
















































































































Автор: Черных В.П. Зименковский Б.С. Гриценко И.С.
Теги: органическая химия химические науки химия учебник органические соединения химические опыты химические реакции химическая наука
ISBN: 5-7768-0310-1
Год: 1998



В. П. Черных, Б. С. Зименковский, И. С. Гриценко
ОРГАНИЧЕСКАЯ
В ТРЕХ КНИГАХ
Книга 3
Гетероциклические и природные соединения
ПОД РЕДАКЦИЕЙ ПРОФЕССОРА В.П.ЧЕРНЫХ
Утверждено Министерством здравоохранения Украины в качестве учебника для студентов фармацевтических вузов и факультетов
^КрвТнсь*в фйр^ат;явтична! А к t д г f i я
Ч. Л.«п -е
Б Г БЛ Г О .LriA
Харьков «Основа» 1998
ББК 24,2я73
449
УДК 547
Рецензенты:
д-р хнм. наук проф. В, Д. Орлов (Харьковский государственный университет), д-р фарм. наук проф, Б. А, Прийменко (Запорожский медицинский университет)
В учебнике рассмотрены главные аспекты строения и реакционной способности гетероциклических соединений. Изложены сведения о важнейших природных соединениях — алкалоидах, белках, нуклеиновых кислотах, жирах, восках, терпенах, каротиноидах и стероидах. Описано строение многих лекарственных препаратов.
Для студентов фармацевтических вузов и факультетов.
Черних В. П. та ш.
449 Органична х!м1я: Пддручник для фарм. ву:«в i фак, У 3 ки. / В, П. Черних, Б, С, Зименковський, I, С, Гриценко. Кн.З, Гетероцикл(чн1 та природш сполуки,— X/, Основа, 1997,- -248 с.
ISBN 5-7768-0310-1 (кн. 3).
ISBN 5-7768-0130-3.
У подручнику розгляпуто основш аспекти будови тк реакгийпо? здатносп гетероцикмчпих сполук, Виклалено В1ломост1 з найважли-внпих при роди их сполук — алкалоццн. бшюв. нуклейювих кислот. жир!в. воск! в, простагландин!в, терпен!в. каротиноцйи та стероиив. Описано будову багатьох л!карських препаратов.
Для студентов зищих фармацевтичних заклад!в оевгги,
4 17°5226-f97° 52 Замов,|е ББК 24 25173
ISBN 5-7768-0310-1 (ки. 3)
ISBN 5-7768-0130
© В. П. Черних, Б. С, Зименковський.
1. С. Гриценко, 1997
ОГЛАВЛЕНИЕ
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ................................8
ГЛАВА 1. КЛАССИФИКАЦИЯ И НОМЕНКЛАТУРА ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ..........................9
1 J. Классификация..........................................9
1.2, Номенклатура...........................................9
Контрольные вопросы и упражнения...........................13
ГЛАВА 2. ГРЕХ- И ЧЕТЫРЕХЧЛЕИНЫЕ ГЕТЕРОЦИКЛИЧЕС-
КИЕ СОЕДИНЕНИЯ С ОДНИМ ГЕТЕРОАТОМОМ (4
2,1, Оксиран и оксетан.....................................14
2,1.1. Способы получения................................14
2.1,2, Физические свойства............................. 15
2.1,3. Химические свойства..............................15
2,1.4. Важнейшие производные оксирана и оксетана........18
2,2, Азнридии и азетидин...................................18
2.2.1. Способы получеЕгия...............................18
2,2,2. Физические свойства..............................19
2,2,3. Химические свойства..............................19
2,2.4, Важнейшие производные азиридина и азетидина......20
Контрольные вопросы и упражнения...........................21
ГЛАВА 3. ПЯТИ- И ШЕСТИЧЛЕННЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ С ОДНИМ И ДВУМЯ ГЕТЕРОАТОМАМИ............................................22
3,1. Ароматичность гетероциклов............................22
3.2. Кислотно-основные свойства гетероциклов...............25
3.3, Неконденснрованные пятичленные гетероциклические соединения с одним гетероатомом.......................26
3,3,1. Способы получения................................26
А. Общие способы получения пиррола, фурана и тиофена.26
Б. Специфические способы получения...................27
3,3.2. Физические свойства..............................28
3.3,3. Химические свойства..............................29
А. Общие химические свойства пиррола, фурана и тиофена. .. 29
Б. Специфические химические свойства пиррола и фурана.33
3,3.4. Методы идентификации пиррола, фурана и тиофена...35
3,3.5. Важнейшие производные пиррола, фурана и тиофена..36
А. Производные пиррола...............................36
Б. Производные фурана................................39
В. Производные тиофеиа...............................42
3
V 3.4. Индол.................................................42
3.4.1, Способы получения...............................42
3.4.2, Физические свойства.............................43
3,4,3. Химические свойства.............................43
3,4.4, Важнейшие производные индола....................45
3,5. Пятичленные гетероциклические соединения с двумя гетеро-атомами....................................................48
7 3.5.1,Пиразол............................................49
А, Способы получения................................49
Б. Физические свойства............................ 50
В, Химические свойства..............................50
Г. Важнейшие производные пиразола...................53
3,5.2, Имидазол........................................55
А. Способы получения................................55
Б. Физические свойства..............................55
В, Химические свойства..............................56
Г. Важнейшие производные имидазола..................57
3,5,3. Бензимидазол....................................58
/ 3.5.4, Тиазол..........................................59
V А, Способы получения.......................................59
Б. Физические свойства..............................59
В. Химические свойства..............................60
Г. Важнейшие производные тиазола....................60
3,5.5. Оксазол.........................................62
3.5.6. Изоксазол.......................................62
3.6. Шестичленные гетероциклические соединения с одним гетероатомом.....................................................63
1 3.6.1. Пиридин.../.......................................63
1 А. Способы получения....................................63
Б, Физические свойства..............................64
В. Химические свойства..............................64
Г. Важнейшие производные пиридина...................70
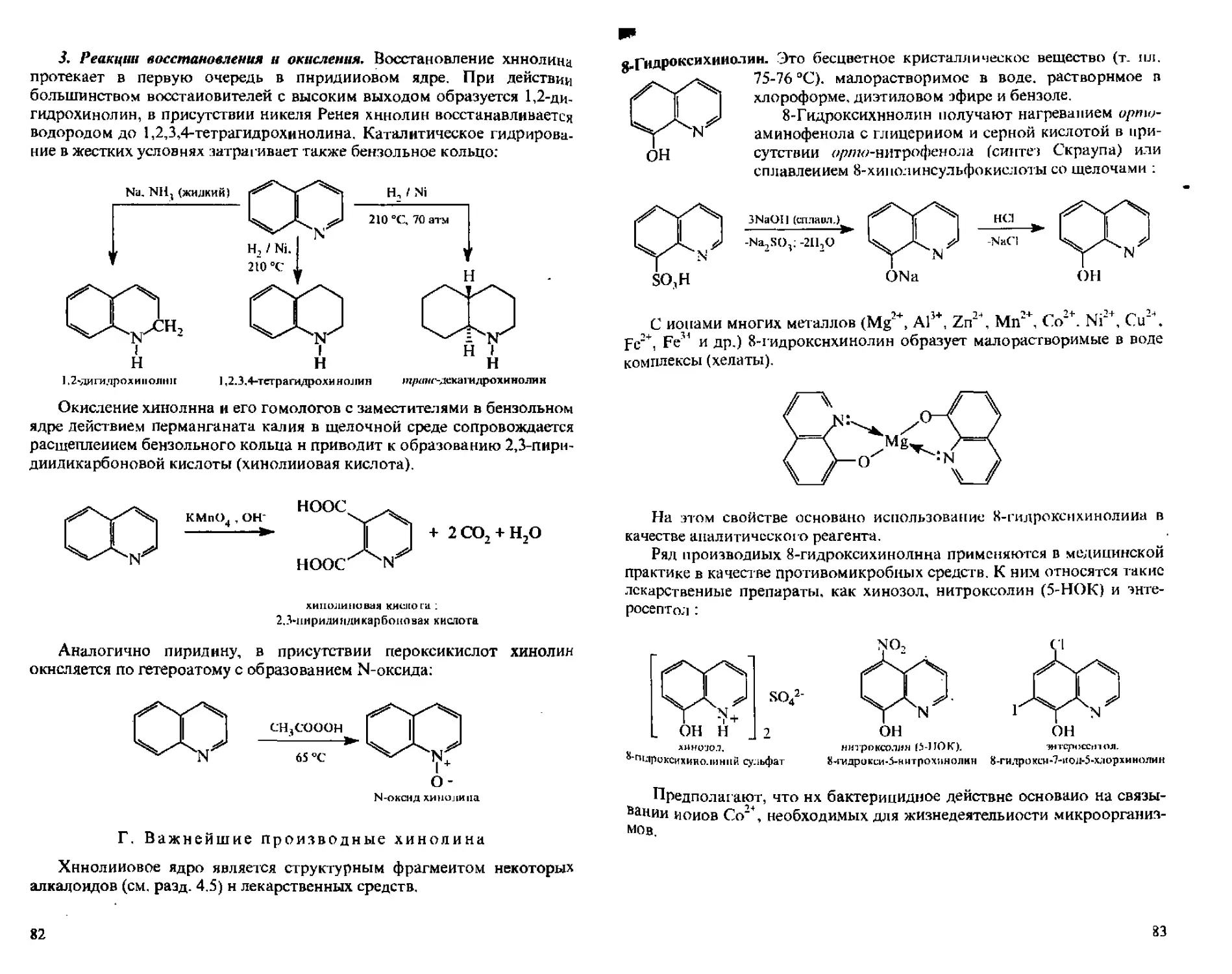
3,6.2, Хинолин....................................... 78
А. Способы получения................................78
Б. Физические свойства..............................80
В. Химические свойства..............................80
Г. Важнейшие производные хинолина...................82
3.6.3, Изохинолин......................................84
А, Способы получения................................84
Б. Физические свойства..............................84
В. Химические свойства..............................84
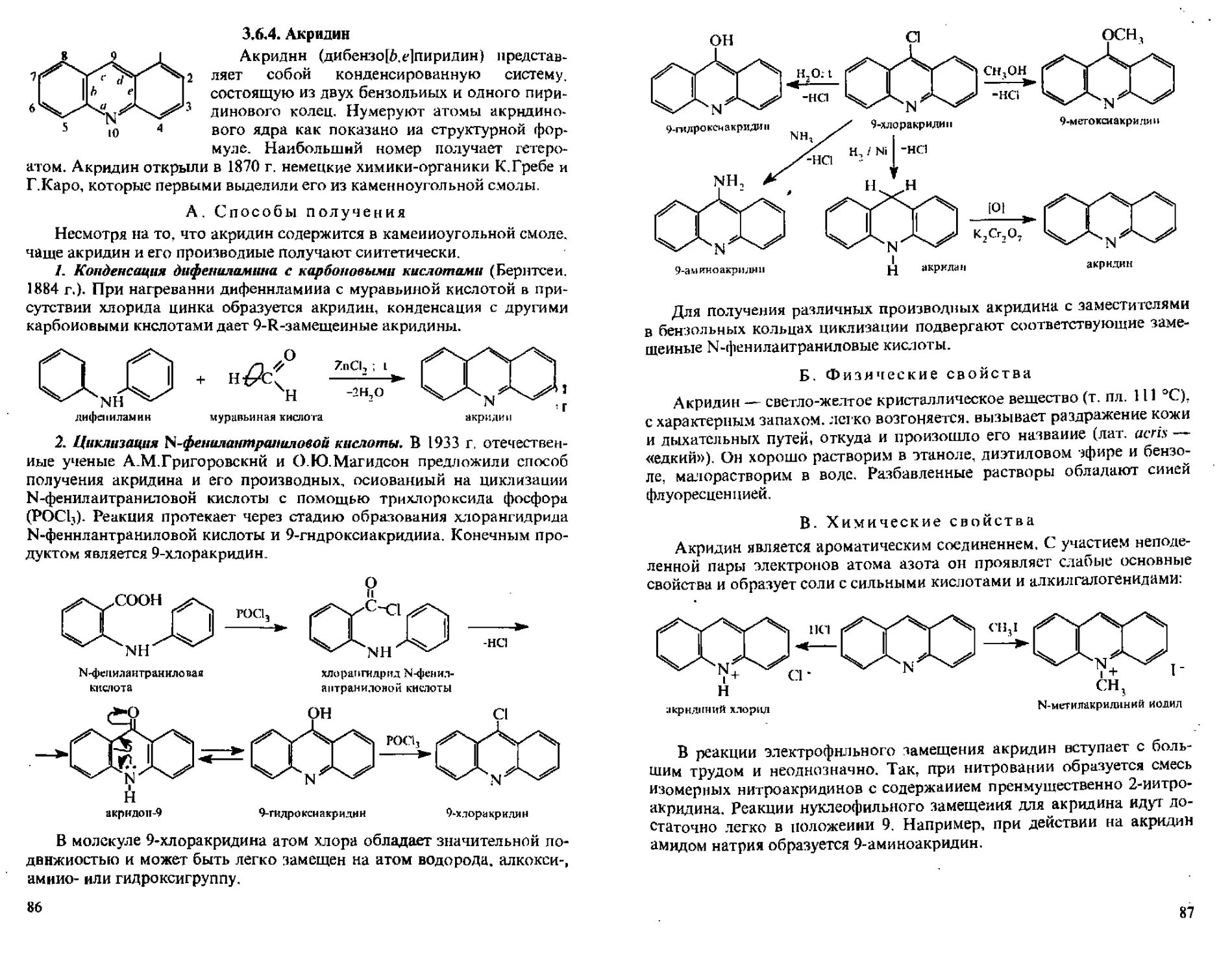
М 3.6.4. Акридин...........................................86
А, Способы получения................................86
Б. Физические свойства..................-...........87
В- Химические свойства..............................87
Г. Важнейшие производные акридина...................89
Z з 7. Шестичлениые гетероциклы с атомом кислорода..........90
3.7.1. а-Пираи и у-пиран...............................90
3.7.2. Кумарин.........................................92
3.7.3. Хромой..........................................93
3.8. Шестичленные гетероциклы с двумя гетероатомами........94
.( 3.8.1. Диазииы........................................94
А, Пиридазин (1,2-диазин)...........................95
Б. Пиримидин (1,3-диазин)...........................96
В. Пиразин (1,4-диазии)............................101
3.8.2. Фенотиазин.....................................102
А. Способы получения...............................102
Б. Физические свойства.............................102
В. Химические свойства......-......................102
3.9. Конденсированные системы гетероциклов.................ЮЗ
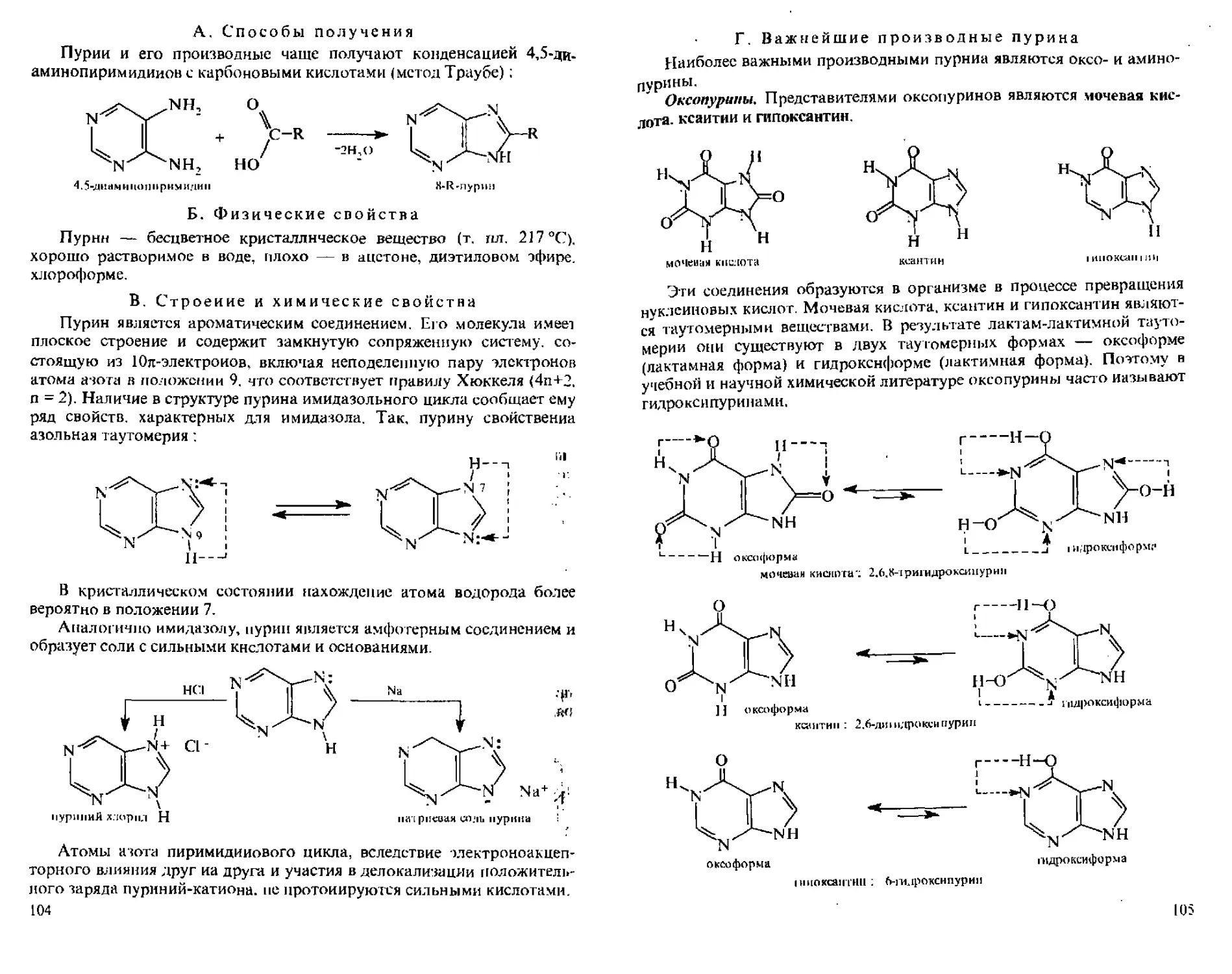
3.9.1. Пурин...........................................ЮЗ
А. Способы получения.............................. 104
Б, Физические свойства.............................104
В, Строение н химические свойства..................104
Г, Важнейшие производные пурина....................105
3.9.2. Птеридин.......................................109
А. Способы получения...............................109
Б. Физические свойства.............................110
В. Химические свойства.............................1 Ю
Г, Важнейшие производные птеридина.................110
3.9,3. Аллоксазии и изоаллоксазнн (флавин)..............................................111
3.10. Семичлеиные азотсодержащие гетероциклы..............112
Контрольные вопросы и упражнения............................ 113
ГЛАВА 4. АЛКАЛОИДЫ........................................115
4.1, Методы выделения из растений.........................115
4.2. Методы обнаружения алкалоидов........................116
4.3. Классификация алкалоидов.............................116
4.4, Алкалоиды группы пиридина и пиперидина...............117
4.4.1. Никотин........................................117
4.4.2. Анабазин.......................................117
4.4.3. Лобелии.........................................117
4.5. Алкалоиды группы хинолина............................118
4,5.1. Хинин........................................... Н8
4.6. Алкалоиды группы изохинолина и фенантренизохинолииа..118
4.6.1. Папаверин.......................................119
4.6.2. Морфин..........................................119
4.6.3. Кодеин........................................ 120
4.7. Алкалоиды группы тропана.............................120
4.7.1. Атропин.........................................121
4,7.2. Скополамин......................................122
4.7.3. Кокаии..........................................122
4.8. Алкалоиды группы индола..............................122
4.8,1, Резерпин........................................123
4.8.2. Стрихнин........................................123
Контрольные вопросы и упражнения..........................124
ГЛАВА 5. УГЛЕВОДЫ.........................................125
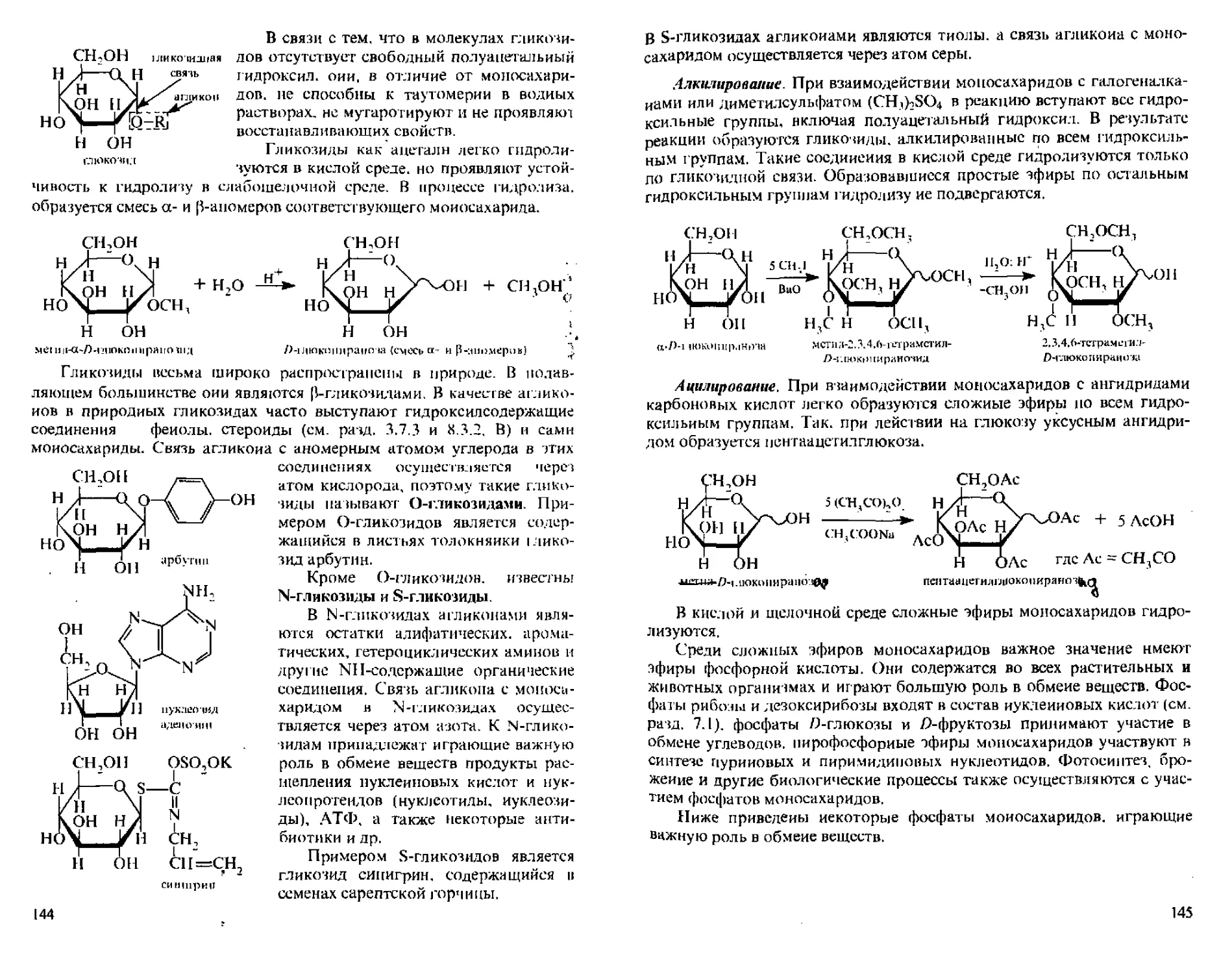
5.1. Моносахариды.........................................126
5,1.1. Классификация и номенклатура....................126
5,1.2, Стереоизомерия..................................127
5,1.3. Строение моносахаридов..........................128
5.1.4. Способы получения...............................135
5.1.5, Физические свойства.............................137
5.1.6, Химические свойства.............................137
А. Реакции с участием открытых форм.................137
Б. Реакции с участием циклических форм..............143
5.1.7. Отдельные представители моносахаридов...........146
5.1.8, Отдельные представители дезокси- и амииосахаров.148
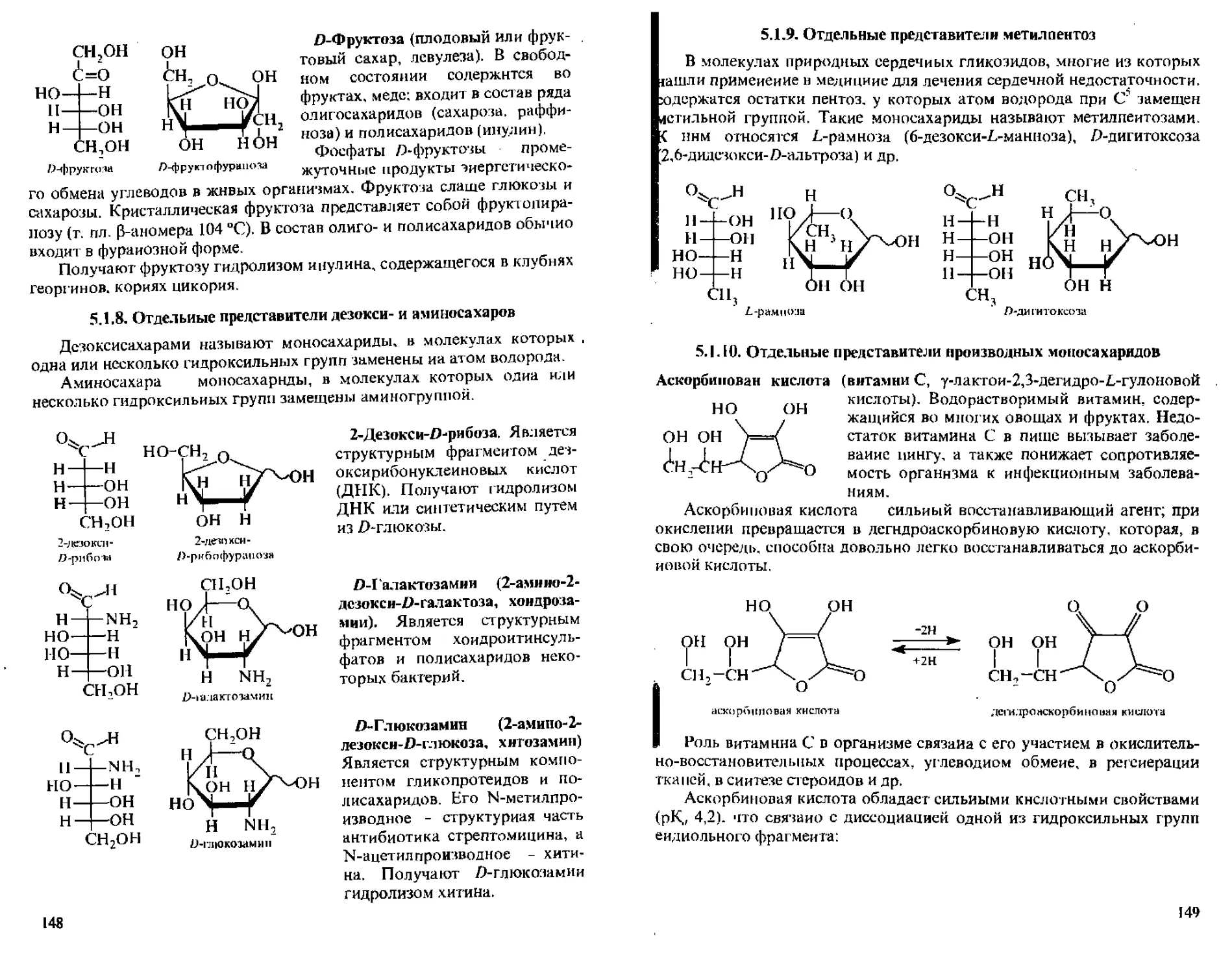
5.1.9. Отдельные представители метилпентоз.............149
5.1.10. От дельные представители производных моносахаридов ..149
5.2, Дисахариды...........................................151
5.2.1. Восстанавливающие дисахариды....................151
5.2.2. Невосстанавливающие дисахариды..................155
5.3, Полисахариды.........................................156
5.3.1. Гомополисахариды................................156
5,3,2. Гетерополисахариды..............................161
Контрольные вопросы и упражнения..........................164
ГЛАВА 6. БЕЛКИ............................................165
6.1, а-Аминокислоты как мономеры белков...................165
6.1.1. Стереоизомерия................................. 168
6.1.2. Физические свойства.............................168
6.1.3. Способы получения...............................169
6.1.4. Химические свойства а-амииокислот...........170
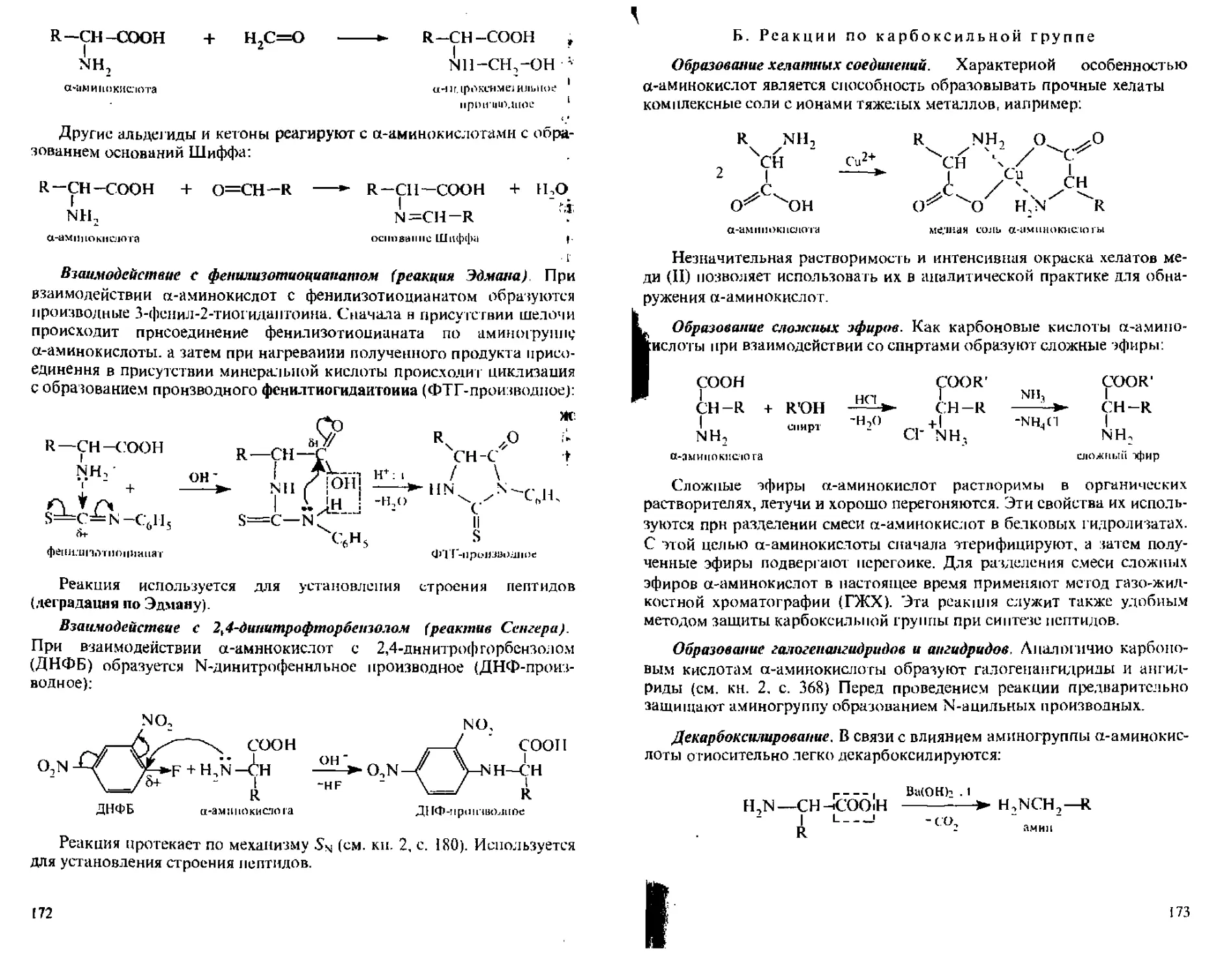
А. Реакции по аминогруппе.......................170
Б. Реакции по карбоксильной группе..............173
6.1.5. Идентификация а-аминокислот.................174
6,2. Строение пептидов и белков.......................174
6.3. Синтез пептидов..................................179
6,4. Сложные белки (протеиды).........................182
Контрольные вопросы и упражнения.................... 183
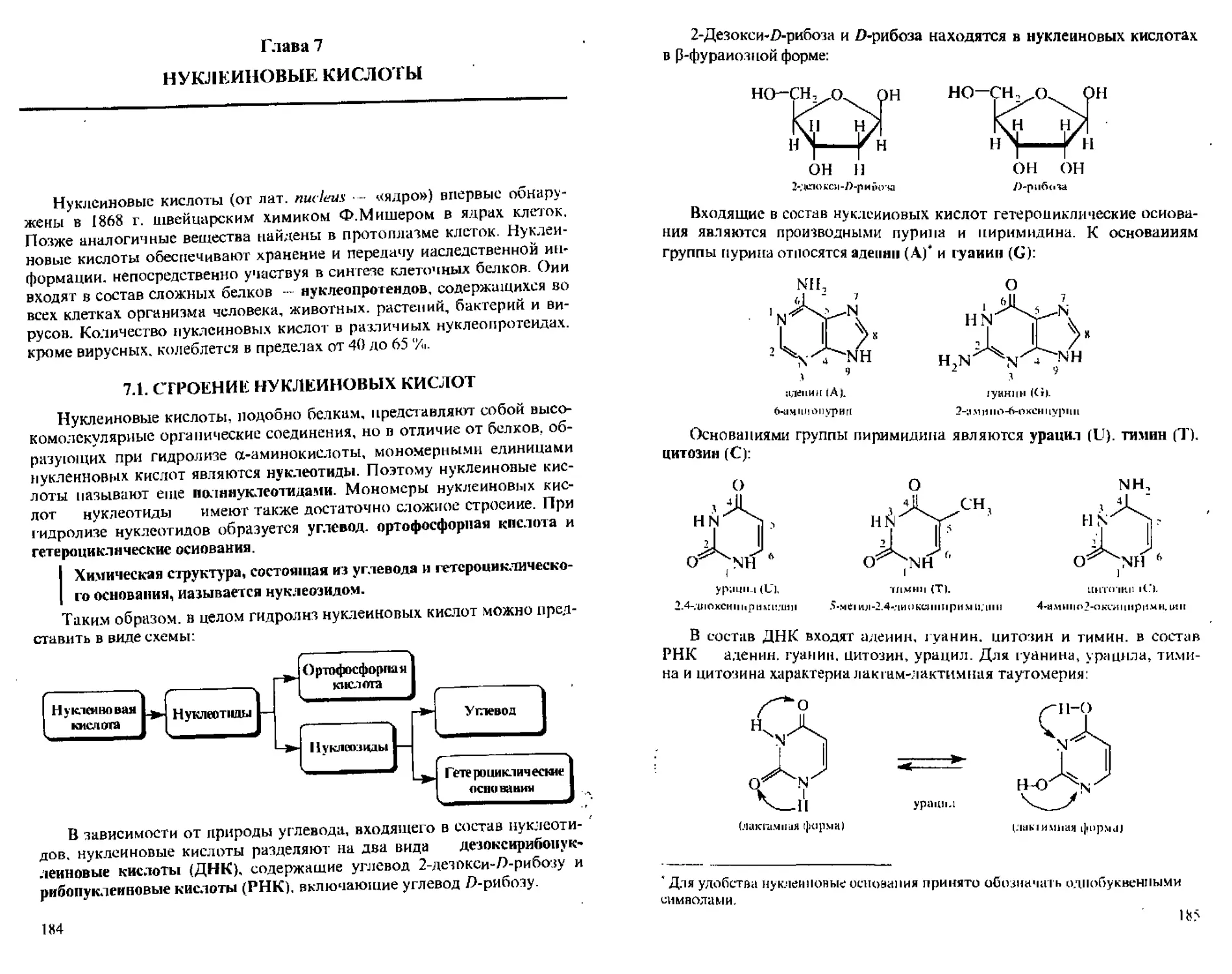
ГЛАВА 7. НУКЛЕИНОВЫЕ КИСЛОТЫ..........................184
7.1. Строение нуклеиновых кислот......................184
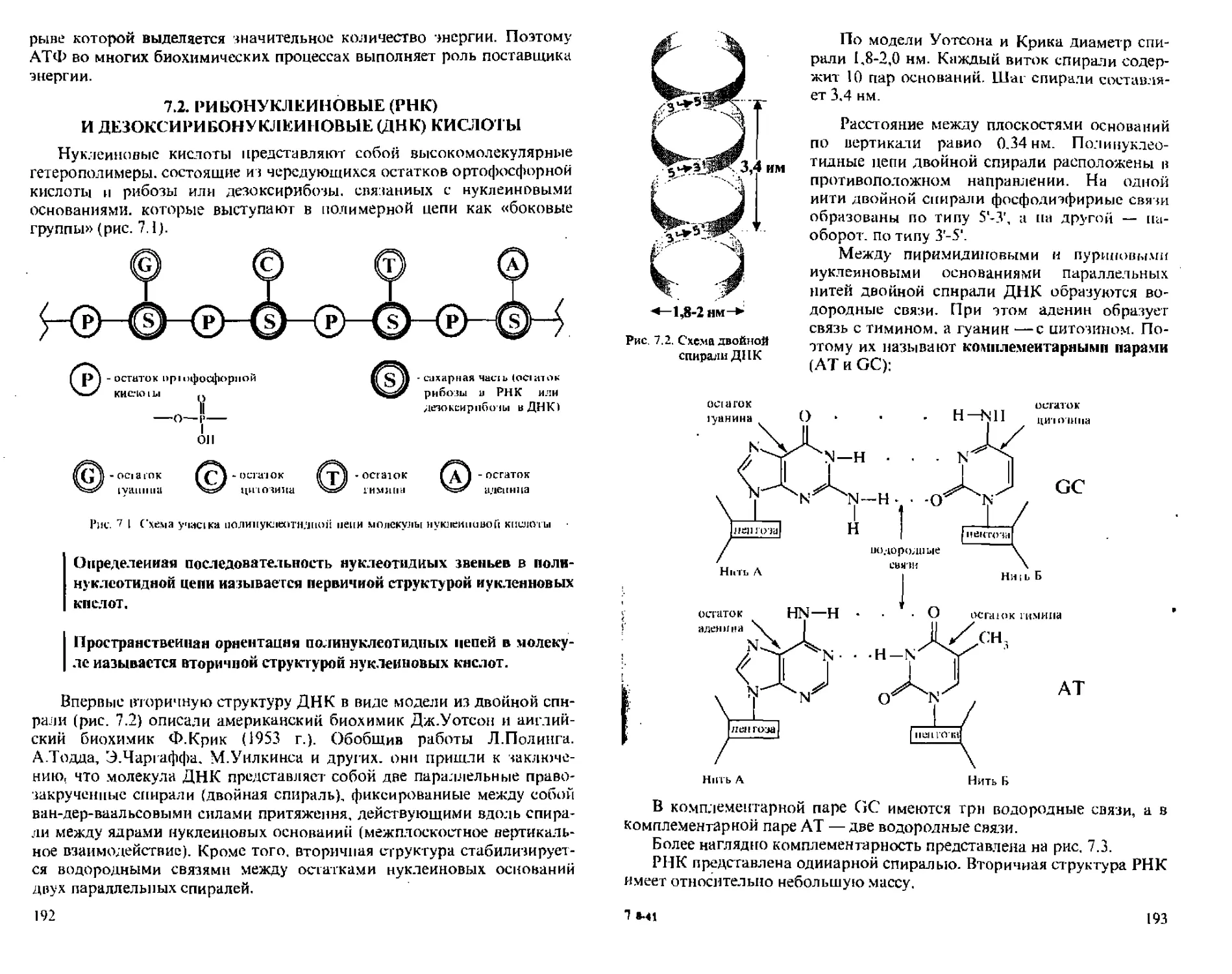
7.2. Рибонуклеиновые (РНК) и дезоксирибонуклеиновые (ДНК) кислоты........................................... 192
Контрольные вопросы и упражнения......................194
ГЛАВА 8. ЛИПИДЫ.......................................196
8.1. Классификация....................................196
8.2. Омыляемые липиды.................................197
8.2.1. Жиры........................................197
А. Номенклатура и изомерия жиров................199
Б. Получение жиров..............................199
В. Физические свойства жиров....................199
Г. Химические свойства жиров....................200
8.2.2. Воски. Твины................................206
8.3. Не омыляемые липиды..............................207
8.3.1, Простагландины..............................207
8.3.2. Изопреноиды.................................209
А. Терпены......................................210
Б. Каротиноиды..................................221
В. Стероиды ................................... 222
Контрольные вопросы и упражнения......................233
СПИСОК ЛИТЕРАТУРЫ.....................................235
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ..................................237
ТЕМАТИКА ВОПРОСОВ К ЭКЗАМЕНУ..........................249
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
Гетероцикличсскими соединениями называют органические вещества, содержащие циклы, в состав которых помимо атомов углерода входят один или несколько атомов других элементов — гетероатомов.
Чаще всего гетероатомами являются азот, кислород н сера, например:
пиридин
фуран
1 и>‘>феп
Гетероциклические соединения широко распространены в природе. Оли входят в состав таких природных веществ, как хлорофилл, гемоглобин, витамины, алкалоиды, нуклеиновые кислоты, ферменты и др. Многие гетероциклические соединения обладаю! высокой биологической активностью, поэтому неслучайно более половины всех лекарственных веществ содержат в своей структуре гетероциклические фрагменты.
В предыдущих разделах мы уже встречались с некоторыми гетероциклическими соединениями, такими, как лактоны (см. кн. 2. с. 440), лактамы (см. кн. 2, с. 463), ангидриды двухосновных карбоновых кислот (см. кн. 2, с. 386. 392) и др. Однако эти гетероциклы легко вступают в реакции, сопровождающиеся раскрытием цикла, и но химическим свойствам более похожи на соответствующие ациклические соединения.
Глава 1 КЛАССИФИКАЦИЯ И НОМЕНКЛАТУРА ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ
1.1. КЛАССИФИКАЦИЯ
Гетероциклические соединения классифицируют в зависимости от размера цикла, природы и числа гетероатомон. а также степени насыщенности цикла.
По размеру цикла различают трех-, четырех-, пяти-, шести- и семичленные гетероциклы.
В зависимости от природы гетероатома 1чл'ероциклические соединения подразделяют на кислород-, азот- и серу с о держащие.
По степени насыщенноеги все гетероциклические соединения классифицируются на насыщенные, ненасыщенные и ароматические.
(.2. НОМЕНКЛАТУРА
Для гетероциклических соединений применяют тривиальные и систематические названия. Тривиальные названия приняты номенклатурой ИЮПАК и в большинстве случаев являются более улотребимыми.
При построении систематических названий гетероциклов номенклатурными правилами ИЮПАК учитывается природа н число гетеро-атомов. а также размер цикла и степень его насыщенности. При зтом природа гетероатома отражается в префиксе, размер цикла — в корне, а степень насыщенности — в суффиксе названия. Для обозначения гетероатомов О. S и N используют префиксы окса- (О), тиа- (S) и аза- (N). Размер цикла обозначается корнями -ир- (трех-), -ет- (четырех-), -ол-(пяти-), -ин- (шести-), -еп- (семичленныи), а степень насыщенности — суффиксами -идин (насыщенный цикл с атомом азота), -ан (насыщенный цикл без атома азота), -ип (ненасыщенный цикл). В названии гетероциклов с максимально возможным числом двойных связей в цикле суффикс не указывается. Для частично гидрированных соединений используют приставки дигидро-, тетрагидро- с указанием номеров атомов, к которым присоединен водород. Если атом водорода присоединен только к одному атому цикла, го в названии указывается номер гидрированного атома и буква Н. В шести- и ссмичлснных азотсодержащих гетероциклах полная насыщенность цикла обозначается приставкой пергидро-. Число гетероатомов одного вида указывается в названии множительными приставками ди-, три-, тетра- и т.д. Если гетероцикл содержит несколько разных гетероатомов, то называют их в определенной последовательности: окса-, тиа-, аза-.
9
При составлении названия в целом допускается ряд упрощений. Ниже приведены примеры составления систематических названий, а также тривиальные названия некоторых гетероциклов.
Трехчленные гетероциклические соединения :
О
оксиран, пилено кси л*
азиридин, ттиленимип*
тинраи.
хгилепсульфид*
Четырехчленные гетероциклические соединения:
тиетан
оксетан
аэетмлин
Пятичленные гетероциклические соединения с одним гетеро а томом:
Пятичленные гетероциклические соединения с двумя гетероатомами:
пиразол*.
1,2-диаюл
NH
Знаком (*) отмечены тривиальные названия. При наличии двух названий чаще применяемое приведено первым.
10
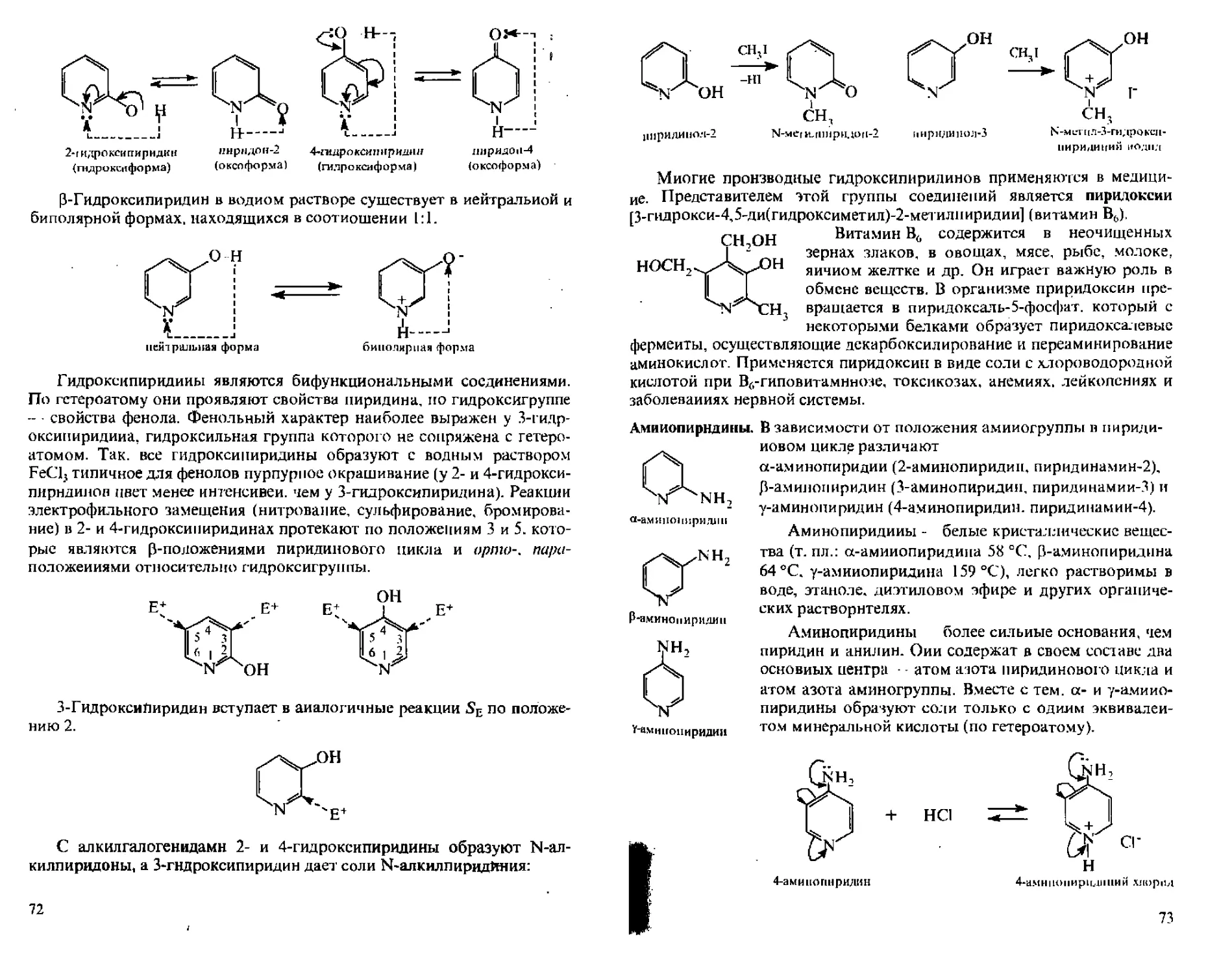
Шести членные гетероциклические соединения с одним гетероатомом:
пиридин*, азин
4Н-пиран
пиперидин*.
пергидроазип
ЩестИчлеииые гетероциклические соединения с двумя гетероатомами:
морфодин*. гетрагидро-1.4-оксазии
пиразин*, 1,4-диазнн
пиридазии*. пиримидин*.
1,2-диазин 1.3-диазин
оксазин*,
4Н-1.4-оксазип
4Н-1,4-1 иазип
пиперазин*.
пергидро-1.4-диазин
Семичлениые гетероциклические соединения с одним гетероатомом:
азепин
оксепип
тненин
Нумерацию атомов в гетероцикле обычно начинают с гетероатома и проводят в том направлении, чтобы заместители получили возможно меньшие номера.
3-м етилп иридии
2-мегилфуран
В пятичленных и шести членных гетероциклах с одним гетероато-мом атомы углерода иногда обозначают греческими буквами а, р, у.
а-метилфуран
р’Метилпиридии
у-аминопиридин
11
В гетероциклах с несколькими равноценными гетероатомами нумерацию проводят таким образом, чтобы гетероатомы получили наименьшие из возможных номеров:
1,2-дназин
1.3-диазин
Если в гетероцикле имеется несколько разных гетероатомов, то нумерацию начинают с того, который в ряду О, S, NH, N расположен левее, и проводят в таком направлении, чтобы остальные гетероатомы получили возможно меньшие номера, например:
1,3-оксазол 1.3-тиазол 1,3-диачоя
Для некоторых гетероциклов существует особый порядок нумерации.
Большую группу составляют гетероциклические соединения с двумя и более конденсированными циклами. Конденсированные гетероциклические системы могут состоять из одного гетероциклического и одного или нескольких бензольных колец, а также нз нескольких гетероциклических ядер. Обычно для таких гетероциклов применяют тривиальные названия, например:
индол
ХИ НОЛИ il
акридин
пурин
Систематические названия конденсированных гетероциклических систем образуют, используя в качестве родоначальных структур тривиальные названия гетероциклов.
При построении названий конденсированных систем, состоящих из одного гетероциклического и одного или двух бензольных ядер, к названию гетероцикла присоединяют префикс бенз- (беизо-) или дибенз-(дибензо-) с указанием буквами а, Л, с, d и т.д. связи гетероцикла, по которой происходит конденсация. Обозначение связей начинают от гетероатома, например:
5 4
бензо^пирндип
дибеню[Л. ^пиридин
12
Если конденсированная система состоит из двух гетероциклов, за основу названия принимают название цикла, большего по размеру; при одинаковом размере — цикла с большим числом гетероатомов, и. наконец, если по первым двум критериям циклы равнозначны, то азотсодержащий цикл имеет предпочтение перед кислородсодержащим, а последний — перед серусодержашим. При помощи букв указывают связь основного цикла, которая является общей для обоих циклов, а при помощи цифр — общую связь второго цикла с основным, напри-
пирр<1ло12,3-/>]п придан
КОНТРОЛЬНЫЕ ВОПРОСЫ И УПРАЖНЕНИЯ
I, Какие органические соединения называются гетероциклическими? Назовите классификационные признаки гетероциклических соединений.
2. Назовите основные номенклатурные системы для гетероциклов. Сформулируйте правила образования систематических названий гетероциклов.
3. Как производят нумерацию атомов в гетероциклах и ^акие существуют способы обозначения положения заместителей в гетероциклических системах?
4. Из каких колец могут состоять конденсированные гетероциклические системы? Как образуют систематические названия конденсированных гетероциклов?
13
Глава 2 ТРЕХ- И ЧЕТЫРЕХЧЛЕННЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ С ОДНИМ ГЕТЕРОАТОМОМ
то
•<й
ГМ
Трех- и четырех членные гетероциклические соелинения с одним гетероатомом можно рассматривать как производные циклопропана и ииклобутаиа, в которых одна группировка —СН2— замещена на гетероатом. Ниже рассмотрены соединения с гетероатомами кислородом и азотом:
О оксиран
оксетаи
аэиридни
NH
азе| ндип
Эти гетероциклические соединения имеют ряд общих методов получения н обладают сходной реакционной способностью, обусловленной наличием в структуре напряженных трех- и четырех членного колец. Этим объясняется их склонность к реакциям присоединения, протекающим с разрывом связи «гетсроатом-углерод». Трехчленные гетероциклы менее устойчивы и более реакционноспособны, чем четы-рехчлеиные.
2,1. ОКСИРАН И ОКСЕТАН
Н,С—СН.
2 \/
о
оксиран, эти лен оксид
н2с—сн2 i
оксеган
2.1.1. Способы получения
I. Общим способом получения оксирана и оксетана является цикли- 1 заиня галогеиоспиртов.
Оксиран и его производные получают циклизацией р-галогеио-спиртов. Реакция протекает при действии концентрированных растворов щелочей: а р
V сн2—сн2 н2с—сн2
|г----L, + NaOH ----------► \/ + NaC + НоО
ОгН _ C1J о
Р-хлор этанол
14
оксиран
Для получения оксетана используют у-галогеноопирты, а 0 Y
СН , —СН2 —СН7 Н,С—СН,
| ---------4, + NaOH — ’ | | - + Naa + Н2О
°Н----------У| н2с—о
у-хлорнропанол оксетан
2. В промышленности оксиран получают в основном окислением этилена кислородом воздуха при температуре 300-400 °C? над серебряным катализатором.
/ Н2С=СН2 + О2
300400° с
Н,с—СН
\/
2.1.2. Физические свойства
Оксиран бесцветный газ с эфирным запахом, т. кип. 10,7 °C. Хорошо растворяется в воде и органических растворителях.
Оксетаи жидкость с т. кип. 47,8 °C. Хорошо растворяется в воде, этиловом спирте и диэтиловом эфире.
2.1.3. Химические свойства
В химическом отношении оксиран и оксетаи являются весьма реакционноспособными соединениями. Это связано с угловым и торсионным напряжением циклов (подобно циклопропану и циклобутаиу), 'а также наличием полярных связей С О- При действии электрофильных и нуклеофильных реагентов происходит разрыв кислород-углеродной связи и присоединение молекулы реагента по месту разрыва цикла. Особенно легко эти реакции проходят в условиях кислотного катализа.
Так, в присутствии серной или фосфорной кислот оксираи легко присоединяет воду и спирты:
н,с—сн, у н+ СН,—СН, + Н,0 I 2 ] 2 он он этилен тли коль
[ </ Н2С^-СНг ! О н+ СН,—СИ2 + с,н5он | - | ОН ОС,Н5 2-:поксюгэнол.
этилцеллоюльв
; Механизм приведенных реакций включает образование оксониево-го соединения (продукт взаимодействия оксирана с кислотой), которое гораздо легче подвергается атаке нуклеофильным реагентом, чем сам оксиран (5+' > S+)
15
оксомиеаое соединение
Аналогичным образом оксиран присоединяет галоген оводороды:
Н.С—СП, X/ + НС1
2-щюругаиол, 'ггиленхлортдрин
Оксиран довольно легко присоединяет сильные нуклеофилы, такие, как аммиак, амины, металлорганические соединения.
При взаимодействии оксирана с аммиаком, в чависисмости от соотношения реагентов, образуются моно-, ди- и триэтаноламины.
Н,С—СН2
NI[4
по—сн2-сн, NH
ПО-СН2“СН/
НО-СН,— сн2—Nil,
71 аполамин, З-амнтяганол
но—сн2-сн но—сн2-сн НО“СН2~ СИ
трин аноламин
и глада ла ми и
Механизм реакции:
н.с— сн2
О' +Ь1Н3
* н,с— сн,
| I
ОН NH,
При действии иа оксиран алифатических аминов аналогично образуется N-алкиламиноэтанолы.
н,с—снТ •"
- \/ “ + *Nrin-lt -----НО—СН,—ОН,—NH—R
О “ к '
• , Гч-алкиламипотпапол
16
С магнийорганическимн соединениями оксиран образует продукты присоединения, которые легко подвергаются гидролизу с образованием соответствующих спиртов.
s+
Н,С----СН
5- 5+
CH3-MgBr
H2C—CH
нон. нт
-MgCOHlBr
BrMgO СН3
лроианолятбромнл-мап1ий
----СН^-СН2-СН2-ОН пропанол-!
В присутствии сильных оснований оксиран полимеризуется с образованием полиэтиленоксида (полиэтиленгликоля).
н,с~сн
Об-
НО^СН,—СН2“О^-Н
цолитгилепоксил
Полиэтиленгликоль, в зависисмости от молекулярной массы, имеет различное агрегатное состояние. До молекулярной массы, равной 400 — это жидкость, хорошо растворимая во многих органических растворителях. Применяется в фармации в качестве растворителя лекарственных веществ, основы для мазей и суппозиториев, а также как связывающее вещество в производстве таблеток.
Оксетан по химическим свойствам сходен с оксираном. Для него, как и для оксирана, характерны реакции присоединения с раскрытием цикла. Одиако меньшая степень напряжения в четырехчленном цикле способствует тому, что эти реакции протекают гораздо медленнее.
Многие реакции оксетана приводят к образованию t.З-дизамешеп- ных пропана
основанне
НОН. н
О
СН2~О
г
НО—СН2—СН2 СН2 СН1 уКра|ИГЬ1Г< фармя»;евтичн1 бутанои-1 I А К t Д И 1 И
БI S Л I О * Е К А 1
.../ ап п
2.1.4. Важнейшие производные оксирана и оксетаиа
Эпихлоргидрин (3-хлор-1,2-эпоксипропан). Бесцветная жидкость с запа-гчл г-и /и хом хлороформа, т. кип. 116,1 °C. Хорошо рас-\ 2 / 2 творим в органических растворителях. Приме-
няется в производстве эпоксидных смол, для получения глицерина и как растворитель эфиров целлюлозы.
Р-Пропиолактон (лактон Р-гидроксипропноиовой кислоты)- Бесцветная
^н3-сн2 °—ч о
жидкость с резким запахом, т. кип. 155 °C. Растворяется в органических растворителях. В воде быстро гидролизуется до Р-гидроксипропионовой КИСЛОТЫ.
p-Пропиолактон легко взаимодействует с аминами и спиртами. Реакции протекают с раскрытием цикла.
О
СН,-СН,-(/
ОН
осн3
СН.ОН CFL—CH. C1LNH, Z?
—г— I 2 I 2 ——fc- сн2—сн2-с
NHCH?
метиловый эфир .1-гпдрок-си ирона и о вой кислоты
о
Р-upon иола кто и
он
меги лам ид З-гидроксинропа-новой кислоты
О---С
р-Пропиолактоь применяется в медицине для стерилизации крови, вакцин и других биологических препаратов.
2.2. АЗИРИДИН И АЗЕТИДИН
н2с—сн2
2 \ / NH
азнрилип, этидепимин
н2^—<рН2
Н2с—NH азегидип. три мет плени мин
2.2.1. Способы получения
Обшнм способом получения азиридина и азетидина .является циклизация галогенам инов в присутствии щелочи.
Азиридин получают циклизацией р-галогенэтиламинов.
а Р
сн.-сн2 NaOH Н2С—СН7
I - | NaOH^ ^ \ / 2 + Naa + н 0
nh2 а NH
0-хлор этилам и и а чиридии
Дчя получения азетидииа используют у-галогеипропиламины.
18
СН,-СИ2-СН2 NaOH Н2с—сн2
| -----► | I + NaBr + Н7О
NH3 Br Н2С—NH
у-бро миро пилами н атетидин
В промышленности азиридин получают взаимодействием 1.2-ди-хлорэтана с аммиаком в присутствии оксида кальция СаО.
а} сн,-сн. н,с—СИ,
ДО I • + NH, — ° » 2 \ / 2 + СаС12 + Н2О
Cl Ct NH
2.2.2. Физические свойства
Азиридин бесцветная жидкость с т. кип. 55 °C. Хорошо растворяется в воде и органических растворителях.
Азетидин бесцветная жидкость с аммиачным запахом, т. кип.
63 °C. Хорошо растворяется в воде и спиртах.
2.2.3. Химические свойства
По химическим свойствам азиридин и азетидии во многом напоми
нают ранее рассмотренные кислородсодержащие гетероциклы оксиран
и оксетаи.
Подобно оксирану и оксетану, для них характерны реакции присоединения, протекающие с раскрытием цикла. Так, азиридиновый цикл раскрывается под действием аммиака, аминов, галоген о водородов.
воды.
h2n— ch?-ch?-nh2
ттандиамнпЧД
R—NH—CH2-CH2-NH2
диамин
но—ch,-ch2-nh2
2-аминоэтанол
Cl—ch2-ch2-nh2
2-хлорланамин
Наряду с этим, азиридин и азетидин представляют собой вторичные циклические амины. Поэтому, в отличие от кислородсодержащих гетероциклов. они проявляют ряд специфических свойств, характерных для вторичных аминов.
Наличие неподеленной пары электронов на атоме азота придает азиридину и азетидииу основные свойства (рКвп+ азиридина 7,48: рКВц азетидина 11,29).
Подобно вторичным аминам, азиридин н азетндин вступают в реакции алкилирования, ацилирования, нитрознрования и другие, например:
Я н
СНл-[
441 О
СНЛ-С-С1 -ИС
O==N-C1 ни грози л хлорид
-НС1
^,N“СН3 О
^N— (*:—сн?
N — N=O
Н-метилазирнлин
N-a нети лази р иди и
М-иичрозоазнрнднн
Эти реакции обычно проводят в присутствии оснований (часто используется избыток триэтиламина) для связывания выделяющегося галогеноводород а или других продуктов кислотной природы, способных раскрывать цикл.
2.2.4. Важнейшие производные азиридина и азетидина
Среди производных азиридина обнаружены вещества, обладающие
выраженной противоопухолевой активностью, на основе которых соз-
• даио ряд противоопухолевых лекарственных препаратов (тиофосфа-мнд, бензотэф, фторбензотэф и др.). Все онн содержат, как правило, остатки фосфорной и тиофосфориой кислот.
тиофосфамид бепэотэф
Из производных азетидииа важное значение имеет азетидинои-2
(Р-лактам). Это внутренний амид Р-амииопропионо-вой кислоты. Его получают термической циклизацией Р-аминопропионовой кислоты. При действии водных растворов кислот и щелочей, аммиака и аминов р-лактамное кольцо раскрывается.
сн2-сн2-с^ -*
NH2 NH2
амид Р-амннопропио-
id вой кислоты
NH; СН2 <j?H2 нон, н*
N-----
н о
сн,-сн,-с*
I “ - X
nh2 он
Р-аминопропионовая кислота
Азетидинои-2 входит в состав антибиотиков группы пенициллина.
ноос
н,с
Н3С
—R
общая формула пенициллинов
20
КОНТРОЛЬНЫЕ ВОПРОСЫ И УПРАЖНЕНИЯ
[ К каким соединениям относятся трех- и четырех членные гетероциклы с одним гетеро атомом:
а) гетероциклическим насыщенным;
б) гетероциклическим ароматическим?
2. Приведите схемы получения оксирана и оксетана н уравнения взаимодействия оксирана с:
а) водой;
б) бромоводородом;
в) этанолом;
г) аммиаком.
Сравните реакционную способность окснраиа и его протонированной формы в реакциях нуклеофильного присоединения.
3. Напишите уравнение реакций получения трех- и четырехчленных азотсодержащих гетероциклов.
Чем объясняется их высокая реакционная способность?
Для азетидииа приведите схемы реакций с:
а) водой;
б)аммиаком;
в) хлороводородом.
4. Приведите схемы реакций азиридина с:
а) хлорэтаном;
б) ацетнлхлоридом;
в) иитрозилхлоридом.
На какие свойства азиридина и азетидина указывают данные реакции и почему их проводят в присутствии триэтиламина?
5, Напишите схемы реакций, которые протекают при действии иа азе-тидинон-2 водного раствора гидроксида иатрия и аммиака. Могут ли антибиотики группы пенициллина существовать в кислой или щелочной среде?
И
21
Глава 3
ПЯТИ- И ШЕСТИЧЛЕННЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ С ОДНИМ И ДВУМЯ ГЕТЕРО А ТОМАМИ
Из большого числа пяти- и шестичлениых гетероциклических соединений с одним и двумя гетероатомами в данном разделе рассматриваются гетероциклы с гетероатомами О, N и S. обладающие ароматическими свойствами. Такие вещества по своей устойчивости и химическим свойствам во многом напоминают бензол и поэтому получили название гетероциклические ароматические или гетероароматические соединения.
К ним относятся:
пятичленные гетероциклы с двумя л-связями
фуран пиррол тиофен пиразол имидазол
гиачои
И Др.
щестичлеиные гетероциклы
пиридин пиридазин пиримидин пиразин
конденсированные гетероциклические системы
ИНДОЛ
хинолин
акридин
3.1. АРОМАТИЧНОСТЬ ГЕТЕРОЦИКЛОВ
7 Как известно, признаком ароматичности соединения является наличие плоской циклической системы, имеющей замкнутую цепь сопряжения, содержащую (4п+2) тг-электронов.
Ароматичность пятичлеиных гетероциклов с двумя тг-связями обусловлена тем, что в сопряжение с л-электроиами двойных связей вступает иеподеленная пара электронов гетероатома О: , N: или S:. В результате образуется замкнутая сопряженная система, в которой число обобщенных электронов отвечает правилу Хюккеля (4п+2).
22
Рис. 3.1. Электронное строение молекулы пиррола
В молекуле пиррола (рнс. 3.1) атомы углерода и атом азота находятся в состоянии луЛгибридизации. За счет sp2~i ибридных орбиталей каждый атом, входящий в состав цикла, образует три ст-связи, расположенные в плоскости кольца. При этом у атомов углерода и атома азота остается по одной иегибридизоваи-ной /7-орбитали, которые расположены параллельно друг другу в плоскости, перпендикулярной плоскости кольца. Каждая из /?-АО атомов углерода имеет одни электрон, а на /7-орбитали атома азота находится неподеленная пара электронов. При
перекрывании р-орбиталей образуется единое шестнэлектроииое облако, охватывающее все атомы цикла.
Атом азота в ^-гибридизации, имеющий электронную конфигурацию, в которой иеподеленная пара электронов занимает негибриди-зоваииую р-атомиую орбиталь, получил название пиррольного-
Аналогично образуется сопряженная система и в других пятичлен-
ных гетероциклах с двумя я-связями. в частности, в молекулах фурана и
фурД1, гиофе11
тиофена. Здесь, как и в пирроле, гетсроатом (:(): и :S:) вносит в ароматический секстет иеподелеиную пару /?-электронов. По аналогии с пирролом, гетероатом, вносящий в п-электронную систему два электрона, зани-
мающих р-атомпую орбиталь, и образующий с другими атомами толь-
ко ст-связи, принято называть гетеро атомом пиррольного типа.
В ряду шестичлениых гетероциклов ароматические свойства характерны для структур, представляющих собой гетероциклические аналоги бензола. Так. в молекуле пиридина (шестичленный гетероцикл с атомом азота, рис. 3.2) все атомы углерода и атом азота находятся в состоянии у/Лгибридизацни. Замкнутая шести-п-элект(х)иная система образуется пятью/7-орбиталями атомов углерода (по одной от каждого) и /7-орбиталью атома азота. То есть, в молекуле пиридина, как и в молекуле бензола, каждый атом цикла вносит в аромати-
ескин секстет по одному /7-электрону.
Неподеленная пара электронов атома азота в молекуле пиридина, в Отличие от молекулы пиррола, занимает л/Лгибридиую орбиталь и не принимает участия в образовании ароматического секстета.
н
11
Рис, 3.2. Электронное строение молекулы пиридина
23
Атом азота в .^^гибридизации, имеющий электронную конфигурацию, в которой неноделенная пара электронов занимает з/Лгибри-дизоваииую орбиталь, и ие участвует в образовании ароматического секстета, получил название пиридинового. Гетероатом такой электронной конфигурации, в данном случае атом азота, еще у ело апо называют гетероатомом пиридинового типа.
Гетероатом пиридинового типа, обладая большей электроотрицательностью по сравнению с углеродом, понижает электронную плотность иа атомах углерода ароматического кольца.
Молекулы гетероциклов с двумя и более гетероатомами, а также конденсированные гетероциклические системны могут включать гетероатомы как пиррольного, так и пиридинового типа.
Деление гетероатомов на атомы пиррольного и пиридинового типа позволило А.Альберту в 1958 г. ввести понятие о л-избыточности и тг-дефицитности гегероароматических соединений.
Гетероциклы, в молекулах которых гетеро атом является донором неподелеииой пары электронов и, следовательно, увеличивает электронную плотность на углеродных атомах ароматического цикла, называют л-избыточиыми.
К ним относят пятичлениые гетероароматические соединения, содержащие гетероатомы пиррольного типа (фуран, пиррол, тиофен и ДР-).
Гетероциклы, в молекулах которых гетероатом понижает электронную плотность иа атомах углерода ароматического кольца, называют л-дефицитиыми.
К я-дефицнтным гетероциклическим сисгемам относят гетероциклы, содержащие гстероатомы пиридинового типа (пиридин, пиримидин, пиразин и др.).
24
3.2. КИСЛО ТНО-ОСНОВНЫЕ СВОЙСТВА ГЕТЕРОЦИКЛОВ
Кислотные н основные свойства гетероциклических соединений обусловлены электронным строением гетероатомов.
В молекуле пиррола нсподеленная пара электронов гетероатома, расположенная на негибридизованной ^-орбитали, принимает участие в образовании тг-элсктронной ароматической системы. Поэтому пиррольный атом азота не способен присоединять протон, т. е. не может быть центром основности. По этой же причине не проявляют основных свойств фуран и тиофен.
Наряду с этим, участие пиррольного атома азота в сопряжении способствует поляризации связи N—Н и тем самым увеличивает подвижность атома водорода, что приводит к появлению у пиррола свойств слабой NH-кислоты. При действии щелочных металлов и сильных оснований (NaOH. NaNFL) происходит замещение атома водорода у пиррольного атома азота на металл.
, NaNH> — О
N амил иа । рия
uiippojiiiai рий
У атома азота пиридинового типа иеподеленная пара электронов находится на sp -гибридной орбитали и не участвует в образовании ароматического секстета. За сч^т этой электронной пары азот пиридинового типа способен присоединять протон, т.с. проявлять осибвные свойства.
+ H2SO4
и иридии иирмДииий тдросул^фаг
Таким образом, кислотные свойства азотсодержащих ароматических гетероциклов обусловлены наличием в их структуре атома азота пиррольного типа, а основные - пиридинового типа.
Гетероциклические соединения, содержащие в своем составе атомы азота пиррольного н пиридинового типов, проявляют амфотерные свойства (пиразол, имидазол, пурин и др.).
IICI
нираюлий хлорид
пиразол
NaNH.
пираюлид-илгрий
25
3.3. НЕКОНДЕНСИРОВАННЫЕ ПЯТИЧЛЕННЫЕ ГЕТЕРО- Н
ЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ С ОДНИМ ГЕТЕРОАТОМОМ
Пятичленныс гетероароматические соединения с одним гетроато*ф мом можно рассматривать как производные бензола, в котором rpyn-J пировка —СН=СН замещена на гетероатом. {
Важнейшими представителями этих гетероциклов являются пиррол» фураи и тиофен; -д
НИррО.'Г.
а:юл
фуран, о кип л
шпфеп.
иол
Названия одновалентных остатков приведенных гетероциклов образуют с помошью суффикса -ил, указывая цифрой или буквой греческого алфавита положение свободной валентности.
а-нпррнл: р-пиррил:
2-пнррил 3-пиррил
а фурил:
2-фурил
Р-фурил: 3-фур ил
п-тиенил p-TiienHT,
2-т испил 3-гиеиил
3.3.1. Способы получения
А. Общие способы получения пиррола, фурана и тиофена
7. Циклизация 7,4-дикирдоиильпых соединений (синтез Паоле-Кнорра). Для получения фурана и его производных на 1,4-дикарбонильные соединения действуют водоотпимающими реагентами (концентрированная H2SO4, Р2О5), для синтеза пиррола н его гомологов действуют аммиаком, для получения тиофена и его производных применяют пеп-тасульфнд фосфора (P2S5).
2. Взаимные превращения фурана, пиррола и тиофена (цикл реакций Юрьева). Реакции взаимных превращений фурана, пиррола и тиофена были открыты советским химиком-органиком Ю.К.Юрьевым в 1936 г, 26
При каталитическом действии оксида алюминия и нагревании (~450°С) фураи в присутствии аммиака превращается в пиррол, а в присутствии сероводорода в тиофен. Под действием воды в этих условиях пиррол и тиофеи образуют фураи. Аналогично, тиофен в присутствии аммиака превращается в пиррол, а пиррол в присутствии H2S — в тиофен.
Следует отметить, что из приведенных реакций с хорошим выходом протекает только превращение фураиа в пиррол и тиофен.
Б. Специфические способы получения
1. Получение пиррола. В незначительных количествах пиррол содержится в каменноугольной смоле. Синтетически пиррол получают нагреванием диаммоиийной соли слизевой кислоты.
ноV он
с—с
Н "
\ /Н
сС
HOZ coo-+nh4
h4n+-ooc чон
диаммонийиая соль слизевой кислоты
----------?---------
-2СО2; -4Н2О: -NH3
Другой метод заключается в перегонке сукцинимида с цинковой пылью
сукцинимид
2Zn
2ZnO
2. Получение фурана. В лабораторных условиях фураи получают сухой перегонкой слизевой кислоты. Реакция протекает через стадии образования дегидрослизевой (а,ос'-фураидикарбоиовон) и пирослизевой (ос-фуранкарбоиовой) кислот.
4ю>
< ОН’
НООС^ \ г-----/ ^СООН -зн2о НООС
СЩНСЙ
слизевая кислота
даидрослизевал кислота
СООН ч
27
пиросинзевая кислота
pi
<4
I*
В промышленности фуран получают нз альдопентоз. При нагревании с в одо отнимающим и средствами альдопеитозы циклизуются, образуя фурфурол, который окислением переводят в пирослизевую кислоту, и далее, путем термического декарбоксилирования в фуран:
-зн2о
и и росли зевая кислота
-со2
3. Получение тиофена. Тиофеи был открыт случайно в 1882 г. профессором Берлинского университета В.Мейером как примесь в бензоле, полученном из каменноугольной смолы. В промышленности тиофен получают в результате парофазной циклизации бутана с серой, а также по реакции Чичибабина при пропускании смеси ацетилена с сероводородом иад катализатором (А12О3).
н н
' Ъ /ХС —с
< н
Н
S\
700°С
-’H:s
HJL-Щ
тиофен
400-5002£ [AL.OJ:
-Н,
НС
НС
СН
+ СН
3.3.2. Физические свойства
Пиррол бесцветная жидкость с запахом, напоминающим запах хлороформа; т. кип. 130 °C; мало растворим в воде, хорошо растворим в этаноле н бензоле. На воздухе темнеет и осмоляется.
Фуран бесцветная жидкость со своеобразным запахом, напоминающим запах хлороформа; т. кип. 32 °C. Нерастворим в воде, хорошо растворяется в этаноле и диэтиловом эфире.
Тиофен бесцветная жидкость со слабым запахом сернистых соединений; т. кип. 84 °C; нерастворим и воде, хорошо растворим в этаноле, эфире и бензоле. Устойчив к высокой температуре. На свету* окисляется. 28
3.3.3. Химические саоистаа
Реакционная способность пиррола, фурана и тиофена определяется наличием в их структуре цикла с тг-электроиоизбыточной ароматической системой (шесть ^-электронов приходится иа пять атомов цикла). Одиако степень ароматичности указанных гетероциклов ниже, чем у бензола, и зависит от природы гетероатома. Поскольку электроотрицательность атома серы меньше электроотрицательности атомов азота и кислорода, доля участия неподелеиной пары электронов атома серы в образовании ароматического секстета молекулы тиофена больше, чем атома азота в пирроле и атома кислорода в фураие. Так, если для бензола энергия сопряжения составляет ~150 кДж/моль, то в ряду пиррол, фураи, тиофен она уменьшается по мере увеличения электроотрицательности гетероатома: тиофен (~130 кДж/моль), пиррол (-110 кДж/моль), фуран (~90 кДж/моль). Поэтому, из приведенных гетероциклов тиофеи по своему химическому поведению в наибольшей степени напоминает бензол, а фуран имеет наименее выраженный ароматический характер. В некоторых реакциях фуран ведет себя как ненасыщенное (диеновое) соединение.
Вследствие электроотрицательности гетероатома в молекулах пиррола, фурана и тиофена, в отличие от бензола, электронная плотность распределена неравномерно, в частности, на атомах углерода в а-поло-жении плотность электронов выше, чем в p-положении, что определяет направленность протекания реакций электрофильного замещения.
А. Общие химические свойства пиррола, фурана и тиофеиа
/. Взаимодействие с минеральными кислотами, В присутствии сильных минеральных кислот пиррол и фуран осмоляются, образуя полимерные продукты темного цвета. Это свойство получило название аци-дофобность, что означает «кислотобоязнь», от лат. acidum — «кислота» и греч. фобос — «страх». Ацидофобность обусловлена присоединением протона, преимущественно к а-углеродному атому цикла, что приводит к нарушению ароматичности кольца. Затем происходит либо разрыв цикла с образованием полимера (наиболее вероятный процесс для фурана), либо полимеризация образовавшейся диеновой структуры, протекающая с сохранением цикла.
О + н+ — О<н ** О<н -
х X н X н
X = О. NH
Введение в фурановое и пиррольное ядро электроиоакцепториых заместителей (-NO3, -СООН. -НС=О) приводит к снижению ацидофобио-сти этих соединений. Тнофен, в отличие от фурана й пиррола, не обладает ацидофобностью, т.к. имеет устойчивую ароматическую структуру. которая не разрушается при действии сильных минеральных кислот.
29
2. Реакции электрофильного замещения. Являясь л-избыточным и ароматическими системами, пиррол, фуран и тиофеи легко вступают в характерные для ароматических соединений реакции электрофильного замещения. Эти реакции протекают значительно легче, чем у бензола. По активности в реакциях с электрофильными реагентами указанные гетероциклы располагаются в ряду пиррол > фураи > тиофеи. В первую очередь замещается атом водорода при а-углеродном атоме и только если это положение занято, замещение протекает в р-положеиии. Такая направленность замещения обусловлена тем, что при участии а-угле-родных атомов образуется более устойчивый су-комплекс, благодаря большей возможности для делокализации положительного заряда.
атака
X = О. S. NH
а-попоженил
атака [З-поло-женин
Нитрование ^Учитывая ацидофобность фурана н пиррола, нитрование их проводят не самой азотной кислотой, а продуктом взаимодействия азотной кислоты с уксусным ангидридом — ацетилнитратом СН?СООЫ02. Тиофен неацидофобен, поэтому его можно пронитровать азотной кислотой в мягких условиях, однако чаще в реакции нитрования тиофеиа также применяют ацетил нитрат. В результат^ нитрования образуются а-нитросоединеиия.
а
О //
4- сн?---С
ono2
сн3соон
X = о. s. NH
2-иичрофуран (X = О):
2-нитропиррол (X = NH): 2-питроч иофен (X = S)
Сульфирование. Для сульфирования пиррола и фурана (ацидофоб-ные вещества) вместо серной кислоты в качестве электрофильного реагента применяют комплекс пиридина с оксидом серы (VI) пиридинсульфотриоксид QHsN-SO;,. Этот сульфирующий реагент был предложен советским химиком А.П.Терентьевым в 1947 г. В процессе реакции образуются а-сульфокислоты.
30
ппридин-сульфогриокснд
пиридин
фуран-2-супьфокислота (X = О): пиррол-2-сул|,фг>кислота (X - NH)
Тиофеи легко сульфируется концентрированной серной кислотой, реакция протекает на холоду, почти с количественным выходом. В этой реакции тиофен значительно более реакционноспособен, чем бензол, который с серной кислотой в указанных условиях нс реагирует. Данную реакцию используют при очистке технического бензола от примеси тиофена,
О Q-SO.H -
тиофен тио фен-2-сульфо кислота
Ацилирование. Для ацилирования фурана и пиррола в качестве электрофильных реагентов используют ангидриды кислот в присутствии кислот Льюиса, чаще SnCU илн ZnCl2. Тиофеи ацилируется не только ангидридами, но и хл оран гидридами карбоновых кислот в присутствии хлорида алюминия. Замещение осуществляется по а-положеиию.
X = о. NH, S
4- СН3СООН
2-апстмлфуран (X = О);
З-ааегилнцррол (X - NH): 2-ашп и;пиофсн (X = S)
Галогенирование. Галогенирование фураиа протекает довольно сложно. Наряду с замещением атомов водорода иа галоген в зависимости от условий проведения реакции образуются также продукты 2,5-присоединения. Пиррол с галогенами реагирует очень легко, образуя тетрагалогенопирролы. Дэя получения моногалогенозамещенных производных пиррола требуются специальные условия. Так, при действии на пиррол сульфурил хлорид a SO2Cl2 происходит постепенное замещение атомов водорода на галоген.
пиррол 2-хлорлиррол 2.5-лихлорпнррол теграхлорпнррол
31
Галогенирование тиофена проводят непосредственным действием галогена (хлора или брома). Реакция идет на холоду с образованием моно-, ди-. три- и тетразаметениых производных тиофена.
нофеп 2-хлоргиофеп 2,5-ЛИХ;юртиифеи геграхлортиофен
Реакция с иодом пропекает медленно в присутствии катализатора HgO,
3. Реакции восстановления. Фуран присоединяет водород при высокой температуре (140°С) и давлении (100-150 атм.) в присутствии катализатора (никель Реиея. палладий) с образованием насыщенного гетероцикла - тетрагндрофурана (оксолана).
фуран
+ 2Н,
тетра nt дрофу pail
По химическому строению тетрагидрофуран представляет собой циклический простой эфир. Это малореакционнеспособное соединение широко используется в органическом синтезе как растворитель.
Присоединение водорода к тиофену в присутствии палладиевого катализатора происходит значительно легче, чем к фурану (при комнатной температуре и давлении 2-4 атм.). В процессе восстановления образуется тетрагидротиофен.
1мофен
Pd
। ei pai и.дроч иофен
Пиррол, в отличие от фурана и тиофена, гидрируется водородом в момент выделения, например, действием цинка в уксусной кислоте. При этом происходит частичное восстановление кольца с образованием ненасыщенного гетероцикла — 2,5-дигидропиррола (пирролина). Полное восстановление пиррольного цикла происходит при гидрировании над платиновым или палладиевым катализатором. В результате образуется тетрагидропиррол(пирролидин)
ПИррОЛИ)!
пирролидин
Пиррол ин и пирролидин являются циклическими аминами и существенно отличаются по химическим свойствам от пиррола.
32
В молекуле пирролина неподеленная .пара электронов не сопряже с тг-электронами двойной связи, поэтому он проявляет свойства амин и непредельных соединений. Пирролидин же относится к насыщеинЕ соединениям — это типичный представитель вторичных циклическ аминов. Пирролидиновый цикл входит в состав многих природных С( динений, таких, как алкалоиды никотин, кокаин, атропин (см.гл.4) и ;
4. Реакции окисления. Фуран й пиррол очень чувствительны к д< ствию окислителей и окисляются уже кислородом воздуха. При окисз иии происходит разрыв гетероциклического ядра и образуются пот мерные соединения. Однако, пропускание смеси фураиа с воздухом н катализатором V2O$ при температуре 320°С приводит к образован! ангидрида малеиновой кислоты.
фуран
[ О J
VA
При окислении пиррола хромовой кислотой образуется имид ь леииовой кислоты.
1QJ
Н:Сг2О4
Тиофен окисляется с большим трудом.
5. Взаимные превращения фурана, пиррола и тиофена. Реакция пр текает при температуре 450|>С в присутствии катализатора А12О3 (с разд. 3.3.1, А).
Б. Специфические химические свойства пиррола и фуран:
1. Пиррол и его производные. Являясь слабой NH-кислот (рК„ ~ 17.5), пиррол взаимодействует с металлическим калием, безвс ным гидроксидом калия, с металлическим натрием и литием в жидк< аммиаке, с амидами калия и натрия, а также магннйоргаиически! соединениями, образуя соли.
пиррм.пмагпий иолил
пиррол
Н2'
2 841
Входящий в состав солей аииои пиррола (пирролид-аниои) представляет собой довольно устойчивую частицу вследствие делокализации отрицательного заряда по пиррольному ядру.
Соли пиррола являются реакционноспособными веществами и широко применяются в органическом синтезе для введения в молекулу пиррола алкильных и ацильных заместителей. Причем направление реакций алкилирования и ацилирования зависит от температуры. При температуре ниже 0°С образуются соответственно N-ал к ил- и Ы-ацил-пирролы. а при нагревании - - а-алкил- и а-ацилпирролы, например;
М-ицегилпиррол
Наряду с хлорангидридами карбоновых кислот, в реакциях ацилирования можно использовать сложные эфиры. Так, при действии этилформиата на пиррилмагний нодид на холоду образуется N-формилпиррол, при нагревании 2-формилпиррол.
^•^ормнлпиррол
В некоторых реакциях электрофильного замещения пиррол напоминает фенол, а его N-металлические производные — фсноксиды щелочных металлов. В частности, пиррол, как и фенол, вступает в реакцию азосочетания.
+ [c6hsXn]ci- ----------
бепзолдиазоний хлорид
беизолаэо пиррол
НС1
Пирролид натрия формулируется в условиях, реакции Райм 'рцмаиа (см- кН- Ъ с- 294),
иирролил натрии 2-форм или и ррол
а также карбоксидируется действием СО2 аналогично реакции Кол Шмитта для фенола (см. кн. 2, с. 445).
пирролид натрия
натриевая соль ниррол-2-карбоновой кислоты
иирро,т-2-карбоио1 кислота
2. Фура» и его производные. Занимая промежуточное положе между ароматическими соединениями н 1,3-диенами, фуран вступа характерную для сопряженных диенов реакцию Дильса-Альдера кн- 2, с, 55). Так, с малеиновым ангидридом он легко образует соот ствующий продукт присоединения.
фуран
1,2,3.б-тегратдро-3,6-
эпокси фталевый ангадрид
3.3.4. Методы идентификации пиррола, фурана и тиофена
Возможность обнаружения гетероциклических соединений с мощью химических методов ограничена.
Для идентификации пиррола и фурана применяют простой и ступный метод — окрашивание сосновой лучинки. Пары пиррола ot Шивают сосновую лучинку, смоченную соляной кислотой, в крае] Чвет, а фурана — в интепенвдо-зелевый.
Качественной реакцией на тиофен служит индофенииоваа реак] смесь изатина с концентрированной серной кислотой даже от еле тиофена окрашивается в синий цвет.
Наряду с этим, пиррол, фуран и тиофен могут быть идентифицр Еаны по физическим константам (температура кипения, показал преломления и др.) н спектральным характеристикам. Поскольку х Ные гетероциклы являются сопряженными системами, они поглощав Ультрафиолетовой области спектра.
2*
В УФ-спектрах наблюдается высокоинтенсивное поглощение t области 180-210 нм нм: для тиофена — 190, фураиа — 200. пиррола —209) и низкоиитенсивное поглощение (за исключением фурана) в области 230-270 нм.
В ПМР-спектрах пиррола, фурана и тиофена сигналы протонов связи С—Н наблюдаются в интервале 6,2-7,3 м.д.
3.3.5. Важнейшие производные пиррола, фурана и тиофеи а
А. Производные пиррола
Пирролидои-2 является лактамом у-аминомасляной кислоты. В промышленности получают взаимодействием бутиролактона с аммиаком.
бутиролактон
NH?
-н2о
Н,С—СН,
Н2^ О
NH .
пирро.-1ид0Н-2
При конденсации пирролидона-2 с ацетиленом образуется N-ви-нилпирролидои-2, который легко полимеризуется, образуя поливииил-пирролидон (ПВП).
иирролилон-2
+ n НОСН П
N-И ИН ИЛИЙ р рол илон-2 ноливинилпирролидол
Низкомолекулярный ПВП (молекулярная масса 12-13 тыс.) образует коллоидные растворы в воде и применяется для приготовления кровезаменителя «Гемодела», среднемолекулярный ПВП (м. м. 35-40 тыс.) используют я фармацевтической промышленности как связывающее средство в производстве таблеток.
При сополимеризации винилпирролидоиа, акриламида и этилакри-лата получают биорастворимый полимер для глазных лекарственных пленок, который обеспечивает продолжительное действие лекарственных веществ (пролонгирующий эффект).
Пролин (пирролиднн-2-карбоновая кислота) и оксипролип (4-гидрокси-пирролидин-2-карбоновая кне-НО* - лота) — это а-аминокислоты
гетероциклического ряда, в которых общий а-аминокислот-иый фрагмент
—N Н—CI 1(—СООН>— включен в пирролидиновый цикл. Пролин имеет один асимметрический атом углерода и поэтому 36
соон NH
окейпролин
соон NH
иролин
уществует в виде двух оптически активных изомеров и одного раце\ Оксипролин содержит два хиральиых центра, а следовательно, к жег существовать в виде двух пар энантиомеров и двух рацемат< д-Продин и L-оксипролип входят в состав белков. Особенно богат ш коллаген.
Норфин кристаллическое вещество темно-красного цвета. По хи^ ческой структуре представляет собой макр /| \ циклическую сопряженную систему, соси
щую из пиррольного (III), пирролипового
/ 7 Ji и двух изопиррольных (II, IV) ядер, связа
Н jv~7| пых между собой метиновыми группа; |1 Ц =СН—. Порфин является ароматическим с<
I динением. Он имеет плоское строение молеь
лы, содержит замкнутую сопряженную сис III// му с числом л-электронов — 26 (11 л-связег
'---/ 2 пары неподелениых электронов при азот
что отвечает правилу Хюккеля (4п+2 при п=6). Производные порфи получили общее название порфирины. В виде комплексов с металла] порфирины входят в состав таких важных природных соединений, к гемоглобин н хлорофилл.
Гемоглобин это красящее вещество крови; содержащееся в 3f троцитах. Он представляет собой сложный белок — хромопротеь состоящий из белка глобина и окрашенной в красный цвет небелков части — гема. По химической структуре гем является комплексом пс фина с Fe (II). При кислотном гидролизе гемоглобина свободный г легко окисляется на воздухе с образованием гемииа, имеющего ту структуру, что и гем, но содержащего Fe (HI).
гем
Строение гемина установил в 1929 г. Г.Фищер и в этом же году 1 осуществлен синтез гемина.
Гем, координационно связанный с глобином (за счет координаг °нной связи между Fe‘+ и имидазольным фрагментом гистидина бель в°й молекулы), образует гемоглобин.
Гемоглобин в организме выполняет роль переносчика кислорода из легких в ткани. При этом, молекула кислорода обратимо реагирует с гемоглобином с образованием оксигемоглобина согласно схеме:
гемоглобин
окси гемоглобин
Некоторые вещества, в частности, оксид углерода (II) и соли синильной кислоты, образуют с гемоглобином более стабильные комплексы, чем кислород, и тем самым блокируют действие гемоглобина. Такие соединения относятся к дыхательным ядам.
Частично гидрированный порфиновый цикл, координационно связанный с магнием, входит в состав зеленого пигмента растений - хлорофилла. Из растений выделены хлорофилл а и хлорофилл 0.
Хлорофиллы содержат 3 асимметрических атома углерода и поэтому обладают оптической активностью. Синтез хлорофиллов был впервые осуществлен в 1960 г. Р.Вудвордом. Хлорофиллы играют важную роль в процессе фотосинтеза, превращая световую энергию солнечных лучей в энергию химических связей.
Витамин В|2 (цианокобаламин). Витамин В[2 был впервые выделен из печени теплокровных животных американским химиком К.Фол-керсом в 1948 г. Однако только в 1956 г. английский химик Д.Кроуфут-38
Ходжкин установила с помощью рентгеноструктурного анализа строение. В основе структуры витамина В[2 лежит макроцикл, сое щий из четырех частично гидрированных пиррольных ядер, в кото атомы азота образуют координационный комплекс с атомом коба/ и цианид-ионом.
О
Н3С
ОН
о
h2n с сн2 cjh2 h2n с с
2 II о Н3С
и г 11
Нзс СН2 C NI сн, CF
н
н сн
H,N С
2 II о
н
CN
NI
СН
сн н
Снз СН, СН
о
Н2С
о=р o chch2hnc!:ch2ch2 ОН о" СН, о вшами!!
о
в
Вследствие наличия цианогрупиы, связанной с кобальтом, витамин называют также нианокобаламином.
В настоящее время витамин В)3 получают в промышленном ? штабе микробиологически. Ои применяется в медицинской пракз для лечения анемий, заболеваний нервной системы и печени,
Б. Производные фурана
Важнейшим производным фураиа является фурфурол (фура карбальдегид) — бесцветная или слегка желтов! а маслянистая жидкость (т. кип. 162 °C), имеющая г «О ятный запах свежеиспеченного ржаного хлеба. Bi у х вые был выделен из отрубей. От лат, furfur «отру н получил свое название,
В промышленности фурфурол получают в б< Ших количествах кислотным гидролизом полисахаридов пеитоза] содержащихся в сельскохозяйственных отходах (соломе, шелухе i солнечника, кукурузных кочерыжках, хлопковых коробочках и др.).
Н Н .-Л /но^_^°п \ н/ / \ \Н
С
н еде онЛ\
и--1 н
фурфурол
альдоле) 11 оза
По химическим свойствам фурфурол во многом сходен с ароматическими альдегидами, в частности, с бензальдегидом. Как ароматический альдегид фурфурол вступает в реакцию Канниццаро,—
фурфурол фурфуриловым Спирт иазриевня соль
иирослилевой кислоты с цианидом калия претерпевает конденсацию типа бензоиновой;
фурфурол фуроин
с аммиаком образует гидрофурфурамид (аналог гидробензамида).
Фурфурол как альдегид окисляется аммиачным раствором оксида серебра, образуя пирослизевую кислоту, восстанавливается в фурфуриловый спирт, присоединяет гидросульфит натрия, с гидроксиламином образует оксим, с фенил гидразином — фенилгидразон и др.
// \\ [ X Х'"сн2он * \ О фурфуриловый А спирт Ь / V c6h5nhnh2 [! ° \ * < .фурфурол NaHS(>? NH,OH 0 XSO3Na ‘ Н:° т.чросульфитиос производное фурфурола ^0 + 4NH3+2 Ag+H,O и ОН н и росли левая кислота zVcH=NNHC6H, Э фепилгидразои фурф^эола /^CH=N-OH оксим фурфурола
40
Кроме реакций по альдегидной группе, для фурфурола характерны реакции по фурановому ядру. Фурфурол легко вступает в реакции Sp, При этом наиболее реакционноспособно положение 5. Вследствие элек-троиоакиепторного влияния альдегидной группы, приводящего к понижению электронной плотности на углеродных атомах фуранового цикла, фурфурол менее ацидофобен, чем фуран.
Нитрование фурфурола проводят концентрированной азотной кислотой в среде уксусного ангидрида. В процессе реакции получают 5-нн-трофурфуролдиацстат, который при гидролизе в присутствии разведенной H2SO4 образует 5-нитрофурфурол.
K.HNO
О N
4. ?снзсоК° 2 н
О
О—C-CHS нон. н+
СН ~ *"O,N
I О 2
I И.
О—С—СН3
фурфурол 5-нитро фурфуролди ацетат 5-нитрофурфурол
5-Нитрофурфурол является исходным веществом для синтеза ряда лекарственных препаратов. Так, при взаимодействии 5-нитрофурфуро-ла с семикарбазидом образуется семикарбазои 5-нитрофурфурола, который применяется в медицине под названием фу ранил ни.
5-нитрофурфурол
H,N—NH -C-NH; семнкарбазид
А х 11
°2N () CH=N-NH-C-NH2
семикарбазои 5-нитрофурфурола, фурацилин
Представителями группы лекарственных препаратов нитрофурано-вого ряда являются также фурадонни и фуразолидои.
фурадолин
I -[ N-(5-i 1Игрофу рфурилидеи) амияо]гидантонн
О
фуразолидои 3-[?4-(5-11итрофурфурилиден) амино] оксазол идо п-2
Препараты нитрофуранового ряда обладают высокой антибактериальной активностью. Оии находят широкое применение в медицине Для лечения гнойных и воспалительных процессов. Особенно ценным свойством этих препаратов является их способность в ряде случаев проявлять эффект против форм возбудителей, устойчивых к сульфаниламидам и антибиотикам.
41
В. Производные тиофена
Биотин (витамин Н). Гетероциклическая часть молекулы биотинМ'
состоит из полностью гидрированных тиофенового и имидазольного колец, а боковая цепь представлена остатком валериановой кислоты. Биотии впервые выделен в 1935 г. из яичного желтка, при этом для получения 1 мг вещества потребовалось 225 кг сухого яично
го желтка.
Особенно богаты биотином почки, печень, горох, бобы, картофель. Биотин входит в состав активного центра ферментов, принимающих участие в синтезе высщих жирных кислот, белков, нуклеиновых кислот и др. При недостатке биотипа в организме развиваются воспалительные заболевания кожи (дерматиты), сопровождающиеся выпадением волос и поражением ногтей.
3.4. ИНДОЛ
Молекула нндола (беизо[ А) пиррола) представляет собой конденси-
рованную гетероциклическую систему, состоящую из пиррольного и бензольного колец. Нумерацию атомов в индоле начинают с гетероатома, атомы углерода в пиррольном цикле обозначают также буквами аир.
3.4.1. Способы получения
Индол содержится в небольших количествах (3-5%) в каменноугольной смоле, откуда может быть выделен в чистом виде.
Из синтетических методов получения индола и его гомологов наиболее важными являются следующие:
1. Циклизация N-формил-о-толуидина. Реакция протекает в присутствии сильного основания (третичного бутилоксида калия нли амида натрия) и относится к реакциям конденсации кротонового типа.
N-форм ИЛ-О-1 п лу ИДИ 11
NaNH,
- н2о
ИЛД0.1
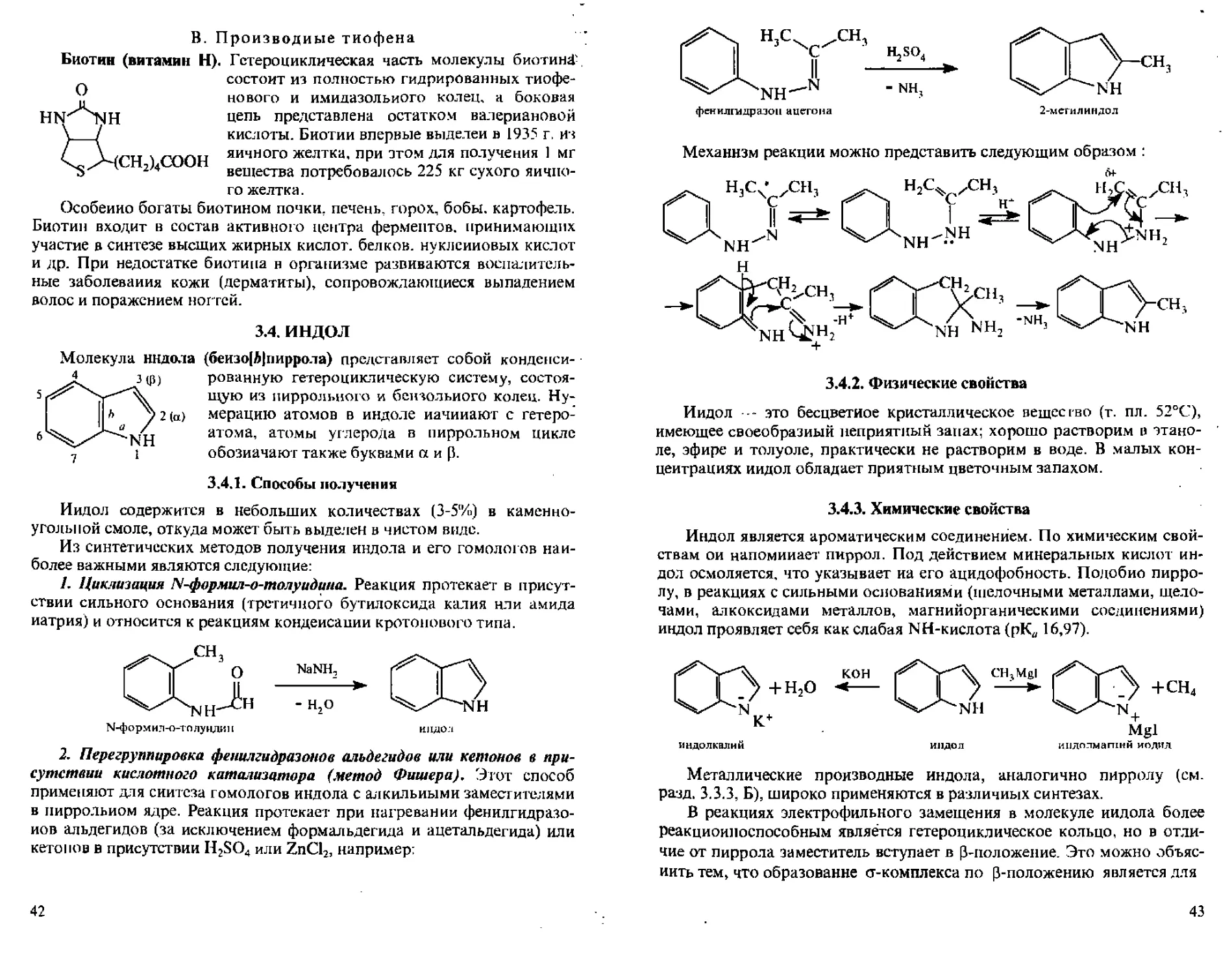
2. Перегруппировка фенилгидразонов альдегидов или кетонов в присутствии кислотного катализатора (метод Фишера). Этот способ применяют для синтеза гомологов индола с алкильными заместителями в пиррольном ядре. Реакция протекает при нагревании фен и л гидразонов альдегидов (за исключением формальдегида и ацетальдегида) или кетонов в присутствии H2SO4 или ZnCI2, например;
42
фенилгадразоц ацетона
H,SO4
- NH?
Механизм реакции можно представить следующим образом :
3.4.2. Физические свойства
Иидол --- это бесцветное кристаллическое вещество (т. пл. 52°С), имеющее своеобразный неприятный запах; хорошо растворим в этаноле, эфире и толуоле, практически не растворим в воде. В малых концентрациях иидол обладает приятным цветочным запахом.
3.4.3. Химические свойства
Индол является ароматическим соединением. По химическим свойствам он напоминает пиррол. Под действием минеральных кислот индол осмоляется, что указывает иа его ацидофобность. Подобно пирролу, в реакциях с сильными основаниями (щелочными металлами, щелочами, алкоксидами металлов, магнийорганическими соединениями) индол проявляет себя как слабая NH-кислота (рКа 16,97).
ИНДОЛ
Металлические производные индола, аналогично пирролу (см. разд, 3.3.3, Б), широко применяются в различных синтезах.
В реакциях электрофильного замещения в молекуле иидола более реакционноспособным является гетероциклическое кольцо, но в отличие от пиррола заместитель вступает в p-положение. Это можно объяснить тем, что образование сг-комплекса по p-положению является для
43
индола более выгодным процессом (в сравнении с a-положением), поскольку в этом случае положительный заряд может быть делокализован без нарушения ароматической системы бензольного ядра.
При атаке электрофильным реагентом сс-положеиия образуется о-комплекс, в котором делокализация положительного заряда может быть осуществлена только с нарушением ароматической системы бензольного кольпа, что энергетически для молекулы не выгодно.
Если p-положение занято, то электрофильное замещение протекает по а-положению. Так, при нитровании индола беи зоил нитратом, сульфировании пиридинсульфотриоксидом, галогенировании хлористым сульфурилом и азосочетании образуются соответствующие р-замещенные.
З-бензолазоинаол
При восстановлении иидола водородом в присутствии платинового катализатора образуется 2,3-дигидроиндол
Pt
2.3-дигилроннлол
44
3.4.4. Важнейшие производные индола
Индоксил (3-гидрокси и идол; 3-оксоиидолин). Желтое кристаллическое вещество с сильным феиольиым запахом; температура плавления 85 °C, растворяется в воде, спиртах, ацетоне, эфире и бензоле. В растворах индоксил существует в двух таутомерных формах — кетонной и енольной (кето-еноль-
яая таутомерия), в кристаллическом состоянии находится в кетоформе (3-оксоиндолии). В промышленности индоксил получают взаимодействием анилина с натриевой солью хлоруксусной кислоты. Образующаяся в процессе реакции натриевая соль N-фенил уксусной кислоты при нагревании (180-200 °C) с амидом натрия превращается в индоксил.
анилин
натриевая соль
N-фсниламиноуксусной кислоты
ИНДОКСИЛ
Индоксил легко вступает в реакции, характерные для карбонильных соединений и фенолов. В щелочной среде индоксил легко окисляется кислородом воздуха, образуя синий краситель нндиго.
1OJ
Индиго. Темно-сииее с
медным отливом кристаллическое вещество; температура плавления 390-392 °C (сразл.), растворяется в хлороформе, нитробензоле, анилине, ледяной уксусной кислоте, не растворяется в воде, спиртах, эфире.
Иидиго один из самых древних органических красителей, отличающийся яркой окраской и высокой светоустойчи-
востью. Оно было известно еще древним египтянам и народам Индии, которые получали его из тропических растений рода indigofera. Синтетическим путем индиго впервые было получено в 1896 году. В настоящее время наибольшее распространение получил способ, основанный На взаимодействии анилина с натриевой солью хлоруксуснон кислоты с
45
последующим окислением образовавшегося индоксила кислородом, воздуха (см. схему получения индиго, с. 45).
Молекула индиго имеет мгрсгнс-етроение и образует между группами С—О и NH две внутримолекулярные водородные связи.
В присутствии восстановителей (глюкоза, дитионат натрия Na2S2O4) синее индиго легко восстанавливается с образованием бесцветного лейкооснования — белого индиго, которое, в отличие от синего, хорошо растворимо в воде. На воздухе очень легко протекает обратный процесс —белое индиго окисляется до синего.
Это свойство индиго используют при крашении тканей. Поскольку синее индиго не растворяется в воде, его вначале восстанавливают Na2S2O4 в белое иидиго (растворимую форму) и этим раствором обрабатывают ткань. Затем иа воздухе происходит окисление белого иидиго в синее и ткань при этом окрашивается в синий цвет.
Метод крашения, при котором краситель образуется на ткани нз бесцветного соединения, называют кубовым крашением, а само индиго относится к кубовым красителям.
При сульфировании индиго концентрированной серной кислотой образуется индиго-5,5'-дисульфокислота, динатрисвая соль которой известна под названием индигокармин.
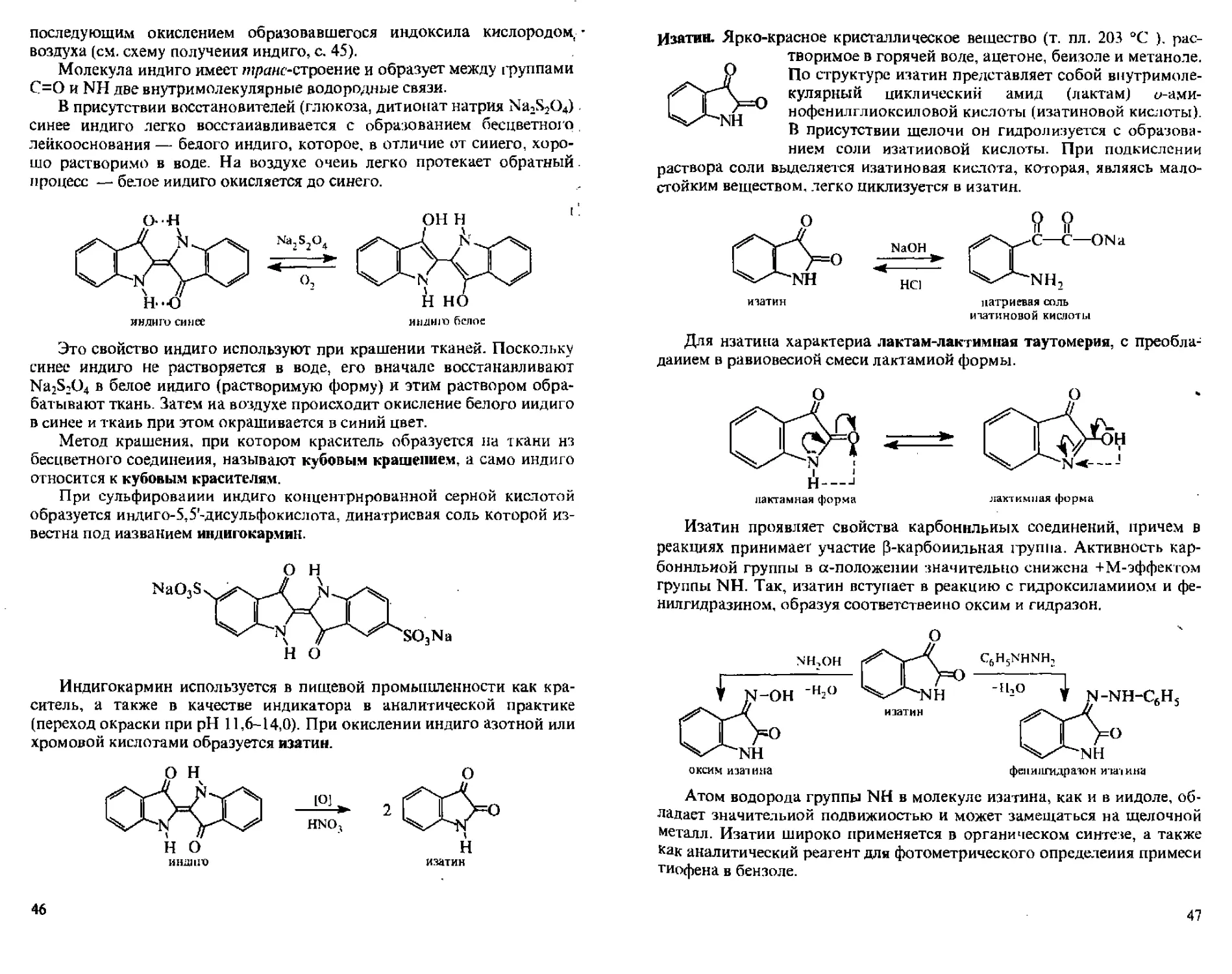
Индигокармин используется в пищевой промышленности как краситель, а также в качестве индикатора в аналитической практике (переход окраски при pH 11,6-14,0). При окислении индиго азотной или хромовой кислотами образуется изатин.
О Н
н о
индию
1OJ
HNOj
изатин
46
Изатин. Ярко-красное кристаллическое вещество (т. пл. 203 °C ), растворимое в горячей воде, ацетоне, бензоле и метаноле. аР По структуре изатин представляет собой виутримоле-\ о кулярный циклический амид (лактам) у-ами-47 нофе нил гл и о кс иловой кислоты (изатиновой кислоты). В присутствии щелочи он гидролизуется с образованием соли изатиновой кислоты. При подкислении раствора соли выделяется изатиновая кислота, которая, являясь малостойким веществом, легко циклизуется в изатин.
NaOH -----►
НС)
итатин натриевая соль
изатиновой кислоты
Для нзатина характерна лактам-лактимная таутомерия, с преобладанием в равновесной смеси лактамной формы.
лактамная форма
Изатин проявляет свойства карбонильных соединений, причем в реакциях принимаег участие р-карбоиильная группа. Активность карбонильной группы в «-положении значительно снижена +М-эффектом группы NH. Так, изатин вступает в реакцию с гидроксил амином и фенил гид разин ом, образуя соответственно оксим и гидразон.
Атом водорода группы NH в молекуле изатина, как и в индоле, обладает значительной подвижностью и может замещаться на щелочной металл. Изатин широко применяется в органическом синтезе, а также как аналитический реагент для фотометрического определения примеси тиофена в бензоле.
47
Триптофан [2-амиио-3-(р-индолил)пропионовая кислота]. Кристалл и-
.-^'Н-СООН nh2
ческое вещество (т. ил. 289 °C), растворимое в горячей воде и спирте, нерастворимое в хлороформе. Триптофан содержит один асимметрический атом углерода и существует в виде двух оптически активных энантиомеров и одного рацемата. Т-Триптофан
является незаменимой а-аминокислотой. входящей в состав белков.
Серотонин [5-гидрокси-3-(0-ямииоэтил)индол]. Кристаллическое вещес-Cli -CH -NH тво (т‘ и51, 207-212 °C), растворимое в НО У 2 2 2 воде, нерастворимое в органических
растворителях. Серотонин является биогенным амином, который играсг важную роль в процессах жизнедеятельности организма. Ои принимает участие в передаче нервных импульсов, вызывает сокращение гладкой мускулатуры внутренних органов и сужение кровеносных сосудов, повышает стойкость капилляров и количество тромбоцитов в крови. В организме образуется из триптофана. С нарушением обмена серотонина связывают появление симптомов шизофрении. В виде соли с адипиновой кислотой серотонин применяется в медицине как антигеморрагичсское средство.
£-Индол нлуксу спая кислота (гетсроауксин). Кристаллическое вещество СИ -СООН С температурой плавления 168 169 °C, рас-Р/ - творимое в воде и этиловом спирте. Гетеро-
р |Tn< ауксин является продуктом окислительного
дезаминирования триптофана. Он оказывает стимулирующее влияние на рост растений (гормон роста) и с этой целью широко применяется в сельском хозяйстве. На основе р-ин дол и л уксусной кислоты создан лекарственный препарат индометацин, обладающий сильным противовоспалительным действием.
2-метнл-5-метокси-1-0г-хпорбензоил}~И11ДОли.1-3-уксуспая кислота
3.5. ПЯТИЧЛЕННЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ С ДВУМЯ ГЕТЕРОАТОМАМИ
Важнейшими представителями большого класса пятичленпых гетероциклов с двумя гетероатомами являются пиразол, имидазол, тиазол, оксазол и изоксазол.
48
пиразол имидазол гиапол оксазол
изоксазол
Поскольку в этих соединениях по крайней мере одшг из двух гете-роатомов является азотом, они получили общее название азолы.
Все приведенные гетероциклы обладают ароматичностью (см. разд. 3.1). Неподеленная пара электронов атома азота пиридинового типа не участвует в образовании ароматического секстета и придает гетероциклам основные свойства. Кроме того, атом азота пиридинового типа, обладая большей электроотрицательностью, чем атом углерода, уменьшает л-электрониую плотность на углеродных атомах цикла и тем самым снижает по сравнению с фураиом, пирролом и тиофеном реакционную способность указанных гетероциклов в реакциях электрофильного замещения.
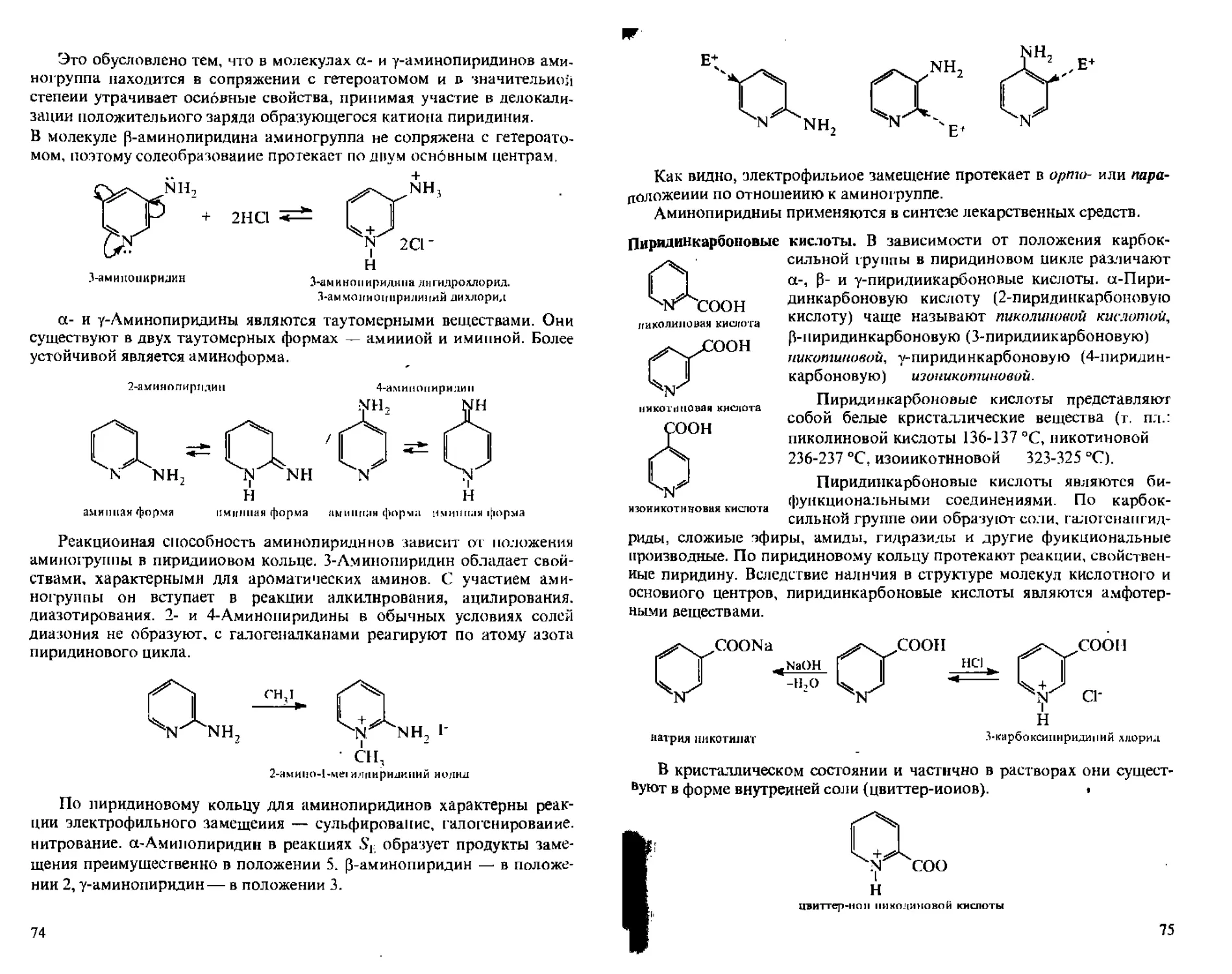
3.5.1. Пиразол
А. Способы получения
В природе пиразол (1,2-диазол) и его производные ие встречаются. Из синтетических способов получения наиболее важными являются :
У. Присоединение диазоалканов к ацетиленам. Реакцию применяют для получения пиразола и его производных. Пиразол по этой реакции получают присоединением диазометана к ацетилену.
СН
СН
ацетилен
диазомегап
2. Взаимодействие гидразина, алкил- или арилгидразинов с 1,3-дикарбонильными соединениями. Этот способ чаще используют для получения гомологов пиразола. Так, при взаимодействии гидразина с ацетилаце-тоном образуется 3,5-диметилпиразол
ацетилацегоп, пен та л дион-2.4
.СН, СН2"С
+ nh2-nh2
о
ZCH3 сн—cz
Н с A 'N XNH
3,5-дим еги л п и р азо л
н2о
49
Б. физические свойства
Пиразол бесцветное кристаллическое вещество со слабым запа-хом пиридина; температура плавления 70 °C, температура кипения 187 °C, хорошо растворяется в воде, этаноле, эфире.
В неполярных растворителях существует в форме димеров и тримеров за счет образования межмолекулярных водородных связей.
три мер пиразоли
В. Химические свойства
1. Кислотность и основность. Кислотио-осиовиые свойства пиразола обусловлены наличием в его структуре атомов азота пиррольного и пиридинового типов (см. разд. 3.1).
За счет атома азота пиридинового типа пиразол проявляет основные свойства (рКвн+ 2,53), за счет азота пиррольного типа слабые кислотные свойства (рКд * 14). Следовательно, пиразол является амфотерным соединением и способен вступать в реакции как с минеральными кислотами, так и со щелочами, образуя при этом соли, например:
I Н
циразолнн хлорид
сг
НС
пиразол
КОИ -----►
" И2°
Соли пиразола довольно устойчивые соединения. Их стабильность обусловлена делокализацией положительного заряда в катионе пиразолия, либо отрицательного заряда в пиразолид-анионе между всеми атомами цикла.
50
пиразодиД-аниои
пиразол
"Н
пиразолий-катион
Наличие в молекуле пиразола подвижного атома водорода КН-группы и основного центра атома азота пиридинового типа является причиной появления прототропной, или так называемой язольноя, таутомерии. Прототропная таутомерия пиразола и его гомологов обусловлена перемещением атома водорода NH-группы к атому азота пиридинового типа.
В результате таутомерных превращений положения 3 и 5 в молекуле пиразола равноценны. Так, З-метиллиразол и 5-метилпиразол являются таутомерными формами одного и того же соединения.
I Н
При этом миграция атома водорода происходит настолько быстро, что выделить индивидуальные таутомеры не представляется возможным. Поэтому в названиях таких соединений наряду с цифрой, указывающей положение заместителя, в скобках приводится цифра, обозначающая возможность отсчета от другого, химически идентичного азота. Так, приведенное выше соединение называют 3(5)-метилпиразол.
2. Реакции с электрофильными реагентами. В силу электро но акцепторного влияния атома азота пиридинового типа реакционная способность пиразола с электрофильными реагентами понижена. При этом направление реакций зависит от природы атакующего реагента и условий их проведения.
Алкилирование и ацилирование пиразола протекает обычно с образованием продуктов N-замещейия. Так, при взаимодействии пиразола с иодметаном в нейтральной или щелочной среде образуется N-метилпиразол. Реакция идет по схеме:
51
пиразол
N-метилпираэолкй иодид
-Hl
N-метилпиразол
Вначале электрофильный реагент CHJ атакует атом азота пиридинового типа молекулы пиразола с образованием соли — N-метилпи-разолий иодида, которая отщепляет HI, образуя конечный продукт реакции. Данная реакция протекает с переносом реакционного центра.
Аналогично происходит ацилирование пиразола.
Реакции пиразола с сильными электрофильными реагентами (нитрование, сульфирование, галогенирование) происходят с образованием продуктов замещения по атому углерода в положении 4 (наиболее удаленное от атомов азота положение). Поскольку пиразол не обладает ацидофобными свойствами, нитрование и сульфирование его проводят концентрированными азотной и серной кислотами соответственно. Обе реакции протекают через стадию образования неактивного катиона пиразолня.
пиразол
4-х;юрпира:ю.т
3. Реакции восстановления. При восстановлении пиразола водородом в момент выделения (C2H5OH+Na) образуется частично гидрированный продукт — пиразолии. Гидрирование в присутствии катализатора приводит к образованию полностью восстановленного производного — пирвзол идина.
лира зояиди».
тетра гидро п ира зол
2Н2
Pi
2Н
С.ЩОН+ Na
пира золин-2, 3,4-дигидроп и разол
52
Пиразолии и пиразолидин являются гораздо более сильными основаниями, чем пиразол. Они обладают свойствами вторичных алифати-
ческих аминов.
!
I, Г. Важнейшие производные пиразола
• Пиразолон-5 (2-пиразолинон-5). Это бесцветное кристаллическое вещес-
тво (т. пл. 165 °C), хорошо растворимое в воде и этиловом спирте, плохо растворяется в эфире и толуоле. Пиразолон-5 является таутомерным веществом и может существовать в СН2-, ОН- и NH- форме
(СН, - форма) пира:юлои-5
(ОН - форма) Зчид рокси пиразол
НС^СН
(NH . форма) пиразолон-3
В указанном равновесии существенно преобладает СН2-форма, поэтому в названии соединения отдают предпочтение пиразолону-5.
Ядро пиразолона-5 входит в структуру ряда лекарственных препаратов, в частности, антипирина, амидопирина н анальгина. В качестве исходного вещества для получения лекарственных препаратов пиразолонового ряда используют 3-метил-1-фенилпиразолон-5. Это соединение было впервые синтезировано в 1883 г. немецким химиком-органиком Л. Кнорром из ацетоуксусного эфира и фени лги др азина :
СН3
сн2 о
£ h2nhn-c6h^ o/z \)С,Н3 -н?°
ацетоуксусный эфир
СН, / 3 сн,-с
О=С ^4
\ /
О HN ебт Чс6н
ад О 3
З-мстил-1-фенил-пиразо лои-5
3-Метнл-1-фепилпиразолон-5, подобно незамещенному пиразолону-5, ' может существовать в трех таутомерных формах:
ад
СН, - форма
. ОН - форма
NH - форма
Установлено, что в неполярных растворителях преобладает СН2-форма, а в водных растворах NH-форма. При взаимодействии 52
З-метил-1 -фенил пиразол о иа- 5 (в NH-форме) с иодистым метилом образуется 2,3-диметил-1-фенилпиразолон-5 (аитипирии).
3-мегил-1-феннппираэолон-5
СН,1
-HI
сн,
СН
с6н5
антипирин;
2,3-димегил-1-феншширазоло11-£
Антипирин бесцветное кристаллическое вещество (т. пл. 114 °C) с горьковатым вкусом, хорошо растворимое в воде. Применяется в медицине как жаропонижающее и болеутоляющее средство.
В молекуле антипирина атом водорода при углероде в четвертом положении пиразолинового цикла обладает значительной подвижностью. При действии азотистой кислоты он легко замещается иа нитрозогруппу. Последующее восстановление образовавшегося 4-нитрозо-антипирина дает 4-аминоантипирин — исходный продукт в синтезе амидопирина и анальгина.
ннтипирип 4-имтрозо антипирин 4-аминаантнпирин
Ниже приведены схемы синтеза амидопирина и анальгина.
Амидопирин получают метилированием 4-амииоантипирина:
с6н5
4-аминоантинирип
2СН?
-2HI
3
амидопирин;
2.3-димегил-4-диметиламиио-1-фепилпиразолон-5
Анальгин синтезируют по схеме:
H2N4 СН3 о*=0'сн, с6н5 4-аминоантнпирин
54
c6h54:h=n cHj
сйн5чн=о (CH3)2SO
н2° ^CHl *
1
сн
О 3 J
4-бенэилмденаминоантипирин i,
. Ъ
I
Н2С=О+ NallSO,
4-метиламиноантипирии
NaO3S-CH2-N
-и, О
,сн3
СН
анальгин:
2.3-диметил-4-метилам ино-l -феиилпи-раэолон-5-Х-мстансульфонат натрия
Амидопирин и анальгин применяются в медицине как жаропонижающие и болеутоляющие средства, причем у амидопирина сильнее выражено жаропонижающее действие, у анальгина -- болеутоляющее.
3.5.2. Имидазол
Q Имидазол (1,3 -ди а зол) является изомером пиразола.
Он представляет собой ароматическую систему, в которой атомы азота (пиррольного и пиридинового типа) нахо-дятся в положении 1,3.
А. Способы получения
Имидазол и его производные чаще всего получают взаимодействие ем 1,2-дикардонилъных соединений, аммиака и альдегидов^
Имидазол синтезируют из глиоксаля, аммиака и формальдегида :
нс—N
// \\
-► НС’ СН + 3 Н,0
Y н
глиоксаль формальдетд
имидапол
з
Б. Физические свойства
Имидазол бесцветное кристаллическое вещество (т. пл. 90 °C, т. кип. 256 °C), хорошо растворимое в воде, этаноле, эфире. В неполярных растворителях имидазол образует межмолекулярные водородные связи, причем, в отличие от пиразола, ассоциаты имеют линейную структуру:
55.
В, Химические свойства
По реакционной способности имидазол имеет много общего с пиразолом. Подобно пиразолу, ои является амфотерным соединением, Проявляя за счет атома азота пиррольного типа слабые кислотные свойства, а азота пиридинового типа - - оснбвные.
ациоп имидазол катион
имндазолнд имидазолин
Одиако, имидазол по сравнению с пиразолом является более сильным основанием (pKRU+ 7,03).
Аналогично пиразолу, для имидазола и его гомологов характерна прототропная (азольная) таутомерия, в результате которой положения 4 и 5 имидазольного цикла являются равноценными :
'н>—1 ।
Следует отметить, что электроноакцепторные заместители (NO2, SO3H, Cl и др.) смешают таутомерное равновесие в сторону 4-замещенного изомера.
Подобно пиразолу ведет себя имидазол и в реакциях с электрофильными реагентами.
Так, реакции алкилирования и ацилирования протекают с участием гетеро атомов.
имидазол
N-метилимида зелий бромид
zCH3
5
N-mci илимидазол
Нитрование и сульфирование идет преимущественно по положение ям 4 и 5 имидазольного цикла. Эти реакции протекают с большим трудом вследствие образования в кислой среде малоактивного катиона имидазолин.
S6
имидазол
имидазол-4-су.чифо кислота
г С бромом в воде,, хлороформе или эфире и иодом в водном растворе щелочи имидазол легко образует 2,4,5-тригалогенпроизводиые
имидазол
Вг
2,4.5-трибромимидазол
ЗНВг
Имидазольный цикл довольно устойчив к действию окислителей '(кислорода, перманганата калия и др.) и восстановителей, Однако, под действием пероксидов происходит разрушение цикла с образованием Ъксамида.
ОО^ ZNH, ;
+ 2Н,О, -------*• V + СО, + 2НгО
Н О NHn
! днями,! щавелевой кисло 1ы.
оксамид
£ Г. Важнейшие производные имидазола
Среди производных имидазола важное значение имеют такие природные соединения как алкалоид пилокарпин, а-амииокислота гисти-’дин и биогенный амин — гистамин.
^Гнстидии [а"амино-р-(нмидазолил-4)-пропиоиовая кислота),
, * В /.-конфигурации входит в состав многих
‘ CHjCH-COOH белков.
\ V* \ nh Хлороводородная соль гистидина применяет-
.. С z ' ся в меДиииие для лечения гепатитов, язвен-
’ NH ной болезни желудка и двенадцатиперстной
кишки.
При ферментативном декарбоксилировании гистидин превращается в гистамин.
гистидин
i hci амн г ।
Гистами и |4-(2,-амииоэтил)ймидазол|. Является биогенным амином, принимающим участие в регуляции жизненнова-СН-гСНп-ЬЛ-Ц жпых функций организма. Обычно гистамин на-а ходится в организме в виде неактивных лабиль-
ных комплексов с белками. При некоторых па-iMi тологических состояниях (ожоги, отморожения,
попадание в организм химических веществ, в том числе и лекарственных препаратов, аллергические заболевания и др.) гистамин высвобождается в свободном виде. Свободный гистамин обладает высокой активностью: вызывает спазм гладкой мускулатуры, расширяет капилляры и увеличивает их проницаемость, усиливает секрецию желудочного сока.
3.5.3. Бензимидазол
Бензимидазол представляет собой конденсированную гетероциклическую систему, состоящую из бензольного и имидазольного колец.
Бензимидазол эго бесцветное кристаллическое вещество (т. пл. 170 °C), хорошо растворимое в воде,
этаноле и других полярных растворителях.
Бензимидазол и его производные получают при нагревании с-фе-нилендиамина (1,2-диаминобензола) с карбоновыми кислотами. Для; получения самого бензимидазола используют муравьиную кислоту.
По химическим свойствам бензимидазол во многом напоминает имидазол. В частности, для него характерны амфотерные свойства, прототропная таутомерия, реакции алкилирования с участием атомов азота. Однако в реакционной способности бензимидазола и имидазола есть и различия. Конденсированное бензольное кольцо приводит к снижению основности бензимидазола (pKWn+5,53) и повышению его кислотности (рК„ 13,2) по сравнению с имидазолом (см, разд. 3.5.2, В). Реакции электрофильного замещения (нитрование, сульфирование) для бензимидазола протекают с большим трудом, и. как правило, в положениях 5 или 6 бензольного кольца.
58
Бензимидазольный цикл входит в состав некоторых природных веществ (витамин В|2), а также синтетических лекарственных препаратов, например дибазола:
дибазол. Г (Л и ft
2-бензилбензимидаэола гидрохлорил
Дибазол обладает сосудорасширяющим, спазмолитическим и гипотензивным действием. Он широко применяется в медицинской практике при спазмах кровеносных сосудов и гладкой мускулатуры внутренних органов.
3.5.4. Тиазол
По химическому строению тиазол (1,3-тиазол) можно л рассматривать как аналог тиофена, в котором группа СН
XI/ в положении 3 замещена на атом азота.
Таутомерные формы
H2I^
с-н
II
.с ч_.. нх fci
хлоруксуспый . тиоформамид апьдешд
-Н^О;
V -HCI
тиазол
А. Способы получения
В природе тиазол в свободном состоянии не найден, но его ядро входит в состав многих природных соединений (витамин В], пенициллины и др.).
Одним из важнейших методов синтеза тиазола и его производных является взаимодействие сс-галогеиозамешенных карбонильных .соединений с амидами тиокислот (синтез Ганча). Сам тиазол получают из хлоруксусиого альдегида и тиоформамида, причем альдегид вступает в реакцию в енольной, а тиоформамид — в тиольной таутомерных формах.
Б. Физические свойства
Тиазол бесцветная жидкость с неприятным запахом (г. кип. 117 °C), хорошо растворимая в воде и органических растворителях.
59
В. Химические свойства
Тиазол является слабым основанием (рКВц+ 2,53). С минеральными кислотами он образует соли тиазол ия, при действии галогекалканов идет алкилирование по атому азота с образованием четвертичных N-алкилтиазо лиевых солей.
(иажлий .хлорид
Наличие в молекуле тиазола атома азота пиридинового типа приводит к снижению электронной плотности в тиазольном кольце, что. с одной стороны, затрудняет протекание реакций электрофильного замещения (нитрование, сульфирование), а с другой создает условия для нуклеофильного замещения. Электрофильное замещение происходит по положению 5, а нуклеофильное -- по положению 2 тиазольного ядра. Так, при нагревании 4~ метилтиазола с амидом натрия образуется 2~амино-4-метилтиазол.
2-амнно-4-метил1 иатол
Тиазол достаточно устойчив к действию восстановителей. В присутствии пероксикислот тиазолы окисляются с образованием N-окси-дов, например:
2,5-днмеппггиазол
СНз-С
о-он перо кси уксусная кислота
N-оксид
2.5-ди метилтиазола
// '
СН3-С f \ он
Г. Важнейшие производные тиазола 2-Аминотиазол — бесцветное кристаллическое вещество (т. пл. 90 °C), хорошо растворимое в воде, этаноле, эфире и хлорофор-Ц ме. 2-Амннотиазол обладает свойствами ароматических
аминов. С участием аминогруппы он вступает в реакции 2 ацилирования, диазотирования, образует продукты конденсации с альдегидами. Одиако, в отличие от ароматических аминов, реакция алкилирования 2-аминотиазола протекает по атому азота тиазольного кольца:
60
и м сн, сн,
СЧ <л«. - <4
2-амиио-З-метиптиаэолнйиолид 2-имино'3-ые1и,тгиааолип
2-Аминотиазол широко применяется в производстве лекарственных средств. Производными 2-аминотиазола являются сульфаниламидные препараты норсульфазол и фталазол, обладающие антибактериальным действием.
Пенициллины группа антибиотиков, продуцируемых различными видами плесневых грибов PenicilHum, и их аналоги полусинтетиче-ского производства.
В основе структуры пенициллинов лежит конденсированная гетероциклическая система, состоящая из тиазолидииового и Р-лактамного колец. Общая формула пенициллинов имеет следующий вид.'
СООН
г--------------, Н С
' тиазолидино вое1 I КОЛЫЮ I
___। 6-лакгамное i
О f КОлыю I
R
В медицинской практике иашли широкое применение такие природные пенициллины как бензилпенициллин (R = С6Н3СН2-), фепокси-метилпенициллин (R = С6Н3ОСН2 -) и др., а также полусинтетические пенициллины — ампициллин [R = СбН5 - CH(NH2)-J, оксациллин
и др. Препараты группы пенициллина являются ценными антимикробными средствами.
61
3.5.5. Оксазол
кт Оксазол (1,3-оксазол) - бесцветная жидкость с т. кип. 0 69 °C. Хорошо смешивается с этанолом и эфиром.
Оксазолы являются ароматическими соединениями.
О Одиако, в результате злектроиоакцепторного влияния атома азота они с трудом вступают в реакции электрофильного замещения. Эти реакции могут протекать в положениях 4 и 5. если оксазольный цикл активирован электроиодоиорными заместителями, такими, как амино- или гидроксигруппа. За счет свободной пары электронов атома азота пиридинового типа оксазолы проявляют слабые основные свойства. Для синтеза оксазолов широко применяют метод пиклодегидратании а-ациламинокетонов в присутствии минеральных кислот, чаще H2SO4:
Cllj-NH
J
н5с^Ч0 (/~~сн.
HC-N
H;so< Ц *
-н,()
а-ацетидамииоирпианан
2.5-ди меги ло ксазол
Среди производных окса: юл а известны вещества, обладающие жаропонижающим, анальгетическим, антибактериальным и снотворным действием.
3.5.6. Изоксазол
I---, Изоксазол (1,2-оксазол) бесцветная жидкость (т. кип,
/С4 р 95 °C), ограниченно растворимая в воде, хорошо растяори-\17^ мая в органических растворителях. Изоксазол обладает
О ароматическими свойствами. Реакции электрофильного замещения (галогенирование, нитрование, сульфирование) протекают преимущественно по положению 4. которое наименее подвержено влиянию гетероатомов. Аналогично оксазолу, изоксазол является слабым основанием (рКВ11+ 1,3).
Общим методом получения изоксазола и его производных является реакция 1,3-дикарбоиильных соединений с гндроксиламииом:
СН / СН,~С
н3с с хо \\ о
ашп илаиегоп
СН;
3 /
нс—с
// \\
.ндГ H3C^CxqzN'
, сн
3 /
сн-с
ЩК-ОН / А \\
—----► Н3С-С N
-що 3 \\ /
о но
оксим ацетилацетона 3.5-лимегилизоксазол
Кольцо изоксазола входит в структуру ряда лекарственных препаратов, в частности, антибиотиков оксациллина и ди клоке а цилли на (см. разд. 3.5.4. Г), противотуберкулезного препарата циклосерина:
62
3.6. ШЕСТИЧЛЕННЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ С ОДНИМ ГЕ ГЕРОА1 ОМОМ
Важнейшими представителями этой группы соединений являются гетероциклы, содержащие в качестве гетероатома атом азота — пиридин, хинолин, иэохннолин, акридин:
пиридин хинолин ичохинолип
акридин
и гетероциклы с атомом кислорода - а-пяран и у-пиран:
3.6.1. Пиридин
По химическому строению пиридин (азии) можно рассматривать как аналог бензола, в молекуле которого группа СН замещена атомом азота.
Для названия производных пиридина осуществляют нумерацию атомов цикла или используют обозначения греческими буквами. Положения 2, 6 назы-
вают а, а1; положения 3,5 — 0. 0'; положение 4 — у.
2-чтилпиридии. З-'ИИЛНИрИДИИ,
а-тгилпиридни р-чтили иридии
4<иилЦ1Ирмдин, у-этилпи ридин
А. Способы получения
Пиридин и его мопометильные производные — а-, 0- и у-пиколины содержатся в небольших количествах в каменноугольной смоле (продукт сухой перегонки каменного угля), из которой их выделяют в индивидуальном виде. Кроме того, существует большое число методов синтеза пиридина и его гомологов, из инх наиболее важные основаны на реакции конденсации альдегидов с аммиаком.
Так, из уксусного альдегида и аммиака при 400 °C в присутствии Катализатора А]2О3 образуется смесь, состоящая главным образом из 2-и 4-метилпиридииов:
63
£ л1’°!
6СН3 С + 2NH3 н ацетальди ид
у-циколип
При нагревании акролеина с аммиаком в основном образуется 0-пиколии:
2 СН2=СН + NH3
Н акролеин
Конденсацией ацетальдегида и формальдегида с аммиаком получают незамещенный пиридин:
О
2СН3-(/ + н
Н~С^ + NH3 н
Используя другие альдегиды и их смеси, можно получить различные алкилпиридины.
Б. Физические свойства
Пиридин бесцветная жидкость (т. кик. 115 °C) с характерным jfe-приятным запахом, смешивающаяся с водой, этанолом и большинством органических растворителей.
В. Строение и химические свойства
По строению пиридин имеет сходство с бензолом. Как и бензол, он является ароматическим соединением, содержащим циклическую бя-электронную сопряженную систему (см, разд. 3.1). Неподеленная пара электронов атома азота не участвует в образовании ароматического секстета и обусловливает основные свойства пиридина. Одиако, в отличие от бензола, в молекуле пиридина электронная плотность распределена неравномерно, что подтверждает сравнительно большой дипольный момент (2,26 Д). Вследствие электроноакцепторного влияния атома азота в пиридиновом цикле на всех углеродных атомах электронная плотность понижена, причем в большей степени в положениях 2, 4 и 6 (а- и у-положения), в меиьшей в положениях 3 и 5 (Р-ио-ложения). То есть пиридин является тг-дефицитпой ароматической системой.
Влияние атома азота иа электронную плотность пиридинового ядра сравнимо с влиянием нитрогруппы на бензольное кольцо в молекуле нитробензола:
64
11 прилип пи! робеи-юл
Характерные для пиридина реакции можно условно разделить на три группы:
— реакции, протекающие с участием гетероатома:
— реакции замещения атомов водорода пиридинового цикла: — реакции восстановления и окисления.
Реакции, протекающие с участием гетсроатома
/. Взаимодействие с кислотами.^ Пиридин является слабым основанием. Основность пиридина (рКпи* 5.25) близка к основности анилина (рКВц* 4,6). Водные растворы пиридина окрашивают красную лакмусовую бумагу в синий цвет. При взаимодействии с сильными минеральными и органическими кислотами, такими, как хлороводородная, бромоводородная. серная и пикриновая пиридин дает хорошо кристаллизующиеся пиридиниевые соли:
пирплннийбромил
Образование соли с пикриновой кислотой используют для идентификации пиридина.
2. Взаимодействие с оксидом серы (VI). С участием неподеленной пары электронов атома азота пиридин сравнительно легко реагирует с оксидом серы (VI), образуя донорно-акцепторный комплекс — пири-Динсульфотриоксид:
+ SO3
so?
пнрилипсульфо'грм ОКСИД
Пиридинсульфотриоксид используется в органическом синтезе как мягкий сульфирующий реагент при сульфировании ацидофобных гетероциклов.
3. Взаимодействие с алкил- и ацилгалогеиидами. При взаимодействии с алкил- и ацилгалогенидами пиридин образует четвертичные соли N-алкил- и N-ацилпиридиния соответственно, В этих реакциях атом азота молекулы пиридина проявляет нуклеофильные свойства, предо-
3 Ml
65
ставляя пару электронов для образования связи с электрофильным атомом углерода молекулы гало ген алкана или галогенангидрида карбоновой кислоты.
Соли N-ацилпиридиния характеризуются высокой реакционной способностью ацильного фрагмента по отношению к нуклеофилам и поэтому являются очень эффективными ацилирующими реагентами.
Реакции замещения атомов водорода пиридинового никла.
Nu-
По пиридиновому кольцу могут протекать реакции электрофильного и нуклеофильного замещения.
/. Реакции электрофильного замещения в пиридиновом цикле идут с большим трудом. Нитрование, сульфирование и галогенирование пиридина происходят только в жестких условиях. Так, нитрование протекает с низким
выходом при нагревании пиридина с нитратом калия в дымяшей серной кислоте при 300 °C. сульфирование происходит при нагревании с олеумом (220-270 °C) в присутствии катализатора сульфата ртути, бромирование возможно при действии брома в олеуме. Электрофильный заместитель направляется в р-положспие цикла:
Реакции алкилирования и ацилирования по Фриделю-Крафтсу для пиридина не характерны. Низкая реакционная способность пиридинового цикла в реакциях электрофильного замещения и ориентация замещения в ^-положение обусловлена электроноакцепторными свойст-
66
вами атома азота, который, снижая электронную плотность на всех атомах углерода цикла, в меньшей степени оказывает влияние на ^-положение, что и предопределяет место атаки электрофильным реагентом. Кроме того, в реакциях с протонными реагентами (нитрование, сульфирование) пиридин образует по гетероатому соли пиридиния, а с галогенами дон орн о-акцепторные комплексы, в которых на атоме азота появляется положительный заряд, а это. в свою очередь, приводит к еще большей дезактивации пиридинового цикла.
пиридиний гидросульфат
N-б ром пир ид и ним бромид
2. Реакции нуклеофильного замещения в результате понижения электронной плотности на атомах углерода пиридинового никла облегчаются. В отличие от бензола, пиридин достаточно легко реагирует с нуклеофильными реагентами, образуя продукты замещения в положениях 2, 4 или 6 (а- и^-п сложения). Из реакций нуклеофильного замещения наиболее известно аминирование по Чичибабину. Оно основано иа взаимодействии пиридина с амидом натрия (NaNHj при нагревании. Лучше всего аминирование протекает в среде жидкого аммиака. В результате реакции образуется 2-аминопиридии.
Дальнейшее аминирование приводит к образованию 2,6-диами-нопиридина. Замещение на аминогруппу атома водорода в у-положе-нии происходит только в том случае, если «-положения уже заняты.
Впервые эту реакцию осуществил русский химик-органик А.Е.Чи-чибабин в 1914 г., поэтому она получила название реакции Чичибаби-на. В дальнейшем оиа была распространена на другие гетероциклические соединения. Механизм реакции:
о-комплекс
Реакция протекает по механизму 5\2. На первой стадии нуклеофильная частица (ILN) атакует пиридиновое кольцо с образованием
3*
67
сг-комплекса. На второй стадии ст-комплекс стабилизируется путем отщепления гидрид-иона (Н*).
Аналогично аминированию протекает гидроксилирование пиридинового цикла. При пропускании паров пиридина над сухим гидроксидом калия при 300-320 °C образуется 2-гидроксипиридин.
кои. I
-н2
2-iiiipu;iiiiiO.'uri кадии
2-гилрокси пиридин, пиридин ол-2
Реакции восстановления и окисления
7. Восстановление. Пиридиновый цикл восстанавливается легче бензольного, В зависимости от природы восстановителя и условий гидрирования образуются разные продукты. Так, при восстановлении пиридина водородом в момент выделения (металлический натрий в этаноле) или водородом над платиновым, палладиевым или родиевым катализатором образуется пиперидин. В достаточно жестких условиях, например, при высокотемпературном каталитическом гидрировании, происходит восстановительное расщепление пиридинового кольца по связи С—N с образованием пентаиамииа-1.
6 н (Na +
1 н„ ————*------>>
Pl Pd. Rh
4 Н, Ni. 1HO°C
пиперидин
CH^CH2-CH2-CH2-CHrNH2 пснтанамии-!
2. Окисление. Пиридиновое кольцо устойчиво к действию окислителей. Алкилпиридины, подобно ал кил бензолам, окисляются достаточно легко, образуя соответствующие пиридинкарбоновые кислоты.
Р-пиколин
ппкотиноваи киежла
Под действием псроксикислот пиридиновый цикл окисляется по атому азота с образованием N-оксидов:
CHjCOOOIl
(Н,О, + СП СООН)
N-оксид гшридпна
+ сн3соон
68
N-оксиды пиридина и его производных довольно легко восстанавливаются до исходных пиридинов. В качестве восстанавливающих агентов чаше всего используют галогениды фосфора (Ill).
N-оксид пиридина, в отличне от пиридина, более активен в реакциях электрофильного замещения. Это обусловлено некоторым электронодонорным эффектом атома кислорода. В результате смешения электронной плотности от атома кислорода в кольцо, на атомах углерода в а- и у-п сложениях молекулы N-оксида пиридина электронная плотность повышена в сравнении с пиридином.
Так. N-оксид пиридина вступает в реакцию нитрования гораздо легче, чем пиридин. При нитровании азотной кислотой или нитратом калия в серной кислоте с высоким выходом образуется N-оксид 4-нитропиридина.
N-оксид 4-ннтропириднни
+ н?о
Поскольку замещеииый N-оксид может быть восстановлен в соответствующий пиридин, эту реакцию используют для получения у-заме-Щеиных пиридина.
N-оксия 4-xj<opr(iipir,-'ijjiia 4-хлорлири,нп<
69
Алкилирование и ацилирование N-оксидов протекает по атому кислорода с образованием солей N-алкокси- и N-ацилоксипиридиния соответственно.
N-метокси пиридиний И О ДИД
чсн3
N-ацетоксипиридлиий хлорид
Реакции N-оксидов с нуклеофильными реагентами протекают аналогично пиридину, т.е. преимущественно в положение 2.
'/ Г. Важнейшие производные пиридина
Пиколины. Пиколинами называют моиометильные производные пири-
дина. Различают а-пиколин (2-метилпиридин), р-пико-лии (3-метил пи ридин) и у-пиколии (4-метилпиридин).
Пиколины являются бесцветными жидкостями с неприятным пиридиноподобиым запахом, хорошо растворимы в воде и органических растворителях. Температура кипения а-пиколина 129.5 °C, р-ииколи-на — 144 °C, у-пиколииа — 145,4 °C.
Химические свойства пиколинов и пиридина сходны. Как и пиридин, мстилпиридины образуют соли с сильными кислотами и алкилгалогснидами, окисляются пероксикислотами до N-оксидов, восстанавливаются водородом в присутствии Pt или Pd с образованием производных пиперидина. Под действием КМпО4. HNO-t или кислорода воздуха в присутствии V2O5 а-, Р-
г-пиколин и у-пиколины окисляются соответственно до пиколиновой, никотиновой и изоникотиновой кислот (см. с. 75).
р -11 и калии
ПИКО1ИНОННЯ кислота
соон
В результате здектроноакцепторпого влияния гетероатома, в молекулах а- и у-пиколииов атомы водорода метильных групп обладают повышенной подвижностью, так как образующиеся в процессе денрото-нирования анионы стабилизируются путем делокализации отрицательного заряда по сопряженной системе, включающей гетероатом:
70
Поэтому а- и у-пиколины. в отличие от p-пиколина, вступают в ре-а[Сц1П° конденсации с альдегидами и кетонами, при действии амида натрия (NaNH2) или фениллития (Cf,H5Li) образуют металл органические соединения, взаимодействием которых е алкил галоген идами получают гомологи пиридина.
a iipii.iiui-2-MCi н.п ттрнп
2-т гили иридии
Пиколины используют в органическом синтезе. a-Пиколины применяют в производстве пестицидов, р- и у-пиколины — для получения никотиновой и изоникотиновой кислот соответственно (см. с. 75).
Гидроксипирцдины (оксипиридины, пиридииолы). В зависимости от по-
он <Х-1ндроксн пиридин
ОН
Рмплро кепии рцдин
ложсния гидроксигруппы в пиридиновом цикле различают
a-гидроксип иридии (2-гидроксипиридин. пиридинол-2). р-гидроксипиридин (3-гилроксипиридип, пиридинол-З). у-гидроксипиридин (4-ГЙДрОКСИПИрИЛИП. пиридинол-4).
Гидроксипиридипы являются белыми кристаллическими веществами, легко растворимыми в этаноле. ацетоне, умеренно — в воде, ограниченно — в диэтиловом эфире и бензоле. Температура плавле-
ния 2-гндроксйпиридина 107 °C. 3-гидроксипириди-на— 130 °C. 4-гидроксипиридина— 151 °C.
о. и у-Гидроксипиридины являются таутомерными соединениями. Они существуют в двух таутомерных формах - гидрокси- и оксаформе. В водном
Растворе эти соединения находятся преимущественно в оксо-, или так называемой пнридоновой, форме, в неполярных растворителях и в газовой фазе преобладает гидроксиформа.
ОН
Y—i’ii/ipoKcminpii/iH и
71
2-г ядро КСИ пир ИДИ II (ni д р о ксн фо р м а)
ни ри дон-2 (оксо форма)
4-ГИир О КСИ 11 и р и д и и (гилрсксиформа)
11ирил0|1-4
(оксоформа)
p-Гидроксипиридин в водном растворе существует в нейтральной и биполярной формах, находящихся в соотношении 1:1.
Гидроксипиридииы являются бифункциональными соединениями. По гетероатому они проявляют свойства пиридина, по гидроксигруппе - свойства фенола. Фенольный характер наиболее выражен у 3-гидр-оксипиридииа, гидроксильная группа которого не сопряжена с гетероатомом. Так. все гидроксипиридины образуют с водным раствором FeClj типичное для фенолов пурпурное окрашивание (у 2- и 4-гидрокси-пирндинон цвет менее ингенсивеи. чем у 3-гидроксипиридина). Реакции электрофильного замещения (нитрование, сульфирование, бромирование) в 2- и 4-гидроксипиридинах протекают по положениям 3 и 5. которые являются p-положениями пиридинового цикла и орто-, пара-положеииями относительно гидроксигруппы.
3-Гидроксил иридии вступает в аналогичные реакции SE по положению 2.
С ал кил галогенидами 2- и 4-гид роксипиридины образуют N-ал-килпиридоны, а 3-гндроксипиридин дает соли N-алкилпиридйния:
72
пириди пол-2
II НрНДИ пол-3
пиридиний lfO;ni/[
Многие производные гидроксипнрилинов применяются в медицине. Представителем этой группы соединений является пиридоксин [3-гидрокси-4,5-ди(гидроксиметил)-2-метил и иридии] (витамин В6),
СН ОН Витамин В6 содержится в неочищенных I 2 зернах злаков, в овощах, мясе, рыбе, .молоке, Яичиом желтке и др. Он играет важную роль в || I обмене веществ. В организме приридоксин пре-вращается в пиридоксаль-5-фосфат. который с некоторыми белками образует пиридоксалевые ферменты, осуществляющие декарбоксилирование и переаминирование аминокислот. Применяется пиридоксин в виде соли с хлороводородной
кислотой при В6-гиповитамнноэе, токсикозах, анемиях, лейкопениях и заболеваниях нервной системы.
Амииопирндины. В зависимости от положения аминогруппы в пириди-
^n^nh,
а-амипоппри,тин
Р-зминаниридии
NH,
У'ам и коп иридии
новом цикле различают а-аминопиридии (2-аминопиридип, пиридинамин-2), 3-аминопиридин (З-аминопиридии, пиридинамии-3) и у-аминопиридин (4-аминопиридин. пиридинамии-4).
Аминопиридииы - белые кристаллические вещества (т. пл.: а-амииопиридина 58 °C, 0-аминопиридина 64 °C. у-амииопиридина 159 °C), легко растворимы в воде, этаноле, диэтиловом эфире и других органических растворителях.
Аминопиридины более сильные основания, чем пиридин и анилин. Оии содержат в своем составе два основных центра атом азота пиридинового цикла и атом азота аминогруппы. Вместе с тем. а- и у-амиио-пиридины образуют соли только с одним эквивалентом минеральной кислоты (по гетероатому).
4-амии<)Г111рид11Н
4-ами|1онир1м1111ИЙ хлорид
73
Это обусловлено тем, что в молекулах а- и у-аминопиридинов аминогруппа находится в сопряжении с гетероатомом и и значительной степени утрачивает основные свойства, принимая участие в делокализации положительного заряда образующегося катиона пиридиния.
В молекуле p-аминопиридина аминогруппа не сопряжена с гетероатомом, поэтомусолеобразоваиие протекает подиум основным центрам,
2НС1
3-ам и но пиридина ди гидрохлорид. 3-ам МОНИ Окирили! ГИЙ ди хлорид
а- и у-Аминопиридины являются таутомерными веществами. Они существуют в двух таутомерных формах - аминной и иминной. Более устойчивой является аминоформа.
2-амияопирпдии
аминная форма иминная форма
4-ами|1Опириди11
ампиная форма иминная форма
Реакционная способность аминопиридннов зависит от положения аминогруппы в пиридиновом кольце. 3-Л.минопиридин обладает свойствами, характерными для ароматических аминов. С участием аминогруппы он вступает в реакции алкилирования, ацилирования, диазотирования. 2- и 4-Аминопиридины в обычных условиях солей диазония не образуют, с гало ген алканам и реагируют по атому азота пиридинового цикла.
2-амиио-1-ме1 илгшридиний иилид
По пиридиновому кольцу для аминопиридинов характерны реакции электрофильного замещения — сульфирование, галогенирование, нитрование. а-Амипопиридин в реакциях образует продукты замещения преимущественно в положении 5. р-аминопиридин — в положении 2, у-аминопиридин — в положении 3.
74
Как видно, электрофильное замещение протекает в орто- или пара-положеиии по отношению к аминогруппе.
Аминопиридниы применяются в синтезе лекарственных средств.
Пиридинкарбоновые
соон ииколипонан кислота
пикопшаван кислота соон
N
нзон и кот н новая кислота
кислоты. В зависимости от положения карбоксильной группы в пиридиновом цикле различают а-, 0- и у-пиридиикарбоновые кислоты. а-Пири-динкарбоновую кислоту (2-пиридинкарбоновую кислоту) чаще называют пиколиновой кислотой, Р-пиридинкарбоновую (3-пиридиикарбоновую) пикотиповой, у-пиридинкарбоновую (4-ииридин-карбоновую) изоникотиновой.
Пиридинкарбоновые кислоты представляют собой белые кристаллические вещества (т. пл.: пиколиновой кислоты 136-137 °C, никотиновой 236-237 °C, изоиикотнновой 323-325 °C).
Пиридинкарбоновые кислоты являются бифункциональными соединениями. По карбоксильной группе оии образуют соли, галогенат ид-
риды, сложные эфиры, амиды, гидразиды и другие функциональные производные. По пиридиновому кольцу протекают реакции, свойственные пиридину. Вследствие наличия в структуре молекул кислотного и основного центров, пиридинкарбоновые кислоты являются амфотерными веществами.
натрия никотинат
сооп
С1-н
3-карбоксипнридиний хлорид
В кристаллическом состоянии и частично в растворах они существуют в форме внутренней соли (цвиттер-иоиов). •
Н цвиттер-ион пиколиновой кислоты
75
При нагревании пиридинкарбоновые кислоты декарбоксилируются, а-Кислоты отщепляют СО? довольно легко, р- и у-кислоты разлагаются иа пиридин и СОт при нагревании со щелочью.
соон
I. он"
+ СО,
итпникптнновая кислота
В результате электроноакцепториого влияния гетероатома пиридинкарбоновые кислоты являются более сильными кислотами, чем бензойная кислота:
Кислота: пиколиновая никотиновая изопикотинояая бензоинах ।
рК(, вводе: 1,50 2,07 1.80 4.17 ;
В молекулах пиколиновой и изоникотиновой кислот карбоксильная группа находится в сопряжении с гетероатомом, поэтому эти кислоты проявляют более сильные кислотные свойства, чем никотиновая кислота,
Пиридипкарбоновые кислоты находят широкое применение в синтезе лекарственных средств. Так, никотиновая кислота и ее амид (никотинамид) известны в медицинской практике как две формы витамина РР (кислота является провитамином, а амид витамином РР). При недостатке витамина РР в организме развивается заболевание кожи -пеллагра. Суточная потребность человека в никотиновой кислоте составляет 20-30 мг и удовлетворяется в основном за счет пищевых продуктов— молока, рыбы, овошей. фруктов, гречневой крупы и др.
У*,№-Диэтиламид никотиновой кислоты в виде 25%-иого водного раствора под названием кордиамин применяют в качестве средства, стимулирующего центральную нервную систему, возбуждающего дыхательный и сосудодвигательиый центры головного мозга.
Амид никотиновой кислоты и N.N-диэтиламид никотиновой кислоты можно получить из никотиновой кислоты.
N.N-ди ииламид никотиновой кислоты
76
На основе производных изоникотиновой кислоты созданы лекарственные препараты изониазид и фтивазид, которые применяют при лечении туберкулеза. Изониазид и фтивазид синтезируют по схеме:
изоникотиио- хлорангидрид этиловый эфир лая кислота изоникотиновой нчоникотиновой
NH2NH,
-CnHs01I
гилриэил изоннкоти-новой кислоты.
и’юниалил
кислоты
кислоты
4-гилрокси-3-мегоксибсгнилидси-гидразнд и юпикотнниврй кислоты, фтивазид
Пиперидин (гексагидропиридин) — это бесцветная жидкость (т. кип. 106 °C) с резким аммиачным запахом, смешивается с водой и | большинством органических растворителей.
> Пиперидин проявляет химические свойства вторичных
Н аминов — образует соли с кислотами, с IINO2 даст N-нит-розопроизводное, вступает в реакции алкилирования и ацилирования по атому азота и др. Как вторичный амин пиперидин является значительно более сильным основанием, чем пиридин (pKnN+ пиперидина в воде составляет 11.22; рКВц+ пиридина — 5,25).
Пиперидиновый цикл является структурным фрагментом алкалоидов лобелина, анабазина (см. разд. 4.4), входит в состав анальгезирую-Щего лекарственного средства — промедола, нейролептического препарата галоперидола и др.
промедол галоперидол
77
3.6.2. Хинолин
Хинол ин(беизо[Л]пирид ин) представляет собой конденсированную гетероциклическую систему, состоящую из пиридинового и бензольного колец. Нумерацию атомов в молекуле хинолина начинают с гетсроатома. атомы углерода в пиридиновом цикле обозначают буквами а. р и у.
А. Способы получения
Хинолнн впервые выделен немецким химиком-органиком Ф.Ф.Рунге в 1834 году из продуктов перегонки каменноугольной смолы. Каменноугольную смолу и в настоящее время используют для получения хи7 нолина и некоторых его метилпроизводиых.
Из способов синтеза хинолина н его производных наиболее важными являются синтез Скраупа и синтез Дебнера-Миллера.
Синтез Скраупа, Реакция основана на взаимодействии анилина и его замещенных в ядре производных, имеющих свободное орто-положение, с глицерином, концентрированной серной кислотой и окислителем при нагревании. В качестве окислителя чаще используют нитросоединение, соответствующее исходномуцамину. Для получения хинолина по методу Скраупа нагревают анилин с глицерином и концентрированной H2SO4 в присутствии окислителя нитробензола:
Uli ИЛИН
'Н-ОН ^н-он н-он
глицерин
Механизм реакции включает три последовательные стадии. На первой стадии глицерин под действием концентрированной серной кислоты подвергается дегидратации с образованием акролеина.
О
H.so4, t //
СН2-^Н-^Н2 ---2----► СН-=СН— + 2 Н2о
ОН ОН он н
акролеин
На второй стадии образующийся акролеин вступает в реакцию с анилином:
3-аннли но пропан аль
78
н он
I н
I н
1,2-литдрохи полип
Вначале происходит нуклеофильное присоединение анилина по месту разрыва активированной двойной углерод-углеродной связи молекулы акролеина. Затем образующийся З-анилинопропаналь в кислой среде подвергается циклизации, превращаясь при этом в 1,2-дигидро-хииолин. Замыкание цикла обусловлено электрофильной атакой карбонильной группой цднщ-положеиия бензольного кольца.
На третьей стадии реакции 1.2-днгидрохинолин окисляется нитробензолом в хинолии.
В процессе окисления нитробензол количественно восстанавливается до анилина, который снова вступает в реакцию по описанному механизму.
При использовании в синтезе Скраупа вместо анилина его замещенных в бензольном ядре со свободным орпю-п сложен нем образуются производные хинолина с заместителями в бензольном ядре. Реакция открыта в 1880 г. австрийским хи.миком-органико.м З.Х.Скраупом.
Синтез Де&иера-Миллера. Данный способ является модификацией синтеза Скраупа и используется для получения производных хинолина с алкильными заместителями в пиридиновом цикле.
Синтез Дебнера-Миллера заключается в нагревании первичного ароматического амииа с альдегидом (способным к кротоновой конденсации) в присутствии хлорида цинка (II), хлороводородной или других кислот. Механизм реакции подобен механизму реакции Скраупа. Па первой стадии протекает кротоновая конденсация двух молекул альдегида с образованием а, p-ненасыщенного альдегида, который взаимодействует далее с ароматическим амином, как и в синтезе Скраупа. Роль окислителя выполняют образующиеся в процессе реакции азоме-тины СЙН5 N=CH R.
//
2 СН—
н
HCI. t
——►
-н,о
//
СИ — СН=СН—
н
кротоновый альдянд
79
NH2 CH.
2-мегш1-1.2-дн1идрохи||олин
- Ч сн, н
3-пни.чинобу iaim.ii.
2-мец(1кицолин
Реакция открыта в 1881 г. О.Дебнером и В,Миллером.
Б. Физические свойства
Хинолин - бесцветная жидкость (т. кип, 237 °C) с весьма неприятным запахом, хорошо смешивается с водой, этанолом, диэтиловым эфиром и другими органическими растворителями, перегоняется с водяным паром,
В, Химические свойства
Хииолин является ароматическим соединением. Его молекула имеет плоское строение и содержит замкнутую сопряженную л-электронную систему из Юл-электронов, удовлетворяющую правилу Хюккеля.
По химическим свойствам хинолин напоминает пиридин. Для пего характерны реакции с участием гетероатома, реакции электрофильного и нуклеофильного замещения атомов водорода хинолинового ядра, а также реакции окисления и восстановления,
I. Реакции по гетсроатому. Наличие в молекуле хинолина атома азота пиридинового типа сообщает соединению основные свойства. Как основание хииолин немного слабее пиридина (рК.П[1+хинолина 4,94; рКВ1/ пиридина в Н:О 5.25).
С участием гетероатома хииолин, аналогично пиридину, образует соли с сильными кислотами, алкил- и ацилгалогеиидами:
80
2. Реакции электрофильного и нуклеофильного замещения. Из-за электроноакцепторного влияния гетероатома в молекуле хинолина электронная плотность понижена в сравнении с бензоаналогом нафталином и распределена неравномерно: в пиридиновом кольце она ниже, чем в бензольном. Поэтому при действии электрофильными реагентами замещение, как правило, идет по бензольному кольцу, а нуклеофильными - по пиридиновому.
Реакции электрофильного замещения в молекуле хинолина протекают преимущественно в положениях 5 и 8. Так. при нитровании нитрующей смесью образуется смесь 5- и 8-нитрохинолинов. сульфирование концентрированной серной кислотой при 220 °C приводит к образованию 8-хинолинсульфокислоты, а при 300 °C термодинамически более предпочтительной 6-хи ноли нсульфо кислоты (в этих условиях 5- и 8-изомеры перегруппировываются в 6-изомер).
6-хицо. in нсульфокисло га
В реакции нуклеофильного замещения хинолин вступает легче, чем пиридин. При этом, как и в кольце пиридина, нуклеофильной атаке подвергается преимущественно положение 2. Так, при действии на хинолин амидом натрия в среде жидкого аммиака образуется 2 аминохинолин, с гидроксидом калия при 280-300 °C хинолин образует 2-гидроксихинолин:
81
3. Реакции восстановления и окисления. Восстановление хинолина протекает в первую очередь в пиридиновом ядре. При действии большинством восстановителей с высоким выходом образуется 1,2-ди-гидрохинолин, в присутствии никеля Ренея хннолин восстанавливается водородом до 1,2,3,4-тетрагидрохинолина. Каталитическое гидрирование в жестких условиях затрагивает также бензольное кольцо:
1Д-дигилрохинолшг
1,2.3.4-тетрагидрохи нолин ш/)«нг--дека1 идрохинолии
Окисление хинолина и его гомологов с заместителями в бензольном ядре действием перманганата калия в щелочной среде сопровождается расщеплением бензольного кольца н приводит к образованию 2,3-пири-диидикарбоновой кислоты (хинолиновая кислота).
КМпО. , он-4 .
ноос
ноос
+ 2СО2 + Н2О
хинолиновая кисло га. :
2,3ч111риди пли карбоновая кислота
Аналогично пиридину, в присутствии пероксикислот хинолин окисляется по гетероатому с образованием N-оксида:
65 °C
сн,актом
о-
N-окснд хинолина
Г. Важнейшие производные хинолина
Хинолиновое ядро является структурным фрагментом некоторых алкалоидов (см. разд. 4.5) н лекарственных средств.
82
^рИдроксихииолин. Это бесцветное кристаллическое вещество (т. пл, 75-76 °C), малорастворимое в воде, растворимое в || ] хлороформе, диэтиловом эфире и бензоле,
I J-L 8-Гидроксихннолин получают нагреванием ирпю-
N аминофенола с глицерином и серной кислотой в нри-ОН сутствии ирпю-нитрофенола (синтез Скраупа) или
сплавлением Й-хинолинсульфокислоты со щелочами :
SO.H
3NaOI] {силам,), -NajSOp -211,0 ‘
ON а
С ионами многих металлов (Mg2+, Al3+, Zn2\ Mn2+, Со2+. Ni2+, Cu2\ Fc:+, Fe31 и Др-) 8-гидрокснхинолин образует мало растворимые в воде комплексы (хелаты).
На этом свойстве основано использование 8-гидрокспхинолииа в качестве аналитического реагента.
Ряд производных 8-гидроксихинолнна применяются в медицинской практике в качестве противомикробных средств, К ним относятся такие лекарственные препараты, как хинозол, нитроксолин (5-НОК) и энте-росептол :
ЭНТСрОССгП ОЛ.
8 -ги лр о кем -7- и од-5 -хл op х и н о ли н
fiLipoKCHXHHO.iHHitii сульфат
нитроксолия (5-JJ0K), 8-гидрикси-5-нитрохинолнн
Предполагают, что нх бактерицидное действие основано на свяэы-Вании иоиов Со"\ необходимых для жизнедеятельности микроорганизмов.
83
3.6.3. Изохииолин
5 4 Изохинолин (бензо[с)пнридин) является изоме-
3 Ром *инолина- Молекула изохннолина. как и хино-I Jr" а Л лина, состоит из конденсированных пиридинового и
- бензольного циклов, но, в отличие от хинолина.
* 1 циклы соединены по связи С -С4 пиридинового
кольца. Нумерацию атомов изохииолинового ядра проводят в соответствии с правилами ИЮПАК указанным способом.
А. Способы получения
Изохииолин содержится в хинолиновой фракции каменноугольной смолы (около 1%). из которой его извлекг1ют в виде соли (гидросульфата). Одним нз распространенных способов синтеза изохинолина и его производных является синтез Бшилера-Напирилъского (1893 г.), основанный па циклизации N-ацильных производных р-фенилэтиламннов в 3,4-днгидронзохинолипы и дальнейшем превращении последних в изохинолины путем каталитического дегидрирования над палладием на угле. Циклизацию N-ацильных производных осуществляют чаще в присутствии P3OS или РОСЦ в ксилоле.
[-К-чамсщснный
3, 4-;1ИП1дроизохинолнн
Г^-лцил-р-фснилэтнлампн
Р2°5
-Н,0
ypi
Б. Физические свойства
Из охи нол и и бесцветное кристаллическое вещество (т. пл.
24,6 °C), растворимое в воде, этаноле, диэтиловом эфире, хлороформе и бензоле.
В. Химические свойства
По химическим свойствам нзохинолии мало отличается от хинолина. За счет гегероатома он проявляет основные свойства и легко образует соли с кислотами, алкил- и ацилгалогеиидами. Как основание изохинолин немного сильнее хинолина (рКвн+ в Н2О изохинолина 5,14; рКВ1]+ в Н2О хинолина 4,94).
84
Реакции электрофильного замещения, как и в ядре хинолина, протекают главным образом в положениях 5 н 8, Нуклеофильное замещение в изохинолииовом цикле происходил преимущественно в положе-
При восстановлении изохинолина, как и в молекуле хинолина, в первую очередь гилрируется пиридиновое ядро. Так, при действии натрием в этаноле или водородом иад никелевым катализатором иэохи-нолин восстанавливается до 1,2,3.4-тетрагидроизохинолина :
н, /м
I, 2. 3, 4 - тетрагидраиаохиноли»
В более жестких условиях гидрированию подвергается и бензольное кольцо.
При окислении изохинолнна щелочным раствором КМпО4 образуется смесь фталевой и 3,4-пиридиндикарбоновой кислот.
х^соон
фгалеиая кислота
ноос
ноос
3,4-п иридии ди карбо! юная
кислот а
Под действием органических нероксикислот изохинолин окисляется по гетероатому, образуя N-оксид.
СН3СХХХ)Н
-СН3(ХХ)Н
N-оксид изолинолииа
Ядро изохинолина является структурным фрагментом молекул алкалоидов изохинолинового ряда — папаверина, морфина, кодеина и ДР. (см. разд, 4,6).
85
3.6.4. Акридин
Акридин (дибензо[6,е]пиридин) представляет собой конденсированную систему, состоящую из двух бензольных и одного пиридинового колец. Нумеруют атомы акридинового ядра как показано иа структурной формуле. Наибольший номер получает гетеро
атом. Акридин открыли в 1870 г. немецкие химики-органики К.Гребе и Г.Каро, которые первыми выделили его из каменноугольной смолы.
А. Способы получения
Несмотря на то. что акридин содержится в каменноугольной смоле, чаще акридин и его производные получают синтетически.
1. Конденсация дифениламина с карбоновыми кислотами (Бернтсеи. 1884 г.). При нагревании дифениламина с муравьиной кислотой в присутствии хлорида цинка образуется акридин, конденсация с другими карбоновыми кислотами дает 9-Я-замещеиные акридины.
дифениламин
муравьиная кислота
-ЗН.О
ZnCI, ; L
акридин
2. Циклизация Ы-фенилантраниловой кислоты. В 1933 г, отечественные ученые А-М. Григорове кнй и О.Ю.Магидсон предложили способ получения акридина и его производных, основанный на циклизации N-фенил антраниловой кислоты с помощью трихлороксида фосфора (РОСЬ). Реакция протекает через стадию образования хлорангидрида N-феннлантраниловой кислоты и 9-гндроксиакридииа. Конечным продуктом является 9-хлоракридин.
N-феиилантраниловая
хлор ап гидрид N-фенил-
-HCI
кнелота
антраниловой кислоты
В молекуле 9-хлоракридина атом хлора обладает значительной подвижностью и может быть легко замещен на атом водорода, алкокси-, амиио- или гидроксигруппу.
86
Для получения различных производных акридина с заместителями в бензольных кольцах циклизации подвергают соответствующие замещенные N-фенилаитраниловые кислоты.
Б. Физические свойства
Акридин — светло-желтое кристаллическое вещество (т, пл. 111 °C), с характерным запахом, легко возгоняется, вызывает раздражение кожи и дыхательных путей, откуда и произошло его название (лат. acris — «едкий»). Он хорошо растворим в этаноле, диэтиловом эфире и бензоле, малорастворим в воде. Разбавленные растворы обладают сиией флуоресценцией,
В. Химические свойства
Акридин является ароматическим соединением. С участием неподе-ленной пары электронов атома азота он проявляет слабые основные свойства и образует соли с сильными кислотами и алкилгалогенидами:
Н
пиридиний хлорид
N-метилакридцннй иодил
В реакции электрофильного замещения акридин вступает с большим трудом и неоднозначно. Так. при нитровании образуется смесь изомерных нитроакридинов с содержанием преимущественно 2-иитро-акридина. Реакции нуклеофильного замещения для акридина идут достаточно легко в положении 9. Например, при действии на акридин амидом натрия образуется 9-аминоакридин.
87
NaNH,
NH} (жидкий)
9-a ми по акр илни
Акридиновое ядро весьма устойчиво к окислению. Под действием дихромата калия в уксуснокислой среде акридин окисляется в акри-дон-9, который является таутомерным веществом и существует в двух формах гидрокси- и оксоформе.
9-гидр окси акридин. акрнлол-9
В присутствии органических пероксикислот акридин окисляется по гетероатому с образованием N-оксида,
CHjCOOOH
-CHjCOOH
При окислении в жестких условиях (КМпО4 в щелочной среде) происходит частичное разрушение акридинового ядра, а продуктом окисления является 2,3-хннолиндикарбоиовая кислота (акридиновая кислота).
КМпО4 (ОН‘)
соон
сооп
2,3-хнпо.111н;1икарбоновая кислота, акридиноьая кислота
Восстановление акридина протекает аналогично антрацену, т.е, по положениям 9 и 10, Так. под действием натрия в спиртовом растворе нли при каталитическом гидрировании акридин превращается в 9,10-дигидроакридин (акридан).
H,/Ni
н н
9,10-ди1хдроакридип, акридин
Г. Важнейшие производные акридина
9-Амйноакридин желтое кристаллическое вещество (т. пл. 236-237 °C), растворимое в этаноле и ацетоне.
9-Аминоакридин - более сильное основание, чем акридин. Он содержит в своем составе два L JL 1 основных центра — атом азота пиридинового типа и атом азота аминогруппы. Однако, вследствие сопряжения аминогруппы с гетероатомом. 9-аминоакрндин. аналогично у-аминопиридину, образует соль только по кольцевому азоту.
НС1
Некоторые производные 9-аминоакридина применяются в качестве лекарственных препаратов, например, акрихин и этакридина лактат (риванол).
11Х-СН —(СН - N(C,Н Л
акрихин.
9-(4-ди7гиламино-1 -метилбутнл-
этакридииа лактат, риванол.
соон I
сн -он
сн3
ам niioJ-2-мстоксн-б-хдоракридина
6.9-лпами1го-2<ггокснакрнлина лактат
ангидро хлор ид
Акрихин проявляет противомалярийное, а этакриднна лактат антисептическое действие.
3.7. ШЕСТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ С А ТОМОМ КИСЛОРОДА
3.7.1. а-Пираи и у-пиран
4
а - пиран
4 (у)
1 у - ниран
а-Пиран (2Н-пиран) и у-пиран (4Н-пиран) представляют собой шестичле[{ные гетероциклические соединения. содержащие в качестве гетсроатома один атом кислорода.
Эти гетероциклы являются структурными изомерами н отличаются друг от друга расположением метиленовой группы относительно гетероатома. В молекуле а-пирана метиленовая группа находится в а-положении, в у-пиране — в у-положении.
В а и у-пираиах отсутствует замкнутая сопряженная система, из-за чего зти вещества не обладают ароматичностью и характеризуются низкой стабильностью. а-Пиран в свободном состоянии не получен.
у-Пиран выделен в виде индивидуального вещества, но является очень неустойчивым, легко разлагающимся на воздухе соединением. Однако,
наряду с этим оке о про из водные пиранов — а-пирон (2Н-пирон) и у-пирои (4Н-пирон) представляют собой достаточно устойчивые ве-
щества.
а-Пирон — бесцветная жидкость (т. кип. 206-209 °C) с запахом свежего сена, у-пироп —• бесцветное кристаллическое вещество ( г. нл. 33 °C).
а- и у-Пироны имеют в своем составе шестичлениый
а - пироп гетероцикл, содержащий атом кислорода и 5 атомов О углерода, находящихся в лу?"-гибридизации. Пеподелен-
Оная пара электронов гетероатома находится в сопряже-з нии с л-электроиами двух двойных связей цикла и карбонильной группой. Делокализацию электронной 2 _
плотности можно представить в виде двух крайних ре-1 зоиансных структур, одна из которых представляет со-
Y ’ш,ро" бой сопряженный диен, а другая — ароматическую систему, подобную пиридину.
Поэтому а- и у-пнроны способны вступать и в реакции, характерные для сопряженных диенов, и в реакции, присущие аренам. а-Пирон вступает преимущественно в реакции первого типа, т. е. этот гетероцикл следует рассматривать скорее как ненасыщенный лактон.
90
По лактонной группировке ) а-пирон вступает в реакции
нуклеофильного присоединения, сопровождающиеся раскрытием цикла. Так, в присутствии щелочен а-пирон легко гидролизуется, под действием аммиака раскрытие цикла сопровождается рецнклизациен с образованием пиридоиа-2.
НОИ <
----->-сн=сн-сн==сн—с; —► хс-сн2-сн=сн-сх (ОН“) I А ОН н ОН
I г 5-0 ксо пентен-2-овая кислота
Ненасыщенный характер а-пироиового цикла подтверждается его способностью к каталитическому гидрированию и к вступлению в реакцию с малеиновым ангидридом по Дильсу-Альдеру как 1,3-диена, у-Пирон, в результате сопряжения непо делен ной пары электронов гетероатома с карбонильной группой, не образует характерных для кетонов производных по карбонильной группе (оксимов, гидразонов, оснований Шиффа) и с трудом вступает в реакции присоединения по месту разрыва двойных углерод-углеродных связей. При взаимодействии с минеральными кислотами (НС1, НСЮ4) или алкилгалогенидами у-пирон образует соли пирилия.
4-мегОксииирилий и о лид
НС]
(в эфире)
4-1-идроксииирилий хлорид
Пирилиевый катион в солях пирилия содержит замкнутую л-сис-тему из шести электронов и, таким образом, подобно бензолу или пиридину, обладает ароматическим характером.
Реакции у-пирона с нуклеофильными реагентами, аналогично а-пирону, как правило, сопровождаются раскрытием цикла по связи О- С. В присутствии аммиака у-пирон превращается ву-пиридон.
Некоторые производные у-пироиа являются природными веществами. К ним относятся хелидоиоваи н мекоиоваи кислоты. Хелидоновая Кислота содержится в траве чистотела (Chelidonium majus), меконовая кислота выделена из мака снотворного (Papa ver somniferum).
91
о
ноос
хелидоновая кислота;
у-пирон-2.6-дикарбонован кислота
о
меконовая кислота;
3-1 мдро кси -y-ii и рол -2,6-дн карбоновая кислота
3.7.2. Кумарии
5^ _4 Кумарин (бензо[/»]п ирон-2) представляет собой
бр з конденсированную гетероциклическую систему.
I l/я состоящую из бензольного и а-пиронового цик-
лов. По строению кумарин является лактоном 8 1 t/ис-о-гидроксикоричной кислоты (кумариновая
кислота). Он встречается во многих растениях. Обладая запахом свежескошенного сеиа, кумарин придает аромат доннику, ясменинку и другим растениям.
Синтетически кумарии получают по реакции Перкина конденсацией салицилового альдегида с уксусным ангидридом в присутствии ацетата натрия.
(СН3СО)2О
салициловый альдегид
CHjCOONa
180 СС
кумирни
Химические свойства кумарина обусловлены наличием в его структуре лактонного и бензольного колец. Реакции с нуклеофильными реагентами протекают по лактонному кольцу и. как правило, сопровождаются раскрытием цикла. Так, при нагревании кумарина с водным раствором NaOH образуется соль одшо-гидроксикоричной кислоты. После подкисления раствора происходит быстрая рециклизация в кумарин.
кумарин .цинатрневая соль
кумариновой кислоты
По бензольному кольцу кумарин вступает в реакции электрофиль? кого замещения (нитрование, сульфирование). Электрофильной атаке в первую очередь подвергается положение 6,
92
Производные кумарина содержатся во многих растениях и обладают фармакологической активностью. Некоторые вещества из i-руппы 4-гидроксикумаринов оказывают выраженное аитикош улируюшее» действие, т. е. понижают свертываемость крови. На их основе созданы антикоагулянтные лекарственные препараты — i геол и кум арин, фепро-марон, синкумар и другие, применяемые для профилактики и лечения тромбозов.
неодикумарин:
ггиловый тфир ди-(4-г1<дракейку мири п11Л-3)уксу слой КИСЛОТЫ
пептилЖумирин
3.7.3, Хромон
Хромон (беизо[Л] пи рои-4) представляет собой конденсированную гетероциклическую систему, состоящую из бензольного и у-пиронового циклов.
Хромон и его производные широко распространены в растительном мире
По химическим свойствам хромон напоминает
у-пирон. Подобно у-пирону, при обработке сухим HCI в зфире хромон образует соль бензопнрилия. под действием водных растворов щелочен происходит раскрытие у-пиронового кольца по связи О—С'2. Важнейшими производными хромона являются флавон (2-фепилхро.мон) и изофлавои (3-феиилхромон):
флниол >’
93
„ . ^г* носятся к обширной группе
Производные флавона и изофдавона о^аионо„дами. В растениях природных соединении, называемых ф-Я флавона _ ф;|авон0лы. широко представлены гидроксипроизвод^ и „ |1атожении
Природные флавонолы всегда содержат i в 11ОЯ0жениях у „ 4- Они 5 и 7. иногда - в положении 3 и. как оравИ^ишми пи1.ментами желтых окрашены в желтый ивет и являются кра>- олин и „„„ цветов. Представителями флавонов являют
гз зилов. Гликозид кверцетина
В растениях они находятся в виде глик" Ь-рамиозы. называют
с дисахаридом, построенным из /Хгдкж^- няют в качестве рутином. В медицинской практике рутин * ства, обладающего Р-витаминной активно^*
3.8. иНХГГИЧЛЕННЫЕ ГЕТЕРОЦИ1^1Ы С ДВУМЯ ГЕ1ЕРО" I
АТОМАМИ
4
пирид;г<ии
пиримидин
пиразин
3.8.1. Диазник1
„ , _п-цчлениЬ]С гетероциклы, со-
Диазинами называют атом'а а,1()та с
держащие в качестве •етероаТ вд„нн
шествует три изомерных диазИ"* 4 Рязии) ’’
пиримидин ПЗ-диазип) и пира>« соедИпения во многом
По строению и свойствам
поминают пиридин. Подобно г*иридину. молекулы иирида-
. *дмеют в своем составе замк-
чина, пиримидина и пиразина у , л.,„. л бл-электронов и обладают
нутую сопряженную систему У*"
} } 3 оделенные пары электро-
ароматическим характером, г1 _______ 1
f в сопряжении и придают
нов атомов азота не участвуй*7
. .. 7 Из-за дезактивирующего
диазинам основные свойств
, „ „ друга, пиридаэип. пиримидин
влияния атомов азота друг на ' 1
_ г 1 ? " основаниями, чем пирн-
н пиразин являются более cjia^s, а
„ * 2,33; пиримидина 1.3; пи-
дни (рКщ|+ в воде пирндазина* гт
ot .... Г По этой причине диазины, разина - 0.6; пиридина -. 5,>' г >,йпых центров, ооразуют соли
несмотря на наличие двух оснн минеральной кислоты.
только с одним эквивалентом
пиримидин
H2SO4
HSO47
11
[гиримидиннй 1 H.ipocyi->фа|
94
Наличие в структуре молекул пиридазина, пиримидина и пиразина двух атомов азота пиридинового типа приводит к значительному понижению электронной плотности на атомах углерода диазинового цикла. Поэтому диазины характеризуются очень низкой реакционной способностью в реакциях 5Е и. наоборот, высокой активностью в реакциях 5м- Реакции электрофильного замещения возможны только тогда, когда диазиновый цикл активирован электронодонорными группами, например, -NH2, -ОН и др.
А. Пиридазни (1,2-диазин)
Пнридазни бесцветная жидкость со слабым запахом (т. кип. 207 °C), растворимая в воде, этаноле, бензоле, дн-этиловом эфире, не растворимая в петролейном эфире. Пиридазин и его производные получают конденсацией гидразина с насыщенными или ненасыщенными 1,4-ди-
карбонильными соединениями.
Преимущество синтезов с ненасыщенными 1,4-дикарбоннльными соединениями состоит в том, что образующийся продукт не требует дальнейшего окисления.
Пиридазин является слабым основанием. С кислотами (хлороводородной, пикриновой) и алкилгалогенидами он образует соли по одному атому азота.
пиридазинни хлорид
М-мегилииридачинин иодид
В реакции электрофильного замещения пиридазин вступает с большим трудом. Осуществить реакции нитрования, сульфирования, галогенирования практически не удается.
Под действием пероксикислот пиридазин окисляется, образуя N-ok-сид;
95
СНдСОООН
-CHjCOOH
N-оксид пн ридазина
Л' ю
При восстановлении пиридаэина натрием в спиртовом растворе или каталитическом гидрировании раскрывается цикл и образуется тетраметилендиамин.
Н->
H3N—СП,—СНП~-СН,— CH.-NH,
теграм^илеплнамни
Производные пиридазина применяют в качестве лекарственных препаратов и гербицидов. Так. пиридазиновый никл содержит молекула лекарственного препарата сульфапнридазина проявляющего антибактериальную активность, и молекула гербицида феиазона используемого для уничтожения сорняков па полях сахарной свеклы.
сульфапирилюин.
3-(||-аминобеп'юлсуяьфам!1до |-6-ме'10кснпирш1азии
феназин.
5-a.MiiHO-2^[)eiiiiJi-4-xjidpnnpn4ajidfiotb3
Б. Пиримидин (1,3-ди азин)
Способы получения. Пиримидин и его производные 4 зN чаще всего получают конденсацией мочевины или тиомо-|б j 2П чевины с 1,3-дикарбонильными соединениями. Так, при взаимодействии мочевины с малоновым зфнром в присутствии этоксида натрия образуется производное пиримидина — барбитуровая кислота, которая существует в оксо- и гидроксиформе.
о о но
С-ОС2Н5 H2N CtJljONa £“NH
н.с + с=о --------►Н,е ,С=О .** нс( ;с-он
С-ОС, В 5 H,N -ЭС,П3ОН “b-NH C=N
о " мочевина О ™«»Ф"РМ« НО Гидрокснформ-
дизеилмалонат барбитуровая khuioih
96
Для получения пиримидина барби гуровую кислоту действием хлороксида или пентаоксида фосфора превращают в 2.4,6-трихлорпирими-дин (в реакцию вступает гидроксиформа), который затем восстанавливают.
Физические свойства. Пиримидин - бесцветное кристаллическое вещество (т. пл. 22.5 °C), легко растворимое в воде, этаноле, диэтиловом эфире.
Химические свойства. По Химическим свойствам пиримидин сходен с пиридином, но, как и другие диазины, характеризуется более низкой реакционной способностью. Несмотря на наличие в молекуле пиримидина двух основных центров, в реакциях с минеральными кислотами он образует соли по одному атому азота:
п прим иднн и ii хлорид
Из-за электроноакцепторного влияния двух агомов азота пиримидин практически ие вступает в реакции электрофильного замещения. Но если пиримидиновый цикл активирован одним или несколькими ; Электронодопорными заместителями (-NH,. -Oil, -SH и др.) возможны реакции электрофильного замещения (нитрование, галогенирование, сульфирование, нитрозирование) преимущественно п положение 5, например:
i
он он
2~амино-4-и|,1рокс111111рнмндин 2-ам1ПЮ-4-гидрокси-5-ни1 рои при миди и
2-а мипо пир и мидин
2-г1МииП-5-брОМ1|1!рИМНД|111
4*41
97
Нуклеофильные реагенты атакуют в .молекулах пиримидина и его
производных электронодефицитные положения 2, 4 и 6. Так, при
действии амидом натрия на 4-метилпиримидин образуются 2-амино~ и
2,4-диамино-б-метилииримидины.
4-меп1лиир[>м1[дин
NH?’
2-амино-4-мс । h.iii ирцмнлии
Нуклеофильное замещение в ряду пиримидинов часто сопровождается раскрытием цикла.
Важнейшие производные пиримидина. Среди производных пиримидина важную биологическую роль выполняют гидрокси- и аминопиримидины. Оли входят в состав нуклеиновых кислот, витаминов, антибиотиков, лекарственных препаратов и др.
Барбитуровая кислота (2, 4, 6-три гидрокси пиримидин) - -- это бесцветное
Н
3.8.1, Б). Поэтому ее можно рассматр лоновой кислоты. Барбитуровая кисл
кристаллическое вещество (т. пл. 245 °C’), малорастворимое в воде и этаноле, хорошо раство-. римое в диэтиловом эфире.
Барбитуровую кислоту получают конденсацией мочевины с малоновым эфиром (см. разд, инать как циклический у рейл ма-ота является таутомерным соеди
нением, в котором одновременно проявляются два вида таутомерии — кето-енольная и лактам-лактимная. Кето-снольная таутомерия обусловлена миграцией атомов водорода метиленовой группы, а лактамлактимная - -атомов водорода групп NH.
кеюнная форма енольная форма
ак|амн.>я форма
лактимная форма
В результате совместного проявления кето-енолыюй и лактам-лак-тимиой таутомерии, барбитуровая кислота существует в двух таутомерных формах — триоксоформе и тригидроксиформе. С помощью рентгеноструктурного анализа установлено, что преобладающим таутомером является триоксоформа.
98
трноксоформа
гришдроксиформа
обшая формула барбитуратов
Барбитуровая кислота является более сильной кислотой, чем уксусная- Ее кислотные свойства обусловлены подвижностью атома водорода, особенно в гидроксиле енольного фрагмента молекулы. Некоторые 5,5-дизнмещенные производные барбитуровой кислоты применяют в медицине в качестве лекарственных препаратов, обладающих снотворным и противосудорожным действием. Эти препараты известны под общим названием барбитураты. К ним относятся барбитал (R и R' = С;Н5), фенобарбитал (R = C2HS. R’ = С6Н5).
бврбамил (R = СгН5, R' = изо-С5Нц) и др.
Так как в молекулах барбитуратов атомы водорода метиленовой группы замещены углеводородными радикалами, для ннх характерна только лактам-лактимная таутомерия. Несмотря на это. барбитураты проявляют слабые кислотные свойства и легко образуют водорастворимые соли с одним эквивалентом гидроксида натрия.
Пиримидиновые основания. К пиримидиновым основаниям относят;
П1МЦН
урацил
(2.4-дигидроксипиримидин), тимин
(2.4-лигидрокси-5-метилпирими-дин) и цитозин
(4-амипо-2-гидроксипиримидин). входящие в состав нуклеиновых кислот. Эти соединения являются таутомерными веществами и существуют в лактамной и лактимной формах.
4*
99
Как правило, в равновесии преобладает лактамная форма. Урацил, тимин и цитозин представляют собой высокоплавкие (т. пл. > 300 °C) бесцветные кристаллические вещества. мало растворимые в
воде, не растворимые в этаноле и диэтилоном эфире. В клетках организма пиримидиновые основания находятся в виде N-гликозидов рибозы или дезоксирибозы (см. разд. 7.1).
Витамин В((тиамин). Витамин В( содержит в своем составе два гетеро-
циклических кольца — пиримидиновое и тиазольное, связанные через метиленовую группу. Полученный из природных источников или путем синтеза витамин В] представляет собой замещенную
аммониевую соль. Витамин В, содержится в дрожжах, пшеничном хлебе. фасоли, горохе, сое. меньше — в картофеле, моркови, капусте. При недостатке витамина В| в организме развивается заболевание бери-бери. получившее распространение в ряде стран Азии и Индокитая, где основным продуктом питания является очищенный рнс.
Витамин В[ — бесцветное кристаллическое вещество, хорошо растворимое в воде. Водные раст воры тиамина в кислой среде выдерживают нагревание до высоких температур без снижения биологической активности. В нейтральной и щелочной средах витамин В, при нагревании бышро разрушается.
В животных тканях свободный витамин Bi содержится в небольших количествах. Физиологически активной формой витамина Bi в живых организмах является кофермент кокарбоксилаза (гиамииднфосфат). входящий в состав ферментов, участвующих в процессах углеводного обмена.
ко карбоксилаза (гиаминдифосфаг)
Оротовая кислота (урацил-6-карбоноваа кислота) — бесцветное кри-
сталлическое вещество (т. пл. 345-347 °C), растворимое в воде и водных растворах щелочей.
Оротовая кислота содержится в животных тканях и растениях. Особенно богаты ею дрожжи, печень и молоко. Оротовая кислота является
100
предшественником в биосинтезе пиримидиновых оснований урацила. цитозина и тимина. В виде калиевой соли (оротат калия) оротовая кислота применяется в медицине как стимулятор обменных процессов при заболеваниях сердца, печени, желчных путей и др.
В. Пира з ни (1.4 -диаз ин)
По физическим свойствам пиразин представляет собой бесцветное кристаллическое вещество (г. пл. 57 °C), со слабым запахом, растворимое в воде, этаноле, диэтиловом эфире. Пиразин и его производные получают конденсацией 1,2-диаминов с 1.2-дикарбон ильными соединениями, например:
I ЛИС KCilJU.
инранш
2.3-ди гидро iiiipanin
Аналогично пи ридазину и пиримидину, пиразин является слабым основанием и образует соли по одному атому азота, в реакции электрофильного замещения вступает с большим трудом, реакции нуклеофильного замещения идут сравнительно легко. При действии на пиразин амидом иатрня в жидкой аммиаке образуется 2-аминопиразин.
NhNH^
2-aMniKitiupa’iiiii
Под действием перокснкислот пиразин окисляется с образованием моно- и ди-М-оксидов.
/ \ СН'СОСЮИ / \ +
N N------------------N N —О
\ / -СН,С'ООН \ /
р. MOito-N-пкснл пиразина
При восстановлении пиразина натрием в этаноле или каталитическом гидрировании образуется гексагндропнразин или пиперазин:
I HJ
(Na + с,н.он )
Пиперазин, в отличие от пиразина, является сильным двукислот-ным основанием. Для него характерны свойства вторичных алифатиче-
101
ских аминов. Соль пиперазина с адипиновой кислотой под названием пиперазина адипинат применяется в медицине как противоглистное средство.
СНуСН-г-СООН
^н^-сн^-соои
пиперазина адипинат
3.8.2. Фенотиазин
Фенотиазин (дибснзо-1.4-тиазин) представляет собой конденсированную гетероциклическую систему, состоящую из 4Н-1,4-тиазннового и двух бензольных циклов. Нумерацию атомов проводят как показано на структурной формуле.
А. Способы получения
Фенотиазин получают нагреванием дифениламина с серой в присутствии хлорида алюминия как катализатора.
дифениламин
AIC1, -H,S
фенотиэ »ин
Б. Физические свойства
Фенотиазин — бесцветное кристаллическое вещество (т. пл. 182 °C), нерастворимое в воде, диэтиловом эфире, хорошо растворимое в горячем этаноле.
В. Химические свойства
По химическим свойствам фснотиазин во многом напоминает вторичные ариламины. Так. будучи вторичным амином, фенотиазин легко вступает в реакции алкилирования и ацилирования по атому азота.
Реакции электрофильного замещения (нитрование, сульфирование, галогенирование) протекают у фенотназина преимущественно в положениях 3 и 7. часто сопровождаясь окислением атома серы.
102
Под действием пероксида водорода или перманганата калия фено-тиазин окисляется по атому серы с образованием фенотиазиндиокси-да-5,5.
Ю1
КМпО4
фено гнгшнлио ксил-5.5
Фенотиазин в прошлом применялся в медицинской практике как антигельминтный препарат. В настоящее время ряд производных феио-тиазина используется в качестве лекарственных средств, обладающих нейролептическим действием (аминазин, этанеразии, трифтазии и др.).
амина ]ип
К производным фенотиазина относился краситель метиленовый синий, который применяется для окрашивания препаратов в микробиологии, а также как антисептическое лекарственное средство в виде 1-3%-ных спиртовых растворов.
С1-
3,9. КОНДЕНСИРОВАННЫЕ СИСТЕМЫ ГЕТЕРОЦИКЛОВ
Важное значение из этой группы гетероциклов имеют широко распространенные рядов.
в природе соединения пуринового и птеридинового
6 7
4%'
3.9.1. Пурин
Пурин (имндазо[4,5-<7)пиримидин) представляет собой конденсированную гетероциклическую систему, состоящую из пиримидинового и имидазольного колец. Исторически сложившаяся нумерация атомов пуринового ядра не отвечает общим правилам нумерации конденсированных систем, но является общепринятой.
1ПЧ
А. Способы получения
Пурии и его производные чаще получают конденсацией 4,5-ди-аминопиримидииов с карбоновыми кислотами (метод Траубе);
^^-лиймниоппрнмидип
НО
H-R-nypini
Б. Физические свойства
Пурнн — бесцветное кристаллическое вещество (т. пл, 217 °C), хорошо растворимое в воде, плохо — в ацетоне, диэтиловом эфире, хлороформе,
В, Строение и химические свойства
Пурин является ароматическим соединением. Его молекула имеет плоское строение и содержит замкнутую сопряженную систему, состоящую из 10л-электроиов, включая неподеленпую пару электронов атома азота в положении 9, что соответствует правилу Хюккеля (4п+2, п = 2), Наличие в структуре пурина имидазольного цикла сообщает ему ряд свойств, характерных для имидазола. Так, пурину свойственна азольная таутомерия:
В кристаллическом состоянии нахождение атома водорода более вероятно в положении 7,
Аналогично имидазолу, пурин является амфотерным соединением и образует соли с сильными кислотами и основаниями.
Атомы азота пиримидинового цикла, вследствие электроноакцепторного влияния друг иа друга и участия в делокализации положительного заряда пуриний-катиона. не протоиируются сильными кислотами. 104
Г. Важнейшие производные пурина
Наиболее важными производными пурниа являются оксо- и амино-рурИНЫ.
Оксопурипы, Представителями оксопуринов являются мочевая кислота. ксаитии и гипоксантин.
моченин кислота
। ип оксан । ни
Эти соединения образуются в организме в процессе превращения нуклеиновых кислот. Мочевая кислота, ксантин и гипоксантин являются таутомерными веществами. В результате лактам-лактимной таутомерии они существуют в двух таутомерных формах — оксоформе (лактамная форма) и гидрокенформе (лактимная форма). Позтому в учебной и научной химической литературе оксопурины часто называют гидроксипуринами.
мочеван кислота*; 2.6,К-1ригидроксипури11
ксантин : 2.6-nHH>;ipoia:nriypHii
гидроксиформа
Iииоксантни ; б-пирокенпурии
105
В кристаллическом состоянии мочевая кислота, ксантин и гипоксантин находятся в оксоформе. В растворах они существуют в виле равновесной смеси таутомерных оксо- и гидроксиформ, в которой преобладает оксоформа.
Наряду с лактам-лактимной таутомерией, у оксопуринов возможна азольная таутомерия, связанная с миграцией атома водорода между атомами азота в имидазольном цикле, например:
аиольцня >аутомерия ипаксаптиин
Мочевая кислота - бесцветное кристаллическое вещество (т. пл. 400 °C), плохо растворимое в воде, этаноле, диэтиловом эфире, растворимое в разбавленных растворах щелочей и глицерине. Мочевая кислота является конечным продуктом обмена пуриновых соединений в организме. Она выделяется с мочой человека в количестве 0.5-1 г в сутки.
Мочевая кислота является двухосновной кислотой (рК(?[ 5.75; рК^ 10.3). С водными растворами щелочей она образует кислые и средине соли.
меню натриевая сель МО'НШОН KJJOlOTU
динатрнеш'я соль мочевой кисяотм
Соли мочевой кислоты называют уратами. Кислые ураты, за исключением солей лития, представляют собой малорастворимые в воде соединения. При некоторых заболеваниях, в частности, подагре, они откладываются в суставах, при почечно-каменной болезни накапливаются в почках в виле почечных камней. Основной составной частью почечных камней является мононатриевая соль мочевой кислоты.
В гидроксиформе мочевая кислота вступает в реакции нуклеофильного замещения, например, с РОСК образует 2.6.8-трихлорпурин.
ОН
I4JC1,
CI
2.6.К-гр и хлорнурии
106
Iff
Вследствие высокой подвижности атомов хлора, 2,6,8-трихлорпу-рИн широко используется в синтезе производных пурина — аденина, гуанина, гипоксантина, ксантина н др. Активность атомов хлора в различных положениях пуринового ядра в реакциях ,S'N неодинакова и уменьшается в ряду 6>2>8, например;
Hi по ксантин
1уани1г
При нагревании мочевой кислоты с азотной кислотой с последующим прибавлением к реакционной смеси аммиака появляется пурпурно-фиолетовое окрашивание, связанное с образованием аммонийной соли пурпурной кислоты, называемой мурекендом. Реакция протекает в несколько стадий. Под действием азотной кислоты мочевая кислота окисляется с образованием смеси аллоксана и диалуровой кислоты, которые, реагируя друг с другом, дают аллоксантин, превращающийся в избытке аммиака в мурексид.
аллоксантин
пурпурная кислота (кстоформа)
107
о
о
о
о
пурпурная кислота (енольная форма)
мурексц.т. пурнурат аммонии
Эта реакция, получившая название мурексидиая реакция, используется для качественного обнаружения мочевой кислоты и других соединений, содержащих пуриновое ядро.
Гипоксантин (6-гидроксипурин) и ксантин (2,6-дигидроксипурни) по химическим свойствам аналогичны мочевой кислоте. Они существуют в двух таутомерных формах — лактамной и лактимной. Подобно мочевой кислоте, эти соединения образуют соли со щелочами. Наряду с этим, гипоксантин и ксантин обладают также слабо выраженными ос-нбвиыми свойствами и образуют соли с сильными минеральными кислотами, т. е. они амфотерны.
хлороводородная соль гипоксантина
натриевая соль гипоксантина
Гипоксантин и ксантин широко распространены в растительном и животном мире. Важное значение в фармации имеют N-метильиые производные ксантина — теофиллин (1,3-диметилксаитии), теобромин (3,7-диметил ксантин) и кофеин (1,3,7-триметилксантин).
Это природные вещества, относящиеся к алкалоидам. Теофиллин содержится в листьях чая, теобромин — в бобах какао, кофеин — в листьях чая и зернах кофе.
Теофиллин, теобромин и кофеин получают из природного сырья или синтетически — метилированием ксантина. По физическим свойствам они представляют собой бесцветные кристаллические всше-
108
стви, легко растворимые в горячей воде, плохо — в холодной. Теофиллин и теобромин являются амфотерными веществами. Их кислотные свойства обусловлены подвижностью атома водорода в NH-^рагменте молекул, основные — наличием пиридинового атома азота N\ Кофеин проявляет только слабые основные свойства, обусловленные наличием атома азота в положении 9. Теофиллин и теобромин обладают мочегонным действием, кофеин оказывает возбуждающее влияние на центральную нервную систему, В медицинской практике кофеин обычно применяется в виде двойной соли с бензоатом натрия — кофеин-беизоата натрия.
Лмшюпурнмы. Важнейшими аминопроизнодиыми пурина являются аденин (6-аминопурии) и гуанин (2-амино-6-гидроксипурин). входящие в состав нуклеиновых кислот в качестве пуриновых основании. Гуанин существует в двух таутомерных формах — лактамной и лактимной. Более устойчива лактамная форма, в виде которой гуаниновый фрагмент находится в нуклеиновых кислотах.
Nil,
Oil
Аденин и гуанин представляют собой бесцветные кристаллические вещества, труднорастворимые в воде, хорошо растворимые в щелочах, Оии образуются при гидролизе нуклеиновых кислот. В организме аденин и гуанин подвергаются дезаминированию с образованием гипоксантина и ксантина, которые окисляются в мочевую кислоту.
3.9.2, Птеридин
Молекула птеридина (пиразипо[2,3-б/]пиримидин) представляет собой конденсированную гетероциклическую систему, состоящую из пиримидинового и пиразинового циклов. Нумерацию атомов осуществляют как показано на структурной формуле.
А. Способы получения
Производные птеридина довольно широко распространены в природе. Впервые птеридины выделены в 1895 г, из пыльцы крыльев бабочек, что нашло отражение в названии (греч. птеро — «крыло»).
Синтетически птеридины чаще получают конденсацией 4,5-диами-нопиримидннов с 1,2-дикарбоиильными соединениями.
I.
1
Ск Н
глиоксаль
и гери.|И и
Б. Физические свойства
Птеридин — кристаллическое вещество (т. пл. 137-138 °C) светло-желтого цвета, хорошо растворимое в воде, этаноле, малорастворимое в диэтиловом эфире и бензоле.
В. Химические свойства
Птеридин является ароматическим соединением. Ядро птеридина устойчиво к действию окислителей, проявляет основные свойства. Вследствие электроноакцеп горного влияния четырех атомов азота пиридинового типа электронная плотность на атомах углерода значительно понижается и ослабляется ароматический характер птеридиновой сисзсмы. Так, птеридин неустойчив к действию кислот и щелочей, которые в зависимости от условий вызывают раскрытие пиримидинового нлн пиразинового циклов, но легче расщепляется пиримидиновое кольцо. Он не вступает в реакции электрофильного замещения. Введение электронодонориых заместителей (NH-, ОН- и др.) в молекулу птеридина увеличивает электронную плотность в ядре и повышает его стабильность. Как слабое основание (рКП11+ 4.12) птеридин протониру-ется по атому азота в положении I.
11
Для птеридиновой системы характерны реакции алкилирования по атомам азота.
Г. Важнейшие производные птеридина
Фолиеваи кислота (витамин Вс). Молекула фолиевой кисло ты включает три структурных фрагмента — птеридиновое ядро, остатки л-ами-нобензойной кислоты и L-глутаминовой кислоты.
фрагмент фрагмент фрагмап
л геридина л-шчнпобеизойной Л-глутаминовой
КИСЛОТЫ КИСЛО гы
фолиевая кислота
Фолиевая кислота впервые выделена в 1938 г. из экстракта печени. В большом количестве она содержится в листьях шпината, моркови и других овощах. Название кислоты связано с выделением ее из листьев шпината (от лат. folium — «лист»), В человеческом организме фолиевая
110
кислота не синтезируется. Потребность организма в этом витамине удовлетворяется за счет поступления с продуктами питания и синтеза микроорганизмами кишечника. Биологическая роль фолиевой кислоты связана не со свободной формой, а с частично восстановленным птеридиновым производным — 5,6,7,8-тетригидрофолиевой кислотой.
Фолиевая кислота стимулирует кроветворение, биосинтез нуклеиновых кислот, белковый и углеводный обмен. Применяется в медицинской практике для лечения некоторых форм анемии.
Фолиевая кислота является стимулятором роста микроорганизмов. Бактериостатическое действие сульфаниламидных препаратов основано на нарушении бносинзеза фолиевой кислоты. Имея структурное сходство с п-амииобензойной кислотой, сульфаниламиды связываются с птеридиновым фрагментом вместо n-амииобензойной кислоты. В результате блокируется последующая конденсация с глутаминовой кислотой и тем самым прекращается биосинтез фолиевой кислоты, что ведет к гибели микроорганизмов.
м-амипобеичойцая кисло та
3.9.3. Аллоксазви н изояллоксазии (флавин)
Аллоксазни представляет собой конденсированную гетероциклическую систему, состоящую из трех циклов — бензольного, пиразинового и гидрированного пиримидинового, в котором два атома углерода находятся в составе карбонильных групп.
Изоаллоксазнн является таутомерной формой аллоксазииа, которая образуется иследствие азольной таутомерии:
Н-----□
Нтоаллоксазин (флавин}
Изоаллоксазин имеет желтую окраску и поэтому называется флавином (от лат. /кг?и.ч — «желтый»). Важным свойством флавина является его способность к восстановлению с образованием бесцветного соединения (лейкосоединение). которое при окислении превращается снова в исходный флавин. В процессе восстановления водород присоединяется к сопряженной системе, включающей атомы азота в положениях 1 и 10.
Ядро флавина входит в структуру рибофлавина (витамин В2),
Название «рибофлавин» отражает наличие в молекуле остатков пя-тиатомиого спирта рибита и флавина. Рибофлавин широко распространен в природе, особенно богаты им дрожжи, бобовые, мясо, яичный желток и др. Витамин Вт входит в структуру окислительных ферментов — флав о протеидов. Действие этих ферментов как переносчиков водорода при окислительно-восстановительных процессах в живых организмах обусловлено способностью флавииа превращаться в лейко-соединепие и наоборот. Отсутствие или недостаток витамина В2 в пище вызывает задержку роста, воспаление слизистых оболочек рта и глаз, нарушение процессов нервной деятельности и др.
3.10. СЕМИЧЛЕНПЫЕ АЗОТСОДЕРЖАЩИЕ ГЕТЕРОЦИКЛЫ
Семичленные гетероциклические соединения с одним атомом азота, содержащие максимальное количество двойных связей в цикле, называют азепинами, аналогичные гетероциклы с двумя атомами азота - -диазепинами. Представителями азецинов и диазепннов являются:
112
Ни один из приведенных гетероциклов до настоящего времени не получен в свободном виде, ио известны многочисленные их производные.
Азепины и диазепины имеют неплоское строение, проявляют свойства полиенов. Вследствие деформации валентных углов семичленные циклы меиее устойчивы, чем шестичлснныс,
Семичлениые азагегероциклы редко встречаются в природе. Повышенный интерес к синтезу зтих гетероциклических систем обусловлен выявлением у некоторых из них транквилизирующего (снимающего перевозбуждение ЦНС, страх, тревогу, напряжение), антидепрессивного, аналептического (повышающего тонус ЦНС) и противосудорожного действия, В медицинской практике широко применяются для лечения заболеваний центральной нервной системы производные бензодиазепина (конденсированная система 1,4-диазепина с бензолом). Эффективными лекарственными средствами бензодиазепинового ряда являются элениум, нитразепам (радедорм), диазепам (седуксен) и др.
ни гргпелам. ралсяорм
лиггипам. седуксен
КОНТРОЛЬНЫЕ ВОПРОСЫ И УПРАЖНЕНИЯ
I. Напишите структурные формулы следующих гетероциклических соединений:
а) 2-бром-4-метилфурана; б) N-метил! шррола;
в) а-тиофенсульфокислоты; г) N-оксида пиридина;
д) 3-метилизохииолина; е) 9-хлоракридина;
ж) мочевой кислоты,
2. Какие гетероциклы называют азолами?
На примере 1,3-диазола объясните явление азольной таутомерии.
3. Какими структурными особенностями обусловлен ароматический характер фурана, пиррола и тиофена?
4, Что такое ацидофобность? Какие гетероциклы являются ацидофоб-ными? Напишите схемы реакций нитрования и сульфирования фурана и индола,
5- Напишите схемы реакций фурфурола со следующими реагентами:
a) NaHSO3: б) гидразином. Какими реакциями можно доказать наличие альдегидной группы в молекуле фурфурола?
ИЗ
/ /
6. Объясните, какое влияние на химические свойства оказывает атом/ азота «пиридинового» типа в молекуле пиразола и имидазола в сравнении с пирролом, 1
7. Напишите таутомерные формы пиразолоиа-5. В основе каких лекарственных препаратов лежит структура пиразолона-5?
8. Отметьте сходство и различие в строении и свойствах пиридина и бензола. Приведите примеры реакций,
9. Приведите схемы реакций, подтверждающих амфотерный характер . имидазола, пиразола и никотиновой кислоты.
10, Напишите схему реакции нитрования пиридина н пиррола и объясните условия проведения реакции в каждом случае.
11, Напишите схемы взаимодействия пиридина, хинолина и изохиио-лина с амидом натрия, К какому типу реакций относится эта рсак-* ция и каков ее механизм?
12. Расположите следующие соединения в порядке уменьшения основности;
а) пиридин; б) пиперидин;
в) хинолин; г) акридин,
13. Расположите в порядке увеличения реакционной способности в реакциях электрофильного замещения: бензол, пиррол, пиридин, хинолин.
14. Охарактеризуйте отношение к действию окислителей:
а) бензола; б) фурана;
в) пиррола; г) пиридина; L
д) хинолина.
15. П риведите схему синтеза 8-гидрокси-хииолина по методу Скраупа.
16. Чем обусловлен кислотный характер барбитуровой кислоты?
Ответ подтвердите схемами соответствующих реакций.
17. Приведите таутомерные формы мочевой кислоты. С помощью какой качественной реакции можно идентифицировать мочевую кислоту? Приведите примеры соединений, дающих эту реакцию. i
.t
1
114
Глава 4 АЛКАЛОИДЫ
Алкалоидами называют группу азотсодержащих органических соединений, преимущественно растительного происхождении, проив-лающих основные свойства и высокую биологическую активность.
Название «алкалоиды» произошло от арабского алкали — «щелочь».
По химическому строению большинство алкалоидов относится к гетероциклическим соединениям.
Алкалоиды широко распространены в растительном мире. Особенно богаты алкалоидами растения семейства маковых (Рарсггегаееае), лютиковых (Ranunculareue), бобовых (Fahaceae) и др. Как правило, в растениях содержится смесь нескольких алкалоидов, имеющих подобную структуру. Так, из опия выделено 22 алкалоида, из коры хинного дерева — 24. из листьев табака — 10. Многие алкалоиды обладают сильным биологическим действием; в больших дозах они часто являются ядами, а в малых дозах— применяются в качестве лекарственных средств. В растениях алкалоиды находятся обычно в виде солеи органических кислот — щавелевой, яблочной, винной, лимонной и др.
По физическим свойствам алкалоиды представляют собой, как правило, бесцветные кристаллические вещества горького вкуса, практически нерастворимые в воде, но хорошо растворимые в органических растворителях - - хлороформе, диэтиловом эфире и бензоле. Соли алкалоидов, напротив, хорошо растворимы в воде и нерастворимы в органических растворителях.
К настоящему времени выделено более 5000 алкалоидов. Большой вклад в их изучение внесли русские и советские химики А.П.Орехов. В, М,Родионов, Н.А.Преображенский, А.А.Вознесенский и др,
4.1. МЕТОДЫ ВЫДЕЛЕНИЯ ИЗ РАСТЕНИЙ
Выделение алкалоидов из растений основано па способности их солей хорошо растворяться в воде, а алкалоидов-оснований — в органических растворителях (хлороформе, ли этиловом эфире, бензоле).
Существует два основных метода выделения алкалоидов: экстракция в виде солей и экстракции в виде оснований. При экстракции в виде солей растительное сырье, содержащее алкалоиды, обрабатывают водой или этиловым спиртом, подкисленными чаще винной кислотой, Все алкалоиды при этом переходят в раствор в виде солей винной кислоты. Для очистки от сопутствующих веществ вытяжку подщелачивают и
115
образовавшиеся основания алкалоидов извлекают органическими растворителями. Операцию очистки (обработка кислотой, а затем щелочью) повторяют несколько раз. После этого растворитель удаляют. а остаток, содержащий сумму алкалоидов, разделяют на индивидуальные вещества.
При экстракции в виде оснований растительный материал обрабатывают раствором щелочи. Чаще для этой цели используют раствор аммиака, гидро карбоната или карбоната натрия. Образующиеся основания алкалоидов экстрагируют органическими растворителями. Далее проводят очистку переведением алкалоидов в соли, а затем в основания, повторяя этот процесс несколько раз.
4.2. МЕТОДЫ ОБНАРУЖЕНИЯ АЛКАЛОИДОВ
Для обнаружения алкалоидов в том или ином объекте применяют общие, так называемые групповые реакции, присущие группе алкалоидов, и специфические реакции, характерные для индивидуальных алкалоидов.
Групповые реакции основаны на способности алкалоидов как оснований образовывать с некоторыми реаген тами труднорастворимыс в воде осадки простых или комплексных солей. Наиболее часто для осаждения алкалоидов применяют раствор иода в иодиде калия (реактив Вагнера), раствор иодида ртути в иодиде калия (реактив Майера), раствор иодида висмута в иодиде калия (реактив Драгендорфа), растворы таннина, пикриновой кислоты и др.
Кроме реакций осаждения, для обнаружения алкалоидов часто используют реакции окрашивания. Так, под действием концентрированных серной, азотной кислот, а также смеси серной и азотной кислот (реактив Эрдмана), смеси сериой кислоты и формальдегида (реактив Марка) и других, многие алкалоиды образуют окрашенные растворы. Реакции окрашивания базируются иа протекании процессов дегидратации, окисления, конденсации и др. В основу этих реакций положены особенности химической структуры алкалоидов, поэтому они могут быть использованы как специфические для определенных групп алкалоидов. Идентификацию конкретных алкалоидов осуществляют с помощью специфических реакций на отдельные функциональные группы, входящие в состав молекулы алкалоида, а также применением физико-химических методов (ИК-, УФ-, ПМР-спектроскопия, масс-спектрометрия и хроматография).
4.3. КЛАССИФИКАЦИЯ АЛКАЛОИДОВ
Для удобства изучения алкалоиды делят на группы. Первоначально, когда химическая структура многих алкалоидов не была установлена, их классифицировали по ботаническому признаку, согласно которому в одну группу объединялись алкалоиды, выделенные из растений определенного рода, например, алкалоиды маковых, мареновых, пасленовых н т.д. и
116
В настоящее время общепринята химическая классификация, в основу которой положена природа гетероцикла, входящего в структуру алкалоида. Согласно этой классификации, алкалоиды делят на следующие основные группы - производные пиридина и пиперидина, хинолина, изохинолина, индола, тропана, пурина и др.
4.4. АЛКАЛОИДЫ ГРУППЫ ПИРИДИНА И ПИПЕРИДИНА
4.4.1. Никотин
Алкалоид никотин {3-[2'-(К-метилпирролидил)]пиридин} содержится в листьях табака (Nicotiana tabacum) в виде 3 / И солей лимонной и яблочной кислот. Его молекула
• 1Л;/ 35. состоит из пиридинового и N-метил пиррол ид ин о-
вого циклов, связанных между собой простой I 1| 2 связью.
3 Никотин бесцветная маслянистая жидкость, 1 смешивающаяся с водой. На воздухе быстро буре-
ет вследствие образования продуктов окисления. Никотин имеет в своем составе асимметрический атом углерода (С-) поэтому он обладает
оптической активностью. Водные растворы никотина вращают плоскость поляризации влево. Никотин очень токсичен, смертельная доза для человека составляет около 40 мг. В небольших количествах
оказывает возбуждающее действие иа вегетативную нервную систему, сужает кровеносные сосуды. Организм постепенно привыкает к этому яду. Являясь очень ядовитым веществом, никотин используется в качестве инсектицида для борьбы с вредителями сельского хозяйства.
4.4.2. Анабазин
Алкалоид анабазин [0-(а'-пиперидил)пиридин] содержится в ежов-никс безлистом (Anabasis aphylla), произрастающем в Средней Азии. Молекула анабазина состоит из связанных простой связью пиридинового и пиперидинового циклов.
Подобно никотину, анабазин очень ядовит, обладает высоким инсектицидным действием. В виде хлороводородной соли анабазин применяется в медицине в качестве средства против
курения. Предполагают, что эффект основан на конкурентном взаимодействии с никотином п области специфических рецепторов.
4.4.3. Лобелии
Алкалоид лобелии [Л-(-)2-бензоилметил-6-(2,-гидрикси-2,-феиилэтил)-1-
4
метилпиперидип] обнаружен в растении лобелии (Lobelia inflata), произрастающего в Северной Америке. По химической структуре он является
117
производным пиперидина. Впервые синтез побед ин а осуществлен в 1929 году. Природный лобелии оптически активен (вращает плоскость поляризации влево). Синтетический лобелии является рацематом и поэтому оптической активностью не обладает. В виде хлороводородной солн лобелии применяется в медицине как стимулятор дыхания при угнетении дыхательного центра. Синтетический лобелии вдвое менее активен, чем природный левовращающий изомер,
4.5. АЛКАЛОИДЫ ГРУППЫ ХИНОЛИНА
К алкалоидам хинолинового ряда относится группа алкалоидов, выделенных из коры хинного дерева. Важнейшим среди них является хинин.
4.5.1. Хинин
молекулы хинина лежит ядро хинолина, свя-
В основе структуры
занное через вторичную спиртовую группу с хинуклидиновым ядром.
Хинии является бесцветным кристаллическим веществом (г. пл. 177 °C) горького вкуса, малорастворимым в воде, оптически активен (вращает плоскость поляризации влево).
Являясь днутретичным двукислотным основанием, хинин образует с
минеральными кислотами два ряда солей. С одним эквивалентом
кислоты солеобразовапие протекает по атому азота хинуклидилового ядра. С двумя эквивалентами минеральной кислоты хинин образует соли по обоим атомам азота.
Хинин специфическое средство для лечения малярии. В акушерской практике его применяют для усиления родовой деятельности. Фармакопейными препаратами являются хинина гидрохлорид, хинина дигидрохлорид и хинина сульфат.
4,6, АЛКАЛОИДЫ ГРУППЫ ИЗОХИНОЛИНА И ФЕНАНТРЕНИЗОХИНОЛИНА
Важное значение из этой группы соединений имеют алкалоиды папаверин, морфии и кодеин, которые широко применяются в качестве лекарственных средств. Основным источником получения указанных алкалоидов является опий, представляющий собой загустевший млечный сок незрелых головок мака (Papaver xomniferum).
118
4.6.1. Папаверин
Папаверин [6,7-диметокси-1 -(3',4’-ди.ме-гокснбецзнл)изохинолнн] впервые был выделен в 1884 г. из опия, а в 1910 г, получен синтетически.
Папаверин бесцветное кристаллическое вещество (т. пл. 147 °C), нерастворимое в воде, растворимое в горячем этаноле н хлороформе. В виде хлороводородной соли папаверин применяется в медицине в качестве спазмолитического и сосудорасширяющего средства.
4.6.2. Морфин
Алкалоид морфин впервые выделен из опия в 1804 г. французским фармацевтом Сегэном. Среди алкалоидов он был открыт первым. Молекула морфина имеет сложное строение. Пал установлением структуры морфина работали многие химики в течение более ста лет и только в 1925 г. английским химиком-оргаииком Р. Робинсоном было установлено строение его молекулы. По химической структуре морфин является производным фенантрен-, изохинолина. Частично гидрированные
ядра фенантрена и изохинолина соединены в молекулы морфина таким образом, что один шестичленный карбоиикл (Ill) является общим и для фенантренового, и для изохинолинового циклов.
Морфин - бесцветное кристаллическое вещество (т. пл. 254 °C), мало растворим в воде, этаноле, диэтиловом эфире, хлороформе, бензоле; оптически активен (вращает плоскость поляризации влево).
Молекула морфина содержит две гидроксильные группы, которые обладают разными свойствами. Гидроксильная группа в положении 3 является феиольиоЙ, так как находится в бензольном ядре, а гидроксильная группа в положении 6 связана с частично гидрированным кольцом и поэтому является вторичной спиртовой, У атома азота находится метильная группа.
В отличие от большинства алкалоидов. морфин обладает не только основными, по и слабыми кислотными свойствами. С минеральными кислотами он образует соли по атому азота, с участием фенольного гидроксила - растворяется в щелочах. Аналогично фенолу, морфин Дает с ИеСЦ синее окрашивание.
В виде хлороводородной соли морфин используется в качестве обезболивающего средства. При длительном применении к нему быстро развивается болезненное пристрастие наркомания. Это свой
119
ство морфина в ряде случаев ограничивают его использование. Диацетильнос производное морфина — героин является наиболее распространенным наркотиком.
По химическому строению кодеин представляет собой метиловый эфир морфина, образованный с участием фенольного гидроксила.
у | Содержание кодеина в опии невели-
11 ко (0,5-1%) поэтому значительную часть
। его производства осуществляют полу-
\ синтетически — путем метилирования
СН3 морфина.
1. л Кодеин представляет собой беснвет-
НС) ное кристаллическое вещество (т. пл.
154-157 °C), горького вкуса, плохо растворимое в воле, хорошо растворимое в этаноле, ди этиловом эфире, оптически активное (вращает плоскость поляризации влево).
В отлнчие от морфина, кодеин не растворим в щелочах и нс дает окрашивания с FeClj. Незначительное изменение структуры молекулы кодеина по сравнению с морфином приводит к изменению фармакологической активности. Болеутоляющее действие кодеина в 6-7 раз ниже, чем морфина. В отличие от морфина, кодеин уменьшает возбудимость
кашлевого центра, что позволяет использовать его в основном как про-тивокашлевое средство. При длительном применении кодеин вызывает к себе привыкание.
4.7. АЛКАЛОИДЫ ГРУППЫ TPO1IAHA
В основе структуры алкалоидов этой группы лежит бициклическая система тропана. состоящая из пирролидинового и пиперидинового колец, имеющих общин атом азота.
т
120
пирролидиновый
ЦИХ.1
трепан
iiitiiepHjHitOBbiH цикл
Алкалоиды группы тропами являются производными аминоспиртов — тропина и скокина, а также гидроксикислоты — экгонина.
СООН
* 1 ронин СКОПИЛ Ж! опии
Главные представители тропановых алкалоидов атропин, скополамин и кокаин.
4.7,1. Атропин
Алкалоид атропин [тропиновый эфир (±)-тро повой кислоты] со-
держится в растениях красавке (Atropu belladonna), дурмане (Datura stramonium) и др.
По химической структуре атропин представляет собой сложный эфир, образованный
спиртом тропином И Громовой кислотой ф-гидрокси-ос-фенилпропионовая кислота).
Громовая кислота содержит асимметрический атом углерода, поэтому * существует в виде двух оптически активных
HOHi^C-CH-COOH изомеров (D- и L-) и оптически неактивной рацемической формы. В молекуле атропина | спирт тропин этерифицировац рацемической
троповой кислотой. Поэтому атропин оптически неактивен.
В растениях содержатся лишь следы атропина. Главной же формой, в которой находится алкалоид в растениях, является гиосциамин, представляющий собой сложный эфир тропина и левовращающего изомера троповой кислоты. При получении из растений гиосциамин подвергают рацемизации нагреванием до 114-116 °C. В этих условиях левовращающий гиосциамин превращается в рацемат, т. е. атропин. Атропин является одним из сильнейших ядов. В очень малых дозах в виде сернокислой солн он применяется в медицине в качестве холинолитического и спазмолитического средства. Кроме того, атропин проявляет мадриатический эффект, т. е. способность расширять зрачок, что широко используется в глазной практике для обследования глазного дна. Атро
121
пин также применяется в качестве противоядия при отравлениях ацетилхолином и фосфорорганическими соединениями.
4.7.2. Скополамин
Алкалоид скополамин [скопиновый эфир (-)-троповой кислоты] со
держится вместе с атропином и
---------\ 9 гиосциамином в растениях, кра-МО N“CH3 \—О—С—СН—СН,—ОН савке, белене, дурмаие. По хи-
-----У мической структуре он имеет Г |1 сходство с атропином. Его мо-
лекула представляет собой сложный эфир, образованный аминоспнртом скопином и троповой кислотой.
Скополамин напоминает атропин и по фармакологическому действию. Он тоже относится к холинолитическим средствам, так же. как атропин, способен расширять зрачок, но мидриатический эффект скополамина мецее продолжительный. В медицине скополамин применяется в виде бромоводородиой соли.
4.7.3. Кокаин
Алкалоид кокаин (метиловый эфир беизоилзкюиина) содержится в листьях кустарника кока (Erythru-xylon соси), произрастающего в Южной Америке. По химической структуре кокаин представляет собой сложный эфир, образованный гидроксикислотой экгонином, метиловым спиртом и бензойной кислотой.
Кокаин оказывает сильное местноаисстезирующес действие. Однако, из-за высокой токсичности и способности вызывать кокаинизм (наркомания) ои имеет ограниченное применение. В виде хлороводородной соли кокаин используют главным образом в качестве местиоа-иестезирующего средства и глазной практике, а также для местного обезболивания слизистых оболочек полости рта, носа, горла, анестезии пульпы зуба.
4.8. АЛКАЛОИДЫ ГРУППЫ ИНДОЛА
Конденсированная система индола входит в состав многих алкалоидов, имеющих, как правило, сложное строение. Большинство алкалоидов группы индола содержит два атома азота, один из которых входит в индольное ядро, а другой находится иа расстоянии двух угле-род-углеродиых связей от P-положения индольного никла. Наибольшую практическую ценность из этой группы представляют алкалоиды резерпин и стрихнин.
122
4.8.1. Резерпин
Алкалоид резерпин содержится в корнях растения раувольфия змеиная (Rauwolfia serpentina), произрастающего в Индии.
Резерпин — желтоватый мелкокристаллический порошок, мало растворим в воде, этаноле и диэтиловом эфире, легко растворим в хлороформе и уксусной кислоте. Резерпин обладает гипотензивным действием (понижает кровяное давление) и оказывает успокаивающее действие на центральную нервную систему. Применяется в медицине преимущественно для лечения гипертонической болезни и психических расстройств.
4.8.2. Стрихнин
Алкалоид стрихнин содержится в семенах тропического растения чилибухи (Strychnos nuxvomica). Он является сложным конденсированным миогояд-ерным соединением, состоящим из семи колец. Из двух атомов азота в молекуле стрихнина основными свойствами обладает только атом азота в положении 19. С участием этого атома азота стрихнин образует соли с кислотами. Азом азота в положении 9 входит в состав лактамного кольца, которое под действием растворов
щелочей размыкается. Из-за наличия в структуре стрихнина двойной связи (С -С“) он способен гидрироваться, обесцвечивать бромную воду и перманганат калия. Стрихнин является очень ядовитым веществом. В малых дозах он действует возбуждающе иа центральную Нервную систему, тонизирует скелетную мускулатуру, мышцу сердца, в больших дозах вызывает судороги. В виде соли с азотной кислотой применяется в качестве тонизирующего средства.
123
КОНТРОЛЬНЫЕ ВОПРОСЫ И УПРАЖНЕНИЯ
1. Какие органические соединения называют алкалоидами?
Приведите их классификацию.
2. Охарактеризуйте методы выделения алкалоидов из растительного сырья.
3. Назовите важнейшие алкалоиды группы пиридина, хииолнпа, нзохинолииа, изохинолилофснаитрена и тропаиа.
4. Чем отличаются по строению алкалоиды никотин и анабазин, морфин и кодеин, атропин и гиосциамин? Приведите структурные формулы указанных алкалоидов.
I 1
I
/
и
н и
124
Глава 5
УГЛЕВОДЫ
Термин «углеводы» предложен в 1844 г. русским химиком К.Г.Шмидтом на основании данных элементного состава первых представителей этого класса соединений, так как было установлено, что их молекулы состоят на атомов углерода, водорода и кислорода, причем соотношение атомов водорода и кислорода такое же. как в молекуле воды [Cx(H2O)y].
Дальнейшее изучение строения этих соединений и открытие веществ с составом, не отвечающим указанной эмпирической формуле, показали, что отнесение их к «гидратам углерода» является лишь формальным, но принятое название «углеводы» сохранилось.
В настоящее время к углеводам относят обширную группу природных и синтетических соединений, являющихся по химическому строению полигидроксильными веществами, содержащими альдегидную или кетопную группы, или образующие их при гидролизе.
Углеводы (сахара) составляют основную массу органических веществ нашей планеты. В растительном и животном мире они широко распространены главным образом в виде различных производных и значительно реже в свободном виде, Углеводы, выполняя различные жизненно важные функции, служат исходным материалом для биосинтеза многих органических соединений Живых организмов.
В природе углеводы образуются в результате фотосинтеза, осуществляемого растениями с участием диоксида углерода, воды и поглощающих солнечный свет пигментов (хлорофилл и др.).
Для растений одни виды углеводов служат строительным материалом, выполняя опорную функцию (целлюлоза), другие источником резервной энергии (крахмал, инулин). Для человека и животных углеводы являются продуктами питания с высокой энергетической ценностью. В организме крахмал, дисахариды, а в некоторых случаях и целлюлоза под влиянием ферментов распадаются с образованием, в основном, глюкозы, которая окисляется в тканях до диоксида углерода и воды с выделением энергии. Избыток глюкозы превращается в гликоген. запасаемый в печени и мышцах. Гликоген снабжает организм глюкозой при выполнении физических нагрузок, а также при недостатке или отсутствии пищи. Углеводы входят в состав гликолипидов, гликопротеидов, нуклеотидов, нуклеозидов и нуклеиновых кислот, составляющих, как известно, основу живой материн.
Углеводы составляют сырьевую базу для текстильной, целлюлозно-бумажной, пищевой, деревообрабатывающей и других отраслей п ро м ы ш ле нн ости.
125
В зависимости от числа моносахаридных единиц, связанных в молекулу. углеводы делятся на простые и сложные.
Простые углеводы, или моносахариды (монозы) не способны гидролизоваться. Сложные углеводы при гидролизе образуют моносахариды. Сложные углеводы, в свою очередь, хотя и условно, классифицируются на олигосахариды, образующие при гидролизе от двух до десяти молекул моносахаридов, и полисахариды (пол позы), гидролизующиеся с образованием более десяти молекул .моносахаридов.
/ 5.1. МОНОСАХАРИДЫ
Моносахариды пред ста вл л юг собой поли гидроксильные соединения, содержащие альдегидную или кетоииую группы. Их еще называют мопозамн или простыми углеводами (сахарами).
Моносахариды, за исключением глюкозы и фруктозы, редко встречаются в природе в свободном виде. В основном они входят в состав олиго- и полисахаридов, гликозидов, гликолипидов, нуклеозидов и других высокомолекулярных соединений.
5.1.1. Классификации н номенклатура
Моносахариды классифицируют с учетом двух признаков характера оксогруппы (альдегидная или кетониая) и длины углеродной цепи.
В зависимости от наличия в структуре моносахаридов альдегидной или кето иной группы, их подразделяют на альдозы и кетозы. В соответствии с числом атомов углерода в молекуле, моносахариды классифицируют иа триозы (CJ, тетрозы (С4). пеитозы (С5). гексозы (С6) и т. д. Моносахариды, в состав которых входит более шести атомов углерода, называют высшими сахарами. Большинство природных моносахаридов являются пентозами и гексозами. Обычно при классификации учитываются одновременно оба классификационных признака, например, альдоле птоза, альдогексоза, кетопентоза, кетогексоза.
H^z>0
2 снон
1 I снон
41 СИОН 5сн2он
альдоле! поза
Н 1 АУ (х
2 СНОП
Зснон
4 I снон
5 I снон
61 СН2ОН альдо гексо ча
сн,он «-
2с=о ч
•’’снон Н
СНОН »’
Ъюн
6СН2ОН
кегО|Сксоча
В названиях моносахаридов, как правило, используют тривиальную номенклатуру. Все тривиальные названия имеют окончание -оза. например, глюкоза, фруктоза, галактоза, рибоза и др. Номенклатура И1ОПЛК в названии углеводов практически ие применяется.
126
5.1.2. Стереоизомерия
Молекулы моносахаридов содержат несколько асимметрических атомов углерода и поэтому они существуют в виде различных пространственных изомеров. Например, альдопентоза имеет 3 асимметрических атома углерода, а следовательно, одной и той же структурной формуле соответствует 8 стереоизомеров (23), альдогексоза содержит 4 асимметрических углеродных атома и может существовать в виде 24= 16 стереоизомеров (см. кн. 1, с. 74).
Для изображения стереоизомеров иа плоскости используют проекционные формулы Фишера (см. ки. 1, с. 73).
Все изомеры моносахаридов подразделяют иа D- и /.-стереохимические ряды, принадлежность к которым определяется по конфигурации асимметрического атома углерода, максимально удаленною от карбонильной группы (для пентоз — С4. для гексоз — С5). Если коифи-[урацня этого хнралыюго атома углерода совпадает с конфигурацией /?-глицеринового альдегида, то моносахарид относят к /?-рялу. если же с конфигурацией L-глицеринового альдегида — то к /.-ряду, например:
г_нч-°_
сн2он
-глицериновый альдегид
н\ с 1 'Н2ОН
н— —он < •=о
но— —и
н — —он н 4
<JH
—он ' [н_- —он '
6( :н,оп ч :н,он
В -ГЛЮКНТЛ I)-фрукт* па
нч
___с _ _
сн,он
f.-i -UHiepiиi<>выii awiein.i
/.-арабино ла
CH,ОН
2 I c-o
HO——H
_JHy1— OH
LhO_T;—Hj
6 CH,OH
Г-сорбоча
IL 1 <O
LUP”—-1* J 'CH,OH
Так, из 16 стереоизомеров альдогексозы 8 относятся к /)-ряду. а 8 — к L-ряду. Представители D-ряда являются оптическими антиподами A-ряда, то есть альдогексоза существует в виде 8 пар энантиомеров. Например, энантиомерами являются D-глюкоза и L-глюкоза.
„о
н— —он
но— —н
н— —он
н— —он
( ?н,он
Д-глюкоза
11. X)
но— —н
н— —он
но— —н
но— —н
сн2он
Гадюки та
L
127
Преобладающее большинство природных моносахаридов относится к D-ряду. Важнейшими природными моносахаридами являются:
IL н il
ч Ч" ч
п— —он н— —он н— —он
н— —он но— —II но— —и
н— —он н— —он НО— L-h
сн,он снэон с н2он
D-рибота Р-ксилоза Д-арабино la
Н. .о н ^о и Л)
( :н,он
но— —н п— —он II— —он ( ‘--О
но н но U но н но н
HU"
LI ГУН н пн но н н он
IT /“irj к r"iLT о пц L1 riTJ
11 О!1 11 СМ 1 Г7 4 VM и СМз
СН2ОН ( ?Н2ОН ( :н.он < 7Н,ОН
D-мапнст D-глюкоэа D-i алак i сгаа />-фрукч оэа
Пространственные изомеры моносахаридов, отличающиеся конфигурацией одного или нескольких атоМов углерода и не являющиеся зеркальными изомерами (энантиомерами) называются диастереомерами (см. кН, 1. с. 74). Так, D-глюкрза и D-галактоза. D-манноза и D-глюкоза, D-маиноза и D-галактоз а/составляют пары диастереомеров. Диастереомеры. различающиеся конфигурацией только одною асимметрического атома углерода, называются эпимерами. Например, D-глюкоза и D-галактоза, а также D-глюкоза и D-маниоза, составляют пары эпимеров. Таким образом, эпимерия является частным случаем диастереоме-рии.
Важнейшим свойством веществ, молекулы которых имеют асимметрический атом углерода или асимметричны в целом, является их способность вращать плоскость поляризованного луча света. Поэтому моносахариды обладают оптической активностью. Вращение пл ос-кости поляризованного света вправо обозначают знаком (+). а влево — знаком (-). Одиако следует помнить, что направление вращения определяется экспериментально и никаким образом не связано с конфигурацией, то есть принадлежностью к D- или Т-ряду. Так, встречающаяся в природе форма D-глюкозы является правовращающей, а природная форма D-фруктозы левовращающей.
5.1.3. Строение моносахаридов
Долгое время в науке господствовало представ ле и не, что моносахариды являются соединениями с открытой углеродной цепью, содержащие в своем составе альдегидную или кегоцную группу и несколько спиртовых гидроксилов. Одиако при более глубоком изучении их строения было установлено, что ряд свойств моносахаридов не согла
128
суется с существовавшими представлениями. Так, оказалось, что моносахариды. являясь альдегидами, не дают некоторых реакций на альдегидную группу. В частности, они не образуют при обычных условиях гидросульфитные производные, не дают окрашивания и фуксинсериис-той кислотой. Прн нагревании моносахаридов со спиртами в присутствии сухого хлороводорода в реакцию вступает только одна гидроксильная группа углевода, хотя, исходя из лииейиой структуры, в утих условиях должно образоваться соединение типа простого эфира по всем гидроксильным группам. Не имело объяснения характерное для .моносахаридов явление мута ротации (от лат. ти«> - «изменяю», rot at io «вращение») изменение величины оптического вращения свежеприготовленных растворов. Для объяснения этих фактов русским химиком А. А. Колли (1870 г.), а затем - немецким химиком Б. Талла i-сом (1883 г.) было высказано предположение о циклическом строении моносахаридов.
Как известно, альдегиды реагируют со спиртами с образованием полуацеталей (см. ки. 2. с. 328).
R-C' + R-0H и
ОН
R-C-OR ч Н
полуацеталь
Аналогично, моносахариды, являясь полигидроксиальдегидами или полигидроксикетоиами. образуют циклические полуацетали в результате внутримолекулярного взаимодействия карбонильной и пространственно сближенной с ней спиртовой групп. Причем, в соответствии с теорией напряжения циклов, наиболее благоприятно взаимодействие, если оно приводит к образованию пяти- или шестичленных циклов. Шестичленный цикл образуется при взаимодействии оксогруппы с гидроксильной группой при С' альдо гексоз или С° кето гекс пз. О и называется пиранозным (от шестичлеиного гетероцикла пираиа).
ОН О
НО-Н
6сн,он
ОН О
5 mi
129
Р-Р-фруктопирано ia D-фру кцш а-Р-фруктОпнрано (а
При взаимодействии оксо группы с гидроксильной группой при С4 альдогексоз или С кетогексоз образуется пятичленный цикл, называемый фуранозным (от пятичленного гетероцикла фурана).
6 6 6
сн,он сн,он СН,ОН
р-Р-глюкофурано:й D-глюкоча а.-/)-1.нокофураП1> ia
СН2ОН (Н,ОН сн,ои
Р-/)-фрукт<ч|>урацо’1а />-фрукто ш а-Р-фруктофургпкш
Внутримолекулярное образование полуацеталя приводит к тому, что углеродный атом карбонильной группы превращается в асимметрический. Этот новый хнральиый центр называется аномерным, а соответствующие ему два новых стереоизомера а- и p-аномерами. Образовавшаяся в процессе циклизации моносахарида гидроксильная груп-130
па при аномерном центре называется полуацетальной или гликозидной, у а-аномера расположение полуацетального гидроксила такое же. как гидроксила «концевого» хирального центра (асимметрического атома углерода, определяющего принадлежность к D- или L-ряду). Таким образом, в проекционных формулах моносахаридов /2-ряда гликозидный гидроксил у а-аномера расположен справа от вертикальной линии углеродной цепи, а у p-аномера слева.
Приведенные выше изображения циклических форм моносахаридов называются формулами Колли-Толленса.
Так как формулы Колли-Толленса громоздки и неудобны для изображения циклических структур, английский химик-органик X.Хеуорс в 20-х годах XX столетия предложил изображать циклические формы моносахаридов в виде плоских многоугольников, расположенных перпендикулярно плоскости рисунка. Химические связи цикла, находящиеся над плоскостью, изображают жирными линиями, а связи, расположенные за плоскостью обычными линиями. Атом кислорода в цикле располагается за плоскостью рисунка, причем в пиранозном цикле в правом углу. Символы атомов углерода в цикле обычно опускаются. Заместители располагаются над и под плоскостью цикла.
iiiipaiiO'iiibiH никл цикл
Эти формулы получили название формул Хеуорса.
Для перехода от формул Колли-Толленса к формулам Хеуорса используют следующие правила:
1. Заместители, расположенные в формуле Колли-Толленса слева от вертикальной линии углеродной цепи, изображаются в формуле Хеуорса над плоскостью цикла, а заместители, расположенные справа под плоскостью. Это означает, что у а-аиомера моносахаридов /2-ряда полуацетальный гидроксил находится под плоскостью цикла, а у [3-аномера - над плоскостью.
2. У альдогексоз /2-ряда в пиранозной форме группа CILOH, а в фуранозной - группа CHfOHiCHjOH всегда располагается нал плоскостью цикла, например:
н----ОН о
НО——н Н—1—он Н —------->
сн;он
и ДЗ-глюкопнрапота
5*
131
полуацетальный гидроксил
X-, ,( i
1HOT-C—H он °
НО—н
н——он
6
сн,он
р-£>-глюко фура пота
Аналогично изображаются с помощью формул Хеуорса пираноз^ ные и фуранозные формы кетогексоз.
а -I.)-фруктами рэпо м
р -О-фруктофурачпш
При изображении рацемической формы а- и Р-аномеров в формуле Хеуорса символ атома водорода у гликозидного атома углерода опускают, а расположение гидроксильной группы обозначают волнистой чертой.
У-глокопираиоча
Таутомерия. Моносахариды являются таутомерными веществами. В кристаллическом состоянии оии имеют циклическое строение. Так, Л-глюкоза. полученная дробной кристаллизацией из этилового спирта иди воды, находится в форме а-/)-глюкопиранозы. В водном растворе циклическая форма под влиянием растворителя превращается через открытую оксоформу в другие циклические формы пиранозные и фуранозные с а- и Р-конфигурацией аномерного центра. Таким образом, в водном растворе моносахариды существуют в виде пяти таутомерных форм открытой, а- и p-пираиозных н а- и Р-фуранозных.
132
P in ко фураноза
р-^-глюкопирапози
Такой вид таутомерии называется цикло-оксо-таутомерией. или коль чато-цеиной.
Переход одной формы н другие происходи т непрерывно. Через определенное время в растворе устанавливается подвижное (динамическое) равновесие, при котором количество всех форм остается постоянным. В равновесной смеси таутомеров альдогексоз преобладают пиранозные формы. Например, равновесная система />глюкозы состоит из 64% (З-Л-глюкопиранозы и около 36% ос-Л-глюкониранозы. Фуранозные формы и открытая присутствуют в очень малых количествах.
Аналогичные таутомерные превращения происходят в водных растворах кетоз. Ниже приведена схема г|мкдп-цлто-таутомерии Л-фруктозы.
Н
а-Р-фрук! «пираноза
u-D -фру к i офу ра п оча
носн 0 он
пн н\| l/CH.OH он и
но—р-н н——он н-|^он
ci LOH
Р-фрукГ|УШ
(3 -D -фру КГО и ира| 10'3
Р -/J -фрук юфу paHO'ia
В равновесной системе таутомеров /)-фруктозы преобладают фуранозные формы.
Способность моносахаридов к цикло-цксо-таутомерии объясняет выявленное задолго до установления их строения явление мута ротации.
133
Мутаротации ( от лат. muto — «изменяю» н rotatio — «вращение») — самопроизвольное изменение величины оптического вращения свежеприготовленных растворов оптически активных соединений.
Например, в свежеприготовленном водном растворе глюкозы наблюдается уменьшение угла оптического вращения с +113° до установления постоянного значения +52,5°. Химической основой злого процесса является 2(мк.7«-окс«-таутомерия. Как известно, кристаллическая глюкоза представляет собой а-/)-глюкоиираиозу, которая имеет удельное вращение [а+113°. При растворении в воде эта форма через альдегидную превращается в другие таутомерные формы, между которыми достигается состояние равновесия. В равновесной системе находятся все 5 таутомерных форм, по преобладает p-D-глюкопираноза. имею->о
шая [а]р +19°. Внешним проявлением этого процесса является постспеи-иое уменьшение угла вращения до установления постоянной величины +52.5°, характерной для равновесной смеси таутомеров.
Конформации моносахаридов. Фуранозные формы моносахаридов имеют практически плоское пространственное строение, а. следовательно. заместители в пятичлен лом цикле вынуждены находиться в нестабильной заслоненной конформации.
Для пиранозных циклов наиболее предпочтительной, отвечающей минимуму энергии, является форма кресла, в которой заместители находятся в конформации, близкой к заторможенной. Поэтому фуранозные формы моносахаридов термодинамически менее выгодны, чем пиранозные.
Следует отмстить, что из двух возможных типов конформации кресла пиранозных форм (JC4 и 4Сф)‘ . более устойчивой является та. у которой максимальное число объемных заместителей (группа ОН и особенно СН2С)Н) находится в экваториальных положениях.
У С1ЦОН сн.он н
н 0Н он он и он
форма кресча 4Cj форма крест
Для большинства альдогексоз D-ряда более предпочтительным является кресло 4С|.
Цифры в верхних и нижних индексах указывают иа номера атомов углерода, находящиеся в верхних и нижних положениях кресла.
134
Для перехода от формулы Хеуорса пиранозного цикла к конформации кресла С, изображают фигуру кресла с атомом кислорода, в которой атом С находи гея в верхнем, а атом С1 в нижнем положениях. Гликозидный гидроксил у а-аномера располагаю! аксиально, у р-ано-мера экваториально. Остальные заместители переносят с учетом иш -или транс- положения их в формуле Хеуорса по отношению к гликозидному гидроксилу, например:
5.1.4. Способы получения
Углеводы широко распространен ел в природе. Они образуются в зеленых частях растений из углекислою газа и воды в процессе фотосинтеза.
пСО-. + 111 ГО --------------
хлорпфн.'т
Спн2поп + пО?
Важнейшим способом получения моносахаридов является кастомный гидролиз природных ди- и полисахаридов. Так. D-глюкозу получают гидролизом крахмала, смесь D-глюкозы с D-фруктозой гидролизом сахарозы и т.д.
135
Синтетические методы применяют, как правило, для получения малодоступных моносахаридов. В большинстве случаев эти методы основаны на превращении моносахаридов, легко выделяемых из природных источников (£>-глюкоза, /^-галактоза. £-маиноза и др.) в другие моносахариды путем укорочения или удлинения углеродной пели.
Распад по Руффу. Это одни из классических методов укорочения углеродной цепи, основанный на окислении моносахаридов.
( 11 1кк ( ,^О HiO^_ <^Oi 1 С ' I т 1 Н ( А '
и о Un 1_1 н он с -о —14
1 ю VI 1 1 Br. HU г! НО— н Г1к? и — —он —он‘
11 н— U11 —он н,ом 11 Un F-e + re г I ОН -со,> гчи - п н—
t 1 ин П LM 1
( :н2он :н2он ( ЗН2ОН ( ?н,он
/J-ицокоча Р-1 люко новей 2-iwi о-Р-1 /ПОКОНОННЯ Р-арабнноза Л
Сначала моносахарид окисляют в мягких условиях до альдоновой кислоты (см. с. 138). Далее кислоту окисляют пероксидом водорода в присутствии солей железа (Ш) до 2-кетоальдо новой кислоты, которая в результате декарбоксилирования превращается в моносахарид, содержащий иа один атом углерода меньше, чем исходный.
Циаигидршювый синтез. Сущность метода заключается в присоединении ниаиоводорода по двойной связи карбонильной группы альдозы с последующим гидролизом образовавшегося гидроксииитрила до гидроксикислоты, которая через стадию лактонизации и восстановления превращается в альдозу, содержащую на один атом углерода больше, чем исходная.
к 0. ОН 0, 1 Ч х 1 д Н. А} 1
с^ снон СН-ОН СН-ОН СН-ОН
СН-ОН сн-он СН-ОН СН-ОН о сн-он
сн-он HCN СН-ОН нон СН-ОН СН-ОН |Н] сн-он
<5н-он ( —* СНОП ' 1 -NH, VH-°H 6н—' 1 ”* £н он 1
СН-ОН СН-ОН СН-ОН СН-ОН сн-он
сн.он СН,ОН СН,ОН СН,ОН сн2он
ajib/ioitKCOiii ГИЛрОКСИПИТрИ: 1 iHjpOKeiiKHCJioia JUlK1! oil а.1ъ;ю гептоза
Циапгидриновый синтез позволяет наращивать углеродную цепь и применяется для получения высших альдоз из низших.
136
5.1.5. Физические свойства
Моносахариды твердые гигроскопичные вещества, легко растворимые в воде, трудно растворимые в этаноле и практически нерастворимые в диэтнловом эфире, бензоле, диоксане. Значительная часть моносахаридов является кристаллическими веществами. Водные растворы моносахаридов имеют, как правило, сладкий вкус и нейтральную реакцию. В растворах молекулы моносахаридов сильно сольватированы, что приводит к образованию вязких «сиропов», из которых процесс кристаллизации в значительной степени замедляется.
Это объясняется, с одной стороны, медленным образованием центров кристаллизации из-за затруднений ориентации молекул моносахаридов в вязких растворах, а с другой стороны, установлением таутомерного равновесия с низкой концентрацией таутомера, наиболее склонного к кристаллизации.
Кристаллы многих моносахаридов состоят из молекул в пиранозной форме. Растворы моносахаридов оптически активны.
5.1.6. Химические свойства
Являясь полигндроксикарбоиильными соединениями, моносахариды проявляют химические свойства карбонильных соединений, многоатомных спиртов, а также циклических полуацеталей.
Химические превращения в ряду моносахаридов можно условно разделить на две группы: реакции с участием открытых форм .моносахаридов и реакции с участием циклических форм.
А. Реакции с участием открытых форм
Восстановление. При восстановлении моносахаридов водородом в присутствии катализатора (никель, палладий), боргидрндом натрия илн амальгамой натрия в разбавленной серной кислоте образуются многоатомные спирты. Из D-глюкозы- при этом получается спирт — D-сорбит, из D-маннозы D-маннит, из D-ксилозы D-ксилит и т. д.
В- *4 сн,он в- 'С*° ( :н,он
Н- —ои и- 4~ он н- -4—он н он
но- — “н ли но- -п—Н но- —1—11 1HI Н0' — —и
н- -он . н- “|—он п- -4-он ‘Ч. н —он
( :н,он CIhOH Н' он н он
СН.ОН сн.он
Р-ксилоза Д-ксилит /?-1 |цоко за Л-а>рбп i
D-Ксилит и D-сорбит кристаллические вещества, сладкие на вкус, применяются при сахарном диабеге как заменители сахара, ^-сорбит является промежуточным продуктом в промышленном синтезе аскорбиновой кислоты из D-глюкозы. При восстановлении ^-фруктозы образуется эквимолекулярная смесь D-сорбита и 0-манпита. что связано с превращением карбонильной группы при С' в асимметрический центр.
J37
сн,он 1 -с=о с н— :н,он —он с но— ?н,он —н
НО- —н [Н]^ НО— —н . но— —н
н- —он * н — —он h н- —он
н —он окон н— ( —он :н,он н-( —он :н2он
Р-фруктлга /)-спрбн г Л>-ман!пп
Окисление. Моносахариды легко окисляются, ио в зависимости от природы окислителя и условий окисления, образуются разные продукты. В кислой и нейтральной среде окисление протекает без разрушения углеродной цепи молекулы, в щелочной среде, как правило, оно сопровождается расщеплением углеродного скелета.
Окисление в кислой и нейтральной среде. При использовании слабых окислителей, например, бромной воды или разбавленной азогной кислоты, альдозы окисляются с образованием одноосновных полиоксикислот, получивших общее название альдоиовые кислоты. Л-гяюказа в этих условиях дает Л-глюконовую кислоту. D-\ алактоза D-галакто новую кислоту н т.д. В кислых растворах образовавшиеся альдоцовые кислоты отщепляют воду и превращаются в соответствующие у-лактоны.
нч н— —он н°\ н— ,^О —он
Вг2 + Н,О rl но— нН 0
ни II — н— ( 11 —он —он ‘,'ОН ни н н+^ Н j
(НВгО) н н— он —он JH2OH -и,о н 11 — ( —он ?н,он
Рз;иоко1а /J-глкжонопая к нс. юга у-лактои Z)-ыкшлиовон кисюгы
Кальциевая соль Л-глюкоиовой кислоты
глюкопат кальция
( 200 - Са2+ применяется в медицине при аллергических заболеваниях, желудочных, кишечных, легоч-
и — —он ных, маточных и носовых кровотечениях.
но н различных заболеваниях кожи, токсических
н — —он • Н20 поражениях печени и др.
н он Кетозы бромной водой ие окисляются.
( 21L0H Сильные окислители, например, концепт-
L 2 рированная HNO3. окисляют в Молекуле аль-
г.чюкоищ кальция доз альдегидную и первичную спиртовую
группы с образованием дикарбоновых оксикислот, получивших общее название альдаровые или сахарные кислоты, Так, Л-глюкоза окисляется
в Л-глюкаровую кислоту, Л-манноза в Л-маинаровую. Л-галакто-за в /)-галактаровую (слизевую) кислоту, например :
138
При
н~с*° н—I—он но—I—н
н-4—он н—|—он сн,он
/>-1ЛЮко*)а
к. HNOj
соон н—I—он
но---н
н—он н—|—он соон
Р-глюкаровая кислота.
D-сахарная кислоа а
окислении кетоз в аналогичных условиях происходит разрыв
углеродной цепи по месту карбонильной группы с образованием дикарбоновой кислоты.
сн2он
с=о соон
НО— —н к. HNO3 но— —н
н— он -СО н— —он
н — —он н — —он
сн.он соон
/)-<|>рукл о за
D-арабппароаая кислота
При селективном окислении в молекуле альдозы первичной спиртовой группы без затрагивания весьма склонной к окислению альдегидной группы образуются уроноаые кислоты. Окислению в таких случаях подвергают моносахариды с защищенной альдегидной группой, например гликозиды (см. ниже):
D-iяюкуронпвая кислота
Уроновые кислоты широко распространены в природе. Оии входят в состав многих полисахаридов. Наряду с А)-глюк у ромовой кислотой широко представлены D-галактуроповая. Л-маннуроноваа и А.-иду-роновая кислоты.
139
:оон
ХЮН
НО/Г Кон ну— Н
D-галакгуриновая юлснота
эн
эн
НО
н рнн
/соон
Кон 11 но Ч—F
эн
D-M'dtniypouoiVM кислота
>н
/.-цлурщтвая киакна
н
Н
I и
D-Галактуроновая кислота является компонентом пектина фруктов. D-маннуроновая кислота содержится в различных морских водорослях, /Э-глюкуро новая и L-идуроновая кислоты входят в состав гепарина и других полисахаридов.
Уро новые кислоты участвуют в процессе выведения из организма токсичных веществ.
Окисление в щелочной среде. Подобно альдегидам, моносахариды окисляются аммиачным раствором оксида серебра (реактив Толленса) и гидроксидом меди (П) в щелочном растворе, или же реактивом Фелинга (см. ки. 2, с, 443). В эти реакции вступают как альдозы, так и кетозы. т.к. в щелочной среде кетозы изомеризуются в альдозы (см. с. 139). С реактивом Толленса осуществляется реакция «серебряного зеркала». С реактивом Фелинга и гидроксидом меди (II) в щелочном растворе образуется оксид меди (И) красио-оранжевого цвета. Моносахариды при окислении в щелочной среде расщепляются до смеси продуктов окисления. Эти реакции являются качественными на альдозы и кетозы.
О
R—С-Н+ [Ag(Nlls)2]OH альдоза реактив Толленса
О
11
R С_Н + Си_+(комплекс) реактив Фелинга
Си;О|
продукты окисления
I продукты
окисления
Превращения моносахаридов иод действием щелочей (эпимеризация). В разбавленных растворах щелочей при комнатной температуре моносахариды подвергаются изомеризации с образованием равновесной смеси моиоз. различающихся конфигурацией углеродных атомов С] и С2. Так. /Э-глюкоза, выдержанная в растворе гидроксида натрия (8103) при 35 °C в течение 4 суток, превращается в смесь, состоящую из D-фруктозы (-28%). D-маннозы (~Э%) и D-глюкозы (-69%.). Аналогичная изомеризация наблюдается у кетоз, например, фруктозы. Изомерные превращения моносахаридов под действием щелочей называют эпимеризацией, так как приводят к образованию эпимеров, например глюкозы и маннозы.
Взаимопревращения в слабощелочной среде протекает через енди-ольную форму, которая образуется в результате миграции к карбонильной группе подвижного водорода при а-ато.ме углерода.
140
1 6+ L-H — —он
но— —н
н— —он
н— —он
сн,он
Л-глюкоча
НО---н
Н^О
ноч рн
но— —н
н — —он
н— —он
сн2он
D-манно ча
н----он
н—|—он
сн2он
ендиольиая форма
сн,он с=о
но-Ч Н1
н — —он
н— —он
сн,он
D-фруктоза
При обратном превращении еидиольвой формы в карбонильную образуется смесь трех моносахаридов.
Образование озазонов. При нагревании моносахаридов с фснил-гидразином в молярном соотношении 1:3 образуются бис-фенилгидра-зоны. получившие название озазоны.
Н ^О
II— —он
но— —н
н — —он
н — —он
сн,он
/З-глюкоза
3 C6H5-NHNH;
-C6H5-NH: ; -NH5 ; -2Н,0
HC.'=N-NH-CfiH5 6=N-NH-C6H, HO—I—H
H---OH
H—OH
CH2OH ota ton
Механизм реакции. Образование озазонов идет в несколько стадий. Вначале молекула моносахарида реагирует с одной молекулой феиил-гидразнна, образуя фенилгидразон, который в результате внутримолекулярной окислительно-восстановительной реакции подвергается перегруппировке в моиоимии 1,2-дикарбонильного соединения. Затем моноимин при взаимодействии с двумя молекулами феиилгидразина превращается в озазон.
Г NH
н ! 1,- ( ^-NH-CbH5 Н</5 ^н-суч, С>АН
н— —он нн>- -ОН о
но- — —н CeH$-NHNJL, но— н но——н
н —он 412О н— -он н—он
н —он н — — -он н он
СН,ОН
П-ГЛЮКОЧИ
СН,ОН снгидразин
сн,он фенилпчдраюн Л-глюкозы
141
HC=NH г=о 2 С6Н? -NHNH,
но— —н
в— —он -Н,О: -NH?
н — —он
( :н.он
MOiioiiMiiii 2-kci «альдегида
HC=N-NH-CHS
I 6 5
C=N-NH-C.HS о э
но— —н
в- —он
н — —он
CH.OH
озазон
Озазоны представляют собой кристаллические вещества желтого цвета, не растворимые в воде. Л-глюкоза, Л-манноза и /9-фруктоза дают один и тот же озазон. Под действием хлороводородной кислоты или при нагревании с бензальдегидом озазоны легко отщепляют две молекулы феиилгидразина, образуя соответствующие озоны (кето но-альдегиды). При восстановлении озонов амальгамой натрия в слабокислых растворах образуются кетозы.
hc=n-nh-c6h5
C=N~NH-C(, но—Нв и—он н-----он
2CtHs41l=N NH С6Н5
СН2ОН
ОЧ.НОН глюкозы
НС-о с~о но н—]—он н—(—он сн.он озон
В 211
CIBOH
С=О Г
—н —он
НО в н—(—он сн.он
П-фруктоза
Таким образом, реакция моносахаридов с фен ил гидразином позволяет осуществить переход от альдоз через озазоиы и озоны к кетозам.
Взаимодействие с гидроксиламином. Альдозы легко вступают в реакцию с гидроксиламипом. образуя оксимы. В присутствии водооз ни-мающиХ средств оксимы могут быть превращены в соответствующие оксинитрилы, которые под действием иоиов серебра отщепляют HCN и образуют оксиальдегиды, содержащие па один атом углерода меньше, чем в исходной альдозе. С помощью этих реакций можно осуществить переход от высших альдоз к низшим.
1L ( н — но— и— н— с р-1 ^0 1ч ( —он н— ОН с —он в — >N н —он ( ^0
н Nikon НО— I1 но —он —► н— ои 11,0 11 AgOIJ н
ин н мм -1ЦО [ т ОН 11 он
2 н ИТОН ( VlIOKO'iU ок ОН 2 Н JH.OH ( СИМ око ОН II 0 ' :н2он - ( нитрил Д-apti он JH.OH биПИ'Ш
142
Внутримолекулярная дегидратация. При нагревании с минеральными кислотами (HCI, H2SO4) пентозы подвергаются внутримолекулярной дегидратации с образованием фурфурола, а гексозы — 5-гидрокси-метнлфурфурола:
/но> Сон\
"7сн _
'bfi’iTof д
— п
альдо па поза
/но* <он\
<н/7н-нс\\н>
НОПХ q[h но(
[_]
альдо 1ексоп(
HCI; t
-ЗИ2О
5-rn;ipoKCiiMCi идфурфурол
Эта реакция позволяет отличить гексозы от пентоз. Фурфурол ласт красное окрашивание с анилином в присутствии хлороводородной кислоты (качественная реакция иа пентозы).
5-Гид рокси,метил фурфурол образует красное окрашивание с резорцином (реакция Селиванова на фруктозу). ч
Б. Реакции с участием циклических форм
Образование гликозидов. Моносахариды, являясь циклическими полуацеталями, реагируют в присутствии кислотного катализатора ио спиртами и фенолами. Реакция протекает с участием полуацетальной гидроксильной группы и приводит к образованию циклических ацеталей, получивших название гликозиды. Независимо от исходной формы моносахарида, в процессе реакции образуется смесь а- и [J-гликозидов.
СН,ОН
Н ОН
СИ,ОН
2( Н -О Н V/н
... 2 .i LZ П >J
HCI (гач) l\OH Н/
-2H,o HO |r OC2H5
H OH
CH, OH
H OH
a-/J-i jiOKOitnpuHOiii тпы-а-Р-глюкониратпид n HJi-p-fl-i.iioMniiipaiiinuji
Названия гликозидов образуют из названий моносахаридов, заменяя суффикс -озл ла -озид. Например: фруктозид. галактозид, рибозид, глюкозид и т.д. В зависимости от размера цикла (пиранозный, фуранозный) гликозиды делятся на пиранозиды и фуранозиды, а- и р-Ано-мерам моносахаридов соответствуют а- и 0-гликозиды. Неуглеводную часть молекулы гликозида называют агликоном. Химическая связь между аномерным атомом углерода моносахарида и агликоном в гликозиде называется гликозидной.
143
сн.он 1 ЛИКО1ИД1(Е1Я
н он
глюкозил
В связи с тем, что в молекулах гликозилов отсутствует свободный полуацетальный гидроксил, оии, в отличие от моносахаридов. не способны к таутомерии в водных растворах, нс мутаротируют и не проявляют восстанавливающих свойств.
Гликозиды как ацетали легко гидролизуются в кислой среде, но проявляют устой
чивость к гидролизу в слабощелочной среде. В процессе гидролиза, образуется смесь а- и [5-апомеров соответствующего моносахарида.
СН.ОН СНпОН
Meiiiji4X-/)'r'iiOKniibip»iiniii;( Л-i люк» и и рано ча (смесь и- и р-аномеров]
Гликозиды весьма широко распространены в природе. В подавляющем большинстве оии являются р-гликозилами. В качестве агликонов в природных гликозидах часто выступают гидроксилсодержащие соединения фенолы, стероиды (см. разд. 3.7.3 и Х.3.2, В) и сами моносахариды. Связь агликона с аномерным атомом углерода в этих
синшрип
соединениях осуществляется через атом кислорода, поэтому такие гликозиды называют О-гликозидами. Примером О-гликозидов является содержащийся В ЛИСТЬЯХ ТОЛОКНЯНКИ 1ЛИКО-зид арбутин.
Кроме ()-гликозидов. известны N-ГЛИКОЗИДЫ и S-гликозиды.
В N-гликозидах агликонами являются остатки алифатических, ароматических, гетероциклических аминов и другие NI 1-содсржашие органические соединения. Связь агликона с моносахаридом в N -гликозидах осуществляется через атом азота. К М-глико-зидам принадлежат играющие важную роль в обмене веществ продукты расщепления нуклеиновых кислот и нуклеопротеидов (нуклеотиды, нуклеозиды), АТФ, а также некоторые антибиотики и др.
Примером S-гликозидов является гликозид сипигрин, содержащийся в семенах сарептской горчицы.
144
В S-гликозидах агликонами являются тиолы, а связь агликона с моносахаридом осуществляется через атом серы.
Алкилирование. При взаимодействии моносахаридов с галогеналка-иами или диметилсульфатом (CHi)iSO4 в реакцию вступают все гидроксильные группы, включая полуацетальный гидроксил. В результате реакции образуются гликозиды, алкилированные по всем гидроксильным группам. Такие соединения в кислой среде гидролизуются только по гликозидной связи. Образовавшиеся простые эфиры по остальным гидроксильным группам гидролизу ие подвергаются.
МСТИ л-2.3.4.6- геграмстил-П-глюкрниранозид
2.3.4.6-тстрам ei и.1-
Р-глюко г । и ра и с ча
Ацилирование. При взаимодействии моносахаридов с ангидридами карбоновых кислот легко образуются сложные эфиры по всем гидроксильным группам. Так. при действии на глюкозу уксусным ангидридом образуется иентаацетилглюкоза.
5 (СН,СО),0
СН_ОАс
Н
<:н,соо№ Лс0
Ас + 5 АсОН
Ас
Н
Н
ОАс Н
где Ас = СИ.СО
шз44*/Э-1.иоконирш1о.!0ф пептаацегилирокоиирлноз^^
В кислой и щелочной среде сложные эфиры моносахаридов гидролизуются.
Среди сложных эфиров моносахаридов важное значение имеют эфиры фосфорной кислоты. Они содержатся во всех растительных и животных организмах и играют большую роль в обмене веществ. Фосфаты рибозы и дезоксирибозы входят в состав нуклеиновых кислот (см. разд. 7.1). фосфаты D-глюкозы и D-фруктозы принимают участие в обмене углеводов, пирофосфориые эфиры моносахаридов участвуют в синтезе пуриновых и пиримидиновых нуклеотидов. Фотосинтез, брожение и другие биологические процессы также осуществляются с участием фосфатов моносахаридов.
Ниже приведены некоторые фосфаты моносахаридов, играющие важную роль в обмене веществ.
145
рибозо-З-фосфат
глюкозо-1-фосфаг о
СН-О-Р-ОН он Н
Н/Н он
но
н
он
он
н
и по ко ю-6-фосфат
о
НО-Р—о-сн
ОН
I
н
НО
он
о
СНг<)-Р-ОН он
он фрук! о ю-1.6-дифосфат
5.1.7. Отдельные представители моносахаридов
СК
V Н----ОН
НО----Н
Н—|—ОН
сн2он 2)-ксп;юза
и
н он
а-D-ксило пи рал оза; .1реиеспый сахар
О. 'Н
н— —он
н— —он
н — —он
сн2он
D-рибоза
он он p-D-рнбофуранози
D-Ксилоза. Является структурным фрагментом полисахарида ксилана. содержащегося в древесине, соломе, подсолнечной шелухе. В состав ксилана входи! в виде nt-D-ксилонираиозы; используется для синтеза ксилита.
D-Рибоза, В 0-фураиозной форме D-рибоза входит в состав РНК. ряда коферментов, гликозидов и антибиотиков.
о. с н—|—он но но-|—н сн2он Л-арабино и
о. JI
С он н он он сн2он D-глюкоза
н
L-арабипопираноза
и— —
но— —
н— —
н— —
сн.он
н он
Р -D-1 люко п при ч и
L-Арабиноза. Содержится в свободном виде в древесине хвойных пород деревьев. Входт в состав растительных гликозидов, полисахаридов растений — арабинанов.
D-Глюкоза (виноградный сахар. декстроза). Широко распространена в природе: в свободном состоянии находится в растениях. меде, крови: входил в состав многих дисахаридов (лактоза, сахароза и др.): полисахаридов
146
(крахмал, клетчатка, гликоген и др.). а-Аномер кристаллизуется из воды. т. пл. 146 °C; p-аномер из пиридина, т. пл. 148-150 °C.
Глюкоза главный источник энергии для большинства организмов. Получают гидролизом крахмала или целлюлозы в присутствии минеральных кислот.
Глюкоза используется в качестве сырья для производства виз амина С и лекарственного препарата глюконата кальция: в медицине применяется в виде растворов для внутривенного введения при гипогликемии. инфекционных заболеваниях, заболеваниях печени и т.д.; является компонентом различных кровезаменителей и противошоковых жидкостей. Под действием ферментов глюкоза подвергается брожению. Известно много видов брожения спиртовое, молочнокислое, маслянокислое. лимоннокислое и др. Наиболее важным из них является спиртовое брожение, которое происходит под влиянием фермента дрожжей зимазы.
дрожжи
С?6НГО6 —---------► 2C,HSOH + 2 СО-,
(знм:па)
Этот вид брожения используют в промышленности для получения этанола, а также в виноделии и пивоварении.
(X л
н — рон
но— —II
но— —н
н— —он
сн,он
D-Галактоза. Входит в состав дисахарида лактозы, содержащейся в молоке, а также некоторых гликозидов и полисахаридов. Полу; чают гидролизом лактозы.
Д-галактст р-7)-i •алактопирапоаа
О. Н
но— —н
но— —н
н— —он
н— —он
( >ГОН
D-манноза
сн. ,он он
н ч!
он но
но н н 11
р п ил нираноча
сн.он с=Ь но—I—н н------он
НО—|—11 с н.он
Ц-еорбоча
Н
ОН н он
a-L-uopf) и и н ра к о за
D-Маниоза. Является структурным фрагментом полисахарида маннана, который содержится в оболочке семян каменного ореха; в свободном состоянии находится в кожуре апельсинов. Получают гидролизом маинана.
L-Сорбоза. Получают при микробиологическом окислении D-сорбита. Этот процесс является важной промежуточной стадией в синтезе витамина С.
147
сн,он i=b НО—4—Н п-----он
Н—он
сн.он П-фруктозн
он
/>-фруктофурапоза
Д-Фруктоза (плодовый или фруктовый сахар, левулеза). В свободном состоянии содержится во фруктах, меде: входит в сослав ряда олигосахаридов (сахароза, раффиноза) и полисахаридов (инулин).
Фосфаты Д-фруктозы промежуточные продукты энергетическо
го обмена углеводов в живых организмах. Фруктоза слаще глюкозы и сахарозы. Кристаллическая фруктоза представляет собой фруктопира-
лозу (т. пл. р-аномера 104 °C). В состав олиго- и полисахаридов обычно входит в фуранозной форме.
Получают фруктозу гидролизом инулина, содержащегося в клубнях
георгинов, корнях цикория.
5.1,8. Отдельные представители дезокси- и аминосахаров
Дезоксисахарами называют моносахариды, в .молекулах которых . одна или несколько гидроксильных групп заменены иа атом водорода.
Аминосахара моносахариды, в молекулах которых одна или несколько гидроксильных групп замещены аминогруппой.
О.
н— —II
н— —он
н— —он
сн3он
2-дезоксн-О-рибош
2-дею кси-
Л-рнбпфурииоз»
Н —I— NH НО но—I— н н—ом сн,он
11 — — NH
но— —н
н— —он
н — —он
сн2он
2-Дезокси-Д-рибоза. Является структурным фрагментом дезоксирибонуклеиновых кислот (ДНК). Получают гидролизом ДНК или синтетическим путем из Д-глюкозы.
н
Д-Галактозамни (2-амнно-2-дсзокси-Д-салактоза, хоидроза-мии). Является структурным фрагментом хоидроитинсуль-фатов и полисахаридов некоторых бактерий.
Д-Глюкозамин (2-амипо-2-лезоксн-Д-глюкоза. хнтозамии) Является структурным компонентом гликопротеидов и полисахаридов. Его N-метилпро-изводное - структурная часть антибиотика стрептомицина, а N-ацетилпроизводное - хитина. Получают Д-глюкозамии гидролиз01М хитина.
148
5.1.9. Отдельные представители метил пентоз
В молекулах природных сердечных гликозидов, многие из которых *ашли применение в медицине для лечения сердечной недостаточности, содержатся остатки пентоз, у которых атом водорода при С замещен цетильной группой. Такие моносахариды называют метил пентозами. К пнм относятся Л-рамноза (6-дезокси-£-манноза), Р-дигитоксоза 2,6-дидезокси-Л-альтроза) и др.
5.1. [0. Отдельные представители производных моносахаридов
Аскорбинован кислота (витамин С, у-лактои-2,3-дегидро-£-гулоновой .
НО ОН кислоты). Водорастворимый витамин, содержащийся во многих овощах и фруктах. Недо-
ОН он статок витамина С в пище вызывает заболе-
вание пингу, а также понижает сопротивляемость организма к инфекционным заболеваниям.
Аскорбиновая кислота сильный восстанавливающий агент; при окислении превращается в дегндроаскорбиновую кислоту, которая, в свою очередь, способна довольно легко восстанавливаться до аскорбиновой кислоты.
аскорбиновая кислота
-2Н
+2Н
дегидрогкжорбиноиая кислота
Роль витамина С в организме связана с его участием в окислитель-
но-восстановительных процессах, углеводном обмене, в регенерации тканей, в синтезе стероидов и др.
Аскорбиновая кислота обладает сильными кислотными свойствами (рК.» 4,2). что связано с диссоциацией одной из гидроксильных групп еидмольного фрагмента:
J49
енднольным фрагмап'
аскорбат- пип
В промышленности аскорбиновую кислоту получают из Л-глюкозы:
1k ^о ( :н,он
н— —он н —он н
но— н 1Н] НО- — н (О] но
11— —он —н —он —► н
н— —он н он
( :н,он сн?он
C1LOH СН, ОН
--ОН С=О
—11 но—I—н [OJ
—он = н—он—*-с=о но—|— н
сн,он сн,он
но---н
СИ,он
1) -глюкоза D-сорбц г /.-сорбоза
С0Ю11 ’ j u--i; с=%:: но-----н/J ;
-► Н-------O[jl_J
2-ктто-А-гул(И1<1вая кислота аскорбиновая кислота
)'
Потребность в витамине С' для человека 50-70 мг в сутки. >
Нейраминовая кислота (3,5-дидсзокси-5-амииононулововая кислота).
Является производным моносахарида кетоионозы но-нулозы. В природе нейраминовая кислота чаще всего встречается в виде N- и О-аци-лированных производных, объединенных общим названием сиаловые кислоты (ацильным фрагментом обычно являются остатки уксусной или гликолевой кислот).
структурные компоненты гликопротеидов.
Сиаловые кислоты специфических веществ крови и тканей, ганглиозидов мозга.
150
5.2, ДИСАХАРИДЫ
Днсахарида.ми называют углеводы, молекулы которых состоят нз двух остатков моносахаридов одинаковой или разной природы, соединенных между собой гликозидной связью.
Являясь О-гликозидами, дисахариды легко гидролизуются в кислой среде с образованием двух молекул моносахаридов. В зависимости от способа образования гликозидной связи, дисахариды разделяют на две группы восстанавливающие и иевосетанавливающие.
5.2.1. Восстанавливающие днсахарнды
СН.ОН ... СН.ОН
В восстанавливающих дисахаридах гликозидная связь образуется за счет полуацетальной (гликозидной) гидроксильной группы одного и любой спиртовой гидроксильной группы (чаще у С.’4) другого моносахарида. При зюм в молекуле остается одна свободная полуацетальная гидроксильная группа, вследствие чего дисахарид сохраняет способность к 1(мон-оксо-таутомерии, и следовательно, обладает восстанавливающими свойствами. В свежеприготовленных растворах таких дн-сахаридов наблюдается явление мутаротации. Представителями восстанавливающих дисахаридов является мальтоза, целлобиоза, лактоза. Мальтоза (солодовый сахар). Молекула мальтозы состоит из двух остатков Л-глюкопиранозы, связанных J ,4-гликозидной связью. При атом остаток глюкозы, аномерный атом углерода которого участвует в образовании гликозидной связи, находится в сх-фор-ме, а остаток глюкозы со свободной полуацетальной гидроксильной группой может иметь «-конфигурацию (а-мильтоза) или p-конфигурацию ((5-мальтоза).
Таким образом, а-малыози
может быть названа как 4-0-(а-/^-глюкопиранозидо)-а-/)-глюкопирано-за. а р-мальтоза 4-0-( а-Л-глюкопираиозидо)-р-Л-глюкопираноза. Мальтоза является восстанавливающим дисахаридом. В растворе существует в нескольких таутомерных формах а- и p-циклической и альдегидной.
сл-1,4-1Лцкои1дпая связь
CILOH СП,ОН
а-фирма
альда-идиая форма
151
[I -<[10 p MH
Растворы мальтозы мутаротируют и дают положительную реакцию с реактивом Толленса и реактивом Фелинга. С участием альдегидной формы мальтоза вступает в характерные для моносахаридов реакции с фенилгидразином, гидроксиламииом. циановодородной кислотой. При окислении в мягких условиях, например, бромной водой, мальтоза превращается в мальтобионовую кислоту:
Г
За счет полуацетального гидроксила мальтоза образует гликозиды :
С участием циклических форм мальтоза, аналогично моносахаридам, образует простые и сложные эфиры по всем гидроксильным группам:
152
Мальтоза содержится в небольших количествах в некоторых растениях, образуется при ферментативном гидролизе крахмала. Она легко растворяется в воде, водные растворы имеют сладкий вкус. В организме человека мальтоза расщепляется ферментом мальтазой до Р-глюкоэы.
Целлобиоза. Молекула целлобиозы,
В-1.4-1 ликвидная свячь
как и мальтозы, состоит из двух остатков Р-Г.'покопиранозы. связанных 1,4-гликозидной связью. Но. в отличие от мальтозы, в молекуле целлобиозы остаток глюкозы, полуацетальный гидроксил которого участвует в образовании гликозидной связи. имеет р-Koj [фигурацию. Остаток же глюкозы со свободной полуацетальной группой.
• аналогично мальтозе, может иметь а- и [^-конфигурацию. В соответствии с ним различают а- и Р-целлобмозу.
Исходя из Химической структуры, а-целлобиоза имеет название — 4-О-(р-/?-глюкопиранозидо)-а-Л-глюкопирано’за. а р-целлобяоза — 4“0-(Р-Р-глюкопиранозидо)-р-Л-глюкопираноза.
Целлобиоза является восстанавливающим дисахаридом. Ее раство
ры проявляют мутаротацию. дают положительную реакцию с реактивами Толленса и Фелинга. При окислении целлобиозы в мягких условиях образуется целлобиоповая кислота. Целлобиоза вступает также во многие другие реакции, свойственные восстанавливающим дисахаридам (см. с. 152).
Целлобиоза и мальтоза имеют разное пространственное строение. В
молекуле мальтозы а-гликозидная связь расположена аксиально, а в молекуле целлобиозы р-гликозидная связь — экваториально.
Такая пространственная структура мальтозы является причиной клубкообразного строения амилозы (составная часть крахмала), а пространственная структура целлобиозы — причиной линейного строения целлюлозы.
153
Целлобиоза является бесцветным кристаллическим веществом, легко растворяется в воде. Ола не расщепляется в организме человека и поэтому не может быть использована в качестве продукта питания.
Лактоза^ (молочный сахар). Молекула лактозы состоит из остатков Л-гала кто пиранозы и Р-глюкопн-ранозы, связанных L4-гликозид? ной связью. В образовании гликозидной свяЗ^ принимает участие полуацетальный гидроксил Z)-ra-лактопиранозы. имеющей р-конфигурацию. Остаток D-глюкони-ранозы может иметь а- и (^конфигурацию. в связи с чем различают а- и р-лактозу. сх-Лактоза может быть названа как 4-О-(0-£)-галак-топиранози до )-a-D-глюкопираноза, а 0-лактоза как 4-О-(0-Р-га-
лактопираиозило)-0-/)-глюкопираноза.
Пространственное строение лактозы подобно строению целлобиозы, т.е. р-гликозидиая связь расположена экваториально.
р-1.4'1 ли ко ныная связь
Лактоза является восстанавливающим дисахаридом. В растворе она существует в нескольких таутомерных формах — альдегидной, а- п р-циклической. В связи с этим растворы лактозы мутаротируют и дают положительную реакцию с реактивом Толлснса и реактивом Фелии! а-154
При окислении лактозы в мягких условиях образуется лактобиоповая кислота. Лактоза содержится в молоке. Она не подвергается спиртовому брожению, обладает в 4-5 раз меньшей сладостью, чем сахароза. При кислотном или ферментативном гидролизе лактозы образуется р-глюкоза и D-галактоза. Лактоза обладает низкой гигроскопичностью, применяется в фармации прн изготовлении порошков и таблеток.
5.2.2. Невосстанавливающис дисахариды
В молекулах невосста и-а вливающих дисахаридов гликозидная связь образуется за счет полуацетальных гидроксильных групп обоих моносахаридов. Такие дисахариды не имеют в своем составе свободного полуацетального гидроксила, поэтому в растворах они существуют только в циклической форме, их растворы ис мутаротируют и не обладают восстанавливающими свойствами. Невосстанавливающие дисахариды не дают реакций по альдегидной группе и гликозидному гидроксилу. Они способны лишь к образованию простых и сложных эфиров. Представителем левосстаиавлнвающих дисахаридов является сахароза.
Сахароза (тростниковый или свекловичный сахар). Молекула сахарозы
aixapoiii
состоит из остатков D-глюкозы и /З-фрукгозы. При этом D-глюкоза входит в состав сахарозы в форме a-D-глюколиранозы, а D-фруктоза в форме p-D-фруктофуранозы. Гликозидная связь между a-D-глюк о пиранозой и [i-D-фруктофуранозой образуется за счет полуацетальных гидроксилов обеих молекул. Исходя из химической структуры, сахарозу можно назвать как a-D-глюкопнра-нозидо-р-D-фру ктофуранозид.
С а хароза п редставляст сабо и бесцветное кристаллическое веще
ство, хорошо растворяется в воде, имеет сладкий вкус. Растворы сахарозы оптически активны [о.]" +66.5°], не мутаротируют п не проявляют
восстановительных свойств.
Под действием минеральных кислот при нагревании сахароза гидролизуется с образованием смеси D-глюкозы и D-фруктозы. При этом Происходит изменение знака удельного вращения, т.е. характерное для сахарозы вращение плоскости поляризации вправо | а2“ +66,5е ] изменяем Ся на левое вращение [ -39,5° ]. В связи с изменением в процессе гидро-
лиза сахарозы знака удельного вращения, гидролиз сахарозы получил Название инверсии. Отсюда, образующаяся в процессе гидролиза смесь Равных количеств /J-глюкозы и D-фруктозы называется инвертным Сахаром. Инвертный сахар является основной составной частью пчели-н°го меда. Причиной инверсии сахарозы является относительно боль
155
шее удельное вращение D-фруктозы плево [ау,и -92 °C ], чем D-глюкозы вправо [а^° +52,5 °C ], поэтому образующаяся при гидролизе смесь обладает левым вращением.
Сахароза содержится в сахарном троеч нике и сахарной свекле (17-20%). из которых ее получают в промышленности. В фармации сахароза применяется для приготовления порошков, сиропов, микстур и др.
5.3. ПОЛИСАХАРИДЫ
К полисахаридам относят соединения, молекулы которых содержал более десяти моносахаридных звеньев, связанных О-гликозшшой связью.
Чаще всего полисахариды состоят из нескольких сотен и даже тысяч моносахаридных остатков, образующих линейные (а) или разветвленные (б) полимерные цепи (рис. 5.1),
Рис. 5.1. Линейные (а) и рач ветвленные (б) полимерные пени полисахаридов
Гликозидные связи в полисахаридах, как правило, образуются за счет гликозидного гидроксила одного и спиртового гидроксила другого моносахаридных остатков. В большинстве своем эти связи возникают между С1 и С", С1 и С3 или О' и Сб.
На конце полисахаридной цепи находится восстанавливающий остаток моносахарида, но поскольку его доля в молекуле ничтожна, то полисахариды с большой молекулярной массой практически не обладают восстанавливающей способностью. Если в состав полисахаридов входят остатки только одного моносахарида, то их называют гомополисахаридами. Полисахариды, состоящие из разных моносахаридных единиц, называют гетерополисахарида мн.
5.3.1. Гомополисяхарнды
Гомополисахариды, построенные из остатков пентоз, называются пентозанами, а из остатков гексоз гексозанами. Общая формула иеи-тозапов (С5НЯО4)П, а гексозанов Подавляющее боль-
шинство природных полисахаридов гексозаны; к ним относятся: крахмал, целлюлоза, гликогеи, декстраны и др.
156
Крахмал. Крахмал служит основным источником резервной энергии в растениях; встречается главным образом в семенах, клубнях, корнях.
Крахмал содержит примерно 20% растворимой в воде фракции, называемой амилозой, и около 80% нерастворимой фракции, называемой амилопектином При постепенном кислотном и ферментативном гидролизе амилоза и амилопектин расщепляются до декстринов (смесь полисахаридов с меньшей молекулярной массой), дальнейших гидролиз которых приводит к мальтозе, а затем к D-глюкозе:
(QH10^5 )п (QjH 10^5 )х ► С12^22^1 J Ц»Н12^(,
крахмал .'1екс;ршгы мальтоза Л-слюкочи
(х < п)
Различие в строении амилозы и амилопектина обусловлено характером гликозидных связей.
Амилоза линейный полимер, в котором D-глюкопираиозныс остатки связаны а-1,4-гликозидной связью; состоит из 200-350 мономерных звеньев:
н ОН н ОН н он н он фрагмент молекулы амилочы
Молекулярная масса амилозы составляет примерно 40000. tie молекулы обладают гибкостью н могут принимать различные пространственные формы. В присутствии комплексообразова гелей. например иода, она может существовать в виде спирали, в каждом витке которой содержится шесть остатков глюкозы. Размер внутренней полости спирали позволяет разместиться в ней молекуле иода, что приводит к образованию окрашенного в синий цвет комплекса. На этом свойства крахмала основано его использование в фармацевтическом анализе в качестве индикатора.
Амилопектин - полимер разветвленной структуры, который может содержать 1000 и более остатков D-глюкозы в молекуле. Молекулярная масса амилопектина достигает 1-6 млн. Все цепи полисахарида основная и боковые — построены однотипно: остатки глюкозы в них соединены а-1.4-гликозидной связью. Боковые ответвления связаны с основной цепью сх-1,6-гликозидной связью. Между двумя соседними точками ветвления основная цепь содержит 20-25 моносахаридных остатков;
157
i|ip;nмет молекулы аминопемина
В связи с наличием большого числа ответвлений молекула амилопектина не способна принимать конформацию спирали и связывает иол лишь в незначительном количестве с образованием красного окрашивания.
Крахмал служит основным источником углеводов в пищевом рационе человека. Фермент амилаза, содержащийся в слюне, расщепляет а-гликозидные связи крахмала до декстринов и частично до мальтозы, дальнейший распад которых до глюкозы происходит в кишечнике. В фармации крахмал используется в производстве таблеток, а также для приготовления присыпок и паст.
Гликогеи (животный крахмал). Если у большинства растений резервным полисахаридом является крахмал, то в животных организмах зту функцию выполняет гликоген. Этот полисахарид снабжает организм глюкозой при повышенных физических нагрузках и в промежутках между приемами пиши.
Гликоген построен аналогично амилопектину, ио представляет собой еще более разветвленную структуру. Связь глюкопирацозиых остатков в основной и боковых цепях а-1.4. а в местах разветвления а-1,6. Между точками ветвления содержится 10-12. реже 2-4 моносахаридных остатков. Молекулярная масса гликогена варьирует и может достигать исскольких миллионов. В отличие от большинства других резервных полисахаридов гликогеи хорошо растворим в воде.
Сильная разветвленность цепей гликогена способствует атаке его ферментами с разных сторон одновременно. Это обстоятельство приводит к чрезвычайно высокой скорости расщепления полисахарида и, следовательно, возможности почти мгновенной мобилизации заключенных в нем энергетических запасов.
Наиболее богаты гликогеном печень и мышцы.
158
Целлюлоза. Целлюлоза — широко распространенный в природе полисахарид, являющийся составной частью оболочек растительных клеток. В состав древесины вхолит от 50 до 70 %, а в состав хлопка до 98 % целлюлозы. Молекула целлюлозы представляет собой линейную цепь, состоящую из остатков />глюкопиранозы, связанных между собой Р-1.4-ГЛИКОЗИДИОЙ связью:
фрагмент молекулы целлюлозы
Молекулярная масса целлюлозы колеблется от 250000 до 1000000 при содержании не менее 1500 остатков глюкозы.
Целлюлоза не растворяется в воде и обычных органических растворителях, но растворяется в аммиачном растворе гидроксида меди (реактив Швейцера) и концентрированном растворе хлорида цинка.
При нагревании с минеральными кислотами целлюлоза гидролизуется по схеме:
целлюлоза
(С6Н10О5)х > С^НззОц
амилоид (х < П)
целлобиоз»
D-глюкоза
Человек и высшие животные не имеют фермента, гидролизующего р-гликозидныс связи целлюлозы, но она является необходимым балластным компонентом нищи, улучшающим пищеварение.
Молекула целлюлозы имеет строго упорядоченную конформацию «жесткого стержня», в которой глюкопирапозные остатки расположены л иней ио.
Такое расположение остатков в пространстве обусловлено тем, что гликозидный атом кислорода и атом кислорода при С4 связаны с пиранозным циклом экваториально. Линейная коиформацня молекулы закрепляется внутримолекулярными водородными связями.
Параллельно уложенные цепи полисахарида удерживаются за счет образования межмолекулярных водородных связей. Благодаря такому
159
строению целлюлоза химически сравнительно инертна (нерастворима в воде, с трудом гидролизуется) и обладает высокой механической прочностью. Целлюлоза не расщепляется ферментами желудочно-кишечного тракта, и поэтому не усваивается, но она является необходимым для нормального питания балластным компонентом пиши.
Важное практическое значение имеют производные целлюлозы. Наличие трех свободных спиртовых групп в каждом глюкозидном остатке целлюлозы дает возможность получать ее сложные эфиры. Так. при обработке целлюлозы смесью азотной и серной кислот образуются нитраты целлюлозы. Свойства и возможности применения этих продуктов зависят от степени нитрования. Смесь моно- и линнтратов называют коллодийной ватой или коллоксилином. Ее используют для изготовления коллодия, применяющегося в медицине для фиксации повязок. Продукт полного нитрования целлюлозы (тринитрат целлюлозы, тринитроклстчатка, пироксилин) является взрывчатым веществом, используемым в производстве бездымного пороха. Большое народнохозяйственное значение имеет диацетат целлюлозы, используемый в производстве ацетатного шелка, а также ксаитогенат целлюлозы, применяемый для получения вискозного волокна и целлофана. Натриевая соль карбоксиметил целлюлозы применяется в производстве лекарственных препаратов.
Декстраны. Декстраны полисахариды бактериального происхождения, построенные из остатков а-/)-глюкопиранозы. Получают их из сахарозы при участии бактерий Leucaiios'loc Hiesenteroides. Основным типом связи в декстранах является а-1,6-гликозидная саязь. а в точках разветвления - - а-1,4-, и «-1,3-гликозидные связи : '
Н ОН Н ОН
фрагмент молекулы деке! рана
Молекулярная масса декстранов составляет несколько миллионов. Частично гидролизованные декстраны (м. м. 40000-800000) использукя в фармации в производстве плазм ©заменителей «полвглюкии» и «реп пол игл юки и». (60
Инулин. Инулин резервный полисахарид, содержащийся в клубнях
нон2с
о
н
сн
н
ОН НА НОН,С о °
ОН Н :
н но
СИ
н
сложноцветных и других растений. Молекула инулина имеет линейное строение и состоит из остатков P-D-фруктофуранозы. связанных 2,1-гликозидными связями, и заканчивается а-Д-глюкопиранозным остатком (как в сахарозе). Молекулярная масса обычно не более 6000.
Инулин получают из клубней георгина экстракцией с горячей водой. Используется для получения D-фруктозы.
Нектшювые вещества. К пектиновым веществам (пектинам) относят полисахариды, в основе строения которых лежит полигалактуроно-вая (центовая) кислота, построенная из остатков а-/)-галакту роковой кислоты, связанных между собой 1.4-гликозидным и связями. Часть карбоксильных групп 11олигалактуроновой кислоты этерифииирована и находится в виде метилового эфира.
Н ОН н он н он н он фрасмеш nojiHiuiaiciypououoii кислош
Водные растворы пектинов способны образовывать прочные гели.
Пектины содержатся практически во всех наземных растениях и некоторых водорослях. Оии нашли широкое применение в пищевой промышленности для изготовления желе, мармелада, т. к. с сахарозой в ' присутствии органических кислот образуют студии.
Некоторые пектиновые вещества обладают противоязвенным действием и являются основой ряда лекарственных препаратов.
5.3.2. Гетерополисахариды
К гетерополисахаридам относят полисахариды, состоящие из остатков разных моносахаридов. К таким полисахаридам принадлежат полисахариды соединительной ткани хондроитиисульфаты, гиалуроновая кислота, гепарин, Все они имеют линейную углеводную цепь и регулярЕго повторяющийся на всем протяжении цепи дисахаридный фрагмент, называемый повторяющимся звеном.
Гиалуроповаи кислота. Является одним из наиболее распространенных полисахаридов соединительной ткани. Содержится в хрящах, пуповине. суставной (синовиальной) жидкости, стекловидном теле. По
бВ-41 161
вторяющимся звеном гиалуроновой кислоты является ZJ-глюкуроновая кислота и К-ацетил-Л-глюкозамин. соединенные Р-1 >3-гликозидной
связью. Связь между дисахаридиыми фрагментами - Р-1.4 :
1ЮВ1ОрЯ1О1цееея звено
СООН СН,ОН СООН СП,он
р-КЗ-к'ипагш.'шая синчь Р-М-гликотиллая связь
фршменг niajjj’pojiouofi кислоты
Молекулярная масса гиалуроновой кислоты колеблется от 1600 до 6400. Этот полисахарид обладает высокой вязкостью, это обеспечивает непроницаемость соединительной ткани для бактерий.
В тканях гиалуроновая кислота связана а комплексе с белком за счет ковалентных связей.
Хондронтннсудьфаты один из главных компонентов хряща. Они содержатся также в коже, сухожилиях, склере, костях. Повторяющимся звеном хондроитинсульфатов является />глкжу ромовая кислота и М-ацетил-Л-галактозамин, содержащий сульфатную группу- Внутри днеахаридного фрагмента связь р-1,3, а .между фрагментами р-1Л-Сульфатная группа образует эфирную связь с гидроксильной группой М-ацетил-/)-галактозамина либо в положении 4 (хондроитин-4-сульфат), либо в положении 6: (хондроитин-6-сульфат).
।юн[ пряюиикся тени
Р-1.3-П»ГИГИ 1Д1ШЯ СВЯ-iti (i-1.4-1 ЛнмОЗнДЛаЯ сия й.
фршмеш молеку.чы хондрой iHii-4-cy.’ib<|iara
Углеводные цепи хондроитинсульфатов содержат до 150 дисаха-ридных остатков, присоединенных в организме ()-гликозидиыми связями к гидроксильным группам остатков аминокислот, входящих в белковую часть молекулы, еще недостаточно изученную.
Гепарин. Вырабатывается в организме человека и животных, содержится в больших количествах в печени, легких; в меньших в ске-летных мышцах, селезенке, сердечной мышце. Повторяющееся звено в структуре гепарина состоит из /Хглюкозамипа и уроновой кислоты.
162
соединенных между собой а-1,4-глнкозндными связями. В качестве уроновых кислот выступают L-идуроновая кислота и. реже, D-глюкуроновая кислота:
Н
/.-илуроноиня кислота
Остатки глюкозамина и £.-идуроновой кислоты в гепарине частично сульфированы. Молекулярная масса гепарина равна 16000-20000. Как и у гиалуроновой кислоты и хондроитинсульфатов, углеводные цепи гепарина связаны в тканях с белковой частью молекулы.
|<еиарнп (QipaiMeiir iio.iHcaxapiijnoii цепи)
Гепарин препятствует свертыванию крови, принимает участие в обмене липидов, жиров и холестерина. Применяется в медицине как антикоагулянтное средство.
Растительные камеди. Являются разветвленными гетеро пол и сахаридам и, содержащими остатки нейтральных моносахаридов (D-галактозы, D-глюкозы, L-рамнозы, L-арабинозы и др.) и уроиовых кислот в виде солей. Камеди выделяются при повреждениях растений в виде вязких жидкостей.
Аравийская камедь (гуммиарабик) включает остатки углеводов ^--арабинозы. D-галактозы, метнлпентозы и D-глюкуроновон кислоты; находит применение в медицине в качестве эмульгаторов при изготовлении эмульсий.
КОНТРОЛЬНЫЕ ВОПРОСЫ И УПРАЖНЕНИЯ
1. С помощью проекционных формул Фншера изобразите строение оптических антиподов (энантиомеров) D-рибозы, D-глюкозы, D-галактозы и D-маннозы.
2. Какие диастереомеры называют эпимерами?
Приведите проекционные формулы Фишера эпимеров D-глюкозы.
3. Дайте определение понятию «аномеры».
Изобразите с помощью перспективных формул Хеуорса пиранозные п фуранозные формы аномеров D-глюкозы и D-фруктозы.
4. Какие виды таутомерии характерны для моносахаридов? Приведите схему таутомерного превращения D-глюкозы в водном > растворе. Объясните явление мутаротации. ; 1
5. Напишите схемы взаимодействия D-глюкозы с:
а) изб. C6II5NHNH2 (/): б) CHJ (нзб.): b)HCN; г)Вг2 (Н2О); .
д) HNO, (кони.), 4
6. Приведите схемы реакций a-D-галактопирапозы с:
а) СН,ОН (Н+): б) (CHjCOfeO (CH3COONa).
К какому функциональному классу относится продукт реакции (а)?
7. Почему фруктоза вступает в реакцию «серебряного зеркала»?
Что происходит с фруктозой в аммиачной (щелочной) среде? .4 Приведите схемы взаимодействия D-фруктозы и D-маниозы с Г* избытком C6HsNHNH2(/).
8. Напишите схемы последовательных реакций получения аскорбиновой кислоты из D-глюкозы.
Укажите кислотный центр в ^олекуле витамина С,
9. Дайте определение понятию «дисахариды».
Что такое восстанавливающие и невосстанавливающие сахара? Почему сахароза не обладает способностью к мутаротации?
Приведите схему реакции, позволяющей отличить мальтозу от сахарозы.
10. Приведите определения понятий полисахариды, гомополисахариды и ге/перопо.и/сахариды. Из каких моносахаридных звеньев построены молекулы следующих гомополисахаридов: амилозы, амилопектина, целлюлозы, гликогена? Что представляют собой декстраны, инулин и пектиновые вещества?
11. К какому типу полисахаридов относятся хондроитинсульфаты, гиалуроновая кислота, гепарин н растительные камеди?
164
Глава 6
БЕЛКИ
Белки основа всего живого. Они являются компонентами всех клеток и тканей живых организмов. К белковым вещеезвам относятся ферменты. некоторые гормоны и др. Наряду с нуклеиновыми кислотами белки представляют собою наиболее сложные из созданных природой биополимеров. Молекулярная масса белков составляет от 5000 до многих миллионов. Белки с низкой молекулярной массой называются пептидами. Мономерными единицами белков и пептидов являются a-а минокнелоты.
6.1. а-АМ ИНОКИ СЛОТЫ КАК МОНОМЕРЫ БЕЛКОВ
В состав большинства белков входит около 25 различных а-ами-нокислот общей формулы R—(NH2)CH—СООН, из которых примерно 20 присутствуют в каждой белковой молекуле. Основные «-аминокислоты, входящие в состав белков, представлены в табл. 6.1.
В номенклатуре а-аминокислот чаще применяют тривиальные названия: глицин, аланин, валин, лейцин и др. В биохимии используют также трехбукве иные сокращения тривиальных названий, например: глицин Гли. алании - Ала. валин Вал, Систематические названия для природных «-аминокислот практически не применяют.
По химической природе остатка, связанного с «.-аминокислотным фрагментом CH(NH2)COOH, а-аминокислоты делятся иа алифатические, ароматические и гетероциклические. В гетероциклических «-аминокислотах продине и оксипролиле «-аминокислотный фрагмент входит в состав гетероцикла:
а-ам]|цок11С!Ю1пыи фрашепт
Таблица б. 1
«-Аминокислоты, входящие в состав белков
Формула Название аминокислоты Трехбуквенное обозначение
1 2 3
Алифатические аминокислоты
сн2-соон NH, Глицин, гликокол, амин «уксусная кислота Гл и
СНз-СН— соон nh. Алании, а-аминопро-пионовая кислота Ала
(СН^СН -сн -соон NIK Валин*, ос-амияоизояале-риаповая кислота Вал
(СН3),СН сн , -сн -соон NH, Лейцин*, а-амипоизо-калроповаякислота Лей
сн ,сн ,сн-сн -соон -1 1 СН3 NH, Изолейцин*, а-амино-р-ме-тилналериаповая кислота Иле
носн2-<рн-соон nh2 Серин, а-амияо-р-гидрокси-пропиоповая кислота Сер
СН.СН-СН-СООН 11 1 он nh. Треонин*. о-амипо-р-гил-роксимасляпая кислота Тре
НООСС1 г-сн -соон NH, Аспарагиновая кислота, амииояитарная кислота Дсп
НООССН,СН,-СН-СООН " 1 nh. Глутаминовая кислота, а-амипоглутаровая кислота Глу
H.NtCH^-^H-COOH NH, Ориитип. а, Й-диам и и о валериановая кислота Орн
Н^(СНД-СН-СООН NH, Лизин*, а, Е-лиамшюканро-новая кислота Лиз
H,NCNI1(CH2),-^H-COOH NH NH, Аргинин, а-амипо-6-гуаии-диновалерианоная кислота Apr
H,NCCH,-CH-COOH 0 NH, Аспарагин, р-амид аспарагиновой кислоты Асн
H,N£CH,CH,-CH-COOH О NH, Глутамин, у-амил глутаминовой кислоты Глн
-
166
Продолжение таблицы 6.1
Г“ 1 2 3
HSCH2-CH-C()OH nh2 Цистеин, a-аминоф-меркап-топропионоваякислота Цис
;сн2-сн-соон NH-. ;сн2-сн-соон nh2 Цисгнн, р,(Зг-литио-бис-а-амипопропиоповая кислота Цис-S-S-Цис
CH3SCH2CI i2 -сн -соон nh2 Метионин*, к-амиио-у-метил-тиомаслялая кислота Мет
Ароматические аминокислоты
О ZCH2-CH-COOH nh2 Фенилаланин* . а-амшю-р-фспилпропионовэя кислота Фен
jO Н<)^ сн,-с:н-соон । nh2 Тнроз ин. и-а м hi ю- р - (и- г ид-роксифс11ил)11ропио11овая кислота Тир
Гетероциклические аминокислоты
сх сн,-сн~соон \ nh2 NH Грнптофан *, а-амино- Р-иплолилпропионовая кислота Три
сн.-сн-соон ^"5 ^н= Nil Гнстцдии, а-амино-р-имидазолил прон ионовая кислота Гис
Q-gogh Пролин, иирролидин-а-кар-боповая кислота Про
но a- Оксипролнн. Р'-ГИДрОКСИ-пирролилип-а-карбоновая кислота Про-ОН
* Незаменимые аминокислоты.
167
В зависимости от числа групп NH2 и СООН в .молекуле различают а-аминокнслоты: моиоампномонокарбоновые (глицин, аланин, валин и др.), моиоаминодикарбоновые (аспарагиновая, глутаминовая кислоты и их амиды) и диамнномонокарбо новые (орнитин, лизин, аргинин, гистидин).
По кислотно-основным свойствам «-аминокислоты делятся на нейтральные (содержащие равное число амино- и карбоксильных групп), кислые (с дополнительной карбоксильной группой) и основные (с дополнительной аминогруппой),
Большинство «-аминокислот образуется в организме (заменимые аминокислоты), но некоторые а-аминокислоты организм человека не способен синтезировать: они поступают в составе белков, вводимых в организм с пищей (незаменимые аминокислоты, табл. 6.1).
6.1,1. Стереоизомерии
Все «-аминокислоты, исключая глицин, содержат хнральный «-углеродный атом и существую! в виде пары энантиомеров. Для обозначения конфигураций при хиральиом ценз ре применяют D. L-систему.
СООН соон
Н—| NH. H,N—|------Н
R R
Л-а-амннокнелот /.-а-ам и повисло ia
«-Аминокислоты, входящие в состав белков животных и человека, имеют /.-конфигурацию. Аминокислоты D-ряда встречаются лишь в небелковых компонентах растений и грибов, а также синтезируются микроорганизмами. Некоторые аминокислоты, например, изолейцин и треопии. содержат по два хиральных центра и могут существовать в виде двух пар энантиомеров; в белках встречаются такие их изомеры:
СООН СООН
H-.N----Н H2N-----Н
Н?С-----Н II----ОН
сн:сн, СН, /.-и чолейшш /.-треонин
Использование а-амниокислот /.-ряда для биосинтеза белков имеет важнейшее значение в формировании их пространственной структуры и проявлении биологической активности.
6.1.2. Физические свойстна
«-Аминокислоты представляют собой кристаллические вещества, не имеющие четких температур плавления и разлагающиеся при температуре выше 200°C. Они не растворимы в неполярных органических 168
растворителях, но заметно растворимы в воде. В кристаллическом состоянии и водных растворах аминокислоты находятся в виде биполярных ионов (цвиттер-ионов, внутренних солей). Возможность образования последних связана с амфотерностью аминокислот, обусловленной наличием в их молекуле кислотной (СООН) и основной (NH2) групп.
В водном растворе «-аминокислоты существуют в виде равновесной смеси, состоящей из цвиттер-ионов, катионной и анионной форм:
H.N — СН-СОО" I
R
ли нон пая форма
+
H.N—CH—COO'
£ -н*
биполярным иоп.
СН—СООН
R.
Kai мин।ши форма
ции [ гер-ион
Положение такого равновесия существенно зависит от pH среды: в снльнокислой среде (pH 1-2) преобладает катионная форма, в сильнощелочной (pH 13-14) анионная. Если раствор аминокислоты поместить в электрическое поле, то в кислой среде молекулы перемещаются к катоду (катионная форма), а в щелочной к аноду (анионная форма). Однако, для каждой аминокислоты существует характерное значение pH, при котором молекулы не перемещаются в электрическом поле. При этом значении pH. называемом изоэлектрической точкой (рЛК аминокислота находится в виде цвиттер-ионов и в целом электро нейтральна. Изоэлектрическая точка зависит от соотношения количеств кислых и основных групп в молекуле: р/ кислых аминокислот имеет значение менее 7, т.к. в кислой среде подавляется ионизация второй карбоксильной группы и, соответственно, р7 основных аминокислот находится в области более 7, т.к. в щелочной среде подавляется протоиирование второй аминогруппы.
6.1.3. Способы получения
Ранее (см. ки. 2, е. 460) были рассмотрены общие методы получения аминокислот, в т.ч. «-аминокислот. В процессе синтеза образуется рацемическая смесь (±)-«-амииокислот, разделение которой на оптические антиподы проводят с помощью химических и ферментативных методов (см. кн. 1,с, 83).
Наиболее широко используемый химический метод расщепления рацематов а-амииокислот основан па образовании диастереомерных солей N-ацильных производных (±)-а-аминокислот с оптически активными основаниями, например, бруцином или стрихнином. Вследствие различной растворимости один из диастереомеров образует осадок, а другой, более растворимый остается в растворе. Разделенные диастереомерные соли затем разлагают до а-аминокислот.
Ферментативный метод расщепления основан на гидролизе N-ацил-а-аминокислот ацилазами или сложных эфиров а-аминокислот эстеразами.
169
Гидролиз белков, а-Аминокислоты получают путем щелочного, кислотною или ферментативного гидролиза белков. При кислотном гидролизе происходят также побочные реакции, например, глутамин н аспарагин гидролизуются до глутаминовой и аспарагиновой кислот, а триптофан разрушается. Щелочной же гидролиз приводит к рацемизации «-аминокислот. Поэтому наиболее широко используется ферментативный метод гидролиза. Разделение а-аминокислот в белковых гидролизатах проводите помощью ионообменной хроматографии.
Микробиологический синтез. Некоторые микроорганизмы в процессе своей жизнедеятельности вырабатывают определенные «-аминокислоты. Эти микроорганизмы выращивают на богатых углеводами средах крахмале, мелассе, патоке и др. Таким образом получаю г аспарагиновую и глутаминовую кислоты, триптофан, лизин н др.
6.1.4. Химические свойства «-аминокислот
Ранее (см, кн. 2. с. 461-464) были рассмотрены химические свойства аминокислот. Здесь представлены реакции, которые используются в анализе а-аминокислот. синтезе пептидов или лежат в основе превращений а-аминокислот в организме,
А. Реакции по аминогруппе
Образование N-ацильпых производных. При взаимодействии «-аминокислот с ангидридами или хлорангндридами карбоновых кислое образуются N-ацнльные производные, которые относительно легко разрушаются до исходных а-аминокислот. В связи с этим реакция ацилирования используется для блокирования (защиты) аминогруппы при синтезе пептидов. В качестве ацилирующих агентов используют бенз-оксикарбонилхлорид (а) или /препг-бутоксикарбоксазид (б):
О R 0
U) СН,О—с a H,N—СН—СООН
w а-амннокиснота а
fl ? fl । «-
CH.O-C-N И -СН -СООН (СН j)?CO-C-N н -сн -соон
1Х-;гМ и ни кислота С ЗиШИЩсННОЙ аминогруппой
Защитную карбобензоксигруппу удаляют каталитическим гидрогенолизом или действием раствора бромоводорода в уксусной кислоте на холоду:
с(н5 О
^h2o-^-nh-сн-соон
н, ( Pd ) : со,
НВг (CHJOOH)
-СлН5СН,Вг: - (X),
H.N — СН-СООН I
R
а-амипокиспота
H,N~СН-СООН
I R
R
170
7 pern-бутоксикарбон ильную группу разрушают действием трифтор-уисусной кислоты:
ft CF,COOH
и C-C-O-C-NM-СН-СООН —---------* R-СН-СООН + Н,С=С
R 'с°2 NH, ЧСН,
Дезаминирование. Пол действием азотистой кислоты «-аминокислоты превращаются в соответствующие «-гидроксикнслоты:
R- СП- СООН + HNO, —►
NH2 а-амипокисио । а
R—СН-СООН + N, + Н,О I
ОН
смздроксмкисло та
Эта реакция применяется в аналитической практике (метод Ваи-Спайка). По объему выделившегося азота определяют количественное содержание «-аминокислоты.
В организме «-аминокислоты подвергаются окислительному дезаминированию. Реакция происходит под действием ферментов оксидаз и окислительного агента кофермента НАД :
R— СН— СООН =->
Ь1Н,
-2Н
R—С—СООН
НО
——R—С—СООН
-NH, [[
О
а-аминокнсппта
а-иминокислота
а-кел окисло i а
Трансаминирование (нереаминировапие). Процесс протекает только в живых организмах. Реакция происходит с участием ферментов трансаминаз и кофермента пиридоксальфосфата между «-амино- и а-кетокислотами и сводится к взаимообмену карбонильной и аминогруппами:
СООН соон СООН СООН
H,N—С—H + с=о С=О + h2n—с—н
СН, сн, СИ, сн, 1 2
1 соон СИ, соон I -соон си2 соон
acnapHiTiiiouHH а-кстоглупфонни I UH UCJICHC у ксу сн ая DiyniMUllUlKIM
кислота киаюта кислота кн a ini а
Взаимодействие с карбонильными соединениями. Формальдегид реагирует с «-аминокислотами в водном растворе с образованием N-гндроксиметильных производных.
Реакция лежит в основе количественного определения «-аминокислот методом формольного титрования по Серенсену.
171
R —CH -СООН + H,C=O -----------------------► R -CH -СООН 9
I 2 I
NH, NH-CH,-OH •*
i a*iiMHii()Kiic.-inTa a-i идрокснмеч ильное
иронию, uioe 1
4 *
Другие альдо иды и кетоны реагируют с ot-ам и но кисло га мн с образованием оснований Шиффа:
R—СН-СООН + o=CH-R ----------* R—СН-СООН + Н2О
NH, N=CH-R '?
а-амннокислога основание Шиффа t
V
Взаимодействие с фенилизотиоииапатом (реакция Эдмана) При взаимодействии «-аминокислот с фенилизотиоцианатом образуются производные З-фснил-2-тиогидаптоина. Сначала н присутствии шелочи происходит присоединение фенилизотионианата по аминогруппе а-аминокислоты. а затем при нагревании полученного продукта присоединения в присутствии минеральной кислоты происходи! циклизация с образованием производного фенил тио гидантоина (ФТ Г-про из водное):
Ж
R—СН-СООН
NH2 +
фе[И1,111ЪТГ1Н>||>1ииат
ОН'
ch-cz t
Н+: i / \
----*^HN >-с'Н
-Н,()
II
S
ФТГ-нроизшышх:
Реакция используется для установления строения пептидов (деградация по Эдману).
Взаимодействие с 214-динитрофторбензолом (реактив Сенгера). При взаимодействии а-амннокислот с 2,4-лннитрофгорбензолом (ДНФБ) образуется N-динитрофеннльное производное (ДНФ-произ-водное):
Д1 Г<1э-г । р<>! i чволюе
Реакция протекает по механизму (см. кн. 2, с. 180). Используется для установления строения пептидов.
[72
Б. Реакции по карбоксильной группе
Образование хелатных соединений. Характерной особенностью а-аминокислот является способность образовывать прочные хелаты комплексные соли с ионами тяжелых металлов, иалример:
R NH
2 ¥
Cu2+
.0
Ск ОН
а-амшнжислога
R NH3
""сн \ / 9
(L сн
"Ч) ^R
медлия соль а-и мин о киста гы
Незначительная растворимость и интенсивная окраска хелатов меди (II) позволяет использовать их в аналитической практике для обнаружения (х-аминокислот.
г Образование сложных эфиров. Как карбоновые кислоты «-аминокислоты при взаимодействии со спиртами образуют сложные эфиры:
СООН 1 СН —R + R’OH спирт
NH,
a-UMwiioKiiciora
HCI
-Н-,0
COOR'
1
СН— R
CI’ NH,
NH
-NH4CI
CWR' I
CH— R
NH: сложный тфир
органических
Сложные эфиры а-аминокислот растворимы в растворителях, летучи и хорошо перегоняются. Эти свойства их используются прн разделении смеси а-аминокислот в белковых гидролизатах. С этой целью а-аминокислоты сначала тарифицируют, а затем полученные эфиры подвергают перегонке. Для разделения смеси сложных эфиров а-аминокислот в настоящее время применяют метод газо-жидкостной хроматографии (ГЖХ). Эта реакция служит также удобным методом защиты карбоксильной группы при синтезе пептидов.
Образование галогепапгидридов и ангидридов. Аналогично карбоновым кислотам а-аминокислоты образуют галогенангидриды и ангидриды (см. кн. 2, с. 368) Перед проведением реакции предварительно защищают аминогруппу образованием N-ацильных производных.
Декарбоксилирование. В связи с влиянием аминогруппы а-аминокислоты относительно легко декарбоксилируются:
ILN—CH-COOiH
2 | L---1
R
Ditto Hb . i
- c o.
амин
173
6.1.5. Идентификация а- аминокисл от >
Ниигидринная реакция. Для обнаружения а-аминокислот используется реакция с нингидрином, в результате которой образуется продукт, окрашенный в сине-фиолетовый цвет с максимумом поглощения при 570 им:
пролук! ст те-фиолетового цвет
Ннигидрииовый реактив применяется в хроматографическом анализе для проявления хроматограмм на бумаге и в тонком слое сорбента, а также для количественного колориметрического определения «-аминокислот.
Ксантопротеиновая реакция. Это реакция с концентрированной азотной кислотой на а-аминокнслоты, содержащие в своей молекуле ароматические циклы. В результате последней происходит нитрование ароматического цикла с образованием нитропроизводного, окрашенного в желтый цвет.
6.2. СТРОЕНИЕ ПЕПТИДОВ И БЕЛКОВ
Вследствие взаимодействия амиио- и карбоксильных групп «-аминокислоты способны к поликонденсации. Образующиеся при этом полиамиды называют пептидами:
HnN-CH-COpH + HJNH-CH-COpH +...+ET|NH-CH-COOH --&
R J R' J R"
---► H.N-CH-CO-NH-CH-CO— NH-CII-COOII k
-Il i *
R R' Rrl гф
пептид
Амидная связь (—CONH—) между двумя а-аминокислотными фрагментами называется пептидной связью. Атом углерода пептидной группы находится в x//-i ибридизованном состоянии. Неподеленная пара электронов атома азота вступает в сопряжение с л-элсктронами карбонильной группы, в результате чего двойная связь С=О несколько удлиняется (124 нм вместо 121 им у обычной связи), а саязь С N несколько укорачивается (132 им вместо 147 нм), и, следовательно, приобретает в значительной мере характер двойной связи, вращение вокруг которой затруднено. Таким образом, электронное строение обуславливает жесткую плоскостную структуру пептидной группы.
174
Рис. ft'.l. Расногюжепие на iuiockolth амиллой (ueinit.uioii) труппы
Наличие пептидной связи в молекулах пептидов и белков подтверждается биуретовой реакцией: при взаимодействии со щелочным раствором сульфата меди образуется фиолетовое окрашивание (см. кн. 2, с. 473).
В зависимости от числа аминокислотных остатков в цепи пептиды деляг на ди-, три- тетрапептиды и т.д. Пептиды с молекулярной массой менее 10000 условно относят к полипептидам, а с м. м. > 10000 к белкам. Поэтому между белками и пептидами трудно провести четкую границу, однако белки имеют более сложную структуру.
При всем огромном разнообразии белков и пептидов в природе строение их полипептид ной (полиамидной) цепи одинаковое. Она состоит из чередующихся пептидных (CONH) и метиноных (СП) групп. На одном конце цени находится аминокислота со свободной аминогруппой (N-концевая аминокислота), а па другом со свободной карбоксильной группой (С-концсваи аминокислота). Пептидные и белковые цепи принято записывать так, чтобы N-концевая аминокислота находилась слева. а С-концевая аминокислота справа:
I ! !-----------7
H2N — <^Н 4-СО—NM 4-р 1 -г-СО—NH -f-CH —СООН k R l~~—yj-1 R.
пептидные святи
N-коплевая аминокислота С-копневая имшнжие.Юта
грнпедтид
Названия пептидов образуются путем последовательного перечисления всех аминокислот, начиная с N-концевой аминокислоты, причем названия аминокислот, кроме последней, приобретают суффикс -нл. В гаком же порядке пишут и сокращенные обозначения:
Г
H,N —СН —СО— N Н —СН —СО— N Н —СН > —СООН ~ I I
СН3 (СН2)2—соон
зланнлглутам илглицпн.
Лла-Глу-Гли
175
Определенную последовательность «-аминокислот, входящих в данную полипептидную цепь, называют первичной структурой пептида или белка. Изменение аминокислотной последовательности приводит к нарушению или исчезновению биологической активности белка. Белки отличаются от пептидов более сложным уровнем структуры. В струк
◄----0,5 нм-----►
турной организации оел-ков. кроме первичной, различают вторичную, третичную и четвертичную структуры.
Вторичной структурой белков называют пространственное расположение (пространственную укладку) атомов основной полипептидной цепи. Различаю! два типа вторичной структуры белков а-спираль и складчатую Р-С трук туру.
а-Спираль имеет пространственную форму, подобную правозакрученной винтовой лестнице (рис.6.2). Т к. она построена из повторяющихся участков (-NH-C“-CO). то размеры ее довольно постоянны. На одни виток спирали приходится примерно 3.6 аминокислотных остатка, что соответствует линейному расстоянию вдоль оси спирали 0,54 нм. Диаметр спирали равен 0.5 нм. Шаг спирали (расстояние между одинаковыми атомами) составляет 0.15 нм.
В формировании спи-
Рис, 6.2. Схема а-спнра.тъной конформации пол и пептидной цени
ральной структуры основную роль выполняют водородные связи, ко-
торые образуются между группами С=О и NH. разделенными тремя аминокислотными остатками. Водородные связи почти параллельны оси спирали, а поскольку каждая группа С=О и NH «-спирали участвует в образовании водородной связи, то это делает конформацию весьма устойчивой.
. 176
Чаще всего полипептидиые цепи в белках, спирализуются не полностью. Например, остатки пролина и оксипролина ие содержат атомов водорода в пептидной группе и. соответственно, не участвуют н образовании водородных связей: полипептидиая цепь иа тгих участках просто изогнута. Изопропильная группа валина также создает стериче-скне препятствия для спнрализацин.
Другим типом вторичной структуры является так называемая складчатая p-структура, в которой отдельные полипептидиые цепи в зигзагообразной конформации уложены параллельно друг другу и связаны между собой многочисленными водородными связями. Если поли пептидные цепи имеют одинаковое направление от N- к С-концу, то образуется параллельная складчатая {^структура, а если противоположное — антипараллельная (рис. 6.3). В ^-структуре боковые группы аминокислотных остатков находятся выше и ниже условной плоскости, проведенной через структуру.
N-kohcu N-конец L N-конец С-конец
-О С\ V R. N Н-' ........ _ Н со. i»H N н * - с< -ОС R Rk ,n н*’ Р- » со.. !»HN н 1 ? GXn " "ОС R sc н со. •V;H N н л-ОС j \ Н’ .-С о. .... ...7< 1 о-с. t Л -N ; •<4> cf i t / Kkc N H.-O < c>r;
H v с o-\-11 \ Il .0 c /' R, N lb- H C O-'r H N H • c < .ОС SR » N Hr/- н co4 •H N ' ,_H .О C Sr 7 H C O-* •H N 4 \ н с O—H N H • •H N H H. °- -i •/•O c R r n h- R4C X H"’O C C>R H C O-H \ 11 •**H N н И С O-” •AO-C’C>r n h- h::”:c c>r н C O -H \ H ; —H \ H H co- ••ЮС. Rr N»-: _Rkc > Ц" 0 c ( ; H c O-H N H , -*H N. J Л C o—
V > i
С-консц С-консц С-консц N-конец
а
Рас 6.3. Параллельный (с;) л анти параллельный (6) участки р-сгруыуры
Полипептидиая цепь, имеющая тот или иной тип вторичной структуры, способна определенным образом скручиваться в пространстве, что и определяет третичную структуру белка, т.е. общую форму поли-
177
пептидной цепи. Третичная структура, кроме водородных связей, стабилизируется ионными (между дополнительными карбоксильными и аминогруппами) и ковалентными (дисульфидные мостики в цистиие) связями, а также гидрофобным взаимодействием (ван-дер-ваальсовы силы притяжения между неполярными боковыми группами аминокислотных остатков).
Третичная структура белка формируется также под влиянием водной среды клетки, что связано со способностью воды гидратировать некоторые гидрофильные боковые группы аминокислотных остатков и смещать вовнутрь белковой молекулы гидрофобные группы.
Четвертичная структура белка относится к макромолекулам, в состав которых входят несколько полипептидных цепей (субъединиц), связанных между собой нековалентными связями.
Для проявления пептидом специфических функций в организме, необходимо воссоздать лишь его первичную структуру, а в случае бачка — воспроизвести все его конформационные особенности.
Особое место в развитии химии белков занимает определение поли-пептидпой структуры гормонов вазопрессина, окситоцина и инсулина. В 1953 г. американский биохимик В. дю Вииьо расшифровал строение гормонов гипофиза окситоцина и вазопрессина. Им установлено, что общим структурным элементом этих гормонов является пептид из девяти аминокислотных остатков с дисульфидной связью — S — S — между четвертым и девятым из них. Указанные гормоны отличаются лишь двумя аминокислотными фрагментами: вместо лейцина и изолейцина в окситоцине (а) вазопрессин (б) содержит аргинин и фенилаланин:
При этом окситоцин вызывает сокращение гладкой мускулатуры, в частности, матки, а вазопрессин поддерживает баланс жидкости в организме.
Десять лет (1943-1953 гг.) понадобилось английскому биохимику Ф.Сенгеру для расшифровки структуры гормона поджелудочной железы инсулина. Им установлено, что молекула состоит из двух поли-пептидиых цепей, связанных между собой дисульфидными мостиками: A-цепь содержит 21 аминокислотный остаток и дополнительную дисульфидную связь, благодаря которой инсулин в пространстве образует петлю, а Б-цепь — 30 остатков (рис. 6.4).
V
178
A
Гли-И.ле-Вал-Глу'
Ци
Цис
Глу
Вал
ис
С^р-Лей-Тир-Глн-Лей-Глу-Асн-Тир-Цис-Асп
Лей—Цис
Гис
Глу
Аси
I Т—
Вал-Фен
I'ли
Лей—Тир—Лей—Вал—Цис—Гли Глу
Гис—-Лей—-‘
______________________] Тли
Ала-Лиз-Про-Тре-Тйр-Фен-Фен
Б
Рие. 6.4. Строение бычьего инсулина
В 1963-1964 гг. были синтезированы обе полипептидные цепи инсулина. Инсулин различных видов животных и человека отличается по строению. Эти структурные различия приходятся на участок 8-10 цепи А. Инсулин регулирует содержание глюкозы в крови, недостаток его в организме вызывает сахарный диабет.
К полипептидам относятся некоторые широко применяемые антибиотики. например, грамицидин С циклический декапептид, используемый для лечения заболеваний, вызванных стрептококками, пневмококками и др. В состав грамицидина С, кроме аминокислотных остатков С-ряда, входят два остатка /Э-фенилалаиина:
Грамицидин С
6.3. СИНТЕЗ ПЕПТИДОВ
В основе синтеза пептидов лежит процесс образования пептидной (амидной) связи между карбоксильной группой одной «-аминокислоты и аминогруппой -- другой. Упрощенно этот процесс можно представить следующей схемой:
Н2 N^HCOjOH + HN НС'Н СООН • » Н2 NCH £со-НN СНСООН
R R ян пентил R1
179
Однако, из-за ди полярной природы «-аминокислот (цвиттер-ионная структура) проведение реакции требует очень высоких температур, что способствует различным нежелательным побочным процессам, например, циклизации с образованием дикетопиперазинов (см. кн. 2. с. 463 и др.). Кроме того, в процессе синтеза возникают сложности, связанные с необходимостью соединять остатки а-аминокислот в определенной последовательности. Например, при взаимодействии глицина и аланина возможно образование четырех дипептидов:
NH, I -
СН2 + боон глнпин
NIL
Н, С-611 боон
*HLNCH:CONHCH(CH,)COOH
Ган-Д-'ш
Н, N СИ (СН, )CON НСН, СООН
Л.пн-Гли
-*► h2nch7conhch,cooh
Гаи-Гаи
*- Н, NCH (СН JCO N Н СН (С Н, )СООН
Лла-Лли
В связи с этим для проведения целенаправленного синтеза следует создать такие условия, при которых одна из аминокислот взаимодействовала бы своей карбоксильной группой, а другая аминогруппой. С этой целью осуществляют защиту функциональных групп (-NH; и -СООН). ие принимающих участия в образовании пептидной связи. Защитные группы выбирают таким образом, чтобы затем каждую из них независимо друг от друга можно было легко удалить, не разрушая прн этом пептидной связи.
Для зашиты аминогрупп используют реакцию ацилирования, чаше всего бензоксикарбонилхлоридом или лдоун-бутоксикирбоксазмдом. Важным свойством карбобензокси- и ш/идп-бутоксикарбонильных групп является то, что они надежно защищают хиральный центр аминокислот от рацемизации. Карбобензоксигруппу удаляют кгпа.1итиче-ским гидрогенолизом, а шреш-бутоксикарбонильную с помощью трифторуксусной кислоты. Для защиты карбоксильной группы используют реакцию этерификации.
Кроме того, с целью повышения эффективности процесса амидирования, осуществляют активацию карбоксильной группы М-чамещенной аминокислоты путем превращения се в хлорангндрид или в смешанный ангидрид (чаще взаимодействием с этилхлорформиатом). Ниже приведена схема синтеза дипептида алаиил-глицииа.
I. Зашита аминогруппы аланина:
CflH5CH2O о=с +
CI
бен зо ней карбо-
NII, н,с-сп °-н'(У^ C„H.CH,O-C-NH-CH-COOH * • I ' ’ II I 18
СООН О сн,
алцНИи М-зашнщеинын аланин г
иш1х:гс)рнл
180
2- Активация карбоксильной группы N-защищенного алаиииа
CI I О=С
OCH,ChH о=^
NH
ос,н5
H.C
щилхлор-формиат
N-jatuHmeiiiibiii аланин
3. Защита карбоксильной группы глицина
о-сн7с.н, । 2 О Э
о=с 1 N Н -СН -С-О-СОС. Н s
I II 11 - 5
сн3 о о
N защищенный аланин с акт и виро ванной карбоксильной [рунной
h,n-ch2-cooh + с,н5он — [ЛИЦИН
h2n-ch2-c-oc2hs
глицин с защищенной карбоксильной группой
4. Образование пептидной связи и снятие защиты
OCH,CAHS I -Од
О=С + H.N-CH,-C-OC.H, ---------
NH-CH-C-O-C-OC2HS О -С,Н5ОН;-00.
II 2 ' О
* c6h5ch2o-c-nh-ch-{-c-nh4cii,-c-oc2h5
I । П <
СН О
Jl_ _ --1
нд)
-СО,
Н, / Pd
ОН"
з
о
о
Н э N -СН -{-СО—Н N {-сн.-С-ОН
2 I • ‘ II
о
СН/--------
Ала-Гли
17 Синтез пептидов согласно приведенной схеме достаточно сложен и ! трудоемок.
: В 1962 г. Б.Меррфилдом был предложен более совершенный метод
получения пептидов, так называемый твердофазный синтез. Сущность Последнего состоит в том. что полипептидпая цепь наращивается на твердом носителе без выделения промежуточных продуктов синтеза, i Пептид, фиксированный на носителе, после каждой стадии тщательно промывают от избытка реагентов и побочных продуктов. Отщепляют Конечный продукт от носителя с помощью смеси бромоводородной и . трифторуксусной кислот.
В качестве твердого носителя используют зерна полимерной смолы, | содержащей хлор метильные (-СН2С1) группы, называемые якорными
181
группами, с которыми реагирует карбоксильная группа N-защищеиной а-амииокислоты. В результате взаимодействия происходит фиксация С-коица будущего полипептида на поверхности носителя. Аминогруппу, как правило, защищают ?»релг-бутоксикарбонильной группой (БОК), которая легко удаляется действием трифторуксусной кислоты. Пептидная связь образуется в присутствии активатора карбоксильной группы — N.N’-дициклогексилкарбодиимида (ДЦГК) C6Hi1N=C=NC6H„. Широкое применение этого вещества связано с легкостью получения, простотой применения, а также скоростью и эффективностью протекания реакции конденсации в его присутствии:
пептид—СН— NH? + HOOC^H-NH-EOK + C\H11-N=C=N-C6Hn *"
r R ДЦГК
N-капец пептидной цепи N-«iiuitinenntiH а-аминокнешна
—> пептид—СН—NH—С—СИ—NH-БОК + C6Hu-NH-J-NH-CfiH(f R О R О
N-N-iw ин клогексинмс’Квина
В настоящее время твердофазный синтез пептидов проводят в спе-циальных синтезаторах, где все этапы осуществляются автоматически с запрограммированной подачей соответствующих а-аминокислот.
6,4. СЛОЖНЫЕ БЕЛКИ (ПРОТЕИДЫ)
В зависимости от структуры белки делят иа простые (протеины) и сложные (протеиды). Последние, кроме белка, содержат в своей структуре химически связанную с ннм простетическую группу— небелковую часть молекулы. По природе простатической группы протеиды делят на липопротеиды, нуклеопротеиды, глико протеиды, хромопротеиды, фосфо-протеиды и металлопротеиды.
Липопротеиды в качестве простетической группы содержат липиды, нуклеопротеиды — нуклеиновые кислоты, гликопротеиды — углеводы, хромопротеиды — пигменты, фосфопротеиды — фосфорную кнепоту, металлопротеиды — металлы.
Существуют также сложные белковые комплексы, в состав которых одновременно входят белки, липиды и углеводы, называемые гликолипопротеидами. Они содержатся в соединительной ткани, клеточных стенках бактерий и др.
В зависимости от пространственной формы молекул белки делят на глобулярные и фибриллярные. Глобулярные белки имеют сферическую или эллипсоидную форму, фибриллярные-— состоят из вытянутых нитевидных макромолекул, называемых п ротенон дам и.
Глобулярные белки (альбумин, глобулин) малоустойчивы к действию температуры, кислот и щелочей, а фибриллярные (белки волос, ногтей, эпидермиса: соединительной, костной, хрящевой ткаии и др.) весьма устойчивы.
182
Под влиянием многих факторов (повышенная температура, изменение pH среды, УФ- и у-излучение и др.) происходит разрушение пространственной формы белков при сохранении первичной структуры. Этот процесс называется денатурацией белка. Денатурация является, как правило, необратимым процессом и приводит к потере биологических функций белков. Примером тепловой денатурации является «свертывание» яичных белков при варке яиц. При денатурации происходит разрыв водородных связей, стабилизирующих пространственную форму белка. Денатурированный белок теряет растворимость, в результате чего первоначальная пространственная форма его ис может быть восстановлена. Денатурация может быть также вызвана образованием нерастворимых солей белков. Это происходит при отравлении солями тяжелых металлов (ртути, свинца и др.). В качестве противоядия в таких случаях применяют белки с повышенным содержанием кис-лоз -пых групп, например, яичный альбумин. Выступая в качестве конкурента, эти белки связывают металлы с образованием нерастворимых солей, которые выводятся из организма.
КОНТРОЛЬНЫЕ ВОПРОСЫ И УПРАЖНЕНИЯ
1. Напишите формулы и назовите по систематической и тривиальной номенклатуре «-аминокислоты, входящие в состав белков.
2. На примере аспарагиновой кислоты напишите схемы реакций с:
а) бензоксикарбонилхлорндом;
б) HNO,;
в) уксусным альдегидом;
г) этанолом.
3. Напишите схему образования пептида аланилглутамилглицииа. Обозначьте пептидные связи.
4. Охарактеризуйте первичную, вторичную, третичную и четвертичную структуры белков.
5. Что такое сложные белки? Приведите классификацию белков в зависимости от природы простетической группы и пространственной формы,
Глава 7
НУКЛЕИНОВЫЕ КИСЛОТЫ
Нуклеиновые кислоты (от лат. nucleus «ядро») впервые обнаружены в 1868 г. швейцарским химиком Ф.Мишером в ядрах клеток. Позже аналогичные вещества найдены в протоплазме клеток. Нуклеиновые кислоты обеспечивают хранение и передачу наследственной информации. непосредственно участвуя в синтезе клеточных белков. Оии входят в состав сложных белков ..нуклеопротеидов, содержащихся во
всех клетках организма человека, животных, растений, бактерий и вирусов. Количество нуклеиновых кислот в различных нуклеопротеидах, кроме вирусных, колеблется в пределах от 40 до 65 %.
7.1. СТРОЕНИЕ НУКЛЕИНОВЫХ КИСЛОТ
Нуклеиновые кислоты, подобно белкам, представляют собой высокомолекулярные органические соединения, но в отличие от белков, образующих при гидролизе а-аминокислоты, мономерными единицами нуклеиновых кислот являются нуклеотиды. Поэтому нуклеиновые кислоты называют еще полинуклеотидами. Мономеры нуклеиновых кислот нуклеотиды имеют также достаточно сложное строение. При гидролизе нуклеотидов образуется углевод, ортофосфорная кпелота и гетероциклические основания.
I Химическая структура, состоящая из углевода и гетероциклического основания, называется нуклеозидом.
Таким образом, в целом гидролиз нуклеиновых кислот можно представить в виде схемы:
В зависимости от природы углевода, входящего в состав нуклеотидов. нуклеиновые кислоты разделяют на два вида дезоксирибонуклеиновые кислоты (ДНК), содержащие углевод 2-дез окси-Л-рибозу и рибонуклеиновые кислоты (РНК), включающие углевод Р-рибозу.
184
2-Дезокси-/)-рибоза и D-рибоза находятся в нуклеиновых кислотах в Р-фураиозиой форме:
2-; ро ксп -/) -р и Гиг м
ОН он
Л'рибоза
Входящие в состав нуклеиновых кислот гетероциклические основания являются производными пурина и пиримидина. К основаниям группы пурина относятся ядепни (А)* и гуанин (G):
6Ч1МИ1|О11урИП
2-н м и 11 о-6-n «сн пурин
Основаниями группы пиримидина являются урацил (U). тимин (Т). цитозин (С):
ураты (L“),
2.4-'UiOKCiiiiiipiiMit’uiii
7IIMHII (Т).
S-mCi nji-2.4-:im)KUiiiiipH м Iijiiii
4-a мин л?-о кем 11 при м н, in it
В состав ДНК входят аденин, гуанин, цитозин и тимин, в состав РНК аденин, гуанин, цитозин, урацил. Для гуанина, урацила, тимина и цитозина характерна лакгам-лактимиая таутомерия:
(лакп1М1к1Я форма)
I.UlKlHMlldM
* Для удобства нуклеиновые основания принято обозначать однобуквенными символами.
185
jaKi нмная форма lyauHiia
пактами ня форма тунцнпа
Наряду с указанными основаниями, в состав нуклеиновых кислот могут входить и другие гетероциклические основания, например, гипоксантин, метильные производные урацнла и гуанина, гидрированные
производные урацила и др.
З-Ы-мсгилураин.т (м’У)
лшидроураиил (УН. )
1-М-мстнягуаиин (м1 Г)
Z-N.N-aiiMC'iiL'iryamui (м; Г )
В нуклеиновых кислотах органические основания связаны N-гликозиднон связью с остатком D-рибозы или 2-дегюксн-Л-рибозы. Гликозидная связь осуществляется за счет полуацетального гидроксила моносахарида (С1') и атома водорода при № в пиримидиновых или № в пуриновых основаниях.
IN-Глнкозилы, состоящие из остатков нуклеиновых оснований и D-рибозы или 2-дезокси-Я-рнбозы, называют нуклеозидами.
В зависимости от природы углеводного остатка различаю! рибонуклеозиды и дезокси рибонуклеозиды.
Для отличия углеродных атомов рибозы и дезоксирибозы от углеродных атомов, входящих в состав пуриновых кислот и пиримидиновых оснований, первый принято обозначать символом «штрих», например С '
186
дезоксирибоза
дезо кси риб о ну клеозид
Названия нуклеозидов образуют аналогично названиям гликозидов. Так, нуклеозид, состоящий из рибозы и урацила, называют 0-у рация рибофуранозидом, нуклеозид из дезоксирибозы и аденина — р~аде-ниндезоксирибофуранозидом и т.д. Однако, чаще применяют названия, которые для рибонуклеозидов образуют из тривиальных названий соответствующих нуклеиновых оснований с окончанием -идии у пирими-. диновых и -озии у пуриновых нуклеозидов, например — аденозин, гуанозин, цитидин, уридин:
уридил цитидин
аденозин
туаиплин
В названиях дезоксирибонуклеозидов дополнительно вводится префикс «дсзоксп-», например, дезоксиадеиозии, дезоксигуанозии, дезоксицитидин. Исключение составляет название нуклеозида, состоящего из дезоксирибозы нтнмнна —тимидин (вместо дезокситимидина).
1 «т
ДВЗОКСИ11И гм дни
дезокси аденозин
лею кем । ya i io зи 11
Являясь N-гликозидам и, нуклеозиды в кислой среде подвергаются-гидролизу. Пуриновые нуклеозиды гидролизуются очень легко, пиримидиновые труднее.
В нуклеиновых кислотах гидроксильная группа у С-'1 * * или С4 пентозного остатка нуклеозида этерифинирована ортофосфорной кислотой.
I Сложный эфир фосфорной кислоты (фосфат) нуклеозида называет-
ся нуклеотидом.
В зависимости от природы пентозы различают рибонуклеотиды и
дезокс нрибопу клеотид ы.
В номенклатуре нуклеотидов используют два подхода. С одной стороны, их рассматривают как сложные эфиры моиофосфаты. а с другой как кислоты (табл. 7.1).
Таблица 7.1
Номенклатура нуклеотидов
Названия нуклеотидов как монофосфатов Названия нуклеотидов как кислот
Адепозин-51-монофосфат (АМФ) 5-адеииловая кислота
Гуапозин-51-моиофосфат (ГМФ) 5‘-гуаииловая кислота
Цитидип-5'-моиофосфат (ЦМФ) 5-цитидиловая кислота
Уридии-5'-монофосфат (УМФ) 5'-уридиловая кислота
Дезоксиале] юзии-5'-монофосфат (ДАМФ) дезоксиалсиилоная кислота
Дезоксигуанозип-51-мопофосфат (ДГМФ) лезоксигуаниловая кислота
Дезокси цитидин-З'-мопофосфат (ДЦМФ) дезокси цитидиловая кислота
Тимиди|[-5'-мо11офосфат (ДТМФ) тимидиловая кислота
При гидролизе нуклеиновых кислот, наряду с пуклеозид-З'-фосфа-тами, образуются также нуклеозид- З-фосф а ты.
Положение остатка фосфорной кислоты определяется местом разрыва фосфолиэфирной связи между соседними нуклеотидами,
Нуклеиновые кислоты представляют собой продукты полимеризации мононуклеотидов. Нуклеотиды связываются в длинные цепи с помощью фосфодиэфирных связей, которые образуются с участием гидроксила при С предыдущего нуклеотидного звена и гидроксила при С5* последующего нуклеотидного звена:
;ti+O Or I
-HO
Мононуклеотиды, их производные и динуклеотиды присутствуют j клетках также в свободном виде и играют важную роль в обмене ве 190
шеств. Во всех тканях организма, наряду с нуклеозид монофосфатами, содержатся ди- и трифосфаты нуклеозидов.
Особенно широко известны аденозин-5'-фосфат (АМФ), аденозин-5-днфосфат (АДФ) и аденозин-5‘-трифосфаг (АТФ),
Эти нуклеотиды способны взаимопревращаться путем фосфорилирования (присоединение одного или двух остатков фосфорной кислоты к АМф), либо дефосфорилирования (отщепление одного или двух остатков фосфорной кислоты от АТФ). При дефосфорнлировании выделяется значительное количество энергии, использующееся в организме для протекания тех или иных биологических процессов, например, в биосинтезе белка.
алепозип монофосфат (АМФ)
адеиознпдифосфат (ЛДФ)
адено ши грпфосфат (АТФ)
Связь Р---О между остатками фосфорной кислоты в молекулах нуклеозидполифосфатов является макроэргнческой связью , при раэ-
Макротргичсскими принято считать те связи, при разрыве которых выделяет-Ся энергия не менее 20 кДж/моль.
191
рыве которой выделяется значительное количество энергии. Поэтому АТФ во многих биохимических процессах выполняет роль поставщика энергии.
7.2. РИБОНУКЛЕИНОВЫЕ (РНК)
И ДЕЗОКСИРИБОНУКЛЕИНОВЫЕ (ДНК) КИСЛОТЫ
Нуклеиновые кислоты представляют собой высокомолекулярные гетерополимеры, состоящие из чередующихся остатков ортофосфорной кислоты и рибозы или дезоксирибозы, связанных с нуклеиновыми основаниями, которые выступают в полимерной цепи как «боковые группы» (рис. 7.1).
сахарная часчь (осипок рибозы и РНК или дезоксирибозы в ДНК)
-остаток оршфосфориой
КИСЛО i и
---О
0С1ЙГПК
I у ш 111 н а
011
- остаток
аденина
Рис. 7 I Схема участка полинуклеотид ной пени молекулы нуклеиновой кислоты
Определенная последовательность нуклеотидных звеньев в полн-нуклсотидной цепи называется первичной структурой нуклеиновых КП слот.
Пространственна и ориентация полинуклсотидиых цепей в молекуле называется вторичной структурой нуклеиновых кислот.
Впервые вторичную структуру ДНК в виде модели из двойной спирали (рис. 7.2) описали американский биохимик Дж.Уотсон и английский биохимик Ф.Крик (1953 г.). Обобщив работы Л.Полинга. А.Тодда, Э.Чаргаффа. М.Уилкинса и других, они пришли к заключению, что молекула ДНК представляет собой две параллельные право-закрученные спирали (двойная спираль), фиксированные между собой ван-дер-ваальсовыми силами притяжения, действующими вдоль спирали между ядрами нуклеиновых оснований (межплоскостное вертикальное взаимодействие). Кроме того, вторичная структура стабилизируется водородными связями между остатками нуклеиновых оснований двух параллельных спиралей.
192
<—1,8-2 нм->
По модели Уотсона и Крика диаметр спирали 1,8-2,0 нм. Каждый виток спирали содержит 10 пар оснований. Шаг спирали составляет 3.4 нм.
Расстояние между плоскостями оснований по вертикали равно 0,34 нм. Пол и нуклеотидные цепи двойной спирали расположены в противоположном направлении. На одной нити двойной спирали фосфодиэфириые связи образованы по типу 5*-3', а пн другой — наоборот. по типу 3-5'.
Между пиримидиновыми и пуриновыми нуклеиновыми основаниями параллельных нитей двойной спирали ДНК образуются водородные связи. При этом аденин образует связь с тимином, а гуанин —с цитозином. Поэтому их называют комплементарными парями (АТ и GC):
Рис. 7.2. Схема двойной спирали ДНК
В комплементарной паре GC имеются три водородные связи, а в комплементарной паре АТ — две водородные связи.
Более наглядно комплементарность представлена на рис. 7.3.
РНК представлена одинарной спиралью. Вторичная структура РНК имеет относительно небольшую массу.
1 Ml
193
Известны три типа РНК: матричная РНК (мРНК), иди информационная РНК. рибосомальная РНК (рРНК) и транспортная РНК (тРНК).
Рис. 7.3. Комплементарные пары нуклеиновых оснований в составе двойной спирали ДНК,
КОНТРОЛЬНЫЕ ВОПРОСЫ И УПРАЖНЕНИЯ
1. Дайте определение каждому из следующих понятий:
а) нуклеозид;
б) нуклеотид;
в) рибозид;
г) дезоксирибозид;
д) рибонуклеиновая кислота:
е) дезоксирибонуклеиновая кислота.
194
2. Напишите структурные формулы следующих соединений:
а) уридин-5‘-монофосфат;
б) дезоксигуаниловая кислота;
Ь в) 2'-адениловая кислота;
В г) аденозиндифосфат;
В д) цитиднн-5‘-монофосфат.
В. Для тимина, цитозина, аденина и гуаиина приведите возможные В таутомерные формы. Почему нуклеиновые основания в ДНК н РНК Г содержатся в оксоформе, а не в гидроксиформе?
: 4. Назовите основные структурные различия между ДНК и РНК. При-I ведите формулы и названия продуктов полного гидролиза ДПК.
; 5. Напишите схему образования ЛТФ (аденоэинтрифосфорной кисло-; ты). Покажите в формуле АТФ макроэргические связи. Назовите все промежуточные продукты.
: 6. Приведите схему гидролиза аденозина, гуанозина и цитидина.
. В каких условиях он протекает?
V. Напишите схему образования уридиловой кислоты и уридин-5'-В дифосфата (УДФ). В состав каких нуклеиновых кислот —ДНК или В РНК входят остатки этих соединений?
К. Напишите схему образования динуклеотида, содержащего остатки В аденина и урацила.
В. Напишите схему образования дииуклеотида. содержащего остатки В цитозина и гуанина.
195
Глива 8
ЛИПИДЫ
Липиды, или жнроподобныс вещества , — обширная группа природных органических соединений, главным образом производных высших алифатических кислот и спиртов.
Липиды содержатся во всех клетках живых организмов и принимают участие в разнообразных физиологических и биохимических процессах. По строению они весьма неоднородны. Однако, все липиды обладают общей особенностью — растворимостью в неполярных растворителях (эфир, хлороформ, углеводороды).
8.1. КЛАССИФИКАЦИЯ
Разное отношение жироподобных веществ к воздействию гидролизующих реагентов положено в основу деления липидов на омыляемые и неомыл яе,мыс:
^Изопреноиды Простагландины]
V___ V
Г Фосф о-] Глико- j I липиды! днпидьН
,--*------1
[ Сложные |
К неомыляемым липидам относят неспособные к гидролизу изопреноиды и простагландины. Омыляемые липиды подразделяют на простые липиды, гидролиз которых приводит к смеси только спиртов и карбоновых кислот (жиры и воски), и сложные липиды, образующие при гидролизе, кроме спиртов и карбоновых кислот, также фосфорную кислоту, моно- или олигосахариды (фосфолипиды и гликолипиды).
’ От грея. Xino? (дивос) — «жир» 196
8.2. ОМЫЛЯЕМЫЕ ЛИПИДЫ
8.2.1. Жиры
Жиры являются сложными эфирами глицерина и высших алифатических кислот, т.е. триацилглнцеринами, или триглицеридами. Их общая формула:
О ch2-o-£-r I - ° сн— o-£-r1
I °
CH2-O-t-R"
Приоритет в установлении строения жиров принадлежит французским химикам М.Э.Шевре-лю, который в 1817 г. выяснил состав и структуру некоторых триглицеридов, и П.Э.М.Бертло. доказавшему в 1854 г. строение жиров путем этерификации глицерина высшими жирными кислотами.
Триацилглнцерины бывают простыми и смещаниыми. Простые включают остатки одинаковых кислот (R - R' = R1'), смешанные — разных. Природные жиры представляют собой главным образом смешанные триацилглицерипы.
По консистенции жиры могут быть твердыми и жидкими. Твердые жиры содержат преимущественно остатки насыщенных высших жирных кислот. В состав жидких жиров. обычно называемых маслами, входят в основном остатки ненасыщенных кислот. Жиры животного происхождения, как правило, — твердые вещества, растительные жиры — жидкие*. В молекулах как растительных, так и животных жиров (за исключением свиного) чаще всего остатки непредельных кислот занимают положение 2.
В состав жиров входят остатки одноосновных жирных кислот, преимущественно с не разветвленной цепью с четным числом (от 4 до 26) углеродных атомов (наиболее часто содержатся ацильные остатки с 16 и 18 атомами углерода).
Наиболее часто в состав жиров человеческого организма входят остатки таких насыщенных высших жирных кислот, как стеариновая и пальмитиновая, ненасыщенных — арахидоновой, олеиновой, линолевой и линоленовой. Насыщенные жирные кислоты поступают в организм с нищей, а также путем биосинтеза. Олеиновая, линолевая, линоленовая и арахидоновая кислоты не образуются в организме человека; оии поступают только с пищей, поэтому их называют незаменимыми.
Тривиальные названия, структурные формулы и некоторые физические константы биологически важных жирных кислот, выделенных из жиров в процессе гидролиза, приведены в табл. 8.1.
Исключения: рыбий жир, являющийся жидкостью, и масло какао —твердое вещество (при обычных условиях).
197
7'аблш(с1 в. /
высшие жирные кпслогьг
Название Формула Температура. °C плавления | кипения
Насыщенные кислоты
Масляная кислота СН3(СН2)2СООН -5.3 164
Капроновая кнсдогх СН/СН2)4СООН .. , -3.2 206
Канрилс&аякислота СН3(СН2)6С’ООН 16.5 240
Каприновая кислота CHjJCH^COOH 31.6 271
Лауриновая кислота СП?(СН2)10СО()П 44.8 299
Миристиновая кислота СЩСНОцСХХШ 54.4 149*
Пальмитиновая кислота СНу(СН?)иСООН 62.9 167*
Стеариновая кислота СН3(СН2)16(ХХ)Н 70.1 184*
Лрахинсвая кислота СН?(СН2)1КСООН 76.1 204*
Ненасыщенные кислоты
Олеиновая кислота Элак/шновдя кислота а-Линолевая кислота Линоленовая кислота Хаульмуг решая кислота Арахидоновая кислота СН(СН,)7СН. СН(СН2)7СООН CHjClt)7CH <Ен(СН2)7СООН СН/СН^СН СНСН.С11 СЩСН,)7СООН снсн2сн3 снсн.сн снсн2сн СП(СН2}7СООН ^^(CHj^COOH СН'ССНДСН СНСНдСН снсн.сн снсн’сн &Н(СН2)3СООН 13.4 и 16.3 51.5 -5.2 -п.з 75.3 —
* При 1 мм рт. ст.
198
А, Номенклатура н изомерия жиров
По систематической номенклатуре ИЮПАК родоначальной структурой в молекуле жнра выступает глицерин. Ацильные остатки жирных кислот перечисляют в начале названия в алфавитном порядке, если необходимо, употребляют множительные приставки ди- и три-. Тривиальные названия жиров образуют из приставок, построенных из названий соответствующих жирных кислот путем опускания части названия кислоты -иловая кислота и добавления суффикса -ил :
СН,-О-СО-С17Н35 сн—о-со~с|7и35 СН2-О-СО-С]7Н35 грнС1'еарся.'и’,'1Ш1срш1.
ipitcrejpHiT
СН,-О-СО-С17П35 СН-О-СО-С17Н33 СП,-О-СО-С,7Н<5 2-njk'o нл-1.3-лиегеа р о I [, г-пш перин: 2-o;i(»juicicapitii
СН-ртО-СО-СнН23 сн- О-СО-С]5Н,| сн,-о-со-с17н35 1-лауреи.г-2-пХ11.\111 rnii'i-3 членро и. 11, tn цер и и:
l-;iaypo-2-nu:n.Mn ruciCiipuir
Изомерия жиров связана в основном е различным взаимным расположением ацильных остатков в структуре гриацилглицерина (структурная изомерия).
Молекулы смешанных триглицеридов, содержащие асимметрический атом углерода, существуют в виде двух оптических изомеров. Для жиров, молекулы которых содержат остатки непредельных кислот. характерна геометрическая изомерия.
Б. Получение жиров
Для синтеза трнацилглицеринов пригодно большинство реакций О-анилирования спиртов (этерификация, взаимодействие глицератов натрия с хлорангидридами кислот и др.), однако, синтетические способы получения жиров из глицерина не имеют промышленного значения ввиду доступности разнообразного природного сырья, К основным методам выделения жиров и масел из предварительно измельченных тканей растений и животных относятся: вытопка, прессование и экстракция органическими растворителями (бензин, трихлорэтилен и др.).
В. Физические свойства жиров
Физические свойства жиров зависят главным образом от строения жирных кислот, образующих их молекулы. Так, температуры плавления жиров, содержащих остатки ненасыщенных кислот, значительно ниже, чем у насыщенных жиров с тем же числом атомов углерода. С увеличением длины углеродных цепей жирных кислот температуры плавления жиров повышаются. Поскольку природные жиры являются смесями трнацилглицеринов. они не имеют четких температур плавления. Большинство жнров плавится при температуре 22-55 °C.
Жиры легко растворимы в углеводородах и их хлорпроизводных, эфирах, кетонах, малорастворимы в этиловом спирте (исключение составляет касторовое .масло, растворимое в этаноле), нерастворимы в во
199
де, однако, в присутствии поверхностно-активных веществ (ПАВ), называемых эмульгаторами жиры образуют высокодисперсные гетерогенные системы типа эмульсий. В частности, эмульгирующее действие белков придаст стабильность эмульсии молочного жира в воде — молоку.
Г. Химические свойства жиров
Как и все сложные эфиры, жиры способны подвергаться гидролизу. При наличии в молекуле триглицерида остатков ненасыщенных кислот он проявляет и свойства алкенов.
1. Гидролиз жиров, Мыла, Детергенты. В результате взаимодействия жиров с водными растворами гидроксидов щелочных металлов образуется смесь глицерина и натриевых (калиевых) солей высших жирных кислот. Указанные соли называют мылами, а реакцию щелочного гидролиза жиров, при которой образуются мыла —омылением* .
О
Clk-O-C-R. сщ-он
О 3NaOir I
СН -O“C“R' ------->- СН~ОН + R-COONa + R’-COONa + R"-COONa
| О I,OH | CH2-O-C-R’‘ ch,-oh
Щелочной гидролиз жиров ускоряется при замене водной среды ни водно-спиртовую. Щелочь служит как реагентом, так и эмульгатором жиров, увеличивая поверхность соприкосновения жировой фазы с гидролизующей средой: спирт понижает вязкость реакционной среды. При температуре 200-225 °C и давлении 2-2.5 МПа гидролиз жиров протекает и без добавления щелочи. В промышленности гидролиз жиров проводят путем их нагревания при обычном давлении с водой в присутствии сульфокислот как эмульгаторов и катализаторов (катализатор Твитчела и контакт Петрова).
Реакцию гидролиза жиров используют в аналитической практике при установлении качества жира, С этой целью определяют так называемое число омыления, которое характеризует общее содержание свободных и связанных в триглицериды жирных кислот.
Число омыления — это количество миллиграммов КОН. которое расходуется при гидролизе 1 г жира. Величина числа омыления чависиз оз относительной молекулярной массы жирных кислот, остатки которых входят в состав жира.
Следует отметить, что в качестве примеси жиры содержат некоторое количество свободных карбоновых кислот, О количественном со
* Термин «омыление» часто применяют и к реакции гидролиза других функциональных производных кислот: всех сложных эфиров, амилов, нитрилов и г,п„ и даже к реакции гидролизы галогенопроизводных.
200
держании этих кислот позволяет судить кислотное число (число нейтрализации). определяемое как количество миллиграммов КОН, необходимое для нейтрализации 1 г жира.
Разность между числом омыления и кислотным числом составляет эфирное число, характеризующее содержание остатков жирных кислот, эфирносвязанных с остатками глицерина.
Смесь чвердых высокомолекулярных жирных кислот, главным образом стеариновой и пальмитиновой, получаемую при гидролизе жиров в кислой и нейтральной средах, называют стеарином. Стеарин используют наряду с парафином для изготовления свечей.
Для получения твердого мыла выделившуюся смесь высших жирных кислот нейтрализуют содой.
Если нейтрализацию смеси высших жирных кислот проводят с помощью поташа (карбоната калия), то образуется калиевое («зеленое») мыло, отличающееся от натриевого жидкой консистенцией.
При нейтрализации жирнокислотной смеси оксидами щелочноземельных и переходных металлов (CaO. MgO. ZnO. Pb() и др.), образуются нерастворимые в воде, так называемые «металлические» мыла, которые применяются в качестве медицинских пластырей (например, простой свинцовый). Молекулы мыла содержат в своей структуре гидрофильный («стремящийся» к молекулам воды) карбоксилат-анионный фрагмент и гидрофобную («избегающую» контакта с водой) протяженную углеродную цепь. В результате такого строения мыла обладают практически одинаковой способностью растворять как гидрофильные (вода, спирты и т.п.), так н липофильные (углеводороды, эфиры и т.п.) вещества. Кроме того, в отличие от молекул натриевых и калиевых солей низших жирных кислот, молекулы мыла способны к агрегации («слипанию» друг с другом) с образованием сферических структур — мицелл. В водной среде мицеллы мыла имеют строение, схематически показанное на рис. 8.1. В мицелле молекулы мыла сцеплены между собой углеводородными «хвостами», а к воде обращены полярными карбоксилатными группами. Из-за участия в образовании множества мицелл молекулы мыла не могут равномерно распределяться средн молекул воды. т.е. мыла не способны к образованию исгипиых водных растворов.
Рис. 8.1. Строение мицеллы мыла в воде
пол яркая ка|хн>ксилитиая группа
Непал ярный углеводородный «хвост»
201
Ila границе раздела водной и газовой (воздушной) фаз, молекулы мыла ориентируются полярными копнами «к воде», а гидрофобными «наружу» (рис. 8,2). При этом они понижают поверхностное натяжение воды, т.е. проявляют так называемые поверхностно-активные свойства. По наличию в молекулах карбоксилатных групп мыла отно-
Рис. 8.2. Ориентация молекул мыла на границе раздела водной л воздушной сред
Описанные свойства мыл обуславливают их моющее действие. Вода, содержащая мыло, благодаря уменьшению своего поверхностного натяжения, приобретает способность проникать в тончайшие поры на отмываемой поверхности. При наличии на этой поверхности частиц веществ, не смачивающихся водой (жиров, восков, нефтепродуктов и пр.) молекулы мыла сцепляются с гидрофобными частицами загрязнений своими углеводородными «хвостами», образуя вокруг таких частиц плотную изолирующую пленку (рис, 83, а), вклиниваются между загрязняющей частицей и очищаемой поверхностью (рис. 83. б) и в конечном итоге отрывают эту частицу, переполи ее во взвешенное состояние в толще водной среды (рис. 83, в).
а б в
Рлс. 8..V Схема моющего дейслвия мыла
Аналогичный механизм имеет процесс солюбилизации не растворимых в воде органических (в том числе лекарственных) веществ.
Более широкое применение мыл в качестве моющих средств ограничено тем, что в «жесткой» воде (с повышенным содержанием иоиов Са’ н Mg+) мыла образуют нерастворимые соли кальция и магния. 202
которые выпадают в виде хлопьев, загрязняющих отмываемые поверхности. На образование этих солей расходуется значительное количество и(ыла. Как соли слабых кислот и сильных оснований мыла вследствие гидролиза создают щелочную среду в водных растворах, обуславливающую раздражающее действие мыл на слизистые оболочки. Указанные недостатки мыл явились причиной создания широкого ассортимента более дешевых и технологичных синтетических заменителей мыл
детергентов.
Детергенты, или синтетические моющие средства, как и мыла, относятся к ПАВ. Их молекулы тоже включают неполярный (гидрофобный) углеводородный фрагмент и полярную (гидрофильную) часть, представленную анионоидной или катионоидной функциональной группировкой. связанной с ионами натрия или минеральной кислоты соответственно. Полярный коней молекулы детергента может быть также образован нейтральным остатком многоатомного спирта или полиэфира. В зависимости от строения полярной части молекулы синтетические моющие средства делят на анионные, катионные и вентральные.
В молекулах анионных детергентов в качестве полярного фрагмента чаще всего содержится бенэолсульфоиатный остаток. Неполярная часть молекулы таких детергентов образована длинноцепочным алкилом, как правило, вторичным и неразветвленного строения.
СН3-(СН2)ЙЧ /=\ _
СН —ртт Н—4 SOjNa
l V. tl ? ____у
и-(дец ил-3)беп -юлсульфо и a i пн t рня
Детергенты с разветвленными углеводородными цепочками неприемлемы в экологическом отношении, поскольку в процессе очистки сточных вод оии не разлагаются микроорганизмами, что приводит к накоплению синтетических моющих веществ в природных водоемах в гибельных для их обитателей концентрациях.
К катионным детергентам относятся прежде всего четвертичные аммониевые соли, например;
СН,— (СНД_ + ’ ст
СН,—(СН,),< 'сн,
N. N-ли мет и л-N. N-лио кгадец и лам м о н и я хло р нд
Примерами нейтральных (неионогенных) детергентов могут служить следующие соединения;
СН2ОН
хон ну| но-(а12-сн,-ок-(сн2)11-сн3
но\—<н О-до лепило ктатг илепгли ко ль
н он
Р-додецил глюкозил
203
В отличие от мыл, синтетические моющие средства ие образуют нерастворимых солен с катионами кальция и магния, а их водные растворы имеют нейтральную реакцию, поскольку детергенты являются либо солями сильных кислот и сильных оснований, либо не содержат групп, способных гидролизоваться в обычных условиях. Важным преимуществом детергентов перед мылами является и то. что их производство не нуждается в использовании в качестве сырья жиров — цепных пищевых продуктов.
2. Окисление жиров. Причиной легкой окислясности жиров кислородом воздуха является наличие двойных связей в молекулах, что приводит к «прогорканию» жиров. При их окислении образуются альдегиды с короткими углеродными цепями, которые обуславливают неприятный запах и вкус «прогоркших» жиров.
О
СН, -О—С—(СН, )7 -сн I II
сн^сн^-сн
О II с-о-сн ----------*---
[ Q о.геинат
R I И
CH?-O-C-R’
о СН?—О—С—(СН->)7—СН—О
г о -н CH.-tCH.h’CH-O ~ с—v—сн
°2 I I О —* Rin
CH.-O-C-R'
ион II --► R-C-O-CH
? /Н
СН.-О-С—(СН2)7—
I ' о
нч
^C-(CH7)7-C}h о
политик аюлии но во го
О иолуалъдешда
II
СН,-О-С—R’
пеларгоновый альдсгнд
Окисление ненасыщенных жиров в мягких условиях (водным раствором перманганата калия) приводит к образованию гликолей. Результатом окисления в более жестких условиях является разрыв углеродного скелета с образованием остатков соответствующих карбоновых кислот с более короткими углеродными цепями.
О
сн; -О-С-(СН2)7 -сн -он
О СН?-(СН,)?-^Н-ОН
с-о-сн 4—:J
I I О.Ю-дипгДроюсн-
R I jj стеарат
CH2-O-C-R’ Hh
КМпО4. Н2О
I Р СН2 -О-С—(СН,)7-СН
?1 I .СН^СНрДн
С-О-СН ------v
I плен inn
II
СН,—о—C-R' о о
км„о,Н1о; О O^-Q^-fCH^-t-OH о
С“О—СИ q азелаинат С—(СН,)-,—СН^
R _Q_____pj НО пслар го новая
- кисчота
204
Для жиров, в составе которых преобладают остатки насыщенных жирных кислот, при окислении характерно образование кетонов,
5. Гидрогенизация жиров. Гидрогеничации подвергаются растительные масла и жиры, получаемые из морских животных (китовый жир и ДР.).
В основу процесса положена реакция присоединения водорода по месту разрыва двойных связей в остатках линолевой, линоленовой, олеиновой и других непредельных кислот в присутствии никелевого или платинового катализаторов при температуре 190-220 °C и давлении 0.2-2.0 МПа (каталитическое гидрирование).
о сн3-сн2-сн=сн-сн,СН о
сн.-о-с—(сн,)7—см=сн-сн,Д!:н сн.-о-^чси.^-сн,
О СН,—(СНД.-СН о
I II ? - 4 II 12 Н I II
СН— О-С—(СН,)-—СН=СН-СН,-СН----------► СН— О-С-(СН,)[5-СН.
I О " Vi. I. р I о
1 II 1 II
СН,-О-С-(СН2)7—СН=СН-(СН,)7-СН3 СН,-О-С-(СН,)]6-СН3
2Ч)-линолсил-ЗЧ)-1ииолс11оил-[-0> олсоидпш нернп трис1еароллыицерип
(жидкий жир - масло) (твердый жир)
Благодаря насыщению углерод-углеродных связей водородом, а также вследствие изомеризации остатков олеиновой кислоты (щ/с-изо-мер, т. пл. 13.4 и 16.3 °C) в элаидиновую (ш/дая-изомер. г, пл. 51.5 °C) под влиянием никелевого катализатора (элвизирование), растительные масла превращаются в твердые жиры.
Промышленные жиры, полученные путем гидрогенизации растительных масел, а также жиров, добываемых из морской фауны, называют саломасами. Пищевые саломасы, имеющие т, пл, 31-33 °C, используются в производстве кондитерских, кулинарных жиров и маргарина. Маргарин — это пищевой жир. представляющий собой смесь гидрогенизированных жнров растительного и животного происхождения с добавлением сливок, вкусовых веществ и отдушек, например, диацетила С113—СО—СО -СП3, ацетоина СП—СНОП—СО—СН, и др. Технические саломасы (т. пл, 39-49 °C) — сырье для получения мыла, стеарина и др.
В результате гидрогенизации у жиров изменяются не только их физические свойства, ио благодаря резкому уменьшению числа двойных связей в их молекулах они приобретают устойчивость к воздействию окислителей и. в частности, кислорода воздуха, т.е. к прогорканию. При этом они сохраняют высокую питательность, характерную для Лучших сортов животных и растительных жиров, но в отличие от животных жиров, продукты гидрогенизации растительных масел не содержат примеси холестерина, избыток которого в организме приводит к атеросклерозу. Количество граммов водорода, необходимое для гидрирования 10 кг жнра. является аналитической характеристикой (число гидрировавня), свидетельствующей о степени ненасыщенности жира.
205
4. Присоединение галогенов. Реакция присоединения галогенов по месту разрыва двойных связей в ряду жиров имеет большое аналитическое значение. Обесцвечивание бромной воды указывает на содержание остатков непредельных кислот в молекуле исследуемою триглицерида. Количество граммов иода, присоединяющегося к 100 г жира, называемое иодиым числом, характеризует степень ненасыщенности жиров.
О СН^СН.Ъ-СН О СНч(СН,)7-СН-1
II • II II “ (
СН.-О-С------(СН2)7—СН СН.-О-С----(СН2)7—СП-1
(j) СН,(СН2)7-СН О СНч(СН2)?-СН-1
сн—о-с-------(СН2)?—СН —сн— о-с----------------(СН2)7—CH-I
<jj> СНЛСН^-СН О CH3(CH2h-CH-1
C1L-O-C------(СН2)7—СН СН2-О-С----(СН2>7—СН-1
триолеин три |9г10-лииг>дстеароил)глниерил
Иодное число варьирует для растительных масел в пределах 100-200, для животных жиров - от 25 (у молочного) до 86 (у конского), для жиров морских млекопитающих и рыб - от 100 (китовый жир) до 193 (тюлений жир).
8.2.2. Воски. Гвины
Воска — это сложные эфиры высших карболовых кислот и высокомолекулярных спиртов, содержащие четное число углеродных атомов в кислотных и спиртовых остатках. Как правило, это насыщенные соединения. Чаще всего в состав восков входит цетиловый (С1бН33ОН) и ми-рициловый (C^H^iOH) спирты.
Воски делят на животные (спермацет, пчелиный воск, ланолин и др.) и растительные (карнаубский воск). К воскам принято относить также озокерит (горный воск) — минеральный ископаемый продукт (т. пл. 58-100 °C), представляющий собой смесь главным образом предельных углеводородов, преимущественно с разветвленными цепями. Применяется в медицине (озокеритолечение), в изготовлении смазок, кремов, гидрофобизации материалов и др.
Температура плавления восков повышается с удлинением углеродной цепи молекул; в связи с этим воски бывают мягкие и твердые.
К мягким воскам относят л а иол ин и спермацет. Ланолин получают путем очистки шерстного воска, который представляет собой смесь сложных эфиров ланолинового спирта (C|2H2iOH), диэфиров а,р-ал-кандиолов (гликолей), высших жирных кислот (C1)f-Ci4), с высоким содержанием свободных высших спиртов и кислот (до 45%). углеводородов (до 18%) и стеринов (до 10%). Шерстный воск вырабатывается кожными железами овец и в большом количестве (5-16%) находится на овечьей шерсти, с которой его смывают на шерстомойных фабриках органическими растворителями. Ланолин (т. пл. 36-42 °C) нерастворим в воде, ио, в отличие от других восков, способен образовывать устой-206
чивые эмульсии даже с двойным по массе количеством воды. Последнее свойство позволяет использовать ланолин в качестве мазевой основы для введения в состав мази водорастворимых лекарственных веществ.
Спермацет, на 98% состоящий из цетнна CtsH^COOCjJHI^. вымораживают из спермацетового масла, получаемого вытапливанием из спермацетового мешка головы кашалота. Спермацет используется в фармации как основа для мазей, кремов; при производстве мыла, свечей и др. Гидрированием спермацетового масла получают цстип-пальмитат воск, более устойчивый к окислению, чем спермацет.
Примером твердого воска является карнаубский воск (покров листьев бразильской восковой пальмы). Он содержит более 80% эфиров высших жирных кислот и высших спиртов. Основная составная часть НО-СН2-(СН?)|1-СОО-СН2-(СН2)|Т1-СН-1, где п = 16-28, m = 30 и 32, Карнаубский воск применяют в качестве компонента полировочных паст, при выделке кожи, в производстве копировальной бумаги.
Пчелиный воск содержит 72% эфиров высших жирных кислот и высших спиртов (33% мирицилпальмитата (?]5Н11(.'ООС111НЬ|). до 13,5% свободных кислот и. 12-12,5% углеводородов. Температура плавления пчелиного воска 62-70 °C. Получают его из пчелиных сот, используют для изготовления искусственной вощины, как компонент для полировочных паст и мазей, косметических препаратов и др.
Теины синтетические продукты, близкие по строению к глицеридам, Они являются неполными простыми эфирами шестиатомного спирта сорбита и полиэтиленгликоля, у которых свободные гидроксильные группы в остатке сорбита этерифицированы высшими жирными кислотами.
8.3. НЕОМЫЛЯЕМЫЕ ЛИПИДЫ
83.1. Простагландины
Исследования в области простагландинов получили широкое развитие с 1933 г., когда было обнаружено физиологически активное вещество биогенного происхождения, стимулирующее гладкую мускулатуру и снижающее кровяное давление. Впоследствии оно было названо простагландином (ПГ), г.к, тогда предполагали, что оно образуется в предстательной железе (glandula prostata). Структура этого простагландина была установлена в 1962 г,, а в 1968 г, впервые удалось осуществить его синтез. Сегодня известно несколько десятков простагландинов. Все они содержат карбоксильную группу и 20 углеродных атомов в молекуле, т,е, их можно рассматривать как производные эйкоза повой кислоты.
tkkowiobuh кислота
207
Однако, в отличие от последней, простагландины имеют в своем составе циклопентановое кольцо, а поэтому их можно рассматривать также как производные простановой кислоты, содержащие от одной д0 трех двойных углерод-углеродиых Связей, одну или две гидроксильные а иногда — карбонильные группы.
В зависимости от наличия двойных связей и заместителей в П5Ь тичлсином цикле и боковых цепях, простагландины обозначаю! буквами А, В, С, Df Е и F.
П ГА П ГВ П ГС 11ГР П ГЕ ИГ!
По числу двойных связей в боковых цепях каждая из указанных групп делится па серин, обозначающиеся индексами. Например, простагландин ПГЕ. содержащий двойную связь в ш/мшс-конфигурации (транс-С1 ?-С|4). обозначается ПГЕ j; имеющий еще одну двойную связь — — ППи:
Простагландин ПГЕ3 содержит еще одну двойную связь Ц(Щ'-С!7-С,!().
В названиях простагландинов D и F ориентацию гидроксильной группы в положении 9 по отношению к углеродной цепи при С обозначают греч. буквами а или 0. Буква а указывает на цис-. ар — на трлпс-конфигурацию:
ПГГ?а (дпнопросг)
Биосинтез простагландинов в организме осуществляется на основ6 пол и не насыщенных жирных кислот, и прежде всего арахидоновой :
208
/=xs/=\^^,cOOH
арахидоновая кисло га
В чрезвычайно малых концентрациях простагландины содержатся в0 всех клетках организма человека и животных, однако наибольшее их количество находится в сперме (до 0,3 мг/г).
Простагландины проявляют целый ряд разнообразных физиологических и фармакологических свойств.
Ученые предполагают, что простагландины регулируют обмен веществ в клетках организма, т.е. являются «клеточными гормонами». Известно, что они влияют на процессы свертывания крови, снижают кровяное давление, угнетают выделение желудочного сока и т,д. Важнейшим свойством простагландинов является их способность стимулировать сокращение гладкой мускулатуры. Наибольшая активность отмечена у простагландинов групп Е. F и А.
В настоящее время приведенные выше простагландины ПГЕ2 и ПП"?и применяются в медицинской практике под названиями динопростом и динопрост соответственно как средства для возбуждения и стимуляции родовой деятельности. Имеются сведения о возможное! и применения проста! ландинов и в качестве бронхолитических, противоязвенных и других средств.
Механизм действия простагландинов до настоящего времени не выяснен. одиако установлено, что ряд противовоспалительных средств тормозят биосинтез и ослабляют физиологическое действие простагландинов. С этим явлением связывают противовоспалительное действие ацетилсалициловой кислоты, индометапипа и др.
8.3.2. Изопреноиды
Изопреноиды — это группа природных соединений, рассматриваемых как продукты превращения изопрена СН^С^СН,)---СН=СН2 (см. ки. 2, с. 59). Структуру изопреноидов имеют некоторые лекарственные средства, витамины, гормоны, душистые вещества и др.
По химической классификации изопреноиды относят к разным классам соединений, но выделены оии в одну группу по биогенетическому признаку, поскольку общим звеном их молекул выступает изопреновый фрагмент. Одиако, биосинтез изопреноидов осуществляется не из свободного изопрена, что вполне выполнимо in -vitro. а происходит с Участием растворимых фосфорилированных производных — изо-пентенил пирофосфа та и его изомера - 3,3-диметил ал л ил пирофосфата
СН.
ИгА /ОК ?н ?н сн; о-р-о-р-он и II о о
и .Юценти шли и р о фосфат
сщ
,сч zch, 9Н 9Н н,с чсн ХО—Р—О~Р—ОН
II II о о 3,3-,[11МС111;Ш.Е|НЛ11 нрлфосфа I
209
К изопреноидам относятся терпены, каротиноиды и стероиды.
При изучении терпенов установлено, что их молекулы построены Из фрагментов изопрена, связанных .между собой но принципу «голова* к хвосту» (изопреновое правило, Л.Ружичка. 1921 г,), например:
сн2=с -&н=сн2 + —сн=с-сн=сн2
СН3 _______________J сщ **
Р сн^с-сн,- сн,-сп-с~сн^сн?
сн? сн3
о ни мен
Хотя это правило и не является строгим, т.е. известны так называемые нерегулярные изопреноиды, образованные по типу «голова к голове» или «хвост к хвосту», оно помогло выяснить строение многих терпенов и родственных им соединений.
А. Терпены
Группа терпенов включает терпеновые углеводороды и их кислородсодержащие производные (спирты, альдегиды и кетоиы), называемые терпеноидами. К терпеновым углеводородам относят ненасыщенные углеводороды состава (C5Hs)n, где п указывает иа число изопреновых фрагментов и обычно колеблется от 2 до 8. Сам изопрен (n = 1) не принято относить к терпенам.
По количеству изопреновых фрагментов (п) терпены делят на:
— монотерпсны (2 изопреновых фрагмента, п = 2);
— сесквитерпены (3 изопреновых фрагмента, п = 3);
— дитерпены (4 изопреновых фрагмента, п = 4);
— тритерпсиы (6 изопреновых фрагментов, п = 6);
— тетратерпеиы (8 изопреновых фрагментов, п = 8);
— политерпеиы (более 8 изопреновых фрагментов, п > 8),
По наличию или отсутствию цикла в молекуле, терпены, кроме того, делят на алифатические (ациклические) и циклические, причем сами циклические, в зависимости от количества циклов, подразделяют на моноциклические, бициклические и трициклические.
В природе терпены встречаются в составе эфириых масел. В отличие от жирных масел, эфирные масла являются летучими и полностью испаряются, ие оставляя жирных пятен. Эфирные масла - источники запахов различных растений. Так. аромат земляники обусловлен содержанием в эфирном масле около 50 различных терпенов, причем некоторых в очень малых количествах.
’ «Головой» принято называть часть молекулы изопрена с метильной группой.
210
^циклические терпены. В основе углеродного скелета ряда ациклн-ческих монотерпенов лежат структуры изомерных димеров изопрена — мирцена и Ранее упомянутого оцнмена :
мирцен: оиимен:
7-метил-3-ме1нле|[Окталиеи-1,6 3,7-ди.метило Krai р иен-1,3.7
Мирней содержится в .эфирных маслах лавра благородного и хмеля обыкновенного, а оцимен — в эфирном масле листьев базилика обыкновенного,
К производным этих моиотерпеновых углеводородов относятся спирты ^раиной и нерол. Это цис-ш/шиг-изомеры. причем гераниолу соответствует цис-форма, а неролу - ш/хшс-форма:
Х^СН2ОН Д.
к к сн.он
lepaiiMOu: нерол;
^йг-.\7-я11метнпоктадиен-2.6-ол-Г /лр0нг-3,7-1-1имеп1ЯОК,га;1ие11-2.6-о;ь1
Эти спирты находятся в гераниевом, розовом и других эф ирных маслах как в свободном виде, так и в виде сложных эфиров. Гераниол бесцветная или светло-желтая жидкость с запахом розы. Нерол отличается от гераниола более тонким ароматом розы.
Нерол в кислой среде легко циклизуется в циклический терпеноид — а-терпинеол;
перол a-i ерпнлео л
Гераниол и нерол применяются как душистые вещества в парфюмерной промышленности. Альдегидами, получаемыми при окислении ГеРаниола и нерола, являются цитрвль А и цнтраль В,
цитраль А;
1/нс-3.7-димегил-2.6-октадиеиаль
цитраль Б;
транс-3,7 -д и мег и j t-2. Ь-о (стадиен аль
211
Природный цитраль является смесью обеих форм и находится s многочисленных эфирных маслах, особенно в лимонном. Это желтая маслянистая жидкость (т. кип. 228-229 °C) с характерным лимоиным запахом. Проявляет антисептическое, болеутоляющее и противовоспалительное действие, в медицине применяется главным образом в глазной практике. В качестве душистого вещества применяется в парфщ. мерной и пищевой промышленности.
Гераниол н цитраль выделяют в окружающую среду рабочие пчелы для привлечения других особей к источнику пиши, поэтому эти вещества называют пищевыми аттрактантами (от лат. aitraho — «притягиваю»). Цитраль же отпугивает муравьев некоторых видов, т.е, является для них репеллентом (от лат. vepello — «отпугиваю»). Природные аттарактаиты и репелленты объединены в группу феромонов --летучих соединений, способных влиять на поведение живых организ-мов. Таких средств «химического общения» известно множество — феромоны тревоги, пищевые, половые и др. Использование ловушек с половыми феромонами, привлекающими самцов насекомых-вредителей сельскохозяйственных культур, позволяет сократить расход ядохимикатов.
Среди разнообразных ациклических терпенов следует еще назвать сесквитерпеновый спирт фарнезол, который обусловливает аромат липы и ландыша, служит аттрактантом для некоторых насекомых, а в виде пирофосфата является полупродуктом в биосинтезе тритерпенового углеводорода сквалена СЗОН5О:
3.7,11-тримегн1щодекатриен-2,6т10-ол-1
Сквален — непосредственный предшественник стероидов в процессе их биосинтеза. Он может быть выделен из дрожжей, семян злаков, оливкового масла. Особенно богат нм жир печени акулы (до 40% п° массе). 212
Мошщиклические терпены. Важнейшими представителями моноцикл ических терпенов являются лимонен и ментан.
Молекула лимонена имеет одни асимметрический атом углерода и существует в виде двух зеркальных изомеров. (-)-Лимонен и его рацемическая форма — ди пентен содержатся в составе скипидара и эфириых масел хвойных пород деревьев, (+)-Лимоиеи находится в померанцевом масле (до 90%), является компонентом апельсинового, бергамотного, тминного и сельдерейного масел. Он же обуславливает запах лимонного масла.
Оптически активные формы лимонена получают из названных эфирных масел.
(±)-Лимонеи (дипеиген) можно получить димеризацией изопрена при нагревании в запаянной ампуле до 150 °C (С.В.Лебедев, 1908-1913 гг.). При 500-700 °C происходит обратный процесс крекинг дипентена с образованием изопрена.
изопрен
150 “С. давление
500-700 °C
(±>лимо1гег1: лицеичен;
мент ада ен-1.8
Возможность как цепной, так н циклической димеризации изопрена наглядно иллюстрирует генетическую взаимосвязь ациклических и циклических моиотерпенов.
Каталитическая гидрогенизация лимонена позволяет получить предельный монотерпеновый углеводород ментан, являющийся родоначальником группы монециклических терпеноидов. Дегидрогенизация лимонена приводит к ароматическому углеводороду н-цимолу .
л-цимоп
лимонен
ментан
Гидратация лимонена в кислой среде протекает по правилу Марковиикова, В результате ее образуется двухатомный спирт терпин.
Цимол содержится в эвкалиптовом и тминном эфирных маслах, откуда его можпо выделить: может быть также получен путем сшггеза из лимонена. а'Пипена.
213
2Н,О
терпин
Лимонен используют как душистое вещество в парфюмерной промышленности, а дипентен — еще н как растворитель для лаков, восков и т.п.
Как упоминалось выше, в основе строения молекул большинства моноцикл ических терпеноидов лежит скелет ментана (1-изопропил-4-метилциклогексаи), Как и лимонен, ментан включает два фрагмента изопрена, однако, в отличие от лимонена, в молекуле ментана отсутствуют двойные связи и, кроме того, последний обнаружен лишь в некоторых эфирных маслах.
Ментан получают гидрированием цимола (н-изопропилметилбен-зола). При этом образуется преимущественно цис-изомер.
мен।ап
Ментан жидкость, растворимая в этаноле, нерастворимая в воде.
К гидроксипроизводным меитана относится терпеноид ментол (ментаиол-3) основной компонент эфирного масла мяты перечной. Имеет три асимметрических центра и поэтому может существовать в виде восьми оптических изомеров.
Наибольшее значение из них имеет (-)-ментол, у которого гидроксильная, метильная и изопропильная группы расположены экваториально.
ОН
Н?С
ОН
СН(СН3)2
ментол, мапанол-3
Левовращающий ментол добывают вымораживанием мятного масла или синтезируют восстановлением ( )-ментона. до 20% которого содержится в эфирных маслах мяты.
мен гол Мен ны
Г В промышленности (±)-ментол получают алкилированием .и-крезо-иа с последующим гидрированием полученного тимола в присутствии катализатора,
CH<CH-OH *H2S°4>
ОН '
.V -кречол
Известны и другие способы
Ментол — летучее кристаллическое вещество (т, пл. 41-43 СС) с сильным мятным запахом, холодящим вкусом, вызывающее характерное чувство холода и покалывания при втирании в кожу. Растворяется в этаноле, ограниченно— в воде, перегоняется с водяным паром.
Проявляет химические свойства вторичных спиртов.
Применяется как слабое антисептическое, успокаивающее и обезболивающее средство в составе целого ряда лекарезвенных препаратов (бороментол, пектусни и мн. др.), 30%-й раствор ментола в его эфире с изопалериаловой кислотой — меитилизовалсрианат — используют при стенокардии как сосудорасширяющий препарат под названием валидол, Благодаря приятному запаху и вкусу ментол нашел широкое применение в кондитерской и парфюмерно-косметической промышленности.
Дигидроксипроизводным ментана является терпеноид терпин (ментандиол-1.8), Это двутретичный двухатомный спирт, существующий в виде цис- и транс-изомеров, В цне-изомере обе гидроксильные группы находятся по одну сторону цикла, в шрдис-изомерс — по разные. Обыкновенный терпии — это 1/ыс-форма,
Терпин можно синтезировать гидратацией лимонена, В промышленности же его получают из пииена, который в большом количестве содержится в скипидаре, В результате этих реакций образуется кристаллогидрат терпина, содержащий одну молекулу воды — терпингидрат.
OH
] • n2o
OH
Терпингидрат (моногидрат ментаидиола-1,8) — это бесцветное кристаллическое вещество (т, пл. 115-117 QC) почти бет запаха, слабогорького вкуса, растворимое в этаноле, мало-растворимое в воде. При нагревании до [00 °C воз гоняется (сублимирует).
Применяется в медицине при хроническом бронхите как отхаркивающее средство. Проявляет также антисептическое н слабое мочегонное действие.
При обработке терпина растворами кислот весьма легко происходит отепление одной молекулы воды и образование ненасыщенных спиртов терпинеолов.
icpuHli a-iepriJJJieo.ii (1 -icpiJHiieo.j
мен i eu-J -ojj-8 mcjij cjj-S-ojj-)
Наиболее широко распространенный в растениях a-тсрнинеол содержит один асимметрический атом углерода, из-за чего существует в двух энантиомерных формах, представленных в составе различных масел: гераниевого — (±)-форма, камфорного — (-)-форма. скипидара (-ь)-форма.
Терпинеолы — иизкоплавкие кристаллические вещества с запахом сирени, что обусловило их применение в парфюмерии.
Бициклические терпены. В основе строения молекул терпеноидов этой группы выступают четыре важнейших представителя циклических терпеновых углеводородов — пинан, камфан (борнан). кд ран и тунам (сабинам):
Пинал:
2,6.6-гриме1илби-
Ц|<кло(3,1. Ijieinan
камфин: борнал:
1.7,7-1 рпмешлби-г[нк.го[2,2, Ijiei 11 ап
карал:
4,7.7-TpiHieinji6H-цик?к>(4,) .Opvirnui
lyiiirir. сабипан:
I -Hit>iipojJwji-4-jneiii ч-бгпыкюр. LBJreKfaji
Чаще других производных этой группы терпенов в природе встречается а-пннен*.
' Существует и р-пинегг, который отличается от а-пииена положением двойной связи.
216
ct-Пииеи находится в различных эфириых маслах, но наибольшее его । количество содержится в терпентинном масле скипнда-
Jl ре (до 75%).
р 3^1 * Получают скипидар перегонкой с водяным паром жи-I । вицы, которая выделяется при надрезах коры («подсочка») хвойных деревьев, главным образом сосны. Иеперегоняе-мый остаток, состоящий из смоляных кислот состава Cj<jHi<)COOH. называется канифолью.
Молекула а-пинена содержит два асимметрических центра и он обладает оптической активностью. Состав терпентинного масла различен и зависит от ботанического вида растения. Может преобладать левовращающий а-пинен из сосны приморской — Pinus pinaster и право вращающий — из сое н ы л ссио й (Pi них sybtestrix).
При нагревании с разбавленными кислотами (азотной, серной) ct-пинен превращается в а-терпянеол и тернии.
а-jeprinnco.j leprfjia
При выдерживании иа воздухе пинен и скипидар окисляются кислородом с образованием пероксида, который можно обнаружить по его окислительным свойствам (по отношению, например, к HI). Затем пероксид превращается в кетон группы пинана— вербеной, содержащийся в некоторых эфирных маслах.
Пинен и, соответственно, скипидар широко £ вербеичп используются в качестве растворителей для лаков и красок. При этом они выполняют роль ускорителя затвердевания (высыхания) красок благодаря присутствию примеси вышеупомянутых пероксидов, стимулирующих псроксидное окисление ненасыщенных цепей высших жирных кислот и образование полимерных соединений из жиров (олифы).
Кроме того, пинен применяют н качестве исходного вещества для получения синтетической камфоры. Скипидар часто применяют для втирания в кожу как раздражающее, обезболивающее и антисептическое средство.
борнеол
Спиртом борнанового (камфанового) ряда является бпрнеол (борниловый спирт, или камфанол-2). Ои имеет три асимметрических атома углерода в молекуле, и поэтому оптические изомеры у него представлены, помимо энантиомеров, парой диастереомеров — борнеолом и изоборнеолом.
217
ОН
изобоппеоа
Н
При кипячении с металлическим натрием в ксилоле борнеол и изоборнеол превращаются друг в друга.
Энантиомерные или рацемические формы борнеолов содержатся во многих эфирных маслах как в свободном состоянии, так и в виде сложных эфиров. Своим названием борнеол обязан борнейскому лавру {Dryobuianops aromatica}^ в выделениях и эфирном
масле которого содержится его правовращающий изомер. (- )-Борнеол получают омылением борнилацетата, который в количестве 30-40% входит в состав пихтового масла (эфирное масло пихты сибирской — Abies sibiriea). Сложные эфиры борнеола можно синтезировать, хотя и с невысокими выходами, путем присоединения органических кислот к а-пинену;
карболопая кислота
<|>ир оориеолп
Борнеолы представляют собой бесцветные кристаллические вещества со специфическим «камфорно-хвойным» запахом, растворимые п спиртах и эфире, практически нерастворимые в воде. Сублимируются ниже температуры плавления.
Борнеолы проявляют химические свойства вторичных спиртов. При окислении хромовой кислотой образуют камфору.
борцео,|
Под действием кислот борнеолы (особенно изоборнеол) легко отшепляют воду и превращаются в камфен. Реакция протекает через стадию перегруппировки образующегося борнил-катиона в изоборннл-катион.
Полученный камфен способен гидратироваться в кислой среде с образованием изоборнеола.
218
камфен
ггюборнеол
Эта реакция является обратной синтезу камфена из борнеола и носит название перегруппировки Вагнера-Меервейна, или камфеновой перегруппировки I рода.
Борнеолы и их эфиры применяют как душистые вещества в парфюмерной, мыловаренной и кондитерской промышленности. Понижают кровяное давление, малотоксичны. (-)-Борнеол и (—)-борнил ацетат, выделяемые из пихтового масла, служат сырьем для получения (-)-камфоры.
Камфора (камфара) — бициклический кетой на основе камфаиа
(бор на на). В струкзуре ее молекул имеется два асимметрических атома углерода, однако вместо четырех ожидаемых оптических изомеров камфора имеет лишь пару антиподов. Отсутствие диастереомеров у камфоры вызвано тем, что асимметрические атомы жестко связаны между собой тремя углеродными цепями, что исключает возможность иной конфигурации только у одного из этих атомов с сохранением прежней конфигурации у другого.
Было установлено, что правовращающий изомер камфоры стсрео-
хнмически соответствует конфигурации Л-глюкозы,
Правовращающую камфору добывают из древесины камфорною дерева Cinnamomum Camphora. произрастающего во Вьетнаме, Китае, на Тайване и в Японии. Его культивируют в тропических районах. Камфора также входит в состав эфириых масел сибирской пихты, базилика, полыни, камфорного шалфея и др. Потребность в камфоре не удовлетворяется только природными ее источниками. Для этого разработаны различные синтетические и полусиитетические методы получения. часто отличающиеся между собой лишь исходным сырьем.
В нашей стране промышленное зиачеиие приобрел метод получения (-)-камфоры из пихтового маслак разработанный Н.В,Вершининым. В
состав пихтового масла входят а-пинеи, камфен, фелландрен и 30-40% борнилацетата. Последний при фракционной разгонке получают, собирая фракцию, кипящую выше 180 °C, подвергают омылению, а образовавшийся (-)-борнеол окисляют хромовой смесью в ( -)-камфору. Промышленный синтез рацемической камфоры нз пинеиа осу-ацествляется по способу В,Е,Тищенко, Синтез протекает в несколько
219
стадий. На первой стадии пинеи превращают (изомеризуют) в камфеи с помощью катализаторов (борнофосфорной кислоты, оксида титана (IV), сульфатов магния или никеля):
Камфора представляет собой бесцветное кристаллическое вещество (т. пл. 178 °C) с резким характерным запахом, летучее (сублимируется), легко перегоняемое с водяным паром. Легко растворяется в спирте, малорастворимо в воде. Флуоресцирует в УФ-свете.
По химическим свойствам камфора - типичный кетой; она образует оксимы, семикарбазоны, фенилгидразоны, даст другие реакции на кетонную группу (см. ки. 2, разд. 15.1.4). Метиленовая группа в «-положении к карбонильной проявляет СН-кислотные свойства, взаимодействуя с галогенами, образует галогенкамфору :
+ НВг
Окисление камфоры азотной кислотой вначале ведет к камфорной кислоте, а затем — к смеси камфановой и камфароновой кислот :
кам фор а
камфорная кислота
СООН камфановая
КИСЛО1Я
СООН
НООС соон
камфар» нова я
кис-ю 1 а
Г^^СООН l^^COOH
В медицине нашли широкое применение как оптически активные, так и рацемическая формы камфоры. Растворы камфоры используют наружно как антисептическое и местнораздражающее средство, подкожно — стимулируют дыхательный и сосудодвигательный центры, обменные процессы в сердечной мышце, препятствуют образованию тромбов и др. Камфора служит исходным веществом для синтеза бромкамфоры.
220
Помимо медицинского применения, камфора широко используется в промышленности как пластификатор в производстве целлулоида, компонент бездымных порохов, является репеллентом против моли, комаров и др, Бромкамфора применяется как успокаивающее средство при заболеваниях центральной нервной системы. Улучшает сердечную деятельность.
Б. Каротиноиды
Каротиноидами называют группу природных пигментов, сходных по строению с каротином (пигмент, впервые выделенный из моркови). Молекулы каротиноидов содержат в своем составе значительное число сопряженных двойных связей, чем и объясняется их окраска. Двойные связи имеют щ/лил-конфигурацию. Большинство каротиноидов относятся к тетратериенам, г.е. их молекулы содержат 40 атомов углерода. Каротиноиды растворимы в растительных и животных жирах, многие из них легко окисляются кислородом воздуха.
Каротин — пигмент желто-красного цвета, содержащийся в большом количестве в моркови, а также молоке, сливочном масле, листьях растений и многих плодах. Появление желто-красной окраски на листьях деревьев осенью обусловлено наличием каротина, который становится заметным вследствие разрушения зеленого пигмента — хлорофилла.
При установлении строения каротина оказалось, что он представляет собой не индивидуальное вещество, а смесь трех изомеров — сс-кя-ротииа, p-каротина и у-каротина. В большинстве случаев в смеси преобладает p-каротин. В химическом отношении молекулы a-, J3- и у-ка-ротинов характеризуются наличием одного нли двух триметилциклогексеновых колец и полиеновой углеродной цепи.
221
&
Все изомеры каротина являются предшественниками витаминов группы Л, т,е, провитаминами. Под влиянием ферментов в организме оии расщепляются по связи С15 CiG с образованием витамина А( ретинола. При этом наибольшую биологическую ценность представляет p-каротнн, из которого при распаде в организме образуется две молекулы витамина А(.
ji - каротин
Молекулы а- и у-каротинов не симметричны, поэтому при ферментативном расщеплении их образуется только по одной молекуле витамина Ар
Витамин А, (ретинол). Витамин А, содержится только в продуктах животного происхождения, Н3С СИ, СН-, СН, Богатыми его источниками
L L СН3ОН являются сливочное масло.
f - яичный желток, печень жи-
I водных и морских рыб. ры-
бий жир, В растениях вита-'“'“з мин Aj не встречается, по
многие из них содержат его предшественник (провитамин) - каротин, из которого в организме человека и животных образуется витамин Аг.
По физическим свойствам — это кристаллическое вещество желтого цвета, т. пл, 63-64 °C, легко растворимое в жирах.
Витамин А| считается фактором роста. При недостатке его в пище наблюдается задержка роста, исхудание, высыхание роговицы глаза (ксерофтальмия), понижается сопротивляемость организма к инфекционным заболеваниям. Ранним признаком А-витамИнной недостаточности является ослабление сумеречного зрения (куриная слепота).
В. Стероиды
К стероидам относят вещества животного и растительного происхождения. в основе структуры которых находится система пиклопента-нонергидрофенаитрена (стерана):
Приведенная в формуле нумера пня атомов углерода не отвечает правилам систематической номенклатуры ииклоалканов, но является общепринятой в химии стероидов.
Входящие в структуру стероидов циклогексановые кольца А. В и С? находятся в конформации кресла, В природных стероидах
222
кольца А и В могут иметь гуис- или транс- сочленения, кольца В и С всегда, а кольца С и D почти всегда имеют л?рл//г-со членение.
шрсшс-еочленение колец Л/В. В/С и C/D
цнс—со'ск'ненис колец Л/В, шрннс-колец В/С и C/IJ
Циклический скелет стероидов является конформационно жесткой системой, поэтому для него не характерны конформационные превраг щен ия.
При написании формул стероидов систему циклопентанопергидро-фснантрена условно изображают плоской.
Незамещенный циклопеитаноперг идрофенантрен имеет 6 асимметрических атомов углерода (узловые атомы С5. С , СУ, С10. Сь, С14) и может существовать в виде 64 стереоизомеров (26 = 64), При наличии заместителей у неузловых атомов углерода, число возможных стереоизомеров значительно возрастает, однако в природе из них встречается лишь небольшое количество.
Для обозначения конфигурации заместителей и атомов водорода при асимметрических центрах циклического скелета стероидов применяют а. 3-систему. Согласно этой системе, конфигурацию заместителей и атомов водорода, расположенных под плоскостью циклического скелета, принято обозначать а-конфигура-цией. а расположенных над плоскостью— ^-конфигурацией. Связи над плоскостью изображают сплошной линией или клином, а под плоскостью — прерывистой линией или
штриховым клином. У большинства природных стероидов при атомах углерода С[0 и С[? находятся «ангулярные» метильные группы, а при С17 — боковая углеродная цепь, имеющие [^-конфигурацию. Кроме того, весьма часто в положении С' находится гидроксильная или карбонильная группа,
В ряду стероидов различают следующие группы соединений: стсри-ны, желчные кислоты, стероидные гормоны, агликоны сердечных гликозидов, агликоны стероидных сапонинов и др.
223
Стерпны (стеролы) представляют собой стероидные спирты, в ос
нове структуры которых находится ядро углеводорода холестана.
В номенклатуре произ
водных холестана принята нумерация атомов углерода, как показано на формуле, Стсрины являются 3-гидрокси-производиыми холестана. Разные стерпны отличаются друг от друга степенью ненасыщенности углеродного скелета (могут содержать
одну или несколько двойных связей), а также разной длиной углеродной цепи радикала у С1, (8-10 атомов углерода). Различают животные стерины (зоостерипы), растительные (фитостерины) и сзерины грибов (микостерины), Зоостерины встречаются в организмах человека и животных, Их углеродный скелет содержит 27 атомов углерода. Фитостерины находятся п растениях, а микостерины — в дрожжевых грибках. В отличие or зоостеринов их молекулы содержат 28 или 29 атомов углерода. Важным представителем зоостеринов является холестерин.
Холестерин (холестсрол. холестен-5-ол-ЗР). Представляет собой произ-
водное холестана. в котором при С' находится гидроксильная группа в 0-конфигурацин, а при С5— двойная связь. Таким образом, холестерин является непредельным спиртом. За счет гидроксильной группы он про
являет химические свойства спиртов, а по двойной связи — вступает в реакции, свойственные алкенам. Холестерин впервые выделен в 1775 г, из желчных камней. Он входит в состав клеточных мембран, в больших
количествах содержится в нервной ткани (головном и спинном мозгу), присутствует в крови и желчи. Нормальное содержание холестерина в крови человека составляет 180-260 мг%. При нарушении холестеринового обмена он откладывается на стенках кровеносных сосудов и способствует развитию атеросклероза. Холестерин яйтяется биогенетиче
ским предшественником стероидных гормонов, желчных кислот, витамина 17-,. В организме холестерин находится как в свободном виде, так н в виде сложных зфиров.
224
В жире морских рыб и животных, а также в липидах кожи человека содержится 7-дегидрохолестерии, который при облучении УФ-светом превращается в витамин Ds (холекальциферол). В процессе изомеризации разрывается связь между С9 и С10,
Поэтому 7-дегидрохолестерии является провитамином Г)?. Близким по строению к витамину D? является витамин ГУ (см, ниже).
Витамины группы D регулируют обмен кальция и фосфора в организме. При недостатке витамина D в рационе детей развивается ia6o-девание рахит.
Эргостерин (эргостерол, 24-метилхолестантриеи-5,7,22-олЭргосте-
три двойные связи в положениях 5, 7 и 22. а также положении 24.
рпн относится к группе микосте-ринов. По химическому строению эргостерин, как и холестерин, представляет собой одноатомный ненасыщенный спирт, По эргостерин, в отличие от холестерина, содержит метильную группу в
Под действием УФ-света эргостерин изомеризуется в витамин D2 (эргокальциферол), В процессе изомеризации происходит разрыв связи между С9 и С10,
Поэтому эргостерин является провитамином D2. Он содержится в большом количестве в дрожжах, откуда н получается. Витамин D2. как и витамин D3. регулирует обмен кальция и фосфора в организме, а следовательно, обладает ангирахитическим действием.
В 6-41
225
Желчные кислоты. Желчные кислоты находятся в желчи человека и животных. Они вырабатываются печенью из холестерина. По химическому строению желчные кислоты являются гидроксипроизводнымс холановой кислоты.
Из желчи человека выделены четыре кислоты, из которых наиболее распространены холевая и дезокеихолевая кислоты.
холевая кислота дечоксихолевцн kucjiOiH
Холевая кислота имеет три спиртовых гидроксила в положениях ? 7 и 12, дезоксихолевая — два в положениях 3 и 12 (отсутствует в подо женин 7). Вес гидроксильные группы имеют a-конфигурацию. а цикдъ А и В — ^'’Сочленение. В желчи желчные кислоты находятся обычи* 226
не в свободном виде, а в виде амидов с аминокислотой глицином NHiCHiCOOH или таурином NH2CH2CH2SO?H, Так, холевая кислота находится в виде гликохолевой и таурохолевой кислот.
rjwiKOXixieiWH кислота
Натриевые и калиевые соли этих кислот, благодаря поверхностно-активным свойствам, способны эмульгировать жиры и тем самым облегчают их переваривание и всасывание.
Стероидные гормоны. К стероидным гормонам относятся кортикостероиды гормоны коры надпочечников и патовые гормоны.
Кортикостероиды вырабатываются в коре надпочечников. По химической природе они являются про-з изводными углеводорода прегнана, По характеру воздействия па процессы обмена кортикостероиды делят на две группы: глюкокортикоиды, регулирующие углеводный обмен, и минералокортикоиды, влияющие на солевой обмен (обмен Na1. К + и воды), глюкокортикоидов являются гидрокор
тизон и кортизон.
8*
227
Эти соединения содержат в положениях С3 кетонную, С17 - гидроксильную и гидроксиацетильиые группы, а в положении С4 двойную связь. Гидро кортизон, кроме того, имеет у Си гидроксильную группу, а кортизон, в отличие от него, содержит при Сп карбонильную группу.
Глюкокортикоиды оказывают активное влияние на углеводы и белковый обмен. Они повышают содержание глюкозы в крови, способствуют накоплению гликогена в печени, вызывают увеличение выделения азота с мочой. Глюкокортикоиды проявляют противовоспалительное и антиаллергическое действие.
Основными представителями минералокортикоидов являются альдостерон и дезоксикортикостерон.
Эти соединения содержат в положениях С3 кето иную. С17 гид-роксиацетильную группы, а в положении С4 двойную связь. Альдостерон. кроме того, имеет в положении С’н гидроксильную группу, а в положении С11 вместо метильной группы содержит альдегидную группу. н:-. основании чего и получил название.
Минералокортикоиды регулируют главным образом обмен натрия, калия хлора и воды Оии способствуют удержанию ионов натрия и хлора в организме и выведению с мочой ионов калия.
Из природных кортикоидов в качестве лекарственных средств применяется гидрокортизон, кортизон и дезокси кортикостерон.
В настоящее время в медицине широко применяются синтетические аналоги кортизона и гидрокортизона преднизон, преднизолон, дексаметазон, триамцинолон и др. Эти соединения более активны, чем природные кортикостероиды, действуют в меньших дозах, оказывают меньшее влияние на минеральный обмен.
ГТрЦДПИ JOJlUir
дексаметаэин
228
Характерной особенностью химического строения дексаметазона и триамцинолона является наличие в их молекулах атома фтора в положении с’.
Половые гормоны. У мужчин половые гормоны вырабатываются в семенниках (тестикулах). у женщин — в яичниках. Мужские половые гормоны называют андрогенами (от греч. андрос — «мужчина»). Женские половые гормоны подразделяют на эстрогены, которые образуются в фолликулах яичников, поэтому их еще называют фолликулярные гормоны, и гестагены (гормоны беременности), образующиеся в желтом теле яичников.
Половые гормоны начинают вырабатываться с периода полового созревания. Под их влиянием происходит формирование вторичных половых признаков. Кроме того, оии принимают участие в регуляции белкового обмена, способствуют улучшению адаптации, повышают устойчивость организма к изменяющимся условиям внешней среды замедляют процессы старения.
Андрогены. Важнейшими андрогенами являются тестостерон и андростерон.
Оба гормона имеют две ангулярные метильные группы. Тестостерон содержит в положениях С5 — кетогруппу. С17 — гидроксильную группу, а в положении С4 — двойную связь. Андростерон имеет в
в положении С — карбонильную группы. Андрогены оказывают влияние на развитие вторичных половых признаков у мужчин, содействуют формированию мужского телосложения.
В настоящее время получены синтетические стероиды, обладающие андрогенной активностью. Примером является метилтестосте-рон.
Эстрогены. Наиболее важными эстрогенами являются эстрон (фолликулин) н эстрадиол.
229
В отличие от андрогенов, в молекулах эстрона и эстрадиола кольцо А является ароматическим и отсутствует ангулярная метильная группа у С10.
В настоящее время получен ряд весьма активных синтетических эстрогенов нестероидиой структуры. К ним относятся синестрол, диэтилстильбестрол, димэстрол и др.
синее ipoji
ди71 иле) нльбесгрол
Преимущество эстрогенов нестероидного строения состоит в том, что они более устойчивы, поэтому их можно применять перорально.
Гестагены. Гормоном желтого тела является прогестерон, По химической структуре прогестерон сходен с мужским половым гормоном тестостероном. Отличие заключается в том. что прогестерон содержит у С17 ацетильную группу, вместо гидроксильной у тестостерона.
tipoieriepOH
Прогестерон способствует протеканию беременности. Под его влиянием прекращается созревание фолликулов и овуляция. Для медицинского применения он получается синтетическим путем. Прогестерон и его аналоги применяют при маточных кровотечениях, бесплодии, недонашивании беременности.
Агликоны сердечных гликозидов. Сердечные гликозиды содержатся в некоторых растениях, таких, как различные виды наперстянки, горицвет весенний, ландыш майский, строфант, олеандр и др. В больших дозах это чрезвычайно ядовитые вещества, а в весьма малых они оказывают кардиотоническое действие (стимулируют деятельность сердечной мышцы). 230
По химической структуре сердечные гликозиды-представляют собой О-гликозиды, в молекулах которых агликон имеет стероидную структуру, а углеводный фрагмент представлен остатками моно-7 ли-, три- или тетрасахарида.
Связь между углеводом и агликоном является 0-гликозидной и осуществляется за счет гидроксильной группы у С’ цнклоцснтанопергид-рофенантренового яд^за. Кроме того, агликоны (гемины) содержат у С14 гидроксильную, у С1 — метильную группы, а у С1' — ненасыщенное лактоииое кольцо.
По характеру лактонного кольца сердечные гликозиды делят на две группы:
карденолиды, содержащие у С1г пятичленное ненасыщенное лактонное кольцо;
— буфадненолиды, имеющие у (' шестичленное ненасыщенное лактонное кольцо.
Наличие ненасыщенных лактонных колец обуславливает неустой-
чивость сердечных гликозидов в щелочных растворах, легко гидроли
зующих лактоны,
В состав углеводной компоненты гликозидов входят широко распространенные в растительном мире D-глюкоза. D-фруктоза, D-ксилоза, D-рамноза, а также встречающиеся только в сердечных гликозидах метилпентозы D-дш итоксоза. D-дигиталоза, D-цимароза. D-фукоза
и др.
231
Сердечные гликозиды группы карденолидов часто содержат в качестве агликонов дигитоксигенин и строфантндин.
Примером сердечного гликозида является п у ри у реа гликозид А, выделенный из листьев наперстянки пурпуровой.
н ОН
В растениях кардеиолиды находятся в виде так называемых первичных (генуинных) гликозидов. т.е, в форме конечных продуктов биосинтеза. После сбора листьев, и особенно в процессе их сушки, под действием протекающих ферментативных процессов происходит «укорачивание» углеводной цепи. В результате образуются вешества, получившие название «вторичные» гликозиды. Так. вторичным гликозидом
232
яуриуреагликозида А является дигитоксин, содержащий в качестве сахарной компоненты три остатка p-ZJ-дигитоксозы. Дигитоксин применяется в медицине при хронической сердечно-сосудистой недостаточности.
Стероидные агликоны сапонинов. Сапонинами (от лат, sapo «мыло») называют группу растительных гликозидов, обладающих высокой поверхностной активностью и вызывающих гемолиз эритроцитов.
Водные растворы сапонинов при встряхивании образуют обильную пену. <
По химической природе сапонины являются О-гликошдами. в которых агликоны (сапогенины) могут иметь стероидную или тритерпеновую структуру. Стероидные сапонины содержатся в растениях семейств лилейных, диоскорейных, норичниковых и др. Большинство агликонов стероидных сапонинов содержит в своем составе спироке-тальную группировку. образующуюся в результате окисления боковой цепи из восьми углеродных атомов у С1г и гидроксильной группы у С,'1”. К сапогепннам стероидной природы относится диосгепин. содержащийся в корневище диоскореи.
лн оа en j I и
Стероидные сапогенины используются в качестве исходного сырья Для получения синтетических стероидных гормонов. Так, диосгенин применяется для синтетического получения гормона беременности прогестерона.
КОНТРОЛЬНЫЕ ВОПРОСЫ И УПРАЖНЕНИЯ
Т Какие органические соединения называют липидами? Приведите их классификацию.
2. Напишите общую формулу жиров. Назовите основные природные источники получения жиров.
3. Напишите уравнение реакции триолеина:
а) с бромом;
б) с подом;
233
в) водородом в присутствии катализатора. Объясните значение этих реакций.
4. Напишите формулы и лайте названия пяти кислотам, остатки которых наиболее часто входят в состав жиров,
5, Напишите схему кислотного гидролиза дипальмитоолеииа.
6, Напишите уравнение реакции щелочного гидролиза три ацил глицерина. в состав которого входят олеиновая, линолевая и пальмитиновая кислоты. На чем основано моющее действие мыла?
7. Напишите схему полного гидрирования триолеина.
К. Приведите структурные формулы моно- и бициклических терпенов. Напишите схему реакции окисления ментола.
9, Отметьте асимметрические атомы углерода в формулах лимонена, пииеиа и камфоры. Сколько оптических изомеров имеет камфора?
10,Напишите схемы качественных реакций, иллюстрирующих непредельные свойства лимонена.
11. Какие соединения образуются нз пинена под действием кислорода воздуха и какой реакцией можно их обнаружить?
12, Напишите схемы реакций камфоры со следующими реагентами:
а) гидросульфитом натрия;
б) гидроксила ми ном;
в) фенилгидразином.
234
СПИСОК ЛИТЕРАТУРЫ
1. Агрономов А. Н„ Шабаров IO. С. Лабораторные работы в органическом практикуме. • М.: Химия. 1974.
2, Гауптман 3.. Грефе 10.. Ре.мане X. Органическая химия: Пер. с нем.! Под рсд. В. М. Потапова, М,: Химия. 1979,
3. Домбровський А. В, Найдаи В. М. Оргагична ximih; навч, nocio-ник. К,: Вища школа. 1992,
4. Иванский В. И. Химия гетероциклических соединений: учеб, пособие для ун-тов, М,г, Высшая школа, 1978,
5, Ингольд К. Теоретические основы органической химии: Пер. с англ. / Под ред. И. П. Белецкой, М.: Мир, 1973.
6. Кам Р.. Дермер О. Введение в химическую номенклатуру: Пер. с англ, / Под ред, В. М, Потапова и Р. А. Лидина, М,: Химия, 1983,
7. Кери Ф.. Сандберг Р. Углубленный курс органической химии: Пер, с англ. В 2 кн. / Под ред. В. М. Потапова, М.: Химия, 1981.
8. Лабораторные работы по органической химии / Под ред, О. Ф, Гинзбурга. А. А. Петрова, М,: Высшая школа. 1974.
9. Марч Дж. Органическая хнмня. Реакции, механизмы и структура. Углубленный курс для ун-тов н хим. вузов; В 4 т. М,: Мир, 1987-1988.
10. Матье Ж., Паника Р. Курс теоретических основ органической химии: Пер. с франц. / Под ред, Л. А. Яновской. М.: Мнр. 1975.
11. Машковский М. Д. Лекарственные средства: В 2 кн. 11-е изд., стереотип. М.: Медицина. 1988.
12. Механизмы реакций в органической химии / П. Сайкс, — 4-е изд. Пер, с англ. / Под ред. В. Ф. Травеня. Мл Химия, 1991,
13. Моррисон Р.. Бойд Р. Органическая химия: Пер. с англ. / Под ред. И. К. Коробицыной. М.: Мир, 1974.
14. Нейланд О. Я. Органическая химия: учеб, для хим. спец, вузов. М.: Высшая школа, 1990,
15. Неницеску К Д. Органическая химия: В 2 ки, Пер, с рум, / Под ред. М, И, Кабачника. - М.: Изд-во иностр, лит., 1962,
16. Несмеянов А. Н., Несмеянов Н. А. Начала органической химии: В 2 кн. 2-е изд,, перераб, -- Мл Химия, 1974,
17. Номенклатурные правила ИЮПАК по химии: В 4 т. Пер. с англ. / Под ред. Л, А. Яновской, М.: ВИНИТИ. 1979-1985,
18, Обща» органическая химия-. В 12 т. Пер. с англ. / Под обшей ред. Д. Бартона. У. Д. Оллиса. М,: Химия. 1981-1988.
19. Орехов А. П. Химия алкалоидов: 2-е изд,, иенр. н доп.
М.: Изд-во АН СССР. 1955.
235
20. (Тальм В. А, Введение в теоретическую органическую химию: учеб, пособие для студ, ун-тов. М,: Высшая школа. 1974.
21. Петров А. А,. Бальян X. В,. Трощенко А, Г, Органическая химия; учеб, для студ. хим.-технол. вузов / Под ред, Л. Л. Петрова. - - 4-е изд., перераб. и доп. М.: Высшая школа. 1981.
22. Потапов В. М. Стереохимия: учеб, пособие для студ. хим. спец, ун-тов. 2-е изд., перепаб. и доп. М.: Химия. 1988.
23. Рено А. Я., Зеленкова В. В. Малый практикум но органической химии, M.I Высшая школа. 1980.
24. Робертс Дж., Касерио М. Основы органической химии: В 2 кн. Пер. с англ.! Под ред. А. П. Несмеянова. М.: Мир. 1968.
25. Руководство к лабораторным и семинарским занятиям по органической химии (В. П. Черных. В, И. Гридасов, И. С. Гриценко и др. Харьков: Основа, 1989.
26, Степаненко Б. Н. Курс органической химии: учеб. ;шя студ. фар-мац. вузов. В 2 ч. - 6-е изд., перераб. и доп. -- М.: Высшая школа. 1981.
27. Теоретические основы органической химии / А. С. Днепровский. Т. И. Темникова: учеб, для спуд. хим. спец, вузов, 2-е изд., псре-раб. Я.; Химия. 1991.
28. Терией А. Современная органическая химия: В 2 т. / Пер. англ, под род. Н. Н. Суворова. М.: Мир, 1981.
29. Тюкавкина Н. А.. Бауков 10. И. Биоорганическая химия: учеб, для студ. мед. ин-тов. 2-е изд., перераб. и доп. М.: Медицина, 1985.
30. Химическая нщиклопедия: В 5 т, / Редкой,: Зефиров Н. С. и др, М,: Большая Российская энцикл., г. 1-4, 1988-1995,
236
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Агликоны 143. 144 сапонинов стероидные 233
- сердечных гликозилов 230-233
Аденин 107. 109, 185
р-Аленипдезоксирибофурапозид. см,
Дезоксиадепозин
Аденозин 144, 187
Аденозиндифосфат 191
Аденозип-З'-монофосфат 189 Аденозин-5'-монофосфат 189 и сл.
Аденозинтрифосфат 191
АДФ. см. Адено шндифоефат
Азепипы 112
Азетпдип 18 псп.
Азетидинон-2 20
Азин, см. Пиридин
Азиридин 18 и сл.
Азолы 49
Акридан 87. 88
Акридин 63. 86 и сл,
Акрилнний хлорид 87
Акридол-9, см. Акридон-9и
9-Гидроксиакридин
Акридоп-9 86. 88
Акрихин 89
Акролеин 64. 78
Аланилглутамнлглипин 175
Аланин 166, 180
Алкалоиды 108, 115 и сл.
Алкиламипоэтанолы 16
Аллоксазин 111
Аллоксан 107
Аллоксантин 107
Альдегид
акриловый, см. Акролеин
- бензойный, см. бензальдегид глицериновый 127
малеиновый, см, Малеындиальде-гид
- муравьиный, см. Формальдегид
- кротоновый 79
- пеларгоновый 204
- салициловый 92
- уксусный, см. Ацетальдегид
- хлоруксуспый 59
- щавелевый. см, /лиокеалъ
Альдозы 126
Альдогексозы 126 и сл,. 143
Альдогептоза 136
Альдопептозы 39, 126 и сл,. 143
Альдостерон 228
Амидопирин 5.3 и сл. Амилоза 157 Амилоид 159 Амилопектин 157. 158 Аминазин 103 9-Амипоакридип 87-89 9-Амипоакрилииий хлорид 89 4-Аминоантипирип 54 2-Ами(ю-5-бромпиримидин 97 2-А миг1()-4-1'илроксипиримиди11 97 2-Амино-4-гидрокси-5-пптро-пиримндин 97 1-Аминоизохиполин 85 а-Амипокислоты 165 исл. заменимые 168
- С-копцевые 175 N-коппевые 175 незаменимые 166-168 с защищенной аминогруппой 170 с защищенной карбоксильной группой 181
2-Аминоттиразин 101
2-Амино-1 -мстилпиридиний иолид 74
2-Амнпо-6-метилпиримнлин 98 2 А м и н о-4 -метнлтин зол 60
2-Амипо-3-метнлтиазо.1Ий иоднд 61 З-Амииопиридппа дигидрохлорид 74 4-Амипопирилнний хлорил 73 Амипопиридипы 67. 69. 73 и сл.
2-Аминопнримидип 97
З-Аминопропанол-1 17 2-Аминотиазол 60 н-Аминофенол ХЗ 2-Амипохииолии 81 2-Аминоэтаиол 16, 19 Ампициллин 61
А МФ. см. Аденозын-5 -мшюфосфат Амфотерные вещества 50,56,58,75 Анабазин 117 Анальгин 53
А игидрил
-малеиновый 33.35
- уксусный 31,41.92.145.152
- - 1.2,3.4-тетрагидро-3.6-эпокси-
237
фталевый 35
Андрогены 229
Андростерон 229
Анилин 45, 78
З-Анил ин обута наль 79
3-Анилинопропапаль 78 Аномерный центр 129.130 Аномеры 130исл, Антипирин 53 и сл.
Антиподы оптические, см. Энантиомеры
Арабинаны 146 /)-Арабиноза 136, 142 L-Арабиноза 127. 128, 146, 163 Л-Арабипопираноза 146 Арбутин 144
Аргинин 166
Аскорбат-ион 150
Аспарагин 166,170
Атропин 121
АТФ. см, Лденозшнприфосфат Аттрактанты 212 Ацетали 144
Ацетальдегид 64, 79
N-Анетилазиридип 20
а-Аиетиламипопропапон 62
А цетилацетоп 49, 62 оксим 62
Ацетилен 28. .36, 49
Ацетил нитрат 30, 44
N-Ацетилпиридиний хлорид 66 Ацетилпирролы 31.34 2-Лцетилтиофси 31
I О-Апетилфенотназин 102
2-Ацетил фура н 31
Ы-Ацетнлхииолиний хлорил 80 Анстонп 205
N-Анет оксипиридиний хлорил 70 Ацидофобность 29 и сл,, 41,43 Апилазы 169
Барбамил, см. барбитураты Барбитураты 99
Барбитал, см. барбитураты Белки 165 и ел.
глобулярные 182
— простые, см. Протеины
- сложные, см. Протеиды
— фибриллярные 182
Бензальдегид 54, 71
Д-Бетилидепаминоаптипирип 54 Бензилпенициллин 61
Бензимидазол 58
238
Бензо ил нитрат 44 I
Бспзоксикарбонилхлорил 170,180 3-Бензол азо и идол 44
Бензолазопиррол 34
Бензоллиазоний хлорид 34
Бензот^ф 20
Биотин 42
Ьииьчера-Папиральского синтез 84
БОК, см, тре.т-Кутоксикирйониль-нан группа
Борнап. см. Камфан
Борнеол 217-220
Борнилацетат 219
Борн ил-катион 218
Брожение спиртовое 147
Бромкамфора 220
З-Бромпиридин 66
N-Бромлиридиний бромид 67 у-Бромпропиламин |9
Бутан 28
Бутанол-! 17 <
Бутиролактон 36
/пропнБутокиикарбиксазил 170. 180 гнретн-Бутокси карбо пильная группа.
182
Буфадиеполиды 231
Ъигнера-Меервейна перегруппировка 219 *
Иагнера реактив 116 J
Вазопрессин 178 f
Валидол 215 Г
Валин 166 t
Ванилин 77 »
Нин-Сиайка метод, см. Реакция дез- f. аминирования
Вербеноп 217 f.
N-Випилпирролидон-2 36 г.
Виноградный сахар, см. D-1 'лкжоза f
Витамин Ah см. Ретинол
Витамин В,, см. Тиамин
Витамин В2, см. Рибофлавин
Витамин В6, см. Пиридоксин
Витамин В|2,см, Цианокобаламии
Витамин BL., см. Кислота фолиевая
Внта мин С, см, Кислота аскорбиновая
Витамин IX. см. Эргокальциферол Витамин D3, см, Хилекальциферол ' Витамин Н, см, биотин г-
Вита м и п Р, с м. Флавонолы :\
Витамин РР, см. Кислота никотино-К -
вая. амид А -
Воски 196,206,207 — горный, см. Озокерит -карнаубский 206,207
— пчелиный 206. 207
— шерстный 206
Вторичная структура — белков 176 - нуклеиновых кислот 192 /ХГалактоза 128.135,147,163 D-Галактозамин 148 a-D-Галактопираноза 135 р-О-Галактогртрапоза 135, 147 Галоперидол 77
Ганча синтез 59
Гексозаны 156 Гексозы 126
Гем 37
Гемин 37
Гемоглобин 37исл.
Гсмодез 36
Гепарин 140, 162, 163
Гераниол 211 Героин 120 Гестагены 229,230 Гетероатомы 8 - пиррол иного типа 23.50 пиридипопого типа 24.50,95
Гетероауксип 48 Гетерополисахариды 161 и сл. 9-Г идроксиакридин 86, 87 Гидрокортизон 227.228 Гидроксикислоты 136 5-Гидроксиметилфурфурол 143 Гидрокси нитрилы 136, 142 Гидроксипиридины 68. 71 цел,. 91 4-Гидроксипирилий хлорид 91 Гидроксихинолипы 81,83 Гидрофурфурамид 40 Гиосциамин 121 Гипоксантин 105. 107-109 - натриевая соль 108 - хлороводородная соль 108 Гистамин 58
Гистидин 57, 167
Гликоген 158
Гликозидный гидроксил 129 и сл. Гликозиды 94, 100, 139. 143исл. Гликокол.см. Глицин Гликолипиды 196 Гликолипопротеиды 182 Гликопротеиды 182 Глиоксаль 55, 101. 109
Глицерин 78. 199 и сл.
Глицин 166,180, 181
Глобин 37 и сл.
Глутамин 166. 170
П-Глюкози 127 и сл,, 151) и сл.
оксим 142
L-Глюкоза 127
/ХГлюкозамин 148. 162. 163
Глюкозил 144
Глюкозо-1-фосфат 146 Глюкозо-6-фосфат 146 Глюкокортикоиды 227, 228
Глюконат кальция 138 a-D-Глюкопираноза 129, 131, 133, 134, 143-147
p-D-Глюкопирапоза 129, 133, 135, 144
а-/)-Глюкофураноза 130, 133 р-Л-Глюкофураиоза 130, 132. 133
ГМФ. см. Гуанозин-5‘-монофосфат Гомополисахариды 156 и сл.
Гормоны 178.179,209,227 коры надпочечников 227-229
- половые 227. 229, 230
Грамицидип С 179
Гуанин 107. 109. 185, 186
Гуанозин I87
Гуа1[озин-5‘-монофосфат 189 Гуммиарабик 163
ДАМФ. см, Де*1оксиалепозин-5'-монофосфат
ДГМФ. см. Дезокгкгуанозин-5 -монофосфат
Дебнера-Миллери синтез 79 7-Дегилрохолестерин 225 Деградация но Эдману, см. .?dnma реакция
Дезоксиаленозии 187, 188
Дезоксиаденозин-51-монофосфат 189 Дезо кси гуанозин 187,188
ДезоксигуанозииФ’-моиофосфат 189 Дезокси кортикостерон 228 Дезоксицитидин 187. 188
Дез окси I in тнди п-5 ‘-мопофосфат 189 2-Дезокси-D- рибоза 148, 184-187 Дезоксирибопуклеозицы 186, 187 Дезокснрибопуклеотиды 188 2-Дезокси-/3-рибофураноза 148 Декагидрохинол и и 82 Дексаметазон 228, 229
Декстраны 160
Декстрины 157
239
Денатурация белка 183 Детергенты, см. Синтетические липощие средства
л-Дефипитные гетероциклы 24. 64 Диазепам [ 13 Дназепниы 112
1.2-Дпазип, см. Ниридазип 1.3-Диазиц, им, Пиримидин 1.4-Дназин, см. Пиразин Диазометан 49 2,4-Диамиио-6-метнлпирим1Шии 98 4.5-ДиаМИ11О11ирим11ДИП 104. 109 Диамины J9 Диастереомеры 128,218,219 Диане гпл 205 Дибазол 59
9Д0“Днп1дроакридин. см, Акридин 2,3-Днгидроипдол 44 2.3-ДиГИДрО[[11р11ЭИМ 101 Дигидроу ранил 186 1.2-Днгнлрохинолин 79, 82 0-Л-Дигиталоза 231 Дигитоксигенин 232 /)-Дигитоксоза 149.231 Днклоксациллин 61
Р-Л-Димарозд 231 3.3-Димстилаллилпирофосфат 209 2-Х.К-Д>1метшпуш1И1< 186 3.5-Димстилизоксазол 62 2,5-Дпмстц.10кса.юл 62 3,5-Диметидпиразол 49 Диметилсульфат 54, 145 2.5-Диметилтиазол 60 2.4-Диимтрофторбеизол 172 Динопрост 208. 209 Дипопростоп 208, 209 Дипукдеотиды 190 Дпосгенил 233 Дипептен, см. Лимонен Дппсптлды 179. 180 Дисахариды 151 чел.
восстанавливающие 151 и сл, певосстанавливающие 155. 156
Дитерпепы 210
Дифениламин 86, 102 2,5-Дихлорпирро.т 31 2.5-Дихдортиофен 32 Дихлорэтан 19
ЫЛ'ЧДнпиклогексилкарболинмид 182
N.N’-Дициклогексилмочевина 182 Диэтаноламин 16
Диэтиламип 76
Диэтнлмалонат 96
Диэтилстильбестрол 230
ДНК, см, Кислоты дезоксирибонуклеиновые
ДНФ- производные 172
Прагеидорфа реактив 116
Древесный сахар, см. D-Kcu:ioiu ДТМФ. см. ‘1'и.иидш1-5,-мон(>ф(н:фат ДЦГК. см, N.N -ДициклогексилкарСю-
OUUMlfd
ДЦМФч см. Дезокснциншдип-З-монофогфат
Енгидразип 141
Животный крахмал, см. Гликоген Жиры 196 и сл.
Изатин 35, 46 и сл, оксим 47
фенилгидразон 47
тг-Избыточные гетероциклы 24
Изоаллоксазип 111
Изоборнеол 218
Изоборпил-катион 218 2’
Изоксазол 48.62
Изолейцин 166, 168
Изониазид 77
И чопепте1гилпирофос'фат 209
Изопрен 209.213
Изопреновое правило 210
Изопреноиды 209 и сл. ;
Изофлавон 93. 94
Изохинолин 63, 84 и сл. г
Изохинолон 85 ’ 1
Изоэлектрическая точка 1р/ > 169 : '
Имидазол 48, 55 пел. J 1
Имидакътил-апиоп 56 5
Имидазолий-катпон 56, 57 *1
а-И минокисл оты 171 2-Имн11о-3-метилтилао.тин 61 Инверсия 155
Инвертный сахар 155 1
Индиго 4.5 и сл. '
Индигокармин 46 '
Индикаторы 46, 157 *
Индоксил 45 4
Индол 42 и сл,
Индолкалий 43 1
Индол магний иодид 43 1
Индометацин 48, 209 г 1
Инсулин 178. 179 iJ
Инулин 148, 161 L1
240
Йод метан 20, 34, 52. 54. 60, 61,66, 70. 73. 80. 87,91.95. 102. 145, 152
Камеди растительные 163
Камфан 216
Камфегг 218. 219
Камфепоння перегруппировка I рола. см. Вагнера-Мееряейни перегруппировка
Камфора 218-221
Канифоль 217
Канниццаро реакция 40
Каран 216
Карбоксимегил целлюлоза 160
Карленолиды 231 Каротин 221.222 Каротиноиды 22 L 222 Катализатор Гвшнчиш и контакт
Петрова 200
Кверцетин, см. Флавонолы Кетогексозы 126, 129. 130. 132. 133 Кетозы I26
а-Кето кислоты 136, 150, 171 Кислота
З'-адепнловая. см. Адено тч-З минофосфит
5'-адениловая, см. Аденипт- ' -моиофосфшп
- адипиновая 102
акридиновая, см, Кне.кнна 2. I-хинолиндцкарбо/нмшя
- н-ам и побег по иная 111 а-амниоглутаровая. см. Кислота глутаминовая о-амипо-б-гуагвгдиионалериало-вая, см. Аргинин а-амипоиювалериапсвая, см. Валин
а-амино шок а про полая, см. Лейцин
a-а м иио-0- гидрок си м асл я и ая. см.
Треонин
«-а.мгшо-0-пирокен пропионовая, см. Серин
а-амипо-р-меркаптопропиопопая, см. Цистеин
а-амнно-Р-метил валерил новая, см. Изолейцнн
«-имипо-у-мстилтиомасляпая, см.
Метионин
а-аминопропиоповая. см, Алинам Р-амипопропиогговая 20
амид 20
— а-амиио-0-фени.тпролионовая, см. Фенилаланин
- аминоуксусная. см, 1 лицин аминияптарпая, см. Кислота аспарагиновая
D-арабипаровая 139 арахидоновая 198. 208, 209 арахиновая 198
— аскорбиновая 149
— аспарагиновая 166. 168, 170, 171 амил, см. Аспарагин барбитуровая 96-99 бензойная 76
/З-галактаровая, см. Кислота слизевая
О-галактонивая 138
— Л-галактуроиовия 139, 140
-- гиалуроновая 161
- 4-гидроксипирролидин-2-карбо-повая 36
--- З-гидроксипронаноная
— - производные 18 гликохоленая 227 глутаминовая 110, 166. 170, 171
у-амид. см. Глутамин
- D-глюкаровая 138.139 - /2-глюконовая 136. 138
--- кильпиевая соль. им, Глюконат кальция
у-лактоп 138
/2-глгокуроповая 139. 140. 163
5'-гуаниловая, см, Гуанозин-5 монофосфит
- легидроаскорбиновая 149 дегидрослизсвая 27
дезоксиадениловая. см. Дезоксиа-денозин-5'-м<нк)ф(нфат
- дезоксигуаниловая. см. Дезокси-гуано ша-5 '-манофосфат дезокспгигтидилоная. см, Дезокси-цитидин-5 -монофвсфат
—•дезоксихолевая 226
- - диалуровая 107
а.с-диаминокапроновая. см. Лизин
А-идурононая 139. 140, 163 изатиновая
натриевая соль 47 иэоникогнповая 70, 75 н сл.
гидразид, см. Изониазид
— - лиловый эфир 77
241
Кислота
5-и:юхиполн11су;ты|ю1Ювая 85 нм и дазпл-4-сульфо новая 57 иидиго-5.5'-дисульфо1говая, см. Индигокармин р-н п дол илу коус! га я, см. / етеро -ауксин индол-З-сульфоповая 44 камфановая 220 камфароповая 220 камфорная 220 каприловая 198 - каприновая 198 - капроновая 198 а-кетоглутаровая 171 2-кето-Л>-гл1оконовая 136 2-кето-Л-гулоповая 150 кумариновая дннатрневаясоль 92 -...лактон, см. Лактоны
- 6-кумаринсульфопонля 93 лауриновая 198
- а-линолетщя 198
..линоленовая 198
— мальтобионовая 152 /)-ман паровая 138 А>-маннуроповая 139, 140 масляная 198
- мекоповая 91,92
-- миристиловая 198
Чниолочная 89
мочевая 105-109 соли. см. Ураты муравьиная 58,86 нейраминовая 150 никотиновая 68. 70. 75 исл.
— амид 76
гидрохлорид 75 N.N-диэтнламид 76 - натриевая соль 75 цвиттер-ион 75 5-океопентсг1-2-овая 91 олеиновая 198. 205 оротовая 100 пальмитиновая 198 нектонам, см. Кислота полигалак-HiypiHHMUtM пеларгоновая 205 псроксиуксуспая 60, 68. 82, 85. 88, 96, 101 пиколиновая 75 и сл.
— пирачо.т-4-сульфоиовая 52
242
3,4-пиридиндикарбоновая 85 '
З-пнридинсульфоиовая 66 j
— пирослизевая 27. 40 1
натриевая соль 40 J
пиррол-2-к a pool юна я 35 1
пирролидип-2-карбоновая. см./ Пролин J
- -полигалактуроновая 161 — простатговая 207, 208 — пурпурная 107,108 — аммонийная соль. см. Мурек-сид
/2-сахарна я 139
сди'левая 27. 138
- - диаммопийпая соль 27
- стеариновая 198
— таурахолевая 227
— тимидиловая, см. Ти.иидкн-5 -монофисфит
тиофен-2-сульфоновая 31
— трифторуксуиная 171
- троповая 121
— уксусная 60
ураиил-6-карбоновая. см. Кисло-та оротовая
5’-уридиловая, см. Уридин-Э -монофосфат
-- N-фепиламииоуксуспая
натриевая соль 45
N-фепилантрапилоаая 86
фолиевая 110
-фталевая 85
фурат 1-2-сульфоновая 31
--- хаульмугровая 198
— хелидоновая 91, 92
- 2.3-хииолиндикарбоповая 88
— хинолиновая 82
- б-хиполнпсульфоиопая 81 8-хицолинсульфоповая 81.83 хлоруксусная
— - натриевая соль 45
— холановая 226
— холевая 226
- целлобиоповая 153
- 5’-цитидиловая. см. Цитидин-5'-монифосфат
--щавелевая
- диамид, см. Оксамид
- тцавелевоуксусная 171
- эйкозановая 207
элаидиноная 205
альдаровые 138
Усислоты
альдоповые 138
высшие жирные 197 и сл.
незаменимые 197
дезоксирибонуклеиновые 184, 185. 192 и сл.
желчные 226. 227 нуклеиновые 184 и сл.
— рибонуклеиновые 184. 192 мел.
— сахарные, см. Кислоты альдаровые сиаловые 150 смоляные 217 уроновые 139
Колени 120
Кокаин 121. 122
Кокарбоксилаза 100
Коллаген 37
Ко. t-iif-Толленси формулы 131
Коллодий 160
Коллодийная вата. см. Коллоксилин
Коллоксилин 160
Кольйе-Н/.нипина реакция 35 Комплементарные пары 193 l Конформации
лисахарилов 153.154
I моносахаридов 134
I поли нуклеотидной цепи ДНК 193
р полииепгилпой пени белков 176 полисахаридов 157,159
стероидов 222. 223
Кортизон 227. 228
Кортикостероиды 227-229
Кофеин 108
Кофермент
НАД* 171
j пиридоксальфосфат 171
| Красители кубовые 46
Крахмал 157. 158
Крашение кубовое 46
.п-Крсзол 215
Ксантин 105. 107-109
Ксилап 146
Л-Ксилит 137
Д-Ксилоза 128. 137. 146
а-ТЭ-Ксилопираиоза 146
Кумарин 92. 93
Лактамы 20. 61
Лактоза 151
Лактоны 90. 93. 136. 138. 149. 231
Ланолин 206
Левулеза, см. D-Фруктоза
Лейцин 166
Лизин 166
Лимонен 213. 214
•- крекинг 213
Липиды 196 и сл. пеомыляемые 196. 207 и сл. омыляемые 196 и сл.
Липопротеиды 182
Лобелии 117
Лутеолин, см. Флавонолы
Магпийоргапичсские соединения 17. 43
Майера реактив 116
Малеиндиалъдегил 95
Малеинимил 33
Мальтаза 153
Мальтоза 151-153. 157
Маннан 147
D-Маннит 138
D-Матгмоза 128. 137, 140-142. 147
а-Л-Маппопираиоза 135
Р-£)-Маппопираноза 135
Маргарин 205
Марки реактив 116
Масла
жирные 197 эфирные 210 и сл.
Ментан 213,214
Ментилизовалерианат 215
Ментол 214. 215
Ментон 215
Мсталлопротеиды 182
Х-Метилазиридин 20
N-Метилакрилиний иодил 87
Метиламин 18
4-МетиламИ1гоантипнрил 55
N-Метил-З-гидрокси пиридин ни иодил 73
Метил-/>-гл юк о пиранозид 139. 144 l-N-Метилгуаиип 186
2-Мстил-1.2-дигидрохинолип 80
Метиленовый синий 103
N-Метилимидазолий бромид 56
Мстилимидазолы 56
2-Метилиндол 43
Метилы ал ьтозид 152
N-Метилпиразолий иодид 52 Метидпиразолы 51 и сл.
N-Метилпиридазипий иодид 95 Метилпиридипы. см. Николины N-Метилпиридипий иодид 66 N-Метилпиридои 66 6-Метил пиримидин 98
243
Метил пирролы 34
Метил тестостерон 229
Метил -2,3.4.6-тетра метил- Л-глюкипиранозид 145
4-Метилтназол 60
N-МетилтиазолнЙ иодил 60
З-М-Метилурацил 186 3-Метил-1-фепилпиризолоп-5 53 1О-Метилфеп0тиазип 102 2-Метилхиполип 80
N-Метилхинолипий иодил 80 Метионин 167 9-Мето кс и акридин 87 N-Метоксипнридиний иодил 70 4-Метоксипирилий иодид 91 З-Метоксипропанол-1 17 М ннералокортикоиды 227-229 Мирнцил пальмитат 207 Мирцен 211 Мицеллы 201
Молочный сахар, см. Лактоза Моноимин 2-кетонльдегида 142 Мононуклеотиды 189, 190 Моносахариды 126 Мопотерпепы 210 и сл. Морфин 119, 120 Мочевина 96 Мурексил 107 Мутарогцция 129. 134. 152. 153 Мыла 200-204 «зеленое» 201 «металлические» 201
Неодикумармп 93
Нерол 211
Никотин 117
Никотинамид, см. Кислота никоти-аовая, амид
Нингидрин 174
Нитразепам 113
Нитробензол 78
Нитрозилхлорид 20 N-Нитрозоазиридип 20 4-Нитрозоантипнрин 54 Нитроизохинолины 85 4-Нитроимидазол 57 З-Нитроинлол 44 Нитроксолин (5-НОК) 83 6-Нитрокумарип 93 4-Нитропиразол 52 З-Нитропнридин 66 4-Ннтропириднп 69 2-Нитропнррол 30
2-Нитротиофен 30 7
о-Нитрофенол 83 ’ f
2-Нитрофураи 30 5-Нитрофурфурол 41 5-Нитрофурфуролдиаиетат 41 Нитрохинолииы 81
Норсульфазол 61
Нуклеотид пол ифосфаты 191 Нуклеозиды 184. 186 и сл.
Нуклеопротеиды 182. 184
Нуклеоз иды 184, 188 и сл.
Озазоны 141
Озокерит 206
Озопы 142
Оксазол 48. 62
Оксамид 57
Оксациллин 61
Оксетап 14исл,
Оксигемоглобин 38
N-Оксид
акридина 88
2,5-ди метилтиазол а 60
изохинолина 85
4-нитропиридина 69 пиразина 101
—пирндазина 96 пиридина 68-70
-хинолина 82
4-хлорпиридииа 69
Оксидазы 171
Оксинитрилы, см. Гидроксинитрилы
Оксипролин 36. 165. 167, 177
Оксиран 14исл.
Окситоцин 178
Олигосахариды 126
Олифа 217
Омыление 200
Оптическая активность 117-12), 128. 134. 155. 169.214. 217-220
Оцимен 210. 211
X\aa:ie-Kuuppu синтез 26
ПАВ. см. Поверхностно-активные вещества
Папаверин 119
ПГ1:., см. Простагландины
П1'1\ см. Простагландины
Пектиновые вещества 161
Пенициллины 20. 61
Псптаацетилглюкопирапоза 145
Пентанамик-! 68
Пептандно! 1-2,4, см. Аиетилацепюн
Пентозаны 39. 156
244
Пентозы J 26
Пептиды 165, 174
Первичная структура пуклеиновых кислот 192 пептидов и белков 176
Перкина реакция 92
Пиколины оЗ. 70 и сл.
Пинан 216
а-Пинен 213. 216 и ст.
ПиперазигГ 101
адипиновокислая соль 102
Пиперидин 68. 77
Пиразин 94. 101
Пиразол 48 и сл.
Пиразолид-аниоп 50
Пиразолидин 52
Пиразол ид-1 [атрий 25
11 иразолий-катион 51.52
Пиразолии хлорил 25. 50
Пиразол и п-2 52
Пиразолкалий 50
Пиразолон-5 53
Пираны 63. 90 нсл.
Пиридазин 94 и сл.
Пиридазиний хлорил 95
Пнриднл-2-мстилпатрии 71
Пиридин 23. 63 и ел.
Пиридиний бромид 65
Пиридинии 1идросулъфат 25. 67 Пиридинолы. см. I идроксипиридины
З-Пиридинолят калия 68
Ииридипсульфотриоксид 30. 44. 65
Пиридоксин 73
Пиридопы, см. Гидриксипиридины
Пиримидин 94. 96 и сл.
Пиримидиний гидросульфат 94
Пирпмидипий хлорил 97
Пиримидиновые основания 99
Пироксилин, см. Трипитроклетчат* ки
Пироны 90исл,
Пиррилмагпий иодид 33, 34
Пиррол 23, 26 и сл.
Пирролидин 32
Пиррол ид калия 33
Пирролидон-? 36
Пирролип 32
Пиррол натрий (пирролид натрия)
25. 33. 35
П иррол-2-сул ьфок пелотн 31
Пластыри медицинские 201
Плодовый сахар, см. П-Фруктаза
Поверхностно-активные вещества
200, 202. 203
Полинипилпирролидон 36
Пол игл юки и 160
Полисахариды 126. 156 и сл.
Политерпены 210
Политтиленоксид 17
Полуацетали 129
Полуацетальный гидроксил, см. Гликозидный гидроксил
Порфин 37
Порфирины 37 и ст.
Прегнан 227
Преднизолон 228
Прогестерон 230
«Прогоркание» жиров 204
Пролин 36. 165, 167. 177
Промедол 77
Пропанол 17
Р-Пропиолактоп 18
Простагландины 196.207-209
Простатическая группа 182
Протеиды 182 _
Протеины 182
Протеноидьг 182
Птеридин 109. 110
Пурин ЮЗ и ст.
натриевая соль 104
Пуриний хлорил 104
Пуриновые основания 109
Пурпуреагликозид А 232
Радедорм, см. Нитразепам Радикалы
пиррола 26
- тиофена 26
- фурана 26
Putcuepit-Тимана реакция 35
/.-Рамноза 149
Распад по Руффу 136
Рацемизация а-амипокистот 170
Реакция
аминирования по Чичибибиму 67
- биуретовая 175
— дезаминирования 171
индофениповая 35
.ксаптопролей новая 174 мурексидпая 107,108 пипгидриииая 174
е переносом реакционного центра 52
Резерпин 123
Реополиглкжин 160
245
Репелленты 212
Ретинол 222
Рибит 112
D-Рибоза 128. 146. 184 и сл.
Рибозо-5-фосфнт 146
Рибонуклеозиды 186
Рибонуклеотиды 188
Рибофлавин 112 p-D-Рнбофураиоза 146 Рияапол 89
РНК. см. Кисмяпырибонуклеиновые
Рутин, см. Флавонолы
Сабинам, см. Туйии
Саломасы 205
Сапонины 233
Сахароза 155, 156
Свекловичный сахар, см. Сахароза
Связь
водородная
внутримолекулярная 46, 176 межмолекулярная 50. 55,159, 193
гликозидная 143. 144. 186
аксиальная 153
экваториальная I53 макрозргическая 191 пептидная 174.181
- фосфодиэфирпая 189. 190
Седуксен, см. Диазепам
Селиванова реакция па фруктозу 143
Семикарбазид 41
Сенгера реактив, см, 2.4-Дм<итро-фпюрбензем
Серин 166
Серотонин 48
Сесквитерпены 210
Синестрол 230
Синигрин 144
Синтезаторы 182
Синтетические моющие средства
203. 204
Скналеи 212
Скипидар 213. 216,217
Складчатая (3-структура, см. Вторичная структура белков
Скопин 121
Скополамин 121, 122
Скраупа синтез 78
Соли пирилия и бенэопирилия 91,93
D-Сорбит 137, 138. 150
/.-Сорбоза 127. 147, 150
а-/.-Сорбопираноза 147
246
Спермацет 207
(1-Спираль. см. Вторичная структура белков
Спирт
— изопропиловый 215 ланолиновый 206 метиловый J 7. 87
- мирициловый 206
фурфуриловый 40
- цетиловый 206
-этиловый 15.143.181
Стеарин 201
Стеран 222
Стерины 224
Стероиды 212, 222 и сл. Стеролы, см. Стерины
а-Стирилпиридин 71
Стрихнин 123. 169
Строфаптидип 232
Сукиинимил 27
Сульфаниламидные препараты 111
Сульфапиридазин 96
Таутомерия азольная 51. 104,106 амшю-иминния 14
а-и у-гидроксипиридинов 71 карботаъ но-ендиолъная 140. 141
кета-енолышя 45. 59. 98
- кольчато-цепная. см. Таутомерия
цикло-окси-
— лик там-лактимная 47. 59, 98.99,
105. 109. 185. 186
З-мстил-1 -фенилпиразолона-5 53 пиразолопа-5 53 цик.чо-оксо- 133, 151
Твердофазный синтез пептидов 18Г
Твииы 207
Теобромин 108 К
Теофилин 108 С
Терпены 210 и сл.
- ациклические 210-212 V
бициклические 216-221 I’
моноцмклическне 213-216 I
Терпин 214.215.217 1 ..
Терпингидрат 215.216 П?
(1-Териинеол 2J 1,216. 217
Тестостерон 229 |! .q
Тетра гидротиофен 32 lj/:
Тстрагилрофуран 32 .;Г(
1,2.3,4-Тстрагилроизохинолип 85!
1,2.3,4-Тстрагидрохинолин 82
2,3,4.6-Тетраметил-/?-глюкопирайФ1’
за 145
Тетраметплепдиамин 96 Тетратерпены 210.221 Тетрахлорпнррол 31 Тетрахлортиофен 32 Тетрозы 126
Тиазол 48, 59 и сл.
Тиазол ий хлорил 60
Тиамин 100
Тимидин I 87. 188
Тими,чип-5'-мопофосфат 189
Тимин 99. 100. 185
Тимол 215
Тиофен 23. 26 и сл.. 47
Тиоформамнл 59
Тиофосфамнд 20
Тирозин 167
Тименса реактив 140. 152. 153
Трансаминазы 171
. Трауие метод 104
Треонин 166. 168
Третичная структура белков 177 Триацил глицерины, см. Жиры 2,4.5-Трибромимидазол 57 Триглицериды, см. Жиры Гринитроклстчатка 160 Триозы 126 Гриолеин 206 Трипептид 175 Триптофан 48, 167, 170
[Тристеарии. ем. Тристеароилглнце-рин
Тристеароил глицерин 199. 205
Трнтерпены 210 2,4,6-Трих.чорпиримилин 97 2.6.8-Трихлорпури11 106 Тр и лги юл ам и 11 16
Трипап 120
Тропин 121
Тростниковый сахар, см. Сахараза Тупаи 216
Углеводы 125
Ураты 106
Урацил 99. 185. 186
Р-Урацилрибофурапозид. см. Уридин
Уридин 187
Уридин-5'-мопофосфат 189
УМФ. см. Уридин-5-лшнофасфат Уридин-5"-мо1!офосфат 189
Фарнсзилпирофосфат 212
Фарнезол 212
Фелинга реактив 140, 152, 153
Феиазон 96
Фенилаланин 167
Феиилгидразип 40,47,53. 14L 142
Фепилгидразон ацетона 43 D-глюкозы 141
о-Фепиленлиамип 58
Фен ил изотиоцианат 172
(Ьенилтиогидмнтоина производные 172
|3-Фепилэтиламин 84 Фенобарбитал, см. барбитураты Феноксиметилпенициллин 61 Фенотиазин 102. 103
Фегютиазипдиоксид-5.5 103 Фепромарон 93 Ферромоны 212
Фишера метод 42
Фл авш ।. ем. Ита.i.wwa3im лейкосоедипение 112
Флавии 93.94
Флавонолы 94
Формальдегид 55.64. 172 N-Формил-р-толуидин 42 Формил пирролы 34, 35 Формальное титрование по Серенсе-пу 171
<Ьос(|юлнпиды 196
Фосфопротеиды 182
Фотосинтез 135
D-Фруктоза 127 и сл.. 138. 139. 141.
142. 148, 161
Ф ру кто:ю -1.6-ди фосфат 146 а-/)-фрукто1 трапеза 130, 132. 133 р-/)-Фруктопирапоза 130. 133 ы-Л-Фруктофураниза 130. 133 |5-/?-Фруктофураноза 130. 132, 133.
148
Фталазол 61
ОПТ-производные, см. Феналпшиги-даппюина производные
Фтивазил 77
[TD-Фукоза 231
Фурадошш 41
Фуразолидон 41 Фуран 23. 26 и сл.
Фурапилин 41
Фуронн 40
Фурфурол 39 и сл.. 143 бисульфитнос произвол!гое 40 оксим 40 фепилгидразон 40
247
Хелаты 83. 173
Хеуорса формулы 131
Хипнп 118
Хшгозол 83
Хинолин 63. 78 и сл.
Хиполиний хлорил 80
Хнпуклндиповоеядро 118
Хитин 148
Хитозамип. см. />-/люкозамии
9-Хлорнкридин 86. 87
Хл о ран гидриды кислот изопикотиповой 77 никотиновой 76 уксусной 20. 34. 66. 70, 80. 102
З-Х.'юриплол 44
Хлорофиллы 38. 135
4-Хлорпира«>л 52
4-Хлорпиридтгп 69
2-Хлорпиррол 31
З-Хлорпропапол-1 15, 17
2-Хлортиофеи 32
2-Хл о ртта н ел 14. 16
Р-Хлор'УГиламнп (2-хлорзтсш-имин} 18. 19
Холекальциферол 225
Холестан 224
Холестерин 205,224
Хоидрозамин. см. D-Гилакпюзалшн
Хондро! 1ти1 «сульфаты 161, 162
Хромон 93
Хромопротеиды 182
Цвиттер-ионы 75. 169
Целлобиоза 151.153. 154. 159
Целлюлоза 159.160
Цетил пальмитат 207
Циапгидриноный синтез 136
Цианокобаламшг 38
Циклопе! гганопергидрофенаитреп, см. Стеран
Цнкдосерин 62
и-Цимол 213.214
Цистеин 167
Цистин 167
Цитидин 187
Цитидн! 1-5'-моиофосфат 189
Цитозин 99. 100. 185
Цитраль 211
ЦМФ. см. Цшнндин-5'-монофосфат
Четвертичная структура белков 178
Число
гидрирования 205
248
иодное 206
кислотное 201
нейтрализации, см. Числа кислотное
омыления 200
эфирное 201
ЧиччбаСита реакции 28. 67
Шиффи основания 172
Эдмона реакция 172
Элаидированне масел 205
Энантиомеры 127. 168. 213. 214, 216,
219
Эптеросептол 83
Эпимеризация 140. 141
Эпимеры 128
Эпихлоргидрин 18
Эргокальциферол 225
Эргостерин 225
Эрдмана реактив 116
Эстеразы 169
Эстрадиол 229. 230
Эстрогены 229. 231)
Эстрон 229.230
Этакридина лактат, см. Риванол
Этапдиамип-1.2 19. 101
Этаноламип. см. 2-Аминиэтаиол
Этапера чип ЮЗ
Этил-а-Л-глюкопиранозил 143
Этил-Ц-Л-глюкот! и рапочил 143 :
Этилен, окисление 15 1
Этиленгликоль 15 *'
Этнлелоксил. см. Оксиран
Этиленхлоргилрин. см. 2-X.iojnma-
Н<М
Этилпирилины 63.71
Этилформиат 34
Этилхлорформиат 180. 181 !
Этплиеллозольв 15 1'
Эфнр
ацетоуксусный 53 4
малоновый, см. Диэши./.маюмат ь
Юрьсш цикл реакций 26 *
Якорные группы 181
а
Е