
















































































































































































































































































































































































































































Текст
Ф. Ю. РАЧИНСКИЙ,
М. Ф. РАЧИНСКАЯ
Техника
лабораторных
работ
под редакцией
ДОКТ. ХИМ. НАУК ПРОФ.
Д. П. ДОБЫЧИНА
Ленинград. «Химия»
Ленинградское отделение
1982
541
Р27
ЗДК 542.2Д7
Рачинский Ф. Ю., Рачинская М. Ф.
Техника лабораторных работ. — Л.: Химия, 1982 — 432 с, ил.
Книга представляет собой практическое руководство по технике лабораторных работ,
проводимых в химических лабораториях различного профиля с применением стандартного
оборудования и стандартных методов исследования. Охвачен широкий круг вопросов — от
мытья и сушки лабораторной посуды до современных физико-химических способов
исследования. Даны описания современных приборов, указания по технике выполнения работ и
рекомендации по технике безопасности. Приводятся примеры изготовления несложной
аппаратуры и приспособлений, повышающих производительность труда.
Предназначена для работников научно-исследовательских, учебных и промышленных
химических лабораторий, а также для учащихся техникумов и студентов вузов.
432 с, 214 рис., 20 табл., библиография 73 названия.
Рецензенты: докт. хим. наук проф. В. В. Некрасов,
докт. хим. наук проф. Ю. С. Шабаров.
п 1801000000—181 «в ' ^ тт v 1noo
050(00—82 ® Издательство «Химия», 1982
Оглавление
От авторов 9
Глава 1. Устройство, оборудование и основные правила работы в
химических лабораториях 11
Планирование и оборудование лаборатории 11
Санитарно-техническое оборудование 12
Водоснабжение и канализация. Газоснабжение. Электротехнические устройства.
Вентиляция.
Установочное лабораторное оборудование (лабораторная мебель) .... 15
Пожароопасность и средства пожаротушения 19
Тушение горящей одежды 2Ь
Средства индивидуальной защиты 21
Первая помощь ,23
Общие правила работы в химических лабораториях 24
Глава 2. Химико-лабораторная посуда 26
Посуда и изделия из стекла ' 26
Соединительные элементы. Краны соединительные и вакуумные. Лабораторная посуда
общего назначения. Обращение с лабораторной стеклянной посудой. Стеклянные изделия
с токопроводящим покрытием. Мерная лабораторная посуда. Проверка мерной посуды.
Правила пользования мерной посудой. Стеклянные дозаторы для жидкостей.
Лабораторная посуда из прозрачного кварцевого стекла 60
Фарфоровая лабораторная посуда 60
Лабораторная посуда и изделия из платины - ... 62
Глава 3. Вспомогательные приспособления и материалы 65
Приспособления для сборки установок 65
Штативы и держатели. Пробки. Резиновые и пластмассовые трубки и зажимы для
трубок.
Приспособления для работы с реактивами 71
Для перенесения из емкости в емкость. Для прокаливании. Для измельчения проб.
Пластмассы как материал для лабораторных изделий . . 73
Асбестовые материалы* 74
Смазки и замазки 75
Глава 4. Химические реактивы 77
Квалификация реактивов и высокочистых веществ 77
Опасные свойства реактивов 79
Хранение реактивов . 82
Обращение с реактивами 83
Щелочные металлы. Огнеопасные жидкости. Хлорная кислота. Ядовитые вещества. Белый
фосфор. Метиловый спирт.
Глава 5. Мытье и сушка лабораторной посуды 89
Способы очистки и моющие средства 89
Некоторые специфические случаи 93
Мытье мерной посуды. Мытье воровок со стеклянными пористыми пластинками.
Устройства для мытья капиллярной посуды.
Сушка , f . 94
3
Глава 6. Дистиллированная и деминерализованная вода 96
Дистиллированная вода - 95
Металлические дистилляторы. Получение бидистиллята. Определение качественных
показателей дистиллированной воды.
Деминерализация пресной воды ионообменным методом 101
Глава 7. Приготовление растворов 104
Некоторые общие сведения о растворах 104
Способы выражения концентрации растворов. Формулы перехода от одних выражений
концентрации растворов к другим. Растворимость.
Техника приготовления растворов 107
Приготовление водных растворов кислот приблизительной концентрации. Приготовление
безводного раствора хлорной кислоты. Приготовление растворов щелочей. Приготовление
водного раствора аммиака. Приготовление рабочих растворов точной концентрации.
Глава 8. Приготовление безводных чистых органических растворителей . .115
Глава 9. Измельчение . , 127
Измельчение вручную . * т 127
Механическое измельчение . 127
Глава 10. Ситовой анализ 129
Сита 129
Методика проведения анализа 131
Ручной рассев. Механический рассев.
Обработка результатов ситового анализа . - ... 134
Глава 11. Перемешивание 136
Сосуды для перемешивания и встряхивания 136
Перемешивание и встряхивание вручную 137
Механическое перемешивание 137
Типы мешалок. Установка мешалки. Приводы для мешалок. Лабораторные
перемешивающие устройства.
Перемешивание барботированием 142
Глава 12. Взвешивание • 143
Классификация весов 144
Общелабораторные равноплечие весы .... . . ... .147
Общелабораторные квадрантные весы .... .... .148
Взвешивание на технических весах 149
Аналитические весы 149
Установка аналитических весов. Правила пользования аналитическими весами. Ошибки
взвешивания и их устранение.
Микро- и ультрамикровесы 156
Торсионные весы ВТ-500 156
Глава 13. Нагревание и охлаждение ... 158
Лабораторные электронагревательные приборы 158
Электропечи сопротивления. Электропечи для элементного микроанализа. Сушильные
электрические шкафы. Термостаты. Приборы для прямого нагревания жидкостей.
Газовые горелки * . 167
Жидкостные горелки 169
Нагревательные бани . * * 169
Воздушные бани. Жидкостные бани. Солевые бани. Металлические бани. Песчаные бани.
Средства и приборы для охлаждения ....... . 173
4
Глава 14. Измерение и регулирование температуры 177
Классификация приборов для измерения температуры 177
Жидкостные термометры расширения* 178
Термометр Бекмана. Общелабораторные ртутные термометры. Термометры на
нормальных конусных шлифах. Технические термометры. Отсчет показаний термометра.
Обращение с термометрами и их проверка.
Термоэлектрические термометры (термопары) 183
Термометры сопротивления * . 185
Термометры излучения 186
Термохимический метод измерения температуры 186
Регулирование температуры 187
Глава 15. Вакуумная техника 190
Правила работы 190
Водоструйные насосы . . 191
Механические вакуумные насосы с масляным уплотнением 193
Диффузионные высоковакуумные насосы 196
Рабочие жидкости — вакуумные масла 197
Глава 16. Измерение и регулирование давления . 199
Классификация приборов для измерения давления 199
Жидкостные манометры 199
Ртутные барометры. Жидкостные манометры для измерения избыточного давления. Ртут-'
ные манометры для измерения вакуума.
Показывающие манометры для измерения избыточного давления .... 205
Вакуумметры для различных диапазонов давления 205
Регулирование давления 207
Глава 17. Фильтрование и центрифугирование : . . .209
Фильтрование . .209
Фильтрование под действием собственного веса жидкости. Фильтрование при нагревании
или охлаждении. Фильтрование при пониженном давлении. Аналитические аэрозольные
фильтры АФА.
Центрифугирование 219
Глава 18. Высушивание . . 223
Осушающие вещества 223
Вещества, образующие гидраты. Вещества, связывающие воду в результате химической
реакции. Вещества, связывающие воду в результате адсорбции.
Высушивание твердых веществ 226
Высушивание жидкостей и растворов 228
Осушение газов 230
Глава 19. Определение влажности 232
Прямые методы 232
Гравиметрический метод. Дистилляционно-азеотропный метод. Денситометрический
(экстракционный) метод. Химический метод..
Косвенные методы 239
Психрометрический метод. Метод определения влажности по точке росы. Гигрометриче-
ский метод. Диэлькометрический метод.
Глава 20. Работа с сжатыми и сжиженными газами ... .... 243
Сосуды Дьюара . .' : . . . 243
Баллоны для газов 244
Эксплуатация баллонов. Отбор жидкости и газа из баллонов.
5
Измерение объема газа 248
Очистка газов 252
Глава 21. Синтез газов 254
Синтез диоксида углерода. Синтез оксида углерода. Синтез диоксида серы. Синтез серо-
водорода. Синтез хлора. Синтез хлористого водорода. Синтез бромистого водорода.
Глава 22. Работа с металлической ртутью 259
Свойства ртути 259
Правила работы со ртутью. Меры безопасности 259
Очистка помещений, загрязненных ртутью . 262
Демеркуризация. Определение содержания паров ртути в воздухе.
Очистка ртути 265
Глава 23. Отбор средних проб 268
Отбор пробы твердых продуктов 268
Отбор пробы жидкостей 269
Отбор пробы газов 270
Газометры. Аспираторы.
Глава 24. Перегонка 273
Простая перегонка при атмосферном давлении 274
Дробная перегонка 277
Ректификация в колонках 277
Перегонка в вакууме . . * 282
Перегонка с водяным паром 284
Глава 25. Возгонка 285
Возгонка при атмосферном давлении .... 286
Возгонка в вакууме 287
Возгонка в токе инертного газа . '. . 288
Глава 26. Выпаривание и концентрирование 289
Выпаривание в открытых сосудах . 289
Выпаривание в закрытых сосудах 291
Вакуумные испарители 291
Глава 27. Экстракция твердых и жидких веществ 294
Экстракция твердых веществ 295
Экстракция веществ из растворов 297
Химическое разделение веществ. Непрерывная экстракция растворов.
Глава 28. Кристаллизация . 300
Выбор растворителя 300
Приготовление раствора для кристаллизации 301
Кристаллизация и отделение кристаллов 302
Глава 29. Хроматография 305
Основные понятия 305
Газовая хроматография ■ 309
Аппаратура для газовой хроматографии. Хроматографические колонки. Детекторы.
Последовательность операций.
Жидкостная хроматография 313
Колоночная хроматография. Инструментальные методы детектирования.
Последовательность операции.
Хроматография на бумаге 318
Основные операции при хроматографировании на бумаге. Одномерная восходящая
хроматография. Двумерная восходящая хроматография'. Круговая (радиальная)
хроматография. Детектирование пятен,
6
Хроматография в тонком слое 323
Пластинки для хроматографирования. Нанесение пробы и проявление хроматограммы.
Комплект оборудования для тонкослойной хроматографии.
Адсорбенты для хроматографии 326
Глава 30. Фотометрия 329
Законы поглощения света. Термины и обозначения 329
Методы измерения поглощения света 330
Расчет концентрации светопоглощающих растворов 331
Фотоколориметрические методы анализа 332
Важнейшие детали фотоколориметров; Лабораторные фотоколориметры.
Спектрофотометрические методы анализа 337
Глава 31. Рефрактометрия . . . . < 339
Показатель преломления 339
Зависимость показателя преломления от температуры. Зависимость показателя
преломления от концентрации.
Рефрактометры 341
Глава 32. Поляриметрия 346
Термины и обозначения 346
Поляриметры и сахариметры 347
Уход за приборами и их хранение 352
Глава 33. Определение рН. Потенциометрия 353
Понятие о водородном показателе 353
Колориметрическое определение рН 354
Индикаторные бумаги. Растворы индикаторов. Буферные растворы. Методы и средства
колориметрического определения рН.
Потенциометрия . ♦ . 360
Электроды для потенциометрии. Приборы для прямой потенциометрии.
Потенциометрическое титрование 367
Глава 34. Определение плотности . . . . , 370
Определение плотности с помощью пикнометров 371
Определение плотности жидкостей. Определение плотности твердого тела.
Определение плотности жидкости ареометрами (денсиметрами) . . . . . 373
Определение плотности гидростатическим взвешиванием 374
Глава 35. Определение вязкости (вискозиметрия) . , 377
Типы вискозиметров 377
Стеклянные капиллярные вискозиметры 378
Определение кинематической вязкости 379
Калибровка вискозиметров. Проведение определения.
Определение динамической вязкости разбавленных растворов полимеров (по
ГОСТ 18249—72) . . 381
Определение условной вязкости 382
Определение водного числа вискозиметра ВУ. Проведение определения.
Глава 36. Определение температуры плавления . , 385
Определение температуры плавления в капилляре 386
Определение температуры плавления на нагреваемой поверхности .... 390
Криоскопичзский метод # ♦ , 390
7
Глава 37. Определение температуры кипения 392
Микрометод определения температуры кипения по Сиволобову -. 392
Определение температуры кипения по ГОСТ 18995.6—73 393
Определение температуры кипения в эбуллиометре 395
Глава 38. Определение температуры вспышки жидких и плавящихся
продуктов 396
Определение температуры вспышки в открытом тигле (по ГОСТ 4333—48) 396
Определение температуры вспышки в закрытом тигле (по ГОСТ 6356—75) 398
Глава 39. Электропитание и измерение электрических величин . . . 399
Источники питания 399
Сетевые источники тока. Химические источники тока.
Измерение электрических величин 406
Приборы непосредственной оценки. Электрические измерения методом сравнения.
Приложения • 411
1. Постоянная влажность 411
2. Приготовление некоторых реагентов 411
Аналитические реагенты. Жидкие поглотители для газов. Реагенты-индикаторы для
детектирования пятен в бумажной и тонкослойной хроматографии.
3. Полезные рецепты и советы 414
Покрытие для деревянного лабораторного стола. Паста для натирания лабораторных сто*
лов и линолеума. Огнезащитные пропитки для спецодежды. Менделеевская замазка. Бу
мага для определения положительного и отрицательного электродов гальванической
цепи. Получение матовой поверхности на стекле. Регенерация серебра. Испытание работы
вентиляции.
Рекомендуемая литература .. .416
Предметный указатель 419
От авторов
Развитие отечественного приборостроения обеспечило
техническое перевооружение химических лабораторий всех профилей.
Все возрастающее значение приобретают в настоящее время
овладение техникой лабораторных работ с использованием
стандартного оборудования и аппаратуры, применение стандартных
методов исследования и определение нормативных показателей
качества материалов. Ознакомление лаборантов со стандартными
оснащением, оборудованием и методами исследования должно
повысить производительность и эффективность их труда.
Ввиду крайнего разнообразия работ, выполняемых в
лабораториях, различающихся по назначению, оборудованию и
оснащенности, мы старались выбрать наиболее характерные, общие для
разнопрофильных лабораторий методики, основанные на
использовании отечественного оборудования и материалов.
В 1941 г. академик Н. Д. Зелинский писал: «В отличие от
физической химии, знающей ряд руководств по лабораторной
технике, органическая химия имеет ряд пособий по методике
проведения синтеза, тогда как лабораторной технике в органической
химии отводятся лишь отдельные главы>. *
С тех пор положение резко изменилось. Опубликованы
прекрасные, но узкопрофильные книги по технике физико-химического
исследования, технике лабораторных работ с преимущественным
использованием зарубежного оборудования. Кроме того, в
большинство учебников по аналитической, органической и физической
химии в настоящее время введены разделы, посвященные
описанию техники эксперимента.
Примером пособия широкого профиля может служить книга
П. И. Воскресенского, которая выдержала 10 изданий, что служит
доказательством методического мастерства автора и большой
потребности в пособии подобного рода. Эта книга
предназначается для младшего и среднего персонала химических лабораторий
(препараторов, лаборантов), преобладающего в химических
лабораториях любого профиля. Как правило, это лица с законченным
общим или специальным средним образованием, а иногда
студенты вечерних и заочных факультетов химических вузов. Все они
должны овладеть техникой работы на основе существующих
нормативных материалов.
В результате развития химической науки и отечественного
приборостроения книга П. И. Воскресенского давно уже
нуждалась в существенной переработке. Отдавая должное уважение
памяти П. И. Воскресенского, мы сделали попытку написать
* Предисловие к книге: Мортон Э. А. Лабораторная техника в органической
химии. М., Госхимиздат, 1941.
9
новое пособие по технике лабораторных работ, включающее
некоторые инструментальные методы анализа. К сожалению,
чтобы сохранить объем книги в разумных пределах, пришлось
отказаться от некоторых тем (ионообменная хроматография, кондук-
тометрия, работа при повышенном давлении и с малыми
количествами веществ и др.)-
Книга знакомит, как правило, с общим устройством и
принципом действия приборов. В тех случаях, когда подробная методика
работы излагается в прилагаемых к прибору инструкциях, дается
соответствующая ссылка.
Мы надеемся, что наше пособие окажется полезным, и будем
благодарны за благожелательную критику и рекомендации.
В заключение выражаем глубокую благодарность
профессорам Ю. С. Шабарову и В. В. Некрасову, канд. хим. наук С. С. Мо-
чалову за ценные критические замечания и полезные советы.
глава i. УСТРОЙСТВО, ОБОРУДОВАНИЕ И ОСНОВНЫЕ
ПРАВИЛА РАБОТЫ
В ХИМИЧЕСКИХ ЛАБОРАТОРИЯХ
ПЛАНИРОВАНИЕ И ОБОРУДОВАНИЕ ЛАБОРАТОРИИ
Химические лаборатории в соответствии с СН 245—71
располагают в отдельных зданиях, специальных пристройках, в верхних
этажах или торцах производственных зданий, изолированно от
других помещений. Они должны иметь естественное освещение.
Высота лабораторного помещения должна быть не менее 3,2 м
[36, с. 9].
Все выходы из рабочих помещений лаборатории должны
сообщаться с линейным коридором здания, имеющим самостоятельный
выход из здания.
Двери лабораторных помещений должны содержаться в
исправном состоянии и свободно открываться наружу, а проходы
не загромождаться.
Химические лаборатории подразделяются на взрывопожаро-
опасные, пожароопасные и взрывоопасные. Большинство
химических лабораторий, в которых проводятся процессы с применением
жидкостей с температурой вспышки паров выше 61 °С и веществ,
способных гореть при взаимодействии с водой или кислородом,
относится к категории пожароопасных помещений. Отдельные
лабораторные помещения могут быть отнесены к взрывопожаро-
опасным (помещения для хранения легковоспламеняющихся
жидкостей, автоклавные комнаты, лаборатории высокого давления,
сероводородные комнаты).
Автоклавную комнату, лабораторию высокого давления и
помещения для работы с взрывоопасными веществами отделяют от
соседних помещений стенами с пределом огнестойкости не менее
2,5 ч*. Из этих лабораторных помещений должны быть
предусмотрены отдельные выходы на лестничную клетку [50; 56, с. 138].
Комнаты, предназначенные для работ с чрезвычайно и
высокоопасными веществами, должны быть изолированы от остальных
помещений лаборатории.
Планирование и оборудование специальных лабораторий и
помещений (автоклавных, ртутных, изотопных, рентгеноструктур-
ных, высоких давлений и др.) определяется специальными
инструкциями.
Ультрацентрифуги, у которых безразмерный фактор разделения
не превышает 5000, имеющие электропривод и выполненные в
одном блоке с пультом управления, можно размещать в обычном
* Под пределом огнестойкости понимают время сопротивления строительных
конструкций воздействию огня до потери ими несущей способности и
устойчивости, до образования сквозных трещин или повышения температуры на
необращенной к огню поверхности более чем на 140—180 °С. Эти пределы опреде*
ляют экспериментально и выражают в часах.
И
лабораторном помещении. Ультрацентрифуги, конструктивно
осуществленные отдельно от пульта управления, следует
размещать в отдельных отсеках первого этажа лаборатории.
Стены, потолки, полы и конструктивные элементы помещений,
в которых работают с ядовитыми и агрессивными веществами,
должны быть облицованы материалами (покрыты составами), не
способными поглощать пары этих веществ и допускающими
очистку, дегазацию и мытье.
Материалы покрытий полов должны обладать минимальной
истираемостью, высокой химической устойчивостью и легко
отмывающейся поверхностью. Этим требованиям отвечают поливинил-
хлоридный линолеум на тканевой основе, релин, пластикат,
бесшовные мастичные, поливинилацетатные, полимерцементные
покрытия. В помещениях для измерения физико-механических
параметров допускается также паркет. В помещениях для работы со
ртутью (гл. 22) рекомендуется настилать глифталевый линолеум
двухгодичной выдержки или пластикат. Материал полов для
изотопных лабораторий по III классу — линолеум или пластикат, а
для лабораторий по II классу — пластикат специальных рецептур
(тип 57-40).
Рабочая площадь определяется как сумма площадей основного
(производственного), вспомогательного и административного
назначения. В рабочую площадь не входят коридоры, тамбуры и
переходы, технические и специальные помещения. Основная
рабочая площадь на одного сотрудника лаборатории должна
составлять 10—12 м 2.
Общее освещение рабочих помещений, в особенности
аналитических, целесообразно осуществлять люминесцентными лампами
дневного света. Минимальная освещенность от одного общего
освещения в горизонтальной плоскости на высоте 0,8 м от пола в
лабораторных помещениях должна быть не ниже 150 лк при
лампах накаливания и 300 лк при люминесцентных лампах.
Кроме общего освещения необходимо местное освещение на
каждом рабочем месте. Оно осуществляется переносными
светильниками, питаемыми от штепсельных розеток. Светильники
должны быть устроены так, чтобы работник мог по желанию
изменить направление светового потока. Местное освещение
должно применяться только вместе с общим. Применение одного
местного освещения запрещается.
САНИТАРНО-ТЕХНИЧЕСКОЕ ОБОРУДОВАНИЕ
Водоснабжение и канализация
Стояки водопровода и горячего водоснабжения, как правило,
располагаются в коридорных нишах. У основания стояков в легко
доступных местах должна устанавливаться запорная арматура.
Разводка воды по помещениям к санитарно-техническим панелям
12
лабораторных столов, вытяжных шкафов и моечных раковин
должна осуществляться по трубам, помещаемым в доступные для
осмотра и ремонта специальные каналы и проемы внутри стен или
пола (в случае установки островных лабораторных столов).
Водопроводные краны в вытяжных шкафах и на. рабочих и
моечных столах располагают так, чтобы исключить возможность
случайного открывания крана. Водопроводные сети должны быть
снабжены вентилями, позволяющими отключить подачу воды как
во всю лабораторию, так и в отдельные ее помещения.
Для отвода лабораторных стоков без предварительной их
обработки обычно служит сеть фекальной канализации.
Местная обработка сильнокислых и сильнощелочных стоков
обязательна, так же как и стоко'в, содержащих токсические
летучие вещества (сероводород, аммиак, цианиды и др.). Недопустимо
сливать в лабораторные раковины органические растворители; их
собирают в бутыль, хранят в вытяжном шкафу до переработки
перегонкой или сжигают в топке котельной. Для сбора стоков,
подлежащих нейтрализации, дегазации или переработке, около
раковин и в вытяжном шкафу устанавливают глиняные
(фаянсовые) бачки вместимостью 10—15 л для раздельного сливания
отработанных растворов. Кислые стоки нейтрализуют карбонатом
натрия, а щелочные — соляной кислотой. В конце рабочего дня
обработанные стоки сливают в канализацию или, в соответствии с
местными условиями, в другое специально отведенное для этой цели место.
Газоснабжение
Для обеспечения работы газовых нагревательных приборов
и стеклодувных горелок используют горючий газ, а для
проведения работ в инертной газовой атмосфере — сжатый азот, гелий
и др. Снабжение горючим газом может осуществляться из общей
газовой сети или с помощью газовых редукторов из автономных
и коллекторных баллонных установок (рамп).
Обеспечение лаборатории газообразным азотом с избыточным
давлением до 0,1 МПа (1 кгс/см2) может осуществляться либо
из баллонов, либо от специальных рамп, к которым присоединяют
баллоны, оборудованные редуцирующими устройствами,
обеспечивающими понижение давления в системе до 0,1 МПа.
Газовая сеть для подачи газа в лабораторные помещения
должна монтироваться из стальных неоцинкованных труб, на
сварке. На вводе газопровода в здание лаборатории снаружи
должно быть установлено устройство для отключения газа.
На ответвлении газопровода от стояка в лабораторию должен
быть установлен бронзовый натяжной запорный кран,
позволяющий отключить подачу газа со стороны коридора. Кран должен
быть окрашен в' красный цвет.
Газопроводы в помещениях должны прокладываться открыто, в
местах, удобных для обслуживания и исключающих- возможность
13
их повреждения, и должны иметь краны, позволяющие включать
отдельные горелки.
Газовая подводка монтируется на санитарно-технических
панелях лабораторных шкафов и столов. Расстояние между отдельно
стоящей санитарно-технической панелью с газовой подводкой и
электрощитом должно быть не менее 500 мм.
Проверять герметичность газовой сети источником открытого
огня запрещается. Неплотности в вентиле баллона или редукторе
и просачивание через них газа обнаруживают, смачивая их
водным раствором мыла (по образованию пузырей в мыльной
пленке).
Присоединение к сети топливного газа резиновых трубок
разрешается только для переносных приборов — лабораторных
горелок и т. п.
Электротехнические устройства
Распределение электроэнергии в лабораторные помещения
должно осуществляться от распределительных щитов,
устанавливаемых открыто, в нишах. При этом должна быть обеспечена
возможность отключения питания каждого помещения
лаборатории (кроме освещения). Помимо этого, необходим один общий
рубильник для включения и выключения всей лабораторной сети.
На каждый лабораторный щиток подается переменный ток
380/220 или 220/127 В.
Электротехнические панели лабораторных столов и вытяжных
шкафов должны иметь подводку переменного (желательно и
постоянного) тока, а также защитные контуры заземления [50; 56,
с. 160].
В помещениях с повышенной пожароопасностью заземление
является обязательным при номинальном напряжении
электроустановки выше 30 В переменного и ПО В постоянного тока. Во
взрывоопасных помещениях заземление выполняется при всех
напряжениях тока.
Электропитание лаборатории с большим и регулярным
расходом постоянного тока целесообразно обеспечивать
централизованно соответствующими выпрямительными установками. При
периодическом использовании постоянного тока напряжением 2, 6, 12 и
24 В лучше пользоваться локальными выпрямителями переменного
тока или переносными аккумуляторными батареями.
Разводка проводов (в скрытом исполнении) осветительной и
силовой сети осуществляется в каждом отдельном случае с учетом
строительных и технологических особенностей лабораторных
помещений.
Приборы мощностью до 800 Вт можно включать в
штепсельные розетки; приборы, потребляющие большую мощность, следует
присоединять к распределительным щитам.
При работе с электрооборудованием и электроприборами необ«
ходимо соблюдать следующие правила.
14
1. Работа должна производиться с исправным
электрооборудованием; неисправности может устранять только специалист-
электрик.
2. Нельзя переносить с места на место включенные в
электросеть приборы, а также ремонтировать электрооборудование,
находящееся под током.
3. В случае перерыва в подаче тока все электроприборы
следует немедленно выключить.
4. Все провода от электроплиток и других электроприборов
должны быть заключены в резиновые трубки.
5. В лабораторных помещениях, где проводятся работы с
горючими и легковоспламеняющимися жидкостями, допускается
применять электронагревательные приборы только с закрытым
нагревателем.
Вентиляция [56, с. 138]
Вентиляция лабораторных помещений предусматривается при-
точно-вытяжная, с механическим побуждением и должна быть
оборудована вентиляционными устройствами для отсоса воздуха
только из вытяжных шкафов. Воздухообмен в лабораторном
помещении должен быть рассчитан таким образом, чтобы
фактические концентрации ядовитых газов, паров и пыли в воздухе не
превышали предельно допустимые концентрации.
В помещениях, где производится работа с особо вредными и
ядовитыми веществами, вентиляционная система должна быть
индивидуальной, не связанной с вентиляцией других помещений.
Кратно'сть воздухообмена — отношение объема воздуха,
подаваемого в помещение или удаляемого из него за 1 час, к объему
помещения, при сохранении комнатной температуры — должна
быть в пределах 4—6. При работе с сильнодействующими
ядовитыми веществами кратность воздухообмена должна быть не менее
10. Наименьшая кратность воздухообмена требуется для весовых
комнат.
Постоянный и эффективный обмен воздуха — обязательное
условие работы в любой химической лаборатории. Это важно не
только в санитарно-гигиеническом отношении, но и в
противопожарном.
УСТАНОВОЧНОЕ ЛАБОРАТОРНОЕ ОБОРУДОВАНИЕ
(ЛАБОРАТОРНАЯ МЕБЕЛЬ)
Выбор размеров основной лабораторной мебели обусловлен
рациональным планированием и площадью лабораторных
помещений, а также профилем работы лаборатории.
Мебель, как правило, расставляется вдоль поперечных стен
помещения в последовательности: вытяжной шкаф, моечная
раковина, пристенный химический стол. При ширине помещения 6 м и
более в середине его обычно устанавливают островные столы. При
15
этом следует учитывать, что проходы между оборудованием,
стенами помещения и мебелью должны составлять не менее 1400 мм.
Существующее лабораторное установочное оборудование
отличается большим разнообразием. Многие организации изготовляют
его самостоятельно.
Разработка типового лабораторного установочного
оборудования в нашей стране осуществляется в основном ГИПРОНИИ АН
СССР [42].
В апреле 1977 г. Госкомитет по гражданскому строительству
и архитектуре при Госстрое СССР утвердил описание новых
унифицированных типовых наборов мебели для оборудования
химических и физических лабораторий ТО 13-88—77 [63]. Все изделия
комплектуются из отдельных конструктивных элементов (сантех-
панелей, выкатных тумб, электрощитов, вентиляционных узлов,
каркасов и т. п.), и это обеспечивает оптимальное использование
площади лабораторных помещений.
В химических лабораториях используются лабораторные
столы двух типов: пристенные и островные [46, с. 12]. Большей
частью они комплектуются коммуникациями для подвода
холодной (а часто и горячей) воды, бытового или баллонного газа,
сжатого азота (а часто и сжатого воздуха) и электрошкафом.
Для стока воды в столах устанавливаются сливные раковины,
а у торцов — моечные раковины.
Поверхности рабочих столов, в зависимости от
соприкасающейся с ними среды, покрывают линолеумом или листами из
пластиков, обладающих высокой химической и термической стойкостью
(асбовиниловые плиты, пластины из фторопласта-4 и др.)-
Наименее химически и термически устойчив линолеум, который
разрушается концентрированными кислотами и растворами щелочей,
а также органическими растворителями. Однако периодическое
натирание линолеума специальной пастой значительно повышает
его устойчивость. Предложен ряд щелоче- и кислотоустойчивых
покрытий: рецепт одного из них приводится в приложении 3.
Столы, на которых производится нагревание открытым огнем
и разгонка нефтепродуктов, должны быть покрыты несгораемым
материалом.
В унифицированный набор типовой лабораторной мебели [63]
включены три типа пристенных столов (СТХ-1, -2, -3) и один
составной островной стол (СТХ-4). Пристенные столы
комплектуются из односторонней санитарно-технической панели,
приставного стола и выкатных тумб. Панель имеет крышку со
встроенными лабораторными раковинами, надстройку с электрощитами и
полками.
Стол СТХ-1 комплектуется одной выкатной тумбой с
мусоросборником и одной раковиной; СТХ-2 — двумя тумбами и двумя
раковинами; СТХ-3 — тремя тумбами и тремя раковинами.
Стол СТХ-4 комплектуется из двухсторонней
санитарно-технической панели, к которой примыкают один стол СТХ-1 и один
стол СТХ-2 по торцам и два стола СТХ-3 по фронту.
16
Лабораторный рабочий стол необходимо содержать в чистоте
и не загромождать рабочую поверхность ненужными в данный
момент приборами, реактивами и посудой.
Если стол покрыт линолеумом, необходимо следить, чтобы на
него не попадали кислоты, щелочи, органические растворители
и т. п., и периодически натирать линолеум мастикой. Под склянки
с веществами, разрушающими линолеум, рекомендуется класть
стеклянные пластины, метлахские или асбестоцементные плитки.
На стол, покрытый линолеумом, не следует ставить горячие
предметы и электронагревательные приборы без
термоизолирующих подставок.
Ящики лабораторного стола следует рационально распределить
для раздельного хранения металлических и стеклянных
предметов, измерительных приборов, фильтровальной бумаги и фильтров,
мелкого лабораторного инструментария и т. п. В тумбах удобно
размещать стеклянную посуду. Во всех случаях дно ящика и
тумбы следует выстилать чистой белой бумагой, а для
стеклянных изделий — еще и ватой. К каждому ящику стола
рекомендуется прикреплять табличку с указанием назначения ящика.
Около рабочих столов и раковин непременно устанавливают
глиняные баки вместимостью 10—15 л для сливания
отработанных растворов, а также корзины для битого стекла, бумаги и
прочего сухого мусора.
По окончании работы, прежде чем уйти из лаборатории,
сотрудник обязан привести в порядок свой рабочий стол.
Устанавливаемые в лабораторных помещениях вытяжные
шкафы [56, с. 138] для работы с вредными веществами должны
иметь верхний и нижний отсосы; объем воздуха, удаляемого через
нижний отсос, должен быть в пределах 30% от общего объема
воздуха, удаляемого через рабочий проем.
Рабочим проемом для вытяжных шкафов следует считать
проем одной дверцы, открытый на высоту ^ 0,40 м, с расчетной
площадью около 0,4 м 2. Для шкафов длиной до 2 м, с одной или
двумя дверцами, за рабочий проем принимают открытый проем,
образуемый одной подвижной дверцей, а для шкафов с тремя и
четырьмя дверцами — открытый проем, образуемый двумя
подвижными дверцами, с площадью <; 0,6 м 2.
Скорость воздуха v в рабочем проеме вытяжных шкафов
должна определяться в зависимости от предельно допустимой
концентрации (ПДК) веществ, с которыми производится работа:
ПДК. мг/мЗ vt м/с
10 0,5
10-0,1 0,7
0,1 1
Работа с токсическими веществами с ПДК < 0,1 мг/м3 должна
осуществляться в специальных вытяжных шкафах с
индивидуальной вытяжной системой*
Скорость потока воздуха в вытяжном шкафу обычно измеряют
ручным крыльчатым анемометром АСО-3.- Он отличается большой
чувствительностью и рассчитан на измерение скорости
воздушного потока порядка 0,3—5 м/с; продолжительность наблюдения
1—2 мин. К прибору прилагаются два графика, с помощью
которых можно, зная разность между конечным и начальным
показаниями стрелок и частное от деления ее на число секунд
наблюдения, определить искомую скорость воздушного потока в метрах на
секунду.
Вытяжные шкафы обеспечиваются коммуникациями для
подвода водопроводной воды, бытового или баллонного газа, сжатого
газа, электроэнергии; для стока воды устанавливают раковины.
Рабочая поверхность щитового химического
вытяжного шкафа [42] покрыта метлахской плиткой. Задняя стенка
двойная из асбестоцементной плиты, облицованная белой
керамической глазурованной плиткой. Шкаф рассчитан на верхний и
нижний отсосы, имеет два отделения для хранения лабораторного
имущества. Два светильника, установленные внутри шкафа,
изготовлены во взрывозащищенном исполнении. Выключатели и
штепсельные розетки расположены вне шкафа.
В унифицированный набор мебели [63] включены тр^типа
шкафов. Они состоят из нижней секции (основания), верхней
секции (рабочей камеры), вентиляционного присоединительного узла,
санитарно-технической панели, выкатных тумб для приборов и
материалов и электрощита.
Воздуховод расположен в задней стенке; к магистральному
воздуховоду шкаф присоединяется через универсальный
вентиляционный узел. В рабочую крышку с покрытием из шлакоситалла
или керамической плитки встроены лабораторные раковины; на
крышке установлена арматура для сантехподводок, управляемая
дистанционно с передней панели, кроме подводок воздуха и
вакуума. Створки рабочей камеры поднимаются вручную,
бесступенчато, с помощью подвесов, скрытых в задней стенке шкафа. На
задней стенке имеются кронштейны для крепления штативной решетки.
Внутренняя облицовка шкафа из стеклотекстолита. Основание
имеет регулируемые по высоте подпятники.
При работе окна вытяжного шкафа поднимают на высоту,
удобную для работы, но не более чем на 7з- Особо опасные
газойли парообразные продукты, образующиеся в процессе
проведения реакции, если они не используются в работе, обязательно
поглощают соответствующими поглотителями (или вымораживают
на выходе из приборов — см. гл. 18). Вытяжные шкафы следует
поддерживать в полной исправности. Пользоваться вытяжными
шкафами с разбитыми стеклами запрещается. Загромождать
вытяжные шкафы посудой, приборами и лабораторным
оборудованием, не связанным с проводимой в данном случае работой, не
разрешается.
Когда в вытяжных шкафах не проводится работа, их дверцы
должны быть опущены до конца.
18
Для оснащения весовых комнат служат столы для
микроаналитических весов на массивных ножках и
консольные большие столы для аналитических весов [46,
с. 21].
Унифицированный набор лабораторной мебели [63] включает
стол для аналитических весов, состоящий из каркаса, рабочей
крышки, встроенной массивной плиты и ящика для разновесов.
Каркас стола выполнен из стальных прямоугольных труб, которые
для увеличения массы забиты металлической дробью. Плита стола
укладывается на песчаную постель. Несимметричное расположение
плиты для установки весов обеспечивает с правой стороны
рабочей крышки место для производства записей.
Для титрования существуют столы с цельным
подстольем, состоящим из столешницы с крышкой и двумя выдвижными
ящиками и подстолья из двух отделений, с надстройкой для пяти
люминесцентных ламп [46, с. 24]. Люминесцентные лампы
расположены горизонтально за матовыми стеклами, что обеспечивает
равномерное распределение света, хорошее просвечивание сосудов
для титрования и безупречный отсчет уровня жидкости по шкале
бюретки. Запасные бутылки с растворами находятся в
защищенном от света шкафу стола. С помощью сжатого воздуха растворы
могут передавливаться в бюретки с автоматически
устанавливаемой нулевой точкой. На рабочем месте расположена магнитная
мешалка для перемешивания титруемого раствора.
Весьма удобная установка для титрования растворов,
требующих эффективного перемешивания и подогрева (УТ),
разработана СКТБСП (г. Клин) [53, с. 237].
ПОЖАРООПАСНОСТЬ И СРЕДСТВА ПОЖАРОТУШЕНИЯ
В химических лабораториях возникает опасность пожара при
несоблюдении мер предосторожности, от неисправности
нагревательных приборов, газопроводов и электропроводки. Часто
происходят возгорания в результате нарушения правил работы с
огнеопасными веществами.
Все работающие в химической лаборатории должны знать
возможные причины возникновения пожаров, способы тушения
различных загораний и владеть первичными средствами огнету-
шения. В коридоре и лабораторном помещении на определенных
местах должны висеть готовые к действию огнетушители. Каждый
сотрудник лаборатории должен знать место расположения
ближайшего аварийного душа, пожарного крана и уметь привести в
действие брандсбойт и огнетушитель.
Наибольшая опасность возникает при проливании на пол
значительных количеств горючих жидкостей, так как при их загорании
обслуживающий персонал и аппаратура мгновенно могут быть
охвачены пламенем. Весьма опасно проливание жидкого
кислорода на масляные поверхности, что влечет за собой загорание со
взрывом.
19
Немалую опасность представляют собой находящиеся в
рабочих помещениях баллоны со сжатыми и сжиженными газами,
которые при сильном нагревании, вследствие повышения давления,
могут взорваться. Поэтому, если очаг загорания в помещении
лаборатории ликвидировать первичными средствами огнетушения
не удается, необходимо пожарной сигнализацией или по телефону
известить пожарную охрану; немедленно выключить газовые
горелки, электронагревательные приборы и вентиляцию; вынести из
помещения все сосуды с огнеопасными веществами и баллоны с
газами; если нет возможности вынести баллоны и быстро
ликвидировать огонь, баллоны следует охлаждать струей воды.
Выбор первичных средств огнетушения определяется природой
воспламеняющегося материала и характером воздействия средств
огнетушения на оборудование и аппаратуру. В каждом помещении
должна быть вывешена инструкция по пожаротушению в
соответствии со спецификой работ в данной лаборатории.
Кошма представляет собой грубошерстное или асбестовое
полотнище; ее подвешивают в свернутом виде в определенном
заметном и доступном месте. Кошмы применяют в случае малой
площади горения.
Сухой песок обычно применяют при загорании небольших
количеств горючих и легковоспламеняющихся жидкостей. Рядом
с ящиком для песка должна находиться лопата. Песок следует
время от времени перемешивать, чтобы он не слеживался.
Вода может служить средством огнетушения при
воспламенении жидкостей, смешивающихся с водой. Воду нельзя применять
для тушения горящих жидкостей, не смешивающихся с водой,
например бензола, эфира. Горение при этом не только не будет лик*
видировано, но даже может усилиться. Многие огнеопасные
органические вещества легче воды, и при соприкосновении с ней
образуют тонкую горящую пленку. При тушении водой площадь
горящей пленки возрастает, и тем опаснее становится пожар.
Огнегасительные свойства воды можно повысить, используя ее
в виде 5—20% раствора MgCl2-6H20, насыщенных растворов
NaCl, CaCl2, Na2C03 и др. Солевые растворы сохраняют в бутылях,
устанавливаемых в рабочем помещении, в определенном и
известном всем работающим месте.
Для улучшения смачиваемости в воду и водные солевые
растворы добавляют ~ 1 % поверхностно-активных веществ (ПАВ).
Водой нельзя тушить электроустановки и электропроводку,
находящиеся под напряжением, а также вещества, способные
вступать с водой в химическую реакцию (щелочные металлы, гидриды
металлов, карбид кальция и др.).
В химических лабораториях используются ручные
огнетушители: пенные, углекислотные и с огнегасительными составами
на основе галогензамещенных углеводородов.
При тушении горючих, в том числе легковоспламеняющихся
веществ особенно эффективны пены — дисперсные системы, в
которых пузырьки газа (С02, воздух) заключены в тонкие оболочки
20
негорючей жидкости (водные растворы солей, кислот). Чтобы об*
разующаяся пена была устойчива во времени, в жидкость, из
которой она образуется, вводят ПАВ.
Пенные огнетушители ОП-5 и ОП-М выпускаются
с массой заряда около 10 кг и временем выхода заряда 55—70 с.
Масса огнетушителей с зарядом 14,5—15,0 кг. Пенные
огнетушители работают очень энергично, но вся лабораторная аппаратура
оказывается при этом забрызганной пеной, вызывающей коррозию;
кроме того, перед использованием необходимо отключить
электрический ток во всем помещении.
Углекислотные огнетушители предназначены для
тушения загораний различных веществ и электроустановок,
находящихся под напряжением не более 1000 В. Имеются огнетушители
типов ОУ-2, ОУ-5, ОУ-8 на 2, 5 и 8 л соответственно. В химических
лабораториях большей частью используется огнетушитель ОУ-5
в настенном исполнении. Продолжительность выпуска заряда
(3,5 кг СОг) около 15 с; длина снежной струи С02 4,5 м; масса
огнетушителя с зарядом 13,5 кг. С помощью огнетушителя типа ОУ
тушение ведут снизу вверх. Образующийся иней С02 не повреждает
аппаратуру, и тушение можно производить не выключая
электрического тока.
Огнетушители с о г н ег а с и тел ьн ы м и составами
на основе бромистого метила ОУБ-3 и ОУБ-6 с содержанием доЗ%
жидкого С02 в 3,5 раза эффективнее углекислотных (с зарядом
такой же массы). Рабочее давление в баллоне огнетушителя
создается сжатым воздухом.
В последнее время появились огнетушители на основе метил-
бромида в сочетании с метиленбромидом («состав 7») и тетрафтор-
бромэтаном (состав «ЖБ»), используемые для тушения самых раз-*
личных загораний.
ТУШЕНИЕ ГОРЯЩЕЙ ОДЕЖДЫ
При возгорании одежды главное —не теряться. Чтобы
потушить на себе пламя, можно обливаться водой или, отбежав от
очага загорания, быстро лечь на пол и, перекатываясь по полу,
погасить горящую одежду. Часто при загорании одежды, человек
сопротивляется попытке оказать ему помощь. В таких-случаях
следует его немедленно повалить на пол и накрыть одеялом или
кошмой, которые не снимают до тех пор, пока не погаснет пламя.
СРЕДСТВА ИНДИВИДУАЛЬНОЙ ЗАЩИТЫ [32; 56, с. 273]
Все работающие в химической лаборатории должны быть
обеспечены средствами индивидуальной защиты постоянного
пользования (халаты, предохранительные перчатки, очки или маски) и
аварийного пользования (противогазы)»
21
Работа в лаборатории должна производиться в халатах из
хлопчатобумажной ткани, застегивающихся спереди. Такой халат
легко сбросить в случае необходимости.
В качестве средств защиты органов дыхания при аварийной
ситуации служат промышленные фильтрующие и изолирующие
противогазы.
Промышленные фильтрующие противогазы
являются средствами защиты органоц дыхания, глаз и кожи лица от
воздействия вредных веществ, содержащихся в воздухе в виде
паров и аэрозолей (пыли, дыма, тумана). В комплект противогаза
входит фильтрующая коробка, резиновая лицевая часть (шлем-
маска) с гофрированной трубкой и сумка.
Размер шлем-маски обозначен на подбородочной части.
Производят два обмера: длину круговой линии, проходящей по
подбородку, щекам и через высшую точку головы, и длину
полуокружности, проходящей от отверстия одного уха к отверстию другого
по лбу через надбровные дуги. Оба результата складывают и
определяют размер шлем-маски:
Суммарный результат Размер
обмеров, см шлем-маски
До 93 О
93-95 1
95-99 2
99—103 3
Более 103 4
Правильно подобранный противогаз (маска нужного номера
и фильтрующая коробка, предназначенная для данной отравленной
среды) защищает органы дыхания при содержании в
воздухе ^18% кислорода и ^0,5% вредных газов [56, с. 297].
Ориентировочные сроки защитного действия фильтрующих коробок
(40—360 мин.) приводятся в прилагаемой к ним инструкции.
Кисло родн о-и золирующие противогазы полностью
изолируют органы дыхания от окружающей среды. Поэтому их
можно применять при недостатке кислорода во вдыхаемом воздухе,
значительных концентрациях вредных веществ и неизвестном их
составе.
Учитывая значительную массу (8—10 кг) и относительную
сложность конструкции изолирующих противогазов, к работе с ними
допускаются лица, признанные медицинской комиссией пригодными
и прошедшие курс обучения.
Для защиты глаз от механических повреждений применяют
открытые очки и маски с прозрачным экраном.
Открытые очки типа ОЗО удобны тем, что имеют широкое
поле зрения, не запотевают и допускают возможность замены
обычных стекол корригирующими.
В защитной маске С-40 экран из органического стекла
удерживается на изголовнике в двух фиксированных положениях:
опущенном (рабочем) и откинутом на 90 ±5°, Масса маски 250 г.
22
От ультрафиолетового излучения глаза защищают очками С-14
(выпускаются с расстоянием между центрами 64, 68 и 72 мм).
Для защиты глаз от электромагнитных длинноволновых
излучений служат о.чки ОРЗ-5 с металлизированными стеклами.
Для защиты рук от агрессивных веществ служат перчатки
резиновые кислотощелочестойкие, резиновые анатомические и рент-
генозащитные (выпускаются размеров 1, 2 и 3).
ПЕРВАЯ ПОМОЩЬ [56, с. 181]
При работе в химической лаборатории наиболее вероятными
несчастными случаями являются порезы стеклом, ожоги
термические и химические, а также ингаляционные поражения парами
токсических веществ.
Доврачебная помощь должна быть оказана пострадавшему
товарищами по работе.
При порезах рук стеклом в первую очередь необходимо
пинцетом, промытым спиртом, удалить из раны видимые осколки
стекла, затем промыть рану 2% раствором лерманганата калия и,
смазав рану 5% раствором иода, забинтовать.
Причинами термических ожогов могут быть
прикосновение к сильно нагретым предметам, воспламенение горючих
жидкостей и сильный электрический разряд. При термических ожогах
рекомендуется вначале делать примочки из 2% раствора перман-
ганата калия или этилового спирта (96%), а затем смазать
обожженный участок мазью от ожогов и наложить повязку.
При химических ожогах кожи необходимо прежде всего
удалить вызвавшее ожог вещество соответствующим
растворителем, а затем пораженный участок обработать этиловым
спиртом.
При ожогах кислотами и фенолом обожженное место обильно
промывают проточной водой, а затем 2% раствором NaHC03; при
ожогах щелочами после обильной промывки проточной водой
промывают обожженное место 2% раствором уксусной или борной
кислоты. При попадании на кожу агрессивных органических веществ
пораженный участок следует быстро промыть этиловым спиртом
(96%), а затем смазать мазью от ожогов.
При химических ожогах глаз необходимо до
обращения в медпункт промыть пострадавшему глаза вначале большим
объемом воды, затем 2% раствором NaHC03 (при попадании
кислоты) или 2% раствором борной кислоты (при попадании
щелочи).
При ингаляционных поражениях пострадавшего
необходимо немедленно вывести (вынести) на свежий воздух,
освободить от стягивающей одежды, создать ему абсолютный покой,
положить на спину, тепло укутать и вызвать врача.
При поражении электрическим током, если
пострадавший остается в соприкосновении с токоведущими частями,
необходимо немедленно выключить ток при помощи пускателя, либо
23
вывернуть предохранительную пробку, или перерубить токопрово-
дящий провод изолированным инструментом. К пострадавшему,
пока он находится под током, нельзя прикасаться незащищенными
руками (без резиновых перчаток). Если пострадавший потерял
сознание, после отключения тока нужно немедленно, не дожидаясь
прибытия врача, применить искусственное дыхание.
Во всех лабораториях в легко доступном постоянном месте
должны быть аптечки с набором необходимых материалов и
медикаментов. Все работающие в лаборатории должны быть обучены
способам оказания первой медицинской помощи. В аптечке
лаборатории должны быть стерильные бинты и вата, 5% спиртовый
раствор иода, 2% раствор гидрокарбоната натрия; мазь от ожогов,
лейкопластырь, 2% раствор перманганата калия, 2% раствор
уксусной кислоты, 2% раствор борной кислоты, этиловый спирт, глазные
пипетки, пинцеты, стеклянные палочки и ножницы.
ОБЩИЕ ПРАВИЛА РАБОТЫ В ХИМИЧЕСКИХ ЛАБОРАТОРИЯХ
[50; 56, с. 136]
К работе в химической лаборатории могут быть допущены
только лица, прошедшие полный инструктаж и обученные
безопасным методам работы. Инструктируемый должен изучить свойства
важнейших химических веществ, с которыми он будет работать;
обращение с лабораторной посудой и основными приборами;
возможные вредности; средства профилактики отравлений и первой
помощи; противопожарный инвентарь и правила пользования им.
Ответственным за соблюдение правил техники безопасности и
пожарной безопасности по лаборатории в целом является
заведующий лабораторией, а по отдельным участкам — руководители работ.
По всем работам, проводимым в лаборатории, заведующий
лабораторией обязан разработать подробные инструкции по технике
безопасности, которые должны находиться на рабочих местах во
всех помещениях лаборатории.
В каждом помещении лаборатории должна быть вывешена
надпись с фамилией сотрудника, ответственного за соблюдение правил
техники безопасности и пожарной безопасности.
При вечерней работе в помещении лаборатории должно быть
не менее двух сотрудников.
В здании лаборатории разрешается хранить запас
легковоспламеняющихся и горючих жидкостей и газов, не превышающий
суточной потребности каждого вида веществ. Хранят этот запас в
специальном помещении (кладовой) или в специальных
металлических ящиках, находящихся в помещении лаборатории.
При работах, связанных с огневым или электрическим нагревом
веществ, оставлять рабочее место без присмотра не разрешается.
При необходимости отлучки работника, даже на
непродолжительное время, источник нагрева должен быть выключен.
Сдавать в мойку посуду из-под концентрированных кислот,
едких и ядовитых продуктов можно только после ее полного освобож-
24
дения и нейтрализации. Эти операции проделывает
непосредственно работник, пользовавшийся посудой.
Хранение и выдача взрывчатых веществ должны производиться
в соответствии со специальными правилами.
Работать с любым прибором разрешается лишь после
детального ознакомления с техническим описанием и инструкцией. Нужно
всегда помнить, что надежность работы прибора и срок его
эксплуатации зависят от правильного его использования.
По окончании работ в лаборатории ответственный сотрудник
обязан убедиться в том, что газовые и водяные краны и общий
вентиль газа в лаборатории закрыты; газовые горелки и другие
огневые приборы потушены; силовая электросеть выключена;
склянки и банки с реактивами и материалами закрыты пробками;
освещение и вентиляция выключены.
ХИМИКО-ЛАБОРАТОРНАЯ ПОСУДА
ПОСУДА И ИЗДЕЛИЯ ИЗ СТЕКЛА [46, с. 30; 53]
Преобладающая часть лабораторных работ осуществляется
в посуде и приборах из специального тонкостенного или
толстостенного прозрачного стекла. Благодаря своей коррозионной стойкости,
твердости, прозрачности и сравнительно небольшому коэффициенту
линейного теплового расширения стекло является ценным
конструктивным материалом для изготовления лабораторной посуды,
приборов и аппаратов. Прозрачность стекла позволяет непосредственно
следить за ходом процесса в реакционном сосуде, а гладкость
поверхности стекла облегчает мытье посуды.
Недостатки стекла —его хрупкость, относительно малая
устойчивость к резким перепадам температуры, а порой и нестойкость
в отношении некоторых агрессивных химических веществ
(концентрированные щелочи, фосфорная, фтористоводородная кислоты и
др.). Почти нет стекол, которые в той или иной степени не
реагировали бы с водой и щелочами.
По составу, а следовательно, и химическим и
физико-химическим свойствам стекла весьма разнообразны. Свойства стекла
зависят кроме состава также от условий варки, формования
(выдувание, прессование, вытягивание и др.) и последующей термической
обработки [60, с. 10].
Основные требования, предъявляемые к лабораторной посуде
и изделиям из стекла — это' термическая и химическая стойкость.
Под термической стойкостью понимают способность
стекла выдерживать без разрушения резкие колебания
температуры. Максимальная разность температур, которую выдерживает
стекло, не разрушаясь, является величиной его термической
устойчивости. Термическая стойкость стеклянных сосудов зависит, в
частности, от толщины стенок. Так, например, термическая стойкость
изделий из чехословацкого стекла «симакс» при толщине стенки
сосуда 1 мм равна 312°С, при 3 мм — 180°, при 10 мм— 100V
По термостойкости стекла принято делить на группы, исходя
из их коэффициентов линейного теплового расширения, в интервале
температур 20—300°С.
Первая группа — стекла с коэффициентом теплового
расширения (70^-90)10"7К""1. К этой группе относятся стекла ХС1 марки
№ 23, тюрингенское (ГДР). Стекла этой группы сравнительно
легкоплавки и склонны к расстекловыванию. При длительном
прогревании в пламени газовой горелки стекло теряет прозрачность,
становится мутным, а по остывании — шероховатым на ощупь.
Расстекловывания можно избежать, если в пламя горелки внести
асбестовый тампон, смоченный насыщенным раствором NaCl.
Соль, оседая на размягченное стекло, возвращает ему
первоначальный вид.
26
Вторая группа—стекла с повышенной термостойкостью, коэф*
фициент теплового расширения которых лежит в пределах (50 -г-
-f- 65) 10~7КМ. К этой группе можно отнести молибденовые стекла,
ДГ-2 («Дружная горка-2»), «сиал» (ЧССР), «иенатерм» (ГДР).
Молибденовые стекла (ЗС-5 и др.) своим названием обязаны
свойству давать вакуумно-плотный спай с металлическим
молибденом. Химически они менее стойки, чем другие
химико-лабораторные стекла, но зато легко поддаются стеклодувной обработке.
Большую химическую и термическую стойкость проявляет стекло
ДГ-2, которое не расстекловывается при обработке в пламени
горелки и легко спаивается с молибденовыми стеклами, стеклами
«сиал» и «иенатерм». Однако «иенатерм» при стеклодувной
обработке необратимо мутнеет.
Третья группа — стекла с высокой термостойкостью и
коэффициентом теплового расширения (38 ч- 49) Ю^К"1. Это
высококремнеземистые малощелочные боросиликатные стекла «пирекс», ТС,
«симакс» (ЧССР), «разотерм» (ГДР).
Четвертая группа — особо высокотермостойкие стекла типа
кварцевого; их коэффициент теплового расширения (5 Ч-7) 10~7К_1.
Под химической стойкостью понимают способность
стекла противостоять разрушающему действию воды, кислот,
щелочей и других химических реагентов.
Химическую стойкость стекла определяют по ГОСТ 21400—75,
которым установлены классы гидролитической стойкости
(водостойкости), кислотостойкости и щелочестойкости стекол.
Согласно ГОСТ 21400—75, для лабораторной посуды,
приборов, аппаратов изготовляются следующие стекла:
ХС1 химически стойкое 1 класса
ХС2 » » 2 »
ХСЗ » » 3 »
ТХС1 термически и химически стойкое 1 класса
ТХС2 > » » 2 »
ТС термически стойкое
Из стекла ХСЗ марок АМК и AM изготовляют толстостенную
посуду, выдувные и прессованные изделия. Стекла других марок
служат для изготовления тонкостенной посуды, приборов,
аппаратов и другого стеклянного оборудования.
Стеклянные трубки изготовляют из стекла ХСЗ марки Л-80
и ХС2 марки № 29 и ТС («пирекс»).
Из химико-лабораторных стекол, производимых в
странах-членах СЭВ, следует отметить чехословацкие стекла «сиал» и
«симакс», немецкие (ГДР) «разотерм», «супремакс», С-20 и иенское
лабораторное стекло [53, с. 8].
Соединительные элементы
В лабораторной практике работа чаще всего начинается со
сборки установок и приборов. Для соединения стеклянных узлов
(а также для герметизации лабораторных сосудов) широко
27
Рис. 1. Нормальные шлифы:
л—муфты; б, в—керны.
Рис* 2. Сферические шлифы:
л —чаша; б —шар.
используются так называемые соединительные элементы на
стандартных взаимозаменяемых шлифах. Поэтому именно с них стоит
начать знакомство со стеклянными лабораторными изделиями.
Преимущества стеклянных соединений на шлифах
заключаются в том, что при работе с ними реакционная масса не
загрязняется, а при поломке поврежденная деталь легко заменяется.
Шлифы устойчивы к действию обычных химических реагентов,
легко уплотняются и позволяют в короткий срок осуществить
сборку разнообразных лабораторных установок.
Из соединительных элементов с взаимозаменяемыми
конусными и сферическими шлифами особо широкое применение нашли
переходы, изгибы, керны, муфты, алонжи и насадки.
За основу конусных (нармальных)
взаимозаменяемых шлифов для лабораторной аппаратуры и посуды взят
усеченный конус с конусностью 1 : 10. Внешнюю деталь называют
шлиф-муфтой, внутреннюю — шлиф-керном (рис. 1). Они
изготовляются со шлифованной (КШ) и нешлифованной (КН)
поверхностью. За размер шлифа принимается отношение наибольшего
диаметра (в мм) к высоте (в мм), например КШ 14/23.
Конусные соединения КШ имеют ряд недостатков:
шероховатая поверхность конуса легко загрязняется, они часто
заклиниваются (их «заедает»), требуют смазывания, а непрозрачность
конусов затрудняет наблюдение за процессом.
Конусные соединения КН прозрачны, обладают большей
механической прочностью, не заклиниваются, могут работать без
смазывания и меньше загрязняются.
Соединения на сферических шлифах (рис. 2)
представляют собой гибкие шарниры, позволяющие поворачивать
соединительные элементы прибора на угол до 20°.
Плотность соединения элементов прибора у сферических
шлифов, состоящих из шара и чаши, больше, чем у конусных. За
размер шлифа принимают диаметр сферы шлифа в миллиметрах.
Перед, началом работы, в процессе сборки прибора, шлифы
рекомендуется смазать равномерным тонким слоем вазелина или
вакуумной смазки. При работе с углеводородами шлиф лучше
28
смазывать силиконовым вазелином, который незначительно
растворим в органических растворителях, а также вязкими
веществами гидрофильного характера (этиленгликоль, полигликоли
или мыло). Смазанные поверхности прижимают друг к другу и
поворачивают несколько раз. При этом шлифы должны
соединяться без усилий (не прибегать к ввинчиванию).
Смазка не только повышает герметичность соединения, но и
облегчает вращение притертых поверхностей, предохраняет от
разъединения и заедания
Заедание может происходить в результате механического
вдавливания внутреннего шлифа во внешний при работе в
вакууме, длительной работы при повышенной температуре, действия
щелочей, кремнийорганических веществ, фосфорной кислоты и
ряда других причин.
«Заевшие» шлифы открывают:
а) механическим пошатыванием внутреннего шлифа;
б) осторожным постукиванием деревянным предметом по
шлифу;
в) нагреванием внешнего шлифа горячей водой, водяным
паром или слабым пламенем спиртовки (горячие шлифы надо
разбирать до их полного охлаждения).
Для разъединения «заклиненных» шлифов также рекомендуют
следующее: готовят раствор из 10 масс. ч. хлоралгидрата, 5 ч.
глицерина, 5 *ч. воды и 3 ч. конц. НС1. Раствор наносят на шлиф
или шлиф погружают в раствор и оставляют на некоторое время
с тем, чтобы раствор проник между притертыми поверхностями.
Рис. 3. Переходы:
а—с одной горловиной; б —с одной горловиной и отводом; в —с одной горловиной изогнутый-
г—с двумя параллельными горловинами; 0 — с двумя горловинами под углом: е—с тпрм^
параллельными горловинами. F я
29
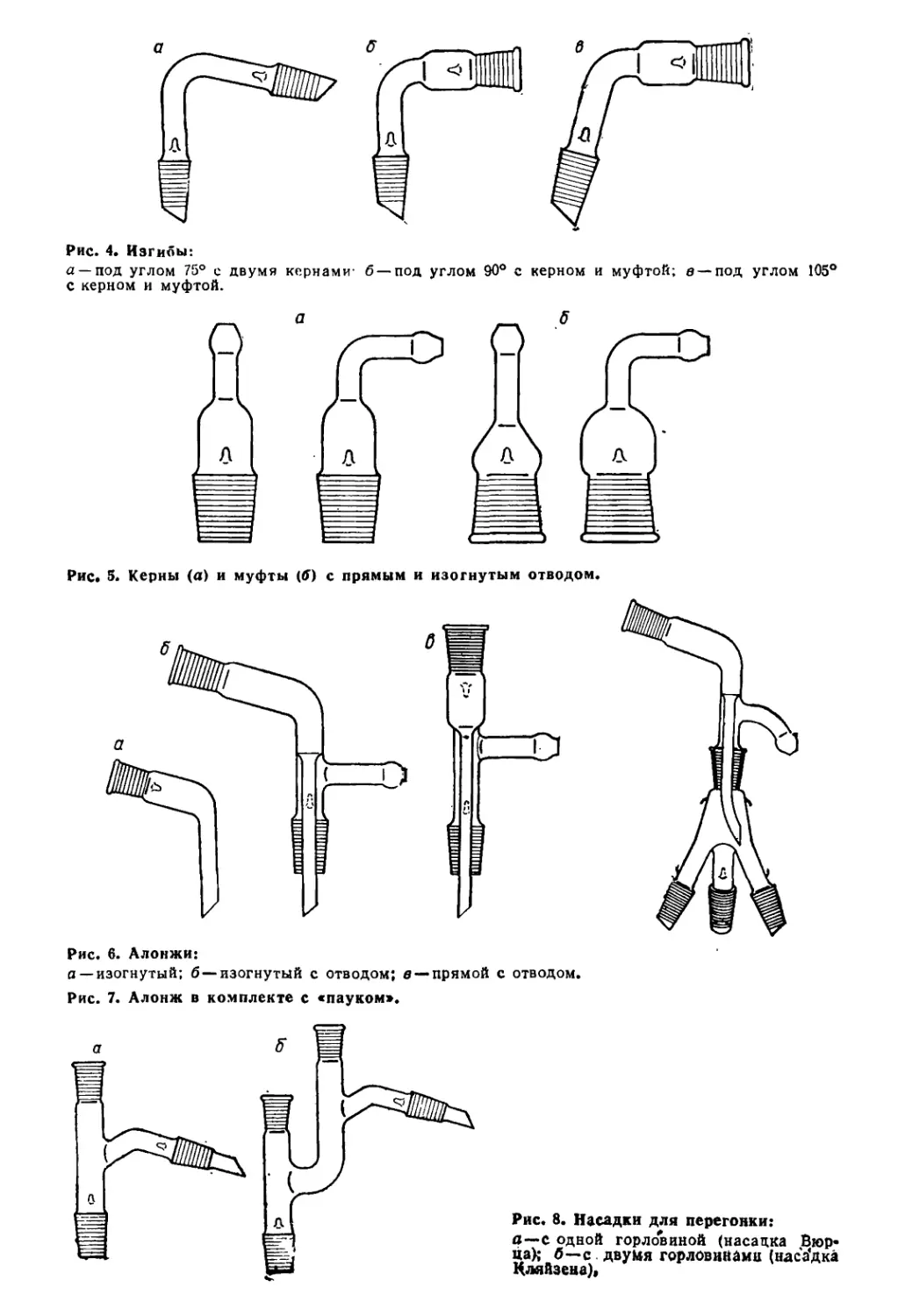
Рис. 4. Изгибы:
а — под углом 75° с двумя кернами: б —под углом 90° с керном и муфтой; в —под углом 105°
с керном и муфтой.
/
Рис. 5. Керны (а) и муфты (б) с прямым и изогнутым отводом.
ЙЩ^
£=о
Рис. 6. Алонжи:
а —изогнутый; б —изогнутый с отводом; в —прямой с отводом.
Рис. 7. Алонж в комплекте с «пауком».
\^Щ^
^Щц^
Рис. 8. Насадки для перегонки:
а—с одной горловиной (насадка Вюр-
ца); б—с двумя горловинами (наса'дка
Нляйзеиа),
Вакуумирование сосуда (прибора) с «заевшим» шлифом облегчает
проникновение раствора, и муфта легко отходит от керна.
При хранении соединительных элементов на шлифах в
собранном виде между шлифами помещают полоски тонкой бумаги,
чтобы предупредить заедание.
Для предохранения собранных конусных шлифов от
разъединения их рекомендуется стягивать металлическими пружинами,
укрепленными на обеих половинках шлифа с помощью
припаянных стеклянных крючков. Для скрепления соединений на
сферических шлифах служат специальные зажимы.
Переходы, прямые и изогнутые (рис. 3), применяются в
.процессе соединения деталей установок для перехода от одного
размера стандартного шлифа к другому.
Для соединения элементов лабораторных установок под
некоторым углом служат стеклянные изгибы под углом 75, 90 и
105°, (рис. 4).
Керны и муфты с прямыми и изогнутыми отводами (рис.5)
используются для соединения реакционных сосудов с сосудами
для поглощения или отвода газов в атмосферу.
Для транспортирования конденсата (жидкости) от
холодильника к приемнику при перегонке используются а л он ж и (рис.6).
У алонжа может быть отвод для присоединения поглотительного
сосуда или резинового шланга для отвода газа.
Соединение холодильника с одним или несколькими
приемниками при перегонке под вакуумом осуществляют с помощью
алонжа «паук» или алонжа в комплекте с «пауком» (рис. 7).
При перегонке веществ широко используются
дистилляций н н ы е насадки (рис. 8). Их выпускают двух типов: с
одной горловиной (насадки Вюрца) для перегонки веществ при
атмосферном давлении и насадки с двумя горловинами для
перегонки веществ под вакуумом (насадки Кляйзена).
Краны соединительные и вакуумные
Выпускаются четыре типа стеклянных соединительных
кранов общего назначения: одно-, двух- и трехходовые и
серповидные.
Необходимо следить за тем, чтобы пришлифованные
поверхности всегда были смазаны и пробка была укреплена резиновым
кольцом так, чтобы она не могла продольно перемещаться в
муфте.
Перед смазыванием кранов их части тщательно очищают
эфиром и слегка нагревают (например, рукой). Смазку двумя
тонкими кольцами наносят деревянной палочкой по окружности
середины верхней и нижней половины пробки. У сравнительно
больших кранов оба кольца смазки соединяют тонкими полосками
смазывающего вещества в местах, наиболее удаленных от
отверстия канала. Затем пробку вставляют в слегка нагретую втулку
так, Чтобы кран оказался открытым, затем с нажимом слегка
31
Рис. 0. Кран вакуумный плоский:
/ — корпус; 2—верхний диск; 3—ось; 4—-защелка; 5 —пружина.
Рис. 10. Крап с фторопластовой пробкой:
J-— муфта; 2—пробка; 5— шайба; 4—кольцо; 5—гайка.
поворачивают пробку то в одну, то в другую сторону, но не
настолько, чтобы кран оказался закрытым. Лишь после того, как
смазка крана распределится совершенно равномерно и между
пришлифованными поверхностями не будет ни одного пузырька
воздуха, пробку можно повертывать кругом.
Вакуумные конусные краны применяются в
различных вакуумных установках и приборах для соединения или
разобщения отдельных частей вакуумной системы.
Пробка крана может быть полой или сплошной. Конусные
вакуумные краны, впаянные в систему и смазанные вакуумной
смазкой, позволяют поддерживать вакуум до Ю-5 мм рт. ст.
Диаметр проходного отверстия крана не должен превышать
внутренний диаметр подсоединенных трубок. Иначе в теле
пробки образуются внутренние карманы, в которых скапливается
смазка и грязь.
Краны вакуумные плоские (фланцевые), одно-,
двух- и трехходовые с проходным отверстием до 5 мм, могут
работать без смазки, они не заклиниваются и позволяют получать
вакуум до 10~~5 мм рт. ст. Кран состоит из двух стеклянных
дисков, тщательно притертых друг к другу (рис. 9). Нижний диск
/ — корпус крана с припаянными трубками, верхний диск 2 —
ручка поворота крана. Оба диска соединяются между собой с
помощью оси 3, пружины 4 и защелки 5. В верхнем диске по
окружности расположены канавки. Величина проходного
отверстия в нижнем диске регулируется поворотом верхнего диска.
В последнее время распространение получили краны с
фторопластовыми (тефлоновыми) пробками (рис. 10).
Краны одно-, двух- и трехходовые с конусностью 1 : 10 и 1:5
применяются в бюретках, делительных зрронках и приборах, когда
требуется производить работу с агрессивными реагентами, а
применение смазки недопустимо. Они обладают значительной
прочностью, химической стойкостью, не требуют смазки, так как
фторопласт пробки действует как смазка.
Для сборки лабораторных установок используются также
трубки соединительные Т- и V-образные с наружным диаметром
25, 40 и 50 мм, а также переходные оливы с разными диаметрами.
32
Лабораторная посуда общего назначения
К стеклянной посуде общего назначения авторы условно
относят воронки для лабораторных работ, дефлегматоры,
капельницы, каплеуловители, колбы, пробирки, промывалки,
холодильники, стаканы лабораторные, стаканчики для взвешивания, хлор-
кальциевые трубки, эксикаторы, чашки стеклянные. Другие виды
стеклянной посуды, приборы и оборудование из стекла
приводятся при описании общих и частных приемов лабораторной работы.
Воронки для лабораторных работ. Стеклянные воронки общего
назначения бывают лабораторные, делительные, капельные и для
фильтрования.
Лабораторные воронки (рис. 11), часто называемые
химическими или простыми воронками, конусообразной формы
(угол конуса 60°), со срезанным длинным концом, служат для
переливания жидкостей из сосуда в сосуд, для фильтрования при
помощи вкладного фильтра, для переноса порошков в колбы и т. д.
Воронки изготовляют из стекла групп ХС2 и ХСЗ.
При переливании жидкости в колбу или бутыль воронку не
следует наполнять до краев. Если воронка плотно прилегает к
горлу сосуда, то переливание затрудняется. В таких случаях
рекомендуется между воронкой и горлом сосуда вложить полоску
бумаги.
Делительные воронки предназначаются для разделения
несмешивающнхся жидкостей при экстрагировании. Они бывают
цилиндрические, конические, грушевидные и шаровидные, негра-
дуированные, градуированные и с боковой трубкой для
выравнивания давления (рис. 12). Все они снабжены притертым
стеклянным спускным краном.
Рис. П. Лабораторные воронки.
Рис. 12. Делительные воронки.
а—цилиндрическая; б—коническая; в—грушевидная; г—с боковой трубкой для
выравнивания давления и шлифами.
2 Зак. 1237 33
Nfk
Рис. 13. Дефлегматоры:
а, б—шариковые; в — елочный; г—с
садкой.
Цилиндрические делительные
воронки выпускаются с
пришлифованными пробкой и стеблем
(спускной трубкой), а
грушевидные и шаровидные — с
пришлифованной пробкой.
В конической делительной
воронке лучше видна граница
раздела жидких фаз. Перед началом
работы герметичность крана
следует проверить наливанием воды или эфира в воронку; при
недостаточной герметичности кран притирают.
Смазывать спускной кран уплотняющими смазками едва ли
целесообразно. Многие экспериментаторы считают, что это
улучшает герметичность только тех кранов, которые хорошо
держат сами по себе. Рано или поздно смазка вымывается
растворителем.
Спускной кран делительной воронки рекомендуется
привязывать или притягивать к корпусу воронки резиновым кольцом.
Полезно также пронумеровать воронки и соответствующие им
стеклянные пришлифованные пробки или привязать пробки к
горлышкам воронок.
Заменять стеклянные пришлифованные пробки резиновыми или
корковыми допустимо лишь в исключительных случаях. При этом
часть пробки, соприкасающуюся с жидкостью в воронке,
необходимо обертывать алюминиевой фольгой.
При работе небольшие делительные воронки осторожно укреп*
ляют в лапке лабораторного металлического штатива, а большие
помещают между двумя кольцами лабораторного штатива, при
этом нижняя часть воронки должна опираться на кольцо, диаметр
которого несколько меньше диаметра воронки, а диаметр
верхнего кольца должен быть несколько больше внешнего диаметра
воронки. Кольца штатива следует обернуть асбестовым шнуром или
надеть на них резиновую трубку.
При заполнении делительной воронки объем разделяемых
жидкостей не должен превышать 2/3 ее общей вместимости.
Капельные воронки цилиндрической формы с
пришлифованными пробкой и стеблем предназначаются для приливания
жидкости в реакционный сосуд небольшими порциями или по
каплям.
В отличие от делительных, капельные воронки изготовляются
из тонкостенного стекла и снабжены более длинным стеблем.
В него подаваемая жидкость поступает из спускного крана через
хорошо видимую узкую короткую трубку. Благодаря этому легко
наблюдать за скоростью подачи жидкости.
34
Часто давление в сосуде, куда добавляется жидкость из
капельной воронки, становится больше атмосферного, и поступление
жидкости из воронки прекращается. Равномерное поступление
жидкости обеспечивается воронкой с боковой трубкой для
выравнивания давления.
Капельные воронки обычно составляют часть прибора.
Поэтому их укрепляют в горле колбы на шлифе. Перед работой шлиф
крана и шлиф керна осторожно смазывают смазкой. Кран должен
открываться легко и без усилия, чтобы смазка не попадала в
трубку воронки или внутрь отверстия крана.
Воронки для фильтрования отличаются от описанных
выше лабораторных удлиненным срезанным концом спускной
трубки, внутренний диаметр которого в верхней части меньше,
чем в нижней, что приводит к ускорению процесса фильтрования.
Они предназначаются в основном для аналитических целей.
Дефлегматоры. Дефлегматоры, или насадки для
дистилляции, применяют при последовательной дробной
(фракционной) перегонке смеси жидкостей, температуры кипения которых
сильно (на 40—50 °С) разнятся между собой. Они бывают самых
разнообразных форм и размеров: елочные, шариковые,
цилиндрические с насадкой (рис. 13). Из них наиболее употребительный —
елочный дефлегматор на шлифах.
Елочные дефлегматоры имеют по окружности трубки
«елочку», т. е. чередующиеся по рядам в шахматном порядке
конусные несквозные наколы по четыре в ряду, расположенные к
оси трубки под углом 30°. Разделяющая способность обычного
елочного дефлегматора невелика; она возрастает с увеличением
длины «елочки».
Елочный дефлегматор шлифованным керном соединяют со
шлиф-муфтой перегонной колбы и осторожно укрепляют в лапке
штатива. Муфту дефлегматора соединяют с насадкой Вюрца (с
одной горловиной), отводную трубку которой соединяют с
холодильником. В муфту насадки вставляют термометр с
пришлифованным керном.
В цилиндрических дефлегматорах с насадкой
широкую часть заполняют стеклянными бусами, кольцами и
спиралями из материалов, на которые пары перегоняющейся жидкости
не действуют. Это зна- а j&gs.
чительно увеличивает Ша»
поверхность охлажде- «8888т
ния и тем самым
способствует лучшему
разделению
перегоняющейся смеси веществ.
Рис. 14. Капельницы:
а—для многократной дозировки
(по Манну); б —с клювиком;
в—для однократной дозировки^
2*
Рис. 15. Каплеуловитсли:
л—с прямой трубкой; б—с отводной трубкой под углом 60°; в—с отводной трубкой под
углом 100°; г—то же, что б, но без шлифов.
При работе с дефлегматорами следует соблюдать
осторожность, так как они легко ломаются.
Капельницы. Для подачи жидкости, например растворов
индикаторов, каплями пользуются капельницами (рис. 14).
Капельницу можно изготовить самому. Для этого к склянке
вместимостью до 50 мл подбирают резиновую пробку, в которую
вставляют вытянутую из стеклянной трубки пипетку. Суженная
часть пипетки должна доходить почти до дна склянки и иметь
внутренний диаметр 1—2 мм. Над пробкой пипетка должна
выступать не менее чем на 1,5—2,0 см. На этот выступающий конец
надевают маленький резиновый баллончик или кусочек резиновой
трубки (3—5 см), верхний конец которой закрывают кусочком
стеклянной палочки.
Каплеуловители. Для улавливания капель, уносимых парами
жидкости при перегонке, а также при конструировании промыва-
лок используют стеклянные каплеуловители (рис. 15). Их
выпускают с конусными шлифами и без них.
При перегонке жидкостей каплеуловитель нижним концом
присоединяют к колбе с кипящей жидкостью, а верхним — к
холодильнику.
Колбы. Отечественная промышленность выпускает колбы круг-
лодонные, конические, плоскодонные, грушевидные, остродонные,
с взаимозаменяемыми конусными и сферическими шлифами и без
них, различной вместимости.
Колбы большинства типов имеют общелабораторное
назначение. Колбы, используемые для перегонки и работы при относитель-
но высокой температуре, изготовляют из термически устойчивого
стекла группы ТС.
Круглодонные колбы с длинным и коротким горлом с
взаимозаменяемыми конусами и без них применяют для
нагревания и перегонки жидкостей, проведения различных препаративных
и аналитических работ, в качестве приемников при простой и
вакуумной перегонке и т. д. Круглодонные колбы с длинным горлом
применяют также для перегонки веществ с водяным паром. Длин-
ногорлые колбы более устойчивы к температурным воздействиям
и толчкам, часто сопровождающим кипение жидкости.
Круглодонные колбы с несколькими
горловинами (рис. 16, в—д) с взаимозаменяемыми конусами и без них
36
служат для перегонки под вакуумом и проведения, например,
препаративных работ, требующих применения мешалки,
холодильника, термометра, капельной воронки.;
Круглодонные колбы удобно ставить в деревянные подставки
или в подставки в виде колец из резины, резиновых трубок и
других материалов.
Плоскодонные колбы, конические (колбы Э р л е н-
мейера рис. 17) и круглые, с взаимозаменяемыми конусами
и без них, предназначаются для аналитических работ, простейших
операций при атмосферном давлении, хранения жидких веществ
и т. д. В качестве приемников их используют только при простой
перегонке. Конические колбы благодаря своей форме обеспечивают
малую поверхность испарения, вследствие чего их используют для
кристаллизации. Плоскодонные колбы не следует применять для
работ, проводимых при высокой температуре и при пониженном
давлении.
Колбы с тубусом (колбы Бунзена) обычно применяют
в качестве приемников при фильтровании при уменьшенном
давлении (рис. 18). Они изготовляются толстостенными, потому что
иначе при фильтровании под вакуумом они могут быть
раздавлены атмосферным давлением.
При работе рекомендуется закрывать колбу полотенцем или
помещать в ящик из толстого картона или жести. Для защиты от
разлетающихся осколков стекла (при возможном взрыве) можно
также наклеивать на наружную стенку колбы липкую прозрачную
ленту, накладывая слой на слой так, чтобы каждый виток
закрывал около половины предыдущего.
К каждой колбе для фильтрования отсасыванием (без
шлифа) следует подобрать несколько резиновых пробок с
отверстиями разных диаметров, соответствующих диаметрам употребляемых^
воронок.
Колбы для фильтрования отсасыванием рассчитаны на
предельное остаточное давление не более 10 мм рт. ст. Поэтому
рекомендуется колбы, еще не бывшие в употреблении, предварительно
тщательно проверить. После внешнего осмотра (отсутствие
царапин, посторонних включений) колбу закрывают пришлифованной
стеклянной или резиновой пробкой, заворачивают полотенцем
{или помещают в, предохранительный ящик) и присоединяют к
Рис. 16. Круглодонные колбы:
Л—с коротким горлом; б —с длинным горлом; в—г—с двумя, тремя и четырьмя горловинам».
8Г
Л 4 f
Рис. 17. Плоскодонная коническая колба (колба Эрленмейера).
Рис. 18. Колбы с тубусом для фильтрования под вакуумом:
а —без шлифа; б—со шлифом и пришлифованной воронкой.
Рис. 19. Грушевидная колба.
вакуум-насосу на 15—20 мин, после чего колбу с манометром
отсоединяют и наблюдают, как долго сохраняется созданное
разрежение. Для работы при пониженном давлении применяют только
проверенные колбы.
Грушевидные колбы (рис. 19) с взаимозаменяемыми
конусами предназначаются в качестве приемников при вакуумной
перегонке.
Остродонные колбы с одной или несколькими
горловинами (рис. 20) с взаимозаменяемыми конусами применяются в
качестве приемников при перегонке и как перегонные колбы в
комплекте с насадками. Кроме того, существуют специальные кругло-
донные (колба Вюрца) и остродонные (колба Кляйзена)
сосуды для перегонки (рис. 21).
Пробирки. Пробирки для лабораторных работ изготовляются*
из прозрачного химико-лабораторного стекла типа ХС1 или ТС и
из медицинского стекла марки HG1 или НСЗ. Пробирки бывают
различной формы, величины и диаметра: простые,
градуированные, а также центрифужные — конические и с отводами. Простые
пробирки (химические) выпускаются с развернутым рантом и без
ранта.
Круглодонные пробирки с взаимозаменяемым конусом КШ 14/23
из стекла групп ХС2 и ХСЗ удобны для хранения препаратов и
проведения некоторых физико-химических работ.
Круглодонные пробирки с боковым отводом и
взаимозаменяемым конусом предназначаются для фильтрования под вакуумом
Небольших объемов жидкостей.
При проведении работы в пробирке реактивы не следует брать
в большом количестве. Заполнять пробирку целесообразно на
11ь—74 объема.
88
Перемешивание жидкости в этом случае осуществляют так:
пробирку держат большим и указательным пальцем левой руки,
а пальцем правой руки ударяют косым ударом по низу пробирки.
Необходимо закрывать пробирку пальцем и встряхивать ее. Если
пробирка наполнена жидкостью больше чем наполовину,
содержимое перемешивают вращательным движением стеклянной палочки.
При нагревании пробирки с содержимым на спиртовке
пробирку следует зажать в держателе и медленно нагревать до
появления пузырьков пара. Затем пробирку держат не в пламени
спиртовки, а около него или над ним; при этом открытый конец
пробирки должен быть обращен в сторону от экспериментатора.
Нагревать пробирки с жидкостью при умеренной температуре, в
особенности маленькие пробирки, следует в горячей воде, налитой
в стеклянный стакан.
Промывалки. Промывалкой называют сосуд, оборудованный
так, что из него можно получить тонкую струю жидкости, с
помощью, которой можно переносить осадки на фильтр и
промывать их. Существуют различные конструкции стеклянных промы-
валок.
Необходимые для работы промывалки можно сделать самому.
Так, промывалкой может служить толстостенная плоскодонная
колба на 100—250 мл, закрытая хорошо промытой резиновой
пробкой с двумя отверстиями (рис. 22, а).
В одно отверстие вставляют короткую трубку, изогнутую под
тупым углом, короткий конец которой находится почти под
пробкой; эта трубка служит для вдувания воздуха. Другая (длинная)
трубка доходит почти до самого дна колбы. Верхний конец ее
загнут под углом 60—70° и соединен отрезком резиновой трубки с
короткой стеклянной трубкой, конец которой оттянут в хорошо
оплавленный капилляр. При работе конец короткой трубки берут
в рот, и, вдувая в колбу, предварительно заполненную водой,
воздух, получают из другой трубки струю воды (жидкости), которую
направляют, например, на стенку воронки, чтобы смыть осадок в
нижнюю часть фильтра. При работе с летучими и ядовитыми
растворами или растворителями на нижний конец короткой изогнутой
Рис. 20. Остродоиные колбы.
Рис. 21. Колбы для перегонки:
л—колба Вгорца; б —колба Кляйзена.
39
Q ff Рис. 22. Промывалки.
под углом трубки надевают клапан
Бунзена*, препятствующий попаданию
паров из промывалки в рот.
При нагревании воды в промывалке
горло колбы обматывают
теплоизоляционным слоем и приподнимают пробку
с трубками. В качестве
теплоизоляционного слоя можно использовать
листовой асбест или асбестовый шнур. При
этом листовой асбест смачивают
вначале водой, а потом плотно обертывают им горло колбы и
обматывают марлевым бинтом.
Стаканы. Лабораторные стаканы представляют собой
тонкостенные цилиндры из стекла группы ТС, различной вместимоспи
Они бывают низкие и высокие, с носиком и без носика.
Стаканы применяются для проведения простейших химических
операций и в качестве вспомогательных сосудов. Нагревать
стаканы с содержимым на голом пламени или на электрической плитке
с открытым нагревом нельзя. Нагревание следует проводить
только через асбестированную сетку или, лучше, на бане с
теплоносителем. Стеклянные стаканы не следует применять для
выпаривания растворов.
Стаканчики для взвешивания (бюксы). Бюксы используются
для взвешивания веществ в условиях защиты от внешней среды.
Выпускаются стаканчики, закрывающиеся пришлифованной
стеклянной пробкой, высокие и низкие, с плечиками и без плечиков
(рис. 23).
Хлоркальциевые трубки. Эти изделия предназначаются для
поглощения влаги и отдельных газов из газового потока при по*
мощи селективных твердых поглотителей. Они выпускаются U-об-
разные (рис. 24) без отводов, с отводами, под резиновую пробку
и с пришлифованной пробкой; прямые с одним шаром без шлифа
и со шлифом; изогнутые под углом 45° со шлифом.
Твердый поглотитель вносят в виде зерен величиной с
горошину, заполняя им трубку на 1—1,5 см ниже отводной трубки
(или до конца трубки). В местах входа и выхода газа, чтобы
укрепить слой поглотителя и предотвратить унос частичек с
потоком газа, помещают небольшой тампон стеклянной ваты. Следует
иметь в виду, что нельзя туго набивать ни вату, ни поглотитель.
Хлоркальциевые трубки присоединяют к реакционному сосуду
при помощи резиновой трубки или соединительным элементом со
шлифом.
* Клапан Бунзена представляет собой отрезок толстостенной резиновой
трубки длиной 5 см, один конец которой плотно закрыт резиновой пробкой.
Другой конец надевают на короткую стеклянную трубку промывалки и лезвием
бритвы делают вдоль трубки глубокий прорез длиной 1,5—2,0 см.
40 %
Холодильники. Стеклянные лабораторные холодильники
предназначаются для конденсации и охлаждения газов и паров.
При перегонке веществ с температурой кипения выше 160°С
применяют воздушные холодильники. Это достаточно
длинные трубки из тонкостенного стекла, диаметром 10—20 мм.
Трубку можно укрепить в горле колбы с помощью просверленной
корковой пробки, но лучше к трубке припаять конусный шлиф-
керн, с помощью которого ее вставляют в шлиф-муфту круглодон-
ной колбы. Полезно также к верхнему концу трубки припаять
шлиф-муфту для соединения сосуда с поглотителем.
При перегонке веществ с температурой кипения до 160°С
обычно применяют нисходящие водяные холодильники с
прямой трубкой (типа Либиха), в которых охлаждающая вода
из водопроводного крана поступает через нижний отвод
холодильной рубашки и выходит через верхний отвод (рис. 25,а).
Необходимо при этом следить, чтобы рубашка была во время работы
заполнена водой и чтобы ток воды не прерывался. При выборе
холодильника следует учитывать: чем ниже температура кипения
перегоняемой жидкости, тем длиннее должен быть холодильник.
При перегонке веществ с температурой кипения до 120 °С
охлаждающим агентом служит проточная вода, а от 120 до 160 °С—
непроточная. При перегонке низкокипящих жидкостей в качестве
охлаждающей жидкости можно пользоваться
также.охлаждающими жидкими смесями (солевые растворы + лед и др.)-
При нагревании летучих жидкостей используют различные
типы обратных холодильников с наружным или внутренним
охлаждением. Холодильники с прямой трубкой применяются
преимущественно в качестве нисходящих; в качестве обратных
холодильников они мало эффективны.
Холодильники Либиха выпускаются без конусов и с
конусами, с длиной охлаждающего кожуха 100, 200, 300, 400 и 600 мм.
Шариковые холодильники с наружным охлаждением
(рис. 25,6) с 4, 5, 6 и 8 шарами используются в качестве обратных
холодильников при нагревании и кипячении жидкостей. Благодаря
большой поверхности охлаждения за счет сферических
выпуклостей на внутренней трубке они значительно короче прямых холо-
и
<z/
Ряс. 23. Бюксы:
а— высокие с внутренней пришлифованной пробкой; б, в—низкие с наружной
пришлифованной крышкой.
Рис. 24. и-обраэыая хлоркальциевая трубка*
41
Рис» 25. Холодильники с прямой трубкой (а) н шариковые (6*).
Рис. 26. Спиральные холодильники:
а— с наружным охлаждением; б—с внутренним охлаждением; в—обратный с внутренним
охлаждением.
дильников, при равной эффективности. Через шариковый холо-
дильник удобно вводить в реакционный сосуд вещества,
способствующие равномерному кипению, приливать жидкости или
пропускать газообразные вещества, вставлять мешалку для
перемешивания реакционной массы.
Опытным путем установлено, что для конденсации паров
метилового и этилового спиртов, этилацетата, бензола, хлороформа
и четыреххлористого углерода при длине муфты холодильника
200 мм достаточно четырех шаров; для полной конденсации паров
диэтилового эфира, ацетона и бромистого этила — шести; для
конденсации паров сероуглерода — восьми. Увеличение числа шаров
обратного холодильника лишено смысла, так как жидкости с
высоким давлением паров (выше, чем у сероуглерода), вообще не
могут полностью конденсироваться при температуре охлаждающей
водопроводной воды.
При пользовании шариковым обратным холодильником следует
регулировать интенсивность кипения жидкости, чтобы
предотвратить «захлебывание» холодильника. Захлебывание является ре-:
зультатом поступления во внутреннюю трубку холодильника
большого количества пара кипящей жидкости, который своим
давлением препятствует непрерывному стеканию конденсата.
Спиральные холодильники с наружным
охлаждением (рис. 26, а) применяются главным образом как прямые,
особенно когда надо сконденсировать пары легколетучих
жидкостей, поступающих сверху вниз. В этом случае перегонную колбу
42
при помощи 75-градусных изгибов соединяют с муфтой
холодильника.
В спиральных холодильниках с внутренним
охлаждением (рис. 26,6) охлаждающая вода протекает через
змеевик. Холодильники этого типа в качестве обратных
эффективнее, чем шариковые.
Обратные холодильники с внутренним
охлаждением (рис. 26, в) используются в качестве нисходящих и
обратных холодильников.
Особый интерес представляет спиральный двухстен-
ный холодильник с внутренним и наружным
охлаждением. Его можно применять в качестве нисходящего
и обратного. Он особенно удобен при длительных операциях, так
как муфта, не охлаждаемая водой, не отпотевает.
Холодильник типа «охлаждающий пальчик»
(рис. 27) используют в качестве дефлегматора при дробной
перегонке и как холодильник при возгонке твердых веществ.
Эксикаторы. Эксикаторы (рис. 28) применяются для
высушивания и хранения веществ, легко поглощающих влагу из воздуха.
Эксикаторы часто применяют также и в качестве гигростатов. Для
этого наполняют нижнюю часть эксикатора смесью веществ,
создающих определенное давление водяного пара и, следовательно,
соответствующую влажность. Эксикаторы подразделяют на обычные
(без крана) и вакуумные (с краном). Внутрь эксикатора, над
суженной частью корпуса, где ставят чашку с осушающим
веществом, помещают фарфоровую эксикаторную вставку с отверстиями.
Высушиваемое вещество находится на вставке, над осушающим
средством. Высушивание осуществляют при комнатной
температуре.
Рис. 27. Пальчиковый холодильник.
Рис. 28. Эксикаторы:
<? —без крана; б—с-краном (вакуумные);
J — крышка; 2 — корпус; 3—вставка; 4—чашка; 5—вакуумный кран,
43
Об осушающих средствах см. гл. 18. Применять в
вакуум-эксикаторах в качестве водопоглощающего средства серную кислоту
запрещается.
Некоторые зарубежные фирмы выпускают вакуум-эксикаторы
с электрическим обогревом. В таких эксикаторах можно вести
высушивание в вакууме при повышенной температуре.
При работе с эксикатором необходимо следить, чтобы
притертые части были слегка смазаны вазелином. При переносе
эксикатора с места на место нужно придерживать крышку.
Если в эксикатор помещают горячий тигель, нагретый бюкс
и т. п., то из-за нагревания воздуха крышка эксикатора может
приподняться, соскользнуть и разбиться. Чтобы этого не
случилось, поместив горячий предмет в эксикатор и накрыв его
крышкой, ее некоторое время притирают, т. е. вращают вправо и влево.
При остывании горячего предмета внутри эксикатора создается
небольшое разрежение, и крышка плотнее пристает к корпусу.
Чтобы открыть эксикатор, нужно сначала сдвинуть крышку в
сторону, после чего она легко снимается.
При работе с вакуум-эксикатором следует, учитывать то
обстоятельство, что величина остаточного давления в нем зависит от
вакуум-насоса, создающего разрежение, и от герметичности
пришлифованных частей. По ГОСТ 6371—73, вакуум-эксикаторы (с
краном) должны сохранять в течение суток предельное остаточное
давление =^6,67 МПа (50 мм рт. ст.).
Впускать воздух в вакуум-эксикатор, в котором создано
разрежение, нужно очень осторожно, так как струя воздуха может
вызвать распыление высушиваемого вещества. Поэтому кран
эксикатора нужно поворачивать очень медленно и поднимать крышку
только после того, как кран будет полностью открыт.
При вакуумировании нового эксикатора следует соблюдать
меры осторожности, так как толстостенный сосуд вследствие
какого-либо дефекта стекла или от сильного толчка может под
вакуумом взорваться. Вакуум-эксикатор следует поместить в
защитный ящик или обернуть полотенцем, а работу производить в
защитных очках.
Обращение с лабораторной стеклянной посудой [32]
При работе со стеклянной посудой следует всегда учитывать
хрупкость стекла. Стеклянную посуду при необходимости
рекомендуется закреплять в зажимах, снабженных амортизирующими
прокладками из эластичной пробки или кусочками не очень
тонких резиновых трубок.
Нужно учитывать, что толстостенные стеклянные изделия хуже
выдерживают резкие перепады температуры, поэтому кипячение
можно вести только в тонкостенной посуде. При этом большое
значение имеет форма сосуда. Круглодонные колбы можно иногда
нагревать даже открытым коптящим пламенем, в то время как для
колб с плоским дном это исключается. Вообще же, стеклянную
посуду запрещается нагревать на открытом огне.
44
Неправильное охлаждение стеклянных изделий при
термической закалке в процессе их изготовления создает внутренние
напряжения, в результате чего изделие через некоторое время может
самопроизвольно дать трещины. При нарушении поверхности
стеклянная посуда теряет стойкость к температурным перепадам и к
механическому удару. Поэтому перед работой следует
тщательно проверять отсутствие царапин на внутренней поверхности
стеклянной посуды.
При переносе стеклянных сосудов с горячей жидкостью
следует пользоваться полотенцем, сосуд при этом нужно держать
обеими руками: одной за дно, а другой за горловину. Большие
химические стаканы с жидкостью нужно поднимать только двумя
руками, так чтобы отогнутые края стакана опирались на
указательные пальцы.
При перемешивании жидкости стеклянной палочкой на кончик
палочки следует надеть кусочек резиновой трубки (в случае
водных растворов) или укрепить кусочек тефлона.
При внесении в тонкостенную посуду твердых веществ, сосуд
следует держать наклонно и осторожно спускать вещество по
стенке сосуда.
В стеклянные ампулы разрешается запаивать
сконденсированные газообразные вещества, имеющие температуру кипения не
ниже 12 °С. Вещества, разлагающиеся при нагревании со взрывом,
запаивать в ампулы запрещается. Ампулы разрешается заполнять
не более чем на 50% их объема. Ампулы перед запаиванием
необходимо охладить ниже температуры кипения помещенного в них
вещества. Во время запаивания нижняя часть ампулы должна
быть погружена в сосуд с хладагентом. Вскрывают ампулы только
после охлаждения, заворачивают их в полотенце, затем делают
надрез напильником или стальной пластинкой на капилляре и
отламывают его. Все операции с ампулами следует проводить, не
вынимая их из защитной оболочки (металлической сетки, трубки),
в вытяжном шкафу, надев защитные очки.
Стеклянные изделия с токопроводящим покрытием [53]
Изделия с токопроводящим покрытием представляют собой
стеклянные сосуды (стаканы, колбы) из термически стойкого
стекла «пирекс,» на наружную поверхность которых нанесена пленка
из двуокиси олова с добавкой веществ, увеличивающих ее
электропроводность. На поверхность пленки нанесен слой изоляционного
лака или глазури.
Наружную поверхность, изолированную кремнийорганическими
лаками, не рекомендуется мыть щелочными растворами, а также
растворами сильных окислителей. К органическим растворителям
глазурь устойчива.
Изделия с токопроводящим покрытием включаются в сеть
переменного тока напряжением 127/220 В через лабораторный
автотрансформатор (ЛАТР).
45
Подача напряжения на токопроводящую пленку
осуществляется через металлические захваты, надеваемые на силикатно-сереб-
ряную шинку, или с помощью проводов, непосредственно
припаянных к силикатно-серебряной шинке.
С токопроводящим покрытием выпускаются трехгорлые кругло-
донные колбы для перегонки и препаративных работ, а также
высокие стаканы для нагрева жидкостей.
Мерная лабораторная посуда [31, 62]
Для измерения объема жидкостей применяют мерные сосуды
с метками, указывающими их вместимость. К мерной посуде
относят бюретки, мерные колбы, пипетки, измерительные цилиндры,
мензурки и градуированные пробирки.
Выпускаемая промышленностью мерная посуда калибрована
(на наливание или выливание), и ее вместимость должна
соответствовать действующим государственным стандартам.
Бюретки, колбы и пипетки, используемые для точных
измерений, калибруются по образцовым мерам вместимости 1 или 2
разряда обычно при 20 °С В соответствии с этим бюретки, мерные
колбы и пипетки изготовляются 1 и 2 классов точности.
Допустимое отклонение для бюреток и градуированных пипеток 1
класса точности равно половине цены наименьшего деления шкалы,
2 класса — цене наименьшего деления шкалы.
Изменение объема мерных сосудов вследствие сжатия или
расширения стекла при изменении температуры незначительны,
что делает возможным пользоваться ею при температуре,
отличающейся от 20 °С на несколько градусов, не делая поправок.
Например, объем литровой колбы, откалиброванной при 20 °С,
будет при 26 °С равен 1000,15 мл.
Бюретки. Бюретки предназначаются для измерения точных
объемов жидкостей при титровании и для других операций. Они
калибруются только на выливание.
Прямые бюретки выпускаются с краном и без него
(рис. 29, а—в). Бюретки без крана — это стеклянные
градуированные трубки, верхний конец которых открыт, а нижний
заканчивается оливой. На оливу надевается затвор, состоящий из
резиновой трубки 6—7 см длиной, в которую предварительно
вставлена стеклянная бусина, закрывающая просвет трубки. Вместо
бусины можно применять металлический пружинный зажим. В
свободный конец резиновой трубки вставляют стеклянную трубку
с оттянутым концом длиной 5—6 см. Отверстие капилляра стек*
лянной трубки должно быть таким, чтобы при открытом затворе
жидкость из бюретки на 25 мл вытекала не менее чем за 24—
45 с, а из бюретки на 50 мл — за 45—55 с. Разжимая зажим
или оттягивая резиновую трубку на бусинке затвора, создают
просвет в трубке, через которую вытекает жидкость.
Наиболее распространенный тип бюреток — прямые с однохо*
довым краном.
46
Рис. 29. Бюретки прямые:
а—с краном; б —с пружинным зажимом; в —с бусиной; г и д— с боковым отводом.
Рис. 30. Микробюретка с запасным резервуаром и подставкой.
Прямые бюретки с дно- и двухходовым спускными
кранами выпускаются также с боковым отводом (рис. 29,2 и д)4
Отвод служит для заполнения бюретки титрованным
раствором из запасной емкости. Бюретки выпускаются в обычном
исполнении и с автоматической установкой нуля.
Микробюретки (рис. 30) предназначаются для измерения
объемов жидкости порядка сотых и десятых долей миллилитра,
для титрования и наливания в пределах полного объема бюретки
или его части.
Микробюретки выпускаются 1 и 2 класса точности. Отклонение
от номинальной вместимости микробюреток при 20 °С на весь
объем для бюреток 1 класса точности ±0,006 мл, для бюреток
2 класса точности ± 0,015 мл при условии вытекания воды при
полностью открытом кране в течение 20—35 с для 1 класса
точности, 15—35 с для бюреток 2 класса точности.
В лабораторной практике большое распространение получилц
бюретки с автоматическим нулем и склянкой (рис. 31). Мениск
поступающего раствора автоматически устанавливается на
нулевой отметке. При создании давления в склянке с помощью
резинового нагнетательного баллона жидкость поднимается по
наружной питающей трубке и заполняет бюретку выше нулевой отметки.
Как только прекратится нагнетание воздуха, избыток жидкости
сливается в склянку через ту же трубку, отверстие которой
находится на уровне нулевой отметки. Выпускаются такие бюретки
2 класса точности.
47
Цена наименьшего деления бюреток зависит от вместимости:
Вместимость, мл Цена деления, Вместимость, мл Цена деления,
мл , мл
1 0,01 25 0,05 и 0,1
2 0,01 50 0,1 и 0,2
5 0,02 и 0,05 100 0,1 и 0,2
10 0,05
Работа с бюретками. Бюретки с одноходовым краном и без
крана с оливой наполняют, наливая жидкость сверху через
маленькую воронку с узкой трубкой, чтобы дать воздуху возможность
свободно выходить из бюретки.
Чтобы заполнить раствором стеклянный кран и кончик (у
бюреток с краном) или наконечник и резиновую трубку (у бюреток с
затвором), бюретку наклоняют и быстро спускают 3—5 мл
раствора. Если не удается этим приемом удалить пузырьки воздуха
из кончика бюретки, поступают следующим образом. При
открытом кране (зажиме) опускают кончик бюретки в небольшой
стакан с раствором и осторожно засасывают резиновым баллоном
Рис. 31. Бюретки с автоматическим нулем и склянкой:
с — со спускным краном; б —без крана;
/ — вертикальная трубка; 2—горизонтальная спускная трубка; 3—наполнительная трубка;
4 — склянка; 5—нагнетательный баллон; 6—спускной кран; 7 — резиновая трубка; 8, 9 —
предохранительные трубки.
48
Рис. 32. Отсчет показаний обычной
бюретки с помощью экрана (а) и бюретки
с цветной полосой (о").
Рис. 33. Мерцая колба с одной меткой.
или шприцем в бюретку
раствор. Пузырьки воздуха при
этом переходят в бюретку.
Закрыв кран или зажим,
наполняют бюретку сверху
приблизительно на 1 см выше
нулевого деления. Затем
осторожно спускают жидкость точно
до нулевой отметки.
Бюретку укрепляют строго вертикально в лапке штатива. Кран
или зажим затвора должны быть с правой стороны; их открывают
или закрывают одной рукой, а другой вращают коническую
колбу для перемешивания титруемого раствора.
Отмеривание жидкости производят всегда от нулевого деления.
При отмеривании объема раствор спускают от нулевого деления
до уровня, находящегося приблизительно на 5 мм выше нужного
деления, выжидают 1 минуту и, приложив кончик бюретки к стенке
сосуда, спускают раствор точно до метки.
Отсчет производят по нижнему краю мениска, за исключением
окрашенных жидкостей, когда отсчет приходится производить по
верхнему краю мениска, что является менее точным. При
установлении положения мениска глаз должен находиться на уровне
поверхности жидкости в бюретке.
Для облегчения отсчета можно пользоваться специальным
экраном из кусочка белого картона, половина которого заклеена
черной бумагой. Картон держат черной половиной вниз позади
бюретки так, чтобы граница черного и белого полей находилась
на 1 мм ниже уровня жидкости. Мениск тогда кажется черным
и резко выделяется на белом фоне (рис. 32,а).
Еще лучше пользоваться бюретками, снабженными на задней
стороне вертикальной синей полоской на белом фоне, что
облегчает отсчет уровня мениска (рис. 32,б).
При работе с растворами едких щелочей и карбонатов
щелочных металлов во избежание «заедания» стеклянных кранов
рекомендуется применять бескрановые бюретки.
Следует иметь в виду, что бюретки калиброваны по воде, и
ими пользуются для отмеривания жидкостей, по вязкости близких
к воде.
Во избежание попадания пыли в раствор рекомендуется
бюретки закрывать сверху опрокинутой пробиркой. Бюретки с
растворами можно эффективно защитить от загрязнений, содержащихся
в воздухе лабораторного помещения, соединяя верхний конец
бюретки со склянкой для жидких промывателей, заполненной на XU
тем же раствором, что и бюретка,
49
Приступая к смазыванию крана бюретки, прежде всего
удаляют старую смазку с крана и кончика бюретки, используя тонкую
проволоку и ватный тампон. Далее на кран тонким равномерным
слоем наносят свежую смазку, но только не силиконовую. Кран
вставляют в муфту бюретки и несколько раз поворачивают.
Мерные колбы. Мерные колбы — плоскодонные с длинными
горлами. Объем, до которого нужно наполнять жидкостью,
ограничен специальной круговой меткой на горле. Вместимость колбы
при 20 °С выгравирована на ее боковой поверхности. Мерные
колбы предназначаются для приготовления растворов определенной
концентрации, разбавления растворов, растворения веществ и т. п.
Их калибруют на наливание или на отливание. Колбы,
предназначенные для наливания, имеют одну кольцевую отметку на
цилиндрической части горла (рис. 33), а калиброванные на
отливание — две.
Выпускаются мерные колбы 1 и 2 класса точности с одной и
двумя метками с пришлифованной пробкой и без пробки.
Для отмеривания точного объема жидкости обычно применяют
мерные колбы, калиброванные по количеству вылитой из них воды.
При приготовлении растворов, концентрация которых
измеряется содержанием растворенного вещества в единице объема
раствора, следует применять мерные колбы, калиброванные на на*
ливание воды.
При приготовлении растворов заданной концентрации
определенное количество вещества в твердом или жидком состоянии или
в виде концентрированного раствора вносят через воронку в
сполоснутую дистиллированной водой (или соответствующим
растворителем в случае неводных растворов) мерную колбу, наполняют
ее более чем на 1/2 водой (растворителем) и тщательно
перемешивают взбалтыванием. Затем добавляют воду (растворитель)
почти до кольцевой метки, закрывают колбу пришлифованной
пробкой и вновь хорошо перемешивают. Наполнение колбы точно до
кольцевой метки производят только после полного растворения
вещества и доведения температуры раствора в колбе
приблизительно до 20 °С.
При наполнении мерных колб их помещают на ровную
поверхность и наполняют жидкостью почти до кольцевой отметки на
колбе. Окончательно уровень жидкости устанавливают
прибавлением нескольких капель ее при помощи стеклянной трубки с
оттянутым концом (или пипетки), так чтобы нижний край мениска
касался верхнего края отметки.
Если в процессе растворения выделяется тепло, то колбу с
раствором охлаждают до комнатной температуры.
При заполнении колбы нужно следить за тем, чтобы вогнутый
мениск в ее шейке был касательным к круговой отметке.
Мерные колбы не предназначены для хранения растворов.
После разбавления в мерной колбе растворы, особенно щелочные,
следует немедленно перелить в стеклянные бутыли, а мерные
колбы вымыть.
50
n
рис. 34. Градуированная и простая
пипетки.
А
25мл |
?5СС
/
и
Рис. 35. Отмеривание объема жидкости
пипеткой.
При сливе жидкости из колбы следует, постепенно наклоняя,
довести ее до вертикального положения горлом вниз. После
прекращения слива сплошной струей необходимо выждать, пока по
каплям стечет жидкость, оставшаяся на стенках колбы. Время
этой выдержки для литровых колб ^ 30 с, для больших — 60 с.
По истечении указанного времени удаляют последнюю каплю
жидкости прикосновением края колбы к внутреннему краю
сосуда, в который сливается жидкость.
Измерительные пипетки. Пипетки предназначаются для
точного отмеривания определенного объема жидкости.
Пипетки (рис. 34) представляют собой стеклянные трубки
различного диаметра, прямые или с грушевидным, шарообразным
либо цилиндрическим расширением посредине. Нижний конец
пипетки слегка оттянут. Пипетки выпускают градуированные и не-
градуированные (с меткой). На расширенной или верхней части
пипетки указывается номинальная вместимость (в мл) и
температура, при которой калибровалась пипетка, а также класс точности.
Пипетки обычно калибруют на выливание.
Выпускаются также микропипетки номинальной вместимостью
0,1 и 0,2 мл с наименьшей ценой деления 0,001 и 0,002 мл.
Пипетки должны быть всегда чисто вымытыми; их следует
держать в особом штативе и закрывать верхнюю часть чистой
фильтровальной бумагой для защиты от пыли. При отсутствии
штатива пипетки можно хранить в высоком стеклянном цилиндре, на
дно которого кладут несколько кружочков фильтровальной
бумаги.
Следует также иметь в виду, что пипетки калиброваны по воде,
и ими надлежит пользоваться для измерения объема жидкостей,
по вязкости близких к воде.
Наполняют и опорожняют пипетки следующим образом.
Опустив кончик пипетки с одной меткой в жидкость, засасывают
резиновым баллоном или шприцем жидкость немного выше верхней
51
метки. Затем быстро закрывают верхнее отверстие пипетки
слегка смоченным указательным пальцем и осторожно ослабляют
нажим пальца на отверстие пипетки так, чтобы нижний край
мениска установился на метке. При этом пипетку держат так, чтобы
метка находилась на уровне глаза (рис. 35). Усилив нажим
пальца, прекращают вытекание жидкости из пипетки. Кончиком
пипетки касаются стенок сосуда, из которого набирают жидкость, и
быстро переносят пипетку к сосуду, в который должна быть влита
жидкость. Держа пипетку вертикально над сосудом, прислонив
кончик («носик») ее к стенке сосуда, ослабляют нажим пальца
на верхний конец пипетки, чтобы уровень жидкости стал медленно
понижаться, пока он не понизится до нижней метки, после чего
усиливают нажим на отверстие пипетки, выжидают 15—25 с и
без стряхивания последней капли отнимают пипетку от стенки
сосуда.
Не допускается выдувать жидкость, оставшуюся в оттянутом
кончике пипетки, и быстро выливать жидкость, так как при этом
некоторая часть жидкости остается на стенках пипетки. Следует
заметить, что время вытекания жидкости зависит от размера
нижнего отверстия носика пипетки и ее вместимости. Размер
отверстия должен быть таким, чтобы вода вытекала из пипетки в еле*
дующие сроки:
Вместимость пипетки, мл 5 10 25 50 100 200
Время вытекания, с 15 20 25 30 40 50
Если отверстие пипетки больше требуемого, то его уменьшают
осторожным оплавлением, а если оно мало, стачивают кончик
наждачной бумагой или мелким напильником.
Наполнение и опорожнение градуированных пипеток проводят
аналогично описанному выше, за исключением того, что жидкости
дают свободно стечь до нужной метки, выжидают 15 с, касаясь
кончиком пипетки внутренней стенки сосуда, и устанавливают
мениск точно на нужной отметке.
Силиконирование пипеток и бюреток [10]. Объем некоторых
водных растворов бывает трудно отмерить, так как на
внутренней стенке сосуда остаются прилипшие капли раствора. В таких
случаях рекомендуется предварительно покрывать внутреннюю
поверхность сосуда тончайшей силиконовой пленкой, не смачиваемой
водой. При этом мениск становится выпуклым.
Для придания гидрофобности чисто вымытую и высушенную
посуду заполняют 2% раствором диметилдихлорсилана
(CHsbSiCb в диэтиловом эфире, выдерживают 1 —1,5 мин, после
чего раствор сливают в склянку (для .последующего
использования), а обработанную посуду оставляют в вытяжном шкафу до
исчезновения запаха и высушивают при 120—140 °С.
В связи с высокой токсичностью диметилдихлорсилана предло*
жено силиконирование мерной посуды 3% растворами полиметил-
силоксановых жидкостей ПМС-200 и ПМС-300 в хлороформе.
Чистую сухую посуду заливают на несколько минут раствором сили*
52
кона, который затем сливают и применяют многократно. Посуду
сушат 2 ч при 180—210 °С. Образующаяся гидрофобная пленка
не смывается водой и не разрушается кислотами, но смывается
при кипячении с 10% раствором NaOH.
Мерные цилиндры. Градуированные мерные цилиндры (рис. 36)
предназначаются для измерения объемов жидкости, наливаемой
или отливаемой в пределах полного объема цилиндра или его части.
Мерные цилиндры калибруют на наливание или на отливание.
Мерные цилиндры выпускаются с носиком и с пришлифованной
пробкой, с пластмассовым или массивным стеклянным
основанием. Цена наименьшего деления зависит от вместимости:
Вместимость, мл 5—100 100—1000 2000
Цена деления, мл 0,1 — 1,0 1 — 10 20
Чтобы отмерить необходимый объем жидкости, ее наливают в.
мерный цилиндр до тех пор, пока нижний край мениска не
достигнет нужного деления.
Мензурки. Мензурки (рис. 37) применяют как для грубых
измерений объема жидкостей, так и для отстаивания мутных
жидкостей (осадок собирается в суженной части).
Калибруют их на отливание.
Наименьшая цена деления мензурок составляет 10% от
номинальной вместимости для мензурок на 50, 100 и 250 мл и 5%
для мензурок вместимостью 500 и 1000 мл.
Градуированные мерные пробирки. Мерные пробирки
(рис. 38, а) предназначаются для проведения в небольшом
масштабе простых химических операций с измерением объема. Их
можно использовать наравне с мерными цилиндрами.
Центрифужные пробирки (рис. 38,6) служат для
одновременного измерения объема осадка и надосадочной жидкости
после центрифугирования взвеси.
Рис. 36. Мерные цилиндры:
а—с носиком; б—с пришлифованной пробкой.
Рис. 37. Мензурки:
а —без ножки; б —коническая с ножкой; в —цилиндрическая с ножкой.
Рис. 38* Градуированные мерные пробирки.
53
Проверка мерной посуды
В практике химических лабораторий иногда приходится
проверять объем пипеток, бюреток и мерных колб. Такая необходимость
возникает при проведении измерений с повышенной точностью,
при сомнениях в стандартности мерной посуды, при получении
мерных изделий из ремонта и т.д.
Проверка мерного сосуда заключается в определении его
истинной вместимости УИСт. В результате проверки находят
поправку, которая представляет собой разность между истинной
вместимостью и номинальной, обозначенной на измеряемом сосуде кном.
ДУ = Кист - Кном
Мерную посуду проверяют, определяя массу дистиллированной
воды (или ртути в случае микропипеток), содержащейся в ней
или вылитой из нее при определенной температуре и
определенном барометрическом давлении. По массе воды, пользуясь
таблицами, рассчитывают истинную вместимость сосуда и пределы
погрешности (отклонение от вместимости, указанной на мерном
изделии).
За единицу объема принимают истинный литр,.т. е. объем,
занимаемый массой воды в 1 кг при 3,98 °С и нормальном
атмосферном давлении 1013 гПа (760 мм рт. ст.).
При проверке мерной посуды можно пользоваться табл. 1,
показывающей, какую массу дистиллированной воды определенной
температуры надо взять при той же температуре воздуха и
нормальном атмосферном давлении, чтобы объем ее соответствовал
1 л при 20 °С.
Данные табл. 1 рассчитаны на нормальное атмосферное
давление. Если давление ниже нормального, то на каждый миллиметр
разницы прибавляют поправку, указанную в таблице
таблица 1. Масса 1 л воды при разных температурах *
Температура воды и
воздуха, °С
Масса 1 л воды, г
Поправка на атмосферное
давление
Температура воды и
воздуха, С°
Масса 1 л воды, г
Поправка на атмосферное
давление
15
997,925
0,00142
19
997,349
0,00140
16
997,798
0,00141
20
997,177
0,00139
17
997,659
0,00141
21
996,995
0,00139
18
997,510
0,00140
22
996,802
0,00138
* Подробные таблицы приведены в ГОСТ 8.234—77.
54
Если давление выше нормального, поправку соответственно
вычитают. Разница между табличной и фактической массой воды
соответствует AV в миллилитрах и долях миллилитра.
Пример расчета. Допустим, что среднее значение массы воды в объеме про*
веряемого мерного сосуда с обозначенной вместимостью VUOm = 25 мл при
19 °С и давлении 750 мм рт. ст. оказалось равным 24,980 г. По табл. 1 находим,
что при 19 °С масса 1 л воды равна 997,349 г, т. е. масса 1 мл воды равна
0,997349 г. Разделив 24,98 на 0,997349, находим, что вместимость мерного сосуда
равна 25,046 мл. Поправка на давление составляет 0,0014-10 = 0,014 мл. При*
бавляем ее к вычисленному объему и получаем УИст = 25,06 мл. Отсюда
AV = U„ct - Vhom = 25,06 - 25,00 = +0,06 мл.
ГОСТ 8.100—73 устанавливает пределы допускаемой погрешности AV
проверяемых мерных сосудов, в зависимости от их класса точности и вместимости.
Перед проверкой мерную посуду тщательно очищают. Ее
считают чистой, если при выливании дистиллированной воды
последняя не собирается на внутренних стенках в виде струек, полос
или капель; внутренняя поверхность стеклянного сосуда должна
оставаться равномерно покрытой тонкой пленкой воды. После
очистки наливные меры высушивают, а отливные непосредственно
перед проверкой смачивают дистиллированной водой.
При проверке пипеток на аналитических весах определяют
массу бюкса или колбы с притертой пробкой, вмещающих па
крайней мере трехкратный объем пипетки. Дистиллированную-
воду для проверки пипеток наливают в большую колбу и держат
ее не менее часа вблизи весов, чтобы вода приняла температуру
воздуха в весовой комнате.
Пипетку с одной меткой наполняют водой, как указано выше,
и спускают воду в бюкс или колбу, придерживаясь ранее данных
рекомендаций. Бюкс закрывают крышкой и взвешивают. Не
выливая воды из бюкса, спускают в него снова полную пипетку воды
и снова взвешивают. Таким же образом поступают и в третий раз.:
Из трех значений массы воды берут среднюю величину. Пользуясь
табл. 1, внося поправку на барометрическое давление, вычисляют
истинную вместимость проверяемой пипетки.
На нижний конец градуированных пипеток надевают
резиновую трубку со стеклянным наконечником и зажимом и проверяют
пипетку так, как указано ниже для бюреток.
Проверку микропипеток осуществляют, большей частью, по мае*
се ртути (марки Р0 или Р1), заполняющей объем пипетки.
Плотность ртути q при комнатной температуре приведена ниже:
U °С р. кг/мз t, °C р, кг/мЗ tt °c р, кг/мз
15 13558,14 19 13548,32 23 13538,51
16 13555,69 20 13545,87 24 13536 06
17 13553,23 21 13543,41 25 13533,61
18 13550,78 22 13540,90 26 13531,16
Значительная плотность ртути позволяет с достаточной
точностью определять вместимость микропипеток. Так, масса ртути
при полной вместимости пипетки 0,1 мл составляет примерно
1350 мг, что позволяет проводить взвешивание на обычных анали-
5S
тических весах. При погрешности взвешивания 1 мг можно
измерять объемы, исчисляемые десятыми долями миллилитра.
По ГОСТ 8234—77, масса ртути в объеме, соответствующем
полной вместимости микропипетки 0,1 мл, должна составлять от
1342 до 1368 мг, а при вместимости в 0,2 мл — от 2682 до 2736 мг.
Если масса отмеренной пипеткой ртути выходит за приведенные
выше пределы, то пипетки бракуют.
Пределы допускаемой погрешности AV для пипеток 2 класса.
•с вместимостью V:
V\ мл
200
100
50
25
20
15
с одной
отметкой
0,2
0,16
0,10
0,08
0,06
0,04
градуированные
—
0,20
0,16
0,10
0,10
0,10*
V, мл
10
5
3
2
0,5
с одной
отметкой
0,04
0,02
0,01
0,01
0,01
градуированных
0,10
0,05
0,01
0,01
0,01
Проверку пипеток с применением ртути следует выполнять в
вытяжных шкафах, скорость движения воздуха в рабочих проемах
которых должна быть 0,5—1 м/с.
Бюретки проверяют по массе воды, вылитой из них от
нулевой отметки до разных отметок, например от 0 до 10 мл, от 0 до
20 мл, при определенной температуре. Приемы и техника работы
те же самые, что и при проверке пипеток. Все отсчеты делают
после полного стекания жидкости со стенок бюретки
Пользуясь табл. 1, находят массу, которую должна иметь вода
при данной температуре и атмосферном давлении и определяют
разницу между номинальной и истинной вместимостью проверяе*
мой бюретки в целом и в отдельных ее отрезках (от 0 до 10 мл,
от 0 до 20 мл и т.д.).
Пределы допускаемой погрешности AV для бюреток 2 класса
при вместимости V:
V, мл AV, мл V, мл ДК, мл ' V, мл AV, мл
200 0,50 25 0,10 3 0,01
100 0,20 10 0,05 2 0,01
50 0,10 5 0,02 1 0,01
Проверка мерных колб производится с учетом особенностей
их калибровки.
Мерные колбы, подготовленные к проверке на отливание,
устанавливают на ровную горизонтальную поверхность и наполняют
дистиллированной водой на несколько миллиметров ниже метки.
После того как содержимое колб примет температуру весовой
комнаты, добавляют пипеткой воду точно до метки. Затем из колбы
выливают воду в заранее взвешенный стакан или коническую колбу.
Дают стечь каплям воды в течение 10—20 с и взвешивают сосуд
с водой. После вычета массы тары получают значение массы воды,
вылитой из мерной колбы. Это определение повторяют три раза и
вычисляют среднюю массу вылитой воды. Пользуясь табл. 1, нахо-
56
дят истинный объем воды, вылитой из проверяемого мерного сосуда
при данной температуре.
Для проверки мерных колб на наливание их после очистки
следует тщательно высушить в сушильном шкафу или подогретым
воздухом, споласкиванием этиловым спиртом или ацетоном, с
последующим продуванием сухим воздухом. Сухую мерную колбу
оставляют на несколько часов у весов и взвешивают с такой
точностью, чтобы ошибка взвешивания не превышала 0,1 % от массы
воды в объеме проверяемой колбы. Затем колбу наполняют
дистиллированной водой до метки, обтирают снаружи сухим полотенцем
и взвешивают вторично.
Пользуясь табл. 1, по массе воды находят вместимость
проверяемой колбы.
Пределы допускаемой погрешности А V для мерных колб 2 класса
при вместимости V:
Vf мл AV, мл V, мл AV. мл V, мл AV, мл
2000 1,00 200 0,20 25 0,06
1000 0,60 100 0,20 10 0,04
500 0,30 50 0,10 5 0,02
250 0,20
Правила пользования мерной посудой
Пользоваться следует только хорошо вымытой посудой. Пипетки
и бюретки перед употреблением споласкивают 2—3 раза
небольшими порциями раствора, который собираются отмеривать.
Всегда следует придерживаться избранного метода
опоражнивания мерной посуды.
По окончании работы пипетки моют дистиллированной водой
(в случае работы с водными растворами) или этиловым спиртом,
прополаскивают 3—5 раз дистиллированной водой, устанавливают
в штатив для пипеток или в сухой стеклянный цилиндр и
прикрывают бумажным колпачком или перевернутой пробиркой для
защиты от пыли.
При наполнении бюреток необходимо следить за тем, чтобы
кончик бюретки был заполнен раствором. По окончании работы
бюретки заполняют титрантом (титруемым раствором) выше
нулевой отметки и верхний конец бюретки присоединяют к промывной
склянке с раствором, которым заполнена бюретка.
Стеклянные дозаторы для жидкостей [25, с. 233]
Дозирование жидкостей — одна из наиболее массовых операций
аналитической лаборатории любого профиля.
Механизации и автоматизации процесса дозирования в
последние годы уделяется все большее внимание. Это стимулировало
создание ряда механизированных ручных, полуавтоматических и
автоматических дозаторов циклического и непрерывного действия.
Конструкции различаются способами фиксации уровня жидкости
в мерном сосуде при его заполнении и типом запорных устройств.
57
^
u
Рис. 39. Дозатор ДР и пользование им.
При точном дозирование, например при отмеривании реагента,
объем которого входит в уравнение для расчета результатов
анализа, к дозаторам предъявляются такие же требования по
точности, как и к бюреткам и пипеткам (0,1—0,2% дозируемой
величины). Вспомогательные жидкие реагенты, объем которых не
оказывает существенного влияния на результаты анализа (привнесении
определенного объема растворителя, кислоты или щелочи для
создания нужной среды, при добавлении буферного раствора и т. п.),
дозируют с меньшей точностью (1—2%).
Стеклянные дозаторы бывают одно- и многопозиционные. Одно-
позиционные предназначаются для взятия одной определенной
порции жидкости, многопозиционные — для взятия регулируемых
мерных порций.
Однопозиционные дозаторы. Для отмеривания постоянных
объемов жидких вспомогательных реагентов удобны
однопозиционные сифонные дозаторы растворов ДР (рис. 39).
Допускаемые отклонения от номинальной вместимости при 20 °С
не должны превышать ±2%. В ЧССР подобные дозаторы получили
название опрокидывающихся пипеток.
Однопозиционные дозаторы типа ДР выпускаются по ГОСТ
6859-77 для дозирования серной кислоты и изоамилового спирта
при определении содержания жира в молоке и молочных продуктах
бутирометром. С помощью подобных дозаторов осуществляется
быстрое отмеривание объемов жидкости, не требующее установки
уровня, так как избыточная жидкость стекает в склянку, к которой
присоединено дозирующее устройство.
Для заполнения дозатора склянку, к которой он присоединен,
наклоняют так, чтобы жидкость вливалась в пипетку через
внутреннее отверстие. Затем склянку приводят в первоначальное
положение. При этом избыток жидкости стекает обратно в склянку.
Наклоняя склянку, отмеренный объем жидкости выливают через
сливное отверстие.
58
Полуавтоматические и автоматические дозаторы. В последние
годы отечественная промышленность освоила производство
полуавтоматических и автоматических дозаторов жидкостей [46, с. 139].
Принцип действия дозатора ДШ-20 понятен из рис. 40. С
поворотом крана 2 жидкость из резервуара 5 заполняет стеклянный
шприц до строго определенного уровня, регулируемого упорным
винтом 4. По достижении установленного уровня последующим
поворотом спускового крана 2 отмеренный объем жидкости сливают.
' К полуавтоматическим жидкостным дозаторам поршневого типа
можно отнести и лабораторную пневматическую
пипетку (рис. 41). Она состоит из стеклянной пипетки / с
делениями и поршневой системы типа медицинского шприца 2,
соединенных резиновой трубкой 4. Раствор, набранный в пипетку,
вытесняется из нее только при перемещении поршня, для герметизации
которого служит слой масла 3. Израсходованный объем раствора
можно отсчитать как по изменению уровня раствора в пипетке, так
и по перемещению поршня, предварительно калиброванного в
единицах объема. Такие пипетки часто используются в качестве
бюреток для титрования растворов.
Автоматические пипетки чаще всего используются для точного
дозирования.
Серийно выпускается лабораторный автоматический
однокомпонентный дозатор ЛАДА. Он предназначен для
дозирования водных растворов, в том числе слабоагрессивных. Два
переключаемых дозирующих элемента поршневого типа снабжены
самоуправляющимися клапанами и электроприводом. Электриче-
Рис. 40. Дозатор полуавтоматический ДШ-20:
/ — штатив; 2 — двухходовой кран; 3 — стеклянный шприц: 4— упорный винт: 5 —резервуар;
АВ—гидростатический напор для заполнения шприца; БВ— напор при опорожнении шприца.
Рис. 41. Лабораторная пневматическая пипетка:
/ — стеклянная пипетка; 2—шприц; 3 —слой масла; 4—резиновая трубка.
59
екая схема дозатора обеспечивает одиночное дозирование,
непрерывное дозирование и дозирование заданного количества доз (от 2
до 10) с цифровой индикацией порядкового номера выдаваемой
дозы. Пользуясь прибором, можно выдавать не менее 10 доз в
минуту.
ЛАБОРАТОРНАЯ ПОСУДА ИЗ ПРОЗРАЧНОГО КВАРЦЕВОГО СТЕКЛА
Кварцевое стекло обладает высокой стойкостью к действию
высоких температур и температурным перепадам. Коэффициент
линейного теплового расширения прозрачного кварца 5 • 10~7 К"1,
т. е. примерно в 15 раз меньше коэффициента расширения
обычного химико-лабораторного стекла. Посуду из кварца можно
нагревать на голом пламени газовой горелки и сразу же охлаждать.
Известны два сорта кварцевого стекла: прозрачный кварц,
получаемый из плавленого кристаллического кварца, и молочно-
матовый (непрозрачный кварц), получаемый из чистого
кремниевого песка. Мутность последнего вызвана большим содержанием
пузырьков воздуха.
Изделия из прозрачного кварца пропускают как видимые, так и
ультрафиолетовые лучи; размягчаются только при 1400°С, однако
они быстро расстекловываются при температуре свыше 1100°С.
Кварцевое стекло обладает наивысшей, по сравнению с другими
-стеклами, химической стойкостью по отношению к воде и кислым
агрессивным средам. Однако при высокой температуре кварц
разрушается щелочами, карбонатами щелочных металлов и оксидами
металлов. При этом изделия при охлаждении растрескиваются.
Посуда из прозрачного кварцевого стекла применяется для
работы с кислыми и нейтральными веществами при температурах
до 1000 °С. Нельзя забывать, что фтористоводородная (плавиковая)
и фосфорная кислоты разрушают кварц.
Следует также иметь в виду, что кварцевое стекло более хрупко,
чем обычное, и его надо охранять от удара.
Из прозрачного кварцевого стекла изготовляют тигли, чаши,
стаканы, колбы, пробирки и др.
ФАРФОРОВАЯ ЛАБОРАТОРНАЯ ПОСУДА [46, с. 51]
Основное преимущество фарфора по сравнению со стеклом —
термостойкость и механическая прочность. Твердый фарфор, из
которого изготовляют лабораторную посуду, без покрытия
глазурью выдерживает нагревание до 1300 °С; фарфор, покрытый
глазурью, размягчается при 1200 °С. Коэффициент линейного
расширения у фарфора примерно такой же, как у стекла «пирекс»..
Тонкостенная фарфоровая посуда выдерживает резкие перепады
температур и поэтому может быть использована для прокаливания
веществ на газовой горелке. Однако изделия из фарфора следует
разогревать постепенно, повышая температуру не резко, разогретый
сосуд не брать холодными щипцами и не ставить на холодную
подставку.
Упаривание в фарфоровой чашке следует осуществлять либо
на водяной или песчаной бане, либо на асбестовой сетке. При
обогревании открытым пламенем перепад температур на границе
фарфор — жидкость настолько велик, что фарфор может дать трещину.
Все фарфоровые изделия, предназначенные для нагревания и
хранения жидкостей, покрывают изнутри глазурью. Сопротивление
фарфоровой посуды действию химических веществ при высокой
температуре зависит прежде всего от качества глазури.
Обычная глазурь по составу примерно соответствует стеклам
с высоким содержанием Si02 и АЬОз. Поэтому ее устойчивость по
отношению к горячим кислотам почти такая же, как у кварцевого
стекла, т. е. концентрированные минеральные кислоты, за
исключением фтористоводородной и фосфорной, при повышенной
температуре на фарфор не действуют. Зато концентрированные растворы
щелочей при нагревании заметно разрушают фарфор.
Фарфоровую посуду преимущественно используют для проведе.-
ния химико-аналитических работ и подсобных операций в
препаративных лабораториях.
Моют ее так же, как стеклянную (см. гл. 5). Тонкостенный
фарфор можно очистить от нерастворимых в минеральных кислотах
органических загрязнений выжиганием.
Для нагревания и проведения простейших операций
выпускаются фарфоровые стаканы. Для выпаривания жидкостей и
кристаллизации веществ служат выпаривательные чаши и кастрюли. Так
как стенки фарфоровых кастрюль сравнительно толсты, нагревать
их нужно равномерно,' иначе они могут лопнуть. Для сплавления,
спекания, озоления и прокаливания служат высокие и низкие
фарфоровые тигли. Тигли Гуча — фарфоровые, с сетчатым дном и
фарфоровой пластинкой (рис. 42) — предназначены для
фильтрования отсасыванием химически активных веществ через асбестовый
вкладыш. Для фильтрования осадков и отделения твердых веществ
от жидкостей в горячем и холодном состоянии под уменьшенным'
давлением служат воронки Бюхнера (рис. 43). Для
нагревания, расплавления и прокаливания веществ с одновременным
улавливанием отходящих газов или подводом газов служат тигли
Розе (рис. 44).
и у ь
Рис. 42. Тигель Гуча.
Рис. 43. Воронка Бюхнера,
Рис. 44. Тигель Розе.
61
ЛАБОРАТОРНАЯ ПОСУДА И ИЗДЕЛИЯ ИЗ ПЛАТИНЫ [46, с. 56]
Лабораторная посуда и изделия из платины находят
применение в химических лабораториях, занимающихся анализом
силикатов, минералов горных пород, электрохимией и др.
Температура плавления платины 1769 °С. Для повышения
твердости и прочности в нее добавляют небольшие количества иридия
или родия.
При комнатной температуре платина стойка ко всем реагентам,
кроме царской водки и брома. Платина медленно реагирует с
горячей концентрированной азотной кислотой и с кипящей серной
кислотой. Под действием расплавленных щелочей и оксидов
металлов, цианидов и сульфидов щелочных металлов платина
корродирует, особенно в присутствии кислорода и окислителей. Платина
реагирует с галогенами при нагревании, а с фосфором, мышьяком»
кремнием, серой, селеном и углеродом лишь при температуре
красного каления.
Платина — драгоценный металл, и поэтому с платиновыми
изделиями следует обращаться очень осторожно и бережно, строго
придерживаясь установленных правил.
Платиновую посуду и изделия разрешается применять:
1) при химическом определении щелочных металлов в
силикатах сплавлением с СаС03 и NH4C1;
2) при сплавлении и спекании веществ с Na2C03 и К2С03 (но
не Li2C03) с целью переведения в раствор сульфатов
щелочноземельных металлов и кислотоупорных оксидов металлов;
3) при получении безводного Na2C03 для установления титра
кислот;
4) при определении Si02 сплавлением силикатов с К2СО3 и
Na2C03;
5) при сплавлении с бурой (борным ангидридом) огнеупорных
оксидов металлов, не растворяющихся в кислотах;
6) при работе с HF;
7) при определении нелетучего остатка в кислотах;
8) при определении галогенов в органических соединениях,
содержащих щелочные металлы;
9) при озолении и определении теплоты сгорания веществ;
10) при электровесовом определении металлов;
11) для потенциометрического, кондуктометрического, амперо-
метрического и полярографического анализа;
12) для арбитражного и микрохимического анализа и в новых
Областях аналитической химии.
Из плдтины изготовляют лабораторную посуду (тигли, чашки,
воронки, стаканы, колбы, пробирки и др.), лабораторные
принадлежности (треугольники для тиглей, лодочки для элементного
анализа, ложки, шпатели и др.), а также электроды для
электродиализа и внутреннего электролиза.
При пользовании платиновой посудой необходимо выполнять
правила обращения с изделиями из платины Г64, с. 216].
62
1. Платина очень мягкий металл, поэтому обращаться с посудой
и изделиями из платины следует осторожно. Аккуратное
обращение предотвращает деформацию изделий, образование прогибов и
царапин. Для выравнивания платиновых тиглей и чашек
используют деревянные формы (болванки) и деревянный молоток.
2. При растворении веществ в платиновой посуде помешивать
нужно стеклянной палочкой с хорошо оплавленным концом.
3. В платиновой посуде нельзя нагревать или сплавлять
следующие вещества:
а) оксиды и гидроксиды щелочных и щелочноземельных
металлов;
б) растворы галогенов и веществ, выделяющих их: царскую
водку, НС1 вместе с окислителями (КМпО^ К2СГ2О7, КС103 и Др.);
в) тяжелые металлы (Pb, Sn, Cd, Bi, Cu, Sb, Fe, Ag) и такие
их соединения, которые могут легко восстановиться до металла;
восстановителем служит углерод, образующийся при сгорании
бумажного фильтра, а также органические вещества, которые могут
находиться в осадках; платина легко сплавляется с этими
металлами, отчего становится рыхлой и хрупкой;
г) соединения фосфора и мышьяка в присутствии
восстановителей; фосфор и мышьяк легко соединяются с платиной при высокой
температуре, что приводит к порче.платиновой посуды;
д) Na2S, смеси Na2SC>4 с углем, силикатов с углем. С платино*
вой посудой нельзя также работать при анализе неизвестных
веществ.
4. Прокаливать платиновые тигли и чашки можно только на
несветящемся (наружном окислительном) пламени газовой горелки
или в электрических муфельных печах. Прокаливание платиновых
тиглей в коптящем, светящемся или внутреннем конусе пламени
горелки (см. гл. 13) не допускается, это приводит к порче изделий
вследствие образования углеродистой платины.
Бумажные фильтры можно сжигать в платиновом тигле только
при условии постепенного нагревания, когда бумага не
воспламеняется, а обугливается, и затем уголь выгорает при низкой
температуре и достаточном доступе воздуха.
5. При прокаливании на горелке тигли вставляют в обычный
фарфоровый треугольник с неповрежденными и не покрытыми
оксидом железа фарфоровыми трубками, но лучше эту операцию
осуществлять на треугольниках из толстой платиновой проволоки.
Учитывая, что горячие платиновые изделия легко сплавляются
с железом, лабораторные щипцы для работы с раскаленными
тиглями должны иметь платиновые наконечники. Нельзя: держа в та-
-ких щипцах тигель, прокаливать его при высоких температурах
длительное время; погружать концы щипцов в кислоту; класть
щипцы непосредственно на стол; брать щипцами за края чашки,
заполненной жидкостью, так как это может привести к разрыву
платины. Однако, если платиновый тигель охладить до
температуры ниже красного каления, то его можно брать щипцами из
никеля или из нержавеющей стали.
63
6. Нельзя нагретые до красного каления тигли резко охлаж<
дать, погружая их в воду. Водой можно охлаждать тигли,
имеющие температуру ниже красного каления.
7. При извлечении твердых веществ, приставших к стенкам
тигля, нельзя очищать их стеклянной палочкой, так как при этом
можно поцарапать металл. Для удаления плава нельзя сминать
стенки тигля, так как при этом легко образуются неровности,
складки и могут появиться трещины.
8. При прокаливании новых платиновых изделий в муфельной
печи рекомендуется ставить их так, чтобы они не соприкасались
друг с другом, так как при температуре свыше 1000 °С может
произойти их слипание.
9. При работе с платиновыми электродами следует соблюдать
общие правила обращения с изделиями из платины; перегибы и
деформация электродов недопустимы.
Платиновая посуда и другие изделия из платины должны быть
всегда чистыми и блестящими.
Грязные тигли и чашки очищают, промывая их в конц. НС1 или
30—40% HNO3. Если при-этом тигель не очищается, то в нем
расплавляют пиросульфат натрия Na2S207 и держат в расплавленном
состоянии при возможно более низкой температуре 5—10 мин.
Расплавленную массу затем выливают в сухой песок, прежде чем она
затвердеет. После этого тигель снова обрабатывают горячей
конц. НС1, а затем дистиллированной водой. Если сплавление
с ЫагЭгОг не дало должного результата, то повторяют операцию
сплавления, но уже с Na2C03. Рекомендуется также очищать
платиновые тигли и чашки плавлением в них хлорида магния-аммония
(MgCl2-2NH4Cl) при 1100—1200 °С. После выщелачивания плава
водой платиновое изделие становится белым и блестящим. После
повторных прокаливаний поверхность платины иногда становится
серой и утрачивает блеск, что связано с начинающимся процессом
рекристаллизации платины. В этих случаях рекомендуется
поверхность платинового тигля или чашки время от времени осторожно
протирать тонким кварцевым песком. Для этого порцию кварцевого
песка, просеянного через сито с отверстиями в 0,1 мм, наносят на
влажный ватный тампон и, слегка прижимая пальцем, полируют
им внутреннюю поверхность тигля или чашки до появления
металлического блеска, после чего промывают НС1 й дистиллированной
водой.
Платиновые электроды перед работой необходимо тщательно
очистить. Для этого их погружают на 10—20 мин в теплую
разбавленную (1:1) HN03, тщательно промывают водопроводной, а
затем дистиллированной водой (погружением в стакан с водой).
После этого электроды погружают последовательно в этиловый
спирт, диэтиловый эфир, а затем сушат несколько минут в
сушильном шкафу или над горелкой, покрытой асбестовой сеткой.
При этом следует помнить, что нельзя прикасаться руками к
рабочим частям электрода. Чистые электроды следует хранить в
вертикальном положении в высоком стеклянном стакане,
64
глава з. ВСПОМОГАТЕЛЬНЫЕ ПРИСПОСОБЛЕНИЯ
И МАТЕРИАЛЫ
ПРИСПОСОБЛЕНИЯ ДЛЯ СБОРКИ УСТАНОВОК
Штативы и держатели
Массивные металлические штативы для крепления посуды и
сборки лабораторных приборов и установок, штативы для хранения
и подогрева пробирок (из дерева, пластмасс и металла), штативы
для пипеток — неотъемлемые части лабораторного инвентаря.
Универсальный лабораторный штатив Бунзена (рис. 45) служит
для закрепления бюреток, холодильников, колб, делительных
воронок и т. д.
Внутренняя часть держателей обычно покрыта пробкой, либо
на губки натянуты отрезки резиновой трубки, чтобы не раздавить
стеклянную посуду. При сильном нагревании держателей
целесообразно аккуратно обмотать губки тонким асбестовым шнуром.
В препаративных лабораториях удобно пользоваться
универсальным штативом (рис. 46). Он состоит из двух опор У, двух
ползунов 2, двух поперечных планок 5, двух штанг 4 с задними
кронштейнами и семи стоек 5. Стойки закрепляются упорными винтами 6.
Раму штатива с ползунами можно передвигать вперед и назад
и закреплять винтами-фиксаторами 7. Поперечные штанги также
перемещаются вниз или вверх, а стойки, все вместе или каждую
в отдельности, можно перемещать по высоте или в любую сторону.
Детали штатива защищены антикоррозионным покрытием. Штатив
комплектуется набором держателей и двойными зажимами.
"\1
-ее
5-
4-
Р
N
Ik
О
*а
г
2
Рис. "45. Лабораторный штатив (Бунзена):
J—3—держатели (малый, средний и большие); 4 — кольца: 5 —вилка.
Рис. 46. Универсальный штатив:
/ — опоры; 2—ползуны; 3—поперечные-планкн; 4 — штанга; 5—стойка; б—упорные винты;
7 — винты-фиксаторы.
3 Зек. 1237
бб
о
Рис. 47. Металлический штатив для
подогрева пробирок.
Для пробирок выпускаются полиэтиленовые штативы на 10, 20
и 40 гнезд, алюминиевые на 12, 24, 48 и 60 гнезд и резиновый на
40 гнезд.
Для подогрева пробирок на водяной бане выпускаются
металлические штативы (рис. 47).
Пробирочные держатели (рис. 48) представляют собой две
пружинящие металлические или деревянные пластины, закрепленные
в деревянной ручке. Применяются при подогревании пробирок
в пламени горелки или в бане с теплоносителем.
Пробки
Кроме укупорки лабораторной посуды и приборов пробки
используют в качестве соединительных элементов при сборке лабора*
торных установок.
Рис. 49. Типы стеклянных пробок со шлифами:
а —массивные; б—пустотелые; в — пустотелые с соединительным краном; г—пустотелые
с внутренним крючком.
Рис. 50. Пробкомялки.
Стеклянные пробки с конусными взаимозаменяемыми
шлифами показаны на рис. 49. У массивных пробок шлиф бывает
и удлиненный и укороченный.
Натуральная пробка обладает упругостью,
значительной термостойкостью, и химической устойчивостью в отношении
паров большинства органических веществ. Однако она мало
устойчива к действию галогенов, сильных окислителей,
концентрированной азотной и серной кислот, а также щелочей.
В зависимости от качества коры пробкового дуба различают
следующие виды пробок: бархатные, полубархатные, средние и
простые. Из прессованной дробленой коры вырабатываются только
пробки среднего вида.
Номера корковых пробок соответствуют следующим размерам
(диаметрам верхнего и нижнего оснований):
Номер
о
1
2
3
Размер,
мм
9X7
ЮХ8
12ХЮ
14X12
Номер
4
5
6
7
Размер,
мм
16ХН
12X16
20X18
22X20
Перед использованием корковых пробок, новых или старых,
полезно повысить их эластичность. Для этого их следует опустить
на 3—4 мин в кипящую воду, а затем в 2% раствор салициловой
кислоты После этого пробки промывают дистиллированной водой
и сушат на воздухе при комнатной температуре. Затем пробки
обжимают пробкомялкой (рис. 50).
Пробку почти целиком вкладывают в подходящее по размерам
углубление и, многократно поворачивая, обжимают верхней частью.
Не нужно сжимать сильно —пробка может растрескаться. В
круглых пробкомялках пробка постепенно закатывается между кругом
и неподвижным полукругом. Если требуется обжать только часть
пробки, ее вставляют на необходимую глубину и придерживают
левой рукой.
До обжимания диаметр корковой пробки должен быть несколько
больше диаметра отверстия посуды. После обжимания пробка при
3* б7
незначительном нажиме должна входить до половины в горло
колбы или отверстие пробирки. Недопустимо с силой вдвигать
пробку в горло колбы. Вставляя пробку, следует держать колбу
левой рукой, предварительно обернув горло колбы полотенцем.
Для сверления пробок выпускаются наборы ручных
сверл — металлических трубок, один конец которых снабжен
ручкой, а другой заточен. Выбранное сверло должно иметь диаметр
немного меньше диаметра заданного отверстия.
Сверлить пробку следует начиная с нижнего торца. Пробку бе*
рут в левую руку, а сверло — в правую. Сверло нужно
равномерно вращать в одном направлении, не прилагая больших усилий,
иначе отверстие, по крайней мере у корковых пробок, будет
неровное, с рваными краями. При сверлении пробка должна опираться
на деревянную дощечку, а не на стол. Просверленные отверстия
должны быть параллельны стенкам горла.
В процессе работы сверла, естественно, тупятся. Для точки сверл
применяют специальный нож (рис. 51). Сверло надевают на
коническую часть, нож прижимают к сверлу, которое держат в левой
руке. Нож вращают правой рукой.
Отжатые и просверленные пробки для повышения химической
стойкости пропитывают парафином, коллодием или полиэтилен-
парафиновой массой.
Парафинирование производят следующим образом: пробки на
15—20 мин погружают в раствор из 100 ч. воды, 50 ч. глицерина
и 30 ч. желатины, нагретый»до 40—50 °С, затем их обмывают водой
и высушивают в сушильном шкафу при 80—90 °С. Высушенные
пробки вносят на несколько минут в расплавленную смесь из
42 ч. парафина и 12 ч. вазелина. Температура смеси должна быть
в пределах ПО—125 °С. При более высокой температуре или более
продолжительной обработке пробки сильно разбухают и
становятся слишком мягкими. По окончании пропитки пробки кладут
остывать нижним торцом кверху.
Полиэтилен-парафиновую массу приготовляют сплавлением при
120 °С 1 ч. полиэтилена и 5 ч. парафина. Расплавленной массой
покрывают поверхность пробки и сразу же вставляют ее в
горловину сосуда. Такое покрытие хорошо противостоит действию кислот
и щелочей, но оно совершенно непригодно при работе с
органическими растворителями [13].
При сборке установки прежде всего нужно укрепить ее части
(холодильник, дефлегматор, термометр, капельную воронку) в от-
ч
м
U
Рис. 51. Затачивание сверла.
63
Рис. 52. Пресс для просечки отверстий в пробках»
верстиях, а уже потом вставить пробку в
горло колбы или в соответствующее
отверстие прибора.
Вставляя в отверстие стеклянную
трубку, нужно держать ее ближе к тому концу,
который вставляется в пробку (концы
трубки должны быть оплавлены). Отверстие в
пробке следует смачивать водой или
глицерином.
Резиновые пробки легко
обрабатываются и обеспечивают достаточную
герметичность сосуда или прибора. Однако
они- разрушаются и теряют эластичность
под действием света, хлористого водорода и сильных окислителей
(галогены, оксиды азота и др.). Кроме того, они легко набухают
под действием паров органических растворителей, в особенности
ароматических углеводородов, и часто являются причиной
загрязнения органических веществ при перегонке. Резиновые пробки,
бывшие хотя бы один раз в работе, уже загрязнены, и повторное
их использование возможно лишь после длительной обработки
кипящей водой.
Чтобы уберечь резиновые пробки от старения, их держат в воде,
глицерине, в 5% растворе карбоната аммония или 3% растворе
фенола.
Для повышения устойчивости резиновых пробок к действию
агрессивных химических веществ их пропитывают парафином при
iOO°C в течение 1 мин, после чего помещают в сушильный шкаф
на проволочную сетку на 30—40 мин при 100—105 °С.
Сверлят резиновые пробки так же, как и корковые, только
сверло не следует вращать в одном направлении.
После каждого полуоборота необходимо менять направление
вращения; при этом нужно извлекать сверло и добавлять в
отверстие глицерин. В противном случае отверстие по мере углубления
суживается.
Выпускается специальный пресс для просечки
отверстий в резиновых пробках (рис. 52).
По окончании сверления вынимают сверло и выбивают из него
стержень пробки. Оставлять стержень пробки в сверле недопустимо.
Просверленную пробку обмывают обильно водой, если смазкой при
сверлении служил глицерин.
Номера резиновых пробок отвечают горловине сосуда,
герметизируемого пробкой. Диаметр верхнего основания на 2—3 мм
превышает диаметр горловины, нижнего — на 2—3 мм меньше. По
ГОСТ 7852—76 выпускаются резиновые пробки следующих
номеров: 5; 7,5; 10; 12,5; 14,5; 16; 19; 21,5; 2.4; 29; 34,5;
40; 45; 50; 60; 85-
69
Пластмассовые пробки, преимущественно
полиэтиленовые, весьма удобны для укупорки, но их нельзя использовать при
монтаже аппаратуры.
Полиэтиленовые пробки выдерживают нагревание до 70—80 °С,
устойчивы по отношению к растворам кислот средних
концентраций и щелочей, но подвергаются порче при действии
концентрированных минеральных,кислот, сильных окислителей, H2S и ССЦ.
Полиэтиленовые пробки выпускаются диаметром 11, 13, 16, 20
и 23 мм.
Резиновые и пластмассовые трубки и зажимы для трубок
Для подвода воды и газа, монтажа лабораторных установок и
приборов, в том числе аппаратуры, работающей при пониженном
и небольшом избыточном давлении, широко используются
резиновые и пластмассовые трубки.
Резиновые технические трубки, мягкие и средней
твердости, тонко- и толстостенные, изготовляются следующих ти-<
пов: кислотощелочестойкие для работы в растворах
кислот и щелочей концентрацией до 20% (исключая азотную и
уксусную кислоты); теплостойкие для работы при температуре
воздуха до 90 °С и в среде водяного пара до 140 °С;
морозостойкие для работы при температуре до —45°С; маслобен-
зостойкие, мягкие, средней и повышенной твердости, для
работы в среде масла или бензина.
Вакуумные резиновые трубки со стенками, толщина
которых равна диаметру отверстия (или больше), можно
применять .при разрежении до 1,3-10~6 Па (10~6 мм рт. ст.) в диапазоне
температур 8—70 °С.
Тонкостенные трубки нельзя использовать при сборке
аппаратуры для работы в вакууме, так как они при разрежении слипаются.
Для подвода воды и газа обычно используют сравнительно
тонкостенные трубки, достаточно эластичные, чтобы их можно
было надеть на стеклянные или металлические трубки. Перед
надеванием резиновую трубку целесообразно смочить изнутри
водой или глицерином. Необходимо, чтобы конец резиновой трубки
был ровно обрезан, а конец стеклянной или металлической трубки
не имел острых граней. Концы стеклянных трубок должны быть
предварительно оплавлены, а металлических — отшлифованы.
Стеклянную трубку большого диаметра следует держать в
непосредственной близости от того конца, на который надевают
резиновую трубку. При работе с тонкостенными трубками
стеклянную и резиновую трубки обматывают полотенцем.
Новые резиновые трубки почти всегда внутри содержат тальк,
который удаляют длительной обработкой струей воды и
разбавленными растворами щелочи.
Если толстостенные резиновые трубки предназначены для
работы со ртутью, то прежде всего необходимо обработать их в
течение 1—2 ч нагретым до 70 °С 20% раствором КОН, а затем в
течение 2 ч горячей дистиллированной водой, чтобы удалить серу. ,
70
Рис. 53. Винтовые (а) и пружинные {б) зажимы для резиновых трубок.
Резиновые трубки следует хранить в помещении, защищенном
от прямых солнечных лучей, при температуре от 0 до +25°С, при
относительной влажности воздуха не более 65%, не менее чем в
1 м от отопительных приборов.
Затвердевшие резиновые трубки применять нельзя, и пытаться
их размягчить не следует.
Пластмассовые трубки изготовляют преимущественно
из пластифицированного поливинилхлорида и полиэтилена.
Для подвода воды и газа к приборам, работающим при
атмосферном давлении, наиболее пригодны медицинские поливинилхло-
ридные трубки.
Поливинилхлоридные шланги менее эластичны, чем резиновые,
но более устойчивы к озону, хлору, галогеноводородам и другим
агрессивным веществам. При кратковременном погружении конца
шланга в кипящую воду (температура размягчения
поливинилхлорида 80 °С) пластичность возрастает настолько, что шланг без
особых усилий надевается на трубку большего диаметра, чем
диаметр шланга.
Полиэтилен обладает меньшей термопластичностью, чем поли-
винилхлорид, и, естественно, надевать подобные шланги на
стеклянные трубки значительно труднее. Температура размягчения
полиэтилена НО—120°С, поэтому приходится осторожно нагревать
конец шланга на слабом пламени горелки, избегая деформации
трубки и ее загорания. Следует также отметить, что по
охлаждении полиэтиленовая трубка прилегает к стеклу не так плотно, как
поливинилхлоридная. Для непродолжительного перекрывания или
уменьшения потока жидкости или газа через шланг чаще всего
используют металлические зажимы, пружинные или винтовые
(рис. 53). В установках, собранных на длительное время, целесо«
образнее использовать стеклянные соединительные краны.
ПРИСПОСОБЛЕНИЯ ДЛЯ РАБОТЫ С РЕАКТИВАМИ
Для перенесения из емкости в емкость
Снимать осадок со стенок сосуда или с фильтров, переносить
осадки, брать твердые реактивы из банки, разрыхлять
слежавшиеся реактивы нужно шпателем.
71
Для переноса небольших количеств твердых веществ из одной
емкости в другую служат фарфоровые ложки.
Пересыпать в колбу твердые кристаллы и порошки удобно с
помощью стеклянной воронки; этой же воронкой можно
пользоваться при переливании очень густьдх, вязких растворов.
Воронки необходимы при работе с посудой, имеющей шлифы.
Для прокаливания
Стальные и никелевые тигли и чашки служат для
прокаливания вспомогательных материалов (например, оксида меди для
аналитических целей), а также в качестве сосудов для песчаных,
масляных и металлических бань. Никель окисляется на воздухе
при повышенной температуре. Поэтому никелевые тигли не могут
служить для прокаливания осадков, которые надо затем
взвешивать. Очень удобны никелевые тигли для сплавления различных
веществ с Na202, NaOH и КОН. Такие тигли могут выдержать
большое число сплавлений. Стальные тигли также можно
применять для сплавления с Na202, но они быстрее изнашиваются.
Следует учитывать, что при последующем растворении сплавов в
кислотах раствор загрязняется железом.
Щипцы для тиглей нужно класть на стол так, чтобы
изогнутые концы их были обращены вверх.
При прокаливании тиглей в пламени газовой горелки их
устанавливают на треугольниках. Фарфоровые трубки
треугольников монтируются на каркасе из проволоки.
Для измельчения проб
По мнению некоторых авторов, фарфоровые ступки и
пестики не следует применять в аналитической практике, так
как испытуемое вещество может оказаться более твердым, чем
фарфор. Для измельчения реактивов их применяют с
осторожностью, так как не исключено попадание фарфоровой пыли в
измельчаемый материал. Более пригодны ступки и пестики из
агата, почти не истирающегося во время измельчения.
Ступки из закаленной стали применяются для
предварительного грубого измельчения. Металлические ступки нельзя
применять, если измельчаемое вещество обладает большей
твердостью, чем металл ступки и если оно может вступать во
взаимодействие с металлом (кислые соли органических и неорганических
кислот и др.)-
В фарфоровых и агатовых ступках, в отличие от
металлических, вещество нельзя толочь, а можно только растирать.
Агатовые ступки нельзя нагревать выше 100 °С — они могут
лопнуть.
Перед пользованием ступки тщательно моют и. сушат в
сушильном шкафу при температуре не выше 100°С. Если внутренние
стенки ступки и пестик нельзя очистить обычным мытьем, в ступ-
7£
ке растирают немного NaCl, через некоторое время соль удаляют
и ступку с пестиком моют водой. Для механической очистки можно
растирать в ступке кварцевый песок.
Измельчаемое вещество насыпают в ступку тонким слоем.
Крупные куски разбивают осторожными ударами пестика, а
затем растирают круговым движением пестика. Неизмельченные
частицы, скапливающиеся в верхних частях внутренней
поверхности ступки, сталкивают пестиком вниз и продолжают истирание.
По окончании работы ступку и пестик нужно хорошо вымыть и
высушить.
ПЛАСТМАССЫ КАК МАТЕРИАЛ ДЛЯ ЛАБОРАТОРНЫХ ИЗДЕЛИЙ
[36, с. 30]
Лабораторные изделия из полимерных материалов, по большей
части обладающих значительной химической стойкостью и малой
теплостойкостью, предназначаются для работ с холодными
средами. В табл. 2 приведены ориентировочные данные о свойствах
наиболее часто используемых полимеров в качестве лабораторных,
изделий и материалов.
Из поливинилхлорида изготовляют некоторые виды
лабораторной посуды, шланги, покрытия для лабораторных столов
и др. Изделия из поливинилхлорида могут применяться при
температурах от —20 до 60 °С. При 70—80 °С поливинилхлорид
переходит в резинообразное состояние, выше 140 °С разлагается с
выделением НС1, а при охлаждении ниже —20 °С становится ломким.
Изделия из полиэтилена высокого давления можно
применять до 60 °С, низкого — до 80 °С. При —30 °С полиэтилен стано-
ТАБЛИЦА 2. Термостойкость и химическая стойкость некоторых
полимеров
Полимер
Поливинилхлорид
Полиэтилен
Политетрафторэтилен (фторо-
шгаст-4, тефлон)
•Полистирол
Полиметилметак-
рилат ,-^^_
Температура
размягчения, °С
80-100
105-180
327
90-120
70-120
Стойкость в различных
средах *
вода
С
с
с
с
с
щелочи
С
с
с
с
НС
кислоты
с
с
с
с
УС
окислители
С
с
с
НС
НС
органические
растворители
УС
НС
С
НС
УС
Растворитель
Циклогексанон
Ароматические
углеводороды при
нагревании
Нет растворителя
Ароматические
углеводороды, кетоны,
альдегиды, эфиры
Толуол, эфиры
♦Принятые обозначения: С —стоек; УС—в определенных условиях подверг
гается действию среды; НС —нестоек.
73
вится твердым. Полиэтилен хорошо выдерживает HF. Даже при
100 °С на него не действует конц. НС1, 40% HF, 30% HN03 и
50% раствор КОН. Стоек к конц. H2SO4 при комнатной
температуре. Набухает в углеводородах и хлорированных углеводородах;
при температуре выше 65 °С растворим в толуоле. Изменяется под
действием галогенов, силиконового масла и конц. H2SO4 при
повышенной температуре.
Из полиэтилена изготовляют мерные цилиндры, стаканы,
воронки, тару, в частности для хранения плавиковой кислоты,
укупорочные пробки, шланги, прокладки и др.
Политетрафторэтилен (тефлон) выдерживает
нагревание до 300°С (только не на открытом огне), а из химических
агентов его разрушают только фтор и расплавленные щелочные
металлы. Из тефлона изготовляют лабораторную посуду
(стаканы, чашки, краны и др.), мешалки, уплотнения, пробки и т. п.
Из тефлоновых трубок и пластин в лабораториях изготовляют
мешалки, затворы для мешалок, ступки; пленками пользуются для
уплотнения соединений.
Имеются сообщения о способе изготовления фторопластовых
выпарных чашек вместимостью до 80 мл, которые успешно
эксплуатировались взамен платиновых чашек и тиглей на
протяжении 2,5 лет [9].
Из полистирола изготовляют тару для хранения
плавиковой кислоты. Пенополистирол используют в качестве
теплоизоляционного материала при низких температурах. При 160—
180 °С он размягчается и деполимеризуется. Благодаря этому из
него можно изготовлять подставки для круглодонных колб. Колбу
нагревают в пламени газовой горелки приблизительно до 200°С
и прижимают дном к блоку пенополистирола — в блоке
образуется углубление. Эту работу следует выполнять в вытяжном шкафу.
АСБЕСТОВЫЕ МАТЕРИАЛЫ
Асбестовый картон, волокнистый асбест, асбестовые шнуры и
асбометаллические сетки применяются в качестве огнезащитного,
термоизоляционного и фильтрующего материала, а также для уц-
лотнения соединений.
Асбестовый картон общего назначения и прокладочный
используется для теплоизоляции до 500 °С.
Волокнистый асбест служит фильтрующим материалом.
Асбест устойчив к щелочам, а при обработке НС1 становится
устойчивым и к кислотам.
Асбестовые шнуры общего назначения,
теплоизоляционные с примесью стеклянных нитей и с повышенной
термоизоляцией предназначаются для теплоизоляции и уплотнения деталей
стеклянных приборов и аппаратов до 400—425 °С.
Асбестовые картон и шнуры крепят на горизонтальной
поверхности раствором силиката натрия, а на приборах — мягкой мед-»
ной проволокой.
74
СМАЗКИ И ЗАМАЗКИ [46, с. 66; 60, с. 27]
Вращающиеся стеклянные детали (оси мешалок, краны, шли-
фы и др.) обычно смазывают маловязкими сортами вазелина.
Чтобы смазка была гуще, вазелин сплавляют с парафином или
ланолином. Смазкой для кранов служит смесь 1 ч. вазелина и 1 ч..
парафина, для эксикаторов рекомендована густая смазка И1
3 ч. ланолина и I ч. желтого вазелина, а также менее вязкая
смазка, состоящая из 2,5 ч. ланолина и 1,5 ч. медицинского вазелина.
Если смазанная часть подвергается действию органических
растворителей, рекомендуется пользоваться смазками на основе
глицерина:
а) смесь из 8 г растворимого крахмала, 2 г сахарозы и 25 г
глицерина осторожно нагревают при перемешивании в
фарфоровой чашке до 140 °С;
б) смесь из 20—30 г декстрина и 35 г глицерина нагревают
при перемешивании до 140°С и выдерживают при этой
температуре, пока расплав не станет прозрачным. При работе с
агрессивными газами, если не вредно присутствие небольшого количества
паров воды, хорошей смазкой могут служить сиропообразная
смазка на основе фосфорной или метафосфорной кислоты. Для
приготовления смазки 10 г НР03 растворяют в 100 мл воды, смешивают
с 2 г Н3ВОз и после полного растворения выпаривают на водяной
бане до объема 25 мл. Затем к сиропу добавляют 1 мл 85% Н3Р04
и 20—30 мин кипятят при помешивании, следя за тем, чтобы
температура не превышала 122°С. По охлаждении смазку помещают
в стеклянную банку с пришлифованной пробкой.
Большое распространение получили смазочные приборные
масла, состоящие из минеральных масел высокой степени очистки и
кремнийорганических веществ (ОКБ-122-3 и ОКБ-122-4 —
маловязкие ОКБ-122-5 и 02Б-122-14— средней вязкости). Эти смазки
обладают низкой температурой застывания (до —70 °С).
Уплотняющую замазку для шлифов готовят так: на водяной
бане сплавляют 50 ч. белого вазелина с 10 ч. парафина,
прибавляют 2—3 ч. невальцованного натурального каучука.
Образовавшуюся однородную массу переливают в банку с притертой
пробкой.
Для уплотнения стеклянных и металлических подвижных
соединений вакуумных установок выпускается смазка вакуумная
(ОСТ 38-0183—75), состоящая из 15% натурального каучука, 20%
церезина и 65% медицинского вазелинового масла *.
Большую часть перечисленных выше смазок в настоящее
время с успехом заменяют силиконовые полимеры. Они не
* Стеклянные шлифы можно уплотнять пленкой из фторопластового уплот-
нительного материала (ФУМ-О) или фторопластовой прокладочной лентой. Это
химически стойкие самосмазывающиеся прокладочные материалы для работы
при температурах от —60 до 250 °С. Кусочек ленты шириной в несколько
миллиметров накладывают равномерно на керн, вставляют его в шлифованную муфту
и поворачивают.
75
смачиваются водой, обладают низким давлением паров, и их
вязкость незначительно изменяется с температурой.
Для склеивания частей приборов, которые обычно не
разбираются, а также для герметизации стеклянной аппаратуры
используют два вида замазок: термопластичные и необратимо
затвердевающие. К замазкам первого рода относят замазку
Менделеева (см. приложение 3), смеси канифоли и пчелиного воска,
шеллак и его смеси. Их легко наносить в нагретом состоянии.
Однако все эти замазки частично или полностью растворимы в
органических растворителях и не могут быть применены при высоких
температурах.
Необратимо затвердевающие замазки, как правило,
выдерживают довольно высокие температуры. Например, замазка,
полученная при растирании 100 г обезвоженного РЬО с 25 мл глице-,
рина выдерживает температуру до 250 °С, не растворяется ни в
кислотах, ни в щелочах.
При размешивании 60% MgCl2 с оксидом магния получается
густая, быстро затвердевающая замазка, так как образуется гидр-
оксохлорид магния.
Твердые и стойкие замазки дают смеси из жидкого стекла и
карбонатов или оксидов кальция, магния, свинца и железа, а
также смеси из жидкого стекла и талька.
Однако в последнее время вместо перечисленных выше
замазок все шире начинают применять замазки на основе
высокомолекулярных материалов, особенно эпоксидных смол.
главам ХИМИЧЕСКИЕ РЕАКТИВЫ
Результативность работы химика в значительной мере зависит
от степени чистоты применяемых веществ. Согласно ГОСТ
13867—68 «Продукты химические. Обозначения чистоты», все
химические продукты подразделяются на четыре группы:
1) сырые продукты — природного происхождения и
полуфабрикаты с большим содержанием примесей;
2) технические продукты, вырабатываемые химическими
предприятиями, с относительно небольшим содержанием примесей;
3) реактивы, предназначаемые для аналитических,
препаративных и иных работ в лабораториях;
4) продукты особой чистоты, качество которых значительно
выше химических реактивов.
В лабораторной практике используются реактивы и особо
чистые вещества, качество которых регламентируется
государственными стандартами и техническими условиями.
Качество химических веществ, используемых в
фармацевтической практике, регламентируется Государственной фармакопеей
(ныне действует десятое издание — ГФ-Х).
КВАЛИФИКАЦИЯ РЕАКТИВОВ И ВЫСОКОЧИСТЫХ ВЕЩЕСТВ
В зависимости от содержания основного вещества и
допустимых примесей для химических реактивов установлены следующие
квалификации [65].
Чистый (ч.) —низшая квалификация реактива. Содержание
основного вещества ^98%; содержание примесей или нелетучего
остатка 0,01—0,5%; остаток после прокаливания — до 0,5%.
Чистый для анализа (ч.д. а.). Эта квалификация ха*
рактеризует аналитическое применение препарата. Содержание
основного вещества ^99%.
Химически чистый (х. ч.). Высшая степень чистоты пре-
парата. Содержание отдельных примесей в пределах ЬЮ-3—
ЬЮ-5 %и нелетучего остатка ^0,1%; остаток после
прокаливания <0,1%.
Высокочистые вещества, подразделяемые на
спектрально-чистые (сп.ч.), эталонной чистоты (в. э. ч.) и особо чистые
(ос. ч.).
Содержание основного вещества и предельное содержание
нежелательных примесей в эталонно-чистых веществах должно
соответствовать марке эталона (в зависимости от его назначения).
Перед символом в. э. ч. стоит цифра, обозначающая общее
процентное содержание лимитируемых примесей, а после символа —
две цифры, разделенные тире; первая означает число
лимитируемых примесей, а вторая — максимальное содержание этих при-
77
месей (в процентах), выраженное как абсолютное значение
показателя степени порядкового множителя. Например, марка
вещества эталонной чистоты, содержащего 99,998% основного
вещества, в котором лимитируется три примеси, причем их общее
содержание не должно превышать 1 • 10'5 %, обозначается «002 в. э. ч.
3—5».
Содержание примесей в особо чистых веществах лимитируется
в пределах 10~5—Ю-10 %.
Если в веществе особой чистоты лимитируются лишь
неорганические примеси, то за символом ос. ч. следуют две цифры, из
которых первая указывает число лимитируемых примесей, а
вторая— их суммарное содержание (в процентах), выраженное как
абсолютное значение показателя степени порядкового множителя.
Например, марка «кремний ос. ч. 21—5» означает, что в кремнии
лимитировано содержание 21 неорганической примеси, суммарное
содержание которых не превышает Ю-5 %.
Если для особо чистых веществ лимитируются только
органические примеси, перед символом ос. ч. ставятся буквы «оп» с
цифрой, обозначающей их предельное содержание. Например, марка
особо чистого вещества с суммарным содержанием органических
примесей 0,001% (Ы0_3%) обозначается «оп-3 ос. ч>.
Марка особо чистых веществ, для которых лимитируются как
неорганические, так и органические примеси, обозначается с
учетом содержания этих примесей. Например, марка вещества, для
которого сумма органических примесей лимитируется величиной
10"4 %, а сумма восьми неорганических примесей составляет 10~5 %,
обозначается «оп-4 ос. ч. 8—5».
Высокочистые вещества используются в новых отраслях техни-
. ки, и в зависимости от области применения к ним предъявляются
особые технические требования.
Химические реактивы классифицируют часто по областям их
преимущественного применения: индикаторы, красители для
микроскопии, для хроматографии (сорбенты, носители, неподвижные
фазы и др.), для люминофоров, для фотографии, для криоскопии,
для спектрального анализа и т. д.
Во многих случаях после названия реактива указывается
область применения реактива, а затем степень его чистоты.
Например: бензол для криоскопии х. ч., судан Ж краситель для
микроскопии ч.д. а., кальция окись для хроматографии ч. д. а. и т. д.
Единой международной квалификации химических реактивов и
высокочистых веществ не существует. Реактивы, поставляемые
странами-членами СЭВ, квалифицируются так, как принято в
СССР.
Согласно ГОСТ 3885—73, реактивы (препараты) должны быть
упакованы в соответствующую потребительскую тару,
герметически упакованы и снабжены стандартной этикеткой.
Для реактива каждой квалификации этикетка на таре должна
быть определенного цвета или на этикетке должна быть цветная
полоса:
78
Квалификация
реактива
Цвет этикетки
Чистый Зеленый
Чистый для анали- Синий
за
Химически чистый Красный
Особо чистый Желтый
Прочие Светло-коричневый
При наличии у реактивов ядовитых, огнеопасных и
взрывоопасных свойств наклеивается отдельная этикетка с надписями
«Огнеопасно» —красная; «Яд» — желтая; «Взрывоопасно» — голубая;
«Беречь от воды» — зеленая.
ОПАСНЫЕ СВОЙСТВА РЕАКТИВОВ [56, с. 139]
Многие химические вещества обладают опасными свойствами,
существенно влияющими на стабильность качественных
показателей препарата, а также способными послужить причиной пожара,
взрыва, отравления или заболевания персонала.
Влагочувствительность. В зависимости от способности
поглощать влагу из воздуха различают малогигрос£опические (нитрат
бария), сильногигроскопические (гидроксиды натрия и калия, ор-
тофосфорная кислота, ацетат натрия, хлорид алюминия) и
расплавляющиеся на воздухе вещества (диметиламин сульфат, ацет-
амидин гидрохлорид).
Поглощение влаги из воздуха может происходить при
негерметичной упаковке реактива.
Взаимодействие с влагой воздуха может носить чисто
физический характер (увлажнение, растворение), но поглощаемая влага
может привести и к необратимой химической реакции. Так,
сульфиды, нитриды, фосфиды щелочных и щелочноземельных
металлов разлагаются водой с образованием токсичных гидридов; гало-
гениды неметаллов (РС13, РС15, S2CI2 и др.) и галогенангидриды
кислот гидролизуются с образованием соответствующих кислот;
щелочные, щелочноземельные и пирофорные металлы
взаимодействуют с водой с выделением водорода (что грозит взрывом).
Светочувствительность. Под действием света некоторые
вещества подвергаются фотохимическим реакциям.
Одни вещества при этом изменяют цвет (аллилбромид, анилин,
соли бромисто- и иодистоводородных кислот, препараты ртути,
салициловой кислоты, фенолы и др.), другие подвергаются
окислению (фурфурол, ксантогидрол) или восстановлению (соединения
серебра).
Действие света может сопровождаться также процессами
изомеризации и фотолиза. Например, малеиновая кислота под
действием света частично превращается в транс-изомер — фумаровую
кислоту, а фотолитический распад трихлорэтилена сопровождается
образованием дихлорацетилена, оксида углерода, фосгена и
хлористого водорода:
2С1СН=СС12 -^ С1С = СС1 + СО + СОС12 + 2НС1
02
79
Теплочувствительность. Значительное изменение температурных
условий при хранении реактива может повлечь за собой как
обратимые (переход в другое агрегатное состояние), так и
необратимые изменения его свойств. Вещества, подвергающиеся
необратимым изменениям при воздействии тепла или холода, называют
термолабильными.
Например, формалин (водный раствор формальдегида) при
температуре ниже 9 °С образует белый осадок параформа,
который при последующем нагревании не переходит в раствор.
Металлическое олово («белое» олово) при —30 °С переходит в
«-модификацию («серое» олово) в виде серого мелкого порошка.
При повышенной температуре многие кристаллогидраты теряют
воду (при 30 °С СаС12-6Н20 переходит в СаС12-4Н20), а некоторые
вещества разлагаются, например карбонат аммония разлагается
при 58 °С по уравнению
(NH4)2C03 —> 2NH3 + C02 + H20
или полимеризуются (непредельные углеводороды, р-аминопропио-
нитрил, олеиновая кислота и др.).
Перечень реактивов, требующих особых условий хранения, и
гарантийные сроки их хранения см. [65].
Пожароопасность. Многие химические вещества
самопроизвольно или при действии внешнего источника зажигания способны к
загоранию, а некоторые — к взрыву.
Пожароопасные вещества условно разделяют на группы [69].
1. Реактивы, способные к разложению со взрывом и в
отсутствие кислорода воздуха. Они способны взрываться не только от
внешнего теплового или электрического источника, но также от
удара, трения, детонации (перхлорат аммония, дипикриламин и его
аммонийная соль, пикриновая кислота, 2,4,6-тринитротолуол,
азиды, многие перекиси и др.).
Для уменьшения взрывоопасное™ некоторые реактивы
рекомендуется сохранять увлажненными. Так, пикриновая кислота
с влажностью более 40% не является взрывоопасным
веществом.
2. Сжатые, сжиженные и растворенные под давлением газы,
например легковоспламеняющиеся водород, метан, ацетилен, оксид
углерода(II), аммиак, метиламин, диметиламин, диборан,
сероводород, циановодород, этиламин, хлористый метил, триметиламин,
трифторэтилен; поддерживающие горение сжатый и жидкий
воздух, сжатый и жидкий кислород, оксид азота(I).
3. Реактивы, выделяющие при взаимодействии с водой
легковоспламеняющиеся газы (Na, К, Са, СаН2, СаС2 и др.). Так,
щелочные металлы при контакте с водой выделяют водород,
самовоспламеняются со взрывом и разбрызгиванием металла. Карбиды,
взаимодействуя с водой, выделяют ацетилен, метан и другие
углеводороды. Карбиды щелочных металлов реагируют с водой со
80
взрывом. Карбиды меди, серебра, ртути способны взрываться от
удара и нагревания.
Гидриды металлов при увлажнении выделяют водород, а
некоторые, например гидрид метилалюминия, воспламеняются.
В присутствии воды способны разлагаться со взрывом амид
натрия, хлорсульфоновая кислота, бромид алюминия.
4. Легковоспламеняющиеся жидкости (ЛВЖ), способные без
предварительного подогревания возгораться от кратковременного
контакта с источником зажигания (спичка, искра и т. п.).
Об огнеопасности органических растворителей принято судить
по температуре вспышки /Всп, т.е. по наименьшей температуре, при
которой пары данного вещества образуют над поверхностью его
смесь с воздухом, вспыхивающую при приближении пламени.
Вещества, температура вспышки которых в закрытом сосуде ниже
61 °С или 65 °С и ниже в открытом, принято относить к
легковоспламеняющимся.
Критерием пожароопасности является также температура
самовоспламенения, т. е. температура, при которой вещество
загорается без постороннего открытого источника огня или электрической
искры.
ЛВЖ по степени пожароопасности подразделяются на три
категории: особоопасные — с £ВСп<;180'С (диэтиловый эфир,
сероуглерод, тетралин); опасные —с £Всп = 18— 23 °С в закрытом сосуде
(ацетон, бензол, диоксан, метилацетат, этилацетат, метиловый и
этиловый спирты и др.); опасные при повышенной температуре —
с /всп = 23—61 °С в закрытом сосуде (многие спирты, изопропил-
бензол, уксусная кислота, метилцеллозольв и др.).
5. Легковоспламеняющиеся твердые реактивы, способные
возгораться от источника огня и распространять горение по
поверхности (красный фосфор, сера, селен, кремний, нитроцеллюлоза,
целлулоид и другие нитросоединения, гексаметилентетрамин, ди-
метилглиоксим, пирокатехин, нафталин и др.).
6. Самовозгорающиеся реактивы, способные при контакте с
воздухом или другими веществами самонагреваться и
самовоспламеняться при нагревании до сравнительно невысоких температур
(пирофорные металлы, многие металлорганические вещества,
белый фосфор, сульфоуголь, катализатор Ренея и др.).
7. Воспламеняющие (окисляющие) реактивы, способствующие
развитию горения. Эти вещества сами по себе не горючи, но,
выделяя кислород, увеличивают интенсивность огня и в смеси с другими
веществами могут вызвать взрыв. К ним относятся перекиси
щелочных и щелочноземельных металлов, азотная кислота и
некоторые ее соли, безводная хлорная кислота,"хлораты, броматы, иода-
ты, перманганаты, соли надсерной кислоты и др.
Ядовитость. Большинство применяемых в химических
лабораториях веществ в той или иной степени ядовиты. Они могут
воздействовать на организм человека в виде газов, паров, жидкостей,
твердых веществ, а также в аэрозольном (пыль, дым, туман)
состоянии.
81
Важным фактором безопасности является надежно работающая
вентиляция, способная снижать в рабочей зоне концентрацию
вредных веществ ниже ПДК. По величине ПДК судят о классе
опасности вещества [56, с. 162].
ХРАНЕНИЕ РЕАКТИВОВ
В лабораторном помещении должны храниться лишь
небольшие запасы химических веществ. Их держат в банках, склянках с
пришлифованными стеклянными пробками или пластмассовыми
крышками с герметизирующими .прокладками из полиэтилена.
На каждом сосуде должна быть этикетка с обозначением, что
в нем находится. Для лучшей сохранности бумажные этикетки
рекомендуется заклеивать прозрачной полиэтиленовой клейкой
лентой. Временные надписи могут быть сделаны карандашом для
стекла: место, на которое намечается надпись, следует слегка
нагреть.
Строго запрещается хранить вещества в сосудах без этикеток
или надписей! Если этикетки нет, применять реактив нельзя. Если
не удается установить состав реактива, его следует уничтожить.
Небольшие количества веществ, выделяющих ядовитые или
раздражающие дыхательные пути пары (бром, олеум,
фтористоводородная и концентрированная соляная кислоты, аммиак, тионил-
хлорид, сульфурилхлорид и др.)» равно как и концентрированных
серной, азотной, уксусной кислот допустимо хранить в вытяжном
шкафу на специально отведенных для них полках.
Запасы подобных веществ следует хранить в вытяжных шкафах
вентилируемых хранилищ, доступ в которые должен быть
ограничен.
Общий запас одновременно хранящихся в каждом рабочем
помещении лаборатории огнеопасных и легковоспламеняющихся
жидкостей не должен превышать суточной потребности, но не
более 2—3 л на одного сотрудника. Склянки, в которых содержится
более 50 мл ЛВЖ, должны храниться в железных ящиках для
горючего с плотно закрывающейся крышкой, со стенками и дном,
выложенными асбестом. Ящик устанавливают на полу вдали от
нагревательных приборов; к нему должен быть удобный подход.
Запрещается хранить горючие жидкости в полиэтиленовой и в
тонкостенной посуде вместимостью более 200 мл. Недопустимо
хранить горючие жидкости в вытяжном шкафу, где производят
работы с нагревательными приборами или рядом с окислителями (С12,
Н202, Вг2, КМп04 и др.).
Щелочные металлы следует хранить под слоем обезвоженного
трансформаторного масла или керосина в стеклянной банке,
плотно закрытой корковой пробкой. Слой масла (керосина) над
металлом должен быть не менее 15—20 мм.
Количество металлического натрия или калия, хранящихся в
лабораторном помещении, не должно превышать однодневной
потребности. При длительном хранении на поверхности металличе-
82
ского калия образуются перекиси, которые при непосредственном
контакте с металлическим калием (например, при разрезании
ножом на воздухе) дают сильные взрывы.
Перекисные соединения следует хранить, как огне- и
взрывоопасные вещества. Температура их хранения должна быть
значительно ниже температуры их разложения. Нестабильные перекиси
хранят при пониженной температуре. Органические перекиси и
гидроперекиси хранят только в емкостях из стекла или
полиэтилена высокого давления.
Реактивы, изменяющиеся под действием света, хранят в желтых
или темных склянках и банках.
Ядовитые вещества (соли ртути (II), алкалоиды, некоторые
эфиры фосфорной кислоты, цианиды) следует хранить в
вентилируемых закрытых и опечатанных шкафах и сейфах.
Об условиях хранения сжиженных и сжатых газов см. гл. 20.
Металлическую ртуть в относительно малых количествах
хранят в толстостенной герметически закрытой стеклянной посуде в
вытяжном шкафу. Надежно и удобно хранение ртути в запаянных
стеклянных толстостенных ампулах по .30—40 мл. Ампулы
рекомендуется укладывать в тонкостенные стальные коробки, В таком
виде ртуть можно держать в лабораторных шкафах вместе с
другими реактивами.
При хранении запасов реактивов в первую очередь следует
организовать раздельное хранение веществ, несовместимых по
свойствам. Например, хлорат калия, перманганат калия, перекись
водорода и другие окислители нельзя хранить рядом с
восстановителями— серой, крахмалом, углем и др.
ОБРАЩЕНИЕ С РЕАКТИВАМИ [56, с. 139]
По назначению реактивы могут быть условно разделены на
общеупотребительные и специальные.
Общеупотребительные реактивы, расход которых может быть
значительным (кислоты, основания, некоторые соли, спирты и
полупродукты для синтетических работ), часто поступают в
лабораторию в крупной расфасовке. Поэтому часто возникает
необходимость отвешивать и отмеривать небольшие массы и объемы
реагентов.
Твердый реактив при хранении может слежаться. Иногда
достаточно потрясти банку, чтобы он рассыпался. В других случаях
приходится разрыхлять верхний слой тщательно очищенным, сухим
фарфоровым или роговым (но не металлическим) шпателем или
стеклянной" палочкой.
При вскрытии банки с реактивом следует сначала очистить
пробку снаружи, затем обтереть ее и горло банки чистым
полотенцем.
При взвешивании необходимо позаботиться о сохранении
чистоты реактива. Нельзя насыпать его прямо на чашку весов или
на бумагу. При взвешивании небольших количеств следует поль-
83
Рис. 54. Металлический стояк для бутылей.
Рис. 55. Приспособление для наклонения бутылей.
Рис. 56. Переливание жидкости из большой бутыли*
зоваться стаканчиками для взвешивания (бюксами).Нельзя брать
реактив руками и возвращать излишки обратно в банку!
Рассыпанный реактив надо немедленно убрать.
Жидкие реагенты взвешивают в склянках, снабженных
притертыми пробками.
Относительно большие количества реактивов следует
взвешивать в чистых, сухих банках со стеклянными и пластмассовыми
пробками. Пересыпать твердые реактивы и препараты из
большой емкости в малую удобно с помощью воронок для порошков;
при этом большую банку медленно вращают и слегка наклоняют,
чтобы вещество высыпалось тонкой струйкой.
Едкие вещества—дымящие концентрированные кислоты (HN03,
H2SO4, НС1), хлорангидриды кислот (SOCl2, PC13 и др.), 25%
раствор аммиака и другие сильнодействующие вещества
(метиловый спирт, дихлорэтан, эфир и др.) —часто поступают в
лаборатории в больших бутылях. Перевозить, переносить и хранить
подобные бутыли без обрешетки или корзины запрещается.
При переливании жидких реактивов из больших бутылей
целесообразно пользоваться металлическими стояками для бутылей
(рис. 54) и приспособлениями, которые дают возможность легко
наклонять бутыли (рис. 55).
Кислоты, щелочи и другие едкие жидкости из больших
бутылей следует разливать при помощи стеклянных сифонов с грушей
(рис. 56) или других нагнетательных средств, желательно в
вытяжном шкафу при включенной тяге.
Случайно разлитый жидкий едкий реагент сразу же засыпают
сухим песком, чтобы он впитал жидкость. Намокший песок
убирают, и это место засыпают известью или карбонатом натрия, если
разлиты кислоты или галогенангидриды, либо заливают 2%
раствором уксусной кислоты, если разлиты щелочи или водный ам«
£4
миак. После такой обработки замывают водой и вытирают насухо.
На рабочем месте необходимо иметь достаточное количество
нейтрализующих средств.
При переливании едких жидкостей обязательно пользоваться
защитными перчатками и очками, а также прорезиненным
фартуком.
Расходные количества концентрированных кислот (HNO3,
H2S04, HC1) должны храниться в толстостенной стеклянной
посуде вместимостью ^ 2 л в вытяжном шкафу на стеклянных или
фарфоровых поддонах. Хранить кислоты можно также в нижней
части вытяжного шкафа, если только туда не вмонтированы сан-
техпроводы, которые в присутствии кислот могут корродировать.
Склянки с дымящей НРТОз следует хранить в специальных ящиках
из нержавеющей стали, выложенных асбестом.
Концентрированные растворы щелочей не поставляются
промышленностью, и их приходится приготовлять в лабораторных
условиях. Для приготовления раствора NaOH, не содержащего
Na2C03, пользуются тем, что Na2C03 нерастворим в
концентрированном растворе NaOH. Для этого 200 г NaOH (ч. д. а.) помещают
в колбу из стекла «пирекс» и растворяют в 200 мл воды,
добавляемой постепенно небольшими порциями. Когда вся щелочь
растворится, останется Na2C03 в виде суспензии, очень медленно
оседающей на дно. Полученный раствор фильтруют через асбестовый
фильтр в фарфоровом тигле с пористым дном в условиях, не
допускающих загрязнения раствора С02 воздуха. Отделить Na2C03
можно также декантацией или центрифугированием.
Концентрированный раствор NaOH хранят в полиэтиленовых бутылях и
склянках.
При работе с жидкими органическими реагентами, растворами
кислот и оснований запрещается засасывание их в пипетки ртом.
Щелочные металлы
При работе с металлическими калием и натрием необходимо
соблюдать особую осторожность, не допуская соприкосновения их
с водой, галогенсодержащими органическими соединениями и
твердым С02 (сухим льдом).
Работу со щелочными металлами выполняют в вытяжном
шкафу, пользуясь защитными очками и резиновыми перчатками.
Запаянные металлические банки с блоком металлического
натрия или калия в парафине (сухая упаковка) надо вскрывать
осторожно, избегая ударов молотком- или другими металлическими
предметами. Блок металлического натрия можна
разрезать ножом и большие куски быстро поместить в
заранее подготовленные толстостенные стеклянные банки с
обезвоженным керосином или трансформаторным маслом. Блок
металлического калия можно разрезать только под слоем обезвоженнога
трансформаторного масла. Банки со щелочными металлами хранят
в металлическом ящике с песком на дне (на случай аварии).
8S
Рис. 57. Пресс для получения проволоки из натрия:
/ — кронштейн; 2—гнездо для патрона; 3 — патрон;
4 — поршень.
Из банки натрий вынимают
лабораторными щипцами, кладут на лист
фильтровальной бумаги, ножом
отрезают нужное количество, срезают
пленку оксида и сейчас же используют
в работе. Обрезки собирают в
отдельную банку из толстого стекла,
заполненную на 7з объема обезвоженным
керосином.
Разрезание
металлического калия и срезание оксидной
•цленки следует производить под слоем обезвоженного
трансформаторного масла, после чего кусочки металла осторожно
переносят в сосуд с безводным диэтиловым эфиром (для освобождения
от масла).
Обрезки металлического калия собирают отдельно от обрезков
металлического натрия. Категорически запрещается накапливать
более 2 г обрезков натрия или калия.
Обрезки щелочных металлов, которые нельзя использовать в
работе, уничтожают либо превращением их в алкоголяты, либо
сжиганием [50]. Чаще всего используют первый способ. Для этого
отходы щелочных металлов небольшими порциями вносят в 80—
85% этиловый спирт. По растворении металла и охлаждении
жидкости вносят новую порцию отходов. Полученный раствор
разбавляют большим объемом воды и сливают в канализацию.
При работе со щелочными металлами нельзя пользоваться
водяными или паровыми банями, а только масляными, песчаными
или воздушными.
В лабораториях органического синтеза часто нужен порош-
кообразн ы й натрий. Чтобы получить порошок, натрий,
предварительно очищенный от оксида, помещают в колбу* с обратным
холодильником и заливают десятикратным (по массе) количеством
толуола или ксилола, высушенного над натрием. Смесь нагревают
на силиконовой бане, пока весь натрий не расплавится. Тогда
нагревание прекращают, колбу закрывают пробкой (желательно
пришлифованной стеклянной) и, завернув ее в полотенце,
энергично встряхивают, чтобы образовались мелкие капельки натрия,
быстро затвердевающие по мере остывания колбы. После полного
охлаждения ксилола его декантируют, а натрий заливают сухим
диэтиловым эфиром или же непосредственно вводят в реакцию.
Для приготовления натриевой проволоки пользуются
специальным прессом (рис. 57). В гнездо пресса 2 вставляют
чистый сухой патрон 3 и помещают в него кусочки натрия, с
поверхности которых предварительно удалена корка. Наполнив
больше половины патрона, вводят в него поршень и плотно придавли-
36
вают кусочки натрия. Затем поднимают поршень, добавляют еще
немного натрия, медленно опускают поршень и с перерывами
вращают винт, пока из патрона не появится проволока, которую
подают в бутыль с бензолом, обезвоженным керосином или
трансформаторным маслом, вплотную подставленную к выходному
отверстию пресса. Выдавив всю проволоку, поднимают поршень и
наливают в патрон этиловый спирт для растворения остатков натрия,
а поршень опускают в стакан со спиртом. Затем патрон и поршень
промывают спиртом и высушивают патрон в сушильном шкафу,
а поршень на воздухе. При длительном контакте с воздухом
натриевая проволока покрывается твердой оксидной пленкой и
вследствие этого перестает действовать.
Огнеопасные жидкости
Из большой группы огнеопасных жидкостей наибольшую
опасность представляют собой органические растворители,
образующие при хранении перекисные соединения (простые-эфиры, диок-
сан, тетрагидрофуран). Перегонять и выпаривать подобные
жидкости разрешается лишь убедившись предварительно в отсутствии в
них перекисей. Но даже при отсутствии перекисей выпаривать
досуха подобные вещества не следует.
Горючие жидкости запрещается выливать в канализацию.
Отработанные горючие жидкости следует собирать в специальную
герметически закрывающуюся тару, которую в конце рабочего дня
удаляют из лабораторного помещения для регенерации или
сожжения.
Хлорная кислота
Безводная хлорная кислота НСЮ4 термически неустойчива.
Уже при комнатной температуре через несколько часов после
приготовления она приобретает окраску, темнеет и, наконец,
становится непрозрачной. В этом состоянии хлорная кислота может
взорваться. Концентрированная НС104 (70%) при обычной
температуре безопасна в обращении, кипит без опасности взрыва, но
если кипящая кислота или ее пары войдут в соприкосновение с
органическими или легко окисляющимися неорганическими
веществами, взрыв может произойти. Поэтому выпаривать растворы с
хлорной кислотой и ее солями необходимо с добавлением азотной
кислоты. Выпаривать следует под сильной вытяжкой. Внутренние
стенки вытяжных шкафов, в которых проводится систематическая
работа с хлорной кислотой и перхлоратами, необходимо тщательно
и часто промывать водой.
Ядовитые вещества [56, с. 143]
При предварительном определении вещества по запаху не еле*
дует наклоняться над сосудом с препаратом и вдыхать пары или
газ полной грудью, а только осторожно направлять пары (газ)'
движением руки к себе.
87
Все работы, связанные с применением ядовитых веществ,
надлежит производить в резиновых перчатках.
Измельчение едких и ядовитых веществ производят в закрытых
ступках и под тягой, надев кроме резиновых перчаток еще и
предохранительные очки.
Остатки ядовитых веществ следует нейтрализовать
соответствующими реагентами (щелочами, хлорактивными веществами
и др.), в соответствии с правилами обезвреживания и
уничтожения вредных веществ [56, с. 151].
Белый фосфор [56, с. 142]
Хранить белый фосфор следует только под слоем воды.
Все работы с белым фосфором следует проводить в вытяжном
шкафу при включенной вентиляции. Аппаратура должна быть
герметичной.
Работающий должен надеть защитные очки и резиновые
перчатки.
В реакционный сосуд фосфор переносят пинцетом. Рабочее
место должно быть оборудовано водопроводным крапом и бачком для
2% раствора CuS04 (нейтрализатора).
Метиловый спирт [56, с. 148]
Применение метилового спирта в лабораторной практике
допускается только в тех случаях, когда он не может быть заменен
другими, менее токсичными веществами. Работать с ним можно
только в вытяжных шкафах, а наливать в емкости сифонами,
заряженными вакуумом, или самотеком. Использовать сифоны с
засасыванием ртом запрещается.
Пролитый метиловый спирт немедленно смывают большими
количествами воды, а освободившуюся из-под него тару немедленно
дважды промывают водой.
Для работы в лабораториях необходимо выписывать не более
суточной потребности метилового спирта. Когда метиловый спирт
израсходован неполностью, остаток следует сдать на склад или
закрыть в несгораемый шкаф, который нужно обязательно опечатать.
Запрещается передавать метиловый спирт из одной лаборатории
в другую. При работе с ним следует руководствоваться «Общими
санитарными правилами по хранению и применению метанола».
глава 5 МЫТЬЕ И СУШКА ЛАБОРАТОРНОЙ ПОСУДЫ
Новая посуда перед использованием должна быть вымыта го-
рячей водой.
Сразу же после освобождения использованной посуды
необходимо обезвредить и удалить из нее остатки веществ. При
обезвреживании и мытье посуды непременно нужно надевать защитные
очки, перчатки, фартук; посуду следует обезвреживать в вытяж~
ном шкафу.
Всегда желательно иметь лабораторную мойку под
вытяжкой или моечныйвытяжной шкаф со столом,
обитым свинцовыми листами, с подводкой горячей и холодной воды
и большой фаянсовой или стальной эмалированной раковиной.
Подобные шкафы производятся для оснащения комплектных
лабораторий стационарного типа. В крайнем случае приходится
удовлетвориться тумбой с лабораторной раковиной.
Для мытья большого количества лабораторной посуды следует
выделять изолированные помещения — моечные, — которые
должны быть, по возможности, расположены в центре обслуживаемых
ими лабораторных помещений.
Допускается устройство мест для мытья посуды в каждом
лабораторном помещении в вытяжном шкафу.
СПОСОБЫ ОЧИСТКИ И МОЮЩИЕ СРЕДСТВА [13, с. 153]
Для выбора способа мытья в каждом отдельном случае
необходимо знать свойства загрязняющих посуду веществ, их
растворимость в холодной и горячей воде, в растворах щелочей и кислот,
способность окисляться с образованием водорастворимых
соединений.
Мытье горячей водой с помощью ершей эффективно
в тех случаях, когда загрязняющие посуду вещества растворимы
в воде. При мытье ершом необходимо следить, чтобы
неосторожным нажатием не проткнуть стенки или дно сосуда. Вынимая ерш
из узких сосудов, не следует спешить, так как быстро
выпрямляющаяся щетина может обрызгать и моющего посуду, и человека,
стоящего рядом.
Вымытую посуду споласкивают два-три раза дистиллированной
водой. Стеклянная посуда считается чистой только в том случае»
если вода равномерно смачивает всю внутреннюю поверхность по*
суды и не оставляет капель на внутренних стенках.
Если после споласкивания в сосуде сохраняются обуглившиеся
массы, то следует попытаться удалить их путем встряхивания с
кусочками влажной фильтровальной бумаги.
89
Рис. 58. Приспособления для
пропаривания посуды:
/—очищаемый сосуд; 2 — трубка
для пара; 5 —трубка для стока
сконденсированной воды.
Мыть посуду
струей водяного
пара — процесс
трудоемкий, но когда требуется
особенно чистая
посуда, ее предварительно
моют обычным
способом и затем
пропаривают с помощью
специальных
приспособлений (рис. 58). Для
равномерного и
спокойного кипения в колбу с
водой (налитой до половины) следует положить «к и п ят и л ь н ы е
камешки».* После пропаривания посуду, не перевертывая,
высушивают.
В исключительных случаях для удаления смолистых, жировых
и других органических веществ, нерастворимых в воде, иногда
используют органические растворители (эфир, изопропи-
ловый спирт, бензин и др.)- При этом следует учитывать
огнеопасность органических растворителей и работу с ними проводить
вдали от огня, в вытяжном шкафу, а загрязненные растворители
собирать каждый в отдельности для последующей регенерации
перегонкой.
Посуду, промытую органическими растворителями, затем моют
водой с мылом (или другими моющими средствами), а потом
чистой водой, после чего обрабатывают хромовой смесью или другим
окислителем.
В последние годы для мытья лабораторной посуды стали
применять ультразвук [25, с. 5], который оказывает
диспергирующее действие на загрязнения; последние легко отстают от стекла
и смываются струей воды. В присутствии моющих средств действие
ультразвука усиливается.
В качестве моющих средст.в применяют
поверхностно-активные вещества, способные адсорбироваться на поверхности
раздела фаз и вследствие этого понижать адгезию (прилипание)
загрязнения. Синтетические моющие средства бытового назначения
находят широкое применение для мытья лабораторной посуды.
Моющими свойствами обладают также соли, гидролиз которых
сопровождается образованием щелочной среды: фосфат натрия
№зР04, гексаметафосфат натрия NaePeOis.
* Так или «к ипелками» называют кусочки необожженного пористого
материала (фарфора, фаянса, пемзы, туфа и т. п.).
90
Хорошим моющим средством является раствор 75—100 г Na2COj
в 1 л воды. Для удаления коксовых остатков рекомендуют посуду
помещать в раствор, содержащий 53 г Na3P04 и 28,5 г олеината
натрия в 470 мл воды.
Для очистки посуды, загрязненной органическими остатками,
используют сильные окислители: соли хромовой и
марганцовой кислот, перекись водорода, «царскую водку», азотную и серную
кислоты и др. [27].
Труднорастворимое загрязнение можно перевести в
водорастворимое. Так, BaS04 при обработке конц. H2SO4
переходит в растворимое соединение Ba(HS04)2; AgCl,
малорастворимый в воде, при обработке водным раствором аммиака образует
комплексное соединение [Ag(NH3)2]Cl, хорошо растворимое в
воде.
Загрязнения основного характера отмывают разбавленными
кислотами, а кислотные — водными растворами аммиака,
карбонатов щелочных металлов или щелочей.
Для мытья посуды чаще всего используют растворы солей
хромовых кислот в серной кислоте (хромовую смесь).
Существует много рецептов приготовления хромовой смеси. При
этом предпочтение отдают натриевым солям хромовой и
двухромовой кислот, обладающим большей растворимостью в воде, чем
калиевые соли.
Ниже приводим некоторые рецепты приготовления хромовой
смеси.
а. К раствору 92 г измельченного ЫагСгС^^НгО в 458 мл воды
при непрерывном перемешивании постепенно добавляют 800 мл
конц. H2S04. Получаемую красно-коричневую жидкость можно
использовать многократно, до перехода окраски в зеленый цвет —
окраска ионов хрома (III).
б. К 100 мл конц. H2SO4 при энергичном перемешивании по-»
степенно добавляют 9,9 г тонкоизмельченного К2СГ2О7.
в. К 100 мл конц. H2S04 при энергичном перемешивании
постепенно добавляют 10 г 50% водного раствора Na2Cr207.
г. Растворяют 15 г тонкоизмельченного К2СГ2О7 в 100 мл
горячей воды. Раствор охлаждают и при непрерывном помешивании
по каплям прибавляют к 100 мл конц. H2SO4 [43].
д. Растворяют 200 г тонкоизмельченного К2СГ2О7 в 1 л конц.
HN03. Азотнокислая хромовая смесь более стойка, чем обычная, а
по моющим свойствам превосходит ее.
Хромовую смесь следует хранить в широком толстостенном
сосуде, который плотно закрывают толстой стеклянной пластинкой,
чтобы избежать выделения крайне едкого и летучего СгОз и
поглощения влаги из воздуха.
Обработку посуды хромовой смесью следует проводить под тя«
гой, в защитных перчатках и очкаху поверх халата надеть
резиновый фартук.
Сначала механически удаляют грубые загрязнения: моют ер«
шами, встряхивают с 2—5% раствором NaOH и обрезками филь-
тровальной бумаги. Не очень загрязненную малогабаритную
посуду хорошо промывают водой и после возможно более полного
удаления воды вносят во взболтанную хромовую смесь. Большей
частью достаточно оставить очищаемый сосуд в холодной смеси на
15—30 мин.
Возможен и другой вариант обработки. Очищаемый сосуд
наполовину наполняют горячим (около 60 °С) раствором хромовой
смеси и, осторожно вращая, смачивают внутренние стенки сосуда.
Через 10—15 мин хромовую смесь сливают обратно в сосуд, где
она хранится, а очищаемый сосуд тщательно промывают
водопроводной водой, а затем споласкивают несколько раз
дистиллированной водой.
Посуду, загрязненную продуктами перегонки нефти (парафин,
керосин, минеральные масла), предварительно очищают
органическими растворителями, затем водным раствором моющих средств
(мыла или стирального порошка) и завершают мытье обработкой
хромовой смесью.
Если посуда загрязнена солями бария, ее нельзя мыть
хромовой смесью, содержащей серную кислоту, так как BaS04 образует
на стенках сосуда трудно удаляемый осадок.
При работе с хромовой смесью (и другими сильными
окислителями) следует избегать попадания в нее легко окисляющихся
метилового и этилового спиртов, так как это приводит к потере
окислительных свойств смеси.
Известные трудности возникают при отмывке посуды
водой от соединений хрома после обработки посуды
хромовой смесью.
Весьма эффективно отмывается посуда от ионов хрома с
помощью комплексонов. Посуду помещают на 2 ч в водный раствор,
содержащий 1% комплексона III (динатриевая соль этилендиамин-
тетрауксусной кислоты, трилон Б, Na-ЭДТА) и 2% NaOH. Затем
посуду ополаскивают водопроводной и дистиллированной водой.
Стеклянные фильтры и другие пористые материалы не
рекомендуется очищать хромовой смесью и растворами КМп04; в этом
случае лучше применять смесь конц. H2S04 и HN03.
При очистке посуды в кислой среде обычно
пользуются 4% раствором КМп04. В сосуд, предварительно вымытый
горячей водой с помощью ерша, наливают раствор КМп04 и
тонкой струей добавляют конц. H2S04 из расчета 3—5 мл на 100 мл
раствора КМп04. При этом происходит разогревание, что
способствует быстрому окислению загрязнений. Отработанный раствор
выливают и повторно не используют.
Если после ополаскивания водой на стенках сосуда
обнаруживается бурый налет Мп02, то его удаляют 5% раствором
щавелевой кислоты или гидросульфита натрия.
Для очистки посуды применяют также раствор,
приготовляемый смешиванием равных объемов насыщенных при комнатной
температуре растворов КМп04 и NaOH. Посуда заливается этой
смесью на 10—15 мин. Бурый налет Мп02 удаляют как указано
92
выше. После этого посуду моют водопроводной водой и
споласкивают 2—3 раза дистиллированной водой.
Эффективным средством является смесь равных объемов 6 н.
раствора НС1 и 5% раствора Н2О2. Эта смесь действует весьма
энергично при 30—40 °С и может быть использована многократно.
Посуду, загрязненную смолистыми
веществами, в зависимости от химической природы последних,
обрабатывают конц. НС1, HN03, H2SO4 или 20—40% раствором NaOH
или КОН при энергичном и длительном встряхивании.
Если посуда загрязнена керосином и другими
нефтепродуктами, водный раствор NaOH или КОН можно
заменить 5—10% раствором Са(ОН)2 — известковым молоком.
После этой обработки посуду моют теплой водой.
В ряде случаев хорошие результаты дает замачивание в
растворе, содержащем стиральный порошок (для хлопчатобумажного
и льняного белья) и аммиак. *
НЕКОТОРЫЕ СПЕЦИФИЧЕСКИЕ СЛУЧАИ
Мытье мерной посуды
Методы и средства мытья мерной посуды регламентируются,
ГОСТ 8.234-77.
Перед мытьем бюреток и других устройств, имеющих
стеклянные краны, следует вынуть кран из муфты, очистить кран и муфту
от смазки диэтиловым эфиром. Затем вновь вставляют кран,
закрепляют его резинкой, и сосуд наполняют 2% мыльным
раствором с кусочками мелко нарезанной бумаги и взбалтывают.
Бюретки можно мыть мыльной водой при помощи ершей на длинной
ручке или длинных стеклянных палочек с надетыми на них
кусочками резиновых трубок. Затем, слив мыльный раствор,
ополаскивают сосуд сначала водопроводной, а затем дистиллированной
водой. Если этого окажется недостаточно, следует подвергнуть 15—
20-минутному действию конц. H2S04 или хромовой смеси. Затем
бюретку снова ополаскивают сначала водопроводной, а затем
дистиллированной водой.
Бюретки удобно очищать смесью этилового • спирта и конц.
HN03 [43]. Бюретку устанавливают в вытяжном шкафу, наливают
3 мл спирта, осторожно по стенкам бюретки приливают 4 мл конц. •
HN03 и закрывают бюретку опрокинутой пробиркой.
Выделяющиеся оксиды азота хорошо очищают стенки. Промывание
водопроводной водой и ополаскивание дистиллированной производят,
не вынимая бюретку из вытяжного шкафа.
При очень сильной загрязненности мерной посуды ее
выдерживают в подкисленном или подщелоченном растворе КМп04 в
течение суток, а затем, после удаления раствора, ополаскивают конц.
НС1 до полного удаления образовавшегося на стенках бурого
налета Мп02 и промывают водой.
93
Мытье воронок со стеклянными пористыми
пластинками
Тотчас же после использования воронки отводную трубку
соединяют резиновым шлангом с водопроводным краном и пускают
воду, чтобы смыть осадок. Через плотные стеклянные фильтры
просасывают воду под уменьшенным давлением. После такой
механической очистки фильтры обрабатывают нагретым растворителем,
подбираемым в зависимости от свойств осадка. Например, в
случае HgS подходит царская водка, в случае AgCl — аммиак или
тиосульфат натрия. В заключение фильтры следует основательно
промыть горячей дистиллированной водой.
Устройства для мытья капиллярной посуды
Для мытья пипеток и бюреток существуют специальные
аппараты.
Аппарат для мытья капиллярной посуды. Аппарат позволяет
механизировать мытье пипеток и бюреток с наружным диаметром
4—6 мм. В двух ваннах на специальных гребенках закрепляется
6 или 12 сосудов. Для наполнения ванн и для наружного
ополаскивания имеется смеситель с душевой головкой.
Растворы моющих средств, холодная и горячая вода, а также
дистиллированная вода для прополаскивания просасываются через
капилляры с помощью водоструйного насоса.
Аппарат должен быть подключен к магистралям холодной и
горячей воды с давлением ^ 0»2 МПа.
Аппарат для мытья пипеток [25]. Этот аппарат позволяет мыть
одновременно до 280 пипеток длиной не свыше 370 мм с
внутренним диаметром ^ 2 мм.
Пипетки сначала отмачивают в кислотном или щелочном
растворе; затем загрузочную корзинку с пипетками вкладывают в
цилиндр аппарата.
Мытье осуществляется за счет непрерывно повторяющегося
цикла заполнения цилиндра водой через резиновый шланг,
подведенный к смесителю, и автоматического сброса воды через
сифонную трубку.
СУШКА
Вымытая посуда должна быть высушена, если только она не
предназначена для работы с водными растворами. Самый
распространенный холодный способ — сушка на колышках или на
решетках. Вымытую посуду надевают на колышки доски или
опрокидывают на решетку, установленную над моечной раковиной и
оставляют до тех пор, пока она не высохнет.
При наличии в лаборатории проводки сжатого воздуха
вымытую посуду можно высушить струей нагретого воздуха,
профильтрованного через слой стеклянной ваты.
94
В отдельных случаях посуду высушивают, ополаскивая этило*
вым спиртом и диэтиловым эфиром, либо ацетоном и эфиром. Для
этого обтирают сосуд снаружи полотенцем, ополаскивают вначале
этиловым спиртом или ацетоном, а затем диэтиловым эфиром;
остатки эфира удаляют продуванием чистым сухим воздухом
вдали от источников огня. Остатки спирта и эфира сливают отдельно
и сохраняют для последующей регенерации.
Мелкие стеклянные сосуды можно высушить в
вакуум-эксикаторе, заполненном высушенным силикагелем.
Горячая сушка проводится в сушильном шкафу при 100—
120 °С. Посуду помещают в шкаф после того, как она некоторое
время постояла перевернутой.
Чтобы посуда не загрязнилась, на выдвижные полки
сушильного шкафа следует положить чистую фильтровальную бумагу. По*
еле отключения сушильного шкафа от сети посуду оставляют
медленно охлаждаться, не вынимая из шкафа, чтобы она не запотела.
При возникновении срочной надобности малогабаритные сосуды
можно вынуть из сушильного шкафа и поместить в эксикатор, где
процесс остывания завершится быстрее. Для ускорения остывания
посуды можно также ее продуть струей сухого воздуха.
глава б ДИСТИЛЛИРОВАННАЯ
И ДЕМИНЕРАЛИЗОВАННАЯ ВОДА
Природная вода всегда содержит различные примеси, от
характера и концентрации которых зависит ее пригодность для тех или
иных целей.
Питьевая вода, подаваемая централизованными хозяйственно-
питьевыми системами водоснабжения и водопроводами, по
ГОСТ 2874—73, может иметь общую жесткость до 10,0 мг-экв/л, а
сухой остаток до 1500 мг/л.
Естественно, что подобная вода непригодна для приготовления
титрованных растворов, для выполнения различных исследований
в водной среде, для многих препаративных работ, связанных с
применением водных растворов, для ополаскивания лабораторной
посуды после мытья и т. п.
ДИСТИЛЛИРОВАННАЯ ВОДА
Метод деминерализации воды дистилляцией (перегонкой)
основан на разности давлений паров воды и растворенных в ней
солей. При не очень высокой температуре можно принять, что соли
практически нелетучи и деминерализованная вода может быть
получена испарением воды и последующей конденсацией ее паров.
Этот конденсат принято называть дистиллированной водой.
Вода, очищенная методом дистилляции в перегонных аппаратах,
используется в химических лабораториях в количествах больших,
чем другие вещества.
По ГОСТ 6709—72, вода дистиллированная — прозрачная,
бесцветная жидкость, не имеющая запаха, с рН = 5,44-6,6 и
содержанием сухого остатка ^ 5 мг/л.
По Государственной фармакопее, сухой остаток в
дистиллированной воде не должен превышать 1,0 мг/л, а рН = 5,0 -f- 6,8.
Вообще требования к чистоте дистиллированной воды по
Государственной фармакопее выше, чем по ГОСТ 6709—72. Так,
фармакопея допускает содержание растворенного аммиака .^ 0,00002%,
ГОСТ < 0,00005%.
Дистиллированная вода не должна содержать
восстанавливающих веществ (органические вещества и восстановители
неорганической природы).
Наиболее четкий показатель чистоты воды — ее
электропроводимость. По литературным данным, удельная электрическая
проводимость идеально чистой воды при 18 °С равна 4,4-Ю-10 См-м-1..
При небольшой потребности в дистиллированной воде
перегонку воды можно осуществить при атмосферном давлении в обычных
установках из стекла.
Однократно перегнанная вода обычно загрязнена С(32, NH3 и
органическими веществами. Если требуется вода с очень низкой
96
проводимостью, то необходимо полностью удалить ССЬ. Для этого
через воду при 80—90 °С в течение 20—30 ч пропускают сильную
струю очищенного от СОг воздуха и затем воду перегоняют при
очень медленном токе воздуха.
Для этой цели рекомендуется применять сжатый воздух из
баллона или засасывать его извне, поскольку в химической
лаборатории он весьма загрязнен. Воздух до подачи в воду пропускают
сначала через промывную склянку с конц. H2SO4, затем через две
промывные склянки с конц. КОН и, наконец, через склянку с
дистиллированной водой. При этом следует избегать применения
длинных резиновых трубок.
Большую часть СОг и органических веществ можно удалить,
если к 1 л перегоняемой воды добавлять около 3 г NaOH и 0,5 г
КМп04 и отбрасывать некоторое количество конденсата в начале
перегонки. Кубовый остаток должен составлять не менее 10—15%
загрузки. Если конденсат подвергнуть вторичной перегонке с
добавлением 3 г KHS04, 5 мл 20% Н3Р04 и 0,1—0,2 г КМп04 на литр, го
это гарантирует полное удаление NH3 и органических загрязнений.
Продолжительное хранение дистиллированной воды в
стеклянной посуде всегда приводит к ее загрязнению продуктами
выщелачивания стекла. Поэтому дистиллированную воду долго хранить
нельзя.
Металлические дистилляторы [25, с, 11; 46, с. 79]
Дистилляторы с электронагревом. На рис. 59 изображен
дистиллятор Д-4 (модель 737). Производительность 4 ± 0,3 л/ч,
потребляемая мощность 3,6 кВт, расход охлаждающей воды до
160 л/ч. Масса аппарата без воды 13,5 кг.
В камере испарения / вода нагревается электронагревателями
3 до кипения. Образующийся пар через патрубок 5 поступает в
конденсационную камеру 7, вмонтированную в камеру 6У через
которую непрерывно протекает водопроводная вода. Из
конденсатора 8 дистиллят вытекает через ниппель 13.
В начале работы водопроводная вода, непрерывно поступающая
через ниппель 12, заполняет водяную камеру б и по сливной
трубке 9 через уравнитель 11 заполняет камеру испарения до
установленного уровня.
В дальнейшем, по мере выкипания, вода будет поступать в
камеру испарения только частично; основная же часть, проходя
через конденсатор, точнее через его водяную камеру 6, будет
сливаться по сливной трубке в уравнитель и далее через ниппель 10
в канализацию. Вытекающая горячая вода может быть
использована для хозяйственных нужд.
Аппарат снабжен датчиком уровня 4, предохраняющим
электронагреватели от перегорания в случае понижения уровня воды ниже
допустимого.
Избыток пара из камеры испарения выходит через трубку,
вмонтированную в стенку конденсатора.
4 Зак. 1237 97
Рис. 59. Дистиллятор Д-4
(модель 737):
/ — камера испарения; 2— кожух;
3 — электронагреватели; 4 — датчик
уровня; 5 —патрубок; 6 — наружная
водяная камера; 7— конденсационная
камера; 8 — конденсатор; 9 — сливная
трубка; 10, 13— ниппеля вытекающей
воды; // — уравнитель; /2 —ниппель
поступающей воды; /4—болт
заземления.
Аппарат
устанавливают на ровной
горизонтальной поверхности и
посредством болта заземле-'
ния 14 присоединяют к
общему контуру заземления, к которому также присоединяют
электрощит.
При первоначальном пуске аппарата пользоваться
дистиллированной водой по прямому назначению можно только после
48-часовой работы аппарата.
Периодически необходимо механически очищать от накипи
электронагреватели и поплавок датчика уровня.
Аналогично устроен дистиллятор Д-25 (модель 784),
производительность которого 25 ± 1,5 л/ч, потребляемая мощность
18 кВт.
В этом аппарате девять электронагревателей—три группы по
три нагревателя. Для нормальной и длительной работы аппарата
достаточно, чтобы одновременно включались шесть нагревателей.
Но для этого требуется периодически, в зависимости от жесткости
питающей воды, производить механическую очистку от накипи
трубки, по которой вода поступает в камеру испарения.
При первоначальном пуске дистиллятора Д-25 пользоваться
дистиллированной водой по прямому назначению рекомендуется
после 8—10 ч работы аппарата.
Значительный интерес представляет аппарат для
получения апирогенной воды для инъекций А-10 (рис. 60).
Производительность 10 + 0,5 л/ч, потребляемая мощность 7,8 кВт,
расход охлаждающей воды 100—180 л/ч.
В этом аппарате в камеру испарения вместе с перегоняемой
водой поступают реагенты для ее умягчения (алюмокалиевые
квасцы A12(S04)3-K2S04-24H20) и для удаления NH3 и
органических загрязнений (КМп04 и Na2HP04).
Раствор квасцов заливают в один стеклянный сосуд
дозирующего устройства, а растворы КМп04 и Na2HP04 — в другой — из
расчета на 1 л апирогенной воды квасцов 0,228 г, КМп04 0,152 г,
Na2HP04 0,228 г.
При первоначальном пуске или при пуске аппарата после
длительной консервации использовать получаемую апирогенную воду
для лабораторных нужд можно только через 48 ч работы аппарата.
98
12
и^-
&
Рис. 60. Аппарат А-Ю для получения апирогенной воды:
/—электронагреватель; 2—камера испарения; 3—конденсатор; 4—датчик уровня; 5—дозатор;
6—стеклянный сосуд; 7—капельница; 8 — уравнитель; 9 — спускной кран.
Перед эксплуатацией металлических дистилляторов с
электронагревом следует проверить правильность включения всех
проводов и наличие заземления. Категорически запрещается включать
эти аппараты в электросеть, не заземлив. При любой
неисправности дистилляторы должны быть отключены от сети.
Качество дистиллированной воды в известной степени зависит
от длительности работы аппарата. Так, при пользовании старыми
дистилляторами в воде могут содержаться хлорид-ионы.
Приемники должны быть из нейтрального стекла и, во
избежание попадания СОг, соединены с атмосферой через хлоркальцие-
вые трубки, наполненные гранулами натронной извести [смесь
NaOH и Са(ОН)2].
Огневой дистиллятор. Дистиллятор ДТ-10 со встроенной топкой
рассчитан на эксплуатацию в условиях отсутствия водопровода и
электроэнергии и позволяет за 1 ч получать до 10 л
дистиллированной воды. Представляет собой цилиндрической формы
конструкцию из нержавеющей стали высотой около 1200 мм,
смонтированную на основании длиной 670 мм и шириной 540 мм.
Дистиллятор состоит из встроенной топки с топочной
фурнитурой, камеры испарения на 7,5 л, камеры охлаждения на 50 л и
сборника дистиллированной воды на 40 л.
Вода в камеры испарения и охлаждения заливается вручную.
По мере расхода воды в камере испарения она автоматически
пополняется из камеры охлаждения.
Получение бидистиллята [53, с. 184]
Однократно перегнанная вода в металлических дистилляторах
всегда содержит небольшие количества посторонних веществ. Для
особо точных работ пользуются повторно перегнанной водой — би-
дистиллятом. Промышленность серийно выпускает аппараты для
4* 99
:. 61. Аппарат для бидистилляции воды.
бидистилляции воды БД-2 и БД-4
производительностью 1,5—2,0 и 4—
5 л/ч соответственно.
Первичная перегонка происходит
в первой секции аппарата (рис. 61).
В полученный дистиллят добавляют
КМп04 для разрушения органических
примесей и переводят его во вторую
колбу, где происходит вторичная
перегонка, и бидистиллят собирают в
приемную колбу. Нагревание
осуществляется с помощью электрических
нагревателей; стеклянные водяные
холодильники охлаждаются
водопроводной водой. Все стеклянные детали
изготовляются из стекла пирекс.
Определение качественных показателей дистиллированной воды
Определение рН. Это испытание производят потенциометриче-
ским методом со стеклянным электродом или — при отсутствии
рН-метра — колориметрическим методом (см. гл. 33).
Пользуясь штативом для колориметрирования (штатив для
пробирок, снабженный экраном), в четыре занумерованные
одинаковые пробирки диаметром около 20 мм и вместимостью 25—
30 мл, чистые, сухие, из бесцветного стекла помещают: в пробирки
№ 1 и 2 —по 10 мл испытуемой воды, в пробирку № 3— 10 мл
буферной смеси, отвечающей рН = 5,4, а в № 4— 10 мл буферной
смеси, отвечающей рН = 6,6. Затем в пробирки № 1 и 3
прибавляют по 0,1 мл 0,04% водноспиртового раствора метилового
красного и перемешивают. В пробирки № 2 и 4 прибавляют по 0,1 мл
0,04% водноспиртового раствора бромтимолового синего и
перемешивают. Воду считают соответствующей стандарту, если
содержимое пробирки № 1 не краснее содержимого пробирки № 3
(рН = 5,4), а содержимое пробирки № 2 не синее содержимого
пробирки № 4 (рН = 6,6).
Определение сухого остатка. В предварительно прокаленной
и взвешенной платиновой чашке выпаривают на водяной бане
досуха 500 мл испытуемой воды. Воду прибавляют в чашку
порциями по мере испарения, а чашку защищают от загрязнения
предохранительным колпачком. Затем чашку с сухим остатком
выдерживают 1 ч в сушильном шкафу при 105—110°С, охлаждают
в эксикаторе и взвешивают на аналитических весах.
Воду считают соответствующей ГОСТ 6709—72, если масса
сухого остатка будет ^2,5 мг.
Определения содержания аммиака и аммонийных солей. В
одну пробирку с притертой стеклянной пробкой вместимостью около
100
25 мл наливают -10 мл испытуемой воды, а в другую—10 мл
эталонного раствора, приготовленного следующим образом: 200 мл
дистиллированной воды помещают в коническую колбу на 250—
300 мл, прибавляют 3 мл 10% раствора NaOH и кипятят 30 мин,
после чего раствор охлаждают. В пробирку с эталонным
раствором прибавляют 0,5 мл раствора, содержащего 0,0005 мг NH4+.
Затем в обе пробирки одновременно прибавляют по 1 мл реактива
на аммиак (см. приложение 2) и перемешивают. Воду считают
соответствующей стандарту, если наблюдаемая через 10 мин
окраска содержимого пробирки будет не интенсивнее окраски
эталонного раствора. Сравнение окраски производят по оси пробирок
на белом фоне.
Проба на восстанавливающие вещества. 100 мл испытуемой
воды доводят до кипения, прибавляют 1 мл 0,01 н. раствора
КМп04 и 2 мл разбавленной (1:5) H2S04 и кипятят 10 мин.
Розовая окраска испытуемой воды должна при этом сохраняться.
ДЕМИНЕРАЛИЗАЦИЯ ПРЕСНОЙ ВОДЫ ИОНООБМЕННЫМ МЕТОДОМ
При деионизации воды последовательно осуществляются
процессы Н+-катионирования и ОН_-анионирования, т.е. замещения
содержащихся в воде катионов на ионы Н+ и анионов на ионы ОН~.
Взаимодействуя друг с другом, ионы Н+ и ОН~ образуют
молекулу Н20.
Метод деионизации позволяет получать воду с более низким
содержанием солей, чем обычная дистилляция, но при этом не
удаляются неэлектролиты (органические загрязнения).
Выбор между дистилляцией и деионизацией зависит от
жесткости исходной воды и расходов, связанных с ее очисткой. В
отличие от дистилляции воды, при деионизации расход энергии
пропорционален содержанию солей в очищаемой воде. Поэтому при
высокой концентрации солей в исходной воде целесообразно
вначале применять метод дистилляции, а затем доочистку осуществить
деионизацией.
Иониты — твердые, практически" нерастворимые в воде и
органических растворителях вещества минерального или органического
происхождения, природные и синтетические. Для целей
деминерализации воды практическое значение имеют синтетические
полимерные иониты — ионообменные смолы, отличающиеся высокой
поглотительной способностью, механической прочностью и
химической устойчивостью.
Деминерализацию воды можно осуществлять
последовательным пропусканием водопроводной воды через колонку катионита в
Н+-форме, затем через колонку анионита в ОН~-форме. Фильтрат
с катионита содержит при этом кислоты, соответствующие солям
в исходной воде. Полнота удаления этих кислот анионитами
зависит от их основности. Сильноосновные аниониты удаляют все
кислоты почти полностью, слабоосновные не удаляют таких
слабых кислот, как угольная, кремневая и борная.
101
Рис. 62. Лабораторная установка для деминерализации воды:
1 — пробка; 2—стеклянная вата; 3 — катионит; 4—трехходовой кран;
5 — пробка; 6 — анионит; 7—сливная трубка.
Если эти кислотные группы допустимы в
деминерализованной воде или их соли отсутствуют в
исходной воде, то лучше применять слабоосновные
аниониты, так как их последующая регенерация
легче и дешевле, чем регенерация сильноосновных
анионитов.
Для деминерализации воды в лабораторных
условиях часто применяют катиониты марок КУ-1,
КУ-2, КУ-2-8чС и аниониты марок ЭДЭ-10П,
АН-1 и др. Иониты, поставляемые в сухом виде,
измельчают и отсеивают зерна размером 0,2—0,4 мм
при помощи набора сит. Затем их промывают
дистиллированной водой декантацией, пока
промывные воды не станут совершенно прозрачны. После
этого иониты переносят в стеклянные колонки
различных конструкций.
На рис. 62 изображена малогабаритная
колонка для деминерализации
воды. В нижнюю часть колонки помещают
стеклянные бусы и поверх них стеклянную вату. Чтобы
между зернами ионитов не попали пузырьки воздуха, колонку
заполняют смесью ионита с водой: Воду по мере ее накопления
спускают, но не ниже уровня ионита. Сверху иониты покрывают
слоем стеклянной ваты и бусами и оставляют под слоем воды на
12—24 ч. Спустив воду с катионита, колонку заполняют 2 н.
раствором НС1, оставляют на 12—24 ч, спускают НС1 и катионит
промывают дистиллированной водой до нейтральной реакции по
метиловому оранжевому. Катионит, переведенный в. Н+-форму,
сохраняют под слоем воды. Аналогично переводят анионит в
ОН~-форму, выдерживая его в колонке после набухания в 1 н.
растворе NaOH. Промывку анионита дистиллированной водой
проводят до нейтральной реакции по фенолфталеину.
Деминерализация относительно больших
объемов воды с раздельным применением ионитных фильтров
может быть осуществлена в более крупной установке. Материалом
для двух колонок высотой 700 и диаметром 50 мм может служить
стекло, кварц, прозрачный пластик. В колонки помещают по 550 г
подготовленного ионита: в одну — катионит в Н+-форме, в другую
анионит— в ОН--форме. Водопроводная вода со скоростью 400—
450 мл/мин поступает в колонку с катионитом, а затем проходит
через колонку с анионитом.
Поскольку иониты постепенно насыщаются, необходимо
контролировать работу установки. В первых порциях фильтрата,
прошедшего через катионит, определяют кислотность титрованием
щелочью но фенолфталеину. После того, как через установку про-
102
пустят около 100 л воды, или она проработает непрерывно 3,5 ч,
следует взять вновь пробу воды из катионитной колонки и
определить кислотность фильтрата. Если наблюдается резкое
уменьшение кислотности, пропускание воды следует прекратить и
провести регенерацию ионитов.
Катионит высыпают из колонки в большую банку с 5%
раствором НС1 и оставляют на ночь. Затем кислоту сливают, катионит
переносят на воронку Бюхнера и промывают дистиллированной
водой до отрицательной реакции на ион С1_ с AgN03. Промытый
катионит снова вводят в колонку.
Анионит регенерируют 5% раствором NaOH, промывают водой
до отрицательной реакции по фенолфталеину, после чего' вновь
заполняют им колонку..
В настоящее время деминерализацию воды большей частью
осуществляют методом смешанного слоя. Исходную воду
пропускают через смесь катионита в Н+-форме и сильно- или
слабоосновного анионита в ОН~-форме. Этот метод обеспечивает
получение воды высокой степени чистоты, но последующая
регенерация ионитов требует больших затрат труда.
Для деионизации воды с-применением смешанных ионитных
фильтров смесь катионита КУ-2-8чС и анионита ЭДЭ-10П в
объемном соотношении 1,25:1 загружают в колрнку диаметром 50 мм
и высотой 600—700 мм. В качестве материала для колонки
предпочтителен плексиглас, а для подводящей и сточной трубок —
полиэтилен.
Один килограмм смеси ионита может очистить до 1000 л
однократно перегнанной воды.
Регенерацию отработанных смешанных ионитов производят
раздельно. Смесь ионитов из колонки переносят на воронку Бюхнера
и отсасывают до получения воздушно-сухой массы. Затем иониты
помещают в делительную воронку такой вместимости, чтобы смесь
ионитов занимала х/\ ее объема. После этого в воронку добавляют
до 3Д объема 30% раствор NaOH и энергично перемешивают.
При этом смесь ионитов благодаря их разной плотности
(катионит 1,1, анионит 1,4) разделяется на слои. После этого катионит и
анионит отмывают водой и регенерируют как указано выше.
В лабораториях, где потребность в глубоко обессоленной воде
превышает 500—600 л/сутки, может быть использован серийно
выпускаемый аппарат Ц 1913. Расчетная производительность
200 л/ч. Пропускная способность деионизатора за межрегенера-
ционный период 4000 л. Масса комплекта 275 кг.
Деминерализатор снабжен системой автоматического
отключения подачи водопроводной воды при понижении ее
электрического сопротивления ниже допустимого значения и поплавковыми
клапанами, позволяющими автоматически удалять воздух из
колонок. Регенерация ионообменных смол производится путем
обработки их непосредственно в колонках раствором NaOH или НС1.
103
глава? ПРИГОТОВЛЕНИЕ РАСТВОРОВ
НЕКОТОРЫЕ ОБЩИЕ СВЕДЕНИЯ О РАСТВОРАХ
Растворы — однородные системы, образованные двумя или
большим числом компонентов. Компонент, содержание которого
в растворе преобладает, обычно называют растворителем;
компонент с меньшим содержанием называют растворенным
веществом.
Способы выражения концентрации растворов
Количественное содержание компонента раствора, отнесенное
к определенной массе или к определенному объему раствора или
растворителя, называется концентрацией этого компонента. При
этом содержание растворенного вещества обычно выражают в
единицах массы, в молях или в эквивалентах.
Процентная концентрация (по массе) — это число
единиц массы растворенного вещества, содержащихся в 100
единицах массы раствора. (Ниже процентная концентрация обозначена
С%.) Так. 20% водный раствор КОН содержит 20 единиц массы
КОН и 80 единиц массы воды.
Молярная концентрация (молярность)
выражается числом молей растворенного вещества в 1 л раствора и
обозначается буквой М или См.
Моль — единица количества вещества. Моль — это количество
вещества системы, содержащее столько молекул, атомов, ионов,
электронов или других структурных единиц, сколько содержится
атомов в 0,012 кг изотопа углерода 12С (6,022-1023). Масса
вещества, содержащаяся в 1 моле данного простого или сложного
вещества, называется мольной массой. Мольная масса вещества,
выраженная в граммах на моль, имеет то же численное значение, что
и его относительная молекулярная масса.
Число молей простого или сложного вещества п находят из
отношения массы т этого вещества в рассматриваемой системе к
его мольной массе М:
п = т/М
Произведение объема раствора, выраженного в миллилитрах,
на его молярность равно числу миллимолей растворенного
вещества.
Эквивалентная концентрация (нормальность)
выражается числом эквивалентов растворенного вещества в 1 л
раствора и обозначается буквами N, H. ИЛИ Сн.
Эквивалентом вещества называется такое его количество,
которое в данной реакции равноценно (эквивалентно) 1 молю ато-
104
мов водорода (1,0079 г). Масса 1 эквивалента называется
эквивалентной массой.
Выражение концентрации растворов в единицах нормальности
значительно упрощает вычисление объемов растворов
количественно реагирующих друг с другом веществ. Эти объемы обратно
пропорциональны их концентрациям, выраженным в единицах
нормальности:
Произведение объема раствора, выраженного в миллилитрах, на
его нормальность равно числу миллиэквивалентов растворенного
вещества.
Концентрацию растворов выражают также через титр, т. е.
массой (в г или мг) вещества, содержащегося в 1 мл раствора, и
обозначают буквой Г. Найденною величину называют титром
по растворенному (рабочему) веществу. В
аналитической практике пользуются также титром по
анализируемому веществу, т. е. массой (в г или мг) анализируемого
вещества, эквивалентной тому количеству реагента, которое
содержится в 1 мл раствора.
Например, титр 0,1 н H2SO4 (эквивалентная масса H2SO4 =
=49,04 г/моль) равен:
49 04 . О 1
шоо —°'004904 г/мл
При титровании этим раствором NaOH титр H2S04,
выраженный по анализируемому веществу NaOH (эквивалентная масса
NaOH = 40,01 г/моль) равен:
«МИ-ОД _ 0>004001 ^
1000
Концентрацию растворов часто выражают в единицах мо-
ляльности — числом молей вещества, растворенного в 1 кг
растворителя. Моляльность обозначают буквой т.
Формулы перехода от одних выражений концентрации
растворов к другим
Примем следующие условные обозначения концентрации:
С% — процентная концентрация по массе;
А — число единиц массы растворенного вещества на 100 единиц
массы растворителя;
Б — масса растворенного вещества в 1 л раствора;
Сн — число эквивалентов растворенного вещества в 1 л раствора
(нормальность);
См — число молей растворенного вещества в 1 л раствора (моляр*
ность);
105
т —число молей растворенного вещества на 1000 г растворителя
(моляльность);
Э —эквивалентная масса растворенного вещества, г/моль;
М — мольная масса растворенного вещества, г/моль;
d — относительная плотность.
Тогда
100А СНЭ СиМ
т=с„э = смуи
/п = -
М ' М
См1000
М (1000 d- Б)
Растворимость [5, с. 38]
Растворимость — величина, характеризующая способность
вещества образовывать с данным растворителем однородную
систему. Количественно растворимость газа, жидкости или твердого
тела в жидком растворителе измеряется концентрацией
насыщенного раствора при данной температуре.
Обычно растворимость твердых и жидких веществ выражают
коэффициентом растворимости, т. е. массой вещества,
растворяющегося при данных условиях в 100 единицах массы
растворителя с образованием насыщенного раствора.
(Насыщенным называется раствор, находящийся в равновесии с избытком
растворяемого вещества.)
Каждой температуре соответствует определенная
растворимость данного вещества в данном растворителе. Сведения о
растворимости приводятся в справочниках.
Растворимость газов в жидкостях повышается с
увеличением давления и, в большинстве случаев, с понижением
температуры.
Растворимость жидких веществ в жидкостях может
быть неограниченной, когда жидкие компоненты смешиваются
друг с другом в любых отношениях (этиловый спирт — вода) и
ограниченной в случае несмешивающихся жидкостей. В последнем
случае расслаивание жидких компонентов системы зависит от
температуры; обычно взаимная растворимость компонентов
возрастает с температурой. Выше некоторой температурной точки,
называемой кр и ти ческой точкой растворимости, взаимная
106
растворимость компонентов системы становится неограниченной
(расслаивания нет).
Растворимость твердых веществ в жидкостях может
изменяться в широких пределах. Обычно она возрастает с
повышением температуры. Однако некоторые вещества не-подчиняются
этому правилу: растворимость их или понижается с повышением
температуры, или повышается только до некоторого предела,
выше которого растворимость уменьшается.
ТЕХНИКА ПРИГОТОВЛЕНИЯ РАСТВОРОВ [31,62]
По точности выражения концентрации растворы делят на
приблизительные, точные и эмпирические.
Растворы кислот и оснований приблизительной
концентрации служат в качестве вспомогательных реагентов при
выполнении аналитических, препаративных и других работ.
Концентрацию подобных растворов рассчитывают либо по степени
разбавления исходных веществ (растворов), либо по массе
вещества (взвешивается на технических весах), растворенного в
известной массе растворителя. Часто приблизительную концентрацию
растворов определяют по величине плотности.
Растворы с точной, заранее установленной
концентрацией, называемые рабочими, стандартными
или титрованными растворами, служат для определения
точной концентрации других растворов.
Концентрации многих растворов вспомогательных веществ
(индикаторы, специфические реактивы и др.) устанавливаются
эмпирически и приводятся в соответствующих прописях.
Независимо от того, какие по точности концентрации
приготовляют растворы, применять следует только чистые исходные
вещества и воду высокой степени очистки, а в ряде случаев (для
растворов NaOH, Na2S203) — очищенную от С02.
Следует иметь в виду, что скорость растворения твердого
вещества зависит от размера его частиц (тонкоизмельченное
растворяется быстрее).
Некоторые вещества не смачиваются водой и плавают на ее
поверхности, образуя тонкую пленку. Для приготовления водных
растворов подобных веществ рекомендуют порошок вначале
облить небольшим количеством этилового спирта (если он инертен
по отношению к компонентам раствора), а уже затем приливать
воду.
Сосуды для растворения и хранения растворов оснований
должны быть снабжены хлоркальциевыми трубками, заполненными
аскаритом или натронной известью, чтобы защитить раствор от
С02. В некоторых случаях растворы следует хранить в атмосфере
инертного газа (N2rC02). Растворы веществ, разлагающихся под
действием света, например AgN03, следует хранить в сосудах из
коричневого стекла или покрытых черным лаком (в крайнем
случае обернутых в черную бумагу)4
107
Приготовление водных растворов кислот приблизительной
концентрации
Водные растворы кислот (H2S04, HC1, HN03) обычно
приготовляют соответствующим разбавлением исходных химически
чистых концентрированных кислот. Разбавление проводят из расчета
на объем, тад< как жидкость всегда легче отмерить, чем взвесить.
Чтобы получить разбавленную кислоту (например, 1:5), к 5
объемам воды прибавляют 1 объем кислоты.
Процентное содержание концентрированных- кислот
контролируют по плотности, определяемой большей частью ареометром
(см. гл. 34). Значения концентрации кислот в зависимости от
плотности см. в справочниках.
Обращаться с концентрированными кислотами следует
осторожно, так как они сильно действуют на кожу, разрушают одежду
и обувь, портят полы и столы. При работе с концентрированными
кислотами необходимо пользоваться резиновыми перчатками и
защитными очками.
При приготовлении разбавленных растворов кислот (в
особенности H2S04) следует приливать кислоту в воду тонкой струей
при непрерывном перемешивании стеклянной палочкой. Если при
этом смесь сильно разогрелась, то ее охлаждают, после чего
приливают следующую порцию кислоты.
Кислоту, попавшую на обувь или одежду, необходимо
незамедлительно смыть большим объемом воды, нейтрализовать
аммиаком или NaHC03 и снова обмыть водой. Кислоту, разлитую
на столе или на полу, засыпают песком, нейтрализуют Na2C03,
СаО, Са(ОН)2, СаСОз и лишь после этого производят уборку.
При приготовлении разбавленных растворов из более
концентрированных или путем смешения растворов разных концентраций,
для расчета соотношения объемов удобно пользоваться так
называемым правилом креста или смешения. Это правило может быть
иллюстрировано схемой получения 5% (по массе) раствора
разбавлением 20% раствора:
Концентрация одного
. из компонентов, %
имеющаяся
требуемая
Разность
Следует
смешать
5-0
20-5
5 частей
15 частей
Правило креста распространяется и на случай, когда
концентрация смешиваемых водных растворов выражена через плотность.
Пусть дан водный раствор плотностью 1,57 г/см3. Нужно из него
108
приготовить раствор с плотностью 1,20 г/см3. По правилу креста
составляем схему:
1,57-
отсюда следует, что нужно смешать 20 см8 раствора с р=1,57 г/см3
с 37 частями по массе воды.
Расчет концентрации по правилу креста не отличается
точностью, и пользоваться этим методом можно лишь для
приготовления растворов приблизительной концентрации.
Приготовление безводного раствора хлорной кислоты
Раствор хлорной кислоты в безводной уксусной кислоте
широко применяют в качестве титранта для кислотно-основного
титрования в неводной среде.
Промышленность выпускает хлорную кислоту различной
концентрации (от 42 до 70%.), чаще всего в виде 57% водного
раствора с плотностью около 1,50.
Избыточную воду из хлорной кислоты удаляют уксусным
ангидридом:
(СН3СО)20 + Н20 —> 2СН3СООН
Предварительно определив содержание воды в хлорной
кислоте, последнюю растворяют в ледяной уксусной кислоте и
рассчитывают, какой объем V\ (в мл) уксусного ангидрида необходим
для удаления из хлорной кислоты избыточной воды:
^ (100 —Л) Ур 102
!~ 100 • 1ьр,
где 100 — А — содержание воды в исходном растворе НС104, %;
V — объем НСЮ4, взятый для приготовления раствора, мл; р —
плотность применяемого раствора НСЮ4, г/см3; pi — плотность
уксусного ангидрида, г/см3; 102 — молекулярная масса уксусного
ангидрида; 18 — молекулярная масса воды.
Определенный объем НС104 V постепенно, при непрерывном
перемешивании, вливают в 800 мл ледяной уксусной кислоты,
прибавляют V{ мл уксусного ангидрида, тщательно перемешивают,
доводят объем раствора ледяной уксусной кислотой до 1 л и снова
перемешивают. Через сутки раствор готов.
Приготовление растворов щелочей
При растворении NaOH или КОН необходимо пользоваться
резиновыми перчатками и защитными очками. Щелочи вызывают
химический ожог кожи, разрушают одежду и обувь. Брать
твердую щелочь руками запрещается.
Водные растворы NaOH и КОН. При растворении твердых
NaOH и КОН в воде происходит сильное разогревание; поэтому
109
насыщенные растворы щелочей приготовляют в термостойкой
стеклянной или, лучше, в фарфоровой посуде, постепенно
добавляя твердую щелочь при перемешивании, чтобы избежать
местного перегрева.
На воздухе NaOH и КОН поглощают воду и С02.
Образующиеся карбонаты цгло растворимы в концентрированном
растворе щелочей и постепенно выпадают в осадок.
Концентрированные растворы щелочей при хранении в
стеклянной посуде разрушают стекло, выщелачивая из него
кремневую кислоту. Поэтому лучше хранить их в сосудах из полиэтилена.
Из концентрированных растворов получают разбавленные
растворы щелочей, концентрацию которых контролируют по
плотности. Ориентировочное значение объемов разбавляемого раствора
щелочи и воды можно рассчитать и по правилу креста.
Приготовление 50% раствора NaOH, не содержащего
карбонатов (по ГОСТ 4517—75), производят следующим образом: в
фарфоровом стакане растворяют при постепенном добавлении и
перемешивании 250 г NaOH в 250 мл дистиллированной воды.
После охлаждения раствор переливают в полиэтиленовый сосуд,
закрывают пробкой и выдерживают 2—3 педели, до полного
осаждения Na2C03. Затем прозрачный раствор спфонируют стеклянной
трубкой и соответственно разбавляют водой, не содержащей С02.
Спиртовый раствор КОН. Растворимость NaOH и КОН в
метиловом спирте выше, чем в этиловом. Однако поскольку метиловый
спирт очень токсичен и огнеопасен, обычно используют этанольные
растворы NaOH и КОН. Растворимость NaOH в этиловом спирте
при 28°С составляет 14,7%, а КОН —27,9%.
Для приготовления раствора КОН применяют этиловый
ректификованный спирт, предварительно очищенный от альдегидов.
Наиболее эффективен следующий способ очистки: раствор из
2 г AgN03 в 5 мл дистиллированной воды вливают в 1200 мл
этилового спирта, находящегося в склянке с притертой пробкой,
и тщательно перемешивают. Отдельно растворяют 5 г КОН в
25 мл горячего этилового спирта, раствор охлаждают и вливают
в спиртовый раствор AgN03. Выпадает осадок Ag20, которому
дают осесть, фильтруют и отгоняют спирт. Этиловый спирт, очи-
щенный этим способом, остается бесцветным несколько лет.
Спиртопый раствор КОН при хранении часто приобретает
слабо-желтую окраску, вызываемую осмолением примесей. Для
приготовления растворов КОН, не окрашивающихся при длительном
хранении, рекомендуют спирт предварительно обработать бутила-
том алюминия (5 г на 1 л спирта). Смеси дают постоять 3—4
недели при комнатной температуре, после чего спирт декантируют
и растворяют в нем КОН.
Приготовление водного раствора аммиака
Поступающий в продажу водный раствор аммиака плотностью
0,901—0,907 г/см3 при 20 °С, содержит 25—27% NH3. Препарат и
его разбавленные растворы вполне пригодны для выполнения
110
Рис. 63. Установка для приготовления раствора аммиака из баллонного газа,
большинства препаративных и вспомогательных лабораторных
работ.
Для аналитических работ ГОСТ 4517—75 рекомендует
приготовлять растворы из баллонного жидкого синтетического аммиака
или из водного аммиака, поступающего в продажу.
Газообразный аммиак вызывает раздражение глаз и слизистой
оболочки носа, тошноту и головные боли. Все работы с аммиаком
должны проводиться в вытяжном шкафу.
Из баллонного аммиака. Собирают установку (рис. 63).
Баллон с аммиаком / устанавливают и закрепляют на подставке 2.
Баллон соединяют с пустой промежуточной склянкой «3, к
которой присоединены две поглотительные склянки 4 с раствором
NaOH для поглощения С02. Аммиак, очищенный от С02, поступает
в приемник 5, где находится дважды перегнанная
дистиллированная вода, не содержащая С02. Насыщение аммиаком проводят
до достижения плотности раствора в приемнике 0,907 г/см3, что
соответствует 25% раствору аммиака.
Для получения более концентрированного раствора приемник
охлаждают водой со льдом в бане 8.
Склянка 6 — брызгоуловитель; склянка 7, содержащая раствор
NaOH, предохраняет от попадания С02 из воздуха в приемник.
Из водного аммиака. 500 мл водного аммиака помещают в
круглодонную колбу вместимостью 1 л и осторожно прибавляют
свежеприготовленную кашицу из 10 г СаО и воды.
Колбу соединяют с вертикально поставленным обратным
холодильником, верхний конец которого закрывают трубкой с
натронной известью, и оставляют раствор в покое на 18—20 ч. Затем
собирают установку (рис. 64). Колбу 2 с водным аммиаком
ставят на водяную баню / так, чтобы холодильник был направлен
вверх под углом 45°, и соединяют верхний его конец через
промежуточную колбу 4 с приемником — колбой 5, содержащей 300—
400 мл воды, и закрытой трубкой с натронной известью 6. При
нагревании водного аммиака на водяной бане газообразный аммиак
поступает в приемник и полностью поглощается водой.
Насыщение аммиаком проводят до достижения плотности раствора в
приемнике 0,907 г/см3, что соответствует 25% раствору аммиака.
111
Рис. 64. Установка для приготовления раствора аммиака из водного аммиака.
Приготовление рабочих растворов точной концентрации
Приготовление раствора из навески стандартного вещества.
Взятую с точностью до 0,0002 г навеску высушенного
стандартного вещества, которая приблизительно соответствует рассчитанной
для получения определенного объема раствора заданной
концентрации, аккуратно переносят в мерную колбу и растворяют в
небольшом объеме дистиллированной воды, не содержащей С02.
Полученный раствор при периодическом взбалтывании разбавляют
водой, доводя объем раствора в мерной колбе несколько ниже
метки. Затем колбу с раствором выдерживают 15—20 мин при 20°С
и осторожно добавляют воду до метки. Колбу закрывают пробкой
и содержимое взбалтывают в течение 15—30 мин.
Зная массу исходного вещества и объем раствора, вычисляют
его концентрацию.
Для упрощения последующих расчетов удобно пользоваться
поправкой на нормальность (или коэффициентом
нормальности) К. Эта поправка представляет собой
отношение нормальности приготовленного раствора к заданной
нормальности раствора, выраженной целыми, десятыми или сотыми
долями нормальности. Например, нормальность приготовленного
раствора оказалась равной 0,1036 н., а заданная 0,1 н. В этом случае
' При умножении объема пошедшего на титрование раствора на
эту поправку К получают эквивалентный объем заданной
концентрации (в данном случае 0,1 н.).
В табл. 3 приведены некоторые твердые стандартные вещества,
с помощью которых точно устанавливается концентрация
наиболее часто применяемых рабочих растворов.
Приготовление растворов из фиксаналов. Фиксаналы, или
стандарт-титры, представляют собой точно отвешенное коли-
112
чество реактива или его раствора, запаянного в стеклянную
ампулу. Как правило, в каждой ампуле содержится 0,1 эквивалента
вещества. При количественном перенесении содержимого подобной
ампулы в мерную колбу на 1 л и доведении объема раствора водой
до метки при 20 °С получаются точно 0,1 н, растворы.
Выпускаются фиксаналы НС1, H2SO4, NaOH, КОН, Na2C03,
NaCl, Na2C204, H2C204-2H20, K2Cr207, K2C204, Na2S203-5H20,
KMn04, AgN03, NH4SCN, KSCN, NaSCN, BaCl2.2H20,
(NH4)2C204-H20, Na2B4O7-10H2O, KC1, K2C03, NH4Cl, I2 и др.
таблица з. Стандартные вещества и рабочие растворы
Рабочий
раствор
Стандартное вещество-
Методика определения
НС1, H2S04
NaOH; КОН
AgN03
KMn04
НСЮ4
Na2S203
Na2B4O710H2O
Эквивалент 190,70
Na2C03
Эквивалент 53,00
Высушивание при 150 °С
(СН2СООН)2 — янтарная
кислота
Эквивалент 59,04
Высушивание при 100 °С
H2NS03H — сульфамино-
вая кислота
Эквивалент 97,10
Высушивание при 100 °С
NaCl
Эквивалент 58,44
Высушивание при 120 °С
Na2C204
Эквивалент 67,00
Высушивание при 105—
120°С
CgH504K — гидрофталат
калия
Эквивалент 201,23
Высушивание при 110-
116°С
Ь (возогнанный)
Эквивалент 126,9
КЮз
Эквивалент 35,67
Навеску ~0,4 г растворяют при
нагревании в 25 мл Н20, охлаждают
и титруют с метиловым
оранжевым до оранжевой окраски
Навеску ~0,1 г растворяют в 25 мл
Н20 и титруют с метиловым
оранжевым до оранжевой окраски
Навеску ~0,12 г растворяют в 30 мл
Н20 и титруют с фенолфталеином
до розовой окраски
Навеску ~0,1 г растворяют в 30 мл
Н20 и титруют с метиловым
оранжевым до желтой окраски
Навеску ~0,1 г растворяют в 20 мл
Н20, добавляют 10 мл HN03
(1 : 10) и титруют с 5 каплями
1 % спиртового раствора дифенил-
карбазона до темно-голубой
окраски
Навеску ~0,13 г растворяют в 50 мл
Н20, добавляют 10 мл H2S04
(1 : 1), нагревают до 80°С и
титруют до розовой окраски
Навеску ~0,5 г растворяют при
нагревании в 50 мл безводной
СНзСООН, добавляют 2 капли
кристаллического фиолетового и
титруют 0,1 н. НС104 до перехода
окраски из синей в зеленую
Навеску ~0,25 г растворяют 10 мл
20% раствора KI и титруют с
раствором крахмала до
обесцвечивания
Навеску ~0,07 г растворяют в 25 мл
Н20, добавляют 2 г KI, 10 мл 1 н.
Н£1 и титруют с раствором
крахмала до обесцвечивания
113
n
Рис* 65. Прибор для приготовления растворов из фиксанала:
/—боек; 2— ампула; 3 — палочка для пробивания.
Фиксаналы рекомендуется применять во всех
случаях, когда требуется быстро приготовить точный
рабочий раствор, не прибегая к взвешиванию.
Вначале теплой водой смывают надпись на
ампуле и хорошо обтирают ее чистым полотенцем. В
мерную колбу вместимостью 1 л вставляют воронку с
вложенным в нее стеклянным бойком (обычно
прилагается к каждой коробке фиксанала), острый конец
которого должен быть обращен вверх (рис. 65).
Ампуле с фиксаналом дают свободно падать так, чтобы
тонкое дно ампулы разбилось при ударе об острый
конец бойка. После этого другим стеклянным бойком
пробивают боковое углубление ампулы и дают со-
вытечь. Не меняя положения ампулы, в
образовавшееся верхнее отверстие вставляют оттянутый в капилляр и
изогнутый вверх конец трубки промывалки и сильной струей
промывают ампулу изнутри. Затем струей воды из промывалки хорошо
промывают наружную поверхность ампулы и воронку с бойком.
Удалив ампулу из воронки, доводят уровень жидкости в колбе до
метки. Колбу плотно закрывают и тщательно перемешивают
раствор.
При пользовании фиксаналом 0,1 н. иода перед вскрытием
ампулы необходимо поместить в мерную колбу 30—40 г KI для
полного растворения иода.
Ампулы с фиксаналами твердых веществ (Н2С204-2Н20, NaCl,
KMn04 и др.) вскрывают так же, как описано выше, но воронка
должна быть совершенно сухая. Когда ампула разбита,
содержимое ее осторожным встряхиванием высыпают в колбу, ампулу и
воронку тщательно промывают дистиллированной водой.
Фиксанал AgN03 при обычных условиях хранения через 2—
3 года темнеет. Фиксаналы большинства других твердых веществ
и кислот могут храниться неопределенно долгое время.
Фиксаналы NaOH, КОН пригодны только в течение 6 месяцев
со дня их выпуска. Помутнение щелочных растворов — признак их
порчи.
Рабочие растворы с точной концентрацией должны быть по
возможности свежеприготовленными. Исключение составляют
растворы КМ1Ю4, титр которых следует устанавливать не ранее чем
через 3—4 дня после их приготовления.
При хранении рабочих растворов следует периодически
проверять их концентрацию. Рабочие растворы щелочей и тиосульфата
натрия следует защищать от действия СОг (хлоркальциевые
трубки с натронной известью или аскаритом).
Сосуды с рабочими растворами должны иметь четкие надписи
с указанием вещества, нормальности, поправочного коэффициента,
даты изготовления и даты проверки концентрации.
П4
держимому
ПРИГОТОВЛЕНИЕ БЕЗВОДНЫХ ЧИСТЫХ
ОРГАНИЧЕСКИХ РАСТВОРИТЕЛЕЙ
Методы очистки органических растворителей зависят от
природы и предназначения растворителя. В большинстве случаев
органические растворители представляют собой индивидуальные
соединения и могут быть охарактеризованы их физико-химическими
показателями. Самая элементарная операция очистки
растворителя— простая или фракционная перегонка. Однако перегонкой
часто не удается освободиться от ряда примесей, в том числе и от
малых количеств воды.
Традиционными методами очистки можно получить
растворитель приблизительно 100% чистоты, С помощью адсорбентов, в
частности молекулярных сит (цеолитов), эта задача решается
более эффективно и с меньшей затратой времени. В лабораторных
условиях для этой цели чаще всего применяют иониты — цеолиты
марок NaA или КА (см. гл. 29).
При приготовлении чистых безводных растворителей следует
особо строго соблюдать меры предосторожности, так как
большинство органических растворителей — горючие вещества, пары
которых образуют с воздухом взрывоопасные смеси, а в
некоторых из них (простые эфиры) при длительном хранении образуются
взрывчатые перекисные соединения. Многие органические
растворители весьма токсичны, как при вдыхании их паров, так и при
действии их на кожу.
Все операции с легковоспламеняющимися и горючими
органическими растворителями должны проводиться в вытяжном шкафу
при работающей вентиляции, выключенных газовых горелках и.
электронагревательных приборах. Нагревать и перегонять
жидкости следует в вытяжном шкафу на предварительно нагретых
банях, заполненных соответствующим теплоносителем. При
перегонке органической жидкости необходимо постоянно следить за
работой холодильника.
Если легковоспламеняющиеся растворители (бензин, диэтило-
вый эфир, сероуглерод и др.) случайно прольются, необходимо
немедленно погасить все источники открытого огня и выключить
электронагревательные приборы (днем обесточить рабочее
помещение). Место, где пролита жидкость, следует засыпать песком,
загрязненный песок собрать деревянным совком и высыпать в
контейнер для мусора, установленный на открытом воздухе.
При высушивании растворителей не следует применять
активные высушивающие средства до тех пор, пока не проведена
предварительная грубая сушка с помощью обычных осушающих
средств. Так, запрещается сушить сырой диэтиловый эфир
металлическим натрием без предварительной его сушки прокаленным
СаС12.
115
При работе с простыми эфлрамн и другими веществами (ди-
этиловый эфир, диоксан, тетрагидрофуран), в процессе хранения
которых могут образоваться перекисные соединения, сначала из
них удаляют. перекиси, а затем перегоняют и осушают.
Перегонять безводные органические растворители надо осторожно. Все
элементы установки для перегонки (перегонная колба,
дефлегматор, холодильник, алонж, приемник дистиллята) предварительно
высушивают в сушильном шкафу. Перегонку проводят без доступа
воздуха, а алонж снабжают хлоркальциевой трубкой,
наполненной аскаритом и плавленым СаСЬ для поглощения С02 и Н20.
Первую порцию дистиллята, служащую для промывки всей
аппаратуры, целесообразно отбросить.
Ниже рассматриваются методы очистки и обезвоживания
наиболее употребительных растворителей [38, с. 591].
Ацетон СН3СОСН3 — бесцветная жидкость; df = 0,7899; /кип=
*=56,24°С; п?£ = 1,3591.Легко воспламеняется. Пары образуют с
воздухом взрывоопасные смеси. Технический ацетон обычно
содержит воду, с которой он смешивается в любых соотношениях.
Иногда ацетон загрязнен метиловым спиртом, уксусной кислотой
и восстанавливающими веществами.
Пробу на присутствие восстанавливающих веществ в ацетоне
проводят следующим образом. К 10 мл ацетона прибавляют 1
каплю 0,1% водного раствора КМп04; через 15 мин при комнатной
температуре раствор не должен обесцветиться.
Для очистки ацетон несколько часов нагревают с безводным
К2СО3 [5% (масс.)] в колбе с обратным холодильником, затем
жидкость переливают в другую колбу с дефлегматором высотой
25—30 см и перегоняют над безводным К2СО3 [около 2% (масс.)]
и кристаллическим КМп04, который добавляют к ацетону до
появления устойчивой фиолетовой окраски на водяной бане. В
полученном ацетоне уже нет метилового спирта, но есть
незначительное количество воды.
Для полного удаления воды ацетон повторно перегоняют над
безводным СаС12. Для этого в 2-литровую круглодонную колбу,
снабженную эффективным обратным холодильником, закрытым
хлоркальциевой трубкой с СаС12, вливают 1 л ацетона, вносят
120 г СаС12 и кипятят на водяной бане с закрытым электрическим
обогревом 5—6 ч. Затем реакционную колбу охлаждают и
переливают ацетон в другую аналогичную колбу со свежей порцией
СаС12 и вновь кипятят 5—6 ч. После этого обратный холодильник
ваменяют на нисходящий, к которому при помощи алонжа,
соединенного с хлоркальциевой трубкой, наполненной СаС12,
присоединяют склянку-приемник, охлаждаемую льдом, и перегоняют
ацетон над СаС12.
Вместо столь длительной и трудоемкой операции, которая
часто приводит к конденсации ацетона, лучше использовать цеолит
NaA. При длительном выдерживании ацетона над этим цеолитом
i[5% (масс.)] достигается абсолютирование ацетона [7].
116
В небольших количествах очень чистый ацетон может быть
получен из аддукта (продукта присоединения) ацетона и Nal,
который уже при слабом нагревании разлагается, выделяя ацетон.
Для этого при нагревании на водяной бане растворяют 100 г Nal
в 440 мл сухого свежеперегнанного ацетона. Образующийся
раствор быстро охлаждают до —3°С, погружая сосуд в смесь льда с
NaCl. Выделившийся твердый аддукт NabC3H60 отделяют на
воронке Бюхнера, переносят в колбу установки для перегонки и
нагревают на водяной бане. При легком нагревании аддукт
разлагается, и освобождающийся ацетон отгоняется. Дистиллят сушат
безводным СаС12 и повторно перегоняют с дефлегматором над
СаС12. Регенерированный Nal может быть вновь применен для
этой же реакции.
Экспрессный способ очистки ацетона от метилового спирта и
восстанавливающих веществ заключается в следующем: к 700 мл
ацетона в колбе вместимостью 1 л приливают раствор 3 г AgNOs
в 20 мл дистиллированной воды и 20 мл 1 н. раствора NaOH.
Смесь встряхивают в течение 10 мин, после чего осадок
отфильтровывают на воронке со стеклянным фильтром, а фильтрат сушат
CaS04 и перегоняют с дефлегматором над СаС12.
Ацетонитрил CH3CN — бесцветная жидкость с характерным
эфирным запахом; df = 0,7828; *КИП = 81,6°С; /г^°= 1,3442. С
водой смешивается во всех отношениях и образует азеотропную
смесь [16% (масс.) Н20] с /КИП=76°С. Хороший растворитель для
ряда органических веществ, в частности хлоргидратов аминов.
Используется также в качестве среды для проведения некоторых
реакций, которые он ускоряет каталитически.
Ацетонитрил — сильный ингаляционный яд и способен
всасываться через кожу.
Для абсолютирования ацетонитрил дважды перегоняют над
P4Oio с последующей перегонкой над безводным К2СО3 для
удаления следов Р4О10.
Можно ацетонитрил предварительно высушить над Na2S04 или
MgS04, затем перемешать с СаН2 до прекращения выделения газа
(водорода) и перегнать над Р4О10 (4—5 г/л). Дистиллят кипятят
с обратным холодильником над СаН2 (5 г/л) не менее 1 ч, затем
медленно перегоняют, отбрасывая первые 5 и последние 10% дис-"
тиллята.
Бензол С6Нб — бесцветная жидкость; df = 0,8790; пл = 5,54 °С;
/кип = 80,10°С; n|J= 1,5011. Бензол и его гомологи — толуол
и ксилолы — широко используются в качестве растворителей, и
среды для азеотропной сушки. Работать с бензолом следует
осторожно из-за его горючести и токсичности, а также из-за
образования с воздухом взрывоопасных смесей.
Пары бензола при многократном воздействии нарушают
нормальную функцию кроветворных органов; в жидком состоянии
бензол сильно всасывается через кожу и раздражает ее.
117
Технический бензол содержит до 0,02% (масс.) воды, немного
тиофена и некоторые другие примеси.
Бензол образует с водой азеотропную смесь [8,83% (масс.)
Н20] с £кип=69,25°С. Поэтому при перегонке влажного бензола
вода практически полностью отгоняется с первыми порциями
дистиллята (мутная жидкость), которые отбрасывают. Как только
начнет перегоняться прозрачный дистиллят, можно считать процесс
осушения завершенным. Доосушение перегнанного бензола обычно
производят прокаленным СаСЬ (в течение 2—3 суток) и
натриевой проволокой.
В холодное время года надо следить за тем, чтобы
перегоняемый бензол не закристаллизовался в трубке холодильника,
омываемой холодной водой (4—5°С).
Бензол и другие углеводороды, высушенные металлическим
натрием, гигроскопичны, т. е. могут поглощать влагу.
Товарный технический бензол содержит до 0,05% (масс.) тио-
.фена C4H4S (/Кип=84,12°С; /Пл=38,3°С), который нельзя отделить
от бензола ни фракционной перегонкой, ни кристаллизацией
(вымораживанием). Тиофен в бензоле обнаруживают следующим
образом: раствор 10 мг изатина в 10 мл конц. H2S04 встряхивают с
3 мл бензола. В присутствии тиофена сернокислотный слой
окрашивается в сине-зеленый цвет.
Бензол очищают от тиофена многократным встряхиванием с
конц. H2SO4 при комнатной температуре. В этих условиях
сульфируется преимущественно тиофен, а не бензол. На 1 л бензола
берут 80 мл кислоты. Первая порция H2S04 окрашивается в сине-
зеленый цвет. Нижний слой отделяют, а бензол встряхивают с
новой порцией кислоты. Очистку ведут до тех пор, пока не будет
достигнуто слабо-желтое окрашивание кислоты. После отделения слоя
кислоты бензол промывают водой, затем 10% раствором Na2C03
и снова водой, после чего бензол перегоняют.
Более эффективный и простой метод очистки бензола от
тиофена— кипячение 1 л бензола с 100 г никеля Ренея * в колбе с
обратным холодильником в течение 15—30 мин.
Еще один способ очистки бензола от тиофена заключается в
\дробной кристаллизации его из этилового спирта. Насыщенный
раствор бензола в спирте охлаждают примерно до —15°С, быстро
отфильтровывают твердый бензол и перегоняют.
Диметилсульфоксид (CH3)2SO — бесцветная сиропообразная
жидкость без выраженного запаха; df= 1,1014; /кип = 189 °С (с
разложением); *пл = 18,45°С; п2£= 1,4770. Смешивается с водой,
спиртами, ацетоном, этилацетоном, диоксаном, пиридином и
ароматическими углеводородами, но не смешивается с
алифатическими углеводородами. Универсальный растворитель для
органических соединений: окиси этилена, гетероциклических соединений,
камфоры, смол, Сахаров, жиров и др. Он растворяет также и мно-
* Никель Ренея — скелетный никелевый катализатор, получаемый путем
выщелачивания алюминия раствором щелочи из сплава никеля с алюминием.
118
гие неорганические соединения, например при 60 °С растворяет
10,6% (масс.) КЖ)3и21,8% СаС12.
Диметилсульфоксид практически не токсичен.
Для очистки диметилсульфоксид выдерживают в течение суток
над активным А1203, после чего дважды перегоняют при давлении
267—400 Па (2—3 мм рт. ст.) над плавленым КОН (или ВаО)
и хранят над цеолитом NaA [7].
Под действием восстановителей диметилсульфоксид
превращается в сульфид (CH3)2S, а под действием окислителей — в сульфон
(CH3)2S02, несовместим с хлорангидридами неорганических и
органических кислот.
#,Л^Диметилформамид HCON (СН3)г — бесцветная
легкоподвижная жидкость со слабым специфическим запахом; df = 0,9445;
/КИП=153°С; п2р = 1,4269. Смешивается в любых отношениях с
водой, спиртом, ацетоном, эфиром, хлороформом, сероуглеродом,,
галогенсодержащими и ароматическими соединениями;
алифатические углеводороды растворяет лишь при нагревании.
Диметилформамид перегоняется при атмосферном давлении
без разложения; разлагается под действием ультрафиолетовых
лучей с образованием диметиламина и формальдегида. Реактив
диметилформамид, кроме метиламина и формальдегида, в качестве
примесей может содержать метилформамид, аммиак и воду.
Диметилформамид очищают следующим образом: к 85 г диме-
тилформамида прибавляют 10 г бензола и 4 мл воды и смесь
перегоняют. Вначале отгоняется бензол с водой и другими примесями*
а затем чистый продукт.
Диэтиловый эфир (С2Нб)20 — бесцветная легкоподвижная лету-
мая жидкость со своеобразным запахом; df~ 0,7135; ^КИП = 35,6°С
п^ = 1,3526. Чрезвычайно легко воспламеняется; пары
образуют с воздухом взрывоопасные смеси. Пары тяжелее воздуха
примерно, в 2,6 раза и могут стелиться по поверхности рабочего
стола. Поэтому необходимо следить за тем, чтобы поблизости (до 2—
3 м) от места работы с эфиром все газовые горелки были
потушены, а электроплитки с открытой спиралью отключены от сети.
При хранении диэтилового эфира под действием света и
кислорода воздуха в нем образуются взрывчатые перекисные
соединения и ацетальдегид. Перекисные соединения являются причиной"
чрезвычайно сильных взрывов, особенно при попытке перегнать
эфир досуха. Поэтому при определении температуры кипения и
нелетучего остатка эфир следует предварительно проверить на
содержание перекисей. При наличии перекисей эти определения
проводить нельзя.
Для обнаружения перекиси в диэтиловом эфире предложены
многие реакции.
1. Реакция с иодидом калия KI. Несколько миллилитров эфира
встряхивают с равным объемом 2% водного раствора KI,
подкисленного 1—2 каплями НС1. Появление коричневого окрашивания
указывает на присутствие перекисей.
119»
2. Реакция с титанилсульфатом TiOS04. Реактив готовят
растворением 0,05 г TiOS04 в 100 мл воды, подкисленной 5 мл
разбавленной H2S04 (1 :5). При встряхивании 2—3 мл этого
реактива с 5 мл испытуемого эфира, содержащего перекисные
соединения, появляется желтая окраска.
3. Реакция с бихроматом натрия Na2Cr207. К 3 мл эфира
прибавляют 2—3 мл 0,01% водного раствора Na2Cr207 и одну каплю
разбавленной H2S04 (1 :5). Смесь сильно взбалтывают. Синяя
окраска эфирного слоя указывает на присутствие перекисей.
4. Реакция с ферротиоцианатом Fe(SCN)2 [7]. Бесцветный
раствор Fe(SCN)2 под действием капли жидкости, содержащей
перекись, окрашивается в красный цвет вследствие образования
ферритиоцианата (Fe2+->Fe3+). Эта реакция позволяет
обнаруживать перекиси в концентрации до 0,001% (масс). Реактив
готовят следующим образом: 9 г FeS04-7H20 растворяют в 50 мл
18% НС1. К раствору в открытом сосуде добавляют
гранулированный цинк и 5 г тиоцианата натрия NaSCN; после исчезновения
красного окрашивания добавляют еще 12 г NaSCN, осторожно
взбалтывают и раствор отделяют декантацией.
Чтобы удалить перекиси, применяют сульфат железа (II). При
взбалтывании 1 л эфира обычно берут 20 мл раствора,
приготовленного из 30 г FeS04-7H20, 55 мл Н20 и 2 мл конц. H2S04.
После промывания эфир встряхивают с 0,5% раствором КМп04 для
окисления ацетальдегида в уксусную кислоту. Затем эфир
промывают 5% раствором NaOH и водой, сушат 24 ч над СаС12 (150—
200 г СаС12 на 1 л эфира). После этого отфильтровывают СаС12
на большом складчатом бумажном фильтре и собирают эфир в
склянку из темного стекла. Склянку плотно закрывают корковой
лробкой со вставленной в нее изогнутой под острым углом хлор-
кальциевой трубкой, наполненной СаС12 и тампонами из
стеклянной ваты. Затем, открыв склянку, быстро вносят в эфир
натриевую проволоку, из расчета 5 г на 1 л эфира.
Через 24 ч, когда перестанут выделяться пузырьки водорода,
добавляют еще 3 г натриевой проволоки на 1 л эфира и спустя
12 ч эфир переливают в колбу для перегонки и перегоняют над
натриевой проволокой. Приемник должен быть защищен хлор-
кальциевой трубкой с СаС12. Дистиллят собирают в склянку из
темного стекла, которую после внесения 1 г натриевой проволоки
на 1 л эфира закрывают корковой пробкой с хлоркальциевой
трубкой и хранят в холодном и темном месте.
Если поверхность проволоки сильно изменилась и при
добавлении проволоки снова выделяются пузырьки водорода, то эфир
следует профильтровать в другую склянку и добавить еще
порцию натриевой проволоки.
Удобный и весьма эффективный способ очистки диэтилового
эфира от перекисей и одновременно от влаги — пропускание эфира
через колонку с активным А120з. Колонки высотой 60—80 см и
диаметром 2—4 см, заполненной 82 г А1203, достаточно для
очистки 700 мл эфира, содержащего значительное количество перекис-
120
ных соединений. Отработанный А1203 легко регенерировать, если
пропустить через колонку 50% подкисленный водный раствор
FeS04-7H20, промыть водой, высушить и провести термическую
активацию при 400—450 °С.
Абсолютный эфир — весьма гигроскопичная жидкость. О
степени поглощения влаги эфиром при его хранении можно судить по
посинению безводного белого порошка CuS04 при внесении его в
эфир (образуется окрашенный гидрат CuS04-5H20).
Диоксан (СНгЬО — бесцветная горючая жидкость со слабым
запахом; d?= 1,03375; \ип = 101,32 °С; /пл= 11,80° С; /г2о0 = 1,4224.
Смешивается с водой, спиртом и эфиром в любых отношениях.
Образует с водой и спиртом азеотропные смеси.
Технический диоксан содержит в качестве примесей ацеталь
этиленгликоля, воду, ацетальдегид и перекиси. Способ очистки
диоксана следует выбирать в зависимости от степени его загряз-
нения, которую определяют, добавляя к диоксану металлический
натрий. Если при этом образуется коричневый осадок, то диоксан
сильно загрязнен; если поверхность натрия изменяется
незначительно, то диоксан содержит мало примесей и его очищают,
перегоняя над натриевой проволокой.
Сильно загрязненный диоксан очищают следующим образом:
0,5 л диоксана, 6 мл конц. НС1 и 50 мл Н20 нагревают на
силиконовой (масляной) бане в токе азота в колбе с обратным
холодильником при 115—120 °С в течение 12 ч.
После охлаждения жидкость встряхивают с небольшими
порциями плавленого КОН для удаления воды и кислоты. Диоксан
образует верхний слой, его отделяют и сушат свежей порцией
КОН. Затем диоксан переносят в чистую перегонную колбу и
нагревают с обратным холодильником над 3—4 г натриевой
проволоки в течение 12 ч. Очистка считается законченной, если
поверхность натрия остается неизменной. Если натрий весь
прореагировал, то необходимо добавить свежую порцию и продолжить
высушивание. Диоксан, не содержащий перекисных соединений,
перегоняют на колонке или с эффективным дефлегматором при
обычном давлении. Очистка диоксана от перекисей проводится
так же, как и очистка диэтилового эфира.
Метиловый спирт (метанол) СНзОН — бесцветная
легкоподвижная горючая жидкость, с запахом, подобным запаху
этилового спирта; df = 0,7928; /кип = 64,51 °С; л* =1,3288.
Смешивается во всех отношениях с водой, спиртами, ацетоном и другими
органическими растворителями; не смешивается с алифатическими
углеводородами. Образует азеотропные смеси с ацетоном (/Кип =
= 55,7 °С), бензолом (/кип = 57,5 °С), сероуглеродом (/кип =
= 37,65 °С), а также со многими другими соединениями. С водой
метиловый спирт не образует азеотропных смесей, поэтому
большую часть воды можно удалить перегонкой спирта.
Метиловый спирт — сильный яд, поражающий
преимущественно нервйую систему и кровеносные сосуды. В организм человека
121
он может поступить через дыхательные пути и кожу. Особенно
опасен при приеме внутрь. Применение метилового спирта в
лабораторной практике допускается только в тех случаях, когда
он не может быть заменен другими, менее токсичными
веществами.
Синтетический абсолютированный метиловый спирт,
выпускаемый промышленностью, содержит лишь следы ацетона и до 0,1%
(масс.) воды. В лабораторных условиях его можно приготовить
из технического СН3ОН, в котором содержание этих примесей
может достигать 0,6 и даже 1,0%. В колбу вместимостью 1,5 л с
обратным холодильником, защищенным хлоркальциевои трубкой
с СаС12, помещают 5 г магниевых стружек, заливают их 60—70 мл
метилового спирта, содержащего не более 1 % воды, прибавляют
инициатор — 0,5 г иода (или соответствующее количество метил-
иодида, этилбромида) и нагревают до растворения последнего.
Когда весь магний перейдет в метилат (на дне колбы образуется
белый осадок), к полученному раствору прибавляют 800—900 мл
технического СН3ОН, кипятят в колбе с обратным холодильником
в течение 30 мин, после чего отгоняют спирт из колбы с
дефлегматором высотой 50 см, собирая фракцию с температурой кипения
64,5—64,7°С (при нормальном давлении). Приемник снабжают
хлоркальциевои трубкой с СаС12. Содержание воды в полученном
таким способом спирте не превышает 0,05% (масс).
Абсолютированный метиловый спирт сохраняют в сосуде, защищенном от
влаги воздуха.
Доосушивание метилового спирта, содержащего 0,5—1% воды,
можно осуществить металлическим магнием и без инициирования
реакции. Для этого к 1 л СН3ОН прибавляют 10 г магниевых
стружек и смесь оставляют в колбе с обратным холодильником,
защищенным хлоркальциевои трубкой с СаС12. Реакция
начинается самопроизвольно, и вскоре спирт закипает. Когда весь магний
растворится, кипение поддерживают нагреванием на водяной бане
еще некоторое время, после чего спирт перегоняют, отбрасывая
первую порцию дистиллята.
Безводный метиловый спирт получают также, выдерживая его
над цеолитом NaA или КА или пропуская через колонку,
заполненную этими молекулярными ситами. Для этого можно
воспользоваться колонкой лабораторного типа [70].
Присутствие ацетона в метиловом спирте устанавливают
пробой с нитропруссидом натрия. Спирт разбавляют водой,
подщелачивают и прибавляют несколько капель свежеприготовленного
насыщенного водного раствора нитропруссида натрия. В
присутствии ацетона появляется красная окраска, усиливающаяся при
подкислении уксусной кислотой.
Для удаления ацетона предложен следующий способ: 500 мл
СНзОН кипятят несколько часов с 25 мл фурфурола и 60 мл 10%
раствора NaOH в колбе с обратным холодильником, а затем
отгоняют спирт на эффективной колонке. В колбе остается смола —
продукт взаимодействия фурфурола с ацетоном.
122
Петролейный эфир, бензин и лигроин. При перегонке легкого,
бензина получают ряд низкокипящих углеводородных фракций,
которые применяют в качестве растворителей. Пары этих
углеводородов оказывают наркотическое действие.
Промышленность выпускает следующие реактивы:
Пределы кипения, °С
Петролейный эфир 30—50; 40—60; 45—70
Бензин 70-90
Лигроин 90—110
Большая летучесть петролейного эфира, бензина и лигроина,,
легкая их воспламеняемость и образование с воздухом взрыво--
опасных смесей требует особой осторожности при работе с ними.
Петролейный эфир, бензин и лигроин не должны содержать,
примеси ненасыщенных и ароматических углеводородов.
Присутствие ненасыщенных углеводородов обычно
устанавливают двумя реагентами: 2% раствором Вг2 в ССЦ и 2% водным
раствором КМп04в ацетоне. Для этого к 0,2 мл углеводорода в,
2 мл ССЦ прибавляют по каплям раствор реагента и наблюдают
за изменением окраски. Проба считается отрицательной, если
обесцвечивается не более 2—3 капель раствора брома или раствора
КМп04.
Ненасыщенные углеводороды можно удалить многократным
30-минутным встряхиванием на механической качалке порции
углеводородов с 10% (об.) конц. H2S04. После встряхивания с
каждой порцией кислоты смеси дают отстояться, затем отделяют
нижний слой. Когда слой кислоты перестанет окрашиваться, углеводо*
родный слой энергично встряхивают с несколькими порциями 2%
раствора КМп04 в 10% растворе H2S04, пока цвет раствора
КМп04 не перестанет изменяться. При этом почти полностью
удаляются ненасыщенные углеводороды и частично —
ароматические. Чтобы полностью удалить ароматические углеводороды,
нужно встряхивать на качалке углеводороды (петролейный эфир и,
др.) с олеумом, содержащим 8—10% (масс.) S03. Склянку с
притертой пробкой, в которой производят встряхивание, заворачивают
в полотенце. После отделения кислотного слоя углеводородную
фракцию промывают водой, 10% раствором Na2C03, снова водой,
высушивают над безводным СаС12 и перегоняют над натриевой
проволокой. Рекомендуется хранить петролейный эфир над CaS04
и перегонять перед употреблением.
Традиционный химический метод очистки насыщенных
углеводородов от ненасыщенных очень трудоемок и может быть заменен
адсорбцией. Примеси многих ненасыщенных соединений удаляются
при пропускании растворителя через стеклянную колонку с
активным А1203 и особенно на цеолитах, например NaA.
Тетрагидрофуран (СН2)40 — бесцветная подвижная жидкость
с эфирным запахом;df = 0,8892; /кип = б6°С; /г2о0= 1,4050.
Растворяется в воде и большинстве органических растворителей.
Образует азеотропную смесь с водой [6% (масс.)Н20], /Кип = 64°С«
123.
Тетрагидрофуран склонен к образованию перекисных соединений,
поэтому обязательно надо проверить наличие в нем перекисей (см.
Диэтиловый эфир). Удалить перекиси можно кипячением с 0,5%
суспензией СигСЬ в течение 30 мин, после чего растворитель
перегоняют и встряхивают с плавленым КОН. Верхний слой тетра-
гидрофурана отделяют, вновь добавляют к нему 16% (масс.)
КОН и кипятят смесь 1 ч в колбе с обратным холодильником.
Затем тетрагидрофуран перегоняют над СаН2 или LiAlH4,
отбрасывают 10—15% головной фракции и оставляют около 10%
остатка в кубе. Головную фракцию и кубовый остаток присоединяют к
техническим продуктам, предназначенным для очистки, а
собранную среднюю фракцию досушивают над натриевой проволокой.
Очищенный продукт хранят без доступа воздуха и влаги.
Хлороформ СНС13 — бесцветная подвижная жидкость с
характерным сладковатым запахом; df= 1,4880; ^кип = 61,15 °С; п2£ =
= 1,4455. Растворим в большинстве органических растворителей;
практически нерастворим в воде. Образует азеотропную смесь
с водой [2,2% (масс.) Н20], /Кип = 56,1 °С. Негорюч и не образует
взрывоопасных смесей с воздухом, но токсичен — действует на
внутренние органы, особенно на печень.
Хлороформ почти всегда содержит до 1% (масс.) этилового
спирта, который добавляют к нему в качестве стабилизатора.
Другой примесью хлороформа может быть фосген, образующийся
при окислении хлороформа на свету.
Пробу на присутствие фосгена выполняют следующим образом:
1 мл 1% раствора я-диметиламинобензальдегида и дифениламина
в ацетоне встряхивают с хлороформом. При наличии фосгена (до
0,005%) через 15 мин возникает интенсивная желтая окраска.
Хлороформ очищают трехкратным встряхиванием с отдельными
порциями конц. H2SO4. На 100 мл хлороформа каждый раз берут
5 мл кислоты. Хлороформ отделяют, промывают 3—4 раза водой,
сушат на СаСЬ и перегоняют.
Очистка хлороформа достигается также медленным
пропусканием препарата через колонку, заполненную активным А1203 в
количестве 50 г на 1л хлороформа.
Хлороформ следует хранить в склянках из темного стекла.
Четыреххлористый углерод ССЦ — бесцветная негорючая
жидкость со сладковатым запахом;df =1,5950; *КИП=76,7°С; Яд =
= 1,4631. Практически нерастворим в воде. С водой образует
азеотропную смесь [4,1% (масс.) Н20], /Кип = 66°С. Растворяет
разнообразные органические соединения. Обладает меньшим, чем
хлороформ, наркотическим действием, но по токсичности
превосходит его, вызывая тяжелые поражения печени.
Четыреххлористый углерод иногда загрязнен сероуглеродом,
который удаляют перемешиванием ССЦ при 60°С в колбе с
обратным холодильником с 10% (об.) концентрированного спиртового
раствора КОН. Эту процедуру повторяют 2—3 раза, после чего
растворитель промывают водой, перемешивают при комнатной
124
температуре с небольшими порциями конц. H2SO4 до тех пор, пока
она не перестанет окрашиваться. Затем растворитель снова
промывают водой, высушивают над СаС12 и перегоняют над Р4О10.
Высушивание СС14 достигается азсотропной перегонкой. Вода
удаляется с первыми мутными порциями дистиллята. Как только
начнет перегоняться прозрачная жидкость, ее можно считать
безводной.
Этилацетат СН3СООС2Н5 — бесцветная жидкость в дриятньщ
фруктовым запахом; df = 0,901; /кип = 77,15°С; ^°= 1,3728.
Образует азеотропную смесь с водой [8,2% (масс.) Н20], *цИп =■
= 70,4 °С.
Технический этилацетат содержит воду, уксусную кислоту и
этиловый спирт. Предложено много способов очистки этилацета-
та. По одному из них этилацетат встряхивают с равным объемом
5% раствора NaHC03 и затем с насыщенным раствором СаС12.
После этого этилацетат сушат К2СО3 и перегоняют на водяной
бане. Для окончательной сушки к дистилляту добавляют 5%
Р4Ою и энергично встряхивают, затем фильтруют и перегоняют
над натриевой проволокой.
Этиловый спирт С2Н5ОН — бесцветная жидкость с
характерным запахом; df = 0,7893; /кип = 78,39°С; п™= 1,3611. Образует
азеотропную смесь с водой [4,4% (масс.) Н20]. Отличается
высокой растворяющей способностью по отношению к самым
разнообразным соединениям и неограниченно смешивается с водой и
со всеми обычными органическими растворителями. Технический
спирт содержит примеси, качественный и количественный состав
которых зависит от условий его получения *.
Выпускаемый абсолютированный спирт, который получают азе-
отропной перегонкой 95% технического спирта с бензолом, может
содержать небольшие количества воды и бензола [до 0,5% (масс.)].
Обезвоживание 95% спирта можно производить длительным
кипячением с прокаленным СаО. На 1 л спирта берут 250 г СаО.
Смесь кипятят в 2-литровой колбе с обратным холодильником,
закрытым трубкой с СаО, в течение 6—10 ч. После охлаждения
колбу присоединяют к установке для перегонки при атмосферном
давлении и отгоняют спирт. Выход 99—99,5% спирта 65—70%,
Более высокими обезвоживающими свойствами обладает оксид
бария ВаО. Кроме того, ВаО способен несколько растворяться
в почти абсолютном спирте, окрашивая его в желтый цвет. По
этому признаку определяют, когда процесс абсолютирования
завершен.
Дальнейшее обезвоживание 99—99,5% спирта можно провести
несколькими методами: с помощью магния (получается этиловый
спирт с содержанием воды не более 0,05%), натрия и диэтилового
эфира щавелевой кислоты.
В круглодонную колбу вместимостью 1,5 л с обратным
холодильником и хлоркальциевой трубкой с СаС12 вливают 1 л 99%
* Методы испытаний этилового спирта приводятся в ГОСТ 5964—67.
125
этилового спирта, после чего небольшими порциями вносят 7 г
натриевой проволоки. По растворении натрия к смеси добавляют
25 г диэтилового эфира щавелевой кислоты, кипятят 2 ч и
отгоняют спирт.
Аналогично получается абсолютный спирт с помощью
диэтилового эфира ортофталевой кислоты. В колбу, снабженную
обратным холодильником и хлоркальциевой трубкой с СаС12, помещают
1 л 95% спирта и растворяют в нем 7 г натриевой проволоки,
после чего прибавляют 27,5 г диэтилового эфира фталевой
кислоты, кипятят смесь около. 1 ч и отгоняют спирт. Если в колбе
образуется небольшое количество осадка, то это доказывает, что
исходный спирт был достаточно хорошего качества. И наоборот,
если выпадает большое количество осадка и кипение
сопровождается толчками, то исходный спирт был недостаточно высушен.
Осушка этилового спирта в настоящее время осуществляется
в аппаратах колонного типа с цеолитом NaA в качестве насадки
[14, 20, 70]. Этиловый спирт, содержащий 4,43% воды, подается
на осушку в колонку диаметром 18 мм с высотой слоя насадки
650 мм при скорости 175 мл/ч. В этих условиях за один цикл
удается получить 300 мл спирта с содержанием воды не выше
0,1—0,12%. Регенерация цеолита производится в колонке в токе
азота при 320 °С в течение 2 ч. При перегонке этилового спирта
рекомендуется применять приборы на шлифах; при этом шлифы
тщательно очищают и не смазывают. Первую часть дистиллята
целесообразно отбросить и перегонку завершить, когда в
перегонной колбе останется немного спирта.
Перегонку спирта рекомендуется проводить над небольшим
количеством никеля Ренея; в этом случае отпадает необходимость
в «кипятильных камешках».
главаэ. ИЗМЕЛЬЧЕНИЕ
Измельчение — механическое разрушение твердых тел с
образованием частиц материала. По размерам образующихся частиц
различают грубое (0,1 — 1 мм), среднее (0,01—0,1 мм) и тонкое
(0,001—0,01 мм) измельчение.
Основная характеристика процесса — степень измельчения,
определяемая отношением среднего размера зерен исходного
материала к среднему размеру частиц измельченного материала.
Измельчение твердых материалов проводится при отборе
средней пробы для анализа, перед приготовлением растворов
кристаллических веществ и т. п. Твердые вещества измельчают вручную
или механически. Выбор способа и средств для измельчения
определяется механическими и химическими свойствами твердого
вещества и требуемой степенью измельчения.
ИЗМЕЛЬЧЕНИЕ ВРУЧНУЮ
Для измельчения небольших проб пользуются металлическими,
фарфоровыми, агатовыми ступками.
Для измельчения образцов минералов при подготовке их к
анализу часто используют истиратель — ступку СМБМ,
снабженную тремя сменными чашами и пестиками из природного
халцедона, яшмы или агата, твердость которых выше 6,5 по шкале Мооса.
В рабочем состоянии ступка герметически закрыта колпаком из
органического стекла, что предотвращает распыление истираемого
материала.
МЕХАНИЧЕСКОЕ ИЗМЕЛЬЧЕНИЕ
Для механического дробления и измельчения твердых материал
лов используются многие типы лабораторных дробилок и
мельниц, которые сконструированы на тех же принципах, что и
большие промышленные установки.
Для дробления крупных кусков руды используют лабораторные
щековые дробилки, а для угля — молотковые, для более
мелкого дробления — валковые.
Грибовидная дробилка предназначена для мелкого
дробления проб металлургического кокса и других материалов
малой прочности. Масса загрузки до 7 кг. Размеры частиц
загружаемого материала до 133 мм, а измельченного—1 мм.
Бисерная мельница предназначена для тонкого измель«
чения (около 0,01 мм) веществ с твердостью не более 4 по шкале
Мооса.
Для механического измельчения небольших (до 50 г) навесок
руд и других сухих материалов перед их химическим анализом
127
предназначен вибрационный измельчитель [38].
Максимальная крупность загружаемого материала до 3 мм,
измельченного—менее 0,01 мм. Прибор снабжен реле времени и
автоматически отключается в заданное время.
Для превращения в тонкий порошок грубо измельченных
твердых и сухих веществ удобны шаровые мельницы. Они
действуют по принципу удара свободно падающих шаров при
вращении барабана. Частота вращения должна быть не более 60—
65 об/мин, чтобы центробежная сила не прижимала шары к
стенке барабана и они свободно перемещались. Размер частиц
измельченного материала —до 0,01—0,02 мм.
Лабораторная шаровая мельница представляет
собой цилиндрический сосуд из твердого фарфора с крышкой,
снабженной резиновым уплотнением. Мельницу заполняют
примерно на 7з объема предварительно измельченным веществом,
вторую треть объема занимают шары из твердого фарфора.
Массу и диаметр шаров подбирают в зависимости от свойств
измельчаемого материала и требуемой тонкости помола. В частности,
диаметр шаров должен быть тем больше, чем крупнее частицы
измельчаемого материала.
Укрепив крышку, мельницу помещают на вращающее
устройство. Для тонкого измельчения обычно необходимо несколько
часов.
Измельченное вещество отделяют от шаров на сите. Более
крупную фракцию снова загружают в мельницу. Во время
работы мельницы необходимо часто отбирать пробы, чтобы
контролировать степень измельчения.
Для тонкого измельчения проб рудных материалов при
лабораторных исследованиях как сухим, так и мокрым способом,
служит шаровая лабораторная мельница 40-МЛ. Объем барабана
7 л; частота вращения 65 об/мин, крупность загружаемых зерен
4—6 мм. Шаровая загрузка: диаметр шаров 19—25 мм; масса —
9 кг.
Для измельчения веществ до частиц размером Ю-3—Ю-4 мм
пользуются коллоидными мельницами. В этих мельницах
можно измельчать только материал, предварительно помолотый в
шаровой мельнице до частиц размером около 0,02 мм.
Коллоидные мельницы работают по принципу удара при
больших скоростях или по принципу истирания вещества между
коническими поверхностями ротора и статора.
Измельчение в коллоидной мельнице всегда проводят в жидкой
среде—в воде для гидрофильных материалов и в органических
жидкостях для гидрофобных.
Для предотвращения коагуляции коллоидных частиц и
облегчения дробления добавляют вещества, действующие как защитные
коллоиды, например поверхностно-активные вещества.
глава ю. СИТОВОЙ АНАЛИЗ
Степень измельчения многих сыпучих и
порошкообразных материалов является одной из важнейших характеристик,
определяющей их технологические качества и облает
практического использования. Гранулометрический
(дисперсный, зерновой) состав наиболее полно характеризует
степень измельчения. Ситовой анализ — один из методов определения
гранулометрического состава порошков и сыпучих материалов —
осуществляется путем механического разделения материала на
фракции с частицами определенной крупности. В ситовом анализе
используют стандартные нормированные тканые проволочные и
шелковые сетки с квадратными отверстиями (ячейками), а
также металлические решетные сетки с пробивными круглыми,
продолговатыми и треугольными отверстиями. Ситовой анализ
применим для материалов с размерами частиц 10—0,04 мм, что
соответствует шкале сит по ГОСТ 3584—73.
Рассев более крупных продуктов на ситах с большими
размерами отверстий (грохотах) называется грохочением.
В ситовом анализе измельченный материал в сухом виде или
в виде взвеси в соответствующей жидкости загружается на сито
с отверстиями известного размера и путем встряхивания,
постукивания, вибрации или другими способами разделяется на две
части: остаток и проход. Остаток R — доля материала,
оставшегося на сите с заданными размерами ячеек, а проход D — доля
материала, прошедшего через данное сито, выраженные в
процентах от общей массы просеиваемого материала. Остаток R часто
обозначают знаком «+», а проход D знаком «—».
Просеивая исследуемый материал через набор нормированных
сит, различающихся величиной отверстий, можно разделить пробу
на несколько фракций, размеры частиц которых ограничены
размерами отверстий используемых в анализе сит. Число фракций,
получаемых при просеивании через набор из п последовательных
сит, составляет п + 1.
Результаты ситового анализа записывают в форме таблиц,
суммарных кривых или диаграмм.
СИТА
Днища сит, применяемых для ситового анализа, представляют
собой сетки с нормированными линейными размерами ячеек.
Номер тканой проволочной сетки соответствует размеру ( в
миллиметрах) стороны ячейки в свету, причем если этот размер менее
1 мм, то в обозначении номера сита опускается запятая перед
десятыми долями миллиметра. Нижняя граница размеров ячеек
проволочных тканых сит находится около 0,04 мм. Очень тонкие
сита могут быть использованы только для анализа хорошо
просеивающихся мелких порошков.
5 Зак. 1237 129
Шелковые сетки, в зависимости от толщины нитей основы и
утка, различают по массе облегченные и утяжеленные (ГОСТ
4403—77). Номер сита с облегченной тканью определяют числом
отверстий на 1 погонный сантиметр по основе и утку. Номер сита
с утяжеленной тканью определяют числом отверстий на 10
погонных сантиметров по основе и утку.
Номер металлических пробивных сит с круглыми отверстиями,
используемых для рассева крупноизмельченных материалов,
соответствует диаметру отверстий в миллиметрах, умноженному на
10 (ГОСТ 21—77).
Выпускаются сетки проволочные тканые с квадратными
ячейками нормальной точности (ГОСТ 6613—73), контрольные и
высокой точности (ГОСТ 3584—73) с размером стороны ячейки в
свету от 0,04 до 2,5 мм. Контрольные сетки предназначены для
контроля различных материалов по размеру частиц при
дроблении, сетки высокой точности — для разделения по размеру зерен
дробленых материалов.
В некоторых странах практикуется нумерация нормированных
тканых сит по числу отверстий на 1 погонный дюйм (25,4 мм). Это
число носит название «меш».
В табл. 4 приведены данные о ситовых шкалах проволочных
тканых сит с квадратными отверстиями.
Ситовая ткань натягивается на круглую или квадратную
обечайку. Круглое сито имеет обычно диаметр 20 см и высоту борта
5 см. Квадратные сита имеют размеры 22X22 см и высоту борта
9 см. При сухом рассеве сито плотно насаживается на поддон,
улавливающий материал прохода; высота поддона обычно равна 3,5 см.
Сверху сито плотно закрывается крышкой, за исключением случаев,
когда просеивание производится при помощи кисточки или мокрым
способом. Поддон при мокром просеивании используется лишь в тех
случаях, в которых требуется определить проход через сито.
таблица 4. Ситовые шкалы
Номер
сита
004
0045
005
0056
0063
0071
0080
009
01
0125
015
020
Размер
отверстий,
мм
0,04
0,045
0,05
0,056
0,063
0,071
0,08
0,09
0,1
0,125
0,15
0,20
Число
отверстий
на 1 см2
20 420
15 252
13 526
10 858
9 428
5 823
5914
4 435
3 906
2 130
1600
918
Число
«меш»
400
—
270 !
—
—
200
—
—
150
115
100
65
Номер
сита
025
0355
04
05
06
07
08
| 09
1,0
1,6
2,0
2,5
Размер
отверстий,
мм
0,25
0,355
0,4
0,5
0,6
0,7
0,8
0,9
1.0
1,6
2,0
2,5
Число
отверстий
на 1 см2
694
400
331
193
139
98
82
64
55
24
16
11
Числз
«меш»
60
42
35
32
—
24
20
—
16
—
9
7
130
Сита, входящие в набор, плотно вставляются одно в другое,
образуя набор сеток с уменьшающимися сверху вниз размерами
ячеек.
Методы определения гранулометрического состава различных
материалов регламентируются стандартами и техническими
условиями. В соответствии с этим выпускаются специальные наборы сит
для ситового анализа отдельных видов материалов (зерна, семян
сельскохозяйственных культур, удобрений, почвы, формовочных
материалов, цемента и др.). В комплект фармакопейных сит
включаются сита шелковые прямоугольные (ГОСТ 4403—77) с размерами
ячеек от 0,1 до 0,315 мм, сито проволочное квадратной формы с
размером отверстий 0,500 мм (ГОСТ 3524—47) и сита
металлические с пробивными отверстиями круглой формы с размерами
отверстий от 1 до 10 мм.
МЕТОДИКА ПРОВЕДЕНИЯ АНАЛИЗА
Ситовой анализ можно проводить ручным и механическими
(машинными) способами. В зависимости от свойств исследуемого
материала применяются сухой или мокрый методы анализа.
При машинном просеивании навеска анализируемой пробы
помещается на сито с наибольшими отверстиями в используемом
наборе стандартных сит. Проход из этого сита падает на следующее,
с меньшими размерами ячеек сито. Такая последовательность
позволяет сита всего набора поставить друг на друга и разделить
пробу по размерам частиц на фракции (классы) в одну рабочую
операцию.
При ручном просеивании пробу чаще всего помещают на
наиболее тонкое сито, а полученный остаток переносят на следующее по
крупности ячеек сито. Целесообразность такой последовательности
заключается в том, что более крупные частицы материала
способствуют процессу ручного просеивания на наиболее тонких ситах.
Анализируемая проба измельченного материала при сухом
рассеве должна быть воздушно-сухой.
Предварительное высушивание пробы до постоянной массы
производят при 105—110°С. Эспериментально найдено, что когда
исследуемый материал недостаточно просушен, данные ситового
анализа мало надежны. Размер навески анализируемого материала,
помещаемой на сито, зависит от площади сита, которую не следует
перегружать. Определяющим является объем просеиваемого
материала, который, при использовании нормированных сит, не должен
превышать 100 см3.
Взвешивание пробы, остатка и прохода производят на
технических весах с точностью до 0,01' г.
Ручной рассев
Сухой способ. Последовательность операций и приемы просева
для различных материалов могут быть разными и обычно
излагаются в специальных технологических инструкциях. Чаще всего
поступают следующим образом.
5* 131
При ручном сухом просеве на круглых ситах сито с поддоном и
крышкой берут одной рукой, наклонив полотно к горизонтальной
плоскости на 10—20°, и ударяют другой рукой примерно 120 раз
в минуту. Около 4 раз в минуту сито располагают горизонтально
и сильно ударяют по обечайке.
При тонких ситах и трудно просеивающемся материале
рекомендуется через каждые 5 минут нижнюю поверхность сита
очищать мягкой кисточкой и опадающие частицы присоединять к
проходу.
Квадратное сито берут в обе руки, держа большие пальцы
сверху, и при изменяющемся наклоне до 20° двигают вперед и назад,
время от времени ударяя сито о ладонь правой и левой руки. Число
встряхиваний, повороты, постукивания и очистка кисточкой такие
же, как и при просеве на круглых ситах.
Продолжительность ручного сухого просева зависит от
плотности, размеров и формы частиц, от объема просеиваемого
материала, интенсивности просева, размеров отверстий сита, площади
закупоренных отверстий сит и влажности воздуха. В случае тонких
сит (004—006) время просева достигает 60—120 мин.
Ручной просев тряской и поколачиванием — самый
обычный способ и применим для ситового анализа большинства
материалов.
При дисперсионном анализе очень тонких порошков (пыли) с
частицами, склонными к слипанию, сита могут забиваться, что
сопровождается значительным уменьшением площади сита. Чтобы
устранить агрегирование частиц, вместе с пробой в сита помещают
латунные штифты длиной 1 см (около 30 г на каждое сито), либо
просев проводят кисточкой. Для этого сито устанавливают
горизонтально на поддоне и, держа под острым углом к полотну мягкую
волосяную кисточку, проводят ею по поверхности просеиваемого
материала так, чтобы избежать пыления.
Сухой ручной просев может считаться оконченным, если при
повторном встряхивании в течение 2 мин масса остатка на сите
уменьшается не более чем на 0,2%. Остаток высыпают в чистый
заранее взвешенный приемник или на лист глянцевой бумаги, сито
очищают с обеих сторон мягкой волосяной щеткой и легким ударом
по обечайке удаляют застрявшие в ячейках сетки частицы, которые
присоединяют к остатку. По окончании рассева каждую фракцию
взвешивают; обычно требуется, чтобы суммарная масса всех
фракций составляла .не менее 98% от массы взятой навески. При
большой точности измерения фракционного состава потери при выпол*
нении ситового анализа рекомендуется разнести по всем
анализируемым фракциям пропорционально их массам. При рассеве для
достоверности обычно выполняют два параллельных анализа. При
этом массы соответствующих фракций не должны различаться
более чем на 1% от массы всей навески.
По действующей фармакопее, ситовой анализ сухим методом
проводят следующим образом: 200 г измельченного материала
помещают на самое крупное (верхнее) сито,и весь комплект встряхни
Ш
вают в течение 5 мин. Затем сита снимают по очереди одно за
другим, после чего каждое сито повторно встряхивают отдельно над
приемником или листом гладкой бумаги. Просеивание считается
законченным, если количество материала, проходящего сквозь сито
при повторном дополнительном встряхивании в течение 1 мин,
составит менее 1% материала, оставшегося на сите. Отсев (проход)
добавляют на верхнее сито оставшегося комплекта сит.
Мокрый способ. Для определения гранулометрического состава
материалов, которые могут приобретать высокие электрические
заряды, склонных к агрегированию при встряхивании или
содержащих большое количество самых мелких фракций, применяется
мокрый способ ситового анализа. Для этого используют жидкость,
хорошо смачивающую частицы просеиваемого материала и не
образующую с ним растворов или химических соединений (вода,
керосин и др.).
Вот, например, одна из наиболее распространенных методик.
В анализируемую пробу, масса которой примерно такая же, что и
при сухом методе рассева, вводят минимальное количество
промывной жидкости и тщательно перетирают до образования густой
кашицы. Разбавленную промывной жидкостью кашицу переносят на
самое грубое сито комплекта, и затем слабой струей жидкости она
промывается через сита с последовательно уменьшающимися
ячейками до тех пор, пока слив не станет прозрачным. Жидкость для
промывания надо подавать на сито осторожно и равномерно. После
промывки сита с остатками материала просушивают при 105—110°С
и остатки взвешивают.
Механический рассев
Механический рассев осуществляют при помощи приборов,
создающих вращательное и колебательное движение сит в
горизонтальной плоскости, качание плоскости сит, вибрацию и
постукивание сит. Механический рассев может осуществляться как при
сухом, так и мокром методе ситового анализа. В последнее время
широко распространился метод пневматического
просеивания. При использовании приборов пневматического и
механического просеивания последнее выполняют в соответствии с
инструкцией, прилагаемой к прибору.
Механический рассев требует значительно меньше времени, чем
ручной, меньше материала и исключает индивидуальные ошибки.
Однако при анализе мелкодисперсных материалов на тонких ситах
рассев затруднен из-за отсутствия в пробе грубых частиц. В таких
случаях рекомендуют сравнивать результаты механического и
ручного рассева.
При механическом рассеве набором сит время просева должно
быть установлено экспериментально для отдельных сит. Например,
нремя просева хорошо просеивающегося материала при размере
отверстий сит 0,04—0,053 мм составляет 20—30 мин, а при 0,071 —
0,16 мм — всего 10—20 мин.
133
Ситовые механические анализаторы выпускаются различных
конструкций и назначений.
Так, для определения гранулометрического состава
формовочных материалов в литейной промышленности выпускается
прибор 028 М. В комплект входят 11 сит с размерами ячеек от
2,5 до 0,05 мм. Частота вращения эксцентрикового вала
300 об/мин, число ударов рычага 180 мин-1. Прибор аналогичного
назначения «Анализатор ситовой» 236-Б-Гр содержит J0
сит в комплекте (размеры отверстий от 1,6 до 0,071 мм).
Прибор позволяет одновременно устанавливать шесть сит. Число
качаний сит 200 мин-1, а число ударов встряхивания 140 мин~1.
Рассев-анализатор РА-5 для разделения муки, крупы и
комбикормов на фракции по крупности имеет четыре ситовых
пакета от двух до восьми сит в каждом. Рассев
лабораторный ЛР-3 для анализа зерна, контроля за процессом очистки и
калибровки семян имеет 4 ситовых пакета, по четыре сита в
каждом; частота вращения сит 220 об/мин.
Для определения тонкости помола цемента по остатку на сите
(ГОСТ 310.2—76) выпускается механическое сито СММ
с контрольными сетками № 02 и 008. Тонкость помола цемента
определяется как остаток на сите № 008 в процентах к
первоначальной массе просеиваемой пробы. Для этой же цели служит
лабораторная установка для пневморассева РП-3 с
контрольными сетками 0063 и 008.
ОБРАБОТКА РЕЗУЛЬТАТОВ СИТОВОГО АНАЛИЗА
Для рядовых ситовых анализов результаты рассева пробы
рекомендуется, в частности, записывать в следующей форме:
Номер
сетки
Размер
отверстий
сита, мм
Масса
фракции
(остатка), г
Остаток
на данном
сите R\,
% (масс.)
Суммарный
остаток /?2t
% (масс.)
Учитывая, что потери при выполнении анализа обычно не
должны превышать 2% от общей массы навески пробы, можно при
обработке полученных результатов принять суммарную массу
всех фракций за 100%.
Содержание остатка Ri на каждом сите вычисляют по форму*
ле:
134
Рис. 66. График зависимости суммарного содержания ионита от размера ячейки сита.
где т\ — масса остатка на данном сите, г; Em — суммарная
масса остатков всех фракций после рассева, г.
Суммарный остаток /?2 для каждого сита рассчитывают,
прибавляя к остатку на данном сите суммарное содержание
остатков, полученных для всех предыдущих сит с большими
отверстиями. По данным таблицы строят график зависимости задержанного
на каждом сите суммарного содержания вещества от размера
ячейки сита, откладывая на оси ординат размер ячейки сита в
миллиметрах, а на оси абсцисс — суммарное содержание
анализируемого вещества и проводя прямую через точки,
соответствующие наибольшим процентам. По построенной прямой определяют
отверстия сит в миллиметрах, задерживающих определенный
суммарный процент вещества, например 40 и 90%. Размер отверстия
сита, задерживающего 90% вещества, называется
эффективным размером зерна и обозначается d9o эфф- Отношение
размера ячейки сита, задерживающего 40% вещества, к
эффективному размеру с?9оэфф называется коэффициентом
однородности К:
д = —
"90 Эфф
Эффективный размер зерна обычно определяют с
погрешностью не более 4%, а коэффициент однородности с погрешность^
не более 5%.
На рис. 66 в качестве примера приведен график зависимости суммарного
содержания ионита от размера ячейки сита.
глава п. ПЕРЕМЕШИВАНИЕ
Перемешивание улучшает контакт между фазами системы,
облегчает их взаимную диффузию, тем самым повышая скорость
реакции. Перемешивание используют для интенсификации
процессов массо- и теплообмена. При реакциях твердых веществ с
жидкостями, двух несмешивающихся жидкостей или жидкости и
газа перемешивание не только ускоряет реакцию, но подчас и
обусловливает возможность ее проведения.
Перемешивание часто бывает необходимым и в однородной
среде, когда один из реагентов прибавляют постепенно, так как
это позволяет избежать местных перегревов и увеличения
концентрации.
Перемешивание проводят вручную и механически (при помощи
устройства, помещаемого внутрь неподвижного реакционного
сосуда, либо периодическим встряхиванием всего сосуда).
Перемешивание встряхиванием применяют, если в ходе процесса не
требуется прибавлять в реакционный сосуд твердые или жидкие
вещества и если процесс протекает без значительного выделения
газов и паров, а также когда применять мешалки затруднительно
по конструктивным соображениям.
Выбор метода перемешивания и аппаратуры для его
осуществления обусловливается, в первую очередь, агрегатным
состоянием перемешиваемых материалов.
СОСУДЫ ДЛЯ ПЕРЕМЕШИВАНИЯ И ВСТРЯХИВАНИЯ
Для проведения процессов при нормальной температуре и
атмосферном давлении, протеканию которых не мешает влага или
воздух, могут быть использованы открытые сосуды (стаканы, ши-
рокогорлые колбы и др.), в которые можно вставить мешалку,
капельные воронки, термометры и другое оборудование.
В случае перемешивания при нагревании и необходимости
предохранять содержимое сосуда от влаги или воздуха, широкогорлые
колбы неудобны. Корковые пробки не обеспечивают
достаточной герметичности сосуда, а резиновые загрязняют реакционную
смесь.
Более удобны колбы с двумя-четырьмя горловинами и
взаимозаменяемыми шлифами, которые позволяют разместить в
центральном горле мешалку, а в других — обратный холодильник и
другое оборудование (рис. 67). Вместо таких колб можно
использовать круглодонные колбы со шлифами и насадками, а для
встряхивания—обычные реакционные сосуды, колбы, банки, бутылки,
делительные воронки. При встряхивании сосуд следует закрывать,
чтобы жидкость не выплеснулась.
136
Рис. 67. Установки для проведения реакции с перемешиванием.
Встряхивание часто используют для ускорения поглощения газа
жидкостями, например при каталитическом гидрировании. Для
этих целей используют специальные сосуды, так называемые утки.
ПЕРЕМЕШИВАНИЕ И ВСТРЯХИВАНИЕ ВРУЧНУЮ
Небольшие объемы жидкостей или твердых веществ с
жидкостями в стакане можно перемешивать стеклянной палочкой. При
перемешивании в колбе ее вращают, придерживая за горло. В
закрытом сосуде перемешивание проводят, встряхивая или
многократно перевертывая сосуд.
Перемешивать вручную можно только маловязкие жидкости.
Если приходится перемешивать вручную в закрытом сосуде
жидкости с низкой температурой кипения, необходимо
придерживать пробку рукой, так как в сосуде в процессе перемешивания
развивается повышенное давление и пробка может выскочить.
При экстрагировании жидкостей с помощью делительной воронки
взбалтывание проводят, придерживая одной рукой нижний кран
воронки, другой — пробку. Время от времени воронку
перевертывают горлом вниз и осторожно открывают кран, уменьшая таким
образом избыточное давление.
МЕХАНИЧЕСКОЕ ПЕРЕМЕШИВАНИЕ
Наиболее распространено механическое перемешивание
посредством разнообразных мешалок из стекла, полиэтилена и
фторопласта, вращающихся в жидкости при помощи электропривода
(или водяной турбины), а также магнитных и вибрационных
мешалок.
137
Типы мешалок
Эффективность перемешивания в значительной степени зависит
от конструкции мешалки. В лабораториях охотно пользуются
мешалками из толстого стеклянного дрота, потому что
непосредственно перед использованием им можно придать любую форму в
зависимости от формы и вместимости реакционного сосуда.
Различают следующие типы лабораторных мешалок.
Лопастные мешалки обычно имеют две или четыре
лопасти, припаянные к стержню под некоторым углом в
вертикальной плоскости в один или несколько рядов. Лопастные мешалки
обычно применяют для перемешивания маловязких и легко
смешивающихся жидкостей, а также в случае смешивания хорошо
растворимых порошкообразных сыпучих материалов с
жидкостями.
Якорные мешалки отличаются тем, что их лопасти
строго повторяют контур днища и стенок реакционного сосуда,
образуя с ними незначительный зазор. Якорные мешалки
применяют для перемешивания вязких жидкостей, а также когда
требуется предупредить образование накипей и осадков на стенках
реакционного сосуда.
Пропеллерные мешалки представляют собой гребной
винт с лопастями. При вращении винта происходит интенсивная
циркуляция жидкости с сильным вихреобразованием,
обеспечивающим энергичное перемешивание. Пропеллерные мешалки
применяют для перемешивания вязких жидкостей, получения суспензий
и эмульсий, поддержания тяжелых частиц во взвешенном
состоянии, интенсивного охлаждения жидкости во время реакции и т. д.
Центробежные мешалки действуют по принципу
всасывания и выброса жидкости. Жидкость, находящаяся в полости
мешалки, выбрасывается в стороны под действием центробежной
силы через боковые отверстия и всасывается обратно через верхние
или нижние отверстия.
Для узкогорлых сосудов часто изготовляют лопастные и пр>
пеллерные мешалки с откидывающимися
лопастями из фторопласта (рис. 68). В спокойном состоянии лопасти
<&
F О
=о
^37
Рис. 68. Мешалк дла узкогорлых сосудов*
138
располагаются отвесно, а при вращении центробежная сила
устанавливает их горизонтально. Такие мешалки эффективны для
вязких жидкостей и суспензий.
Для перемешивания в узких цилиндрических сосудах можно
пользоваться спиральными мешалками, а также
мешалками вертикального действия.
Установка мешалки
Обычно мешалку монтируют следующим образом: стержень
мешалки помещают в стеклянную трубку (подшипник). Эту
трубку вставляют в пробку, которую зажимают в лапу (держатель)
лабораторного штатива. Верхний конец мешалки соединяют с
валом электродвигателя.
Если сосуд не должен сообщаться с атмосферой, ввод стержня
мешалки в сосуд необходимо уплотнить. В этих случаях
применяют мешалки с затворами. Наиболее простой способ
уплотнения состоит в следующем: мешалку помещают в длинную
направляющую трубку (рис. 69, а), нижний конец которой
находится ниже уровня жидкости, а верхний — выше уровня
конденсации паров в обратном холодильнике. Способ применим лишь
для работы с высококипящими жидкостями при атмосферном
давлении. При очень энергичном перемешивании уровень жидкости
в центре сосуда вследствие образования воронки может
опуститься ниже конца стеклянной трубки, и затвор перестанет
действовать.
Широко распространен и другой способ уплотнения мешалок:
на верхний конец направляющей стеклянной трубки плотно надет
отрезок вакуумной резиновой трубки (сальник). В трубку через
сальник, слегка смазанный вазелином или глицерином, вводят
стержень мешалки. Такое уплотнение обеспечивает герметичность
собранного прибора (рис. 69,6) при работе в вакууме с
остаточным давлением 667—2000 Па (5—15 мм рт. ст.).
Для герметизации часто пользуются также жидкостными
(ртутными, глицериновыми или с вазелиновым маслом) затворами
(рис. 69,0 и г), которые можно применять только при работе под
атмосферным давлением. Они состоят из собственно затвора с
пробкой или шлифом и вставной трубки.
Ртутный затвор может уравновешивать лишь малые разности
давлений и пригоден для работы при разрежениях до 4 кПа
(30 мм рт. ст.) и при избыточном давлении до 13,3 кПа (100 мм
рт. ст.). Частота вращения мешалки не должна быть большой,
иначе ртуть будет разбрызгиваться. Чтобы этого не произошло,
целесообразно покрыть поверхность ртути вазелиновым маслом.
Ртутные затворы нельзя применять при работе с ацетиленом, так
как при этом возможно образование взрывчатого ацетиленида
ртути.
Для перемешивания в вакууме можно пользоваться мешалкой
типа КПГ, ось которой на протяжении 3—5 см пришлифована
139
Рис. 69. Уплотнения для мешалок:
с—затвор из перемешиваемой жидкости; б —резиновый сальник; е, г—жидкостные затворы.
к направляющей стеклянной трубке. Такую мешалку можно
изготовить из стеклянного медицинского шприца с полым стеклянным
поршнем, вместимостью 5 мл. Цилиндр аккуратно обрезают
вблизи места, где вставляется игла, а поршень — с обоих концов.
В трубке, изготовленной из поршня, закрепляют вал мешалки при
помощи замазки, противостоящей действию паров реакционной
смеси. Ось вала должна очень точно совпадать с осью поршня.
Для этого пользуются просверленными резиновыми пробками,
которыми фиксируют вал, пока замазка не затвердеет. После
затвердения замазки пробки удаляют и выступающий конец
мешалки центрируют.
Центрирование мешалки в сложных установках представляет
некоторые трудности. Необходимо, чтобы применяемый для
крепления мешалки и прибора штатив был неподвижен. При
пользовании обычными штативами (например, Бунзена) следует
привинтить их к столу или утяжелить основание металлическими
брусками и закрепить верхний конец штатива, чтобы стержень
не колебался.
Стержень мешалки должен быть совершенно прямым; со
шкивом привода он соединяется посредством отрезка вакуумной
резиновой трубки. Внутренний диаметр направляющей стеклянной
трубки должен быть ненамного больше диаметра стержня
мешалки.
Приводы для мешалок
В качестве приводов для лабораторных мешалок чаще всего
используют электродвигатели малой мощности, преимущественно
однофазные, работающие от сети напряжением 120/220 В.
Мешалки можно закреплять непосредственно на выступающем
конце вала электродвигателя при помощи вакуумной резиновой
трубки (вал двигателя и мешалки должны составлять одну пря*
НО
мую) или приводить их в движение при помощи различного типа
передач.
Перед включением мешалки ее предварительно прокручивают
рукой, чтобы убедиться в том, что ее не заклинивает и при
вращении она не касается стенок сосуда или термометра.
Частота вращения электродвигателя регулируется с помощью
лабораторных автотрансформаторов, предназначенных для
плавного регулирования напряжения однофазного переменного тока
промышленной частоты 50 Гц.
Лабораторные перемешивающие устройства
Мешалки. Лабораторная мешалка МЛ-2 (рис. 70)
предназначена для работ, требующих интенсивного
перемешивания. Электродвигатель обеспечивает плавную регулировку
частоты вращения мешалки до 4000 об/мин. Привод соединен с
пропеллерной мешалкой гибким валом; при вертикальной установке
двигателя гибкий вал заменяется на жесткий.
Для перемешивания и подогрева относительно небольших
объемов жидкостей в химических лабораториях широко пользуются
магнитной мешалкой. Движение стержню, помещенному в
сосуд с жидкостью передается с помощью магнитного поля ротора
и может быть как вращательным, так и прямолинейным возвратно-
поступательным.
Чтобы предотвратить коррозию и химическое взаимодействие с
жидкостями, стержень заключен в герметическую полиэтиленовую
или стеклянную оболочку. Интенсивность перемешивания зависит
от скорости вращения магнитного ротора, длины перемешивающего
ШМ'
С^
tf-
Рис. 70. Мешалка МЛ-2 с гибким (а) и жестким (б) валом:
1 — пропеллер; 2—зажимный патрон; 3 — головка; 4 —зажимная гайка; 5—двигатель.
141
стержня и от вязкости перемешиваемой жидкости. В комплект
магнитной мешалки (ММ-ЗМ) входят перемешивающие стержни,
представляющие собой стеклянные трубки диаметром 5—8 мм и
длиной 20—40 м, наполненные железными опилками и запаянные
с обоих концов. Стержни помещают в сосуд, под дном которого
вращается постоянный магнит. Дно сосуда должно быть плоским,
так как при круглом дне между внутренней поверхностью сосуда и
концами перемешивающего стержня возникает сильное трение.
Прибор включают в сеть только при подсоединенном шнуре
заземления. Максимальная частота вращения магнитного ротора
1400 об/мин. Объем перемешиваемой жидкости до 1,5 л.
Приборы для встряхивания. Наиболее распространенный тип
лабораторного прибора для встряхивания — универсальные
аппараты типа АВУ. В этих аппаратах встряхиваемый сосуд
перемещается по горизонтали. Аппарат АВУ-бп работает от сети
переменного тока напряжением 220 В. Число колебаний платформы
в минуту регулируется в пределах 100—225.
Для смешивания жидкостей и получения эмульсий и суспензий
служит также вибромешалка «Вибратор В-1», выпускаемая
серийно. Максимальная нагрузка на вибратор — 4 колбы по 500 мл.
Зажимы вибратора позволяют устанавливать колбы с диаметром
горла от 19 до 38 мм. Вибратор может работать при
асимметричной нагрузке. Частота встряхивания регулируется от 30 до 60 Гц
при помощи плавной регулировки оборотов двигателя. Вибрация
возникает в результате вращения закрепленной на валу двигателя
неуравновешенной массы, создающей при вращении центробежную
силу. Находящаяся в колбах жидкость при определенных оборотах
интенсивно перемешивается. Вибрация гасится на резиновых
амортизаторах и не передается на корпус прибора.
Для встряхивания используют также электронный
вибратор, который имеет приставные детали для крепления бутылок,
пробирок, длительных воронок и др. Прибор снабжен регулятором
частоты колебания.
ПЕРЕМЕШИВАНИЕ БАРБОТИРОВАНИЕМ
Жидкости можно перемешивать, пропуская через них под
небольшим давлением ток сухого чистого воздуха или азота.
Подобное перемешивание (барботирование) можно эффективно
осуществлять, используя газопромыватели. При барботировании
следует регулировать скорость поступления воздуха или азота,
чтобы избежать разбрызгивания перемешиваемой жидкости.
глава 12 ВЗВЕШИВАНИЕ
Лабораторные весы различаются по назначению, конструкции,
диапазону взвешивания и по другим характеристикам.
Методы взвешивания делятся на две принципиально различные
группы — метод сравнения с мерой и метод непосредственной
оценки. По методу сравнения с мерой массу груза принимают
равной массе сравниваемых с ним гирь (простое взвешивание)
или вычисляют как сумму значений массы гирь и показаний
весов (точное взвешивание). Метод непосредственной
оценки заключается в определении массы груза по отсчетному
устройству весов без применения гирь.
В большинстве современных лабораторных весов используется
дифференциальный метод взвешивания, при котором большая
часть измеряемой массы тела (свыше 99%) уравновешивается
гирями или противовесом (нулевой метод), а оставшаяся малая
разность между массой взвешиваемого тела и массой гирь измеряется
по углу отклонения коромысла от исходного положения
равновесия (непосредственный метод) с помощью отсчетных шкал.
Лабораторные весы характеризуются рядом параметров.
Главные из них следующие.
1. Предельно допустимая нагрузка, в диапазоне
которой погрешность показаний находится в установленных
пределах. Нельзя выходить за пределы предельно допустимой
нагрузки, на которую рассчитана данная модель весов. Слишком
большая нагрузка может вызвать остаточные деформации в
коромысле, что приведет к порче весов.
2. Допустимая погрешность показаний —
предельная разность между действительным значением массы
взвешиваемого груза и показаниями весов. Значение погрешности
характеризует правильность результатов взвешивания в стандартных
условиях и не может быть меньше неисключенных погрешностей
гирь, применяемых при взвешивании и аттестации весов.
3. Допустимая вариация (непостоянство) —
показаний— предельно допустимая разность показаний весов при
неоднократном взвешивании одного и того же груза в стандартных
условиях с применением одних и тех же гирь. Значение вариации
характеризует воспроизводимость результата взвешивания и, в
значительной степени, точность взвешивания.
4. Чувствительность — предельное отношение
приращения отклонения указателя весов к приращению измеряемой
величины. Чувствительность определяется числом делений шкалы, на
которое отклоняется стрелка весов, когда на одну из чашек весов
помещен груз массой 1 мг. Выражают чувствительность в
делениях шкалы на миллиграмм или обратной величиной.
143
С увеличением нагрузки на чашки чувствительность весов
уменьшается, т. е. чем больше масса взвешиваемого объекта, тем
слабее весы реагируют на изменение массы.
5. Цена деления — значение деления отсчетных устройств.
Часто цена деления согласуется со значением допустимой
погрешности или вариацией показаний весов.
6. Быстродействие — возможная производительность
работы на весах, т. е, возможное число взвешиваний в единицу
времени.
КЛАССИФИКАЦИЯ ВЕСОВ
По назначению лабораторные весы делятся на технические
(общелабораторные), аналитические и специальные, а гири — на
гири общего пользования и специальные [46, с. 132].
Наибольшие пределы взвешивания технических весов
находятся в диапазоне 20 г — 50 кг. Наиболее распространены весы,
имеющие нагрузку 0,2—5 кг, с ценой деления 0,05—0,1 г.
Аналитические весы применяют для макро- и
микрохимических анализов при взвешивании высшей и высокой точности.
В зависимости от предельно допустимой нагрузки и цены деления
аналитические весы делятся на следующие группы:
Предельно Цена
допустимая деления
нагрузка
Большегрузные весы 1—5 кг 1—5 мг
Весы для макроанализа 100—200 г 0,05—0,1 мг
Микроаналитические ве- ^ 20 г ^ 0,01 мг
сы
Ультрамикроапалитиче- < 1 г 0,01 — 1 мкг
скне весы
Специальные весы служат для определения величин,
зависящих от массы (весовые влагомеры, весы для измерения
магнитной восприимчивости и др.).
Весы аналитической группы относятся к 1 и 2 классу точности,
технические весы — к 3 и 4 классам. Усредненная приведенная
погрешность взвешивания для весов I класса — 0,0001%; 2 класса —
0,0005%; 3 класса —0,001%; 4 класса — 0,01%.
Гири общего лабораторного пользования делятся на четыре
класса. Гири 1 и 2 классов предназначены в основном для
аналитических весов, 3 и 4 классов — для технических.
По характеру перемещения подвижной системы весы
подразделяются на безрычажные и рычажные. В безрычажных
весах подвижная система перемещается возвратно-поступательно
вертикально, поэтому гири для уравновешивания взвешиваемого
груза применять нельзя. При пользовании безрычажными весами
пригоден лшиь метод непосредственной оценки результатов
взвешивания.
Рычажные весы характеризуются поворотом подвижной
системы вокруг неподвижной или условно неподвижной оси. Они
144
бывают гирные (с накладными или встроенными гирями) и без-
гирные. Весы со встроенными гирями производительнее и удобнее,
но в них затруднен контроль действительных значений массы
гирь.
Рычажные весы различаются по типу опор весового рычага
и подвесок. Наиболее распространенной жесткой опорой является
подушка, по которой острой гранью перекатывается призма. Весы
с такими опорами получили название призменных. Призмен-
ные весы делятся на равноплечие, двухпризменные (одночашеч-
ные) и квадрантные [64а].
Равноплечие весы представляют собой, в основном,
рычаг первого рода, в котором расстояния от приложения сил до
точки опоры равны (рис. 71). Если на левую чашку весов
поместить груз, имеющий массу Mi, то для возвращения стрелки Р в
первоначальное положение потребуется поместить на правую
чашку некоторое количество гирь (с известной массой). Когда
установится равновесие, моменты сил, действующие на левую и правую
части коромысла в точках, на которые опираются чашки, на
расстоянии 1\ и /2 от этих точек до точки опоры, будут равны: /Vi=
=F2l2-
Так как /i=/2, то, следовательно, при достижении равновесия
F} = f2t Возникновение сил F\ и F2 связано с притяжением Землей
тел, находящихся на чашках весов. Сила F{ определяет
притяжение к Земле тела с массой Mi, т. е. его вес. Единица веса —
ньютон (Н). Ньютон равен силе, сообщающей телу массой 1 кг
ускорение 1 м/с2 в направлении действия силы. Вес тела G связан с
его массой соотношениями: G = Mg, где М — масса тела, a g—
ускорение свободного падения. Единица массы — килограмм (кг).
Из изложенного выше следует, что весы — это приборы для
определения массы, а не веса.
Равноплечие коромысловые весы показаны на
рис. 72. Равновесное положение ненагруженных весов называют
нулевой точкой, нагруженных — точкой равновесия.
Чтобы защитить ребра призм коромысла от повреждений и
быстрого изнашивания, все движущиеся части весов могут быть
приподняты и ребра призм отделены от пластинок, с которыми
Рис. 71. Принцип взвешивания на равноплечих весах.
Рис. 72. Равноплечие коромысловые весы:
/, 2—подушки; 3 —грузоприемная призма; 4—опорная призма; 5—рычаг; 5 —микрошкала;
7 — груз; 8 — подвеска; 9—гири.
145
они соприкасаются. Приспособление, служащее для поднятия
коромысла и сережек, носит название арретира (изолира).
Когда весами не пользуются и когда накладывают на чашки
взвешиваемые предметы и разновесы, весы должны быть арретированы.
До недавних пор на коромысло равноплечих аналитических
весов наносились на одинаковом расстоянии друг от друга V-об-
разные впадины рейтерной шкалы (рис. 73), в которые
специальным устройством устанавливали гирю-рейтер в 10 или 5 мг.
Передвижением рейтера по коромыслу можно было определить массу
с точностью до десятых долей миллиграмма.
В современных призменных весах имеются успокоители
колебаний стрелки весов — демпферы. В демпферных весах за
нулевую точку и точку равновесия принимают деление шкалы,
против которого останавливается стрелка. У весов, которые не имеют
демпферов, эти точки определяют методом качаний. Этот метод
основан на измерении 3—5 последовательных отклонений стрелки.
Первые 2—3 колебания после включения весов не принимают во
внимание, а последующие 5 отклонений стрелки в одну и другую
сторону фиксируют с точностью до десятых долей по шкале.
Нулевую точку рассчитывают, например, следующим образом.
Отклонения влево: —3,4 и —2,8; среднее значение —3,1.
Отклонение вправо: +4,0, +3,5 и 3,0, среднее значение +3,5.
Найдем сумму отклонений: +3,5 + (—3,1) = 0,4.
Найдем нулевую точку: +0,4 : 2 = +0,2.
Точность демпферных весов того же порядка, что и точность
обычных весов.
Двухпризменные (одночашечные) весы
изображены на рис. 74. В исходном положении все встроенные гири
нагружены на подвеску и рычаг уравновешен противовесом. Поместив
на грузоприемную чашку груз при помощи специального
механизма гиреналожения, с рейки снимают такое число встроенных
гирь, чтобы их суммарная масса приблизительно соответствовала
массе груза. Разница между массой груза и массой снятых гирь
определяется по показаниям отсчетного устройства.
Двухпризменные одночашечные весы используются преимущественно в
качестве аналитических весов. Достоинства этой конструкции весов
заключаются в том, что работа всегда ведется при постоянной
нагрузке на коромысло, а в этом случае постоянны и
чувствительность весов и точность взвешивания.
-^*мул*лнулл/*лл^
Рис. 73. Коромысло с рейтерной шкалой.
146
Рис. 74. Двухпризменные весы.
Обозначения позиций 1—9 см. рис. 72; 70 —рейка; // —противовес.
Рис. 75. Квадрантные весы.
Обозначения позиций 1-8 см. рис. 72: 9—„струнка*.
Квадрантные весы или весы с верхним расположением
грузоприемной чаши (рис. 75) являются разновидностью двух-
призменных.
Рычажные весы с опорами на упруго-деформируемых
элементах выпускаются для взвешивания небольших масс. К ним
относятся торсионные весы и пружинные рычажные ультра*
микровесы.
ОБЩЕЛАБОРАТОРНЫЕ РАВНОПЛЕЧИЕ ВЕСЫ
Общелабораторные равноплечие весы — технические весы
преимущественно 3 и 4 классов точности — служат для взвешивания
относительно больших масс. Они могут быть заключены в
остекленную витрину и снабжены гиревым механизмом со встроенными
гирями, а могут быть подвешены на стойку, закрепленную на
подставке, без гиревого механизма. Простейшим типом
равноплечих весов с двумя чашками являются ручные, или аптечные,
весы.
Технохимические весы типа ВЛТ-200г (Т-200) и
В Л Т- 1кг (Т-1000) представлены на рис. 76. При взвешивании
поворотом ручки арретира весы приводят в рабочее положение.
Допустимая погрешность для весов ВЛТ-200г ±60 мг, для
ВЛТ-1кг ±200 мг.
Более совершенные технохимические весы типа ВЛР-1кг
состоят из равноплечего коромысла со стрелкой, колонки с опорной
подушкой, изолирующего устройства и двух грузоприемных
чашек, подвешенных на концевых призмах коромысла. Весы
снабжены масляным успокоителем колебаний коромысла и
устройством для механического гиреналожения встроенных гирь (от 10
до 990 мг).
Перед взвешиванием необходимо убедиться по уровню, что
весы правильно установлены. В случае необходимости с помощью
винтовых ножек весы устанавливают строго горизонтально. Затем
нужно проверить отклонение стрелки и добиться полного
совмещения ее с контрольным штрихом циферблата шкалы.
147
Рис. 76. Технохимические весы:
/ — чашка; 2 —регулятор; «3 —коромысло;
4—серьга; 5 —отвес; 6 — стрелка; 7—-шкала; 8 — ручка
арретира.
В последние годы
равноплечие технические двухчашечные
весы типа ВЛТ были
значительно модернизированы и
осуществлен серийный выпуск ряда
новых моделей весов типа ВЛР,
3 класса точности (с
погрешностью ±10 мг),
грузоподъемностью 1, 10, 20 и 50 кг, ценой
деления шкалы 10 мг.
Весы ВЛР помещены в застекленный футляр с дверцами на
двух боковых сторонах. На верхнем конце колонки находится
подушка, на которую опирается ребром средняя призма
коромысла. У основания колонки укреплен масляный успокоитель.
В концах коромысла в специальных седлах закреплены призмы,
на которые навешиваются серьги с ^рузоприемными чашками. На
планку, скрепленную с правой серьгой, при помощи гиревого
механизма навешиваются и снимаются кольцевые гири (от 100 до
900 мг и от 10 до 90 мг), связанные с большим и малым лимбом.
На середине коромысла укреплена стрелка, а внизу колонки
расположена шкала, по которой проверяется равновесие весов.
Под основанием весов смонтировано изолирующее устройство
(арретир). Открывать и закрывать арретир надо осторожно,
плавным вращением маховичка в тот момент, когда стрелка весов
проходит мимо нулевого деления шкалы.
ОБЩЕЛАБОРАТОРНЫЕ КВАДРАНТНЫЕ ВЕСЫ
В последние годы большое распространение получили
квадрантные весы, которые выгодно отличаются быстротой действия.
Это двухпризменные весы с верхним расположением чашки.
Успокоитель колебаний магнитный. Имеются оптическое устройство
и экран, по которому производится отсчет результатов
взвешивания. Наложение и съем накладных гирь осуществляется ручкой,
расположенной на металлическом корпусе весов. Весы
включаются в сеть переменного тока через встроенный трансформатор,
укрепленный под витриной весов.
Квадрантные весы предназначаются для определения массы
различных веществ и материалов при проведении лабораторных
технических анализов и препаративных работ.
Принцип действия весов основан на уравновешивании момента
сил, создаваемого измеряемой массой, отклонением квадранта и
встроенными гирями.
В настоящее время выпускаются шесть модификаций
лабораторных квадрантных весов 4 класса ВЛКТ и ВЛК с пределами
взвешивания от 160 до 10000 г.
148
Рис. 77. Лабораторные квадрантные весы 4 класса ВЛКТ.
Обозначения позиций см. рис. 78.
Весы ВЛКТ (рис. 77) имеют меха- у^Р^ВЩРд
низм компенсации тары, который позво- ^=?^SsLjffl
ляет повысить производительность взве- ШИЩЩрЩЩI
шиваиия и предназначен для установки ^^^^^^^й|
шкалы на нулевую отметку после разме- ^^^^^^S^II 11||И1
щения тары на чашке весов. 1Щ||1Щ1^Щ^
Значение измеряемой массы тела, ^^^^^S=^--||Hr
находящегося на чашке весов, находят И|ИИШР§§Й/^
суммированием показаний на оптической ^Г^^^^^ш^
шкале и на счетчике. Число сотен или Щ
тысяч граммов отсчитывается по
счетчику, в окне которого появляются цифры 0, 1, 2, 3 и 4 в
зависимости от массы гирь, снятых с подвески.
ВЗВЕШИВАНИЕ НА ТЕХНИЧЕСКИХ ВЕСАХ
Весы устанавливают на прочных устойчивых столах в рабочем
помещении лаборатории строго вертикально по отвесу. Перед
взвешиванием проверяют, правильно ли установлены весы, после
чего опускают арретиром коромысло и наблюдают колебания
стрелки по нижней шкале. Если стрелка отклоняется от нуля на
одно и то же число делений вправо и влево, весами можно
пользоваться. В ином случае равновесие весов достигается с помощью
балансировочных гаек коромысла.
Взвешиваемую массу помещают на левую площадку весов,
гири граммового набора — на правую, а гири миллиграммового
набора навешиваются гиревым механизмом.
Массу вещества лучше определять методом двойного
взвешивания, который заключается в следующем: на левую
чашку весов помещают взвешиваемый предмет, а на правую —
гири до установления стрелки весов на нулевую отметку шкалы.
После этого взвешиваемый предмет переносят на правую чашку,
а гири — на левую. Если одна из чашек будет перевешивать
другую, то, добавляя или снимая разновески, снова устанавливают
точку равновесия. Действительная масса взвешиваемого предмета
равна среднему арифметическому результатов этих двух
взвешиваний. По окончании взвешивания с чашек весов удаляют
взвешиваемый предмет, убирают гири и разновески, укладывая их в
установленном порядке в футляр.
АНАЛИТИЧЕСКИЕ ВЕСЫ
Еще в большей степени, чем технические и технохимические
весы, подвергались в последние годы модернизации весы
аналитической группы. Вместе с тем во многих химических
лабораториях еще успешно используются равноплечие р ей тер ные весы
14£
без демпферов — весы периодического качания, не
снабженные встроенными гирями. Особенность работы на весах
периодического качания заключается в определении их нулевой точки.
Коромысло, освобожденное от арретира, начинает совершать
постепенно затухающие колебания. Нулевую точку и точку
равновесия определяют методом многократных отклонений стрелки
коромысла. Перед определением нулевой точки рейтер должен быть
снят с коромысла, если нуль находится в центре коромысла, или
установлен на нуле, если нуль на левом конце коромысла.
Чтобы определить чувствительность весов, устанавливают
точку равновесия при различных нагрузках. Для этого после
установления нулевой точки помещают на коромысло рейтер (при
арретированных весах) так, чтобы он показывал 1 мг, опускают
арретир и определяют точку равновесия.
Например, если нулевая точка весов равна + 0,2 деления, а точка
равновесия при нагрузке на правую чашку в 1 мг + 3,8 делений, то чувствительность
весов находят, помещая на обе чашки последовательно по 5, 10, 20, 30, 40, 50 и
100 г. Полученные результаты наносят на график.
При пользовании рейтерны ми демпферными весами
определение массы с помощью встроенных или накладных гирь
производят только до 10 или 5 мг (т. е. до массы рейтера).
Дальнейшее уравновешивание производят с помощью рейтера, который
устанавливают только до ближайшего к равновесию целого
миллиграмма. Если не требуется точность взвешивания, превышающая
0,1 мг, десятые доли миллиграмма находят передвижением
рейтера по коромыслу. При необходимости более точного
взвешивания рейтер устанавливают как и в предыдущем случае, а
десятые и сотые доли миллиграмма находят по разности между
нулевой точкой и найденной точкой равновесия на основании ранее
определенной чувствительности весов для данной нагрузки.
Пусть, например, нулевая точка весов равна + 0,5; точка павновесия весов
при нагрузке 14,3300 г на правой чашке и рейтера на делении 3 мг равна + 2,0;
чувствительность весов при нагрузке в 14,5 г равна 4 делениям на 1 мг.
очевидно, что взвешиваемый предмет полностью не уравновешен. Если рейтер
будет перенесен на деление 4 мг, то точка равновесия сдвинется на 4 деления
влево, т. е. окажется равной —2,0. Чтобы точка равновесия совпала с нулевой
точкой (+0,5), рейтер надо передвинуть на ' fl ' = 0,38 делений, т. е. на
0,38 мг. Следовательно, масса взвешиваемого предмета окажется равной 14,3300 г
(на чашке весов) +0,00038 г (показание рейтера) — 14,33038 г.
Во многих лабораториях используются двухчашечные
равноплечие лабораторные аналитические весы ВЛА-200 г-М (АД-200)
с такими основными характеристиками: наибольшая допустимая
нагрузка 200 г; диапазон измерения по оптической шкале ±10 мг;
погрешность из-за иеравноплечности коромысла не более 2 мг.
Управление гирями производится посредством лимбов. При
вращении малого лимба происходит навешивание или снятие
десятков миллиграммов, при вращении большого — сотен
миллиграммов. Лимбы вращаются независимо друг от друга. Включение и
150
выключение весов производится ручкой, надетой на валик арре*
тира, вынесенного на переднюю стенку основания.
В настоящее время промышленность выпускает, главным
образом, двухчашечные равноплечие весы типа ВЛР, например весы
класса точности ВЛР-200г и ВЛР-20г. Весы ВЛР-20г,
заменяющие полумикроаналитические весы ВЛМ-20г-М, отличаются
большой чувствительностью и меньшими габаритными размерами.
На базе весов ВЛР-200г с электронной приставкой выпускаются
электронные весы ВЛЭ-200г.
Технические данные весов ВЛР-200г (рис. 78) и весов ВЛР-20г
приведены ниже:
Предел взвешивания, г
Диапазон измерения по
оптической шкале, мг
Цена деления оптической
шкалы, мг/дел
Диапазон механического гире-
наложения, мг
Время успокоения колебаний
коромысла, с
ВЛР-200г
200
10
0,1
0-900
25
ВЛР.20г
20
1
0,01
0-90
30
При пользовании весами типа ВЛР-200г, в первую очередь,
включают осветитель в сеть, после чего, не открывая дверок
шкафа весов, осторожно поворачивают до отказа диск арретира.
Автоматически загорающаяся при этом электрическая лампочка
освещает на экране вейтографа увеличенное изображение
микрошкалы, прикрепленной к стрелке весов. Если весы не нагружены,
нуль шкалы должен точно совпадать с вертикальной чертой на
Рис. 78. Лабораторные равноплечие весь: типа ВЛР-200г:
У —чашки; 2 — арретир; 3—-ручки устройства накладки гирь; 4 — цифрозой указатель и
световая шкала массы.
Рис. 79. Лабораторные двухпризменные одночашечные весы ВЛДП-ПЭг:
/ — чашка; 2—ручка установки нуля; 3 — ручт устройства накладки гирь; 4 — цифровой
указатель и световая шкала массы.
151
экране (риской). В ином случае совпадение достигается
вращением регулировочного винта, находящегося снаружи на нижней
доске весов над диском арретира. Затем груз помещают на
левую чашку весов, а на правую — граммовые гири из набора гирь
к весам; при этом находят массу числа целых граммов.
Закрывают дверцу шкафа; поворачивая малый лимб с десятыми долями
грамма, совмещают неподвижный указатель с различными
цифрами диска. При каждом повороте диска необходимо
предварительно арретировать весы. Установив число десятых долей
грамма, находят сотые доли грамма с помощью большого лимба.
Далее диск арретира поворачивают до отказа и, после прекращения
колебаний стрелки коромысла, делают отсчет положения
вертикальной линии по шкале на экране. Крупные деления этой
шкалы, соответствующие миллиграммам, обозначены цифрами со
знаком «+» или «—». Плюс показывает, что величину сделанного
отсчета нужно прибавить к массе помещенных на весы
разновесок, а минус — вычесть
После окончания взвешивания записывают результат, снимают
с весов взвешенный предмет и разновески. Чтобы освободить
коромысло от встроенных гирь, вращают дисковые рукоятки до тех
пор, пока неподвижный указатель не совместится с нулевым
делением обоих дисков.
Кроме равноплечих аналитических весов типа ВЛР
промышленность выпускает одноплечие весы 2 класса типа ВЛДП-ЮОг
(рис. 79). Принцип взвешивания на двухпризменных весах
основан на уравновешивании момента, создаваемого грузом, и
момента, получаемого при снятии с подвески встроенных гирь.
Коромысло весов представляет собой неравноплечий рычаг; на
коротком плече закреплено седло с грузоприемной призмой, а на
длинном — отсчетная шкала. На грузоприемную призму
коромысла подушкой опирается серьга, к которой жестко прикреплена
планка для наложения встроенных гирь. Для снятия и
наложения встроенных гирь служит гиревой механизм. Одновременно
со снятием гирь в трех левых окнах экрана высвечивается
значение их массы (в г). При точном взвешивании коромысло
успокаивают с помощью воздушного демпфера; при предварительном —
масляного. Ручка ввода весов в рабочее положение находится
с левой стороны весов. Предварительное взвешивание
осуществляют поворотом ручки от оператора, точное — на оператора.
Нулевое положение шкалы при предварительном взвешивании
регулируют ручкой, находящейся с правой стороны весов, вверху;
при точном взвешивании — ручкой внизу. Механизм
предварительного взвешивания предназначен для определения массы
встроенных гирь. Для снятия отсчета по шкале на экране имеется
отсчетная отметка в виде двух параллельных штрихов.
Результат взвешивания определяется суммой показаний от-
счетной шкалы, счетчиков гиревого механизма и делительного
устройства. Диапазон взвешивания от 0 до 100 мг. Цена
наименьшего деления шкалы 0,05 мг. Погрешность взвешивания ±0,065 мг.
152
Установка аналитических весов
Установка аналитических весов начинается с выбора помеще*
ния и организации рабочего места химика. Помещение для
установки весов 1 и 2 классов должно состоять из весовой комнаты
и препараторской. Одно из условий, предъявляемых к весовой
комнате,— полная изолированность ее от смежных лабораторных
помещений.
Для весовой комнаты выбирают светлое сухое помещение. Же*
лательно, чтобы оно было расположено на первом этаже, окнами
на северную сторону. В весовой комнате должна поддерживаться
постоянная температура — около 20 °С. Весы нужно предохранять
от воздействия тепловых и воздушных потоков, а также от
сырости, пыли, вредных газов и сотрясений. Чтобы уменьшить
влияние воздушных и тепловых потоков, рекомендуется закрыть
плотными шторами окна и двери. Окна должны быть снабжены
двойными рамами и плотно замазаны; окна и форточки открывать
нельзя. Проветривать весовое помещение рекомендуется
вентилятором, и лишь тогда, когда не ведется взвешивание. Пол
рекомендуется покрыть линолеумом, который легко очищается от
пыли и является плохим проводником тепла.
Весы следует устанавливать в горизонтальном положении на
особо прочных постаментах, предохраняющих весы от всяких
сотрясений. Не рекомендуется переносить весы с места на
место.
Аналитические весы с предельной нагрузкой 100 г и больше
рекомендуют устанавливать на консольный стол, состоящий из
бетонной плиты, свободно лежащей на амортизирующих
резиновых или пенопластовых прокладках в обвязке стола, покоящейся
на двух металлических кронштейнах, прикрепленных к
капитальной стене.
Полумикроаналитические весы целесообразно устанавливать
на столе с массивными ножками. Стол состоит из массивной
крышки, в раму которой вложен войлок, железобетонная
мозаичная плита и линолеум.
Лампы в весовой комнате должны достаточно освещать
шкалу весов и, вместе с тем, не нагревать коромысла. Лучше всего
устанавливать лампы дневного света.
В весовой комнате следует вывесить таблицу с основными
правилами обращения с весами.
Необходимо тщательно следить за чистотой весовой комнаты.
По окончании взвешивания весы рекомендуется покрывать
чехлами.
На консольный стол или полку на кронштейнах, где установ*
лены весы, нельзя ничего ставить. Слева от стола (полки) целе*
сообразно иметь передвижной столик для эксикатора со
взвешиваемым веществом и для производства записей.
163
Правила пользования аналитическими весами
1 Нагрузка на чашках весов не должна превышать
наибольшей для данного типа весов. Взвешивают только сидя против
весов, опираясь руками на крышку стола. Взвешиваемый предмет
берут пинцетом, щипцами или чистой бумагой и помещают на
середину левой чашки. Химические вещества взвешивают в
стеклянной посуде (бюкс, ампула). Нельзя помещать химические
вещества непосредственно на чашку весов или производить
взвешивание на листочке бумаги.
2. Взвешиваемый предмет должен иметь ту же температуру,
что и весы. Поэтому перед взвешиванием следует вещество
выдерживать в эксикаторе вблизи весов в течение 20—30 мин. Если
при взвешивании над весами включают лампу, то сделать это
надо за 10—15 мин до начала работы.
3. Прибавлять или убавлять взвешиваемое вещество следует
только вне шкафа весов. Если взвешиваемое вещество просыпано
на чашку весов или на дно шкафчика, надо немедленно вымести
его кисточкой.
4. Гири следует помещать на правую чашку весов таким
образом, чтобы они находились в центре чашки. Брать гири следует
пинцетом с костяными (пластмассовыми) наконечниками.
5. Когда взвешиваемое вещество или гирьки кладут на чашку
весов или снимают с них, весы должны быть арретированы.
6. Перед каждым взвешиванием следует проверять, а если
нужно, то и устанавливать их нулевую точку. Во время
наблюдения за отклонением стрелки весов дверцы шкафа должны быть
закрыты.
7. При уравновешивании взвешиваемого предмета начинают
с больших разновесок и затем переходят к более мелким.
Следует всегда пользоваться наименьшим числом разновесок,
например, брать разновеску в 2 г, а не две разновески по 1 г.
На чашке весов разновески должны лежать в определенном
порядке; мелкие разновески не следует класть друг на друга.
Большие разновески надо помещать в центре чашки, чтобы она не
качалась.
Ошибки взвешивания и их устранение
Ошибки при точном взвешивании могут происходить от
различных причин: от неравноплечести весов; от взвешивания в
воздухе, а не в пустоте; от изменения массы тел в процессе
взвешивания вследствие колебаний температуры, влажности и давления
воздуха; от неточных значений масс гирь; от инструментальных
погрешностей.
Ошибки от неравноплечести весов большей частью
возникают при методе простого взвешивания на весах
периодического качания. Однако поправки на неравноплечесть не всегда
требуются. Так, при определении процентного состава вещества
1[в % (масс.)], когда взвешивание анализируемого вещества и
154
его весовой формы производят на одних и тех же весах и когда
взвешиваемые вещества помещены на одну и ту же чашку,
относительная ошибка при обоих взвешиваниях будет примерно
одинаковой. Но когда требуется определить абсолютную массу
предмета с точностью, превышающей 0,1 мг, приходится прибегать к
методам взвешивания, исключающим поправки на неравнопле-
честь, например к методу замещения.
Метод замещения по Борда заключается в
следующем. Измеряемую массу помещают на правую чашку весов и
уравновешивают любой тарной массой на левой чашке.
Определяют положение равновесия Е\. Затем с правой чашки снимают
измеряемую массу, не снимая тары с левой, и накладывают
вместо снятой массы гири в таком количестве, чтобы получить
возможность отсчета по шкале, и определяют положение
равновесия Е2. Результат измерения равен массе наложенных гирь плюс
отсчет по шкале и определяется по формуле (Е{ — E2)Sy где
S — чувствительность весов.
Метод замещения, предложенный Д. И.
Менделеевым, состоит в том, что на одну из чашек помещают гири в
количестве, соответствующем предельной нагрузке весов, и
уравновешивают весы тарным грузом. Взвешиваемое тело помещают
на чашку с гирями и снимают такое количество гирь, чтобы весы
пришли в положение исходного равновесия. Значение массы
взвешиваемого тела определяется как алгебраическая сумма массы
снятых с чашки гирь и показаний по шкале весов. Этот метод
положен в основу принципа действия двухпризменных
одноплечих весов.
Ошибки, вызванные взвешиванием в воздухе,
вытекают из общеизвестного физического закона, что каждое тело,
погруженное в жидкость (газ), теряет в своем весе столько,
сколько весит вытесненная им жидкость (газ). Все тела,
следовательно, в воздухе весят меньше, чем в пустоте. Обычное
взвешивание в воздухе приводило бы к правильному результату, если
бы гири теряли в своей массе столько же, сколько теряет
взвешиваемое тело. Однако аналитический разновес обычно
изготовляют из нержавеющей стали (р = 8,0 г/см3) или из латуни (р =
= 8,4 г/см3), а миллиграммовые разновески — из алюминия (р =
= 2,7 г/см3). Если плотность взвешиваемого тела меньше
плотности гирь, то тело вытесняет больше воздуха, чем гири и,
следовательно, в воздухе оно весит меньше, чем в пустоте. Величина
ошибки обычно не превышает 0,04—0,05%.
Ошибки, вызванные изменением массы тел в
процессе взвешивания, могут происходить вследствие
поглощения или потери влаги, испарения летучих веществ, изменения
температуры, невнимательности и неаккуратности экспериментатора.
Эти ошибки могут быть устранены взвешиванием веществ по раз*
ности в герметически закрываемой стеклянной посуде малого объ*
ема. При взвешивании по разности положение нулевой точки мож«
но не учитывать.
155
Погрешности массы гирь зависят от степени
точности подгонки их массы к номинальному значению, погрешности
аттестации и от необратимых изменений массы в
межпроверочном периоде, в основном из-за коррозии. Ошибки, связанные с
неточностью масс используемых гирь, можно устранить сличением
их с массой образцовых гирь на тех весах, на которых будут с
ними работать.
МИКРО- И УЛЬТРАМИКРОВЕСЫ
Для особо точных измерений небольших масс при проведении
физико-химических исследований и микроанализов используют
точные рычажные и безрычажные весы различных конструкций.
Выпускаются рычажные пружинные весы с предельной
нагрузкой от 20 до 100 мг, с ценой деления 10~7 — 10~5 мг (ВЛУ-20мг
и ВЛУ-ЮОмг). Принцип действия этих весов основан на
уравновешивании момента, создаваемого измеряемой массой,
закручиванием кварцевой растяжки. По конструкции это весы на растяжках
с равноплечим коромыслом и нулевым методом взвешивания.
Коромысло помещено в специальный контейнер, предохраняющий
его от действия воздушных потоков и одновременно служащий
теплораспределителем. Чашка с взвешиваемым грузом выносится
из колонки коромысла в боковой отсек витрины манипулятором,
который сблокирован с механизмом открывания и закрывания
колонок. Отсчет результатов измерения производится по шкале
измерительного лимба, цена деления которой составляет 0,00032 мг
(ВЛУ-20мг) и 0,0005 мг (ВЛУ-ЮОмг). Время успокоения
колебаний коромысла около 1,5 мин.
Микроаналитические весы ВЛМ-1г предназначаются для
взвешивания драгоценных камней и металлов, а также для различных
веществ при микрохимических анализах повышенной точности.
Весы имеют равноплечие коромысла с двумя подвесками и
чашками. Полное механическое гиреналожение осуществляется двумя
гиревыми механизмами. Весы снабжены механизмом выноса
левой чашки. Диапазон измерения по
оптической шкале ± 1 мг. Цена деления оптической
шкалы 0,01 мг. Погрешность взвешивания
±0,07 мг.
ТОРСИОННЫЕ ВЕСЫ ВТ-500
Для быстрого определения массы очень
малых количеств веществ часто используют
торсионные (пружинные) весы (рис. 80).
Рис. 80. Торсионные весы ВТ-500:
1 — уровень; 2 — опорные винты; 3 — коромысло; 4 —
закрепительный рычаг; 5 —указатель массы; б —рычаг натяжения;
7—указатель равновесия; 8—предохранительная крышка;. 9—крючок
коромысла.
156
Они отличаются от квадрантных тем, что грузоприемная чашка в
них заключена в витрину и снабжена арретирным
приспособлением. Торсионные весы выпускаются с различными пределами
взвешивания. В лабораторной практике часто используют модель
ВТ-500. Предельно допустимая нагрузка весов 500 мг, а
наименьшая— 10 мг. Абсолютная погрешность показаний в любой
отметке шкалы не более ± 1 мг.
Измерительный элемент в торсионных весах — пружина,
натяжением которой при закручивании уравновешивается взвешиваемая
проба. Угол закручивания пружины пропорционален массе
взвешиваемой пробы, поэтому шкала весов проградуирована в единицах
массы.
При пользовании торсионными весами ВТ-500 их устанавливают
по уровню 1 посредством опорных винтов 2, после чего
освобождают коромысло 3 передвижением вправо закрепительного рычага 4.
Указатель массы 5 устанавливают на нуль с помощью рычага
натяжения 6. При таком положении весов указатель равновесия 7
перекрывает черту равновесия или же приводится в это положение
тарировочной головкой, находящейся на оборотной стороне весов
в центре. Затем закрепляют коромысло, передвигая
закрепительный рычаг влево до отказа, и приступают к взвешиванию. Для
этого открывают предохранительную крышку #, на крючок
коромысла 9 подвешивают взвешиваемый груз и вновь закрывают
крышку. Коромысло освобождают, передвигая рычаг 4 вправо.
Поворачивая рычаг 6 влево, перемещают указатель 5 до тех пор, пока
указатель 7 не установится точно на черте равновесия. В таком
положении указатель 5 показывает на шкале величину массы
измеряемого груза. После взвешивания закрепляют коромысло,
передвигая влево рычаг 4, открывают крышку 8У снимают с крючка
груз и закрывают крышку. Рычаг 6 перемещают вправо,
устанавливают указатель 5 на нуль — и весы готовы для следующего
взвешивания.
глава is. НАГРЕВАНИЕ И ОХЛАЖДЕНИЕ
Подводом или отводом тепла достигаются перегонка,
возгонка, образование и затвердевание расплавов, вымораживание,
кристаллизация и т. п. Нагревание и охлаждение являются
важнейшими средствами регулирования скорости и направления
протекания реакций. Охлаждение необходимо для получения, выделения и
хранения нестойких веществ, легко изменяющихся при комнатной
температуре, а также для растворения газообразных веществ в
жидкостях, получения сжиженных газов и т. п.
Здесь рассматриваются общие приемы нагревания и
охлаждения и оборудование для выполнения этих операций. Специальные
приемы нагревания и охлаждения описаны в других главах.
ЛАБОРАТОРНЫЕ ЭЛЕКТРОНАГРЕВАТЕЛЬНЫЕ ПРИБОРЫ [12; 46, с. 80]
Наибольшее распространение в лабораторной практике
находят электрические нагревательные приборы. В отличие от приборов
с другими источниками нагрева, они имеют следующие
преимущества: точная регулировка количества подводимой тепловой
энергии и легкость автоматического ^поддержания температуры;
равномерный нагрев всего рабочего пространства; отсутствие в
рабочем пространстве продуктов горения; простота обслуживания.
По способу преобразования электрической энергии в тепловую
лабораторные нагревательные приборы преимущественно
относятся к приборам сопротивления. Действие таких приборов
основано на выделении теплоты при прохождении тока по
нагревательному элементу, выполненному из проводников с большим
омическим сопротивлением. Количество теплоты, выделяемой в
нагревателе, и, следовательно, его температура, возрастают тем
значительнее, чем больше сила тока, протекающего по проводнику, и
чем больше сопротивление проводника.
Для каждого нагревательного прибора, в зависимости от
материала нагревателя и его сечения, а также от условий теплоотдачи,
существует предел силы тока, превышать который не следует во
избежание перегрева нагревателя.
Мощность нагревателей рассчитана на разогрев и поддержание
максимального температурного режима в рабочем пространстве.
При необходимости установить более низкую температуру
количество подводимой к нагревателю энергии должно быть уменьшено
регулятором напряжения.
Питание электронагревательных приборов производится от
сети переменного тока напряжением 127/220 В.
Различаются два типа нагревателей: открытый,
расположенный в рабочем пространстве, и закрытый,— вне рабочего
пространства.
158
Характер теплообмена между нагревателями и нагреваемым
материалом зависит от типа прибора и его температурного
режима. В высокотемпературных приборах при повышении
температуры рабочего пространства тепловая энергия передается главным
образом излучением нагревателей или стенок рабочего
пространства, и, частично, путем конвекции и теплопроводности.
В низкотемпературных приборах (сушильных шкафах и
термостатах) материал нагревается, главным образом, благодаря
конвекции движущегося нагретого воздуха. Поэтому для
равномерного нагрева должна быть обеспечена циркуляция воздуха внутри
рабочего пространства.
В печах (электрошкафах), в рабочем пространстве которых
создан вакуум, теплопередача конвекцией отсутствует, и
материал нагревается только излучением.
В жидкостных термостатах равномерный теплообмен
обеспечивается принудительной циркуляцией с помощью мешалок,
имеющих привод.
Электропечи сопротивления
Лабораторные печи сопротивления подразделяются на
камерные (СНОЛ), трубчатые (СУОЛ) и шахтные (СШОЛ). К
электропечам сопротивления относятся также сушильные электрошкафы
типа СНОЛ.
Основные технические показатели электропечей
сопротивления— размеры рабочего пространства, максимальная
температура нагрева и пределы диапазона регулирования температуры.
Для обеспечения безопасности работающих в лаборатории
металлические части электроустановок и корпуса
электрооборудования должны быть заземлены. Работа с незаземленными
электропечами запрещается. Каждый заземляемый элемент установки
должен быть присоединен к заземлителю с помощью отдельного
ответвления. Последовательное включение в заземляющий
проводник нескольких заземляемых частей установок запрещается.
Нельзя использовать для заземления водопроводную сеть или сеть
парового отопления.
В настоящем разделе будут рассмотрены лишь некоторые
электронагревательные приборы, наиболее часто используемые в
химических лабораториях.
Электропечи сопротивления камерные
лабораторные типа СНОЛ предназначаются для проведения
различных термических процессов и аналитических работ в
стационарных условиях до 900—1100 °С.
Печи состоят из металлического корпуса, в верхней части
которого находится рабочая камера, в нижней — пусковая и
контрольно-регулировочная аппаратура. Рабочее пространство электропечей
образуется разъемными огнеупорными фасонами. Пространство
между фасонами и корпусом заполнено теплоизоляцией. В па-
159
зах фасонов установлены четыре спиральных нагревателя,
соединенных последовательно между собой. Контроль и регулирование
температуры осуществляются показывающим и регулирующим
прибором, который связан с термопарой, установленной в
нагревательной камере.
Современные образцы печей типа СНОЛ выгодно отличаются
от ранее выпускавшихся муфельных печей более точной
автоматической регулировкой температуры (± 10 °С).
Электропечи сопротивления трубчатые лабораторные типа
СУОЛ предназначены для проведения различных термических
процессов в лабораторных условиях при температуре до 1250 °С.
Электропечь состоит из камеры нагрева и пульта управления.
Рабочее пространство образовано внутренней поверхностью
керамической трубы, на наружную поверхность которой намотан
проволочный нагреватель из сплава высокого омического
сопротивления, обмазанный термостойкой массой. Чтобы нагреватель не
контактировал с материалом теплоизоляции, блок нагревателя
вставляется в другую керамическую трубу большего диаметра.
Теплоизоляция электропечи двухслойная, выполнена из
кремнеземистых матов и перлитовой крошки. Корпус электропечи
выполнен из листовой стали и с внутренней стороны обложен
асбестовыми прокладками. Для предохранения от случайного
прикосновения к горячему корпусу электропечь снабжена защитным
фальшкожухом.
Автоматическое поддержание и регулирование температуры в
диапазоне 400—1250 °С, с точностью ±10°С осуществляется
показывающим и регулирующим милливольтметром от термопары,
установленной в пространстве между нагревателем и защитной
керамической трубой.
Электропечи сопротивления шахтные
лабораторные типа СШОЛ предназначены для плавки металлов и
термообработки различных материалов при температуре до 1250°С
в лабораторных условиях. Электропечи состоят из прямоугольного
корпуса, выполненного из
тонколистовой стали, в котором
размещены камеры нагрева и
блок управления. Точность
автоматического
регулирования температуры ±2°С.
Малогабаритные
тигельные печи являются
разновидностью электропечей
типа СШОЛ и служат для
тех же целей. О температуре
Рис. 81. Электропечь для микроанализа
типа СУОЛ:
J—рабочая камера; 2—керамические
фасоны; 5—металлический шток; 4— чугунная
подставка.
160
тигельной печи (в °С) можно судить приближенно по цвету
нагретого муфеля:
Начало красного каления Около 520
Темно-красное каление » 700
Ярко-красное каление » 950
Желтое каление » 1100
Ослепительно-белое каление » 1500
Электропечи для элементного микроанализа
Электропечи сопротивления трубчатые лабораторные типа
СУОЛ предназначены для элементного микроанализа
органических соединений и служат для нагрева кварцевых трубок и
сожжения находящихся в них органических соединений.
Электропечи входят в комплект приборов для элементного
микроанализа органических соединений АКОМ-5 для определения
углерода и водорода (ПМУВ); углерода, водорода и фтора
(ПМУВФ); кислорода и галогенов (ПОМКГ); азота по Дюма
(ПМА).
Электропечь для микроанализа типа СУОЛ
представлена на рис. 81. Температура в печи регулируется вручную
при помощи регулятора напряжения. Контроль температуры в
рабочей камере осуществляется посредством термопары ПП-1,
вводящейся через торцовую стенку электропечи в спиральный
нагреватель, и показывающего прибора.
В табл. 5 приведены технические данные трубчатых
электропечей для элементного микроанализа.
таблица 5. Трубчатые электропечи типа СУОЛ для микроанализа
Максимальная температура 1200 °С.
Печи
Разъемные
СУОЛ-0,15.0,6/12-МР
СУОЛ-0,15.1,1/12-МР
СУОЛ-0,15.1,4/12-МР
СУОЛ-0,15.2/12-МР
СУОЛ-0,25.1,1/12-МР
Неразъемные
СУОЛ 0,15.1,1/12 М
СУОЛ 0,15.2/12 М
Потребляемая
мощность,
\ Зт
540 ±50
625 ±60
675
950
600
350
350
! Рабочее
пространство
диаметр,
мм
!5
15
15
15
25
15
15
длина,
мм
60
110
140
200
НО
110 1
200
Масса,
кг
2,8
4.0
4,1
4,6
4,0
8,9
4,2
6 Зак. 1237
161
Сушильные электрические шкафы
Сушильные электрические шкафы предназначены для сушки
различных материалов, определения влажности твердых
материалов, суховоздушной стерилизации стеклянной и металлической
посуды и других работ, требующих нагревания до 100—350 °С и
автоматической регулировки температуры с точностью ±2—3 °С.
Они представляют собой теплоизолированные металлические
камеры прямоугольной или круглой формы с полками для
просушиваемых материалов.
Обогрев сушильных шкафов производится нагревателями
открытого или закрытого типов.
Тепло от нагревателя открытого типа к высушиваемому
материалу передается, в основном, конвекцией, поэтому материал
следует располагать на полках, оставляя пространство для
беспрепятственной и равномерной циркуляции воздуха. Подвод
сухого воздуха в сушильный шкаф и удаление продуктов испарения
происходит через отверстия, имеющие регулировочные заслонки.
Во всех сушильных шкафах заданная температура
поддерживается автоматически с помощью терморегулятора. Нагреватель
сушильных электрических шкафов рассчитан на рабочее
напряжение 220 В. Включение на большее напряжение не допускается.
Шкаф сушильный электрический круглый
модели 2В-151 представлен на рис. 82. Пределы регулирования
температуры от 40 до 200 °С с точностью ±1 °С.
Рабочая камера обогревается основным нагревателем,
намотанным на наружной поверхности камеры на изолирующей
прокладке из листового асбеста, и дополнительным нагревательным
элементом, расположенным на
задней стенке камеры. Пространство
между корпусом и рабочей
камерой заполнено стекловатой.
Для автоматического
поддержания температуры шкаф снабжен
терморегул ирующим устройством,
которое состоит из механической
и электрической частей. Действие
механической части основано на
использовании различного
температурного удлинения алюминиевой
лодочки и стального стержня.
Верхний конец стального стержня
прикреплен к лодочке, а нижний
Рис 82. Электрический сушильный круглый шкаф
2В-151:
/ — корпус; 2 — подставка; 3—рабочая камера; 4
—металлическая оправа термометра; 5 —нагреватель;
6 —алюминиевая лодочка; 7 —стальной стержень;
в —контактная пластина; 9 — пробка.
К заземлению *К се/тщ
162
Рис. £3. Сушильно-стерилизационный шкаф ШСС-80п:
/ — корпус; 2 — подставка; В—пульт управления.
воздействует на контактную пластину,
размыкая контакты при повышении
температуры в рабочей камере.
Замыкание контактов при понижении
температуры в рабочей камере
происходит под действием пружины. Для
удобства регулировки верхний конец
стержня выведен в отверстие
наружного корпуса, которое закрывается
пробкой. В нижней части рабочей
камеры имеется отверстие для притока
воздуха.
Электрическая часть терморегулируюшего устройства
расположена в подставке шкафа. На передней стенке подставки
расположены сигнальная лампа, выключатель, предохранители и
ручка терморегулятора со шкалой. Шкала служит для ориентировки
при настройке работы шкафа на требуемую температуру. В
процессе работы можно установить, какое деление шкалы
соответствует определенной температуре в рабочей камере (по
наружному термометру). Сигнальная лампа зажигается при нагревании
шкафа.
Шкаф сушильно-стерилизационный ШСС-80п
представлен на рис. 83. В корпусе шкафа расположена рабочая
камера объемом в 80 л, в которой установлены полки для
обрабатываемых предметов и нагревательные элементы. Дверь корпуса,
закрывающая рабочую камеру, снабжена теплоизоляцией и
уплотнением. В верхней части корпуса находятся втулки для установки
термометров. Во втулках имеются отверстия для выхода паров в
процессе сушки.
В пульте управления смонтированы электронный
терморегулятор на транзисторах и схема коммутации нагревательных
элементов. В задней стенке кожуха пульта имеется люк для доступа к
предохранителям. В верхней части лицевой панели пульта
расположена розетка для подключения электроконтактного термометра.
На лицевой панели находятся кнопки включения и выключения
шкафа, кнопка ускоренного нагрева и сигнальные лампы.
При сушке материалов, для которых температура нагрева
ограничена из-за разложения вещества, а также при
необходимости ускорить процесс сушки, применяются электрические
шкафы, в рабочем пространстве которых создается и поддерживается
вакуум механическим вакуум-насосом.
Для сушки и нагревания различных материалов в вакууме
при температуре от 80 до 200 °С используется вакуум-шкаф
ВШ-0,035М (рис. 84). Рабочая камера нагревается переменным
током напряжением 220 В. Нагревательный элемент намотан на
внешней поверхности рабочей камеры, закрывающейся крышкой,
6* 163
Рис* 84. Вакуумный сушильный шкаф ВШ-0,035М:
/ — корпус; 2—подставка; 3— стрелочный вакуумметр; 4—термометр; 5 — патрубок; 6 —
крышка; 7—трубка; «—терморегулятор; 5 —сигнальная лампа.
Рис. 85. Схема установки вакуумного сушильного шкафа:
/ — сушильный шкаф; 2—вакуумный насос ВН-461; 3 — ловушка; 4 — соединительная муфта;
5— хомут в сборе; 5 —трубка отсоса; 7—впускной кран; 5—вакуумметр; 9—термометр.
имеющей уплотнение из резины. Место соприкосновения резины
с рабочей камерой охлаждается проточной водой через трубку.
Расход воды составляет 50—60 л/ч. Термометр устанавливается
в снабженный вакуумным уплотнением патрубок. Остаточное
давление в рабочей камере замеряется стрелочным вакуумметром,
шкала которого выполнена в относительных единицах. При
остаточном давлении 1,33 кПа (10 мм рт. ст.) стрелка вакуумметра
должна находиться около цифры 1. По окончании работы воздух
вводится в рабочую камеру игольчатым краном, расположенным в
ее центре. Температура регулируется автоматически с помощью
терморегулятора дилатометрического типа. Схема установки
шкафа в лаборатории изображена на рис. 85.
Термостаты
Термостаты —электронагревательные приборы, в рабочем
пространстве которых с высокой точностью поддерживается
определенная температура. В зависимости от среды, заполняющей
термостат, они подразделяются на воздушные и жидкостные.
Воздушные термостаты предназначены для
проведения различных работ в интервале температур 20—80 °С. Рабочее
пространство воздушного термостата, в которое загружаются
исследуемые материалы, представляет собой прямоугольную камеру,
часто имеющую водяную рубашку. Температура внутри
термостата регулируется нагревом водяной рубашки, в которой
размещаются водонепроницаемые электронагреватели. Это обеспечивает
равномерное распределение температуры по всей поверхности
стенок камеры термостата. Заданная температура с точностью
+0,1 °С поддерживается терморегулятором.
164
Жидкостный термостат представляет собой бачок,
наполненный рабочей жидкостью (вода, масло). Теплота
подводится нагревателем, а отводится холодильником, расположенными в
жидкости. Постоянная температура в рабочем пространстве
обеспечивается перемешиванием жидкости пропеллерной мешалкой,
приводимой в движение электродвигателем, и поддерживается
автоматически с помощью терморегулятора.
Аппараты и сосуды, в которых нужно поддерживать
постоянную температуру, погружают в рабочую жидкость. Можно также
подавать рабочую жидкость по трубкам из термостата в другие
аппараты и сосуды для поддержания в них постоянной
температуры (например, в рефрактометрах, вискозиметрах и т.п.).
Электрический термостат ЗЦ-1125М с водяной
рубашкой состоит из каркаса, внутренней и наружной дверей,
блока управления и подставки. Внутри каркаса установлена
рабочая камера с водяной рубашкой. Объем воды в рубашке 96 л.
В нижней части водяной рубашки расположены
электронагреватели. Устанавливается термостат на подставке из стальных труб.
Термостат снабжен терморегулятором. Диапазон автоматически
регулируемых температур 28—55 °С, точность ±0,5 °С.
Ультратермостат УТ-15 представлен на рис. 86. Режим
работы автоматический. Объем рабочего сосуда не менее 15 л.
Диапазон рабочих температур 0—150 °С, точность ±0,5 °С.
Для работы при температуре ниже температуры проточной
водопроводной воды в комплект ультратермостата входит
специальное устройство — охладитель. В качестве хладоносителя
используются лед или твердый диоксид углерода («сухой лед»).
165
Рабочая жидкость охлаждается за счет перекачивания ее через
змеевик, установленный в сосуде с хладоносителем.
Состав рабочей жидкости зависит от заданной температуры
термостатирования:
Рабочая жидкость
От 0 до 5—8 °С 30% раствор глицерина
в воде
От 5—8 до 75—80 °С Дистиллированная вода
От 75—80 до 150 °С 80% раствор глицерина в
воде
Корпус ультратермостата должен быть надежно заземлен.
Категорически запрещается включение ультратермостата в сеть
постоянного тока.
За работой ультратермостата должен осуществляться
периодический контроль.
Для термостатирования небольших объемов жидкости
выпускаются малогабаритные термостаты МТ-1,2 и МТ-0,3. Рабочим
сосудом микротермостата МТ-0,3 (рис. 87) служит
стеклянный стакан объемом 300—500 см3 с нанесенной на наружную
поверхность токопроводящей прозрачной пленкой — нагревательным
элементом. Для подведения напряжения (220 В) к пленке на
стакан нанесены две контактные полосы. Рабочая жидкость в
стакане перемешивается с помощью электромагнитной мешалки.
Терморегулирующее устройство позволяет устанавливать
следующие рабочие температуры: 37 ±г 0,5, 56 ± 0,6 и 85 ± 1,5 °С.
Термостатирование жидкости может быть достигнуто также с
помощью погружного устройства УТП-1, состоящего из
блока нагрева и перемешивания и блока управления. Блок
нагрева и перемешивания — узел с центробежным насосом, витыми
трубчатыми нагревательными элементами и датчиком
температуры. Блок управления — комплекс узлов и деталей
терморегулятора, смонтированный на дюралюминиевом шасси. Диапазон
устанавливаемых и автоматически регулируемых температур 25—85 °С.
Допускаемая погрешность в диапазоне 25—55 и 55—85 °С равна
±0,1 и ±0,25 °С соответственно. Вместимость рабочих сосудов,
используемых для погружного устройства, 5—25 л.
Приборы для прямого нагревания жидкостей
Прямое нагревание жидкостей большей частью
осуществляется с помощью закрытых электрических нагревателей
(электроплитки, колбонагреватели, электрокипятильники погружные).
Электрическая плитка обладает большой
теплоемкостью, а передача тепла от нагревательной спирали к жидкости
происходит через несколько поверхностей раздела, так что
температура регулируется со значительным запаздыванием и
периодически колеблется около заданного значения. Диапазон
колебаний несколько уменьшается с увеличением количества
нагреваемой жидкости, но и в этих случаях составляет ±2—5 °С.
166
В химических лабораториях большей частью используются
одноконфорочные настольные электроплитки с закрытым
нагревательным элементом и переключателем мощности, например
ПКЭ-800/3 и «Экран».
Электроплитки, как и другие настольные
электронагревательные приборы, следует ставить на асбестированную прокладку или
на лист теплоизолирующего материала.
Жидкость в толстостенном сосуде или в сосуде из материала,
плохо проводящего тепло, нельзя нагревать на электроплитке*
В таких случаях часто используют погружные
электрокипятильники бытового типа ВПМ или ВПН номинальной
мощности 0,3; 0,6 и 1 кВт. Они имеют меньшую теплоемкость,
нежели обычные электроплитки, и передача тепла жидкости
осуществляется лучше.
Погружной электрокипятильник нельзя включать в сеть, если
он не погружен в жидкость, потому что в воздухе температура
нагревателя может сильно возрасти, и он перегорит. Нередко
случается, что при длительном нагревании воды и водных
растворов нарушается изоляция между нагревателем и металлическим
кожухом, поэтому при погружении руки в сосуд с жидкостью
может произойти поражение электрическим током.
Электрокипятильник можно вынимать из жидкости, только отключив его от сети.
В последнее время выпускают колбы с прямым
электрообогревом. Нагревательный элемент встроен в дно колбы
и присоединяется непосредственно к источнику тока.
Прямой нагрев жидкости может быть также осуществлен
теплоизлучателями. Когда требуется не очень
интенсивное нагревание (например, при перегонке диэтилового эфира), в
качестве теплоизлучателей можно применять электрические
лампы накаливания.
Для обогревания перегонных и реакционных колб применяют
инфракрасные излучатели. Подобные излучатели
применяются также для высушивания твердых тел (см. гл. 18) и
испарения жидкостей. В зависимости от конструкции лампы и
расстояния между ней и нагреваемым предметом можно варьировать
температуру от 40 до 200 °С. Применение инфракрасных
излучателей особенно удобно при работе с огнеопасными веществами.
Для нагревания веществ в круглодонной стеклянной посуде
применяют электрические колбонагреватели с закрытыми и
открытыми нагревателями. Они имеют конусообразное
углубление в середине. По поверхности конуса расположена
нагревательная спираль, почти целиком погруженная в керамику.
ГАЗОВЫЕ ГОРЕЛКИ
Для выполнения ряда операций (прокаливание, термическая
обработка стекла и др.) часто используется нагрев природным
горючим газом, главная составная часть которого — метан. При
отсутствии газопровода с природным горючим газом, в качестве
167
отопительного газа применяют смесь газовых нефтяных
фракций— пропана-бутана, поставляемых в сжиженном виде в
баллонах. Теплота сгорания смеси пропан-бутан приблизительно в
два раза больше теплоты сгорания природного газа. Температура
пламени газа зависит от его состава и полноты сгорания.
Температуру можно регулировать подачей воздуха (кислорода).
Непосредственное нагревание стеклянных сосудов открытым
пламенем газовой горелки применяется очень редко ввиду малой
термической стойкости стекла и неравномерности нагревания. При
этом из-за местных перегревов возможно частичное разложение
нагреваемых веществ. Иногда вещества в стеклянной посуде
нагревают газовой горелкой, устанавливая сосуды на асбестирован-
ные сетки или тонкие листы асбеста. Открытым пламенем обычно
пользуются при прокаливании веществ в кварцевой, фарфоровой
или металлической посуде.
В лаборатории обычно применяют газовые горелки трех типов
(рис. 88). Горелки типа I (Бунзена) и II (Теклю) различаются
только устройством регулировки подсоса воздуха. Значительно
эффективнее как природный газ, так и пропан-бутановая смесь
сгорают в горелках типа III(горелка Меккера).
В горелках типа I и II наибольшая температура пламени во
внешней части (зона окисления) и наименьшая — в нижней
внутренней части (зона восстановления). Максимальная температура
нагрева для горелок типа III выше, так как горячая сетка
катализирует горение и сосредоточивает его вблизи головки.
Отечественная промышленность в настоящее время выпускает
усовершенствованную модель газовой горелки типа Бунзена для
работы с природным газом и универсальную горелку для работы
с различными видами отопительных газов.
Воздух
Газ
Рис. 88. Различные типы лабораторных горелок.
Тип I: / — цилиндрическая заслонка для регулировки воздуха; 2—камера смешения;
3—инжектор.
Тин II: /—диск с винтовой нареакой для регулировки подачи воздуха; 2 — винт для
регулировки подачи газа; 3—инжектор; 4—камера смешения.
Тип. III: /—отверстие для засоса воздуха; 2 — камера смешения; 5—труба для
принудительной подачи воздух*; *—инжектор; *—г*ловка с сеткой.
168
Рис. 89. Насадки к газовым горелкам.
Х1'
Для получения пламени различной формы горелки снабжают
насадками (рис. 89), При проведении простейших операций со
стеклом пользуются паяльной горелкой.
При зажигании газовой горелки сначала, закрыв доступ
воздуха, подносят к ней зажженную спичку, затем медленно
открывают газовый кран и регулируют доступ воздуха таким образом,
чтобы получилось несветящееся пламя. При несоблюдении
рекомендуемого порядка зажигания возможен «проскок» пламени—■
газ горит на месте ввода воздуха в горелку. В результате
«проскока» горелка сильно нагревается, что может привести к
расплавлению и воспламенению газоподводящей резиновой трубки. В таких
случаях нужно немедленно закрыть газовый кран и только после
того, как остынет горелка, зажечь ее вновь. Зажигать газовые
горелки разрешается только спичками или другой горящей
горелкой. Перенос пламени бумагой запрещается.
ЖИДКОСТНЫЕ ГОРЕЛКИ
В последние годы жидкостные горелки — бензиновые и
керосиновые— не используются в практике химических лабораторий.
Исключение составляют стеклянные и металлические спиртовые
лампочки (спиртовки).
Спиртовые стеклянные лампочки выпускаются двух
типов: СЛ-1 со стеклянным колпачком и СЛ-2 с фенопластовым
колпачком и подставкой из стальной проволоки. Спиртовая
металлическая лампочка состоит из стального корпуса и таганчика,
фитиль закрывается пластмассовым колпачком. Номинальная
вместимость спиртовок 100 мл.
Обращение со спиртовками несложно. Необходимо следить за
тем, чтобы у спиртовой лампочки был хорошо притертый
колпачок, предохраняющий спирт от испарения, и исправный фитиль,
сделанный не из марли или ваты, а из некрученых хлопчатобу*
мажных нитей. Фитиль вставляется в жестяную трубку
светильника так, чтобы не забивая трубки плотно, он тем не менее
заполнял ее и не мог сам по себе спускаться в резервуар спиртовки.
НАГРЕВАТЕЛЬНЫЕ БАНИ
В тех случаях, когда необходимо вести нагревание в течение
длительного времени при определенной температуре, обычно
применяют различные нагревательные бани с теплоносителем. В
зависимости от теплоносителя, различают бани воздушные,
жидкостные, металлические, солевые, песчаные, паровые.
169
Рис. 90. Воронка Бабо для воздушной бани:
/—внутренняя стенка из черной жести; 2 — ребра из асбеста; 3—желез-
ная пластинка с асбестом; 4—отверстия для выхода горячего воздуха.
Воздушные бани
Из всех теплоносителей, используемых в нагревательных
банях, воздух обладает наименьшей теплопроводностью, и переход
тепла от горячего воздуха к обогреваемому предмету
относительно мал. Для передачи больших количеств тепла воздушную баню
необходимо нагревать до высокой температуры, но это может
вызвать местные перегревы.
В качестве воздушной бани в лабораториях, оснащеяных
газовыми горелками, применяют так называемые воронки Бабо
(рис. 90). Горячие газы проходят снизу через щели между
асбестовыми полочками и нагревают колбу, опирающуюся на полоски
асбеста. Горизонтальная жестяная пластинка защищает колбу от
открытого пламени. Помещая воронку Бабо в вертикальный
цилиндр, можно добиться постоянной температуры бани.
Когда требуется не очень сильное нагревание (перегонка ди-
этилового эфира, ацетона и др.), можно применить воздушную
баню, в которой нагревание осуществляется обычной электрической
лампой накаливания. Прибор для нагревания можно изготовить
из жестяного конического сосуда.
Жидкостные бани
В качестве теплоносителей для жидкостных бань используют
воду и водные солевые растворы, минеральные масла, силиконовое
масло, глицерин, три-о-крезилфосфат, дибутилфталат, фосфорную
кислоту и др. Теплоноситель помещают в металлические кастрюли
или специальные медные (латунные) цилиндрические или
конические сосуды, закрываемые сверху набором металлических колец
(конфорок). Это дает возможность подобрать нужный диаметр
отверстия бани, когда необходимо погрузить нагреваемый сосуд в
жидкий теплоноситель.
Водяная баня пригодна для нагревания веществ почти до
температуры кипения воды (до 100 °С) и для перегонки
жидкостей, кипящих не выше 80 °С.
Растворяя в воде некоторые соли (NaCl, СаС12 и др.)» можно
значительно повысить температуру нагревания. Например, при
заполнении бани насыщенным раствором NaCl можно повысить
температуру до 108°С, а 50% раствор СаС12 позволяет доводить ее
до 130°С. Водные растворы солей обладают серьезными
недостатками: они усиливают коррозию металлических стенок бань, а при
охлаждении часто затвердевают, при этом стеклянный нагреваемый
сосуд может быть раздавлен, если его не вынули из бани до ее
охлаждения. Нагревание на водяной или водно-солевой бане
сосудов со щелочными металлами категорически запрещается.
170
Водяные бани бывают одно- и многоместные с электрическим
или газовым обогревом и часто имеют устройства для
автоматического регулирования постоянного уровня воды, действующие по
принципу сообщающихся сосудов.
Для нагревания в интервале температур 100—240°С
применяют масляные бани. В качестве теплоносителя чаще всего
применяют минеральное масло с температурой воспламенения не
ниже 300 °С. Необходимо, чтобы масла были стойкими в требуемом
интервале температур, нетоксичными, прозрачными,
легкодоступными, с возможно более низким давлением пара, малой вязкостью,
высокой удельной теплоемкостью. Масла не должны вызывать
коррозию металлов. Этим требованиям в наибольшей степени
отвечает цилиндровое масло-52 («Вапор»). Часто применяемое в
лабораторной практике вазелиновое масло сильно дымит уже при
200 °С и при длительном пользовании окрашивается в желтый или
коричневый цвет и становится вязким. Даже «Вапор» со временем
становится вязким, поэтому его необходимо менять через
некоторое время.
Более эффективным теплоносителем является силиконовое
масло. Силиконовую баню можно нагревать до 300 °С без заметного
изменения цвета и вязкости масла при длительном пользовании.
Нагревание на масляной бане следует проводить в вытяжном
шкафу, так как пары масла неприятны и вредны. В масляную
баню необходимо поместить контрольный термометр, на котором
красной чертой отмечена температура, выше которой нагревать
опасно. Баню заполняют маслом до половины объема, так как при
нагревании масло расширяется и избыток масла может
перелиться через край и воспламениться. Нагреваемый сосуд помещают в
баню таким образом, чтобы уровень вещества в сосуде был на
одном уровне с маслом.
При нагревании необходимо следить за тем, чтобы в масло не
попала вода или другая низкокипящая жидкость. Уже от
нескольких капель воды горячее масло начинает сильно пениться,
разбрызгиваться и может воспламениться. Опасность воспламенения
паров масла можно уменьшить, прикрывая баню двумя
полукольцами, вырезанными из асбестового картона по размеру бани. По
окончании нагревания сосуд вынимают из горячей бани, дают
маслу стечь со стенок и кусочками фильтровальной бумаги или ватой
снимают оставшееся на поверхности сосуда масло.
Часто в жидкостных банях в качестве теплоносителя
используют глицерин. Глицерин кипит при 290°С, очень гигроскопичен и
растворим в воде. Глицериновые бани можно нагревать до
200°С; при более высокой температуре он начинает разлагаться с
образованием акролеина, вызывающего слезотечение и кашель.
Глицерин легко смывается с посуды водой.
Три-о-крезилфосфат, кипящий при 410 °С (с
разложением), более устойчив, чем минеральные масла и глицерин.
Три-о-крезилфосфат безопасен в пожарном отношении и может
быть нагрет до 300 °С без заметного изменения цвета и свойств.
171
В качестве теплоносителя для жидкостных бань в интервале
температур от —10 до +180°С рекомендуют этиленгликоль,
а в интервале от 0 до 250°С—триэтиленгликоль.
Этиленгликоль и триэтиленгликоль растворяются в воде, не вызывают
коррозию и бесцветны.
Солевые бани
Для нагревания до температур, превышающих 300 °С,
используют расплавленные соли.
Солевая баня, заполненная смесью 48,7% (масс.) NaNC>3 и
51,3% KN03, применима в интервале температур от 230 до 500 °С,
а баня, заполненная смесью 40% NaN02, 7% NaN03 и 53%
KNOs,—от 150 до 500 °С.
При работе с солевыми банями нужно учитывать следующее:
нагреваемые сосуды должны быть из термически устойчивого
стекла; сосуды погружают в жидкий расплав, а вынимают из еще
горячего расплава; попадание горючих органических веществ в
расплав нитратов и нитритов вызывает мгновенное воспламенение,
причем горение может принять характер взрыва.
Металлические бани
Металлические бани используют для тех же целей, что и
солевые. В качестве теплоносителей используют низкоплавкий сплав
Вуда (50% Bi, 25% Pb, 12,5% Sn и 12,5% Cd) с гпл=65,5°С или
сплав Розе (50% Bi, 25% Pb и 25% Sn) с /ПЛ=94°С.
Металлические бани обладают сравнительно низкой температурой плавления,
а высокая теплопроводность металлов способствует очень быстрой
передаче тепла.
Недостатки металлических теплоносителей: высокая плотность
и окисление при повышенной температуре в присутствии воздуха
(образующиеся оксиды загрязняют поверхность нагрева и
ухудшают теплоотдачу).
Металлические бани используют главным образом для
нагревания небольших колб; сосудом для бани может служить
железная чашка. По окончании нагрева колбу и термометр вынимают
из еще горячего сплава (прежде чем он затвердеет). Работа с
металлическими банями должна производиться в вытяжном шкафу.
Песчаные бани
Для осторожного нагревания до высокой температуры часто
пользуются песчаными банями. В металлическую чашку, дно
которой покрыто слоем сухого мелкозернистого песка, помещают
нагреваемый сосуд и засыпают его со всех сторон песком (часто по
ошибке пытаются поместить сосуд в уже наполненную песком
баню). Песчаную баню нагревают пламенем газовой горелки или
электрической плиткой.
172
Передача тепла при толстом слое песка недостаточна, поэтому
полусферическая форма песчаной бани для круглодонных колб
предпочтительнее плоской формы. Песчаные бани прогреваются
неравномерно, вследствие чего при работе с ними трудно
поддерживать постоянную температуру. Ценное преимущество песчаных
бань — химическая индифферентность; они значительно безопаснее
масляных бань.
В последнее время в качестве твердых проводников тепла для
нагревательных бань предложены различные разновидности
графита, обладающие высокой жаростойкостью наряду с тепло-
и электропроводностью.
СРЕДСТВА И ПРИБОРЫ ДЛЯ ОХЛАЖДЕНИЯ
При перегонке, экстракции и ряде других процессов газы и
пары охлаждают при помощи разнообразных холодильников
(см. гл. 2).
Для охлаждения поверхности нагретых колб проточной водой
служат свинцовые кольцевые трубки (рис. 91). Если пары
необходимо охладить до температуры ниже температуры
проточной воды, то воду заменяют другим охлаждающим агентом.
Для охлаждения до —20 °С можно использовать обычные
водяные холодильники, через которые пропускают
концентрированный раствор NaCl, этиловый спирт или другую жидкость, которую
предварительно охлаждают, пропуская жидкость через
спиральную трубку, погруженную в сосуд Дьюара с охладительной
смесью.
Низкую температуру охлаждающей смеси можно получить при
помощи твердого диоксида углерода — «сухого льда» (до —80 °С)
или жидкого азота (до —195,8 °С). Жидким воздухом и в
особенности кислородом, как правило, редко пользуются, они образуют
с горючими веществами взрывоопасные смеси.
Охлаждающие смеси обычно готовят в стеклянном сосуде
Дьюара.
Лед, вследствие высокой теплоты плавления,— ценное
охлаждающее средство. Хорошее охлаждение достигается при тесном
контакте охлаждаемо^ поверхности со льдом. Для этого куски
льда разбивают деревянном молотком в полотняном мешочке до
зерен величиной в горошину и добавляют небольшое количество
воды, чтобы получилась кашица.
Рис. 91. Свинцовая кольцевая трубка для охлаждения
поверхности колб:
с —свинцовая кольцевая трубка с отверстиями;
б—охлаждение колбы проточной водой.
178
Водные растворы, где допустимо небольшое разбавление,
можно быстро охладить, добавляя кусочки чистого льда
(полученного из дистиллированной воды) непосредственно в реакционную
смесь.
Если вещество необходимо охладить до температуры несколько
ниже 0°С, то применяют смесь льда (снега) с некоторыми
электролитами (соли, кислоты). При контакте электролитов со льдом
сначала возникают небольшие количества концентрированных
водных растворов, которые замерзают при более низкой
температуре, чем чистая вода. Это приводит к ускоренному таянию
льда, что сопровождается поглощением тепла. В табл. 6
приводятся данные о предельно низких температурах охлаждающих
смесей электролитов со льдом (снегом).
При отсутствии льда охлаждающую баню можно приготовить,
используя высокие теплоты растворения некоторых солей в воде.
Так, например, 42,9% раствор NaN03 понижает температуру до
—5,3°С; 60% раствор KSCN —до —23,7°С; 37,5% раствор
NH4N08 —до —13,6°С.
В качестве охлаждающей смеси можно использовать раствор
Na2SO4-10H2O в конц. НС1. При содержании 39% (масс) соли
температура понижается до —8,1 °С; 50,22% соли — до —12,2°С;
63% соли —до —15,3°С.
При растворении 5 масс. ч. Na2SO4-10H2O в 4 ч. 66%
раствора H2S04 предельно низкая температура достигает —17°С.
Известно, что испарение низкокипящих жидкостей за счет
тепла окружающей среды сопровождается интенсивным охлаждением.
Жидкий аммиак (/Кип=—33,35°С) при выливании из баллонов
в сосуд Дьюара испаряется так быстро, что охлаждается ниже
температуры кипения. После этого жидкий аммиак можно долго
сохранять в открытом сосуде Дьюара в вытяжном шкафу и
использовать для охлаждения веществ до —30 °С. Жидкий аммиак
таблица 6. Охлаждающие смеси со льдом (снегом)
Вещество
NaCi
СаС12-6Н20
КС1
NH4C1
KN03 + NH4CI
NH4NO3 + NaN03
NH4CI + NaCl
KNO3+NH4SCN
NH4NO3 + NaCl
66% раствор H2S04
Массовые
льда
100
100
100
100
100
100
100
100
100
100
90,1
81,2
69,0
52,2
части
вещества
33
81
123
30
25
13,5 + 26
25 + 55
20 + 40
2+ И2
41,6 + 41,5
9,9
18,8
31,0
47,8
Предельно низкая
температура, °С
—21,3
-21,5
-40,3
-10,9
— 15,4
-17,8
-25,8
-30,0
-34,1
-40,0
-19,0
-25,0
-31,0
-37,0
174
Рис. 92. Устройство для передавливания жидкого азота:
/, 2—сосуды; 3, 4—трубки
при соприкосновении с кожей вызывает сильные
ожоги. Особенно опасно попадание аммиака в
глаза. Смеси аммиака с воздухом взрывоопас
ны.
В качестве охлаждающего средства широкое
распространение в химических лабораториях
получил твердый диоксид углерода («сухой лед»). Твердый С02
возгоняется при —78,8 °С. Для непосредственного охлаждения
один «сухой лед» непригоден, так как он плохо проводит тепло
и не позволяет достигнуть хорошего контакта со стенками
охлаждаемого сосуда. Кроме того, твердый С02 во влажном воздухе
быстро обволакивается слоем льда, поэтому твердый С02
используют в смесях с некоторыми органическими растворителями, что
значительно повышает теплообмен.
Твердый С02 в настоящее время поступает в продажу в виде
блоков или больших кусков. Их измельчают деревянным молотком
и небольшими порциями переносят деревянной ложкой в сосуд
Дьюара, куда заранее внесен безводный растворитель (ацетон,
этиловый спирт, диэтиловый эфир).
«Сухой лед» добавляют при перемешивании до образования
кашицы; при этом смесь не должна вспениваться. «Сухой лед»
можно получить, используя баллон с жидким С02. Для этой цели
к выпускному патрубку баллона привязывают прочный
парусиновый мешок и по возможности полностью открывают вентиль
перевернутого вниз горлом баллона. Газ с сильным шипением
стремительно выходит в мешок; в это время нужно сильно ударять по
мешку, чтобы на его стенках не отлагался твердый диоксид
углерода, который может закупорить поры ткани. Твердый С02
сохраняют в сосуде Дьюара.
Следует иметь в виду, что в холодной смеси твердого С02 с
растворителем (/» —70°С) заметно конденсируются пары воды,
и время от времени растворитель следует обезвоживать.
В последнее время получили распространение
низкотемпературные бани на основе жидкого азота (£Кип=—195,8 °С). Жидкий
азот сохраняют в металлических сосудах Дьюара. Переливать
жидкий азот из металлического сосуда Дьюара в стеклянный
меньшей вместимости рекомендуют путем передавливания газом, как
показано на рис. 92.
В горло сосуда 1 вставляют резиновую пробку с двумя
трубками 3 и 4. Слегка зажимая пальцем трубку 4, повышают
давление в сосуде /. За счет повышения давления жидкий азот
передавливается в сосуд 2.
Жидкий азот хранят в сосудах Дьюара, горло которых
негерметично прикрывают войлочной крышкой. Следует следить за тем,
свободно ли газ выходит из сосуда. На ночь сосуд Дьюара с жидким
азотом рекомендуют вообще не закрывать, так как намерзающие
175
ТАБЛИЦА 7. Низкотемпературные бани на основе жидкого азота
Жидкость
Анилин
Тетрахлорэтан
о-Ксилол
Бромбензол
Ацетонитрил
Хлорбензол
Предельно
низкая
температура, °С
—6
-22
-29
-30
-41
-45
Жидкость
1
ж-Ксилол
Трихлорметан
Бутилацетат
Этилацетат
Нитроэтан
1
Предельно
низкая
температура, °С
-50
-63
-77
-84
-90
Жидкость
Гептан
Метанол
Этилбромид
Пентан
Изонентан
Предельно
низкая
температура, °С
-91
-98
-119
-131
— 160
на войлоке пары воды могут создать герметичную пробку, что
повлечет за собой повышение давления и разрушение сосуда.
Металлические сосуды Дьюара устойчивы к быстрому
изменению температуры, которое для стеклянных сосудов нередко бывает
причиной разрушения. Если необходимо перелить жидкий азот в
теплый стеклянный сосуд Дьюара, его следует предварительно
охладить током холодного газообразного азота, затем — небольшим
количеством жидкого. Лишь после этого можно переливать
жидкий азот. Следует также избегать переливания жидкого азота
через край стеклянного сосуда Дьюара, так как это может привести
к растрескиванию обратного спая и взрыву сосуда. При работе с
сосудом Дьюара необходимо надевать защитные очки.
Низкотемпературные бани на основе жидкого азота получают,
вливая жидкий азот в растворитель и перемешивая до тех пор,
пока не образуется вязкая масса. Температуру бани можно
поддерживать постоянной за счет периодического добавления
жидкого азота, следя за тем, чтобы смесь оставалась вязкой.
В табл. 7 приведены данные о предельно низкой температуре
бань из смесей азота с некоторыми органическими жидкостями.
В лабораторной практике часто возникает необходимость в
хранении некоторых веществ при низкой температуре, а в ряде
случаев и в получении небольших количеств льда. Для этих целей,
как правило, используются бытовые электрические холодильники.
глава и. ИЗМЕРЕНИЕ И РЕГУЛИРОВАНИЕ ТЕМПЕРАТУРЫ
Измерять температуру практически возможно лишь методом
сравнения нагретости двух тел, причем степень нагретости одного
предполагается известной.
В настоящее время применяют две температурные шкалы;
термодинамическую и международную практическую, введенную как
обязательную с 1 января 1971 г. В термодинамической шкале
Кельвина начальная точка отсчета — точка абсолютного нуля
(точка, лежащая на 273,16° ниже точки плавления льда — 0,01 °С).
Единственная экспериментальная точка в этой шкале — тройная
точка воды, при которой все три фазы воды (твердая, жидкая и
газообразная) находятся в равновесии, и для которой установлено
числовое значение 273,16 К. По размеру кельвин равен градусу
Цельсия.
На основе термодинамической шкалы для практических целей
установлена международная практическая температурная шкала.
Эта шкала основана на 11 постоянных (реперных) точках
температуры, которые служат для калибрования термометров и
термопар [13].
Температура по термодинамической и международной шкале
может быть выражена как в Кельвинах (К), так и в градусах
Цельсия (°С), в зависимости от начала отсчета на шкале. Соотношение
между абсолютной температурой Т, выраженной в Кельвинах, и
температурой t, выраженной в градусах Цельсия:
Г = * + 273,15
КЛАССИФИКАЦИЯ ПРИБОРОВ ДЛЯ ИЗМЕРЕНИЯ ТЕМПЕРАТУРЫ
[46, с. 156]
Температуру измеряют с помощью термометров. По принципу
действия термометры, в соответствии с ГОСТ 13417—76, делятся
на следующие группы: дилатометрические, манометрические,
электрические (термоэлектрические и термометры сопротивления), пи<
рометры — термометры излучения.
По методам измерения температуры приборы можно разделить
на две группы: контактные и бесконтактные. К бесконтактным от*
носятся пирометры.
В последние годы выявилась еще одна группа средств для
ориентировочного измерения температуры поверхности нагретых тел
контактным методом — термохимические, изменяющие окраску при
изменении температуры.
177
ЖИДКОСТНЫЕ ТЕРМОМЕТРЫ РАСШИРЕНИЯ
Чаще всего в химических лабораториях применяют
жидкостные термометры расширения.
Для измерения температуры от —35 до +350°С используют
ртутные термометры; при помощи ртутных термометров из
кварцевого стекла, заполненных азотом под давлением до 2—7 МПа
(газонаполненные термометры), можно измерять температуру до
650 °С.
Для измерения температур ниже —30°С применяют
жидкостные нертутные термометры, наполненные этиловым спиртом
(от +65 до —70 °С), толуолом (от +90 до —90 °С), петролейным
эфиром или пентаном (от +20 до —190°С). Их часто называют
«минусовыми». Выбор рабочей жидкости определяется диапазоном
измеряемых температур. Учитывая высокое давление паров
нертутных термометрических жидкостей, капилляры термометров
заполняют азотом под давлением. Следует иметь в виду, что для
определения температуры такими термометрами требуется больше
времени, чем в случае ртутных, так как эти жидкости обладают
значительно меньшей теплопроводностью и большей вязкостью,
чем ртуть. Поскольку нертутные жидкости, в отличие от ртути,
смачивают поверхность стекла, охлаждать термометры можно только
медленно, погружая сначала лишь шарик, иначе возможна
значительная ошибка.
Точность измерения при умеренно низких температурах
составляет ±0,5—1°. Термометры со смачивающими жидкостями
рекомендуется хранить в вертикальном положении. В случае разрыва
столбика жидкости, его соединяют, постукивая по термометру
пальцем, но ни в коем случае не нагревая термометр.
Жидкостные термометры по конструкции делятся на
палочные— из массивных толстостенных трубок, на внешней
поверхности которых нанесена температурная шкала, и шкальные —
со шкальной пластинкой из молочного стекла, помещенной сзади
капилляра. По назначению и области применения жидкостные
термометры делятся на лабораторные и технические.
И те и другие могут быть как общего, так и специального
назначения.
Допускаемая погрешность показаний ртутных термометров
зависит, главным образом от интервала измеряемых температур и
цены деления шкалы. Так, пределы погрешности термометров при
цене деления 1 °С составляют:
Интервалы измеряемых
температур, °С: 0—100 100—200 200—300 300—400 400-500
Погрешность, ± °С: 1 2 3 4 5
В зависимости от цены деления шкалы и размеров, термометры
делятся на группы. Жидкостные термометры расширения
общелабораторного назначения образуют группу ТЛ. В настоящем
руководстве будут рассмотрены лишь наиболее часто применяемые
термометры лабораторные (химические) типа ТЛ.
178
Рис. 93. Термометр Бекмана:
а —общий вид; б —верхний резервуар.
Термометр Бекмана
Дифференциальный ртутный термометр с
вложенной внутрь шкальной пластинкой из
молочного стекла предназначен для
измерения с большой точностью в узком пределе
температур (не превышающем 5°С) в
интервале температур от —20 до +150°С.
Шкала термометра разделена всего на 5—
6°С с делениями в 0,01 °С, что позволяет
проводить измерения с точностью до 0,002 °С.
В верхней части термометра (рис. 93)
находится резервуар с запасом ртути. В
нижней части также имеется резервуар для
ртути. Оба резервуара соединены капилляром,
что дает возможность изменять объем ртути
в рабочем (нижнем) резервуаре. Изменяя
объем ртути в рабочем резервуаре, можно
«настроить» термометр так, чтобы его
показания отвечали требуемому интервалу
температур. Если температура в процессе
эксперимента понижается, то термометр настраивают
так, чтобы в начале измерения мениск ртути
находился в верхней градуированной части
капилляра. При измерении повышения
температуры мениск ртути устанавливают в
нижней части капилляра.
Для настройки термометра его
переворачивают верхней, расширенной частью вниз и,
слегка постукивая пальцем по нему,
переводят каплю ртути в расширение верхней изогнутой трубки.
Затем переворачивают термометр и опускают в слегка подогретую
воду. Столбик ртути, поднимающийся из резервуара, должен
соединиться с ртутью, находящейся в верхней части термометра.
После этого нижний резервуар нагревают в жидкостной бане,
снабженной вспомогательным термометром, до температуры на
2—3°С выше той, которую нужно будет измерить, и легким
постукиванием разрывают столбик ртути в месте соединения капилляра
с верхним расширением.
Общелабораторные ртутные термометры
Ртутные химические термометры типа ТЛ-2 с
вложенной внутрь оболочки шкальной пластинкой из молочного
стекла предназначены для измерения температуры в пределах от —30
до +350 °С; цена деления шкалы 1 °С.
Л^
179
Ртутные газонаполненные высокоградусные
термометры типа ТЛ-3 изготовлены из массивной капиллярной
трубки с нанесенными на ее поверхность делениями шкалы и
цифрами, без эмалевой полоски. Термометры предназначаются для
измерения температуры в пределах 0—400, 0—500 и 0—600°С; цена
деления шкалы 2°С.
Для точных измерений применяют наборы из восьми
нормальных или образцовых шкальных термометров
типа ТЛ-4 для измерения температуры в пределах от —30 до
+350°С; цена деления 0,1 °С.
Комплект из четырех ртутных термометров по Аллину
типа ТЛ-5 предназначен для измерения температуры в пределах
30—300 °С; цена деления 0,5 °С.
Комплект из восьми укороченных термометров по
Аншютцу предназначен для измерения температуры в
малогабаритных сосудах от —30 до +360 °С, с ценой деления шкалы
0,5 °С.
Ртутные термометры типа ТЛ-19 предназначены для точных
измерений температуры в пределах от 10 до 35 ± 0,2°С, а ТЛ-20 —
от 35 до 60±0,1°С.
Более точны специальные термометры с очень
большим и длинным резервуаром для ртути. Для высокоточных
измерений температуры в лабораторных условиях используют наборы
равноделенных палочных термометров типа TP-I и ТР-П. Набор
TP-I состоит из 15 термометров, рассчитанных на измерение
температуры в пределах от 0 до 4, от 4 до 8 °С и так далее до 60 °С;
цена деления 0,01 °С. Набор ТР-П состоит из 10 термометров,
предназначенных для измерения температуры 2 пределах 55—65,
65—75 °С и так далее до 155 °С; цена деления 0,02 °С.
О ртутных термометрах специального назначения см. [46].
Термометры на нормальных конусных шлифах
Для измерения температуры в лабораторных узкогорлых
сосудах и аппаратах с конусными взаимозаменяемыми шлифами
предназначены два типа ртутных термометров с вложенной шкалой на
нормальных конусных шлифах: ТЛ-33 со шлифом КШ-14 в
верхней части и ТЛ-50 с КШ-14 в нижней части.
Термометры ТЛ-33 погружают в измеряемую среду полностью.
Пределы измеряемой температуры термометрами ТЛ-33 от —30
до 200 °С.
Термометры ТЛ-50 подразделяются на четыре группы:
Диапазоны измерения, °С Диапазоны измерения, °С
I группа От —5 до 100 III группа От —60 до 150
II группа От —30 до 250 IV группа От —200 до 360
Технические термометры
Технические термометры (прямые и угловые) изготовляются
обычно с вложенной шкальной пластинкой (рис. 94). В отличие от
1*0
лабораторных (химических) термометров, их резервуар большего
объема и капилляр большего диаметра, поэтому градусные
деления у них крупные, а столбик ртути более заметен.
Отсчет показаний термометра
Правильный отсчет показаний термометра возможен лишь тог*
да, когда в нагреваемую зону погружен не только резервуар с
ртутью, но и весь выступающий столбик ртути.
Стекло и ртуть нагреваются неравномерно, и поэтому
показания термометра при погружении только его нижней части в
нагретое вещество несколько отличаются от истинных значений. Чтобы
получить точные данные, в измеренную величину U вводят
поправку К, которую вычисляют по формуле:
К в па (ti — /2)
где п — число градусов в выступающем столбике; а — коэффици*
ент расширения стекла (для палочных термометров а=0,000168 и
для трубчатых а=0,000158); t2 — средняя температура
выступающего столбика ртути.
Вместо t2 измеряется температура воздуха на уровне середины
выступающего столбика ртути с помощью вспомогательного
укороченного термометра, прикрепленного так, чтобы его конец касался
середины выступающего столбика (рис. 95).
Рис. 114. Технические стеклянные ртутные термометры:
а —прямой; б—угловые.
Рис. 95. Поправка на выступающий столбик термометра.
1SI
Поправка на выступающий столбик ртути зависит от
температуры:
Измеряемая
температура, Поправка, °С
°С
До 100 1
До 200 3-5
От 250—350 6-10
При отсчете показаний термометра по шкале глаз должен
находиться на одной линии с уровнем ртути.
Обращение с термометрами и их проверка [13]
После того как температура измерена, термометру дают
остыть, затем его вытирают, кладут в футляр и убирают в место,
отведенное для хранения термометров.
Если нижняя часть термометра запачкана смолистыми
веществами, то термометр следует вытереть кусочком ваты, смоченной
органическим растворителем (спирт, ацетон, эфир). С
термометрами следует обращаться очень осторожно. Нельзя нагревать их
выше максимальной температуры, указанной на шкале.
Правильность показаний термометра следует время от
времени проверять, сравнивая их с нормальным эталонным
термометром, которым не пользуются для рядовых работ, а хранят для
проверки лабораторных приборов. Для этой цели проверяемый и
эталонный термометры соединяют вместе резиновыми колечками
так, чтобы их ртутные баллоны находились как можно ближе один
к другому. Затем их помещают в жидкостный ультратермостат
так, чтобы столбики ртути несколько выступали из жидкости. До
100°С в качестве термостатной жидкости применяют воду, а
выше (до 250—300°С)—цилиндровое или силиконовое масло.
По результатам проверки строят график, где по оси ординат
откладывают температуру по эталонному термометру, а по оси
абсцисс — наблюдаемую температуру по проверяемому
термометру.
Чаще всего термометр проверяют, определяя температуры
кипения или плавления химически чистых веществ, подобранных
таким образом, чтобы охватить как можно более широкие интервалы
температур.
Нулевую точку по шкале Цельсия проверяют при погружении
термометра в дистиллированную воду в смеси со льдом. Для
этого в стакан вместимостью 300—400 мл помещают тонко растертый
лед, полученный при замораживании дистиллированной воды.
Деревянной палочкой делают углубление и, вставив термометр так,
чтобы нулевая точка шкалы несколько выступала изо льда,
уплотняют лед, выдерживают 7—10 мин и снимают показания.
Ледяная масса должна иметь вид густой кашицы (нельзя допускать,
чтобы куски льда плавали в воде). Перед отсчетом показаний
слегка постукивают по термометру деревянной палочкой; отсчет
повторяют несколько раз.
182
Рис. 96. Устройство для проверки термометра по точке кипения воды.
Для проверки термометра по точке кипения воды
может быть использовано простое устройство (рис.
96). В колбу вставляют широкую стеклянную
трубку с оттянутым нижним концом. Трубку помещают в
стеклянный кожух, чем достигается хорошая
термоизоляция. Проверяемый термометр вставляют через
верхнюю пробку кожуха, а конденсат отводят через
трубку в нижней пробке. Термометр должен быть
погружен таким образом, чтобы отсчет можно было
производить через стекло.
В расширение трубки с термометром помещают
слой битого стекла, чтобы брызги воды не попадали
на шарик термометра. Применяют только
дистиллированную воду, в которую для равномерного кипения вносят
несколько кусочков пористого фаянса или фарфора («кипелки»).
Доведя воду в колбе до кипения, выдерживают термометр до тех
пор, пока мениск ртути не установится на постоянном уровне.
При этом не следует забывать, что температура кипения воды
зависит от атмосферного давления:
Я, кПа 98,42 99,09 100,42 101,08 101,75 102,4 103,08
'кип, °С 99,25 99,44 99,63 99,81 100,0 100,18 100,55
Для проверки термометров могут быть использованы
химически чистые жидкости и металлы, температуры кипения и плавления
которых приводятся в справочной литературе. Проверка
термометра по точкам плавления или затвердевания чистых металлов
может быть выполнена так, как указано в гл. 36 и 37.
ТЕРМОЭЛЕКТРИЧЕСКИЕ ТЕРМОМЕТРЫ (ТЕРМОПАРЫ)
Принцип действия термоэлектрических термометров основан
на термоэлектрическом эффекте, заключающемся в том, что в замк*
нутой цепи из двух или нескольких разнородных металлов или
полупроводников возникает термоэлектродвижущая сила
(т. э. д. с), если места соединения (спая) проводников имеют
разную температуру. Термопара состоит из двух различных
металлических проводов, сваренных или спаянных одними концами
(«горячий» спай), а другими концами соединенных с гальванометром.
Места скрепления обоих проводов с проводниками цепи называют
«холодными» спаями. Если горячий спай помещен в среду с более
высокой температурой, чем холодный, который для большей
точности измерения помещают в сосуд с постоянной температурой
(лучше всего 0°С), то на концах проводников возникает разность
потенциалов (рис. 97). Разность потенциалов неодинакова для
разных пар металлов и будет тем выше, чем больше разность
температур горячего и холодного спаев.
# 183
Каждая термопара в цепи с данным гальванометром должна
быть предварительно отградуирована. Градуировку начинают с
установки гальванометра на нуль. Для этого оба спая (горячий
и холодный) погружают в пробирки с вазелиновым маслом,
которые помещают в сосуд Дьюара, наполненный тающим чистым
льдом, и устанавливают гальванометр на нуль.
Для температур порядка 0—350 °С рекомендуется калибровать
термопару по эталонному стеклянному ртутному термометру. Для
этого горячий спай термопары и эталонный термометр помещают
в пробирку с вазелиновым маслом и погружают в
ультратермостат, меняя температуру термостатной жидкости. После
5—10-минутной выдержки записывают показания гальванометра и
термометра.
Для температур выше 350°С рекомендуется калибровать
термопару по температурам кипения высококипящих веществ или
температурам плавления веществ, применяемых для проверки
термометров. В этом случае для измерения температуры горячий спай
термопары помещают в массивную металлическую гильзу так,
чтобы спай не касался металла гильзы, и производят отсчет
показаний гальванометра. Регистрируют соответствие показаний
гальванометра данной температуре и строят график зависимости
т. э. д. с. от температуры.
Непременное условие измерения температуры до 100 °С —
идентичность т. э. д. с. пары проводов, соединяющих термопару с
гальванометром, и термопары.
При пользовании градуированной термопарой не следует
менять гальванометр, иначе придется градуировать термопару
снова. Время от времени повторно проверяют градуировку
гальванометра.
В ГОСТ 3044—74 приведены градуировочные таблицы для
термоэлектрических термометров при температуре свободных
концов 0°С.
Термопары могут быть выполнены как из благородных, так и
из неблагородных металлов. К первой группе относятся стан*
а£
Соединительные
прод од о 3 <5"
">
Рис. 97. Схема монтажа термопары:
i—горячий спай; 2 —компенсационные провода; 3, 4—холодные спаи; 5—термостат; б —галь«
ванометр.
*»ис. 98. Схема дифференциальной термопары:
/, 2 — ветви термопары; 3 — промежуточная ветвь; 4 —гальванометр; Л, Д —спаи.
184
дартные, выпускаемые промышленностью термопары платино-
родий-платиновые (ТПП) и платинородий-платинородиевые
(ТПР), а ко второй — хромель-алюмелевые (ТХА) и хромель-ко-
пелевые (ТХК).
Кроме того, в лабораторной практике часто используют
нестандартные термопары: медь-константановые, железо-кон-
стантановые, медь-копелевые и железо-копелевые, которые
обязательно градуируют.
Термопары, стандартные и нестандартные, изготовляют
обычно в виде проволоки, которая изолирована фарфоровыми бусами
или трубками.
В последнее время распространение получили
полупроводниковые термопары с т. э. д. с. в 5—10 раз больше, чем т. э. д. с.
термопар из металлов и металлических сплавов.
Термоэлектродами в этих термопарах служит проволока из сплавов ZnSb
и CdSb.
Для измерения небольших разностей температур или для
получения больших значений т. э. д. с. при одной и той же
температуре применяют дифференциальные термопары и термобатареи.
Дифференциальная термопара (рис. 98) состоит из двух
ветвей одного и того же металла 1У 2 и проводника 3 из другого
металла или сплава. Спаи А и В помещают в места, разность
температур которых нужно измерить. Показания гальванометра 4
пропорциональны разности температур спаев Л и В. При этом
отпадает необходимость поддержания постоянной температуры
холодного спая. Нужно лишь следить, чтобы температура в обеих
точках присоединения проводников, идущих к гальванометру,
была одинаковой.
Термобатареи представляют собой несколько
последовательно соединенных термопар.
ТЕРМОМЕТРЫ СОПРОТИВЛЕНИЯ
Принцип действия термометров сопротивления основан на
изменении электрического сопротивления материалов с
температурой. В проводниковых термометрах сопротивления электрическое
сопротивление увеличивается с повышением температуры, в
полупроводниковых — уменьшается.
Проводниковые термометры сопротивления большей
частью изготовляют из платины и меди и применяют для
измерения температуры в пределах от —200 до +500°С.
Полупроводниковые термометры сопротивления — термисторы
состоят из оксида меди и оксида марганца и применяются для
измерения температуры в пределах от —200 до +200 °С.
Чувствительный элемент платиновых термометров
представляет собой спираль, расположенную в капиллярах керамических
трубок, заполненных дополнительно керамическим порошком,
который служит изолятором. Чувствительный элемент медных
термометров— бескаркасная безындукционная намотка из медной
1S5
Рис. 99. Термометр сопротивления:
/—термометр; 2—источник тока; 3 —известное сопротивление;
4 —гальванометр; 5 —передвижной контакт.
проволоки, покрытая фторопластовой пленкой.
2 К намотке припаяны два вывода.
Чувствительный элемент помещают в тонкостенную
металлическую гильзу, засыпают керамическим
порошком и герметизируют.
В СССР серийно выпускаются платиновые
термометры сопротивления (ТСП) для
температур от —200 до +650°С и медные
термометры сопротивления (ТСМ) для температур
от —50 до +180°С.
В качестве измерительного прибора используют измерительные
мосты, отградуированные по температурной шкале Цельсия, лого-
метры и потенциометры.
Измерительная схема, применяемая для проводникового
термометра сопротивления, изображена на рис. 99. Температуру по
этой схеме определяют на основании величины переменного
сопротивления, которое нужно ввести, чтобы стрелка гальванометра
вернулась в нулевое положение.
ТЕРМОМЕТРЫ ИЗЛУЧЕНИЯ
Термометры излучения, или пирометры,— приборы,
измеряющие температуру по тепловому излучению тел без
непосредственного контакта чувствительных элементов с измеряемой средой-
При температурах до 500°С нагретое тело испускает лучи, не
воспринимаемые человеческим глазом. При более высоких
температурах возрастает энергия излучения волн определенной яркости
и увеличивается полное излучение энергии (радиация). Эти
явления используют для измерения температуры нагретых тел.
В оптических термометрах сравнивается яркость
нагретого выше 600—800 °С тела с яркостью нити накала
электрической лампочки, интенсивность излучения которой в зависимости
от температуры известна. Яркость оценивается визуально или с
помощью фотоприемников — фотоэлементов и фотоумножителей.
ТЕРМОХИМИЧЕСКИЙ МЕТОД ИЗМЕРЕНИЯ ТЕМПЕРАТУРЫ
Термохимический метод — сравнительно новый, но уже
вошедший в практику контактный метод быстрого приближенного
измерения температуры поверхности твердых тел в пределах 60—
600 °С с точностью до ±10°С Для этой цели пользуются
термоиндикаторами— карандашами, красками и термоиндикаторной
бумагой.
Термохимический метод основан на изменении фазового
состава, вызывающего изменение цвета или яркости свечения
компонентов термоиндикаторов.
№
Термоиндикаторные карандаши изготовляют на
основе термоиндикаторных паст и получают методом литья. Марка
карандаша указывает на температуру перехода окраски (исходная
окраска и окраска после воздействия температуры).
Термоиндикаторные карандаши хранят в закрытых коробках в сухом,
прохладном и защищенном от света месте.
Термоиндикаторные краски на основе синтетических
смол применяются в виде спиртовых растворов. Их наносят на
металлическую поверхность кистью или пульверизатором; время
высыхания нанесенного слоя 20—25 мин. Термоиндикаторными
красками измеряют температуру в пределах 85—600 °С.
Термоиндикаторные бумаги (ТБ) выпускаются
10 марок. Номер марки отвечает ориентировочной температуре
перехода окраски (в °С): ТБ-60, ТБ-70, ТБ-80, ТБ-90, ТБ-100,.
ТБ-110, ТБ-120, ТБ-130, ТБ-140, ТБ-150.
РЕГУЛИРОВАНИЕ ТЕМПЕРАТУРЫ [4, 33]
Терморегуляторы — приспособления, позволяющие
поддерживать в нагреваемых приборах (установках) температуру,,
постоянную в узких пределах,— бывают различных систем и видов.
Простейшие терморегуляторы — контактные жидкостные
термометры, в которых расширяющаяся термометрическая жидкость при
достижении заданной температуры замыкает или размыкает
контактное устройство, управляющее нагревом.
В качестве терморегуляторов для лабораторных приборов и
установок часто используются ртутные стеклянные
электроконтактные термометры.
При регулировании температуры в пределах от —30 до 300°С
первичными приборами служат стеклянные ртутные
электроконтактные термометры с погружаемой нижней частью и шкальной
пластинкой, вложенной внутрь оболочки. Электроконтактные
термометры могут работать в цепях постоянного и переменного тока.
Они изготовляются двух типов: ТЗК — с заданным постоянным
контактом и ТПК — с подвижным рабочим контактом, прямые и
угловые, изогнутые под углом 90°.
Термометры ТЗК служат для поддержания заданной, вполне
определенной для данного термометра температуры. Замыкание
или размыкание электрической цепи между впаянными в
капиллярную трубку контактами происходит вследствие расширения или
сжатия ртути при нагревании или охлаждении нижней части
термометра. Для термометров ТЗК в интервале от —30 до +300°С
может быть несколько заданных значений температур (одно, два
или три).
Термометры ТПК служат для поддержания любой температуры
рабочего интервала. Замыкание и размыкание электрической цепи
между концом подвижного контакта и контактом, впаянным в
капилляр термометра, можно регулировать при помощи специального
магнитного устройства, которым снабжен термометр.
187
При замыкании цепи срабатывает реле и размыкает контакты
в цепи электрического нагревательного прибора, который
включается в сеть самостоятельно. Нагреватель отключается от сети, и
нагрев прекращается. Температура снижается до тех пор, пока
ртутный столбик не опустится и не разомкнет контакты
термометра. В это время замыкаются контакты реле в цепи
электрического нагревателя, и температура поднимается до момента
замыкания контактов термометра. Таким образом, при помощи
контактного термометра поддерживается постоянная температура среды.
Не рекомендуется перегревать термометр относительно
температуры контактирования более чем на 1 °С, так как это может
привести к разрыву ртутного столбика при последующем понижении
температуры. Если ртутный столбик разорвался, его можно
соединить, осторожно подогревая резервуар со ртутью до тех пор, пока
ртуть не поднимется в расширенную часть капилляра. При этом
подвижной контакт (вольфрамовую нить) надо поднять до предела
вверх, а термометр держать вертикально, ртутным резервуаром
вниз.
Предварительную настройку контактного термометра
производят по верхней шкале, следя, чтобы овальная гайка нижним
обрезом была установлена на штрихе заданной температуры
контактирования. После этого по нижней шкале проверяют положение конца
подвижного контакта относительно штриха. При необходимости
производят дополнительную регулировку и закрепляют магнитное
приспособление стопорным винтом.
Терморегуляторы, как правило, не включают непосредственно
в цепь нагревательного устройства, так как они не рассчитаны на
большую силу тока. Поэтому прибегают к помощи специальных
устройств, которые могут замыкать или размыкать цепь тока
значительной силы при подводе к ним сравнительно слабых
электрических сигналов.
Реле-устройство, включающее или выключающее на основе
внешних импульсов силовую цепь при помощи вспомогательной
цепи тока [33].
Известно очень много конструкций реле, действие которых
основано на самых разнообразных физических явлениях. В
лабораторной практике чаще всего используются электромагнитные реле.
u.„j===_Q.
Рис* 100. Схема включения электромагнитного реле;
а—нагреватель включен; б — нагреватель отключен; / — контактный термометр с подвижным
контактом; 2 — вспомогательная цепь; 3—электромагнит; 4—контакт; 5—главная цепь;
$—электронагреватель; 7—столбик ртути.
188
На рис. 100 приведена схема включения электромагнитного
реле, регулирующего нагревание жидкостной бани. Пока
температура бани не достигнет определенной величины и столбик ртути 7
не коснется контакта /, ток через цепь 2 не проходит, и ртуть в
переключателе 4 замыкает цепь 5, в результате чего действует
нагреватель 6. Как только ртуть в термометре коснется подвижного
контакта 1, цепь замкнется и электромагнит 3, через который теперь
уже проходит ток, притянет металлическую оболочку контакта 4,
тот наклонится и, разрывая поверхность ртути, тем самым
разомкнет нагревательную цепь 5.
Важнейшим элементом электромагнитного реле является
ртутный переключатель. Замыкание и размыкание
электрических цепей в установках различного назначения имеет место при
определенных углах поворота переключателя или под воздействием
внешнего магнитного поля.
Ртутные стеклянные переключатели выпускаются
промышленностью в большом ассортименте. Для лабораторных установок
весьма удобен двухконтактный ртутный переключатель ПР-5.
Переключатель работает в цепи постоянного или переменного тока при
нагрузке 15 мА и напряжении 1,5 В.
глава is. ВАКУУМНАЯ ТЕХНИКА
Вакуум —состояние газа (пара) при давлениях ниже
атмосферного. Понятие «вакуум» применяется только для газа (пара),
заключенного в объем, ограниченный стенками сосуда.
Весь диапазон от атмосферного до наименьшего досягаемого
давления делится на области низкого — до 665 Па (5 мм рт. ст.),
среднего —до 0,133 Па (Ю-3 мм рт. ст.), высокого — до 1,33 • К)-5 Па
(10~7 мм рт. ст.) и сверхвысокого — до 1,33- 10~10 Па (10~12 мм
рт. ст.) вакуума. Сверхвысокий вакуум, как правило, не
используется в практике работы химических лабораторий; он применяется
только при выполнении специальных работ. Мы ограничимся
описанием лишь некоторых источников получения вакуума.
Вакуум создают при помощи различных типов насосов [46,
с. 92]. Характерные параметры вакуумного насоса: предельное
(минимальное) давление, создаваемое насосом; производительность
насоса—количество газа, удаляемого в единицу времени; скорость
откачки—отношение производительности к давлению, измеренному
во впускном сечении насоса. По выпускному давлению все
вакуумные насосы делятся на три группы: выбрасывающие
откачиваемый газ в атмосферу, требующие предварительного разрежения и
такие, в которых откачиваемый газ не выводится наружу, а
связывается внутри самого насоса. Если насос требует
предварительного разрежения, то последовательно с ним устанавливается фор-
вакуумный насос.
Водоструйные и механические насосы, служащие для создания
низкого и среднего вакуума, можно использовать непосредственно
при атмосферном давлении, тогда как диффузионные
(пароструйные) насосы можно применять только при откачивании паров из
предварительно вакуумированного пространства.
ПРАВИЛА РАБОТЫ f 15]
Работы под вакуумом должны выполняться в вытяжном шкафу
на противнях; по месту работ следует устанавливать прозрачные
предохранительные экраны. Работающие должны пользоваться
защитными очками и перчатками.
Стеклянные и кварцевые сосуды, предназначенные для работы
под вакуумом, должны быть предварительно испытаны на
максимальное разрежение; при этом испытуемый сосуд следует обернуть
полотенцем или надеть на него металлическую сетку.
Запрещается:
а) создавать вакуум в перегонной колбе, наполненной горячей
жидкостью. После окончания вакуумной перегонки необходимо
охладить перегонную колбу, а затем наполнить прибор воздухом
(инертным газом) и после этого разобрать его;
190
б) допускать работу механического насоса без заземления
электродвигателя и ограждения приводных ремней;
в) устранять дефекты и чистить наружные поверхности во время
работы механического насоса.
ВОДОСТРУЙНЫЕ НАСОСЫ
Для получения разрежения, не превышающего 665—1330 Па
(5—10 мм рт. ст.), обычно применяют водоструйные насосы
различных конструкций, действующие по принципу инжектора.
Основной элемент конструкции водоструйного насоса — сопло,
из которого с большой скоростью вытекает струя воды. В
пространстве, окружающем сопло, создается разрежение, и окружающий
воздух увлекается в направлении течения струи.
Водоструйные насосы могут применяться повсюду, где имеется
достаточное давление воды в водопроводной системе (^ 196,2 кПа,
т. е. 2 ат). При этом предельный вакуум, создаваемый насосом,
зависит как от конструкции насоса, так и от температуры
вытекающей воды. Предельный вакуум не может превышать давление
паров воды при данной температуре:
Температура, °С: 4 6 8 10 12 14 16 18
Давление паров воды
в кПа 0,81 0,93 1,06 1,00 1,40 1,58 1,81 2,05
в мм рт. ст. 6,1 7,0 8,0 9,2 10,5 11,9 13,6 15,4
Поэтому зимой с помощью водоструйного насоса хорошей
конструкции можно достигнуть разрежения 665—931 Па, а летом лишь
1330—2660 Па.
Водоструйные насосы бывают стеклянные, пластмассовые и
металлические.
Стеклянные водоструйные насосы разнообразны
по конструкции. Большей частью их изготовляют в стеклодувных
мастерских по индивидуальным заказам. Остаточное давление,
создаваемое насосом заводского изготовления (рис. 101) при
давлении воды не более 196,2 кПа и температуре 8 ± 1 °С не более
1330 Па (10 мм рт. ст.); время создания остаточного давления в
объеме 1 л не более 6 мин. Нижняя трубка, через которую вода
вытекает из насоса, достаточна широка, что не создает излишнего
сопротивления и уменьшает опасность «захлебывания» и
неравномерной работы насоса.
Водоструйные стеклянные насосы могут служить не только для
разрежения, но и для нагнетания газов (воздуха). Подобный
насос, известный под названием насоса Ветцеля, представлен
на рис. 102. Такой насос, установленный в линии водопровода с
выходным напором воды не менее 196,2 кПа, должен давать в
откачиваемом сосуде предельный вакуум около 2660 Па и давление
нагнетаемого газа (воздуха) не менее 392,4 Па при температуре
воды в водопроводе не выше + 16 °С. Время создания остаточного
давления 2660 Па в объеме 1 л не более 10 мин. Обычный водо-
191
струйный насос может быть также легко приспособлен для
нагнетания воздуха (рис. 103).
В последние годы промышленность освоила производство
пластмассовых водоструйных насосов марки КМ-1230,
позволяющих получать предельный вакуум 1995—3325 Па, при
избыточном давлении воды в водопроводной сети 196,2—245,3 кПа.
Время создания остаточного давления в объеме 1 л не более
7—8 мин. Эти насосы в работе очень удобны. Они практически
не ломаются, и при засорении сопла ржавчиной их проще чистить,
чем стеклянные.
Металлические водоструйные вакуумные
насосы в работе во многом лучше стеклянных. Серийного выпуска
подобных насосов пока нет; они изготовляются по
индивидуальным заказам.
Для нормальной работы водоструйных насосов очень важно,
чтобы напор воды, вытекающей из водопроводной сети, был
постоянным. Производительность насоса зависит не только от
температуры, но и от скорости истечения воды. Расход воды обычно
достигает 180—200 л/ч. Скорость откачивания нельзя существенно
повысить, увеличивая приток воды, тем не менее водоструйные
насосы не следует присоединять к кранам водопроводной сети с
диаметром меньше чем 10 мм.
Насосы прикрепляют к водопроводному крану через разборный
ниппель или к крану со съемным ниппелем с помощью короткой
толстостенной резиновой трубки, которую в двух-трех местах
укрепляют жестяными кольцами или мягкой проволокой (железной,
медной). Резиновую трубку рекомендуется обмотать несколькими
рядами изоляционной ленты.
Рис. 101. Водоструйный насос.
Рис. 102. Водоструйный насос Ветцеля:
/, $,— трубки; 2, 4 —грушевидные расширения; 5, 7—тубусы; б—кожух; в—сифонообразная
трубка.
Рис. 103. Приспособление водоструйного насоса для нагнетания.
192
Л аппаратуре
Вакуум
Рис. 104. Предохранительная склянка.
Рис. 105. Предохранительное приспособление:
1—цилиндрик; 2—шлиф.
На боковой отросток насоса надевают вакуумную резиновую
трубку, с помощью которой присоединяют предохранительную
трехгорлую склянку, снабженную краном для впуска воздуха,
коротким манометром и патрубком, соединенным с вакуумируемым
сосудом (рис. 104). Вследствие падения давления воды или
внезапного сужения поперечного сечения при засорении сопла может
произойти всасывание воды в откачиваемый сосуд. Если вода
начнет поступать в предохранительную склянку, сосуд, из которого
откачивают газ (воздух), нужно осторожно отключить и, не
закрывая кран водопровода, дать насосу работать некоторое время
вхолостую; при этом вода из предохранительной склянки
полностью удалится.
Вместо предохранительной склянки часто используют
предохранительные приспособления, включаемые между водоструйным
насосом и вакуумируемым сосудом (рис. 105). Когда вода из
водоструйного насоса потечет по направлению к сосуду, цилиндрик /
всплывет и прижмется к шлифу 2; тем самым дальнейшее
поступление воды прекратится.
При работе с водоструйным насосом вакуум регулируют,
впуская воздух через кран предохранительной склянки, а не закрывая
водопроводный кран, который должен быть открыт все время
полностью. По окончании работы сначала надо медленно впустить
воздух в систему и только затем перекрыть воду.
При закупорке сопла стеклянного насоса твердыми частицами,
поступающими вместе с водой из водопроводной сети, насос
демонтируют и очищают, пуская струю воды в обратном
направлении или растворяют ржавчину соляной кислотой. При очистке
забитого сопла стеклянного насоса проволокой, прибор может
разбиться.
МЕХАНИЧЕСКИЕ ВАКУУМНЫЕ НАСОСЫ С МАСЛЯНЫМ УПЛОТНЕНИЕМ
Механические вакуумные насосы с масляным уплотнением
работают за .счет периодического изменения объема в рабочей
7 Зак. 1237 193
камере, куда поступает откачиваемый газ, который сжимается
в ней и выбрасывается в атмосферу. Все модели механических
вакуумных насосов построены на одном принципе, хотя и
различаются конструктивно.
Уплотнителем (рабочая жидкость) во всех насосах служат
масло, кремнийорганические жидкости или высококипящие эфиры
(см. ниже). Все зазоры механического насоса находятся в
рабочей жидкости, препятствующей обратному притоку или натеканию
газов.
Предельный вакуум насоса определяется давлением паров
используемой рабочей жидкости и примесями; качеством
уплотнения; величиной вредного пространства, из которого газ
механически не выбрасывается при работе насоса; растворимостью
откачиваемого газа в рабочей жидкости; герметичностью корпуса и
сальников вала.
Механические вакуумные насосы могут быть поршневыми и
вращательными. Поршневые насосы в настоящее время в
химических лабораториях не используются. Вращательные насосы
широко применяются в лабораторной практике для вакуумирова-
ния больших объемов сосудов до разрежения примерно 133—1,33
Па, для вакуумирования эксикаторов, для вакуумных перегонок и
других работ в области среднего вакуума. При откачке
агрессивных газов и паров воды происходит коррозия поверхностей насоса
и осмоление масла. При увлажнении масло постепенно
превращается в водомасляную эмульсию и предельное давление
насоса увеличивается.
Для уменьшения предельного давления изготовляются также
двуступенчатые механические насосы. При этом первая ступень
служит насосом предварительного разрежения для второй. Со све-
жезаполненным масляным вакуумным насосом можно достичь
вакуума 1,33—0,133 Па.
Существуют три основных типа масляных вращательных
насосов: пластинчато-роторные, пластинчато-статорные и
золотниковые.
Пластинчато-роторные насосы с масляным
уплотнением предназначены для откачки воздуха, химически неактивных
газов, паров и парогазовых смесей, не воздействующих на
материалы конструкции и рабочую жидкость насосов.
Пластинчато-роторные насосы типа НВР откачивают конденсируемые пары и
парогазовые смеси с предельным парциальным давлением паров
воды на выходе 2,34 кПа. Насосы небезопасны для откачки газов
с содержанием кислорода больше, чем в воздухе при нормальных
условиях [21% (об.)].
Работа пластинчато-роторного одноступенчатого насоса
происходит следующим образом (рис. 106). При вращении ротора
лопатки скользят по внутренней поверхности цилиндра. Полость,
образованная цилиндром, ротором и торцовыми крышками,
делится лопатками на полость всасывания А и полость выхлопа Б. При
вращении ротора объем полости А периодически увеличивается,
194
благодаря чему в нее поступает газ из откачиваемой системы;
объем полости Б периодически уменьшается, в ней происходит
сжатие. Сжатый газ выбрасывается через клапан.
В двухступенном исполнении выход первой ступени соединен
каналом со входом второй ступени.
Уплотнение между полостями А и Б достигается при помощи
масляной пленки.
Вакуумные механические насосы с масляным уплотнением
пластинчато-роторного типа изготовляются и поставляются агре-
гатированными с электродвигателем на общей плите.
Портативный пластинчато-роторный насос НВР-025Д создает
предельное остаточное давление 1,33 Па; количество масла,
заливаемого в насос (ВМ-6 или ВМ-1) 0,86 л.
Пластинчато-статорный двухступенчатый вакуум-на->
сое ВН-461М создает предельное остаточное давление 0,665 Па
(5-Ю"3 мм рт. ст.) и служит для откачки сухих и нейтральных
газов, не вступающих в активную реакцию с металлами, а также
для создания форвакуума при работе с высоковакуумными
диффузионными насосами. Рабочая жидкость — масло ВМ-4;
количество масла, заливаемого в насос 2,3 л.
Во многих лабораториях в качестве форвакуумного насоса
используют насос РВН-20, обладающий быстротой действия 2,6 л/с»
Количество масла ВМ-4, заливаемого в насос 0,5 л.
Масляные насосы создают более высокий вакуум, чем
водоструйные. Однако опыт показывает, что в химических
лабораториях ими пользуются для получения разрежения на несколько
порядков ниже их паспортных данных. Это объясняется тем, что при
продолжительной работе в масло насоса попадают пары летучих
растворителей, влага и продукты термического разложения
органических веществ. Давление пара рабочей жидкости при этом
повышается и предельный вакуум ухудшается. Поэтому масляный
насос нельзя использовать для отгонки летучих растворителей.
.Вход
Выход
ъ.к
w
|=л
41/Г
vry
Рис. 106. Принцип действия пластинчато-роторного насоса:
1—цилиндр; 2—ротор; 3 —лопатки; 4 — пружины; А—полость всасывания; £—полость выхлопа.
Рис. 107. Вымораживающие ловушки.
7*
195
Растворители, когда это необходимо, должны сначала
отгоняться в вакууме водоструйного насоса, и лишь после этого
аппаратуру для перегонки можно присоединять к масляному насосу.
Для предохранения рабочей жидкости масляного насоса от
попадания нежелательных веществ между вакуумируемой
аппаратурой и масляным насосом помещают поглотительные колонки
или вымораживающие ловушки. Поглотительные
колонки (см. гл. 18) наполняют крупнозернистым активным углем,
силикагелем или плавленой щелочью (КОН или NaOH). При
высокой влажности откачиваемых паров плавленая щелочь легко
спекается, и пропускная способность поглотительной колонки
резко ухудшается. Этого можно избежать, если перемешать
плавленую щелочь с кусочками необожженной керамики.
Поглотительные колонки с наполнител-ем ухудшают предельный вакуум в
аппаратуре. Этим недостатком не обладают вымораживающие
ловушки различной конструкции (рис. 107). Вымораживающие
ловушки погружают в сосуд Дьюара, наполненный смесью
ацетона с «сухим льдом» или жидким воздухом. В последнем случае
ловушки должны быть изготовлены из кварца или меди.
При сборке аппаратуры для работы с масляным насосом
следует по возможности применять короткие соединения из
толстостенного вакуумного резинового шланга и особое внимание
уделять проверке герметичности соединений и установки в целом.
ДИФФУЗИОННЫЕ ВЫСОКОВАКУУМНЫЕ НАСОСЫ
Для получения высокого вакуума применяются диффузионные
насосы. Рабочий вакуум, достигаемый диффузионными насосами
ниже 0,133 Па; предельный вакуум составляет около 1,33«Ю-6 Па.
В диффузионных (струйных) вакуумных стеклянных и
металлических насосах для создания вакуума используют кипящую
ртуть, высококипящие углеводороды, кремнийорганические
вещества и другие жидкости с высокой температурой кипения.
Пары жидкости доходят до сопла, где они захватывают
молекулы газа (пара), поступающие из откачиваемой емкости.
Диффузионные насосы изготовляют по многоступенчатой схеме, т. е.
пары рабочей жидкости из верхнего сопла передают молекулы газа
(пара) в следующее сопло и т.д. На выходе из насоса газы
откачивают масляным форвакуумным насосом в атмосферу. Пары
рабочей жидкости при этом конденсируются на стенках
диффузионного насоса, охлаждаемых снаружи, и возвращаются обратно в
сборник.
Масляные диффузионные насосы имеют некоторые
особенности, отличающие их от ртутных. Необходимо строго следить за
тем, чтобы рабочая жидкость не перегревалась, так как при этом
могут образоваться летучие продукты разложения.
В химических лабораториях часто используются
малогабаритные стеклянные паромасляные и парортутные насосы (рис. 108).
Паромасляный насос СДН-1, в сочетании с форвакуум-
196
Рис. 108. Диффузионный пароструйный масляный (а) и ртутный (б) насосы:
/ — колба; 2—отвод к эвакуируемой системе; 3—сопло; 4—холодильник {а), дефлегматор (6*);
5 —отвод к форвакууму; 6 — трубка для возврата ртути.
ным насосом марки ВН-461М (или РВН-20) со скоростью откачки
5—7 л/с обеспечивает создание предельного вакуума 1,33• 10~3—»
1,33-10~4 Па. Объем заливаемой рабочей жидкости составляет
30—40 мл. Парортутный стальной насос ДРН-10-1М со
скоростью откачки 7 л/с обеспечивает создание предельного
вакуума 2,66-Ю-5 Па. Объем заливаемой ртути равен 130 мл.
Более удобны стальные диффузионные пароструйные ртутные
насосы (двух- и трехступенчатые). Они безопасны в обращении и
допускают применение более энергичного нагревания, что
увеличивает скорость откачки. Поэтому предварительного разрежения
удается добиться даже водоструйным насосом. Часто сначала
быстро создают разрежение с помощью масляного насоса, а по*
том переходят на отсасывание водоструйным насосом.
Парортутный металлический насос Н-10Р со
скоростью откачки 15—20 л/с обеспечивает достижение предельного
вакуума 1,33-Ю-5 Па. Объем заливаемой ртути 400 мл.
РАБОЧИЕ ЖИДКОСТИ - ВАКУУМНЫЕ МАСЛА [46, с. 97]
Вакуумные масла получают в результате вакуумной перегонки
смесей высококипящих природных и синтетических жидкостей,,
Природные масла получают путем вакуумной
дистилляции нефтяных продуктов. Это неоднородные по составу жидкости,
состоящие из смеси углеводородов различного химического
состава и молекулярной массы, со средним давлением пара при 20 °С
1,33- Ю-6—1,33- Ю-7 Па.
Масла ВМ-1 и ВМ-2 представляют собой дистилляты
медицинского вазелинового масла, получаемые путем однократной высоко-
197
вакуумной дистилляции сырья. Масло ВМ-3 получают из масел
кавказских и восточных нефтей путем выделения узкой фракции
при разгонке сырья в высоковакуумной дистилляционной
аппаратуре. Масло ВМ-4, получаемое из машинного масла, имеет
сравнительно широкий фракционный состав. Масло ВМ-5 представляет
собой сверхвысоковакуумную рабочую жидкость; его получают
путем двухкратной дистилляции медицинского вазелинового масла.
Масло ВМ-6 получается из машинного масла, обладает более
узким фракционным составом, чем масло ВМ-4. Масло ВМ-7
изготовляется из турбинного масла 46.
Масла ВМ-4 и ВМ-6 предназначаются в качестве рабочих
жидкостей для механических насосов с масляным уплотнением,
ВМ-1, ВМ-2, ВМ-5, ВМ-7 — для высоковакуумных диффузионных
пароструйных насосов.
Применяемые для вращательных насосов масла должны
обладать определенной вязкостью, не слишком большой при комнатной
температуре, чтобы не затруднять запуск насоса. С другой
стороны, при рабочей температуре насоса порядка 60—70°С масло
должно сохранять вязкость, достаточную для того, чтобы в зазорах
не нарушалась целостность уплотнения.
Для высоковакуумных насосов применяются также кремний-
органические и эфирные рабочие жидкости.
Кремнийорганические жидкости — соединения,
молекулы которых состоят из чередующихся атомов кремния и
кислорода с присоединенными углеводородными радикалами по
свободным связям кремния. Эти жидкости обладают повышенной
термической и химической стойкостью. Даже после длительной
работы в насосах при периодическом попадании воздуха они не
образуют смолистых налетов на внутренних деталях насосов.
Масла ВКЖ-94А и ВКЖ-94Б— узкие фракции полиэтилсило-
ксановой жидкости. Давление пара при 20 °С около 1,33- Ю-6 Па;
молекулярная масса 700—750; плотность при 20 °С 0,97 г/см3.
Масло ПФМС-2 — узкая фракция полифенилметилсилоксано-
вой жидкости обладает очень высокой термоокислительной
стойкостью. Давление пара при 20 °С 0,665-10"6—0,931-Ю^4 Па.
глава 16. ИЗМЕРЕНИЕ И РЕГУЛИРОВАНИЕ ДАВЛЕНИЯ
Давлением называют физическую величину, равную отношению
силы, действующей на элемент поверхности нормально к ней, к
площади этого элемента.
Различают абсолютное и избыточное давление. Абсолютным
(полным) называют давление, отсчитываемое от абсолютного нуля,
т. е. истинное давление. Оно может быть как выше, так и ниже
атмосферного. Если абсолютное давление ниже атмосферного, его
называют остаточным. Разность между атмосферным и
остаточным давлением называют вакуумом или разрежением.
Избыточное давление представляет собой разность между
абсолютным давлением и давлением окружающей среды.
Основная единица давления по Международной системе
единиц (СИ) — паскаль (Па). Паскаль равен давлению, вызываемому
силой 1Н (1 ньютон), равномерно распределенной по нормальной
к ней поверхности площадью 1 м2, т. е. 1 Па = 1 Н/м2.
Соотношения между паскалем и некоторыми внесистемными
единицами представлены ниже:
1 атм = 101325 Па
1 ат =98066,5 Па
1 мм рт.ст. = 133,322 Па
1 мм вод. ст. = 9,807 Па
1 бар= 105 Па
КЛАССИФИКАЦИЯ ПРИБОРОВ ДЛЯ ИЗМЕРЕНИЯ ДАВЛЕНИЯ [55]
Приборы для измерения давления и разрежения обычно
классифицируют по роду измеряемой величины и по принципу
действия. Деление по первому принципу представлено ниже:
Прибор Измеряемая величина
Манометр Абсолютное и избыточное
давление
Вакуумметр Разрежение (вакуум)
Мановакуумметр Избыточное давление и вакуум
Барометр Барометрическое давление
атмосферного воздуха
г По второму принципу приборы подразделяются на жидкостные»
деформационные и электрические манометры.
ЖИДКОСТНЫЕ МАНОМЕТРЫ
Жидкостные стеклянные манометры широко применяются в
лабораториях для измерения барометрического давления атмосфер
його воздуха, вакуума, разности давлений, для градуировки и
проверки приборов других систем.
1М
Жидкостные манометры — самые простые и точные приборы
для измерения давления. Верхний предел измеряемых
величиной МПа (2кгс/см2).
Ртутные барометры
Для измерения атмосферного или близкого к нему давления в
открытом пространстве применяют барометры [23],
показывающие абсолютное давление воздуха.
Ртутный барометр (рис. 109) состоит из вертикальной
стеклянной трубки, наполненной ртутью; верхний конец трубки
запаян, а нижний опущен в чашку с ртутью. В верхней части
трубки образуется вакуум. При увеличении давления воздух давит на
поверхность ртути в чашке и часть ртути входит в трубку, а при
понижении давления происходит обратное. Трубка заключена в
оправу, в верхней части которой имеется вертикальный прорез,
позволяющий видеть мениск ртути. В пределах этого прореза на
оправе нанесена шкала в миллиметрах ртутного столба.
Дополнительно имеется подвижная шкала-нониус, позволяющая измерять
давление с точностью до десятых долей миллиметра. Перед
отсчетом давления необходимо слегка постучать пальцем по защитной
оправе барометра, чтобы мениск ртути принял нормальную форму,
и установить при помощи винта нулевое деление нониуса на одной
линии с верхним мениском ртутного столба.
Пример. На барометре, изображенном на рис. 108, давление в целых числах
равно 762 мм рт. ст. К нему следует прибавить столько десятых долей
миллиметра, сколько делений по нониусу приходится до первого
совпадения одного из них с делениями основной шкалы.
В нашем примере — это 7-е деление, следовательно
атмосферное давление равно 762 -+• 0,7 = 762,7 мм рт. ст.
Ртутные барометры устанавливают в
помещениях вдали от дверей и окон, укрепляя их
на контрольной стене во избежание
сотрясения.
При измерении давления барометром
необходимо ввести поправки на температуру и силу
тяжести. На практике пользуются готовыми
таблицами поправок, прилагаемых к барометру.
Барометры имеют еще инструментальную
поправку, которая приводится в паспорте
прибора.
Точность определения давления обычными
ртутными барометрами составляет 13,3 Па (0,1 мм
рт. ст.).
Рис. 109. Ртутный барометр:
1 — шкала; 2 —винт; 3—термометр; 4—чашечка с ртутью; 5—шкала
с нониусом*
ш
№
й
1 I мм | '
eoJ
■w-Э
-40-
70-
0-
■20-
60 н
80
^Ш
ЗЕ
о
Рис. ПО. Жидкостный манометр.
Рис. Ш. Ртутный вакуумметр.
Жидкостные манометры для измерения избыточного
давления
Для измерения давления больше атмосферного выпускаются
стеклянные U-образные ртутные манометры с
пределами измерения 100 и 160 мм рт. ст., а также U-образные
жидкостные манометры, заполненные водой,
силиконовым маслом, спиртом, этилфталатом, с пределами измерения 100,
160, 250, 400, 600, 1000 мм жидкостного столба.
Жидкостные манометры представляют собой открытую с обеих
сторон стеклянную U-образную трубку, укрепленную на
деревянном штативе со шкалой, расположенной между ветвями трубки
(рис. 110) .Трубку манометра заполняют запирающей жидкостью
до нулевой отметки шкалы. Левый конец трубки присоединяют
к установке, в которой измеряется давление, а правый
сообщается с атмосферой. Под действием измеряемого давления жидкость
в трубке перемещается из одного колена в другое. Когда
измеряемое давление уравновесится гидростатическим давлением столба
жидкости, перемещение жидкости прекратится. Поскольку
давление в системе выше атмосферного, высота столба жидкости в
правом колене больше, чем в левом. Разность высот равна величине
столба жидкости, измеряемой по градуированной шкале.
Ртутные манометры для измерения вакуума [15, 18]
Для измерения низкого вакуума (более 133 Па) наиболее рас-»
пространен — U-образный стеклянный ртутный ва«
куумметр (рис. 111). В U-образной манометрической трубке
находится ртуть, полностью заполняющая левое колено. Когда
вакуумметр будет присоединен к вакуумируемой установке, ртуть в
левом колене начнет опускаться и остановится, когда разность
уровней в обоих коленах будет соответствовать давлению в системе.
201
В вакуумметрах заводского изготовления манометрическая
трубка сужена в месте нижнего изгиба, чтобы уменьшить скорость
движения ртути и предупредить сильный удар в запаянный конец
трубки при быстром впускании воздуха в манометр.
Иногда манометрические трубки изготавливают и заполняют
ртутью в лаборатории. Манометр монтируют на деревянной
подставке либо присоединяют его к установке для вакуум ирования
и шкалу с делениями укрепляют непосредственно на манометре.
Перед заполнением манометрической трубки ртутью ее необходимо
вымыть разбавленной азотной кислотой, водой и тщательно
высушить; ртуть должна быть также тщательно очищена и осушена
(см. гл. 22).
Предложено много методов заполнения манометра ртутью.
По одному из них [36, с. 141], заполнение проводят следующим
образом: встряхиванием переводят в запаянное колено часть ртути
и при помощи вакуум-насоса осторожно откачивают воздух из
трубки, держа ее почти в горизонтальном положении. Когда пузырьки
воздуха будут удалены, ртуть осторожно нагревают в вакууме
спиртовой или газовой горелкой до кипения, непрерывно
встряхивая трубку. После дегазации добавляют новую порцию ртути и
повторяют эту операцию до тех пор, пока в правом колене уровень
ртути не поднимется на 20—25 мм выше изгиба.
По другому способу, при помощи отрезка вакуумной резиновой
трубки к манометру присоединяют почти встык стеклянный шар
со ртутью, соединенный с масляным вакуумным насосом (рис. 112).
Вакуумировав систему до предельного давления, создаваемого
масляным насосом, закрывают кран и, поднимая шар, переливают
часть ртути в трубку манометра. Затем манометр переворачивают
сгибом вверх и снаружи осторожно нагревают пламенем горелки
до тех пор, пока ртуть не закипит и из нее не удалятся пузырьки
воды и воздуха. После этого в манометр подливают еще ртути,
прогретой предварительно в шаре, и повторяют операцию кипячения.
Наконец, заполняют и согнутую часть манометра, переворачивают
манометр в нормальное положение и добавляют нужное
количество ртути.
Рис. 112. Наполнение ртутью закрытого манометра.
Рис. 113. Видоизменение закрытого колена вакуумметра:
о —сужение; 0—кран; в —кран и расширение яод краном,
202
Затруднения, связанные с заполнением вакуумметра ртутью,
могут быть значительно уменьшены, если закрытое колено
манометрической трубки не запаяно, а имеет хорошо пришлифованный
стеклянный кран (рис. ИЗ). Для заполнения вакуумметра чистую
ртуть с помощью водоструйного насоса засасывают в
манометрическую трубку при открытом верхнем кране. Кран закрывают,
когда ртуть попадает в расширение над ним. Затем ртуть с
большой осторожностью нагревают в вакууме для удаления остатков
адсорбированного воздуха.
Если манометр правильно заполнен ртутью и не загрязнен
воздухом или парами веществ, то при присоединении его к
масляному насосу ртуть в обоих коленах должна находиться на
одинаковом уровне. Если внутрь закрытого конца манометра попадет
воздух или пары веществ, то может возникнуть большая
погрешность.
Точность измерений давления таким манометром составляет
66,5—133 Па (0,5—1,0 мм рт. ст.).
При выполнении работ под вакуумом до впуска воздуха в
эвакуированную систему необходимо закрывать кран манометра,
иначе ртуть загрязняется. Кран следует ненадолго открывать
только непосредственно при снятии показаний.
Если в результате неосторожного обращения вода из
водоструйного насоса или жидкость, с которой производилась
работа в вакууме, попадет в манометр, его нужно разобрать,
тщательно вымыть, высушить и вновь заполнить сухой ртутью.
Перед заполнением манометра сухой ртутью, чтобы
предотвратить загрязнение ртути, рекомендуется прокипятить резиновый
шланг в 5% растворе Na2C03, тщательно промыть его
дистиллированной водой, затем спиртом и высушить струей сухого чистого
воздуха.
Уравнительный (подвижный) сосуд со ртутью следует
поднимать осторожно, чтобы ртуть не попала в аппаратуру или насос.
Масса ртути для заполнения манометра обычно значительна
(2,5—6 кг), потому необходимо очень осторожно обращаться с
прибором и строго соблюдать правила работы со ртутью (см.
гл. 22).
Для измерения давления в диапазоне 1330—0,0133 Па (10—
10~4 мм рт. ст.) часто применяют компрессионный
ртутный манометр Мак-Леода, принцип действия которого
основан на закономерном уменьшении известного объема газа при
сжатии под действием ртути. Благодаря этому давление газа
достигает заметной величины, а исходный объем газа, равный 100—
300 мл, сокращается обычно в 104—105 раз. Наиболее
употребительны измерительные баллоны шарообразной или грушевидной
форм объемом в 100 мл.
Предложено много конструкций компрессионного манометра;
две модели приведены на рис. 114. Чувствительность манометра
Мак-Леода зависит от отношения объемов измерительного сосуда
и измерительного капилляра (от степени сжатия),
203
Исходное ,
положение
кистановке 6 рис- ,,4« Компрессионный ртутный манометр
^ й* Мак-Леода:
"Ю || а —простой; б —укороченный;/ — баллон; 2 —за«
паянный капилляр; 3 — соединительная трубка;
4 —боковой открытый капилляр; 5 —ловушка;
6 — резервуар для ртути; 7 —подвижная
подставка для резервуара; Я—отвод; 9» /0
—манометрические трубки; И, 12 — краны.
Увеличивая объем измери*
тельного сосуда при
одновременном уменьшении объема
капилляра, можно достигнуть любой
степени точности измерения
высокого вакуума. На практике точ-
По/1П'№0Н//Р
Триизмер&ш ность показания манометра
ограничивается массой ртути,
препятствующей увеличению объема
измерительного сосуда, и
диаметром измерительного
капилляра. Если диаметр капилляра
меньше 0,5 мм, ртуть прилипает
к стенкам и капилляр легко
забивается.
Манометры Мак-Леода
применяют для абсолютных
измерений и калибровки вакуумметров других типов. Однако ими нельзя
измерять давление паров углеводородов, воды и ртути, так как эти
вещества при сжатии конденсируются и адсорбируются на
стеклянных стенках прибора.
При пользовании простым манометром Мак-Леода поступают
следующим образом: через отвод 8 манометр присоединяют к
установке. Резервуар с ртутью 6 опускают так, чтобы ртуть вытекла
из баллона 1 и капилляра 2. После того как вакуум в системе
установится, резервуар 6 поднимают на такую высоту, чтобы
уровень ртути в боковом, открытом капилляре 4 оказался на высоте
верхнего конца запаянного капилляра 2. Объем воздуха,
оставшегося в капилляре 2, пропорционален первоначальному остаточному
давлению. Давление, в соответствии с законом Бойля-Мариотта,
может быть вычислено по формуле:
р .. ААУ2
V,
где V\ — обьем баллона 1 вместе с капилляром; V2 — объем
воздуха в капилляре 2 после сжатия; Д/г— разность уровней ртути в
капиллярах.
Обычно манометры снабжаются шкалой, прокалиброванной
для прямого отсчета давления по величине Д/г.
В некоторых случаях в паспорте манометра приводится
постоянная капилляра К, измеряемая объемом на единицу длины
капилляра. В этом случае давление может быть рассчитано по фор«
муле:
• ч . Р — Kbh
204
Этот метод позволяет ностроить линейную шкалу зависимости
разности уровней ртути от измеряемого давления для данного
манометра. Зная разность уровней ртути Ah, точный объем V\ и
площадь сечения капилляра S, измеряемый вакуум можно также
вычислить по формуле:
На основе этих вычислений можно заранее изготовить
нелинейную квадратичную шкалу для данного манометра. Линейная
шкала удобнее для измерения более высоких давлений, а
квадратичная— для более низких.
ПОКАЗЫВАЮЩИЕ МАНОМЕТРЫ ДЛЯ ИЗМЕРЕНИЯ
ИЗБЫТОЧНОГО ДАВЛЕНИЯ
Для измерения избыточного давления применяются манометры
с одновитковой и многовитковой трубчатыми пружинами.
Показывающие лабораторные манометры изготовляют с одновитковой
трубчатой пружиной. Последняя представляет собой полую
металлическую трубку овального сечения, изогнутую по дуге и закрытую
с одного конца. Второй конец пружины впаян в штуцер,
соединяющий манометрическую трубку с установкой, в которой измеряется
давление. Под действием давления трубчатая пружина меняет
форму своего сечения, в результате чего ее запаянный конец
перемещается пропорционально измеряемому давлению (трубка
разгибается).
Для измерения давления до 5 МПа {50 кгс/см2) трубки
изготовляют из латуни и бронзы, для более высоких давлений — из
стали.
Показывающие манометры выпускают в круглом корпусе
диаметром от 40 до 250 мм с верхними пределами измерений от 0,1
до 160 МПа. Нижний предел у всех манометров равен нулю.
Корпуса манометров, предназначенных для измерения давления
различных газов, окрашиваются в соответствующие цвета: для
кислорода— в голубой, водорода — в темно-зеленый, ацетилена—в
белый, хлора и фосгена — в серовато-зеленый, горючих газов —
в красный, а остальных негорючих газов — в черный.
ВАКУУММЕТРЫ ДЛЯ РАЗЛИЧНЫХ ДИАПАЗОНОВ ДАВЛЕНИЯ
Диапазон давлений, измеряемых вакуумметрами, значителен
1013—1,33- 10"12 Па (760—10~13 мм рт. ст.), поэтому в настоящее
время применяют различные манометры, каждый из которых имеет
свой диапазон измеряемого давления.
Термопарный манометр типа В Т-3 предназначен
для индикации давления в диапазоне 665—66,5 Па (5—0,5 мм
рт. ст.) и для измерения давления в диапазоне 13,3—0,133 Па
'v(10~l — 10~3 мм рт. ст.). Прибор состоит из измерительной установки
205
и термопарного манометрического преобразователя (лампы) ЛТ-2
или ЛТ-4М. Измерительная установка обеспечивает питание
манометрического преобразователя, измерение тока подогревателя
и т. э. д. с.
В диапазоне индикации давления вакуумметр работает только
с преобразователем ЛТ-2, работающим в режиме постоянства
т. э. д. с, в диапазоне измерения давления — с преобразователями
ЛТ-2 или ЛТ-4М, работающими в режиме постоянства тока
нагревателя.
Отсчет давлений производится по градуировочным графикам,
приведенным в приложении к выпускному аттестату, и инструкции
по эксплуатации прибора.
Принцип действия термопарного манометрического
преобразователя давления основан на зависимости теплопроводности газа
от давления. Температура нагревателя определяет
электродвижущую силу термопарного преобразователя. Если в преобразователе,
вакуумно соединенном с обследуемым объемом, ток нагревателя
поддерживать постоянным, то т. э. д. с. термопарного
преобразователя будет определяться давлением окружающего газа, так как
изменение температуры нагревателя зависит от теплопроводности
окружающего газа. Следовательно, при понижении давления
теплопроводность газа уменьшится, температура нагревателя
увеличится, и возрастет т. э. д. с. термопары.
Ионизационно-термопарный вакуумметр
ВИТ-2 имеет два диапазона измерений: 26,6—0,133 Па
(2. Ю-1 —Ю-3 мм рт. ст.) и 0,133— 1,33-10~5 Па (10"3 —
— 10~7 мм рт. ст.). Точность измерения давлений в области
высокого вакуума не очень велика, и порой определяется лишь
порядок величины давления.
Прибор состоит из датчика, соединяемого с вакуумной
системой, и отдельного блока электрического питания и измерения.
Показания прибора зависят от вида газа, и он требует
предварительной градуировки по каждому газу.
Принцип действия ионизационно-термопарных приборов
основан на том, что при создании потока электронов в разреженном
газе будет происходить ионизация и между двумя электродами,
к которым подводится электрическое напряжение, возникнет
ионный ток. Сила ионного тока при прочих равных условиях будет
пропорциональна плотности газа, а следовательно, при
определенной температуре пропорциональна его давлению.
Вакуумметр радиоизотопный ВР-4 предназначен
для измерения давления газов в диапазоне давлений 1,33-10~2—
— 1013 Па (10~4— 760 мм рт. ст.) как в лабораторных, так и в
производственных условиях.
Вакуумметр ВР-4 состоит из измерительной установки с
выносным каскадом и радиоизотопного преобразователя МР-8.
Принцип действия преобразователя основан на свойстве а-ча-
стиц ионизировать газ, в результате чего образуется ионный ток,
пропорциональный давлению газа.
206
Рис. 115. Кран с капилляром для
регулирования давления.
=ЧР
Плутоний-238, используемый в преобразователе в качестве
радиоактивного вещества, не обладает проникающим излучением^
Прибор безопасен в эксплуатации. Вскрывать преобразователь
категорически запрещается.
РЕГУЛИРОВАНИЕ ДАВЛЕНИЯ
Приспособления, при помощи которых можно регулировать
давление в вакуумируемой системе, по характеру действия могут
быть разделены на две группы [5, 36]:
1) регулирующие давление путем периодического впуска
воздуха в систему, когда разрежение превышает заданную величину;
2) регулирующие давление путем периодического включения
и выключения вакуум-насоса.
Простое и достаточно эффективное приспособление для
регулирования давления путем впуска воздуха в вакуумированную
систему представлено на рис. 115. На шлифованной поверхности
крана имеются риски, начинающиеся у отверстий и постепенно
сходящие на нет. Скорость впуска воздуха регулируется длиной
и диаметром капилляра.
Для регулирования вакуума наиболее распространены мано-
статы, в основе действия которых лежит гидростатический
принцип (рис. 116). Кран 1 остается открытым до тех пор, пока не
будет достигнуто нужное разрежение, затем его закрывают. Если
в процессе работы давление в системе повышается, то в
промывную склянку 2 через слой жидкости поступит такое количество
газа, что разность давлений снова окажется постоянной. Нужная
Остановка
Вакуум-насос'
Рис. Ив. Гидростатический манометр:
7 —кран; 2—промывная склянка с нижним тубусом; 5—уравнительный сосуд.
Рис. 117. Ртутные контактные маностаты:
а — O-образный; б —U-образныя.
207
разность давлений устанавливается путем регулирования высоты
положения уравнительного сосуда 3.
В качестве маностатной жидкости лучше всего пользоваться
дибутилфталатом, силиконовым маслом, маслом для
вакуум-насосов.
Ртутные маностаты контактного действия с
помощью реле замыкают или размыкают электрическую цепь, что
сопровождается включением или выключением электродвигателя
вакуумного насоса. Одна из моделей изображена на рис. 117, а.
Маностат состоит из замкнутой О-образной стеклянной трубки с
краном вверху и с боковым отводом, присоединяемым к вакууми-
руемой системе. Нижняя часть трубки наполнена ртутью, которая
постоянно соприкасается с одним нижним контактом и почти
достигает второго контакта. Вначале устанавливают требуемое
разрежение при открытом кране маиостата, после чего кран закры<
вают. При увеличении вакуума ртуть в правом колене
поднимается и замыкает электрическую цепь, в результате чего реле
отключает питание электродвигателя вакуум-насоса.
Маностат другой конструкции (рис. 117,6) представляет собой
манометрическую трубку с небольшим резервуаром, который
соединен краном с нижней частью открытого колена трубки и
трехходовым краном с верхней частью открытого колена и с
атмосферой. Один из контактов введен в нижний изгиб трубки, а
другой— в закрытое колено на середине расстояния между уровнями
ртути в обоих коленах. В этом положении маностат установлен на
нуль. Открывая нижний кран, можно спустить некоторое
количество ртути из первого колена в резервуар и, таким образом,
установить маностат на желаемое остаточное давление, при
достижении которого электрическая цепь будет разомкнута. Для
уменьшения требуемого остаточного давления резервуар маностата,
включенного в эвакуированную систему, соединяют при помощи
трехходового крана с атмосферой, в результате чего ртуть в
правом колене поднимается на нужную высоту,
глава 17. ФИЛЬТРОВАНИЕМ ЦЕНТРИФУГИРОВАНИЕ
ФИЛЬТРОВАНИЕ [13, с. 424; 36, с. 179]
Фильтрование — процесс отделения взвешенных твердых
частиц в жидкостях или газах. Жидкость или газ с находящимися
в них частицами твердого вещества пропускают через пористый
материал (фильтр), размеры пор которого столь малы, что
частицы твердого тела не проходят сквозь фильтр. Размеры пор
определяют способность фильтра задерживать твердые частицы
различной крупности, а также его производительность, т. е.
количество жидкости, которое может быть отделено в единицу
времени.
На процесс фильтрования влияют вязкость жидкости и
разность давлений по обе стороны фильтра. Чем выше вязкость
жидкости, тем труднее ее фильтровать. Так как вязкость жидкости
понижается с повышением температуры, то горячие жидкости
легче фильтровать, чем холодные. Фильтрование вязких жидкостей
часто можно облегчить, разбавляя их растворителем, который по
окончании фильтрования можно легко отогнать. Чем больше
разность давлений, тем выше скорость фильтрования. Поэтому
фильтрование часто проводят при уменьшенном или избыточном
давлении. При фильтровании под давлением студнеобразных осадков
последние плотно прилегают к фильтру, поры которого легко
забиваются, и фильтрование прекращается.;
Если размер частиц твердой фазы меньше размера пор
фильтра, отфильтровать взвесь не удается. Так, обычные бумажные
фильтры не задерживают мелкодисперсные частицы многих
коллоидных растворов. В таких случаях перед фильтрованием
коллоидный раствор нагревают или к нему добавляют электролит,
что приводит к коагуляции (укрупнению частиц и образованию
осадка).
Когда цель фильтрования — получение прозрачного фильтрата,
а не чистого осадка, для лучшего отделения мелкодисперсных
частиц от жидкости к последней прибавляют небольшое количество
порошкообразного активного угля, взбалтывают и фильтруют.
Фильтрование смесей, содержащих вещества, которые забивают
поры фильтра и образуют на нем вязкие слои, часто облегчается
добавлением мелкого кварцевого песка, инфузорной земли,
асбестового волокна, целлюлозной (бумажной) массы.
Фильтрование можно проводить различными способами, в за*
висимости от характера фильтруемых жидкостей и свойств
твердой фазы (осадка), которую нужно отделить от жидкости или
газа.
Если твердая фаза смеси легко осаждается, то большую часть
ее можно удалить перед самим фильтрованием путем декантации.
209
Рис. 118. Декантация при помощи
сифона:
а —наполнение сифона под
давлением; б —проведение декантации на-
сасыванием жидкости в сифон.
Декантация —
наиболее простой метод
разделения твердой и жидкой
фаз — основана на том,
что при отсутствии
перемешивания твердое
вещество оседает на дно
сосуда и прозрачная жидкость
может быть отделена
сливанием с отстоявшегося
осадка. Иногда
декантацию можно использовать и для разделения двух твердых веществ
с различной плотностью. Для промывания труднорастворимых
твердых веществ часто используют декантацию с помощью сифона
(рис. 118). Промывание декантацией значительно более
эффективно, чем промывание осадка на фильтре, где жидкость обычно
не проникает равномерно между частицами твердого вещества.
Фильтрование под действием собственного веса
жидкости
Этот способ фильтрования обычно используется в тех
случаях, когда отфильтрованная твердая фаза не нужна (удаление
механических загрязнений из растворов), или когда жидкая фаза
может быть полностью удалена многократной обработкой осадка
соответствующим растворителем.
Обычное фильтрование применяют, когда приходится
фильтровать горячие концентрированные растворы или растворы
кристаллических веществ в летучих растворителях. При
фильтровании подобных растворов в вакууме растворитель испаряется под
фильтром, который резко охлаждается и забивается
выделяющимися кристаллами.
В качестве фильтрующего материала, в основном,
используют различные сорта фильтровальной бумаги, готовые бумажные
обезжиренные и беззольные фильтры.
Фильтровальная бумага для непосредственного использования
выпускается двух марок: ФНБ — быстрой фильтрации с
размером пор 3,5—10 мкм и ФНС — средней скорости фильтрации с
размером пор 1—2,5 мкм. Зольность бумаги этих марок — до
0,2 о/0.
Для изготовления беззольных и обезжиренных бумажных
фильтров выпускается фильтровальная бумага трех марок:
ФОБ — быстрой фильтрации; ФОС — средней фильтрации;
ФОМ — медленной фильтрации.
210
Ж
уК
ж
Готовые бумажные фильтры круглой формы обезжиренные
(с желтой лентой) и беззольные выпускаются различного
диаметра в пачках по 100 шт. Выбор размера фильтра зависит от
массы отделяемого твердого вещества, а не от объема
фильтруемой жидкости.
Беззольные фильтры для лабораторных работ различаются
по разделительной (задерживающей) способности. Это различие
определяется по цвету бумажной ленты, которой оклеивают
упаковку. Приняты следующие обозначения: белая лента — быстро
фильтрующие, красная — средне фильтрующие, синяя —
медленно фильтрующие, предназначенные для фильтрования
мелкозернистых осадков (типа BaS04).
Выбор марки фильтра в каждом отдельном случае зависит от
свойств отделяемого твердого вещества. Очень плотными
фильтрами следует пользоваться только тогда, когда это действительно
необходимо.
Фильтровальную бумагу и готовые фильтры нельзя
использовать для фильтрования концентрированных растворов сильных
кислот или щелочей, так как при этом снижается механическая
прочность фильтров.
Бумажные фильтры бывают простые и складчатые
(плоеные). Для изготовления простого гладкого фильтра
круглый кусок фильтровальной бумаги определенного размера
складывают в четыре раза и обрезают ножницами так, чтобы
получился сектор круга. Зависимость диаметра фильтра от
диаметра стеклянной воронки для фильтрования представлена ниже:
Диаметр воронки, мм 35 45 55 70 80 100 150 200
Диаметр бумажного
фильтра, мм 55 70 90 ПО 125 150 240 320
Гладкий фильтр должен плотно прилегать к стенкам воронки,
в особенности в верхней части. Для этого рекомендуется при
складывании фильтра сгибать полукруг не по средней линии, а
по близкой к ней параллельной линии.
Сложенный фильтр помещают в воронку (заполнять его
осадком можно не более чем на 7з или V2), смачивают его
дистиллированной водой и заполняют водой носик (трубку)
воронки. Для этого фильтр приподнимают и быстро опускают. Края
фильтра должны быть на 5—10 мм ниже края воронки. Мокрый
фильтр осторожно прижимают к воронке. Фильтрование
начинают немедленно, чтобы носик воронки оставался заполненным
жидкостью. Нельзя наполнять воронку раствором более чем на
3Д объема. Кончик носика должен касаться внутренней стенки
стакана с фильтратом, чтобы предотвратить разбрызгивание.
Простые гладкие фильтры обычно используются в
аналитических лабораториях для фильтрования разбавленных растворов.
Фильтрование значительно ускоряется при пользовании
складчатыми фильтрами. Эти фильтры легко изготовить
(рис. 119). Складки фильтра не должны подходить вплотную к
211
J I г I гт
1 ' V
6 7 6 9 W
Рис. П9. Изготовление складчатого (плоеного) фильтра.
его центру, иначе бумага в центре фильтра может прорваться.
Готовый фильтр вставляют в воронку так, чтобы он прилегал к
ее стенкам. Если воронка имеет угол больше или меньше 60°,
фильтр подгоняют к ней, изменяя положение второго сгиба.
Необходимо, чтобы фильтр имел достаточно острый конец,
фильтровальная бумага не была испорчена многократным сгибанием.
Прежде чем поместить приготовленный фильтр в воронку, его
разворачивают и перегибают так, чтобы внешняя сторона
фильтровальной бумаги оказалась на внутренней стороне фильтра.
Правильно уложенный в воронку фильтр смачивают фильтруемой
жидкостью или дистиллированной водой.
При фильтровании горячих растворов и употреблении
воронок большого диаметра верхушка фильтра может прорваться. Для
устранения этой опасности в большую воронку вставляют
маленькую или специальную дырчатую фарфоровую вставку, а
лучше всего фильтровать через два сложенных вместе складчатых
фильтра.
Оборудование для фильтрования при атмосферном давлении
и комнатной температуре несложно и состоит из воронки,
фильтра, приемника и штатива. Для фильтрования горячих
насыщенных растворов твердых веществ применяют широкие укороченные
воронки, а для быстрого фильтрования больших объемов
жидкостей— рифленые воронки, неровные стенки которых в
сочетании с гладкими фильтрами увеличивают эффективную
фильтрующую поверхность. Воронку укрепляют в кольце, присоединенном
к лабораторному штативу или вставляют ее непосредственно в
горло колбы — приемника фильтрата. В последнем случае
необходимо помещать под воронку полоску фильтровальной бумаги,
чтобы воздух, вытесняемый фильтратом, мог выходить из колбы.
Часто фильтрование бывает затруднено, если между бумажным
фильтром и стенкой воронки образуется прослойка воздуха
(воздушный карман). Чтобы этого избежать, внутри воронки создают
небольшое избыточное давление: воронку накрывают смоченным
по краям куском фильтровальной бумаги и перевернутой
воронкой такого же диаметра. Через трубку верхней воронки при по*
мощи резиновой груши нагнетают воздух и тем самым
устраняют воздушный карман.
212
Для ускорения фильтрования удлиняют трубку воронки: к
носику резиновой трубкой подсоединяют стеклянную трубку того
же (или немного меньшего) внутреннего диаметра. Через
некоторое время вся трубка заполняется столбом фильтрата,
создающим разрежение.
Сильнощелочные растворы и растворы фтористоводородной
кислоты рекомендуется фильтровать через воронку из
пористого полиэтилена. Для изготовления подобной воронки
(рис. 120) используют две стеклянные воронки, из которых
внешнюю закрывают в месте сужения пробкой, а внутреннюю в том
же месте заплавляют. Смесь полиэтиленового порошка и мелко-
измельченного хлорида натрия в массовом соотношении 1 :4
помещают между стенками воронок и выдерживают в сушильном
шкафу при 130—150 °С. Время от времени внутреннюю воронку
поворачивают при надавливании, чтобы равномерно нанести
полужидкую массу на внутреннюю поверхность внешней воронки.
После охлаждения внутреннюю воронку вынимают, извлекают
пробку из трубки внешней воронки и спекшуюся массу
промывают теплой водой для удаления хлорида натрия.
Скорость фильтрования прямо пропорциональна
гидростатическому давлению фильтруемой жидкости, поэтому в процессе
фильтрования больших объемов жидкостей выгодно
поддерживать постоянный уровень жидкости на фильтре. На рис. 121
изображены простые самодельные устройства для автоматического
доливания жидкости на фильтр. Сосуд с жидкостью закрывают
чистой резиновой пробкой, снабженной трубкой для поступления
жидкости и трубкой для поступления воздуха. Уровень нижнего
конца трубки для поступления воздуха определяет уровень
жидкости на фильтре. Если уровень понижается, то воздух проникает
внутрь сосуда и выдавливает жидкость на фильтр. В результате
уровень жидкости на фильтре повышается, а доступ воздуху
внутрь сосуда закрывается.
Рис. 120. Приготовление фильтра из пористого полиэтилена:
J — воронка с заплавленным дном; 2 — воронка; 3 — пористый полиэтилен; 4 — пробка.
Рис. 121. Приборы для непрерывного фильтрования:
а—с бутылью и сифоном; б—с делительной воронкой; в—с перевернутой конической колбой*
213
Фильтрование при нагревании или охлаждении
Фильтрование при нагревании проводят, когда
необходимо очистить горячие концентрированные растворы от
примесей, профильтровать вязкие растворы, а также растворы,
содержащие легко кристаллизующиеся при обычной температуре
вещества.
В первую очередь нужно тщательно выбрать сорт
фильтровальной бумаги, размер фильтра и воронки, с тем чтобы ускорить
процесс. Прежде чем налить горячий раствор на фильтр, воронку
с вложенным фильтром подогревают, пропуская через фильтр
некоторое количество горячего чистого растворителя или пары
растворителя, если его нагреть в бане до кипения. В последнем
случае воронку накрывают часовым стеклом. Перед
фильтрованием растворитель из приемника выливают, чтобы он не
разбавлял фильтрат. На фильтре следует поддерживать высокий
уровень жидкости для ускорения фильтрования.
Воронку с фильтром можно обогревать также металлической
воронкой для горячего фильтрования (рис. 122, а) или воронкой,
между двойными стенками которой пропускают горячую воду,
водяной пар или горячий воздух (рис. 122, б). Подогрев можно также
осуществить погружением электронагревателя в фильтруемый
раствор, если в последнем не содержатся вещества, реагирующие
с металлом.
Для равномерного нагревания стеклянной лабораторной
посуды применяют также вязаные чехлы (колпаки) с
электрообогревом. Они обычно изготовляются из тонкой стеклянной нити и
содержат гибкий нагревательный элемент в виде тонкой
проволоки или спирали [5].
Фильтрование при охлаждении может быть
проведено в воронке, охлаждаемой льдом, или в воронке, между
двойными стенками которой пропускают охлажденный солевой
раствор.
Рис. 122. Воронки для горячего фильтрования:
а — металлическая; б — стеклянная.
214
Фильтрование при пониженном давлении
Фильтрование при пониженном давлении позволяет достиг*
нуть более полного отделения твердого вещества от жидкости и
увеличить скорость процесса.
Аппаратура для фильтрования под вакуумом состоит из
устройства для фильтрования, приемника, водоструйного насоса и
предохранительной склянки.
При фильтровании больших количеств веществ чаще всего
используют дырчатые фарфоровые или щелевидные стеклянные
цилиндрические воронки Бюхнера, вставленные в конические
колбы для фильтрования под вакуумом с тубусом; последние
присоединяются к водоструйному насосу через предохранительную
склянку. Необходимо, чтобы размер воронки соответствовал
количеству отфильтрованного твердого вещества, которое должно
полностью покрывать поверхность фильтра. Однако слишком
толстый слой осадка затрудняет отсасывание и последующее его
промывание.
Фильтр для воронок Бюу.лера представляет собой круглый
лист фильтровальной бумаги, помещаемый на дырчатую
перегородку воронки. Диаметр фильтра должен быть немного
меньше, чем диаметр перегородки. На большие воронки Бюхнера
обычно кладут друг на друга два фильтра. Чтобы подогнанный
бумажный фильтр достаточно плотно прилегал к дырчатой
перегородке воронки, его предварительно смачивают на воронке
растворителем и равномерно прижимают к ней. Затем, после
удаления растворителя, наливают в воронку фильтруемую смесь и
отсасывают.
В случае водных растворов небольшие количества воды,
используемой для смачивания фильтра, не имеют значения. В тех
же случаях, когда присутствие воды недопустимо, влажный
фильтр, добившись плотного его прилегания, промывают
этиловым спиртом или ацетоном, а затем растворителем, присутствие
которого в фильтрате допустимо. Фильтровальная бумага,
смоченная органическим растворителем, не пристает к воронке так
хорошо, как при смачивании водой.
Воронки Бюхнера укрепляются в конических колбах при
помощи резиновых пробок или толстых плоских кусков резины,
закрывающих горло колбы сверху; последние удобны тем, что не
могут быть втянуты в колбу при отсасывании во время
фильтрования.
Для полного отделения маточного раствора осадок на
фильтре отжимают плоской поверхностью стеклянной пробки, или
толстостенным цилиндром с плоским дном до тех пор, пока
жидкость не перестанет капать. При этом необходимо следить, чтобы
на поверхности толстого слоя осадка не образовывались
трещины, так как это ведет к неполному отсасыванию маточного pre*
твора и загрязнению осадка. Чтобы удалить остатки маточного
раствора, осадок промывают на фильтре небольшими порциями
215
растворителя при атмосферном давлении. Когда осадок на
фильтре пропитается растворителем, вновь подключают вакуум для
отсасывания.
При фильтровании с отсасыванием в качестве фильтрующего
материала кроме обычных бумажных фильтров применяют
фильтры из синтетического волокна. Так, фильтры
из поливинилхлоридного или полиэфирного волокна устойчивы к
действию кислот и щелочей, но разрушаются органическими
растворителями.
Для отделения трудно фильтрующихся липких осадков нередко
применяют асбестовую массу, которую можно уплотнить на
воронке для отсасывания или тигле Гуча.
Асбестовую массу готовят следующим образом: в фарфоровой ступке
асбест растирают с конц. НС1, переносят массу в стакан и кипятят
20—30 мин в вытяжном шкафу. Затем разбавляют массу 20—
30-кратным объемом дистиллированной воды, фильтруют на
воронке Бюхнера и промывают водой до исчезновения кислой
реакции в фильтрате. Затем массу высушивают при 100—120 °С и
прокаливают в муфеле. Прокаленный асбест взбалтывают с
водой до получения однородной массы, переносят на фильтрующую
пластину воронки или тигля Гуча, отсасывают и уплотняют.
Чрезвычайно удобны для фильтрования воронки, тигли и
газовые фильтры со впаянной пластинкой из спекшегося
стеклянного порошка. Стеклянные фильтры служат для отделения
твердых веществ от жидкостей при фильтровании и экстракции,
для удаления частиц тумана из газов, для барботирования
(распределения) газов в жидкостях. Стеклянные фильтры, однако,
неудобны в тех случаях, когда требуется количественное
выделение осадка, так как полностью снять осадок с фильтра трудно.
Они не пригодны для фильтрования очень концентрированных
горячих растворов щелочей и карбонатов щелочных металлов.
Пористость пластинок стеклянных фильтров и их обозначен
ния часто менялись. По ГОСТ 9775—69 класс фильтра зависит
от размера пор (табл. 8).
ТАБЛИЦА 8 Области применения стеклянных фильтров
Класс
ПОР 500
ПОР 250
ПОР 160
ПОР 100
ПОР 40
ПОР 16
ПОР 10
ПОР 3,0
ПОР 1,6
ПОР 1,0
Размер пор,
мкм
250-500
160—250
100—160 ;
40-100
16-40
10-16
3—10
1,6-3,0
1,0-1,6
До 1,0
Области применения
Распыление газов в жидкостях
То же ч
Фильтрование грубозернистых и студнеобразных
осадков
Фильтрование кристаллических осадков
Фильтрование мелкокристаллических осадков
Аналитические работы; фильтрование BaS04 итд
Фильтрование коллоидных растворов
То же
Отделение бактериальных взвесей
То же
216
\У
Q
V
Y
Рис. 123. Типы фильтрующих воронок и тиглей:
а —без шлифов типа ВФ; б— с конусными шлифами типа ВФКШ; в—с отводом и шлифами
типа ВФОКШ; г—обратные типа ВФО; д —тигли фильтрующие типа ТФ.
Типы стеклянных воронок и тиглей с пористыми фильтрами
представлены на рис. 123.
Кроме стеклянных изделий с фильтрами для жидкостей
изготовляются также изделия с фильтрами для фильтрования и
промывания газов.
Выпускаются также фильтрующие воронки с
термостатированной трубкой и термостатированной рубашкой (рис. 124). Во*
ронки с электроподогревом предназначаются для
фильтрования в нагретом состоянии кристаллизующихся и вязких при
комнатной температуре растворов и суспензий. Подогрев
фильтрующей воронки до 130 °С исключает застывание раствора, и
фильтрование протекает быстро.
Основной элемент фильтрующей воронки с электроподогревом
и термостатированной трубкой — стеклянный фильтр диаметром
40 мм со впаянной тонкостенной стеклянной трубкой, куда
заключен электронагреватель мощностью 30 Вт. Воронки выпуска*
ются с фильтрами, размер которых составляет 40, 100, 160 мкм.
В фильтрующей воронке с
подогревом термостатирование
обеспечивается проточным
теплоносителем. Объем воронки над
фильтром с термостатированной
трубкой — 80 мл, с термостати-
руемой рубашкой — 58 мл.
Рис. 124. Фильтрующие воронки с
электрообогревом:
а —с термостатированной трубкой; б—с
термостатированной рубашкой.
<&С)
21Т
Рис. 125. Отсасывание в инертной атмосфере:
в —с погружной обратной воронкой; б —с воронкой Бюхнера.
Для отделения жидкости от твердого вещества применяют
обратную погружную фильтрующую воронку
(рис. 123, г). Фильтр погружают в жидкость, и фильтрат поступает
в приемник, с которым фильтр соединен. С помощью этого
устройства удобно проводить фильтрование при пониженной
температуре, поддерживая низкую температуру фильтруемой смеси
посредством охлаждающей бани.
Чтобы отделить малые количества веществ, пользуются
воронкой со стеклянным «гвоздиком», который покрывают круглым
кусочком фильтровальной бумаги. Для этого конец стеклянной
палочки размягчают в пламени горелки и затем расплющивают его,
прижимая к ровной горизонтальной поверхности металлической
пластинки. Необходимо, чтобы фильтр плотно прилегал к
«гвоздику», а края фильтра на 1—2 мм загибались вдоль стенки
воронки. Приемником фильтрата служит пробирка для
фильтрования (с боковым отводом).
Для фильтрования веществ с низкой температурой плавления
или хорошо растворимых при комнатной температуре, пользуются
разрежением при охлаждении. В случае малых
количеств осадка воронку и раствор предварительно охлаждают в
холодильном шкафу. В других случаях воронку Бюхнера
встраивают в склянку с отрезанным дном, заполняя последнюю льдом или
охлаждающей смесью.
При фильтровании в атмосфере инертного газа
пользуются установками, изображенными на рис. 125.
Аналитические аэрозольные фильтры АФА
Для исследования и контроля аэрозолей, содержащихся в
воздухе или других газах, служат фильтры АФА. Фильтры АФА
состоят из отдельного или наклеенного на опорное кольцо
фильтрующего элемента и защитных бумажных колец с выступами.
В качестве фильтрующего элемента используется
фильтрующий материал ФП (фильтр Петрянова) из ультратонких
218
полимерных волокон (ацетилцеллюлозы, перхлорвинила,
полистирола). Рабочая поверхность круглого сечения фильтра 3, 10, 20
и 160 см2.
ЦЕНТРИФУГИРОВАНИЕ [36, с. 179]
Центрифугирование — один из методов разделения
неоднородных систем (жидкость — жидкость, жидкость — твердые
частицы) в роторах под действием центробежных сил. Центрифугиро*
вание выгодно применять, если фильтруемые вещества забивают
поры фильтра, портятся при соприкосновении с фильтрующим
материалом или мелкодисперсны. -
Центрифугирование производят в особых аппаратах,
называемых центрифугами. Основная часть центрифуги — ротор,
вращающийся с большой скоростью.
Типы центрифуг многочисленны; их подразделяют прежде
всего по величине фактора разделения. Он равен отношению
ускорения центробежного поля, развиваемого в центрифуге, к
ускорению силы тяжести. Фактор разделения — величина
безразмерная. Разделяющее действие центрифуги возрастает
пропорционально фактору разделения.
Фактор разделения у выпускаемых отечественной промышлег -
ностью центрифуг с электрическим приводом варьируется в
пределах от 1600 до 300 000, а частота вращения ротора — от 1000
до 50000 об/мин [46, с. 106].
Неоднородные системы в центрифугах разделяют либо
отстаиванием, либо фильтрованием. В зависимости от этого центрифуги
бывают со сплошным ротором либо с дырчатым, покрытым фйль*
трующим материалом.
Центрифугирование отстаиванием производят для освпления
жидкости, содержащей взвешенные твердые частицы, или же для
осаждения твердой фазы. Оно слагается из осаждения твердой
фазы, уплотнения осадка и выделения надосадочной жидкости.
В лабораторной практике применяют различные типы центра
фуг: с ручным или электрическим приводом, настольные
(переносные), передвижные и стационарные. По величине фактора
разделения центрифуги подразделяются на обычные (с
фактором разделения <3 500), суперцентрифуги и ультрацентрифуги
(с фактором разделения ^3500). Обычные центрифуги
используют преимущественно для разделения низкодисперсных
(крупность >10—50 мкм) суспензий различной концентрации.
Суперцентрифуги, в основном, применяют для разделения эмульсий и
высокодисперсных суспензий (крупность <10 мкм). Для
разделения и исследования высокодисперсных систем и
высокомолекулярных соединений распространены аналитические и
препаративные ультрацентрифуги с фактором разделения > 100 000.
Аналитические центрифуги используются для определения молекулярной
массы и степени полимеризации высокомолекулярных
соединений, препаративные—для выделения веществ из растворов,
219
Рис. 126. Положение
пробирок в угловом роторе (а)
и в роторе с качающимися
стаканами (б):
/ — поверхность во время
вращения; 2 —осадок.
которые в обычных условиях находятся в коллоидном состоянии
или в виде неразделимых суспензий (белки, нуклеиновые кислоты,
полисахариды).
Ротор ультрацентрифуги вращается, как правило, в
вакуумной камере при охлаждении (рефрижераторные центрифуги) t
Скорость и время вращения ротора, а также температурный
режим центрифугирования контролируются электронными
устройствами.
Обрабатываемый раствор помещают в специальный сосуд,
который затем вращают с высокой скоростью на роторе
центрифуги. При этом компоненты смеси под действием центробежной
силы распределяются слоями на различную глубину (в
соответствии с массами частиц); наиболее тяжелые частицы
прижимаются ко дну сосуда.
При пользовании пробирочными малогабаритными
переносными центрифугами с ручным или
электрическим приводом суспензию помещают в стеклянные или
пластмассовые пробирки, которые вращаются вокруг главной оси, будучи
подвешенными на цапфах. Пробирочные центрифуги для
периодического разделения малых количеств вещества могут быть двух
типов. В одних пробирки удерживаются цапфами на роторе и
принимают горизонтальное положение при вращении, в других
они жестко укреплены под определенным углом к оси вращения
(угловые роторы).
На рис. 126 показано положение пробирок при
центрифугировании в угловом роторе и в роторе с качающимися стаканами.
После остановки центрифуги прозрачную жидкую фазу (ф у-
гат) сливают или отбирают при помощи пипетки. Осадок
промывают и снова центрифугируют. Если из пробирки нужно
извлечь максимальное количество осадка, то фугат отбрасывают, а
осадок высушивают в вакуум-эксикаторе, не вынимая его из
центрифужной стеклянной пробирки.
При пользовании пробирочными центрифугами пробирки из
толстостенного стекла или из синтетического материала
вставляют в предохранительные металлические стаканы. Дно
стеклянных пробирок предохраняют прокладками из резины. Стеклянные
пробирки можно наполнять до половины объема, а пробирки из
22Q
синтетических материалов при больших частотах вращения
ротора (5000 об/мин) следует наполнять почти доверху для того,
чтобы они под действием центробежной силы не деформировались.
Для обеспечения безопасности работы необходимо очень точно
уравновешивать пробирки с центрифугируемой суспензией.
Нарушение равновесия при больших частотах вращения может
привести к повреждению ротора. Учитывая, что летучие
растворители при центрифугировании могут испаряться, пробирки лучше
закрывать пробками.
Роторы лабораторных пробирочных центрифуг, за
исключением ручных, помещены в предохранительные металлические
чехлы (крышки), чтобы не возникала угроза опасности для
работающих, если пробирка со стаканом сорвется с подвесок.
Необходимо строго соблюдать указания, приводимые в
заводской инструкции для данной центрифуги, нельзя превышать
частоту вращения ротора, указанную в инструкции. Приводить
центрифугу в движение можно лишь при закрытой предохранительной
крышке; открывать крышку разрешается только после полной
остановки центрифуги.
Ручная центрифуга РЦ-4. Эта центрифуга предназначена для
разделения жидкостей различной плотности или для отделения
от жидкостей взвешенных или взмученных в них частиц.
Основные части центрифуги: чугунный корпус, внутри которого
смонтированы шестерни (червячная передача), пробиркодержатель,
рукоятка и струбцина. На шарнирных подвесках пробиркодержате-
ля имеются четыре гильзы, изготовленные из карболита.
Жидкости и твердые частицы, обладающие различной плотностью, при
вращении распределяются в разных местах пробирки. Разделение
можно проводить одновременно в четырех пробирках. За один
оборот рукоятки пробиркодержатель делает восемь оборотов. Для
работы центрифуга крепится зажимом на крышке лабораторного
стола или на специальной подставке.
Лабораторная настольная центрифуга ЦЛН-2. Центрифуга
ЦЛН-2 работает с ротором углового типа РУ 6ХЮ.
Максимальный объем центрифугируемого материала 60 см3. Частота
вращения ротора 3000—8000 об/мин; интервал частоты вращения,
регулируемый переключателем, равен 1000 оборотов. Фактор
разделения достигает 5 500. Время разгона ротора до максимальной
частоты вращения 10 мин; время торможения не более 8 мин.
Время непрерывной работы 60 мин; минимальный обязательный
перерыв 15 мин. Рабочая камера центрифуги закрывается
крышкой с самозакрывающимся устройством. Масса
центрифуги 8 кг.
При работе с центрифугой ЦЛН-2 запрещается: работать без
заземления; увеличивать частоту вращения свыше 8000 об/мин;
работать с открытыми крышками ротора и центрифуги; работать
со стеклянными пробирками'при частоте вращения ротора свыше
4000 об/мин; размещать заполненные центрифугируемым
материалом пробирки не диаметрально противоположно,
221
Разность массы диаметрально расположенных пробирок,
заполненных центрифугируемым материалом, не должна
превышать 0,5 г. Плотность жидкости, разделяемой в пробирках из
полимерных материалов, не должна быть более 2 г/см3, в
стеклянных пробирках — не более 1,5 г/см3.
Угловая малогабаритная центрифуга ЦУМ-1. Центрифуга
имеет ротор — крестовину для одновременного центрифугирования
жидкостей в четырех пробирках вместимостью по 25 мл, четырех
по 10 мл и восьми по 5 мл. Частота вращения ротора от 2000 до
8000 об/мин регулируется ступенчато. Фактор разделения
достигает 6000. Время разгона ротора 8—10 мин. Центрифуга
снабжена электрическими часами, дающими возможность устанавливать
время центрифугирования от 0 до 60 мин, с последующим
автоматическим торможением. Масса центрифуги 16 кг.
глава is. ВЫСУШИВАНИЕ
Под высушиванием (осушением) обычно понимают удаление
воды или остатков растворителя из жидкого, твердого или
газообразного вещества.
Высушивание можно проводить физическими методами,
обычно используемыми для разделения и очистки веществ (испарение,
вымораживание, экстракция, азеотропная перегонка, дистилляция,
сублимация и др.)» а также с помощью осушающих реагентов [5,
с. 58; 45, с. 9].
При выборе способа осушения следует учитывать агрегатное
состояние вещества, его химические свойства, содержание воды
или другого вещества, которое надо удалить при сушке, и
требуемую степень осушения.
ОСУШАЮЩИЕ ВЕЩЕСТВА
Химические осушающие реагенты можно разделить по
способам связывания ими воды на три основные группы.
1. Вещества, образующие с водой гидраты. Это безводные соли
(СаСЬ, К2СО3) или низшие гидраты, переходящие при контакте с
водой в устойчивые высшие гидраты [Mg(C104)2-2H20].
2. Вещества, поглощающие воду в результате химической
реакции, например некоторые металлы (Na, Ca) и оксиды (Р4Ою,
СаО).
3. Вещества, поглощающие воду за счет физической
адсорбции, например активный оксид алюминия, силикагель, цеолиты.
Вещества, образующие гидраты
Хлорид кальция СаС12 наиболее часто используется как
наполнитель осушающих трубок и колонок при сушке газов, как
поглотительный реагент в эксикаторах и для непосредственного
осушения многих органических жидкостей.
Хлорид кальция применяют в порошкообразном или
прокаленном виде. Порошкообразный безводный СаС12 содержит, как
правило, небольшое количество основной соли Са(ОН)С1. Хлорид
кальция — осушитель средней эффективности. Мало эффективен
для осушения НС1, НВг, HI, Br2, S03 и совершенно непригоден
для осушения аммиака и аминов, с которыми образует
комплексные соединения. Хлорид кальция можно употреблять
неоднократно, если его после каждого использования регенерировать
прокаливанием.
Концентрированная серная кислота H2S04—эффективный
реагент для осушения газов, с которыми H2S04 не реагирует
223
(Н2, 02, N2, С12, СН4, С2Н6, СО, НС1, N20 и др.). Запрещаете*
применять серную кислоту в вакуум-эксикаторах в качестве во-
допоглощающего средства [56, с. 148].
Конц. H2S04 — довольно сильный окислитель, особенно при
нагревании. Она окисляет HI и частично НВг (но не НС1) до
свободных галогенов. Поэтому ее нельзя использовать для
осушения этих веществ, а также H2S, РНз, AsH3, HCN, непредельных
углеводородов, аммиака, аминов. Осушающая эффективность
H2S04 резко снижается по мере постепенного разбавления ее
водой. Так, 95,1% кислота проявляет уже значительно меньшую
эффективность, чем 98,3% кислота. Конц. H2S04 иногда содержит
S02. Поэтому перед осушением газов нужно нагреть кислоту до
появления дыма, при этом S02 полностью удаляется.
Перхлорат магния (ангидрон) Mg (СЮ4)2 —
высокоэффективный осушающий реагент, может служить для осушения
большинства газов.
Ангидрон применяется для поглощения паров воды в
элементном анализе органических веществ при определении содержания
водорода, а также для определения абсолютной влажности
воздуха. По эффективности высушивания ангидрон не уступает
оксиду фосфора (V), выгодно отличаясь от последнего тем, что
применяется в виде зерен, не спекается при поглощении паров воды
и не образует в колонке каналов.
Перхлорат магния поступает в продажу также и в виде три-
гидрата Mg(C104)2*3H20, который по осушающему действию
сопоставим с конц. H2S04.
При использовании перхлоратов следует иметь в виду, что
сильные минеральные кислоты и кислотные оксиды разлагают их
с выделением свободной хлорной кислоты, способной взрываться
дри взаимодействии с осушаемым газом. Поэтому нельзя
последовательно соединять поглотительный сосуд с Mg(C104)2 и про-
в ыватель с конц. H2S04.
Карбонат калия безводный (плавленый поташ) К2СО3
применяют для осушения жидкостей и растворов веществ в
органических растворителях, когда можно не опасаться щелочности
реагента (осушение органических оснований, спиртов и т. д.). В
лабораторных условиях осушитель готовят непродолжительным
нагреванием товарного карбоната калия на металлической
сковороде.
Сульфат натрия безводный Na2S04 — относительно
малоэффективный осушитель. Его применяют для осушения растворов
органических веществ в неполярных растворителях (бензол, ди-
этиловый эфир и др.). Получают прокаливанием Na2SO410H2O на
металлической сковороде.
Сульфат магния безводный MgS04 — более эффективный и
емкий осушитель, чем безводный Na2S04. Получают
прокаливанием MgS04-7H20 при 210—250 °С
224 .
Сульфат кальция безводный Ca2S04 по осушающей
эффективности сходен с конц. H2S04. Применяют для осушения газов и
жидкостей, а также для наполнения эксикаторов.
Гидроксиды натрия и калия NaOH и КОН применяют для
наполнения поглотительных трубок, колонок (при осушении газов)
и эксикаторов, а также для непосредственного осушения некото*
рых органических жидкостей. Плавленый NaOH для осушения
газов столь же эффективен, как и гранулированный СаСЬ.
Эффективность плавленого КОН во много раз больше эффективности
NaOH.
Гидроксиды щелочных металлов часто используются для
одновременного поглощения Н20 и С02.
Вещества, связывающие воду в результате химической реакции
Оксид фосфора (V) Р4О10 — исключительно эффективный
осушающий реагент, однако очень неудобен в обращении. Под
действием паров воды порошок Р4О10 превращается в тягучую клейкую
массу, покрытую непроницаемой вязкой пленкой, что создает
большое сопротивление току газа. Поэтому Р4О10 обычно наносят
на стеклянную или асбестовую вату, стеклянные бусинки или
кусочки прокаленной пемзы. Пемзу нагревают в фарфоровой чашке
до 100°С и затем смачивают конц. Н3РО4. Затем на пемзе при
перемешивании распределяют оксид фосфора. В результате
образуются удобные в обращении гранулы реагента.
С галогенами (за исключением фтора) оксид фосфора не
реагирует. С сухими HF, HC1 и НВг образует оксигалогениды и ме-
тафосфорную кислоту:
Р40ю + 2НС1 —v 2НР03 + 2Р02С1
Оксиды кальция и бария СаО и ВаО рекомендуются
исключительно для осушения низших спиртов, в которых образующиеся
гидроксиды кальция и бария нерастворимы.
Натрий — весьма эффективный реагент для осушения
углеводородов, простых эфиров и др. Поверхность металла быстро
покрывается слоем гидроксида, и дальнейшее осушение
замедляется. Поэтому стремятся вносить металл с возможно большей
удельной поверхностью, например в виде тонкой проволоки. Нат*
рий можно применять для осушения жидкостей, содержащих лишь
незначительное количество воды.
Гидрид кальция СаН2 — очень эффективный осушающий
реагент. Его реакция с водой протекает необратимо в широком
интервале температур.
Гидрид лития-алюминия UAIH4 — один из наиболее
эффективных осушающих реагентов. Его применяют только для полного
удаления следов влаги из органических жидкостей.
Вещества, связывающие воду в результате адсорбции
Преимущество сорбентов заключается в том, что они
доступны, большей частью химически инертны по отношению к осушаю-
8 Зак. 1237 225
щему газу, не создают значительного сопротивления току газа
(при использовании их в зерненом виде) и легко регенерируются
нагреванием в токе сухого воздуха.
Крупнозернистый активный оксид алюминия (алюмо-
гель)— более эффективный осушающий реагент, чем
силикате л ь.
По осушающей активности цеолиты намного превосходят
алюмогель и силикагель. Цеолиты некоторых марок интенсивно
поглощают пары воды даже при 100 °С, а аммиак при 250—
300СС, когда силикагель полностью теряет активность. Так,
например, цеолит марки КА адсорбирует при обычной температуре
преимущественно молекулы воды. При 70 °С 1 см3 таблетирован-
ного цеолита КА удерживает 62—85 мг Н20. Свойства этих сор«
бентов приведены в гл. 29.
ВЫСУШИВАНИЕ ТВЕРДЫХ ВЕЩЕСТВ
Процесс высушивания твердых веществ большей частью
основан на испарении влаги, которое может быть проведено при
комнатной температуре или при нагревании. Влага испаряется в том
случае, когда давление паров воды над поверхностью твердого
осушаемого вещества превышает парциальное давление паров
воды в окружающей газовой фазе. Давление паров воды в
осушаемом веществе резко возрастает с увеличением температуры.
Поэтому высушивание стараются осуществлять при повышенной
температуре. Снизить парциальное давление паров воды в
газовой фазе можно применением вакуума или осушением с помощью
веществ, эффективно поглощающих влагу из газовой фазы.
Многие твердые негигроскопичные вещества можно
высушивать па открытом воздухе при обычной температуре. Влага
с поверхности вещества будет испаряться до тех пор, пока не
установится равновесие между давлением водяных паров в
испытуемом веществе и в воздухе. Для ускорения процесса, если это
допустимо, высушивание проводят при движении воздуха или
перемешивании материала. Толщина слоя высушиваемого
материала не должна превышать 1—2 см. В результате высушивания
на воздухе получают воздушно-сухой продукт с весьма
неравномерным содержанием остаточной влаги. Часто высушивание на
воздухе предваряет высушивание другими методами.
Высушивание твердых веществ на воздухе лучше всего проводить на
фильтрокерамических пластинках; при высушивании на
фильтровальной бумаге продукт загрязняется ее волокнами.
Осушаемое на воздухе вещество целесообразно покрывать
фильтровальной бумагой, чтобы защитить его от пыли и
механических загрязнений. Кроме того, надо учитывать фотохимическое
действие освещения на продукт. Так, многие бромиды при
высушивании на воздухе желтеют под действием света.
Твердые вещества, устойчивые термически, могут быть
высушены в сушильных шкафах. В сушильных шкафах нельзя
226
удалять летучие вещества, например остатки летучих
органических растворителей, так как смесь паров растворителя с
воздухом может взорваться при контакте с проволочной спиралью
нагревателя, и нельзя высушивать низкоплавкие вещества.
При высушивании мелкокристаллических веществ на их
поверхности может образоваться плотная корка, значительно
снижающая скорость осушения. В этих случаях осушаемое вещество
в процессе сушки следует многократно перемешивать. Вещества,
легко разлагающиеся или изменяющиеся при нагревании до
100°С, следует сушить в вакуум-сушильных шкафах.
В последнее время в лабораторной практике стали применять
сушильные установки [37], в которых в качестве источника тепла
используют инфракрасные лампы. Инфракрасные лучи с
длиной волны 1000—3000 нм обладают достаточной проникающей
способностью и не вызывают химических изменений в осушаемом
веществе. Сушка происходит при более низкой температуре и
быстрее, чем при обычном нагревании веществ. Приборы для
высушивания материалов инфракрасным облучением выпускаются
серийно. Потребляемая мощность лампы 500 Вт. Время
высушивания навески в 3 г от 5 до 10 мин. Вначале включают лампу, и в
центр освещенного круга помещают резервуар термометра.
Регулируя высоту рефлектора, создают требуемую температуру
для осушения вещества. После этого в центр освещенного круга
помещают сосуд с осушаемым веществом на установленное
время.
Высушивание твердых веществ воздухом, осушаемым
химическими реагентами, в лабораторных условиях осуществляется в
эксикаторах. Осушающий реагент подбирают в зависимости
от химических свойств высушиваемого вещества. Чаще всего на
дно эксикатора помещают безводный СаСЬ, Mg(C104h, P4O10,
плавленый КОН, силикагель, цеолиты. Для удаления остатков
углеводородных растворителей в качестве заполнителя для
эксикатора применяют парафиновые стружки или полоски
фильтровальной бумаги, пропитанные расплавленным парафином.
В эксикаторе водяные пары перемещаются вследствие
диффузии или конвекционных токов и поэтому высушивание
происходит медленнее, чем в токе воздуха. Для ускорения процесса при
комнатной температуре используют вакуум-эксикаторы.
Вакуум создается обычно водоструйным насосом. В тех случаях,
когда малые количества вещества необходимо осушить в вакууме
при повышенной температуре, применяют прибор, называемый
«осушительным пистолетом» (рис. 127). В реторту 4
помещают поглотитель влаги (Р4О10, СаС12, адсорбенты). В колбу 3
наливают до половины объема жидкость с определенной
температурой кипения и вносят несколько «кипятильных камешков».
В сосуд 1 в фарфоровой лодочке 5 вносят высушиваемое вещество.
Кран реторты соединяют с вакуум-насосом. Жидкость в колбе 3
нагревают до кипения. Горячие пары омывают сосуд /,
конденсируются в холодильнике и вновь стекают в колбу <3. Через некото-
8* 227
Рис. 127. «Осушительный пистолет» ПСВ:
У —сосуд; 2 — обратный холодильник; 3 — колба; 4— реторта;
5—фарфоровая лодочка.
рое время в сосуде / устанавливается
температура, равная температуре паров
применяемой жидкости.
В качестве теплоносителя обычно
применяют негорючие жидкости: хлороформ
(^кип = 61 °С), трихлорэтилен (tKllu = 86 °С),
воду (^ип = 100 °С), тетрахлорэтилен (/Кип=
= 120 °С), трихлорэтан (/кип=146 °С).
Твердое вещество (осадок) можно
обезвоживать экстракцией
растворителем, который смешивается с водой, но в котором осадок не
растворяется или очень плохо растворяется. Например, для быстрого
высушивания осадков применяют ацетон, метиловый или этиловый
спирт, эфир. Высушивание влажных кристаллических осадков
может быть выполнено одним из следующих приемов.
1. Высушиваемое вещество помещают в коническую колбу с
пришлифованной стеклянной пробкой, куда прибавляют
соответствующий растворитель в таком количестве, чтобы над осадком
был слой растворителя в несколько сантиметров. Колбу
закрывают и энергично встряхивают около 1 мин, после чего дают
отстояться 15—20 мин. Затем осторожно сливают растворитель и
заменяют его свежей порцией. Растворитель меняют 3—4 раза, после
чего осадок переносят на воронку с пористым дном (воронка
Бюхнера), отфильтровывают при разрежении и, если осушаемое
вещество негигроскопично, высыпают на керамическую пористую
плитку, покрывают листом фильтровальной бумаги и оставляют
на воздухе (или под тягой) до полного испарения растворителя.
Гигроскопические вещества досушивают в вакуум-эксикаторе или
в вакуум-сушильном шкафу.
2. Высушиваемое вещество помещают на воронку с пористым
стеклянным дном и понемногу поливают высушивающей
жидкостью (растворителем). Затем воронку присоединяют к установке
для отсасывания и отфильтровывают растворитель. Отключив
установку от источника вакуума, осадок на фильтре разрыхляют
стеклянной палочкой или фарфоровым шпателем, вновь
приливают растворитель, дают осадку постоять под слоем
растворителя 10—15 мин, после чего вновь подключают установку к
источнику вакуума. Фильтруют до тех пор, пока не перестанет
чувствоваться запах растворителя. Когда это достигнуто, отключают
вакуум, а обезвоженный осадок помещают в банку.
ВЫСУШИВАНИЕ ЖИДКОСТЕЙ И РАСТВОРОВ [7. 14,20,70]
Жидкости, содержащие относительно большие количества
воды, высушивают в два этапа: сначала физическими методами, а
228
КЪахддь
линии
затем доосушивают с помощью химических осушающих
реагентов и адсорбентов.
Некоторые органические жидкости, содержащие воду, можно
предварительно осушить высаливанием — прибавлением к
ним электролита, не растворяющегося в органическом
растворителе, но растворяющегося в воде. Происходит разделение
жидкости на два слоя. Водный слой может быть отделен, а
органический— доосушен и очищен дистилляцией. Вещество, которым
проводят высаливание, может быть добавлено в твердом виде или в
виде концентрированного водного раствора; например при помощи
NaCl можно удалить большую часть воды из водного раствора
метилэтилкетона.
Жидкости, не образующие с водой раздельно кипящих (азео-
тропных) смесей, часто удается осушить фракционной
перегонкой на эффективной колонке. Условие успешного проведения
осушения — достаточно большая разница температур кипения
осушаемой жидкости и воды. Этим методом, например, можно
получить почти сухой метиловый спирт, доосушение которого
достигается с помощью химических осушающих средств
(металлический кальций, амальгама алюминия) и на цеолите КА.
Если осушаемое вещество очень плохо растворяет воду, но
образует с ней двойные или тройные азеотропные смеси, то его
можно осушить, отогнав небольшую часть его вместе с водой. До
тех пор, пока отгоняется бинарная смесь, дистиллят остается
мутным.
В сочетании с азеотропной отгонкой осушение можно
проводить методом экстракции. К осушаемой жидкости
прибавляют такое количество не смешивающегося с водой
органического растворителя, чтобы отделился водный слой, после чего
остаток воды из раствора органического растворителя удаляют
азеотропной перегонкой.
Осушение органических жидкостей чаще всего проводят при
их непосредственном контакте с осушающим
реагентом. Осушитель, образующий с водой концентрированные
растворы (СаС12, К2СО3, КОН), прибавляют к осушаемому
веществу частями, а образующийся раствор осушающего реагента
в воде отделяют в делительной воронке. По окончании
высушивания жидкость отделяют от твердого осушающего реагента
фильтрацией.;
В случае водных растворов термически нестойких веществ
применяют лиофильную сушку. Принцип проведения лио-
фильной сушки весьма прост. Водный раствор полностью
замораживают в тонком слое и выдерживают в вакууме 1,33—266 Па
(0,01—2 мм рт. ст.). При этом давлении вода быстро испаряется
(возгоняется) и замороженный раствор постепенно охлаждается.
Удаляемые водяные пары улавливают в охлаждаемых ловушках
или при помощи адсорбентов. Лиофильная сушка не
сопровождается вспениванием, приводит к образованию мелкокристал-
лического продукта повышенной растворимости, предохраняет
229
продукт от окислительного действия кислорода воздуха и
сохраняет биологическую активность осушаемых веществ.
Для осушения органических жидкостей широко используются
адсорбенты — алюмогель и цеолиты. Вместе с водой
адсорбенты поглощают и многие другие загрязнения. Так, например,.
цеолит СаА может быть использован для избирательного
поглощения полярных веществ (Н20, H2S и др.) из неполярных
жидкостей. Цеолит NaA применяют для глубокой осушки различных
фракций нефти и многих продуктов нефтехимического синтеза.
ОСУШЕНИЕ ГАЗОВ
Газы осушают химическими реагентами и вымораживанием.
При большой скорости газа равновесие насыщенных водяных
паров над осушителем, как правило, не успевает установиться.
Степень осушения газа зависит от свойств осушителя, толщины слоя
и величины поверхности осушителя, соприкасающейся с газом.
Осушение газов твердыми реагентами проводят обычно в
поглотительных устройствах (абсорберах), изображенных
на рис. 128, и в сосудах для твердых промывателей — склянке
Тищенко (рис. 129, а). При наполнении поглотительных устройств
необходимо обеспечить равномерное распределение реагента, с тем
чтобы в нем не образовались каналы. Для того чтобы укрепить
слой осушителя и предотвратить унос его частичек с газом, в
поглотительные устройства в местах входа и выхода газа
помещают небольшие тампоны стеклянной ваты. После наполнения
поглотительных устройств следует убедиться, не создается ли в них
слишком сильного сопротивления току осушаемого газа. Если
это так, то наполнение повторяют с большими кусками
осушающего реагента или же смешивают осушитель с кусками пемзы
или пористого фосфора.
Для осушения газов конц. H2S04 используют сосуды для
жидких промывателей (рис. 129). При этом необходимо
обеспечить хороший контакт газа с осушающим реагентом и
следить за тем, чтобы капельки реагента не уносились током газа.
Это достигается подбором высоты осушающего слоя и скорости
газа. Сосуды для жидких промывателей можно включать по два
последовательно.
Рис. 128. Поглотительные
устройства для осушения
газов химическими
реагентами:
а — горизонтальная
осушительная трубка; б, г —
хлоркальциевые трубки;
в — утка для сушки газа
над слоем Р4О10; д —
осушительная колонка.
-32
Ш
W
230
Рис. 129. Сосуды для жидких промывателей:
л —склянка Тищенко; б—с распыляющей насадкой; в—с изогнутым газопромывателем.
Рис. 130. Колонка с самоорошением:
/, 2 — трубки для ввода газа; 3 — насадка; 4 — трубка.
Эффективные приборы для промывки газов —
поглотительные колонки с орошаемой насадкой из обрезков
стеклянных трубок, стеклянных колец или шариков. Преимущество
колонок с орошаемой насадкой проявляется в том, что не
приходится создавать заметного избыточного давления для
прохождения газа.
На рис. 130 изображена поглотительная колонка с
самоорошением для очистки газа. Газ проходит в трубку 1,
Дополнительный поток газа поступает в трубку 2. Увлекая в тройнике
капельки жидкости, газ гонит их цепочкой по трубке 4 вверх. Выходя
из узкого отверстия над насадкой 3, пузырьки газа лопаются и
разбрызгивают жидкость по насадке. Стекающая жидкость
отделяется от газа в приемнике и снова возвращается в цикл. Трубку
4, в которой поднимается цепочка пузырьков, делают узкой, так
как в противном случае цепочка будет рваться.
Для высушивания газов (паров) наибольшее значение имеют
адсорбенты (оксид алюминия, силикагель, цеолиты).
Безводный силикагель, содержащий немного хлорида кобальта, окрашен
в синий цвет, а при насыщении влагой становится розовым.
Таким образом, по внешнему виду сорбента, находящегося в
осушительной колонке, можно судить о его пригодности для
дальнейшего высушивания.
Высокой степени высушивания газов можно достигнуть
вымораживанием, т. е. охлаждением их до низкой
температуры. При вымораживании газ пропускают через трубку,
погруженную почти до дна сосуда, который помещен в охлаждающую
баню.
231
глава 19 ОПРЕДЕЛЕНИЕ ВЛАЖНОСТИ
В лабораторных условиях влажность можно определить как
прямыми, так и косвенными методами. К прямым относятся
гравиметрический, азеотропно-дистилляционный, денситометриче-
ский и химический методы, а к косвенным — психрометрический, по
точке росы, сорбционный, кулонометрический и диэлькометриче-
ский. В настоящем разделе будут рассмотрены лишь некоторые
из них.
ПРЯМЫЕ МЕТОДЫ
Гравиметрический метод
Возможны два варианта определения влажности твердых
веществ гравиметрическим методом: термическая сушка и
поглощение влаги осушителями. В первом варианте о содержании
влаги в твердом веществе судят по уменьшению массы образца после
продолжительной воздушно-тепловой сушки в
сушильном шкафу до постоянной массы как при
обычном, так и при пониженном давлении. Часто высушивание
твердого вещества в сушильном шкафу при обычном давлении
производят при 105—115 °С. Метод применим для определения
влажности веществ, устойчивых к повышенной температуре и
действию кислорода и относительно легко отдающих воду при
нагревании.
Эффективность высушивания твердых веществ нагреванием
зависит от состояния, в котором вода находится в веществе.
Многие вещества, особенно тонкоизмельченные, адсорбируют на
поверхности так называемую гигроскопическую влагу.
Некоторые твердые вещества содержат внутри мелких пор своих
частиц (или кристаллов) так называемую
окклюдированную (сорбированную) влагу. Гигроскопическая и
окклюдированная влага объединяются под общим названием несте-
хиометрическая влага; при 100—130°С и атмосферном
давлении полностью удалить ее не удается.
Стехиометрическая влага— это
кристаллизационная влага. Многие твердые вещества существуют в
форме кристаллогидратов, устойчивых на воздухе в определенных
границах влажности последнего. Количество кристаллизационной
влаги, участвующей в образовании данного кристаллогидрата
строго определено (например, CuS04-5H20, Na2S04- IOH2O). Для
определения содержания кристаллизационной влаги необходимо
по справочникам или опытным путем выяснить, при какой
температуре данное вещество теряет свою кристаллизационную воду*
232
В коллоидных системах связанная вода удаляется высушиванием
при 100—125 °С лишь частично. Более полное удаление воды
достигается высушиванием твердых веществ в вакуум-су-
шильном шкафу до постоянной массы при остаточном
давлении 2,66—3,99 кПа (20—30 мм рт. ст.) и при 50—70 °С.
Некоторые твердые материалы сушат ускоренным методом,
нагревая их при более высокой температуре, что сокращает
время высушивания до 40 мин. Для экспрессного определения
влажности ряда материалов часто применяют инфракрасное
облучение (см. гл. 18).
При определении влажности твердых материалов
высушиванием возникают погрешности, которые следует учитывать.
Высушивание органических веществ часто сопровождается
наряду с потерей влаги потерей летучих продуктов; одновременно
происходит поглощение кислорода, а иногда и термическое
разложение вещества. Удаление связанной воды при высушивании
часто происходит лишь частично.
Техника определения влажности твердых веществ методом
термической сушки при атмосферном давлении сводится к
следующему. Навески измельченного вещества помещают в
предварительно взвешенный бюкс. Высота слоя исследуемого вещества
должна быть не более 5—10 мм. Бюкс с навеской взвешивают
(как и пустой) на аналитических весах с точностью до 0,0002 тА
Взвешенную навеску в бюксе с открытой крышкой помещают на
полку предварительно нагретого до 105 °С сушильного шкафа и
шкаф закрывают. (Нельзя помещать бюкс на дно шкафа.)
Периодически следят за температурой высушивания по термометру,
помещенному в верхнее отверстие шкафа. Желательно, чтобы
шарик термометра был на одном уровне с бюксом. Во время
высушивания образца отверстие шкафа для выхода паров должно
быть открыто. В сушильном шкафу можно одновременно сушить
несколько навесок, если только влажность их не сильно разнится.
Когда намеченное время высушивания истечет (2—4 ч), бюкс
закрывают крышкой и помещают в эксикатор, содержащий
СаС12. Затем переносят эксикатор в весовую комнату и ставят
около весов для охлаждения. Через 20—30 мин открывают
эксикатор и взвешивают бюкс. Затем бюкс снова помещают в
сушильный шкаф и с открытой крышкой выдерживают 30—40 мин,
охлаждают и взвешивают. Когда масса бюкса с навеской
перестанет убывать, определение заканчивают и вычисляют
процентное содержание влаги (массовую долю воды) в испытуемом
образце.
Определение влажности твердых материалов. Для
определения процентного содержания влаги в образцах древесных стру*
жек предназначены весы ВЛВ-100г. Они же могут быть
использованы и для определения влажности других термически
стойких твердых материалов. Это квадрантные весы с
механизмом компенсации тары, оборудованные сушильной камерой с
терморегулятором. Массу определяют по отсчетной шкале, счет-
233
чикам гиревого механизма и делительного устройства, а содержа-
ние влаги — по прилагаемой к прибору таблице. Масса
исследуемого образца должна быть не менее 15 г; наибольший предел
взвешивания 100 г; диапазон измерения влажности 0—12%
(масс).
Для ускоренного определения влажности зерна служит
вакуум-тепловая установка ОВЗ-1М. Основные
элементы установки: термостат с терморегулятором, вакуум-насос
ВН-461, аналитические весы, размалывающее устройство.
Точность измерения влажности 0,1%; предельное остаточное
давление 1,06 кПа (8 мм рт. ст.); предельная температура 130°С;
точность регулирования температуры ±0,5°С; продолжительность
проведения анализа \ ч.
Для ускоренного определения влажности формовочных
материалов служит прибор, состоящий из станины с расположенным
на ней реле времени и электродвигателем для вращения стола.
К станине крепится стойка, на которой смонтирован кожух.
В верхней части кожуха находятся патрон с термоизлучающей
инфракрасной лампой и окна для охлаждения цоколя и
параболического зеркала лампы и отвода испаренной влаги. Расстояние
между нижней плоскостью термоизлучающей лампы и
высушиваемыми образцами регулируется подъемом или опусканием
кожуха. Выключение лампы и привода стола происходит
автоматически после срабатывания реле времени. При мощности
термоизлучающей лампы в 500 Вт средняя продолжительность
высушивания образца 3—5 мин. Весы в комплект прибора не входят.
Определение влажности газов (воздуха). Анализируемый газ
пропускают через сосуд, заполненный твердым
поглотителем влаги (см. гл. 18). Сосуд с поглотителем взвешивают до
и после пропускания через него определенного объема газа. Па
разности масс находят содержание влаги в измеренном объеме
газа.
Поглотитель влаги не должен взаимодействовать с
анализируемым газом, что может привести к изменению массы. Время
контакта поглотителя с газом зависит от осушающей способности
поглотителя, длины поглотительного слоя и скорости потока. Me*
тод применим для измерения больших концентраций влаги в га*
зах.
Дистилляционно-азеотропный метод [49]
Метод азеотропной отгонки воды с растворителем
(бензол, толуол, четыреххлористый углерод, тетрахлорэтан и др.)
заключается в том, что испытуемое вещество кипятят с избытком
растворителя, не смешивающегося с водой, но образующего с ней
азеотропную смесь. Смесь паров растворителя и воды
конденсируют с помощью обратного холодильника, а конденсат собирают
в приемник-ловушку. Если растворитель легче воды, используют
приемник-ловушку Дина — Старка, а если тяжелее — приемник
234
Рис. 131. Приемники-ловушки:
л—Дина — Старка; б — Александера.
Рис. 132. Аппарат типа АКОВ-10:
1 — холодильник; 2 — приемник-ловушка; 3 —колба круглодонная; 4—распылительная трубка.
Александера (рис. 131). В приемнике-ловушке конденсат
разделяется на два слоя: органический растворитель и воду. Избыток
растворителя стекает обратно в перегонную колбу. Дистилляцию
продолжают до извлечения последних следов воды, о чем судят
по наступающей прозрачности дистиллята и прекращению
изменения объема воды в приемнике-ловушке. Измеряя объем воды,
отогнанной из известной массы осушаемого вещества, можно
судить о содержании в нем влаги. На этом основан стандартный
метод определения содержания воды в нефтяных, пищевых и
других продуктах. Для этих целей выпускается аппарат АКОВ-10
(рис. 132) с приемниками-ловушками Дина — Старка без крана
и с краном, вместимостью 2, 10 и 25 мл.
Дистилляционный метод может служить лишь для
приближенного определения влажности жидких и твердых материалов,
содержащих более 1 —10% (масс.) воды.
Содержание воды дистилляционно-азеотропным
методом определяют следующим образом: навеску испытуемого
вещества, содержащую не более 5—8 г воды, помещают в круг-
лодонную колбу, предварительно промытую и высушенную в
сушильном шкафу, приливают 100 мл безводного бензола (или
другого растворителя) и присоединяют к приемнику-ловушке с
обратным холодильником. Колбу нагревают на водяной (или
водно-солевой) бане с закрытым электрическим нагревом и ведут
отгонку азеотропной смеси так, чтобы из трубки обратного
холодильника в ловушку падали 2—4 капли в секунду. Перегонку
прекращают, как только объем воды в приемнике-ловушке
перестанет увеличиваться и верхний слой растворителя станет
совершенно прозрачным. Если в конце перегонки в трубке обратного
235
холодильника задерживаются капли воды, то их снимают
стеклянной распылительной трубкой. После того как колба охладит-»
ся, прибор разбирают и измеряют объем отогнанной воды.
Денситометрический (экстракционный) метод
Денситометрический метод основан на определении плотности
жидкого экстракта, полученного при извлечении влаги из
исследуемого образца твердого материала водопоглощающей
жидкостью (диоксан, этиловый спирт). По значению плотности
бинарной смеси находят ее состав, пользуясь заранее построенным
калибровочным графиком.
Экстрагентом может служить вещество, хорошо
растворяющееся в воде или смешивающееся с ней во всех отношениях и
обладающее плотностью, отличной от плотности воды. Экстрагент
не должен вступать во взаимодействие с компонентами
исследуемого вещества. Метод применим для определения влажности
веществ, содержащих более 1 % воды.
Химический метод
Определение влажности реактивом Фишера. Реактив Фишера,
представляющий собой раствор возогнанного иода, безводного
пиридина и сухого диоксида серы в абсолютном метиловом
спирте, применяют для определения влажности твердых, жидких и
газообразных веществ. В качестве растворителя обычно
используют безводный метиловый спирт. Если испытуемое вещество
нерастворимо в метиловом спирте, навеску тонкоизмельченного
вещества взбалтывают или настаивают с определенным
количеством метилового спирта, после чего смесь титруют реактивом
Фишера. В качестве растворителей могут также быть использованы
ледяная уксусная кислота и безводный хлороформ (при анализе
жиров, масел и Др.)- При определении влажности следует
вводить поправку на растворитель. Метод неприменим к
соединениям, реагирующим с компонентами реактива Фишера
(восстановители, карбонаты и гидрокарбонаты щелочных металлов,
карбонильные соединения и др.).
Взаимодействие реактива Фишера с водой протекает в две
стадии:
1) I2 + S02 + H20 + 3C5H5N —► 2C5H5N.Hi4-C5H5N.SO3
пиридин
2) C5H6N. S03 + CH3OH —> C5H5N.HSO4CH3
Метиловый спирт служит не только растворителем, но и ком*
понентом, взаимодействующим с сульфотриоксидом пиридина а
образованием пиридиновой соли метилсерной кислоты.
Метод Фишера относится к числу титриметрических методов
анализа и отличается универсальностью, высокой
чувствительностью и точностью, применим для анализа в широком диапазоне
236
Рис. 133. Прибор для визуального титрования
воды реактивом Фишера:
/ — резервуар (бутыль) с реактивом Фишера; 2
—груша; 3—хлоркальциевые трубки; 4~бюретка; 5—тит-
ровальная колба; 6—магнитная мешалка.
влагосодержаний. Обычно конец
титрования определяют визуально
или электрометрическим способом;
в последнее время появились и
автоматические титраторы влаги.
Метод прямого визуального
определения содержания воды по Фишеру
положен, например, в основу стандартного
метода анализа окиси этилена (ГОСТ
7568—64).
Этапы работы: приготовление
стандартных растворов воды в метиловом спирте;
приготовление реактива Фишера; определение титра реактива Фишера;
проведение испытания.
Стандартные растворы Н20 в СИ3ОН с содержанием Н20 ^ 1 г/л (для
окиси этилена марок А и Б) и <6 г/л (для марки В) готовят следующим об-,
разом: в две мерные колбы вместимостью по 500 мл помещают соответственно
0,5 и 3 г дистиллированной воды, взвешенной с точностью до 0,0002 г и
выдерживают в термостате при 20 °С. В колбы приливают до метки абсолютированный
СН3ОН с температурой 20 °С Полученные растворы сохраняют в запасных
емкостях 1 микробюреток для титрования (рис. 133). Растворы защищают от
влаги воздуха, закрывая бюретки хлоркальциевыми трубками с СаС12. Далее,
готовят реактивы Фишера I и II.
Чтобы приготовить реактив I с титром, равным 0,006 г/мл, 84,7 г свеже-
возогнанного иода растворяют в 269 мл сухого пиридина *. После полного
растворения иода в колбу наливают 556 мл абсолютированного СН3ОН. Реактив II
готовят растворением 25,4 г иода в 81 мл сухого пиридина. После полного
растворения иода в колбу доливают 900 мл абсолютированного СН3ОН. Через
2 суток реактивы I и II охлаждают в бане с льдом. Затем к реактиву I
осторожно приливают 15 мл диоксида серы (получение S02 см. гл. 21), а к
реактиву II — 13,5 мл.
Реактивы Фишера годны к употреблению по истечении суток после их
приготовления. Допускается применение реактива Фишера заводского изготовления.
Реактив Фишера хранят в сухом месте в темных или обернутых черной бумагой
стеклянных бутылях, плотно закрытых стеклянной пробкой.
Титр реактивов I и II определяют следующим образом. При помощи
микробюреток в конические колбы вместимостью 50 мл с притертыми пробками
отбирают три пробы по 5 мл стандартного раствора Н20 в СН3ОН и три пробы
по 6 мл СН3ОН, взятого для приготовления стандартного раствора Н20 в СН3ОН.
Пробы титруют соответствующим реактивом Фишера (I или II) до перехода
желтой окраски раствора в красновато-коричневую. Во время титрования
стеклянную пробку колбы заменяют резиновой, через которую проходит носик
микробюретки с титраитом (реактивом) и трубка с СаС12. Расхождение между
параллельными результатами титрования допускается не более чем на 0,2 мл.
Титр реактива Т (в г/мл) вычисляют по формуле:
т 5G
~ 500 (V -V0
* Сухой пиридин готовят, высушивая пиридин в течение 10 дней над
плавленым КОН и затем перегоняя; собирают фракцию с температурой кипения
114—117 °С.
237
где (/ — навеска Н20, взятая для приготовления стандартного раствора Н20 в
СН3ОН, г; V — объем реактива Фишера, пошедший на титрование стандартного
раствора Н20 в СН3ОН, мл; V\ — объем реактива Фишера, пошедший на
титрование СН3ОН, мл; 500 — объем мерной колбы, взятой для приготовления
стандартного раствора Н20 в СН3ОН, мл; 5 — объем стандартного раствора Н20
в СН3ОН, мл.
Титр реактива Фишера в течение первой недели после его приготовления
проверяют один раз в сутки, в последующее время — один раз в неделю.
При определении содержания воды в окиси этилена в коническую колбу
вместимостью 50 мл отбирают из микробюретки 5 мл абсолютированного СН3ОН.
Пипеткой, промытой изнутри несколько раз окисью этилена, вносят в метиловый
спирт 5 мл предварительно охлажденной окиси этилена (опуская кончик пипетки
в спирт). Колбу закрывают пробкой, энергично перемешивают и тотчас же
титруют реактивом Фишера до глубокой красно-коричневой окраски. В тех же
условиях титруют 5 мл СН3ОН.
Содержание воды в окиси этилена [в % (масс.)] вычисляют по формуле:
^_ (У— УО Г-100
Х 0,8972
где V — объем реактива Фишера, пошедший на титрование, окиси этилена в
5 мл СН3ОН, мл; V\ — объем реактива Фишера, пошедший на титрование 5 мл
СН3ОН, мл; У2— объем окиси этилена, взятой для определения, мл; Т — титр
реактива Фишера (по Н20), г/мл; 0,89 — плотность окиси этилена при 5 °С, г/мл.
Конечную точку титрования реактивом Фишера в настоящее
время большей частью определяют физико-химическими
методами. Для электрометрического и визуального определения
конечной точки титрования выпускается настольный прибор ПМВ,
нижний предел измерений которого 5 • 10—3 % Н20. Вместимость
прилагаемых к прибору пипеток: 2 ± 0,005 и 5 ± 0,01 мл.
Для полуавтоматического титрования реактивом Фишера с
установлением конечной точки титрования по заданному
потенциалу служит титр а тор «Влага», нижний предел измерений
которого Ю-4 % Н20. Титратор предназначен для определения
содержания влаги в органических растворителях. Титратор
Л Т В - 3 7 5, служащий для определения малых содержаний
влаги в органических растворителях, действует по принципу кулоно-
метрического и амперометрического титрования.
Титраторы выпускаются в комплекте с автоматическими
бюретками. В прилагаемых инструкциях приводится описание
приборов и правил работы с ними.
Определение влажности реакцией с гидридом кальция. Один
из методов химического определения влажности жидкостей, не
реагирующих с щелочноземельными металлами и их гидрокси-
Дами, основан на взаимодействии гидрида кальция с водой,
содержащейся в анализируемой жидкости, с последующим
измерением объема выделившегося при этом водорода:
СаН2 + 2Н20 = Са (ОН)2 + 2Н2
Примером этого метода может служить определение содержания воды в
полиэтилсилоксановых жидкостях. Массовая доля воды в этих жидкостях по
ГОСТ 13004—77 не должна превышать 0,01%.
В реакционную колбу 1 прибора (рис. 134) помещают 100 ±1 г
анализируемой жидкости. В боковой отросток колбы помещают около 1 г измельчен-
238
Cart
Рис. 134. Прибор для определения
массовой доли воды:
1—реакционная колба с отводом для СаНг;
2—склянка Дрекселя; 3 — одноходовой кран;
4—трехходовой кран; 5 —бюретка; 6 —
уравнительная склянка.
ного в порошок гидрида кальция.
Реакционную колбу поворачивают так,
чтобы СаН2 попал в жидкость, и
тщательно перемешивают содержимое
колбы. Выделившийся водород
собирают в газовую бюретку 5,
постепенно опуская уравнительную склянку 6.
Колбу встряхивают каждые 10—
15 мин Отсчет показаний бюретки
производится через 15 мин после
встряхивания колбы. Определение
считают законченным, когда два отсчета, сделанные через 15 мин, совпадают.
Массовую долю воды х (в %) вычисляют по формуле:
V • 273 (Р - р) - 0,000804
Х~~ 101300 (273 + t) m
где У —объем Н2, выделившегося при анализе, мл; Р — барометрическое
давление, Па; р — давление паров Н20 при температуре анализа, Па; 0,000804 —
коэффициент для пересчета объема Н2 (в мл), приведенного к нормальным
условиям, на массу Н20, г; t — температура, при которой измерен объем Н2, °С;
т — навеска анализируемой жидкости, г.
КОСВЕННЫЕ МЕТОДЫ [21]
Все косвенные методы определения влажности основаны на
влиянии влажности на ту или иную физическую или физико-
химическую величину, более удобную для измерения и
дальнейших преобразований.
Психрометрический метод
Для характеристики влажности воздуха применяются
следующие величины: парциальное давление водяного пара (в Па);
абсолютная влажность — количество водяного пара в 1 м3
воздуха (в г/м3); относительная влажность — отношение давления
водяного пара, содержащегося в воздухе, к максимальному
давлению пара при данной температуре (в %)• Относительную
влажность воздуха обычно определяют с помощью психрометров
Августа и Ассмана.
Психрометр Августа состоит из двух одинаковых
ртутных термометров, с ценой деления до 0,2 °С, укрепленных рядом
на особом штативе. Резервуар одного из термометров обернут
марлей или батистом, опущенным в стаканчик с водой. С
поверхности смачиваемого («влажного») термометра вода испаряется
тем сильнее, чем оуше воздух. «Влажный» термометр показывает
более низкую температуру, чем «сухой». По разности показаний
239
Рис. 135. Аспирационнын
психрометр Ассмана:
1, 2 — термометры; 3 —
пружина заводного
механизма; 4 — защитные планки;
5—прорези; 6 —
аспираторная головка; 7
—стержень для подвешивания
прибора; 8 — вентилятор;
9 — щиток для защиты
вентилятора; 10 — пипетка
для воды; //
—воздухопроводная трубка;^?
—защитная трубка.
сухого и влажного термометров, пользуясь данными
психрометрической таблицы, прилагаемой к прибору, находят
относительную влажность воздуха.
Показания влажного термометра зависят от скорости
движения воздуха в непосредственной близости от него. Скорость
испарения воды с поверхности влажного термометра повышается при
увеличении скорости движения воздуха. Поэтому психрометр
Августа используется только для приближенных определений
влажности воздуха в закрытых помещениях, где скорость движения
воздуха мала. Психрометр устанавливают на расстоянии 1,5—•
1,6 м от пола, ограждая от источников лучистой энергии.
Продолжительность наблюдения 10—15 мин.
Аспирационный психрометр Ассмана
представлен на рис. 135. При вращении вентилятора в прибор всасывается
воздух, который обтекает резервуары термометров, проходит по
воздухопроводной трубке 11 к вентилятору и выбрасывается им
240
наружу через прорези 5. Сухой термометр покажет температуру
потока воздуха, а показания влажного термометра будут меньше,
так как он охладится вследствие испарения воды с поверхности
батиста.
Влажность воздуха определяется по разности показаний
сухого и влажного термометров с помощью психрометрической
таблицы, прилагаемой к прибору.
Еще более совершенной конструкцией обладает аспирацион-
ный психрометр с электродвигателем,
установленным в аспирационной головке. Основные технические
характеристики психрометра аналогичны характеристикам прибора Ассмана,
за исключением механизма аспирации.
Метод определения влажности по точке росы
Предельное содержание паров воды в любом газе зависит от
температуры: чем она ниже, тем меньше воды содержится в
газовой фазе. Отсюда следует, что определенной влажности газа
соответствует строго определенная температура, при которой
появляются следы жидкой фазы воды. Поэтому удобной характеристикой
влажности воздуха (неконденсируемых газов) может служить
температура начала конденсации водяных паров — точка росы.
Зависимость влажности от точки росы представлена ниже:
Точка росы»
°С
0
-10
—20
-30
Влажность,
% (об.)
0,602
0,256
0,102
0,0371
Точка росы,
°С
-40
-50
-60
-70
Влажность,
% (об.)
0,0127
0,00388
0,00106
0,00046
Среди различных приборов, используемых для определения
влажности воздуха по точке росы, относительно прост
конденсационный влагомер (рис. 136). Анализируемый воздух
(газ) пропускают через стеклянную камеру / и следят за
появлением конденсата (росы) на полированной поверхности зеркаль-
N "V2 5 б
N
ЧУ
Рис. 136. Конденсационный влагомер:
/ — камера; 2—зеркальце; 3—металлический
стержень; 4—сосуд Дьюара; 5—термопара;
6 — милливольтметр.
Рис. 137. Гигрометр.
241
на 2. Температуру зеркальца постепенно понижают, погружая
металлический стержень 3 в сосуд Дьюара 4 с охлаждающей смесью
из «сухого льда» и этилового спирта. В момент помутнения
поверхности зеркальца определяют температуру с помощью
термопары 5 и гальванометра 6. После этого повышают температуру
зеркальца, медленно уменьшая глубину погружения стержня в
охлаждающую смесь, и фиксируют момент исчезновения
конденсата (помутнения). Среднее значение этих двух отметок
температуры считают точкой росы.
Гигрометрический метод
Для непосредственного определения относительной влажности
воздуха применяют волосяные гигрометры, принцип действия
которых основан на зависимости удлинения волоса от относительной
влажности: при изменении влажности от 0 до 100%
относительное удлинение волоса достигает 2,0—2,5%.
Волосяной гигрометр (рис. 137) представляет собой
металлическую рамку, посередине которой вертикально натянут
светлый обезжиренный волос. Верхний конец волоса укреплен
неподвижно, а нижний перекинут через блок и слегка натягивается
небольшим грузом. К блоку прикреплена стрелка, которая в
зависимости от изменения длины волоса перемещается вдоль шкалы,
укрепленной горизонтально посередине металлической рамки и
градуированной, в процентах относительной влажности. Точность
отсчета по шкале ±1%, продолжительность наблюдения 20—
30 мин. Чувствительность волоса со временем меняется, а поэтому
показания гигрометра следует периодически проверять по аспира-
ционному психрометру.
Кроме человеческих волос, применяют животные пленки
толщиной 5—30 мкм или синтетические пленки, например из
целлофана.
При определении влажности воздуха в помещении гигрометр
подвешивают на стене вдали от источников тепла.
Диэлькометрический метод
В основе этого метода лежит тот факт, что диэлектрическая
проницаемость (ДП) большинства веществ намного меньше
соответствующего значения для воды. Анализ проводят на установках
для измерения ДП в ячейках, параметры которых находят по
эталонным жидкостям с известной величиной ДП [49]. Этим методом
может быть проанализирована любая бинарная смесь воды и
органического растворителя. Метод принят в качестве стандартного
при определении влажности брома (ГОСТ 454—70). Широкое
распространение диэлькометрический метод получил при
определении влажности зерна и муки (влагомеры зерна АФИ-1, ИВЗ),
изделий целлюлозно-бумажной и других отраслей
промышленности.
242
глава». РАБОТА СО СЖАТЫМИ И СЖИЖЕННЫМИ
ГАЗАМИ
Газы — важные реагенты для препаративных и аналитических
работ (хлор, водород, окись этилена, сероводород, аммиак и др.)»
вспомогательные средства для выполнения работ в инертной
атмосфере (азот, аргон, диоксид углерода), для хроматографии
(азот, гелий, аргон), для создания низких температур (жидкий
азот, жидкий кислород и др.).
Сжатые и многие сжиженные газы хранят и транспортируют в
металлических баллонах. Небольшие количества сжиженных
газов (фосген, бутадиен, окись этилена и др.) иногда хранят в
запаянных стеклянных ампулах.
Сжатые газы, критическая температура которых ниже
комнатной (Н2, N2, 02, Аг, Не), находятся в баллонах в газообразном
состоянии. Количество сжатого газа в баллоне пропорционально
давлению газа и объему баллона. При отборе части газа
давление в баллоне снижается.
Давление внутри баллона со сжиженными газами (С12, С0С12,
С02, S02, СзНв, С4Н10 и др.) сохраняется постоянным до тех пор,
пока не испарится вся жидкость.
Сжиженные газы, используемые в качестве криогенных средств
(N2, 02 или воздух), доставляются в лаборатории в металлических
сосудах Дьюара. Газы в баллонах часто содержат примеси,
предельное содержание и методы определения которых приводятся в
соответствующих ГОСТах и ТУ.
СОСУДЫ ДЬЮАРА
Сосуды Дьюара предназначены для хранения и
транспортировки сжиженных газов, работ с охлаждающими смесями, хранения
веществ при низких температурах, выполнения различных
исследований в области низких температур.
Сосуды Дьюара представляют собой двухстенные сосуды
цилиндрической или шаровой формы, стеклянные или металлические,
у которых пространство между стенками вакуумировано.
Теплообмен в сосудах Дьюара, в основном, происходит за счет
теплопроводности вдоль стенок сосуда и лучеиспускания. Теплообмен
за счет теплопроводности можно снизить, применяя сосуды с
узким горлом и высоковакуумную изоляцию, а также уменьшая
остаточное давление в межстеночном пространстве. Снижение
теплообмена за счет лучеиспускания в стеклянных сосудах
достигается тем, что внутренние стенки сосудов посеребрены или
покрыты медной пленкой, чтобы зеркальная поверхность отражала
лучи,
243
Сосуды Дьюара бывают цилиндрические и шаровой формы,
узко- и широкогорлые, различной вместимости.
Цилиндрические стеклянные сосуды лабораторного назначения
выпускаются на деревянной или пластмассовой подставке,
вместимостью от 250 до 3700 см3, шарообразные — в защитном
проволочном или металлическом кожухе, вместимостью от 500 до 5000 см3.
Вместимость металлических сосудов 5, 15, 25 и 50 л.
Металлические сосуды Дьюара имеют на средней части
круговую полосу: для азота — черную, с надписью желтого цвета
«Азот»:, для кислорода — голубую с надписью черного цвета
«Кислород»; для аргона — черную с надписью желтого цвета
«Аргон»; для воздуха — черную с надписью белого цвета «Воздух».
При работе с металлическими сосудами Дьюара следует
руководствоваться действующей «Инструкцией по эксплуатации и
хранению металлических сосудов Дьюара» [56, с. 154].
Металлические сосуды Дьюара наполняют жидкими газами
через металлическую воронку с трубкой, которая должна быть
значительно длиннее горловины сосуда. В сосуде не должно быть
влаги и посторонних предметов. Сжиженный газ необходимо
заливать осторожно во избежание выброса жидкости из горловины.
Запрещается: закрывать пробками горловины сосудов Дьюара,
протирать внутреннюю поверхность горловины ветошью и другими
обтирочными материалами, а также использовать сосуды в
качестве тары для других веществ; хранить органические вещества
вблизи сосуда Дьюара, в котором содержится жидкий кислород;
проводить операции с жидкими воздухом и кислородом руками,
загрязненными маслом; курить и пользоваться открытым огнем в
помещении, где находятся заполненные жидким кислородом
сосуды Дьюара; применять для охлаждения чистый жидкий
кислород, не разбавленный жидким азотом в объемном соотношении
1:3.
Жидкие газы при попадании на тело человека вызывают
обморожение. При работе с сжиженными газами необходимо
пользоваться прорезиненными или кожаными рукавицами.
БАЛЛОНЫ ДЛЯ ГАЗОВ [64; 56, с. 153]
Баллоны для хранения и транспортировки сжатых и
сжиженных газов представляют собой цилиндрические сосуды,
изготовленные из стальных бесшовных труб, со слегка выпуклым днищем
(рис. 138). В горловине баллона находится запорный вентиль с
боковым штуцером. Вентиль открывается и закрывается
поворотом маховичка, надетого на шпиндель (стержень).
Баллоны различаются по типу резьбы запорного вентиля:
вентили баллонов с водородом, этиленом, пропаном и некоторыми
другими горючими газами имеют левую резьбу, с остальными
газами (С02, 02, N2 и др.) —обычную правую.
На кольцо горловины надевают колпак, защищающий вентиль
баллона от грязи и повреждений. Колпак имеет два отверстия для
пломбировки баллона.
244
Рис. 138. Газовые баллоны:
а —простой; б —с сифоном;
/ — корпус; 2—сифонная трубка; 3—вентиль; 4—колпак; 5 — пяткабаллона.
Рис. 139. Баллон для хлора:
а — разрез баллона; б —перевернутый баллон в стойке;
J—корпус; 2—сифонная трубка; 3—вентиль; 4—колпак; 5 — горловина.
Вентиль вмонтирован в штампованный корпус с боковым
штуцером для присоединения редуктора и заглушкой, закрывающей
отверстие штуцера и предохраняющей его от засорения и
повреждения резьбы. Вентиль кислородного баллона ввинчивают в
горловину баллона на водной замазке из глета, не содержащего
жировых веществ, на фольге или жидком натриевом стекле.
Несколько иначе устроены баллоны для хлора и фосгена.
Вентиль этих баллонов имеет сифонную трубку, доходящую почти до
дна баллона (рис. 139). При работе баллон перевертывают вверх
дном, и тогда по сифонной трубке в вентиль поступает
газообразный хлор или фосген. К вентилю присоединяют газовый редуктор,,
позволяющий регулировать давление выпускаемого газа.
В зависимости от газа баллоны окрашивают снаружи в
различные цвета, делают соответствующие надписи и наносят отличи*
тельные цветные полосы в соответствии с правилами [54].
На верхней сферической части каждого баллона должны быть
отчетливо нанесены клеймением следующие данные: товарный
знак завода-изготовителя; номер баллона; фактическая масса
порожнего баллона; дата (месяц и год изготовления) и год следую-*
щего освидетельствования баллона; рабочее давление Р; пробное
давление П; вместимость баллона; клеймо ОТК
завода-изготовителя; номер стандарта для баллонов вместимостью свыше 55 л.
На баллонах вместимостью до 5 л паспортные данные могут
быть выбиты на пластинке, припаянной к баллону, либо нанесены,
эмалевой или масляной краской.
Эксплуатация баллонов
Баллоны эксплуатируют в соответствии с «Правилами
устройства и безопасной эксплуатации сосудов, работающих под
давлением» [54]. Эти правила распространяются на все виды сосудов^
(баллоны, реакторы, автоклавы), работающие под избыточным
245-
давлением свыше 0,07 МПа. Лица, имеющие непосредственное
отношение к эксплуатации баллонов, должны быть обучены этим
правилам, и их знания следует периодически проверять.
Согласно правилам, запрещается наполнять газом баллоны,
у которых истек срок периодического освидетельствования, нет
установленных клейм, неисправны вентили, поврежден корпус,
окраска и надписи не соответствуют требованиям. Никогда не
следует снижать давление газа в баллоне до атмосферного.
Остаточное давление газа в отработанном баллоне должно быть не менее
0,05 МПа.
Газы из баллонов в емкость с более низким давлением
выпускают только через редуктор, предназначенный исключительно для
данного газа; окраска редуктора должна соответствовать окраске
баллона с газом. При неисправности вентиля баллон с газом
возвращают на завод или наполнительную станцию с надписью
«полный, неисправен вентиль».
Наполненные баллоны с насаженными башмаками хранят в
вертикальном положении в специальных стойках или прикрепляют
с помощью железных хомутов к рабочему столу или вытяжному
шкафу. Баллоны малого объема могут быть укреплены на рабочем
столе с помощью штатива. При падении баллона может
произойти взрыв, особенно при минусовых температурах, когда ударная
вязкость стали понижается и она делается хрупкой. Переносить
баллоны можно только на носилках или специальных лямках-
креплениях. Запрещается переносить баллоны на руках.
Прочность баллонов может нарушиться из-за переполнения их
сжатыми и особенно сжиженными газами, что приводит к
повышению давления выше допустимого значения, поэтому количество
заполняющих баллоны газов строго регламентируется по массе и
давлению.
Особые требования предъявляются к баллонам с ацетиленом,
который может разлагаться со взрывом в результате повышения
давления и температуры, а также в присутствии инициаторов
разложения. Чтобы уменьшить опасность взрыва, баллоны заполняют
пористой массой (уголь, пемза), пропитанной ацетоном, в котором
растворим ацетилен.
Баллоны с газами рекомендуется устанавливать вне здания
в специальных металлических шкафах. При такой установке
баллонов газ к рабочим местам подается по медным трубам, а
ацетилен— по стальным бесшовным трубам.
В одном металлическом шкафу (или в одном помещении)
запрещается размещать баллоны с кислородом и горючим газом.
В помещении лаборатории запрещается хранить более одного
баллона, наполненного горючим газом.
Баллоны, наполненные газами, должны быть удалены от
электрических щитков и отопительных батарей на расстояние не менее
1 м и защищены от действия солнечных лучей, так как повышение
температуры газа в баллоне приводит к резкому повышению
давления и может быть причиной разрыва баллона. Запрещается ра-
246
ботать с жидким воздухом или кислородом в помещениях, где горят
газовые горелки, включены электроприборы, искрящее
оборудование и другие источники воспламенения.
В помещении лаборатории производить какой-либо ремонт
арматуры баллонов со сжатыми и сжиженными газами запрещается.
Перед работой с наполненным баллоном необходимо тщательно
проверить, не истек ли срок очередного освидетельствования
баллона, соответствуют ли окраска и надписи на баллоне действующим
правилам Госгортехнадзора, нет ли видимых повреждений на
корпусе баллона, исправен ли вентиль, нет ли следов жира или масла
на баллоне, нет ли пропуска газа из запорного вентиля.
Отбор жидкости и газа из баллонов [36, с. 622]
Отбор сжиженного газа из баллона осуществляется довольно
просто. Баллон с газом перевертывают вентилем вниз и наклоняют
его так, чтобы жидкость могла вытекать через открытый вентиль.
При этом следует учитывать, что низкокипящие жидкости
интенсивно испаряются и сильно охлаждаются.
В ряде случаев баллоны со сжиженными газами оборудованы
специальной трубкой, позволяющей отбирать жидкости из баллона,
находящегося в горизонтальном положении. При открытии вентиля
сжиженный газ выталкивается из баллона давлением паров и
может быть подан по трубке в нужный сосуд.
Перед отбором из баллона сжатого газа сперва следует
установить и укрепить баллон. Затем отворачивают колпак (разводным
ключом) и снимают его с баллона. Внимательно осматривают
вентиль баллона, нет ли на нем следов масла, не просачивается ли газ
и не повреждена ли резьба бокового штуцера.
Неплотности в вентиле баллона проверяют, смачивая вентиль
мыльной водой. Если вентиль неисправен, образуются пузыри.
Категорически запрещается применять для проверки открытое пламя.
Отбор проб газа из баллонов должен производиться через
редуктор (мембранный вентиль) или через запорный игольчатый
вентиль. Схема присоединения редуктора к баллону приведена на
рис. 140.
Большинство запорных вентилей и редукторов изготовляют из
бронзы. При работе с аммиаком и ацетиленом, реагирующим
с медью, используют железные вентили.
Между резьбой запорного вентиля баллона и накидной гайкой
редукционного вентиля помещают уплотняющие прокладки. Для
баллонов с Н2, N2, C02 и другими не вызывающими коррозии
газами используют кожаные, резиновые или фибровые прокладки.
При работе с вызывающими коррозию газами — Cl2, HC1, S02,
NH3 — применяют асбестовые уплотнения. Для кислородных
баллонов уплотняющие прокладки должны изготовляться из
негорючих материалов.
До присоединения редуктора рекомендуется провести продувку
вентиля; для этого открывают вентиль быстрым поворотом махо-
247
Рис. 140. Присоединение редуктора к баллону:
1 — горловина баллона; 2 — вентиль; 5 —маховичок; 4—накидная гайка; 5 —манометр
высокого давления; 6 — редуктор; 7 — регулировочный винт; Я — манометр низкого давления; 9 —
запорный кран.
вичка на *Д оборота против часовой стрелки и тотчас закрывают.
При продувке вентиля экспериментатор должен стоять сбоку или
сзади бокового штуцера вентиля. Затем осматривают накидную
гайку редуктора и прокладку в ней, ослабляют регулировочный
винт редуктора, поворачивая его налево, и присоединяют редуктор
к вентилю баллона, надежно закрепляя накидную гайку ключом.
Закрепив редуктор, начинают медленно и осторожно открывать
вентиль баллона, поворачивая маховичок против часовой стрелки.
Когда манометр высокого давления на редукторе покажет
давление газа в баллоне, то с помощью регулировочного винта редуктора
выпускают газ и устанавливают ток нужной скорости или нужное
рабочее давление (по показанию манометра низкого давления).
Скорость тока газа легко контролируется с помощью счетчика
пузырьков— небольшого стеклянного сосуда, в котором газ пробуль-
кивает через жидкость (например, склянка Тишенко с конц. H2S04
или с 50% раствором КОН в случае NH3). Счетчик пузырьков
присоединяют к редуктору при помощи вакуумной резиновой трубки.
По окончании работы вентиль баллона плотно закрывают, дают
снизиться давлению в манометре высокого давления редуктора и
только тогда закрывают выход газа из редуктора, поворачивая
регулировочный винт влево.
ИЗМЕРЕНИЕ ОБЪЕМА ГАЗА [46, с. 112]
В лабораторной практике объем газа измеряют с помощью
газовых бюреток, газометров, реометров, ротаметров и газовых часов.
Для точного измерения относительно небольших объемов газа
(до 100 мл) применяют газовые бюретки. Это градуирован-
248
ные стеклянные трубки, на одном конце которых имеется кран.
К другому концу бюретки с помощью резиновой трубки
присоединяется уравнительная склянка, заполненная запирающей
жидкостью (раствор соли, ртуть). Перед отсчетом объема газа в
бюретке, через несколько минут после отбора пробы, давление газа
в бюретке уравнивают с атмосферным. Для этого уравнительную
склянку подносят вплотную к бюретке и устанавливают в таком
положении, чтобы поверхность жидкости в бюретке и уравнительной
склянке была на одном уровне.
При отсчете показаний бюретки глаз наблюдателя должен быть
на одном уровне с установленным уровнем запирающей жидкости.
Отсчет в бюретке с ртутным затвором производят по верхнему
мениску, а с водным — по нижнему.
Поскольку объем газа изменяется в зависимости от
температуры и давления, одновременно с ним измеряют эти величины.
Объем газа обычно приводят к нормальным условиям, т. е.
к температуре 0°С и давлению 1013 гПа (760 мм рт. ст.) по
уравнению:
VP 273
0 ~~ 760 (273 + О
где V — объем газа при температуре опыта; Р — давление, при
котором измерен объем газа.
Если газ содержит пары воды, или находился в сосуде над
водой или водным раствором, его объем к нормальным условиям
приводят по уравнению:
_ У (Р-Я,) 273
0 "" 760 (273 + /)
где Pi —давление паров воды при температуре опыта.
Чтобы устранить ошибки при измерении объема газа, обусловь
ленные изменением температуры и атмосферного давления, приме*
Рис. 141. Газовая бюретка с компенсационным устройством:
7 — манометр; 2, 3—краны; 4 —компенсационная трубка; 5—напорная склянка.
Рис. 142. Градуированный газометр с напорной воронкой:
/—манометр; 2, 3, 4—краны; 5—напорная воронка.
249
"-=С^~
рис. 143. Реометры:
а—с диафрагмой; б—с капилляром.
V^7
няют бюретки с ко м-
пенсационной
трубкой и водяным
манометром (рис. 141).
Все отсчеты
производятся при одной и той же
высоте жидкости в обоих
коленах манометра 1.
Если компенсационная трубка 4 во время измерения не сообщается
с атмосферой, то изменение атмосферного давления не влияет на
давление в компенсационной трубке и все измерения объема газа
производятся при одном давлении; изменение объема газа в
бюретке, обусловленное изменением температуры, уравновешивается
изменением давления воздуха в компенсационной трубке.
Компенсационная трубка, по размеру близкая к размеру
бюретки, помещается в муфту рядом с бюреткой. Нижний конец трубки
запаян, верхний открыт. В трубку вводят около 1 мл
дистиллированной воды. Жидкость (подкрашенная вода) в обоих коленах
манометра устанавливают на одном уровне. Одно колено
присоединяют к открытому концу компенсационной трубки, другое — к
бюретке. Для проведения отсчета объема газа поворотом крана 2
колено манометра сообщают с воздухом, а бюретку — с
манометром. Осторожным поднятием или опусканием напорной склянки 5
жидкость в обоих коленах манометра доводят до одного уровня и
перекрывают краны манометра и бюретки. В бюретку набирают
пробу газа и с помощью напорной склянки уравнивают давление
в бюретке с атмосферным, после чего сообщают бюретку с
манометром. Поднятием или опусканием напорной склянки доводят
жидкость в обоих коленах манометра до одного уровня и
производят отсчет.
Газометры, предназначаемые для отбора и хранения газов
(см. гл. 23), также могут быть использованы для измерений объема
газа.
При измерении объема газа градуированными газометрами
с напорной воронкой (рис. 142) отсчет производят после
приведения объема газа к атмосферному давлению, которое устанавливают
по манометру /. При повышенном давлении выпускают часть воды
через кран 3, при пониженном добавляют воду из воронки,
открывая кран 4.
С увеличением вместимости газометра точность измерения
объема газа снижается.
Дозирование газов часто осуществляют путем измерения
скорости движения газового потока в единицу времени. Для этой цели
используются реометры и ротаметры.
Стеклянные лабораторные реометры
выпускаются двух модификаций: РКС — со съемным и постоянным капил-
250
лярами и РДС — с диафрагмой (рис. 143). При пропускании газа
через капиллярную трубку реометра на концах ее создается
разность давлений, отмечаемая жидкостным дифференциальным
манометром, одно колено которого включено в систему до
капилляра, а другое — после капилляра. С увеличением скорости
пропускания газа увеличивается разность уровней жидкости в
манометрической трубке.
Пределы измерений, л/мин
Реометр
РКС
РДС
0-0,06; 0-0,10; 0-0,25;
0-0,40; 0-0,60 и 0—1,00
0—4; 0-6; 0—10; 0-16;
0-25; 0-40; 0-60 и 0-100
Капилляр реометра перед употреблением должен быть програ-
дуирован по тому газу, для измерения которого он предназначен.
В первую очередь заполняют манометрическую трубку реометра
жидкостью (водой, дибутилфалатом, парафиновым маслом и др.).
Чем меньше плотность жидкости, наполняющей реометр, тем
последний чувствительнее и тем меньший объем газа можно измерять
с его помощью. Чистый капилляр присоединяют к манометрической
трубке и реометр включают в систему, состоящую из емкости с
газом, манометра и газометра, заполненного водой (рис. 144). Газ,
предназначенный для градуировки, пропускают с постоянной
скоростью через реометр и избыток выводят через кран 7 в вытяжную
систему.
Когда установится постоянная скорость газа, поворотом крана 7
соединяют реометр с газометром и включают секундомер. Воду,
вытесняемую из газометра, сливают через кран 8, чтобы давление
^
Рис. 144. Калибрование реометра:
/ — манометр; 2—бутыль с тубусом; 5—реометр; 4—регулятор давления; 5—уравнительная
трубка; 6—тройник; 7—трехходовой кран; 8% 9 — краны.
Рис. 145. Ротаметр:
/—вертикальная трубка; 2—волчок.
251
в газометре в течение всего опыта было равно атмосферному.
Через определенные промежутки времени записывают показания
реометра и объем вытекшей воды.
Каждый капилляр градуируют на 3—4 различные скорости и
определение повторяют 3—4 раза. По окончании градуировки
строят градуировочную кривую, выражающую зависимость
скорости газового потока от высоты столба жидкости в манометре.
Для измерения скорости потока газа применяются также
ротаметры с различными диапазонами измерений. Ротаметр
(рис. 145) представляет собой стеклянную трубку, сужающуюся
книзу и тщательно отшлифованную внутри. Газ поступает в
ротаметр снизу и выходит в верхней его части. В зависимости от
скорости газового потока волчок, находящийся внутри трубки,
поднимается на определенную высоту.
Для измерения больших объемов газа применяют
барабанный газовый счетчике жидкостным затвором типа ГСБ-400
(газовые часы). Недостаток газовых часов заключается в том,
что они не пригодны для работы с агрессивными газами и газами,
хорошо растворимыми в воде, так как изготовляются из металла,
а запирающей жидкостью в них служит вода.
ОЧИСТКА ГАЗОВ [36, с, 318]
Чаще всего газ очищают от примесей, пропуская его через по-»
глотительные сосуды, содержащие соответствующие жидкие и
твердые поглотители. Эффективность поглощения компонентов газовой
смеси жидкими поглотителями существенно зависит от поверхности
Рис. 146. Прибор Гемпеля:
а —измерительная часть; б —простая пипетка; в —сложная пипетка;
1 —бюретка; 2 —уравнительная трубка; 3 —резиновая трубка; 4, 8—капилляры; 5, /4—за*
жимы; капиллярная трубка; 7—резиновая трубка; 9, 10, 12, /3—шары; //— трубка.
252
и времени соприкосновения газа с поглотителем. Для увеличения
поверхности соприкосновения используют центробежную мешалку,
через отверстие в оси которой подводится газ, распределяющийся
в жидкости.
Особенно тесный контакт между жидкостью и газом
достигается при проведении тонкого вспенивания. Газ
продавливают через газопромыватель со стеклянной пористой пластинкой
в жидкость, содержащую пенообразователь (0,1% раствор
натриевой соли желчной или гидрокоричной кислоты, 0,1% раствор
NaOH, 0,1% раствор СН3СООН). Подбором пенообразователя и
его концентрации можно изменить устойчивость пены.
При анализе газов в качестве поглотительных сосудов
применяют пипетки Гемпеля (рис. 146) с двумя и четырьмя шарами
(для жидких поглотителей), с одним и тремя шарами (для твердых
поглотителей). Пользуясь уравнительной склянкой 2, набирают
в газовую бюретку 1 пробу газа, отмеряют объем взятой пробы,
затем газ переводят в пипетку Гемпеля. Непоглощенную часть
газовой пробы возвращают в бюретку, где измеряют ее объем. По
разности объемов до и после поглощения находят объем
поглощенного компонента.
Для очистки газов пропусканием через слой твердого
поглотителя применяют те же устройства, что и для осушения газов*
глава2Г. СИНТЕЗ ГАЗОВ
Синтез газов осуществляют взаимодействием твердого вещества
с жидкостью, взаимодействием двух жидких веществ или
термическим разложением вещества [24, 47, 58].
Качество синтезируемых газов зависит от чистоты исходных
веществ, а также от формы и размеров применяемых приборов.
Форму и размеры приборов обычно выбирают в зависимости от
характера реакций, лежащих в основе метода получения газа,
требуемого количества газа и степени его чистоты.
Приборы для получения газов должны быть изготовлены из
термически и химически устойчивых сортов стекла. Необходимо, чтобы
соединения отдельных частей были герметичны; по возможности
следует избегать соединений резиновыми или пластмассовыми
трубками.
Одно из основных требований при выборе прибора —
минимальная величина «вредных» объемов, из-за которых необходимо долго
пропускать через прибор получаемый газ для удаления из него
воздуха.
Удобен и весьма универсален лабораторный прибор для
получения газов, образующихся при взаимодействии жидкостей с
твердыми веществами (Н2, С02, H2S) — стеклянный аппарат
Киппа (рис. 147). Твердое вещество в реакторе 4 помещается на
вкладыше 5 из винипласта; вкладыш имеет три отверстия
диаметром 1,5—2 мм для стекания жидкости из реактора в нижнюю часть
аппарата. Тубус реактора, служащий для отвода выделяющегося
газа, соединен с краном отводной трубки 2.
Аппараты Киппа выпускаются вместимостью (по шару
реактора) 500, 1000 и 2000 мл из толстостенного стекла ТХС1 или
ТХС2.
Для подготовки аппарата к работе вынимают резиновую пробку
3 из тубуса реактора 4 и вводят в реактор кусочки твердого
реагента, например, мрамора (объем реагента не должен превышать
2/5 объема реактора). Пользоваться порошком не рекомендуется,
так как в этом случае газ выделяется бурно и возможен его прорыв
через воронку 1 аппарата. Затем пробку 3 вставляют в тубус
реактора и при открытом кране отводной трубки 2 через воронку /
наливают жидкость (например, разбавленный раствор НС1) в таком
количестве, чтобы реактор был заполнен наполовину.
Образующийся газ пропускают через отводную трубку в течение 7—10 мин
для вытеснения воздуха из аппарата. При закрытом кране
отводной трубки 2 выделяющийся газ выталкивает жидкость через
нижнюю часть аппарата в воронку /; при этом жидкость не
контактирует с твердым веществом и газ не выделяется. При получении
агрессивных или дурно пахнущих газов воронку 1 закрывают
жидкостным затвором с нейтрализующим раствором. По окончании
254
Рис. 147. Аппарат Киппа для получения газа:
/ — воронка; 2 —кран отводной трубки; 3, 5 —пробки; 4—реактор; 5—вкладыш.
Рис. 148. Прибор для получения газа:
/, 2—трехходовые краны; 3 — ртутный затвор; 4 — кран.
промывания аппарата выделяющимся газом газоотводную трубку
с краном 2 соединяют с тем прибором, в который требуется
пропускать газ.
Аппарат периодически очищают. Для этого осторожно при
закрытом кране отводной трубки вынимают стеклянную пробку 6 и
через нижний тубус сливают жидкость в заранее подготовленный
сосуд, приоткрывая кран отводной трубки. Затем споласкивают
аппарат водой, которую наливают через воронку 1.
Несколько изменив аппарат, можно получить газ, практически
не загрязненный воздухом. В приборе (рис. 148) предусмотрено
соединение среднего и верхнего шаров трубкой с двумя
трехходовыми кранами 1 к 2. Это простое приспособление позволяет, при
установившейся работе прибора, заполнять все три шара чистым
газом. При соответствующих положениях кранов 1 и 2 можно
отбирать газ как из среднего, так и из верхнего шара. Для
регулирования давления в приборе служит ртутный затвор 3. В средний шар
загружают кусочки твердого вещества и из прибора полностью
удаляют воздух, заполняя прибор и трубки свежепрокипяченной
водой. Затем кран 4 соединяют с сосудом, содержащим жидкий
реагент, и с помощью водоструйного насоса, присоединенного
к трубке крана /, отсасывают воду из среднего шара до тех пор,
пока уровень жидкости в нижнем шаре не достигнет твердого
реагента; при этом выделяется газ. Далее переключают краны и
отсасывают воду из верхнего шара, пока жидкость не поднимется
до половины объема шара. Снова переключают краны и засасывают
жидкий реагент в средний шар; при этом газ начинает бурно
выделяться. Краны переключают так, чтобы жидкость, вытесняемая
газом, выливалась через ртутный затвор. Затем, пользуясь обоими
кранами, регулируют давление газа в приборе.
255
В настоящем разделе рассматриваются наиболее доступные и
проверенные в лабораторной практике методы получения С02, СО,
S02, H2S, С12, НС1, НВг.
Синтез диоксида углерода
Диоксид углерода получают термическим разложением
гидрокарбоната натрия NaHC03 при 100—120 °С или взаимодействием
мрамора СаСОз с НС1:
2NaHC03 —► Na2C03 + C02 + Н20
СаС03 + 2НС1 —► СаС12 + С02 + Н20
Термическое разложение NaHC03 проводят в закрытой с одного
конца стеклянной тугоплавкой трубке, соединенной последовательно
с промывной склянкой, наполненной стеклянной ватой для
задержания пыли, и промывной склянкой с конц. H2S04 для осушения
С02. Трубку полностью заполняют кусочками NaHC03 величиной
с горошину, откачивают воздух водоструйным насосом и нагревают
с закрытого конца.
Получение С02 взаимодействием мрамора с НС1 [1 об. ч. НС1
(р = 1,19 г/см3) и 1 об. ч. дистиллированной воды] проводят в
аппарате Киппа.
Небольшие кусочки мрамора предварительно очищают. Для
этого мрамор помещают в фарфоровую чашку, заливают
разбавленной НС1 (1:2), перемешивают 5—10 мин, сливают кислоту,
ополаскивают мрамор водой и кипятят в течение нескольких
часов в дистиллированной воде.
Синтез оксида углерода
Наиболее целесообразный способ получения СО — обезвожива*
ние концентрированной муравьиной кислоты, что достигается
осторожным (по каплям) добавлением конц. H2S04 к охлаждаемой
льдом НСООН, либо холодной НСООН к конц. Н3Р04, нагретой
до80°С:
НСООН 5г± СО + Н20
Равномерное и непрерывное выделение СО достигается
нагреванием смеси конц. Н3Р04 и НСООН (1:1) при 80 °С.
Образующийся СО содержит примесь паров воды и кислоты, которые
можно удалить промыванием раствором КОН и высушиванием конц,
H2S04.
Синтез диоксида серы
Диоксид серы получают взаимодействием насыщенного водного
раствора сульфита натрия Na2S03 или пиросульфита натрия
Na2S20s с конц. H2S04:
Na2S03 + H2S04 —> Na2S04 + S02 + H20
Na2S205 + H2S04 —v Na2S04 + 2S02 + H20
256
8 колбу, снабженную капельной воронкой с капиллярной
отводной и газоотводной трубками, помещают насыщенный
водный раствор Na2S03 или Na2S205 и прибавляют по каплям конц.
H2SO4. Выделяющийся газ пропускают через две промывные
склянки с конц. H2S04 и сжижают в сосуде, охлаждаемом смесью
льда и NaCl.
Синтез сероводорода
Сероводород высокой степени чистоты образуется при
нагревании концентрированного раствора гидросульфида магния
Mg(HS)2:
Mg (HS)2 —* MgS + H2S
Гидросульфид магния получается при смешивании конц.
растворов MgCU и NaHS (мольное соотношение 1:2):
MgCl2 + 2NaHS —> Mg(HS)2 + 2NaCi
В качестве реакционного сосуда может служить круглодонная
колба вместимостью 1 л с резиновой пробкой, имеющей два
отверстия, через которые проходят капельная воронка
вместимостью 250 мл и газоотводная трубка. Из капельной воронки
вначале приливают 250 мл насыщенного раствора MgCb, затем
столько же дистиллированной воды и наконец 250 мл
насыщенного раствора NaHS. При незначительном нагревании (40—60 °С)
реакционной колбы начинается выделение H2S равномерным
током. Выход реакции около 80%, считая на NaHS.
Синтез хлора
Хлор высокой степени чистоты получают реакцией между НС1
(3 об.ч. конц. НС1 и 1 об.ч. дистиллированной воды) и твердым
бихроматом калия К2СГ2О7, протекающей при нагревании:
K2Cr207+14HC1 —* 2KCI + ЗС12 + 2СгС13 + 7Н20
Твердый измельченный К2СГ2О7 помещают в круглодонную
колбу, снабженную делительной воронкой и газоотводной
трубкой. Колбу устанавливают в нагревательную жидкостную баню
(солевую) и при нагревании приливают из капельной воронки НС1.
Интенсивность выделения СЬ регулируют нагреванием бани. Хлор
осушают конц. H2S04 и СаСЬ.
Синтез хлористого водорода
Легко регулируемый ток хлористого водорода получают
прибавлением чистой конц. соляной кислоты к конц. H2SO4 или
наоборот.
Для получения относительно небольших количеств НС1 можно
использовать толстостенную колбу для фильтрования под
вакуумом, снабженную пробкой и воронкой, оттянутый конец которой
доходит почти до дна колбы. Склянку наполняют конц. H2SO4,
9 Зак. 1237 257
*s
Рис, 149. Приборы для получения хлористого (а) и
бромистого (6") водорода:
/ — реакционная емкость; 2 —колонка; 3—делительная
воронка; 4 — отводная трубка; 5 —кран.
а воронку — соляной кислотой. Из
капельной воронки постепенно добавляют
соляную кислоту к серной; при этом следует
предварительно заполнить оттянутый конец
отводной трубки капельной воронки
соляной кислотой, чтобы обеспечить
гидростатическое давление, необходимое для
вытекания кислоты у дна сосуда.
Равномерного выделения НС1 можно
также достигнуть при приливании конц.
H2SO4 к кашице, состоящей из смеси тонкоизмельченного NaCl
и конц. НС1.
Относительно большие количества НС! целесообразно получать
в приборе, изображенном на рис. 149, а. При закрытом кране 5
в склянку / наливают конц. соляную кислоту, а из делительной
воронки 3 постепенно приливают конц. H2SO4. Газообразный НС1
поднимается в колонку навстречу току H2SO4 и выходит через
боковую трубку 4. Отработанную кислоту периодически выпускают
из склянки / через тубус с краном. Хлористый водород осушают
конц. H2S04 или СаСЬ.
1
Синтез бромистого водорода
Бромистый водород получают гидролизом бромидов фосфора,
образующихся при взаимодействии фосфора и брома, или
действием брома на тетралин (тетрагидронафталин). Последний
способ более удобен и обеспечивает получение сухого бромистого
водорода:
СюНц + 4Вг2 «= С10Н8Вг4 + 4ЬВг
Для этого используют прибор, изображенный на рис. 149,0.
В колбу /, снабженную делительной воронкой 3 и колонкой с
кольцами Рашига 2, загружают 200—250 мл тетралина и 3—5 г
железных опилок. Тетралин предварительно высушивают над
Na2S04 и перегоняют при атмосферном давлении и 206—208 °С.
Реакционную колбу с тетралином помещают на силиконовую
баню. К кипящему тетралину из делительной воронки постепенно
добавляют бром. Образующийся НВг и пары тетралина
поднимаются по колонке 2У где тетралин конденсируется, а бромистый
водород выходит из прибора. Колонка 2 соединена с капельной
воронкой 3 трубкой, уравнивающей давление, что позволяет
осуществить непрерывную подачу брома. Бромистый водород
пропускают через промывную склянку с тетралином (для улавливания
паров брома) и осушают СаВг2,
268
глава 22 РАБОТА С МЕТАЛЛИЧЕСКОЙ РТУТЬЮ
Металлическая ртуть, обладая очень ценными свойствами, на*
шла широкое применение в лабораторной практике [57].
Пары ртути и многие ее соединения весьма токсичны.
Хроническое отравление парами ртути (хронический меркуриализм)
может наступить даже при длительном пребывании человека в
помещении, где были разбиты ртутные термометры. Поэтому при
работе со ртутью необходимо изучить правила работы с нею,
исключающие опасность острого или хронического отравления.
СВОЙСТВА РТУТИ
Ртуть — серебристо-белый металл с синеватым оттенком;
/кип = 356,7 °С; /зам = —39 °С; q = 13,55 г/см3.
Ртуть — химически стойкий элемент. Чистая ртуть в сухом
воздухе не изменяется; медленно окисляется кислородом лишь при
повышенных температурах. Ртуть хорошо растворяет почти все
металлы, а также многие оксиды металлов. При соприкосновении
с металлами, резиновыми трубками и многими другими
материалами ртуть сильно загрязняется. Чистая ртуть при выливании на
поверхность образует круглые блестящие легкоподвижные капли-
При комнатной температуре ртуть медленно реагирует с
разбавленными растворами HN03:
6Hg + 8HN03 —* 3Hg2 (N03)2 + 2NO + 4H20
Значительное число посторонних металлов, растворенных в
ртути, при достаточно тонком дроблении последней, окисляются
и переходят в водный раствор в виде солей. Многие посторонние
металлы при контакте с Hg2(N03)2 вытесняют ртуть из соли и
переходят в раствор. Например:
Hg2+ + Zn —* Zn2+ + 2Hg
Загрязнение ртути металлами (образование амальгамы) может
произойти при соприкосновении ее с металлическими частями
приборов, в которых она находится, поэтому металлические части,
контактирующие со ртутью, покрывают масляной краской или лаком.
ПРАВИЛА РАБОТЫ СО РТУТЬЮ. МЕРЫ БЕЗОПАСНОСТИ [56, с. 77, 146]
Работа со ртутью может производиться в специальных и в
общих лабораторных помещениях. В этих помещениях должны
быть вывешены правила внутреннего распорядка, включающие
специальные пункты по безопасным способам работы со ртутью
и ртутными приборами и аппаратами, а также по мерам личной
профилактики.
9* 259
Стены и потолки специально оборудованных изолированных
помещений для работы со ртутью должны быть ровными,
гладкими и обработаны специальными составами для придания им ртуте-
непроницаемости. Дверные полотна, подоконники, оконные рамы,
рабочая мебель, деревянные части оборудования, стойки приборов
и вытяжные шкафы должны быть обработаны составами для
защиты древесины от сорбции паров ртути (нитроклей, нитроэмали,
нитролаки, масляные краски на натуральной олифе).
В специальных помещениях поверхности стен, потолка, мебели,
коммуникации необходимо 1 раз в месяц мыть теплой водой и
1 раз в квартал — с применением средств химической
демеркуризации.
В общих лабораторных помещениях разрешается проводить
работы с переносными или стационарными приборами и аппаратами,
в которых ртуть хорошо изолирована. Все приборы с ртутным
заполнением должны быть установлены на эмалированных или
окрашенных масляной краской металлических противнях в
вытяжных шкафах, снабженных верхним и нижним отсосами.
При работе с открытой ртутью вентиляция вытяжного шкафа
должна включаться за 15—20 мин до начала работы и не должна
выключаться в течение 30 мин после окончания работы.
В лабораторных помещениях, где проводится работа со ртутью,
температура воздуха не должна быть выше 18 °С.
В затворах раковин и по ходу канализационной сети должны
быть установлены ловушки для улавливания металлической ртути
из сточных вод.
В специальных помещениях запрещается пользоваться мягкой
или обитой тканью мебелью, шторами, гардинами и т. п.
Под рабочей поверхностью рабочих и лабораторных столов
не должно быть ящиков и шкафов. Лабораторная мебель должна
обеспечить удобство работы, возможность легко собрать пролитую
ртуть, применять химические демеркуризаторы, а также
исключить скопление ртути в щелях, неровностях рабочих поверхностей,
попадание ртути на пол при разливе ее по рабочей поверхности.
Столы и вытяжные шкафы должны быть снабжены по краям
возвышающимися бортами. Рабочую поверхность столов и шкафов
покрывают линолеумом.
Ртуть должна выдаваться сотрудникам только с разрешения
руководителя лаборатории, в количестве не превышающем
дневную потребность.
Приборы и установки с ртутным заполнением нельзя
располагать непосредственно у дверей, проходов, оконных проемов,
ориентированных на юг или юго-запад, вблизи нагретых
поверхностей.
К работам со ртутью и ее препаратами работники допускаются
только после предварительного инструктажа и проверки знания
соответствующей инструкции. Работающие со ртутью должны
строго выполнять меры личной гигиены: работать только в
спецодежде (наглухо застегнутом халате, шапочке или косынке). Сред-
260
ствами индивидуальной защиты органов дыхания (противогаз
марки «Г») следует пользоваться лишь при авариях, связанных с
разливом больших количеств ртути, при проведении работ с
нагретой ртутью, при выходе из строя системы местной вытяжной
вентиляции. Манипуляции с открытой ртутью (очистка,
дистилляция, заполнение приборов) необходимо проводить только в
хлорвиниловых или тонких резиновых перчатках над поддоном внутри
вытяжных шкафов при работающей вентиляции. Категорически
запрещается брать ртуть незащищенными руками или отсасывать ее
ртом! После окончания работ перчатки, прежде чем снять их с
рук, следует тщательно вымыть с мылом.
В помещениях, где производится работа со ртутью,
запрещается хранить и принимать пищу и воду. Перед приемом пищи
необходимо снять спецодежду, тщательно вымыть руки и лицо
теплой водой с мылом и прополоскать рот 0,025% раствором КМп04.
В лаборатории ртуть должна храниться в вытяжном шкафу в
небьющейся посуде или толстостенной стеклянной посуде с
притертыми пробками, установленной в амортизационном футляре
на металлических поддонах. В небольшом количестве (20—30 мл)
ртуть может храниться в запаянных стеклянных ампулах в
лабораторных шкафах. Ампулы должны быть заключены в плотные
футляры (пластмассовые или металлические), чтобы ртуть не
разлилась при случайном повреждении ампул.
Отработанную ртуть хранят в толстостенной посуде с
притертыми пробками под слоем подкисленного водного раствора КМп04.
Сосуд должен быть установлен на металлический поддон в
вытяжном шкафу. Запрещается выливать ртуть в канализацию!
Ртутные приборы с открытой поверхностью ртути должны
размещаться внутри вытяжных шкафов как во время эксплуатации,
так и во внерабочее время.
Приборы или аппаратуру наполняют ртутью только на
металлических или пластмассовых поддонах (под наблюдением
ответственного лица) и, как правило, внутри вытяжного шкафа.
Заполнение сосудов ртутью следует производить через воронку с
оттянутым капилляром и лить по стенкам сосуда.
Пролитая ртуть должна быть немедленно и тщательно собрана
в герметичный толстостенный стеклянный сосуд с подкисленным
раствором КМп04.
Приборы с ртутным заполнением после окончания циклов
работ или приборы, нуждающиеся в ремонте, необходимо освободить
от ртути, обработать конц. HN03 с последующим
прополаскиванием водой и 0,1% раствором 12 в 2% растворе KI.
При перегонке ртути в вакууме для предупреждения
загрязнения парами ртути масла насосов необходимо в цепь перед
масляным насосом включить патрон (колонку) с поглотителем паров
ртути (активный диоксид марганца или активный уголь,
пропитанный иодом). Активный уголь, пропитанный иодом, готовят
следующим образом: в стеклянную ампулу вместимостью 250—300 мл
261
загружают 100 г сухого активного березового угля БАУ и 10 г
возогнанного иода. Ампулу запаивают и помещают на 24 ч в
термостат, нагретый до 100—105 °С. По охлаждении ампулу
вскрывают и содержимое переносят в банку с притертой пробкой,
К выхлопным отверстиям ртутных насосов нужно присоединять
фильтр с активным диоксидом марганца или выводить выхлоп
в трубопроводы вытяжной вентиляции. Для контроля проскока
паров ртути из выхлопа через поглотитель подвешивают ртутные
реактивные бумажки.
В помещениях, где работают с открытой поверхностью ртути,
необходимо периодически определять содержание ее паров в
воздухе. Если концентрация паров ртути выше предельно допустимой
(ПДК), следует немедленно прекратить работу и провести
дегазацию помещения. Работы в данном помещении могут быть
возобновлены только после того, как содержание паров ртути не будет
превышать ПДК (0,01 мг/м3).
ОЧИСТКА ПОМЕЩЕНИЙ, ЗАГРЯЗНЕННЫХ РТУТЬЮ
Если ртуть случайно разольется в лаборатории, следует
немедленно ее собрать. Чтобы ртуть не распространилась по помещению,
собирать капли ртути начинают с периферии загрязненного
участка пола по направлению к центру. Капли вначале собирают при
помощи железных эмалированных совков и переносят в приемник
из толстостенного стекла, предварительно заполненный
подкисленным раствором КМп04 (1 г КМп04 на 1 л воды и 5 мл конц.
НС1).
Видимые капли можно собрать механическим путем
(засасыванием с воздухом). Удобно пользоваться также пипетками,
изображенными на рис. 150. Верхняя часть пипетки
присоединяется при помощи вакуумной резиновой трубки к водоструйному
насосу или резиновой груше. Засасываемая ртуть накапливается в
расширенной части пипетки (баллончике). При переворачивании
пипетки, после отсоединения вакуума, ртуть
выливается в подготовленный сосуд.
Для сбора ртути можно пользоваться
также поглотительной склянкой
Тишенко, на один тубус которой
надевают длинную вакуумную резиновую трубку
с присоединенным толстостенным
стеклянным капилляром, а другой тубус
присоединяют к водоструйному насосу.
Весьма эффективен сбор мелких капель
ртути с помощью пластинок (или кис*
Рис. 150. Пипетки для собирания ртути:
а—специальные; б—из обычной пипетки;
/ — капиллярная трубка; 2—баллончик; 5—верхний конец
пипетки для присоединения к вакуум-насосу.
262
точек) из белой жести, предварительно натертых ртутью.
Приставшие к амальгамированной поверхности капельки ртути
стряхивают в сосуд с подкисленным раствором КМп04.
Отдельные капельки размером до 0,5—1 мм могут быть
эффективно удалены с поверхности при помощи пасты,
представляющей собой смесь Мп02 и 5% раствора HC1 в отношении 1 : 2.
Пасту накладывают толстым слоем на обрабатываемую
поверхность на 1,5 ч, после чего этот слой с прилипшими капельками
ртути удаляют эмалированной металлической пластинкой. Капли
ртути стряхивают в приемник, заполненный подкисленным раствором
КМп04.
Ежедневно в процессе работы и по окончании ее необходимо
тщательно очищать рабочее место и аппаратуру от мельчайших
капель ртути.
Демеркуризация
Даже тщательная уборка видимых ртутных загрязнений не
всегда достаточна, поскольку мелкие капельки ртути могут
задерживаться на неровностях загрязненной поверхности, попадать
в щели и трещины. Йоэтому часто возникает необходимость в
демеркуризации химическими способами.
Демеркуризация достигается окислением ртути в каломель
Hg2Cb подкисленным раствором КМп04:
2КМп04 + 16НС1 =2КС1 + 2МпС!2 + 5С12 + 8Н20
2Hg + Cl2 = Hg2Cl2
Надежный способ демеркуризации — обработка 10—12%
(считая на безводную соль) раствором FeCl3. Этот раствор оказывает
сильное химическое и эмульгирующее действие на металлическую
ртуть. При энергичном перемешивании металлической ртути с
раствором FeCh капельки ртути теряют свою подвижность,
деформируются и превращаются в мелкий серый порошок. В результате
окислительно-восстановительной реакции эмульгированная ртуть
переходит в оксиды и хлориды, которые легко удаляются. После
удаления видимых капель ртути на загрязненную поверхность
наносят раствор FeCb из расчета 0,5 л раствора на 1 м2 площади.
Поверхность, покрытую раствором FeCl3, несколько раз протирают
мягкой кистью или щеткой и оставляют до полного высыхания на
1—2 суток. После этого обработанную поверхность очищают,
промывают сначала мыльной, а затем чистой водой.
Поверхности, загрязненные ртутью, рекомендуется обрабатывать
4—5% водным раствором хлорамина Б (C6H6S02NNaCl -3H20)
или 4—5% раствором дихлорамина Б (C6H5S02NC12) в четырех-
хлористом углероде. Через 8—11 ч обработанные поверхности
обильно смачивают 4—5% раствором полисульфида натрия Na2S*.
Спустя 8—10 ч после вторичной обработки поверхность тщательно
промывают водой.
263
Хлорамины и полисульфид как окислители переводят ртуть в
одновалентное (каломель) и двухвалентное соединение (полисуль*
фид ртути):
C6H5S02NC12 + 3Hg —> C6H5S02NHg + Hg2Cl2
C6H6S02NHg + Na2SJC —-► C6H5S02NNa2 + HgS*
Полисульфид натрия готовят нагреванием до 105 °С смеси
сульфида натрия Na2S-9H20 с тонкоизмельченной серой в массовом
соотношении 85 : 15.
Эффективная демеркуризация достигается обработкой 2,5%
водным раствором реагента, содержащего 5—20% этилендиаминтетра-
уксусной кислоты (ЭДТА, комплексон III) и 80—95% тиосульфата
натрия Na2S203-5H20. Раствором покрывают загрязненную
поверхность. После того как обработанная поверхность высохнет, ее
очищают и промывают водой.
Демеркуризации необходимо подвергать также посуду и
стеклянную аппаратуру, которые использовались при работе со ртутью.
Определение содержания паров ртути в воздухе
Анализ воздуха рабочей зоны на содержание паров ртути
должен производиться не реже 1 раза в две недели при помощи
бумажных индикаторов.
Индикацию паров ртути можно осуществить реактивной
бумагой, пропитанной иодидом меди(1) Cu2I2 или сульфидом селена
SeS. Для приготовления индикаторной бумаги с Cu2I2
фильтровальную бумагу пропитывают 5% водным раствором CuS04-5H20,
выдерживают ее на воздухе и затем опрыскивают из пульверизатора
10% раствором KI:
2CuS04 + 4KI —> Cu2I2+I2 + 2K2S04
Побуревшую бумагу опускают в 10% раствор Na2S203, в
котором бумага белеет:
I2 + 2Na2S203 —> 2NaI + Na2S406
Затем индикаторную бумагу промывают водой, высушивают и
нарезают полосками шириной 1 см и длиной 5—6 см.
Индикатор помещают в места возможного выделения паров
ртути в рабочую зону на уровне дыхания работающего. Если через
4 ч при 15 °С бумажки не приобретают палево-розового цвета, то
содержание паров ртути в воздухе ниже ПДК.
Индикаторную бумагу с SeS готовят следующим образом:
фильтровальную бумагу смачивают 10% водным раствором А1С13, а
затем раствором H2Se03. Мокрую бумагу помещают в
вакуум-эксикатор с двумя отводными трубками. Через одну трубку,
доходящую до дна эксикатора, пропускают ток сероводорода, а другая
служит для выхода избыточного газа, нейтрализуемого 10%
раствором щелочи. Бумага приобретает светло-желтый цвет за счет
образующегося сульфида селена, который под действием паров
264
ртути разлагается и темнеет вследствие образования черного
сульфида ртути HgS.
В последние годы сконструированы анализаторы паров
ртути, обладающие большой селективностью и достаточной
чувствительностью. Например, анализатор «Меркурий» предназначен
для количественного определения содержания паров ртути в
присутствии посторонних примесей, а также для обнаружения
локальных скоплений металлической ртути. Действие прибора основано
на фотоэлектрическом поглощении парами ртути излучения
ртутной лампы с длиной волны 253,7 нм. Прибор позволяет определять
содержание паров ртути в воздухе в диапазонах 0 — 0,005 мг/м3 и
О — 0,25 мг/м3. Время анализа измеряется минутами.
Если содержание паров ртути в лабораторном помещении пре*
вышает ПДК, необходимо: 1) не теряя времени, тщательно
осмотреть и очистить лабораторные столы, шкафы и пол от капель
ртути; 2) оторвать плинтуса от стен, осмотреть поверхность и при
обнаружении ртути, вкрапленной в штукатурку за плинтусом,
удалить этот участок; 3) снять неплотно прилегающие к полу участки
покрытий, если предполагается, что под ними имеются скопления
ртути.
После проведения демеркуризации окна, двери, рамы и мебель,
радиаторы, пол и поверхность стен, покрытых масляной краской,
необходимо промыть мыльной горячей водой при помощи щеток.
Затем помещение проветривают и через 5 дней проводят повторно
анализ воздушной среды; если концентрация паров ртути все же
окажется выше предельно допустимой, то очистку помещения
следует повторить.
ОЧИСТКА РТУТИ
Очистка ртути заключается в удалении механических
примесей, органических загрязнений, следов посторонних металлов,
влаги. Для некоторых работ требуется металлическая ртуть с малым
содержанием посторонних металлов, что достигается перегонкой
ее в вакууме.
В лабораторных условиях ртуть от механических примесей
можно очистить фильтрованием (продавливанием) через замшу или
бумажный фильтр.
Приспособление для фильтрации через замшу состоит
из узкой железной трубки, оканчивающейся цилиндрическим
расширением с винтовой нарезкой и навинчивающейся муфтой с
отверстием посредине. В расширение вкладывают несколько кусков
замши и зажимают их муфтой. В трубку наливают ртуть, которая
продавливается через замшу и стекает мелкими каплями в
приемник.
Для очистки ртути фильтрацией через бумажный
фильтр в стеклянную воронку с оттянутым концом помешают
двойной бумажный фильтр, кончик которого прокалывают тонкой
булавкой (диаметром ^0,5 мм). Одновременно закрывают месго
265
прокола оттянутой стеклянной палочкой. В воронку наливают
немного ртути (^2/з объема) и слегка перемещают вверх палочку,
закрывающую место прокола фильтра. Когда в воронке остается
около у4 первоначального объема ртути, отверстие в фильтре
закрывают палочкой и в воронку наливают следующую порцию
ртути. Остаток из фильтра переносят в толстостенный стеклянный
герметичный сосуд, в котором накапливают загрязненную ртуть
для последующей очистки фильтрацией.
Ртуть, отфильтрованную от механических примесей, собирают
в ампулу или толстостенный стеклянный сосуд.
Очистка ртути от механических примесей и оксидов может
быть также проведена в делительной воронке, снабженной
хорошо притертым спускным краном. Налитой ртути дают
отстояться, осторожно приоткрывают кран и собирают вытекающую
ртуть. В воронке остается часть загрязненной ртути, которую
очищают по мере накопления.
Заслуживает внимание метод очистки ртути проточной
водой. Ртуть помещают в толстостенный стакан (до !/з объема), за*
полняют его водой и в течение 1—2 мин пускают в стакан сильную
струю воды из водопровода, которая уносит с собой мелкие
частицы ртути. Поэтому необходимо стакан со ртутью поместить в
сосуд-отстойник (например, эксикатор или кристаллизатор), где будут
собираться капельки ртути. Вода вымывает загрязнения, и
поверхность ртути приобретает зеркальный блеск. Промытую ртуть
переносят в сухую делительную воронку, прибавляют 20—30 мл
этилового спирта и энергично встряхивают. Операцию повторяют в
Рис. 151. Прибор для химической очистки ртути:
J —стеклянная колонка; 2—сифонная трубка; 3 —воронка с согнутой спускной трубкой.
Рис. 152. Прибор для перегонки ртути:
/ — трубка для заливания ртути; 2 — колба; 3 — трубка для просасывания воздуха; 4 —
холодильник; 5 —трубка, соединяющая холодильник с ампулой для ртути; б —ампула для отгб-
няемой ртути.
266
чистой делительной воронке. Затем ртуть встряхивают с 10—15 мл
диэтилового эфира в делительной воронке, помещают в широко-
горлый сосуд и оставляют в вытяжном шкафу до исчезновения
запаха эфира.
От органических загрязнений ртуть очищают обработкой
органическими растворителями с последующим тщательным
удалением остатков последних в вакууме.
При диспергировании в растворе Hg2(N03)2, содержащем HN03,
или в разбавленной HN03 ртуть освобождается от значительного
числа металлов, загрязняющих ее.
Простейший прибор для химической очистки ртути
представлен на рис. 151. В колонку 1 вначале помещают небольшое
количество чистой ртути, служащей затвором, после чего колонку
заполняют 5% раствором Hg2(N03h в 7—8% растворе HN03.
Вытекая из воронки 5, ртуть разбивается на мелкие капли, которые
впоследствии стекают вниз. После 4—5-кратного пропускания
через раствор, а затем 4—5-кратного через дистиллированную воду
получают ртуть достаточной чистоты. Капли воды с поверхности
ртути удаляют при помощи полоски фильтровальной бумаги.
Для очистки ртути водным раствором HN03 в делительную
воронку с пришлифованной пробкой, вместимостью 2—3 л,
помещают 50—100 мл ртути и 1,5 л 10% водного раствора HN03,
нагретого до 50 °С, закрывают воронку пробкой и энергично
встряхивают 15—20 мин. Затем раствор HN03 сливают и процесс
повторяют до тех пор, пока поверхность ртути не станет блестящей.
После этого ртуть промывают несколько раз дистиллированной
водой. Профильтрованная ртуть при встряхивании в чистом
стеклянном сосуде не должна оставлять следов на его стенках.
Для получения ртути высокой степени чистоты, после очистки
механическим и химическим способами, ее необходимо
перегнать. Наиболее часто перегонку проводят при 2,66—3,99 кПа
(20—30 мм рт. ст.) и температуре около 200 °С, в токе сухого
азота или сухого воздуха. Перегонку ртути при пониженном давлении
можно осуществить в установке, изображенной на рис. 152. В
колбу 2 через трубку / вносят порцию очищенной ртути и трубку /
запаивают. Затем через трубку 3 пропускают пузырьки сухого
азота и создают разрежение. Колбу 2 помещают в нагревательную
баню с силиконовым маслом.
Пары ртути, конденсирующиеся в холодильнике 4, стекают по
трубке 5 в ампулу б, которую по окончании перегонки отпаивают.
глава 23 ОТБОР СРЕДНИХ ПРОБ
Основное условие выполнения операции отбора проб — пред-*
ставительность отобранной пробы: она должна сохранить
средний состав анализируемого твердого, жидкого или
газообразного вещества.
Приемы отбора средней пробы зависят от агрегатного
состояния и степени однородности вещества. Наиболее просто отбирать
пробы газов и смешивающихся жидкостей, труднее всего — пробы
крупнозернистых и крупнокусковых твердых веществ.
Неоднородность природных твердых тел (руда, каменный уголь и
др.)—когда определяемый компонент составляет лишь небольшую часть
пробы, а отдельные компоненты обладают различной плотностью—
осложняет процесс отбора пробы. Неоднородность проявляется
также в виде сегрегации — расслаивания материала при
перевозке, тряске и пересыпании. При этом мелкие куски располагаются
ближе к нижней части тары, а крупные — к поверхности. Поэтому
перед отбором пробы анализируемый материал всегда следует
хорошо перемешивать. Чтобы средняя проба отражала состав
твердого продукта, необходимо сделать несколько частных проб,
отбираемых по правилам, приводимым в ГОСТах или ТУ для
конкретного вида продукта.
ОТБОР ПРОБЫ ТВЕРДЫХ ПРОДУКТОВ [67]
Размер пробы зависит от крупности кусков исследуемого
продукта. Чем крупнее куски, тем больше масса отбираемой пробы.
Например, для кусковых материалов масса первичной пробы
составляет 1 —10 кг и более, а для порошкообразных и сыпучих
материалов — 0,2—0,Б кг. Если материал рассыпан относительно
тонким слоем на большой площади, то пробы нужно брать в
нескольких точках в шахматном порядке.
Пробы сыпучих тел можно отбирать ручным способом и
механическими пробоотборниками. Примером пробоотборников
сыпучих тел могут служить пробоотборники зерна и семян (амбарный,
мешочный, клеверный щупы).
Известные трудности возникают при отборе проб металлов.
Анализируемый металл может быть в виде изделия, слитков
(чушек) или же в расплавленном состоянии. В большинстве случаев
металлы — кусковой материал, причем неоднородными могут быть
не только отдельные куски, но и разные места одного и того же
куска или отливки. Правила отбора проб конкретных видов
металлов и изделий из них установлены соответствующими ГОСТами.
Пробы от отдельных слитков; кусков или изделий отбирают
сверлением, строганием или фрезерованием. Перед отбором пробы
исследуемую поверхность очищают от загрязнений и окалины.
268
Из первичной пробы нужно отобрать меньшую по массе
лабораторную, что достигается путем сокращения
первичной пробы одновременно с перемешиванием и измельчением
ее. Измельченный и тщательно перемешанный материал первичной
пробы чаще всего сокращают квартованием. Для этого
собранный в конусную кучу материал расплющивают, надавливая на
вершину конуса каким-либо плоским предметом. Конус
превращается в круглый или квадратный пласт, имеющий одинаковую
толщину во всех точках. Пласт делят на четыре сектора, два
противоположных сектора отбрасывают, а два оставшихся
перемешивают, вновь собирают в конус и повторяют квартование до тех
пор, пока не достигнут необходимой массы пробы, определяемых
соответствующими нормами разделки пробы.
Для сокращения проб применяют также делительные
аппараты (сократители проб).
После последнего сокращения и перемешивания пробы
коническую кучу расплющивают, как для сокращения, и делят на 16—
20 равных квадратов взаимно перпендикулярными линиями. Из
середины квадратов в шахматном порядке при помощи
пластмассового или фарфорового совочка отбирают порции вещества по всей
глубине слоя.
Первичные пробы металлов смешивают и измельчают в ступке
из закаленной стали до тех пор, пока проба не пройдет сквозь
сито № 09—1. Затем пробу высыпают на лист белой плотной
бумаги, тщательно перемешивают, разделывают, как указано выше,
и измельчают. Проба должна проходить через сито № 015.
Отобранную лабораторную пробу помещают в две стеклянные
банки с притертыми пробками, одну отправляют в лабораторию,
другую хранят определенный срок на случай проверки. Пробы
снабжают этикеткой, на которой должны быть указаны
наименование продукта, сорт, марка, завод-изготовитель, откуда взята
проба, время отбора пробы, подпись лица, отбиравшего пробу.
ОТБОР ПРОБЫ ЖИДКОСТЕЙ [68]
Перед отбором проб в производственных условиях горячие
жидкости охлаждают. Жидкости, находящиеся в сосудах
(аппаратах) под давлением, предварительно переводят в промежуточные
емкости. После снижения давления их сливают в пробоотборники,
конструкции которых зависят от вида анализируемой жидкости и
условий отбора пробы.
Для отбора и взвешивания проб летучих, агрессивных и
ядовитых жидкостей служат пипетки ПК с одним краном и П-2К с
двумя кранами (рис.153).
Отбор проб воды из водоисточников допускается бутылью
и специальными приборами — батометрами.
При отборе проб из мелкой тары жидкость перемешивают.
Пробы отбирают с помощью длинной стеклянной трубки или
стеклянного сифона. Трубку последовательно погружают в
269
4 У •Н Рис. 153. Пипетки с одним (а) и двумя кранами (Ф) для от-
I L Д бора и взвешивания проб жидкости.
жидкость на разные глубины, верхнее
отверстие трубки закрывают, вынимают и
сливают пробы жидкости в чистую посуду,
затем тщательно взбалтывают.
Для отбора жидкости из баков и
мелкой тары служит также
шприц-пробоотборник (объем рабочей полости
120 см3).
При отборе проб из больших
резервуаров необходимо учитывать, однородна или
неоднородна жидкость. В последнем случае
среднюю пробу составляют из трех частных проб, отобранных на
разных уровнях жидкости: на расстоянии 0,5 м от поверхности,
0,5 м от дна и в середине толщи жидкости.
Из неглубоких резервуаров пробы берут при помощи
стеклянных трубок-пипеток, из глубоких — специальными пробо*
отборниками.
Пробу жидкости, текущей по трубопроводу, отбирают через
специальный кран, устанавливаемый на трубопроводе. Пробоот-
борный кран соединен с тремя трубками, загнутые концы
которых направлены своими отверстиями навстречу текущей
жидкости. Кран привинчивается к патрубку трубопровода так, что
трубки входят внутрь трубопровода. Пробы отбирают через
определенные промежутки времени.
Средняя проба жидкости составляется путем смешивания
частных проб.
ОТБОР ПРОБЫ ГАЗОВ [25, с. 88]
Отбор проб газов из газопроводов и аппаратов производится
через пробоотборную трубку, которая устанавливается
на газопроводе. Сосуд для отбора пробы при помощи вакуумной
резиновой трубки присоединяют к пробоотборной трубке. При
этом пробу можно отбирать вытеснением затворной жидкости из
сосуда, продуванием сосуда исследуемым газом или засасыванием
газа в сосуд. Выбор способа отбора зависит от того, находится
ли газ в системе при избыточном, атмосферном давлении или при
разрежении.
При избыточном давлении отбор пробы проводят первыми
двумя способами. Для этого используются газометры с напорной
воронкой, аспираторы и газовые пипетки. Пробы газа,
находящегося под разрежением или при атмосферном давлении, отбирают
путем засасывания. Независимо от метода отбора пробы объем
ее должен быть достаточным для проведения анализа выбранным
методом. Вид сосудов для отбора проб газов определяется в
зависимости от природы компонентов анализируемого газа и объема
пробы,
270
Отбор проб воздуха на промышленных предприятиях и методы
их анализа подробно изложены в работе [51, с. 295—310].
Газометры
В лабораторной практике применяют газометры с жидкостным
затвором и сухие (рис. 154). Серийно выпускаются лишь
газометры с жидкостными затворами вместимостью 5000, 10 000 и
20 000 мл.
В качестве затворной жидкости может быть использована вода,
насыщенные водные растворы NaCl, Na2S04, 50% раствор
глицерина, вазелиновое масло. Затворная жидкость не должна
реагировать с компонентами анализируемой пробы и сколько-нибудь
значительно их растворять. Для уменьшения потерь анализируемого
газа вследствие растворения в затворной жидкости последнюю
рекомендуется предварительно насыщать исследуемым газом. При
заполнении газометра затворной жидкостью необходимо добиться
полного удаления воздуха из сосуда.
Газометр с жидкостным затвором может служить
также и в качестве аспиратора для просасывания газа через
сосуды с поглотительным раствором (см. ниже). В этом случае кран 4
закрывают, к крану 2 присоединяют поглотительный сосуд и,
регулируя скорость вытекания затворной жидкости краном 3,
пропускают газ. При использовании газометра в качестве аспиратора
производят его градуировку сливанием воды в мерный цилиндр через
кран 3 при открытом кране 2 и закрытом 4. При этом необходимо
газометр установить строго вертикально.
На полоске миллиметровой бумаги, наклеенной на газометр,
делают, в зависимости от вместимости газометра, пометки через
каждые 100—200 мл. Градуировку повторяют два-три раза По
окончании градуировки проставляют цифры, соответствующие
полученным объемам, и полоску бумаги для предохранения от влаги
покрывают тонким слоем расплавленного парафина.
Газометры с жидкостным затвором легко смонтировать
самостоятельно, если имеются склянки с тубусом соответствующей
вместимости.
Сухие газометры со шлифом
или с резиновой пробкой
используются для засасывания или просасывания
газа после того, как воздух был
откачан вакуумным насосом. Поэтому
они подлежат проверке на
герметичность. Для этого сухой газометр
одним краном присоединяют к ртутному
манометру, другим — к вакуумному
Рис. 154. Газометры:
а —с жидкостным затвором; б—сухой;
J — напорная воронка; 2> $, 4 — краны.
271
(И
Е
й
м
г
-5
t
1ро
Ущ
г
и
-10
г
-5
Z
рр|
1—iTo|
и
и
1-
-ад
Hi
1—сг]
F5f
-о,ч
S
Рч
Hi
-
И1
Ы
© ©
4
©
"^=7
(У)
3^
Рис. 155. Аспиратор для
отбора проб воздуха:
/ — колодка для
подключения прибора в сеть;
2 — тумблер; 3 —
предохранитель; 4 — разгрузочный
клапан; 5 —ротаметры;
6—ручка вентилей
ротаметров; 7
—присоединение к фильтрам; 8 —
клеммы для заземления.
насосу. После откачивания воздуха насос отключают и отмечают
уровень ртути в манометре. Постоянство уровня ртути в течение
10—15 мин свидетельствует о герметичности газометра.
В сухие газометры обычно отбирают газ (воздух), содержащий
компоненты, легко растворимые в воде или взаимодействующие
с водой. Кроме того, сухие газометры применяют в том случае,
когда газ не сразу поступает на анализ и хранится более суток.
Аспираторы
Для отбора проб воздуха используют аспираторы. Одна из
моделей аспираторов изображена на рис. 155. Пробы воздуха
отбираются путем просасывания воздуха через специальные
фильтры с определенной скоростью (^50 л/ч). Одновременно
отбираются 4 пробы. Зная скорость и время просасывания воздуха, легко
рассчитать объем воздуха, прошедшего через фильтр или
поглотитель (при замене фильтра сосудом с жидким или твердым
поглотителем).
Аспиратор целесообразно использовать при определении
запыленности воздуха производственных помещений, а также для
отбора проб газа, находящегося под уменьшенным или атмосферным
давлением.
Отбор пробы из системы, давление в которой близко к
атмосферному, производится отсасыванием газа в пипетки.
Разрежение, необходимое для этого, создается опусканием напорной
склянки.
Аспирацию анализируемого газа (воздуха) через жидкие по*
глотительные среды проводят с помощью водяных аспираторов
различной вместимости, одновременно измеряющих объем прохо*
дящего газа.
272
ГЛАВА24. ПЕРЕГОНКА
Перегонка, или дистилляция,— процесс разделения
бинарных и многокомпонентных жидких смесей на отдельные
компоненты. При этом можно рассматривать в отдельности два процесса:
переход жидкости в парообразное состояние и конденсацию пара.
Жидкость закипает, когда давление ее насыщенного пара
становится равным внешнему давлению. Температура кипения
жидкости изменяется с изменением давления. При перегонке чистого
индивидуального вещества температура кипения при
определенном давлении постоянна, так как состав жидкости и
образующегося пара одинаков. В случае перегонки смеси жидких веществ,
компоненты которых смешиваются друг с другом в любом
соотношении, состав пара и жидкости все время изменяется в
результате обогащения газовой фазы более летучим компонентом.
Пар, обогащенный более летучим компонентом, конденсируется.
В результате получается жидкость (дистиллят) с тем же
составом, что и пар. При перегонке дистиллята вначале образуется
пар, еще более обогащенный легкокипящим компонентом.
В результате подобной многократной перегонки можно
практически разделить смесь на чистые компоненты. В этом заключается
сущность метода разделения смеси жидких веществ путем
дробной (фракционной) перегонки. Разделение смеси веществ
методом дробной перегонки тем легче, чем больше разница между
составом жидкости и составом пара. Однако методом дробной
перегонки нельзя разделить азеотропные (нераздельно кипящие)
смеси.
Дробная перегонка — чрезвычайно длительный процесс даже
для разделения смеси, состоящей из двух веществ. При большем
числе компонентов возникают еще большие трудности, которые
могут быть устранены перегонкой в колонке для точного
фракционирования. В результате этого процесса образуется
ряд отграниченных друг от друга фракций, кипящих в узких
температурных пределах.
Перегонку можно проводить не только при атмосферном, но и
при повышенном и уменьшенном давлении. Перегонка при
уменьшенном давлении применяется для разделения компонентов высо-
кокипящих жидкостей, которые при температуре кипения под
атмосферным давлением разлагаются или изменяются. Желаемая
температура кипения достигается путем соответственного
уменьшения остаточного давления.
Для очистки или разделения веществ, особенно термически
малостойких, мало растворимых в воде, или легко отделяющихся
от воды, часто используют перегонку с насыщенным или пере*
гретым водяным паром.
273
ПРОСТАЯ ПЕРЕГОНКА ПРИ АТМОСФЕРНОМ ДАВЛЕНИИ [5, с. НО]
Простая перегонка сводится к частичному испарению кипящей
жидкой смеси, полному отводу и конденсации образовавшихся при
этом паров. Как правило, простую перегонку применяют для
разделения терм-ически устойчивых жидких веществ, сильно
отличающихся по своим температурам кипения; для отделения жидкости от
растворенных в ней твердых веществ; для концентрирования
растворов; для очистки сжиженных газов и т. п.
Простейший прибор для перегонки при
атмосферном давлении состоит из колбы Вюрца, к горлу которой припаяна
трубка для отвода паров кипящей жидкости в холодильник, или из
отдельной колбы, снабженной насадкой Вюрца, термометром,
нисходящим холодильником, алонжем и приемником (рис. 156). В
качестве перегонной колбы можно использовать круглодонную,
остроконечную или грушевидную колбы. Колбу Вюрца или отдельную
перегонную колбу выбирают такой вместимости, чтобы
перегоняемая жидкость занимала не более 2/3 объема. Для перегонки высоко-
кипящих жидкостей следует пользоваться короткогорлыми колбами.
Небольшие объемы жидкостей лучше перегонять из остроконечных
колб, оттянутая форма которых позволяет вести отгонку до
минимального остатка. Наиболее пригодна грушевидная форма
перегонной колбы, обеспечивающая почти постоянную поверхность
испарения при уменьшении содержимого колбы.
Стеклянную насадку Вюрца выбирают так, чтобы номер КШ
керна соответствовал номеру КШ муфты перегонной колбы. Для
конденсации пара применяют обычный нисходящий холодильник.
Длина холодильника должна быть тем больше, чем ниже
температура кипения жидкости.
Пары твердых при комнатной температуре веществ не должны
охлаждаться в холодильнике до температуры затвердевания. Во
избежание этого рекомендуется холодильник периодически
прогревать теплой (горячей) водой.
При перегонке жидкостей, кипящих выше 150 °С, применяют
только воздушные холодильники. Жидкости, кипящие в пределах
а 6 ft
Рис. 156. Приборы для простой перегонки:
с —с колбой Вюрца без шлифов; б —с круглодонной колбой и насадкой на шлифах.
274
Рис. 157. Прибор для перегонки высококипящнх веществ.
Рис* 158. Стеклянные колба Фаворского (а) и колба Арбузова (6").
200—300 °С, перегоняют без холодильника, функцию которого в
этом случае может выполнять отводная трубка насадки
перегонной колбы (рис. 157).
При перегонке низкокипящих веществ приемную колбу
помещают в ледяную или охладительную баню.
Перегонку веществ вредных для здоровья (сероуглерод, тио-
нилхлорид и др.), следует выполнять в вытяжном шкафу.
Устанавливать термометр в горло перегонной колбы нужно так,
чтобы ртутный шарик его находился не менее чем на 5 мм ниже
нижнего края отверстия отводной трубки в шейке колбы Вюрца или
дистилляционной насадки. В этом случае термометр хорошо
омывается парами жидкости, что обеспечивает правильное измерение
температуры кипения. На конце шарика термометра должна
удерживаться капля конденсата; ее отсутствие указывает на то, что пар
в колбе перегрет и, следовательно, регистрируемая теглпература не
соответствует истинной температуре кипения.
В качестве приемника дистиллята может служить коническая
склянка или круглодонная колба. Приемник присоединяют к
холодильнику с помощью алонжа с отводной трубкой, служащей для
отвода несконденсировавшихся вредных или опасных паров газов
в безопасное место или в промывную склянку с соответствующим
поглотителем (нейтрализатором).
Если дистиллят чувствителен к влаге или диоксиду углерода,
к отводной трубке алонжа присоединяют хлоркальциевую трубку,
заполненную СаСЬ, натронной известью или аскаритом. Если
перегоняемое вещество чувствительно к кислороду, то перегонку ведут
в токе сухого азота. При этом приемную колбу следует
присоединять к поглотительной склянке, заполненной раствором,
поглощающим кислород.
Когда установка собрана, в колбу вливают перегоняемую
жидкость и для равномерного кипения вносят кусочки необожженного
фарфора, кирпича, а также заплавленные с одной стороны
стеклянные капилляры, которые помещают в колбу открытым концом
275
книзу. Сильный перегрев может сопровождаться «взрывным»
кипением, разбрызгиванием и перебрасыванием перегоняемой жидкости.
При длительном кипячении все количество адсорбированного
«кипелкой» воздуха может оказаться израсходованным, и жидкость
начинает кипеть неравномерно. Тогда необходимо повторно ввести
«кипелки» (или капилляры), предварительно охладив жидкость
ниже температуры кипения, иначе жидкость может внезапно
бурно вскипеть.
Перегонную колбу нагревают газовой горелкой через
асбестовую сетку, на закрытой электрической плитке или с помощью кол-
бонагревателя, на водяной, воздушной, масляной или
металлической бане. Нагревание на бане обеспечивает равномерность нагрева,
предотвращает перегревы и связанные с ними нарушения процесса
кипения. Температура бани не должна превышать температуру
кипения перегоняемого вещества больше чем на 20—30 °С.
Нагревание проводят так, чтобы перегонка проходила не
слишком быстро. При слишком интенсивном кипении в результате
перегрева паров в колбе создается повышенное давление, и
измеряемая температура не соответствует температуре кипения данной
фракции при атмосферном давлении. Скорость перегонки
считается нормальной, если из холодильника стекает 30—35 капель
конденсата в минуту.
При перегонке индивидуальных веществ температура кипения
постоянна в течение всего процесса, лишь к концу перегонки она
может подняться на несколько градусов. Непрерывное повышение
температуры кипения свидетельствует о том, что отгоняется смесь
веществ.
Для увеличения эффективности разделения смеси, когда
простая перегонка не позволяет добиться нужных результатов,
пользуются перегонной колбой с дефлегматором.
Дефлегматоры часто называют фракционирующими насадками;
некоторые из них послужили прообразом современных
ректификационных колонок.
В дефлегматоре за счет охлаждения наружным воздухом часть
паров перегоняемой смеси конденсируется, причем конденсат
(флегма) содержит преимущественно менее летучий компонент,
а пары — более летучий.
Когда стекающий конденсат соприкасается с поднимающимися
вверх парами кипящей жидкости, между ними происходит
взаимодействие, приводящее к дополнительной конденсации менее
летучего компонента и испарению легколетучего.
Для разделения смеси жидкостей, имеющих относительно
близкие температуры кипения, обычные дефлегматоры практически
малоэффективны. В практике для повышения эффективности
простых и вакуумных перегонок используют дефлегматоры различных
конструкций и колбы Фаворского и Арбузова (рис. 158).
При простой перегонке под атмосферным давлением боковая
горловина колбы Фаворского служит для загрузки
перегоняемой жидкости, выгрузки остатка от перегонки, внесения «кипелок»,
276
пропускания тока инертного газа и установки термометра. При
вакуумной перегонке, чтобы предотвратить бурное вскипание
жидкости, в боковую горловину вставляют капиллярную трубку для
перемешивания жидкости пузырьками воздуха или инертного газа.
Колба Арбузова представляет собой перегонную колбу,
спаянную с шариковым дефлегматором. На дно каждого шара
такого дефлегматора помещен стеклянный шарик, что значительно
повышает разделительную способность дефлегматора.
ДРОБНАЯ ПЕРЕГОНКА [36, с. 210]
При простой перегонке нельзя добиться полного разделения
смеси, а можно лишь выделить отдельные фракции, причем первая
будет обогащена более летучим компонентом, а последняя —
менее летучим. Средняя, промежуточная, фракция будет состоять из
смеси низко- и высококипящих компонентов. При дробной
перегонке процессы испарения и конденсации многократно повторяются;
дистиллят каждый раз становится исходным материалом для
следующего процесса. В результате концентрируется низкокипящий
компонент.
Дробной перегонке предшествует пробная перегонка, в результате которой
узнают величину всего интервала кипения смеси. Пусть этот интервал равен
90—135°С. Предположим, что разделяемая смесь состоит из двух веществ с
температурами кипения 90 и 135 °С. Интервал между температурами кипения
делят на три равные части. При первичной перегонке собирают фракции: I —
в пределах 90—105°С, II—105—120°С и III — 120—135°С. Фракцию I
перегоняют вторично до тех пор, пока термометр не покажет 105°С. При этой
температуре перегонку прерывают и прибавляют к остатку в колбе фракцию II
и снова доводят жидкость до кипения. То, что перегонится до 105°С, собирают
в тот же приемник. Когда температура достигнет 105°С, меняют приемник и
отгоняют жидкость до 120 СС. Снова прерывают перегонку, прибавляют к
остатку фракцию III, нагревают и, когда температура достигнет 120 °С, меняют
приемник и собирают дистиллят, перегоняющийся при 120—125 °С. После вторичной
разгонки оказывается, что количество вещества в I и III фракциях увеличилось,
а во II фракции значительно уменьшилось.
Нередко дробную перегонку целесообразно проводить при
уменьшенном давлении, особенно в тех случаях, когда компоненты
смеси имеют близкие температуры кипения и относятся к разным
классам соединений. Иногда различие в температурах кипения
таких веществ в вакууме может быть значительно большим, чем
при атмосферном давлении.
РЕКТИФИКАЦИЯ В КОЛОНКАХ [34, 59]
Ректификация — многократное испарение и конденсация —
реализуется в колонках при противотоке пара и жидкости с частичным
возвратом (флегмой) дистиллята при установившихся массо- и
теплообмене. В ректификационных колонках в условиях известного
температурного перепада по всей длине колонки создается
последовательный ряд фазовых равновесий между конденсатом,
стекающим обратно в перегонную колбу, и поднимающимся вверх паром.
При этом высококипящий компонент частично конденсируется из
паровой фазы, а низкокипящий — испаряется из флегмы.
277
Разделение компонентов на колонке должно происходить в
условиях адиабатического процесса. Тепловые потери приводят к
нарушению состояния равновесия, при этом пар конденсируется
вдоль стенок колонки. Разделение протекает тем полнее, чем
больше поверхность соприкосновения между флегмой и паром.
Для увеличения поверхности соприкосновения пара и флегмы
в лабораторных условиях применяют так называемые насадоч-
ные колонки, представляющие собой стеклянные или
кварцевые трубки с насыпной насадкой из одиночных стеклянных или
проволочных витков. Поверхность соприкосновения пара и
жидкости в колонке велика, поэтому облегчается теплообмен и
улучшается разделение компонентов.
Флегма, возвращаясь в перегонную колбу, обогащается менее
летучим компонентом, а газовая фаза, поступающая вверх, —
более летучим.
Скорость установления равновесия фаз в колонке зависит от
ее конструкции. За установлением равновесия в колонке обычно
следят по показанию термометра. В момент заполнения колонки
парами термометр показывает некоторую температуру, которая со
временем понижается до определенного уровня. Достижение этого
уровня означает, что установилось равновесие, т. е. пары
максимально для данной колонки обогатились наиболее летучим
компонентом.
Важный фактор точного фракционирования на колонке—флег-
мовое число, измеряемое долей конденсата, возвращаемого в
колонку для орошения, т. е. конденсата, стекающего из головки
колонки в ректифицирующую трубку с насадкой. Величину флегмо-
вого числа обычно регулируют при помощи выходного крана по
числу капель флегмовой жидкости и числу капель отбираемого
дистиллята так, чтобы до тех пор, пока продукт получается в
чистом виде, отбираемое в единицу времени количество его
поддерживалось примерно постоянным. Однако, как только количество
отгоняемого вещества уменьшается или температура кипения
повышается, что указывает на начало отгонки более высококипящей
составной части смеси, скорость отбора дистиллята следует сильно
уменьшить.
Конденсация паров, прошедших через ректификационную
колонку, и разделение конденсата на флегмовую жидкость, стекающую
обратно из колонки в перегонную колбу, и отбираемые порции
дистиллята, происходят в головке колонки. Если в головке
задерживается только часть паров, конденсат которых идет на орошение
колонки, а другая часть паров конденсируется в холодильнике и
отбирается в виде фракции дистиллята, то такие головки называют
головками частичной конденсации. В лабораторных
колонках аналитического и препаративного назначения
преимущественно применяются головки полной конденсации, в
которых конденсируются все пары, а конденсат специальным
устройством разделяется на флегмовую жидкость и
дистиллят,
278
Во время перегонки одного компонента температура кипения
держится постоянной, затем скачкообразно повышается вследствие
появления в дистилляте другого компонента и опять держится на
постоянном уровне, соответствующем температуре кипения этого
компонента. Во время «скачка» температуры получаются
различные количества промежуточных фракций. Колонка считается тем
эффективней, чем меньше количество промежуточных фракций.
Рабочая мощность колонки оценивается количеством
паров и жидкостей, проходящих противотоком через колонку, не
вызывая ее «захлебывания», т. е. заполнения центральной трубки
избытком флегмы. Каждая колонка имеет некоторую оптимальную
скорость перегонки, при которой она работает наиболее
эффективно. При выборе колонки следует учитывать также и то, что часть
конденсата расходуется на орошение внутреннего пространства
колонки. Эта неиспользованная часть продукта не отбирается в виде
дистиллята. Количество перегоняемой жидкости должно быть по
меньшей мере в 20 раз больше количества конденсата, орошающего
колонку.
Сравнительную разделительную эффективность колонок для
ректификации условно оценивают числом теоретических
тарелок (ЧТТ). Другая величина, характеризующая
эффективность ректификационной колонки — высота,
эквивалентная одной теоретической тарелке (ВЭТТ), которая
получается делением высоты колонки на число теоретических
тарелок.
В лабораторной практике пользуются разнообразными типами
перегонных колонок. Во многих лабораториях особенно широко
используются колонки с насыпной насадкой из одиночных
витков стеклянной спирали или обрезков металлической
проволочной спирали. Один из вариантов подобных стеклянных колонок
[36 с. 210] представлен на рис. 159.
Стеклянные детали для сборки лабораторных колонок точной
ректификации при атмосферном и пониженном давлении с высотой
ректифицирующей части 400 и 1100 мм выпускаются серийно [46,
с. 104]. Общая длина центральной трубки с головкой полной
конденсации 840 и 1620 мм. Объем перегоняемой жидкости 10—50 и
50—500 мл. Соединение деталей установки выполнено на
нормальных взаимозаменяемых шлифах. Общий вид колонок, собранных из
стеклянных деталей, представлен на рис. 160.
Ректификационная стационарная
лабораторная установка РУТ предназначена для разделения
различных веществ в непрерывном или периодическом процессе. На
установке можно производить ректификацию как при уменьшенном,
так и при атмосферном давлении. Установки выпускаются с
колонками тарельчатого типа трех типоразмеров РУТ-20, РУТ-25 и
РУТ-55. Для разделения больших объемов органических веществ
при атмосферном и пониженном давлении выпускается
стационарная лабораторная установка с насадочной колонкой
УПФ. Для разделения нефтяных фракций углеводородных смесей
279
Рис. 159. Ректификационная колонка:
/ — конденсатор; 2 —кран для отбора дистиллята; 3—холодильник; 4 — изогнутый переход;
5 —хлоркальциевая трубка; 5 —трубка для термометра; 7 —соединительная трубка; 5
—отводная трубка; 9 —верхний счетчик капель (счетчик орошения); 10 — центральная трубка с насадкой;
// — обогревающая трубка рубашки; 12—изолирующая трубка рубашки; 13—расширение
центральной трубки с сетчатым колпачком; 14 — нижний капельник; /5 —хвостовик; 16—колба
(куб); 17 — колбонагреватель; 18, /9—автотрансформаторы; 20, 2/ —амперметры;
22—трехходовой кран с полулунным ходом.
Рис. 160. Колонки точной ректификации при атмосферном давлении (а) и в вакууме (б),
и сложных смесей других органических соединений в лаборатор*
ных условиях изготовляется аппарат четкой
ректификации АЧР-2.
В соответствии с конструкцией применяемой колонки в
значительной степени меняется и техника перегонки. Однако имеются
общие операции проведения перегонки на насадочных колонках,
краткое описание которых приводим ниже.
Когда из стеклянных деталей собрана колонка (согласно
прилагаемой инструкции), ее следует промыть и просушить. Для
этого в колбу для перегонки помещают этиловый спирт, нагревают
колбу при закрытом кране для отбора проб дистиллята и
одновременно пускают воду, охлаждающую головку колонки. Затем
устанавливают такой режим обогрева, чтобы число капель,
падающих в колонку из головки в единицу времени, было в 2—2,5
раза меньше, чем число капель, падающих из колонки в колбу,
а температура рубашки была на 5—10 °С ниже температуры
кипения спирта. При этом колонка «захлебывается». В результате
этого из насадки вытесняется воздух и вся она смачивается флег-
280
мой. Уменьшая нагрев перегонной колбы, дают стечь избытку
флегмы в течение 15—20 мин, после чего, сохраняя установленный
обогрев колонки, усиливают нагрев колбы, пока в головке вновь
не соберется столб жидкости. Целесообразно держать колонку в
состоянии «захлебывания» 5—10 мин и чтобы объем жидкости над
насадкой составлял 10—25% объема насадки. «Захлебывание»
повторяют 2—3 раза, затем устанавливают режим разгонки и
заставляют колонку работать 30—60 мин при полном орошении,
отбирают несколько раз по 0,5—1,0 мл дистиллята, чтобы промыть
кран и отводные трубки, после чего выключают нагрев колбы.
Когда вся флегма стечет из колонки, колбу отсоединяют,
прокачивают через колонку сухой воздух с помощью водоструйного
насоса, присоединяемого к отводной трубке головки, из которой на
это время выпускают воду. После выполнения описанных
операций колонка готова к работе.
Перегоняемое вещество помещают в колбу для перегонки. Если
температуры кипения компонентов смеси не превышают 150 °С,
перегонку предпочитают проводить при атмосферном давлении, при
более высоких температурах — при пониженном: 133 Па (1 мм
рт. ст.).
Перегонную колбу предпочтительнее всего нагревать на
жидкостной бане с регулируемым обогревом. Включив нагрев
перегонной колбы, устанавливают режим обогрева колонки, как было
описано выше, и 2 раза дают колонке «захлебнуться». После
этого устанавливают требуемый режим обогрева колбы и рубашки.
При полном орошении колонку выдерживают около 1 ч для того,
чтобы установилось равновесие фаз. Далее опытным путем
устанавливают оптимальный режим работы колонки.
При работе с колонками, у которых отсутствует обогрев,
режим работы определяется только скоростью испарения жидкости.
Отбор дистиллята можно проводить непрерывно и
периодически. При непрерывном отборе слегка открывают кран для
отбора дистиллята у головки полной конденсации так, чтобы
установить заранее заданное флегмовое число (1:10, 1:20, 1:30).
Температуру измеряют при каждом отборе дистиллята.
Если в приемнике собирается индивидуальное вещество,
признаком чего служит постоянство температуры паров в головке в
течение некоторого времени, величину флегмового числа можно
уменьшить, увеличивая скорость отбора дистиллята. При переходе
от фракции к фракции флегмовое число вновь увеличивают,
уменьшая скорость отбора дистиллята. Это называется «отжимом
фракции».
При периодическом отборе дистиллята на 1—5 с
полиостью открывают кран для отбора дистиллята, спускают
небольшое количество последнего в приемник и закрывают кран. Так
как при этом удаляется часть легколетучего компонента, то
равновесие в колонке нарушается, и температура пара в головке
начинает увеличиваться. Когда кран вновь закрывают, в колонке
постепенно устанавливается состояние равновесия, и температура
281
пара при этом понижается. Когда температура установится, вновь
отбирают пробу дистиллята. Температуру и объем дистиллята
отсчитывают непосредственно в момент отбора очередной порции
дистиллята.
При ректификации в вакууме головку колонки
снабжают вакуум-приемником.
ПЕРЕГОНКА В ВАКУУМЕ
Некоторые вещества не могут быть перегнаны при
атмосферном давлении ввиду частичного или полного разложения их при
температуре кипения. Такие вещества можно перегнать при
условии, если давление в перегонной колбе будет понижено настолько,
чтобы температура кипения вещества оказалась ниже температуры
его разложения. Например, вещества, кипящие с разложением
при 350 °С и 101 кПа (760 мм рт.ст.), можно перегнать без
разложения при 160—210 °С и 1,33 кПа (10 мм рт.ст.) или при 100—
130 °С и 1,33 Па (0,01 мм рт.ст.).
При вакуумной перегонке вещества в меньшей степени
подвержены действию кислорода. В ряде случаев, как было указано
выше, вакуумная перегонка способствует разделению азеотропных
смесей.
Прибор для перегонки в вакууме (рис. 161) состоит
из перегонной колбы, приспособления для ее нагрева, термометра,
холодильника, приемника конденсата или приспособления для
смены приемников, источника вакуума и приспособления для
измерения и регулирования давления.
В качестве перегонной колбы могут быть использованы колбы
Кляйзена, Фаворского и Арбузова, а также круглодонные или
остродонные колбы на шлифах с дистилляционнои насадкой или
холодильником Кляйзена.
Форма и вместимость перегонной колбы определяются
объемом и температурными пределами кипения перегоняемой
жидкости. Объем последней не должен превышать 1/2 объема колбы.
Высококипящие жидкости следует перегонять из колб с низко-
припаянной и широкой
боковой отводной
трубкой.
В центральное
горло колбы Кляйзена или
дистилляционнои
насадки Кляйзена для
п р е ду пр е ж д е н и я
«в з р ы в н о г о» в с к и -
п а н и я вставляют тон-
кооттянутый стеклян-
Рис. 161. Прибор для перегонки
в вакууме:
1—дистилляционная насадка
Кляйзена; 2 — алонж изогнутый
на шлифах с отводом.
282
Рис. 162. Насадка Аншютца и Тиле:
/, 2, 3—краны; 4—приемник конденсата; 5—измерительный
цилиндр алонжа.
К вакуум£
ный капилляр, доходящий почти до
дна колбы. О пригодности капилляра
судят, продувая через него воздух в пробирку
с диэтиловым эфиром. Хороший капилляр
даже при небольшом избыточном давлении
пропускает лишь мелкие отдельные
пузырьки, образующие в жидкости тонкую
цепочку. После подключения прибора к
вакуум-насосу через жидкость должны
проходить очень мелкие пузырьки газа
(воздуха), обеспечивающие перемешивание перегоняемой жидкости и
равномерность кипения.
В боковое горло перегонной колбы или насадки помещают
термометр.
В случае пользования колбой Кляйзена без шлифов капилляр
и термометр вставляют через кусочки резиновой вакуумной
трубки, обеспечивающие должную герметичность.
Значительные трудности возникают при перегонке в вакууме
пенящихся жидкостей. Чтобы избежать переброса
жидкости, рекомендуется заполнять боковое горло колбы или насадки
отрезками стеклянных трубок, стеклянной ватой или же вставлять
вместо термометра второй стеклянный капилляр. Пенящиеся
жидкости перегоняют при незначительном заполнении перегонной
колбы. Пенообразование часто вызывается примесями,
содержащимися в перегоняемой жидкости; при повторной перегонке пена,
как правило, не образуется.
Перегонную колбу обычно нагревают на силиконовой бане —
для жидкостей, кипящих ниже 200 °С, или металлической — для
высококипящих жидкостей. Температуру бани контролируют
термометром. Дистиллят охлаждают так же, как и при перегонке
при атмосферном давлении.
Чтобы собирать несколько фракций дистиллята
отдельно, не отключая вакуум, пользуются различными устройствами.
К наиболее простым из них относится алонж «паук» (см. гл. 2).
Отводы «паука» соединяют с приемниками, объем которых
соизмеряют с ожидаемым объемом фракции. В качестве приемников
применяют круглодонные или остродонные колбочки или
пробирки со шлифами; шлифы смазывают чистым касторовым маслом
или глицерином. Смазка обеспечивает хорошее скольжение и
предотвращает заклинивание шлифов.
Устройство, позволяющее менять любое число приемников во
время перегонки, представлено на рис. 162. Пользование
подобным приемником для сбора низкокипящих жидкостей исключает
возможность контакта паров фракций дистиллята, находящихся в
отдельных приемниках, и тем самым предотвращает взаимное
загрязнение фракций.
283
Применение для создания вакуума водоструйного насо-
с а не требует дополнительных приспособлений для поглощения
паров перегоняемой жидкости; нужно только предохранять
приемник и перегонную колбу от попадания воды, для этого между
насосом и прибором перегонки помещают предохранительную
склянку. Если используют масляный вакуумный насос, то между
перегонным прибором и насосом должна находиться
поглотительная или вымораживающая система.
Вакуумная перегонка взрывоопасна. Мелкие осколки стекла,
образующиеся при взрыве вакуумированного сосуда,
представляют большую опасность, особенно для глаз. Поэтому при
проведении работ под вакуумом необходимо надевать защитные очки или
пользоваться защитной маской.
Перед началом вакуумной перегонки следует проверить
герметичность системы при минимальном давлении без применения
нагрева.
Запрещается создавать вакуум в перегонной колбе,
наполненной горячей жидкостью. После отгонки растворителя при
атмосферном давлении следует сначала охладить содержимое колбы,
а затем включить вакуум-насос.
Стеклянные сосуды, например вакуум-эксикаторьь в которых
создано уменьшенное давление, должны быть закрыты чехлом или
обернуты легкой прочной тканью, чтобы предотвратить разлетание
осколков (при взрыве сосуда). Переносить вакуумированные
сосуды с места на место не рекомендуется.
При окончании вакуумной перегонки нефтепродуктов
включать насос и сообщать систему с воздухом следует, если
температура нефтепродукта не менее чем на 50 °С ниже температуры его
самовоспламенения.
ПЕРЕГОНКА С ВОДЯНЫМ ПАРОМ
Перегонку с водяным паром применяют для выделения,
очистки и разделения веществ, мало растворимых в воде и
обладающих достаточным давлением паров при температуре кипения
воды. Если вещества нерастворимы в воде, давление паров смеси
равно сумме парциальных давлений паров каждого из
компонентов. Вследствие этого температура кипения вещества при
перегонке с водой всегда ниже температуры кипения воды при
данном давлении. Поэтому перегонка с водяным паром позволяет
отгонять вещества, которые при обычной перегонке в той или иной
степени разлагаются.
Если вещества растворимы в воде, давление их паров в
присутствии воды понижается. Например, масляная кислота,
растворяющаяся в воде хуже, чем муравьиная, с водяным паром
перегоняется лучше, чем муравьиная, несмотря на то, что
температура кипения муравьиной кислоты 101 °С, а масляной—162 °С.
Перегонку с водяным паром можно проводить как при
атмосферном давлении, так и в вакууме. Подводимый водяной пар
должен быть насыщен или перегрет. Перегретый водяной пар по-
284
Рис. 163. Перегонка с водяным
паром:
/ — медный паровик; 2
—перегонная колба; 3 — холодильник; 4—
приемник дистиллята; 5 —
трубка для водяного пара; б
—соединительная трубка; 7 —
предохранительная трубка; 8» 9 —
нагреватели.
зволяет отгонять вещества с довольно низким давлением паров.
Для перегревания водяного пара пользуются
пароперегревателями. Они обычно представляют собой металлическую или
стеклянную спираль, нагреваемую газовыми горелками или
нагревательными банями, в которой насыщенный водяной пар
превращается в перегретый.
Перегонка с водяным паром производится на установке,
представленной на рис. 163. Колбу располагают наклонно, чтобы
летящие вверх брызги не попадали в отводящую пары трубку.
Пар, поступающий из паровика, очень влажен, и перегонная
колба может быстро заполниться водой, поэтому между паровиком
и перегонной колбой устанавливают водоотделитель.
Для перегонки с водяным паром в вакууме в качестве
парообразователя используют перегонную колбу, присоединяя ее к
вакуумной линии. Тонкий капилляр, помещенный в колбу, позволяет
регулировать расход водяного пара.
В процессе перегонки с водяным паром перегонную колбу
следует подогревать, а горло колбы теплоизолировать асбестовым
шнуром. Если из холодильника начнут выходить несконденсировав-
шиеся пары дистиллята, нужно уменьшить подачу пара или
увеличить подачу охлаждающей воды в холодильник.
При перегонке твердые вещества часто
затвердевают в холодильнике. Кристаллы целесообразно расплавить в
процессе перегонки, прекращая временно подачу охлаждающей
воды в холодильник. Если температура плавления твердого
вещества выше температуры пара, то по окончании перегонки это
вещество выталкивают из холодильника длинной стеклянной
палочкой или извлекают подходящим растворителем, из которого оно
хорошо кристаллизуется.
При перегонке с водяным паром иногда образуются
устойчивые эмульсии. В этом случае перегнанный продукт
может быть выделен выслаиванием или экстрагированием
органическим растворителем.
О конце перегонки веществ, нерастворимых в воде, судят по
отгону прозрачной воды, не содержащей маслянистых примесей.
При перегонке веществ, растворимых в воде, о ходе перегонки
судят по физическим, химическим или другим свойствам дистиллята.
285
ГЛАВА 25. ВОЗГОНКА
Возгонка (сублимация) представляет собой операцию,
при которой в нагреваемой части прибора кристаллическое
вещество испаряется, а затем, минуя жидкую фазу, конденсируется с
образованием кристаллов в охлаждаемой части.
Возгонка происходит при температуре более низкой, чем темпе*
ратура плавления вещества и, конечно, ниже температуры его
кипения.
Процесс можно проводить под атмосферным давлением, при
пониженном давлении, а также в токе инертного газа.
Часто возгонка служит превосходным средством очистки
твердого органического вещества от смолистой примеси, отделить
которую путем кристаллизации очень трудно, а прибегнуть к
перегонке невозможно, так как вещество разлагается при температуре
кипения и даже плавления.
Преимущество очистки твердых веществ возгонкой по
сравнению с кристаллизацией заключается в большем выходе чистого
вещества и простоте аппаратуры, а также в том, что очищенное
вещество свободно от загрязнений, которые даже при тщательной
кристаллизации попадают в конечный продукт (волокна
фильтровальной бумаги, мелкие частицы угля, остатки растворителя).
Возгонка используется также для очистки твердых
неорганических веществ, обладающих значительным давлением паров (As203,
NH4C1, I2).
Вопрос о том, отдавать ли предпочтение возгонке или
кристаллизации, необходимо решать в каждом конкретном случае
отдельно, в зависимости от свойств очищаемого твердого вещества.
ВОЗГОНКА ПРИ АТМОСФЕРНОМ ДАВЛЕНИИ
Характер образующегося сублимата в значительной степени
зависит от температуры конденсации паров очищаемого вещества.
Чем ниже температура охлаждающего устройства (холодильника),
тем тверже и мельче образующиеся кристаллы. Наилучшая форма
кристаллов сублимата достигается в том случае, когда температура
конденсирующей поверхности лишь немного ниже температуры
плавления вещества.
Для возгонки небольших количеств вещества часто пользуются
двумя часовыми стек л а м и одинаковой величины,
пришлифованными друг к другу. На нижнее стекло помещают возгоняемое
вещество, между стеклами зажимают продырявленный в
нескольких местах кружок фильтровальной бумаги, чтобы образующиеся
кристаллы сублимата не падали на нижнее нагретое стекло.
Нижнее стекло подогревают на песчаной бане или очень осторожно
маленьким пламенем спиртовки на асбестовой сетке, верхнее
охлаждают кусочками влажной фильтровальной бумаги.
286
а
гг
в
Вода
«4. г"
чх^ га
t | t w
Рис. 164. Устройства для возгонки при атмосферном давлении!
а, б—охлаждение проточной водой; в —воздушное охлаждение.
Рис. 165. Прибор для возгонки малых количеств веществ в вакууме.
Рассмотрим часто проводимую в лабораториях операцию
очистки иода возгонкой. Иод перед возгонкой смешивают с KI и Саб
и тщательно растирают. Обычно на 6 масс. ч. иода берут 1 ч. KI и
2 ч. СаО.
Самое простое устройство для возгонки небольших
количеств иода — тонкостенный стакан, поставленный на песчаную
баню так, чтобы дно его было погружено в песок на 1—2 см.
Стакан сверху накрывают часовым стеклом, причем выпуклая сторона
его должна быть обращена внутрь стакана. При осторожном
нагревании иод возгоняется, и на стекле оседают игольчатые
кристаллы, которые собирают при помощи стеклянного или
фарфорового шпателя.
Повторная возгонка приводит к получению препарата иода
высокой степени чистоты.
Для очистки веществ возгонкой используют различные
устройства (рис. 164). При относительно невысоких температурах
возгонки применяют водяное охлаждение, а при высоких обычно
достаточно воздушного. Так, например, для возгонки иода иногда
используют прибор, состоящий из двух пришлифованных
полусферических частей, из которых верхняя служит холодильником.
ВОЗГОНКА В ВАКУУМЕ
К возгонке в вакууме прибегают тогда, когда возгоняемые
вещества малолетучи. Возгонка в вакууме протекает быстрее и более
Эффективна, чем возгонка при обычных условиях.
Приборы для возгонки в вакууме весьма просты. Простейший
прибор может быть изготовлен из пробирки для отсасывания и
Рис. 166. Прибор для возгонки в вакууме с
горизонтальной конденсационной трубкой:
/ — колба с веществом; 2 — конденсационная
трубка; 3 —головка с краном.
btXXAXXXX*-] I Vi*££*rf
287
обычной пробирки с подводящей и отводящей трубками для воды
(рис. 165). Пробирка с водой выполняет в данном случае роль
холодильника. Боковой отвод пробирки для отсасывания
присоединяют к водоструйному или масляному насосу через предохрани*
тельную склянку.
Часто используют прибор, изображенный на рис. 166. Его
помещают в воздушную баню так, чтобы снаружи оставалась головка
с краном и часть конденсационной трубки. Когда часть вещества
возгонится и кристаллы осядут в виде кольца на выступающей
части трубки 2, прибор немного выдвигают из воздушной бани и
продолжают возгонку. При многократном повторении этого приема
можно собрать отдельные порции сублимата и по температуре
плавления определить их идентичность. Во время разборки прибора
рыхлый сублимат часто отстает от конденсационного устройства
(или трубки) и падает на дно прибора, в котором еще содержится
неочищенное вещество. В этом случае возгонку нужно повторить.
ВОЗГОНКА В ТОКЕ ИНЕРТНОГО ГАЗА
Если возгонка при обычном давлении протекает медленно, что
вызывается медленным отводом образующихся паров в
охлаждаемую часть прибора, то процесс выгодно проводить в токе инертного
газа (N2, C02 и т. п.), который уносит пары вещества из
обогреваемого пространства. Одновременно создается защитная от воздуха
атмосфера. Инертный газ рекомендуется подогревать до
температуры, немного превышающей температуру возгонки, чтобы
конденсация паров не начиналась прежде, чем они достигнут
охлаждающей поверхности. Следует, однако, иметь в виду, что при этом
способе возгонки получается очень рыхлый сублимат.
ГЛАВА26 ВЫПАРИВАНИЕМ КОНЦЕНТРИРОВАНИЕ
При работе как с водными, так и неводными растворами
нередко возникает необходимость в выделении растворенного
вещества или повышении его концентрации в растворе. Например,
концентрирование растворов для кристаллизации как правило, —
обязательная операция, так как для растворения вещества всегда
используется большее количество растворителя, чем это необходимо
для приготовления насыщенного раствора.
Выпаривание и перегонка основаны на одном и том же
принципе— разделении веществ вследствие их различной летучести.
Однако эти операции различаются по технике проведения.
Основная цель перегонки — получение дистиллята, а выпаривания —
выделение нелетучего остатка или повышение концентрации раствора.
Обычная перегонная аппаратура неудобна для выпаривания в тех
случаях, когда необходимо количественно выделить твердый
остаток, и мало пригодна для отгонки воды при низких температурах,
особенно от веществ, которые при этом пенятся.
Выпаривание и концентрирование растворов можно проводить
при комнатной, повышенной температуре или при температуре
кипения растворителя как при атмосферном, так и при пониженном
давлении. Скорость испарения жидкости зависит не только от
температуры и давления, но и от площади поверхности испарения,
интенсивности перемешивания и скорости газа, омывающего
поверхность испаряющейся жидкости.
При выпаривании и концентрировании растворитель может либо
улетучиваться, либо улавливаться.
ВЫПАРИВАНИЕ В ОТКРЫТЫХ СОСУДАХ
Наиболее упрощенный способ упаривания — это упаривание
при кипячении, которое проводят в открытых сосудах. Этот способ
обычно применяют при работе с водными растворами
неорганических веществ. Во избежание перегрева жидкости и внезапного ее
вскипания в сосуд для выпаривания помещают «кипелки». Если
из раствора в процессе упаривания начинает выделяться твердое
вещество, то «кипелки» перестают действовать. Перегрев жидкости
в этом случае можно предотвратить, пропуская через жидкость
ток газа или перемешивая ее.
В качестве сосудов для выпаривания используют плоские
стеклянные или кварцевые, фарфоровые или платиновые чашки,
вместимость которых зависит от количества выпариваемого раствора.
При концентрировании малых объемов раствора могут быть
применены фарфоровые или платиновые тигли. Реже используют
стеклянные, фарфоровые или кварцевые стаканы.
Ю Зак. 1237 289
При выпаривании в чашках или стаканах на водяной бане
следует предохранить раствор от попадания загрязнений из воздуха.
Для этой цели используют предохранительную воронку
для собирания паров типа ВСП (рис. 167). Воронку
укрепляют над чашкой. Пары жидкости попадают в воронку и частично
выходят через ее отвод, частично конденсируются на стенках.
Образующаяся жидкость стекает в желобок, а из него по резиновой трубке —
в приемник для конденсата. В качестве предохранительной
воронки можно применять тзкже обычную воронку, диаметр которой
несколько больше чашки для выпаривания. Воронку с коротко
отрезанной трубкой укрепляют над чашкой в несколько наклонном
положении; при этом конденсат стекает с опущенного края.
Ускорить процесс выпаривания и одновременно защитить
раствор от загрязнения можно с помощью воронки,
приспособленной для отсасывания воздуха водоструйным насосом с поверхности
испаряемой жидкости.
При выпаривании водных растворов твердых веществ часто
наблюдается явление ползучести. Вещество «ползет» по стенкам
сосуда, в котором испаряется растворитель, и даже выходит за края
сосуда. Нежелательная ползучесть — следствие неравномерного
обогревания раствора, когда верхняя часть чашки (стакана) почти
не нагревается. Для предотвращения ползучести рекомендуется
К вакуумной
линии
Рис. 167. Воронка для собирания паров.
Рис. 168. Прибор для выпаривания в
вакууме:
1 — перегонная колба; 2 — насадка;
3—холодильник; 4 — приемная колба;
5—капельная воронка; б—капиллярная
трубка.
290
пользоваться приспособлением из двух фарфоровых чашек,
внутренняя из которых несколько меньше внешней и более плоская.
Выпариваемый раствор наливают во внутреннюю чашку, а
наружную подогревают. При этом края внутренней чашки нагреваются
быстрее, чем дно, и сначала начинает просыхать верхняя кромка
выпавшей корочки кристаллов, а затем нижняя, что препятствует
ползучести вещества.
ВЫПАРИВАНИЕ В ЗАКРЫТЫХ СОСУДАХ [53, с. 189]
Если выпариваемый раствор можно нагревать до кипения при
обычном давлении, то пользуются установкой, применяемой для
обычной перегонки. Если нагревание до температуры кипения
нежелательно, то упаривание проводят с отсасыванием паров в токе
сухого газа при температуре несколько ниже температуры кипения.
Когда нагревание до относительно высокой температуры может
привести к разложению растворимого вещества, выпаривание
проводят при уменьшенном давлении.
В вакууме выпаривание протекает при относительно более
низкой температуре, скорость выпаривания выше и исключается
опасность загрязнения вещества пылью и влагой из воздуха.
Для выпаривания в вакууме, особенно пенящихся жид*
костей, удобен прибор (рис. 168), который можно легко собрать
из деталей приборов для вакуумной перегонки. Прибор позволяет
в мягких условиях получать сухие остатки из различных элюатов.
Прибор присоединяют к источнику вакуума и при достижении
заданного разрежения непрерывно прибавляют в обогреваемую
колбу / испаряемую жидкость. Пары растворителя проходят по
наклонной насадке 2, конденсируются во внутренней полости
холодильника 3, и конденсат стекает в приемную колбу 4. Кран
позволяет регулировать скорость отсоса. Воздух (азот) в систему
поступает через капиллярную трубку 6. Испаряемая жидкость
непрерывно подается из капельной воронки 5.
Перегонную колбу при упаривании сильно пенящихся
жидкостей полезно всю погрузить в нагревательную баню. Если
позволяют условия эксперимента, то пенообразование можно
предотвратить прибавлением нескольких капель октилового или децилового
спирта.
ВАКУУМНЫЕ ИСПАРИТЕЛИ [46, с. 103]
Для выпаривания растворов в вакууме большое
распространение получили приборы, работающие по принципу пленочного
испарения на стенках непрерывно вращающейся колбы с испаряемым
раствором.
Подобные роторные вакуум-испарители
предназначаются для концентрирования растворов в вакууме, упаривания
растворов термолабильных и пенящихся веществ, получения чистых
растворителей, высушивания сыпучих веществ и для других работ.
10* 29L
Рис. 169. Роторный испари*
тель:
1—водяная баня;
2—вращающаяся колба для упаривания;
3 — мотор и уплотнение;
4—холодильник; 5—приемник
дистиллята.
Один из таких роторных испарителей схематически изображен на
рис. 169. Испаряемый раствор по узкой внутренней трубке попадает
в колбу 2, нагреваемую на водяной бане / и вращающуюся с
частотой 20—140 об/мин. Вращение осуществляется при помощи
механизма 3, находящегося в кожухе. Соединительные трубки, одна
из которых вращается на конусном шлифе относительно
неподвижной другой трубки, выполнены из тефлона. Пары растворителя
конденсируются в холодильнике 4, Баню нагревают с помощью
нагревателя с электрообогревом; температура жидкого
теплоносителя (воды, масла) поддерживается автоматически. Конденсат через
спиральный холодильник, охлаждаемый водой или рассолом,
собирается в приемник. Испарительная вращающаяся колба
заполняется через питательный кран периодически или непрерывно.
Выпускаются два типа лабораторных ротационных
испарителей: ИР-1М и ИР-10. Настольный аппарат ИР-1М состоит
из привода, испарителя, бани, блока управления, станины и
держателя. Вместимость испарительной колбы может быть от 50 до
1000 мл. Частота вращения испарительной колбы 20—140 об/мин.
Остаточное давление не превышает 3,99 кПа (30 мм рт. ст.). Баня
водяная или масляная с
электронагревателем. Температура бани регулируется в
пределах 15—100 °С.
Если необходимо быстро отгонять или
испарять большие объемы растворителя,
в лабораториях часто применяют ц и р к у *
ляционный вакуумный
испаритель типа АЦВ
производительностью 1, 5, 10 и 20 л/ч. Принцип действия
испарителя (рис. 170) основан на
непрерывной циркуляции. Жидкость,
предназначенная для упаривания, подается в
Рис. 170. Испаритель циркуляиионно-выпарной типа АЦВ:
1 — генератор пара; 2—сосуд для упариваемой жидкости;
5—нагреватель; 4— колба испарителя; 5— холодильник.
292
оба колена испарителя. В трубках теплообменника 3, через
который пропускают из ультратермостата горячую воду или водяной
пар из генератора У, испаряемая жидкость поступает в сосуд 4>
где она растекается по стенке. Часть воды испаряется и
конденсируется затем в холодильнике 5, а упаренный раствор стекает во
второе колено. После упаривания до минимального объема
водоструйный насос отключают и концентрированный раствор сливают
через нижний кран.
Кроме аппарата АЦВ отечественная промышленность
выпускает испаритель циркуляционный ИЦ и аппарат
экстракционно-у парной АУЭЛ производительностью 2 и
5 л/ч. Преимущество этих аппаратов заключается в том, что все
узлы и детали, соприкасающиеся с рабочими растворами,
выполнены из стекла и соединены между собой на конусных и
сферических шлифах. Это дает возможность работать с агрессивными
жидкостями, а также концентрировать нестойкие и пенящиеся
жидкости.
глава 27. ЭКСТРАКЦИЯ ТВЕРДЫХ И ЖИДКИХ ВЕЩЕСТВ
Экстракция — процесс разделения смеси твердых или жидких
веществ с помощью избирательных растворителей (экстрагентов).
Физическая сущность экстракции состоит в переходе извлекаемого
вещества из жидкой или твердой фазы в фазу жидкого экстрагента
при их взаимном соприкосновении.
Экстракция органическими растворителями — один из наиболее
эффективных и универсальных методов разделения,
концентрирования и очистки веществ [3, 30].
В простейшем случае для экстракции растворенного вещества
из воды к водному раствору прибавляют органический
растворитель (экстрагент), практически не смешивающийся с водой, и
смесь взбалтывают для ускорения распределения растворенных
веществ между двумя жидкостями. По установлении межфазного
равновесия, что обычно достигается после взбалтывания в течение
2—5 мин, взбалтывание прекращают, и жидкие фазы вновь
расслаиваются.
При экстракции в делительной воронке нижний слой жидкости
можно аккуратно слить и таким образом разделить два
растворенных вещества. Если плотность экстрагента больше плотности воды,
то он образует нижний слой, а если меньше — верхний.
Предположим, что исходный водный раствор содержал два
растворенных вещества А и Б. Если сродство органического
растворителя к одному из них намного больше, чем сродство к нему воды,
то это вещество полностью или почти полностью перейдет из
водной фазы в органическую. Распределение растворенного вещества
между двумя жидкими фазами определяется законом
распределения: согласно ему отношение концентрации вещества, которое
растворено в двух несмешивающихся и находящихся в равновесии
жидких фазах, при определенной температуре — величина
постоянная, называемая концентрационным коэффициентом
распределения Dc:
ис — l^Jopr/l^jBOfl
Если разность Dc экстрагируемых веществ в двух
несмешивающихся растворителях достаточно велика, то их можно разделить
простой однократной экстракцией; в ином случае применяют
многократную простую или же д р о б н у ю экстракцию.
В отличие от большинства других методов очистки веществ
экстрагирование органическими растворителями из водных растворов
чаще всего осуществляют при относительно невысокой температуре,
а иногда при охлаждении, что особенно удобно при работе с
термически нестойкими веществами. Эффективность экстракции в
системе твердое вещество — жидкость определяется прежде всего
растворимостью и скоростью перехода твердого вещества в
растворитель.
294
Растворимость твердого экстрагируемого вещества можно
регулировать, подбирая растворитель с таким расчетом, чтобы в
раствор переходило преимущественно очищаемое вещество, а
загрязнения оставались в твердой фазе. Скорость перехода вещества из
твердой фазы в раствор определяется, в основном, скоростью
проникновения экстрагента в твердую фазу, диффузии экстрагируемого
вещества в жидкости и удаления вещества с поверхности твердое
тело — жидкость (влияние температуры и перемешивания).
В отличие от системы жидкость — жидкость, на границе
твердой и жидкой фаз равновесия практически не наступает. Основная
задача, возникающая при экстракции твердых веществ (смол,
жиров, растительных продуктов), заключается в максимальном
ускорении приближения системы к состоянию равновесия, что
достигается увеличением поверхности твердой фазы (измельчение),
постоянной подачей свежего растворителя на границу фаз,
перемешиванием или противотоком.
В настоящем разделе рассматриваются лишь простейшие
методы экстракции, обычно используемые в практике работы
препаративных и аналитических лабораторий. Более подробные сведения
о методах экстракции (дробной, противоточной) можно найти в
рекомендуемой литературе.
ЭКСТРАКЦИЯ ТВЕРДЫХ ВЕЩЕСТВ [36, с. 375]
Выделение и очистка твердого вещества может осуществляться
однократной или многократной экстракцией.
При однократной экстракции твердое тонкоизмельчен-
ное вещество при перемешивании нагревают с растворителем,
(экстрагентом) в колбе с обратным холодильником, после чего
экстракт отфильтровывают, испаряют растворитель и выделившееся
твердое вещество очищают перекристаллизацией.
При многократной экстракции экстрагируемый
материал помещают непосредственно в экстракционный сосуд (насадку),
снабженный промежуточным дном из пористого стекла, либо в
специальный патрон из фильтровальной бумаги. Пары растворителя
из перегонной колбы поступают в обратный холодильник, и
сконденсированный растворитель попадает на экстрагируемое вещество
в патроне. Когда экстракционный сосуд наполнится до сгиба
сливной трубки, экстракт сбрасывается по сифону в перегонную колбу,
и процесс экстрагирования повторяется. В подобных экстракторах
непрерывного действия экстракт на поверхности экстрагируемого
вещества все время обновляется.
При экстракции твердых веществ, смол, масел и растительных
продуктов органическими растворителями часто применяют
аппарат Сокслета (рис. 171). Эффект экстракции в аппаратах Сок-
слета зависит, главным образом, от конструкции насадки.
Выпускаются лабораторные стеклянные насадки для экстракции
твердых веществ с конусными шлифами типа НЭТ, со впаянным
стеклянным фильтром (типа НЭТФ) и со вставным стеклянным
вкладышем (типа НЭТВ), с конусными шлифами (рис. 172).
295
Рис. 1/1. Аппарат Сокслета:
1— колба; 2— насадка для экстракции; 3—обратный холодильник.
Рис. 172. Насадки для экстракции твердых веществ:
а — НЭТ; б —НЭТФ; в — НЭТВ.
На лабораторном штативе укрепляют колбу с
соответствующим номером КШ муфты, присоединяют насадку и обратный
холодильник с КШ 45/40 и проверяют герметичность соединений
установки.
Размер насадки выбирают так, чтобы патрон с экстрагируемым
веществом заполнял приблизительно 2/з ее объема. Навеску экс-
трагируемого вещества помещают в стеклянный вставной
вкладыш насадки НЭТВ (при малой массе навески) или в патрон
из фильтровальной бумаги — при использовании насадок НЭТ и
НЭТФ. Патроны с твердым веществом вводят в насадки и
наливают растворитель до тех пор, пока он не начнет стекать по
сифонной трубке в колбу. Когда растворитель стечет полностью, его
добавляют еще один раз, а затем присоединяют обратный
холодильник, пускают в него холодную воду, погружают колбу в
водяную баню и начинают нагревать. Продолжительность
нагревания устанавливается опытным путем.
Если экстрагируемое вещество окрашено, то окончание
экстрагирования определяется моментом, когда жидкость в насадке
станет бесцветной. Если экстрагируемое вещество бесцветно,
можно периодически отбирать пробы раствора из насадки при помощи
капиллярной трубки, которую вводят через холодильник. Пробу
испаряют на часовом стекле. Если на стекле не образуется
матового пятна или пленки, экстракцию можно считать законченной.
Разбирая установку, следует прежде всего отключить холодильник
от водопроводной сети, затем осторожно снять холодильник и дать
296
стечь экстрагенту из насадки в колбу, для чего насадку наклоняют
так, чтобы жидкость переливалась через отводную трубку.
Полученный экстракт выпаривают до минимального объема и
завершают испарение в вакуум-эксикаторе.
Учитывая, что аппарат Сокслета часто работает много часов
подряд, а растворитель — горючая жидкость, необходимо
обеспечить безопасный обогрев; наиболее надежна воздушная баня с
ламповым нагревом.
Экстрагирование твердых веществ водой или водными
растворами принято называть выщелачиванием. Обычно
выщелачивание или водную экстракцию проводят в стеклянном или
фарфоровом стакане при энергичном перемешивании. Затем,
прекратив перемешивание, дают жидкости отстояться, сливают
прозрачную жидкость на бумажный фильтр, а к нерастворившейся
части вещества снова добавляют растворитель и повторяют
операцию до полного извлечения нужного вещества. Окончание
экстракции определяют по специфической реакции извлекаемого
вещества.
ЭКСТРАКЦИЯ ВЕЩЕСТВ ИЗ РАСТВОРОВ [45, с. 175]
Простейший вид экстракции — встряхивание раствора
вещества в определенном растворителе с другим растворителем, не
смешивающимся с первым. Растворитель, служащий экстрагентом,
должен лучше растворять экстрагируемое вещество, чем
растворитель, из которого это вещество извлекается.
Для экстракции органических веществ из водных растворов
пользуются делительной воронкой, в которой смешивают водный
раствор вещества (около 1/2 объема воронки) с экстрагентом (7б—
7з объема воронки). Воронку закрывают пробкой и энергично
взбалтывают в течение нескольких секунд, придерживая верхнюю
лробку одной рукой, а кран на спускной трубке — другой. Затем
делительную воронку переворачивают краном вверх и, осторожно
открывая кран, выпускают образующиеся пары.
Если экстрагентом служит диэтиловый эфир, давление паров
которого при комнатной температуре довольно велико, в
делительной воронке создается значительное избыточное давление. После
выравнивания давления кран делительной воронки закрывают,
встряхивание и периодическое выпускание паров продолжают до
тех пор, пока газовое пространство над жидкостью в делительной
воронке не будет заполнено парами растворителя и давление не
перестанет меняться. Тогда приступают к более
продолжительному и энергичному встряхиванию. По окончании встряхивания
делительную воронку укрепляют в штативе, приоткрывают пробку
и дают жидкости полностью разделиться на два слоя, которые
должны быть совершенно прозрачны. Далее открывают кран и
спускают водный слой (если плотность экстрагента меньше
плотности воды) в приемную емкость. Слой органической жидкости
через верхнее отверстие делительной воронки выливают в колбу с
притертой пробкой.
297
Водный раствор снова переносят в делительную воронку,
добавляют новую порцию растворителя и повторяют процесс
несколько раз.
Для того чтобы определить, закончен ли процесс экстракции,
небольшое количество последней порции экстракта выпаривают
на часовом стекле и высушивают в вакуум-эксикаторе над
Mg(C104)2.
Полученные вытяжки собирают в одну колбу и сушат с
помощью соответствующего осушающего средства (см. гл. 18). После
сушки вытяжку фильтруют, затем удаляют растворитель
(отгонкой, выпариванием в вакууме), а остаток очищают путем
кристаллизации, сублимации или перегонки.
Часто, особенно при экстракции водных щелочных растворов,
образуются эмульсии, которые с большим трудом разделяются
на два слоя. В этих случаях разделение слоев может быть
достигнуто одним из следующих приемов: 1) через жидкость в
делительной воронке продувают ток воздуха; 2) водный слой
насыщают NaCl; 3) добавляют несколько капель
поверхностно-активного вещества, понижающего поверхностное натяжение (например,
октилового спирта).
Если вещество значительно лучше растворяется в воде, чем
в органическом растворителе, экстракция путем взбалтывания в
делительной воронке не дает хороших результатов, и тогда
приходится применять непрерывную экстракцию.
Химическое разделение веществ
Из раствора смеси веществ в органическом растворителе
можно выделить компоненты, используя различие их химических
свойств. Это достигается встряхиванием раствора в делительной
Рис. 173. Прибор для экстракции веществ из растворов легким (а) и тяжелым (6~)
растворителем:
У —круглодонная колба; 2—пароотводная трубка; 3—воронка; 4—пробирка с боковым отводом»
Рис. 174. Насадки для экстракции веществ из растворов тяжелым растворителем — НЭР (а)
и легким растворителем —НЭРВ (6*).
298
воронке с водным раствором кислоты, если нужно отделить
вещество основного характера (амины, аминокислоты, аминоспирты),
либо водным раствором Ыа2СОз или NaHC03, если нужно отделить
вещество кислотного характера. Эту операцию выполняют так же,
как и обычную экстракцию, хотя она основана не на различной
растворимости вещества в разных растворителях, а на его
химических свойствах.
Непрерывная экстракция растворов
С помощью экстракторов непрерывного действия — перко-
ляторов — можно извлекать вещество из раствора небольшим
количеством растворителя. Растворитель при этом испаряют в
колбе, а пары его конденсируют в обратном холодильнике.
Конденсат в виде мелких капель, проходя через раствор, постепенно
обогащается извлекаемым веществом и стекает через перелив
обратно в колбу. Перколяторы, в зависимости от того, легче или
тяжелее растворитель экстрагируемого раствора, различаются
конструктивно.
На рис. 173, а изображен простейший прибор, применяемый
для экстракции жидкости более легким растворителем.
Растворитель нагревают в колбе 1. Пары растворителя через трубку 2
попадают в обратный холодильник, и конденсат стекает в воронку
3\ через трубку воронки растворитель попадает в нижнюю часть
пробирки 4 и, поднимаясь на поверхность раствора, извлекает
растворенное в нем вещество. Экстракт через боковую трубку
стекает в колбу /.
Прибор для экстракции из растворов тяжелым растворителем
изображен на рис. 173,6.
Насадки для непрерывного экстрагирования веществ из
растворов выпускаются двух типов (рис. 174): НЭР—для
экстрагирования тяжелым растворителем; НЭРВ — для экстрагирования
легким растворителем.
299
глава 28. КРИСТАЛЛИЗАЦИЯ
Очистка твердых химических веществ кристаллизацией
основана на различной растворимости основного вещества и
загрязняющих его примесей в соответствующем растворителе или смеси
растворителей. Для кристаллизации необходимо создать условия,
при которых из пересыщенного раствора могли бы выпадать
кристаллы очищаемого вещества. Примеси нередко уменьшают
скорость и полноту образования кристаллов при охлаждении
насыщенных растворов. Это объясняется тем, что последние образуют
стойкие адсорбционные комплексы, затрудняющие ориентацию
частиц растворенного вещества на поверхности кристаллов. Поэтому
не следует приступать к перекристаллизации сырого продукта да
тех пор, пока не были испытаны другие способы его очистки. При
любой кристаллизации основной задачей должно являться
получение продукта в возможно более чистом виде [66].
Перекристаллизация состоит из нескольких стадий:
приготовления раствора; фильтрации горячего раствора; охлаждения
раствора; кристаллизации; отделения кристаллов от маточного
раствора; удаления следов растворителя» Часто перед фильтрованием
горячий раствор обрабатывают активным углем с целью его
обесцвечивания и удаления коллоидных взвесей. В ряде случаев
необходимая очистка твердого вещества достигается лишь в
результате многократной кристаллизации.
ВЫБОР РАСТВОРИТЕЛЯ [5, с. 137]
При выборе растворителя следует руководствоваться
следующими правилами.
Очищаемое твердое вещество должно плохо растворяться в
выбранном растворителе на холоду и хорошо — при нагревании.
При этом загрязняющие примеси должны обладать, по
возможности, лучшей растворимостью в выбранном растворителе.
«Подобное растворяется в подобном», т.е. вещества хороша
растворимы в растворителях, обладающих строением молекул,
химически сходным со строением молекул растворяемого вещества.
Так, вещества с ионной структурой хорошо растворяются в
растворителях с высокой диэлектрической проницаемостью (вода,
спирт), а полярные соединения лучше растворяются в полярных
растворителях (вода, спирты, сложные эфиры, кетоны и др.)» чем
в неполярных (бензол, дихлорэтан и др.)-
Растворитель не должен химически взаимодействовать с
растворенным веществом.
Практически выбор растворителя производится следующим
образом. В ряд химических пробирок помещают по 0,1 г тонко-
измельченного вещества, подлежащего кристаллизации, прибав-
300
ляют в каждую пробирку по 1 мл растворителя —
дистиллированную воду, метиловый спирт, этиловый спирт, ледяную усусную
кислоту, хлороформ, бензол, ацетон — и встряхивают пробирки.
Растворитель считается непригодным для кристаллизации, если
навеска вещества растворяется на холоду или при слабом нагревании.
Если в 1 мл растворителя большая часть навески твердого
вещества не растворилась на холоду, пробирки закрывают
пробками со вставленными в них стеклянными трубками длиной 120—
150 мм и нагревают смесь до кипения. При неполном растворении
навески твердого вещества прибавляют еще 0,5 мл растворителя
и вновь кипятят. Так поступают до тех пор, пока навеска
полностью не растворится. Растворитель считается непригодным для
кристаллизации, если 0,1 г твердого вещества не растворяется в
3 мл растворителя при кипячении.
При полном растворении вещества горячий раствор
профильтровывают в другую пробирку, пользуясь маленьким складчатым
фильтром и воронкой с коротко отрезанной трубкой. Прозрачный
фильтрат охлаждают (холодной водой, водой со льдом, смесью
лед — соль). Если при этом образуются кристаллы, то это
свидетельствует о правильном выборе растворителя. Определив
приблизительно количество использованного растворителя,
устанавливают количественное отношение вещество : растворитель.
Если в результате предварительных испытаний одинаково
пригодными оказались два растворителя, то предпочтение отдают
тому, который менее огнеопасен, менее токсичен и более удобен
для работы. Например, следует по возможности избегать
применения сероуглерода, так как он ядовит, исключительно огне- и
взрывоопасен. Применение диэтилового эфира также
нежелательно, так как он способен ползти по стенкам сосуда, вследствие чего
на них выделяется неочищенное вещество.
Часто, смешивая растворители, удается получить раствор, в
котором происходит кристаллизация очищаемого вещества.
Обычно это наблюдается в тех случаях, когда вещество очень легко
растворяется в одном растворителе, но не выделяется из него
при охлаждении раствора, а в другом не растворяется. В
подобных случаях растворяют очищаемый продукт в первом
растворителе и при кипячении осторожно прибавляют горячий второй
растворитель, пока не наступит слабое помутнение раствора. Затем
добавляют первый растворитель до исчезновения помутнения,
фильтруют горячий раствор и дают смеси охладиться до комнатной
температуры.
Для такой кристаллизации чаще всего используют следующие
пары растворителей: этиловый спирт и вода, этиловый спирт и
бензол, вода и ледяная уксусная кислота, ацетон и вода.
ПРИГОТОВЛЕНИЕ РАСТВОРА ДЛЯ КРИСТАЛЛИЗАЦИИ [45, с. 137]
Взвешенное количество измельченного твердого вещества
помещают в коническую колбу, снабженную обратным
холодильником. В колбу вносят длинные стеклянные капилляры, достигающие
301
запаянным концом до середины горлышка колбы. После этого в
колбу вливают несколько меньшее количество растворителя, чем
вычисленное по данным предварительных проб, и нагревают смесь
до кипения на бане с теплоносителем. Кипящий раствор должен
быть прозрачным. Если вещество полностью не растворится,
прекращают нагревание и добавляют через холодильник
дополнительную порцию растворителя. Если и после этого количество
нерастворившегося вещества не уменьшится, то его следует
отфильтровать через складчатый обогреваемый фильтр,
предварительно смоченный горячим растворителем.
Часто твердый неочищенный продукт содержит окрашенные и
коллоидно-дисперсные загрязнения. В этом случае к раствору
добавляют 1—2% активного осветляющего угля типа ОУ-3 и
кипятят с обратным холодильником 10—15 мин. Перед добавлением
осветляющего угля следует несколько охладить раствор, так как
адсорбент может интенсифицировать процесс кипения и вызвать
энергичное взрывообразное вскипание.
Адсорбционная способность активного угля убывает в ряду
растворителей: вода — этиловый спирт — метиловый спирт
—уксусная кислота — ацетон — хлороформ.
Горячий раствор фильтруют с отсасыванием через воронку для
горячего фильтрования. Процесс следует проводить возможно
быстрее, чтобы вещество не кристаллизовалось на фильтре.
При работе с легколетучими растворителями фильтрование с
отсасыванием приводит к испарению растворителя и
кристаллизации вещества на фильтре. В этих случаях можно фильтровать
через воронку для горячего фильтрования без отсасывания. Для
уменьшения испарения растворителя накрывают воронку часовым
стеклом (выпуклой стороной вниз).
КРИСТАЛЛИЗАЦИЯ И ОТДЕЛЕНИЕ КРИСТАЛЛОВ
Для получения хороших кристаллов необходимо дать
раствору медленно охладиться в полном покое, предохраняя раствор
от попадания пыли. Колбы с горячим раствором нельзя закрывать
герметично, так как при охлаждении они могут лопнуть под
давлением воздуха. Если компоненты раствора чувствительны к влаге и
диоксиду углерода, то колбу закрывают хлоркальциевой трубкой с
хлоридом кальция и натронной известью.
Часто при попадании горячего фильтруемого раствора в
холодный приемник наблюдается быстрое выделение плохо
образованных кристаллов. В этом случае профильтрованный раствор в
приемнике необходимо снова нагреть до растворения кристаллов и
оставить медленно охлаждаться. Для более полного выделения
кристаллов из маточного раствора часто охлаждают его при помощи
охлаждающих смесей или же ставят сосуд с раствором в
холодильник. Охлаждение, уменьшая растворимость вещества, облегчает
кристаллизацию. Однако понижение температуры может и
уменьшить скорость роста кристаллов, что особенно заметно в случае вяз-
302
ких жидкостей. Резкое снижение температуры фильтрата вызывает
внезапное пересыщение раствора и ведет к образованию
мелкокристаллического, а иногда даже аморфного осадка.
Мелкокристаллические вещества, у которых общая поверхность
больше, чем у крупнокристаллических, в большей степени
адсорбируют загрязнения.
Охлаждение до очень низких температур не всегда приводит к
образованию кристаллов, в особенности при кристаллизации из
сиропообразной среды или же среды, содержащей смолы. Такие
жидкости при сильном охлаждении становятся иногда настолько
вязкими, что образование кристаллов невозможно.
У органических соединений очень велика склонность к
образованию пересыщенных растворов. При внесении затравки — того
же самого или изоморфного ему вещества — пересыщение обычно
ликвидируется. Таким путем создаются ядра
кристаллизации определенного и желательного типа. Если в раствор,
пересыщенный относительно нескольких соединений, ввести в качестве
затравки кристаллы одного из них, то кристаллизуется вещество
того же типа, а другие соединения останутся в растворе.
Начало кристаллизации можно также вызвать, царапая стенки
сосуда с пересыщенным раствором стеклянной палочкой.
Поверхность становится шероховатой, и частицы растворенного вещества
быстрее ориентируются на ней, чем на гладкой поверхности.
По своим результатам энергичное перемешивание
пересыщенного раствора с небольшим количеством органического
растворителя, в котором твердое вещество нерастворимо (или мало
растворимо), сходно с нацарапыванием. Вязкая масса, покрытая
небольшим количеством растворителя, растирается в фарфоровой ступке
до образования кристаллов. Эта операция иногда длится более 1 ч.
Скорость кристаллизации довольно часто бывает мала. В
отдельных случаях кристаллы образуются через несколько недель и даже
месяцев.
Опытные экспериментаторы утверждают, что никогда нельзя
преждевременно выбрасывать маточные растворы. Вещества с
низкой температурой плавления в процессе кристаллизации часто
выделяются в виде масла, которое затвердевает только при
продолжительном охлаждении, адсорбируя при этом первоначальные
загрязнения. В таком случае раствор разбавляют чистым
растворителем, нагревают до растворения масла и снова медленно
охлаждают. Эту операцию следует повторять несколько раз до
образования хорошо кристаллизующегося осадка.
Выкристаллизовавшееся вещество отделяют от маточного
раствора фильтрацией под уменьшенным давлением, применяя
обогреваемую воронку со стеклянным фильтром или воронку Бюхнера.
После отсасывания маточного раствора отключают вакуумную
установку, кристаллы заливают небольшим количеством чистого
холодного растворителя, перемешивают стеклянной палочкой, снова
подключают вакуумную установку и отсасывают раствор. После
двух- или трехкратной промывки кристаллы отжимают на фильтре
303
плоской стороной стеклянной пробки, а затем переносят в
кристаллизатор или на фильтровальную бумагу.
Отделение маслянистых загрязнений и остатков маточного
раствора от кристаллов может быть также достигнуто с помощью
пластин из неглазурованного фарфора. Кристаллический осадок
размещают на пористой поверхности пластины и отжимают его.
К осушенному веществу приливают по каплям промывную
жидкость (растворитель); при этом кристаллическое вещество на
пластинке несколько раз перемешивают шпателем.
Кристаллы высушивают, в зависимости от их свойств, на
воздухе, между листами фильтровальной бумаги, в вакуум-эксикаторе
с поглотителем или подвергают термической сушке. Вещества,
устойчивые к действию воздуха и температуры, можно сушить в
сушильном или вакуум-сушильном шкафу при температуре
примерно на 50° ниже температуры плавления твердого вещества.
Два или несколько веществ разделяют дробной
кристаллизацией, используя их различную растворимость. Для дробной
кристаллизации используют ряд методов: медленное испарение
растворителя, постепенное добавление разбавителя, в котором один из
компонентов смеси нерастворим, и ступенчатое охлаждение.
Для медленного испарения растворителя стакан
с раствором, накрытый часовым стеклом, оставляют стоять при
комнатной температуре либо помещают в вакуумный эксикатор над
поглотителем, способным поглощать пары данного растворителя.
Периодически, по мере выделения однородных по внешнему виду
кристаллов, маточный раствор переносят в другой стакан и
продолжают дробную кристаллизацию.
При кристаллизации постепенным добавлением
разбавителя после удаления каждой порции кристаллов маточный
раствор нагревают и охлаждают для кристаллизации новой
порции вещества. Эта процедура повторяется до тех пор, пока все
твердое вещество не будет выделено в виде нескольких порций
кристаллов.
Для дробной кристаллизации ступенчатым
охлаждением применяют растворители с низкой (до — 78 °С)
температурой замерзания (метиловый и этиловый спирты, ацетон, диэтиловый
эфир и др.). Охлаждением до 0°С выделяют первую порцию
кристаллов, после чего маточный раствор можно охладить до более низ*
кой температуры ( — 20, — 40 °С) и окончательно до — 78 °С.
Каждую порцию кристаллов собирают и анализируют отдельно, после
чего производят их раздельную перекристаллизацию из
соответствующего растворителя.
Повторные кристаллизации проводят до тех пор, пока не
получатся чистые вещества с постоянной температурой плавления.
304
г л а в а 29 ХРОМАТОГРАФИЯ *
ОСНОВНЫЕ ПОНЯТИЯ
Хроматографией называют метод разделения веществ,
основанный на различном взаимодействии компонентов смеси с подвижной
и неподвижной фазами в динамических условиях. В процессе хро-
матографирования подвижная фаза (жидкость или газ),
содержащая анализируемую пробу, перемещается через неподвижную. При
этом происходит многократное повторение актов взаимодействия,
что обусловливает высокую эффективность разделения веществ
с близкими свойствами. Во всех случаях компоненты разделяемой
смеси распределяются между подвижной и неподвижной фазами,
и их перемещение по колонке или по листу бумаги протекает с
различной скоростью. В результате достигается разделение.
Подвижную фазу, проходящую через неподвижную, часто
вызывают элюентом; растворенные вещества, покидающие
неподвижную фазу вместе с элюентом, — элю атом и, наконец, процесс
перемещения разделяемых компонентов образца вместе с
элюентом — элюированием.
В колоночной хроматографии обычно проводят элюирование
до тех пор, пока разделенные компоненты смеси не выйдут один за
другим из колонки. Фракции элюата, в которых содержатся
разделенные компоненты, собирают отдельно и анализируют
соответствующими аналитическими методами.
В современных хроматографах — приборах для проведения
хроматографического разделения и анализа — для определения
состава и содержания разделенных компонентов смеси после выхода
из колонки установлены детекторы.
Детекторы регистрируют изменение физических свойств
элюата; их показания можно с помощью самописца записывать
автоматически, получая при этом кривую элюирования — хрома-
тограмму. Каждый пик на такой кривой (рис. 175)
соответствует отдельному компоненту, а площадь каждого пика
характеризует относительное содержание данного компонента. Основными
характеристиками пиков являются удаленность их центров тяжести
от точки, отмечающей момент ввода образца в хроматограф,
а также ширина.
Положение пиков можно характеризовать либо временем
выхода соответствующих компонентов образца, либо удерживаемым
объемом Vr.
Степень хроматографического разделения обычно оценивают
отношением расстояния между максимумами пиков двух веществ
к сумме их полуширин. Для оценки в равной мере важны как
расхождение пиков, так и их уширение.
* Глава написана совместно с канд. физ.-мат. наук В. В. Нестеровым.
305
LL v*. ZZ
LZL_
Ввод
Пробы,
Рис. 175. Параметры выходной кривой элюирован!
Идентификацию осуществляют хроматографированием
стандартных веществ (реперов) и отождествлением их
удерживаемых объемов с удерживаемыми объемами компонентов
исследуемой смеси. Так, при сопоставлении хроматограммы
анализируемой смеси спиртов (рис. 176) с хроматограммой смеси
2-фенилэтилового и бензилового спиртов видно, что в
анализируемой смеси содержатся оба эти спирта.
Для количественного определения разделенных компонентов
зависимость величины сигнала детектора (т. е. отклонения
показаний самописца или прибора индикатора) от концентрации
компонента устанавливают калибровкой. При калибровке фиксируют
изменение высоты пика или его площади от количества
стандартного вещества, подаваемого в колонку. Высоту пика от вершины до
нулевой линии измеряют линейкой. Площади пиков определяют
несколькими способами: планиметром, электронным интегратором,
взвешиванием бумаги, вырезанной из хроматограммы по контурам
кривой, образующей пик, и умножением высоты пика на его
ширину (на расстоянии, равном половине высоты от основания).
Ширину узких пиков можно измерить с помощью специальной
измерительной лупы.
Хроматографические методы используются как в аналитических,
так и в препаративных целях. В первом случае с их помощью
определяют состав смесей, контролируют чистоту препаратов,
идентифицируют вещества, очищают их от примесей и т. п. В качестве
препаративного метода разделения хроматографию используют
для выделения и очистки природных веществ или продуктов
химических реакций.
Существует много вариантов хроматографического анализа.
Хроматографические методы различают по агрегатному состоянию
фаз, между которыми происходит разделение компонентов смеси,
по методике эксперимента и механизмам разделения.
В газовой хроматографии применяют системы газ —
жидкость и газ — твердое вещество. Жидкостная
хроматография включает системы жидкость — жидкость и жидкость —
твердое вещество.
По механизму разделения хроматографические методы обычно
подразделяются по типу взаимодействия компонентов смеси с сор-
306
бентом: адсорбционная хроматография — на
поверхности сорбента, распределительная — в объеме сорбента.
Метод разделения, в котором в качестве твердой фазы
используются ионообменные материалы, называют ионообменной
хроматографией.
Осадочная хроматография основана на химических
реакциях хемосорбента с компонентами смеси растворенных
веществ с образованием новой фазы ^осадка [44].
Вэксклюзионной (гель-проникающей, ситовой)
хроматографии используется способность материалов с
контролируемой пористостью «сортировать» и разделять компоненты
смеси в соответствии с размером и формой их молекул.
Разделение компонентов смеси может осуществляться в
колонках (колоночная хроматография) и без них. К
неколоночным методам относятся бумажная и тонкослойная
хроматография.
Процесс переноса растворенных веществ подвижной фазой
через неподвижную называют проявлением. (Не следует путать
проявление с опрыскиванием тонкослойных хроматограмм
реагентами, которые образуют с бесцветными компонентами окрашенные
производные и таким образом раскрывают присутствие
разделенных компонентов. Этот процесс часто также называют проявлением,
хотя его лучше называть обнаружением, выявлением или
детектированием.)
Проявление можно вести тремя различными методами: элюиро-
ванием, фронтальным анализом, вытеснением. Наиболее широко
используется элюирование.
При фронтальном анализе раствор исследуемой смеси
веществ непрерывно подают к одному концу неподвижной фазы и
заставляют ее продвигаться к другому концу. Если исследуемый
раствор состоит из смеси компонентов А и Б в несорбируемом
растворителе В, то первым из слоя начинает вытекать чистый
растворитель В. После насыщения слоя сорбента менее сорбирующимся
компонентом А из колонки будет вытекать раствор А в paCTBOpH-
tf? 25
Время, мим
Рис. 176. Экспериментальная хроматограмма смеси спиртов (а) и смеси 2-фенилэтилового
и бензилового спиртов (о").
307
теле В. Наконец, когда сорбент насытится веществом Б, из
колонки будет вытекать раствор компонентов А и Б , т. е. смесь
исходного состава. Таким образом, фронтальным методом можно получить
в чистом виде только менее сорбируемый компонент А.
Фронтальный метод применяется для очистки от примесей, если
эти примеси сорбируются значительно лучше, чем очищаемое
вещество. Для аналитических целей он неприменим.
При элюировании (или элюентном методе хрома-
тографирования) исследуемую смесь (например, компонентов А и
Б) растворяют в подходящем растворителе и вводят в верхнюю
часть колонки или в начальный участок бумажной полоски.
При этом компоненты А и Б практически не разделяются или
разделяются в очень малой степени. Если через неподвижную
фазу пропускать подвижную (растворитель или смесь растворителей),
которая слабее удерживается твердой фазой, чем компоненты А и
Б, то последние будут вымываться, т. е. перемещаться вниз по
колонке (слою, бумаге). Скорость этого перемещения зависит от
концентрационных коэффициентов распределения компонентов,
называемых в современной хроматографии факторами емкости К:
концентрация вещества в неподвижной фазе
концентрация вещества в элюате
Чем меньше К растворенного компонента смеси, тем быстрее
он движется вниз по колонке (слою, бумаге). Даже при невысоком
различии коэффициентов распределения компоненты А и Б
постепенно разделяются, а их зоны перекрываются все меньше по мере
перемещения по колонке. Чистые вещества А и Б собирают по
выходе из колонки (слоя, бумаги).
В колоночной хроматографии объем элюента, необходимый для
извлечения из колонки максимальной концентрации вещества,
называют объемом удерживания VR.
Время от момента ввода пробы в колонку до момента выхода
из нее максимальной концентрации определяемого вещества
называют временем удерживания tf*.
Объем удерживания Vr равен произведению времени
удерживания на объемный расход элюента со при температуре и давлении
колонки:
Интересно, что в любом хроматографическом анализе
существует минимальный удерживаемый объем, с которым может
появиться хроматографический пик. Этот объем Vo (иногда его
называют свободным, или «мертвым») характерен для
химического соединения, никак не взаимодействующего с неподвижной
фазой, и равен объему межчастичного пространства в хроматогра-
фической колонке. Очевидно, что для компонентов разделяемой
смеси всегда имеет место соотношение VR > V0 (рис. 175).
Ввытеснительной хроматографии небольшую пробу
смеси помещают на одном конце слоя неподвижной фазы и
пропускают растворитель В, который удерживается твердой фазой сильнее,
-308
чем компоненты А и Б. Растворитель В вытесняет сорбированные
вещества А и Б и проталкивает их по всему слою неподвижной
фазы. Если компонент Б удерживается неподвижной фазой сильнее,
чем компонент А, то Б вытесняет А и последний движется вниз по
колонке первым; при этом образуются зоны от чистого А до чистого
Б и от чистого Б до чистого В. Вытеснительный метод обычно
используют для разделения больших количеств веществ и получения
чистых соединений.
ГАЗОВАЯ ХРОМАТОГРАФИЯ [28, 48, 61]
В этом методе разделения подвижной фазой служит газ (гелий,
азот), протекающий через неподвижную фазу.
Различают две принципиальные разновидности газовой
хроматографии: в системе газ — твердое вещество (адсорбционная
хроматография) и в системе газ — жидкость (газо-жидкостная
хроматография). В первом случае разделение происходит за счет адсорбции
веществ на поверхности твердого адсорбента, которым
наполнена хроматографическая колонка. Во втором случае
анализируемая газовая смесь проходит через колонку, наполненную твердым
носителем (определенной степени зернения), на поверхность
которого нанесен тонкий слой нелетучей жидкости. Эффективность
разделения в газо-жидкостной хроматографии определяется не
процессами сорбции — десорбции газа, а степенью растворения
газообразных компонентов анализируемого вещества в жидкой нелетучей
пленке. В качестве жидкой фазы в газо-жидкостной хроматографии
используют вазелиновое масло, силиконовое масло, эфиры фтале-
вой кислоты, трикрезилфосфат и др. В качестве твердых носителей
применяют инертные вещества с развитой поверхностью, но малой
микропористостью, чтобы исключить адсорбцию газа на
поверхности. Наибольшее распространение получили каолин, диатомиты,
тефлон и др.
При хроматографии в системе газ — адсорбент используют
приемы фронтальной, элюентной и вытеснительной хроматографии;
в системе газ — жидкость применяют только метод элюирования.
В настоящее время методами газовой хроматографии можно
выполнять качественные и количественные определения компонентов
смесей органических и неорганических газообразных, жидких и
твердых веществ, давление паров которых превышает 0,133—
133 Па, перегоняющихся без разложения в области температур до
400—500 °С. Основными достоинствами газовой хроматографии
являются высокая чувствительность и разделяющая способность,
скорость, точность и высокая степень автоматизации анализа.
Аппаратура для газовой хроматографии [46, с. 247]
Принципиальная схема аналитического лабораторного
газового хроматографа представлена на рис. 177.
Установка и стабилизация скорости потока и очистка
газа-носителя и дополнительных газов (если они необходимы для питания
309
детектора), а также измерение скорости потока газа выполняются
системой подготовки газов 1.
Особенно важное значение имеет установка и стабилизация
расхода газа-носителя, оказывающего непосредственное влияние на
параметры удерживания и размеры пиков. Для этих целей
используют совокупность нескольких элементов, основными из которых
являются дроссель, регулятор давления и регулятор расхода.
С помощью дросселя регулируют расход (объемную скорость)
газа, изменяя аэродинамическое сопротивление канала, по которому
этот газ течет. Если питание газового хроматографа
осуществляется от индивидуального баллона газом, то дроссель обеспечивает
и необходимый постоянный расход газа. В общем же случае
используют регулятор давления, стабилизирующий давление на
входе в хроматографическую колонку.
В некоторых случаях, например при программировании
температуры колонки, необходимо поддерживать постоянный расход
газа-носителя через колонку, когда ее сопротивление изменяется в
процессе анализа. Для этой цели используют регулятор расхода.
Он реагирует на изменение сопротивления колонки изменением
сопротивления входящего в его состав регулирующего дросселя,
так что суммарное сопротивление остается неизменным и расход
не меняется.
Величины газовых потоков измеряют с помощью
мыльно-пленочных измерителей по времени прохождения мыльной пленкой
известного объема калиброванной стеклянной трубки.
Дозирующее устройство 2 позволяет вводить в поток
газа-носителя непосредственно перед колонкой определенное количество
анализируемой смеси в газообразном состоянии. Поток
газа-носителя вносит анализируемую пробу в колонку 3, где осуществляется
разделение смеси. Составляющие ее компоненты вместе с
газом-носителем подаются в детектор 4, который преобразует разницу в
физических или физико-химических свойствах бинарных смесей
компонент— газ-носитель по сравнению с чистым газом-носителем в
электрический сигнал. Величина сигнала зависит как от природы
компонента, так и от содержания его в анализируемой смеси.
Необходимые температурные режимы колонки, детектора и
дозирующих устройств достигаются помещением их в термостат,
управляемый терморегулятором 5.
Сигнал детектора, преобразованный усилителем 7,
записывается в виде хроматограммы автоматическим потенциометром 8.
Рис. 177. Принципиальная схема газового
хроматографа:
/—система подготовки газов; 2 —
дозирующее устройство; 3 — колонка; 4 — детектор;
5—терморегулятор; 6 — блок питания
детектора; 7 — усилитель; 8 — регистратор.
Газовые связи показаны двойной чертой,
электрические —одинарной. Термостатируе-
мые элементы заключены в пунктирный
контур.
)(ии|Е
310
Количественная обработка хроматограмм может производиться
вручную или с помощью интегратора, автоматически
фиксирующего площадь пика и время удерживания.
Существует несколько типов дозаторов. Для дозирования
газообразных смесей используют газовые краны-дозаторы,
позволяющие включать калиброванную емкость, предварительно
заполненную анализируемой газовой смесью, в поток газа-носителя,
который переносит дозу в виде газовой «пробки» в хроматографи-
ческую колонку. Жидкие смеси вводят в колонку специальными
шприцами объемом 0,1—50 мкл через термостойкое резиновое
уплотнение испарителя. (Испаритель представляет собой
нагреваемый до определенной температуры металлический блок с
каналом для ввода и испарения жидкой пробы. В канал подается поток
предварительно нагретого газа-носителя. С одной стороны канал
закрыт пробкой из термостойкой резины, с другой присоединена
хроматографическая колонка.) Твердые вещества чаще всего
вводят в виде раствора. При этом растворитель должен не только
хорошо растворять твердые компоненты пробы, но и образовывать
пик, не мешающий измерению пиков анализируемых компонентов.
Хроматографические колонки
Различают три основных типа аналитических колонок — наса-
дочные (набивные), микронасадочные и капиллярные. Ввиду
простоты приготовления наибольшее распространение получили наса-
дочные колонки.
У насадочной колонки внутренняя полость заполнена
инертным твердым носителем, покрытым тонкой пленкой
нелетучего растворителя (неподвижной фазы) или твердым активным
веществом (адсорбентом). Эти колонки изготавливают из
металлических (нержавеющая сталь, медь), стеклянных или
фторопластовых трубок с внутренним диаметром 2—10 мм (чаще 2—4 мм) и
длиной 0,5—3,0 м. Им придают спиральную, U-образную или
зигзагообразную форму. Микронасадочные колонки
отличаются от насадочных только диаметром трубок (0,8—1,0 мм).
Форма колонок практически не влияет на эффективность
разделения и определяется размерами термостата.
В капиллярных колонках неподвижная фаза не
заполняет всю внутреннюю полость, а покрывает лишь внутреннюю
поверхность трубки, остающейся по существу полой и после нанесения
неподвижной фазы.
Заполнение прямых и U-образных колонок сыпучим материалом
не представляет трудностей. Наполнитель насыпают в колонку
(через воронку) и осторожным постукиванием добиваются
равномерного распределения его по длине колонки. Важно, чтобы столбик
наполнителя не имел разрывов и чтобы отсутствовали местные
пустоты. В спиралеобразные колонки рекомендуется насыпать
наполнитель порциями с одного конца. Медленно поворачивая колонку
311
по окружности спирали при одновременной ее вибрации с помощью
касания с вращающимся эксцентриком, насаженным на ось
моторчика, переводят наполнитель в другой конец колонки. Операцию
повторяют до полного заполнения. Оптимальная плотность
наполнения составляет примерно 0,3 г/см3.
При заполнении капиллярных колонок раствор жидкой фазы в
летучем растворителе (эфире, пентане) продавливают из сосуда
с двумя отверстиями с помощью сжатого газа (азота) через
колонку до появления жидкости на противоположном конце. Затем
сосуд отсоединяют и продувают колонку газом до полного удаления
паров растворителя.
Детекторы
В газовой хроматографии наибольшее распространение
получили ионизационно-пламенный детектор и катарометр (детектор по
теплопроводности).
Катарометр измеряет различие в теплопроводности чистого
газа-носителя и его смеси с веществом, выходящим из колонки.
Поэтому чувствительность тем выше, чем больше теплопроводность
анализируемого вещества отличается от теплопроводности газа-
носителя. Подавляющая часть органических веществ имеет низкую
теплопроводность, и для их анализа целесообразно использовать
газы-носители с возможно более высокой теплопроводностью.
Этому требованию удовлетворяют водород и гелий, но на практике
водород ввиду его взрывоопасное™ применяется значительно реже
гелия.
Ионизационно-пламенный детектор представляет
собой камеру, в которой поддерживается водородное пламя,
являющееся источником ионизации. В камеру вводят необходимые
для поддержания пламени водород и воздух; водород в смеси с
Газом-носителем подается в детектор через канал горелки, а
воздух— через другой канал и распределяется равномерно
диффузором. Горелка является одним из электродов, она изолирована от
корпуса и соединена с источником стабилизированного
напряжения. Второй электрод, называемый часто коллектором, расположен
над горелкой. Во внешнюю цепь включен электрометр,
измеряющий ток между электродами.
Поскольку в пламени чистого водорода число ионов мало,
сопротивление межэлектродного газового пространства очень
велико (1013—10м Ом) и ток детектора весьма мал (10~12—КН1 А).
Этот ток, возникающий за счет ионизации примесей в
газе-носителе, водороде и воздухе, является постоянным фоновым током
детектора. При внесении с газом-носителем из колонки
анализируемых органических веществ число ионов в пламени резко
увеличивается, сопротивление пламени падает, и во внешней цепи
детектора регистрируется соответствующее возрастание ионного
тока.
312
Последовательность операций
Вначале устанавливают постоянную скорость подачи
газа-носителя в хроматограф, нагревают детектор и колонку до
определенной постоянной температуры, проверяют и настраивают цепь
детектора. После этого вводят в колонку образец. В зависимости
от концентрации и числа разделяемых компонентов объем
вводимого газа-носителя колеблется от 1 до 10 мл, а объем жидкого
образца — от 0,1 до 10 мкл. В системе ввода поддерживается
такая температура, чтобы образец практически мгновенно испарялся.
Вместе с газом-носителем введенный образец в парообразном
состоянии поступает в колонку. Колонка поддерживается в
нагретом состоянии, но ее температура должна быть несколько ниже
температуры кипения компонентов разделяемой смеси.
Отдельные компоненты образца в порядке их элюирования в
колонке поступают в детектор, с помощью которого
устанавливают наличие и количественное содержание каждого из них.
Метод газовой хроматографии не позволяет автоматически
идентифицировать компоненты смеси по форме пика. Кривая
элюирования показывает только число компонентов разделенной
смеси.
ЖИДКОСТНАЯ ХРОМАТОГРАФИЯ
Жидкостная распределительная хроматография является
аналогом газо-жидкостной. Разделение веществ также основано на
различном их распределении между неподвижной и подвижной
фазами.
В жидкостной распределительной хроматографии могут иметь
место две системы фаз: неподвижная водная фаза (силикагель,
на котором имеется слой воды)—подвижная органическая фаза;
органическая неподвижная фаза — неорганическая подвижная
фаза. В качестве неподвижной органической фазы используют
гранулированные полимеры — полистирол, тефлон и другие
материалы, способные сорбировать и удерживать на поверхности
органические растворители. Используя такую систему, можно разделять
как органические, так и неорганические вещества.
В жидкостной адсорбционной хроматографии в качестве
неподвижной фазы обычно используют твердые активные адсорбенты с
частицами малого размера: оксид алюминия, силикагель, цеолиты,
целлюлозу, тальк и другие, а в качестве подвижной —
органические растворители и их смеси.
Эффективность разделения зависит от различия в сродстве
разделяемых компонентов к поверхности неподвижной фазы и
элюенту.
Колоночная хроматография
Прежде чем перейти к описанию современных
инструментальных методов жидкостной хроматографии, рассмотрим
классическую схему разделения на хроматографической колонке [2].
313
Простейшие колонки изображены на рис. 178. Колонки
заполняют гранулированным сорбентом таким образом, чтобы он
образовывал столбик равномерной плотности. Для этого слой
адсорбента механически уплотняют, или вносят адсорбент в виде
однородной суспензии (из делительной воронки, снабженной эффективной
мешалкой), или же вносят его в колонку, наполненную на 2/з
объема растворителем. Во время загрузки растворитель должен
каплями выливаться из нижнего конца колонки. Анализируемый
раствор вводят в колонку и разделяют компоненты элюированием
соответствующей подвижной фазы. Элюент через колонку
внутренним диаметром 1 см пропускают со скоростью примерно 1 мл/мин.
Фракции, содержащие обычно несколько миллилитров элюата,
собирают вручную или с помощью автоматического коллектора
фракций и определяют в них содержание веществ
соответствующим аналитическим методом. Время выделения одного
компонента составляет приблизительно 30 мин, т. е. для элюирования
одного компонента требуется не менее 25 мл элюента.
В современных жидкостных хроматографах элюент пропускают
через колонку со скоростью в 100 раз выше, чем в обычной
колоночной хроматографии.
При возрастании скорости элюента длина хроматографической
колонки увеличивается, а для быстрого установления равновесия
рис. 17S. Приборы для адсорбционной хроматографии:
Колонки для хроматографирования: а —при слабом отсасывании (с открытым кондом—для
извлечения адсорбента); б—при повышенном цавлении;гв —в обычных условиях; г —с
восходящим потоком жидкости; 1 — адсорбент; 2 — стеклянная вата; 3 —резиновая пробка;
4-—растворитель; 5**-Напорный сосуд с растворителем; Я —патрубок для выхода жидкости.
314
Рис. 179. Блок-схема
колоночного хроматографа:
/ — резервуар с элюентом;
2 — насос или другой
источник давления; 3 — манометр;
4 — устройство для ввода
пробы; 5 —колонка; 5
—коллектор фракций; 7—-детектор;
8—расходомер;
$—регистратор (самописец).
между неподвижной и подвижной фазами размер частиц твердого
носителя должен быть очень мал.
Для пропускания элюента через плотно упакованную колонку
с должной скоростью используют насос высокого давления. Объем
раствора исследуемого образца составляет несколько
микролитров, а масса образца измеряется в микрограммах. Образец вводят
в верхнюю часть колонки с помощью шприца. Пройдя через
колонку, элюент и растворенное в нем вещество попадают в
детектор. Малые объем и масса образца позволяют получить узкие и
четкие хроматографические пики.
На рис. 179 показана схема типичного колоночного
хроматографа.
Элюент из резервуара 1 подается с помощью насоса 2 через
устройство для ввода пробы 4 на вход колонки 5. Давление на
входе в колонку контролируется манометром 3. Проходя через
дозирующее устройство, элюент захватывает анализируемую
пробу. Компоненты пробы выходят из колонки в виде разбавленного
раствора в элюенте и поступают либо в коллектор фракций 6,
либо в детектор 7 и затем в измеритель расхода 8. Сигнал
детектора регистрируется на ленте самописца 9.
Существуют два основных способа ввода образца в колонку:
с помощью шприца, когда образец наносят непосредственно в центр
колонки на слой сорбента и более распространенный — с помощью
крана-дозатора.
Длина и диаметр колонок влияют на степень разделения и
способы их заполнения. Например, набивая колонки с внутренним
диаметром 2 мм, трудно достичь хорошего уплотнения слоя. В то
же время длинные колонки, как правило, удобнее согнуть или
свернуть кольцом. Это следует сделать до заполнения.
Прямые металлические колонки длиной до 1 м заполняют по
следующей методике.
1. Закрепляют соединительное звено (адаптер) на нижнем
конце колонки. Пористый фильтр будет удерживать сорбент.
2. Насыпают в колонку такое количество сухого сорбента,
чтобы до уплотнения длина слоя была около 2 см.
3. Опускают в колонку металлический пруток или шток с теф-
лоновым наконечником. Шток должен быть плотно пригнан к
колонке и вместе с тем свободно в ней двигаться.
4. Утрамбовывают слой набивки, осторожно постукивая
нижней частью колонки о пол, так чтобы шток или трамбовочный
пруток спрессовал слой неподвижной фазы.
315
SL
Подвижная фаз&
U
Ы 7- Ы 8 U*»ВСлиЗ
5. Несколько раз осторожно постукивают по стенкам колонки,
чтобы слой лучше уплотнился.
6. После того, как завершено уплотнение первой порции
сорбента, т.е. не наблюдается никакого дальнейшего спрессовывания,
шток вынимают, добавляют еще 2 см сорбента и повторяют
операцию до тех пор, пока колонка не будет заполнена полностью.
После приобретения некоторого опыта заполнение колонки
длиной 1 м занимает около 15 мин. Таким способом рекомендуется
производить упаковку сорбента с размером частиц 2^20 мкм. Для
более тонких материалов эта методика непригодна; в настоящее
время для них используются суспензионные методы.
В случае мягких (например, набухающих) сорбентов колонку
заполняют 20—30% суспензией сорбента в элюенте, непрерывно
прокачивая ее при малом давлении из сосуда, где обеспечивается
эффективное перемешивание. Прокачивание осуществляют до
установления постоянного давления; обычно это 20—30-кратный объем
заполняемой колонки.
В случае жестких частиц сорбента, наоборот, 10—15%
суспензию (в метиловом спирте, диоксане) прокачивают с максимально
возможной скоростью (под максимальным давлением), иногда
даже подают мгновенно — метод «впрыска» или «взрыва>.
Инструментальные методы детектирования
Автоматические непрерывные коллекторы фракций
(пробоотборники) карусельного или линейного типа обеспечивают
отбор проб элюата. По мере заполнения приемника жидкостью,
вытекающей из колонки до определенного объема или массы,
автоматически подается следующий приемник. Однако небходимость
в коллекторах фракций практически отпадает при наличии
проточного чувствительного детектора.
Наибольшее распространение получили два детектора:
дифференциальный рефрактометрический и спектрофотометрический.
Рефрактометрический детектор обеспечивает
непрерывную запись изменения показателя преломления элюата на
выходе из колонки. Он наиболее универсален, так как практически
всегда элюат и элюент имеют разные показатели преломления.
При применении спектрофотометрического детектора
измеряется поглощение элюатом падающего светового потока,
длина волны которого может меняться от 200 до 700 нм.
Широкое распространение получили ультрафиолетовые
фотометры, измеряющие поглощение на одной длине волны
(обычно 254 нм), поскольку многие органические соединения
содержат ароматические группировки и интенсивно поглощают
лучистую энергию в этой области спектра.
В жидкостной хроматографии чрезвычайно важно непрерывно
измерять скорость потока элюента, так как надежный
качественный и количественный анализ затруднен, если скорость потока че
постоянна.
316
Рис. 180. Стеклянное устройство для создания линейных
градиентов элюента.
В современных хроматографах
используются, в основном, три типа измерителей потока.
Счетчик капель регистрирует каждую
вытекающую из колонки каплю,
пролетающую между фотоэлементом и источником
света. При наборе заданного числа капель на
ленте самописца делается отметка, т. е. на хрома-
тограмму как бы наносится масштаб
объемного расхода элюента во времени.
Пузырьковый отметчик времени измеряет время,
за которое пузырек воздуха, введенный в поток элюента, проходит
расстояние между двумя рисками в калиброванной стеклянной
трубке, установленной на выходе из детектора. В сифонном
расходомере используется объемный метод: подвижная фаза
собирается в калиброванный сосуд до определенного объема
(обычно 5 см3) и затем сосуд освобождается сифонированием. На
ленте самописца автоматически отмечается каждый акт сифониро-
вания.
В жидкостной хроматографии широко используется так
называемое градиентное элюирование, т. е. непрерывное
изменение состава элюента в процессе проявления хроматограммы.
Так, если какой-то элюент один компонент вымывает очень
быстро, а другие очень медленно, то, чтобы ускорить вымывание
последних компонентов, желательно изменить состав элюента.
Используя несложный прибор (рис. 180), позволяющий программировать
состав элюента, можно смешивать два растворителя, постепенно
меняя состав смеси таким образом, чтобы вначале преобладал
один растворитель, а затем другой.
Последовательность операций
Навеску образца 0,05—5 мг растворяют в элюенте из расчета
0,5—5 мг/мл. Раствор должен быть оптически прозрачным и не
содержать коллоидных взвесей. Удалить их можно фильтрованием
через воронку с пористым фильтром с размером пор 5—10 мкм.
Раствор не должен содержать избыточных количеств воздуха —
для этого пробу вакуумируют с помощью водоструйного насоса.
Хроматограф включают за 1—2 ч до начала работы и в процессе
выхода его на рабочий режим проверяют наличие растворителя
в сосудах для градиентного элюирования, отсутствие течей в
жидкостных коммуникациях, исправную работу нагревателей.
Критерием готовности прибора является ровная и устойчивая нулевая
(базовая) линия на самописце при «холостом» протекании
элюента через колонку и детектор. В момент ввода образца в колонку
на ленте самописца делается отметка, которая служит началом
отсчета при последующей идентификации разделенных
компонентов.
флШША ЭлюенгП&
Камер?
смешивание
Кнасосуц колонка
317
ХРОМАТОГРАФИЯ НА БУМАГЕ
Бумажная хроматография является одним из видов
жидкостной распределительной хроматографии.
В качестве носителя неподвижной жидкой фазы здесь
применяется специальная фильтровальная бумага высокой степени
чистоты и равномерной плотности. Считают, что неподвижной фазой
служит постоянно присутствующая в целлюлозе вода. Подвижной
фазой является органический растворитель или смесь
растворителей.
Техника хроматографии на бумаге сводится к тому, что каплю
раствора, содержащую 5—50 мкг исследуемой смеси веществ,
наносят в виде точек на полоску или лист бумаги и высушивают
горячим воздухом. Затем бумагу помещают в закрытый сосуд, в
котором бумага непрерывно смачивается соответствующим
растворителем — элюентом. Растворитель может двигаться в
горизонтальной плоскости, подниматься снизу вверх (восходящая
хроматография), сверху вниз (нисходящая
хроматография) или движение растворителя может происходить от
центра бумаги к периферии (круговая, или радиальная,
хроматография).
Восходящая, нисходящая и круговая хроматография по
технике выполнения может быть одномерной и двумерной. Для
получения двумерных хроматограмм хроматографирование производят
дважды во взаимно перпендикулярных направлениях: после
проявления хроматограммы одним элюентом хроматограмму
поворачивают на 90° и вторично проявляют другим элюентом.
Для оценки хроматографического поведения веществ в
определенных условиях эксперимента используют фактор Rf (от ratio of
fronts — отношение фронтов), который равен отношению
расстояния, пройденного веществом, к расстоянию, пройденному
растворителем. Обычно для расчета Rf выбирают точку в центре пятна
(рис. 181).
Абсолютные значения величин Rf трудно установить заранее,
так как они изменяются в зависимости от условий проведения
эксперимента. Однако они полезны для предварительной
идентификации соединений, а при условии сравнения Rf исследуемого
вещества и стандартного образца в одинаковых условиях
достигается достоверная идентификация. Для этой цели на стартовую
линию одной и той же бумажной полоски наносят три пробы: одну
с исследуемым веществом, вторую со стандартным образцом и
третью — со смесью равных количеств исследуемого и
стандартного веществ. Количество хроматографируемого материала
должно быть одинаковым во всех трех образцах. Если исследуемое
4 3 2 i
318
Рис. 181. Бумажная хроматограмма после
обработки реагентом-индикатором:
1 — линия старта; 2—центр пятна одного
из компонентов исследуемой смеси; 3 — центр
пятна второго компонента; 4 — линия фронта
растворителя.
вещество и стандартный образец идентичны по составу и строению,
все три пятна после детектирования имеют одинаковую окраску
и значения Rf\ хроматограмма смеси в этом случае дает одно
пятно.
Метод хроматографии на бумаге находит широкое применение
для целей идентификации отдельных компонентов простых и
сложных смесей, для качественного и полуколичественного их анализа,
а также для установления степени чистоты препаратов.
Основные операции при хроматографировании на бумаге
Хроматографическое разделение веществ на бумаге состоит из
следующих основных операций: подготовка бумаги и
растворителей; нанесение пробы; хроматографирование и детектирование
пятен на хроматограмме.
Бумага для хроматографических качественных анализов
выпускается двух марок: «М» — бумага медленной впитываемости, № 1
и 2; «С»—бумага средней впитываемости, № 3 и 4. Бумага
представляет собой 100% сульфатную целлюлозу, предгидролизную,
кордную. Содержание воды около б—7%. Обычная бумага
гидрофильна. Однако в отдельных случаях, например при разделении
смесей органических веществ, нерастворимых в воде, необходимо
ее превратить в гидрофобную;
Для этого бумагу на 45 мин помещают в ацетилирующую
смесь, состоящую из 90 мл уксусного ангидрида, 10 мл петролей-
ного эфира и 8—10 капель конц. H2S04. Затем ее вынимают и
тщательно промывают в проточной воде в течение 15 мин,
оставляют на 10—15 мин в дистиллированной воде, а затем промывают
и высушивают в вакуум-эксикаторе над СаС12.
Из бумаги вырезают соответствующего размера
прямоугольные полосы или сворачивают из листа цилиндр, скрепляемый
канцелярскими скрепками, для двумерной хроматографии.
Размеры полосок и цилиндра обусловливаются размерами
камеры для хроматографирования, необходимой длиной перемещения
разделяемых веществ и числом наносимых проб. Затем, отступя
от нижнего края полосы бумаги, проводят простым карандашом
линию старта, которая при хроматографировании должна
быть на 20—30 мм выше уровня растворителя в камере для
хроматографирования. На этой линии, на расстоянии 20—25 мм от
краев бумаги, отмечаются кружками диаметром около 3 мм места,
на которые наносят затем капли исследуемого раствора.
Для круговой хроматографии линией старта служит
окружность диаметром 20—30 мм, проведенная из центра круга
(см. рис. 183).
Затем бумагу выдерживают в камере для
хроматографирования, на дно которой налита подвижная фаза (растворитель или
смесь растворителей). Если в качестве неподвижной фазы
применяют водные растворы нелетучих веществ (например, буферные
растворы), то бумагу пропитывают этим раствором, высушивают,
319
а перед применением выдерживают 10—16 ч в камере для хромато-
графирования.
Для пропитывания бумаги труднолетучими растворителями
(формамид, пропиленгликоль и т. д.) их разбавляют метиловым
спиртом, а затем бумагу погружают на 1—2 с в эту смесь,
обжимают между листами фильтровальной бумаги и выдерживают 15—
20 мин на воздухе. =1
Если бумага предназначена для хроматографирования
аминокислот, ее вначале отмывают в 0,3 н. НС1, затем нейтрализуют
0,5 н. NH4OH, промывают дистиллированной водой до
отрицательной реакции по фенолфталеину, пропитывают 0,1% фосфатным
буферным раствором с рН = 7,0 -=-7,5 и высушивают на воздухе.
Подготовка растворителей заключается в приготовлении смеси
с взаимной насыщенностью подвижной и неподвижной фазы.
Обычно для этого смесь, взятую в определенном соотношении,
встряхивают в делительной воронке. После отстаивания органическую
фазу наливают в лодочку (при нисходящей хроматографии) или
в чашку (при восходящей). Водную фазу помещают в хромато-
графический сосуд, чтобы обеспечить насыщение атмосферы
камеры парами обеих фаз.
Нанесение пробы на бумагу проводят до или после
пропитывания ее неподвижной фазой. Каплю раствора (0,5—1 мг в 1 мл)
наносят из капиллярной пипетки, легко прикасаясь ею к
отмеченным местам на линии старта. Раствор должен заполнять только
отмеченный кружок, и образующееся пятно должно иметь
диаметр не более 5 мм. После высыхания пятна снова наносят каплю
раствора и повторяют это несколько раз (после высыхания
предыдущей капли) с таким расчетом, чтобы в целом нанести 0,01—
0,05 мл раствора.
Затем бумажную полоску высушивают и применяют ее для
нисходящей или восходящей хроматографии.
Одномерная восходящая хроматографии
Осуществление восходящего варианта хроматографии не
требует сложного оборудования, и исключена опасность перетекания
растворителя. Как только фронт растворителя подойдет к
верхнему концу бумаги, хроматографирование само собой прекращается.
Ближайший к линии старта конец бумаги с нанесенными
пробами погружают на 20—30 мм в подвижную фазу, закрепляют
бумагу и, закрыв камеру, дают растворителю медленно
распространяться вдоль бумаги. В отдельных случаях, когда значения
Rf компонентов смеси резко различаются, хроматографирование
можно заканчивать, как только растворитель пройдет 100—150 мм
от линии старта. Затем бумагу вынимают, карандашом отмечают
на ней верхнюю границу фронта растворителя и подсушивают.
Условия сушки зависят от летучести растворителя, свойств
разделяемых веществ и реагента, применяемого для детектирования
пятен.
320
Рис. 182. Прибор для восходящей одномерной хроматографии на бумаге:
/ — стеклянный цилиндр; 2 — резиновая или пришлифованная стеклянная
пробка; 3—стеклянный стержень для подвешивания бумаги; 4 — буманг-
ная полоска; 5—зажимы; 6—растворитель.
На рис. 182 представлена камера для
восходящей хроматографии. К пробке 2 цилиндра 1
прикреплен стеклянный стержень <?, служащий для
подвешивания полоски хроматографической бумаги
4, вертикальное положение которой достигается с
помощью зажимов 5. На дно цилиндра помещают
подвижную фазу-растворитель 6, Пробка 2
герметизируется вазелином или консистентной смазкой.
Камеру, в которой происходит хроматографиро- *
вание, плотно закрывают и устанавливают в
термостат. Колебания температуры на время хроматографирования не
должны превышать ±1,5°С.
Двумерная восходящая хроматография
Простое устройство для проведения восходящей двумерной
хроматографии состоит из стеклянной кюветы, устанавливаемой
под стеклянным колпаком на плоской подставке. В кювету с
растворителем помещают цилиндр, сшитый из квадратного
(200 X 200, 300 X 300 или 400 X 400 мм) листка
хроматографической бумаги, с нанесенной и высушенной пробой. (Пробу наносят
в нижнем левом углу квадрата в 50 мм от краев).
После того как фронт подвижной фазы достигнет верхнего края
бумаги, хроматографирование прекращают, бумагу высушивают
и поворачивают на 90° против часовой стрелки. При этом место
нанесения капли исследуемого вещества окажется справа, а линия,
по которой происходил подъем зон, — внизу.
В таком положении бумагу помещают в другую кювету с
другой системой растворителей и снова хроматографируют. После
достижения фронтом новой подвижной фазы верхнего края бумаги
хроматографирование прекращают, бумагу высушивают и
образовавшиеся зоны обнаруживают соответствующим реактивом.
Нисходящая хроматография применяется реже, и ее
методическое оформление подобно восходящему варианту, только элюент
движется сверху вниз.
Круговая (радиальная) хроматография
При круговой хроматографии вместо пятен на хроматограмме
образуются концентрически расположенные кольца. Камерой для
хроматографирования может служить небольшой эксикатор. Диаметр
бумажного круга, в центр которого наносят пробу, должен на 20—
30 мм превышать диаметр узкой части эксикатора. Насыщенный
11 Зак. 1237 321
Рис. 183. Прибор и бумажный диск
для круговой хроматографии:
А — место нанесения капель; / — бу*
мажный кружок; 2 — фитиль для
подачи растворителя; 3 —
растворитель.
неподвижной фазой растворитель наливают на дно эксикатора.
Бумажный круг, по радиусу которого вырезают полоску
(«фитиль») шириной 20 мм, помещают над узкой частью эксикатора,
фитиль отгибают и опускают в растворитель (рис. 183). Скорость
поступления растворителя к центру круга можно регулировать,
изменяя ширину фитиля. Хроматографирование производят при
герметически закрытом эксикаторе и, по возможности, при
постоянной температуре. После того как растворитель дойдет почти до
края бумажного круга, хроматографирование заканчивают, круг
вынимают, высушивают и пятна детектируют. Круговую
хроматографию на бумаге можно осуществить и в чашках Петри.
Детектирование пятен
Выбор метода детектирования зависит от химических свойств
хроматографируемых веществ [17],
Расположение пятен бесцветных веществ на хроматограмме
обнаруживают обычно при помощи реактивов, которые превращают
разделенные компоненты смеси в окрашенные соединения. В ряде
случаев положение вещества на хроматограмме можно обнаружить
при рассматривании хроматограмм в затемненной комнате
в" ультрафиолетовом свете, под действием которого одни вещества
начинают светиться, другие образуют темные пятна вследствие
поглощения света.
После высушивания (на воздухе или в сушильном шкафу) хро-
матограмму опускают на несколько секунд в сосуд с раствором
индикатора или опрыскивают им из пульверизатора. При этом
бумажный цилиндр следует развернуть. Для выявления окраски
пятен хроматограмму после опрыскивания помещают на 5 мин в су*
шильный шкаф, нагретый до 90—100 °С, после чего пятна
очерчивают карандашом.
В ряде случаев для сохранения окраски уже проявленную
хроматограмму опрыскивают вторым реагентом или вводят
соответствующее соединение в первоначальный раствор реагента-индика*
тора. Например, при детектировании пятен аминокислот нингидри-
ном образуются пурпурные пятна, выцветающие при комнатной
температуре в течение одного дня. Если в исходный нингидриновый
реактив ввести ацетат кадмия или после детектирования нингидри-
ном опрыскать пятна раствором ацетата никеля, окраска сохра*
няется пять лет»
322
В последние годы разработан и выпускается комплект аппарат
туры для бумажной хроматографии, позволяющий
автоматизировать нанесение пробы и хроматографирование при любой заданной
температуре в интервале 10—40 °С. Специальное программное
устройство позволяет начинать и заканчивать процесс хроматогра-
фирования в заранее заданное время.
ХРОМАТОГРАФИЯ В ТОНКОМ СЛОЕ
Тонкослойная хроматография является одним из видов
распределительной жидкостной хроматографии. Быстрота разделения
смесей, относительная простота и универсальность обеспечили
быстрое распространение этого метода, а использование
гранулированного сорбента дает ему преимущества перед бумажной
хроматографией из-за меньшего размывания пятен в направлении разделения.
Пластинки для хроматографирования
Разделение смесей производят на пластинках, покрытых тонким
слоем адсорбента (силикагеля, оксида алюминия, полиамида,
целлюлозы). Адсорбционный слой с пленкой растворителя,
удерживаемого сорбентом, служит неподвижной фазой, а подвижной—-
индивидуальные растворители или смеси растворителей.
Техника хроматографирования в тонких слоях состоит в
подготовке пластинок, нанесении пробы, проявлении хроматограммы и
детектировании пятен. Положение пятна разделенного вещества
описывается, так же как и в бумажной хроматографии, посредством
измерения величины Rf, т. е. путем сопоставления с Rf стандартных
веществ; вещества идентифицируются способом «свидетелей».
Тонкослойная хроматография применяется для идентификации
веществ, определения чистоты вещества, качественного и
количественного анализа веществ и их смесей, а в последнее время и для
препаративных целей.
Существуют две модификации тонкослойной хроматографии:
с закрепленным и с незакрепленным слоем адсорбента.
Тонкий слой адсорбента распределяют на подложке — пластинке
из стекла, алюминиевой фольги или пластмассы. Чаще всего
используют стеклянные пластинки (их легко очищать и можно
использовать многократно) толщиной 2—4 мм, размером 3X8,
2,5 X 7,5; 5 X 15; 10 X 20; 20 X 20 см.
Пластинки промывают проточной водой, погружают на 24 ч
в сосуд с 1 % раствором моющего вещества, после чего тщательно
промывают дистиллированной водой и высушивают, предохраняя
от пыли. Брать пластинки следует за ребра, не касаясь
поверхности. Хранить пластинки можно в эксикаторах или полиэтиленовых
мешочках. Использованные пластинки для повторного применения
сначала очищают от слоя адсорбента, после чего их промывают и
высушивают, как указано выше.
Для приготовления пластинок с закрепленным слоем
адсорбента применяют массу, состоящую из адсорбента,
связующего материала и воды. В качестве связующего используют гипс
И* 323
CaS04-Y2H2C) или крахмал. Иногда в состав массы для
идентификации пятен разделяемых соединений добавляют флуоресцирующие
вещества.
В случае применения гипса смесь из 5 г адсорбента (силикагеля
или оксида алюминия), 0,2 г гипса и 12 мл воды растирают в
фарфоровой ступке до однородной консистенции. Полученную массу
наносят на подготовленную стеклянную пластинку и разравнивают
при помощи специального устройства, обеспечивающего толщину
слоя 0,15—0,25 мм по всей поверхности. В случае применения
крахмала 28,5 г тонкоизмельченногои просеянного через сито № 0085
силикагеля, 1,5 г растворимого рисового крахмала и 54 мл
дистиллированной воды нагревают при перемешивании до тех пор, пока
раствор не начнет загустевать. Затем при постоянном
перемешивании смесь охлаждают, размешивают с 20 мл дистиллированной
воды и равномерно наносят на пластинки.
Пластинку с закрепленным слоем высушивают на воздухе в
горизонтальном положении, после чего уже в вертикальном положении
активируют нагреванием в течение 30 мин в сушильном шкафу при
110 °С и хранят в эксикаторе над СаС12 или Mg(C104)2. В процессе
сушки необходимо оберегать пластинки от загрязнения пылью или
от поглощения паров летучих соединений, которые могут
находиться в воздухе.
При нанесении тонкого слоя адсорбента на предметные стекла
рекомендуется поступать следующим образом. Готовят суспензию
из 44 г А1203 и 1,6 г гипса в смеси 70 мл хлороформа и 30 мл
метилового спирта или же из 30 г силикагеля и 1,5 г гипса в смеси
50 мл хлороформа и 50 мл метилового спирта. Суспензию тщательно
перемешивают встряхиванием. Затем два предметных стекла,
сложенных плоскостями, погружают в нее, медленно вынимают и дают
стечь жидкости. После этого стекла разделяют, высушивают и
хранят в эксикаторе над СаС12. Если в поверхностном слое адсорбента
появились трещины, пластинки непригодны к работе.
Очень удобны готовые пластинки «Силуфол» с тонким слоем
А1203 на подложке из алюминиевой фольги размером 150 X 150 мм
и пластинки «Сата D-0», «Сата DS-0» с тонким слоем силикагеля,
размером 100 X 100 и 200 X 200 мм, поставляемые странами СЭВ.
Пластинки с незакрепленным слоем
подготавливают следующим образом. Адсорбент насыпают на стекло (лучше
матовое) и разравнивают валиком, снимая при этом избыток.
Валиком обычно служит стеклянная палочка диаметром 10 мм с
надетыми на концы кусочками резиновой трубки. Расстояние между
утолщенными концами трубки должно быть на 10—12 мм меньше
ширины применяемой стеклянной пластинки. Толщину стенок
трубки подбирают так, чтобы при накатывании образовался слой
толщиной примерно 1 мм. Некоторые авторы рекомендуют распылять
суспензию тонкоизмельченного адсорбента специальным
распылителем по поверхности пластинки.
Пластинки с незакрепленным слоем приготавливают непосред*
ственно перед употреблением.
324
Нанесение пробы и проявление хроматограммы
Раствор пробы наносят микропипеткой в 10—20 мм от края
пластинки в виде точек или коротких черточек. В случае
незакрепленного слоя необходимо следить, чтобы конец пипетки не касался
слоя адсорбента.
В одну точку на пластинке можно наносить до 1 мг раствора
пробы. Затем растворителю дают испариться и пластинки
переносят в камеру для проявления. Пластинки с закрепленным слоем
можно проявлять в наклонном или вертикальном положении;
пластинки с незакрепленным слоем обычно ставят под углом 10—15°.
Камерой для проявления хроматограмм методом восходящей
хроматографии может служить стеклянный сосуд с плоским дном,
закрывающийся пришлифованной крышкой или стеклом. На дно
камеры наливают столько растворителя, чтобы пластинка
погружалась в него на 3—5 мм. Сначала помещают пластинку в слегка
наклоненную камеру так, чтобы растворитель не смачивал
стартовую полосу. Через 5—10 мин камеру возвращают в вертикальное
положение. Для лучшего насыщения к стенке сосуда по всей
высоте камеры прикрепляют смоченный растворителем лист
фильтровальной бумаги, доходящий до дна сосуда.
При работе с закрепленным слоем адсорбента фронт
продвижения растворителя на пластинке не должен превышать 100—150 мм.
После того как фронт растворителя продвинется на 100—150 мм
(на это требуется 15—90 мин), пластинку вынимают из камеры,
отмечают карандашом линию фронта, высушивают и детектируют
пятна, опрыскивая поверхность пластинки общими или
специфическими реагентами [17].
В качестве общих реагентов для детектирования органических
соединений используют конц. H2S04, раствор Na2Cr04 в H2S04,
1 % раствор иода в метиловом спирте и др. Преимущество
тонкослойной хроматографии перед бумажной состоит в том, что
перечисленные общие реактивы не разрушают слой адсорбента, как
разрушают бумагу. Весьма 4acfo в качестве универсального
реагента для детектирования пятен используют пары иода. Пластинку
помещают в закрытую банку (эксикатор), содержащую несколько
кристалликов иода. Пары иода растворяются в большинстве
органических соединений, которые становятся заметными в виде
коричневых пятен на бледно-желтом фоне. Пятна, обнаруженные с
помощью иода, на воздухе быстро обесцвечиваются. Можйо замедлить
обесцвечивание и повысить чувствительность реакции, если
проявленную пластинку опрыскать раствором 7,8-бензофлавона (0,3 г
7,8-бензофлавона в 95 мл этилового спирта + 5 мл 30% H2S04).
Этот реагент дает интенсивное голубое окрашивание с иодом.
Существует и другой универсальный способ детектирования
разделенных веществ. При приготовлении пластинок для тонкослой-
ной хроматографии к адсорбенту прибавляют неорганический
фосфор и таким образом получают флуоресцирующие пластинки.
Поскольку органические вещества чаще всего гасят флуоресценцию,
325
то в ультрафиолетовом свете они обнаруживаются в виде темных
пятен. Если вещества сами флуоресцируют, то под действием
ультрафиолетового излучения они обнаруживаются в виде
светящихся пятен на темном фоне.
Разработаны также методы количественного определения
веществ в хроматографических пятнах непосредственно на пластинке.
Для этих целей используются денситометры или флуориметры.
Комплект оборудования для тонкослойной хроматографии
Оборудование для тонкослойной хроматографии КТХ-01
отечественного производства предназначено для качественного анализа
многокомпонентных смесей органических и неорганических
веществ, а также для препаративного получения веществ,
составляющих эти смеси. Оборудование поставляется в трех вариантах:
в виде полного (КТХ-01-1), среднего (КТХ-01-2) или малого
(КТХ-01-3) комплектов, различающихся наличием тех или иных
устройств. В комплект КТХ-01 входит оборудование для
приготовления хроматографических пластин, для нанесения пробы, для
проведения тонкослойной хроматографии, для детектирования
пятен химическими методами и для обнаружения их в
ультрафиолетовом свете.
АДСОРБЕНТЫ ДЛЯ ХРОМАТОГРАФИИ
Адсорбенты, применяемые в хроматографии, кроме большой
удельной поверхности, достигающей сотен квадратных метров на
грамм, должны обладать избирательностью, химической и
каталитической инертностью в отношении компонентов разделяемой смеси
и подвижной фазы. Используемые в хроматографии адсорбенты
должны быть, по возможности, монодисперсными (с одинаковым
размером частиц), и состав их должен быть стандартным, что
гарантирует воспроизводимость эксперимента.
Из адсорбентов, применяемых в хроматографии, назовем
главнейшие: оксид алюминия, силикагели, силохромы, цеолиты и поли-
сорбы.
Оксид алюминия — мелкокристаллический порошок амфотер-
ного характера с удельной поверхностью 230—280 м2/г.
Адсорбционные свойства А1203 в сильной степени зависят от способа его
приготовления, обработки и качества исходных материалов.
Гигроскопичен. При увлажнении адсорбционная активность
снижается. Оптимальной активности можно достигнуть нагреванием
А1203 при 400—450 °С в течение 5—6 ч. При хранении в отсутствие
благи не теряет активности. Выпускается промышленностью: 1-й
и 2-й активности (марки А-1 или А-2); по Брокману; оксид
алюминия кислый, нейтральный и основной для тонкослойной
хроматографии.
Силикагель — высушенный гель кремневой кислоты пористого
строения с развитой внутренней поверхностью, кислого характера.
326
Твердые хрупкие полупрозрачные или меловидные зерна от белого
и желтоватого до коричневого цвета, с удельной поверхностью
.'5(Ю—700 м2/г — в зависимости от способа получения. Обладает
резко выраженной гидрофильностью, в связи с чем широко
применяется в качестве осушителя. Сорбирует пары многих органиче-»
с к их соединений.
В зависимости от режима приготовления и формы зерен си-
лмкагели разделяются на кусковой — с зернами произвольной
формы — и гранулированный. Промышленность выпускает большое
число различных марок крупнопористых (КСК, ШСК, МСК, АСК)
I! мелкопористых (КСМ, ШСМ, МСМ, АСМ) силикагелей,
различающихся составом, пористостью и зернением. Особый интерес
представляет силикагель марки АСМ с радиусом пор 1,5 нм и
величиной зерен 0,2—0,5 мм. Кроме силикагелей заводского
изготовления, осуществляется полупромышленное производство.
Специальный набор из семи марок силикагелей, по 0,25 кг каждого,
выпускается для хроматографии [46, с. 375].
Силохромы — зерна пористого диоксида кремния высокой
химической чистоты. Они характеризуются весьма однородным
распределением пор по размерам, высокой механической и термической
устойчивостью. Применяются для разделения высококипящих
неполярных и слабополярных веществ. Силохромы могут также быть
использованы в качестве носителей для разделения сильно
адсорбирующихся веществ. Выпускаются марки С-80 и С-120 с удельной
оверхностью 60—100 и 100—140 м2/г соответственно.
Цеолиты (молекулярные сита)—мелкопористые гидратирован-
ые алюмосиликаты щелочных и щелочноземельных металлов, при-
одные или синтетические. Наибольшее применение нашли синтети-
еские цеолиты марок КА, NaA, CaA, CaX, NaX (первые буквы
шачают преобладающий в цеолите катион, последняя — тип цео-
пта А или X). Поры цеолитов представляют собой сферические
•)лости с диаметром 1,14 нм, соединенные друг с другом более
узкими отверстиями (их называют окнами).
Для цеолита типа А в калиевой форме (марка КА)
эффективный диаметр окон около 0,3 нм, у натриевой формы (марка NaA) —
химерно 0,4 нм, а у кальциевой (СаА) —около 0,5 нм.
У цеолитов марки X эффективный ^диаметр окон значительно
■ >льше. Так, для СаХ он близок к 0,8 нм, а для NaX — около 0,9 нм.
.жим образом, основное различие между цеолитами типов А и X
■ включается в размерах окон, соединяющих полости.
Особенностью адсорбционных свойств цеолитов является их м о -
лкулярно- ситовое действие. Адсорбироваться в пер-
.'чной пористой структуре могут только те молекулы, диаметр ко-
>рых меньше эффективного диаметра окон. Более крупные моле-
лы практически не адсорбируются. Таким образом, при адсорбции
■ и цеолитах происходит как бы отсеивание более мелких молекул
и г более крупных. Емкость таких молекулярных сит значительно
больше, чем емкость других адсорбентов.
327
Синтетические цеолиты общего назначения предназначаются
для глубокой осушки и тонкой очистки газов и жидкостей, а также
для разделения газообразных и жидких смесей.
Цеолиты КА, NaA, CaA, NaX, CaX выпускаются в виде
таблеток, гранул или шариков различных размеров.
Мелкокристаллические цеолиты формуют, применяя до 15—20% связующих (обычно
глин), после чего подвергают кратковременной (2—6 ч)
термической обработке при 550—600 °С.
Промышленность выпускает набор синтетических цеолитов из
четырех марок (NaA, NaX, CaA и CaX) no 0,25 кг каждого.
Полисорбы — сополимеры стирола и дивинилбензола,
используемые в хроматографии спиртов, жирных кислот, альдегидов, кето-
нов, нитрилов, а также для определения примеси воды в органа*
ческих жидкостях.
Выпускаются полисорбы марок 4, 4Т, 10 и др.
Пористые стекла — пористый продукт, получаемый
выщелачиванием боронатровой фазы из фазоразделенного натровоборосп-
ликатного стекла. Отличаются однородностью размеров пор.
Для гель-хроматографии выпускается набор мелкозернистых
стекол (МПС) с разными размерами пор: МПС-250 ГХ, МПС-750 ГХ,
МПС-1150 ГХ, МПС-1600 ГХ и МПС-2000 ГХ. Цифры означают
преобладающий диаметр пор (в ангстремах), а буквы ГХ —
назначение (гель-хроматография).
глава зо. ФОТОМЕТРИЯ
Фотометрический метод анализа [8, 53] основан на
избирательном поглощении электромагнитных излучений различных
участков спектра однородной системы *. Каждая однородная система
обладает способностью избирательно поглощать излучения
определенных длин волн, причем количество поглощенной энергии
пропорционально концентрации поглощающего вещества в
растворе. Поэтому фотометрический анализ при условии использования
монохроматических излучений называют методом
адсорбционной спектрофотометрии.
Степень монохроматичности потока излучения определяется
интервалом длин волн, который выделяется данным монохромато-
ром (светофильтром, призмой или дифракционной решеткой) из
сплошного потока электромагнитного излучения.
В зависимости от используемой аппаратуры в фотометрическом
анализе различают фотоколориметрические и спектрофотометри-
ческие методы анализа. Фотоколориметрические мето-
д ы, в которых измеряется светопоглощение окрашенных
растворов, используют сравнительно несложную аппаратуру и при этом
обеспечивают достаточную точность измерений (Д = ±1-т-2
отн.%) и широко применяются в концентрационном анализе
(определение концентрации растворов). В большинстве
фотоколориметров монохроматизация осуществляется с помощью светофильтров.
В спектрофотометрических методах применяют
более сложные приборы — спектрофотометры, позволяющие
проводить анализ как окрашенных, так и бесцветных соединений по
избирательному поглощению монохроматического света в видимой
(К = 400 -г- 700 нм), ультрафиолетовой (К = 200 Ч- 400 нм) или
ближней инфракрасной (Л, = 700~-1500 нм) областях спектра.
Ввиду того, что спектр поглощения каждого поглощающего
вещества имеет вполне определенную форму, спектрофотометр может
быть применен как для количественного, так и качественного
анализа химических соединений.
ЗАКОНЫ ПОГЛОЩЕНИЯ СВЕТА. ТЕРМИНЫ И ОБОЗНАЧЕНИЯ
Интенсивность монохроматического светового потока,
прошедшего через слой окрашенного раствора, уменьшается по сравнению
с первоначальной величиной в зависимости от концентрации
окрашенного вещества и толщины слоя раствора. Эти условия выра-
* Если система неоднородна (коллоидные растворы, взвеси), то при взаимо-
действии излучения с веществом кроме поглощения будет иметь место и
рассеяние лучистой энергии. На этом основаны методы нефелометрии и тур-
бидиметрии, которые не рассматриваются в настоящей главе.
329
жаются уравнением объединенного закона Бугера — Ламберта —
Бера:
где /о и / — соответственно начальная интенсивность светового
потока и интенсивность его после прохождения через раствор; С —
концентрация поглощающего раствора; ел — коэффициент
поглощения при длине волны К; I — толщина слоя раствора.
Если концентрация раствора выражена в молях на литр, а
толщина поглощающего слоя — в сантиметрах, то постоянную
е^ называют молярным коэффициентом' поглощения
(эксти н кции). Он зависит от длины волны падающего света,
природы растворенного вещества и температуры раствора.
Молярный коэффициент поглощения отражает индивидуальные свойства
окрашенных соединений и является их важной характеристикой.
Для разных веществ молярный коэффициент поглощения имеет
различное значение.
Величину lg-у- называют оптической плотностью
поглощающего вещества и обозначают буквой D. Оптическая
плотность может иметь любые положительные значения, однако
современные приборы позволяют определять оптическую плотность,,
не превышающую 2—3.
Отношение -г- = Т характеризует пропускание или про-
зрачность раствора. Величина пропускания Т может изменять*
ся от 0 до 1 или от 0 до 100%. Величину пропускания Г,
отнесенную к толщине поглощающего слоя / = 1 см, называют
коэффициентом пропускания.
Оптическая плотность и пропускание связаны между собой
соотношением D = 2 — lg Г (если Т выражено в процентах). При
соблюдении основного закона светопоглощения оптическая
плотность раствора прямо пропорциональна молярному коэффициенту
поглощения, концентрации поглощающего вещества и толщине слоя
раствора:
D = екС1
При графическом изображении зависимости оптической
плотности от концентрации (при постоянной толщине слоя /)
получается прямая линия, которая проходит через начало координат при
отсутствии поглощения света растворителем.
МЕТОДЫ ИЗМЕРЕНИЯ ПОГЛОЩЕНИЯ СВЕТА
Найти абсолютные значения интенсивности светового потока
до (/о) и после (/) прохождения его через раствор практически
очень затруднительно. Поэтому при измерении поглощения
излучений обычно сравнивают два световых потока: один проходит
через испытуемый раствор, а другой через определенный стандарт*
330
ный раствор или через растворитель (нулевой раствор,
поглощение которого условно принимается равным нулю).
Сравнение можно проводить визуально или посредством
фотоэлектрических приборов, в которых приемником излучений служат
фотоэлементы. Визуально можно лишь констатировать наличие
сходства или различия в окраске, но оценить количественно
степень различия ее невозможно.
Интенсивность окраски двух сравниваемых растворов
выравнивают, изменяя концентрацию (метод разбавления, метод
стандартных серий и метод колориметрического титрования), или толщину
поглощающего слоя, или интенсивность светового потока
(Последнее возможно в том случае, если более интенсивный поток
ослабить при помощи измерительной диафрагмы.)
Изменение ширины щели диафрагмы, находящейся на пути
одного из двух сравниваемых световых потоков, может быть скор-
релировано поворотом отсчетного барабана, отградуированного в
величинах оптической плотности D или пропускания Г.
РАСЧЕТ КОНЦЕНТРАЦИИ СВЕТОПОГЛОЩАЮЩИХ РАСТВОРОВ
При работе с приборами, позволяющими непосредственно
измерять оптическую плотность D, для расчета концентрации
испытуемых растворов можно применять следующие методы.
1. Графический метод, основанный на построении
калибровочного графика в координатах оптическая плотность —
концентрация.
Для построения калибровочного графика измеряют поглощение
серии окрашенных растворов известной, но различной
концентрации, оптические плотности которых охватывают требуемый
интервал. С этой целью применяют стандартный раствор определяемого
вещества. Тщательно отмеряют пипеткой определенные части этого
раствора, добавляют к ним соответствующий реагент и соблюдают
условия максимального развития окраски (время выдержки,
температура). После этого каждый раствор разбавляют в мерной
колбе до определенного объема и измеряют поглощение при
выбранной длине волны. График зависимости поглощения света от
концентрации поглощающего вещества обычно представляет собой
прямую линию, тангенс угла наклона которой равен
коэффициенту пропускания Т или молярному коэффициенту поглощения е*.
При построении калибровочного графика результаты измерений
вначале наносят в виде 5—8 точек, различающихся по
концентрации не менее чем на 30%, а затем проводят прямую линию либо
через эти точки, либо как можно ближе к ним. Это ведет к
усреднению и уменьшению ошибок, вызванных неточностями
приготовления и измерения поглощения стандартных растворов.
Фотометрическую реакцию анализируемого образца проводят в тех же
условиях, что и для стандартных растворов. Измерив поглощение
раствора образца, можно по калибровочному графику определить
его концентрацию.
331
2. Если заранее известно, что испытуемые растворы
подчиняются законам поглощения излучений, то приготовляют два
раствора— эталонный, концентрация которого Сэ известна, и
испытуемый (его концентрация Сх) и определяют их оптические плотности
D3 и Dx. Концентрацию испытуемого раствора вычисляют по
формуле:
г D*C*
3. Если заранее известно значение молярного коэффициента
поглощения при данной длине волны монохроматического света
еь то, зная толщину поглощающего слоя (толщину слоя кюветы),
концентрацию испытуемого раствора вычисляют по формуле:
ФОТОКОЛОРИМЕТРИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
Фотоколориметрические -методы определения концентрации
вещества [64, 71] основаны на сравнении поглощения или
пропускания света стандартным и исследуемым окрашенными растворами.
Под колориметрией имеют в виду методы определения
концентрации окрашенных веществ в растворе по поглощению света. Если
определяемый компонент бесцветен, то при помощи химической
реакции переводят его в окрашенное соединение, после чего
инструментальным или визуальным способом измеряют
интенсивность окраски.
В визуальных методах используют несложные приборы
непосредственного сравнения интенсивности окрасок испытуемого рас*
твора с серией стандартных растворов, полученных путем
последовательного разбавления раствора известной концентрации,
содержащего одно и то же окрашенное вещество. Визуально
сравниваемые растворы должны быть налиты в пробирки или цилиндры
одинакового размера и освещены от источника с рассеянным
светом. В другом варианте визуального метода изменяют толщину
слоя жидкости, через который проходит свет, и этим добиваются
одинаковой интенсивности окраски испытуемого и стандартного
растворов. Для этого используют цилиндры Ген ер а — стеклянные
градуированные цилиндры одинакового диаметра, снабженные у
основания кранами для сливания жидкости.
Важнейшие детали фотоколориметров
Фотоэлементы. В отличие от визуальных методов колориметри-
рования в фотоколориметрии степень поглощения света
определяется не глазом, а при помощи колориметров с фотоэлементами
(ФЭК). Фотоэлемент преобразует световую энергию, проходящую
через исследуемый окрашенный раствор, в электрическую. Сила
332
возникающего фототока (общая чувствительность фотоэлемента)
зависит от длины волны падающего на фотоэлемент света и от
температуры.
В приборах для фотометрического анализа в основном нашли
применение селеновые, сурьмяно-цезиевые и кислородно-цезиевые
фотоэлементы.
Селеновые фотоэлементы пригодны для измерений толь-
ко в видимой части спектра. Они обладают высокой
чувствительностью, и при их применении не требуется усиливать возникающий
фототок, измерение которого возможно обычным стрелочным
гальванометром. Селеновые фотоэлементы нельзя применять для
измерения в узких участках спектра, так как используемые в этих
случаях узкополосные светофильтры значительно ослабляют световые
потоки.
Сурьмяно-цезиевые и кислородно-цезиевые
фотоэлементы обладают небольшой чувствительностью,
поэтому возникающие фототоки необходимо усиливать.
Сурьмяно-цезиевые фотоэлементы применимы в ультрафиолетовой и видимой
областях спектра, а кислородно-цезиевые — в видимой и ближней
инфракрасной областях.
Измерять оптическую плотность раствора следует при длине
волны, при которой наблюдается максимальное поглощение света.
Светофильтры. Область максимального поглощения света пря
фотометрическом анализе выделяют при помощи светофильтров,
устанавливаемых на пути световых потоков перед поглощающими
растворами.
Светофильтры — жидкие или твердые среды, обладающие
избирательным пропусканием излучения в достаточно узком интервале
длин волн. В качестве светофильтров используют окрашенные
растворы некоторых веществ, окрашенные оптические стекла,
интерференционные светофильтры и диспергирующие призмы;
последние характеризуются более высокой степенью монохроматизации,
чем светофильтры. Ширина пропускания определенного
спектрального участка (линейная дисперсия) для светофильтров колеблется
от 100 до 20—40 нм; в призменных и дифракционных приборах
линейная дисперсия колеблется от 0,5 до 2 нм.
Кюветы. Они представляют собой прямоугольные или
цилиндрические сосуды из стекла или кварца с определенным
расстоянием между стенками (у прямоугольных кювет) — или между
крышками — у цилиндрических.
Стеклянные кюветы пропускают все лучи видимого света;
кварцевые— не только видимые, но и ультрафиолетовые и частично
инфракрасные лучи.
Поверхность кюветы, на которую падает световой поток, и
поверхность, через которую выходит непоглощенная часть светового
потока, должны быть строго параллельны. В зависимости от
интенсивности окраски раствора для измерения выбирают кювету с
большей или меньшей толщиной слоя. Следует иметь в виду, что
наименьшая ошибка измерения получается при D = 0,3-Ь 0,5. Поэтому
333
нужно так подобрать кюветы, чтобы вести измерения в этом
интервале оптической плотности.
В наборах кювет для фотометрирования имеется, как правило,
по две пары кювет с одинаковой толщиной слоя жидкости.
Рабочие поверхности кювет должны быть чистыми. Перед
заполнением кювет их следует тщательно промыть дистиллированной
водой, затем ополоснуть исследуемой жидкостью и только после
этого, непосредственно перед измерением, заполнить светопогло-
щающим раствором.
Лабораторные фотоколориметры [46, с. 219]
Фотоэлектрические лабораторные колориметры
предназначаются для определения светопропускания или оптической плотности
жидких окрашенных растворов и твердых тел, а также
светопропускания взвесей, эмульсий и коллоидных растворов.
С помощью современных фотоколориметров можно измерять
коэффициенты пропускания или оптическую плотность в
спектральной области 300—1000 нм. Все фотоколориметры имеют
обязательно следующие элементы схемы: осветитель, светофильтры, кюветы,
фотоэлементы, регулируемые сопротивления и гальванометры.
Различают два основных метода измерения тока фотоэлемента:
прямое измерение и нулевой метод. В последнем случае ток
фотоэлемента, на который падает поток света, прошедший через
кювету с исследуемым раствором, компенсируется при помощи щелевой
диафрагмы или иным способом. Гальванометр при этом
используется не для измерения тока, а только как нуль-прибор.
Фотоэлектрический колориметр ФЭК-М. Колориметр имеет
стеклянную оптику, прозрачную только для лучей видимого участка
спектра. Источником излучения служит лампа накаливания
(вольфрамовая лампа), дающая излучение в видимой части спектра.
Прибор снабжен четырьмя светофильтрами с полушириной
пропускания 80—100 нм и поэтому пригоден только для
концентрационного анализа. В основу конструкции прибора (рис. 184) положен
принцип уравнивания интенсивности
двух световых потоков с помощью ще-
J 2 t 2' У левой диафрагмы.
4 ' Dfl ® И ' \ Измерение интенсивности световых
потоков проводится с помощью двух
селеновых фотоэлементов 9 и 9',
соединенных между собой и со
стрелочным гальванометром 14 по дифферен-
Рис. 184. Принципиальная схема фотоколориметра
ФЭК-М:
/ — источник света; 2 и 2' — конденсоры; 3 и 3' —
зеркала; 4 и 4'— светофильтры; 5 и 5'\ 6 и 6'—кюветы
с растворами; 7 и 7'— линзы; 8 и 8' — призмы; 9 и 9Г —
селеновые фотоэлементы; 10 и // — фотометрические
клинья; /2—щелевая диафрагма; /<?—отсчетный
барабан; /4—гальванометр.
334
циальной схеме таким образом, что при равенстве потока лучей
стрелка гальванометра стоит на нуле.
Световые потоки от источника излучения 1 направляются на
зеркала 3 и 3', затем проходят через светофильтры 4 и 4' в
кюветы с растворами 6 и 6' и попадают на селеновые фотоэлементы 9
и 9'. Перед фотоэлементами на пути левого светового потока
помещены круговые фотометрические клинья 10 и 11 для ослабления
светового потока, падающего на фотоэлемент, а на пути правого
потока — щелевая диафрагма 12, связанная с отсчетным барабаном
13. На отсчетных барабанах имеется две шкалы: оптических
плотностей D и коэффициентов светопропускания Т.
Измерения оптической плотности растворов производят при
помощи правого и левого барабанов. Шкала оптической плотности
левого барабана проградуирована от 0 до 2 (Т = 100 4-0%)-
Шкала оптической плотности правого барабана имеет пределы
измерений 0,00—0,52; точность измерений наибольшая на участке 0,15—
0,52 (по шкале светопропускания 70—30%). Работа с правым
барабаном имеет ряд преимуществ.
Измерение проводят 3—5 раз и находят среднее значение
оптической плотности.
Для нахождения концентрации анализируемого раствора
пользуются калибровочной (градуировочной) кривой. Определив
значение оптической плотности анализируемого раствора, находят на
оси ординат точку, соответствующую данному значению D, из
которой проводят линию, параллельную оси абсцисс до пересечения ее
с калибровочной кривой, а из точки пересечения опускают
перпендикуляр на ось абсцисс и по точке пересечения с ней находят
процентное содержание определенного вещества.
Фотоколориметр-нефелометр ФЭК-60. Прибор предназначен для
измерения концентрации жидких сред (прозрачных окрашенных
растворов, взвесей, эмульсий, коллоидных растворов).
Концентрация веществ определяется по ослаблению
проходящего через слой жидкости пучка излучения, которое может быть
вызвано поглощением в растворе или рассеянием во взвеси
(эмульсии). Прибор может служить как колориметром
(концентрационный), так и нефелометром.
Прибор укомплектован сменными фотоэлементами: сурьмяно-
цезиевым и кислородно-цезиевым. Спектральная область работы
прибора — видимая и ближняя инфракрасная часть спектра (360—
1000 нм). Из девяти пар светофильтров одна предназначена для
измерений в ближней ультрафиолетовой, пять — в видимой и три —
в ближней инфракрасной области. Колориметр-нефелометр ФЭК-60
является однофотоэлементным прибором, в основу которого
положен принцип уравнивания интенсивности двух световых
модулированных потоков при помощи переменной щелевой диафрагмы.
Принципиальная оптическая схема колориметра-нефелометра
ФЭК-60 представлена на рис. 185.
Два пучка лучей от источника света / проходят через две
симметричные оптические системы, каждая из которых состоит из
335
Рис. 185. Оптическая схема
колориметра-нефелометра
ФЭК-60:
/—источник света;
2—конденсоры; 3—щелевые
диафрагмы; 4 и 7 —зеркала; 5 и
6 — линзы; 5—фотоэлемент;
0 — призма; 10 — кюветы; // —
светофильтры; 12 —
вращающийся цилиндр.
конденсора 2,
щелевой диафрагмы 3,
зеркал 4 и 7 и линз 5
и 6. Световые потоки,
правый и левый,
модулируются в проти-
вофазе с частотой
350 Гц посредством вращающегося цилиндра 12.
Модулированные световые потоки, пройдя через светофильтры
11 и кюветы 10, направляются призмой 9 на общий приемник
излучения— фотоэлемент 8 и возбуждают переменный электрический
ток, пропорциональный разности интенсивности правого и левого
световых потоков. Фототок усиливается четырехкаскадным
усилителем. Выходной сигнал переменного тока выпрямляется фазовым
детектором, к которому непосредственно подключен
измерительный прибор — микроамперметр.
Интенсивности модулированных потоков излучений —
относительного (нулевого) и измеряемого — регулируют с помощью
щелевых диафрагм 3. Одна из них, расположенная на пути правого
светового потока, является измерительной. Она связана с отсчетным
барабаном, проградуированным в значениях оптической плотности
D и пропускания 7Y Максимальное раскрытие щелевой диафрагмы
соответствует 100% пропускания, а полное закрытие — нулю.
Щелевая диафрагма, расположенная на пути левого светового потока,
является компенсационной и шкалы не имеет.
Измерения проводятся со сменными парными светофильтрами
//, выделяющими узкие спектральные участки в видимой или
ближней инфракрасной области спектра. Для измерений в видимой
области спектра используют сурьмяно-цезиевый фотоэлемент, а в
инфракрасной — кислородно-цезиевый.
Питание источника света, фотоэлементов и усилителя
производится от сети переменного тока через питающее устройство,
обеспечивающее стабилизацию напряжения с точностью ±0,5%.
Первоначальная установка фотоколориметра, его включение
в сеть при помощи тумблера на панели стабилизатора, проверка
правильности установки осветителя и смена фотоэлементов
производятся согласно инструкции, прилагаемой к прибору. Измерения
можно начинать только спустя 15—20 мин после включения
прибора; за это время прогреется электросхема и установится
стабильный режим ее работы,
336
" nff ,
^еч
-12
ю
\$^
?Ш
гСМэ
10
Если в процессе работы заменяют светофильтр, то до измерения
нужно выждать 1—2 мин. Следует иметь в виду, что вчачале
целесообразно уравнивать световые потоки при малой чувствительности
прибора, что достигается включением в оба плеча нейтральных
светофильтров из прилагаемого комплекта. Затем устанавливают
«электрический нуль» прибора. Для этого закрывают
измерительную и компенсационную диафрагмы и добиваются нулевого
положения микроамперметра. Правый барабан устанавливают на
Т = 100% (или D = 0), включают светофильтр на пути излучения,
после чего на пути правого светового потока помещают кювету с
испытуемым раствором; левый поток остается свободным.
Вращением левого барабана устанавливают стрелку микроамперметра на
нуль. После этого поворотом рукоятки вводят в правый поток
кювету с нулевым раствором и вращением правого барабана
восстанавливают нарушенное равновесие и приводят стрелку
микроамперметра к нулю. Отсчет величины D производят по шкале диафрагмы.
СПЕКТРОФОТОМЕТРИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
Спектрофотометрия широко применяется для установления
связи между спектрами поглощения различных веществ и их
химическим строением и составом, а также для количественного опре^
деления веществ.
Абсорбционная спектрометрия основана на тех же законах све-
топоглощения, что и фотоколориметрические методы, однако, в
отличие от последних, в ней используется поглощение
монохроматического света с очень узким интервалом длин волн (1—2 нм). Это
значительно увеличивает чувствительность и точность
количественного анализа окрашенных растворов, поглощающих свет в
видимой области спектра, а также «бесцветных» для глаза растворов,
которые поглощают излучение в ультрафиолетовой (200—400 нм)
или ближней инфракрасной области спектра.
Спектрофотометры [46, с. 229] подразделяются на
регистрирующие и нерегистрирующие. В регистрирующих приборах
результаты всех
измерений автоматически
записываются на
специальном бланке,
имеющем вид сетки.
Нерегистрирующие
спектрофотометры обычно
Рис. 186. Оптическая схема
спектрофотометра СФ-16:
/ — источник света; 2 — зеркало-
конденсор; 3 — зеркало; 4—моно-
хроматор; 5 —призма; 6 —
объектив; 7 — выходная щель; 8 —
светофильтр; 9 —кювета;
/0—фотоэлемент.
'337
зключают источник излучения, монохроматор, приемник излучения
й отсчетное устройство. Количественные измерения пропускания
производятся сравнением сигналов приемника при попеременной
установке в световой пучок образца и эталона. При измерениях
поглощения светового потока жидкостями обычно пользуются
двумя идентичными кюветами, одна из которых заполняется
исследуемым раствором, а другая (пустая или наполненная растворителем)
играет роль эталона, пропускание которого принимают за 100%, а
оптическую плотность считают равной нулю.
К нерегистрирующим спектрофотометрам с кварцевой оптикой
относятся модели СФ-4, СФ-4А, СФ-16, обеспечивающие
возможность производить измерения, помимо видимой и ближней
инфракрасной, также в ультрафиолетовой области спектра.
Кнерегистрирующим спектрографам со стеклянной
оптикой относится модель СФ-5, используемая для измерений
только в видимой и ближней инфракрасной области спектра.
Нерегистрирующие спектрофотометры имеют одинаковую
оптическую схему, но несколько различаются электрическими схемами
и методикой измерений.
Принципиальная оптическая схема спектрофотометра СФ-16,
с пределами измерения оптических плотностей 0—2 и пропускания
100—0, 10—0, 100—90% представлена на рис. 186. Свет от
источника / попадает на зеркало-конденсор 2, которое направляет пучок
лучей на плоское зеркало 3, поворачивающее лучи на 90° и
направляющее их на входную щель монохроматора 4. Зеркальный
объектив 6 направляет параллельный пучок лучей на призму 5, которая
разлагает его в спектр и возвращает его обратно на объектив 6.
Луч, прошедший призму под углом близким к углу наименьшего
отклонения, попадает на выходную щель 7, расположенную под
входной. Поворачивая призму вокруг оси, можно получить на
выходе монохроматора лучи различных длин волн. Выходящий из
монохроматора пучок света проходит фильтр 8У кювету с
исследуемым раствором 9 и попадает на фотоэлемент 10. Фототок,
возникающий в фотоэлементе, передается на усилитель постоянного
тока. Усиленный ток попадает на милливольтметр.
Спектрофотометр СФ-16 относится к однолучевым приборам,
поэтому в процессе измерений на пути потока излучения
устанавливаются поочередно «нулевой» и испытуемый образцы.
Происходящие при этом изменения интенсивности излучения, падающега
на фотоэлемент, вызывают изменение напряжения в системе
усилителя, которое компенсируется путем изменения напряжения на
потенциометре, связанном с отсчетным устройством.
Включение прибора в сеть производится согласно прилагаемой
к нему инструкции, в которой также даются указания
относительно техники работы с ним.
338
глава 3i РЕФРАКТОМЕТРИЯ
При пересечении лучом света границы раздела двух
прозрачных сред направление луча изменяется, луч преломляется. Это
явление носит название рефракции, т. е. преломления света.
Угол а, образованный направлением падающего луча света с
нормалью (рис. 187), называется углом падения, а угол р,
образованный направлением преломленного луча с продолжением этой
нормали,— углом преломления. Согласно закону преломления света,
для оптически однородных сред отношение синусов углов падения
и преломления есть величина постоянная:
sin a
sin p
Показателем преломления п также называют
отношение скорости распространения света в воздухе к скорости
распространения света в испытуемом веществе (растворе). Это важная
константа, позволяющая уточнить химическую природу вещества,
определить степень его чистоты, а также определить
концентрацию растворов.
Рефрактометрия — это совокупность методов
физико-химического исследования жидкостей, минералов и растворов,
основанных на измерении их показателей преломления. Основными
достоинствами рефрактометрии являются быстрота измерений,
малый расход вещества и высокая точность (около 0,01 %).
Приборы, служащие для измерения показателя преломления,
называются рефрактометрами.
ПОКАЗАТЕЛЬ ПРЕЛОМЛЕНИЯ
Из сравнительно большого числа методов наибольшее
распространение получил метод определения показателя преломления
по предельному углу преломления или полного внутреннего
отражения. Когда луч света падает из среды с показателем
преломления п\ в среду с показателем преломления я2, то между углом
падения а и углом преломления (J существует зависимость:
—^-5в— или пх sin р = п2 sin a (l)
Sin р fl2
Пусть луч света падает на границу раздела двух сред I n II
(рис. 187), и пусть среда / оптически плотнее среды //. В этом
случае угол падения меньше угла преломления. Если угол падения
приближается к своему предельному значению 90°, то и угол
преломления может стать равным 90°. В этом случае луч света не
входит во вторую среду, а скользит по поверхности раздела фаз.
При дальнейшем увеличении угла падения луч отражается от
среды //. Это явление называется полным внутренним отражением,
ЗЭ9
Рис. 187. Полное внутреннее отражение света:
а —луч, падающий под углом а, преломляется под.
углом 0 и входит в среду /; b — луч, падающий под
предельным углом, преломляется под углом 90°;
с —луч, падающий под углом, большим, чем
предельный угол, не входит в среду /.
а угол падения, при котором оно
наступает — предельным углом
падения ф. В этом случае уравнение (1)
примет вид:
П\ sin 90Q = rt2 sin ф
Так как sin 90° = 1, то п\ = п2 sin ср. Если показатель
преломления одной среды п2 известен, то достаточно измерить
предельный угол ф, чтобы определить показатель преломления
анализируемой среды п\.
Важной деталью рефрактометров, основанных на определении
предельного угла, является измерительная призма из оптического
стекла с точно известным показателем преломления. Поэтому
каждый рефрактометр пригоден для измерения показателей
преломления только в определенном диапазоне их значений.
При рассматривании вышедших из измерительной призмы
лучей, близких к предельному, поле зрения трубы оказывается
разделенным на освещенную и темные части, граница между
которыми соответствует предельному лучу.
Показатель преломления зависит от длины волны излучения.
Лучи разных длин волн преломляются по-разному. Зависимость
показателя преломления света в веществе от длины волны света
называют дисперсией света или рефракционной
дисперсией.
В качестве меры дисперсии принята разность показателей
преломления для спектральных линий водорода С (656,3 нм) и
F (486,1 нм), охватывающих среднюю часть видимого спектра,
называемая средней дисперсией (nF — пс).
Зависимость показателя преломления от температуры
чВлияние температуры на показатель преломления
определяется двумя факторами: изменением числа частиц вещества в
единице объема и зависимостью поляризуемости от температуры.
Для большей части жидкостей показатель преломления
уменьшается примерно на 0,00015 при увеличении температуры на 1 °С.
Поэтому, чтобы можно было делать измерения с точностью до
четвертого десятичного знака, жидкие образцы необходимо термо-
статировать с точностью ±0,2 °С. Показателю преломления
придают два индекса: верхний, обозначающий температуру, и
нижний— длину волны. Например, nD20 означает, что измерение
выполнено при 20 °С и длине волны желтой линии D, спектра натрия
(589,3 нм).
340
Зависимость показателя преломления от концентрации
Во многих случаях показатель преломления бинарных
растворов линейно изменяется с составом раствора. Зависимость
показателя преломления растворов от концентрации
устанавливается эмпирически для каждого отдельного вещества, методом
построения калибровочной кривой. Готовят серию растворов
известных концентраций, измеряют их показатели преломления №
строят калибровочный график в координатах концентрация —
показатель преломления.
Концентрацию двухкомпонентных растворов можно также
вычислить, пользуясь формулой:
х~ F
где х — концентрация раствора, %(масс); п — показатель
преломления раствора; п0 — показатель преломления растворителя
при той же температуре; F—фактор, равный величине прироста
показателя преломления при увеличении концентрации на 1°/о
(устанавливается экспериментально).
Если разница в показателях преломления составляющих
раствор компонентов равна примерно 0,1, то точность определения:
концентрации может составить сотые доли процента.
РЕФРАКТОМЕТРЫ
Рефрактометры различаются диапазонами измерения и
источниками света. Если для освещения используется белый свет, в
состав прибора входят часто также призмы для компенсации
различия в длине волны. Благодаря этому можно определять
показатель преломления при длине волны желтой линии D спектра
натрия, проводя измерения при дневном свете или при свете лампы
накаливания.
Из многих типов рефрактометров, предназначаемых для
непосредственного измерения показателя преломления жидких и
твердых веществ по предельному углу преломления или полного
внутреннего отражения, их средней дисперсии и для определения
концентрации растворов, рассмотрим как основные два
отечественных рефрактометра типа Аббе — рефрактометр УРЛ и
рефрактометр ИРФ-22.
Универсальный лабораторный рефрактометр УРЛ.
Рефрактометры выпускают трех моделей, предназначенных:
модель I—для измерения показателя преломления в пределах
Мл = 1,2 ч-1,7, средней дисперсии и концентрации сухих веществ
по сахарозе в пределах 0 -f- 95%;
модель II — для измерения показателя преломления в пределах
nD = 1,65 -г- 2,1 и средней дисперсии;
модель III — для измерения процентного содержания белка в
пределах 0-1-33% и сухого обезжиренного остатка молока (СОМО)
341
fit— fe^s.
1 2 J ^
Рис. 1S8. Оптическая схема рефрактометра УРЛ:
/ — источник света; 2 и 3— конденсоры; 4 — призма осветительная; 5 — призма измерительная;
6 — призма прямого зрения; 7 — призма преломляющая; 8—объектив зрительной трубы; 9 —
перекрестие сетки; 10—шкала; // — окуляр зрительной трубы.
Рис. 189. Общий вид рефрактометра УРЛ:
1 — корпус; 2 —-окуляр; 3—дисперсионный компенсатор; 4—осветитель; 5—термометр;
б—верхняя призма; 7—выключатель.
в молочных продуктах с высоким (до 38%) содержанием сухого
вещества.
Предел допускаемой погрешности измерений по шкале
Пй =Ь 0,0001, по другим шкалам ±0,1%.
На рис. 188 приведена оптическая схема рефрактометра. Ис*
следуемый раствор помещают между плоскостями двух призм —
осветительной 4 и измерительной 5. От источника света /
конденсорами 2, 3 луч света направляется на входную грань
осветительной призмы, затем проходит тонкий слой исследуемого
вещества и плоскости измерительной призмы.
Лучи, предельные и преломленные под различными углами и
вышедшие затем из измерительной призмы через вторую ее грань,
пройдя через призмы дисперсионного компенсатора 6 и
преломляющую призму 7, фокусируются объективом 8 зрительной трубы в ее
поле зрения, образуя светлую и темную части поля, разделенные
прямой границей. Грань осветительной призмы матирована,
рассеянный на ней свет переходит в жидкость под всевозможными
углами (от 0 до 90°). Если показатель преломления жидкости
меньше показателя преломления материала призмы, то лучи
преломляются под углами от нуля до предельного. В окуляре
зрительной трубы // при этом наблюдается граница света и тени,
перекрестие сетки 9 и шкала 10. Положение этой границы
светотени зависит от величины предельного угла преломления ф,
который в свою очередь зависит от показателя преломления
испытуемой жидкости. Это дает возможность проградуировать шкалу
рефрактометра по показателям преломления или, соответственно,
по концентрации раствора,
342
Отсчет по шкале производится после устранения спектральной
окраски границы светотени в положении пересечения границей
светотени центра перекрытия сетки.
Конструктивно рефрактометр УРЛ (рис. 189) состоит из двух
основных частей: верхней — корпуса 1 и нижней — основания. К
корпусу крепятся камеры: верхняя и нижняя. Нижняя камера,
заключающая в себе измерительную призму, жестко закреплена на
корпусе; верхняя камера, заключающая в себе осветительную
призму 6, соединена шарниром с нижней и может поворачиваться
относительно нее.
Нижняя и верхняя камеры имеют окна, закрывающиеся
пробкой. На штуцере нижней камеры подвижно укреплен осветитель
4, свет от которого может быть направлен в одно из окон камеры.
Каждая камера оборудована двумя штуцерами для подвода и
вывода термостатирующей жидкости (воды). При помощи резиновых
трубок штуцеры соединяются с каналами, расположенными
внутри камер.
Для контроля температуры измеряемого вещества служит
термометр 5, укрепленный на штуцере нижней камеры с помощью-
накидной гайки. Со стороны передней крышки корпуса видна
шкала рефрактометра.
На оси прибора укреплены окуляр 2 для наблюдения границы
светотени и рукоятка для совмещения ее с перекрестием сетки;
лимб дисперсии 3 для устранения окрашенности наблюдаемой в-
окуляр границы светотени; механизм наведения (находится
внутри корпуса), который вместе с рукояткой может поворачиваться
вдоль шкалы.
На корпусе имеется закрытое пробкой отверстие для ввода
ключа и установки нулевой точки.
Внутри основания расположены понижающий трансформатор,,
предохранитель и весь электрический монтаж. На передней
стенке основания находится выключатель осветителя, а на боковой
стенке — шнур с вилкой для подвода питания от сети.
Подготовка к измерен и ю. Рефрактохметр устанавливают
в удобное для измерений положение, укрепляют термометр и
присоединяют камеру к термостатирующей установке так, чтобы
вода поступала в верхнюю часть камеры и выходила из штуцера,
в котором установлен термометр. Температуру в камере
поддерживают на уровне 20±0,1°С. При этой температуре проверяют-
и устанавливают правильность нулевой точки.
В моделях I и III нулевую точку устанавливают по
дистиллированной воде. Для этого верхнюю камеру открывают,
промывают дистиллированной водой или спиртом поверхности
измерительной и осветительной призм и насухо вытирают их салфеткой
или ватой. Затем оплавленным концом стеклянной палочки
наносят на плоскость измерительной призмы 1—2 капли
дистиллированной воды. Закрывают верхнюю камеру и, смещая осветитель,
направляют луч света в окно верхней камеры. Затем, перемещая
343.
Рис. 100. Поле зрения трубы рефрактометра ИРФ-22.
рукоятку с окуляром вдоль шкалы вверх и вниз,
вводят в поле зрения границу светотени.
Резкость границы светотени, штрихов шкалы
и перекрестия сетки по глазу наблюдателя
устанавливают вращением гайки окуляра; окрашенность
границы светотени устраняют вращением рукоятки дисперсионного
компенсатора.
Границу светотени подводят к центру перекрестия сетки. При
этом граница светотени должна находиться на делении 1,3329
шкалы nD и 0% шкалы сухих веществ. Если граница светотени
не совпадает со значениями, приведенными выше, то установку
нулевой точки производят следующим образом: центр перекрестия
устанавливают по шкале на деление нулевой точки, откручивают
гайку на корпусе прибора и. вращением ключа, прилагаемого к
прибору, подводят границу светотени к центру перекрестия сетки,
т. е. к нужному делению шкалы. Установку нулевой точки
проверяют 2—3 раза путем смещения рукояткой границы светотени и
вновь подводят ее к перекрестию сетки.
Проверка и установка нулевой точки в модели II производится
при помощи контрольной призмы, прилагаемой к прибору,
следующим образом; на большую полированную грань призмы наносят
2—3 капли иммерсионной жидкости (монобромнафталина; ло =
= 1,66) и этой гранью ее устанавливают на измерительную
призму камеры так, чтобы малая полированная грань была обращена
к осветителю и чтобы жидкость распространялась равномерно
по толщине слоя. После установки призмы верхнюю часть камеры
закрывают до упора на пружину. Затем перемещают осветитель
и одновременно наблюдают в окуляр за границей светотени к
центру перекрестия сетки; если при этом граница светотени
пройдет через деление шкалы, соответствующее показателю
преломления контрольной призмы, то нулевая точка установлена
правильно.
Измерение показателя преломления
прозрачных жидкостей и растворов производят аналогично измерению
показателя преломления дистиллированной воды при установке
нулевой точки. Показатель преломления невязких растворов
измеряют на моделях I и II как в проходящем свете (свет направляется
в окно верхней камеры), так и в отраженном (свет направляется
в окно нижней камеры). Направление света определяется опытным
путем.
Для работы в проходящем свете при исследовании темных
растворов модель I снабжена красным светофильтром, который
надевают на осветитель взамен диафрагмы. Для измерения вязких
темных растворов в модели I предусмотрена призма, на гипотенуз-
ную (входную) грань которой наносят исследуемый раствор. Этой
гранью призму устанавливают на измерительную призму прибора
344
таким образом, чтобы большая полированная грань была
обращена к осветителю. Верхнюю камеру закрывают, осветитель и окуляр
устанавливают в нужное положение и производят отсчет.
Измерение показателя преломления твердых
тел можно производить на моделях I и II. Методика измерения
аналогична измерению показателя преломления с помощью
контрольной призмы при установке нулевой точки. Исследуемый
образец должен быть оптически однородным. Конфигурация его
может быть любой при условии наличия двух взаимно
перпендикулярных полированных плоскостей, из которых одна должна быть
равна по своим размерам входной грани измерительной призмы;
толщина образцов должна быть не менее 0,15 мм.
Для веществ, показатель преломления которых не превышает
1,65, в качестве иммерсионной жидкости применяют монобромнаф-
талин (л^=1,66), а для имеющих высокие показатели
преломления— иммерсионную жидкость с/г^0 = 1,939, прилагаемую к
прибору. Эта иммерсионная жидкость ядовита, и обращаться с ней
следует осторожно.
Измерение содержания «белка» и «СОМО» в молоке, а также
средней дисперсии, производят по инструкции, прилагаемой к
прибору.
Рефрактометр ИРФ-22. Конструктивные особенности
рефрактометра ИРФ-22 делают производство измерений удобным и менее
утомительным.
Скрытая в корпусе прибора и вращающаяся вместе с призмен-
ным блоком стеклянная шкала подсвечивается зеркалом и
проектируется специальной оптической системой в поле зрения трубы.
Таким образом в поле зрения трубы видны одновременно
граничная линия, крест, деления шкалы и визирный штрих шкалы
(рис. 190). Целые, десятые, сотые и тысячные доли значения
показателя преломления отсчитывают по шкале, а десятичные — на
глаз, с точностью до 0,0002.
Пределы измерений, возможности применения, техника работы
и правила обращения для рефрактометра ИРФ-22 совершенно
Такие же, как у описанного выше рефрактометра УРЛ,
глава 32. ПОЛЯРИМЕТРИЯ
Поляриметрия — метод физико-химического анализа,
основанный на измерении вращения плоскости линейно поляризованного
света оптически активными веществами. Большая часть оптически
активных веществ — это органические соединения с ассимметрич-
ным атомом углерода, т. е. с таким, единицы сродства которого
насыщены четырьмя различными заместителями. Соединения
асимметричных атомов четырехвалентного олова, серы, селена, кремния
и пятивалентного азота также оптически активны.
ТЕРМИНЫ И ОБОЗНАЧЕНИЯ
Если через слой оптически активного вещества или его раствор
пропускать поляризованный луч света, то плоскость поляризации
вышедшего луча оказывается повернутой на некоторый угол,
называемый углом вращения плоскости поляризации
или углом вращения а.
В зависимости от природы вещества, вращение плоскости
поляризации может иметь различное направление и величину.
Если плоскость поляризации вращается вправо от наблюдателя,
к которому направлен свет, проходящий через оптически активное
вещество (по движению часовой стрелки), то эещество называют
правовращающим и перед названием ставят знак «Ч-» или D;
если же вращение плоскости поляризации происходит влево, то
вещество называют левовращающим и перед названием ставят
знак «—» или L.
Величина угла вращения зависит от природы оптически
активного вещества, толщины слоя вещества, через который проходит
свет, температуры и длины волны света. Угол вращения прямо
пропорционален толщине слоя. Влияние температуры связано, главным
образом, с изменением плотности растворов и, в большинстве
случаев, незначительно. Обычно определение оптического вращения
проводят при 20 °С и при длине волны максимально
соответствующей желтой линии D спектра натрия (589,3 нм).
Для сравнительной оценки способности различных веществ
вращать плоскость поляризации вычисляют так называемое
удельное вращение плоскости поляризации
монохроматического света, вызванное слоем вещества толщиной
в 1 дм, при температуре t и концентрации активного вещества
1 г/см3. Если определение удельного вращения проводят при 20 °С
и длине волны линии D спектра натрия, то его обозначают знаком
Кг
Для жидких индивидуальных веществ удельное вращение
определяют по формуле;
346
где а — измеренный угол вращения, градусы; I — толщина слоя
жидкости, дм; d — относительная плотность жидкости.
Для растворов удельное вращение определяют по формуле:
20_ OjJOO^
Wd
feme С—концентрация раствора, г растворенного активного веще*
ства на 100 см3 раствора.
Удельное вращение зависит от природы растворителя и концен-
трации раствора. При замене растворителя может измениться не
только величина угла вращения, но и его направление. Во многих
случаях удельное вращение постоянно лишь в определенном
интервале концентраций. Поэтому, приводя значение удельного
вращения, необходимо указывать растворитель и концентрацию
испытуемого раствора.
В интервале концентраций, в котором удельное вращение
постоянно, можно по углу вращения рассчитать концентрацию
вещества в растворе по формуле:
Полезно построить график зависимости концентрации активного
вещества от угла вращения плоскости поляризации стандартными
растворами различной концентрации. При помощи этого графика
в дальнейшем по полученному значению угла вращения определяют
концентрацию раствора.
Поляриметрические измерения имеют широкое практическое
применение. На основании определения знака и величины
вращения плоскости поляризации можно судить о химическом строении
и пространственной конфигурации оптически активных веществ,
делать порой выводы о механизме реакции.
Поляриметрический метод давно уже является основным
методом контроля в сахарной промышленности— по углу вращения
плоскости поляризации света определяют содержание сахара
в растворе.
ПОЛЯРИМЕТРЫ И САХАРИМЕТРЫ
Поляриметр круговой СМ. Устройство и принцип
действия. Поляриметр СМ схематически изображен на рис. 191. Свет
от источника 9 проходит последовательно через поляризационное
устройство 7, поляриметрическую трубку 6, анализатор с устрой^
ством 5, поворачивающим плоскость поляризованного луча, и по«
падает в зрительную трубу 8.
Поляризационное устройство состоит из осветительной линзы,
поляризатора и кварцевой пластинки, расположенной симметрично
относительно поляризатора. Поляризатор и кварцевая пластинка
находятся в определенном положении и крепятся жестко к оправе.
347
Рис. 191. Поляриметр круговой СМ:
1 — неподвижный лимб; 2—лупа; 3 — муфта;
4—нониусы; 5 — фрикцион для вращения
анализатора; 6—поляриметрическая трубка;
7—поляризатор; S—зрительная труба;
9—осветитель.
Основной рабочей частью
прибора является головка
анализатора, состоящая из
неподвижного лимба /,
вращающихся одновременно
фрикциона 5 и двух нониусов 4,
анализатора и зрительной трубы 8.
В трубу 8 при измерении вкладывают поляриметрическую
трубку 6 с исследуемым веществом (раствором). Во избежание
проникновения постороннего света, вырез в трубе закрывается
вращающейся шторкой. Зрительная трубка служит для наблюдения
тройного поля зрения и состоит из объектива и окуляра. Движением
муфты 3 окуляр устанавливают на резкость изображения тройного
поля зрения. В раковине окуляра находятся две лупы 2, которые
позволяют, не меняя положение головы, отсчитывать угол
вращения нониуса относительно шкалы лимба. На лимбе / нанесена
градусная шкала от 0 до 360°. Внутри лимба на подвижной втулке,
связанной с анализатором, нанесены два нониуса 4,
расположенные диаметрально. Нониусы имеют по 20 делений ценой 0,05°. При
больших углах вращения пользуются обоими нониусами и
результатом измерения считают среднее значение из полученных отсчетов
по первому и второму нониусам.
Поляриметрическая трубка 6 изготовляется из стекла. На
трубке имеется выпуклость, необходимая для сбора пузырьков
воздуха. На концах трубки укреплены металлические наконечники,
на которые навертываются крышки, прижимающие покровные
стекла. Между крышками и покровными стеклами имеются
резиновые прокладки, предохраняющие от образования натяжений
в стекле при завертывании крышек.
Осветитель 9 состоит из патрона, прикрепленного к кронштейну.
Для регулирования освещения патрон можно перемещать вдоль
кронштейна, а сам кронштейн перемещать по стойке вверх, вниз
и вокруг нее. Источником света служит матовая лампа
накаливания мощностью 25 Вт. Свет от лампочки проходит через специально
подобранные светофильтр и поляроиды, в результате чего максимум
спектрального распределения пучка соответствует желтой линии
натрия.
Проведение измерений. Пустую поляриметрическую
трубку вставляют в зрительную трубу, закрывают шторкой,
включают осветитель в сеть и наблюдают в окуляр освещенность
тройного поля. Если крайние поля неравномерно освещены, то
перемещением осветителя добиваются их равномерного освещения. После
установки осветителя определяют начальное положение анализа-
348
тора. Перемещением муфты вдоль оси добиваются резкого
изображения разделяющей линии тройного поля, наблюдаемого в окуляр.
Плавно вращая анализатор с помощью фрикциона, добиваются
равной затемненности изображения тройного поля (рис. 192),
видимого в окуляр, которое определяет начальное положение
анализатора. После установки на равную затемненность тройного поля
через лупу с помощью нониуса лимба производят отсчет.
Начальное положение не обязательно должно совпадать с нулевым
делением градусной шкалы лимба.
Установку начального положения анализатора и отсчет делений
градусной шкалы лимба следует повторить не менее 5 раз и
показанием прибора считать среднее значение от полученных отсчетов.
После этого наполняют поляриметрическую трубку исследуемым
раствором, для чего, отвинтив крышку с одного конца трубки,
наполняют ее в вертикальном положении прозрачным раствором
(мутный раствор фильтруют) до появления в верхнем конце трубки
выпуклого мениска. Затем сбоку надвигают покровное стекло,
накладывают резиновую прокладку и завинчивают крышку. При этом
необходимо следить за тем, чтобы в трубке не оставалось^
пузырьков воздуха. Наружные стороны покровных стекол должны быть
прозрачными и без следов жидкости, которая удаляется
фильтровальной бумагой.
Наполненную поляризационную трубку вставляют в зрительную
трубу и закрывают шторкой. Перемещением муфты 3
устанавливают на резкость разделяющие линии тройного поля. Затем, плавно
вращая анализатор с помощью фрикциона 5, добиваются
равномерной затемненности изображения тройного поля и производят
отсчет.
Порядок отсчета следующий: определяют, на сколько полных
градусов повернут нуль нониуса по отношению к лимбу, затем
определяют число делений от нуля нониуса до штриха нониуса,
совпадающего с градусным штрихом лимба, и умножают
полученное число делений на 0,05°. Полученный результат прибавляют
к первому. Разность отсчетов, соответствующих фотометрическому
равновесию поля с оптически активным веществом и без него, равна
углу вращения плоскости поляризации данного раствора.
Установку на равную затемненность тройного поля и отсчет
необходимо производить не менее 5 раз.
Если поляриметр имеет кожух для термостатирования
поляризационной трубки, то перед
началом испытания раствора через
кожух в течение 10 мин
пропускают воду из термостата при
температуре 20±0,1°С. В случае
отсутствия кожуха следует работать
Рис. 192, Начальное изложение нониуса (а)
после установки поля зрения на равную
затемненность {б) при введенной трубке для
жидкости, без раствора.
□
349
14 /5 16 /7
*: f— Щ ^
Л? 12 11 tO 9 6 7 6 5" 4 S g ;
Рис. 193. Оптическая схема сахариметра СУ-3:
/ — источник света; 2—матовое стекло или светофильтр; 3— конденсор; 4 — поляризатор; 5 и
б—защитные стекла; 7 и 9—кварцевые клинья; 8—стеклянный контрклин; /0—анализатор;
11 — объектив; 12—окуляр; /3—лупа; /4 —нониус; 15—шкала; 16—защитное стекло; 17—отра*
жательная призма.
в помещении при 20±3°С. При определении удельного вращения
индивидуальной жидкости ее выдерживают в термостате при
20 ± 0,1 °С в течение 30 мин.
Сахариметр универсальный СУ-3. В сахариметрах, в отличие от
поляриметров, для компенсации поворота плоскости
поляризованного луча исследуемым раствором используют специальные
кварцевые клинья, соответственно положению которых по шкале
отсчитывают значение угла вращения.
Шкала сахариметров градуирована в градусах Международной
сахарной шкалы (°S). Один градус сахарной шкалы соответствует
0,26 г сахарозы в 100 см3 раствора, если измерение производится в
поляриметрической трубке длиной 200 мм при 20 °С. Сто градусов
сахарной шкалы (100°S) соответствуют 34,62° угловым.
Устройство и принцип действия. Оптическая схема
универсального сахариметра СУ-3 изображена на рис. 193.
Свет от источника / проходит через матовое стекло 2,
предназначенное для рассеивания света (вместо него в оптическую систему
может быть введен светофильтр). Далее световой поток проходит
конденсорную линзу 5, попадает в поляризатор 4 и выходит из него
плоскополяризованным. За поляризатором стоят два защитных
стекла 5 и 6У между которыми установлена поляриметрическая
кювета с исследуемым раствором. Подвижный кварцевый клин 7,
стеклянный контрклин 8 и неподвижный кварцевый клин 9
образуют кварцевый компенсатор, который компенсирует вращение
плоскости поляризации. За анализатором 10 расположена
зрительная трубка, состоящая из двухлинзового объектива 11 и окуляра 12,
которая сфокусирована на выходную грань поляризатора 4. При
помощи зрительной трубки можно рассмотреть в увеличенном виде
линию раздела поля зрения прибора.
Свет от электролампы освещает также шкалу 15 и нониус 14
с помощью отражательной призмы 17 и защитного стекла 16,
рассеивающего свет. Цифры и деления нониуса и шкалы
рассматриваются в увеличенном виде под лупой 13, состоящей из двух линз.
По нулевому делению нониуса фиксируют значение шкалы,
соответствующее состоянию одинаковой освещенности обеих половин поля
зрения,
350
Источником света является электролампа А-10 (15 Вт, 12 В),
читаемая от сети переменного тока 220 В через понижающий
трансформатор 12 В, встроенный в основание прибора.
Проведение измерений. Сахариметр должен быть
установлен на столе в темном лабораторном помещении со стенами,
окрашенными в черный цвет, что повышает чувствительность глаза
наблюдателя.
Перед началом измерений прибор необходимо заземлить при
помощи винта заземления, включить в сеть и установить на нуль.
Установка на нуль производится при отсутствии в камере
поляриметрической кюветы. Вращая рукоятку кремальерной передачи,
устанавливают однородность освещения обеих половин поля зрения.
При этом нулевые деления шкалы и нониуса должны совпадать
(рис. 194). В противном случае перемещают нониус до совмещения
его нулевого деления с нулевым делением шкалы. После проверки
нулевой точки шкалы можно непосредственно приступить к
измерениям.
В камеру прибора вкладывают поляриметрическую кювету с
испытуемым раствором. При этом изменяется однородность
освещения обеих половин поля зрения. Вращая рукоятку кремальерной
передачи, уравнивают освещенность обеих половин поля зрения и
производят отсчет показаний с точностью до 0,1 деления шкалы
при помощи нониуса. Отсчет показаний повторяют 5 раз.
Результатом считают среднее арифметическое пяти измерений.
Отсчет показаний при помощи нониуса поясняется рисунками.
На рис. 195 слева показано положение шкалы и нониуса, соответствующее
отсчету +11,8 °S (нуль нониуса расположен правее нуля шкалы на 11 полных
делений, и в правой части нониуса с одним из делений шкалы совмещается
восьмое деление нониуса). На том же рисунке справа показано положение
шкалы и нониуса, соответствующее отсчету —3,2 °S (нуль нониуса расположен
левее нуля шкалы на три полных деления шкалы, и в левой части нониуса с
одним из делений шкалы совмещается второе деление нониуса).
Для определения массового процента сахарозы в исследуемом
растворе следует отсчитанные по шкале сахариметра градусы
сахарной шкалы умножить на переводной коэффициент 0,260 и
разделить на плотность исследуемого раствора.
Сахариметр СУ-3 комплектуется поляриметрическими трубками
длиной 100, 200 и 300 мм. Трубки длиной 100 мм применяют для
окрашенных растворов и при расчетах результатов анализа
значение, полученное по шкале прибора, увеличивают в два раза. Трубки
^...j.iiji.-ijnli|i.i.|.i.. ....,...l,iAl,.l».|i.^l„I.,ltlM<<l.1i.,«jgwi г i,,,,],,,,!,,,.!,,,,! % l....l.,J„.,l,.,,l
stn \1П П Л. «л/ sis-» ' „„ ' .i. < » ' Unit к .1 111 II III i ill nil lllll I 1 II lllllll lllllr
1.0 , Q . 10 10 0 10
11 r 111 iiiiln 111 mi il
40
иикрла.
2o' g0 m » /li,;,,5,,"i,,,i,,,:i:,>!i ]»"r'"i,,,,i"" ..ит^
"V* 10 20 10 -0+ 10
Рис. 194. Шкала и нониус сахариметра.
Рис. 195. Положение шкалы и нониуса сахариметра при 4-11.8 °S (слева) и —3,2 °S (справа).
351
длиной 400 мм применяют при исследовании растворов малой
концентрации и при расчете значения по шкале прибора уменьшают
в два раза.
К прибору для проверки правильности показаний прилагается
контрольная трубка с двумя нормальными кварцевыми
пластинками на -40 и +100°S.
Точность показаний сахариметра с помощью контрольной трубки
необходимо проверять при установившейся температуре 20 °С.
В случае проверки прибора при температуре, отличающейся от
20 °С, производят пересчет по формуле:
а/ = а20 [1 + 0,000143 (/ - 20)]
где <Х2о и at — вращательная способность кварцевой пластинки
при 20 °С и температуре измерения t, соответственно, °S.
Расхождение в показаниях проверяемого прибора и контрольной
трубки при выверенной нулевой точке является погрешностью
данного прибора.
УХОД ЗА ПРИБОРАМИ И ИХ ХРАНЕНИЕ
После окончания работы приборы и принадлежности к ним
следует тщательно протереть мягкой неворсистой салфеткой; на
приборы надеть чехол, а принадлежности уложить в футляр.
Хранить приборы следует в сухом и чистом помещении при
температуре воздуха 10—35 °С и относительной влажности 30—80%.
В воздухе помещения не должно быть вредных примесей.
Не допускается разбирать приборы и чистить оптические
детали, расположенные внутри приборов.
Защитные стекла чистят при помощи деревянной палочки с
намотанным на нее тонким слоем гигроскопической ваты, соблюдая
осторожность, чтобы не поцарапать полированные поверхности
стекол.
г л aba зз ОПРЕДЕЛЕНИЕ рН. ПОТЕНЦИОМЕТРИЯ
ПОНЯТИЕ О ВОДОРОДНОМ ПОКАЗАТЕЛЕ
Вода, являясь очень слабым электролитом, плохо проводит
электрический ток, но все же обладает измеримой
электропроводностью, которая является результатом ее, пусть незначительной,
диссоциации:
Н20 ^zt Н+ + ОН-
Ионы водорода с молекулами воды образуют гидратированные
протоны НзО+, называемые ионами гидроксония. Однако ради
удобства обычно обозначают ионы водорода в виде Н+.
Вода — типично амфотерное соединение: она образует в
равных количествах ионы водорода, являющиеся носителями
кислотных свойств, и ионы гидроксила — носители щелочных (основных)
свойств. Применяя закон действия масс, константу равновесия
(диссоциации) можно выразить уравнением:
[Н+ПОН-1
л [Н20]
где [Н+] и [ОН-]—концентрация ионов водорода и гидроксила,
моль/л.
Поскольку вода в очень малой степени диссоциирует на ионы,
то концентрацию молекул воды [Н20] в момент установления
динамического равновесия можно считать постоянной величиной,
равной начальной концентрации 1000/18,015 = 55,5 моль/л. В
разбавленных водных растворах концентрацию воды можно считать такой
же. В этом случае константа электролитической диссоциации воды
примет вид:
55,5 К — [Н+]- [ОН"] *=Kw
Величина Kw называется ионным произведением воды
(константой автопротолиза воды) и является величиной
постоянной в воде, разбавленных водных растворах кислот, щелочей,
солей и других соединений при постоянной температуре. При 25 °С
Kw = 1,008- Ю-14. В идеально чистой воде и в строго нейтральных
разбавленных растворах электролитов концентрация водородных
ионов равна концентрации гидр.оксильных ионов: [Н+] == [ОН-].
Отсюда следует, что в нейтральной среде при 25 °С
[Н+] = [ОН-] = ^/К^ = дЛ,008.10-14 =, 1,004. 10-7 моль/л
В кислой среде [Н+]>[ОН~], а в щелочной среде, напротив,
[ОН-]>[Н+].
Для выражения концентрации водородных ионов была
введена величина рН, численно равная отрицательному десятичному
логарифму концентрации ионов водорода в единицах нормальности:
pH--1g[H+J
12 Зак. 1237 353
Величина рН получила название водородного
показателя.
Однако теория сильных электролитов вводит некоторую
поправку в определение рН. Известно, что в водных растворах
сильные электролиты полностью диссоциируют, но под влиянием
электростатических сил притяжения и отталкивания происходит
интенсивное влияние противоположно заряженных ионов. Это взаимное
влияние тем сильнее, чем выше концентрация ионов. В этих
условиях реакция среды определяется эффективной концентрацией
ионов, так называемой активностью ионов.
Активность ионов водорода аи+ — это величина
концентрации ионов Н+, уменьшенная под влиянием
взаимодействия этих ионов в растворе:
где fa — коэффициент активности.
Заменяя концентрацию Н+-ионов величиной активности,
получим:
рН = —lgaH+ и рОН = —lgaOH-
Повышение активности ионов водорода приводит
соответственно к понижению активности ионов тидроксила, так как
произведение этих активностей должно быть постоянной величиной при
данной температуре. Чтобы упростить вычисления, часто не
учитывают коэффициенты активности. Для разбавленных растворов
вводимая при этом погрешность не очень велика.
При определении рН необходимо учитывать температуру
раствора, так как рН нейтральной точки зависит от температуры.
Например, при О °С для нейтральной точки pH=-lg л/1,139 • 10~15=*
= 7,472, а при 50 °С рН = - lg VM74 . 1(Г14 = 6,631.
КОЛОРИМЕТРИЧЕСКОЕ ОПРЕДЕЛЕНИЕ рН [64, с. 24]
Колориметрический метод определения рН растворов основан
на свойстве кислотно-основных индикаторов изменять свою окраску
в зависимости от активности ионов водорода в определенном
интервале рН. Колориметрическое определение рН производят путем
сравнения окраски испытуемого раствора и ряда буферных
растворов (растворов со строго определенной величиной рН)
при одном и том же содержании индикатора. Но вначале
определяют приближенную величину рН испытуемого раствора с
помощью индикаторных бумажек, или последовательного ряда
индивидуальных или универсальных индикаторов.
Индикаторные бумаги
Индикаторные бумаги представляют собой полоски хромато-»
графической бумаги, пропитанные индивидуальными или смешан*
354
ТАБЛИЦА 9. Индикаторные бумаги
Наименование бумаги
Метиловая оранжевая
Конго красная
Лакмоидная синяя
Лакмусовая красная
Лакмусовая нейтральная
Лакмусовая синяя
Куркумовая
Фенолфталеиновая
Интервал рН
перехода
окраски
3,1-4,4
3,0—5,2
4,0—6,4
6,0—8,0
6,0—8,0
6,0—8,0
8,0-^-10,2
8,0-9,6
Окраска индикатора
в кислой среде
Красная
Синяя
»
Бледно-розовая
Красная
»
Желтая
Белая
в щелочной среде
Желтая
Красная
»
Синяя
»
»
Красно-коричневая
Красная
ными кислотно-основными индикаторами («универсальные») *
Определять рН при помощи индикаторной бумаги возможно
только в растворах с не очень высокой концентрацией солей и в
отсутствие сильных окислителей.
Индикаторная бумага выпускается в виде книжечек по 100
полосок; она очень чувствительна к свету, влажности и действию
паров веществ кислотного или основного характера. При
правильном хранении индикаторные бумаги не теряют химических свойств
в течение 2—4 лет.
Для определения рН полоску реактивной бумаги на несколько
секунд погружают в испытуемый раствор, либо наносят
испытуемый раствор на бумагу стеклянной палочкой, либо прикладывают
полоску бумаги к влажной пробе.
В табл. 9 приведены данные об отечественных индикаторных
бумагах.
Более широкое распространение получили универсальные
индикаторные бумаги. Они представляют собой полоски хромато-
графической бумаги, пропитанные смесью кислотно-основных
индикаторов с разными интервалами перехода окраски. При
изменении значения рН в широких пределах происходят заметные на
глаз изменения окраски бумажек.
Универсальная индикаторная бумага «Рифан» предназначена
для определения рН воды и водных растворов в пределах рН =
= 1,0-4-10. Она представляет собой листочки хроматографической
бумаги размером 10X85 мм, на которые нанесены цветная
полоса индикатора и цветные полосы, служащие шкалой для сравнения.
Для определения рН листочек бумаги погружают в испытуемый
раствор так, чтобы все цветные полосы были одинаково смочены
жидкостью. Затем его извлекают и немедленно сравнивают цвет
индикаторной полосы (средняя без цифр) с цветной шкалой на
полоске бумаги, имеющей цифровые обозначения рН.
Тождественность окраски индикаторной полосы с маркированной полосой
цветной шкалы указывает величину рН.
Индикаторная бумага «Рифан» позволяет с достаточной
точностью оценить значения рН водных растворов и суспензий, так
12*, 355
ТАБЛИЦА 10. Индивидуальные индикаторы
Индикатор
Метиловый оранжевый
Метиловый красный
Лакмус красный
Феноловый красный
Фенолфталеин
Растворитель
Вода
»
Спирт
Концентрация,
% (масс.)
0.1
0.2
0.2
0.1
0.1
Интервал рН
перехода окраски
3,1—4,4
4,2-6,2
6,0-8,0
6,8—8,0
8,0-9,6
как отдельные ступени шкалы выражают разницу в основном
значении 0,3—0,4 единиц рН.
Индикаторная бумага «Фан» (Phan), поставляемая
Чехословакией, позволяет еще с большей точностью определять величины
рН в интервале L—14.
Растворы индикаторов
Для ориентировочной оценки значения рН можно также
непосредственно применить индивидуальные или смешанные
(универсальные) индикаторы.
К отдельным пробам испытуемого раствора (5—10 мл)
прибавляют по 1—2 капли каждого из индивидуальных
индикаторов, указанных в табл. 10.
По изменению окраски судят о приближенном значении рН.
Приводим состав и способ приготовления одного из
универсальных индикаторов. В 50 мл 96 % этилового спирта
растворяют 0,01 г фенолфталеина, 0,02 г метилового красного, 0,03 г
метилового желтого, 0,04 г бромтимолового синего, 0,05 г
тимолового синего. К полученному раствору добавляют 0,1 н. раствор
NaOH до появления желтой окраски.
К 1 мл испытуемого раствора добавляют 1 каплю
универсального индикатора и наблюдают изменение окраски: красная —
рН = 2; оранжевая — рН =4,0; желтая — рН = 6,0; зеленая —
рН =8; синяя — рН = 10.
Буферные растворы
Буфер — это соединение или смесь веществ, введение которых
в раствор способствует поддержанию постоянного значения рН.
Буферный раствор препятствует изменению рН, которое может
происходить при разбавлении раствора или добавлении к нему
сильной кислоты или сильного основания.
Буферные растворы, однако, сохраняют постоянство рН лишь
до прибавления некоторого определенного количества кислоты (или
щелочи). Если концентрация добавленной кислоты (щелочи)
превышает предел буферной емкости данного раствора, то рН
резко изменяется»
356
За единицу буферной емкости условно принимают емкость
такого раствора, для изменения рН которого на единицу требуется
введение 1 эквивалента сильной кислоты (щелочи) на 1 литр.
Буферная емкость зависит от концентрации компонентов
буферного раствора. Чем раствор концентрированнее, тем больше
его буферная емкость. Разбавление буферного раствора не влияет
заметно на изменение рН, но значительно отражается на
буферной емкости.
Буферные растворы используются в качестве рН-стандартов
для калибровки рН-метров, для колориметрического определения
рН и других целей.
Изготовляют буферные растворы из реактивов, выпускаемых
специально для рН-метрии. В соответствии с ГОСТ 10170—62,
шкала рН основана на воспроизводимых значениях рН пяти
образцовых (реперных) буферных растворов (табл.
11). Они выпускаются в виде стандарт-титров (ампул),
рассчитанных на приготовление 1000 мл буферного раствора
каждого наименования.
Кроме пяти образцовых буферных растворов, можно
использовать также 0,1 н. раствор НС1, значение рН которого равно 1,10
в диапазоне температур 0—50 °С.
При отсутствии стандарт-титров для рН-метрии можно
готовить образцовые буферные растворы из реактивов квалификации
х. ч. или ч.д.а. следующим образом.
1. 0,05 М раствор тетраоксалата калия, содержащий
12,7 ±0,02 г КНС204-Н2С204-2Н20 в 1 л при 20 °С. Тетраоксалат
калия перекристаллизовывают дважды из воды и сушат на
воздухе при температуре не выше 58 °С.
2. Насыщенный при 25 °С раствор гидротартрата калия. Тонко-
измельченный КНС4Н4О6 встряхивают некоторое время с
дистиллированной водой при 25 °С. Нерастворившуюся часть кристаллов
отфильтровывают.
ТАБЛИЦА и. Значения рН образцовых буферных растворов
в зависимости от температуры
Температура, °С
10
15
20
25
30
40
50
60
0,05 М р-р
тетраоксалата калия
, 1,67
1,67
1,68
1,68
1,69
' 1,70
1,71
1,73
Насыщенный
при 25 °С р-р
гидротартрата
калия
3,56
3,55
3,54
3,55
3,57
0,05 М р-р
гидрофталата
калия
4,00
4,00
4,00
4,01
4,01
4,03
4,06
4,10
0,025 М р-р
дигидрофос-
фата +0,025 М
гидрофосфата
натрия
6,92
6,90
6,88
6,86
6,84
6,84
6,83
6,84
0,01 М р-р
тетрабората
натрия
9,33
9,27
9,22
9,18
9,14
9,07
9,01
8,96
357
ТАБЛИЦА 12. Стандартные серии буферных растворов
А. Область рН= 1,2 -т- 2,2; 50 мл 0,2 М КС1 + а: мл 0,2 М НС1
рн
X
1,2
64,6
1.4
41,5
1 I»6
26,3
1 *>8
16,6
1 2,0
10,6
2,2
6,7
Б. Область рН = 2,2 -т- 3,8; 50 мл 0,2 М КНСвН404 + х мл 0,2 M HC1
рн
X
2,2
39,6
2,4
, 32,95
2,6
26,42
2,8
20,32
3,2
14,70
3,4
9,9
1 3,6
5,97
3,8
2,62
В. Область рН = 4,0 ~ 6,2; 50 мл 0,2 М КНС8Н404 + х мл 0,2 М КОН
X
4,0
0,4
4,2
3,7
4,4
7,5
4,6
12,15
4,8
17,70
5,0
23,25
6,2
29,95
5,4
36,45
5,6
39,85
5,8
43,0
6,0
43,5
Г. Область рН = 5,8 -т- 8,0; 50 мл 0,2 М Na2HP04 -f х мл 0,2 М КОН
рн
X
5,8
3,72
6,0
5,7
6,2
8,6
6,4 1
12,6
6,6
17,8
6,8
23,65
7,0
29,63
7,2 1
35,0
7,4
39,5
7,6
42,8
7,8
45,2
8,0
46,8
Д. Область рН = 7,8+- 10,0; 50 мл 0,2 М Na2B407 • 10 Н20 + х мл 0,2 М КОН
рН|
7,8
2,61
8,0
3,97
8,2
5,90
8,4
8,5
8,6
12,0
8,8
16,3
9,0
21,3
9,4
26,7
9,6
32,0
9,8
40,7
10,0 1
43,9
3. 0,05 М раствор гидрофталата калия, содержащий 10,21 г
КНС8Н404 в 1 л; 0,2 М раствор — 40,846 г КНС8Н404 в I л при
20 °С. Гвдрофталат калия перекристаллизовывают дважды из воды
и сушат при 110±5°С.
4. 0,025 М раствор дигидрофосфата калия и 0,025 М раствор
гидрофосфата натрия, содержащий 3,40±0,01 г КН2Р04 и
3,65±0,01 г Na2HP04 в 1 л при 20 °С. Оба реактива дважды
перекристаллизовывают из воды и сушат до постоянной массы:
Na2HP04 при 130, а КН2Р04 при 110°С.
5. 0,01 М раствор тетрабората натрия, содержащий 3,81±0,01г
Na2B407-10Н2О в 1 л при 20°С. Тетраборат натрия
перекристаллизовывают дважды из воды при температуре не выше 55 °С.
Соль высушивают до постоянной массы в эксикаторе над смесью
влажного NaCl и сахара.
Стандартные серии буферных растворов в более узком
интервале рН = 1,2ч- 10 с шагом через 0,2 единицы рН готовят путем
смешения 0,2 М образцовых буферных растворов с 0,2 М
растворами НС1 или КОН в соотношениях, приведенных в табл. 12.
Образцовые буферные растворы и стандартные серии буферных
растворов должны храниться в склянках из химически стойкого
стекла или полиэтилена. В тартратных растворах может вырасти
плесень, и ее рост сопровождается увеличением рН на 0,01—0,1
единицы рН. Этот раствор следует менять или добавлять к нему
тимол (6—8 кристаллов на 1 л), предохраняющий раствор от за-
358
плесневения на срок до двух месяцев. Остальные буферные
растворы устойчивы в течение месяца. Хранение буферных растворов
в холодильнике значительно удлиняет срок их годности.
Методы и средства колориметрического определения рН
Колориметрическое определение рН водных растворов обычно
основывается на предположении, что рН двух одинаково
окрашенных индикатором растворов равны, если активность ионов
водорода и температура обоих растворов одинаковы. Буферные
стандартные растворы должны иметь приблизительно ту же
ионную силу, что и исследуемые растворы.
При измерении рН в неводных средах и смешанных
растворителях (водно-спиртовые, водно-ацетоновые" растворы) следует
иметь в виду, что измерения, сделанные в сравнении с водными
стандартными буферными растворами, имеют только
относительное значение в связи с изменением интервалов перехода окраски
индикаторов и изменением рН нейтральной точки.
Для определения рН воды и разбавленных водных растворов
колориметрическим методом выпускаются специальные наборы и
аппараты, в частности аппараты Михаэлиса: ММ-1 для
проб объемом 0,5 мл; ММ-2 — для проб объемом 5 мл.
Индикаторами служат: а-динитрофенол (рН = 2,8-f-4,4); у-динитрофенол
(рН = 4,0 -т- 5,4); п-динитрофенол (рН = 5,4 -f- 7,0) и ж-динитрофе-
нол (рН = 6,8 -г- 8,4), а также универсальный индикатор для
предварительного определения рН в пределах 3—8. К универсальному
индикатору прилагается цветная шкала. Каждому индикатору
соответствует серия окрашенных буферных растворов, причем
рН каждого раствора отличается от предыдущего на 0,2
единицы рН.
При определении рН слабоокрашенных водных растворов
используется принцип сложения окраски раствора сравнения с
собственной окраской испытуемого раствора. Для этой цели в
комплект прибора включен компаратор (рис. 196). Он представляет
собой деревянный брусок, в котором сделаны три пары
параллельных углублений, достаточно широких, чтобы вставить туда
пробирки. Перпендикулярно этим углублениям проходят три канала
меньшего диаметра, которые пересекают насквозь стенки каждой
пары широких углублений. Через эти каналы можно
наблюдать пробирки с содержимым в проходящем
свете. Стенки углублений и каналов окрашены в
неотражающий черный цвет. В одно из центральных
углублений помещают пробирку с испытуемым
раствором плюс индикатор, а в другое — с
дистиллированной водой. В передние боковые углубления
помещают пробирки с буферными растворами
Рис. 196, Компаратор.
359
плюс индикатор, а позади них — пробирки, наполненные
только окрашенным или мутным испытуемым раствором.
Сравнение окрасок производят на фоне экрана из молочного стекла.
Если окраска испытуемого раствора совпадает с окраской какого-
либо эталона, то значение рН испытуемого раствора то же самое,
что и эталона (буферного раствора). Если нет эталона с
совпадающей окраской, то подбирают два эталона, окрашенных несколько
сильнее и слабее, чем испытуемый раствор. Величину рН находят
как среднее между этими двумя эталонами..
ПОТЕНЦИОМЕТРИЯ [1]
Потенциометрия — электрохимический метод анализа,
заключающийся в измерении электродного потенциала и нахождении
зависимости между его величиной и концентрацией (точнее,
активностью) потенциалоопределяющего компонента в растворе.
Потенциал всякого электрода можно измерить только по отношению к
какому-либо постоянному потенциалу другого электрода.
Поэтому необходимо составить гальванический элемент
(электролитическую ячейку) из двух полуэлементов с электродами,
между которыми возникает разность потенциалов. Потенциал
одного электрода (электрод сравнения,
вспомогательный электрод) постоянен и заранее известен. Потенциал
другого электрода (индикаторный, измерительный
электрод) зависит от активности (концентрации) ионов водорода.
Электродвижущая сила (э.д.с), возникающая между
электродами, равна алгебраической разности между их потенциалами.
Электроды для потенциометрии
В качестве стандартного электрода сравнения повсеместно
принят нормальный водородный электрод, потенциал
которого условно приравнен нулю при любой температуре и при
давлении газообразного водорода 1013 гПа (760 мм рт. ст.).
Однако из-за трудоемкости приготовления водородного электрода и
необходимости соблюдать постоянство концентрации ионов
водорода в процессе измерения рН на практике используют другие
электроды сравнения.
Наиболее распространенными электродами сравнения
являются каломельный и проточный хлорсеребряный электроды.
Потенциал каломельного электрода, который состоит
из небольшого количества ртути, покрытой тонким слоем
каломели Hg2Cl2 и погруженной в раствор хлорида калия (рис. 197),
зависит только от концентрации хлорида калия и температуры.
Имеются три различных каломельных электрода:
насыщенный, в котором водный раствор насыщен КС1; нормальный —
заполненный 1 н. раствором КО, и децинормальный — заполненный
0,1 н. раствором КС1. При 20 °С потенциалы этих трех электродов
по отношению к нормальному водородному электроду равны
соответственно 0,2444, 0,2816 и 0,3340 В.
360
Рис. 107. Каломельный электрод: s~*^
/ — контакт; 2— подвижное резиновое кольцо, закрываю- f/^\\
щее отверстие для заполнения; 3 — ртуть и паста Hg2CIj; || ||
4 — насыщенный раствор КС1; 5—асбестовое волокно. U L*
Рис. 198. Стеклянный электрод: 1§7
/ — контакт; 2—0,1 М раствор HCI; 3—Ag + AgCl; 4—тон- \J~~4
кая стеклянная мембрана.
Хлорсеребряный электрод сравнения аналогичен
каломельному, только вместо металлической ртути в нем
используется металлическое серебро, а вместо каломели — хлорид серебра.
Если используется насыщенный раствор КС1, то потенциал хлор-
серебряного электрода при 25 °С равен 0,1988 В. Потенциал
отечественных хлорсеребряных электродов сравнения ЭВЛ-1МЗ,
ЭВЛ-1М1, ЭХСВ-1 [46, с. 280] относительно нормального
водородного электрода при 20 °С равен 0,201 ± 0,003 В.
Электрод сравнения соединяют с измерительными электродами
через электролитический ключ — трубку, заполненную
насыщенным раствором КС1 и заканчивающуюся пористой
перегородкой.
Возникшую между электродами э.д.с. можно измерить
различными способами. Наибольшее распространение получил метод
непосредственного измерения с помощью рН-метров (прямая по-
тенциометрия).
В качестве индикаторных электродов используют различные
электроды, потенциал которых является функцией активности
ионов водорода. Практически чаще всего используют стеклянные
мембранные электроды.
На рис. 198 показан один из таких электродов. Тонкостенная
колба из специального стекла, обладающего высокой
чувствительностью к активности ионов водорода в растворе, прикреплена к
нижней части стеклянной трубки. Внутри колбы находится 0,1 М
раствор соляной кислоты. В него погружена серебряная
проволока, покрытая слоем AgCl. Проволока проходит через трубку,
заполненную смолой; ее верхний конец обеспечивает контакт с
внешней цепью.
Потенциал стеклянного электрода представляет собой разность
потенциалов на обеих сторонах стеклянной мембраны. Он зависит
только от активности ионов водорода в растворе и в широкой
области рН не зависит от присутствия в растворе других ионов.
361
/Г\
\т\
Преимущества стеклянного электрода таковы: наблюдаемые
э.д.с, измеренные по отношению к электроду сравнения, не
подвержены влиянию присутствующих окислителей или
восстановителей; электрод можно применять в окрашенных, мутных или
коллоидных растворах; время установления равновесного потенциала
измеряется долей минуты; точные результаты могут быть
получены при пользовании им в незабуференных растворах, так как
ток, проходящий через цепь, крайне незначителен; измерение рН.
можно проводить в небольшом объеме жидкости.
Однако стеклянные электроды очень хрупки, чувствительны к
щелочным растворам; кроме того, из-за неодинаковых свойств
внутренней и внешней поверхности стекла мембраны возникают
также потенциалы, отличающиеся друг от друга. Эта разность
потенциалов носит название- потенциала асимметрии.
В случае тонкостенных электродов из мягкого стекла потенциал
асимметрии может составлять несколько милливольт, а в случае
толстостенных электродов из тугоплавкого стекла — десятки
милливольт. Для уменьшения потенциала асимметрии стеклянный
электрод вымачивают и хранят в воде или в 0,1 н. растворе НС1.
Стеклянные электроды, обладающие высокой избирательностью
(селективностью) по отношению к каким-либо ионам, называют
электродами с электродной функцией по данному виду
ионов [19]. Изменяя состав стекла, из которого изготовляется
нижняя часть электрода, получают ионоселективные
солевые мембранные индикаторные электроды,
избирательно реагирующие на изменение активности катионов (К+,
Na+, Ag+, NHJ и др.).
При изготовлении анионоселективных электродов в твердый
материал, который запрессовывают в нижнюю часть стеклянной
трубки, вводят нерастворимое соединение, содержащее ионы,
которые нужно определять (F-, С1~, Br~, I-, S2-).
Приборы для прямой потенциометрии [41; 46, с. 278; 64, с. 47]
Методы прямой потенциометрии основаны на определении
активности (концентрации) отдельных веществ-участников элек^
тродной реакции по экспериментально измеренной э.д.с. цепи или
потенциалу индикаторного (измерительного) электрода.
Наиболее широкое применение метод прямой потенциометрии
находит для определения активности ионов водорода (рН
растворов), а также различных катионов и анионов с использованием
ионоселективных мембранных электродов.
При потенциометрическом определении рН используются
приборы (рН-метры, ионометры), устройство которых позволяет
непосредственно получать величины рН и рХ, где рХ —
отрицательный десятичный логарифм активности (концентрации)
анализируемого катиона или аниона. Эти приборы позволяют также измерять
окислительно-восстановительные потенциалы растворов.
362
Для измерения окислительно-восстановительных потенциалов,
выражаемых в вольтах или милливольтах, применяют платиновый
индикаторный электрод и хлорсеребряный электрод сравнения.
Для измерения активности (концентрации) одно- и двухвалентных
катионов и анионов в растворах используют электродную систему
из ионоселективных мембранных измерительных электродов, хлор-
серебряных электродов сравнения и измерительного
преобразователя.
Промышленность выпускает рН-метры двух основных типов:
нулевого (потенциометры) и с прямым отсчетом (ламповые
вольтметры). В последние годы рН-метры со стеклянным измерительным
электродом и непосредственным отсчетом результатов измерений
получили наибольшее распространение.
Стеклянные электроды имеют очень высокое электрическое
сопротивление. По этой причине измерение рН и другие потенцио-
метрические измерения с помощью стеклянных электродов
проводятся с использованием электронных приборов, специально
предназначенных для таких высоких сопротивлений. Потенциал
стеклянного электрода накладывается на сетку электрометрической
электронной лампы с высоким сопротивлением, а электрод
сравнения соединяется с катодом этой лампы. Увеличение силы тока
в лампе регистрируется измерительным прибором, откалиброван-.
ным в единицах рН.
Все рН-метры требуют периодической настройки. Для этого
измерительный электрод и электрод сравнения погружают в
буферный раствор с точно известным рН и регулируют прибор таким
образом, чтобы стрелка измерительного прибора показывала точное
значение рН буфера. В первые несколько дней эксплуатации
прибора или нового стеклянного электрода настройку рН-метра
следует производить по буферным или контрольным растворам
каждый день. При последующей работе с прибором настройку можно
производить один раз в три дня.
При температуре исследуемого раствора 0—40 °С настройку
прибора производят по буферным растворам с 20 и 50 °С, а при
15—100° температура буферного раствора должна быть 20 и 80 °С.
Значения рН при этом должны составлять:
Исследуемый раствор 1—4 4—9 9—14
Буферный раствор 1,68 6,88 9,22
Электроды перед погружением в буферный или контрольный
раствор необходимо тщательно промыть дистиллированной водой,
а остатки воды с них удалить фильтровальной бумагой.
Измерения, проводимые с помощью ионоселективных
электродов, требуют построения калибровочного графика, выражающего
зависимость измеряемого потенциала от активности анализируемых
ионов в стандартных условиях.
Если концентрация посторонних ионов в исследуемом растворе
мала, на графике можно откладывать концентрацию выбранных
ионов, а не их активность.
363
Рис. 199. Схема действия
рН-метра:
Слева измерительный электрод»
справа — вспомогательный.
Электродвижущая
сила электродных си*
стем, применяемых для
измерений рН, зависит
от температуры.
Современные рН-метры
снабжены температурным
компенсатором,
устанавливаемым вручную
или автоматически. С
помощью
температурной компенсации в рН-
метре устраняются изменения э. д. с. в зависимости от температуры.
Элементарная схема, поясняющая общий принцип действия
рН-метра, приведена на рис. 199. Электродвижущая сила Ех,
развиваемая электродной системой, сравнивается с падением
напряжения на сопротивлении R, через которое протекает ток /вых
оконечного каскада усилителя. Падение напряжения £/ВЫх на
сопротивления R противоположно по знаку электродвижущей силе Ех,
и на вход усилителя подается напряжение UBX — EX—UBbtx =
s= Ex — /вых R. Напряжение UBX преобразуется в переменное на*
пряжение, которое затем многократно усиливается и вновь
преобразуется в постоянное. Это напряжение управляет током /ВЫх
оконечного каскада усилителя. При достаточно большом коэффициенте
усиления напряжение £/вых мало отличается от э. д. с. ЕХу и
благодаря этому ток, протекающий через электроды в процессе
измерения э. д. с, весьма мал. Ток /ВЫх, протекающий через
сопротивление R, пропорционален э. д. с. электродной системы, т. е. рН
анализируемого раствора.
Устройство, подготовка к работе и порядок работы на
различных рН-метрах излагаются в прилагаемых к этим приборам
инструкциях и паспортах.
В настоящем разделе кратко описаны лишь некоторые
лабораторные рН-метры, выпускаемые в настоящее время
промышленностью [46, с. 277].
Лабораторный рН-метр— милливольтметр рН-121.
Показывающий прибор, градуированный в единицах рН и милливольтах,
для определения величин рН, pNa, pAg, pK, pNH4 и окислительно-
восстановительных потенциалов; может также использоваться в
качестве высокоомного нуль-индикатора и милливольтметра. Его
можно использовать для ручного и автоматического потенциометри-
ческого титрования (с блоком автоматического титрования БАТ-15).
С его помощью можно производить измерения как методом отбора
864
проб с использованием прилагаемой к прибору термостатированной
ячейки или ячейки для микроизмерений, так и непосредственно
в лабораторных установках.
Пределы измерения величины рН — от 1 до 14 с диапазонами 1 —
14; 1—4; 4—9; 9—14. Основная погрешность измерений 0,05
единицы рН.
Температурная компенсация ручная и автоматическая от 0 до
100 °С. Питание от сети переменного тока напряжением 220 В.
Усилитель постоянного напряжения питается от полупроводникового
стабилизатора напряжения компенсационного типа на
полупроводниковых элементах. Для питания измерительной схемы в блоке
питания имеются два стабилизированных источника питания,
имеющие малый коэффициент пульсации.
Электродом сравнения в приборе рН-121 служит проточный
хлорсеребряный электрод ЭВЛ-1МЗ, один для всех видов потен-
циометрических измерений. Его потенциал при 20 °С равен 202±
±2 мВ; область рабочей температуры 0—100 °С. Гарантийный срок
хранения и эксплуатации электрода 18 месяцев. Электрод хранят
закрытым пробкой и колпачком или погруженным в насыщенный
раствор КО.
В качестве индикаторного электрода для измерения величины
рН служат стеклянные электроды ЭСЛ-43-07 и ЭСЛ-63-07.
Диапазон прямолинейной водородной характеристики электрода ЭСЛ-
43-07 при 25 °С от 0 до 12 рН, а при наибольшей рабочей
температуре (40 °С) от 0 до 10 рН. Поэтому этим электродом следует
пользоваться для измерений рН в пределах 0—10 рН при
температуре анализируемой среды от 0 до +40 °С.
Электрод ЭСЛ-63-07 используется для измерений рН в
пределах 0—14 единиц, при температуре анализируемой среды от +25
до +100°С.
рН-метр — милливольтметр состоит из преобразователя и
подставки (рис. 200). На лицевой панели преобразователя (прибора)
располагаются органы управления, показывающий прибор и органы
заводской настройки и регулировки. На шкале показывающего
прибора имеются оцифровки, соответствующие диапазонам
измерения рН (э. д. с). Блок измерения прибора обеспечивает его
настройку для работ на
различных диапазонах
измерения и коррекцию
показаний при
изменении температуры
исследуемого раствора от
0 до +100°С.
Подставка предназначена для
крепления электродов
Рис. 200. рН-метр —
милливольтметр рН-121:
! — преобразователь; 2—подстав-
365
и установки сосудов с анализируемым или буферным" раствором.
При необходимости на подставке укрепляется также
электромагнитная мешалка, органы управления которой (тумблер включения,
ручка регулирования скорости и направления вращения) вынесены
на переднюю панель прибора. На подставке закреплены два
кронштейна. Высота их установки может регулироваться в зависимости
от вида измерений — в стакане, в термостатированной ячейке или
в ячейке для измерений рН в малом объеме раствора.
Термостатированная ячейка представляет собой
пластмассовый стакан со штуцерами, к которым подсоединяются
резиновые шланги от лабораторного ультратермостата. В термо-
статируемую ячейку устанавливается внутренний стеклянный
стакан с контролируемым раствором.
Ячейка для микроизмерений представляет собой
стакан с крышкой, в которой имеются три отверстия: для установки
вспомогательного электрода, термометра и электрического ключа.
Стакан заполняется насыщенным раствором КО.
Электролитический ключ имеет форму цилиндра со сферическим дном. Впаянная
в нижней части асбестовая нить обеспечивает связь полой части
ключа с раствором КС1 в стакане. В электролитический ключ
помещают контролируемый раствор (1—2 мл) и чувствительный
элемент измерительного электрода. При этом следует отрегулировать
положение электродов, чтобы измерительный электрод прилегал
к внутренней сферической поверхности электролитического ключа,
а вспомогательный был погружен в раствор КО на 30—40 мм.
При измерении рН растворов в стакане или в
термостатированной ячейке нижний конец электрода сравнения должен быть
погружен на несколько миллиметров ниже, чем измерительный
стеклянный электрод, во избежание удара последнего о дно стакана. На
одном из кронштейнов закрепляют держатели с отверстиями для
установки электродов, термометра и автоматического
термокомпенсатора. На штативе имеется винт заземления; при эксплуатации
прибора он должен быть соединен с зажимом заземления на
задней панели прибора. В зависимости от вида и условий измерений рН
следует выбрать необходимые электроды. Измерительные
электроды подключают к гнездам с надписью «изм» на панели прибора
непосредственно или с помощью переходного штекера.
При измерении отсчет величин рН и э. д. с. по шкале прибора
производят при заданной температуре, после того как показания
прибора примут установившееся значение. Измерение производят
несколько раз и берут среднее из 3—5 показаний.
При эксплуатации прибора следует иметь в виду, что буферные
растворы при многократном применении могут менять рН.
Для выполнения измерений рН-метр устанавливают на рабочем
месте и проверяют механический нуль показывающего прибора.
При" необходимости коррекции нуля отверткой устанавливают
стрелку на начальную отметку. Затем присоединяют провод
заземления к зажиму заземления и включают прибор в сеть 220 В, 50 Гц.
При этом должна загореться контрольная лампочка на панели
366
прибора. Прибору дают прогреться в течение 25—30 мин и ручкой
с надписью «Температура раствора» устанавливают значение
температуры контролируемого раствора. После этого корректируют
шкалу рН прибора по стандартным буферным растворам
(включая 0,1 н. раствор НС1). Для этого в стакан или термостатируемую
ячейку наливают стандартный буферный раствор, опускают в него
измерительный стеклянный электрод и хлорсеребряный электрод
сравнения и переключателем устанавливают показывающий прибор
на соответствующий диапазон измерений. Затем ручкой
потенциометра «Калибровка» устанавливают стрелку показывающего
прибора на деление шкалы, отвечающее значению рН стандартного
буферного раствора при данной температуре. По окончании
калибровки выключают прибор (контрольная лампочка гаснет) и
отключают его от сети. Электроды осторожно вынимают, промывают
дистиллированной водой и оставляют погруженными в 0,1 н. НС1 —
измерительный электрод и в насыщенный раствор КС1 — электрод
сравнения.
При измерении рН помещают электроды в сосуд с испытуемым
раствором, присоединяют их к соответствующим клеммам,
устанавливают автокомпенсатор температуры, включают прибор в сеть и
спустя 25—30 мин отмечают показания стрелки по шкале 1 —14.
Далее устанавливают переключатель на соответствующий диапазон
измерений и производят отсчет показаний прибора.
Переносный портативный специализированный прибор рН-222.
Предназначен для контроля кислотности молока и молочных
продуктов в лабораторных и цеховых условиях.
Измерение э. д. с. электродной системы производится с помощью
высокоомного усилителя постоянного тока, выполненного на элект*
рометрических лампах и транзисторах. Питание прибора
осуществляется от аккумуляторов или сухого элемента «Сатурн». Для
подзарядки аккумуляторов в комплекте прибора имеется зарядное
устройство. От других рН-метров отличается наличием
специальной температурной компенсации, благодаря которой показания его
не изменяются при изменении температуры исследуемого продукта
и соответствуют значению рН при 20 °С. Настройка производится
по одному фосфатному буферному раствору (рН = 6,88) при 20 °С.
Пределы измерения рН 3—8; диапазоны 3—4, 4—5, б—7, 7—8.
Погрешность измерения ±0,03—0,05 единиц рН. Выбор
необходимого диапазона измерения осуществляется переключателем,
расположенным на лицевой панели прибора.
В комплект рН-метра кроме обычных стеклянных электродов
(с шарообразной мембраной) входят копьевидные электроды для
измерения рН вязких продуктов.
ПОТЕНЦИОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ [64, с. 42; 71, с. 34]
Потенциометрическое титрование основано на определении точки
эквивалентности в реакциях нейтрализации,
окисления-восстановления, осаждения и в комплексообразовании по результатам
367'
потенциометрических измерений. Вблизи точки эквивалентности
происходит резкое изменение (скачок) потенциала индикаторного
электрода.
В объемном (титриметрическом) анализе
неизвестное количество вещества в контролируемом растворе
определяют по расходу титрующего раствора (т и т р а н т а). Объем
израсходованного титранта точно известной концентрации позволяет
рассчитать количество определяемого вещества в том случае, когда
точно установлена эквивалентная точка титрования
(окончание реакции). Конечную точку титрования фиксируют либо
визуально (по изменению окраски добавленного индикатора,
появлению мути и т. п.), либо измерением физико-химических свойств
титриметрической системы (изменение потенциала электродной
системы, проводимости, оптических свойств и т. п.).
Для потенциометрического титрования собирают цепь из
индикаторного электрода и электрода сравнения в анализируемом
растворе. Титрант приливают из бюретки порциями при непрерывном
перемешивании. После добавления каждой порции титранта
выдерживают 1—2 мин и измеряют э. д. с. цепи.
В начале титрования титрант добавляют относительно большими
порциями, в конце титрования — по 0,1 и 0,05 мл.
Вблизи точки эквивалентности (конечной точки титрования)
наблюдается наибольшее изменение электродного потенциала
(скачок потенциала).
Еще более резко изменяется отношение изменения потенциала
А£ к соответствующему изменению объема титранта AV. Точку
эквивалентности обычно определяют по максимальному значению
величины Д£/Д1Л
В табл. 13 в качестве примера представлены результаты титрования 50 мл
0,1 н. раствора НС1 0,1 н. раствором NaOH.
Величина скачка в эквивалентной точке при потенциометриче-
ском титровании кислот и оснований зависит от силы кислоты и
основания, а также от концентрации раствора. С уменьшением силы
кислоты (основания) и концентрации раствора скачок уменьшается.
ТАБЛИЦА 13. Изменение Ai?/AV в процессе титрования
Добавлено 0,1 н.
NaOH, мл
0
45,0
49,0
49,5
49,9
49,95
50,00
50,05
50,10
Потенциал
электрода, мВ
57
132
174
191
232
249
408
566
583
Д£, мВ
z
75
41
17
41
17
159
158
17
AV\ мл
45,0
4,0
0,5
0,4
0,05
0,05
0,05
0,05
Д£/ДК
z
1,66
10,00
34,00
102,00
340
3180
3160
340
368
Для нахождения эквивалентной точки титрования строят гра*
фик в координатах Д£/ДV — V. При титровании смеси кислот
различной силы появится несколько скачков.
При титровании определенного объема раствора V\ неизвестной
концентрации содержание вещества в растворе, выраженное в
эквивалентах на литр, легко вычислить по формуле:
где N — нормальность и V — объем титранта; Nx — нормальность
и V\ — объем испытуемого раствора.
Потенциометрическое титрование в лабораторных условиях
можно проводить вручную, с использованием любого типа рН-метра
и обычных бюреток, а также с помощью полуавтоматиче-.
ских титраторов, автоматически прекращающих подачу
титранта к моменту окончания титрования. При использовании
титраторов все подготовительные операции для анализа проводятся
вручную.
Для полуавтоматического титрования выпускаются также тит-
р о графы — лабораторные приборы, автоматически
регистрирующие на диаграмме всю кривую титрования в выбранных
координатах.
Выпускается также специальное электронное
регистрирующее устройство — блок автоматического титрования
БАТ-15. В комплексе со специальной бюреткой и рН-метром-мил-
ливольтметром типа рН-121 оно составляет установку для
полуавтоматического титрования.
Специальная бюретка-дозатор служит для автоматического
прибавления титранта в контролируемый раствор с регулярной
скоростью и автоматической остановкой в точке эквивалентности.
Принцип действия установки заключается в следующем.
Напряжение, пропорциональное э. д. с. электродной системы, с выхода
рН-метра— милливольтметра подается на вход БАТ-15, где
сравнивается с напряжением, установленным на датчике конечной точки
титрования. Разность этих напряжений поступает на вход
усилителя. На его выходе включено бесконтактное электронное реле,
управляющее работой электромагнитного клапана. При открытом
клапане титрующий раствор подается в ячейку, в которой
перемешивание осуществляется магнитной мешалкой. При равенстве
напряжений реле отключает питание клапана, который, закрываясь,
пережимает резиновую трубку и прекращает подачу титрующего
раствора. Прибор имеет реле времени выдержки от 0 до 40 с.
Относительная погрешность метода не превышает ±1%. Питание
прибора от сети переменного тока напряжением 220 В.
главам. ОПРЕДЕЛЕНИЕ ПЛОТНОСТИ
Определение плотности газов, жидкостей и твердых веществ
осуществляется с целью исследования свойств веществ,
идентификации и определения степени их чистоты, определения концентрации
двухкомпонентных растворов спиртов, кислот и оснований.
Плотность однородного вещества р —физическая величина,
равная отношению массы т вещества к занимаемому им объему V:
p = m/V
Единицей плотности в Международной системе единиц (СИ)
является килограмм на кубический метр; в единицах СГС
плотность выражается в граммах на кубический сантиметр.
Относительная плотность вещества — величина, равная
отношению его плотности к плотности некоторого другого вещества при
определенных физических условиях. Такими стандартными
веществами служат вода при температуре 3,98°С и нормальном
атмосферном давлении (760 мм рт. ст., или 1013 гПа) или сухой воздух
при 20 °С и нормальном атмосферном давлении:
d = р/ро
где р — плотность данного вещества, р0 — плотность стандартного
вещества.
Относительная плотность — безразмерная величина.
Относительную плотность жидкости принято относить к
температуре 20°С и к плотности воды при 3,98°С (4°С). В этом случае
относительная плотность обозначается d2±.
В тех случаях, когда плотность жидкости по условиям опыта
определяют не при 20 °С, а при другой температуре /, ее значение
d\ может быть пересчитано на нормальное значение по формуле:
df=*df4 + a(t-20)
где d\ — относительная плотность исследуемой жидкости при
температуре испытания t°C; а — средняя температурная поправка на
1 °С, находимая по табл. 14.
ТАБЛИЦА 14. Температурные поправки при определении плотности
Относительная
плотность
0,810—0,820
0,820-0,830
0,830-0,840
0,840-0,850
0,850-0,860
Поправка а
0,000752
0,000738
0,000725
0,000712
0,000699
Относительная
плотность
0,860—0,870
0,870-0,880
0,880—0,890
0,890-0,900
0,900-0,910
Поправка а
0,000686
0,000673
0,000660
0,000647
0,000633
Относительная
плотность
0,910—0,920
0,920-0,930
0,930-0,940
0,940-0,950
Поправка а
0,000620
0,000607
0,000594
0,000581
370
ПЖ4
Рис 201. Типы пикноМетров:
ЛГ—для определения плотности газа; ПЖ1 — Для жидкостей, с узкой трубкбй; ПЖ2 — ЛЛЯ
жидкостей, с меткой; ПЖЗ—для жидкостей, с капилляром; ПЖ4—для микроопределения
жидкостей; ЛГ—для твердых тел.
Относительная плотность является одной из важнейших физико-
химических характеристик веществ (особенно жидкостей), наряду
с температурой плавления и кипения.
Плотность веществ определяют с помощью пикнометров,
ареометров и гидростатических весов [5, с. 247].
ОПРЕДЕЛЕНИЕ ПЛОТНОСТИ С ПОМОЩЬЮ ПИКНОМЕТРОВ
Пикнометрами можно определять плотность газов, жидкостей
и твердых тел [13, с. 618]. Это стеклянные тонкостенные сосуды
с меткой на горловине или с капиллярным отверстием в пробке,
закрывающей горловину пикнометра. Пикнометры для определения
плотности газов имеют несколько иную форму (рис. 201).
Определение плотности жидкостей
Высушенный до постоянной массы и охлажденный до
комнатной температуры пикнометр взвешивают с точностью до 0,0002 г,
заполняют при помощи маленькой воронки дистиллированной водой
немного выше метки (пикнометры типа ПЖ1, ПЖ2 и ПЖ4) или
доверху (пикнометр типа ПЖЗ). В пикнометре ПЖЗ вода
выступает из капилляра, и избыток ее осторожно удаляют
фильтровальной бумагой. Пикнометр закрывают пробкой и выдерживают 20 мин
в водяном термостате, в котором поддерживают постоянную
температуру воды 20 °С с точностью ±0,1 °С. При этой температуре
уровень воды в пикнометре типа ПЖ1 или ПЖ2 доводят до метки при
помощи капиллярной трубки или свернутой в трубку полоски
371
фильтровальной бумаги. Пикнометр снова закрывают пробкой и
выдерживают в термостате еще 10 мин, проверяя положение
мениска по отношению к метке. Затем пикнометр вынимают из
термостата, вытирают снаружи мягкой тканью досуха, оставляют под
стеклом аналитических весов в течение 20 мин и взвешивают с
точностью до 0,0002 г. Потом его освобождают от воды,
высушивают, споласкивая последовательно этиловым спиртом и диэти-
ловым эфиром, удаляют остатки эфира просасыванием сухого
чистого воздуха и заполняют испытуемой жидкостью, после чего
производят те же операции, что и с дистиллированной водой.
Плотность испытуемой жидкости р2о, в г/см3, вычисляют по
формуле:
(т2 — т) 0,99823
Р2оШВ ™ ™
где т — масса пустого пикнометра, г; т\ — масса пикнометра с
дистиллированной водой, г; т2 —масса пикнометра с испытуемой
жидкостью, г; 0,99823 — значение плотности воды при 20 °С, г/см3.
Определение плотности твердого тела
Чаще всего взвешивают тело и пикнометр ПТ со
вспомогательной жидкостью, налитой в него до требуемого уровня при
определенной температуре, опускают тело в пикнометр с жидкостью,
устанавливают жидкость на первоначальном уровне при той же
температуре и взвешивают пикнометр с телом и жидкостью.
В качестве вспомогательной жидкости используют главным
образом воду. Если испытуемое твердое тело растворимо в воде или
взаимодействует с ней, то применяют другую жидкость (толуол,
ксилол, бензин, керосин, спирт), причем предварительно ее
плотность определяют описанным выше способом.
Испытуемое вещество вносят в пикнометр в виде порошка или
крупных кристаллов. Для лучшего проникновения жидкости в
капиллярные пустоты твердого тела рекомендуется присоединить
пикнометр, содержащий испытуемое вещество и вспомогательную
жидкость, к вакуумной системе и выдержать при пониженном
давлении 30—40 мин.
Возможен и другой порядок определения. В качестве примера приводим
определение плотности огнеупорных материалов но ГОСТ 2211—65.
Плотность огнеупоров определяют как отношение массы материала к ее
объему без пор.
Пробу, измельченную до крупности зерна 0,063 мм, высушивают при
110±5°С до постоянной массы. Навеску материала 5—8 г засыпают в
предварительно взвешенный пикнометр для твердых веществ вместимостью 25 мл.
Пикнометр с пробой взвешивают, затем до !/2 объема наполняют
вспомогательной жидкостью. Пикнометр, частично заполненный вспомогательной
жидкостью и испытуемым веществом, подвергают вакуумированию ^ 30 мин.
Такой же обработке под вакуумом подвергают и вспомогательную жидкость,
необходимую для дополнительного заполнения пикнометра. После отключения
вакуума пикнометр осторожно дополняют дегазированной вспомогательной
жидкостью и помещают в термостат на ^ 30 мин. Температура в термостате
должна быть 20±0,1°С при насыщении пробы водой и 20 it 0,2 °С при использова-
372
нии ксилола и толуола. Затем уровень жидкости в пикнометре доводят точно
до метки, закрывают пикнометр пробкой, вынимают его из термостата; обтирают
v взвешивают.
Массу высушенного пикнометра, а также пикнометра, заполненного
вспомогательной жидкостью, определяют заранее. Плотность пробы р, в г/см3,
вычисляют с точностью до 0,001 г/см3 по формуле:
"*Рж
Р =
т ■
(тх — т2)
где т — масса пробы, г; т{ — масса пикнометра с пробой и жидкостью, г; т2 —
масса пикнометра с жидкостью, г; рж — плотность вспомогательной жидкости при
20 °С, г/см3 (для воды р = 0,998 г/см3).
Плотность вспомогательной жидкости вычисляют по формуле:
_ (т2 — тэ) 0,998
рж т{ — тъ
где гпх — масса сухого пикнометра, г; тг — масса пикнометра с водой, г; т2 —-
масса пикнометра с жидкостью, г.
ОПРЕДЕЛЕНИЕ ПЛОТНОСТИ ЖИДКОСТИ АРЕОМЕТРАМИ
(ДЕНСИМЕТРАМИ)
Применение ареометров (денсиметров) [46, с. 146] для опреде*
ления относительной плотности жидкостей основано на законе
Архимеда.
Ареометр (рис. 202) представляет собой стеклянную трубку,,
расширенная (нижняя) часть которой заполнена балластом —
чистой и сухой металлической дробью, залитой слоем смолы с
температурой плавления не ниже 80 °С. Благодаря балласту во время
измерения плотности ареометр находится в
вертикальном положении. На верхней части
нанесена шкала. Чем меньше плотность
жидкости, тем глубже погружается в нее
ареометр, поэтому верхние деления шкалы
соответствуют наименьшей, а нижние — наибольшей
плотности. Отсчет показаний производится по
нижнему мениску.
Стеклянные ареометры общего назначения,
предназначаемые для измерения плотности
жидкости, выпускаются со шкалами,
градуированными в единицах плотности, а ареоме-
тры для измерения содержания веществ в
двухкомпонентных растворах — со шкалами,
градуированными в процентах растворенного
вещества (по объему или по массе).
Выпускаются также ареометры
специального назначения: для нефти, для морской
воды, молока, для определения концентрации
Рис. 202. Ареометры общего назначения!
в —без шара; б—с шаром.
37а
сухих веществ в сахаросодержащих веществах (сахарометры), для
определения крепости водно-спиртовых растворов (спиртометры)
и др. На шкалах специальных ареометров нанесены деления,
показывающие концентрации раствора в процентах (по объему или по
массе).
Сифонные ареометры используют в частности для измерения
плотности электролита в аккумуляторах. Ареометр состоит из
стеклянного сосуда цилиндрической или грушевидной формы. На
верхнюю часть сосуда плотно насаживается резиновый шар, на
нижнюю— резиновая трубка. Внутри стеклянного сосуда помещен
маленький ареометр. Сжав предварительно резиновый шар, опускают
резиновую трубку в аккумулятор. При разжимании шара в
стеклянный сосуд всасывается электролит в количестве достаточном,
чтобы ареометр мог свободно плавать.
При измерении плотности испытуемую жидкость помещают
в стеклянный цилиндр для ареометров и при температуре жидкости
20 °С осторожно опускают в нее чистый сухой ареометр, на шкале
которого предусмотрена ожидаемая величина плотности. Ареометр
не выпускают из рук до тех пор, пока не станет очевидным, что он
не тонет; при этом необходимо следить, чтобы ареометр не касался
стенок и дна цилиндра. Отсчет производят через 3—4 мин после
погружения по делению на шкале ареометра, соответствующему
нижнему мениску жидкости (при отсчете глаз наблюдателя должен
быть на уровне мениска).
В. случае определения плотности темноокрашенных жидкостей.
отсчет допускается производить по верхнему мениску.
Определение ареометром плотности летучих веществ не
допускается, так как при энергичном испарении снижается температура
жидкости.
ОПРЕДЕЛЕНИЕ ПЛОТНОСТИ ГИДРОСТАТИЧЕСКИМ ВЗВЕШИВАНИЕМ
При погружении тела последовательно в разные жидкости оно
вытеснит равные по объему, но разные по массе количества этих
жидкостей. Массы этих объемов прямо пропорциональны
плотностям жидкостей.
Следовательно, тело массой т в воде будет иметь кажущуюся
массу mi, а разность т — т\ будет равна массе вытесненной этим
телом воды. В другой жидкости тело будет иметь кажущуюся
массу Ш2, а разность т — тг будет равна массе вытесненной
жидкости.
Если для определения плотности применять тело определенной
массы и объема (стеклянный поплавок), а в качестве «другой»
жидкости — исследуемую, то относительную плотность последней
легко вычислить по формуле:
р т — Ш\
При использовании гидростатических весов Мора — Вестфаля,
прокалиброванных с учетом определенного объема стеклянного
•374
Рис. 203. Гидростатические весы и гири-рейтеры:
i-стойка; 2-коромысло; 5-стрелка; 4-сережки; 5-поплавок; 5-указатель; 7-установоч-
ный винт; 5—шкала; 0—прижимной винт; /0—цилиндр.
поплавка, результат определения плотности не нужно вычислять,
по формуле; его сразу отсчитывают по взятому разновесу для
достижения равновесия весов.
Гидростатические весы (рис. 203) состоят из неравноплечего
коромысла 2, опирающегося призмой на подушку, которая заделана
в вилке раздвижной колонки У, скрепляемой прижимным винтом
9 и снабженной установочным винтом 7. На одном плече коромысла
жестко укреплен постоянный противовес 3 с указателем 6 и
шкалой S, а на другом, при помощи серьги 4, к грузоприемной призме
подвешен на тонкой металлической проволоке поплавок 5 с
впаянным в него термометром. Плечо коромысла, несущее
грузоподъемную призму, разделено на 10 равных частей углубленными наре
зами, на которые навешивают специальные гири-рейтеры. Для на*
ливания испытуемой жидкости к весам прилагается специальный,
стеклянный цилиндр 10. Набор разновесов состоит из нескольких
рейтеров: два из них .имеют массу, равную массе воды,
вытесненной данным поплавком при 20 °С; другие разновески в 10, 100 и
1000 раз меньше.
Определение плотности производят на проверенных весах. Для
этого металлические части весов тщательно протирают, а поплавок
и проволоку промывают этиловым спиртом и диэтиловым эфиром
и просушивают продуванием воздуха.
После этого, не касаясь поплавка и проволоки руками, пинцетом
подвешивают их на крючок коромысла весов. С помощью
установочного винта 7 колонку с коромыслом устанавливают в
равновесие. Колонка при этом должна быть установлена строго
вертикально. После установления равновесия весов с подвешенным
поплавком в воздухе наливают в стеклянный цилиндр
дистиллированную воду с температурой точно 20 °С и опускают поплавок
375
в воду так, чтобы в воде был не только весь поплавок, но и часть
проволоки (около 15 мм).
При опускании поплавка в воду нужно следить за тем, чтобы
он находился в середине цилиндра, а не прикасался к стенке. При
этом равновесие весов нарушится и плечо коромысла с поплавком
поднимется. Для восстановления равновесия навешивают на 10-е
деление коромысла (на крючок) гирю-единицу. Если равновесие
не наступает, то коромысло приводят в равновесие с помощью
самой маленькой гири, навешивая ее на 1-е, 2-е, 3-е или 4-е деление,
если гиря-единица несколько легче, чем нужно; если гиря-единица
несколько тяжелее, чем нужно, то ее навешивают на 9-е деление,
а самую маленькую — на 9-е, 8-е, 7-е или 6-е деление.
Установленную таким образом величину ошибки в пределах ±0,0004
учитывают при помощи отклонений коромысла, производя отсчет при
определении плотности испытуемой жидкости при тех же самых
отклонениях указательной стрелки.
Если ошибка превышает ±0,0004, то весы подлежат ремонту.
После проверки весов поплавок и цилиндр высушивают.
В чистый сухой цилиндр осторожно наливают испытуемую
жидкость, пока в нее не погрузится поплавок и около 15 мм проволоки,
на которую он подвешен. При этом равновесие весов нарушится
и плечо с поплавком поднимется. На коромысло постепенно
навешивают гири, начиная с самой крупной, пока не наступит
равновесие.
Температуру жидкости измеряют или по термометру, впаянному
в поплавок, или дополнительным термометром. По достижении
равновесия весов и установлении температуры записывают так
называемую «видимую» плотность испытуемой жидкости, начиная
с гири-единицы.
«Видимую» плотность р' пересчитывают в действительную
плотность р по формуле:
р£0= (0,99823 - 0,0012)^ f 0,0012
где 0,99823 —значение плотности воды при 20 °С; 0,0012
—значение плотности воздуха при 20 °С и барометрическом давлении
760 мм рт. ст. (1013 гПа).
глАВАзз. ОПРЕДЕЛЕНИЕ ВЯЗКОСТИ (ВИСКОЗИМЕТРИЯ)
Вязкость, внутреннее трение — свойство жидкостей и газов
оказывать сопротивление перемещению одного их слоя относительно-
другого. Вязкость жидкостей легче всего обнаруживается при их
переливании или помешивании.
Количественно вязкость характеризуется значением величины,
называемой динамической вязкостью или коэффи*
ц иен том внутреннего трения и обозначаемой т) или jj,.
Характерной особенностью этого вида трения является то, что оно
наблюдается не на границе твердого тела и жидкости, а во всем
объеме жидкости.
Единицей динамической вязкости в Международной системе
единиц (СИ) является паскаль-секунда (Па-с). Паскаль-секунда
равна динамической вязкости среды, касательное напряжение в
которой при ламинарном (упорядоченном) течении и при разности
скоростей слоев, находящихся на расстоянии 1 м по нормали к
направлению скорости, равной 1 м/с, составляет 1 Па*.
Кинематическая вязкость равна отношению
динамической вязкости среды к ее плотности при той же температуре:
£ v = ч/Р
Единицей кинематической вязкости в СИ является квадратный
метр в секунду (м2/с). При кинематической вязкости 1 м2/с
динамическая вязкость среды плотностью 1 кг/м3 равна 1 Па-с**.
Часто пользуются также величиной относительной, или
условной, вязкости (ВУ) —отношением вязкости данной жидкости к
вязкости воды при той же температуре (см. ниже).
Широкий диапазон значений вязкости, а также необходимость
измерять вязкость в условиях низких или высоких температур и
давлений обусловливает большое разнообразие методов
определения вязкости и конструкции вискозиметров.
ТИПЫ ВИСКОЗИМЕТРОВ [46, с. 149]
В зависимости от способа измерения вискозиметры
подразделяются на капиллярные (визкозиметры истечения), шариковые,
ротационные, вибрационные и ультразвуковые.
При пользовании капиллярными вискозиметрами
измеряется время истечения известного количества (объема)
жидкости сквозь капиллярные трубки определенного диаметра.
* Соотношение между паскалем-секундой и бытующей еще в литературе
единицей системы С ГС пуазом (П): 1П = 0,1 Па-с.
** Соотношение между квадратным метром в секунду и встречающейся еще в
литературе и ГОСТах единицей системы СГС стоксом (Ст): 1Ст — 10~4 м2/с.
377
Стеклянные капиллярные вискозиметры чаще других
используются в практике химических лабораторий.
При пользовании шариковыми вискозиметрами
измеряется скорость падения шарика в исследуемой жидкости — она
тем меньше, чем больше вязкость жидкости,
В ротационных вискозиметрах измеряется крутящий
момент или угловая скорость вращения одного из двух соосных
тел, в зазоре между которыми находится испытуемая жидкость.
Область измерения вязкости 0,5—106 Па «с. Они широко
используются для определения вязкости высокомолекулярных жидкостей
и растворов полимерных соединений.
Измерение вязкости вибрационными
вискозиметрами основано на зависимости амплитуды колебаний тела в
исследуемой жидкости от ее вязкости.
Ультразвуковыми вискозиметрами измеряют
скорость затухания колебаний магнитострикционного материала,
помещенного в исследуемую жидкость.
Независимо от конструкции вискозиметра, определение
вязкости следует проводить в условиях строгого термостатирования.
СТЕКЛЯННЫЕ КАПИЛЛЯРНЫЕ ВИСКОЗИМЕТРЫ
Для измерения вязкости прозрачных жидкостей служат
вискозиметры ВПЖ-1, ВПЖ-2, типа Пинкевича, ВПЖМ, а для
непрозрачных — ВНЖ (рис. 204).
Кинематическая вязкость жидкости v равна произведению
времени т истечения через капилляр определенного ее объема на по-
впж-1
ВПЖ-2 типа Пинкедичъ ВПЖМ
ВНЖ
Рис. 204. Вискозиметры стеклянные капиллярные:
/, 2 — колена прибора; 3 —отводная трубка; 4-6 — расширения (резервуары); 7—капилляр;
Я — пипетка; 0 —пробирка-муфта; 1Q—приемник; М — метки.
37$
стоянную вискозиметра С.. Постоянная С не зависит от
температуры и определяется только геометрическими размерами
вискозиметра*.
Для определения постоянной вискозиметра пользуются
эталонными жидкостями с известной кинематической вязкостью. Измеряя
иремя истечения определенного объема эталонной жидкости
определяют постоянную вискозиметра: С = v/т сСт/с.
Вискозиметры выпускаются с разными капиллярами, причем
диаметр капилляра резко сказывается на постоянной вискозиметра.
В каждом наборе имеется по девять вискозиметров, диаметры
внутренних капилляров которых варьируются в пределах 0,34—
5,5 мм, что соответствует значения^ С = 0,003^-30 сСт/с. Набор
вискозиметров типа Пинкевича состоит из 11 вискозиметров с
диаметрами капилляров от 0,4 до 4,0 мм.
В качестве эталонной жидкости при калибровке вискозиметров
для маловязких жидкостей может служить свежеперегнанная
дистиллированная вода, кинематическую вязкость которой принимают
равной 1,0067 сСт/с при 20 °С и 0,89748 сСт/с при 25 °С.
По существующему положению каждый капиллярный вискози*
метр заводского изготовления должен снабжаться паспортом, в ко-»
тором указана его постоянная. Так, вискозиметры ВПЖ-1, ВПЖ-2,
ВНЖ выпускаются со значением постоянной С: 0,003; 0,01; 0,03;
0,1; 0,3; 1; 3; 10 и 30 сСт/с. Постоянная вискозиметров типа ВПМЖ
составляет 0,01; 0,03; 0,1; 0,3; 1 и 3 сСт/с.
ОПРЕДЕЛЕНИЕ КИНЕМАТИЧЕСКОЙ ВЯЗКОСТИ
Методики определения кинематической вязкости практически
наиболее распространены.
Калибровка вискозиметров
Новые вискозиметры, а также вискозиметры, находящиеся давно
в работе, следует периодически подвергать проверочной калибровке.
Калибровка заключается в определении времени протекания
через вискозиметр эталонной жидкости. Перед выполнением
работы вискозиметр промывают последовательно петролейным
эфиром, хромовой смесью, водопроводной и дистиллированной водой,
спиртом и диэтиловым эфиром, после чего продувают чистым,
сухим воздухом.
Пусть для калибровки выбран вискозиметр типа ВПЖ-1
(рис. 204). На отводную трубку 3 надевают резиновый шланг,
соединенный с грушей, и, зажав пальцем колено 2, переворачивают
вискозиметр, опускают отверстие колена 1 в сосуд с эталонной
жидкостью, засасывают ее в вискозиметр с помощью резиновой груши
* Стеклянные вискозиметры, которыми оснащены современные лаборатории,
пока характеризуются постоянной, выраженной в сантистоксах в секунду;
1 сСт/с= Ю-6 м2/с
379
или водоструйного насоса до метки М2, следя за тем, чтобы в
расширениях 4 и 5 не образовалось разрывов жидкости. Затем
колено 1 вынимают из жидкости и снимают шланг с отводной
трубки 3.
На колено / надевают резиновую трубку; вискозиметр
погружают в жидкостной термостат так, чтобы расширение 4 оказалось
в жидкости, и укрепляют строго вертикально с помощью зажима
на штативе. Другим зажимом укрепляют термометр, шарик
которого должен быть на одном уровне с серединой капилляра 6.
В термостате устанавливают температуру 20 ± 0,2 °С и
вискозиметр выдерживают при этой температуре 10—15 мин.
Затем грушей или насосом, присоединенными к резиновой
трубке, засасывают жидкость в колено 1 примерно до 1/3 его высоты,
следя, чтобы не образовалось разрывов жидкости или пузырьков
воздуха. Прекратив засасывание, дают жидкости стекать в
расширение 5 и наблюдают опускание уровня жидкости. Как только
уровень вытекающей жидкости коснется метки М\, включают
секундомер; когда уровень жидкости коснется метки М2,
останавливают секундомер. Записав время истечения жидкости, повторяют
определение не менее четырех раз. Затем вискозиметр моют, сушат,
вновь заполняют эталонной жидкостью и вновь производят не
менее четырех определений.
Если разность между средним временем двух опытов не
превышает 0,3%, то находят среднее арифметическое времени
истечения т эталонной жидкости в обоих опытах и вычисляют
постоянную вискозиметра:
С= Гэталона сСт/с
Тэталона
Проведение определения
Определяют время протекания через вискозиметр испытуемой
жидкости точно так же, как при калибровке поступали с
эталонной. Следует лишь иметь в виду, что время предварительной
выдержки вискозиметра с испытуемым веществом в термостате
следует увеличивать с повышением температуры проведения
испытания (от 10 мин при 20 °С до 20 мин при 100 °С).
Среднюю арифметическую величину времени истечения
жидкости в вискозиметре определяют с точностью до 0,1 с и вычисляют
кинематическую вязкость (в сантистоксах) по формуле:
где С—постоянная вискозиметра, сСт/с; т — среднее
арифметическое время истечения жидкости, с; g — ускорение силы тяжести
в месте измерения вязкости, см/с2 (можно принять #/980,7=1, если
дополнительная погрешность 0,02% не имеет значения); К —
коэффициент, учитывающий изменение гидростатического напора
жидкости в результате расширения ее при нагревании; для ВПЖ-1
380
К=\\ для ВПЖ-2 и ВПЖ-4 К = 1 ± 0,00004 Д/; для ВНЖ
/С— 1 ±0,000087 Д/; для ВПЖМ К= 1 ± 0,000074 М (М —
разность между температурой жидкости при заполнении
вискозиметра и при определении вязкости).
ОПРЕДЕЛЕНИЕ ДИНАМИЧЕСКОЙ ВЯЗКОСТИ
РАЗБАВЛЕННЫХ РАСТВОРОВ ПОЛИМЕРОВ (ПО ГОСТ 18249—72)
Концентрацию раствора полимера выбирают так, чтобы
отношение времени истечения раствора т ко времени истечения
растворителя то составляло 1,2—1,6. В соответствии с этим
подбирают вискозиметр.
Величина навески полимера, выбор растворителя, его объем
и условия растворения указываются в стандартах или технических
условиях на данный полимер.
При определении вязкости на вискозиметрах типа ВПЖ-2
приготовляют растворы четырех концентраций, на вискозиметре
ВПЖ-1—одной концентрации; растворы меньших концентраций
получают разбавлением в самом вискозиметре. Для этого в
вискозиметр наливают 13—16 мл раствора, измеряют время истечения,
после чего последовательно добавляют измеренный объем
растворителя и перед каждым последующим измерением времени
истечения тщательно перемешивают. Концентрацию разбавленного
раствора А\ вычисляют по формуле:
где А—концентрация раствора полимера, залитого в
вискозиметр, г/мл; V — объем раствора в вискозиметре, мл; V\ — объем
добавленного растворителя, мл.
Вискозиметр типа ВПЖ-2 заполняют чистым растворителем
или раствором так же, как описано выше. Отклонения
температуры термостатирования не должны превышать при комнатных
температурах ±0,05 °С, при повышенных ±0,15 °С. Уровень тер-
мостатирующей жидкости должен быть на 3—4 см ниже верхнего
конца колена вискозиметра.
После 15-минутного термостатирования вискозиметра с
растворителем или раствором полимера определяют время истечения
растворителя то или растворов различных концентраций т. При
этом за результат принимают среднее арифметическое не менее
трех определений, расхождение между которыми не должно
превышать 0,4 с. '
Динамическая вязкость разбавленных растворов г\ или
растворителя т)о (в сантипуазах) вычисляют по формулам:
Л = Срт; Ло ^ сРото
где С — постоянная вискозиметра, сСт/с; р, р0 — плотность
раствора полимера или растворителя при температуре испытания, г/см8;
381
HJJjtf
Рис. 205. Вискозиметр для определения условной
вязкости:
/ — латунный резервуар; 2—внешний сосуд; 5—выходное
отверстие; 4—термометр; 5—штифты; 5—стержень;
7—мешалка; 8—трубка; S—установочные винты;
/0—треножник.
т, то — время истечения раствора или
растворителя, с.
ОПРЕДЕЛЕНИЕ УСЛОВНОЙ ВЯЗКОСТИ
Метод определения условной
вязкости применяется для нефтепродуктов,
лакокрасочных материалов и ряда
других вязких жидкостей, вязкость которых
нельзя определить с помощью
стеклянных капиллярных вискозиметров. Для
ряда нефтепродуктов вязкость
нормируется в условных единицах.
Условной вязкостью называют отношение времени
истечения из вискозиметра типа ВУ 200 мл испытуемого продукта
при температуре испытания ко времени истечения 200 мл
дистиллированной воды при температуре 20 °С, являющемуся
постоянной (водным числом) прибора. Величина этого отношения
выражается как число условных градусов.
Водное число вискозиметра должно контролироваться
лабораторией организации, которой принадлежит прибор.
Условная вязкость при температуре £ обозначается знаком ВУ*.
Прибором для измерения условной вязкости служит
вискозиметр типа ВУ-4 (рис. 205). Он состоит из резервуара 1 с
трубкой 8 в его дне. Резервуар помещают в сосуд 2, служащий
водяной или масляной баней. Резервуар закрывают крышкой с двумя
отверстиями. В одно из отверстий вставляют деревянный
стержень 6, который закрывает трубку 8; в другое помещают
термометр 4. Внутри резервуара i, на равном расстоянии от дна,
прикреплены три заостренных изогнутых вверх под прямым углом
штифта 5. По ним устанавливают уровень наливаемой в резервуар
исследуемой жидкости, и, кроме того, эти штифты служат для
установки прибора в горизонтальное положение. Во внешнем
резервуаре помещены мешалка 7 и термометр со шкалой от 10 до
110°С и ценой деления 1 °С.
Прибор устанавливают на железном треножнике 10, на двух
ножках которого имеются установочные винты 9. Для подогрева
термостатирующей жидкости в сосуде 2 к треножнику прикреп*
ляется газовая горелка или прибор снабжают
электрообогревательным устройством с терморегулятором.
Для измерения объема^ вытекающей из вискозиметра
жидкости к прибору прилагается специальная мерная колба, калибро*
ванная при 20 °С на 200 мл.
382ч
Определение водного числа вискозиметра ВУ
Внутренний резервуар промывают последовательно петролей-
ным или диэтиловым эфиром, этиловым спиртом и
дистиллированной водой и высушивают воздухом. Затем вискозиметр вставляют
ножками в прорези треножника и закрепляют зажимными
винтами. Выходное отверстие 3-закрывают чистым стержнем 6. Во
внутренний резервуар вискозиметра 1 наливают профильтрованную
дистиллированную воду до уровня, при котором острия трех
штифтов 5 едва лишь выдаются над зеркальной поверхностью воды;
температура воды должна быть 20±0,2°С.
Водой такой же температуры заполняют и внешний сосуд 2.
Мерную колбу подставляют под сточную трубку 8 внутреннего
резервуара и, приподняв стержень б, спускают всю воду из
резервуара в колбу, не замеряя времени ее истечения; при этом водой
наполняется и вся трубка 8, на нижнем конце которой повисает
капля воды. Опустив конец стержня 6 в выходное отверстие <?,
вновь осторожно выливают воду из колбы в резервуар по
стеклянной палочке; опорожненную колбу держат 1—2 мин над
резервуаром в опрокинутом положении и затем вновь подставляют под
сточную трубку.
Воду во внутреннем резервуаре и внешнем сосуде тщательно
перемешивают: в первом — вращением крышки вокруг стержня 5,
во втором — мешалкой 7. Убедившись, что температура воды в
обоих резервуарах равна 20°С и в течение 5 мин отклонение
температуры не превышает ±0,2 °С, приподнимают коротким
движением стержень 6, пуская одновременно секундомер, и
наблюдают вытекание воды из резервуара, В момент, когда нижний
край мениска достигнет кольцевой метки на колбе, останавливают
секундомер. Наблюдение времени истечения 200 мл
дистиллированной воды повторяют не менее 4 раз. Для стандартного
вискозиметра время истечения 200 мл воды при 20 °С должно быть
равным 51±1 с.
Проведение определения
Перед каждым определением резервуар 1 промывают и
высушивают.
Сточное отверстие закрывают стержнем 6 и наполняют
внутренний резервуар испытуемой жидкостью, предварительно
подогретой несколько выше заданной температуры определения.
Уровень налитой жидкости должен быть немного выше остриев
штифтов 5. у
В сосуд 2 наливают воду (при определении вязкости до 80 °С)
или вазелиновое масло, нагретые несколько выше заданной
температуры определения.
Подняв немного стержень 5, дают стечь избытку испытуемой
жидкости, с тем чтобы острия всех трех штифтов лишь едва
заметно выдавались над уровнем жидкости.
3&?
Установив вискозиметр, закрывают его крышкой и под
сточное отверстие ставят чистую специальную мерную колбу на
200 мл, в то же время осторожно вращая вокруг стержня крышку
прибора, в которую вставлен термометр.
Когда термометр будет показывать точно заданную
температуру определения, следует выждать 5 мин, быстро вынуть
стержень и одновременно пустить секундомер. Когда жидкость в
мерной колбе дойдет точно до метки, секундомер останавливают и
отсчитывают время истечения с точностью до 0,2 с.
Условную вязкость при температуре / в условных градусах
вязкости вычисляют по формуле:
где xt — время истечения из вискозиметра 200 мл испытуемого
продукта при температуре /, с; тн^о — водное число
вискозиметра, с.
глАвлзб ОПРЕДЕЛЕНИЕ ТЕМПЕРАТУРЫ ПЛАВЛЕНИЯ
Температурой плавления вещества называют
температуру равновесия фаз твердое вещество — жидкость во время
процесса плавления. Определить эту температуру можно в процессе
плавления или в процессе застывания расплава, так как, если
исключено переохлаждение, то температура застывания
совпадает с температурой плавления. Различные наименования
применяют для того, чтобы показать, каким методом было
проведено определение температуры равновесия фаз твердое тело —
жидкость: при нагревании наблюдают температуру плавления, при
охлаждении — температуру затвердевания.
Точное определение фазового равновесия может быть
успешным лишь тогда, когда нагревание или охлаждение идет настолько
медленно, что действительно устанавливается необходимое
равновесие. Определение точки плавления имеет особо важное значение'
при работе с органическими веществами, однако им пользуются
также и при работе с неорганическими препаратами.
В связи с высокими температурами плавления неорганических
веществ, определение обычными методами с капиллярами,
погружаемыми в нагретые до определенной температуры бани, в
большинстве случаев не удается. Определение температуры плавления
неорганических веществ выполняют большей частью так
называемым тигельным методом, наблюдая скорость охлаждения расплава
при помощи погруженной в него термопары.
Одни исследователи считают, что температуре плавления
отвечает момент появления жидкой фазы. По мнению других, точкой
плавления следует считать момент полного исчезновения твердой
фазы, то есть наступление полной прозрачности расплава при
условии медленного нагревания.
Часто под температурой плавления подразумевают интервал
температур между появлением первых капель жидкости и полным
переходом твердого вещества в жидкое состояние. Для чистых
индивидуальных веществ "этот интервал измеряется долями градуса.
Точнее определить температурный интервал плавления можно
путем повторного расплавления образца после его застывания. О
приближении расплавления узнают по стеканию расплавившихся
частиц, которые обыкновенно при застывании высоко поднимаются по
стенкам капилляра под действием капиллярных сил.
При наличии примеси точка плавления вещества понижается.
Это дало повод предположить, что смеси веществ должны
плавиться при более низкой температуре, чем составляющие их
индивидуальные вещества. Отсутствие депрессии температуры
плавления смеси исследуемого вещества со стандартными рассматривается
как доказательство их идентичности (или их полной взаимной не-.
растворимости). Этим пользуются для идентификации химических
13 Зак. 1237 385
соединений. Для этого смешивают в равных количествах (по 0,05
или 0,1 г) исследуемое вещество и химически чистое стандартное
вещество (вещество сравнения) и определяют температуру
плавления смеси. Если проба смешения плавится при той же
температуре, что и каждый компонент в отдельности, то
идентичность исследуемого вещества со стандартными считается
доказанной. Если же проба смешения плавится при более низкой
температуре, чем каждый компонент в отдельности, то это значит,
что исследуемое вещество не идентично стандартному.
Определение температуры плавления пробы смешения —
наиболее широко используемый и легко применимый критерий'
идентичности, которым, однако, следует пользоваться с осторожностью.
Наличие депрессии указывает на различие веществ, но отсутствие
депрессии не всегда является свидетельством их идентичности.
Отсутствие депрессии наблюдается иногда в случае двух сравнительно
сложных соединений, имеющих незначительные структурные
различия.
Полезные сведения по определению температуры плавления
можно найти в литературе, например [11, с. 105].
ОПРЕДЕЛЕНИЕ ТЕМПЕРАТУРЫ ПЛАВЛЕНИЯ В КАПИЛЛЯРЕ
Из всех методов определения температуры плавления
органических соединений этот — наиболее распространенный.
Техника его исполнения несложна. Небольшое количество тонко
растертого и хорошо высушенного препарата помещают в
запаянный с одного конца тонкостенный стеклянный капилляр длиной
45—50 мм и диаметром 1,0—1,2 мм для бесцветных и 0,8—1,0 мм
для окрашенных веществ. (Для воскообразных и волокнистых
веществ можно брать капиллярные трубки несколько большего
размера.) Наполненный капилляр осторожно бросают 10—15 раз в
стеклянную трубку высотой 800 мм и диаметром 15—20 мм,
поставленную вертикально на часовое стекло, до уплотнения вещества в
слой высотой около 2 мм. В случае определения трудно
уплотняемого вещества капилляр набивают при помощи другого
капилляра, меньшего диаметра. Оплавлять открытый конец капилляра
недопустимо, так как при этом попадает внутрь конденсационная вода.
Температуру плавления гигроскопических и возгоняющихся
веществ определяют в капиллярах, запаянных с обоих концов; при
этом капиллярная часть должна быть целиком погружена в
нагревательную баню (нагревательный блок).
Капилляр закрепляют на термометре резиновым колечком
(кольцо отрезают от подходящего по размерам резинового шланга),
медной проволокой или приклеивают верхний конец каплей серной
кислоты. Проба вещества должна находиться на уровне шарика
термометра.
При переходе из твердого состояния в жидкое в обычных
условиях нагревания в капилляре можно наблюдать следующие
явления: усадка вещества — столбик вещества меняет свою форму,
386
сжимаясь и отставая от стенок капилляра, без видимого перехода
в жидкое состояние; отпотевание — на внутренней поверхности
капиллярной трубочки появляются капельки жидкости, вещество
спекается, не теряя своей связности; частичное
плавление—в капилляре наряду с твердыми частицами образуется
мениск жидкости по всему сечению капилляра. После этого при
несколько более высокой температуре наступает состояние полного
расплавления.
Температуру плавления большей частью считывают с термо»
метра, когда вещество полностью расплавилось, образовав
прозрачный расплав.
Чтобы избежать довольно больших и порой недостаточно
надежных поправок на выступающий столбик ртути, рекомендуется
применять укороченные термометры из набора ТЛ-6 (по Аншютцу).
Многие органические вещества плавятся с разложением
(появление окраски, выделение газа). Температура разложения обычно
нерезко выражена и часто не может быть точно воспроизведена.
Определение температуры плавления в капиллярах может быть
выполнено очень точно, если в предварительном опыте определить
приблизительно температуру плавления вещества и затем
капилляр с веществом поместить в прибор, нагретый до температуры
на несколько градусов ниже этого приблизительного значения.
В качестве обогревательной жидкости (теплоносителя)
используют концентрированную серную кислоту, парафиновое масло,
силиконовое масло. Серную кислоту, применение которой требует
большой осторожности, можно использовать при определении
температуры плавления вплоть до 250 °С. Смесь конц. H2SO4 (7 масс, ч)
и K2SO4 (3 ч.) после 5-минутного кипячения при энергичном
перемешивании образуют прозрачную жидкость, которую можно
использовать в качестве теплоносителя до 320°С (смесь
гигроскопична, и ее следует предохранять от увлажнения).
Предложено много конструкций приборов для определения
температуры плавления капиллярным методом [5, с. 205].
Отечественная промышленность выпускает два типа таких приборов.
Прибор ПТОП. Этот прибор (рис. 206) состоит из круглодонной
колбы / вместимостью 100—150 мл с горлом длиной 90 мм и
диаметром 30 мм. В колбу наливают на 2/3 ее объема бесцветной *
конц. H2SO4 или бесцветного силиконового масла. В горло колбы
вставляют специальную пробирку 3 длиной 150 мм и диаметром
около 15 мм с приплавленными к ней четырьмя отростками 4 на
расстоянии 30 мм от ее верхнего края. Над одним из отростков
в пробирке имеется отверстие 5 диаметром 2—3 мм.
Пробирку вставляют в колбу так, чтобы между дном пробирки
и колбы оставалось 10—15 мм. Пробирку закрывают корковой
пробкой 6 со вставленным в нее укороченным термометром, из
набора ТЛ-6 (по Аншютцу), нижний конец которого должен
* Если конц, H2SO4 приобретает в процессе работы буроватый оттенок, то
для обесцвечивания к ней прибавляют несколько кристаллов KNOs или NaNOs-
13* 387
J:
Рис. 206. Прибор ПТП для определения температуры плавления:
J — круглодонная колба; 2—капилляр с испытуемым веществом; 3 — пробирка; 4—отростки
для удержания пробирки; 5—отверстие; б —пробка со вставленным термометром.
Рис. 207. Прибор ПТП для определения температуры плавления:
/ — номограмма; 2— стакан для капиллярных трубок; 3—вольтметр; 4—осветитель с
рефлектором; 5 — константановая спираль; 6 — блок-нагреватель; 7—термометр; 8 — капилляр с
испытуемым веществом; 9 — оптическое устройство; /0 —регулятор напряжения.
находиться на несколько миллиметров выше дна пробирки. При
определении температуры плавления веществ, плавящихся ниже
170°С, применяют порожнюю пробирку; для веществ с
температурой плавления выше 170 °С в пробирку наливают конц. H2SO4
или силиконовое масло так, чтобы ртутный резервуар термометра
был погружен в нее. Для веществ с температурой плавления 250—
320 °С пробирку и колбу наполняют упомянутой выше смесью
H2S04 и K2S04.
Содержимое колбы нагревают до температуры на 10—15 °С
ниже предполагаемой температуры плавления препарата, измеряя
температуру в колбе термометром, после чего в пробирку помещают
укороченный термометр с капилляром так, чтобы ни термометр,
ни капилляр не касались дна и стенок пробирки. Затем
продолжают нагревать прибор, повышая температуру со скоростью 0,5 °С
в минуту. Если вещество в процессе определения температуры
плавления разлагается, то скорость нагрева увеличивают до 2—3 °С
в минуту или капилляр с препаратом перемещают в прибор,
предварительно нагретый до температуры приблизительно на 5°С
ниже предполагаемой температуры плавления.
Началом плавления считают появление первой капли
расплавленного препарата или появление мениска в капилляре, а концом—
888
момент полного расплавления препарата. Обе температуры
отмечают и считают интервалом температуры плавления препарата.
Прибор ПТП. В отличие от описанного выше, в приборе ПТП
(рис. 207) предусмотрен электронагрев капилляра с пробой.
Прибор предназначен для определения температуры плавления веществ
в диапазоне 20—360 °С при регулируемых скоростях нагрева 1, 2,
4, 6, 8 и 10 °С в минуту.
Основной частью прибора является блок-нагреватель 6\ он
состоит из двух сосудов из термостойкого стекла, вставленных один
в другой. На внутренний сосуд навита бифилярно константановая
спираль 5.
В блок-нагреватель устанавливают термометр 5, к которому
крепят капилляры 8.
Прибор снабжен номограммой 7, позволяющей при
определенной фиксированной глубине погружения термометров установить
напряжение, необходимое для заданной скорости нагревания.
Для удобства наблюдения за плавлением вещества и шкалой
термометра на приборе установлены оптическое приспособление
9 с фокусировкой и два осветителя 4 с рефлекторами.
Исследуемый образец тонкоизмельченного сухого вещества
помещают в капиллярную трубку §, запаянную с одного конца,
которая устанавливается так, чтобы столбик вещества находился на
уровне середины ртутного резервуара термометра. Затем
включают прибор в сеть.
По номограмме 1 определяют напряжение, соответствующее
необходимой скорости нагрева; включают нагреватель 6 и
осветители. При помощи ручки регулятора 10, следя за показанием
вольтметра 3, устанавливают необходимое напряжение. За процессом
плавления наблюдают при помощи оптического приспособления 9,
а за показаниями термометра — с помощью дополнительной
лупы.
В виду некоторого влияния температуры окружающей среды и
движения воздуха вокруг блока-нагревателя 6, необходима
дополнительная коррекция скорости нагрева, которая осуществляется
вручную регулятором напряжения 10.
Медные блоки. Температуру плавления высокоплавких веществ
(>300°С) лучше всего определять нагреванием капилляров в
медных блоках. Термометр помещают в горизонтальный канал блока,
на поверхности которого находится ряд углублений,
расположенных на равных расстояниях. Система каналов под острым углом
друг к другу позволяет рассматривать капилляр как в проходящем,
так и в отраженном свете от источника, установленного за
отверстием канала. Выходные отверстия каналов во избежание
движения воздуха закрыты стеклом. После предварительного грубого
определения температуры плавления образца термометр выдвигают
из блока настолько, чтобы можно было только отсчитать
ожидаемую температуру. Проба вещества устанавливается в углублении,
ближайшем к шарику термометра.
389
ч^/
Рис, 208. Прибор Баумана—Фрома для определения температуры
кристаллизации:
/ — насадка; 2— пробковая пластинка; £—-толстостенный сосуд; 4 — термо*
f метр; 5—мешалка; 6 — пробирка.
ОПРЕДЕЛЕНИЕ ТЕМПЕРАТУРЫ ПЛАВЛЕНИЯ
НА НАГРЕВАЕМОЙ ПОВЕРХНОСТИ
При определении температуры плавления легко
разлагающихся при нагревании веществ наиболее
точные результаты могут быть получены при
максимальном сокращении времени от момента начала
нагревания вещества до его плавления. Это
достигается применением различного типа блоков для
плавления на открытой поверхности. Одним из
таких блоков является нагреватель Кофлера.
Нагреватель представляет собой металлический
брусок длиной около 370 мм и шириной около 40 мм,
в котором посредством одностороннего
электрического обогрева создан перепад температур. Благодаря
форме бруска и использованию двух металлов с
различной теплопроводностью его температурный градиент
приближается к линейному. При помощи этого нагревателя можно
определять температуру плавления веществ в интервале 50—
250 °С. Для отсчета температур служит металлическая шкала с
ценой деления 2°С. Вмонтированный в прибор стабилизатор
исключает влияние колебаний напряжения в сети и обеспечивает
всегда одинаковое нагревание.
Исследуемое вещество насыпают непосредственно на
хромированную поверхность нагревателя. Вскоре можно увидеть более или
менее четкую границу жидкой и твердой фазами. Пределы
точности определения!—2°С.
W
КРИОСКОПИЧЕСКИЙ МЕТОД
Очень точно определить температуру плавления можно крио-
скопическим методом — по кривым охлаждения. Этот метод
позволяет установить не только температуру плавления, но и процентное
содержание примеси. При кристаллизации расплавленного вещества
в отсутствие переохлаждения температура остается почти
постоянной, что связано с выделением скрытой теплоты кристаллизации.
Недостатком этого метода является то, что для этого требуется
значительно больше испытуемого вещества, чем при определении
в капиллярах.
Определение температуры кристаллизации органических
реактивов и препаратов в пределах 20—100 °С по кривой охлаждения
проводят в приборе Баумана-Фрома (рис. 208).
Прибор Баумана-Фрома состоит из стеклянного
толстостенного сосуда 5, нижняя часть которого имеет диаметр 20 мм, а
верхняя — 50 ммГ В верхнюю часть помещают насадку 1 в виде
390
стаканчика. В дне насадки имеются два отверстия для
термометра 4 и мешалки 5. Последние свободно подвешены в отверстиях
на пробковых пластинках 2. Цилиндрический сосуд с насадкой
помещают в широкую пробирку 6 диаметром 40—45 мм. Весь
прибор помещают в стакан вместимостью 500 мл.
Испытуемый препарат помещают в сосуд 3 и расплавляют,
погружая в стакан с водой, нагретой до температуры на 15—20 °С
выше предполагаемой температуры кристаллизации. Слой
расплавленного препарата должен быть высотой 25—30 мм.
Сосуд с расплавленным препаратом вынимают из воды, насухо
вытирают, вставляют в пробирку 6 и закрепляют на штативе. В
расплавленный препарат погружают термометр 4 и мешалку 5 так,
чтобы они не касались ни дна, ни стенок сосуда 3 и чтобы ртутный
резервуар термометра был полностью погружен в испытуемое
вещество.
Испытуемое вещество в приборе охлаждают на 2—3°С ниже
предполагаемой температуры кристаллизации и осторожно
помешивают, не касаясь дна и стенок прибора.
В момент кристаллизации вещества температура
самопроизвольно повышается (в этот момент прекращают перемешивание)
и, достигнув определенного максимума, остается на этом уровне
в течение некоторого времени, а затем вновь начинает понижаться.
Высшую точку подъема температуры принимают за темпера*,
туру кристаллизации.
ГЛАВА37 ОПРЕДЕЛЕНИЕ ТЕМПЕРАТУРЫ КИПЕНИЯ
Температура кипения веществ, в отличие от температуры
плавления, очень сильно зависит от давления, и ее точное определение
связано со многими трудностями.
Под температурой кипения обычно подразумевают температуру
жидкости, при которой давление ее насыщенного пара над плоской
поверхностью раздела фаз равно внешнему давлению.
При незначительном отклонении давления насыщенных паров
от нормального выполняется правило:
ЛГ==С0(760-р)Г
где ДТ — отклонение температуры кипения от нормальной, К;
Со — эмпирический коэффициент; р — давление, при котором была
измерена температура Г, мм рт. ст.
Коэффициент Со имеет следующие значения:
Вещества с очень низкими температурами
кипения 0,00014
Большинство веществ 0,00012
Вода, спирты, карбоновые кислоты 0,00010
В большинстве случаев температуру кипения определяют при
помощи термометров, погруженных в паровую фазу. Температура
кипения, определяемая в парах, всегда ниже истинной, так как пар
несколько переохлаждается и требуется время для достижения и
установления теплового равновесия фаз жидкость — пар. Поэтому
при перегонке любой жидкости первые капли дистиллята
отгоняются при более низкой температуре. Наоборот, к концу перегонки,
когда в колбе остается мало жидкости, в результате изменения ее
состава за счет уменьшения содержания легколетучих примесей,
а также за счет перегрева дистиллят отгоняется при более высокой
температуре. В связи с этим Государственная фармакопея
определяет температуру кипения как интервал между начальной и
конечной температурами кипения при нормальном давлении (760 мм
рт. ст., или 1013 гПа). При этом начальной температурой кипения
считают температуру, при которой в приемник перегоняются первые
пять капель жидкости, а конечной — температуру, при которой
в приемник переходит 95% жидкости.
Но строго говоря, температурой кипения жидкости является
такая температура, которую показывает термометр,
соприкасающийся одновременно с жидкой и паровой фазами; в этом случае
температура измеряется в условиях равновесия (36, с. 710].
МИКРОМЕТОД ОПРЕДЕЛЕНИЯ ТЕМПЕРАТУРЫ КИПЕНИЯ
ПО СИВОЛОБОВУ
Этот метод может быть использован для ориентировочного
определения температуры кипения.
392
ТАБЛИЦА 15. Поправки для приведения наблюдаемой температуры
кипения к нормальному давлению
Температурные пределы
кипения, °С
| 10-30
i 30-50
| 50-70
1 70-90
1-
Поправка,
°С на каждый
мм рт. ст.
0,035
0,038
0,040
0,042
Температурные пределы
кипения, °С
90—110
110-130
130—150
150-170
Поправка,
°С на каждый
мм рт. ст.
0,045
0,047
0,050
0,052
Температурные пределы
кипения, °С
170—190
190-210
210—230
230—250
Поправка,
"С на каждый
мм рт. ст.
0,054
0,057
0,059
0,062
Маленькую стеклянную пробирку длиной 50 мм и диаметром
3 мм наполняют испытуемой жидкостью на высоту 7—10 мм и
помещают в нее запаянный с одного конца капилляр (длиной
55 мм, диаметром 1—1,2 мм) так, чтобы его открытый конец был
погружен в жидкость.
Пробирку резиновым или металлическим кольцом прикрепляют
к термометру, помещают в прибор для определения температуры
плавления (типа ПТОП) и нагревают. За 10—15 °С до
предполагаемой температуры кипения скорость нагревания уменьшают до 1 °С
в минуту. Вблизи точки кипения из капилляра начинают выделяться
отдельные пузырьки воздуха, число которых очень быстро
увеличивается, а затем появляется непрерывная цепочка маленьких
пузырьков пара испытуемой жидкости. Этот момент считают точкой
кипения вещества и отмечают показания термометра. Определение
повторяют несколько раз, применяя каждый раз новую
капиллярную трубку и новую порцию испытуемого вещества.
Окончательным результатом считают среднее арифметическое из всех
определений. Эту величину, приведенную к нормальному давлению,
принимают за температуру кипения.
Для этого сначала наблюдаемое давление приводят к
температуре 0°С, вычитая из показаний барометра:
2 мм рт. ст. при температуре окружающей среды 13—20 °С
3 мм рт. ст. » » » » 21— 28 °С
4 мм рт. ст. » » » » 29—35 °С
Затем, пользуясь табл. 15, в значение наблюдаемой температуры
кипения вносят поправку на каждый миллиметр ртутного столба
разности между нормальным давлением (760 мм рт. ст.) и
наблюдаемым, приведенным к 0°С.
При давлении ниже 760 мм рт. ст. поправку прибавляют, в
противном случае — вычитают.
ОПРЕДЕЛЕНИЕ ТЕМПЕРАТУРЫ КИПЕНИЯ ПО ГОСТ 18995.6—73
Прибор для определения температуры кипения (рис. 209)
состоит из круглодонной колбы 1 с отростком, к которому
присоединяют с помощью корковой пробки или на шлифе обратный
393
Ряс. 209. Прибор для определения температуры кипения по ГОСТ 18995.6—73:
7 —круглодонная колба с отростком; 2—термометр; 3 — насадка; 4 — баня.
Рис. 210. Эбуллиометр:
а —прибор; б —кольцевая стеклянная газовая горелка; /—резервуар; 2—отверстие для
внесения испытуемой жидкости; 3 — счетчик капель; 4—отверстие для соединения с холодильником:
5—пробирка, обвитая стеклянной спиралью.
холодильник. В колбу вставляют с помощью корковой пробки или
шлифа насадку 3 с отверстиями, а в нее помещают термометр 2 на
корковой пробке. Прибор помещают в баню 4 с жидким
теплоносителем или обогревают электронагревателем с закрытой спиралью.
Испытуемую жидкость слоем высотой 10—15 мм наливают
в колбу, на дно которой ставят несколько капилляров, запаянным
концом вверх, вставляют насадку так, чтобы расстояние от уровня
жидкости до нижних отверстий в насадке было 25 мм. Внутрь
насадки помещают термометр. При этом термометр должен полностью
находиться в парах испытуемой жидкости, чем исключается
необходимость внесения поправки на выступающий столбик ртути.
Прибор соединяют с обратным холодильником.
При определении температур кипения ниже 170°С через
рубашку холодильника пропускают воду; для более высококипящих
жидкостей применяют воздушное охлаждение. При нагревании
шарообразной части колбы пары испытуемой жидкости проходят через
отверстия внутрь насадки и омывают термометр.
Интенсивность нагрева регулируют так, чтобы с конца
холодильника падало 20—30 капель в минуту.
За температуру кипения принимают приведенное к нормальному
давлению значение температуры, которая оставалась постоянной
в течение 5—8 мин.
394
ОПРЕДЕЛЕНИЕ ТЕМПЕРАТУРЫ КИПЕНИЯ В ЭБУЛЛИОМЕТРЕ
Определение температуры кипения в эбуллиометре считается
наиболее точным методом для индивидуальных веществ с
температурой кипения ниже 200 °С.
Эбуллиометр (рис. 210) состоит из резервуара /, в который
через отверстие 2 наливают испытуемую жидкость. К внутренней
стенке резервуара для равномерного кипения жидкости припаяно
толченое стекло. Нагрев резервуара осуществляют при помощи
нихромовой спирали, подключенной к лабораторному
автоматическому трансформатору или при- помощи стеклянной кольцевой
газовой горелки, которая охватывает нижнюю часть резервуара.
Интенсивность нагрева измеряют количеством капель,
падающих в минуту со счетчика 3. Над счетчиком капель находится
отверстие 4 для соединения с обратным холодильником. В верхней
части прибора находится также ячейка 5, представляющая собой
впаянную в прибор пробирку, обвитую стеклянной спиралью.
Ячейку заполняют ртутью и вставляют в нее с помощью корковой
пробки термометр так, чтобы ртутный резервуар был полностью
погружен в ртуть. Поверхность ртути покрывают слоем
вазелинового масла и следят за тем, чтобы ртуть находилась под слоем
масла.
Все части прибора, за исключением счетчика капель и нижней
части резервуара У, обматывают асбестовым шнуром во избежание
охлаждения прибора.
Все соединения частей прибора должны быть пришлифованы.
Прибор помещают в кожух из листовой жести или алюминия.
В кожухе должно быть застекленное смотровое окно. Работу
проводят в вытяжном шкафу.
Определение выполняют следующим способом. В прибор через
отверстие 2 наливают 50 мл испытуемой жидкости, закрывают
притертой стеклянной пробкой, подводят воду в холодильник и
начинают обогрев. Кипящая жидкость и ее пары омывают ячейку 5 и
попадают на счетчик капель 3. Как*только со счетчика начинают
капать капли, нагрев регулируют так, чтобы в минуту падало 60 —
80 капель. Через 10 мин после начала кипения температуру
измеряют через каждые 2 мин. Наблюдение прекращают, когда
результаты последних пяти измерений совпадают в пределах 0,1 °С
глава38. ОПРЕДЕЛЕНИЕ ТЕМПЕРАТУРЫ ВСПЫШКИ
ЖИДКИХ И ПЛАВЯЩИХСЯ ПРОДУКТОВ
Температурой вспышки называется самая низкая температура
вещества, при которой в условиях специальных испытаний
образующиеся над его поверхностью пары или газы способны
вспыхивать в воздухе от постороннего источника зажигания;
устойчивого горения вещества при этом не возникает. Температура вспышки
является экспресс-параметром, ориентировочно показывающим
температурные условия, при которых горючее вещество
становится огнеопасным в открытом сосуде или при разливе. Температуру
вспышки горючих веществ используют при классификации
жидкостей по воспламеняемости и степени пожарной опасности.
Температура вспышки зависит от природы вещества и наличия
примесей легкоиспаряющихся компонентов. Некоторое влияние
на температуру вспышки оказывают атмосферное давление и
влажность воздуха. С повышением давления температура
вспышки повышается примерно на 0,0345 °С на каждый миллиметр
ртутного столба. Температура вспышки в значительной степени
зависит от методики определения и конструкции прибора. Различают
два метода определения температуры вспышки: в открытом тигле
и закрытом тигле.
ОПРЕДЕЛЕНИЕ ТЕМПЕРАТУРЫ ВСПЫШКИ В ОТКРЫТОМ ТИГЛЕ
(ПО ГОСТ 4333-48)
Прибор состоит из железного тигля диаметром 63—65 мм,
высотой 46—48 мм со стенками толщиной 1 мм. Тигель помещают
в металлическую песчаную баню высотой 45—55 мм и диаметром
95—105 мм, установленную на штативе, на котором укреплен
термометр ТН-2 на 360 °С с ценой деления 1 °С
(рис. 211). Баня нагревается газовой горелкой.
Зажигательное устройство представляет собой
стеклянную трубку с оттянутым концом
диаметром 1 мм, соединенную с источником
горючего газа. Для защиты от движения воздуха
прибор во время определения окружают
железным щитом.
Тигель промывают бензином, высушивают
и подогревают на пламени газовой горелки.
После охлаждения его ставят в песчаную баню,
на дне которой находится слой прокаленного
песка толщиной 5—8 мм. Пространство между
тиглем и баней засыпают песком, так чтобы его
Рис. 211. Прибор для определения температуры вспышки в
открытом тигле,
396
и и и
Рис. 212. Приборы для определения температуры вспышки в закрытом тигле:
а—ПВНЭ: /—металлический стакан с электронагревательным элементом; 2 — металлический
кожух на треноге, заполненный внутри асбестом или другим теплоизоляционным материалом;
8—тигель и крышка с укрепленными на ней деталями и газовой зажигательной лампочкой.
б — ПНВО: / — металлический (чугунный) сосуд на треноге с сеткой; 2 —металлический
колпак; 3—тигель и крышка с укрепленными на ней деталями, фитильной зажигательной л:м-
почкой и термометром; 4 — бензиновая горелка.
уровень на 12 мм не доходил до края тигля. Испытуемый
продукт, предварительно обезвоженный, наливают в тигель, так
чтобы уровень жидкости отстоял от края тигля на 12 мм для веществ
с температурой вспышки ^220 °С или 18 мм при температуре
вспышки >220°С.
Термометр устанавливают строго вертикально, причем так,
чтобы шарик его находился посредине столба жидкости в тигле.
Под песчаной баней устанавливают горелку и нагревание
регулируют так, чтобы температура поднималась на 10°С в минуту. За
40 °С до ожидаемой температуры вспышки скорость нагревания
уменьшают до предполагаемой температуры вспышки, и через
каждые 2°С проносят на расстоянии 10—14 мм поверхности
жидкости пламя зажигательного устройства. Время проведения
пламенем от одного края тигля до другого 2—3 с. За вспышку
принимают момент появления перебегающего и быстро исчезающего
синего пламени над всей поверхностью испытуемой жидкости.
Температура, соответствующая этому моменту, является
температурой вспышки.
При повторном определении берется новая порция испытуемого
вещества. Расхождение между параллельными определениями не
должно превышать ±4°С для веществ с температурой вспышки
^150°С и ±6°С для других веществ.
397
ОПРЕДЕЛЕНИЕ ТЕМПЕРАТУРЫ ВСПЫШКИ В ЗАКРЫТОМ ТИГЛЕ
(ПО ГОСТ 6356—75)
Приборы для определения температуры вспышки в закрытом
тигле в интервале 20—275 °С изготовляются двух типов: типа
ПВНЭ — с электрическим нагревом (рис. 212) и типа ПВНО —
с огневым нагревом.
Прибор состоит из металлического закрытого тигля 4, который
помещен в чугунный стакан 1 с электронагревательным элементом;
последний вмонтирован в заполненный асбестом металлический
кожух 2. Тигель с внутренней стороны имеет метку для указания
уровня налива испытуемой жидкости. Крышка тигля 3 снабжена
заслонкой с двумя отверстиями, гнездом для термометра,
зажигательным приспособлением, пружинным рычагом и мешалкой с
гибкой передачей. Поворотом пружинного рычага открываются
заслонки и наклоняется в паровое пространство тигля зажигательное
приспособление.
Прибор устанавливают в помещении, где отсутствует резкое
движение воздуха. Снимают с прибора термометр, крышку с
мешалкой и вынимают тигель. Испытуемую жидкость наливают в
тигель до метки, устанавливают его на место и закрывают
крышкой. В крышке укрепляют термометр, проверяют, работают ли
мешалка, пружинный рычаг, и зажигают фитиль зажигательного
устройства. Затем нагревают прибор, повышая температуру на
1 °С в минуту при непрерывном перемешивании для веществ с
температурой вспышки <50°С; для веществ с предполагаемой
температурой вспышки 50—150 °С скорость нагрева при
периодическом перемешивании в периоды между включениями
зажигательного устройства 5—8°С в минуту; для веществ с более высокой
температурой вспышки скорость нагревания 10—12 °С в минуту.
За 30 °С до ожидаемой температуры вспышки скорость
нагревания уменьшают до 2°С/мин.
Испытание на вспыхивание испытуемой жидкости проводят
через 1—2°С, для чего на 1 с поворачивают пружинный рычаг и
наблюдают, не появилось ли синее быстро исчезающее пламя над
поверхностью жидкости. Получив первый результат, продолжают
нагревание и через 1—2°С повторяют зажигание. Если вспышки
не происходит, испытание считают неправильным и повторяют его
снова со свежей порцией испытуемой жидкости.
Расхождение между параллельными определениями не должно
превышать 1— 2°С
глава 39. ЭЛЕКТРОПИТАНИЕ
И ИЗМЕРЕНИЕ ЭЛЕКТРИЧЕСКИХ ВЕЛИЧИН
Для питания измерительных схем и приборов необходимы
источники стабилизированного постоянного и переменного тока, которые
должны удовлетворять различным требованиям в отношении
выходного напряжения, мощности, стабильности и др. В большинстве
химических лабораторий постоянный ток получают путем
выпрямления переменного тока сети и с помощью автономных источников
питания — химических источников тока.
ИСТОЧНИКИ ПИТАНИЯ
Сетевые источники тока
Напряжение переменного тока промышленной частоты (50 Гц),
подводимого в лабораторию, может быть 127, 220 и 380 В,
Индикацию напряжения можно выполнить сигнальной
неоновой лампочкой, свечение которой зависит от напряжения
тока. Так, порог зажигания неонового индикатора МИН-1
составляет 90 В; верхний предел индикации 500 В.
Электроэнергия, поступающая из сети переменного тока,
стабилизируется, трансформируется до необходимой величины
напряжения и выпрямляется.
Для стабилизации напряжения переменного тока
большей частью используют электромагнитные и феррорезонанс-
ные стабилизаторы. Например, для поддержания неизменным
переменного напряжения на входных зажимах приемников энергии
часто пользуются электромагнитными стабилизаторами типа С-0,09
и С-0,16 с номинальной мощностью 0,09 и 0,16 кВт соответственно.
Напряжение стабилизированного тока 127 ±5 В; ток нагрузки
0,71—0,73 А.
Для питания цепей электроизмерительных приборов и установок
можно применять стабилизатор переменного напряжения П71М,
особенно в случаях, когда необходимо иметь высокостабильное
напряжение. Номинальное стабилизированное напряжение 220 В; ток
нагрузки 1,7 А.
Стабилизатор переменного напряжения СЭМ-1 используется
для питания источников света в оптико-механических приборах.
Напряжение стабилизированного тока 220 ±2 В.
Если требуются низкие напряжения при малых токах,
необходимое понижение сетевого переменного напряжения
осуществляют с помощью лабораторных трансформаторов.
Наиболее простой способ получения постоянного тока —
преобразование переменного тока с помощью
выпрямителей. Последние все больше вытесняют электромашинные
преобразователи переменного тока в постоянный.
399
Основной элемент выпрямителей — электрический вентиль, по
типам которых выпрямители разделяют на электронные
(кенотронные) и полупроводниковые. Наибольшее распространение получили
полупроводниковые выпрямители (селеновые, кремниевые и
германиевые) .
Полупроводниковые выпрямители, в особенности селеновые,
обладают рядом преимуществ и чаще других используются в
химических лабораториях при относительно больших токах нагрузки и
относительно малых напряжениях.
В качестве примера приводим данные о селеновом выпрямителе
типа ВСА-А10, который используется для зарядки аккумуляторных
батарей напряжением 6 и 12 В, емкостью до 135 А-ч. Выпрямитель
может быть также использован как источник постоянного тока
для работы на активную нагрузку. Питание от сети переменного
тока 127/220 В.
Напряжение (соответственно ток), снимаемое с сетевого
выпрямителя, должно быть постоянным и не зависимым от
отбираемой мощности и от колебаний сетевого напряжения. В известной
мере этим требованиям отвечают стабилизатор напряжения
постоянного тока П136М2 и источник калиброванного напряжения
П137. Последний прибор предназначен для питания цепей
электроизмерительных приборов, а также для измерения постоянного тока
в диапазоне 2—300 В.
Выпускаемые стабилизированные выпрямителя постоянного
тока типа ВС и ВСП обеспечивают выходное напряжение 3—200 В,
с максимальным током нагрузки 0,2—10 А.
В лабораторной практике находят применение разнообразные
устройства для зарядки аккумуляторов, которые
могут быть использованы и как источники стабилизированного
постоянного тока для работы на активную нагрузку, например
выпрямитель зарядный В31—5А и малогабаритный универсальный
выпрямитель ВЗУ.
В последние годы появилось большое число унифицированных
блоков питания, которые используют в качестве составных
частей (сублоков) устройств электропитания приборов
стабилизированным постоянным током. Каждый блок питания состоит из
функциональных узлов: выпрямителя с фильтром, стабилизатора
напряжения и узла защиты от короткого замыкания (для блоков
с выходным напряжением 27 и 50 В).
Одним из таких блоков питания является модель 2БП2,
Напряжение питающей сети переменного тока 380/220 В. Номинальное
выходное стабилизированное напряжение постоянного тока 6,3; 10
и 27 В. Номинальный ток нагрузки 4 и 2 А.
Химические источники тока
Химическими источниками тока называют устройства, с
помощью которых свободная энергия пространственно разделенных
окислительно-восстановительных реакций превращается в
электрическую энергию,
400
Процесс превращения химической энергии в электрическую в
химическом источнике тока называется разрядом. Химические
источники тока, используемые в лабораторной практике, по
характеру работы делятся на две группы: гальванические элементы и
аккумуляторы.
К гальваническим элементам, группе первичных
химических источников тока, относятся все устройства, которые
допускают лишь однократное использование заключенных в них
активных веществ, принимающих участие в токообразующих
электрохимических процессах; их разряд (отдача электрической
энергии) может быть осуществлен в один или несколько приемов.
Полностью разряженный гальванический элемент к дальнейшей
работе не пригоден. Гальванические элементы бывают с жидким
электролитом и сухие, содержащие невыливающийся электролит.
Химические источники тока, работоспособность которых после
их разряда может быть восстановлена путем заряда, т. е.
пропусканием постоянного тока в направлении, обратном направлению
тока при разряде, называют аккумуляторами. В отличие от
гальванических элементов, аккумулятор сам энергию не
производит, он ее накапливает при заряде (электрическая энергия
превращается в химическую) и отдает при разряде (химическая энергия
превращается в электрическую). Электрод, который при разряде
аккумулятора был анодом, при заряде становится катодом.
ТАБЛИЦА 16. Важнейшие электрохимические системы
первичных элементов и аккумуляторов
Гальванические элементы
Марганцево-цинковый МЦ с
солевым электролитом
Марганцево-кислородно-цинко-
вый ВМЦ
Марганцево-цинковый МЦ со
щелочным электролитом
Ртутно-цииковый РЦ
Медио-магниевый МХМ
Кислотный (свинцовый)
Никель-кадмиевый НК
Никель-железный НЖ
Серебряно-цинковый
1 + Мп021 NH4C1 | Zn -
+ Mn021 NH4C1 | Zn -
o2
+ Mn021 KOH I Zn -
+ HgO|KOH|Zn -
+ Cu2Cl2|MgCl2|Mg -
Аккумуляторы
| - Pb | H2S041 Pb02 +
- Cd | KOH | NiO(OH) +
- Fe | KOH | NiO(OH) +
- Zn | KOH 1 Ag20 +
1,5-1,8
1,5
1,5
1,34
1,75
2,1
1,36
1,40
1,25
232
250
240
255
410
175
220
279
459
* Условная величина энергии, получаемой с 1 кг массы активных веществ, считая
коэффициент их использования равным единице (100%).
14
401
ТАБЛИЦА 17. Основные параметры химических источников тока
Параметр
Электродвижущая
сила (э. д. с.)
Начальное
напряжение
Емкость
Продолжительность
работы
Гарантийный срок хра-
нечия
Определение параметра
Разность потенциалов между
выводами химического источника
тока при разомкнутой внешней цепи
Разность потенциалов между
выводами химического источника под
нагрузкой
Количество электричества,
отдаваемое (потребляемое) химическим
источником тока при его разряде
(заряде)
Время работы химического
источника тока при разряде его до
обусловленного конечного
напряжения при подключении внешней
цепи, имеющей установленное
сопротивление
Время до начала эксплуатации, в
течение которого элемент
сохраняет обусловленную часть
емкости, или продолжительности
работы
Единица измерения
Вольт (В)
Вольт (В)
Ампер-час (А • ч)
или ватт-час
(В-ч)
Час (ч), число
циклов
заряд-разряд
Месяц
Совокупность веществ, участвующих в токообразующей
реакции, называется электрохимической системой.
Химические источники тока делятся на типы в зависимости от
электрохимической системы. Электрохимическая система источников тока
имеет свое условное обозначение (табл. 16). В этом обозначении
между двумя вертикальными черточками пишется формула
электролита, а слева и справа — формулы активных веществ,
принимающих участие в токообразующих процессах.
Работа химических источников тока характеризуется рядом
параметров, которые зависят от электрохимической системы и
конструкции (табл. 17).
Практически химический источник тока перестает работать до
того, как полностью израсходовались активные вещества. Часть их
расходуется на бесполезные процессы, которые называют
саморазрядом. Процесс саморазряда сопровождается выделением
газообразных водорода или кислорода.
Конструктивное исполнение химических источников тока может
быть самым различным, но все они состоят из двух электродов,
разделенных слоем электролита (жидкого или загущенного).
Химические источники тока можно соединять последовательно,
когда требуется получать высокое напряжение, и параллельно,
когда необходимо иметь большой ток. Для повышения напряжения
или емкости (продолжительности работы) или того и другого
одновременно, элементы и аккумуляторы соединяют электрическими
контактами в батареи. Различают аккумуляторные батареи и
батареи элементов,
402
таблица 18. Первичные элементы и батареи
Гарантийный срок хранения 12 месяцев.
Элемент (батарея)
Элемент 343 «Орион»
» А-314 «Карат»
» А-316 «Квант»
» А-332 «Ореол»
» А-336 «Свет»
» А-343 «Салют»
» А-343 «Мир»
Батарея 3336 «Планета-1»
» «Рубин-1»
Начальное
напряжение.
В
1,5-1,55
1,50
1,50
1,50
1,50
1,50
1,50
3,9
4,5
Разрядное
сопротивление,
Ом
5
40
40
5
5
5
5
15
100
Продолжительность
работы, ч
15
25
45
6
6
12,5
15
7
120
f Сухие элементы и батареи. Они предназначены для питания
различных устройств в тех случаях, когда необходим автономный
и портативный источник постоянного тока небольшой мощности.
Выпускают элементы и батареи, готовые к действию
(заряженные), содержащие электролит, и резервные (наливные), которые
приводятся в действие путем контакта электродов с электролитом
или перевода электролита в рабочее состояние.
По конструктивной форме элементы подразделяются на
прямоугольные, цилиндрические и галетные (плоские).
На каждом элементе (батарее) заводского изготовления
указаны номер ГОСТ или ТУ, условный номер или наименование,
номинальное напряжение, гарантийный срок хранения, дата
изготовления и полярность выводов («+» и «—»).
Цилиндрические элементы сухие и прямоугольные батареи
марганцево-цинковой системы с солевым или .щелочным
электролитом часто используются в лабораторной практике (табл. 18).
Аккумуляторы. По типу электролита аккумуляторы
подразделяются на кислотные (свинцовые) и щелочные —
никель-кадмиевые (НК) и никель-железные (НЖ).
Кислотный аккумулятор состоит из сосуда
(стеклянного, эбонитового, пластмассового или деревянного, выложенного
листовым свинцом), пластин-электродов, сепараторов и
электролита. Решетка пластин отлита из свинца с добавкой веществ,
повышающих твердость свинца (Sb, Sn, Sr).
Сепараторы — тонкие прокладки из пористого материала (типа
мипоры) служат для предупреждения замыкания между
положительными и отрицательными пластинами. Ячейки решетки
заполнены пастообразной активной массой. Сосуд закрывается крышкой
с тремя отверстиями: по краям для вывода полюсных штырей, а
среднее — для заливки электролита; оно плотно закрывается
металлической пробкой на резьбе.
Щелочные аккумуляторы состоят из электродов,
собранных в комплекте положительных и отрицательных пластин,
стального сосуда и ряда деталей (выводы, пробка и др.). Внутри
14* 403
пластин заключена активная масса в виде отдельных пакетов
(ламелей); между пластинами проложены эбонитовые'палочки.
В никель-кадмиевых аккумуляторах крайние положительные
пластины соединены со стенкой металлического сосуда,
следовательно, корпус является положительным полюсом. В никель-железных
аккумуляторах корпус металлического сосуда соединен с
отрицательным полюсом.
Кислотные и щелочные аккумуляторы, как правило,
выпускаются в сухом виде, и поэтому для эксплуатации их требуется
заполнить соответствующим электролитом и вслед за этим зарядить.
Кислотные аккумуляторы при хранении в сухом виде с закрытыми
пробками не портятся; щелочные рекомендуется хранить в сухом
виде не более двух месяцев и притом обязательно с закрытыми
пробками.
Электролиты для аккумуляторов. Электролитом
для кислотных аккумуляторов служит 35—40% раствор
химически чистой H2S04 в дистиллированной воде (р = 1260 — 1300 кг/м3).
При правильном уходе за аккумулятором электролит не требует
замены на протяжении 12—16 месяцев. Гарантированная емкость
щелочных аккумуляторов обеспечивается при применении
составного электролита из раствора КОН (р = 1190-^ 1210 кг/м3) с
добавкой 20 ±1 г/л LiOH-H20. В качестве составного электролита
можно также применить раствор NaOH (р = 1170-г- 1190 кг/м3) с
добавкой 10 г/л LiOH-H20. В щелочных аккумуляторах
электролит подлежит периодической замене через 8—10 месяцев.
При приготовлении электролита, как кислотного, та-к и
щелочного, имеет место разогревание. Наливать в аккумуляторы
горячий электролит нельзя, надо дать ему остыть до комнатной
температуры. В аккумулятор наливают электролит через воронку в
таком количестве, чтобы уровень раствора был по крайней мере
на 10—12 мм выше пластин. О высоте этого уровня можно
судить, опуская в отверстие для пробки узкую стеклянную трубку.
Закрыв верхний конец трубки пальцем, трубку осторожно
вынимают и отмечают высоту уровня жидкости в ней. Затем
отверстие в крышке аккумулятора плотно закрывают.
Ввиду того что электролит частично впитывается в пластины,
через некоторое время нужно долить электролит и измерить его
плотность. Для замера плотности при температуре близкой к
20 °С кроме обычного ареометра часто используют сифонный
ареометр (см. гл. 34).
Электролит сменяют через каждые 100—150 зарядно-разряд-
ных циклов, руководствуясь заводской инструкцией. Перед
сменой электролита аккумуляторы разряжают. Старый электролит
выливают, энергично встряхивая аккумулятор (батарею) для
удаления грязи из сосуда и промывают дистиллированной водой.
Промытые аккумуляторы следует немедленно залить
свежеприготовленным электролитом, дать постоять 2 ч, затем замерить
плотность раствора в аккумуляторе, довести ее до требуемой
величины и закрыть аккумуляторы пробками,
404
Приведение аккумуляторов в рабочее
состояние. Для приведения аккумуляторов в рабочее состояние масса
электродов должна быть отформована. Процесс
электрохимического формования осуществляется путем заряда и разряда
аккумулятора. Формование никель-железных аккумуляторов
заканчивается после двух зарядно-разрядных циклов, для
никель-кадмиевых — через 3—4, для кислотных — через 6—8 циклов.
В заводских инструкциях по эксплуатации аккумуляторов
предусматриваются нормальные зарядные и разрядные режимы, при
которых гарантируется надежная работа аккумуляторов. Для
заряда аккумулятор подключают к внешнему источнику постоянного
тока плюсом к плюсу и минусом к минусу. Конечные напряжения
заряда и разряда являются нормированными величинами (ГОСТ
9241—71).
Конечное напряжение разряда для кислотных аккумуляторов
при нормируемых режимах разряда составляет 1,7—1,8 В, а для
щелочных 1 В. Конечное напряжение заряда составляет для
кислотных аккумуляторов 2,6—2,7 В, а для щелочных 1,56—2,05 В.
Критерием окончания заряда является совокупность
признаков: напряжение остается постоянным в течение часа; плотность
электролита в течение этого времени не изменяется; происходит
сильное газообразование.
При заряде аккумулятора нельзя допускать повышения
температуры электролита выше 40—45 °С, так как емкость
аккумулятора при этом резко уменьшается.
Причины порчи аккумуляторов. Основными причина-*
ми порчи кислотных аккумуляторов являются саморазряд,
достигающий около 1% его емкости в сутки, и сульфатация —
отложение на поверхности пластин Na2S04, плохо проводящего ток.
Быстрый саморазряд может возникнуть из-за коротких замыканий
м^жду пластинками вследствие выпадения активной массы из
пластин, а также из-за чрезмерных токов при заряде и разряде,
Сульфатация является результатом несвоевременного заряда
разряженных аккумуляторов. Особенно бурно происходит сульфата*
ция пластин при переплюсовке аккумулятора, т. е.. вследствие
неправильного подключения аккумулятора к источнику постоянного
тока при заряде. Об определении полярности электродов см. при*
ложение 3.
Порча щелочных аккумуляторов вызывается в основном
перегреванием электролита, зарядом током больше нормированного,
переплюсовкой, образованием карбонатов и закупоркой клапанов
пробок.
Правила ухода за аккумуляторами.
1. Не допускать при разряде уменьшения напряжения аккуму*
ляторов ниже нормируемых величин. Замерять напряжение мож-<
но аккумуляторным пробником типа М209 или надежным
вольтметром.
2. Не допускать коротких замыканий, в особенности у
кислотных аккумуляторов,
405
3. Не превышать нормированных значений тока как при
заряде аккумуляторов, так и при их разряде, т. е. при питании
электрической цепи.
4. Следить за емкостью аккумуляторов. Уменьшение емкости
у свинцовых аккумуляторов — первый признак сульфатации
пластин, у щелочных — необходимости замены раствора
электролита.
5. При длительном перерыве в эксплуатации аккумуляторов
необходимо: щелочные аккумуляторы зарядить и хранить
заполненными (2—2,5 месяцев), а кислотные, после полного разряда,
слив электролит, промыть дистиллированной водой и хранить с
закрытыми пробками в сухом виде.
6. Внимательно следить за исправностью клапанов в пробках
щелочных аккумуляторов, а у свинцовых — чтобы не засорилось
отверстие в пробке, служащей для выхода газа.
7. Следить за чистотой крышек и клемм аккумуляторов. Для
удаления налетов и кристаллических осадков следует пользовать*
ся деревянной палочкой и куском ткани. Полезно крышки
смазать вазелином.
8. При заряде аккумуляторов надо вывертывать пробки.
Через 10—15 мин после окончания заряда пробки завертывают и
тряпкой удаляют с крышек раствор, выплескивающийся обычно
в конце заряда вследствие сильного газообразования. Нельзя к
пробкам аккумуляторов подносить огонь, так как под их
крышками находится гремучий газ.
ИЗМЕРЕНИЕ ЭЛЕКТРИЧЕСКИХ ВЕЛИЧИН
Методы и средства измерения электрических величин весьма
разнообразны. Менее точные измерения (с погрешностью около
0,1%) осуществляются приборами непосредственной оценки.
Более точные измерения выполняют с помощью приборов сравнения
определяемой величины с образцовой мерой.
Приборы непосредственной оценки электрических величин —
показывающие приборы — используются также и для косвенного
определения некоторых неэлектрических величин, как например,
определения температуры термометром сопротивления,,
освещенности при помощи люксметра и т.д.
Приборы непосредственной оценки
Эти приборы дают значение измеряемой величины,
определяемое по предварительно проградуированным счетным
приспособлениям (шкалы, диаграммы). Их назначение определяется
измеряемой величиной (ток, напряжение и др.). Они характеризуются
классом точности, системой (определяемой принципом действия
прибора), диапазоном измерений и условиями эксплуатации.
Показывающие измерительные приборы (стрелочные, со
световым указателем и цифровые) выпускаются для измерений в
целях переменного тока, постоянного тока или в цепях переменного
и постоянного тока.
406
Выбор соответствующего измерительного прибора должен
быть сделан на основе справочных данных.
Лабораторные и переносные показывающие
электроизмерительные приборы обычно имеют несколько пределов измерений,
что достигается с помощью встроенных в прибор переключателей.
На лицевой панели размещаются зажимы для включения прибора
в схему, переключатели и букса корректора для установки
указателя на отметку механического нуля.
Шкалы многих приборов класса точности 0,1; 0,2; 0,5 и 1,0 со
стрелочным указателем снабжены зеркальными антипараллакс-
ными устройствами. Приборы со световым указателем
погрешности от параллакса не имеют.
Гальванометры — электроизмерительные приборы
высокой чувствительности для измерения малых значений тока,
напряжения или количества электричества. Гальванометры также
служат нулевыми приборами для определения с большой точностью
отсутствия тока в замкнутой электрической цепи или нулевой
разности потенциалов между двумя точками в ее параллельных
ветвях.
В лабораторной практике применяются гальванометры
постоянного и переменного тока. Наиболее распространены
гальванометры постоянного тока магнитоэлектрической измерительной
системы — стрелочные для нулевых измерений в мостовых и
компенсационных системах и зеркальные для индикации и измерения
тока, напряжения, заряда магнитного потока, сравнения токов в
двух независимых цепях.
Амперметры — приборы для измерения электрического
тока, включаемые последовательно в цепь этого тока. В
зависимости от значений номинальных токов различают амперметры,
миллиамперметры и микроамперметры.
Значение электрического тока, измеряемого амперметром,
определяется по отклонению подвижной системы амперметра. Для
измерения постоянного тока применяют главным образом
амперметры магнитоэлектрической системы — одно- и многопредельные.
Благодаря конструкции переключателя можно переключать
пределы измерения без разрыва электрической цепи.
Для измерения переменного тока также применяют
многопредельные амперметры.
Вольтметры—приборы для измерения напряжения между
двумя точками электрической цепи, подключаемые параллельно
приемнику энергии.
Для уменьшения ошибки измерения вольтметр должен
потреблять возможно меньший ток, т. е. вольтметр тем лучше, чем
больше его собственное внутреннее сопротивление.
Для выполнения различных измерений служат переносные
вольтметры разных систем и классов точности, с различным
внутренним сопротивлением, одно- и многопредельные,
различные по чувствительности и роду тока и разнообразные по
конструкции.
407
Электрические измерения методом сравнения
В практике химических лабораторий часто пользуются
нулевым методом и з м е р е н и я, основанным на приведении к
нулю разности измеряемой и известной величины, так что их
совокупное действие на нулевой прибор (гальванометр) полностью
уравновешивается.
Примерами нулевого метода измерения являются
компенсационный метод определения напряжения малых постоянных токов
и мостовой метод измерения сопротивлений.
Точность нулевого метода определяется классом точности
образцовых мер и чувствительностью нулевого прибора
(гальванометра).
Компенсационный метод измерения напряжений- Сущность
метода заключается в том, что к измеряемому напряжению
подключают в противоположном направлении точно известное
напряжение (образцовая мера). Компенсирующее напряжение
снимают с образцового потенциометра.
Потенциометры (компенсаторы) — приборы
сравнения, служащие для измерения компенсационным методом э.д.с,
напряжений (реже токов) или величин, функционально с ними
связанных. В этих приборах в процессе измерения производится
сравнение измеряемой величины или ее части с электрической
мерой, например с э.д.с. нормального элемента.
Различают потенциометры постоянного и переменного тока,
одно- и многопредельные.
В практике химических лабораторий используются большей
частью потенциометры постоянного тока с двумя рядами
измерительных декадных переключателей, с диапазонами измерения
0,01 мкВ—2,121111 В и универсальный потенциометр
постоянного тока УПЛ-80-2 с тремя верхними пределами измерения:
2,511110; 0,11110 и 0,011111 В.
В качестве меры э.д.с. для проверки и градуировки
электроизмерительных приборов в лабораторных и цеховых условиях
служат нормальные элементы.
Нормаль н'ы е элементы — обратимые гальванические
элементы, дающие при постоянных температурах и давлении
устойчивые э.д.с, значения которых могут быть точно воспроизведены.
В зависимости от концентрации электролита различают
насыщенные и ненасыщенные нормальные элементы.
В насыщенных нормальных элементах (НЭ-65 и Х-482)
положительным электродом служит ртуть, покрытая пастой из Hg2S04
(деполяризатор), растертого со ртутью и кристаллами CdS04»8H20, a
отрицательным — амальгама кадмия (10% Cd и 90% Hg).
Электролитом служит насыщенный раствор CdS04, подкисленный 0,03 н.
H2S04 и содержащий избыток кристаллов CdS04«8H20.
В насыщенных элементах (Х-485 и Х-487) электролитом
служит насыщенный при 4 °С водный раствор CdS04 (т.е. при ком*
натной температуре ненасыщенный).
408
Рис. 213. Мост Уитстона:
/ — передвижной контакт; 2—гальванометр; а и б—части реохорда; #-~известное
сопротивление; Л —измеряемое сопротивление.
Рис. 214. Уравновешенный измерительный мост.
Э.д.с. нормальных элементов зависит от температуры;
нормальной температурой для этих стандартных мер считается 20 °С.
Нормальный элемент Х-485 класса точности 0,005; э.д.с. при
20 °С равна 1,01880—1,01960 В. Рабочий диапазон температур
17—23 °С.
Нормальный элемент Х-487 класса точности 0,001; термостати-*
рован; его э.д.с. при 20 °С равна 1,01854—1,01873 В.
Мостовой метод измерения сопротивления. Метод измерения
электрических сопротивлений постоянному или переменному току
при помощи измерительных мостов находит широкое применение
в измерениях физических величин, функционально связанных с
электрическим сопротивлением (удельная проводимость и
температурный коэффициент сопротивления — при постоянном токе,
емкость, частота и др.— при переменном).
Измерительные мосты — приборы для измерения
электрического сопротивления постоянному или переменному току
методом сравнения с образцовым сопротивлением.
В простейшем случае измерение сопротивлений
осуществляется при помощи мостика сопротивления, построенного по
принципу моста Уитстона (рис. 213). При измерении скользящий
контакт 1 потенциометра перемещают до тех пор, пока стрелка
гальванометра 2 не станет на нуль. В этом положении неизвестное
сопротивление относится к известному сопротивлению, как длина
участка реохорда а к длине участка Ъ\
. X = Ra/b
Большинство измерительных мостов построено по принципу
сравнения с образцовым сопротивлением в схеме замкнутого
четырехугольника (рис. 214). Сопротивления h—/4, образующие
замкнутый четырехугольник, называются плечами моста. В одну
из диагоналей моста включается источник питания, в другую
(измерительную) индикатор нуля (##). При соотношении плеч
409
l\h = hU напряжение на измерительной диагонали равно нулю и
ток в ИН отсутствует — мост находится в равновесии. Зная
сопротивления трех плеч моста, можно определить неизвестное
сопротивление. Такие измерительные мосты называют
уравновешенными.
В качестве мер электрического сопротивления используют
меры постоянного значения—измерительные катушки
электрического сопротивления и меры ступенчато-переменного значения —
магазины сопротивлений. Последние служат в качестве
регулируемых мер сопротивления в цепях постоянного тока (тип Р 362)
и в цепях постоянного и переменного тока (тип Р 517 М).
Для измерения емкости и тангенса угла диэлектрических
потерь в цепях переменного тока применяют мост измерительный
Р 5026.
Приложения
1. ПОСТОЯННАЯ ВЛАЖНОСТЬ
В замкнутом пространстве (например, в эксикаторе) можно поддерживать
постоянную влажность, применяя некоторые кристаллогидраты, водные растворы
солей и кислот. Относительная влажность воздуха (газа) над твердой фазой
или раствором Л (в %) вычисляется по формуле:
А = -^- - 100
Р2
где pi — давление водяного пара, /?2 — давление насыщенного водяного пара
при той же температуре. Ниже приводятся значения pi при 20 °С.
Твердая фаза
Ca(N03)2-4H20
СаС12-6Н20
CuS04'5H20
Mg(N03)2 • 6H20
LiCl • Н20
К2С03 • 2Н20
Na2S04 • ЮН20
Na2HP04 • 12Н20
Na2C03 • ЮН20
Na2S203 • 5Н20
ри гПа
12,8
7,5
22,7
12,5
3,5
10,0
21,5
22,0
21,3
18,0
А, %
55
32
98
55
15
44
93
95
91
78
Насыщенный
раствор
K2S04
kno3
К2СЮ4
КС1
NaN03
NaCi
NH4N03
H2S04
10% (масс.)
20% »
30% >
40% »
50% »
Pi. Ша
23,1
22,2
20,4
20,1
17,5
17,5
14,7
22,08
20,23
16,93
12,65
7,91
A. %
99
95
88
86
75
75
63
94,7
86,8
72,6
54,2
33,9
2; ПРИГОТОВЛЕНИЕ НЕКОТОРЫХ РЕАГЕНТОВ
Аналитические реагенты
Бромная вода. Насыщенный водный раствор брома. Приготовляют
встряхиванием 3 мл брома со 100 мл дистиллированной воды. Сохраняют в склянке
оранжевого стекла с притертой пробкой, в защищенном от света месте. На дне
склянки должен находиться слой жидкого брома. Перед употреблением жидкость
встряхивают и после отстаивания сливают прозрачный слой.
Известковая вода. Одну часть (по массе) СаО гасят 5 частями воды,
переносят кашицу в бутыль, прибавляют 15 частей воды, сильно взбалтывают и
оставляют на 4—5 ч, после чего надосадочную жидкость сливают и отбрасывают.
Остаток обливают 5 частями холодной воды, взбалтывают, закрывают бутыль
пробкой и оставляют в прохладном месте на 3—4 дня, время от времени
взбалтывая.
Для употребления известковую воду по мере надобности сливают и
фильтруют. Содержание Са(ОН)2 в прозрачном фильтрате 0,15—0,17%.
Раствор крахмала. 2 г растворимого крахмала и 10 мг Hgl2 (консервируй
ющее вещество) растирают в ступке с 5 мл дистиллированной воды, кашицу
медленно вносят при энергичном перемешивании в 200 мл кипящей воды. Кп-
пятят в течение 2—3 мин до получения слегка опалесцирующей жидкости,
охлаждают и переливают в склянку с притертой пробкой. Раствором можно
пользоваться 5—10 дней.
411
Реактив Несслера на аммиак. Растворяют 30 г KI в 20 мл Н20 и прибав*
ляют 22,5 г иода; после полного растворения иода прибавляют 30 г
металлической ртути и встряхивают до тех пор, пока окраска иода не исчезнет и ртуть
не растворится полностью.
Раствор пробуют с крахмалом на содержание свободного иода. Если синяя
окраска не появляется, иод отсутствует, то прибавляют несколько капель
раствора иода в иодиде калия до появления в пробе синего окрашивания, после
чего раствор разбавляют водой до 200 мл и добавляют 975 мл 10% раствора
КОН.
Жидкие поглотители для газов
Поглощаемый газ.
Ацетилен и
углеводороды ряда ацетилена
Водород
мышьяковистый
фосфористый
хлористый
Кислород
Сероводород
Серы диоксид и другие
кислые газы
Углерода
диоксид
оксид
Этилен и его гомологи
.1
Поглотитель
Тетраиодомерку-
роат калия
K2[HgI4]
Ацетат кадмия
Селенистая
кислота
Гидроксид калия
Пирогаллол
Три ацетил гид рок-
сигидрохинон
Дитионит натрия
Na2S204
Ацетат кадмия
Гидроксид калия
» »
» »
Сульфат меди (I)
с (J-нафтолом
Хлорид меди (I) в
аммиачной среде
Бромная вода
Серная кислота
Сульфованадие- i
вый реактив 1
Приготовление
поглотительного раствора
23 г Hg2I2 и 30 г KI растворяют
в 100 мл дистиллированной
воды; перед применением
реактив подщелачивают раствором
кон
80 г Cd(CH3COO)2 растворяют в
100 мл воды и добавляют 3—
4 капли ледяной уксусной
кислоты
80 г H2Se03 растворяют в 100 мл
воды
30—35% водный раствор КОН
56 г пирогаллола растворяют в
100 мл воды и добавляют
260 мл 33% водного раствора
кон
40 г триацетилгидроксигидрохи-
нона растворяют в 200 мл 38%'
водного раствора КОН
Смешивают свежеприготовленные
растворы: 1) 50 г Na2S204 в
250 мл воды; 2) 30 г NaOH
в 40 мл воды
См. Мышьяковистый водород
См. Хлористый водород
» » »
» » »
К взвеси 20 г Cu20 в 200 мл
конц. H2SO4 и 25 мл воды
прибавляют 25 г Р-нафтола
32 г Си2С12 растворяют в ПО мл
25% водного раствора NH4Cl и
добавляют 80—100 мл 25%
раствора NH3
20% водный раствор КВг,
насыщенный Вг2
84% раствор H2S04
1 г V2O5 растворяют при
нагревании в 100 мл конц. H2SO4
412
Реагенты-индикаторы для
Реагент
Анисовый альдегид
Бромтимоловый голубой
8-Гидроксихинолин
2,4-Динитрофенилгидразин
Драгендорфа реактив
Нингидрин
Нитрат серебра + флуорес-
цеин
Флуоресцеинат натрия
детектирования пятен в бу
Детектируемые соединения
Углеводы
Карбоновые кислоты
Фенолы, енолы
Неорганические катионы
Альдегиды, кетоны
Алкалоиды, органические
основания
Аминокислоты, аминоса-
хара, аминофосфатиды
Галогенид-ионы
Ароматические и
гетероциклические соединения
жной и тонкослойной хроматографии
Приготовление реагента
Наблюдаемый эффект
0,5 мл реагента + 0,5 мл конц. H2S04+
+ 9 мл С2Н5ОН + несколько капель
СНзСООН. Опрыскивают и
нагревают 20—30 мин при 100—П0°С
0,1—0,5% раствор в С2Н5ОН
(подщелоченный)
1 % водный раствор
0,5% раствор в 60% С2Н5ОН. Вначале
хроматограмму выдерживают в
парах NH3, затем обрабатывают
реагентом
0,5% раствор в 2 н. НС1
Два раствора:
а) 1,7 г BiON03 в 80 мл конц.
H2S04 + 20 мл Н20;
б) 40 г KI в 100 мл Н20.
Перед употреблением смешивают 5 мл
раствора «а», 5 мл раствора «б» с
20 мл ледяной СН3СООН и 70 мл
н2о
0,3% раствор нингидрина в бутаноле,
содержащий 3% СНзСООН.
Опрыскивают и нагревают 10 мин при
125 °С
а) 1 % водный раствор AgN03 +
. + NH4OH до растворения
осадка;
б) 0,1% раствор флуоресцеина в
С2Н5ОН
Раствор 50 мг флуоресцеината натрия
в 100 мл 50% раствора СН3ОН
Голубые пятна
Желтые пятна
Пятна различных цветов
В ультрафиолетовом свете
пятна различных цветов
Пятна от красного до
желтого цвета
Осадок
Голубые пятна
В ультрафиолетовом свете
флуоресцирующие пятна
То же
Реактив Несслера хранят в склянке из темного стекла, плотно закрыв сухой
резиновой пробкой, в помещении, где в воздухе нет NH3. Нельзя реактив
хранить в склянке с притертой пробкой, так как пробки заедают.
Твердый комплекс крахмал — мочевина. В небольшом стакане расплавляют
40 г мочевины (tnjl = 137,2 °С), добавляют 10 г растворимого крахмала и
перемешивают до образования жидкой массы. По охлаждении затвердевшую массу
растирают в ступке и хранят в стеклянной банке. Комплекс легко растворим
в воде, устойчив при длительном хранении и применяется в иодометрии в виде
порошка.
Бесцветный тионилхлорид. 50 мл SOCl2 смешивают с 10 мл хинолина,
отгоняют тионилхлорид при атмосферном давлении, дистиллят смешивают с 20 мл
льняного масла и смесь подвергают перегонке.
3. ПОЛЕЗНЫЕ РЕЦЕПТЫ И СОВЕТЫ
Покрытие для деревянного лабораторного стола
Свежевыструганное, ничем не пропитанное и не шпаклеванное дерево
покрывают сначала раствором 400 г C6H5NH2HC1 и 60 г NH4Cl в 2500 мл воды.
Раствор наносят горячим при помощи обыкновенной волосяной кисти. Когда
дерево просохнет, его покрывают горячим раствором 400 г CuS04-5H20 и 200 г
КСЮз в 2500 мл воды. Когда дерево просохнет после пропитки вторым
раствором, его снова покрывают первым раствором. Эту операцию повторяют
четырежды. В результате поверхность стола оказывается покрытой темно-зеленым
кристаллическим порошком, который счищают наждачной бумагой, после чего
дерево пропитывают несколько раз смесью скипидара с олифой.
Ласта для натирания лабораторных столов и линолеума
Деревянную поверхность лабораторного стола или линолеум рекомендуется
раз в 10 дней покрывать пастой, состоящей из 10 масс. ч. воска, 20 ч. церезина,
0,9 ч. скипидара и 70—ВО ч. бензина. Тонкий слой пасты наносят на дерево или
линолеум кистью или тряпкой. После высыхания слой растирают щеткой для
натирки полов.
Огнезащитные пропитки для спецодежды из бумажной, льняной и смешанной ткани
а) 120 г (NH4)2HP04 и 90 г (NH4)2S04 в I л воды;
б) 300 г мочевины и 150 г (NH4)3P04 в 1 л воды.
Пропитку производят при 50—60 °С в течение 5—10 мин, после чего ткань
или готовое изделие прополаскивают холодной водой и высушивают на
воздухе.
Менделеевская замазка (мягкая)
Сплавляют 8 масс. ч. воска и 30 ч. канифоли, нагревают смесь до 150—
200 СС, добавляют при перемешивании 10 ч. прокаленной и просеянной мумии
(Fe203) и 1 ч. льняного растительного масла. Нагревание продолжают до
образования вполне однородной жидкой массы, застывающей при 45 °С. Употребляют
замазку в расплавленном состоянии.
Бумага для определения положительного и отрицательного электродов
гальванической цепи
Фильтровальную бумагу пропитывают смесью из равных объемов двух
растворов:
а) 1 г фенолфталеина в 100 мл 95%-ного этилового спирта;
б) 5 г NaCl в 100 мл воды.
Затем бумагу высушивают на воздухе.
Перед употреблением бумагу смачивают водой, затем прикасаются к ней
одновременно проводниками, присоединенными к обоим электродам. В месте
соприкосновения бумаги с проводником^ соединенным с катодом( появляется
малиново-красное пятно.
414
Получение матовой поверхности на стекле
Смесь из 10 г BaS04, Ю г NH4F и 12 г HF тщательно растирают в ступке,
а затем кисточкой наносят на чистую поверхность стекла. Дают стеклу
высохнуть на воздухе, а затем промывают водой.
Регенерация серебра
Отработанные растворы и осадки, содержащие серебро, собирают в бутыль
с 10% раствором НС1. После отстаивания осадка AgCl надосадочную жидкость
сливают декантацией. Осадок кипятят с гранулированным цинком в 10%
растворе НС1, периодически добавляя НС1 до полного растворения цинка.
Выделившееся серебро отфильтровывают, промывают дистиллированной водой,
растворяют при нагревании в 13—16% растворе HNO3 и осаждают AgCl добавлением
НС1. Осадок AgCl отделяют, выдерживают в течение суток под слоем 13—16%
HNO3, вновь отделяют, промывают водой, размешивают с водой, подкисленной
(по конго) H2SO4, и смешивают с гранулированным цинком (23,5 г Zn на 100 г
AgCl). Смесь постепенно разогревается, большая часть цинка растворяется, а
порошок серебра выпадает на дно сосуда. Осадок отделяют, промывают водой
на воронке Бюхнера и обрабатывают 10% раствором H2S04 для удаления цинка.
После этого порошок серебра промывают водой до отрицательной реакции на
сульфат-ион и высушивают при 80—100°С.
Испытание работы вентиляции
Для испытания работы вентиляции можно пользоваться малотоксичными
дымообразующими средствами. Быстрота удаления дыма -- показатель
эффективности действия вентиляции.
а. В одну фарфоровую чашку наливают 25% раствор NH3, в другую —
конц. НС1. Чашки устанавливают рядом в работающий вытяжной шкаф и
наблюдают, насколько быстро удаляется аэрозоль NH4C1.
б. В высокий стеклянный стакан наливают 200 г 50% раствора HN03 и
100 мл 40% формалина. Стакан устанавливают в вытяжном шкафу рядом с
фарфоровой чашкой, наполненной 25% раствором NH3. •
Рекомендуемая литература
1. Агасян П. £., Николаева К. Р. Теория и практика потенциометрии и потен-
циометрического титрования. М., Изд-во МГУ, 1972. 138 с.
2. Айвазов Б. В. Практическое руководство по хроматографии. М., Высшая
школа, 1968. 279 с.
3. Аксельрод Г. Л., Лысянский В. Л1 Экстрагирование. М., Химия, 1974. 254 с.
4. Алексеев Н. Г., Прохоров В. А., Чмутов л. В. Современные электронные
приборы и схемы в физико-химическом исследовании. М., Химия, 1971.
494 с.
5. Берлин А. Я. Техника лабораторной работы в органической химии. М.,
Химия, 1973. 350 с.
6. Берлинер М. А. Измерение влажности. М., Энергия, 1973. 400 с.
7. Брек Д. Цеолитовые молекулярные сита. М., Мир, 1976. 764 с.
8. Булатов М. И., Калинкин И. П. Практическое руководство по
фотоколориметрическим и спектрофотометрическим методам анализа. Л., Химия, 1976.
345 с.
9. Бунаков Э. А. — Изготовление химической посуды из фторопласта. Зав. лаб.,
1973, т. 39, № 3, с. 288.
10. Бычко-Токовой Г. Г. — Силиконирование лабораторной посуды при помощи
полиметилсилоксановых жидкостей. Лаб. дело, 1973, № 10, с. 629—639.
11. Вейганд К. Методы эксперимента в органической химии. Ч. I и II. М., ИЛ,
1950.
12. Веселовский В. С, Шманенко И. В., Носичев Е. В. Нагревательные приборы
в лабораторной практике. М., Госхимиздат, 1951. 230 с.
13. Воскресенский П. И. Техника лабораторных работ. М., Химия, 1973. 719 с.
14. Гельман И. Э. и др. — Осушка изопропилового спирта на цеолите NaA. Хим.
и технол. топлив и масел, 1972, № 6, с. 16—19.
15. Гладков Л. Л., Милованова Р. А. Учебная лаборатория вакуумной техники.
М., Атомиздат, 1971. 277 с.
16. Гриднев А. С, Мандрохлебов В. Ф. — Солевые генераторы влажного
воздуха. Приборы и системы управления, 1974, ч 11, с. 22.
17. Губен— Вейль. Методы органической химии. Т. 2. Методы анализа. М.,
Госхимиздат, 1963. 961 с.
18. Гуляев М. Л., Ерухин А. В. Измерение вакуума. М., Изд-во стандартов, 1967,
146 с.
19. Гурьев И. Л. Потенциометрия С ионоселективными электродами. Горький,
"Изд-во ГГУ, 1978. 99 с.
20. Ермаков В. #., Гуцыков В. В., Дмитровский Л. Л. — Глубокая осушка
этанола на цеолитах. Зав. лаб. 1975, т. 41, № 1, с. 34.
21. Иващенко В. Е. Методы и приборы для измерения относительной влажности
' воздуха. М., НИИТЭХИМ, 1977. 37 с.
22. Иоффе Б. В. Рефрактометрические методы химии. Л., Химия, 1970. 395 с.
23. Карпуша В. £., Чернов Б. С. Измерение атмосферного давления. Л., Гидро-
•метеоиздат, 1973. 271 с.
24. Карягин Ю. В., Ангедов И. И. Чистые химические вещества. М., Химия,
1974, 407 с.
25. Кац Л. М., Канторович Л. С. Руководство по приборам и оборудованию для
медико-биологических исследований. Л., Медицина, 1976. 254 с.
26. Кибардин С. Л., Макаров К А. Тонкослойная хроматография в органической
химии. М., Химия, 1978. 124 с.
27. Кирсанов Г. П. — Мойка и обезжиривание лабораторной посуды. Лаб. дело,
1972, №7, с-507.
28. Киселев А. В., Яшин Я. И. Адсорбционная газовая и жидкостная
хроматография. М., Химия, 1979. 287 с.
29. Комаров В. С. Адсорбенты и их свойства. Минск, Наука и техника, 1977.
248 с,
416
30. Коренман И. М. Экстракция в анализе органических веществ. М., Химия,
1977. 196 с.
31. Коростелев Я. Я. Реактивы и растворы в металлургическом анализе. AL,
Металлургия, 1977. 400 с.
32. Костин И. В. Техника безопасности работы в химической лаборатории. М.,
Изд-во МГУ, 1966. 344 с.
33. Косткевич Б. В. Электронные приборы для измерения и регулирования тем*
пературы. М., Оборонгиз, 1953. 218 с.
33а. Коузов Я. А. Основы анализа дисперсного состава промышленных пылей
и измельченных материалов. Л., Химия, 1974. 280 с.
34. Крель Э. Руководство по лабораторной ректификации. М., ИЛ, 1960.
631 с.
35. Лабораторное оборудование для дробления и измельчения. Каталог. Л., Ме-
ханобр, 1977. 116 с.
36. Лабораторная техника органической химии/Под ред. Б. Кейля. М., Мир,
1966. 718 с.
37. Лебедев Я. Я. Сушка инфракрасными лучами. Л., Госэнергоиздат, 1955,
232 с.
38. Лидерман И, С., Зудашкин И. А. Измерение влажности сыпучих материалов,
М., ЦНИИТЭИприборостроения, 1970. 204 с.
39. Луке Г, Экспериментальные методы в неорганической химии. М., Мир, 1965.
465 с.
40. Лурье Л. Л. Хроматографические материалы. Справочник. М., Химия, 1978.
439 с.
41. Макаров Л. К-, Свердлин В. М. Приборы для измерения рН. Л., Энергия,
1970.. 90 с.
42. Мебель и оборудование лаборатории. Каталог-справочник. Серия 811.
Инвентарный номер 127425. М., ГИПРОНИИ АН СССР, 1973.
43. Мейке В. Л. Руководство для препараторов химико-аналитических
лабораторий. М., Госгеолиздат, 1956. 160 с.
44. Морозов Л. Л. Хроматография в неорганическом анализе. М., Высшая школа,
1972.
45. Мортон Э. Л. Лабораторная техника в органической химии. М., Госхимиздат,
1941. 214 с.
46. Мусакин Л. Я., Рачинский Ф, Ю., Суглобова К. Д. Оборудование
химических лабораторий. Справочник. Л., Химия, 1978. 474 с.
47. Мюллер Г., Гнаук Г. Газы высокой чистоты. М., Мир, 1968. 233 с.
48. Набиванец Б. И., Мазуренко Е. Л. Хроматографический анализ. Киев, Вища
школа, 1979. 263 с.
49. Ничиговский Г. Ф. Определение влажности химических веществ. Л., Химия,
1977. 197 с.
50. Охрана труда в научных учреждениях АН СССР/Под ред. Г. Г. Чехмачева..
М., Наука, 1972. 575 с.
51. Перегуд Е. А. Санитарно-химический контроль воздушной среды. Л., Химия,
1977. 336 с.
52. Пешкова В. М.$ Громова М. И. Методы абсорбционной спектроскопии в
аналитической химии. М., Высшая школа, 1976. 279 с.
53. Правдин П. В. Лабораторные приборы и оборудование из стекла. М.,
Химия, 1978. 306 с.
54. Правила устройства и безопасной эксплуатации сосудов, работающих под
давлением. М., Металлургия, 1975, 104 с.
55. Приборы для измерения давления. Номенклатурный справочник. М.,
ЦНИИТЭИприборостроения, 1977. 56 с.
56. Пряников В. //., Родионова А. И. Техника безопасности и промышленная
санитария. Справочник. Т. 2. М., Химия, 1979. 319 с.
57. Пугашевич П. Я. Техника работы со ртутью в лабораторных условиях. М.,
Госхимиздат, 1961. 142 с.
58. Рапопорт Ф. М.у Ильинская А. Л. Лабораторные методы получения чистых
газов. М., Госхимиздат, 1963. 419 с.
59. Розенгарт М. И. Техника лабораторной перегонки и ректификации. М.,
Госхимиздат, 1951. 194 с.
417
60. Руководство по препаративной неорганической химии/Под ред. Г. М. Брауэ*
pa. M., ИЛ, 1956. 895 с.
61. Столяров Б. В., Савинов И. М., Витенберг А. Г. Руководство к
практическим работам по газовой хроматографии. Л., Химия, 1978.
62. Сусленикова В. М., Киселева Е. К. Руководство по приготовлению
титрованных растворов. Л., Химия, 1978. 179 с.
63. Установочное лабораторное оборудование (лабораторная мебель). Каталог-
справочник. Инвентарный номер 136356. М., ГИПРОНИИ АН СССР, 1978.
64. Ушакова Н. #., Николаева Е. Р., Моросанова С. А. Пособие по
аналитической химии. Количественный анализ. М., Изд-во МГУ, 1978. 219 с.
64а. Феоктистов В. Г. Лабораторные весы. М., Изд-во стандартов, 1979. 196 с.
65. Фрайштат Д. М. Реактивы и препараты. Хранение и перевозка. М., Химия,
1977. 418 с.
66. Хамский Е. В. Кристаллизация из раствора. М., Наука, 1967. 105 с.
67. Хан Г. А. Опробование и контроль на обогатительных фабриках. М.,
Металлургия, 1954. 217 с.
68. Хан Г. А., Анфимова Е. А. Опробование сырья и продуктов промышленности.
М., Гостехиздат, 1953.
69. Юдин К. А. Техника безопасности при работе с химическими веществами.
М., Профиздат, 1964. 144 с.
70. Юзифович В. И. и др. — Лабораторная установка для адсорбционной
осушки органических жидкостей. Хим. и технол. топлив и масел. 1972 № 1,
с. 59—61.
71. Юинг Г. В. Инструментальные методы химического анализа. М., Госхим-
издат, 1960, 504 с.
Предметный указатель
Абсорберы 230
Адсорбенты 231, 261
для хроматографии 313, 323, 326—
328
Азот жидкий, применение для
охлаждения 175, 176
Азота соединения, оптическая
активность 346
Аккумуляторы 401, 403—406
кислотные 403
переплюсовка 405
правила ухода 405, 406
приведение в рабочее состояние
405
причины порчи 405
сульфатация 405
щелочные 403, 404
электролиты 404
Активность
водородных ионов 354
оптическая 346
Алонжи стеклянные 30, 31
Алюминия амальгама, осушающее
действие 229
Алюминия оксид
как сорбент для хроматографии
313, 326
осушающее действие 225, 231
Алюмогель см. Алюминия оксид
Аммиак
использование для охлаждения
174
приготовление водного раствора
111, 112
Амперметры 407
Ампулы стеклянные 45
Анализ
дисперсионный пыли 132
/ объемный 368
ситовой см. Ситовой анализ
титриметрический 236, 368
Анализатор ситовой 134
Ангидрон см. Магния перхлорат
Анемометр 18
Аптечка лабораторная 24
Ареометры 373, 374
Арретир 146
Асбест 74 см. также Материалы
асбестовые
Аспираторы 272
Ацетон 116
обезвоживание 116
очистка 116
проба на присутствие
восстанавливающих веществ 116
Ацетонитрил 117
обезвоживание 117
токсичность 117
Баллоны газовые 244
для фосгена и хлора 245
Бани
водно-солевые 170,
водяные 170, 171
глицериновые 171
жидкостные 170—172, 189
масляные 171
металлические 172
на основе жидкого азота 176
песчаные 172, 173
солевые 172
три-окрезилфосфатные 171
Барботирование 142
Бария оксид, осушающее действие 225
Барометр ртутный 200
Батареи химических источников тока
402, 403
Батометры 269
Бензол 118
обезвоживание 118
очистка 118, 123
проба на тиофен 118
проба на ненасыщенные
углеводороды 123
токсичность 118
Бидистилляция воды 99, 100
Бромистый водород, синтез 258
Бромная вода, приготовление 411
Бумага
гидрофобизация 319
для хроматографии 319
индикаторная для определения
рН 354 ел.
полюсов гальванической цепи 414
термоиндикаторная 187
фильтровальная 210
Бутыли, приспособления для работы 84
Буферные растворы см. Растворы
Бюксы 40, 41
Бюретки 46
газовые 248, 249
микро- 47
мытье 93, 94
отсчет показаний 49
правила работы 48—50
проверка 55, 56
прямые 46, 47
с автоматическим нулем и склян*
кой 47, 48
силиконирование 52, 53
цена деления 48
419
Вакуум 190, 199
высокий 196, 197
правила работы 190
Вакуум-испарители 291—293
Вакуумметр (ы) 205, 206
ионизационно-термопарный 206
радиоизотопный 206
U-образный стеклянный ртутный
201, 202
Вакуумные масла 197, 198
Вакуум-перегонка см. Перегонка в
вакууме
Вакуум-шкаф сушильный 163, 164
Вакуум-эксикаторы 43, 44, 227
Вентили газовых баллонов 244, 245
Вентиляция лабораторных помещений
15
весовых 153
испытание 415
Весы лабораторные
аналитические 144, 149 ел.
правила пользования 154
установка 153
аптечные 147
безрычажные 144
быстродействие 144
вариация показаний 143
ВЛВ-ЮОг 233
гидростатические 374—376
двухпризменные (одночашечные)
146, 147, 151, 152
демпферные 146, 150
квадрантные 147—149
классификация 144
микро- 156
нулевая точка 145, 150
одноплечие 152
погрешность 143
предельно допустимая нагрузка
143
призменные 145
равноплечие 145, 150, 151
коромысловые 145
общелабораторные 147, 148
ручные 147
рейтерные 149, 150
рычажные 144, 145
технические, пределы взвешивания
144
технохимические 147, 148
торсионные (пружинные) 147, 156,
157
точка равновесия 145, 150
точность 143, 144, 146
ультра микро- 156
цена деления 144
чувствительность 143
Взвешивание 143 ел.
методы 143
Борда 155
двойное 149
Взвешивание
методы
Менделеева 155
ошибки 154, 155
«Взрывное» вскипание, предупреждение
275, 276, 282
Вибратор электронный 142
Вибромешалка 142
Вискозиметрия 377 ел.
Вискозиметры
вибрационные 378
водное число 382
ВУ 382—384
калибровка 379, 380
капиллярные 377—379
ротационные 378
ультразвуковые 378
Влага
гигроскопическая 232
нестехиометрическая 232
окклюдированная 232
сорбированная 232
Влагомер 241, 242
Влажности определение 232 ел.
бинарных смесей воды с
органическим растворителем 242
брома, стандартный метод 242
воздуха
гигрометрами 242
психрометрами 239—241
газов 234
по точке росы 241
денситометрический метод 236
дистилляционно-азеотропный метод
234-236
диэлькометрический метод 242
жидкостей 234—236
реакцией с СаНг 238
зерна 234, 242
косвенные методы 239—242
муки 242
нефтяных продуктов, стандартный
метод 235
окиси этилена, стандартный метод
237
пищевых продуктов, стандартный
метод 235
полиэтилсилоксановых жидкостей
238
стружек 233
твердых веществ
весами ВЛВ-ЮОг 233
гравиметрическим методом 232
денситометрическим методом 236
реактивом Фишера 236
термической сушкой 232, 233
экстракционным методом 236 ч
химические методы 236—239
целлюлозно-бумажных изделий 242
экстракционный метод 236
Влажность постоянная 411
420
Вода
анионирование 101
апирогенная, аппарат для
получения 98, 99
бидистилляция 99, 100
бромная, приготовление 411
деионизация 101
деминерализация 101—103
дистиллированная 96 ел.
определение рН 100
определение содержания
аммиака и аммонийных солей 100,
101
определение сухого остатка 100
проба на восстанавливающие
вещества 101
требования к чистоте 96
хранение 97
электрическая проводимость 96
для пожаротушения 20
, зависимость температуры кипения
от атмосферного давления 183
известковая, приготовление 411
ионное произведение 353
как стандартное вещество при
определении относительной
плотности 370
катионирование 101
плотность при различных
температурах 54
тройная точка 177
Водородный показатель
определение 100, 353 ел.
приборы 362—367
Водоснабжение лабораторий 12, 13
Возгонка 286—288
в вакууме 287
в токе инертного газа 288
малолетучих веществ 287, 288
Воздухообмен, кратность 15
Вольтметры 407
Воронка (и)
Бабо 170
для горючего фильтрования 214
из пористого полиэтилена 213
- предохранительная ВСП 290
со стеклянными пористыми
пластинками, мытье 94
стеклянные 72
делительные 33
для фильтрования 35
капельные 34, 35
лабораторные (химические) 33
фарфоровая Бюхнера 61, 215
фильтрующие 216, 217
обратные погружные 218
с электрообогревом 217
Вращение плоскости поляризации 346.
347
Вспенивание тонкое 253
Время удерживания 308
Встряхивание 136
вручную 137
механическое 142
Выпрямители электрического тока 399,.
400
Выпаривание 289 ел.
агрессивных жидкостей 293
пенящихся жидкостей 291, 293
сосуды 61, 289
Высушивание 223 ел. см. также Сушка
газов
в абсорберах 230
вымораживанием 231
осушающими реагентами 230>.
231
жидкостей
высаливанием 229
осушающими реагентами 229
фракционной перегонкой 228
экстракцией 229
осадка 228
пиридина 237
растворов термически нестойких.
веществ 229
твердых веществ
в вакуум-сушильных шкафах 226
в сушильных шкафах 226, 233
в эксикаторах 43, 44, 227
до постоянной массы 233
ИК-облучением 227
на открытом воздухе 226
экстракцией 228
физические методы см. Возгонка,,.
Выпаривание, Дистилляция,
Отгонка азеотропная,
Экстракция
формовочных материалов 234
Вытяжные шкафы 17, 18
Выщелачивание 297
Вязкости определение 377 ел. см.
также Вискозиметры
Вязкость
динамическая 377
единицы измерения 377
кинематическая 377
условная 382
«Газовые часы» 252
Газометры 250, 251, 271
Газы
измерение объема 248 ел.
очистка 252
растворимость 106
сжатые и сжиженные 80х 243 с/ц
меры предосторожности при
работе 244 ел.
отбор из баллонов 247
Газопроводы 13, 14
проверка герметичности 14
Газопромыватели 142
42k
Газоснабжение 13, 14
Гальванометры 407
Гигрометр 241, 242
Гидрофобизация
бумаги 319
бюреток, пипеток см. Силикониро-
вание
Гири к лабораторным весам 144
гидростатическим 375
погрешность массы 156
рейтеры 146, 375
Горелки
газовые 167
Бунзена 168
Меккера 168
насадки 169
паяльные 169
Теклю 168
жидкостные 169
Гранулометрический состав 129
приборы для определения 134
Грохоты 129
Давление
абсолютное 199
единицы измерения 199
избыточное 199
4 классификация приборов для
измерения 199
остаточное 199
регулирование 207, 208
Декантация 210
Демеркуризация 263 ел.
Демпферы 146
Денсиметры см. Ареометры
Держатели пробирочные 66
Детектирование пятен в
хроматографии
бумажной 322
индикаторы 413
тонкослойной 325
Детекторы для хроматографирования
305, 312, 316
Дефлегматоры 43
елочные 34, 35
цилиндрические с насадкой 34, 35
шариковые 34
Диметилсульфоксид 118, 119
очистка 119
совместимость 119
токсичность 119
#,ЛГ-Диметилформамид 119
очистка 119
Диоксан 121
Дисперсионный состав см.
Гранулометрический состав
Дисперсия света 340
Дистиллят 273
Дистиллятор (ы) 97—99
Дистилляция см. также Перегонка
воды 96, см. также Вода дистилли*
рованная
422
Диэтиловый эфир 119
обезвоживание 120
очистка от перекисей 120, 121
проба на содержание перекисей
119
хранение 119
Дозаторы
для жидкостей 57—60
автоматические однокомпонент-
ные 59
однопозиционные сифонные 58
полуавтоматические 59
для хроматографов 311
Дробилки 127
Едкий натр, приготовление водного
раствора 110
Едкое кали, приготовление растворов
109, ПО
«Заедание» шлифов 29
Зажимы для резиновых трубок 71
. Замазка (и)
для герметизации стеклянной
посуды
необратимо затвердевающие 76
термопластичные 76
для шлифов уплотняющая 75
Менделеева 76
приготовление 414
на основе эпоксидных смол 76
Запас реактивов в лаборатории 82, 88
Затвор ртутный 139
«Захлебывание»
ректификационных колонок 279
холодильников 42
Зерновой состав см.
Гранулометрический состав
Известковая вода, приготовление 411
Изгибы стеклянные 30, 31
Излучатели инфракрасные 16?
Измельчение 127, 128
вручную 127
механическое 127, 128
приспособления 72, 73
степень 129
тонкое 128
Измельчитель вибрационный 128
Индикаторы
для детектирования пятен в
хроматографии 413
для рН-метрии 354—356, 359
паров ртути в воздухе 264, 265
Индикация напряжения электрического
тока 399
Инфракрасные лампы 167, 227
Иод, очистка возгонкой 287
Иониты 101
регенерация 103
синтетические полимерные 101
Ионное произведение воды 353
Ионометры 362
Источники тока
сетевые 399, 400
химические 400—406
основные параметры 402
саморазряд 402
Калий, разрезание 86
Калия
гидроксид, осушающее действие
225
карбонат, осушающее действие 224
Кальций металлический, осушающее
действие 223
Кальция
гидрид
использование при определении
влажности 238, 239
осушающее действие 225
оксид, осушающее действие 225
сульфат, осушающее действие 225
хлорид, осушающее действие 223
Канализация 12, 13
Капельницы 35, 36
Каплеуловители 36
Карандаши термоиндикаторные 187
Катарометр 312
Квартование пробы 269
Кварцевая посуда см. Посуда из
прозрачного кварцевого стекла
Керны стеклянные 30, 31
«Кипятильные камешки» («кипелки»)
90, 183
Кипячение, сосуды 44
Кислота (ы)
правила работы 108
приготовление растворов 108
хлорная 87
приготовление безводного
раствора 109
хранение 85
Клапан Бунзена 40
Ключ электролитический 361
Колбонагреватели 167
Колбы с прямым электрообогревом
167
Колбы стеклянные
Арбузова 275, 277
Бунзена 37
Вюрца 38, 39
грушевидные 38
Кляйзена 38, 39, 282
круглодонные 36, 37
мерные 49—51
проверка 56—57
остродоиные 38, 39
плоскодонные 37
с токопроводящим покрытием 45,
46
Колбы
с тубусом 37
Фаворского 275, 276
Эрленмейера 37
Коллекторы фракций 316
Колонки
осушительные 230
поглотительные 196, 231
для паров ртути 261
с самоорошением 231
ректификационные
«захлебывание» 279
насадочные 278—280
рабочая мощность 279
точного фракционирования 273-
точной ректификации 280, 281
эффективность 279
хроматографические 314
заполнение 311, 313—315
насадочные 311
Колориметр-нефелометр 335
Компаратор 359
Компенсаторы см. Потенциометры
Концентрация раствора
моляльная 105
молярная 104
процентная 104
эквивалентная 104
Концентрирование растворов 289—295
Кошма 20
Коэффициент
внутреннего трения см. Вязкость
динамическая
нормальности 112
однородности 135
поглощения молярный 330
пропускания 330
распределения компонентов
концентрационный 294, 308 см.
также Фактор емкости
растворимости 106
сопротивления температурный,
измерение 409
экстинкции 330
Кран(ы)
вакуумные 32
пробоотборный 270
смазка 31
с фторопластовыми пробками 32
соединительные 31, 32
Краски термоиндикаторные 187
Кратность воздухообмена 15
Крахмал—мочевина, твердый комплекс,
приготовление 414
Крахмал, раствор 411
Кремния диоксид как сорбент для
хроматографии 327
Криогенные средства 243
Кристаллизация 300 ел.
выбор растворителя 300, 304
дробная 304
423
Кристаллизация
затравка 303
маточные растворы 303
отделение кристаллов 303, 304
получение хороших кристаллов 302,
303
ядра 303
Критическая точка растворимости 106,
107
Лампочки спиртовые 169
Лигроин
очистка 123
проба на ненасыщенные
углеводороды 123
Лития-алюминия гидрид, осушающее
действие 225
Ловушки вымораживающие 195, 196
Ложки фарфоровые 72
Магния
сульфат, осушающее действие 224
перхлорат, осушающее действие
224
Манометр (ы)
для измерения избыточного
давления 205
жидкостные 199 ел.
U-образные 201
ртутные
для измерения вакуума ?01—205
методы заполнения 202, 203
компрессионные Мак-Леода 203,.
204
U-образные 201, 202
термопарный 205, 206
Маностаты 207, 208
Маска защитная 22
Масла
вакуумные 197, 198
смазочные приборные 75
Материалы асбестовые 74, 216
Мебель лабораторная 15 ел.
Мельницы 127, 128
Мензурки 53
«Мертвый» объем 308
Металлы щелочные см. Щелочные
металлы
Метанол см. Метиловый спирт
Метиловый спирт 88, 121
обезвоживание 122, 229
очистка от ацетона 122
проба на ацетон 122
токсичность 121
Метилэтилкетон, высушивание 229
«Меш» 130
Мешалки
вертикального действия 139
вибро- 142
лабораторные 141, 142
. лопастные 138
Мешалки
магнитные 141
приводы 140, 141
пропеллерные 138
с затворами 139
с откидывающимися лопастями 138
спиральные 139
уплотнение 139, 140
установка 139
центрирование 141
центробежные 138
якорные 138
Микробюретки 47
проверка 55
Микровесы 156
Микропипетки, проверка 55
Микротермостат 165, 166
Мойка лабораторная под вытяжкой 89
Моляльность 105
Молярность 104
Мосты измерительные 409
Муфты стеклянные 30, 31
Нагревание 158 ел.
в пробирке 39
Напряжение электрического тока
индикация 399
компенсационный метод измерения
408
стабилизация 399
Насадка (и)
Аншютца 283
дистилляционные
Вюрца 30, 31
Кляйзена 30, 31
для экстракции 296, 298
к газовым горелкам 169
Тиле 283
фракционирующие см.
Дефлегматоры
Насосы
водоструйные 191
Ветцеля 191, 192
металлические вакуумные 192
пластмассовые 192
стеклянные 191, 192
высоковакуумные диффузионные
паромасляные 196, 197
парортутные 196, 197
механические вакуумные
вращательные 194
двуступенчатые 194
пластинчато-роторные 194, 195
пластинчато-статорные 195
поршневые 194
ртутные,, поглощение паров Hg из
выхлопных отверстий 262
Натрий
осушающее действие 225
получение порошка д проволоки
86, 87
424
Натрия
гидроксид, осушающее действие
225
сульфат, осушающее действие 224
Нейтрализация стоков см. Стоки
Несслера реактив, приготовление 412,
414
Никель Ренея 118
Нитрат серебра, хранение растворов
107
Нормальность 104, 105
коэффициент 112
Объем удерживания 308
Огнестойкость, предел 11
Огнетушители ручные 20, 21
Ожоги 23
Оливы переходные 32
Оптически активные вещества 346
Освещение рабочих помещений 12
Осушающие реагенты 223 ел.
«.Осушительный пистолет» 227
Отбор проб 268 ел.
газов 270—272
сжатых и сжиженных из
баллонов 247, 248
жидкостей 269, 270
из аппаратов под давлением 269
металлов 268
сыпучих тел 268, 269
Отгонка азеотропная 229, 234
<Ютжим» фракции 281
Отметчик времени пузырьковый 317
Охлаждающие смеси 173, 174
Охлаждение
жидким азотом 175, 176
жидким аммиаком 174
кольцевыми трубками поверхности
колб 173
льдом 173
«сухим льдом» 175
Очки защитные 22, 23
Пароперегреватели 285
Пенополистирол, лабораторные
изделия 74
Первая помощь 23, 24
Перегонка 273 ел.
в вакууме 282 ел.
меры предосторожности 284
ртути 261
веществ термически малостойких
273, 282
воды 96 см. также Вода
дистиллированная
высококипящих жидкостей 274, 275
дробная 273, 277 см. также
Перегонка фракционная
пенящихся жидкостей 283
при атмосферном давлении 274 ел,
ртути 267
Перегонка
с водяным паром 284, 285
насыщенным 273
фракционная 229, 273
Перекисные соединения, хранение 83
Перемешивание 136 ел.
барботированием 142
в вакууме 139, 141
в пробирке 39
вручную 45, 137
механическое 137 ел.
Переходы стеклянные 29, 31
Перколяторы 299
Перчатки
для работы со ртутью 261
резиновые 23
Петролейный эфир
очистка 123
проба на присутствие
ненасыщенных углеводородов 123 "
хранение 123
Печи электрические см. Электропечи
сопротивлений
Пикнометры 371—373
Пипетка (и)
Гемпеля 252, 253
для отбора проб жидкостей 269,
270
для собирания ртути 262
измерительные 51, 52
проверка 55
силиконирование 52, 53
лабораторная пневматическая 59
мытье 94
Пиридин, высушивание 237
Пирометры 186
Пластинки для хроматографии 323, 324
Пластмассовые лабораторные изделия
73, 74
Платиновая посуда см. Посуда из
платины
Плитка электрическая 166, 167
Плотность
единицы измерения 370
определение 370 ел.
ареометрами 373, 374
гидростатическим взвешиванием
374—376
жидкости 371—373
пикнометрами 371—373
твердого тела 372—376
относительная 370
температурные поправки 370
Плотность оптическая 330
Поглотительные устройства 230 см.
также Колонки поглотительные
Поглощение света, методы измерения
330
Пожароопасность 19 ел.
веществ 80, 81
425
Показатель преломления света 339
зависимость
от длины волны излучения 340
от концентрации 341
от температуры 340
измерение
для дистиллированной воды 343
для прозрачных жидкостей 344
для твердых тел 345
Покрытие
полов 1т2
столов лабораторных 16, 414
токопроводящее 45
«Ползучесть» при выпаривании 290
Поливинилхлорид, лабораторные
изделия 73
Поливинилхлоридные шланги 71
Полимеры
как неподвижная фаза для
хроматографии 313
определение вязкости растворов
381
Полисорбы 328
Полистирол, лабораторные изделия 73,
74
Политетрафторэтилен, лабораторные
изделия 73, 74
Полиэтилен
лабораторные изделия 73, 74
пористый 213
Полиэтиленовые шланги 71
Поляриметрия 346 ел.
Поляриметры 347 ел.
Помещения лабораторные
вентиляция 15
испытание 415
весовые 153
для работы со ртутью 260
планировка 12
Поправка на нормальность 112
Посуда
из платины 62—64
области применения 62
правила обращения 63, 64
термостойкость 62
химическая стойкость 62
из прозрачного кварцевого стекла
60
области применения 60
термостойкость 60
химическая стойкость 60
мерная 49—51, 53, 93, 94
мытье 89 ел.
мерной 93, 94
моющими средствами 90
окислителями 90
органическими растворителями
90, 92
перманганатом калия 92
с помощью ершей 89
струей водяного пара 90
Посуда
мытье
ультразвуком 90
хромовой смесью 91
общего назначения 33 ел.
стеклянная см. Стеклянная посуда
сушка 94, 95
фарфоровая 60, 61
термостойкость 60
химическая стойкость 61
Поташ см. Калия карбонат
Потенциал
асимметрии 362
окислительно-восстановительный,
измерение 363
скачок 368
Потенциометрия 360 ел.
Потенциометры 408
Правило креста 108, 109
Представительность пробы 268
Приборы оптические, уход и хранение
352
Приемники-ловушки 235
Проба смешения при определении
температуры плавления 386
Пробирки
градуированные 38, 53
мерные 53
центрифужные 38, 53
Пробки
к делительным воронкам 34
натуральные (корковые) 67
парафинирование 68
пластмассовые 69, 70
резиновые 69
предохранение от старения 69
просечка отверстий прессом 69
сверление 68, 69
.стеклянные 66, 67
Пробкомялка 67
Пробоотборники 268—270, 316
Пробоотборные трубки 269, 270
Пробоотборный кран 270
Пробы
отбор см. Отбор проб,
Пробоотборники
представительность 268
твердых продуктов 268
квартование 269
размер 268
сокращение 269
Проводимость удельная, измерение 409
Прокаливание, приспособления 72
Промывалки 39, 40
Просев
механический 133, 134
пневматический 133, 134
ручной
мокрый 133
сухой 131—133
426
Противогазы
выбор 22
для работы со ртутью 261
кислородно-изолирующие 22
промышленные фильтрующие 22
Психрометр
Августа 239
аспирационный с
электродвигателем 241
Ассмана аспирационный 240, 241
Пыль, дисперсионный анализ 132
рН см. Водородный показатель
Разделение см. Выпаривание,
Дистилляция, Кристаллизация, Перегонка,
Ректификация, Хроматография,
Экстракция
Разрежение 199
Рассев-анализатор 134
Раствор (ы)
аммиака ПО—112
буферные для рН-метрии 354, 356—
359
буферная емкость 356, 357
образцовые 357, 358
стандартные серии 358
хранение 358
двухкомпонентные, определение
концентрации по показателю
преломления 341
для кристаллизации, приготовление
301, 302
измерение
окислительно-восстановительного потенциала 362
кислот, приготовление 108
концентрация, способы выражения
104, 105
концентрирование 289—293
крахмала, приготовление 411
насыщенный 106
нитрата серебра, хранение 107
общие сведения 104 ел.
поглотительные, приготовления 412
полимеров, определение вязкости
381
приготовление 107 ел.
заданной концентрации 50
из навески стандартного
вещества 112
из фиксаналов 112—114
светопоглощающие, расчет
концентрации 331
тартратные, предупреждение за-
плесневения 358
титрованные (рабочие,
стандартные) 107
точной концентрации 112—114
хлорной кислоты 109
хранение 107
щелочей 85, 109, НО
Растворимость 106
газов 106
жидких веществ 106
коэффициент 106
критическая точка 106, 107
твердых веществ 107
Растворитель(и)
для кристаллизации 30Q, 301
для полимеров 73
определение 104
органические 90
методы очистки 115 ел.
обезвоживание 115 ел.
Расходомер сифонный 317
Реактивы
влагочувствительность 79
воспламеняющие (окисляющие) 81
выделяющие
легковоспламеняющиеся газы 80
запас в лаборатории 82, 88
классификация по чистоте 77 ел.
легковоспламеняющиеся 81
маркировка 77, 78
пожароопасность 80, 81
правила работы 83—85
приспособления для работы 71—73
разрыхление 71
самовозгорающиеся 81
светочувствительность 79
теплочувствительность 80
термолабильные 80
Фишера см. Фишера реактив
хранение 82, 83
ядовитость 81, 82
Реле электромагнитное 188
Ректификация 277 ел.
Реперы для хроматографирования см.
Стандартные вещества
Реометры 250—252
Рефрактометрия 339 ел. см. также
Показатель преломления
Рефрактометры 341 ел.
Рефракция 339
Ротаметры 251, 252
Ртуть 259 ел.
очистка 266, 267
пары
индикация 264, 265
поглотитель 261
правила безопасности при работе
259 ел. j
пролитая, собирание 262, 263
хранение 83, 261
Руда, дробление 127
Сахариметры 350 ел.
Сборка установок 68 см. также
Пробки, Трубки, Шлифы
Сверла для пробок 68
Серебро, регенерация из отработанных
растворов и осадков 415
427
Серная кислота, осушающее действие
223
Сероводород, синтез 257
Серы диоксид, синтез 256
Силикагель
как сорбент для хроматографии
313, 326, 327
осушающее действие 226, 231
Силиконирование бюреток, пипеток 52,
53
Силохромы 327
Сито (а)
металлические пробивные 130
механическое 134
тканые
нумерация 130
размер ячеек 129
Ситовой анализ 129 ел.
методика проведения 131 ел.
механический (машинный) 131,133,
134
мокрый 133
обработка результатов 134, 135
приборы 134
ручной 131—133
сухой 131—133
фармакопейный 132
Смазка (и)
вакуумная 75
для кранов 31, 75
для шлифов 28, 283
на основе глицерина 75
Смолы ионообменные 101
Сопротивление электрическое,
мостовой метод измерения 409
'Состав ""гранулометрический
(дисперсионный, зерновой) 129
Сосуд Дьюара 173, 243, 244
Спаи термопар, «горячий» и
«холодный» 183
Спектрографы 338
Спектрофотометрия 329, 337, 338
Спецодежда 22
огнезащитная пропитка 414
Спиртовки 169
Сплавы легкоплавкие 172
Стабилизаторы электрического тока
399
Стаканчики для взвешивания 40, 41
Стаканы лабораторные 40
с токопроводящим покрытием 45
Стандартные вещества
для определения относительной
плотности 370
для определения температуры
плавления 385, 386
для хроматографирования 306
Стандарт-титры см. Фиксаналы
•Стекло(а)
для лабораторной посуды 27
защитные оптических приборов,
чистка 352
Стекло(а)
нанесение матового слоя 415
стойкость
термическая 26
химическая 27
Стеклянная посуда 26 ел.
мерная 46 ел.
проверка 54 ел.
общего назначения 33 ел.
правила работы 44, 45, 57
с токопроводящим покрытием 45
Степень измельчения 129
Стоки
нейтрализация 13
сбор 13
Столы
для микроаналитических весов 19
для титрования 19
консольные большие для
аналитических весов 19
лабораторные 16
паста для натирания 414
покрытия 16, 414
с цельным подстольем 19
Стояк металлический для бутылей 84
Ступки 72
Сублимация см. Возгонка
«Сухой лед» 165, 173, 175
Сушка см. также Высушивание
в вакууме 163, 164
лиофильная 229
Счетчик
газовый барабанный 252
капель 317
Тарелки теоретические 279
Температура
измерение 177 ел.
регулирование 187, 188
Температура вспышки 81
определение
в закрытом тигле 397, 398
в открытом тигле 396, 397
Температура застывания 385
Температура затвердевания 385
Температура кипения
воды, зависимость от атмосферного
давления 183
определение 392 ел.
ориентировочное 392, 393
приборы 392, 393
поправки для приведения к
нормальному давлению 393—395
Температура кристаллизации 391
Температура плавления, определение
. 385 ел.
криоскопическое 390, 391
легко разлагающихся веществ 390
органических соединений
в капилляре 386, 387
приборы 387—390
428
Температура самовоспламенения 81
Теплоизлучатели 167
Термисторы 185
Термобатареи 185
Термоиндикаторы 187
Термолабильные вещества 80
Термометр (ы)
Бекмана 179
излучения 186
лабораторные 178
«минусовые» 178
на нормальных конусных шлифах
180
оптические 186
отсчет показаний 181, 182
палочные 178
проверка показаний 182, 183
расширения жидкостные 178 ел.
ртутные 178—180
Аллина 180
газонаполненные
высокоградусные 180
образцовые шкальные 180
погрешность показаний 178
химические 179
специальные 180
укороченные 180
сопротивления
полупроводниковые 1S5
проводниковые 185, 186
термоэлектрические см. Термопары
технические 178, 180, 181
шкальные 178
электроконтактные 187
Термопары
градуировка 184
дифференциальные 185
нестандартные 185
полупроводниковые 185
стандартные 184, 185
Терморегуляторы 187
Термостат (ы)
воздушные 164
жидкостной 165
микро- 165, 166
с водяной рубашкой 165
ультра- 165
Тетрагидрофуран 123
очистка от перекисей 124
хранение 124
Тефлон см. Политетрафторэтилен
Тигельные щипцы 72
Тигли
металлические для прокаливания
72
фарфоровые 61
Гуча 61, 216
Розе 61
фильтрующие 2 К), 217
Тионилхлорнд бесцветный,
приготовление 414
Тиосульфат натрия, хранение 114
Титр 105
Титрант 368 -
Титраторы 238, 369
Титрование
автоматическое 369
потенциометрическое 367—369
установки 19
эквивалентная точка 368, 369
Титрографы 269
Тищенко склянка, использование
для осушения газов 231
для собирания пролитой ртути 262
Тонкое вспенивание 253
Тонкое измельчение 128
Трансформаторы 399
Треугольники фарфоровые 72
Триэтиленгликоль 172
Тройная точка воды 177
Трубки
кольцевые свинцовые для
охлаждения 173
пластмассовые 71
пробоотборные 269, 270
резиновые 70
соединительные 32
хлоркальциевые 40, 41
Тушение горящей одежды 21
Углерода диоксид, синтез 256
Углерода оксид, синтез 256
Углерод четыреххлористый 124
обезвоживание 125
очистка 124, 125
токсичность 124
Угол вращения плоскости поляризации
346
Ультрамикровесы 147, 156
Ультратермостат 165
Ультрацентрифуги 11, 219
Установки
для титрования 19
поглотительные 40 см. также
Колонки
сборка см. Сборка установок
Фактор
емкости 308
разделения 219
Фиксаналы 112—114
для рН-метрии 357
прибор для приготовления
растворов 114
хранение 114
Фильтр (ы)
асбестовые 216
аэрозольные аналитические 218, 219
бумажные
беззольные 211
простые гладкие 211
складчатые 211, 212
429
Фильтр(ы)
для ртути 265, 266
из синтетического волокна 216
Петрянова 218, 219
стеклянные 216
Фильтрование 209
в атмосфере инертного газа 218
горячих растворов 212
под действием собственного веса
жидкости 210 ел., 265, 266
при нагревании 214
при охлаждении 214
при пониженном давлении 37, 61,
215
сильнощелочных растворов 213
фтористоводородной кислоты 213
Фишера реактив, использование для
определения влажности 236—238
Флегма 276
Флегмовое число 278
Фосфор белый 88
Фосфора оксид, осушающее действие
225
Фотоколориметрия 329, 332 ел.
Фотоколориметры 334—337
Фотометрия 329 ел.
Фотометры ультрафиолетовые 316
Фугат 220
Хлор, получение 257
Хлористый водород, синтез 257
Хлорная кислота 87
Хлороформ 124
очистка 124
проба на присутствие фосгена 124
токсичность 124
хранение 124
Холодильники 173
бытовые электрические 176
стеклянные
водяные 41—43
воздушные 41
Либиха 41
нисходящие 43
обратные 41—43
прямые 40, 41
спиральные 42, 43
типа «охлаждающий пальчик» 43
шариковые 41, 42
Хранение реактивов 82, 83
Хроматограмма 305—307
Хроматограф
газовый 309—311
колоночный 315
Хроматография 305 ел.
адсорбенты 323, 326—328
адсорбционная 307, 309
бумажная 307, 318 ел.
ввод (нанесение) пробы
в газовый хроматограф 311
Хроматография
ввод (нанесение) пробы
в колонку 315
на бумагу 320
на пластинку 325
время удерживания 308
газовая 306, 309 ел.
газо-жидкостная 309
детектирование 307, 322, 325, 413
детекторы 305, 312, 316
жидкостная 306, 313 ел.
заполнение колонок 311, 312, 314,
315, 316
идентификация компонентов 306.
318
измерение потока элюента 316, 317
количественное определение
компонентов 306
колоночная 307, 313 ел.
«мертвый» объем 308
объем удерживания 308
осадочная 307
проявление 307
вытеснением 308
фронтальным анализом 307
элюированием 308
распределительная 307
свободный объем 308
ситовая 307
твердые носители 309, 318
тонкослойная 307, 323 ел.
пластинки 323, 324
фазы 309, 313, 318, 319, 323
Хромовая смесь -
приготовление 91
хранение 91
Целлюлоза как сорбент для
хроматографии 313
Центрифуги 219
лабораторные настольные 221
пробирочные переносные 220
ручные 221
типы 219
угловые малогабаритные 222
Центрифугирование 219 ел.
Цеолиты
как сорбенты для хроматографии
313, 327
осушающее действие 226, 230, 231
Цилиндры мерные 53
Чашки для прокаливания 72
Четыреххлористый углерод 124
обезвоживание 125
очистка 124, 125
токсичность 124
Число
водное вискозиметра 382, 383
теоретических тарелок 279
флегмовое 278
430
Шкаф(ы)
вакуум-сушильные 163, 164
вытяжные 17, 18
моечный вытяжной 89
сушильно-стерилизационный 163
сушильный круглый 162
Шланги 71
Шлем-маска противогазная, выбор раз*
мера 22
Шлифы стеклянные
«заедание» 29
конусные (нррмальные)
взаимозаменяемые 28
скрепление соединений 31
смазка 28, 29
сферические 28
Шпатель 71
Штатив
Бунзена 65
для пробирок 66
универсальный 65
Щелочи
приготовление растворов 85, 109,
110
хранение 85, ПО, 114
Щелочные металлы 85—87
правила работы 85, 86
уничтожение обрезков 86
хранение 82, 85
Щипцы для тиглей 72
Эбуллиометр 394, 395
Эксикаторы
вакуумные 43
с электрическим обогревом 44
обычные 43, 227
Экстракция 294 ел.
как способ высушивания 228, 229
многократная 294, 295
непрерывная 299
однократная 294, 295
определение окончания процесса
296, 298
подбор экстрагента 296
разделение образовавшихся
эмульсий 298
химические методы 298, 299
Электроды для потенциометрии 360—
362
вспомогательный см. сравнения
Электроды для потенциометрии
измерительный см. индикаторный
индикаторный 360, 362, 363
каломельный 360, 361
нормальный водородный 360
сравнения 360, 363
стеклянный 361, 363
хлорсеребряный 361
Электроды платиновые 64
Электрические величины, измерение
406 ел.
методом сравнения 408—410
непосредственное 406, 407
нулевым методом 408 ел.
Электрокипятильники погружные 167
Электролиты для аккумуляторов 404
Электрооборудование 14, 15
Электропечи сопротивления
для микроанализа 160, 161
камерные 159, 160
малогабаритные тигельные 160
оценка температуры по цвету
нагретого муфеля 161
шахтные 160
Электропитание 399 ел.
Электрохимические системы источников
тока 401
Элементы гальванические 401, 403
нормальные 408
Элюат 305
Элюент 305
Элюирование 305, 308
градиентное 317
Эмульсии, разделение 298
Этанол см. Этиловый спирт
Этикетки 81, 114
Этилацетат 125
обезвоживание 125
очистка 125
Этиленгликоль 172
Этиловый спирт 125
обезвоживание 125, 126
очистка от альдегидов ПО
Эфир петролейный см. Петролейный
эфир
Эффективный размер зерна 135
Ядовитые вещества 81, 82, 87, 88
измельчение 88
нейтрализация 88
хранение 83
ФОМА ЮРЬЕВИЧ РАЧИНСКИИ
МАРГАРИТА ФОМИНИЧНА РАЧИНСКАЯ
Техника
лабораторных
работ
Редакторы: Н. Р. Либерман, Л, Н. Исаева
Техн. редактор Д. Д. Некрасова
Корректор Б. Н. Тамаркина
Переплет художника Б. Н. Осенчакова
ИБ № 684
Сдано в набор 03.08.81. Подписано в печать 29.01.82.
М-30227. Формат бумаги 60X90'/ie- Бумага тип № 2.
Гарнитура литературная. Печать высокая. Усл.
печ. л. 27,0. Усл. кр.-отт. 27,0. Уч.-изд. л. 31,0.
Тираж 30 000 экз. Зак. 1237. Цена 1 р. 90 к. Изд. № 1328.
Ордена „Знак Почета'* издательство „Химия",
Ленинградское отделение, 191186, Ленинградг Д-186,
Невский пр., 28.
Ленинградская типография № 2 головное предприя*
тие ордена Трудового Красного Знамени
Ленинградского объединения «Техническая книга» им.
Евгении Соколовой Союзполиграфпрома при
Государственном комитете СССР по делам издательств,
полиграфии и книжной торговли.
198052, г. Ленинград, Л-Б2, Измайловский
проспект, 29.