



























































Автор: Черных В.П.
Теги: органическая химия химия учебник органические соединения химические опыты химические реакции химическая наука
ISBN: 5-7768-0131-1
Год: 1993




Текст
В.П.Черных, Б.С.Зименковский, И,С.Гриценко
ОРГАНИЧЕСКАЯ ХИМИЯ
В ТРЕХ КНИГАХ
Книга!
Основы строения органических соединений
Под редакцией профессора В. П. Черных
Утверждено Министерством здравоохранения Украины в качестве учебника
для студентов фармацевтических вузов t-*— - - -г фануЛ£УйТ№”""1 I УкраТгч-- : а ф*рчи• гвтичц-а I А К »Д С И I Я | ij. Хар«!в
!
„I ।
Видавництво «Основа» при Хэркгвському державному ун'|верситет!
1993
ББК 24.2
449
УДК 547
Рецензенты: д-р хим. наук проф. В. Д. Орлов (Харьковский го^ судэрственный университет), д-р фарм. наук проф, Б. А. Приймепко (Запорожский медицинский институт)
Черних В. П, та in,
Ч 49 Органична х!м1‘я / В. П. Черних, Б, С. Зимепковський, I. С. Гриценко: Шдручник для фарм. вуз!в i факультетов. У 3 кп.: Кн. 1. Осноаи будови оргашчних сполук.— X. ! Вид-во «Основах- при Харк. ун-т!, 1993,— 144 с.: 1л.
ISBN 5-7768-0131-1 (кп. 1)
ISBN 5-7768-0130-3
У п!дручнику внкладено фундаменталья! загальнотеоретячн! положения органично! xiMii э опнсом рсакшйноТ эдаткост) найважли-В1шнх клас!в оргаш'чних речовин.
Для студентов фармацсвтичпих вуз1в 1 факультетов.
Ч <2О-^2?2±Э.2- Замоине ББК 24.2
226-93
ГПдручник
Черних Валентин Петрович Зименковський Борис Семенович Гриценко 1ввн Семенович
ОРГАН1ЧНА Х1М1Я
У трьох книгах
Книгв 1
Основи будови орган!чних сполук
(на рос1йськ(й мов1)
Редактор А. Л. Амева
Художник обкладинкй О. Г. Круглое
Художшй редактор Т, П. Короленко Техшчний редактор Л, Т. бна Коректор 1. П. Сич
Эдано до овладения 20.08.92. П1дпвсаио до друку 04.11.92, Формат 60X90/1». Патр друк. № 2, Гар«1тура,л1терагурм"а. Друк високий, Умов. друк. арк. 9.
Умов. фарбо-в!дб. 9,25, 'Обл1к.-вид,г¥р^, 10. Вид. № 2225. Зям. 2-253, Замовне.
Видавництво «Основа» Ари Харк(вському державному университет/, 310003 Харк!в, мяАдая Идудддоя. . __
Харк1вськя книжкой в фабрика 1м. МГн,* Фрунзе. 310057, Харк1п. вул, Донецъ* Закаржевського. 6/8.
ISBN 5-7768-0131-1 (кн. 1) ® £ с.' зйХкиШ.
ISBN 5-7768*0130-3 И, С, Гриценко, 1993
ОГЛАВЛЕНИЕ
Предисловие.................................................... • .
ГЛАВА 1. ВВЕДЕНИЕ
1 1. Предмет органической химии..................................
1.2. Краткий обзор истории развития органической химии ..........
]’з" Развитие теоретических представлений о строении органических соединений ........................
1.4. Способы изображения органических молекул ........
Контрольные вопросы и упражнения.............................
ГЛАВА 2. КЛАССИФИКАЦИЯ И НОМЕНКЛАТУРА ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
2.1. Классификация органических соединений.................... 14
2.2. Номенклатура органических соединений..................... 18
Контрольные вопросы и упражнения.......................... 25
ГЛАВА 3. ХИМИЧЕСКАЯ СВЯЗЬ
3.1. Типы химических связей . . .............................. 27
3.2. Кванте помех эпические основы теории химической связи , . . , 32
3.2.1. Гибридизация атомных орбиталей....................... 35
З.Й.2. Ковалентные а- и л-евпэн ............................. 39
3.2.3. Основные характеристики ковалентных связей............. 42
Контрольные вопросы и упражнения.......................... 4fi
ГЛАВА 4. ВЗАИМНОЕ ВЛИЯНИЕ АТОМОВ В ОРГАНИЧЕСКИХ СОЕДИНЕНИЯХ
4.1. Индуктивный эффект .......................................... 48
4,2. Мезомерный эффект (эффект сопряжения) 50
4.3, Совместное проявление индуктивного и мезомерного эффектов заместителей ....................................................... 54
4.4. Сверхсопряжение (гиперконъюгация) ; , 56
4.5. Пространственные препятствия сопряжению................. . , 57
4.6. Способы изображения распределения электронкой плотности в молекулах. Понятие о резонансе...................................... 58
Контрольные вопросы н упражнения .............................. 60
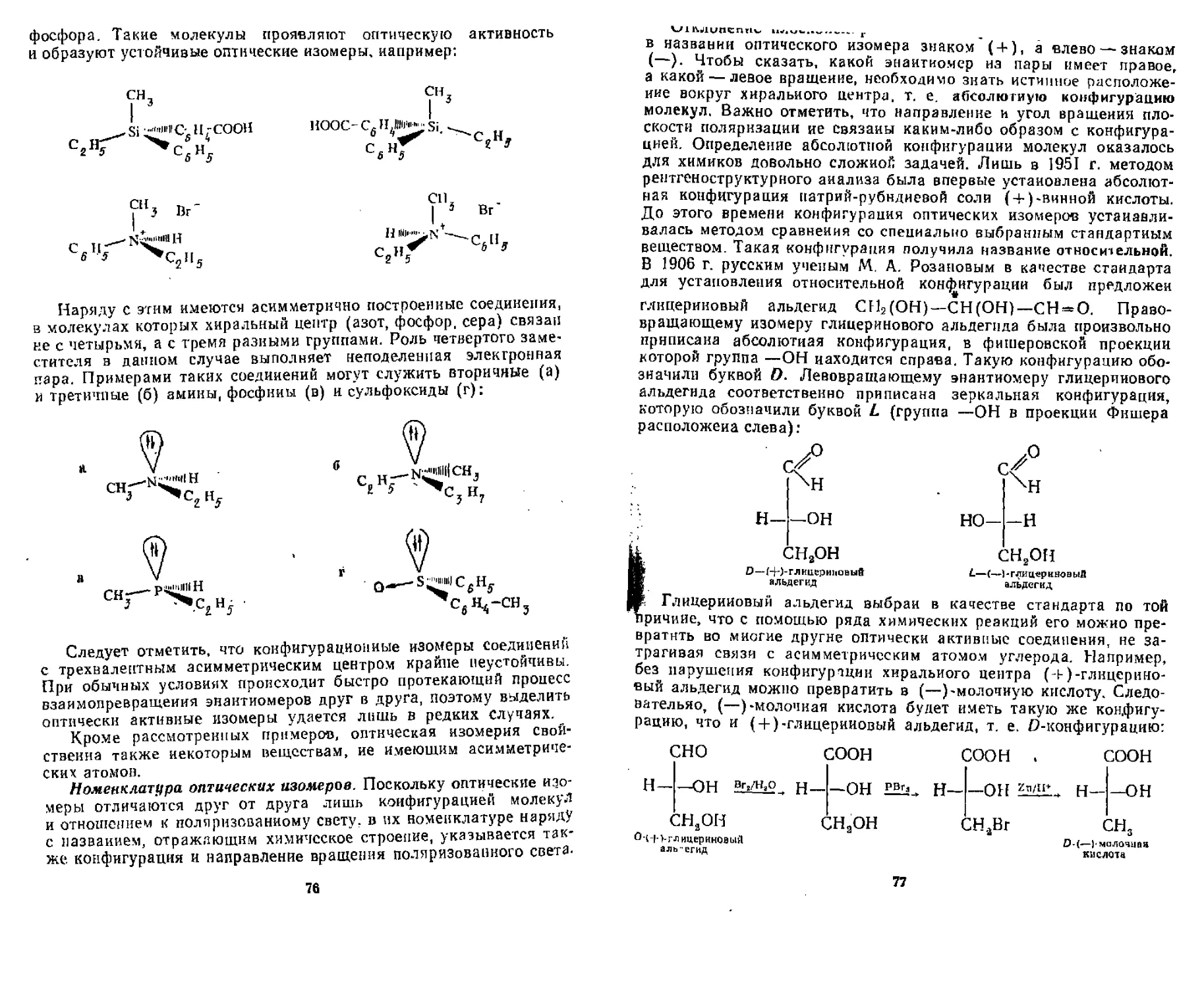
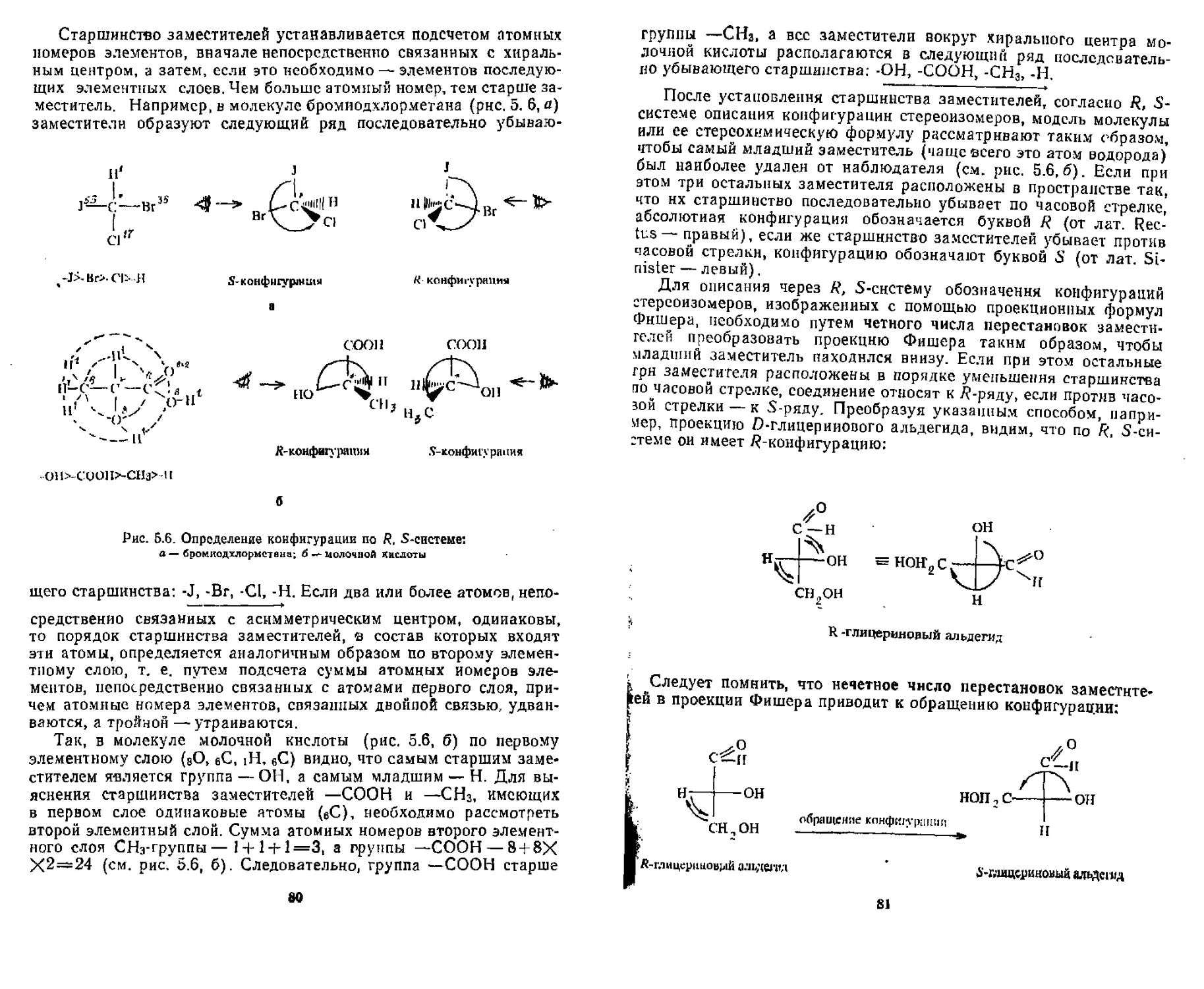
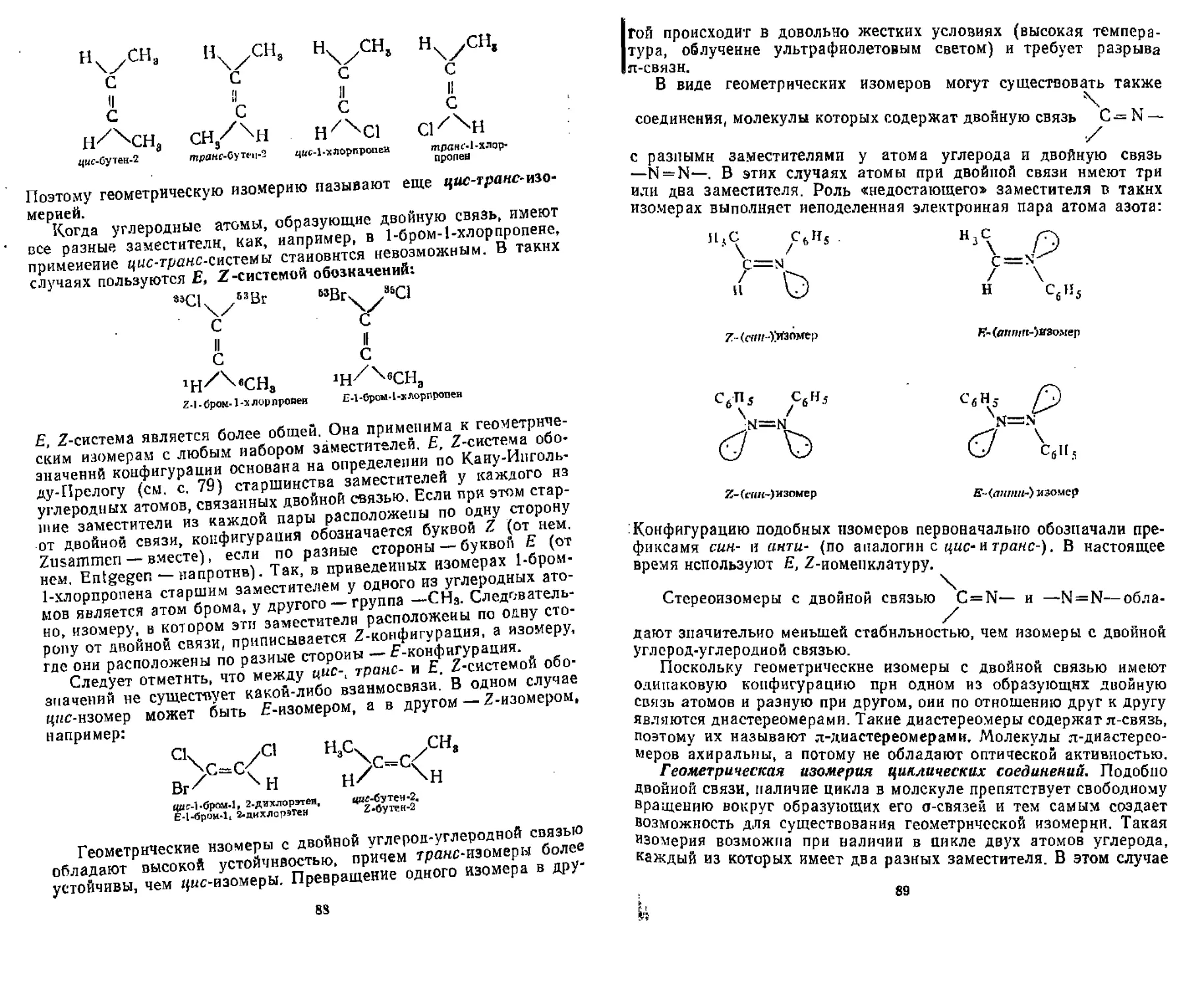
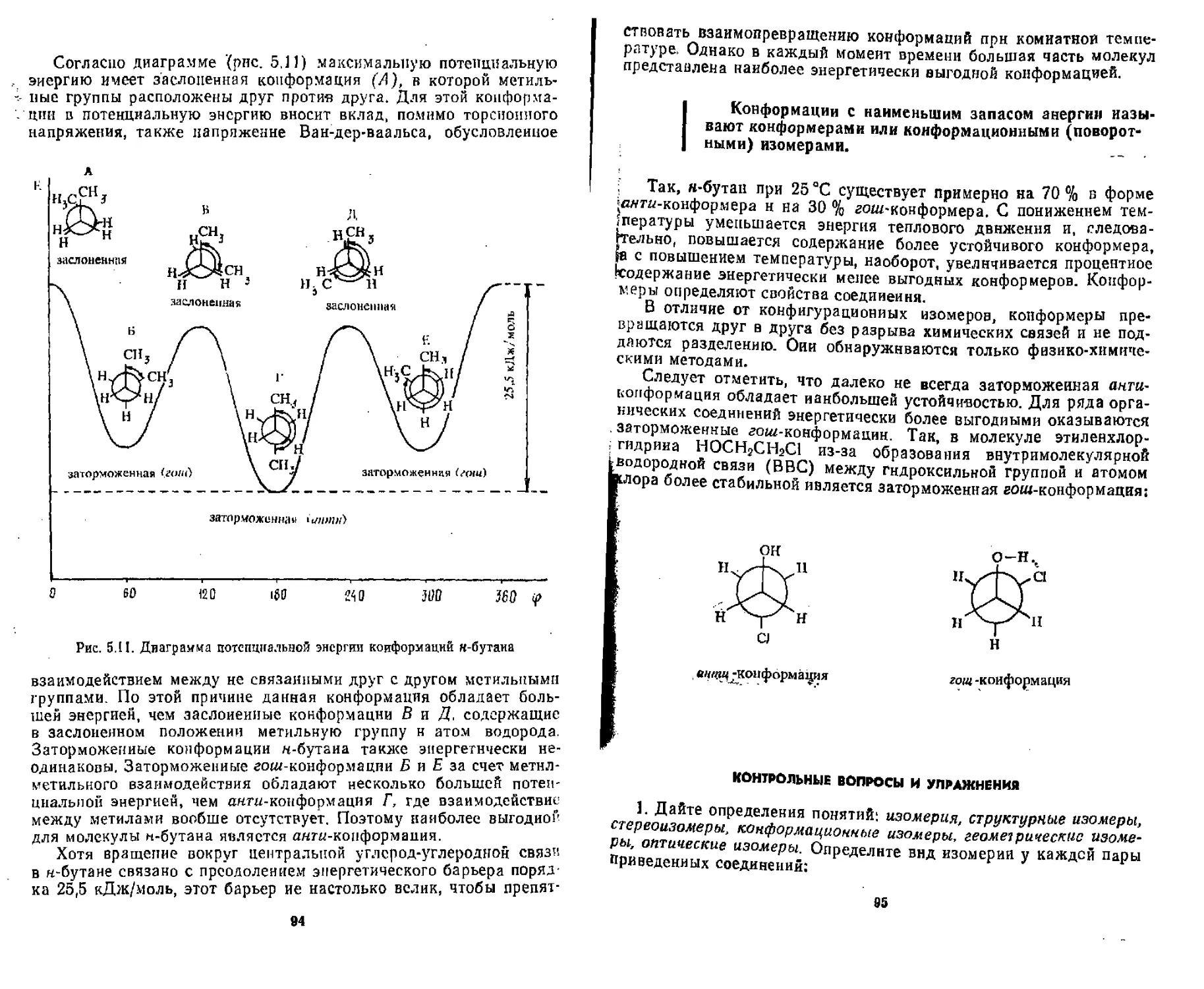
ГЛАВА 5, ИЗОМЕРИЯ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ. ПРОСТРАНСТВЕННОЕ СТРОЕНИЕ МОЛЕКУЛ
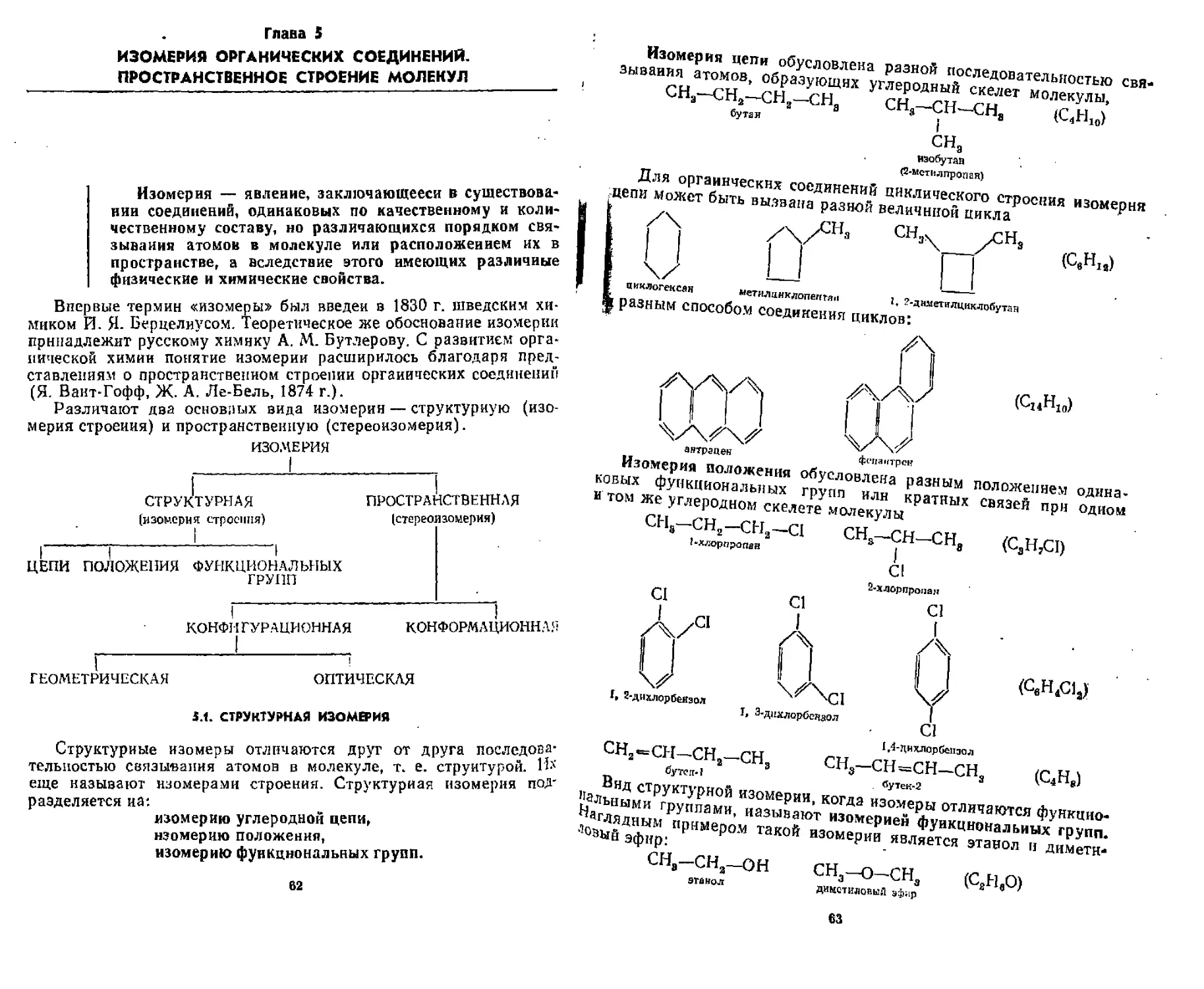
53- Структурная изомерия ......... 62
5 .2. Пространственная изомерия (стереоизомерия) 64
Roe Способы изображения пространственного строения . . . . , 65
9ПТнчсская изомерия . . .................................... 68
5 91 I еометРи,’еская изомерия .................................. 86
-^информационная (поворотная) изомерия...................... 91
Контрольные попроси и упражнения................................95
3
ГЛАВА 6. КИСЛОТНОСТЬ И ОСНОВНОСТЬ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
6.1, Кислотность и основность по теории Бренстеда.............. 98
6,1.1. Типы органических кислот................................ 99
6,1.2, Типы органических оснований 101
6.2. Кислоты и основания Льюиса............, ,............... 103
Контрольные вопросы и упражнения............................ Ю5
Г Л А В А 7. МЕТОДЫ УСТАНОВЛЕНИЯ СТРОЕНИЯ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
7.1, Химические методы........................................ 107
7-2. Физические (инструментальные) методы .................... 107
7.2.1. Электронная спектроскопия ............................. 109
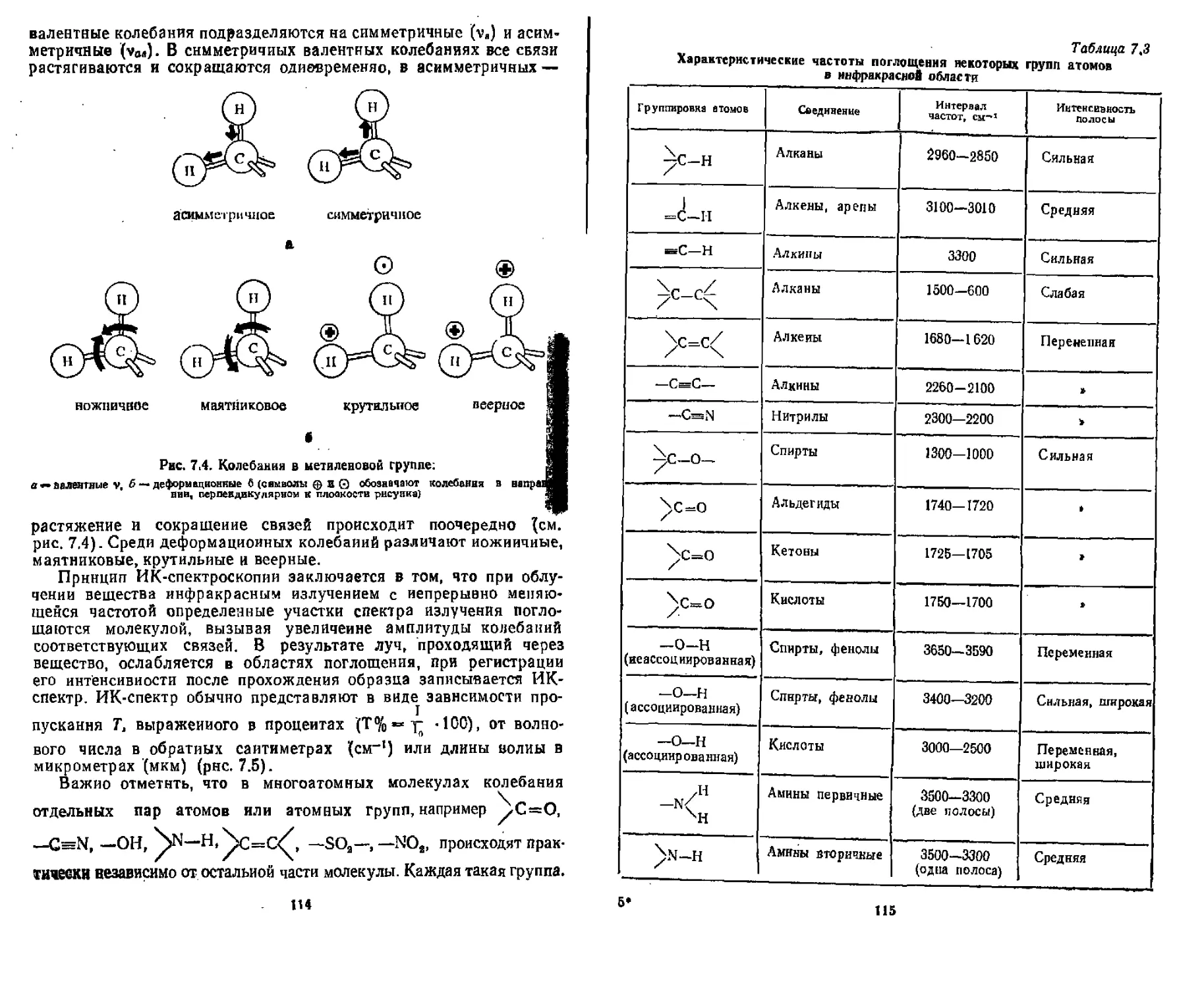
7,2.2, Инфракрасная спектроскопия , . ,....................... ИЗ
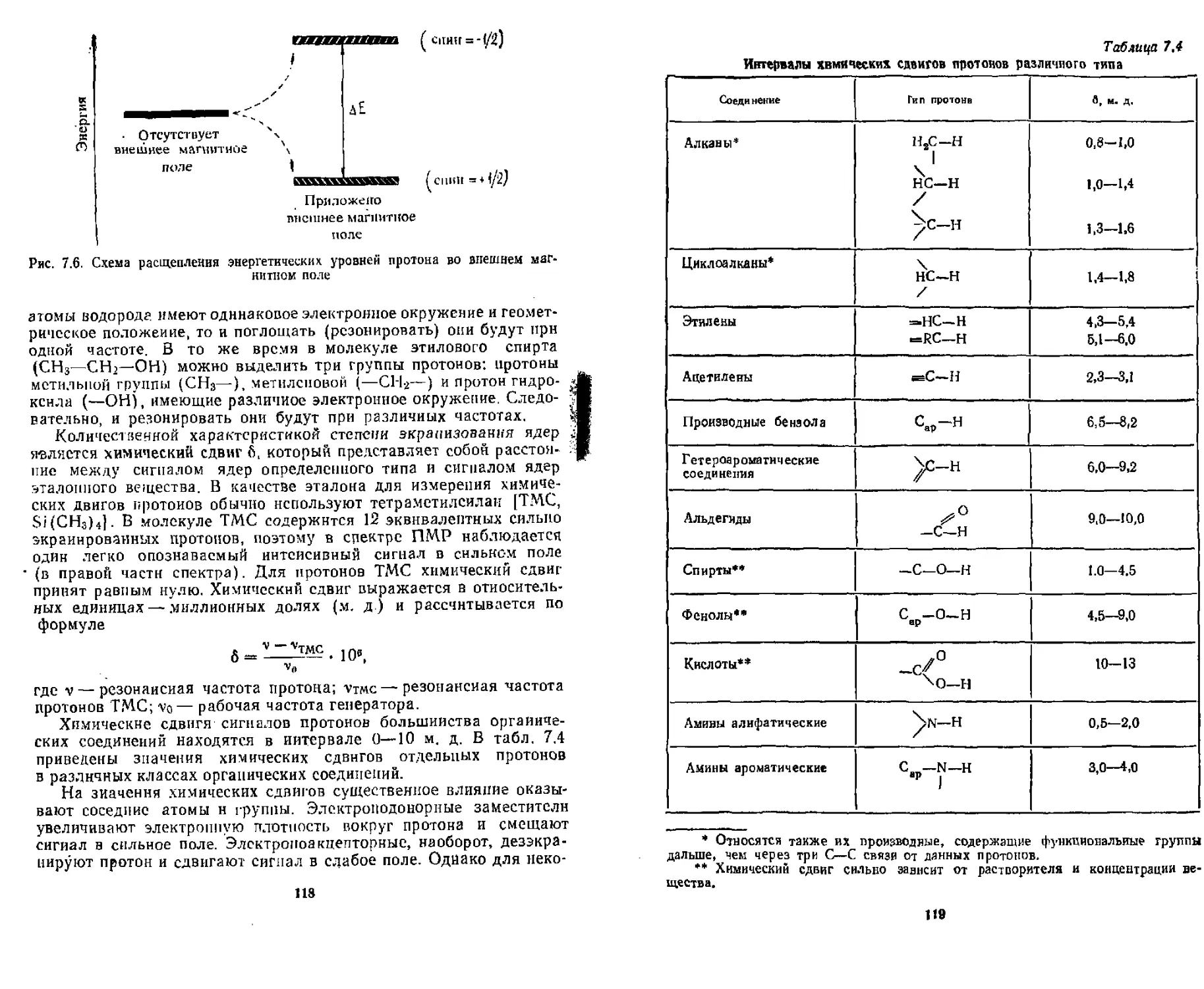
7,2.3. Спектроскопия ядерпого магнитного резонанса............ 117
7.2.4. Масс-спектрометрия................................. . . 121
7.2.5. Дифракционные методы........................... . , . , 123
Контрольные вопросы и упражнения......................... 123
ГЛАВА В. ОСНОВЫ ТЕОРИИ РЕАКЦИЙ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
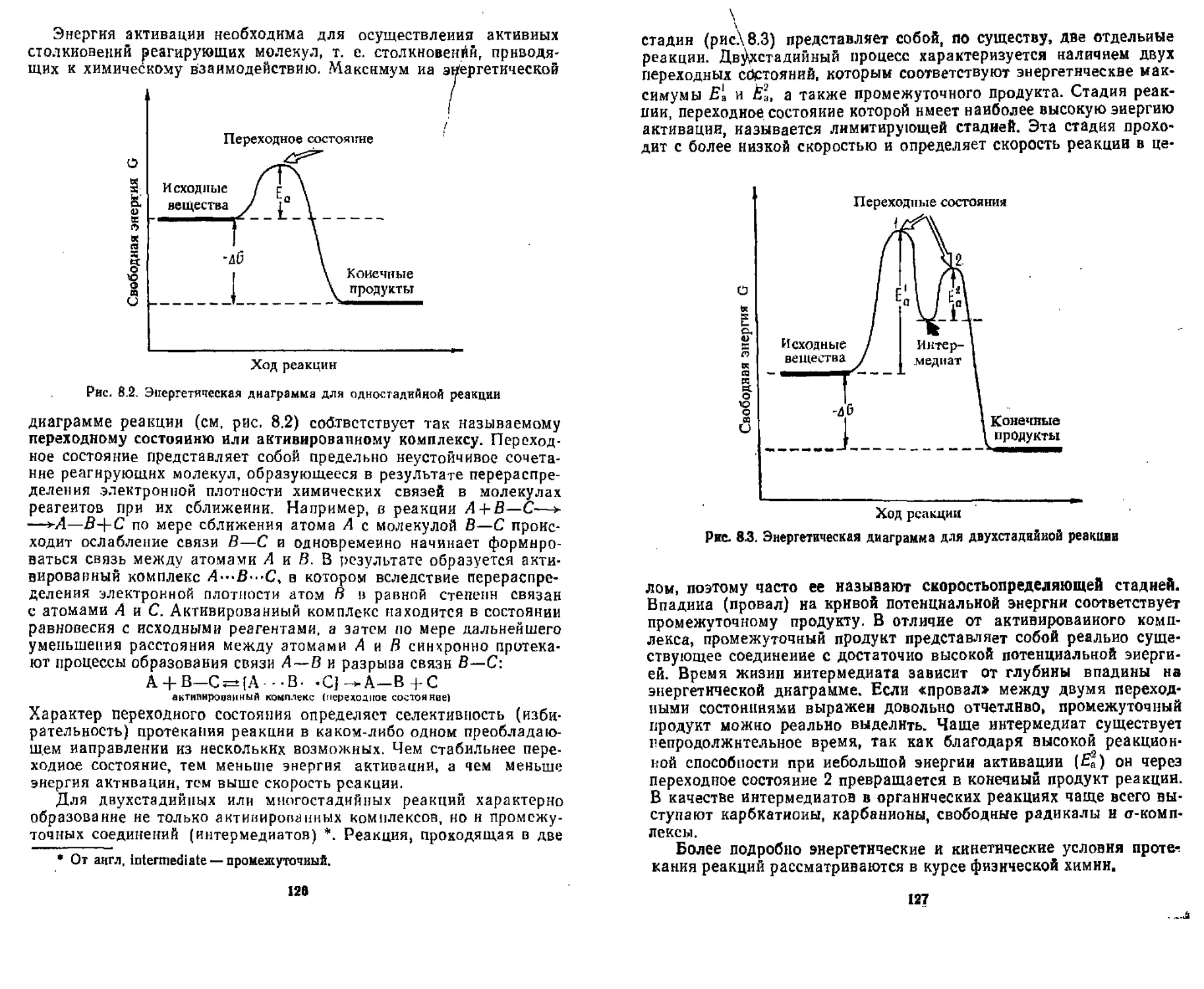
8.1. Энергетические условия протекания реакций................ 125
8.2. Понятие о механизме реакции............................. 128
8.3. Типы механизмов реакций................................. 128
8.4. Типы органических реакции................................ 130
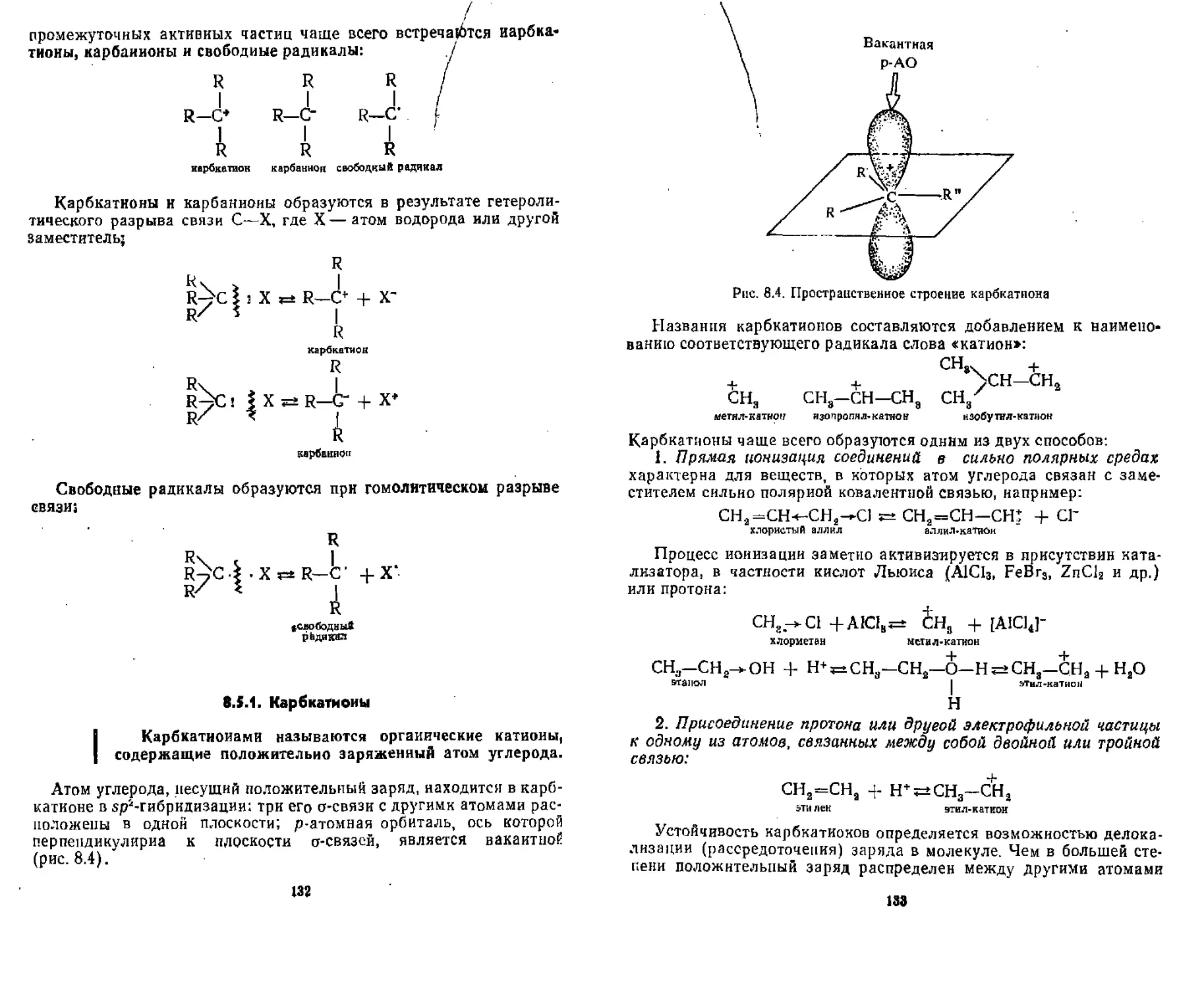
8.5. Промежуточные активные частицы........................ 131
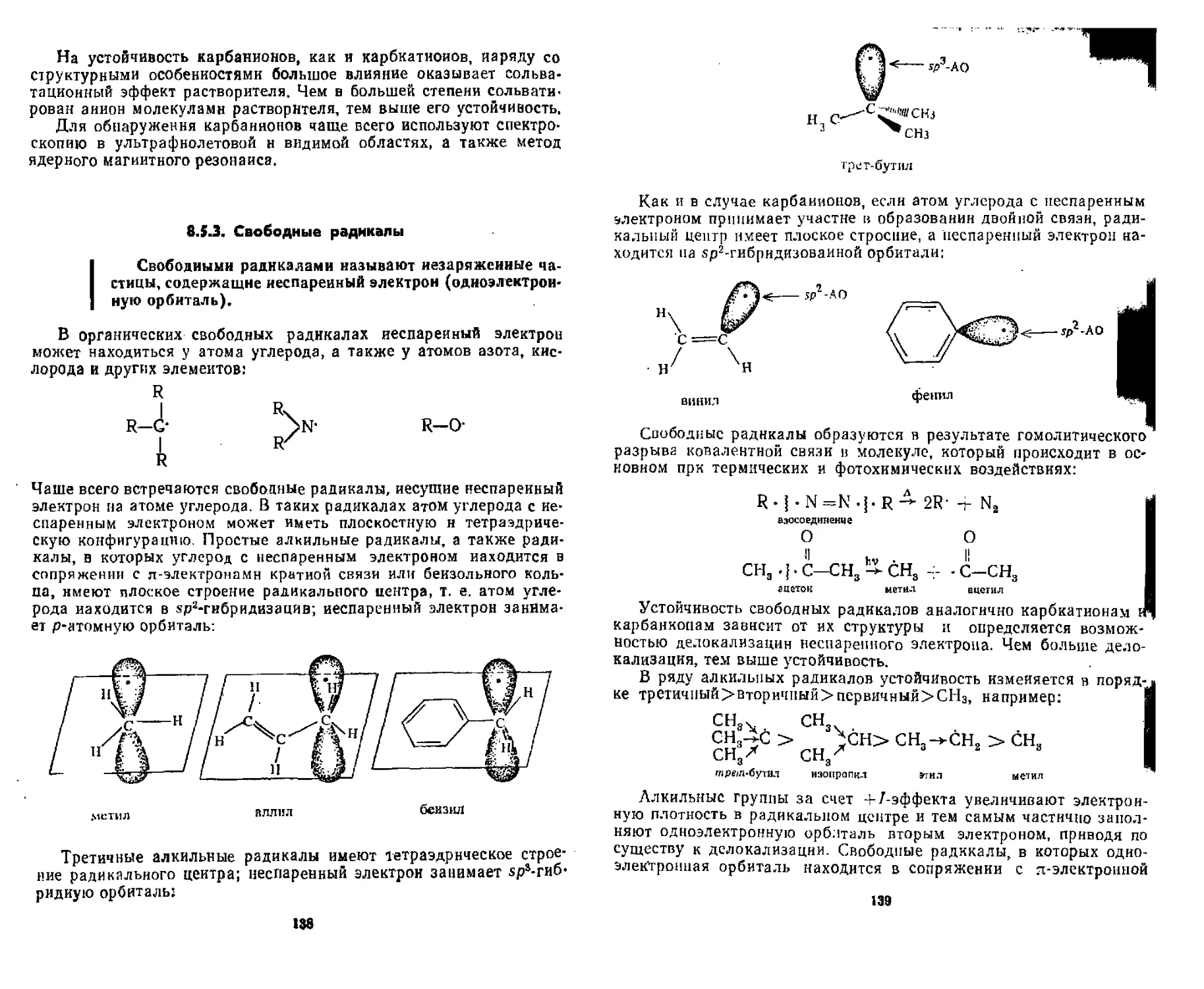
8 5.1. Карбкатионы ........................................ 132
8.5. 2. Карбанионы . . ...................................... 135
8.5. 3. Свободные радикалы ........................ ........ 138
Контрольные вопросы и упражнения........................... НО
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ.......................................... 142
ПРЕДИСЛОВИЕ
Органическая химия претерпевает период бурного развития, и се преподаяние требует поиска новых подходов. В предлагаемом учебнике изложены основы современной органической химии с учетом опыта преподавания этой дисциплины в Украинской фармацевтической академии и Львовском медицинском институте. ,
Учебник построен по принципу изложения фундаментальных общетеоретических положений органической химии с последующим описанием реакционной способности важнейших классов органических веществ. В самостоятельные разделы выделены гетероциклические и природные соединения.
В общетеоретическом разделе изложены основные теоретические положения органической химии, а именно: природа химической связи, взаимное влияние атомов в органических молекулах, изомерия, пространственное строение и геометрия молекул, кислотно-основные свойства органических веществ, физические методы установления строения соединений, энергетика, механизмы органических реакций, строение промежуточных активных частиц и др.
В основу изложения материала важнейших классов положена классификация органических соединений по функциональным группам, что позволяет избежать дублирования материала и способствует формированию у студента более целостных представлений о реакционной способности органических соединений.
Авторы акцентируют внимание на ряде аналитических реакций, лежащих в основе фармакопейных методов идентификации лекарственных веществ, и на значении прикладных аспектов органической химии в современной фармации.
Большое внимание уделено гетероциклическим соединениям, поскольку значительная часть лекарственных средств содержит в своей структуре гетероциклические ядра. О природных соединениях даны лишь общие сведения, необходимые для дальнейшего, более подробного изучения данной группы веществ в курсах биохимии, фармакогнозии, фармацевтической и токсикологической химии. Материал изложен с позиции квантовО'механических представлений, для многих реакций приведены механизмы их протекания,
Учебник издается в трех книгах: «Основы строения органических соединений», «Углеводороды и их функциональные производные», «Гетероциклические и природные соединения».
Студент, овладевший материалом, изложенным в учебнике, должен приобрести прочные знания современных основ органической химии и умение работать с оригинальной литературой. Издание представляет также определенный интерес для аспирантов, преподавателей и других специалистов, работающих в области органической химии и фармации.
Авторы выражают благодарность преподавателям кафедры органической химии Украинской фармацевтической академии Т, А, Колесниковой, Л. А. Шем--КУ- Г. М. Солельник, В. И. Гридасову, Е. Л. Снитковскому, С. Н. Коваленко, также преподавателям кафедры органической и биоорганической химии Львов-„ °™ медицинского института 3. Я. Паращук, М. М. Орлинскому, В. А, Муэы-а_ Эмее-Ми сен ко, Н, Е. Плевачук, О, В. Гаврил юк, И. Д. Ком арице
а -> ’£стие в подготовке материала, совеп1Т1?РЫ ЙуДут пРизнательны читателям за критические замечания, полезные ы и пожелания относительна содержания и оформления учебника,
Глава 1 ВВЕДЕНИЕ
1.1. ПРЕДМЕТ ОРГАНИЧЕСКОЙ ХИМИИ
Органическая хнмня как самостоятельная наука сформировалась в начале XIX в. Название «органическая» предложено шведским химиком И. Я. Берцелиусом в 1808 г. Оно произошло от слова «организм», поскольку в то время предметом изучения органической химии являлись вещества, образующиеся только в живых Организмах якобы с участием особой «жизненной силы» (виталистические воззрения). И хотя последующее развитие показало ошибочность виталистического взгляда па происхождение органических веществ, первоначальное название сохранилось, ио смысл его принципиально изменился. По признаку наличия во всех органических соединениях атома углерода немецкий химик X. Гмелин в 1848 г. дал определение органической химии как химии соеди-мений углерода. Однако, как известно, существует ряд соединений углерода, относящихся к неорганическим веществам, иапрнмер СО, СО21 солн угольной н цианистоводородной кислот, Учитывая это, другой немецкий химик — К. Шорлеммер предложил более точное определение органической химии: наука, изучающая углеводороды и их производные.
Современная органическая химия — одна из обширных областей естествознания. Выделение ее в самостоятельный раздел химической науки вызвано большим количеством и многообразием органических соединений, существенным различием их реакционной способности, а также исключительной важностью органической химии в жизни человека и общества.
В настоящее время известно свыше пяти миллионов органических веществ, тогда как неорганических — лишь около 700 тысяч, Каждый год получают примерно 200 тысяч новых органических соединений.
Органические вещества играют важную роль в жизнедеятельности растительных и животных оргаггизмов, являясь основой проявления жизни. Достижения органической химии широко используются в повседневной жизни человека — это пластмассы и синтетический каучук, красящие и взрывчатые вещества, искусственные волокна и топливо, лекарственные вещества. В системе фармацевтического образования большое значение придается изучению органической химии, поскольку около 90 % лекарственных препаратов составляют органические соединения.
6
1.1, КРАТКИЙ ОБЗОР ИСТОРИИ РАЗВИТИЯ ОРГАНИЧЕСКОЙ ХИМИИ
Развитие органической химии в историческом аспекте можно условно разделить на четыре периода: эмпирический (середина XVII— конец XVIII в.), аналитический (конец XVIII — середина XIX в.), структурный (вторая половина XIX — начало XX в.) и современный (начало XX в,— до наших дней).
Эмпирическому периоду предшествовал этап многовекового накопления фактического материала в процессе практической деятельности человека. Так, в Египте и Индии издавна было развито искусство крашения тканей природными красителями, Древние римляне и египтяне умели варить мыло. С глубокой древности люди использовали процесс брожения для получения спиртных напитков. Немало природных веществ применялось в древией медицине.
В средние века (VIII—XVI вв.) — период господства алхимии— знания об органических веществах развивались крайне медленно. Некоторый шаг вперед в это время был сделан лишь благодаря совершенствованию методов перегонки: выделены некоторые эфирные масла, получен винный спирт, ацетон.
В первоначальный период развития химии исследователи не замечали различий между веществами, выделенными нз неживой природы и полученными нз растений и животных. Однако в дальнейшем стали обращать внимание на различную устойчивость и химическое поведение веществ живой и неживой природы, что послужило причиной разделения химии во второй половине XVII в. на минеральную и химию растений и животных. В это время органическая химия начала формироваться как химия растении и животных (эмпирический период), К концу XVIII в, были достигнуты заметные успехи в получении индивидуальных органических веществ: из растений выделены винная, лимонная, яблочная, молочная, слизевая кислоты (К. Шееле, 1769—1785 гг,) и другие вещества, получен ряд органических соединений животного происхождения — мочевина (И, Руэль, 1773 г.), мочевая кислота (К. Шееле) и др. Однако исследования в эмпирическом периоде проводились еще без четких теоретических представлений, методом проб и ошибок.
В копие XVIII в. и первой половине XIX в. основное внимание химиков было сосредоточено па изучении качественного н количественного состава полученных веществ (аналитический период). В это время французский химик Л. Лавуазье установил, что вещества и растительного, я животного происхождения могут содержать углерод, Кроме того, в начале XIX в, обнаружен ряд растительных и животных веществ (щавелевая кислота, жиры я лр.). Полученный материал позволил сделать первые теоретические обобщения. Поскольку, как показали исследования, между химией растений и животных принципиальной разницы пет, в этот период формируется общее понятие «органическое вещество» и возникает термин «органическая химия» (Й. Я. Берцелиус,
7
1808 г,). Однако многие ученые того времени, в том числе и И. Я, Берцелиус, имели виталистические воззрения. Они полагали, что органические вещества присущи только живым организмам н образуются под влиянием особой «жизненной силы». Первый сокрушительный удар ио витализму был нанесен в 1828 г., когда немецкому химику Ф. Вёлеру удалось получить синтетическим путем мочевину. В письме к своему учителю Берцелиусу Велер тогда писал; «Я должен Вам заявить, что могу делать мочевину, не нуждаясь прн этом в почках и вообще в животных, будь это человек илн собака». Вскоре последовал и ряд других блестящих исследований — синтез уксусной кислоты, осуществленный Г, Кольбе (1845 г.), жиров — М. Бертло (1854 г.), сахаристого вещества — А. М. Бутлеровым (186] г.) и др. Успешно развивающийся органический синтез продемонстрировал ошибочность и необоснованность виталистических взглядов. Эти достижения аналитического периода способствовали утверждению материалистической точки зрения на единство природы и создали предпосылки для выработки прочной теоретической основы органической химии.
Крупнейшим событием в развитии органической химии явилось создание в 60-х гг. XIX в. великим русским ученым А. М. Бутлеровым теории химического строения органических соединений. С этого времени начинается структурный период бурного развития органической химии, в течение которого она окончательно сформировалась как самостоятельная наука, Первостепенное значение приобретает органический синтез, с помощью которого получены многие, не найденные в природе вещества. В это время было установлено строение многих природных соединений, сделан ряд важных открытий. В конце XIX—начале XX в. органический синтез начинает внедряться в химическую промышленность, Появляются отрасли по производству синтетических красителей, лекарственных
средств, взрывчатых веществ.
Современный период развития органической химии характеризуется значительно возросшими масштабами органического синтеза, широким применением в органической химии физических методов и вычислительной техники, все большим внедрением методов квантово-механической теории. Это позволило получить качественно новые представления о строении органических соединений, выяснить механизмы химических взаимодействий.
Из современной органической химии выросли и успешно развиваются такие самостоятельные направления, как химия гетероциклических соединений, химия высокомолекулярных соединений, антибиотиков, гормонов, витаминов, химия элементооргамических соединений,
Значительный вклад в развитие органической химии внесли многие выдающиеся русские и советские ученые: А. М. Бутлеров, И. Н, Зинин, В. В, Марковников, Е, Е. Вагнер, Н. Д. Зелинский, А. Е. Фаворский, А, Е. Чичибабин, С. В. Лебедев, М. Г. Кучеров, А. Е. Арбузов, А, Н. Несмеянов.
8
1.3. РАЗВИТИЕ ТЕОРЕТИЧЕСКИХ ПРЕДСТАВЛЕНИЯ О СТРОЕНИИ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Возникновение первых теорий о строении органических соединений приходится на аналитический период истории органической химии, В начале XIX в. при исследовании органических соединений химики обратили внимание на то, что в ряде химических превращений отдельные группы атомов, так называемые радикалы, в неизменном виде переходят из одного вещества в другое (1815 г., Ж- Гей-Люссак; 1832 г., Ф, Велер и Ю. Либих). Основываясь на созданной в 1812 г. Й, Я. Берцелиусом электрохимической теории, утверждавшей, что все химические соединения построены из электроположительных и электроотрицательных атомов или атомных групп, удерживаемых за счёт электростатических сил, ряд ученых создали первую теорию в органической химии — теорию радикалов (Ю. Либих, Ф. Велер, Й. Я. Берцелиус).
Авторы этой теории считали, что если радикалы не изменяются при химических превращениях, то подобно тому, как неорганические вещества состоят из атомов, органические—состоят из радикалов.
В 1833—1834 гг. французский химик Ж. Дюма, изучив действие хлора на органические соединения, установил, что в органических радикалах атомы водорода могут замещаться на хлор, т. е. радикал может изменяться. Эти исследования нанесли сокрушительный удар по теории радикалов.
В 40-х гг. XIX в. теорию радикалов сменила более совершенная теория тнлов, основоположником которой является французский химик Ш. Жерар. В отличие от теории радикалов, акцентирующей внимание в химических превращениях на неизменяемой части молекулы — радикале, теория типов появилась в результате обобщения наблюдений за изменяемой частью молекулы (то, что сегодня мы называем функциональной группой). Эти наблюдения легли h основу классификации органических соединений по типам химических превращений. Первоначально были выделены аналоги (типы) воды, хлористого водорода, аммиака, водорода, затем появился тип метана.
Тип воды
Тип хлороводорода
Тип аммиака
н1о
Н1и
вода щ
CIJ
хлороводород н
Н N н)
%Н»]0 тнловый спирт
СНА
Cl J хлористый метил СН81 Н н
металамин
СНА н J метан
СаН5
С2Н5
ди этиловый эфир
с3НА
Cl J
хлористый этил
сн3 сна н
Тип водорода
аммиак
Н\ н) водород
диметиламин
С,Н5| н )
этан
N
N
Приведенные типические формулы показывают, например, что при замещении в молекуле воды одного атома водорода на остаток С^Нз образуется этиловый спирт, двух—диэтиловый эфир и т. д.
К середине XIX в. по мере накопления большого экспериментального материала теория типов уже была не в состоянии объяснить многие факты. Пытаясь спасти теорию, ее сторонники вводили новые типы. Часто одно и то же соединение приходилось относить к нескольким типам и обозначать различными типическими формулами,
Для дальнейшего развития органической химии требовалась новая, более совершенная теория, Создание такой теории, заложившей научные основы органической химии, принадлежит великому русскому ученому А. М. Бутлерову, Используя открытие немецкого химика Ф, Кекуле о четырехвалентности атома углерода (1857 г.) и шотландского химика А. Купера о способности атомов углерода соединяться в длинные цепи (1858 г,), А. М, Бутлеров создал теорию химического строения органических соединений, основные принципы которой изложил в докладе «О теории химического строения» на Международном съезде естествоиспытателей и врачей в Шпейере 19 сентября 18G1 г.
Основные положения теории химического строения состоят в следующем:
1. Входящие в состав молекулы органических соединений атомы связаны друг с другом в строго определенном порядяе, согласно кх валентности. Последовательность связывания атомов в молекуле называется химическим строением,
2. Свойства вещества зависят не только от того, какие атомы и в каком количестве входят в состав молекулы, ио и от того, в какой последовательности они связаны между собой, т. е, от химического строения молекулы,
3. Образующие молекулу атомы или группы атомов, связанные непосредственно или через другие атомы, оказывают взаимное влияние друг на друга, от чего зависит реакционная способность молекулы.
4, Изучив реакционную способность вещества, можно установить его строение и, наоборот, по строению вещества судить о его свойствах.
Теория химического строения А. М, Бутлерова позволила не только систематизировать накопившийся на то времи в органической химии огромный материал, но и предсказать существование новых соединений, а также указать пути их синтеза, По значимости ее можно сравнить с периодической системой элементов Д. И, Менделеева. Блестящим подтверждением теории стало получение в 1867 г. А, М. Бутлеровым предсказанного им изобутана.
ю
Теория химического строения А. М. Бутлерова является важнейшей частью теоретического фундамента органической химии. Дальнейшему развитию теории посвящены работы ученика А, М, Бутлерова — В. В, Марковннкова, который установил закономерности взаимного влияния атомов в молекулах.
В 1874 г. теорию химического строения дополнила теория пространственного расположения атомов в молекулах (стереохимическая теория). Ее авторы—голландский химик Я. Вант-Гофф и французский химик Ж. Ле-Бель — независимо друг от друга пришли к выводу о тетраэдрической направленности связей атома углерода в пространстве (четыре валентности атома углерода направлены к углам тетраэдра, в центре которого находится углеродный атом).’
Свое дальнейшее развитие теория химического строения получила с введением в органическую химию электронных представлений, В 1916 г. американским ученым Г. Льюисом была предложена электронная теория химической саязи (так называемая теория электронных пар), согласно которой химическая связь в органических соединениях представлена парой электронов, выделяемых по одному каждым из связываемых атомов. Кроме того, Г. Льюис высказал предположение, что электронная пара, участвующая в образовании химической связи, может смещаться к одному из атомов. Эта мысль оказалась чрезвычайно важной и была положена в основу теории электронных смещений, В работах Р, Робинсона (1922 г.), а позднее К. Ингольда (1926—1934 гг.) были введены и развиты представления о смещении электронов в простых связях (индуктивный эффект) и кратных (мезомерный эффект). Теория электронных смещений получила довольно широкое распространение в органической химии, поскольку она позволяет установить зависимость между электронным строением и реакционной способностью органических соединений. Новым этапом в развитии теории химического строения явилось применение в 30-х годах нынешнего столетия в органической химии квантовой механики, В это время разработаны квантово-механические методы описания структуры молекул — метод молекулярных орбиталей (Дж. Леннард-Джопсои, Р, Малликен, Ф. Хунд, 1928— 1932 гг.) и метод валентных связей (Л, Полинг, Дж, Слейтер, 1931—1934 гг.). Используй метод валентных связей, Л, Полинг разработал теорию резонанса, которая позволила объяснить многие свойства ароматических систем. С помощью метода молекулярных орбиталей Э, Хюккель дал объяснение устойчивости ароматических систем и сформулировал теоретически обоснованное правило, позволяющее предсказать, будет система ароматической или пет (правило Хюккеля), Интенсивному развитию квантово-мехапических исследований способствовало появление в 60-х гг, 5<Х в. электронно-вычислительной техники.
11
1.4. СПОСОБЫ ИЗОБРАЖЕНИЯ ОРГАНИЧЕСКИХ МОЛЕКУЛ
СТРУКТУРНАЯ ФОРМУЛА
Структурная (графическая) формула отражает природу атомов, входящих в состав молекулы, их число и последовательность связывания, а также тип связей между ними. Химические связи в структурной формуле условно принято обозначать чертой:
н н н
J I I н—с—с—с— н
Н I н н—с—н
I н мзобутан
циклопропан этилен
ацетон
Для удобства записи часто используют сокращенную структурную формулу, в которой часть связей не обозначается, а приводятся лишь те, которые необходимы для однозначного описания структуры молекулы;
СН8-СН-СНЭ или (CH3)2CHCHS
CHS иэобутав СНа—СНа г____________!
или СНЙ—СНа—СН3
сна циклопропан
СН2=СНа СН3—С—СН8 или сн3сосн3
этилен |]
о
ацетон
Существует также упрощенный способ написания структурных формул, заключающийся в том, что углеродный остов молекулы изображают только посредством валентных связей без обозначения атомов углерода и связей С—Н:
NH
бутан циклопропан анилин фуран
Этот способ широко используется для изображения молекул карбоциклических и гетероциклических соединений.
12
МОЛЕКУЛЯРНАЯ ФОРМУЛА
Молекулярная (брутто-) формула показывает, какие атомы и каком количестве входят в состав молекулы. В качестве пример, приведены структурная и молекулярная формулы метанола:
СН3ОН СН4О
структурная формула молекулярная формула
При составлении молекулярной формулы прежде всего указы вают число атомов углерода и водорода, а затем в алфавитно: порядке (по латинским названиям элементов)—число остальпы элементов, входящих в состав молекулы. Например, для хлор уксусной кислоты С1СН2СООН молекулярная формула имеет вн С2НзС1Оа.
В отличие от структурных формул, молекулярные формул! не дают однозначного ответа о строении вещества. Одной и то же молекулярной формуле могут соответствовать два и боле соединений. Так, молекулярную формулу С2Н6О имеет этиловы спирт — С2Н5ОН и диметиловый эфир — СН3—О—СН3.
Из приведенных вариантов изображения молекул более широк применяют структурные формулы, которые позволяют е пемощы определенной символики показать распределение элекгроино плотности в Молекуле, выделить реакционные центры и описат предлагаемый механизм реакции. Недостаток структурных форму заключается в том, что они не отражают истинного расположены атомов в пространстве. Более полное представление о строеин молекул дают стереохимические формулы и молекулярные модел (см. подразд. 5.2.1).
КОНТРОЛЬНЫЕ ВОПРОСЫ И УПРАЖНЕНИЯ
1. Охарактеризуйте четыре основных периода в истории развития органической химии, В чем заключены наиболее характер пые черты современного этапа в ее развитии?
2, Сформулируйте основные положения теории химическое строения А. М. Бутлерова, В чем ее преимущества перед предтш ствовавшими теориями? Каковы были пути ее дальнейшего ра; вития?
3. Назовите основные способы изображения органически молекул, С помощью соответствующих формул покажите: а) качс ственный и количественный элементный состав; б) химическс строение для следующих органических веществ; пропан; цикле гексан; пентеи-1; хлорбензол; я-бутиловый спирт; диэтиловый эфи
4. Перечислите основные преимущества структурных форму перед брутто-формулами, Приведите все возможные структурнь формулы и названия органических веществ, которым соответств; ют следующие молекулярные формулы: CiHeJ CsHi2; С3Н7С C3HSO.
13
Глава 2 КЛАССИФИКАЦИЯ И НОМЕНКЛАТУРА ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
2.1. КЛАССИФИКАЦИЯ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Важнейшими классификационными признаками органических соединений являются строение углеродной цепи и природа функциональной группы.
Классификация по строению углеродной цепи
В завнсимоств от структуры углеродного скелета органические соединения классифицируют следующим образом*.
ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
АЦИКЛИЧЕСКИЕ ЦИКЛИЧЕСКИЕ
КАРБОЦИКЛИЧЕСКИЕ ГЕТЕРОЦИКЛИЧЕСКИЕ
АЛИЦИКЛИЧЕСКИЕ АРОМАТИЧЕСКИЕ
Все органические соединения делятся на два типа: ациклические и циклические. К ациклическим (их еще называют алифатическими) относят вещества с открытой (незамкнутой) цепью, По строению углеводородного остова молекулы различают алифатические соединения предельные (насыщенные) и непредельные (ненасыщенные) . Предельные содержат только простые углерод-углеродные связи, непредельные имеют кратные (двойные и тройные) углерод-углеродиые связи:
СН3-СНа—СООН предельное алифатическое соединение
СН2^СН^С1 сна—с=сн
непредельные алифатические соединения
Циклические органические соединения содержат в своей структуре замкнутые цепв атомов — циклы. В зависимости от природы атомов, входящих в цикл, их разделяют па карбоциклические
14
и гетероциклические, В молекулах карбоциклических соединений циклы состоят только нз\томов углерода:
Н,С--СН»
I
Н2С сн—сн3
nh2
I с hc/Vh нс^сн сн
карбоциклические соединения
В гетероциклических соединениях циклы, наряду с атомами углерода, содержат атомы других элементов (чаще N, О, S). Гетероциклические соединения могут быть насыщенными, ненасыщенными н ароматическими:
НХ-------СНа
Н2Сч JcHa о
насыщенный гетероцикл
Н2С|---Г.СН
Нвс1^2СН о
ненасыщенный гетероцикл
ароматический гетероцикл
Группа карбоциклов объединяет два ряда органических веществ, существенно различающихся по химическим свойствам — алициклические и ароматические.
Для ароматических соединений характерно наличие ароматической системы, Родоначальником ароматических соединений является бензол:
СН hc/Vh hcI^Jch сн
К алициклическим соединениям относят ряд карбоциклов, ие обладающих ароматическим характером. Диалогично алифатическим по степени насыщенности алициклические соединения делят на предельные и непредельные:
HaCj---jCHB
HjjC'^CH—СООН
СНа предельное алициклическое соединение
сна
непредельное алициклическое соединение
15
В пределах каждого из названных рядов органические вещества распределяются по классам. Соединения, молекулы которых состоят только из атомов углерода и водорода, образуют класс углеводородов, При замещении в углеводородах одного или нескольких атомов водорода на функциональную группу образуются другие классы органических соединений.
Классификация по природе функциональной группы
Функциональная группа — структурный фрагмент молекулы, определяющий ее химические свойства *. Например, свойства карбоновых кислот определяются главным образом присутствием карбоксильной группы —СООН, спиртов — наличием гидроксила —ОН и т. д. По природе функциональной группы различают разнообразные классы органических соединений, основные из которых приведены па рис. 2,1,
По количеству и однородности функциональных групп органические соединения делят на монофункциональные, полифупкцио-нальные н гетерофункциональные (см, рис. 2.1),
Монофункциональные содержат одну функциональную группу, полифункциональные — несколько одинаковых, а гетерофуикцио-нальиые — несколько различных групп:
санБон
монофункциональное соединение
СН8—NHa
I
СНа—NHa тюлифу нкцим ta льное соединение
СООН
V-0H гатеро функциональное соединение
Соединения определенных классов объединяются в гомологические ряды. Гомологический ряд — это ряд соединений, в котором каждый последующий представитель отличается от предыдущего на группу —СНг—, называемую гомологической разностью. Например, соединения, образующие класс предельных одноатомных спиртов, можно расположить в виде гомологического ряда:
СН3ОН —метанол,
С2Н5ОН —этанол,
С3Н7ОН — пропанол,
С4Н9ОН —бутанол и т. д,
Все классы органических соединений тесно взаимосвязаны. Существует множество путей перехода от одних классов к другим Через превращения функциональных групп.
• В понятие «функциональная группа» условно не включаются углеводород. Выв заместители, например винил, фенил.
‘ .1 ?в
2.2. НОМЕНКЛАТУРА ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Номенклатура органических соединений складывалась на протяжении всего периода развития органической химии. В историческом аспекте следует выделить три основные номенклатурные системы: тривиальную, рациональную н международную (ИЮПАК).
Тривиальная номенклатура
Первые названия, которые давали органическим соединениям, были случайными. Они, как правило, отражали способы получения веществ (пирогаллол — продукт пиролиза галловой кислоты), их отличительные свойства (флуоресцеин — флуоресцирует) или природный источник, из которого соединении впервые выделялись (муравьиная кислота, лимонная и др.), Такие названия составляют тривиальную номенклатуру. Тривиальные названия органических соединений прочно укоренились и многие нэ них до сих пор общепризнанны. Особенно широко их применяют в химии природных и гетероциклических соединений.
Рациональная номенклатура
Это первая номенклатура, в которой стали учитывать в иазва-t нии вещества его строение. В основе рациональных названий используется принцип деления органических соединений иа гомологические ряды. Согласно рациональной номенклатуре все вещества в определенном гомологическом ряду рассматриваются как производные простейшего представителя данного ряда, в частности у алканов — метана*, у алкенов — этилена и т, д., например:
сн, сн,1сн,1
метан 1 1
метилметаи
СНя-!-СНв4-СНв
I----J
дяметнлметан
Однако для названия сложных химических структур рациональная номенклатура оказалась непригодной. В настоящее время применение этой номенклатуры ограничено.
Международная номенклатура (ИЮПАК)
Основы международной номенклатуры заложены в 1892 г. на международном конгрессе химиков в Женеве (женевская номенклатура). В 1930 г, на X конгрессе химиков в Льеже она была
* Название «метан» присваивают наиболее замещенному атому углерода.
„ 18
Таблица 2.t
Основные функциональные группы, расположенные в порядке уменьшения старшинства
Класс веществ Функциональная группа* Обозначение в префиксе 1 Обозначение I в суффиксе
Карбоновые КИ5ЛОТЫ —соон —(C) ООН карбокси— —карбоновая кислота —овая кислота
Сульфокислоты —SOBH сульфо— —сульфоновая кислота
Сложные эфиры - COOR —(C)OOR R-оксикарбонил— —карбоксилат —оат
Хлорангидриды С^'С1 ,0 клорформил— —карбон и л хлорид —оилхлорид
Амиды -с€° XNHa -<н, карбамоил— —карбоксамид —амид
Гидразиды А) \VHNH2 ,О ~(С)\ , v — —карбогидразид —огидразид
Нитрилы 1 i I циано— —карбонитрил —оиитрил
Альдегиды -с<° ,О -к формил— — карбальдегид —аль
Кетоны оксо— —он
Спирты, фенолы —он гидрокси— —ол
Тиолы —SH меркапто— —тиол
Амины -NH, амино— —амин
•Взятый в скобки атом углерода входит в состав главной углеродной цепи.
19
несколько усовершенствована и дополнена (льежская номенклатура) н, наконец, иа XIX конгрессе Международного союза теоретической и прикладной химии в 1957 г. были разработаны правила современной номенклатуры, известной под названием «номенклатура ИЮПАК» * .
Номенклатурные правила ИЮПАК признаны во всех странах мира н позволяют дать однозначное название любому органическому соединению.
Номенклатура ИЮПАК предусматривает несколько вариантов образования названий органических соединений, нз которых наиболее широко применяются заместительный и радикало-функциональный.
Заместительная номенклатура. При образовании названий но заместительной номенклатуре органические соединения рассматривают как производные простейших углеводородов, в молекуле которых один или несколько атомов водорода замещены на другие атомы пли группы атомов, называемые заместителями.
При составлении названия по заместительному варианту номенклатуры ИЮПАК прежде всего определяют, какие функциональные группы входят в состав соединения, и выбирают средн них старшую, если она имеется (табл. 2.1). Если в соединении имеется одна такая группа, она считается старшей; если две и более, старшей из них является расположенная в табл. 2.1 выше. В названии органического вещества старшая функциональная группа обозначается в суффиксе, а все остальные — в префиксе. Согласно номенклатурным правилам ИЮПАК некоторые функциональные группы не рассматривают по старшинству и обозначают в названии всегда в префиксе (табл. 2.2).
Таблица 2.2
Функциональные группы, обозначаемые только в префиксе
Функциональная группа Обозначение в префиксе Ф ункцион аль на я группа Обозначение в префиксе
— F фтор- -OR R-ОкСИ-
— о хлор- -SR R-TJiO-
— Вг бром- — N =0 нитрозо-
-J иод- — нитро-
’• IUPAC—International Union of Pure and Applied Chemistry — Международный союз теоретической и прикладной химии.
20
После определения старшей группы устанавливают так называемую родоиачальиую структуру соединения. Родоначальная структура — это структурный фрагмент молекулы, лежащий в основе названия, В ациклических соединениях родопачальной структурой является главная углеродная цепь, в карбоциклических и гетероциклических — цикл:
В качестве главной углеродной цепи выбирается та, которая, содержит максимальное число (в порядке убывающей значимости): функциональных групп, приведенных в табл. 2.1; кратных (двойных и тройных) связей; атомов углерода; заместителей.
Заместителем называют любой атом или группу атомов, которые не входят в родопачальную структуру. Понятие заместитель включает в себя функциональную группу и радикал.
Радикал * представляет собой остаток молекулы углеводорода, образующийся в результате удаления одного или нескольких атомов водорода. Свободную валентность в радикалах обозначают чертой. По количеству свободных валентностей различают одно-, двух- и трехвалентные радикалы:
СН3—СН2— СН8—СН= — СН2—СН2— СН8—С=
одновалентный двухвалентные радикалы трехвалентный
радикал радикал
В зависимости от того, у какого атома углерода (первичного, вторичного или третичного) находитсн свободная валентность, различают первичные, вторичные н третичные радикалы. Первичным называется атом углерода, непосредственно связанный только с одним атомом углерода, вторичный — с двумя, третичный — с тремя:
третичный атом углерода вторичный атом углерода
СН8—СН-СНЯ-СН, I снэ
| первичные атомы углерода
* Следует отличать от понятия «свободный раднкалж (частица с кеспарен-ныы апектроном), например СНа,
21
сн8—сн2—сна—сна первичный радикал
СНЭ-СН2—СН—снэ
вторичный радикал
сн3 I сн3—с—сна
третичный радикал
В табл. 2.3 приведены структурные формулы и названия часто встречающихся углеводородных радикалов.
Таблиц* 2.3
Наиболее употребительные названия некоторых углеводородных радикалов
Структурная формула Название Структурная формула Название кВ
СН3 — СН,-СН2-сн3—сна—сна— снэ—сн—снэ сн3-сн2-сн2—сна (СН3)3СН—СН2— сн3—сн—сн2—сна (СНа)эС-сна=сн— 3 2 f снЕ=сн—снг— 3 2 1 СНЭ—сн=сн— метил этил пропил изопропил н-бутил нзобутнл flflio/j-бутил трет-бутил ВИНИЛ, этенил аллил, проион-2-ил пропеп-1-ил СН=С— 3 2] СН=С—СН2— -С1Ц- —СН,—сн2— —СН2-СНа—сн3— СН3—СН-сн3-^_сн3 Свна-CBHS-CH2- сен6-сп= СаН6— этинил Я пр опин-2-м Лч метилен этилен триметилен этилиден изопропили-ДСН фенил оензил бензилиден бснзилидио
Определив родоначальную структуру, проводят нумерацию ее атомов таким образом, чтобы старшая группа получила по возможности менылин номер. Если соединение не имеет старшей группы, предпочтение при нумерации (возможно меиьшие номера) отдают положениям кратных связей, а в их отсутствие — заместителям.
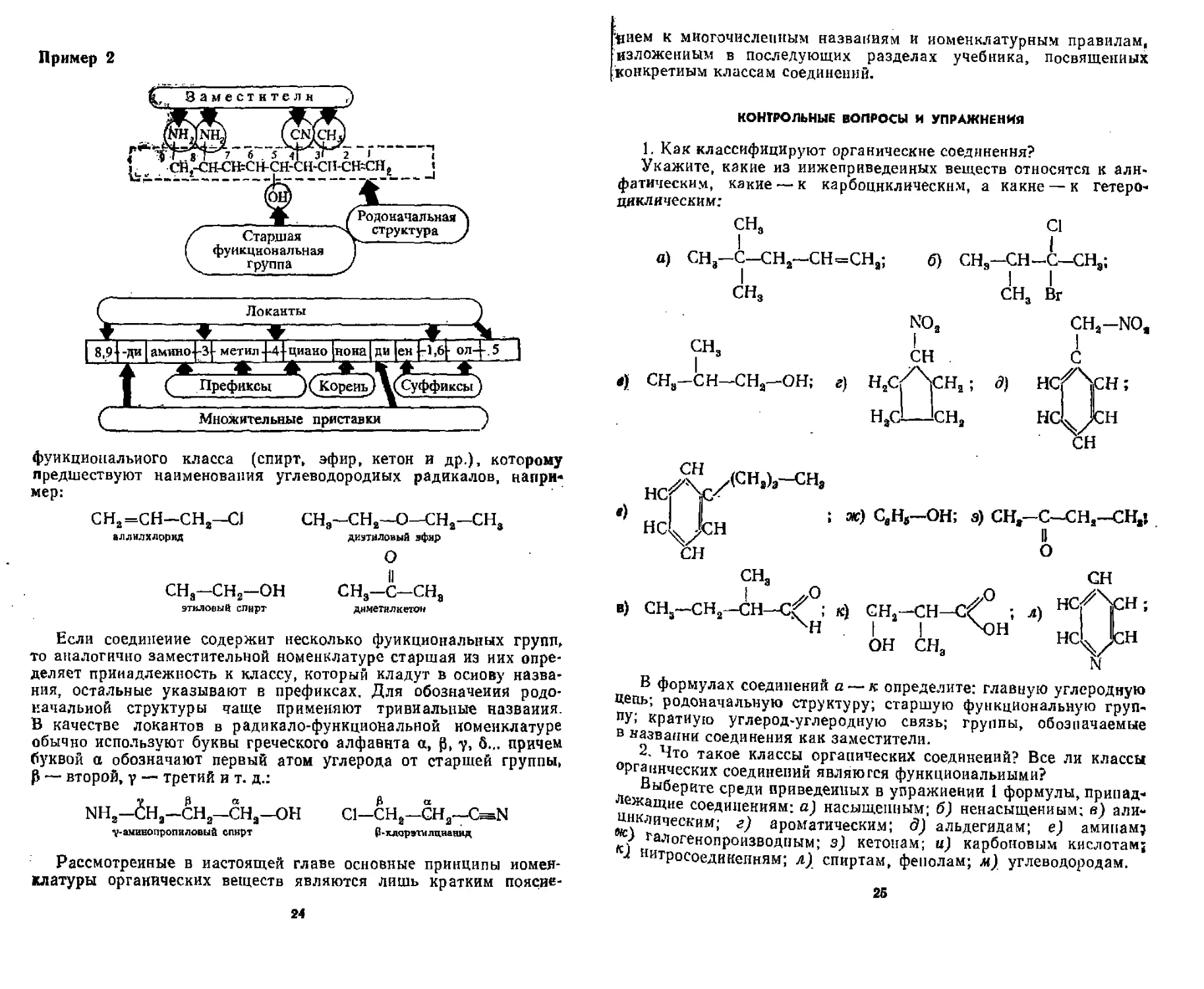
Наконец, составляют название соединения в целом, соблюдая следующую последовательность: вначале указываются в алфавитном порядке функциональные группы, кроме старшей, и углеводородные радикалы (префикс), затем приводят название родоначальной структуры (корень), в ковце указываются кратные связи и старшая функциональная группа (суффикс):
22
Название
префикс + корен ь + суффикс |
♦ ♦ _
Функциональные Родоначалиная Старшая
группы, структура функциональная
кроме старшей ; (главная углеродная группа \
углеводородные радикалы ( в алфавитном порядке) цепь или цикл) кратные связи
Двойная связь в названии обозначается суффиксом -ен-, тройная----ин. Положение заместителей и кратных связей указывают
цифрами или буквами (локантами), которые помещают перед префиксом и после суффикса, При наличии в молекуле нескольких одинаковых заместителей или кратных связей для их обозначения применяют множительные приставки: ди- (два), трн- (три), тетра- (четыре), пента- (пять) н г. д.
Примеры названий органических веществ по заместительной номенклатуре
долачальиая структура
|СНгСН-СП-1
3.
JlOKili ПЫ
гидрокси
метил бутаи [ овая кислота
Радикало-функциональная номенклатура. Для некоторых органических соединений номенклатура ИЮПАК, наряду с заместительным, предусматривает радикало-фупкцнональный вариант образования названий, В основе этих названий лежит название
23
Пример 2
Локанты
8,9 -ди амино- -3 - метил - -4 циано нона ди |ен -1,6
Множительные приставки
функционального класса (спирт, эфир, кетон и др.), которому предшествуют наименования углеводородных радикалов, например:
СН2=СН—СНа—CI аллнлхлорцд
сна—сна—ОН
этиловый спцрт
снэ—сн2—о—сна—сн8 ДИЭТИЛОВЫЙ эфир
о и сн3—с—сна
диметилкетон
Если соединение содержит несколько функциональных групп, то аналогично заместительной номенклатуре старшая из них определяет принадлежность к классу, который кладут в основу названия, остальные указывают в префиксах. Для обозначения родо-начальной структуры чаще применяют тривиальные названия. В качестве локантов в радикало-функциональной номенклатуре обычно используют буквы греческого алфавита а, 0, у, б... причем буквой а обозначают первый атом углерода от старшей группы, 0 — второй, у — третий и т. д.:
NH„—£на—СНЭ—СНа—ОН в в д я
-V-аминопропиловый спирт
В а
С1—СНЙ—CH,—C=N я Я
₽ - клорэтилцнанид
Рассмотренные в настоящей клатуры органических веществ
главе основные принципы иомея-являются лишь кратким поясие-
24
цпем к многочисленным названиям и номенклатурным правилам, изложенным в последующих разделах учебника, посвященных конкретным классам соединений.
КОНТРОЛЬНЫЕ ВОПРОСЫ И УПРАЖНЕНИЯ
1. Ках классифицируют органические соединения?
Укажите, какие из нижеприведенных веществ относятся к алифатическим, какие — к карбоциклическим, а какие — к гетеро* циклическим:
СН3 С1
I I
а) СН3-С-СН2—СН=СНа; б) СНЭ—СН-С—СН8;
СН3 СН3 Вг
NOa си3 сн
в) СН8-СН—СНа—ОН; В) Нас/\сНа; д) Н#О----------------------------'сна
ОН
С
HcZ\cH; hcL Jch
ж) СаНв—ОН; 3) СН,—С—СНа—СНа;
I) О
В формулах соединений а — к определите: главную углеродную Цепь; родоначальную структуру; старшую функциональную группу; кратную углерод-углеродную связь; группы, обозначаемые в названии соединения как заместители.
2. Что такое классы органических соединений? Все ли классы органических соединений являются функциональными?
Выберите среди приведенных в упражнении 1 формулы, принадлежащие соединениям: а) насыщенным; б) ненасыщенным; в) алициклическим; г) ароматическим; д) альдегидам; е) аминам; галогенопроизводным; з,) кетонам; и) карбоновым кислотам; J нитросоединениям; л) спиртам, фенолам; я) углеводородам.
25
Укажите в формулах соединений 1а— к первичные, вторичные; третичные и четвертичные атомы углерода.
3. Дайте определение понятию функциональная группа. К соединениям а — к, формулы которых даны в упражнении 1, примените классификацию органических веществ по количеству и однородности функциональных групп.
Выделите в указанных формулах радикалы; метил, этил, этилен, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, цикло-пентнл, бензил, фенил, винил.
4. Что такое гомологический ряд и гомологическая разность?
Приведите структурные формулы и химические названия простейших представителей гомологических рядов, к которым относятся соединения, обозначенные в упражнении 1 буквами а, б,е— и.
5. Какие основные номенклатурные системы сложились в процессе развития органической химии?
Напишите структурные формулы соединений по их названиям н определите, по какой номенклатурной системе образованы эти названия: а) бромистый аллил; б) хлорбензол; в) изопропилиоднд; г) метиловый спирт; д) винный спирт; е) бутанол-2; ж). изобу-тнловый спирт; з) трнметилнитрометан; а) анилин; к) метаиамин; л) формальдегид; м) пропаиаль; н) уксусная кислота,
6. Сформулируйте основные правила образования названий органических соединений по заместительной и радикало-функцио-11 альНой номенклатурам ИЮПАК,
Объясните, какие ошибки допущены в названиях следующих соединений:
а)
г)
(CH,)SCH-CSH,; б) II изопропил этап [| |
3-прогтлбснзол
О NH,
II I
СН3-С~СН2-СНа-ОН ; д) J З-оксобутаиол-L I
и
I
SO8H +-сульфоаиилин
в) СН2—СН,;
ОН ОН дигидроксиэта»
,о
₽)
1 хн соон
2-карбоксиэтаналь
СН2—СНа—CH2—NHa;
ОН
3-гидр0кеипропаиамин
С1 Вг
з) СН3—СН—СН3; «) СН3—С—СН
NH, Cl J
2-нвтролропаи [-бромиод-З-днхлорпропан
Приведите правильные клатуре ИЮПАК.
названия этих соединений по иомен-
26
Глава 3 ХИМИЧЕСКАЯ СВЯЗЬ
Изучение органической хнмни значительно упростилось благодаря развитию теории химической связи. На основании существующих представлений о природе химической связи н электронной структуре молекул стало возможным объяснить реакционную способность органических соединений, попять и запомнить фактический материал химии.
3.1. ТИПЫ ХИМИЧЕСКИХ СВЯЗЕЙ
В 1916 г. немецкий ученый В, Коссель и американский ученый Дж. Н, Льюис предложили электронную теорию химической связи, которая явилась важным этапом в развитии теории строения. Согласно электронной теории, химическая связь рассматривается как результат взаимодействия внешних электронных оболочек (валентных электронов) атомов. Учитывая химическую инертность благородных газов, нх внешние электронные оболочки (для гелия — двухэлектронная, для неона, аргона и т. п.— восьмиэлектронная) , считают наиболее устойчивыми к взаимодействию. Стремление атомов других элементов связываться друг с другом обусловлено неустойчивостью нх валентных оболочек. Поэтому каждый атом, образуя химическую связь, принимает, отдает илн обобществляет валентные электроны таким образом, чтобы его внешняя электронная оболочка соответствовала конфигурации инертных газов. Такой принцип заполнения валентных оболочек получил название «октетное правило».
В зависимости от способа образования различают два основных типа химической связи: ионную и ковалентную,
Ионная связь образуется между атомами, которые значительно отличаются по электроотрицательности. Электронная конфигурация инертного газа достигается в данном случае путем переноса электрона к более электроотрицательному атому. В результате образуются противоположно заряженные иоиы, которые электростатически притягиваются друг к другу. Поэтому ионную связь называют еще электровалентной:
Na* + :Cl;-»-Na+Cr t нонггая связь
В приведенном примере ион натрия обладает электронной конфигурацией неона» а ион хлора — конфигурацией аргона.
27
Соединения с ионной связью имеют высокие температуры плавления, хорошо растворяются в полярных растворителях, в водных растворах диссоциируют на ионы. Их растворы и расплавы проводят электрический ток.
Ковалентная связь является основным типом химической связи в органических соединениях. Она образуется между атомами, равными или близкими по электроотрицательности. Электронная конфигурация инертного газа в данном случае достигается за счет обобществления валентных электронов и формирования одной нли нескольких общих электронных пар:
с • f 411
Щ) с Ди (HJ \
Н — с—н
ковалентная связь
В результате обобщсстиления валентных электронов углерода н водорода образуется четыре электронные пары, которые являются общими для данных атомов. Углеродный атом при этом приобретает конфигурацию неона, а атомы водорода — конфигурацию гелия. Каждая из обобществленных пар электронов составляет ковалентную связь, которая обозначается в виде валентного штриха (—).
При образовании ковалентной связи между атомами с одинаковой электроотрицательностью общая электронная пара располагается симметрично по отношению к центрам обоих атомов, Такая связь называется ковалентной неполярной:
я.с0сна " J A q
Н,с—сн3
. ковалентная неполярная связь
Если в образовании ковалентной связи участвуют атомы с различной электроотрицательностью, общая пара электронов смещается к атому с большей электроотрицательностью, В этом случае связь называется ковалентной полярной:
н3 С$С1
$+ &
Н3 с— С1
йЬваленгняя полярная связь
Полярную ковалентную связь изображают в виде стрелки направленной к более электроотрицательному атому. Буквой С £греч, «дельта») условно обозначают дробные (частичные) заряды
на атомах, Символ 6+ отражает пониженную, а б"—повышенную электронную плотность.
В зависимости от числа общих электронных пар, возникающих между атомами при образовании связи, различают простые и кратные ковалеитпые связи. Простые представлены одной, кратные — двумя или тремя парами электронов:
простая кратная
ковалентная связь ковалентная связь
Атомы азота, кислорода, серы, галогенов и некоторых других элементов при образовании ковалентных связей формируют октет-ную оболочку обобществлением ие всех внешних электронов. Часть электронов не участвует в образовании химических связей. Этн электроны называют необобществленными, неподеленными или п-электропами:
Донорно-акцепторная связь представляет собой разновидность ковалентной связи и отличается от последней только способом образования. Если ковалентная связь образуется путем обобществления пары электронов по одному от каждого атома, то допор-ио-акцепторная — за счет двух электронов, предостаиляемых одним из атомов. При этом атом, поставляющий пару электронов в общее пользование, называют донором, а атом, принимающий электроны,— акцептором. Донор должен иметь пару неподеленпых электронов. Акцептором может быть протон или другой атом, у которого не хватает до образовании октета двух электронов,
По донор ио-акцептор ному механизму, например, протекает образование иона аммония
Н Н
н—hJr*—н
I н
донор акцептор
Поскольку пара электронов азота ушла на образование связи с Протоном и стала общей для двух атомов, азот приобретает поло
22
жительный заряд. Образовавшаяся в ионе аммония по донорноакцепторному механизму ковалентная связь ничем ие отличается от трех остальных связен.
Донорно-акцепторную связь называют еще координационной, поскольку при ее образовании происходит процесс координации неподеленной электронной пары.
Частным случаем донорио-акцепторной связи является семнпо-ляриая связь, Она образуется при взаимодействии атомов, имеющих неподелениые электронные пары (доноры), с электронейтраль-ными частицами, которые содержат секстет электронов (акцепторы), В результате образования координационной связи атом-донор приобретает положительный заряд, а атом-акцептор—• отрицательный. В итоге эти два атома оказываются связанными двояко: ковалентной связью и ионной. Ниже приведены примеры образования семпполярной связи в молекулах оксида триметнл-амнна и нитробензола;
СНзЧ СНзЧ+ _ СН3хч+ -
CH3->N: О: -CH9^N:6: es CH3^N—О СН/‘ " СН/ ' сня/
триметаламин оксид триметплзмияг
Ч-’О’ 4-
C,H5-N=O4-O: ^С,!1,Д
витрозобензол
нитробензол
Несмотря на то, что в семнполярных связях наряду с ковалентной связью имеется и ионное взаимодействие, соединения, построенные по этому типу, ие проводят электрический ток. Семиполяриую связь принято обозначать следующим образом:
+
R—N< или R—N<f X)- ХО
К донорно-акцепториому типу относятся связи в комплексных соединениях.
Водородная связь образуется в результате электростатического взаимодействия между активными атомами водорода в молекуле и атомами с неподеленной электронной парой (—О—, —N—, —F—, реже —S—, —С1:) в этой же или в другой молекуле. Активными называют атомы водорода, связанные в молекуле сильнополярной ковалентной связью, например —CU-H, —S+-H
и др. Графически водородную связь обозначают тремя точкамш
эодородпая связь
Энергия водородной связи (10—40 кДж/моль) невелика по сравнению с энергией ковалентной связи (340—360 кДж/моль).
зо
Различают внутримолекулярные и межмолекулярные видирид-ные связи. Внутримолекулярные водородные связи (ВВС) возникают в пределах одной молекулы с образованием пяти-, шестн-или семичлепных хелатообразпых структур (от лат, chela — клешня):
Н
о-хлорфенФТ
салициловый альдегид
Межмолекулярые водородные связи (MBQ возникают между двумя или несколькими молекулами с образованием димеров или ассоциатов:
СНа—С 0
3
димер уксусной кислоты
с2нБо—н.. .о—н.. -о—н.. .о-н
I I I
СаН& С2Н5 с2н5
ассоциат йтилового спирта
Наличие водородных связей оказывает влияние па физические '(температура кипения н плавления, растворимость, вязкость, спектральные характеристики) и химические свойства органических соединений. МежмолекулярЕ1ая водородная связь способствует повышению температуры кипения, а часто и температуры плавления вещества.
Например, за счет образования ассоциатов температура кипения этилового спирта С2Н5ОН (78 °C) значительно выше, чем у диметилового эфира СН3—О—СН3 (—24 СС), имеющего одинаковую с ним молекулярную массу, но не способного образовывать водородную связь. Аналогично более высокая температура плавления лп?та-нитрофенола (97 °C) и пара-нитрофенола (114°С) по сравнению с орто-нитрофенолом (45 °C) объясняется образованием межмолекулярной водородной связи:
О
с-Ентрофенол П. пл. 45 °C)
31
Образование водородной связи между растворенным веществом и растворителем (если это возможно) значительно увеличивает растворимость вещества,
Водородпаи связь, особенно внутримолекулярная, оказывает влияние на конформацию молекул и скорость протекания химических реакций.
Установить наличие водородной связи можно с помощью методов ИК-, КР-, и ЯМР-спектроскопии. Например, в инфракрасных спектрах гндрокснлсодержащцх соединений (спирты, фенолы) наблюдается полоса поглощения свободной ОН-группы в области 3650—3590 см”', если же эта группа участвует в образовании водородной связи, полоса поглощения проявляется в области 3400— 3200 см-‘.
Водородные связи играют важную роль в протекании различных биохимических процессов в организме, в частности они определяют пространственную структуру белков, полисахаридов, участвуют в образовании двойной спирали ДНК и др.
3.2, КВАНТОВО-МЕХАНИЧЕСКИЕ ОСНОВЫ ТЕОРИИ ХИМИЧЕСКОЙ СВЯЗИ
Современная теория химической связи базируется на квантово-механическом рассмотрении молекулы как системы из электронов и атомных ядер. Квантово-механический подход к изучению химических систем основан на решении волнового уравнения Шредингера /ЛР=£’ЧГ, где Я — оператор Гамильтона, Е— полная энергия системы, 'У (греч. «пси») — волновая функция.
Уравнение Шредингера описывает энергию молекулы любого соединения в соответствии с волновой функцией, характеризующей распределение электронной плотности в поле атомных ядер. Решение уравнения сводится к нахождению волновой функции V, зависящей от пространственных координат и спиновых переменных всех электронов, Квадрат модуля волновой функции РУ)2 определяет вероятность обнаружения электрона в «точке» пространства, а сама функция 'F описывает орбиталь.
Атомные орбитали
Атомной орбиталью (АО) называют область пространства, внутри которой вероятность нахождения электрона максимальна.
Согласно квантовой теории состояние любого электрона в атоме описывается четырьмя квантовыми числами; п, I, т, s. Первые три характеризуют атомную орбиталь: главное квантовое число п определяет энергетический уровень орбнтадн, побочное квантовое число I — геометрическую форму орбитали, магнитное квантовое число т— ориентацию различных атомных орбиталей в пространстве. Спиновое квантовое число s описывает вращение электрона вокруг собственной осн.
32
Из курса неорганическом химии студентам известны геометрические формы s-, р-> d-атомных орбиталей (рис. 3J).
Рис. 3.1. Геометрическая форма р- и rf-атомных орбиталей
Атомные орбитали s-типа имеют сферическую симметрию. Для электронов в p-состоянии существует три одинаковых по энергии ЛО гантелеобразиой формы, отличающихся друг от друга ориентацией в пространстве: рх, ру. Р* (рис. 3,2).
Av-орбиталь
Р>- орбиталь pL~ орбиталь
Рис. 3.2, Ориентация р-атомных орбиталей по осям координат
Каждая р-орбнталь имеет узловую область, в которой вероятность нахождения электрона равна пулю. Существует пять атомных орбиталей d-тина с более сложной геометрической формой.
Согласно принципу Паули на одной орбитали может находиться не более двух электронов, и эти электроны должны иметь противоположные спины (условно записывается Ifjl).
Заполнение атомных орбиталей электронами осуществляется в порядке возрастания энергий: ls<; 2s <С -Р <3s<; 3pC4$<;3d й т. д. Атомная орбиталь, не занятая электронами, называется вакантной (условно обозначается □ ).
В соответствии с правилом Гуяда заполнение орбиталей с одинаковой энергией (так называемых вырожденных орбиталей) происходит в таком порядке, что вначале каждая орбиталь заселяется одним электроном, и эти электроны должны иметь параллельные спины; затем происходит окончательное заполнение орбитали аторым электроном.
2
2-253
S3
В табл. 3.1 приведены электронные конфигурации элементен, наиболее часто встречаемых в органической химии.
Таблица 3.1
Строение электронных оболочек атомов водорода, углерода, азота и кислорода
Элемент Тип орбиталей Электронная конфигурация
is 2s 2px Spy
II 4 И
С fl N f t Is® 2sa 2p3
N t! tl t t t Js2 2s3 2ps
О’ fl tl n t + Is2 2s3 2p*
Ковалентная связь. Для описания ковалентной связи с позиций квантовой механики используют два основных метода: молекулярных орбиталей (МО) и валентных связей (ВС). Оба метода представляют собой математические приемы приближенного решения уравнения Шредингера применительно к молекуле.
Согласно методу ВС (теория резонанса) ковалентная связь рассматривается как результат перекрывания атомных орбиталей, .несущих по одному электрону с противоположными спинами. При этом атомные орбитали сохраняют свою Индивидуальность, а пара электронов локализуется в пространстве между ядрами атомов. Применение метода ВС для описания молекулы сводится к составлению максимального числа так называемых резонансных структур и рассмотрению реальной молекулы как гибрида данных структур. Резонансными или граничными называются структуры, отличающиеся только расположением электронов, Например, молекулу водорода Н—Н можно представить как гибрид граничных структур Н+Н- ** Н-Н+ ( <-> —обозначение резонанса). Теория резонанса применяется в основном для качественной оценки реакционной способности органических соединений.
В методе МО тоже исходят из допущения, что ковалентная связь образуется в результате перекрывания одиоэлектропных АО. Однако, в отличие от метода ВС, атомные орбитали здесь не сохраняют своей индивидуальности, а вместо иих образуется такое же количество орбиталей нового типа, называемых молекулярными орбиталями (МО). Из двух АО образуется две МО, из четырех — четыре, из шести — шесть и т. д, Молекулярные орбитали отличаются от атомных тем, что электронные облака окружают ядро уже не одного атома, а двух илн нескольких. Для образования МО необходимо, чтобы атомные орбитали имели близкие энергии.
34
одинаковую симметрию относительно липин связи в молекуле и достаточную для перекрывания протяженность в пространстве. На рис. 3.3 представлена схема образования ковалентной связи в молекуле водорода. Как видно, перекрывание двух атомных орбита-лей приводит к образованию двух молекулярных орбиталей. Одна из них имеет более низкую энергию, чем исходные АО, и называется связывающей орбиталью, другая обладает более высокой
Рис. 3.3. Схема образования ковалентной связи II— Н
энергией, чем образующая ее АО, я называется разрыхляющей, или антисвязывающей, орбиталью. Заполнение молекулярных орбиталей электронами происходит аналогично заполнению атомных, т. е. по принципу Паули и в соответствии с правилом Гунда. Вначале электроны занимают более низкие по энергии связывающие орбитали, что приводит к образованию химической связи между атомами. Молекулярная разрыхляющая орбиталь в основном состоянии остается вакантной, Ее заполнение электронами происходит прн возбуждении молекулы, что ведет к разрыхлению связи и распаду молекулы па атомы. Таким образом, согласно методу А1О образование ковалентной снязи представляется как переход двух электронов с атомных орбиталей на связывающую молекулярную орбиталь.
Наряду со связывающими и разрыхляющими МО существуют иесвяэывающие молекулярные орбитали, иа которых располагаются иеподелениые пары электронов.
3.2.1. Гибридизация атомных орбиталей
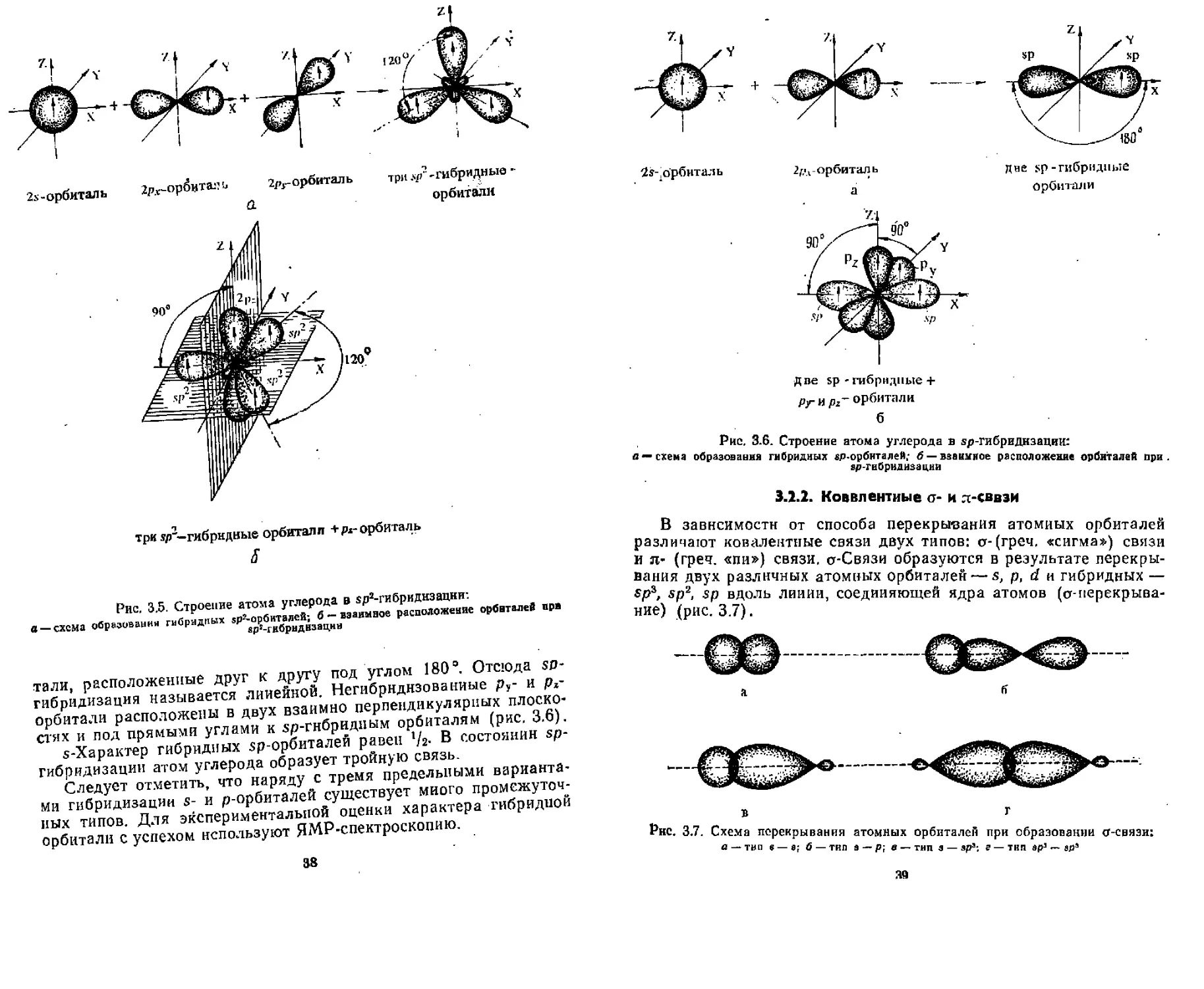
Согласно квантово-механической теории химической связи число образуемых атомом ковалентных связей определяется количеством одноэлектронных атомных орбиталей, т, е. количеством
неспареииых электронов. Однако в действительности атомы некоторых элементов образуют большее число ковалентных связей по сравнению с количеством неспаренных электронов в основном состоянии. Так, атом углерода в основном состоянии имеет д-аа неспареииых электрона, но, как известно, в органических соединениях ои образует четыре ковалентные связи. Это можно объяснить, если представить, что один из 2s-электронов переходит на вакант-
В таком состоянии, называемом возбужденным, атом углерода имеет четыре одиоэлектрониые орбитали, Поскольку валентные орбитали углерода неравноценны (одна — а, а три — р), и ковалентные связи, образованные с их участием, не должны быть эквивалентными. В действительности же, например, в молекуле метана СН4 все четыре ковалентные связи С—Н равноценны. Для объяснения этого факта в квантово-механической теории химической связи введено математически обоснованное понятие о гибридизации атомных орбиталей. Сущность гибридизации заключается в том, что из нескольких различных по форме и близких по энергии атомных орбиталей образуется такое же число одинаковых по форме и энергии гибридных орбиталей, Нвпрвмер, при взаимодействии одной 5- и трех р-атомных орбиталей образуется четыре ар3-гибрндные орбитали. По форме гибридная орбиталь отличается как от S-, так и от р-орбиталей, представляя собой несимметричную объемную восьмерку: Гибридные орбяталп явля-
ются атомными орбиталями, но возникают они только в процессе образования химической связи и не отражают реальной структуры свободного атома. В результате перекрывания большей доли гибридной орбитали с атомными орбиталями других атомов образуется ковалентная связь. По сравнению с негибридизопанпымя гибридные орбитали более выгодны геометрически и в результате большего перекрывания с орбиталями других атомов образуют более прочные сяязи.
Для атома углерода характерны три вида гибридизации с участием s- и р-орбиталей: sp3-, sp3- и sp-гпбридизация, Каждому из этих видов соответствует определенное валентное состояние атома.
зр^Гибридизация углерода (первое валентное состояние). При 5р3-гибриднзации из одной з- и трех р-орбиталей образуется четы
ЗЯ
ре качественно новые, равноценные $р3-гибридные орбитали, направленные в пространстве под углом 109с28' (от центра правильного тетраэдра к его вершинам). Поэтому s/Агибриднзацию называют еще тетраэдрической,
Исходя из схемы, прнведевной на рис. 3,4, можно условно представить, что доля s-облака а гибридных $р3-орбиталях равна ’Д. В первом валентном состоянии атом углерода образует только простые ковалентные связи,
Рис, 3.4, Схема образования и расположение в пространстве гибридных ^-орбиталей
sp2 Гибридизация углерода (второе валентное состояние), sp2-Гибридизация осуществляется в результате взаимодействия одной s- и двух р-орбиталей (рх, ру). В результате образуется три эквивалентные зр2-гибрндные орбитали, которые лежат в одной плоскости под углом 120°, Поэтому зр2-гибриднзация называется еще тригональной. Оставшаяся негибридизоваппой рг-орбиталь расположена в плоскости, перпендикулярной к плоскости гибридных орбиталей (рис. 3.5),
Условно доля s-облака в гибридных $р2-орбиталях равна 73. Атом углерода в $р2-гибридизации образует двойную связь,
sp-Гибриднзация углерода (третье валентное состояние), sp-Гибридизация возникает в результате слияния одной $- п одной Р-орбиталей (рх). При этом образуются две sp-гибридпые орби-
37
zl
три л/л-гибридные орбитали
три я/г—гибридные орбиталп +р*- орбиталь
Рис, 3,5. Строение атома углерода в ур2-гибриднзацни: а — схема образования гибридных вр^орбиталей; б — взаимное расположение орбиталей при врг-гибриднзации
тали, расположенные друг к другу под углом 180°. Отсюда SP-гибридизация называется линейной. Негибрнднзованиые ру- и рх~ орбитали расположены в двух взаимно перпендикулярных плоскостях и под прямыми углами к зр-гнбридпым орбиталям (рис, 3,6).
s-Характер гибридных sp-орбиталей равен Vs- В состоянии sp-гибридизации атом углерода образует тройную связь.
Следует отметить, что наряду с тремя предельными вариантами гибридизации s- и р-орбиталей существует много промежуточных типов. Для эйсперименталыюй оценки характера гибридной орбитали с успехом используют ЯМР-спектроскопию.
38
2 s- .орбиталь
2^-орбиталь
дне sp-гибридные орбитали
Две sp - гибридные + Рг и рг орбитали б
Рис, 3.6. Строение атома углерода в sp-гибриднзации:
а —схема образования гибридных зр-орбнталей,- б —взаимное расположение орбиталей при. яр-гибридизации
3.2.2. Коввлентиые а- и л-сввзи
В зависимости от способа перекрывания атомных орбиталей различают ковалентные связи двух типов: а-(греч, «сигма») связи и я- (греч. «пи») связи, а-Связи образуются в результате перекрывания двух различных атомных орбиталей — s, р, d и гибридных — sp\ sp2, sp вдоль линии, соединяющей ядра атомов (а-перекрыва-ние) (рис. 37).
гг
в г
Рнс. 3.7. Схема перекрывания атомных орбиталей при образовании ч-свяэн: а — тип в — в; б — тип а — р; в — тип з — зр’; е — тип sp3 <- вра
3Q
Возникающие при этом молекулярные орбитали называют соответственно о-МО (связывающая молекулярная орбиталь) и а*-МО (разрыхляющая молекулярная орбиталь) (см, рис. 3,3). Максимальная электронная плотность при а-перекрывапни находится на оси, соединяющей два ядра.
В большинстве органических соединений о-связи образуются преимущественно за счет перекрывания гибридных spb и sp-op-биталей, которые являются более выгодными энергетически и обеспечивают более эффективное перекрывание (рис, 3.8). В молекуле
(г перекрывание
Рис. 3.8. Образование ст-связей в молекуле этана
этапа ст-связи С—Н образуются в результате осевого s—^-перекрывания, а a-связь С—С — за счет зрэ — $рэ-перекрывания орбиталей. Поскольку максимальная электронная плотность при о-пере-крывании сосредоточена в пространстве между ядрами атомов, о-связь обладает большой прочностью.
Наряду с перекрыванием атомных орбиталей вдоль оси, соединяющей ядра атомов, существует так называемое боковое пере-крывание атомных орбиталей. В нем принимают участие только параллельно расположенные р-атомпые орбитали. Боковое перекрывание р-орбпталей, называют л-перекрыванием, а образующуюся связь — л-связью (рис. 3.9).
В результате л-перекрываиия двух р-атомных орбиталей (рис. 3.9) образуются две молекулярные л-орбитали, одна из которых называется л-свизывающей МО, другая — л*-разрыхляющей МО. В основном состоянии оба электрона занимают связывающую л-орбиталь, что приводит к образованию л-связи. Максимальная электронная плотность л-связи сконцентрирована в двух областях — выше м ниже оси, соединяющей ядра атомов. Поэтому л-связь является менее прочной, чем ст-связь. Образуется л-связь только между атомами, которые находятся в spa- и sp-гибридном состоянии (рис. 3,10).
В молекуле этилена (рис. 3.10) атомы углерода находятся в $р2-гнбрндизацни. я-Перекрываиие трех гибридизованиых орбита-
40
Л- орбиталь ( снязываюи
Рис. 3,9, Схема перекрывания атомных орбиталей при образовании я
а б
Рис, 3.!0. Образование л-связн в молекуле этилена
лей каждого из атомов углерода дает уже рассмотренные о-с (две С—Н и одну С—С). л-Перекрынаиие двух негибридиэова* р-орбнталей дает углерод-углеродную л-связь, В итоге м& атомами углерода образуется двойная связь, которая представ собой сочетание а- и л-связей. При этом л-сэязь располот* в плоскости, перпендикулярной к плоскости о-связи.
Аналогично образуется я-связь между атомами, находяицг в sp-гибридизованном состоянии,
В молекуле ацетилена (рис. 3.11) углеродные атомы нахоД£ е sp-гнбриднзации и образуют между собой тройную связь, г
41
рая состоит из одной о- и двух я-связей. о-Связь возникает за счет перекрывания sp-гибридизованных орбиталей, а четыре р-орби-тали образуют две л-связи, расположенные во взаимно перпендикулярных плоскостях.
Рис. 3.11. Образование л-свяэей в молекуле ацетилена
Как уже говорилось, в зависимости от количества электронных пар, принимающих участие в образовании химической связи между двумя атомами, различают простые и кратные ковалентные связи. Простые связи, их еще называют одинарными, представлены всегда a-связью. В состав кратных связей наряду с п-связью входит также п-связь. Кратные связи различают двойные и тройные.
3.2.3. Основные характеристики ковалентных связей
Длина связи. Длиной связи называют расстояние между центрами связанных атомов в молекуле. Для определения длины связи применяют такие основные методы: дифракцию рентгеновских лучей, дифракцию электронов и спектроскопические методы. Поскольку атомы в молекуле находятся в постоянном колебательном движении, длины связей являются приближенными величинами. В табл, 3.2 приведены длины наиболее распространенных ковалентных связей.
В химической литературе длины связей чаще приводятся в ангстремах (А) или нанометрах (им). В международной системе единиц (СП) в качестве единицы длины связи применяют пикометры, сокращенно пм (1 нм = 10 А — 1 000 пм). Как видно из приведенных данных (табл. 3.2), длина связи зависит от природы и типа гибридизации атомов, образующих связь. Чем больше s-характер атомов углерода, тем прочнее связь между ними. Чаще всего это объясняют тем, что с увеличением s-характера гибридной орбитали она сильнее удерживается ядром,
42
Длины химических связей
Т аблица 3.2
Связь Длина Связь Длина
О А НМ пм О л нм им
С3рэ-Н 1,11 0,111 111 C=N 1.16 0,116 116
С8?—С^Я 1,54 0,154 154 С- F 1,39 0,139 139
С-'--С2 1.34 0,134 134 С—С1 1,78 0,178 178
cs^-c ®₽ г С) ₽ 1,20 1,43 0,120 0,143 120 143 С—Вг С—J 1,93 2,14 0,193 0.214 193 214
с=о 1,21 0,121 121 о—н 0,96 0.096 96
С—N 1.47 0,147 147 N—Н 1 01 0.101 101
C=N 1,28 0,128 128
Энергия связи. Энергией связи называют энергию, выделяющуюся при образовании химической связи между двумя свободными атомами, или же энергию, которую необходимо затратить на разъединение двух связанных атомов. Образование связи всегда сопровождается выделением, а ее разрыв — затратой определенного количества энергии. Энергия, необходимая для гомолитического (А- VВ) расщепления отдельной связи г. молекулу называется энергией диссоциации связи. Поскольку энергия диссоциации зависит от структуры молекулы, для характеристики энергии связи обычно используют средине значения энергии диссоциации. Энергию связи выражают в килоджоулях па моль (кДж/моль) или в килокалориях на моль (ккал/моль). Значение энергии связи служит мерой ее прочности. В табл. 3.3 приведены энергии связей, наиболее распространенных в органических соединениях,
Таблица 3t3
Энергии связей
Связь Энергия, кДж/моль (ккал/моль) при 25 “С Связь Энергия, кДж/моль {ккал/моль) при 25 °C
CjpS—Н 414 (99) Ce=N 890 (213)
S’p3 Qsp3 347 (83) С—F 427 (102)
^4Pa=Cspa 610 (146) С“~С1 339 (81) |fl
836 (200) С—Вг 284 (68) |
С—о 368 (88) С—I 213(51)
с=о 724 (J73) О—н 464 (111) Ц
С—к 305 (73) N— Н 389 (93)
C=N 598 (143) S—н 339 (83)
Сравнение данных табл. 3.2 и 3.3 показывает, что с увеличен нием s-характера связи ее длина уменьшается, а прочность воз-Растает. Двойные связи короче и прочнее соответствующих оди-Парных, а тройные — двойных. Но вместе с тем энергия двойной
43
связи меньше удвоенной, а тройной — утроенной энергии одинарной связи. Это означает, что ст-связь прочнее л-связи. Проследив за элементами в пределах группы периодической системы (например, связь С—Hal), можно увидеть, что при движении сверху вниз длина связи увеличивается, а прочность уменьшается.
Полярность связи. Полярностью связи называют неравномерное распределение электронной плотности связи, обусловленное различной электроотрицательностью атомов. Электроотрицательность — это способность атома притягивать электроны. Она зависит от эффективного заряда ядра атома и вида гибридизации атомных орбиталей. Наиболее известна шкала электроотрицательности, составленная американским химиком Л. Полингом (табл. 3.4)
Таблица ЗА
Электроотрицательность атомов по шкале Полинга
Атом н S С 3 J Г 3 '-'SP В г С1 : N 1 С5Р О । F
Электро-отри цате-льиость 2.1 2.5 2.5 2.6 2,8 2,8 3.0 з.о 3,2 3;5 4,0
Как видно из табл. 3.4, электроотрицательность атома углерода, равная 2,5, характерна для £р3-гибридизоваипого состояния. При увеличении s-характера орбиталей электроотрицательность возрастает; Csp2 — 2,8; Cfip — 3,2,
Полярная ковалентная связь образуется между атомами с различной электроотрицательностью, а также между атомами с одинаковой электроотрицательностью, ио которые в свою очередь связаны с атомами, имеющими другую электроотрицательность (см. подразд. 4J);
а+ а—
Н£+С1 Н3С^СНа->С1
хлорметан хлорэтап
В молекуле хлорэтапа атом хлора поляризует не только связь С—С1, но и связь С—С.
Количественно полярность связи выражается значением дипольного момента, который обозначают буквой р, (греч, «мю»). Дипольный момент равен произведению расстояния между «центрами тяжести» положительных и отрицательных зарядов на их значение. Обычно дипольный момент молекул выражают в единицах дебая (Д). В единицах СИ его приводят в аттокулоннанометрах (аК-нм). 1 Д=3,33564• I(НаК-нм. Дипольный момент — векторная величина. Для большинства ковалентных связей дипольный мо* мент равен 0—3 Д, сильно полярные связи имеют 4—7 Д, ионные — свыше 10 Д. Молекулу, состоящую из трех и более атомов, рассматривают как систему нескольких диполей. Измерить дипольный мо
44
мент каждой отдельной связи в такой молекуле невозможно. Можно измерить только суммарный дипольный момент молекулы (результирующий диполь), который представляет собой сумму векторов дипольных моментов отдельных связей. Зиая дипольный момент молекулы и значение углов между направлением связей, можно определить дипольные моменты отдельных связей путем разложения по правилу параллелограмма. Суммарный дипольный момент молекулы зависит не только от числа и природы полярных связей, ио и от взаимного расположения нх в пространстве. Например, молекула тетрахлорметаиа (СО*) содержит четыре связи С—С1 с дипольным моментом 1,46 Д каждая, а в целом соединение не имеет диполя (ц, —0), так как происходит взаимная компенсация дипольных моментов отдельных связей. Молекула же хлорметана (СН3—CI) имеет дипольный момент 1,86 Д, обусловленный в основном полярностью связи С—С1.
Наличие диполя оказывает существенное влияние иа физические и химические свойства вещества. Например, температуры кипения полярных соединений выше, чем неполярных. Полярные вещества хорошо растворяются в полярных растворителях, а неполярные, как правило,— в неполярных.
Полярность химических связей определяет тип химического взаимодействия (тип реакции) и является количественной характеристикой реакционной способности вещества.
Поляризуемость связи. Под «поляризуемостью» понимают легкость, с которой смещаются электроны связи под влиянием внешних воздействий (электрическое поле, реагирующая частица и др.). Другими словами, поляризуемость — это способность электронного облака связи к поляризации при действии внешнего электрического поля или электрически заряженных частиц. В результате внешних воздействий происходит деформация электронного облака связи, возрастает ее полярность (увеличивается дипольный момент). Следует отличать понятия «поляризуемость» и «полярность» связи Если полярность обусловлена различной электроотрицательностью связанных атомов, то поляризуемость определяется степенью подвижности электронов связи. Полярность — это статическое явление, а поляризуемость — динамическое. Поляризуемость ие всегде согласуется с полярностью. Так, полярность углерод-галоидно{ связи в ряду С—F>C—С1>С—Вг>С—J уменьшается, а ее поля ризуемость, наоборот, увеличивается.
При сравнении поляризуемости о- и гс-связей необходимо отме тить, что л-связн поляризуются гораздо легче, чем ст-связи, по скольку л-электронная плотность находится дальше от атомиы: ядер. Как и полярность, поляризуемость влияет на реакционную способность веществ, ио ее вклад значительно больше.
Направленность связей. Ковалентные связи имеют определен ную направленность в пространстве. Электронные пары, образую Шие химические связи, стремятся занять такое пространственно положение относительно друг друга, чтобы силы электростатиче ского отталкивания между ними были минимальными. Углы межд
45
направлениями связей в молекуле называют валентными углами. Значение валентного угла зависит от состояния гибридизации атомных орбиталей н природы атомов, образующих связи. Так, углы между связями С—Н в метане составляют 109e28z. Однако это справедливо только в тех случаях, когда атом углерода связан с четырьмя одинаковыми заместителями. В большинстве же случаев наблюдаются небольшие отклонения от угла правильного
(н н н \
111 1
Н—С—С—С—н 1 111 /
Н Вг Н /
валентный угол С—С—Вг составляет 114,2°. Аналогично незначительное отклонение от валентных углов 120° и -180° наблюдается в соединениях с атомом углерода в sp2- н sp-гибридизации.
КОНТРОЛЬНЫЕ ВОПРОСЫ И УПРАЖНЕНИЯ
1. Приведите электронные конфигурации элементов: С, О, N, S, С1, Укажите, сколько валентных электронов имеет каждый из этнх элементов? Объясните, почему углерод в органических соединениях имеет валентность, равную четырем?
2. Запишите приведенные ниже соединения в виде структур Льюиса, обозначив (где необходимо) неподеленные пары электронов: Вг2> CjHfjj С2Н2; СзНв; СН2О; CH3NH2; CH3CI.
3. Дайте определение понятию «гибридизация». Какие виды гибридизации возможны для атома углерода в органических молекулах? Приведите примеры и укажите форму и пространственную! направленность атомных орбиталей в каждом случае. J
4. Определите вид гибридизации атомов в следующих соединен инях: В
СС14; СО2; CHa-C^N; СН2-СН-СН-СН2;
.О /===\
СНа—с\^си: СН3—NH2; Н3С-С=С—СНэ; ^-СН3.
5. Дайте определение основным типам химической связи. Укажите, какие типы химической связи имеются в молекулах приведенных соединений: СН3—СН2—Вг; СН8—C^N; CHS~CH—СН3;
>O“Na+
CH3—NOa; СНЭ—ОН; СНЯ—NH—NH2; [CHsNHal+J-; H’C^Q J /O-NH+
CHB—
6. Дайте определения о- и л-связям. Укажите, какие из связей в молекулах приведенных соединений относятся к о-, какие —к
46
л-тииу. Укажите атомные орбитали, участвующие -в образовании этих связей:
\ С1
СН8^СН^СН—СН3; СН3—teCH;
СНЧ—NH—CH; CH4—C~N
3 й 3
7, Диэтиловый эфир С2Н5—О—С2Нэ и н-бутиловый спирт С4Н9ОН имеют одинаковую молекулярную массу. Объясните, почему температура кипения спирта (118СС) значительно выше, чем эфира (35 СС)?
8. Дайте определение понятиям «полярность» и «поляризуемость» связи. Объясните причину появления дробных или целых зарядов в соединениях, структурные формулы которых приведены ниже:
а) Н->Вг;
а—
6-1-
в) СНа-ЮН;
Д)
снз—N-H
ОН'; е)
6- Й4-НС^Сч-Н
Н
О
Н J
Глава 4 ВЗАИМНОЕ ВЛИЯНИЕ АТОМОВ В ОРГАНИЧЕСКИХ СОЕДИНЕНИЯХ
Идея о взаимном влиянии атомов иа реакционную способность органических соединений впервые высказана А. М. Бутлеровым <в теории химического строения. Дальнейшее развитие она получила в теории электронных смещений (электронных эффектов). Согласно современным представлениям, реакционная способность органических соединений определяется характером распределения электронной плотности в молекуле и подвижностью (поляризуемостью) электронов ковалентных связей.
В органических соединениях различают два вида электронных смещений: смещение элеитронной плотности по цепи о-связен — индуктивный эффект; смещение по системе п-связей — мезомер-нын эффект.
4.1. ИНДУКТИВНЫЙ ЭФФЕКТ
Как уже отмечалось, при образовании ковалентной связи между двумя атомами с одинаковой электроотрицательностью электронная пара в равной мере принадлежит связанным атомам (неполярная связь). Если же атомы имеют разную электроотрицательность, электронная плотность ковалентной связи смещается к более электроотрицательному атому (полярная связь), В сложных органических молекулах полярная ковалентная связь оказывает влияние на соседние связи: приводит к изменению их электронного состояния, вызывая поляризацию:
6"+ й'х б |-4 3 2 1
НэС—СНа—СН3—СНЭ Н.С^СН2-н-СН2^СН2^С(
U > dfa lb *
«-бутан, Ц = 0 1-хларбутзн, ц. =£ О
Например, в молекуле я-бутана все углерод-углеродные связи неполярны, электронная плотность распределена симметрично и молекула не имеет дипольного момента. Введение в молекулу н-бутана атома хлора (1-хлорбутаи) приводит и поляризации не только связи С—С1, но и соседних углерод-углеродных связей. Атом хлора, имея большую электроотрицательность, чем углерод, оттягивает электроны о-связи С—С1 в свою сторону. В результате такого смещения на атоме хлора появляется частичный отрицательный заряд (6_), а на углеродном атоме — равный по значении: положительный заряд (б+). Понижение электронной плотности на С1 частично компенсируется оттягиванием в его сторону электро-
48
нов с соседнего атома углерода, что приводит к поляризации связи С2—С1 н возникновению частичного положительного заряда иа атоме С2. Тот, в свою очередь, вызывает поляризацию связи С3—С2 н т. д. По мере удаления связи дробный заряд на атомах углерода уменьшается: Таким образом, поляризация
связи С—С1 вызывает электронную асимметрию молекулы н появ-
• леиие дипольного момента.
I Передача электронного влияния заместителя вдоль цепи о-связей называется индуктивным (индукционным) эффектом.
Обозначается индуктивный эффект буквой Направление смещения электронной плотности о-связей изображают прямой стрелкой (-* **).
Действие индуктивного эффекта наиболее сильно проявляется на двух ближайших о-связях. Из-за слабой поляризуемости о-связей индуктивный эффект через 3—4 связи затухает.
В зависимости от направления электронного влияния заместителя различают индуктивный эффект положительный (+/) и отрицательный (—/). В качестве стандарта для оценки направления индуктивного эффекта заместителя принят индуктивный эффект атома водорода, который из-за небольшого дипольного момента связи С—Н считается равным нулю.
Заместители, притягивающие к себе электроны о-свя-зи в большей степени, чем атом водорода, проявляют отрицательный индуктивный эффект (—1), а заместители, отталкивающие от себя электроны связи сильнее атома водорода, проявляют положительный индуктивный эффект (+/).
Под притяжением и отталкиванием имеется в виду разница
д“
d+
R-^CH2
-/-эффект
[_А j-_ OR2rNR3rNO2,-CS N,
-с yF.-ci-Br.-j, - он
-Cz 7OR,-CZ , -SR. XOR
-OH, -NH2;i-C=CR;-Ar*
- CH=CR 2
R ~ сн2-{ н !
d г-1
RСН Г-' В J
стандарт /-0
* Аг — ароматический радикал,
** Aik — алкильные группы.
40
в положении электронов связи, обусловленная различной электроотрицательностью атома водорода и заместителей.
Выше приведены некоторые важные заместители, расположенные в порядке уменьшения —1- или +/-эффектов по отношению к водороду.
Отрицательный индуктивный эффект заместителя, как правило, тем больше, чем выше электроотрицательность атома, связанного с углеродной цепью. Наибольший отрицательный индуктивный эффект проявляют группы, несущие положительный заряд. При прочих равных условиях sp-гибридизованные атомы обладают большей электроноакцепторной способностью, чем $р2-атомы, а те, в свою очередь, большей, чем зр3-атомы. Этим объясняется отрицательный индуктивный, эффект остатков непредельных и ароматических углеводородов:
пропев
-/- эффект
•/-эффект
толуол
SP1 Д________
СИ,—fcs.CHJ 7
•/-эффект
иронии
Наибольший положительный индуктивный эффект проявляют заместители, несущие отрицательный заряд. Среди алкильных групп большими электронодонориыми свойствами обладают третичные радикалы, затем вторичные и, наконец, первичные:
СН3
СН3^-С— >
1
сн3
сн3
\СН— > сна^сн4^сн2-сна— -/ О А л А
с2н/
В ряду первичных алкильных радикалов положительный индуктивный эффект возрастает с увеличением углеродной цепи: СШ9->С3Н7->С2Н5->СН3-,
Таким образом, на основании изложенного можно сделать следующие выводы:
1. Индуктивный эффект проявляется всегда при наличии в молекуле атомов с различной электроотрицательностью.
2. Индуктивный эффект распространяется только через и-свя-зи и всегда в одном направлении.
3. Индуктивный эффект затухает в цепи через 3—4 п-связи.
4.2. МЕЗОМ ЕР НЫ И ЭФФЕКТ (ЭФФЕКТ СОПРЯЖЕНИЯ)
Иным образом, в отличие от индуктивного эффекта, происходил передача электронного илияння заместителя по сопряженной смете ме rt-связей.
Сопряженной называется система, состоящая из чередующихся простых и кратных связей, или же когда рядом с углеродным ато'
50
мом, образующим кратную связь, находится втом с неподелеииой *парой р-электронов или атом, имеющий вакантную р-орбиталь. Например:
; /ч
.СНв=СН—СН«СН, II СН-СН—С1 сн,=сн—сна
4 Я и За 15 <3
бутадявн-1, 3 хлористый винил аллильный катион
\/
бензол
Сопряженные системы делятся иа системы с открытой и замкиу-. той цепью.
В сопряженных системах имеет место дополнительное перекры-; ванне л- и р-орбиталей, которое называют сопряжением. Различа-‘ ют (рис. 4.1.) л, л-сопряжепие (перекрывание двух л-орбиталей) р, л-сопряжение (перекрывание р-орбиталн с п-орбиталыо). Со-спряжение возможно лишь в случае параллельности осей симметрии ^взаимодействующих орбиталей.
л,Jt-coпряжение делокализованная
вакантная р-орбиталь
4.1, Сопряженные свстемы бутадиепа-1,3, хлористого винила и аллильного катиона
R1
В результате сопряжения образуется единая л-электроииая система н происходит перераспределение (делокализация) л-элек-троиной плотности (ряс 4Л).
Сопряжение является для молекулы энергетически выгодным процессом. Ойо сопровождается уменьшением энергии системы и приводит к повышению термодинамической устойчивости молекулы.
Процесс передачи электронного влияния заместителя по сопряженной системе л-связей называют мезомерным эффектом (М) или эффектом сопряжения (С). Мезомериый эффект проявляется лишь в том случае, когда заместитель включен в сопряженную систему молекулы.
Например, аминогруппа в молекуле анилина и альдегидная группа в акролеине входят в сопряженную систему, и, следовательно, проявляют мезомериый эффект, а в молекуле бензиламина аминогруппа мезомерным эффектом не обладает.
NH2| f ___J
анилин
СПз=СН-;С.
бензиламин
акролеин
Различают мезомериый эффект заместителя положительный (+Л4) и отрицательный (—Л)).
(Положительный мезомериый эффект проявляют заместители, подающие электроны в сопряженную систему.
К иим относят атомы, содержащие неподелеииые пары электронов или отрицательный заряд, а также атомные группы, имеющие на первом атоме неподелеииые электронные пары или отрицательный заряд.
I Отрицательный мезомериый эффект проявляют заместители, оттягивающие иа себя электронную плотность сопряженной системы.
—Л4-эффектом обладают заместители, первый атом которых несет положительный заряд, а также атомные группы, в которых первый атом связан кратной связью с более электроотрицательным атомом, чем он сам.
Направление смещения электронной плотности я-связей и ие-поделенных пар электронов изображают изогнутой стрелкой (<-*), начало которой указывает, какие электроны смещаются, а конец — связь или атом, к которым оии смещаются.
Ниже приведены некоторые заместители, расположенные в порядке уменьшения +Л1- или —М-эффектов.
62
Наибольший отрицательный мезомериый эффект проявляют заместители, несущие положительный заряд. —/И-эффект ненасыщенных группировок тем больше, чем больше разность электроотрицательностей атомов, связанных кратной связью. Наибольшим положительным мезомерным эффектом обладают атомы, несущие отрицательный заряд. + /Vl-эффект заместителей, содержащих атомы с неподеленпымн парами электронов тем больше, чем меньше в пределах периода электроотрицательность атома, несущего непо-деленную электронную пару, например!—NH2>—ОН>—F. В пределах группы периодической системы ф-Л^-эффект заместителей ослабевает сверху вниз (—F>—С1>—Вг), что объясняют большей выгодностью при перекрывании близких по размерам р-орбиталей.
Различают четыре основных типа проявлении мезомерного эффекта и органических соединениях,
1. Взаимодействие заместителя, обладающего Ч-М-эффектом с п-электроиной системой молекулы!
+м~ эффект
винидметиловый эфир
фенол
2. Взаимодействие заместители, с л-электронной системой молекулы:
обладающего —М-эффектом
Л Гг—ж-1
CH2=CH-fc^Ni
-М-эффект ' акрилонитрил
д-М-эффект '. нитробензол
S3
3. Взаимодействие двух заместителей с 4-Л1-эффектом и —М-эф-фектом, связанных о-связыо:
СН3-!-С $ OHj-f- М-эффект ~М- эффект уксусная кислота
4. Взаимодействие заместителей, обладающих +Л!-эффектом и —М-эффектом через я-электроиную систему молекулы:
’Н^Ы
|
Ч-М-эффект ~М- эффект
4-аминобсизальдегид
В отличие от индуктивного эффекта, передача элеитронпого влияния заместителя по сопряженной системе происходит на значительно большее расстояние, практически не затухая.
4.3. СОВМЕСТНОЕ ПРОЯВЛЕНИЕ ИНДУКТИВНОГО И МЕЗОМЕРНОГО ЭФФЕКТОВ ЗАМЕСТИТЕЛЕЙ
Ранее отмечалось, что индуктивный эффект проявляется в органических соединениях всегда при наличии в молекуле атомов с разной электроотрвцательностыо. Мезомерный же эффект проявляется лишь в том случае, когда атомы с разной электроотрицательностью входят в сопряженную систему. Поэтому включенные в сопряженную систему заместители вызывают поляризацию связей за счет индуктивного и мезомерного эффектов. Эти эффекты могут совпадать и не совпадать по па правлению:
СНо*£сН-к:Л1 р I __ __1
-I-эффект
-М-эффект
акрилонитрил
фенол
Для большинства заместителей индуктивный и мезомерный эффекты совпадают по направлению. У заместителей, содержании* атомы с неподелеиными электронными парами, индуктивны#
54
И КаК Vbl Apyi др/ia.
а Поскольку я-связи поляризуются легче по сравнению с о-свя-I аями, более выраженное влияние на поляризацию молекул оказы-1 вает мезомерный эффект, т. е. мезомерный эффект заместителя, как правило, больше его индуктивного эффекта. Исключение составляют лишь атомы галогеиов (й, Ci, Вг, J), для которых и статическом (нереагирующем) состоянии молекулы 4- М-эффект меньше —/-эффекта, ио в процессе реакции (динамическое состоя-
|ние) и для них 4- М-эффект больше —/-эффекта.
Заместители, повышающие электронную плотность в молекуле, называют электронодоиорными, а заместители, понижающие eet— алектроноакцепторнымн.
В результате совместного проявления индуктивного и мезомер--ного эффектов заместителя происходит альтернирование (чередование) поляризации в сопряженной системе по значению зарядов:
сн >= сн -* сн сн А а ‘ 3 2 I
1-хлорбутадиен-1,3
Так, в молекуле 1-хлорбутадйена-1,3 в результате —/-эффекта . атома хлора, который больше, чем 4-Л1«эффект( все атомы углерода приобретают дробный положительный заряд. Но благодаря 4-М-эф-фекту хлора на атомах О и С4 электронная плотность несколько увеличивается. И несмотря иа то, что в итоге все атомы углерода несут дробный положительный заряд, в положениях С2 и С4 он меньше, чем в С1 и С3, т. е. происходит чередование поляризации по значению заряда *.
Аналогичное явление наблюдается в системах с замкнутой цепью сопряжения;
В молекуле анилина —/-эффект аминогруппы вызывает понижение электронной плотности на всех атомах углерода беизоль-ного кольца. Однако так как 4--эффект электронной лары атома азота больше —/-эффекта, это приводит в целом к повышению
* Знак б++ обозначает, что в этом положенив электронная плотность ниже, в положении со знаком б^,
58
,у атомах кольца и особенно электронной плотности на Углер^“СХоДит альтернирующая поля-в положениях 2, 4 и 6, т. е. р
ризация. К1ТПЙ „еПью сопряжения указывают
Обычно в молекулах с о™р«™ « Ые на концевых втомах только частичные заряды, соср ых сопряженных c"CTcHev сопряженной системы, а в за У максимальные заряды на атомах.
$
системах —
вг,я- сопряЖёшЛ’
рис. 4.2. Схема перекрывания о-орбиталей связи С — Нс л-орбиталыо кратной связи в молекуле пропена
s
бензальдегид
б"
д"
ОН
фезол
н
акролеин
4.4.
СВЕРХСОПРЯЖВНИМГИП^КОНЪЮГАЦИЯ!
-.Ир, .^-сопряжением в моле-
Наряду с обсужденным ранее л,л ний имеет место особым кулах некоторых органическихп ^пряжением, или гиперконь-вид сопряжения, нэзывэемый 'верхе л-™ веществ, и которых югацней. Сверхсопряжение харак и е один атом ВОДорода, атом углерода, несущий по кРайне1 связан с ненасыщенной группой.
н _ н
Н-2с^сН=^СН2 t н н н
-с3-с. н
пропан аль
„Л1, толуол
пропен '
пинаний электронная плотность ff-связей
В молекулах таких соединении енной группировкой, смеща-С—Н углерода, связанного с смешение электронов назЫ-
ется в'сторону кратной связино изображают изогнутой иают сверхсопряжением и схема стрелкой. ^яинческой теории сверхсопряжение рас-
С позиций кваптово-механнческ а.орбиталей связей С—Н
сматривают как частичное перекрыв нсходит (^-сопряжение с я-орбиталями кратной связи, т. •
Трис. 4.2). сильнее, чем больше
Эффект сверхсопряжения тем анном с ненасыщенной сл атомов водорода прн углероде, стемой:
[ау- > .
S6
В результате сверхсопряжения эти атомы водорода активируются (протонируются) и проявляют повышенную реакционную способность.
4.5. ПРОСТРАНСТВЕННЫЕ ПРЕПЯТСТВИЯ СОПРЯЖЕНИЮ
Наличие сопряженной системы является обязательным, ио не единственным условием для сопряжения. Важную роль в сопряжении играет пространственное расположение взаимодействующих орбиталей. Как уже отмечалось, мезомерпый эффект проявляется лишь в том случае, когда все атомы сопряженной системы лежат в одной плоскости или близко к ней, т. е. когда наблюдается условие параллельности осей симметрии участвующих в сопряжении орбиталей. Если это условие не выполняется, сопряжение не происходит нлн эффективность его значительно снижается (рис. 4.3).
а б
Рис. 4.3. Пространственное расположение р-орбиталев си пряженной системы: а — сопряжение невозможно; б — сопряжение осуществляется
Одной из причин нарушения эффекта сопряжения заместители Являются пространственные (стернческие) препятствия. Они воз-
57
никают при наличии в молекуле у соседних с заместителем атомов объемных групп. В результате стерических факторов заместитель выходит из плоскости сопряженной системы, что частично или полностью нарушает перекрывание р-орбиталей:
' /4’3.97 Д
Д-3.17Д .
Так, в молекуле нитробензола иитрогруппа и бензольное кольцо расположены в одной плоскости, что обеспечивает сопряжение. В молекуле же 2, 4, 6-трибромнитробензола нитрогруппа в результате пространственных препятствий со стороны заместителей и положениях 2 н 6 вынужденно выведена из плоскости бензольного кольца. При этом осн р-орбмталей атомов азота и углерода кольца располагаются под углом по отношению друг к другу, что затрудняет их перекрывание, а следовательно, и сопряжение.
Влияние пространственных препятствий на проявление эффекта сопряжения заместителя можно обнаружить методом дипольных моментов. В приведенном примере молекула нитробензола имеет значительно больший дипольный момент (3,97 Д), чем молекула 2, 4, 6-трибромпнтробепзола (3,17 Д). При сопоставлении дипольных моментов необходимо иметь в энду, что расположенные в цеитросимметричиых положениях бензольного кольца одинаковые заместители не влияют на дипольный момеит молекулы.
4.6. СПОСОБЫ ИЗОБРАЖЕНИЯ РАСПРЕДЕЛЕНИЯ ЭЛЕКТРОННОЙ ПЛОТНОСТИ в МОЛЕКУЛАХ. ПОНЯТИЕ О РЕЗОНАНСЕ
Структурные формулы достаточно однозначно описывают строение органических соединений лишь с локализованными химическими связями, т. е. когда электроны связи разделены (локализованы) между двумя ядрами. Смещение электронной плотности в локализованных связях молекулы изображают в структурной формуле прямой стрелкой (->)
СН,,->СН,-^С1 а л
хлорэтеэ
В молекулах с сопряженными системами связей одна нлЯ несколько связывающих орбиталей принадлежат уже не двум атомам, а охватывают несколько атомных ядер. Такую связь назЫ' вают делокализованной. Используемые в структурных формула*
68
для обозначения химических связен черточки не отражаю! рсало-иое положение электронов в делокализованных связях.
Для изображения распределения электронной плотности в сопряженной системе молекулы с помощью структурных формул используют два способа. Один из них состоит в том, что соединение изображается одной структурной формулой, в которой изогнутой стрелкой (">) указывается направление смещения р- или л-элек-троиов:
СН2-СП —СН —сн2
СН-С л
ОС- IU
2. э
4 -литроанилин
бутадиен-] ,3
этилацетат i
Идея второго способа принадлежит американскому ученому Л. Полингу и носит название метода резонансных структур. Сущность его заключается в том, что делокализации электронов в сопряженной системе изображается с помощью нескольких структурных формул, так называемых резонансных (предельных, граничных) структур, отличающихся друг от друга только распределением электронных пар между ядрами атомов. Истинное электронное строение молекулы не соответствует ни одной из данных структур, а является промежуточным между ними, т. е. реальная молекула рассматривается как гибрид резонансных структур. Взаимосвязь предельных структур изображают двухсторонней стрелкой (**). Например, делокализацию электронов в молекуле фенола можно изобразить в виде следующих структур:
ОН ОН ОН ОН
I I! !i II
Резонансные структуры не существуют реально, это лишь способ описания делокализации электронов в молекуле. Вклад каждой структуры в истинное строение молекулы пропорционален ее Устойчивости, т. е. чем стабильнее граничная структура, тем вклад ее значительнее. Относительная устойчивость резонансных структур определяется несколькими факторами.
Более устойчивой считается структура, в которой: а) содержится большее число ковалентных связей
сн8=сн—сн^сн, +* ск-сн-сн—сн9, т „ , * в ’
*е-структура (А) стабильнее, чем (Б);
59
б) отрицательный заряд расположен на более электроотрицательном атоме (нли положительный зарнд на атомах с низкой электроотрицательностью)
ZO + ZO“ _ уО*
СНа=СН—СС о СНа—СН=С< О СН2-СН=С< »
\н \н \н
ДБ В
т, е. структура '(А) вносит основной вклад; структура (Б) стабильнее, чем (В);
в) меньше расстояние между разноименными зарядами
NHa—СН=О NH2—СН—О о Кна=сн—О,
А Б В
т. е. структура (А) вносит основной вклад; структура (Б) —значительный, а структура (В) — незначительный.
Энергия истинной молекулы всегда меньше энергии, рассчитанной для любой из резонансных структур. Разница между энергиями реальной молекулы и наиболее устойчивой граничной структуры называется энергней резонанса. Энергия резонанса тем больше, чем больше количество резонансных структур и чем больше число ннзкоэнергетичиых структур с равной или близкой энергией. Энергия резонанса является мерой стабилизации молекулы, обусловленной делокализацией электронов в сопряженных системах.
Несмотря иа то что метод резонанса более наглядно описывает делокализацию электронной плотности в сопряженных системах, его применение затруднено необходимостью написания большого числа граничных структур. Это особенно неудобно при составлении уравнений реакций, поэтому в дальнейшем, как правило, делокализация электронов в сопряженных связях будет изображаться с помощью изогнутых стрелок. Метод же резонанса будет использован только для качественной оценки строения и реакционной способности отдельных органических соединений.
КОНТРОЛЬНЫЕ ВОПРОСЫ И УПРАЖНЕНИЯ
1. Дайте определения понятиям: индуктивный эффект (I)! мезомерный эффект (А1). Приведите примеры заместителей, проявляющих а) +/-эффект; б) —/-эффект.
2. Как изменяется электроотрицательность атомов углерода в зависимости от вида гибридизации? Укажите электроиоакцеп-торные группы в следующих соединениях: а) СН3—СН2—СН =СНг-
б) СНа—С=СН; в) СН3—СН3—
60
3. Изобразите распределение электронной плотности в молекулах приведенных соединений;
,0
a) CHs^CHa-CHa-Nfo;
б) СН3—СН2—ОН;
сн,
в) СН,—СН,—СН,—Ci; г) GH,=CH—С—СН,; д) СН,—C^N.
' сн,
4. Дайте определение понятиям: сопряженная система, сопряжение. В приведенных соединениях укажите сопряженную систему
и виды сопряжения:
а) СНа=СН—СН—СНа; б) СНа—СНа—СН=СН—СН=СНа;
в)
СНЭ—СН=СН—С
г) СНа=СН-б-СН8;
Д) е) ^-СНа-СНя
5. Какие атомы и атомиые группы способны проявлять: а) + Л4-эффекТ; б) —/-эффект?
6. С помощью стрелок, используемых для обозначения индуктивного и мезомерного эффектов, покажите распределение электронной плотности в следующих молекулах! a) CHa=sCH— СН=СН—CI;
/---z°
б) СНа=»СН—СН=СН—О=Ы; в) <f ;
х==-< хо
г) С1--СНа—-(СНа)а—
7. Что такое резонансные структуры? Перечислите факторы, которые определяют относительную устойчивость резонансных струитур. Приведите резонансные структуры для молекулы анилина.
8. Дайте определение понятию энергия резонанса. От чего зависит значение энергии резонанса?
ei
Глава 5 ИЗОМЕРИЯ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ. ПРОСТРАНСТВЕННОЕ СТРОЕНИЕ МОЛЕКУЛ
Изомерия — явление, заключающееся в существовании соединений, одинаковых по качественному и количественному составу, но различающихся порядком связывания атомов в молекуле или расположением их в пространстве, а вследствие этого имеющих различные физические и химические свойства.
Впервые термин «изомеры» был введен в 1830 г. шведским химиком й. Я- Берцелиусом. Теоретическое же обоснование изомерии принадлежит русскому химику А. М. Бутлерову. С развитием органической химии понятие изомерии расширилось благодаря представлениям о пространственном строении органических соединении (Я. Вант-Гофф, Ж. А. Ле-Бель, 1874 г.).
Различают два основных вида изомерин — структурную (изомерия строения) и пространственную (стереоизомерия).
ИЗОМЕРИЯ
СТРУКТУРНАЯ ПРОСТРАНСТВЕННАЯ
(изомерия строения) (стереоизомерия)
I Г~ “
ЦЕПИ ПОЛОЖЕНИЯ ФУНКЦИОНАЛЬНЫХ
ГРУПП
КОНФИ ГУРАНИОННАЯ КОНФОРМАЦИОННАЯ
1
ГЕОМЕТРИЧЕСКАЯ ОПТИЧЕСКАЯ
1.1. СТРУКТУРНАЯ ИЗОМЕРИЯ
Структурные изомеры отличаются друг от друга последовательностью связывания атомов в молекуле, т. е. структурой. И*' еще называют изомерами строения. Структурная изомерия поД' разделяется иа:
изомерию углеродной цепи, изомерию положения, изомерию функциональных групп.
62
Изомерия цепи обусловлен „.—мшиисгью
эываиия атомов, образующих углеродный скелет молекулы, СН3-СН2—СН„—СНа ГН и - г
и Й 0
бутан
а разной последовательностью свя
(С.Н10)
3
сня—сн—сня
5* о
сн3
нэобутан
(2-мстилпропаи)
циклического строения изомерия величиной цикла
СН.,4 -ГН
(C,HIt)
• циклогексан метиланклопекгли я разным способом соединения
1 • г-днметидцнклобутан циклов:
(С14Н10)
антрацен фс-цактрен
Изомерия положения обусловлена разным ковых функциональных групп или кратных и том же углеродном скелете молекулы
СНа—СН,—СИ,—С1
0 £ А
t --
положением одина-связей при одном
(С3Н?С1)
СН^СН—СИ t я
t-хлорпропан
С!
<СвН4С1а)
1,4-днхлорбепзол
СИ—СН—СН—СН V «
бутен-3
(С4Н8)
^крумурной изомерии, когда изомеры отличаются функциональными группами, называют изомеоией Л’»>"и-
лсвын эфир:
СНВ—СНа—он
этанол
CH2-=CH-CHS—СН3 буте re-1
Вид структурной I--- t ________ _ ___
гт.„ .rJllJJOBn, называют изомерией функциональных групп, агдядным примером такой изомерии является этанол и диметн-
СН3-О-СН3 (СаНвО)
ДИМСТИЛОВЫЙ эфир
63
Изомерия цепи и положения проявляются» как правило, в пределах одного класса органических соединений, а в случае изомерин функциональных групп изомеры принадлежат к разным классам.
В некоторых случаях два структурных изомера могут находиться в состоянии динамического равновесия друг с другом. Такое явление называют таутомерией, а структурные изомеры — таутомерами:
таутомеры
О он
сн,—с—сн. 5=г=^±:сна-с=сн, ацетон
(СвНвО)
5.2. ПРОСТРАНСТВЕННАЯ ИЗОМЕРИЯ [СТЕРЕОИЗОМЕРИЯ]
Вещества, имеющие одинаковый состав и порядок связывания атомов в молекуле, но отличающиеся друг от друга их расположением в пространстве, называют пространственными изомерами, нли стереоизомерами.
Для характеристики пространственных различий в стереоизомерии используют понятия конфигурация и конформация,
(Конфигурацией называют то или иное относительное расположение атомов молекулы в пространстве.
Например, в молекуле метана СН4 атом углерода имеет тетраэдрическую конфигурацию. Его пространственная модель представляет правильный тетраэдр, в центре которого находится углеродный атом, а в вершинах—атомы водорода. Угол между связями метана, равный 109°28\ называют нормальным или тетраэдрическим (рис. 5.1). Если углерод связан с разными замести
Н
б
180а
Рис. 5.1. Конфигурация атома углерода: а — тетраэдрическая; б — плоскостная; в — линейная
телями, углы между связями незначительно отклоняются от ноР' мальиого. Так, в молекуле трихлорметана СНС13 валентный угоЛ С1—С—С1 равен 112°, В этилене Н^С^СНз атом углерода имееТ плоскостную конфигурацию (все атомы лежат в одной плоскости»
«4
валентный угол 120"), в ацетилене — линейную (атомы расположены лииейво1 валентный угол 180°).
Понятие конформация отражает более тонкие овобенности пространственного строения молекул.
Конформацией называют различное пространственное расположение атомов или атомных групп в молекулах определенной конфигурации, обусловленное вращением вокруг о-сэязей.
Например, в молекуле этана НзС—СНд в результате вращения воируг углерод-углеродной связи меняется пространственное расположение одной метильной группы относительно другой, Прн этом молекула принимает множество конформаций.
Органические соединения, отличающиеся друг от друга только конфигурацией молекул (без учета возможных конформаций), называют конфигурационными изомерами. Различают конфигурационные изомеры оптические и геометрические.
Стереоизомеры, имеющие различное пространственное расположение атомов или атомных групп, обусловленное вращением вокруг простой углерод-углеродиой связи, называют конформационными изомерами.
5.2.1. Способы изобретения пространственного строения
Для изображения простран стае иного строения органических соединении используют молекулярные модели и стереоформулы.
Молекулярные модели. Более наглядное представление о пространственном строении молекулы дают молекулярные модели. Обычгго применяют три основных типа моделей: шаростержневые, скелетные (модели Драндинга) и полусферические (модели Стю-арта-Бриглеба).
В исаростержкевых моделях молекул атомы представлены разноцветными шариками, а химические связи—стержнями (рис. 5.2). Шарики связаны друг с другом стержнями с учетом взаимного расположения атомов в пространстве. Шаростержпе-вые модели удобны для рассмотрения валентных углов и вращения вокруг простых связей, однако они не отражают относительных размеров атомов и межатомных расстояний в молекуле.
Модели Драйдинга, в отличие от шаростержневых, ограничиваются изображением лишь скелета молекулы, т. е. химических связей между атомами (см. рис. 5.2), причем межатомные расстояния в этих моделях пропорциональны истинным (в масштабе 0Л нм=2,5 см).
Полусферические модели Стюарта-Бриглеба изображают реальные молекулы с учетом пространственного расположения атомов, межатомных расстояний н размеров атомов (см. рис. 5.2). В молена* Стюарта-Бриглеба атомы представлены разноцветными, Этично срезанными шариками, радиус которых пропорционален
2-253
3
65
Рис Б.2. Модели молекул этана (слева) и этилена (справа): а — шаростержневые; б -г- ДраИднвга; а — полусферические (Стюарта — Бриглсба)
ваи-дер-ваальсоеу радиусу * атома, а расстояние от центра шарика до поверхности среза — ковалентному радиусу **. Атом угле рода в состоянии лр3-гибридизации изображают в виде шаре
•Ван-дер-ваальсов радиус атома г„ равен половине расстояния между двумя несвязанными атомами при их максимальном сближении.
•* Ковалентный радиус атома ги равен половине длины ковалентной связи между одинаковыми атомами.
66
К* четырьмя, в sp2— с тремя, в sp— с двумя срезамл, атом водорода представляют шаром с одним срезом и т. д. При сборке моделей шарики соединяются между собой по плоскостям срезов. Несмотря и а то, что полусферические-модели наиболее удачно отражают реальные молекулы, они не пригодны для рассмотрения валентных углов между атомами и вращения вокруг простых связей.
Стереоформулы. Для изображения пространственного строения органических соединений на плоскости (например, на листе бумаги нли доске) используют стереохимические и перспективные формулы, а также проекционные формулы Ньюмена.
В стереохимических формулах химические связи, расположенные в плоскости чертежа, изображают обычной чертой; связи, находящиеся над плоскостью — жирным клниом нли жирной чертой, а расположенные под плоскостью — штриховым клином или пунктирной линией:
этаи
Перспективные формулы описывают пространственное строение на плоскости с учетом рассмотрения молекулы вдоль одной нз углерод-углеродных связей. По внешнему виду они напоминают лесопильные козлы:
этан
и-бутан
При построении проекционных формул Ньюмена молекулу рас* матривают в направлении одной С—С-связи таким образом, что-вькатомы’ образующие данную связь, заслонили друг друга. Из оранной пары ближний к наблюдателю атом углерода изобража-точкой, а дальний — окружностью. Химические связи ближнего з*
67
атома углерода с другими атомами представляют линиям;!, берущими начало от точки в центре круга, а дальнего — от окружности:
этап п-бутан
Ни один из приведенных способов изображения пространственного строения не является универсальным. Стереохимические формулы чаще применяют для описания стереохимических аспектов протекания реакций, перспективные формулы и проекции Ньюмена — в основном для изображения конформаций молекулы.
Существуют также проекционные формулы Фишера, которые применяют обычно для изображения ва плоскости пространственного строения оптических изомеров (см. с. 73).
5.2.2. Оптическая изомерия
Оптическая активность и хиральность молекул. Для характеристики оптической изомерии важное значение имеют такие понятия, как оптическая активность и хиральность молекул.
Оптической активностью называют свойство вещества вращать плоскость поляризации поляризованного света. Если луч обычного света, в котором, как известно, электромагнитные колебания происходят в разных плоскостях, перпендикулярных к направлению его распространения, пропустить через призму Николя, то выходящий свет будет плоскополяризованиым. В таком луче электромагнитные колебания совершаются только в одной плоскости. Эту плоскость называют плоскостью поляризации (рис. 5.3).
При прохождении поляризованного света через оптически активное вещество плоскость поляризации поворачивается па определенный угол Gt вправо нли влево (см. рис. 5.3). Если вещество отклоняет плоскость поляризации вправо (при наблюдении навстречу лучу), его называют правовращающим, если влево — левовра-
* Призма Николя — призма, изготовленная из двух кристаллов яслаядского шпата (СаСО3), склеенных канадским бальзамом,
6S
IТричма Николя Плоскость поляризаций
t Обычный свет Поляризованный Оптически активное j свет вещество
Нис 5,3. Схема образования поляризованного света и вращения плоскости поля-В риэации оптически активным веществом
щающим. Правое вращение обозначают знаком ( + ), левое — знаком (—).
Угол вращения а зависит от природы оптически активного вещества, толщниы слоя оптически активной среды, через которую проходит поляризованный свет, и его длины волны. Дли растворов угол а зависит также от природы растворителя и концентрации оптически активного вещества. В меньшей степени оптическое вращение подвержено влиянию температуры,
, Для сравнительной оценки оптической активности различных соединений используют значение удельного вращения [а]. Удельное вращение является константой оптически активного вещества. Оно характеризует оптическую активность раствора с концентрацией оптически активного вещества 1 г/мл при толщине слоя 1 дм.
Удельное вращение вычисляют по одной из приведенных формул:
для веществ в растворе [а] = —;
для жидких веществ [а] — ,
I ' р
где а — измеренный угол вращения, град.; I — толщина слоя, дм; с — концентрация оптически активного вещества, г/100 мл раствора; р — плотность жидкого вещества.
Оптическую активность измеряют с помощью приборов, называемых поляриметрами. Обычно определение проводят при температуре 20 СС и длине волны D-линии спеитра натрия (589,3 км). Соответствующее зиачеиие удельного вращения обозначают [а] д.
Обязательным условием для проявления органическим соединением оптической активности является асимметрия (отсутствие симметрии) его молекул. Поскольку молекула представляет собой
69
трехмерное образование, ее строение можно рассматривать с точки зрения симметрии геометрических фигур. Основными элементами симметрии являются плоскость, центр и ось симметрии.
Плоскость симметрии — это воображаемая плоскость, которая проходит через молекулу, разделяя ее на две зеркально равные части (ркс, 5.4, а).
Рис. 5.4. Элементы симметрии органических молекул: а — днхлорметапа; б — 113-дифтор.2,4-дихлорциклобутава; в —хлороформа
Центром симметрии называют воображаемую внутри молекулы точку, равноудаленную от одинаковых атомов, расположенных иа прямой, проходящей через эту точку (рис. 5.4, б).
Осью симметрии называется проходящая через молекулу во обряжаемая ось, при повороте вокруг которой иа угол 360°//i (п — целое число, равное 2, 3, 4 и т. д.) молекула совмещается со своим исходным положением. Число п определяет порядок осн симметрии. Если rz=2, 3 и т. д., ось симметрии называют соответственно осью второго, третьего и т, д. Порядка. Например, молекула хлороформа (рис. 5.4, в) совмещается со своим исходным положением при каждом повороте вокруг связи С—Н на угол 36073— --120°. Следовательно, проходящая через молекулу вдоль связи С—Н ось называется осью симметрии третьего порядка.
Различные объекты, в том числе и молекулы, не имеющие плоскости, центра и оси симметрии, обладают свойством не совмещаться со своим зеркальным изображением. Это свойство называют хиральностью (от греч. — рука), а молекулы им обладающие— хирлльиыми. Наглядным примером хиральности могут служить левая и правая руки, которые являются зеркальным отра* жением друг друга, но вместе с тем их нельзя совместить прй
70
любом способе наложения. По этой причине перчатка с левой руки не пригодна для правой и наоборот.
Молекулы, имеющие хотя бы один элемент симметрии, всегда идентичны со своим зеркальным изображением и называются ахиральными. Хиральность молекулы можно легко установить путем построения модели молекулы и модели ее зеркального изображения с последующим их совмещением. Если модели ие совмещаются — молекула хиральпа, если совмещаются — ахиральна. Такой же вывод можно сделать и на основе стереохимических формул молекул по наличию или отсутствию элементов симметрии, чаще нсего плоскости симметрии:
С1
gr ^"Сцт///Ц
ЧКиральные молекулы (плоскость симметрии отсутствует)
ахиральные молекулы
(имеется
плоскость симметрии)
нзс
2-хлорбутан
бромиодхлорметан
хлороформ
бромхлорметан
Хиральность молекул является обязательным условием для проявления веществом оптической активности.
Соединения с одним асимметрическим атомом углерода. Одной из причин возникновения хиральности органических молекул является наличие в их структуре 5р3-гибридизованиого атома углерода, связанного с четырьмя разными заместителями. Такой атом углерода называется хиральиым, или асимметрическим. Часто для него применяют более общее название — хиральиый центр* В структурных формулах асимметрический атом углерода принято обозначать звездочкой — С *;
Н
I сн3—с*—санв
С1 2-хлорбутан
н
сня—с*—соон
он
молочная кислота
Молекулы, содержащие один асимметрический атом углерода, существуют в виде двух изомеров, относящихся друг к другу как Хиральный предмет к своему зеркальному изображению. Такие Гомеры называют энантиомерами (рис. 5.5),
7i
Рис, 55. Модели энантиомерных молекул бром иодх лор метана
Зеркальное изображение хиральной молекулы образуется, если поменять местами любые дна заместителя у асимметрического атома углерода. Энантиомеры очень похожи друг на друга, ио тем не менее не тождественны. Онн имеют одинаковый состав и последовательность связывания атомов в молекуле, но отличаются относительным расположением их в пространстве, т. е. конфигурацией. Если, например, в изомере а переход от атома иода к хлору через атом брома направлен по часовой стрелке, то в изомере б аналогичный переход имеет противоположное направление (см. ряс. 5,5). В том, что эти молекулы разные, можно легко убедиться при наложении нх моделей друг на друга (см. рис. 5.5, е).
Энантиомеры одинаковы по физическим и химическим свойствам. Существенное отличие нх состоит в том, что они по-разному относятся к поляризованному свету, а именно; вращают плоскость поляризации па один и тот же угол, ио в противоположных направлениях (если один вращает влево, то другой — на такой же угол вправо). Поэтому их еще называют оптическими изомерами или оптическими антиподами. Кроме того, энантиомеры с различной скоростью реагируют с другими хкральиыми соединениями и, как правило, обладают разной физиологической активностью.
Смесь равных количеств энантиомеров называют рацемической. Такая смесь нс обладает оптической активностью, так как одинаковое по значению, но противоположное по направлению вращение взаимно компенсируется. Для обозначения рацемической смеси перед названием соединения ставится символ (±).
Изображение оптических изомеров на плоскости. Для нзобра* жения пространственного строения оптических изомеров на плоскости могут быть использованы стереохимические формулы. Например, энантиомеры бутанола-2, изображенные с помощью стереохимических формул, имеют следующий вид:
СН, I *
CiHJ
си, I
И ЦМ^С — нет
сгН
3
72
Одиако стереохимические формулы не удобны для описания пространственного строения молекул с несколькими асимметрическими атомами. Поэтому чаще зсего оптические изомеры'изображают на плоскости с помощью проекционных формул Фишера. Для получения проекционной формулы Фишера необходимо руководствоваться определенными правилами расположения тетраэдрической модели молекулы в пространстве. Вначале модель молекулы располагают таким образом, чтобы главная углеродная цепь была ориентирована вертикально, причем вверху находился тот сс конец, с которого согласно номенклатуре И1ОПАК начинали нумерацию атомов. Затем модель ориентируют в пространстве так, чтобы асимметрический атом углерода находился в плоскости чертежа, заместители, расположенные горизонтально, были над плоскостью, а расположенные вертикально — за плоскостью чертежа. При проецировании такой модели иа плоскость получают проекционную формулу Фишера, в которой связи, находящиеся за плоскостью, изображают вертикальными линиями, а расположенные над плоскостью — горизонтальными. Асимметрический атом углерода при этом расположен ® точке пересечения вертикальной и горизонтальной линий и обычно пе обозначается символом.
Используя для наглядности клиновидную форму изображения связей, получение проекционной формулы Фишера одного нэ энантиомеров бутаиола-2 можно представить следующим образом:
СН,
I
Г и—С^Н
С2Н£ ^ОН
СН, s 110 С —=
СН3 НО —-----Н
С2П5 проекция Фишера
Аналогичные приемы применяют при построении проекций Фншера для молекул, содержащих несколько асимметрических атомов углерода:
псС?>н .
C--OII 1 ... т г
сно
НО^С-^вН
СН2ОН
( :но
ЙО VI
пл II
ГК/
CHgOH
Чтобы убедиться, являются ли молекулы идентичными или раз-Ыми, необходимо проверить, совмещаются вх проекционные фор-Улы при наложении нли нет. Согласно правилам обращения про-
73
екциоиные формулы Фишера нельзя выводить из плоскости чертежа, а также поворачивать в плоскости на 90°, хотя поворот иа 180° в плоскости чертежа разрешается. Как и в реальной молекуле, перемена местами в формуле Фишера двух любых заместителей при асимметрическом атоме углерода приводит к формуле оптического антипода:
СН8 но—н
меняют местами
Н в ОН
СНа н—он
СаН5
энантиомеры бутанола-2
Сан5
Соединения с несколькими асимметрическими атомами углерода, Если молекула имеет несколько асимметрических атомов углерода, число возможных изомеров увеличивается. Общее количество стереоизомеров в молекуле, содержащей несколько асимметрических атомов углерода с разным набором заместителей, можно рассчитать по формуле .V = 2" , где jV — количество изомеров, п— число асимметрических атомов углерода. Так, при наличии в молекуле двух неэквивалентных асимметрических атомов углерода число изомеров равно 2~ = 4; когда содержится три, количество стереоизомеров равно 23 = 8 и т. д.
Например, хлоряблочиая кислота НООС—СНОН—СНС1—СООН имеет два асимметрических атома углерода и существует в виде четырех пространственных изомеров:
Н
С1
СООН СООН СООН СООН
—он но—н 1 н—он но н
-н Н С1 Н С) С1—н
СООН I СООН П СООН HI СООН ! iv
В том, что молекулы I—IV неидеитичиы, легко убедиться, используя метод наложения. Поскольку приведенные изомеры ие имеют плоскости симметрии, все они обладают оптической активностью. Изомеры 1, II, а также III, IV представляют собой зеркальное изображение друг друга, т. е. являются энантиомерами. Как уже отмечалось, энантиомеры обладают одинаковыми физическими н химическими свойствами, имеют равное по значению, ио противоположное по знаку удельное вращение. O/i на ко энантиомеры I, II отличаются по физико-химическим характеристикам от энантиомеров III, IV.
Стереоизомеры 1, III и I, IV, а также II, III и 11, IV не являются зеркальным изображением друг друга. Они имеют одинако
74
вую конфигурацию при одном асимметрическом атоме углерода, но 'разную при другом. Такне стереоизомеры называют диастереомерами. В отличие от энантиомеров, диастереомеры обладают разными физическими и химическими свойствами, имеют разное по значению удельное вращение.
Когда молекула содержит два асимметрических атома углерода с одинаковым набором заместителей, число стереоизомеров уменьшается с четырех до трех. В данном случае один из изомеров имеет плоскость симметрии, поэтому он совместим со своим зер-. кальным изображением. Этот изомер называется мезоформон.
i Примером сказанного может служить винная кислота V *СН(ОН)-СООН
♦СН(ОН)—СООН, имеющая только три изомера—два энантиомера : и одну мезоформу:
30011 СООН СООН
НО— - н Н— —ОН II- -ОН плоскость “ симметрии
н- 1 -он ХОН НО— 1 —н ХЮН н— —он ХОН
энантиомеры мезо форма
Если для меэоформы изобразить формально оптический антипод, то, повернув его проекцию на 180° в плоскости чертежа, получим первоначальную структуру. Это свидетельствует о том, что данные стереоизомеры идентичны:
ХЮН I ( 1 ХОН
н- —он i но— —н
н— —он Т но— —н
ХОН 1 зоон
мем форма
Молекула мезоформы имеет плоскость симметрии, следовательно, она ахиральна, а поэтому не обладает оптической активностью. Мезоформа является типичным примером, когда молекула, имеющая несколько хиральпых центров, в целом может быть ахиральной. Каждый из энантиомеров винной кислоты по отношению к мезоформе является диастереомером.
Оптически активные соединения с другими асимметрическими атомами. Помимо углерода молекула может иметь в качестве асимметрического центра и другие атомы, связанные с четырьмя разными заместителями. Это, в частности, атомы кремния, азота,
75
фосфора. Такие молекулы проявляют оптическую активность и образуют устойчивые оптические изомеры, например:
с2н;
Г3
Si -™ис- н.-соон
о 5
СИ, п -| 5 Вг
С И-'КЧГ’Н «•* ^С2И3
СНг
13
ноос- с, и .ton'll1" ' sj __
fi 14
сЛнГ * J о 5
СП,
I, Н rNii^N —. с2»Г
Вг'
СЛ1.
О J
Наряду с этим имеются асимметрично построенные соединения, в молекулах которых хиральный центр (азот, фосфор, сера) связан не с четырьмя, а с тремя разными группами. Роль четвертого заместителя з данном случае выполняет неподеленпая электронная пара. Примерами таких соединений могут служить вторичные (а) и третичные (б) амины, фосфины (в) и сульфоксиды (г):
а
С H-N^IICH,
И Э ~с Й
Следует отметить, что конфигурационные изомеры соединений с трехвалептным асимметрическим центром крайне неустойчивы. При обычных условиях происходит быстро протекающий процесс взаимопревращения энантиомеров друг в друга, поэтому выделить оптически активные изомеры удается лишь в редких случаях.
Кроме рассмотренных примеров, оптическая изомерия свойственна также некоторым веществам, ие имеющим асимметрических атомов.
Номенклатура оптических изомеров. Поскольку оптические изомеры отличаются друг от друга лишь конфигурацией молекул и отношением к поляризованному свету, в их номенклатуре наряду с названием, отражающим химическое строение, указывается также. конфигурация и направление вращения поляризованного света.
76
VlKJluntntlv ---r
в названии оптического изомера знаком '( + ), а влево —знаком (—). Чтобы сказать, какой энантиомер из пары имеет правое, а какой — левое вращение, необходимо знать истинное расположение вокруг хиральиого центра, т. е. абсолютную конфигурацию молекул. Важно отметить, что направление и угол вращения плоскости поляризации ие связаны каким-либо образом с конфигурацией. Определение абсолютной конфигурации молекул оказалось для химиков довольно сложной задачей. Лишь в 1951 г. методом рентгеноструктурного анализа была впервые установлена абсолютная конфигурация патрий-рубндиевой соли ( + )-винной кислоты. До этого времени конфигурация оптических изомеров устанавливалась методом сравнения со специально выбранным стандартным веществом. Такая конфигурация получила название относительной. В 1906 г. русским ученым М. А. Розановым в качестве стандарта для установления относительной конфигурации был предложен глицериновый альдегид СН2(ОН)—СН(ОН)—СП —О. Правовращающему изомеру глицеринового альдегида была произвольно приписана абсолютная конфигурация, в фишеровской проекции которой группа —ОН находится справа. Такую конфигурацию обозначили буквой О. Левовращающему энантиомеру глицеринового альдегида соответственно приписана зеркальная конфигурация, которую обозначили буквой L (группа —ОН в проекции Фишера расположена слева):
Н---ОН НО---Н
(СНаОН сн2он
D—(+)- г л ниерииовый L— (—1 - глицер инов ыЯ
альдегид альдегид
Глицериновый альдегид выбран в качестве стандарта по той ричиие, что с помощью ряда химических реакций его можно превратить во многие другие оптически активные соединения, не затрагивая связи с асимметрическим атомом углерода. Например, без нарушения конфигурации хиральиого центра ( !-)-глицериновый альдегид можно превратить в (—)-молочную кислоту. Следовательно, (—)-молочная кислота будет иметь такую же конфигурацию, что и ( + ) -глицериновый альдегид, т. е. D-конфигурацию:
СНО
СООН СООН . СООН
Н-----ОН Вг^° . Н------ОН РВт* г
СНаОН СНаОН
0(-|->-гл ицериноаый аль-егид
Н----ОН н—он
СНЛЗг сн,
в «э
D-(—) молочно» кислота
77
Этот пример свидетельствует о том, что молекулы с одинаковой конфигурацией необязательно должны вращать плоскость поляризованного света в одном направлении.
Аналогичным образом, независимо от знака вращения, конфигурация других оптически активных соединений косвенно отнесена к D- или L-глнцериновому альдегиду или соответственно к й~ нли L-стереохимическому ряду.
Позже экспериментальным путем было установлено, что произвольно приписанная ( + )- и (—)-глицериновому альдегиду конфигурация соответствует истинному расположению заместителей в пространство, т. е. абсолютной конфигурации этих веществ. Это удачное совпадение позволило избежать путаницы в химической литературе, так как установленная для различных соединений относительная конфигурация приобрела силу абсолютной.
D, L-система обозначения конфигурации оптических изомеров применима только для структурно родственных глицериновому альдегиду соединений. К пим относят вещества с такой конфигурацией хирального центра, когда в проекции Фишера с одной стороны от вертикальной линии расположен атом водорода, а с другой — группа ОН, КНз, ХО?, атом галогена и др. Если указанные заместители находятся справа, молекула имеет D-конфигурацию, а если слеаа — L-конфигурацню:
СООН СООН СНЯ
о
Н-----ОН НаЫ--------------Н Н------------CI
сн,соон сн„ сан5
D-яблсчная кислота L-алании D-5-хлорбутан
Применительно к соединениям с несколькими хиральными центрами принадлежность изомеров к D- или Л-стереохимическому ряду устанавливают по конфигурации асимметрического атома углерода с наибольшим порядковым номером, т. е. по ннжиему асимметрическому атому углерода в проекции Фишера:
о
н
О
н-
—ОН
-н
—ОН
но—
н—
но-
н—
—н н— -он н—
—он НО— —н н—
-ОН
—ОН!
снаон £-треста
СН2ОН
Л-рибоза
СН2ОН
D-глюкоза
78
Исключением иэ этого правила являются а-амииокпслоты, а-гидроксикислоты, винная кислота, в которых принадлежность к D- или t-ряду устанавливают по конфигурации а-углеродиого атома (верхнего асимметрического атома углерода в проекции Фишера)!
( ХЮН СООН ( зоон
! H2N- —н Н— —он но— -н i I
н- —он но- —н н- —он
с :ня с ,оон ЗНа
L« треонин D-винная кислота £-2, 3-дигидроксн
масляная кислота
Пространственное положение одинаковых заместителей в молекулах с двумя соседними асимметрическими атомами углерода принято обозначать в названии изомера приставками трео- и эра-тро-. Трео-изомерами называют вещества, в проекциях Фишера которых одинаковые заместители при асимметрических атомах углерода расположены по разные стороны, эритро-изомерами — по одну сторону:
( ХЮН СООН СООН соон
н— —он но—н н он но—н
Н- -С1 С1—н а—н Н С1
( ХЮН СООН соон соон
D -эритро - хлор- L -5 ритро-хлор- О-трео-хлор- £-трео-хлор •
яблочная кислота яблочная кислота яблочная кислота яблочная кислоте
D, L-система обозначения конфигурации, несмотря на широкое использование, имеет ограниченную область применении. Иногда соединение содержит такие заместители вокруг асимметрического центра, что бывает просто невозможно каким-либо образом Сравнить его конфигурацию с глицериновым альдегидом.
В последнее время в органической химии все шире стала применяться предложенная Каиом, Иигольдом и Прелогом более универсальная номенклатурная система обозначения абсолютной Конфигурации оптических изомеров — так называемая /?, S-спстема.
I/?, Л-Систсма осиоваиа на определении направления последовательного убывания старшинства заместителей, связанных с асимметрическим атомом углерода*
79
Старшинство заместителей устанавливается подсчетом атомных номеров элементов, вначале непосредственно связанных с хираль-ным центром, а затем, если это необходимо — элементов последующих элементных слоев. Чем больше атомный номер, тем старше заместитель. Например, в молекуле бромиодхлорметана (рнс. 5. б, а) заместители образуют следующий ряд последовательно убываю-
X-конфигурация а
К конфигурнния
.•ОН>-СиОН*-СИз> ч
/{-конфигурация ^-конфигурация
б
Рис. 5.6. Определение конфигурации по R, S-системе: а — бромиодхлорметана; б — молочной кислоты
щего старшинства: -J, -Вг, -Cl, -Н. Если два или более атомов, непосредственно связанных с асимметрическим центром, одинаковы, то порядок старшинства заместителей, а состав которых входят эти атомы, определяется аналогичным образом по второму элементному слою, т. е. путем подсчета суммы атомных номеров элементов, непосредственно связанных с атомами первого слоя, причем атомные номера элементов, связанных двойной связью, удваиваются, а тройной — утраиваются.
Так, в молекуле .молочной кислоты (рис. 5.6, б) по первому элементному слою (8О, вС, 1Н, 6С) видно, что самым старшим заместителем является группа — ОН, а самым младшим — Н. Для выяснения старшинства заместителей —СООН и —СНз, имеющих в первом слое одинаковые атомы (еС), необходимо рассмотреть второй элементный слой. Сумма атомных номеров второго элементного слоя СНз-группы—14-1 + 1=3, а группы —СООН — 8 + 8Х \2=24 (см. рис. 5.6, б). Следовательно, группа —СООН старше
80
группы —СНз» а вес заместители вокруг хиралыюго центра молочной кислоты располагаются в следующий рад последовательно убывающего старшинства: -ОН, -СООН, -СНа, -Н.
После установления старшинства заместителей, согласно R, S-системе описания конфигурации стереоизомеров, модель молекулы или ее стереохимическую формулу рассматривают таким образом, чтобы самый младший заместитель (чаще всего это атом водорода) был наиболее удален от наблюдателя (см. рис. 5.6,6). Если при этом три остальных заместителя расположены в пространстве так, что нх старшинство последовательно убывает по часовой стрелке, абсолютная конфигурация обозначается буквой R (от лат. Rectvs— правый), если же старшинство заместителей убывает против часовой стрелки, конфигурацию обозначают буквой S (от лат. Sinister — левый).
Для описания через R, S-снстему обозначения конфигураций стереоизомеров, изображенных с помощью проекционных формул Фишера, необходимо путем четного числа перестановок заместителей преобразовать проекцию Фишера таким образом, чтобы младший заместитель находился внизу. Если при этом остальные грн заместителя расположены в порядке уменьшения старшинства по часовой стрелке, соединение относят к 7?-ряду, если против часовой стрелки — к S-ряду. Преобразуя указанным способом, например, проекцию О-глицерииового альдегида, видим, что по R, S-системе он имеет /^-конфигурацию:
R -глицериновый альдегид
I Следует помнить, что нечетное число перестановок заместнте-|ей в проекции Фишера приводит к обращению конфигурации:
обращение конфигурации
НОИ , С
б'-глмцериновьщ альдегид
81
Особую трудность представляет преобразование проекционных формул для соединений с несколькими хиральиыми центрами. В таких случаях R, S-система допускает использование иепреобра-зоваяных проекционных формул. При этом, если в проекционной формуле Фишера младший заместитель расположен справа или слева от асимметрического центра, для описания конфигурации по R, S-системе применяется обращенное правило, а именно — уменьшение старшинства заместителей по часовой стрелке придает хиралыюму центру S-конфигурацию, а против часовой стрелки — ^-конфигурацию. Например, из четырех стереоизомеров 3-бром-бутапола-2 первый имеет (2R, 3/?)-, второй— (23, 3S)-, третий — (2S, 37?)-, четвертый—(2R, 3S)-конфигурацию, которая обозначается в виде приставки к систематическому названию:
(2/?, ЗЯ;-изомер
I
(25, 35)-изомер
11
(25, .72?) -изомер
Ш
<2Rr 3S)-изомер
IV
Рацемические формы. В индивидуальном виде органически соединения с хнральными молекулами образуются лишь в резуль тате сложных биохимических процессов, протекающих в живы организмах с участием ферментов. При синтетических методах и получения, как правило, образуются равные количества леновр; щающего и правовращающего энантиомеров, составляющих тг называемую рацемическую форму. Рацемическая форма ие обл; дает оптической активностью. В газообразном и жидком агрегатнь состояниях, а также в растворах рацемическая форма представлЯ’ собой идеальную смесь равного числа энантиомерных молекул, п этому она имеет те же свойства, что и индивидуальные энантиомер (температура кипения, показатель преломления, ИК-спектры).
В твердом состоянии рацемическая форма может существова в виде смеси зеркально идентичных кристаллов инднвидуальи энантиомеров, получившей название рацемическая смесь, илн пр» ставлять собой молекулярное соединение, кристаллы которого i строены яз строго чередующихся молекул лево- и правовращг щего изомера. Такая рацемическая форма называется рацеми ским соединением, или рацематом. Рацематы отличаются от ин видуальных энантиомеров физическими свойствами (температ;
82
плавления, растворимость) и спектральными характеристиками (ИК-спектры в твердом состоянии). Так, например, температура плавления (+) -винной и (—)-винной кислоты равна 170°C, а рацемическая винная кислота (виноградная) плавится при температуре 204—206 °C.
Причиной образования рацемической формы при возникновении хирального центра и молекуле являются тонкие различия в пространственном строении ахиральных молекул.
Если ахиральная молекула содержит при $р3-гибрндизованиом атоме углерода четыре иля три одинаковых заместителя, то эти заместители эквивалентны, т. е. нх невозможно отличить друг от -друга какими-либо физическими и химическими методами. Такие заместители имеют одинаковое пространственное расположение, их называют гомотопными.
Когда прн 5р3-гибридизовапиом атоме углерода имеется два одинаковых заместителя, их пространственное положение может быть в одних случаях эквивалентным, в других — неэквивалентным. В симметричном окружении такие два заместителя являются эквивалентными, т. е. гомотопными. Так, в молекуле дихлорметана СНаС12 атомы водорода имеют симметричное окружение, поэтому замещение любого нз них на другой заместитель, например, атом брома, приводит к идентичным продуктам.
гомотопные атомы
бромдихлорметан
бромдихлорметан дихлорметан
В несимметричном окружении два одинаковых заместителя имеют неэквивалентное пространственное положение. При замене одного из пнх на новую группу образуется продукт, пространственно не идентичный тому, который получается при замещении другого. Эти продукты по отношению друг к другу являются энантиомерами, Атомы или группы в молекуле, замощение которых приводит к образованию энантиомеров, называются энантиотопными. Так, в молекуле фторхлорметаиа СН2ЕС1 атомы водорода имеют несимметричное окружение, а вследствие этого — неэквивалентное пространственное положение. Они энантиотопны, поскольку при замещении одного яз иих, например, бромом образуется один энантиомер, тогда как прн замене другого — другой энантиомер.
83
. |а
Вг IlwyC-—
F*^
зпантиотопные
Вг2
Л1*
фтор х л орм ст ;ш
Вг
К- бромфторхлорметан
S -бромфторхлорметан
Эквивалентными и эпаитиотопиымн могут быть не только заместители при хр3-гнбридизованном атоме углерода, но и стороны двойной связи 5р2-гибрндизовапиого атома углерода в молекуле. Например, для формальдегида (рис. 5.7), имеющего симметричное окружение карбонильной группы, атака ахиральным реагентом двойной связи с любой стороны приводит к одному и тому же продукту. Это свидетельствует о том, что стороны двойной связи в молекуле эквивалентны. В ацетальдегиде (см. рис. 5.7) карбо-
Идентичяые молекулы Энантиомерные молекулы
Рис, 5.7. Эквивалентные стороны двойной связи в формальдегиде (I) и энантио-топпые стороны в ацетальдегиде (II)
пильная группа имеет несимметричное окружение, поэтому при атаке ахиральным реагентом с двух разных сторон образуются пространственно не идентичные продукты. По отношению друг к другу они являются энантиомерами. Такие две стороны двойно:-' связи в молекуле называются эпаитиотопными.
84
При взаимодействии с ахиральными реагентами энантиотопные заместители или стороны а молекуле подвергаются атаке с одинаковой скоростью, поэтому в результате реакции образуются равные количества энантиомеров, т. е. рацемическая форма. Хиральными реагентами, включая и хиральные биохимические агенты типа ферментов, энантнотопные атомы, группы или стороны атакуются с разной скоростью, что приводит к образованию преимущественно или исключительно одного энантиомера. Реакции, протекающие с образованием исключительно или преимущественно одного из возможных стереоизомеров, называются стереоселективными.
Молекулы, содержащие энаитиотопные атомы, группы или стороны, называют прохиральными, так как замена одного из энантио-топиых заместителей группой, отличающейся от трех остальных, а также присоединение новой группы по месту разрыва двойной связи приводит к образованию хиральной молекулы.
Методы разделения рацемических форм на энантиомеры. Для разделения рацемических форм ва составляющие их энантиомеры применяют механический, биохимический и химический методы.
Механический метод можно использовать в тех случаях, когда образующие рацемическую форму энантиомеры кристаллизуются из раствора раздельно. При этом кристаллы лево- и правовращающего изомеров отличаются друг от друга по внешнему виду, как хиральный предмет отличается от своего зеркального изображе? иия. Такие кристаллы могут быть разделены механически. Этим методом в 1848 г. Л. Пастер впервые разделил на энантиомеры натрийаммоииевую соль виноградной кислоты. Поскольку раздельная кристаллизация энантиомеров свойственна лишь некоторым рацемическим формам, метод механического разделения имеет ограниченное применение.
Биохимический метод основан на избирательном потреблении некоторыми микроорганизмами какого-либо одного из двух энантиомеров рацемической формы. В результате другой изомер может быть выделен из остатка в чистом виде. Этим методом в 1857 г. Л. Пастер с помощью плесневого грибка Penicillium glaucum из рацемической винной кислоты (виноградной) получил (—)-винную кислоту. Применение биохимического метода разделения ограничено необходимостью поиска подходящего микроорганизма, а также тем, что не всякое вещество может потребляться микроорганизмами. Кроме того, один из энантиомеров в процессе разделения пропадает. Однако биохимический метод используют для промышленного получения L-a-аминокнслот.
Химический метод основан на превращении энантиомеров рацемической формы в диастереомеры с последующим их разделением и выделением индивидуальных энантиомеров. Диастереомеры получают взаимодействием рацемической формы с какнм-лнбо оптически активным реагентом. Разделение рацемических форм химическим методом можно представить в общем виде следующей схемой:
88
оптически активный реагент
рацемическая форма
С
С
смесь диастереомеров'
индивидуальные энантиомеры
При взаимодействии рацемической формы (А+В) с оптически активным реагентом (С) образуется смесь диастереомеров (А-С) и (Б-С), которые в отличие от энантиомеров обладают разными физическими свойствами (температура плавления или кипения, растворимость и т. д.), на чем и основано их разделение. Для разделения твердых диастереомеров чаще всего используют разную растворимость (метод кристаллизации), для жидких — разную температуру кипения (метод перегонки). В последнее время широкое распространение получили хроматографические методы разделения диастереомеров. Действием и а индивидуальные диастереомеры (А-С) и (В-С) других реагентов получают индивидуальные энантиомеры (А) и (В).
Практически наиболее часто диастереомеры получают реакцией солеобразования, т. е. на смесь энантиомерных кислот действуют оптически активным основанием, а на смесь энантиомерных оснований — оптически активной кислотой.
Кроме рассмотренных методов разделения рацемических форм применяются также хроматографические методьь
5.2.3. Геометрическая изомерия
Геометрическими изомерами называют вещества, имеющие од1[" наковый состав и последовательность связывания атомов п молекулах, ио разное расположение заместителей в пространстве отно-сительио плоскости двойной связи или плоскости цикла.
86
Появление данного вида изомерии обусловлено невозможностью свободного вращения вокруг двойной связи в молекуле и о-связей, образующих циклы *.
Геометрическая изомерия наблюдается в органических соединениях с двойной связью и алициклических соединениях.
Геометрическая изомерия соединений с двойной связью. Она наиболее распространена среди органических соединенин,^молс-, культ которых содержат двойную углерод-углеродную связь JC—С). При этом геометрическая изомерия возможна лишь в том случае, когда у каждого из углеродных атомов, образующих двойную связь, : находится два разных заместителя, например:
Н. Н
Clz ХС1
I, 2-дихлор5тсн
С1 ,Н
>С-С< У хВг
2-бром - 1-иод-1-хлорэтен
Молекулы таких соединений могут существовать в виде двух пространственных изомеров, отличающихся друг от друга расположением заместителей относительно плоскости двойной связи (рнс, 5.8).
i
Плоскость двойной связи
Рис. 5.8. Геометрические изомеры 1,2-дихлорэтена
Для обозначения конфигурации геометрических изомеров используют ({uc- транс- и Z, f-систему. Цис-транс-система обозначений конфигурации имеет ограниченное применение. Ее можно использовать только тогда, когда связанные Двойной связью углеродные атомы имеют одинаковые заместители. Если одинаковые заместители расположены по одну сторону от плоскости двойной связи — конфигурацию обозначают цис~, если по разные стороны — ?ранс~:
* Вокруг о-связей цикла в зависимости от его размера возможны конформационные повороты, но в целом циклы представляют собой жесткие образования,
87
НЧу,СНэ с
II с
0
^ис-бутен-2
II
С
СН,/\н
транс-Су теп-2
С
quc-1-хлорпропеи
Н^уСН, с
II с а^н траноЬхлор-пропен
Поэтому геометрическую изомерию называют еще цис-тране-изомерией.
Когда углеродные атомы, образующие двойную связь, имеют все разные заместители, как, например, в 1-бром-1-хлорпропене, применение цас-транс-снстемы становится невозможным. В таких случаях пользуются Е, Z-системой обозначений:
3дС1^^63Вг
С
II С
II
С ш/^всн. а £ -1 -бром I -я лорп ролен
Z-1 - бром-1 -х лорпройен
Е, /-система является более общей. Она применима к геометрическим изомерам с любым набором заместителей. Е, /-система обозначений конфигурации основана на определении по Каиу-Ипголь-ду-Г1релогу (см. с. 79) старшинства заместителей у каждого из углеродных атомов, связанных двойной связью. Если при этом старшие заместители из каждой пары расположены по одну сторону от двойной связи, конфигурация обозначается буквой / (от нем. Zusanrimen — вместе), если по разные стороны — буквой Е (от нем. Entgegen— напротив). Так, в приведенных изомерах 1-бром-1-хлорпропена старшим заместителем у одного из углеродных атомов является атом брома, у другого — группа —СНд. Следовательно, изомеру, в котором эти заместители расположены по одну сторону от двойной связи, приписывается /-конфигурация, а изомеру, где они расположены по разные стороны — £-конфигурация.
Следует отметить, что между транс- и Е. Z-системой обозначений не существует какой-либо взаимосвязи. В одном случае ционзомер может быть £-изомером, а в другом — Z-изомером, например:
н,сх ул, н/С“С\н
Чаг-Ъбром-1, 2-дихлорэтеи, «$аг-бутен-2,
Е-1-бром4. 3-дихлорэтен Z*6yrf!H-2
Геометрические изомеры с двойной углерод-углеродной связью обладают высокой устойчивостью, причем транс-изомеры более устойчивы, чем /|цс-изомеры. Превращение одного изомера в дру
88
Гой происходит в довольно жестких условиях (высокая температура, облучение ультрафиолетовым светом) и требует разрыва л-связн.
В виде геометрических изомеров могут существовать также соединения, молекулы которых содержат двойную связь С-= N — с разными заместителями у атома углерода и двойную связь —N = N—. В этих случаях атомы при двойной связи имеют три или два заместителя. Роль «недостающего» заместителя в таких изомерах выполняет иеподеленная электронная пара атома азота:
/ \
Н С6н5
7. (аггтп-Уязомер
с6п5 с6н5 \ /
г-(снк-)изомер
изомер
: Конфигурацию подобных изомеров первоначально обозначали префиксами сип- и анти- (по аналогии с цис- и транс-). В настоящее время используют Е, Z-иомепклатуру.
Стереоизомеры с двойной связью C«N— и —N=N—обладают значительно меиыцей стабильностью, чем изомеры с двойной углсрод-углеродиой связью.
Поскольку геометрические изомеры с двойной связью имеют одинаковую конфигурацию прн одном из образующих двойную связь атомов и разную при другом, они по отношению друг к другу являются диастереомерами. Такие диастереомеры содержат л-связь, поэтому их называют л-ди а стерео мер а ми. Молекулы л-диастерсо-меров ахиралъны, а потому не обладают оптической активностью.
Геометрическая изомерия циклических соединений. Подобно двойной связи, наличие цикла в молекуле препятствует свободному вращению вокруг образующих его о-связей и тем самым создает возможность для существования геометрической изомерии. Такая изомерия возможна при наличии в цикле двух атомов углерода, каждый из которых имеет два разных заместителя. В этом случае
89
два заместителя у разных углеродных атомов могут быть расположены по одну сторону от плоскости цикла * — ч«^-изомер, или по разные стороны — транс-изомер:
:1{нс-циклоиропан-
/1,2г дикарбоновая кислота
Я1да/^циклопропан-
1,2-дикарбоиовая кислота
/^с-1,3-диметилциклогсксан , транс-1,3-димет и л циклогексан
Для обозначения конфигурации геометрических изомеров циклических соединений Е, Z-система не применяется.
В отличие от соединений с двойными связями, в циклических соединениях геометрическая изомерия неразрыино связана с оптической, поскольку замещенные атомы углерода в цикле являются асимметрическими.
Если в цикле у разных углеродных атомов имеются одинаковые пары заместителей, т. е. асимметрические атомы углерода равноценны, то подобно винной кислоте (см. с. 75) число оптических изомеров уменьшается от четырех до трех. £(ис-изомер в этом случае имеет плоскость симметрии, а потому совместим со своим зеркальным изображением и не обладает оптической активностью (мезоформа). Транс-изомер не имеет плоскости симметрии и существует в виде двух энантиомеров, например;
энантиомеры циклопропан- форма циклопропан—
1,2-дикарбонояой кислоты
1,2-дикарбоновой кислоты
• В действительности циклы, начиная с четырехчленного и больше, не имеют плоского строения, но при рассмотрении вопросов изомерии этим пренебрегают-
90
Если замещенные углеродные атомы в цикле не равноценны, как, например, в 2-бутилциклопропан-.1 -карбоновой кислоте, то количество стереоизомеров соответствует формуле W=2n и равно четырем. В данном случае и цнс-изомер, и транс-изомер существуют в виде двух энантиомеров:
энантиомеры цис-2- бутилциклопропан-1-карбдновой кислоты
энантиомеры
>пр>1лс~2-бутш1цик.топроиаи-
1-карббновой кислоты
Геометрические изомеры имеют разные физические свойства > (температура плавления и кипения, растворимость и т, д.), спектральные характеристики н химические свойства. Такое различие в свойствах позволяет довольно легко установить их конфигурацию «. с помощью физических и химических методов.
: 5.2.4. Конформационная (поворотная) изомерия
г
Конформационная изомерия обусловлена вращением отдельных фрагментов молекулы вокруг одинарных связей. В результате вращения молекула может принимать различные пространственные формы, называемые конформациями. Например, молекула этапа вследствие вращения вокруг углерод-углеродной связи может принимать бесконечное множество конформаций, каждая из которых характеризуется определенным значением потенциальной энергии. Две крайние конформации называют заслоненной и заторможенной.
Для представления конформаций на плоскости используют формулы Ньюмена и перспективные формулы:
заслоненная конформация этана заторможенная конформация этана
В заслоненной конформации этана атомы водорода метильных групп, если смотреть вдоль связи углерод-углерод, расположены Друг за другом. В заторможенной — атомы водорода одной метиль-
91
вой группы максимально удалены от атомов водорода другой. Между заслоненной и заторможенной конформациями молекула в процессе вращения принимает множество скошенных конформаций. Поворот фрагментов молекулы вокруг одинарной связи характеризуется торсионным (двугранным) углом ф (рис. 5.9).
Рис. 5.9. Торсионный (двугранный) угол <р: а—• перспективная проекция; 6 — ньюменооскяя проекция
Изменение потенциальной энергии £ молекулы этана в зави-симости от торсионного (поворотного) угла ф изображено иа рис. 5.10. Максимальную потенциальную энергию имеет заслонен-
Заторможенные конформатщи
D 3D !2D JBD 240 300 360 град
Рис. Ь.10. Диаграмм* потенциальной энергии конформаций этана
92
ная конформация. При переходе от заслоненной конформации к заторможенной энергия постепенно уменьшается и становится минимальной в заторможенной конформации. Поскольку эта конформация для молекулы энергетически наиболее выгодна» большую часть времени молекула находится в заторможенной конформации. Прн дальнейшем вращении энергия снова увеличивается и дости-.гает максимума в следующей заслоненной конформации. В конечном итоге при обороте на ЗбО” молекула трижды принимает заслоненную и трижды заторможенную конформации.
Энергетическая неравноценность различных конформаций объясняется существованием в молекуле так называемого торсионного напряжения (напряжения Питцера)» которое обусловлено взаимодействием (отталкиванием) электронных облаков противостоящих 'связей. В заслоненной конформации противостоящие связи макси-,мально сближены, поэтому взаимодействие между ними нанболь- шее. По мере удаление противостоящих связей друг от друга торсионное напряжение уменьшается и становится минимальным в заторможенной конформации. Разность энергий заслоненной и заторможенной конформаций называется энергетическим барьером нращеиня. Для этаиа энергетический барьер невелик, он составляет около 12 кДж^моль и легко преодолевается молекулой при обычных температурах за счет энергии теплового движения.
При наличии у атомов, связанных одинарной связью, объемных заместителей, наряду с торсионным напряжением в молекуле возникает напряжение Вап-дер-ваальса. Это напряжение обусловлено । взаимным отталкиванием заместителей при сближении на рас-' стояние, приблизительно равное сумме их ван-дер-ваальсовых радиусов. Такое напряжение имеет место, например, в молекуле jp-бутана.
К При вращении вокруг С2—С3-связи в я-бутане возможны четыре крайние ковформации» из которых две заторможенные и две заслоненные:
СП3
fff/fiut- taui-
Заторможенные конформации
Засяоиеняые конфирмации
Заторможенная конформация, в которой метильные группы (объемные заместители) максимально удалены друг от друга (торсионный угол равен 180°), получила название онти-конформацни. Заторможенная конформация с торсионным углом между объемными группами, равным 60е, называется гош-конформацией.
ал
Согласно диаграмме (рис, 5.11) максимальную потенциальную .. энергию имеет заслоненная конформация (7I), в которой метиль--> пые группы расположены друг протия друга. Для этой конформа-- цпи в потенциальную энергию вносит вклад, помимо торсионного напряжения, также напряжение Ван-дер-ваальса, обусловленное
затор.чожиниди ынтн}
Ю 120 iW 2^0 JOO 360 ip
Рис. 5.11. Диаграмма потенциальной энергии конформаций к-бутаиа взаимодействием между не связанными друг с другом мстилышмн группами. По этой причине данная конформация обладает большей энергией, чем заслоненные конформации В и Д, содержащие в заслоненном положении метильную группу н атом водорода. Заторможенные конформации н-бутаиа также энергетически неодинаковы, Заторможенные гонг-конформации Б и Е за счет метнл-метильного взаимодействия обладают несколько большей потенциальной энергией, чем ангц-конформация Г, взаимодействие между метилами вообше отсутствует. Поэтому наиболее выгодной для молекулы м-бутана является аят«-копформаиия.
Хотя вращение вокруг центральной углерод-углеродной связи в н-бутане связано с преодолением энергетического барьера порядка 25,5 кДж/моль, этот барьер ие настолько велик, чтобы препят
94
ствовать взаимопревращению конформаций при комнатной темпе-ратуре> Однако в каждый момент времени большая часть молекул представлена наиболее энергетически выгодной конформацией.
Конформации с наименьшим запасом анергии называют конформерами или конформационными (поворотными) изомерами. . _ .
Так, я-бутан при 25 °C существует примерно на 70 % в форме ранта-конформера н на 30 % гош-конформера. С понижением температуры уменьшается энергия теплового движения и, следова-ггельно, повышается содержание более устойчивого конформера, FJ с повышением температуры, наоборот, увеличивается процентное «содержание энергетически менее выгодных конформеров. Конформеры определяют свойства соединен ня.
В отличие от конфигурационных изомеров, конформеры превращаются друг в друга без разрыва химических связей и не поддаются разделению. Оии обнаруживаются только физико-химическими методами.
.2ЛеДует °™етить, что Далеко не всегда заторможенная анти-ь г формация обладает наибольшей устойчивостью. Для ряда органических соединений энергетически более выгодными оказываются
‘ *?ш'К0НФ°РмаЦин. Так, в молекуле этиленхлор-[ ь нисн2СН3С1 из-за образования внутримолекулярной ^«Дородной связи (ВВС) между гидроксильной группой и атомом р олее стабильной является заторможенная гош-конфор мадия:
он
н н н
гош -конформация
н Y н
CJ
Л'^^ЦгКонформация
КОНТРОЛЬНЫЕ ВОПРОСЫ И УПРАЖНЕНИЯ
1. Дайте определения понятий: изомерия, структурные изомеры, стереоизомеры, конформационные изомеры, геометрические изомеры, оптические изомеры. Определите вид изомерии у каждой пары приведенных соединений:
95
,('Hi C2H5 c=c <=c
XC1 If CHj
ty CH3CII2C.H2OII С11зСН(ОН)СНз
Cif\ с:»з С'гЩ УН
Д1 с»сх > iC±eC4
СНдх 41 п СПз ЧСНз
СООН ’ СООН
СНЭ СПз
С2Н5
н ч L
*- с
Пз(Х ""Вг
2. Какие способы существуют для изображения пространственного строения органических соединений? Определите, какие виды стсреоформул были использованы в упражнении 1.
3. Объясните явление таутомерии. Напишите таутомерные формы для соединений г из упражнения 1.
4. Используя соединения е (упражнение 1), объясните, в чем состоит отличие понятий конформация и конфигурация.
5. Какие вещества называют оптически активными? Объясните, какие из соединений в упражнении 1 оптически активны. Какая константа характеризует оптическую активность вещества и как она определяется?
6, Какими правилами руководствуются при построении проекций Фишера? Постройте проекционные формулы Фишера для соединений а и л. указанных в упражнении 1, и обозначьте их конфигурацию по D, L-системе.
7. Напишите проекционные формулы Фишера всех возможных стереоизомеров:
а) 2-аминопропаповой кислоты;
б) 2-бром-З-хлсрбутапа;
в) 3, 4-дибромгексана,
Отметьте среди них пары энантиомеров, диастереомеров, мезо форму, D-, L-, эритро- н грео-изомеры.
90
8. Сформулируйте правила R, S-поменклатуры для обозначения конфигурации энантиомеров. Назовите по R, S-системе соединения е в упражнении 1.
9. Среди приведенных соединений укажите хиральные и прохиральные молекулы: а) СН3СН2СН = СН2; б) СН3СН2С1; в) СНзСС12СН3; г) СН3СНС1СН3; д) СНзСН(ОН)СН2СН3.
10. Обозначьте конфигурацию геометрических изомеров, имеющихся в упражнении 1, по цис-, транс- и Е, /-системам, Для каких геометрических изомеров применима цис-, транс-, а для каких—• Е, /-система обозначения конфигураций?
11. С помощью соответствующих формул изобразите заторможенную и заслоненную конформации молекулы хлорэтака. Какому положению па энергетической кривой соответствуют эти конформации? Какая из конформаций наиболее иыгодна энергетически н почему?
12. Какие виды взаимодействий и молекуле влияют на устойчивость конформаций? Приведите примеры конформеров.
4 2.283
Глава 6 КИСЛОТНОСТЬ И ОСНОВНОСТЬ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Для оценки кислотности н основности органических соединений в современной органической химии используют две теории—протонную (протолитическую) теорию Бренстеда и электронную теорию Лыоиса.
6,1. КИСЛОТНОСТЬ И ОСНОВНОСТЬ ПО ТЕОРИИ БРЕНСТЕДА
Согласно теории Бренстеда кислотой называют любое вещество, способное отдавать протон, а основанием — вещество, способное присоединять протон, т. е., по Бренстеду, кислота является донором, а основание — акцептором протона.
Отсюда теория получила название протонной или протолитической. Для взаимодействия с протоном основание должно иметь лсподелепную пару электронов или л-молекулярную орбиталь.
Кислотность и основность являются относительными свойствами вещества. Кислотный характер может проявляться лишь в присутствии основания, и, наоборот, основный характер — только в присутствии кислоты. В целом кислотно- оснбвный процесс состоит в переносе протона от кислоты к основанию и может быть представлен следующей схемой!
А—Н + В » А“ + В—Н кислота основание сопряженное сопряженная основание кислота
Кислота А—Н, отдав протон, превращается в основание А~, которое называют сопряженным основанием данной кислоты. Основание же В, присоединив протон, переходит в сопряженную кислоту В—Н.
Кислота А—Н и основание А-, а также основание В н кислота +
В—Н являются сопряженными кислотно-основными парами.
Многие органические соединения могут одновременно обладать свойствами и основания, и кислоты. Такие вещества называются амфотерными.
Мерой силы кислоты А—Я является константа кислотности Ка, которая обычно определяется по отношению к стандартному основанию — воде; А—*Н -|- Н2О^ А" + Н3О+.
В достаточно разбавленном растворе рассчитывается по формуле
к Лл“1 (Н»°Ч
~ [A—IT]
98
Чем больше значение Ка, тем сильнее кислота. Как правило, константы кислотности очень малы; например, для уксусной кислоты Ка при 25 °C равна 1,76-10'5. Оперировать такими малыми числами неудобно, поэтому в практической работе чаще пользуются величинами рКа. где рК0=^—Так, рКа уксусной кислоты равна 4,75. Чем меньше значение рК0, тем сильнее кислота.
Подобно кислотам, силу оснований количественно выражают константой основности Кь. Константа основности основания В j в воде определяется нз равновесия
S. - +
В+Н2О5=В—Н + НО-
_ |вн] {НО-)
I " IBJ
-Чем больше Кь, тем сильнее основание. Как и в случае с кислотами, для удобства силу оснований выражают обычно величиной рКъ, где рКб=—lgK&. Прн этом чем меньше рКл, тем сильнее соответствующее основание. Однако чаще всего силу оснований оценивают константой кислотности сопряженной основанию В кислоты ВН, обозначаемую как рК вн- Чем больше значение рКви, тем сильнее основание.
6.1.1. Тилы органических кислот
В зависимости от природы кислотного центра атома (элемента, г которым связан атом водорода, обусловливающий кислотные свойства), органические кислоты подразделяются на четыре основных типа.
1. ОН-Кислоты: карбоновые кислоты, спирты, фенолы. Н2О и пругне соединения, содержащие гидрокенгруппу.
2. SH-Ккслоты: тиолы, тиоловые кислоты и другие соединения : SH-группой.
3. NH-Кислоты: амины, амиды кислот, имиды.
4. СН-Кислоты: соединения, содержащие сильно полярные 3— Н-связи.
Сила кислот определяется устойчивостью образующихся после тгшепления протона сопряженных оснований (анионов). Чем устойчивее сопряженное основание, тем сильнее кислота. Устойчивость ке аниона обусловлена степенью делокализации отрицательного ‘гряда и зависит от ряда факторов: природы кислотного центра 'О, S, N, С); характера заместителя, связанного с кислотным «штром; природы растворителя.
При равных других факторах устойчивость анионов, а слсдова-ельно, н кислотность возрастают с увеличением электроотрица-'ельности и поляризуемости атомов кислотного центра. Поскольку 1 Престелах периода периодической системы электроотрицательность ‘Томов возрастает слева направо (поляризуемость не изменяется), 1
99
то ОН-кислоты сильнее соответствующйх NH-кислот, а те, в свою очередь, сильнее СН-кислот, например:
уксусная жнсюга рКу4,7
z°
СИ.-С,^ ^Н1Н|
ацпкмкд
рКу15,1
сп~ <4--„
ацетон
рК=20,0
В пределах группы периодической системы электроотрицательность атомов уменьшается сверху вниз, но увеличивается их объем, а следовательно, возрастает поляризуемость, т. е. возможность делокализации внешнего электронного облака. Это способствует повышению стабильности аниона и приводит к возрастанию кислотности. Поэтому SH-кислоты обладают большей кислотностью, чем ОН-кяслоты, например:
СН3—СН2—ОН СН3—СНа—SH
этанол <рКд=18.0} этантиол (рКд =» 10,5)
Таким образом, в зависимости от природы кислотного центра органические кислоты с одинаковыми радикалами можно расположить по возрастанию кислотности в следующий ряд: СН-кнслоты< >Ш-кнслоты<ОН-кислоты< SH-кислоты.
В пределах отдельного типа кислот кислотность зависит от строения радикала, связанного с кислотным центром. Алкильные радикалы, благодаря 4-/-эффекту, увеличивают электронную плотность в кислотном центре и тем самым дестабилизируют аннон, что приводит к уменьшению кислотности. Ароматические радикалы, наоборот, повышают устойчивость аниона за счет делокализации отрицательного заряда и способствуют увеличению кислотных свойств, например:
СН3—*ОН метанол (рК»1б,0
фенол ( рК,=» 9,9 }
Заместители, введенные в алифатические и в ароматические радикалы, оказывают влияние на кислотность вследствие прояэ ления ими электронных эффектов — индуктивного и мезомериогс При этом электронодоиорные заместители (-{-/; 4-Л1-эффект) пс ннжают кислотность, а электроноакцепторные {—1; — М-эффект) " увеличивают ее. Так, введение в молекулу фенола (рКя — 9,9 электроноакцепториой группы —NO2 приводит к повышению ки< лотности (у 4-нитрофенола рКа**7,1), введение же электроподоно! иой группы —NH2 понижает кислотность (у 4-амнпофенола рКд:
]0,7), В алифатическом ряду наиболее сильное влияние на кисло
100
ность оказывают заместители, ближе расположенные к кислотному центру, например:
С1х-СНв—СООН Ch-CHa—СНа—СООН Ск-СНа—СН2 -СН2—СООН мояохлоруксугная р-хлорпропиоНомап у-хлормасляная
кислота (рК<з=2Л6) кислота (|>KlJ-=4,1) кислота (рКи=4,5)
Наряду с природой кислотного центра н строением радикала» значительное влияние на проявление кислотных свойств оказывает растворитель. Влияние растворителя определяется его диэлектрической проницаемостью е и способностью сольватировать растворенные частицы. Чем выше диэлектрическая проницаемость растворителя и сольватационный эффект, тем стабильнее ноны в растворе. При равных прочих условиях сольватация аниона протекает тем сильнее, чем меньше его размер и менее делокализован в нем заряд. Таким образом, влияние сольватационного эффекта растворителя и влияние заместителей иа кислотность противоположны друг другу. Наиболее эффективным растворителем является вода, обладающая высокой диэлектрической проницаемостью (s;-80 при 20 °C) и способностью к сольватации растворенных частиц.
6.1.2, Типы органических оснований
Как отмечалось ранее, согласно протонной теории в роли основания может выступать любое вещество, способное присоединять протон, Для образования химической связи с протоном основание должно иметь неподелеиную пару электронов или «-молекулярную орбиталь.
В зависимости от природы основного центра (атом с неподслен-пой парой электронов илн электроны л-связи) органические основания подразделяются па n-основания и «-основания.
В n-основанинх центром основности является атом с пеяоделеп-ной парой электронов. По природе центра основности п-оскования классифицируют на следующие:
аммониевые (центр основности—N<^, =N—, aN)‘, к ним относятся амины, азометины (RCH=®NR), нитрилы (R—C^N), азотсодержащие гетероциклы;
оксониевые (центр основности —С)—, =0); относятся спирты, простые эфиры, альдегиды, кетоны, сложные эфиры, амиды кислот и др.;
сульфонневые (центр основности —S—); относятся тиоспирты '(R—SH), тиоэфнры (R—S—R).
При взаимодействии любого из п-оснований с протоном в качестве сопряженной кислоты образуется соответствующий катион
В Н+ ?=* В— Н п-основа ине сопряженная
КИСЛОТЕ
В «-основаниях центром осиовггости являются электроны л-свя-8И. К ним относятся алкены, алкадиены, арены. По сравнению в
101
л-основаннями это очень слабые основания. В процессе взаимодействия протона с я-основаннем происходит частичное перекрывание s-орбитали протона со связывающей л-МО основания, в результате чего образуется так называемый л-комплекс;
По аналогии с кислотами сила оснований зависит от ряда факторов: природы основного центра; характера заместителя, сая-занного с основным центром; природы растворителя. При равных других факторах с увеличением электроотрицательности атома основного центра, в пределах одного н того же периода, иеиоделеи-на.я пара электронов удерживается прочнее, а следовательно, основность соединения уменьшается, Поэтому оксониевые основания слабее аммониевых.
В пределах группы периодической системы с увеличением поляризуемости атома основного центра усиливается делокализация неподеленной электронной пары и соответственно уменьшается основность соединения. Благодаря этому сульфониевые основания слабее оксониевых. Еще более слабыми основными свойствами обладают л-основания, в которых электронная пара, присоединяющая протон, не является свободной.
Таким образом, в зависимости от природы основного центра органические основания можно расположить по возрастанию основности в следующий ряд: л-основання<сульфониевые<оксоиие-вые< аммониевые.
Большое влияние ла основность органических соединений оказывает природа заместителя, связанного с основным центром. Элек-тронодоноркые заместители повышают электронную плотность в основном центре и приводят к увеличению основности; электроноакцепторные, наоборот, снижают электронную плотность, а следовательно, уменьшают основность. Например, за счет электронодонорного влияния алкильных групп основность алифатических аминов значительно выше, чем ароматических, где в результате сопряже-
СНз—>NH2
NHj
В'Н " 4,58
102
ния неподеленной пары атома азота с л-электронной системой кольца бензольное кольцо проявляет электроноакцепторный характер: Влияние растворителя на основность определяется главным образом эффектом сольватации, Как и н случае с кислотами, сольватационный эффект растворителя и электронные эффекты заместителей оказывают на основность противоположное воздействие.
6,2, КИСЛОТЫ И ОСНОВАНИЯ ЛЬЮИСА
Жесткие и мягкие кислоты и основания. В 1923 г. американский ученый Дж. Н. Льюис предложил электронную теорию кислот и оснований, которая ие противоречит теории Бренстеда, но в отличие от нее является более общей. Согласно этой теории
Основанием считается любая частица (атом, молекула или анион), способная отдавать электронную пару для образования ковалентной связи, а кислотой — любая частица (атом, молекула, катион), способная принимать пару электронов с образованием ковалентной связи.
То есть, по Льюису, основание является донором, а кислота — акцептором пары электронов. Из приведенного определения видно, что основания Льюиса тождественны основаниям Бренстеда. Од-па ко кислоты Льюиса охватывают более широкий круг органических соединений.
(Кислотой Льюиса считается любая частица, имеющая вакантную орбиталь.
Если в теории Бренстеда кислота—это донор протона, то согласно теории Льюиса сам протон является кислотой, поскольку имеет вакантную орбиталь. Таким образом, в представлении электронной теории кислота Бренстеда является соединением, которое образует кислоту Льюиса. Поэтому согласно теории Льюиса к кис-чотам относят не только соединения, отщепляющие протон (протонные кислоты), но и другие вещества, имеющие вакантную орбиталь з способные принимать пару электронов (апротонные кислоты). Кислотами Льюиса, например, являются такие соединения, как ВЕз, AlCh, FeCl3, SbCl3, ZnCl2( HgCl2 и др.
Кислотно-основный процесс по Льюису состоит в образовании ювалентиой связи между основанием н кислотой за счет электрон-»ой пары основания н вакантной орбитали кислоты. Так, основана Льюиса, имеющие неподелеииые пары электронов, образуют кислотами Льюиса п-комплексы:
СНа-*С1: хлормстая (основание Льюиса)
А1С13
хлорид алюминии
(кислота Льюиса)
СНЭ—С1—А1С13 -> СНЭ [A1C1J
л- комплекс
103
Основания Льюиса, содержащие в своей структуре я-свяэь, образуют с кислотами Льюиса л-комплексы:
Вг +
бензол катион брома 31 -комплекс
Легкость протекания кислотно-основной реакции определяется силой кислоты и основания, а также жесткостью или мягкостью кислоты и основания. Представление о жестких и мягких кислотах и основаниях (ЖМКО), введенное Пирсоном, по существу явля--, ется дальнейшим развитием льюисовской теории. Согласно кон-; цспции Пирсона кислоты и основания Льюиса делятся на жесткий и мягкие. ;
К жестким кислотам относятся кислоты Льюиса, в которых атомы-акцепторы имеют малый объем н несут высокий положительный заряд, а следовательно, обладают высокой электроотрицательностью и инзкой поляризуемостью (Н"1", Li+, Na+, К+, Mg2+( Саа+ +
Mn2+, Al3+, Fe3+, А1С13, R—С'=0 и др.). Нижняя свободная молекулярная орбиталь (НСМО) в жестких кислотах имеет низкую энергию.
К мягким кислотам относятся кислоты Льюиса, в которых атомы-акцепторы имеют большой объем и несут низкий положительный заряд, а поэтому обладают низкой электроотрицательностью н высокой поляризуемостью (Cu+, Ag+, Hg2+, Pt2+, I2, Br2 и др.). НСМО в мягких кислотах имеет высокую энергию.
К жестким основаниям относятся основания Льюиса, в которых атомы-доноры имеютвысокуго электроотрицательность и низкую поляризуемость (Н2О, ОН', F“, С1“, СН3СОО“, SO<—, СОз^, NOT, R—ОН, R—О“( R—О—R, NH3, R—NH2, NH2NH2, NH2 и др.), Верхняя занятая молекулярная орбиталь (ВЗМО) в жестких основаниях обладает низкой энергией.
К мягким основаниям относятся основания Льюиса, в которых атомы-доноры имеют низкую электроотрицательность и высокую поляризуемость (RSH, RS", R—S—R, HS“, J~, CN“, R—CN, СЙН4, CeHe, H’, R" н др.). ВЗМО в мягких основаниях обладает высокой энергией.
Исходя нэ общего положения о том, что более эффективно протекает взаимодействие между орбиталями с близкими энергиями, жесткие кислоты преимущественно реагируют с жесткими основаниями, а мягкие кислоты — с мягкими основаниями (принцип ЖМКО).
Следует отметить, что понятия «жесткие» и «мягкие» кислоты и основания не связаны с понятиями «сильные» и «слабые» кислоты и основания. Так, мягкое основание Н~ и жесткое — С2Н-О" являются сильными, а мягкое основание HS” и жесткое СП3СОО" являются слабыми основаниями,
104
КОНТРОЛЬНЫЕ ВОПРОСЫ И УПРАЖНЕНИЯ
1. Дайте определения понятиям кислота и основание согласно теориям Бренстеда и «Пыонса.
Приведите пример кислотно-основного взаимодействия.
2. В приведенных ниже реакциях укажите сопряженные кислот* но-осибвные пары;
a) н + Н0 Н *=* нв°+ + снзсосг
б) СН NH2 + HOH^CH3NHa 4-ОН~
3, Применительно к реакциям а и б, уравнения которых даны н упражнении 2, приведите формулы для расчета силы кислот и оснований.
4. Перечислите факторы, -влияющие на силу кислот и оснований.
5. Укажите кислотные и основные центры в следующих соединениях:
CHeNH,;CH8OH; C2Hfi—SH; GHS—О-СНа; СН3-С— NHa; И
О
СН3-С-СН31 сна«сна.
К какому типу кислот и оснований относятся приведенные сочинения?
; 6. Сравните силу кислот в следующих парах: а) этанол !С2НбОН) и этанамнн '(C2H5NH2); б) этанол и этантиол ifC-HsSH): танол и фенол ^СвН5ОН),
Глава 7
МЕТОДЫ УСТАНОВЛЕНИЯ СТРОЕНИЯ ОРГАНИЧЕСКИХ СОЕДИНЕНИИ
Прежде чем приступить к изучению строения органического соединения, химик должен выделить нз реакционной смеси или природных источников индивидуальное вещество в чистом виде и провести оценку его чистоты.
Для выделения и очистки органических соединений обычно используют перекристаллизацию, перегонку, экстракцию, хроматографию и другие методы. Для оценки чистоты определяют физические константы (температура плавления или кипения), хроматографические характеристики, показатель преломления и др. Подробное описание методов выделения, очистки и доказательства индивидуальности органических соединений приведено в практических руководствах по органической химии.
После получения вещества в химически чистом виде устанавливают его строение (структуру), т. е. определяют природу и число атомов, входящих в состав молекулы, последовательность их связывания, расположение в пространстве и тип химической связи между ними,
Существует два основных подхода к установлению строения органических соединений. Если исследуемое вещество было ранее изучено, для доказательства его структуры определяют физические константы и спектральные характеристики, которые сравнивают с литературными данными. Если же органическое соединение получено впервые, сначала его подвергают качественному и количественному элементному анализу, т. е. устанавливают, какие элементы и в каком количестве входят в состав (см. практические руководства по органической химии). Затем определяют молекулярную массу вещества, Для этой цели применяют криоскопический (по понижению температуры замерзания) и эбулиоскопический (по повышению температуры кипения) методы, которые рассматриваются и курсе физической химии. В настоящее время для определения молекулярной массы широко используют метод масс-спектрометрии.
На основании молекулярной массы и данных элементного анализа устанавливают молекулярную формулу (брутто-формулу) -вещества. Наконец, определяют структуру углеродного скелета, природу и положение функциональных групп, устанавливают определенные фрагменты молекулы и расположение атомов в простраи-
юв
стве, Для этих целей используют химические и физические (инстру-\ ментальные) методы. На. основании полученных данных выводят структурную или стереохимическую формулу.
7.1. ХИМИЧЕСКИЕ МЕТОДЫ
Применение химических методой для установления строения органических соединений основано на использовании качественных реакций, позволяющих определить структуру углеродного остова молекулы, обнаружить функциональные группы, кратные связи и т. д. Например, наличие в соединении альдегидной группы обнаруживают реакцией «серебряного зеркала», двойную связь определяют по обесцвечиванию бромной воды. Иногда с целью идентификации проводят деструкцию (расщепление) углеродного скелета молекулы или получают различные производные, которые определяют путем сравнения с известными веществами. Все эти реакции рассматриваются в разделах, характеризующих реакционную способность соответствующих классов.
7.2. ФИЗИЧЕСКИЕ (ИНСТРУМЕНТАЛЬНЫЕ) МЕТОДЫ
В последние годы наряду с химическими методами для исследования строения органических соединений широко используются физические (инструментальные) методы. В ряде случаев инструментальные методы играют решающую роль при доказательстве структуры вещества. Благодаря физическим методам зачастую получают такую информацию о строении, которую не дают химические методы. Например, только с помощью физических методов можно определить расстояния и углы между атомами в молекуле, нх взаимное расположение в пространстве, внутримолекулярные и межмолекулярпые взаимодействия. Преимущество инструмеи-[•’«льных методов над химическими состоит также в том, что в прессе анализа исследуемое вещество, как правило, подвергается абым воздействиям, ие изменяется и не расходуется, что позво-ет использовать их для изучения строения нестойких и малостой-х веществ, решать структурную задачу при наличии небольшого личества вещества. С одним и тем же образцом можно провести рию различных измерений.
К важнейшим физическим методам исследования строения иосятся спектральные — инфракрасная (ИК-) спектроскопия, ектроскопия комбинационного рассеяния (КР), ультрафиолетовая у'Ф*) спектроскопия и спектроскопия в видимой области спектра, Щектросколия ядерного магнитного резонанса (ЯМР), масс-юектрометрия и дифракционные методы (рентгенография, электро-гография, нейтронография). Каждый отдельный метод имеет ограниченные возможности и однозначно не может решить структурную адачу. Поэтому чаще для изучения структуры используют ком
107
1
плекс физических методов. Наилучшне результаты при таком подходе дает совместное использование ядерного магнитного резонанса, инфракрасной, ультрафиолетовой спектроскопии и масс-спектрометрии,
В основу спектральных методов положено свойство органических молекул поглощать электромагнитное излучение в определенном диапазоне электромагнитного спектра. Электромагнитное излучение можно охарактеризовать несколькими взаимосвязанными параметрами; энергией £, длиной волны X, частотой v или волновым числом VJ
£ =s /iv = hev; X ; -v = — ; v = ~-,
где с — скорость света; h — постоянная Планка.
Между энергией и частотой, или волновым числом существует прямая зависимость, а между энергией и длиной волны — обратная, т. е. чем выше энергия излучения, тем меньше его длина волны, больше частота и волновое число.
Энергия излучения обычно выражается -в электроновольтах (эВ), частота — в герцах (Гц) нли мегагерцах (МГц) *, длина волны— в нанометрах (нм), волновое число — в обратных сантиметрах (см-1).
В зависимости от длины волны электромагнитное излучение подразделяется на несколько областей: рентгеновскую, ультрафиолетовую (УФ), видимую, инфракрасную (ИК), микроволновую и радиочастотную (табл. 7.]).
Области электромагнитного спектра
Таблица 7L
Название области Границы длин волн
Рентгеновская Ю'э — Ю нм t
Ультрафиолетовая (УФ): дальняя 10 — 200 нм
ближняя 200 — 400 нм uj ?•
Видимая 400—800 нм к £ Е о
Инфракрасная (ИК): ближняя 0.8 — 2,5 мкм g "
средняя 2,5 — 50 мкм
дальняя сО — 1000 мкм
Микроволновая 0,1 — 100 см
Радиочастотна я • 1МГц-10вГх 108 | 1 — 10О0 м
Поглощение молекулой электромагнитного излучения происходит квантами (hv). При этом молекула переходит из основного
энергетического состояния в возбужденное. Следует отметить, что органические молекулы взаимодействуют с излучением избирательно. Молекула поглощает только те кванты электромагнитного излучения, энергия которых соответствует разности энергий (ДЕ) двух квантовых состояний молекулы — основного (Е\) и возбужденного (Е2): ДЕ=Е2—E{=h\\
Поглощенная энергия в ультрафиолетовой и видимой областях
спектра используется молекулой па возбуждение электронов. Спектроскопия, изучающая электронные переходы в молекулах, называется электронной спектроскопией. Энергия нифра-
красного излучения расходуется веществом на возбуждение колебаний атомных групп в молекуле и является предметом изучения инфракрасной спектроскопии (ИК). Радиочастотное излучение вызывает изменение спинового состояния атомных ядер, которое измеряется спектроскопией ядерного магнитного резонанса.
В реальных условиях поглощение веществом энергии излучения происходит не строго при определенной длине волны, соответствующей энергии перехода молекулы в возбужденное
^макс
Л макс
Рис. 7.1- Основные характеристики спектральной полосы;
Хмакс — положение максимума;
—интенсивность поглощения в максимуме}
состояние, а в некотором интервале длин волн — в виде так называемой спектральной полосы
А 1/2 — полуширина
(рис. 7.1). Для характеристики
спектральных полос используют три основных параметра: положение максимума (Л-макс или vMaKc), интенсивность поглощения в максимуме 8mjkc) и полуширину (Д 1/2) —ширину полосы поглощения ПрН Гмзкс/2.
Необходимо отметить, что переходов в молекуле, а следовательно, н полос поглощения может быть большое число. В совокупно* стн спектральные полосы образуют спектр поглощения соедннв* ния. Каждому веществу присущ характерный тольяо для негб
спентр, что позволяет отличать соединения друг от друга.
7.2.1. Электронная спектроскопия
Электронная спектроскопия изучает спектры поглощения в ультрафиолетовом (ультрафиолетовая спектроскопия) и видимом (спектроскопия в видимой области) диапазонах электромагнит-
109
ного излучения. Спектры поглощения в ультрафиолетовой н видимой областях обусловлены электронными переходами в молекулах, поэтому их еще называют электронными спектрами. При поглощении молекулой энергии, соответствующей энергетической разнице двух ее электронных состояний, электроны, образующие о- н л-связи, а также неподелеииые электроны (я-электроны) возбуждаются и переходят на более высокие энергетические уровни. Согласно теории молекулярных орбиталей электронные переходы рассматривают как перемещение электронов со связывающих о-и л- и несвязывающих n-МО иа разрыхляющие о*- и л*-МО. При этом возможны четыре типа электронных переходов:- ст-хт*, И —> CF*, П -> Л*.
Переходы о-*-о* наблюдаются в органических соединениях, содержащих только 0-электроны (ст-связи), например в алканах и цнкло-алканах. Если в состав органического соединения входят гетероатомы с неподеленнымн парами электронов (N. 0, S, галогены и другие), поглощение электромагнитного излучения может быть обусловлено как ст-хт*, так и п ст*-переходами. В молекулах с кратными связями наряду с ст-* ст* п n-хт*-переходамн наблюдаются
и л-> л*-переходы. Как видно нз рис. 7.2, переходы ст->ст* н л-*о* требуют большой энергии, Они происходят при поглощении излучения в дальней ультрафиолетовой области. Переходы же я-и л*
tf*M0h.....-.....— — - -—г-1
яЫо)
} возбужденное состояние молекулы
щ мо)
МО)
V основное состояние
| молекулы
Рас. 7.2, Энергетическая диаграмма молекулярных орбиталей и типы электрож* ных переходов в молекулах ' J
110
электронов на разрыхляющие л*-МО иизкоэнергетнчны и проявляются, как правило, в виде поглощения в блнжней ультрафиолетовой (200—*400 нм) и видимой (400—800 нм) областях.
Наряду с энергией поглощения, электронные спектры характеризуются интенсивностью поглощения е. Интенсивность поглощения определяет вероятность электронного перехода каждого типа, т, е. переходы, совершающиеся с большей вероятностью, имеют большую интенсивность поглощения, и наоборот. С большой вероятностью происходят электронные переходы между МО одного типа симметрии (о->о* и л-кд*). Такие переходы являются разрешенными по симметрии и имеют большую интенсивность. Переходы типа п->о* и п-*л* явлиютси запрещенными по симметрии и происходите малой вероятностью, а следовательно, имеют малую интенсивность поглощения.
Поскольку современные спектрофотометры пе позволяют измерять поглощение в дальней ультрафиолетовой области в силу ряда причин (ограниченная прозрачность материала кювет и оптических деталей спектрофотомера, поглощение излучения воздухом), метод электронной спектроскопии используется в основном для анализа органических соединений, содержащих кратные связи. Атомная группировка, содержащая одну или нескольких кратных связей, называетси хромофором. Наличие в структуре вещества хромофора обусловливает избирательное поглощение излучения в ближией ультрафиолетовой и видимой областях, которое связано с л->л*-или п-^л*-переходами. Различают хромофоры изолированные и сопряженные, к первым относят rpvnnnpoBKH с одной кратной связью, например X?=C<f, —teC—, \c=N, —C^N, —N=^N—,
' o ' ' ।
—N=0, —, ко вторым — системы из сопряженных кратных
связей \с=СН—CH=C<f, ^С-СН—С=О, 11^ и др.
Z 4 7 I W
В табл, 7.2 приведены характеристики поглощения некоторых изолированных и сопряженных хромофоров.
Как видно из табл, 7.2, в молекулах с сопряженными хромофорами поглощение происходит в более длинноволновой области и с большей интенсивностью, чем в аналогичных системах с изолированными хромофорами. Максимум поглощения хромофора в определенных пределах может изменяться в зависимости от его окружения, т. е. от строения молекулы. К увеличению длины волны и повышению интенсивности поглощения приводит включение в систему хромофора так называемых ауксохромов — групп, содержащих гетероатом с иеподелеинымн парами электронов, напрямер —ОН, —NH2, —SH (см. табл. 7.2).
Особую структуру электронных спектров имеют ароматические соединения, где сопряженная система замкнута в цикл. Так, бензол
111
Таблица 7.2
Параметры электронных спектров некоторых органических соединений
Соединение* Хромофор ^ыакс* нм е1иакс Тип нор слада
нас=сна ^с=с<^ 162 10000 л->л*
П2С=СН—сн=сн2 х 1 I 4 217 20900 Л~*Л*
1 ! с н НгС-СН-^./С‘ » 1 ->С,Н, /с=с\ 231 4200 л->л*
сн3-с==сн —с^с— 18G 450 л->л*
СН3^С< 9 хн ^>с=о 160 293 20600 12 * * к к t t К с !
>0 H8C=CH-C<f \п ^С-С—С--О z 1 1 230 327 12600 40 п->л*
снз—с—сна 11 О ^)С=О 187 271 1000 16 я е t t К с
183 46000 Л-»-Л*
11 203 7400
255 220 л->л'к
га lii U \—/ 234 286 8300 1500 л-»-л* я«+я*
*Пунктирпой линией отмечены ауксохромы.
имеет три полосы поглощения, обусловленные л->п*-переходами, наиболее длинноволновая из которых имеет ЛМаквв255 им (е = 220). Введение в бензольное кольцо различных заместителей приводит к сглаживанию тонкой структуры и небольшому смещению полосы поглощения в длинноволновую область.
В целом для характеристики спектральных изменений хромофора, вызванных модификацией структуры или заменой растворителя, используют специальную терминологию: смещение полосы поглощении в сторону больших длин волн называют батохромиым сдвигом; смещение полосы поглощения в сторону меньших длин волн — гипсохромным сдвигом; увеличение интенсивности погло-
112
щепия — гиперхромным эффектом; уменьшение интенсивности поглощения — гипохромным эффектом.
Электронные спектры обычно измеряют в разбавленных растворах, применяя растворители, которые не поглощают выше 200 нм. Этим условиям отвечают насыщенные углеводороды, вода, спирты, простые эфиры и др. При исследовании электронных спектров измеряется оптическая плотность раствора 1g, где /0— интенсив
ность светового потока, падающего на вещество; / — интенсивность потока, прошедшего через вещество. Согласно закону Бугера—Ламберта — Бера D = «г/, где с — концентрация вещества в растворе, моль/л; I — толщина слоя раствора, см; е — коэффициент пропорциональности, так называемый «молярный коэффициент поглощения».
Молярный коэффициент поглощения е служит мерой поглощательной способности молекулы н является величиной, характеристичной для каждого вещества.
Электронный спектр поглощения, полученный автоматически или записанный по точкам, представляет собой график, где по оси абсцисс
Рис. 7.3. Электронный спектр поглощения антрацена
откладывается энергия излучения
(обычно длина волны нли волновое число), а по оси ординат — молярный коэффициент поглощения 8 (рис. 7.3).
Электронная спектроскопия используется для идентификации соединений, исследования строения и количественного ана-
лиза.
7.2.2. Инфракрасная спектроскопия
Инфракрасная спектроскопия (ИК) основана иа свойстве органических веществ поглощать излучение инфракрасного диапазона электромагнитного спектра. При взаимодействии органических молекул с инфракрасным излучением (4000—400 см~1) происходит возбуждение их колебательных движений и переход на более высокий колебательный уровень.
Различают два основных типа колебаний в молекулах — валентные и деформационные (рис. 7.4). Колебания, которые происходят вдоль осн связи атомов (растягивание—сокращение связи), называются валентными (обозначаются v). Колебания, обусловленные изменением валентных углов (атомы отклоняются от оси связи), называются деформационными (обозначаются 6). В свою очередь,
5 г-263
113
валентные колебания подразделяются на симметричные (va) и асимметричные (vw). В симметричных валентных колебаниях все связи растягиваются и сокращаются одновременно, в асимметричных —
асимметричное
симметричное
маятниковое
ножничное
веерное
крутильное
Рис. 74. Колебания в метиленовой группе;
а *• йвлентные v, б — деформационные € (символы @ И Q обозначают колебания в напр нии, перпендикулярном к плоскости рисунка}
растяжение и сокращение связей происходит поочередно {см. рис. 7,4). Среди деформационных колебаний различают ножничные, маятниковые, крутильные и веерные.
Принцип ИК-спектроскопни заключается в том, что при облучении вещества инфракрасным излучением с непрерывно меняющейся частотой определенные участки спектра излучения поглощаются молекулой, вызывая увеличение амплитуды колебаний соответствующих связей. В результате луч, проходящий через вещество, ослабляется в областях поглощения, при регистрации его интенсивности после прохождения образна записывается ИК-спектр. ИК-спектр обычно представляют в виде зависимости пропускания 7\ выраженного в процентах (Т%~ц -100), от волнового числа в обратных сантиметрах (см-1) или длины волны в микрометрах (мкм) (рнс. 7.5).
Важно отметить, что в многоатомных молекулах колебания отдельных пар атомов или атомных групп, например О, —G==N, — ОН, ^>N—Н, >с=С<^, —SO2—,—NOe, происходят практически независимо от остальной части молекулы. Каждая такая группа.
Н4
Таблица 7t3
Характеристические частоты поглощения некоторых групп атомов в инфракрасной области
Группировка атомов Сеединение Интервал частот, см-11 Интенсивность полосы
j~C-H Алканы 2960—2850 Сильная
1 =С-И Алкены, арены 3100—3010 Средняя
&С—н Алкины 3300 Сильная
/ п /IX 1 Алканы 1500-600 Слабая
/С=С\ Алкены 1680-1620 Переменная
—С=С— Алкины 2260-2100
—CsN Нитрилы 2300—2200
i о J Спирты 1300-1000 Сильная
^>с=о Альдегиды 1740-1720 »
^с=о Кетоны 1725—1705 >
/с==о Кислоты 1750—1700 »
—0-Н (не ассоциированна я) Спирты, фенолы 3650—3590 Переменная
-О—Н (ассоциированная) Спирты, фенолы 3400—3200 Сильная, широкая
—О—Н (ассоциир ованная) Кислоты 3000—2500 Переменная, широкая
-<и хн Амины первичные 3500—3300 (две полосы) Средняя
>N-H Амины вторичные 3500-3300 (одна полоса) Средняя
115
Длина ВОЛНЫ, мкм
Частота, см"?
Рис. 7.5. Инфракрасный спектр пропионитрила имеет собственную, ей присущую частоту колебаний, которая не-| значительно изменяется при переходе от одного вещества к дру-| тому. Такие частоты и соответствующие им полосы поглощения получили название характеристических, В табл. 7.3 приведены характеристические частоты для рнда часто встречающихся групп. Нахождение в спектре характеристических частот является главной задачей функционально-структурного анализа вещестиа по его спектру.
ИК-спектры органических соединений можно измерить в газообразном состоянии (для летучих веществ), в жидком (для жидкостей), в твердой фазе (для кристаллических) и в растворе. В качестве растворителей чаще используют четыреххлористый углерод, хлороформ, сероуглерод, тетрахлорэтилен и др. Вода как растворитель в ИК-спектроскопии не используется, поскольку кюветы ИК-спектрометров изготавливаются из растворимых солей (КВг, NaCl, UF).
Интерпретацию ИК-спектров начинают, как правило, с высокочастотной области (3700—2900 см-1), в которой располагаются полосы валентных колебаний связей С—Н, О—Н, \1\т—Н, -N<f .
Эти полосы обычно интенсивны, причем положение и контур полос групп NH и ОН зависят от участия их в образовании водородных связей. Возникновение -водородных связей приводит к смещению максимумов поглощения в низкочастотную область (вправо) и к увеличению ширины полос.
Звтем проводят отнесение полос поглощения в области 2500— 1900 см-1, которую называют областью тройных связей. Здесь проявляются полосы поглощения связей С^С н С=\\
Важное значение при анализе ИК-спектров имеет область 1900—1300 см-1, называемая областью двойных связей. В ней наблюдаются полосы поглощения весьма важных функциональных групп и структурных фрагментов: С = О, C = N, N = N, NO2, С=С алкенов, С=С ароматических соединений и др. Здесь поглощают
116
карбонильные соединения, карбоновые кислоты н их производные, ннтросоединении, гетероциклы н др, Область менее 1300 см~! содержит большое число полос, многие из которых трудно отнести к определенному типу колебаний, поскольку они обусловлены колебаниями углеродного скелета всей молекулы. Характер спектра в этой области существенно изменяется даже при незначительных изменениях в структуре соединении. Данная область строго индивидуальна для каждого органического вещества, поэтому ее называют областью «отпечатков пальцев».
Метод ИК-спектроскопин широко применяется для установления строения и идентификации органических соединений, доказательства идентичности веществ, изучения внутри- и межмолекулярных взаимодействий, осуществления контроля за ходом реакций.
7.2.3. Спектроскопия ядериого магнитного резонанса
Спектроскопия ядерного магнитного резонанса (ЯИР) основана на свойстве ядер некоторых атомов, находясь во внешнем магнитном поле, поглощать излучение в диапазоне радиочастот.
Важно отметить, что поглощение энергии радиочастотного излучения характерно только для ядер, обладающих магнитным моментом. К ним относятся ядра с нечетной суммой протонов и нейтронов, т. е. имеющие спиновое квантовое число, не равное нулю—'Н, 13С, 19F, 31Р и др. Такне ядра ведут себя во внешнем магнитном поле как магниты.
В настоящее время в органической химии широко применяется спектроскопия ЯМР на протонах (‘Н), называемая протонным магнитным резонансом (ПМР).
Сущность спектроскопии ПМР состоит в том, что при помещении органического вещества в магнитное поле обладающие магнитным моментом ядра атомов водорода (протоны) ориентируются осью вращения вдоль направления силовых линий магнитного поля. При этом возможны две ориентации магнитного момента протонов относительно направления внешнего магнитного поля — по направлению поля (параллельная ориентация, спин= -4- 1/2) и против направления поля (антипараллельиая ориентация, спин--—1/2), Первой ориентации отвечает состояние с более низкой энергией, чем второй (рис. 7.6). Следовательно, большая часть протонов обычно занимает низшее энергетическое состояние. Переход их на более высокий энергетический уровень происходит при поглощении веществом радиочастотного излучения с энергией, равной энергетической разности (ДЕ) уровней, и сопровождается переориентацией спина (спин +1/2 меняется на —1/2). Поглощенная энергия фиксируется на спектрограмме в виде пика (сигнала).
Окружающая протон электронная оболочка частично заслоняет {экранирует) его от воздейстння внешнего магнитного поля. Поэтому протоны, имеющие различное электпоииое окружение, совершают энергетические переходы и поглощают излучение при различных частотах. Если, например, в молекуле метана (СН4) все
117
паоЕЕЕ тоииэ ( спин = -1/2)
——- < д£
Отсутствует \ вне ш нее м аг нит н ое \
поле 1 ,
ВДЕИзхдоакк! (спин = + i/2)
Приложено внешнее магнитное
поле
Рис. 7.6. Схема расщепления энергетических уровней протона во внешнем магнитном поле
атомы водороде имеют одинаковое электронное окружение и геометрическое положение, то и поглощать (резонировать) они будут прн одной частоте. В то же время в молекуле этилового спирта (СН3—СНэ—ОН) можно выделить три группы протонов: протоны метильной группы (СНз—), метиленовой (—CH-j—) и протон гидроксила (—ОН), имеющие различное электронное окружение. Следовательно, и резонировать они будут при различных частотах.
Количественной характеристикой степени экранирования ядер является химический сдвиг 6, который представляет собой расстояние между сигналом ядер определенного типа и сигналом ядер эталонного вещества. В качестве эталона для измерения химических двигов протонов обычно используют тетраметилсилан [ТМС, Si(CH3)4], В молекуле ТМС содержится 12 эквивалентных сильно экранированных протонов, поэтому в спектре ПМР наблюдается один легко опознаваемый интенсивный сигнал в сильном поле (в правой части спектра). Для протонов ТМС химический сдвиг принят равным нулю. Химический сдвиг выражается в относительных единицах — .миллионных долях (м. д ) и рассчитывается по формуле
6 . ю°,
где v — резонансная частота протона; ттмс — резонансная частота протонов ТМС; vo— рабочая частота генератора.
Химические сдвиги сигналов протонов большинства органических соединений находятся в интервале 0—10 м. д. В табл. 7.4 приведены значения химических сдвигов отдельных протонов в различных классах органических соединений.
На значения химических сдвигов существенное влияние оказывают соседние атомы н группы. Электроподонорные заместители увеличивают электронную плотность вокруг протона и смещают сигнал в сильное поле. Электровозкцепторные, наоборот, дезэкра-нируют протон и сдвигают сигнал в слабое поле. Однако для неко-
118
Таблица 7,4
Интервалы химических сдвигов протонов различного типа
~ в 1 — — Соединение Гип протоне а, м. д.
Алканы* Н2С—Н 1 нс—н ^с—н о.е-1,0 1,0—1,4 1,3-1,6
Циклоалканы* нс-н 1,4—1,8
Этилены ГЕ I 1 1 о о X at Л и 4,3—5,4 БА—6,0
Ацетилены еС-Н 2,3—3,1
Производные бензола С„-Н 6,5—8(2
Гете роар ома ги ч еские соединения )с-н 6,0—9,2
Альдегиды о X X} 9,0—10,0
Спирты** _С—О—н 1.0—4.5
Фенолы** с —0—Н ар 4,5—9,0
Кислоты** -с/° ^0—н 10—13
Амины алифатические >N—Н 0,5—2,0
Амины ароматические C.P-N-H 3,0—4,0
* Относятся также их производные, содержащие функциональные группы дальше, чем через три С—С связи от данных протонов.
** Химический сдвиг сильно зависит от растворителя и концентрации вещества.
119
торых соединений наблюдаются существенные отклонения от этой зависимости вследствие значительного влияния кольцевых токов, которые возникают в ненасыщенных группировках, особенно в ароматических системах и соединениях с тройной связью. Такне токи могут как дезэкранировать протоны (ароматические соединения), так и экранировать их (соединения с тройной связью),
Большое значение при решении структурной задачи с помощью спектров ПМР имеет интенсивность сигналов, которая определяется площадью под резонансной кривой я является величиной, пропорциональной количеству протонов определенного типа. В спектре ПМР интенсивность записывается в виде кривой, делающей в области каждого сигнала ступеньку, высота которой пропорциональна площади соответствующего сигнала (рис. 7.7). Отношение высот ступенек на этой кривой показывает соотношение количества протонов различных типов.
Рис. 7.7, ПМР-спектр метилпропноната (Ja6 — константа спин-спинового взаимодействия)
Таким образом, количество сигналов в спектре ПМР показывает, сколько групп эквивалентных протонов содержится в образце; Химический сдвиг указывает на принадлежность протонов к определенной группировке, а интенсивность сигнала определяет количество протонов в каждой группировке.
Ценную информацию о строении органического соединения дает форма (структура) сигналов спектра ПМР. Протоны одного типа, достаточно удаленные от других протонов в молекуле, дают резонансный сигнал в виде узкого пика с одним максимумом (синглетный сигнал). Если протоны различных типов (имеющие разное электронное окружение или геометрическое положение) разделены дву-
Н Н
\ ZH 11
мя или тремя ковалентными связями , —С-С-, то резо-
7 хн I I
нянснын сигнал проявляется в спектре ис в виде синглета, а расщепляется на ряд линий различной интенсивности, расположенных; на равном расстоянии друг от друга. Такой сигнал называют муль- ’
типлетом, а явление — спин-спиновым взаимодействием. Спин-спи-Новос взаимодействие обусловлено взаимным влиянием магнитных полей соседних протонов и наблюдается только в том случае, когда протоны нс эквивалентны и расстояние между ними не превышает двух-трех ковалентных связей. Расстояние между ближайшими линиями мультиплета измеряется в герцах и носит название константы спин-спинового взаимодействия (/).
Мультиплетность (Л4), т. е. степень расщепления сигнала, зависит от количества соседних протонов и определяется по формуле где п — количество соседних протонов. В приведенном на рис. 7.7 спектре ПМР метилпропнопата протоны группы — ОСНа не вступают в спин-спиновое взаимодействие и проявляются в виде синглета (в), три эквивалентных протона группы —СНз имеют два соседних протона и в результате спин-спинового взаимодействия (п-11—3) дают сигнал в виде триплета (а). Протоны метиленовой группы (СНг), взаимодействуя с тремя протонами СНз-группы (га+1 = 4), проявляются в спектре в виде квартета (б).
В более сложных случаях, когда расщепление сигнала вызвано спин-спипоным взаимодействием с двумя или более группами неэквивалентных протонов, мультиплетность сигнала определяется произведением мультиплетностей, обусловленных каждой из этих групп в отдельности.
Мультиплетность и константа спин-спинового взаимодействия имеют важное значение для структурного анализа. Мультиплетность указывает на число протонов вблизи от данного. Копстанта спин-спинового взаимодействия зависит от природы химической связи и пространственного расположения взаимодействующих протонов, что позволяет применять спектроскопию ПМР для изучения стереохимических особенностей молекул.
7.2.4. Масс-спектрометрия
Масс-спектрометрия относится к деструктивным* методам анализа. Она основана на ионизации молекул исследуемого вещества и регистрации спектра масс образовавшихсв ионов. Существует несколько методов ионизации, ио наиболее распространен в настоящее время так называемый метод электронного удара, когда вещество в газовой фазе подвергается бомбардировке пучком ускоренных электронов. В этих условиях первоначально из нейтральной молекулы (М.) выбивается один электрон и образуется положительно заряжепый нон — молекулярный ион (катион-радикал М*), который затем претерпевает ряд последовательных распадов с образованном более мелких положительно заряженных ионов (фрагментарных ионов) и нейтральных частиц: М -j- е-> М* фрагментарные
—2е
ионы 4- нейтральные частицы.
После ускорения в сильном электростатическом поле поток положительно заряженных ионов дифференцированно разделяется
* Деструкция — разрушение.
121
в переменном магнитном поле в зависимости от отношения нх массы к заряду (т/е) и регистрируется в виде спектра. Ввиду того, что обычно заряд иона равен единице, величина т/е является мерой массы частиц. В масс спектре каждый положительно заряженный ион проявляется в виде отдельного сигнала (пика), положение которого определяется массой иона (точнее отношением массы к заряду), а интенсивность (высота) сигнала пропорциональна количеству попов с данной массой (рнс. 7.8).
£ 400
Б о к
Я 50-о к а)
£ GO-
CH— с=сп-сн3 I
С({3
55
(М-СН3)
ш-
2723
ро-
зо
70 (М*)
>' 1 U---т------------------1----.
50 60 /0 60 т/е
Рис. 7.8. Масс-спектр 2-мети л бутена-2 при ионизации в л ект ровным ударом (70 эВ)
При низкой энергии пучка электронов (порядка 10 эВ) наиболее интенсивный пик в спектре, как правило, соответствует молекулярному иону исходной молекулы. При более высоких энергвях ионизации (обычно 70 эВ) интенсивность пика молекулярного иона падает за счет его дальнейшего распада. Направления распада молекулярного иона и последующие распады фрагментарных ионов определяются строением молекулы, поэтому масс-спектр характеристичен для каждого соединения. В целом распад ионов подчиняется обычным для органических реакций закономерностям и определяется местом локализации заряда и стабильностью образующихся при распаде частиц. Отношение массы к заряду для молекулярного иона соответствует молекулярной массе исследуемого вещества.
Масс-спектрометрию используют для установления структуры органических соединений, нх идентификации и определения молекулярной массы веществ. Высокая чувствительность метода, а также то, что для получения результата достаточно небольшого количества вещества (вплоть до Ю~12 г), позволяет широко применять масс-спектрометрию в судебной экспертизе.
122
7.2.5. Дифракционные методы
Дифракционные методы исследования строения основаны на изучении распределения интенсивности рассеянного веществом излучения. Как правило, в этих методах используют рентгеновские лучи (рентгенография), ускоренные электроны (электронографии) или нейтроны (нейтронография). В результате интерференции (сложение волн) рассеянного излучения получается дифракционная картина—система максимумов и минимумов интенсивности, которую фиксируют на фотопленке в виде пятен с различной степенью затемнения или регистрируют другим способом. Расположение дифракционных максимумов и их интенсивность зависят от строения анализируемого вещества.
Рентгеноструктурный анализ. Рентгеноструктурный анализ (рентгенография) — метод исследования пространственного расположения атомов в молекуле, основанный па изучении дифракции рентгеновских лучей, имеющих длины воли, соизмеримые с межатомными расстояниями. С помощью рентгеноструктурного анализа исследуют главным образом вещества в кристаллической форме. Рентгеновские лучи рассеиваются в результате взаимодействия с электронными оболочками атомов вещества. На основании исследования дифракционной картины (рентгенограммы) строят карты электронной плотности молекулы, соединяя точки с одинаковой электронной плотностью непрерывной линией. Из этих же данных рассчитывают межатомные расстояния, валентные утлы и строят пространственную модель молекулы.
Электронография. Метод основан на явлении дифракции ускоренных электронов па ядрах атомов. Анализ дифракционной картины (элсктропограммы) позволяет установить расположение атомов, межъядериые расстояния и валентные углы. Использование электронографии для больших органических молекул затруднено из-за сложности расшифровки дифракционной картины.
КОНТРОЛЬНЫЕ ВОПРОСЫ И УПРАЖНЕНИЯ
1. Какие методы используются при выделении и очистке: а) твердых (кристаллических и аморфных) органических веществ; б) органических жидкостей?
2. Назовите важнейшие инструментальные методы установления строения органических соединений. В чем нх преимущества перед химическими методами решения структурной задачи?
3. Какое физическое свойство органических веществ используется в спектральных методах исследования их строения? Укажите диапазон длни волн электромагнитного излучения, прошедшего через изучаемое вещество, который анализируется в спектроскопии: а) электронной; б) инфракрасной; в) ЯМР.
4. Что такое характеристическая полоса поглощения? Какие параметры спектральных полос используются для их характеристики?
123
5. К каким обратимым изменениям электронной структуры молекулы приводит поглощение ею кванта УФ-излучения?
Какие особенности в строении молекул органических веществ позволяет выявить УФ-спектроскопия?
6. Какие задачи из нижеперечисленных можно решать с помощью ИК-. а какие — с помощью УФ-спектроскопии;
а) подтверждение присутствия в молекуле гидрокси-, амино-или нитрогрупп при 5р3-гибридизоваппом атоме углерода;
б) подтверждение наличия двойной или тройной углерод-углеродной связи;
в) доказательство существовании в структуре молекулы тех или иных хромофоров, сопряженных систем;
г) контроль за скоростью протекания реакция замещения атомов водорода в ароматическом ядре на нитро-, сульфо-группу или галогены;
д) выяснение способности атомов в молекулах к образованию водородных связей?
7. На каком участке ИК-спсктра и за счет каких процессов, происходящих в молекуле, проявляют себя; а) кратные связи; б) связи атомов водорода с углеродом, азотом и кислородом?
8. Как осуществляется резонанс радиочастотного излучения с молекулой органического вещества? Опишите основные параметры ЯМР (ПМР)-спектра Соединения. Какова роль каждого из них в решении структурной задачи с помощью ПМР-спектроскопни?
9. В чем состоит сущность метода масс-спектрометрии? Каковы его возможности при установлении строения и идентификации органических соединений?
10. Какие дифракционные методы исследования структуры органических веществ Вам известны? В чем они заключаются и какова область применения каждого из них?
Глава в ОСНОВЫ ТЕОРИИ РЕАКЦИЙ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
8.1. ЭНЕРГЕТИЧЕСКИЕ УСЛОВИЯ ПРОТЕКАНИЯ РЕАКЦИЙ
Возможность протекания той или иной реакции определяется не только химической природой реагирующих молекул, ио и рядом других факторов, среди которых важное значение имеют энергетические. Поскольку любая система стремится, как известно, к состоянию с возможно меньшей потенциальной энергией, реакция может проходить лишь при условии, если свободная энергия исходных веществ Gнсх больше свободной энергии конечных продуктов бКО)1 реакции, т. е. когда изменение свободной энергии Д(? отрицательно (рис. B.I).
Однако отрицательная величина ДО является необходимым, но недостаточным условием для самопроизвольного протекания реакции. Обычно иа пути к продуктам реакции молекулам исходных веществ приходится преодолевать энергетический барьер, называемый энергией активации Ь\ (рис. 8.2).
12о
Энергия активации необходима для осуществления активных столкновений реагирующих молекул, т. е. столкновений, приводящих к химическому взаимодействию. Максимум иа энергетической
Ход реакции
Рис. 8.2. Энергетическая диаграмма для одностадийной реакции
диаграмме реакции (см, рис. 8.2) соответствует так называемому переходному состоянию или активированному комплексу. Переходное состояние представляет собой предельно неустойчивое сочетание реагирующих молекул, образующееся в результате перераспределения электронной плотности химических связей в молекулах реагентов при их сближении. Например, в реакции А + В—С-—> 1—>Л—В-\-С по мере сближения атома А с молекулой В—С происходит ослабление связи В—С и одновременно начинает формироваться связь между атомами Л и В. В результате образуется акти-вироваг?иый комплекс А •••/?--С, в котором вследствие перераспределения электронной плотности атом В в равной степени связан с атомами А и С. Активированный комплекс находится в состоянии равновесия с исходными реагентами, а затем но мере дальнейшего уменьшения расстояния между атомами Л и В синхронно протекают процессы образования связи А—В и разрыва связи В—С:
А4-В—С^[А---В- В j-C
активированный комплекс (переходное состояние) Характер переходного состояния определяет селективность (избирательность) протекания реакции в каком-либо одном преобладающем направлении из нескольких возможных. Чем стабильнее переходное состояние, тем меньше энергия активации, а чем меньше энергия активации, тем выше скорость реакции.
Для двухстадийпых или многостадийных реакций характерно образование ие только активированных комплексов, но и промежуточных соединений (интермедиатов) *. Реакция, проходящая в две
* От англ, intermediate — промежуточный.
120
\
стадии (рис.\8.3) представляет собой, по существу, две отдельные реакции. Дв^хстадийный процесс характеризуется наличием двух переходных состояний, которым соответствуют энергетнческве максимумы Е'а и й, а также промежуточного продукта. Стадия реакции, переходное состояние которой имеет наиболее высокую энергию активации, называется лимитирующей стадией. Эта стадия проходит с более низкой скоростью и определяет скорость реакции в це-
Рис. 8.3. Энергетическая диаграмма для двухстадийиой реакции
лом, поэтому часто ее называют скоростьопределяющей стадией. Впадина (провал) на кривой потенциальной энергии соответствует промежуточному продукту. В отличие от активированного комплекса, промежуточный продукт представляет собой реально существующее соединение с достаточно высокой потенциальной энергией. Время жизни интермедиата зависит от глубины впадины на энергетической диаграмме. Если «провал» между двумя переходными состояниями выражен довольно отчетливо, промежуточный продукт можно реально выделить. Чаще интермедиат существует непродолжительное время, так как благодаря высокой реакционной способности при небольшой энергии активации (Еа) он через переходное состояние 2 превращается в конечный продукт реакции. В качестве интермедиатов в органических реакциях чаще всего выступают карбкатионы, карбанионы, свободные радикалы и ст-комп-лексы.
Более подробно энергетические и кинетические условия протекания реакций рассматриваются в курсе физической химии.
127
, -Ji
8.2, ПОНЯТИЕ О МЕХАНИЗМЕ РЕАКЦИИ /
Общие схемы химических реакций, с помощью которых описывают превращения органических соединений, не отражают в полной мере реально протекающих при взаимодействии процессов. Большинство органических реакций проходит, как, правило, в несколько элементарных стадий, совокупность которых составляет механизм реакции. Иначе говоря, механизмом реакции называют общий путь, по которому осуществляется переход от исходных веществ к конечным продуктам реакции. Установление механизма реакции представляет довольно сложную задачу, решением которой занимается преимущественно физическая органическая химия. Для доказательства механизма реакции применяют химические методы (метод меченых атомов, изотопный метод, кинетические и стереохимические методы) и физические методы. Лучшим доказательством механизма реакции является выделение из реакционной среды промежуточных продуктов или обнаружение их с помощью физических методов. Механизм реакции должен объяснять все экспериментально полученные данные и включать сведения о том, каким образом, в какой последовательности н какие связи разрываются в ходе реакции, как образуются промежуточные и конечные продукты, из каких элементарных стадий состоит реакция, каковы относительные скорости каждой стадии, и т. д. Несмотря на все это, механизм реакции всегда следует рассматривать как гипотезу с достаточной степенью достоверности. Нет ни одного механизма, который был бы известен полностью. С появлением новых фактов механизм может уточняться и даже изменяться.
8.3. ТИПЫ МЕХАНИЗМОВ РЕАКЦИЙ
При рассмотрении типов механизмов реакций для удобства условно принято одно из реагирующих веществ называть субстратом, а другое — атакующим реагентом,
В зависимости от способа разрыва связей в атакующем реагенте и субстрате различают три типа механизмов реакций: гомолитический (радикальный), гетеро литический (ионный) и пернциклический (молекулярный).
Гомолитическим или свободнорадикальным называют механизм, в котором при разрыве связей в реагирующих молекулах у каждого из образующихся фрагментов остается по одному электрону. Такие частицы называют радикалами: А- $ -В->А‘ В' Свободнорадикальный механизм обозначают символом «R».
Гетер олитическим, или ионным, называют механизм, в котором при разрыве связей в реагирующих молекулах оба электрона остаются на одном нз образующихся фрагментов. Такие частицы называют н о и а м и:
А:1 В -V + В+
В зависимости от электронной природы атакующего реагента реакции, протекающие по ионному механизму, делят на нуклеофильные (символ «N») и электрофильные (символ «Е»). В нуклео-
128
филъных реакциях атакующим реагентом является нуклеофил (Nu_), в электрофильных — электрофил (Е+).
| \Нуклеофильными * (ищущими ядра) называют ре-
агенты, которые отдают электронную пару при образовании химической связи с субстратом.
К нуклеофильным реагентам относят: молекулы, содержащие одну или несколько иепод елейных пар электронов; ионы, несущие отрицательный заряд (анионы); молекулы, имеющие центры с повышенной электронной плотностью.
Ниже приведены важнейшие нуклеофильные реагенты.
1, Нейтральные молекулы, имеющие неподелспные пары электронов; NH„, R—NH„, R„NH, RSN, H26, R—OH, R—6—R.
iir e'd'O'ar f
2. Анионы:
OH’, CN~, RO-, NH£, R—COO“, RS', SH_, Cl". Br, J-, HSO3, CNS“.
3. Соединения, содержащие центры с повышенной электронной плотностью:
;С=
Нуклеофилы способны образовывать ковалентную связь с субстратом, атакуя в его молекуле центры с пониженной электронной плотностью.
I Электрофильными ** (ищущими электроны) называют реагенты, принимающие электронную пару от субстрата при образовании с ним химической связи.
К электрофильным реагентам относят катионы и нейтральные молекулы, имеющие вакантную орбиталь или центры с пониженной электронной плотностью. Типичными электрофильными реаген* тами являются:
а)
б)
нейтральные молекулы, имеющие вакантную орбиталь: SO3, кислоты Льюиса (AlCls, FeBr3, SnCI<, BF3 и др.);
катионы: протон (Н+), ионы металлов (Меп+), арилдназоний-ионы (Ar-N:), протонированый трнокенд серы (HSOg), нит-роний-иои (NO ), нитрозоний-ион (NO+) и др.;
молекулы, имеющие центры с пониженной электронной плотностью: галогенопронзводные углеводородов (R->Hal), соединения с карбонильной группой
R ’Й?0 rJc?0 R SC^° R бС^° R н „о
R-C\h, R-CXr, R-C\oh , R-Cx^, R-C\Hal и др..
галогены*** (С1а, Вгв, Ja).
* От лат. nucleus —ядро и греч. (рЛих («филиа») — дружба, любовь.
** От греч qXpxTpnv — «электрон» и <piA.ict —дружба, любовь.
*** Галогены проявляют электрофильные свойства под влиянием катализатора или л-электронав атакуемого реагента, вызывзднцих поляризацию связи в молекуле .
129
i
Электрофильные реагенты способны образовывать ковалентную сбязь с субстратом, атакуя в его молекуле центры с /повышенной электронной плотностью.
Электрофильные и нуклеофильные реакции, подобно процессам окисления и восстановления, неразрывно связаны между собой, т. е. в зависимости от того, какое нз реагирующих веществ принимается за атакующий реагент, а какое за субстрат, одну к ту же реакцию можно назвать как электрофильной, так и нуклеофильной. Однако чаще всего субстратом считают вещество с более сложной структурой или вещество, молекулы которого представляют атом углерода для образования новой связи.
Кроме гомо- и гетеролитнческих известны реакции, протекающие по так называемому пернциклическому (молекулярному) механизму. Молекулярный механизм характеризуется одновременным (согласованным) разрывом и образоваяием связей в реагирующих молекулах. Молекулярные реакции протекают без образования ионов или радикалов, они сопровождаются синхронным перемещением электронов в субстрате и реагенте. Типичным примером молекулярной реакции является присоединение диенов к алкеиам:
3.4. ТИПЫ ОРГАНИЧЕСКИХ РЕАКЦИЯ
Огромное количество органических реакций можно разделить на несколько основных типов.
1. Реакции присоединения (обозначаются символом «4»):
у^А------В
X У
Они характерны для соединений, имеющих кратные связи между атомами углерода, углерода и кислорода, углерода и азота, азота и азота, а также для соединений, содержащих атомы с иеподелен-ными электронными парами и вакантными орбиталями.
Реакции присоединения могут происходить по следующим возможным механизмам:
а) электрофильное присоединение (символ Де);
б) нуклеофильное присоединение (An);
в) свободнорадикальное присоединение (Ar);
130
г) молекулярное (синхронное) присоединение.
2. Реакции замещения (обозначаются символом «5»): А—В-\-Н-Х—У-+А—4+B—У. Они характерны для всех классов органических соединений и могут протекать по следующим механизмам:
а) электрофильное замещение (символ Se);
б) нуклеофильное замещение (SN);
в) свободнорадикальное замещение (Sr).
3. Реакции отщепления (элиминирования) (обозначаются символом «£»):
А----В -> А-=В + X—У
I I
X У
От органических соединений чаше всего отщепляются такие вещества, как вода, галогеноводороды, аммиак. Реакции отщепления характерны для галогепопроизводных углеводородов, спиртов, галогене-, гидрокск- и аминокислот.
4. Перегруппировки:
А----В -> А---В
1 I
X X
. Перегруппировки включают переход (миграцию) отдельных атомов или групп от одного фрагмента молекулы к другому. К ним весьма склонны непредельные соединения.
5. Реакции окисления и восстановления сопровождаются изменением степени окисления атома углерода, являющегося реакционным центром. Процессы окисления и восстановления неразрывно связаны между собой. Однако при классификации органических реакций окисление или восстановление рассматривается по отношению к органическому веществу.
По количеству молекул, принимающих участие в стадии, определяющей скорость реакции, различают моиомолекулярные и бимолекулярные реакции, которые обозначают цифровыми индексами 1 и 2 соответственно. В лимитирующей (самой медленной) стадии мономолекулярной реакции принимают участие молекулы одного реагента, в бимолекулярной — молекулы двух реагентов.
8.5. ПРОМЕЖУТОЧНЫЕ АКТИВНЫЕ ЧАСТИЦЫ
Превращение исходных веществ в продукты реакции в большинстве случаев протекает по сложному механизму, включающему образование промежуточных активных частиц (интермедиатов)’. Обычно это короткоживущие частицы, которые быстро превращаются в более устойчивые молек\лы. Однако некоторые интермедиаты обладают достаточно высокой стабильностью и могут быть выделены из реакционной среды в свободном состоянии. В качестве
1ST
/ промежуточных активных частиц чаще всего встречается иарбка-тноны, карбанионы и свободные радикалы: /
R R R /
R—С* R—С" R—С* t
I I I '
R R R
карбкатион карбанион свободный радикал
Карбкатионы н карбанионы образуются в результате гетероли-тического разрыва связи С—X, где X — атом водорода или другой Заместитель;
карбанион
Свободные радикалы образуются при гомолитическом разрыве связи;
«свободный рЬдйКИЛ
8.5.1. Карбкатионы
(Карбкатионами называются органические катиоиы( содержащие положительно заряженный атом углерода.
Атом углерода, несущий положительный заряд, находится в карбкатионе в ар^-гибридизации: три его о-связи с другими атомами расположены в одной плоскости; р-атомная орбиталь, ось которой перпендикулярна к плоскости о-связсй, является вакантной (рис. 8.4).
132
Вакантная
Рис. 8.4. Пространственное строение карбкатиона
Названия карбкатионов составляются добавлением к наименованию соответствующего радикала слова «катион»:
СН8Ч 4-+ + >СН—сна
сня сн8—сн—сна сн/
«етнл-катнон изопропил-катион нзобутнл-катнон
Карбкатионы чаще всего образуются одним из двух способов: 1. Прямая ионизация соединений в сильно полярных средах характерна для веществ, в которых атом углерода связан с заместителем сильно полярной ковалентной связью, например: сна=сн<-снй-<] сн2=сн-сн: + сг
х.таристый аллил аллил-катион
Процесс ионизации заметно активизируется в присутствии катализатора, в частности кислот Льюиса (A1CI3, FeBr3, ZnCl2 и др.) или протона:
СН3.-+С1 + А1С1в5=ь СНа + [AIC1J-х лорнет а и метил-катион
СН.-СН2^ОН 4- Н+^СНа-СН2—О—Н^СНЯ—СНа + НаО 1 U 3S (I dle
этанол | этнл-катион
н
2. Присоединение протона или друеой электрофильной частицы к одному из атомов, связанных между собой двойной или тройной связью:
СН2=СНа 4 Н+^СН3-СН3 эти лен этил-катион
Устойчивость карбкатиоков определяется возможностью делокализации (рассредоточения) заряда в молекуле. Чем в большей степени положительный заряд распределен между другими атомами
133
в молекуле, том более устойчивым является катион. Степень рас-средато’чения заряда в карбкатионе обусловлена его строением. Электрояодонорные группы увеличивают электронную плотность е катионном центре, а следовательно, способствуют рассредоточению
положительного заряда в катионе, оказывая на него тем самым стабилизирующее влияние. Например, за счет увеличения + /-эффекта и эффекта гиперконъюгании со стороны алкильных групп устойчивость алкильных карбкатионов возрастает при переходе от первичного к третичному;
+ + СН8Ч + СНдЧ^^.
снэ < снэ~>сн2 < ;сн < сп34с сн,/ сна^
метил-катион этмл-катион изопропил- ' трет-бутил-кятион катион
Если положительно заряженный атом углерода связан с атомом углерода в ^-гибридизации или гетероатомом, имеющим недоделанную пару электронов (О, N, галоген), устойчивость карбкатиона значительно повышается вследствие делокализации заряда по сопряженной системе:
CH.^/CH, rtCH2^CHa eCHS4~ сн, сн сн сн
аллил-катиои
СН3О-СН2 О CHsO=CHa
метоксиметил-катион
Аналогичным образом происходит делокализация заряда в карбкатионах, положительно заряженный атом углерода которых находится в сопряжении с я-электроииой системой бензольного кольца. Возможность такой делокализации приводит к более сильному возрастанию устойчивости карбкатиона:
беиаил-катион
Электроноакцепторные заместители, наоборот, увеличивают электронный дефицит в катионном центре, тем самым дестабилизируя карбкатион. Так, хлорметил-катиоп СН2С1 в результате —/-эффекта со стороны атома хлора менее устойчив, чем метил-d-катион СН3.
Наряду со структурными особенностями большое влияние на стабильность карбкатионов оказывают внешние факторы, в частности сольватация катионов молекулами растворителя, т. е. образование вокруг каждого катиона оболочки из молекул растворителя. Чем в большей степени сольватирован катион, тем выше его устойчивость. При прочих равных условиях сольватационный эф-
134
\
фект тем выше, чем меньше размер иона и чем меиее делокализован в нем заряд. Сольватная оболочка в значительной степени экранирует заряд катиона и ослабляет его взаимодействие с другими ионами, препятствуя таким образом взаимной нейтрализации разноименно заряженных ионов. Сольватационная способность растворителя определяется значением диэлектрической проницаемости е. Наибольшей сольватационной способностью обладает вода (в=80) и серная кислота (к=84).
Поскольку карбкатионы чаше всего являются короткоживущими промежуточными частицами, которые при небольшой энергии активации вступают в дальнейшие химические превращения, обнаруживают их в основном с помощью инструментальных методов — ЯМР- н УФ-спектроскопии,
8.5.2. Карбанионы
Карбанионами называют органические анионы, содержащие отрицательно заряженный атом углерода, т. е. трехвалентный атом углерода с неподелениой парой олектроиов:
В зависимости от структуры аниона анионный центр может иметь тетраэдрическое или плоскостное строение.
Тетраэдрическое строение характерно для насыщенных карбанионов и карбанионов, в которых анионный центр не находится в сопряжении с л-электронами кратной связи. В таких карбанионах отрицательно заряженный атом углерода находится в $р3-гибриди-вации, а свободная электронная пара занимает 5рэ-гнбрндную орбиталь:
R
Карбанионы, в которых иеподеленная пара электронов анионного центра находится в сопряжении с л-электронами кратной связи, имеют плоское строение, При этом отрицательно заряженный
135
атом углерода находится в $р3-гибридизации, а свободная электронная пара занимает р-атомную орбиталь:
Плоскую структуру имеют также карбанионы, в которых анионный центр содержит двойную или тройную связь. В таких карбанионах свободная электронная пара занимает sp3- или зр-гибридиую орбвталь соответственно:
-*— sp-A.0
Названия карбанионов составляются добавлением к наименованию соответствующего радикала слова «анион»:
СН3 СНЭ—СН3—СН2 (С6Н5)ЭС-
метнл-аиион пропил-анион трнфенил мстил-аиион
Карбанионы образуются при действии сильных оснований на соединения с достаточно полярной связью С—Н.
Св^5\
СвН8^С-Н + NaOH₽(CeH6)sC’ + Na+ + Н2О,
С0Н5^ трифенилметил-аыион < -
трифенклметац
а также в результате ионизации металлоорганических соединений/
C8H6MgBr *=± СН3—СН2 + Mg Вг этил магии я бромид этил-анисы ;
Устойчивость карбанионов определяется возможностью делока-^ лизацни отрицательного заряда. Чем в большей степени заряд рассредоточен между другими атомами в молекуле, тем более устойчив карбанион. Степень делокализации отрицательного заряда определяется прежде всего строением карбаниона. К структурным факторам, определяющим устойчивость, относятся состояние гиб- • ридизации атома углерода анионного центра, а также природа и число заместителей, с которыми он связан. При равных других условиях устойчивость карбанионов возрастает с увеличением s-характера гибридной орбитали, которую занимает неподеленная пара электронов анионного центра:
СН3-СНа-СН2 (sp8) <СНа-СН=^СН (sp3) <СН3—С=С (sp) пропил-вннон пропенил-а нн он пропинм л-аг|иоп
138
Иначе говоря, устойчивость карбанионов возрастает с увеличением электроотрицательности атома углерода анионного Центра.
Существенное влияние на устойчивость карбанионов оказывают электронная природа и число заместителей, с которыми связан анионный центр. Электроиодоиорные заместители повышают электронную плотность в анионном центре и тем самым дестабилизируют карбанион. Так, устойчивость простых алкил анионов уменьшается В ряду
снч и
СН
сн
метил-апион
эти л-анион
и зо пропил-анион
Электроноа кцепторные заместители (—/-эффект) оттягивают электроЕшую плотность с анионного центра (делокализуют отрицательный заряд) и, следовательно, повышают устойчивость карбаниона. Например, 2,2,2-трифторэтнл-аниои СНг—CF3 в результате электроноакцепторного влияния со стороны атомов фтора более устойчив, чем этил-анион СНз—СН3.
Устойчивость карбанионов значительно повышается вследствие сопряжения неподеленной пары электронов анионного центра с л-электронами кратных связей или л-электронной системой бензольного кольца. В этом случае отрицательный заряд делокализуется по всей сопряженной системе:
Еще более устойчивы карбанионы, в которых анионный центр сопряжен с кратной связью углерод — кислород:
О 0~ (О
- II ~ I , ____11 ,
R—СН—С—R R—СН-С—R R—СН—C-R
I II
Поскольку атом кислорода обладает большей электроотрицательностью, чем углерод, а следовательно, способен сильнее удерживать отрицательный заряд, вклад структуры II в резонансный гибрид больше вклада структуры I. Поэтому карбанионы такого типа называют енолят-анионами.
137
На устойчивость карбанионов, как и карбкатионов, наряду со структурными особенностями большое влияние оказывает сольватационный эффект растворителя. Чем в большей степени сольватирован анион молекулами растворителя, тем выше его устойчивость, Для об[1аруження карбанионов чаще всего используют спектро-скопню в ультрафиолетовой и видимой областях, а также метод ядерного магнитного резонанса.
8.5.3. Свободные радикалы
Свободными радикалами называют незаряженные частицы, содержащие иеспареиный электрон (одиоэлектрои-ную орбиталь).
В органических свободных радикалах иеспареиный электрон может находиться у атома углерода, а также у атомов азота, кислорода и других элементов:
R
। Ч
R—С* >N- R—О-
I RZ
R
Чаше всего встречаются свободные радикалы, несущие иеспареиный электрон на атоме углерода. В таких радикалах атом углерода с не-спаренным электроном может иметь плоскостную н тетраэдрическую конфигурацию. Простые алкильные радикалы, а также радикалы, в которых углерод с неспаренным электроном находится в сопряжении с л-электронамн кратной связи или бензольного кольца, имеют плоское строение радикального центра, т. е. атом углерода находится в 8р2-гибридизацнв; иеспареиный электрон занимает р-атомную орбиталь:
метил
аллил
бензил
Третичные алкильные радикалы имеют тетраэдрическое строение радикального центра; неспаренный электрон занимает 5р3-гиб* ридиую орбиталь:
138
Ф-АО
M к-
’ СНз
трет-бут ил
Как и в случае карбанионов, если атом углерода с неспаренным электроном принимает участие в образовании двойной связи, радикальный центр имеет плоское строение, а иеспареиный электрон находится на 5р2-гибридизоваиной орбитали:
разрыва ковалентной связи и молекуле, который происходит в основном прк термических и фотохимических воздействиях:
R.|.N=N.|.rA 2R- -J- Na
азосоединенне О о
сн3 ф с—сн3 ™ сн3 - с-сн3
ацетон метил ацетил
Устойчивость свободных радикалов аналогично карбкатионам
карбанкопам зависит от их структуры и определяется возможностью делокализации неспарепного электрона. Чем больше дело-
кализация, тем выше устойчивость.
В ряду алкильных радикалов устойчивость изменяется в порядке третичиый>вторичпый>первичный>СНэ, например: |
СН
ЗСН> СН3->СН2 > СН8 I
сн/ 1
изопропил
СН.
СН/ mpein -бутил
Этил
метил
Алкильные группы за счет +/-эффекта увеличивают электронную плотность в радикальном центре и тем самым частично заполняют одноэлектронную орбиталь вторым электроном, приводя по существу к делокализации. Свободные радикалы, в которых одио-электронная орбиталь находится в сопряжении с л-электронной
139
системой кратных связей, являются более устойчивыми, так как я данном случае делокализация иеспареииого электрона происходит по всей сопряженной системе:
сшч хсн2
СН
СН2 СН1 СП“
\н ~ ен
аллил
бензил
Несмотря на то что бензильные и аллильяые радикалы более устойчивы, чем простые алкильные, все они обладают высокой реакционной способностью и существуют только как промежуточные частицы. Наряду с этим имеются и долгоживущие свободные радикалы. Например, радикал трифенил метил (CflH5)3C сохраняется в свободном состояний в растворе при комнатной температуре длительное время.
Поскольку свободные радикалы имеют неспареииый электрон; оии обладают магнитным моментом и являются парамагнитными частицами. Это свойство используют для их обнаружения. Метод, основанный на измерении магнитных свойств неспареняых элеитро-иов, получил название электронного парамагнитного резонанса (ЭПР). Этот метод аналогичен ядериому магнитному резонансу; только вместо ядериого спина в нем фиксируют спин электрона.
КОНТРОЛЬНЫЕ ВОПРОСЫ И УПРАЖНЕНИЯ
1. Назовите энергетические условия, необходимые для протекания химических реакций. Дайте определения понятиям «энергия активации», «переходное состояние», «промежуточное соединение (интермедиат)», «лимитирующая стадия реакции».
2. Охарактеризуйте основные типы химических реакций. Определите тип каждой из следующих реакций и обозначьте соответствующим символом, в ионных реакциях укажите нуклеофил и электрофил:
a) CH.-CH- + CI, СНЯ-СН.-С1 + НС1
б) Je + CH3CI з=4 CH3J + cie
в) CH8NHa + НС1 [СН3ХНДС1-
г) CflHe + Вгг F-^*r’ СйН5Вг 4- НВг
д) СН. = СНа + НОН'^' СН8-СНаОН
140
е) СН —СН—СН8 —> СНа-СН-СНа 4- HCI
' а । ° спнит. раствор
С1
U. „ т, Д, кат. + зна-------*-
о
3) Н-С—Н -I- Ag2O -> НСООН + 2Ag|
и) сна—</° + HCN СН,—СН—CN Н он
о+ ,0 .-О
к) сн,-с/ + СН.ОН Щ? сн,—с/' + Н,0
\он 4 осн,
^1
3. Как классифицируют механизмы органических реакций? Используя уравнения реакций, приведенных в задании 2, укажите, какие из них протекают по гомолитическому, а какие — по гетеро-лнтическому механизму?
4. Дайте определения понятиям карбкатион, карбанион свободный. радикал. Назовите частицы:
Н®; ОН®; СНа-СН2е; СН2=СН-СНйе; СНЭО-СНГ; С6Н5-СН?
СН8Ч
(С6Н6)ЯС-; СН3-СН2—СН2; СН8-С^СЭ; (СН3)3С®; ^СН® ’ СН/
5. Рассмотрите электронное и пространственное строение следующих промежуточных активных частиц:
СНГ, iCHf; СН3—СНа; СвН8—СНа; СН2=СН—СНЙ;
О
СНа—CII—C—-СН8; (СвН6)8Се.
Назовите эти частицы.
6. Какие факторы определяют устойчивость карбкатионов? Расположите следующие карбкатионы: метил, пропил-, втор-бутил-, беизил-; трет-пентил-, хлирметил-катиои в порядке увеличения их устойчивости.
7. Перечислите структурные факторы, влияющие иа устойчивость карбанионов. Приведите примеры наиболее и наименее устойчивых карбанионов.
8. От чего зависит степень делокализации иоспарепиого электрона в свободных радикалах? Объясните, какой радикал более устойчив: пропил-, аллил- или трифенилметил- и почему?
предметный указатель
Активированный комплекс 126, 127, см. Переходное состояние Алифатические соединения 14, см. Ациклические соединения Алициклические соединения 14, 15,87 Альтернирование поляризации 55, 56 Ароматические системы 11, 15 — соединения 15, 111 Атом углерода — валентные состояния (гибридизация) 36, 37, 41, 44, 46. 50, 66, 67, 7], 83, 84, 132, 134, 135, 136, 138 — возбужденное состояние 36 — основное состояние 36
— первичный, вторичный, третичный 21 — хиральный (асимметрический) 71, 73—75, 77—79, 90
— электроотрицательность 44, см. Электроотрицательность атомов
Ауксохромы 111
Ахиральные молекулы 71, 75, 83 Ациклические (алифатические) соединения 14 Волновое число 108 Гстерофункциопальиые органические соединения 16, 17 Гетероциклические соединения 14,15 Гипсрконъюгацин 56, 57, см. Сверхсопряжение
Главная углеродная цепь 21 Гомологический ряд 16 Гомотопные (эквивалентные) атомы или заместители 83, 84
Граничные структуры 34, 59, 60, см. Резонансные структуры Диастереомеры 75, 85, 86, 89 Заместители 20, 21, 23, 24, 49, 50, 52— 55. 57, 71, 74-76, 78-83, 87-90,99 100—102, 112, 118, 134, 136, 137 Изомерия 62 — углеродной цепи 62—64 — геометрическая (*{ыотраноиаоме-рия) 62, 86—89
— конфигурационная 62
— конформационная (поворотная) 62, 91 — оптическая 62, 68, 76 — положения 62—64
— пространственная (стереоизомерия) 62, 64, 74
— структурная (изомерия строения) 62, 63
— функциональных групп 62—64 Изомеры 62
— геометрические 65, 86, 88, 89
— конфигурационные 65, 95
— конформационные (поворотные) 65,95 — оптические 65, 68, 72, 74—78.. 90, (см. также Энантиомеры}
— структурные 62, 64
— пространственные 64, 74, 75, 87
— трео-, эритро- 79
~ цис-, транс- 90, 91
Интенсивность
— поглощения 111, 115
— сигналов 120
Интермедиаты (промежуточные продукты) 126, 127, 131
Карбанионы 127, 132, 135, 137, 138
Карбкатионы 127, 132—135, 138
Карбоциклические соединения 14,15
Квантовые числа 32
Кислотный центр 99—101
Кислоты 98
— жесткие, мягкие 104
— Льюиса (апротонные) 103, 133
— органические, типы 99
— сопряженные 98, 101
Классификация органических соедине-
ний 14. 16
Колебания в молекулах 113
Комплекс
— активированный 126, 127, см. Переходное состояние
— п- 103
— л- 102, 104
— о- 127
Константа
— кислотности 98, 99
— основности 99
— спин-спинового взаимодействии 121
Конфигурация 64
— абсолютная 77, 78, 81
— линейная 65
— молекул 65, 76—78
— относительная 77—78
— плоскостная 64, 130
— системы обозначения
---D, L- 77—79
---£, Z- 87—90
---&S- 79—82
— — син-анти- 89
---цис-транс 87, 89, 90
— тетраэдрическая 64, 138
— элементов электронная 34
— энантиомеров 72, 76—79, 81
Конформация 65
— заторможенная 91—95
---анти, гот 93—95
— заслоненная 91—94
— молекул 32, 65, 68, 91, 95
— скошенная 92
Конформеры 65, 95, см. Изомеры ин-
формационные
Локанты 23, 24
Масс-спектрометрия 106—108, 121
Мезоформа 75
Методы
— валентных связей (ВС) 11,34
142
— деструктивные 121
— дипольных моментов 68
— дифракционные 107, 123
— криоскопический 106
— молекулярных орбиталей (МО) 11, 34, ПО
— резонансных структур 59
— спектральные 107, 108
— эбулиоскопический 106
— электронного парамагнитного резонанса (ЭПР) 140
Модели
— Драйдинга 65, 66
— молекулярные 13, 65, 71, 73, 81
— полусферические 65, 67, см. Модели Стюарта — Бриглеба
— Стюарта — Бриглеба 65, 67
— шарастержневые 65, 66
Молярный коэффициент поглощения 113
Монофункциональные органические соединения 16, 17
Мультиплетность 121 Напряжение — Ван-дер-Ваальса 93 — торсионное (Питцера) 93 Насыщенные соединения 14, 15, см. Предельные соединения Нейтронография 107, 123 Ненасыщенные соединения 14, 15, см. Непредельные соединения Непредельные соединения 14, 15 Номенклатура органических соединений — женевская 18 — заместительная 20, 23, 24 — международная (ИЮПАК) 18,20 — радикало-функциональная 23, 24 — рациональная 18 —тривиальная 18 Оптическая активность 68, 69, 72, 74, 75. 77, 82
Оптические антиподы (оптические изомеры) 65, 68, 72, 74—78, 90, см. Изомеры оптические Оптическая плотность 113 Орбитали — атомные (АО) 32—35, 39, 40 ----гибридизация 36 ----d- 33, 39 ----р- 33—42, 51, 53, 57, 58, 132,133, 136, 138 ----$- 36—42, 136 ----зр. зз( 36—40, 102 ----jp2. 36—38, 40, 50, 136, 139 ----ар3- 36, 37, 39, 40, 50, 135. 138 — вакантные 33, 36. 51, 103, 129, 133 — вырожденные 33 — молекулярные (МО) 34, 35, 40 ----антисвязывающие см. Орбитали молекулярные разрыхляющие
— — иесвяэывающие 35, НО
---л- 39, 40, 51, 56, 57, 98, 102, ПО,
---разрыхляющие.35, 40, 110
---связывающие 35, 40, 58, ПО
---о. 39. 40, 56. 57, ПО, 111
Основания 98
— жесткие, мягкие 104
— Льюиса 103
— типы органических 10)
— сопряженные 98, 99
Основный центр 102
Переходное состояние 126, 127
Плоскость поляризации 68. 72, 77, 78
Полифункциональные органические соединения 16, 17
Полосы поглощения спектральные 109
— характеристические 116
Поляризованный свет 68, 72, 76
Правило
— Гунда 33, 35
— октетное 27
— Хюккеля 11
Предельные соединения 14, 15
Призма Николя 68
Принцип
— ЖМКО 106
— Паули 33
Промежуточные активные частицы
131, 135, 140
Пропускание 114
Прохиральные молекулы 85
Радикалы 21, 22, 24, 50
— свободные 21, 128, 132, 138, 140
Рацемат 72
Рацемическая форма 82, 83, 85, 86 — методы разделения 85, 86
Рацемическая смесь 72, 82
Реагенты
— атакующие 128, 129
— ахиральные 84, 85
— н укл еофи льн ые 129
— хиральиые 85, 86
— электрофильные 129
Реакции
— бимолекулярные 131
— замещения 131
— механизм 128
---гетеролитический (ионный) 128
---гомолитический (свободнорадикальный) 128
---перициклический (молекулярный) 128, 130
—• мономолекулярпые 131
— нуклеофильные 128, 130
— окисления а восстановления 131
— отщепления (элиминирования) 131
— перегруппировки 130
— присоединения 130
— селективность(избирательность) 126
— скоростьопределяюшая (лимити руюшая) стадия 127, 131
— стереоселективные 85
143
— типы 130
— электрофильные 128, 130
Резонанс 58
Резонансные (предельные, граничные) структуры 34, 59, 60
Рентгенография (рентгеноструктурный анализ) 77, 107, 123
Родоначальная структура соединения
21, 22, 24
Сверхсопряжение 56, 57
Сдвиг
— батохромный 112
— гипсохромный 112
— химический 118—120
Сопряжение 51, 52, 56—58, 139
Сопряженные системы 50—52, 55—60,
111, 137, 140
Спектроскопия
— инфракрасная (ИК) 32, 107—109, 113, 114, 117
— комбинационного рассеяния (КР) 32, 107
— протонного магнитного резонанса (FIMPJ 117
— ультрафиолетовая (УФ) 107, 108, 135, 138
— электронная 109, 111, 113
— ядерного магнитного резонанса
(ЯМР) 38, 107-109, 117, 135, 138
Стереоизомеры 64 , 74, 75, 85, 87, 91, см. Изомеры пространственные
Стереоформулы 65, 67
Субстрат 128
Таутомерия 64
Таутомеры 64
Теория
— квантовая (квантово-механическая) 11, 32, 34, 36, 56
— кислот и оснований
---протонная (протолитическая теория Бренстеда) 98
--- электронная (апротонная теория
Льюиса) 98, 103
— радикалов 9
— резонанса 11, 34
— стереохимическая И
— типов 9, 10
— химического строения Бутлерова 10,11,48
— химической связи электронная 11, 27
— электронных смещений 11, 48
Торсионный (двугранный) угол 92
Удельное вращение 69, 74
Формулы
— графические 12, 58, 59, 107, см. Формулы структурные
— молекулярные (брутто) 13, 106
— перспективные 67, 68, 91, 92
— проекционные Ньюмена 67, 91, 92
— проекционные Фишера 68, 73 74
77—79, 81, 82 ’ ’
— стереохимические 13, 67, 68, 71—73. — структурные 12, 58, 59, 107 Функциональные группы 16, 17, 19, 20—23, 62—64, 106, 107 Химическая связь — водородная 30—32. 95 —делокализованная 58, 59 — донорно-акпелюрная 29, 30 —ионная (электровалентная) 27 — ковалентная 27—30, 34—36, 48, ЮЗ. ----двойная 14, 23, 34, 42, 43, 84-90, 107, 133, 136, 139
----дипольный момент 44, 48, 49,58 ----длила 42—44
----кратная 14, 22, 23, 29, 42, 53, 56, 111, 130, 135, 137, 138, 140
----направленность 45
----л- 40—42, 45, 48, 50, 52, 55, 89, 101, 104, ПО
----поляризуемость 45, 48 ----полярность 28, 44, 48 ----простая (одинарная) 29,37,42—
44, 91, 92, 95
----о. 40—42, 45, 48—50, 54—56, 65, 87, 89, ПО, 132
----тройная 14, 23, 41, 43, 133, 136 ----энергия 30, 43
— координационная 30
— локализованная 58
— семиполярная 30 — типы 29
Хиральность 68, 70—72
Хиральные молекулы 70, 71, 82, 85 ХиральныЙ (асимметрический) центр 71, 76—83
Хромофоры 111
Циклические соединения 14, 89, 90 Частоты характеристические 116 Эквивалентные заместители 83, 84, см. Гомотопные атомы или заместители Электронография 107, 123 Электроотрицательность атомов 44 Элементы симметрии молекул 70,71 Энантиомеры (см. также Изомеры оптические) 71—77, 82—86, 90 Энантиотопные атомы (группы) 83 Энергетический барьер вращения 92* Энергия —активации 125—127 — поглощения излучения 109, 1П — резонанса 60 — свободная 125—127 Эффект — гиперхромпый 113 — гипохромный 113 — индуктивный 11, 48—50, 54, 55,100, — мезомериый (эффект сопряжения) 11, 48, 52-55, 57, 58, 100
— сверхсопряжения (гмперконъюга-цни) 56, 134
— сольватационный 101, 103, 134, 138
144