонильной группы
до метиленовой приводит к соответствующему угле-
водороду, характер фрагментации которого позволяет
установить строение углеродного скелета.
1.6.4.2. Циклические кетоны
Пик молекулярного иона в масс-спектрах цикличе-
ских кетонов обычно выражен довольно отчетливо.
Как и в алифатических кетонах, первичное расще-
пление циклических кетонов происходит по сосед-
ству с карбонильной группой, образовавшийся ион
подвергается дальнейшему распаду на фрагменты.
Основной пик в спектрах циклопентанона и цикло-
гексанона (рис. 1.22) соответствует m/z 55. Механизм
в обоих случаях одинаков: гидридный сдвиг в первич-
ном радикале приводит к сопряженному вторичному
радикалу с последующим образованием резонансно-
стабилизированного иона с m/z 55. Появление других
отчетливых пиков с m/z 83 и 42 в спектре циклогек-
санона объясняется на рис. 1.22.
1.6.4.3. Ароматические кетоны
Пик молекулярного иона ароматических кетонов
имеет заметную интенсивность. Фрагментация
Рис. 1.22. Масс-спектр с ионизацией ЭУ циклогексанона
1.6. Маа-спектры некоторых классов органических соединений
41
арилалкилкетонов происходит по связи в Р-положении
по отношению к кольцу с образованием характери-
стического фрагмента ArC=O+ (m/z 105, если Аг =
фенил), который обычно является максимальным.
Потеря СО этим фрагментом дает «арильный» ион
(с m/z 77 в случае ацетофенона). Разрыв связи, со-
седней с кольцом, дает фрагмент RC=O+, причем он
не так важен, хотя иногда увеличивается при наличии
электроноакцепторных групп (уменьшается в присут-
ствии электронодонорных групп) в иара-положении
ароматического кольца.
Если алкильная цепь содержит три или более ато-
мов углерода, происходит разрыв связи С-С через
одну от карбонильной группы, сопровождающийся
переносом водорода. По такому же механизму через
циклическое переходное состояние с элиминирова-
нием алкена и образованием устойчивого иона рас-
падаются алифатические кетоны.
Масс-спектр несимметричного диарилкетона -
и-хлорбензофенона - показан на рис. 1.23. Пик мо-
лекулярного иона (m/z 216) достаточно заметен,
а интенсивность пика М+2 (33.99% относительно
интенсивности пика молекулярного иона) указывает
на присутствие в молекуле хлора (см. обсуждение
табл. 1.5 и рис. 1.29 в разделе 1.6.16).
Так как интенсивность пика m/z 141 составляет
примерно треть от интенсивности пика m/z 139, они
относятся к одному хлорсодержащему фрагменту.
То же можно сказать о фрагментах, дающих пики
m/z 111 и 113.
Схема приводящей к основным пикам фрагментации
изображена на рис. 1.23. Пик С1-АгС=О+ имеет более
высокую интенсивность, чем С1-Аг+, а АгС=О+ интен-
сивнее, чем пик Аг+ (преимущественный Р-распад).
Однако, если рассматривать пики [фрагмент+2] хлор-
замещенных осколков, то различие в интенсивностях
между С1-АгСО+ и АгСО+ или между С1-Аг+ и Аг+
невелико. Индукционный (электроноакцепторный)
и резонансный (электронодонорный) эффекты атома
хлора в иара-положении примерно сбалансированы,
как и в реакциях ароматического электрофильного
замещения.
1.6.5. Альдегиды
7.6.5.7. Алифатические альдегиды
Пик молекулярного иона алифатических альдегидов
обычно имеет заметную интенсивность. Разрыв свя-
зей С-Н и С-С, следующих за атомом кислорода,
приводит к пикам ионов М-1 и M-R (m/z 29, СНО+).
Пик М-1 удобен для идентификации даже альдеги-
дов с длинной цепью; однако пик с m/z 29 в аль-
дегидах с С4 и выше обусловлен углеводородным
ионом С2Н5+.
В альдегидах, содержащих четыре и более атомов
углерода, происходит разрыв а,Р-связи С-С по схеме
Мак-Лафферти, что приводит к основному пику с m/z
44,58 или 72... в зависимости от a-заместителя. Обра-
зовавшийся через циклическое переходное состояние,
42
/. Маа-спектрометрия
Рис. 1.24. Масс-спектр с ионизацией ЭУ нонилового альдегида
в соответствии со схемой 1.7 при Y=H, резонансно-
стабилизированный ион показан на схеме 1.20.
Лафферти (схема 1.21). Разветвление на а-углеродном
атоме увеличивает вероятность такого распада.
Схема 1.20
В альдегидах с нормальной цепью распознаются
другие специфические, полезные для диагностики
пики М-18 (отщепление воды), М-28 (отщепление
этилена), М-43 (отщепление СН2=СН-Ое) и М-44 (от-
щепление СН2=СН-ОН). Перегруппировки, приводя-
щие к этим фрагментам, получили свое объяснение
(см. Budzikiewicz et al., 1967). По мере удлинения
цепи преобладающей становится углеводородная
схема фрагментации (m/z 29,43, 57, 71,...). Эти осо-
бенности видны в спектре нонилового альдегида
(рис. 1.24).
1.6.5.2. Ароматические альдегиды
Ароматические альдегиды характеризуются интенсив-
ными пиками молекулярного иона и М-1 (Аг—С=О+),
который всегда интенсивен, а иногда может быть
интенсивнее пика молекулярного иона. Ион М-1
(С6Н5-СО+, если Аг-фенил) отщепляет СО, давая фе-
нильный фрагмент (m/z 77), который, в свою очередь,
элиминирует ацетилен, образуя ион С4Н3+ (m/z 51).
1.6.6. Карбоновые кислоты
1.6.6.1. Алифатические кислоты
Пик молекулярного иона в масс-спектре монокар-
боновой кислоты с неразветвленной цепью мало-
интенсивен, но обычно все же заметен. Наиболее
характеристичным (иногда максимальным) является
пик с m/z 60, обусловленный перегруппировкой Мак-
Схема 1.21
В кислотах с короткой цепью пики ионов М - ОН
и М - СО2Н хорошо заметны; они соответствуют
разрыву связей, следующих за карбонильной груп-
пой. Спектры кислот с длинными цепями состо-
ят из двух серий пиков, обусловленных разрывом
углерод-углеродных связей с сохранением заряда
либо на кислородсодержащих фрагментах (m/z 45,
59, 73, 87,...), либо на алкильных фрагментах (m/z
29,43, 57, 71, 85,...). Как обсуждалось ранее, угле-
водороды также дают последовательность пиков
с m/z 27, 28; 41,42; 55, 56; 69, 70; ... В итоге, за ис-
ключением пика перегруппировки Мак-Лафферти,
спектр кислоты с длинной цепью похож на серию
«углеводородных» кластеров с интервалом 14 а. е. м.
Однако в каждом кластере имеется характерный
пик Показанный на рис. 1.25 масс-спектр
декановой кислоты хорошо иллюстрирует эти по-
ложения.
Двухосновные кислоты обычно имеют низкую ле-
тучесть, и для увеличения давления паров их пре-
вращают в сложные эфиры. С успехом используют
триметилсилильные эфиры.
1.6. Маа-спектры некоторых классов органических соединений
43
1.6.6.2. Ароматические кислоты
Пик молекулярного иона в масс-спектрах аромати-
ческих кислот интенсивен. Другие характерные пики
возникают при потере ОН (М-17) и СО2Н (М-45).
Если в орто-положении имеются водородсодержащие
группы, то, как показано на схеме 1.22, происходит
потеря воды (М-18). Это один из примеров общего
«орто-эффекта», наблюдаемого в случае образования
заместителями шестичленного переходного состояния,
благоприятствующего потере нейтральных молекул
Н2О, ROH или NH3.
Z = ОН, OR, NH,; Y = CHj, О, NH
Схема 1.22
1.6.7. Сложные эфиры карбоновых кислот
1.6.7.1. Алифатические сложные эфиры
Пик молекулярного иона в масс-спектре метилово-
го эфира алифатической кислоты с прямой цепью
обычно хорошо заметен. Даже в спектрах восков
пик молекулярного иона различим. Пик молекуляр-
ного иона в диапазоне m/z от 130 до 200 малоин-
тенсивен, но становится несколько интенсивнее за
его пределами. Наиболее характерный пик обуслов-
лен обычной перегруппировкой Мак-Лафферти (на
схеме 1.23 показана перегруппировка для сложного
эфира) и разрывом связи С-С, отстоящей от С=О-
группы на одну связь. В спектре метилового эфира
алифатической кислоты, не имеющей разветвления
у а-атома углерода, наблюдается интенсивный пик
иона с m/z 74, который является основным для слож-
ных эфиров с нормальной цепью от С6 до С26. Часто
можно идентифицировать спиртовой остаток и (или)
a-заместитель, если обнаружен пик, получающийся
при таком распаде.
RO СН2
Схема 1.23
В общем случае при разрыве связи, следующей
за карбонильной группой в сложном эфире, могут
возникнуть четыре иона.
О
R— С—OR'
Пик иона R+ выделяется в масс-спектрах сложных
эфиров с короткой цепью, но его интенсивность бы-
стро уменьшается с увеличением длины цепи, и пик
44
1. Маа-спектрометрия
m/z
Рис. 1.26. Масс-спектр с ионизацией ЭУ метилоктаноата
становится едва различимым в спектре метилгек-
саноата. Пик иона R-C=O+ в сложных эфирах рас-
познается чрезвычайно легко. В метиловых эфирах он
наблюдается при М-31. В метилацетате это основной
пик, а в метиловом эфире кислоты С26 его интен-
сивность еще составляет 4% основного пика. Ионы
[OR']+ и [C(=O)OR']+ обычно не имеют большого
значения. Пик последнего отчетливо наблюдается,
когда R' = СН3 (см. пик с m/z 59 на рис. 1.26).
Сначала рассмотрим масс-спектры сложных эфи-
ров, в которых доминирует кислотная часть молеку-
лы. Типы фрагментации метиловых эфиров кислот
с неразветвленной цепью можно описать так же,
как фрагментацию свободных кислот. Разрыв каж-
дой связи С-С дает алкильную серию (m/z 29, 43,
57,...) и серию кислородсодержащих ионов CnH2w_iO2+
(m/z 59,73, 87,...). Таким образом, формируются угле-
водородные кластеры пиков с интервалами 14 а. е. м.,
в каждом из них содержится характерный пик иона
CJ12n-\O2+- Пик (m/z 87), формально соответствующий
иону [СН2СН2СООСНз]+, всегда интенсивнее, чем его
гомологи, но причина этого не установлена. Однако
ясно, что ионы СлН2л_^О2+ не всегда возникают при
простом разрыве связей.
На рис. 1.26 представлен спектр метилоктаноата.
На этом спектре проявляется одно ранее упомянутое
затруднение (раздел 1.5.2.1), касающееся использова-
ния пика М+1 для определения молекулярной фор-
мулы. Измеренная интенсивность пика М+1 равна
12%, а вычисленное значение - 10.0%. Измеренное
значение завышено из-за ион-молекулярной реак-
ции, которая происходит при работе с относительно
большими количествами образца, необходимыми для
обнаружения слабого пика молекулярного иона.
Теперь рассмотрим масс-спектры сложных эфиров,
в которых доминирует спиртовая часть молекулы.
Эфиры жирных спиртов (за исключением метиловых)
элиминируют молекулу кислоты тем же путем, каким
спирты элиминируют воду. Можно привести схему,
подобную описанной ранее для спиртов и включаю-
щую перенос одного атома водорода к спиртовому
атому кислорода эфира. Альтернативный механизм
включает перенос водорода к карбонильному атому
кислорода (перегруппировка Мак-Лафферти).
Потеря уксусной кислоты по изложенному меха-
низму содержащими ацетатную группу стероидами
происходит так быстро, что пик молекулярного иона
часто не удается зарегистрировать. Стероидные систе-
мы необычны тем, что при свободной гидроксильной
группе часто дают значительный пик молекулярного
иона, даже если соответствующие ацетаты - нет.
Эфиры длинноцепочечных спиртов идентифициру-
ются по пикам с m/z 61,75 или 89,... образующимся
при элиминировании алкильной части в виде алкена
и переносе двух атомов водорода на кислородсо-
держащий фрагмент, который по существу является
протонированной карбоновой кислотой.
В общем случае эфиры двухосновных кислот
ROOC(CH2)„COOR дают узнаваемые пики моле-
кулярных ионов. Интенсивные пики находятся для
[ROOC(CH2)/JC=O]+ и для [ROOC(CH2)w]+.
7.6.72. Бензильные и фенильные сложные эфиры
Бензилацетат (а также фурфурилацетат и другие по-
добные ацетаты) и фенилацетат элиминируют ней-
тральную молекулу кетена (схема 1.24), что часто
дает максимальный пик.
m/z 108
Схема 1.24
Конечно, для бензилацетата характерны пики ионов
с m/z 43 (СН3=О+) и m/z 91 (С7Н7+).
1.6. Маа-спектры некоторых классов органических соединений
45
1.6.7.3. Сложные эфиры ароматических кислот
В масс-спектрах метиловых эфиров ароматических
кислот довольно интенсивным является пик молеку-
лярного иона (ArCOOR, R=CH3). С увеличением раз-
мера спиртовой части молекулы интенсивность пика
молекулярного иона быстро падает практически до
нуля уже для С5. Основной пик получается в результа-
те отщепления OR, а отщеплением -COOR объясняет-
ся появление другого характерного пика. В метиловых
эфирах это соответственно пики М-31 и М-59.
С увеличением длины алкильной части молеку-
лы становятся важными три типа разрыва связей:
1) перегруппировка Мак-Лафферти, 2) перегруппи-
ровка двух атомов водорода с элиминированием ал-
лильного радикала и 3) удержание положительного
заряда алкильной группой.
Хорошо известная перегруппировка Мак-Лафферти
приводит к пику (АгСООН)+ в масс-спектре арома-
тической кислоты. Другой пик возникает по второму
распространенному механизму образования протони-
рованной ароматической кислоты (АгСООН2)+. Третий
тип распада дает алкильный катион R+.
Соответственно, из рр/ио-замещенных бензоатов
элиминируется ROH благодаря общему орто-эффекту,
описанному выше для ароматических кислот. В масс-
спектре метилсалицилата максимальный пик нахо-
лится при m/z 120; этот ион далее теряет СО, давая
интенсивный пик иона с m/z 92.
Интенсивный доминирующий пик иона с массой
149 наблюдается в масс-спектрах всех сложных эфи-
ров фталевой кислоты, начиная с диэтилового эфира.
Этот пик незначителен в диметиловом или метилэ-
тиловом эфире фталевой кислоты, а также в эфи-
рах изофталевой и терефталевой кислот, которые
дают ожидаемые ионы М - R, М - 2R, М - CO2R
и М - 2CO2R. Поскольку сложные эфиры фтале-
вой кислоты с длинноцепочными спиртами широко
применяются в качестве пластификаторов, нередко
интенсивный пик иона с m/z 149 указывает на за-
грязнение. Вероятно, этот фрагмент (по существу
протонированный фталевый ангидрид) образуется
при расщеплении двух сложноэфирных групп, при
этом происходит перемещение двух атомов водорода,
а затем еще одного атома водорода с последующим
элиминированием воды.
1.6.8. Лактоны
Пик молекулярного иона пятичленных циклических
лактонов довольно заметен, но его интенсивность
уменьшается, когда имеется алкильный заместитель
С4. Легкий разрыв боковой цепи при С4 (правила 3 и 8,
раздел 1.5.4 ) дает интенсивный пик иона М - Aik.
Основной пик (m/z 56) у-валеролактона (рис. 1.27)
и такой же интенсивный пик в масс-спектре бутиролак-
тона возникают при элиминировании ацетальдегида.
Опыты с мечеными соединениями показали,
что в у-валеролактоне пик с m/z 56 дает ион С4Н8+.
В масс-спектре этого соединения имеются и дру-
гие интенсивные пики: m/z 27 (С2Н3+), 28 (С2Н4+),
29 (С2Н5+),41 (С3Н5+),43 (С3Н7+) и 85 (С4Н5О2+, потеря
метильной группы). В масс-спектре бутиролактона
имеются интенсивные пики ионов с m/z 27, 28, 29,
41 и 42 (С3Н6+).
1.6.9. Амины
1.6.9.1. Алифатические амины
Пик молекулярного иона первичного алифатическо-
го амина имеет нечетное значение массового чис-
ла и обычно крайне малоинтенсивен, а в аминах
46
/. Маа-спектрометрия
с длинной цепью или в сильно разветвленных его
вообще нельзя обнаружить. Доминирующий пик часто
обусловлен разрывом углерод-углеродной связи, со-
седней с атомом азота (а, 0-правило 8, раздел 1.5.4);
для первичных аминов, неразветвленных у а-атома
углерода, это пик иона с m/z 30 (CH2NH2+). Такое рас-
щепление приводит к максимальному пику в масс-
спектрах первичных, вторичных и третичных аминов,
не имеющих разветвления у а-углеродного атома.
Потеря самого большого радикала (-R" на схеме 1.25)
предпочтительно происходит от а-углеродного атома.
R R R
•+ Z —R" Z
I^NyC^-R' —<—>H2N—
R" R' R'
Схема 1.25
Пик М-1 обычно наблюдается, когда у а-угле-
родного атома нет разветвления. При этом проис-
ходит распад того же типа, который был отмечен
выше для спиртов. В аминах он выражен отчетливее
из-за лучшей резонансной стабилизации фрагментно-
го иона менее электроотрицательным по сравнению
с атомом кислорода атомом азота.
Первичные амины с неразветвленной цепью дают го-
мологические серии пиков с постепенно уменьшающейся
интенсивностью (разрыв 8-связи происходит немного
легче, чем соседних) с m/z 30,44,58,..., получающиеся
при разрыве все более удаленных от атома азота связей
С—С, причем заряд остается на азотсодержащем фрагмен-
те. Эти пики сопровождаются сериями углеводородного
типа: С„Н2агИ, С„Н2„ и Таким образом, появляются
характеристические кластеры с интервалами 14 а. е. м.;
каждый кластер содержит пик, обусловленный ионом
C„H2n+2N+. Из-за очень легкого расщепления, приводя-
щего к максимальному пику, пики ионов с большими
массами, как правило, малоинтенсивны.
В процессе распада аминов с более длинной
цепью образуются фрагменты, имеющие цикличе-
ское строение. Показанный на схеме 1.26 фрагмент
дает шестичленный цикл; обычно образуются также
и пятичленные циклы.
Пик иона с m/z 30 является хорошим, хотя и не
окончательным аргументом в пользу первичного амина
с неразветвленной цепью. Дальнейший распад иона,
первоначально образовавшегося из вторичного или
третичного амина, приводит к пикам с m/z 30, 44,
58, 72,.... Этот процесс подобен описанному выше
для алифатических спиртов и простых эфиров и
так же усиливается (приводя к пикам повышенной
интенсивности) при наличии разветвления у одного
из а-углеродных атомов.
Распад сложных эфиров аминокислот происходит
по обеим углерод-углеродным связям (пунктирные
линии в формуле) рядом с атомом азота, причем
предпочтительным является отщепление карбалкокси-
группы (-COOR'). Фрагмент алифатического амина
(+NH2=CHCH2CH2R) распадается далее, давая пик
с m/z 30.
NH,
R'OOC-rC-rCH2CH2R
1.6.9.2. Циклические амины
В противоположность ациклическим аминам пики мо-
лекулярных ионов циклических аминов обычно интен-
сивны, если только нет заместителя в а-положении;
например, пик молекулярного иона пирролидина ин-
тенсивный. Первичный разрыв связей рядом с ато-
мом азота приводит либо к отрыву а-атома водорода,
что дает интенсивный пик М-1, либо к размыканию
цикла. При размыкании цикла, за которым следует
отщепление этилена, образуется •CH2-+NH=CH2 {m/z
43, максимальный пик); из этого иона при потере
атома водорода получается CH2=+N=CH2 {m/z 42).
В масс-спектре N-метилпирролидина также имеется
пик иона C2H4N+ {m/z 42), по-видимому, образую-
щийся не одним путем.
Подобным образом в масс-спектре пиперидина
появляется интенсивный пик молекулярного иона
и пик иона М-1 (максимальный). Размыкание цикла
и следующие за ним последовательные превращения
приводят к серии характеристических пиков ионов
с m/z 70, 57, 56, 44, 43, 42, 30, 29 и 28. Заместители
отрываются от места сочленения с циклом (правило
6, раздел 1.5.4).
1.6.9.3. Ароматические амины (анилины)
В масс-спектрах ароматических моноаминов наблюда-
ется интенсивный пик молекулярного иона (нечетное
массовое число). Отрыв одного атома водорода от
аминогруппы дает пик М-1 умеренной интенсивно-
сти, а отрыв нейтральной молекулы HCN и последую-
щий отрыв атома водорода - заметные характерные
пики ионов с m/z 66 и 65 соответственно.
1.6. Маа-спектры некоторых классов органических соединений
47
Выше отмечалось, что расщепление алкиларило-
вых простых эфиров происходит с перегруппировкой,
включающей разрыв связи ArO-R, т. е. определяет-
ся циклом, а не атомом кислорода. В случае алкил-
ариламинов доминирует разрыв, показанный на схе-
ме 1.27, т. е. процесс расщепления контролируется
гетероатомом.
m/z 106
Схема 1.27
1.6.10. Амиды
1.6.10.1. Алифатические амиды
В масс-спектрах моноамидов с неразветвленной
цепью пик молекулярного иона обычно различим.
Преобладающий тип фрагментации зависит от длины
ацильной части молекулы, а также от числа и длины
алкильных групп, присоединенных к атому азота.
Максимальный пик (m/z 59 H2NC(=OH+)CH2*) во
всех первичных амидах с неразветвленной цепью,
более длинной, чем в амиде пропионовой кислоты,
получается в результате перегруппировки Мак-
Лафферти. При разветвлении у а-углеродного атома
(СН3 и т. д.) возникают гомологические пики ионов
с m/z 73 или 87,....
Первичные амиды дают интенсивный пик иона
с m/z 44 в результате разрыва связи R-CONH2 (ион
O=C=+NH2); это максимальный пик в первичных ами-
дах Cj-Сз и амиде изомасляной кислоты. Пик уме-
ренной интенсивности с m/z 86, получающийся при
разрыве у,6-связи С-С, возможно, связан с циклиза-
цией (схема 1.28).
m/z 86
Схема 1.28
Вторичные и третичные амиды со свободным ато-
мом водорода у у- атома углерода ацильной части
молекулы и метильными группами у атома азота
дают доминирующий пик, обусловленный перегруп-
пировкой Мак-Лафферти. Если же алкильные группы
содержат два или более атомов углерода, а ациль-
ная часть короче, чем С3, то преобладает другой тип
расщепления. Это расщепление N-алкильной группы
в 0-положении относительно атома азота и расще-
пление связи карбонильный углерод-азот с миграци-
ей а-атома водорода в ацильной части молекулы (с
выбросом нейтральной молекулы кетена) приводит
к образованию +NH2=CH2 (m/z 30).
1.6.10.2. Ароматические амиды
Типичным примером является масс-спектр
бензамида (рис. 1.1). Потеря фрагмента NH2 молеку-
лярным ионом приводит к образованию резонансно-
стабилизированного бензоильного катиона, который,
в свою очередь, распадается с образованием фениль-
ного катиона. Другой путь фрагментации приводит
к умеренно интенсивному пику с m/z 44.
1.6.11. Алифатические нитрилы
Пики молекулярных ионов алифатических нитрилов
(за исключением ацетонитрила и пропионитрила)
либо малоинтенсивны, либо вообще отсутствуют,
но пик М + 1 обычно обнаруживается по его по-
ведению при увеличении давления в камере напуска
(раздел 1.5.2.1). Малоинтенсивный, но полезный для
идентификации пик иона М-1 возникает при потере
а-атома водорода с образованием устойчивого иона
RCH=C=N+.
Максимальный пик в масс-спектре нитрилов
С4-С9 с неразветвленной цепью наблюдается при
m/z 41. Это пик иона CH2=C=N+-H, который получа-
ется в результате миграции водорода в шестичленном
переходном состоянии, подобной перегруппировке
Мак-Лафферти. Однако этот пик нельзя уверенно
использовать для идентификации, поскольку и фраг-
мент С3Н5 (m/z 41) присутствует во всех молекулах,
содержащих углеводородную цепь.
В масс-спектрах нитрилов с неразветвленной цепью
из восьми и более углеродных атомов пик иона с
m/z 97 является характеристическим и интенсивным
(иногда максимальным). Предложен изображенный
на схеме 1.29 механизм образования этого иона.
Схема 1.29
Простой разрыв каждой связи С-С (за исключением
соседней с атомом азота) дает характеристические
48
/. Маа-спектрометрия
серии гомологических пиков с четными массовы-
ми числами, уменьшающиеся по интенсивности по
мере возрастания длины цепи (m/z 40, 54,68, 82,...).
Они обусловлены ионами (CH2)„C=N+. Сопутствую-
щие им пики те же, что обычно характерны для
углеводородов.
1.6.12. Нитросоединения
1.6.12.1. Алифатические нитросоединения
В масс-спектрах пик молекулярного иона (с нечетным
массовым числом) алифатического мононитросое-
динения либо малоинтенсивен, либо отсутствует (за
исключением низших гомологов). Главные пики обу-
словлены углеводородными фрагментами вплоть до
М - NO2. Присутствие нитрогруппы обнаруживается
по интенсивному пику иона с m/z 30 (NO+) и менее
интенсивному пику иона с массой 46 (NO2+).
1.6.12.2. Ароматические нитросоединения
В масс-спектрах ароматических нитросоединений
(нечетное массовое число для одного атома азота)
пик молекулярного иона достаточно интенсивен. За-
метные пики обусловлены элиминированием ради-
кала NO2 (в нитробензоле максимальный пик М-46)
и нейтральной молекулы NO с перегруппировкой,
приводящей к образованию фенокси-катиона (М-30);
оба пика полезны для идентификации. Отщепление
молекулы ацетилена от иона М-46 объясняет по-
явление интенсивного пика М-72; при отщеплении
СО от иона М-30 возникает пик М-58. Для иденти-
фикации используется пик с m/z 30, обусловленный
ионом NO+.
Каждый из изомерных о-, м- и и-нитроанилинов
дает интенсивный пик молекулярного иона (с чет-
ным массовым числом). В их масс-спектрах на-
блюдаются характерные пики, получающиеся
в результате двух последовательностей превра-
щений. Первая заключается в отщеплении группы
NO2 (М-46), и образовании иона m/z 92, который
теряет HCN и порождает ион с m/z 65. Вторая
последовательность превращений включает поте-
рю молекулой группы NO (М-30) с образованием
иона с m/z 108, из которого в результате потери
СО получается ион с m/z 80.
Если не учитывать различий в интенсивностях,
то все три изомера имеют очень похожие спектры.
При отрыве атома кислорода масс-спектры мета-
и пара-соединений содержат небольшой пик иона
с m/z 122, в то время как орто-соединения отщепля-
ют ЮН, давая небольшой пик иона с m/z 121 (как
показано на схеме 1.30).
Схема 1.30
1.6.13. Алифатические нитриты
В масс-спектрах алифатических нитритов с одним
атомом азота пик молекулярного иона (с нечетным
массовым числом) либо малоинтенсивен, либо во-
обще отсутствует. Пик иона с m/z 30 (NO+) всегда
интенсивен и часто является максимальным. Всем
нитритам, не имеющим разветвления у а-атома угле-
рода, присущ интенсивный пик с m/z 60 (CH2=+ONO);
это соответствует разрыву углерод-углеродной свя-
зи рядом с группой ONO. Наличие разветвления
у а-углеродного атома может быть установлено по
пикам ионов с m/z 74, 88 или 102,.... Отсутствие ин-
тенсивного пика иона с m/z 46 позволяет отличать
нитриты от нитросоединений. Отчетливо видны угле-
водородные пики; их распределение и интенсивности
зависят от строения углеродной цепи.
1.6.14. Алифатические нитраты
В масс-спектрах алифатических нитратов с одним
атомом азота пик молекулярного иона (нечетное мас-
совое число) либо малоинтенсивен, либо вообще от-
сутствует. Характерный (часто максимальный) пик
получается при разрыве связи С-С рядом с группой
ONO2 с потерей самой тяжелой алкильной группы,
связанной с а-атомом углерода. Характерен также
пик иона NO2+ с m/z 46. Как и в масс-спектрах али-
фатических нитритов, пики ионов углеводородных
фрагментов имеют заметную интенсивность.
1.6.15. Соединения серы
Вклад изотопа 34S (4.4%, см. табл. 1.3 и рис. 1.28)
в пик М + 2, а часто и в пик с массой (фрагмент + 2)
позволяет легко распознать серосодержащие соеди-
нения. Гомологические серии серосодержащих фраг-
ментов имеют на четыре единицы массы больше,
чем серии углеводородных фрагментов. Число ато-
мов серы можно определить по величине вклада
изотопа 34S в пик М + 2. Масса атома (или атомов)
серы, присутствующего в молекуле, вычитается из
молекулярной массы. Например, молекулярная масса
1.6. Маа-спектры некоторых классов органических соединений
49
Й
о
о
х
д
S
X
8
X
S
174 М+ 100.00%
175 М+1 11.33%
176 М+2 4.67%
174-1
т—'—,“"4
Рис. 1.28. Масс-спектр с ионизацией ЭУ ди-н-пентилсульфида
диизопентилдисульфида равна 206, и молекула со-
держит два атома серы. Поэтому формула для остатка
молекулы находится из массы 142, т. е. 206 - (2x32).
1.6.15.1. Алифатические тиолы (меркаптаны}
Пик молекулярного иона алифатических тиолов, за ис-
ключением высших третичных тиолов, обычно на-
столько интенсивен, что можно точно измерить пик
М+2. Вообще говоря, типы расщепления напоминают
распад спиртов. Разрыв а,0-связей С-С рядом с груп-
пой SH приводит к появлению характеристического
иона CH2=SH+ (m/z 41}. Сера в качестве стабилизато-
ра такого фрагмента хуже азота, но лучше кислорода.
При разрыве 0,у-связи наблюдается пик иона с m/z 61,
интенсивность которого равна приблизительно полови-
не интенсивности пика иона с m/z 47. Разрыв у,5-связи
приводит к небольшому пику иона с m/z 75, а разрыв
5,8-связи — к пику иона с m/z 89, который более ин-
тенсивен, чем пик иона с m/z 75; вероятно, ион с m/z 89
стабилизируется циклизацией:
Аналогично спиртам, первичные тиолы элиминируют
H2S, давая интенсивный пик иона М-34; образовав-
шийся ион отщепляет этилен. Таким образом возни-
кают гомологические серии М - H2S - (СН2=СН2)„.
Вторичные и третичные тиолы распадаются
у а-углеродного атома с потерей самой большой
группы, давая характерные пики ионов М - СН3,
М - С2Н5, М - С3Н7,.... Однако пик иона с m/z 41 мо-
жет проявляться также во вторичных и третичных
тиолах как перегруппировочный. Во вторичных тиолах
обычно присутствует пик иона М-33 (отрыв HS).
В меркаптанах с длинной цепью фрагментация,
характерная для углеводородов, накладывается на
фрагментацию, присущую тиолам. Как и в спиртах,
алкенильные пики (т. е. m/z 41,55,69,...) по величине
такие же или еще больше, чем алкильные пики (m/z
43,67,71,...).
1.6.15.2. Алифатические сульфиды
Пик молекулярного иона алифатических сульфидов
обычно достаточно интенсивен для точного измерения
пика М+2. Обычно схемы распада этих соединений
похожи на распад эфиров. Происходит разрыв одной
из а,0-связей углерод-углерод, причем предпочтителен
отрыв самой большой группы. Эти первоначально об-
разовавшиеся ионы распадаются далее с переносом
водорода и элиминированием алкена. Для сульфидов
наблюдаются те же стадии, что и для алифатических
эфиров (схема 1.31); в итоге получается ион RCH=SH+
(в качестве примера см. рис. 1.28).
Схема 1.31
Для сульфидов, не имеющих разветвления
у 5-атомов углерода, этим ионом является CH2=SH+
с m/z 47, и его интенсивность может привести к пу-
танице с таким же ионом, получающимся из тио-
ла. Однако отсутствие пиков М - H2S или М - HS
в масс-спектре сульфидов позволяет их отличать.
В спектрах всех сульфидов, за исключением тре-
тичных, присутствует пик иона с m/z 61, интенсив-
ность которого меняется от умеренной до сильной (см.
расщепление алкилсульфида, рис. 1.28). Если имеется
а-метильная группа, то пик с m/z 61 относится к иону
CH3CH=SH+, появляющемуся в результате двойного
расщепления. Первичные метилсульфиды распадаются
по а,0-связи, образуя ион CH3-S+=CH2 с m/z 61.
50
1. Маа-спектрометрия
Правдоподобное объяснение появления интенсив-
ного пика иона с m/z 61 в масс-спектре сульфида
с неразветвленными цепями предлагается на схеме
1.32.
R
Схема 1.32
Сульфиды дают характеристический ион при
разрыве связи С-S с сохранением заряда на ато-
ме серы. Образующийся ион RS+ дает пики с m/z
32+СН3, 32+С2Н5, 32+С3Н7... Появление пика с m/z
103 объясняется предпочтительным образованием
циклического перегруппировочного иона (схема 1.33).
Эти особенности иллюстрируются спектром ди-н-
пентилсульфида (рис. 1.28).
m/z 103
1.6.1 б. Галогенсодержащие соединения
В масс-спектрах соединений с одним атомом хлора
благодаря присутствию иона с изотопом 37С1 возни-
кает пик М+2, интенсивность которого равна при-
близительно одной трети интенсивности пика моле-
кулярного иона (см. табл. 1.4). Соединения с одним
атомом брома демонстрируют пик М+2, почти равный
по интенсивности пику молекулярного иона и относя-
щийся к молекулярному иону с изотопом 81Вг. В масс-
спектре соединения, содержащего два атома хлора, или
два атома брома, или один атом хлора и один атом
брома, в дополнение к пику М+2 будет проявляться
отчетливый пик М+4, обусловленный молекулярными
ионами, содержащими два атома тяжелых изотопов.
Вообще говоря, число атомов хлора и (или) атомов
брома в молекуле можно определить по числу пиков,
больших по массе по сравнению с молекулярным
ионом. Так, три атома хлора в молекуле дают пики
М+2, М+4 и М+6; в полихлорированных соединениях
пик с самой большой массой может быть настолько
малоинтенсивным, что его можно не заметить.
Относительные интенсивности пиков молекулярно-
го иона, М+2, М+4 и т. д. были вычислены Бейноном
и соавторами (1968) для содержащих хлор и бром
соединений (другие атомы не учитывались). Часть
этих результатов представлена в табл. 1.5 в несколько
модифицированном виде. По ней можно определить,
какая имеется комбинация атомов хлора и брома.
Схема 1.33
Как и в случае соответствующих простых эфи-
ров, в масс-спектрах сульфидов с длинными цепями
может преобладать расщепление, характерное для
углеводородов; пики С„Н2„ оказываются особенно
заметными. В сульфидах с разветвленными цепями
разрыв в месте разветвления может уменьшать от-
носительную интенсивность характеристических
пиков сульфидов.
1.6.15.3. Алифатические дисульфиды
В масс-спектрах дисульфидов, по крайней мере до
С10, пики молекулярных ионов интенсивны. Главный
пик обусловлен разрывом одной из связей С-S с со-
хранением заряда на алкильном фрагменте. Другой
главный пик возникает при таком же расщеплении с
переносом водородного атома и образованием фраг-
мента RSSH, на котором находится заряд. Остальные
пики, очевидно, возникают при разрыве связи между
атомами серы без перегруппировки и с миграцией
одного или двух атомов водорода с образованием,
соответственно RS+, RS+-1 и RS+-2.
Таблица 1.5. Интенсивности изотопных пиков (в % от-
носительно пика молекулярного иона) для различных
комбинаций атомов брома и хлора
Атомы галогенов М+2 М+4 М+б М+8 М+10 М+12
С1 32.6
С12 65.3 10.6
С13 97.8 31.9 3.5
С14 131.0 63.9 14.0 1.2
С15 163.0 106.0 34.7 5.7 0.4
С16 196.0 161.0 69.4 17.0 2.2 0.1
Вг 97.9
Вг2 195.0 95.5
Вг3 293.0 286.0 93.4
ВгС1 130.0 31.9
ВгС12 163.0 74.4 10.4
Вг2С1 228.0 159.0 31.2
1.6. Масс-спектры некоторых классов органических соединений
51
Рис. 1.29. Расчетные наборы пиков М, М+2, М+4, ... для соединений
с различными комбинациями атомов хлора и брома. (Интенсивность пиков в %
относительно пика молекулярного иона.)
Необходимо заметить, что в табл. 1.5 изотопные
вклады даются в процентах от пика молекулярного
иона. На рис. 1.29 графически показаны наборы пи-
ков в области молекулярного иона для соединений,
содержащих атомы хлора и брома.
В соответствии с данными табл. 1.5, интенсив-
ность пика М+2 в масс-спектре и-хлорбензофенона
(рис. 1.23) составляет около одной трети интенсив-
ности пика молекулярного иона (m/z 218). Ранее было
упомянуто, что рядом с хлорсодержащими фрагмента-
ми с m/z 141 и 113 наблюдаются пики [фрагмент+2]
соответствующей интенсивности.
Хотя использование изотопных сигналов полез-
но в случае ароматических галогенпроизводных,
оно ограничено малой интенсивностью пика моле-
кулярного иона большинства алифатических галоген-
производных более чем с шестью атомами углерода
в неразветвленных и разветвленных цепях. Однако
галогенсодержащие фрагменты в монохлоридах или
монобромидах распознаются по отношению интен-
сивностей пиков [фрагмент + 2] и фрагмента. В по-
лихлор- или полибромсоединениях эти пики (фраг-
мент + изотоп) образуют отчетливые мультиплетные
серии (рис. 1.30). Необходимо всегда иметь в виду
100:
50 :
СС13
117-| [-119
СС12
82 q
Г 84
40 50 60 70 80 90 100 110
m/z
|—121
Г123
il 11111111111111111111111111111111111111111
120 130 140 150 160
Рис. 1.30. Масс-спектр с ионизацией ЭУ четыреххлористого углерода (СС14)
0
52
1. Маа-спектрометрия
возможность совпадения других фрагментных ионов
с одним из изотопных фрагментов, что приводит к на-
рушению характеристических отношений.
Ни фтор, ни иод не имеют более тяжелых изотопов.
1.6.16.1. Алифатические хлорпроизводные
Пик молекулярного иона детектируется только у низ-
ших монохлоридов. Фрагментация молекулярного
иона направляется атомом хлора, но в гораздо мень-
шей степени, чем в случае кислород-, азот- и серо-
содержащих соединений. Разрыв углерод-углеродной
связи, соседней с атомом хлора, в монохлоридах с не-
разветвленной цепью объясняет появление слабого
пика иона с m/z 49 СН2=С1+ и, конечно, изотопного
пика с m/z 51.
Разрыв связи С-С1 приводит к появлению неболь-
шого пика С1+ и пика R+, который весьма заметен
для низших хлоридов, но очень мал, если в цепи
содержится более пяти атомов углерода.
Хлориды с неразветвленной цепью, содержащей
более шести атомов углерода, дают ионы С3Н6С1+,
С4Н8С1+ и С5Н10СГ. Из них наиболее интенсивный
пик принадлежит иону С4Н8С1+ (иногда это основной
пик); его устойчивость можно объяснить образованием
пятичленной циклической структуры (схема 1.34).
Схема 1.34
Отрыв НС1 происходит, возможно, путем 1,3-
элиминирования, при котором образуется ион М-36,
дающий пик умеренной или низкой интенсивно-
сти.
В общем случае масс-спектр алифатического моно-
хлорида в значительно большей степени похож на
спектр углеводорода, чем спектры соответствующих
спирта, амина или тиола.
1.6.16.2. Алифатические бромиды
Все замечания для алифатических хлоридов
полностью применимы к соответствующим бромидам.
1.6.16.3. Алифатические иодиды
Среди всех алифатических галогенидов алифатические
иодиды дают самый интенсивный пик молекулярного
иона. Поскольку у иода только один изотоп, у него нет
отличительных изотопных пиков. Присутствие атома
иода можно иногда установить на основании измере-
ний интенсивностей изотопных пиков, которые имеют
подозрительно низкую по отношению к молекулярной
массе интенсивность, а также из некоторых других
обособленных пиков; для полииодсоединений харак-
терен большой интервал между главными пиками.
Фрагментация иодидов в основном напоминает
таковую хлоридов и бромидов, но пик иона С4Н81+
не столь интенсивен, как соответствующий пик хло-
рида или бромида.
1.6.16.4. Алифатические фториды
Из всех алифатических галогенидов алифатические
фториды дают наименее интенсивный пик моле-
кулярного иона. У фтора только один изотоп, и его
определение в полифторсоединениях основано, как и в
случае иодидов, на подозрительно низкой интенсивно-
сти изотопных пиков по отношению к интенсивности
пика молекулярного иона, на специфическом интервале
между пиками и на характеристических пиках. Из них
выделяется пик с m/z 69, который обусловлен ионом
CF3+ и является максимальным для всех перфторпро-
изводных. Наблюдаются заметные пики с m/z 119,169,
219,...; эти массы отличаются на величину инкремента
CF2. Устойчивые ионы C3F5+ и C4F7+ дают большие
пики с m/z 131 и 181. В масс-спектрах перфторирован-
ных соединений часто наблюдается пик М - F. Разрыв
углерод-углеродной а,0-связи в монофторидах не имеет
такого значения, как в других моногалогенидах, но бо-
лее важным становится разрыв связи С-Н у а-атома
углерода. Такая картина является следствием высокой
электроотрицательности атома фтора и объясняется по-
ложительным зарядом на а-атоме углерода. Вторичный
карбониевый ион, образовавшийся при отрыве атома
водорода, как показано на схеме 1.35, стабильнее пер-
вичного карбониевого иона, возникающего при отрыве
алкильного радикала.
’R-CH2-FJ + R—CH—F
I-------------» CHj—F
Схема 1.35
1.6.16.5. Бензилгалогениды
Пик молекулярного иона бензилгалогенидов обычно
определяется без труда. Образование бензильного (или
тропилиевого) иона при потере галогенида (правило 8,
раздел 1.5.4) преобладает даже над разрывом 0-связи
алкильного заместителя. Замещенный фенильный ион
(разрыв a-связи) дает заметный пик в случае поли-
замещенного цикла.
1.6.16.6. Ароматические галогениды
Пик молекулярного иона арилгалогенидов распозна-
ется легко. В масс-спектрах соединений, в которых
Литература
53
галоген X присоединен непосредственно к кольцу,
наблюдается интенсивный пик М - X.
1.6.17. Гетероароматические соединения
Пик молекулярного иона в масс-спектрах гетероаро-
матических соединений и их алкилированных произ-
водных достаточно интенсивен. В алкилпроизводных
пиридина, как и в случае алкилбензолов, обычно про-
исходит разрыв связи в P-положении по отношению
к кольцу; легкость разрыва этой связи зависит от ее
положения (см. ниже).
Заряд молекулярного иона локализован на гете-
роатоме, а не на л-системе кольца, что удовлетво-
рительно объясняет происходящую фрагментацию.
Данная схема предложена Джерасси (Budzikiewicz
et. al., 1967).
В пятичленных гетероароматических системах (фу-
ране, тиофене и пирроле) наблюдается сходная кар-
тина распада кольца. Первой стадией в этих случаях
является разрыв связи углерод - гетероатом, после
чего происходит потеря либо нейтральной молекулы
ацетилена, либо радикальных фрагментов. Так, в масс-
спектре фурана имеются два основных пика: С3Н3+
(m/z 39) и НОО+ (m/z 29). Для тиофена их три: С3Н3+
(m/z 39), HC=S+ (m/z 45) и C2H2S+ (m/z 58). В масс-
спектре пиррола их также три: С3Н3+ (m/z 39), HC=NH+
(m/z 28) и C2H2NH+ (m/z 41). Пиррол элиминирует
нейтральную молекулу HCN, давая интенсивный пик
с m/z 40. Основным пиком для 2,5-диметилфурана
является пик с m/z 43 (СН3С=О+).
Разрыв углерод-углеродной P-связи в алкилпири-
динах (схема 1.36) зависит от положения заместителя
в кольце, и наиболее выражен, если алкильная группа
находится в положении 3. Алкильная группа в по-
ложении 2, содержащая более трех атомов углерода,
может подвергаться перегруппировке с миграцией
водорода к атому азота цикла.
Схема 1.36
Подобный разрыв связей присходит в пиразинах,
так как все заместители в кольце находятся в орто-
положении по отношению к одному из атомов азота.
Литература
Общая
Бейнон Дж. Масс-спектрометрия и ее применение орга-
нической химии. Пер. с англ., Москва, Мир, 1964.
Beynon, J. Н., and Brenton, A. G. (1982). Introduction to Mass
Spectrometry. Cardiff: University of Wales Publications.
Beynon, J. H., Saunders, R. A., and Williams A. E. (1968).
The Mass Spectra of Organic Molecules. New York:
Elsevier.
Biemann, K. (1962). Mass Spectrometry, Applications to
Organic Chemistry. New York: McGraw-Hill.
Budzikiewicz, H., Djerassi, C., and Williams, D. H. (1967).
Mass Spectrometry of Organic Compounds. San Francisco:
Holden-Day. (Есть русское издание: Будзикевич Г., Уильямс
Д., Интерпретация масс-спектров органических соеди-
нений. Пер. с англ., Москва, Мир, 1966.)
Budzikiewicz, Н., Djerassi, С., and Williams, D. Н. (1964).
Structure Elucidation of Natural Products by Mass
Spectrometry, Vols. I and II. San Francisco: Holden-Day.
Chapman, J. R. (1993). Practical Organic Mass Spectrometry,
2nd ed. New York: Wiley. (Есть русское издание: Чепмен
Дж., Практическая органическая масс-спектрометрия. Пер.
с англ., Москва, Мир, 1988.)
Constatin, Е., Schnell, A., and Thompson, М. (1990). Mass
Spectrometry. Englewood Cliffs, NJ: Prentice-Hall.
Davis, R., and Frierson, M. (1987). Mass Spectrometry.
New York: Wiley (самоучитель).
Hamming, M., and Foster, N. (1972). Interpretation of
Mass Spectra of Organic Compounds. New York: Academic
Press.
Hoffmann, E.D., and Stroobant, V. (2002). Mass Spectrometry:
Principles and Applications. John Wiley and Sons, Ltd.
Howe. I., Williams, D. H., and Bowen, R. D. (1981).
Mass Spectrometry — Principles and Application. New
York: McGraw-Hill.
McLafferty, F. W, and Turecek. A. (1993). Interpretation of
Mass Spectra, 4th ed. Mill Valley, CA: University Scientific
Books.
McLafferty, F. W, and Venkataraghavan, R. (1982). Mass
Spectral Correlations. Washington, DC: American Chemical
Society.
McNeal, C. J., Ed. (1986). Mass Spectrometry in the Analysis
of Large Molecules. New York: Wiley.
54
1. Маа-спектрометрия
Middleditch, В. S.. Ed. (1979). Practical Mass Spectrometry.
New York: Plenum Press.
Milne, G. W. A. (1971). Mass Spectrometry'. Techniques and
Applications. New York: Wiley-Interscience.
Rose, M., and Johnston, R. A. W. (1982). Mass Spectrom-
etry for Chemists and Biochemists. New York: Cambridge
University Press.
Shrader, S. R. (1971). Introduction to Mass Spectrometry.
Boston: Allyn and Bacon.
Smith, R. M. (1998). Understanding Mass Spectra'. A Basic
Approach. Wiley-Interscience.
Watson, J. T. (1985). Introduction to Mass Spectrometry. 2nd
ed. New York: Raven Press.
Williams, D. H. (June 1968-June 1979). Mass Spectrometry.
A Specialist Periodical Report, Vols. I-V. London: Chemi-
cal Society.
(1980) Advances in Mass Spectrometry; Applications in Organic
and Analytical Chemistry. New York: Pergamon Press.
Mass Spectrometry Reviews. New York: Wiley, 1982 to date.
Mass Spectrometry Bulletin. Aldermaston, England, 1966 to
date.
Таблицы данных и атласы масс-спектров
American Petroleum Institute Research Project 44 and
Thermodynamics Research Center (formerly MCA Research
Project). (1947 to date). Catalog of Selected Mass Spectral
Data. College Station, TX; Texas A & M University, Dr.
Bruno Zwohnski, Director.
Beynon, J. H., and Williams. A. E. (1963). Mass and Abun-
dance Tables for Use in Mass Spectrometry. Amsterdam:
Elsevier.
(1992). Eight Peak Index of Mass Spectra, 4th ed. Boca
Raton, FL: CRC Press.
Heller, S. R., and Milne, G. W. EPA/NIH Mass Spectral
Search System (MSSS). A Division of CIS. Washington,
DC: U.S. Government Printing Office. Интерактивная
поисковая система, содержащая спектры более
32 000 соединений. Поиск осуществляется на основе
интенсивности пиков, как было предложено Биманном,
и вероятности совпадений.
McLafferty, F. W. (1982). Mass Spectral Correlations, 2nd
ed. Washington, DC: American Chemical Society.
McLafferty, F. W, and Stauffer, D. B. (1988). The Wiley/
NBS Registry of Mass Spectral Data (7 volumes). New York:
Wiley-Interscience.
McLafferty, F. W, and Stauffer, D. B. (1992). Registry of Mass
Spectral Data, 5th ed. New York: Wiley (220000 спектров).
McLafferty, F. W, and Stauffer, D. B. (1991). The Important
Peak Index of the Registry of Mass Spectral Data, 3 Vols.
New York: Wiley.
Специальные монографии
Ardrey, R. E. (2003). Liquid Chromatography-Mass Spec-
trometry: An Introduction. New York: John Wiley & Sons.
Ashcroft, A. E. (1997). Ionization Methods in Organic Mass
Spectrometry. London: Royal Society of Chemistry.
Cech, N. B., and Enke, C. G. (2002). Practical implica-
tions of some recent studies in electrospray ionization
fundamentals. Mass Spectrometry Reviews, 20, 362-387.
Cole, R. B. Ed. (1997). Electrospray Ionization Mass Spec-
trometry. New York: Wiley-Interscience.
Harrison, G. (1992). Chemical Ionization Mass Spectrometry,
2nd ed. Boca Raton. FL: CRC Press.
Kinter, M., and Sherman, N. E. (2000). Protein Sequencing
and Identification Using Tandem Mass Spectrometry. New York:
Wiley-Interscience.
Linskens, H. F, and Berlin, J. Eds. (1986). Gas Chromatogra-
phy-Mass Spectrometry. New York: Springer-Verlag.
March, R. E., and Hughes. R. J. (1989). Quadrupole Storage
Mass Spectrometry. New York: Wiley-Interscience.
McLafferty, F. W. (1983). Tandem Mass Spectrometry, 2nd ed.
New York: Wiley-Interscience.
Message, G. M. (1984). Practical Aspects of Gas Chromato-
graphy-Mass Spectrometry. New York: Wiley.
Siuzdak, G. (1996). Mass Spectrometry for Biotechnology.
New York: Academic Press.
Waller, G. R., Ed. (1972). Biochemical Applications of Mass
Spectrometry. New York: Wiley-Interscience.
Waller, G. R., and Denner, О. C, Eds. (1980). Biochemical
Applications of Mass Spectrometry, First Suppl. Vol. New York:
Wiley-Interscience.
Дополнительная литература
Лебедев A.T. Масс-спектрометрия в органической
химии. Москва, «Бином. Лаборатория знаний», 2003.
Заикин В. Г., Варламов А. В., Микая А.И., Простаков
Н. С., Основы масс-спектрометрии органических соеди-
нений. Москва, МАИК «Наука/интерпериодика», 2001.
Терентьев П. Б. Масс-спектрометрия в органической
химии, Москва, «Высшая школа», 1979.
Упражнения
55
Упражнения
1.1. Используя данные таблицы 1.4, рассчитайте точ-
ные массы приведенных выше молекул (а-п).
1.2. Определите индексы водородной ненасыщенности
для приведенных выше соединений.
1.3. Напишите структуры молекулярных ионов для
каждого из соединений (a-и), указывая, где это
возможно, локализацию катион-радикала.
1.4. Предскажите главные направления фрагмента-
ции и перегруппировки для приведенных выше
соединений. В каждом случае опирайтесь на пра-
вила из раздела 1.5.4.
1.5. Для каждой схемы фрагментации или перегруп-
пировки из упражнения 1.4 покажите детальный
механизм, используя в соответствующих случаях
стрелки с одной или двумя «засечками».
1.6. Подберите точные значения масс к следующим
масс-спектрам (А-Ч). Обратите внимание, что два
соединения имеют равные точные массы; для них
необходимо рассмотреть масс-спектры с ХИ:
(а) 56.0264, (б) 73.0896, (в) 74.0363, (г) 89.0479,
(д) 94.0535, (е) 96.0572, (ж) 98.0736, (з) 100.0893,
(и) 102.0678, (к) 113.0845, (л) 114.1043, (м) 116.0841,
(н) 116.1206, (о) 122.0733, (п) 122.0733, (р) 126.1041,
(с) 138.0687, (т) 150.0041, (у) 152.0476, (ф) 156.9934,
(х) 161.9637, (ц) 169.9735, (ч) 208.0094.
1.7. Определите, входят ли в состав соединений, масс-
спектры которых представлены (А-Ч), следующие
гетероатомы: S, С1, Вг.
1.8. Для каждого из соединений, для которых рассчи-
таны точные массы и представлены масс-спектры
А-Ч, определите молекулярную формулу. Имейте
в виду гетероатомы, которые были определены
в упражнении 1.7.
1.9. Определите индексы водородной ненасыщенно-
сти для каждой из формул, полученных в упраж-
нении 1.6.
1.10. Составьте список основных пиков и пиков мо-
лекулярных ионов для каждого из масс-спектров
ЭУ (А-Ч).
1.11. Выберите три иона (кроме молекулярного) из
каждого масс-спектра ЭУ (А-Ч, кроме спектра 3)
и определите молекулярные формулы отрываю-
щихся от молекулярного иона и остающихся
фрагментов. Укажите, какие ионы получаются
в результате перегруппировки.
Ov
Упражнение 1.6(A-BJ
1. Маа-спектрометрия
Упражнение 1.б(Г-Ж)
И Интенсивность, % Jq Интенсивность, %
Упражнения
58
1 Массхпектрометрия
57-1
50 :
55 60 65 70 75 80 85 90 95 100 105
43-1
« 100:
К
§ ----:
£ 50 :
® 1 _____________ 72-, *>1
I I |'| I I II 11 I I | П1'1 | I I I I 11 I 11 | I I I I | I I I I | 11 11 I I I Г I | I I I I | 1 I I I 11 I 11 | I I I I | I I I f I I I I I | 11 I I I TTI I | I I I I ] I 11 I | I 11 I I I I I I I
40 45 50 55 60 65 70 75 80 85 90
m/z
И (ХИ, газ-реагент - метан)
90-1
100:
50 :
119-1
130q
1111 Р 1'111111111111111ГГ| 1111'| Г111 I/| 111111111111111111111111111П 11111II111111111111111111Г111111111111111111 III [I Г11111111
75 80 85 90 95 100 105 ПО 115 120 125 130 135
m/z
Упражнение 1.6(3-14)
Упражнение 1.6(К-Н)
60
1. Маа-спектрометрия
О
100:
94-|
!
д
50 :
40-|
S
Г42
53-1
67-1
40
45
50
55 60
65
I I I 11 I I I 11 I I 11 I 11 I I I I 11 ГI I I I I Г[11 I I | I I Г 11 г ггтр I П | I г
70 75
80
85
90 95
m/z
Р
m/z
Упражнение 1.6(О-С)
Упражнения
61
С (ХИ, газ-реагент - метан)
100 :
111
129-1
209-1 г211
191-1 г 193
239
237-1
o"i—1—Д'—1—гД—।—I—।—।—।—।—।—।—।—।—।—Дт—।—। Ф1 ।—1—।—।—гД
100 150 200
m/z
m/z
Упражнение 1.б(С-Ф)
62
/. Маа-спектрометрия
ф (ХИ, газ-реагент-метан)
1Г7-|
10б”Е
507 "-|
I I I I ГI I I I I I I I | I I I I [ ГI I I | I I I I I I /1 I | I I I I I
130 140 150 160
m/z
m/z
Упражнение 1.б(Ф-Ч)
Приложение А
63
Приложение А. Точные величины масс (М) для молекулярных формул,
содержащих углерод, водород, азот и кислород1)
м М M M
12 H4N2 32.0375 cao 46.0419 CH3N2O 59.0246
С 12.0000 СН4О 32.0262 47 ch5n3 59.0484
13 33 hno2 47.0007 адо2 59.0133
сн 13.0078 но2 32.9976 СН3О2 47.0133 садмо 59.0371
14 H3NO 33.0215 CH5NO 47.0371 адм2 59.0610
N 14.0031 34 48 садо 59.0497
СН2 14.0157 Н2О2 34.0054 о3 47.9847 c3h9n 59.0736
15 38 H2NO2 48.0085 60
HN 15.0109 С3Н2 38.0157 h4n2o 48.0324 CH2NO2 60.0085
СН3 15.0235 39 CH4O2 48.0211 ch4n2o 60.0324
16 QHN 39.0109 49 сад 60.0563
О 15.9949 С3Н3 39.0235 h3no2 49.0164 адо2 60.0211
h2n 16.0187 40 52 садмо 60.0450
сн4 16.0313 C,H2N 40.0187 C4H4 52.0313 ади2 60.0688
17 С3Н4 40.0313 53 СзН8О 60.0575
НО 17.0027 41 C3H3N 53.0266 с5 60.0000
H3N 17.0266 chn2 41.0140 c4h5 53.0391 61
18 c2h3n 41.0266 54 CH3NO2 61.0164
Н2О 18.0106 c3H5 41.0391 C2H2N2 54.0218 ch5n2o 61.0402
24 42 C3H2O 54.0106 ch7n3 61.0641
Q 24.0000 N3 42.0093 c3h4n 54.0344 адо2 61.0289
26 CNO 41.9980 СД 54.0470 садмо 61.0528
CN 26.0031 CH2N2 42.0218 55 62
ад 26.0157 садо 42.0106 C2H3N2 55.0297 СН2О3 62.0003
27 садм 42.0344 C3H3O 55.0184 CH4NO2 62.0242
CHN 27.0109 ад 42.0470 c3h5n 55.0422 ch6n2o 62.0480
27.0235 43 C4H7 55.0548 адо2 62.0368
28 HN3 43.0170 56 63
n2 28.0062 CHNO 43.0058 QO2 55.9898 HNO3 62.9956
СО 27.9949 CH3N2 43.0297 c,h2no 56.0136 CH5NO2 63.0320
ch2n 28.0187 садо 43.0184 C,H4N2 56.0375 64
ад 28.0313 садм 43.0422 CAO 56.0262 ад 64.0313
29 ад 43.0548 CAN 56.0501 65
hn2 29.0140 44 C4H8 56.0626 C4H3N 65.0266
сно 29.0027 N2O 44.0011 57 ад 65.0391
CH3N 29.0266 со2 43.9898 CANO 57.0215 66
ад 29.0391 ch2no 44.0136 C:H,N, 57.0453 c4h4n 66.0344
30 ch4n2 44.0375 CAO 57.0340 ад 66.0470
NO 29.9980 садо 44.0262 c3h7n 57.0579 67
H2N2 30.0218 QH6N 44.0501 CA 57.0705 c3h3n2 67.0297
СН2О 30.0106 ад 44.0626 58 С4Н3О 67.0184
ch4n 30.0344 45 CH2N2O 58.0167 c4h5n 67.0422
ад 30.0470 CH3NO 45.0215 ch4n3 58.0406 ад 67.0548
31 CH5N2 45.0453 C2H2O2 58.0054 68
HNO 31.0058 адо 45.0340 CANO 58.0293 адм2 68.0375
h3n2 31.0297 садм 45.0579 QHA 58.0532 С4Н4О 68.0262
СН3О 31.0184 46 c3H6o 58.0419 C4H6N 68.0501
ch5n 31.0422 no2 45.9929 CAN 58.0657 ад 68.0626
32 СН2О2 46.0054 C4Ht0 58.0783 69
О2 31.9898 ch4no 46.0293 59 C3H3NO 69.0215
h2no 32.0136 ch6n2 46.0532 chno2 59.0007 c3h5n2 69.0453
° С разрешения из [J.H.Beinon, Mass Spectrometry and its Application to Organic Chemistry, Amsterdam, 1960]. В колонке,
«Массы» указаны точные значения молекулярных масс, вычисленные для молекулярных формул с использованием масс наи-
более распространенных изотопов каждого из элементов; масса наиболее распространенного изотопа углерода равна 12.0000.
Молекулярные формулы включают только углерод, водород, азот и кислород.
64
1. Масс-спектрометрия
Приложение А (продолжение)
M M М M
с4н5о 69.0340 76.0399 c4h9n2 85.0767 c3h8no2 90.0555
c4h7n 69.0579 ед«М2О 76.0637 С,Н9О 85.0653 c3h„,n2o 90.0794
ед, 69.0705 C3H8O2 76.0524 C5H„N 85.0892 c4H.„o2 90.0681
70 c5h2n 76.0187 ед,3 85.1018 <ед 90.0470
СгНЛ 70.0406 c6H4 76.0313 86 91
С3Н2О2 70.0054 77 ед2м2о2 86.0116 ед3о4 91.0031
C3H4NO 70.0293 CH3NO3 77.0113 ед4Ы3О 86.0355 ед5мо, 91.0269
с3ед 70.0532 QHsO, 77.0238 ед6м4 86.0594 c2h7n2o2 91.0508
с4н6о 70.0419 C2H7NO2 77.0477 c3h4no2 86.0242 c2h9n3o 91.0746
c4h8n 70.0657 C6H5 77.0391 c3h6n2o 86.0480 ед7о3 91.0395
ед10 70.0783 78 c3h8n3 86.0719 C3H9NO2 91.0634
71 c2H6o3 78.0317 С4Н6О2 86.0368 C6H5N 91.0422
C2H3N2O 71.0246 c5h4n 78.0344 C4H8NO 86.0606 ед7 91.0548
C2H5N3 71.0484 ед 78.0470 c4h„,n2 86.0845 92
С3Н3О2 71.0133 79 с,н10о 86.0732 C2H4O4 92.0109
C3H5NO 71.0371 C5H5N 79.0422 C5Ht2N 86.0970 «едко, 92.0348
c3h7n2 71.0610 ед 79.0548 ед.4 86.1096 ед«н2о2 92.0586
С4Н7О 71.0497 80 87 едо, 92.0473
c4h9n 71.0736 C3H2N3 80.0249 ед,м4 87.0672 ед4о 92.0262
c3H„ 71.0861 c4h4n2 80.0375 С3Н3О3 87.0082 «еды 92.0501
72 C5H4O 80.0262 c3h5no2 87.0320 ед8 92.0626
C2H2NO2 72.0085 c5h6n 80.0501 c3h7n2o 87.0559 93
C2H4N2O 72.0324 <ед 80.0626 c3h9n3 87.0798 ед,о4 93.0187
QH.N, 72.0563 81 c4H7o2 87.0446 Qh.no., 92.0426
с3н4о2 72.0211 c3h3n3 81.0328 c4h9no 87.0684 едм2 93.0453
C3H6NO 72.0449 c4h5n2 81.0453 c4h,,n2 87.0923 ед5о 93.0340
ОД№ 72.0688 ед5о 81.0340 с,нпо 87.0810 c„h7n 93.0579
c4H8o 72.0575 c5h7n 81.0579 c5h13n 87.1049 ед9 93.0705
c4h1()n 72.0814 ед. 81.0705 88 94
C5H12 72.0939 82 c2h4n2o2 88.0273 ед6о4 94.0266
73 c3h4n3 82.0406 (ед^о 88.0511 c4h4n3 94.0406
QHjNO, 73.0164 c4h4no 82.0293 qh8n4 88.0750 c5h4no 94.0293
C2H,N2O 73.0402 c4h6n2 82.0532 ед4о3 88.0160 c5h(1n2 94.0532
c2h7n3 73.0641 ед6о 82.0419 C3H6NO2 88.0399 едо 94.0419
C3H5O2 73.0289 c5h8n 82.0657 C3H8N2O 88.0637 c6h8n 94.0657
C3H7NO 73.0528 ед>о 82.0783 C3H,ON3 88.0876 едю 94.0783
c3h9n2 73.0767 83 С4Н8О2 88.0524 95
C4H9O 73.0653 C3H,N3 83.0484 C4HioNO 88.0763 C4H5N3 95.0484
C4HnN 73.0892 C4H3O2 83.0133 c4h12n2 88.1001 c5h5no 95.0371
74 C4H5NO 83.0371 С5Н|2О 88.0888 c5h7n2 95.0610
C2H2O3 74.0003 c4h7n2 83.0610 89 ед7о 95.0497
C^NO, 74.0242 C,H7O 83.0497 c2h5n2o2 89.0351 c6h9n 95.0736
QH6N2O 74.0480 c5h9n 83.0736 ед7м,о 89.0590 ед,, 95.0861
74.0719 ед.. 83.0861 c2h9n4 89.0829 96
c3H6o2 74.0368 84 С3н,о3 89.0238 c4h6n3 96.0563
C3H8NO 74.0606 еди, 84.0563 C3H7NO2 89.0477 c,h4o2 96.0211
c3h10n2 74.0845 ед4о2 84.0211 c3h9n2o 89.0715 c5h6no 96.0449
c4h10o 74.0732 c4h„no 84.0449 C3H,,N3 89.0954 c5h8n2 96.0688
75 c4h8n2 84.0688 едо2 89.0603 ед8о 96.0575
C2H3O3 75.0082 С5Н8О 84.0575 C4H„NO 89.0841 C6H1(>N 96.0814
C2H5NO2 75.0320 ед.л 84.0814 ед. 89.0391 ед,2 96.0939
C2H7N2O 75.0559 ед12 84.0939 90 97
C2H9N3 75.0798 85 c2h4no, 90.0191 C3H,N4 97.0515
C3H7O2 75.0446 C,H5N2O 85.0402 c2h6n2o2 90.0429 c4h5n2o 97.0402
C3H9NO 75.0684 C>H7N3 85.0641 C;H.N,O 90.0668 ед,о2 97.0289
76 C4H,O2 85.0289 QH.oN, 90.0907 C5H7NO 97.0528
C2H4O3 76.0160 C4H7NO 85.0528 едо. 90.0317 c5h9n2 97.0767
Приложение А
65
Приложение А (продолжение)
M M M M
С6Н9О 97.0653 102 c4h„no2 105.0790 c4h6n4 110.0594
QH..N 97.0892 QHeN.O 102.0542 CeH5N2 105.0453 c5h6n2o 110.0480
C?HI3 97.1018 C3H4NO3 102.0191 QHjO 105.0340 c5hrn3 110.0719
98 c3h6n2o2 102.0429 QH.N 105.0579 C6H6O2 110.0368
СэН4М3О 98.0355 C3HrN3O 102.0668 QH, 105.0705 c6hrno 110.0606
C3H,N4 98.0594 C3H,ON4 102.0907 106 c6h10n2 110.0845
C4H4NO2 98.0242 ОДО, 102.0317 C2H4NO4 106.0140 С^НюО 110.0732
СДМгО 98.0480 C4H8NO2 102.0555 106.0379 110.0970
c4h8n3 98.0719 c4h10n2o 102.0794 C2H8N3O2 106.0617 c8h14 110.1096
С5Н6О2 98.0368 c4h12n3 102.1032 QH.oN.O 106.0856 111
C5H8NO 98.0606 CsH10O2 102.0681 C3H6O4 106.0266 C4H5N3O 111.0433
C5H10N2 98.0845 c5h12no 102.0919 c3h8no3 106.0504 c4h7n4 111.0672
СбН.оО 98.0732 c5h14n2 102.1158 CjHjoN202 106.0743 c5h5no2 111.0320
QH12n 98.0970 СбН.4О 102.1045 c4H10o3 106.0630 CsH7N2O 111.0559
с?н14 98.1096 СцНй 102.0470 C6H4NO 106.0293 c5h9n3 111.0789
99 103 106.0532 СбН7О2 111.0446
C3H5N3O 99.0433 C2H5N3O2 103.0382 c,H6o 106.0419 СбЩЮ 111.0684
CjH7N4 99.0672 q,h7n4o 103.0621 QHgN 106.0657 C6HnN2 111.0923
C4H3O3 99.0082 C,H3O4 103.0031 G.H.0 106.0783 C/HjjO 111.0810
C4H5NO2 99.0320 C,H5NO3 103.0269 107 QHnN 111.1049
c4h7n2o 99.0559 СзН7К2О2 103.0508 QHjNC^ 107.0218 c8h15 111.1174
c4h9n3 99.0798 C3H9N3O 103.0746 QH7N2O3 107.0457 112
c5h7o2 99.0446 103.0985 QHsNjO, 107.0695 C,H4N4O 112.0386
C5H5.NO 99.0685 C4H7O3 103.0395 C3H7O4 107.0344 c4h4n2o2 112.0273
C5HnN2 99.0923 C4H9NO2 103.0634 c3h9no3 107.0583 c4h6n3o 112.0511
QHUO 99.0810 C4HnN2O 103.0872 C5H5N3 107.0484 c4h8n4 112.0750
QHjjN 99.1049 c4h13n3 103.1111 C6HsNO 107.0371 C5H4o3 112.0160
C,HI5 99.1174 C5HnO2 103.0759 QH7N2 107.0610 QHeNOj 112.0399
100 CSH13NO 103.0998 C7H7O 107.0497 C5H8N2O 112.0637
CjH4N4O 100.0386 QHjN 103.0422 c7h9n 107.0736 C5HioN3 112.0876
CjH.NjOj 100.0273 QH7 103.0548 СцНц 107.0861 СбН8О2 112.0524
CjHeNjO 100.0511 104 108 QHjoNO 112.0763
C3HRN4 100.0750 CzH4N2O3 104.0222 QHeNO. 108.0297 112.1001
C4H,O3 100.0160 QH6N3O2 104.0460 c2h8n2o3 108.0535 112.0888
c4h6no2 100.0399 QH^O 104.0699 C3H8O4 108.0422 QHuN 112.1127
c4h8n2o 100.0637 C3H4O4 104.0109 C4H|N4 108.0437 СцН16 112.1253
c4h10n3 100.0876 сдаю. 104.0348 c5h4n2o 108.0324 113
c5H8o2 100.0524 C5H8N2O2 104.0586 C5H6N3 108.0563 c3h5n4o 113.0464
C5H1(1NO 100.0763 C3H10N3O 104.0825 QH4O2 108.0211 c4h5n2o2 113.0351
c5h12n2 100.1001 C3H12N4 104.1063 C6H6NO 108.0449 c4h7n3o 113.0590
QHi2O 100.0888 c4H8o3 104.0473 c6h8n2 108.0688 c4h,n4 113.0829
c6h14n 100.1127 C4H10NO2 104.0712 QHgO 108.0575 QHjOj 113.0238
QH16 100.1253 c4h12n2o 104.0950 C7H10N 108.0814 CjH7NO2 113.0477
101 C,H12O2 104.0837 CeH12 108.0939 C5H>n2o 113.0715
CjH3NO3 101.0113 c6h4n2 104.0375 109 CsHnNj 113.0954
c3h,n2o2 101.0351 CzH.O 104.0262 C2H7NO4 109.0375 CgHOz 113.0603
C,H7N3O 101.0590 QH6N 104.0501 c4h5n4 109.0515 QHnNO 113.0841
c3h9n4 101.0829 C„H8 104.0626 c5h5n2o 109.0402 CgH13N2 113.1080
C4H5O3 101.0238 105 C5H7N3 109.0641 QHnO 113.0967
C4H7NO2 101.0477 C2H5N2O3 105.0300 c6H5o2 109.0289 c,h15n 113.1205
C4H9N2O 101.0715 CzH7N3O2 105.0539 C6H7NO 109.0528 QHp 113.1331
c4h„n3 101.0954 QHoN.O 105.0777 C6H9N2 109.0767 114
C5H,O2 101.0603 ОДО4 105.0187 c7h9o 109.0653 CjHgN.0 114.0542
CsHnNO 101.0841 C3H7NO3 105.0426 QHnN 109.0892 c4h4no3 114.0191
CsH13N2 101.1080 C3H.,N2O2 105.0664 C8H13 109.1018 c4h<,n2o2 114.0429
СбН13О 101.0967 ОНиЫэО 105.0903 110 c4h8n3o 114.0668
C6H1SN 101.1205 C4H9O3 105.0552 c4h,n3o 110.0355 c4h10n4 114.0907
66
1. Маа-спектрометрия
Приложение А (продолжение)
M M м M
CjHftOj 114.0317 C4HgN2O2 117.0664 C4HgO4 120.0422 QHeNO 123.0684
C5H8NO2 114.0555 C4HnN3O 117.0903 C4H10NO3 120.0661 QH>iN2 123.0923
QHjoNjO 114.0794 c4h13n4 117.1142 c4h12n2o2 120.0899 ад.о 123.0810
CjH12N3 114.1032 C5H9O3 117.0552 c5h4n4 120.0437 QHnN 123.1049
C6H10O2 114.0681 адпыо2 117.0790 c5h12o3 120.0786 ад.5 123.1174
c6h]2no 114.0919 C5H13N2O 117.1029 c6h4n2o 120.0324 124
QHi4N2 114.1158 c5h15n3 117.1267 адн3 120.0563 Q,H8N2O4 124.0484
C7H14O 114.1045 C6H13O2 117.0916 QHgNO 120.0449 c4h4n4o 124.0386
C7H16N 114.1284 c6h15no 117.1154 QHgbk 120.0688 c,h4n2o2 124.02^3
С,н18 114.1409 QH.N 117.0579 <адо 120.0575 ады3о 124.0511
ад 114.0470 ад 117.0705 CgH10N 120.0814 CsHgN4 124.0750
115 118 ад2 120.0939 c6H4o3 124.0160
c3h5n3o2 115.0382 ад4н3о3 118.0253 121 адью, 124.0399
СзН7М4О 115.0621 ады4о2 118.0491 QH5N2O4 121.0249 C6HgN2O 124.0637
C4HsNO3 115.0269 c3h4no4 118.0140 c2h7n3o3 121.0488 QH10N3 124.0876
c4h7n2o2 115.0508 c3h6n2o3 118.0379 121.0726 ад8о2 124.0524
C4H9N3O 115.0746 c3h8n3o2 118.0617 CjH7NO4 121.0375 QHwNO 124.0763
C4HnN4 115.0985 c3h10n4o 118.0856 адм2о3 121.0614 ад12к2 124.1001
ад7о3 115.0395 адо4 118.0266 C3HnN3O2 121.0852 ад 124.0062
CjH^NOz 115.0634 C4H8NO3 118.0504 ад>о4 121.0501 ад^о 124.0888
C5HnN2O 115.0872 C4H10N2O2 118.0743 C4HnNO3 121.0739 CgHI4N 124.1127
c5h13n3 115.1111 c4hI2n3o 118.0981 c5h5n4 121.0515 ад.б 124.1253
C6HnO2 115.0759 c4hI4n4 118.1220 C6H5N2O 121.0402 125
C6H,3NO 115.0998 CsHI0O3 118.0630 CgH7N3 121.0641 C4H3N3O2 125.0226
C6H,5N2 115.1236 C5H12NO2 118.0868 ад5о2 121.0289 c4h5n4o 125.0464
QHisO 115.1123 c5h14n2o 118.1107 C,H7NO 121.0528 c5h5n2o2 125.0351
QHnN 115.1362 C6H14O2 118.0994 ад>м2 121.0767 ад7н3о 125.0590
ад7 115.0548 ади2 118.0532 адо 121.0653 CsHsbU 125.0829
116 адо 118.0419 CgH.iN 121.0892 ад5о3 125.0238
ады4о2 116.0335 CgHgN 118.0657 ад3 121.1018 CgH7NO2 125.0477
ад,ы2о3 116.0222 ад0 118.0783 122 (адЫгО 125.0715
c3h6n3o2 116.0460 119 адм2о4 122.0328 CgHnN, 125.0954
c3h8n4o 116.0699 qh5n3o3 119.0331 адм3о3 122.0566 адА 125.0603
адо4 116.0109 qh7n4o2 119.0570 адоМ4о2 122.0805 QHnNO 125.0841
адыо3 116.0348 адыо4 119.0218 CjH8NO4 122.0453 c,h13n2 125.1080
C4HgN2O2 116.0586 CjH7N2O3 119.0457 СэНщЫгОэ 122.0692 ад13о 125.0967
C4H10N3O 116.0825 ад,ьг3о2 119.0695 С4Н10О4 122.0579 QHisN 125.1205
c4hi2n4 116.1063 C3HnN4O 119.0934 ады4 122.0594 <ад7 125.1331
ад8о3 116.0473 адо4 119.0344 C6H4NO2 122.0242 126
c5h10no2 116.0712 c4h,no3 119.0583 ады2о 122.0480 ады4о2 126.0178
CsH12N2O 116.0950 c4h„n2o2 119.0821 ад8ы3 122.0719 c4H4N3o2 126.0304
c5h14n3 116.1189 c4h13n3o 119.1060 адо2 122.0368 адм4о 126.0542
CeH12O2 116.0837 c5Hno3 119.0708 QHgNO 122.0606 CsH4NO3 126.0191
адмью 116.1076 C5H13NO2 119.0947 QHioN, 122.0845 с5адо2 126.0429
C6H16N2 116.1315 c6h5n3 119.0484 QHWO 122.0732 C5HgN3O 126.0668
C7H4N2 116.0375 C7H5NO 119.0371 ад2ы 122.0970 CjH10N4 126.0907
QHieO 116.1202 c7H7N2 119.0610 ад4 122.1096 адо3 126.0317
CgHgN 116.0501 ад7о 119.0497 123 CgHgNO, 126.0555
ад 116.0626 C8H9N 119.0736 C2H7N2O4 123.0406 c6h10n2o 126.0794
117 ад. 119.0861 C,H,N3O3 123.0644 c6h12n3 126.1032
QH5N4O2 117.0413 120 ад»мо4 123.0532 С^НщОг 126.0681
CjH3NO4 117.0062 QHgNA 120.0410 CjHjNjO 123.0433 ад12мо 126.0919
c3h5n2o3 117.0300 Q.h8n4o2 120.0648 qh7n4 123.0672 QHuNj 126.1158
CjH7N3O2 117.0539 адью4 120.0297 c6h5no2 123.0320 QHhO 126.1045
CjH,N4O 117.0777 QHgNjOj 120.0535 c6h7n2o 123.0559 QH16N 126.1284
адо4 117.0187 СзН10Н3О2 120.0774 адм3 123.0798 ад18 126.1409
c4h7no3 117.0426 C,H12N4O 120.1012 ад7о2 123.0446 127
Приложение А
67
Приложение А (продолжение)
M M M M
C3H3N4O2 127.0257 C«H19N 129.1519 c4h10n3o2 132.0774 CgHgNO 134.0606
c4h5n3o2 127.0382 C^N 129.0579 c4h12n4o 132.1012 CgH,^ 134.0845
c4h7n4o 127.0621 CiqH9 129.0705 C5H8O4 132.0422 С9Н10О 134.0732
c5h5no3 127.0269 130 c5h10no3 132.0661 c,h12n 134.0970
c5h7n2o2 127.0508 C,H4N3O3 130.0253 C5H12N2O2 132.0899 Ci0H|4 134.1096
QHcJSO 127.0746 c,h6n4o2 130.0491 C5H14N3O 132.1138 135
C5H„N4 127.0985 c4h4no4 130.0140 CsH16N4 132.1377 СзН7М2О4 135.0406
C6H7O3 127.0395 c4h6n2o3 130.0379 c6h4n4 132.0437 c3h9n3o3 135.0644
C6H9NO2 127.0634 c4h8n3o2 130.0617 C6H12O3 132.0786 C^HnN^ 135.0883
QHMN2O 127.0872 c4h10n4o 130.0856 CeH14NO2 132.1025 C4H9NO4 135.0532
C6H13N3 127.1111 C5H6O4 130.0266 c6h16n2o 132.1264 C4HuN2O3 135.0770
C7H„O2 127.0759 c5h8no3 130.0504 c7h9n, 132.0563 C4H|3N3O2 135.1009
QH^NO 127.0998 c5h10n2o2 130.0743 C?H16O2 132.1151 c5h3n4o 135.0308
C7H15N2 127.1236 c5h12n3o 130.0981 CBH6NO 132.0449 C5H„O4 135.0657
CeH15O 127.1123 c5h14n4 130.1220 c8h8n2 132.0688 CSHI3NO3 135.0896
q,h17n 127.1362 c6H10o3 130.0630 C,H8O 132.0575 c6h5n3o 135.0433
c,h19 127.1488 C6H12NO2 130.0868 C,H10N 132.0814 c6h7n4 135.0672
128 c6h14n2o 130.1107 Ci0Hi2 132.0939 c7h5no2 135.0320
C3H4N4O2 128.0335 C6H16N3 130.1346 133 C7H7N2O 135.0559
C4H4N2O3 128.0222 QH4N3 130.0406 C3H5N2O4 133.0249 135.0798
c4h^n3o2 128.0460 C7H,4O2 130.0994 c3h7n3o3 133.0488 Q,H7O2 135.0446
c4h8n4o 128.0699 CjH^NO 130.1233 C3H9N4O2 133.0726 СвНДЧО 135.0684
QH4O4 128.0109 130.1471 c4h7no4 133.0375 CgH„N2 135.0923
C5H,NO3 128.0348 C8H6N2 130.0532 c4h,n2o3 133.0614 C.HnO 135.0810
C5H8N2O2 128.0586 csH18o 130.1358 c4h„n3o2 133.0852 C<)H13N 135.1049
C;H10N3O 128.0825 c,h8n 130.0657 c4h13n4o 133.1091 CioH15 135.1174
C5H12N4 128.1063 CioHio 130.0783 CsH9O4 133.0501 136
QHgO3 128.0473 131 C5HnNO3 133.0739 C3H8N2O4 136.0484
QHkjNO, 128.0712 C3H3N2O4 131.0093 C5H13N2O2 133.0978 C3H10N3O3 136.0723
СбН^О 128.0950 CjH5N3O3 131.0331 c5h15n3o 133.1216 C3H12N4O2 136.0961
c6h14n3 128.1189 c,h7n4o2 131.0570 c6h5n4 133.0515 c4hI0no4 136.0610
С7Н12О2 128.0837 c4h5no4 131.0218 C6H13O3 133.0865 c4hI2n2o3 136.0848
C7H14NO 128.1076 c4h7n2o3 131.0457 C6H15NO2 133.1103 c5h2n3o2 136.0147
C7H16n2 128.1315 c4h„n3o2 131.0695 C7H,N2O 133.0402 c5h4n4o 136.0386
QH16o 128.1202 c4hun4o 131.0934 C7H7N3 133.0641 c5h]2o4 136.0735
CeHjgN 128.1440 C5H7O4 131.0344 CgH7NO 133.0528 CeH4N2O2 136.0273
C^o 128.1566 C5H9NO3 131.0583 c8h9n2 133.0767 C6H6N3O 136.0511
CioHg 128.0626 C5HuN2O2 131.0821 C,H,O 133.0653 QHgN, 136.0750
129 c5h13n3o 131.1060 C,HnN 133.0892 СДО, 136.0160
C3H3N3O, 129.0175 c5h15n4 131.1298 CioH13 133.1018 c7h6no2 136.0399
c3h5n4o2 129.0413 C6HIIO3 131.0708 134 C^HgNjO 136.0637
c4h5n2o3 129.0300 C6Hi3NO2 131.0947 C3H6N2O4 134.0328 С.НюИз 136.0876
c4h7n3o2 129.0539 c6h15n2o 131.1185 C3HgN3O3 134.0566 CgH8O2 136.0524
c4h9n4o 129.0777 c6hI7n3 131.1424 QHioN^ 134.0805 CgHI0NO 136.0763
QH5O4 129.0187 c7h5n3 131.0484 C4H8NO4 134.0453 CgHI2N2 136.1001
C5H7NO3 129.0426 c7hi5o2 131.1072 C4H1cN2O3 134.0692 C9H!2O 136.0888
C5H,N2O2 129.0664 c,h17no 131.1311 C4H12N3O2 134.0930 c9h14n 136.1127
C5HuN3O 129.0903 cbh7n2 131.0610 c4h14n4o 134.1169 QoHjg 136.1253
CjH13n4 129.1142 C,H7O 131.0497 C5H10O4 134.0579 137
Q,h9o3 129.0552 C9H9N 131.0736 C5H12NO3 134.0817 137.0563
C6H„NO2 129.0790 CioHn 131.0861 c5h14n2o2 134.1056 QHhNjO, 137.0801
QH13N2O 129.1029 132 c6h4n3o 134.0355 c4h„no4 137.0688
C6H13N3 129.1267 C3h4N2O4 132.0171 QH6N4 134.0594 c5h3n3o2 137.0226
C?H13O2 129.0916 C,H6N3O3 132.0410 C6H14O3 134.0943 c5h5n4o 137.0464
C7H15NO 129.1154 c3h8n4o2 132.0648 C7H6N2O 134.0480 137.0351
C?H17N2 129.1393 c4h6no4 132.0297 C7H8N3 134.0719 c6h7n3o 137.0590
CgH17O 129.1280 c4h8n2o3 132.0535 C8H6O2 134.0368 c6h9n4 137.0829
68
?. Масс<пектрометрия
Приложение А (продолжение)
M M M M
С7Н5О3 137.0238 c6h8n2o2 140.0586 CKHI6NO 142.1233 CW4, 144.0688
c7h7no7 137.0477 QH.oNjO 140.0825 Q,h)8n2 142.1471 С^НгнО 144.1515
QH^O 137.0715 c6h12n4 140.1063 g,h6n2 142.0532 C|(>H8O 144.0575
QHhNj 137.0954 C,H,O3 140.0473 G.H.kO 142.1358 C„,HION 144.0814
QH9O, 137.0603 C7H,„NO2 140.0712 c,h2(,n 142.1597 CUH12 144.0939
QHuNO 137.0841 c7h12n2o 140.0950 cI(,h8n 142.0657 145
QHI3N2 137.1080 c,h14n3 140.1189 c1((H22 142.1722 C4H,N2O4 145.0249
g,h13o 137.0967 QH12O2 140.0837 C||H|<> 142.0783 c4h7n3o3 145.0488
C,HISN 137.1205 c8hI4no 140.1076 143 c4h9n4o2 145.0726
C10H17 137.1331 c8hI6n2 140.1315 C4H3N2O4 143.0093 c,h7no4 145.0375
138 QH16O 140.1202 c4h5n,o, 143.0331 QH^O, 145.0614
c3h10n2o4 138.0641 c,hI8n 140.1440 c4h7n4o2 143.0570 c5h„n,o2 145.0852
c5h4n3o2 138.0304 CI()H6N 140.0501 c,h,no4 143.0218 c5h13n4o 145.1091
CjH^O 138.0542 Ci()H2o 140.1566 c,h7n2o3 143.0457 C6H9O4 145.0501
c6h4no, 138.0191 c„H8 140.0626 QH9N3O2 143.0695 C6HHNO3 145.0739
c6h6n2o2 138.0429 141 C,H,,N4O 143.0934 СбН,3К2О2 145.0978
csh8n3o 138.0668 C4H3N3O. 141.0175 q,h7o4 143.0344 C6H„N3O 145.1216
c6h10n4 138.0907 C4H5N4O. 141.0413 c6h9no3 143.0583 c6hI7n4 145.1455
C7H6O, 138.0317 c5h3no4 141.0062 c6h„n2o2 143.0821 c7h5n4 145.0515
C7HSNO2 138.0555 c5h5n2o3 141.0300 c6hI3n3o 143.1060 C7H13O3 145.0865
QHkjNjO 138.0794 c5h7n3o2 141.0539 QHL5N4 143.1298 C7H,,NO2 145.1103
c7h12n3 138.1032 c,h9n4o 141.0777 C,HtlO3 143.0708 QHnNjO 145.1342
СнНщОг 138.0681 СбН5О4 141.0187 c7hI3no2 143.0947 145.1580
QH,2NO 138.0919 CJ^NO, 141.0426 C7H,5N2O 143.1185 ChH5N2O 145.0402
C«H14N2 138.1158 C6H9N2O, 141.0664 c7h,7n3 143.1424 c8h7n3 145.0641
C,H14O 138.1045 QHhN.,0 141.0903 CkH15O2 143.1072 C8H)7O2 145.1229
C,H16N 138.1284 c6h13n4 141.1142 QHI7NO 143.1311 C8H|9NO 145.1467
C10H18 138.1409 C/HijO., 141.0552 QHI9N2 143.1549 C)H7NO 145.0528
139 QHhNO, 141.0790 g,h7n2 143.0610 c,h9n2 145.0767
C4H3N4O2 139.0257 C7H„N2O 141.1029 g,hI9o 143.1436 C((,H9O 145.0653
c5h,n2o3 139.0144 c7h15n, 141.1267 qh21n 143.1675 CihHhN 145.0892
c5h5n3o2 139.0382 c8h13o2 141.0916 Cl0H7O 143.0497 СцН|3 145.1018
c5h7n4o 139.0621 ChHi5NO 141.1154 c,„h9n 143.0736 146
c6h5no3 139.0269 c8hI7n2 141.1393 C„H„ 143.0861 C4H6N2O4 146.0328
C6H7N2O2 139.0508 C9HI7O 141.1280 144 c4h8n,o, 146.0566
C6h9n3O 139.0747 CoHI9N 141.1519 C4H4N2O4 144.0171 c4hI0n4o2 146.0805
QH»n4 139.0985 cI()h7n 141.0579 c4h6n,o, 144.0410 c,h8no4 146.0453
C7H7O3 139.0395 C1(,H2, 141.1644 c4h8n4o2 144.0648 c,h1(,n2o, 146.0692
C7H9NO2 139.0634 C„H9 141.0705 c5h6no4 144.0297 c,hI2n3o2 146.0930
C7HnN2O 139.0872 142 C5H8n2o3 144.0535 C,H14N4O 146.1169
C7H13N3 139.1111 c4h4n3o3 142.0253 c5h,„n3o2 144.0774 СбН|()О4 146.0579
СцНнО2 139.0759 c4h6n4o2 142.0491 c5hI2n4o 144.1012 СбН|2НО3 146.0817
QH.jNO 139.0998 c5h4no4 142.0140 c6H8o4 144.0422 СбН14М2О2 146.1056
c8h15n2 139.1236 c5h6n2o3 142.0379 C6Hh.NO., 144.0661 СбН16Н,О 146.1295
c,h3n2 139.0297 C,H8N,O2 142.0617 c6h12n2o2 144.0899 QHeN, 146.0594
C9H15O 139.1123 c5hI0n4o 142.0856 СбН|4Н,О 144.1138 С7Н14О3 146.0943
c,h17n 139.1362 c6H6o4 142.0266 C6H,6N4 144.1377 C^NO, 146.1182
CioH|9 139.1488 CeH8NO3 142.0504 C?H |2O, 144.0786 146.1420
C„H7 139.0548 G,h1un2o2 142.0743 C7HI4NO2 144.1025 c^o, 146.0003
140 C6H12N3O 142.0981 c7h16n2o 144.1264 CkH6N2O 146.0480
C4H4N4O2 140.0335 c6hI4n4 142.1220 c7h,8n, 144.1502 c8h8n3 146.0719
c5h4n2o3 140.0222 C7H|(1O3 142.0630 CKH6N, 144.0563 C8H|8O2 146.1307
c5h6n3o2 140.0460 C7Hi2NO2 142.0868 C8H .eO2 144.1151 С.НбО, 146.0368
c5h„n4o 140.0699 C7H,4N2O 142.1107 C8H18NO 144.1389 CHtNO 146.0606
C6H4O4 140.0109 C7HI6N, 142.1346 chh2„n2 144.1628 G,H1(>N2 146.0845
C6H6NO3 140.0348 CXHI4O2 142.0994 C4H6NO 144.0449 C|(>H|()O 146.0732
Приложение А
69
Приложение А (продолжение)
FM FM FM FM
c10hI2n 146.0970 C5Hi5N3O2 149.1165 C9HI3NO 151.0998 c6h10n4o 154.0856
С,.нм 146.1096 C6H5N4O 149.0464 c,h15n2 151.1236 С,Н6О4 154.0266
147 QH13O4 149.0814 CiiJijsO 151.1123 QHgNO., 154.0504
C4H7N2O4 147.0406 C6HI5NO3 149.1052 cioh17n 151.1362 C7H10N2O2 154.0743
C4H9N3O3 147.0644 С7Н5М2О2 149.0351 CtlH|9 151.1488 154.0981
C4HuN4O2 147.0883 QH.NjO 149.0590 152 154.1220
C5H9NO4 147.0532 сдд4 149.0829 C4H|2N2O4 152.0797 CgHi<>O3 154.0630
C5HnN2O3 147.0770 СвН5Оз 149.0238 c5h4n4o2 152.0335 CgH12NO2 154.0868
QH13N3O2 147.1009 QH7NO2 149.0477 c6h4n2o_, 152.0222 CgHI4N2O 154.1107
C5H15N4O 147.1247 CgH,N2O 149.0715 QH6N3O2 152.0460 C«H16N3 154.1346
СбНпО4 147.0657 CgHnN3 149.0954 C6H8N4O 152.0699 СчН14О2 154.0994
C6H13NO3 147.0896 СД9О2 149.0603 C,H6NO3 ' 152.0348 c9h16no 154.1233
C6HI5N2O2 147.1134 СдНцЫО 149.0841 c7h8n2o2 152.0586 C.H.gN, 154.1471
QhI7n3o 147.1373 CoH13N2 149.1080 C7H1(IN3O 152.0825 Ск)Н18О 154.1358
C7H5N3O 147.0433 О<Д1зО 149.0967 QHI2N4 152.1063 Ck)H20N 154.1597
QH7n4 147.0672 cioh15n 149.1205 CgHgO3 152.0473 C„HgN 154.0657
QHI5O3 147.1021 OjH17 149.1331 CgHl0NO2 152.0712 СцН22 154.1722
C7H17NO2 147.1260 150 CgH12N2O 152.0950 C12H|(1 154.0783
CgH5NO2 147.0320 C4H10N2O4 150.0641 CgHl4N3 152.1189 155
CgH7N2O 147.0559 C4H12N3O3 150.0879 C9HI2O2 152.0837 C5H3N2O4 155.0093
CgH,N3 147.0798 c4h14n4o2 150.1118 g,h14no 152.1076 C5H5n3o3 155.0331
C,H7O2 147.0446 c5h12no4 150.0766 Qh16n2 152.1315 c,h7n4o2 155.0570
сддчо 147.0684 c5h14n2o3 150.1005 c10h16o 152.1202 c6h5no4 155.0218
c^„N2 147.0923 c6h4n3o2 150.0304 C10H,gN 152.1440 c6h7n2o3 155.0457
СюНиО 147.0810 C6h<,n4o 150.0542 C„HfiN 152.0501 c6h9n3o2 155.0695
C|oH13N 147.1049 QH14O4 150.0892 СцН20 152.1566 QHnN4O 155.0934
c„H15 147.1174 QH6N2O2 150.0429 C12Hg 152.0626 155.0344
148 C7H8N3O 150.0668 153 C^HoNOj 155.0583
C4HgN2O4 148.0484 QHI0N4 150.0907 C5H3N3O3 153.0175 155.0821
C4HI0N3O3 148.0723 CgH6O3 150.0317 C5H5N4O2 153.0413 c7hI3n3o 155.1060
C4HI2N4O2 148.0961 CgH8NO2 150.0555 QHsN2O3 153.0300 CgH„O3 155.0708
C5H10NO4 148.0610 CgH10N2O 150.0794 c6h7n3o2 153.0539 C8H13NO2 155.0947
QH12N2O3 148.0849 CgHI2N3 150.1032 CftH9N4O 153.0777 CgHl5N2O 155.1185
C5H,6N4O 148.1325 СэНюОг 150.0681 C7H5O4 153.0187 CgHl7N3 155.1424
C6H4N4O 148.0386 C,Hi2NO 150.0919 C7H7NO3 153.0426 С^Н^Ог 155.1072
QH12O4 148.0735 QH14n2 150.1158 c7h9n2o2 153.0664 G)H17NO 155.1311
C6H,4NO3 148.0974 QoHi40 150.1045 QHhNjO 153.0903 c,h19n2 155.1549
QH16N2O2 148.1213 CI0H16N 150.1284 153.1142 c10h7n2 155.0610
c7h6n3o 148.0511 СцН18 150.1409 CgH9O3 153.0552 C10H|9O 155.1436
c7h8n4 148.0750 151 CgH„NO2 153.0790 cI()h2In 155.1675
C7H,6O3 148.1100 C4H„N2O4 151.0719 C«H13N2O 153.1029 c„H7o 155.0497
c8h6no2 148.0399 C4H13N3O3 151.0958 C8H15N3 153.1267 CnHsN 155.0736
CgH8N2O 148.0637 C5H3N4O2 151.0257 C9H13O2 153.0916 C„H23 155.1801
CgHI0N3 148.0876 c5h13no4 151.0845 QHijNO 153.1154 cI2H„ 155.0861
QHgOj 148.0524 c6h3n2o3 151.0144 C»H„N2 153.1393 156
QH.qNO 148.0763 QH5N3O2 151.0382 CtoHl70 153.1280 156.0171
QH12n2 148.1001 c6h7n4o 151.0621 c10hI9n 153.1519 CsH^O, 156.0410
C10H|2O 148.0888 QHjNOj 151.0269 c„h7n 153.0579 C,HgN4O2 156.0648
QoHI4N 148.1127 151.0508 ChH2i 153.1644 C6H6NO4 156.0297
CnH16 148.1253 CjHgNjO 151.0746 CI2H9 153.0705 QHgNA, 156.0535
149 QHnN4 151.0985 154 CgH^o, 156.0774
C4H9N2O4 149.0563 CgH7O3 151.0395 C5H4N3O3 154.0253 cftH12N4o 156.1012
C4H„N3O3 149.0801 CgHNOj 151.0634 QHgN.O, 154.0491 QHgO, 156.0422
C4HI3N4O2 149.1040 CgHuN2O 151.0872 CftH4NO4 154.0140 QHh.no, 156.0661
C5H„NO4 149.0688 CgHI3N3 151.1111 c6h6n2o3 154.0379 c7hI2n2o2 156.0899
CJit3N,O, 149.0927 C,H„O7 151.0759 QHxN.O, 154.0617 c,h14n,o 156.1138
70
/. Масс-спектрометрия
Приложение А (продолжение)
M M M M
QH16N4 156.1377 158.1056 C7H14NO3 160.0974 c„hI0n4 162.0907
156.0786 QHjeNjO 158.1295 c7h16n2o2 160.1213 СвН18О3 162.1256
C8H14NO2 156.1025 C7H,8N4 158.1533 С,Н18М3О 160.1451 C,H6O3 162.0317
c8h16n2o 156.1264 QH6N4 158.0594 160.1690 С,Н8МО2 162.0555
QH18N3 156.1502 C8H14O3 158.0943 QW 160.0511 CsHuNjO 162.0794
156.0563 C8H16NO2 158.1182 C8H8N4 160.0750 c,h12n3 162.1032
C,Hl6O2 156.1151 crh18n2o 158.1420 C«H16O3 160.1100 QoH|o02 162.0681
C9H18NO 156.1389 C8H2ON3 158.1659 C8H18NO2 160.1338 c10hi2no 162.0919
C,H20N2 156.1628 c,h6n2o 158.0480 C8h20n2o 160.1577 CieH14N2 162.1158
QoHNO 156.0449 c,h8n3 158.0719 c9h6no2 160.0399 C„H14O 162.1045
c10h8n2 156.0688 158.1307 C9H8N2O 160.0637 CnH16N 162.1284
CioH200 156.1515 qh20no 158.1546 C^oN, 160.0876 Ci2H18 162.1409
QoH^N 156.1753 CioH6o2 158.0368 СдНгоОг 160.1464 163
cnH8o 156.0575 c10h8no 158.0606 c.0H8o2 160.0524 163.0719
CUHION 156.0814 CioHioN2 158.0845 CI0H10NO 160.0763 C5Hl3N3O3 163.0958
Q1H24 156.1879 C1()H22O 158.1672 CioH12N2 160.1001 C5H15n4o2 163.1196
cl2H12 156.0939 СцН10О 158.0732 СцН12О 160.0888 CgHI3NO4 163.0845
157 CnH12N 158.0970 CnH14N 160.1127 CgH15N2O3 163.1083
c5h5n2o4 157.0249 C12H14 158.1096 C12H16 160.1253 C6H17N3O2 163.1322
c5h7n3o3 157.0488 159 161 c,h5n3o2 163.0382
c5h9n4o2 157.0726 c5h7n2o4 159.0406 C5H9N2O4 161.0563 c,h7n4o 163.0621
c6h7no4 157.0375 c5h,n3o3 159.0644 C5H„N3O3 161.0801 163.0970
C6H9N2O3 157.0614 C5HuN4O2 159.0883 C5H13N4O2 161.1040 QHnNOj 163.1209
С6НцМ3О2 157.0852 C6H9NO4 159.0532 CeHnNO4 161.0688 QHsNOj 163.0269
c6hI3n4o 157.1091 C6HnN2O3 159.0770 C6H13N2O3 161.0927 163.0508
слд 157.0501 QH13N3O2 159.1009 C6HI5N3O2 161.1165 CeH9N3O 163.0746
GHuNCb 157.0739 C6H15N4O 159.1247 C6H17N4O 161.1404 QHhN, 163.0985
C7H13N2O2 157.0978 QH„O« 159.0657 C7HSN4O 161.0464 C,H7O3 163.0395
c7h15n3o 157.1216 C7H13NO3 159.0896 CgH5N2O2 161.0351 CjHeNOj 163.0634
QHhN, 157.1455 159.1134 C8H7N3O 161.0590 C.HnN.O 163.0872
C8H5N4 157.0515 C7H17N3O 159.1373 c8h9n4 161.0829 C,H13N3 163.1111
C8H13O3 157.0865 C8H,N3O 159.0433 CeH17O3 161.1178 CjoHnOa 163.0759
CgH15NO2 157.1103 q,h7n4 159.0672 CgHI9NO2 161.1416 C10H13NO 163.0998
qh„n2o 157.1342 CgH15O3 159.1021 С,Н5О3 161.0238 C1OHI5N2 163.1236
QH19N3 157.1580 CRH17NO2 159.1260 G>H7NO2 161.0477 cnHI5o 163.1123
C,H5N2O 157.0402 C8H19N2O 159.1498 c,h,n2o 161.0715 c„h17n 163.1362
c9h7n3 157.0641 c8h21n3 159.1737 C,HnN3 161.0954 C)2Hi9 163.1488
CeH17O2 157.1229 c.,h5no2 159.0320 C10H9O2 161.0603 164
c9h19no 157.1467 c9h7n2o 159.0559 C10HnNO 161.0841 c5H12N2o4 164.0797
c,h21n2 157.1706 c>h9n3 159.0798 cioh13n2 161.1080 C5H14n3o3 164.1036
c10h7no 157.0528 C9H|9O2 159.1385 CnH13O 161.0967 c5hI6n4o2 164.1275
C,oH9N2 157.0767 GJl^NO 159.1624 c„h15n 161.1205 c6h4n4o2 164.0335
C10H21O 157.1593 C|()H7O2 159.0446 c12h17 161.1331 QH14NO4 164.0923
C,oH23N 157.1832 cI0h9no 159.0684 162 c6h16n2o3 164.1162
CnH9O 157.0653 C10HnN2 159.0923 C5HI0N2O4 162.0641 c7h6n3o2 164.0460
CnHnN 157.0892 СнНпО 159.0810 c5h12n3o3 162.0879 С,н8м4о 164.0699
C12H13 157.1018 C„HI3N 159.1049 c5h14n4o2 162.1118 С,н|6о4 164.1049
158 c12Ht5 159.1174 c6h,2no4 162.0766 CgHgNO, 164.0348
C5H6N2O4 158.0328 160 c6h14n2o3 162.1005 QHgNA 164.0586
c5h8n3o3 158.0566 c5h8n2o4 160.0484 c6h16n3o2 162.1244 C8H10N3O 164.0825
c5h10n4o2 158.0805 Qh10n3o3 160.0723 c6h18n4o 162.1482 C8H12n4 164.1063
c6h8no4 158.0453 C5Hi2N4O2 160.0961 c7h6n4o 162.0542 CgHgOs 164.0473
C6H10N2O3 158.0692 QH10NO4 160.0610 С.НиО, 162.0892 c9h10no2 164.0712
C^H12N3O2 158.0930 c6h12n2o3 160.0848 QH^NO, 162.1131 164.0950
c6h14n4o 158.1169 c6h14n3o2 160.1087 C?H18N2O2 162.1369 c,h14n3 164.1189
C7HI0O4 158.0579 c6h16n4o 160.1325 c„h6n2o2 162.0429 c10H12o2 164.0837
C7H12NO3 158.0817 c7h12o4 160.0735 CgHeNjO 162.0668 c10h14no 164.1076
Приложение А
71
Приложение А (продолжение)
M M M M
c10h16n2 164.1315 C7H7N2O3 167.0457 C8HhNO3 169.0739 c7hI3n3o2 171.1009
спн16о 164.1202 c7h9n3o2 167.0695 c8h13n2o2 169.0978 c7h,5n4o 171.1247
C„H18N 164.1440 QHhN.O 167.0934 ChH15N3O 169.1216 C8HUO4 171.0657
Q2H20 164.1566 CrH7O4 167.0344 169.1455 c8h13no3 171.0896
165 CkH9NO, 167.0583 CyHl3O3 169.0865 ChH15N2O2 171.1134
C5H13N2O4 165.0876 167.0821 CyHl5NO2 169.1103 c8h,7n3o 171.1373
c5h15n3o3 165.1114 CKHI3N3O 167.1060 CyH17N2O 169.1342 Q.H.yNy 171.1611
c6h5n4o2 165.0413 CsHi5N4 167.1298 c9h19n3 169.1580 CyH,N3O 171.0433
c6h15no4 165.1001 CyHnO3 167.0708 C1oH7N3 169.0641 CyH7N4 171.0672
c,h5n2o3 165.0300 c,h13no2 167.0947 C|()H17O2 169.1229 CyH|5O3 171.1021
СуН7м3о2 165.0539 QHijNjO 167.1185 ClflHl9NO 169.1467 C9H17NO2 171.1260
QH^O 165.0777 CyH17N3 167.1424 C|OH2|N2 169.1706 C9H|yN2O 171.1498
CKHSO4 165.0187 QoHis02 167.1072 c„h7no 169.0528 CyH21N3 171.1737
C8H7NO3 165.0426 c10h]7no 167.1311 c„h9n2 169.0767 c„,h7n2o 171.0559
CaHQN7O? 165.0664 CwH19N2 167.1549 c„H21o 169.1593 CI()H9N3 171.0798
CsHuN3O 165.0903 c„h7n2 167.0610 c„h23n 169.1832 C|()HI9O2 171.1385
CsH13N4 165.1142 СцН19О 167.1436 CI2H9O 169.0653 c1()h21no 171.1624
C9H9O3 165.0552 CltH21N 167.1675 CI2H„N 169.0892 Ck)H23N2 171.1863
c,h„no2 165.0790 cI2h9n 167.0736 c,2h25 169.1957 C„H7O2 171.0446
QH13N2O 165.1029 c12h23 167.1801 C13H13 169.1018 CnHyNO 171.0684
c,h15n3 165.1267 c13Hn 167.0861 170 CnHnN, 171.0923
C10H13O2 165.0916 168 C6H6N2O4 170.0328 C„H23O 171.1750
C10Ht5NO 165.1154 WA 168.0171 c6h8n3o3 170.0566 ChH^N 171.1988
cI0h17n2 165.1393 c6h6n3o3 168.0410 c6hI0n4o2 170.0805 C|2HUO 171.0810
c„H17o 165.1280 C6H8N4O2 168.0648 c7h8no4 170.0453 c,2h13n 171.1049
cnH19N 165.1519 C7H6NO4 168.0297 c7h10n2o3 170.0692 CI3H15 171.1174
c12h7n 165.0579 C7H8N2O3 168.0535 C7H|2N3O2 170.0930 172
CI2H21 165.1644 CyH10N3O2 168.0774 c,h14n4o 170.1169 c6h8n2o4 172.0484
cI3H9 165.0705 c7hi2n4o 168.1012 CKH,0O4 170.0579 C6H1(IN3O3 172.0723
166 C8H8O4 168.0422 Q,HI2NO3 170.0817 CaH12N4O2 172.0961
C5H14N2O4 166.0954 c8h10no3 168.0661 C„H14N2O2 170.1056 QH.oNOy 172.0610
QH4n3o3 166.0253 168.0899 CkHI6N3O 170.1295 C7HI2N2O3 172.0848
c6h6n4o2 166.0491 C8H14N,O 168.1138 QHwn4 170.1533 СуНиад 172.1087
CyH6N2O3 166.0379 168.1377 c,h6n4 170.0594 QH.eNyO 172.1325
cyH8N3o2 166.0617 CyH|2O3 168.0786 CyH14O3 170.0943 C8H12O4 172.0735
CyHI0N4O 166.0856 c,h14no2 168.1025 c,h16no2 170.1182 C8H14NO3 172.0974
QH6O4 166.0266 c,hI6n2o 168.1264 CyH18N2O 170.1420 c8h16n2o2 172.1213
c8h«no3 166.0504 QH1#N, 168.1502 CyH2UN3 170.1659 c8h18n3o 172.1451
CgH|oN202 166.0743 C10H,6O2 168.1151 C10H6N2O 170.0480 QH^Ny 172.1690
QH12N3O 166.0981 c10h18no 168.1389 C1(iH8N3 170.0719 CyH6N3O 172.0511
QH14N4 166.1220 CioH2oN2 168.1628 CI0HihO2 170.1307 CyH8N4 172.0750
CyHI0O3 166.0630 C„H8N2 168.0688 c10h2(1no 170.1546 CyH|AO3 172.1100
c,h12no2 166.0868 Снн2по 168.1515 c10h22n2 170.1784 CyHl8NO2 172.1338
c,h14n2o 166.1107 c„h22n 168.1753 c„h8no 170.0606 CyH2(,N2O 172.1577
C,H16N3 166.1346 C12H8O 168.0575 ChH10N2 170.0845 CyH22N3 172.1815
C10Hl4O2 166.0994 c12hI0n 168.0814 C„H22O 170.1671 c1(,h6no2 172.0399
C10H16NO 166.1233 C12H24 168.1879 c„h24n 170.1910 c1()h8n2o 172.0637
Ch>HI8N2 166.1471 C13H12 168.0939 Ci2HwO 170.0732 Ck>H|()N3 172.0876
ChHihO 166.1358 169 C12H12N 170.0970 CjoHyoOy 172.1464
c„h20n 166.1597 C6H5N2O4 169.0249 С12Н26 170.2036 C.pH^NO 172.1702
c12h8n 166.0657 c6h7n3o3 169.0488 C13H14 170.1096 Ck)H24N2 172.1941
C12H22 166.1722 c6h9n4o2 169.0726 171 C||H8O2 172.0524
С13Н10 166.0783 C7H7NO4 169.0375 C6H7N2O4 171.0406 CnH10NO 172.0763
167 C7H9N2O3 169.0614 C6HyN3O3 171.0644 c„h12n2 172.1001
QHjNjO, 167.0331 CyHnN3O2 169.0852 CaH„N4O2 171.0883 C„H24O 172.1828
c6h7n4o2 167.0570 C7H,3N4O 169.1091 CyHyNOy 171.0532 C|2H)2O 172.0888
C7HsNO4 167.0218 cbh9o4 169.0501 C7H„N2O3 171.0770 cI2h14n 172.1127
72
1. Маа-а1ектрометрия
Приложение А (продолжение)
M M M M
С,зН16 172.1253 CnH12NO 174.0919 CnH12O2 176.0837 CI2HmN 178.1597
173 cuh14n2 174.1158 c„h14no 176.1076 CI3H8N 178.0657
c6h9n2o4 173.0563 Ci2H14O 174.1045 ChH16N2 176.1315 C|3H22 178.1722
C6HItN3O3 173.0801 Cl2H16N 174.1284 CuHigO 176.1202 СиНю 178.0783
QHI3N4O2 173.1040 С|зН18 174.1409 C,2Hl8N 176.1440 179
C7H,,NO4 173.0688 175 C13H2O 176.1566 C6HI5N2O4 179.1032
C7HI3N2O3 173.0927 C6HhN2O4 175.0719 177 c6hI7n3o3 179.1271
C7H15N3O2 173.1165 CgHI3N3O3 175.0958 CgH13N2O4 177.0876 C^NjO-, 179.0331
C7H17N4O 173.1404 C6H15N4O2 175.1196 C6HI5N3O3 177.1114 QJWA 179.0570
C8H13O4 173.0814 C7HI3NO4 175.0845 c6h17n4o2 177.1353 QHpNO, 179.1158
CgH15NO3 173.1052 C7H15N2O3 175.1083 СДОД 177.0413 CgH5NO4 179.0218
C8H17N2O2 173.1291 C7H17N3O2 175.1322 C7H)5NO4 177.1001 CgH7N2O3 179.0457
c8h19n3o 173.1529 C7H19N4O 175.1560 CzH.^O, 177.1240 CgH9N3O2 179.0695
c«h21n4 173.1768 CgH7N4O 175.0621 C7HI9N3O2 177.1478 CgHnN4O 179.0934
C9H7N3O 173.0590 CgH15O4 175.0970 CgH5N2O3 177.0300 179.0344
ЭДД 173.0829 CgHl7NO3 175.1209 c«h7n3o2 177.0539 G>H9NO3 179.0583
c.H17o3 173.1178 C8H19N2O2 175.1447 QH9N4O 177.0777 179.0821
QH^NO, 173.1416 CgH21N3O 175.1686 CgH17O4 177.1127 C,H13N3O 179.1060
C9H21N2O 173.1655 C9H5NO3 175.0269 CgHt9NO3 177.1365 179.1298
CtoHs03 173.0238 175.0508 C9H7NO3 177.0426 СюНцО3 179.0708
C10H7NO2 173.0477 C9H9N3O 175.0746 c9h9n2o2 177.0664 CtoH|3N02 179.0947
CioHqN^O 173.0715 175.0985 QHhNjO 177.0903 Ci0H15N2O 179.1185
Cl0HnN3 173.0954 C9H19O3 175.1334 C»HI3N4 177.1142 c,„hI7n3 179.1424
CioH2102 173.1542 c9h21no2 175.1573 C10H9O3 177.0552 CiiHi5O2 179.1072
c10h23no 173.1781 СщН7О3 175.0395 C1(,H„NO2 177.0790 c„hi7no 179.1311
C„H9O2 173.0603 C10H9NO2 175.0634 CioHj3N20 177.1029 c„hI9n2 179.1549
СцНиЫО 173.0841 C10HtlN2O 175.0872 cioh15n3 177.1267 С1гН|9О 179.1436
c„h13n2 173.1080 CI0H13N3 175.1111 C„H13O2 177.0916 c12h21n 179.1675
C)2H13O 173.0967 СцНцОг 175.0759 c„h15no 177.1154 CI3H,N 179.0736
C12Ht5NO2 173.1205 CUH13NO 175.0998 c„h17n2 177.1393 C13H23 179.1801
CI3H17 173.1331 CnH15N2 175.1236 C12H17O 177.1280 C14H„ 179.0861
174 Ci2H]5O 175.1123 c,2hi9n 177.1519 180
C6H|0N2O4 174.0641 c12h,7n 175.1362 C13H21 177.1644 C6Hl6N2O4 180.1111
C6H12N3O3 174.0879 C13H3O 175.0184 178 C7H6n3o3 180.0410
c6h14n4o2 174.1118 C|3H19 175.1488 C6Hl4N2O4 178.0954 QHgbLA 180.0648
C7H12NO4 174.0766 176 C6H16N3O3 178.1193 CgHgNO, 180.0297
гинкго, 174.1005 он12к2о4 176.0797 C6HI8N4O2 178.1431 CgH8N2O3 180.0535
C7H16N3O2 174.1244 C6H14N3O3 176.1036 C7H6N4O2 178.0491 CgHI0N3O2 180.0774
C7HI8N4O 174.1482 c6h16n4o2 176.1275 C7H,6NO4 178.1080 CgH12N4O 180.1012
C7HI6N4O 174.1244 C7H14NO4 176.0923 C7Hi8N2O3 178.1318 C,HgO4 180.0422
CgH6N4O 174.0542 C7H16N2O3 176.1162 Q,h6n2o3 178.0379 C,HI0NO3 180.0661
C8H14O4 174.0892 C7H18N3O2 176.1400 C8H8N3O2 178.0617 C9Hj2N2O2 180.0899
C«H1(,NO3 174.1131 QH^N.O 176.1639 C8H10N4O 178.0856 GJHuNjO 180.1138
CgH18N2O2 174.1369 CgH6N3O2 176.0460 CgHl8O4 178.1205 c,h16n4 180.1377
CgH^NgO 174.1608 CgHgN4O 176.0699 QHgO, 178.0266 C10H|2O3 180.0786
CgH22N4 174.1846 CgHt6O4 176.1049 C,HgNO3 178.0504 c10h14no2 180.1025
C9H6N2O2 174.0429 CgHI8NO3 176.1287 C9H10N2O2 178.0743 CioHi6N20 180.1264
c,h10n4 174.0907 CgH20N2O2 176.1526 c,hI2n3o 178.0981 CioHI8N3 180.1502
С^Н^Оз 174.1256 C,H6NO3 176.0348 g,h14n4 178.1220 СцН)6О2 180.1151
CJlzoNOj 174.1495 . C9H8N2O2 176.0586 СюН10О3 178.0630 C„H,gNO 180.1389
c9h22n2o 174.1733 QH.oNjO 176.0825 Ci0HI2NO2 178.0868 CnH^N, 180.1628
C1(1H6O3 174.0317 c9h]2n4 176.1063 C10Hi4N2O 178.1107 C12H8N2 180.0688
CioH8N02 174.0555 C9H20O3 176.1413 Cl0H16N3 178.1346 C12H20O 180.1515
CioH(0N20 174.0794 C10HgO3 176.0473 CtiHi4O2 178.0994 Ci2H22N 180.1753
QoH^N, 174.1032 Ck)H10NO2 176.0712 C„H16NO 178.1233 C,3HgO 180.0575
c.oH22o2 174.1620 CioH12N20 176.0950 CiiH18N2 178.1471 C,3HI0N 180.0814
cnH10o2 174.0681 C,oH,4N3 176.1189 c12H18o 178.1358 С,3Н24 180.1879
Приложение А
73
Приложение А (продолжение)
M M м м
^14^12 180.0939 c13h12n 182.0970 CuH22NO 184.1702 cI0h8n3o 186.0668
181 Q3H26 182.2036 CnH24H2 184.1941 CioHtoN, 186.0907
181.0249 C14H14 182.1096 C12HsO2 184.0524 QoH|803 186.1256
с7н7н3о3 181.0488 183 C12HI0HO 184.0763 С1оНгоК02 186.1495
СДДА 181.0726 C7H7N2O4 183.0406 C12Ht2H2 184.1001 CjpH^b^O 186.1733
c8h7no. 181.0375 одад 183.0644 C12H24O 184.1828 C\oH24N3 186.1972
c8h9n2o3 181.0614 C7HnN4O2 183.0883 Сдан 184.2067 CuH8NO2 186.0555
qh„n3o2 181.0852 сдаю. 183.0532 CI3HI2O 184.0888 ChH10N2O 186.0794
csh13n4o 181.1091 Q,HuN2O3 183.0770 c13h14n 184.1127 cuh12n3 186.1032
сда4 181.0501 CsH13N3O2 183.1009 184.2192 СцН^Ог 186.1620
<^H„NO3 181.0739 CsH15N4O 183.1247 cl4H16 184.1253 CnH24NO 186.1859
C9H13N2O2 181.0978 сдао. 183.0657 185 C\iH26N2 186.2098
C,H15N3O 181.1216 C,Hl3NO3 183.0896 C7H9N2O4 185.0563 Ci2H10O2 186.0681
сдан. 181.1455 183.1134 ' сдан3о3 185.0801 c12h12no 186.0919
Q0H13O3 181.0865 ОДЛО 183.1373 сдан,о2 185.1040 Ci2H14N2 186.1158
c10h15no2 181.1103 c,h19n4 183.1611 сдано. 185.0688 186.1985
C10H17N2O 181.1342 c10h7n4 183.0672 C8H13H2O3 185.0927 C„H14O 186.1045
C10H19N3 181.1580 Ci0Hi5O3 183.1021 QH15N3o2 185.1165 CI3HI6N 186.1284
c„h7n3 181.0641 Ci0H17NO2 183.1260 сдан,о 185.1404 C14Hj8 186.1409
cnH17o2 181.1229 С10Н1Д2О 183.1498 С,Н13О, 185.0814 187
CnH19NO 181.1467 C10H21N3 183.1737 сдано, 185.1052 C7HnN2O4 187.0719
Q1H21N2 181.1706 CnH7N2O 183.0559 сдаи2о2 185.1291 187.0958
c„h7no 181.0528 CnH,N3 183.0798 QH^NjO 185.1529 c7h15n4o2 187.1196
c12h,n2 181.0767 CnH19O2 183.1385 c»h21n. 185.1768 сдамо. 187.0845
с.дао 181.1593 CnH21NO 183.1624 c10h7n3o 185.0590 Q,HI5N2O3 187.1083
c12h23n 181.1832 СцН23Н2 183.1863 сдан. 185.0829 C8H17N3O2 187.1322
C13H9O 181.0653 C12H7O2 183.0446 Ck)H17O3 185.1178 CgH^O 187.1560
C13HnN 181.0892 C12H9HO 183.0684 C-io^29N02 185.1416 C,H7N4O 187.0621
C13H25 181.1957 C12H„H2 183.0923 C10H21H2O 185.1655 CsHjjO, 187.0970
C14HI3 181.1018 СдаО 183.1750 C1OH23H3 185.1894 сдано, 187.1209
182 сдан 183.1988 CnH,H2O 185.0715 СдН^ЫгОг 187.1447
C7H6N2O4 182.0328 Ci3HtlO 183.0810 C„HnN3 185.0954 сдан3о 187.1686
c,h8h3o3 182.0566 C13H13N 183.1049 СцН21О2 185.1542 сдай. 187.1925
C7HlnH,O2 182.0805 183.2114 c,,h23no 185.1781 c,0h7n2o2 187.0508
QHgNO, 182.0453 сда5 183.1174 СцН^Нг 185.2019 Ct0H9N3O 187.0746
CjH10N2O3 182.0692 184 C12H9O2 185.0603 C10HnN4 187.0985
CjH12N3O2 182.0930 C7H8H2O, 184.0484 C12HnNO 185.0841 C\oH]903 187.1334
crh14n,o 182.1169 c7h1((h3o3 184.0723 C12H13H2 185.1080 CioH2iN02 187.1573
C9H10O4 182.0579 C7H12N4O2 184.0961 C^H^O 185.1906 C|qH23N2O 187.1811
C^NOj 182.0817 C8H1UHO4 184.0610 c12h27n 185.2145 C\oH2sN3 187.2050
C,H14H2O2 182.1056 CgH12N2O3 184.0848 C13H13O 185.0967 C„H7O3 187.0395
QH16N3O 182.1295 CsH14N3O2 184.1087 c13h15n 185.1205 CnHsNOj 187.0634
сдан. 182.1533 сдан,о 184.1325 с,да 185.1331 C„HnN2O 187.0872
C,oH6N4 182.0594 C»H12O4 184.0735 186 CnHI3N3 187.1111
C10H14O3 182.0943 c9h14no3 184.0974 C7H1qH2O, 186.0641 CUH23O2 187.1699
c10h16no2 182.1182 C,H16H2O2 184.1213 C7H12n3o3 186.0879 CnH25NO 187.1937
C|0H18H2O 182.1420 сдан3о 184.1451 c7h„n,o2 186.1118 Ci2Hu02 187.0759
QoHmNj 182.1659 c,h20n4 184.1690 сдано. 186.0766 c12h13no 187.0998
CnH8N3 182.0719 C10H6H3O 184.0511 CsH14N2O3 186.1005 C\2H1sN2 187.1236
C„H18O2 182.1307 c10h8n4 184.0750 сдан3о2 186.1244 C13Hl5O 187.1123
C.jH^O 182.1546 CioHieQj 184.1100 сдаМдО 186.1482 c13h17n 187.1362
C11H22N2 182.1784 C10Ht8HO2 184.1338 сда4о 186.0542 Ci4H19 187.1488
c12h8no 182.0606 CioH2qN20 184.1577 сдао. 186.0892 188
Ci2H10H2 182.0845 CjqH^H, 184.1815 C»HI6NO3 186.1131 C7H12N2O4 188.0797
C^H^O 182.1671 CnH8H2O 184.0637 СДиКзОз 186.1369 C7H14n3o3 188.1036
c12h24n 182.1910 CnHI0N3 184.0876 186.1608 сдан4о2 188.1275
QjHioO 182.0732 СцНиОг 184.1464 cI0h6n2o2 186.0429 сдано. 188.0923
74
7. Маа-спектрометрия
Приложение А (продолжение)
M M M M
CrH16N2O3 188.1162 190 cI4h9n 191.0736 C13H2IO 193.1593
QjH18N3O2 188.1400 C7H14N2O4 190.0954 cI4h23 191.1801 c13h23n 193.1832
C8H20N4O 188.1639 190.1193 c,5H„ 191.0861 CI4H9O 193.0653
C9H6N3O2 188.0460 C?HI8N4O2 190.1431 192 c14hun 193.0892
c,hsn4o 188.0699 c8h6n4o2 190.0491 192.1111 C14H25 193.1957
QH16O4 188.1049 c8hI6no4 190.1080 C7H18N3O3 192.1349 C15HI3 193.1018
C,H18NO3 188.1287 CkH18N2O3 190.1318 C7H20N4O2 192.1588 194
C9H2()N2O2 188.1526 c«h2()n3o2 190.1557 СДИД 192.0410 194.1267
C9H22N3O 188.1764 csh22n4o 190.1795 C8h8n4O2 192.0648 C8HbN2O4 194.0328
g,h24n4 188.2003 cwso. 190.0617 CbH18NO4 192.1236 c8h8n3o3 194.0566
C1()H8N2O2 188.0586 CJI.oN.O 190.0856 C8H2))N2O3 192.1475 C8H1()N4O2 194.0805
C]oHioN30 188.0825 C9H18O4 190.1205 CJW 192.0297 QHnNO, 194.0453
CioH12N4 188.1063 c9h20no3 190.1444 GjHgNjOj 192.0535 C9H|0N203 194.0692
QoH2003 188.1413 C9H22N2O2 190.1682 C9H1()N3O2 192.0774 C9H|2N3O2 194.0930
^10^22^О2 188.1651 C10H„NO3 190.0504 C9H,2N4C) 192.1012 GjHhN.O 194.1169
c10h24n2o 188.1890 C10HI0N2O2 190.0743 C9H2(1O4 192.1362 C|<>H|()O4 194.0579
cnH8o3 188.0473 CiqHi2N3O 190.0981 Cl0H8O4 192.0422 C|0H|2NO3 194.0817
СцН10М)2 188.0712 CioH14N4 190.1220 C|oHleN03 192.0661 CinH14N2O2 194.1056
CltH12N2O 188.0950 Q oH22o3 190.1569 C|0H|2N202 192.0899 C10Hl6N3O 194.1295
cuh14n3 188.1189 СцН|0О3 190.0630 c10h14n3o 192.1138 C|oH|8N4 194.1533
СцН24О2 188.1777 CnH12NO2 190.0868 cioh16n4 192.1377 СцН|4О3 194.0943
C12HI2O2 188.0837 q,h14n2o 190.1107 C]|HI2O3 192.0786 c,,h16no2 194.1182
c12h14no 188.1076 C.,H16N3 190.1346 c,.h14no2 192.1025 C||Hi8N2O 194.1420
Ci2H|6N2 188.1315 CJ2H14O2 190.0994 ChHi6N2O 192.1264 ChH^N-j 194.1659
cl3H16o 188.1202 C12HIftNO 190.1233 c„h18n3 192.1502 c12H8N3 194.0719
C13HI8N 188.1440 C12HikN2 190.1471 C12H16O2 192.1151 C|2H18O2 194.1307
C14H20 188.1566 QsHihO 190.1358 c12h18no 192.1389 C^Ha.NO 194.1546
189 C13H20N 190.1597 Ci2H20N2 192.1628 C|2H22N2 194.1784
C7H13N2O4 189.0876 Ci4H22 190.1722 c13h8n2 192.0688 cI3h8no 194.0606
c7h15n3o3 189.1114 CisHjo 190.0783 СвНгоО 192.1515 c13h10n2 194.0845
C7HI7n4o2 189.1353 191 c13h22n 192.1753 C13H22O 194.1671
QHI5NO4 189.1001 C7H15N2O4 191.1032 cI4h10n 192.0814 c,3h24n 194.1910
ChH17N2O3 189.1240 C7H17n3o3 191.1271 Ci4H24 192.1879 C]4H|0O 194.0732
C8H19N3O2 189.1478 C7H19n4o2 191.1509 CisH|2 192.0939 c14h,2n 194.0970
ckh21n4o 189.1717 c8h7n4o2 191.0570 193 C|4H26 194.2036
189.0539 CsH17NO4 191.1158 C7H17N2O4 193.1189 clsHl4 194.1096
QH.N.O 189.0777 C8H19N2O3 191.1396 C7HI9n3o3 193.1427 195
C9H17O4 189.1127 c«h21n3o2 191.1635 C«H7N3O3 193.0488 C„H7N2O4 195.0406
c,h19no3 189.1365 СуН^Оз 191.0457 c«h9n4o2 193.0726 195.0644
C,H21N2O2 189.1604 С9Н9М3О2 191.0695 c8h19no4 193.1315 C.HnN.O, 195.0883
c,h23n3o 189.1842 QHnN.O 191.0934 С)Н7МО4 193.0375 C7H9NO4 195.0532
c,0h7no3 189.0426 191.1284 G»H9N2O3 193.0614 СЛн^О, 195.0770
C10H9N2O2 189.0664 QHjjNO, 191.1522 193.0852 195.1009
CwHnN3O 189.0903 C10H7O4 191.0344 qfl13N4O 193.1091 СЛ15Н4О 195.1247
C10H13N4 189.1142 c10h9no3 191.0583 C10H9O4 193.0501 c10Hno4 195.0657
Ci„H2iO3 189.1491 QoHhN202 191.0821 Cl0HnNO3 193.0739 CI(1H13NO3 195.0896
CI()H23NO2 189.1730 C10H13N3O 191.1060 C|(>H|3N2O2 193.0978 Ck)H|5N2O2 195.1134
СцН9О3 189.0552 cl()H15N4 191.1298 cI0h15n3o 193.1216 c10hi7n3o 195.1373
CnHnNO2 189.0790 СцНцО3 191.0708 cI0hI7n4 193.1455 C|0HI9N4 195.1611
c„h13n2o 189.1029 cnH13NO2 191.0947 СцН|3О3 193.0865 c„h7n4 195.0672
c>,h15n3 189.1267 ChH15N2O 191.1185 C„HI5NO2 193.1103 СцН15О3 195.1021
CI2H]3O2 189.0916 c„h17n3 191.1424 c„h17n2o 193.1342 C„HI7NO2 195.1260
C12Hi5NO 189.1154 Ci2Hi5O2 191.1072 c„h19n3 193.1580 c„h19n2o 195.1498
C12HI7N2 189.1393 c12h17no 191.1311 C]2H17O2 193.1229 c„h21n3 195.1737
C13H17O 189.1280 C12H]9N2 191.1549 Ci2HI9NO 193.1467 cI2h7n2o 195.0559
c13hI9n 189.1519 С1зН]9О 191.1436 c12h2In2 193.1706 c12h9n3 195.0798
C]4H2| 189.1644 c13h21n 191.1675 c13h9n2 193.0767 CJ2HI9O2 195.1385
Приложение Л
75
Приложение А (продолжение)
M M M M
c12h21no 195.1624 CUH17O3 197.1178 c,h15n2o3 199.1083 200.2254
C12H23N2 195.1863 CuHl9NO2 197.1416 c,h17n3o2 199.1322 CbH12O2 200.0837
c13h9no 195.0684 ChH21N2O 197.1655 C,H19N4O 199.1560 c13h14no 200.1076
CI3HnN2 195.0923 СцН2зМ3 197.1894 cI0h7n4o 199.0621 c13h16n2 200.1315
C13H23O 195.1750 C12H9N2O 197.0715 QoHi504 199.0970 С1зН28О 200.2141
c13h25n 195.1988 C12HuN3 197.0954 c10h17no3 199.1209 c14H16o 200.1202
Ci4HnO 195.0810 C12H21O2 197.1542 C10H19N2O2 199.1447 c14h18n 200.1440
CMH13N 195.1049 c12h23no 197.1781 QoH21N30 199.1686 C15H2O 200.1566
C14H27 195.2114 C12H25N2 197.2019 CioH23N4 199.1925 201
Q5H15 195.1174 C13H9O2 197.0603 CuH7N2O2 199.0508 C8Hi3N2O4 201.0876
196 C13HUNO 197.0841 CnH9N3O 199.0746 c8h15n3o3 201.1114
c8h8n2o4 196.0484 cuh13n2 197.1080 CnHnN4 199.0985 c8h17n4o2 201.1353
c8h10n3o3 196.0723 197.1906 CnH19O3 199.1334 C^HbNC^ 201.1001
CgH12N4O2 196.0961 C13H27N 197.2145 CnH21NO2 199.1573 C9Hi7N2O3 201.1240
C^oNO, 196.0610 C14H13O 197.0967 ChH^NjjO 199.1811 C9Hj9N3O2 201.1478
QHl2N2O3 196.0848 c14h15n 197.1205 CnH25N3 199.2050 C^N.O 201.1717
c,h14n3o2 196.1087 сын29 197.2270 C12H9NO2 199.0634 Ci0H7N3O2 201.0539
c,h16n4o 196.1325 197.1331 С12НцМ2О 199.0872 c10h9n4o 201.0777
QoHi204 196.0735 198 c12h13n3 199.1111 CioHi704 201.1127
c10h14no3 196.0974 C8H]0N2O4 198.0641 C]2H23O2 199.1699 Ci0Hi9NO3 201.1365
QoH16N202 196.1213 C^H12N3O3 198.0879 c12h25no 199.1937 CwH21N2O2 201.1604
CioHi8N30 196.1451 c8h14n4o2 198.1118 C12H27N2 199.2176 201.1842
CioH2ON4 196.1690 C9H12NO4 198.0766 ^1зНцО2 199.0759 CioH2sN4 201.2081
c„h8n4 196.0750 C9H14n2o3 198.1005 C13H13NO 199.0998 ChH7NO3 201.0426
СцН1бО3 196.1100 C9H16n3o2 198.1244 c13h15n2 199.1236 CuH9N2O2 201.0664
СцН18ЫО2 196.1338 198.1482 C13H27O 199.2063 CnHnN3O 201.0903
CnH20N2O 196.1577 C10H14O4 198.0892 c13h29n 199.2301 CnH13N4 201.1142
CiiH22N3 196.1815 C10H16NO3 198.1131 c14h15o 199.1123 QiH2iO3 201.1491
C12H8N2O 196.0637 QoH18n2o2 198.1369 c14h17n 199.1362 ChH23NO2 201.1730
Q2H10N3 196.0876 CioH2oN30 198.1608 Q5H19 199.1488 CiiH2sN2O 201.1968
^i2H20O2 196.1464 CiqH22N4 198.1846 200 CnH27N3 201.2207
c12h22no 196.1702 cuh8n3o 198.0668 C8H12N2O4 200.0797 Ci2H9O3 201.0552
Ci2H24N2 196.1941 C„H1oN4 198.0907 Qh14n3o3 200.1036 Ci2HnNO2 201.0790
C13H8O2 196.0524 QjH18O3 198.1256 C«H16N4O2 200.1275 C12H13N2O 201.1029
C13H10NO 196.0763 СцН20МО2 198.1495 G>H14NO4 200.0923 c12h15n3 201.1267
CisH12N2 196.1001 ChH22N2O 198.1733 C9H16n2o3 200.1162 С1гН25О2 201.1855
C13H24O 196.1828 cnH24N3 198.1972 C9H18n3o2 200.1400 C12H27NO 201.2094
c13h26n 196.2067 c12h8no2 198.0555 QH^O 200.1639 С1зН1зО2 201.0916
Q4H12O 196.0888 C12H10N2O 198.0794 c10h8n4o 200.0699 c13h15no 201.1154
c14h14n 196.1127 c12h12n3 198.1032 СюН1бО4 200.1049 Ci3H17N2 201.1393
Q4H28 196.2192 C12H22O2 198.1620 CiqH18NO3 200.1287 c14h17o 201.1280
C15H16 196.1253 c12h24no 198.1859 QoH2oN202 200.1526 c14h19n 201.1519
197 Ci2H26N2 198.2098 Ci&H22N3O 200.1764 CisH2i 201.1644
CsH9N2O4 197.0563 Q3H10O2 198.0681 Ci0H24N4 200.2003 202
QHnN3O3 197.0801 c13h12no 198.0919 ChH8N2O2 200.0586 c8h14n2o4 202.0954
Q?h13n4o2 197.1040 c13h14n2 198.1158 CnH10N3O 200.0825 c8h16n3o3 202.1193
C^NO, 197.0688 С1зН26О 198.1985 CnH12N4 200.1063 C8Hi8N4O2 202.1431
C9H13N2O3 197.0927 CbH^N 198.2223 C|iH20O3 200.1413 202.0491
C9H15n3o2 197.1165 c14h14o 198.1045 CnH22NO2 200.1651 C^eNO, 202.1080
197.1404 c14h16n 198.1284 CnH24N2O 200.1890 C9H18N2O3 202.1318
С1оН1з04 197.0814 C14H3O 198.2349 cnN26N3 200.2129 202.1557
C10H15NO3 197.1052 198.1409 Ci2H8O3 200.0473 202.1795
CiqH17N2O2 197.1291 199 C12H10NO2 200.0712 CiqH8N3O2 202.0617
C10H19N3O 197.1529 C8HnN2O4 199.0719 c12h12n2o 200.0950 QoHioN40 202.0856
CioH21N4 197.1768 199.0958 c12h14n3 200.1189 QoHi804 202.1205
CuH7N3O 197.0590 Qh15n4o2 199.1196 С1гН24О2 200.1777 CioH2oN03 202.1444
CnH^ 197.0829 GJIbNCXi 199.0845 c12h26no 200.2015 СюНгг^Ог 202.1682
76
1. Маа-спектрометрия
Приложение А (продолжение)
FM FM FM FM
c10H24N3o 202.1921 C8H20N4O2 204.1588 C|3H21N2 205.1706 c„h19n4 207.1611
QoH26N4 202.2160 ОДЛзО, 204.0410 c14h9n2 205.0767 C|2H15O3 207.1021
c„h8no3 202.0504 c,h8n4o2 204.0648 C14H2IO 205.1593 c12h17no2 207.1260
CuH10N2O2 202.0743 c,h18no4 204.1236 c14h23n 205.1832 C|2Hj9N2O 207.1498
CuH12N3O 202.0981 С^НгоНгОз 204.1475 C15H9O 205.0653 C12H21N3 207.1737
c„h14n4 202.1220 C9H22N3O2 204.1713 C15HnN 205.0892 c13h9n3 207.0798
СцН^Оз 202.1569 204.1952 C15H25 205.1957 C13H19O2 207.1385
СцН24МО2 202.1808 CioH8N203 204.0535 c16H13 205.1018 c13h21no 207.1624
CnH^O 202.2046 CioH10N302 204.0774 206 Ci3H23N2 207.1863
С1гН10Оз 202.0630 c10h12n4o 204.1012 C8H18N2O4 206.1267 c14h9no 207.0684
C17H12NO7 202.0868 CioH2004 204.1362 C8H20N3O3 206.1506 C14HhN2 207.0923
C|2H14N2O 202.1107 СюН^ЬЮз 204.1600 CfiH22N4O2 206.1744 207.1750
c12h16n3 202.1346 СюН^ЫгОг 204.1839 c<,h6n2o4 206.0328 c14h25n 207.1988
Ci2H26O2 202.1934 СцН8О4 204.0422 ОД^Оз 206.0566 C15HnO 207.0810
С1зН14О2 202.0994 C„H10NO3 204.0661 C9HiqN4O2 206.0805 cI5h13n 207.1049
CI3H16NO 202.1233 CnH12N2O2 204.0899 C^oNO, 206.1393 CisH27 207.2114
Ci3Hi8N2 202.1471 c„h14n3o 204.1138 C9H22N2O3 206.1631 c16H15 207.1174
СмН18О 202.1358 CnH16N4 204.1377 C10H8NO4 206.0453 208
CmH^N 202.1597 СцН24О3 204.1726 C|oHioN203 206.0692 C8H20N2O4 208.1424
Ci5H22 202.1722 C12H12O3 204.0786 CinHi2N3O2 206.0930 208.0484
203 c12h14no2 204.1025 c10h14n4o 206.1169 ОД^Оз 208.0723
QHI5N2O4 203.1032 c12h16n2o 204.1264 CiqH22O4 206.1518 C9H|2N4O2 208.0961
CsHi7N3O3 203.1271 c12h18n3 204.1502 СцН10О4 206.0579 CioH10N04 208.0610
C8H19N4O2 203.1509 Ci3H16O2 204.1151 CnH12NO3 206.0817 C10H12N2O3 208.0848
C9H7N4O2 203.0570 c13h18no 204.1389 CnH14N2O2 206.1056 C10H14N3O2 208.1087
C9H17NO4 203.1158 CbH^ 204.1628 CnH16N3O 206.1295 c10h16n4o 208.1325
C9H19N2O3 203.1396 С^НэдО 204.1515 ChH18N4 206.1533 СцН12О4 208.0735
Q^21N3O2 203.1635 C^H^N 204.1753 C12H14O3 206.0943 CnH14NO3 208.0974
QH^N.O 203.1873 c15h10n 204.0814 C12H16NO2 206.1182 CjiH16N2O2 208.1213
C10H7N2O3 203.0457 c15h24 204.1879 C12H18N2O 206.1420 CnH18N3O 208.1451
CiqH9N3O2 203.0695 Ci6H12 204.0939 C12H20N3 206.1659 СцН20Ы4 208.1690
C10HnN4O 203.0934 205 c13h8n3 206.0719 c12h8n4 208.0750
CioH1904 203.1284 QH17n2o4 205.1189 С1зН18О2 206.1307 Ci2H16O3 208.1100
CioH21N03 203.1522 C8H19N3O3 205.1427 C13H20NO 206.1546 Cj2H18NO2 208.1338
CioH23N202 203.1761 QjH21N4O2 205.1666 c13h22n2 206.1784 Ci2H20N2O 208.1577
C10H25N3O 203.1999 C9H7N3O3 205.0488 Ci4H10N2 206.0845 Cj2H22N3 208.1815
СцН7О4 203.0344 205.0726 C14H22O 206.1671 c13h8n2o 208.0637
CllH9NO3 203.0583 C9H19NO4 205.1315 c14h24n 206.1910 C13H1ON3 208.0876
QiHhN2O2 203.0821 C9H2j N2O3 205.1553 C15H10O 206.0732 Ci3H20O2 208.1464
CnH13N3O 203.1060 C9H23N3O2 205.1791 c15h12n 206.0970 c13h22no 208.1702
CnH15N4 203.1298 c10h7no4 205.0375 Cl 5^26 206.2036 CbH24N2 208.1941
CnH23O3 203.1648 C10H9N2O3 205.0614 c16H14 206.1096 c14h10no 208.0763
CnH^NO, 203.1886 QoHhN302 205.0852 207 ChH12N2 208.1001
C12HnO3 203.0708 CioH]3N40 205.1091 C«H19N2O4 207.1345 Ci4H24O 208.1828
c12h13no2 203.0947 QqH21O4 205.1440 c8h21n3o3 207.1584 c14h26n 208.2067
c12h15n2o 203.1185 СюН^ЫОз 205.1679 c9h7n2o4 207.0406 c15h12o 208.0888
c12h17n3 203.1424 CnH9O4 205.0501 едМзОз 207.0644 c15h14n 208.1127
С1зН15О2 203.1072 СцНнЫОз 205.0739 CgHnN4O2 207.0883 CisH28 208.2192
Cl3H17NO 203.1311 CiiH13N2O2 205.0978 C,H21NO4 207.1471 C16H16 208.1253
С1зН19Ь12 203.1549 C11H15N3O 205.1216 c10h9no4 207.0532 209
C14H19O 203.1436 CnH17N4 205.1455 СдоНцЬ^Оз 207.0770 C9H9N2O4 209.0563
c14h21n 203.1675 Ct2H13O3 205.0865 C10H13N302 207.1009 ОДЛОз 209.0801
c15h9n 203.0736 » c12h15no2 205.1103 C10H15N4O 207.1247 C9H13N4O2 209.1040
Q5H23 203.1801 C12H17N2O 205.1342 СцНнО4 207.0657 c10hhno4 209.0688
204 ^12^1^3 205.1580 CnH13NO3 207.0896 CioH13N203 209.0927
QH16N2O4 204.1111 C13H17O2 205.1229 CiiH15N2O2 207.1134 CioH15N302 209.1165
204.1349 c13h19no 205.1467 CnH17N3O 207.1373 C10H17N4O 209.1404
Приложение А
77
Приложение А (продолжение)
M M M M
СцН13О4 209.0814 Ci6H18 210.1409 Ci3H8O3 212.0473 QoH18N203 214.1318
C„H,5NO3 209.1052 211 Cl3HI0NO2 212.0712 Ci(>H2oN302 214.1557
ChH17N2O2 209.1291 C9H„N2O4 211.0719 c„hi2n2o 212.0950 ci0h22n4o 214.1795
C.,H19N3O 209.1529 C,H13N3O3 211.0958 c,3h14n3 212.1189 ChH8N3O2 214.0617
c„h2in4 209.1768 c,h15n4o2 211.1196 C13H24O2 212.1777 cnH10N4o 214.0856
cI2h9n4 209.0829 c10h13no4 211.0845 Ci3H26NO 212.2015 СцН18О4 214.1205
C12H17O3 209.1178 CioH15N203 211.1083 C,3H28N2 212.2254 CnH20NO3 214.1444
C12Hl9NO2 209.1416 C]0H17N3O2 211.1322 CI4H12O2 212.0837 СцН^МгОг 214.1682
C12H21N2O 209.1655 C10Hl9N4O 211.1560 c14h14no 212.1076 cnH24N3o 214.1921
CI2HmN3 209.1894 c„h7n4o 211.0621 c14h16n2 212.1315 c„h26n4 214.2160
CnH^O 209.0715 СцН15О4 211.0970 Ci4H28O 212.2141 c12h8no3 214.0504
C13H„N3 209.0954 c„h17no3 211.1209 CuH^N 212.2380 Ci2H]()N2O2 214.0743
Q3H21O2 209.1542 CnH19N2O2 211.1447 C15Hl6O 212.1202 C]2H12N3O 214.0981
C13H23NO 209.1781 cnH21N3o 211.1686 C15Ht8N 212.1440 c12h14n4 214.1220
c„h2sn2 209.2019 C„H23N4 211.1925 C15H32 212.2505 2^22^3 214.1569
C14H9O2 209.0603 C^H^O 211.0746 С[бН20 212.1566 C12H24NO2 214.1808
c,4h„no 209.0841 C12HnN4 211.0985 213 Ci2H26N2O 214.2046
c,4h13n2 209.1080 C12H19O3 211.1334 C9H,3N2O4 213.0876 СпНгдЫэ 214.2285
с,4нио 209.1906 Ci2H21NO2 211.1573 C,HI5N3O3 213.1114 C13H1()O3 214.0630
c14h27n . 209.2145 c12h23n2o 211.1811 Qh17n4o2 213.1353 c13h12no2 214.0869
C15HI3O 209.0967 C^H^N, 211.2050 ClnH,,NO4 213.1001 c13h14n2o 214.1107
C|4HI5N 209.1205 CI3H9NO2 211.0634 ClnHt7N2O3 213.1240 CI3H16N3 214.1346
C,.H24 209.2270 211.0872 C]0H19N3O2 213.1478 С13Н2бО2 214.1934
C16H,7 209.1331 C„HI3N3 211.1111 C10H21N4O 213.1717 cI3h28no 214.2172
210 С13НгзО2 211.1699 c„h7n3o2 213.0539 СвНэоМг 214.2411
C»HI0N2O4 210.0641 Cl3H25NO 211.1937 C,,H9N4O 213.0777 Ci4Hi4O2 214.0994
ЗДЛО, 210.0879 C13H27N2 211.2176 QiHi7O4 213.1127 c14h16no 214.1233
c^iI4n4o2 210.1118 C14HnO2 211.0759 CltHl9NO3 213.1365 CI4H]8N2 214.1471
c10h12no4 210.0766 c14h13no 211.0998 ChH2iN2O2 213.1604 CisHjgO 214.1358
CtoH14N203 210.1005 c14h15n2 211.1236 CnH^NjO 213.1842 C15H2ON 214.1597
Cl0H16N3O2 210.1244 Q4H27O 211.2063 CnH25N4 213.2081 cl6H22 214.1722
c10h18n4o 210.1482 c14h29n 211.2301 C]2H9N2O2 213.0664 215
c„HI4o4 210.0892 C15H15O 211.1123 C12HhN3O 213.0903 215.1032
C„H16NO3 210.1131 c15h17n 211.1362 Ci2H13N4 213.1142 C9HI7N3O3 215.1271
CiiH|8N2O2 210.1369 c15H31 211.2427 C(2H2IO3 213.1491 215.1509
Q.W 210.1608 C16H19 211.1488 C,2H23NO2 213.1730 QoH7N402 215.0570
CnH^N, 210.1846 212 C,2H25N2O 213.1968 c10h17no4 215.1158
C12H„N3O 210.0668 C9H12N2O4 212.0797 CnH^N, 213.2207 Ck)H19N2O3 215.1396
C12H1oN4 210.0907 C9H14n3o3 212.1036 C13H9O3 213.0552 C10H2iN3O2 215.1635
C,2H18O3 210.1256 C9H16n4o2 212.1275 cI3huno2 213.0790 ^10^23^0 215.1873
C,,H2„NO2 210.1495 Cl0H14NO4 212.0923 Ci3H)3N2O 213.1029 cnH7N2o3 215.0457
С|2НИ^О 210.1733 CioH16N203 212.1162 C13Hi5N3 213.1267 ChH^O, 215.0695
C12H24N3 210.1972 QoHi8N302 212.1400 C„H25O2 213.1855 CnHnN4O 215.0934
c13h„no2 210.0555 QoH2()N40 212.1639 CI3H27NO 213.2094 CnH19O4 215.1284
c13hI0n2o 210.0794 CnH8N4O 212.0699 Ci3H29N2 213.2332 CnH21NO3 215.1522
Ci3H12N3 210.1032 CiiHi6O4 212.1049 C14HI3O2 213.0916 CI1H23N2O2 215.1761
СвНцОг 210.1620 CnH18NO3 212.1287 cI4h15no 213.1154 ChH25N3O 215.1999
c13h24no 210.1859 СцН20Ы2О2 212.1526 C14Hi7N2 213.1393 chh27n4 215.2238
c13h26n2 210.2098 СцН^^О 212.1764 C14H29O 213.2219 c12h9no3 215.0583
C14HiuO2 210.0681 c„h24n4 212.2003 CI5H17O 213.1280 Q2HI1N2O2 215.0821
CI4Hi2NO 210.0919 Ci2h8n2o2 212.0586 cI5h19n 213.1519 C12H13N3O 215.1060
c14h14n2 210.1158 Ci2H10N3O 212.0825 c16H21 213.1644 c12h15n4 215.1298
C14H26O 210.1985 C12HI2N4 212.1063 214 Ci2H23O3 215.1648
CI4H»N 210.2223 C12H20O3 212.1413 c,hI4n2o4 214.0954 ^12^25^02 215.1886
C15H14O 210.1045 С^Н^ЬЮг 212.1651 СЛ.^зОз 214.1193 Ci 2Н27^2О 215.2125
c,5h16n 210.1284 C12H24N2O 212.1890 С<»Н|8М4О2 214.1431 Ci2H29N3 215.2363
QsHso 210.2349 c12h26n3 212.2129 c10h16no4 214.1080 Ci3HmO3 215.0708
78
7. Маа-спектрометрия
Приложение А (продолжение)
M M M M
c13h13no2 215.0947 c„h7no4 217.0375 C14H22N2 218.1784 C„H16N4O 220.1325
Ct3H15N2O 215.1185 ChH9N2O3 217.0614 Q5H10N2 218.0845 Ci2Hi2O4 220.0735
Cl3HI7N3 215.1424 QiHhN3O2 217.0852 Q5H22O 218.1671 c12h14no3 220.0974
С]4Н15О2 215.1072 CuHI3N4O 217.1091 c15h24n 218.1910 C12H16N2O2 220.1213
CmH17NO 215.1311 QiH2iO4 217.1440 Ci6H10O 218.0732 C12Hi8N3O 220.1451
C14H19N2 215.1549 c„h23no3 217.1679 c16h12n 218.0970 ^12^2oN4 220.1690
C15H19O 215.1436 C11H25N2O2 217.1917 C16H26 218.2036 c13h8n4 220.0750
c15h21n 215.1675 CnH27N3O 217.2156 C17H14 218.1096 С1зН16О3 220.1100
c,6h23 215.1801 C12H9O4 217.0501 219 Ci3Hi8NO2 220.1338
216 C12HuNO3 217.0739 C9H19N2O4 219.1345 C13H20N2O 220.1577
c9h16n2o4 216.1111 C12H13N2O2 217.0978 C9H21N3O3 219.1584 ^13^22^ 220.1815
Cs>h18n3o3 216.1349 c12h15n3o 217.1216 C9H23N4O2 219.1822 c14h10n3 220.0876
C9H20N4O2 216.1588 c12h17n4 217.1455 CwH7N2O4 219.0406 Ci4H20O2 220.1464
CiqH8N4O2 216.0648 > C12H25O3 217.1804 СюН9?43О3 219.0644 c14h22no 220.1702
CioH18N04 216.1236 C12H27NO2 217.2043 CioHnN402 219.0883 Q4h24n2 220.1941
Ci0H20N2O3 216.1475 С1зН13О3 217.0865 CioH2]N04 219.1471 c15h10no 220.0763
Ci0H22N3O2 216.1713 c13h15no2 217.1103 CioH23N203 219.1710 CisH12N2 220.1001
Ci0H24N4O 216.1952 c13h17n2o 217.1342 Ci0H25N3O2 219.1948 C15H24O 220.1828
c„h8n2o3 216.0535 Ci3H19N3 217.1580 СПН^О4 219.0532 c15h26n 220.2067
ChH10N3O2 216.0774 C14H17O2 217.1229 СцНцМ2О3 219.0770 C16H12O 220.0888
CuH12N4O 216.1012 Ci4HI9NO 217.1467 CnH13N3O2 219.1009 c16h14n 220.1127
СцН2()О4 216.1362 c14h21n2 217.1706 c„h15n4o 219.1247 ^16^28 220.2192
СцН22МО3 216.1600 c15h9n2 217.0767 C11H23O4 219.1597 C17H16 220.1253
CiiH24N2O2 216.1839 Q5H21O 217.1593 ChH25NO3 219.1835 221
CuH26N3O 216.2077 Q5H23N 217.1832 С1гНцО4 219.0657 C9H2 j N2O4 221.1502
CnH28N4 216.2316 C16HnN 217.0892 c12h13no3 219.0896 СоН^зОз 221.1741
C12H8O4 216.0422 С1бН25 217.1957 Ci2H15N2O2 219.1134 Ck)H9N2O4 221.0563
Ci2Hl0NO3 216.0661 Q7H13 217.1018 c12h17n3o 219.1373 CioHnN303 221.0801
Ci2H12N2O2 216.0899 218 C12HJ9N4 219.1611 QoHj3N402 221.1040
c12h14n3o 216.1138 C9H18N2O4 218.1267 C13H15O3 219.1021 ^-'10^23^O4 221.1628
c12h16n4 216.1377 СЛЖ 218.1506 c13h17no2 219.1260 CnHnNO4 221.0688
Ci2H24O3 216.1726 ^9^22^4О2 218.1744 C13H19N2O 219.1498 QiH13n2o3 221.0927
Ci2H26NO2 216.1965 QoH8N303 218.0566 Ci3H2iN3 219.1737 ChH15N3O2 221.1165
c12h28n2o 216.2203 CioH10N402 218.0805 c14h9n3 219.0798 c„h17n4o 221.1404
C13H12O3 216.0786 Ci0H20NO4 218.1393 C14H19O2 219.1385 C12Hi3O4 221.0814
c13h14no2 216.1025 CioH22N203 218.1631 c14h21no 219.1624 c12h15no3 221.1052
c13h16n2o 216.1264 C]oH24N302 218.1870 c14h23n2 219.1863 Ci2Hj7N2O2 221.1291
c13h18n3 216.1502 Ci0H26N4O 218.2108 c15h9no 219.0684 C12H19N3O 221.1529
Ci3H28O2 216.2090 cuh8no4 218.0453 C15HnN2 219.0923 c12h21n4 221.1768
Ci4H16O2 216.1151 . CiiHioN203 218.0692 C15H23O 219.1750 c13h9n4 221.0829
c14h18no 216.1389 CuH12N3O2 218.0930 c15h25n 219.1988 C]3H17O3 221.1178
Ci4H20N2 216.1628 CnH14N4O 218.1169 c16Hno 219.0810 Ci3H19NO2 221.1416
C15H20O 216.1515 СцН22О4 218.1518 C16Hl3N 219.1049 CnH21N2O 221.1655
c15h22n 216.1753 CuH24NO3 218.1757 C16H27 219.2114 c13h23n3 221.1894
C16H10N 216.0814 ChH26N2O2 218.1996 c17h15 219.1174 CmH^O 221.0715
Ci6H24 216.1879 C12H10O4 218.0579 220 C14HhN3 221.0954
C17H12 216.0939 C12H12NO3 218.0817 C9H20N2O4 220.1424 C14H2iO2 221.1542
217 Ci2Hi4N2O2 218.1056 C9H22N3O3 220.1662 c14h23no 221.1781
C9H17N2O4 217.1189 c12h16n3o 218.1295 C9H24N4O2 220.1901 £14^25^ 221.2019
C9H19N3O3 217.1427 C12H]8N4 218.1533 CioH8N204 220.0484 CisH9O2 221.0603
C9H21N4O2 217.1666 С^Оз 218.1883 QoHioN303 220.0723 c15huno 221.0841
c10h7n3o3 217.0488 СвН14О3 218.0943 QoHi2N402 220.0961 c15h13n2 221.1080
C10H9N4O2 217.0726 c13h16no2 218.1182 CioH22N04 220.1549 CisH25O 221.1906
C10H19NO4 217.1315 Ci3H18N2O 218.1420 СкЛ^^Оз 220.1788 c15h27n 221.2145
C10H21N2O3 217.1553 C13H2ON3 218.1659 CnH10NO4 220.0610 c16H13o 221.0967
C10H23N3O2 217.1791 ^14^18O2 218.1307 СцН12Ы2О3 220.0848 c16h15n 221.1205
C10H25N40 217.2030 c14h20no 218.1546 CnH14N3O2 220.1087 C16H29 221.2270
Приложение Л
79
Приложение А (продолжение)
M M M M
СпН17 221.1331 cI4h9no2 223.0634 c„h21n4o 225.1717 CI4H28NO 226.2172
222 c14h„n2o 223.0872 c12h9n4o 225.0777 Ci4H30N2 226.2411
C9H22N2O4 222.1580 c14hI3n3 223.1111 Cj2H17O4 225.1127 QsHi4O2 226.0994
CioHI()N204 222.0641 C14H23O2 223.1699 C12H19NO3 225.1365 c15h16no 226.1233
CioH12n3o3 222.0879 c14h25no 223.1937 C12H21N2O2 225.1604 C15H18N2 226.1471
Cu>H14N4O2 222.1118 C14H27N2 223.2176 C|2H23N3O 225.1842 CisH^O 226.2298
c„hi2no4 222.0766 С15НцО2 223.0759 C|2H25N4 225.2081 c15h32n 226.2536
ChHi4N2O3 222.1005 c15h13no 223.0998 c13h9n2o2 225.0664 C16H18O 226.1358
ChH16N3O2 222.1244 Ci5H27O 223.2063 C13HnN3O 225.0903 C16H2()N 226.1597
C„H1kN4O 222.1482 cI5h29n 223.2301 C13H13N4 225.1142 c16H34 226.2662
C„N3O3 221.9940 сцн15о 223.1123 C13H21O3 225.1491 C]?H22 226.1722
C12Hi4O4 222.0892 cI6h17n 223.1362 C13H23NO2 225.1730 227
Ci2HI6NO3 222.1131 c16H3) 223.2427 C13H25N2O 225.1968 Ck)H15N2O4 227.1032
C|2Hi8N2O2 222.1369 C17H19 223.1488 C13H27N3 225.2207 CioH17N303 227.1271
c12h20n3o 222.1608 224 C14H9O3 225.0552 CK)H|9N4O2 227.1509
C12HkN4 222.1846 CioHi2N204 224.0797 C14H„NO2 225.0790 chh17no4 227.1158
C13H8N3O 222.0668 C|(jH14N3O3 224.1036 c14h13n2o 225.1029 ChH21N3O2 227.1635
C13HioN4 222.0907 Ck)H16N4O2 224.1275 c14h15n3 225.1267 CuH23N4O 227.1873
^i3H18O3 222.1256 c„h14no4 224.0923 Ci4H25O2 225.1855 c12h7n2o3 227.0457
C.jHa.NO, 222.1495 CnHI6N2O3 224.1162 c14h27no 225.2094 C12H9N3O2 227.0695
C,3H22N2O 222.1733 ChHI8N3O2 224.1400 c)4h29n2 225.2332 C12HhN4O 227.0934
CbH^Nj 222.1972 C„H2(№O 224.1639 C15H|3O2 225.0916 Ci2Ht9O4 227.1284
cI4h10n2o 222.0794 c12h8n4o 224.0699 cI5h15no 225.1154 c12h21no3 227.1522
c14hI2n3 222.1032 ^12^1бО4 224.1049 cI5h17n2 225.1393 Ci2H23N2O2 227.1761
С|4Н2гО2 222.1620 C12H18NO3 224.1287 c15h29o 225.2219 c12h25n3o 227.1999
c14h24no 222.1859 Cl2H2()N2O2 224.1526 cI5h31n 225.2458 c12h27n4 227.2238
c14h»n2 222.2098 C12H22N3O 224.1764 c16H17o 225.1280 c13h9no3 227.0583
C15H1002 222.0681 Ci2H24N4 224.2003 c16h19n 225.1519 Ci3HuN2O2 227.0821
c15h12no 222.0919 С1зН8Ы2О2 224.0586 cI6H33 225.2584 C13H13N3O 227.1060
Ci5H14N2 222.1158 Ci3H10N3O 224.0825 Cj7H21 225.1644 c13h15n4 227.1298
C.sH^O 222.1985 c13hI2n4 224.1063 226 c13h25no2 227.1886
c15hmn 222.2223 Cl3H20O3 224.1413 C]()H|4N2O4 226.0954 c13h27n2o 227.2125
C16H14O 222.1045 Ci3H22NO2 224.1651 CioH16N303 226.1193 ^i3H29N3 227.2363
C16HI6N 222.1284 C13H24N2O 224.1890 C|()H1KN4O2 226.1431 C14HnO3 227.0708
222.2349 C13H26N3 224.2129 chh16no4 226.1080 C14H13NO2 227.0947
c1ano 221.9980 C14H|0NO2 224.0712 c(1hI8n2o3 226.1318 C14H15N2O 227.1185
CnHllt 222.1409 c14h12n2o 224.0950 Q !H2()N3O2 226.1557 c14h17n3 227.1424
223 C14H14N3 224.1189 cnH22N4o 226.1795 C|4H27O2 227.2012
C|(>H||N2O4 223.0719 C14H24O2 224.1777 C12H8N3O2 226.0617 c14h29no 227.2250
C10H13N3O3 223.0958 c14h26no 224.2015 C|2H|()N4O 226.0856 C15HisO2 227.1072
Cl0H15N4O2 223.1196 cI4h28n2 224.2254 C12H18O4 226.1205 c15hI7no 227.1311
CnH13NO4 223.0845 ^15Hi2O2 224.0837 Q2H2()NO3 226.1444 c15h19n2 227.1549
C„Hl5N2O3 223.1083 c15h14no 224.1076 CI2H22N2O2 226.1682 C15H31O 227.2376
c„h17n3o2 223.1322 C15H16N2 224.1315 c12h24n3o 226.1929 c15h33n 227.2615
c„hI9n4o 223.1560 CjsH^O 224.2141 c12h26n4 226.2160 C16H19O 227.1436
c,2h7n4o 223.0621 с15н^ 224.2380 C|3H1()N2O2 226.0743 c16h21n 227.1675
C,,H15O4 223.0970 c16H16o 224.1202 cI3h12n3o 226.0981 C17H23 227.1801
C,2H17NO3 223.1209 c16h18n 224.1440 c13h14n4 226.1220 228
Ci2H19N2O2 223.1447 c16H32 224.2505 c13h22o3 226.1569 Cn)H16N2O2 228.1111
Ci2H21N3O 223.1686 Ci7H2() 224.1566 c13h24no2 226.1808 C|()H18N3O3 228.1349
C12H23N4 223.1925 225 c13h26n2o 226.2046 CioH20N402 228.1588
C13H9N3O 223.0746 C|()Hi3N2O4 225.0876 Ci3H28N3 226.2285 ChH8N4O2 228.0648
c13h„n4 223.0985 Си)Н15Ы3О3 225.1114 С14Н1()О3 226.0630 ChH18NO4 228.1236
C|3H]9O3 223.1334 C10Hl7N4O2 225.1353 cl4H12NO2 226.0868 QiH20N2O3 228.1475
c13h21no2 223.1573 c„hI5no4 225.1001 c14h14n2o 226.1107 QiH22n3o2 228.1713
Ci3H23N2O 223.1811 CuH17N2O3 225.1240 c14h16n3 226.1346 c„h24n4o 228.1952
C13H25N3 223.2050 ChH19N3O2 225.1478 Ci4H26O2 226.1934 Ci2H8N2O3 228.0535
80
1. Маа-спектрометрия
Приложение А (продолжение)
M M . M M
cI2h12n4o 228.1012 c14h15no2 229.1103 231.1822 CnH^ 232.1690
С12Н20О4 228.1362 c14h17n2o 229.1342 CnH7N2O4 231.0406 СвНэдОэ 232.2039
Ci 2H22N О3 228.1600 c14h19n3 229.1580 CuH9N3O3 231.0644 Q4Hi6O3 232.1100
CI2H24N2O2 228.1839 C14H29O2 229.2168 CiiHiiN4O2 231.0883 Ci4H18NO2 232.1338
C12H26N3O 228.2077 cI4h31no 229.2407 ChH21NO4 231.1471 c14h20n2o 232.1577
c12hmn4 228.2316 C15H17O2 229.1229 СцН^^Оэ 231.1710 C14H22N3 232.1815
С1зН8О4 228.0422 c15h19no 229.1467 CiiH25N3O2 231.1948 c15h10n3 232.0876
C13H10NO3 228.0661 CisH2iN2 229.1706 c„h27n4o 231.2187 CisH2o02 232.1464
Ci3Hi2N2O2 228.0899 C16H21O 229.1593 c12h9no4 231.0532 c15h22no 232.1702
c13h14n3o 228.1138 C.eH^N 229.1832 C12HhN2O3 231.0770 c15h24n2 232.1941
С1зН24О3 228.1726 C17H9O 229.0653 Ci2Hi3N3O2 231.1009 C16H10NO 232.0768
c13h26no2 228.1965 ' c17HnN 229.0892 C12H15N4O 231.1247 C16H12N2 232.1001
СлзНгкЫгО 228.2203 QeHi3 229.1018 231.1597 c16H24o 232.1828
CI3H30N3 228.2442 230 c12h25no3 231.1835 c16h26n 232.2067
Ci4Hi2O3 228.0786 QoHieN204 230.1267 Ci2H27N2O2 231.2074 c17hI2o 232.0888
c14h14no2 228.1025 CioH20N303 230.1506 C12H29N3O 231.2312 c17h14n 232.1127
c14hI6n2o 228.1264 Cj0H22N4O2 230.1744 С1зНцО4 231.0657 СпНгв 232.2192
c14h18n3 228.1502 CuH8N3O3 230.0566 C13H13NO3 231.0896 CieH16 232.1253
228.2090 CiiH10N4O2 230.0805 Ci3H15N2O2 231.1134 233
c14hwno 228.2329 CuH20NO4 230.1393 c13h17n3o 231.1373 С1оН2зМ303 233.1741
Q4H32N2 228.2567 ChH22N2O3 230.1631 C13H19N4 231.1611 Q()H25n4o2 233.1979
QsHi6O2 228.1151 ChH24N3O2 230.1870 Cj4H15O3 231.1021 c„h9n2o4 233.0563
c15h18no 228.1389 CnH^N.O 230.2108 c14h17no2 231.1260 CnHnN3O3 233.0801
QsH20N2 228.1628 c12h8no4 230.0453 c14h19n2o 231.1498 ChH^NO, 233.1628
c15h32o 228.2454 Ci2HiqN2O3 230.0692 c14h21n3 231.1737 СцН25М2О3 233.1866
Ci6H20O 228.1515 Q2Hi2N3O2 230.0930 231.0798 СцН27М3О2 233.2105
c16h22n 228.1753 c12h14n4o 230.1169 CisH19O2 231.1385 C12HnNO4 233.0688
c17h10n 228.0814 С1гН22О4 230.1518 c15h21no 231.1624 С1гН13М2О3 233.0927
Ci7H24 228.1879 c12h24no3 230.1757 231.1863 C12HisN3O2 233.1165
228.0939 c12h26n2o2 230.1996 c16h9no 231.0684 c12h17n4o 233.1404
229 230.2234 Cl6HnN2 231.0923 ^-'i2H25O4 233.1753
CioHi7N204 229.1189 Q2H30N4 230.2473 c16h23o 231.1750 C12H27NO3 233.1992
C10H19N3O3 229.1427 С1зНюО4 230.0579 C17HnO 231.0810 CbHi3O4 233.0814
CwH2iN4O2 229.1666 С1зН12МО3 230.0817 c17h13n 231.1049 c13h15no3 233.1052
CuH7N3O3 229.0488 С1зН14М2О2 230.1056 Q7h27 231.2114 Ci3H]7N2O2 233.1291
ChH9N4O2 229.0726 C13H16N3O 230.1295 231.1174 c13h19n3o 233.1529
CnH19NO4 229.1315 c13h18n4 230.1533 232 c13h21n4 233.1768
QiH2jN2O3 229.1553 С1зН26О3 230.1883 CiqH20N2O4 232.1424 c14h9n4 233.0829
CiiH23N3O2 229.1791 Cl3H28NO2 230.2121 СюН^МзОэ 232.1662 C14H17O3 233.1178
CuH25N4O 229.2030 С^Н^О 230.2360 CioH24N402 232.1901 C14H19NO2 233.1416
229.0614 С14Н14О3 230.0943 QiH8N2O4 232.0484 c14h21n2o 233.1655
Ci2HnN3O2 229.0852 c14h16no2 230.1182 CiiHh)N3O3 232.0723 c15h9n2o 233.0715
c12h13n4o 229.1091 c14h18n2o 230.1420 ChH12N4O2 232.0961 C15H„N3 233.0954
C12H21O4 229.1440 C14H20N3 230.1659 ChH22NO4 232.1549 QsH2iO2 233.1542
c12h23no3 229.1679 СмНадОг 230.2247 СцН24М2О3 232.1788 c15h23no 233.1781
Ci2H25N2O2 229.1917 QsHi8O2 230.1307 СцН261Ч3О2 232.2026 233.2019
Ci2H27N3O 229.2156 c15h20no 230.1546 ChH^O 232.2265 С1бН9О2 233.0603
Q2h29n4 229.2394 230.1784 C12H10NO4 232.0610 C16HnNO 233.0841
C13H9O4 229.0501 Ci6HioN2 230.0845 Ci2Hi2N2O3 232.0848 C16H13N2 233.1080
C13HnNO3 229.0739 Ci6H22O 230.1671 C12H14N3O2 232.1087 C16H25O 233.1906
C13H13N2O2 229.0978 c16h24n 230.1910 c12h16n4o 232.1325 Cl6H27N 233.2145
c13h15n3o 229.1216 C17Hl0O 230.0732 Q2h24o4 232.1675 c17H13o 233.0967
c13h17n4 229.1455 c17h12n 230.0970 ^12^26^О3 232.1914 c17h15n 233.1205
СвН^Оэ 229.1804 Ci?H26 230.2036 СвНгв^Ог 232.2152 CnH29 233.2270
CnH27NO2 229.2043 QsHj4 230.1096 Ci3Hi2O4 232.0735 C18H17 233.1331
C13H29N2O 229.2281 231 C13H14NO3 232.0974 234
C13H31N3 229.2520 C10H19N2O4 231.1345 CbH16N2O2 232.1213 Ch)H22N2O4 234.1580
Ci4Hi3O3 229.0865 C10H21N3O3 231.1584 c13h18n3o 232.1451 Ci0H24N3O3 234.1819
Приложение А
81
Приложение А (продолжение)
M M M M
C10H26N4O2 234.2057 ClsHI3N3 235.1111 Ci2H2iN4O 237.1717 С^НэоМг 238.2411
СцН10Ы2О4 234.0641 Ci5H23O2 235.1699 С13Н^4О 237.0777 С1бН14О2 238.0994
CnH12N3O3 234.0879 c15h25no 235.1937 C13H17O4 237.1127 C16H16NO 238.1233
CnH14N4O2 234.1118 c15h27n2 235.2176 C13H19NO3 237.1365 c16h18n2 238.1471
c„h24no4 234.1706 C16HnO2 235.0759 Q3h21n.2o2 237.1604 238.2298
ChH26N2O3 234.1945 C16H13NO 235.0998 C13H23N3O 237.1842 cl6H32N 238.2536
CI2H12NO4 234.0766 ' CI6H15N2 235.1236 c13h25n 237.2081 СпН18О 238.1358
c,2h14n2o3 234.1005 C16H27O 235.2063 C14H9N2O2 237.0664 C17H2oN 238.1597
C12H16N3O2 234.1244 c16h29n 235.2301 C14HhN3O 237.0903 CnH.4 238.2662
c12h18n4o 234.1482 C17H15O 235.1123 C14H13N4 237.1142 Ci8H22 238.1722
C12H26O4 234.1832 c17h]7n 235.1362 Ci4H21O3 237.1491 239
C13H14O4 234.0892 C17H31 235.2427 Ci4H23NO2 237.1730 CnHI5N2O4 239.1032
CI3HI6NO3 234.1131 Cl 8^19 235.1488 C14H25N2O 237.1968 ChH17N3O3 239.1271
Cj3H18N2O2 234.1369 236 C14H27N3 237.2207 CnH19N4O2 239.1509
CbH^O 234.1608 CioH24n2o4 236.1737 Ci5H9O3 237.0552 c12h17no4 239.1158
c13h22n4 234.1846 CnH12N2O4 236.0797 C15HnNO2 237.0790 Ci2H19N2O3 239.1396
c14h10n4 234.0907 CiiH]4N3O3 236.1036 c15h13n2o 237.1029 Ci2H21N3O2 239.1635
C14H18O3 234.1256 ChH16N4O2 236.1275 c15h15n3 237.1267 Ci2H23N4O 239.1873
CI4H2()NO2 234.1495 Ci 7H7N 3O3 236.0096 C]5H25O2 237.1855 C13H9N3O2 239.0695
cI4h22n2o 234.1733 C12H4N4O2 236.0335 c15h27no 237.2094 C13HuN4O 239.0934
c14h24n3 234.1972 c12h14no4 236.0923 C15H29N2 237.2332 Ci3H19O4 239.1284
c15hI0n2o 234.0794 Ci2H16N2O3 236.1162 С1бН13О2 237.0916 c13h21no3 239.1522
c15h12n3 234.1032 Ci2H18N3O2 236.1400 c16h15no 237.1154 c13h23n2o2 239.1761
C15H22O2 234.1620 Ci2H20N4O 236.1639 c16h17n2 237.1393 Q3H25N3O 239.1999
C,5H24NO 234.1859 c13h8n4o 236.0699 С1бН29О 237.2219 Ci3H27N4 239.2238
c15h26n2 234.2098 CbH16O4 236.1049 C16H31N 237.2458 C14H9NO3 239.0583
C16H10O2 234.0681 c13h18no3 236.1287 C17H17O 237.1280 Ci4HnN2O2 239.0821
c16h12no 234.0919 Ci3H20N2O2 236.1526 c17h19n 237.1519 C14H13N3O 239.1060
Ct6H14N2 234.1158 Cj3H22N3O 236.1764 спн33 237.2584 c14h15n4 239.1298
СиНзбО 234.1985 Ci3H24N4 236.2003 C18H21 237.1644 Ci4H23O3 239.1648
cI6H28N 234.2223 C14H10N3O 236.0825 238 Cj 4H25N O2 239.1886
c17h16n 234.1284 C14H12N4 236.1063 CnH14N2O4 238.0954 C14H27N2O 239.2125
СрНда 234.2349 СиНгоОэ 236.1413 CUH16N3O3 238.1193 Ci4H29N3 239.2363
QgHig 234.1409 c14h22no2 236.1651 CnH18N4O2 238.1431 Ci5HnO3 239.0708
235 Ci4H24N2O 236.1890 C12H16NO4 238.1080 c15h13no2 239.0947
^10^23^2^4 235.1659 c14h26n3 236.2129 Ci2H]8N2O3 238.1318 c15h15n2o 239.1185
QoHzsNjOs 235.1897 Ci5HwNO2 236.0712 Ci2H20N3O2 238.1557 c15h17n3 239.1424
c„h„n2o4 235.0719 c15h12n2o 236.0950 c12h22n4o 238.1795 Ci5H27O2 239.2012
C„H13N3O3 235.0958 c15h14n3 236.1189 Ci3H8N3O2 238.0617 c15h29no 239.2250
CnH15N4O2 235.1196 Ci5H24O2 236.1777 Ci3H10N4O 238.0856 c15h31n2 239.2489
cuh25no4 235.1784 c15h26no 236.2015 Q3H18O4 238.1205 С1бН15О2 239.1072
CI2H13NO4 235.0845 C15H28N2 236.2254 Ci3H20NO3 238.1444 c16h17no 239.1311
Ci2HI5N2O3 235.1083 C16H12O2 236.0837 . C13H22N2O2 238.1682 Ci6H19N2 239.1549
C12Hi7N3O2 235.1322 c16h14no 236.1076 Cj3H24N3O 238.1921 c16H31o 239.2376
c12h19n4o 235.1560 Ci6H16N2 236.1315 c13h26n4 238.2160 C16H33N 239.2615
C1JH15O4 235.0970 236.2141 Ci4H10N2O2 238.0743 Ci7H19O 239.1436
CI3H17NO3 235.1209 Q6H30N 236.2380 C14H12N3O 238.0981 c17h21n 239.1675
C)3H19N2O2 235.1447 c17H16o 236.1202 Ci4H14N4 238.1220 c17h35 239.2740
c13h21n3o 235.1686 c17h18n 236.1440 C14H22O3 238.1569 Q8h23 239.1801
c13h23n4 235.1925 CnH32 236.2505 C14H24NO2 238.1808 240
c14h9n3o 235.0746 Ci8H20 236.1566 c14h26n2o 238.2046 ChH16N2O4 240.1111
c14h„n4 235.0985 237 238.2285 ChH18N3O3 240.1349
C|4H19O3 235.1334 ChH13N2O4 237.0876 Ci5H10O3 238.0630 ChH20N4O2 240.1588
C14H21NO2 235.1573 CnH15N3O3 237.1114 Cl5H12NO2 238.0868 Ci2H8N4O2 240.0648
C14H23N2O 235.1811 CnH17N4O2 237.1353 c15h14n2o 238.1107 Ci2H18NO4 240.1236
С14НИК3 235.2050 C12H15NO4 237.1001 c15h16n3 238.1346 Ci2H20N2O3 240.1475
c15h9no2 235.0634 Ci2H17N2O3 237.1240 CisH26O2 238.1934 С12^22^3О2 240.1713
cI5h„n2o 235.0872 C12H19N3O2 237.1478 c15h28no 238.2172 Ci2H24N4O 240.1952
82
/. Маа-спектрометрия
Приложение А (продолжение)
'M M M M
Ci3H8N2O3 240.0535 c15h17n2o 241.1342 c17h24n 242.1910 c12h24n2o3 244.1788
Q3H10N3O2 240.0774 CisH19N3 241.1580 QeHioO 242.0732 Ci2H26N3O2 244.2026
cI3h12n4o 240.1012 C15H29O2 241.2168 c18h12n 242.0970 Ci2H28N4O 244.2265
Q3H20O4 240.1362 c15h31no 241.2407 Q8H26 242.2036 c13h10no4 244.0610
c13h22no3 240.1600 CisH33N2 241.2646 Ci9H14 242.1096 Ci3H12N2O3 244.0848
C13H24N2O2 240.1839 C16H17O2 241.1229 243 c13h14n3o2 244.1087
Ci3H28N4 240.2316 c16h19no 241.1467 CnH19N2O4 243.1345 C13H16N4O 244.1325
Cj4H8O4 240.0422 c16h21n2 241.1706 cnH21N3o3 243.1584 Ci3H24O4 244.1675
Ci4H10NO3 240.0661 c16H33o 241.2533 СпН^Ь^Ог 243.1822 cI3h26no3 244.1914
Ci4H12N2O2 240.0899 c16h35n 241.2771 c12h7n2o4 243.0406 C]3H28N2O2 244.2152
c14h14n3o 240.1138 CI7H2IO 241.1593 C12H9N3O3 243.0644 CbH^O 244.2391
c14h16n4 240.1377 c17h23n 241.1832 C12HnN4O2 243.0883 CnH32N4 244.2629
ChH24O3 240.1726 Q8H25 241.1957 c12h21no4 243.1471 СМН12О4 244.0735
Ci4H26NO2 240.1965 242 c12h23n2o3 243.1710 C14H14NO3 244.0974
CI4H«N2O 240.2203 СцН18М2О4 242.1267 243.1948 Ci4H16N2O2 244.1213
CuH^Nj 240.2442 CnH20N3O3 242.1506 C12H27N4O 243.2187 c14h18n3o 244.1451
C15Hi2O3 240.0786 CnH22N4O2 242.1744 c13h9no4 243.0532 Ci4H20N4 244.1690
c15h14no2 240.1025 c12h8n3o3 242.0566 C13HhN2O3 243.0770 ChH28O3 244.2039
c15h16n2o 240.1264 Ci2H1()N4O2 242.0805 Ci3H13N3O2 243.1009 СмНэоЬЮг 244.2278
c15h18n3 240.1502 C12Ha)NO4 242.1393 c13h15n4o 243.1247 Ci4H32N2O 244.2516
CisH2XO2 240.2090 Ci2H22N2O3 242.1631 243.1597 CisH16O3 244.1100
CISH30NO 240.2329 C12H24N3O2 242.1870 c13h25no3 243.1835 CisH18NO2 244.1338
Ci5H32N2 240.2567 c12h26n4o 242.2108 c13h27n2o2 243.2074 c15h20n2o 244.1577
C]fjH16O2 240.1151 c13h8no4 242.0453 c13h29n3o 243.2312 Ci§H22N3 244.1815
C16H20N2 240.1628 Ci3H10N2O3 242.0692 c13h31n4 243.2551 CisH32O2 244.2403
c16h18no 240.1389 C,3H12N3O2 242.0930 Ci4HhO4 243.0657 C16H10N3 244.0876
Ci6H32O 240.2454 CI3Hi4N4O 242.1169 c14h13no3 243.0896 С1бН20О2 244.1464
с16н^ 240.2693 C13H22O4 242.1518 C14H15N2O-> 243.1134 c16h22no 244.1702
Ci7H2()O 240.1515 c13h24no3 242.1757 c14h17n3o 243.1373 Cj6H24N2 244.1941
c17h22n 240.1753 C„H26N2O2 242.1996 c14h19n4 243.1611 c17h1()no 244.0763
с17н36 240.2819 242.2234 C14H27O3 243.1961 c17h12n2 244.1001
C18H24 240.1879 C13H30N4 242.2473 C14H29NO2 243.2199 c17h24o 244.1828
241 Cj4H|0O4 242.0579 c14h31n2o 243.2438 c17h26n 244.2067
C„HI7N2O4 241.1189 c14h12no3 242.0817 c14h33n3 243.2677 Cj8H12O 244.0888
ChH19N3O3 241.1427 C14H14N2O2 242.1056 CisH15O3 243.1021 c18h14n 244.1127
CnH21N4O2 241.1666 c14h16n3o 242.1295 c15h17no2 243.1260 Ci8H28 244.2192
c12h19no4 241.1315 c14h18n4 242.1533 c15h19n2o 243.1498 Ci9H16 244.1253
C12H21N2O3 241.1553 ChH26O3 242.1883 c15h21n3 243.1737 245
C12H23N3O2 241.1791 C14H28NO2 242.2121 CisH31O2 243.2325 CnH21N2O4 245.1502
Ci2H25N4O 241.2030 242.2360 c15h33no 243.2564 CnH23N3O3 245.1741
С1зНцМ3О2 241.0852 c14h32n3 242.2598 C16Hi9O2 243.1385 CnH25N4O2 245.1979
c13h13n4o 241.1091 С13Н|4О3 242.0943 c16h21no 243.1624 C12H9N2O4 245.0563
C13H21O4 241.1440 C15HI6NO2 242.1182 c16h23n2 243.1863 Ci2HnN3O3 245.0801
Ci3H25N2O2 241.1679 c15h18n2o 242.1420 C^H^O 243.1750 Ci2H13N4O2 245.1040
c13h25n2o2 241.1917 CisH2ON3 242.1659 c17h25n 243.1988 Ci2H23NO4 245.1628
c13h27n3o 241.2156 CisH30O2 242.2247 Ci8HnO 243.0810 Ci2H25N2O3 245.1866
c13h29n4 241.2394 c15h32no 242.2485 c18h13n 243.1049 Ci2H27N3O2 245.2105
C14HhNO3 241.0739 C15H34N2 242.2724 Ci8H27 243.2114 C12H29N4O 245.2343
Ci4H13N2O2 241.0978 C16HI8O2 242.1307 C19H15 243.1174 c13HnNO4 245.0688
c14h15n3o 241.1216 c,6h2()no 242.1546 244 c13h13n2o3 245.0927
c14h17n4 241.1445 C,6H22N2 242.1784 CnH20N2O4 244.1424 Q3h15n3o2 245.1165
Ci4H25O3 241.1804 242.2611 ChH22N3O3 244.1662 c13h17n4o 245.1404
c14h27no2 241.2043 С]бН18О2 242.1307 c„h24n4o2 244.1901 Ci3H25O4 245.1753
C14H29N2O 241.2281 c16h20no 242.1546 c12h8n2o4 244.0484 c13h27no3 245.1992
c14h31n3 241.2520 242.1784 Ci2Hi0N3O3 244.0723 CbH29N2O2 245.2230
CisH13o3 241.0865 c^o 242.2611 c12h12n4o2 244.0961 C13H31N3O 245.2469
c15h15no2 241.1103 C17H22O 242.1871 Ci2H22NO4 244.1549 Ci4H13O4 245.0814
Приложение А
83
Приложение А (окончание)
M M M M
cI4h15no3 245.1052 ^16^26^2 246.2098 C12Hi4N3O3 248.1036 Ci5H25N2O 249.1968
c14h17n2o2 245.1291 Ci7H10O2 246.0681 Ci2H16N4O2 248.1275 c15h27n3 249.2207
C14H19N3O 245.1529 c17h12no 246.0919 c12h26no4 248.1863 c16huno2 249.0790
c14h21n4 245.1768 c17HI4N2 246.1158 Ci2H28N2O3 248.2101 c16h13n2o 249.1029
C14H29O3 245.2117 C17H26O 246.1985 C13H14NO4 248.0923 c16h15n3 249.1267
C14H31NO2 245.2356 C17H28N 246.2223 Ci3H16N2O3 248.1162 С1бН25О2 249.1855.
CisH17O3 245.1178 C18H14O 246.1045 CisHi8N3O2 248.1400 c16h27no 249.2094
c15h19no2 245.1416 c18h16n 246.1284 C] 3^20^0 248.1639 c16h29n2 249.2332
CisHj^O 245.1655 ^18^30 246.2349 Ci3H28O4 248.1988 Ci7H13O2 249.0916
C15H23N, 245.1894 Ci9Hi8 246.1409 Ci4H16O4 248.1049 c17h15no 249.1154
c16h9n2o 245.0715 247 Ci4H20N2O2 248.1526 c17h17n2 249.1393
CI6HnN3 245.0954 C^N^ 247.1659 C14H22N3O 248.1764 C17H29O 249.2219
Q6^21O2 245.1542 QiH2sN3O3 247.1897 Ci4H24N4 248.2003 C17H31N 249.2458
c1(,h23no 245.1781 ChH27N4O2 247.2136 c15h10n3o 248.0825 Ci8H17O 249.1280
СиНиЛ, 245.2019 C12HnN2O4 247.0719 C15H12N4 248.1063 c18h19n 249.1519
C17HnNO 245.0841 C12H13N3O3 247.0958 CisH20O3 248.1413 Ci8H33 249.2584
c17h13n2 245.1080 Ci2H15N4O2 247.1196 Ci5H22NO2 248.1651 Ci9H21 249.1644
C17H25O 245.1906 c12h25no4 247.1784 c15h24n2o 248.1890 250
c17h27n 245.2145 C12H27N2O3 247.2023 c15h26n3 248.2129 C„H26N2O4 250.1894
Ci8h13o 245.0967 C12H29N3O2 247.2261 c16h10no2 248.0712 C12H14N2O4 250.0954
c18h15n 245.1205 cI3h13no4 247.0845 c16h12n2o 248.0950 Ci2H16N3O3 250.1193
C18H29 245.2270 Ci3H15N2O3 247.1083 c16h14n3 248.1189 Ci2H18N4O2 250.1431
Q9H17 245.1331 C13H17N3O2 247.1322 Ci6H24O2 248.1777 c13h16no4 250.1080
246 c13h19n4o 247.1560 c16h26no 248.2015 CnH18N2O3 250.1318
CnH22N2O4 246.1580 C13H27O4 247.1910 Ci6H2eN2 248.2254 CbH20N3O2 250.1557
CnH24N3O3 246.1819 Ci3H29NO3 247.2148 Ci7Hi2O2 248.0837 CnH22N4O 250.1795
СцН26М4О2 246.2057 Ci4H15O4 247.0970 c17h14no 248.1076 C14H10N4O 250.0856
C12H10N2O4 246.0641 c14h17no3 247.1209 C17H16N2 248.1315 Ci4H20NO3 250.1444
C12H12N3O3 246.0879 C14H19N2O2 247.1448 C17H28O 248.2141 СмН^МгОг 250.1682
C12H14N4O2 246.1118 c14h21n3o 247.1686 C17H3oN 248.2380 C14H24N3O 250.1921
c12h24no4 246.1706 247.1925 C18H16O 248.1202 c14h26n4 250.2160
^12^26^2O3 246.1945 C15H9N3O 247.0746 c18h18n 248.1440 Ci5H10N2O2 250.0743
Ci2H2eN3O2 246.2183 c15hun4 247.0985 c18h32 248.2505 c15h12n3o 250.0981
С12Н^4О 246.2422 CisH19O3 247.1334 C19H20 248.1566 c15h14n4 250.1220
c13h12no4 246.0766 c15h21no2 247.1573 249 CisH22O3 250.1569
CbH14N2O3 246.1005 c15h23n2o 247.1811 Cl 1H25N2O4 249.1815 c15h24no2 250.1808
Ci3H16N3O2 246.1244 c15h25n3 247.2050 ChH27N3O3 249.2054 CbH^O 250.2046
C13H18N4O 246.1482 C16HnN2O 247.0872 C12H13N2O4 249.0876 CisH28N3 250.2285
Ci3H26O4 246.1832' C16H13N3 247.1111 Ci2H15N3O3 249.1114 С1бН10О3 250.0630
Ci3H28NO3 246.2070 С1бН23О2 247.1699 Ci2H17N4O2 249.1353 c16h12no2 250.0868
С1зНэоЫ202 246.2309 c16h25no 247.1937 С\2Н27М C4 249.1941 c16h14n2o 250.1107
C14H14O4 246.0892 С1бН27Ь12 247.2176 c13h15no4 249.1001 C16H16N3 250.1346
c14h16no3 246.1131 C17HnO2 247.0759 Ci3H17N2O3 249.1240 C16H26O2 250.1934
Ci4H18N2O2 246.1369 c17h13no 247.0998 C13H19N3O2 249.1478 C^NO 250.2172
C14H20N3O 246.1608 Q7h15n2 247.1236 C13H21N4O 249.1717 CieHjoh^ 250.2411
246.1846 C17H27O 247.2063 c14h9n4o 249.0777 Ci7H14O2 250.0994
СмНадОэ 246.2196 c17h29n 247.2301 Ci4H17O4 249.1127 c17h16no 250.1233
c15h10n4 246.0907 Ci8H15O 247.1123 c14h19no3 249.1365 Ci7H18N2 250.1471
CisH18O3 246.1256 c18H17bT 247.1362 C14H21N2O2 249.1604 Ci7H30O 250.2298
C15H20NO2 246.1495 Ci8H31 247.2427 c14h23n3o 249.1842 c17h32n 250.2536
С^Н^Ь^О 246.1733 ^19H19 247.1488 c14h25n4 249.2081 Ci8H18O 250.1358
c15h24n3 246.1972 248 c15h9n2o2 249.0664 c18h20n 250.1597
^-'16^1()N2O 246.0794 СцН24Ы2О4 248.1737 Ci5HnN3O 249.0903 Ci8H34 250.2662
c16h12n3 246.1032 СцН26М3О3 248.1976 c15h13n4 249.1142 Ci9H22 250.1722
С1бН22О2 246.1620 ChH28N4O2 248.2214 CisH21O3 249.1491
c16h24no 246.1859 C12H12N2O4 248.0797 C^H^NO, 249.1730
84
1. Маа-спектрометрия
Приложение Б. Фрагментные ионы
Все перечисленные фрагментные ионы несут заряд +1.
Рекомендуется использовать совместно с приложением
В. Эта таблица предназначена для подсказки возмож-
ных вариантов при первоначальном рассмотрении и не
претендует на полноту. В качестве дополнения можно
рекомендовать Приложение П в книге (Hamming, Foster,
1972), таблицу А-7 в книге (McLafferty, 1993), и данные
по спектрам высокого разрешения (McLafferty, 1982).
m/z Ионы*
14 сн2 70 C5H10
15 СН3 71 C5H11,C3H7C=O
16 О 72 C2H5C(=O)CH2 + н, СзН7СНМН2,
17 он (CH3)2N=C=O, CjHjNHCHCHj
18 H2O,NH4 и изомеры
19 F,H3O 73 Гомологи 59,(CH3)3Si
26 C=N,C2H2 74 CH2CO2CH3 + н
27 ед 75 со2ед5 + 2н, <едсо2 + гн,
28 QH4, СО, N2 (воздух), CH=NH сн28ед, (ch3)2csh, (сн3о)2сн,.
29 <ед,сно (CH3)2SiOH
30 CH2NH2,NO 76 ед(едху)
31 СН2ОН,ОСН3 77 ед^сда)
32 О2 (воздух) 78 CjHs + H
33 SH,CH2F 79 ед5 ч-гн,79^
34 H2S 80 CH3SS+Н,Н79Вть,
35 36 35С1Ь Н»СР QCH-Q
39 ед
40 CH2C=N, Аг (воздух)
41 СзН5, ch2c=n + н, ед2мн Н СН2
42 ед,ед2о ГУ
43 CjH7, СН3С=О, QHjN 81
44 СН2С(=О)Н + Н, CH3CHNH2, СО2 (воздух) О CHj, CjHj
45 NH2C=O, (CH3)2N СН3СН(ОН), СН2СН2ОН, СН2ОСН3, С(=О)ОН 1 82 (CH2)4CSN, СбН10, C3SCl2b
46 no2 83 c^oFcy»,^ >
47 CH2SH,CH3S s
48 CH3S + H
49 сн235аь 85
51 CH2F2, C4H3 С > ’ .еди>едс=о,с1з5с1Р2ь
53 с4н5
54 55 ch2ch2c^n с4н7,сн2=снс=о 86 C3H7C(=O)CH2 + H, C4H9CHNH2 и изомеры
56 C4H8 87 С3Н7СО2, гомологи 73,
57 58 C4H9, C^HjC—0 CH3C(=O)CH2 + H.QHjCHNHj, (CH3)2NCH2, QHjNHCH,, QHjS 88 СН2СН2СО2СН3 сн2со2ед5 + н
59 (сн3)2сон, сн2оед5, со2сн3, NH2C(=O)CH2 + H, CH3OCHCH3, CH3CHCH2OH, QHjCHOH 89 со2ед + гн, (| ]
60 CH2CO2H + H, ch2ono
61 CH3CO2 + 2H, CH2CH2SH, CH2SCH3
65 CsHS 90 || J ,CHjCHONO2
66 91 (Од)сн2, (ед)сн + н, (ед)с + гн, (CH2)435Clb,(C6H5)N
67 ед
68 69 ch2ch2ch2c^n C5H,. CF3, CH3CH=CHC=O, CH2=C(CH3)C=O 92 || ^-амедод + н
Приложение Б
85
Приложение Б (окончание)
m/z Ионы°
93
СД,,(С6Н5)О,
С=О
94
(С6Н5)О + Н,
95 ОС=0
О
96 (CH2)5C=N
97
119 CF3CF2,(C6H5)C(CH3)2>
СН3СН(С6Н4)СН3, СОСОН^СНз
99
c^QHuO,
122 С6Н5СО2 + Н
123 F(C6H4)C=O, СбН5СО2 + 2Н
125 QHjSO
127 I
128 HI
100 C4H9C(=O)CH2 + H,C5HnCHNH2
101 СО2С4Н9
102 СН2СО2С2Н7 + Н
103 СО2С4Н9 + 2Н, CsHnS, СН(ОСН2СН3)2
104 QHjCHONOz
105 С6Н5С=О,С6Н5СН2СН2,С6Н5СНСНз
106 СбНзЫНСЕЪ
107 СбН5СН2О,НО(С6Н4)СН2,С2Н479Вг'5
131 C3F5,C6H5CH=CHC=O
135 (СН2)479ВгЛ
138 СО2(СбН4)ОН + Н
139 35C1(C6H4)C=O'S
141 СН21
147 (CH3)2Si=O—Si(CH3)3
108
с=о
154 (ОД),
109
“ Ионы, обозначенные «фрагмент + иН» (и = 1,2,3,.. .)> образуются в результате
перегруппировки, включающей перенос водорода.
6 Приведен только наиболее распространенный изотоп.
86
/. Маа-спектрометрия
Приложение В. Наиболее распространенные нейтральные фрагменты
Этот перечень предлагает варианты и не претендует на
полноту. Его следует использовать в сочетании с при-
ложением Б. Все указанные фрагменты отщепляются
в виде нейтральных частиц. В качестве дополнения
можно рекомендовать таблицу 5-19 в книге (Hamming,
Foster, 1972) и таблицу А-5 в книге (McLafferty, 1993).
М Возможные фрагменты
1 Н-
2 2Н-
15 СН3-
16 О (ArNO2, аминоксиды, сульфоксиды); -NH2 (карбоксамиды, сульфонамиды)
17 НО-
18 Н2О (спирты, альдегиды, кетоны)
19 F-
20 HF
26 СН=СН, -CH=N
27 СН2=СН-, HC=N (ароматические нитрилы, N-гетероциклы)
28 СН2=СН2, СО, (хиноны) (HCN + Н)
29 СН3СН2-, (этилкетоны, АгСН2СН2СН3), • СНО
30 NH2CH2-, СН2О (АгОСН3), NO (ArNO2),
31 • ОСН3 (метиловые сложные эфиры), • СН2ОН, CH3NH2
32 СН3ОН, S
33 HS • (тиолы), (• СН3 и Н2О)
34 H2S (тиолы)
35 С1-
36 НС1,2Н2О
37 Н2С1 (или НС1 + Н)
38 СзН2, QjN, F2
39 СзН3, HQN
40 СН3С=СН
41 СН2=СНСН2-
А
42 СН2=СНСН3, СН2=С=О, Н2С-----СН2, NCO, NCNH2
О О
II II
43 С3Н7 • (пропилкетоны, АгСН2—С3Н7), СН3С • (метилкетоны, CH3CG, где G = различные функциональные
группы), СН2 = СН— О •, (СН3- и СН2= СН2), HCNO
44 СН2=СНОН, СО2 (сложные эфиры, ангидриды), N2O, CONH2, NHCH2CH3
45 СН3СНОН, СН3СН2О - (этиловые сложные эфиры), СО2Н, CH3CH2NH2
46 (Н2О and СН2=СН2), СН3СН2ОН, -NO2 (ArNO2)
47 CH3S-
48 CH3SH, SO (сульфоксиды), O3
49 -CH2C1
51 -CHF2
52 C4H4, СУ^
53 C4H5
54 CH2=CH—CH=CH2
55 CH2=CHCHCH3
Приложение В
87
Приложение В (окончание)
м Возможные фрагменты
56 СН2=СНСН2СН3, СН3СН=СНСН3,2СО
57 С4Н9* (бутилкетоны), CjHsCO (этилкетоны, EtC=OG, G = различные структ. фрагменты)
58 •NCS, (NO + СО), СН3СОСН3, С4Н10 Н О О 1 II И А
59 снэос-, ch3cnh2, ZA
60 СзН7ОН, СН2=С(ОН)2 (ацетаты-эфиры)" Н 1 S-
61 ch3ch2s-,ZA
62 (H2S и СН2=СН2)
63 •СН2СН2С1
64 С3Н4, S2, SO2 сн3 1 л
68 сн2=с—сн=сн2
69 CF3", С5Н9-
71 QHjj- О
73 СН3СН2ОС-
74 С4Н,ОН
75 С6н3
76 c6H4,cs2
77 СбН5, CS2H
78 С6Н6, CS2H2, CsH4N
79 Br, CjHjN
80 HBr
85 •CC1F2
100 cf2=cf2
119 CF3—CF2-
122 C6H5COOH
127 I-
128 HI
Перегруппировка Мак-Лафферти.
2. Инфракрасная спектроскопия
2.1. Введение
Инфракрасное (ИК) излучение относится к той ча-
сти электромагнитного спектра, которая находится
между видимой и микроволновой областями. В ор-
ганической химии наибольшее применение нашла
только его ограниченная часть между 4000 и 400 см-1.
Определенный интерес представляют ближняя ИК-
область (14 290-4000 см-1) и дальняя (длинноволно-
вая)-700-200 см-1.
Из последующего краткого теоретического введения
станет ясно, что даже очень простая молекула может
дать чрезвычайно сложный спектр. Химик-органик
пользуется этим, когда сравнивает спектр неизвестного
вещества со спектром заведомо известного образца.
Совпадение всех полос - надежное доказательство
идентичности. Невероятно, чтобы два соединения,
за исключением энантиомеров, могли бы дать оди-
наковые ИК-спектры.
Хотя ИК-спектр характеризует молекулу в целом,
оказывается, что некоторые группы атомов погло-
щают при определенной частоте (или вблизи нее)
независимо от структуры остальной части молекулы.
Положение этих полос, которые называют характери-
стическими, настолько постоянно, что по ним можно
судить о структурных элементах молекулы. Обширные
таблицы характеристических частот позволяют связать
многие полосы ИК-спектра с определенными струк-
турными элементами (функциональными группами),
входящими в состав молекулы. Мы будем опираться
на эти табличные данные при решении задач.
Так как в процессе идентификации мы используем не
только ИК-спектроскопию, но и другие методы, то де-
тального анализа спектра не требуется. В соответствии
с общим замыслом книги теория ИК-спектроскопии
будет представлена лишь в объеме, достаточном, чтобы
выполнить основную задачу - определить структуру
молекулы, используя ИК-спектры в сочетании с дан-
ными других спектроскопических методов.
Возросшее значение ИК-спектроскопии, используе-
мой химиками-органиками в своей экспериментальной
работе, доказывает число опубликованных книг, по-
священных (полностью или частично) обсуждению
приложений этого метода (см. список литературы
в конце главы). Нет недостатка и в библиографии,
охватывающей все аспекты ИК-спектроскопии, и в
атласах спектров. Среди справочных изданий наи-
более популярны атласы Садтлера (Sadder, 1994)
и Олдрича (Aldrich, 1989).°
2.2. Теория
Инфракрасное излучение с волновыми числами ме-
нее 100 см-1 при поглощении органической молеку-
лой преобразуется в энергию вращения. Поглощение
квантовано, поэтому вращательный спектр молекул
состоит из дискретных линий.
Излучение в интервале 10 000-100 см-1 при поглоще-
нии преобразуется органической молекулой в энергию
колебаний. Это поглощение также квантовано, но ко-
лебательный спектр состоит не из линий, а из полос,
поскольку каждый колебательный переход сопровожда-
ется изменениями вращательных состояний. Именно
такие колебательно-вращательные полосы, особенно
те, которые проявляются между 4000 и 400 см-1, будут
рассматриваться в дальнейшем. Частота или длина
волны, при которой наблюдается полоса поглощения,
зависят от относительных масс атомов, силовых по-
стоянных связей и геометрии молекулы.
Положение полос в спектрах обозначается в данном
издании через волновые числа (v), которые измеряют
в обратных сантиметрах (см-1); эта единица прямо про-
порциональна энергии колебания, а шкалы современ-
ных спектрометров линейны относительно см-1. В бо-
лее старой литературе использовали длины волн (X),
которые измеряли в микрометрах (1 мкм = 10-6 м;
прежнее название - микрон). Длины волн и волновые
числа связаны следующим соотношением:
см-1 = 104/мкм
Заметим также, что волновые числа (v) иногда на-
зывают «частотами». В принципе, это неправильно,
° Доступные для отечественного читателя издания приведе-
ны в списке дополнительной литературы. - Прим, перев.
2.2. Теория
89
так как волновые числа (v в см-1) равны 1 • 104/Х
в мкм, а частоты (v в Гц) равны с/Х в см, где с -
скорость света (ЗЮ10 см/с). Символ v читается
«ню с чертой». В настоящей книге используются
спектры в линейных относительно см-1 шкалах,
если не указано другое. В дальнейшем будет пока-
зано (см. рис. 2.6), что спектры, зарегистрированные
в шкалах мкм и см-1, существенно различаются по
внешнему виду.
Интенсивности полос выражаются либо через про-
пускание (Г,%), либо через оптическую плотность
(А). Пропускание - это отношение энергии излуче-
ния, пропущенного образцом, к энергии излучения,
падающего на образец. Оптическая плотность - это
десятичный логарифм величины, обратной пропуска-
нию: А = lg(l/7). Химики-органики обычно обозначают
интенсивности полос в полуколичественных терминах
(с. - сильная, ср. - средняя, сл. - слабая, английские
обозначения s, m, w соответственно).
Молекулярные колебания подразделяются на
два типа: валентные (stretching) и деформацион-
ные (bending). Валентное колебание - это такое
ритмичное движение вдоль связи, когда межатом-
ное расстояние периодически увеличивается или
уменьшается. Деформационное колебание может
заключаться в изменении валентного угла (т.е. угла,
образованного связями у общего атома) или в дви-
жении группы атомов по отношению к остальной
части молекулы без смещения атомов относительно
друг друга внутри этой группы. Например, крутиль-
ные (twisting), маятниковые (rocking) и торсион-
ные (torsional) колебания происходят с изменением
валентных углов в системе внутренних координат
молекулы.
В ИК-спектрах наблюдаются только такие колеба-
ния, которые приводят к периодическому изменению
дипольного момента молекулы. Переменное электри-
ческое поле, возникающее при изменении распре-
деления зарядов в процессе колебаний, связывает
колебание молекулы с осциллирующим электрическим
полем электромагнитного излучения.
Число степеней свободы многоатомной молекулы
равно сумме степеней свободы составляющих ее
индивидуальных атомов. Каждый атом имеет 3 сте-
пени свободы, которые соответствуют декартовым
координатам (x,y,z), необходимым для описания его
положения относительно других атомов в молекуле.
Поэтому молекула из п атомов имеет Зп степеней
свободы. Для нелинейной молекулы 3 координаты
описывают вращательное движение и 3 координа-
ты - поступательное движение молекулы как целого;
остающиеся Зп - 6 координат описывают основные
(нормальные) колебания. Линейные молекулы имеют
Зп - 5 степеней свободы, так как в этом случае для
описания вращения требуются только 2 координаты.
Основные колебания происходят без изменения
положения центра тяжести молекулы. Три основ-
ных колебания нелинейной трехатомной молекулы
воды изображены в верхней части рис. 2.1. Отметим,
что взаимодействующие («связанные») симметрич-
ное и антисимметричное валентные колебания очень
близки по частоте и сильно отличаются от ножничных
(scissoring) деформационных колебаний.
Молекула СО2 линейна, состоит из трех атомов
и поэтому имеет 4 основных колебания [(3-3) - 5],
как показано в средней части рис. 2.1. Симметричное
валентное колебание (1) неактивно в ИК-спектре,
так как во время этого колебания дипольный момент
молекулы не изменяется. Деформационные колеба-
ния (3) и (4) эквивалентны. Они имеют одинаковую
частоту и называются дважды вырожденными.
Различные валентные и деформационные колебания
для фрагмента АХ2, являющегося частью молекулы,
например группы СН2 в молекулах углеводородов,
показаны в нижней части рис. 2.1. В этом случае
правило (Зп - 6) не применимо, поскольку группа
СН2 является только частью молекулы.
Теоретическое число основных колебаний (частот
поглощения) наблюдается редко, так как, с одной сто-
роны, число полос увеличивается за счет обертонов
(частот, кратных данной частоте) и составных частот
(сумма двух разных частот). С другой стороны, тео-
ретическое число полос уменьшается за счет сле-
дующих факторов.
1. Часть основных частот не попадает в область
4000-400 см1.
2. Некоторые из основных полос являются такими
слабыми, что не наблюдаются.
3. Частоты некоторых основных колебаний настоль-
ко близки, что сливаются и проявляются в виде
одной полосы.
4. Вырождение нескольких частот в высокосиммет-
ричных молекулах.
5. Некоторые из основных колебаний отсутствуют
из-за того, что в процессе колебания не изменя-
ется дипольный момент молекулы.
Используя закон Гука, можно сделать прибли-
зительное отнесение частот валентных колебаний.
Два атома и связь между ними рассматриваются
как гармонический осциллятор, состоящий из двух
масс, соединенных пружинкой. Следующее уравне-
ние, выведенное из закона Гука, связывает частоту
90
2. Инфракрасная спектроскопия
колебания с массами атомов и силовой постоянной
связи.
-= 1 I f
2пс у (МхМу)!(Мх 4- Му)
где V - волновое число (см-1), с-скорость света (см/с),
/-силовая постоянная связи (дин/см), Мх и Му- со-
ответственно массы атомов х и у (г).
Для одинарной связи значение / около 5105 дин/см.
Для двойной связи оно приблизительно в два раза
больше и для тройной связи - в три раза (см. табл. 2.1).
Силовую постоянную / можно рассматривать
как «жесткость» связи. Величина силовой посто-
янной может коррелировать с такими свойствами,
как порядок связи и ее прочность. Так как часто-
та колебания прямо пропорциональна квадратному
корню из силовой постоянной, частота понижается
с уменьшением прочности связи.
Применение данного уравнения к валентным ко-
лебаниям С-Н (если для массы углерода исполь-
зуется значение 2.1010-23 г, а для массы водорода
1.67-10-24 г) дает частоту 3032 см-1 (в гармоническом
приближении). В действительности валентные ко-
лебания С-Н в метильной и метиленовой группах
наблюдаются в области 2960-2850 см-1. Расхожде-
ние вызвано тем, что при расчете не учитывается
влияние атомов, соседних со связью С-Н. Частоты
поглощения в ИК-области обычно используются для
вычисления силовых постоянных связей.
Для отнесения частот валентных колебаний С-Н
часто используется дейтерирование, в результате
которого происходит смещение полос поглощения.
Таблица 2.1. Вычисленные на основе закона Гука
области поглощения валентных колебаний некото-
рых связей
Тип связи Силовые постоянные f, дин/см Области поглощения, см-1
Вычисл. Эксперим.
С-0 5.0-105 1113 1300-800
С-С 4.5-105 1128 1300-800
C-N 4.9105 1135 1250-1000
с=с 9.7-105 1657 1900-1500
с=о 12.1-105 1731 1850-1600
с=с 15.6-105 2101 2150-2100
C-D 5.0-105 2225 2250-2080
С-Н 5.0-105 3032 3000-2850
О-Н 7.0-105 3553 3800-2700
Приведенное выше уравнение можно использовать
для оценки изменения частоты валентных колеба-
ний в результате дейтерирования. Приведенная масса
для двухатомного фрагмента С-Н равна
МСМН/(МС+А/Н). Так как МС»А/Н, то приближенно
можно записать МсМп!Мс или Мн. При замене во-
дорода в группе С-Н на дейтерий С-D этот член
становится равным MD; частота, по закону Гука,
обратно пропорциональна квадратному корню из
массы изотопа водорода. Вычисленное отношение
частот валентных колебаний С-Н к С-D должно быть
равно V2. Если это отношение гораздо меньше V2,
то можно предположить, что это колебание нель-
зя рассматривать как простое валентное колебание
С-Н; скорее всего его можно отнести к смешанно-
му колебанию, возникающему при взаимодействии
других колебаний.
Приближенные оценки, основанные на законе Гука,
можно сделать путем расчета частот валентных ко-
лебаний для некоторых типов связей, как показано
в табл. 2.1.
Что бы аппроксимировать частоты валентных
колебаний на основании закона Гука, необходимо
оценить относительные вклады не только атомных
масс, но и прочности связей. Например, сравнение
частот валентных колебаний связей С-Н и F-Н только
с использованием атомных масс приводит к непра-
вильному выводу, что частоты колебаний связи F-H
ниже, чем связи С-Н. Однако, увеличение силовой
постоянной слева направо вдоль второго и третьего
периодов периодической системы элементов оказывает
большее влияние, чем увеличение массы. Поэтому
группа F-Н поглощает при более высокой частоте
(4138 см-1), чем группа С-Н (3040 см-1).
Функциональные группы, имеющие большой ди-
польный момент, дают сильные полосы поглощения
в ИК-области.
2.2.1. Взаимодействие колебаний
Если два осциллятора не очень сильно отличаются по
частоте и связаны друг с другом через общий атом,
то они редко ведут себя независимо. Это происходит
из-за того, что между осцилляторами существует меха-
ническое взаимодействие. Например, молекула диокси-
да углерода, содержащая две связи С=О (см. рис. 2.1)
с общим атомом углерода, имеет два основных валент-
ных колебания - антисимметричное и симметричное,
различающиеся по частоте. Симметричное валентное
колебание представляет собой одновременное рас-
тяжение или сокращение связей С=О, т. е. колебание
двух связей в фазе. Поглощение наблюдалось бы при
больших длинах волн, чем для карбонильной группы
2.2. Теория
91
н2о
а
Симметричное
валентное, vs ОН,
3652 см'1
Антисимметричное
валентное, Vgs ОН,
3756 см"1
Ножничное (scissoring)
деформационное, 6S ОН,
1596 см"1
© © G
(1) Симметричное
валентное, vs СО2,
1340 см1
(2) Антисимметричное
валентное, Vas СО2,
2350 см"1
(4) Ножничное (scissoring)
деформационное, Ss СО2,
(3) Ножничное (scissoring) 665 см"1
деформационное, Ss СО2,
665 см"1
в
Симметричное
валентное, vs СН2,
-2853 см"1
Антисимметричное
валентное, СН2,
-2926 см"1
Плоскостное деформационное
или ножничное (scissoring), 6S СН2,
-1465 см"1
Внеплоскостное
деформационное
или веерное (wagging),
соСН2, 1350-1150 см"1
Внеплоскостное
деформационное
или крутильное (twisting),
тСН2, 1350-1150 см"1
Плоскостное
деформационное
или маятниковое (rocking),
рСН2, -720 см"1
Рис. 2.1. Формы колебаний молекулы воды (я). Формы колебаний линейной
молекулы СО2 (б). Формы колебаний группы СН2 (в) (знаки + и - обозначают
движение атомов перпендикулярно плоскости страницы)
92
2. Инфракрасная спектроскопия
в алифатических кетонах. Симметричное валентное
колебание не изменяет дипольный момент молекулы
(ц) и поэтому «неактивно» в ИК-спектре, но наблю-
дается в спектре комбинационного рассеяния около
1340 см-1. (В спектре комбинационного рассеяния ин-
тенсивность зависит от поляризуемости связи, а не от
изменений дипольного момента.) При антисимметрич-
ном валентном колебании две связи С=О колеблются
в противофазе: когда одна связь С=О растягивается,
другая - сокращается. Поскольку антисимметричное
валентное колебание вызывает изменение дипольного
момента, оно активно в ИК-спектре; полоса поглоще-
ния (2350 см-1) наблюдается в области более высоких
частот (коротких волн), чем для карбонильной группы
в алифатических кетонах.
<—о=с=о—> <—о=с=о<—
Симметричное Антисимметричное
колебание колебание
fJL = 0 =# 0
Эта разница в частотах поглощения карбонильной
группы в молекуле диоксида углерода обусловлена
сильной механической связью между осцилляторами,
т. е. взаимодействием колебаний. В противоположность
этому две кетонные карбонильные группы, разделенные
одним или большим числом атомов углерода, имеют
нормальное поглощение в области 1715 см-1, характер-
ное для изолированной карбонильной группы, так как
заметному взаимодействию препятствует внедренный
между ними атом (или атомы) углерода.
Механическим взаимодействием колебаний объ-
ясняется наличие двух полос валентных колебаний
N-H в области 3497-3077 см-1 в спектрах первичных
аминов и первичных амидов, двух полос валентных
колебаний С=О в области 1818-1720 см-1 в спектрах
ангидридов карбоновых кислот и имидов и двух полос
валентных колебаний С-Н в области 3000-2760 см-1
для метиленовых и метильных групп.
Широко используемые характеристические часто-
ты нередко включают связанные колебания. Спектры
спиртов имеют интенсивную полосу в области между
1260 и 1000 см-1, которая обычно обозначается как
«полоса валентного колебания С-О». В спектре ме-
тилового спирта эта полоса находится при 1034 см"1,
а в спектре этилового спирта-при 1053 см-1. Раз-
ветвление и ненасыщенность в этих соединениях
изменяют характеристическое поглощение (см. обсуж-
дение спектров спиртов). Очевидно, что валентное
колебание связи С-О не является изолированным,
а взаимодействует с антисимметричным колебанием
всей группы С-С-О.
Колебания, обусловленные деформацией валентного
угла, часто подобным образом взаимодействуют с ва-
лентными колебаниями. Так, частоты внеплоскостных
деформационных колебаний С-Н кольца в аромати-
ческих молекулах зависят от числа соседних атомов
водорода в кольце; колебательное взаимодействие
между атомами водорода испытывает влияние дефор-
мации углерод-углеродной связи кольца, к которой
присоединены эти атомы водорода.
Взаимное влияние валентных и деформационных
колебаний можно проиллюстрировать на примере
вторичных ациклических амидов, которые существуют
преимущественно в тиране-конформации: взаимодей-
ствие деформационного колебания N-H и валентного
колебания C-N приводит к интенсивной полосе по-
глощения в области 1563-1515 см-1.
Связь колебаний может быть эффективной только
при следующих условиях:
1. Взаимодействующие колебания должны относить-
ся к одному типу симметрии.
2. Сильное взаимодействие между валентными ко-
лебаниями требует общего атома между груп-
пами.
3. Взаимодействие колебаний групп максимально
в случае близости их частот.
4. Валентные и деформационные колебания взаимо-
действуют в том случае, когда растягивающаяся
связь образует одну из сторон изменяющегося
угла.
5. Для взаимодействия между деформационными
колебаниями необходима общая связь.
6. Взаимодействие колебаний становится пренебре-
жимо малым, если группы разделены одним или
большим числом атомов углерода, а колебания про-
исходят во взаимно перпендикулярных плоскостях.
Взаимодействие между двумя основными колеба-
ниями приводит к двум новым колебаниям с часто-
тами выше и ниже, чем наблюдаемые в отсутствие
взаимодействия. Основные колебания и обертоны или
составные частоты также могут взаимодействовать.
Такое взаимодействие известно как резонанс Ферми.
Примером резонанса Ферми является поглощение
диоксида углерода. Мы уже упоминали, что полоса
симметричного валентного колебания СО2 проявля-
ется в спектре КР при 1340 см-1. В действительности
наблюдаются две полосы: одна при 1286 см-1, другая
при 1388 см-1. Расщепление возникает в результате
взаимодействия между основным валентным коле-
банием С=О при 1340 см-1 и первым обертоном де-
формационного колебания. Основное деформацион-
2.2. Теория
93
Рис. 2.2. ИК-Спектр чистого циклогептанона
ное колебание проявляется при 666 см-1, а первый
обертон-при 1334 см-1.
Резонанс Ферми - обычное явление в ИК-спектрах
и спектрах КР. Для его возникновения требуется
принадлежность колебательных уровней к одному
типу симметрии, а расположение взаимодействующих
групп в молекуле должно допускать механическую
связь.
Одним из примеров резонанса Ферми в органиче-
ских структурах является «дублетный» вид валентного
колебания карбонильной группы циклических кетонов
при достаточно высоком разрешении спектрального
прибора. На рис. 2.2 приведен спектр циклогепта-
нона при обычном разрешении. Полоса валентных
колебаний карбонильной группы при 1709 см-1 про-
является в виде синглета. На рис. 2.3 показана об-
ласть «карбонильного поглощения» циклопентанона
в четырех разных растворителях при соответствую-
щем разрешении. Наблюдающиеся дублеты полосы
Волновые числа (см1)
1740 1780 1740 1780 1740 1780 1740 1780
Рис. 2.3. ИК-Спектры циклопентанона в разных
растворителях: раствор в четыреххлористом
углероде (0.15 моль/л) (а); в сероуглероде
(0.023 моль/л) (б); в хлороформе (0.025 моль/л) (в);
жидкое состояние (тонкая пленка) (г). Спектральная
ширина щели 2 см-1
карбонильной группы возникают по причине ее ре-
зонанса Ферми с обертоном или составной частотой
а-метиленовой группы.
2.2.2. Водородная связь
Водородная связь может возникать в любой системе,
содержащей доноры протонов (группы Х-Н) и акцеп-
торы протонов (группы Y:), при условии, что возмож-
но эффективное перекрывание 5-орбитали водорода
с р- или л-орбиталью акцепторной группы. Атомы
X и Y: электроотрицательны, причем у Y: имеется
неподеленная пара электронов. Донорами протонов
в органических молекулах обычно являются карбок-
сильные, гидроксильные, амино- или амидогруппы.
Акцепторами протонов обычно служат атомы кис-
лорода, азота и галогенов. Ненасыщенные группы,
такие как этиленовый фрагмент С=С, также могут
выступать в роли акцепторов протонов.
Прочность водородной связи максимальна, когда
группа-донор протонов и ось орбитали неподе-
ленной пары коллинеарны. Прочность водородной
связи уменьшается при увеличении расстояния
между X и Y.
Водородная связь изменяет силовые постоянные
обеих групп; тем самым изменяются частоты как
валентного, так и деформационного колебаний. По-
лосы валентных колебаний группы Х-Н смещаются
в сторону более низких частот (более длинных волн),
причем интенсивность и ширина полосы обычно уве-
личиваются. Частота валентного колебания акцеп-
торной группы, например С=О, также понижается,
но в меньшей степени, чем группы-донора протонов.
Когда образуется водородная связь, частота дефор-
мационного колебания Н-Х, как правило, сдвигается
в сторону более коротких длин волн; этот сдвиг менее
выражен, чем для валентных колебаний.
94
2. Инфракрасная спектроскопия
Возникновение межмолекулярных водородных
связей приводит к ассоциации двух или более моле-
кул одного и того же или разных соединений. Меж-
молекулярная водородная связь может приводить
к образованию димеров (как это наблюдается для
карбоновых кислот) или полимерных молекулярных
цепочек, которые образуются, например, в нераз-
бавленных образцах или концентрированных рас-
творах одноатомных спиртов. Внутримолекулярные
водородные связи возникают тогда, когда в одной
и той же молекуле имеются протонодонорная и про-
тоноакцепторная группы и нет пространственных
затруднений для перекрывания орбиталей, напри-
мер образуются пяти- или шестичленные циклы.
Степень межмолекулярных и внутримолекулярных
взаимодействий зависит от температуры. Влияние
концентрации растворов на меж- и внутримоле-
кулярную водородную связь существенно разли-
чается. Полосы, обусловленные межмолекулярной
водородной связью, обычно исчезают при низких
концентрациях (менее 0.01 моль-л1 в неполярных
растворителях). Внутримолекулярная водородная
связь представляет собой «внутренний» эффект,
который сохраняется даже при очень низких кон-
центрациях.
Изменение частоты поглощения при переходе от
поглощения «свободной» гидроксильной группы
к связанной характеризует прочность водородной
связи. На прочность связи влияют: напряжение коль-
ца, геометрия молекулы, относительная кислотность
и основность донорных и акцепторных групп. Вну-
тримолекулярная связь при одних и тех же связан-
ных группах прочнее в случае шестичленного цикла,
чем для циклов меньшего размера. Самые прочные
водородные связи образуются при стабилизации цикла
в результате резонанса.
Влияние водородных связей на частоты валент-
ных колебаний гидроксильных и карбонильных
групп обобщено в табл. 2.2;. ИК-спектр циклогек-
силкарбинола в области валентных колебаний О-Н
иллюстрирует влияние концентрации в инертном
растворителе (см. рис. 2.14).
Важным аспектом образования водородной связи
является взаимодействие между функциональными
группами растворителя и растворенного вещества.
Если растворенное вещество полярно, то в услови-
ях регистрации ИК-спектра необходимо указывать
использованный растворитель и концентрацию рас-
творенного вещества.
23. Аппаратура
2.3.1. Диспергирующий ИК-спектрометр
Многие годы ИК-спектры поглощения получали, про-
пуская излучение через пробу и проводя сканирование
по спектру посредством диспергирующего устройства,
например хорошо известной дифракционной решетки.
Сканирование осуществляли, разворачивая решетку,
спектры поглощения регистрировали в виде зависи-
мости интенсивностей от волновых чисел.
Таблица 2.2. Частоты валентных колебаний при образовании водородных связей
Прочность связи Х-Н—У: Межмолекулярные водородные связи Внутримолекулярные водородные связи
Уменьшение частоты, см-1 Классы органических соединений Уменьшение частоты, см"1 Классы органических соединений
VOH vc=o VOH Vc=o
Слабая Средняя 300* 15б Спирты, фенолы и межмолеку- лярные водородные связи между гидроксильными и карбонильными группами <100* 100- 300* 10 50 1,2-Диолы, а- и большин- ство Р-гидроксикетонов, о-хлорфенолы, о-алкоксифенолы 1,3-Диолы, некоторые р-гидроксикетоны, Р-гидроксиаминосоединения, нитросоединения
Сильная >500‘ 3 506 Димеры RCO2H >300* 100 о-Г идроксиарилкетоны, о-гидроксиарилкислоты, р-дикетоны, трополоны
а Сдвиг частоты относительно «свободных» валентных колебаний.
6 Только для соединений, где могут быть валентные колебания группы С=О.
2.3. Аппаратура
95
Рис. 2.4. Оптическая схема двухлучевого ИК-спектрофотометра
На рис. 2.4 показана сложная оптическая схема
двухлучевого диспергирующего спектрофотометра.
Излучение источника разделяется на два пучка, один
из которых проходит через кювету с пробой, а другой
через кювету сравнения. Эти пучки затем объединя-
ются вращающимся секторным зеркалом М7 в один
луч переменной интенсивности. Различие интенсив-
ностей поглощения в каналах образца и сравнения
уравнивается аттенюатором (специальной диафрагмой)
в канале сравнения. Таким способом учитывается
поглощение растворителя в кювете с образцом, и в
результате в спектре наблюдаются только полосы
поглощения растворенной пробы.
2.3.2. ИК-Спектрометр с преобразованием
Фурье (интерферометр)
Инфакрасная спектрометрия с преобразованием Фурье,
имеющая ряд преимуществ, получила широкое развитие
в последнее десятилетие. Поток излучения, содержащего
весь интервал частот (например, 4000-400 см-1), разде-
ляется полупрозрачным зеркалом на два луча (рис. 2.5).
Один из них имеет постоянную длину пути, а другой за
счет отражения от подвижного зеркала-переменную.
Возвратно-поступательное движение зеркала В из-
меняет разность двух оптических путей, что дает кар-
тину с переменной интенсивностью, называемую ин-
терферограммой. Преобразование Фурье превращает
эту интерферограмму из зависимости интенсивности
от разности хода в зависимость от волнового числа.
Плавное и непрерывное движение поршня с закреплен-
ным на нем зеркалом В изменяет длину луча (создает
разность хода). Преобразование Фурье, выполняемое
последовательно по точкам на всем интервале интер-
ферограммы, дает полный ИК-спектр. Прохождение
этого излучения через образец подвергает последний
действию широкого интервала энергий. В принципе,
фурье-анализ одного прохождения излучения через
образец дает полный ИК-спектр соединения.
Ифракрасная спектроскопия с преобразованием
Фурье (ИК-ФП, IR-FT) имеет ряд преимуществ.
Так как в этом случае монохроматор не используется,
через пробу одновременно проходит весь частотный
интервал излучения, что сокращает время регистра-
ции спектра (выигрыш Фельджета). Спектрометры
с преобразованием Фурье могут иметь очень высокое
разрешение (<0.001см-1). Представление результатов
ИК-измерений в цифровом виде облегчает обраще-
ние с ними: результаты нескольких сканирований
Привод зеркала
I Поршень
Подвижное зеркало В
•-------
Источник
излучения
\\\\\\\\\\\
Неподвижное
зеркало А
Делитель
светового
пучка
Объединенный
(интерферирующий)
пучок
Кювета с образцом
Компьютер
Детектор
Аналого-цифровой
преобразователь
Регистрация
спектра
Рис. 2.5. Схема спектрометра с преобразованием
Фурье
96
2. Инфракрасная спектроскопия
можно суммировать, усреднять, понижая тем самым
уровень шума, и получать прекрасные спектры очень
маленьких проб. Отсюда следует возможность со-
вместного использования ИК-ФП и хроматографии
(ВЭЖХ и ГХ). Как в любом управляемом компьюте-
ром спектрометре, спектры индивидуальных веществ
и растворителей можно вычитать из спектров смесей.
Доступны обычные возможности обработки спек-
троскопических данных в цифровом виде; например,
спектр можно представлять либо в линейной шкале
волновых чисел, либо длин волн.
Некоторые производители предлагают на рынок
ИК-ФП-спекгрометры, соединенные с ГХ, с помощью
которых можно получать спектры фракций, выходя-
щих из капиллярной колонки на уровне нанограммо-
вых количеств. ИК-Спектры паров веществ похожи
на спектры при высоком разбавлении в неполярном
растворителе: концентрационно-зависимые полосы сме-
щены в область более высоких частот по сравнению
со спектрами концентрированных растворов, тонких
пленок или твердого состояния (см. Aldrich, 1989).
2.4. Подготовка пробы
Можно получать ИК-спектры веществ в газообраз-
ном, жидком и твердом состояниях. Спектры газов
или низкокипящих жидкостей можно регистриро-
вать при напуске образца в вакуумированную кю-
вету. Газовые кюветы имеют длину от нескольких
сантиметров до 40 м. Кюветное отделение серийных
ИК-спектрофотометров не приспособлено для кювет
длиной более 10 см; в этом случае большая длина
поглощающего слоя достигается при помощи оптики
многократного отражения.
Жидкости можно исследовать в индивидуальном
состоянии или в растворах. Обычно чистая жидкость
помещается между солевыми пластинками без раз-
делительной прокладки. Сдавливая жидкий образец
между плоскими пластинками, получают пленку тол-
щиной 0.01 мм или менее, пластинки удерживаются
вместе капиллярными силами. При этом требуется
1-10 мг пробы. Обычно толстые слои чистых жид-
костей поглощают слишком сильно, что не позволяет
получать качественные спектры. Летучие жидкости
исследуются в герметических кюветах с очень тон-
кими прокладками. В случае образцов, растворяющих
пластинки из хлорида натрия, используют хлорид
серебра или ZnSe, устойчивые к действию воды.
Растворы помещают в кюветы толщиной 0.1-1 мм.
Разборные кюветы требуют объемов 0.1-1 мл для
0.05-10%-ных растворов. В пучок сравнения поме-
щается компенсирующая кювета, содержащая чистый
растворитель. Полученный таким путем спектр являет-
ся спектром растворенного вещества, за исключением
тех областей, в которых поглощение растворителя
велико. Например, толстый слой образца четыреххло-
ристого углерода сильно поглощает около 800 см-1.
Компенсация для этой полосы не эффективна, так как
из-за сильного поглощения излучения в обоих пучках
сигнал в приемнике не возникает.
Выбранный растворитель должен быть сухим и до-
статочно прозрачным в интересующей нас области.
Когда необходимо зарегистрировать полный спектр,
используют несколько растворителей. Наиболее рас-
пространенной парой растворителей можно считать
четыреххлористый углерод (СС14) - сероуглерод (CS2).
Четыреххлористый углерод относительно прозрачен
при частотах свыше 1333 см-1, в то время как се-
роуглерод дает слабое поглощение ниже 1333 см-1.
Следует избегать таких комбинаций растворителя
и растворенного вещества, между которыми возмож-
ны реакции. Например, сероуглерод нельзя исполь-
зовать в качестве растворителя для первичных или
вторичных аминов. Аминоспирты с сероуглеродом
и четыреххлористым углеродом взаимодействуют
медленно. Спектры поглощения некоторых раство-
рителей и иммерсионных масел для приготовления
паст приведены в приложении А.
Твердые вещества обычно исследуют в виде паст,
прессованных таблеток или в виде осажденных сте-
кловидных пленок. Пасты приготовляют тщательным
растиранием 2-5 мг твердого вещества в полирован-
ной агатовой ступке. Растирание продолжается после
добавления одной или двух капель иммерсионного
масла. Чтобы не было сильного рассеяния излучения,
размер взвешенных частиц должен быть менее 2 мкм.
Пасту раздавливают между двумя окнами и исследуют
в виде тонкой пленки. В качестве иммерсионной среды
обычно используется вазелиновое масло (нуйол, смесь
высококипящих углеводородов). В тех случаях, когда
полосы поглощения вазелинового масла (углеводоро-
да) перекрываются с исследуемым спектром, можно
использовать полностью галогенированный (F, С1)
полимер, гексахлорбутадиен-1,3 или полностью фто-
рированные углеводороды. Совместное использование
паст как с вазелиновым маслом, так и с полностью
галогенированными углеводородами дает возмож-
ность получать спектры во всей области от 4000 до
250 см-1 без мешающих полос.
Методика прессования таблеток основана на том,
что сухой порошкообразный бромид калия (или гало-
гениды других щелочных металлов) при прессовании
образует прозрачные таблетки. Пробу (0.5-1.0 мг)
исследуемого вещества тщательно измельчают и пере-
мешивают с веществом матрицы (приблизительно
2.5. Интерпретация спектров
97
100 мг сухого порошкообразного бромида калия).
Смешивание и измельчение могут проводиться в по-
лированной агатовой ступке, более эффективно в не-
большой вибрационной шаровой мельнице, а также
методом лиофилизации. Смесь прессуют в прозрачные
таблетки в специальной пресс-форме при давлении
700-1000 кгс/см2. Качество спектра зависит от степени
перемешивания и размера взвешенных частиц (2 мкм
или меньше). С микрооосветителем можно использо-
вать микродиски диаметром 0.5-1.5 мм. Эта методика
позволяет исследовать образцы с массой до 1 мкг.
Часто в спектрах, полученных методом прессования
таблеток, проявляются полосы около 3448 и 1639 см-1,
связанные с присутствием адсорбированной воды.
Повышенные требования к процессу получения хо-
роших таблеток приводят к тому, что многие избегают
использовать эту методику. Поэтому более приемлема
методика прессования с помощью мини-пресса, упро-
щающего процедуру. Смесь КВг с образцом помеща-
ют в полость удлиненной гайки, в которую с одной
стороны завернут на небольшую глубину болт. Встав-
ляют второй болт и при его затягивании создается
давление. Когда болты откручивают, в гайке, которая
теперь служит кюветой, остается таблетка.
Осажденные пленки полезны только тогда, когда ис-
следуемое вещество можно осадить из раствора либо
выморозить из расплава в виде микрокристаллов или
стекловидной пленки. Поликристаллические пленки
обычно очень сильно рассеивают свет. Специфическая
ориентация кристаллов может приводить к спектрам,
отличающимся от спектров беспорядочно ориентиро-
ванных частиц, которые присутствуют в пастах или
таблетках. Методика осажденных пленок особенно
полезна для получения спектров смол и пластиков.
Следует обращать внимание на то, чтобы из про-
бы был полностью удален растворитель откачкой
в вакууме или слабым нагреванием.
В настоящее время для получения качественных
спектров твердых веществ независимо от толщины
пробы применяют методику, известную как нару-
шенное полное внутреннее отражение (НПВО) или
спектроскопия внутреннего отражения. Методика
состоит в том, что ИК-излучение, которое претер-
певает внутреннее отражение от поверхности про-
зрачной среды с высоким показателем преломления,
проникает на некоторую глубину по другую сторо-
ну отражающей границы, возвращается обратно в
прозрачную среду и далее зеркалом направляется в
спектрометр. Если материал (т. е. образец) с более
низким показателем преломления, чем прозрачная
среда, находится в контакте с отражающей поверх-
ностью, то световая волна проникает в образец на
глубину нескольких микрометров, давая его спектр
поглощения. В другом варианте методика позволяет
создать многократное внутреннее отражение вдоль
поверхности образца, сокращенно метод МНПВО.
Методика многократного внутреннего отражения
дает спектры, интенсивность которых сравнима со
спектрами поглощения.
В общем, случае лучшие, т. е. наименее искаженные,
спектры получаются для разбавленных растворов в
неполярном растворителе. Неполярные соединения в
конденсированной фазе (т. е. чистая жидкость, паста,
таблетка из КВг или тонкая пленка) по существу
дают те же самые спектры, что и в неполярных
растворителях К сожалению, полярные соединения
часто нерастворимы в неполярных растворителях,
и их спектры приходится получать или в конден-
сированной фазе, или в полярном растворителе; в
последнем случае между растворенным веществом
и растворителем могут возникать межмолекулярные
взаимодействия (водородные связи).
Определенное внимание при работе необходимо
уделять солевым кюветам и пластинкам. Прежде все-
го необходимо работать с сухими, не содержащими
влаги образцами. Оптических поверхностей нельзя
касаться пальцами. Окна необходимо предохранять
от загрязнения силиконовой смазкой, которую трудно
удалять и которая имеет сильные полосы поглощения..
2.5. Интерпретация спектров
При интерпретации ИК-спектров нет жестких правил.
Однако прежде чем приступать к этой работе, нуж-
но добиться, чтобы спектр удовлетворял следующим
требованиям.
1. Спектр должен быть достаточно интенсивным
и хорошо разрешенным.
2. При съемке спектра нужно использовать доста-
точно чистый образец.
3. Спектрофотометр должен быть прокалиброван
так, чтобы волновые числа или длины волн на-
блюдаемых полос соответствовали их истинным
значениям. Калибровку спектрометра необходимо
выполнять по подходящим стандартам, таким как
полистирол или пары воды.
4. Должен быть описан метод подготовки пробы.
При работе с раствором должны быть указаны
растворитель, концентрация и толщина кюветы.
Точный расчет колебаний сложной многоатомной
молекулы невозможен, поэтому ИК-спектр нужно ин-
терпретировать на основе эмпирического сравнения
98
2. Инфракрасная спектроскопия
спектров и экстраполяции данных по более простым
молекулам. На многие вопросы, возникающие при
интерпретации ИК-спектров, можно ответить при
изучении данных, получаемых из масс-спектров
(гл. 2) и спектров ЯМР (гл. 3-6).
Многочисленные ИК-спектры органических молекул
обобщены в таблицах характеристических групповых
частот, которые приведены в приложении Б. Многие
групповые частоты изменяются в довольно широких
интервалах, что вызвано сложными взаимодействия-
ми колебаний внутри молекулы. Однако ряд полос
поглощения может отвечать и простым колебаниям.
Например, полосы, обусловленные валентными коле-
баниями групп С-Н, О-Н и С=О, остаются в узких
областях спектра. Важные детали структуры молекулы
можно обнаружить по точному положению полосы
поглощения внутри этих ограниченных областей. Сме-
щения положения полос и изменения их контуров,
сопровождающие изменения окружения молекулы,
также могут подтверждать важные детали структуры.
При предварительном рассмотрении спектра наибо-
лее информативными являются две области: 4000-1300
и 909-650 см"1. Высокочастотная часть спектра назы-
вается областью функциональных групп. В этой части
спектра наблюдаются характеристические частоты
валентных колебаний таких функциональных групп,
как ОН, NH, С=О и др. Отсутствие полос в областях,
связанных с поглощением какой-либо функциональ-
ной группы, обычно используется как доказательство
отсутствия этих групп в молекуле. При такой интер-
претации следует проявлять осторожность, так как
некоторые структурные особенности могут настолько
ослабить и уширить полосу, что она не будет за-
метна. Например, внутримолекулярная водородная
связь в енольной форме ацетилацетона приводит
к появлению слабой широкой полосы ОН, которая
может маскироваться другими полосами. Отсутствие
поглощения в области 1850-1540 см"1 исключает
структуры, содержащие карбонильную группу.
Основные колебания функциональных групп S-H
и С=С дают слабые полосы в высокочастотной обла-
сти, которые весьма важны для структурного анализа.
В других, более сложных областях спектра подобные
слабые полосы не имели бы такого значения. В высо-
кочастотной области нередко проявляются обертоны
и составные частоты более длинноволновых полос.
Эти полосы характеризуются малой интенсивностью,
за исключением тех случаев, когда наблюдается резонанс
Ферми. Сильные полосы колебаний углеродного скелета
(скелетные колебания) для ароматических и гетероарома-
тических соединений попадают в область 1600-1300 см”1.
Отсутствие сильных полос поглощения в области
900-650 см"1 обычно указывает на неароматическую
структуру. В спектрах ароматических и гетероароматиче-
ских соединений в этой области проявляются интенсив-
ные полосы поглощения, связанные с внеплоскостньгми
деформационными колебаниями С-Н и деформацион-
ными колебаниями кольца, которые часто коррелируют
с характером замещения. Широкие умеренной интенсив-
ности полосы поглощения в низкочастотной области
подтверждают наличие димера карбоновой кислоты,
а также амино- или амидной группировок, внеплоскост-
ные деформационные колебания которых находятся
в этой области. Если низкочастотную область продлить
до 1000 см"1, то в нее попадут полосы, характерные
для алкеновых структур.
Промежуточную область спектра 1300-900 см"1
обычно называют областью «отпечатков пальцев».
Поглощение в этой области часто имеет сложный
вид, причем некоторые полосы обусловлены взаи-
модействием колебаний. Эта часть спектра очень
информативна, когда исследуется совместно с дру-
гими областями. Например, если в высокочастотной
области спектра спиртов или фенолов проявляется
поглощение группы О-Н, то положение полосы
поглощения С-С-О в области 1260-1000 см”1 ча-
сто позволяет отнести поглощение группы О-Н
к спиртам или фенолам совершенно определен-
ной структуры. Каждый тип молекул имеет свое
характерное поглощение в этой промежуточной
области спектра.
Любые выводы, сделанные на основе анализа отдель-
ной характеристической полосы, должны подтверждать-
ся, по возможности, поглощением в других областях
спектра. Например, отнесение карбонильной полосы
к альдегидной группе подтверждается появлением по-
лосы или пары полос в области 2900-2695 см"1, от-
носящихся к валентным колебаниям С-Н альдегидной
группы. Подобным образом отнесение карбонильной
полосы к эфирной группе должно подтверждаться по-
явлением сильной полосы в области 1300-1100 см”1,
относящейся к валентным колебаниям С-О.
Соединения сходного строения при обычных усло-
виях могут давать практически идентичные спектры,
но отличия в области отпечатков пальцев можно за-
метить либо при растянутой вертикальной шкале,
либо для очень большой пробы (при этом основные
полосы будут зашкаливать).
Например, ИК-спектры пентана и гексана при
обычных условиях по существу одинаковы и их
можно отличить только на приборе с очень высокой
чувствительностью.
При сравнении спектров в области «отпечатков
пальцев» или в случае, когда важна форма полос,
следует помнить о существенном изменении общего
вида спектра при переходе от спектра, линейного
2.5. Интерпретация спектров
99
Рис. 2.6. ИК-Спектры одной и той же пробы полистирола, (а) Шкала линейна по
отношению к волновым числам, см-1, (6) шкала линейна по отношению к длинам
волн, мкм
относительно волновых чисел, к спектру, линейному
относительно длин волн (рис. 2.6).
Нужно признать, что размер полной диаграммы груп-
повых характеристических частот может устрашить
(приложение Б). При отнесении полос в ИК-спектре
помогают следующие рекомендации и упрощенная
диаграмма характеристических частот (рис. 2.7).
Первый совет носит негативный оттенок: не пред-
принимайте лобовой прямой атаки на ИК-спектр.
Поищите доказательства присутствия или отсутствия
обычных функциональных групп с характеристиче-
ским поглощением. Начните с групп ОН, С=О и NH,
так как прямой ответ на уровне «да/нет» обычно
возможен с помощью диаграммы на рис. 2.7. Ответ
«да» для любой из этих групп значительно сужает
круг поиска. Определенный ответ внесет вклад в по-
лученную из масс-спектра молекулярную формулу
(гл. 1) и будет отправной точкой при анализе спектров
ЯМР (гл. 3-6). Эти спектры другого типа определяют
дальнейшие шаги в ходе расшифровки ИК-спектра.
На рис. 2.7 показана диаграмма характеристических
частот распространенных функциональных групп.
В разделе 2.6 представлена более подробная информация
о характеристическом поглощении.
2.6. Характеристические групповые
частоты органических молекул
Таблица характеристических групповых частот
дана в приложении Б. Спектральные интервалы,
выбранные для групповых частот, были приняты
после исследования большого числа соединений,
в которых имеются эти группы. Хотя интервалы
определены довольно хорошо, точное значение ча-
стоты или длины волны, где поглощает эта группа,
зависит от ее окружения в молекуле и физического
состояния пробы.
Этот раздел главы посвящен общему рассмотрению
характеристических групповых частот и их связи со
структурой молекул. Обсуждение главных классов мо-
лекул или основных функциональных групп сопрово-
ждается примерами ИК-спектров с отнесением наиболее
важных полос.
100
2, Инфракрасная спектроскопия
* Свободная ОН-группа: узкая полоса средней интенсивности; ОН, связанная водородной связью: сильная и широкая полоса.
Рис. 2.7. Упрощенная диаграмма характеристических частот некоторых
распространенных функциональных групп (принятые обозначения: s - сильная,
m - средняя, w - слабая, sh - узкая, Ьг - широкая)
2.6.1. Нормальные алканы (парафины)
Спектры нормальных алканов (парафинов) можно интер-
претировать на основе четырех колебаний - валентных
и деформационных колебаний связей С-Н и С-С. Под-
робный анализ спектров низших членов ряда парафи-
новых углеводородов позволил установить связь между
положением полос и определенными типами колебаний.
Не все возможные полосы в ИК-спекгре насыщенного
углеводорода играют одинаковую роль при определении
структуры молекул. Деформационные колебания связей
С-С наблюдаются при очень низких частотах (ниже
500 см-1) и выходят за пределы обычных спектров,
используемых химиками-органиками. Полосы, относя-
щиеся к валентным колебаниям С-С, малоинтенсивны
и проявляются в широком интервале 1200-800 см-1 (не
являются характеристическими); как правило, они не
имеют существенного значения для идентификации.
Наиболее важными являются характеристические
полосы, обусловленные валентными и деформацион-
ными колебаниями связей С-Н. Для целей иденти-
фикации структурных групп крутильные и веерные
колебания находят ограниченное применение. Это свя-
зано с их низкой интенсивностью, нестабильностью
положения и из-за сильного взаимодействия с коле-
баниями остальной части молекулы.
Формы (типы) колебаний насыщенных углеводо-
родов являются общими для многих органических
молекул. Хотя частоты валентных и деформацион-
ных колебаний С-Н метильных и метиленовых групп
в углеводородах остаются приблизительно постоян-
ными, присоединение группы СН3 или СН2 к неугле-
родному атому, карбонильной группе или аромати-
ческому кольцу может заметно их смещать.
Спектр додекана, представленный на рис. 2.8, типи-
чен для углеводородов с неразветвленной цепью.
2.6.1.1. Валентные колебания С-Н
Поглощение, обусловленное валентными колебаниями
связи С-Н в алканах, обычно наблюдается в области
3000-2840 см-1. Полосы валентных колебаний С-Н яв-
ляются одними из наиболее стабильных в спектре.
Метильные группы. Исследование большого числа
насыщенных углеводородов, содержащих метильные
группы, показало, что всегда наблюдаются две разли-
чающиеся полосы при 2962 и 2872 см-1. Первая-ре-
зультат антисимметричного (as) валентного колебания,
в котором две связи С-Н метильной группы растя-
гиваются, в то время как третья сжимается (vas СН3).
Вторая полоса обусловлена симметричными (s) ва-
лентными колебаниями (vs СН3), когда все три связи
С-Н растягиваются или сжимаются в фазе. Наличие
2.6. Характеристические групповые частоты органических молекул
101
Рис. 2.8. ИК-Спектр додекана
Валентные колебания С-Н (см-1): 2953 (vas СН3), 2870 (vs СН3), 2922 (vas СН2),
2853 (vs СН2);
деформационные колебания С-Н (см-1): 1464 (6S СН2), 1450 (6as СН3), 1379 (б5 СН3);
маятниковое колебание группы СН2: 724 см-1 (р СН2)
нескольких метильных групп в молекуле приводит
к увеличению интенсивности соответствующих полос.
Метиленовые группы. Антисимметричные ва-
лентные колебания (v^ СН2) и симметричные валент-
ные колебания (vs СН2) проявляются соответственно
вблизи 2926 и 2853 см-1. Положение этих полос из-
меняется не более чем на ± 10 см-1 в алифатических
и ненапряженных циклических углеводородах. Частота
валентных колебаний метиленовой группы повыша-
ется, если она является частью напряженного цикла.
2.6.1.2. Деформационные колебания С-Н
метильных групп
В метильной группе могут проявляться два деформа-
ционных колебания. Первое из них-это симметричное
деформационное колебание, при котором деформация
связей С-Н происходит в фазе (I). Второе - антисим-
метричное деформационное колебание, при котором
деформация связей С-Н происходит не в фазе (II).
В случае I связи С-Н движутся подобно закрываю-
щимся лепесткам цветка, в случае II один лепесток
открывается, а два других закрываются.
Симметричное деформационное колебание (8S СН3)
проявляется около 1375 см-1, а антисимметричное
(5as СН3)- около 1450 см-1.
Полоса антисимметричного колебания обычно
перекрывается полосой ножничного колебания ме-
тиленовой группы (см. ниже). Однако в соединениях,
подобных диэтилкетону, наблюдаются две различ-
ные полосы, так как полоса ножничного колеба-
ния группы СН2 сдвинута в сторону низких частот
к 1439-1399 см-1 и имеет повышенную интенсивность
из-за соседства с карбонильной группой.
Если метильная группа связана с другим угле-
родным атомом, то положение полосы поглощения
около 1375 см-1, возникающей при симметричном
деформационном колебании связей С-Н метильной
группы, очень устойчиво. Интенсивность этой полосы
выше, чем интенсивность полосы антисимметричного
деформационного колебания метильных групп или
ножничных колебаний метиленовых групп.
Метиленовые группы. Деформационные колеба-
ния связей С-Н в метиленовых группах уже были
показаны схематически на рис. 2.1. Четырем дефор-
мационным колебаниям даны следующие названия:
ножничные, веерные, маятниковые и крутильные.
В спектрах углеводородов полоса ножничных ко-
лебаний (<5S СН2) имеет почти постоянное положение
и проявляется около 1465 см-1 (см. рис. 2.8).
Для парафиновых углеводородов с неразветвлен-
ной цепью из семи и более атомов углерода полоса
маятниковых колебаний (р СН2), при которых все
метиленовые группы качаются в одной фазе, нахо-
дится около 720 см-1. В спектре твердого образца
эта полоса может проявляться в виде дублета. У низ-
ших представителей н-алканов она может несколько
смещаться в сторону более высоких частот.
Поглощение углеводородов, обусловленное крутиль-
ными и веерными колебаниями метиленовых групп,
наблюдается в области 1350-1150 см-1. Эти полосы
обычно значительно слабее, чем те, которые наблю-
102
2. Инфракрасная спектроскопия
Рис. 2.9. ИК-Спектр 2,2,4-триметилпентана (изооктана).
Валентные колебания С-Н (см. рис. 2.8); деформационные колебания С-Н (см. рис. 2.8).
Видны перекрывающиеся дублеты для л?рел?-бутильной и изопропильной групп
в области 1400-1340 см-1.
Обратите внимание на отсутствие полос(ы) маятниковых колебаний метиленовых
групп в области 1000-800 см-1 на рис. 2.8
даются в результате ножничных колебаний. Серии
полос в этой области, связанные с метиленовыми
группами, характерны для твердых образцов кислот
с длинной цепью, амидов и эфиров.
2.6.2. Углеводороды с разветвленной цепью
В общем случае изменения, наблюдающиеся в спек-
тре углеводорода при его разветвлении, являются
результатом изменений в скелетных валентных ко-
лебаниях и в деформационных колебаниях метиль-
ных групп; последние наблюдаются ниже 1500 см-1.
На рис. 2.9 показан ИК-спектр 2,2,4-триметилпентана,
который типичен для разветвленного алкана.
2.6.2.1. Валентные колебания С-Н
при третичном атоме углерода
Полосы поглощения, связанные с колебаниями этого
типа, малоинтенсивны и обычно маскируются полосами
других колебаний алифатических групп С-Н. В углево-
дородах это поглощение наблюдается около 2890 см-1.
2.6.2.2. Деформационные колебания С-Н
в геминальных метильных группах
Структурные фрагменты, в которых две метильные
группы присоединены к одному атому углерода,
имеют отчетливое поглощение в области деформа-
ционных колебаний С-Н. Изопропильная группа дает
сильный дублет с пиками почти равной интенсивно-
сти в интервалах 1385-1380 и 1370-1365 см-1, трет-
Бутильная группа дает две полосы деформационных
колебаний С-Н: одну в области 1395-1385 см-1,
а другую около 1370 см-1. В wpew-бутильном ду-
блете длинноволновая полоса более интенсивна.
Если группа (СН3)2С находится не на конце угле-
родной цепи, то дублет наблюдается, как правило,
в той же области, что и для изопропильной и трет-
бутильной групп. Появление дублета полос в спек-
тре соединений, содержащих группы (СН3)2С, объ-
ясняется взаимодействием между симметричными
деформационными колебаниями двух метильных
групп (присоединенных к общему атому углерода),
происходящими в фазе и противофазе.
Маятниковые колебания метильных групп в изопро-
пильных и wpew-бутильных фрагментах дают слабые
полосы. Эти колебания чувствительны к массе молеку-
лы, взаимодействуют со скелетными валентными коле-
баниями и обычно менее надежны, чем деформационные
колебания С-Н. Эти полосы для изопропильной группы
наблюдаются при 922-919 см-1, а для wpew-бутильной
группы-при 932-926 см-1.
2.6.3. Циклоалканы
2.6.3.1. Валентные колебания С-Н
Полосы валентных колебаний метиленовых групп
в ненапряженных циклических полиметиленовых
структурах в большинстве случаев те же, что и для
ациклических насыщенных углеводородов. Увеличе-
ние напряженности цикла постепенно повышает ча-
стоты валентных колебаний связей С-Н. Кольцевые
метиленовые и метановые группы в моноалкилци-
клопропанах поглощают в области 3100-2990 см-1.
2.6.3.2. Деформационные колебания С-Н
Циклизация уменьшает частоту ножничного колебания
группы СН2. Циклогексан поглощает при 1452 см-1, в то
2.6. Характеристические групповые частоты органических молекул
103
100 -
Н2С=СН-(СН2)9СН3
4000 3000 2000 1000
Волновые числа (скг1)
Рис. 2.10. ИК-Спектр додецена-1.
Валентные колебания С-Н (см. рис. 2.8). Обратите внимание на полосу
3082 см-1 валентного колебания С-Н в алкеновом звене. Валентное колебание
С=С проявляется при частоте 1648 см"1, см. приложение В, табл. В-1.
Внеплоскостное деформационное колебание С-Н 1000 см"1, (в алкеновом звене)
915 см"1. Маятниковое колебание метиленовой группы 730 см"1
время как н-гексан-при 1468 см-1.Циклопентан погло-
щает при 1455 см-1, а циклопропан-при 1442. см-1. Этот
сдвиг частот часто позволяет наблюдать в этой области
отдельно полосы для метиленовых и метильных групп.
2.6.4. Алкены
Наличие двойной связи приводит к появлению в ИК-
спектрах углеводородов нескольких новых типов коле-
баний: валентных колебаний ОС, валентных колеба-
ний С-Н, когда атом углерода находится в алкеновом
звене, плоскостных и внеплоскостных деформаци-
онных колебаний алкеновых связей С-Н. Спектр на
рис. 2.10 типичен для углеводорода с двойной связью
на конце цепи.
2.6.4.1. Валентные колебания связи С=С
в несопряженных линейных алкенах
Валентные колебания связи ОС несопряженных ал-
кенов обычно дают полосы средней или малой ин-
тенсивности при 1667-1640 см-1. В монозамещенных
алкенах наблюдаются полосы винильного поглощения
умеренной интенсивности около 1640 см-1. Дизаме-
щенные гараис-алкены, три- и тетраалкилзамещенные
алкены поглощают при (или вблизи) 1670 см-1, диза-
мещенные z/wc-алкены и винилиденовые производные
поглощают около 1650 см-1.
Полоса поглощения симметричных дизамещенных
гаранс-алкенов или тетразамещенных алкенов может
быть очень слабой или вообще отсутствовать, цис-
Алкены, которые менее симметричны, чем транс-
алкены, имеют более сильную полосу поглощения.
Обычно двойные связи в середине цепи вследствие
своей псевдосимметричности поглощают слабее,
чем терминальные двойные связи.
Аномально высокая частота полосы поглощения на-
блюдается для групп -CH=CF2 и -CF=CF2. Первая дает
полосу около 1754 см-1, а вторая-около 1786 см-1.
При присоединении хлора, брома или иода частота
полосы поглощения, наоборот, уменьшается.
Циклоалкены. Поглощение двойной связи в нена-
пряженной системе циклогексена практически такое
же, как и для z/wc-изомера ациклической системы.
Валентные колебания двойной связи взаимодейству-
ют с валентными колебаниями соседних одинарных
углерод-углеродных связей. При уменьшении угла а
это взаимодействие уменьшается.
а
Оно становится минимальным при угле 90° в ци-
клобутене (1566 см1). В циклопропеновой структуре
взаимодействие снова становится значительным, и ча-
стота полосы поглощения возрастает до 1641 см-1.
Замещение атома водорода алкильной группой по-
вышает частоту полосы поглощения двойной связи
в напряженных циклических системах. Так, в спектре
циклобутена эта полоса находится при 1566 см-1, а в
спектре 1-метилциклобутена-при 1641 см"1.
Полоса поглощения внешней (экзоциклической)
двойной связи смещается в сторону больших частот
с уменьшением размера цикла. Метиленциклогексан
поглощает при 1650 см-1, а метиленциклопропан-
при 1781 см-1.
104
2. Инфракрасная спектроскопия
Рис. 2.11. ИК-Спектр изопрена.
Валентное колебание С-Н (=С-Н) 3090 см-1; валентные колебания сопряженной
системы С=С-С=С: симметричное 1640 см-1 (слабая), антисимметричное
1601 см-1 (сильная); деформационные внеплоскостные колебания С-Н:
992 и 899 см-1 (см. приложение В, табл. В-1, винильная группа)
Сопряженные системы. Валентные колебания
двойных связей в сопряженных диенах, не имею-
щих центра симметрии, взаимодействуют, давая
две полосы валентных колебаний С=С. В спектре
несимметричного сопряженного диена, например
пентадиена-1,3 наблюдаются полосы поглощения
около 1650 и 1600 см-1. Симметричная молекула
бутадиена-1,3 дает только одну полосу поглощения
вблизи 1600 см-1, которая появляется в результате
антисимметричного валентного колебания; симме-
тричное валентное колебание в ИК-спектре неактив-
но. ИК-Спектр изопрена (рис. 2.11) иллюстрирует
указанные особенности.
Сопряжение двойной связи с ароматическим коль-
цом увеличивает интенсивность полосы валентных
колебаний двойной связи алкенового фрагмента около
1625 см"1.
Полоса поглощения двойной связи, сопряженной
с карбонильной группой, сдвигается приблизитель-
но на 30 см-1 в низкочастотную область; интенсив-
ность поглощения при этом повышается. В s-цис-
структурах1) интенсивность алкенового поглощения
может сравняться с полосой поглощения карбониль-
ной группы. 5-Аиранс-Структуры имеют более слабое
поглощение, чем s-z/wc-структуры.
Кумулированные алкены. Системы с кумули-
рованными двойными связями, например в алленах
(>С=С=СН2), поглощают около 2000-1900 см-1. По-
глощение является результатом асимметричных ва-
0 Обозначения s-цис- и s-транс- используют для описания
геометрии 1,3-диена относительно центральной одинарной
(single, 5) связи. - Прим, перев.
лентных колебаний фрагмента С=С=С. Эта полоса
может рассматриваться как крайний случай погло-
щения экзоциклической двойной связи С=С.
2.6.4.2. Валентные колебания связей С-Н в алкенах
Любые полосы валентных колебаний связей С-Н,
находящиеся выше 3000 см-1, относятся к арома-
тическим, гетероароматическим, ацетиленовым или
алкеновым системам. В этой области расположены
также полосы валентных колебаний С-Н малых ци-
клов, (циклопропан) и галогенированных алкильных
групп. На частоту и интенсивность полос валентных
колебаний С-Н алкеновых соединений влияет ха-
рактер замещения. Для структур, в которых может
проявляться взаимодействие валентных колебаний,
при достаточном разрешении наблюдается несколько
полос. Например, винильная группа дает три близко-
лежащие полосы валентных колебаний С-Н. Две из
них возникают в результате симметричных и анти-
симметричных валентных колебаний концевых связей
С-Н, а третья - в результате валентного колебания
одной связи С-Н при другом атоме углерода.
2.6.4.3. Деформационные колебания связей С-Н в алкенах
Деформационные колебания связей С-Н в алкенах
могут происходить либо в плоскости двойной связи,
либо перпендикулярно к ней. Каждая связь С-Н может
совершать эти колебания либо в фазе, либо в противо-
фазе по отношению к колебаниям других связей.
Отнесения частот были сделаны лишь в случае
нескольких наиболее характерных и надежных пло-
скостных деформационных колебаний. Поглощение
винильной группы при 1416 см-1 обусловлено ножнич-
2.6. Характеристические групповые частоты органических молекул
105
Рис. 2.12. ИК-Спектр гептина-1.
Валентное колебание =С-Н 3314 см-1; валентные колебания алкильных С-Н
2960-2860 см-1 (см. рис. 2.8), валентное колебание С=С 2126 см-1, деформационные
колебания С-Н (1450-1360 см-1): 1463 см-1 (б5 СН2), 1450 см-1(ба5, СН3); обертон
деформационного колебания 6as =С-Н 1247 см-1; основное деформационное
колебание =С-Н 637 см-1
ным колебанием концевой метиленовой группы. Ма-
ятниковые колебания связей С-Н z/wc-дизамещенных
алкенов наблюдаются в той же области.
Наиболее характерными типами колебаний алкенах
являются внеплоскостные деформационные колебания
С-Н в области 1000-650 см-1. Эти полосы обычно
самые интенсивные в спектрах алкенов. Самыми
надежными для интерпретации являются полосы
винильной и винилиденовой групп, а также транс-
дизамещенных алкенов. Примеры характерных для
алкенов полос поглощения представлены в табл. В-1
и В-2 приложения В.
В алленовых структурах наблюдается сильная
полоса поглощения около 850 см-1, относящаяся
к веерным колебаниям группы =СН2. Можно также
наблюдать первый обертон этой полосы.
2.6.5. Алкины
В ИК-спектрах ацетиленовых углеводородов (алки-
нов) проявляются валентные колебания связей двух
типов: С=С и С-Н. Для ацетилена и его монопроиз-
водных характеристична также полоса поглощения
деформационных колебаний С-Н. На рис. 2.12 пред-
ставлен ИК-спектр типичного алкина с концевой
тройной связью.
2.6.5.1. Валентные колебания связи С=С
Слабая полоса валентных колебаний связей С=С аце-
тиленовых соединений наблюдается в области 2260-
2100 см-1. Для самого ацетилена и его симметрично
замещенных производных валентные колебания С=С
в ИК-спектре не проявляются из-за симметрии моле-
кулы. В ИК-спектрах монозамещенных алкинов эта
полоса наблюдается в области 2140-2100 см-1. Диза-
мещенные алкины, в которых заместители различны,
поглощают около 2260-2190 см-1. Если заместители
близки по массе либо оказывают одинаковое мезо-
мерное или индуктивное влияние, то полосы могут
быть настолько слабыми, что не будут проявляться
в ИК-спектре. Из-за асимметрии концевая тройная
связь дает более сильную полосу, чем внутренняя
(псевдосимметрия). Интенсивность полосы валент-
ных колебаний С=С увеличивается при сопряжении
с карбонильной группой.
2.6.S.2. Валентные колебания связей С-Н
Валентные колебания связей С-Н в спектрах моно-
замещенных алкинов проявляются в области 3333—
3267 см-1. Эта интенсивная полоса уже, чем полосы
колебаний связанных водородными связями групп
ОН или NH, которые наблюдаются в той же области.
2.6.5.3. Деформационные колебания связей С-Н
Деформационные колебания С-Н алкинов и их моно-
замещенных производных дают сильную широкую
полосу поглощения в области 700-610 см-1. Пер-
вый обертон деформационного колебания С-Н про-
является в виде слабой широкой полосы в области
1370-1220 см-1.
2.6.6. Моноядерные ароматические углеводороды
Наиболее интенсивные и информативные полосы
в спектрах ароматических соединений наблюдают-
ся в области низких частот между 900 и 675 см-1.
106
2. Инфракрасная спектроскопия
Рис. 2.13. ИК-Спектр о-ксилола.
Валентные колебания ароматических С-Н: 3017 см~1; валентные колебания
метильных С-Н: 2970, 2940, 2878 см-1; обертоны или составные полосы:
2000-1667 см-1 (см. рис. 2.16); валентные колебания С-С кольца: 1605, 1497,
1466 см-1; плоскостные деформационные колебания С-Н: 1050, 1019 см-1;
внеплоскостное деформационное колебание С-Н: 741 см"1
Эти сильные полосы поглощения возникают в ре-
зультате внеплоскостных деформационных колебаний
связей С-Н в кольце. Полосы плоскостных колебаний
проявляются в области 1300-1000 см-1. Скелетные
колебания, включая колебания С-С цикла, поглощают
в областях 1600-1585 и 1500-1400 см-1. Полосы ске-
летных колебаний часто проявляются в виде дублетов
в зависимости от природы заместителей в кольце.
Полосы валентных колебаний С-Н в ароматических
соединениях наблюдаются между 3100 и 3000 см-1.
Слабые полосы составных частот и обертонов про-
являются в области 2000-1650 см-1, вид этих полос
характеризует тип замещения в кольце. Полосы обер-
тонов и составных частот очень слабые, поэтому их
легче наблюдать для толстого слоя образца. Спектр,
показанный на рис. 2.13, типичен для ароматических
(бензольных) соединений.
2.6.6.1. Внеплоскостные деформационные
колебания связей С-Н
Внеплоскостные деформационные колебания соседних
атомов водорода в бензольном кольце, происходящие
в фазе, сильно взаимодействуют между собой. Поэто-
му положение этих полос поглощения характеризует
число соседних атомов водорода в кольце. Эти по-
лосы часто интенсивны и наблюдаются в области
900-650 см-1.
Отнесения полос внеплоскостных деформацион-
ных колебаний С-Н в спектрах замещенных бензо-
лов приведено на диаграммах характеристических
групповых частот (приложение Б). Эти отнесения
обычно надежны для алкилзамещенных бензолов,
но при интерпретации спектров соединений, в ко-
торых полярные группы связаны непосредственно
с кольцом, например нитробензолов, ароматических
кислот или их амидов и эфиров, необходимо соблю-
дать осторожность.
Полоса поглощения, которая часто проявляется
в спектрах замещенных бензолов около 600-420 см-1,
относится к внеплоскостными деформационными
колебаниями кольца.
2.6.7. Полиядерные ароматические соединения
Полиядерные ароматические соединения, подобно
моноядерным, имеют характеристические полосы по-
глощения в трех областях спектра.
Валентные колебания связей С-Н и скелетные ко-
лебания имеют полосы поглощения в той же области,
что и в моноядерных ароматических соединениях.
Наиболее характеристичные полосы поглощения
полиядерных ароматических соединений связаны
с внеплоскостными деформационными колебаниями
связей С-Н, расположенными в области 900-675 см-1.
Эти полосы могут коррелировать с числом соседних
атомов водорода в кольцах. Например, в ПК-спектрах
большинства 0-замещенных нафталинов наблюдаются
три полосы поглощения, обусловленные внеплоскост-
ными колебаниями С-Н; они соответствуют одному
изолированному и двум соседним атомам водорода
одного кольца и колебаниям четырех соседних атомов
водорода другого кольца.
В спектрах а-замещенных нафталинов наблюдается
полоса для трех соседних атомов водорода в обла-
сти 810-785 см-1 и для четырех соседних атомов
водорода в области 780-760 см-1. В ИК-спектрах
2.6. Характеристические групповые частоты органических молекул
107
Таблица 2.3. Внеплоскостные деформационные ко-
лебания С-Н в р-замещенных нафталинах
Тип замещения Область поглощения, см-1
Изолированный атом водорода 862-835
Два соседних атома водорода 835-805
Четыре соседних атома водорода 760-735
Р-замещенных нафталинов наблюдаются 2 полосы:
при 862-835 см-1 для изолированной связи С-Н
и 835-805 см-1 для двух соседних атомов водорода.
Кроме того, имеется полоса 760-735 см-1 для четырех
соседних атомов водорода. В результате деформаци-
онных колебаний могут появляться дополнительные
полосы (см. табл. 2.3). Положение полос для нафта-
линов и других полиядерных ароматических соеди-
нений с большим числом заместителей можно найти
в литературе (см. Colthup et. al., 1990; Conley, 1972).
2.6.8. Спирты и фенолы
Характеристические полосы, наблюдаемые в спектрах
спиртов и фенолов, обусловлены валентными коле-
баниями связей О-Н и С-О. Эти колебания чувстви-
тельны к образованию водородных связей. Валентные
колебания С-О и деформационные колебания О-Н не
являются независимыми, так как они взаимодействуют
с колебаниями соседних групп.
2.6.8.1. Валентные колебания связи 0-Н
Не участвующая в водородной связи («свободная»)
гидроксильная группа спиртов и фенолов дает до-
вольно сильную полосу поглощения в области 3700-
3584 см-1. Узкие полосы «свободной» гидроксильной
группы наблюдаются только в газовой фазе, сильно
разбавленных растворах в неполярных растворителях
или для пространственно затрудненных ОН-групп.
Возможность образования межмолекулярных водород-
ных связей увеличивается с концентрацией раствора,
что вызывает появление дополнительных полос при
более низких частотах (3550-3200 см-1) за счет умень-
шения интенсивности полосы «свободной» гидрок-
сильной группы. Этот эффект иллюстрирует рис. 2.14.
На нем показаны полосы поглощения в области ва-
лентных колебаний О-Н для двух различных кон-
центраций циклогексилметанола в четыреххлористом
углероде. Для сравнений подобного рода необходимо
изменять толщину кюветы и концентрацию таким об-
разом, чтобы для каждой концентрации в пучке света
находилось одинаковое число поглощающих моле-
кул. Полоса при 3623 см-1 обусловлена мономером,
Рис. 2.14. ИК-Спектр в области валентных
колебаний О-Н раствора циклогексилметанола
в СС14:
а - концентрация 0.03 моль л-1, толщина кюветы
0.406 мм;
б - концентрация 1.00 моль л-1, толщина кюветы
0.014 мм
см-1
а широкая полоса около 3333 см ^«полимерными»
структурами.
Сильная внутримолекулярная водородная связь об-
разуется в о-гидроксиацетофеноне. В ИК-спектре этого
соединения около 3077 см-1 наблюдается широкая,
не слишком интенсивная не зависимая от концен-
трации раствора в неполярном растворителе полоса
(рис. 2.15).
Наоборот, спектр и-гидроксиацетофенона
Т. д.
содержит узкую полосу «свободной» ОН-группы при
3600 см-1 в случае разбавленного раствора в СС14
108
2. Инфракрасная спектроскопия
СМ’1
Рис. 2.15. Участок ИК-спектра о-гидроксиацетофенона.
а - концентрация раствора 0.03 моль-л-1, толщина
кюветы 0.41 мм;
б- концентрация раствора 1.0 моль л"1, толщина
кюветы 0.015 мм
и широкую полосу около 3100 см-1, соответствующую
образованию межмолекулярной водородной связи,
в случае индивидуального вещества. В таких соеди-
нениях, как 2,6-ди-гареги-бутилфенол, где стерические
Таблица 2.4. Валентные колебания фрагмента С-С-0
в ИК-спектрах вторичных спиртов
Вторичный спирт Полоса поглощения, см1
Бутанол-2 1105
3 -Метил бутанол-2 1091
1-Фенилэтанол 1073
Бутен-3-ол-2 1058
Дифенилметанол 1014
факторы препятствуют образованию водородных
связей, полоса связанной гидроксильной группы
не проявляется даже в спектре чистого, неразбав-
ленного вещества.
2.6.8.2. Валентные колебали связи С-0
Валентные колебания связи С-О в спиртах
(рис. 2.16 и 2.18) и фенолах (рис. 2.17) дают силь-
ную полосу в области 1260-1000 см-1.
Валентные колебания связи С-О взаимодей-
ствуют с валентными колебаниями соседней связи
С-С; поэтому в первичных спиртах это колебание
лучше относить к антисимметричному валентному
колебанию фрагмента С-С-О. Форма колебания
усложняется при разветвлении и при наличии
двойной связи в а,0-положении по отношению
к гидроксильной группе. Эти эффекты для ряда
вторичных спиртов (чистые образцы) обобщены
в табл. 2.4:
3000 Z 2000 1000
Волновые числа (см1)
Рис. 2.16. ИК-Спектр бензилового спирта.
Валентное колебание группы О-Н, связанной водородной связью: 3329 см"1;
валентные колебания С-Н: ароматические 3100-3000 см-1, метиленовые 2940-
2860 см"1; обертоны или составные полосы: 2000-1667 см-1; валентные колебания
С-С ароматического кольца 1501,1455 см-1, перекрываются с ножничными
колебаниями СН2 около 1471 см-1; деформационное колебание О-Н, вероятно,
усиленное плоскостным деформационным колебанием С-Н: 1209 см"1; валентное
колебание С-О первичного спирта (см. табл. 2.5): 1023 см"1; внеплоскостное
колебание ароматических С-Н: 745 см"1; деформационное колебание С-С
ароматического кольца: 707 см"1
2.6. Характеристические групповые частоты органических молекул
109
Рис. 2.17. ИК-Спектр фенола (расплав).
Широкая полоса валентного колебания групп О-Н, связанных межмолекулярными
водородными связями: 3244 см-1; валентные колебания ароматических С-Н:
3052 см"1; обертоны или составные частоты: 2000-1667 см"1; валентные колебания
С-С ароматического кольца: 1601,1501,1478 см"1; плоскостные деформационные
колебания: О-Н 1378 см"1; валентные колебания С-О: 1231 см"1; внеплоскостные
деформационные колебания С-Н: 815, 753 см"1; внеплоскостные деформационные
колебания С-С ароматического кольца: 699 см"1; широкая полоса внеплоскостного
деформационного колебания группы О-Н, связанной водородной связью: около
650 см"1
Области поглощения различных типов чистых об-
разцов спиртов приведены в табл. 2.5.
Пасты, таблетки или расплавы фенолов поглоща-
ют при 1390-1330 и 1260-1180 см-1. Эти полосы
возникают в результате взаимодействия между де-
формационными колебаниями О-Н и валентными
колебаниями С-О. Низкочастотная полоса сильнее.
В растворах обе полосы сдвигаются в сторону низ-
ких частот. Спектр фенола на рис. 2.17 был полу-
чен в расплаве для демонстрации высокой степени
ассоциации.
2.6.8.3. Деформационные колебания связи 0-Н
Плоскостные деформационные колебания связи О-Н
наблюдаются в обычной области 1420-1330 см-1.
В первичных и вторичных спиртах плоскостные
деформационные колебания О-Н взаимодействуют
с веерными колебаниями С-Н, давая две полосы: око-
ло 1420 см-1 и около 1330 см-1. Для идентификации
эти полосы не имеют большого значения. Третичные
спирты, в которых нет такого взаимодействия, дают
в этой области одну полосу, ее положение зависит от
прочности водородной связи.
Рис. 2.18. ИК-Спектр 2-метилбутанола-1.
Полоса валентного колебания группы О-Н, связанной межмолекулярной
водородной связью: 3337 см"1; валентные колебания С-Н: 3000-2800 см"1
(см. рис. 2.8), валентные колебания С-О:Ю54 см"1
110
2. Инфракрасная спектроскопия
Таблица 2.5. Валентные колебания спиртовой груп-
пы С-О
Тип спирта Область поглощения, СМ"1
(1) Насыщенный третичный (2) Вторичный, 1205-1124
высокосимметричный (1) Насыщенный вторичный (2) а-Ненасыщенный или 1124-1087
циклический третичный (1) Вторичный, а-ненасыщенный (2) Вторичный, алициклический с 5- или 6-членным циклом (3) Насыщенный первичный (1) Третичный с несколькими ненасыщенными группами в а-положении (2) Вторичный ди-а-ненасыщенный (3) Вторичный а-ненасыщенный и а-разветвленный (4) Вторичный, алициклический с 7- или 8-членным циклом (5) Первичный, а-ненасыщенный и (или) а-разветвленный 1085-1050 <1050
В спектрах спиртов и фенолов в жидком состоянии
проявляется широкая полоса поглощения в области
769-650 см-1, обусловленная внеплоскостными дефор-
мационными колебаниями связанной гидроксильной
группы.
2.6.9. Простые эфиры, эпоксиды и пероксиды
2.6.9.1. Валентные колебания С-О
Характеристические полосы поглощения простых
эфиров в ИК-области обусловлены валентными ко-
лебаниями группы С-О-С. Поскольку ее колебания
не сильно отличаются от колебаний системы С-С-С,
можно ожидать совпадения областей этих валентных
колебаний. Однако колебания системы, содержащей
атом кислорода, приводят к большему изменению
дипольного момента, поэтому ИК-полосы поглоще-
ния эфиров более интенсивны. Валентные колебания
С-О-С в эфирах, как и валентные колебания С-О
в спиртах, взаимодействуют с другими колебаниями
молекулы. Спектр анизола на рис. 2.19 типичен для
арилалкильного простого эфира.
В спектрах алифатических простых эфиров наи-
более характеристичной полосой поглощения яв-
ляется сильная полоса в области 1150-1085 см-1,
обусловленная антисимметричными валентными
колебаниями С-О-С и обычно появляющаяся около
1125 см-1. Полоса симметричных валентных колеба-
ний, как правило, слабая, и она лучше проявляется
в спектре комбинационного рассеяния.
Группа С-О-С в шестичленном цикле погло-
щает при той же частоте, что и в ациклических
Рис. 2.19. ИК-Спектр анизола.
Валентные колебания ароматических С-Н: 3067, 3030, 3005 см-1; валентные
колебания С-Н метильной группы: 2950,2843 см-1; область обертонов и составных
частот: 2000-1667 см-1; валентные колебания С-С ароматического кольца:
1601,1501 см"1; антисимметричные валентные колебания С-О-С: 1254 см"1;
симметричные валентные колебания С-О-С: 1046 см"1; внеплоскостные
деформационные колебания С-Н: 784, 761 см"1; внеплоскостные деформационные
колебания С-С: 699 см"1
2.6. Характеристические групповые частоты органических молекул
111
простых эфирах. С уменьшением размера цикла
полоса антисимметричного валентного колебания
С-О-С постепенно сдвигается в сторону меньших
частот (более длинных волн), в то время как полоса
симметричного валентного колебания С-О-С (пуль-
сационное колебание цикла) сдвигается в сторону
больших частот.
Разветвление цепи у углеродного атома, сосед-
него с атомом кислорода, приводит к расщеплению
полосы С-О-С. В спектре изопропилового эфира
в области 1170-1114 см-1 проявляется триплетная
структура, причем главный максимум находится при
1114 см"1.
В спектрах арилалкиловых эфиров полоса анти-
симметричного валентного колебания С-О-С прояв-
ляется в области 1275-1200 см-1, а полоса симметрич-
ного валентного колебания - около 1075-1020 см-1.
В виниловых эфирах сильная полоса поглощения,
обусловленная антисимметричным валентным колеба-
нием С-О-С, наблюдается в области 1225-1200 см-1,
а сильная полоса симметричного валентного коле-
бания-в области 1075-1020 см-1. Такое смещение
полосы антисимметричных колебаний в арилалки-
ловых и виниловых эфирах удовлетворительно объ-
ясняется резонансом, который приводит к усилению
связи С-О.
Полоса валентных колебаний связи С=С в про-
стых виниловых эфирах наблюдается в области 1660-
1610 см-1. Эта полоса имеет большую интенсивность
по сравнению с полосой валентных колебаний С=С
в алкенах. Часто она проявляется в виде дублета,
что связано с поглощением поворотных изомеров.
Вследствие копланарности га/ганс-изомера в нем
достигается максимальный резонанс, что заметно
уменьшает двоесвязность алкенового фрагмента.
Стерические затруднения в z/wc-изомере ослабляют
резонанс.
В результате веерных колебаний группы =С-Н
в алкенах с терминальной двойной связью в их спек-
трах появляются две полосы: одна около 1000 см-1,
другая около 909 см-1. В спектрах виниловых эфи-
ров эти полосы из-за резонанса смещены в сторону
меньших частот.
и /н
R—О—С=С
Н
Веерное колебание концевой
группы СН2, 813 см’1
Веерное колебание группы
СН в трлнс-положении, 960 см’1
В спектрах алкил- и арилпероксидов наблюдает-
ся полоса поглощения фрагмента С-С-О в области
1198-1176 см-1. Ацил- и ароилпероксиды дают две
полосы поглощения карбонильных групп в области
1818-1754 см-1. Появление двух полос объясняется
механическим взамодействием между валентными
колебаниями этих карбонильных групп.
Полоса симметричного валентного, или пульса-
ционного, колебания эпоксидного кольца, когда все
связи в цикле растягиваются или сжимаются в фазе,
наблюдается около 1250 см-1. Другая полоса, относя-
щаяся к антисимметричному валентному колебанию,
при котором связь С-С растягивается одновременно
с сокращением связи С-О, проявляется в области
950-810 см-1. Третья полоса, называемая «полосой
12 мкм», проявляется в области 840-750 см-1. По-
лоса валентных колебаний С-Н эпоксидного кольца
наблюдается в области 3050-2990 см-1.
2.6.10. Кетоны
2.6.10.1. Валентные колебания группы С=0
Кетоны, альдегиды, карбоновые кислоты, эфиры кар-
боновых кислот, лактоны, галогенангидриды и анги-
дриды кислот, амиды и лактамы дают сильную полосу
поглощения в области 1870-1540 см-1, обусловленную
валентными колебаниями группы С=О. Относительно
постоянное положение этой полосы, высокая интен-
сивность и отсутствие в этой области мешающих по-
лос делают ее наиболее узнаваемой в ИК-спектре.
Положение полосы валентных колебаний груп-
пы С=О в указанном интервале определяется сле-
дующими факторами: 1) физическим состоянием, 2)
электронным влиянием и влиянием масс соседних
заместителей, 3) сопряжением, 4) наличием водород-
ных связей (внутри- и межмолекулярных), 5) напря-
жением цикла. Анализ этих факторов дает большую
информацию об окружении карбонильной группы.
При обсуждении этих эффектов обычно за «нор-
мальную» частоту поглощения карбонильной груп-
пы принимается частота 1715 см-1, проявляющаяся
в спектрах чистого (без растворителя) образца на-
сыщенного алифатического кетона. Например, в ПК-
112
2. Инфракрасная спектроскопия
4000 3000 „ z к 2000 1000
Волновые числа (см )
Рис. 2.20. ИК-Спектр ацетона.
Валентные колебания С-Н: метильная группа vas 2995, vs 2918 см-1; метиленовая
группа vas 2964 см-1; валентные колебания С=О: 1715 см-1; деформационные
колебания СН3: 6as 1422 cm‘1,6s 1360 см-1; валентные и деформационные
колебания С-СО-С: 1213 см-1.
спектрах ацетона и циклогексанона наблюдается по-
лоса 1715 см-1. Изменения в окружении карбонильной
группы могут либо повышать, либо понижать частоту
поглощения по сравнению с «нормальной». Типичный
спектр кетона приведен на рис. 2.20.
В спектрах растворов в неполярных растворителях
частота полосы поглощения карбонильной группы
повышена по сравнению со спектрами чистого ве-
щества. Полярные растворители понижают частоту
поглощения. В среднем эти эффекты не превышают
25 см-1.
Замещение алкильной группы насыщенного алифа-
тического кетона гетероатомом (G) смещает полосу
карбонильной группы. Направление смещения зависит
от того, какой эффект преобладает: индуктивный (а)
или резонансный (б):
°>-б
К
(б)
Индуктивный эффект уменьшает длину связи С=О
и тем самым увеличивает ее силовую постоянную
и частоту поглощения. Резонансный эффект увели-
чивает длину связи С=О и уменьшает частоту по-
глощения.
Поглощение некоторых классов соединений, содержа-
щих карбонильную группу, представлено в табл. 2.6.
Сопряжение с двойной углерод-углеродной связью
С=С приводит к делокализации л-электронов обеих
ненасыщенных групп. Делокализация л-электронов
карбонильной группы уменьшает двоесвязность С=О,
в результате полоса поглощения наблюдается при
более низких частотах (более длинных волнах). Сопря-
жение с алкеновой или фенильной группой смещает
полосу поглощения в область 1685-1666 см1. Допол-
нительное сопряжение может вызвать дальнейшее
небольшое уменьшение частоты. Эффект сопряжения
иллюстрирует рис. 2.21.
Стерические эффекты, которые уменьшают ко-
планарность сопряженной системы, ослабляют со-
пряжение. В отсутствие стерических затруднений
сопряженная система будет стремиться принять пло-
скую конформацию. Так, а,0-ненасыщенные кетоны
могут существовать в s-цис- и 5-^/7анс-конформациях.
Когда присутствуют обе формы, наблюдается погло-
щение каждой из них. Поглощение бензальацетона
в сероуглероде может служить одним из примеров:
при комнатной температуре в нем присутствуют обе
(s-цис- и s-транс) формы.
s-цис 1699 см"1
Поглощение связи С=С, находящейся в сопряже-
нии с карбонильной группой, проявляется при более
низких частотах, чем изолированной двойной связи.
Интенсивность полосы поглощения сопряженной
двойной связи в 5-г/г/с-системе выше, чем изолиро-
ванной.
2.6. Характеристические групповые частоты органических молекул
113
Таблица 2.6. Частоты валентных колебаний карбо-
нильной группы в различных соединениях RC(=O)G
Преобладает индукционный эффект заместителя G
G v (С=О), см-1
С1 1815-1785
F -1869
Вг 1812
ОН (для мономера) 1760
OR 1750-1735
Преобладает резонансный эффект заместителя G
G v (С=О), см-1
nh2 1695-1650
SR 1720-1690
Межмолекулярные водородные связи между кето-
ном и гидроксилсодержащим растворителем, таким
как метиловый спирт, вызывают небольшое умень-
шение частоты поглощения карбонильной группы.
Например, чистый образец метилэтилкетона поглощает
при 1715 см-1, а его 10%-ный раствор в метаноле-
при 1706 см-1.
Р-Дикетоны обычно существуют в виде смеси ке-
тонной и енольной таутомерных форм. В спектрах
енольной формы сопряженных кетонов обычная по-
лоса поглощения не проявляется. Вместо нее наблю-
дается широкая полоса в области 1640-1580 см-1,
более интенсивная, чем нормальная карбонильная
полоса. Такая интенсивная и смещенная полоса яв-
ляется результатом образования внутримолекуляр-
ной водородной связи и резонансной стабилизации
енольной формы.
С R"
R'
В жидком ацетилацетоне при 40 °C содержится 64%
енольной формы, которая поглощает при 1613 см-1.
Кетоформа и небольшое количество енольной фор-
мы, не связанной водородной связью, могут вызвать
появление двух полос с центром около 1725 см-1.
Предполагается также, что причиной появления этого
дублета может быть взаимодействие между карбо-
нильными группами кетоформы. Валентные колебания
О-Н енольной группы проявляются в виде широкой
слабой полосы при 3000-2700 см-1.
В спектрах а-дикетонов, у которых карбониль-
ные группы формально сопряжены, наблюдается
одна полоса поглощения вблизи частоты соответ-
ствующего монокетона. Биацетил поглощает при
1718 см-1, а бензил (дибензоил) - при 1681 см-1.
Сопряжение в а-дикетонах не проявляется, и группы
С=О в а-дикетонах не взаимодействуют, как и со-
ответствующие группы в ангидридах карбоновых
кислот (см. раздел 2.6.16).
В хинонах обе карбонильные группы находятся
в одном цикле и поглощают в области 1690-1655 см-1.
При увеличении цепи сопряжения, когда карбониль-
ные группы входят в разные циклы, полоса погло-
щения смещается в область 1655-1635 см-1.
Рис. 2.21. ИК-Спектр ацетофенона.
Обертон валентного колебания С=О 3352 см-1, удвоенная частота валентного
колебания С=О 1686 см-1, частота понижена по сравнению с частотой С=О
в ацетоне (см. рис. 2.20) из-за сопряжения с фенильной группой
114
2. Инфракрасная спектроскопия
В спектрах ациклических а-хлоркетонов наблюда-
ются две полосы поглощения, обусловленные поворот-
ной изомерией. Когда атом хлора расположен вблизи
атома кислорода, его отрицательное поле отталкивает
неспаренные электроны атома кислорода, увеличивая
тем самым силовую постоянную связи С=О. Эта кон-
формация дает высокочастотную полосу поглощения
1745 см-1, другая конформация, в которой карбониль-
ный кислород и атом хлора удалены друг от друга,
дает полосу 1725 см-1. В жестких молекулах, подобных
монокетостероидам, а-галогенирование приводит либо
к экваториальному, либо к аксиальному замещению.
В экваториальном положении атом галогена оказывается
вблизи карбонильной группы, и «эффект поля» вызы-
вает увеличение частоты валентного колебания С=О.
В спектре изомера, в котором атом галогена находится
в аксиальном положении и удален от карбонильной
группы, такого сдвига частоты не наблюдается.
сх
В циклических кетонах валентный угол t,C=O
С
влияет на частоту полосы поглощения карбонильной
группы. На валентное колебание связи С=О, безу-
словно, воздействуют валентные колебания соседних
связей С-С. В ациклических кетонах и в кетонах
с шестичленным циклом рассматриваемый угол равен
-120°. В напряженных циклах, где угол меньше 120°,
взаимодействие с валентными колебаниями связей
С-С увеличивает энергию, необходимую для воз-
буждения валентного колебания С=О, и тем самым
увеличивает частоту соответствующей полосы по-
глощения. Циклогептанон поглощает при 1709 см-1,
циклогексанон - при 1715 см-1, циклопентанон - при
1751 см-1, а циклобутанон - при 1775 см-1.
2.6.10.2. Валентные и деформационные
колебания фрагмента С-С(=О)-С
В области 1300-1100 см-1 в спектрах кетонов на-
блюдаются полосы поглощения умеренной интен-
сивности, обусловленные валентными колебаниями
фрагмента С-С-С и деформационными колебаниями
в группе С—С (—О)—С. Поглощение может представлять
собой мультиплет. Для алифатических кетонов эта
полоса наблюдается в области 1230-1100 см-1; арома-
тические кетоны поглощают в более высокочастотной
части спектрального интервала, принятого в качестве
характеристического для этой группы.
2.6.11. Альдегиды
Спектр октаналя, иллюстрирующий особенности
поглощения, типичные для альдегидов, показан на
рис. 2.22.
2.6.11.1. Валентные колебания связи С=0
Полоса поглощения карбонильной группы в спектрах
альдегидов находится в области несколько более вы-
соких частот, чем в соответствующих метилкетонах.
В алифатических альдегидах она находится около
1740-1720 см-1. Поглощение альдегидной группы реа-
гирует на структурные изменения таким же образом,
как и поглощение кетонной группы. Электроотри-
цательные заместители у а-атома углерода увели-
чивают частоту поглощения карбонильной группы.
Уксусный альдегид поглощает при 1730 см1, а три-
хлоруксусный альдегид-при 1768 см1. Сопряжение
с ненасыщенными группировками, например в а,Р-
ненасыщенных или ароматических альдегидах, по-
нижает частоту поглощения карбонильной группы.
Рис. 2.22. ИК-Спектр октаналя.
Валентные колебания С-Н в алифатической части: 2980-2860 см-1 (см. рис. 2.8);
валентные колебания С-Н альдегидной группы: 2715 см-1; обычная частота
валентного колебания С=О в альдегиде: 1728 см~1; деформационное колебание
С-Н альдегидной группы: 1381 см-1.
2.6. Характеристические групповые частоты органических молекул
115
100 -
50 -
1--1--1--1--1--г
1000
3000 „ , 1 2000
Волновые числа (см-1)
Рис. 2.23. ИК-Спектр гексановой кислоты.
Широкая полоса валентных колебаний О-Н: 3300-2500 см-1; валентные колебания
С-Н (см. рис. 2.8): 2967,2874,2855 см-1, перекрываются с широкой полосой
валентных колебаний О-Н; валентное колебание карбоксильной группы С=О,
характерное для димерного состояния: 1717 см-1; плоскостное деформационное
колебание С-О-Н: 1424 см-1; валентное колебание С-О в димере: 1301 см-1;
внеплоскостное деформационное колебание О-Н: 946 см-1.
4000
а,Р-Ненасыщенные и ароматические альдегиды погло-
щают в области 1710-1685 см-1. Внутримолекулярная
водородная связь смещает полосу поглощения в сто-
рону низких частот (в случае салицилового альдеги-
да до 1666 см-1). В спектре глиоксаля (простейшего
а-диальдегида), как и в спектрах а-дикетонов, имеется
только одна полоса поглощения карбонильной группы
без смещения относительно нормального положения
полосы поглощения моноальдегида.
2.6.11.2. Валентные колебания связи С-Н
В спектрах большинства альдегидов полосы валент-
ных колебаний связи С-Н альдегидной группы рас-
положены в области 2830-2695 см-1. В этой области
часто наблюдаются две полосы умеренной интенсив-
ности. Появление двух полос объясняется резонансом
Ферми между основным колебанием С-Н альдегид-
ной группы и первым обертоном деформационного
колебания С-Н альдегидной группы, которое обычно
проявляется около 1390 см-1. Для альдегидов, у кото-
рых полоса деформационного колебания С-Н заметно
смещена относительно 1390 см-1, наблюдается только
одна полоса в области валентного колебания С-Н.
Некоторые ароматические альдегиды с сильными
электроотрицательными группами в ортио-положении
могут поглощать даже при 2900 см-1.
Полоса поглощения средней интенсивности около
2720 см-1 в сочетании с полосой поглощения карбо-
нильной группы является хорошим доказательством
наличия альдегидной группы.
2.6.12. Карбоновые кислоты
2.6.12.1. Валентные колебания группы 0-Н
В жидком или твердом состоянии и в растворах
в СС14 при концентрациях выше 0.01 моль-л-1 кар-
боновые кислоты благодаря сильным водородным
связям существуют в виде димеров.
/°-н-Ч
о^-н—о о—н—о
+ -
Исключительная прочность водородных связей
объясняется большим вкладом ионной резонансной
структуры. Из-за сильного связывания валентные
колебания свободной гидроксильной группы (око-
ло 3520 см-1) наблюдаются только в очень разбав-
ленных растворах в неполярных растворителях или
в газовой фазе.
Для димеров карбоновых кислот характерна очень
широкая интенсивная полоса валентных колебаний
связи О-Н в области 3300-2500 см-1 с центром
около 3000 см-1. Более слабые полосы валентных
колебаний С-Н обычно накладываются на широкую
полосу связи О-Н. Тонкая структура, наблюдаемая со
стороны низкочастотного края широкой полосы О-Н,
обусловлена обертонами и составными частотами
основных полос, проявляющихся при более низких
частотах. Спектр типичной алифатической карбоновой
кислоты представлен на рис. 2.23.
116
2. Инфракрасная спектроскопия
Другие соединения с сильными водородными свя-
зями, например Р-дикетоны, также поглощают в об-
ласти 3300-2500 см"1, но их поглощение обычно
менее интенсивно. Кроме того, полосы валентных
колебаний группы С=О в соединениях, подобных
Р-дикетонам, смещены в сторону низких частот по
сравнению с карбоновыми кислотами.
Карбоновые кислоты могут связываться межмоле-
кулярными водородными связями с простыми эфира-
ми, например с диоксаном, тетрагидрофураном или
с другими растворителями акцепторами протонов.
Спектры, зарегистрированные в таких растворителях,
имеют полосу поглощения связанной гидроксильной
группы около 3100 см-1.
2.6.12.2. Валентные колебания группы (=0
Полосы валентных колебаний карбонильной группы
кислот гораздо интенсивнее, чем полосы валентных
колебаний кетонной карбонильной группы. Мономе-
ры насыщенных алифатических кислот поглощают
около 1760 см-1.
Димеры карбоновых кислот центросимметричные;
в ИК-спектре проявляется полоса поглощения толь-
ко антисимметричного валентного колебания С=О.
Полоса поглощения группы С=О, связанной водо-
родными связями и ослабленной за счет резонанса,
смещается в область более низких частот по срав-
нению с мономером. Карбонильная группа в диме-
ризованных насыщенных алифатических кислотах
поглощает в области 1720-1706 см1.
Внутримолекулярная водородная связь понижает
частоту валентного колебания карбонильной группы
в большей степени, чем межмолекулярная. Напри-
мер, салициловая кислота имеет полосу поглощения
при 1665 см-1, в то время как и-гидроксибензойная
кислота-при 1680 см-1.
Ненасыщенные группировки в сопряжении с кар-
бонильной группой кислоты лишь в незначительной
степени уменьшают частоту (увеличивают длину вол-
ны) полосы поглощения как мономерной, так и ди-
мерной форм. В спектрах а,Р-ненасыщенных кислот
и кислот с сопряженными арильными заместителями
наблюдается полоса поглощения димера в области
1710-1680 см"1. При удлинении цепи сопряжения за
пределы а,Р-положения происходит очень небольшое
дополнительное длинноволновое смещение полосы
поглощения карбонильной группы.
Замещение в a-положении электроотрицательны-
ми группами, такими как галогены, приводит к не-
которому увеличению частоты поглощения С=О
(на 10-20 см"1). В спектрах кислот с галогенами
в a-положении, полученных в жидком состоянии или
в растворе, из-за поворотной изомерии проявляются
двойные полосы карбонильной группы (эффект поля).
Полоса с более высокой частотой соответствует кон-
формации, в которой атом галогена и карбонильная
группа сближены в пространстве.
2.6.12.3. Валентные колебания связи С-О
и деформационные колебания связи 0-Н
В спектрах карбоновых кислот проявляются две по-
лосы около 1320-1210 и 1440-1395 см"1, возникающие
в результате валентных колебаний С-О и деформа-
ционных колебаний О-Н соответственно. Эти полосы
отражают взаимодействие между валентными колеба-
ниями связи С-О и плоскостными деформационны-
ми колебаниями группы С-О-Н. Более интенсивная
полоса (для димеров в области 1315-1280 см"1) рас-
сматривается как полоса валентных колебаний С-О
и в спектрах жирных кислот с длинной цепью обычно
проявляется в виде дублета. Полоса деформацион-
ных колебаний С-О-Н около 1440-1395 см"1 имеет
умеренную интенсивность и наблюдается в той же
области, что и ножничные колебания группы СН2,
соседней с карбонильной группой.
Одна из характеристических полос в спектрах ди-
мерных карбоновых кислот возникает в результате
внеплоскостных деформационных колебаний связан-
ной гидроксильной группы. Эта полоса проявляется
в спектре около 920 см"1 и характеризуется большой
шириной и средней интенсивностью.
2.6.13. Карбоксилат-анион
Карбоксилат-анион имеет две сильно взаимодействую-
щие углерод-кислородные связи с длиной, промежу-
точной между С=О и С-О.
О
—с-
чо
Карбоксилат-ион дает две полосы: сильную полосу
антисимметричных валентных колебаний в области
1650-1550 см"1 и более слабую полосу симметричных
валентных колебаний около 1400 см"1.
Превращение карбоновой кислоты в соль мож-
но использовать для идентификации. Обычно это
делают, добавляя третичный алифатический амин,
например триэтиламин, к раствору карбоновой
кислоты в хлороформе (в СС14 реакция не идет).
В спектре образовавшейся соли наряду с «аммоние-
вой полосой» в области 2700-2200 см"1 видны две
характеристические полосы карбоксильной группы.
Полоса валентных колебаний О-Н, разумеется, ис-
чезает. В ИК-спектре бензоата аммония (рис. 2.24)
проявляются указанные особенности.
2.6. Характеристические групповые частоты органических молекул
117
Рис. 2.24. ИК-Спектр бензоата аммония.
А: валентные колебания N-H и С-Н, 3600-2500 см"1; Б: валентные колебания С-С кольца,
1600 см"1; В: антисимметричное валентное колебание карбоксилат-аниона -COj, 1550 см"1;
Г: симметричное валентное колебание карбоксилат-аниона -COj, 1385 см"1
2.6.14. Сложные эфиры и лактоны
В ИК-спектрах сложных эфиров и лактонов на-
блюдаются две сильные характеристические полосы
поглощения, относящиеся к валентным колебаниям
связей С=О и С-О. Интенсивная полоса валент-
ных колебаний карбонильной группы наблюдает-
ся при более высоких частотах (коротких волнах),
чем в спектрах обычных кетонов. В силу электро-
ноакцепторной природы соседнего атома кислорода
(индуктивный эффект) силовая постоянная связи
С=О увеличивается. Однако области, характерные
для поглощения карбонильной группы в сложных
эфирах, могут перекрываться с соответствующими
областями в кетонах нормального строенияя. Отли-
чительным признаком сложных эфиров и лактонов
является наличие сильной полосы валентных коле-
баний связи С-О в области, где для кетонов наблю-
даются более слабые полосы. Карбонильные полосы
эфиров и лактонов могут перекрываться с полосами
кислот, но кислоты отличаются наличием полосы
валентных и деформационных колебаний группы
О-Н, а также способностью к образованию солей.
В спектрах сложных эфиров положение карбониль-
ной полосы зависит от ближайшего окружения этой
группы таким же образом, как и в случае кетонов.
Спектр фенилацетата (рис. 2.25) демонстрирует наи-
более важные и характерные полосы поглощения
сложных эфиров.
2.6.14.1. Валентные колебания группы С=0
Полоса поглощения группы С=О насыщенных али-
фатических сложных эфиров (за исключением фор-
миатов) находится в области 1750-1735 см-1. Поло-
сы поглощения С=О формиатов, а,Р-ненасыщенных
сложных эфиров и бензоатов находятся в области
1730-1715 см-1. Дальнейшее увеличение цепи сопря-
жения практически не влияет на частоту поглощения
карбонильной группы.
В спектрах винильных или фенильных сложных
эфиров с ненасыщенной группой по соседству с груп-
пой С-О- наблюдается заметное увеличение частоты
поглощения карбонильной группы наряду с пониже-
нием частоты группы С-О. Винилацетат поглощает
при 1776 см-1, а фенилацетат - при 1771 см-1.
Замещение а-атомов водорода на галоген приводит
к увеличению частоты валентного колебания С=О.
Этиловый эфир трихлоруксусной кислоты поглощает
при 1770 см-1.
В оксалатах и а-кетоэфирах, как и в а-дикетонах,
либо нет взаимодействия между двумя карбониль-
ными группами, либо оно мало, и в этом случае на-
блюдается нормальная полоса поглощения в области
1755-1740 см-1. В спектрах Р-кетоэфиров, где может
происходить енолизация, полоса поглощения про-
является около 1650 см-1, потому что эфирная кар-
бонильная группа связывается водородной связью
с енольной гидроксильной группой.
Карбонильная полоса в спектрах насыщенных
8-лактонов (шестичленные циклы) наблюдается в той
118
2. Инфракрасная спектроскопия
Рис. 2.25. ИК-Спектр фенилацетата.
Валентные колебания ароматических С-Н: 3075,3052 см-1; валентное колебание
С=О: 1771 см-1, эта частота выше, чем частота валентных колебаний нормальной
сложноэфирной группы (1740 см"1, см. табл. 2.6) из-за сопряжения фенильной
группы с атомом кислорода фенольного фрагмента, сопряжение арильной
или другой ненасыщенной группы с карбонильной группой понижает частоту
валентных колебаний С=О по сравнению с нормальной (например, бензоаты
поглощают около 1724 см"1); валентное колебание С-С кольца: 1601 см-1;
5as СН3 1493 см"1; 6S СН3 1378 см"1; валентное колебание ацетатной группы
С(=О)-О: 1223 см"1; антисимметричное валентное колебание О-С-С: 1200 см~1
же самой области, что и в случае несопряженных
эфиров с неразветвленной цепью. Ненасыщенные
группировки в a-положении по отношению к кар-
бонильной группе уменьшают частоту карбонильной
полосы, а ненасыщенные группировки в а-положении
к группе -О- увеличивают ее.
1720 см-1 1760 см"1
а-Пироны часто дают две полосы поглощения
карбонильной группы в области 1775-1715 см-1,
возможно, вследствие резонанса Ферми.
Насыщенные у-лактоны (пятичленные циклы) по-
глощают при более высоких частотах, чем эфиры или
5-лактоны: 1795-1760 см-1; у-валеролактон поглощает
при 1770 см-1. Введение двойной связи в молекулу
у-лактона влияет на положение полосы поглощения
карбонильной группы точно так же, как в 5-лактонах.
1800 см"1 1750 см"1
В ненасыщенных лактонах, в которых двойная
связь находится по соседству с -О-, наблюдается
сильная полоса поглощения связи С=С в области
1685-1660 см-1.
2.6.14.2. Валентные колебания связи С-О
«Валентные колебания связи С-О» в сложных эфирах
фактически состоят из двух взаимодействующих анти-
симметричных колебаний: С-С(=О)-О и О-С-С, причем
первое намного важнее. Эти полосы наблюдаются в об-
ласти 1300-1000 см"1. Соответствующие симметричные
колебания имеют меньшее значение. Валентные коле-
бания С-О не так надежны для корреляций спектр -
структура, как валентные колебания С=О.
Полоса С-С(=О)-О в ИК-спектрах насыщенных
эфиров, за исключением ацетатов, имеет высокую ин-
тенсивность и расположена в области 1210-1163 см1.
Она часто шире и интенсивнее, чем полоса валентных
колебаний С=О. В спектрах ацетатов насыщенных
спиртов эта полоса проявляется при 1240 см-1. Ви-
нил- и фенилацетаты поглощают при более низкой
частоте (1190-1140 см-1) (см., например, рис. 2.25).
Валентное колебание группы С-С(=О)-О в эфирах
а,Р-ненасыщенных кислот дает ряд полос в области
1300-1160 см-1. Эфиры ароматических кислот силь-
но поглощают в области 1310-1250 см-1. Валентные
колебания такого же типа в лактонах проявляются
в области 1250-1111 см-1.
Полоса О-С-С сложных эфиров первичных спиртов
(валентные колебания «спиртового» фрагмента С-О)
2.6. Характеристические групповые частоты органических молекул
119
Рис. 2.26. ИК-Спектр 4-гексилбензоилхлорида.
Валентные колебания ароматических С-Н: 3036 см-1; валентные колебания С-Н:
2936,2866 см-1; валентные колебания С=О: 1779 см-1 (см. табл. 2.6), положение этой
полосы очень слабо зависит от сопряжения, ароилхлориды идентифицируют по
полосе 1748 см-1, обусловленной резонансом Ферми полосы валентных колебаний
С=О с обертоном полосы 884 см-1
наблюдается около 1164-1031см-1, а для эфиров вто-
ричных спиртов - около 1100 см-1. В ароматических
эфирах первичных спиртов эта полоса проявляется
вблизи 1111 см-1.
Метиловые эфиры жирных кислот с длинной цепью
дают три полосы с максимумами при 1250, 1205
и 1175 см-1. Полоса при 1175 см-1 самая сильная.
2.6.15. Галогенангидриды кислот
2.6.15.1. Валентные колебания группы С=0
В спектрах галогенангидридов кислот в области валент-
ных колебаний карбонильной группы наблюдается силь-
ная полоса поглощения. Несопряженные хлорангидриды
поглощают в области 1815-1785 см-1. Ацетилфгорид в га-
зовой фазе поглощает около 1869 см-1. Карбонильная
полоса сопряженных галогенангидридов кислот имеет
пониженную частоту, потому что резонанс уменьшает
силовую постоянную связи С=О. Хлорангидриды аро-
матических кислот дают сильную полосу поглощения
в области 1800-1770 см-1. Слабая полоса в области 1750—
1735 см-1, наблюдающаяся в спектрах ароилхлоридов,
возможно, является результатом резонанса Ферми между
колебаниями С=О и обертоном основного колебания,
с волновым числом 884 см-1. Типичный спектр хлоран-
гидрида ароматической кислоты приведен на рис. 2.26.
2.6.16. Ангидриды карбоновых кислот
2.6.16.1. Валентные колебания группы С=0
В спектрах ангидридов кислот в области валентных
колебаний карбонильной группы наблюдаются две
полосы. Они возникают в результате симметрично-
го и антисимметричного валентных колебаний С=О.
Насыщенные ациклические ангидриды поглощают
около 1818 и 1750 см-1. В спектрах сопряженных
ациклических ангидридов наблюдаются полосы при
1775 и 1720 см-1; уменьшение частоты вызвано ре-
зонансом. Высокочастотная полоса более интенсивна.
Пятичленные циклические ангидриды из-за на-
пряжения цикла дают полосу поглощения с более
высокой частотой (меньшей длиной волны), чем аци-
клические ангидриды: янтарный ангидрид поглощает
при 1865 и 1782 см-1. Из двух карбонильных полос
пятичленных циклических ангидридов более интен-
сивной является полоса с низкой частотой (длин-
новолновая).
2.6.16.2. Валентные колебания связи С-О
Другие сильные полосы проявляются в спектрах ан-
гидридов в результате валентных колебаний фрагмен-
та С-С(=О)-О-С(=О)-С. Несопряженные ангидриды
с неразветвленной цепью поглощают около 1047 см-1.
В спектрах циклических ангидридов наблюдаются
полосы в областях 952-909 и 1299-1176 см-1. Полоса
валентных колебаний С-О уксусного ангидрида на-
ходится при 1125 см-1.
Спектр ангидрида бензойной кислоты, показанный на
рис. 2.27, типичен для ароматического ангидрида.
2.6.17. Амиды и лактамы
В спектрах всех амидов проявляется полоса погло-
щения карбонильной группы, известная как полоса
120
2. Инфракрасная спектроскопия
Рис. 2.27. ИК-Спектр ангидрида бензойной кислоты.
Валентные колебания ароматических С-Н: 3067,3013 см-1; взаимодействующие
антисимметричное и симметричное валентные колебания С=О:
1779 и 1717 см-1 соответственно (см. табл. 2.6); валентное колебание
С-СО-О-СО-С: 1046 см’1
«Амид I». Ее положение зависит от степени ассо-
циации посредством образования водородных связей
и тем самым от физического состояния соединения.
Первичные амиды дают две полосы валентных
колебаний связи N-H, являющиеся результатом симме-
тричных и антисимметричных валентных колебаний
N-H. Вторичные амиды и лактамы дают только одну
полосу валентных колебаний N-H. Как и в случае
валентных колебаний О-Н, частота валентных колеба-
ний N-H уменьшается при образовании водородных
связей, хотя и в меньшей степени. Области, в кото-
рых проявляются валентные колебания связей N-H
и О-Н, перекрываются, из-за чего иногда невозможно
сделать однозначное отнесение структуры.
В спектрах первичных и вторичных амидов, а также
многих лактамов имеется полоса или полосы в об-
ласти 1650-1515 см-1: это так называемая область
полосы «Амид II», обусловленной главным образом
деформационными колебаниями групп NH2 или NH.
Эта полоса включает взаимодействие между дефор-
мационными колебаниями N-H и другими основными
колебаниями и требует тиране-конформации.
Широкие полосы средней интенсивности в об-
ласти 800-666 см-1 обусловлены внеплоскостными
веерными колебаниями N-H.
Спектр амида акриловой кислоты, показанный на
рис. 2.28, типичен для первичного амида ненасы-
щенной кислоты.
2.6.17.1. Валентные колебания связи N-H
В разбавленных растворах в неполярных растворите-
лях первичные амиды дают две полосы поглощения
умеренной интенсивности, отвечающие антисим-
метричным и симметричным валентным колеба-
ниям связей N-H. Эти полосы проявляются около
3520 и 3400 см-1 соответственно. В спектрах твердых
образцов эти полосы вследствие образования водо-
родных связей наблюдаются при 3350 и 3180 см-1.
В ИК-спектрах вторичных амидов, которые су-
ществуют главным образом в тиране-конформации,
валентные колебания свободной группы N-H, на-
блюдаемые в разбавленных растворах, проявляются
в области 3500-3400 см-1 соответственно. В более
концентрированных растворах и в твердых образцах
свободная группа N-H проявляется в виде ряда полос
в области 3330-3060 см-1. Мультиплетные полосы
обусловлены тем, что амидная группа может связы-
ваться водородными связями, образуя димеры с s-цис-
конформацией и полимеры с s-тиранс-конформацией:
\ Z“H\ zR' к
У /N~ Н— О=С\
N С —О=С N—Н—
R'Z Н—G \ R R-Z
s-цыс-конформация s-тлраяс-конформация
2.6.17.2. Валентные колебания группы С=0 (полоса «Амид Ь>)
Благодаря резонансному эффекту (см. раздел 2.6.10.1)
полоса поглощения карбонильной группы амидов
проявляется при меньших частотах, чем «нормальная»
полоса карбонильной группы. Положение этой полосы
зависит от тех же факторов, связанных с окружением
карбонильной группы, что и в случае других соеди-
нений.
2.6. Характеристические групповые частоты органических молекул
121
Рис. 2.28. ИК-Спектр акриламида.
Валентные колебания N-H первичного амида, связанной водородными связями:
антисимметричные 3352 см-1, симметричные 3198 см-1; перекрывание валентного
колебания С=О и полосы Амид I: 1679 см"1 (см. табл. 2.6); деформационное
колебание N-H (полоса Амид II): 1617 см"1; валентное колебание C-N: 1432 см"1;
широкая полоса деформационного внеплоскостного колебания N-H: 700-600 см"1
Твердые образцы первичных амидов (за исключе-
нием ацетамида, для которого полоса поглощения кар-
бонильной группы наблюдается при 1694 см"1) дают
сильную полосу Амид I вблизи 1650 см"1. Если амид
исследуют в разбавленном растворе, полоса проявляется
при более высокой частоте (около 1690 см"1). В более
концентрированных растворах карбонильная полоса за-
нимает промежуточное положение, зависящее от степени
ассоциации посредством водородных связей.
Простые вторичные амиды с открытой цепью
в твердом состоянии поглощают около 1640 см"1.
В разбавленном растворе частота полосы Амид I мо-
жет повышаться до 1680 и даже до 1700 см"1 в случае
анилидов. В анилидной структуре бензольное кольцо
и карбонильная группа конкурируют за неподеленную
электронную пару атома азота.
Положение полосы карбонильной группы в тре-
тичных амидах не зависит от физического состояния
вещества, так как образование водородных связей
с другими третичными амидными группами невоз-
можно. Полоса поглощения карбонильной группы
проявляется в области 1680-1630 см”1. На область
поглощения третичных амидов в растворе влияет об-
разование водородных связей с растворителем: так,
Ы,Ы-диэтилацетамид поглощает при 1647 см"1 в ди-
оксане и при 1615 см”1 в метаноле.
Электроноакцепторные группы, связанные с ато-
мом азота, повышают частоту поглощения, так как
они успешно конкурируют с атомом кислорода за
электронную пару азота, увеличивая тем самым си-
ловую постоянную связи С=О.
2.6.17.3. Деформационные колебания связи N-H
(полоса «Амид II»)
Все первичные амиды в разбавленных растворах
дают резкую полосу поглощения Амид II, возникаю-
щую в результате деформационных колебаний связи
N-H при несколько более низкой частоте, чем ча-
стота полосы карбонильной группы. Интенсивность
полосы Амид II составляет от половины до одной
трети интенсивности полосы поглощения С=О.
В вазелиновом масле и в таблетках с КВг полоса
проявляется в интервале 1655-1620 см”1 и обычно
перекрывается с полосой Амид I. В разбавленных
растворах полоса наблюдается при более низкой ча-
стоте 1620-1590 см"1 и обычно проявляется отдельно
от полосы Амид I. В спектрах концентрированных
растворов может появляться ряд полос вследствие
наличия как свободных, так и ассоциированных
молекул. Природа радикала R в первичных амидах
R-C(=O)-NH2 мало влияет на положение полосы
Амид II.
Вторичные ациклические амиды в твердом состо-
янии дают полосу Амид II в области 1570-1515 см”1.
В разбавленных растворах эта полоса наблюдается
в области 1550-1510 см"1. Данная полоса обуслов-
лена взаимодействием между деформационными
колебаниями N-H и валентными колебаниями C-N
в структурном фрагменте C-N-Н. Вторая, более
слабая полоса около 1250 см"1 также возникает
в результате взаимодействия между деформаци-
онными колебаниями N-H и валентными колеба-
ниями C-N.
122
2. Инфракрасная спектроскопия
2.6.17.4. Другие колебательные полосы
Полоса валентных колебаний связи C-N в первичных
амидах наблюдается около 1400 см"1. Широкая полоса
средней интенсивности в области 800-666 см"1 в спек-
трах первичных и вторичных амидов появляется в ре-
зультате внеплоскостных веерных колебаний N-H.
В лактамах со средним размером кольца амидная
группа вынуждена находиться в s-i/wc-конформации.
В спектрах лактамов в твердом состоянии сильная
полоса валентных колебаний N-H проявляется при
3200 см"1. Эта полоса заметно не смещается при
разбавлении раствора, так как s-i/wc-форма остается
в ассоциированном состоянии и при относительно
низких концентрациях.
2.6.17.5. Валентные колебания группы С=0 в лактамах
Полоса поглощения карбонильной группы лактамов
с циклами, содержащими шесть и более атомов, про-
является около 1650 см"1. Пятичленные (у) лактамы
поглощают в области 1750-1700 см"1. Четырехчлен-
ные (Р) неконденсированные лактамы дают полосу
поглощения в области 1760-1730 см"1. Конденсация
лактамного цикла с другим циклом обычно повышает
частоту на 20-50 см"1.
В спектрах большинства лактамов отсутствует
полоса около 1550 см"1, которая характерна для
5-Аиранс-конфигурации ациклических вторичных
амидов. Внеплоскостные веерные колебания N-H
в лактамах дают широкую полосу поглощения в об-
ласти 800-700 см"1.
2.6.18. Амины
Спектр типичного первичного алифатического
диамина приведен на рис. 2.29.
2.6.18.1. Валентные колебания связи N-H
В спектрах первичных аминов, исследуемых в раз-
бавленных растворах, проявляются две слабые по-
лосы поглощения: одна около 3500, а другая около
3400 см"1. Эти полосы относятся соответственно
к антисимметричным и симметричным валентным
колебаниям «свободной» аминогруппы. В спектрах
вторичных аминов наблюдается одиночная слабая
полоса в области 3350-3310 см"1. Полосы валентных
колебаний смещаются в сторону меньших частот при
образовании водородных связей. В случае ассоции-
рованной аминогруппы полосы менее интенсивные
и часто более узкие, чем соответствующие полосы
гидроксильной группы. Алифатические первичные
амины (без растворителя) дают полосы поглощения
в областях 3400-3300 и 3330-3250 см"1. Полосы
поглощения первичных ароматических аминов рас-
положены при несколько более высоких частотах
(меньших длинах волн). В спектрах жидких образцов
первичных и вторичных аминов у полосы валентных
колебаний N-H со стороны низких частот обычно
появляется плечо, обусловленное обертоном полосы
деформационных колебаний N-H, усиленным резо-
нансом Ферми. Для третичных аминов в этой области
спектра поглощение не наблюдается.
оо
--1--1-1--1--1--1-1--1--1-1--П--1--1-1--1--1--1-1--1--1-1--1--1--1-1--1--1-1--1--1--1-1--1--1-1
4000 3000 2000 1000
Волновые числа (см1)
Рис. 2.29. ИК-Спектр 2-метилпентандиамина-1,5.
Две полосы взаимодействующих валентных колебаний групп N-H, участвующих
в водородных связях в первичном амине: антисимметричное 3368 см-1,
симметричное 3291 см"1 (плечо около 3200 см"1 обусловлено резонансом Ферми
с обертоном полосы 1601 см"1); валентные колебания С-Н: 2928,2859 см"1;
деформационное колебание N-H (ножничное): 1601 см-1; 6S СН2 (ножничное):
1470 см"1; валентные колебания связей C-N: 1069 см"1; веерное колебание N-H
(чистый образец): -900-700 см"1
2.6. Характеристические групповые частоты органических молекул
123
2.6.18.2. Деформационные колебания связей N-H
Деформационные (ножничные) колебания N-Н первич-
ных аминов наблюдаются в области 1650-1580 см"1.
Интенсивность этой полосы изменяется от средней
до высокой, а в случае ассоциации молекул она не-
много смещается в сторону более высоких частот.
В спектрах вторичных алифатических аминов редко
удается определить полосу деформационных колеба-
ний N-Н, в то время как вторичные ароматические
амины поглощают при 1515 см"1.
Жидкие образцы первичных и вторичных аминов
дают широкую полосу поглощения средней или высо-
кой интенсивности в области 909-666 см"1, обуслов-
ленную веерными колебаниями N-Н. Положение этой
полосы зависит от степени ассоциации посредством
образования водородных связей.
2.6.18.3. Валентные колебания связи C-N
В ИК-спектрах первичных, вторичных и третичных
алифатических аминов в области 1250-1020 см"1 про-
являются полосы поглощения несопряженной связи
С-N средней или низкой интенсивности. Эти полосы
обусловлены валентными колебаниями С-N, взаимо-
действующими с валентными колебаниями сосед-
них связей в молекуле. Положение полос поглощения
в этой области зависит от класса амина и типа за-
мещения а-углеродного атома.
Ароматические амины дают сильную полосу валент-
ных колебаний связи С-N в области 1342-1266 см"1.
Полоса поглощения возникает при более высоких
частотах (коротких волнах), чем соответствующая по-
лоса С-N в спектрах алифатических аминов, потому
что силовая постоянная связи С-N увеличивается
при резонансе с кольцом.
Отнесения характеристических интенсивных полос
валентных колебаний С-N в спектрах ароматических
аминов приведено в табл. 2.7.
2.6.19. Соли аминов
2.6.79.7. Валентные колебания связи N-H
В спектрах иона аммония находится сильная широ-
кая полоса поглощения в области 3300-3030 см"1,
Таблица 2.7. Валентные колебания связи C-N
в ароматических аминах
Ароматические амины Область поглощения, см-1
Первичные 1340-1250
Вторичные 1350-1280
Третичные 1360-1310
обусловленная валентными колебаниями N-Н (см.
рис. 2.24). В области 2000-1709 см"1 возникает со-
ставная полоса.
В спектрах солей первичных аминов наблюда-
ется сильная широкая полоса поглощения между
3000 и 2800 см"1, обусловленная антисимметрич-
ными и симметричными валентными колебаниями
группы NH3+. Кроме того, имеются составные полосы
средней интенсивности в области 2800-2000 см"1,
наиболее характерна полоса около 2000 см"1. Соли
вторичных аминов сильно поглощают в области
3000-2700 см"1 с мультиплетными полосами, рас-
пространяющимися до 2273 см"1, полоса средней
интенсивности может возникнуть около 2000 см"1.
Соли третичных аминов поглощают при больших
длинах волн, чем соли первичных и вторичных аминов
(2700-2250 см"1). Соли четвертичных ионов аммония
не имеют валентных колебаний N-H.
2.6.79.2. Деформационные колебания связей N-H
В ИК-спектре иона аммония NH4+ наблюдается силь-
ная широкая полоса деформационного колебания око-
ло 1429 см"1. Группа NH3+ соли первичного амина
поглощает в областях 1600-1575 и 1550-1504 см"1.
Эти полосы обусловлены антисимметричными и сим-
метричными деформационными колебаниями NH3+
подобно соответствующим полосам группы СН3. Соли
вторичных аминов поглощают около 1620-1560 см"1.
Полосы деформационных колебаний N-Н солей тре-
тичных аминов слабые и не имеют практического
значения.
2.6.20. Аминокислоты и их соли
Аминокислоты встречаются в трех формах.
1. Свободная аминокислота (цвиттер-ион).0
I
-с-со2-
NH3+
2. Гидрохлорид (или другая соль).
—С—СО2Н
NH3+ СГ
0 Ароматические аминокислоты не являются цвиттер-
ионами. Например, и-аминобензойная кислота
СООН
124
2. Инфракрасная атектроскопия^
3. Натриевая соль (или соль другого катиона).
I
—С—СО2- Na
NH2
Свободные первичные аминокислоты характери-
зуются следующими полосами поглощения (боль-
шинство работ было выполнено с а-аминокислотами,
но относительное положение амино- и карбоксильной
групп, по-видимому, не имеет большого значения):
1. Широкая сильная полоса валентных колебаний
групп NH3+ в области 3100-2600 см-1. За счет
многочисленных составных полос и обертонов
область поглощения распространяется примерно
до 2000 см-1. Эта область обертонов обычно вклю-
чает характерную полосу около 2222-2000 см-1,
относящуюся к комбинации антисимметричного
деформационного и крутильного колебаний груп-
пы NH3+. Крутильное колебание проявляется око-
ло 500 см-1. Полоса при 2000 см-1 отсутствует,
если атом азота в аминокислоте замещен.
2. Слабая полоса антисимметричных деформа-
ционных колебаний NH3+ в области 1660—
1610 см"1 и довольно сильная полоса симме-
тричных деформационных колебаний в области
1550-1485 см"1. о
3. Карбоксилат-анион —Сч- имеет сильное по-
чо
глощение около 1600-1590 см"1 и более сла-
бое - около 1400 см"1. Эти полосы появляются
в результате соответственно антисимметричных
и симметричных валентных колебаний СО2".
На рис. 2.30 приведен спектр аминокислоты лей-
цина с отнесением частот в соответствии с тремя
указанными выше пунктами.
Гидрохлориды аминокислот дают следующую
картину поглощения.
1. Широкая сильная полоса в области 3333-2380 см"1,
являющаяся результатом наложения полос валент-
ных колебаний групп О-Н и NH3+. Поглощение
в этой области характеризуется мультиплетной
тонкой структурой со стороны меньших частот.
(±i - Лейцин
СН3 О
I о
сн3- сн - сн2 - сн - с -б
NHS
C6H13NO2 ММ 131.18, т. пл. 332°С (разл.) (лит.), вазел. масло
Длина волны (мкм)
2.5 3 4 5 6 7 8 9 10 12 15 20 30 40
0.00
0.05
I I I I I I I I I I I 1 I I I I I » I I I I I I I I 1 1 I I I I I 1 I I I 1111 n 11111 11111111111111111 11111 I I 11 11111| t И tlllllliml I I 11 11 Hll I 1 I 1111 III I 1111 I
о
Б 0.20
с;
с
к 0.30
S
Ф 0.40
$0.50
с аво
о
аво
1.00
zoo
“4000 3500 3000 2500 2000 1800 1600 1400 1200 1000 800 600 400 200
Волновые числа (см'1)
© SADTLER RESEARCH LABORATORIES.
PHILADELPHIA. РА. 1В1О4 U.SA.
Рис. 2.30. ИК-Спектр (±)-лейцина. А: широкая (-NH3+) полоса валентных колебаний
N-H, 3100-2000 см-1, уширенная составной полосой при 2140 см-1 и другими
составными полосами и обертонами; Б: валентное колебание алифатических
связей С-Н (перекрытое полосой валентных колебаний N-H), 2967 см-1;
В: антисимметричное деформационное колебание связей N-H группы NH3+,
1610 см-1; Г: антисимметричное валентное колебание карбоксилатной группы СО2~,
1580 см-1; Д: симметричное деформационное колебание N-H группы NH3+,
1505 см-1; Е: симметричное валентное колебание карбоксилатной группы СО2”,
1405 см-1; Ж: торсионное колебание группы NH3+, 525 см-1
2.6. Характеристические групповые частоты органических молекул
125
100:
& 90 :
<D "
д ___-
О 80 :
& -
Т--1-1--1-1--1-1--1--1-1--1-1--г
3000 . 2000
Волновые числа (см-1)
Рис. 2.31. ИК-Спектр а-метилбензилцианида.
Валентные колебания ароматических С-Н: 3067,3030 см-1; валентные колебания
алифатических С-Н: 2990,2944 см-1; валентные колебания C=N: 2249 см-1;
внеплоскостные деформационные колебания ароматических С-Н: 761 см-1
Т—I—I—'—'—«—I
1000
2. Слабая полоса антисимметричных деформа-
ционных колебаний группы NH3+ около 1610-
1590 см-1 и сравнительно сильная полоса сим-
метричных деформационных колебаний группы
NH3+при 1550-1481 см’1.
3. Сильная полоса при 1220-1190 см-1, обусловленная
валентными колебаниями группы С-С(=О)-О.
4. Интенсивная полоса поглощения карбонильной
группы при 1755-1730 см-1 для гидрохлори-
дов а-аминокислот и при 1730-1700 см-1 для
гидрохлоридов других аминокислот.
В спектрах натриевых солей аминокислот наблюда-
ются «обычные» валентные колебания связи N-H
в области 3400-3200 см-1. Полосы, характерные
для карбоксилат-аниона, проявляются около 1600—
1590 см-1 и около 1400 см-1.
2.6.21. Нитрилы
ИК-Спектры нитрилов (R-C=N) характеризуются
полосами малой или средней интенсивности ва-
лентных колебаний тройной связи. Алифатические
нитрилы поглощают около 2260-2240 см"1. Электро-
ноакцепторные атомы, такие как кислород или хлор,
присоединенные к а-углеродному атому (относи-
тельно группы C=N), понижают частоту полосы
поглощения до 2240-2222 см"1 и увеличивают ее
интенсивность. Спектр типичного нитрила показан
на рис. 2.31.
2.6.22. Изонитрилы (R-N+=C_),
цианаты (R-O-C=N),
изоцианаты (R-N=C=O),
тиоцианаты (R-S-C=N)
и изотиоцианаты (R-N=C=S)
Эти группы поглощают в области 2280-2000 см'1
благодаря валентным колебаниям тройных связей или
кумулированных двойных связей.
2.6.23. Соединения, содержащие группу -N=N
Валентное колебание группы N=N для симметрич-
ного тирянс-азосоединения в ИК-спектре запрещено,
но оно проявляется в спектре комбинационного рас-
сеяния в области 1576 см"1. Несимметричные пара-
замещенные азобензолы с электронодонорными заме-
стителями имеют полосы поглощения при 1429 см”1.
Эти полосы слабые, так как связь неполярна.
2.6.24. Ковалентные соединения, содержащие
связи азот-кислород
Нитросоединения, нитраты и нитрамины содержат
группу NO2. Каждый из этих классов соединений
поглощает в результате антисимметричных и сим-
метричных валентных колебаний группы NO2. Ан-
тисимметричные колебания дают сильную полосу
в области 1661-1499 см"1; симметричные колебания
проявляются в области 1389-1259 см”1. Точное поло-
126
2. Инфракрасная спектроскопия
жение полос зависит от заместителей и ненасыщен-
ных группировок по соседству с группой NO2.
2.6.24.7. Валентные колебания группы N=0
в нитросоединениях
В нитроалканах полосы поглощения наблюдаются
около 1550 и 1372 см"1. Сопряжение понижает частоту
обеих полос, в результате чего они проявляются в об-
ластях 1550-1500 и 1360-1290 см-1. Присоединение
электроотрицательных групп к а-углеродному атому
нитросоединений увеличивает частоту полосы анти-
симметричного колебания NO2 и уменьшает часто-
ту полосы симметричного колебания. Хлорпикрин
C13CNO2 поглощает при 1610 и 1307 см-1.
Ароматические нитрогруппы поглощают прибли-
зительно при тех же частотах, что и сопряженные
алифатические нитросоединения. Взаимодействие
между внеплоскостными деформационными ко-
лебаниями группы NO2 и внеплоскостными де-
формационными колебаниями связей С-Н кольца
не позволяет определять тип замещения в арома-
тических нитросоединениях по низкочастотной
области спектра. Валентные колебания С-N аро-
матических нитросоединений проявляются око-
ло 870 см-1. Спектр нитробензола, показанный
на рис. 2.32 с отнесением частот, иллюстрирует
предыдущее обсуждение.
Симметричные колебания группы NO2 в арома-
тических системах, содержащих электронодонорные
группы (например, аминогруппы) в орто- и пара-
положениях по отношению к NO2, из-за сильного
резонанса смещаются в сторону более низких частот
и имеют повышенную интенсивность. и-Нитроанилин
поглощает при 1475 и 1310 см-1.
Положение полос симметричных и антисимме-
тричных валентных колебаний NO2 нитраминов
N—NO2 и валентных колебаний NO нитрозами-
нов дано в приложении Б.
Нитраты. В спектрах органических нитратов про-
являются полосы поглощения, обусловленные валент-
ными колебаниями связей N-0 группы NO2 и проме-
жуточного звена О-N. Антисимметричные валентные
колебания группы NO2 приводят к сильному поглоще-
нию в области 1660-1625 см-1. Интенсивные полосы,
вызванные симметричными колебаниями, проявляют-
ся в области 1300-1255 см-1. Валентные колебания
промежуточного звена N-0 связаны с поглощением
около 870-833 см-1. Полоса поглощения, наблюдае-
мая при более низких частотах около 763-690 см-1,
возникает, вероятно, в результате деформационных
колебаний NO2.
Нитриты. В ИК-спектрах нитритов проявляются
две сильные полосы валентных колебаний группы
N=O. Полоса в области 1680-1650 см-1 принадлежит
транс-изомеру, а полоса в области 1625-1610 см"1 от-
носится к z/мс-изомеру. Полоса валентных колебаний
Рис. 2.32. ИК-Спектр нитробензола.
Валентные колебания ароматических С-Н: 3113,3082 см-1; антисимметричное
валентное колебание группы NO2 в ArNO2: 1532 см-1; симметричное валентное
колебание группы NO2 в ArNO2: 1355 см-1; валентное колебание группы C-N
в ArNO2: 853 см-1. Низкочастотные полосы играют незначительную роль при
определении характера замещения в кольце, так как поглощение в этой
области обусловлено взаимодействием внеплоскостных деформационных
колебаний NO2 и С-Н. Невозможность использования внеплоскостных колебаний
в низкочастотной области для получения структурной информации характерна
для ароматических соединений с сильнополярными заместителями
2.6. Характеристические групповые частоты органических молекул
127
Рис. 2.33. ИК-Спектр 1,6-гександитиола.
Валентные колебания алифатических С-Н: 2936,2859 см-1; умеренно слабая полоса
валентного колебания S-H: 2558 см-1; валентное колебание C-S: 730 см"1
связи N-O проявляется между 850 и 750 см-1. По-
лосы поглощения нитритов - одни из самых сильных
полос, наблюдаемых в ИК-спектрах.
Нитрозосоединения. Первичные и вторичные али-
фатические нитрозосоединения обычно неустойчивы
и перегруппировываются в оксимы либо димеризу-
ются. Третичные и ароматические нитрозосоединения
достаточно устойчивы и существуют в газовой фазе
или в разбавленных растворах в виде мономеров, а в
чистых образцах без растворителя в виде димеров.
Полоса поглощения N=O мономерных третичных
алифатических нитрозосоединений находится в об-
ласти 1585-1539 см-1; ароматические мономеры по-
глощают между 1511 и 1495 см-1.
В приложении Б полосы валентных колебаний
N—>О димерных нитрозосоединений указаны отдельно
для цис- и гаранс-димеров, а также для алифатических
и ароматических соединений. В приложении Б при-
ведены также полосы поглощения нитрозаминов.
2.6.25. Органические соединения серы
2.6.25.1. Валентные колебания связи S-Н в тиолах
Полоса валентных колебаний группы S-Н алифатиче-
ских тиолов и тиофенолов (жидких или в растворе)
проявляется в области 2600-2550 см-1. Она малоин-
тенсивна, и ее трудно обнаруживать в разбавленных
растворах и в тонких слоях (жидких пленках). Однако
она полезна для определения группы S-Н, так как
в эту область полосы поглощения других групп поч-
ти не попадают. На рис. 2.33 представлен ИК-спектр
1,6-гександитиола, на котором хорошо видна эта по-
лоса. Она может маскироваться сильными полоса-
ми карбоксильной группы, попадающими в эту же
область. Водородные связи с участием группы S-H
гораздо слабее, чем для групп О-Н и N-H.
Полоса поглощения группы S-Н тиокарбоновых
кислот находится в той же области, что и у меркап-
танов и тиофенолов.
2.6.25.2. Валентные колебания связей С-S и С=5
в сульфидах
Полосы валентных колебаний, относящиеся к связи
С-S, наблюдаются в области 700-600 см-1. Эти по-
лосы из-за низкой интенсивности и изменяющегося
положения малопригодны для определения структуры.
Дисульфиды. Полоса валентных колебаний свя-
зи S-S очень слабая и попадает в область между
500 и 400 см-1.
Тиокарбонильные соединения. Алифатические
тиоальдегиды или тиокетоны существуют в виде три-
мерных циклических сульфидов. Арилалкилтиокетоны
могут находиться либо в мономерной, либо в три-
мерной формах, в то время как диарилтиокетоны,
подобные тиобензофенону, существуют только в виде
мономеров. Группа C=S менее полярна, чем группа
С=О, и характеризуется значительно более слабой
связью. В результате этого интенсивность полосы
понижается, а ее положение смещается в сторону
низких частот, где вероятнее эффекты взаимодействия
с другими колебаниями. Поэтому идентификация
затрудняется и становится неопределенной.
Соединения, которые содержат тиокарбонильную
группу, поглощают в области 1250-1020 см-1. Тиобен-
зофенон и его производные имеют полосу поглощения
умеренной интенсивности в области 1224—1207 см-1.
Поскольку эта полоса поглощения проявляется в той
же области, что и полосы валентных колебаний связей
128
2. Инфракрасная спектроскопия
С-О и С-N, между этими колебаниями в отдельной
молекуле возможно сильное взаимодействие.
В спектрах соединений, в которых группа C=S
присоединена к атому азота, наблюдается полоса по-
глощения в обычной области валентных колебаний
C=S. Ряд других полос в широкой области спектра от
1563 до 700 см-1 может быть отнесен к колебаниям,
связанным с взаимодействием между валентными
колебаниями C=S и C-N.
Тиокетоны, которые способны к енолизации, су-
ществуют в виде таутомерных систем тиокетон-тио-
енол; поглощение в таких системах обусловлено ва-
лентными колебаниями S-Н. Тиоенольный таутомер
этилтиобензоилацетата
имеет широкую полосу при 2415 см-1, обусловленную
валентными колебаниями участвующей в водородном
связывании группы S-H.
2.6.26. Соединения, содержащие
связи сера - кислород
2.6.26.1. Валентные колебания фрагмента 5=0
в сульфоксидах
Алкил- и арилсульфоксиды в жидком состоянии и в
растворе имеют сильное поглощение в области 1070-
ЮЗО см-1. В случае диметилсульфоксида (ДМСО) оно
наблюдается при 1050 см-1. Сопряжение незначитель-
но понижает эту частоту, в то время как частота ко-
лебания связи С=О при сопряжении уменьшается за-
метно. Диаллилсульфоксид поглощает при 1047 см-1.
Метилфенилсульфоксид и метилциклогексилсульфок-
сид поглощают при 1055 см-1 в разбавленном рас-
творе в четыреххлористом углероде. Сульфоксидная
группа может образовывать водородные связи; в рас-
творах в метаноле и хлороформе при переходе от
разбавленного раствора к чистой жидкости полоса
немного смещается в сторону низких частот. Частота
колебания связи S=O увеличивается при введении
электроотрицательного заместителя.
Сульфоны. В спектрах сульфонов наблюдают-
ся сильные полосы поглощения в областях 1350-
1300 и 1160-1120 см"1. Эти полосы обусловлены
антисимметричными и симметричными валентными
колебаниями группы SO2 соответственно. Образование
водородных связей приводит к поглощению около
1300 и 1125 см"1. В растворе в СС14 или в твердом
состоянии часто наблюдается расщепление высоко-
частотной полосы.
Сульфонилхлориды. В ИК-спектрах сульфо-
нилхлоридов наблюдается интенсивное поглощение
в областях 1410-1380 и 1204-1177 см"1. Увеличение
частоты по сравнению с сульфонами происходит
благодаря присутствию электроотрицательного ато-
ма хлора.
Сульфонамиды. В ИК-спектрах растворов суль-
фонамидов наблюдается сильное поглощение при
1370-1335 и 1170-1155 см"1. В случае твердых об-
разцов эти частоты понижаются на 10-20 см"1, вы-
сокочастотная полоса уширяется и обычно имеет
несколько дополнительных максимумов.
В спектрах первичных сульфонамидов в твердом
состоянии имеются сильные полосы валентных коле-
баний N-Н при 3390-3330 и 3300-3247 см"1. Вторич-
ные сульфонамиды поглощают около 3265 см"1.
Сульфонаты, сульфаты и сульфокислоты.
Интервалы частот, в которых проявляются полосы
антисимметричных (более высокочастотных, т. е.
коротковолновых) и симметричных валентных ко-
лебаний связей S=O этих соединений, перечислены
в табл. 2.8..
Спектр типичного алкиларилсульфоната приве-
ден на рис. 2.34. Фактически в ИК-спектрах всех
сульфонатов полосы антисимметричных валентных
колебаний проявляются в виде дублета. Спектры ал-
кил- и арилсульфонатов различаются весьма незначи-
тельно. Электронодонорные группы в шря-положении
арилсульфонатов вызывают смещение полос погло-
щения в сторону более высоких частот.
Узкие интервалы поглощения сульфокислот, при-
веденные в табл. 2.8, относятся только к безводным
формам. Такие кислоты легко гидратируются; гидраты
дают полосы поглощения в области 1230-1120 см"1,
которые, вероятно, являются результатом взаимодей-
ствия сульфоната с ионом гидроксония.
Таблица 2.8. Частоты валентных колебаний связи
S=O в сульфонатах, сульфатах, сульфокислотах и со-
лях сульфокислот
Классы соединений Частоты валентных колебаний, см"1
Сульфонаты (ковалентные) Сульфаты (органические) Сульфокислоты Соли сульфокислот 1372-1335, 1195-1168 1415-1380, 1200-1185 1350-1342, 1165-1150 -1175,-1055
2.6. Характеристические групповые частоты органических молекул
129
Рис. 2.34. ИК-Спектр этилового эфира л-толуолсульфокислоты.
А: антисимметричное валентное колебание SO2, 1355 см-1; Б: симметричное
валентное колебание SO2, 1177 см-1; В: сильные полосы валентных колебаний
S-O-C, 1000-769 см"1
2.6.27. Органические галогенсодержащие
соединения
Сильное поглощение галогенированных углеводо-
родов обусловлено валентными колебаниями связи
углерод - галоген.
Поглощение группы С-С1 алифатических
соединений наблюдается в широкой области между
850 и 550 см-1. Если несколько атомов хлора при-
соединено к одному атому углерода, то полоса обычно
более интенсивна и находится в высокочастотной
части указанной области. Четыреххлористый угле-
род имеет интенсивную полосу при 797 см-1. Часто
наблюдаются первые обертоны интенсивных основ-
ных полос. Бромированные соединения поглощают
в области 690-515 см-1, иодпроизводные - в области
600-500 см-1. Для соединений, содержащих группу
СН2Х (Х=С1, Вг, I), в области 1300-1150 см"1 наблю-
даются интенсивные полосы, относящиеся к веерным
колебаниям группы СН2.
Фторсодержащие соединения имеют сильные по-
лосы поглощения валентных колебаний связи C-F
в широкой области между 1400 и 1000 см"1. В спек-
трах монофторалканов проявляется сильная полоса
в области 1100-1000 см"1. По мере увеличения числа
атомов фтора в алифатической молекуле вид полос
усложняется, причем появляются интенсивные муль-
типлетные полосы во всей области, характерной для
поглощения С-F. Группы CF3 и CF2 дают интенсивные
полосы поглощения в области 1350-1120 см"1.
Хлорбензолы поглощают в области 1096-1089 см"1.
Положение полосы внутри этой области зависит от
типа замещения. Арилфториды поглощают в области
1250-1100 см"1. В спектрах монофторпроизводных
бензола проявляется сильная узкая полоса поглоще-
ния около 1230 см"1.
2.6.28. Соединения кремния
2.6.28.7. Колебания связей Si-H
В ИК-спектрах соединений, содержащих связи Si-H,
наблюдаются валентные (-2200 см"1) и дефомацион-
ные (800-950 см"1) колебания Si-H. Присоединение
к атому кремния электроотрицательной группы повы-
шает частоты валентных колебаний группы Si-H.
2.6.28.2. Колебания связей SiO—H и Si-0
Валентные колебания группы ОН фрагмента SiOH
проявляются в той же области, что и для спиртов
(3700-3200) см"1, интенсивная полоса валентного ко-
лебания Si-О находится в интервале 830-1110 см"1.
Характер поглощения зависит от прочности водо-
родных связей, как и в спиртах.
2.6.28.3. Валентные колебания связей Si—Hal
Полосы поглощения связи Si-F проявляются в об-
ласти 800-1000 см"1.
Полосы валентных колебаний Si-Cl находятся ниже
666 см"1.
130
2, Инфракрасная спектроскопия
2.6.29. Соединения фосфора
2.6.29.7. Валентные колебания связей Р=0 и Р-0
Полосы поглощения этих связей приведены в при-
ложении Г, табл. Г-1.
2.6.30. Гетероароматические соединения
ИК-Спектры гетероароматических соединений обу-
словлены в основном теми же типами колебаний, ко-
торые наблюдаются для ароматических соединений.
2.6.30.7. Валентные колебания связей С-Н
В спектрах гетероароматических соединений, таких
как пиридины, пиразины, пирролы, фураны и тиофе-
ны, валентные колебания связей С-Н проявляются
в области 3077-3003 см-1.
2.6.30.2. Частоты валентных колебаний связей N-H
В спектрах гетероароматических соединений, содержа-
щих, группу N-Н, наблюдается полоса поглощения в об-
ласти 3500-3220 см-1, обусловленная валентными колеба-
ниями N-Н. Положение полосы поглощения в пределах
этой основной области зависит от степени водородного
связывания, а следовательно, от физического состояния
образца и полярности растворителя. В спектрах разбав-
ленных растворов пиррола и индола в неполярных рас-
творителях наблюдается резкая полоса около 3495 см-1,
а в концентрированных растворах - уширенная полоса
около 3400 см-1. В растворах промежуточных концен-
траций могут наблюдаться обе полосы.
2.6.30.3. Валентные колебания кольца (скелетные полосы)
Валентные колебания кольца проявляются в основном
в области между 1600 и 1300 см-1. Поглощение свя-
зано с растяжением и сжатием всех связей в кольце
и взаимодействием между этими валентными колеба-
ниями. Вид полос и их относительная интенсивность
зависят от типа замещения и природы заместителя.
В спектре пиридина (рис. 2.35) в указанной области
наблюдаются четыре полосы, и в этом отношении он
очень похож на монозамещенный бензол. Фураны,
пирролы и тиофены также имеют в этой области от
двух до четырех полос.
2.6.30.4. Внеплоскостные деформационные колебания
связей С-Н
Вид полос поглощения, обусловленных внеплоскост-
ными деформационными колебаниями С-Н (у-СН)
в ИК-спектрах гетероароматических соединений,
определяется числом соседних атомов водорода,
колеблющихся в фазе. Полосы поглощения внепло-
скостных колебаний С-Н и деформационных коле-
баний кольца алкилпиридинов представлены в табл.
Д-1 приложения Д.
Данные о положении полос внеплоскостных дефор-
мационных колебаний СН (у-СН) и деформационных
колебаний кольца трех пятичленных гетероаромати-
ческих циклов приведены в табл. Д-2 приложения
Д. В интервалы частот, указанные в этой таблице,
попадают полосы поглощения гетероароматических
соединений как с полярными, так и с неполярными
заместителями в цикле.
Рис. 2.35. ИК-Спектр пиридина.
Валентные колебания ароматических С-Н: 3090-3000 см-1; валентные колебания
С-С и С-N ароматического кольца (скелетные полосы): 1600-1430 см-1;
внеплоскостные деформационные колебания С-Н: 753,707 см"1; картину
поглощения в низкочастотной области для замещенных пиридинов см.
в приложении Д, табл. Д-1
Литература
131
Литература
Введение в ИК-спектроскопию и теория
Bellamy, L. J. (1975). The Infrared Spectra of Complex
Molecules, 3rd ed. London: Chapman and Hall; New York:
Halsted-Wiley. (Есть русское издание: Беллами Л., Ин-
фракрасные спектры сложных молекул. Пер. с англ.,
М.: ИЛ, 1963.)
Беллами Л., Новые данные по ИК-спектрам сложных
молекул. М.: Мир, 1971.
Coleman, Р.А. (1991). Practical Sampling Techniques for
Infrared Analysis. Boca Raton, FL: CRC Press.
Colthup, N.B., Daly, L.H., and Wiberley, S.E. (1990/ In-
troduction to Infrared and Raman Spectroscopy, 3rd ed.
New York and London: Academic Press.
Conley, R.T. (1972). Infrared Spectroscopy, 2nd ed. Boston:
Allyn and Bacon.
Durig, J.R. (1985). Chemical, Biological, and Industrial
Applications of Infrared Spectra. New York: Wiley.
Ferraro, J.R., and Basile, L.J., Eds. (1985). Fourier Trans-
form Infrared Spectroscopy, 4 vols. New York: Academic
Press. Прикладные вопросы.
George, W.O., and McIntire, P. (1986). Infrared Spectroscopy.
New York: Wiley.
Griffiths, P.R., and DeHaseth, J.A. (1986). FTIR. New York:
Wiley.
Herres, W. (1987). HRGC-FTIR, Capillary GC and FTIR
Spectroscopy'. Theory and Applications. New York: Huthig.
Mattson, J.S., Ed. (1977). Infrared, Correlation and Fourier
Transform Spectroscopy. New York: Marcel Dekker.
Nakanishi, K. and Solomon, P. H. (1977). Infrared Absorption
Spectroscopy-Practical, 2nd ed. San Francisco: Holden-Day
(Есть русское издание: Наканиси К. Инфракрасные
спектры и строение органических соединений. Пер.
с англ., М.: Мир, 1965.)
Roeges, N.P.G. (1984). Guide to Interpretation of Infrared
Spectra of Organic Structures. New York: Wiley.
Smith, B.C. (1995). Fundamentals of Fourier Transform
Infrared Spectroscopy. Boca Raton, FL: CRC Press.
Szymanski, H.A. Ed. (1969, 1970). Raman Spectroscopy:
Theory and Practice, Vols. 1.2. New York: Plenum Press.
Szymanski, H.A. (1989). Correlation of Infrared and Raman
Spectra of Organic Compounds. Hertillon Press.
Szymanski, H.A. (1964). IR Theory and Practice of Infrared
Spectroscopy. New York: Plenum Press.
Szymanski, H.A. (1964-1967). Interpreted Infrared Spectra,
Vols. I—III. New York: Plenum Press.
Szymanski, H.A. (1963). Infrared Band Handbook.
New York: Plenum Press.
Атласы ИК-спектров, справочники,
специальная литература
Aldrich Library of FTIR Spectra (1989), 3 Vols. Milwaukee,
WI: Includes vapor-phase spectrum Vol. 3
ASTM-Wyandotte Index, Molecular Formula List of Com-
pounds, Names and References to Published Infrared
Spectra. Philadelphia, PA: ASTM Special Technical Pub-
lications 131 (1962) and 131-A (1963). Содержит око-
ло 57 ООО соединений; включает спектры в ближней
и дальней ИК-областях.
Bentley, F.F., Smithson, L.D., and Rozek, A.L. (1988).
Infrared Spectra and Characteristic Frequencies - 700 to
300 cm1, A Collection of Spectra, Interpretation and
Bibliography. New York: Wiley-Interscience.
Brown, C.R., Ayton, M.W., Goodwin, T.C., and Derby, TJ.
(1954). Infrared-A Bibliography. Washington, DC: Library
of Congress, Technical Information Division.
Catalog of Infrared Spectrograms, American Petroleum
Institute Research Project 44. Pittsburgh, PA: Carnegie
Institute of Technology.
Catalog of Infrared Spectrograms. Philadelphia: Sadder
Research Laboratories, PA. 19 104. Спектры указаны
по названиям и специальным индексам (Spec-Finder),
в которых обозначены интервалы волновых чисел для
главных полос. Это помогает быстро идентифицировать
неизвестные соединения.
Catalog of Infrared Spectral Data, Manufacturing Chemists
Association Research Project, Chemical and Petroleum
Research Laboratories. Pittsburgh, PA: Carnegie Institute
of Technology, to June 30, 1960; College Station, TX:
Chemical Thermodynamics Properties Center, Agriculture
and Mechanical College of Texas, from July 1, 1960.
Craver, C., Ed. (1982). Desk Book of Infrared Spectra, 2nd
ed. Kirkwood MO: Coblenz Society. (900 спектров)
Dobriner, K, Katzenellenbogen, E.R., and Jones, R.N. (1953).
Infrared Absorption Spectra of Steroids-An Atlas, Vol. 1.
New York: Wiley-Interscience.
Documentation of Molecular Spectroscopy (DMS), Lon-
don: Butterworths Scientific Publications, and Weinheim/
Bergstrasse. West Germany: Verlag Chemie GMBH,
in cooperation with the Infrared Absorption Data Joint
Committee, London, and the Institut fur Spectrochemie
and Angewandte Spectroskopie, Dortmund. (Спектры
представлены на кодированных картах; изданы так-
же аналогичные карты с аннотациями статей по
ИК-спектроскопии.).
Dolphin, D., and Wick, А.Е. (1977) Tabulation of Infrared
Spectral Data. New York: Wiley.
Финч А., Гейтс П., Редклиф К., Диксон Ф, Бентли Ф.,
Применение длинноволновой ИК-спетроскопии в химии.
Пер. с англ., М.: Мир, 1973.
132
2. Инфракрасная спектроскопия
Flett, М. St. С. (1969). Characteristic Frequencies of Chemi-
cal Groups in the Infrared. New York: American Elsevier.
Hershenson, H.H. (1959,1964). Infrared Absorption Spectra
Index. New York and London: Academic Press. (Два тома
охватывают данные 1945-1962 гг.)
Infrared Band Handbook. Supplements 1 and 2; (1964).
New York: Plenum Press, 259 pp.
Infrared Band Handbook. Supplements 3 and 4. (1965).
New York: Plenum Press.
Infrared Band Handbook. 2nd rev. ed. (1970). New York:
IFI/ Plenum Press. Two volumes.
IRDC Cards: Infrared Data Committee of Japan (S. Mizushima).
Haruki-cho, Tokyo: Handled by Nankodo Co.
Katritzky, A.R., Ed. (1963). Physical Methods in Heterocyclic
Chemistry, Vol. II. New York and London: Academic
Press.
Lin-Vien, D., Colthup, N.B., Fately, W.G., and Grasselli, J.G.
(1991). Handbook of Infrared and Raman Characteristic
Frequencies of Organic Molecules. New York: Academic
Press.
Loader, E.J. (1970). Basic Laser Raman Spectroscopy.
London, New York: Heyden-Sadtler.
Maslowsky, E. (1977). Vibrational Spectra of Organometallic
Compounds. New York: Wiley-Interscience.
Ministry of Aviation Technical Information and Library
Services, Ed. (1960). An Index of Published Infra-Red
Spectra, Vols. 1 and 2. London: Her Majesty’s Stationery
Office.
NRC-NBS (Creitz) File of Spectrograms. Issued by Na-
tional Research Council-National Bureau of Standards
Committee on Spectral Absorption Data, National Bureau
of Standards, Washington 25, DC. (Спектры представлены
на картах.)
Nyquist, R.A. (1984). Interpretation of Vapor-Phase Infrared
Spectra. Philadelphia: Sadtler-Heyden.
Porro, T.J., and Pattacini, S.C. (1993). Spectroscopy 8(7),
40-47. (Подготовка образцов.)
Sadtler Standard Infrared Grating Spectra. (1972). Philadel-
phia: Sadder Research Laboratories, Inc. (26 000 спектров
в 26 томах.)
Sadtler Standard Infrared Prism Spectra. (1972). Philadel-
phia: Sadtler Research Laboratories, Inc. (43 000 спектров
в 43 томах.)
Sadtler Reference Spectra-Commonly Abused Drugs IR &
UV Spectra. (1972). Philadelphia: Sadtler Research Labo-
ratories, Inc. (600 ИК-спектров, 300 УФ-спектров.)
Sadtler Reference Spectra-Gases & Vapors High Resolution
Infrared. (1972). Philadelphia: Sadtler Research Laboratories,
Inc. (150 спектров.)
Sadtler Reference Spectra-Inorganics IR Grating. (1967).
Philadelphia: Sadtler Research Laboratories, Inc. (1300 спек-
тров.)
Sadtler Reference Spectra - Organometallics IR Grating.
(1966). Philadelphia: Sadtler Research Laboratories, Inc.
(400 спектров.)
Sadtler Digital FTIR Libraries. (1994). Philadelphia: Sadtler
Research Labs. (100 000 оцифрованных спектров.)
Socrates, G. (1994). IR Characteristic Group Frequencies.
New York: Wiley.
Stewart, J.E. (1965). Far infrared spectroscopy. In S.K.
Freeman (Ed.), Interpretive Spectroscopy. New York:
Reinhold, p. 131.
Tichy, M. (1964). The determination of intramolecular
hydrogen bonding by infrared spectroscopy and its ap-
plications in stereochemistry. In R.R. Raphael (Ed.), Ad-
vances in Organic Chemistry: Methods and Results, Vol.
5. New York: Wiley-Interscience
Дополнительная литература
Смит А. Прикладная ИК-спектроскопия. Пер. с англ.
М.: Мир, 1982.
Преч Э., Бюльманн Ф., Аффольтер К., Определение
строения органических соединений. Таблицы спектраль-
ных данных. Пер. с англ. М.: Мир, БИНОМ. Лаборатория
знаний, 2006.
Накамото К., ИК-спектры и спектры КР неорганиче-
ских и координационных соединений. Пер. с англ. М.:
Мир, 1991.
Грибов Л.А., Баранов В.И., Эляшберг М.Е., Безэталонный
молекулярный спектральный анализ. М.: УРСС, 2002.
Браун Д., Флойд А., Сензбери М. Спектроскопия орга-
нических веществ. Пер. с англ. М.: Мир, 1992.
Шагидуллин RP., Мухамедов Ф.С., Нигматулина Р. Б.,
Виноградова В.С., Чернова А.В., Атлас ИК-спектров
фосфорорганических соединений. М.: Наука, 1977
Шиманко Н.А., Шишкина Н.А. Инфракрасные и уль-
трафиолетовые спектры поглощения ароматических
эфиров. М.: Наука, 1987.
Большаков Г.Ф. Инфракрасные спектры аренов. Ново-
сибирск: Наука, 1989.
Чумаевский Н.А. Колебательные спектры элементо-
органических соединений элементов IVE и VE групп.
М.: Наука, 1971.
Свердлов Л.М., Ковнер М.А., Крайнов Е.П. Колеба-
тельные спектры многоатомных молекул. М.: Наука,
1970.
Упражнения
133
Упражнения
2.1. Объясните, почему в ИК-спектрах бензони-
трила и фенилацетонитрила в одном случае
наблюдается полоса средней интенсивности
при 2940 см-1, а в другом в области 3000-
2500 см-1 поглощения нет.
2.2. Выберите соединение, которое наилучшим
образом удовлетворяет одному из следующих
наборов полос (a-и) в ИК-спектрах (см-1).
Каждый набор полос состоит их нескольких
наиболее важных полос для одного из при-
веденных ниже соединений.
Бензамид Дифенилсульфон
Бензойная кислота Муравьиная кислота
Бензонитрил Изобутиламин
Бифенил 1-Нитропропан
Диоксан
а. 3080 (w), 3000-2800 поглощение отсутству-
ет, 2230 (s), 1450 (s), 760 (s), 688 (s);
б. 3380 (m), 3300 (m), 3200-3000 поглощение
отсутствует, 2980 (s), 2870 (m), 1610 (m),
-900-700 (b);
в. 3080 (m), поглощение отсутствует 3000-2800,
1315 (s), 1155 (s);
r. 2955 (s), 2850 (s), 1120 (s);
д. 2946 (s), 2930 (m), 1550 (s), 1386 (m);
e. 2900 (b,s), 1720 (b,s);
ж. 3030 (m), 730 (s), 690 (s);
з. 3200-2400 (s), 1685 (b,s), 705 (s);
и. 3350(s), 3060 (m), 1635 (s);
s - сильная, m - средняя,
w - слабая, b - широкая.
В упражнениях 2.3-2.6 приведите в соответствие
названия веществ из каждого списка и ИК-спектры.
Укажите необходимые для идентификации характе-
ристические полосы в каждом спектре.
Спектры для упражнений 2.3 - 2.8 мож-
но найти в формате PDF на сайте Wiley
www.wiley.com/college/silverstein.
2.3. Спектры A-D
Циклогексадиен-1,3
Дифенилацетилен
Октен-1
Пентен-2
2.4. Спектры E-I
Бутилацетат
Бутирамид
Изобутиламин
Лауриновая кислота
Пропионат натрия
2.5. Спектры J-M
Аллилфениловый эфир
Бензальдегид
о-Крезол
л/-Толуиловая кислота
2.6. Спектры N-R
Анилин
Азобензол
Оксим бензофенона
Бензиламин
Гидрохлорид диметиламина
2.7. Определите структуру соединения с брутто-
формулой C2H3NS по его ИК-спектру (S).
2.8. Представьте доказательства образования еноль-
ной формы 2,4-пентандиона (соединение Т).
Объясните появление двух полос в области
1700-1750 см-1, полосы 1650 см-1 и очень
широкой полосы с рядом максимумов 3400-
2600 см-1 (полосы 3000-2900 см-1 обусловле-
ны валентными колебаниями С-Н).
2.9. Для каждого из следующих ИК-спектров (А-Ч)
перечислите функциональные группы, которые
а) присутствуют, б) отсутствуют. Масс-спектры
этих соединений представлены в гл. 1 (упраж-
нение 1.6).
134
2. Инфракрасная атектроскопия
Спектр А
g 85 Ё
i —
| 80 j
О =
Е 75 Ё
70
--1--1--1--1--1--1--1--г
4000
3000 „ . .к 2000 1000
Волновое число (см ')
Спектр Б
Спектр В
Упражнение 2.9(А-В)
Упражнения
135
Спектр Г
Спектр Д
Спектр Е
Упражнение 2.9(Г-Е)
136
2. Инфракрасная спектроскопия
Упражнение 2.9(Ж-И)
Упражнения
137
Спектр К
Спектр Л
Спектр М
Упражнение 2.9(К-М)
138
2. Инфракрасная спектроскопия
Спектр О
100:
о
S
X
S
£
с
о
Оч
к
т
4000
1--1-1--1--1-1--1--1-1--1-1--1--г
3000 2000
Волновое число (см-1)
1000
Упражнение 2.9(Н-П)
Упражнения
139
100 =
8
--1--1-1--1--1-1--1--1-1--1-1--1--1-г
3000 2000
Волновое число (см-1)
т—।—।—।—।—i—।
1000
Спектр T
Упражнение 2.9(P-T)
140
2. Инфракрасная мектроскопия
СпектрФ
Упражнение 2.9(У-Х)
Упражнения
141
Спектр Ч
Упражнение 2.9(Ц,Ч)
142
2. Инфракрасная спектроскопия
Приложение А. Области прозрачности растворителей и иммерсионных масел
Волновое число (см‘1)
3600 3200 2800 2400 2000 1800 1600 1400 1200 1000 800 600
Растворители3 Сероуглерод Хлористый метилен Хлороформ Четыреххлористый углерод Тетрахлорэтилен 1 3400 1 3000 1 2600 1 2200 — — - — ——
— — —— — — — - —
Бромистый метилен Бромоформ Иммерсионные масла6 Вазелиновое масло Гексахл орбутад иен Фторированное — - — — — — — - ——
— — —-—- — —
вазелиновое масло
а В областях спектра, которые не выделены, пропускание растворителя больше 25% при толщине поглощающего
слоя 1 мм.
6 Для иммерсионных масел не выделенные области спектра соответствуют прозрачности тонкой жидкой пленки.
Приложение В
143
Приложение Б. Диапазоны характеристического поглощения органических
соединений0
СМ"1 36 00 3200 2800 2400 2000 1800 1600 1400 1200 1000 800
Алканы Алкены Винил с/ 1 3400 1 3000 m 1 2600 1 2200 m г Tl r n w
m m —
m s s
m w s
Цис Ы Винилиден Тризамещенные /У Тетразамещенные // Сопряженные cj-lj Кумулированные ^с=С=СН2 Циклические Алкины Монозамещенные Дизамещенные Моноядерные ароматические соединения Бензол Монозамещенные 1,2-Дизамещенные 1,3-Дизамещенные 1,4-Дизамещенные 1,2,4-Тризамещенные 1,2,3-Тризамещенные 1,3,5-Тризамещенные Спирты и фенолы Свободная группа ОН Связанная слабой внутримолекулярной водородной связью Связанная сильной внутримолекулярной водородной связью Связанная межмолекулярной водородной связью Насыщенные третичные 1 Высокосимметричные вторичные] Насыщенные вторичные 1 а-Ненасыщенные или J циклические третичные а-Ненасыщенные вторичные 1 алициклические вторичные (5- или 6-членные циклы) Насыщенные первичные J а-Ненасыщенные третичные ] а-Ненасыщенные и а-разветвленные вторичные Ди-а-ненасыщенные вторичные » Алициклические вторичные (7- или 8-членные циклы) а-Разветвленные и/или a-ненасыщенные первичные J m jn s
m m m s
— m — w w — IT 1
m w,
< m m 1 1 1 1 К К II II II II II II II s w | IT 1 w — — s
w w s m — m m j sff _ss 1 _s > s6
— — m m m m __Q? m
— — m rr i s __ m s 1 1—1 1 1 1 1 1 1 1 1 1 i i imi i 1 1 1 1 1 1 1 1 * 1 1
s s
'В 1 3 1 I I 1 ; со; со 700-345 1 704-350 Osh 1 9 sh —
br
s Ьг
— s s — —
s
s
3400 3000 2600 2200
I I I I I I I I I__________________________________|_________|_________|_________|_________|_________|_________
СМ-1 3600 3200 2800 2400 2000 1800 1600 1400 1200 1000 800 6'
а Области поглощения показаны сплошными линиями, s - сильная, m - средняя, sh - узкая, Ьг - широкая. Двойное буквенное
обозначение над линией показывает, что могут наблюдаться две полосы.
6 Деформационные колебания кольца.
144
2, Инфракрасная спектроскопия
Приложение Б (продолжение)
СМ'1 3600 3200 2800 2400 2000 1800 1600 1400 1200 1000 800 600
Ацетали Кетали3 Простые эфиры Алифатические Ароматические (Аг—О—СН2) Виниловые Эпоксиды Алкил- и арилпероксиды Ацил- и ароилпероксиды Карбонильные соединения Кетоны6 Диалкил (—СН2СОСН2—) Ароматические (сопряженные) Енолы 1, З-д и кето но в о-Гидроксиарилкетоны Альдегиды6 Алифатические Ароматические (сопряженные) Карбоновые кислоты® Димеры® Карбоксилат-ион Сложные эфиры Формиаты Ацетаты Другие несопряженные сложные эфиры Сопряженные сложные эфиры Ароматические сложные эфиры I 3400 I 3000 I 2600 I 2200 — s s s s m —
s s m
- m I m br 'br — s JS — m m m m m rv, ПЛ m —
— m (ду! s — s £ s < s г m L nr n m m < — m
S s
s s
_s _s
_s m
3400 3000 2600 2200 I I I I I I I I I I I I I I I I
CM'1 3600 3200 2800 2400 2000 1800 1600 1400 1200 1000 800 600
а Три полосы, иногда четыре для кеталей, пять полос для ацеталей.
6 В сопряженных соединениях частота валентных колебаний С=О такая же, как в ароматических.
в В сопряженных соединениях частота валентных колебаний С=О понижена (1710-1680 см-1).
Полоса валентных колебаний О-Н (3300-2600 см-1) очень широкая.
Приложение Б
145
Приложение Б (продолжение)
СМ'1 3600 3200 2800 2400 2000 1800 1600 1400 1200 1000 800 600
Лактоны р-Лактоны у-Лактоны 5-Лактоны Хлорангидриды Алифатические Ароматические Ангидриды Ациклические (несопряженные) Ациклические (сопряжённые) Циклические (несопряженные) Циклические (сопряженные) Амиды Первичные Растворы Твердое состояние Вторичные Растворы Твердое состояние Третичные Лактамы Растворы Твердое состояние 5-членные циклы 6- или 7-членные циклы Амины Первичные Алифатические Ароматические Вторичные Алифатические Ароматические Третичные Алифатические Ароматические Соли аминов Первичные Вторичные Третичные Ион аммония Г 3400 1 3000 1 2600 1 2200 S s s
s s
s s
< m
"s w m
s s
c s
JS, s s
s s £ s
m m_ s m m br m br
m m s
m s
m m s
s
m
i 71 w
s
<
m w m w — — — — n r n i m s 1 br
m br
w m n n
w s
r i
s
s m 1 m m
s — — m — — — —
m
s — m c
3400 3000 2600 2200
- I---------1___I____I____I____I____I____I___I_________I_________I________I_________I_________I________I_________I
СМ'1 3600 3200 2800 2400 2000 1800 1600 1400 1200 1000 800 600
146
2. Инфракрасная атектроскопия
Приложение Б (продолжение)
СМ’1 3600 3200 2800 2400 2000 1800 1600 1400 1200 1000 800 600
Нитрилы (RCN) Алифатические Ароматические Карбодиимиды Изонитрилы (RNC) Алифатические Ароматические Изоцианаты (RNCO) Тиоцианаты (RSCN) Изотиоцианаты (RNCS) Алифатические Ароматические Нитросоединения Алифатические Ароматические Сопряжённые Нитрамины Нитрозоамины Газообразные Жидкие Нитраты (RONO2) Нитриты (RONO) Нитрозосоединения (RNO) Алифатические димеры (транс) Алифатические димеры (цис) Ароматические димеры (транс) Ароматические димеры (цис) Алифатические мономеры Ароматические мономеры Соединения серы Тиолы, тиофенолы, тиокислоты Тиокарбонильная группа C=S (не связанная с азотом) C=S (связанная с азотом) Сульфоксиды Сульфоны Сульфонилхлориды Первичные сульфонамиды (в твердом состоянии) Вторичные сульфонамиды (в твердом состоянии) Сульфонаты - 1 3400 1 3000 1 2600 1 2200 m
rn
s s
s
s
sJdi w
— — s s s s m m m
— s s s_ s m m s —
- — s s s < s s > s — s s
Г -s
W — s c s >s — —
— m — m m s —
s £
-- SS — s H 1 1 1 1 1 1 1 1 1 1 s s —
— s s s —
3400 3000 2600 2200 I I I I I I I I I I I I I I I I
CM’1 3600 3200 2800 2400 2000 1800 1600 1400 1200 1000 800 600
Приложение Б
147
СМ-1 3600 3200 2800 2400 2000 1800 1600 1400 1200 1000 800 600
Галогенпроизводные 1 3400 1 3000 1 2600 1 2200 c c
—СН2С1 — о — о r*
—СН2Вг — — s — 5
—С 21 — s — s
—cf2— — s q s
—CF3 — s —
—ch=cf2 — s — s —
—cf=cf2 — s s s
Арилфториды — —
Арилхлориды — s —
Соединения кремния
SiH — — - — S —
SiH2 — — —»- —
SiH3 — —
SiCH3 — 1/y s __ — —
SiCH2 — г П —
SiC6H5 — m m —
SiO Алифатические — s —
SiOCH3 — — — — — — — _m _ s — .
SiOCH2CH3 — m s - —
SiOC6H5 —
SiOSi — - — —
SiOH — m s —
SiF — s —
SiF2 — s m —
SiF3 — s m
Соединения фосфора
PH — n n —
PH2 m — m s
PCH3 — m s —
PCH2— — m
PC6H5 — rn m \ у
Alk3P=O — s —
Ar3P=O — s
(RO)3P=O — s —
P—0—CH3 — m — m j > 5 r——
P—0—CH2CH3 — m m m m i Tl J > 5
p-oc6H5 — s — —
P—0—P — s w
P—0—H — s s br — > —
0 II 1—» — s — — s —
II D nu t -
1 МП <одмс> ип/
s - сильная, m - средняя, w - слабая, v - переменная
3400 3000 2600 2200
. I________I___I___I___I___I___I___I___I________I_______I_______I_______I________I_______I_______I
СМ"1 3600 3200 2800 2400 2000 1800 1600 1400 1200 1000 800 600
148
2 Инфракрасная спектроскопия
Приложение В. Поглощение алкенов
Таблица В-1. Поглощение алкеновых фрагментов0
И Н
/С=С\
R Н
Винил
1648-1638 см'1
995-985 см'1 (s)e
915-905 см'1 (s)
Н Н
/С=С\
R R
цис
1662-1626 см'1 (v)
730-665 см'1 (s)
R Н
/С=СС
Н R
транс
1678-1668 см'1 (v)
980-960 см'1 (s)«
Винилиден
1658-1648 см'1 (т)
895-885 см'1 (s)
Тризамещенные
1675-1665 см'1 (w)
840-790 см'1 (т)
Тетразамещенные
1675-1665 см'1 (очень слабая
или отсутствует)
°s - сильная, m - средняя, w - слабая, v - переменная.
бЭта полоса имеет интенсивный обертон.
вЭта полоса встречается около 1000 см-1 в сопряженных /иранс-тирднс-системах, таких как эфиры сорбиновой кислоты.
Таблица В-2. Частоты валентных колебаний связей С=С в циклических и ациклических системах (см-1)
Кольцо0 или цепь н с 'с=< н чс н СН3 чс СНзх с' сн3 чс с
с' Л=с\ с хс=сн2
Цепь цис Цепь транс 1661 1676 1681 1672 1661
Трёхчленный цикл 1641 1890 1780
Четырёхчленный цикл 1566 1685 1678
Пятичленный цикл 1611 1658 1686 1657
Шестичленный цикл 1649 1678 1685 1651
Семичленный цикл 1651 1673
Восьмичленный цикл 1653
Во всех циклах сдержатся i/i/c-замещенные двойные связи.
Приложение Д
149
Приложение Г. Поглощение фосфорорганических соединений
Таблица Г-1. Валентные колебания связей Р=О и Р-0
Группа Положение (см”1), интенсивность0 Полосы (cm~1),vp_o
Валентные колебания Р=О Фосфиноксиды Алифатические Ароматические Фосфаты (эфиры) 6 Р-ОН Р-О-Р Р-О-С (алифатич.) Р-О-С (ароматич.) -1150 -1190 1299-1250 1040-910 (s) 1000-870 (s) -700 (w) 1050-970 (s)° 830-740 (s)a 1260-1160 (s) 994-855 (s)
а s - сильная, w - слабая.
6 Электроотрицательные алкоксильные группы повышают частоту валентного колебания Р=О в сложных эфирах
по сравнению с оксидами.
в Может проявляться в виде дублета.
г Может отсутствовать.
Приложение Д. Поглощение ароматических гетероциклических соединений
Таблица Д-1. Полосы у-СН и деформационных колебаний кольца ((З-кольцевые) пиридинов0 (в см-1)
Положение заместителя Число присоединенных атомов Н у-СН Р-кольцевые
2- 4 781-740 752-746
3- 3 810-789 715-712
4- 2 820-794 775-709
"Обозначения у и р объяснены в тесте (см. раздел 2.6.30.4); см. также (Katritzky, 1963).
Таблица Д-2. Характеристические полосы у-СН или деформационных (3-кольцевых колебаний фуранов,
тиофенов и пирролов
Гетероцикл Положение заместителя Фаза у-СН или р-кольцевые колебания0, см-1
Фуран 2- в СНС13 -925 -884 835-780
2- Жидкая 960-915 890-875 780-725
2- Твердая 955-906 887-860 821-793 750-723
3- Жидкая 885-870 741
Тиофен 2- в СНС13 -925 -853 843-803
3- Жидкая 755
Пиррол 2-ацил Твердая 774-740 -755
Обозначения у и Р объяснены в тесте (см. раздел 2.6.30.4); см. также (Katritzky, 1963).
3. Спектроскопия ЯМР 1Н
3.1. Введение
Спектроскопия ядерного магнитного резонанса (ЯМР)
по сути является одним из методов абсорбционной
спектрометрии подобно ИК- и УФ-спектроскопии.
В соответствующих условиях в магнитном поле об-
разец может поглощать электромагнитное излучение
в радиочастотной (РЧ) области, причем частоты погло-
щения определяются свойствами образца. Поглощение
обусловлено наличием определенных ядер в молекуле.
График в координатах частота поглощения - интенсив-
ность сигнала представляет собой спектр ЯМР. На-
стоящая глава относится к спектроскопии протонного
резонанса (ЯМР 1Н).
При соответствующем владении основами тео-
рии интерпретация спектра ЯМР обычно доступна
в существенно больших деталях, чем интерпрета-
ция ИК- или масс-спектра. Предлагаемое изложение
вполне достаточно для решения конкретной задачи:
идентификации органических соединений с исполь-
зованием другой спектральной информации. Список
литературы приведен в конце главы.
3.2. Теория
3.2.1. Магнитные свойства ядер
Мы начнем с описания магнитных свойств ядер.
Все ядра несут заряд. В некоторых ядрах этот заряд
«вращается» относительно оси ядра, и это вращение
ядерного заряда генерирует магнитный диполь вдоль
оси (рис. 3.1). Угловой момент вращающегося заряда
может быть описан его спиновым квантовым чис-
лом I. Эти числа имеют значения 0, 1/2, 1,3/2 и т.д.
(7=0 означает отсутствие спина). Характеристическая
величина генерируемого диполя выражается в вели-
чинах ядерного магнитного момента ц.
Основные характеристики некоторых ядер, вклю-
чая спин ядра 7, приведены в гл. 6, приложение А.
Спин ядра можно предсказать, зная атомную массу
и атомный номер, как это показано в табл. 3.1.
Спектры некоторых ядер могут быть получены
легко (например, ядер }Н, fH, k3C, )5N, £9F, ^|P), по-
скольку они имеют спин 1/2 и однородное сфериче-
ское распределение заряда (рис. 3.1). Из указанных ядер
наиболее широко используются в спектроскопии ЯМР
протоны (1Н) (настоящая глава) и углерод 13С (гл. 4).
Ядра со спиновым числом I, равным 1 или больше,
имеют несферическое распределение заряда, Эта асим-
метрия описывается электрическим квадрупольным
моментом, который, как мы увидим позже, влияет на
время релаксации, а следовательно, на ширину линии
сигнала и взаимодействие с соседними ядрами. В тер-
минах квантовой механики спин ядра I определяет
число ориентаций, которое может приобретать ядро во
внешнем однородном магнитном поле в соответствии
с формулой 21 + 1. В этой главе мы рассматриваем
протон, спин которого 7 равен 1/2.
Рис. 3.1. Вращающийся заряд протона генерирует
магнитный диполь
Таблица 3.1. Типы ядерных спинов (/) для
различных комбинаций атомной массы и атомного
номера
Спин Атомная масса Атомный номер Пример ядра
Полуцелый Нечетная Нечетный |НС/г), ?Н(*/г), 75N(!4), '№), ?|РС/2)
Полуцелый Нечетная Четный I6’c(*/2),I870(’/2),^siC/2)
Целый Четная Нечетный ?Н(1), ’74N(1), ’5°В(3)
Нулевой Четная Четный ^(0)A«o(0),ftS(0)
3.2. Теория
151
3.2.2. Возбуждение ядер со спином Уг
Для ядер со спином Уг во внешнем магнитном поле
(рис. 3.2) имеется два энергетических уровня и неболь-
шой избыток населенности на нижнем уровне (Na > 7Vp)
в соответствии с распределением Больцмана. Эти со-
стояния обозначаются как аир или 54 и - 54. Разность
энергий двух уровней (АЕ) записывается как
АЕ = (Ау/2л)В0
где h - постоянная Планка. Это выражение просто
утверждает, что разность энергий АЕ пропорциональ-
на Bq, поскольку остальные величины (Л, у и тс) - кон-
станты. Величина Bq характеризует напряженность
магнитного поля.
Как только возникла система двух уровней, можно
вводить энергию в виде радиочастотного излучения
с частотой (vj, чтобы возбудить переходы между
этими уровнями энергии в постоянном магнитном
поле BQ. Фундаментальное уравнение ЯМР, связываю-
щее прикладываемую частоту (v ) с величиной на-
пряженности магнитного поля, записывается в виде
V= (у/2я)Во,
поскольку
АЕ = hv.
Частота облучения находится в мегагерцевом
диапазоне (МГц). Для протонов при величине поля
Bq, равной 2.35 Тл, частота облучения равна 100 МГц.
При увеличении поля в п раз во столько же возрас-
тает и частота резонанса. При соотношении частоты
и поля, равном (у/2л), система находится в резонан-
се; протон поглощает энергию, переходит на более
высокий энергетический уровень, и можно записать
спектр. Отсюда и возникает название спектроскопия
ядерного магнитного резонанса. Постоянная у назы-
вается гиромагнитным отношением и является фун-
даментальной ядерной постоянной. Это коэффициент
пропорциональности между магнитным моментом
р и спином ядра Г.
у= 2itp/hl.
Радиочастотная энергия может вводиться либо
в режиме непрерывной развертки в некотором диа-
1) Обозначение В (магнитная индукция или плотность
потока) заменяет величину Н (напряженность магнит-
ного поля). В системе СИ единица измерения В тесла
(Тл) заменяет единицу гаусс (Гс): 1 Тл = 104 Гс. Едини-
ца частоты герц (Гц) заменяет число циклов в секунду.
МГц = 106 Гц.
Рис. 3.2. Два энергетических уровня протона
в магнитном поле Во. N - населенности спинов
на верхнем (Л/а) и нижнем (Л/р) уровнях энергии.
Магнитное поле (Во) направлено вверх параллельно
ординате, напряженность поля Во возрастает слева
направо. Чем больше Во, тем больше величина ДЕ
пазоне частот (continuous-wave (CW) или непрерыв-
ный режим), либо в виде короткого радиочастотного
импульса, содержащего весь набор частот (импульс-
ный режим). Эти два способа соответствуют двум
разным типам спектрометров ЯМР.
В спектрометрах стационарного типа (CW-типа)
образец помещают в магнитное поле и облучают
путем медленной развертки частоты в требуе-
мом диапазоне частот. Спектр получают, записы-
вая поглощение энергии как функцию частоты.
В импульсном спектрометре образец помещают
в магнитное поле и облучают мощным импуль-
сом радиочастотной энергии, содержащим широкий
диапазон частот, достаточный для облучения всего
требуемого диапазона. Этот импульс возбуждает
все ядра образца одновременно. Непосредственно
вслед за импульсом ядра начинают возвращаться
в основное состояние и излучают поглощенную
энергию. Детектор собирает эту энергию в виде
сигнала спада свободной индукции (ССИ или FID,
free induction decay), который является суммой
излучения всех ядер в зависимости от времени.
Информация, содержащаяся в ССИ как функция
времени, с помощью преобразования Фурье (FT)
превращается в результирующий спектр, который
является функцией частоты.
Задача состоит в том, каким образом приложить
радиочастотную электромагнитную энергию к про-
тонам, ориентированным в постоянном магнитном
поле, и как затем измерить энергию, поглощенную
протонами при переходе в более высокое спино-
вое состояние. Это можно объяснить в терминах
классической механики, если представить протон
152
3. Спектроскопия ЯМР
Рис. 3.3. Классическое представление протона,
прецессирующего в магнитном поле Во, по аналогии
с прецессией вращающегося волчка
как частицу, вращающуюся во внешнем магнитном
поле. Магнитная ось протона прецессирует вокруг
оси z постоянного магнитного поля BQ подобно тому
как прецессирует под действием гравитации волчок,
ось вращения которого отклонена от перпендикуляра
(рис. 3.3).
Ансамбль эквивалентных протонов, прецессирую-
щих со случайной фазой вокруг оси z (т.е. вокруг
направления постоянного магнитного поля Во), порож-
дает суммарную макроскопическую намагниченность
в направлении оси z, но не в плоскости ху (рис. 3.4).
Когда частота прикладываемого высокочастотного
(ВЧ) поля (vj равна частоте прецессии эквивалентных
протонов (называемой в классической физике лар-
моровой частотой vL, в МГц), достигается состояние
ядерного магнитного резонанса, и основное уравнение
ЯМР может быть записано в виде
VL = V, = (у/2л)В0
Это уравнение применимо к ансамблю изолиро-
ванных протонов (см. раздел 3.5).
Далее необходимо повернуть намагниченность
Мо по направлению к горизонтальной плоскости ху
в стационарной декартовой системе координат и из-
мерить результирующую компоненту намагниченности
в этой плоскости. Электромагнитное ВЧ-излучение
v, прикладывается таким образом, чтобы его магнит-
ная компонента Вх составила прямой угол с главным
магнитным полем Во и вращалась вместе с ансамблем
прецессирующих протонов. Это осуществляется с по-
мощью ВЧ-осциллятора, ось которого (по условию
направленная вдоль координаты х) перпендикулярна
оси главного магнитного поля Во. Такой осциллятор
будет генерировать непрерывное (CW) осциллирую-
щее магнитное поле Въ направленное вдоль оси х.
Осциллирующее магнитное поле можно разложить
на две компоненты, вращающиеся в противополож-
ных направлениях (рис. 3.5). Одна из этих компо-
нент вращается в том же направлении, в котором
прецессирует вектор намагниченности протонов;
а компонента, вращающаяся в противоположном
направлении, неэффективна.
При изменении частоты осциллятора (у) (т.е. когда
осуществляется, как говорят, «развертка» частоты)
частота вращающегося магнитного поля попада-
ет в резонанс с ларморовой частотой прецесии
(т.е. совпадут меняющаяся частота Vj и частота vL для
прецесии протонов). Это приведет к когерентности
Рис. 3.4. Ансамбль прецессирующих ядер приводит к появлению макроскопической
намагниченности Мо в направлении постоянного магнитного поля Во
3.2. Теория
153
Рис. 3.5. Осциллирующее магнитное поле можно разложить на две компоненты
В, , вращающиеся в противоположных направлениях; В1с и В1сс обозначают
вращение по часовой стрелке и против часовой стрелки соответственно
(совпадению) фаз и вызовет отклонение общей на-
магниченности Мо по направлению к горизонтальной
плоскости (рис. 3.6, а и б). Магнитную компоненту,
возникающую в плоскости ху, можно детектировать
с помощью приемной катушки, размещенной в этой
плоскости. Отклонение вектора намагниченности
от направления вдоль оси z (т.е появление компо-
ненты в плоскости ху) может происходить, когда
частота V! близко подходит к частоте vL , однако
максимальную интенсивность сигнала получают при
частоте осциллятора, точно совпадающей с частотой
прецессии.
Часто в спектрах ЯМР обнаруживаются небольшие
«боковые» сигналы от вращения (рис. Э.7),^ симме-
трично расположенные по обе стороны от интенсив-
ного сигнала поглощения; эти полосы обусловлены
остаточной неоднородностью магнитного поля и де-
фектами вращающейся ампулы. Их легко распознать
вследствие их симметрии и потому, что их расстояние
° Одна из важных проблем в спектроскопии ЯМР - это
однородность магнитного поля. Дело в том, что реальный
образец имеет макроскопические размеры (объем порядка
0.1-1 см3). Поэтому в разных точках образца напряжен-
ность магнитного поля может слегка различаться. Допу-
стим, что это различие составляет величину ДВ0. Тогда и
различия в частотах резонанса в разных участках спектра
могут достигать величины Avj = (у/2л)ДВ0. Величину не-
однородности обычно оценивают в относительных еди-
ницах. Например, неоднородность в 10-8 означает 10-8 от
величины приложенного поля, и если это поле составляет,
допустим, 2.35 Тл и резонансная частота для протонов
равна соответственно 100 МГц, то неоднородности в
10-8 будет соответствовать неоднородность 2.35-10-8 Тл
или 1 Гц в единицах частоты. Для уменьшения неодно-
родности часто используют простой способ - вращение
цилиндрической ампулы вокруг продольной оси, что при-
водит к усреднению неоднородности и фактически к ее
уменьшению - Прим, перев.
от сигнала поглощения равно скорости вращения,
которая составляет обычно 10-20 Гц.
Осцилляции, которые обнаруживаются в низкоча-
стотной области интенсивных острых сигналов в спек-
трах с непрерывной разверткой частоты, называются
«звоном» или «вигглями» (wiggles) (рис. 3.8). Они
являются «биениями» частот, возникающими при
прохождении через сигнал поглощения, и обычно
встречаются в старой литературе.
Самым большим недостатком спектрометров ста-
ционарного типа является то, что запись полного
спектра (т.е. в области 0-14 м.д.) занимает несколько
минут. Отношение сигнал/шум можно было бы увели-
чить при использовании метода накопления сигналов.
Известно, что при N прохождениях спектра сигнал
растет пропорционально 7V, а шум пропорционально
ylN и таким образом отношение сигнал/шум растет
пропорционально ^N. Это значит, что увеличение от-
ношения сигнал/шум в 10 раз потребует 100-крагного
прохождения спектра или нескольких часов приборно-
го времени. Таким образом, о широкой применимости
стационарного метода ЯМР для исследования систем
при ограниченных количествах вещества (например,
для разбавленных растворов, газов) или для мало-
чувствительных ядер, например 13С (ядра, примерно
в 6000 раз менее чувствительного, чем 1Н), речь идти
не может. Спектрометры этого типа могут быть ис-
пользованы для исследования чистых жидкостей или
очень концентрированных растворов.
До сих пор мы описывали взаимодействие между
общей намагниченностью Мо ансамбля идентичных
протонов в постоянном однородном магнитном поле
Bq и осциллирующим радиочастотным полем Vj (в
действительности с одной из двух круговых компо-
нент генерируемого магнитного поля Вх (см. рис. 3.5)).
Для получения спектра производят развертку частоты
осциллятора Vi во всем диапазоне частот протонов (ме-
154
3. Спектроскопия ЯМР 'Н
а
Рис. 3.6. а,б - Осциллятор генерирует вращающуюся компоненту возбуждающего
магнитного поля В,. Общая намагниченность Мо отклоняется от оси z и
создает вектор М, который прецессирует вокруг оси z, генерируя компоненту
намагниченности в горизонтальной плоскости, в - Продольная релаксация
(т.е. возвращение) вектора М к положению Мо происходит по сворачивающейся
спирали за время Tv Аналогично происходит и поперечная релаксация
(т.е. исчезновение поперечных компонент (в плоскости ху)) со временем Т2.
Используется лабораторная (неподвижная) декартова системе координат
тод частотной развертки); в альтернативном подходе
(магнитная развертка) частоту осциллятора можно удер-
живать постоянной и производить развертку поля Во-
Каждый отдельный вид протонов должен входить в ре-
зонанс один за другим. Этот принцип записи спектров
называют непрерывной (CW в английской литературе)
спектроскопией, и его применяли в спектрометрах на
начальном этапе развития спектроскопии ЯМР. Непре-
рывная развертка еще используется в некоторых спек-
трометрах низкого разрешения, но в настоящее время
этот метод практически полностью вытеснен импульсной
фурье-спекгроскопией ЯМР. Однако, поскольку непрерыв-
ный метод легче для понимания, мы начали обсуждение
с него, используя привычную стационарную декартову
систему координат (оси х, у, z). Теперь мы перейдем
к рассмотрению импульсной фурье-спектроскопии
и вращающейся системы координат.
Развитие импульсного метода было стимулировано
главным образом необходимостью повышения чув-
ствительности для регистрации спектров ЯМР 13С
(гл. 4). Более высокая чувствительность достигается
одновременным возбуждением всех интересующих
нас ядер (в данной главе протонов), чтобы затем
собрать одновременно все сигналы (рис. 3.9, а).
Рис. 3.7. Сигнал чистого хлороформа с боковыми
полосами от вращения с частотой 6 Гц (а) и 14 Гц (б)
(С разрешения из книги F. Bovey, NMR Spectroscopy,
New York, Academic Press, 1969)
Рис. 3.8. Звон (или «виггли»), видимые после
прохождения через резонанс в спектрах
с непрерывной разверткой. Направление записи -
слева направо (т.е. в сторону уменьшения частот
резонанса)
3.2. Теория
155
а
ВЧ- импульс в = 90°
Выборка (/2)
Релаксационная
задержка (7?d)
Сигнал
отсутствует
Положительный
сигнал
Отрицательный сигнал
Сигнал
отсутствует
Отрицательный
сигнал
Положительный
сигнал
Время выборки (/2)
в
Рис. 3.9. а - Последовательность импульсов в стандартном эксперименте по
ЯМР (0-радиочастотный импульс варьируемой длительности); б - связь
длительности импульса (0) и ожидаемого сигнала; в - вращение сигнала
в плоскости ху после 90°-ного импульса
156
3. Спектроскопия ЯМР 'Н
При этом однократное прохождение спектра стано-
вится очень коротким (секунды), и в определенном
смысле импульс может быть описан как «мгновен-
ное прохождение». Короткий (микросекундный, мкс)
мощный радиочастотный импульс с центральной
частотой v15 прилагаемый вдоль оси х, генериру-
ет целый требуемый диапазон частот и произво-
дит в значительной степени тот же самый эффект,
что и сканирующий осциллятор: он отклоняет вектор
намагниченности Мо по направлению к плоскости
ху (обычно на 90!), но делает это для всех протонов
одновременно (рис 3.9, б). Сигналы намагниченности
детектируются практически немедленно вслед за
импульсом в плоскости ху и собираются компью-
тером (после аналого-цифрового преобразования)
в течение периода времени, который называется
временем выборки (рис. 3.9, в). В течение этого
периода сигнал (вектор М, который прецессирует
вокруг оси z), детектируется как проекция на ось у.
Например, непосредственно после импульса вектор
М располагается вдоль оси у, и сигнал максимален;
через какой-то промежуток времени вектор М по-
ворачивается до совпадения с осью х. Проекция
на ось у в этот момент равна 0, при продолжении
поворота вектор М доходит до оси -у (минимум).
Это вращение вокруг оси z продолжается до тех
пор, пока намагниченность М не вернется к своему
равновесному значению (раздел 3.2.3).
В результате получают так называемый сигнал
ССИ (сигнал спада свободной индукции) (в ан-
глийской литературе FID - free induction decay),
который может быть описан как затухающее си-
нусоидальное колебание (примеры см. в разделе
3.2.3). Зарегистрированные сигналы представляют
собой разность между прикладываемой частотой
Vi и ларморовой частотой vL каждого протона. За-
тем ССИ с помощью преобразования Фурье, про-
изводимого компьютером, превращают в обычный
спектр ЯМР. Поскольку времена релаксации для
протонов обычно имеют величину порядка не-
скольких секунд, можно быстро повторять им-
пульсы с накоплением сигнала и таким образом
существенно увеличивать отношение сигнал/шум
и соответственно чувствительность спектрометра.
Некоторые ядра 13С, не связанные с протонами, ко-
торые обеспечивают продольную релаксацию (со
временем TJ, требуют применения гораздо более
длительных интервалов между импульсами, чтобы
дать таким ядрам возможность отрелаксировать.
Отсутствие соответствующих интервалов приво-
дит к ослаблению сигнала и неточному измерению
площади пика (т.е. к ошибкам в оценке количества
ядер, вызывающих данный сигнал, см. раздел 4.3.1).
3.2.3. Релаксация
В предыдущем разделе мы описали в терминах клас-
сической механики отклонение вектора ядерной на-
магниченности (Af0) по направлению к плоскости
ху\ теперь необходимо рассмотреть, каким образом
Мо возвращается к оси z.
Существуют два «релаксационных» процесса.
Спин-решеточная, или продольная, релаксация, ха-
рактеризуемая временем Тъ включает перенос энергии
от «возбужденных» протонов на окружающие прото-
ны, дающие резонанс на соответствующих частотах.
На рис. 3.6, в показано исчезновение ху-компоненты по
механизму Тх, в то время как общая намагниченность
возвращается к оси z по скручивающейся спирали.
Спин-спиновая, или поперечная, релаксация, харак-
теризуемая временем Т2, включает перенос энергии
между прецессирующими протонами, который приво-
дит к расфазировке для отдельных спиновых пакетов,
уширению линий и исчезновению сигнала (рис. 3.10).
Обозначение Т2* используется для описания всех
факторов, вносящих вклад в исчезновение сигнала
за счет поперечной релаксации. Это время включает
в себя как собственно Т2 (время, характеризующее
истинную спиновую динамику), так и влияние не-
однородностей магнитного поля, которое обычно
преобладает.
Для малых молекул времена Т\ и Т2 обычно близки
по величине; соотношение между Тх и Т2 иллюстри-
рует следующая схема:
Как только включается Г2-релаксация, она умень-
шает М в плоскости ху за счет переноса энергии на
ось z . Для больших молекул (и других ядер) времена
релаксации Тх обычно гораздо больше, чем Т2.
Для протонов в обычных невязких растворах
времена релаксации Т2 достаточно длинные, чтобы
получались узкие линии, а времена релаксации Т\ до-
статочно короткие, чтобы интенсивности линий были
пропорциональны числу вносящих в них вклад про-
тонов. Таким образом, относительное число протонов
различного типа в спектре может быть определено
путем измерения площадей сигналов.
3.2. Теория
157
Рис. 3.10. Один спин, вращающийся в плоскости ху с той же частотой,
что и вращающаяся система координат, и со временем релаксации Т2.
Т2-Релаксация вызовет расфазировку или рассеяние намагниченности в плоскости
ху, что приведет к уширению линии и спаду сигнала. Величина Т2 частично
определяется неоднородностью магнитного поля
Для ядер 13С и 15N, однако, необходимо учитывать
7^-релаксацию, так как времена релаксации Тх мо-
гут быть существенно большими, чем для протонов,
и изменяться в широком диапазоне. Далее мы дадим
несколько общих предварительных сведений, более
подробное обсуждение можно найти в гл. 4 и 6.
Времена релаксации Тх для ядер 13С и 15N подвер-
жены влиянию диполь-дипольных взаимодействий
с присоединенными к ним протонами и, в меньшей
степени, с другими соседними протонами. В рутин-
ном спектре 13С и 15N большие времена Тх приво-
дят только к частичному восстановлению сигнала,
и поэтому следует вводить задержки между после-
довательными импульсами (см. рис. 4.2, а). Таким
образом, мы видим, что релаксация Тх тесно связана
с интенсивностью сигнала.
Время Т2, напротив, определяет ширину линии
в соответствии с принципом неопределенности
Гейзенберга, который утверждает, что произведение
неопределенности интервала частоты и неопределен-
ности интервала времени не может быть меньше
определенной величины
На рис. 3.16. (см. далее) проиллюстрировано влия-
ние Т2 на уширение линии с помощью добавления
к образцу небольшого количества железа. Совершенно
очевидно, какой эффект это добавление производит на
ширину линий в частотном представлении спектра,
и эффект проявляется во временном представлении
(ССИ). Как мы упоминали выше, Т2 определяет вре-
мя, в течение которого намагниченность М остается
в плоскости ху, поэтому если поперечная релаксация
происходит достаточно быстро (Г2 мало и наблюда-
ется широкий сигнал ЯМР), то спад ССИ до нуля
происходит гораздо быстрее, чем при медленной
поперечной релаксации (при большом Т2 и узком
сигнале ЯМР).
Предположим, что суммарная общая намагничен-
ность Mq (вектор, выделенный жирной линией на
рис. 3.12 и расположенный вдоль оси z) состоит из
трех индивидуальных намагниченностей, характери-
зующих три сорта протонов в химическом соеди-
нении. При наложении 90°-]
из этих намагниченностей
•ного импульса каждая
находится в резонансе
Av А/ > 1.
Если AZ (т.е. Г2*) мало, то Av велико, и линия
уширяется. Время Г2*, основным вкладом в вели-
чину которого является неоднородность магнитного
поля, является, таким образом, главным фактором,
определяющим ширину линии, поскольку Г2* всегда
меньше, чем Тх или Т2. Величину Т2 можно изме-
рить, определяя ширину линии на половине высоты
(рис. 3.11).
V
Рис. 3.11. Ширина линии на полувысоте
А ^1/2 = (зтсТЪ)1
Скорость
релаксации (R) = 1/ Т2
158
3, Спектроскопия ЯМР 1Н
со своей частотой в импульсе, и все их векторы
вращаются одновременно вокруг оси у. Каждая из
компонент намагниченности (тонкие векторы) на-
чинает теперь прецессировать со своей собственной
ларморовой частотой с одновременной релаксацией
по механизмам Тх и Т2. Возникает практическая про-
блема-трудно точно измерить абсолютные частоты,
которые изменяются, скажем, в интервале 5000 Гц
вокруг центральной частоты импульса, равной, напри-
мер, 300 000 000 Гц (vj, называемой также несущей
частотой. Например, указанные три сорта протонов,
вносящих вклад в намагниченность, имеют ларморовы
частоты (vj 300 002 000,300 000 800 и 299 999 000 Гц.
Эта проблема разрешается путем измерения разно-
сти между каждой ларморовой частотой и несущей
частотой, которая прикладывается в середине спек-
трального окна. Разность частот составляет 2000 Гц,
800 Гц и -1000 Гц, т.е. две частоты выше, чем не-
сущая частота, и одна - ниже.
Чтобы упростить визуализацию, неподвижная де-
картова система координат заменяется на систему
координат, вращающуюся вокруг оси z с частотой,
равной несущей частоте (vj. Эта ситуация аналогична
той, в которой находится наблюдатель, вращающийся
на карусели, к которой жестко привязана собственная
система координат.
Как было сказано выше, разностные частоты про-
являются в виде отдельных сигналов ССИ с посте-
пенно затухающими интенсивностями.
Спад свободной индукции и импульсные
фурье-спектры были описаны в разделе 3.2.2. Сейчас
будет очень поучительно посмотреть на оба «спектра»,
хотя такие важнейшие темы, как химические сдвиги
и спин-спиновое взаимодействие, еще не обсуждались.
И ССИ, и фурье-преобразованный спектр действи-
тельно являются спектрами, хотя ССИ (спектр во
временном представлении) должен быть с помощью
математической процедуры - преобразования Фурье-
превращен в спектр в частотном представлении.
Протоны в ацетоне СН3-(С=О)-СН3 химически эк-
вивалентны. По мере протекания релаксации вслед за
импульсом сигнал детектируется в виде одной экс-
поненциально спадающей синусоиды (временное
представление, ССИ) с частотой, равной разности
между несущей частотой (vj и ларморовой часто-
той (vL), протонов. Осуществляемое компьютером
фурье-преобразование переводит спектр в частотное
представление и дает синглет (рис. 3.13, а) с химиче-
ским сдвигом 5 2.05 м.д. относительно произвольного
стандарта при 8 0.00 (см. раздел 3.4). Синглет представ-
ляет собой привычный спектр протонов ацетона.
Рис. 3.12. Вращающаяся система координат. Общая намагниченность М (после
90°-ного импульса) имеет три компоненты с ларморовыми частотами vL1,
vL2 и vL3 (т.е. три различных протона). Система координат вращается с частотой
Vt (возбуждающий импульс). Сразу вслед за импульсом компоненты прецессируют
относительно vv причем компоненты vL1 и vL2 выше по частоте, чем возбуждающий
импульс, a vL3 - ниже
3.3. Техника эксперимента и подготовка образца
159
0.4
м.д.
0.2 0.4 0.6 0.8 4.0 3.5 3.0 2.5 2.0 1.5
Рис. 3.13. а - ССИ и протонный фурье-спектр ацетона, б - ССИ и протонные
фурье-спектры этилацетата. Оба спектра зарегистрированы в растворе CDCI3 при
300 Мгц
В отличие от эквивалентных протонов метильных
групп ацетона группы СН3 и СН2 в этилацетате (СН3-
(С=О)-ОСН2СН3) отличаются друг от друга. Поэтому
ССИ (Рис. 3.13, б) состоит из суперпозиции трех
синусоид и биений (интерференции). Фурье-спектр
состоит из трех раздельных групп сигналов. Слева
направо (рис. 3.13, б) мы видим квартет, синглет и три-
плет, которые появляются вследствие «химических
сдвигов» и «спин-спинового взаимодействия» (см.
разделы 3.4 и 3.5).
3.3. Техника эксперимента
и подготовка образца
3.3.1. Техника эксперимента
Начиная с 1953 г., когда появился первый коммерче-
ский спектрометр ЯМР, использовали спектрометры
с постоянными магнитами и электромагнитами с ве-
личиной поля 1.41; 1.87; 2.20 и 2.35 Тл, что отвечает
частотам 60,80,90 и 100 МГц соответственно для на-
блюдения протонов (обычный способ характеристики
спектрометра ЯМР).
Гонка за мощностью, стимулированная необходи-
мостью повышения разрешения и чувствительности,
привела к широкому использованию в настоящее
время приборов с рабочей частотой 300-600 МГц.
Все спектрометры с рабочей частотой выше 100 МГц
имеют в своей основе сверхпроводящие магниты
(соленоиды) и работают в импульсном режиме.
Другими основными требованиями, помимо силь-
ного поля и высокой стабильности отношения поле/
частота, является однородность поля и компьютерный
интерфейс.
Образец (в рутинных исследованиях - это раствор
в дейтерированном растворителе в стеклянной ампуле
внешним диаметром 5 мм) помещают в датчик, ко-
торый содержит передающую и приемную катушки
и турбинку для вращения ампулы вокруг ее верти-
кальной оси, чтобы усреднить неоднородности поля.
Спектрометр ЯМР стационарного типа (рис. 3.14)
состоит из управляющей консоли, магнита (обычно
используют электромагниты или постоянные маг-
ниты) и двух ортогональных проволочных катушек
(передающей и приемной катушки), которые служат
антеннами для ВЧ-излучения. Одна катушка соеди-
нена с ВЧ-передатчиком, другая катушка является
ВЧ-усилителем (приемная катушка) и соединена
с электронным детектором.
На рис. 3.15. показано устройство спектрометра
со сверхпроводящим магнитом. Отметим, что в элек-
тромагнитах ампула вращается под прямым углом
к оси z, которая расположена горизонтально, тогда
160
3. Спектроскопия ЯМР 1Н
Высокочастотный
передатчик
Усилитель
низкой частоты
Генератор
развертки
Осциллограф
Рис. 3.14. Схема спектрометра ЯМР непрерывного типа. Ампула перпендикулярна
оси z магнита: А - ампула, Б - катушка передатчика, В - катушки развертки,
Г - катушка приемника, Д - магнит
как в сверхпроводящих магнитах ампула распола-
гается в сердечнике соленоида и вращается вокруг
оси z, которая расположена вертикально. Приемная
и передающая катушки связаны между собой через
ядра образца (протоны в данной гл.).
Протонный спектр, получаемый путем сканирования
в непрерывном или импульсном режиме в посто-
янном магнитном поле, состоит из серии сигналов,
площади которых пропорциональны числу протонов,
которые они представляют. Площади сигналов изме-
ряют с помощью электронного интегратора, который
рисует серию ступенек, их высоты пропорциональ-
ны площадям сигналов (см. рис. 3.26). Ч Подсчет
количества протонов с помощью интегрирования
полезен для определения молекулярной формулы,
Химически различные протоны поглощают радиочастот-
ную энергию при очень слабо различающихся частотах -
различие составляет максимально 5000 Гц при частоте
спектрометра 300 МГц (см. раздел 4.7). Использование
спектрометров ЯМР для нужд органической химии на-
чиналось с экспериментов на фирме Вариан, где впервые
удалось получить три сигнала для химически различных
групп протонов в этаноле СН3СН2ОН, площади пиков со-
относились как 3:2:1 [J.T. Arnold, S.S. Dharmatti, and М. Е.
Packard,/. Chem. Phys., '19, 507 (1951)].
обнаружения скрытых сигналов, контроля чистоты
образца и проведения количественного анализа. По-
ложения сигналов (химические сдвиги, раздел 3.4)
измеряют в единицах частоты относительно сигнала
стандарта.
3.3.2. Чувствительность в экспериментах по ЯМР
Чувствительность в импульсных ФП-экспериментах
задается величиной отношения сигнала к шуму (S/N):
где S/N- отношение сигнал / шум, N- число спинов
в системе (концентрация образца), Т2 - время по-
перечной релаксации (определяет ширину линии),
уехс - гиромагнитное соотношение возбуждаемого
ядра, ydet - гиромагнитное соотношение детектируе-
мого ядра, ns - число прохождений, Во - внешнее
магнитное поле, Т - температура образца.
Примечание: в обычном эксперименте ЯМР !Н
Уехс и Ydet ~ характеристики одного и того же ядра.
Чтобы улучшить отношение S/N для менее чувстви-
тельного ядра может быть полезно возбудить один
3.3. Техника эксперимента и подготовка образца
161
Отверстие магнита
Жидкий азот------
Компьютер
Жидкий гелий
Рис. 3.15. Схема спектрометра ЯМР с преобразованием Фурье и сверхпроводящим
магнитом. Датчик параллелен оси z магнита, который охлаждается жидким гелием,
окруженным жидким азотом в большом сосуде Дьюара
Датчик
Выходные
устройства
и устройства
для хранения
информации
тип ядер, а детектировать другой тип с более высо-
ким гиромагнитным отношением в одном и том же
эксперименте. Так и поступают в инверсных экс-
периментах, которые описаны в гл. 5 и 6.
Обычный образец для регистрации протонного
спектра ЯМР на спектрометре с рабочей частотой
300 МГц содержит около 10 мг вещества примерно
в 0.5 мл растворителя в стеклянной ампуле с внешним
диаметром 5 мм. Существуют микродатчики, в кото-
рые можно помещать ампулы с внешним диаметром
1.0; 2.5 и 3 мм, и они обеспечивают более высо-
кую чувствительность. При благоприятных условиях
в микроампуле 1 мм (объемом 5 мкл) можно получить
спектр от 100 нг соединения средней молекулярной
массы на спектрометре с частотой 600 МГц.
Создание криогенно охлаждаемых датчиков суще-
ственно уменьшило количество образца, требуемое
для записи спектра ЯМР 1Н. Тепловой шум датчика
и электроники первой ступени усилителя являет-
ся преобладающим шумом в экспериментах ЯМР.
Эти новые усовершенствованные датчики встроены
в усилитель первой ступени и ВЧ-катушки, которые
охлаждаются до температуры порядка 20 К. Такие
криогенные датчики имеют повышенное отноше-
ние S/N, равное примерно 4 отношениям S/N для
стандартных датчиков. Совершенно очевидно для
пользователей, что спектрометры с наиболее силь-
ными полями дают наилучшую чувствительность.
Чтобы получить одинаковое отношение S/N при
фиксированной концентрации (N), на спектрометре
300 МГц необходимо иметь в 2.8 раза более высо-
кую концентрацию, чем на спектрометре 600 Мгц,
в соответствии с простым расчетом
S/N =
^боо _ ?|б00 ~ 2 8
Л^зоо V 300
Следовательно, при очень ограниченном количестве
образца обычно наилучшим вариантом является ис-
пользование наиболее сильных из возможных полей
и криодатчик с наименьшим диаметром. Криокапил-
лярный проточный микродатчик, который позволяет
работать с несколькими нанограммами материала
в 1 мкл растворителя, обладает наивысшей чувстви-
тельностью для образцов, доступных в ограниченных
количествах.
3.3.3. Выбор растворителя
Идеальный растворитель не должен содержать про-
тонов. Кроме того, желательно, чтобы растворитель
был инертным, низкокипящим и недорогим. Для со-
временных приборов необходимы дейтерированные
растворители, поскольку стабилизация поля BQ маг-
нита осуществляется с помощью сигнала дейтерия
растворителя. Прибор имеет дейтериевый «канал»,
который постоянно изменяет и подстраивает поле
Во к частоте дейтерированного растворителя. Эта про-
цедура автоматического удерживания нужной часто-
162
3. Спектроскопия ЯМР 1Н
ты (точнее, условий резонанса) называется захватом
или «локом» (от английского lock). Типичный сигнал
ЯМР !Н имеет ширину от 0.1 до нескольких герц
по сравнению с 300000000 Гц (для спектрометров
300 МГц), поэтому очевидно, что поле Во должно быть
очень стабильным и однородным.
Сигнал дейтерия используется также для «шим-
мирования» поля BQ. Шиммированием называют
традиционную для специалистов ЯМР процедуру
улучшения однородности магнитного поля, которую
осуществляют с помощью специальных встроенных
в прибор маленьких электромагнитных катушек (на-
зываемых шиммами), которые корректируют главное
магнитное поле (Во) таким образом, чтобы однород-
ность его была наивысшей точно в центре образ-
ца. Большинство из современных приборов имеют
20-30 электромагнитных шиммирующих катушек;
они контролируются компьютером и могут настраи-
ваться в автоматическом режиме.
Дейтерохлороформ (CDC13) используется всегда,
когда позволяют обстоятельства, в действительности,
в большинстве случаев. Маленький узкий протон-
ный сигнал при 7.26 м.д. от примесного хлороформа
(СНС13) мешает в очень редких случаях. Для очень
разбавленных образцов доступен CDC13 100%-ной
изотопной чистоты. Список обычных коммерчески
доступных растворителей с указанием положения
сигнала примесной протонной формы (например,
СНС13 в CDC13) приведен в приложении Ж.
Следы ферромагнитных примесей приводят к ка-
тастрофическому уширению сигналов поглощения
вследствие сильного уменьшения времени релакса-
ции Т2. Обычными источниками ухудшения одно-
родности являются частички загрязнений из водо-
проводной воды, стальных волокон, никеля Ренея
и частички от металлических шпателей и наполни-
телей колонок (рис. 3.16). Эти загрязнения можно
удалить фильтрованием.
Рис. 3.16. Влияние микроскопических парамагнитных частиц на спектр
протонного магнитного резонанса октаацетата целлобиозы. Верхний спектр
записан в присутствии частиц, внизу - после их удаления
3.4. Химический сдвиг
163
Много неприятностей могут доставить следы
обычных лабораторных растворителей. В прило-
жении 3 приведен список растворителей, обычно
присутствующих в образцах как примеси. Другими
источниками загрязнений могут быть смазки и пла-
стификаторы (в частности, фталаты). Растворители
для ЯМР следует хранить в эксикаторе.
3.4. Химический сдвиг
В соответствии с основным уравнением ЯМР (см.
раздел 3.2.2)
v,= (у/2л)В0,
где V] - прилагаемая частота, BQ - плотность потока
постоянного магнитного поля и (у/2л) - константа
для всех протонов (т.е. частиц с одинаковым гиро-
магнитным отношением), следует ожидать появления
только одного сигнала ЯМР. К счастью, ситуация не
столь проста. Протон в молекуле экранирован в очень
малой степени его электронным облаком, плотность
которого изменяется в зависимости от химического
окружения протона. Это изменение приводит к разли-
чиям в положениях химического сдвига д ля химически
различных протонов. Спектроскопия ЯМР высокого
разрешения использует эту способность различать
каждое индивидуальное поглощение
Базисное уравнение ЯМР для всех протонов мо-
дифицируется теперь в уравнение для ансамбля эк-
вивалентных протонов в молекуле
VefF = (у/2л)В0(1 - О).
Символ а используется для обозначения «константы
экранирования», величина которой пропорциональна
степени экранирования протона его электронным об-
лаком. При данной величине Во эффективная резо-
нансная частота меньше, чем частота облучения vP
Электроны под воздействием магнитного поля враща-
ются и, вращаясь, генерируют собственное магнитное
поле, направленное противоположно прилагаемому
полю, а следовательно, создают «экранирующий» эф-
фект (рис. 3.17). Этот эффект объясняет диамагнетизм,
присущий всем органическим веществам. В случае
соединений с неспаренным электроном парамагне-
тизм, обусловленный электронным спином, много-
кратно превосходит диамагнетизм циркулирующих
спаренных электронов.
Степень экранирования зависит от плотности цир-
кулирующих электронов, и, прежде всего, в очень
грубом приближении степень экранирования про-
тона при атоме углерода зависит от индуктивного
эффекта других групп, присоединенных к тому же
А Ядро А
I I / Циркулирующие
электроны
Ш'^'^Мапштные
силовые линии
Во
Рис. 3.17. Диамагнитное экранирование ядра
циркулирующими электронами. Стрелки |||
показывают направление постоянного магнитного
поля Во. Циркулирующие электроны создают
электрический ток, однако направление тока
условно показано как поток положительного заряда
атому углерода. Разность положения сигнала данного
протона и положения сигнала протона стандарта
называется химическим сдвигом данного протона.
Теперь мы имеем представление о том, что про-
тоны в «разном» химическом окружении имеют
различные химические сдвиги. И наоборот, прото-
ны в «одинаковом» химическом окружении имеют
один и тот же химический сдвиг. Что же, однако,
мы имеем в виду, говоря «разный» и «одинаковый»?
Интуитивно кажется очевидным, что химически раз-
личные метиленовые группы в С1СН2СН2ОН име-
ют различные химические сдвиги и что протоны
в каждой из метиленовых групп имеют один и тот
же химический сдвиг. Однако это может быть не
столь очевидным, например, для индивидуальных
протонов метиленовых групп в С6Н5СН2СНВгС1,
которые в действительности не имеют одинаковых
химических сдвигов. Здесь мы будем рассмотривать
только очевидные случаи и отложим более строгое
обсуждение эквивалентности химических сдвигов
до раздела 3.8.
Наиболее удобным веществом для стандарта яв-
ляется тетраметил сил ан (ТМС).
СН3
Н3С—Si—СН3
СН3
Это вещество имеет несколько преимуществ: оно
химически инертно, летуче (т. кип. 27 °C), раство-
римо в большинстве органических растворителей,
его молекула симметрична. Это соединение дает
интенсивный, узкий, синглетный сигнал в спектре
ЯМР *Н, и его протоны «экранированы» в большей
степени, чем протоны почти всех органических
164
3. Спектроскопия ЯМР ’Н
300 МГц
3000 2700 2400 2100 1800 1500 1200 900 600 300 Оу (Гц)
10 98765 --- - --
Более низкие частоты
большее экранирование
экранированные
Более высокие частоты
меньшее экранирование
дезэкранированные
600 МГц
“6000 5400 4800 4200 3600 3000 2400 1800 1200 600 0 v (Гц)
10 9876543210 6(м.д.)
Рис. 3.18. Шкалы ЯМР для рабочих частот 300 и 600 МГц. Относительно малое
количество органических соединений дают сигналы справа от сигнала ТМС.
Эти более низкие частоты обозначаются отрицательными числами справа
(не показаны на рисунке)
соединений. В тех случаях, когда растворителем
служит вода или оксид дейтерия, можно исполь-
зовать ТМС в концентрическом капилляре в каче-
стве «внешнего стандарта» или водорастворимую
соль - 2,2-диметил-2-силапентан-5-сульфонат натрия
(ДСС, (СНз)з81СН2СН2СН28ОзНа) в качестве вну-
треннего стандарта (0.015 м.д.).
Исторически, а теперь уже по договоренности, сиг-
нал стандарта (ТМС) помещают на правом краю спек-
тра и принимают его за нуль шкалы либо в герцах,
либо в 3-шкале (определенной ниже). Положительные
значения в герцах или 3 увеличиваются от ТМС на-
лево, отрицательные значения возрастают направо.1)
Посмотрим на шкалы ЯМР, приведенные на
рис. 3.18, и по договоренности поместим сигнал
° Эта договоренность, однако, послужила основанием для
конфликта с практикой на раннем этапе развития ЯМР,
когда увеличение напряженности магнитного поля (поле-
вая развертка) происходило слева направо при постоянной
частоте и напряженность магнитного поля сообщалась в
единицах резонансной частоты. Эта «сильнопольная раз-
вертка» означала, что резонансные частоты возрастают
слева направо в противоречии с принятой 3-шкалой.
Чтобы разрешить этот конфликт, была введена т
(тау)-шкала (т = 10-3), что нашло свое отражение в
литературе в период с конца 50-х и до 70-х г.г. XX в.,
в особенности в английской литературе. Следует тща-
тельно проверять химические сдвиги, опубликованные
в этот период, на соответствие шкал (см., например,
Bovey, 1967).
т-Шкала исчезла с развитием приборов с частотной
разверткой и импульсной фурье-спектроскопии ЯМР, ко-
торая по существу является мгновенной частотной «раз-
верткой». Термины «сильнопольный» и «слабопольный»
сейчас используются наравне с терминами соответственно
«экранированный» (более низкие значения 3 или справа
на шкале) или «дезэкранированный» (более высокие зна-
чения 5 или слева на шкале).
ТМС при нуле на правом краю шкалы. Химические
сдвиги можно сообщать в безразмерных единицах
(обозначаемых 3 или м.д.), начиная с нулевого хи-
мического сдвига для ТМС. Химические сдвиги не
зависят от рабочей частоты используемого спектро-
метра. Это достигается делением химического сдвига
сигнала, выраженного в герцах, на рабочую частоту
спектрометра, выраженную в мегагерцах. Допустим,
что мы имеем протонный сигнал на частоте 1200 от
ТМС (рис. 3.18) для частоты 300 МГц. Эта частота
будет соответствовать химическому сдвигу в безраз-
мерных единицах равному
1200 Гц
300Ю6Гц
= 54 или 4 м.д.
Если перейти к спектрометру с рабочей частотой
600 МГц (рис. 3.18), то теперь тот же протонный
сигнал будет находиться на частоте 2400 (от ТМС),
что соответствует тому же химическому сдвигу.
2400 Гц
600 Ю6 Гц
= 54 или 4 м.д.
Для растяжки шкалы химических сдвигов следу-
ет использовать максимально сильные доступные
магнитные поля. Это очевидно из рис. 3.18 и 3.19;
на последнем показано, как увеличение внешнего
магнитного поля при регистрации спектра ЯМР
акрилонитрила приводит к увеличению разделения
сигналов.
Для объяснения закономерностей химических
сдвигов весьма плодотворной является концепция
электроотрицательности заместителей (табл. 3.2).
Эта концепция говорит о том, что электронная плот-
ность вокруг протонов в метильных группах ТМС
высока (кремний менее электроотрицателен, чем угле-
3.4. Химический сдвиг
165
род), и эти протоны поэтому сильно экранированы
(табл. 3.3). Поскольку углерод более электроотрица-
телен, чем водород, сигналы поглощения протонов
групп СН4, RCH3, R2CH2 и R3CH будут располагаться
справа налево в спектре (см. приложение А, диа-
грамма А.1).
Используя концепции электроотрицательности
и кислотности протона, можно сделать ряд полез-
ных оценок химических сдвигов. Это демонстрируют,
например, разумные величины химических сдвигов,
приведенные в табл. 3.3, которые определены на осно-
вании только одной электроотрицательности.
Однако протоны ацетилена, сигнал которых рас-
положен при 1.80 м.д., оказываются более экраниро-
ванными, чем протоны этилена (8 5.25), т.е. выпадают
из этой закономерности; а тот факт, что альдегидный
протон в ацетальдегиде дает сигнал при 9.97 м.д.
определенно требует учета других факторов, помимо
электроотрицательности. Для объяснения этих и дру-
гих явных аномалий, таких как неожиданно большое
дезэкранирование в бензольном кольце (8 7.27 для
протонов в бензоле), мы будем использовать пред-
ставление о диамагнитной анизотропии химических
связей.
Начнем с ацетилена. Его молекула линейна, и трой-
ная связь симметрична относительно оси. Если эта
связь направлена вдоль внешнего магнитного поля,
Таблица 3.2. Электроотрицательность некоторых
элементов по Полингу
Н (2.1)
Li (1.0) Be (1.5) В (2.0) С (2.5) N(3.0) 0(3.5) F (4.0)
Na(0.9) Mg (1.2) Al (1.5) Si (1.8) P(2.1) S (2.5) Cl (3.0)
Br (2.8)
К2.5)
Таблица 3.3. Закономерные изменения химических
сдвигов протонов в соответствии с изменением
электроотрицательности
Соединение б Соединение б
(CH3)4Si 0.00 CH3F 4.30
(СН3)2О 3.27 RCO2H -10.80
то л-электроны связи могут циркулировать под пря-
мым углом к внешнему полю, и таким образом они
будут индуцировать свое собственное поле, направ-
ленное противоположно внешнему полю. Так как про-
тоны располагаются вдоль магнитной оси, силовые
линии магнитного поля, индуцированного циркули-
рующими электронами, будут действовать так, чтобы
60 МГц
симулированный
Нв Нх
\ /
с=с
н/ XCN
100 МГц
симулированный
6.4 6.3 6.2 6.1 6.0 5.9 5.8 5.7 5.6 5.5 5.4 5.3 м.д.
Рис. 3.19. Теоретически рассчитанные спектры ЯМР 1Н акрилонитрила на частотах
60,100 и 300 МГц; для сравнения приведен экспериментальный спектр (в CDCI3)
на рабочей частоте 300 МГц
166
3. Спектроскопия ЯМР 1Н
Силовые линии индуцированного
магнитного поля-^
Циркулирующие л-электроны
Рис. 3.20. Экранирование алкиновых протонов.
экранировать протоны (рис. 3.20), и сигнал ЯМР бу-
дет смещен вправо по сравнению с его положением,
предсказываемым на основе электроотрицательности.
Разумеется, только небольшая часть беспорядочно
движущихся молекул ориентирована вдоль магнитного
поля, но химический сдвиг, усредненный по всем мо-
лекулам, будет определенно испытывать воздействие
ориентированных таким образом молекул.
Этот эффект основан на диамагнитной анизотро-
пии, которая выражается в том, что экранирование и
дезэкранирование зависят от ориентации молекулы
по отношению к внешнему магнитному полю. Анало-
гичные аргументы могут быть привлечены для объ-
яснения неожиданного дезэкранирования альдегидного
протона. В этом случае эффект внешнего магнитного
поля максимален вдоль оси, перпендикулярной к оси
связи С=О (т.е. в плоскости страницы на рис. 3.21)
Геометрия такова, что альдегидный протон, который
находится над плоскостью страницы, оказывается в
дезэкранирующей части индуцированного магнитного
поля. Те же самые аргументы могут быть, по крайней
мере частично, привлечены для объяснения довольно
большого дезэкранирования алкеновых протонов.
Еще одним примером диамагнитной анизотропии
является так называемый «эффект кольцевого тока»,
который ответственен за сильное дезэкранирование
протонов бензольного кольца. Этот эффект проде-
Рис. 3.21. Дезэкранирование альдегидных протонов
Силовые линии
индуцированного
магнитного поля
Циркулирующие
электроны
Рис. 3.22. Эффекты кольцевого тока в бензоле
монстрирован на рис. 3.22. Из рисунка ясно также,
что протоны, расположенные над или под плоскостью
кольца, должны быть дополнительно экранированы. И
действительно, это было обнаружено для некоторых
из метиленовых протонов в 1,4-полиметиленбензолах.
Все кольцевые протоны ацетофенона дезэкрани-
рованы вследствие эффекта кольцевого тока. Кроме
того, ор/ио-протоны дезэкранированы в большей сте-
пени (мета, пара 8 -7.40; орто 8 -7.85) вследствие
дополнительного дезэкранирующего эффекта карбо-
нильной группы. На рис. 3.23 карбонильная связь
и бензольное кольцо компланарны. Если молекула
ориентирована таким образом, что внешнее магнит-
ное поле BQ перпендикулярно плоскости молекулы,
циркулирующие л-электроны связи С=О экраниру-
ют конические зоны выше и ниже этой связи и де-
зэкранируют боковые зоны, в которых находятся
ор/ио-протоны. Оба ор/ио-протона дезэкранированы
в одинаковой степени, так как можно нарисовать
другую, равнонаселенную конформацию, в которой
находящийся «по левую руку» ордио-протон попадает
в зону дезэкранирования конуса анизотропии. В ни-
тробензоле этот эффект проявляется еще сильнее.
Очень яркие примеры экранирования и дезэкра-
нирования за счет кольцевого тока дают некоторые
аннулены. При -60°С протоны снаружи от кольца
Рис. 3.23. Зоны экранирования (+)
и дезэкранирования (-) в ацетофеноне
3.4. Химический сдвиг
167
Рис. 3.24. Дезэкранирование экваториального
протона в жестком шестичленном цикле
[18]-аннулена сильно дезэкранированы (8 9.3), а вну-
три кольца - сильно экранированы (8 -3.0, т.е. более
экранированы, чем протоны ТМС).
[18]-Аннулен
Демонстрация такого кольцевого тока является хо-
рошим доказательством планарности и ароматичности,
по крайней мере при низких температурах. По мере
повышения температуры сигналы уширяются вследствие
медленных конформационных превращений. Пример-
но при ПО °C появляется единственный усредненный
сигнал приблизительно при 8 5.3, что обусловлено
быстрым взаимопревращением конформаций кольца,
которое приводит к усредненному химическому сдвигу.
В противоположность удивительным анизотропным
эффектам циркулирующих л-электронов, о-элекгроны
связи С-С в циклогексане производят небольшой
эффект. Например, ось связи С-С в циклогексане
является осью конуса дезэкранирования (рис. 3.24).
Это позволяет дать объяснение наблюдаемому фак-
ту: в жестком шестичленном цикле экваториальный
протон практически всегда сдвинут в область слабо-
го поля на 0.1-0.7 м.д. по сравнению с аксиальным
протоном при том же атоме углерода. Аксиальные
и экваториальные протоны при С] ориентированы
одинаково по отношению к связям С!~С2 и С2-С6,
но экваториальный протон находится внутри конуса
дезэкранирования связи С2-С3 (и С5-С6).
Большие таблицы и диаграммы химических сдви-
гов, приведенные в приложениях, позволяют сделать
полезный вывод: химические сдвиги протонов в ор-
ганических соединениях разбиваются, грубо говоря,
на 8 областей, как показано на рис. 3.25.
Алициклические соединения
р-Замещенные алифатические
соединения
Алкины
а-Монозамещенные
алифатические соединения
g-Дизамещенные алифатические соединения
Алкены
Ароматические и
гетероароматические соединения
Альдегиды
I I I I I I I I I I I
10 98765432108
м.д.
Рис. 3.25. Обобщенные области химических сдвигов. Некоторые альдегиды,
некоторые енолы и большинство карбоновых кислот дают сигналы при б > 10
168
3. Спектроскопия ЯМР 1Н
Чтобы продемонстрировать использование неко-
торых данных, приведенных в приложениях, пред-
скажем химические сдвиги протонов в бензилаце-
тате. В приложении А на диаграмме А. 1 находим
химический сдвиг группы СН3, составляющий 8
~2.0. Из таблицы Б.1 мы видим, что СН2-группа
дает сигнал при 8 -5.07. В приложении Г на диа-
грамме Г. 1 находим сдвиг ароматических протонов
при 8 -7.2. В спектре бензилацетата (рис. 3.26) на-
блюдаются 3 узких сигнала при (слева направо)
8 1.96,8 5.00 и 8 7.22; причем «ступени интеграла»
находятся в соотношении 3:2:5, что соответствует
группам СН3, СН2 и пяти кольцевым протонам. О)
Все сигналы являются синглетами. Это означает,
что группы СН3 и СН2 изолированы, т.е. нет про-
тонов при соседних атомах углерода, с которыми
они могли бы взаимодействовать (см. раздел 3.5).
Однако возникает проблема с кажущимся сингле-
том, относящимся к протонам бензольного кольца,
которые на самом деле, конечно, не являются хими-
чески эквивалентными (см. раздел 3.8.1) и должны
взаимодействовать друг с другом. При более высо-
ком разрешении мы увидели бы мультиплет, а не
кажущийся синглет. И действительно, на растянутой
вставке видны частично разрешенные сигналы.
Подчеркнем еще раз, что применение концепций
электроотрицательности (индуктивный эффект) и де-
локализации электронов в сочетании с представле-
ниями о диамагнитной анизотропии позволяет как
понять, так и приближенно предсказывать химиче-
ские сдвиги. Для иллюстрации ниже представлено
несколько примеров.
1. В а,р-ненасыщенном кетоне благодаря резонанс-
ному эффекту дезэкранирован р-протон.
/з / /
н3с-с /СН3 Н3С-С+ /СНз
р—с; <—> ,с=с;
/° Ч / \
но но-
а-протон 5 ~ 6.2
Р-протон 8 ~ 6.8
2. В замещенном виниловом эфире атом кислорода
дезэкранирует а-протон вследствие индуктивного
эффекта и экранирует p-протон вследствие резо-
нансного эффекта.
Н3С—С=С^О—СН
3 л |
н
а-протон 5 ~ 6.2
Р-протон 5 ~ 4.6
Приведенные выше приближенные вели-
чины были рассчитаны по данным прило-
жения Г. Для сравнения: алкеновые протоны
wpawc-гексена-З поглощают при 8 5.4.
3. Сдвиги орто-, мета- и лрря-протонов в заме-
щенных ароматических соединениях коррелиру-
ют с электронной плотностью и с направлением
атаки электрофильных реагентов (приложение Г,
диаграмма Г.1). Например, орто- и ияря-протоны
фенола экранированы вследствие высокой элек-
тронной плотности, которая также проявляется
в преобладании орто- и лрря-замещения при
взаимодействии с электрофильными реагента-
ми. И напротив, орто- и лрря-протоны в нитро-
бензоле дезэкранированы, причем ор/ио-протоны
в большей степени (см. рис. 3.23).
Поскольку инкременты химических сдвигов при-
близительно аддитивны, можно для расчета химиче-
ских сдвигов кольцевых протонов в полизамещенных
бензолах использовать величины инкрементов для
монозамещенных бензолов, приведенные в приложе-
нии (диаграмма Г.1). Инкременты химических сдвигов
для кольцевых протонов л/-диацетилбензола, напри-
мер, рассчитываются следующим образом.
«Ступеньки интеграла» - расстояния по вертикали меж-
ду горизонтальными линиями интегральной кривой - про-
порциональны числу протонов, представляемых каждым
данным отдельным сигналом поглощения (синглетом или
мультиплетом). Эти ступени пропорциональны площадям
соответствующих сигналов и дают только отношения, а
не абсолютные количества протонов в соответствующих
группах.
Инкременты химических сдвигов - это разность
химических сдвигов замещенного производного
и бензола (8 7.27). Таким образом, для заместителя
3.5. Спин-спиновое взаимодействие, мультиплеты, спиновые системы
169
Рис. 3.26. Спектр ЯМР 1Н бензилацетата в CDCI3 рабочая частота (300 Мгц)
СН3С=О (строка 26 диаграммы Г.1) opwo-инкремент
составляет +0.63, а мета- и ларя-инкременты имеют
величину +0.28 ( + означает сдвиг к более высоким
частотам по сравнению с 8 7.27). Протон С-2 име-
ет два орто-замести теля; протоны С-4 и С-6 эк-
вивалентны и имеют орто- и иоря-заместители.
Тогда инкремент для С-2 имеет величину +1.26,
для С-4 и С-6 +0.91 и для С-5 +0.56. Инкременты,
рассчитанные из экспериментального спектра, имеют
величины +1.13,+0.81 и +0.20 соответственно. Такое
согласие является вполне приемлемым0).
Бесспорно, что протонная спектроскопия ЯМР (как
и спектроскопия ЯМР 13С, см. гл. 4) является мощным
средством для распознавания типов замещения в аро-
матических производных. Другим мощным средством
является двумерная спектроскопия ЯМР (см. гл. 5).
3.5. Спин-спиновое взаимодействие,
мультиплеты, спиновые системы
3.5.1. Простые и составные мультиплеты
первого порядка
Мы получили набор сигналов поглощения, представля-
ющих протоны в различных химических окружениях,
° Расчеты для ортио-дизамещенных соединений дают
менее удовлетворительные результаты вследствие
пространственных и других взаимодействий между
оря?о-заместителями.
причем площадь под каждым сигналом (полученная
путем интегрирования) пропорциональна числу про-
тонов, которые он представляет. Теперь рассмотрим
спин-спиновое взаимодействие. Это явление может
быть описано как взаимодействие спинов протонов
через электроны связи. Согласно принципу Паули,
электроны, связывающие два ядра, спарены, т.е. их спи-
ны антипараллельны. В магнитном поле имеется опре-
деленная тенденция для каждого из ядер спаривать
свой спин со спином одного из связывающих электро-
нов таким образом, чтобы большинство из них были
антипараллельными; это соответствует устойчивому
состоянию. Обычно спин-спиновое взаимодействие
распространяется очень слабо далее трех связей, если
только это не напряженные циклы (такие как малые
циклы), мостиковые системы, делокализованные си-
стемы связей (в ароматических или ненасыщенных
структурах) или система из четырех связей, находя-
щихся в конфигурации W (раздел 3.14). Спин-спиновое
взаимодействие через две связи называется геминаль-
ным, через три связи - вицинальным’.
Н—С—Н Н—С—С—Н
Геминальное Вицинальное
спин-спиновое спин-спиновое
взаимодействие взаимодействие
через 2 связи (2J) через 3 связи (V)
Предположим, что два вицинальных протона нахо-
дятся в совершенно различном химическом окружении.
Каждый протон будет давать сигнал, и эти сигналы
будут расположены далеко друг от друга, однако спин
170
3. Отекпуюскопия ЯМР^7/
rh rS
Рис. 3.27. Спин-спиновое взаимодействие между
протонами с сильно различающимися химическими
сдвигами
Рис. 3.29. «Центр тяжести», а не линейная средняя
точка характеризует положение химического сдвига
(в случае «низких» значений отношения Av/J)
Рис. 3.28. Система из двух протонов со
спин-спиновым взаимодействием; разность
химических сдвигов последовательно уменьшается
от а к д при достаточно большой величине J (10 Гц)
каждого протона испытывает слабое воздействие двух
ориентаций другого протона, осуществляющееся че-
рез электроны связи, и поэтому сигнал поглощения
оказывается дублетом (рис. 3.27). Разность частот
в герцах между компонентами в дублете пропорцио-
нальна эффективности взаимодействия и характери-
зуется константой спин-спинового взаимодействия J,
которая не зависит от величины внешнего магнитного
поля то время как химические сдвиги обычно
покрывают интервал 3600 Гц при рабочей частоте
прибора 300 Мгц, величины констант спин-спинового
взаимодействия между протонами редко превышают
20 Гц (см. приложение Е).
Пока разность химических сдвигов в герцах (Av)
гораздо больше, чем константа спин-спинового взаимо-
действия (произвольно можно принять, что Av/J > 8),
в спектре проявляются простые сигналы с виде двух
дублетов. По мере того как отношение Av/J становится
меньше, дублеты приближаются друг к другу, интен-
сивность двух внутренних линий возрастает, а двух
внешних линий-уменьшается (рис. 3.28). Положение
химического сдвига для каждого из протонов не будет
уже соответствовать центру дублета, как на рис. 3.27,
а находится в «центре тяжести» (рис. 3.29) и опреде-
ляется без расчета с невысокой точностью.
Число связей между взаимодействующими ядрами (про-
тонами в данной главе) обозначается верхним индексом
при J. Например, для Н-С-Н это 2J, для Н-С-С-Н - 3J,
для Н“С=С—С-Н - 4J. Двойные и тройные связи рас-
сматриваются как одна связь.
3.5. Спин-спиновое взаимодействие, мультиплеты, спиновые системы
171
Рис. 3.30. Спин-спиновое взаимодействие между
группами СН и СН2 с различными химическими
сдвигами
Спектр (г) на рис. 3.28 (воспроизведен на рис. 3.29),
который состоит из двух дублетов с неравными ин-
тенсивностями линий, легко может быть ошибочно
принят за квартет (см. рис. 3.34). Однако для истин-
ного квартета увеличение рабочей частоты спектро-
метра не будет приводить к превращению его в два
дублета. Поскольку, по мере сближения химических
сдвигов, интенсивности внешних линий в спектре
будут продолжать уменьшаться, их интенсивность мо-
жет стать столь малой, что они станут незаметными,
и внутренние линии могут быть ошибочно приняты
за дублет. В пределе внешние линии исчезают и воз-
никает синглет, соответствующий двум протонам .
На этом этапе мы можем предсказать спектр про-
стого соединения, если нам известна его структу-
ра. Рассмотрим метановую и метиленовую группу
в соединении
OR
RO-C-CH2-Ph
Н
в котором единственный метановый протон нахо-
дится в совершенно ином окружении, чем два мети-
леновых протона. Оба протона группы СН2 равным
образом взаимодействуют с метановым протоном.
На рис. 3.30 мы видим триплет и дублет, далеко от-
стоящие друг от друга, с соотношением интегральных
интенсивностей 1:2 соответственно. Дублет появляет-
ся вследствие расщепления сигнала группы СН2 на
протоне СН. Триплет является результатом последо-
вательных расщеплений сигнала СН на двух про-
тонах группы СН2; сигналы перекрываются, так как
константы спин-спинового взаимодействия равны
(рис. 3.31).
Рис. 3.31. Триплет, обусловленный
последовательными расщеплениями
В простых мультиплетах первого порядка число
линий определяется числом взаимодействий с сосед-
ними протонами, имеющими одни и те же (или очень
слабо различающиеся) константы спин-спинового
взаимодействия. Соседними являются геминальные
или вицинальные протоны, т.е. протоны, отстоящие на
две или три связи; дальние константы спин-спинового
взаимодействия обычно гораздо меньше по величине
(см. раздел 3.14). Как мы уже видели, один протон
вызывает расщепление сигнала в дублет, два прото-
на с одинаковым спин-спиновым взаимодействием
приводят к триплету. Отсюда мультиплетность равна
п + 1, где п - число соседних протонов, имеющих оди-
наковые константы спин-спинового взаимодействия
с данным протоном. Общая формула, справедливая
для подсчета числа линий при взаимодействии с п
ядрами со спинами I, записывается в виде Ini + 1,
где I - спин ядра (см. раздел 3.2.1). Относительные
интенсивности линий в простых мультиплетах первого
порядка также зависят от п. Мы видели, что в ду-
блете (п = 1) это соотношение равно 1:1, в триплете
(п = 2) - 1:2:1, в квартете (п = 3) - 1:3:3:1 и т.д.
Требования, которые должны быть выполнены, что-
бы наблюдался простой мультиплет первого порядка,
можно суммировать следующим образом.
• Отношение Av/J должно быть больше пример-
но 8; Av - расстояние в герцах между средними
точками мультиплетов, связанных спин-спиновым
172
3. Спектроскопия ЯМР JH
п Мультиплетность Относительная интенсивность Спины
Ъш расщепления
О Синглет (s)
1
1 Дублет (d)
2 Триплет (t)
3 Квартет (q)
4 Квинтет
U/4. .6./4. Л.
5 Секстет
6 Септет
R ^6. Л5^2(к Л
7 Октет
8 Нонет 1 8 28 56 70 56 28 8 1
10. ^5
n=l | ! t
I
III : tit
III til • ttl ttt
Рис. 3.32. Треугольник Паскаля. Относительные интенсивности линий
в мультиплетах первого порядка; л - число соседних ядер со спином !6
(например, протонов), имеющих одинаковые константы спин-спинового
взаимодействия
взаимодействием, J - константа спин-спинового
взаимодействия.
• Число линий в мультиплете равно п + 1 ,где п -
число соседних протонов, имеющих одинаковые
константы спин-спинового взаимодействия с дан-
ным протоном.
• Расстояния в герцах между линиями простого
мультиплета первого порядка равны константам
спин-спинового взаимодействия.
• Простые мультиплеты первого порядка симме-
тричны относительно центра, и наиболее интен-
сивная линия (линии) находятся в центре (см.
треугольник Паскаля, рис. 3.32).
Сложный мультиплет первого порядка отличается
от простого мультиплета первого порядка тем, что он
содержит несколько констант спин-спинового взаи-
модействия. Требование «Av/J должно быть больше
примерно 8» остается справедливым, однако соотно-
шение интенсивностей линий в сложном мультиплете
первого порядка не подчиняется треугольнику Па-
скаля. Пример такого мультиплета будет представлен
далее на рис. 3.37, где на растянутых участках спектра
видно, что мультиплет состоит из квартета дублетов;
представление этого мультиплета в виде «палочек»,
показанное в тексте, состоит из последовательности
двух простых мультиплетов. Впрочем, необходима
некоторая осторожность с этими «палочковыми»
представлениями. Обсуждение на более сложном
уровне проведено в разделе 3.5.5
После приобретения некоторого опыта становит-
ся очевидным, что правило отношения Av/J = 8 не
является строгим, однако по мере уменьшения этого
отношения интерпретация мультиплета становится
все более приблизительной. Гонка за мощностью,
упомянутая в разделе 3.3, чрезвычайно успешно
служила (и служит) увеличению этого отношения.
В результате упрощаются спектры, а следовательно,
появляется возможность идентификации более слож-
ных молекул. Этот эффект наглядно продемонстри-
рован на рис. 3.33, где показано изменение спектра
при увеличении рабочей частоты спектрометра от
60 до 300 и далее до 600 МГц. Исследовали сле-
дующее соединение:
С1-СН2-СН2-О-СН2-СН2-С1
На частоте 600 МГц спектр состоит из двух про-
стых триплетов первого порядка. На частоте 300 МГц
происходит некоторое возмущение спектра, и в нем
появляется несколько слабых дополнительных линий.
На частоте 60 МГц сигналы с очевидностью пере-
крываются, и появляются дополнительные линии.
(Совмещение растянутых мультиплетов возможно
потому, что во всех спектрах использовалась 8-шкала.)
Отношение Av/J составляет 12 при 600 МГц (бес-
спорно, спектр первого порядка), 6 при 300 МГц
(еще легко распознается спектр первого порядка,
3.5. Спин-спиновое взаимодействие, мультиплеты, спиновые системы
173
3.95 3.90 3.85 3.80 3.75 3.70 3.65 3.60 3.55 3.50 3.45 м.д.
Рис. 3.33. Спектры ЯМР ’Н ди(хлорэтилового) эфира в CDCI3,
зарегистрированные на рабочих частотах 60 (а), 300 (6) и 600 МГц (в)
если пренебречь дополнительными расщепления-
ми). На частоте 60 МГц перекрывание мультипле-
тов и появление дополнительных линий описывается
как возмущения «более высокого» (чем первый) по-
рядка. Если обратиться к приложению А, то видно,
что заместители С1 и OR обладают практически оди-
наковым дезэкранирующим эффектом, что и делает
регистрацию спектра на приборе с частотой 60 МГц
рискованным предприятием. В качестве еще одного
примера влияния частоты спектрометра рекомендуем
обратиться к рис. 3.19, где приведены теоретически
рассчитанные спектры акрилонитрила для трех раз-
личных частот.
3.5.2. Спиновые системы первого порядка
Спиновая система состоит из мультиплетов ядер,
которые взаимодействуют друг с другом и не взаи-
модействуют с ядрами вне этой спиновой системы.
При этом нет необходимости, чтобы каждый муль-
типлет внутри системы обнаруживал спин-спиновые
взаимодействия с каждым из остальных мультипле-
тов. Спиновая система изолируется от других спи-
новых систем гетероатомами или четвертичными
атомами углерода (атомами углерода, к которым не
присоединены атомы водорода). Мультиплеты могут
иметь вид простых мультиплетов первого порядка,
составных мультиплетов первого порядка или муль-
типлетов более высокого порядка. В спиновой системе
через изолирующие атомы могут проявляться дальние
спин-спиновые взаимодействия, однако константы
этого взаимодействия обычно малы, что приводит
только к уширению линий.
Спектр на частоте 600 МГц (рис. 3.33) состоит
из простой спиновой системы первого порядка.
Аналогичный спектр с небольшими возмущениями
мы видим и на частоте 300 МГц. Спектр на частоте
60 МГц соответствует спиновой системе более вы-
сокого порядка. Спектры сложных спиновых систем
могут состоять из одного или более мультиплетов
высокого порядка; в этом случае трудно определить
все величины 8, константы спин-спинового взаимо-
действия и мультиплетности при простом рассмо-
трении спектра.* 2)
Следует отметить, что здесь дано весьма упрощенное
толкование сложных спектров ЯМР. Существует полная и
точная теория спектров ЯМР, основанная на технике спи-
нового гамильтониана (см. например, книги Дж. Попла и
др., X. Гюнтера и Н. М. Сергеева), позволяющая провести
расчет спектра любой сложности. - Прим, перев.
2) Константы спин-спинового взаимодействия могут быть
положительными и отрицательными. Впрочем, знак кон-
станты не проявляется в спектрах первого порядка, поэто-
му мы не будем учитывать знаки констант при анализе
таких спектров.
174
3. Спектроскопия ЯМР 1Н
Можно отметить, что химические сдвиги обычно
приводят с двумя знаками после запятой. Всегда ли,
однако, эти цифры достоверны? Это, разумеется, почти
всегда так для рабочей частоты 600 МГц, как для
триплетов первого порядка на рис. 3.33. При частоте
60 МГц, однако, химические сдвиги перекрывающихся
мультиплетов не могут быть точно измерены при
простом осмотре спектра. При рабочей частоте между
300 и 60 МГц попытки измерить химические сдвиги
родственных соединений позволяют получить только
приближенные оценки, но не величины с точностью
до сотых.
3.5.3. Номенклатура Попла для спиновых систем
Чтобы формализовать описание спектров сложных
спиновых мультиплетов, Дж. Попл ввел номенклатуру
спиновых систем1). Мультиплеты отождествляются
с «группой ядер», каждая из которых обозначается
заглавной буквой алфавита. Если отношение Av/J до-
статочно велико, мультиплеты рассматриваются как
хорошо разделенные (а взаимодействующие группы
ядер как слабо связанные); тогда результирующая
спиновая система обозначается буквами алфавита,
далеко отстоящими друг от друга, например АХ. Если
это отношение невелико, используют набор близких
букв алфавита, например АВ. Если три ядра слабо
взаимодействуют между собой, то они обозначаются
буквами АМХ. Если между первыми двумя ядрами
связь сильная, а третье ядро слабо связано с первым
двумя ядрами, спиновая система обозначается как
АВХ. Число ядер в группе обозначается нижним ин-
дексом; причем если группа состоит только из одного
ядра, подстрочный индекс не используется.
Любая группа ядер, изолированная от всех других
групп, является спиновой системой. Вот некоторые
примеры спиновых систем первого порядка: АХ (два
дублета), А2Х (дублет, триплет), А2Х2 (два триплета),
А3Х2 (триплет, квартет). Почему это так - очевидно.
Например, в спиновой системе А2Х группа А2 должна
расщепляться в мультиплет из (п + 1) линий (где
п - число соседних ядер: поскольку в группе X одно
ядро (п = 1), сигнал А расщепляется в дублет. Соот-
ветственно, два протона в группе А2 обусловливают
появление триплета для сигнала ядра X.
В случае двух или большего числа групп ядер
возникают усложнения, связанные с наличием не-
скольких констант спин-спинового взаимодействия.
Так, нитропропан может быть описан как спиновая
1) Дж. Попл, В. Шнейдер, Г. Бернстейн. Спектры ядерно-
го магнитного резонанса высокого разрешения. М.: Мир,
1962.
система А3М2Х2, поскольку влияние группы NO2 при-
водит к хорошему разделению трех групп ядер:
СН3—СН2—СН2—NO2
Аз М2 Х2
Группы А и X взаимодействуют с группой М,
но группа А не взаимодействует с группой X. Этот
случай анализируется в разделе 3.8.1.1 после изло-
жения необходимого материала. В данный момент
мы только отметим, что спектр состоит из двух три-
плетов с несколько различающимися константами
спин-спинового взаимодействия и секстета со слегка
уширенными линиями.
В молекуле стирола
все винильные протоны образуют слабо связанную
систему (для частоты 600 МГц, см. раздел 3.10).
Как было упомянуто выше, близко расположенные
в алфавите буквы используются для описания спи-
новых систем не первого порядка: АВ, А2В, АВС,
А3В2С2 и т.д. Эти спиновые системы относят к систе-
мам высщих порядков, и их нельзя интерпретировать
при простом обзоре спектра.
Кроме этих спиновых систем есть еще системы,
содержащие «магнитно неэквивалентные» протоны.
Они достаточно распространены и характеризуются
одной неприятной особенностью: они не могут стать
системами первого порядка при любом усилении
магнитного поля. Такие спиновые системы обсуж-
даются в разделе 3.12.
Имеются коллекции расчетных спектров, которые
можно использовать для интерпретации сложных спек-
тров первого порядка (см. Wiberg, 1962; Bovey, 1988).
Спектры, соответствующие этим спиновым системам,
можно также воспроизвести с помощью специальных
программ либо на автономных компьютерах, либо
непосредственно с помощью компьютеров, входящих
в комплектацию современного спектрометра ЯМР
(см., например, программу NMRSIM, поставляемую
фирмой Bruker BioSpin).
3.5.4. Другие примеры простых спиновых
систем первого порядка
Для большего понимания простых спиновых систем
первого порядка в рамках номенклатуры Попла мы
рассмотрим следующие примеры.
3.5. Спин-спиновое взаимодействие, мультиплеты, спиновые системы
175
тмс
I.............г
7 6 5 4 3 2 1 0 м.д.
Рис. 3.34. Спектр ЯМР 1Н этилбензола в CDCI3 при рабочей частоте 600 МГц.
Этильному фрагменту соответствуют триплет группы СН3 и квартет группы СН2
Спектр этилбензола, (рис. 3.34) состоит из двух
спиновых систем, которые изолированы друг от друга
четвертичным атомом углерода, не имеющим при себе
протонов (атом Са ароматического кольца). Спиновая
система, соответствующая этильной группе СН3СН2,
состоит из хорошо разделенных триплета и квартета;
это спиновая система типа А3Х2, как это следует из
мультиплетности сигналов и соотношения интеграль-
ных интенсивностей трех групп сигналов 5:2:3 (слева
направо), где к этильной группе относятся два сигнала
в сильном поле (в правой части спектра). Большая
величина отношения Av/J обусловлена дезэкранирую-
щим эффектом бензольного кольца, в результате чего
сигнал группы СН2 достаточно сильно смещается
в слабые поля.
Бензольное кольцо, которое имеет ось симметрии,
содержит два opwo-протона, два .медиа-протона и один
лара-протон, и сигналы всех этих протонов находятся
в области, характерной для поглощения протонов
бензольного кольца. Внимательный студент, следуя
концепции эквивалентности химических сдвигов (см.
раздел 3.8.3) и описанию спиновых систем в номен-
клатуре Попла (раздел 3.5.3), описал бы эту систе-
му как А2В2С. Этот студент мог бы пойти дальше
и предсказать, что при еще более высоких полях
получилась бы спиновая система первого порядка
А2М2Х. И действительно, в более высоких полях
будет увеличиваться разность химических сдвигов
протонов кольца (в Гц), но, как будет объяснено
в разделе 3.9, все равно не удастся получить спектр
первого порядка. Коротко говоря, проблема состоит
в том, что ни орто-, ни лдедиа-протоны не являются
«магнитно эквивалентными».
Можно отметить одну не очень значительную,
но часто встречающуюся особенность. Обратите
внимание на небольшое уширение линий кварте-
та. Это уширение является результатом небольшо-
го спин-спинового взаимодействия через «изоли-
рующий» углеродный атом; т.е. существует очень
маленькая дальняя константа спин-спинового
взаимодействия (между протонами группы
СН2 и ордио-протонами кольца), которую можно
попытаться обнаружить при записи спектра с бо-
лее высоким инструментальным разрешением (т.е.
с меньшей шириной спектральной линии).
Спектр изопропильной группы в изопропилбензоле
на рис. 3.35 соответствует простой спиновой системе
первого порядка. Сигнал метанового протона имеет
вид септета вследствие спин-спинового взаимодей-
ствия с шестью эквивалентными соседними про-
тонами двух групп СН3, и все эти константы имеют
одну и ту же величину. Две химически эквивалентные
(вследствие свободного вращения) группы СН3 дают
дублет интенсивностью в шесть протонных единиц
вследствие взаимодействия с единственным мета-
новым протоном. Интерпретация спектра кольцевых
протонов имеет те же особенности, что и в случае
этилбензола (см. рис. 3.34).
176
3. Спектроскопия ЯМР ?//
7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 м.д.
Рис. 3.35. Спектр ЯМР ]Н кумола (изопропилбензола) в CDCI3 при рабочей частоте
300 МГц. Изопропильный фрагмент легко распознается по характерному дублету
(шесть протонов от двух групп СН3) и септету метинового протона
3.5.5. Анализ спектров первого порядка
Обычные «линейчатые диаграммы» являются удоб-
ным средством для интерпретации, если мы владеем
какой-либо информацией о спектральных параметрах.
Однако спектр первого порядка, который получается
в результате взаимодействия одного протона с груп-
пой СН3 (J = 7 Гц) и еще с двумя различными прото-
нами с различными константами спин-спинового взаи-
модействия (J = 4 Гц и J= 12 Гц) (рис. 3.36), было бы
трудно расшифровать с помощью «линейчатой диа-
граммы», если мы предварительно не знаем констант
спин-спинового взаимодействия. Манн1) предложил
рутинную процедуру для построения таких «линей-
чатых диаграмм». Ниже приводится краткое, слегка
модифицированное описание этой процедуры для
анализа спектра, возникающего при взаимодействии
одного протона с тремя указанными выше группами
протонов.
1. Запишем интенсивности сигналов, выразив их
целыми числами непосредственно под центром
каждого сигнала спектра, приняв интенсивность
самой крайней линии за 1. В спектрах первого
порядка интенсивности должны распределяться
° См., например, Mann В.Е., J. Chem. Educ., 1995, 72, 614.
симметрично относительно центра (уровень а).
Если это не так, то это спектр высшего порядка.
2. Сумма интенсивностей линий на уровне а, долж-
на быть равна 2", где п - число протонов, кото-
рые связаны с данным протоном спин-спиновым
взаимодействием. В данном примере п = 5,
что соответствует взаимодействию с тремя эк-
вивалентными протонами и двумя неэквивалент-
ными соседними протонами, как было сказано
выше.
3. Нарисуем вертикальные линии (отрезки прямых)
непосредственно под центром каждой линии
спектра, т.е. под каждой величиной интенсив-
ности. Все эти вертикальные линии рисуются
сейчас равной интенсивности (уровень б на
рис. 3.36) .
4. Числа интенсивностей двух крайних линий на
каждом конце спектра определяют мультиплет-
ность на уровне б. В настоящем примере соот-
ношение интенсивностей 1 : 1 свидетельствует
о присутствии серии дублетов. Соотношение ин-
тенсивностей 1 : 2 говорило бы о присутствии
серии триплетов 1:2:1, 1:3 - серии квартетов
1:3:3:1 и т.д. согласно треугольнику Паскаля
(см. рис. 3.32). Отметим, что некоторые дублеты
3.5. Спин-спиновое взаимодействие, мультиплеты, спиновые системы
177
Рис. 3.36. Константы спин-спинового взаимодействия в мультиплете первого
порядка определяются из дублетов с расщеплениями 4 и 12 Гц и двух квартетов
с расщеплениями 7 Гц
имеют интенсивности 3:3, но отношение остается
по-прежнему 1:1.
5. Расстояние в герцах между двумя первыми ли-
ниями на уровне б дает величину константы
спин-спинового взаимодействия для этого уров-
ня. Все дублеты, разумеется, имеют одну и ту же
константу спин-спинового взаимодействия, со-
впадающую с расщеплением для концевой пары
сигналов. Хотя имеется перекрывание дублетов,
не составляет никакого труда отсортировать их,
руководствуясь равными константами и соотно-
шением 1:1 парных линий. В соответствии с мас-
штабом шкалы определяем, что расстояние между
линиями дублетов составляет 4 Гц.
6. На уровне в объединим дублеты (4 Гц) уров-
ня б «стяжками» и припишем этим «стяжкам»
интенсивности, равные интенсивностям первой
линии каждой пары на уровне б. Эти «стяжки»
становятся теперь линиями мультиплетов уров-
ня в: в данном примере двух перекрывающихся
квартетов (J = 7 Гц) в соответствии с соотно-
шением 1:3 первых двух линий. В свою очередь
«стяжкам» этих квартетов приписывается число
интенсивности первой линии каждого квартета.
«Стяжки» становятся теперь линиями мульти-
плета уровня г - в данном примере дублетом
(J = 12 Гц). Отметим, что на каждом уровне ли-
нии следует проверять на центросимметричность
интенсивностей и равные КССВ.
7. В итоге мы на самом деле находим все три кон-
станты спин-спинового взаимодействия и можем
теперь представить в точности характер муль-
типлетного расщепления. Взаимодействие с од-
ним из протонов дает дублет с константой J =
12 Гц. Каждая линия этого дублета расщепле-
на в квартет (J = 7 Гц) за счет взаимодействия
с вицинальными протонами группы СН3 на
уровне в. Каждая линия квартета далее расще-
плена в дублет за счет взаимодействия с другим
СН-протоном, давая набор дублетов (J = 4 Гц)
уровня б. Вся схема основывается на числах
интенсивностей первых двух линий и расстоя-
нии в герцах между этими линиями. Эти числа
интенсивностей определяют тип мультиплета на
каждом уровне линий; расстояния в герцах дают
величины КССВ на каждом уровне.
Следует отметить, что случайная вырожденность
линий-полное или частичное совпадение - могут
создать проблемы при отнесении чисел интенсив-
ности. Некоторые примеры даны в работе1). Отметим
различие между перекрыванием и совпадением ли-
ний. На рис. 3.36 дублеты перекрываются, но это не
вызывает никаких трудностей. На рис. 3.37 концевые
линии трех триплетов совпадают. Поэтому кажущиеся
интенсивности неправильны на уровне а и правильны
на уровне б. Ошибочность отнесения обнаруживается
потому, что две внешних линии с обеих сторон на-
l) Mann В Е J. Chem. Educ., 1995, 72, 614.
178
3. Спектроскопия ЯМР
Рис. 3.37. Совпадение линий, возникающее в триплете триплетов с КССВ 5 и 10 Гц
ходится в соотношении 1:2 и должны быть частью
триплета 1:2:1. После вычитания интенсивностей
крайних линий внешних триплетов из крайних ли-
ний центрального триплета получаем соотношение
интенсивностей 2:4:2 (вместо исходного 3:4:3)
З.б. Протоны у атомов кислорода,
азота и серы.
Обменивающиеся протоны
Протоны, присоединенные непосредственно к атомам
кислорода, азота или серы, отличаются от протонов,
присоединенных к атомам углерода, следующими осо-
бенностями.
1. Они способны к обмену.
2. Они способны к образованию водородной связи.
3. Атомы водорода, присоединенные к азоту, пре-
терпевают частичное или полное подавление
спин-спинового взаимодействия под влиянием
электрического квадрупольного момента ядра
14N, которое имеет спин, равный 1.
Диапазоны химических сдвигов таких протонов
приведены в приложении Д. Эти сдвиги сильно зави-
сят от концентрации, температуры и растворителя.
3.6.1. Протоны у атома кислорода
3.6.1.1. Спирты
Сигналы гидроксильных протонов в спиртах распола-
гаются между 8 ~0.5 и -4.0 в зависимости от кон-
центрации. Изменение растворителя и температуры
также влияет на положение сигнала.
Зависимость химических сдвигов от концентрации,
температуры и полярности растворителя объясняется
наличием в этих системах межмолекулярных водо-
родных связей. Водородное связывание уменьшает
электронную плотность вокруг гидроксильного про-
тона, и поэтому протонный сигнал смещается к более
высоким частотам. Уменьшение концентрации в не-
полярном растворителе приводит к разрыву таких
водородных связей, и сигнал появляется при более
низких частотах-молекулы спирта становятся менее
«полимерными». Увеличение температуры произво-
дит аналогичный эффект.
—О—Н— О—Н— О—Н—
I I I
R R R
Внутримолекулярные водородные связи в мень-
шей степени подвержены влиянию их окружения,
чем межмолекулярные. И действительно, сигнал
енольного гидроксильного протона в р-дикетонах,
например, очень слабо зависит от концентрации
или растворителя, хотя его можно сместить к бо-
лее низким частотам нагреванием. Спектроскопия
ядерного магнитного резонанса является мощным
методом изучения водородных связей.
Сигнал гидроксильного протона в этаноле обычно
выглядит как синглет вследствие быстрого протон-
3.6. Протоны у атомов кислорода, азота и серы. Обменивающиеся протоны
179
1110 1100 1090 1080 Гц
н3с-сн2-он
в CDC13
4.5 4.0 3.5 3.0 2.5 2.0 1.5
Рис. 3.38. Спектр ЯМР 1Н СН3СН2ОН в CDCI3 при рабочей частоте 300 МГц
после стояния в течение ночи на воздухе при комнатной температуре. Сигнал
СН2 уширен за счет остаточного спин-спинового взаимодействия с ОН, сигнал
которого также слегка уширен. Поглощенная влага приводит к увеличению
интенсивности сигнала гидроксильного протона
ного обмена (рис. 3.38). При обычных условиях, т.е.
под воздействием воздуха, света и водяных паров,
в дейтерохлороформе появляются кислотные примеси,
которые катализируют быстрый обмен гидроксильного
протона1). Протон уже не связан с атомом кислорода
индивидуальной молекулы достаточно долго, чтобы
проявилась его связь с метиленовыми протонами;
поэтому спин-спиновое взаимодействие не проявля-
ется. Протон ОН-группы проявляется в спектре как
синглет, протоны группы СН2 - как квартет, группы
СН3 - как триплет.
Скорость обмена можно уменьшить понижением
температуры, использованием разбавленных растворов
либо обработкой растворителя безводным карбонатом
натрия или безводным оксидом алюминия с последую-
Дейтерохлороформ (CDC13) в маленьких хорошо за-
крытых стеклянных емкостях фирмы Aldrich является
настолько чистым, что в спектре СН3СН2ОН, регистри-
руемом в течение нескольких часов, сигнал протона груп-
пы ОН выглядит как триплет. При выдерживании этого
образца в течение 24 ч на воздухе сигнал превращается
в синглет (рис. 3.38). Высокое разбавление, которое по-
зволяют использовать современные спектрометры, также
способствует сохранению вицинального спин-спинового
взаимодействия для ОН-протона.
щим фильтрованием через высушенную стеклянную
вату, помещенную в капилляр, непосредственно перед
регистрацией спектра. В этих условиях ОН-протон
взаимодействует с протонами группы СН2, и полез-
ная информация становится доступной: синглетный
сигнал ОН указывает на третичный спирт, дублет-
ный - на вторичный и триплетный - на первичный.
Использование сухого дейтерированного диметил-
сульфоксида (ДМСО-сЦ) или дейтероацетона приводит
к тому же эффекту, что и описанная выше процедура.
Кроме того, образование водородной связи между
растворенным веществом и растворителем вызывает
в этом случае смещение сигнала к более высоким
частотам и вследствие этого полезное разделение
с перекрывающимися сигналами других протонов
(рис. 3.39).
Из рис. 3.39 и приведенной далее схемы обра-
зования мультиплета очевидно, что протоны груп-
пы СН2 этанола взаимодействуют как с протоном
ОН-группы, так и с протонами СН3-группы. Схема
отражает некоторое перекрывание квартета дублетов.
КССВ протонов группы СН2 с протоном ОН равна
5 Гц, с протонами группы СН3 - 7 Гц. При изображе-
нии таких схем обычно лучше начинатьь с большей
константы.
180
3. Спектроскопия ЯМР ?//
Рис 3.39. Спектр ЯМР ’Н СН3СН2ОН, записанный в сухом
дейтеродиметил сульфокси де при рабочей частоте 300 МГц. Слева направо
расположены сигналы, относящиеся к группам ОН, СН2 и СН3. Маленький сигнал
при 2.5 м.д. относится к примеси flMCO-d5 в ДМСО-с16 (см. приложение Ж)
который далее при высоких скоростях обмена пре-
вращается в синглет (рис. 3.38).1}
Диол может давать два раздельных сигнала для
двух гидроксильных протонов; в этом случае ско-
рость обмена в герцах гораздо меньше, чем расстояние
в герцах между этими сигналами. По мере увеличе-
ния скорости обмена (например, при каталитическом
действии следов кислоты) два сигнала поглощения
уширяются и затем сливаются, давая один широкий
сигнал; в этой точке скорость обмена в герцах (k)
примерно равна удвоенному первоначальному рас-
стоянию между сигналами в герцах. По мере увели-
чения скорости обмена широкий сигнал сужается.
Относительное положение каждого сигнала зависит
от силы водородной связи каждого из гидроксиль-
ных протонов; пространственная затрудненность об-
разования водородной связи приводит к смещению
сигнала вправо.
При промежуточных скоростях обмена мульти-
плет гидроксила сливается, образуя широкий сигнал,
Вода как примесь может обменивать протоны с дру-
гими способными к обмену протонами, что приводит к
появлению усредненного сигнала, находящегося между
сигналами вовлеченных в обмен протонов.
3.6. Протоны у атомов кислорода, азота и серы. Обменивающиеся протоны
181
Спектр ПМР соединения, содержащего быстро
обменивающиеся протоны, может быть упрощен,
и сигналы обменивающихся протонов удалены просто
встряхиванием раствора с избытком тяжелой воды
(D2O) или регистрацией спектра в растворе в тяже-
лой воде, если соединение в ней растворимо. Сиг-
нал образующейся HDO обычно появляется между
4.5 и 5 м.д. в неполярных растворителях и вбли-
зи 3.33 м.д. в ДМСО (см. приложение Д). Раствор
в CDC13 или СС14 в ампуле для ЯМР с крышкой
можно сильно встряхнуть в течение нескольких се-
кунд с 1-2 каплями D2O и оставить смесь до четкого
разделения слоев (или центрифугировать). Верхний
водный слой не мешает съемке.
Ацетилирование или бензоилирование гидроксиль-
ной группы смещает сигнал протонов группы СН2ОН
первичных спиртов примерно на 0.5 м.д. влево, а про-
тонов группы СНОН вторичных спиртов примерно
на 1.0-1.2 м.д. Такое смещение подтверждает при-
сутствие первичного или вторичного спирта.
З.6.1.2. Вода
Помимо проблем, связанных с обменом, что уже
было рассмотрено выше, вода является неизбежной
примесью, которая подчиняется «закону бутерброда»
и накладывается на критически важные сигналы. Вода
в виде объемных образований (суспензированных ка-
пель или пленки на стенках ампулы) дает сигнал при
~4.7 м.д. в CDCI3 (сигнал смешанного изотопомера
воды HOD; он же возникает при экспериментах по
обмену, о которых рассказывалось в разделе 3.6.1.1).
Растворенная (мономерная) вода поглощает при
8 -1.55 в CDCI3 и может быть серьезной помехой
в критической области спектра в разбавленных рас-
творах^. Использование C6D6 (растворенная вода при
0.4 м.д.) снимает это осложнение. Таблица положений
сигнала воды в обычных дейтерированных раство-
рителях приведена в приложении 3.
3.6.1.3. Фенолы
Поведение фенольного протона напоминает поведе-
ние протона в молекуле спирта. Сигнал фенольного
протона обычно представляет собой острый синглет
(быстрый обмен, спин-спиновое взаимодействие от-
сутствует); который в зависимости от концентрации,
растворителя и температуры находится в диапазо-
не от 8 -7.5 до -4.0 и располагается обычно левее
сигнала спиртового протона. Карбонильная группа
в opwo-положении смещает сигнал фенольного ги-
дроксила в область примерно 10-12 м.д. вследствие
Webster F.X., Silverstein R. М. (1985) Aldrichimica Acta
18 (No 3), 58.
образования внутримолекулярной водородной связи.
Так, в о-гидроксиацетофеноне сигнал ОН-группы рас-
положен при 8 12.05 и практически не зависит от
концентрации. Гораздо более слабая внутримолекуляр-
ная водородная связь в о-хлорфеноле объясняет из-
менение его химического сдвига (8 -6.3 для 1 М рас-
твора и -5.6 при бесконечном разбавлении), которое
значительно по сравнению с о-оксиацетофеноном,
но мало по сравнению с фенолом.
3.6.1.4. Енолы
Обычное таутомерное равновесие кето-формы
и енольной формы в ацетилацетоне описано в разделе
3.8.3.1 (см. рис. 3.45). Енольная форма преобладает
над кето-формой при описанных условиях.
Обычно мы не записываем енольную форму для
ацетона или кето-форму фенола, хотя их крошечные
количества присутствуют в равновесии, однако обе
формы ацетилацетона обнаруживаются в спектре
ПМР, поскольку равновесие является достаточно
медленным в шкале времени ЯМР, и енольная форма
стабилизирована внутримолекулярной водородной
связью. Енольная форма ацетона и кето-форма фенола
не имеют такой стабилизации; кроме того, арома-
тическая резонансная стабилизация фенола сильно
благоприятствует енольной форме. Отметим сильное
дезэкранирование енольного протона на рис. 3.45.
(см.также приложение Д).
Обычно в спектрах ЯМР а-дикетонов, таких как
2,3-бутандион, обнаруживаются сигналы только
кето-формы. Однако, если енольная форма а-дикетона
стабилизирована водородной связью, как в приве-
денных ниже циклических а-дикетонах, в спектрах
ЯМР появляются сигналы только стабилизированной
енольной формы.
5 7.43 8 6.53
3.6.1.5. Карбоновые кислоты
Карбоновые кислоты в неполярных растворителях
существуют в виде стабильных связанных водородной
связью димеров даже при высоком разбавлении. Кар-
боксильный протон дает сигнал в характеристической
области -13.2-10 м.д, и его положение слабо зависит
от концентрации. Полярные растворители частично
182
3. Спектроскопия ЯМР 1Н
разрушают димер, что приводит к соответствующему
смещению сигнала.
Форма сигнала для данной кислоты при комнат-
ной температуре может изменяться от узкого сиг-
нала до очень широкого в зависимости от скорости
обмена. Карбоксильный протон обменивается с про-
тонами воды и спиртов (или гидроксильной группы
гидроксикислоты) достаточно быстро, чтобы давать
синглет, усредненное положение которого зависит
от концентрации. Тиольные и енольные протоны не
претерпевают быстрого обмена с карбоксильными
протонами, и поэтому наблюдаются индивидуальные
сигналы для каждого из соединений.
3.6.2. Протоны у атома азота
Преобладающий изотоп1) азота 14N имеет спин, равный
1, и в соответствии с формулой 21 + 1 сигнал протона,
присоединенного к атому азота или к атому углерода,
соседнему с азотом, должен быть расщеплен в три-
плет с равными интенсивностями. Имеются, однако,
два фактора, которые усложняют картину: скорость
обмена протона при атоме азота и электрический
квадрупольный момент ядра 14N (см. раздел 3.2.1).
Протоны у атома азота могут быть вовлечены
в быстрый, с умеренной скоростью или медленный
обмен. Если обмен быстрый, то происходит устра-
нение спин-спинового взаимодействия атома азота
с протоном (протонами) NH и протонами у соседних
атомов углерода. Сигнал протона NH представля-
ет собой в этом случае острый синглет, и соседние
СН-протоны также не расщепляются на протоне
NH. Такая картина наблюдается для большинства
алифатических аминов2).
При промежуточной скорости обмена NH
спин-спиновое взаимодействие протона с азотом
устраняется частично, в результате возникает ши-
рокий сигнал NH. Сигналы соседних СН-протонов
не расщепляются на протоне NH. Такая ситуация
характерна для N-метил-иа/га-нитроанилина.
° Спектры ЯМР на ядрах 15N обсуждаются в гл. 6.
2) Взаимодействие H-C-N-H в нескольких аминах наблю-
дали при условии полного удаления (с помощью сплава
Na-K) следов воды. Таким образом эффективно подавля-
ется протонный обмен в шкале времени ЯМР.
[K.L. Henold, Chem. Соттип., 1340 (1970).]
При малых скоростях обмена сигнал протона NH
остается широким, поскольку электрический квадру-
польный момент ядра азота индуцирует умеренно
эффективную спиновую релаксацию и, соответственно,
промежуточное время жизни спиновых состояний
ядра азота. Протон поэтому «видит» три спиновых
состояния ядра азота (спин равен 1), которые из-
меняются с умеренной скоростью, и протон отве-
чает тем, что дает широкий сигнал, который может
исчезнуть в шумах базисной линии. В этом случае
наблюдается спин-спиновое взаимодействие сосед-
них протонов с протоном NH. Такова картина для
пирролов, индолов, первичных и вторичных амидов
и карбаматов (рис. 3.40).
Отметим, что существует протон-протонное взаи-
модействие H-N-C-H через три связи (С-Н, C-N
и N-Н), а взаимодействие между ядром азота и про-
тонами у соседнего атома углерода пренебрежимо
мало. Протон-протонное взаимодействие наблюдается
только на сигнале, соответствующем водороду у атома
углерода, сигнал протона NH остается сильно уши-
ренным вследствие квадрупольного взаимодействия.
В спектре этил-М-метилкарбамата (рис. 3.40)
NH-протон дает широкий сигнал с центром около
4.70 м.д., а сигнал N-CH3 при 8 2.78 расщеплен в ду-
блет (J = ~5 Гц) на NH-протоне. Протонам этокси-
группы соответствуют триплет при 8 1.23 и квартет
при 8 4.14.
Протоны алифатических и циклических аминов
дают сигналы в области ~6 3.0-0.5; ароматические
амины поглощают в области ~8 5.0-3.0 в CDC13 ( см.
приложение Д), и поскольку в них может происходить
образование водородных связей, этот сдвиг зависит
от концентрации, растворителя и температуры. Про-
Таблица 3.4. Классификация аминов по
спектрам ЯМР растворов их аммониевых солей
в трифторуксусной кислоте
Тип амина- предшественника Структура аммониевой соли Мультиплетность сигнала метиленового фрагмента
Первичный c6h5ch2nh3+ Квартет (рис. 3.41)
Вторичный c6h5ch2nh2r+ Триплет
Третичный c6h5ch2nhr2+ Дублет
Источник: Anderson W.R., Jr., and Silverstein R.M., Anal. Chem.,
37, 1417 (1965).
3.6. Протоны у атомов кислорода, азота и серы. Обменивающиеся протоны
183
А
1250 1210 840 Гц 800 380 Гц 340
II
h3c-nh-c-o-ch2-ch3
6 5 4 3 2 1 м.д.
Рис. 3.40. Спектр ЯМР ]Н этил-М-метилкарбамата в CDCI3 при рабочей частоте
300 Мгц
тоны NH в амидах, пирролах и индолах дают сиг-
налы в области ~6 8.5-5.0; влияние концентрации,
растворителя и температуры на положение сигнала
обычно меньше, чем в случае аминов. Неэквивалент-
ность протонов у атома азота в первичном амиде
и метильных групп в М,М-диметиламидах вызва-
на медленным вращением вокруг связи N-C(=O)
вследствие вклада резонансной формы “O-C=N+
(раздел 3.8.3.2 и табл. 3.4).
Протоны у атома азота в солях аминов обменива-
ются с умеренной скоростью; они обнаруживаются
Рис. 3.41. Спектр ЯРМ ’Н а-метиленовой группы
первичного амина в CF3CO2H при рабочей
частоте 100 МГц (см. первую строку табл. 3.4)
как широкие сигналы ~8 8.5-6.0 и демонстрируют
спин-спиновое взаимодействие с протонами у со-
седних атомов углерода (J ~7 Гц).
Использование трифторуксусной кислоты как одно-
временно протонирующего агента и растворителя ча-
сто позволяет классифицировать амины на первичные,
вторичные или третичные. Это проиллюстрировано
в табл. 3.4, где показано, что число протонов у атома
азота определяет мультиплетность сигналов прото-
нов метиленовых групп в солях (рис. 3.41). Иногда
широкие сигналы +NH, +NH2 и +NH3 имеют вид трех
широких горбов. Эти горбы отражают расщепление
на ядре азота (J ~ 50 Гц). При хорошем разрешении
иногда можно наблюдать расщепление каждого из
горбов вследствие спин-спинового взаимодействия
с протонами у соседних атомов углерода (J ~ 7 Гц),
однако это расщепление легче наблюдать на более
острых сигналах протонов а-СН. Поведение прото-
нов фрагмента H-N-C-H суммировано в табл. 3.5Ч
Химические сдвиги некоторых классов протонов,
присоединенных к атому 14N, приведены в прило-
жении Д.
° С разрешения доктора Дональда Диттмера (Университет
Сиракуз).
184
3. Спектроскопия ЯМР 1Н
Таблица 3.5. Влияние скорости обмена протона NH на спин-спиновое взаимодействие
Скорость обмена NH
Быстрый Промежуточный Медленный
Влияние на сигнал N-H Острый синглет Широкий синглет Широкий синглет
Влияние на сигнал Спин-спиновое Спин-спиновое Есть спин-спиновое
с-н взаимодействие отсутствует взаимодействие отсутствует взаимодействие
3.6.3. Протоны у атома серы
Сульфгидрильные протоны (SH) обычно находятся
в состоянии медленного обмена, так что при комнат-
ной температуре обнаруживаются расщепления на этих
протонах с константой порядка 8 Гц. Эти протоны не
вступают в быстрый обмен с протонами гидроксильных,
карбоксильных или енольных групп той же или другой
молекулы, и таким образом, их можно видеть в виде от-
дельных пиков. Однако встряхивание раствора в тяжелой
воде в течение нескольких минут приводит к замещению
водородов сульфгидрильных групп на дейтерий. Диапазон
поглощения для алифатических сульфгидрильных про-
тонов составляет от 8 2.5 до 8 0.9, а для ароматических
сульфгидрильных протонов-от 8 3.6 до 8 2.8. Влияние
температуры, концентрации и типа растворителя изменяет
химические сдвиги внутри этих диапазонов.
3.6.4. Протоны вблизи от атомов хлора,
брома и иода
Сигналы этих протонов не обнаруживают явной
мультиплетности от взаимодействия с ядрами 35С1,
37С1,79Вг, 81Вг, 1271, так как последние имеют большие
квадрупольные моменты, что приводит к быстрой ре-
лаксации этих ядер и исчезновению соответствующих
спин-спиновых расщеплений. Например, в спектре
СН3СН2С1 наблюдаются узкие сигналы триплета (СН3)
и квартета (СН2) без каких-либо признаков дополни-
тельных расщеплений от ядер хлора.
В принципе при очень хорошем разрешении можно
наблюдать небольшие различия в химических сдвигах
протонов, расположенных через две связи от ядер хлора,
вызванные различиями колебательных характеристик изо-
топов 35С1 и 37С1 (см. F. A. L. Anet and М. Kopelevich, J.
Amer. Chem. Soc., 109, 5870 (1987)). Аналогичные слабые
изотопные сдвиги на ядрах 13С наблюдаются от пар изо-
топов 35С1,37С1 и 79Вг,81 Вг (см. W. Т. Raynes, М. Grayson,
N. М. Sergeyev and N. D. Sergeyeva, Chem. Phys. Letters,
226, 433 (1994); N. M. Sergeyev, P. Sandor, N. D. Sergeyeva
and W. T. Raynes, J. Magn. Res., Ser. A, 115, 174 (1995)). -
Прим, nepee.
3.7. Константы спин-спинового
взаимодействия протонов
с другими важными ядрами
(19F, D,31P,29Si и В * * * * 13 * * * * *С)
3.7.1. Константы взаимодействия протонов
с ядрами 19F
Поскольку ядро 19F имеет спин Vi, константы H-F
подчиняются тем же правилам мультиплетности,
что и константы Н-Н. В целом константы Н-F по-
крывают несколько больший диапазон, чем константы
Н-Н (см. приложение Е), и, кроме того, константы
Н-F могут распространяться через большее число
связей, чем константы Н-Н.
В спектре ЯМР !Н фторацетона (CH3)-C(=O)-CH2F
в CDC13 на частоте 300 МГц (рис. 3.42) группа СН3 об-
наруживается как дублет при 8 2.2 с небольшим рас-
щеплением (J 4.3 Гц), вызванным спин-спиновым
взаимодействием протонов с ядром 19F через четыре
связи (H-C-C(O)-C-F). Дублет при 8 4.75 (J48 Гц)
соответствует значительно большему геминальному
взаимодействию Н-F через две связи. Ядра 19F имеют
почти такую же чувствительность, как и ядра !Н,
и могут легко наблюдаться при соответствующих
частотах (см. раздел 6.3).
3.7.2. Константы взаимодействия протонов
с ядрами дейтерия
Атомы дейтерия (2Н или D) обычно вводят в мо-
лекулы, чтобы распознать отдельные группы или
чтобы упростить спектр ЯМР 1Н. Нужно также
учитывать, что в небольшом количестве дейте-
рий встречается в любом органическом соедине-
нии (природное содержание изотопа 2Н составляет
—0.015% ). Дейтерий имеет спин 1 и небольшой ква-
друпольный момент. Ядра дейтерия обнаруживают
спин-спиновое взаимодействие с протонами. Констан-
3.7. Константы спин-спинового взаимодействия протонов с другими важными ядрами (19F, D, 31Р, 29Si и 13С)
185
Рис. 3.42. Спектр ЯМР ’Н фторацетона в CDCI3 при рабочей частоте 300 МГц
ты J(H-D) примерно в 6.5 раз меньше соответствую-
щих констант J(H-H) (в соответствии с формулой
J(H-D) = (7d/Yh) ЛН-Н) = (1/6.5) J(H-H)).
Предположим, что протоны у а-атома углерода в ке-
тоне (заместители X и Y не содержат протонов)
О
у Р « ||
X—СН2— СН2—СН2— С—Y
замещены на дейтерий, что дает следующее соеди-
нение
О
у Д « ||
X—СН,—СНг— CD2— с—Y
Спектр недейтерированного соединения состоит
из триплета для а-протонов, квинтета для Р-протонов
(в предположении, что константы для Р-метиленовой
группы с а- и у-метиленовыми группами равны)
и триплета для у-протонов. Для дейтерированного
соединения сигнал для а-протонов вообще будет
отсутствовать, сигнал для Р-протонов при не очень
высоком разрешении будет выглядеть как слегка уши-
ренный триплет, а сигнал у-протонов не изменится.
Фактически при очень хорошем разрешении каждая
линия сигнала триплета Р-протонов будет выглядеть
как очень тесный квинтет с константой 3JHCCD равной
~1 Гц, поскольку величина мультиплетности равна
2п1 +1 = 2x2x1 + 1=5 (л-число ядер дейтерия,
взаимодействующих с Р-протонами).
Большинство коммерческих дейтерированных
растворителей имеют уровень дейтерирования
98-99%, поскольку этого достаточно для большин-
ства практических целей, а более высокий уровень
дейтерирования приведет к существенному удоро-
жанию продуктов. Например, дейтерированный ди-
метилсульфоксид (CD3)2S=O содержит в виде при-
меси соединение CD2H-(S=O)-CD3. Это соединение
обнаруживается в спектре ЯМР !Н в виде тесного
квинтета 1:2:3:2:1 (с константой J - 2 Гц ) в соот-
ветствии с правилом 2nl + 1 (см. приложение Ж).
Поскольку ядро дейтерия обладает электрическим
квадрупольным моментом, в спектре ЯМР на ядрах
2D наблюдают уширенные линии1).
В рутинной практике (для целей идентификации синте-
зированных соединений, анализа смеси продуктов и т.д.)
наиболее важное значение имеет спектроскопия ЯМР !Н.
Однако и спектры ЯМР 2Н даже при природном содер-
жании дейтерия находят важное применение в качестве
метода сертификации продуктов (вин, масел, соков и т.д.)
(см., например, обзор М. L. Martin, G.J.Martin. In NMR:
Basic Principles and Progress. Vol. 23, 1990. P. 1). - Прим,
nepee.
186
1 йтектроскопия ЯМР ’Я
3.7.3. Взаимодействие протонов с ядрами31Р
Ядро 31Р имеет 100%-ное природное содержа-
ние и спин !6. Правила расщеплений для взаимо-
действия ядер фосфора с протонами те же самые,
что и для протон-протонных взаимодействий. Кон-
станты спин-спинового взаимодействия велики
(Vp-h -200-700 Гц, 2«7НС-р 0.5-20 Гц) (см. приложе-
ние Е) и могут наблюдаться более чем через четыре
связи. Сигналы от ядер 31Р наблюдаются при соот-
ветствующих величинах частоты и напряженности
магнитного поля.
3.7.4. Взаимодействие протонов с ядрами 29Si
Активный в ЯМР изотоп 29Si имеет естественное со-
держание 4.7% и спин !6. Два других немагнитных
изотопа кремния (28Si и 30Si) имеют природное со-
держание 92.28 и 3.09% соответственно. Величина
2J(29Si~CH) составляет примерно 6 Гц. В спектре
соединений, содержащих группу Si-CH3 (например,
тетраметилсилана), наряду с основным интенсивным
синглетом, вызванным группами 28Si-CH3, можно ви-
деть дублет сигналов с небольшой интенсивностью,
обусловленный взаимодействием 29Si-CH3. Этот ду-
блет располагается на расстоянии ±3 Гц от централь-
ного сигнала. Можно также наблюдать «сателлитный»
дублет 13С-Н малой интенсивности на расстоянии
± 59 Гц (см. раздел 3.7.5). Сигналы от ядер 29Si на-
блюдаются при соответствующих величинах частоты
и напряженности магнитного поля (см. гл. 6).
3.7.5. Взаимодействие протонов с ядрами 13С
Изотоп |3С имеет естественное содержание 1.1%
и спин !4. Сигналы протонов, непосредственно связан-
ных с ядром 13С, расщепляются в дублет с большой
константой спин-спинового взаимодействия, равной
примерно 115-270 Гц для взаимодействия 13С-Н (кон-
станта через одну связь ^сн)- В группе СН3-СН2,
например, преобладает изотопомер 12СН3-12СН2, но со-
держится также небольшое количество изотопомеров
13СН3-12СН2 и 12СН3 -13СН2. Поэтому сигнал прото-
нов группы 13СН3 расщеплен в дублет в результате
взаимодействия с ядром 13С (константа J -120 Гц),
причем каждая линия дублета расщеплена в триплет
на протонах группы 12СН2 (J -1 Гц), как это показано
ниже на схеме. Эти «сателлиты 13С» («спутники 13С»)
имеют малую интенсивность вследствие малого со-
держания групп 13СН3; они обычно обнаруживаются
в виде сигналов, симметрично расположенных относи-
тельно интенсивного центрального сигнала 12СН3 (на-
пример, триплет 12СН3, показанный далее).
J13CH3-12CH2~7 Гц
J13СН3-12СН2 ~7 Гц
3.8. Эквивалентность по
химическому сдвигу
Понятие эквивалентности по химическому сдвигу
является центральным в спектроскопии ЯМР. В обо-
значениях Попла (см. раздел 3.5) ядра, имеющие оди-
наковый химический сдвиг, образуют группу в рас-
сматриваемой спиновой системе. Ядра с одинаковым
химическим сдвигом иногда называют изохронными
ядрами.
При рассмотрении структуры молекулы воз-
никает вопрос, эквивалентны ли входящие в ее
состав ядра по химическому сдвигу или нет (и
если ядра имеют одинаковый химический сдвиг,
то их объединяют в одну и ту же группу). Ответ
можно сформулировать следующим образом: ядра
эквивалентны по химическому сдвигу, если они
«взаимозаменяемы» (т.е переходят друг в друга
при выполнении операции симметрии или в ре-
зультате процесса быстрого химического обмена).
Это довольно широкое определение предполагает
ахиральное окружение (растворитель или реагент).
Ахиральность присуща большинству распростра-
ненных растворителей. Вообще под взаимозаме-
няемостью (или взаимоналагаемостью) двух мо-
лекул (или объектов) мы будем понимать полное
совпадение этих двух объектов друг с другом при
мысленном их совмещении.
Сначала мы рассмотрим операции симметрии,
а затем - быстрые процессы химического обмена
(раздел 3.8.3).
3.8.1. Определение эквивалентности
по химическому сдвигу при
использовании операций симметрии
Существует три типа операций симметрии, соотнося-
щихся с определенными элементами симметрии: вра-
щение вокруг осей симметрии п-го порядка (CJ, отра-
жение в плоскости симметрии (а) и инверсия в центре
3.8. Эквивалентность по химическому сдвигу
187
симметрии (i)'\ Говоря более строго, операции сим-
метрии можно разделить всего на два вида - Сп и Sn,
причем второй символ относится к вращению вокруг
зеркально-поворотной оси симметрии. Можно показать,
что операция Sj - это то же самое, что и операция ст,
а операция S2 - это то же самое, что и операция /; более
высокие порядки операции Sn (операции 53 и более вы-
сокие) встречаются очень редко. Мы будем использовать
операции С„, о и i. Нижние индексы показывают число
поворотов, необходимых для выполнения поворота на
360°. Таким образом, операция Q - это вращение на
360°, С2- на 180° и т.д. Обозначение 5j соответствует
повороту на 360° с последующим отражением в пло-
скости, перпендикулярной к оси, S2 - это поворот на
180° с последующим отражением и т.д.
Прежде чем рассмотреть, как три общих операции
симметрии влияют на обмен ядер внутри молекулы,
зададим другой вопрос. Как убедиться в том, что опе-
рации симметрии приводят к взаимному обмену по-
ложений ядер? Ответ достаточно прост. Постройте
две модели молекулы: одну перед проведением опе-
рации, а вторую - после проведения операции. Если
эти модели неразличимы, то рассматриваемые ядра
обмениваются своими местами, или, другими сло-
вами, эти ядра взаимозаменяемы. Затем переходите
к «трехмерным» изображениям или, еще лучше, -
к мысленному трехмерному представлению.
Конечно, операция идентичности (вращение на 360°
вокруг оси СО не имеет особого смысла. Элементы
симметрии - это оси, плоскости и центры.
3.8.1.1. Обмен позициями при вращении вокруг простой
оси симметрии (CJ
Протоны в дихлорметане (рис. 3.43, структура а) об-
мениваются позициями при повороте на 180° вокруг
простой оси симметрии лежащей в плоскости стра-
ницы. Такой обмен (С2 типа) показывает, что про-
тоны гомотопны. Они также обмениваются своими
положениями при проведении операции отражения
о (см. следующий раздел) в вертикальной плоскости,
перпендикулярной к плоскости листа, но операция
С2 младше (по рангу операций симметрии) и более
предпочтительна. Гомотопные протоны эквивалентны
по химическому сдвигу как в хиральном, так и в ахи-
ральном окружении, т.е. в растворах и при контакте
с реагентами. Рисунки и модели молекул неразли-
чимы до и после проведения операции симметрии.
При выполнении операции Сп молекула рассматрива-
ется как жесткий (точечный) объект, причем никаких
изменений в углах связей не допускается. В случае
° Операции симметрии должны относиться ко всей мо-
лекуле в целом.
операции С2 одна половина молекулы обменивается
местами с другой.
3.8.1.2. Обмен позициями при отражении
в плоскости симметрии (о)
Плоскость симметрии существует, если одна половина
молекулы является зеркальным отражением другой
половины. Протоны в хлорфторметане (рис. 3.43,
структура б) обмениваются местами только при от-
ражении в плоскости, проходящей через центральный
атом углерода вертикально к плоскости листа, других
элементов симметрии нет. Протоны являются зер-
кальным отражением друг друга (они энантиотопны),
причем эквивалентность по химическому сдвигу спра-
ведлива только для ахиральных растворителей или
при контакте с ахиральными реагентами. Хиральные
растворители или хиральные реагенты могут вызвать
различие в химических сдвигах для пары энантитоп-
ных протонов (см. раздел 3.17).
Метиленовые протоны пропионовой кислоты
(рис. 3.43, в) обмениваются местами при отраже-
нии в плоскости симметрии, которая совпадает
с плоскостью страницы. Других элементов симме-
трии нет. Протоны энантитопны и имеют одинако-
вый химический сдвиг только в ахиральных средах.
Рисунки и модели таких молекул неразличимы до
и после операции симметрии.
3.8.1.3. Обмен при инверсии в центре симметрии (i)
Центр симметрии существует в том случае, если пря-
мая, проведенная от данного ядра (или группы ядер)
через выбранный центр при продолжении пересекает
такое же ядро на том же расстоянии от центра. Такую
ситуацию мы встречаем в молекуле дифтордихлор-
циклобутана (рис. 3.43, г).
Инверсия эквивалента операции S2, состоящей из
поворота на 180° вокруг зеркально-поворотной оси
симметрии с последующим отражением в плоскости
симметрии, перпендикулярной к этой оси. Таким об-
разом, инверсия включает отражение, поэтому, строго
говоря, обменивающиеся ядра должны быть зеркаль-
ным отражением друг друга. Это условие не обяза-
тельно для ядер или для ахиральных групп, но оно
является обязательным при обмене позициями двух
хиральных групп. Таким образом, /^-группа обмени-
вается позицией с 5-группой, однако две /^-группы
(или две 5-группы) не обмениваются между со-
бой. Конечно, те же ограничения действительны
для обмена позициями при отражении в плоскости
симметрии (раздел 3.8.1.2). При выполнении обеих
операций обменивающиеся ядра или группы энан-
титопны и имеют одинаковые химические сдвиги
только в ахиральном окружении.
188
3. Спектроскопия ЯМР ]Н
Рис. 3.43. Примеры взаимного обмена протонов при действии операций
симметрии.
а - гомотопные протоны (операция поворота С2);
б, в - энантиотопные протоны (операция отражения о);
г - энантиотопные протоны (операция инверсии /);
д - диастереотопные протоны в хиральной молекуле (символ * обознает
хиральный центр) (см. раздел 3.12.1);
е, ж, з,и, к - диастереотопные протоны в ахиральных молекулах
(е - 3-гидроксиглутаровая кислота, ж - глицерин, з - лимонная кислота,
и - диэтилацеталь, к - циклический ацеталь бензальдегида, 2-фенил-1,3-диоксолан).
Смысл штрихов в обозначениях атомов Н объясняется в разделе 3.9
3.8.1.4. Обмен позициями при проведении операций
симметрии отсутствует
Если геминальные протоны в группе СН2 не об-
мениваются своими положениями при выполнении
операций симметрии, то такие протоны называются
диастереотопными. Каждый протон имеет свой хи-
мический сдвиг (исключая случайные совпадения).
Диастереотопные геминальные пары протонов связа-
ны друг с другом геминальными константами (через
две связи). В принципе, каждый из двух геминальных
протонов должен обнаруживать различные константы
спин-спинового взаимодействия с другими соседни-
ми ядрами (хотя это различие не всегда обнаружи-
мо). Группа СН2, состоящая из пары диастереотоп-
ных протонов, показана на рис. 3.43 (структура д),
хиральный центр отмечен звездочкой, но наличие
хирального центра не обязательно для появления
диастереотопии (см. рис. 3.43, структуры е-к). Ахи-
ральная молекула 3-гидроксиглутаровой кислоты
(рис 3.43, ё) имеет плоскость симметрии, перпенди-
кулярную к плоскости страницы и проходящую через
центральный атом углерода. Через эту плоскость два
протона На и два протона Нь обмениваются своими
позициями, и эти протоны энантиотопны. Поскольку
нет плоскости симметрии, проходящей между двумя
протонами каждой группы СН2, то протоны а и b в
каждой СН2-группе диастереотопны. Идеализирован-
ный спектр первого порядка для диастереотопных
протонов 3-гидроксиглутаровой кислоты представлен
ниже на схеме.
Следует обратить внимание на то, что константы
Jab и Jba соответствуют одной и той же геминальной
константе между диастереотопными протонами На
и Нь. В то же время константы Jac и Jbc соответ-
ствуют двум различным константам взаимодействия
протонов На и Нс, а также Нь и Нс соответственно.
3.8. Эквивалентность по химическому сдвигу
189
Рис. 3.44. Молекулы, содержащие метку: а - эквивалентные молекулы,
6 - энантиомеры, в - диастереомеры
Гомомерные модели
Гомотопные атомы
Энантиомерные модели
Энантиотопные атомы
Диастереомерные модели
Диастереотопные атомы
Приведенные на рис. 3.43 ахиральные молекулы
(3-гидроксиглутаровая кислота, глицерин, лимон-
ная кислота, диэтилацеталь, циклический ацеталь,
см. рис. 3.43 структуры е, ж, з, и, к соответственно)
содержат диастеротопные метиленовые протоны.
На основании приведенного выше обсуждения
можно сделать вывод, что диастереотопные протоны
нельзя поместить в одну группу, потому что они не-
эквивалентны по химическому сдвигу. Однако нередко
диастереотопные протоны проявляются как протоны,
эквивалентные по химическому сдвигу, при работе
с не очень высокими магнитными полями и при ис-
пользовании обычных растворителей. Однако такое
случайное совпадение химических сдвигов может
быть выявлено с помощью измерений с большими
частотами или при смене растворителя. Пример диа-
стереотопии метильных групп см. на рис. 3.55.
Диастереотопные протоны (или другие лиганды)
находятся в структурно эквивалентных позициях,
т.е они не различаются по характеру связывания (по
связанности). Например, в структуре д на рис. 3.43 ге-
минальные протоны одинаковы по характеру связы-
вания, но они не взаимозаменяемы (т.е не существует
такой операции симметрии, которая может переве-
сти их друг в друга). Таким образом, эти протоны
диастереотопны. В то же время протон при атоме
С-3 имеет характер связывания, отличный от такового
для протонов при атоме С-2, в этом случае термин
«диастереотопный» не используется.
Студенты из курсов стереохимии должны быть
знакомы с терминами для обозначения соотношений
между стереоизомерными молекулами: гомомерные мо-
лекулы (взаимоналагаемые молекулы), энантиомерные
молекулы (являющиеся зеркальными отображениями
друг друга), диастереомерные молекулы (стереоизоме-
ры, которые не являются зеркальными отображения-
ми друг друга). Эти знакомые термины аналогичны
введенным выше терминам «гомотопные», «энанти-
топные», «диастереотопные», которые используются
для ядер или групп ядер в молекуле.
3.8.2. Определение эквивалентности
химических сдвигов с помощью метки
(или замещения)
Как отмечалось в разделе 3.8.1, четкое понимание
концепции химической эквивалентности и ее связи
с элементами симметрии и операциями симметрии
является очень важным для интерпретации спек-
тров ЯМР. Одним из способов решения вопроса об
эквивалентности химических сдвигов в отдельной
группе ядер является введение «метки» или опера-
ции замещения1), которая заключается в следующем.
Рисуют два одинаковых изображения одного и того
же соединения. На одном из изображений для одного
из атомов водорода ставят метку (или замещают его
на какой-либо другой атом). На другом изображении
аналогичным образом помечают или замещают другой
атом водорода. Получаемые в результате изображения
(или модели) могут быть по отношению друг к другу
гомомерами, энантиомерами или диастереоомерами.
Атомы Н будут соответственно гомотопными, энан-
’) Ault. А.(1974) J.Chem. Educ., 51, 729.
190
3. Спектроскопия ЯМР
тиотопными или диастереотопными. Пример, приве-
денный на рис. 3.44, иллюстрирует этот метод.
В первом примере модели являются взаимона-
лагаемыми (т. е. гомомерами), во втором - не взаи-
моналагаемыми зеркальными отображениями (т. е.
энантиомерами), в третьем - не зеркальными отобра-
жениями (т. е. диастереомерами). Заметим, что метки
являются постоянными, т. е. Н и(н)- это различные
типы атомов. В другом подходе один из протонов
в каждой структуре может быть заменен на Z - лю-
бое ядро, не присутствующее в молекуле.
3.8.3. Эквивалентность химических сдвигов,
обусловленная быстрым
взаимопревращением структур
Если химические структуры могут претерпевать вза-
имное превращение, то результат будет зависеть от
температуры, катализатора, растворителя и концен-
трации. Рассмотрим четыре системы, предположив,
что концентрация фиксирована и катализатор отсут-
ствует.
3.8.3.1. Кето-енолыюе равновесие
Взаимопревращение таутомеров в ацетилацетоне
(рис. 3.45) при комнатной температуре является до-
статочно медленным, так что можно наблюдать сиг-
налы обеих форм, т.е. имеем два спектра. Константу
кето-енольного равновесия можно определить пу-
тем измерения относительных интегральных интен-
сивностей сигналов СН3 кето- и енольной формы.
При повышении температуры скорость взаимного
превращения увеличивается, что приведет к по-
лучению одного «усредненного» спектра. В этом
случае достигается эквивалентность химических
сдвигов для всех взаимопревращающихся прото-
нов. Отметим, что шкала времени ЯМР имеет тот
же самый порядок величины, что и выраженная
в герцах разность химических сдвигов превращаю-
щихся друг в друга протонов, т.е. величину порядка
10 -103 Гц. Процессы, протекающие с более высо-
кими скоростями, будут приводить к усредненным
сигналам. Отметим также, что енольный протон
ОН дезэкранирован по отношению к спиртовому
протону ОН вследствие того, что енольная форма
существенно стабилизирована внутримолекулярной
водородной связью
1.5 м.д.
Рис. 3.45. Спектр ЯМР 1Н ацетилацетона в CDCI3 при рабочей частоте 300 МГц
и 32°С. Соотношение енол - кетон определено с помощью интегрирования двух
сигналов СН3
3.8. Эквивалентность по химическому сдвигу
191
3.8.3.2. Взаимопревращение структур с «частично
двойной связью» (заторможенное вращение)
При комнатной температуре образец чистого ди-
метилформамида (CH3)2NC(=O)H дает два сигнала
СН3 (2.85 и 2.94 м.д.) вследствие малой скорости
заторможенного вращения вокруг «частично двойной
связи». При температуре около 123°С скорость обме-
на двух групп СН3 становится достаточно высокой,
чтобы произошло слияние этих двух сигналов.
3.8.3.3. Взаимопревращение в результате инверсии
циклов
При комнатной температура циклогексан существует
в виде быстро переходящих друг в друга взаимона-
лагаемых конформаций кресла.
В этих переходящих друг в друга структурах
аксиальный протон становится экваториальным
протоном (и наоборот), и спектр состоит из одного
«усредненного» сигнала. По мере понижения тем-
пературы сигналы уширяются, и при достаточно
низкой температуре появляются два сигнала: один,
соответствующий аксиальному протону, второй -
экваториальному. Иными словами, при комнатной
температуре аксиальный и экваториальный прото-
ны становятся химически эквивалентными вслед-
ствие быстрого взаимного превращения. При очень
низких температурах они становятся химически
неэквивалентными: действительно, в каждой «вы-
мороженной» кресловидной конформации протоны
в каждой из групп СН2 являются диастереотопны-
ми, однако при комнатной температуре скорость
инверсии кресла достаточно велика, и происходит
усреднение химических сдвигов этих геминаль-
ных протонов.
Метилциклогексан при комнатной температуре
существует как смесь быстро переходящих друг
в друга аксиального и экваториального конформе-
ров. Эти конформеры не взаимоналагаемы, и при
низкой температуре существует спектр каждого из
конформеров в отдельности.
В конденсированных циклогексановых кольцах,
например стероидах, циклы «заморожены» при ком-
натной температуре, и аксиальные и экваториальные
протоны в каждой из групп СН2 являются химически
неэквивалентными.
3.8.3.4. Вращение вокруг простой связи
в насыщенных цепях
Эквивалентность химических сдвигов протонов
СН3 возникает как следствие быстрого вращения во-
круг простой углерод-углеродной связи даже в отсут-
ствие элементов симметрии. На рис. 3.46, а показаны
ньюменовские проекции трех заторможенных ротаме-
ров молекулы, содержащей метильную группу, которая
присоединена к другому лр5-атому углерода, имеюще-
му четыре различных заместителя, т.е. к хиральному
центру. В каждом отдельном ротамере ни один из
протонов группы СН3 не может быть переведен в дру-
гой с помощью какой-либо операции симметрии (не
считая тривиальной операции CJ. Однако протоны
быстро меняют свои положения. Время пребывания
протона в каком-либо одном ротамере мало (порядка
10"6 с), поскольку энергетический барьер вращения
вокруг простой связи С-С невелик. Химический сдвиг
метильной группы является средним значением для
химических сдвигов трех протонов. Иначе говоря,
каждый протон может быть переведен в любой другой
с помощью операции вращения вокруг связи С-С.
Поэтому, если не метить протоны, ротамеры оказы-
ваются неразличимыми.
Таким же образом три вращающиеся метильные
группы >и/?ею-бутильной группы являются химиче-
ски эквивалентными за исключением редких случаев
пространственной затрудненности. Как метильная,
так и т/7вт-бутильная группы описываются моделью
симметричного волчка.
Заторможенные ротамеры 1 -бром-2-хлорэтана
(рис. 3.46, б), в принципе, являются разными струк-
турами, поскольку они несовместимы друг с дру-
гом. Однако в анюи-ротамере протоны На и Нь,
как и протоны Нс и Hd, химически эквивалентны
(энантиотопны) вследствие наличия плоскости сим-
метрии; в этом случае, следовательно, имеется два
набора энантиотопных протонов. В гоы/-ротамерах
элементы симметрии отсутствуют, и для любого из
гош-ротаметров протоны На и Нь, как и протоны Нс
и Hd, оказываются химически неэквивалентными (если
бы удалось выделить такой ротамер как отдельное
вещество). Однако вследствие быстрого взаимного
обмена между ротамерами происходит усреднение
химических сдвигов в обоих парах протонов (т.е
химические сдвиги для протонов На и Нь и для
протонов Нс и Hd будут попарно совпадать). Строго
говоря, в данном случае мы обсуждаем спиновую
систему АА'ХХ', однако, как будет объяснено в раз-
деле 3.9, ее можно приближенно рассмотреть как
систему А2Х2. Вообще говоря, если протоны могут
поменяться местами с помощью операции симметрии
в одном из ротамеров, то они оказываются химиче-
192
3. Спектроскопия ЯМР 7//
Рис. 3.46. а - Ньюменовская проекция заторможенных ротамеров молекулы,
содержащей метильную группу и асимметрический центр; б - 1-бром-2-хлорэтан;
в - 1-бром-1,2-дихлорэтан
ски эквивалентными (энантиотопными) вследствие
быстрого обмена за счет вращения1).
Рассмотрим метиленовую группу, соседнюю с хи-
ральным центром, как в 1-бром-1,2-дихлорэтане
(рис. 3.46, в). Протоны На и Нь химически неэкви-
валентны, поскольку их положения нельзя взаим-
но обменять ни в одном из конформеров; у моле-
кулы нет оси, плоскости, центра симметрии или
зеркально-поворотной оси. Хотя здесь и происходит
быстрое вращение вокруг простой связи углерод-
углерод, протоны группы СН2 не переходят при этом
друг в друга; и поэтому усредненные химические
сдвиги протонов На и Нь различаются. Наблюдатель
может заметить это различие до и после вращения
метиленовой группы - протоны в каждом из ротаме-
ров диастереотопны. Итак, в данном случае имеем
спиновую систему АВХ.
Обсуждение ротамеров частично заимствовано из ра-
боты [Silverstein R.M., and LaLonde R.T. (1980). J. Chem.
Educ., 57, 343].
3.9. Магнитная эквивалентность
(эквивалентность спин-спинового
взаимодействия)9
В дополнение к изложенным выше требованиям к спи-
новым системам первого порядка далее мы рассмо-
трим концепцию «магнитной эквивалентности» (или
«эквивалентности спин-спинового взаимодействия»)
и сравним ее с концепцией химической эквивалент-
ности (или эквивалентности химически сдвигов).
Если два химически эквивалентных ядра (например,
два протона) в одной группе ядер имеют одинаковые
константы спин-спинового взаимодействия с любым 2 * * * * *
2) Превосходное изложение вопроса о стереоизомер-
ных соотношениях групп в молекулах можно найти в
обзорах [Mislow К., and Raban М. (1967). In Topics in
Stereochemistry, Vol. I, p. 1; Jennings W.B. (1975) Chem.
Rev., 75, 307]. Эти авторы предпочитают термин «эквива-
лентность спин-спинового взаимодействия».
3.9. Магнитная эквивалентность (эквивалентность спин-спинового взаимодействия)
193
другим протоном в другой группе химически экви-
валентных ядер, то эти ядра будут также «магнит-
но эквивалентными», и к ним применимы обычные
обозначения Попла: А2, В2, Х2 и т.д. В противном
случае эти ядра будут магнитно неэквивалентными
и будут обозначаться следующим образом: АА', ВВ',
XX' и т.д. Другими словами, два химически эквива-
лентных протона являются магнитно эквивалентными,
если они расположены симметрично по отношению
к каждому протону спиновой системы. Магнитная
эквивалентность, безусловно, предполагает наличие
химической эквивалентности. Иначе говоря, не сле-
дует проверять ядра на магнитную эквивалентность,
если они не являются химически эквивалентными.
Часто встречающаяся магнитная неэквивалент-
ность в ароматических кольцах зависит от числа,
типа и расположения заместителей.
Рассмотрим протоны в иорл-хлорнитробензоле
(рис. 3.47). В нем имеется ось симметрии, проходя-
щая через заместители, вследствие чего возникают две
группы химически эквивалентных протонов, которые
мы можем предварительно обозначить как А2 и Х2.
Однако два протона в группе А2 (как и в группе Х2)
не являются магнитно эквивалентными, и коррект-
ным обозначением для них будет соответственно АА'
и XX'. Это спиновая система не первого порядка,
в обозначениях Попла она записывается как АА'ХХ'.
В мультиплетах, соответствующих таким спиновым
системам, не выполняются правила треугольника Па-
скаля для соотношения интенсивностей в мультипле-
тах первого порядка (см. рис. 3.32), и расстояния (в
герцах) между линиями не соответствуют константам
спин-спинового взаимодействия. Такие спектры не ста-
новятся спектрами первого порядка при повышении
напряженности магнитного поля. Как обозначить такую
спин-систему - АА'ХХ' или АА'ВВ', - определяется
отношением Av/J (где Av - химический сдвиг между
ядрами А и X (или В), a J- некая средняя константа
спин спинового взаимодействия между ядрами групп
А и X (или В). При низких магнитных полях эти
спектры становятся «обманчиво простыми». В эпоху
распространения спектрометров на 60 МГц студенты
учились распознавать иарл-дизамещение по картинке,
напоминающей спектр системы АВ (четыре главных
линии, см. рис 3.28). Симметричное ppwo-дизамещение,
как в о-дихлорбензоле, также приводит к четырех-
спиновой системе типа АА'ХХ' или АА'ВВ' (т. е.
имеет место магнитная неэквивалентность для двух
пар протонов), однако спектр выглядит существенно
сложнее (рис. 3.48) и не напоминает спектр системы
АВ. Аналогичные закономерности свойственны и для
гетероароматических колец.
Три изомерных дифторэтилена (1,1-дифторэтилен,
1,2-г/мс-дифторэтилен и 1,2-тиранс-дифторэтилен)
дают еще один пример химически эквивалентных,
но магнитно неэквивалентных ядер. Для всех трех
изомеров четыре ядра описываются спиновыми си-
стемами типа АА'ХХ'.
1,1 1,2-z/wc i,2-транс
В каждой системе протоны и атомы фтора об-
разуют две группы химически эквивалентных ядер,
но, поскольку эти ядра в каждой группе магнитно
8.20 8.18 8.16 8.14 8.12 м.д. 7.56 7.54 7.52 7.50 7.48 м.д.
Рис. 3.47. Спектр ЯМР ’Н лара-хлорнитробензола в CDCI3 при рабочей
частоте 300 Мгц
194
3. Спектроскопия ЯМР *//
неэквивалентны, соответствующие им спектры не
являются спектрами первого порядка. Отметим,
что экспериментальные спектры ЯМР ]Н и 19F
для каждой из этих трех систем попарно совпада-
ют. Кроме того, каждый из спектров имеет центр
симметрии. Теория показывает, что в общем случае
спектр системы АА'ХХ' содержит 20 линий, причем
спектр группы АА' совпадает со спектром группы
XX', и каждый содержит 10 линий; эти 10 линий
располагаются симметрично относительно центра.
Освоившись с понятиями химической и магнитной
эквивалентности, вернемся к структуре к на рис. 3.43;
соответствующий этой структуре спектр приведен
на рис. 3.49.
Рис. 3.49. Спектр ЯМР 1Н 2-фенил-1,3-Диоксолана в CDCI3 при рабочей частоте 300 МГц
3.10. Жесткие системы АМХ, АВХ и АВС с тремя константами спин-спинового взаимодействия
195
Структура к напоминает структуры е-и тем, что про-
тоны в каждой из групп СН2 диастереотопны вследствие
наличия асимметрического центра. Структура к отлича-
ется, однако, тем, что 1) группы СН2 находятся в кольце
с ограниченной гибкостью и 2) протоны групп СН2
взаимодействуют друг с другом. Возникает вопрос
о магнитной эквивалентности, а именно: одинаковы
ли константы спин-спинового взаимодействия протонов
группы «а», которые являются, несомненно, химически
эквивалентными, с одним из протонов группы «Ь». Со-
вершенно очевидно, что протон, помеченный, например,
как На, имеет разные константы с протонами Нь и Нь-,
поскольку для взаимодействия с протоном Нь это будет
геминальная константа, а для взаимодействия с про-
тоном Нь, - вицинальная. Следовательно, и в данном
случае мы имеем спиновую систему типа АА'ВВ'.
В открытых цепях конформационно подвижных
соединений типа
имеется две группы протонов, взаимодействующих друг
с другом. Группы Z и Y не содержат никаких элементов
хиральности и никаких протонов, которые взаимодей-
ствовали бы с двумя указанными группами. Полярность
заместителей Z и Y определяет разность химических
сдвигов этих двух групп (т.е. Av). Если бы эта молекула
была жесткой, ее следовало бы рассматривать как спи-
новую систему АА'ВВ' или АА'ХХ' в зависимости от
отношения Av/J, и при достаточно низких температурах
это описание было бы достаточно точным. Однако для
таких молекул при комнатной температуре (если толь-
ко нет условий для существенной предпочтительности
какой-то из конформаций) значения J очень близки, и на
практике возникают спектры, напоминающие спектры
типа А2Х2 или А2В2. Спектр «слабо связанной» системы
А2Х2 должнен состоять из двух триплетов, а «сильно
связанной» системе А2В2 должен соответствовать слож-
ный спектр не первого порядка. Далее мы будем рас-
сматривать эти подвижные системы как системы типа
А2Х2 или А2В2, а не АА'ВВ' или АА'ХХ'.
3.10. Жесткие системы АМХ,
АВХ и АВС с тремя константами
спин-спинового взаимодействия
В разделе 3.5 мы обсудили простую спиновую си-
стему первого порядка типа АХ. По мере того как
отношение Av/J уменьшается, два дублета сближа-
ются, и происходит изменение интенсивностей ли-
ний, приводящее к соотношению, характерному для
системы АВ, но никаких дополнительных линий не
возникает. При переходе от системы А2Х к системе
А2В, однако, появляются новые линии, и в резуль-
тате имеем спектр не первого порядка: константы
спин-спинового взаимодействия уже не совпадают
с измеряемой разностью частот линий (см. рассчитан-
ные спектры в книгах Bovey, 1988; Wiberg, Nist, 1962).
Перейдем к рассмотрению систем АМХ, АВХ и АВС
и начнем с жестких систем. Хорошей отправной
точкой является стирол, жесткая винильная группа
которого дает спектр первого порядка типа АМХ
при рабочей частоте 600 МГц (рис. 3.50).
Отнесение спектра для соединения с известной
структурой является в действительности существенно
более легкой задачей, чем интерпретация спектра
неизвестного соединения, однако оно достаточно
поучительно.
Поскольку в стироле происходит свободное вра-
щение вокруг связи между бензольным кольцом
и двойной связью, в молекуле возможны две пло-
скости симметрии. Для стирола следует рассматривать
две возможные конформации. В одной конформа-
ции винильная группа и кольцо копланарны и ле-
жат в плоскости страницы и все протоны находятся
в плоскости симметрии, а следовательно, не взаимо-
заменяемы. В другой конформации винильная группа
и бензольное кольцо перпендикулярны друг другу.
В любом случае винильные протоны лежат в пло-
скости симметрии.
Винильная система АМХ состоит трех групп,
и каждая группа содержит один протон.
Химические сдвиги таковы: X = 5 6.73, М = 5 5.75,
А = 5 5.25;
AvXm = 588 Гц, AvXa = 888 Гц, Avam = 300 Гц;
Ам = 17 Гц, JXA = 11 Гц, Jam = 1-0 Гц;
отношения Av/J : ХМ = 35, ХА = 88, МА =300.
Каждой группе соответствует дублет дублетов (см.
палочковые диаграммы на рис. 3.50). Протон X наи-
более сильно дезэкранирован бензольным ядром,
а протон А - наименее сильно. Протон X через
двойную связь взаимодействует с протоном М, и эта
/иранс-константа спин-спинового взаимодействия
имеет наибольшую величину; с несколько меньшей
константой он взаимодействует с i/мс-протоном А.
В результате получается дублет дублетов с центром
при 5 6.73 с двумя большими константами.
Протон М взаимодействует, разумеется, с протоном
X и геминально с протоном А с очень маленькой
константой. В результате получается дублет дубле-
тов с центром при 5 5.75 с одной большой и одной
очень маленькой константой.
196
3. Спектроскопия ЯМР *//
АА’ ВВ’ С
4460 4450 4440 4430 4420 4410 4400 4390 4380 4370 4360 4350 Гц
Рис. 3.50. Спектр ЯМР 7Н стирола в CDCI3 при рабочей частоте 600 МГц
Протон А имеет г/г/с-взаимодействие через двой-
ную связь с протоном X с несколько меньшей (по
сравнению с транс-) константой спин-спинового
взаимодействия. Протон А связан геминально с про-
тоном М очень маленькой константой, уже упоми-
навшейся ранее. В результате наблюдается дублет
дублетов с центром при 5 5.25 с одной большой
(несколько меньшей, чем транс-) и одной очень
маленькой геминальной константой спин-спинового
взаимодействия.
В конформации молекулы с перпендикуляр-
ной плоскостью симметрии ортпо-протоны кольца,
как и .метпа-протоны, взаимозаменяемы. Однако, по-
скольку протоны ни в одной из групп не являются
магнитно эквивалентными, мы имеем спиновую систему
АА'ВВ'С. Это система высокого порядка. Тем не менее
удается выделить три группы сигналов с соотношением
интенсивностей 2:2:1. При рабочей частоте 600 Мгц
можно произвести отнесение химических сд вигов (слева
направо): 8 7.42 (2), 8 7.33 (2) и 8 7.26 (1).
Отношение интегральных интенсивностей
(2:2:1:1:1:1 слева направо) позволяет отнести сиг-
нал при 8 7.26 к «ара-протону. Ортла-протоны (т.е
протоны А и А') дезэкранированы в большей степе-
ни (чем протоны В и В') вследствие диамагнитного
эффекта винильной двойной связи (см. рис. 3.21 для
сравнения с аналогичным, но еще более сильным
эффектом связи С=О).
3.11. Конформационно подвижные
линейные системы с открытой
цепью. Виртуальное
взаимодействие
В этом разделе будут рассмотрены некоторые наибо-
лее часто встречающиеся спиновые системы и при-
ведены примеры спектров, которые обычно вызывают
затруднения у студентов.
3.11. Конформационно подвижные линейные системы с открытой цепью. Виртуальное взаимодействие
197
3.11.1. Несимметричные цепи
3.11.1.1. 1-Нитропропан
Как было упомянуто в разделе 3.9, большинство
соединений с открытой цепью, за исключением слу-
чаев очень сильной пространственной затруднен-
ности, являются конформационно подвижными при
комнатной температуре; константы спин-спинового
взаимодействия в каждой группе усредняются, и ядра
в них становятся практически химически эквивалент-
ными. Так, при 300 МГц при комнатной температу-
ре спектр 1-нитропропана описывается как спектр
спин-системы А3М2Х2, а не А3ММ'ХХ', и при его
интерпретации применимы правила первого порядка
(см. рис. 3.51)
Протоны Х2 сильно дезэкранированы из-за влия-
ния группы NO2, протоны М2 - в меньшей степени,
а протоны А3 - лишь незначительно. В этой системе
появляются две константы спин-спинового взаимо-
действия (JAX и JMX), очень близкие по величине,
но не равные в точности. На практике при рабочей
частоте 300 МГц спектр протонов М2 оказывается
обманчиво простым и представляет собой слегка
уширенный секстет (иА + пх + 1 = 6). При более
высоком разрешении можно наблюдать 12 линий:
(«А + 1)(«х + 1) = 12.
Сигналы групп А3 и Х2 представляют собой трипле-
ты со слегка различающимися константами. Система
описывается как слабо связанная, и можно мысленно
выделить мультиплеты для анализа.
3.11.1.2. 1-Гексанол
Для сравнения рассмотрим спектр 1-гексанола, заре-
гистрированный при рабочей частоте 300 и 600 МГц
(рис. 3.52). На первый взгляд, в спектре, зарегистри-
рованном при 300 МГц, трехпротонный триплет при
5 0.87 кажется не соответствующим группе СН3 по-
скольку интенсивности линий отклоняются от соот-
ношения 1:2:1, характерного для мультиплетов первого
порядка. Кроме того, как видно из растяжки, он являет-
ся «заполненным», несмотря на то, что отношение Av/J
для группы СН2-СН3 достаточно велико и равно 13.
Проблема заключается в том, что метиленовые
группы В2, С2 и D2 являются сильно связанными,
соответствующий им сигнал в спектре представляет
собой слабо разрешенный мультиплет. Эти группы ра-
ботают как «конгломерат» спинов при спин-спиновом
взаимодействии с метильной группой, которая фор-
мально связана только с соседней метиленовой груп-
пой. Это явление называется «виртуальным взаимо-
действием» (как будто проявляется спин-спиновое
взаимодействие с группой С2).
Рис. 3.51. Спектр ЯМР ]Н 1-нитропропана в CDCI3 при рабочей частоте 300 МГц
198
3. Спектроскопия ЯМР 1Н
3.5 3.0 2.5 2.0 1.5 1.0 м.д.
Рис. 3.52. Спектр ЯМР ’Н 1-гексанола в CDCI3 при рабочей частоте 600 МГц (а),
300 МГц (б). Шкала на спектрах имеет одинаковый масштаб в м.д., поэтому на
врезках значения в герцах при 600 МГц вдвое превышают значения при 300 МГц
При рабочей частоте 600 МГц отношение Av/J
группы СН2~СН3 составляет 26, и мультиплет, со-
ответствующий этой группе, является триплетом
первого порядка. Мультиплеты В, С и D остаются
перекрывающимся мультиплетом при 300 МГц, однако
мультиплет М оказывается достаточно дезэкраниро-
ванным ОН-группой при 300 МГц и выглядит как
искаженный квинтет (расщепление на протонах D и
X), содержащий несколько дополнительных линий.
При 600 МГц наблюдается квинтет первого порядка.
В обоих спектрах сильно дезэкранированный три-
плет соответствует сигналу метиленовых протонов
X, расщепленному взаимодействием с метиленовыми
протонами М. Однопротонный синглет соответству-
ет протону ОН. Чтобы предотвратить образование
межмолекулярной водородной связи, к раствору
в CDC13 добавляли следы и-толуолсульфокислоты.
Необходимость в этом возникает из-за высокой чи-
стоты CDC13 (см. раздел 3.6.1.1), а также потому,
что при работе с сильными магнитными полями ис-
пользуют растворы низкой концентрации.
3.11.2. Симметричные цепи
3.11.2.1. Диметилсукцинат
Заслуживают внимания симметричные конформаци-
онно подвижные диэфиры с открытой цепью. Ди-
метилсукцинат дает, разумеется, четырехпротонный
синглет.
МеООС—СН,—СН2— СООМе
Aj А2
3.11.2.2. Диметилглутарат
Спектр ПМР диметилглутарата при рабочей ча-
стоте 300 МГц соответствует спиновой системе
ХХ'АА'Х"Х"', поскольку, например, протоны цен-
тральной метиленовой группы магнитно неэквива-
лентны. Однако экспериментальный спектр выглядит
как квинтет (для группы А) и триплет (для группы X),
3.11. Конформационно подвижные линейные системы с открытой цепью. Виртуальное взаимодействие
199
и упрощенно система может быть записана как А2Х4.
Менее электроотрицательные заместители (чем группа
СООМе) могут привести к спектру типа А2В4.
МеООС—СН2—СН2— СН,—СООМе
X, Д2 X,
3.11.2.3. Диметиладипат
Пойдем далее, добавим еще одну группу СН2 в цепь.
Получим молекулу диметиладипата с двумя идентич-
ными метиленовыми парами протонов А2Х2. В этой
системе можно ожидать появление двух триплетов:
более дезэкранированного (для групп СН2 при ато-
мах С2 и С5) и менее дезэкранированного (для групп
СН2 при атомах С3 и С4).
Однако спектр, зарегистрированный при рабочей
частоте 600 МГц, оказывается совершенно неожидан-
ным (рис. 3.53). Это никак не спектр первого порядка,
хотя отношение Av/J для группы А2Х2 составляет
примерно 21, если предположить, что J имеет вели-
чину около 7 Гц. Если мы снова нарисуем структуру
диметиладипата (см. рис. 3.53), то увидим, что труд-
ность заключается в двух центральных СН2-группах
(отмеченных как 3 и 4), протоны которых являются
химически эквивалентными и сильно связаны друг
с другом; так как Av равно нулю, а константа J равна
примерно 7 Гц (т.е. Av/J = 0/7 = 0) - определенно
ситуация не первого порядка. Ситуация еще более
осложняется, если учесть, что протоны групп 3 и 4 не
являются магнитно эквивалентными, поэтому про-
тоны группы 3 обозначены НА, а группы 4 - НА'.
Рассмотрим вопрос о магнитной эквивалентности
протонов в группах 3 и 4 более детально. В част-
ности, зададимся вопросом, одинаковым ли образом
взаимодействуют протоны этих СН2-групп с прото-
ном Нх из группы 2. Ответ будет «нет», поскольку
константа спин-спинового взаимодействия НАНХ -это
константа через три связи, а константа НАИХ - это
взаимодействие через четыре связи, и различие этих
констант очень велико. Аналогичным образом протоны
НА и НА' неодинаково взаимодействуют с протоном Нх'
из группы 5. В обозначениях Попла эта система явля-
ется спиновой системой ХХ'Х"Х'"АА'А"А'". Эта си-
стема характеризуется двумя химическими сдвигами
А и X и шестью константами спин-спинового взаи-
модействия (исключая константы более чем через
три связи, в частности между протонами при атомах
углерода С2 и С4 ). Шесть констант спин-спинового
взаимодействия-это четыре различные вицинальные
константы JAX (а также равные ей константы JA”X",
Лх'> А'"Х"')> Лх'> Ла "> Ла'" и две геминальных
и JAAr. Впрочем, несмотря на сложность формально-
го описания такой системы, интерпретация спектра
ЯМР !Н может оказаться не очень сложной, и все
нюансы могут свестись к различию двух геминаль-
ных констант типа XX' и АА' и двух вицинальных
констант типа АХ и АА.
200
3. Спектроскопия ЯМР 1Н
Необходимо прояснить один возможный источ-
ник недоразумений. Часто утверждается, что хими-
чески эквивалентные протоны взаимодействуют друг
с другом, но это спин-спиновое взаимодействие не
проявляется в спектре в виде расщепления линий.
Это утверждение недостаточно корректно и спра-
ведливо только для систем первого порядка. Если
же в дело вступает магнитная неэквивалентность,
система перестает быть системой первого порядка,
и в спектре наблюдается расщепление линий.
3.11.3. Менее симметричные цепи
3.11.3.1. З-Метилглутаровая кислота
Ряды менее симметричных соединений с открытой
цепью можно получить присоединением заместите-
ля к центральному углеродному атому (см. рис. 3.43,
структуры е, ж, з, и). Рассмотрим, например, 3-метил-
глутаровую кислоту. Из-за заместителя в положении
3 нет плоскости симметрии, проходящей через цепь
в плоскости листа, и, как это обсуждалось в разде-
ле 3.8.1, протоны в каждой из метиленовых групп
не являются взаимозаменяемыми, и, следовательно,
они диастереотопны.
Эта молекула дает другой пример виртуального
взаимодействия (см. раздел 3.11.1.2). Вновь при взгля-
де на структуру можно ожидать появление спектра
первого порядка, по крайней мере при рабочей ча-
стоте 300 МГц (см. рис. 3.54). Другими словами,
отношение Av/J для группы
СН2
Н3С—СН
СН,
кажется удовлетворяющим правилам первого порядка,
и вполне разумно ожидать появление чистого дублета
для СН3-группы.
Трудность состоит в том, что группы СООН
дезэкранируют группы СН2 и поэтому уменьшают
отношение Av/J для группы СН2-СН; виртуальное
взаимодействие приводит к уширенному, искаженно-
му «дублету» для группы СН3 при рабочей частоте
300 МГц. Аналогичный искаженный «триплет» был
2.4 2.3 2.2 2.1 2.0 1.9 1.8 1.7 1.6 1.5 1.4 1.3 1.2 1.1 1.0 0.9 0.8 0.7 м.д.
Рис. 3.54. Спектр ЯМР ’Н 3-метилглутаровой кислоты в D2O при рабочей частоте
300 МГц (а) и 600 МГц (6). Протон группы СООН участвует в обмене с D2O, поэтому
его сигнал не виден
3.12. Хиральность
201
показан на рис. 3.52. Имея некоторый опыт, можно
научиться распознавать такие искажения. Однако пере-
крывающиеся сигналы фрагмента СН2-СН-СН2 не
поддаются интерпретации при 300 МГц.
При рабочей частоте 600 МГц (см. рис. 3.54,6) группа
СН3 представлена в спектре чистым дублетом, возни-
кающим вследствие спин-спинового взаимодействия
с протоном СН, сигнал которого уже более не пере-
крывается с мультиплетами групп СН2. Протон СН
взаимодействует практически одинаково с семью со-
седними протонами; это означает, что соответствующий
ему мультиплет должен состоять из 8 линий. И действи-
тельно это так, однако восьмой сигнал замаскирован
сигналом одного из мультиплетов группы СН2, каждый
их которых представляет собой дублет дублетов, хотя
и несколько искаженных. Как упоминалось выше, про-
тоны каждой из групп СН2 диастереотопны и, следо-
вательно, имеют два различных химических сдвига.
Протоны каждой из групп СН2 взаимодействуют друг
с другом (геминальное спин-спиновое взаимодействие)
и с СН-протоном (вицинальное спин-спиновое взаимо-
действие), причем геминальная константа больше по
величине; отсюда возникает два мультиплета, имеющие
вид дублета дублетов.
Итак, при рабочей частоте 300 МГц наблюдает-
ся спектр не первого порядка: мультиплеты двух
СН2-групп и группы СН сильно перекрываются
и не могут быть проанализированы без использо-
вания расчетных программ. При 600 МГц мульти-
плеты достаточно хорошо разделены и могут быть
проанализированы вручную, несмотря на некоторое
перекрывание и искажение интенсивностей линий.
При 300 МГц в обозначениях Попла имеем систему
А2В2СХ3, которая переходит в систему A2G2MX3 при
600 МГц. Отметим, что при выборе букв имеются
определенные допуски. Ядра для сильно связанных
спиновых систем выбирают из близких букв алфави-
та, а для слабо связанных систем используют более
отдаленные друг от друга буквы алфавита.
3.12. Хиральность
Химики-органики, особенно работающие в области
природных соединений, никогда не должны забывать
о хиральности при интерпретации спектров ЯМР.
Эта тема была затронута в разделе 3.6. Здесь достаточ-
но дать формальное определение и краткое объясне-
ние: хиральность является необходимым и доста-
точным условием для существования энантиомеров.^
Cahn R.S., Ingold С., and Prelog V. (1966). Angew. Chem.,
Int. Ed. Engl., 5, 385.
Это строгое определение можно дополнить сле-
дующим полезным комментарием. Энантиомеры
являются несовместимыми зеркальными отображе-
ниями. Самый прямой критерий хиральности мо-
лекулы-это невозможность совместить исходную
структуру с ее зеркальным отображением. Если же
они совместимы, то молекула ахиральна. Наиболее
общей особенностью хиральных молекул является
наличие асимметрического центра, который называют
также стереогенным центром. Хиральная молекула
не имеет никаких элементов симметрии, кроме, воз-
можно, простой оси или осей. Примеры можно най-
ти на рис. 3.43. (структура д) и в гл. 7 в решенной
задаче 7.6. Для ясности рассмотрим человеческую
руку, которая не имеет никаких элементов симме-
трии. Левая и правая руки являются несовместимыми
зеркальными отображениями (т.е. энантиомерами).
Термин «хиральность» происходит от греческого
слова %etp, что значит рука.
3.12.1. Один хиральный центр. Ипсенол
Обычный углеродный хиральный центр имеет четыре
различных заместителя, как в 3-гидроксибутановой кис-
лоте (соединение д на рис. 3.43). Этот хиральный центр
обозначается как R в соответствии с хорошо известным
правилом приоритета заместителей; в энантиомерном
соединении хиральный центр имеет обозначение S.
Оба энантиомера, как и рацемат в ахиральном раство-
рителе, дают один и тот же спектр ЯМР. Поскольку
есть асимметрический центр, элементы симметрии
отсутствуют, и метиленовые протоны диастереотопны.
Часто наблюдают неэквивалентность химических
сдвигов метильных групп изопропильного фрагмента,
близкого к асимметрическому центру; этот эффект
наблюдали даже при удалении метильных протонов на
семь связей от асимметрического центра. Метильные
группы в терпеновом спирте 2-метил-6-метилен-7-
октен-4-оле (ипсеноле) химически неэквивалентны
(рис. 3.55). Они диастереотопны, но чтобы избежать
суперпозиции сигналов, необходимо использовать
сильные магнитные поля.
Поскольку сигнал каждой из неэквивалентных
метильных групп расщеплен в результате взаимо-
действия с вицинальным СН-протоном, мы ожидаем
увидеть два раздельных дублета. При рабочей частоте
300 МГц, к сожалению, можно видеть классический
триплет, который обычно считается свидетельством
присутствия группы СН3-СН2; в этом случае ее при-
сутствие не согласуется ни со структурной формулой,
ни с данными интегрирования. При более высоком
разрешении удается расщепить среднюю линию на
две и получить два дублета.
202
3. Спектроскопия ЯМР '//
1950 Гц
1550 Гц
юСН2
Н2С, 6 7 сн
Is II
СН4 8СН2
НС) зСН2
JCH.
Н3С 2 сн
9 1
1150 Гц
13
_______ V к____
' ' ' ' I ' ' ' ' I
750 Гц
100% Бензол-ёб
20% Бензол-ёб/80% CDC13
/
7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5
Рис. 3.55. Спектр ЯМР 1H 2-метил-6-метилен-7-октен-4-ола (ипсенола) в CDCI3 при
рабочей частоте 300 МГц. «Титрование» дейтеробензолом. Образец был подарен
Phero Tech, Inc., Vancouver, ВС, Canada
hi. a 'iL j _JjUL_
• I • • ’ I ‘ • I • • I • 100% cdc1j
Чтобы устранить совпадение внутренних линий,
что является причиной появления кажущегося трипле-
та, используют очень эффективный метод «титрования»
дейтеробензолом1), который дает убедительное свидетель-
ство существования двух дублетов при составе смеси
20% C6D6 / 80% CDC13, причем оптимальный резуль-
тат достигается при составе смеси 50:50 (см. рис. 3.55).
При 600 МГц видны два индивидуальных дублета.
Sanders and Hunter, В.К. (1993), Modern NMR
Spectroscopy, 2nd ed. Oxford: Oxford University Press,
p. 289.
Отметим, что наличие асимметрического центра
объясняет тот факт, что протоны каждой из двух ме-
тиленовых групп также являются диастереотопными.
В результате интерпретация спектра для такой доста-
точно простой структуры становится затруднительной.
Соотношения интегральных интенсивностей сиг-
налов ПМР слева направо таковы: 1 : 4 : 1 : 1: 1 :
2 : 1 : 1 : 6; это соответствует поглощению 18 про-
тонов и молекулярной формуле С10Н8О, однако на-
блюдаются удивительные расхождения. Шестипро-
тонная ступенька интеграла при 8 0.92 отвечает
описанным выше протонам двух диастереотопных
метильных групп, однопротонная ступенька инте-
грала при 8 3.82 с несколькими различающимися
константами спин-спинового взаимодействия может
хорошо представлять протон СНОН (см. приложе-
ние А, диаграмму А.1). Пока все хорошо. Однако
в структуре имеется четыре СН2-группы, но только
одна ступенька интеграла соответствует двум про-
тонам, да и она вводит в заблуждение.
На этой стадии интерпретации спектра уместно
вспомнить, что молекула состоит из трех спиновых
систем, изолированных друг от друга в точке С-6
(см. раздел 3.5.2). Алкильную систему составляют
3.12. Хиральность
203
протоны Н-1, Н-2, Н-3, Н-4, Н-5 и Н-9; алкеновая
система состоит из протонов Н-7 и Н-8; вторая алке-
новая система состоит из протонов Н-10. Алкильной
системе соответствуют мультиплеты в правой части
спектра, а алкеновым - в левой. Полезно еще раз на-
помнить, что протоны алкильных групп СН2 будут
диастереотопными в присутствии асимметрического
центра; то же самое относится и к протонам алке-
новой группы =СН2.
Кажется достаточно очевидным, что диастереотоп-
ные протоны каждой алкильной группы СН2 встре-
чаются парами. Одна пара (Н-3) дает сигналы при
5 1.28 и 5 1.42; другая пара (Н-5)-при 5 2.21 и 5
2.48. Мультиплет (с интенсивностью, соответствую-
щей двум протонам) при 5 1.8 обсудим позже.
Протоны Н-5 дают сигналы при более высоких
частотах, поскольку они дезэкранированы как груп-
пой ОН, так и группой С=СН2, тогда как протоны
Н-3 дезэкранированы только группой ОН. Протоны
Н-5 имеют геминальную константу спин-спинового
взаимодействия, и каждому из них отвечает также
вицинальная константа; поэтому мы имеем дублет
дублетов (первого порядка) для каждого из прото-
нов Н-5. Протоны Н-3 взаимодействуют геминально
и вицинально с двумя другими протонами. В резуль-
тате протоны Н-3 дают (при 300 МГц) два слож-
ных мультиплета. В пользу интерпретации спектров
в рамках первого порядка для мультиплета протонов
Н-5 свидетельствует большое значение (в герцах) Av
между двумя мультиплетами Н-5.
Загадочный мультиплет при 5 1.8 можно теперь
идентифицировать как наложение сигналов силь-
но связанного протона Н-2 и протона группы ОН,
что может быть подтверждено нагреванием, сменой
растворителя или встряхиванием раствора с D2O, что-
бы сместить или удалить сигнал группы ОН (см.
раздел 3.6.1.1). Разбавление смещает этот сигнал
к более низким частотам. Очень слабый сигнал при
5 2.05 является сигналом примеси.
Можно заранее ожидать, что сигнал алкенового
протона Н-7 будет находиться в наиболее высокоча-
стотной области спектра. Он имеет транс (J = 18 Гц)
и цис (J = 10.5 Гц) константы с протонами Н8 через
двойную связь и дает дублет дублетов (см. прило-
жение Е).
Область 1585-1525 Гц содержит сигналы протонов
Н-8 и Н-10. Слева находится дублет с расщеплением
18 Гц (линии 1585 и 1567 Гц), относящийся к одному
из протонов Н-8, который связан Аи/7акс-константой
спин-спинового взаимодействия через двойную связь
с протоном Н-7.
При 1546 и 1525 Гц находятся два индивидуальных
сигнала (не дублеты) протонов Н-10. Очень маленькая
геминальная константа приводит к небольшому уши-
рению, зазубренные края свидетельствуют о наличии
дальнего спин-спинового взаимодействия. Отметим,
что высота правого сигнала кажется завышенной.
Поскольку мы уже нашли сигнал протона Н-8, со-
держащий трянс-константу, поищем соответствующий
дублет, содержащий z/wc-константу. К сожалению, у нас
остался по-видимому только один сигнал примерно
при 1538 Гц, однако мы можем быстро сообразить,
что вторая линия дублета скрыта под подозритель-
но большим сигналом справа. Исследование спектра
этого диена при рабочей частоте 600 МГц см. спектр,
приведенный ниже) позволяет явно обнаружить этот
дублет протона Н-8 с расщеплением 10.5 Гц (т.е.
содержащий г/ис-КССВ), что подтверждает выска-
занное выше предположение. Отметим, насколько
усложнился спектр при наличии хирального центра.
3.12.2. Два асимметрических центра
В 1,3-дибром-1,3-дифенилпропане метиленовая группа
находится между двумя идентичными асимметрически-
ми центрами (рис. 3.56). Для изомера (17?,37?) (одного из
рацемической пары) протоны На и Нь, как и протоны
Нс и Hd, эквивалентны вследствие наличия оси сим-
метрии С2. В изомере (15,37?) (жезо-форма) попытка
вращения вокруг оси С2 приводит к отличающейся
структуре. Однако протоны На и Нь энантиотопны,
потому что они могут быть переведены друг в друга
при отражении в плоскости симметрии, показанной
перпендикулярно плоскости страницы. Протоны Нс
и Hd, однако, не могут быть переведены друг в друга,
поскольку они лежат в плоскости симметрии; они диа-
стереотопны.
В (17?,37?)-изомере протоны На и Нь магнитно не-
эквивалентны, поскольку они по-разному взаимодей-
ствуют с протонами Нс и Hd; протоны Нс и Hd также
магнитно неэквивалентны, так как они по-разному
взаимодействуют с протонами На и Нь. Однако по-
скольку величины J приблизительно усредняются
204
3. Спектроскопия ЯМР 1Н
(1/?,3/?)-1,3-Дибром-1,3-дифенилпропан
(15,3/?)-1,3-Дибром-1,3-дифенилпропан
Рис. 3.56. Два изомера 1,3-дибром-1,3-дифенилпропана.
В (1/?,3/?)-изомере протоны На и Нь химически
эквивалентны, как и протоны Нс и Hd; в (15,3/?)-изомере
протоны На и Нь химически эквивалентны, а протоны Нс
и Hd - нет
при свободном вращении, эту систему можно рас-
сматривать как спиновую систему А2Х2, и спектр
будет состоять из двух триплетов. В изомере (15,37?)
константы Jad = Jbd и Jac = Jbc; таким образом, в этой
молекуле протоны На и Нь магнитно эквивалентны.
Вопрос о магнитной эквивалентности протонов Нс
и Hd не возникает, поскольку они химически неэк-
вивалентны. Получаем спиновую систему АВХ2.
В рассмотренном выше соединении оба асимме-
трических центра имеют одинаковые заместители.
Рис. 3.57. Графическое представление уравнения
Карплуса: зависимость вицинальной константы
спин-спинового взаимодействия от двугранного угла ф
В противном случае не будет никаких элементов симме-
трии, и никакие протоны не будут взаимозаменяемыми.
3.13. Вицинальное и геминальное
спин-спиновое взаимодействие
Спин-спиновое взаимодействие между протонами
при соседних атомах углерода зависит, прежде всего,
от двугранного угла ф между плоскостями Н-С-С'
и С-С'-Н'. Этот угол можно представить наглядно
с помощью проекции Ньюмена для двух соседних
атомов углерода или с помощью перспективной
проекции, приведенной на рис. 3.57, на котором
показан график зависимости вицинальной константы
спин-спинового взаимодействия от двугранного угла.
Карплус0 подчеркивал приближенный характер рас-
четов этой зависимости, которые не учитывают такие
факторы, как электроотрицательность заместителей,
валентные утлы 0 (<Н-С-С' и <С-С'-Н') и длины свя-
зей. Оценку двугранных углов, исходя из измеренных
величин констант спин-спинового взаимодействия,
можно надежно проводить только при сравнении род-
ственных соединений. Такие корреляции оказались
очень полезными для циклопентанов, циклогексанов,
углеводов и полициклических систем. В циклопен-
танах наблюдаемая величина константы составляет
примерно 8 Гц для вициальных z/wc-протонов и около
О для вицинальных транс-протонов, что находится
в согласии с соответствующими углами 0 и 90° соот-
ветственно. В замещенных циклогексанах и пиранозах
кольцо имеет предпочтительную конформацию кресла;
в табл. 3.6 представлены величины, которые подчи-
няются уравнению Карплуса и позволяют определять
двугранные углы между заместителями, исходя из
величин констант 3<7.
Отметим, что при двугранном угле, равном 90°,
вицинальная константа спин-спинового взаимодей-
ствия практически равна нулю. Это было источником
недоразумений при попытках согласования предпо-
лагаемых структур с их спектрами ЯМР.
D Karplus М. (1959). J. Chem. Phys., 30, 11.
3.14. Дальнее спин-спиновое взаимодействие
205
Таблица 3.6. Расчетные и экспериментальные константы спин-спинового
взаимодействия (J) в циклогексанах в зависимости от двухгранных углов
Двугранный угол Расчетное значение константы J, Гц Экспериментальное значение константы Л Гц
Аксиально-аксиальное 180° 9 Аксиально-экваториальное 60° 1.8 Экваториально-экваториальное 60° 1.8 8-14 (обычно 8-10) 1-7 (обычно 2-3) 1-7 (обычно 2-3)
Модифицированное уравнение Карплуса можно
применить к вицинальному спин-спиновому взаи-
модействию в алкенах. Предсказание большей ве-
личины тирянс-константы (ф = 180°) по сравнению
с г/мс-константой (ф = 0°) оказывается справедливым.
r/wc-Константа в ненасыщенных циклах уменьшается
с уменьшением размера цикла (увеличением угла
между связями) следующим образом: в циклогексе-
нах 3J = 8.8 - 10.5, в циклопентенах 3J= 5.1 - 7.0,
в циклобутенах 3J = 2.5 - 4.0 и в циклопропенах
3J = 0.5 - 2.0 (все в герцах).
Константа спин-спинового взаимодействия через
две связи (геминальная константа) зависит от валент-
ного угла Н-С-Н, как показано на рис. 3.58. Эта за-
висимость очень чувствительна к другим влияниям
и должна использоваться с осторожностью. Однако
она оказывается полезной для характеристики мети-
е (градусы)
Рис. 3.58. Корреляция Карплуса для геминального
спин-спинового взаимодействия. Величина 7НН Для
группы СН2 как функция угла Н-С-Н. Отметим,
что нулевое значение константы достигается при
величине угла, равной примерно 125°
леновых групп в конденсированных циклогексановых
кольцах (имеющих приблизительно тетраэдрическую
конфигурацию, V-12 -18), метиленовых групп в ци-
клопропане (2J ~5) и концевой метиленовой группы
=СН2 (2J ~0 -3). Электроотрицательные заместители
уменьшают геминальную константу спин-спинового
взаимодействия, тогда как sp2- или зр-гибридизованные
атомы углерода увеличивают ее.
Геминальная константа спин-спинового взаимо-
действия обычно отрицательна, но это необходимо
учитывать только при расчетах. Отметим, что геми-
нальные константы проявляются в обычных спек-
трах только при условии, что метиленовые протоны
диастереотопны.
Следует иметь в виду, что помимо величины углов
на константы спин-спинового взаимодействия оказы-
вает влияние множество других факторов, поэтому
неудивительно, что уравнение Карплуса выполняется
не всегда. Прямой «отсчет» угла по величине кон-
станты 2J очень рискован.
3.14. Дальнее спин-спиновое
взаимодействие
Константы протон-протонного спин-спинового взаимо-
действия более чем через 3 связи (3J) имеют обычно
величину менее 1 Гц, однако более сильное дальнее
спин-спиновое взаимодействие может встретиться в та-
ких жестких структурах, как алкены, алкины, аромати-
ческие соединения, гетероароматические соединения
и в напряженных циклах (малых или мостиковых).
Аллильное (Н-С-С=С-Н) спин-спиновое взаимодей-
ствие имеет типичную величину 1.6 Гц. Спин-спиновое
взаимодействие через 5 связей сопряженной системы
бутадиена равно 1.3 Гц. Спин-спиновое взаимодействие
в сопряженных полиалкинах может иметь заметную ве-
личину даже через 9 связей. тиетя-Константа в бензоль-
ном кольце имеет величину 1-3 Гц, иара-константа -
0-1 Гц. В пятичленных гетероароматических кольцах
константы спин-спинового взаимодействия между
206
3. Спектроскопия ЯМР ?//
протонами в положениях 2 и 4 составляют 0-2 Гц.
Константа VAB в бицикло[2.1.1.]гексане имеет вели-
чину около 7 Гц.
Эту необычно большую константу спин-спинового
взаимодействия объясняют W-образной конформацией
(в виде зигзага) четырех о-связей между протонами
НА и Нв.
Дальнее
W-взаимодействие
При лучшем разрешении маленькие константы
спин-спинового взаимодействия более чем через 3 свя-
зи становятся видны как уширение линий, например
в случае бензильных СН2- и СН3-групп на рис. 3.34 и в
задаче 7.2 соответственно (см. приложение Е)
3.15. Селективное подавление
спин-спинового взаимодействия.
Двойной резонанс
Сильное облучение протона (или группы эквива-
лентных протонов) на их резонансной частоте в дан-
ной спиновой системе приводит к исчезновению
спин-спинового взаимодействия этого протона с со-
седними протонами. Например, в случае 1-пропанола
при облучении различных протонов получаются сле-
дующие результаты
Облучается
Н3С—СН— СН2ОН------> Н3С—CHj— СН2ОН
Триплет Секстет Триплет Триплет Квартет
Облучается
Н3С—СН— СНгОН > Нзс—СН—СН2ОН
Синглет Синглет
Облучается
н3с—сн2— сн2он—> н3с—сн2— СН2ОН
Триплет Триплет
Таким образом, мы располагаем мощным методом
определения взаимной связи протонов и, следователь-
но, методом отнесения протонных сигналов. Более
того, даже картина перекрывающихся сигналов может
быть упрощена при удалении какой-то константы.
На рис 3.59 показан эффект селективного подавле-
ния спин-спинового взаимодействия при облучении
соседних протонов в З-бутен-1-оле.
Так, в спектре на рис. 3.59, а слева направо располо-
жены следующие сигналы: мультиплет Н-1 (триплет
вида 1:2:1 при 3.65 м.д с константой расщепления
6 Гц), далее мультиплет Н-2 при 2.38 м.д. (триплет ду-
блетов с константами 6 и 3 Гц) и мультиплет Н-4 при
1.95 м.д. (триплет с константой 3 Гц). Имеется также
сигнал ОН-группы (при 2.03 м.д.). При интегрирова-
нии эти четыре сигнала (Н-1, Н-2, ОН и Н-4) дают
ступени, пропорциональные 2:2:1:1 соответственно
(интеграл не показан).
В спектре на рис. 3.59, б протон Н-1 облучается,
и триплет дублетов для протона Н-2 сливается просто
в дублет с константой 3 Гц. Этот дублет возникает
в результате взаимодействия протонов Н-2 и Н-4.
В спектре на рис 3.59, в облучается протон Н-2, а сиг-
налы протов Н-1 и Н-4 коллапсируют в синглеты.
В спектре на рис. 3.59, г облучается протон Н-4,
при этом триплет дублетов для протона Н-2 коллап-
сирует в триплет с константой 6 Гц.
Общий смысл информации, получаемой из экспери-
ментов по селективному подавлению спин-спинового
взаимодействия, заключается в установлении взаи-
модействий между отдельными протонами, а затем
последовательности связей атомов углерода.
Можно также упростить форму перекрывающихся
сигналов, если селективно облучать отдельные со-
седние протоны, тем самым выявляя мультиплетность
и КССВ в оставшемся мультеплете.
Другие более современные методы установле-
ния связей между протонами, основанные на дву-
мерной спектроскопии ЯМР, обсуждаются в гл. 5.
Нужно, однако, отметить, что селективное подавление
спин-спинового взаимодействия по-прежнему оста-
ется весьма полезным методом.
3.16. Ядерный эффект Оверхаузера,
разностная спектроскопия,
пространственная сближенность
протонов
Как описано в разделе 3.15, можно сильно облучать
какой-то из протонов на его резонансной частоте (v2)
и детектировать потерю мультиплетности за счет
207
3.16. Ядерный эффект Оверхаузера, разностная спектроскопия, пространственная сближенность протонов
г । но-сн2-сн2-с=сн 1 1 2 3 4 1 Облучение
Облу^ [ение
в 1
J L 11
Об! [учение ।
б . 1 А
3.7 3.6 3.5 3.4 3.3 3.2 3.1 3.0 2.9 2.8 2.7. 2.6 2.5 2.4 2.3 2.2 2.1 2.0 м.д.
Рис. 3.59. а - Спектр ЯМР ’Н З-бутин-1-ола в CDCL3 при рабочей частоте 300 Мгц.
б - Облучение протонов при С-1, в - Облучение протонов при С-2, г - Облучение
протонов при С-4. Облучение может приводить к заметному изменению
химических сдвигов соседних протонов
устранения взаимодействия со всеми ядрами, кото-
рые связаны с облучаемым протоном спин-спиновым
взаимодейсвием. Детектирование такого спектра про-
водится с помощью обычного датчика. Дополнитель-
ный канал для подавления (декаплер) дает сильную
селективную импульсную мощность на резонансной
частоте облучаемого протона.
В разделе 3.15 описывается «развязка» протонов,
связанных внутримолекулярными спин-спиновыми
взаимодействиями. В разделе 4.2.1 будет обсуждаться
широкополосное подавление спин-спинового взаимо-
действия 13С-1Н, которое позволяет при регистрации
спектров ЯМР 13С не только освободиться от констант
13С-Н, но также приводит к дополнительному усиле-
нию интенсивностей сигналов 13С вследствие ядер-
ного эффекта Оверхаузера (ЯЭО или NOE-Nuclear
Overhauser Effect). Впрочем, NOE обнаруживается
и при подавлении спин-спинового взаимодействия
между протонами, однако в этом случае его величина
существенно меньше.
Как спиновая «развязка», так и NOE являются
примерами двойного резонанса, когда использует-
ся дополнительный канал, позволяющий облучать
одно ядро на частоте v2, а детектировать эффект
облучения на других ядрах на частоте При этом
мы заинтересованы в определении пространствен-
ной близости протонов внутри данной молекулы
с помощью передающегося через пространство
NOE, который можно вызвать при использовании
существенно более слабого облучения на частоте
у2, чем при подавлении спин-спинового взаимодей-
ствия. Протон, который пространственно близок
к облучаемому протону (несущественно, связаны
эти протоны спин-спиновым взаимодействием или
нет), испытывает воздействие NOE. Если он свя-
зан спин-спиновым взаимодействием, то и остается
связанным (по крайней мере частично), поскольку
облучение слабое по сравнению с тем, которое
нужно для его полного подавления. Поляризация
(изменение в населенностях энергетических уров-
208
3. Спектроскопия ЯМР ]Н
Рис. 3.60. Разностная спектроскопия NOE для соединения III (CDCI3, рабочая
частота 300 мГц), а - Обычный спектр ЯМР 1Н; б - усиление сигнала протона
Н-4 при облучении метильной группы С-3, в - усиление сигналов обоих протонов
Н-4 и Н-б при облучении метильной группы С-5. Обратите внимание
на существенное усиление режима записи сигналов в разностных спектрах (х128)
по сравнению с обычным спектром (х4)
ней) при слабом облучении приводит (через про-
странство) к росту населенности высокоэнергетиче-
ского уровня для соседнего необлученного протона.
Избыток населенности подвержен релаксации со
временем Т\ . Это приводит к росту интенсивности
сигнала соседнего протона. Для очень больших
молекул становятся важными и другие факторы,
и в результате может происходить уменьшение
интенсивности.
Расстояния, определяемые в этих экспериментах,
малы, и эффект быстро уменьшается с ростом рас-
стояния (он обратно пропорционален шестой степе-
ни расстояния). Обычно наблюдаемые эффекты не
превышают 20%. Для увеличения чувствительности
используют так называемую разностную спектроско-
пию. При этом обычный спектр0 ЯМР !Н вычитается
из спектра, полученного при облучении отдельного
протона; в результате остаются только те сигналы,
интенсивность которых возросла. При этих условиях
измеряемый эффект можно ожидать для протонов,
удаленных на расстояния до 4 А (0.4 нм)* 2). Напри-
мер, расстояние между 1,3-диаксиальными протонами
° При этом облучается пустой участок спектра.
2) В обычных моделях Дрейдинга 1 см = 0.4 А (0.04 нм).
в циклогексане в форме кресла составляет ~2.6 А.
Эта процедура является мощным средством для рас-
познавания разных изомеров (см., например, рис. 3.60).
Впрочем, при проведении этих оценок следует
соблюдать определенные меры предосторожности
(более подробно см. в книгах Дероума, 1992; Sanders,
Hunter, 1993; Claridge, 1999). Так, облучающий гене-
ратор выключается при проведении выборки, чтобы
избежать возможной интерференции. Эта предосто-
рожность очень важна, поскольку NOE медленно
восстанавливается при облучении и затем сохраняется
в течении выборки, в то время как эффекты подавле-
ния спин-спинового взаимодействия устанавливают-
ся мгновенно. Далее, поскольку скорость релаксации
весьма чувствительна к присутствию парамагнитных
примесей, то растворенный кислород (являющийся
бирадикалом) должен быть удален путем нескольких
циклов замораживание - откачка - размораживание
или путем пробулькивания через раствор азота или
аргона. После этого ампула с образцом должна быть
запаяна.
Разностная спектроскопия NOE позволила опре-
делить тип замещения для природного соединения
(см. далее на схеме), которое предполагалось в виде
структур I или II. Для этого дополнительно доступ-
ный продукт III (диметильный гомолог изучаемого
3.17. Заключение
209
соединения) был исследован с помощью разностной
спектроскопии NOE (рис. 3.60).
Облучение метильной группы у С-5 привело в ре-
зультате к усилению сигналов протонов Н-4 и Н-6, в то
время как облучение метильной группы у С-3 при-
вело к увеличению интенсивности только сигнала
Н-4. Поскольку химический сдвиг сигнала метильной
группы природного соединения был почти иденти-
чен сдвигу метильной группы у С-3 соединения III,
структура изучаемого природного соединения соот-
ветствует структуре I.
На рис 3.60, а приведен обычный спектр ЯМР !Н .
Спектр на рис. 3.60, б показывает результат облучения
метильной группы у С-3, при этом увеличивается
только сигнал протона Н-4. Спектр на рис. 3.60, в
показывает результат облучения метильной группы
у С-5. При этом возрастают сигналы ароматических
протонов Н-4 и Н-6, поскольку оба они близки к ме-
тильной группе у С-5. Впрочем, некоторые детали
спектров могут показаться странными, но помните,
что это разностные спектры. Обычный спектр вы-
читается из спектра с облучением; при вычитании
и усилении облучаемый пик возвращается в поло-
жение «вверх», а необлучаемые пики вообще не де-
тектируются. Показанные на рис. 3.60 коэффициенты
усиления *4, *128 относятся к вертикально показан-
ным уровням чувствительности для левых частей
разорванных спектров по отношению к правым ча-
стям, для которых усиление х 1. При этом обычный
спектр дан с коэффициентом 4, а разностные спектры
даны с существенно большими усилениями (128)
Смысл этой нормировки в том, чтобы удерживать
все сигналы на удобной высоте.1)
Облучение ОН-группы приводит к усилению
сигналов соседних протонов по механизму NOE.
Однако обычный быстрый обмен гидроксильных
протонов должен быть замедлен либо применени-
ем сухих растворителей (дейтерированного ацетона
или дейтерированного диметилсульфоксида), либо
просто охлаждением раствора в CDC13. Любой их
этих способов сдвигает также сигнал группы ОН
влево, и это смещение можно контролировать, чтобы
избежать перекрывания сигналов. И конечно, этот
прием используется для идентификации пиков ОН
(см. раздел 3.6.1.1).
Распознавание тризамещенных Е- и Z-изомеров
при двойной связи является нетривиальной задачей.
Использование NOE для этой цели иллюстрируется
в задаче 7.4
3.17. Заключение
Освоение спектроскопии ЯМР !Н позволяет перейти
к освоению других типов спектроскопии ЯМР (на-
пример, ЯМР 13С). Мы начали с описания магнитных
свойств ядер, особенно выделяя ядра со спином !6.
Последовательное развитие спектроскопии ЯМР !Н
привело к появлению спектроскопии ЯМР высоко-
го разрешения и созданию коммерчески доступных
спектрометров ЯМР. Родоначальники спектрометров -
спектрометры ЯМР стационарного типа (CW) - сей-
час почти полностью заменены на более мощные
и разносторонние импульсные спектрометры ЯМР
с фурье-преобразованием.
Интерпретация спектров ЯМР !Н основана на
рассмотрение трех видов информации: на данных
интегрирования, на значениях химических сдви-
гов отдельных сигналов и на величинах констант
спин-спинового взаимодействия. Интегрирование
сигналов дает соотношение протонов в отдельных
группах. Величины химических сдвигов позволя-
ют связать сигнал с типом химического окруже-
ния. Важным является понимание эквивалентно-
сти отдельных протонов на основании данных по
химических сдвигам. Константы спин-спинового
взаимодействия позволяют связать пары ядер друг
с другом. Спин-спиновые взаимодействия позволяют
определить типы соседних ядер. В последующих
главах книги обсуждаются другие виды спектроско-
пии ЯМР (гл. 4, 5 и 6) и задачи с использованием
ЯМР (гл. 7 и 8).
° См. работу [R. Е. Charlton, F. X. Webster, A. Zhang,
С. Schal. D.Liang, I. Sreng, W.L.Roelofs. Proc. Natl. Acad.
Sci.'W, 10202 (1993)] для более подробных сведений.
210
3. Спектроскопия ЯМР ’Н
Литература
Учебные издания и монографии
Abraham, R.J., Fisher, L, and Loftus, P. (1989). Introduction
to NMR Spectroscopy, 2nd ed. London - New York: Wiley.
Akitt, J.W. (1992). NMR and Chemistry: An Introduction
to Modern NMR Spectroscopy, 3rd ed. London: Chapman
and Hall.
Asahi. (1997). Handbook of Proton-NMR Spectra and Data.
New York: Academic Press.
Atta-ur-Rahman. (1986). Nuclear Magnetic Resonance.
New York: Springer-Verlag.
Atta-ur Rahman, and Choudhary, M. (1995) Solving Problems
with NMR Spectroscopy. New York: Academic Press.
Ault, A., and Dudek, G.O. (1976). NMR, An Introduction
to Proton Nuclear Magnetic Resonance Spectrometry.
San Francisco: Holden-Day.
Becker, E.D. (1999). High Resolution NMR: Theory and
Chemical Applications, 3rd ed. New York: Academic Press.
Berger, S., and Braun, S. (2004). 200 and More NMR Experi-
ments: A Practical Course. New York: Wiley; (July 9,2004)
Bovey, F.A. (1988). Nuclear Magnetic Resonance
Spectroscopy. 2nd ed. New York: Academic Press.
Braun, S., Kalinowski, H., and Berger, S. (1999). 150 and
More Basic NMR Experiments: A Practical Course. 2nd
ed., New York: Wiley.
Breitmaier, E. (2002). Structure Elucidation by NMR in
Organic Chemistry: A Practical Guide. 3rd Revised ed.,
New York: Wiley.
Cavanagh, J., Fairbrother, W., Palmer III, A., and Skelton,
N. (1995). Protein NMR Spectroscopy: Principles and
Practice. New York: Academic Press.
Claridge,T.D.W. (1999). High-Resolution NMR Techniques
in Organic Chemistry. Elsevier Science. Oxford, UK.
Croasmun, W.R., and Carlson, R.M.K. (1994). Two-Dimensional
NMR Spectroscopy: Applications for Chemists and
Biochemists, 2nd Ed., Fully Updated and Expanded to Include
Multidimensional Work. New York: Wiley.
Дероум Э. (1992) Современные методы ЯМР для хи-
мических исследований, Пер. с англ. Москва, Мир.
Фаррар Т., Беккер Э. (1973). Импульсная фурье-
спектроскопия ЯМР. Пер. с англ. Москва, Мир.
Farrar, Т.С. (1987). An Introduction to Pulse NMR
Spectroscopy. Chicago: Farragut Press.
Freeman, R. (1998). Spin Choreography: Basic Steps in High
Resolution NMR. Oxford University Press on Demand.
Friebolin, H. (1993). Basic One- and Two-Dimensional
Spectroscopy, 2nd ed. New York: VCH.
Friebolin, H. (1998). Basic One- and Two-Dimensional NMR
Spectroscopy, 3rd Revised Ed., New York: Wiley.
Fukushima, E., and Roeder, S.B.W. (1986). Experimental
Pulse NMR a Nuts and Bolts Approach. Perseus Publishing.
Boulder, CO.
Grant, D.M., and Harris, R.K. (2002). Encyclopedia of Nuclear
Magnetic Resonance, Advances in NMR. New York: Wiley.
Gunther, H. (1995). NMR Spectroscopy: Basic Principles,
Concepts, and Applications in Chemistry, 2nd Ed. New York:
Wiley. (Есть русский перевод: Гюнтер X., (VNA) Введение
в курс спектроскопии ЯМР. Москва, Мир.)
Homans, S. W. (1992). Л Dictionary of Concepts in NMR.
Clarendon Press.
Ноге, P.J., Wimperis, S., and Jones, J. (2000). NMR: The
Toolkit. Oxford University Press.
Jackman, L.M., and Stemhell. S. (1969). Applications of
NMR Spectroscopy in Organic Chemistry, 2nd ed. New York:
Pergamon Press.
Macomber, R.S. (1988). NMR Spectroscopy — Essential
Theory and Practice. New York: Harcourt.
Macomber, R.S. (1995). A Complete Introduction to Modem
NMR Spectroscopy. New York: Wiley-Interscience.
Martin, G.E., and Zektzer, A.S. (1988). Two-Dimensional
NMR Methods for Establishing Molecular Connectivity:
A Chemist s Guide to Experiment Selection, Performance,
and Interpretation. New York: Wiley.
Munowitz, M. (1988). Coherence and NMR. New York:
Wiley-Interscience.
Nakanishi, K. (1990). One-Dimensional and Two-Dimensional
NMR Spectra by Modern Pulse Techniques. University
Science Books. Herndon, VA.
Neuhaus, D.N., and Williamson, M.P. (1992). The Nuclear
Overhauser Effect in Structural and Conformational
Analysis. New York: VCH.
Paudler, W.W. (1987). Nuclear Magnetic Resonance.
New York: Wiley.
Pihlaja, K., and Kleinpeter, E. (1994). Carbon-13 NMR
Chemical Shifts in Structural and Stereochemical Analysis,
VCH Publishing.
Roberts, J.D. (2000). ABCs of FT-NMR. University Science
Books. Herndon, VA.
Sanders, J.K., and Hunter, B.K. (1993). Modern NMR
Spectroscopy, 2nd ed. Oxford: Oxford University Press.
Schwartz, L.J. (1988). A Step-by-Step Picture of Pulsed
(Time-Domain) NMR. J. Chem. Ed. 65, 752-756.
Shaw, D. (1987). Fourier Transform NMR Spectroscopy.
Amsterdam: Elsevier.
Shoolery, J .N. (1972). A Basic Guide to NMR. Palo Alto,
CA: Varian Associates.
Williams, K.R., and King, R.W. (1990). J. Chem. Educ. 67,
A125. (см. ссылки в ней на предыдущие работы.) The
Fourier Transform in Chemistry - NMR.
Yoder, С.-H., and Schaeffer, C.D., Jr. (1987). Introduction to
Multinuclear NMR. Menlo Park, CA: Benjamin Cummings.
Упражнения
211
Van de Ven, F. J. M. (1995). Multidimensional NMR in
Liquids: Basic Principles and Experimental Methods.
New York: Wiley.
Weber, U., and Thiele, H. (1998). NMR-Spectroscopy: Modem
Spectral Analysis New York: Wiley.
Каталоги спектров, спектральных данных
и специальные темы
Bovey, F.A. (1967). NMR Data Tables for Organic
Compounds, Vol. 1. New York: Wiley-Interscience.
Brugel, W. (1979). Handbook of NMR Spectral Parameters,
Vols. 1-3. Philadelphia: Hoyden.
Chamberlain, N.E. (1974). The Practice of NMR Spectroscopy
with Spectra—Structure Correlation for 1H. New York:
Plenum Press.
Kiemle, D.J., and Winter, W.T. (1995). Magnetic Spin
Resonance. In Kirk-Othmer Encyclopedia of Chemical
Technology, 4th ed. Vol. 15. New York: Wiley.
Morrill, T.G, Ed. (1988). Lanthanide Shift Reagents in
Stereochemical Analysis. New York: VCH.
Pouchert, C.J., and Behnke, J. (1993). Aldrich Library of
13C and !H FT-NMR Spectra, 300 MHz. Milwaukee, WI:
Aldrich Chemical Co.
Упражнения
3.1. Для каждого из химических соединений, приве-
денных далее (см. структуры а-п), предложите
обозначения спиновых систем (используя по не-
обходимости номенклатуру Попла) с учетом хи-
мической эквивалентности протонов, магнитной
эквивалентности, энантиотопии и диастереотопии.
3.2. Для каждого из указанных выше соединений
предскажите ожидаемые химические сдвиги про-
тонов. Укажите на источник информации (таблицу
или приложение), который вы использовали для
этих предсказаний.
3.3. Нарисуйте условные спектры ЯМР !Н для указан-
ных выше соединений. Где это возможно, предпо-
ложите спектры (мультиплеты) первого порядка.
3.4. Определите структурные формулы для соеди-
нений, спектры ЯМР !Н которых приведены на
рисунке (спектры А-Ч). Спектры записаны на ра-
бочей частоте 300 МГц для растворов в CDC13.
Масс-спектры этих соединений даны в гл. 1
(упражнение 1.6), а ИК-спектры-в гл. 2 (упраж-
нение 2.9).
3.5. После того как определите структуры по спектрам
в упражнении 3.4, рассчитайте отношения Av/J
для соответствующих связанных мультиплетов
в спектрах А, Д, Е, Ж, 3, И, Л, М, С, X.
Pretsch, Е.. Clerc, Т., Seihl, J., and Simon, W. (1989). Spectra
Data for Structure Determination of Organic Compounds,
2nd ed. Berlin: Springef Verlag.
Sadtler Collection of High Resolution Spectra. Philadelphia:
Sadtler Research Laboratories.
Sasaki, S. (1985). Handbook of Proton-NMR Spectra and Data,
Vols. 15. New York: Academic Press (4000 спектров).
Varian Associates. (1962,1963). High Resolution NMR
Spectra Catalogue, Vol. 1, Vol. 2. Palo Alto, CA: Varian.
Wiberg, K.B, and Nist, B.J. (1962). The Interpretation of
NMR Spectra. New York: Benjamin.
Дополнительная литература
Сергеев H. М., Спектроскопия ЯМР для химиков орга-
ников, М.: Изд-во МГУ, 1981.
Ноге Р. J. Nuclear Magnetic Resonance, in Oxford Chemistry
Primers, Oxford University Press (1995).
Попл Дж., Шнейдер В., Бернстейн Г. Спектры ядерного
магнитного резонанса высокого разрешения. Пер. с
англ. М.: Мир, 1962.
Преч.Э., Бюльманн Ф., Аффольтер К. Определение строе-
ния органических соединений. Таблицы спектральных
данных. Пер. с англ. М.: Мир, БИНОМ, 2006.
3.6. Для каждой из структур, установленных в упраж-
нении 3.4, опишите все спиновые системы (ис-
пользуя, где это нужно, номенклатуру Попла)
с учетом химической эквивалентности протонов,
магнитной эквивалентности, энантиотопии и диа-
стереотопии.
3.7. Изобразите спектры следующих спиновых систем
(достаточно изобразить спектр в форме палочек):
АХ, А2Х, А3Х, а2х2, а2х3, а3х2.
3.8. Изобразите спектры следующих спиновых систем
(достаточно изобразить спектр в форме палочек):
АМХ, А2МХ, А3МХ, А2МХ2, А2МХ3, А3МХ3. Счи-
тайте, что ядро А не связано с ядром X и что
•Лм=^мх-
3.9. Изобразите спектры следующих спиновых систем
(достаточно изобразить спектр в форме палочек):
АМХ, А2МХ, А3МХ, А2МХ2, А2МХ3, А3МХ3. Счи-
тайте, что ядро А не связано с ядром X и что
JAM = 10 Гц и Jmx = 5 Гц.
3.10. Для трех симулированных спиновых систем (см.
с. 217-219) нарисуйте палочковую диаграмму для
каждого мультиплета первого порядка во всех
спиновых системах. После того как получено
расщепление для одного мультиплета в спиновой
системе, два других мультиплета могут быть ис-
пользованы для проверки.
212
3. Спектроскопия ЯМР ]Н
е
'"\и*
ьГТЪ,
н
п
Упражнение 3.1 (а-п)
Упражнения
213
Спектр В
Упражнение 3.4(А-Г)
214
3. Спектроскопия ЯМР ’Н
2.7 2.6 2.5 2.4 2.3 2.2 2.1 2.0 1.9 1.8 1.7 1.6 1.5 1.4 1.3 1.2 1.1 1.0 0.9 0.8 м.д.
Упражнение 3.4(Д-3)
Упражнения
215
' I ’ ' ' ' П1 * 1 1 I ' ' ’ ' I ' ' 1 1 I ' ' 1 ' J ' ' ’ ’ I ' ' ; ' I ' ' ' 1 I 1 ' ' I ' ' ' ' I 1 ' 1 ' I 1 * 1 ' I
7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 м.д.
Спектр Л
Спектр М
Упражнение 3.4(И-М)
216
3. Спектроскопия ЯМР 1Н
Спектр П
1 протон (х32)
i~/~
6.0
4.5
4.0 м.д.
Упражнение 3.4(Н-Р)
Упражнения
217
Спектр Т
Спектр У (ненасыщенный кетон)
I.........| г,т,-1 . ! . , . , . . , | .-П
1520 1510 1500 Гц
I —•
1.5 м.д.
"I ' 1 1 1 I 1 ' * Ч ' ' "1 1 I 1 1 1 1 'I
5.0 4.5 4.0 3.5 3.0
I 1 1 1 1 ’ т-
2.5 2.0
Упражнение 3.4(С-Ф)
218
3. йтектроскопия ЯМР ’Н
Спектр X
I ' ' ' ' I ' ' ' ' I 1 ' ' ' I ' ।............, । । ।
7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 м.д.
Упражнение 3.4(Х-Ч)
Упражнения
219
°a1)JvuiAa
1605 1600 1595 1590 1585 1580 1575 1570 1565
5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 м.д.
Упражнение 3.10(A)
220
3. Спектроскопия ЯМР^'//
1ТХ.............................
385 380 375 370 365 360 355 350 345 340 335 Гц
I I
2 vUU
1120 1115 1110 1105 1100 1095 1090 1085 1080 Гц
...................
1605 1600 1595 1590 1585 1580 1575 1570 1565 Гц
Упражнение 3.10(B)
Упражнения
221
• jJjULLLa
385 380 375 370 365 360 355 350 345 340 335 Гц
1120 1115 1110 1105 1100 1095 1090 1085 1080 Гц
•, , кJULJUJULa,
1605 1600 1595 1590 1585 1580 1575 1570 1565 Гц
а б в
5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 м.д.
Упражнение 3.10(B)
222
3. Спектроскопия ЯМР 1Н
Приложение А. Химические сдвиги протонов в алифатических соединениях
Диаграмма АЛ.Химические сдвиги протонов в алифатических соединениях
у атомов углерода, соседних (a-позиция) с функциональными группами Y (M-Y)
I М = метил
8 М = метилен
! М = метин
£
.4 .2 5 .8 .6 .4 .2 4 .8 .6 .4 .2 3 .8 .6 .4 .2 2 .8 .6 .4 .2 1 .8 .6 .4 .2 О
m-ch2r • • 1
м-с=с о о
м-с=с 1
М —Ph • о ।
M-F o ~r
M-CI • • о о I
М-Вг • • j 1
M-I • • о о 1
М-ОН
М-OR
M-OPh • • о о I
M-OC( = O)R • • о о I
М—OC(=O)Ph • • ( I
M-OC(-O)CF3 о о I
M-OTs* • • о о I
M-C( = O)H
M-C(=O)R • • о о 1
M-C(“O)Ph о о
M-C( = O)OH • • о о 1
M-C(«O)OR • • о о
M-C( = O)NR2 > о ► о
M-C=N • • о о 1
M-nh2 • • о о 1
m-nr2 • • i 1
M-NPhR о о
M-N+R3 о о I
M-NHC( = O)R о о 1
m-no2 SI
.4 .2 5 .8 .6 .4 .2 4 .8 .6 .4 .2 3 .8 .6 .4 .2 2 .8 .6 .4 .2 1 .8 .6 .4 .2 О
Приложение А
223
б
.4 .2 5 .8 .6 .4 .2 4 .8 .6 .4 .2 3 .8 .6 .4 .2 2 .8 .6 .4 .2 1 .8 .6 .4 .2 О
M-N=C • • 0 0 1
M-N=C=O 0 0
M-O-CEN
M-N=C=S 0 9
M-S-CEN • • 1
M-O-N=O
M-SH • • T
M-SR • • 1
M-SPh 1
M-SSR 0 0
M-SOR 0 0
M-SO2R 0 0
M-SO3R
m-pr2
M-P*CI3 I
M-P( = O)R2
M-P(=S)Rj
.4 .2 5 .8 .6 .4 2 4 .8 .6 .4 .2 3 .8 .6 .4 .2 2 .8 .6 .4 2 1 .8 .6 .4 .2 О
224
j. Спектроскопия ЯМР ’Я
Приложение А (продолжение)
Диаграмма А.2. Химические сдвиги протонов в алифатических соединения у атомов углерода, отстоящих на
1 атом от функциональной группы У ( 0-положение) (фрагмент M-C-Y)
I М = метил
8 М = метилен
: М = метин 6
.7 .6 .5 .4 .3 .2 .1 2 .9 .8 .7 .6 .5 .4 .3 .2 .1 1 .9 .8 .7 .6 .5 .4 .3 .2 .1 О
м-с-сн2 • • I
м-с-с=с • • 0 0
м-с-с=с
M-C-Ph 0 0 I
M-C-F • • 0 0 I
M-C-CI • • I
M-C-Br • 0 • 0
M-C-I J fl
м-с-он • •
M-C-OR • •
M-C-OPh 0 0
M-C-OC( = O)R
М—С—OC( = O)Ph • • 0 0 I
M-C-OC( = O)CF3 0 0
M-C-C( = O)H 0 0
M-C-C( = O)R • • 0 0 I
M-C-C( = O)Ph 0 0 I
M-C-C( = O)OR I
M-C-C( = O)NR2
M-C-CEN 0 0 I
M-C-NRj 0 0 I
M-C-NPhR
M-C-NR3 • • 0 0
M-C-NHC( = O)R
m-c-no2 0 0
M-C-SH • ( • (
M-C-SR I
.7 .6 .5 .4 .3 .2 .1 2 .9 .8 .7 .6 .5 .4 .3 .2 .1 1 .9 .8 .7 .6 .5 .4 .3 .2 .1 О
Приложение Б
225
Приложение Б. Влияние на химические сдвиги протонов двух
или трех непосредственно присоединенных заместителей
Y—СН2—Z и Y—СН—Z
W
Химический сдвиг протонов метиленовой группы
с двумя заместителями может быть рассчитан с по-
Таблица Б.1. Инкременты для отдельных заместителей
Заместитель Y или Z Инкремент (a) Заместитель Y или Z Инкремент (a)
—H 0.34 —OC(=O)R 3.01
—CH3 0.68 —OC(=O)Ph 3.27
—c—c 1.32 —C(=O)R 1.50
—c=c 1.44 —C(=O)Ph 1.90
—Ph 1.83 —C(=O)OR 1.46
CF2 1.12 -C(=O)NR2(H2) 1.47
—CF3 1.14 —C=N 1.59
—F 3.30 -NR2(H2) 1.57
—Cl 2.53 —NHPh 2.04
—Br 2.33 —NHC(=O)R 2.27
—I 2.19 -N3 1.97
—OH 2.56 —no2 3.36
—OR 2.36 —SR(H) 1.64
—OPh 2.94 —OSO2R 3.13
Таблица Б.2а. Инкременты заместителей
для метиновых протонов
Группа Инкремент (a)
—F 1.59
—Cl 1.56
—Br 1.53
—no2 1.84
—nh2 0.64
—NH3+ 1.34
—NHCOR 1.80
—OH, —OR 1.14
—OAr 1.79
—OCOR 2.07
—Ar 0.99
—C=C 0.46
—C^C 0.79
—C=N 0.66
—COR, —COOR, —COOH 0.47
—conh2 0.60
—COAr 1.22
—SH, —SR 0.61
—SO2R 0.94
—R 0
мощью инкрементов замещения, приведенных в табл.
Б.1. Правило Шулери1) утверждает, что сумма ин-
крементов для двух групп прибавляется к величине
0.23 (химический сдвиг протонов в СН4).
8(Y-CH2-Z) = 0.23 + oY + oz
Например, химический сдвиг протонов в группе
СН2 для соединения С6Н5СН2Вг рассчитывается с по-
мощью величин, приведенных в табл. Б.1
0.23
Орь= 1.83
РВг = 2.33
8 = 4.39 (экспериментальное
значение равно 8 4.43)
Инкременты, первоначально предложенные Шуле-
ри, были впоследствии пересмотрены и расширены
(см. табл. Б.1). Наблюдаемые и расчетные химические
сдвиги были проверены, и для 62% образцов отклоне-
ния были в пределах ±0.2 м.д., для 92%-в пределах
±0.3 м.д., для 96% -в пределах ±0.4 м.д. Для 99%
образцов отклонения были в пределах ±0.5 м.д.* 2)
Таблица Б. 1 содержит инкременты для нескольких
функциональных групп по данным работы [Friedrich
Е.С., and Runkle К. G. (1984) J. Chem. Educ. 61, 830.]
Обратите внимание на то, что химические сдвиги
метильных групп могут быть рассчитаны с исполь-
зованием инкрементов для заместителя Н (0.34). На-
пример, химический сдвиг для СН3Вг будет рассчиты-
ваться как химический сдвиг для группы Н-СН2-Вг.
Таблицы Б.2а, Б.26, и Б.2в. Корреляции
для химических сдвигов метиновых протонов
Таблица Б.2а содержит инкременты 3) для заместите-
лей X, Y, Z для случая тризамещенных систем
Д CHXYZ = 2.50 + ох +aY +oz
Эта схема удовлетворительно работает, если по крайней
мере два заместителя являются электроотрицательными
° Shoolery J.N. Varian Technical Information Bulletin, Vol 2,
No. 3. Palo Alto, CA, Varian Associates (1959).
2) Данные из работ [Friedrich E.C., and Runkle K. G. (1984).
J. Chem. Educ., 61, 830; (1986) 63, 127].
3) С разрешения из работы [Bell H.M., Bowles D.B., and
Senese F. (1981). Org. Magn. Reson., 16, 285].
226
3. Спектроскопия ЯМР ]Н
Таблица Б.26. Наблюдаемые химические сдвиги
метиновых протонов в изопропильных производных
(CK3)2CHZ (CH3)2CHZ
Z 6, М.Д. (набл.) z б, М.Д. (набл.)
н 1.33 HO 3.94
Н3С 1.56 RO 3.55
R 1.50 C6H5O 4.51
ХСН2 1.85 R(H)C(=O)O 4.94
R(H)C(=O) 2.54 СбН5С(=О)О 5.22
С6Н5С(=О) 3.58 F3CC(=O)O 5.20
R(H)OC(=O) 2.52 ArSO2O 4.70
R2(H2)NC(=O) 2.44
СбН5 2.89 R(H)S 3.16
R2(H2)C=CR(H) 2.62 RSS 2.63
R(H)C=C 2.59
N=C 2.67 F 4.50
Cl 4.14
R2(H2)N 3.07 Br 4.21
R(H)C(=O)NH 4.01 I 4.24
o2n 4.67
группами. Другими словами, только один заместитель
может быть алкильной группой (R). При этих условиях
отклонения для такой схемы расчета не превышают
0.2 м.д. Например, для соединения
OEt
I
СН3—СН—OEt
химический сдвиг метикового протона рассчитыва-
ется следующим образом:
8 = 2.50 + 1.14 + 1.14 + 0.00 = 4.78
Экспериментальное значение равно 4.72.
Таблицы Б.26 и Б.2в используются совместно для
протонов метиновых групп, которые замещены по
крайней мере двумя алкильными группами (или дру-
гими группами с низкой полярностью). Было пред-
ложено соотношение (см. Friedrich Е.С. and Runkle
K.G. J. Chem. Educ., 61, 830 (1984); 63, 127 (1986)):
ScHXYZ = S(CH3)2CHZ + toy
где заместители X и Y - алкильные группы или другие
заместители с низкой полярностью. Заместитель Z мо-
жет иметь широкий диапазон полярности. Соотношение
утверждает, что химический сдвиг протона метановой
группы с по крайней мере двумя группами низкой поляр-
ности эквивалентен химическому сдвигу изопропильного
метанового протона плюс корректирующая поправка.
Наблюдаемые химические сдвиги метиновых про-
тонов в изопропильных производных даны в табл.
Б.26. Поправки Дху приведены в табл. Б.2в.
Таблица Б.2в. Корректирующие поправки для
метиновых протонов в случае заместителей
с низкой полярностью.
Открытые
цепи
с метиновым
протоном
Дху Циклические
системы
с метиновым
протоном
сн3—сн—сн3
Z
СН3—СН—R
Z
I
R—СН—R
Z
I
сн3—сн—СН2Х
Z
I
сн3—сн—сн=сн2
Z
I
сн3—сн—С6Н5
Z
I
R—СН—С6Н5
+0.40 экваториал. Н +0.25
0.00
0.00
+1.15
+0.90
Следующий пример иллюстрирует совместное ис-
пользование таблиц Б.26 и Б.2в в случае групп СН3,
СН=СН2 и С6Н5 в качестве заместителей. Наиболее
полярный заместитель всегда обозначается как Z.
Z Z
6 X—СН—Y = 8 СН3—СН—СН=СН2 =
с6н5
= 8 СН3—СН—СН3 + Дху
По данным табл. Б.26, для группы
СН3-СН(С6Н5)-СН3 5 = 2.89.
По данным табл. Б.2в, Дху = 0.00 для СН3;
Дху = 0.40 для группы СН=СН2.
Таким образом, 8 для метанового протона в со-
единении СН3-СН(С6Н5)-СН=СН2 определяется по
следующей формуле:
б = 2.89 + 0.00 + 0.40 = 3.29
(экспериментальное значение 8 = 3. 44).
Приложение В
227
Приложение В. Химические сдвиги протонов в алициклических
и гетероциклических системах
Таблица В.1. Химические сдвиги в алициклических системах
Таблица В.2. Химические сдвиги в гетероциклических системах
R
Н
4.75-4.90
3.80
О 1
228
3. Спектроскопия ЯМР ’Н
Приложение Г. Химические сдвиги протонов в ненасыщенных и ароматических
системах
(см. табл. Г.1).
рассчитываются следующим образом
8Н 5.25 + 2гем + Zyuc + ZmpaHC
Например, химические сдвиги алкеновых протонов
в соединении
Ha ГбН5гем OR 1.35 -1.28 0.07 5.25 0.07
8 5.32
Hb ORaeM 1.18 5.25
с и <-'6rL5mpaHC -0.10 1.08
1.08 8 6.33
С6Н5 PQHg
/С=С\
На Нь
Табл. ГЛ. Инкременты заместителей (Z) в замещенных этиленах
Заместитель R Z Заместитель R z
гем цис транс гем цис транс
—H 0 0 0 н
—Aik —Aik циклически CH2O, CH2I 0.44 0.71 0.67 -0.26 -0.33 -0.02 -0.29 -0.30 -0.07 —c=o N 1.03 0.97 1.21
—CH2S —CH2C1, —CH2Br 0.53 0.72 -0.15 0.12 -0.15 0.07 —c=o 1.37 0.93 0.35
—ch2n —c=c 0.66 0.50 -0.05 0.35 -0.23 0.10 Cl —c=o 1.10 1.41 0.99
—C^N 0.23 0.78 0.58 —OR, R: алифат. 1.18 -1.06 -1.28
—c=c 0.98 -0.04 -0.21 —OR, R: сопряж.6 1.14 -0.65 -1.05
—C=C сопряж.^ 1.26 0;08 -0.01 —OCOR 2.09 -0.40 -0.67
—c=o 1.10 1.13 0.81 —Ar 1.35 0.37 -0.10
—C=O сопряж.^ 1.06 1.01 0.95 —Cl 1.00 0.19 0.03
—COOH 1.00 1.35 0.74 —Br zR 1.04 0.40 0.55
—COOH сопряжЛ 0.69 0.97 0.39 —N алифат. R R 0.69 -1.19 -1.31
—COOR 0.84 1.15 0.56 —N R: сопряж.^ 2.30 R -0.73 -0.81
—COOR сопряжЛ 0.68 1.02 0.33 —SR —so2 1.00 1.58 -0.24 1.15 -0.04 0.95
Aik циклич. означает, что двойная связь является частью кольца R || .
6 Фактор Z для сопряженных заместителей используется в том случае, если
заместитель или двойная связь еще сопряжены с какой-то другой группой.
Источник'. Pascual С., Meier J., Simon W., Helv. Chim. Acta, 49, 164 (1966).
Приложение Г
229
Таблица Г.2. Химические сдвиги в разных алкенах
Таблица Г.3. Химические сдвиги алкиновых протонов
HC=CR 1.73-1.88 нс=с—сон 2.23
НС=С—C=CR 1.95 нс=сн 1.80
НС=С—Ph 2.71-3.37 нс^с—ch=cr2 2.60-3.10
Таблица Г.4. Химические сдвиги протонов в конденсированных ароматических системах
230
3. Спектроскопия ЯМР^ '//
Диаграмма Г.1. Химические сдвиги протонов в монозамещенных бензолах
9 .8 .6 .4 .2 8 Я .6 .4 2 7 Л .6 .4 2 6 6
Бензол" •
СН3 (отр)
СНзСН, (ошр)
(СН3)3СН(отр)
(СНэ)эС о,т,р • • a
С=СН2 (отр) • •
С^СН о, (тр) • • •
Phenyl о, т, р • • • •
CF3 (отр) •
СНаС1(отр) •
CHClj (отр) •
СС1Э о, (тр) • •
СН2ОН(отр) •
CHjOR (отр) •
СНаОС(=О)СНз (отр) •
CH2NH2 (отр) •
F т,р,о • a
С1 (отр)
Вг о, (рт) •
I °»Р»т • «
ОН т,р,о • • •
OR т, (ор) •
ОС(—О)СНз т,р,о • • •
OTsb (тр), о • •
СН(=О)о,р,т • • •
С(=О)СНз о, (тр) • •
С(=О)ОНо,р,т a •
C(=O)OR о, р, т • • •
С(в0)С1 о, р, т • • • • •
C=N(omp) • •
NHa т,р,о • • • •
N(CH»),m(op) • • a
NHC(=O)R о,т,р e • •
КНзо(тр) • •
NOj o,p/n • • •
SR (отр) •
N=C=O (отр) a
9 Я .6 .4 2 8 Л .6 .4 2 7 Я Я .4 2 6 6
а Протоны бензола дают резонанс при 7.27 м.д. От этого значения
и отсчитываются инкременты, как это показано в конце раздела 3.4
6 OTs - и-толуолсульфонилокси-группа
Приложение Д
231
Таблица Г.5. Химические сдвиги протонов в гетероароматических циклах
Таблица Г.б. Химические сдвиги протонов в группах НС=О, H-C=N и HC(OR)3
RCH=O PhCH=O 9.70 9.98 HC(=O)OR HC(=O)NR2 8.05 8.05 RCH=NOH цис RCH=NOH транс 7.25 6.65
RCH=CHCH=O 9.78 HC(OR)3 5.00 r. Ж JL J n H H no2 6.05
Приложение Д. Протоны, на которые влияет водородная связь
(протоны у гетероатомов)0
5 17 16 15 14 13 12 11 10 9 8 7 6 5 4 3 2 1 О
Протон Класс соединений
ОН Карбоновые кислоты h —
Сульфоновые кислоты —1
Фенолы н- —
Фенолы (с внутримолекулярной Н-связью) 1— — —1
Спирты Ь bDN/ ► н
Енолы (циклические а-дикетонов) 1 1
Енолы ф-дикетонов) 1— — -1
Енолы ф-кетоэфиров) в DMSI ч 'f' в ацето не
Вода5 Н н н •
Оксимы —
NH2 и NHR Алифатич. и циклич. амины — -ч
Ароматич. амины
Амиды 1— —
Уретаны 1— — -1
Амины в трифторуксусной кислоте 1— —
SH Алифатические тиолы |—|
Тиофенолы 1— н
S 17 16 15 14 13 12 И 10 9 8 7 6 5 4 3 2 1 О
° Растворитель CDC13. Химические сдвиги зависят от концентрации
(в пределах указанного диапазона).
6 См. раздел 3.6.1.2.
232
3. Спектроскопия ЯМР ]Н
Приложение Е. Константы спин-спинового взаимодействия
Тип Ль. Гц Jab (типичные) Тип Ль. Гц Jab (типичные)
На 0-30 12-15 на /н> С-С 6-12 10
СН,
СН0—СНЬ (свободное вращение) 6-8
7
/СН»
<с=с;
0-3
1-2
СН-С-СНЬ
0-1
О
ах-ах
/СИ.
£=с\
Нь
'с=с:
Н„
4-10
7
6-14
8-10
ax-eq
0-5
2-3
0-5
2-3
снд
0-3
1.5
и, /снь
С=с
C=CHfl—СНЬ=С
0-3
2
eq-eq
(цис или транс)
цис 5-10
транс 5-10
Н,
Нь
К
цис^-12
транс 2-10
сн-с=снь
—сн0—с=с—снь—
9-13
3-х членный
4-х членный
5-х членный
6-х членный
7-х членный
8-х членный
2-3
10
0.5-2.0
2.5-4.0
5.1-7.0
8.8-11.0
9-13
10-13
2-3
(цис или транс)
Н
ъ
(цис или транс)
CHfl—ОНЬ (нет обмена)
О
,сн-снь
О
С=СН-СНЬ
Ио
/С=С\
нь
Иа
'С=С\
Нь
цис 7 -13 транс 4-9 н/‘
ЧТ"*'
4-10 1-3 5 2-3 Н?4 ЧТ'"' Н
5-8 12-18 6 17 4 6^s К ):
4п -пЗ
0-3
0-2
>2
5'
Нь
Нь
6
4
2.5
J (орто) 6-10 9
J (мета) 1-3 3
J (пара) 0-1 -0
7(2-3) 5-6 5
7(3-4) 7-9 8
7(2-4) 1-2 1.5
7(3-5) 1-2 1.5
7(2-5) 0-1 1
7(2-6) 0-1 ~0
7(2-3) 1.3-2.0 1.8
7(3-4) 3.1-3.8 3.6
7(2-4) 0-1 ~0
7 (2-5) 1-2 1.5
Приложение Е
233
Приложение Е (продолжение)
Тип Ль. Гц Jab (типичные) Тип Ль. Гц 7аЬ (типичные)
7(2-3) 4.9-6.2 5.4 Протон—углерод-13 4|| ||3 7(3-4) 3.4-5.0 4.0 (см. таблицы 5.17 и 5.18) ^(2-4) 1.2-1.7 1.5 Протон-фтор S J(2-5) 3.2-3.7 3.4 44_81 4li—ii3 7<1-3) 2"3 а 5|1 JJ2 7(2-3) 2-3 /\ тГ 7(3-4) 3-4 Fo 1 7(2-4) 1-2 7(2-5) 1.5-2.5 XCHe—CF6 Jx J (4-5) 4-6 7 । 3-25 Т I? J (2-5) 1-2 \ 1 1 °-4 6^ JL J (2-4) 0-1 /CH-C-CF, N J (4-6) 2-3 1 1 4|| ii1 J 0-5) 3-4 zc-c\ J(2-4) ~0 НЛ F6 S J (2-5) 1-2 1-8 Ho zc=c\ Fb 12-40 F Ai o6-10 [1 1 m5-6 p2 О || ay 4.3 aH3C—C—CHjFy p-y 48
Протон—фосфор
о ^рн 630-707
(СНз)зР 2.7
(СНз)зР=0 13.4
(СН3СН2)зР 0.5 (HCCP) 13.7 (HCP)
(СН3СН2)зР=0 11.9 (HCCP) 16.3 (HCP)
о II
CH3P(OR)2 10-13
о 1II
CH3CP(OR)2 15-20
СН3ОР (OR)2 10.5-12
P[N(CH3)2]3 8.8
O=P[N(CH3)2]3 9.5
Источник', данные фирмы Вариан (воспроизводится с разрешения).
234
3. Спектроскопия ЯМР ]Н
Приложение Ж. Химические сдвиги и мультиплетности остаточных протонов
в коммерчески доступных дейтерированных растворителях
(Merck Со., 1пс.)в
Соединение6 Молекулярный вес бн (мультиплет)
Ацетон-ё6 64.117 2.04 (5)
Ацетонитрил^ 44.071 1.93 (5)
Бензол-ё6 84.152 7.17 (Вг)
Вода дейтериров. 20.028 4.63 (отн. DSS)e 4.67 (отн. TSP)e
Гексаметилфосфорамид-б18 197.314 2.53 (2x5)
Гексафторацетон дей- териров 198.067 5.26 (1)
Глим-дю 100.184 3.40 (m) 3.22 (5)
Диглим-d )4 148.263 3.49 (br) 3.40 (br) 3.22 (5)
Диметилсульфоксид^6 84.170 2.49 (5)
М,Ы-Диметилформамид-б2 80.138 8.01 (br) 2.91 (5) 2.74 (5)
я-Диоксан-dg 96.156 3.53 (m)
Дихлорметан-б2 86.945 5.32 (3)
1,2-Дихлорэтан^4 102.985 3.72 (br)
Диэтиловый эфир-d 10 84.185 3.34 (m) 1.07 (m)
Изопропиловый спирт-dg 68.146 5.12 (1) 3.89 (br) 1.10 (br) 8.71 (br)
Соединение6 Молекулярный вес бн (мультиплет)
Метанол^4 36.067 4.78 (1) 3.30 (5)
Нитробензол^ 128.143 8.11 (br) 7.67 (br) 7.50 (br)
Нитрометан^ 64.059 4.33 (5)
Пиридин-б5 84.133 7.55 (br) 7.19 (br)
Тетрагидрофуран-dg 80.157 3.58 (br) 1.73 (br)
Толуол-dg 100.191 7.09 (m) 7.00 (br) 6.98 (m) 2.09 (5)
Трифторуксусная кислота-d 115.030 11.50 (1)
2,2,2-Трифторэтанол^3 103.059 5.02 (1) 3.88 (4x3)
Уксусная кислота-д4 64.078 11.53 (1) 2.03 (5)
Хлороформ-d 120.384 7.26 (1)
Циклогексан-di 2 96.236 1.38 (br)
Этанол^6 (абс.) 52.106 5-19 (1)
3.55 (br)
1.11 (m)
а Остаточный протон - это один из протонов любого типа в соединении, в котором все осталь-
ные протоны заменены на дейтерий. Например, дейтерированная уксусная кислота име-
ет остаточные протоны двух типов CD2H-COOD и CD3-COOH. Протон CD2H, связанный
спин-спиновым взаимодействием с двумя атомами дейтерия, имеет химический сдвиг при
5 2.03 с мультиплетностью 5 (т.е. 2nl = 2><2х 1 + 1=5). Карбоксильный протон-это синглет
с 5 11.53
6 Для некоторых растворителей атомарная чистота (в ат. % D) достигает 99.96% («100%-ная»
чистота)
в DSS (ДСС) - натриевая соль З-триметилсилил-1-пропансульфоновой кислоты, TSP -
3-триметилпропионат-2,2,3,3-d4 натрия. Оба стандарта используются в водных растворах.
Приложение 3
235
Приложение 3. Химические сдвиги наиболее распространенных растворителей
в следовых количествах
Протон Мульти- плетность CDCI3 (CD3)2CO (CD3)2SO QD6 CD3CN CD3OD d2o
Остаточный пик растворителя 7.26 2.05 2.50 7.16 1.94 3.31 4.79
Н2О S 1.56 2.84° 3.33° 0.40 2.13 4.87
Ацетон сн3 S 2.17 2.09 2.09 1.55 2.08 2.15 2.22
Ацетонитрил сн3 S 2.10 2.05 2.07 1.55 1.96 2.03 2.06
Бензол СН S 7.36 7.36 7.37 7.15 7.37 7.33
тирети-Бутилметиловый эфир ссн3 S 1.19 1.13 1.11 1.07 1.14 1.15 1.21
осн3 S 3.22 3.13 3.08 3.04 3.13 3.20 3.22
wpew-Бутиловый спирт сн3 S 1.28 1.18 1.11 1.05 1.16 1.40 1.24
он® S 4.19 1.55 2.18
Гесаметилфосфорамид сн3 d, 9.5 2.85 2.59 2.53 2.40 2.57 2.64 2.61
н-Гексан сн3 t 0.88 0.88 0.86 0.89 0.89 0.90
сн2 m 1.26 1.28 1.25 1.24 1.28 1.29
2,6-Ди-тирети-бутил-4- АгН s 6.98 6.96 6.87 7.05 6.97 6.92
метилфенол он® s 5.01 6.65 4.79 5.20
АгСН3 s 2.27 2.22 2.18 2.24 2.22 2.21
АгС(СН3)3 s 1.43 1.41 1.36 1.38 1.39 1.40
Диглим СН2 m 3.65 3.56 3.51 3.46 3.53 3.61 3.67
СН2 m 3.57 3.47 3.38 3.34 3.45 3.58 3.61
ОСН3 s 3.39 3.28 3.24 3.11 3.29 3.35 3.37
Диметилацетамид СН3СО s 2.09 1.97 1.96 1.60 1.97 2.07 2.08
NCH3 s 3.02 3.00 2.94 2.57 2.96 3.31 3.06
NCH3 s 2.94 2.83 2.78 2.05 2.83 2.92 2.90
Диметилсульфоксид СН3 s 2.62 2.52 2.54 1.68 2.50 2.65 2.71
Диметилформамид СН s 8.02 7.96 7.95 7.63 7.92 7.97 7.92
СН3 s 2.96 2.94 2.89 2.36 2.89 2.99 3.01
сн3 s 2.88 2.78 2.73 1.86 2.77 2.86 2.85
1,2-Диметоксиэтан сн3 s 3.40 3.28 3.24 3.12 3.28 3.35 3.37
сн2 s 3.55 3.46 3.43 3.33 3.45 3.52 3.60
Диоксан сн2 s 3.71 3.59 3.57 3.35 3.60 3.66 3.75
Дихлорометан сн2 s 5.30 5.63 5.76 4.27 5.44 5.49
1,2-Дихлорэтан сн2 s 3.73 3.87 3.90 2.90 3.81 3.78
Диэтиловый эфир сн3 t, 7 1.21 1.11 1.09 1.11 1.12 1.18 1.17
сн2 q,7 3.48 3.41 3.38 3.26 3.42 3.49 3.56
Метанол сн3 s6 3.49 3.31 3.16 3.07 3.28 3.34 2.34
он s6 1.09 3.12 4.01 2.16
Нитрометан сн3 s 4.33 4.43 4.42 2.94 4.31 4.34 4.40
н-Пентан сн3 t, 7 0.88 0.88 0.86 0.87 0.89 0.90
сн2 m 1.27 1.27 1.27 1.23 1.29 1.29
Пиридин СН(2) m 8.62 8.58 8.58 8.53 8.57 8.53 8.52
СН(3) m 7.29 7.35 7.39 6.66 7.33 7.44 7.45
СН(4) m 7.68 7.76 7.796 6.98 7.73 7.85 7.87
2-Пропанол сн3 d, 6 1.22 1.10 1.04 0.95 1.09 1.50 1.17
СН sep, 6 4.04 3.90 3.78 3.67 3.87 3.92 4.02
Силиконовая смазка® сн3 s 0.07 0.13 0.29 0.08 0.10
236
3. Спектроскопия ЯМР 'Н
Приложение 3 (продолжение)
Протон Мульти- плетность CDCI3 (CD3)2CO (CD3)2SO QD6 CD3CN CD3OD d2o
Смазкаг СН3 m 0.86 0.87 0.92 0.86 0.88
сн2 br s 1.26 1.29 1.36 1.27 1.29
Тетрагидрофуран сн2 m 1.85 1.79 1.76 1.40 1.80 1.87 1.88
СН2О m 3.76 3.63 3.60 3.57 3.64 3.71 3.74
Толуол СН3 s 2.36 2.32 2.30 2.11 2.33 2.32
СН (о/р) m 7.17 7.1-7.2 7.18 7.02 7.1-7.3 7.16
СН (т) m 7.25 7.1-7.2 7.25 7.13 7.1-7.3 7.16
Триэтиламин СН3 t, 7 1.03 0.96 0.93 0.96 0.96 1.05 0.99
СН2 q,7 2.53 2.45 2.43 2.40 2.45 2.5 2.57
Уксусная кислота СН3 s 2.10 1.96 1.91 1.55 1.96 1.99 2.08
Хлороформ СН s 7.26 8.02 8.32 6.15 7.58 7.9
Циклогексан сн2 s 1.43 1.43 1.40 1.40 1.44 1.45
Этанол сн3 t, 7 1.25 1.12 1.06 0.96 1.12 1.19 1.17
сн2 q, 7d 3.72 3.57 3.44 3.34 3.54 3.60 3.65
он sa.e 1.32 3.39 4.63 2.47
Этилацетат СН3СО s 2.14 2.07 2.07 1.58 2.06 2.12 2.19
СН2СН3 q> 7 2.46 2.45 2.43 1.81 2.43 2.50 3.18
СН2СЯ3 t, 7 1.06 0.96 0.91 0.85 0.96 1.01 1.26
Этиленгликоль СН 3.76 3.28 3.34 3.41 3.51 3.59 3.65
Этилметилкетон СН3СО s 2.14 2.07 2.07 1.58 2.06 2.12 2.19
СН2СН3 q,7 2.46 2.45 2.43 1.81 2.43 2.50 3.18
СН2СН3 t, 7 1.06 0.96 0.91 0.85 0.96 1.01 1.26
° Для этих растворителей скорость межмолекулярного обмена достаточно мала, так что обычно
наблюдается сигнал от HDO, он обнаруживается при 5 2.81 и 3.30 в ацетоне и ДМСО со-
ответственно. В ацетоне этот сигнал является триплетом (1:1:1) с константой 2JHD = 1 Гц.
6 В некоторых случаях (см. сноски а и в) может наблюдаться КССВ между протонами групп
СН3 и ОН (J = 5.5 Гц).
в Пол и( диметил си л оксан). Его растворимость в ДМСО слишком низка, чтобы можно было
наблюдать видимые сигналы.
г Длинноцепочечные, линейные алифатические углеводороды. Их растворимость в ДМСО
слишком мала, чтобы можно было наблюдать достаточно интенсивные сигналы.
д В некоторых случаях (см. сноску а) может наблюдаться КССВ между протонами групп
СН2 и ОН (J = 5 Гц).
е Сигналы обменивающихся протонов не всегда обнаруживаются.
ж Для CD3CN протон ОН-группы наблюдается как мультиплет при 5 2.69 и обычно на мети-
леновом сигнале наблюдается дополнительная константа.
Источник'. Н.Е. Gottlieb, V. Kotlyar, A. Nudelman, J. Org. Chem., 62, 7512 (1997).
Приложение И
237
Приложение И. Химические сдвиги протонов аминокислот в D20
о
Аргинин (Arg) (R)
О
Аспарагин (Asn) (N)
Аланин (Ala) (А)
Аспарагиновая (Asp) (D)
кислота
Цистеин (Cys) (С)
О О
МНз
Глутамин (Gin) (Q)
Глутаминовая (Glu) (Е)
кислота
Глицин (Gly) (G)
Гистидин (His) (Н)
Изолейцин (Due) (I)
Лейцин (Leu) (L)
1.70 1.87
Лизин (Lys) (К)
Метионин (Met) (М)
2.07,2.32
Пролин (Pro) (Р) Фенилаланин (Phe) (F)
О
Серин (Ser) (S)
Н
Триптофан (Trp) (W)
Тирозин (Туг) (Y)
0.97
О
Валин (Vai) (V)
Треонин (Thr) (Т)
4. Спектроскопия ЯМР 13С
4.1. Введение1*
Если бы перед химиками стояла проблема выбора
ядра на ранней стадии развития спектроскопии ЯМР,
то большинство химиков-органиков предпочло бы
спектроскопию на ядрах углерода, а не водорода.
Прежде всего, именно углеродный скелет является
основой строения всех органических соединений.
Но проблема состояла, конечно, в том, что большую
часть скелетных ядер углерода составляют ядра 12С,
которые нельзя изучать с помощью ЯМР. И приходит-
ся ориентироваться только на небольшие количества
ядер 13С.
Между методами спектроскопии ЯМР !Н и 13С
имеется достаточно много различий, что оправдывает
посвящение им отдельных глав. Если хорошо понят
материал гл. 3 (спектроскопия ЯМР !H), то основной
материал данной главы, посвященной ЯМР 13С, будет
усваиваться быстрее.
Ядро 12С магнитно неактивно (спиновое число
равно 0), однако ядро 13С, как и протон, имеет спин
54. Поскольку природное содержание изотопа 13С со-
ставляет только 1.1 %, а чувствительность ядра 13С
составляет лишь 1.6% от чувствительности протона
(т.е. более чем в 60 раз слабее), общая чувствитель-
ность метода ЯМР 13С равна —1/5700 от чувствитель-
ности ПМР* 2).
4.2. Теория
Теоретические основы спектроскопии ЯМР уже были
изложены в гл. 3. Здесь мы рассмотрим только неко-
торые аспекты, связанные с отличием спектроскопии
ЯМР 13С от спектроскопии ЯМР 1Н.
° При изложении материала в этой главе предполагается
знакомство с гл. 3.
2) Вследствие низкого природного содержания 13С вероят-
ность нахождения по соседству двух таких ядер очень
мала, поэтому, нет необходимости учитывать константы
спин-спинового взаимодействия 13С-13С.
• При регистрации спектров ЯМР 13С с полным
подавлением спин-спинового взимодействия
с протонами все сигналы являются синглетами,
если только в молекуле нет других магнитно ак-
тивных ядер (например, 2Н, 31Р или 19F).
• Сигналы 13С распределены по значительно боль-
шему диапазону химических сдвигов по сравне-
нию с диапазоном для ядер !H.
• В обзорных рутинных спектрах интенсивности
сигналов 13С не коррелируют с числом ядер угле-
рода из-за больших времен релаксации и ядер-
ного эффекта Оверхаузера (ЯЭО, NOE).
• Природное содержание ядер 13С существенно
меньше, чем протонов: чувствительность ядер 13С
также существенно ниже, чем у ядер !H. Таким
образом, требуется большее количество образца
и большее время регистрации спектра.
• Для данного дейтерированного растворителя сиг-
налы ЯМР !Н и 13С имеют разную мультиплет-
ность.
На первый взгляд, может показаться, что некоторые
из перечисленных положений сильно снижают ценность
спектров ЯМР 13С. Однако различные изобретательные
находки превратили спектроскопию ЯМР 13С в мощный
метод, что и будет показано в этой главе. В действи-
тельности «паралельная» интерпретация спектров ЯМР
’Н и 13С дает взаимно дополняющую информацию.
4.2.1. Подавление спин-спинового
взаимодействия с протонами
Как отмечалось в разделе 3.7.5, ядра 13С не обна-
руживают констант в спектрах ЯМР (не считая
13С-сателлитов) из-за низкого природного содержания
изотопа 13С (1.1%). Однако в спектрах ЯМР 13С расще-
пления на протонах, конечно, оказывают существенное
влияние на форму спектра. Природное содержание
ядер !Н более 99%, поэтому каждое ядро 13С связано
спин-спиновым взаимодействием с каким-то числом
4.2. Теория
239
---мммрм.-.-пг.р . . |.|М....|.. I I | Т О I I I I I I | I I I Г .r.jr. Г !.|1.|.|.
140 130 120 ПО 100 90 80 70 60 50 40 30 20 10 м.д.
Рис. 4.1. Спектры ЯМР 13С холестерина в CDCI3 на частоте 150.9 МГц
без подавления (а) и с полным подавлением (б) спин-спинового
взаимодействия с протонами
протонов. Поскольку константы V^C^H) (через одну
связь) велики (110-320 Гц), а также заметную вели-
чину имеют геминальные и вицинальные константы
(V^C-'H) и Ц^СЯН)) (0-60 Гц), в спектрах ЯМР
13С обнаруживаются сложные перекрывающиеся муль-
типлеты, которые довольно трудно интерпретировать.
В качестве примера на рис. 4.1, а приведен спектр
ЯМР 13С холестерина. На самом деле интерпретация
спектров ЯМР 13С без подавления спин-спинового
взаимодействия с протонами весьма сложна, так как
нужно выявить все возможные взаимодействия для
каждого ядра углерода 13С с протонами этой системы.
Для решения этой проблемы была предложе-
на важная методика - регистрация спектров ЯМР
13С с одновременным полным подавлением спин-
спинового взаимодействия с протонами (иначе го-
ворят «с широкополосной развязкой от протонов»).
Такое подавление достигается облучением протонов
в широком диапазоне частот с помощью специального
широкополосного генератора частот или посредством
метода «развязки» составными импульсами (метод
CPD-Composite Pulse Decoupling)^. На рис. 4.1 б по- В *
В методе CPD используется непрерывная последова-
тельность составных импульсов.
казан спектр ЯМР 13С холестерина с подавлением
спин-спинового взаимодействия; в нем присутствуют
27 сигналов, причем каждый сигнал соответствует
одному из 27 неэквивалентных углеродных атомов
холестерина. Чтобы не забегать вперед слишком силь-
но, отложим интерпретацию спектров ЯМР 13С до
разделов 4.3 и 4.7.
Стандартная импульсная программа для при реги-
страции спектров ЯМР 13С с полной «развязкой» от
протонов показана на рис. 4.2, а. Последовательность
включает релаксационную задержку (/?J (см. раздел
4.2.3), ВЧ-импульс (0) и выборку сигнала (/2). Про-
тонный канал действует для подавления констант
^С^Н, а мощный и короткий (порядка нескольких
микросекунд) импульс возбуждает резонанс всех ядер
13С одновременно. Поскольку несущая частота на-
ходится слегка вне положения точного резонанса,
каждое ядро 13С дает свой ССИ, представляющий
собой экспоненциально спадающую синусоидаль-
ную функцию.
На рис. 4.2, б показан сигнал ССИ для спектра
ЯМР 13С холестерина с «развязкой» от протонов, а на
рис. 4.2, в - растянутая небольшая часть этого сигнала.
Сложная форма ССИ является результатом наложения
большого числа синусоидальных колебаний (наложение
240
4. Спектроскопия ЯМР 13С
0.2 0.4 0.6 0.8 1.0 1.2 1.4 1.6 1.8 2.0 2.2 2.4 2.6 2.8 с
Рис. 4.2. а - Стандартная импульсная последовательность для регистрации
спектров ЯМР 13С с полным подавлением спин-спинового взамодействия
с протонами (^-релаксационная задержка, в- переменный угол импульса,
t2 - время выборки), б - Сигнал ССИ, представляющий собой синусоидальную
функцию, для холестерина (ЯМР 13С на частоте 150.9 МГц в CDCI3).
в - Растянутый участок сигнала ССИ
сигналов приводит к их интерференции и, в частно-
сти, к характерным биениям при наложении близких
частот). Сигнал создается посредством серии им-
пульсов, повторяющихся выборок сигнала (сканов)
и релаксационных задержек. Фурье-преобразование,
проводимое с помощью компьютера, превращает сиг-
нал ССИ во временном представлении в спектр в при-
вычном для нас частотном представлении. Именно
этот окончательный спектр для резонансной частоты
ЯМР 13С 150.9 МГц (раствор в CDC13) показан на
рис. 4.1, б.
В результате при отсутствии других (кроме про-
тонов) магнитных ядер в системе (таких как 2Н, 31Р,
19F) для каждого неэквивалентного атома углерода
возникает узкий сигнал (за исключением редких слу-
чаев совпадений химических сдвигов). Посмотрите на
спектр ЯМР 13С холестерина с «развязкой» от прото-
нов, приведенный на рис. 4.1, б, и убедитесь, насколько
он прост по сравнению со спектром без подавления
взаимодействия с протонами (рис. 4.1, а). Однако не-
обходимо учесть, что при «развязке» от протонов
утрачивается полезная информация о мультиплетности
сигналов. В частности, прямые константы ^(^С-1!!)
в спектре без «развязки» должны расщеплять сигнал
в мультиплет, состоящий из (п + 1) линий (где п -
число непосредственно присоединенных протонов).
Например, на рис. 4.1, а сигнал метильной группы
(при 11 м.д.) представляет собой квартет (1:3:3:1),
а метановый сигнал (при 122 м.д.)-дублет (1:1).
Более полное обсуждение мультиплетов можно найти
в разделах 4.2.5 и 4.3. Впрочем, имеются методики
(например, последовательность DEPT, см. раздел 4.6),
которые дают эту информацию в существенно более
простой форме.
4.2.2. Шкала химических сдвигов
Как и в случае спектров ЯМР !Н, шкала частот
в спектрах ЯМР 13С превращается в безразмерную
шкалу химических сдвигов (выраженных в едини-
цах 8, м.д.). В обзорных (рутинных) спектрах ЯМР
13С диапазон химических сдвигов обычно составляет
220 м.д. в слабое поле от сигнала ТМС, и это при-
мерно в 20 раз больше диапазона химических сдвигов
4.2. Теория
241
I------1 I I 1 I---------------1------1-----1-------1-----1 I----------1-------1-----1------1-----1------1------r I----------1------1------1 *3
200 /ВО /60 /40 /20 /00 80 60 40 20 О C
MEZZZao Альдегиды
L ...
Ароматические соединения
Сопряженные алкены ^м?7777///л~ ~ и ~| Концевые
метиленовые группы
I-----1 )СН-0
М диапазон химических
сдвигов 13С
□ диапазон химических
сдвигов 1Н
ZZ] область перекрывания
-снго
Химические
сдвиги 1Н и 13С
□= -О-СН,
Хн-ыС,-снг-Ксм3-г<
-с-с-н i---1
)СН -cf,-CHt-C^ CH3-Ct° I !/////!
Рис.4.3. Сравнение химических сдвигов ядер ]Н и 13С
протонов (~10 м.д.). Вследствие большого диапазона
химических сдвигов и узости спектральных линий
совпадение химических сдвигов от неэквивалентных
атомов углерода весьма маловероятно, и примеси лег-
ко идентифицируются. Часто даже смеси веществ
дают полезную информацию (см. приложение Б,
где приведен обширный список распространенных
примесей). Например, с помощью спектров ЯМР !Н
довольно трудно анализировать стереоизомеры, в то
же время эти изомеры, как правило, дают различаю-
щиеся сигналы в спектрах ЯМР 13С.
Для определения частоты магнитного резонанса ис-
пользуется фундаментальное выражение (у = (у/2л)Б0),
позволяющее определить частоту при известном маг-
нитном поле. Например, для спектрометра с частотой
резонанса протонов 600 МГц получается частота ЯМР
13С, равная 150.9 МГц, т.е. частоты ЯМР !Н и 13С
соотносятся приблизительно как 4:1. Это отношение
прямо связано с отношением гиромагнитных посто-
янных, которые равны 26.753 и 6.728 (в единицах
107 рад Тл-1 с-1) для ядер !Н и 13С соответственно
(см. приложение А к гл. 6). Как дань истории, спек-
трометры часто называют по их протонной частоте
независимо от того, на каких ядрах работают.
Рисунок 4.3 подтверждает идею о параллельности
(симбатности) химических сдвигов *Н и 13С, впрочем,
можно заметить и некоторые отклонения, которые
не так легко объяснить и которые в целом требу-
ют других доказательств (см. раздел 4.7)!) В целом,
корреляции химических сдвигов (например, с элек-
троотрицательностью) лучше работают для ядер *Н,
чем для ядер 13С.
4.2.3. Спин-решеточная релаксация (время 1\)
В отличие от спектров ЯМР !Н, где интегрирование
сигналов дает нам отношение протонов в отдельных
группах, интегрирование сигналов 13С в рутинных
спектрах не коррелирует с числом атомов углерода.
(Здесь под рутинными спектрами мы понимаем спек-
тры, записанные в некоторой обобщенной манере,
с минимальными релаксационными задержками (для
ускорения эксперимента) и достаточно короткими вре-
менами выборки, приводящими к ограничениям по
° Например, доказательств, основанных на теоретических
расчетах констант экранирования. В последнее время рас-
четы методами ab initio или DFT (теория функционала
плотности) стали весьма популярны. - Прим, персе.
242
4. Спектроскопия ЯМР 13С
разрешению и к эффектам насыщения.) Два главных
фактора определяют проблему интенсивности в спек-
трах ЯМР 13С.
• Времена спин-решеточной релаксации (Тх) (вре-
мена продольной релаксации) изменяются в ши-
роких пределах для различных типов атомов
углерода.
• Усиление сигналов за счет ЯЭО проявляется
только для атомов углерода с непосредствен-
но присоединенными протонами (т.е. для СН,
СН2 и СН3-групп) (раздел 4.2.4).
При регистрации спектров ЯМР 13С в импульс-
ном режиме возникают две технические проблемы:
оптимизация чувствительности спектрометра (чтобы
использовать минимальное количество образца) и вы-
полнение условия пропорциональности интенсивности
сигнала количеству ядер данного типа (что могло бы
позволить использовать интенсивности сигналов для
целей количественного анализа). Для оптимизации
соотношения сигнал/шум используют не 90°-ные
импульсы (казалось бы, дающие максимальную чув-
ствительность, поскольку эти импульсы полностью
поворачивают вектор намагниченности в плоскость
ху), а импульсы с оптимальным углом (называемым
углом Эрнста по имени первооткрывателя метода им-
пульсной фурье-спекгроскопии-лауреата Нобелевской
премии по химии, 1992 г.), который находится из урав-
нения cosO = exp^Tr/TJ, где ТТ- период повторения,
а Тх - время релаксации. Для малых молекул обычно
используют 45°-ные импульсы и достаточно короткие
задержки между импульсами (3-4 с). Для молекул
большего размера, которые обычно имеют короткие
времена релаксации, релаксационные задержки мо-
гут быть существенно короче. При использовании
импульсного режима с многократным повторением
возбуждения сигнала ССИ нужно обращать внима-
ние на то, что из-за слишком частого повторения
импульсов возбужденная спиновая система может
не успевать отрелаксировать за время между двумя
последовательными импульсами. В результате каждый
последующий ССИ становится меньшим по интенсив-
ности (эффект насыщения), и постепенно возбуждение
становится неэффективным. При этом интенсивность
сигнала становится непропорциональной количеству
ядер данного сорта, поскольку для разных ядер в связи
с различием времен релаксации эффекты насыщения
будут оказывать различное влияние. Чтобы мини-
мизировать эти эффекты насыщения, рекомендуется
выбирать интервал между импульсами или период
повторения Тт > 5 Тх (где Тх - максимальное время
релаксации для системы ядер с разными временами
релаксации). Поскольку интервал между импульса-
ми состоит из очень короткого импульса (порядка
10-3—10-5 с) и выборки сигнала, длящейся порядка
времени Т2, то для удовлетворения условия Тг > 5 Тх
вводится релаксационная задержка между последо-
вательными ВЧ-импульсами. Для протонов (см. раз-
дел 3.2.3) времена релаксации Тх и Т2 сравнительно
малы, поэтому указанное правило Тт > 5 Тх проще
удовлетворить, и при этом интенсивности сигналов
более точно оказываются пропорциональными числу
протонов. Для ядер 13С времена релаксации могут
оказаться очень велики (например, для четвертичных
атомов углерода, в частности для групп С=О, они
могут достигать величин порядка 102 с). Тогда для
выполнения правила Тх > 5 Тх могут потребоваться
задержки между импульсами, составляющие десятки
минут, что приведет к резкому увеличению времени
эксперимента и в целом существенно нивелирует
преимущества импульсной методики.
Иногда необходимо измерить времена релаксации
Ть чтобы правильно поставить эксперимент (т.е. вве-
сти необходимые интервалы между импульсами и
не потерять слабые сигналы в шуме и получить
количественные данные). Для определения времен
релаксации Тх очень часто используют метод «инвер-
сия-восстановление» (см. рис. 4.4). В общем случае
время Тх уменьшается при увеличении числа про-
тонов, непосредственно присоединенных к данному
атому углерода. Другими словами, четвертичные атомы
дают сигналы 13С минимальной интенсивности (что
показано на рис. 4.4, а), и одновременно сигналы этих
углеродных ядер наиболее медленно восстанавлива-
ются в экспериментах по методу «инверсия-восста-
новление». Впрочем, часто довольно трудно различить
атомы углерода СН, СН2 и СН3 только лишь на основе
их времен релаксации Ть поскольку на последние влия-
ют и другие факторы (кроме числа непосредственно
присоединенных протонов). Значения времен релакса-
ции Тх для групп СН3 занимают диапазон в несколько
секунд, а для четвертичных атомов углерода они могут
превышать минуту. Рекомендуется вводить задержку
между импульсами, равную 5 Тх (Тх - максимальное
время релаксации для группы изучаемых ядер угле-
рода), и этого правила следует придерживаться при
проведении количественного анализа.
На рис. 4.4, а показана последовательность импуль-
сов, используемая в методике «инверсия-восстанов-
ление». Эта последовательность включает следующие
шаги: 1) релаксационную задержку (для полного вос-
становления намагниченности), 2) 180°-ный импульс,
3) переменный временной интервал (Ат), 4) 90°-ный
импульс, 5) выборку (при одновременной «развязке»
от протонов).
42. Теория
243
180%
Релаксационная\ I
задержка (Rd) I
*Н: ---------Ц_
90%
«Развязка » включена
Рис. 4.4. а - Импульсная последовательность метода «инверсия-восстановление»
для измерения Tv б - Малые задержки Ат, в - большие задержки Ат. г - Набор
данных, полученных по описываемой методике, для диэтилфталата при 75.5 МГц
(отнесение сигналов в спектре диэтилфталата обсуждается в разделе 4.3)
На рис. 4.4, б общая ядерная намагниченность
MQ представлена в виде вектора, направленного
вверх и показанного жирным шрифтом. Первый
180°-ный импульс инвертирует намагниченность на
180° (вращает вектор намагниченности по часовой
стрелке) и направляет ее вектор вдоль оси -z. Далее
дается короткий временной интервал Ат, в течение
которого происходит уменьшение длины вектора Af0
244
4. Спектроскопия ЯМР 13С
(т.е. возвращение к исходному положению вдоль оси
+z) вследствие релаксации. После этого действует
90°-ный импульс, который поворачивает вектор на-
магниченности по часовой стрелке и направляет
его вдоль оси -у. В этот момент приемник по оси
z считывает сигнал как отрицательный и несколько
уменьшенный по интенсивности.
На рис. 4.4, в интервал Дт был увеличен, так что
вектор намагниченности релаксирует дальше по
направлению восстановления. На самом деле век-
тор MQ проходит нулевую точку и, как показано на
этом рисунке, уже смотрит вверх (вдоль оси +z),
хотя он и уменьшен по амплитуде (по сравнению
с равновесным значением). Затем 90°-ный импульс
поворачивает этот вектор по часовой стрелке, и он
регистрируется как положительный сигнал.
На рис. 4.4, г в спектре ЯМР 13С диэтилфтала-
та последовательность «инверсия-восстановление»
повторяется 8 раз (см. раздел 4.4, где дано полное
отнесение сигналов в спектре) с постепенно воз-
растающей задержкой Ат. Видно, что различные
сигналы имеют разное время прохождения нуле-
вой точки, что указывает на их разные времена ре-
лаксации Тх. Как отмечалось выше, сигнал группы
С=О при 167 м.д. характеризуется самой медленной
релаксацией (т.е. самым длинным временем восста-
новления в этих экспериментах). Этот сигнал про-
ходит нулевую точку где-то между седьмым и вось-
мым спектрами, в то время как сигнал метильной
группы при 13 м.д. инвертирует между четвертым
и пятым спектрами. Время может быть рассчитано
по формуле Тх = Znull/ln(2) (здесь /пи11-время прохож-
дения нулевой интенсивности), которая дает оценки
времени достаточно хорошие для большинства
практических задач.
4.2.4. Ядерный эффект Оверхаузера
Мы уже описывали ядерный эффект Оверхаузера
для протонов в разделе 3.16, а теперь рассмотрим
гетероядерный ЯЭО, который возникает при широко-
полосной «развязке» от протонов при регистрации
спектров ЯМР 13С (см. рис. 4.1, б). Общий эффект
ЯЭО при этом состоит в усилении интенсивностей
сигналов тех ядер 13С, которые непосредственно связа-
ны с протонами. Это усиление вызвано нарушениями
в населенностях спиновых уровней (по сравнению
с предсказываемыми на основании закона Больцмана).
Общий эффект увеличения интенсивностей зависит от
теоретического максимума и от типа релаксации. Тео-
ретический максимум (т.е. максимальное увеличение
интенсивностей) равен (1/2)(ун/Ус)> и он достижим,
когда доминирует механизм дипольной релаксации
(т.е. влиянием других каналов релаксации, в частно-
сти квадрупольной или на парамагнитных примесях,
можно пренебречь). При регистрации спектров ЯМР
13С с полным подавлением спин-спинового взаимо-
действия с протонами величина усиления может ока-
заться близкой к (1/2)(ун/ус) (т.е. равной 1.98). Таким
образом, может быть получено общее трехкратное
увеличение интенсивности сигнала, так как усиление
вследствие ЯЭО прибавляется к исходной интенсив-
ности (т.е. 1+1.98 = 2.98).
Фактические усиления в результате ЯЭО могут ле-
жать в интервале от 0 (какое-либо усиление отсутствует)
до 1.98 (почти трехкратное усиление сигнала) в за-
висимости от механизма релаксации для конкретного
ядра. В реальности атомы углерода, не имеющие непо-
средственно присоединенных протонов (четвертичные
атомы углерода), не показывают усиления за счет ЯЭО,
так как для них практически отсутствует дипольная
релаксация. Для небольших органических молекул ди-
польная релаксация 13С-!Н является весьма эффектив-
ным механизмом релаксации, что приводит к сильному
(до 3 раз) увеличению интенсивности сигнала.
Общий эффект состоит в существенном уменьше-
нии времени, необходимого для регистрации спектра
с «развязкой» от протонов (по сравнению со спектром
без «развязки»). Кроме того, вклад в интенсивность
возрастает (хотя и нелинейно) с числом непосред-
ственно присоединенных протонов. Ядра 13С без
таких протонов характеризуются сигналами низкой
интенсивности (см. рис. 4.1, б). Эти искажения интен-
сивностей затрудняют их использование как меры
количества атомов углерода данного типа.
4.2.5. Константы спин-спинового
взаимодействия 13С-1Н (/сн)
Константы спин-спинового взаимодействия
(или величины JCH), по крайней мере на началь-
ной стадии интерпретации спектров, менее важны,
«Развязка» включена «Развязка» выключена
(Rd)
13С: ----------
Рис. 4.5. Импульсная последовательность для
прерываемой «развязки» от протонов
(/^-релаксационная задержка, в - изменяемый угол
импульса, t2 - время выборки)
4.2. Теория
245
чем протон-протонные константы JHH в спектроско-
пии ЯМР 1Н. Рутинные спектры ЯМР 13С обычно
регистрируют с подавлением спин-спинового взаи-
модействия с протономи, т.е. константами 13С-!Н
жертвуют, чтобы быстрее получить спектр (или если
количество образца ограниченно). В результате спектр
получается сравнительно простым и не содержит
сложных перекрываний сигналов.
Для спектра ЯМР 13С холестерина (см. рис. 4.1, а)
константы 13С-!Н были сохранены. Причем удалось сэ-
кономить значительное время, благодаря тому что ис-
пользовался не обычный метод непрерывной широкопо-
лосной развязки, а специальная методика-прерываемая
протонная «развязка» (gated proton decoupling). Принцип
прерываемой «развязки» можно пояснить следующим
образом. Чтобы сохранить по крайней мере часть ЯЭО
и одновременно сохранить константы ^С^Н, широко-
полосная протонная «развязка» включается только в пе-
риод релаксационной задержки, а затем выключается
на период короткой выборки (рис. 4.5). Таким образом,
ЯЭО (медленный процесс), который происходит в тече-
ние довольно длительного периода релаксации (обыч-
но несколько секунд), утрачивается только частично за
короткое время выборки. Установление спин-спинового
взаимодействия и соответствующих констант (быстрый
процесс) происходит мгновенно, и эти константы со-
храняются в течении периода выборки. В результате
получается спектр с константами 13С-!Н, в котором по
крайней мере частично сохраняется ЯЭО. Таким спосо-
бом можно сэкономить время при накоплении сигналов.
Мы уже демонстрировали полезность констант спин-
спинового взаимодействия на рис. 4.1, а, где хорошо вид-
ны некоторые константы Ц^СНН). Некоторые важные
данные по значениям констант Ц^СНН) приведены
в табл. 4.1.
Константы через одну связь (VCH) находятся
в диапазоне 110-320 Гц, и они растут с увеличени-
ем 5-характера связи С-Н (т.е. в ряду этан, этилен,
ацетилен). Кроме того, константы 13С-!Н растут при
наличии у данного атома углерода электроотрицатель-
ных заместителей. Константы 13С-!Н увеличиваются
также при искажении валентных углов (например,
в ряду циклогексан, циклобутан, циклопропан). За-
метные константы 13С-!Н возникают также между
атомами углерода и протоном, разделенными двумя
и даже тремя связями (константы V^C^H)). Неко-
торые константы через две связи (2JCH) перечислены
в табл. 4.2.
В случае 5р3-гибридизованных атомов углерода
константы 13С-!Н через три связи (3JCH) сравнимы
с константами через две связи. Однако в ароматиче-
ских системах константы 3JCH существенно больше
Таблица 4.1. Некоторые константы 17сн
Соединение
Константа Vch, Гц
СНд CH3CH3 СНзСН2СН3 (СНз)зСН 125.0 124.9 119.2 114.2
^-н 123.0
СУН 128.0
PhCH3 129.0
ch3nh2 133.0
134.0
н
ROCH3 140.0
CH3OH 141.0
СН3С1 150.0
СН3Вг 151.0
£>—Н 161.0
(СНзОЬОЬ 162.0
СН2С12 178.0
t>-H 180.0
н
205.0
СНС13 209.0
СНзСН=С(СН3)2 148.4
сн2=сн2 156.2
ед 159.0
(Vй 160.0
с=с=с—н 168.0
170.0
н
СНз£Н=О 172.4
\ У~н 178.0
NH2CH=O 188.3
=COH(OR) 195.0
СН3СНХ, X=halogen 198.0
[^-н 238.0
CH=CH 249.0
C6H5C=CH 251.0
HC=N 269.0
246
4. Спектроскопия ЯМР 13С
Таблица 4.2. Некоторые константы 27сн
Соединение
Константа 2JCH, Гц
sp3
СНзСНэ -4.5
СНзСС13 5.9
RC(=O)CHj 6.0
СН3СН=О 26.7
*С6Н6 1.0
сн2=сн2 2.4
с=с(сн3)н 5.0
(СН3)2С=О 5.5
сн2=снсн=о 26.9
сн=сн 49.3
с6н5ос=сн 61.0
* 3JCH = 7.6 Гц (>2 J)
констант 2<7СН. Например, в бензоле константа, а кон-
станта 2JCH = 1.0 Гц (см. табл. 4.2).
Константы спин-спинового взаимодействия между
13С и другими ядрами (например, 31Р, 19F, 2D) можно
наблюдать в спектрах с «развязкой» от протонов.
Некоторые данные по этим константам приведены
в табл. 4.3.
4.2.6. Чувствительность
Ядра 13С характеризуются низким уровнем природ-
ного содержания (1.1%), и они имеют существенно
меньшую чувствительность, чем протоны. Поэтому
для достижения достаточного высокого отношения
сигнал/шум необходимо использовать большие ко-
личества образца и большие времена накопления.
Давайте вновь обратимся к величине чувствитель-
ности в импульсном эксперименте ЯМР, где величина
отношения сигнал/шум определялась формулой (см.
раздел 3.3.2):
SIN =
Заметим, что отношение сигнал/шум (S/N) растет
пропорционально величине Vns, где ns - число по-
вторений импульсной последовательности. Это со-
отношение не очень важно в спектроскопии ЯМР
!Н, где достаточно хорошее отношение сигнал/шум
может быть получено для образца массой от не-
скольких микрограммов до 1 мг. Как указывалось
ранее, метод ЯМР * 13С примерно в 6000 раз менее
чувствителен, чем ЯМР !Н. Следовательно, для ЯМР
13С требуется большое количество образца (7V), бо-
лее сильные магнитные поля (Во) (или даже другие,
более чувствительные датчики ЯМР, например крио-
датчики) или большее число прохождений (ns) при
накоплении сигнала.
В большинстве случаев единственной реальной
возможностью увеличения отношения сигнал/шум
является увеличение числа прохождений. Причем
увеличение отношения S/N, например, в 10 раз тре-
бует увеличения числа прохождений в 100 раз (и,
таким образом, при этом существенно увеличивает-
ся время эксперимента). Другое решение, о котором
речь пойдет в разделе 5.4.2, состоит в проведении
Таблица 4.3. Константы взаимодействия ядра 13С с ядрами 19F, 31Р и D
Соединение ’Л Гц 2ЛГц 3ЛГц 4ЛГц
CH3CF3 271
CF2H2 235
cf3co2h 284 43.7
c6h5f 245 21.0 7.7 3.3
(C4H9)3P 10.9 11.7 12.5
(СН3СН2)Р+ВГ 49.0 4.3
(С6Н5)Р+СН3Г 88.0 10.9
lJ для СН3=52 Гц
С6Н5(Р=О)(ОС2Н5)2 143 7.1 Ссор) 6-9 Сссор)
(С4Н5)3Р 12.4 19.6 6.7
CDC13 31.5
CD3(C=O)CD3 19.5
(CD3)2SO 22.0
C6D6 25.5
4.2. Теория
247
эксперимента по передаче более высокой чувстви-
тельности от одного высокочувствительного ядра
(например, !Н) к другому менее чувствительному
ядру (например, ядру 13С).
Обычный (рутинный) спектр ЯМР 13С на частоте
75.5 МГц, как правило, требует 10 мг образца, рас-
творенного в 0.5 мл дейтерированного растворителя
и помещенного в ампулу с внешним диаметром 5 мм.
Образцы с массой порядка 100 мкг могут измеряться
в более тонких ампулах (в частности, с диаметром
2.5 мм), в которых достигается существенно более
высокая чувствительность (см. раздел 3.3).
4.2.7. Растворители
Образцы для измерений спектров ЯМР 13С обычно
представляют собой растворы в CDC13, причем для
обеспечения сигнала внутреннего стандарта исполь-
зуется добавка ТМСЧ Список часто используемых
дейтерированных растворителей приведен в прило-
жении А.
Для данного дейтерированного растворителя его
сигналы в спектрах ЯМР !Н и 13С существенно раз-
личаются по мультиплетности. Например, небольшое
количество остаточного СНС13 в дейтерированном
коммерческом хлороформе (CDC13) дает небольшой
синглетный сигнал около 7.26 м.д. Сигнал ЯМР !Н
при этом не расщепляется, поскольку соседнее ядро
12С магнитно неактивно, а изотопы хлора (35С1 и 37С1)
обладают сильными квадрупольными эффектами,
вызывающими устранение спиновых расщепле-
ний за счет взаимодействия !Н и ядер 35С1 и 37С1
(см. раздел 3.7). Небольшое количество изотопа 13С
в остаточном хлороформе недостаточно, чтобы вы-
звать появление заметных 13С-сателлитных сигналов
в спектре ЯМР !Н. Для другого частого используемого
растворителя - дейтерированного диметилсульфок-
сида (ДМСО-с16) - в протонном спектре наблюдается
квинтет (1:2:3:2:1) линий при 2.49 м.д. от остаточного
(«недодейтерированного») ДМСО (т.е. ДМСО-с^ или
HD2C-S(=O)-CD3).
Такой характер мультиплетности вызван тем,
что два соседних ядра дейтерия (со спином 1) могут
находиться в пяти возможных состояниях (с про-
екциями -2, -1,0, +1, +2), причем вероятности этих
и В настоящее время для современных спектрометров
ТМС, как правило, не добавляют. В качестве опорного
сигнала для измерения химического сдвига используются
сигналы дейтерированного растворителя. Впрочем, спектр
вещества представляется в 5-шкале (относительно ТМС)
и для этого используются значения химических сдвигов
растворителей, приведенные в приложении А.
состояний равны 1, 2, 3, 2 и 1 соответственно. Рас-
щепление !H-2D может быть рассчитано по формуле
JfH-^D) = (Уг/Ун) Анн)- Учитывая, что геминальная
константа J(1H-1H) для метильной группы должна
быть порядка -10 + -15 Гц, соответствующая констан-
та JfH-^D) должна быть порядка -1.5...-2 Гц, и она
будет проявляться в спектрах ЯМР !Н как расстояние
между линиями мультиплета. Заметим, что ядра дей-
терия в соседней CD3-rpynne ДМСО-с15 практически
не взаимодействуют с протоном СВ2Н-группы (кон-
станта через четыре химических связи).
В спектре ЯМР 13С растворитель CDC13 дает три-
плет вида 1:1:1 при 77 м.д. В данном случае при-
месь небольшого количества остаточного СНС13 не-
существенна, так как этой примеси обычно немного
(1-2%). Кроме того, в спектре ЯМР 13С между цен-
тром сигналов дейтерированного и недейтерирован-
ного хлороформа должен наблюдаться небольшой
изотопный сдвиг (порядка 0.1-0.2 м.д.), причем
сигнал CDC13 оказывается в более сильном поле
(справа в спектре). Триплет в спектре CDC13 вы-
зван спин-спиновым взаимодействием ядра 13С с од-
ним ядром дейтерия, где возникают три проекции
-1, 0 и +1 с одинаковыми вероятностями, отсюда
и интенсивности компонент 1:1:1. Для случая дей-
терированного ДМСО-с16 расщепление в спектрах
ЯМР 13С в результате взаимодействия с тремя со-
седними ядрами дейтерия приводит к септету вида
1:3:6:7:6:3:1 при 39.7 м.д., так как три спина 1 могут
давать семь возможных проекций (-3, -2, -1, 0, +1,
+2,+3) с вероятностями 1:3:6:7:6:3:1. На приведенной
ниже диаграмме показаны расщепления сигналов на
ядрах дейтерия в группах CD, CD2 и CD3, что, по су-
ществу, является аналогом треугольника Паскаля для
спина 1 (ср. с рис. 3.32).
Пример
CDC13
CD2C12
(CD3)2SO
Замещение протона на атом дейтерия при данном
атоме углерода приводит к драматическим эффектам
в спектрах ЯМР 13С. Во-первых, возникает небольшой
изотопный сдвиг (сигнал фрагмента 13C-D находится
в более сильном поле, чем сигнал фрагмента 13С-Н).
Во-вторых, появляется расщепление сигнала ядра
13С на соседних ядрах дейтерия, причем константа
спин-спинового взаимодействия 13C-D может быть
248
4. Спектроскопия ЯМР 13С
рассчитана по формуле J(13C-D) = (Yd/YhVch, и пРи
константе «7СН, равной 120-200 Гц (см. табл. 4.2),
соответствующая константа JCD будет составлять
20-30 Гц. В-третьих, для ядра углерода, соседнего
с дейтерием, понижается вклад дипольной релакса-
ции (13С-1Н) и существенно возрастает время спин-
решеточной релаксации Тх. Наконец, в-четвертых, из-за
отсутствия соседнего протона1) уменьшается (либо
полностью исчезает) ЯЭО 13С-!Н. В качестве примера
проявления части этих эффектов можно рассмотреть
спектр ЯМР 13С смеси бензолов С6Н6 и C6D6. В этом
спектре наблюдается триплет от С6Э6при 128.0 м.д.
с расщеплением порядка 25 Гц и синглет от бензола
С6Н6 при 128.5 м.д. (см. табл. 4.12 и приложение А).
4.3. Интерпретация спектра ЯМР 13С
диэтилфталата
Пока мы не обсуждали химические сдвиги для от-
дельных классов соединений (см. раздел 4.7). Однако
уже сейчас можно поговорить об отнесении сигна-
лов в спектре ЯМР 13С диэтил фталата на основе тех
знаний, которые у нас уже имеются, т.е. используя
интенсивности сигналов, константы спин-спинового
взаимодействия 13С-!Н и оценки ожидаемых хими-
ческих сдвигов с использованием аналогий из спек-
тров ЯМР !Н (см. гл. 3). Интенсивности сигналов под-
вержены влиянию времен релаксации Тх и величин
ЯЭО, и, следовательно, интенсивности не связаны
непосредственно с числом атомов углерода. Однако
в ряде случаев это можно использовать при отнесе-
нии сигналов. Так, при рассмотрении спектра ЯМР
13С можно легко распознать сигналы четвертичных
атомов углерода по их низкой интенсивности по
сравнению с другими сигналами спектра. Например,
интенсивности сигналов при 167 м.д. и 132 м.д. отне-
сены к сигналам группы С=О и четвертичного атома
углерода ароматического цикла (отмеченного цифрой
1 на рис. 4.6, а} соответственно. Спин-решеточная
релаксация для ядер 13С, как правило, определяет-
ся диполь-дипольными взаимодействиями ядер 13С
с протонами. Известно, что скорость релаксации про-
порциональна величине 2иГсн, где гсн - расстояние
i 1
между ядром 13С и z-тым протоном, и суммирование
ведется по всем протонам системы. Отсюда очевидно,
что чем ближе расположен протон к данному ядру
’) Это также объясняет относительно низкие интенсив-
ности сигналов дейтерированных растворителей.
углерода, тем сильнее его влияние на релаксацию ядра
13С. В частности, максимальный эффект убыстрения
релаксации создают непосредственно связанные про-
тоны, а атомы углерода без протонов характеризуются
медленной релаксацией и, соответственно, большими
временами релаксации Тх. Это обстоятельство вместе
с небольшим ЯЭО (ЯЭО может вообще отсутствовать)
приводит к уменьшению интенсивности соответствую-
щих сигналов. Следовательно, можно легко определить
карбонильные группы (кроме, формильной), ширильные
группы, алкеновые и алкиновые атомы углерода без
протонов и другие четвертичные углеродные атомы.
Поскольку молекула диэтилфталата (С12Н14О4) име-
ет ось симметрии (и плоскость симметрии), спектр
ЯМР 13С с подавлением ССВ с протонами содержит
шесть пиков (см. рис. 4.6, а). В этом спектре все сиг-
налы - синглеты (так как устранено взаимодействие
с протонами), поэтому перекрывания сигналов не на-
блюдается. Рассмотрим химические сдвиги сигналов
и отметим сходство со шкалой химических сдвигов
*Н относительно расположения сигналов (см. рис.
4.3 и приложение В). Например, следует размещать
сигналы алкильных групп в правой (сильнопольной)
части спектра ЯМР 13С, причем СН2-группы оказы-
ваются менее экранированы, чем группы СН3 (т.е.
сигналы СН2-групп располагаются левее сигналов
СН3-групп). Таким образом, разумно предположить,
что группа из трех сигналов в слабых полях отно-
сится к трем типам ароматических атомов углерода
(отмеченных цифрами 1,2,3). Слабый сигнал в самых
слабых полях (при 167 м.д.) относится к группе С=О.
Уменьшенный по интенсивности сигнал в аромати-
ческой области при 132 м.д., как уже указывалось,
относится к четвертичному атому углерода 1.
На рис. 4.6, б показан спектр ЯМР 13С без подавления
ССВ с протонами. В этом спектре обнаруживаются
константы КССВ 1JCH, и по мультиплетности сигналов
можно определить число протонов, непосредственно
связанных с определенным атомом углерода, что хо-
рошо согласуется с нашим отнесением. В самом деле,
в правой части спектра мы видим квартет СН3-группы
при 14 м.д. (применяется правило М = и+1, где п -
число протонов, связанных с данным атомом углерода,
М - мультиплетность сигнала) и левее при 62 м.д. -
триплет СН2-группы. Далее расположены кластер из
двух дублетов двух ароматических СН-групп и синглет
ароматического атома углерода без протонов (отмечен
цифрой 1). И, наконец, в слабопольной области виден
синглет группы С=О.
На рис. 4.6, в мы видим спектр с растяжкой для
квартета метильной группы, каждый сигнал которого
теперь выглядит как триплет с КССВ 2JCH вследствие
расщепления на двух протонах расположенной ря-
4.3. Интерпретация спектра ЯМР ,}С диэтилфталата
249
Г"......I........I........I........I........I.....'"I.......................... ।........।........j........,.......।.........|..и....!........,.......
170 160 150 140 130 120 ПО 100 90 80 70 60 50 40 30 20 м.д.
СН2
Рис. 4.6. Спектры ЯМР 13С (на частоте 150.9 МГц) диэтилфталата в CDCI3:
а - с подавлением ССВ с протонами, б - без подавления ССВ с протонами,
в-е - растянутые фрагменты спектра б
250
4. Спектроскопия ЯМР 13С
дом СН2-группы. Эта КССВ 2</сн намного меньше,
чем КССВ 7СН.
На рис. 4.6, г показан растянутый триплет СН2-
группы, каждый пик которого теперь является кварте-
том с КССВ 2<7СН в результате взаимодействия с тремя
протонами группы СН3. На рис. 4.6, д приводится
растянутый дублет из правой части группы сигна-
лов в области ароматических атомов углерода; он
отмечен цифрой 2 на рис. 4.6, б. Каждый пик этого
дублета расщепляется взаимодействием с соседними
протонами ароматического кольца в сложный муль-
типлет с КССВ 2JCH и 3JCH.
На рис. 4.6.е в области 19630-19800 Гц показан
дублет дублетов, соответствующий еще одному атому
углерода (3) ароматического цикла. Каждый пик этого
дублета расщепляется еще в один дублет за счет спин-
спинового взаимодействия с соседними протонами
ароматического кольца. На этом же рисунке (рис. 4.6, ё)
приводится растянутый синглет ароматического атома
С1. Этот синглет не расщепляется с большой КССВ
1J, так как соответствующий атом углерода не имеет
связанных с ним протонов, однако он расщепляется
в сложный мультиплет с небольшими КССВ 2J и 3J.
Отнесение сигналов ароматических атомов угле-
рода С2 и СЗ можно попытаться сделать на основе
аддитивности влияния заместителей. Так, химический
сдвиг атома С2 определяется влиянием заместителей
-С(=О)ОС2Н5, находящихся к нему в орто- и мета-
положениях, а химический сдвиг атома СЗ определя-
ется влиянием заместителей -С(=О)ОС2Н5, находя-
щихся к нему в мета- и ияря-положениях. Используя
данные табл. 4.12 (см. раздел 4.7.4), можно рассчи-
тать химические сдвиги для сигналов атома С2 (5 =
129.6 м.д.) и СЗ (8 = 133.2 м.д.). Экспериментальные
химические сдвиги этих атомов углерода составляют
128.5 и 131.2 м.д. Таким образом, более предпочтите-
лен следующий вариант отнесения: 8(С2) = 128.5 м.д.
и 8(СЗ) = 131.2 м.д. Если попытаться спрогнози-
ровать вид протонного спектра для ароматических
протонов диэтилфталата, то используя аддитивные
влияния по данным схемы Г.1 (из приложения Г гл.З),
получим для Н2 8=8.0 м.д. и для НЗ 8 = 7.60 м.д.,
следовательно, сигналы протонов меняются места-
ми по отношению к сигналам атомов углерода, т.е.
8(Н2) > 8(НЗ), а 8(С2) < 8(СЗ).
4.4. Количественный анализ
спектров ЯМР 13С
Для количественной спектроскопии ЯМР 13С необ-
ходимо, чтобы выполнялись два условия. Во-первых,
при определении структуры соединения, очевидно,
полезно знать, обусловлен ли данный сигнал толь-
ко одним типом химически эквивалентных атомов
или несколькими. Во-вторых, количественный анализ
смесей, состоящих из двух и более веществ, требует,
чтобы площадь пиков была пропорциональна числу
атомов углерода, дающих этот сигнал.
Спектроскопия ЯМР 13С с широкополосным пода-
влением ССВ с протонами, как правило, не годится
для количественного анализа по двум причинам:
• Для ядер 13С характерны большие времена ре-
лаксации Гь поэтому они не всегда приходят
к равновесному больцмановскому распределению
за время задержки между импульсами. Таким об-
разом, сигналы могут не достигнуть полной ам-
плитуды (см. раздел 4.2.3).
• Ядра 13С различаются по величине ядерного
эффекта Оверхаузера (ЯЭО, см. раздел 4.2.4), и,
следовательно, интенсивности сигналов откло-
няются от простой пропорциональности числу
ядер данного типа.
В разделе 4.2.5 мы уже обсудили методику пре-
рываемой «развязки» от протонов. Цель этой мето-
дики состояла в достижении высокого уровня ЯЭО,
чтобы получить спектр без развязки от протонов за
минимальное время. В данном разделе мы обсудим
метод инверсной прерываемой «развязки» от про-
тонов: «развязка» выключается, когда начинается
релаксационная задержка, и включается во время
выборки (Z2) (рис. 4.7). Цель состоит в том, чтобы
сохранить постоянным и на низком уровне значение
ЯЭО. Это возможно благодаря тому, что ЯЭО не
успевает достигнуть максимального значения за от-
носительно короткий период выборки. В результате
получаем спектр, состоящий из синглетов, интен-
сивности которых соответствуют числу ядер 13С,
дающих этот сигнал. Не забывайте, что мы должны
учитывать время 1\ и вводить релаксационную за-
держку (/?J.
jp «Развязка» выключена «Развязка» включена
13С:
Рис. 4.7. Импульсная последовательность метода
инверсной прерываемой «развязки» от протонов.
(Rd- релаксационная задержка, в- переменный угол
импульса, t2 - выборка)
4.4. Количественный анализ спектров ЯМР 13С
251
0.12 0.17
170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 20 м.д.
Рис. 4.8. Спектры ЯМР 13С диэтилфталата в CDCI3 при резонансной
частоте 150.9 МГц. а - Обычный спектр с полным подавлением ССВ
с протонами и с релаксационной задержкой Rd < Тх. б, в - Спектры, полученные
с помощью методики инверсной прерываемой «развязки» от протонов при
Rd < Л (6) и Rd > 5 Г, (в). Числа над каждым сигналом показывают его
интегральную интенсивность
Рисунок 4.8 демонстрирует влияние Тх и ЯЭО
на интенсивности пиков. На рис. 4.8, а представлен
обычный спектр ЯМР 13С с подавлением ССВ с про-
тонами, причем Rd< Тх; отметим, что интенсивность
сигнала метильной группы в этом спектре принята за
единицу, а интенсивности сигналов показаны в виде
численных значений сверху. Как видно, наблюдаются
сильные искажения интенсивностей, и этот спектр
нельзя применять для количественных расчетов вслед-
ствие влияния эффектов Тх и ЯЭО. На рис. 4.4, г
(см. раздел 4.2.3) протонированные ароматические
атомы углерода имеют самые маленькие времена
Тх, что объясняет повышенные значения интегралов
для этих сигналов. Спектр на рис. 4.8, б был полу-
чен при использовании методики инверсной пре-
рываемой «развязки» от протонов. Отметим, что при
устранении влияния ЯЭО интенсивности сигналов
атомов углерода, непосредственно связанных с про-
тонами, будут искажаться в меньшей степени, чем
интенсивности сигналов четвертичных углеродных
атомов. На рис. 4.8, б Rd все еще меньше Тх. Рису-
нок 4.8, в показывает, что при использовании более
длинных релаксационных задержек (Rd >5 Л) и при
минимизации ЯЭО с помощью методики инверсной
прерываемой «развязки» от протонов интегральные
интенсивности сигналов в спектре ЯМР 13С почти
не искажаются (т.е. могут использоваться в количе-
ственном анализе). Основное препятствие заключается
в том, что приходится использовать длительную за-
держку до выборки, потому что величина Rd должна
по меньшей мере в пять раз превышать время Тх,
кроме того, требуется намного больше повторений для
достижения неискаженной интенсивности сигналов.
Время, необходимое для получения достаточно силь-
ного сигнала, можно уменьшить, если использовать
специальные парамагнитные добавки. Довольно рас-
пространенный способ-добавление к исследуемому
веществу в «каталитических количествах» ацетил-
252
4. Спектроскопия ЯМР пС
ацетоната хрома(Ш), который представляет собой па-
рамагнитное вещество, чьи неспаренные электроны
стимулируют перенос спина, что приводит к пони-
жению времен релаксации Тх и Т2 (и к некоторому
уширению линий).
4.5. Химическая эквивалентность
Определение, данное для химической эквивалентно-
сти протонов, применимо также для атомов углерода:
атомы являются химически эквивалентными, если они
взаимозаменяемы с помощью операций симметрии
или с помощью механизма быстрого обмена. Нали-
чие в молекуле эквивалентных атомов углерода (т.е.
атомов углерода с точно совпадающими химическими
сдвигами) приводит к несовпадению числа сигналов,
присутствующих в спектре, и действительного числа
атомов углерода в молекуле.
Таким образом, ядра 13С метильных групп в тпрет-
бутиловом спирте (рис. 4.9) являются эквивалент-
ными в результате быстрого вращения, как и про-
тоны метильных групп, которые тоже эквивалентны.
В спектре ЯМР 13С Аи/?вАи-бутилового спирта видны
два сигнала, интенсивность одного из них намного
больше, чем другого; пик четвертичного атома угле-
рода намного меньше по интенсивности, чем пик
атомов углерода метильных групп.
Молекула 2,2,4-триметил-1,3-пентандиола хиральна;
заметим, что группы СН3(я) и СН3(б) неэквивалентны
и дают два сигнала для частоты ЯМР 13С 75.5 МГц
(рис. 4.10). Две метильные группы (в) и (в') также
неэквивалентны, но вследствие совпадения их хи-
мических сдвигов они дают один общий пик (при
частоте 75.5 МГц). Использование спектрометра
с более высокой резонансной частотой (150.9 МГц)
позволяет увидеть два отдельных сигнала для этих
метильных групп.
В разделе 3.8.3.2 мы уже говорили о том, что при
комнатной температуре СН3-протоны в молекуле
(CH3)2NCH=O дают разные сигналы, но при тем-
пературе 123 °C эти протоны становятся химически
эквивалентными (т.е. с одинаковыми химическими
сдвигами). Разумеется, для сигналов ЯМР 13С на-
блюдается аналогичная картина.
4.6. Спектры DEPT
Раньше в спектроскопии ЯМР 13С стремились
полностью устранить взаимодействия 1Н-13С с по-
мощью облучения протонов во всем диапазоне хи-
мических сдвигов протонов. Однако это решение вос-
принималось со смешанными чувствами. С одной
стороны, упрощение спектра (т.е. стремление получить
отдельные, изолированные сигналы) для каждого от-
дельного атома углерода в молекуле на самом деле яв-
ляется весьма заманчивой задачей. С другой стороны,
информация о константах спин-спинового взаимодей-
ствия 1Н—13С является весьма ценной и явно полезна
при определении структуры соединений. В течение
нескольких лет проводилось много экспериментов
по извлечению этой полезной информации при со-
хранении простоты спектра с полной «развязкой».
Имеет смысл упомянуть о двух типах эксперимен-
тов, потому что ранее они были популярны и сейчас
встречаются в старых литературных источниках. Более
того, некоторые из терминов, используемые и по на-
стоящее время, появились в результате проведения
таких экспериментов.
Первый из этих, теперь уже устаревших, экспе-
риментов называется спектром с неполным подавле-
(СН3)3СОН
—] . । . । . । , । . । . । , । . । . ।
180 160 140 120 100 80 60 40 20 м.д.
Рис. 4.9. Спектр ЯМР 13С mpem-бутилового спирта в CDCI3; полное подавление ССВ
с протонами; резонансная частота 75.5 МГц
4.6. Спектры DEPT
253
Г 1 ' ' I ' 1 ' ' I ' ' ' ' I ' ' 1 Ч ' ’ ' ’ I ’ ' ' ' I ' ' ' * Г ' ‘ ' Г ' 1 ' I ‘ ' 1 ' I ' ' ' ' Г ' I ' 1 1 ' Г‘ 1 ' 1 Г 1 1 1 Г 1 1 1 Г 1 1 1 I
85 80 75 70 65 60 55 50 45 40 35 30 25 20 15 10 м.д.
Рис. 4.10. Спектры ЯМР 13С 2,2,4-триметил-1,3-пентандиола в CDCI3; полное
подавление ССВ с протонами; резонансная частота 75.5 МГц, (а) и 150.9 МГц (6)
нием ССВ с протонами (с неполной «развязкой»)
или спектром «офф-резонанса». В этом эксперименте
проявляются прямые взаимодействия ^-^С (т.е. каж-
дый атом углерода показывает взаимодействие только
с протонами, связанными с ним непосредственно).
Для этого положение частоты декаплера устанав-
ливается в стороне от центра частотного диапазона
химических сдвигов протонов (отсюда термин «офф-
резонанс»). Таким образом, для четвертичного атома
углерода (нет связанных с ним протонов) получается
синглет, для СН-группы (метиновый углерод) - ду-
блет, для углерода СН2-группы-триплет и для атома
углерода СН3-группы - квартет. (Напомним, что муль-
типлетность (М) определяется по формуле Л/=и+1,
где п - число протонов, непосредственно связанных
с данным атомом углерода.) Этот эксперимент стал
активно использоваться на практике (и по настоя-
щее время используется), что позволяет провести
отнесение сигналов атомов углерода по их «мульти-
плетности» («s», «d», «t» или «q»). Мы используем
эти описания мультиплетности независимо от того,
применяем ли мы это эксперимент или нет.
Стоит упомянуть и о другом эксперименте, который
также устарел, - это тест на присоединенные про-
тоны (Attached Proton Test, APT). Этот эксперимент
основывается на различии в величине взаимодействий
КССВ 1Н-13С для метановой, метиленовой и метиль-
ной групп. Подбирая определенные задержки в им-
пульсной последовательности (последовательность
тут не приводится), в таких спектрах сигналы чет-
вертичного и метиленового атомов углерода можно
повернуть вверх, а метанового и метильного - вниз.
Так как фазировка производится произвольно, то на-
правление сигналов может быть обратным. Возмож-
ность различать типы углеродных атомов по величине
соответствующих констант взаимодействия и привела
к появлению эксперимента DEPT, который стал так
популярен в настоящее время.
Последовательность DEPT (неискаженное усиле-
ние переноса поляризации, Distortion Enhancement by
Polarization Transfer) стала более предпочтительной
при определении числа протонов, непосредственно
связанных с конкретным ядром 13С. Эксперимент
DEPT можно провести за достаточно разумное время
и он применим для малых образцов; фактически он
в несколько раз более чувствителен, чем обычный
эксперимент ЯМР 13С. В настоящее время DEPT стал
традиционной методикой во многих лабораториях,
и в этой книге широко используется в упражнениях.
Отличительной особенностью в последовательности
DEPT является применение переменного протонного
импульса с углом в (рис. 4.11), в одних экспериментах
254
4. Спектроскопия ЯМР J3C
Рис. 4.11. Импульсная последовательность
эксперимента DEPT. Значение J для КССВ С-Н
обычно составляет 145 Гц,
в- переменный угол импульса, Rd - релаксационная
задержка, t2 - выборка
применяются 90°-ные импульсы, в других - 135°-ные
импульсы.
На рис. 4.12 приведен спектр DEPT ипсенола.
На рис. 4.12, а показан «главный спектр» - обычный
спектр ЯМР 13С с «развязкой» от протонов; на рис.
4.12, б спектр DEPT-1350, где сигналы СН3- и СН-
групп направлены вверх, а сигналы СН2-групп - вниз.
На рис. 4.12, в приведен спектр DEPT-900, где обна-
руживаются только сигналы атомов СН. Четвертич-
ные атомы углерода 13С вообще не регистрируются
в спектрах DEPT. Теперь мы можем перейти к интер-
претации сигналов 13С на «главном спектре» (рис.
4.12, а) и, используя два других спектра (рис. 4.12,
б, в), определить к какому типу (СН3, СН2, СН или
четвертичный) относится данный атом углерода. Луч-
ше всего интерпретацию спектров начать с поиска
сигналов, которые присутствуют в спектре ЯМР 13С
с «развязкой» от протонов, но отсутствуют в спектрах
DEPT. Таким способом можно легко идентифици-
ровать четвертичные атомы углерода (не имеющие
протонов). Следующий шаг - взглянуть на спектр
DEPT и определить СН2-группы; это просто сде-
лать, так как сигналы этих групп направлены вниз.
И, наконец, последний шаг - идентифицировать ато-
мы углерода СН3- и СН-групп, сопоставляя спектры
DEPT-135° и DEPT-900. DEPT-900 содержит сигналы
только атомов СН, таким образом, сигналы, оставшие-
ся без отнесения в спектре DEPT-1350,-это и есть
сигналы СН3-групп.
Чтобы проиллюстрировать, как спектры DEPT
упрощают интерпретацию, вернемся к спектрам ип-
сенола на рис. 4.12. Начиная с пика в слабопольной
части спектра (т.е. начиная с левого конца «главного
140 130 120 110 100 90 80 70 60 50 40 30 20 10 м.д.
Рис. 4.12. а - Стандартный спектр ЯМР 13С (75.5 МГц) с «развязкой» от протонов
для раствора ипсенола в CDCI3. б - Спектр DEPT-1350: сигналы СН3- и СН-групп
направлены вверх, а СН2-группы - вниз, в - Спектр DEPT-900: присутствуют только
пики СН-групп
4.7. Классы химических соединений и их химические сдвиги
255
спектра»), отметим, что этого сигнала нет в спектрах
DEPT, следовательно, этот пик в «главном спектре»
(рис. 4.12, а) относится к четвертичному атому угле-
рода (т.е. к углеродному атому, не имеющему про-
тонов). Следующий сигнал на рис. 4.12, а, правее
уже рассмотренного нами сигнала, - это СН-сигнал,
так как в обоих спектрах DEPT он направлен вверх.
Еще правее видны следующие два сигнала на рис.
4.12, а - это сигналы СН2-групп, так как в спектре
DEPT-135° они направлены вниз. Следующий сигнал
(двигаясь слева направо по спектру) после триплета
растворителя CDC13 - это, очевидно, еще один сигнал
СН-группы. Затем идут два сигнала СН2-групп, по-
том снова сигнал СН-группы. И, наконец, последние
два сигнала в спектре относятся к группам СН3,
так как в спектре DEPT-135° они направлены вверх,
а в спектре DEPT-900 вообще отсутствуют. В итоге,
получается следующая картина (при движении по
спектру слева направо):
678 10 453219
С СН СН2 СН2 растворитель СН СН2 СН2 СН СН3 СН3
Интерпретация спектра DEPT требует некоторого
опыта, но ее результаты весьма полезны. Теперь мы
не только можем определить число атомов углерода
и водорода, но и установить по спектру, сколько же
атомов водорода в молекуле непосредственно связа-
но с каждым конкретным атомом углерода. Однако
существует разница между подсчетом числа прото-
нов в протонном спектре (см. рис. 3.55, где приведен
спектр ЯМР !Н для подтверждения) и в спектре DEPT,
так как ОН-протоны не регистрируются в спектре
DEPT, а также в нем не видны протоны, связанные
с ядрами 15N, 33S, 29Si и 31Р. Но никаких трудностей не
возникнет при проведении корреляции спектра DEPT
с со спектром ЯМР 1Н. В самом деле поразительно,
как далеко отстоят друг от друга алкеновые и алка-
новые системы в обоих спектрах.
4.7. Классы химических соединений
и их химические сдвиги
В этом разделе мы обсудим наиболее распространен-
ные классы органических соединений и их характер-
ные химические сдвиги. Как уже отмечалось ранее,
в рутинной практике диапазон химических сдвигов
13С составляет около 220 м.д.
Мы уже говорили об этом, но повторимся еще раз:
порядок расположения химических сдвигов классов
соединений в спектроскопии ЯМР 13С и имеет не-
которое сходство, так что некоторые закономерности из
спектров ЯМР !Н можно перенести на спектры ЯМР
13С. Более того, для обоих типов спектров (13С и !Н)
полезны уравнения аддитивности для замещенных
соединений (см. разделы 4.7.1 и 4.7.6). Химические
сдвиги 13С в основном зависят от типа гибридизации
атома углерода, от электроотрицательности заместите-
лей и диамагнитной анизотропии (в меньшей степе-
ни); на вид обоих спектров сильно влияют различные
эффекты от растворителей. На химический сдвиг 13С
влияют даже заместители, расположенные достаточ-
но далеко от изучаемого атома углерода (вплоть до
5-положения, т.е через четыре о-связи); в бензольном
кольце химические сдвиги 13С зависят от положения
заместителей в кольце {орто, мета или пара}. Хими-
ческие сдвиги 13С могут также значительно сместиться
вправо (от отношению к исходному) под действием
у-гом/-эффекта (см. раздел 4.7.1). При разбавлении ис-
следуемого раствора также может произойти смещение
сдвига вправо на несколько м.д. Напротив, при при-
менении полярных растворителей химический сдвиг
может сместиться влево (от исходного значения) в ре-
зультате появления водородных связей.
Как и в других видах спектрометрии, отнесение
пиков проводят на основе сигнала эталонного соеди-
нения. В литературе приводятся разные эталоны для
различных классов соединений. Начинают с того, что,
используя таблицы химических сдвигов 13С (см. рис.
4.3 и приложение В), определяют класс, к которому
относится данное органическое соединение; далее
для изучаемого соединения по положению сигнала
внутри этой области сдвигов определяют более част-
ные детали в структуре вещества. Однако значениям
для химических сдвигов из этих таблиц нельзя до-
верять абсолютно, так как на практике эти величины
могут слегка отличаться вследствие применения раз-
ных растворителей и разных концентраций вещества.
Например, сигнал группы С=О ацетофенона при ис-
пользовании в качестве растворителя CDC13 появится
на 2.4 м.д. левее, чем при применении СС14; анало-
гичные эффекты будут наблюдаться и для других
атомов углерода ацетофенона, но эти сигналы сме-
стятся несколько меньше (на 0.0-1.1 м.д.). Более того,
во многих более ранних работах использовали разные
эталонные соединения, величины их сдвигов затем
были скорректированы относительно ТМС (в м.д.).
Спектры ЯМР 13С могут использоваться для распозна-
вания типа замещения в ароматических соединениях.
Если, например, в кольце имеются два одинаковых (ахи-
ральных) заместителя, то только на основе симметрии,
при условии достаточно хорошего разрешения в спектре,
можно различать орто-, мета- и ия/ю-изомеры. пара-
Изомер имеет две поворотные оси симметрии и две
плоскости симметрии; орто- и jwewa-изомеры имеют
256
4. Спектроскопия ЯМР 13С
одну поворотную ось и одну плоскость симметрии,
но в тиел/ш-изомере элементы симметрии проходят че-
рез два атома. Существует также плоскость симметрии
в плоскости кольца в каждом соединении, которая не
влияет на атомы углерода кольца.
В спектре ЯМР 13С для ияра-изомера в аромати-
ческой области будут два пика, для ори/о-изомера -
три, а для jwewa-изомера - четыре. Сигналы четвер-
тичных атомов углерода будут намного меньше по
интенсивности, чем сигналы незамещенных атомов
углерода в кольце.
Аддитивность инкрементов химических сдвигов
будет показана в разделе 4.7.6.
4.7.1. Алканы
4.7.1.1. Неразветвленные (линейные)
и разветвленные алканы
Из общей корреляционной диаграммы химических
сдвигов отдельных классов органических веществ
(приложение В) мы знаем, что незамещенные алкано-
вые группы дают сигналы в области от 0 до 60 м.д.
Таблица 4.4. Параметры для расчета химических
сдвигов 13С неразветвленных (линейных)
и разветвленных углеводородов
Атомы 13С Аддитивный параметр (А), м.д.
а 9.1
0 9.4
Y -2.5
5 0.3
е 0.1
1° (3°)а -1.1
1° (4°)а -3.4
2° (3°)° -2.5
2° (4°) -7.2
3° (2°) -3.7
3° (3°) -9.5
4° (1°) -1.5
4° (2°) -8.4
а Обозначения 1°(3°) и 1°(4°) относятся к СН3-группам,
связанным с R2CH- или К3С-группами соответственно;
2°(3°) - к RCH2-rpynne, связанной с Р2СН-группой, и т.д.
(Метан дает сигнал при -2.5 м.д.) Внутри этой об-
ласти мы можем предсказать химические сдвиги для
отдельных атомов 13С в неразветвленных или развет-
вленных углеводородах, используя данные табл. 4.4
и приведенную ниже формулу.
В этой таблице приведены аддитивные параметры
(А) для расчета химических сдвигов 13С в углево-
дородах: (а-эффект: +9.1 м.д., 0-эффект: +9.4 м.д.,
у-эффект: -2.5 м.д., 5-эффект: +0.3 м.д., е-эффект:
+0.1 м.д.) и поправки, связанные с разветвлением
в цепи. Для наглядности рассчитаем сдвиги для ато-
мов углерода в 3-метилпентане (в скобках указаны
экспериментальные значения):
+ 19.3 (18.6)
б!
сн3
1 2 з| 4 5
СН3—СН2—СН—СН2—СН3
+ 11.3 + 29.5 + 36.2
(+ 11.3) (+ 29.3) (+ 36.7)
Для расчета химических сдвигов 13С используют
уравнение
5 = -2.5 + ЪпА,
где 5-предсказываемый химический сдвиг для атома
углерода, Л - аддитивный параметр (см. табл. 4.4), п -
число атомов углерода для каждого типа замещения
( -2.5 - это сдвиг сигнала ЯМР 13С для метана). Так,
у атома С-1 в 3-метилпентане есть соседние 1 а-, 1 0-,
2 у- и 1 5-атомы углерода.
5j = -2.5 + (9.1x1) + (9.4x1) + (-2.5x2) + (0.3x1) =11.3
Атом С-2 в 3-метилпентане имеет соседние 2 а-,
2 0- и 1 у-атомы углерода. Атом С-2 является 2°-угле-
родом, связанным с 3°-углеродом (2° (3°) = -2.5).
52 = -2.5 + (9.1x2) + (9.4x2) + (-2.5x1) + (-2.5x1) = 29.5
У атома С-3 в 3-метилпентане соседние 3 а- и 2 0-
атомы углерода. Это атом типа 3°, связанный с двумя
2°-атомами (3° (2°) = -3.7). Таким образом,
53 = -2.5 + (9.1x3) + (9.4x2) + (-3.7x2) = 36.2
Атом С-6 в 3-метилпентане имеет соседние 1 а-,
2 0- и 2 у-атомы углерода, это атом типа 1°, при-
соединенный к атому 3° (1° (3°) = -1.1).
56 = -2.5 + (9.1x1) + (9.4x2) + (-2.5x2) + (-1.1x1) = 19.3
Результаты таких расчетов хорошо согласуются
с экспериментальными значениями. В литературе
4.7. Классы химических соединений и их химические сдвиги
257
Таблица 4.5. Химические сдвиги 13С для некоторых
неразветвленных (линейных) и разветвленных
алканов (в м.д. относительно ТМС)
Соединение С-1 С-2 С-3 С-4 С-5
Метан -2.3
Этан 5.7
Пропан 15.8 16.3
Бутан 13.4 25.2
Пентан 13.9 22.8 34.7
Гексан 14.1 23.1 32.2
Гептан 14.1 23.2 32.6 29.7
Октан 14.2 23.2 32.6 29.9
Нонан 14.2 23.3 32.6 30.0 30.3
Декан 14.2 23.2 32.6 31.1 30.5
Изобутан 24.5 25.4
Изопентан 22.2 31.1 32.0 11.7
Изогексан 22.7 28.0 42.0 20.9 14.3
Неопентан 31.7 28.1
2,2-Диметилбутан 29.1 30.6 36.9 8.9
3-Метил пентан 11.5 29.5 36.9 18.8 (3-СН3)
2,3-Диметил бутан 19.5 34.3
2,2,3-Триметилбутан 27.4 33.1 38.3 16.1
2,3 - Диметил пентан 7.0 25.3 36.3 14.6 (3-СНз)
приводится1) и другой способ расчета химических
сдвигов 13С. у-Сдвиг сигналов 13С в более низкие
частоты, возникающий под влиянием у-атома угле-
рода, объясняется стерическим воздействием у-гош-
эффекта, однако он не проявляется в спектрах ЯМР !Н.
Это, например, объясняет смещение сигнала в более
сильные поля для аксиального метильного замести-
теля в конформационно жестком циклогексановом
кольце по сравнению с экваториальной метильной
группой, а также сдвиг в более сильные поля для
у-углеродного атома кольца. В таблице 4.5 перечис-
лены химические сдвиги некоторых неразветвленных
(линейных) и разветвленных алканов.
4.7.1.2. Влияние заместителей в алканах
Данные таблицы 4.6 демонстрируют эффекты заме-
стителей на сдвиги ЯМР 13С для неразветвленных
и разветвленных алканов. Изменение химических
сдвигов а-атома углерода коррелирует с электро-
отрицательностью заместителя (за исключением
брома и иода)2). Влияние на 0-атом углерода до-
статочно постоянно для всех заместителей, кроме
карбонильной, циано- и нитро-групп. Сдвиг сигнала
вправо для у-атома углерода происходит (как было
и выше) из-за стерических затруднений, вызванных
гом/-взаимодействиями. Если Y = N, О и F, то также
происходит сдвиг сигнала вправо, если Y находится
в awww-конформации, стабилизированной сверхсо-
пряжением.
гош анти
В таблице 4.6 перечислены инкременты для функ-
циональных групп, которые необходимо прибавить
к величине химического сдвига соответствующе-
го алкана, приведенного в таблице 4.5. Например,
можно рассчитать химические сдвиги 13С для
3-пентанола.
У Р а & У
СН3—СН2— СН—сн2 — сн3
он
Заместитель ОН присоединяется к цепи неразвет-
вленного пентана «сбоку» (а не на конце); обозначим
атом углерода, к которому прикреплена эта группа,
как а-углерод. Этот атом углерода соответствует в моле-
куле пентана атому С-3, для которого химический сдвиг
составляет 34.7 м.д. (см. табл. 4.6). К этой величине
необходимо прибавить инкремент +41, это значение
инкремента для ОН-группы, присоединенной «сбоку»
к а-углероду 3-пентанола (см. табл. 4.6: 2-я колонка,
12-я строка). Следовательно, химический сдвиг для
этого атома углерода будет равен 75.7 (расчетная
величина). Аналогично рассчитываются химические
сдвиги для 0- и у-атомов углерода. Все три расчетные
величины хорошо согласуются с экспериментальными
данными (см. табл. 4.14) (все значения в м.д.).
Расчет Эксперимент (см. табл. 4.14)
Са 34.7 + 41 = 75.7 73.8
Ср 22.8 + 8 = 30.8 29.7
Су 13.9 - 5 = 8.9 9.8
Аналогично можно рассчитать сдвиги 13С для
пентанола-1, но учесть, что в этом случае группа
° Lindeman L.P. and Adams J.Q. Anal. Chem., 43, 1245
(1971).
2) См. раздел 3.4, табл. 3.2 (Электроотрицательность не-
которых элементов по Полингу).
258
4. Спектроскопия ЯМР пС
Таблица 4.6. Инкременты заместителей (в м.д.) при
замещении атома Н на группу У в алканах.
(Заместитель У концевой или боковой)0
У ct P Концевой Y Y У 1 У P P Боковой
Y a 0 У
Концевой Боковой Концевой Боковой
сн3 9 6 10 8 -2
сн=сн2 20 6 -0.5
с=сн 4.5 5.5 -3.5
соон 21 16 3 2 -2
СОСУ 25 20 5 3 -2
COOR 20 17 3 2 -2
СОС1 33 28 2
conh2 22 2.5 -0.5
COR 30 24 1 1 -2
сно 31 -2
Фенил 23 17 9 7 -2
ОН 48 41 10 8 -5
OR 58 51 8 5 -4
OCOR 51 45 6 5 -3
nh2 29 24 11 10
NH3+ 26 24 8 6 -5
NHR 37 31 8 6 -4
nr2 42 6 -3
NR3+ 31 5 -7
no2 63 57 4 4
CN 4 1 3 3 -3
SH 11 11 12 11 -4
SR 20 7 -3
F 68 63 9 6 -4
Cl 31 32 11 10 -4
Br 20 25 11 10 -3
I -6 4 11 12 -1
а Этот инкремент добавляется к величине химического
сдвига соответствующего атома углерода из табл. 4.5 или
к величине химического сдвига, рассчитанного с помощью
табл. 4.4.
Источник: Wehrli F.W., Marchand А.Р., and Wehrli S. (1983).
Interpretation of Carbon-13 NMR Spectra. 2nd ed. London:
Heyden.
ОН является «концевой»; атом углерода, к которо-
му присоединена эта функциональная группа, также
будет а-атомом углерода.
Таблица 4.7. Химические сдвиги циклоалканов
(в м.д. относительно ТМС)
С3Н6 -2.9 С7Н14 28.4
С4Н8 22.4 С8Н16 26.9
С5Н10 25.6 с9н18 26.1
СбН12 26.9 СюН20 25.3
4.7.1.3. Циклоалканы и насыщенные гетероциклические
соединения
В таблице 4.7 приведены химические сдвиги СН2-
групп моноциклических алканов. Из общего ряда вы-
деляется циклопропан, химический сдвиг которого
смещен сильно вправо относительно других цикло-
алканов; аналогичная картина наблюдается и в про-
тонном спектре этого соединения.
Скелет каждого кольца имеет свой собственный
набор параметров для расчета химических сдвигов
13С, но мы не будем детально останавливаться на этом.
Используя данные, приведенные в табл. 4.6, можно
провести приблизительную оценку сдвигов сигналов
для колец с заместителями. Одним из удивительных
эффектов, наблюдаемых в жестких циклогексановых
системах, является сдвиг вправо, вызванный у-гош-
стерическим напряжением. Таким образом, аксиальная
метильная группа при атоме углерода С-1 вызывает
смещение сигналов атомов С-3 и С-5 на несколько
м.д. в сильные поля.
В таблице 4.8 представлены химические сдвиги 13С
для насыщенных гетероциклических соединений.
4.7.2. Алкены
^//-Гибридизованные атомы углерода алкенов дают сиг-
налы в диапазоне ~110-150 м.д., если заместителями
являются алкильные группы. Двойные связи довольно
слабо влияют на значения химических сдвигов sp3-
гибридизованных атомов углерода в молекуле, что мож-
но продемонстрировать на следующем примере:
25.4
сн3 сн3
Н3С—с—сн2— с=сн2
30.4 | 52.2 143.7 114.4
сн3
31.6
24.7
сн3 сн3
Н3С—с—сн,—сн—сн3
29.9 I 53.5 25.3
сн3
30.4
Метильный сигнал
в пропене находится при
18.7 м.д., а в пропане-при 15.8 м.д. В (7)-2-бутене
4.7. Классы химических соединений и их химические сдвиги
259
химический сдвиг метильной группы составляет
12.1 м.д. вследствие у-эффекта, в то время как в (£)-2-
бутене он равен 17.6 м.д. (Для сравнения, химический
сдвиг метильного сигнала в бутане равен 13.4 м.д.)
Обратите внимание на у-эффект для одной из двух
геминальных метильных групп в 2-метил-2-бутене
(табл. 4.9).
Как правило, концевые =СН2-группы дают сигналы
в более сильных полях по сравнению с внутренними
=СН-группами, а сигналы фрагментов (Z)-CH=CH-
находятся в более сильных полях по сравнению с их
(£)-аналогами. Можно рассчитать приблизительные
химические сдвиги, используя специальные поправки:
Таблица 4.8. Химические сдвиги 13С
(в м.д. относительно ТМС) для насыщенных
гетероциклических соединений
Незамещенные
О
2—40.6
а 10.6
₽ 7.2
У -1.5
а' -7.9
₽' -1.8
У' -1.5
Z (1/мс)-поправка -1.1
24.9
27.7
69.5
Здесь а, Р, у относятся к заместителям с той же сто-
роны от двойной связи алкена, что и интересующий
Замещенные
О
/ \ 47.6
473
18.1
Таблица 4.9. Химические сдвиги 13С (в м.д. относительно ТМС) для алкенов и циклоалкенов
18.7 136.2
123.2
27.4 113.3
124.6
133.3 17.9
126.0
13.4 140.2
17.6
13.7
13.7 36.1 114.3
22.3
138.5
115.9
132.7 123.2
14.0 20.5
12.3
14.0
14.0 31.4 138.7
25.8
123.7
22.4 33.7 114.5
29.4
12.6
137.2 124.0
13.7 35.3 125.1
23.2
17.7
131.2
14.5 20.6
14.4 131.3
74.8
213.5
117.5
115.9
38.0
131.7
137.2
137.3
20.5
114.4 129.5 17.2
137.8 133.2
116.5 13™=™А
18.0
109.3
16.9
12.4
109.8
12.8
130.2
13.0
19.6
128.3
127.4 123.1
149.3
131.4
22.5
12.4
25.3
144.5
20.5
132.5
21.2
112.9
11.7
42.1
144.9 31.2
197 14.6
131.6) 1266
30.2 | 137.2
22.1
14.5
23.6
107.1
124.5
22.1
26.9
25.6
126.1
124.6
22.3
48.0
126.4 137.1
128.7
UD.Z 113 3
128.0 1264
128.7
260
4, Спектроскопия ЯМР 13С
нас атом углерода, а а', 0', у' - к заместителям с дру-
гой стороны от двойной связи.
Эти поправки прибавляются к величине 123.3 м.д.
(это химический сдвиг 13С для этилена). Напри-
мер, значения химических сдвигов для атомов
С-3 и С-2 в (7)-3-метил-2-пентене можно рассчитать
следующим образом:
СН3Н
Да I I а'
Н3С—CTL—С=С—СН3
35 4 Т 2 1 3
СН3Н
Д' а' I | а
н3с—сн2—с=с—сн3
5 4 2 3 2 1 3
ных заместителей при алкеновом атоме углерода не
существует. Химические сдвиги 13С для виниловых
эфиров можно рассчитать на основе электронной
плотности одной из резонансных структур
н2с=с^0)—сн3<—>н2с—с=о—СН,
Н " Н "
84.2 153.2
Аналогично это можно сделать и для
а, P-ненасыщенных кетонов.
&3 = 123.3 + (2x10.6) + (1x7.2) + (1х(-7.9)) - 1.1 = 142.7 мд.
6г = 123.3 + (1x10.6) + (2х(-7.9)) + (1x1.8) - 1.1 = 115.2 мд.
Измеренные величины для атомов С-3 и С-2 это-
го соединения равны 137.2 м.д. и 116.8 м.д. соот-
ветственно, т.е. они достаточно хорошо согласуются
с расчетными значениями.
Атомы углерода, непосредственно присоединенные
к С=С-группе, в (7)-стереоизомере более экранирова-
ны (на 4-6 м.д.), чем в (£)-стереоизомере (см. табл.
4.9). Алкеновые атомы углерода в полиенах рассчиты-
ваются подобно алкановым углеродным заместителям,
расположенным с одной стороны от двойных связей.
Таким образом, при расчете химического сдвига атома
С-2 в 1,4-пентадиене атом С-4 рассматривают как
Р-5/?5-атом углерода.
В таблице 4.9 перечислены типичные представи-
тели алкенов. Простого правила для учета поляр-
Подобные процедуры применяются и для расчета
протонных сдвигов для таких соединений. В табли-
це 4.10 приведены химические сдвиги 13С для не-
которых замещенных алкенов.
Центральный атом углерода (=С=) в алкилзаме-
щенных алленах дает сигнал в области 200-215 м.д.,
в то время как концевые атомы углерода в группе
С=С=С - в диапазоне 75-97 м.д.
4.7.3. Алкины
572-Гибридизованные атомы углерода алкинов, име-
ющих в качестве заместителей только алкильные
группы, дают сигналы в области приблизитель-
Таблица 4.10. Химические сдвиги 13С (в м.д. относительно ТМС) для замещенных алкенов
115.0
122.0
176.0
88.8
136.0 192.1
153.2
^^0^55.1
84.2
107.7
137.8 117.5
4.7. Классы химических соединений и их химические сдвиги
261
Таблица 4.11. Химические сдвиги 13С (в м.д.) алкинов
Соединение С-1 С-2 С-3 С-4 С-5 С-6
1 -Бутин 71.9 86.0
2-Бутин 3.3 74.6
1-Гексин 68.1 84.6 18.2 30.7 21.9 13.5
2-Гексин 2.7 73.7 76.9 19.6 21.6 12.1
3-Гексин 14.5 12.6 80.9
но 65-90 м.д. по шкале химических сдвигов 13С
(табл. 4.11). з/АГибридиз о ванные атомы углерода,
непосредственно связанные с фрагментом С=С, име-
ют химические сдвиги, отличающиеся на 5-15 м.д.
(смещены в сильные поля) относительно сдвигов
соответствующих алканов. Концевая группа =СН
дает сигнал, сильно смещенный вправо по сравне-
нию с внутренней группой =CR. Алкиновые атомы
углерода, к которым присоединены полярные группы,
дают сигналы в диапазоне 20-95 м.д.
Полярные резонансные структуры объясняют
химические сдвиги алкиниловых эфиров, которые
аналогичны сдвигам виниловых эфиров (см. раздел
3.34).
23.2 89.4 28.0 88.4
НС=С—н3с—с=с-о
н^с—сн3 хсн3
релируют с константами Гаммета (о) заместителей.
Сдвиги для рр/ио-атомов углерода не так легко пред-
сказать, отклонения могут достигать 15 м.д. Сигналы
.метия-атомов углерода смещаются ненамного (всего
на несколько м.д.) для ароматических соединений
с одним заместителем.
Ароматические атомы углерода с заместителями
можно отличить от ароматических атомов углерода
без заместителей по изменению интенсивности сиг-
нала (как мы уже упоминали, интенсивность сигналов
четвертичных атомов углерода сильно уменьшается
по сравнению с другими атомами углерода); т.е. из-за
отсутствия протонов у четвертичного атома углерода
увеличивается время Т\ и уменьшается ЯЭО.
В таблице 4.12 приведены инкременты для рас-
чета химических сдвигов атомов углерода кольца
для наиболее распространенных монозамещенных
ароматических соединений, которые необходимо при-
бавить к химическому сдвигу 13С сигнала бензола
(а также перечислены сдвиги атомов углерода для
углеродсодержащих заместителей). Сдвиги атомов
углерода в полизамещенных ароматических соеди-
нениях можно приблизительно рассчитать, используя
принцип аддитивности инкрементов. Например, рас-
считаем сдвиг для атома С-2 в дизамещенном соеди-
нении (4-хлорбензонитриле), добавляя инкременты
для CN-группы (+3.6), находящейся в ррио-положении
по отношению к атому С-2, и для атома Cl (+1.0), на-
ходящегося в .метия-положении по отношению к С-2:
128.5 + 3.6 + 1 = 133.1 м.д.
4.7.4. Ароматические соединения
Бензол дает сигнал 13С с химическим сдвигом
128.5 м.д. (растворитель CDC13). Заместитель смещает
сигнал связанного непосредственно с ним ароматиче-
ского атома углерода примерно на + 35 м.д. Конден-
сированные полиядерные ароматические соединения
имеют следующие химические сдвиги 13С (в м.д.):
Нафталин: С-1 128.1; С-2 125.9; С-4а 133.7
Антрацен: С-1 130.1; С-2 125.4; С-4а 132.2;
С-9 132.6
Фенантрен: С-1 128.3; С-2 126.3; С-3 126.3; С-4 122.2;
С-4а 131.91); С-9 126.6; С-10а 130.Р
Химический сдвиг ароматического атома углеро-
да, напрямую связанного с заместителем, зависит
от электроотрицательности последнего (с учетом
поправки на магнитную анизотропию); сдвиги для
яяря-атомов углерода ароматического кольца кор-
CN CN
С1 С1
1 II III
я Я с v * 'н * В (в ф cd 5 га $ и * В и 5 Я 5 и Si
S га у га Q- 2 гс £ S гс Q-
о о- v о. ф О Q. s о °- 2
1- Е Я Е С 1- Е С Н с Е
< о о. о а < о а < о g
= = Л, с А с Л
1 -18.0 -16.6 1 -16.6 4 -2.0
2 4.6 5.1 2 3.6 3 1.0
3 0.8 1.3 3 0.6 2 0.2
4 10.7 10.8 4 4.3 1 6.4
]) Отнесение спорно.
262
4. Спектроскопия ЯМР 13С
Таблица 4.12. Инкременты для расчета химических сдвигов атомов углерода
кольца для монозамещенных ароматических соединений (соответствующий
инкремент необходимо прибавить к величине сдвига бензола 128.5 м.д.). Сдвиги
атомов углерода заместителей (в м.д. относительно ТМС)а.
Заместитель (у атома С-1) Инкременты для ароматических атомов С Хим. сдвиги атомов С заместителей (м.д. относит. ТМС)
С-1 С-2 С-3 С-4
Н 0.0 0.0 0.0 0.0
сн3 9.3 0.7 -0.1 -2.9 21.3
СН2СН3 15.6 -0.5 0.0 -2.6 29.2 (СН2); 15.8 (СН3)
СН(СН3)2 20.1 -2.0 0.0 -2.5 34.4 (СН); 24.1 (СН3)
С(СН3)3 22.2 -3.4 -0.4 -3.1 34.5 (С); 31.4 (СН3)
сн=сн2 9.1 -2.4 0.2 -0.5 137.1 (СН); 113.3 (СН2)
с=сн -5.8 6.9 0.1 0.4 84.0 (С); 77.8 (СН)
С6Н5 12.1 -1.8 -0.1 -1.6
СН2ОН 13.3 -0.8 -0.6 -0.4 64.5
СН2О(С=О)СН3 7.7 ~0.0 ~0.0 ~0.0 20.7 (СН3); 66.1 (СН2); 170.5 (С=О)
он 26.6 -12.7 1.6 -7.3
осн3 31.4 -14.4 1.0 -7.7 54.1
ос6н5 29.0 -9.4 1.6 -5.3
О(С=О)СН3 22.4 -7.1 -0.4 -3.2 23.9 (СН3); 169.7 (С=О)
(С=О)Н 8.2 1.2 0.6 5.8 192
(С=О)СН3 7.8 -0.4 -0.4 2.8 24.6 (СН3); 195.7 (С=О)
(С=О)С6Н5 9.1 1.5 -0.2 3.8 196.4 (С=О)
(C=O)F -5.6 1.8 0.7 6.7
(С=О)ОН 2.9 1.3 0.4 4.3 168
(С=О)ОСН3 2.0 1.2 -0.1 4.8 51.0 (СН3); 166.8 (С=О)
(С=О)С1 4.6 2.9 0.6 7.0 168.5
(C=O)NH2 5.0 -1.2 0.0 3.4
C=N -16 3.6 0.6 4.3 119.5
nh2 19.2 -12.4 1.3 -9.5
N(CH3)2 22.4 -15.7 0.8 -11.8 40.3
NH(C=O)CH3 11.1 -9.9 0.2 -5.6
no2 19.6 -5.3 0.9 6.0
N=C=O 5.7 -3.6 1.2 -2.8 129.5
F 35.1 -14.3 0.9 -4.5
Cl 6.4 0.2 1.0 -2.0
Br -5,4 3.4 2.2 -1.0
I -32.2 9.9 2.6 -7.3
CF3 2.6 -3.1 0.4 3.4
SH 2.3 0.6 0.2 -3.3
SCH3 10.2 -1.8 0.4 -3.6 15.9
SO2NH2 15.3 -2.9 0.4 3.3
Si(CH3)3 13.4 4.4 -1.1 -1.1
а Источник'. Ewing D.E. (1979). Org. Magn. Reson., 12,499 (приведено 709 значений химических
сдвигов для монозамещенных бензолов).
4.7. Классы химических соединений и их химические сдвиги
263
Таблица 4.13. Химические сдвиги атомов углерода гетероароматических
соединений (в м.д. относительно ТМС)
Соединение С-2 С-3 С-4 С-5 С-6 Заместитель
Фуран 142.7 109.6
2-Метилфуран 152.2 106.2 110.9 141.2 13.4
Фуран-2-карбальдегид 153.3 121.7 112.9 148.5 178.2
Метил-2-фуроат 144.8 117.9 111.9 146.4 159.1 (С=О); 51.8 (СН3)
Пиррол 118.4 108.0
2-Метилпиррол 127.2 105.9 108.1 116.7 12.4
Пиррол-2-карбальдегид 134.0 123.0 112.0 129.0 178.9
Тиофен 124.4 126.2
2-Метилтиофен 139.0 124.7 126.4 122.6 14.8
Тиофен-2-карбальдегид 143.3 136.4 128.1 134.6 182.8
Тиазол 152.2 142.4 118.5
Имидазол 136.2 122.3 122.3
Пиридин 150.2 123.9 135.9
Пиримидин 159.5 157.4 122.1 157.4
Пиразин 145.6
2-Метилпиразин 154.0 141.8° 143.8° 144.7° 21.6
а Отнесение спорно.
4.7.5. Гетероароматические соединения
Для расчетов химических сдвигов углеродных
атомов в гетероароматических соединениях были
предложены различные усложненные схемы. Обыч-
но сигнал атома С-2 в кислород- и азотсодержащих
кольцах сдвинут в слабые поля относительно сиг-
нала атома С-3. Было обнаружено сильное влияние
растворителя и pH раствора. В таблице 4.13 приве-
дены значения сдвигов 13С для некоторых пяти- и
шестичленных гетероциклических соединений (для
чистых жидкостей).
4.7.6. Спирты
Замена протона в алкане на ОН-группу смещает
сигнал в слабые поля на 35-52 м.д. для атома С-1,
на 5-12 м.д. для углерода С-2 и в сильные поля на
0-6 м.д. для атома С-3. В таблице 4.14 приведены
химические сдвиги 13С некоторых ациклических (не-
циклических) и алициклических спиртов. Для обна-
ружения спиртовой группы полезно использовать
реакцию ацетилирования, при этом сигнал атома
С-1 сместится в слабые поля на 2.5-4.5 м.д., сигнал
атома С-2 - в сильные поля на такую же величину,
а 1,3-диаксиальное взаимодействие вызывает неболь-
шое (около 1 м.д.) смещение в слабые поля сигнала
С-3. Данные табл. 4.14 можно использовать для расче-
тов химических сдвигов 13С спиртов, если применять
метод, описанный ранее.
4.7.7. Эфиры, ацетали, эпоксиды
Алкокси-заместитель вызывает существенно большее
смещение в слабые поля сигнала атома С-1 (примерно
на 11 м.д.) по сравнению с гидрокси-заместителем.
Этот эффект связан с атомом и С-Г, который является
0-атомом углерода по отношению к атому С-1. Атом
кислорода здесь принимается за «а-С» по отношению
к атому углерода С-1.
2 1 Iх
сн3—сн2—он сн3—сн2—о—сн3
17.6 57.0 14.7 67.9 57.6
Отметим также, что «у-эффект» (смещение сиг-
нала вправо) для атома С-2 вполне объясним теми
же причинами. И наоборот, этокси-группа действу-
ет на ОСН3-группу (сравните с СН3ОН). В табли-
це 4.15 приведены химические сдвиги 13С некоторых
эфиров.
В ацеталях атом углерода, связанный с дву-
мя атомами кислорода, дает сигнал 13С в области
88-112 м.д. Сигнал оксирана (эпоксида) находится
при 40.6 м.д.
264
4. Спектроскопия ЯМР 13С
Таблица 4.14. Химические сдвиги 13С спиртов (в м.д. относительно ТМС)
Алкильные атомы углерода в арилалкиловых эфи-
рах дают сдвиги, подобные сдвигам диалкиловых
эфиров. Отметим, что сильно смещенный вправо
сигнал ароматического ортио-атома углерода объяс-
няется делокализацией электрона, как и в виниловых
простых эфирах.
4.7.8. Галогенсодержащие соединения
Эффекты, вызванные присутствием атома галогена в мо-
лекуле, довольно сложны. Даже наличие одного атома
фтора в CH3F приводит к значительному смещению сиг-
нала влево по сравнению с сигналом СН4, что объясня-
ется электроотрицательностью атома фтора. Атом хлора
как заместитель (в СН3С1, СН2С12, СНС13, СС14) приводит
к аналогичному эффекту (смещает сигнал сильно влево),
что также можно объяснить электроотрицательностью
Таблица 4.15. Химические сдвиги 13С эфиров, ацеталей
и эпоксидов (в м.д. относительно ТМС)
4.7. Классы химических соединений и их химические сдвиги
265
хлора. Однако для брома и иода возникает «эффект тяже-
лого атома». Например, по мере увеличения числа атомов
брома сигнал 13С сначала смещается влево, т. е. в сла-
бые поля (ср. химические сдвиги СН4 (-2.3 м.д.), СН3Вг
(10.0 м.д.) СН2Вг2 (21.4 м.д.)), но потом вправо (ср. хи-
мические сдвиги 13ССНВг3(12.1 м.д.) и СВг4(-28.5 м.д.)).
Для атома углерода в СН31 также наблюдается сильное
смещение сигнала вправо по сравнению с углеродным
сигналом СН4. Для атома углерода С-2 смещение сигнала
влево уменьшается в ряду I > Вг > Е Атомы О и Вг вы-
зывают у-гош-экранирование для атома С-3, но это не
характерно для атома иода, возможно, вследствие низкого
содержания гом/-ротамера. В таблице 4.16 показаны эти
закономерности.
Химические сдвиги 13С галогенсодержащих соедине-
ний сильно зависят от природы растворителя. Напри-
мер, при растворении иодэтана в циклогексане сигнал
С-1 иодэтана имеет сдвиг -6.6 м.д., а при использовании
ДМФА этот же сигнал наблюдается при -0.4 м.д.
4.7.9. Амины
Концевая NH2-rpynna, связанная с алкильной цепочкой,
смещает сдвиг атома С-1 влево примерно на 30 м.д.,
атома С-2 - влево на 11 м.д. и атома С-3 - вправо на
4.0 м.д. по сравнению с аналогичными сигналами ал-
канов. NH3+-Ipynna вызывает чуть меньшее смещение
сигналов. N-Алкилирование приводит к увеличению
сдвига сигнала углеродного атома С-1 в слабые поля.
В таблице 4.17 приведены химические сдвиги 13С
некоторых ациклических и алициклических аминов
(гетероциклические амины см. в табл. 4.8).
4.7.10. Тиолы, сульфиды, дисульфиды
Поскольку электроотрицательность серы меньше, чем кис-
лорода, серосодержащие заместители вызывают меньшие
смещения сигналов. В таблице 4.18 приведены данные
для некоторых тиолов, сульфидов и дисульфидов.
4.7.11. Функциональные группы,
содержащие углерод
Спектроскопия ЯМР 13С позволяет проводить прямое
наблюдение углеродсодержащих функциональных групп;
Таблица 4.16. Химические сдвиги 13С
(в м.д. относительно ТМС) галогенсодержащих
алканов (чистые жидкости)
Соединение С-1 С-2 С-3
СН4 -2.3
CH3F 75.4
СН3С1 24.9
СН2С12 54.0
СНС13 77.5
СС14 96.5
СН3Вг 10.0
СН2Вг2 21.4
СНВг3 12.1
СВг4 -28.5
СН31 -20.7
СН212 -54.0
СН13 -139.9
С14 -292.5
ch3ch2f 79.3 14.6
СН3СН2С1 39.9 18.7
СН3СН2Вг 28.3 20.3
СН3СН21 -0.2 21.6
СН3СН2СН2С1 46.7 26.5 11.5
СН3СН2СН2Вг 35.7 26.8 13.2
СН3СН2СН21 10.0 27.6 16.2
в приложении В приводятся сдвиги этих групп. С помо-
щью протонной спектроскопии ЯМР такие группы нельзя
обнаружить, за исключением, пожалуй, группы СН=О.
4.7.11.1. Кетоны и альдегиды
Атомы углерода в соединениях R200 и RCH=O
имеют характерные сдвиги. Ацетон дает сигнал при
203.3 м.д., а ацетальдегид-при 199.3 м.д. Алкильные
заместители при а-атоме углерода вызывают смещение
сигнала группы С=О в слабые поля на 2-3 м.д., если
только не доминируют стерические эффекты. Замена
СН3-группы в ацетоне или ацетальдегиде на фениль-
ную группу вызывает смещение сигнала группы С=О
вправо (в ацетофеноне С=О-группа дает сигнал при
195.7 м.д., а в бензальдегиде - при 190.7 м.д.); ана-
логичное смещение пика вправо наблюдается и для
а,Р-незамещенных соединений (сравните, акролеин
дает сигнал при 192.1 м.д., а пропионовый альдегид -
при 201.5 м.д.). Возможно, что делокализация заряда
в бензольном кольце или по двойной связи вызывает
дефицит электронной плотности на карбонильном
атоме углерода.
266
4. Спектроскопия ЯМР 13С
Таблица 4.17. Химические сдвиги 13С (в м.д. относительно ТМС) ациклических
и алициклических аминов в CDCI3
Н3С—NH2
26.9
4,4
25.6
Таблица 4.18. Химические сдвиги 13С (в м.д. относительно ТМС)
тиолов, сульфидов и дисульфидов
Соединение C-1 C-2 C-3 Соединение C-1 C-2 C-3
CH3SH 6.5 (CH3CH2)2S 25.5 14.8
CH3CH2SH 19.8 17.3 (CH3CH2CH2)2S 34.3 23.2 13.7
CH3CH2CH2SH 26.4 27.6 12.6 (CH3CH2CH2CH2)2SH 34.1 31.4 22.0
CH3CH2CH2CH2SH 23.7 35.7 21.0 CH3SSCH3 22.0
(CH3)2S 19.3 CH3CH2SSCH2CH3 32.8 14.5
4.7. Классы химических соединений и их химические сдвиги
267
Таблица 4.19. Химические сдвиги 13С (в м.д. относительно ТМС) группы С=О
и других атомов углерода некоторых кетонов и альдегидов
Таблица 4.20. Химические сдвиги 13С (в м.д. относительно ТМС) группы С=О
и других атомов углерода некоторых карбоновых кислот, сложных эфиров,
лактонов, хлорангидридов, карбаматов и нитрилов
178.1
Н3С—СООН
20.6
168.0
С13С—СООН
89.1
о
ci69-5
163.0
F3C—СООН
115.0
181.5
Н3С—COO Na+
bD2O
33.8
С1
34.1)—СООН
/ 184.8
18.8
131.9
128.0
СООН
173.2
NH2
О
176.5
17.2 515 СООН
о
66.8
ll^^O.O О'
20.4
166.0 соосн3
1130.2 515
.129.9
128.7
169.4 СООН
130.3
128.7
134.0
28.4
о
О
о
171.7
Р
136.7
О
о
31.1
—С—NH2
25.5 174.3
го.о^О-З О
14.1
164.3
О
II 51.1
44mOZ
33.9
129.9
128.7 || 52.0
О
о
о
142.4
135.0
97.4
О 167.3
170.3 О
о
о
167.2 соосн3
1132.8 523
39.5
61.0
F3C 158.1 О
114.1.
О167^С1
о
о
36.2
н
18.8
—C=N
1.3 117.7
10.8
106 120.8
149.3}
н
132.8 133.0
о
lnh2
166.3
N—С 162.4
H2N157.8° 145
н
(101.2
CN
117.6
н
150.2}
118.7
Н
(100.9
17о CN 132,о
173 116.0
268
4. Спектроскопия ЯМР 13С
Для циклоалканонов, в частности для циклопен-
танона, химический сдвиг смещен влево. В таблице
4.19 приведены химические сдвиги групп С=О не-
которых кетонов и альдегидов. Вследствие сильного
влияния растворителей значения этих сдвигов в раз-
личных литературных источниках могут отличаться
на несколько м.д. Замена СН2-группы в алканах на
С=О-группу вызывает смещение сигнала а-атома
углерода влево (примерно на 10-14 м.д.), а Р-атома
углерода - вправо (для ациклических соединений
примерно на несколько м.д.). В спектре ЯМР 13С
без подавления ССВ с протонами сигнал формаль-
дегида (СН=О) представляет собой дублет.
4.7.11.2. Карбоновые кислоты, сложные эфиры, хлоран-
гидриды, ангидриды, амиды и нитрилы
Группы С=О карбоновых кислот и их производных
дают сигналы в области 150-185 м.д. На карбоновые
кислоты сильно влияет разбавление и природа рас-
творителя; появление анионов в растворе смещает
сигнал сильно влево. В целом, влияние заместителей
и делокализация электронов аналогичны таковым для
кетонов. Нитрилы дают сигнал в области 115-125 м.д.
В амидах алкильные заместители у атома азота вы-
зывают незначительное (всего на несколько м.д.) сме-
щение вправо сигнала группы С=О (см. табл. 4.20).
4.7.11.3. Оксимы
Четвертичный атом углерода простых оксимов дает
сигнал в области 145-165 м.д. Спектроскопия ЯМР 13С
позволяет различить в данном случае (Е)- и (Z)-
изомеры, так как для группы C=N наблюдается сдвиг
в сильные поля для стерически более напряженной
формы. Кроме того, сдвиг для более стерически затруд-
ненного заместителя (син по отношению к ОН-группе)
находится правее, чем для менее затрудненного.
ОН
N
Литература
Общая
Abraham, R.J., Fisher, J., and Loftus, P. (1992). Introduction
to NMR Spectroscopy. New York: Wiley.
Beckmann, N. (1995). Carbon-13 NMR Spectroscopy of
Biological Systems. New York: Academic Press.
Breitmaier, E. (2002). Structure Elucidation by NMR in
Organic Chemistry: A Practical Guide. John Wiley & Sons;
3rd Revision edition. New York: Wiley.
Breitmaier, E., and Vbelter, W. (1987). Carbon-13 NMR
Spectroscopy. 3rd ed., New York: VCH Publishers.
Claridge.T.D.W. (1999). High-Resolution NMR Techniques in
Organic Chemistry. Elsevier Science.
Farrar, T.C. (1987). An Introduction to Pulse NMR
Spectroscopy. Chicago: Farragut Press.
Kalinowski, H.O., Berger, S., and Braun, S. (1988). Carbon-
13 NMR Spectroscopy. New York: Wiley.
Levy, G.C., Lichter, R.L., and Nelson, G.L. (1980). Carbon-13
Nuclear Magnetic Resonance for Organic Chemists, 2nd ed.
New York: Wiley. (Есть русский перевод: Леви Г., Нельсон
Г. Г., Руководство по ядерному магнитному резонансу
углерода-13. Пер. с англ. (1975). Москва, Мир.)
Muller, К., and Pregosin, P.S. (1976). Fourier Transform
NM/EA Practical Approach. New York: Academic Press.
Paudler, W.W. (1987). Nuclear Magnetic Resonance.
New York, Wiley.
Pihlaja, K., and Kleinpeter, E. (1994). Carbon-13 NMR
Chemical Shifts in Structural and Stereochemical
Analysis. VCH Publishing
Sanders, J.K. and Hunter, B.K. (1993). Modern NMR
Spectroscopy, 2nd ed. Oxford: Wiley.
Shaw, D. (1984). Fourier Transform NMR Spectroscopy, 2nd
ed. Amsterdam: Elsevier.
Wehrli, F.W., Marchand, A.P., and Wehrli, S. (1988). Interpretation
of Carbon-13 NMR Spectra, 2nd ed. New York: Wiley.
Каталоги спектров, базы данных по спектрам
Bates, R.B., and Beavers, W.A. (1981). С-13 NMR Problems.
Clifton, NJ: Humana Press.
Breitmaier, E., Haas, G., and Voelter, W. (1979). Atlas
of C-13 NMR Data, Vols. 1-3. Philadelphia: Heyden
(3017 соединений).
Bremser, W., Ernst, L., Franke, B., Gerhards, R., and Hardt,
A. (1987). Carbon-13 NMR Spectral Data (microfiche), 4th
ed.New York: VCH Publishers (58 108 табулированных
спектров для 48 357 соединений).
Упражнения
269
Fuchs, P.L., and Bunnell, C.A. (1979). Carbon-13 NMR-Based
Organic Spectral Problems. New York: Wiley.
Johnson, L. E, and Jankowski, W. C. (1972). Carbon-13 NMR
Spectra, a Collection of Assigned, Coded, and Indexed
Spectra. New York: Wiley.
Pouched, C.J., and Behnke, J. (1993). Aldrich Library of 13C
and !H FT-NMR Spectra, 300 MHz. Milwaukee, WI: Aldrich
Chemical Co.
Pretsch, E., Clem, T., Seihl, J., and Simon, W. (1981). Spectra
Data for Structure Determination of Organic Compounds.
Berlin: Springer-Verlag.
Sadtler Research Lab. 13C NMR Spectra. Philadelphia: Sadtler
Research Laboratories.
Упражнения
4.1. Для приведенных ниже соединений
найдите все химически эквивалентные атомы
углерода.
4.2. Предскажите химические сдвиги каждого атома
углерода в тех же соединениях. Укажите источ-
ники (таблицы и приложения), которые вы при
этом использовали.
4.3. Изобразите спектры ЯМР 13С с «развязкой» от
протонов и DEPT для тех же соединений (можно
обозначать сигналы черточками).
4.4. Подтвердите структуру и сделайте отнесение
всех сигналов в спектрах ЯМР 13С (см. спектры
3.4А-3.4Ч). Все спектры ЯМР 13С были зареги-
стрированы при 75.5 МГц в CDC13. Эти струк-
туры вы уже определяли в упражнении 3.4 по
соответствующим спектрам ЯМР 1Н. Их масс-
спектры были приведены в гл. 1 (см. упражнение
1.6), а ИК-спектры - в гл. 2 (см. упражнение 2.9).
4.5. Предскажите число сигналов в спектре ЯМР 13С
для следующих соединений:
С1 С1 С1
13 I 13 I 13 I
D—С—Cl D—С—Cl D—С—D
Cl D D
4.6. Объясните следующие спектры 13C/DEPT (см.
спектры 4.6А - 4.6Е). Подтвердите структуры
и сделайте отнесение всех сигналов 13С. Укажите
источники (таблицы и приложения), которые вы
использовали при работе.
4.7. Какие элементы симметрии характерны для мо-
лекул о-, м- и и-диэтилфталатов? Сколько неэк-
вивалентных атомов углерода и атомов водорода
в каждом из этих соединений? Изобразите (схе-
матично) для каждого из них спектры ЯМР 13С
с «развязкой» от протонов и спектры 13C/DEPT.
270
4. Спектроскопия ЯМР^
Спектр А
200 180 160 140 120 100 80 60 40 20 м.д.
Спектр Б
___________Ш_1________________________________ 111 II____________________
। ' ' ' • । ' ' 1 । ' ' ' I ' ' ' । ' ' ' г ' ' ' । । г • г ' ' I ' ' ' ' I ’ ' 1 I 1 1 । г । 1 ' 1 I ‘ 1 1 1 |
85 80 75 70 65 60 55 50 45 40 35 30 25 20 15 10 м.д.
Спектр В
I I I I I I I I I I I I I I I I I I I I I
131 м.д.
I I I ~ III ..................
140 130 120 110 100 90 80 70 60 50 40 30 20 10 м.д.
Упражнение 4.4(А-В)
Упражнение
271
Спектр Г
I I • • • I " ' I " '' I " " I " " I " '' I ' " ' I ' " ' I " " I ' I' " ' I " " Г’’"1’г Г"'' Г" " I " " I ’1 ‘1 I " " I ' ‘ ' 11
95 90 85 80 75 70 65 60 55 50 45 40 35 30 25 20 15 10 м.д.
Спектр Д
200 180 160 140 120 100 80 60 40 20 м.д.
Спектр Е
180 160 140 120 100 80 60 40 20 м.д.
Упражнение 4.4(Г-Е)
272
4. Спектроскопия ЯМР^ ПС
Спектр Ж
95 90 85 80 75 70 65 60 55 50 45 40 35 30 25 20 15 10 м.д.
Спектр 3
________________I___ш________________________________________________________________________
" " I " " I " " Г " ' I " " I 1 " ' I 1 ' " 'I.Г 1 ' ' I ' ' ' ' I 1 ' ' ' I " " I " " I ' 11 1 I 1 1 ' ' I " ' V ' 1 ' I ' 1 ' ' 1 Г1~" I " " I
95 90 85 80 75 70 65 60 55 50 45 40 35 30 25 20 15 10 м.д.
Спектр И
1 I " " I " " I " " Г ’’’T ' " I ' ' ' ' I ' ‘ " I " " Г ' г' I.. ‘ । । ।। j
95 90 85 80 75 70 65 60 55 50 45 40 35 30 25 20 15 10 м.д.
Упражнение 4.4(Ж-И)
Упражнения
273
Спектр К
_____________I________________L__1__J_________________________in _______
""''i..।.....i......i......i....i.....i.....i.....i.....i.....i......i......i
170 160 150 140 130 120 110 100 90 80 70 60 м.д.
Спектр Л
Спектр М
...............................................I............................ II.......
170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 20 м.д.
Упражнение 4.4(К-М)
274
4. Спектроскопия ЯМР ,3С
Спектр Н
—)--------1-------1--------1-----•---г-
180 160 140 120 100
20
Спектр О
170 160 150 140 130 120 110 100 90 80 70 60 м.д.
Упражнение 4.4(Н-П)
Упражнения
275
Спектр Р
155 150 145 140 135 130 125 120 115 110 105 100 95 90 85 80 м.д.
Спектр С
I . . I tlL
180 160 140 120 100 80 60 40 20 м.д.
Спектр Т
Упражнение 4.4(Р-Т)
276
4. Спектроскопия ЯМР ,3С
Спектр У
I_________________L—1_________I________ 1111
।—•—।——।—•—।——।—•—।—•—।—• । 1 । • ।— । —।
200 180 160 140 120 100 80 60 40 20 м.д.
Спектр Ф
I 1 । 1 I • I । • । • । • । • । ।
180 160 140 120 100 80 60 40 20 м.д.
Спектр X
г " 1 I " " I " " I " " I " " I " " I " " I " " I 11111 " " ।...।...।......I " " I 11 "I " " I " 111 1" 11
155 150 145 140 135 130 125 120 115 ПО 105 100 95 90 85 80 м.д.
Упражнение 4.4(У-Х)
Упражнения
Спектр Ц
J I щ
"I....I.......I.......I.......I......I'......Ч"".....I......I......I.......I.......................|.....»|><....|
150 140 130 120 ПО 100 90 80 70 60 50 40 30 20 м.д.
Спектр Ч
—!---,----!---,---1----,---1---,---1---,---1----,---1-
200 180 160 140 120 100 80
60
т
40
20
м.д.
Упражнение 4.4(Ц, Ч)
278
4. Спектроскопия ЯМР ,3С
Спектр А
C5HioBr2
' 1 1 1 I 1 1 1 1 Г 11 1 I — I ' ' ' | ' 1 1 ' | ' ' ' ' I ' ' ' ' | ' ' ' ' | ' ' | ' ' 1 ' | г' ' ' Г г ' ' ' г ' ' ' 1 ' ' ' |
75 70 65 60 55 50 45 40 35 30 25 20 15 10 5 м.д.
Спектр Б
СзНзОг
__. „ i____,,_
I—•—।——I—•—I—1—I—•
132 130 128 м.д.
170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 20 м.д.
Спектр В
c12h27n
• I • • • • I • • • • I • • • • I • • • • I • • • • I • • I • • • I • • • • I • • I • • I • • • I • • • • I • • • • I • • • I • • • I
75 70 65 60 55 50 45 40 35 30 25 20 15 10 5 м.д.
Упражнение 4.б(А-В)
Упражнения
279
Спектр Г
170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 20 м.д.
Спектр Д
С7Н12О
___1______________________1________________________м_______________и__1________
200 180 160 140 120 100 80 60 40 20 м.д.
Спектр Е
С8Н14О
82 80 78 м.д.
I I I • • • • I I • • • I I • ' I • ' I ' 1 1 1 I • I Г Г • • I
85 80 75 70 65 60 55
50 45 40 35 30 25 20
'Т’
15
гтт
10
м.д.
Упражнение 4.б(Г-Е)
280
4. Спектроскопия ЯМР
Приложение А. Химические сдвиги 13С, константы спин-спинового взаимодействия
и мультиплетность сигналов растворителей, часто используемых
в спектроскопии ЯМР
Структура Название 8, м.д. •Ас-D» Мульти- плетность
CDC13 Хлороформ-Ji 77.0 32 Триплет
CD3OD Метанол-б/4 49.0 21.5 Септет
CD3SOCD3 дмсо-б/6 39.7 21 Септет
0
II DCN(CD3)2 ДМФА-Д7 30.1 21 Септет
35.2 21 Септет
167.7 30 Триплет
C6D6 Бензол-с4 128.0 24 Триплет
D2C—cd2
/ \ D,C CD, ТГФ-с/8 25.2 20.5 Квинтет
0 67.4 22 Квинтет
.°.
d2c cd2
I I Диоксан-с/8 66.5 22 Квинтет
d2c. cd2
0
D n A n Пиридин-^ 123.5 (С-3,5) 25 Триплет
135.5 (С-4) 24.5 Триплет
149.2 (С-2,6) 27.5 Триплет
D^N^D Септет
0 29.8 (метил) 20 Септет
CD3CCD3 Ацетон-^ 206.5 (карбонил) <1 Септет^
CD3CN Ацетонитрил-^ 1.3 (метил) 32 Септет
118.2 (CN) <1 Септет^
CD3NO2 Нитрометан-с/з 60.5 23.5 Септет
CD3CD2OD Этанол-^ 15.8 (С-2) 19.5 Септет
55.4 (С-1) 22 Квинтет
(CD3CD2)2O Эфир-dio 13.4 (С-2) 19 Септет
64.3 (С-1) 21 Квинтет
[(CD3)2N]3P=O ГМФА-^8 35.8 21 Септет
CD3CO2D Уксусная кислота-^ 20.2 (С-2) 20 Септет
178.4 (С-1) <1 Септет^
CD2Q2 Дихлорметан-с/2 53.1 29 Квинтет
° Интенсивности в триплете 1:1:1, в квинтете 1:2:3:2:1, в септете 1:3:6:7:6:3:1.
6 Неразрешенный (дальнее ССВ).
Источник: Breitmaier Е., and Voelter W. Carbon-13 NMR Spectroscopy. 3rd ed., New York: VCH,
p. 109 (С разрешения Also Merck Co., Inc.)
Приложение Б. Химические сдвиги 13C обычных растворителей как примесей
CDCI3 (CD3)2CO (CD3)2SO QD6 CD3CN CD3OD d2o
Сигналы растворителей 77.16±0.06 29.84±0.01 206.26±0.13 39.52±0.05 128.06±0.02 1.32±0.02 118.26±0.02 49.00±0.01
Ацетон СО 207.07 205.87 206.31 204.43 20.743 299.67 215.94
СН3 30.92 30.60 30.56 30.14 30.91 30.67 30.89
Приложение Б
281
Приложение Б (продолжение)
CDCI3 (CD3)2CO (CD3)jSO C6D6 CD3CN CD3OD D2O
Ацетонитрил CN 116.43 117.70 117.91 116.02 118.26 118.06 119.68
сн3 1.89 1.12 1.03 0.20 1.79 0.85 1.47
Бензол СН 128.37 129.15 128.30 128.62 129.32 129.34
wpew-Бутанол с 69.15 68.13 66.88 68.19 68.74 69.40 70.36
СН3 31.25 30.72 30.38 30.47 30.68 30.91 30.29
тирети-Бутилметиловый ОСН3 49.45 49.35 48.70 49.19 49.52 49.66 49.37
эфир С 72.87 72.81 72.04 72.40 73.17 74.32 75.62
ССН3 26.99 27.24 26.79 27.09 27.28 27.32 26.60
Гексаметилфосфорамид СН3 36.87 37.04 36.42 36.88 37.10 37.00 36.46
н-Гексан сн3 14.14 14.34 13.88 14.32 14.43 14.45
СН2(2) 22.70 23.28 22.05 23.04 23.40 23.68
СН2(3) 31.64 32.30 30.95 31.96 32.36 32.72
2,6-Ди-тирети-бутил-4- С(1) 151.55 152.51 151.47 152.05 152.42 152.85
метилфенол С(2) 135.87 138.19 139.12 136.08 138.13 139.09
СН(3) 125.55 129.05 127.97 128.52 129.61 129.49
С(4) 128.27 126.03 124.85 125.83 126.38 126.11
СН3Аг 21.20 21.31 20.97 21.40 21.23 21.38
СН3С 30.33 31.61 31.25 31.34 31.50 31.15
С 34.25 35.00 34.33 34.35 35.05 35.36
Диглим СН3 59.01 58.77 57.98 58.66 58.90 59.06 58.67
СН2 70.51 71.03 69.54 70.87 70.99 71.33 70.05
СН2 71.90 72.63 71.25 72.35 72.63 72.92 71.63
Диметилацетамид СН3 21.53 21.51 21.29 21.16 21.76 21.32 21.09
СО 171.07 170.61 169.54 169.95 171.31 173.32 174.57
NCH3 35.28 34.89 37.38 34.67 35.17 35.50 35.03
NCH3 38.13 37.92 34.42 37.03 38.26 38.43 38.76
Диметилсульфоксид СН3 40.76 41.23 40.45 40.03 41.31 40.45 39.39
Диметилформамид СН 162.62 162.79 162.29 162.13 163.31 164.73 165.53
СН3 36.50 36.15 35.73 35.25 36.57 36.89 37.54
сн3 31.45 31.03 30.73 30.72 31.32 31.61 32.03
1,2-Диметоксиэтан 59.08 58.45 58.01 58.68 58.89 59.06 58.67
71.84 72.47 17.07 72.21 72.47 72.72 71.49
Диоксан сн2 67.14 67.60 66.36 67.16 67.72 68.11 67.19
Дихлорметан сн2 53.52 54.95 54.84 53.46 55.32 54.78
1,2-Дихлорэтан сн2 43.50 45.25 45.02 43.59 45.54 45.11
Диэтиловый эфир сн3 15.20 15.78 5.12 15.46 15.63 15.46 14.77
сн2 65.91 66.12 62.05 65.94 66.32 66.88 66.42
Метанол СН3 50.41 49.77 58.59 49.97 49.90 49.86 49.50
Нитрометан СН3 62.50 63.21 63.28 61.16 63.66 63.08 63.22
н-Пентан сн3 14.08 14.29 13.28 14.25 14.37 14.39
СН2(2) 22.38 22.98 21.70 22.72 23.08 23.38
СН2(3) 34.16 34.83 33.48 34.45 34.89 35.30
Пиридин СН(2) 149.90 150.67 149.58 150.27 150.76 150.07 149.18
СН(3) 123.75 124.57 123.84 123.58 127.76 125.53 125.12
СН(4) 135.96 136.56 136.05 135.28 136.89 138.35 138.27
2-Пропанол СН3 25.14 25.67 25.43 25.18 25.55 25.27 24.38
сн2 64.50 63.85 64.92 64.23 64.30 64.71 64.88
Силиконовое масло СН3 1.04 1.40 1.38 2.10
Смазка СН2 29.76 30.73 29.20 30.21 30.86 31.29
4. Отектроскопия ЯМР^ °f
282
Приложение Б (окончание)
CDCI3 (CD3)2CO (CD3)2SO C6D6 CD3CN CD3OD d2o
Тетрагидрофуран CH2 25.62 26.15 25.14 25.72 26.27 26.48 25.67
CH2O 67.97 68.07 67.03 67.80 68.33 68.83 68.68
Толуол CH3 21.46 21.46 20.99 21.10 21.50 21.50
co) 137.89 138.48 137.35 137.91 138.90 138.85
CH(o) 129.07 129.76 128.88 129.33 129.94 129.91
CH(w) 128.26 129.03 128.18 128.56 129.23 129.20
CH(p) 125.33 126.12 125.29 125.68 126.28 126.29
Триэтил амин CH3 11.61 12.49 11.74 12.35 12.38 11.09
CH2 46.25 47.07 45.74 46.77 47.10 46.96
Уксусная кислота co 175.99 172.31 171.93 175.82 173.21 175.11 177.21
CH3 20.81 20.51 20.95 20.37 20.73 20.56 21.03
Хлороформ CH 77.36 79.19 79.16 77.79 79.17 79.44
Циклогексан CH2 26.94 27.51 26.33 27.23 27.63 27.96
Этанол CH3 18.41 18.89 18.51 18.72 18.80 18.40 17.47
CH2 58.28 57.72 56.07 57.86 57.96 58.26 58.05
Этилацетат CH3CO 21.04 20.83 20.68 20.56 21.16 20.88 21.15
co 171.36 170.96 170.31 170.44 171.68 172.89 175.26
CH2 60.49 60.56 59.74 60.21 60.98 61.50 62.32
CH3 14.19 14.50 14.40 14.19 14.54 14.49 13.92
Этиленгликоль CH2 63.73 64.26 62.76 64.34 64.22 64.30 63.17
Этилметилкетон CH3CO 29.49 29.30 29.26 28.56 29.60 29.39 29.49
co 209.56 208.30 208.72 206.55 209.88 212.16 218.43
CH2CH3 36.89 36.75 35.83 26.36 37.09 37.34 37.27
CH2CH3 7.86 8.03 7.61 7.91 8.14 8.09 7.87
Источник: Н.Е. Gottlieb, V. Kotlyar, A. Nudelman, J. Org. Chem., 62, 7512 (1997)
Приложение В. Диапазон химических сдвигов 13С для различных классов соединений
R- Н или алкильный заместитель
Y-полярный заместитель 220 200 180 160 140 120 100 80 60 40 20 0 -20
-сн3 1 -CHj 1 -CH 1 1 — c— 1 Алициклические углеводороды C3He C4He to СюНзд Алкены Н2С= С- R Н2С = С— У C=C-C=C-R Аллены С«С = С Алкины С=С —R С5С-У Ароматические соединения Ar - R Аг —У
»С —R • Н2С =
1 1
1 1
= С = = с
= с- НС= ,R
Гетероароматические соединения
Спирты С-ОН
Простые эфиры С—О—С
Приложение В
283
Приложение В (продолжение)
220 200 180 160 140 120 100 80 60 40 20 0 -20
Ацетали, кетали О-С-О Галогенсодержащие соединения C-F,_3 С-СЦ-4 С - Br1-4 с ц_4 Амины C-NR2 Нитросоединения C-NO2 Тиолы, сульфиды C-S-R Сульфоксиды, сульфоны C-SO-R, C-SOj-R Насыщенные альдегиды RCHO а, р-Ненасыщенные альдегиды R-C«C-CH=O Насыщенные кетоны R2C=O а, р-Ненасыщенные кетоны R-C=C-C*O Насыщенные карбоновые кислоты RCOOH Соли RCOO” а, р-Ненасыщенные карбоновые кислоты R-C»C-COOH Насыщенные сложные эфиры R-COOR' а, Р-Ненасыщенные сложные эфиры R-C=C—COOR'
r -28.5
-292.5
220 200 180 160 140 120 100 80 60 40 20 0 -20
Ангидриды (RCO2)O Амиды RCONH2 Нитрилы R—ON Карбаматы R2ONOH Оксимы R2NCOOR' Изоцианаты R—N=C“O Цианаты R—О—ON Изотиоцианаты R—N=OS Тиоцианаты R-S-ON — — - —
— — — —— —
——— — — — —— — - —
284
4. Спектроскопия ЯМР пС
Приложение Г. Химические сдвиги ЯМР 13С для некоторых природных соединений
112.9
16.0
137.2 . 58-7
39.7
26.6
ОН
124.5
124.9
Мирцен
131.2
17.4 25.5
Гераниол
27.2
Линалоол
120.8
30.6
23.8
133.2
30.9
28.0
149.7
20.5 108.4
Лимонен
119.6
116.5
22.2
131.5
28.5
120.7
23.1
24.8
140.9
34.0
20.6
а-Терпинен
25.9 <
44.6
71.9,
ЖГ
а-Терпинеол
27.1
128.7
СНО 189.4
123.1
132.9
25.3
цис- Цитраль
(Нераль, цитраль Ь)
21.7
22.1
Пулегон
Ментол
38.7
19.5
20.0
36,9 30.1
Норборнан
9.7 О
Камфора
23.0
а-Пинен
105.9
Р-Пинен
32.6
38.4
17.5
О
195.5
25.2
270 140.7
130.7
133.4
31.8 19'7
Р-Ионон
37.5
НО
18.8
СНз.358
12.0м35-8’
28.3
71.3
42.4
36.4
24.1
4О.о1_?9^_
28.0
- 24.3
42.4
56.9
32.3
36.5
141.2
21.2
19.4
CHJ50.5
56.5
22.5
22.8
Н
32.0
121.3
Холестерин
41.4
123.6
148.5
Никотин
Кокаин
P-D-Глюкоза
100.9
143.0
О
Д165.2
^NH
Ъ-527
N O
Н
Урацил
5. Корреляционная спектроскопия ЯМР. 2D ЯМР
5.1. Введение
В главах 3 и 4 (знакомство с которыми подразумева-
ется) изложены сведения об эффективных методах,
позволяющих установить структуру органических
соединений, - спектроскопии ЯМР на ядрах !Н и 13С.
Особенно эффективны эти методы становятся в том
случае, когда они сочетаются с информацией, извлека-
емой из ИК- и масс-спектров. Методики ЯМР, обсуж-
даемые в гл. 3 и 4, объединяют под общим названием
«одномерные методы ЯМР». Чтобы расширить наши
возможности, мы снова обратимся к ЯМР. В качестве
примеров будут использованы четыре соединения:
ипсенол (см. гл. 3), кариофилленоксид (эпоксид се-
сквитерпена), лактоза (0-дисахарид), а также малень-
кий пептид (валин-глицин-серин-глутамат, VGSE).
Структуры этих соединений показаны на рис. 5.1.
Эти соединения демонстрируют большое
разнообразие типов и структурных особенностей.
Два терпеноида обладают характерными изопрено-
идными углеродными скелетами. Оба соединения
имеют диастереотопные метиленовые и метильные
группы. Лактоза-это 0-1,4-сочлененный дисахарид
галактозы и глюкозы, причем глюкоза является вос-
станавливающим остатком, который при равновесии
в водных растворах существует в виде обоих аноме-
ров. Тетрапептид (VGSE) содержит четыре различ-
ных аминокислотных остатка (валин-глицин-серин-
глутаминовая кислота) и может служить моделью
биополимеров (например, белков или нуклеиновых
кислот). Интерпретация сигналов ЯМР этих соедине-
ний с использованием только одномерных спектров
ЯМР может вызвать определенные трудности.
В этой главе мы обсудим корреляционную спек-
троскопию ЯМР, причем будем использовать указан-
ные соединения в качестве примеров. Большинство
(но не все) полезных экспериментов относятся к ка-
тегории двумерного (2D) ЯМР. Наш подход будет
заключаться в представлении спектров отдельно для
каждого соединения в виде некоторого логического
ОН
I
ртт С—О
сн3 он сн2
НхГ СН2 СН—СН3 СН2 СНг
НО | H,N—СН—CH-N—CILC-i-N—СН—C-HN—СН—С—ОН
А II :н АНН || :н ||
Н3С сн3 о о о о
Ипсенол Валин (V) Глицин (G) Серин (S) Глутаминовая (Е)
Тетрапептид VGSE кислота
Кариофилленоксид Лактоза (0-аномер)
Рис. 5.1. Структуры четырех соединений, используемых в данной главе в качестве
примеров
286
5. Корреляционная спектроскопия ЯМР. 2D ЯМР
ряда спектров. Большая часть общих положений и
понятий для каждого типа эксперимента будет дана
при обсуждении спектров ипсенола. Остальные будут
вводиться при обсуждении более сложных соединений
по мере необходимости. Поэтому сначала детально
рассмотрим случай ипсенола. Другие соединения
рассматриваются отдельно или вообще не рассма-
триваются.
Корреляция - это концепция, которая не является
для нас совершенно новой. Например, спектр ЯМР !Н
этилбензола (см. рис. 3.34) показывает четкий три-
плет и четкий квартет для метильной и метиленовой
групп соответственно. Эти две группы «коррелиру-
ют» между собой в том смысле, что индивидуальные
спины внутри обеих групп связаны между собой
(скалярной связью). Правила спектров первого по-
рядка помогли нам проинтерпретировать эти взаи-
модействия. Спин-спиновое взаимодействие между
протонами - это только один из видов корреляций,
которые будут рассмотрены далее.
Мы уже проделали много работы по интерпрета-
ции спектра ипсенола при использовании спектров
ЯМР !Н (см. гл. 3), но мы сможем это сделать лучше
при использовании корреляций - быстрее и более на-
дежно. Другие перечисленные соединения (кариофил-
ленокси д, лактоза и пептид VGSE) слишком сложны
для проведения полного анализа при использовании
одномерных спектров ЯМР !Н и 13С. Но прежде чем
приступить к рассмотрению отдельных методик, сна-
чала коротко остановимся на импульсных последо-
вательностях и фурье-преобразовании (ФП).
5.2. Теоретические положения
Напомним, чтобы получить обычный спектр ЯМР !Н
или 13С в импульсном эксперименте, используется
импульсная последовательность, показанная на рис.
5.2, которая включает достижение равновесия в маг-
нитном поле, ВЧ-импульс и выборку сигнала. Эта по-
следовательность повторяется, пока не будет получено
удовлетворительное отношение сигнал/шум. Затем
с помощью фурье-преобразования (ФП) сигнала спа-
да свободной индукции (ССИ) получают обычный
сигнал в частотном представлении.
На рис. 5.2. можно обнаружить несколько харак-
терных особенностей. Заметим, что отдельно пока-
зан канал !Н и канал 13С. Эти каналы соответствуют
оборудованию, связанному с облучением и выборкой
сигнала ядер, участвующих в эксперименте. Сразу по-
сле достижения равновесия используется импульсная
последовательность для получения одномерных (1D)
протонных спектров, состоящая из импульса (л/2)х
0
Релаксационная _
задержка (Rd) I
«Развязка» включена
Релаксационная
задержка (Rd)
13С: ------------
Рис. 5.2. а - Импульсная последовательность для
стандартного 1D спектра ЯМР ]Н. б- Импульсная
последовательность для стандартного с «развязкой»
спектра ЯМР 13С. Угол в обычно равен (п/2)х
(90°-ный импульс вдоль оси х); величина Rd- период
установления равновесия в магнитном поле перед
следующим импульсом
(0, задержки и выборки сигнала, длящейся порядка
нескольких секунд (рис. 5.2, а). Заметим также, что ка-
нал 13С (он не показан на этом рисунке) неактивен
во время экспериментов на протонах (обычно канал
не показывают, если он не используется).
На рис. 5.2, б представлена импульсная после-
довательность для эксперимента на ядрах13С. По-
следовательность импульсов такая же, как и для
ПМР на рис. 5.2, а. Спин-спиновое взаимодействие
протонов с ядрами 13С подавляется с помощью об-
лучения протонов во время эксперимента, т.е. во все
время эксперимента включена «развязка» от прото-
нов. В других экспериментах «развязка» для данного
ядра может включаться и выключаться в соответствии
с импульсами и задержками в другом канале (для
другого ядра). Этот процесс называется импульсной
(или прерываемой) развязкой (gated decoupling) (см.
разделы 4.2.5 и 4.4).
Стоит рассмотреть кратко (см. гл. 3 и 4), что проис-
ходит с вектором намагниченности MQ для единичного
спина при такой импульсной последовательности.
Будем использовать вращающуюся систему координат.
В системе координат, вращающейся с ларморовой
частотой, вектор Мо сохраняет компоненту вдоль
оси z (период установления равновесия, см. рис. 5.2).
Импульс л/2 (0, 90°) поворачивает вектор Мо и на-
правляет его вдоль оси у. При рассмотрении про-
цесса во вращающейся системе координат вектор
намагниченности представляется нам стационарным,
52. Теоретические положения
287
(л/2)х
Rd I
AZ1
(л/2)х| Выборка (fi)
Рис. 5.3. Прототип импульсной последовательности
2О-ЯМР. Инкрементная задержка At7 и время
выборки t2 после фурье-преобразования переходят
в частоты v2 и Ут соответственно. Импульс fn/2)x
представляет собой 90°-ный импульс вдоль оси х.
Интервал порядка нескольких микросекунд,
Г2 - нескольких секунд
хотя амплитуда этого вектора уменьшается (из-за
процессов релаксации с временами Тх и Т2). Вернув-
шись на минуту к лабораторной системе координат,
мы увидим, что вектор намагниченности фактически
нестационарен: он вращается в плоскости ху вокруг
оси z с частотой Лармора. Этот вращающийся век-
тор генерирует ВЧ-сигнал, который детектируется
как ССИ в эксперименте ЯМР. Вектор намагничен-
ности вскоре вернется к направлению вдоль оси z,
завершится процесс релаксации, и после этого по-
следовательность импульсов может быть повторена.
В простом одноимпульсном эксперименте применя-
ется импульс л/2, так как он дает наиболее сильный
сигнал. Импульс с углом несколько большим или
меньшим, чем 90°, оставляет некоторую компоненту
сигнала вдоль оси z или - z, и только компонента
вектора по оси у д&ет сигнал.
Теперь рассмотрим многоимпульсные эксперименты
и двумерный ЯМР. Что в точности означает термин
«размерность» в ЯМР? Обычный спектр есть график
зависимости интенсивности (в произвольных едини-
цах) от частоты (в 8-единицах). И хотя это двумерный
(плоский) график, такие спектры называются одно-
мерными. Важно помнить, что частотная ось, с кото-
рой так удобно работать, получается из временной
развертки (в течение времени выборки) с помощью
фурье-преобразования. Таким образом, эксперимен-
тально абсцисса в ID-эксперименте-это время.
Фактически так называемые 2В-спектры ЯМР яв-
ляются трехмерными графиками (т.е. ЗО-спекграми).
Еще одно измерение всегда относится к интенсив-
ности. Два измерения, упоминаемые в 2В-спектрах
ЯМР, соответствуют частотам. Это приводит к необ-
ходимости двух фурье-преобразований под прямым
углом друг к другу по двум независимым временным
осям, что дает две ортогональные частотные оси.
В простом одноимпульсном эксперименте имеется
только один временной фактор, который оказыва-
ет влияние на спектр, а именно время выборки t2.
Рассмотрим теперь многоимпульсный эксперимент,
в котором за периодом установления равновесия сле-
дуют два импульса с разделяющим их временным
интервалом, причем завершающим импульсом явля-
ется (л/2)-импульс выборки. Таким образом, мы ввели
время эволюции между импульсами. Если теперь мы
будем изменять время эволюции (Zj в серии экспе-
риментов и собирать результирующие ССИ по всем
экспериментам, то получим основу для 2В-спектров
ЯМР. Последовательные ФП этих ССИ дадут набор
спектров, где интенсивности пиков будут изменяться
синусоидально. Эта первая серия ФП дает в резуль-
тате вторую частотную ось (v2), которая получается
из времени выборки t2 каждого ССИ. Затем данные
смещают (поворачивают) на угол 90° и проводят вто-
рое ФП под углом 90° к первой серии преобразова-
ний. Эта вторая серия ФП дает в результате первую
частотную ось (vj, функцию времени эволюции Zb
которое изменялось (по инкрементам) в импульсной
последовательности для каждого нового ССИ.
Можно предложить простой прототип 2В-экспе-
римента, который помог бы прояснить некоторые
идеи такого эксперимента и в то же время мог бы
служить «заготовкой» для других более полезных
2В-экспериментов. В этом простом случае импульс-
ная последовательность (рис. 5.3) состоит из релак-
сационной задержки (7?Д (л/2)-импульса, временного
переменного интервала (AZb период эволюции), второго
(л/2)-импульса, используемого для выборки, и самой вы-
борки (Z2). Эта последовательность (отдельный экспери-
мент) повторяется определенное число раз (каждый раз
давая отдельный ССИ) с возрастающим интервалом tx.
Мы используем для этого эксперимента простое
соединение - ацетон (СН3-(С=О)-СН3), - чтобы
избежать усложнений, вызванных спин-спиновым
взаимодействием. На рис. 5.4, мы видим, что после
первого (л/2)-импульса вдоль оси х (т.е. (л/2)х), на-
магниченность MQ поворачивается так, что становится
направлена вдоль оси у (М). Эволюция в течение
времени tx для вектора намагниченности спинов эк-
вивалентных протонов ацетона показана во вращаю-
щейся системе координат. При таком рассмотрении
мы пренебрегаем спин-решеточной релаксацией,
но учитываем поперечную релаксацию с постоянной
времени Т2. Если частота Лармора (v2) больше частоты
вращающейся системы координат, то вектор М пре-
цессирует по часовой стрелке в плоскости ху и за
промежуток времени tx поворачивается на угол 2wZb
В соответствии с тригонометрическими соотноше-
ниями, у-компонента вектора М равна М cos^nr^),
а х-компонента -М sinp^n^).
Спустя время tx импульс выборки (л/2)х повора-
чивает у-компоненту намагниченности вниз вдоль
288
5. Корреляционная спектроскопия ЯМР. 2D ЯМР
Рис. 5.4. Эволюция во вращающейся системе координат для протонов ацетона
показана в течение временного интервала t1z который следует за первым
импульсом. Второй импульс и выборка дают сигнал, возникающий только из
х-компоненты вектора М; амплитуда этого сигнала изменяется синусоидально
с изменением ^.Интервал составляет от нескольких микросекунд до нескольких
миллисекунд, t2 - порядка секунд. Частота прецессии протонов выше, чем частота
вращения системы координат
Рис. 5.5. а - Расположенные друг за другом 22 «спектра» ацетона, для которых
параметр г, изменяется инкрементно (с постоянным шагом), б - «Проекция»
системы спектров (а), показывающая синусоидальное поведение максимумов
и минимумов в зависимости от времени tve - Результирующая интерферограмма,
представляющая собой срез, параллельный оси и проходящий через вершины
пиков системы графиков (а)
5.2. Теоретические положения
289
ось-z; таким образом, эта компонента вообще не дает
вклада в сигнал ЯМР. В то же время х-компонента
остается неизмененной (в плоскости ху), и сигнал
этой компоненты записывается как сигнал ССИ.
Фурье-преобразование этого ССИ приведет к пику
с частотой v2 и амплитудой М $т(2тгМх). Если по-
вторять этот «эксперимент» много раз (например,
1024 или 210), каждый раз увеличивая время tx на
одну и ту же величину, получится 210 сигналов ССИ.
Последовательные фурье-преобразования каждого из
них дают серию «спектров», каждый спектр содер-
жит единственный сигнал с частот v2 и амплитудой
М sm(2jcvtx). На рис. 5.5, а 22 из 1024 спектров рас-
положены друг за другом; видно, что амплитуда пика
ацетона изменяется синусоидально как функция
Таким образом, мы установили одну из частотных
осей (v2) для нашего прототипа 2В-спектра.
Перед тем как установить вторую ось, проведем
небольшой подсчет. Напомним, что любой из полу-
ченных спектров фактически является оцифрован-
ным множеством точек. Предположим, что каждый
из этих 1024 спектров состоит из 1024 точек. Таким
образом, получается квадратная матрица данных. Если
мысленно повернуть это множество спектров, мож-
но провести вторую серию фурье-преобразований
данных, ортогональных к первому ряду преобразо-
ваний. Перед тем как провести эти преобразования,
посмотрим более внимательно на рис. 5.5, а.
При повороте спектров на рис. 5.5, а так, как будто
мы смотрим на них по направлению оси v2, эти дан-
ные будут выглядеть как некие «проекции», показан-
ные на рис. 5.5, 6. На вершинах первых семи спек-
тров на рис. 5.5, а изображены маленькие кружочки.
Эти кружочки на семи максимумах снова показаны
на рис. 5.5, б (и на рис. 5.5, в), так что мы можем
следить за процессом вращения и проектирования.
Если снова изобразить в виде графика одну колонку
данных с рис. 5.5, а (возьмем колонку, которая соот-
ветствует максимуму (и минимуму) пика ацетона),
то результат будет выглядеть как типичный сигнал
ССИ или как спектр во временном представлении
(рис. 5.5, в). Действительно, этот спектр является функ-
цией интервала tx импульсной последовательности.
Чтобы как-то различать их, данные, полученные в ре-
альном времени (в виде функции величины /2), будем
называть сигналом ССИ, а данные, восстановленные
по точкам в виде функции tx - интерферограммой.
Проведем теперь вторую серию фурье-преобразова-
ний для каждой из 1024 интерферограмм, чтобы
получить, буквально говоря, преобразование преоб-
разований. Конечным продуктом будет 2В-спектр;
однако перед нами возникает проблема визуализации
полученных результатов. Один из способов состо-
ит в изображении набора спектров друг за другом,
как на рис. 5.5, а. Изображение такого типа, показанное
в левой части рис. 5.6, дает ощущение трехмерности.
Рис. 5.6. Фурье-преобразование серии сигналов ССИ, аналогичных тому, которое
показано на рис. 5.5, в, приводит к спектру в частотном представлении, в виде
контура и в форме пика. На контурном графике показана также проекция,
параллельная F2
290
5. Корреляционная спектроскопия ЯМР. 2D ЯМР
Обратите внимание, что две оси теперь помечены как
оси F2 и F1, что согласуется с дальнейшим текстом
и что используется повсеместно. Для этого спектра
подобный вид изображения вполне удовлетворителен,
поскольку нет пиков, которые были бы заслонены.
Таким образом, достаточно одного ракурса.
Для более сложных спектров данные обычно пред-
ставляют в виде ряда контуров, подобно тому как
на топографических картах изображают вершины
и впадины. Этот тип изображения показан справа на
рис. 5.6. Проекции, часто включаемые в спектры 2D,
можно сравнить с «тенью» пика при его «освещении»;
очевидно, что такая проекция является 1 D-спектром.
Часто эти проекции помещают рядом с фактическим
спектром 1D, который был получен отдельно. Если нет
отрицательных пиков (как, например, в фазочувстви-
тельном варианте метода COSY, не рассматриваемом
в этой книге), мы будем использовать такой способ
представления без каких-либо комментариев.
5.3. Корреляционная спектроскопия
Наблюдательный читатель уже, наверное, понял,
что описанный выше эксперимент, который пред-
ставляет собой вариант частотной модуляции, не дает
никакой дополнительной информации по сравнению
с обычным спектром ЯМР !Н ацетона. Фактически,
мы просто получили красивую картинку. В ней видны
все элементы типичного двумерного корреляционного
эксперимента, и можно проследить поведение вектора
суммарной намагниченности для ацетона с использо-
ванием простой векторной модели. Теперь перейдем
от этой импульсной последовательности в общему
формату для всех 2О-экспериментов. Если мы заме-
ним первый (л/2)-импульс неким обобщенным «им-
пульсом», содержащим один или более импульсов
и сопутствующие задержки, и заменим второй (л/2)-
импульс выборки обобщенным «импульсом выбор-
ки», также содержащим один или более импульсов
и соответствующие задержки, мы получим общую
импульсную последовательность для двумерных кор-
реляционных экспериментов, показанную на рис. 5.7.
С использованием простой векторной модели и эле-
ментарной тригонометрии можно описать результат
того, что происходит внутри каждого «ящика». Однако
на данный момент давайте пренебрежем тем, что про-
исходит внутри «ящиков», и сконцентрируемся на том,
что происходит в периоды времени tx и t2.
Во всех 2О-экспериментах во время выборки мы ре-
гистрируем сигнал как функцию времени t2<i однако
этот сигнал модулирован как функция от времени
tx. Эксперимент с ацетоном, показанный выше, - это
простой пример, так как намагниченность испытыва-
ет одинаковую модуляцию как в период времени tx,
так и в период времени t2. Для эксперимента, в кото-
ром намагниченность одинаково модулируется в пе-
риоды tx и t2, результирующие пики будут такими,
как будто частота V! равна частоте v2j в терминоло-
гии 2О-ЯМР, такой эксперимент дает диагональные
пики. Чтобы получить из 2О-экспериментов полез-
ную информацию, нужны эксперименты, в которых
намагниченность эволюционирует с одной частотой
в период времени tx и с другой частотой в период
времени t2. В этом случае эксперимент даст пики,
в которых частоты Vj и v2 различны; такие пики мы
Релаксацион-
ная задержка
Обобщенный
импуль:
(с)
Обобщенный
импульс
выборки
(с)
Повторяется п раз
Рис. 5.7. Обобщенная импульсная последовательность для экспериментов по 2D-
ЯМР. Сигнал детектируется во время выборки t2 и модулируется инкрементно по
времени таким образом, в спектре 2D возникают кросс-пики
5.3. Корреляционная спектроскопия
291
называем недиагональными или кросс-пиками. Чтобы
интерпретировать 2О-спектры ЯМР, необходимо знать
две вещи. Во-первых, каким частотам соответствуют
оси? Одна ось (v2) всегда представляет детектируемое
ядро в период выборки (Z2). Другая ось (уД которая
зависит от времени Zb может представлять то же самое
ядро (например, в методе COSY), или какое-
то другое ядро (например, в методе ‘Н-^С COSY,
также называемом HMQC или HETCOR), или кон-
станту спин-спинового взаимодействия (например,
в /-разрешенной спектроскопии, не рассматриваемой
в этой главе). Во-вторых, нужно знать, как намагни-
ченности связаны в периоды времени tx и t2; тогда
можно понять происхождение кросс-пиков.
Если вернуться к прототипу 2В-эксперимента
и применить эту импульсную последовательность
к системе АХ, то появится возможность понять смысл
введения пошагового времени tx. Хотя мы можем точно
математически описать, как спины ведут себя в тече-
ние этого времени, мы не можем показать наглядно
с помощью векторных диаграмм, как эта эволюция
происходит. (Математическое описание системы
требует квантовой механики и решения с помощью
матрицы плотности, что выходит за рамки данной
книги.) После первого (л/2)-импульса, система мо-
жет быть описана как сумма двух членов; причем
каждый член содержит спин только одного из двух
протонов. Во течение времени tx спины прецессиру-
ют под влиянием как химического сдвига, так и вза-
имной константы спин-спинового взаимодействия.
Взаимная константа взаимодействия вызывает эффект
изменения некоторых спиновых членов в произве-
дениях, содержащих компоненты намагниченности
обоих ядер. Далее второй (л/2)-импульс заставляет
спины (которые прецессировали под влиянием как
химического сдвига, так и констант спин-спинового
взаимодействия) перераспределять намагниченность
между всеми спинами (в данном случае есть только
один другой спин), с которыми связан рассматривае-
мый спин. Это перераспределение намагниченности
детектируется по оси /2, таким образом, частота, детек-
тируемая по оси /2, оказывается амплитудно модули-
рованной как функция других спинов (опять в данном
случае есть только один другой спин), с которыми
данный спин связан в период времени Zb что при-
водит к появлению кросс-пика, который связывает
взаимодействующие ядра. Поскольку намагниченность
перераспределяется одинаково в обоих направлениях
(т.е. от А к X и от X к А), кросс-пики (по крайней
мере в этом эксперименте) будут симметрично рас-
положены относительно диагонали. Это описание
спинов, прецессирующих и смешивающихся в пе-
риод времени Zb а также их перераспределение во
время импульса выборки (и детектирование в период
времени /2) очень трудно представить без картинок.
Из-за этих трудностей импульсные последователь-
ности детально не поясняются.
5.3.1. Корреляции ’Н-’Н: COSY
Наш простой 20-эксперимент-это фактически очень
важный эксперимент, иногда называемый COSY (от
слов Correlation SpectrocopY), его мы будем назы-
вать COSY, чтобы показать, какие параметры
в данном случае коррелируют.^ Импульсная после-
довательность 1Н—1Н COSY-это ни что иное, как по-
следовательность, которая уже была описана на рис.
5.3: два протонных (л/2)-импульса, разделенных пе-
риодом эволюции tx, который разбивается по шагам
(т.е. по инкрементам) с последующим периодом вы-
борки Z2.
Реальные импульсные последовательности, исполь-
зуемые во всех современных спектрометрах, более
сложные, чем идеализированные модели, которые
описаны в этой книге. Во многих спектрометрах
используется методика известная как «фазовое ци-
клирование», при этом фаза ВЧ-импульса изменяется
регулярным образом («циклически») для каждого
инкремента tx. Эти фазовые циклы являются важными
экспериментальными факторами, которые помогают
устранить артефакты и другие особенности квадра-
турного детектирования. Мы не будем рассматривать
фазовое циклирование в деталях, так как это не влияет
на понимание и интерпретацию экспериментов.
Интересующийся читатель может обратиться к кни-
ге Клариджа (Т. Claridge, см. список литературы)
для уточнения этих и других параметров экспери-
ментов в 20-спектроскопии ЯМР. Еще один фактор,
которым мы также будем пока пренебрегать, - это
использование «градиентов». Краткое обсуждение
этих экспериментов и их практического значения
приведено в конце данной главы.
При описании 20-экспериментов для спиновой
системы АХ (см. выше) мы выяснили, что в течение
времени tx взаимно связанные спины прецессируют
под действием химических сдвигов обоих ядер
Многие читатели догадываются, что число акронимов
(сокращений) для 20-экспериментов ЯМР все время
увеличивается вместе с расширением возможностей 2D-
экспериментов. Мы не пытаемся в этой главе ни энцикло-
педически полно толковать эти акронимы, ни описывать
соответствующие эксперименты. Однако будут рассмотре-
ны некоторые достаточно важные эксперименты, чтобы
читатель имел возможность интерпретировать большую
часть 20-экспериментов, реально встречающихся на прак-
тике. Сами акронимы перечислены в указателе.
292
5. Корреляционная спектроскопия ЯМР. 2D ЯМР
и таким образом дают пики, у которых частота
V! не равна частоте v2. В общем случае спектры
типа COSY дают недиагональные пики
(или кросс-пики) для всех протонов, связанных
спин-спиновым взаимодействием; проще говоря,
кросс-пики связывают взаимодействующие прото-
ны. В некотором смысле эксперимент по 2О-ЯМР
можно представить себе так, как будто одновре-
менно проводятся все нужные эксперименты по
спиновой «развязке», чтобы обнаружить взаимные
связи протонов. Конечно, в экспериментах 1Н—1Н
COSY никакая «развязка» не проводится, и такой
20-эксперимент не следует понимать просто как
замену экспериментов по подавлению гомоядерного
ССВ (см. раздел 3.15).
5.4. Ипсенол: спектры COSY
Начнем обсуждение 2О-ЯМР с рассмотрения спек-
тров !Н—’Н COSY ипсенола, спирта класса монотер-
пенов. Это соединение уже рассматривалось ранее
в разделах 3.12.1 и 4.6. В качестве напоминания на
рис. 5.8. приведены типичный 1 D-спектр ЯМР !Н
ипсенола для рабочей частоты 300 МГц и структура
этого соединения.
Контурное представление простого спектра COSY
ипсенола показано в верхней части рис. 5.9. Такое
изображение спектра типично; ось F2 находится внизу
(или вверху) вместе со шкалой протонных сдвигов,
которая располагается, как обычно, справа налево.
Ось F1 показана справа (или слева) с протонной шка-
лой, направленной сверху вниз. Протонный спектр
показан справа вдоль шкалы F1 для удобства (вместо
того чтобы давать плохо разрешенные проекции);
заметим, что этот 1 D-спектр не является частью спек-
тра 1Н-’Н COSY, и был добавлен позже. Из правого
верхнего угла к левому нижнему углу проходит «диа-
гональ» - серия поглощений, для которых частоты
V! и v2 равны; эти диагональные пики не дают новой
информации по сравнению с обычным 1 D-спектром
ЯМР 1Н. По обеим сторонам симметрично от диаго-
нали располагаются (по крайней мере теоретически)
так называемые кросс-пики. Впрочем, эта симметрия
в спектрах часто бывает неидеальной.
Прежде чем пытаться детально обсудить спектры
Щ-’Н COSY и структуру ипсенола, нужно упомянуть
еще одно важное экспериментальное усовершенство-
вание, которое уменьшает «хаос» вдоль диагонали.
И хотя этот спектр можно интерпретировать без
такого усовершенствования, в некоторых случаях
(например, для кариофилленоксида) это улучшение
дает серьезный эффект.
5.4.1. Ипсенол: спектр Ж-1)) COSY
с двухквантовой фильтрацией
При простом добавлении третьего импульса л/2,
следующего сразу вслед за вторым (л/2)-импульсом
в простой импульсной последовательности COSY
(если более ничего не изменять), мы получим им-
пульсную последовательность для очень популярного
эксперимента по ’Н-’Н COSY с двухквантовой филь-
трацией (DQF-COSY) (рис. 5.10). Назначение третьего
(л/2)-импульса состоит в том, чтобы удалить (или
«отфильтровать») простые одноквантовые переходы,
чтобы оставались только двухквантовые переходы
(или переходы более высокого порядка). В практиче-
ском смысле двухквантовый фильтр будет выбирать
только такие системы, которые содержат по крайней
мере пару спинов (минимум системы АВ или АХ);
таким образом, сигналы метильных групп (для сингле-
тов, не связанных с другими ядрами спин-спиновым
взаимодействием) будут существенно подавлены. Воз-
можна фильтрация и более высокого порядка (чем
двухквантовая), но она, как правило, не используется.
Например, в эксперименте COSY с трехквантовой
фильтрацией будут выбираться только системы, кото-
рые содержат три или более спинов, так что спиновые
системы АВ и АХ (как и ядра, не связанные никаким
взаимодействием, дающие в спектрах синглеты) будут
исключаться.
Спектр ’Н-*Н DQF-COSY ипсенола можно найти
в нижней части рис. 5.9. Заметим, что спектр кажется
более чистым, особенно вдоль диагонали, что суще-
ственно облегчает задачу интерпретации спектра.
Спектры DQF-COSY выглядят существенно лучше,
все спектры типа COSY, приводимые в этой книге,
являются спектрами с двухквантовой фильтрацией.
Поскольку мы начинаем интерпретацию спектра
DQF-COSY (рис. 5.9), напомним, что этот спектр
показывает корреляцию между связанными прото-
нами. Правильное начальное отнесение или удачная
«точка входа» в спектре COSY (а также других
типах корреляционных спектров) - это один из за-
логов успешного извлечения информации из таких
спектров. Структура ипсенола позволяет выбрать
несколько таких точек входа: например, возьмем
метиковый протон рядом с группой ОН при 3.83 м.д.
Если мы начнем с диагонали и проследуем либо
направо, либо вверх (поскольку спектр симметричен
относительно диагонали, направление движения несу-
щественно), мы попадаем на четыре недиагональных
пика (кросс-пика). Нарисовав линии через эти пики
под прямыми углами вверх относительно одного
выбранного направления, мы найдем (по диагонали)
химические сдвиги четырех связанных протонов.
54. Ипсенол: спектры ,Н-Ч[ COSY
293
Спектр ЯМР !Н (300 МПф
Спектры ЯМР 13С и DEPT (75.5 МПд)
140 130 120 110 100 90 80 70 60 50 40 30 20 м.д.
Рис. 5.8. Спектры ЯМР 1Н,13С и спектры DEPT ипсенола (рабочая частота для
протонов 300 МГц, раствор в CDCI3). Структура ипсенола приведена вместе
с нумерацией ядер, которая используется в тексте
294
5. Корреляционная спектроскопия ЯМР. 2D ЯМР
Рис. 5.9. Спектр COSY ипсенола (300 МГц, простая методика COSY) (вверху), спектр
DQF-COSY ипсенола (внизу)
5А. Ипсенол: спектры ’Н-Ч1 COSY
295
*Н:
Rd
Рис. 5.10. Импульсная последовательность
для ’Н-’Н COSY с двухквантовой фильтрацией
(DQF-COSY). 0 обозначает 90°-ные импульсы, а б -
это фиксированная задержка порядка нескольких
микросекунд
Быстрая проверка структуры ипсенола обнаружи-
вает, что указанный метановый протон примыкает
к двум парам диастереотопных метиленовых групп,
другими словами, протон при 3.83 м.д. связан с че-
тырьмя неэквивалентными протонами, и эти четыре
протона соответствуют протонам в двух примыкаю-
щих метиленовых группах.
Мы могли бы продолжить отслеживать корреляцион-
ные маршруты от этих четырех протонов, а читателю
предлагается сделать это в конце данного раздела. Вме-
сто этого мы выберем другую, также очень полезную
точку отсчета: метановые протоны изопропильной
группы при 1.8 м.д. Мы снова начинаем с диагона-
ли, и на этот раз выясняется, что метановые протоны
изопропильной группы коррелируют с тремя четкими
резонансными сигналами. Две из этих трех корреляций
соответствуют двум протонам одной из диастереотоп-
ных метиленовых групп, которая также коррелирует
с метановым спиртовым протоном. В дополнение,
мы находим корреляцию с двумя перекрывающи-
мися метильными дублетами при 0.93 м.д. Конечно,
эта корреляции полностью согласуются со структурой;
и в самом деле, при рассмотрении только этих двух
протонов (т.е. сигналов при 3.83 и 1.8 м.д.) мы уже
установили корреляции (также называемые «связан-
ностями») для более чем половины молекулы.
Далее мы рассмотрим два протона при атоме
С-5 метиленовой группы со сдвигами 2.48 и 2.22 м.д.
Мы уже видели, что они связаны с метановой спир-
товой группой (вы можете и должны это проверить
с помощью метиленовых протонов), и мы видим,
что они также связаны друг с другом.* 2) Кроме того,
° Мы вернемся к обсуждению диастереотопии метиле-
новых групп в следующем разделе, когда обратимся к
спектрам COSY или HMQC.
2) Геминальные метиленовые протоны (при зр3-гибридизо-
ванных атомах углерода) всегда связаны друг с другом, и
их константа взаимодействия (2JHH) всегда весьма велика
(см. приложения к гл. 3).
видно, что слабые кросс-пики от обоих метиленовых
протонов указывают на корреляцию с олефиновым
протоном при 5.08 м.д.. Эта корреляция вызвана
дальними константами взаимодействия (констан-
той 4JHH, константой взаимодействия через четыре
связи) метиленовых протонов с г/нс-протоном сосед-
ней двойной связи. Очень приятно обнаружить эту
корреляцию, поскольку это позволяет найти ’Н-’Н
связанность с изолированной диеновой спиновой
системой. В отсутствие этих дальних корреляций
подобные изолированные спиновые системы могут
быть «привязаны» с помощью последовательностей
типа НМВС или INADEQUATE, которые описываются
ниже. Далее читателю предлагается самостоятельно
завершить анализ корреляций в спектре DQF-COSY
ипсенола. Корреляции можно найти для всех протонов,
за исключением гидроксильного протона, который
участвует в быстром обмене.
5.4.2. Детектирование по ядрам углерода:
«С-’Н COSY (HETCOR)
Эксперимент ^С-^Н COSY (HETCOR) устанавливает
соотношение ядер 13С с непосредственно связанны-
ми протонами; т.е. выявляются константы через одну
связь (Vch). Частотные оси Fl (vj и F2 (v2) соответ-
ствуют разным ядрам, так что в данном случае нет
диагонали и нет симметрии.
Импульсная последовательность для этого экс-
перимента, часто называемая последовательностью
HETCOR (HETeronuclear CORrelation - гетероядер-
ная корреляция), приведена на рис. 5.11, а. В тече-
ние периода эволюции (ZJ большая гетероядерная
константа через одну связь (^сн) используется для
переноса поляризации, и поэтому детектируются
только ядра 13С, к которым присоединены атомы
водорода. Для получения максимального эффекта
переноса поляризации после периода прибавляется
фиксированная задержка А! = 1/(2/сн). Короткая за-
держка (А2) между заключительным 13С-импульсом
и началом выборки представляет собой период ре-
фокусировки, так что линии 13С не имеют противо-
положных фаз и поэтому не компенсируют друг
друга, если используется «развязка» от протонов.
Оптимальное время рефокусировки (А2) зависит от
того, принадлежит ли ядро 13С к группе СН, СН2 или
СН3. Обычно используется некая компромиссная
величина времени А2 = 1/(3JCH).
Ось Fl (vj, которая получается из инкремент-
ных задержек представляет собой ось протонов.
Ось F2 (vj, которая получается за время Z2, являет-
296
5. Корреляционная спектроскопия ЯМР. 2D ЯМР
АЛ
180
Выборка (Л)
90°
_1 1 _________________________
2^ | 3 J «Развязка» включена
180°
A/j
«Развязка» включена
б
90°
Рис. 5.11. Импульсные последовательности для
экспериментов HETCOR (а) и HMQC (6). Типичное
значение константы исн равно 145 Гц
ся осью 13С. Таким образом, считывающий импульс
л/2 поступает в канал 13С, и выбираемый в период
t2 сигнал ССИ представляет собой сигнал ядер 13С.
Наконец, в случае идеального эксперимента резо-
нансы ядер углерода должны быть синглетами (а
не мультиплетами):1) в протонном канале в момент
выборки используется широкополосная «развязка»
с помощью составных импульсов (метод CPD -
composite pulse decoupling), поэтому сигналы ядер
углерода, получаемые из каждого ССИ, являются син-
глетами. Напомним, что в случае 2В-эксперимента,
корреляция возникает в период времени и, сле-
довательно, «развязка» от протонов не включается
в течение этого периода.
5.4.3. Детектирование по протонам:
]Н -13С COSY (HMQC)
Исторически эксперимент HMQC (Heteronuclear
Multiple Quantum Correlation - многоквантовая
гетероядерная корреляция) предшествовал экспе-
рименту HETCOR. В методиках проведения этих
экспериментов имеется много различий, однако
самое существенное состоит в том, что в экспери-
]) При наличии КССВ через одну связь сигнал метильного
атома углерода будет выглядеть как квартет, метиленово-
го - как триплет и т.д.
менте HETCOR детектируются ядра 13С, а в экс-
перименте HMQC - протоны. Поскольку протоны
и углерод-13 существенно различаются по отно-
сительному содержанию и чувствительности, в на-
стоящее время более предпочтителен метод HMQC.
Преимущество обратных экспериментов над пря-
мыми экспериментами состоит в том, что в об-
ратных экспериментах детектируется ядро с более
высоким значением величины у (обычно это ядро
!Н), что обеспечивает более высокую чувствитель-
ность.
Как отмечалось в гл. 3, абсолютная чувствитель-
ность эксперимента ЯМР дается следующей фор-
мулой:
S/N = (NT^fyMl2nsm)IT,
где Уехси Ydet- это гиромагнитные отношения для 13С
в случае эксперимента с прямым детектированием,
в то время как в случае эксперимента по обратному
детектированию эти величины являются гиромагнит-
ными отношениями для протонов. Замена гиромаг-
нитных отношений приводит к увеличению чувстви-
тельности в (у1Н/у13с)5/2 раз, т.е. примерно в 30 раз для
эксперимента HMQC по сравнению с методом
прямого детектирования HETCOR. Недостатком экс-
перимента по корреляции через химический сдвиг
с инверсным детектором является то, что сильные
сигналы от протонов, не связанных непосредственно
с ядрами 13С, должны быть подавлены, если же это не
удается сделать, возникает проблема динамического
диапазона, и при этом, как правило, требуется про-
ведение дополнительного фазового циклирования.
Введение импульсного градиента поля в ЯМР высоко-
го разрешения существенно улучшает проблему пода-
вления сигналов протонов, присоединенных к атомам
изотопа 12С. Подавление является почти идеальным
без использования какого-либо дополнительного фа-
зового циклирования, что значительно улучшает ка-
чество спектра и приводит к существенной экономии
времени эксперимента.
Импульсная последовательность для эксперимента
HMQC приведена на рис. 5.11, б. Имеет смысл обсу-
дить три особенности этой импульсной последователь-
ности. Ось Fl (vj), которая возникает из инкрементных
задержек tX9 - это ось ядер углерода. Ось F2 (v2),
получаемая в течение времени t2, - это ось протонов.
Таким образом, считывающий импульс л/2 идет по
каналу !Н, и принимаемый ССИ в период времени
t2 представляет собой сигнал ядер !H. Наконец, «раз-
вязка» методом GARP (использующим низкую мощ-
ность и представляющим собой вариант широкопо-
лосной «развязки» CPD) используется для подавления
5.4. Ипсенол: спектры 'Н-’Н COSY
297
ССВ с ядрами с большим диапазоном химических
сдвигов. Метод GARP (Globaly optimized Alternating
phase Rectangular Pulses; метод прямоугольных им-
пульсов, глобально оптимизированных с помощью
чередующейся фазы) - это метод широкополосной
«развязки», применяемый в углеродном канале во
время выборки, так что получаемые сигналы протонов
не расщепляются в дублеты из-за взаимодействия
с ядрами 13С. Напомним, что эксперимент HMQC
обращен на связь 1Н-13С, что проявляется в виде
13С-сателлитов в спектре !Н (при природном со-
держании 1.1%, см. раздел 3.7.5).
5.4.4. Ипсенол: спектры HETCOR и HMQC
В верхней части рис. 5.12 приведен спектр HETCOR
ипсенола, а в нижней - спектр HMQC этого же со-
единения. Оба спектра представлены одинаковым
образом, за исключением того что оси поменялись
местами. Так, в спектре HETCOR ось F2 снизу соот-
ветствует углеродной шкале, а ось F1 справа-это про-
тонная шкала, а в спектре HMQC эти оси меняются
местами. Теоретически информационное содержание
обоих спектров идентично; и в самом деле, мы об-
наруживаем те же самые корреляции. Единственное
реальное различие, выявляемое при сравнении двух
экспериментов, - это различие в цифровом разреше-
нии по оси ядер углерода:
Давайте познакомимся со спектрами HMQC путем
анализа спектра HMQC ипсенола (рис. 5.12, нижняя
часть). Сразу видно, что тут нет диагонали, а также
отсутствует симметрия, поскольку оси F1 и F2 соот-
ветствуют различным ядрам. В данном представлении
ось F1 (ось углерода) показана слева по вертикали,
а ось F2 (протоны) - по горизонтали. Напротив этих
осей (сверху для оси F2 и справа для оси F1) рас-
положены соответствующие 1 D-спектры, которые
даются для удобства (они не являются частью ре-
альных 2В-спектров). Интерпретация этого спектра
очевидна. Мы можем начать с любого атома углерода
и мысленно вести горизонтальную линию, пока она
не пересечет кросс-пик.1) Другая линия мысленно
проводится перпендикулярно к первой линии, и на
ней находится протон или несколько протонов, с ко-
торыми есть корреляция.
Для каждого атома углерода есть только три воз-
можности. Если проведенная линия вообще не встре-
' Мы могли бы начать с оси протонов и получили бы
точно такие же результаты. В случае перекрывания в про-
тонном спектре не всегда можно найти все надлежащие
начальные точки. Для углеродной оси перекрывание обыч-
но не представляет серьезной проблемы.
чает никакие кросс-пики, то этот атом углерода не
имеет непосредственно связанных протонов. Если
линия встречает только один кросс-пик, то атом угле-
рода связан с 1,2 или 3 протонами; если присоеди-
нены два протона, то либо они имеют одинаковый
химический сдвиг, либо случайно два сигнала на-
ложились друг на друга. Если горизонтальная линия
пересекает два кросс-пика, то это особый случай
диастереотопных протонов, находящихся в метиле-
новой группе. Большая часть этой информации нами
уже была получена из спектров DEPT (см. раздел
4.6); в действительности спектр HMQC должен рас-
сматриваться вместе со спектром DEPT (там, где это
возможно).
В молекуле ипсенола присутствует четыре ме-
тиленовых группы, и все они включают пары диа-
стереотопных протонов. Резонансы для двух из этих
четырех метиленовых групп наблюдаются в спектре
13С при 41 и 47 м.д. Отметим, с какими протонами
эти углеродные атомы коррелируют, и сравним ре-
зультат с данными спектров COSY. Как и ожидалось,
результаты подтверждают наше отнесение, сделанное
из спектров COSY, и это помогает построить проч-
ную основу для отнесения. Два других метиленовых
атома углерода обнаруживаются при более высо-
ких частотах в олефиновой области, а кросс-пики
HMQC для этих углеродных резонансов помогают
выяснить характер перекрывания сигналов в про-
тоном спектре.
Молекула ипсенола содержит две метильных груп-
пы, проявляющихся в виде «триплета» при 0.93 м.д.
в протонном спектре. Однако более внимательный
анализ показывает (см. врезку), что этот триплет на
самом деле является парой частично совпадающих
дублетов. Прежде чем переходить к следующему
разделу, рассмотрим такой вопрос: можно ли отне-
сти олефиновые метиленовые углеродные сигналы
на основе объединенной информации из спектров
COSY и HMQC?
5.4.5. Ипсенол: детектирование по протонам;
гетероядерные корреляции через
дальние константы ’Н-’Ч (метод НМВС)
Для эксперимента HMQC, описанного выше, нам нуж-
но было исключить дальние (т.е. через две и три свя-
зи) протон-углеродные константы и в то же время
сохранить прямые (т.е. через одну связь) константы,
дающие корреляции в 2О-эксперименте. Напротив,
в эксперименте НМВС (Heteronuclear Multiple Bond
Coherence - гетероядерная когерентность через не-
сколько связей), который также основан на протоном
298
5. Корреляционная спектроскопия ЯМР. 2D ЯМР
2он HMQC (300 МГЦ)
СН2=
7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 м.д.
F2
Рис. 5.12. Спектры ипсенола: HMQC (внизу) и HETCOR (вверху)
5.4. Ипсенол: спектры 'Н-’Н COSY
299
детектировании1), используются именно константы че-
рез две и три связи, что дает очень эффективный (хотя
иногда трудно интерпретируемый) спектр. По су-
ществу мы косвенно получаем углерод-углеродные
(хотя и не прямые 13С-13С) корреляции, и, кроме
того, мы теперь можем «видеть» или коррелировать
четвертичные атомы углерода с соседними прото-
нами. Поскольку сохраняются как константы 2JCH,
так и константы 3JCH, интерпретация может оказаться
затруднительной; следует быть последовательными
и не забывать о корреляциях HMQC.
Интерпретация спектров НМВС требует опреде-
ленной маневренности, так как мы не всегда находим
то, что ожидаем найти. В частности, в зависимости от
типа гибридизации атома углерода и других факторов
некоторые из корреляций через две связи (2JCH) или
через три связи (3JCH) могут случайно отсутствовать.
Вдобавок к этой путанице иногда обнаруживаются
корреляции через четыре связи (VCH)! Вариации
в корреляциях вызваны изменениями величин кон-
стант 2Jch» 3*Zch и 4*Zch-
Для интересующихся читателей импульсная после-
довательность для эксперимента по НМВС показана
на рис. 5.13. Время задержки 1/(27) может быть опти-
мизировано для различных типов констант. Типич-
ное значение величины J предполагает, что средняя
дальняя константа JCH составляет 8 Гц.
Спектр НМВС ипсенола (рис. 5.14) похож на спектр
HMQC ипсенола с двумя очевидными различиями:
имеется существенно больше корреляций и исчез-
ли корреляции через одну связь (HMQC). (Спектр
разделен на пять частей, так что разрешение до-
статочно высокое, чтобы позволить видеть все кор-
реляции.) Интерпретация для ипсенола очевидна.
Но для начала обратим внимание на один общий
артефакт: появление сателлитов 13С от интенсивных
протонных пиков, особенно от сигналов метильных
групп. Если следовать параллельно протонной оси
(F2) при значении сдвига 23 м.д. по оси углерода
(F1), обнаружится кросс-пик при примерно 0.93 м.д.
(протонный), который действительно соответствует
протонному сигналу. В то же время мы находим два
«кросс-пика», которые не коррелируют ни с одним
из протонов на оси F2. Это сателлиты 13С и они
п Существует эксперимент, аналогичный НМВС, с детекти-
рованием по углероду, называемый экспериментом COLOC
(Correlated spectroscopy for LOng range Couplings - корре-
ляционная спектроскопия для дальних констант); он был
предложен ранее, чем рассматриваемый тут эксперимент.
Спектры COLOC в настоящее время используются срав-
нительно редко, и мы не будем давать примеры подобных
спектров.
90° 180° | Выборка (t2)
‘К -----------------S-------I
90° 90°
“С:__________| " |_______________
Рис. 5.13. Импульсная последовательность
для эксперимента НМВС. Константа J выбрана
в соответствии с дальними константами (2JCH и 3JCH),
обычно составляющими 8 Гц
должны быть исключены. Резонанс 13С другой ме-
тильной группы демонстрирует то же явление; пики
сателлитов отмечены на рис. 5.14 стрелками.
Мы можем начать с любого из резонансов - угле-
родного или протонного - и получим эквивалентные
результаты. Мы будем использовать ось углерода
как начальную точку, поскольку по ней обычно су-
щественно меньше перекрывание. Например, линия,
проведенная параллельно оси протонов при примерно
68 м.д. на углеродной оси (атом углерода рядом с ОН-
группой), пересекает пять кросс-пиков; ни одна из
пяти корреляций не соответствует непосредственно
присоединенному протону (^сн) при 3.8 м.д. Четыре
кросс-пика соответствуют двум парам диастереотоп-
ных протонов в метиленовых группах (при 2.48,2.22,
1.45 и 1.28 м.д.),и они соответствуют константам 2JCH
(или константам через две связи). Пятое пересечение
(константа 3JCH) коррелирует этот углеродный атом
(при 68 м.д.) с изопропильным метиковым протоном
(при 1.82 м.д.), который связан с углеродным атомом
в P-положении. Другой углеродный атом в Р-положении
не связан с протоном, так что мы не имеем корреляции
с ним от спиртового атома углерода. Таким образом,
мы получили косвенные связанности углерода с двумя
а-утлеродными атомами и с одним из двух Р-углеродов.
Другой полезный пример можно найти при про-
ведении линии от углеродного резонанса при 41 м.д.
Это атом С-5 метиленовой группы; прежде всего от-
метим, что отсутствуют корреляции с непосредственно
присоединенными протонами при 2.48 и 2.22 м.д. Име-
ется только один а-углеродный атом, который связан
с одним (или более) протоном; его соответствующие
корреляции обнаруживаются с протоном у метанового
атома С-4 спиртовой группы при 3.83 м.д.2) Имеются
2) Другой а-атом углерода при С-6, который был р-атомом
углерода в первом примере, также не показывает корре-
ляций в спектре HMQC. Читатель должен показать, что
имеются полезные корреляции с этим атомом углерода
на рис. 5.14.
300
5. Корреляционная а>ектроскопия ЯМР. 2D ЯМР
7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 м.д.
F2
Рис. 5.14. Спектр НМВС ипсенола. Для наглядности спектр разбит на пять частей.
Стрелки в верхней части указывают на сателлиты 13С. Корреляции показаны
линиями
5.5. Кариофилленоксид
301
три Р-атома углерода, и все они связаны с протона-
ми. Метиленовый атом С-3 показывает косвенные
корреляции для обоих диастереотопных протонов
при 1.45 и 1.28 м.д. Алкеновый метановый протон
при С-7 дает кросс-пик при 6.39 м.д., как и протоны
олефиновой метиленовой группы, присоединенной
к С-6, при 5.16 и 5.09 м.д. Поскольку атом С-6 яв-
ляется четвертичным, эксперимент НМВС дает нам
возможность «видеть сквозь» эти обычно недоступные
точки в молекуле. Другие отнесения мы оставляем
читателю в качестве упражнения; хорошая стартовая
точка-четвертичный углерод с химическим сдвигом
примерно 143 м.д. (атом С-6).
5.5. Кариофилленоксид
Структура кариофилленоксида существенно более
сложная, и это достойный пример для проверки ме-
тодов, которые мы только что использовали в случае
ипсенола. Спектры ЯМР 1Н, 13С и DEPT этого соеди-
нения приведены на рис. 5.15. В качестве помощи при
обсуждении спектров DQF-COSY кариофилленоксида
выделим следующие моменты в описании протонно-
го спектра: три метильных синглета при 0.98, 1.01,
1.19 м.д., два олефиновых сигнала «дублета» (с ма-
ленькими геминальными олефиновыми константами)
при 4.86 и 4.97 м.д. и резонансные сигналы от 13-ти
остальных протонов, дающих мультиплеты между
0.9 и 3.0 м.д. И хотя нам структура известна, невоз-
можно отнести эти протоны, пока мы не сделаем одно
или несколько произвольных допущений. О
5.5.1. Кариофилленоксид: спектр DQF-COSY
Спектр DQF-COSY кариофилленоксида представлен
на рис. 5.16. Проблема состоит в том, что нет хорошей
стартовой точки. Отметим, что это утверждение не
очевидно. Без исходной точки невозможно провести
многие очевидные корреляции (нарисованные для
удобства), которые видны для данной структурной
формулы. Поэтому наш подход будет заключаться
в регистрации некоторых корреляций, которые мы
действительно видим, и затем ожидании дополни-
тельной информации (например, спектры HMQC),
° Например, можно предположить, что аллильный метико-
вый протон в голове моста должен быть в более слабых
полях, и поэтому отнести дублет дублетов при 2.86 м.д. к
этому протону (что неверно). Методы, описанные в этой
главе, позволят провести эти отнесения без выдвижения
слабо аргументированных предположений.
прежде чем пытаться преобразовать эти корреляции
в структуру.
Экзоциклические олефиновые метиленовые про-
тоны показывают очевидные взаимные COSY корре-
ляции. Кроме того, видны слабые кросс-пики между
олефиновыми протонами при 4.86 и 4.97 м.д. и диа-
стереотопной метиленовой группой (2.11 и 2.37 м.д.)
и квартетом при 2.60 м.д. соответственно. Эти взаи-
модействия напоминают о дальних аллильных кон-
стантах, которые мы видели в ипсеноле; можно было
бы отнести эти корреляции к диастереотопным ме-
тиленовым протонам при С-7 и к метановому про-
тону при С-9. Однако мы пока не будем торопиться
и вернемся к этой проблеме позже.
Если взглянуть на часть этого спектра COSY в са-
мых сильных полях, то обнаруживается неожиданное
взаимодействие. Кажется, что либо один, либо оба
метильных синглета показывают константы связи
с сигналами при 1.65 и 2.09 м.д. Это кажущееся
противоречие может быть разрешено путем вни-
мательного анализа метильных синглетов в районе
0.98 м.д. Здесь наблюдается необычно высокопольный
мультиплет, частично закрытый метильными сингле-
тами, который мы вначале не заметили. Неожиданные
находки такого рода весьма типичны для корреляци-
онных спектров; как частично, так и полностью скры-
тые сигналы обычно обнаруживаются в 2О-спектрах
(см. ниже спектр HMQC). Прежде чем продолжить
обсуждение спектров кариофилленоксида, рассмо-
трим корреляции ’Н-^С и как корреляции
взаимосвязаны с корреляциями
5.5.2. Кариофилленоксид: спектр HMQC
Спектр COSY кариофилленоксида легче понять, если
при интерпретации использовать информацию, из-
влекаемую из спектра HMQC (рис. 5.17). Из спек-
тра DEPT (см. рис. 5.15) мы уже знаем, что карио-
филленоксид дает три сигнала метильных атомов
углерода (при 16.4, 22.6, и 29.3 м.д.), шесть сигна-
лов от метиленовых атомов углерода (при 26.6,29.2,
29.5,38.4,39.1 и 112.0 м.д.), три сигнала метиновых
атомов углерода (при 48.1, 50.1 и 63.0 м.д.) и три
сигнала четвертичных атомов углерода (при 33.3,
59.1 и 151.0 м.д.).
Отнесение олефиновых метиленовых групп (как
протонов, так и 13С), а также трех метильных групп
(протонов и углеродов) тривиально, и эти отнесения
соответствуют нашему предыдущему обсуждению.
Больший интерес и пользу представляет отнесение
трех метиновых протонов: дублет дублетов при
2.86 м.д. (коррелирует с сигналом углерода при
63.0 м.д.), кажущийся квартет при 2.60 м.д. (корре-
302
5. Корреляционная атектроскопия ЯМР. 2D ЯМР
Спектр ЯМР 'Н (600 МГЦ)
। • । ‘ । ‘ ' 1 । • 1 • • • I 1 1 I । । ' 1 I 1 '
5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 м.д.
Спектр ЯМР 13С и спектр DEPT (150.9 MIU)
150 140 130 120 110 100 90 80 70 60 50 40 30 20 м.д.
Рис. 5.15. Спектры ЯМР 'Н и 13С, а также спектр DEPT кариофилленоксида.
Показанная нумерация атомов используется в тексте
5S. Кариофилленоксид
303
Спектр COSY (600 МП1)
F2
Fl
Рис. 5.1 б. Спектр DQF-COSY кариофилленоксида. Нижняя часть спектра
представляет собой растянутый спектр с проведенными линиями корреляций
и обозначенными отнесениями
304
5. Корреляционная спектроскопия ЯМР. 2D ЯМР
13 5 9 7 6 3,7’
LI______1_____
1 10,2,10’ 2’ 6' 12
1 10,2,10’ 2’ 6' 12 3' I 15,14
Спектр HMQC (600 МПф
15,14
5.0 м.д. 2.8 2.6 2.4 2.2 2.0 1.8 1.6 1.4 1.2 1.0 м.д.
F2
Рис. 5.17. Спектр HMQC кариофилленоксида. Вместо обычного спектра ЯМР 13С на
врезке показан спектр DEPT 135 для большей ясности
5.5. Кариофилленоксид
305
лирует с сигналом углерода при 48.0 м.д.) и кажу-
щийся триплет при 1.76 м.д. (коррелирует с сигна-
лом углерода при 50.1 м.д.). По спектру COSY и на
основании известной структуры мы теперь можем
отнести все три сигнала метиновых протонов и ис-
пользовать эту информацию снова в спектре COSY,
чтобы установить другие корреляции.
На основании корреляции через дальнюю ал-
лильную константу, найденную в спектрах COSY,
мы можем подтвердить предварительное отнесение,
сделанное ранее. Дублет дублетов при 2.86 м.д. от-
носится к метиновому протону эпоксидного кольца,
и его химический сдвиг может быть объяснен на
основе дезэкранирования за счет эпоксидного атома
кислорода. Другой метановый протон (в голове мо-
ста, примыкающий к гем-диметильной группе) соот-
ветствует мультиплету при 1.76 м.д. С учетом этих
отнесений мы могли бы снова вернуться к спектрам
COSY, но вместо этого сделаем сначала отнесение
для метиленовых протонов. Знание этих отнесений
поможет нам ускорить анализ спектров COSY.
Начиная с высокопольной части спектра ЯМР 13С,
можно сделать следующие отнесения: метиленовый
атом углерода при 26.6 м.д. коррелирует с сигналами
протонов при 1.45 и 1.63 м.д., метиленовый атом
углерода при 29.2 м.д. коррелирует с протонными
резонансами при 2.11 и 2.37 м.д., метиленовый атом
углерода при 29.5 м.д. коррелирует с протонными
резонансами при 1.33 и 2.23 м.д., метиленовый атом
углерода при 38.4 м.д. коррелирует с протонными
резонансами при 0.96 и 2.09 м.д., метильный атом
углерода при 39.1 м.д. коррелирует с протонными
резонансами при 1.62 и 1.68 м.д.; олефиновые ме-
тиленовые группы мы уже отнесли ранее.
Таким образом, затратив небольшие усилия, мы от-
несли химические сдвиги всех протонов в карио-
филленоксиде и провели корреляцию этих сдвигов
с резонансами спектра ЯМР 13С; мы сгруппировали
диастереотопные протоны для каждой из метилено-
вых групп и получили три отдельных точки входа
для спектра COSY, хотя до этого мы не имели ни
одной такой точки. Теперь мы готовы вернуться
к анализу спектра COSY кариофилленоксида и про-
вести отнесение корреляций с учетом рассматривае-
мой структуры.
Растянутая часть от 0.8 до 3.0 м.д. спектра DQF-COSY
кариофилленоксида приведена в нижней части рис.
5.16. На этом рисунке показаны линии, связывающие
протон-протонные корреляции, чтобы упростить обсуж-
дение. Связанности в спектре COSY позволяют скон-
струировать фрагменты структуры или, как в данном
случае, подтвердить структурные фрагменты. Для поиска
корреляций с атомами С-5, С-6 и С-7 начнем с протона
Н-5 при 2.87 м.д. Этот протон показывает кросс-пики
с двумя резонансами при 1.32 и 2.24 м.д. Из спектра
HMQC мы знаем, что это сигналы диастереотопных
протонов, и они могут быть отнесены к протонам
Н-6 и Н-6'. Протоны, присоединенные к атому С-6,
дают корреляции с протонами при 2.11 и 2.37 м.д.; мы
относим эти протоны, которые также являются диа-
стереотопными, к атому С-7 при 29.2 м.д. Протоны
при С-7 взаимодействуют друг с другом, как и про-
тоны при С-6.
Другие корреляции также очевидны. Спиновая си-
стема при С-5, С-6, С-7 изолирована, поэтому нужно
выбрать другую точку входа. Можно начать с ал-
лильного метанового протона в голове моста (Н-9)
при 2.60 м.д. Мы уже отмечали дальние аллильные
взаимодействия. Кроме того, обнаруживаются три
других взаимодействия, которые можно отнести с по-
мощью спектра HMQC. Одна из корреляций относится
к метиновому протону при 1.76 м.д., который соот-
ветствует Н-1. Две другие корреляции находят два
диастереотопных протона (снова из спектра HMQC)
при 1.62 и 1.68 м.д.; мы идентифицируем их как
протоны Н-10 и Н-1 О'. Протоны при атоме С-10 не
связаны с какими-либо еще протонами, и мы не на-
ходим никаких других корреляций для них.
Протон Н-1 обнаруживает константу связи с обоими
протонами у С-2 при 1.45 и 1.63 м.д. Взаимодействие
между протонами Н-1 и Н-2' при 1.63 м.д. является
слабым. Оба протона у С-2 связаны с обоими про-
тонами у С-3 при 0.95 и 2.06 м. д., и можно найти
соответствующие кросс-пики. Таким образом, мы уста-
новили косвенные связи от атома С-10 через С-9,
С-1 и С-2 до С-3. Спектр HMQC оказал неоценимую
помощь в интерпретации. Однако многие вопросы
остаются нерешенными. Мы не нашли никаких корре-
ляций для трех четвертичных атомов углерода и трех
метильных групп. Оба спектра HMQC и COSY под-
тверждают структуру кариофилленоксида, но они
не исключают других возможных структур.
5.5.3. Кариофилленоксид: спектры НМВС
Спектр НМВС для кариофилленоксида (рис. 5.18)
позволяет полностью подтвердить его структу-
ру с помощью проявляющихся связанностей через
косвенные спин-спиновые взаимодействия. Ана-
лиз структуры кариофилленоксида показывает,
что должно быть 87 кросс-пиков; причем это число
получается из рассмотрения каждого из 15 углерод-
ных атомов и подсчета числа протонов с различными
химическими сдвигами в а- и 0-положениях. Чтобы
выявить все эти взаимодействия, необходимо быть
очень внимательным.
306
5. Корреляционная спектроскопия ЯМР. 2D ЯМР
Спектр НМВС (600 МПф
7 6 3,7’
1 10,2,10* 2* 6* 12 3’ II 15,14
Рис. 5.18. Спектр НМВС кариофилленоксида
F2
5.5. Кариофилленоксид
307
Таблица 5.1. Корреляции в спектре НМВС для кариофилленоксида
Атом углерода С-1 С-2 С-3 С-4 С-12 С-5 С-б С-7 С-8 С-13 С-9 С-10 С-11 С-15 С-14
Протон М.Д. 50.1 26.6 38.4 59.1 16.4 63.0 29.5 29.2 151 112 48.0 39.1 33.3 22.6 29.3
Н-1 1.76 HCfl а р р а р а р р
Н-2 1.45 а НС а р ₽ р р
Н-2' 1.63 а НС а р р р р
Н-3 0.95 р а нс а р р
Н-3' 2.06 р а НС а р р
Н-12 1.19 р а НС р
Н-5 2.86 р а р НС а р
Н-6 1.28 р а НС а р
Н-6' 2.23 р а НС а р
Н-7 2.11 Р а НС а р р
Н-7' 2.37 Р а НС а р р
Н-13 4.86 Р а НС р
Н-13' 4.97 Р а НС р
Н-9 2.6 а р Р а р НС а р
Н-10 1.43 р р а НС а р р
Н-10' 1.47 р р а НС а р р
Н-15 0.98 р Р а НС р
Н-14 1.01 р Р а р НС
а НС - непосредственно связанные протон и атом углерода, которые не видны в спектре
НМВС.
Один из способов учета этих данных сводится
к составлению таблицы, в которой по одному на-
правлению (например, по столбцам) перечисляют-
ся углеродные сигналы, а по другому направлению
(соответственно, по строкам таблицы) - протонные
сигналы. Так, в табл. 5.1 углеродные сигналы пере-
числены сверху (по столбцам), а протонные сигналы
даны сбоку (по строкам). Нумерация атомов для
кариофилленоксида в табл. 5.1 соответствует нуме-
рации, которая дана на рисунках.
Наш подход к данному спектру ничем не отли-
чается от подхода, который был использован при
рассмотрении любых других спектров. В данном
случае легче начинать с протонной оси и отслежи-
вать кросс-пики с сигналами 13С, как это показано
в табл. 5.1. Если мы начнем с углеродной оси, то,
конечно, должны получить те же самые резуль-
таты. Если начать с левого верхнего угла табли-
цы с протона Н-1 при 1.76 м.д., мы прежде всего
увидим, что протон Н-1 связан с атомом С-1 при
50.1 м.д.; этот результат мы уже получали из спек-
тра HMQC. Двигаясь вдоль строки, мы находим
все восемь ожидаемых взаимодействий; каждое
взаимодействие отмечено значком а или 0 в за-
висимости от типа дальней связи: а используется
для атомов углерода через две связи от данного
протона (константа 2JCH) , а 0 - для атомов угле-
рода через три связи (константа 3JCH). Конечно,
в самом спектре нет различий между этими двумя
типами констант; мы отмечаем их таким образом
просто для удобства подсчета. Каждое из взаимо-
действий для протона Н-1, указанное в табл. 5.1,
можно найти в спектре.
У атома С-2 имеются два непосредственно связанных
протона, которые обозначены как протоны Н-2 и Н-2'.
Эти протоны имеют разные химические сдвиги; мы
все же ожидаем, что они проявляются в основном
одинаковым образом в спектрах НМВС. Таким об-
разом, у нас есть полезная независимая проверка от-
несений, сделанных по спектру НМВС, для каждой
пары диастереотопных протонов в кариофилленок-
сиде. Для протона Н-2 при 1.45 м.д. мы получаем
те же пять корреляций, что и находим для протона
Н-2' при 1.63 м.д. При более внимательном анализе
спектра и соответствующей таблицы мы находим,
что получили исключительно детальную информа-
308
5. Корреляционная спектроскопия ЯМР. 2D ЯМР
цию, которая может быть расшифрована при после-
довательном и тщательном анализе.
Относительно четвертичных атомов углерода
нужно сделать одно важное замечание. До сих пор
у нас не было прямых корреляций для таких ато-
мов углерода. Мы также не могли «видеть сквозь»
гетероатомы, такие как кислород, азот, сера и др.
Константы через две и три связи в спектрах НМВС
дают нам возможность получить новую важную
информацию. Например, атом углерода С-4 в карио-
филленоксиде при 59.1 м.д. не имеет присоединен-
ных протонов, поэтому он виден только в спектре
ЯМР 13С; и мы знаем, что это четвертичный атом
углерода, по спектрам DEFT. Если поглядеть в табл.
5.1 на колонку атома С-4, мы найдем четыре корре-
ляции через две связи и четыре корреляции через
три связи. Спектр НМВС полностью подтверждает
эти ожидания, что дает прямое доказательство по-
ложения атома С-4 в данной молекуле.
5.6. Корреляции ,3С-”С: INADEQUATE
На основании косвенных связанностей углерод-угле-
род эксперимент НМВС дает возможность устано-
вить скелет органического соединения, но это уто-
мительный процесс, потому что заранее не известно,
результатом какого взаимодействия (через две или
три связи) являются эти корреляции. 20-Эксперимент
INADEQUATE (Incredible Natural Abundance DoublE
QUAntum Transfer Experiment, что можно перевести
как эксперимент по двухквантовому переносу при
природном содержании углерода) завершает набор
«основных» корреляций через связи; мы уже рас-
смотрели такие эксперименты, как COSY (который
дает протон-протонные взаимодействия), HMQC
(HETCOR) (дает протон-углеродные взаимодействия
через одну связь), НМВС (дает протон-углеродные
взаимодействия через две и три связи); и теперь
рассмотрим INADEQUATE (дает прямое углерод-
углеродное взаимодействие, т.е. через одну связь).
Для выяснения структуры органических соединений
этот эксперимент, безусловно, является самым убеди-
тельным и легким для интерпретации, в нем не воз-
никает никаких неясностей, двусмысленностей, и не
бывает никаких исключений из правил. Прочитав по-
следнее утверждение, пессимистически настроенный
читатель может задать вопрос: в чем же «секрет»?
На самом деле секрет заключается в чувствитель-
ности. Вернемся опять к гл. 4, в которой говорилось,
что вероятность того, что любой из атомов углерода
окажется именно атомом 13С составляет около 0.01.
Таким образом, вероятность того, что два соседних
атома углерода будут оба атомами 13С (два независи-
мых события), равна 0.01 х 0.01 (или 0.0001); другими
словами, это 1 молекула из 10000!
Задача поиска связанностей 13С-13С, на первый
взгляд, кажется невыполнимой, но здесь может помочь
двухквантовая фильтрация. Возвращаясь к эксперимен-
там DQF-COSY, вспомним, что двухквантовая фильтра-
ция устраняет все простые одноквантовые переходы,
которые в данном случае соответствуют изолированным
атомам 13С; во время выборки детектируются только
те переходы, которые соответствуют системам двух
спинов (системы типа АВ и АХ) и системам более
высокого порядкаЧ Основная экспериментальная про-
блема, с которой можно столкнуться, - это количество
образца: предполагается, что соединение достаточно
растворимо в растворителе, обеспечивающем систему
стабилизации сигнала (lock). Для низкомолекулярных
соединений (с молекулярной массой менее 500) при
условии использования современного спектрометра
высокого разрешения необходимо растворить при-
мерно 50-100 мг исследуемого вещества в 0.5 мл
дейтерированного растворителя.
Этот эксперимент можно представить как угле-
родный аналог эксперимента DQF-COSY, в котором
обе оси F1 и F2 были бы осями ядер 13С. Теорети-
чески это возможно, но практически эксперимент
INADEQUATE проводят немного иначе, чтобы до-
стичь полной двухквантовой фильтрации. Спектр
INADEQUATE кариофилленоксида приведен на
рис. 5.19; видно, что ось F2 - это обычная (хоро-
шо нам знакомая) углеродная ось, которая связана,
со временем выборки t2. Ось F1 выглядит необычно
и требует некоторых пояснений.
За время эволюции tx мы получаем частоты, которые
не являются химическими сдвигами взаимодействующих
ядер, как это было в экспериментах DQF-COSY. Напро-
тив, это сумма смещений частот от несущей частоты
передатчика для связанных ССВ ядер. Эти смещения
возникают за время и поскольку осуществляется
двухквантовая фильтрация, в спектрах INADEQUATE
существенный вклад в интенсивности кросс-пиков бу-
дут вносить только двухспиновые системы типа АВ
и АХ. Соответствующий подбор экспериментальных
параметров в импульсной последовательности позволяет
выделить большие прямые константы (Vcch и таким
образом, увидеть только корреляции между непосред-
ственно связанными атомами углерода. Обычно ось
F1 приводится в герцах, и она в два раза больше оси
F2, которая приводится в м.д.
Следуя той же логике, вероятность трехспиновой систе-
мы для необогащенного образца составляет 1 на 1000000.
5,6. Корреляции INADEQUATE
309
F2
Рис. 5.19. Спектр INADEQUATE кариофилленоксида. Для удобства показаны линии
корреляций
5.6.1. Спектр INADEQUATE кариофилленоксида
На рис. 5.19 представлен двумерный спектр
INADEQUATE кариофилленоксида. Кросс-пики или
корреляции в системе координат F1 и F2 для данной
системы АХ можно обнаружить в точках с координа-
тами (vA+vx, Уд) и (va+vx, Vx). Кросс-пики представляют
собой дублеты с расщеплениями, равными константам
взаимодействия (^сс) (см. растянутый фрагмент спек-
тра в нижней части рис. 5.19). Середина линии, свя-
зывающей два дублета, имеет координаты {(vA+vx)/2,
(vA+vx)}; таким образом, набор средних точек для
всех пар дублетов дает диагональную линию спек-
тра. Это очень важное наблюдение, потому что оно
может быть использовано, чтобы отличить реальные
кросс-пики от ложных сигналов и других артефактов.
310
5. Корреляционная спектроскопия ЯМР. 2D ЯМР
Лучше поняв, что такое ось F1 и диагональ, мы можем
продолжить расшифровку спектра. В таблице 5.1 пере-
числены химические сдвиги 13С для соответствующих
атомов углерода (нумерация атомов углерода в молекуле
приводилась ранее); при дальнейшем рассмотрении
мы будем придерживаться этой нумерации. Установить
взаимодействия в высокочастотной части спектра на
рис. 5.19 можно быстро и легко. Углеродный сигнал
в самой слабопольной части спектра с химическим
сдвигом 151.0 м.д. соответствует атому С-8. Спускаясь
вертикально вниз от этого пика к оси F2, мы встретим
три дублета кросс-пиков. Эти три пика «связаны» го-
ризонтально с тремя соответствующими кросс-пиками
для сигналов при 112.0,48.0 и 29.2 м.д. Самый слабо-
польный из этих трех сигналов (при 112.0 м.д.) можно
отнести к атому углерода С-13, два других сигнала от-
носятся к атомам С-7 при 29.2 м.д. и С-9 при 48.0 м.д.
Отметим, кстати, что атом углерода С-13 с химическим
сдвигом 112.0 м.д. имеет только один кросс-пик, и он
соответствует связи с углеродом С-8 (151.0 м.д.).
Чтобы разобраться в низкочастотной области спектра,
эту область растянули (она приведена на рис. 5.19 вни-
зу). Более высокое разрешение на этом фрагменте по-
зволяет легко увидеть дублетную структуру сигналов.
Проследим за взаимодействиями для каждого атома
углерода, приведенного на этом увеличенном фрагменте
спектра. Атом углерода С-11 с химическим сдвигом
33.3 м.д. - это четвертичный атом, поэтому видны
четыре кросс-пика. Таким образом, устанавливается
связь атома С-11 с атомами углерода С-15 (22.6 м.д),
С-14 (29.3 м.д.), С-10 (39.1 м.д.) и С-1 (50.1 м.д.).
Прежде чем завершить обсуждение, отметим,
что в спектре INADEQUATE кариофилленоксида есть
одна особенность, которую стоит проанализировать.
Сигналы атомов углерода 6 и 7 кариофилленоксида
в спектре ЯМР 13С почти перекрываются друг с дру-
гом и с сигналом метильного атома углерода С-14:
их химические сдвиги равны 29.5 (С-6), 29.2 (С-7)
и 29.3 (С-14) м.д. (см. табл. 5.1). Поскольку в молекуле
кариофилленоксида атомы С-6 и С-7 связаны друг
с другом, они должны давать корреляции в спектре
INADEQUATE, но вместо системы АХ получается
система АВ с отношением Av/J много меньше 10.
Поэтому будут отсутствовать два дублета, средние
точки которых лежат на диагонали, но должен поя-
виться мультиплет АВ (см. гл. 3), сам попадающий на
диагональ. На рис. 5.19 (внизу) мы просто видим один
сигнал (вероятно, центральная часть АВ-квартета),
что соответствует общему кросс-пику от прямого
взаимодействия между атомами С-6 и С-7; этот сиг-
нал располагается прямо на диагональной линии.
Другие взаимодействия, которые можно обнаружить
на нижнем фрагменте, оставим для самостоятельного
рассмотрения читателям (в качестве упражнения).
Мы же подведем итоги этого раздела:
• Двумерные эксперименты INADEQUATE отра-
жают на спектре прямые взаимодействия между
атомами углерода, давая возможность однозначно
построить схему углеродного скелета изучаемого
соединения.
• Двумерные эксперименты INADEQUATE имеют
ограниченное применение из-за их чрезвычайно
низкой чувствительности.
5.7. Лактоза
Структура 0-аномера лактозы была приведена на рис.
5.1. При расшифровке спектров ЯМР !Н и 13С (рис. 5.20)
лактозы возникает ряд сложностей, которые преодоли-
мы посредством корреляционной спектроскопии ЯМР.
В растворе лактоза находится в виде равновесной сме-
си а- и 0-аномеров. Эти два диастереомера являются
эпимерами, так как различаются конфигурацией только
одного из десяти стереоцентров. Кроме того, протоны
двух сахарных остатков в каждом диастереомере не
связаны друг с другом благодаря наличию гликозид-
ного кислородного атома и образуют изолированные
спиновые системы. Аналогичная ситуация наблюдается
для всех олиго- и полисахаридов.
Мы не будем много времени тратить на обсуж-
дение одномерных спектров, отметим только неко-
торые из характерных особенностей. Резонансные
сигналы аномерных протонов можно обнаружить при
4.45,4.67 и 5.23 м.д., а сигналы аномерных атомов
углерода-при 91.7,95.6 и 102.8 м.д. Наличие трех
аномерных протонов и атомов углерода объясняется
тем, что а- и 0-аномеры остатка глюкозы дают два
набора резонансных сигналов, в то время как остаток
галактозы, которая существует здесь только в 0-форме,
дает один набор сигналов в протонном и углеродном
спектрах. Другие сигналы в обоих спектрах, особенно
в протонном, очень сложны вследствие достаточно
сильного перекрывания.
5.7.1. Спектр DQF-COSY лактозы
Спектр DQF-COSY (рис. 5.21) лактозы богат корреля-
циями, при этом легко обнаружить точку входа в спектр.
На этом рисунке, как и на других, использованы сле-
дующие обозначения: п - номер положения атома угле-
рода или протона, символ Gn относится к сигналам от
остатков галактозы, символами ал или 0л обозначены
соответственно сигналы, относящиеся к остаткам а- и 0-
глюкозы. Каждый из аномерных протонов, связанных
5.7. Лактоза
311
Спектр ЯМР *Н (600 МГЦ)
a-форма лактозы (а-аномер) ОН
Г"""П......I....I.....I..."Т"тп,"1....1.....1....1.....1.....1....1....1.....1гтт,,,п.""I"—....................I....I.....I
5.3 5.2 5.1 5.0 4.9 4.8 4.7 4.6 4.5 4.4 4.3 4.2 4.1 4.0 3.9 3.8 3.7 3.6 3.5 3.4 м.д.
Спектр ЯМР 13С (150.9 МГЦ)
Рис. 5.20. Спектры 1Н, 13С и DEPT лактозы. В растворе лактоза представляет собой
равновесную смесь аномеров
312
5. Корреляционная спектроскопия ЯМР. 2D ЯМР
F1
М7Т 5.2 5.0 4.8 4.6 4.4 4.2 4.0 3.8 3.6 3.4 м.д.
м.д. , , , , , , , । , ।
3.4-
3.6-
3.8-
4.0-
4.2-
4.4-
4.6-
I
Спектр COSY (600 Mlty
в
I
и * М1
Р2
-3.4
-3.6
G2
a2
-4.0
-4.2
-4.4
-4.6
-3.8
G1
Р1
4.8-
-4.8
5.0-
5.2-
-5.0
-5.2
HDO
F1
al
4.0 3.9 3.8 3.7 3.6 3.5 3.4 3.3 3.2 м.д.
F2
Рис. 5.21. Спектр DQF-COSY лактозы. Обозначения протонных сигналов см.
в тексте. Для пояснения приведены отнесения и проведены корреляционные
линии
5.8. Эстафетный перенос когерентности: метод TOCSY
313
с атомом С-1 в цикле сахара, дает одну и только одну
корреляцию с соответствующим протоном при атоме
углерода С-2. Например, аномерный протон с химиче-
ским сдвигом 4.67 м.д. дает корреляцию с протоном
(очевидно, присоединенным к атому углерода С-2) при
3.29 м.д. Этот протон при атоме С-2 с химическим сдви-
гом 3.29 м.д. дает корреляцию с протоном при атоме
С-3, химический сдвиг которого равен 3.64 м.д.
Дальше продолжать аналогичные рассуждения до-
вольно трудно вследствие сложного перекрывания
сигналов. На растянутом фрагменте (см. нижнюю
часть рис. 5.21) разными прерывистыми линиями
обозначены корреляции для разных остатков сахаров
молекулы лактозы. Рекомендуем читателю проследить
за этими корреляциями, но будьте при этом предель-
но аккуратны. Ниже мы снова будем анализировать
лактозу, но в других экспериментах, которые позволят
снять перекрывание сигналов.
5.7.2. Спектр HMQC лактозы
На рис. 5.23 спектр HMQC лактозы показан в виде трех
частей, чтобы минимизировать перекрывание сигналов
и дать хорошее разрешение вдоль углеродной оси спек-
тра. На этом спектре, как и на спектре COSY, приведено
отнесение. Эти отнесения были проведены с помощью
информации, которую мы не смогли пока получить. (В
конце обсуждения лактозы станет ясно, что эти отнесения
правильны и понятно, как они были сделаны.) Перекры-
вание сигналов не столь сильное, и поэтому некоторые
отнесения все же можно сделать достаточно легко. Этот
спектр (HMQC) полезен тем, что позволяет нам опреде-
лять химические сдвиги большинства протонов, сигналы
которых перекрываются в одномерном спектре.
5.7.3. Спектр НМВС лактозы
Принимая во внимание некоторые сложности, связан-
ные с лактозой, можно ожидать, что спектр НМВС
лактозы будет сложным, с перекрыванием сигналов.
Спектр, приведенный на рис. 5.24, не подтверждает
этих ожиданий. Читателю предлагается проанализи-
ровать данный спектр, который для облегчения задачи
дополнен отнесением.
Одна важная корреляция в этом спектре выделена на
растянутом фрагменте спектра (см. врезку): показана
корреляция между атомом С-4 глюкозы (от обоих а-
и Р-аномеров) и протоном при атоме С-1 галактозы.
Это взаимодействие через три связи чрезвычайно
важное, потому что оно показывает, что связь между
атомом углерода С-1 галактозы и атомом углерода
С-4 глюкозы через гликозидный мостик существует
на самом деле. Отсутствие перекрывания в этом фраг-
менте позволяет нам сделать однозначные выводы.
Очень вероятно, что существует и обратная корреляция
между атомом С-1 галактозы и протоном при атоме
С-4 глюкозы (для обоих аномеров), однако в данном
случае мы не можем сделать однозначных выводов
из-за перекрывания сигналов протона G3 (который
также взаимодействует с атомом углерода С-1 галак-
тозы) с сигналами протонов а4 и Р4.
5.8. Эстафетный перенос
когерентности: метод TOCSY
До настоящего момента мы в основном пытались до-
биться с помощью корреляционных экспериментов
того, чтобы спины эволюционировали за время под
влиянием прямого взаимодействия ядерных спинов.
Мы видели, что эксперименты COSY, HMQC, НМВС
и INADEQUATE позволяют получить подробную ин-
формацию о структурах таких соединений, как ипсенол,
кариофилленоксид и лактоза. В этом разделе мы рассмо-
трим другой метод для получения данных о корреляциях
и применим его к таким молекулам, как углеводы, белки
и нуклеиновые кислоты (которые содержат изолирован-
ные протонные спиновые системы).
Наша цель состоит в том, чтобы добиться перено-
са намагниченности спинов, напрямую друг с другом
не связанных, что позволит нам увидеть корреляции
между ядрами, непосредственно не взаимодействую-
щими, но входящими в одну спиновую систему. Такой
эксперимент называется TOCSY (Totally Correlated
SpectroscopY); мы рассмотрим его в двух вариантах: 1D
и 2D. Импульсная последовательность для 2D-TOCSY на-
поминает прототип двумерного эксперимента, но вместо
второго импульса л/2 вставляется «период смешивания»,
во время которого намагниченность «запирается» по
оси у (рис. 5.22). Чтобы лучше понять этот эксперимент,
можно пренебречь деталями, связанными со «спиновым
захватом», и сконцентрировать внимание на периоде
<—------->
Спиновый
захвату
Выборка (/2)
Рис. 5.22. Импульсная последовательность для
двумерного эксперимента TOCSY
314
5, Корреляционная спектроскопия ЯМР. 2D ЯМР
Рис. 5.23. Спектр HMQC лактозы. Объяснения обозначений см. в тексте.
Для простоты приводится отнесение
5.8. Эстафетный перенос когерентности: метод TOCSY
315
Рис. 5.24. Спектр НМВС лактозы. Пояснения для врезки см. в тексте
316
5. Корреляционная спектроскопия ЯМР. 2D ЯМР
F1
Рис. 5.25. Двумерный спектр TOCSY р-лактозы. Для простоты приводятся отнесения
для некоторых сигналов и проведены корреляционные линии
смешивания. Во время периода смешивания намагни-
ченность переносится от одного спина к соседнему,
а затем к следующему соседнему и т. д. Чем длиннее
период смешивания, тем дальше происходит перенос
намагниченности; теоретически он может распростра-
ниться через всю спиновую систему
Внешне двумерный эксперимент TOCSY очень по-
хож на эксперимент COSY. Оси F1 и F2 являются
протонными; диагональ этого спектра содержит ин-
формацию, которую можно получить из одномерного
спектра, и даже кросс-пики имеют аналогичный внеш-
ний вид. Различие заключается в том, что кросс-пики
в спектре COSY являются результатом взаимодействия
спинов, в то время как кросс-пики в спектре TOCSY
являются результатом эстафетного (последователь-
ного) переноса намагниченности. При длительном
времени смешивания в спектре TOCSY все спины
внутри спиновой системы кажутся взаимодействую-
щими. Чтобы оценить преимущества метода TOCSY,
рассмотрим этот эксперимент на примере дисахарида
лактозы, молекула которого включает три изолиро-
ванные (т.е. отдельные) спиновые системы.
5.8.1. Двумерный спектр TOCSY лактозы
На рис. 5.25 приведен двумерный спектр TOCSY
лактозы. Время смешивания в этом двумерном спек-
тре TOCSY было достаточно длительным, и за это
время произошел более или менее полный перенос
магнитной когерентности через спиновую систему
каждого из сахаридных остатков. Сравним этот спектр
со спектром COSY лактозы, показанным на рис. 5.21,
и отметим их сходства и различия.
Например, мы можем найти все протонные сиг-
налы (и определить их химические сдвиги) для
а-аномера глюкозы, начиная с его аномерного
протона при 5.23 м.д. На рисунке отмечены его
корреляции с другими протонами. Мы не можем
провести однозначное отнесение протонных сиг-
налов только на основе этого одного спектра (хотя
оно приведено на рисунке 5.25), но мы можем ис-
пользовать данные, полученные из спектра COSY.
Аналогичные выкладки можно сделать и для двух
других аномерных сигналов лактозы, но предоста-
вим это читателю.
5.9. HMQC-TOCSY
317
5.8.2. Одномерный спектр TOCSY лактозы
Каждый двумерный эксперимент имеет свой одномер-
ный аналог, и мы склонны считать одномерные экспе-
рименты менее эффективными; часто это так и бывает.
Если снова вернуться к эксперименту COSY, то можно
сказать, что гомоядерная «развязка» могла бы дать нам
аналогичную информацию. В экспериментах с «развяз-
кой» мы выделяем какой-то прогонный сигнал, облучаем
его и сравниваем полученный результат с исходным
(без «развязки») протонным одномерным спектром.
Аналогичное действие происходит и в одномерных
экспериментах TOCSY, часто их называют НОНАНА
(HOmonuclear Hartmann-HAnn - гомоядерная спек-
троскопия Хартмана-Хана). Мы выделяем протонный
сигнал и облучаем его; затем подбираем соответствую-
щее время смешивания для переноса намагниченности,
во время которого мы применяем метод «спинового
захвата», и, наконец, регистрируем одномерный спектр.
На таком спектре будут регистрироваться только те сиг-
налы, для которых происходит перенос намагниченно-
сти. И наоборот, все другие сигналы, находящиеся вне
данной спиновой системы, не появятся.
Еще более эффективно было бы запустить серию
одномерных экспериментов TOCSY, в которых время
смешивания периодически увеличивалось при по-
стоянном облучении данного протона. Чтобы проил-
люстрировать такие эксперименты, мы облучали ано-
мерный протон (при 4.67 м.д.) в 0-аномере глюкозы,
входящем в лактозу, и провели серию экспериментов,
где время смешивания изменялось в диапазоне от
20 до 400 мс. На рис. 5.26 приведены результаты
этой серии экспериментов.
В случае времени смешивания 20 мс обнаружи-
вается только сигнал 02-протона в виде триплета
с химическим сдвигом 3.29 м.д. Если время сме-
шивания составляет более 40 мс, на спектре уже
отчетливо виден 03-протон (появляется еще один
триплет), и начинает появляться 04-протон. В экс-
перименте, где время смешивания уже 80 мс, сигнал
от 04-протона лучше виден, увеличение времени сме-
шивания до 120 мс приводит к появлению в спектре
05-протона. При времени смешивания порядка 400 мс
происходит перенос когерентности через всю спи-
новую систему; на спектре появляется интенсивный
Н-5 сигнал, а также сигналы протонов 06 от диа-
стереотопных метиленовых групп. На таком спектре
(где время смешивания 400 мс) показаны сигналы
06- и 06'-протонов с разными константами спин-
спинового взаимодействия с протоном 05. Однако
эксперименты с применением длительного времени
смешивания имеют свой недостаток: разрешение на
таких спектрах снижается, что может привести к по-
тере некоторых сигналов. Но существует возможность
снизить вероятность потери сигналов, если задать
достаточно большое число ССИ. На рис. 5.26 при-
ведены также аналогичные спектры при облучении
аномерного протона галактозы и аномерного протона
а-глюкозы (при времени смешивания 400 мс).
Эксперименты TOCSY (как одномерные, так и дву-
мерные) широко применяются к разным спиновым
системам, но они особенно эффективны при рас-
шифровке перекрывающихся сигналов. В частности,
одномерные эксперименты TOCSY интересны тем,
что дают возможность «пройти» по всей спиновой
системе, постепенно увеличивая время смешивания.
5.9. Метод HMQC-TOCSY
Предложено много разных «гибридных» двумерных
корреляционных экспериментов, которые объединяют
в себе особенности двух более простых двумерных
экспериментов. Примером может служить весь-
ма популярный и полезный метод HMQC-TOCSY.
В спектрах, получаемых этим методом, видны взаи-
модействия через одну связь Щ-^С (прямые взаимо-
действия) (HMQC), но показаны эти корреляции для
всей спиновой системы (TOCSY). Такой эксперимент
упрощает сложные спектры углеводов и белков и по-
зволяет быстро сделать отнесение сигналов.
5.9.1. Спектр HMQC-TOCSY лактозы
На рис. 5.28 приведен спектр HMQC-TOCSY лактозы,
и отмечены все сигналы протонов и ядер углерода.
Этот спектр сильно напоминает спектр НМВС, но кор-
реляции совершенно другие. Данный спектр очень
полезен и интересен тем, что расшифровку спектра
можно начать не только с протонной оси (F2), но и с
углеродной (F1). Если начать с протонной оси, а имен-
но с сигнала аномерного протона а-глюкозы (а1) при
5.23 м.д, и вертикально двигаться дальше, то можно
обнаружить шесть корреляций от шести атомов угле-
рода этого глюкозного кольца. Но если вспомнить
«простой» спектр HMQC лактозы, на нем видна толь-
ко одна корреляция для этого протона. Аналогично
аномерный протон 0-аномера глюкозы при 4.67 м.д.
также дает шесть корреляций для атомов углерода
соответствующего глюкозного кольца в лактозе.
Однако корреляции аномерного протона галактозы
(4.46 м.д) показывают только четыре взаимодействия
вдоль углеродной оси. Этот результат для по аномер-
ного протона галактозы согласуется с одномерным
спектром TOCSY(см. рис. 5.26), который показывает,
что не происходит переноса когерентности дальше
318
5. Корреляционная аектроскопия ЯМР 2D ЯМР
Время смешивания 400 мс, облучается al-протон
аб
________________________________________________________________
Время смешивания 400 мс, облучается Gl-протон
Время смешивания 400 мс, облучается pi-протон
Время смешивания 80 мс
Время смешивания 40 мс
Время смешивания 20 мс
Спектр ЯМР ’Н (600 МПа) ft
_____________________________________________________________________Да
I........I........'I.........I 1 1 ....I.........I.........I..........I........I
3.9 3.8
3.7 3.6 3.5 3.4 3.3 м.д.
Рис. 5.26. Серия одномерных экспериментов TOCSY для 0-лактозы с постепенно
увеличивающимся временем смешивания. Пояснения см. в тексте. В нижней части
рисунка для удобства приведен фрагмент спектра ЯМР 1Н вместе со шкалой
химических сдвигов
5.11. Тетрапептид (валин-глицин-серин-глутаминовая кислота) (VGSE)
319
протона Н-4 (G4). Если начать с протона Н-4 (G4,
3.93 м.д.), обнаруживаются все шесть корреляций.
В качестве задания, попытайтесь проделать анало-
гичные выкладки, начиная не с протонной, а с угле-
родной оси и двигаясь горизонтально влево, и найти
корреляции HMQC-TOCSY с протонами. Проще всего
обнаружить сигналы аномерных атомов углерода,
но стоит найти и все другие сигналы.
5.10. Метод ROESY
В главе 3 (раздел 3.16) описан эксперимент, осно-
ванный на использовании ядерного эффекта Овер-
хаузера (ЯЭО), который дает информацию о взаи-
модействии соседних атомов водорода, но не через
углеродный скелет молекулы, а через пространство.
Краткая информация, изложенная в разделе 3.16,
необходима для дальнейшего обсуждения. Экспе-
римент ROESY (Rotating-frame Overhauser Effect
SpectroscopY - спектроскопия эффекта Оверхаузера
во вращающейся системе координат) является по-
лезным двумерным аналогом других экспериментов,
основанных на ЯЭО. Такие эксперименты можно
применять для молекул разного размера, в отличие
от спектроскопии NOESY (Nuclear Overhauser Effect
SpectroscopY - спектроскопия ядерного эффекта
Оверхаузера), которая не используется при анализе
небольших по размеру молекул. Метод NOESY при-
меняется преимущественно для изучения биологи-
ческих макромолекул. Но и эксперименты NOESY,
и эксперименты ROESY дают корреляции протонов,
находящихся близко друг от друга в пространстве,
обычно на расстоянии 4.5 А и менее.
Эксперименты ROESY дают протон-протонные корре-
ляции (через пространство), но похожие взаимодействия
можно увидеть и в экспериментах COSY. Фактически
сигналы из спектров COSY (спин-спиновые взаимодей-
ствия) присутствуют и в спектрах ROESY; эти сигналы
COSY лишние, и их необходимо убрать. Иногда ослож-
нения возникают также из-за TOCSY-подобного перено-
са магнитной когерентности между ядрами, связанными
спин-спиновым взаимодействием. На рис. 5.27 приве-
дены импульсные последовательности для двумерных
экспериментов NOESY и ROESY. Разница между ними
заключается в том, что в методе ROESY используется
«спиновый захват» в течении времени смешивания (тт).
5.10.1. Спектр ROESY лактозы
На рис. 5.29 приведен спектр ROESY лактозы. В верх-
ней части рисунка изображен общий вид спектра,
в нижней части - растянутые фрагменты области
90r I Выборка (й)
Рис. 5.27. Импульсная последовательность для
двумерных экспериментов NOESY (а) и ROESY (б)
аномерных сигналов. Оба глюкозных остатка про-
интерпретировать легко. а-Аномерный протон дает
только одну корреляцию, то же самое можно уви-
деть и в спектре COSY. Напомним, что, по определе-
нию, для а-аномера гидроксильная группа при атоме
С-1 находится в аксиальном положении, а протон
при атоме углерода С-1 (al)-в экваториальном по-
ложении. В экваториальном положении невозможно
взаимодействие через пространство, и поэтому этот
протон не виден.
У другого аномера глюкозы, наоборот, аномерный
протон (pl) находится в аксиальном положении. Глюко-
за находится преимущественно в конформации «крес-
ла», при этом протоны занимают все другие аксиальные
положения. Таким образом, возникают диаксиальные
взаимодействия (взаимодействия с другими аксиальны-
ми протонами) между протонами Н-1 (pi) и Н-3 (рЗ),
а также между протонами Н-1 (Р1) и Н-5 (Р5). Спектр
ROESY показывает три взаимодействия типа COSY для
аномерного протона pi (при 4.67 м.д.). Конечно, при-
сутствует взаимодействие между протонами Н-1 (pl)
и Н-2 (Р2); и диаксиальные ЯЭО-взаимодействия
протона Н-1 (pl) с протонами Н-3 (РЗ) и Н-5 (Р5)
совершенно очевидны.
5.11. Тетрапептид (валин-глицин-
серин-глутаминовая кислота)
(VGSE)
Белки или полипептиды - это полимеры или олиго-
меры, которые состоят из остатков разных (их более
20) аминокислот (главным образом а-аминокислот),
320
5. Корреляционная спектроскопия ЯМР. 2D ЯМР
Рис. 5.28. Спектр HMQC-TOCSY лактозы. Для простоты приводится отнесение
сигналов
5.7?. Тетрапептид (валин-глицин^^^ кислота} (VGSE)
321
F1
Fl
Fl
Спектр ROESY (600 МПд)
м.«.
G2
4.45-
м.д. .
4.65-
4.70-
G5 G3
II |< INIBNII it!
06 аб 06’ 84 05
ct5«4
03 05
Ilia
02
01
G1
F2
Рис. 5.29. Двумерный спектр ROESY лактозы (вверху) и растянутые фрагменты
спектра для трех аномерных корреляций (внизу)
322
5. Корреляционная спектроскопия ЯМР. 2D ЯМР
связанных между собой амидными (пептидными)
связями. Тетрапептид является хорошим примером,
на котором можно рассмотреть большинство основ-
ных особенностей настоящих полипептидов, и с ним
достаточно легко работать в отличие от полипептидов.
В природе существует большое число олигопептидов,
но в настоящее время с применением специального
автоматического оборудования для синтеза получено
множество небольших пептидов.
Структура небольшого пептида, используемого
в этой главе в качестве примера, была приведена
на рис. 5.1. Данный пептид состоит из четырех свя-
занных между собой аминокислотных остатков; на-
чиная с N-конца, это валин, глицин, серин и глутами-
новая кислота (VGSE). Из одномерного спектра ЯМР
(рис. 5.30) видно, что аминокислоты, образующие
это соединение, относительно простые, что позво-
ляет быстро обнаружить их индивидуальные сиг-
налы. Атомы каждой аминокислоты пронумерованы
и сделано отнесение как протонных, так и углерод-
ных сигналов. Для облегчения понимания отнесение
указано на этом рисунке, хотя оно было выполнено
не только на основе этих спектров, привлекались
и другие данные.
Чтобы понять интерпретацию спектров, необходи-
мо учитывать, в каких экспериментальных условиях
они зарегистрированы. Как правило, чтобы добиться
хорошей растворимости и исключить разложение об-
разца, пептид растворяют в водном буфере. Как уже
говорилось в гл. 3, почти всегда для изучения с по-
мощью спектроскопии ЯМР соединение растворяют
в дейтерированном растворителе. Последний необхо-
дим для получения сигнала частотной стабилизации
(так называемого «лока» или захвата). Однако если
образец пептида растворить в буфере из тяжелой
воды (D2O), все протоны, склонные к обмену, вклю-
чая амидные протоны, могут замещаться на дейте-
рий, и мы эти протоны не увидим. Поэтому были
найдены оптимальные условия: образец растворяли
в смеси 95% Н2О и 5% D2O. Заметим, что в этом
случае в спектре появляются сигналы от трех амид-
ных протонов в области от 8.0 до 9.0 м.д. Это очень
важный момент, так как если образец растворили
бы в чистой D2O, протонные сигналы от этих трех
амидных групп в спектре отсутствовали бы.
5.11.1. Спектр COSY тетрапептида VGSE
На рис. 5.31 приведен спектр DQF-COSY тетрапеп-
тида VGSE. Как и в случае лактозы, можно найти
несколько хороших точек входа в спектр, особенно
это касается трех амидных протонов. Три из четырех
аминокислотных остатка можно легко исследовать,
используя в качестве точки входа в спектр именно эти
три протона от амидных групп. Четвертый аминокис-
лотный остаток можно изучить, если начать с сигнала
метильных групп при 1.0 м.д. (имеется в виду валин,
содержащий метильные группы, которых нет в трех
других аминокислотных остатках). Проследите за кор-
реляциями, которые обозначены линиями на рис. 5.31.
Будьте осторожны, не перепутайте валин и серин.
5.11.2. Спектр TOCSY тетрапептида VGSE
На рис. 5.32 изображен двумерный спектр TOCSY
тетрапептида VGSE. Как и лактоза, этот пептид содер-
жит несколько изолированных протонных спиновых
систем, в данном случае таких систем четыре. В ка-
честве примера «тотальной корреляции» рассмотрим
корреляции между протонами остатка глутаминовой
кислоты, они обозначены на рисунке пунктирными
линиями. Как и в спектре COSY, начинать интерпрета-
цию проще с амидных протонов. Можете ли вы найти
подходящую точку входа в спектр для изучения остат-
ка валина, который не имеет амидных протонов?
В некоторых отношениях информация из спектра
COSY VGSE дополняет спектр TOCSY, но в дру-
гих отношениях эта информация бывает излишней.
Оба спектра позволяют сделать отнесение всех про-
тонных сигналов этого тетрапептида, хотя и разными
путями. Какой из этих двух спектров, на ваш взгляд,
позволяет более просто и однозначно сделать отне-
сение и разобраться в структуре в данном случае?
5.11.3. Спектр HMQC тетрапептида VGSE
Спектр HMQC тетрапептида VGSE (рис. 5.33) по сравне-
нию со спектрами кариофилленоксида и лактозы кажется
более простым. Действительно, в молекулы пептида все-
го 10 атомов углерода имеют присоединенные протоны,
в спектре видны корреляции от девяти атомов углерода.
На самом деле этих корреляций десять, в чем можно
убедиться по растянутой метильной области спектра.
Обобщим уже полученную нами выше информацию
и приступим к рассмотрению спектра HMQC.
Начнем анализ спектра HMQC с углеродного
сигнала при 43 м.д. Как видно из спектра DEPT,
это метиленовая (СН2) группа; отметим, что протоны
этой группы дают в спектре ЯМР !Н мультиплет
с центром при 4.04 м.д. В спектре DQF-COSY (см.
рис. 5.31) мультиплет при 4.04 м.д. коррелирует только
с одним амидным протоном при 8.75 м.д. Таким об-
разом, эта метиленовая группа должна принадлежать
остатку глицина; более того, глицин не может быть
концевой аминокислотой, так как концевая амино-
кислота должна иметь свободную аминогруппу.
5.11. Тетрапептид (валин-глицин-серин-глутаминовая кислота) (VGSE)
323
Спектр ЯМР 'Н (600 МПд)
GNH
SNH ENH
5200 5100 5000 4900 Гц
S2
"'I.......I....।।................г.п
2700 2600 2500 2400 Гц
V4,5
"Г"""
600 Гц
1400 1300 1200 Гц ?Н
5 С=О
4 СН3 ОН 4 сн2
I 5 I I 2
ЗСН-СНз зсн, зсн2
I ; 2 1 . I 21 . I 2
H2N— CH-C-;-N— CH2C-:-N— CH-C--N— СН-С— ОН
2 |р: н II :Н 2 || !Н 2 ||1
О О О О
Валин (V) Тлицин (G) Серин (S)
’ Г' * 1 2 1 1 I ' 1 1 ' Г ' 1 ' Г 1 1 ' Г ' 1 ' Г ' ' 1 I ' 1 ' 1 Г 1 ' I ' ' ' ’ I ’ ' I ' ' ' ' I • ' ' I.| | ।|
8.5 8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 м.д.
Спектр ЯМР 13C/DEPT (150,9 МПф
S3
V2 S2
VI
178 176 174 172 м.д.
Рис. 5.30. Спектры ЯМР 1Н, 13С и DEPT тетрапептида VGSE в смеси 95% Н2О и 5% D2O
G2
1 Г ' 1 ' Г ’ 1 1 Г ' Г ' 1 1 Г 1 ' ' Г 1 1 1 Г 1 1 1 Г1 ' '-П — -1
60 55 50 45 40 35 30 25 м.д.
324
5. Корреляционная спектроскопия ЯМР. 2D ЯМР
Рис. 5.31. Спектр DQF-COSY тетрапептида VGSE. Для простоты приведено
отнесение некоторых сигналов и проведены корреляционные линии
Для анализа сигналов остатка серина можно на-
чать с углеродного сигнала при 62 м.д. Спектр DEPT
свидетельствует о наличии еще одной метиленовой
группы, а спектр HMQC показывает, что эта группа
коррелирует с протонами при 3.85 м.д. Сигналы этих
протонов перекрываются с еще одним протоном. Тща-
тельно проводя линию в спектре DQF-COSY (лучше
это видно в спектре TOCSY), находим корреляцию
с протоном при 4.48 м.д. Этот протон дает корреляцию
в спектре HMQC с углеродным атомом, сигнал которого
находится при 56 м.д., и в спектре COSY корреляцию
с амидным протоном при 8.40 м.д. Как и глицин, серин
не может быть концевой аминокислотой.
Валин - единственная аминокислота в данном те-
трапептиде VGSE, которая имеет метильные группы;
в спектре DEPT наблюдаются две метильные группы
с сигналами при 17 и 18 м.д., можно начать рассмо-
трение спектра HMQC именно с них. Оба метильных
атома углерода коррелируют с одним дублетом при
1.01 м.д. Отметим, что химические сдвиги протонов
этих метильных групп совпали случайно, так как
они диастереотопны. (Интегрирование этого дублета
в протонном спектре, приведенном на рис. 5.30, дает
шесть протонов, что подтверждает наши предполо-
жения.) Протоны метильных групп взаимодействуют
с метиновым протоном валина V3 с сигналом при
2.20 м.д. (COSY). Возвращаясь к спектру HMQC,
заметим, что метиновый протон V3 коррелирует
с углеродным сигналом при 31.5 м.д. Метиновый
протон изопропильной группы (с химическим сдви-
гом 2.20 м.д.), согласно спектру COSY, коррелирует
с еще одним метиновым протоном V2, который дает
мультиплет при 3.85 м.д., частично перекрывающийся
с каким-то другим сигналом. Этот протон V2 в спек-
тре HMQC дает корреляцию с углеродным сигналом
при 59 м.д. Хотя это и трудно увидеть, но данный
5.11. Тетрапептид (валин-глицин-серин-глутаминовая кислота) (VGSE)
325
Спектр TOCSY (600 МПд)
Рис. 5.32. Двумерный спектр TOCSY тетрапептида VGSE. Для простоты приведено
отнесение сигналов и проведены некоторые корреляционные линии
метановый сигнал далее ни с чем не коррелирует
(т.е. не расщепляется ни на одном из амидных про-
тонов); следовательно, валин-концевая аминокислота.
Отнесение сигналов для последней аминокислоты
(глутаминовой кислоты) можно начать с углеродного
сигнала при 54 м.д. Спектр HMQC подсказывает,
что этому атому углерода соответствует протон
при 4.21 м.д. (этот протон дает в спектре сложный
мультиплет). Спектр COSY позволяет обнаружить
корреляции этого протона с протонами, химические
сдвиги которых равны 1.93 и 2.11 м.д. Нетрудно уви-
деть, что эти два протона являются диастереотоп-
ными: в спектре HMQC они коррелируют с одним
и тем же атомом углерода (при 27 м.д.). Оба эти
диастереотопных протона коррелируют с триплетом
при 2.37 м.д. (см. спектр COSY), т.е. с протонами
второй метиленовой группы глутаминовой кислоты.
Углеродный сигнал этой второй метиленовой группы
располагается в спектре HMQC при 31 м.д. Ценную
информацию можно получить из спектра COSY: сиг-
нал метанового протона при 4.21 м.д. расщепляется
из-за взаимодействия с амидным протоном с хими-
ческим сдвигом 8.10 м.д.
Для отнесения всех протонов и почти всех атомов
углерода (за исключением карбонильных) в тетра-
пептиде VGSE спектр HMQC является отличным до-
полнением к спектрам DQF-COSY и TOCSY. Однако
этот набор спектров не позволяет нам установить
и подтвердить порядок следования аминокислотных
остатков в тетрапептиде, что является чрезвычайно
важной задачей.
5.11.4. Спектр НМВС тетрапептида VGSE
На рис. 5.34 изображен спектр НМВС тетрапепти-
да VGSE. Чтобы повысить разрешение кросс-пиков,
326
5. Корреляционная спектроскопия ЯМР, 2D ЯМР
Спектр HMQC (600 МПф
Рис. 5.33. Спектр HMQC тетрапептида VGSE. Для простоты приведено отнесение
сигналов
спектр разбит на четыре части. Для отнесения сигналов
карбонильных групп в спектре ЯМР 13С и для опреде-
ления порядка следования аминокислотных остатков в
пептиде можно начать с любого конца молекулы. Ранее
мы установили, что валин-N-концевая аминокислота,
потому что ее амидная группа не имеет пептидной свя-
зи. При анализе валинового остатка пептида логично
начать с установления положения сигнала его карбо-
нильной группы. Можно предположить, что сигнал при
170.1 м.д. как раз и соответствует карбонильному атому
углерода этого аминокислотного остатка, так как явно
видны корреляции с протонами валина V2 и V3 при
атомах углерода С-2 и С-3 соответственно. Более того,
этот карбонильный атом углерода коррелирует также
с метильной группой глицина (G2) и амидным про-
тоном глицина (G NH).
Две последние корреляции (и вообще подобные
корреляции) чрезвычайно важны для установления
аминокислотной последовательности белка или пепти-
да. До сих пор мы рассматривали корреляции в преде-
лах одного аминокислотного остатка, но эксперимен-
ты НМВС позволяют выявлять корреляции между
аминокислотными остатками в белке. Продолжите
дальше аналогичные рассуждения самостоятельно
и выясните порядок следования двух оставшихся
аминокислот (серина и глутаминовой кислоты). Об-
ратим внимание на тот факт, что в углеродном спек-
тре сигналы карбонильных атомов углерода глицина
и серина перекрываются.
5.11.5. Спектр ROESY тетрапептида VGSE
Закончим обсуждение тетрапептида VGSE сравнени-
ем корреляций амидных протонов в спектре ROESY
с соответствующими взаимодействиями в спектрах
COSY и TOCSY. На рис. 5.35 показаны сопоставимые
фрагменты этих трех спектров. Мы уже ранее виде-
ли эти спектры COSY и TOCSY и показали, как они
вместе со спектром HMQC могут использоваться
для отнесения сигналов в пределах аминокислотных
остатков, образующих белок. Корреляции ROESY по-
могают подтвердить информацию, полученную из
спектра НМВС, относительно последовательности
аминокислот в белке.
Кросс-пики, выделенные квадратиками, иллюстри-
руют взаимодействие через пространство между
5.17. Тетрапептид (валин-глицин^^ кислота^ (VGSE}
327
Спектр НМВС (600 МП<)
F2
F2
Рис. 5.34. Спектр НМВС тетрапептида VGSE. Для простоты приведено отнесение
сигналов и проведены важные корреляционные линии
328
5. Корреляционная спектроскопия ЯМР. 2D ЯМР
Спектр COSY (600 МП<) Спектр TOCSY (600 Mlh) Спектр ROESY (600 MIX)
Рис. 5.35. Сравнение спектров DQF-COSY, 2D-TOCSY и ROESY, показывающее
взаимодействия амидных протонов
амидным протоном одной аминокислоты в пептиде
и С-2-протоном другой, соседней с ней, аминокисло-
ты. Таким образом, амидный протон глицина (G NH)
коррелирует с С-2-протоном валина (V2), амидный
протон серина (S NH) коррелирует с С-2-протоном
глицина (G2), а амидный протон глутаминовой кис-
лоты (Е NH) коррелирует с С-2-протоном (а также
с С-3-протонами) серина (S2 и S3). Форма представ-
ления данных, использованная на рис. 5.35, заметно
упрощает интерпретацию и делает сравнение более
содержательным.
5.12. Спектроскопия ЯМР
с использованием
градиентного поля
Хорошо разработанная область в ЯМР - это спектро-
скопия ЯМР с использованием импульсных гради-
ентов поля (PFG NMR, Pulsed Field Gradients NMR,
ЯМР с импульсным полевым градиентом). Ирония
состоит в том, что в течение многих лет тратилось
очень много усилий для получения как можно более
однородного и стабильного магнитного поля. В на-
стоящее время большинство спектрометров ЯМР
высокого разрешения обычно укомплектованы спе-
циальными катушками, которые позволяют быстро
добиться градиентов магнитного поля вдоль одной
из трех взаимно ортогональных осей. Эти градиенты
магнитного поля вводятся в импульсную последова-
тельность и широко применяются в различных иссле-
дованиях. Спектрометр с рабочей частотой 600 МГц.
который использовался для регистрации спектров,
приведенных в этой книге, оснащен трехосевым гра-
диентным датчиком.
В этой главе мы кратко рассмотрим градиенты,
потому что они часто применяются в корреляцион-
ных экспериментах. Мы обсудим общие аспекты: для
более детального знакомства и анализа множества
приложений этой методики обратитесь к обзору
Прайса (Price, 1996).
В разделе 5.3.1 мы упоминали термин «фазовое
циклирование», это важная составляющая любой
импульсной последовательности. Опуская детальное
рассмотрение понятия фазового циклирования (см.
Литература
329
Claridge, 1999), мы можем сразу назвать один из не-
достатков этой методики-это время. В корреляци-
онных экспериментах, для того чтобы получить один
ССИ, необходимо просуммировать от 4 до 64 фазовых
циклов. Если отношение сигнал/шум недостаточно
хорошее, то один и тот же цикл повторяется до тех
пор, пока мы не получим достаточно хороший сиг-
нал, необходимый для исследования. На эти фазовые
циклы затрачивается достаточно много приборного
времени; это одна из причин, почему двумерные экс-
перименты такие длительные.
В эксперименте PGF импульсная последователь-
ность может быть перестроена (видоизменена) та-
ким образом, что фазовое циклирование будет силь-
но сокращено или вообще полностью исключено
из нее. Так, в двумерном эксперименте при условии
достижения оптимального отношения сигнал/шум
каждый импульс можно сохранить в виде одного
ССИ, что ведет к значительной экономии приборного
времени; т.е. эксперимент, который раньше занимал
несколько часов (иногда даже целый день), теперь
может быть проведен в течение нескольких минут.
Обнадеживает также тот факт, что хотя эксперимент
проводится несколько иначе, результаты, а следова-
тельно, и интерпретация остаются теми же.
Хотя интересы в области градиентого ЯМР в основ-
ном связаны с тем, что обычные эксперименты мо-
гут быть проведены быстрее и эффективнее, все же
наиболее перспективным является использование
градиентов в экспериментах, которые дают принци-
пиально новую информацию. Установлено, что гра-
диенты позволяют усовершенствовать такие методы,
как магнитная томография и магнитно-резонансная
микроскопия, а также дают возможность лучше по-
давить сигнал растворителя в спектре (особенно это
касается водных растворов), благодаря градиентам
появилась совершенно новая область исследований -
измерения диффузии.
Литература
Atta-ur, Rahman, and Choudhary, M. (1995). Solving Problems
with NAIR Spectroscopy. New York: Academic Press.
Becker, E. (1999). High Resolution NMR: Theory and Chemical
Applications. 3rd ed. New York: Academic Press.
Berger, S., and Braun, S. (2004). 200 and More NMR
Experiments: A Practical Course. Weinheim, Germany: VCH
Verlagsgesellschaft MBH.
Braun, S., Kalinowski, H., and Berger, S. (1999). 150 and
More Basic NMR Experiments: A Practical Course. 2nd ed.
New York: Wiley.
Brey, W., (Ed.). (1988). Pulse Methods in ID and 2D Liquid-
Phase NMR. New York: Academic Press.
Cavanagh, J., Fairbrother, W., Palmer III, A., and Skelton, N.
(1995). Protein NMR Spectroscopy: Principles and Practice.
New York: Academic Press.
Chandrakumar, N., and Subramanian, S. (1987). Modern
Techniques in High Resolution FT NMR. New York: Springer-
Verlag.
Claridge,T.D.W. (1999). High-Resolution NMR Techniques in
Organic Chemistry. New York: Elsevier Science.
Croasmun, W. R., and Carlson, R. M. K. (1987). Two-
Dimesional NMR Spectroscopy. 2nd ed. New York: VCH.
Croasmun,W.R., and Carlson, R.M.K. (1994). Two-Dimesional
NMR Spectroscopy: Applications for Chemists and Biochemists,
Fully Updated and Expanded to Include Multidimensional
Work. 2nd ed. New York: Wiley.
Дероум Э. (1992). Современные методы ЯМР для хими-
ческих исследований. Москва: Мир.
Эрнст Р.Р., Боденхаузен Дж., Вокаун А. (1990). ЯМР в одном
и двух измерениях. Пер. с англ. Москва: Мир.
Farrar, Т.С. (1987). An Introduction to NMR Spectroscopy.
Chicago: Farragut.
Фаррар T., Беккер Э. (1973). Импульсная и фурье-
спектроскопия ЯМР. Пер. с англ. Москва: Мир.
Freeman, R. (1998). Spin Choreography: Basic Steps in High
Resolution NMR. Oxford: University Press on Demand.
Friebolin, H. (1998). Basic One- and Two-Dimensional NMR
Spectroscopy. 3rd revised ed New York: Wiley.
Fukushima, E., and Roeder, S.B.W. (1986). Experimental
Pulse NMR: A Nuts and Bolts Approach. New York: Perseus
Publishing.
Gunther, H. (1995). NMR Spectroscopy: Basic Principles,
Concepts, and Applications in Chemistry. 2nd ed. New
York: Wiley.
Hoch, J.C., and Stem, A. (1996). NMR Data Processing.
New York: Wiley.
Ноге, P.J. (1995). Nuclear Magnetic Resonance (Oxford
Chemistry Primers, 32). Oxford: Oxford University Press.
Ноге, P.J., Wimperis, S., and Jones, J. (2000). NMR: The
Toolkit. Oxford: Oxford University Press.
Kessler, H.. Gehrke, M., and Griesinger, C. (1988). Two-
Dimensional NMR Spectroscopy. Angew. Chem, Int. Ed.
Engl., 27,490-536.
Macomber, R.S. (1995). A Complete Introduction to Modern
NMR Spectroscopy. New York: Wiley-Interscience.
Martin, G.E., and Zektzer, A.S. (1988). Two-Dimensional
NMR Methods for Establishing Molecular Connectivity:
330
5. Корреляционная спектроскопия ЯМР. 2D ЯМР
A Chemists Guide to Experiment Selection, Performance,
and Interpretation. New York: Wiley.
Mitchell, T.N., and Costisella, B. (2004). NMR - From Spectra
to Structures: An Experimental Approach. New York:
Springer Verlag.
Munowitz, M. (1988). Coherence and NMR. New York:
Wiley-Interscience.
Nakanishi, K. (1990). One-Dimensional and Two-Dimensional
NMR Spectra by Modern Pulse Techniques. New York:
University Science Books.
Pihlaja, K., and Kleinpeter, E. (1994). Carbon-13 NMR
Chemical Shifts in Structural and Stereochemical Analysis.
Weinheim, Germany: VCH Publishing.
Price, W.S. (1996). Gradient NMR. In G.A. Webb (Ed.). Annual
Reports on NMR Spectroscopy. Vol. 32. London: Academic
Press.
Sanders, J.K.M., and Hunter, B.K. (1993). Modem NMR Spec-
troscopy. 2nd ed. Oxford: Oxford University Press.
Schraml, J., and Bellama, J.M. (1988). Two-Dimensional NMR
Spectrometry. New York: Wiley.
Van de Ven, F.J.M. (1995). Multidimensional NMR in
Liquids: Basic Principles and Experimental Methods.
Weinheim, Germany: Wiley-VCH.
Williams, K.R., and King, R.W. (1990). The Fourier transform
in chemistry - NMR. J. Chem. Ed., 67, 125-138.
Упражнения
5.1. Для соединений из упражнения 3.3 (а-п) нари-
суйте следующие спектры: COSY, HMQC, НМВС
и INADEQUATE. Разберитесь в обозначениях осей
F1 и F2. Предполагается, что экспериментальные
условия те же самые, что и в упражнении 3.3.
5.2. Установите все корреляции для ипсенола в спек-
тре DQF-COSY, приведенном на рис. 5.9. Укажите
тип взаимодействия (геминальное, вицинальное
или более дальнее взаимодействие).
5.3. Закончите отнесение сигналов для ипсено-
ла в спектре НМВС, приведенном на рис. 5.14.
Для облегчения работы вы можете построить та-
блицу, аналогичную табл. 5.1.
5.4. Идентифицируйте соединение с брутто-формулой
С6Н10О по его спектрам ЯМР: !Н, 13C/DEPT, COSY
и HMQC. Покажите все корреляции.
5.5. Идентифицируйте соединение с брутто-формулой
С10Н10О по его спектрам ЯМР !Н, 13C/DEPT, COSY
и HMQC. Покажите все корреляции.
5.6. Идентифицируйте соединение с брутто-формулой
C8H9NO2 по его спектрам ЯМР !Н, 13C/DEPT,
COSY, HMQC и INADEQUATE. Покажите все
корреляции.
5.7. Установите все связанности для атомов угле-
рода кариофилленоксида, используя спектр
INADEQUATE, изображенный на рис. 5.19.
5.8. Идентифицируйте соединение с брутто-формулой
С10Н18О по его спектрам ЯМР !Н, 13C/DEPT,
HMQC, НМВС и INADEQUATE.
5.9. Определите как можно больше корреляций для
лактозы, используя двумерный спектр TOCSY на
рис. 5.25. Сравните ваши результаты для фраг-
мента глюкозы с результатами, полученными из
одномерного спектра НОНАНА, приведенного на
рис. 5.26.
5.10. Для рафинозы дана структурная формула, а так-
же спектры ЯМР !Н, 13C/DEPT, COSY, 1D-TOCSY,
2D-TOCSY, HMQC, HMQC-TOCSY, НМВС
и ROESY. Подтвердите структуру, сделайте от-
несение сигналов для всех протонов и атомов
углерода, укажите как можно больше корреляций.
5.11. Для стигмастерина дана структурная форму-
ла, а также спектры ЯМР: !Н, 13C/DEPT, COSY,
HMQC и НМВС. Подтвердите структуру, сделайте
отнесение сигналов для всех протонов и атомов
углерода, укажите как можно больше корреляций.
5.12. Для трипептида, содержащего аминокислоты ли-
зин, серин и треонин, даны спектры ЯМР !Н, 13С/
DEPT, COSY, 2D-TOCSY, HMQC, НМВС и ROESY.
Установите последовательность аминокислот, сде-
лайте отнесение сигналов для всех протонов и
атомов углерода. Укажите как можно больше кор-
реляций.
Упражнения
331
Спектр ЯМР *Н (600 МГЦ)
Jl
1300 Гц 1250 Гц 1150 Гц
Жц
1050 1000 Гц
JW1L
860 840 820 Гц
I ' I 1 I ’ I
620 600 580 Гц
I.....'I" ""I......I|"TTT1 ' "I ...|......"I""...I"......[""Г""!,,,,...........|....,t| I...I ...Ц...|
2.3 2.2 2.1 2.0 1.9 1.8 1.7 1.6 1.5 1.4 1.3 1.2 1.1 1.0 м.д.
Спектр ЯМР l3C/DEPT (150.9 МГц)
J________________________________________________I 111 11__________________
220 200 180 160 140 120 100 80 60 40 20 м.д.
Упражнение 5.4
332
5. Корреляционная спектроскопия ЯМР. 21) ЯМР
м _ 2.4 2.2 2.0 1.8 1.6 1.4 1.2 1.0
М.д.
1.0-
1.2-
1.4-
F1
ГА 1.6-
1.8-
2.0-
2.2-
* 11 # к
И* *
* »Я* * I
* $* в •
М.Д.
1.0
1.2
1.4
1.6
1.8
2.0
2.2
HMQC (600 МГЦ)
Упражнение 5.4
Упражнения
333
Спектр ЯМР ’Н (600 МПф
ш
I ' ' 1 1 I ' ' I 1 ' г
V
4800
4750 Гц 4450
1750
1700 Гц
1550
4400 Гц
1500 Гц
4350
4300 Гц
1250
1200 Гц
8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 м.д.
Спектр ЯМР 13C/DEPT (150.9 МПф
200 180 160 140 120 100 80 60 40 м.д.
Упражнение 5.5
334
5. Корреляционная спектроскопия ЯМР. 2D ЯМР
COSY (600 MDO
8 7 6 5 4 3 2 м.д.
.1.........।.........।..........।.........।..........।..........।
М.Д.
F2
HMQC (600 МПС
80-
F1
40-
60-
8.2 8.0 7.8 7.6 7.4 7.2 м.д.
100-
120-
I
140-
- 40
- 60
80
-100
-120
-140
7 6 5 4 3
2
м.д.
F2
Упражнение 5.5
Упражнения
335
Спектр ЯМР *Н (600 МВД
5480 5470 Гц 5200 5190 Гц 4910 4900 Гц
4370 4360 Гц 2590 2580 Гц 790 780 Гц
I 1 ' ‘ I Г ' । • I ' I ' I 1 1 1 1 Г 1 1 • I ' ' I • I ' ' ' I • • I • I ' I ‘ I ' ' I I
9.0 8.5 8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 м.д.
Спектр ЯМР 13C/DEPT (150.9 MRj)
160 150 140 130 120 110 100 90 80 70 60 50 40 30 м.д.
Упражнение 5.6
336
5. Корреляционная спектроскопия ЯМР. 2D ЯМР
F2
Упражнение 5.6
Упражнения
337
INADEQUATE (150.9 MDj)
Гц
-250007 -20000- -150007 -IOOOO7 -50007 Fl O7 50004 100007 15000- 20000- 250007 • в 1 11 Il 7 -25000 - -20000 ? -15000 7 -10000 7 -5000 7 0 7 5000 - 10000 - 15000 7 20000 7 25000
' 1 ' 1 ' 1 ' 1 ’ 1 ' 1 ' 1 ' 1 160 140 120 100 80 60 40 20 М.Д.
F2
F2
338
5. Корреляционная спектроскопия ЯМР. 2D ЯМР
Спектр ЯМР *Н (600 МПф
। । • 1 । I । • • । • । • • • । • । • • । ।
5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 м.д.
Спектр ЯМР 13C/DEPT (150.9 МПф
150 140 130 120 110 100 90 80 70 60 50 40 30 20 м.д.
Упражнение 5.8
Упражнения
339
F2
Упражнение 5.8
340
5. Корреляционная спектроскопия ЯМР. 2D ЯМР^
НМВС (600 МГЦ)
Упражнение 5.8
Упражнения
341
INADEQUATE (150.9 МПд)
F2
Упражнение 5.8
342
5. Корреляционная спектроскопия ЯМР, 2D ЯМР
Спектр ЯМР *Н (600 МГЦ)
11 MkLjAv-Л
4.1 4.0 3.9 3.8 3.7 3.6
3.5 м.д.
HDO
jy
jwMjL
5.4 5.3 5.2 5.1 5.0 4.9 4.8 4.7 4.6 4.5 4.4 4.3 4.2 4.1 4.0 3.9 3.8 3.7 3.6 м.д.
Спектр ЯМР 13C/DEPT (150.9 МПф
74.0 73.5 73.0 72.5 72.0 71.5 71.0 70.5 70.0 69.5 69.0 м.д.
Упражнение 5.10
Упражнения
343
Спектры ID TOCSY (600 МП1)
Время смешивания 300 мс с облучением
сигнала при 5.40 м.д.
Время смешивания 40 мс
Время смешивания 80 мс
Время смешивания 20 мс
JL .
4.05 4.00 3.95 3.90 3.85 3.80 3.75 3.70 3.65 3.60 3.55 3.50 м.д.
Время смешивания 300 мс с облучением
сигнала при 4.97 м.д.
_________
Спектры ID TOCSY (600 МПх)
Время смешивания 80 мс .
Время смешивания 40 мс
Время смешивания 20 мс
Спектр ЯМР ’Н (600 МПф
‘ I ‘ I • I • • I • I I • I • I • • I ‘ • I ‘ • I
4.05 4.00 3.95 3.90 3.85 3.80 3.75 3.70 3.65 3.60 3.55
• ।
3.50
1 ।
м.д.
Упражнение 5.10
344
5. Корреляционная спектроскопия ЯМР. 2D ЯМР^
Спектры ID TOCSY (600 МПд)
Время смешивания 80 мс
Время смешивания 40 мс
Время смешивания 20 мс
3.50
м.д.
4.05 4.00 3.95 3.90 3.85 3.80 3.75 3.70 3.65 3.60
Спектр ЯМР !Н (600 МПд)
Упражнение 5.10
Упражнения
345
COSY (600 МПд)
Упражнение 5.10
346
5. Корреляционная спектроскопия ЯМР. 2D ЯМР
5.4 5.2 5.0 4.8 4.6 4.4 4.2 4.0 3.8 3.6 м.д. Спектр ID TOCSY (600 МГц)
F2
Упражнение 5.10
Упражнения
347
Лл\Лл.
HMQC (600 МПд)
5.5 м.д. 4.1 4.0 3.9 3.8 3.7 3.6 3.5 м.д.
F2
Упражнение 5.10
348
5. Корреляционная спекпцзоскопия ЯМР. 2D ЯМР
HMQC- TOCSY (600 MHO
Упражнение 5.10
Упражнения
349
Упражнение 5.10
350
5. Корреляционная спектроскопия ЯМР. 2D ЯМР
м.д.
F2
Упражнение 5.10
Упражнения
351
Спектр ЯМР *Н (600 МГЦ)
3200 3100 Гц 2200 2100
j IL Ik A.
3200 3100 Гц 2200 2100 1222
aAmUk jLjll
•"I.I...I ..I..I... "‘I..I.I
1300 Гц 1100 1000 Гц
1000 900 Гц 700 600 Гц
СН3//,.^
Ч1Н
600 500 Гц
HOI
' НН
5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 м.д.
Спектр ЯМР 13C/DEPT (150.9 Mill)
""'i......।.......।.......।.......।........।......।.......।.......।.......।.......। * * ' *'' ।.।.....।
140 130 120 110 100 90 80 70 60 50 40 30 20 м.д.
Упражнение 5.11
352
5. Корреляционная спектроскопия ЯМР. 2D ЯМР
F1
Упражнение 5.11
Упражнения
353
HMQC (600 МГЦ)
5.5 м.д. 3.6 м.д. 2.2 2.0 1.8 1.6 1.4 1.2 1.0 0.8 0.6 м.д.
F2
Упражнение 5.11
354
5. Корреляционная спектроскопия ЯМР. М ЯМР^
НМВС (600 МП1)
dlL J.
„ „ 5.5 м.д. 3.6 м.д. 2.2 2.0 1.8 1.6 1.4 1.2 1.0 0.8 0.6 м.д.
М.Д.
15
20
25 —
30
35
F1 =
40
45
50 '----------------------------------------------------------------------------
55
60
65
70
м.д. _____
130 —
140 “
5.5 м.д. 3.6 м.д. 2.2 2.0 1.8 1.6 1.4 1.2 1.0 0.8 0.6 м.д.
F2
Упражнение 5.11
Упражнения
355
Спектр ЯМР ’Н, 600 Mill
при 0°С в смеси D2O/H2O
1600 Гц 900 800 700 600 Гц
8.5 8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 м.д.
Спектр ЯМР 13C/DEPT (150.9 Mill)
„лw-мк.а.д мл*-.!
Упражнение 5.12
356
5. Корреляционная а^ектроскопия ЯМР М ЯМР
Упражнение 5.12
Упражнения
357
Упражнение 5.12
358
5. Корреляционная спектроскопия ЯМР. 2D ЯМР^
Спектры COSY (600 МГЦ) TOCSY (600 МГЦ) ROESY (600 МГЦ)
9.0 8.5 8.0 7.5 м.д. 8.5 8.0 7.5 м.д. 8.5 8.0 7.5 7.0 м.д.
F2 F2 F2
HMQC (600 МГЦ)
F2
Упражнение 5.12
Упражнения
359
НМВС (600 МПд)
F2
Упражнение 5.12
6. Спектроскопия ЯМР на других важных ядах со спином Уг
6.1. Введение
В предыдущих трех главах было показано, что экс-
перименты ЯМР с применением ядер !Н и 13С чрез-
вычайно полезны для химиков-органиков. Однако
спектроскопия ЯМР не ограничивается применением
только этих двух ядер. Действительно, существует
120 других ядер со спиновым числом (7) больше
нуля, и следовательно, теоретически их можно ис-
пользовать в экспериментах ЯМР. Из этих 120 ядер
31 относится к дипольным, т.е. их спиновое число
равно 1/2 (/=1/2).
В приложении А перечислены все магнитно актив-
ные ядра с указанием некоторых полезных данных.
Имеет смысл потратить немного времени и сравнить
некоторые ядра, приведенные в приложении А, с ядра-
ми !Н и 13С (которые также указаны). Во-первых,
можно обнаружить, что многие элементы имеют более
одного магнитно активного изотопа. В таблице 6.1 по-
вторяется небольшая часть приложения А (для рабо-
чей частоты 300 МГц по ядрам !Н) и дополнительно
приведены диапазоны полезных химических сдвигов.
В таблице 6.1 перечислены ядра, которые обсуждаются
в этой главе. В качестве примера рассмотрим водород
и возможности его применения. Водород имеет три
изотопа: 1Н (протий), 2Н (дейтерий) и 3Н (тритий);
в экспериментах ЯМР используются все изотопы,
каждый из них имеет ряд своих преимуществ. Ядра
!Н и 3Н имеют спиновое число !4 и относительно
высокую чувствительность, но у изотопа 3Н при-
родное содержание практически равно нулю, кроме
того, он радиоактивен. Тритий, следовательно, может
наблюдаться только в случае, если он специально ис-
кусственно введен в интересующую молекулу, в то
время как протий присутствует повсеместно, и мы
уже познакомились с его использованием.
Дейтерий имеет спин 1 и очень низкое природное
содержание;1) он не радиоактивен. Дейтерий очень
удобен для изучения механизмов реакций в орга-
Ядра со спином более !4 не включены в рассмотрение.
Отметим, что при определенных условиях можно зареги-
стрировать спектр образца, содержащего ядра дейтерия.
нической химии и биохимии. Его низкое природное
содержание позволяет пренебрегать им во время
наблюдения других ядер (’Н и 13С), тем не менее
на современных приборах существует возможность
наблюдения спектров ЯМР 2Н при природном со-
держании этого изотопа.
В таблице 6.1 указаны также резонансные частоты
для каждого ядра при магнитном поле 7.0463 Тл, что со-
ответствует резонансной частоте 300 МГц для ядра !Н.
В таблице также перечислены полезные стандартные
соединения для отсчета химического сдвига и диапазоны
химических сдвигов в миллионных долях. Данные по
химическим сдвигам других ядер (т.е. не ядер !Н и 13С)
нужно использовать с большой осторожностью, так как
для большинства этих ядер используются различные
соединения в качестве эталонов химического сдвига,
и могут возникать различные поправки.
Отметим для начала, что наша цель весьма скром-
ная, так как здесь мы встречаемся с такой обшир-
ной областью, как многоядерный ЯМР. В разделе
3.7 мы уже видели, как другие ядра, обладающие
магнитными моментами (особенно ядра, имеющие
спин 1/2), проявляются в протонных спектрах. Крат-
ко рассмотрим спектроскопию ЯМР для четырех
ядер со спином 1/2, которые наиболее важны для
органической химии (включая химию природных
соединений и фармацевтических препаратов), био-
химии и химии полимеров. Приведем несколько
простых примеров и кратко рассмотрим наиболее
важные экспериментальные условия и ограничения
для следующих четырех ядер: 15N, 19F, 29 *Si и 31Р.
Теоретические основы уже были изложены в
гл. 3 и 4. Такие понятия, как спин, константа спин-
спинового взаимодействия, ядерный эффект Овер-
хаузера (ЯЭО), фурье-преобразование (ФП) и другие,
могут применяться без изменений и для этих ядер.
Понятие химического сдвига мы также будем исполь-
зовать без изменений, но его необходимо применять
с осторожностью и помнить, что это понятие все же
было введено для ядер !Н и 13С (за исключением
некоторых случаев, которые будут отмечены ниже).
В настоящее время применение спектроскопии ЯМР
на других ядрах (помимо !Н и 13С) для характери-
6.2. Ядерный магнитный резонанс на ядрах 15N
361
Таблица 6.1. Полезные данные ЯМР для ядер, обсуждаемых в этой главе
Изотоп Спиновое число Природное содержание, % Чувствительность Резонансная частота (МГц) при напряженности магнитного поля 7.0463 Тл Стандартное соединение Диапазон химических СДВИГОВ, М.Д.
Относительная0 Абсолютная6
'н 1/2 99.98 1.00 1.00 300.00 Si(CH3)4 0-10
2Н 1 1.5-10"2 9.6510-3 1.45-10-6 46.051 Si(CD3)4 0-10
Зн 1/2 0 1.21 0 319.990 Si(CT3)4 0-10
|3С 1/2 1.108 1.59-10-2 1.7610'4 75.432 Si(CH3)4 0-220
l4N 1 99.63 LOl-lO’3 1.01-1 о-3 21.671 |4МН3(ж.)“ 0-900
l5N 1/2 0.37 1.04-10’3 3.85-10-6 30.398 |5КН3(ж.)’ 0-900
,7О 5/2 3.7-102 2.91-10-2 1.08-10“5 40.670 Н2О -50-1700
19р 1/2 100 0.83 0.83 282.231 CFC13 -280-276
29Si 1/2 4.7 7.84-10“3 3.69-10’4 59.595 Si(CH3)4 -380-80
3,р 1/2 100 6.63-10"2 6.63-10-2 121.442 85%-ная Н3РО4 -480-270
а При постоянном поле для равного числа ядер.
6 Рассчитывается из относительной чувствительности и природного содержания.
в При 25 °C.
стики и идентификации органических соединений
стало обычной практикой. Использование других ядер
в экспериментах ЯМР может помочь в различных
областях, начиная с определения неизвестных соеди-
нений, содержащих азот, и кончая более сложными
вопросами стереохимии и изучением механизмов
реакций. Хотя мы ограничим обсуждение только
четырьмя ядрами (15N, 19F, 29Si и 31Р), не надо за-
бывать о возможности применения и других ядер.
Фактически, мы стремимся расширить наши пред-
ставления относительно огромных потенциальных
возможностей, предоставляемых ЯМР и периодиче-
ской системой элементов.
6.2. Ядерный магнитный резонанс
на ядрах 15N
Помимо углерода и водорода двумя наиболее важ-
ными элементами в органической химии являются
кислород и азот. Присутствие одного из этих эле-
ментов говорит о том, что в соединении присутству-
ют функциональные группы, и для таких веществ
широко используется ИК-спектроскопия. Не будем
преуменьшать возможности этого метода, но он име-
ет ряд ограничений, особенно для такого элемента,
как азот. Что касается кислорода, то, как видно из
табл. 6.1, эксперименты ЯМР можно проводить только
для одного из его изотопов (17О); спиновое число для
17О равно 5/2, впрочем, это ядро редко используется
в исследованиях ЯМР, поскольку его природное со-
держание очень низкое (0.04 %) и сигналы ЯМР 17О
наблюдаются с большим трудом (из-за очень дли-
тельного накопления). Наиболее распространенное
ядро кислорода (16О) имеет спин равный нулю, и не
дает сигналов ЯМР.
Азот имеет два магнитно активных изотопа: 14N
и 15N. Поскольку соединения азота чрезвычайно
важны в органической химии О, особенно в химии
природных соединений, фармакологии и биохимии,
оба изотопа широко используются в исследованиях
методом ЯМР.
Если снова обратиться к табл. 6.1, то выяснится,
что оба изотопа азота не очень удобны для исследо-
ваний ЯМР. Природное содержание изотопа 14N со-
ставляет более 99%, он имеет спин 1 и, следователь-
но, электрический квадрупольный момент. Но у этого
ядра низкая чувствительность: кроме того, спектры
оказываются сильно уширенными вследствие быстрой
квадрупольной релаксации. В дальнейшем мы не будем
рассматривать спектроскопию ЯМР для этого ядра.
Другой изотоп азота (15N) также имеет низкую от-
носительную чувствительность, а из-за очень низкого
Не считая различных азотсодержащих функциональных
групп, с которыми мы хорошо знакомы из курса органи-
ческой химии, с помощью спектров ЯМР изучались целые
классы органических N-содержащих соединений, таких
как алкалоиды, пептиды, белки и нуклеиновые кислоты.
Причем нуклеиновые кислоты наиболее перспективны для
изучения методом ЯМР, так как они содержат не только
азот, но и фосфор (см. раздел 6.5).
362
6. Спектроскопия ЯМР на других важных ядах со спином Уг
природного содержания еще и чрезвычайно низкую
абсолютную чувствительность. Но благодаря приме-
нению современной аппаратуры проблему чувстви-
тельности можно преодолеть (используя образцы,
обогащенные изотопом 15N, или с помощью косвен-
ного определения): мы сфокусируем внимание на
изотопе 15N, так как его спиновое число равно Уг,
и спектры ЯМР этого ядра характеризуются довольно
узкими линиями.
Для успешного проведения экспериментов ЯМР на
ядрах 15N необходимо учесть два важных эксперимен-
тальных фактора. Ядро 15N имеет очень низкую скорость
релаксации; Тх для него составляет более 80 с. Таким
образом, необходимо либо применять длительные за-
держки между импульсами в используемой импульсной
последовательности, либо обеспечить какой-то допол-
нительный канал спиновой релаксации. Довольно рас-
пространенным приемом является использование до-
бавок ацетилацетоната хрома(Ш) в «каталитических»
количествах. Это парамагнитное вещество, неспаренные
электроны которого достаточно эффективно стимулируют
перенос спина. В случае, если значение Тх неизвестно (и
не планируется его измерение), необходимо тщательно
продумать импульсные задержки и импульсные углы,
иначе резонансный сигнал (или сигналы) будет накапли-
ваться очень медленно или вообще может не появиться.
Теперь более подробно рассмотрим другой экспе-
риментальный фактор, который ранее уже обсуждался
в гл. 3 и 4. Речь идет о ядерном эффекте Оверхаузера
(ЯЭО). Вспомним, что обычно мы проводим экспе-
рименты ЯМР 13С с подавлением ССВ с протонами,
при этом сигналы от всех ядер углерода являются
синглетами. Кроме того, интенсивность углеродных
сигналов увеличивается (это наблюдается для атомов
углерода, связанных с протонами, т.е. в группах
СН, СН2 и СН3). Это увеличение интенсивности
обусловлено ЯЭО; изменение интенсивности сигна-
лов возникает в результате изменения (иначе говоря,
поляризации) населенностей спиновых уровней по
сравнению с тем, которое ожидается на основании
больцмановского распределения. Степень увеличения
интенсивности сигнала зависит от двух факторов:
максимально возможное увеличение интенсивности
сигнала равно половине отношения гиромагнитных
отношений двух ядер (т.е. величине (l/2)(yx/yA), где
А - регистрируемое ядро, а X - облучаемое ядро).
Впрочем, реальное увеличение интенсивностей сиг-
налов пропорционально относительному вкладу ди-
польной релаксации в общую релаксацию
ядер 13С. Для экспериментов ЯМР 13С с «развязкой»
от протонов максимальное увеличение интенсивно-
стей в результате ЯЭО определяется из соотноше-
ния (1/2)(ун/У13с)- Поскольку ун = 26.7522 (в единицах
107 рад-с-1-Тл-1), а увс = 6.7282 (в тех же единицах),
то величина (1 /2)(ун/у»зс) равна 1.98. Заметим, что от-
ношение гиромагнитных отношений можно выра-
зить с помощью частот ЯМР, т.е. (Ун/у,3с) = (рн/г,3с)>
где vH и р1зс - резонансные частоты для ядер !Н
и 13С соответственно (в одном и том же поле). Ис-
пользуя данные табл. 6.1, где приведены резонансные
частоты для ядер !Н и 13С в поле 7.0463 Тл, равные
300 и 75.43 МГц соответственно, также получим,
что (1/2)(ун/у»зс) составляет 1.98. Поэтому в целом
интенсивность утраивается, поскольку к исходной
интенсивности сигнала добавляется почти удвоенная
интенсивность вследствие эффекта Оверхаузера.
Реальное увеличение интенсивностей для системы
13С~1Н может быть любым в диапазоне от нуля до
1.98, и для каждого конкретного случая это увеличе-
ние определяется механизмом релаксации. Для атома
углерода, не имеющего непосредственно связанных
протонов (т.е. для четвертичного атома углерода),
интенсивность сигнала не искажается, так как при
этом практически отсутствует вклад в релаксацию
ядра 13С дипольной релаксации 13С-1Н. Для неболь-
ших и средних по размеру органических молекул
релаксация ядер углерода, имеющих непосредствен-
но связанные протоны, содержит ощутимый вклад
дипольной релаксации 13С-1Н, что может приво-
дить к увеличению интенсивности сигнала вплоть
до 200%.
Аналогичные рассуждения для ядра 15N приводят
к совершенно другому результату. Гиромагнитное
отношение для 15N очень маленькое и отрицатель-
ное (у = -2.712 в единицах 107 рад-с-1-Тл-1). Таким
образом, оказывается, что максимальное увеличение
интенсивности сигнала вследствие ЯЭО (при «раз-
вязке» от протонов) для ядра 15N определяется со-
отношением yH/(2)yN (т.е. 26.753/2-(-2.712)), которое
равно -4.93). Следовательно, фактически происходит
существенное уменьшение интенсивности сигнала,
и может оказаться так, что вместо обычной полярно-
сти сигнала (направления вверх), мы будем наблюдать
отрицательную полярность (сигнал ориентирован
вниз). Условно, вместо сигнала с интенсивностью
+1 можем получить сигнал с интенсивностью око-
ло -4. Еще раз отметим, что аналогичные оценки
можно получить из соотношения частот ЯМР ядер
1Н и 15N (для поля 7.0463 Тл эти частоты равны
300 и (-) 30.398 МГц, при этом знак «-» для частоты
резонанса ядра 15N поставлен условно, поскольку
прямой эксперимент по наблюдению ЯМР не по-
зволяет определить знак гиромагнитного отношения,
вследствие того что возбуждаются обе возможные
частоты, с вращением вектора намагниченности
в обоих направлениях-по и против часовой стрелки).
6.2. Ядерный магнитный резонанс на ядрах 15N
363
В большинстве случаев для ядер со спином Уг
и положительным гиромагнитным отношением
происходит увеличение интенсивностей сигналов
вследствие ЯЭО при «развязке» от протонов, в то
время как для ядер со спином Уг и отрицательным
гиромагнитным отношением интенсивности сигналов
уменьшаются.
Итак, для ядра 11 * * * 15N максимальное искажение
интенсивности сигнала определяется величиной
-4.93 (т.е. в диапазоне от +1 до -3.93). На самом
деле если механизм дипольной релаксации 15N-!H
является основным, интенсивность сигнала занижается
примерно в четыре раза по сравнению со случаем,
когда отсутствует развязка от протонов. Однако так
как дипольная релаксация 15N-!H является только
одним из многих возможных релаксационных меха-
низмов для ядра 15N, то развязка от протонов может
привести к ЯЭО в диапазоне от нуля до -4.93 (что
соответствует изменению интенсивности сигнала в
диапазоне от +1 до -3.93). Любопытно, что для ЯЭО
точно равного -1.0 вообще на спектре не наблюда-
ется сигналов! (ЯЭО полностью «съедает» обычный
сигнал ЯМР!) При наблюдении спектров ЯМР ядра
15N (как и в случае наблюдения спектров ЯМР 13С)
«развязка» от протонов является обычной практи-
кой (это позволяет освободиться от всех возможных
спин-спиновых расщеплений, вызванных константами
13С-!Н или 15N-!H). Таким образом, всегда необхо-
димо помнить об ЯЭО, который часто на практике
искажает интенсивности сигналов.
Вернемся к рассмотрению спектров ЯМР 15N.
Как мы уже говорили выше, благодаря применению
современных приборов можно достаточно просто
получить спектр ЯМР 15N, несмотря на то, что этот
изотоп имеет очень низкое природное содержание
и на порядок более низкую чувствительность по срав-
нению с ядром 13С. В настоящее время чаще всего
в качестве внешнего стандарта (эталона химическо-
го сдвига) для экспериментов ЯМР 15N используют
жидкий аммиак0), хотя можно использовать и другие
соединения, например нитрат аммония, азотную кис-
11 Иногда в качестве внутреннего стандарта использо-
вался нитрометан, причем его химический сдвиг 15N при-
нимался за нуль. Однако при этом химические сдвиги для
большинства органических азотсодержащих соединений
получались отрицательные. При использовании жидкого
аммиака в качестве внешнего стандарта отпадает необ-
ходимость в отрицательных значениях, поскольку прак-
тически все ядра 15N дезэкранированы по сравнению с
1?NH3. Однако с жидким аммиаком трудно работать, поэто-
му обычно прибавляют 380 м.д. к величине химического
сдвига относительно нитрометана и тем самым получают
значение относительно жидкого аммиака.
лоту, нитрометан. При рассмотрении литературных
данных можно достаточно точно скорректировать
шкалу химических сдвигов, если учитывать исполь-
зуемый эталон химического сдвига.
Мы уже знакомы со шкалой химических сдви-
гов, ранее введенных для ядер !Н и 13С. Азот (как
и углерод) относится ко второму ряду периодической
системы элементов и характеризуется в некоторой
степени аналогичными электронными влияниями.
В первом приближении можно считать, что хими-
ческие сдвиги 15N азотсодержащих соединений сим-
батны с химическими сдвигами ядер 13С. Диапазон
химических сдвигов 15N для обычных органических
соединений составляет 500 м.д., что в два раза больше,
чем для ядра 13С.2) На рис. 6.1 приведены диапазоны
химических сдвигов для наиболее распространенных
азотсодержащих соединений. Относительно широкие
диапазоны химических сдвигов для отдельных клас-
сов соединений и более узкие резонансные линии
15N приводят к тому, что вероятность случайного
перекрывания сигналов в спектрах ЯМР 15N, в от-
личие от спектров ЯМР 13С, очень низкая. Как и в
случае спектроскопии ЯМР 13С, можно рассчитать
параметры для заместителей в пределах конкрет-
ного класса соединений, причем по величине эти
параметры подобны таковым в ЯМР 13С.
Несмотря на некоторое сходство с углеродом, не-
обходимо помнить, что азот все-таки имеет и свои от-
личительные особенности, в том числе свои значения
химических сдвигов, характерные только для него.
Две наиболее важные особенности азота связаны
с наличием у его атома неподеленной электронной
пары.
Эта электронная пара сильно влияет на химические
свойства азотсодержащих соединений, а также на
чувствительность химических сдвигов азота к окру-
жению атома. Обнаружено, что часто положение сиг-
нала на шкале химических сдвигов сильно зависит
от растворителя; аналогичный эффект в углеродных
спектрах проявляется в меньшей степени. Влияние
неподеленной пары электронов на химические сдвиги
азота может возникнуть также из-за протонирова-
ния. Любопытно, что мы не можем сказать, в каком
направлении сместится тот или иной сигнал при
протонировании, однако можно охарактеризовать на-
правление и величину этого смещения в зависимости
от типа азотсодержащего соединения.
2) Диапазоны химических сдвигов для обоих ядер (и 13С,
и 15N) на самом деле больше, чем указано здесь. Однако
химические сдвиги вне этих пределов встречаются редко
и не обсуждаются в данной книге (см. рис. 6.1)
364
6, Спектроскопия ЯМР на других важных ядах со спином Уг
900 800 700 600 500 400 300 200 100 0
1 АЛИФАТИЧЕСКИЕ АМИНЫ Первичные Вторичные Третичные АЛИФАТИЧЕСКИЕ Первичные АММОНИЕВЫЕ ИОНЫ В1^ичные Третичные ДИМЕТИЛ ЕНАМИНЫ ИОНЫ АНИЛИНИЯ АНИЛИНЫ ДЕКАГИДРОХИНОЛИНЫ, ПИПЕРИДИНЫ, ЦИКЛИЧЕСКИЕ ЕНАМИНЫ АМИНОФОСФИНЫ ГУАНИДИНЫ МОЧЕВИНЫ, КАРБАМАТЫ, ЛАКТАМЫ АМИДЫ Первичные Вторичные Третичные ТИОМОЧЕВИНЫ ТИОАМИДЫ НИТРАМИНЫ ИНДОЛЫ, ПИРРОЛЫ ГИДРАЗОНЫ, ТРИАЗЕНЫ ИМИДЫ НИТРИЛЫ, ИЗОНИТРИЛЫ ПИРРОЛОПОДОБНЫЕ АТОМЫ АЗОТА ДИАЗОГРУППА ИОНДИАЗОНИЯ ПИРИДИН ПИРИДИНОПОДОБНЫЕ АТОМЫ АЗОТА ИМИНЫ оксимы АЗОКСИГРУППА НИТРОГРУППА АЗОГРУППА НИТРОЗОГРУППА Ar-NO 1 S-NO 1 1 1 1 1 1 1 Имино Амино no2 -n< -H=N-N. -N=N-N Имино Амино — Изо Концевая Внутренняя Концевой — — Внутренний Пиридин Бензофуразан Фуразан Пиразин Т1иримидин Имидазол d а 1 г N-N=0
1 1 1 1 1 1 1 1 1
900 800 700 600
500 400 300 200 100
Рис. 6.1. Диапазоны химических сдвигов для различных азотсодержащих
соединений. (Из книги Levy, Lichter, 1979)
На рисунке 6.2, а приведен спектр ЯМР 15N
формамида с «развязкой» от протонов. Этот спектр
похож на спектр ЯМР 13С данного соединения, где так-
же наблюдаетсясинглет. Сходство со спектрами ЯМР
13С будет наблюдаться на протяжении всей этой гла-
вы, но только при условии использования полной
«развязки» от протонов. Другая характеристика, за-
служивающая внимания, - это направленность сиг-
нала. Подавление ССВ с протонами приводит к тому,
что интенсивность сигнала уменьшается вследствие
отрицательного ЯЭО. Тем не менее в экспериментах
с фурье-преобразованием фазировка ССИ проводится
произвольно, и можно «сфазировать» пик либо вверх,
либо вниз; направленность пика в спектре является
произвольной, за исключением тех случаев, когда на
одном спектре некоторые сигналы направлены вверх,
а другие - вниз (например, в экспериментах DEPT).
Частично двойной характер связи в формамиде при-
водит к соответствующему смещению сигнала азота
(вследствие дезэкранирующего эффекта).
На рисунке 6.2,6 приведены спектры ЯМР 15N
этилендиамина с «развязкой» от протонов и без
«развязки». Во-первых, можно отметить, что сигнал
в данном случае смещен в область более сильного
поля по сравнению с формамидом. На врезке показано
расщепление сигнала в результате ССВ с протона-
ми. Прежде чем анализировать эту часть рисунка,
необходимо сделать несколько важных замечаний.
Константы спин-спинового взаимодействия (КССВ)
15NelH через одну связь (прямые константы), че-
рез две связи и через три связи хорошо изучены,
в то время как более дальние константы возникают
только при наличии в промежутках между взаимо-
действующими ядрами л-связей. Существуют табли-
цы, где перечислены величины и знаки КССВ, но их
детальное рассмотрение выходит за рамки данной
62. Ядерный магнитньш резонанс на ядрах Щ
365
I 1 I ' I ' I ' I ' I ' I ' I ' I 1 I ' I ' I ' I ' I ' I ' I
328 326 324 322 320 318 316 314 312 310 308 306 304 м.д.
320 300 280 260 240 220 200 180 160 140 120 100 80 60 м.д.
Рис. 6.2. а - Спектр ЯМР * 15N (рабочая частота 30.4 МГц) формамида
в CDCI3 с «развязкой» от протонов (внешний стандарт NH3). б - Спектр ЯМР
15N (30.4 МГц) этилендиамина в CDCI3 с «развязкой» от протонов; на врезке
спектр без «развязки» от протонов, в - спектр ЯМР 15N (30.4 МГц) пиридина
в CDCI3 с развязкой от протонов, г - Спектр ЯМР 15N (30.4 МГц) хинина в CDCI3 с
развязкой от протонов
366
6. Отектроскопия ЯМР на других важных ядах со спином А
ОН
I
5С=О
I
4 СН3 ОН 4 СН2
I 5 | | 2
зсн-сн3 зсн, зсн,
| . 21, I 21 , I 2
H2N—СН-СЧ-N—CH2-C-t-N—ch-c-i-n—сн-с—он
2 II1: Н II :н 2 II :н 2 щ
0 0 0 0
Валин (V) Глицин (G) Серин (S) Глутаминовая
Спектр HSQC 1Н- 15N (600 МПд) кислота (Е)
Проекция спектра HSQC 1H-15N (600 МГЦ) на ось 15N
।..........।...........।............।............।...........।............।............।.........................
170 160 150 140 130 120 110 100 90 м.д.
Спектр ЯМР 15N (60.8 МПд, время регистрации 15 ч)
rW’lFW'IPlpI^^
...।.......।.......। ।.।...।.......।.......।.......।.......<
170 160 150.140 130 120 110 100 90 м.д.
Рис. 6.3. Спектр HSQC ]H-15N и его проекция для тетрапептида (его структура
показана на рисунке) в разбавленном растворе. Для регистрации двумерного
спектра потребовался 1 ч приборного времени. Внизу показан спектр, полученный
при попытке зарегистрировать в течение 15 ч одномерный спектр ЯМР 15N
6.3. Спектроскопия ЯМР на ядрах 19F
367
книги. Отметим только, что значения констант ’JNH
изменяются в диапазоне от 75 до 135 Гц, констант
2Jnh-ot 0 до 20 Гц и констант 3Jnh-ot 0 до 10 Гц.
Вернемся к врезке на рис. 6.2,6: там виден квинтет
с очень небольшой КССВ около 2-3 Гц. Следова-
тельно, прямая константа ’JNH не видна вследствие
быстрых обменных процессов с участием протонов
NH, а константы 2JNH и 3JNH примерно одинаковы. За-
метим, что для гетероядерных систем (как в данном
случае при взаимодействии протонов и азота 15N) при
анализе расщеплений применимы правила спектров
первого порядка, поскольку разделяющая эти ядра
разность частот (по существу частот резонанса) со-
ставляет миллионы герц.
На рисунке 6.2, в приведен спектр ЯМР 15N (с
«развязкой» от протонов) пиридина, имеющего аро-
матический атом азота. Итак, мы показали достаточ-
ное количество примеров и можем сказать, что не
существует каких-либо необычных особенностей,
которые нельзя было бы объяснить, все подчиняет-
ся общим закономерностям. Поскольку в этой гла-
ве мы не ставили своей целью систематизировать
всю информацию по химическим сдвигам из тысячи
опубликованных литературных источников, а только
хотели «приоткрыть дверь» в спектроскопию ЯМР
на ядрах, отличных от углерода и водорода, на этом
мы собираемся закончить с ЯМР 15N. В заключение
приведем спектр ЯМР 15N хинина (рис. 6.2, г) - хо-
рошо изученного природного алкалоида.
Молекула хинина содержит два атома азота, и мы
без труда можем сделать отнесение сигналов. Но если
бы мы выделили хинин впервые как новое неиз-
вестное природное соединение, то нам пришлось
бы применить для его идентификации специальную
методику, основанную на совместном изучении не-
скольких спектров, включая гетероядерный ЯМР;
спектроскопию ЯМР 15N можно применять парал-
лельно с масс-спектрометрией, ИК-спектроскопией
и спектроскопией ЯМР на других ядрах. Более того,
вспомнив переход от простых спектров ЯМР ’Ни 13С
к их корреляциям (см. гл. 5), можно задать вопрос:
существуют ли другие спектры ЯМР 15N, помимо
спектров с «развязкой» и без «развязки» от протонов?
Это риторический вопрос; очевидно, что также воз-
можны корреляционные эксперименты с применением
ядра 15N, и они широко применяются на практике.
На рис. 6.3 приведен пример использования мето-
дики HSQC ’H-15N применительно к тетрапептиду
(валин-глицин-серин-глутаминовая кислота), о кото-
ром уже говорилось в гл. 5. (Методика HSQC дает
спектр, похожий на спектр HMQC; однако в спектре
HSQC линии получаются менее широкими вдоль
оси F1, и следовательно, вдоль этой оси получается
большее разрешение.) Спектр регистрировали для
разбавленного раствора соединения (5 мг вещества
растворяли в 0.5 мл смеси 95% Н2О/ 5% D2O). Этот
спектр косвенно дает химические сдвиги всех трех
атомов азота амидных групп. Внизу на этом рисунке
приводится проекция из двумерного спектра на соот-
ветствующую ось 15N. Регистрация этого двумерного
спектра потребовала 1 ч приборного времени, в то
время как попытка получить хороший одномерный
спектр ЯМР 15N не увенчалась успехом, сигнал на
этом одномерном спектре ЯМР 15N не появился
даже при накоплении его в течение 15 ч. Как уже
обсуждалось в гл. 5, инверсные методики обладают
более высокой чувствительностью; так, инверсная
методика HSQC ’H-15N в 310 раз [(Y’h/Y,5n)5/2] более
чувствительна по сравнению с обычными методи-
ками.
В биохимии для анализа методом ЯМР образцы
белков готовят специальным способом: в ходе син-
теза в аминокислотные остатки вводят изотоп 15N.
Так как природное содержание этого изотопа всего
0.368%, это позволяет увеличить чувствительность
эксперимента примерно в 270 раз; а если проводить
эксперимент HSQC, то чувствительность повышается
в 84 000 раз по сравнению с обычными неинверсными
методами. Таким образом можно получить спектр
хорошего качества для белков с молекулярной массой
вплоть до 50 кДа.
Для иллюстрации на рис. 6.4 приведен спектр
HSQC ’H-15N миоглобина; полный анализ этого
спектра здесь мы проводить не будем. Однако от-
метим, что миоглобин содержит 153 аминокислотных
остатка, и его анализ с помощью простого спектра
ЯМР ’Н невозможен вследствие перекрывания сигна-
лов амидных протонов. Но эти резонансные сигналы
хорошо разделяются вдоль оси 15N (ось F1) благо-
даря корреляции ’H-15N. Амидная область в спектре
ЯМР 15N белков находится в диапазоне от ПО до
150 м.д., т.е. составляет примерно 40 м.д.
6.3. Спектроскопия ЯМР на ядрах 19F
Спектроскопия ЯМР 19F имеет большое значение
с исторической точки зрения. У фтора существует
только один стабильный изотоп 19F (т.е. его природное
содержание 100%), и, как видно из табл. 6.1, фтор яв-
ляется идеальным ядром для исследований ЯМР. Чув-
ствительность для ядра 19F составляет приблизительно
0.83 от чувствительности для ядра ’Н; этот факт объ-
ясняет, почему спектроскопия ЯМР 19F развивалась
одновременно со спектроскопией ЯМР ’Н. Литератур-
ные данные по химическим сдвигам и КССВ с уча-
стием ядер фтора теперь уже устарели, и использовать
их нужно осторожно, только после сравнения старого
химического сдвига с новыми опубликованными дан-
ными, потому что раньше часто в качестве внешнего
стандарта использовалась трифторуксусная кислота
(CF3COOH). В настоящее же время в качестве ре-
перного соединения в спектроскопии ЯМР 19F при-
меняется трихлорфторметан (CFC13); это инертное
летучее соединение дает в спектре ЯМР 19F синглет,
принимаемый за 0 м.д. (относительно него CF3COOH
дает сигнал при -8.5 м.д.).
К спектроскопии ЯМР 19F необходим другой подход
по сравнению со спектроскопией ЯМР 15N, и мы его
изложим в этом разделе. В органических соединени-
ях фтор является одновалентным и может замещать
водород. Природных же фторсодержащих соединений
фактически не существует, поэтому спектроскопия
ЯМР 19F касается синтезируемых веществ. Подавление
Рис. 6.5. Диапазоны химических сдвигов для различных фторсодержащих
соединений
6.4. Спектроскопия ЯМР на ядрах 29Si
369
ССВ с протонами в спектрах ЯМР 19F не вызывает
значительных искажений в интенсивностях сигналов
из-за ЯЭО, и не существует других факторов, вызы-
вающих эти искажения. В действительности спектры
ЯМР 19F обычно регистрировались с использованием
спектрометров стационарного типа (как и спектры
ЯМР 1Н). Многие данные по химическим сдвигам
и КССВ для ядер 19F публиковались параллельно
с данными по 1Н. На рис. 6.5 приведены диапазоны
химических сдвигов для различных фторсодержащих
соединений.
В главе 3 уже приводился спектр ЯМР !Н фтор-
ацетона, чтобы показать влияние фтора на вид спектра
ЯМР !Н. Напомним, что сигналы протонов и метиль-
ной, и метиленовой групп в данном случае расще-
пляются в дублеты взаимодействием с атомом фтора,
но с разными значениями КССВ. На рис. 6.6 можем
увидеть аналогичный эффект от протонов для сигнала
атома фтора фторацетона. На этом рисунке показа-
ны спектры ЯМР 19F, *Н и 13С. Конечно, в спектре
ЯМР 19F с «развязкой» от протонов для атома фтора
мы видим только синглет, напоминающий синглеты
в спектрах ЯМР 13С с «развязкой» от протонов. Однако
в спектрах без «развязки» от протонов мы видим,
что фтор взаимодействует с двумя группами протонов,
давая триплет с большой КССВ (что соответствует
геминальной константе JHF порядка 40-50 Гц); далее
каждая линия триплета расщепляется в квартет за счет
спин-спинового взаимодействия фтора с протонами
метильной группы через четыре связи. На каждом
спектре для удобства приведены численные значе-
ния констант спин-спинового взаимодействия. Сно-
ва подчеркнем, что совместное изучение спектров
ЯМР 19F, *Н и 13С дает более убедительную и более
полную информацию, чем любой из этих спектров
в отдельности.
На рис. 6.7 приведены спектры ЯМР 19F, а также
спектры ЯМР *Н и 13С ароматического фторсодер-
жащего соединения, а именно фторбензола. Заметим,
что спектры ЯМР 19F даются с «развязкой» и без
«развязки» от протонов. Для фтора в спектре с «раз-
вязкой» от протонов мы видим синглет, а в спектре
без «развязки» от протонов наблюдается сложный
мультиплет. В этом соединении фтор по-разному
взаимодействует с орто-, мета- и ияря-протонами.
Соответствующие константы спин-спинового взаи-
модействия ^-^F (через три, четыре и пять связей)
приведены в приложении Е к гл. 3.
Довольно заманчиво было бы рассмотреть про-
тонный спектр данного соединения, как мы сделали
для фторацетона. Однако в данном случае спиновая
система более сложная (система типа AA'GG'MX,
где X - атом фтора, М - протон в иора-положении
к атому фтора). Из-за наличия в бензольном коль-
це заместителя (атома фтора) возникает зеркальная
симметрия. Протоны А и А' являются магнитно не-
эквивалентными, они по-разному взаимодействуют
с протонами G и G'. Аналогично, протоны G и G'
также магнитно неэквивалентны, и они по-разному
взаимодействуют с протонами А и А'.
Отметим, что ранее случаи магнитно неэквивалент-
ных групп ядер приводились для ияра-замещенных
бензолов (система АА'ММ') (см. раздел 3.9). Слож-
ность спектров таких систем не исчезает с увеличе-
нием напряженности магнитного поля. В некоторых
случаях увеличение поля приводит даже к усложне-
нию спектра. Эта ситуация существенно отличается
от поведения тех систем с магнитной неэквивалент-
ностью, где увеличение напряженности магнитного
поля приводит к постепенному переходу к спектрам
первого порядка.
Химические сдвиги для ядер 19F довольно трудно
предсказывать, и мы не будем пытаться предлагать
никаких прогностических моделей (алгоритмов).
В таблице 6.2 представлены эмпирические данные
по химическим сдвигам 19F для различных фтор-
содержащих соединений. Одна из причин, почему
химические сдвиги для 19F трудно предсказывать
и объяснять, заключается в том, что для ядер 19F
существенный вклад в экранирование вносит не толь-
ко диамагнитная составляющая (связанная с общей
электронной плотностью около данного атома фто-
ра), но и парамагнитное экранирование, вызванное
поляризацией электронного окружения.
6.4. Спектроскопия ЯМР на ядрах 29Si
Кремний, как и фтор, не встречается в природных
органических соединениях. Кремнийсодержащие ор-
ганические вещества, однако, все больше и больше
используются химиками, специализирующимися в об-
ласти органического синтеза и химии полимеров.
Среди стабильных изотопов кремния (28Si, 29S, 30Si)
только ядро 29Si имеет магнитный момент; его при-
родное содержание составляет 4.7%. Мы уже стал-
кивались с этим ядром при рассмотрении спектра
ЯМР !Н тетраметилсилана (ТМС); ТМС дает острый
интенсивный синглет, по бокам которого обнаружи-
ваются два сателлитных сигнала, каждый из них со-
ставляет по интенсивности 2-3% от интенсивности
синглета ТМС, расстояние между этими сателлитами
равно около 6 Гц, и это расщепление соответствует
константе КССВ (VS1H) (см. раздел 3.7.4).
Из таблицы 6.1 видно, что чувствительность ядра
29Si в два раза выше, чем ядра 13С. Гиромагнитное
-224.4 -224.6 -224.8 м.д.
-224.4 -224.6 -224.8 м.д.
Спектр ЯМР ХН (300 МГц)
4.3 Гц
48 Гц
I ' I 1 I ' I ' I ' I 1 I • I • I • I I • I —I—•—I—•—।—1—।
200 180 160 140 120 100 80 60 40 м.д.
Рис. 6.6. Спектры ЯМР 19F (282.4 МГц) фторацетона в CDCI3 с «развязкой» и без «развязки» от
протонов. Показаны спектры 1Н и 13С с соответствующими расщеплениями сигналов от 19F
6.4. Отектроскопия ЯМР^ на ядрах 2^$[
371
। 1 ' । 1 । ' I 1 ' • । • । ’
7.45 7.40 7.35 7.30 7.25
Спектр ЯМР 13С (75.5 MRj)
7.20 7.15 7.10 7.05 м.д.
7.9 Гц 21 Гц
11 JUL
245 Гц
3.1 Гц
' I ' ' ' 1 I ' ' ' 1 I" ' ' ' ' I ' ' ' ' 1 1 1 ' । • • । । ' । ' ' । 1 । .
165 160 155 150 145 140 135 130 125 120 115 м.д.
Рис. 6.7. Спектры ЯМР 19F (282.4 МГц) фторбензола в CDCI3 с «развязкой» и без
«развязки» от протонов. Спектры ЯМР ’Н и 13С: видны расщепления за счет
дальних взаимодействий с ядром 19F (пояснения см. в тексте)
372
6. Спектроскопия ЯМР на других важных ядах со спином 16
Таблица 6.2. Химические сдвиги для различных
фторсодержащих соединений
Соединение Химический сдвиг, м.д.в
CFC13 (реперное соединение) 0.0
CF2C12 -8.0
CF3C1 -28.6
CFBr3 7.4
CF2Br2 7.0
CF3Br 7.0
CFH3 -271.9
CF2H2 -143.4
CF3H -78.6
cf4 -62.3
c4f8 -135.15
C5F10 -132.9
(CF3)2CO -84.6
CF3C(O)OH -76.5
CF3C(O)OCH3 -74.2
CF3COOEt -78.7
(CF3)3N -56.0
CH2FCN -251.0
fch=ch2 -114.0
f2c=ch2 -81.3
f2c=cf2 -135.0
C6F6 -164.9
c6h5f -113.5
w-C6H4F2 -106.0
c6h5cfh2 -207
C6H5C(O)OCF3 -73.9
C6H5C(CF3)2OH -74.7
c6h5cf3 -63.7
F2 (простое вещество) 422.9
SF6 57.4
SiF4 -163.3
HF (водный) -204.0
KF (водный F") -125.3
а В большинстве литературных источников знак химическо-
го сдвига изменен на противоположный (т.е. отрицатель-
ные сдвиги представляются положительной величиной).
отношение для 29Si (ysi) отрицательное (-5.319) (в еди-
ницах 107 рад с-1 Тл-1), так что если регистрировать
обычный спектр с «развязкой» от протонов, то снова
будет происходить искажение интенсивностей сиг-
налов из-за отрицательного ЯЭО для 29Si вследствие
дипольной спиновой релаксации. В данном случае
максимальный ЯЭО будет составлять -2.51. Эта ситуа-
ция даже хуже, чем для ядра 15N, потому что только
ЯЭО в диапазоне от -2.01 до максимального значения
(-2.51) действительно приводит к увеличению интен-
сивности сигнала (в данном случае в отрицательном
направлении). Все другие значения ЯЭО на самом
деле приводят к уменьшению интенсивности сигналов
по сравнению со случаем, когда отсутствует развязка
от протонов. Таким образом, необходимо точно со-
блюдать экспериментальные условия, чтобы добить-
ся максимальной интенсивности сигнала (избежать
искажения), главным образом потому, что ядро 29Si
может иметь большие времена релаксации.
Химические сдвиги ядер 29Si для наиболее рас-
пространенных органических соединений намного
меньше, чем химические сдвиги ядер 13С. Умень-
шение диапазона химических сдвигов, вероятно,
является следствием отсутствия кратных связей
у кремния, поскольку (как в случае углерода 13С)
именно .^-гибридизация атома приводит к дезэкра-
нированию ядра. На рис. 6.8 приведены диапазоны
химических сдвигов для различных кремнийсодер-
жащих соединений.
На рисунке 6.9, а приведен спектр ЯМР 29Si те-
траметилсилана (ТМС) с «развязкой» от протонов,
на врезке изображен фрагмент спектра без «развязки»
от протонов. В качестве эталона химического сдвига
для регистрации спектров ЯМР 29Si используют ТМС.
Спектр без «развязки» от протонов вызывает боль-
шой интерес, потому что в ТМС ядро 29Si связано
с 12 эквивалентными протонами. Исходя из правила
для определения мультиплетности сигналов (и+1),
мы должны увидеть мультиплет, состоящий из 13 ли-
ний. Отчетливо видны 9 линий, но если присмотреть-
ся, то можно разглядеть также две более слабые линии
(10 и 11); еще две (линии 12 и 13) совсем не видны,
так как они настолько слабые, что теряются в шуме.
На рисунке 6.9, б приведены спектры ЯМР 29Si
триэтилсилана с «развязкой» и без «развязки» от
протонов. На спектре с «развязкой» от протонов
виден синглет, слегка смещенный относительно ТМС.
В триэтилсилане имеется протон, непосредственно
связанный с атомом кремния, поэтому возникает боль-
шая прямая константа (^щ) около 175 Гц. Еще есть
меньшие по величине геминальная и вицинальная
КССВ, которые приводят к появлению двух одина-
ковых сложных мультиплетов.
Последний пример - это спектры ЯМР 29Si
1,1,3,3-тетраэтилдисилоксана с «развязкой» и без «раз-
вязки» от протонов (рис. 6.9, в). Данное соединение
доступно в готовом виде, и оно широко используется
для синтеза различных кремнийсодержащих поли-
меров. Перед использованием соединения в синтезе
добросовестный химик должен его проанализировать
разными методами, возможно, даже с привлечением
6.5. Спектроскопия ЯМР на ядрах },Р
373
RnSiH4-n
4 3 2 1
-- — —— Me„Si(Sis)4-n
4-----------------------------------------------------3 2 1
— — . — — MenSi(0Si=)4-n
-------------- (H0)„Si(0Si»)4-„
4<------------>0
------- Si(OSH„(OAl=)4-n
------------------------ SiLe
SiX4 ________________LBr—I F_________________________________►
и т.д. до -346 м.д.
--------------------------------------------------- MSi
Кремнийсодержащие соединения переходных металлов
I I I I I I I I I I I
50 0 -50 -100 -150 -200
М.Д.
Рис. 6.8. Диапазоны химических сдвигов 29Si для различных кремнийсодержащих
соединений. (Из Bruker Almanac 1995)
спектров ЯМР 29Si. Спектр ЯМР 29Si с «развязкой»
от протонов, приведенный на рис. 6.9, позволяет уви-
деть два сигнала, вероятно, соответствующие двум
соединениям кремния. По-видимому, сигнал меньшей
интенсивности (5-10%) отвечает какой-то примеси;
таким образом, спектр позволяет оценить чистоту
реагента. Химический сдвиг имеет отрицательное
значение, что характерно для веществ, в которых
атом кремния связан с атомом кислорода.
Спектр этого соединения без развязки от протонов
позволяет обнаружить большую прямую КССВ (V^hX
которая составляет примерно 215 Гц. Расщепления
за счет геминальной и вицинальной КССВ имеют
более сложный характер. Попробуйте провести интер-
претацию этого спектра самостоятельно, это может
быть вам полезно для интерпретации спектров ЯМР
29Si продуктов реакций.
6.5. Спектроскопия ЯМР на ядрах 31Р
Последнее ядро (из четырех), которое мы рассмотрим
в этой главе, - это ядро 31Р, единственный природный
изотоп фосфора. Данное ядро вызывает большой ин-
терес у химиков-органиков, поскольку они с давних
пор используют фосфорсодержащие реагенты как
неорганические, так и органические (фосфины, фос-
фиты, фосфониевые соли, фосфорные илиды и т.д.).
Это ядро интересно и для биохимиков, спектроскопию
ЯМР 31Р они используют для исследования нуклеи-
новых кислот, которые содержат фосфатные эфирные
группы, а также для исследования небольших моле-
кул, таких как АТФ, АДФ и т.д.
Эксперименты по спектроскопии ЯМР 31Р доволь-
но просты; ядро 31Р имеет спин !4 и положитель-
ное гиромагнитное отношение (10.840 в единицах
107 рад с-1Тл-1). ЯЭО в спектрах ЯМР 31Р с «раз-
вязкой» от протонов положителен с максимальным
значением 1.23. Спектроскопия ЯМР 31Р развивается
уже давно и хорошо представлена в литературе. Дан-
ные по химическим сдвигам 31Р достоверны, потому
что как раньше, так и по настоящий момент внешним
стандартом в спектроскопии ЯМР 31Р, относительно
которого приводятся сдвиги всех остальных веществ,
служит 85%-ная фосфорная кислота (Н3РО4) (см.
спектр на рис. 6.10, а). Диапазон химических сдвигов
для ядер 31Р довольно большой, и обобщения для
отдельных классов фосфорсодержащих соединений
необходимо проводить с большой осторожностью.
В действительности на основании только химических
сдвигов 31Р невозможно сказать, в каком валентном
состоянии находится данный атом фосфора. Однако не
все так плохо, поскольку существует много надежных
опубликованных данных по химическим сдвигам 31Р.
В приложении Е к гл. 3 представлены характерные
константы спин-спинового взаимодействия 1Н-31Р.
В настоящее время доступны и более полные данные
374
6. Спектроскопия ЯМР на других важных ядах со спином 'А
Спектры ЯМР ,29Si (59.6 МПф
СН3
сн2
H3C-CH2-Si-CH2-CH3
Н
-50
-100 Гц
4
3
2
1
0
-1
-2
-3
-5
м.д.
I . . I . I I I I . -I I I I I I I . I I-
-150 -200 Гц -350 -400 Гц
Рис. 6.9. а - Спектры ЯМР 29Si (59.6 МГц) тетраметилсилана (ТМС)
в CDCI3 с развязкой и без развязки от протонов. В спектре надежно видны только
9 сигналов в мультиплете. Всего же должно наблюдаться 13 сигналов, таким
образом, две пары внешних пиков не видны, поскольку они теряются в шуме, б -
Спектры ЯМР 29Si (59.6 МГц) триэтилсилана в CDCI3 с «развязкой» и без «развязки»
от протонов, в - Спектры ЯМР 29Si (59.6 МГц) 1,1,3,3-тетраэтилдисилоксана
в CDCI3 с «развязкой» и без «развязки» от протонов
6.5. Спектроскопия ЯМР на ядрах 31Р
375
Спектры ЯМР 31Р (121.5 МПО с « развязкой» от протонов
Н3РО4
140 130 120 ПО 100 90 80 70 60 50 40 30 20 10 О М.Д.
(СбН5)зР
'"I.....I.....I......г ....,......,.....,........... ,...р........|.....,|.....|......
140 130 120 ПО 100 90 80 70 60 50 40 30 20 10 О М.Д.
(С6Н5О)3Р
140 130 120 ПО 100 90 80 70 60 50 40 30 20 10 0 м.д.
(С2Н5О)зР
I.....I......I....."I...................|............ |......,......,......|......,.....|......j......|.....|
140 130 120 ПО 100 90 80 70 60 50 40 30 20 10 0 м.д.
Рис. 6.10. Спектры ЯМР31Р (121.5 МГц) с «развязкой» от протонов 85%-ной
фосфорной кислоты с небольшой добавкой D2O (о), трифенилфосфина в CDCI3 (б),
трифенилфосфита в CDCI3 (в), триэтилфосфита в CDCI3 (г)
376
6. Спектроскопия ЯМР на других важных ядах со спином А
по константам спин-спинового взаимодействия 1Н-31Р
и другим константам с участием фосфора.
На рисунке 6.10, б показан спектр ЯМР 31Р трифе-
нилфосфина с «развязкой» от протонов. Это широко
распространенный реагент в органическом синтезе.
Собственно, это обычный спектр, в котором виден син-
глет от одного атома фосфора в трифенилфосфине. Не-
обычно то, что для трех соединений (см. рис. 6.10, а, б и
рис. 6.11, а) сигналы ЯМР 31Р размещаются на шкале
химических сдвигов в диапазоне менее 10 м.д., хотя
атомы фосфора в них существенно различаются по
валентному состоянию, по присоединенным замести-
телям и т.д. У нас возникли бы большие трудности
при попытке дать объяснение этим рисункам; было бы
глупо пытаться предсказать соответствующие хими-
ческие сдвиги 31Р. В табл. 6.3 приведены химические
сдвиги 31Р для ряда фосфорсодержащих соединений.
На рисунке 6.10, в, г приведены два спектра
ЯМР 31Р с «развязкой» от протонов для двух фос-
фитов (трифенилфосфита и триэтилфосфита).
Как и следовало ожидать, оба соединения дают
узкие синглеты. Мы их привели, чтобы проиллю-
стрировать тот факт, что несмотря на отсутствие
каких-либо прогностических алгоритмов для опре-
деления химических сдвигов, можно получить по-
лезную информацию. Химические сдвиги 31Р для
этих соединений не перекрываются и могут быть
использованы для аналитических задач. Собственно
сами спектры ЯМР 31Р не могут дать много ин-
формации, но совместное использование с други-
ми спектрами помогает сделать выводы о составе,
структуре соединения и его стереохимии.
На рисунке 6.11 приведен набор спектров ЯМР диэ-
тилхлорфосфата: спектры ЯМР 31Р (с «развязкой» и без
«развязки» от протонов), спектр ЯМР *Н и спектр ЯМР
13С. В спектре ЯМР 31Р без «развязки» от протонов
наблюдается квинтет, указывающий на то, что нет за-
метного взаимодействия между ядром 31Р и протонами
метильных групп. Однако изучение соответствующих
протонных спектров (с «развязкой» и без «развязки» от
ядер фосфора) позволяет обнаружить, что в действи-
тельности сигналы метильных групп представляют
собой триплет дублетов с КССВ VfH-31?) через три
связи, составляющей около 1 Гц. Таким образом, мож-
но сделать вывод, что расщепление в 1 Гц в спектре
ЯМР 31Р мы не видим, но оно проявляется в некотором
дополнительном уширении линий в спектре ЯМР 31Р.
Четыре же протона двух метиленовых групп эквива-
лентны, и они взаимодействуют с атомом фосфора
с одинаковой константой V(1H-31P), давая квинтет,
который мы и видим.
Таблица 6.3. Химические сдвиги некоторых
фосфорсодержащих соединений по данным Broker
Almanac (1995)
Соединения фосфора(Ш) Химический Соединения Химический СДВИГ, М.Д."
сдвиг, м.д.в фосфора(У)
РМе3 -62 Ме3РО 32.6
PEt3 -20 Et3PO 48.3
Р(я-Рг)3 -33 [Ме4Р]+ 24.4
Р(/-Рг)3 19.4 [РО4]3- 6
Р(и-Ви)3 -32.5 pf5 -80.3
Р(/-Ви)3 -45.3 РС15 -80
P(s-Bu)3 7.9 MePF4 -29.9
P(/-Bu)3 63 Me3PF2 -158
PMeF2 245 Me3PS 59.1
РМеН2 -163.5 Et3PS 54.5
РМеС12 192 [Et4P]- 40.1
РМеВг2 184 [PS4]3- 87
PMe2F 186 [PF6]- -145
РМе2Н -99 [PC14]+ 86
РМе2С1 -96.5 [pci6]- -295
РМе2Вг -90.5 Me2PF3 8
а Относительно сигнала 85%-ной фосфорной кислоты.
Сигналы метиленовых протонов в спектре ЯМР 1Н.
на первый взгляд, кажутся очень сложными. Однако
внимательный анализ показывает, что метиленовые
протоны в диэтилхлорфосфате диастереотопны (см.
рис. 3.42), и, следовательно, они являются химически
неэквивалентными. При этом обнаруживается силь-
ное спин-спиновое взаимодействие с геминальной
константой 2J(H-H), что приводит к существенно-
му усложнению спектра. Анализ спектров в рамках
спектров первого порядка невозможен.
6.6. Заключение
В этой главе мы попытались кратко ознакомить вас
с ЯМР на некоторых других ядрах, привели несколько
примеров их спектров. Особый акцент был сделан на
совместном использовании этих спектров с другими
методами, в частности на одновременном сопостав-
лении спектров ЯМР на нескольких ядрах. Следует
признать, что нет необходимости становиться «экс-
пертом» по значениям химических сдвигов этих (или
других) ядер и значениям их КССВ. Но не стоит огра-
ничиваться применением только этих четырех ядер,
поскольку нередко может быть полезен ЯМР и на
ядрах других элементов периодической таблицы.
6.6. Заключение
377
Спектр ЯМР 31Р (242.9 MIX) без «развязки»
Спектр ЯМР 31Р (242.9 MIX)
с «развязкой» от протонов
от протонов
(СгН5)2Р(О)С1
I.......I........I........I.......I......‘ • I
5.8 5.7 5.6 5.5 м.д.
Спектр ЯМР 'Н (600 MIX)
с «развязкой» от ядер 31Р
। 1 । • । i ' । • । • । •
4.32 4.30 4.28 4.26 4.24 4.22 м.д.
।..........।.................................।..........।
5.8 5.7 5.6 5.5 м.д.
1.44 1.42 1.40 1.38 1.36 1.34 м.д.
Спектр ЯМР !Н (600 MIX)
без «развязки» от ядер 31Р.
в л А
। । • । • । • ।—•—।—•—।—•
4.32 4.30 4.28 4.26 4.24 4.22 м.д.
Спектр ЯМР *4: (150.9 MIX)
JL
15.8 15.6 м.д.
I ' ’ ' ' I ’ ’ ' ' I ‘ ' I.। । f - j (
65 60 55 50 45 40 35 30 25 20 15 м.д.
Рис. 6.11. Спектры ЯМР диэтилхлорфосфата в CDCI3:3,P (121.5 МГц) с «развязкой»
и без «развязки» от протонов (о), ’Н с «развязкой» от 31Р (б) и без «развязки» от 3,Р
(в), 13С без «развязки» от 31Р (г)
378
6. Спектроскопия ЯМР на других важных ядах со спином 'А
Литература
Andreis, М., and Koenig, J. L. (1995). Application of nitro-
gen-15 NMR to polymers. Adv. Polym. Sci., 124, 191-237.
Argyropoulos, D. S. (1995). Phosphorus-31 NMR in wood
chemistry: a review of recent progress. Res. Chem. Interme-
diates, 21,373-395.
Berger, S., Braun, S., and Kalinowski, H. O. (1992). NMR-
Spektroskopie von Nichtmetallen. Stuttgart: Georg Thieme
Verlag.
Band 1: Grundlagen, 17O-, 33S- and 129Xe-NMR Spek-
troskopie.
Band 2: 15N-NMR Spektroskopie.
Band 3: 31P-NMR Spektroskopie.
Band 4: 19F-NMR Spektroskopie.
Brey, W., (Ed.,) (1988). Pulse Methods in ID and 2D Liquid-
Phase NMR. New York: Academic Press.
Buchanan, G. W. (1989). Applications of nitrogen-15 NMR
spectroscopy to the study of molecular structure, stereochem-
istry and binding phenomena. Tetrahedron, 45(3), 581-604.
Chandrakumar, N., and Subramanian, S. (1987). Modern
Techniques in High Resolution FT NMR. New York: Springer-
Verlag.
Chesnut, D. B., and Quin, L. D. (1999). The theoretical de-
termination of phosphorus NMR chemical shielding. Adv.
Mol. Struct. Res., 5, 189-222.
Coleman, B. (1983). Applications of silicon-29 NMR spec-
troscopy. In P. Laszlo, Ed. NMR Newly Accessible Nuclear, 2,
197-228. New York: Academic.
Dungan, C. Fl., and Van Wazer, J. R. (1970). Compilation
of Reported F-19 NMR Chemical Shifts. New York: Wiley-
Interscience.
Emsley, J.W., and Phillips, L. (1971). Fluorine chemical shifts.
In J. W. Emsley, J. Feeney, and L. H. Sutcliffe, Eds., Progress in
Nuclear Magnetic Resonance Spectroscopy, Vol. 7. Oxford:
Pergamon Press.
Everett, T. S. (1988). The correlation of multinuclear spectral
data for selectively fluorinated organic compounds. J.
Chem. Educ., 65, 422-425.
Fluck, E., and Heckmann, G. (1987). Empirical methods for
interpreting chemical shifts of phosphorus compounds. Meth.
Stereochemical Anal., 8 (Phosphorus-31 NMR Spectrosc.
Stereochem. Anal.), 61-113.
Fushman, D. (2003). Determination of protein dynamics
using 15N relaxation measurements. Meth. Principles Med.
Chem., 16, 283-308.
Gorenstein, D. G. (1983). Nonbiological aspects of phos-
phorus-31 NMR spectroscopy. Prog. Nucl. Magn. Resonance
Spectrosc, 16(1), 1-98.
Gorenstein, D. G. (1984.) Phosphorus-31 NMR. London:
Academic.
Gorenstein, D. G. (1992). Advances in phosphorus-31 NMR.
In R. Engel, Ed. Handbook of Organophosphorus Chemistry.
New York: Dekker.
Harris, R. K. (1983). Solution-state NMR studies of Group IV
elements (other than carbon). NATO ASI Series, Series C:
Mathematical and Physical Sciences, 103 (Multinucl. Ap-
proach NMR Spectrosc), 343-359.
Holmes, R. R., and Prakasha, T. K. (1993). Cyclic oxyphospho-
ranes. Phosphorus-31 chemical shift correlations. Phophorus,
Sulfur and Silicon and the Related Elements, 80, 1-22.
Hom, H. G. (1973). NMR spectra and characteristic frequen-
cies of compounds containing nitrogen-sulfur-fluorine bonds.
Fluorine Chem. Rev., 6, 135-192.
Hulst, R., Kellogg, R. M., and Feringa, B. L. (1995). New meth-
odologies for enantiomeric excess (ее) determination based
on phosphorus NMR. Recueil des Travaux Chimiques des
Pays-Bas, 114, 115-138.
Hutton, W. C. (1984). Two-dimensional phosphorus-31 NMR.
In D. G. Gorenstein, Ed. Phosphorus-31 [Thirty-One] NMR.
Orlando: Academic.
Karaghiosoff, K, and Schmidpeter, A. (1988). The chemical
shift of two-coordinate phosphorus. II. Heterocycles. Phos-
phorus and Sulfur and the Related Elements, 36, 217-259.
Laszlo, P., Ed. (1983). NMR of Newly Accessible Nuclei,
Vol. 1: New York: Academic Press.
Levy, G. C, and Lichter, R. L. (1979). Nitrogen-15 Nuclear
Magnetic Resonance Spectroscopy. New York: John Wiley
&Sons.
Lichter, R. L. (1983). Nitrogen nuclear magnetic resonance
spectroscopy. NATO ASI Series, Series C: Mathematical and
Physical Sciences, 103 (Multinucl. Approach NMR Spec-
trosc), 207-244.
Majoral, J. -P, Caminade, A. -M., and Igau, A. (1994). Eluci-
dation of small ring and macrocycle structures using
31P NMR. In L. D. Quin and J. G. Verkade, Eds., Phosphorus-
31 NMR Spectral Properties in Compound Characterization
and Structural Analysis. New York: VCH pp. 57-68.
Marek, R., and Lycka, A. (2002). 15N NMR spectroscopy in
structural analysis. Curr. Org. Chem., 6(1), 35-66.
Martin, G. J., Martin, M. L., and Gouesnard, J. P. (1981). 15N-
NMR Spectroscopy, Vol. 18. Berlin: Springer-Verlag.
Mason, J. (Ed.) (1987). Multinuclear NMR. New York:
Plenum Press.
Mason, J. (1983). Patterns and prospects in nitrogen NMR.
Chemistry in Britain, 19(8), 654-658.
McFarlane, W. (1987). Special experimental techniques in
phosphorus NMR spectroscopy. Methods in Stereochemi-
cal Anal, 8 (Phosphorus-31 NMR Spectrosc. Stereochem.
Anal.), 115-150.
Mooney, E. F. (1970). An Introduction to I9F NMR Spec-
troscopy. London: Hoyden.
Упражнения
379
Quin, L. D., and Verkade, J. G., (Eds.) (1994). Phospho-
rus-31 NMR Spectral Properties in Compound Character-
ization and Structural Analysis. New York: VCH Publishers.
Randall, E. W., and Gillies, D. G. (1971). Nitrogen nu-
clear magnetic resonance. In J. W. Emsley, J. Feeney,
and L. H. Sutcliffe, Eds., Progress in Nuclear Magnetic
Resonance Spectroscopy, Vol. 6. Oxford: Pergamon Press,
pp. 119-174.
Rao, B. D. N. (1983). Phosphorus-31 nuclear magnetic
resonance investigations of enzyme systems. Biol. Magn.
Resonance, 5, 75-128.
Roberts, J. D. (1979). Use of nitrogen-15 nuclear magnetic
resonance spectroscopy in natural-product chemistry. Jap.
J. Antibiotics, 32(Suppl.) SI 12-121.
Schilf, W., and Stefaniak, L. (2000). Application of 14N and 15N
NMR for determination of protonation site and hydrogen
bond strength in nitrogen organic compounds. In Atta-ur-
Rahman, Ed., New Adv. Anal. Chem., Amsterdam: Harwood
Academic Publishers.
Schiller. J., and Arnold. K. (2002). Application of high reso-
lution 3IP NMR spectroscopy to the characterization of the
phospholipid composition of tissues and body fluids - a meth-
odological review. Med.. Sci. Monitor, 8(11), 205-222.
Schraml, J., and Bellama, J. M. (1976). Silicon-29 nuclear
magnetic resonance. In F. C. Nachod, J. J. Zuckerman,
and E. W. Randall, Eds. Determination Org. Struct. Phys.
Methods, 6, 203-269. New York: Academic Press.
Tebby. J. C. (1987). General experimental techniques and
compilation of chemical shift data. Methods in Stereochemi-
cal Anal., 8 (Phosphorus-31 NMR Spectrosc. Stereochem.
Anal.), 1-60.
Webb, G. A. (Ed.). (1981). Annual Reports on NMR Spec-
troscopy, Vol. 11B: Nitrogen NMR Spectroscopy. London;
Academic Press.
Webb, G. A., and Witanowski, M. (1985). Nitrogen NMR and
molecular interactions. Proc. - Indian Acad. Sci. Chem. Sci,
94(2), 241-290.
Witanowski, M., Stefaniak, L., and Webb, G. A. (1986). Nitrogen
NMR spectroscopy. Annu. Rep. NMR Spectrosc., 18, 1-761.
Witanowski, M., and Webb, G. A. (1973). Nitrogen NMR.
New York: Plenum.
Yeagle, P. L. (1990). Phosphorus NMR of membranes. Biol.
Magn. Resonance, 9, 1-54.
Упражнения
6.1. Установите структуру соединения с молекулярной
формулой C5H12N2, исходя из информации, полу-
ченной из спектров ЯМР !Н, 13С, DEPT и 15N.
Спектр ЯМР 15N без развязки от протонов не
приводится, так как он не дает полезной инфор-
мации.
6.2. Соединение, обсуждаемое в этой задаче, являет-
ся распространенным реагентом, который широко
используется в органическом синтезе; его моле-
кулярная формула C6H15SiCl. Приведены спектры
ЯМР ’Н, 13С, DEPT и 29Si (с «развязкой» от про-
тонов).
6.3. Определите структуру фосфорсодержащего
соединения с молекулярной формулой С19Н18РВг
по спектрам ЯМР !Н, 13С, DEPT и 31Р (с «раз-
вязкой» и без «развязки» от протонов).
6.4. Определите структуру фторсодержащего соеди-
нения, для которого приведены масс-спектры,
ИК-спектры, а также спектры ЯМР !Н, 13С, DEPT
и 19F.
380
6. Спектроскопия ЯМР на других важных ядах со спином У2
Спектры ЯМР 13C/DEPT (75.5 МГЦ)
130 120 110 100 90 80 70 60 50 40 30 20 м.д.
Спектр ЯМР 15N (30.4 МГЦ)
I—•—I—’—I—•—I—•—I—•—I—'—I—•—I—'—I—'—I—•—I—•—I—'—I—1—I
240 220 200 180 160 140 120 100 80 60 40 20 м.д.
Упражнение 6.1
Упражнения
381
Спектр ЯМР ’Н (300 МПд)
1.3 1.2 1.1 1.0 0.9 0.8 0.7 0.6 0.5 0.4 0.3 0.2 0.1 м.д.
Спектры ЯМР 13C/DEPT (75.5 МПд)
35 30 25 20 15 10
I.........। '
0 -5 м.д.
Спектр ЯМР 29Si (59.6 МПд)
55 50 45 40 35 30 25 20 15 10 5 м.д.
Упражнение 6.2
382
6. Спектроскопия ЯМР на других важных ядах со спином У2
Спектр ЯМР ’Н (300 МПд)
Спектры ЯМР 13C/DEPT (75.5 МГЦ)
Спектр ЯМР 31Р с «развязкой» от протонов (121.5 МГЦ)
-I-.-j-.-j-.--!-,-!-,--j-.-]-.--j-г—п,--j--,-г
28 26 24 22 20 18 16 14 12 10 8
"I 1 I 1 I 1 I
6 4 2 м.д.
Упражнение 6.3
Упражнения
Масс-спектр
100 Е
50
ИК-Спектр
’Т1
100
175 п
244-1
127-1
, 150
m/z
205 q
-Н
200
Спектр ЯМР 19F (282.4 МГЦ)
Длина волны (см"1)
Спектр ЯМР ’Н (300 МПд)
Упражнение 6.4
Приложение А. Свойства магнитно активных ядер
Изотоп Спин Природное содержание Магнитный момент р, pN Гиромагнитное отношение Yr Ю7 рад с-1 Тл"1 Квадрупольный момент Q, фм2 Резонансная частота в поле 2.35 Тл Эталонное соединение Относительная чувствительность
Q протон □ УГЛОРОД
’Н 1/2 99.9885 4.837353570 26.7522128 — 100.000000 Me4Si 1 5.87 X 103
2Н 1 0.0115 1.21260077 4.106 62791 0.286 15.350609 (CD3)3Si 1.11 X 10-6 6.52 X IO-3
Зн 1/2 — 5.159714367 28.5349779 — 106.663974 Me4Si-tj — —
3Не 1/2 1.37 X КГ4 -3.685154336 -20.3801587 — 76.179437 Не(газ) 6.06 x 10~7 3.56 X IO-3
6Li 1 7.59 1.1625637 3.9371709 -0.0808 14.716086 LiCl 6.45 X 10~4 3.79
7Li 3/2 92.41 4.20407505 10.3977013 -4.01 38.863797 LiCl 0.271 1.59 X 103
9Ве 3/2 100 -1.520136 -3.759666 5.288 14.051813 BeSO4 1.39 X IO"2 81.5
1ОВ 3 19.9 2.0792055 2.8746786 8.459 10.743658 BF3.Et2O 3.95 X IO”3 23.2
”В 3/2 80.1 3.4710308 8.5847044 4.059 32.083974 BF3.Et2O 0.132 7.77 X 102
,3С 1/2 1.07 1.216613 6.728284 — 25.145020 Me4Si 1.70 X IO-4 1
14N 1 99.632 0.57100428 1.9337792 2.044 7.226317 CH3NO2 1.00 X 10"3 5.9
,5N 1/2 0.368 -0.49049746 -2.71261804 — 10.136767 MeNO2 3.84 X IO"6 2.25 x IO’2
,7О 5/2 0.038 -2.24077 -3.62808 -2.56 13.556457 D2O 1.11 X IO-5 6.50 X IO-2
19р 1/2 100 4.553333 25.18148 — 94.094011 CC13F 0.834 4.90 X 103
21Ne 3/2 0.27 -0.854376 -2.11308 10.155 7.894296 Ne (газ) 6.65 X IO-6 3.91 X 10~2
23Na 3/2 100 2.8629811 7.0808493 10.40 26.451900 NaCl 9.27 X IO-2 5.45 x 102
KMg 5/2 10.00 -1.01220 -1.63887 19.94 6.121635 MgCl2 2.68 X IO-4 1.58
27 Al 5/2 100 4.3086865 6.9762715 14.66 26.056859 A1(NO3)3 0.207 1.22 X 103
29Si 1/2 4.6832 -0.96179 -5.3190 — 19.867187 Me4Si 3.68 X 10'4 2.16
31Р 1/2 100 1.95999 10.8394 — 40.480742 H3PO4 6.65 X IO"2 3.91 X 102
33s 3/2 0.76 0.8311696 2.055685 -6.78 7.676000 (NH4)2SO4 1.72 X IO'5 0.101
35С1 3/2 75.78 1.061035 2.624198 -8.165 9.797909 NaCl 3.58 X IO-3 21
37С1 3/2 24.22 0.8831998 2.184368 -6.435 8.155725 NaCl 6.59 X IO'4 3.87
39к 3/2 93.2581 0.50543376 1.2500608 5.85 4.666373 KC1 4.76 X IO’4 2.79
С°к) 4 0.0117 -1.4513203 -1.5542854 -7.30 5.802018 KC1 6.12 X IO"7 3.59 X IO-3
С’к) 3/2 6.7302 0.2773961 0.68606808 7.11 2.561305 KC1 5.68 X IO’6 3.33 X IO-2
43Са 7/2 0.135 -1.494067 -1.803069 -4.08 6.730029 CaCl2 8.68 X IO"6 5.10 x IO"2
45Sc 7/2 100 5.3933489 6.5087973 -22.0 24.291747 Sc(NO3)3 0.302 1.78 X 103
47Ti 5/2 7.44 -0.93294 -1.5105 30.2 5.637534 TiCl4 1.56 X IO'4 0.918
49Т1 7/2 5.41 -1.25201 -1.51095 24.7 5.639037 TiCl4 2.05 X IO-4 1.2
(soy) 6 0.25 3.6137570 2.6706490 21.0 9.970309 VOC13 1.39 X IO"4 0.818
51у 7/2 99.75 5.8380835 7.0455117 -5.2 26.302948 VOC13 0.383 2.25 X 103
53Сг 3/2 9.501 -0.61263 -1.5152 -15.0 5.652496 K2CrO4 8.63 X IO’5 0.507
55Мп 5/2 100 4.1042437 6.645 2546 33.0 24.789218 KMnO4 0.179 1.05 X 103
57Fe 1/2 2.119 0.1569636 0.8680624 — 3.237778 Fe(CO)5 7.24 x IO’7 4.25 X IO-3
5,Со 7/2 100 5.247 6.332 42.0 23.727074 K3[Co(CN)6] 0.278 1.64 X 103
61Ni 3/2 1.1399 -0.96827 -2.3948 16.2 8.936051 Ni(CO)4 4.09 X IO’5 0.24
«Си 3/2 69.17 2.8754908 7.1117890 -22.0 26.515473 [Cu(CH3CN)4][C1O4] 6.50 X IO"2 3.82 X 102
6. Спектроскопия ЯМР на других важных ядах со спином А
«Си 3/2 30.83 3.07465 7.60435 -20.4 28.403693 [Cu(CH3CN)4][C1O4] 3.54 X IO'2 2.08 X 102
67Zn 5/2 4.10 1.035556 1.676688 15.0 6.256803 Zn(NO3)2 1.18 x IO"4 0.692
(69Ga) 3/2 60.108 2.603405 6.438855 17.1 24.001354 Ga(NO3)3 4.19 x IO"2 2.46 X IO2
7,Ga 3/2 39.892 3.307871 8.181171 10.7 30.496704 Ga(NO3)3 5.71 x IO’2 3.35 X 102
73Ge 9/2 7.73 -0.9722881 -0.9360303 -19.6 3.488315 (CH3)4Ge 1.09 x IO"4 0.642
75 As 3/2 100 1.858354 4.596163 31.4 17.122614 NaAsF6 2.54 x IO"2 1.49 X IO2
17Se 1/2 7.63 0.92677577 5.1253857 — 19.071513 Me2Se 5.37 x IO’4 3.15
(79Br) 3/2 50.69 2.719351 6.725616 31.3 25.053980 NaBr 4.03 x IO’2 2.37 x IO2
81Br 3/2 49.31 2.931283 7.249776 26.2 27.006518 NaBr 4.91 x IO’2 2.88 X IO2
83Kr 9/2 11.49 -1.07311 -1.03310 25.9 3.847600 Кг(газ ) 2.18 x 10’4 1.28
(85Rb) 5/2 72.17 1.6013071 2.5927050 27.6 9.654943 RbCl 7.67 x IO’3 45
87Rb 3/2 27.83 3.552582 8.786400 13.35 32.720454 RbCl 4.93 X 10’2 2.90 X IO2
87Sr 9/2 7.00 -1.2090236 -1.1639376 33.5 4.333822 SrCl2 1.90 x IO 4 1.12
89y 1/2 100 -0.23801049 -1.3162791 — 4.900198 Y(NO3)3 1.19 x IO"4 0.7
91Zr 5/2 11.22 -1.54246 -2.49743 -17.6 9.296298 Zr(C5H5)2Cl2 1.07 x IO’3 6.26
93Nb 9/2 100 6.8217 6.5674 -32.0 24.476170 K[NbCl6] 0.488 2.87 X 103
95Mo 5/2 15.92 -1.0820 -1.7510 -2.2 6.516926 Na2MoO4 5.21 X 10“4 3.06
(97Mo) 5/2 9.55 -1.1050 -1.7880 25.5 6.653695 Na2MoO4 3.33 X IO"4 1.95
9/2 - 6.2810 6.0460 -12.9 22.508326 NH4TcO4 — —
"Ru 5/2 12.76 -0.7588 -1.2290 7.9 4.605151 K4[Ru(CN)6] 1.44 x IO'4 0.848
101Ru 5/2 17.06 -0.8505 -1.3770 45.7 5.161369 K4[Ru(CN)6] 2.71 x 10~4 1.59
,03Rh 1/2 100 -0.1531 -0.8468 - 3.186447 Rh(acac)3 3.17 x IO"5 0.186 §
lOSpd 5/2 22.33 -0.7600 -1.2300 66.0 4.576100 K2PdCl6 2.53 x IO’4 1.49
(107Ag) 1/2 51.839 -0.19689893 -1.0889181 - 4.047819 AgNO3 3.50 x 10~5 0.205 $ c:
109Ag 1/2 48.161 -0.22636279 -1.2518634 — 4.653533 AgNO3 4.94 x 10~5 0.290 гъ
(niCd) 1/2 12.80 -1.0303729 -5.6983131 - 21.215480 Me2Cd 1.24 x IO"3 7.27
n3Cd 1/2 12.22 -1.0778568 -5.9609155 — 22.193175 Me2Cd 1.35 x IO’3 7.94
(113In) 9/2 4.29 6.1124 5.8845 79.9 21.865755 In(NO3)3 1.51 x IO'2 88.50
115In 9/2 95.71 6.1256 5.8972 81.0 21.912629 In(NO3)3 0.338 1.98 x IO3
(115Sn) 1/2 0.34 -1.5915 -8.8013 — 32.718749 Me4Sn 1.21 x IO'4 0.711
(117Sn) 1/2 7.68 -1.73385 -9.58879 — 35.632259 Me4Sn 3.54 x IO"3 20.8
119Sn 1/2 8.59 -1.81394 -10.0317 — 37.290632 Me4Sn 4.53 x IO’3 26.6
121Sb 5/2 57.21 3.9796 6.4435 -36.0 23.930577 KSbCl6 9.33 x IO'2 5.48 X 102
(123Sb) 7/2 42.79 2.8912 3.4892 -49.0 12.959217 KSbCl6 1.99 X 10“2 1.17 x IO2
(I23Te) 1/2 0.89 -1.276431 -7.059098 — 26.169742 Me2Te 1.64 x IO’4 0.961
,25Te 1/2 7.07 -1.5389360 -8.5108404 — 31.549769 Me2Te 2.28 x IO"3 13.40
,27I 5/2 100 3.328710 5.389573 -71.0 20.007486 KI 9.54 x IO'2 5.60 x 10z
129Xe 1/2 26.44 -1.347494 -7.452103 — 27.810186 XeOF4 5.72 x IO"3 33.60
13,Xe 3/2 21.18 0.8931899 2.209076 -11.4 8.243921 XeOF4 5.96 x IO’4 3.50
,33Cs 7/2 100 2.9277407 3.5332539 -0.343 13.116142 CsNO3 4.84 x 10“2 2.84 x 102
(135Ba) 3/2 6.592 1.08178 2.67550 16.0 9.934457 BaCl2 3.30 x IO4 1.93
137Ba 3/2 11.232 1.21013 2.99295 24.5 11.112928 BaCl2 7.87 X IO"4 4.62
l38La 5 0.09 4.068095 3.557239 45.0 13.19430 LaCl3 8.46 X IO"5 0.497
139La 7/2 99.91 3.155677 3.8083318 20.0 14.125641 LaCl3 6.05 x IO’2 3.56 x IO2
141pr 5/2 100 5.0587 8.1907 -5.89 30.62 — — — IM
143Nd 7/2 12.2 -1.208 -1.4570 -63.0 5.45 - - - CO 1Л
Приложение А (окончание)
Изотоп Спин Природное содержание Магнитный момент р, pN Гиромагнитное отношение у, 107 рад с-1 Тл-1 Квадрупольный момент Q, фм2 Резонансная частота в поле 2.35 Тл Эталонное соединение Относительная чувствительность
p протон P углерод
l45Nd 7/2 8.3 -0.7440 -0.8980 -33.0 3.36 —
l47Sm 7/2 14.99 -0.9239 -1.1150 -25.9 4.17 — —
l49Sm 7/2 13.82 -0.7616 -0.9192 7.4 3.44 — — —
151Eu 5/2 47.81 4.1078 6.6510 90.3 24.86 — —
153Eu 5/2 52.19 1.8139 2.9369 241.2 10.98 — — —
155Gd 3/2 14.80 -0.33208 -0.82132 127.0 3.07 — — —
157Gd 3/2 15.65 -0.4354 - 1.0769 135.0 4.03 — —
159Tb 3/2 100 2.6000 6.4310 143.2 24.04 — —
16lDy 5/2 18.91 -0.5683 -0.9201 250.7 3.44 — —
'«Dy 5/2 24.90 0.7958 1.2890 264.8 4.82 — —
165Ho 7/2 100 4.7320 5.7100 358.0 21.34 — —
l67Er 7/2 22.93 -0.63935 -0.77157 356.5 2.88 — —
l69Tm 1/2 100 -0.4011 -2.2180 — 8.29 — —
l7lYb 1/2 14.28 0.85506 4.7288 — 17.499306 — — —
l73Yb 5/2 16.13 -0.80446 -1.3025 280.0 4.821 — —
175Lu 7/2 97.41 2.5316 3.0552 349.0 11.404 — — —
l76Lu 7 2.59 3.3880 2.16844 497.0 8.131 — — —
177Hf 7/2 18.60 0.8997 1.0860 336.5 4.007 — 2.61 x 10~4 1.54
179Hf 9/2 13.62 -0.7085 -0.6821 379.3 2.517 — 7.45 x 10“5 0.438
l81Ta 7/2 99.988 2.6879 3.2438 317.0 11.989600 КТаС16 3.74 x IO’2 2.20 X 102
183W 1/2 14.31 0.20400919 1.1282403 — 4.166387 Na2WO4 1.07 x 10*5 6.31 x IO’2
(,85Re) 5/2 37.4 3.7710 6.1057 218.0 22.524600 KReO4 5.19 x IO’2 3.05 x 102
187Re 5/2 62.6 3.8096 6.1682 207.0 22.751600 KReO4 8.95 x 10’2 5.26 x 102
,87Os 1/2 1.96 0.1119804 0.6192895 — 2.282331 OsO4 2.43 x 10"7 1.43 x 10‘3
189Os 3/2 16.15 0.851970 2.10713 85.6 7.765400 OsO4 3.95 x IO"4 2.32
C’4r) 3/2 37.3 0.1946 0.4812 81.6 -1.718 - 1.09 X 10’5 6.38 x IO"2
193Ir 3/2 62.7 0.2113 0.5227 75.1 -1.871 - 2.34 x 10~5 0.137
i’Spt 1/2 33.832 1.0557 5.8385 — 21.496784 Na2PtCl6 3.51 x 10"3 20.7
'"Hg 1/2 16.87 0.87621937 4.8457916 — 17.910822 Me2Hg 1.00 x 10"3 5.89
197 Au 3/2 100 0.191271 0.473060 54.7 -1.729 - 2.77 x IO’5 0.162
“’Hg 3/2 13.18 -0.7232483 -1.788769 38.6 6.611583 (CH3)2Hg 1.97 X IO"4 1.16
(2O3T1) 1/2 29.524 2.80983305 15.5393338 — 57.123200 T1(NO3)3 5.79 x IO"2 3.40 X 102
205yj 1/2 70.476 2.8374709 15.6921808 — 57.683838 T1(NO3)3 0.142 8.36 X 102
20’pb 1/2 22.1 1.00906 5.58046 — 20.920599 Me4Pb 2.01 x 10’3 11.8
209Bi 9/2 100 4.5444 4.3750 -51.6 16.069288 Bi(NO3)2 0.144 8.48 X 102
255U 7/2 0.72 -0.4300 -0.5200 493.6 1.8414 - - -
Примечания: Ядра, заключенные в скобки, не самые удобные для наблюдения спектров ЯМР данного элемента.
Источник: R. К. Harris, Е. D. Becker, S. М. Cabral de Menezes, R. Goodfellow, and P. Granger, (2001). NMR nomenclature. Nuclear spin prop-
erties and conventions for chemical shifts. Pure. Appl. Chem., 73, 1795-1818.
386________________6. Спектроскопия ЯМР на других важных ядах со спином %
7. Задачи и решения
7.1. Введение
Студент, приступающий к спектрометрической иден-
тификации, всегда задает вопрос: «С чего начать?».
Доброжелательный преподаватель мягко, но непре-
клонно отвечает «Начинайте с определения молеку-
лярной формулы». Почему? Просто потому, что это
самая доступная и полезная информация, для полу-
чения которой оправданы любые усилия. Молеку-
лярная формула несет в себе общее представление
о молекуле, т.е. о числе и типах составляющих ее
атомов, что дает индекс водородной ненасыщенно-
сти - суммарное число циклов, двойных и тройных
связей (см. раздел 1.5.3).
Определение молекулярной формулы начинается
с выявления пика молекулярного иона (см. раздел
1.5). Предположим обычную ситуацию, когда масс-
спектрометр высокого разрешения недоступен. До-
пустим, что пик с наибольшим m/z (за исключением
изотопных пиков) является молекулярным и его ин-
тенсивность достаточна для измерения относительных
интенсивностей изотопных пиков, при этих условиях
можно точно установить наличие и определить число
атомов S, Вг и С1. Далее нужно проанализировать
масс-спектр с точки зрения выявления надежно рас-
познаваемых фрагментов. Если масса молекулярного
иона нечетная, то молекула содержит нечетное число
атомов азота.
Трудности часто начинаются с неопределенно-
сти выбора пика молекулярного иона. Во многих
лабораториях химическая ионизация использует-
ся в качестве рутинного дополнения к ионизации
электронным ударом. Для решения более сложных
задач, конечно, желателен доступ к масс-спектрометру
высокого разрешения.
Обычные функциональные группы принято опре-
делять методом ИК-спектроскопии. Особо отметим
валентные колебания С-Н, О-Н, N-Н, а также при-
сутствие или отсутствие ненасыщенных функцио-
нальных групп.
Располагая этой информацией, исследуйте про-
тонный спектр ЯМР для ее подтверждения и выбора
следующих шагов. Если спектр позволяет, определите
по интегралам группы химически эквивалентных про-
тонов и их число. Проанализируйте картину спин-
спинового взаимодействия первого порядка и ха-
рактеристические химические сдвиги. С помощью
спектров l3C/DEPT определите число групп СН3, СН2,
СН и С. Разность между числами протонов, опреде-
ленными по интегралу и по спектру 13C/DEPT, дает
число протонов, связанных с гетероатомами.
Перекрывание сигналов в спектре ПМР - явление
обычное, но в спектрах ЯМР 13С при использовании
приборов с высоким разрешением абсолютное совпа-
дение сигналов неэквивалентных ядер происходит до-
вольно редко. Теперь, используя приложение А к гл. 1,
можно подобрать наиболее вероятную молекулярную
формулу(ы); далее для их сравнения определим для
каждой индекс водородной ненасыщенности. Кроме
затруднений, вызванных неразрешенными или пере-
крывающимися сигналами, из-за наличия элементов
симметрии молекулы могут возникать расхождения
между выбранной молекулярной формулой и числом
протонов и ядер 13С. Эту информацию можно ис-
пользовать для осмысления молекулярной структуры.
Решение задач способствует формированию у сту-
дентов собственных подходов. Чтобы привить навыки
практического использования новейших методов, в не-
которых задачах иногда дается больше информации,
чем необходимо для их решения. В других задачах
встречаются такие затруднения, которые воспроиз-
водят реальные условия. Общая стратегия решения
состоит в сопоставлении спектров и выявлении наи-
более очевидных особенностей. Далее на основании
данных одного из спектров предположить структуру,
а другие спектры привлечь для ее подтверждения
или опровержения и, если необходимо, изменить
структуру. Такой подход обладает эффектом синер-
гизма-общая информация превышает информацию
суммы отдельных частей.
При доступном в настоящее время высоком раз-
решении многие спектры ЯМР являются спектрами
первого порядка (по крайней мере приблизительно)
и могут анализироваться визуально с учетом под-
сказок, которые дают масс-спектры и ИК-спектры.
388
7. Задачи и решения
Тем не менее перечитывание разделов 3.8-3.12 может
напомнить об осмотрительности.
В качестве примера рассмотрим два близких по
структуре соединения.
А В
В обоих кольцах происходят быстрые конформаци-
онные переходы, но только в соединении А протоны
каждой из СН2-групп совершают обмен, что приводит
к эквивалентности их химических сдвигов (энантиото-
пия). У молекулы А имеется совпадающая с плоскостью
страницы плоскость симметрии, относительно кото-
рой и происходит обмен протонов. В спектре ПМР
соединения А можно предсказать появление слева на-
право следующих сигналов: Н-5, триплет, два протона;
Н-3, триплет, два протона; Н-4, квинтет, два протона (в
предположении, что константы спин-спинового взаи-
модействия почти равны). При среднем разрешении
прибора наблюдается спектр первого порядка.
Молекула В в плоской конформации не имеет
элементов симметрии. Атом С-5 является хиральным,
и протоны каждой СН2-групп образуют диастеретоп-
ную пару. Сигнал каждого из протонов этой пары
имеет собственное значение химического сдвига.
Сигналы протонов Н-4, расположенных рядом с хи-
ральным центром, отчетливо разделены, но сигналы
Н-3 и при 300 МГц не разрешаются. Каждый из
протонов диастереотопной пары вступает в геми-
нальное взаимодействие с соседним протоном и в
вицинальное взаимодействие с другой величиной
константы спин-спинового взаимодействия, что при-
водит к появлению сложных мультиплетов.
При установлении структуры всегда нужно иметь
в виду возможность появления хирального центра.
Польза двумерных спектров станет более очевидной
при решении задач, помещенных в гл. 7 и 8. Пока не
предложены возможные структуры или фрагменты,
зачастую нет необходимости в детальной интерпрета-
ции. Особенности спектров, предсказанные для этих
структур или фрагментов, сравнивают с наблюдае-
мыми спектрами и, изменяя структуру, приводят ее
в соответствие со спектрами.
Эти рекомендации иллюстрируются представлен-
ными ниже задачами с разборами решений; задачи
расположены в порядке увеличения их сложности.
Задачи на отнесение в гл. 8 также расположены
в порядке увеличения сложности, что дает студенту
необходимые навыки.
Большинство студентов получают удовольствие от
решения этих усложняющихся задач. По мере анализа
спектров они начинают ощущать красоту химических
структур. Удачи в расследовании! Помните о хираль-
ности, диастереотопии, возможных спин-спиновых
взаимодействиях, двугранных углах, близких 90°.
и магнитной неэквивалентности.
И, наконец, каковы требования к доказательству
правильности структуры? В конечном счете это со-
впадение всех имеющихся в распоряжении спектров
со спектрами чистого индивидуального вещества,
зарегистрированными при одинаковых условиях на
тех же приборах. Очевидно, что возможны и опреде-
ленные компромиссы. Соответствие опубликованным
спектрам или спектральным данным приемлемо для
публикации, но неприемлемо для нового соединения,
которое в таком случае должно уже быть синтези-
ровано.
Существуют компьютерные программы для моде-
лирования спектров ПМР1). Если можно выполнить
точные измерения химических сдвигов сигналов
и констант спин-спинового взаимодействия всех
протонов, то расчетный спектр совпадет с реальным.
Во многих случаях некоторые из спиновых систем
будут первого порядка. Если этого нет, то можно вы-
полнить разумные оценки химических сдвигов и кон-
стант спин-спинового взаимодействия посредством
итерационной процедуры, которая будет приближать
оцениваемые величины к реальному спектру при
условии, что идентификация правильна.
Примерная последовательность логических и шагов
в процессе полного решения учебной задачи:
1. Показать, как получена молекулярная формула.
2. Вычислить индекс водородной ненасыщенно-
сти.
3. Выполнить отнесение характеристических полос
в ИК-спектре.
4. Провести отнесение сигналов всех протонов
в спектре ПМР.
5. Провести отнесение сигналов всех атомов угле-
рода в спектре DEPT ЯМР 13С.
6. Провести, по возможности, расчеты или оценки
величин Av/J.
7. Объяснить, по возможности, мультиплетность
сигналов.
1) Спектры ЯМР можно рассчитать либо на компью-
тере современного спектрометра, либо на персональном
компьютере. Например, см. программу NMR-SIM, разра-
ботанную Bruker BIOSPIN, Billerica, МА. см. также гл. 3
(раздел 3.5.3).
Задача 7.1. Решение
389
8. Найти все корреляции в двумерных спектрах.
9. Найти подтверждения структуры в масс-спектре
с ионизацией электронным ударом.
10. Рассмотреть возможные изомеры.
В каждой задаче этой главы сначала показаны
молекулярная структура и спектры, а затем следует
обсуждение. Чтобы избавить читателя от лишнего пере-
листывания страниц, на большей части из представ-
ленных спектров изображены структурные формулы.
Такое расположение материала поощряет студентов
самостоятельно находить экспериментальные связи
между структурой молекулы и хорошо известными
особенностями спектров. С учетом данного вступления
последующее обсуждение будет более полезным.
Задача 7.1. Решение
Все указывает на небольшой размер молекулы. В масс-
спектре с ЭУ нет пиков с m/z более 69, но это не пик
молекулярного иона, так как следующий ближайший
пик с m/z 55 соответствует предполагаемой потере
массы 14 а.е.м. В масс-спектре с ХИ основной пик m/z
71 можно отнести к псевдомолекулярному иону М+1.
Таким образом, молекулярную массу этого соединения
принимаем за 70 а.е.м. Широкая полоса валентных
колебаний О-Н в области 3350 см-1 и сильная поло-
са валентных колебаний С-О около 1049 см-1 в ИК-
спектре указывают на спирт.
В спектре ЯМР *Н наблюдаются классические
мультиплеты первого порядка. Слева направо сигналы
с интегральными интенсивностями таковы: триплет (2),
синглет (1), триплет дублетов (2), триплет (1), что ука-
зывает на шесть атомов водорода. В спектре ЯМР
DEPT 13С четыре сигнала атомов углерода располага-
ются слева направо следующим образом: С, СН, СН2,
СН2. Такое расхождение в числе протонов означает,
что один из них связан с гетероатомом. Сигнал около
2.68 м.д. в спектре ПМР относится к гидроксильной
группе и объясняет разницу между числом протонов,
проявляющихся в спектре ПМР, и числом протонов,
определенным из спектра ЯМР 13С.
Теперь предположение о том, что m/z 70 относится
к молекулярному иону, становится вполне достоверным.
Принимаем в качестве молекулярной формулы С4Н6О,
для которой степень водородной ненасыщенности равна
двум. Выбор имеется следующий: две двойных связи,
одна двойная связь и кольцо, два кольца и одна тройная
связь. Рассмотрим эти возможности по порядку.
Две двойные связи. Попадает ли какой-либо из
сигналов протона или атома углерода в обычную об-
ласть для алкенов? Внимательное чтение гл. 3 и 4 от-
вергает эту возможность и оставляет выбор между
кольцом и тройной связью.
Исключить кольца только на основании химических
сдвигов затруднительно, но спин-спиновые взаимо-
действия в этом случае было бы трудно объяснить.
Рассмотрим тройную связь.
Тройная связь идентифицируется на основе хи-
мических сдвигов протонов и 13С. Далее зададимся
вопросом о положении этой связи: находится она на
конце цепи или в середине. Другими словами, есть
ли в молекуле алкиновый протон?
Н—С=С-R или R—С=С—R'
Спектр ЯМР 13С дает недвусмысленный ответ на
этот вопрос. В нем наблюдаются два сигнала в об-
ласти алкиновых атомов углерода. Пик при 70 м.д.
примерно такой же высоты, как пики СН2, но пик при
81.2 м.д. значительно менее интенсивен, что говорит
об отсутствии связи с протоном. Пик около 70 м.д.
в спектре ЯМР 13С DEPT указывает на СН-группу.
Теперь можно записать два структурных фрагмента:
Н-С=с- и —СН2—ОН
Введение недостающей группы СН2 завершает
структуру молекулы:
Н—С^С—СН2—СН2—ОН
Эта структура полностью согласуется со спектрами
ЯМР !Н и 13С DEPT. В спектре ПМР превосходно
наблюдается дальнее спин-спиновое взаимодействие
через тройную связь (от Н-4 до Н-2), которое рас-
щепляет компоненты триплета в дублеты.
Возвращаясь к ИК-спектру, можно заметить
сильную полосу валентных колебаний Н-С= при
3294 см-1, перекрывающуюся с полосой О-Н. В об-
ласти 640 см-1 наблюдается интенсивная полоса
деформационных колебаний группы =С-Н, а по-
лоса валентных колебаний ОС проявляется при
2117 см-1 в виде слабой, но различимой полосы.
Некоторые из интенсивных пиков в масс-спектре
отнести трудно, так как в молекуле имеется две близко
расположенные функциональные группы. Хотя это
и тривиально, проверку отнесений сигналов прото-
нов и их мультиплетное™ можно предложить для
студентов в качестве упражнения. Точно так же про-
верку отнесений в спектре ЯМР 13С DEPT можно
оставить для студентов.
390
7. Задачи и решения
Масс-спектр с ХИ (газ-реагент — метан)
Задача 7.1 А
Задача 7.1. Решение
391
Спектр ЯМР *Н (600 МПх)
3.7 3.6 3.5 3.4 3.3 3.2 3.1 3.0 2.9 2.8 2.7 2.6 2.5 2.4 2.3 2.2 2.1 2.0 м.д.
Спектры ЯМР 13С и DEPT (150.9 МП<)
। • • । । । • । 1 । • । । • । • । • । । । • । ।
85 80 75 70 65 60 55 50 45 40 35 30 25 20 м.д.
Задача 7.1 Б
392
7. Задачи и решения
Задача 7.2. Решение
Относительно сильный пик с m/z 140 в масс-спектре
целесообразно выбрать в качестве молекулярного,
так как более тяжелых фрагментов нет, а пик с m/z
125 отвечает потере группы СН3. Поскольку 140 —
число четное, в молекуле может содержаться 0,2,4...
атомов азота, для начала предположим его отсутствие.
Очень низкие интенсивности пиков М+1 и М+2 ис-
ключают присутствие атомов S, С1 и Вг.
Сильная полоса при 1716 см-1 в ИК-спектре свиде-
тельствует о наличии карбонильной группы. Две рез-
кие полосы при 1647 см-1 и 1620 см-1 указывают на
присутствие одной или более углерод-углеродных
двойных связей (С=С), возможно, сопряженных (см.
раздел 2.6.4.1).
Из спектра ЯМР *Н следует, что в молекуле име-
ется шесть типов протонов, интенсивности сигналов
которых изменяются по шкале слева направо в от-
ношении 1:2:1:2:3:3, что дает в сумме 12 протонов.
В спектре ЯМР 13С насчитывается восемь пиков (в
предположении, что одному типу атомов углерода
соответствует один пик), из спектра 13С DEPT, счи-
тая слева, определяем следующие фрагменты: С=О
(подтверждается ИК-спектром), СН, СН, СН, СН, СН2,
СН3, СН3. На основе приведенной информации мож-
но записать брутто-формулу С8Н12О с молекулярной
массой 124, которая на 16 единиц меньше, чем m/z
140 для молекулярного иона. Не соответствует ли
эта разница массе второго атома кислорода в мо-
лекулярном ионе?
Это действительно так. Химический сдвиг атома
углерода СН2-группы при 60 м.д. свидетельствует о на-
личии структурного фрагмента -(С=О)ОСН2- (см. табл.
4.20). Химический сдвиг карбонильного атома углерода
в спектре 13С (168 м.д.) соответствует сложному эфиру
карбоновой кислоты. Из сказанного следует, что уточ-
ненная молекулярная формула С8Н12О2 с показателем
ненасыщености по водороду, равным трем.
Крайний триплет справа в спектре ЯМР !Н
принадлежит метильной группе, непосредственно
связанной с дезэкранированной группой СН2 (ква-
друплет). Спектр COSY подтверждает это отнесе-
ние. Таким образом, мы приходим к следующему
структурному фрагменту на одном конце молекулы:
-(С=О)ОСН2СН3.
В спектре 13С DEPT в области от -119 до -145 м.д.
наблюдаются четыре пика алкеновых групп СН.
При -18.5 м.д. наблюдается сигнал оставшейся груп-
пы СН3, в спектре ЯМР !Н она проявляется в виде
дублета при -1.8 м.д., что указывает на ее связь
с одной из четырех групп СН.
Кажется, что на этом этапе у нас имеются осно-
вания для предположения о молекулярной структу-
ре: уже есть структурный фрагмент одного конца,
четыре группы СН с концевой метильной группой,
не забываем также о двух двойных связях. С неко-
торой осторожностью мы предлагаем следующую
структуру:
О
СН3—СН=СН—СН=СН—С—О—СН^—СН3
65 4 321 78
Теперь используем комбинированные результаты
ЯМР ’Н и 13С DEPT. В спектрах *Н мультиплеты
первого порядка обычно разрешаются хорошо, а муль-
типлеты более высоких порядков разрешаются плохо.
В данном спектре имеется пять мультиплетов первого
порядка и два перекрывающихся мультиплета более
высокого порядка.
Протоны этильной группы проявляются в виде
триплета при -1.2 м.д. и дезэкранированного ква-
друплета около -4.1 м.д. Другая метильная группа
проявляется в виде дублета при -1.8 м.д., возникаю-
щего при спин-спиновом взаимодействии с одним
из четырех алкеновых протонов СН. Прежде чем
интерпретировать мультиплеты высоких порядков,
обратимся к двумерным спектрам.
В спектре COSY видно спин-спиновое взаимодей-
ствие одной из двух СН-групп, дающих перекрываю-
щиеся сигналы Н-5 и Н-4 с центром около -6.1 м.д.
(обозначены Н-5 и Н-4) с группой СН3, дающей,
дублет при -1.8 м.д. Это подтверждает высказанное
ранее предположение о терминальном положении
данной группы СН3. Протон Н-5, кроме того, взаимо-
действует с протоном Н-4 соседней СН-группы, кото-
рый, в свою очередь, взаимодействует с СН-группой
Н-3 с сигналом при -7.2 м.д. Несколько уширенный
дублет при -5.7 м.д. (обозначенный Н-2) возника-
ет в результате взаимодействия с Н-3 и дальнего
спин-спинового взаимодействия. Сказанное можно
обобщить следующим образом:
О
СН3—СН=СН—СН=СН—С—О—СН2—СН3
1.8 6.1 7.2 5.7 4.1 1.2 м.д.
654321 78
На основании отнесения всех сигналов протонов
и прямых корреляций между сигналами атомов угле-
рода и связанных с ними протонов в спектре HMQC
можно отнести сигналы всех атомов углерода, кро-
ме четвертичного (последнее тривиально в данном
случае). Интересный пример корреляционного соот-
Задача 7.2. Решение
393
Масс-спектр
&
Й
О
о
к
S
о
К
р
100:
50 :
411
о
Н Н II
6 /С^4^.С^2
Н3С 5
НН9-
Этиловый эфир
сорбиновой кислоты
ч СН2 8
С 3 С 1 О' 7 'СН3
67-1
Г97
125-1
140-1
ш-!
40 50 60 70 80 , 90 100 110 120 130 140
m/z
ИК-Спектр
Волновое число, см1
Спектр ЯМР *Н (600 МП<)
—I—,—,—,—,—I—,—,—,
3 4,5 2 7 6 8
। • • । • • । । • • । • • । • • • • । • • । • 1 • • । • • । • ' । • । • ।
7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 м.д.
Задача 7.2А
Спектр ЯМР с гомоядерной «развязкой» (600 МПд)
3700 3680 3660 3640 3620 Гц
1____,___I_____,__I___.____I___.____I___.____I___.___
Облучается Облучается
н3с-сн=сн-сн=сн—С-ОС2Н5
J\J ЦхМлЛл.
л.ЛЛ._1л
Облучается
I О
н3с-сн=сн-сн=сн—с-ос2н5
Облучается
н3с-сн=сн-сн=сн—с-ос2н5
О
Спектр ЯМР ’Н (600 MIU)
М.Д.
о
н II
4 С<2 Ji
Спектры ЯМР 13С и DEPT (150.9 МПд)
Н
6/С^.4 С^2Х СН2 8
Н3С 5 С З Т1О'7 'СН3
н н
Этиловый эфир
сорбиновой кислоты
U
CDC13
I 1111___________________________। ____________L_
....1 3 5 4 2 7 6 8
170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 м.д.
Задача 7.2Б
Задача 1.2. Решение
395
6
F2
7|
4,5 2
4 I
3
М.Д. л
Спектр COSY (600 МПд)
Спектр HMQC (600 МПд)
20-
- 20
40-
- 40
60-
- 60
80-
F1
100-
120-
140-
160-
7
6
150 ф-
6.2
м.д.
4
130-=
140-
-100
-120
• ••НН
-140
6.1
м.д.
80
-160
5 4
F2
3
2
1
м.д.
----------------7
---- CDC13
2
- 4
Задача 7.2В
396
7. Задачи и решения
ношения перекрывающихся сигналов двух протонов
Н-4 и Н-5 изображен на врезке в спектре HMQC.
Несмотря на то что эти сигналы в спектре ЯМР
!Н перекрываются, в спектре HMQC они хорошо
разрешены, так как и в спектре 13С они разрешены.
Остается один важный вопрос о конфигурациях
двойных связей: Z (цис) или Е (транс). Ответ на него
дает измерение константы J для олефиновых про-
тонов. Очевидной отправной точкой является дублет
Н-2, который возникает при взаимодействии с Н-3.
Величина J составляет около 16 Гц, что попадает
в область для Е-конфигурации двойных связей (см.
приложение Е к гл. 3).
Сложные перекрывающиеся мультиплеты
Н-4 и Н-5 не слишком привлекательны для анализа.
Протон Н-3 проявляется в виде пары дублетов, возни-
кающих в результате взаимодействия с Н-2 с транс-
константой 16 Гц и с Н-4 через одинарную связь
с константой 10 Гц. К сожалению, константу взаи-
модействия протонов при двойной связи С4-С5 из-
мерить непросто. Заслуживает внимания результат
подавления спин-спинового взаимодействия (см. раз-
дел 3.15). Облучение на резонансной частоте протона
Н-6 значительно упрощает картину перекрывания
сигналов Н-5 и Н-4, а с низкочастотного края муль-
типлета остается несколько искаженный дублет с J
16 Гц. Облучение на частоте Н-3 также упрощает этот
мультиплет, но теперь дублет с J 16 Гц появляется
с высокочастотной стороны. Одновременное облуче-
ние на двух частотах (Н-3 и Н-6) приводит к двум
дублетам с J 16 Гц. Распределение интенсивностей
в этих дублетах неидеально из-за малой величины
отношения Av/J. Теперь не остается сомнений, что обе
двойные связи имеют Е-конфигурации.
Задача 7.3. Решение
Сигнал средней интенсивности с m/z 150 в масс-
спектре, несомненно, принадлежит молекулярному
иону; появление основного пика с m/z 135 разумно
объяснить потерей СН3-группы. Распределение изо-
топных пиков в области молекулярного иона под-
тверждает отсутствие атомов S, С1 или Вг. Предпо-
ложим, что четный молекулярный пик указывает на
отсутствие атома N. Если это так, то, в соответствии
с приложением А к гл. 1, молекулярные формулы мо-
гут быть следующие: С6Н14О4, С8Н6О3, С9Н10О2 или
С10Н14О. В ИК-спектре выделяется широкая интенсив-
ная полоса валентных колебаний ОН при 3464 см-1.
Возникает вопрос о присутствии или отсутствии аро-
матического кольца. Положительный ответ на этот
вопрос дают сигналы в области ароматичности в спек-
трах ЯМР ’Н и 13С. Сильные полосы в области 1600—
600 см-1 в ИК-спектре и пики с m/z 77 и 91 в масс-
спектре подтверждают это заключение. Если есть
ароматическое кольцо, то возникает вопрос о месте
гидроксильной группы: присоединена она к кольцу
с образованием фенола или нет?
В спектре ЯМР !Н наблюдается семь типов прото-
нов, сигналы которых, если двигаться по шкале слева
направо, имеют следующие интегральные интенсив-
ности: 1:1:1:1:1:3:6. В сумме получается 14 протонов.
Дублет от шести протонов при 1.25 м.д., вероятно,
обусловлен двумя эквивалентными метильными
группами изопропильного остатка, метановый про-
тон проявляется в виде септета при 3.2 м.д.
В спектре ЯМР 13С наблюдается 9 сигналов,
но один из них (23 м.д.) подозрительно интенси-
вен, и так как он коррелирует с дублетом шести
протонов в спектре HMQC, мы делаем вывод о том,
что два сигнала метильных групп перекрываются,
откуда следует, что в молекуле 10 атомов углерода.
Сигналы в спектре 13С DEPT слева направо распола-
гаются в следующем порядке: С, С, С, СН, СН, СН,
СН, СН3(х2), СН3; к этому еще добавляется группа
ОН. Для массы молекулы 150 а.е.м. наиболее при-
емлемой оказывается молекулярная формула С10Н14О
с индексом водородной ненасыщенности, равным
четырем. Степень ненасыщенности полностью со-
ответствует ароматическому кольцу - три двойных
связи и один цикл. В спектре ЯМР 13С наблюдаются
сигналы в ароматической и алифатической областях.
В области ароматических сигналов имеется три
слабых сигнала четвертичных атомов углерода и три
более интенсивных сигнала, обусловленных связанны-
ми с протонами атомами углерода. Сигнал при 153 м.д.
относится к самому дезэкранированному атому угле-
рода, соединенному с ОН-группой (см. табл. 4.12).
В области алифатических сигналов в спектре ЯМР
1Н, где проявляются метильная и изопропильная группы,
наблюдаются (слева направо по шкале) септет одного
протона (СН), синглет трех протонов (СН3) и дублет
шеста протонов. Этот дублет возникает при взаимо-
действии двух идентичных СН3-групп и СН-группы,
что подтверждает наличие изопропильного заместителя.
Теперь займемся размещением двух алкильных
заместителей по отношению к гидроксильной группе
и сделаем это, анализируя химические сдвиги и кон-
станты спин-спинового взаимодействия трех кольце-
вых протонов. Предположим, что сигнал с 5 6.6 м.д.
относится к ор/ио-протону по отношению к ОН-группе
(см. диаграмму Г. 1 приложения к гл. 3). Так как этот
сигнал является уширенным синглетом, то рядом нет
атома водорода, но есть в лгетия-положении с констан-
той спин-спинового взаимодействия слишком малень-
Задача 7.3. Решение
397
кой, чтобы сигналы разрешились. Так как в спектре
наблюдается только один протон в ор/ио-положении
по отношению к заместителю ОН, то другое орто-
положение должно быть занято либо метильной, либо
изопропильной группой.
Дублет резких полос с 8 7.1 м.д. и J около 8 Гц
обусловлен ароматическим протоном с одним орто-
спин-спиновым взаимодействием, Так как сигналы
узкие, лге/иа-взаимодействие отсутствует. Химический
сдвиг этого сигнала указывает на л/е/иа-положение
протона по отношению к ОН-группе; алкильные
группы оказывают небольшое влияние на химиче-
ский сдвиг (см. диаграмму Г.1 приложения к гл. 3).
Дублет уширенных полос с 5 6.75 м.д. принадлежит
протону в «ара-положении по отношению к группе
ОН, взаимодействующему с орто- и л/е^а-протонами,
в последнем случае слабо. Теперь нужно сделать
выбор между структурами I и II.
Спектр COSY подтверждает предложенные от-
несения и показывает дальнее взаимодействие (4J)
протонов метильной группы с протонами Н-4 и Н-6.
Интересно отметить, что такого же дальнего взаимо-
действия не наблюдается для изопропильного протона
СН и протона Н-3, возможно, из-за высокой мульти-
плетности этого сигнала и размытости кросс-пика,
что делает его невидимым. лге/ла-Взаимодействие
(4J) между Н-6 и Н-4 и opwa-взаимодействие (3J)
между Н-4 и Н-3 для протонов ароматического кольца,
как и следовало ожидать, проявляется в спектре !Н.
Структура I (тимол) теперь становится предпочти-
тельной. Отметим, что дальнее взаимодействие между
заместителем СН3 и протонами Н-4 и Н-6 в спектре
ЯМР *Н не наблюдается.
Спектр HMQC отражает спин-спиновое взаимо-
действие 7СН. Химические сдвиги ароматических
незамещенных атомов углерода можно оценить из
данных табл. 4.12 в гл. 4. Эти значения увеличиваются
в порядке С-6, С-4, С-3. Спектр HMQC подтверждает
эту последовательность для Н-6, Н-4, Н-3. Сигналы
незамещенных ароматических атомов углерода могут
коррелировать с надежно отнесенными сигналами
алифатических протонов. Остаются пока не отнесе-
ными химические сдвиги замещенных ароматических
атомов углерода.
Спектр НМВС позволяет проводить корреляцию
между изолированными спиновыми системами про-
тонов, которые связаны через «изолирующие» мо-
стиковые атомы, например О, S, N или четвертичные
атомы углерода.
т
с—о—с с—с—с
Н R Н
3J 3J
Даже в молекуле среднего размера количество
констант спин-спинового взаимодействия 2JCH и 3</сн
может обескураживать. С чего начать?
Зададимся таким важным вопросом, как опреде-
ление положения алкильных заместителей в арома-
тическом кольце. Спектр COSY отражает дальнее
спин-спиновое взаимодействие метильных протонов
с протонами в ор/ио-положении, чего не наблюдает-
ся для изопропильной группы. Это взаимодействие
подтверждается при проведении в спектре НМВС
вертикальной линии от изопропильного септета вниз,
которая в четырех местах пересекает сигналы, связы-
вающие этот СН-протон с С-8,9 (2J), C-2 (2J), C-3 (3J)
и C-1 (V) в структуре тимола. Это выглядит довольно
убедительно. В качестве избыточной информации от-
метим, что в спектре НМВС сигналы протонов ме-
тильного заместителя коррелируют с С-6 (V), С-4 (V)
и С-5 (2<7). Сигналы шести протонов изопропильной
группы коррелируют с С-7 (V) и C-2 (3J). Интересно
отметить корреляции Н-8 с С-9 (3<7) и Н-9 с С-8 (3J).
Польза спектра НМВС заключается в корреля-
ции сигналов четвертичных атомов углерода С-1,
С-5 и С-2 с уже отнесенными сигналами протонов. Ранее
выполненное отнесение С-1 на основании его химиче-
ского сдвига надежно, но отнесения С-5 и С-2 нужно
еще подтвердить и корреляциями. Предлагаем выпол-
нить это студентам в качестве упражнения.
На примере приведенных выше корреляций были
показаны взаимодействия через связи, включающие
четвертичные атомы углерода. Наконец, два последних
замечания: 1) на спектре НМВС имеется четыре
контура (отмечены стрелками), отражающие спин-
спиновые взаимодействия с большими константами
1JCH, которые полностью не подавляются. Эти дублеты
СН остаются, вероятно, из-за большого расстояния
между сигналами протонов, и ими можно пренебречь.
2) Нужно отметить корреляции протона гидроксиль-
ной группы ОН с С-6, С-2 и С-1. Корреляции с про-
тонами ОН могут быть очень полезными, но в спектре
НМВС наблюдаются редко, так как обычно сигналы
слишком широки для их детектирования.
398
7. Задачи и решения^
bJ О О м QO ^1 QO
--1-1--1-1--1--1-1--1-1--1-1--1-1--1-1--1-1--1-1--1-1--1-1--1-1--1--1-1--1-1--1-1--1-1--1-1
4000 3000 2000 1000
Волновое число, см'1
Спектр ЯМР ’Н (600 МПд)
7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 м.д.
Задача 7.3А
Задача 7.3. Решение
399
Спектры ЯМР 13С и DEPT (150.9 МП<)
CDCI3 * 1
_____L
1 5 2 3 4 6 7 8,9
I"''..।.....।........।....।.......।.....।......।......।......।......।......।.......।......।.....।......।
150 140 130 120 110 100 90 80 70 60 50 40 30 20 м.д.
Задача 7.3Б
400
7. Задачи и решения
Спектр HMQC (600 МПд)
7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 м.д.
F2
Задача 7.3В
Задача 7.4. Решение
401
Задача 7.4. Решение
Вполне вероятно, что малоинтенсивный пик с m/z
154 в масс-спектре принадлежит молекулярному иону
(интенсивности сигналов в отмеченной области умно-
жены на 10).
Интенсивность пика с m/z 139, образующегося при
потере метильной группы, также невелика. Внима-
тельный читатель отметит пик М-18 с m/z 136 и не-
медленно подтвердит это наблюдение интенсивной
широкой полосой ОН при 3321 см-1 в ИК-спектре;
полоса 1003 см-1, вероятно, относится к валентным
колебаниям С-О. Вновь, как и в задаче 7.3, возни-
кает вопрос: спирт или фенол данное соединение?
Ароматическое оно или нет?
Очень низкая интенсивность пика молекулярного
иона совместно с потерей воды подсказывает, но не
доказывает, что данное вещество, скорее, спирт,
чем фенол. В этом месте имеет смысл допустить
возможность того, что основной пик (m/z 69) обуслов-
лен фрагментом С5Н9+ и возникает непосредствен-
но из молекулярного иона по преимущественному
механизму. Если это так, то исследуемая молекула
содержит хотя бы одну двойную связь.
Спектры 13С DEPT показывают, что имеется 10 не-
эквивалентных атомов углерода, 17 присоединенных
к ним атомов водорода, и они располагаются по
шкале слева направо в следующем порядке: С, С,
СН, СН, СН2, СН2, СН2, СН3, СН3, СН3. Первые четы-
ре атома с очень высокой вероятностью относятся
к олефиновым. Если учесть гироксильную группу,
то в первом приближении молекулярная формула,
согласующаяся с молекулярным ионом с m/z 154,
имеет вид С10Н18О. Индекс ненасыщенности по
водороду равен двум и указывает на две двойные
связи, что подтверждает четыре алкеновых атома
углерода.
Теперь, используя информацию из спектров ЯМР,
можно установить полную структуру. Сначала при-
меним традиционный подход, а потом используем
современные двумерные методики ЯМР. Отметим,
что стереохимию этой молекулы невозможно уста-
новить, используя только обычные спектры и ЯМР
!Н и 13С.
Начнем со спектра ЯМР ’Н. Интегрирование сиг-
налов слева направо по шкале дает следующее соот-
ношение: 1:1:2:2:2:6:3:1, что соответствует 18 атомам
водорода в молекулярной формуле. Этот результат
можно записать в другом виде: СН, СН (алкеновые),
СН2 (дезэкранированная ОН), СН2, СН2, СН3, СН3 (поч-
ти перекрывающиеся), СН3, ОН. Из спектра 13С DEPT
видно, что два атома углерода не связаны с атомами
водорода. Этот же спектр указывает на три разные
метильные группы, в то время как в спектре ЯМР
!Н сигналы Н-9 и Н-10 перекрываются даже при
рабочей частоте 600 МГц. Однако при сильном рас-
тяжении шкалы химических сдвигов видно, что они
перекрываются не полностью, и проявляется дальнее
спин-спиновое взаимодействие.
Из спектра 13С DEPT следует, что гидроксильная
группа соединена с метиленовой, сигнал которой
в спектре ’Н имеет вид дублета при 4.15 м.д. с тонкой
структурой (из-за дальнего взаимодействия с Н-10).
Так как в данной структуре только метиновые группы
алкеновые (см. также 13С DEPT), соединение должно
быть аллиловым спиртом. Относительное дезэкрани-
рование и отсутствие вицинального спин-спинового
взаимодействия для трех метильных групп подсказы-
вает нам поместить их на алкеновые атомы углерода.
В результате получаются две возможные структуры
аллиловых спиртов:
Н С Н Н3С Н
113 \ I 31 I
С=С—СН2—ОН или — С=С—СН2— ОН
Н3С
Структура слева-молекула без свободных валент-
ностей, и мы ее отвергаем в качестве спиртового
«фрагмента». Фрагмент справа кажется правдопо-
добным.
Н3С Н Н3С Н
I I 3| I
Нзс—С=С— и —с=с—сн2—ОН
Другой «фрагмент» искомой молекулы можно
сконструировать из второй двойной связи с двумя
метильными группами, не имеющими вицинального
взаимодействия, т.е. находящимися в геминальном по-
ложении, и метикового протона алкенового фрагмента.
Этот фрагмент показан выше слева. Теперь остается
соединить эти два фрагмента недостающими метилено-
выми группами, чтобы получить следующую структуру.
Н3С Н Н3С Н
II II
н3с—с=с—сн2—сн2—с=с—сн2—он
Это соединение - дважды ненасыщенный терпе-
новый спирт. Можно принять, что его стереохимия
совпадает со стереохимией обычного терпена:
402
7. Задачи и решения
В этой структуре нет хирального центра. Име-
ется совпадающая с плоскостью страницы пло-
скость симметрии, поэтому протоны метиленовых
групп взаимозаменяемы (энантиотопны). Протоны
Н-4 в результате спин-спинового взаимодействия
с двумя протонами Н-5 проявляются в виде ис-
каженного триплета. Протоны Н-5 дополнительно
взаимодействуют с Н-6 с несколько отличающейся
константой спин-спинового взаимодействия и дают
искаженный квадруплет. Вклад в это искажение вно-
сит и малая величина отношения Av/J для Н-4 и Н-5.
Обе метильные группы Н-8 и Н-9 лежат в плоскости
симметрии, поэтому не взаимозаменяемы.
Сказанное выглядит убедительно, но доказательства
структуры основаны исключительно на химических
сдвигах и на характере спин-спинового взаимодей-
ствия. Этого недостаточно, и однозначный результат
может дать только детальный анализ данных дву-
мерных экспериментов.
Лучший способ установления структуры основан
на извлечении богатой информации из двумерных
спектров и в меньшей степени из одномерных спек-
тров. Мы получаем молекулярную формулу, как уже
сделали это выше, отмечая присутствие гидроксиль-
ной группы на основании ИК-спекгра и подтверждая
спектрами ЯМР 13С DEPT и !Н. Затем обращаемся
к анализу двумерных спектров. Диастереотопия про-
тонов доказывается спектром HMQC, если обнару-
живается корреляция сигналов двух протонов с раз-
ными химическими сдвигами с одним сигналом 13С.
В данном случае таких диастереотопных корреляций
не наблюдается.
Информация о связанности сигналов из спектра
COSY наиболее обнадеживающая, и начинать удоб-
нее с него. Для удобства пики на главной диагона-
ли пронумерованы. Отправной точкой для анализа
данных COSY является связанная с группой ОН
метиленовая группа с 5 4.15 м.д. (Н-1). (Если нуж-
но убедиться что этот пик относится к связанной
с ОН метиленовой группе, то подтверждение можно
найти в спектрах HMQC и 13С DEPT). Очевидны
корреляции, обусловленные вицинальным взаимо-
действием с алкеновым метиновым протоном (Н-2)
(5 5.41 м.д.) и дальним взаимодействием с метиль-
ной группой (Н-10) (5 1.68 м.д.). Откуда мы знаем,
что мультиплет с 5 5.41 м.д. относится к метановому
протону? Используя естественную взаимосвязь спек-
тров, наблюдаем корреляцию мультиплета протонов
с 5 5.41 м.д. с углеродным сигналом при 123 м.д.
в спектре HMQC, а спектр 13С DEPT, в свою очередь,
подтверждает, что сигнал при 123 м.д. обусловлен
метиновой группой. Подобным образом сигнал с 5
1.68 м.д. коррелирует с сигналом метильного атома
углерода в спектре HMQC.
Из спектра COSY видно также слабое дальнее
взаимодействие между метильными протонами
Н-10 и метиленовыми Н-4 с сигналом с 5 2.11 м.д.
(Студенту предлагается подтвердить, что мульти-
плет с 5 2.11 м.д. обусловлен метиленовым фраг-
ментом, перейдя от анализа спектра COSY к HMQC
и 13С DEPT.) Другую (последнюю для Н-4) кор-
реляцию - между Н-4 и Н-5 - распознать трудно,
поскольку кросс-пики находятся почти на диагона-
ли. Сигнал метиленовой группы Н-5 коррелирует
с сигналом другого алкенового метинового атома
водорода (Н-6) с 5 2.03 м.д. Для Н-6 наблюдаются
еще две корреляции, отражающие дальние взаимо-
действия с метильными группами Н-8 и Н-9. Дру-
гие корреляции в спектре COSY не проявляются.
Гидроксильные протоны из-за быстрого обмена
кросс-пиков, естественно, не дают.
К данному моменту мы отнесли сигналы всех
протонов, но пока не смогли различить метильные
группы Н-8 и Н-9. Так как отнесение сигналов всех
протонов известно, то задача «привязки» этих сиг-
налов к спектру 13С посредством спектра HMQC
тривиальна. С четвертичными атомами углерода
С-3 и С-7 протоны не связаны, поэтому для них
в спектре HMQC корреляций нет. Для корреляций
четвертичных атомов углерода можно использовать
спектр НМВС, но в данной задаче мы используем
взаимосвязи атомов углерода.
Спектр INADEQUATE устанавливает связи между
соседними атомами 13С. Это самый мощный метод
установления строения. В конце концов все структу-
ры органических соединений состоят в основном из
углеродных цепей и колец. На спектре INADEQUATE
проведены пунктирные линии, связывающие атомы
углерода, между которыми имеется корреляция. Нач-
нем с анализа трех атомов углерода метильных групп
С-8, С-9 и С-10. Мы видим, что атом С-10 связан
с С-3, а С-8 и С-9 связаны с алкеновым атомом угле-
рода С-7. Эти связи подтверждают отнесение С-10,
но мы пока не можем различить атомы С-8 и С-9.
Эти отнесения будут сделаны в следующем абзаце.
Если продолжить рассмотрение сигнала С-3, то про-
являются еще две связи: одна с алкеновым атомом
углерода С-2 (см. врезку на спектре) и другая с али-
Задача 7.4. Решение
403
Масс-спектр
10
50 60 70 80 90 100 ,110 120 130 140 150
ИК-Спектр "**
О\ О ОО О Ю ^1 СЛ CQ
—|—I—I—I—I—|—I—I—I—I—|—I—1—I—I—|—I—I—I—I—|—I—I—I—I—|—I—I—I—I—|—I—I—I—I—|—Г
4000 3000 2000 1000
Волновое число, см*1
Спектр ЯМР !Н (600 МПд)
|........I........I..... .......| | . ....|.........|........|.......... I ' гггги . . |..|
Задача 7.4А
404
7. Задачи и решения
F2
Спектры ЯМР 13С и DEPT (150.9 МПф
CDC13
____I_I_____________I__
3 7 62
"Г".I...I.|||(...........
140 130 120 ПО 100 90 80 70
1 4 5,9 8,10
I.....।.......।.......।......।.......।
60 50 40 30 20 м.д.
Задача 7.4Б
Задача 7.4. Решение
405
Задача 7.4В
406
7. Задачи и решения
Спектр INADEQUATE (600 Mill)
140 130 120 110 100 90 80 70 60 50 40 30 20 мд.
F2
Задача 7.4Г
Задача 7.4. Решение
407
Разностный спектр ЯМР с ЯЭО (600 МПд)
Облучается Н8
Облучается Н1
Облучается Н2
Облучается Н6
9,10 8
Г' ' -Гг, , . , . г, . j , , ..
1.7 1.6 м.д.
Облучается Н8
Облучается Н1
Облучается Н9
Облучается НЮ
2 б
...............।.......।.......I.......I
5.4 5.3 5.2 м.д.
10 9 8
।..........। 1 ‘ ' 1...।...........
1.7 1.6 м.д.
Задача 7.4Д
408
7. Задачи и решения
фатическим атомом углерода С-4. Анализ остальных
корреляций и их связи со структурой предлагается
выполнить студентам самостоятельно в качестве
упражнения.
До сих пор остаются нерешенными два вопро-
са: стереохимия двойной связи между атомами
С-2 и С-3, а также отнесения сигналов метильных
групп С-8 и С-9. Разностная спектрометрия с ис-
пользованием ядерного эффекта Оверхаузера (ЯЭО)
описана в разделе 3.16. Этот одномерный эксперимент
выявляет пространственную близость ядер
по усилению соответствующих сигналов благодаря
ЯЭО. Вычитание стандартного спектра ЯМР !Н из
спектра с ЯЭО дает разностный спектр, в котором
остаются только усиленные сигналы.
Проблема, с которой мы сталкиваемся при анализе
данной молекулы, состоит в поиске спектроскопи-
ческих различий между тризамещенными двойными
связями с £- и Z-конфигурациями и является не-
тривиальной. Это относится и к ранее упомянуто-
му поиску различия между метильными группами
Н-8 и Н-9. Для надежного результата мы сопоставим
спектры двух геометрических изомеров с двойной
связью С-2 - С-3: Е-изомера (гераниола) и Z-изомера
(нерола).
В верхней части рисунка приведены спектр ЯМР
!Н гераниола и разностный спектр, возникающий при
облучении ключевых групп протонов, а в нижней
части страницы - аналогичные спектры для геометри-
ческого изомера гераниола - нерола. При облучении
гераниола электромагнитным излучением с резо-
нансной частотой алкенового метинового протона
Н-2 не наблюдается усиления сигнала метильной
группы Н-10; и наоборот, облучение с частотой
Н-10 не приводит к усилению сигнала в спектре
ЯЭО протона Н-2. Следовательно, эти две группы
находятся по разные стороны двойной связи и ге-
раниол является Е-изомером. Это отнесение под-
тверждается тем, что облучение протонов аллильной
метиленовой группы Н-1 сопровождается усилением
в результате ЯЭО сигнала метильного протона Н-10;
таким образом, они расположены по одну сторону
от двойной связи. Так как сигналы метильных групп
Н-9 и Н-10 в протонном спектре перекрываются,
они подвергаются облучению одновременно и в
спектре ЯЭО видно усиление сигнала алкенового
метинового протона Н-6. Проверьте результат об-
лучения на частоте сигнала метинового протона Н-6.
Нередко в реальных исследованиях, особенно ка-
сающихся природных соединений, геометрические
изомеры в чистом виде отсутствуют (хотя в прин-
ципе их можно синтезировать). В учебных целях
здесь представлены спектры для нерола (см. с. 405).
В данном случае облучение алкенового Н-2 приводит
к усилению сигнала метильной группы Н-10, на осно-
вании чего мы делаем заключение о Z-конфигурации
двойной связи. Отнесения сигналов метильных групп
Н-8 и Н-9 предлагается студентам выполнить само-
стоятельно.
Возвращаясь к масс-спектру, теперь можно отнести
фрагментный пик (основной) с m/z 69 к аллильному
распаду алкена. Обычно использование такой схемы
распада для локализации двойной связи вызывает со-
мнение, но в случае гераниола разрыв бис-аллильной
связи между атомами С-4 и С-5 приводит к стаби-
лизированному фрагменту с m/z 69. Пример анало-
гичного аллильного разрыва представлен в разделе
1.6.1.2 для Р-мирцена с образованием фрагментов
с m/z 69 и 67.
Задача 7.5. Решение
Пик иона с m/z 149 в масс-спектре возникает в резуль-
тате потери молекулой СН3-группы (М-15) и указы-
вает на то, что ион с m/z 164 является молекулярным.
Отсутствие пика (М+2) свидетельствует об отсут-
ствии атомов С1, Вг и S. В ИК-спектре наблюдается
интенсивная полоса при 1697 см-1, подтверждающая
вместе с пиком 208 м.д. в спектре ЯМР 13С (в области
кетонов) наличие в молекуле группы С=О. В спектре
ЯМР !Н интегральные интенсивности соотносятся
как 1:1:2:2:2:2:3:3 (что соответствует 16 протонам).
В спектре ЯМР 13С наблюдаются сигналы 11 неэк-
вивалентных атомов углерода. Примем в качестве
пробной молекулярной формулы, согласующейся
с сигналом молекулярного иона с m/z 164, СнН16О.
Индекс водородной ненасыщенности равен четырем.
В спектре 13С DEPT слева направо проявляются сиг-
налы следующих групп: С, С, С, СН, СН, СН2, СН2,
СН2, СН2, СН3, СН3.
Три из четырех степеней ненасыщенности мож-
но отнести непосредственно на основании спектра
ЯМР 13С. Мы уже отмечали, что по величине хи-
мического сдвига карбонильный сигнал относится
к кетону. Четыре алкеновых сигнала в спектре ЯМР
13С отвечают двум двойным связям. В качестве пред-
положения отнесем четвертую единицу ненасыщен-
ности к циклу.
Из спектров 13С DEPT мы получили информа-
цию о молекулярном составе, что является важным
шагом в расшифровке структуры. Обратимся теперь
к двумерным спектрам, чтобы соединить фрагмен-
ты молекулы. По мере необходимости будем пере-
ходить от одного типа спектров к другому. Сигнал
более экранированной из двух метильных группп с
Задача 7.5. Решение
409
Н
Спектр ЯМР ХН (600 МГц)
Л " . ' Л л
I • I • I 1 . . I . . . . I . . . . 1 . I .... I ... .
3200 3150 Гц 1750 1700 Гц 1450 1400 Гц
5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 м.д.
Задача 7.5А
410
7. Задачи и решения
F2
Спектры ЯМР 13С и DEPT (150.9 МПд)
CDC13
1 ' 1 ' 1 ' 1 ' 1 1 1 200 180 160 140 120 100 80 Задача 7.5Б г ’ 1 ' 1 60 40 м.д.
Задача 7.5. Решение
411
Спектр HMQC (600 МПд)
м.д.:
205-
2104
215 I • । • । ‘ । • 1 । ' 1 1 1 । 1 1 1 1 । • । 1 । । ।
6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 м.д.
F2
Задача 7.5В
412
7. Задачи и решения
F2
Задача 7.5Г
Задача 7.6. Решение
413
5 0.93 м.д., который коррелирует с метильным атомом
углерода с сигналом 15 м.д. в спектре HMQC, явля-
ется хорошим началом. Эта метильная группа взаи-
модействует с метиленовой группой с сигналом с 5
2.12 м.д., как доказывает сильный кросс-пик в спек-
тре COSY. Углеродный сигнал этой группы в спектре
HMQC имеет величину химического сдвига 21 м.д.
Эта метиленовая группа коррелирует также с одной
из алкеновых метиновых групп в спектре COSY (при
5 5.33 м.д.). Сигнал метиленовой группы 5 2.12 м.д.
является квинтетом почти первого порядка.
Другая алкеновая метановая группа с сигналом
при при 5 5.18 м.д. коррелирует в спектре COSY
с первой алкеновой метановой группой (5 5.33 м.д.),
что подтверждает присутствие дизамещенной двойной
связи. Кроме того, метановая группа с сигналом при
5 5.18 м.д. взаимодействует с заметно дезэкраниро-
ванной метиленовой группой с сигналом 5 2.88 м.д.
Этот сигнал представляет собой уширенный дублет,
и другого вицинального взаимодействия на нем не
наблюдается. В соответствии со спектром COSY,
на этой метиленовой группе проявляется еще не рас-
смотренное нами дальнее взаимодействие с двумя
другими группами. Одна из них метиленовая груп-
па, а другая - метильная. Пока получается пент-2-
енильный фрагмент, который должен быть присоеди-
нен к одному из четвертичных алкеновых атомов
углерода. Уширенный синглет с 2.01 м.д. отвечает
метильной группе, связанной с другим четвертичным
алкеновым атомом углерода.
Если рассмотреть остальные фрагменты на осно-
вании данных спектра 13С DEPT (кетонная карбо-
нильная группа, две метиленовых группы, два чет-
вертичных алкеновых атома углерода.) и учесть,
что есть еще одна степень водородной ненасыщен-
ности для кольца, становится ясно, что следует по-
строить ненасыщенный пятичленный циклический
кетон. Дальнейший анализ позволит упорядочить
перечисленные структурные элементы. Во-первых,
спектр COSY указывает на то, что две метиленовые
группы (с 5 2.30 и 2.45 м.д.) находятся в вициналь-
ной связи, т. е. расположены по соседству. Во-вторых,
химический сдвиг одного из четвертичных атомов
углерода в спектре 13С составляет 170 м.д.; столь
сильное дезэкранирование можно объяснить толь-
ко сопряжением с карбонильной группой. Одна из
очевидных структур показана ниже. Спектр НМВС
(см. корреляции для карбонильного атома углерода)
подтверждает циклическую структуру, а отсутствие
асимметрического атома углерода (имеется совпа-
дающая с плоскостью страницы плоскость сим-
метрии) объясняет отсутствие диастереомерных
метиленовых групп.
___ Н
СН2 сн2—сн3
сн3
Прежде чем закончить анализ, проверим, суще-
ствуют ли структурные изомеры, которые необхо-
димо учесть при определении структуры? Да, если
поменять местами два заместителя (пент-2-енильную
и метильную группы), то новая структура также будет
соответствовать экспериментальным данным. И снова
сложный спектр НМВС разрешает проблему. Напри-
мер, метильный заместитель в кольце, имеющий сиг-
нал при 17 м.д., коррелирует только с метиленовой
группой с 5 2.45 м.д. Эта метиленовая группа нахо-
дится в Р-положении по отношению к карбонильной,
что полностью подтверждает предложенную выше
структуру. Имеются ли другие корреляции в спек-
тре НМВС, которые подтверждают эту структуру?
Предлагаем студентам самостоятельно выполнить
эти отнесения.
Задача 7.6. Решение
Последняя из рассмотренных в этой главе задач со-
вершенно отличается от разобранных ранее, поэтому
наш подход также изменяется. Данное соединение
является трипептидом, а расшифровка структуры
пептидов предполагает два разных уровня решения.
Первый - определение числа индивидуальных ами-
нокислотных остатков и их идентификация, второй -
последовательность их соединения (секвенирование).
Обе задачи не являются простыми, так как число
распространенных аминокислот более 20, а природа
пептидной связи такова, что они могут быть соеди-
нены в любом порядке.
Прежде чем переходить к решению, имеет смысл
обсудить некоторые методические вопросы. Масс-
спектр этого трипептида получен методом ЖХ-МС с
ионизацией электрораспылением (см. гл. 1). Электро-
распыление (ЭР) является «мягким» методом ио-
низации (подобно химической ионизации), который
подавляет или ограничивает фрагментацию и уве-
личивает интенсивность пиков псевдомолекулярных
ионов (в зависимости от числа зарядов z на ионе).
Эксперименты по ЯМР были выполнены для рас-
творов в смеси 95% Н2О и 5% D2O при 0° С. При-
чины использования таких растворителей и детали
экспериментов объяснены в разделе 5.11.
414
7. Задачи и решения
Спектр ЖХ-МС с ЭР дает М+1 с m/z /Л7, что со-
ответствует молекулярной формуле C12H22N6O6. По-
лучение этой формулы мы не рассматриваем, так как
в приложении А к гл. 1 приведены данные только до
250 а.е.м. Малоинтенсивный пик с m/z 369 (М+23)
обусловлен ионами натрия, которые всегда присут-
ствуют в водных растворах. Хотя фрагментация не-
значительна, наблюдаемые в масс-спектре фрагменты
могут оказаться весьма полезными для опытного ис-
следователя; некоторые из путей распада показаны
на масс-спектре (7.6А).
Для идентификации трех аминокислот использу-
ем информацию из спектров ЯМР !Н , 13С DEPT,
COSY, TOCSY и HMQC. Химические сдвиги протонов
аминокислот представлены в приложении И к гл. 3.
Пептиды являются хиральными молекулами, и все
метиленовые группы диастереотопны, даже группы
глицина. (Метиленовая группа в свободном глицине
энантиотопна.) Начинать анализ пептидов (и других
соединений, построенных из отдельных структурных
единиц, таких как олиго- и полисахариды) нужно со
спектра TOCSY. В двумерном варианте TOCSY по-
казывает корреляции между всеми спинами спиновой
системы, а корреляции со спинами вне этой системы
отсутствуют. В трипептиде три отдельные спиновые
системы, и их легко найти в спектре TOCSY Протон
N-Н с сигналом при 8.95 м.д. коррелирует с двумя
резонансами (3.96 и 4.13 м.д.),что выявляет спиновую
систему. Если перенести эту информацию на спектр
HMQC, то выяснится, что эти протонные резонансы
коррелируют с одним атомом углерода при 41.9 м.д.
(т.е. диастереотопной метиленовой группой). Эта ами-
нокислота идентифицируется как остаток глицина,
а корреляция с протоном N-Н амидной группы по-
зволяет сделать вывод, что остаток глицина не яв-
ляется N-концевым. (Этот вывод будет подтвержден
с использованием других методов.)
Другая спиновая система становится очевидной,
если начать с общепринятой отправной точки -
сигнала при N-H 8.26 м.д. Есть три корреляции
с этим сигналом для всех четырех фрагментов
этой спиновой системы. Если опять перенести эту
информацию прямо в спектр HMQC, можно найти
соответствующие углеродные сигналы. Конечно,
корреляция с резонансом N-Н отсутствует. Сигнал
протона при 4.41 м.д. коррелирует с резонансом
атома углерода при 52.4 м.д., а спектр 13С DEPT
подтверждает, что это сигнал метиновой группы.
Оставшиеся протоны дают сигналы 2.58 и 2.72
м.д., которые коррелируют с одним сигналом атома
углерода при 38.6 м.д. Спектр 13С DEPT подтверждает,
что это сигнал метиленовой группы. Этот остаток
идентифицируется как аспарагиновая кислота, и опять
можно сделать заключение, что это не N-концевой
остаток.
Отправной точкой для анализа последней спиновой
системы является сигнал протона N-Н при 7.35 м.д.
Эта спиновая система включает еще четыре протон-
ных резонанса; спектр HMQC и спектры 13С DEPT
указывают, что они соответствуют одной метиновой
и трем метиленовым группам. Таким образом, этот
аминокислотный остаток является аргинином.
Спектр COSY можно использовать для подтверж-
дения полученных результатов, хотя необходимости
в этом нет. Спектры COSY и TOCSY не являются
излишними и взаимно дополняют друг друга
по крайней мере в одном важном аспекте. Хотя
спектр TOCSY показывает все спины в спиновой
системе, он не отражает реальные спин-спиновые
взаимодействия. Например, поскольку протон N-H
с сигналом при 8.26 м.д. (в остатке аспарагиновой
кислоты) показывает корреляцию только с метиновой
группой в спектре COSY, то можно с уверенностью
заключить, что эта группа N-Н участвует в пептидном
связывании, а метановая группа является альфа- или
асимметрическим атомом углерода в аминокислотном
остатке. (Глицин является тривиальным случаем
и здесь не рассматривается.) Протон N-Н с сигналом
при 7.35 м.д., отнесенный нами к остатку аргинина,
коррелирует со всеми спинами в спектре TOCSY,
но обнаруживает спин-спиновое взаимодействие
только с метиленовой группой при 2.24 м.д. в спектре
COSY. Это наблюдение позволяет сделать два вывода,
которые недоступны для спектра TOCSY. Во-первых,
протон N-Н не взаимодействует с а-атомом углерода
аргинина и поэтому должен относиться к N-H
гуанидиновой группы, а не а-аминогруппы. Во-вторых,
аргининовый остаток должен быть N-терминальным,
так как в спектрах COSY и TOCSY нет корреляции
между метановым протоном с сигналом при 4.13 м.д.
и протоном N-Н. При обсуждении аминокислотной
последовательности мы подтвердим второе
утверждение.
Суммарная информация, полученная из спектров 13С
DEPT, COSY, TOCSY и HMQC, позволяет выполнить
отнесения сигналов всех протонов в спектре !Н за
исключением тех, которые претерпевают быстрый
обмен (т.е. протоны карбоксильных групп и свободных
аминогрупп), и сигналов всех не четвертичных атомов
углерода в спектре 13С. Необходимости в отнесении
быстро обменивающихся протонов нет, а сигналы
четвертичных атомов углерода будут отнесены при
последующем обсуждении (см. с. 418).
Вторая главная цель состоит в «секвенировании»
пептида, т. е. определении порядка расположения ами-
нокислот. Двумя мощными методами из арсенала ЯМР
Задача 7.6. Решение
415
Спектр ЖХ-МС (с ЭР)
й
и
о
ж
и
к
и
Ж
ь
к
S
100
90
80
70
60
50
40
30
20
1-112
10
Аспарагиновая
кислота (D)
C^NO/
Масса: 132
О
Глицин (G)
C2H3NO**
Масса: 57
о.
. U 2 II ;
H2N—СН-СЧ-N—CH-C-rN—СН-С—ОН
I 1 ! Н I 1 1 Н 1 1
Н
Аргинин (R)
C6H13N4O*
Масса: 157
О
M-(G,D)
1571 I
150
ЗСН2
4CH2
5CH2
6NH
3471 М+7
C12H22N6°6
Масса: 346
7C=NH
nh2
M-(D) 232-л
214 q
200
m/z
I 1
3CH2
4С=О
250
ОН
288 q
300
М-16
330-1
М+23
369 -|
'll’'11 1 '| । । । 'i |
350
Задача 7.6А
416
7. Задачи и решения
Спектр ЯМР *Н (600 МГЦ)
AspNH
Arg 5
GlyNH
ArgNH(6)
Arg 2 Gly2
Gly2‘
Asp 2
У\
5200 5000
4800 4600
Гц
2600 2400
2200
2000
Гц
Asp
Arg3
Arg 4
1800 1600 1400 1200 1000
Гц
О
О
о,
- .. ; - II :
h2n—ch-c-:-n—ch-c-;-n—ch-c—oh
1 1 ' H I 1 ' H 1
H
I i :
зсн2
I 1
ЗСН2
4СН2
5СН2
Arg-Gly-Asp
6NH
7C=NH
NH2
2 1
Ч исло протонов
4С=О
ОН
Л___ML
2 11 2 2
9.0 8.5 8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5
М.д.
Спектры ЯМР и DEPT (150.9 МПф
T
1 1 ' ‘ 1 1 1 1 1 1 ' 1 ' 1 I ' ' ' ' I ' ' ' ' I ' ' ' I ' I ' ' I ' ' ' ' I ' ' ' I
175 170 165 160 м.д. 50 45 40 35 30 25 м.д.
Задача 7.6Б
Задача 7.6. Решение
417
Fl
Fl
Спектр COSY (600 MIX)
Спектр TOCSY (600 MIX)
Задача 7.6В
418
7. Задачи и решения
Спектр ROESY (600 МГЦ)
Спектр COSY (600 МГЦ) Спектр TOCSY (600 МГЦ) Спектр ROESY (600 МГЦ)
F2
Задача 7.6Г
Задача 7.6. Решение
419
Спектр НМВС (600 МПд)
4.5 4.0 3.5 3.0 2.5 2.0 м.д.
Arg 4
Arg 3
Asp 3
Arg 5
Gly2
Asp 2
Arg3
F2
9.5 9.0 8.5 8.0 7.5 7.0 м.д. 4.5 4.0 3.5 3.0 2.5 2.0 м.д.
F2
Задача 7.6Д
420
7. Задачи и решения
являются НМВС и ROESY (или NOESY). Вспом-
ним, что НМВС показывает дальние спин-спиновые
взаимодействия ^-^С (через 2 связи с 2</сн и через
3 связи с 3«/сн). Для решения задачи секвенирования
этот эксперимент показывает корреляцию между со-
седними аминокислотами, поскольку «видит» через
амидный (пептидный) карбонил амидный протон N-H.
Это упражнение позволит нам провести отнесение
карбонильных групп.
Эксперимент со спектром ROESY облегчает
секвенирование на основании пространственных
корреляций между соседними аминокислотными
остатками. Мы ожидаем найти взаимодействия че-
рез пространство одного аминокислотного протона
N-Н с присоединенным аминокислотным а- или С-2-
протоном(ами). Данные любого из этих экспериментов
должны быть достаточными для решения задачи, а в
совокупности они дают окончательное подтверждение.
Полный спектр ROESY показан в верхней
части иллюстрации 7.6Г. Кросс-пики этого спектра
отражают корреляции в спектре COSY и в спектре
ЯЭО. Для облегчения сравнения область, интересная
для секвенирования, выделена в рамку и показана
вместе с соответствующими спектрами COSY
и TOCSY в нижней части страницы. Протон N-H
глицина коррелирует с соседней метиленовой
группой в обоих спектрах (COSY и TOCSY)
и дополнительно с протоном Н-2 аргинина в спектре
ROESY. Эта корреляция показывает, что глицин
связан с аргинином. Эту корреляцию можно считать
неубедительной из-за перекрывания сигналов
Н-2 аргинина и одного из протонов Н-2 глицина.
В этом случае желательно подтверждение (см. ниже)
данного заключения.
Другая связь между аминокислотными остатками
устанавливается при анализе взаимодействия между
протоном N-Н аспарагиновой кислоты и двумя
протонами Н-2 глицина, выявляемого посредством
ЯЭО. В этом случае нет перекрывания сигналов, и
корреляция подтверждает показанную на иллюстрации
7.6А последовательность аминокислот. Интересно
отметить корреляцию через ЯЭО в остатке
аспарагиновой кислоты между протоном N-H
и только одним из двух диастереотопных метиленовых
протонов (Н-3). Это селективное взаимодействие
указывает на заторможенное вращение и позволяет
пространственную дифференциацию и отнесение
сигналов двух диастереотопных протонов.
Найденную последовательность аминокислот и от-
несение четвертичных атомов углерода подтверждает
спектр НМВС (7.6Д, здесь рассматриваем его нижнюю
часть). До подтверждения аминокислотной последова-
тельности проведено отнесение сигнала четвертичного
атома углерода С-7 в аргинине. Отнесение сигнала
этого атома проводится путем корреляции между
сигналом протона N-Н (Н-6) в аргинине и сигналом
четверичного атома углерода при 156.5 м.д.
Анализ спектра НМВС в области карбонильных
атомов углерода затруднен из-за отсутствия цифро-
вого разрешения по оси F1. Единственным спосо-
бом улучшить разрешение вдоль оси F1 является
увеличение в эксперименте числа ССИ, что имеет
серьезные практические ограничения. Напомним,
что в эксперименте НМВС получается хорошее раз-
решение сигналов протонов по оси F2. Пунктирные
линии, проведенные на врезках, помогают прояснить
корреляции.
Корреляция сигнала протона N-Н глицина с сигналом
карбонильной группы аргинина (С-1) подтверждает
связывание аргинин-глицин. Отнесение сигнала
карбонильной группы аргинина, расположенного
около 170 м.д., подтверждается его корреляцией
с сигналом метиновой группы Н-2 аргинина.
Другая связь между аминокислотными остатками
подтверждается корреляцией сигнала протона N-H
аспарагиновой кислоты с карбонильной группой (С-1)
глицина; положение этого сигнала точно определяется
из корреляций с глициновыми диастереотопными
метиленовыми протонами. Такая последовательность
аминокислотных остатков в этом трипептиде под-
тверждена двумя независимыми методами.
Упражнения
В следующих упражнениях для двух соединений по-
казаны и спектры и структуры. Студентам предлагается
«объяснить» эти структуры, т.е. подтвердить на осно-
вании спектроскопических данных, и выполнить от-
несения сигналов всех протонов и атомов углерода.
Упражнения
421
Масс-спектры
И VV...........Ж
? .!. X У IX
о
S
i
I
4000
I I I I”-1 I ГП I I I I I | г
3000 2000
VO
Г-Г-Г
1000
-]—.—,—.—.—]—.—.—.—.—I—.—.—.—,—[-
3.5 3.0 2.5 2.0
Спектры ЯМР 13C и DEPT (150.9 МПф
З-Гидроксиметил-3-метилоксетан
CH2OH
---CH3
О
80 75 70 65 60 55 50 45 40 35 30 25 20 мд.
Задача 7.7A
422
7. Задачи и решения
м.д.
4.5 4.0
3.0 2.5 2.0
м.д.
2.0-
2.5-
F1
з.о-
4.0-
4.5-
М.Д.
20
304
404
F1 50
60
70
80
Задача 7.7Б
З-Гидроксиметил-З-метилоксетан
СН2ОН
СН3
О
F2
г 20
- 30
-40
450
-60
-70
г 80
4.5 4.0 3.5 3.0 2.5 2.0 1.5 м.д.
F2
-2.0
-2.5
-3.0
-3.5
-4.0
-4.5
Спектр COSY (600 МПд)
Спектр HMQC (600 МПд)
Упражнение
423
Масс-спектр
I 221“|
Й 100 Е
О
о т
S -------
Д 50 Е 97-1 137-1
Спектр ЯМР !Н (600 МГЦ)
А 1111 Д Д.л. /Л м''Д_
| > | - т т , | т т т т | Т Т Т Т | I | I I Т Т | т Т Т , —г 1 Т 1 Т 1 1 1 т г-
2350 2300 Гц 1200 1150 Гц 1050 1000 Гц 900 850 Гц
Задача 7.8А
424
7. Задачи и решения
Спектры ЯМР 13С и DEPT (150.9 МГЦ)
Спектр HMQC (600 МП|)
F1
20
30
40
50
11 м
4.0 м.д. 1.9 1.8 1.7 1.6 1.5 1.4 1.3 1.2 1.1 1.0 0.9 0.8 м.д.
F2
Задача 7.8Б
Упражнения
425
Спектр HMQC (600 МПд)
М дАА» Да Liu 11 1 ч.
4.0 3.9 м.д. 2.0 1.8 1.6 1.4 1.2 1.0 0.8 м.д.
F2
Задача 7.8В
426
7. Задачи и решения
Спектр INADEQUATE (150.9 МП|)
Гц J __П LLd L LLL_
(—)-Амброксид 1
-7000- СН3/ г0 * 1 j 4 -7000
-60004 ^СНз ! 1 । 1 I | 4 -6000
-5000- Нзс снз 4 -5000
-4000- 4-4000
i 1 1 ।
1 1
-3000 - 4 -3000
1 1
-20004 1 1 - -2000
1 1
-10004 1 1 4 -1000
Р 1
1 °
1000 4 4 1000
2000 4 4 2000
3000 4 1 к 4 3000
40004 4 4000
50004 4 5000
60004 | 1 4 6000
70004 4 7000
' I ' ' ' ' I ' " 1 I ' " ' I " " I " ' ' I " ' ' I • ' • I.. ' .. ’ ' ‘ 1 I | •
80 75 70 65 60 55 50 45 40 35 30 25 20 15 м.д.
F2
Задача 7.8Г
8. Контрольные задачи
8.1. Введение
Глава 7 была построена так, как будто авторы искали
решение задач. В действительности это не так. Авто-
ры, разумеется, знали искомые структуры. И студен-
ты также неизбежно видят перед собой структуры,
когда читают описание решения задач. В результате
они получают опыт скорее в соотнесении спектров
со структурами, чем опыт анализа спектров. Тем не
менее и такая практика, без сомнения, полезна.
Задачи из главы 8 дадут навык предсказания,
а не описания структур (за исключением группы
задач в конце главы), но решение нескольких пер-
вых простых задач должно поддержать студентов,
как и задачи в конце каждой главы. Авторы наде-
ются, что уверенность придет по мере преодоления
трудностей.
Спектры ЯМР были зарегистрированы на рабочих
частотах 300 или 600 Мгц для протонов и 75.5 или
150.9 Мгц для углерода; в качестве растворителя ис-
пользовали CDC13, если не указано иное. При 600 Мгц
все 2В-Спектры были зарегистрированны с гради-
ентами (за исключением спектров INADEQUATE).
ИК-Спектры были получены для чистых жидкостей
(т.е. без растворителей), если не указаны другие усло-
вия. Масс-Спектры были измерены методом ГХ-МС
с ионизацией электронным ударом, если не указано
иное. Для получения масс-спектров с химической
ионизацией использовался метан.
Задачи, приведенные в настоящей главе, охватывают
различные классы соединений и располагаются по
порядку от легких к умеренно трудным и трудным.
Ясно, что эти определения относительны. Для начи-
нающего студента ни одна из задач не будет легкой.
С приобретением опыта многие из умеренно сложных
задач покажутся легкими, особенно при использова-
нии 2В-спекгров. В первой группе задач приведены
масс-спектр, ИК-спектр, спектры ЯРМ !Н и 13C/DEPT.
Вторая группа задач помимо перечисленных спек-
тров включает еще некоторые 2В-спектры. В третьей
группе задач приведена структура соединения, и для
этих задач дано большее число 2В-спекгров.
Цель задач третьей группы - научить студентов
работать с более сложными структурами. По сути
эти задачи сводятся к подтверждению структуры
и отнесению сигналов.
Для полного решения большинства задач, включая
стереохимию (конечно, за исключением абсолютной
конфигурации), информации более чем достаточно.
При решении тех задач, в которых недостаточно дан-
ных для определения стереохимии, студент должен
указать возможные стереоизомеры и предложить пути
для их распознавания. Дополнительные упражнения
для студентов можно найти на сайте http://www.wiley.
com/college/silverstein/.
И в заключение можно процитировать Уоллеса
Стегнера: «Легко быть мудрым в прошлом, труд-
но - в настоящем».0
° Stegner, W. (1954). Beyond the Hundredth meridian.
Houghton: Miffin. Reprint, Penguin Books, 1992, Chapter 3.
428
8. Контрольные задачи
Масс-спектр
68п
ИК-Спектр
Спектры ЯМР 13С и DEPT (75.5 МПд)
J__________________________________________________________I__________________|__
. | I | ! | - | ! | I | I | . | I | I |
200 180 160 140 120 100 80 60 40 м.д.
Задача 8.1
8. Контрольные задачи
429
Масс-спектр
100:
о
Я
Я
г
&
О
50 :
I
00
4000
Спектр ЯМР !Н (300 МПд)
О V© ОО
О 4^ W
Lh <1 ч©
I | I I 1 I | I I I I | Г
3000 2000
Волновое число, см1
1000
jUuL Jt jJ
2150 2140 Гц 1960 1950 Гц 1930 1920 Гц
£
I ' 1 ' ' I • ' ' I ' ' ' ..................I | ' |........ । < . . . ............
7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 м.д.
Спектры ЯМР 13С и DEPT (75.5 МПд)
190 180 170 160 150 140 130 120 110 100 90 80 70 60 50 40 м.д.
Задача 8.2
430
8. Контрольные задачи
Масс-спектр
4000 3000 2000 1000
Спектры ЯМР 13С и DEPT (75.5 МГЦ)
CDC13
78.0 77.5 77.0 76.5 м.д.
I IIL I,
95 90 85 80 75 70 65 60 55 50 45 40 35 30 25 20 м.д.
Задача 8.3
8. Контрольные задачи
431
Масс-спектр
100
о
S
о
90
80
70
4000
Спектр ЯМР !Н (300 MIU)
Ох
оо ь-
00 чо
3000 2000
Волновое число, см1
1000
4.35 4.30 4.25 4.20 4.15 4.10 4.05 4.00 3.95 3.90 3.85 3.80
М.Д.
Спектры ЯМР 13С и DEPT (75.5 МГЦ)
75
70
65
60
50
45
м.д.
Задача 8.4
432
8. Контрольные задачи
Масс-спектр
I loo]
Я :
д
s ____:
g 50 Е 39-1
43 -]
трт'
40
ИК-Спектр
m/z %М
126 (М) 100.0
127 (М+1) 8.2
128 (М+2) 4.9
50 60 70 80 90 100 110 120 130
m/z
Волновое число, см1
Спектр ЯМР !Н (300 MIX)
jL
—I---•-I- —I-------1-r-
2300 2280 Гц 2140 2120 Гц
Г ’ • 1 I I ' 1 1 I | 1 | ( ' - | - | ' • । । • I’ * 1 |
7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 м.д.
Спектры ЯМР 13C и DEPT (75.5 MIX)
190 180 170 160 150 140 130 120 110 100 90 80 70 60 50 40 м.д.
Задача 8.5
8. Контрольные задачи
433
Волновое число, см
Спектр ЯМР (300 МП<)
дЛ аЛаАлЛ^,
2260
2250 2240 2230 2220 2210 2200
Гц
zt
1590 1580 1570 Гц
JUL
"I.....'"I"'....
640 630 620 Гц
Л
I ' • ' • I • • I • I • ' I • • • I I ' ' • I ' ' ' • I • ' ' • I ' ' ' ' I ' I I
7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 м.д.
Спектры ЯМР 13С и DEPT (75.5 МПд)
........[........t.......,........,........|........t........,.........।........।.........t
140 130 120 110 100 90 80 70 60 50 40 30 м.д.
Задача 8.6
434
8. Контрольные задачи
Масс-спектр
< 100 Е
О -
81-1
109-]
124 п
50 60 70
I | 1 ТП|ТТГ|-| ГГП | П -Г I | I I I Г | I I 1-1 ] I I I 1 | I
90 100 ПО 120
ИК-Спектр
I I | I I I Г|~1 т
80 ,
m/z
—I—I—I—I—I—I—I—I—I—I—I—I—I—I—I—I—I—I—I—I—I—I—I—I—I—I—I—I—I—I—I—I—I—I—I
4000 3000 2000 1000
Спектр ЯМР *11 (300 МПд)
Волновое число, см-1
_____।_______лА _________________________________
I I ' 1 I • I ' I I ‘ 1 I ' ' I ' ' I ' I
7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 м.д.
Спектры ЯМР 13С и DEPT (75.5 МГЦ)
I.......г.........।..........।.........।.........।.....'•I.........।........।.........।.........।........।
150 140 130 120 110 100 90 80 70 60 50 м.д.
Задача 8.7
8. Контрольные задачи
435
Масс-спектр
00 ОО С/l СО о СО 00 40
СП К-* 4^ 00 о о___________________________________
Т | I I I I I ! ”1 I I | I I I I |
2000 1000
Спектр ЯМР *Н (300 MIX)
Волновое число, см1
Спектры ЯМР 13С и DEPT (75.5 MIX)
Задача 8.8
436
8. Контрольные задачи
Масс-спектр
100 z
аГ
S
50 :
о
ЧО
оо
4000 3000 2000
. Волновое число, см-1
Спектр ЯМР !Н (300 МПх)
1000
Спектры ЯМР 13С и DEPT (75.5 MDi)
33.0 32.5 32.0 М.Д.
....I.............|...up-..I................ I....|......[I.....
170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 м.д.
Задача 8.9
8. Контрольные задачи
437
Масс-спектр
4000 3000 2000 1000
Волновое число, см"1
Спектр ЯМР !Н (300 МПд)
Спектры ЯМР 13С и DEPT (75.5 МПд)
85 80 75 70 65 60 55 50 45 40 35 30 25 20 м.д.
Задача 8.10
438
8. Контрольные задачи
Масс-спектр
ж _____
й 100:
о -*
о
к
ю
S
о
К
i
S
162-1
63-1
г 164
50
98-1
73-]
50 60 70
ИК-Спектр
126-1
I Г128
1111 П 1111 ITjl 11 I |~4 111 11111111111ТГ| 111Т[~Г1 111 111111111111111111 11111111 111111111 -I
80 90 100 110 120 130 140 150 160
m/z
ттт
100-
о
s
!
I
о
50 -
1 I
СП О СП
ЬЭ ко 00
Ь- о СП
--1-1-1--I--1-1-1-1--1-1-1--1-1-1--1-1-1-1--1-1-1--1-1-Г-
4000 3000 2000
„ <1»*П 1жт лпа к<п. \ Волновое число, см-1
Спектр ЯМР *Н (300 МГЦ)
1000
Спектры ЯМР 13С и DEPT (75.5 МГЦ)
155 150 145 140 135 130 125 120 115 м.д.
Задача 8.11
8. Контрольные задачи
439
Масс-спектр
____
123-1
95-1
£ 100 Е
о -
О Z
ИК-Спектр
80 90
m/z
I I I I I I I I I I I I I I I I I I I I I I I I I I
100 110 120
138-1
h । и 111111111U111
130 140
Спектр ЯМР ’Н (300 МПд)
Спектр ЯМР 19F (282.4 МПд)
без «развязки» от !Н
11Ш1 >
с «развязкой» от ’Н
2400 2390 2380 Гц 2140 2130 Гц
I " " Г ' Г " I • • I • I • I ' ' " I " " I " " I " " I ‘ I.. I
8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 м.д.
' ' I ' ....... ' I.......
-104.4 -104.5 м.д.
Спектры ЯМР 13С и DEPT (75.5 МПд)
130 128 126 124 122 120 118 м.Д.
190 180 170 160 150 140 130 120 110 100 90 80 70 60 50 40 м.д.
Задача 8.12
440
8. Контрольные задачи
Масс-спектр
50 55 60 65 70 75 80 85 90 95
ИК-Спектр
4000 3000 2000 1000
Волновое число, см*1
Спектры ЯМР 13С и DEPT (75.5 МПд)
। 1 1 । । • । • • । • । । । 1 • । • । • ...
165 160 155 150 145 140 135 130 125 120 115 мд.
Задача 8.13
8. Контрольные задачи
441
Масс-спектр
£ ----J
§ 100:
82-1
50
ИК-Спектр
138-1
90 100 ПО 120 130 140
m/z
671
1111111111111111111111111
60 70 80
Спектры ЯМР 13С и DEPT (75.5 MIU)
—I----’----1-----’---1-----’---1-----'---1-----'---1-----1---1-----'----1----’----1----'----1
200 180 160 140 120 100 80 60 40 м.д.
Задача 8.14
442
8. Контрольные задачи
Масс-спектр
& 54-|
50 60 70 80 90 , 100 110 120 130 140 150
ИК-Спектр
Спектры ЯМР 13С и DEPT (75.5 МПд)
I____________________________________________________________LJ________1__
“I......г.....।......I......।......।......।......।......।......।......।.....।
120 110 100 90 80 70 60 50 40 30 20 м.д.
Задача 8.15
8. Контрольные задачи
443
Масс-спектр
106-1
о
X
0
S
и
X
о
К
S
100 =
50
771
137-|
50
ИК-Спектр
60 70 80 , 90 100 110 120 130 140
m/z
/ ЛГЛ1Л Л
I I ' ' I I ' I 1 ' ' ' I ' ' ' 1 I ' ' I ' I • I 1 1 ' ' I ' ' ' ' I I
7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0
' I 1 1 ।
1.5 м.д.
Спектры ЯМР 13С и DEPT (75.5 Mlty
М.Д.
140 130 120 110 100
Задача 8.16
444
8. Контрольные задачи
Масс-спектр
50 60 70 80 90 100 110 120
ИК-Спектр Z
4000 3000 2000 1000
Спектры ЯМР 13С и DEPT (75.5 МПф
165 160 155 150 145 140 135 130 125 120 115 ПО м.д.
Задача 8.17
8. Контрольные задачи
445
Масс-спектр
f 100Ё
О
163-1
178-|
100 , 150
m/z
t—1—г
50
ИК-Спектр
I I I I | I 1““1 I | I I I I | I I I I | I I I I | I I Г~1 | г
4000 3000 2000 1000
Волновое число, см1
Спектр ЯМР !Н (300 MDj)
I.....I ' ' ' .. т | .-т......Г ' ' '
2150 2100 Гц 1000 950 Гц 450 400 Гц
Г...... | | ......| . . . . । . . . . । , -г г- г......... । , , . । , . . . |
7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 м.д.
Спектры ЯМР 13С и DEPT (75.5 МГЦ)
150 140 130 120 110 100 90 80 70 60 50 40 30 20 м.д.
Задача 8.18
446
8. Контрольные задачи
Масс-спектр
ё
§
п
S
о
К
S
100 Е
78-1
106-1
121-|
50
51-1
60
70
80 90
m/z
100
110
120
50
ИК-Спектр
3000 2000 1000
9.0 8.5 8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 м.д.
Спектры ЯМР 13С и DEPT (75.5 МПд)
190 180 170 160 150 140 130 120 110 100 90 80 70 60 50 40 м.д.
Задача 8.19
8. Контрольные задачи
447
Масс-спектр
108-1
123-|
100 Е
о
я
в
S
о
К
о
50
40
53-1
50
60
70
80-1
80
т~1 I I I I I I [TI 1 I | I I I I | I I I 1 | I I I I | Г1 1 I | Т T-ITJ гг
, 90 100 110 120
m/z
ИК-Спектр
4000 3000 2000 1000
S
~ -лк ж» кт „ х волновое число,
Спектр ЯМР ЖН (300 МГЦ)
jt _л_ л
..I I...................... I I
2060 Гц 2020 Гц 1820 Гц
I ‘ I ' ' .........I ........Г’" • • • । • • • । । । ।
7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 м.д.
Спектры ЯМР 13С и DEPT (75.5 МПф
Задача 8.20
448
8. Контрольные задачи
Масс-спектр
85 -I
103-1
х20
ХИ (газ-реагент — метан)
159-1
ИК-Спектр
ibo7'
, 100
m/z
100J
158-1 ____=
50 j
Т 1 Г-Ц- '---1 ।
150 150
187-|
I I I I II
m/z
50 :
4000
т—।—।—।—[—।—।—।—।—।—।—।—।—।—।—।—।—।—।—।—।—।—।—।—।—।—।—।—।—।
3000 2000 1000
Спектр ЯМР ГН (300 МПф
Волновое число, см1
। I I I I । I I > I р-
3.5 3.0
—I—• ’ । • • •—Г"
2.5 2.0 1.5
________
1.0 м.д.
Спектры ЯМР 13С и DEPT (75.5 МГц)
60
80
40
160
100
200
140
120
180
м.д.
Задача 8.21
8. Контрольные задачи
449
Масс-спектр
119-1
100 z
о
х
0
X
о
X
I
S
50
91-i
100
120
60
70
110
100
90
80
70
о
§
50
ИК-Спекгр
80 90
m/z
1000
3000 2000
Волновое число, см'1
4000
Спектр ЯМР *Н (300 МПх)
Ж
7.4 7.3 7.2 7.1 7.0 6.9 6.8 6.7 6.6 6.5 6.4 6.3 6.2
М.Д.
Спектры ЯМР 13С и DEPT (75.5 МГЦ)
155 150 145 140 135 130 125 120 115 м.д.
Задача 8.22
450
8. Контрольные задачи
Масс-спектр
х20
183-1 г 185
100 =
155-1 г 157
200-1
198-1
'• I 1 1 1
150
m/z
200
264-1
262-1
260-1
250
100
ИК-Спектр
т | I 1 I I | I I I I | I I г I |
2000 1000
Спектр ЯМР ’Н (300 МПх)
Волновое число, см’1
Спектры ЯМР 13С и DEPT (75.5 МПф
190 180 170 160 150 140 130 120 110 100 90 80 70 60 50 40 м.д.
Задача 8.23
К Контрольные задачи
451
ИК-Спектр
Спектры ЯМР 13С и DEPT (75.5 МПд)
180 170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 м.д.
Задача 8.24
452
8. Контрольные задачи
Спектр ЯМР !Н (300 МПд)
Волновое число, см'1
' • ' ' I................................................ ' ' ' ' I...............................................................I......................... . . . ( . . . . । - . . . । . . . . , .
2050 2040 Гц 1090 1080 Гц 615 610 605 Гц
I 1 ' ’ ' I ’ 1 ' 1 I ’ 1 ‘ ’ I ь ' ' ' I ' ' “ ' I ’ ' ‘ I ' ' ' ' I ‘ ' I ' 1 ’ I ’ 1 ' ’ I 1 1 ' 1 I 1 • 1 ।
7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 м.д.
Спектры ЯМР 13С и DEPT (75.5 МПд)
Задача 8.25
8. Контрольные задачи
453
Масс-спектр
£
о
к
д
S
«
8
л
S
М
156-1
100 Е
63 -1
50 Е
98-|
121 q
50 60 70 80
ИК-Спектр
90 100 , 110 120 130 140 150
m/z
М+2
г 158
111111111
160
। । । । |' I ।—।—г- ।—।—।—।—।—।—।—।—г ।—। । ।—•—।—г ।—।—।—I—I—I—I—|
4000 3000 2000 1000
, Волновое число, см-1
Спектр ЯМР !Н (300 МГЦ)
Спектры ЯМР 13С и DEPT (75.5 МГЦ)
Задача 8.26
454
8. Контрольные задачи
Масс-спектр
4000 3000 2000 1000
, Волновое число, см'1
Спектр ЯМР ТН (300 МГЦ)
Спектры ЯМР 13С и DEPT (75.5 МПд)
170 160 150 140 130 120 110 100 90 80 70 60 50 40 м.д.
Задача 8.27А
8. Контрольные задачи
455
Спектр COSY (300 МГц)
Спектр HETCOR (75.5 МПф
Задача 8.27Б
456
8. Контрольные задачи
Масс-спектр
ОО
1—I—г-i- ।—| I I I । |
1000
--1-1-1-1-1-1-1--1-1-1--Г—I-1-1-1-1-1-1-1-1-г
4000 3000 2000
- Волновое число, см1
Спектр ЯМР (600 МПд)
Спектры ЯМР 13С и DEPT (150.9 МПд)
40
35
50
30
25
60
55
45
м.д.
Задача 8.28А
8. Контрольные задачи
457
F2
Задача 8.28Б
458
8. Контрольные задачи
Масс-спектр
О
к
0
s
о
к
100 Е
50
45 55-1
67-1
г 69
811
95 -1
110-1
50 60 70 80 90 100 1
ИК-Спектр m/z
128-1
[-113
ti I I | i т I I | I I I I | I I I | I
0 120 130
100:
о 2
S --
|5°:
VV
о
00
ю
1—г
4000 3000 2000
Волновое число, см-1
Спектр ЯМР ’Н (300 МПд)
Ch >-
1000
Л A ]\J[_ At
1540 Гц
А
5.0 4.5 4.0
1140 Гц 500 480 Гц 440
2.0
м.д.
Спектры ЯМР 13С и DEPT (75.5 МГц)
20 м.д.
130 120
110 100
Задача 8.29А
8. Контрольные задачи
459
Задача 8.29Б
460
8. Контрольные задам
Масс-спектр
&
к
И
S
к
е
к
S
100 Е
69 п
88 -|
100
50 z
° 50
ИК-Спектр
100:
<D
S
о
4000
ХИ (газ-реагент — метан)
143-i 177 n Г179 Т7Г
I 159~! 1.11941
* I I *1 "I1 I *1 I I I 11 I I I
100 , 150
m/z
230 240 , 250 260
m/z
I
\О О Os
1-1--1-1-1--1-1-1--1-1-1--1-1-1--1
3000 2000
Волновое число, см’*
00
со
00
' 1 I 1
1000
Спектр ЯМР (300 МГЦ)
Спектры ЯМР 13С и DEPT (75.5 МПд)
170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 м.д.
Задача 8.30А
8. Контрольны^ задачи
461
Задача 8.30Б
462
8. Контрольные задачу
Масс-спектр
I Г I I ”| "1 "I" 'I I | I I I I | I- I I I | I-1 I I | I I Г' I | г
4000 3000 2000 1000
~ 3680 3660 Гц 3300 3280 Гц 3220 3200 Гц 2760 2740 Гц
—। . । । । -| . । .—। . । . । . । . । . । . । , । . । . । , ,
7.4 7.2 7.0 6.8 6.6 6.4 6.2 6.0 5.8 5.6 5.4 5.2 5.0 4.8 м.д.
Спектры ЯМР 13С и DEPT (150.9 МГЦ)
1
160 150 140 130 120 110 100 90 80 70 м.д.
Задача 8.31 А
8. Контрольные задачи
463
F2
Задача 8.31 Б
464
8. Контрольные задачи
Масс-спектр
Й
О
о
и
5
Я
е
я
S
100
50
55-] 67-|
1111111Г1111111 if 14
50 60
70
80
95 -|
х20
ИК-Спектр
110-1
109-|
121-|
139-1
136-|
154-1
90 100 110 120 130 140 150
m/z
1 г ---------- । - — г г .................................I 1
2400 Гц 1350 Гц 1150 Гц 1050 Гц 950 Гц
На врезках цена деления шкалы 10 Гц
—I 1 т т т I ' т т ' I т т 1 т I т т т 1 I т т т т I т т т т | т
4.0 3.5 3.0 2.5 2.0 1.5 м.д.
Спектры ЯМР 13С и DEPT (150.9 МПф
I I I I < I I |-r-. > , I > - T-П) . . . . ( 1 , . . , | I .. . . ! . . . . ! . . . . ! . . . . ! . . . . J . . . . ,
75 70 65 60 55 50 45 40 35 30 25 20 15 м.д.)
Задача 8.32A
8. Контрольные задачи
465
Спектр COSY (600 МПд)
Спектр HMQC (600 МГц)
F2
Задача 8.32Б
466
8. Контрольные задачи
Масс-спектр
—।—।—।—।—।—।—।—।—।—।—।—।—।—।—।—।—।—।—।—।—।—।—।—।—।—।—।—।—।—।—।—।—।—।
4000 3000 2000 1000
80
60
180
160
140
120
100
40
20
м.д.
Задача 8.33А
8. Контрольные задачи
467
F2
Задача 8.33Б
468
8. Контрольные задачу
Масс-спектр
ГМ; ..................................
11 1 1
ср \о -Р^ СО о СР
СО to СП 00 СП СР
to О Ь- -о
--т—т-Т-I-f-Т-Т-Т-Т-|-Т-Т-т-Т-|-Т-ТГ—ИТ-Т-|-Т-Т-Т-Т-|Т-Т-Т Т 1 Т Т Т Т-
4000-3000----------2000-1000
Спектры ЯМР 13С и DEPT (150.9 МГЦ)
,’ГГГ,,"Г"Т.....I......."1.......1.......1.......1.......1........1.......1........1.......1........1
130 120 ПО 100 90 80 70 60 50 40 30 20 м.д.
Задача 8.34А
8. Контрольные задачи
469
Спектр HMQC (600 МГц)
5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 м.д.
F2
Задача 8.34Б
470
8. Контрольные задачи
Масс-спектр
СР
--1--1--1-1--1--1--1-1--1--1-1--1--1-1--1--1--1-1--1--1-1--Г-^1-1--1--1--1-1--1--1-1--1--1-1--}
4000 3000 2000 1000
Спектры ЯМР 13С и DEPT (150.9 МГЦ)
।..........I
220
. । • • • । • • । • • । • • • । । • । • i • • i • • i • • • i • ।
м.д. 55 50 45 40 35 30 25 20 15 М.Д.
Задача 8.35A
8. Контрольные задачи
471
F2
Задача 8.35Б
472
8. Контрольные задачи
Спектр INADEQUATE (150.9 МПд)
Задача 8.35В
8. Контрольные задачи
473
Масс-спектр
о
К
m
S
X
g
S
431
1001 70-1
хЗО
50 j 551
40 50 60 70
115—। 130-|
I 11 1111111 I I I | I I I I | I I I I | I I I I | I I I I | I I I 11 г I г I | I I I I | I I I I 111 I
ИК-Спектр
80 , 90 100 110 120 130
m/z
100-
о “
X
i -
&—:
a so
с
4000
1 I 1 '
3000
2000
1000
Спектр ЯМР *Н (600 МГЦ)
Волновое число, см-1
jlL JIl
2350 Гц 2300 Гц 1000 Гц 850 Гц 700 Гц 550 Гц
На врезках цена деления шкалы 10 Гц
т—•—
М.Д.
Спектры ЯМР 13С и DEPT (150.9 МПд)
170 160 150 140 130 120 ПО 100 90 80 70 60 50 40 30 20 м.д.
Задача 8.36А
474
8. Контрольные задачи
Спектр HMQC (600 МПд)
F2
Задача 8.36Б
8. Контрольные задачи
475
Масс-спектр
ИИ
100 Е
о
д
0
S
о
К
о
ь
К
S
50 Е
69 -|
83 “I
ИК-Спектр
m/z
151 (М-1)
152 (М)
153 (М+1)
154 (М+2)
%м
37.5
100.0
12.7
5.2
152-1
50 60 70 80 90 , 100 ПО 120 130 140 150
m/z
Т I-—I I | I I I Г-—j I Г”—I I | I I I I | I I Г I | I I I 1 | I I I I |
4000 3000 2000 1000
, Волновое число, см-1
Спектр ЯМР (600 МГЦ)
4680 Гц 4560 Гц 4260 Гц 1520 Гц 740 720 Гц 600 Гц
7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 м.д.
Спектры ЯМР 13С и DEPT (150.9 МГЦ)
J_________________________। II _______________________________I______________________________——
—. | Т | Т | Т | тг—|] т | Т | Т | Т | тг—
180 160 140 120 100 80 60 40 20 м.д.
Задача 8.37А
476
8. Контрольные задачи
F2
Задача 8.37Б
8, Контрольные задачи
477
Масс-спектр
ё ЮО :
о -
4000 3000 2000 1000
Волновое число, см-1
Спектр ЯМР ’Н (600 МГЦ)
3940 3920 Гц 3880 3860 Гц 3720 Гц 1280 Гц 320 300 Гц
1г~Г___________~______
।..।..।..।..।...।1 ..।.।.।..।. 1
987654321 м.д.
Спектры ЯМР ,3С и DEPT (150.9 МП<)
171.5 171.0 м.д.
170 160 150 140 130 120 ПО 100 90 80 70 60 50 40 30 м.д.
Задача 8.38А
478
8. Контрольные задачи
7 6 5 4 3 2 1 м.д. Спектр COSY (600 МПд)
F2
Задача 8.38Б
8. Контрольные задачи
479
F2
Задача 8.38В
480
8. Контрольные задачи
Масс-спектр
___
123-1
100 Е
83-|
Я
0
S
к
н
я
S
50
41
Г5569-1
1,1
151-1
95-| 110-|
50
100 150
m/z
232-1
230 п
ИК-Спектр
4000 3000 2000 1000
Спектры ЯМР 13С и DEPT (150.9 МПд)
20.0 19.9 19.8 м.д.
, , ( J Jll, II
200 180 160 140 120 100 80 60 40 20 м.д.
Задача 8.39А
8. Контрольные задачи
481
Задача 8.39Б
482
8. Контрольные задачи
F2
Задача 8.39В
8. Контрольные задачи
483
Масс-спектр
Спектры ЯМР 13С и DEPT (150.9 МП<)
45
50
25 м.д.
Задача 8.40А
484
8. Контрольные задачи
F2
Задача 8.40Б
8. Контрольные задачи
485
Масс-спектр
100:
67-|
82-|
о
я
S
о
К
р
50
54-i
100
ХИ (газ-реагент — метан)
Г155
Найденная методом
ХИ ММ = 155.0711
S
50
ИК-Спектр
m/z
100
100:
о
S
50 :
о
а
4000
50
110-J
1831 195 -|
160 170 180 190
m/z
Г '
ij S! to V
1000
Спектр ЯМР *Н (600 МПд)
3000 2000
Волновое число, см-1
3.3 3.2 3.1 3.0 2.9 2.8 2.7 2.6 2.5 2.4 2.3 2.2 2.1 2.0 1.9 1.8 1.7 1.6 1.5 1.4 м.д.
Спектры ЯМР 13С и DEPT (150.9 МПд)
М.Д.
170 160 150 140 130 120 ПО 100
Задача 8.41 А
486
8. Контрольные задачи
F2
Задача 8.41 Б
8. Контрольные задачи
487
Масс-спектр
—।—।—।—।—।—।—।—।—।—।—।—।—।—।—।—।—।—।—।—।—।—।—।—।—।—।—।—।—।—।—।—।—।—।—।
4000 3000 2000 1000
Спектр ЯМР ХН (600 МПд)
Волновое число, см1
Спектры ЯМР 13С и DEPT (150.9 МПд)
Задача 8.42А
488
8. Контрольные задачи
7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 м.д. Спектр COSY (600 МПд)
F2
Задача 8.42Б
8. Контрольные задачи
489
7.5 7.4 7.3 7.2 7.1 м.д. 4.5 4.0 3.5 3.0 м.д.
Задача 8.42В
490
8. Контрольные задачи
Масс-спектр
г43
хЗО
к
«
S
о
К
100
85-|
131-|
S
50
95-, 1091
182 q г-185
200-|
100
m/z
150
200
50
ИК-Спектр
—I--1-1-1-1-1-1-1-1-1-1-1-1-1-1-1-1-1-1-1-1-1-1-1-1-1-1-1-1-1-г
4000 3000 2000 1000
« Волновое число, см1
Спектр ЯМР 'Н (600 МГц)
JiL
--'—I——I——I —I—'—I—‘—I—-
—I ' ' ' ' Г"
4.0 3.5
—। . . . । , , . , ] . . ,—,—]—,—.—,—,—।
3.0 2.5 2.0 1.5 м.д.
Спектры ЯМР 13С и DEPT (150.9 МГЦ)
170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 м.д.
Задача 8.43А
8. Контрольные задачи
491
F2
Задача 8.43Б
492
8. Контрольные задачи
Спектр НМВС (600 МГЦ)
F2
Задача 8.43В
8. Контрольные задачи
493
Масс-спектр
О 1ООЁ
О
121-1
136-1
149-| 1781
т—। ' । । । ' । ।—L|—iiii—।—Ц-
150
5 5° •
Ё -Е 65П 93-|
S : | |
ь-1--1--1-Г-**Г-1-[“—I--1-Г^1---]-1--
50 100
ИК-Спектр
I I I I | I I I I | г
4000 3000
ж Ггг
2000 1000
Спектр ЯМР !Н (600 МПд)
Волновое число, см-1
Спектры ЯМР 13С и DEPT (150.9 МПд)
1 200 1 180 "1 160 । 140 1 1 120 । 100 1 80 । 60 1 1 40 1 М.Д.
Задача 8.44А
494
8. Контрольные задачи
м.д.
И
-2
'-3
-4
-5
-6
-7
-8
Спектр COSY (600 МПд)
Спектр HMQC (600 МПд)
F2
Задача 8.44Б
8. Контрольные задачи
495
Спектр НМВС (600 МГЦ)
8.0 7.5 7.0 м.д. 3.0 2.5 2.0 1.5 1.0 м.д.
Задача 8.44В
496
8. Контрольные задачи
Масс-спектр
125-|
1----1 “ *
1---1--г
150
168-|
т--1—|т
Спектр ЯМР ГН (600 МГЦ)
Волновое число, см1
uiki—jaJ
I'"'.....j. r
12 м.д. 4.5 4.0 3.5 3.0 2.5 2.0 1.5 м.д.
Спектры ЯМР 13C и DEPT (150.9 МПд)
.....I......I.....I.....I.....I......I.....I.....I.......I....I.....I......I..."I.......I.....I.....
180 170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 м.д.
Задача 8.45A
8. Контрольные задачи
497
5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 м.д.
Спектр COSY (600 МПд)
Спектр HMQC (600 МПд)
F2
Задача 8.45Б
498
8. Контрольные задачи
Спектр НМВС (600 МГЦ)
—ЛЛ_
F2
Задача 8.45В
8. Контрольные задачи
499
ИК-Спектр
100 =
о
S
Я
л
50 :
W
о
£
4000
Спектр ЯМР ГН (600 МПд)
JL
1000
3000 2000
Волновое число, см1
Спектры ЯМР 13С и DEPT (150.9 МПд)
140 130 120 110 100 90 80 70 60 50 40 30 м.д.
Задача 8.46А
500
8. Контрольные задачи
F2
Задача 8.46Б
8. Контрольные задачу
501
Спектр INADEQUATE (150.9 MDi)
-150007
-10000-
-50004
Fl 04
5000-
10000-
15000-
-15000
-10000
-5000
О
5000
10000
15000
F2
Задача 8.46В
502
8. Контрольные задачи
Масс-спектр 105
ХИ (газ-реагент — метан)
277-1
136-1
р154
50 100 150 200
ИК-Спектр
11111111111111
250
Найденная методом
ХИ ММ = 277.1083
, 300
m/z
—г
4000
1 “Г -г' т
3000
1—I—I—I—I—I—I—I—I—I—I—I—I—I—I—I—I
2000 1000
Волновое число, см-1
Спектр ЯМР ’Н (600 МГц)
Задача 8.47А
8. Контрольные задачи
503
Задача 8.47Б
504
8. Контрольные задачи
Спектры ЯМР 13С и DEPT (150.9 МГЦ)
130 129 М.Д.
Jll
77.5 77.0 м.д.
180 170 160 150 140 130 120 110 100 90 80 70 60 50 40 м.д.
Спектр HMQC (600 МПд)
М.Д.
40-
50-
60 4
70-
Fl i
80-Ё
90 *:
100-
110-5
120-
130 4
О к.
8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 м.д.
F2
Задача 8.47В
8. Контрольные задачи
505
Спектр НМВС (600 МПх)
.. I_____________________________1_____U1L
8.2 8.0 7.8 7.6 7.4 м.д. 5.0 4.5 4.0 3.5 3.0 2.5 м.д.
F2
Задача 8.47Г
506
8. Контрольные задачи
Спектр ЯМР *Н в D2O (600 МПф
3460 3440 Гц 2800 2780 Гц 2720 Гц 2280 2260 Гц
_лЛЛл_ УЧ ЛМ_ ЧУУ
2220 2200 Гц 2160 2140 Гц 1920 1900 Гц 1440 1420 Гц
_лМААу _Л_
— । । 2“ --1-•-1-•--1—•—।—•—।—•—।—•—1 ч.।....""i"^.।.।.
1260 1240 1Ц 1о8о Ю60 Гц 920 900 880 860 Гц 560 550 540 530 Гц
HDO
5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 м.д.
Задача 8.48А
8. Контрольные задачи
507
Задача 8.48Б
508
8. Контрольные задачи
Спектры ЯМР 13С и DEPT (150.9 МПц)
q qqq
s s ssds.sdd t
•i......i.......i.......i.......i.......i......i.......i.......i.......i.......।.......i.......i.......i.......i.......1
160 150 140 130 120 110 100 90 80 70 60 50 40 30 мд.
Спектр HMQC (600 Mill)
6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 м.д.
F2
Задача 8.48В
510
8. Контрольные задачи
Масс-спектр
382-1
100 Е
О
Ж
д
S
О
К
о
К
S
50
Г56
266-1
245 q
2161 I.
292-|
337
50
ИК-Спектр
100
200 , 250
т/г
300
350
100:
150
to tb 22
ОО оо
S
§§
50 :
и й
О
0
У
—I—I—I—I—I—I—I—I—I—I—I—I—I—I—I—I—I—I—I—I—I—I—I—I—I—I—I—I—I—I—I—I—I—I—I
4000 3000 2000 1000
Волновое число, см1
Спектры ЯМР 13С и DEPT (150.9 МГЦ)
150 140 130 120 110 100 90 80 70 60 50 40 30 20 м.д.
Задача 8.49А
8. Контрольные задачи
511
Задача 8.49Б
512
8. Контрольные задачи
Задача 8.49В
514
8. Контрольные задачи
Масс-спектр (с ЭР)
100
488-j
М+7
о
к
s
о
К
о
К
S
129-1
244п
г147 A L..A
I I l'| I I 1’Т |'Г1 I I | Г1 I I П I I | I I |‘ I I гг
150 200 250 m/z
М+23
510-1
500
Спектр ЯМР !Н (600 МПд)
при 0° С в смеси 5% D2O + 95% Н2О
О
О
СН3
О
О
HN—СН-С—N—СН-С--N-CH-C—N
Н
сн2
он
н
сн2
I
с=о
С—ОН
О
373-1
355-1 413-]
I I I I I I I I Г Г Г I | I11 I I '| I I I* I | I I I I ) I I I I' | I I I
300 350 400 450
он
СН2
СН2
СН2
н2с—nh2
5100 5000 Гц
1 1 2
2 1 1
Л- . AjM
2 2
2300 2200 Гц
1800 1700
2800 2700 Гц
Гц
1 3 2 1 112 2
• ।...।.....।.... 1...... 11' I..I'' ..
1400 1300 1200 Гц 1100 1000 900 Гц
I 1 I I • 1 • I • • 1 । • I ' ' ' Г ' ' ' Г ' ‘ ' I I • • • I ' • ' I ' • • ' I • • ' • I • • ' • I ' ‘ I I
8.5 8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 м.д.
Задача 8.50А
8. Контрольные задачи
515
Задача 8.50Б
516
8. Контрольные задачи
Спектр TOCSY (600 МГЦ)
Задача 8.50В
8. Контрольные задачи
517
Спектр COSY (600 МПд) Спектр TOCSY (600 МПд) Спектр ROESY (600 МПд)
F2 F2 F2
Задача 8.50Г
518
8. Контрольные задачи
Спектры ЯМР 13С и DEPT (150.9 МПд)
F2
Задача 8.50Д
8. Контрольные задачи
519
Масс-спектр
| fool
к -
« z
S :
| 5(П
121-1
161-1
50
ИК-Спектр
100 Е
:
g 90 =
I ]
I 80^
Q.
92-1
100
-I—г
m/z
189-]
133 q
1 | г
150
I
2000 1000
—г
4000
3000
Спектры ЯМР 13С и DEPT (150.9 МПО
190 180 170 160 150 140 130 120 ПО м.д.
Задача 8.51 А
520
8. Контрольные задачи
Задача 8.51 Б
8. Контрольные задачу
521
F2
Спектр HSQC (600 МПф
F2
Задача 8.51В
522
8. Контрольные задачи
Масс-спектр 7 4
50 100 150
ИК-Спекгр
10.0 9.5 9.0 8.5 8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 м.д.
Спектры ЯМР 13С и DEPT (150.9 МП<)
I___________________________________________________________________________I.1.UI 1.1.
Задача 8.52А
8. Контрольные задачи
523
F2
Задача 8.52Б
524
8. Контрольные задачи
Спектр НМВС (600 МПд)
F2
Задача 8.52В
8. Контрольные задачи
525
Масс-спектр
____
л 100 :
о :
120-1
50
761
92-1
222-1
194-1
165-1
I I
т । ।—। ,JI* ।—।—। । г—Ч | I I । Ду
150 200
' I 1
100
I ‘I "Г
m/z
Спектры ЯМР 13С и DEPT (150.9 МГЦ)
185 180 175 170 165 160 155 150 145 140 135 130 125 120 115 ПО
м.д.
Задача 8.53А
526
8. Контрольные задачи
Задача 8.53 Б
8. Контрольные задачи
527
Спектр НМВС (600 МПд)
F2
Задача 8.53В
528
8. Контрольные задачи
4000 3000 2000 1000
Волновое число, см"1
Спектр ЯМР *Н (600 МПд)
Задача 8.54А
8. Контрольные задачи
529
Спектр COSY (600 МПд)
С
Задача 8.54Б
530
8. Контрольные задачи
Спектры ЯМР 13С и DEPT (150.9 МПд)
' г Г ' '~ГТ
41.5 41.0 м.д.
11111111111111111111111-| 111111111 |Т11Г111111111 ।. 11 гр 1111 । рч-1 I 1111 р it. ..... р .. гг.... р । IT rr-ri | 11 I 11 .. 111111.. । ।....... i 111..
180 170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 20 м.д.
Спектр HMQC (600 МГЦ)
Задача 8.54В
8. Контрольные задачи
531
Спектр НМВС (600 МПд)
7.0 6.5 6.0 5.5 5.0 м.д. 2.4 2.2 2.0 1.8 1.6 1.4 1.2 м.д.
F2
Задача 8.54Г
532
& Контрольные задачи
Масс-спектр
133 -I
хЗО
О
я
и
я
о
к
е
я
S
100 Ё
77-1
50
611
89п
117-1
105-1
161-|
50
ИК-Спектр
100
,150
m/z
50 z
1--1-1-1--1-1--1-1-1--1-1-1--г
3000 2000
М-1
203-j 219 -j 233 “| 247 q
-i—ph—।—i——।—гА—।—Н-р
200 250
1000
Волновое число, см'1
Спектр ЯМР 29Si (150.9 МПд)
с «развязкой» от гН
1 I 1 1 1 I 1 1 1 1 I 1 1 1 1 I 1 1 1 1 I 1 ' I 1 1 —Т1 -' I I • | • ] |
3.5 3.0 2.5 2.0 1.5 1.0 0.5 0.0 м.д. -4 -6 м.д.
Спектры ЯМР 13С и DEPT (150.9 МПд)
75 70 65 60 55 50 45 40 35 30 25 20 15 10 5 0 м.д.
Задача 8.55А
8. Контрольные задачи
533
F2
Задача 8.55Б
534
8. Контрольные задачи
Задача 8.55В
8. Контрольные задачи
535
Масс-спектр
« ----J
й 100 Ё
о т
о
X I
д ----
5 50 Е
а _
О
S
ИК-Спектр 50
671
82-| 94-1
100
124-1
140-|
150
m/z
200
289-1
272-|
1 I ’ ' ' ' । '
250
_________________________________________________________________о\___________
I I I I I I I I I I I I I I I I I I I I I I I I I I I I I I I I I I I
4000 3000 2000 1000
Спектр ЯМР в D2O (600 МПд) Волновое число, см 1
609—
4450 4400 Гц 3000 Гц 2500 Гц 2350 Гц
2250 Гц 1400 1350 Гц 1150 Гц 940 920 900 Гц
N— СН3 HDO
7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 м.д.
Задача 8.56А
536
8. Контрольные задачи
Задача 8.56Б
8. Контрольные задачи
537
Спектры ЯМР 13С и DEPT (150.9 MDi)
170 160 150 140 130 120 110 100 90 80 70 60 50 40 м.д.
Задача 8.56В
538
8. Контрольные задачи
Спектр НМВС (600 МПф
7.5 7.4 м.д. 5.0 4.8 4.6 4.4 4.2 4.0 м.д. 2.5 2.0 1.5 мд.
F2
Задача 8.56Г
8. Контрольные задачи
539
Масс-спектр
iod
42
384-1
g 50 Е
353-|370-|
269-1
Г'1*1 I'"'|'Т I "I 7 | "Г 1*1 t"| Г'1
250 300
Спектр ЯМР *Н (600 МГЦ) Волновое число, см-1
। . . , , । , • । . . . . । . , 1 । . । . . . . । . . . . । I I < I । > < । '
6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 м.д.
Задача 8.57А
540
8. Контрольные задачи
F2
Задача 8.57Б
8. Контрольные задачи
541
Спектры ЯМР 13С и DEPT (150.9 МПф
—I---•---1---'---1--- ----1---•--1---•---1---•---1........1....~г" TH-.....
150 148 м.д. 114 112 м.д. 55.9 55.8 м.д.
-I.....I......I.....I.....Г """"I.....'I.....I.....f.....I.....I.....I......I.....I.....I......
170 160 150 140 130 120 ПО 100 90 80 70 60 50 40 м.д.
7.0 6.5 6.0 5.5
5.0 4.5 4.0 3.5 3.0 2.5 м.д.
F2
Задача 8.57В
542
8. Контрольные задачи
Задача 8.57Г
8. Контрольные задачи
543
Масс-спектр
312-1
100:
о
£
s
о
К
(D
X
S
50 Е
297-1
281-]
254-1
239-|
399-1
371-1
356-1
50 100 150 200 , 250 300 350 400
m/z
ИК-Спектр
4000 3000 2000 1000
Спектры ЯМР 13С и DEPT (150.9 MDi)
180 170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 м.д.
Задача 8.58А
544
8. Контрольные задачи
F2
Задача 8.58Б
8. Контрольны^ задачи
545
Спектр НМВС (600 МГц)
8.5 8.0 7.5 7.0 м.д. 4.5 4.0 3.5 3.0 2.5 2.0 м.д.
F2
Задача 8.58В
Предметный указатель
«Азотное» правило 26
Аннулены 167
Боковые сигналы от вращения (в ЯМР) 153-154
Больцмана распределение 151, 244, 362
Бомбардировка быстрыми атомами (ББА, FAB) 11, 15, 23
Быстрый обмен 167, 182, 190
Взаимозаменяемые атомы 187, 189, 201
Взаимопревращение структур с частично двойной связью 191,
364
Влияние растворителя 112, 178, 255, 268
Водородная связь 107-125, 178-182, 190, 198, 255
Водородной ненасыщенности показатель (индекс) 28, 29, 388
Волновое число 88, НО
Вращающаяся система координат (в ЯМР) 158, 286
Вращение вокруг простых связей в насыщенных цепях 191
Временное представление спектра ЯМР 158, 288, 289
Время эволюции 287
Времяпролетная масс-спектрометрия (TOF) 16, 17, 23
Газ-реагент 14
Гамма-гош-эффект 257
Гетероядерная когерентность через несколько связей
(НМВС) 297-301, 305-308, 313
Гетероядерная корреляция (HETCOR) 291, 295-297, 308,
455, 459, 461
Гидроксильные протоны в ЯМР ’Н 178-181
Гиромагнитное отношение 151, 160, 362, 369, 372, 373,
384-386
Главный пик 11
Гомомерные молекулы 189
Гомотопные атомы 187, 189
Гомоядерная спектроскопия Хартмана-Хана 317
гом/-Ротамеры 191
Гука закон 89, 90
Дальтон (Да) 11
Двойная фокусировка 21
Двойной резонанс 206
Двугранный угол 204-205
Двумерные методы ЯМР 285-329
Двухквантовая фильтрация в методе *Н - *Н COSY 292, 308
Двухквантовый перенос при природном содержании углерода
(INADEQUATE) 308-310, 402
Дезэкранирование 166, 167, 173
Дейтерий 184, 361
Дейтерированные растворители 161, 162, 185, 234, 246
Диагональные пики в методе COSY 290
Диамагнетизм 163
Диамагнитная анизотропия 166-168, 255
Диастереомерные молекулы 189
Диастереотопные группы 188-190
Длина волны 88, 99
Дочерние ионы 24
Единичное разрешение в масс-спектрометрии 12, 21, 25,
27, 28
Задержка между импульсами 157, 362
Заторможенное вращение 191, 416
Звон (вигли) 153-154
Зеркальное отражение 187, 201
Зеркально-поворотная ось симметрии 187
Изотопные пики 26, 27, 50-52
Изотопы
дейтерий 184, 361
протий 361
соотношение 13С/,2С 27
тритий 361
14N и I5N 361-367
Изохронные ядра 186
Импульсная масс-спектрометрия 17, 24
Импульсная последовательность 155, 240, 243, 250, 254,
286-299, 319
Импульсная спектрометрия с преобразованием Фурье 17,
23, 24, 95, 151, 152, 158, 239, 287
Импульсные градиенты поля (PFG) в ЯМР 328, 329
Инверсия циклов 191
Инверсные (обратные) методы ЯМР 296, 367
Индукция магнитная (Во) 20, 21, 152, 155, 163, 170, 241, 246
Интегрирование сигналов в ЯМР 160, 168, 241, 387
Интенсивность сигналов 156, 160, 161, 168, 241, 387
Предметный указатель
547
Интерферограмма 24, 95, 156, 288, 289
Интерферометр 95
Инфракрасные спектры газов 96, 131
Инфракрасный спектрометр
диспергирующий 94
с преобразованием Фурье (IR-FT) 95
Ионизации методы (в масс-спектрометрии) 13-20
Ионизационная камера 12
Ионизация
при атмосферном давлении (API) 17, 22
химическая (ХИ, CI) 13-15
электронным ударом (ЭУ, EI) 12, 13
электрораспылением (ЭР, ESI) 17-20
Ион-молекулярные взаимодействия 27
Ионная ловушка 22-24
Квадратурное детектирование в ЯМР 291
Квадрупольные масс-спектрометры 21
Квази молекулярные ионы 14, 15, 31, 390, 415
Кето-енольное равновесие 190
Колебаний взаимодействие 90
Колебания молекулярные
валентные 89-92
деформационные 89, 91, 92
ножничные 89, 91
Колебательные спектры 88, 89 сл.
Количественные измерения в ЯМР 13С 250
Коллектор ионов 12
Кольцевого тока эффект 166
Константа экранирования 163
Константы спин-спинового взаимодействия
!Н - 13С через 2 и 3 связи 245-250
с 15N 182, 364, 367
с 31Р 233, 376
Контурное представление 2О-спектра 290
Корреляционная спектроскопия ЯМР 290
для дальних констант (COLOC) 299
13С - 13С (INADEQUATE) 308-310
COSY 291-296, 301, 310
Корреляция 286
Кросс-пики в ЯМР 291, 292
Круговая частота 24
«Кулоновский взрыв» 18
Кулоновское отталкивание 18
Ларморова частота 152, 156, 158, 287
Магнитный диполь 150
Магнитный момент 150, 151, 360, 361, 384-386
Масса/заряд отношение 11, 20
Масс-спектрометрия с преобразованием Фурье 17, 23, 24
Матричная лазерная десорбционная ионизация (MALDI) 17,
23
Миллионные доли (м.д.) в ЯМР 163-169
Многоквантовая гетероядерная корреляция (HMQC) 296,
297, 301, 305, 313, 314, 317, 322, 326
Многократного внутреннего отражения метод 97
Многоимпульсные эксперименты 287
Модуляция как функция времени tx 290
Молекулярная масса 12, 15, 28, 63-83
Молекулярная формула И, 21-28
Молекулярное вращение 88
Молекулярные колебания 88, 89 сл.
Молекулярный ион 11, 13, 15
в спектрах высокого разрешения 27
Мониторинг конкретного иона 23
Мультиплетность для сигналов 13С 238, 247, 252, 253, 280
Мультиплетность и относительные интенсивности сигна-
лов 169-178
Набор спектров как способ представления 2Э-спектра
ЯМР 288-290
Намагниченность общая 152-158, 287
Недиагональные пики (кросс-пики) 291, 292
Неискаженное усиление переноса поляризации 252-255
Непрерывная развертка в ЯМР (CW) 151-154, 160
Ньюмена проекции 192
Область функциональных групп (в ИК-спектрах) 98-130
Обмен ОН-протонов 178, 179, 182
Обмен позициями
при вращении вокруг оси симметрии Сп 187
при действии операций симметрии 186, 187
при инверсии в центре симметрии 187
при отражении в плоскости симметрии 187
хиральных групп 187
Обменивающиеся ядра 187
Оптическая плотность 89
Оси F1 и F2 в 2Э-ЯМР 296
Основные колебания 89, 92
Отпечатков пальцев область в ИК-спектре 98
Отщепление воды (в масс-спектрометрии) 37, 38
Парамагнетизм 163
Парамагнитные вещества 208, 251, 362
Паскаля треугольник 172, 177, 193, 247
Пасты 96, 109, 121
Перегруппировка Мак-Лафферти 31, 42-47
Перегруппировки 31, 42-47
Перенос когерентности 313
Плоскость симметрии 187, 196, 248, 388, 402
548
Предметный указатель
Поворотная ось симметрии 187, 192, 248 Спиновый захват 313, 317, 319
Полевая десорбция (ПД, FD) 15 Спин-решеточная релаксация (время 1\) 242, 248, 287
Полное подавление спин-спинового взаимодействия с про- Спин-спиновое взаимодействие
тонами 238, 239 виртуальное 197
Попла система обозначений 174, 175, 186, 193, 199-201 вицинальное 169, 201, 204
Примеси 166, 179, 184, 235, 236, 241, 280-282 геминальное 169, 201, 205
Принцип неопределенности Гейзенберга 157 дальнее 171, 203, 205, 389
Пропускание (7) 89 1Н _ вс 297-301, 305-308, 313
Радиочастота 151 для W-образной конформации 206 подавление 206
«Развязка» от протонов 206 с протонами 206
инверсная прерываемая 250 протонов с D 184, 185
неполная («офф-резонанс») 253 протонов с 13С 186, 244-246
прерываемая 245, 286 протонов с ,9F 184
составными импульсами (CPD) 239, 296 протонов с 31Р 186
Разделение ионов 11, 12 протонов с 29Si 186
Размерность спектра (в ЯМР) 287 протон-протонное (подавление) 206, 396
Разностная спектроскопия на основе ЯЭО 206-209, 408 слабое и сильное 170-177
Распад, индуцированный столкновениями (CID) 24 углерод - протоны, константы 238, 245, 246
Распылитель 17 углерод - углерод 308
Расфазировка спинов 156 Спутники (сателлиты) 13С 186, 238, 297, 299
Резонансная частота 163, 164, 206, 239-241, 361 Степени свободы многоатомной молекулы 89
Релаксация поперечная (спин-спиновая, дипольная) 156, 160, 287 Таблетка (с КВг) 96, 109, 120
продольная (спин-решеточная) 156, 208, 241, 250, 251, 362 «Родительский» ион 24 Ротамеры (конформации) 191, 192 Тандемная масс-спектрометрия 24
Таутомеров взаимопревращение 190 Точка входа (в COSY) 292 Тритий (3Н) 361
Сближение пространственное атомов 207-209, 319, 408 Угловой момент 150
Сверхпроводящие магниты 24, 159 Усиление сигналов благодаря ЯЭО 244, 363, 369, 372
Селективное облучение ядер 206, 396, 408 для 29Si 372
Сигналы ЯМР !Н ОН-протонов 178-180 Фазовое циклирование 291, 296, 328
HOD 181 Сильно связанные колебания 106, 116 Сильно связанные спины 174, 195 Симметрии операции и элементы 186-188, 255 Ферми резонанс 92, 93 Ферромагнитные примеси 162 Фрагментация 11, 13, 25, 29-31 Фрагментные ионы 11, 14, 84
Слабо связанные спины 174, 195, 196 Слабопольный и сильнопольный 164 Смешивания период (время) 313, 316, 317 Фурье преобразование 152, 158, 240, 286-289 Центр симметрии 104, 187
Спад свободной индукции (ССИ, FID) 152, 156, 239, 240 Спектроскопия комбинационного рассеяния 92, 93 Частично двойная связь 191, 364
Спектроскопия ЯМР с преобразованием Фурье 151-161 Частота
с использованием градиентного поля 328, 329 ларморова 152, 156, 158, 286
Спиновая релаксация 182, 362, 372 прецессии 152, 153, 288
Спиновое число 150, 151, 171, 182, 183, 238, 247, 361, 384-386 прикладываемого ВЧ-поля 152, 159, 160, 163
Спиновые системы 171-173, 176, 177, 197, 202, 211 эффективная 163
первого порядка 173-178 Частотное представление спектра 152, 158, 240
Предметный указатель
549
Частотные оси Vj и v2 в 2D-AMP 287-289
Четвертичные атомы углерода 299, 301, 308
Эквивалентность
магнитная (спин-спинового взаимодействия) 192-195
химическая (по химическому сдвигу) 186-192
Экранирование 163
Эксперименты ЯМР
COSY *Н - *Н 291-295, 301, 310, 312, 313
DEPT 252-255
HETCOR 295-298
НМВС 297, 299-301, 305-308, 313, 315, 325-327
HMQC 296-298, 301, 304, 305, 313, 314, 322, 324-326
HMQC-TOCSY 317
INADEQUATE 308-310, 402
NOESY 319, 420
ROESY 319, 321, 326, 328, 420
TOCSY (ID) 317
TOCSY (2D) 313, 316, 322, 414
Электрический квадрупольный момент 150, 182-184, 247
Электроотрицательность 128, 165, 168, 255-257, 264, 265
Энантиомеры 87, 189, 201
Энантиотопы 187, 188, 203, 388
Эрнста угол 242
Эстафетный перенос когерентности 313, 316, 317
Эталонные (стандартные) соединения 255, 363, 368, 373,
384-386
для 29Si 372
Эффект тяжелого атома 269
Эффективная частота 163
Ядерный магнитный момент 150, 151, 247, 360, 361, 384-386
Ядерный магнитный резонанс
на ядрах 19F 367-369
на ядрах 15N 361-367
на ядрах 31р 373-376
на ядрах 29Si 369-373
Ядерный эффект Оверхаузера (NOE, ЯЭО) 206-209, 242,
244, 248, 250, 251
для 29Si 369, 372
Оглавление
Предисловие к русскому изданию............................................................ 5
Предисловие 7
Предисловие к первому изданию........................................................... 10
1. Масс-спектрометрия...................................................................... 11
1.1. Введение........................................................................... 11
1.2. Техника эксперимента............................................................... 11
1.3. Методы ионизации................................................................... 13
1.3.1. Методы ионизации в газовой фазе............................................ 13
1.3.1.1. Ионизация электронным ударом........................................ 13
1.3.1.2. Химическая ионизация................................................ 13
1.3.2. Методы десорбционной ионизации............................................. 15
1.3.2.1. Полевая десорбция................................................... 15
1.3.2.2. Бомбардировка быстрыми атомами ..................................... 15
1.3.2.3. Плазменная десорбционная ионизация ................................. 16
1.3.2.4. Лазерная десорбционная ионизация.................................... 17
1.3.3. Методы ионизации при испарении.............................................. 17
1.3.3.1. Масс-спектрометрия с термораспылением............................... 17
1.3.3.2. Масс-спектрометрия с электрораспылением............................. 17
1.4. Анализаторы масс................................................................... 20
1.4.1. Магнитные секторные масс-спектрометры....................................... 20
1.4.2. Квадрупольные масс-спектрометры............................................. 21
1.4.3. Масс-спектрометр с ионной ловушкой.......................................... 22
1.4.4. Времяпролетный масс-спектрометр............................................. 23
1.4.5. Масс-спектрометр с преобразованием Фурье.................................... 23
1.4.6. Тандемная масс-спектрометрия................................................ 24
1.5. Интерпретация масс-спектров с ионизацией электронным ударом........................ 25
1.5.1. Идентификация пика молекулярного иона....................................... 26
1.5.2. Определение молекулярной формулы............................................ 26
1.5.2.1. Молекулярный ион и изотопные пики в масс-спектрах низкого разрешения ... 26
1.5.2.2. Молекулярный ион в спектре высокого разрешения...................... 27
1.5.3. Использование молекулярной формулы. Показатель водородной ненасыщенности ... 28
1.5.4. Фрагментация................................................................ 29
1.5.5. Перегруппировки............................................................. 31
1.6. Масс-спектры некоторых классов органических соединений............................. 32
1.6.1. Углеводороды ............................................................... 32
1.6.1.1. Насыщенные углеводороды............................................. 32
1.6.1.2. Алкены (олефины).................................................... 34
1.6.1.3. Ароматические и алкилароматические углеводороды..................... 35
1.6.2. Гидроксилсодержащие соединения.............................................. 36
1.6.2.1. Спирты.............................................................. 36
1.6.2.2. Фенолы.............................................................. 37
Оглавление 551
1.6.3. Простые эфиры............................................................... 38
1.6.3.1. Алифатические эфиры и ацетали....................................... 38
1.6.3.2. Ароматические эфиры................................................. 39
1.6.4. Кетоны...................................................................... 39
1.6.4.1. Алифатические кетоны................................................ 39
1.6.4.2. Циклические кетоны.................................................. 40
1.6.4.3. Ароматические кетоны................................................ 40
1.6.5. Альдегиды................................................................... 41
1.6.5.1. Алифатические альдегиды............................................. 41
1.6.5.2. Ароматические альдегиды............................................. 42
1.6.6. Карбоновые кислоты.......................................................... 42
1.6.6.1. Алифатические кислоты............................................... 42
1.6.6.2. Ароматические кислоты............................................... 43
1.6.7. Сложные эфиры карбоновых кислот............................................. 43
1.6.7.1. Алифатические сложные эфиры......................................... 43
1.6.7.2. Бензильные и фенильные сложные эфиры................................ 44
1.6.7.3. Сложные эфиры ароматических кислот.................................. 45
1.6.8. Лактоны..................................................................... 45
1.6.9. Амины....................................................................... 45
1.6.9.1. Алифатические амины................................................. 45
1.6.9.2. Циклические амины................................................... 46
1.6.9.3. Ароматические амины (анилины)....................................... 46
1.6.10. Амиды...................................................................... 47
1.6.10.1. Алифатические амиды................................................ 47
1.6.10.2. Ароматические амиды................................................ 47
1.6.11. Алифатические нитрилы...................................................... 47
1.6.12. Нитросоединения............................................................ 48
1.6.12.1. Алифатические нитросоединения...................................... 48
1.6.12.2. Ароматические нитросоединения ..................................... 48
1.6.13. Алифатические нитриты...................................................... 48
1.6.14. Алифатические нитраты...................................................... 48
1.6.15. Соединения серы............................................................ 48
1.6.15.1. Алифатические тиолы (меркаптаны)................................... 49
1.6.15.2. Алифатические сульфиды............................................. 49
1.6.15.3. Алифатические дисульфиды........................................... 50
1.6.16. Галогенсодержащие соединения............................................... 50
1.6.16.1. Алифатические хлорпроизводные...................................... 52
1.6.16.2. Алифатические бромиды.............................................. 52
1.6.16.3. Алифатические иодиды .............................................. 52
1.6.16.4. Алифатические фториды.............................................. 52
1.6.16.5. Бензилгалогениды .................................................. 52
1.6.16.6. Ароматические галогениды .......................................... 52
1.6.17. Гетероароматические соединения............................................. 53
Литература.............................................................................. 53
Общая 53
Таблицы данных и атласы масс-спектров............................................. 54
Специал ьные монографии........................................................... 54
Дополни тельная литература........................................................ 54
Упражнения.............................................................................. 55
Приложение А.Точные величины масс для молекулярных формул,
содержащих углерод, водород, азот и кислород............................................ 63
Приложение Б. Фрагментные ионы.......................................................... 84
Приложение В. Наиболее распространенные нейтральные фрагменты 86
552
Оглавление
2. Инфракрасная спектроскопия 88
2.1. Введение............................................................................. 88
2.2. Теория............................................................................... 88
2.2.1. Взаимодействие колебаний...................................................... 90
2.2.2. Водородная связь.............................................................. 93
2.3. Аппаратура........................................................................... 94
2.3.1. Диспергирующий ИК-спектрометр ................................................ 94
2.3.2. ИК-Спектрометр с преобразованием Фурье (интерферометр)........................ 95
2.4. Подготовка пробы..................................................................... 96
2.5. Интерпретация спектров............................................................... 97
2.6. Характеристические групповые частоты органических молекул............................ 99
2.6.1. Нормальные алканы (парафины)................................................. 100
2.6.1.1. Валентные колебания С-Н.............................................. 100
2.6.1.2. Деформационные колебания С-Н метильных групп....................... 101
2.6.2. Углеводороды с разветвленной цепью........................................... 102
2.6.2.1. Валентные колебания С-Н при третичном атоме углерода................. 102
2.6.2.2. Деформационные колебания С-Н в геминальных метильных группах........ 102
2.6.3. Циклоалканы...................................................................102
2.6.3.1. Валентные колебания С-Н...............................................102
2.6.3.2. Деформационные колебания С-Н..........................................102
2.6.4. Алкены........................................................................103
2.6.4.1. Валентные колебания связи С=С в несопряженных линейных алкенах ......103
2.6.4.2. Валентные колебания связей С-Н в алкенах.............................104
2.6.4.3. Деформационные колебания связей С-Н в алкенах.........................104
2.6.5. Алкины........................................................................105
2.6.5.1. Валентные колебания связи С=С .......................................105
2.6.5.2. Валентные колебания связей С-Н........................................105
2.6.5.3. Деформационные колебания связей С-Н...................................105
2.6.6. Моноядерные ароматические углеводороды .......................................105
2.6.6.1. Внеплоскостные деформационные колебания связей С-Н ...................106
2.6.7. Полиядерные ароматические соединения..........................................106
2.6.8. Спирты и фенолы...............................................................107
2.6.8.1. Валентные колебания связи О-Н ........................................107
2.6.8.2. Валентные колебани связи С-О..........................................108
2.6.8.3. Деформационные колебания связи О-Н....................................109
2.6.9. Простые эфиры, эпоксиды и пероксиды...........................................110
2.6.9.1. Валентные колебания С-О...............................................110
2.6.10. Кетоны.......................................................................111
2.6.10.1. Валентные колебания группы С=О ......................................111
2.6.10.2. Валентные и деформационные колебания фрагмента С-С(=О)-С.............114
2.6.11. Альдегиды....................................................................114
2.6.11.1. Валентные колебания связи С=О .......................................114
2.6.11.2. Валентные колебания связи С-Н .......................................115
2.6.12. Карбоновые кислоты...........................................................115
2.6.12.1. Валентные колебания группы О-Н ......................................115
2.6.12.2. Валентные колебания группы С=О ......................................116
2.6.12.3. Валентные колебания связи С-О и деформационные колебания связи О-Н ... 116
2.6.13. Карбоксилат-анион............................................................116
2.6.14. Сложные эфиры и лактоны ...................................................117
2.6.14.1. Валентные колебания группы С=О ......................................117
2.6.14.2. Валентные колебания связи С-О....................................... 118
2.6.15. Галогенангидриды кислот......................................................119
2.6.15.1. Валентные колебания группы С=О ......................................119
Оглавление 553
2.6.16. Ангидриды карбоновых кислот.......................................................119
2.6.16.1. Валентные колебания группы С=О ..........................................119
2.6.16.2. Валентные колебания связи С-О............................................119
2.6.17. Амиды и лактамы...................................................................119
2.6.17.1. Валентные колебания связи N-H ...........................................120
2.6.17.2. Валентные колебания группы С=О (полоса Амид I)...........................120
2.6.17.3. Деформационные колебания связи N-Н (полоса «Амид II»)....................121
2.6.17.4. Другие колебательные полосы..............................................122
2.6.17.5. Валентные колебания группы С=О в лактамах................................122
2.6.18. Амины.............................................................................122
2.6.18.1. Валентные колебания связи N-H ...........................................122
2.6.18.2. Деформационные колебания связей N-H......................................123
2.6.18.3. Валентные колебания связи C-N ...........................................123
2.6.19. Соли аминов.......................................................................123
2.6.19.1. Валентные колебания связи N-H........................................... 123
2.6.19.2. Деформационные колебания связей N-H......................................123
2.6.20. Аминокислоты и их соли ...........................................................123
2.6.21. Нитрилы...........................................................................125
2.6.22. Изонитрилы (R-N+=C~), цианаты (R-O-C=N), изоцианаты (R-N=C=O),
тиоцианаты (R-S-C=N) и изотиоцианаты (R-N=C=S)...........................................125
2.6.23. Соединения, содержащие группу -N=N................................................125
2.6.24. Ковалентные соединения, содержащие связи азот-кислород............................125
2.6.24.1. Валентные колебания группы N=O в нитро соединениях.......................126
2.6.25. Органические соединения серы......................................................127
2.6.25.1. Валентные колебания связи S-Н в тиолах ..................................127
2.6.25.2. Валентные колебания связей С-S и C=S в сульфидах.........................127
2.6.26. Соединения, содержащие связи сера - кислород......................................128
2.6.26.1. Валентные колебания фрагмента S=O в сульфоксидах.........................128
2.6.27. Органические галогенсодержащие соединения.........................................129
2.6.28. Соединения кремния................................................................129
2.6.28.1. Колебания связей Si-H....................................................129
2.6.28.2. Колебания связей SiO-H и Si-0............................................129
2.6.28.3. Валентные колебания связей Si-Hal........................................129
2.6.29. Соединения фосфора................................................................130
2.6.29.1. Валентные колебания связей Р=О и Р-О.....................................130
2.6.30. Гетероароматические соединения....................................................130
2.6.30.1. Валентные колебания связей С-Н...........................................130
2.6.30.2. Частоты валентных колебаний связей N-H...................................130
2.6.30.3. Валентные колебания кольца (скелетные полосы) ...........................130
2.6.30.4. Внеплоскостные деформационные колебания связей С-Н ......................130
Литература....................................................................................131
Введение в ИК-спектроскопию и теория......................................................131
Атласы ИК-спектров, справочники, специальная литература...................................131
Дополнительная литература.................................................................132
Упражнения....................................................................................133
Приложение А. Области прозрачности растворителей и иммерсионных масел ........................142
Приложение Б. Диапазоны характеристического поглощения органических соединений .... 143
Приложение В. Поглощение алкенов .............................................................148
Приложение Г. Поглощение фосфорорганических соединений........................................149
Приложение Д. Поглощение ароматических гетероциклических соединений...........................149
554
Оглавление
3. Спектроскопия ЯМР ПН....................................................................150
3.1. Введение...........................................................................150
3.2. Теория.............................................................................150
3.2.1. Магнитные свойства ядер.....................................................150
3.2.2. Возбуждение ядер со спином !4...............................................151
3.2.3. Релаксация..................................................................156
3.3. Техника эксперимента и подготовка образца..........................................159
3.3.1. Техника эксперимента........................................................159
3.3.2. Чувствительность в экспериментах по ЯМР.....................................160
3.3.3. Выбор растворителя .........................................................161
3.4. Химический сдвиг...................................................................163
3.5. Спин-спиновое взаимодействие, мультиплеты, спиновые системы........................169
3.5.1. Простые и составные мультиплеты первого порядка.............................169
3.5.2. Спиновые системы первого порядка............................................173
3.5.3. Номенклатура Попла для спиновых систем......................................174
3.5.4. Другие примеры простых спиновых систем первого порядка......................174
3.5.5. Анализ спектров первого порядка.............................................176
3.6. Протоны у атомов кислорода, азота и серы. Обменивающиеся протоны...................178
3.6.1. Протоны у атома кислорода...................................................178
3.6.1.1. Спирты..............................................................178
З.6.1.2. Вода ...............................................................181
3.6.1.3. Фенолы..............................................................181
3.6.1.4. Енолы ..............................................................181
3.6.1.5. Карбоновые кислоты..................................................181
3.6.2. Протоны у атома азота.......................................................182
3.6.3. Протоны у атома серы........................................................184
3.6.4. Протоны вблизи от атомов хлора, брома и иода................................184
3.7. Константы спин-спинового взаимодействия протонов
с другими важными ядрами (19F, D,31Р, 29Si и 13С) .....................................184
3.7.1. Константы взаимодействия протонов с ядрами 19F...........................184
3.7.2. Константы взаимодействия протонов с ядрами дейтерия.........................184
3.7.3. Взаимодействие протонов с ядрами 31Р .......................................186
3.7.4. Взаимодействие протонов с ядрами 29Si.......................................186
3.7.5. Взаимодействие протонов с ядрами 13С .......................................186
3.8. Эквивалентность по химическому сдвигу..............................................186
3.8.1. Определение эквивалентности по химическому сдвигу
при использовании операций симметрии...............................................186
3.8.1.1. Обмен позициями при вращении вокруг простой оси симметрии (С„)......187
3.8.1.2. Обмен позициями при отражении в плоскости симметрии (а).............187
3.8.1.3. Обмен при инверсии в центре симметрии (/)...........................187
3.8.1.4. Обмен позициями при проведении операций симметрии отсутствует...... 188
3.8.2. Определение эквивалентности химических сдвигов с помощью метки
(или замещения)....................................................................189
3.8.3. Эквивалентность химических сдвигов, обусловленная быстрым
взаимопревращением структур....................................................... 190
3.8.3.1. Кето-енольное равновесие............................................190
3.8.3.2. Взаимопревращение структур с «частично двойной связью»
(заторможенное вращение).....................................................191
3.8.3.3. Взаимопревращение в результате инверсии циклов......................191
3.8.3.4. Вращение вокруг простой связи в насыщенных цепях....................191
3.9. Магнитная эквивалентность (эквивалентность спин-спинового взаимодействия)..........192
Оглавление
555
3.10. Жесткие системы АМХ, АВХ и АВС
с тремя константами спин-спинового взаимодействия....................................195
3.11. Конформационно подвижные линейные системы с открытой цепью.
Виртуальное взаимодействие...........................................................196
3.11.1. Несимметричные цепи......................................................197
3.11.1.1. 1-Нитропропан....................................................197
3.11.1.2. 1-Гексанол.......................................................197
3.11.2. Симметричные цепи .......................................................198
3.11.2.1. Диметилсукцинат..................................................198
3.11.2.2. Диметилглутарат..................................................198
3.11.2.3. Диметиладипат....................................................199
3.11.3. Менее симметричные цепи..................................................200
3.11.3.1. З-Метилглутаровая кислота ...................................... 200
3.12. Хиральность....................................................................201
3.12.1. Один хиральный центр. Ипсенол............................................201
3.12.2. Два асимметрических центра...............................................203
3.13. Вицинальное и геминальное спин-спиновое взаимодействие.........................204
3.14. Дальнее спин-спиновое взаимодействие...........................................205
3.15. Селективное подавление спин-спинового взаимодействия. Двойной резонанс.........206
ЗЛб.Ядерный эффект Оверхаузера, разностная спектроскопия,
пространственная сближенность протонов............................................206
3.1/. Заключение.....................................................................209
Литература...........................................................................210
Учебные издания и монографии.....................................................210
Каталоги спектров, спектральных данных и специальные темы........................211
Дополнительная литература........................................................211
Упражнения...........................................................................211
Приложение А. Химические сдвиги протонов в алифатических соединениях.................222
Приложение Б. Влияние на химические сдвиги протонов двух
или трех непосредственно присоединенных заместителей .............................225
Приложение В.Химические сдвиги протонов в алициклических
и гетероциклических системах .....................................................227
Приложение Г. Химические сдвиги протонов в ненасыщенных и ароматических системах . . . 228
Приложение Д. Протоны, на которые влияет водородная связь (протоны у гетероатомов) ... 231
Приложение Е. Константы спин-спинового взаимодействия................................232
Приложение Ж. Химические сдвиги и мультиплетности остаточных протонов
в коммерчески доступных дейтерированных растворителях (Merck Со., Inc.) ..........234
Приложение 3. Химические сдвиги наиболее распространенных растворителей
в следовых количествах............................................................235
Приложение И. Протонные химические сдвиги аминокислот в D2O..........................237
4. Спектроскопия ЯМР 13С ...............................................................238
4.1. Введение........................................................................238
4.2. Теория..........................................................................238
4.2.1. Подавление спин-спинового взаимодействия с протонами......................238
4.2.2. Шкала химических сдвигов..................................................240
4.2.3. Спин-решеточная релаксация (время Г]).....................................241
4.2.4. Ядерный эффект Оверхаузера................................................244
4.2.5. Константы спин-спинового взаимодействия 13С-*Н (JCH)......................244
4.2.6. Чувствительность..........................................................246
4.2.7. Растворители..............................................................247
556
Оглавление
4.3. Интерпретация спектра ЯМР 13С диэтилфталата.......................................248
4.4. Количественный анализ спектров ЯМР 13С............................................250
4.5. Химическая эквивалентность........................................................252
4.6. Спектры DEPT......................................................................252
4.7. Классы химических соединений и их химические сдвиги...............................255
4.7.1. Алканы......................................................................256
4.7.1.1. Неразветвленные (линейные) и разветвленные алканы......256
4.7.1.2. Влияние заместителей в алканах ....................................257
4.7.1.3. Циклоалканы и насыщенные гетероциклические соединения..............258
4.7.2. Алкены......................................................................258
4.7.3. Алкины......................................................................260
4.7.4. Ароматические соединения....................................................261
4.7.5. Гетероароматические соединения..............................................263
4.7.6. Спирты......................................................................263
4.7.7. Эфиры, ацетали, эпоксиды....................................................263
4.7.8. Галогенсодержащие соединения................................................264
4.7.9. Амины.......................................................................265
4.7.10. Тиолы, сульфиды, дисульфиды................................................265
4.7.11. Функциональные группы, содержащие углерод..................................265
4.7.11.1. Кетоны и альдегиды................................................265
4.7.11.2. Карбоновые кислоты, сложные эфиры, хлорангидриды,
ангидриды, амиды и нитрилы...................................................268
4.7.11.3. Оксимы .......................................................... 268
Литература.............................................................................268
Общая..............................................................................• 268
Каталоги спектров, базы данных по спектрам.........................................268
Упражнения.............................................................................269
Приложение А. Химические сдвиги 13С, константы спин-спинового взаимодействия
и мультиплетность сигналов растворителей, часто используемых в спектроскопии ЯМР . . . 280
Приложение Б.Химические сдвиги 13С обычных растворителей как примесей..................280
Приложение В.Диапазон химических сдвигов 13С для различных классов соединени...........282
Приложение Г. Химические сдвиги ЯМР 13С для некоторых природных соединений.............284
5. Корреляционная спектроскопия ЯМР. 2D ЯМР...............................................285
5.1. Введение..........................................................................285
5.2. Теоретические положения...........................................................286
5.3. Корреляционная спектроскопия .....................................................290
5.3.1. Корреляции ’Н-’Н: COSY......................................................291
5.4. Ипсенол: спектры ’Н-’Н COSY.......................................................292
5.4.1. Ипсенол: спектр 'Н-'Н COSY с двухквантовой фильтрацией......................292
5.4.2. Детектирование по ядрам углерода: 13С-*Н COSY (HETCOR)......................295
5.4.3. Детектирование по протонам: ’Н -13С COSY (HMQC).............................296
5.4.4. Ипсенол: спектры HETCOR и HMQC .............................................297
5.4.5. Ипсенол: детектирование по протонам; гетероядерные корреляции
через дальние константы *Н-13С (метод НМВС)..................................297
5.5. Кариофилленоксид..................................................................301
5.5.1. Кариофилленоксид: спектр DQF-COSY..........................................301
5.5.2. Кариофилленоксид: спектр HMQC .............................................301
5.5.3. Кариофилленоксид: спектры НМВС ............................................305
5.6. Корреляции 13С-13С: INADEQUATE....................................................308
5.6.1. Спектр INADEQUATE кариофилленоксида.........................................309
Оглавление
557
5.7. Лактоза......................................................................310
5.7.1. Спектр DQF-COSY лактозы................................................310
5.7.2. Спектр HMQC лактозы....................................................313
5.7.3. Спектр НМВС лактозы....................................................313
5.8. Эстафетный перенос когерентности: метод TOCSY................................313
5.8.1. Двумерный спектр TOCSY лактозы.........................................316
5.8.2. Одномерный спектр TOCSY лактозы........................................317
5.9. Метод HMQC-TOCSY ............................................................317
5.9.1. Спектр HMQC-TOCSY лактозы .............................................317
5.10. Метод ROESY.................................................................319
5.10.1. Спектр ROESY лактозы..................................................319
5.11. Тетрапептид (валин-глицин-серин-глутаминовая кислота) (VGSE)................319
5.11.1. Спектр COSY тетрапептида VGSE.........................................322
5.11.2. Спектр TOCSY тетрапептида VGSE........................................322
5.11.3. Спектр HMQC тетрапептида VGSE.........................................322
5.11.4. Спектр НМВС тетрапептида VGSE.........................................325
5.11.5. Спектр ROESY тетрапептида VGSE........................................326
5.12. Спектроскопия ЯМР с использованием градиентного поля........................328
Литература........................................................................329
Упражнения........................................................................330
6. Спектроскопия ЯМР на других важных ядах со спином 14..............................360
6.1. Введение.....................................................................360
6.2. Ядерный магнитный резонанс на ядрах 15N......................................361
6.3. Спектроскопия ЯМР на ядрах 19F...............................................367
6.4. Спектроскопия ЯМР на ядрах 29Si..............................................369
6.5. Спектроскопия ЯМР на ядрах 31Р...............................................373
6.6. Заключение...................................................................376
Литература........................................................................378
Упражнения........................................................................379
Приложение А. Свойства магнитно активных ядер.....................................384
7. Задачи и решения .................................................................387
7.1. Введение.....................................................................387
Задача 7.1. Решение ..............................................................389
Задача 7.2. Решение ..............................................................392
Задача 7.3. Решение ..............................................................396
Задача 7.4. Решение ..............................................................401
Задача 7.5. Решение...............................................................408
Задача 7.6. Решение ..............................................................413
Упражнения........................................................................420
8. Контрольные задачи............................................................... 427
8.1. Введение.................................................................... 427
Предметный указатель..................................................................546
Учебное издание
Серия: «Методы в химии»
Сильверстейн Роберт
Вебстер Фрэнсис
Кимл Дэвид
СПЕКТРОМЕТРИЧЕСКАЯ ИДЕНТИФИКАЦИЯ
ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Ведущий редактор канд. хим. наук Т. И. Почкаева
Редактор канд. хим. наук Е. Э. Григорьева
Художник Н. А. Новак
Технический редактор Е. В. Денюкова
Компьютерная верстка: К. А. Мордвинцев
Подписано в печать 29.08.11. Формат 60x90/8.
Усл. печ. л. 70. Тираж 700 экз. Заказ Р-1020.
Издательство «БИНОМ. Лаборатория знаний»
125167, Москва, проезд Аэропорта, д. 3
Телефон: (499)157-5272
e-mail: binom@Lbz.ru, http://www.Lbz.ru
При участии ООО «ЭМПРЕЗА»
Отпечатано в полном соответствии с качеством
предоставленного электронного оригинал-макета
в типографии филиала ОАО «ТАТМЕДИА» «ПИК «Идел-Пресс».
420066, г. Казань, ул. Декабристов, 2.
E-mail: idelpress@mail.ru
ВУЗОВСКАЯ И ПРОФЕССИОНАЛЬНАЯ ЛИТЕРАТУРА
ХИМИЯ
ИМЕЕТСЯ В ПРОДАЖЕ
Пентин Ю. А. Основы молекулярной спектроскопии /
Ю. А. Пентин, Г. М. Курамшина. — 2008. — 398 с. :
ил. — (Методы в химии).
В учебном пособии, написанном преподавателями кафедры
физической химии химического факультета МГУ, изложены
теоретические основы важнейших спектроскопических ме-
тодов исследования строения молекул: ИК, УФ, КР, ЯМР,
ЯКР, ЭПР, ФЭС и др. Обсуждаются природа взаимодействия
электромагнитного излучения с веществом, теория групп
и современные вычислительные возможности квантовой
химии. Рассмотрены возможности и ограничения каждого
метода, показаны схемы интерпретации спектров, приведены
необходимые сведения о приборах и методиках эксперимента.
Книга хорошо иллюстрирована, содержит упражнения и спи-
сок дополнительной литературы.
Для студентов химических и других естественно-научных
факультетов и вузов, а также для специалистов, которым при-
менение спектральных методов необходимо в практической
деятельности.
j ИЗДАТЕЛЬСТВО
«БИНОМ
Лаборатория знаний»
125167, Москва, проезд Аэропорта, д. 3
Телефон: (499)157-5272
e-mail: binom@Lbz.ru, http://www.Lbz.ru
Оптовые поставки:
(499)174-7616, 171-1954, 170-6674
Р. Сильверстейн, Ф. Вебстер, Д. Кимл
СПЕКТРОМЕТРИЧЕСКАЯ ИДЕНТИФИКАЦИЯ
ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
ик
г-0
мс
13с
1Н
,Я БЙНОМ I