
























































































































































































































































































































































































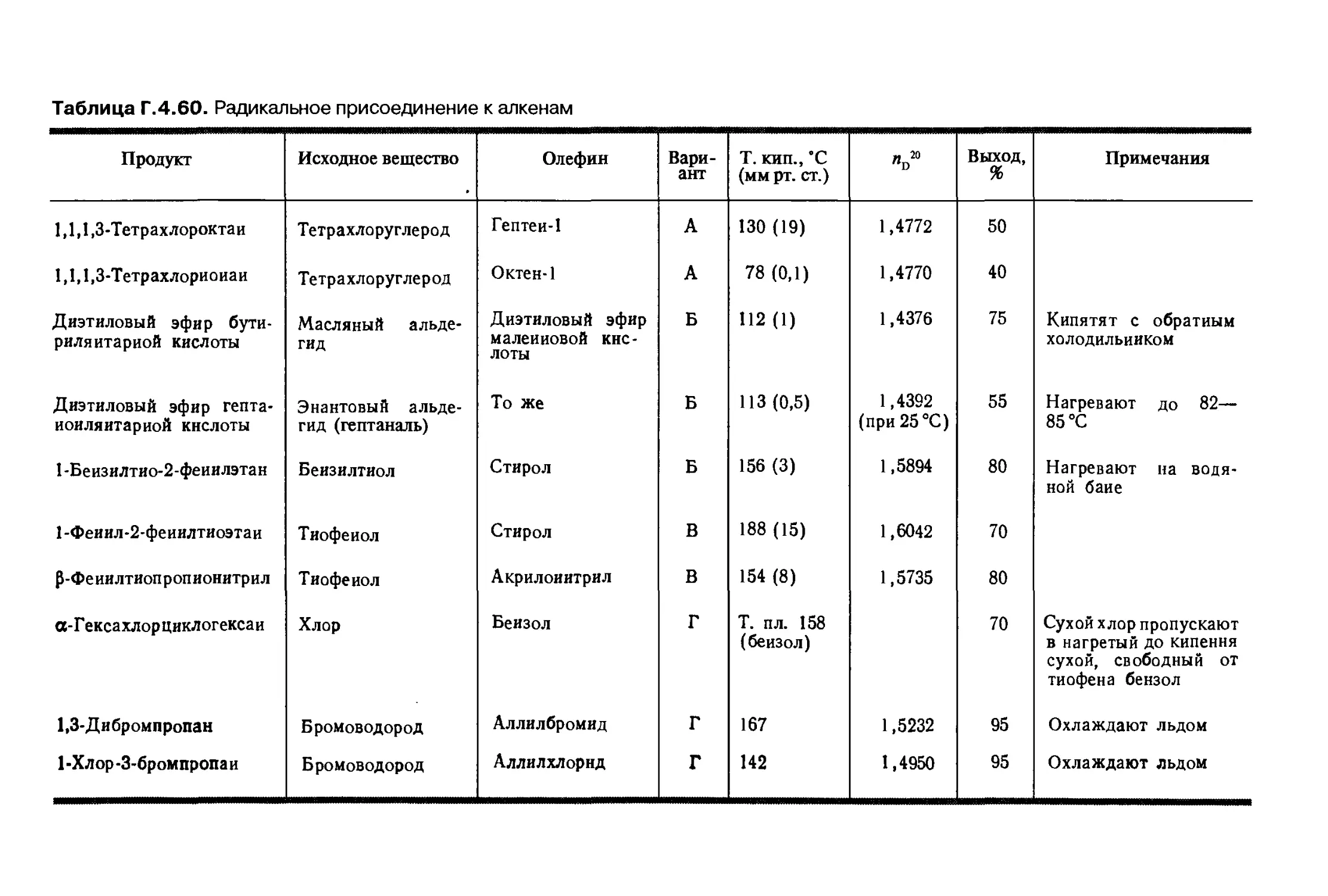


























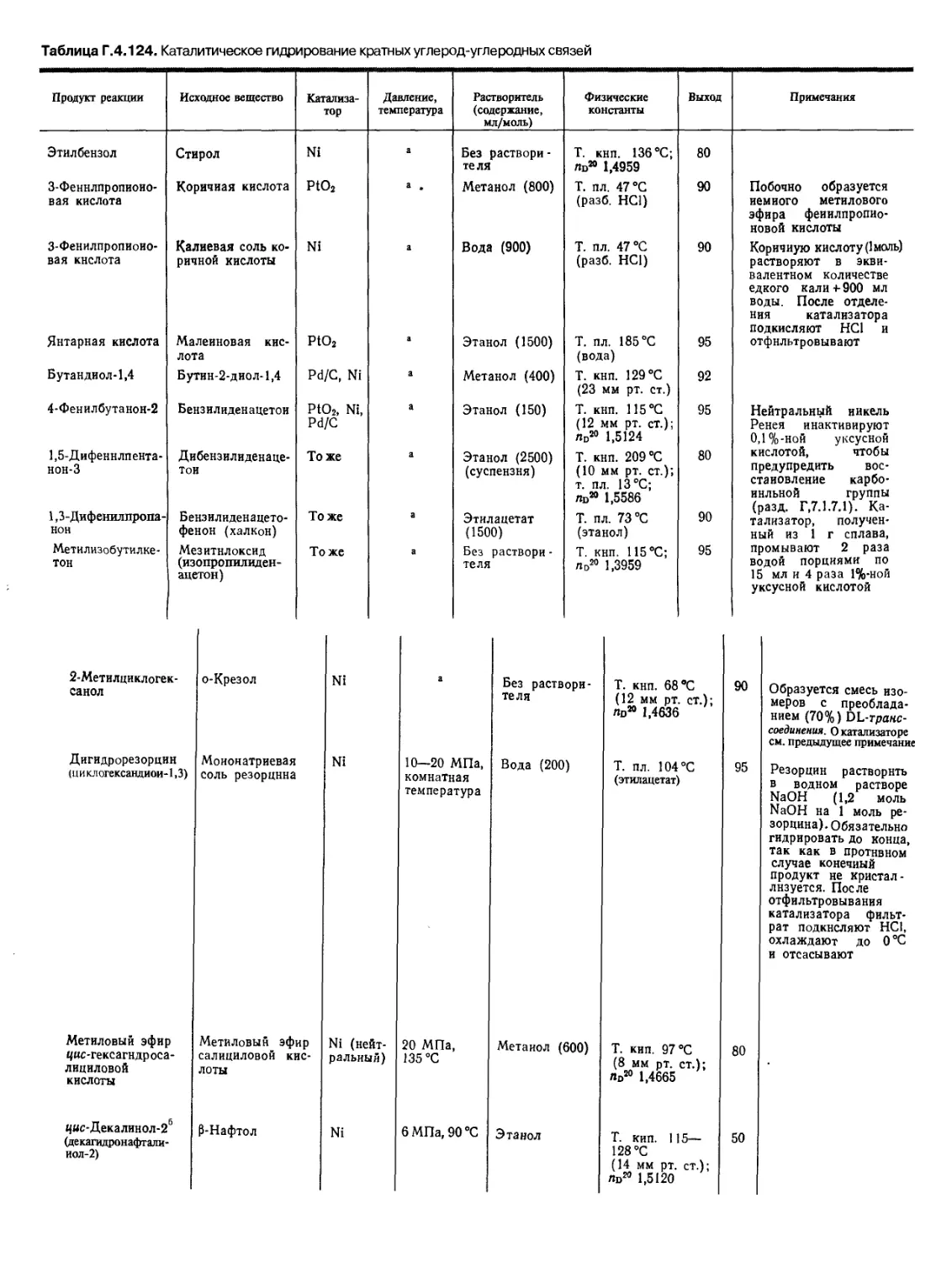

















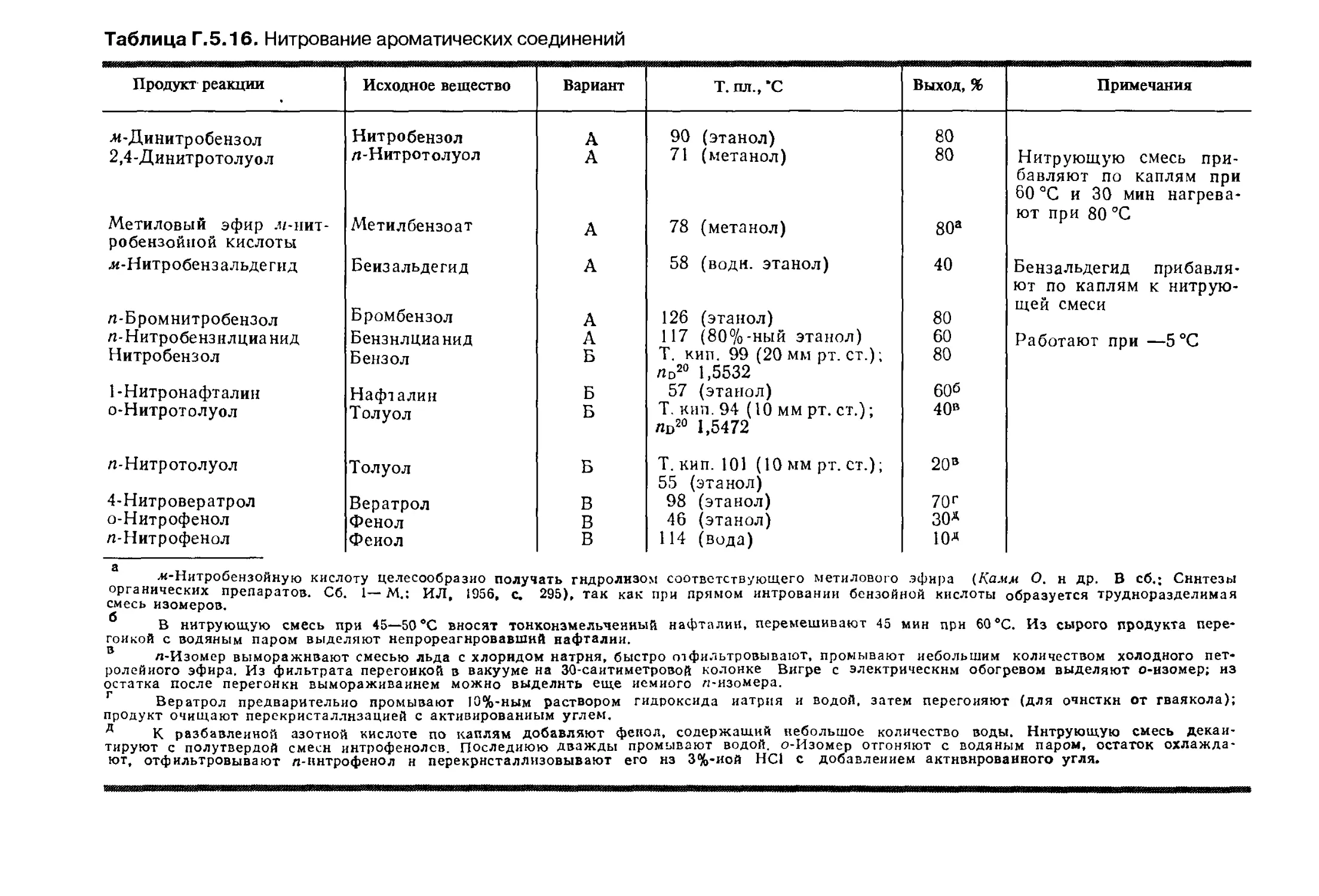






































































Автор: Беликова Н.А. Гришина Г.В.
Теги: органическая химия химия практикум органический синтез переводная литература издательство мир учебное пособие для студентов
ISBN: 978-5-03-003805-6
Год: 2008




Organisch-chemisches Grundpraktikum
Von
Heinz G O. Becker
Werner Berger
Gunter Domschke
Egon Fanghanel
Jiirgen Faust
Mechthild Fischer
Frithjof Gentz
Karl Gewald
Reiner Gluch
Roland Mayer
Klaus Miiller
Dietrich Pavel
Hermann Schmidt
Karl Schollberg
Klaus Schwetlick
Erika Seiler
Gunter Zeppenfeld
22., vollstandig iiberarbeitete und aktualisierte Auflage
Von
Rainer Beckert
Egon Fanghanel
WolfD. Habicher
Peter Metz
Dietrich Pavel
Klaus Schwetlick
WILEY-
VCH
WILEY-VCH Verlag GmbH & Co. KGaA
ЛУЧШИЙ
ЗАРУБЕЖНЫЙ
УЧЕБНИК
В двух томах
4-е издание
Перевод с немецкого
д-ра хим. наук Н. А. Беликовой и профессора, д-ра хим. наук Г. В. Гришиной
Допущено УМ О по классическому университетскому образованию
в качестве учебного пособия для студентов высших учебных заведений,
обучающихся по специальности ВПО 020101.65 — химия
БИНОМ
Москва 2008
УДК 547
ББК24.2
0 64
Авторы: X Беккер, Р. Беккерт, В. Бергер, К. Гевальд, Ф. Генц,
Р. Глух, Г. Домшке, Э. Зайлер, Р. Майер, П. Мец, К. Мюллер,
Д. Пафель, Э. Фаигхенелъ, Ю. Фауст, М. Фишер, В. Хабихер,
К. Шветлик, Г. Шмидт, К. Шолъберг, Г. Цеппенфельд
Органикум: В 2-х т. Пер. с нем. 4-е изд. — М.: Мир, 2008. —
О 64 Т. 1 - 504 с. ил.
ISBN 978-5-03-003805-6
В учебном издании, написанном авторским коллективом из Германии,
подробно рассмотрены все особенности работы в практикуме по органической
химии, включая лабораторное оборудование и методики синтеза.
Том 1 включает описание лабораторного оборудования и приемов работы,
краткие теоретические основы органической химии, а также описание препа-
препаративных методов синтеза (методики проведения реакций радикального и нук-
леофильного замещения, элиминирования, присоединения, замещения в аро-
ароматическом кольце).
Для студентов, аспирантов и преподавателей университетов и химико-тех-
химико-технологических вузов, а также научных сотрудников.
УДК 547
ББК24.2
Редакция литературы по химии
Originaly published in the German language by
WILEY-VCH Verlag GmbH, Pappelalle 3,
D-69469 Weinheim, Federal Republic of
Germany, under the title «Schwetlick et al.:
Organikum».
ISBN 978-5-03-003805-6 (русск.) © 2004 WILEY-VCH Verlag GmbH, Weinheim
ISBN 978-5-03-003807-0 © перевод на русский язык, оформление,
ISBN 5-527-31148-3 (нем.) издательство «Мир», 2008
ОТ ПЕРЕВОДЧИКОВ
ДВАДЦАТЬ ВТОРОГО ИЗДАНИЯ
Прошло уже 50 лет, как группа молодых энергичных химиков, в основном сот-
сотрудников Дрезденского и Л ейпцигского университетов, при участии и под руко-
руководством проф. X. Беккера и проф. Р. Майера написали чрезвычайно удачное по
содержанию и композиции учебное пособие по синтетической органической
химии «Organikum». За прошедшие с тех пор годы вышло в свет 22 немецких из-
издания, причем материал книги постоянно обновлялся и пересматривался в сто-
сторону его расширения. Изменялся и авторский коллектив. Книга получила приз-
признание не только в Германии, но и в других странах, в том числе и в Советском
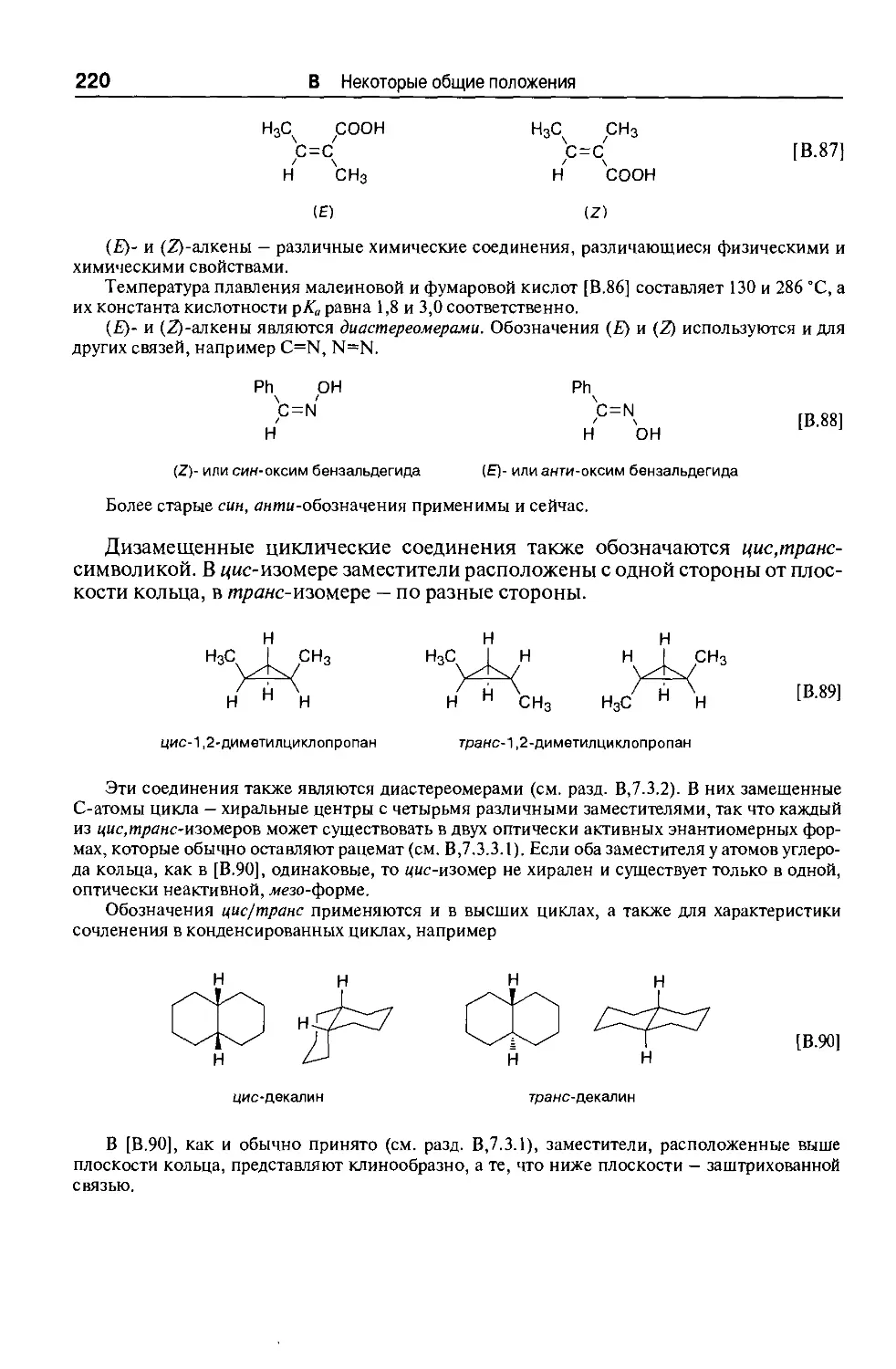
Союзе (России). Общий тираж всех изданий в мире составляет более 4 млн эк-
экземпляров. Первый перевод на русский язык вышел под названием «Общий
практикум по органической химии» в 1965 г. (М: Мир), потом были сделаны
переводы 15-го (Органикум. Практикум по органической химии. — М.: Мир,
1979 г.) и 18-го (Органикум. — М.: Мир, 1992 г.) изданий.
Настоящее издание D-е на русском языке) выполнено с 22-го оригинально-
оригинального издания. Книга в значительной степени обновлена; это касается описания
приемов работы, лабораторного оборудования и самого ценного в этой книге —
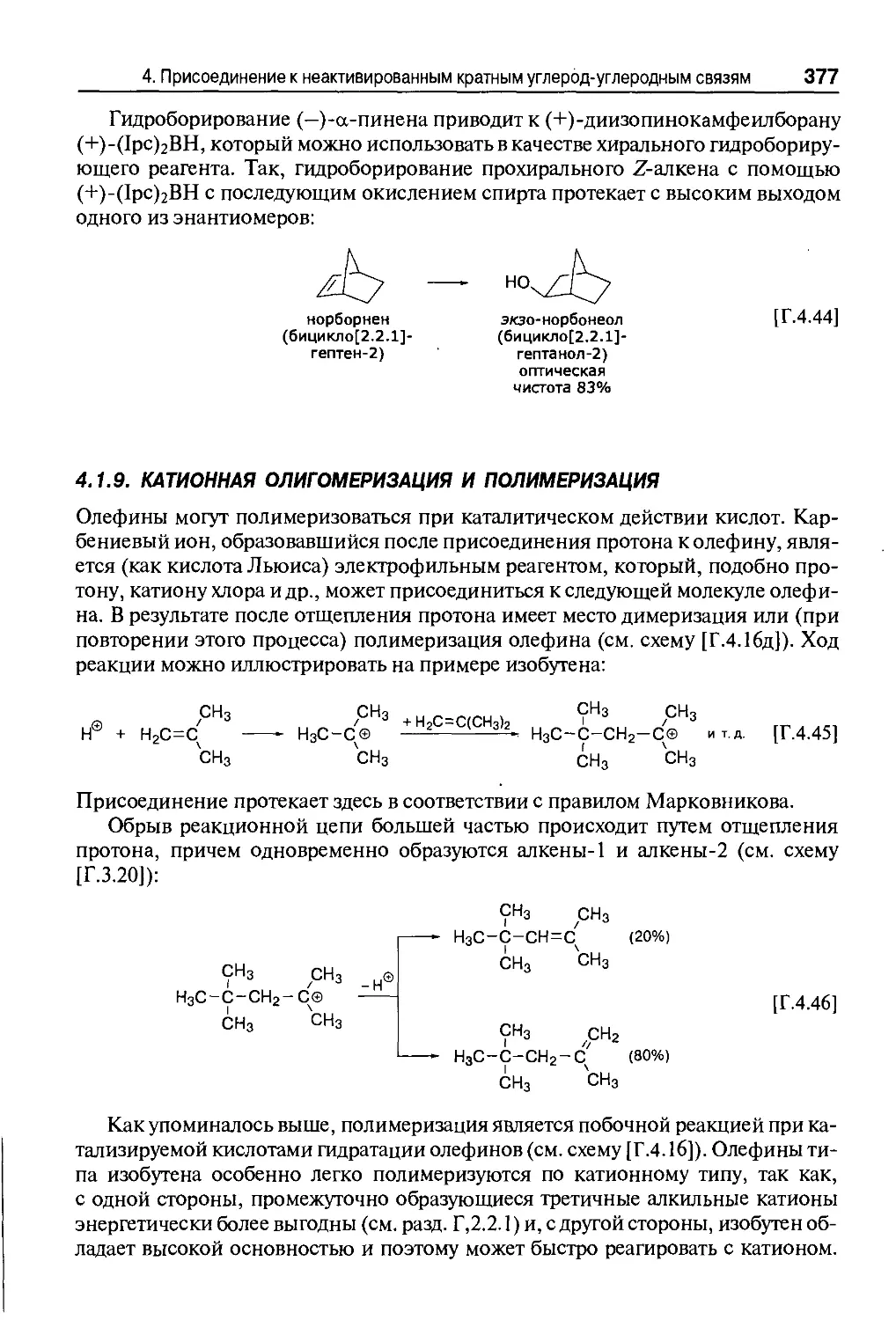
методик синтеза органических соединений. Уделено больше внимания стерео-
химическим аспектам органической химии, а также новым методам спектраль-
спектрального анализа.
Мы надеемся, что настоящее издание «Органикума» будет встречено с боль-
большим интересом как студентами и аспирантами, так и преподавателями хими-
химических вузов и научными сотрудниками профильных и смежных научных
учреждений.
Н. А. Беликова,
Г. В. Гришина,
С. С. Грюнер,
П. Б. Терентъев
ПРЕДИСЛОВИЕ К ДВАДЦАТЬ ВТОРОМУ ИЗДАНИЮ
Предыдущее 21-е издание «Органикума» было с большим удовлетворением встре-
встречено студентами и преподавателями многих университетов и институтов, а также
химиками, работающими в научно-исследовательских учреждениях и в промыш-
промышленности. Это побудило нас подготовить через четыре года новое издание, в кото-
котором мы устранили замеченные ошибки. В некоторых разделах были актуализиро-
актуализированы литературные ссылки и к общим методикам проведения синтезов добавлены
указания по мерам предосторожности при работе с опасными веществами.
Мы будем рады, если читатели нашей новой книги не оставят наш труд без
внимания, как это происходило и раньше в течение 42 лет с момента первой публи-
публикации книги. Мы надеемся получить ценные критические замечания, а также
описание новых методов синтетической органической химии. После подобных
контактов с коллегами по специальности и студентами мы в последующих изданиях
сделаем все, чтобы сохранить актуальность и полезность нашей книги, но все-таки
без изменения принятых в ней основных методических принципов. Мы благодарим
всех, кто до сих пор помогал нам в этом стремлении. Особая благодарность г-ну Ул ь-
риху Хаугу за совместную работу при актуализации данных по опасным веществам.
Сердечно благодарим также издательство за плодотворную совместную
работу и внимание к нашим пожеланиям.
Дрезден, январь 2004 г. Авторы
ПРЕДИСЛОВИЕ К ДВАДЦАТЬ ПЕРВОМУ ИЗДАНИЮ
Один рецензент любезно написал о двадцатом издании «Органикума»: «Это
так же свежо и актуально, как и прежде». В 21-м издании книга была опять
обновлена и улучшена, так что и сегодня «Органикум» может в полной мере
считаться незаменимым справочником не только для студентов, но и для
дипломированных химиков.
Предлагаемое 21-е издание, согласно укоренившейся традиции, было также
переработано и дополнено. Мы привели новые общие методики хлорирования
бензальдоксимов N-хлорсукцинимидом, получения алкилтиоцианатов, синтеза
арилиодидов из солей диазония и нескольких палладий-катализируемых реак-
реакций, вакер-окисления, реакций Хека и Соногаширы, а также написали новую
главу о способах металлирования заместителей в ароматических соединениях.
Переработан и обновлен материал, описывающий оборудование и приборы.
Мы благодарим читателей и своих коллег, которые способствовали улучше-
улучшению книги, делая ценные замечания. Особенно это касается проф., д-ра Клауса
Рюгера, который предоставил нам обширный литературный материал по
новейшим методам синтеза медицинских препаратов.
Дрезден, весна 2000 г. Авторы
ВВЕДЕНИЕ
В ЛАБОРАТОРНУЮ ПРАКТИКУ
1. ОБОРУДОВАНИЕ И ПРИЕМЫ, ИСПОЛЬЗУЕМЫЕ ПРИ
ПРОВЕДЕНИИ РЕАКЦИЙ В ОРГАНИЧЕСКОЙ ХИМИИ
1.1. СОРТА СТЕКЛА, СПОСОБЫ СОЕДИНЕНИЯ СТЕКЛЯННЫХ ДЕТАЛЕЙ
Наиболее употребительный материал для изготовления химической лаборатор-
лабораторной посуды и деталей различных установок для проведения экспериментов —
это стекло.
Простые изделия из стекла, например пипетки, бюретки, некоторые ампулы
и др., часто изготавливают из мягкого АР-стекла (силикатного), которое
обладает средней устойчивостью к воде (WB ЗI. Оно примерно соответствует
старому тюрингскому стеклу, которое и сегодня применяется в лаборатории под
названием стекло Хазельбахера (виртулан); из него изготавливают простые
части приборов, например мешалки, трубки для ввода газов, пипетки, капил-
капилляры при кипячении и др. Этот материал легко обрабатывается, например, в
пламени паяльной или бунзеновской горелки, но из-за малой устойчивости к
изменению температуры такое стекло непригодно для изготовления
термически устойчивых приборов, таких как перегонные колбы, холодильники
и т. д.
Для изготовления пипеток из стекла для реакционныю колб в продаже
имеется специальное стекло под названием дюробакс (WB 1).
Большинство приборов для препаративной работы изготавливается из
дурана, боросиликатного стекла. Его можно сплавить со всеми другими
боросиликатными сортами стекла, например с пирексом, симаксом или стеклом
разотерм. Дурановое стекло обладает высокой устойчивостью к воде (WB 1) и
относительной термостойкостью. Для работы под давлением используют
автоклавы, ампулы, нагреваемые в бомбе (ср. разд. А. 1.8.1), и др. стеклянные
приборы, изготовленные из толстостенного дурана.
Иенское приборное боросшшкатное стекло 20 (Jenaer Gerateglas 20) отличается
хорошей устойчивостью к воде, щелочам и кислотам, характеризуется относи-
WB (от нем. Wasserbestandigkeit) — устойчивость к воде. — Прим. перев.
А Введение в лабораторную практику
Рис. А.1. Типы шлифов.
а — плоские (например, у эксикаторов); б — цилиндрические (например, у мешалок (см. рис. А.6));
в — конические (НШ 29); г— шаровые; д — конические с резьбовым уплотнением.
тельно малым коэффициентом расширения (всего в восемь раз больше, чем у
кварцевого стекла); оно достаточно термостойко: посуда и детали из этого стекла
выдерживают нагревание до 190 °С. Поэтому оно нашло широкое применение для
изготовления посуды и приборов, подвергающихся нагреванию (перегонные кол-
колбы, холодильники, дефлегматоры и т. д.). Более высокая стоимость этих приборов
окупается их более длительным сроком службы. Однако это стекло уступает дурану
в случае работы под повышенным давлением.
Если к термостойкости прибора предъявляются особые требования, то ис-
используется кварцевое стекло или плавленый кварц. Плавленый кварц — молоч-
но-мутное, непрозрачное кварцевое стекло; оно гораздо дешевле прозрачного.
При очень высокой термостойкости (температура размягчения выше 1400 °С)
кварцевое стекло также устойчиво к перепаду температур, поскольку имеет
очень малый коэффициент расширения E,8 • 10~7 см/К).
Изделия из кварцевого стекла дороги, так как кварц очень трудно подверга-
подвергается обработке. Обычное стекло непрозрачно для ультрафиолетовых лучей, и в
тех случаях, когда необходимо облучение ультрафиолетовым светом, следует
применять отдельные части приборов из кварцевого стекла (например, ультра-
ультрафиолетовые лампы, вводимые непосредственно в реакционную смесь).
Сорта стекла можно различить по различной окраске в местах спая или на
срезах. Во многих случаях употребляется цветная маркировка (Gerateglas 20 име-
имеет черную полосу) или же изделия имеют вытравленное клеймо (например,
перегонные колбы и другая посуда).
Отдельные части приборов могут быть спаяны1. Однако применение прибо-
приборов больших размеров, изготовленных на спаях, ограничено лишь специальны-
специальными целями (например, работа в высоком вакууме). Для препаративных целей
части приборов обычно соединяют при помощи стеклянных шлифов, наиболее
употребительные типы которых изображены на рис. АЛ.
Лабораторная посуда, как правило, снабжена стандартными взаимозаменя-
взаимозаменяемыми коническими шлифами {нормальные шлифы, НШ).
Соединение на шлифах осуществляется при помощи шлифа-муфты (внеш-
(внешний шлиф) и шлифа-керна (внутренний шлиф). Наибольший диаметр и длина
шлифа обозначаются соответствующими цифрами, например НШ 29/32,
НШ 29/42 (удлиненный шлиф используется для работы в вакууме), НШ 14,5/23,
НШ 45/50 и т. д.
1 Стекло спаиваемых частей приборов должно иметь одинаковый коэффициент расшире-
расширения (эти части должны быть изготовлены из одного сорта стекла). Если прибор (или его часть)
не подвергается термическому воздействию, то для коэффициентов расширения стекол,
используемых для изготовления спаиваемых деталей, допустимо различие не более чем на 10%.
1. Оборудование и приемы
3=1
Рис. А.2. Переходные муфты.
Кроме лабораторных приборов с нормальными шлифами существуют
также приборы, снабженные стандартными резьбовыми соединениями
(рис. АЛ, д).
Шлифы разных размеров можно соединять при помощи переходных муфт
(рис. А.2).
Применение стандартных шлифов позволяет за короткое время собирать
довольно сложные приборы по принципу детского конструктора.
При работе с коническими шлифами необходимо соблюдать следующие
правила:
а) муфта и керн должны быть из одинакового сорта стекла, в крайнем слу-
случае стекло для муфты может иметь больший коэффициент расширения;
б) обе части конического шлифа следует соединять друг с другом путем лег-
легкого поворачивания муфты и керна относительно друг друга;
в) по возможности надо исключить попадание смолообразующих, полиме-
ризующихся и сильнощелочных веществ на пришлифованную поверх-
поверхность шлифового соединения.
При сборке больших и сложных лабораторных установок применяются
шаровые шлифы, так как они обеспечивают гибкое соединение отдельных час-
частей установки. Гибкость системы в случае конических шлифов может быть дос-
достигнута лишь при использовании большого числа следующих друг за другом
шлифов, в то время как шаровые шлифы позволяют легко собрать подобную
систему. Кроме того, шаровые шлифы легко разъединяются. При избыточном
давлении часто бывает трудно добиться герметичности прибора на шаровых
шлифах, зато такие шлифы отлично подходят для вакуумной аппаратуры.
Шаровые шлифы дороже конических.
Перед началом работы в вакууме шлифы надо смазать жиром или смазкой.
Наносить смазку надо всегда тонким слоем, для того чтобы реакционная смесь
или дистиллят не загрязнялись из-за растворения в них избытка смазки. Лучше
всего смазку наносить на середину смазываемой поверхности шлифа, а затем
распределять ее, равномерно поворачивая керн в муфте. Правильно смазанный
шлиф должен выглядеть прозрачным!
В качестве смазок используют: вазелин для кранов, плоских шлифов
(эксикаторы) и конических шлифов при работе с нормальным давлением; апие-
зоновые смазки различных типов, а также силиконовые пасты различной вязкос-
вязкости — для работ в вакууме. Для многих работ удобны водорастворимые смазки,
которые легко удаляются. При больших колебаниях температур (от —40 °С до
+200 °С) успешно используют KWS-смазку. Особенной устойчивостью по
отношению к химически агрессивным средам обладают пасты из полихлор-
трифторэтилена или политетрафторэтилена.
«Заевшие» (неразъединяемые) шлифы часто не удается разнять вращением
путем поворота одной части шлифа относительно другой. В этом случае, удер-
удерживая керн и муфту разными руками, следует раскачивать части шлифа, делая
10 А Введение в лабораторную практику
движения подобные тем, когда пытаются сломать палку, или же следует слегка
нагревать (до -70 °С) муфту на коптящем пламени газовой горелки (при этом
керн должен оставаться по возможности холодным). Заевший шлиф можно
разъединить также, обстукивая его деревянным молоточком (так открывают
склянки со стеклянными пробками).
Соединения на пробках по сравнению со стеклянными шлифами имеют мень-
меньшее значение. Резиновые пробки и шланги неустойчивы к действию галогенов,
сильных кислот и т. д. и набухают в присутствии органических растворителей.
Для работ с хлором, бромоводородом, фосгеном, озоном и другими агрессивны-
агрессивными веществами целесообразно применять шланги из поливинилхлорида или по-
полиэтилена. Такой шланг легко надевается на конец стеклянной трубки после
непродолжительного нагревания в кипящей воде.
При сборке лабораторных установок резиновые пробки удобно заменять
специальными резьбовыми соединениями (рис. АЛ, д). С помощью конических
уплотнений (предпочтительно из химически стойкого тефлона) термометры,
капельные воронки, подводящие трубки и т. п. можно плотно зафиксировать в
реакционной колбе на нужной глубине.
1.2. ХИМИЧЕСКАЯ ПОСУДА
В лабораториях органической химии применяется та же химическая посуда, что и
в лабораториях неорганической химии: пробирки, стаканы, плоскодонные и ко-
конические колбы (колбы Эрленмейера) и др. Для полумикросинтезов используют
короткие и широкие пробирки (имеющие диаметр -15 мм и длины -60—80 мм),
мерные пробирки. Для работы с низкокипящими или огнеопасными растворите-
растворителями непригодны стеклянные стаканы, так как из них жидкости легко испаряют-
испаряются. Более подходящими для этого являются колбы Эрленмейера (с нормальными
шлифами, которые могут быть легко закрыты пробками).
При работе под уменьшенным давлением нельзя применять плоскодонную посу-
посуду (при этом возникает опасность раздавливания внешним давлением!).
При перегонке в качестве перегонных колб и приемников употребляют круглодон-
ные, грушевидные и остроконечные колбы. Последние особенно хороши в качестве
перегонных колб при работе с полумикроколичествами вещества, так как из таких
сосудов можно отогнать жидкость почти до конца (без потерь, без остатка;
рис. А.59). Сложные реакции проводятвдвух-, трех- и четырехгорлых колбах (рис. А.4).
Следует выработать привычку надписывать карандашом вес пустой колбы на
специально вытравленном месте.
1.3. ХОЛОДИЛЬНИКИ
При осуществлении реакций между органическими веществами компоненты
обычно необходимо нагреть, часто в том или ином растворителе.
Для того чтобы избежать потерь низкокипящих компонентов, реакционные
сосуды снабжают обратными холодильниками, где пары охлаждаются и конденсат
1. Оборудование и приемы
11
а б в г д е ж
Рис. А.З. Типы холодильников.
а и б — воздушные холодильники; в — холодильник Либиха; г — шариковый холодильник;
д — змеевиковый холодильник; е — холодильник Штеделера; ж — холодильник Димрота;
з — интенсивный холодильник; и — погружной холодильник (охлаждающий палец).
возвращается в реакцию. При перегонке конденсат отводится с помощью нисхо-
нисходящих холодильников. Типы наиболее часто применяемых холодильников изоб-
изображены на рис. А.З.
Простейшим холодильником является воздушный (рис. А.З, а). Он годится
только для работы с высококипящими жидкостями (ст. кип. >150 °С), посколь-
поскольку эффективность воздуха как охлаждающего средства невелика. Воздушный
обратный холодильник может представлять собой простую трубку, однако в
этом случае при конденсации жидкости возникает преимущественно ламинар-
ламинарный поток и вещество может легко «выбрасываться» из верхнего конца трубки,
т. е. холодильник такого типа малоэффективен. Более удобны шариковые воз-
воздушные холодильники (рис. А.З, б). Такие холодильники нашли применение при
проведении полумикросинтезов, когда количество отводимого тепла невелико и
для конденсации даже низкокипящих веществ воздушное охлаждение оказыва-
оказывается вполне достаточным. (В этом случае при необходимости холодильник мож-
можно обмотать влажной фильтровальной бумагой.) Прямой воздушный холодильник
(рис. А.З, а) можно использовать в качестве нисходящего при не слишком боль-
большой скорости перегонки для веществ с температурой кипения > 150 "С.
Особой формой воздушного холодильника является мечеобразная насадка к
саблевидной колбе (рис. А.58), где холодильник совмещен с приемником.
Холодильник Либиха (рис. А.З, в) применяется преимущественно в качестве
нисходящего до температуры -160 °С. Охлаждающим средством для веществ с
температурой кипения < 120 "С служит в нем проточная вода, а в интервале
120—160 °С — непроточная. В качестве обратного такой холодильник малоэф-
малоэффективен, так как имеет малую охлаждающую поверхность и характеризуется
12 А Введение в лабораторную практику
ламинарным течением паров; с этой целью он применяется только в случае от-
относительно высококипящих соединений (т. кип. >100 °С). На наружной пове-
поверхности обратного холодильника конденсируется атмосферная влага, которая
через капиллярные течи в шлифе может попадать внутрь перегонной колбы,
поэтому шлифы на холодильнике и колбе следует тщательно смазывать. Реко-
Рекомендуется также на холодильник выше шлифа надевать манжету («воротник»)
из сухой фильтровальной бумаги.
При работе с более высококипящими жидкостями в местах спая / (рис. А.З)
могут возникать напряжения, приводящие к растрескиванию. Поэтому холо-
холодильники Либиха нельзя изготовлять из нетермостойкого стекла, например из
тюрингского.
Шариковый холодильник (рис. А.З, г) используется исключительно как обрат-
обратный. Поскольку этот холодильник имеет шаровидные расширения, ток паров в
нем турбулентный; охлаждающее действие такого холодильника значительно
выше, чем у холодильника Либиха. Однако на внешней его поверхности также
конденсируются влага и в местах спая 1 также могут возникнуть все те же опас-
опасные напряжения со всеми вытекающими отсюда последствиями.
Змеевиковый холодильник (рис. А.З, д) никогда не следует использовать как
обратный, так как конденсат, который недостаточно хорошо стекает по сгибам
змеевика, может быть выброшен из холодильника, что может послужить причи-
причиной несчастного случая. Змеевиковый холодильник, установленный вертикаль-
вертикально, является наиболее эффективным нисходящим холодильником, особенно
для низкокипящих веществ. Змеевиковый холодильник нельзя устанавливать
наклонно (почему?).
Модификация змеевикового — холодильник Штеделера (рис. А.З, е), в кото-
котором охлаждающий сосуд может быть заполнен смесью льда с поваренной солью,
твердой углекислотой с ацетоном и т. д. Такой холодильник можно применять
для конденсации веществ, кипящих при очень низких температурах.
Холодильник Димрота (рис. А.З, ж) — очень эффективный обратный холо-
холодильник. Его также используют в качестве нисходящего, если можно пренебречь
относительно большими потерями дистиллята на змеевике. Спаи змеевика с ру-
рубашкой 1 находятся вне зоны с большим перепадом температур, поэтому, при-
применяя такой холодильник при работе с жидкостями, кипящими ниже 160 °С,
можно не опасаться осложнений. Поскольку внешней «рубашкой» холодильни-
холодильника является воздух при комнатной температуре, на внешней поверхности такого
холодильника не конденсируется атмосферная влага (см. выше). Правда, низко-
кипящие вещества могут «ползти» по внутренней стороне рубашки и тем самым
«проскакивать» зону охлаждения. Холодильник Димрота поэтому не подходит в
качестве обратного для сравнительно низкокипящих веществ, например для
эфира. У верхнего открытого конца холодильника на подводящих воду штуцерах
легко конденсируется атмосферная влага, поэтому для защиты от влаги выход
холодильника закрывают хлоркальциевой трубкой (рис. А.4, а).
Эффективным холодильником является конструкция, сочетающая принци-
принципы холодильников Либиха и Димрота (рис. А.З, з). Его охлаждающее действие
вполне достаточно для конденсации низкокипящих растворителей (эфир), од-
однако на внешней рубашке этого холодильника конденсируется атмосферная
влага. Поскольку интенсивные холодильники очень дороги, к ним не следует
1. Оборудование и приемы 13
прибегать без особой необходимости. Кроме того, надо обратить внимание на
то, что большой объем циркулирующей воды очень утяжеляет такой холодиль-
холодильник, и поэтому его необходимо укреплять при сборке прибора особенно
тщательно.
Погружной холодильник — охлаждающий палец (рис. А.З, и) — это обратный
холодильник особой формы (его можно специально не закреплять в системе ох-
охлаждения) используется прежде всего в приборах для полумикрометодов. Если
«охлаждающий палец» введен в реакционный сосуд на пробке (как это показано
на рис. А.4, ё) или на резиновом шланге, прибор не может быть герметичным.
Необходимо постоянно следить, чтобы через рубашку холодильника не прекра-
прекращалась циркуляция воды, так как отключение холодильника может привести к
пожарам и взрывам!
Особенно следует обращать внимание на то, что набухание прокладок (саль-
(сальников) водопроводных кранов очень часто может препятствовать циркуляции
воды через холодильник. Если в лабораторную установку включено такое цен-
ценное оборудование, как, например, ртутный или масляный диффузионный на-
насос, на пути воды, протекающей через холодильник, всегда надо монтировать
детектор-предохранитель, связанный с нагревателем. Такие приборы имеются в
продаже.
1.4. СТАНДАРТНОЕ ОБОРУДОВАНИЕ ДЛЯ ПРОВЕДЕНИЯ РЕАКЦИЙ
В ОРГАНИЧЕСКОЙ ХИМИИ
Приборы для проведения реакции обычно собирают из отдельных деталей со
стандартными шлифами; наиболее распространенные представлены на рис. А.4.
Прибор, изображенный на рис. А.4, а, используется для проведения реак-
реакций, в которых реагенты смешиваются перед началом опыта. Этот же прибор
применяется и при перекристаллизации веществ (разд. А,2.2.2). Хлоркальциевая
трубка на выходе из холодильника необходима в тех случаях, когда реакционную
смесь надо защитить от влаги. Перед использованием следует проверить, будет
ли проходить через хлоркальциевую трубку воздух (продуть!). В реакционную
колбу надо не забыть положить кипятильники (разд. А, 1.7.2).
Дву- и трехгорлые колбы — это наиболее часто употребляемая посуда при про-
проведении химических реакций в препаративных целях. Их применение позволя-
позволяет одновременно осуществлять несколько операций, например пропускать газ и
в то же время кипятить реакционную смесь, вставив обратный холодильник
(рис. А.4, б), или постепенно прибавлять жидкость при перемешивании1 и так-
также кипятить смесь (обратный холодильник!) (рис. А.4, в) и т. д. С помощью
насадки Аншютца A) трехгорлую колбу можно переоборудовать в четырехгор-
лую (рис. А.4, г), что позволяет проводить реакции, требующие добавления жид-
жидкости к перемешиваемой или нагреваемой с обратным холодильником смеси,
1 На рис. А.4, в изображена мешалка с цилиндрическим шлифом; другие типы мешалок
и их крепление см. вразд. АД.5.1 и А,1.5.2.
14
А Введение в лабораторную практику
г д е ж
Рис. А.4. Приборы для проведения реакций.
а-ж — см. текст; 1 — насадка Аншютца; 2 — трехгорлая насадка.
измеряя в то же время температуру внутри колбы. Трехгорлая насадка 2 изоб-
изображена на рис. А.4, д. Такая насадка часто очень удобна, колба при этом снаб-
снабжается несколькими параллельно расположенными горлами. У небольших
колб параллельное расположение горл затрудняет монтаж приборов, которые
должны иметь также мешалку и обратный холодильник, и поэтому более
1. Оборудование и приемы
15
удобно наклонное расположение горл
(рис. А.4, в).
Для измерения температур приме-
применяется термометр на шлифе, длина ко-
которого должна соответствовать размеру
колбы, либо следует использовать спе-
специальные направляющие на шлифах,
которые позволяют изменять глубину
опускания термометра. В некоторых
случаях допустимо использование тер-
термометров без шлифа1.
Для полумикросинтезов использу-
используют посуду со шлифами меньшего диа-
диаметра (НШ 14,5). При проведении по-
полумикросинтезов можно отказаться от
применения многогорлых колб. В этом
случае вполне пригодны обычные кол-
колбы с насадкой Аншютца или трехгор-
лой насадкой 2 (рис. А.4, д), поскольку
можно добавить реагенты через холо-
холодильник и не контролировать темпера-
температуру внутри реакционной смеси (рас-
(расход и перенос тепла незначительны,
достаточно наблюдать за температурой внешней нагревательной бани). Для пе-
перемешивания при проведении этих синтезов особенно удобно использовать
магнитную мешалку (разд. А, 1.5.1).
В случае вязких растворов или при реакциях, сопровождающихся выделени-
выделением твердых веществ, удобен прибор, изображенный на рис. А.5. Он включает
реакционную колбу 1 с плоским шлифом и головку, которая кроме плоского
шлифа имеет еще четыре конических шлифа (НШ 14,5), два из которых направ-
направлены вертикально (их можно использовать для мешалки и термометра или
капельной воронки). Не только шлиф для мешалки, но также для термометра и
капельной воронки устроены так, чтобы можно было регулировать глубину
расположения (см. рис. А.8, а); прибор снабжен шлифованными гильзами и
зажимами для регулирования глубины опускания.
На рис. А.4, е, ж изображена дешевая, просто изготавливаемая посуда для
полумикросинтезов, которая нашла широкое применение в лабораториях.
Пользуясь такой посудой, легко собрать два простых прибора для нагревания с
обратным холодильником (рис. А.4, е и А.4, ж). Прибор на рис. А.4, ж особен-
особенно удобен, если нужно отогнать вещество из раствора.
Для того чтобы избежать возникновения напряжений и поломки собранного
прибора, необходимо следить, чтобы на зажимах были пробковые прокладки,
Рис. А.5. Прибор с плоскими шлифами.
1 — реакционная колба.
1 Применяют термометры без шлифа на корковой, резиновой или поливинилхлоридной
пробках, а также так называемый палочный термометр с удлиненным термометрическим
капилляром (рис. А.4, г).
16 А Введение в лабораторную практику
а на захватах лапок — резиновые наконечники. Муфты на штативе надо закреп-
закреплять всегда открытой частью вверх.
При креплении колб со шлифами зажимы и лапки (круглые!) следует закреп-
закреплять настолько, чтобы не вызвать деформации шлифа (колба удерживается благо-
благодаря наличию утолщения в верхней части шлифа). По таким же причинам нельзя
слишком жестко крепить приборы больших размеров. Приборы, изображенные
на рис. А.4, надо всегда монтировать на одном штативе. Если для более сложного
прибора это невозможно, то для его сборки лучше использовать штативную стен-
стенку, на которой отдельные стойки жестко связаны между собой. Мешалки, дефлег-
дефлегматоры и т. д. необходимо закреплять в строго вертикальном положении.
1.5. ПЕРЕМЕШИВАНИЕ И ВСТРЯХИВАНИЕ
В гетерогенных системах в целях лучшего смешивания компонентов необходи-
необходимо производить перемешивание и встряхивание. Если надо перемешать две нес-
мешивающиеся жидкости, то мешалка должна проходить через границу их раз-
раздела. Для гомогенных реакций перемешивание часто также необходимо, напри-
например, для того, чтобы быстро и равномерно распределить добавляемое вещество
по всему объему раствора либо чтобы избежать местного перегрева или локаль-
локальных повышений концентраций.
1.5.1. ТИПЫ МЕШАЛОК
Поставляются мешалки различного назначения и разных размеров. В зависи-
зависимости от природы перемешиваемых веществ материалом, из которого изго-
изготовлена мешалка, может быть стекло, политетрафторэтилен (тефлон), нержа-
нержавеющая сталь и другие металлы, в случае необходимости с покрытием (поли-
винилхлорид или полиэтилен). Для средне- и сильновязких растворов
используются мешалки с U-образной лопастью (рис. А.6, а). Такая форма
позволяет эффективно перемешивать реакционную смесь вблизи стенок
реакционного сосуда. Мешалки пропеллерного типа (рис. А.6, б и д) создают
аксиальный поток, приводящий к хорошему перемешиванию реакционной
смеси. Они используются как стандартные мешалки для перевода во взвешен-
взвешенное состояние легких твердых веществ при скоростях перемешивания (враще-
(вращения) от средних до высоких. В качестве модификации последних используют-
используются мешалки со сменными насадками-лопастями (в), которые используются в
основном для перемешивания в реакционных сосудах с выпуклым дном. Тур-
Турбинные мешалки (г) используются при скоростях перемешивания от средних
до высоких. Для перемешивания в реакционных сосудах с узким горлышком
используются мешалки с маленькими пропеллерами (б, д), лучшие результа-
результаты однако дают мешалки центрифужного типа, крылья которых при увеличе-
увеличении частоты вращения поднимаются (е). Для измельчения расплавленного
натрия можно использовать мешалки Гершберга (ж), которые, в случае боль-
большого размера насадки, позволяют собрать прибор для эффективного диспер-
диспергирования.
1. Оборудование и приемы
17
г д
Рис. А.6. Типы мешалок.
ж
а — мешалка с U-образной насадкой; б — пропеллерная насадка;
в — пропеллерная мешалка со сменной насадкой;
г — турбинная насадка; д — лопастная мешалка;
е — лопастная мешалка, раскрывающаяся под действием центробежной силы;
ж — мешалка Гершберга.
Такие мешалки различных размеров (как для небольших реакционных колб, так и для боль-
больших емкостей, применяемых в промышленности) используются главным образом для работы
с двухфазными системами (жидкость — жидкость или жидкость — твердое тело). Они состо-
состоят из приводного элемента и стальной трубки (статора), на которой крепится волнообразная
насадка (ротор). Эта движущаяся волнообразная насадка предназначена для диспергиро-
диспергирования смесей (жидкость — жидкость или твердое вещество — жидкость) и представляет собой
деталь в виде металлического решета или крылышек из трубки из нержавеющей стали.
Очень высокие скорости вращения (до 40 000 об./мин) приводят к эффективному перемеши-
перемешиванию.
Для взаимодействия твердых суспендированных веществ в растворителях, например, при
получении реактивов Гриньяра, а также для реакций восстановления со щелочными металла-
металлами можно рекомендовать активирование с использованием ультразвука (ультразвуковая баня
или сонотрон).
Магнитная мешалка (рис. А.7, рис. Г.4.125) позволяет осуществлять переме-
перемешивание в полностью закрытых сосудах и во многих моделях совмещено с наг-
нагревательным устройством. Она состоит из вращаемого с помощью мотора маг-
магнита, который вызывает движение железного стерженька в реакционной колбе;
железный стерженек заключен в стеклян-
стеклянную или тефлоновую трубку. Магнитные
мешалки применяют при гидрировании,
при работе в высоком вакууме и в других
случаях. При перемешивании малых
количеств реагентов также отдается
предпочтение этому типу мешалок. Одна-
Однако перемешивающий стерженек мешал-
мешалки должен хорошо прилегать ко дну
колбы. Поэтому магнитные мешалки
используют только в сосудах с плоским
дном (конические колбы, стаканы и
т.д.).
Очень часто, особенно при полумик-
росинтезах, для перемешивания бывает
достаточно барботировать через реакци-
реакционную смесь (инертный) газ.
Рис. А.7. Нагревательное устройство
с магнитной мешалкой.
18
А Введение в лабораторную практику
1.5.2. КРЕПЛЕНИЕ МЕШАЛОК В ПРИБОРЕ И ПРИМЕНЯЕМЫЕ УПЛОТНЕНИЯ
Для крепления мешалки обычно используется затвор с цилиндрическим шли-
шлифом, и такие мешалки обозначаются аббревиатурой KPG (от нем. kerngezogene
Prazisions-Glasgerate; рис. А.8, а). Такая
Г: мешалка состоит из прецизионного ва-
вала и точно пришлифованной к валу
трубки (±0,01 мм), оканчивающейся
шлифом. Для поддержания в норме ис-
истирания стекла и продления его срока
службы используется имеющаяся в на-
наличии специальная смазка. Поскольку
мешалки с цилиндрическим шлифом
при высоких оборотах сильно нагрева-
нагреваются, скорость вращения не должна
привышать 600 об./мин.
Для работы в вакууме, а также при
небольшом давлении следует ис-
использовать специальную мешалку с
винтовым затвором и уплотняющим
Внутренний
подшипник
| Винтовой
затвор
ПТФЭ-уплотнение
Рис. А.8. Типы затворов для мешалок,
а — с цилиндрическим шлифом для
KPG-мешалки (см. текст);
б — самоцентрирующийся стеклянный затвор
с внутренним подшипником.
элементом из тефлона. Соответствую-
Соответствующий затвор держит давление до
10~2 мбар при скоростях вращения до
800 об./мин. На рис. А.8, б приведено устройство, состоящее из трех частей с
внутренним подшипником. В случае перекоса вращающегося вала внутренний
подшипник поворачивается вместе с ним, что позволяет избежать блокировки
мешалки.
Мотор
1.5.3. МОТОРЫ
Как правило, мешалки приводятся в движение электромоторами. Скорость вра-
вращения мотора следует регулировать при помощи реостата или регулировочного
трансформатора. Перед включением мешалку прокручивают вручную, чтобы
убедиться, что она не «заедает» и при движении не касается стенки сосуда или
термометра. Все зажимы должны закреплять при-
прибор прочно, но не создавать напряжений. При ис-
использовании мешалки с цилиндрическим шли-
шлифом ее гильзу следует закрепить дополнительным
Вакуумный зажимом> так как ПРИ вращении она может из-за
шланг трения прийти в движение и выскочить из горла
колбы. Для того чтобы направляющая мешалки
не выскочила из шлифа, необходимо чтобы не-
неподвижно закрепленный мотор находился на од-
одной прямой с валом мешалки. Для этого их соеди-
соединяют с помощью двух кусочков вакуумного
Рис. А.9. Крепление мешалки шланга и стеклянной палочки (рис. А.9). Кроме
к мотору. того, используются гибкие соединения, напри-
Стеклянная
палочка
1. Оборудование и приемы 19
мер, из неопренового каучука, которые способны соединять мотор и вал мешал-
мешалки путем завинчивания.
Необходимо обратить внимание на то, что электроприборы обычно не снабжены
защитой от искрения, и поэтому при работе с легковоспламеняющимися вещест-
веществами (например, водородом, диэтиловым эфиром и др.) мешалку следует приво-
приводить во вращение при помощи водяной или воздушной турбины.
Рекомендуется использовать моторы с гибким приводом, позволяющим
пространственно разделить мотор и реакционный сосуд.
1.5.4. ВСТРЯХИВАНИЕ
В лабораторной практике встряхивание имеет гораздо меньшее значение по
сравнению с перемешиванием. Оно применяется преимущественно при работе
при повышенном давлении, например, в автоклавах (разд. А, 1.8.2), для переме-
перемешивания тяжелых осадков, таких, как цинковая пыль или амальгама натрия, в
покрывающей их жидкости, а также при работе с полумикроколичествами
веществ (например, при пробирочных опытах). В последнем случае, если реак-
реакционную смесь кипятят, вообще можно не прибегать к какому-либо дополни-
дополнительному встряхиванию.
Для длительного встряхивания используют специальные аппараты, однако
при этом не всегда можно нагревать или охлаждать реакционную смесь. Сосуды
для встряхивания надо очень тщательно закреплять!
1.6. ВВЕДЕНИЕ ГАЗОВ В РЕАКЦИЮ И ИХ ДОЗИРОВАНИЕ
Количество газов определяют по объему или массе. Объем газов измеряют либо
при помощи проградуированных сосудов (мерных цилиндров, газометров), либо
при помощи дозировочного насоса или газовых часов. Чаще всего пользуются
так называемыми жидкостными газовыми часами с водяным наполнением, в
которых газовый поток вращает барабан, связанный с отсчетным устройством.
Приборами для косвенного определения количеств газов служат реометры и
ротаметры. В реометрах (рис. АЛО, а) сужение капилляра на пути газового по-
потока вызывает перепад давления, пропорциональный количеству протекающего
газа и измеряемый при помощи параллельно подключенного U-образного
манометра. Прибор фадуируют по известным количествам протекающего газа и
строят диафамму зависимости перепада давления Ар от количества газа, пропус-
пропускаемого в единицу времени. Такая диафамма пригодна только для газа, по кото-
которому произведена фадуировка.
Выпускаемые промышленностью ротаметры (рис. А. 10, б) имеют разные ди-
диапазоны измерений. Градуированная трубка ротаметра к низу сужается, поэто-
поэтому разной скорости течения газа соответствует положение вращающегося
волчка на определенной высоте.
Количество газа можно также найти, определяя увеличение массы реакцион-
реакционного сосуда или уменьшение массы газового баллона (взвешиванием с точ-
точностью до десятых долей фамма), если газ расходовался в больших количествах.
20
А Введение в лабораторную практику
Капиляр
а
Ар
Рис. А. 10. Приборы для измерения расхода газа,
а — реометр; б — ротаметр.
Легко конденсируемые газы (аммиак, сероводород) перед дозированием
сжижают. Далее сжиженный газ, масса которого точно известна, испаряют и
пропускают через склянку-счетчик пузырьков; затем пары поступают в реакци-
реакционный сосуд.
При подаче газов в реакционную смесь конец газоподводящеи трубки обычно
погружен в жидкость; при этом для газов, которые интенсивно поглощаются раст-
раствором, возникает опасность засасывания жидкости в газоподводящую трубку.
Поэтому перед приборами, в которые вводится газ, всегда надо устанавливать
пустую предохранительную склянку (например, промывалку), размеры которой
должны быть больше объема реакционной смеси. Точно так же предохранитель-
предохранительный сосуд необходим и после приборов для получения газов (или после газовых
баллонов).
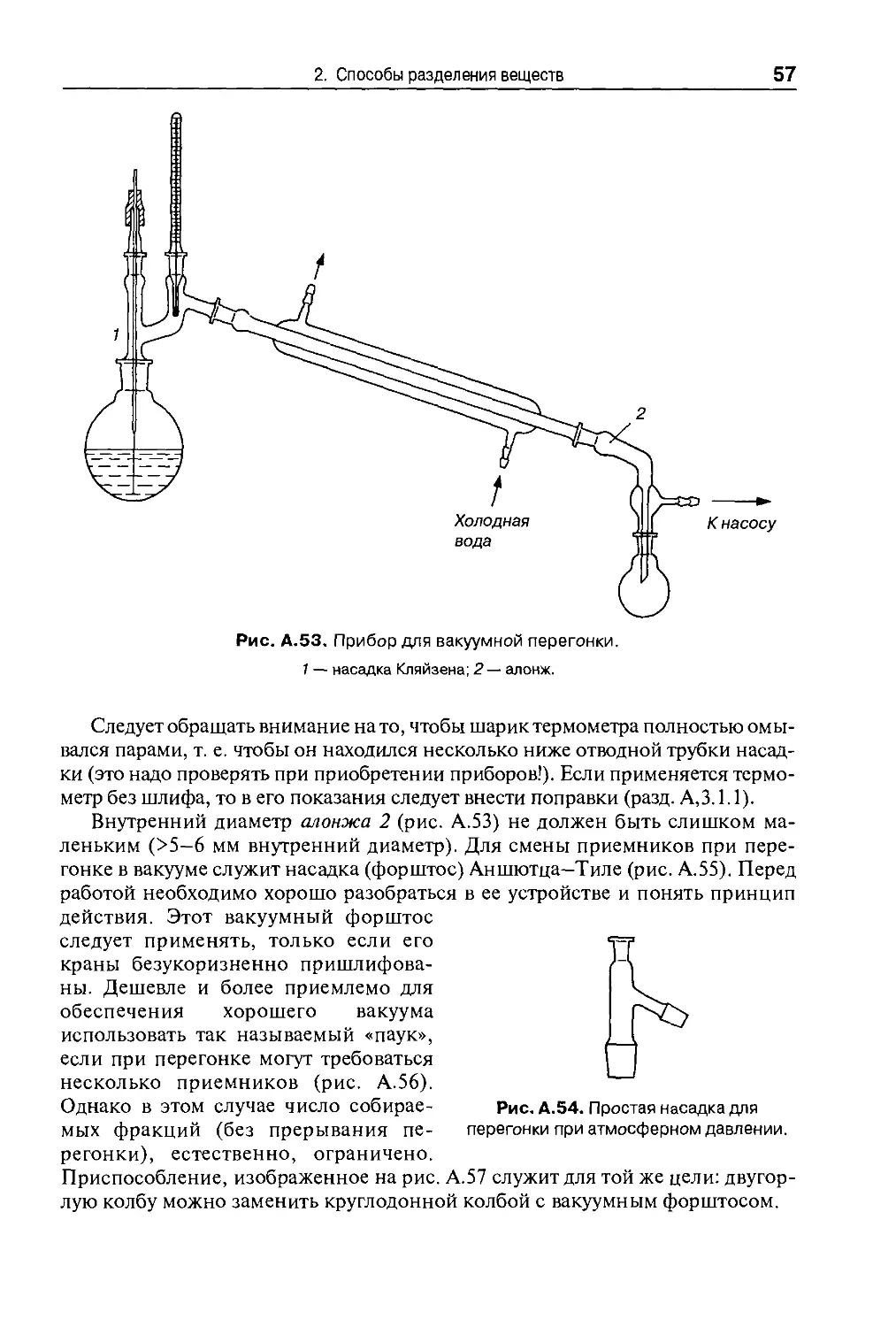
Схема стандартной системы для ввода газа приведена на рис. А. 11.
Баллон
с газом
, Предохранительные склянки
Жидкостный
клапан
Склянка для
очистки газа
Рис. А.11. Схема прибора для ввода газа.
Сосуд для
проведения
реакций
1. Оборудование и приемы
21
Стеклянная
палочка
Т-образная
трубка
О
Рис. А. 12. Промывная склянка
со стеклянной пористой пластиной.
Рис. А. 13. Приспособления для введения
газа в реакционную смесь при выпадении
в ней осадка.
Для хорошо поглощаемых газов можно предупредить засасывание растворов
из реакционного сосуда, помещая конец газоподводящей трубки над поверх-
поверхностью жидкости. При этом скорость поглощения газов можно повысить,
интенсивно перемешивая жидкость. Если требуется равномерное распределе-
распределение газа, например при тщательной промывке или для увеличения скорости его
поглощения, газ пропускают через промывную склянку со стеклянной пористой
пластиной (фриттой; рис. А. 12).
Если при введении газа образуются твердые вещества, что может быть обус-
обусловлено уже тем, что газовый поток, выходящий из газопроводящей трубки, вы-
вызывает испарение растворителя, выпадающий осадок может легко закупорить
выходное отверстие газоподводящей трубки. Эту опасность можно уменьшить,
расширив конец этой трубки. Для этой цели обычно достаточно закрепить на
конце газоподводящей трубки с помощью резинового шланга (если последний
не разрушается реакционной смесью) прямую пустую хлоркальциевую трубку и
через эту систему вводить газ (рис. А. 13, а).
Используя прибор, схема которого изображена на рис. А.13, 5 можно прочи-
прочищать закупоривающуюся газоподводящую трубку, не открывая прибора.
Во многих случаях рекомендуется установить на пути газа устройство,
предотвращающее повышение давления (предохранитель). Это совершенно
необходимо, если газ вводится через капилляр, например при вакуумной
перегонке в атмосфере инертного газа. Простейшей формой такого предохра-
предохранителя является клапан Бунзена. Он состоит из отрезка резиновой трубки,
на которой бритвенным лезвием сделан тонкий продольный надрез
(длиной 1-2 см).
22
А Введение в лабораторную практику
Рис. А. 14. Счетчик пузырьков.
Рис. А. 15. Крепление насадки
металлическими пружинами к горлу
промывной склянки для высушивания
или очистки газов.
Приспособление, изображенное на рис. А. 11 {жидкостный клапан), позволя-
позволяет надежнее обнаруживать удаление газа через клапан. Подбирая подходящие
жидкости (вода, растворы солей, силиконовые масла) и высоту их столба, можно
точно поддерживать определенное повышенное давление в замкнутых приборах.
Любая система для введения газов должна иметь приспособление, позволя-
позволяющее контролировать поток газа. Если не используют промывную склянку с
жидкостью, реометр, ротаметр и др., то на пути газа ставят простой счетчик
пузырьков (см. рис. А. 14).
Вся аппаратура для проведения реакции в потоке газа должна быть тщательно
проверена перед использованием. Особенно опасными бывают неправильно
присоединенные промывные склянки, так как при пропускании газа их содержи-
содержимое (например, концентрированная серная кислота) может быть «выброшено» из
прибора. Между промывными склянками с щелочами и кислотами в схему необ-
необходимо всегда включать пустую склянку. Все промывные склянки должны быть
плотно закрыты и для предотвращения случайного открывания снабжены метал-
металлическими (из проволоки) пружинами (рис. А. 15). Кроме того, необходимо, чтобы
реакционный сосуд свободно сообщался с атмосферой, тогда в нем не возникнет
избыточное давление. Заполненные хлоркальциевые трубки всегда следует
проверять, пропускают ли они газ.
1.7. НАГРЕВАНИЕ И ОХЛАЖДЕНИЕ
1.7.1. НАГРЕВАТЕЛИ. ТЕПЛОПЕРЕДАЧА. НАГРЕВАТЕЛЬНЫЕ БАНИ
Реакционные сосуды нагревают на газовых горелках, водяным паром или при
помощи электрических приборов. При выборе нагревателя учитывают степень и
скорость нагревания, а также указания в методике синтеза.
При помощи газовых горелок Теклю и Бунзена можно достигнуть быстрого
нагревания до относительно высоких температур.
В качестве электрических нагревателей, предназначенных для прямого наг-
нагревания круглодонных колб, применяют инфракрасные лампы или колбонагрева-
тели полукруглой формы из стекловолокна с вплетенньши электрическими спира-
1. Оборудование и приемы
23
Асбестовая
пластинка
Проволочная
' стенка
Рис. А. 16. Воздушная баня.
Подача воды
Резиновый шланг
Передвижная стеклянная
трубка
Сток воды
Рис. А.17. Регулятор уровня воды
для водяной бани.
лями. Устроенные таким же образом нагревательные элементы в форме лент
используют также и для нагревания трубок.
Прямое нагревание на пламени газовой горелки или на электрической плит-
плитке может приводить к местным перегревам, поскольку в этих случаях трудно
поддерживать заданную температуру и нельзя ее автоматически регулировать.
Кроме того, согласно правилам техники безопасности, нагревание горючих
жидкостей на открытом пламени запрещается1.
Всех этих неудобств можно избежать при использовании нагревательных
бань. В качестве теплопроводящей среды в банях применяют воздух, воду, расп-
расплавы солей, органические жидкости или сплавы.
Простейшая воздушная баня получается, если между открытым пламенем и
нагреваемой колбой помещают асбестовую сетку.
Гораздо лучше, если воздушная баня изготовлена в виде сосуда из иенского
стекла (рис. А. 16). При таком способе передача тепла осуществляется с незначи-
незначительной инерцией, хотя при этом нельзя передавать большие количества тепла.
При перегонке с использованием такой бани можно все время наблюдать за
процессом кипения. Сверху стеклянный сосуд, применяемый в качестве воз-
воздушной бани, следует прикрывать подогнанными асбестовыми пластинками.
Горючие или термически нестабильные вещества следует нагревать над асбесто-
асбестовой сеткой или с помощью воздушной бани без использования открытого пламени.
Песчаные бани обладают очень большой тепловой инерцией и ими трудно ре-
регулировать температуру. Их следует заменять по возможности другими типами
нагревательных бань.
Наиболее удобны жидкостные бани, позволяющие осуществлять очень рав-
равномерное нагревание. Для нагревания до 100 °С обычно применяют водяные бани.
Вследствие их незначительной тепловой инерции они позволяют очень точно
поддерживать заданную температуру. Регулятор уровня воды в бане (рис. А.17)
должен быть всегда подключен к водопроводу.
1 Электроплитки с открытыми спиралями с точки зрения техники безопасности также
относятся к нагревателям с открытым пламенем.
24 А Введение в лабораторную практику
Водяные бани нельзя использовать при работе с металлическим калием или
натрием, гидридами металлов или другими веществами, которые активно реаги-
реагируют с водой!
Для нагревания до -250 °С используются силиконовые бани. Они обладают
относительно большой тепловой инерцией. Особенно надо следить за тем, что-
чтобы в такие бани не попадала вода, так как в противном случае масло при нагре-
нагревании начинает пениться и разбрызгиваться. Обратные холодильники поэтому
должны всегда иметь около нижнего конца манжету из фильтровальной бумаги.
После окончания работы сразу обтирают поверхность колбы, удаляя еще горя-
горячее силиконовое масло.
Во многих случаях более удобными оказываются гликолевые бани (полиэти-
ленгликоль, триэтиленгликоль, диэтиленгликоль, этиленгликоль). Попадание в
такую баню воды не вызывает опасных явлений, а оставшийся на поверхности
колбы гликоль легко смывается водой. Эти бани используют при нагревании до
150—200 °С (температура зависит от применяемого гликоля). Однако при более
высоких температурах они тоже сильно «дымят», и работать с ними следует толь-
только в вытяжном шкафу.
Для достижения температур > 100 °С универсальными являются бани с теплопро-
водящей средой из легкоплавких сплавов {металлические бани) — сплава Вуда или
сплава Розе (т. пл. 71 и 95 °С). Эти материалы обладают высокой теплопроводностью,
что позволяет осуществлять быстрый и очень равномерный обогрев. Недостатками
их являются высокая цена и при больших размерах бани большая масса.
Бани прочно закрепляют на штативе на такой высоте, чтобы их можно было
при необходимости легко опустить и убрать с прибора. Большие бани устанав-
устанавливают на треногах.
Для нагревания жидкостных бань применяют газовые горелки или (предпоч-
(предпочтительно) регулируемые электроплитки.
При необходимости длительного нагревания реакционной смеси преимуще-
преимущество имеют нагревательные бани со ступенчатой регулировкой температуры,
обладающие интегрированной системой контроля температуры с помощью
электронно-регулируемого термостата.
Для контроля за температурой в баню всегда вводят термометр (из металли-
металлических и парафиновых бань его необходимо удалять до затвердевания расплава).
Для получения водяного пара в лаборатории пользуются обыкновенными круг-
лодонными колбами с пароотводом и вертикальной трубкой или лучше специ-
специальными медными парообразователями. Однако этот способ получения пара
используется преимущественно при перегонке с водяным паром (см. рис. А.80).
1.7.2. НАГРЕВАНИЕ ОГНЕОПАСНЫХ ЖИДКОСТЕЙ
Огнеопасные жидкости могут быть нагреты с использованием открытого пламени
только в исключительных случаях, например, если количество вещества в реакцион-
реакционном сосуде мало. Из открытых сосудов их перегоняют только в неопасных количест-
количествах и только в закрытом вытяжном шкафу в отсутствие источников открытого пламе-
пламени. Даже небольшие количества огнеопасных жидкостей не следует выпаривать в
сушильном шкафу. Как правило, огнеопасные жидкости можно нагревать лишь с ис-
использованием закрытой аппаратуры, снабженной холодильником, с использовани-
1. Оборудование и приемы
25
ем электрических нагревательных устройств или жидкостных бань. Источники тепла
должны в любой момент легко удаляться, для чего подходят регулируемые по высо-
высоте лабораторные подставки.
При нагревании больших количеств огнеопасных жидкостей следует предприни-
предпринимать особые меры безопасности, например использовать металлические сосуды.
Если необходимо нагревание в стеклянных сосудах, рекомендуется установить под
аппаратурой емкость, устланную стекловолокном или асбестом.
При растрескивании реакционного сосуда или при выливании огнеопасной
жидкости, температура которой выше, чем температура ее воспламенения, сле-
следует немедленно удалить все источники воспламенения, помещение проветрить,
а вылившуюся жидкость собрать и удалить.
Работа с эфиром, дисульфидом углерода и другими низкокипящими и легко-
легковоспламеняющимися веществами требует особых мер предосторожности.
С большими количествами огнеопасных веществ, взрыв паров которых может
привести к разрушениям, разрешается работать только в специально оборудо-
оборудованных помещениях. В таких лабораториях должны отсутствовать любые источ-
источники тепла, способные вызвать воспламенение, и установлены лишь взрывобе-
зопасные электрические приборы, в т. ч. моторы для мешалок и т. д.
При нагревании жидкостей выше температуры кипения может произойти пе-
перегрев, а затем взрывообразное вскипание. Этого весьма опасного явления во
многих случаях можно избежать, применяя «кипятильники» (небольшие кусочки
обоженного неглазурованного фарфора и т. д.), которые ни в коем случае нельзя
бросать в уже нагретую до кипения жидкость. Каждый кипятильник используют
только один раз, так как при охлаждении жидкость заполняет все его поры и он
теряет свою действенность. При нагревании в вакууме регулируют кипение,
пропуская газ через капилляр (разд. А.2.3.2.2).
1.7.3. ОХЛАЖДАЮЩИЕ СРЕДСТВА
При выборе охлаждающего средства принимают во внимание, какую температу-
температуру необходимо поддерживать и какое количество тепла надо отвести. Для этой
цели наибольшее применение нашла вода как дешевое средство, имеющее высо-
высокую теплоемкость. Если в приборе надо охлаждать колбу, то ее помещают в
большую воронку, на кончик которой одет нисходящий шланг, опущенный в
раковину; на колбу подается водопроводная вода.
Лед для охлаждения тонко измельчают (мельница для льда). Для улучшения
теплопередачи готовят кашицу из льда с небольшим количеством воды.
При помощи смеси льда с поваренной солью можно добиться охлаждения при-
примерно до —20 "С. Для приготовления такой смеси тонкоизмельченный лед
смешивают с технической поваренной солью ('/з по
массе).
Добавляя твердую углекислоту («сухой лед») к ме-
метиловому спирту, ацетону или другим подходящим
растворителям (диэтиловый эфир запрещен!), мож-
можно достичь температуры —78 °С. Для достаточного
охлаждения твердую углекислоту надо брать в избыт-
избытке, так как хладоемкость такой смеси не очень боль-
большая. Охладительную смесь готовят в сосуде Дьюара
(рис. А.18), чтобы уменьшить обмен тепла с окружа-
окружающей средой. Рис. А.18. Сосуд Дьюара.
26 А Введение в лабораторную практику
Сухой лед следует хорошо измельчить в металлической (но не фарфоровой)
ступке (защитные очки и резиновые перчатки!). Следует быть осторожным при
добавлении растворителей из-за сильного вспенивания. Сосуды Дьюара во
избежание взрыва надо либо обмотать снаружи асбестовым шнуром или иной
тканью, либо поместить в проволочный или деревянный ящик. Особенно чувстви-
чувствителен у сосуда Дьюара верхний край.
Если охлаждающее действие такой смеси недостаточно, используют жидкий
азот (охлаждение до —196 °С). Сосуд Дьюара перед заполнением следует очень
тщательно высушить. Жидкий воздух или жидкий азот, обогащающиеся по ме-
мере стояния кислородом, нельзя использовать для охлаждения органических ве-
веществ, так как при этом возникает возможность воспламенения.
Точное термостатирование при низких температурах (примерно до —80 °С)
обеспечивается низкотемпературными термостатами (криостатами). С помощью
электронно-регулируемого ротационного насоса, обладающего высокой устойчи-
устойчивостью к перемене температур, к реакционным сосудам и вспомогательным
устройствам подается для охлаждения специальная охлаждающая жидкость.
Для хранения веществ при пониженных температурах в течение длительного
времени служат холодильные шкафы (холодильники). В холодильных шкафах на ве-
веществах может конденсироваться влага, выделяющиеся из веществ агрессивные
газы могут вызвать коррозию холодильника, а пары органических растворителей —
вызвать взрыв, поэтому в холодильные шкафы разрешается ставить только хорошо
закупоренные сосуды. Последние должны быть снабжены четкими надписями.
Хранение огнеопасных жидкостей в холодильных шкафах допускается только в
том случае, если внутри охлаждающего пространства нет электротехнических
устройств, либо имеются только взрывобезопасные устройства. Охлаждающее
пространство должно быть изолированным от электротехнических и пожаро-
пожароопасных устройств.
1.8. РАБОТА ПОД ДАВЛЕНИЕМ
Если требуется проводить реакцию при температуре более высокой, чем темпе-
температура кипения вводимых в реакцию компонентов, или если необходима повы-
повышенная концентрация газообразного вещества (например, при гидрировании,
см. разд. Г,4.5.2), то работу проводят в герметичном приборе под давлением. Для
опытов с небольшими количествами веществ при невысоких избыточных давле-
давлениях используют запаянные трубки; в случае больших количеств и высоких дав-
давлений применяют автоклавы, в которых давление должно постоянно контроли-
контролироваться, а газ может вводиться под давлением.
Обычная лабораторная посуда не годится для работы при повышенных давлениях.
Если по окончании взаимодействия давление в системе возвращается к нормальному,
то для проведения реакций иногда можно использовать специальные стеклянные
сосуды.
1. Оборудование и приемы 27
1.8.1. ТОЛСТОСТЕННЫЕ АМПУЛЫ ДЛЯ РАБОТЫ ПОД ДАВЛЕНИЕМ
Запаянные толстостенные ампулы из стекла дуран позволяют работать при дав-
давлении 20—30 атм и максимальной температуре 400 °С.
Реакционную смесь помещают в такую ампулу через воронку с длинным гор-
горлом. Три четверти ампулы должны оставаться незаполненными. Затем на пламе-
пламени газовой горелки с кислородным дутьем оттягивают толстостенный капилляр,
запаивают его (лучше всего поручить эти операции стеклодуву) и оттянутый запа-
запаянный конец осторожно и медленно охлаждают. Запаянную ампулу помещают в
железный кожух, частично заполненный песком, так, чтобы верхний конец
ампулы выступал из кожуха на 1—2 см. Кожух с ампулой устанавливают в печь
так, чтобы открытый конец кожуха был обращен в сторону специальной ловуш-
ловушки для осколков, укрепленной на стене. Температура обогрева должна регулиро-
регулироваться автоматически. Окружающие предметы предохраняют от повреждений с
помощью защитного экрана. По окончании реакции ампулу оставляют в печи
до полного охлаждения, затем вместе с железным кожухом вынимают из печи
(открытый конец кожуха нельзя направлять в свою сторону!) и осторожно нагре-
нагревают верхний запаянный конец ампулы, выходящий из защитного кожуха,
острым пламенем паяльной горелки (защитные очки!). Если в ампуле давление
повышено, в стекле в размягченном месте появляется отверстие и избыточные
газы удаляются. Вскрытие ампулы также лучше всего поручить стеклодуву.
Реакции в запаянных ампулах разрешается проводить только в специально пред-
предназначенных для этого помещениях при строгом соблюдении всех вышеуказан-
вышеуказанных требований. Запаянные ампулы до вскрытия нельзя извлекать из защитного
кожуха или выносить из помещения для работы с ними. Перед работой необходи-
необходимо выяснить по справочным таблицам упругость паров применяемых раствори-
растворителей и учитывать эти значения наряду с соответствующими упругостями образу-
образующихся газов, оценивая давление внутри ампулы во время реакции.
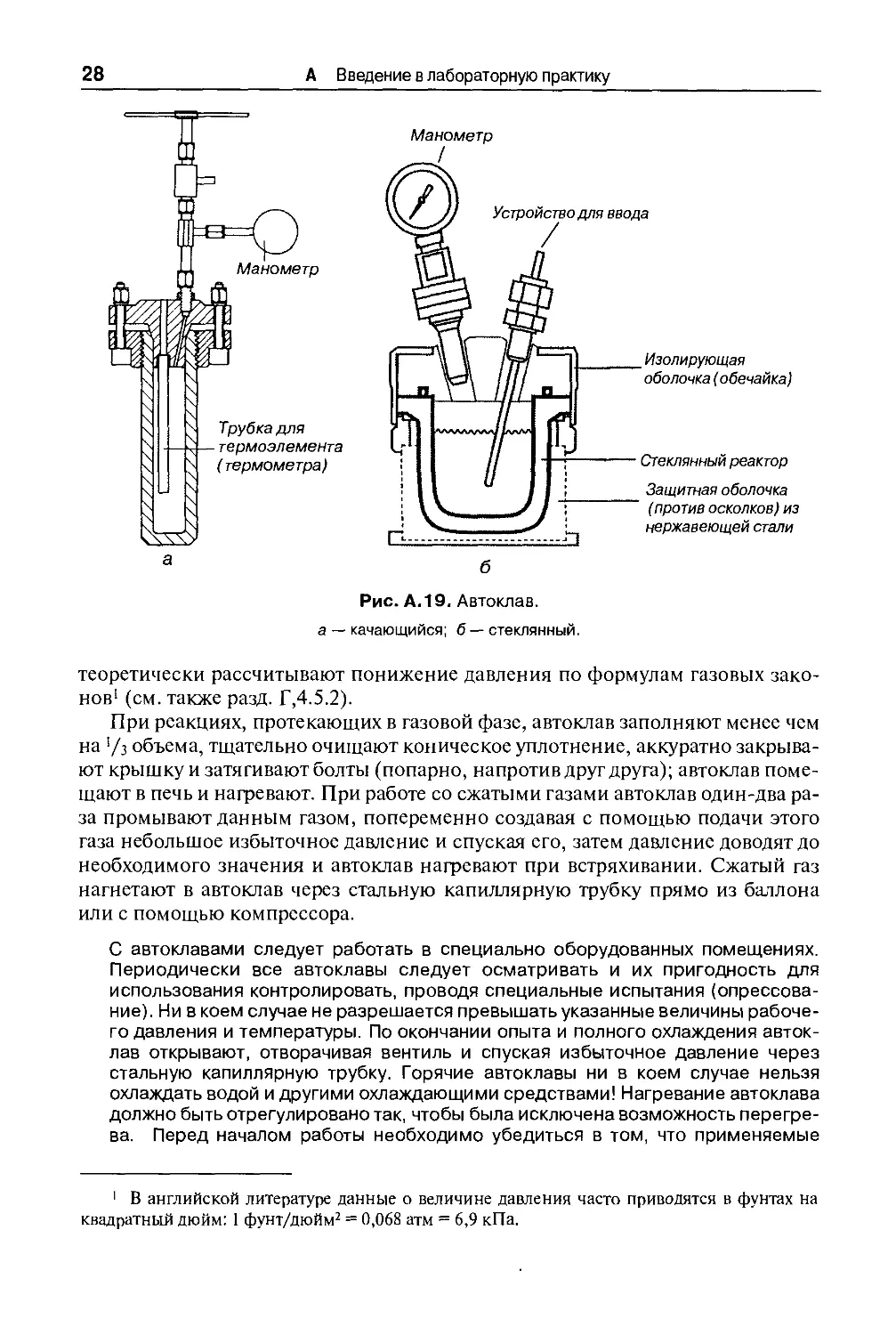
1.8.2. АВТОКЛАВЫ
Широкое применение для разнообразных целей в лабораториях получил
качающийся автоклав (рис. А. 19, а) (емкость 1 л, максимальное рабочее давле-
давление 35 МПа C50 атм), максимальная рабочая температура 350 °С, изготовляется
из стали типа V2A или V4A). Автоклав нагревается в автоматической электри-
электрической печи. Он должен легко выниматься из печи. Корпус и крышка автоклава
соединяются болтами; наиболее пригодно коническое уплотнение между крыш-
крышкой и корпусом автоклава.
На рис. А. 19, а автоклав снабжен манометром и имеет трубку для термомет-
термометра. С манометрической трубкой связан вентиль, еще один находится у специаль-
специально высверленного отверстия (не изображенного на рисунке). Содержимое ав-
автоклава может перемешиваться специальной мешалкой. Особенно удобны
автоклавы с магнитной мешалкой, в которых сердечник мешалки, находящийся
внутри автоклава, приводится в движение сильным электромагнитом, располо-
расположенным снаружи. Мешалки с сальниковым уплотнением требуют очень
тщательного ухода и гораздо менее удобны для работы в лабораториях.
До начала эксперимента необходимо выяснить величину ожидаемого при
реакции давления. При реакциях с газами (например, при гидрировании)
28
А Введение в лабораторную практику
Манометр
Манометр
I
Устройство для ввода
Трубка для
термоэлемента
(термометра)
Изолирующая
оболочка (обечайка)
Стеклянный реактор
Защитная оболочка
(против осколков) из
нержавеющей стали
Рис. А. 19. Автоклав,
а — качающийся; б — стеклянный.
теоретически рассчитывают понижение давления по формулам газовых зако-
законов1 (см. также разд. Г,4.5.2).
При реакциях, протекающих в газовой фазе, автоклав заполняют менее чем
на '/з объема, тщательно очищают коническое уплотнение, аккуратно закрыва-
закрывают крышку и затягивают болты (попарно, напротив друг друга); автоклав поме-
помещают в печь и нагревают. При работе со сжатыми газами автоклав один-два ра-
раза промывают данным газом, попеременно создавая с помощью подачи этого
газа небольшое избыточное давление и спуская его, затем давление доводят до
необходимого значения и автоклав нагревают при встряхивании. Сжатый газ
нагнетают в автоклав через стальную капиллярную трубку прямо из баллона
или с помощью компрессора.
С автоклавами следует работать в специально оборудованных помещениях.
Периодически все автоклавы следует осматривать и их пригодность для
использования контролировать, проводя специальные испытания (опрессова-
ние). Ни в коем случае не разрешается превышать указанные величины рабоче-
рабочего давления и температуры. По окончании опыта и полного охлаждения авток-
автоклав открывают, отворачивая вентиль и спуская избыточное давление через
стальную капиллярную трубку. Горячие автоклавы ни в коем случае нельзя
охлаждать водой и другими охлаждающими средствами! Нагревание автоклава
должно быть отрегулировано так, чтобы была исключена возможность перегре-
перегрева. Перед началом работы необходимо убедиться в том, что применяемые
1 В английской литературе данные о величине давления часто приводятся в фунтах на
квадратный дюйм: 1 фунт/дюйм2 = 0,068 атм = 6,9 кПа.
1. Оборудование и приемы
29
вещества не взаимодействуют с металлом, из которого изготовлен корпус ав-
автоклава. Например, сталь V2A при нагревании неустойчива к действию галоге-
новодородных, муравьиной и уксусной кислот, галогенид-ионов и к действию
окислителей при обычных условиях1.
При работе с небольшими давлениями и загрузками имеет смысл пользо-
пользоваться стеклянными автоклавами (рис. А. 19, б). Такой реактор из стекла с объе-
объемом от 100 мл подходит для проведения химических реакций в водной или орга-
органической среде при предельных давлениях и температурах до 10 бар/100 °С или
6 бар/150 °С. Содержимое автоклава можно удобно перемешивать с помощью
магнитной мешалки.
1.8.3. БАЛЛОНЫ ДЛЯ ГАЗОВ
Газы, имеющие наибольший спрос, поступают в продажу в стальных баллонах;
баллоны для различных газов отличаются друг от друга по цвету корпуса, а так-
также резьбой на штуцере для вентиля. Характеристика баллонов для некоторых
газов представлена в табл. А.20.
Таблица А.20. Характеристика стальных баллонов для некоторых газов
Газ
Водород
Монооксид углерода
Амины (моно-, диметиламин и др.)
Углеводороды
Кислород
Азот
Хлор
Диоксид серы
Фосген
Диоксид углерода
Аммиак
Ацетилен
Цвет корпуса
баллонаа
Красный
»
»
»
Голубой
Зеленый
Серый
»
»
»
»
Желтый
Направление резьбы
штуцера вентиля
Левая
»
»
»
Правая
»
»
»
»
»
»
Специальный вентиль
а Иногда баллоны маркируются только одной полосой (кольцевой) соответствующего цвета.
Стальные газовые баллоны следует оберегать от нагревания; их прочно закреп-
закрепляют в вертикальном положении у стены с помощью цепочки или хранят в гори-
горизонтальном положении. Отбор газа следует производить только при помощи
редуктора.
Баллоны для газов из-за опасности их разрушения (взрыва) при пожаре сле-
следует устанавливать вне помещения лаборатории. На рабочее место газы следует
подавать с помощью хорошо закрепленных прочных (металлических) трубок.
1 Подробную информацию по этому вопросу см.: Ullmanns Encyklopadie der Technischen
Chemie, 4. Aufl. — Verlag Chemie, Weinheim/Bergstr., 1972, Bd. 3. S. 14ff. C. Aufl. Bd. 16, S. 260ff).
30
А Введение в лабораторную практику
Если монтаж (установка) газовых баллонов невозможен по техническим причи-
причинам, то их следует размещать либо в проветриваемых и теплоизолированных шка-
шкафах, либо после окончания работы перемещать в безопасное место.
Газовые баллоны с очень ядовитыми, ядовитыми, опасными для здоровья
или канцерогенными газами, которые необходимы при работе, должны на-
находиться в условиях постоянного откачивания окружающего воздуха (венти-
(вентиляции), например под тягой. Для таких газов следует использовать, по воз-
возможности, небольшие баллоны.
Газовые баллоны следует обезопасить от опрокидывания, например, с
помощью цепей, обручей (круговых) или фиксирующих устройств (стоек).
Газовые баллоны следует защищать от сильного нагревания (например, от
действий горячих предметов, солнечного излучения), а также от ударов, особен-
особенно, при сильных морозах.
Газовые баллоны следует использовать лишь при наличии подходящих
(паспортных) редукционных вентилей.
:?¦:)
Предохранительный клапан
Запорный
вентиль
б
Рис. А.21. Редукторы для баллонов с газами,
а — игольчатый вентиль; б — редукционный вентиль.
1. Оборудование и приемы 31
Принципиальное устройство игольчатого вентиля изображено на рис. А.21, а.
Он может употребляться как редукционный вентиль для любого газа (кроме аце-
ацетилена). При помощи редукционного вентиля, изображенного на рис. А.21, б,
можно регулировать подачу газа и установить постоянный поток газа. Вентиль
открывают, поднимая конус вентиля (закручивая установочный винт, см.
рис. А.21, б), при закрытом запорном вентиле; при этом манометр малого давле-
давления показывает небольшое избыточное давление. Необходимый расход газа уста-
устанавливают, осторожно открывая запорный вентиль.
Арматура, манометр, уплотнения и т. п. для сжатых газов, являющихся сильными
окислителями (например, кислород, закись азота), должны быть свободны от
масла, жира и глицерина. Эти детали также не следует протирать содержащими
масла тряпками или касаться жирными пальцами. Остаток растворителя, кото-
который служит для удаления жировых загрязнений, следует удалять потоком не
содержащего масел воздуха.
В случае кислорода следует использовать манометр, маркированный голу-
голубым цветом и на котором имеется надпись «Кислород! Содержать свободным от
масла и жира».
У вентилей кислородных баллонов винтовую резьбу на штуцерах не разреша-
разрешается смазывать жирами, так как это может привести к взрывам.
Для полумикроколичественных синтезов пригодны небольшие (содержание газа в преде-
пределах 200—450 г) баллоны со сжатыми газами, которые удобно устанавливать около тяги с соот-
соответствующей аппаратурой. Список имеющихся в продаже газов можно найти в химических ка-
1.9. РАБОТА ПОД УМЕНЬШЕННЫМ ДАВЛЕНИЕМ
Вакуум применяется в лаборатории для различных целей, важнейшими из них
являются перегонка и возгонка под уменьшенным давлением, высушивание,
фильтрование (с отсасыванием) и, наконец, теплоизоляция.
Сосуды Дьюара (рис. А.18), предназначенные для хранения охлаждающих
смесей, сухого льда, сжиженного воздуха, представляют собой тонкостенные,
вакуумированные (давление <10~5 мм рт. ст.) стеклянные сосуды, посеребрен-
посеребренные изнутри. Теплопроводность сильно разреженных газов очень мала, и поэ-
поэтому сосуды Дьюара превосходят по своим теплоизолирующим свойствам все
остальные приспособления. По принципу устройства сосудов Дьюара изготав-
изготавливаются рубашки для ректификационных колонок (посеребренные изнутри
вакуумированные рубашки).
Проведение в вакууме перегонки (разд. А,2.3.2.2), возгонки (разд. А,2.4),
высушивания (разд. АД.10.3) и отсасывания (разд. А,2.1) рассмотрены в соотве-
соответствующих разделах.
1.9.1. СОЗДАНИЕ ВАКУУМА
Для практических целей различают следующие интервалы давления: небольшой
вакуум A-760 мм рт. ст.), умеренный вакуум @,001-1 мм рт. ст.) и высокий
(глубокий) вакуум (<10~3 мм рт. ст.).
32 А Введение в лабораторную практику
Для создания пониженного давления в лаборатории применяют чаще всего
водоструйные и ротационные масляные насосы.
Водоструйный насос требует довольно большого расхода воды A л на 0,6 л
отсасываемого газа). Вакуум водоструйного насоса ограничен упругостью паров
воды (8— 15 мм рт. ст. в зависимости от температуры).
Из соображений экономии воды водоструйные насосы все в большей степе-
степени заменяются на электрические мембранные насосы. Последние работают без
использования масла, не требуют для работы воду, что не приводит к ее значи-
значительному расходованию. Поскольку мембранные насосы производятся с
использованием устойчивых к коррозии материалов, они не чувствительны к
действию агрессивных веществ и конденсатов. Эвакуируемые растворители
собираются во встроенных приемниках (сепараторах) и могут быть в конце
работы отброшены или после переработки использованы вновь. В продаже име-
имеются насосы с производительностью от 2 до 11 м3/ч, при этом достигается ваку-
вакуум от 80 до 2 мбар (от ~60 до 2 мм рт. ст.).
Ротационный вакуумный насос работает по принципу сжатия газа в простран-
пространстве, состоящем из двух частей, в котором эксцентрически расположенный
ротор сжимает всасываемый газ, который затем выбрасывается (в атмосферу).
Такие ротационные насосы, по преимуществу, использующие для уплотнения
масло, чувствительны к агрессивным и легкоконденсируемым жидкостям. Уста-
Установка газобалластного вентиля или охлаждаемой ловушки (рис. А.26, б, запол-
заполнение: сухой лед/этанол или жидкий азот) минимизирует попадание этих меша-
мешающих жидкостей в масло.
При работе с агрессивными (приводящими к коррозии) веществами
или легкоконденсируемыми парами имеет смысл использовать гибрид-
гибридный насос, в котором мембранный насос перманентно эвакуирует
маслосодержащее составляющее ротационного насоса. При таком соединении
с помощью ротационного насоса можно достичь вакуума до 10~4 мбар
(~10ммрт. ст.).
Для достижения высокого вакуума (<10~4 кПа; <10~3 мм рт. ст.) используют
масляные или ртутные диффузионные насосы. Устройство и эксплуатация
таких насосов, а также способы измерения давления при высоком вакууме
рассматриваются в соответствующей специальной литературе.
Для достижения и поддержания в приборе пониженного давления, которое всегда несколь-
несколько выше максимального разряжения, создаваемого насосом, например при дистилляции и при
перегонке на ротационном испарителе, применяются различного типа маностаты. Удобными и
повсеместно используемыми являются регулируемые с помощью электронных устройств прибо-
приборы, оборудованные вентилем для сброса давления. С их помощью можно просто и быстро дос-
достичь оптимального для работы вакуума и затем его регулировать. Рабочая область этих маноста-
тов лежит между 0,1 и 100кПа(от~1 до 750 мм рт. ст.).
Очень просто (но для большинства целей достаточно точно) можно регули-
регулировать давление в интервале 10—760 мм рт. ст. без маностата, если впускать не-
небольшие количества воздуха через кран склянки Вульфа (рис. А.25). Регулирова-
Регулирование количества поступающего воздуха облегчается, если на отверстии крана
сделать насечку (рис. А.22). Достаточно тонкая регулировка давления может
осуществляться с помощью обычного металлического винтового зажима: через
1. Оборудование и приемы
33
Рис. А.22. Насечка на кране
для тонкой регулировки.
Рис. А.23. Укороченный вакуумметр.
резиновую трубку надо пропустить тонкую проволоку и, ослабляя и затягивая
зажим, оставить небольшое отверстие для воздуха.
1.9.2. ИЗМЕРЕНИЕ ДАВЛЕНИЯ В ВАКУУМНЫХ СИСТЕМАХ
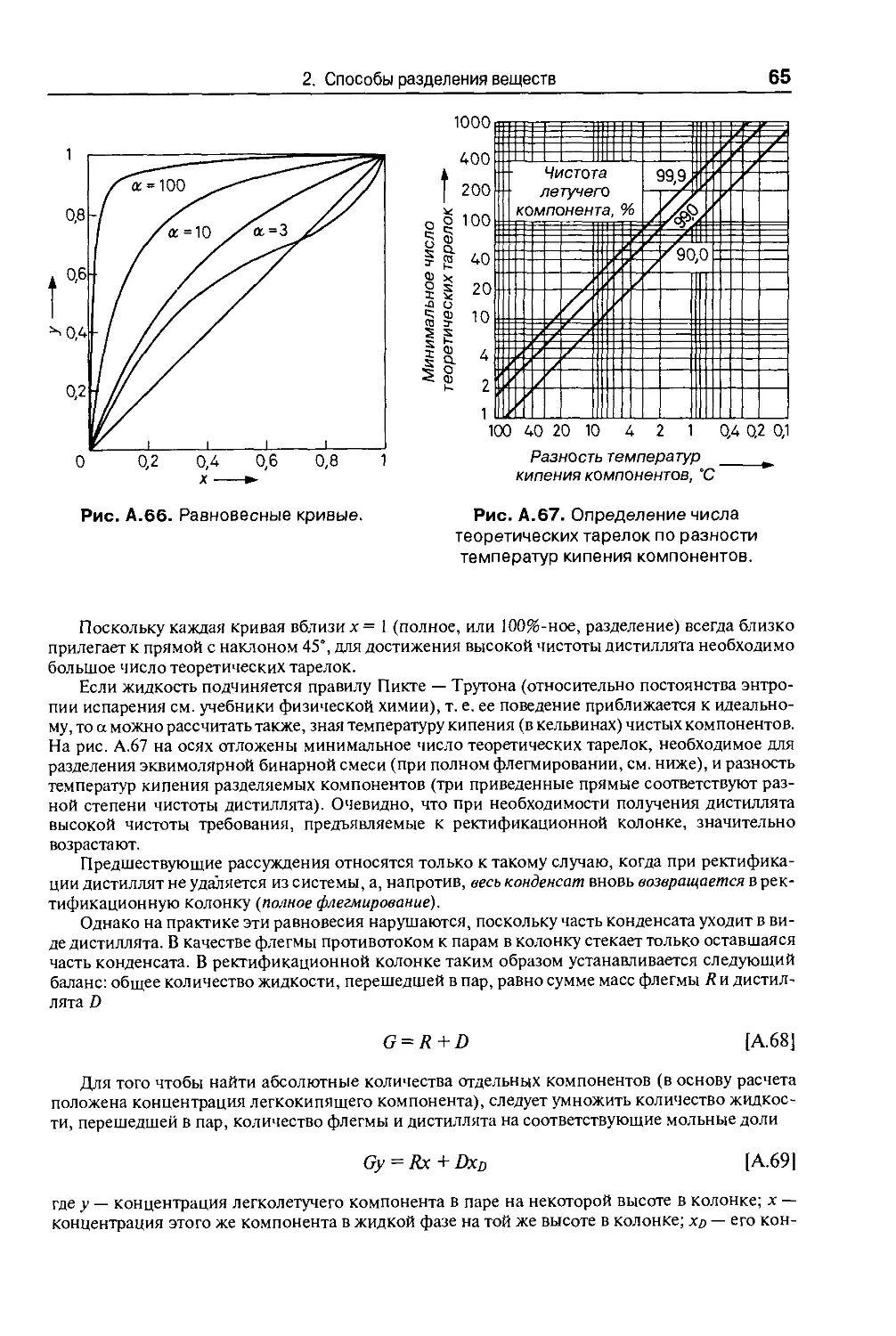
Для измерения давления в интервале 1—200 мм рт. ст. служат укороченные
ртутные вакуумметры (рис. А.23), которые для защиты от разбрызгивания рту-
ртути при поломке, делают с двойными стенками. Точность измерений составля-
составляет +0,5 мм рт. ст. Однако, если внутрь запаянного конца манометра попадут
воздух или пары, точность понижается и получаемые результаты могут быть
совершенно ошибочны. Поэтому необходимо следовать правилу — открывать
кран манометра только при снятии показаний. Простой способ проверить, не
загрязнен ли манометр воздухом или летучими веществами, состоит в том, что
его соединяют с масляным насосом и создают давление <0,2 мм рт. ст. При
этом ртуть в обоих коленах должна нахо-
находиться на одинаковом уровне. Загрязнения
легко обнаруживаются по появлению «от-
«отрицательного» давления.
Для измерения давлений 1—10 мм рт. ст.
используют компрессионные вакуумметры.
Наиболее известен манометр Мак-Леода.
Принцип его действия можно объяснить на
примере вполне пригодного для большин-
большинства целей укороченного вакуумметра Геде
(рис. А.24). При горизонтальном положении
в измерительном пространстве 1 поддержи-
поддерживается давление, равное давлению в приборе.
При повороте вакуумметра на 90° в положе-
положение, изображенное на рисунке, ртуть (масса
Рис. А.24. Вакуумметр Геде.
34
А Введение в лабораторную практику
которой определена с достаточной точностью) сжимает находящийся в объе-
объеме 1 газ, т. е. объем / уменьшается. По шкале прибора (в единицах давления)
можно определить первоначальное давление газа. При измерении давления ваку-
вакуумметром Геде во время отсчета нельзя снимать вакуум в приборе. Компрессион-
Компрессионные вакуумметры показывают действительное давление только в том случае,
когда в приборе нет паров, конденсирующихся при комнатной температуре.
Ртуть для манометра следует периодически очищать. При этом необходимо
соблюдать правила техники безопасности при обращении с ртутью (см. часть Е).
Точное определение рабочего давления (вакуума) достигается использованием не содержа-
содержащих ртути электронных приборов для измерения вакуума. Это обеспечивается их прочностью,
компактностью конструкции и коррозионной устойчивостью. Использование этого типа уст-
устройств подходит для измерений давления вплоть до 0,1 Па A0~3 мм рт. ст.).
1.9.3. РАБОТА ПОД ВАКУУМОМ
Приборы для работы под вакуумом должны быть собраны так, чтобы потери
давления в них были незначительны и можно было полностью использовать
мощность вакуумного насоса. Поэтому в вакуумной системе должно быть, воз-
возможно меньше деталей небольшого сечения (это относится к вакуумным шлан-
шлангам, кранам с узкими отверстиями, узким насадкам, форштосам, плотно запол-
заполненным ректификационным колонкам и др.). Необходимо обратить особое
внимание на то, что при вакуумной перегонке (или возгонке в вакууме) следует
применять только круглодонные колбы, так как плоскодонные могут под ваку-
вакуумом лопнуть.
Водоструйный насос разрешается присоединять к прибору только через предох-
предохранительную склянку (склянка Вульфа), чтобы избежать переброса воды из насо-
насоса в манометр или прибор (например, при внезапном падении давления воды в
водопроводе).
Целесообразно присоединять манометр параллельно прибору через склянку
Вульфа (рис. А.25). Перед выключением водоструйного насоса всегда необходи-
необходимо впустить воздух в вакуумную систему через кран на склянке Вульфа или на
манометре.
Прибор
Манометр
Водоструйный
насос
Склянка Вульфа
Рис. А.25. Схема установки для работы при уменьшенном давлении.
1. Оборудование и приемы
35
Вакуумметр
ь-
Трехходовой
кран
¦ Прибор
, Охлаждаемая
ловушка
\ Предохранительный
сосуд
Масляный
насос
Рис. А.26. Схема установки для создания вакуума (а) и используемая в ней
ловушка (б), охлаждаемая в сосуде Дьюара.
Схема очень простой установки для создания вакуума приведена на
рис. А.26. Между масляным насосом и прибором находятся предохранительный
сосуд объемом 1 л и охлаждаемая ловушка. Если во время вакуумной перегонки
пришлось очень сильно нафеть перегонную колбу, то перед заполнением ваку-
вакуумной системы воздухом надо дождаться полного остывания нафетой колбы.
Внезапный впуск воздуха в нагретый прибор может привести к взрыву образую-
образующейся при этом смеси паров перегоняемого вещества с воздухом!
Еще раз следует подчеркнуть, что при всех видах работ под уменьшенным давле-
давлением [перегонка, возгонка, высушивание (в вакуум-эксикаторах!), отсасывание,
работа с сосудами Дьюара и ректификационными колонками] совершенно необ-
необходимо надевать защитные очки!
1.10. ВЫСУШИВАНИЕ
Средство считается удовлетворительным, если при хорошей эффективности
высушивания также обладает значительной осушающей емкостью.
Максимальная эффективность осушителя определяется упругостью водяных
паров над ним (табл. А.27). Гидраты приведенных в таблице осушителей, обра-
образующиеся по мере присоединения воды, обладают меньшей эффективностью,
чем исходные безводные соединения (ср. данные о перхлоратах магния в табли-
таблице). Чем больше воды может принять осушитель при сохранении достаточной
эффективности, тем выше его осушающая емкость.
Такие вещества, как фосфорный ангидрид, серная кислота, хлорид кальция,
сульфаты магния и натрия, удовлетворяют обоим требованиям и поэтому при-
применяются наиболее часто. Сульфат кальция, обладающий высокой эффектив-
эффективностью, имеет, однако, лишь незначительную осушающую емкость.
36
А Введение в лабораторную практику
Таблица А.27. Упругость водяных паров над некоторыми осушителями при темпера-
температуре 20 'С
Осушитель
Р4О10
Mg(C104J (ангидрон)
Mg(C104J • ЗН2О (дегидрит)
КОН (плавленый)
А12О3
CaSO4 (дриерит)
H2SO4 (конц.)
Силикагель
NaOH (плавленый)
СаО
СаС12
CuSO4
Упругость водяных паров,
з • ю-6
7 • Ю-5
з • ю-4
з • ю-4
4 • Ю-4
5 • Ю-4
7 • Ю-4
8 • Ю-4
0,02
0,03
0,03
0,2
кПа (мм рт. ст.)
B ¦ 10-5)
E • Ю-4)
@,002)
@,002)
@,003)
@,004)
@,005)
@,006)
@,15)
@,2)
@,2)
A,3)
1.10.1. СУШКА ГАЗОВ
Для сушки газов с применением твердых осушителей применяют осушитель-
осушительные колонки (рис. А.28, а). Чтобы предотвратить слипание таких аморфных
осушителей, как фосфорный ангидрид, колонки наполняют предварительно
приготовленной смесью осушителя с асбестовым или стеклянным волокном
либо с пемзой. Эффективнее и экономнее использование осушающих батарей
(рис. А.28, б), установленных вертикально на стене лаборатории. Газ через
счетчик пузырьков пропускают через две или несколько стеклянных трубок,
наполненных осушителем. При этом может быть использовано и несколько
различных осушителей.
Химически индифферентные газы обычно сушат, пропуская их через про-
промывные склянки с концентрированной серной кислотой. При этом надо обяза-
обязательно включать в схему прибора предохранительные склянки (ср. рис. А. 11),
снабженные специальными приспособлениями от случайного открывания
Стеклянная
вата
Осушитель
Стеклянная
вата
Рис. А.28. а — осушительная колонка; б — осушающая батарея.
1. Оборудование и приемы 37
(рис. А. 15). Предохранительные склянки могут представлять собой промывал-
ки, снабженные барботером (с пористой пластиной) (см. рис. А. 12).
Низкокипящие газы сушат, вымораживая воду и другие конденсирующиеся
примеси в охлаждаемой ловушке (рис. А.26, б). При этом достигается очень вы-
высокая степень осушки (табл. А.29). Для охлаждения ловушки применяют смесь
сухого льда с метиловым спиртом или жидкий азот (разд. А, 1.7.3).
Таблица А.29. Содержание водяных паров в газах при различных температурах
Температура, °С
+20
0
-10
-70
-100
Парциальное давление
водяных паров, кПа (мм рт. ст.)
2,3 A7,5)
0,6 D,6)
0,1 @,77)
3 • Ю-4 @,002)
1 ¦ Ю-6 A • Ю-5)
Для защиты от атмосферной влаги приборы закрывают трубками; в качестве
наполнителей таких трубок применяют хлорид кальция, натронную известь или
другие подходящие осушители. Осушитель удерживают в трубке, закрепляя его
с обеих сторон тампонами из стеклянной ваты. Перед повторным использова-
использованием трубок следует проверять, сохранили ли они способность пропускать газ
(не забиты ли).
1.10.2. ОБЕЗВОЖИВАНИЕ ЖИДКОСТЕЙ
Осушители для определенных классов органических веществ указаны в табл. А.31.
Способы очистки и обезвоживания наиболее распространенных растворителей
приведены в части Е.
Растворители можно высушивать подходящими молекулярными ситами. По
статическому методу растворитель оставляют на ночь с молекулярным ситом.
Предпочтительнее динамический метод, заключающийся в пропускании раство-
растворителя через колонку, наполненную молекулярными ситами. Следует предус-
предусмотреть возможность нагрева и вакуумирования колонки. Тогда заполняющие
ее молекулярные сита можно будет регенерировать путем нагревания в вакууме
до указываемой изготовителем температуры. По охлаждении колонки через нее
пропускают сухой воздух (предварительно прошедший через трубку с перхлора-
перхлоратом магния — ангидроном).
Металлический натрий в качестве осушителя применяют в виде проволоки,
получаемой с помощью пресса для натрия и вводимой непосредственно в жид-
жидкость. Кусочки натрия перед загрузкой в пресс освобождают от корки (защит-
(защитные очки!). После работы пресс тщательно очищают сначала спиртом, а затем
водой.
Эффективный метод осушки углеводородов и эфирных растворов — кетиль-
ная сушка, при которой используется металлический натрий в присутствии
небольшого количества бензофенона в аппарате с циркулирующим растворите-
растворителем (рис. А.ЗОа). Выясните, какие реакции при этом протекают (разд. Г,7.3.3)!
38
А Введение в лабораторную практику
К вакуумному
насосу
Рис. А.ЗОа. Аппарат для кетильной сушки. Рис. А.306. Сушильный пистолет.
Применение азеотропной перегонки для обезвоживания жидкостей описано
в разд. А,2.3.5.
Для сушки растворов неизвестных веществ следует использовать химически
индифферентные осушители, например сульфат магния или натрия. В осушае-
осушаемый раствор всыпают тонко измельченный осушитель и оставляют стоять, вре-
время от времени энергично встряхивая. Обезвоживание жидкостей, содержащих
много воды, производят в несколько стадий (почему?), добавляя свежие порции
осушителя к декантированной с уже «отработанного» осушителя жидкости. Эту
операцию повторяют, до тех пор пока не исчезнут признаки изменения осуши-
осушителя (хлорид кальция остается гранулированным и не липнет ко дну сосуда при
взбалтывании осушаемой жидкости, сульфат меди — бесцветным, пентаоксид
фосфора не слипается в крупные комки).
1.10.3. СУШКА ТВЕРДЫХ ВЕЩЕСТВ
Из твердых веществ перед их идентификацией (т. е. определением физико-хи-
физико-химических констант и перед проведением химического анализа) должны быть
удалены следы воды и органических растворителей.
Из негигроскопичных твердых веществ легколетучие примеси можно уда-
удалять, распределив вещество тонким слоем на листе фильтровальной бумаги или
на обожженных и неглазурованных глиняных плитках. Вещества, не разлагаю-
разлагающиеся при нагревании, можно сушить в сушильном шкафу. Гораздо лучше для
высушивания веществ использовать эксикаторы, а при повышенных температу-
1. Оборудование и приемы 39
pax — так называемые «пистолеты», в которых вещество нагревается парами
кипящей жидкости (рис. А. 306). Для ускорения высушивания в эксикаторах и
сушильных пистолетах обычно поддерживают вакуум («откачивают»).
Иногда при работе с вакуумными эксикаторами они взрываются, поэтому перед
включением насоса их следует обернуть полотенцем или другой тканью. Между
водоструйным насосом и эксикатором с серной кислотой обязательно должна
быть установлена предохранительная склянка Вульфа (разд. А, 1.9.3).
Трубка для впуска воздуха в эксикатор (а также для «откачки», т. е. вакууми-
рования) оканчивается внутри эксикатора капилляром, открытый конец кото-
которого должен быть обращен вверх или же отгорожен кусочком картона, иначе
струя воздуха может разбросать осушитель или высушиваемое вещество.
В качестве осушителей применяют фосфорный ангидрид или серную кисло-
кислоту, которые способны поглощать кроме влаги пары спиртов и кетонов (весьма
распространенные растворители). Следы углеводородов (гексан, бензол, лигро-
лигроин) поглощаются кусочками парафинированной бумаги, помещенной в писто-
пистолет. Наряду с этими веществами хорошо адсорбирует остаточные количества
паров растворителей силикагель, который поэтому удобен для наполнения
эксикаторов.
Для того чтобы избежать разбрызгивания серной кислоты при ее примене-
применении в качестве осушителя, в нижнюю часть эксикатора помещают стеклянные
кольца, кольца Рашига и т. д. Для поглощения газов кислого характера часто
помещают в эксикаторы также чашечки с гидроксидом калия. Серную кислоту
нельзя применять для высушивания в вакууме (среднем и высоком) или при
повышенных температурах.
1.10.4. НАИБОЛЕЕ РАСПРОСТРАНЕННЫЕ ОСУШИТЕЛИ
В табл. А.31 приведены наиболее часто употребляемые осушители и области их
применения. Следует обратить внимание на то, что газы и жидкости, как описа-
описано в разд. А, 1.10.1 и А, 1.10.2, обычно непосредственно контактируют с осушите-
осушителями. Даже при правильном выборе последних можно иногда ожидать бурной
реакции с примесями (см. часть Е, сушка тетрагидрофурана). Поэтому следует
взять за правило в сомнительных случаях с помощью проб с малыми количест-
количествами вещества убеждаться в пригодности выбранного способа сушки.
1.11. РАБОТЫ В МИКРОМАСШТАБЕ
Под микросинтезами подразумевают работы с веществами в количестве от 15 до
150 мг. В данном учебнике это понятие применяется также для количеств от 150
до 500 мг.
Большинство прописей, приведенных в «Органикуме», можно использовать
и для микросинтезов, только, как правило, время реакций обычно будет при этом
существенно короче. Количество растворителей, которые при этом покажутся
слишком малыми, можно без опасности увеличить. Но разбавление не следует
повышать слишком сильно, иначе скорость реакции из-за этого существенно
Таблица А.31. Наиболее распространенные осушители и их применение
Осушитель
РАо
СаН2
H2SO4
Натронная известь,
СаО и ВаО
NaOH, КОН
К2СО3
Натрий
Осушаемые вещества
Газы нейтрального и кислотного характера, аце-
ацетилен, сероуглерод, углеводороды, галогенопро-
изводные углеводородов, растворы кислот (экси-
(эксикатор, «пистолет»)
Благородные газы, углеводороды, кетоны, тет-
рахлоруглерод, диметилсульфоксид, ацетонит-
рил, сложные и простые эфиры
Газы нейтрального и кислотного характера (экси-
(эксикатор, промывные склянки)
Газы нейтрального и основного характера, ами-
амины, спирты, простые эфиры
Аммиак, амины, простые эфиры, углеводороды
(эксикатор)
Ацетон, амины
Простые эфиры, углеводороды, третичные амины
Вещества, для которых
применение данного
вещества недопустимо
Основания, спирты, прос-
простые эфиры, НС1, FH
Вещества кислотного ха-
характера, спирты, аммиак,
нитросоединения
Ненасыщенные соедине-
соединения, спирты, кетоны, осно-
основания, H2S, HI
Альдегиды, кетоны, веще-
вещества кислотного характера
Альдегиды, кетоны, веще-
вещества кислотного характера
Вещества кислотного ха-
характера
Хлорпроизводные углево-
углеводородов (осторожно! опас-
опасность взрыва), спирты и
другие вещества, реагирую-
реагирующие с натрием
Примечания
Расплывается; при высушивании га-
газов осушитель необходимо смеши-
смешивать с наполнителями (ср. разд.
АД.10.1)
Осушаемые газы загрязняются водо-
водородом. При сушке растворителей
необходимо обеспечить возмож-
возможность выхода газа
Не применяют для высушивания ве-
веществ в вакууме при повышенных
температурах
Особенно подходят для сушки газов
Расплывается
Расплывается
При разложении остатков использо-
использованного в качестве осушителя ме-
металлического натрия соблюдайте ос-
осторожность! Разлагать только спир-
спиртом (с водой взрывается!)
СаС12
Mg(C104J
Na2SO4, MgSO4
Силикагель
Молекулярные сита
(алюмосиликаты
натрия и кальция)
Парафиновые углеводороды, олефины, ацетон,
простые эфиры, нейтральные газы, НС1 (эксика-
(эксикатор)
Газы, в том числе аммиак (эксикатор)
Сложные эфиры, растворы веществ, чувстви-
чувствительных к различным воздействиям
(Эксикатор)
Газы (до 100 °С), органические растворители
(эксикатор)
Спирты, аммиак, амины
Легко окисляющиеся орга-
органические жидкости
HF
Ненасыщенные углеводо-
углеводороды, полярные неоргани-
неорганические молекулы в газовой
фазе
Дешевый осушитель; содержит при-
примеси основного характера
Особенно удобен в качестве осуши-
осушителя при аналитических работах
Поглощает остаточные количества
растворителей
Превосходное средство для высуши-
высушивания растворителей; способ регене-
регенерации: нагревают в вакууме при
150—300 °С; значительная осушаю-
осушающая емкость
42
А Введение в лабораторную практику
Проволока из
нержавеющей стали
Ватная пробка
Рис. А.32. Стеклянная пипетка и пипетка
с фильтром по Пастеру.
понизится, что в отдельных случаях может привести к нежелательным эффек-
эффектам. Выходы, приведенные в таблицах, обычно не будут достигнуты, так как при
очень малых количествах уже небольшие остатки веществ в аппаратуре могут су-
существенно понизить выход.
Для работ с очень малыми количествами требуется специальная аппаратура
и техника.
Особенному обращению с несколькими миллилитрами жидких веществ сле-
следует научиться. Микроколичества жидкостей никогда не переливают, но пере-
переносят пипеткой. Простые стеклянные пипетки (так называемые пипетки Пасте -
ра, рис. А.32) можно легко сделать самим из стеклянной трубки. Трубку длиной
12—15 см оттягивают с одного конца в тонкий капилляр так, чтобы стенки были
одинаковыми и ровными. С другой стороны конец трубки немного оттягивают
или выдувают небольшую оливу, чтобы хорошо держался резиновый шланг, на-
надетый на нее. Полезно пипетки калибровать, примерно на содержание 0,5, 1,0,
1,5 и 2,0 мл. Для этого из маленького мерного цилиндра наливают соответству-
соответствующее количество и отмечают положение жидкости, например, клейкой лентой
или фольгой.
Из пипетки Пастера легко можно приготовить пипетку с фильтром (рис. А.32).
Для этого в более широкое отверстие пипетки помещают маленький кусочек ва-
ваты и продвигают его стеклянной палочкой до сужения. Затем кусочком стальной
проволоки проталкивают эту вату до конца капилляра. При этом обращают вни-
внимание на то, чтобы кусочек ваты заполнил только 2—3 мм нижнего конца капил-
капилляра, и не был вдавлен слишком плотно1. Если же его нужно извлечь, его
вымывают этанолом или чистым гексаном.
Автоматические пипетки позволяют быстро, надежно и воспроизводимо
дозировать жидкости. Их применение необходимо тогда, когда объем реагентов
нужно точно измерить.
1 Слишком плотный тампон мешает отсасыванию жидкости.
1. Оборудование и приемы
43
Рис. А.ЗЗ. а — микроколбочки C-5 мл). Колпачок с отверстием и уплотнителем либо с колечком;
б — закрепление шлифа клеммой;
в — воротничковые насадки Хикмана и Хикмана — Хинкле.
Для работы с твердыми веществами необходим микрошпатель, примерно
такой, какой употребляется в зубоврачебной практике.
В микропрепаративной практике наряду с круглодонными колбами особенно
удобными показали себя грушевидные колбочки на 5 и 10 мл. Как и большинство
других приспособлений для микрохимических работ они снабжены кроме шли-
шлифов наружной нарезкой из стекла (рис. А.ЗЗ, а). Благодаря этому их можно зак-
закрывать снаружи винтообразной крышкой. Обычно отверстие в колпачке закры-
закрывается уплотнителем, например, из силиконового каучука. Колбочки, конечно,
можно закрывать и обычным шлифом, если вместо уплотнителя положить ко-
колечко. Для сосудов со шлифом без наружной нарезки, можно пользоваться и
специальными клеммами (рис. А.ЗЗ, б). Они обычно дешевле и также действен-
действенны.
Кроме грушевидных колб с одинаковой толщиной стенок имеются и колбоч-
колбочки с плоским наружным дном и тем же самым наружным диаметром (рис. А.ЗЗ, а).
Эти колбочки можно также эвакуировать, особенно выгоден перенос тепла при
нагревании на металлических блоках с соответствующими отверстиями; эти кол-
колбочки кроме того легко ставить на ровные подставки. Очень маленькие колбочки
подобного рода (~0,1 мл содержимого) служат для хранения проб в аналитических
исследованиях.
Либиховские и воздушные холодильники, а также двугорлая насадка соответ-
соответствуют обычным, только в уменьшенных размерах. То же можно сказать и о
газоотводных и осушительных трубках (ср. рис. А.34, а).
Для обычных перегонок применяют аппаратуру, состоящую из грушевидной
колбы с магнитной мешалкой и воротничковой насадкой с боковым отводом и
насаженным обратным холодильником (воздушным или Либиха). Воротничко-
вая насадка имеется в простом изготовлении (по Хикману) или с боковой запи-
запираемой отводной трубкой (Хикмана — Хинкле; рис. А.ЗЗ, в), которая в больши-
большинстве случаев наиболее удобна. Термометр вставляют сверху. Шарик ртути тер-
термометра должен находиться сразу под краем воротничка (рис. А.34, б). Боковой
отвод насадки закрывается колпачком, через который идет канал к самому ниж-
нижнему концу воротничка. Этим путем с помощью шприца для впрыскивания
предгон может быть полностью отсосан. Также имеется возможность прервать
перегонку, если температура паров почти достигла желаемого значения. Откры-
Открывают боковой отвод, отсасывают пипеткой количественно предгон (остатки
предгона можно с помощью фена отправить со стенок воротничка насадки в
44 А Введение в лабораторную практику
воротничок). Снова запирают боковой отвод и перегоняют основное количест-
количество. Его затем отбирают так же, как было описано выше. Для перемешивания
используют магнит как он показан на рис. А.34, а. Насаживают винт для поворо-
поворота (рис. А.34, б), если ожидают, что различие в температурах кипения предгона и
основного вещества невелико.
Фракционная перегонка может быть осуществлена в зависимости от требуемо-
требуемого разделения в аппаратах с вращающейся лентой (рис. А.34, б) или на специаль-
специальных колонках, например с вращающейся лентой и магнитом, содержащемся в
тефлоновом винте, который движется благодаря магнитной мешалке. Продаж-
Продажная колонка с вращающейся лентой, как на рис. А.34, в, имеет 12 теоретических
тарелок при 1000—1500 об./мин, меньшая по размеру — около 6 теоретических
тарелок (ср. разд. А,2.3.3). На рис. А.34, в показана перегонная насадка, которая
позволяет благодаря вращению увеличивать скорость перегонки. Перегонять
следует по возможности медленно, чтобы колонка не захлебывалась. При пра-
правильной установке мотора и скорости вращения флегма без проблем должна
стекать обратно в колбу.
Температура в колонке не должна быть существенно выше 120 °С. При бо-
более высоко кипящих жидкостях с помощью маностата устанавливают соответ-
соответствующий вакуум, который должен быть включен только тогда, когда движется
мешалка. При перегонке малых количеств важно, чтобы для измерения темпе-
температуры внутри использовать чувствительный термометр, иначе он будет пока-
показывать заниженные значения. В качестве нагревателей можно использовать ма-
маленькие масляные бани или металлические блоки, которые помещаются на
магнитной мешалке, снабженной нагревателем.
Для жидкостно-жидкостной экстракции и для отделения жидкостей использу-
используют не делительные воротки, но взбалтывают смесь в грушевидной колбе, закры-
закрытой шлифом. Фазы разделяются, пипеткой полностью отсасывают нижнюю фазу
и затем переносят во вторую грушевидную колбочку. Часто имеет смысл приме-
применять при этом пипетку с фильтром (см. рис. А.32), особенно если отбирают фазу с
веществом (например, при экстракции водной фазы растворителем, который тя-
тяжелее воды). Взбалтывание и отсасывание повторяют многократно. Длительные
жидкостно-жидкостные экстракции осуществляют в обычных экстракторах с со-
соответственно уменьшенными размерами.
Перекристаллизацию твердых веществ в микромасштабе осуществляют так
же, как в больших количествах. Сперва вещество растворяют в подходящем раст-
растворителе, фильтруют путем засасывания в пипетку с фильтром, выдавливают
раствор через ватный тампон в грушевидную колбу, следя за тем, чтобы в горячем
растворе не наступала кристаллизация, и оставляют медленно охлаждаться, на-
например, помещают колбочку в водяную или масляную баню, заранее нагретую
до температуры кипения растворителя. Из-за своей большой теплоемкости баня
охлаждается очень медленно, что нередко способствует образованию хороших
кристаллов. Создать условия для увеличения времени кристаллизации возмож-
возможно, если водяную или масляную баню заменить на сосуд Дьюара с горячей жид-
жидкостью. При значительном количестве кристаллов их собирают на воронке Хир-
ша (рис. А.38), промывают небольшим количеством растворителя (добавляют по
каплям; часто достаточно одной капли) и, наконец, сушат в пистолете. Пере-
Перекристаллизацию проводят до постоянной температуры плавления вещества.
1. Оборудование и приемы
45
Масляная
баня
Рис. А.34.
а — аппаратура для сбора газов; б — перегонная аппаратура с винтовым поворотом и
с воротничковой насадкой; в — перегонная аппаратура с вращающейся лентой и вакуумной рубашкой.
Очень маленькие количества твердых веществ освобождают от растворителя в
трубочке Крэга, снабженной тефлоновой насадкой (рис. А.35, а; объем маточного
раствора с веществом от 2 до 3 мл). Ее заполняют суспензией вещества в используе-
используемом растворителе до максимального уровня так, чтобы эта взвесь не доходила до на-
начала шлифа; закрывают трубочку Крэга тефлоновой насадкой (пробкой), на которую
одевают сверху центрифужную пробирку (конец тефлоновой насадки, т. е. держатель
пробки, должен почти касаться дна центрифужной пробирки, но не упираться в не-
него!). Перед тем как одеть центрифужную пробку, в дырку в тефлоновой насадке (см.
рис.) пропускают проволоку, концы которой потом надо вынуть из центрифужной
пробирки и повесить вдоль ее внешних стенок. Этот собранный приборчик помеща-
помещают в центрифугу и центрифугируют при малых оборотах, чтобы конец тефлоновой
насадки не согнулся; предпочтительно использовать тефлоную насадку с не слишком
тонким концом (держателем). Трубочка Крэга на входе имеет не очень совершенный
шлиф, поэтому при центрифугировании через соединение трубочки Крэга с тефло-
тефлоновой насадкой может проходить только растворитель. По окончании центрифугиро-
центрифугирования трубочку Крэга, закрытую тефлоновой насадкой, вытягивают из центрифуж-
центрифужной пробирки за концы проволоки. Тефлоновую пробку (насадку) открывают и осво-
освобожденное от растворителя твердое вещество собирают микрошпателем.
При некоторых превращениях образуются газообразные продукты, которые
можно уловить в приборе, изображенном на рис. А.34, а. Отводная капиллярная
трубка для газа гарантирует малый мертвый объем. Газ можно собирать в цили-
цилиндре с запорным колпачком и с помощью шприца для инъекций использовать
для анализа. Если в реакциях образуются жидкости в количестве меньшем
46
А Введение в лабораторную практику
a U
Тефлоновая насадка
Дырка для протягивания
проволоки
б
Рис. А.35. а — трубочка Крэга для перекристаллизации;
б — ГХ-трубочка с колбочкой в центрифужной пробирке.
100 мкл, их перегонять уже нельзя. Для очистки тогда используют препаратив-
препаративную газовую хроматофафию (ГХ). Очищенный продукт можно центрифугиро-
центрифугировать из ГХ-трубочки в соответствующую колбочку (рис. А.35, б). Трубочку и
колбочку фиксируют в центрифужной пробирке ватой.
2. СПОСОБЫ РАЗДЕЛЕНИЯ ВЕЩЕСТВ
2.1. ФИЛЬТРОВАНИЕ И ЦЕНТРИФУГИРОВАНИЕ
Для отделения твердых частиц от жидкости в простейшем случае можно слить
{декантировать) жидкость с осадка. Однако при этом невозможно достигнуть
полного отделения жидкости, и при необходимости для получения чистого твер-
твердого вещества следует прибегнуть к фильтрованию или центрифугированию.
Обычно для фильтрования используют воронку, в которую вложен смоченный
бумажный фильтр (складчатый фильтр). При отделении крупнодисперсных осад-
осадков затруднений не возникает, однако высокодисперсные частицы часто не задер-
задерживаются на фильтре. Если первые порции фильтрата
оказываются мутными, их надо повторно профильтровать
через тот же фильтр. В отдельных случаях перед фильтро-
фильтрованием к разделяемой смеси добавляют так называемые
вспомогательные средства (кусочки фильтровальной бума-
бумаги, асбест, кизельгур, активированный уголь); при этом
также облегчается отделение осадков, которые забивают
поры фильтра. Само собой разумеется, что этот способ
применим только тогда, когда основную ценность предс-
представляет фильтрат, а осадок отбрасывается.
Когда же целевым продуктом является кристалличес-
кристаллический осадок, обычное фильтрование мало пригодно.
В этом случае используют фильтрование с отсасыванием
через бумажные фильтры. При больших количествах ве-
веществ осадок отфильтровывают на воронке Бюхнера в
Рис. А.36. Колба комплекте с колбой Бунзена (рис. А.36). Вакуумирование
Бунзена с воронкой отсасывающей склянки обязательно производится через
Бюхнера. предохранительную склянку Вульфа.
2. Способы разделения веществ
47
Рис. А.37. Фильтр со
стеклянной пористой
пластиной (фриттой).
Размер воронки, применяемой для фильтрования с
отсасыванием, выбирается в соответствии с количест-
количеством осадка: кристаллы должны полностью покрывать
поверхность фильтра, однако не слишком толстым сло-
слоем, так как в противном случае это затрудняет полное
удаление жидкости при отсасывании и промывании.
Хорошо подогнанный круглый бумажный фильтр
предварительно смачивают на воронке растворите-
растворителем, который затем отсасывают; после этого через во-
воронку пропускают разделяемую смесь. При отсасыва-
отсасывании необходимо поддерживать такое пониженное
давление, чтобы обеспечить достаточную скорость
фильтрования. Осадок на фильтре отжимают при по-
помощи плоской широкой стеклянной пробки от склянки, до тех пор пока не пе-
перестанет капать маточный раствор. Необходимо следить, чтобы поверхность
осадка на фильтре не растрескалась, так как это может быть причиной неравно-
неравномерного и неполного удаления маточного раствора, а также может привести к
загрязнению осадка примесями из маточного раствора в результате испарения
растворителя. Для удаления остатков маточного раствора влажные кристаллы
промывают на фильтре небольшими порциями растворителя — того же самого
или другого подходящего, в котором кристаллы малорастворимы. Иногда про-
промывную жидкость следует охладить. Это надо сделать еще перед началом фильт-
фильтрования. При промывании к осадку на фильтре сначала добавляют очередную
порцию растворителя, а затем уже подключают вакуум для отсасывания.
По окончании промывания осадок отжимают и отсасывают жидкость как
можно полнее; затем осадок высушивают. Предвари-
Предварительно высококипящий растворитель часто вытесня-
вытесняют из осадка более низкокипящим (для экономии
времени высушивания); с этой целью выбирают та-
такой растворитель, в котором осадок тоже малораство-
малорастворим (высококипящие углеводороды вытесняют лиг-
лигроином, высшие спирты — этанолом, уксусную кис-
кислоту — эфиром).
В присутствии сильных щелочей и кислот, ангид-
ангидридов, окислителей и других веществ, разрушающих
обычную фильтровальную бумагу, осадки фильтруют
через пористые стеклянные фильтры с различным раз-
размером пор (№ 2 и 3) (рис. А.37). Эти фильтры можно
употреблять вместо бумажных фильтров и во всех дру-
других случаях1.
При работе с малыми количествами веществ для
фильтрования используют воронки Хирша в комплек-
комплекте с отсасывающими пробирками (рис. А.38). Для
Рис. А.38. Прибор для
фильтрования малых
количеств веществ
(воронка Хирша).
1 При приготовлении образцов для аналитических целей применение стеклянных фильт-
фильтров обязательно, так как бумажные могут служить источником загрязнения целлюлозными
волокнами. — Прим. перев.
48
А Введение в лабораторную практику
Рис. А.39. Фильтрование
на «гвоздике».
отделения очень малых количеств удобны воронки со
стеклянным «гвоздиком» (рис. А.39), который изго-
изготовляют из тонкой стеклянной палочки (конец па-
палочки нагревают в пламени горелки до размягчения и
расплющивают). Бумажный фильтр должен плотно
прилегать к гвоздику и не иметь загнутых кверху кра-
краев. Для уменьшения потерь фильтрата в пробирку для
отсасывания помещают пробирку меньшего диамет-
диаметра и собирают в нее малые количества фильтрата.
В случае низкоплавких или хорошо растворимых
при комнатной температуре твердых кристаллов ис-
используют фильтрование с отсасыванием при низких
температурах. Для этого при малых количествах
осадка предварительно охлаждают в холодильном
шкафу воронку и раствор. В других случаях исполь-
используют воронку Бюхнера и склянку с отрезанным дном
(рис. А.40). Острые края склянки должны быть зак-
закруглены или оплавлены. Для охлаждения применяют
лед или охлаждающие смеси (разд. А, 1.7.3).
Для фильтрования горячих растворов, что необ-
необходимо почти при каждой перекристаллизации, ис-
используют воронки с водяным обогревом; обычно вода в
них нагревается перед фильтрованием открытым
пламенем (затем его следует обязательно погасить)
(рис. А.41, а); применяются также воронки, оберну-
обернутые шлангом, через который подается горячий пар
для обогрева (рис. А.41, б). Воронка Бюхнера для
горячего фильтрования, обогреваемая паром, изоб-
изображена на рис. А.41, в. В продаже бывают также во-
воронки для горячего фильтрования (с пористым стек-
стеклянным фильтром), снабженные электрическим обогревателем. «Хвостик» та-
таких воронок должен быть как можно более коротким и широким, так как иначе
легкокристаллизующиеся вещества могут его быстро закупорить.
Часто, однако, отсасывание горячих растворов оказывается невозможным,
так как при пониженном давлении (как это имеет место при отсасывании) испа-
Рис. А.40. Фильтрование
при охлаждении.
О \ Г
Рис. А.41. Воронки для горячего фильтрования,
а — с водяным обогревом; б — обогреваемая паром; в — обогреваемая паром воронка Бюхнера.
2. Способы разделения веществ
49
Фильтровальная
бумага
Рис. А.42. Удаление растворителя
после центрифугирования.
Рис. А.43. Испарение растворителя
из центрифужной пробирки после
центрифугирования.
рение растворителя увеличивается, что приводит к закупорке пор фильтра и
отверстий в воронках Бюхнера выпадающим осадком. Поэтому горячие раство-
растворы отсасывают лишь при небольших разрежениях.
Вместо фильтрования в лабораторной практике нашло применение центри-
центрифугирование; особенно это относится к случаям, если необходимо без потерь от-
отделить малые количества осадка или если последний забивает поры фильтра.
Обычно для препаративной работы используются седиментационные центри-
центрифуги со скоростью вращения 2000—3000 об./мин. Наиболее распространенные
модели центрифуг имеют четыре гнезда для сосудов емкостью 150 мл каждый.
Суспензию помещают в центрифужные (а не обычные лабораторные!) стакан-
стаканчики и выравнивают их массу, переливая содержимое из одного стаканчика в
другой. Если после центрифугирования осадок достаточно прочно удерживает-
удерживается на дне пробирки, то жидкость сливают; затем взмучивают осадок с неболь-
небольшим количеством растворителя и повторно центрифугируют. Большую часть
оставшегося растворителя удаляют с помощью кусочка фильтровальной бумаги
(рис. А.42). Остаток растворителя откачивают, присоединив центрифужную
пробирку (рис. А.43) к вакуумному насосу. Эту операцию проводят медленно и
осторожно, отсасывая пары растворителя и подогревая, если нужно, пробирку
на бане.
2.2. ПЕРЕКРИСТАЛЛИЗАЦИЯ
Важнейшим методом очистки твердых веществ служит перекристаллизация.
Для этого готовят насыщенный раствор продукта, который надо очистить, в
подходящем растворителе (при нагревании); горячий раствор отфильтровывают
от нерастворимых примесей и фильтрат охлаждают. Выпадающие при этом
кристаллы, как правило, более чистые, чем исходный продукт.
2.2.1. ВЫБОР РАСТВОРИТЕЛЯ
Вещество должно плохо растворяться в растворителе на холоду и хорошо раст-
растворяться при нагревании. При этом загрязняющие примеси должны по возмож-
возможности быть более растворимыми, чем основное вещество. Иногда применяют и
50
А Введение в лабораторную практику
такие растворители, в которых загрязняющие примеси растворимы в очень
небольшой степени и поэтому либо выпадают в осадок в первую очередь, либо
вообще не переходят в растворимое состояние. Однако в этих случаях достаточ-
достаточно чистое вещество удается получить только после многократной перекристал-
перекристаллизации. При перекристаллизации неизвестного вещества, когда и раствори-
растворитель, и его количество, необходимое для перекристаллизации, неизвестны,
следует провести предварительные опыты с малыми количествами вещества в
пробирках. Первоначально при выборе растворителя руководствуются старым
правилом, установленным прежде всего на соединениях более простого строе-
строения: «подобное растворяется в подобном», т. е. соединение хорошо растворимо
в растворителях, химически и структурно близких ему. Для подбора растворите-
растворителя можно воспользоваться следующими качественными данными:
Классы соединений
Углеводороды
Галогеноуглеводороды
Простые эфиры
Амины
Сложные эфиры
Нитросоединения
Нитрилы
Кетоны
Альдегиды
Фенолы
Амины
Спирты
Карбоновые кислоты
Сульфоновые кислоты
Соли
Гидрофобные свойства
Гидрофильные свойства
Хорошо растворимы
в следующих растворителях
Углеводороды, эфир,
галогеноуглеводороды
Сложные эфиры
Спирты, диоксан,
уксусная кислота
Спирты, вода
Вода
Само собой разумеется, что растворитель не должен химически взаимодей-
взаимодействовать с растворенным веществом.
В отдельных случаях для перекристаллизации можно использовать смеси
растворителей (например, вода — спирт, вода — диоксан, хлороформ — петро-
лейный эфир). Их состав в каждом отдельном случае надо предварительно под-
подбирать.
2.2.2. ПРОВЕДЕНИЕ ПЕРЕКРИСТАЛЛИЗАЦИИ
Вещество с растворителем нагревают, соблюдая все меры предосторожности
(разд. А, 1.7.2); растворитель берется в меньшем количестве, чем это необходимо
для полного растворения вещества. Обычно кривая растворимости вблизи точ-
точки кипения растворителя резко поднимается вверх, поэтому при перекристал-
перекристаллизации всегда следует доводить раствор до кипения. Затем через обратный
холодильник осторожно добавляют такое количество растворителя, чтобы при
2. Способы разделения веществ 51
кипячении все вещество полностью растворилось. Если для перекристаллиза-
перекристаллизации применяются огнеопасные жидкости, следует погасить все горелки с откры-
открытым пламенем! Нельзя забывать, что кипятильники уже не выполняют своей ро-
роли, если раствор охлаждается ниже температуры кипения (например, при добав-
добавлении новой порции растворителя) (разд. А, 1.7.2). Если предварительными
опытами установлено, что нерастворимые примеси остаются в осадке, не надо
добавлять слишком много растворителя, добиваясь прозрачности раствора.
Необходимо выработать привычку взвешивать вещество, подвергаемое
очистке (перекристаллизации), измерять объем растворителя, чтобы количе-
количественно оценить процесс перекристаллизации и иметь возможность его вос-
воспроизвести.
При применении смесей растворителей сначала растворяют вещество в не-
небольшом количестве одного растворителя, который является лучшим из компо-
компонентов смешанного растворителя, затем к раствору медленно при нагревании
добавляют другой растворитель, хуже растворяющий данное вещество, до тех
пор пока осадок, появляющийся в месте падения капли второго растворителя,
еще будет растворяться. Если суммарный объем раствора все же слишком мал,
надо еще раз прилить небольшое количество «хорошего» растворителя, а затем
повторить добавление «плохого». Однако надо следить (в особенности начина-
начинающему химику) за тем, чтобы количество растворителя было не слишком вели-
велико. Иногда удобен обратный порядок растворения (постепенное добавление хо-
хорошего растворителя к суспензии вещества в плохом растворителе).
Если необходимо (разд. А,2.6.1), после растворения вещества раствор обес-
обесцвечивают, добавляя измельченный активированный или животный уголь
(в количестве 1/20—1/50 массы растворенного вещества), или добиваются проз-
прозрачности раствора путем внесения туда мелко нарезанных кусочков фильтро-
фильтровальной бумаги, кизельгура и т. д.
Перед добавлением осветляющих адсорбентов раствор следует немного охла-
охладить, так как эти вещества могут интенсифицировать процесс кипения и привес-
привести к энергичному, взрывообразному вскипанию. Из активированного угля выде-
выделяется много воздуха, который вызывает вспенивание.
Полученный раствор еще раз быстро доводят до кипения и фильтруют горя-
горячим (разд. А,2.1). Сосуд с фильтратом закрывают и оставляют для охлаждения.
Для полноты осаждения кристаллов из фильтрата сосуд либо помещают в холо-
холодильный шкаф, либо ставят на лед или в охлаждающую смесь.
Органические вещества очень склонны к образованию пересыщенных раство-
растворов. При внесении в такие растворы затравки — кристалла того же самого или
изоморфного ему соединения — пересыщение обычно ликвидируется. Трение
стеклянной палочкой о стенку сосуда также вызывает образование центров
кристаллизации, на которых и происходит кристаллизация.
Скорость кристаллизации довольно часто бывает мала, и поэтому кристалли-
кристаллизация из охлажденного раствора может заканчиваться только через несколько
часов. Иногда кристаллические осадки образуются через несколько недель или
даже месяцев. Поэтому никогда нельзя преждевременно выбрасывать маточные
растворы.
52
А Введение в лабораторную практику
2.2.3. КРИСТАЛЛИЗАЦИЯ ИЗ РАСПЛАВА
Органические вещества образуют не только пересыщенные растворы, они так-
также легко дают переохлажденные расплавы. Так, низкоплавкие вещества при
температурах ниже их точек плавления довольно часто выделяются из растворов
в виде густых сиропов или масел. В таких случаях добавляют небольшие количе-
количества растворителей и полученный раствор очень медленно охлаждают (напри-
(например, оставляют в остывшей водяной бане). При растворении такого соединения
раствор ни в коем случае нельзя нагревать выше температуры плавления; повы-
повышение температуры необходимо прекратить по крайней мере за -10 "С до точки
плавления. Кристаллизацию можно вызвать трением стеклянной палочкой о
стенки сосуда; можно также несколько капель раствора растирать на шерохова-
шероховатой стеклянной пластинке или же добавить легколетучий растворитель на часо-
часовом стекле.
При отгонке растворителя твердые органические вещества при температурах
ниже их точек плавления часто тоже выделяются в виде маслянистых жидкос-
жидкостей. Кристаллизация таких веществ — нередко довольно трудная задача. Про-
Процессы зародышеобразования и роста кристаллов зависят от температуры в раз-
различной степени. Согласно правилу Таммана, скорость образования зародышей
максимальна при температуре на -100 "С ниже температуры плавления, а ско-
скорость кристаллизации — на 20—50 °С (рис. А.44):
\
2
\
i
1 \
/1 )
1/
S \3
ч. \
1 1
Si
-60 -40 -20 0 20 40 60 80 100 120
Температура, °С >¦
Рис. А.44. Температурная зависимость скорости образования зародышей G),
вязкости B) и скорости кристаллизации C).
Для того чтобы кристаллизация происходила при оптимальной температуре,
вещество для образования зародышей выдерживают несколько часов при темпе-
температуре на -100 "С ниже предполагаемой температуры плавления, а затем повы-
повышают температуру на -50 °С.
Часто образование зародышей и кристаллизацию задерживают растворен-
растворенные примеси. К замедлению кристаллизации может, в частности, привести
смазка для шлифов, увлеченная с веществом или раствором, поэтому смазку на
шлифы следует наносить очень тонким слоем, а в некоторых случаях шлифы
лучше вообще не смазывать.
2. Способы разделения веществ 53
Если перекристаллизация не дает желательных результатов, вещество следу-
следует подвергнуть очистке, используя другие методы (фракционная перегонка, хро-
хроматография, распределение между двумя фазами). В присутствии известных
примесей некристаллизующееся масло целесообразно несколько раз промыть
специальными реактивами. Так, кислоты удаляются путем промывания раство-
раствором соды, амины — кислотами, альдегиды — раствором гидросульфита натрия.
2.3. ПЕРЕГОНКА И РЕКТИФИКАЦИЯ
Перегонка служит важнейшим методом разделения и очистки жидкостей. В прос-
простейшем случае перегонка заключается в нагревании жидкости до кипения с пос-
последующей конденсацией паров в виде дистиллята в холодильнике. Так как при
этом происходит перемещение только одной фазы, а именно пара, то говорят о
прямоточной перегонке. Если же часть сконденсированного пара (так называемая
флегма) стекает навстречу восходящему пару и постоянно возвращается в колбу,
то такой процесс называют противоточной перегонкой. Противоточная перегонка,
или ректификация, осуществляется в ректификационных колонках.
2.3.1. ЗАВИСИМОСТЬ ТЕМПЕРАТУРЫ КИПЕНИЯ ОТ ДАВЛЕНИЯ
Давление паров жидкости сильно возрастает с температурой. Когда оно стано-
становится равным внешнему, жидкость кипит. Зависимость давления паров от тем-
температуры выражается уравнением Клаузиуса — Клапейрона.
dlnp AyH
= [А.45]
йТ ЯП
гдер — давление паров, AVH— молярная энтальпия испарения; Л — газовая пос-
постоянная. При интегрировании уравнения получают
AVH
1
RT
[А.46]
При этом допускается, что АуНпе зависит от температуры.
Зависимости логарифма давления паров от обратной температуры (в абсо-
абсолютной шкале, т. е. в Кельвинах) графически изображаются линиями, близкими
к прямым (рис. А.47). Наклон прямой определяется величиной молярной
энтальпии испарения. Для веществ, сходных по своему химическому строению,
температуры кипения которых довольно близки, различие в наклоне прямых
невелико.
Для приблизительной оценки температуры кипения с помощью таких зави-
зависимостей при определенном внешнем давлении достаточно знать температуру
кипения при каком-либо другом известном давлении.
Для грубой оценки может служить следующее эмпирическое правило: при
уменьшении внешнего давления вдвое температура кипения понижается на
-15 "С. Так, вещество с температурой кипения 180 °С при нормальном давле-
давлении (-100 кПа, или 760 мм рт. ст.) будет кипеть при 50 кПа C80 мм рт. ст.) при
температуре ~ 165 "С, а при 25 кПа A90 мм рт. ст.) — около 150 "С.
54
А Введение в лабораторную практику
1
Я)
I
§
133,3A000)
106,7 (800)
101,3 G60)
80,0 F00)
53,3 D00)
40,0 C00)
26,7 B00)
Z0,0 A50)
13,3 A00)
10,7 (80)
8,0 F0)
5,3 D0)
4,0 C0I
2,7 B0)
2.0 A5)
1,3 A0)
1.1 (8)
0,8 F )
0,5 D)
0,4 C)
0,3 B)
0,1 A!
@,8)
@,61
@,4)
@,3)
@,2)
@,1)
7
20 40 60 80 100 120 140 160 180 220 260 300
I I I
7
Z
7
I
7
7
г
I
L
I
I
I
I
L
I
0 20 40 60 8 100 120 HO 160 180 220 260 300
Температура, °с >-
Рис. А.47. Зависимость температуры кипения от давления.
1 — диэтиловый эфир; 2 — ацетон; 3 — бензол; 4 — вода; 5 — хлорбензол; 6 — бромбензол;
7 — анилин; 8 — нитробензол; 9 — хинолин; 10 — додеканол; 11 — триэтиленгликоль;
12 — дибутилфталат; 13 — тетракозан; 14 — октакозан.
2. Способы разделения веществ 55
2.3.2. ПРОСТАЯ ПЕРЕГОНКА
2.3.2.1. Физические основы процесса разделения
При перегонке бинарной смеси парциальные давления рд и рв обоих компонентов в паровой
фазе (предполагается, что пары подчиняются законам идеальных газов; это допущение хоро-
хорошо выполняется для веществ сходного химического строения, прежде всего для гомологов)
определяются как
рА = Р\ хА
(закон Рауля) [А.48]
Рв = Рв хв
где /д и Рв — упругости паров чистых компонентов А и В, ха и хв — мольные доли компонен-
компонентов А и В в жидкой фазе.
Поскольку в бинарной смеси хв = 1— хА, отношение парциальных давлений в паровой
фазе определяется уравнением
Рв 1-Ха
га.49]
Парциальное давление в паровой фазе рА и рв, кроме того, связано с общим давлением па-
пара р через мольные доли обоих компонентов в паровой фазе у а и ув
Ра = РУа, Рв = РУв = р(\ ~Уа) [А. 50]
Подстановка этих выражений в уравнение [А.49] дает
У а Ра ха
1-Ха
[А.51]
Обычно принято опускать индекс при обозначении мольных долей у и х более летучего
компонента. Отношение упругостей паров чистых компонентов называют относительной
летучестью а. Тогда уравнение [А.51] принимает вид
у х
= а [А.52]
1-у 1-х
где
Ра
— = а
Рв
Уравнение [А.52] описывает зависимость между относительными концентрациями легко-
кипящего компонента в парах и в жидкости. Пар и жидкость различны по составу только тог-
тогда, когда a > 1. И лишь в этом случае возможно разделение компонентов при перегонке. В то
же время обогащение паров летучим компонентом тем больше, чем больше а, т. е. чем больше
различаются упругости паров чистых компонентов. При помощи уравнения [А.52] можно оце-
оценить обогащение дистиллята легколетучим компонентом при однократном испарении.
Если разделяемые соединения отличаются по своей летучести незначитель-
незначительно, то их невозможно удовлетворительно разделить при однократном испарении
и конденсации, т. е. путем простой перегонки. В таких случаях процесс испаре-
испарения и конденсации следует повторить многократно. При помощи ректификаци-
ректификационных колонок многократное повторение можно осуществить в одном рабочем
процессе (фракционированная перегонка, ректификация, см. разд. А,2.3.3).
56 А Введение в лабораторную практику
Для начинающего химика часто бывает неясно, когда необходима перегонка
на ректификационной колонке. Разделительная способность при простой пря-
прямоточной перегонке, как правило, переоценивается. Можно руководствоваться
следующим эмпирическим правилом: ректификационная перегонка должна
применяться в тех случаях, когда температуры кипения разделяемых веществ
отличаются менее, чем на 80 °С.
2.3.2.2. Проведение простой перегонки
Такую перегонку целесообразно применять для жидкостей с температурой ки-
кипения от 40 до 150 °С, так как при температуре выше 150 °С многие соединения
уже заметно разлагаются, а жидкости с температурой кипения <40 °С нельзя пе-
перегонять без значительных потерь в обычных приборах.
Если жидкость кипит выше 150 °С, ее перегоняют под уменьшенным давле-
давлением. Во многих случаях для этого вполне достаточно вакуума водоструйного
(~1,1—2,0 кПа, или 8—15 мм рт. ст.) или ротационного масляного (~10-3—ДО кПа,
или 0,01—1 мм рт. ст.) насоса (ср. разд. А,1.9).
Некоторые соединения выдерживают только непродолжительное нагрева-
нагревание и поэтому, даже если они кипят при нормальном давлении ниже 150 °С, их
надо также перегонять при небольшом разрежении (например, метилвинилке-
тон, см. разд. Г,3.1.6).
Л При вакуумной перегонке надевайте защитные очки (см. также разд. А, 1.9)!
На рис. А.53 изображен прибор для вакуумной перегонки, собранный из
обычных стеклянных деталей на стандартных шлифах. Этот прибор (без капил-
капилляра для регулировки кипения) применим и для перегонки при атмосферном
давлении.
В качестве перегонных сосудов в лаборатории обычно употребляют кругло-
донные колбы. Количество жидкости в перегонной колбе в вакууме не должно
превышать половины общего объема колбы, а при перегонке при атмосферном
давлении — 2/з ее объема. Это учитывают при выборе размера перегонной кол-
колбы. Однако слишком большие колбы не надо брать, так как в конце перегонки в
них остается большое количество перегоняемого вещества.
Для нагревания колб применяют нагревательные бани (разд. А, 1.7.1) или
специальные колбы с внутренним электрообогревом. Категорически запре-
запрещено нагревать колбы на асбестовых сетках или голым пламенем, так как
такое нагревание может привести к местному перегреву. При выборе холо-
холодильника следует учитывать температуру кипения, теплоту парообразования
перегоняемой жидкости и необходимую скорость перегонки (более подробно
см. разд. А, 1.3).
Перегонную колбу соединяют с холодильником с помощью специальных наса-
насадок. При перегонке в вакууме применяют насадку Кляйзена 1 (рис. А.53), ее мож-
можно использовать также и при перегонке при атмосферном давлении. Более прос-
простая насадка для перегонки при атмосферном давлении1 изображена на рис. А.54.
Насадка Вюрца. — Прим. перев.
2. Способы разделения веществ
57
Рис. А.53. Прибор для вакуумной перегонки.
1 — насадка Кляйзена; 2 — алонж.
Следует обращать внимание на то, чтобы шарик термометра полностью омы-
омывался парами, т. е. чтобы он находился несколько ниже отводной трубки насад-
насадки (это надо проверять при приобретении приборов!). Если применяется термо-
термометр без шлифа, то в его показания следует внести поправки (разд. А,3.1.1).
Внутренний диаметр алонжа 2 (рис. А.53) не должен быть слишком ма-
маленьким (>5—6 мм внутренний диаметр). Для смены приемников при пере-
перегонке в вакууме служит насадка (форштос) Аншютца-Тиле (рис. А.55). Перед
работой необходимо хорошо разобраться в ее устройстве и понять принцип
действия. Этот вакуумный форштос
следует применять, только если его
краны безукоризненно пришлифова-
пришлифованы. Дешевле и более приемлемо для
обеспечения хорошего вакуума
использовать так называемый «паук»,
если при перегонке могут требоваться
несколько приемников (рис. А.56).
Однако в этом случае число собирае- Рис. А.54. Простая насадка для
мых фракций (без прерывания пе- перегонки при атмосферном давлении,
регонки), естественно, ограничено.
Приспособление, изображенное на рис. А.57 служит для той же цели: двугор-
двугорлую колбу можно заменить круглодонной колбой с вакуумным форштосом.
58
А Введение в лабораторную практику
Рис. А.55. Насадка Аншютца-Тиле.
Рис. А.56. «Паук» для сбора фракций
при вакуумной перегонке.
В качестве приемников при перегонке при атмосферном давлении также
лучше всего использовать круглодонные колбы. Предварительно взвешивают
несколько пустых колб и их массу записывают чернилами по стеклу или обыч-
обычным карандашом на вытравленном для маркировки месте.
Вещества, затвердевающие при комнатной температуре, перегоняют в сабле-
саблевидных колбах (рис. А.58). Однако такие колбы имеют недостаток: в них можно
собрать только одну фракцию. Гораздо удобнее использовать воздушный холо-
холодильник с простым приспособлением, изображенным на рис. А.57; при этом
следует избегать кранов и сужений в трубках системы. Вещество, застывшее в
холодильнике, расплавляют, осторожно нагревая светящимся пламенем газовой
горелки или с помощью электролампы с рефлектором (отражателем).
Для перегонки небольших количеств веществ, как это имеет место при полу-
микросинтезах или в аналитической практике, можно использовать прибор,
изображенный на рис. А.59. Для предупреждения вскипания жидкости при пе-
перегонке малых количеств веществ, даже если процесс осуществляется под ваку-
вакуумом, достаточно в перегонную колбу бросить кипятильники, опустить в жид-
жидкость лучинку или комочек стеклянной ваты.
Перед выполнением перегонки жидкость в перегонной колбе взвешивают,
чтобы по окончании работы, определив также массу фракций и остатка, соста-
Кнасосу
Рис. А.57. Приемник для вакуумной
перегонки, пригодный также в случае
перегонки веществ, застывающих при
комнатной температуре.
Рис. А.58. Саблевидная колба.
2. Способы разделения веществ 59
Рис. А.59. Прибор для вакуумной перегонки малых количеств веществ.
вить количественный баланс перегонки. При вакуумной перегонке в приборе
сначала надо создать необходимый вакуум и только затем приступить к нагрева-
нагреванию перегонной колбы (по окончании перегонки сначала прекращают нагрева-
нагревание, а затем уже осторожно снимают вакуум).
Для предотвращения перегрева и последующего бурного вскипания в еще
холодную жидкость помещают два-три кусочка обожженного неглазурованного
фарфора (кипятильники, см. разд. А, 1.7.2). Если перегонка была приостановле-
приостановлена, перед ее возобновлением следует добавить свежие кипятильники.
При вакуумной перегонке вскипание жидкости предотвращается при помо-
помощи капилляра. Его вытягивают по возможности из тонкой и достаточно механи-
механически прочной трубки, нагревая ее на несветящемся пламени; затем капилляр
повторно оттягивают на маленьком пламени. При продувании воздуха через
изготовленный таким образом капилляр, конец которого погружен в ацетон, пу-
пузырьки воздуха должны выходить из него медленно и подниматься к поверхнос-
поверхности каждый отдельно.
Капилляр вводят в перегонную колбу через насадку Кляйзена (ср. рис. А.53)
или через горло многогорлой колбы (рис. А.73) и закрепляют там, используя
стеклянную трубку со шлифом (керном) и надетый на нее кусочек резинового
шланга или резиновой пробки. В разд. А, 1.6 описана методика работы в том слу-
случае, когда необходимо через капилляр пропускать не воздух, а инертный газ
60
А Введение в лабораторную практику
Впаянная
трубка
с дырками
Рис. А.60. Приспособление для
разрушения пены.
а Ь'Ь с
Объем дистиллята, мл —•>
Рис. А.61. Кривая перегонки.
(чаще всего азот). Если надо провести перегонку при обычном давлении в
атмосфере инертного газа, капилляр заменяют трубкой для введения газа и про-
пропускают через перегоняемую жидкость медленный поток газа.
Если для вакуумной перегонки применяют колбу с магнитной мешалкой, то
необходимость в капилляре отпадает.
При вакуумной перегонке сперва устанавливают необходимый вакуум, а затем
нагревают колбу (в конце перегонки сперва удаляют нагрев, а затем осторожно
спускают вакуум).
Некоторые жидкости сильно пенятся при перегонке. Пенообразованию в
водных растворах можно воспрепятствовать, прибавив к перегоняемой жид-
жидкости несколько капель октилового спирта или силиконового масла (антаф-
рон). При устойчивом пенообразовании во второе горло насадки Кляйзена
вместо термометра вводят капилляр. Встречный поток воздуха разрушает
пузырьки пены. Между колбой и насадкой можно поставить специальный
пеногаситель (рис. А.60).
Скорость перегонки регулируется обычно таким образом, чтобы в 1 с стекало
не более одной-двух капель дистиллята. При простой перегонке полезно результа-
результаты представить в виде кривой кипения, т. е. в виде зависимости объема дистилля-
дистиллята (в миллилитрах) от температуры кипения (рис. А.61). С этой целью применяют
градуированный приемник (мерный цилиндр, форштос Аншютца — Тиле); для
построения графика необходимо примерно 20 точек.
Если предварительно необходимо отогнать большое количество растворите-
растворителя, то кривую кипения надо снимать, начиная с точки а (на рис. А.61), т. е. с то-
того момента, когда начинается рост температуры кипения. В этот же момент ме-
меняют приемник. Когда отгонка промежуточной фракции (между точками а и Ь)
закончится, начинает перегоняться основное вещество (между точками b и с);
при этом объем промежуточной фракции тем больше, чем ближе температуры
кипения разделяемых веществ. О других факторах, определяющих объем проме-
промежуточной фракции, см. разд. А,2.3.3.
2. Способы разделения веществ 61
При перегонке чистых соединений основная фракция (интервал Ъ—с) гонит-
гонится почти при постоянной температуре. В конце перегонки этой фракции темпе-
температура обычно слегка (на 1—2 °С) увеличивается, так как пары могут немного пе-
перегреваться. Если фракция собрана в более широком температурном интервале,
следует вновь повторить перегонку, но уже используя ректификационную
колонку.
Иногда окончание перегонки одной фракции и переход к следующей можно
заметить визуально по появлению неоднородности дистиллята в приемнике.
Однако начало перегонки новой фракции часто бывает трудно определить
однозначно. В этом случае надежнее увеличить число фракций (например, на
объемы a—b'w b'—b). Эти фракции отмечают на кривой, а после измерения конс-
констант (показателя преломления, плотности, температуры плавления), если дан-
данные для них совпадают, объединяют.
По окончании перегонки определяют массу всех фракций и остатка в пере-
перегонной колбе.
2.3.2.3. Отгонка растворителей
В результате многих препаративных работ получается раствор соединения в лег-
кокипящем растворителе, из которого вещество выделяется после отгонки раст-
растворителя. Эту операцию целесообразно всегда осуществлять при помощи водя-
водяной или паровой бани, поскольку большинство органических растворителей
очень огнеопасно (разд. А, 1.7.2). Преимущество такого нагрева состоит также в
том, что вещество не подвергается излишнему термическому воздействию.
В конце отгонки растворителя температура кипения раствора сильно возрастает
(закон Рауля, уравнение [А.48]), причем настолько резко, что даже легкокипя-
щие растворители (спирт, бензол, эфир) при нагревании на водяной бане не
полностью удаляются из оставшейся в перегонной колбе высококипящей фрак-
фракции. Для их удаления применяют легкий вакуум и по мере уменьшения количе-
количества растворителя в растворе вакуум постепенно увеличивают, чтобы добиться
необходимой скорости испарения. Если получаемый продукт чувствителен к
нагреванию, то даже первые порции растворителя надо отгонять под уменьшен-
уменьшенным давлением. При отгонке больших количеств легкокипящего растворителя
под уменьшенным давлением надо использовать эффективный холодильник, а
приемник дополнительно охлаждать льдом или смесью льда с поваренной
солью.
В тех случаях, когда после отгонки растворителя продукт также надо перего-
перегонять, остаток переносят в перегонную колбу меньшего размера, смывая неболь-
небольшим количеством растворителя.
Можно сразу работать с колбами меньшего размера. При этом перегоняемый
раствор добавляют в перегонную колбу из капельной воронки, находящейся в
одном из горл насадки Кляйзена, по мере испарения растворителя.
Для испарения растворителя и концентрирования раствора часто применяют
роторный вакуум-испаритель (рис. А.62). Роторный испаритель позволяет уда-
удалять растворитель быстро и в мягких условиях. Испарение происходит в вакууме
из тонкой пленки жидкости, находящейся на внутренней стенке колбы и посто-
постоянно обновляющейся благодаря вращению колбы. Для компенсации теплоты
62
А Введение в лабораторную практику
Рис. А.62. Роторный
вакуум-испаритель.
испарения колбу подогревают на водяной
бане. Для того чтобы предотвратить пено-
образование, сначала приводят во враще-
вращение колбу, затем устанавливают вакуум и
лишь после этого начинают нагревать водя-
водяную баню.
Если нужно отогнать очень большие ко-
количества растворителя, то раствор из запас-
запасного сосуда можно засасывать во вращаю-
вращающуюся колбу через холодильник, осущес-
осуществляя соединение с помощью устойчивого к
действию растворителя- шланга. В отличие
от обычной перегонки в вакууме, выпадаю-
выпадающие осадки работе роторного испарителя
не мешают.
Внимание! Низкокипящие растворители в большинстве роторных испарителей
с водяным охлаждением конденсируются не полностью. Для полной регенера-
регенерации растворителя, а также для предотвращения его попадания в канализацию
приемник надо дополнительно охлаждать льдом или охладительной смесью
(см. разд. А, 1.7.3).
Существует множество конструкций роторных испарителей. В некоторых конструкциях
после отгонки растворителей предусмотрено также проведение ректификации, экстракции
твердых веществ, возгонки, лиофильного высушивания и сушки твердых веществ.
2.3.2.4. «Короткая» перегонка. Перегонка с шарообразными трубками
Для очистки чувствительных к нагреванию и высококипящих веществ применя-
применяют так называемую «короткую» перегонку: в аппарате расстояние между поверх-
поверхностями испарителя и конденсатора очень мало и перегнанное вещество образу-
образует тонкую пленку жидкости. Обычно работают в вакууме.
Рис. А.63. Аппарат для перегонки с шарообразными трубками.
1 — источник нагрева; 2 — перегоняемое вещество; 3 — приемники для дистиллята;
4 — привод с вакуумным присоединением.
2. Способы разделения веществ 63
Модификация этого метода — перегонка с шарообразными трубками,
рис. А.63. Перегоняемое вещество и конденсат находится во вращающихся стек-
стеклянных шарах. Можно применять вакуум. Этот метод превосходно подходит для
отделения жидкостей и низкоплавких твердых веществ от полимерных приме-
примесей и смолообразных загрязнений и для отделения плохо отгоняемых раствори-
растворителей. В продаже имеются модели для различных количеств веществ, от полу-
микромасштаба до пилотных установок с регулируемыми ИК. воздушными
банями, что позволяет осуществлять равномерное перенесение тепла.
2.3.3. РЕКТИФИКАЦИЯ
Ректификация — это фракционная перегонка с использованием ректификаци-
ректификационных колонок. Ее применяют в тех случаях, когда однократная простая пере-
перегонка не приводит к разделению смеси, т. е. обычно если разность температур
кипения отдельных компонентов меньше 80 °С (ср. разд. А,2.3.2.1).
2.3.3.1. Физические основы ректификации
При испарении бинарной смеси, содержащей Х\ мольных долей более летучего компонента,
концентрация этого компонента в паровой фазе возрастает в соответствии с уравнением [А.52]
до у\ мольных долей, т. е.
У\ Х\
-=а [А.64а]
l-y. l-
При полной конденсации пара этого состава концентрация летучего компонента, естест-
естественно, сохраняется таковой и в дистилляте, в результате образуется новая жидкая фаза соста-
состава х2 = у\:
У\ конденсация Хг Х\
— > = а [А.646]
1-Х2
Если испарять полученную жидкость во второй раз, то состав образующегося при этом
пара будет отвечать содержанию этого компонента у2~.
Уг *г Х[
= a = а2 [А.64в]
1-уг 1-х2 1-xi
Для л-кратного повторения процесса испарения и конденсации получим
= а" — [А.64г]
1
\-у„
При этом достигается более эффективное разделение.
Процесс многократного испарения и конденсации (ректификация) осуществляется в рек-
ректификационных колонках, в которых пар и жидкость движутся противотоком. Проще всего
это понять на примере тарельчатой колонки, в которой на каждом отрезке (тарелке) в извест-
известной степени происходит новая перегонка (рис. А.65, б).
Теоретической тарелкой (а также теоретической ступенью разделения) называют некото-
некоторую (условную!) единицу колонки, где обогащение легколетучим компонентом соответствует
термодинамическому равновесию между паром и жидкостью (уравнение [64а]). Любая реаль-
реальная тарелка колонки не достигает эффективности одной теоретической тарелки.
64
А Введение в лабораторную практику
10
Q8
т °'6
^0,4-
OZ-i
О 0,2 0,4 0,6 0,8 1,0
к >¦
а б
Уг /
1
1
1
1
1
1
Рис. А.65. Принципиальная схема процесса ректификации.
а — графическое определение теоретического числа тарелок;
б — схема, дающая представление о тарелках.
Число теоретических тарелок, необходимых для разделения бинарной смеси, выражается в
уравнении [А.64г] показателем степени, который можно рассчитать, решив уравнение относи-
относительно и для смеси исходного и желаемого состава.
Если а = 1, уравнение [А.52] приводит к прямой у = х, которая проходит через начало ко-
координат и имеет тангенс угла наклона, равный 1 (рис. А.65, а и А.66). Для а > 1 получаются
кривые, причем их кривизна увеличивается с ростом а (равновесные кривые). На рис. А.66
представлены три такие кривые. Кроме них приведена еще кривая с перегибом (S-образная
кривая). Очевидно, что в точке пересечения последней с прямой а = 1 разделение при помо-
помощи перегонки окажется невозможным; речь идет о кривой для азеотропной смеси. Этот слу-
случай нельзя описать уравнением [А.52], поскольку оно составлено в предположении, что веще-
вещества ведут себя как идеальные. Более подробно об азеотропной перегонке см. разд. А,2.3.5.
Изменение концентрации вещества в процессе ректификации, соответствующее уравне-
уравнениям [А.64а — А.64г], можно установить и графически с помощью диаграммы х/у, т. е. равно-
равновесной кривой (рис. А.65, а).
При испарении бинарной смеси исходного состава х\ получается пар состава у\, при охлаж-
охлаждении которого образуется конденсат того же самого состава х2. Этот конденсат (Хг) при повтор-
повторном испарении дает пар состава у^, а после конденсации новый конденсат состава хз. Таким
образом, состав жидкость — пар меняется «ступенчато», каждый «шаг» ступеньки находится
между прямой с углом наклона 45° (поскольку у„ = х„+\) и равновесной кривой; в конце концов
ступеньки поднимаются к заданному (желательному) составу дистиллята. Число ступенек как
раз равно числу теоретических тарелок, необходимому для разделения смеси. Очевидно, что их
число тем меньше, чем большую кривизну имеет равновесная кривая, т. е. чем больше а.
2. Способы разделения веществ
65
Рис. А.66. Равновесные кривые.
l!
11
1000
400
200
100
40
20
10
КОМ
2 Z
/
ПС
\
мента
// ,
-/-
/
%
с
/
/
Hi
=7$
^90,0
--/
А
1
100 40 20 10 4 2 1 0,4 0.2 0,1
Разность температур ^
кипения компонентов, °С
Рис. А.67. Определение числа
теоретических тарелок по разности
температур кипения компонентов.
Поскольку каждая кривая вблизи х = 1 (полное, или 100%-ное, разделение) всегда близко
прилегает к прямой с наклоном 45°, для достижения высокой чистоты дистиллята необходимо
большое число теоретических тарелок.
Если жидкость подчиняется правилу Пикте — Трутона (относительно постоянства энтро-
энтропии испарения см. учебники физической химии), т. е. ее поведение приближается к идеально-
идеальному, то а можно рассчитать также, зная температуру кипения (в Кельвинах) чистых компонентов.
На рис. А.67 на осях отложены минимальное число теоретических тарелок, необходимое для
разделения эквимолярной бинарной смеси (при полном флегмировании, см. ниже), и разность
температур кипения разделяемых компонентов (три приведенные прямые соответствуют раз-
разной степени чистоты дистиллята). Очевидно, что при необходимости получения дистиллята
высокой чистоты требования, предъявляемые к ректификационной колонке, значительно
возрастают.
Предшествующие рассуждения относятся только к такому случаю, когда при ректифика-
ректификации дистиллят не удаляется из системы, а, напротив, весь конденсат вновь возвращается в рек-
ректификационную колонку (полное флегмирование).
Однако на практике эти равновесия нарушаются, поскольку часть конденсата уходит в ви-
виде дистиллята. В качестве флегмы противотоком к парам в колонку стекает только оставшаяся
часть конденсата. В ректификационной колонке таким образом устанавливается следующий
баланс: общее количество жидкости, перешедшей в пар, равно сумме масс флегмы R и дистил-
дистиллята D
= R+D
[А.68]
Для того чтобы найти абсолютные количества отдельных компонентов (в основу расчета
положена концентрация легкокипящего компонента), следует умножить количество жидкос-
жидкости, перешедшей в пар, количество флегмы и дистиллята на соответствующие мольные доли
Gy = Rx + DxD
[А.69]
где у — концентрация легколетучего компонента в паре на некоторой высоте в колонке; х —
концентрация этого же компонента в жидкой фазе на той же высоте в колонке; хд — его кон-
66
А Введение в лабораторную практику
центрация в дистилляте. Поделив уравнение [А.68] на [А.69], получим
Rx DxD
У =
R + D R+D
[А.70а]
После умножения числителя и знаменателя дробей на 1/D и введения флегмового числа
v = R/D получают уравнение
vx
1 + V 1 + V
[А.706]
Графически функция у изобразится прямой с тангенсом угла наклона v/( I + v), отсекающей на
оси ординат отрезок Хд/A + v).
При графическом определении числа теоретических тарелок эту прямую используют
вместо прямой с наклоном 45° на рис. А.65, а. «Строить» ступеньки для процесса разделения
при этом следует, как показано на рис. А.65, между этой «рабочей» прямой и равновесной кри-
кривой. Эти соотношения воспроизведены на рис. А.71.
Концентрация хо является заданной величиной (это желательная степень чистоты дистил-
дистиллята). Рабочая прямая исходит из точки на прямой с наклоном 45 °С с абсциссой, равной Хо;
наклон рабочей прямой определяется флегмовым числом. На рис. А.71 изображены три случая.
Прямая, отсекающая на оси ординат отрезок А2, проходит через точку на равновесной кри-
кривой с абсциссой хв. В этом случае придется построить бесконечно большое число ступенек.
Таким образом, при таком флегмовом числе для разделения необходимо бесконечно большое
число тарелок. Такое флегмовое число называют минимальным флегмовым числом.
Рабочая прямая, отсекающая отрезок на оси ординат А^, вообще не позволяет получить
обогащение исходной смеси состава хв до xD. Рабочая прямая с ординатой Ах, напротив, харак-
характеризует практически реализуемый случай (см. ступенчатую линию).
Ясно, что для разделения необходимо тем меньшее число ступеней, чем больше флегмовое
число, т. е. чем меньший отрезок отсекается на оси ординат. При бесконечно большом флегмо-
флегмовом числе наклон рабочей прямой приближается к наклону 45°, причем такая прямая одновре-
одновременно определяет минимальное число тарелок для соответствующего разделения. В пределах
обеих границ (минимальное число тарелок и минимальное флегмовое число) недостающее число
тарелок можно компенсировать повышением флегмового числа и наоборот.
Q2 0,4 0,6 0,8 1,0
Рис. А.71. Графическое определение числа теоретических тарелок при
одновременном отборе дистиллята.
2. Способы разделения веществ
67
4 8 12
Объем дистиллята, мл —*¦
Рис. А.72. Кривые перегонки бензол-толуольной смеси.
На рис. А.72 показано, как происходят описанные процессы при разделении
перегонкой смеси толуола с бензолом (разность температур кипения ~30 °С).
Кривая кипения / получена при простой перегонке без ректификационной ко-
колонки. Эффективность разделения смеси при такой перегонке можно прирав-
приравнять к эффективности разделения в колонке с одной теоретической тарелкой.
При этом ни один из компонентов нельзя выделить в чистом виде. Эффектив-
Эффективность разделения на ректификационной колонке (примерно с 12 теоретически-
теоретическими тарелками, флегмовое число '/ю) соответствует кривой 2. Влияние флегмово-
го числа на разделение становится ясным из сопоставления кривых 2 и 3. Кривая
3 получена на такой же колонке, но весь пар, конденсирующийся в головке
колонки, полностью отбирается в виде дистиллята.
2.3.3.2. Проведение ректификации
Прибор для ректификации состоит из следующих частей (рис. А.73): колбы для
испарения жидкости (куб колонки); ректификационной колонки; головки
колонки (в этой части осуществляется измерение температуры, конденсация
паров и разделение конденсата на флегму и дистиллят); приемника (при работе
под уменьшенным давлением необходимо использовать устройство для отбора
фракций под вакуумом, например форштос Аншютца — Тиле).
Помимо уже упомянутых тарельчатых колонок (рис. А.65, б) применяются
также полые трубки и их разновидности: колонки Вигре (рис. А.73); колонки, за-
заполненные специальными насадками (рис. А.74, А.75); колонки с вращающими-
вращающимися массообменными устройствами (роторные колонки) (рис. А.34, б и А.34, в).
Массо- и теплообмен между жидкой и паровой фазами, необходимый для ректи-
ректификации, тем интенсивнее (т. е. эффективность колонки тем выше), чем больше
поверхность соприкосновения обеих фаз.
Тип колонки выбирают, учитывая особенности разделяемой системы, а так-
также количество перегоняемого продукта и величину давления, при котором
должна выполняться перегонка.
Эффективность разделения зависит от относительной летучести компонен-
компонентов (а). В первом приближении трудность поставленной задачи можно оценить
по разности температур кипения разделяемых соединений (ср. рис. А.67), по
68
А Введение в лабораторную практику
Рис. А.73. Прибор для ректификации.
1 и 2 — см. текст, с. 72.
Рис. А.74. Ректификационная колонка
по Фишеру (поперечный разрез).
7 — кольцевая щель для поднимающегося пара;
2— винтообразные пазы (канавки) внутренней трубки;
3 — винтообразные пазы наружной трубки
(в противоположную сторону относительно
внутренней трубки).
Рис. А.75. Типы насадок.
а — кольца Рашига; б — брауншвейгские спиральки; в — седловидная насадка;
г— рулончики из проволочной сетки.
2. Способы разделения веществ 69
концентрации компонентов смеси, а также учитывая желательную чистоту дис-
дистиллята. При выборе ректификационной колонки необходимо ясно представ-
представлять себе ход равновесной кривой (рис. А.66).
Размер ректификационной колонки должен соответствовать количеству
перегоняемой жидкости. Совершенно ясно, что 10 мл смеси не следует перего-
перегонять в колонке диаметром 50 мм.
Однако даже на колонке диаметром 10 мм, по своей эффективности пригодной для разде-
разделения данной смеси, иногда можно выделить только небольшую часть высококипящего ком-
компонента, так как колонка «удерживает» слишком много жидкости. В этом случае говорят, что
колонка имеет слишком большой рабочий объем. Под последним понимают количество ве-
веществ (пара и жидкости) в работающей колонке, находящееся между поверхностью жидкости
в колбе и холодильником. Однако удерживаемую в колбе и колонке часть высококипящего
компонента можно перегнать, если добавить в перегонную колбу вещество, температура кипе-
кипения которого намного выше температуры кипения удерживаемого колонкой компонента и
которое не образует с ним азеотропной смеси.
Величина рабочего объема колонки сказывается и на четкости разделения
исходной смеси. Эмпирически установлено правило, по которому количество
каждого компонента, подлежащего выделению в чистом виде из исходной сме-
смеси, должно быть по меньшей мере в 10 раз больше рабочего объема колонки. Для
перегонки малых количеств веществ и для аналитических целей поэтому приме-
применяют колонки с возможно меньшим рабочим объемом (полые трубки, колонки
Вигре, колонки с вращающейся лентой; см. табл. А.76).
Падение давления в колонках для перегонки в вакууме должно быть по воз-
возможности минимальным, так как давление в перегонной колбе не может быть
ниже давления в колонке. Например, если падение давления в колонке состав-
составляет 1 кПа G,5 мм рт. ст.), а в конденсационной головке поддерживают давление
0,1 кПа/0,75 мм рт. ст.), то в перегонной колбе оно будет составлять 1,1 кПа
(8 мм рт. ст.). Соединения, чувствительные к нагреванию, могут при этом разла-
разлагаться.
В табл. А.76 дана характеристика наиболее важных видов колонок, использу-
используемых в лабораторной практике. Эффективность колонки оценивается значени-
значениями ВЭТТ (высота, эквивалентная одной теоретической тарелке; эта условная
величина соответствует высоте колонки в сантиметрах между соседними тарел-
тарелками). ВЭТТ приведенных колонок зависит от их пропускной способности1:
у большинства типов колонок ВЭТТ возрастает (т. е. эффективность колонок
падает) с повышением пропускной способности. При определенной пропуск-
пропускной способности флегма может не стекать в перегонную колбу, а удерживаться в
колонке потоком поднимающихся ей навстречу паров. Колонка «захлебывает-
«захлебывается». Естественно, что при этом ректификация невозможна.
Пропускная способность всех колонок в вакууме ниже, так как объем пара
определенного количества вещества, а тем самым и скорость движения пара
обратно пропорциональны давлению. Колонка захлебывается уже при меньших
нагрузках, чем при атмосферном давлении.
1 Нагрузкой (или пропускной способностью) называют количество жидкости, которое
испаряется в единицу времени из перегонной колбы. Оно равно суммарному количеству дис-
дистиллята и флегмы.
Таблица А.76. Характеристика некоторых типов ректификационных колонок
Тип колонки
Полая трубка
Колонка Вигре (рис. А.73)
Колонка, заполненная
стеклянными шариками
диаметром 3 мм
Колонка с седловидной
(фарфоровой) насадкой
(рис. А.75, в) размером
4х4мм
6х 6мм
Колонка, заполненная
кольцами Рашига размером
4,5 х 4,5 мм (рис. А.75, а)
Диаметр,
мм
24
6
6
24
12
12
24
30
30
24
24
24
Пропускная
способность,
мл/ч
400
115
10
510
294
54
100-800
400
400
600
500
400
ВЭТТ, см
15
15
1,7
11,5
7,7
5,4
6,0
5,3
8,2
8,2
7,6
7,0
Примечания
Рабочий объем и падение давления незначительны. Хорошо подхо-
подходит для вакуумной перегонки и перегонки полумикроколичеств. Не-
Небольшая эффективность. Чрезвычайно низкая пропускная способ-
способность, и поэтому высокая эффективность достигается только с очень
большим трудом. Эффективность понижается с увеличением пло-
площади поперечного сечения (почему?)
Подобна полым трубкам, но, поскольку поверхность больше, облада-
обладает несколько лучшей эффективностью, ббльшими рабочим объемом и
падением давления. Пригодна для вакуумной и полумикроперегонок
Высокая пропускная способность при нормальном давлении. Эффек-
Эффективность в достаточно широком интервале не зависит от пропускной
способности. Большой рабочий объем. Непригодна для вакуумной и
полумикропере гонки
Для перегонки в условиях грубого вакуума более пригодна, чем колон-
колонки с другими упомянутыми здесь насадками (меньшее сопротивление
потоку паров). Высокая пропускная способность. Большой рабочий
объем
Среди колонок с насадками имеет наименьшую эффективность.
Малопригодна для перегонки в вакууме. Большой рабочий объем
Колонка с насадкой из спи-
спиралек (рис. А.75, б) размером
2х 2мм
4х 4мм
Трубчатая колонка с коль-
кольцевой щелью (рис. А.74)
Колонка с вращающейся
лентой (рис. А.34, в)
Колонка с рулончиками из
проволочной сетки
Тарельчатая колонка
(рис. А.65, б)
24
24
5-10
15-30
18-25
500
500
700
300
200
50-200
500
500
1,95
2,86
1,7
0,8
0,6
1,9-2,7
1,2-3
2-3
Высокая эффективность, средняя пропускная способность, большое
падение давления, большой рабочий объем
Высокая разделяющая способность и пропускная. Малое падение дав-
давления. Хорошо пригодна для полумикро- и вакуумперегонок
Для перегонки в аналитических целях и полумикроперегонки. Рабо-
Рабочий объем и падение давления очень невелики. Хорошо подходит для
перегонки в вакууме
Хорошие эффективность и пропускная способность
Высокая пропускная способность, большой рабочий объем и падение
давления. Для перегонки больших количеств веществ (>1 л) при нор-
нормальном давлении
72 А Введение в лабораторную практику
При проведении ректификации в вакууме, кроме того, необходимо следить,
чтобы давление в процессе перегонки оставалось постоянным. Для этого ис-
используют маностаты (ср. разд. АД.9.1).
Оптимальная производительность колонки достигается при адиабатичес-
адиабатическом режиме работы, когда потери тепла за счет конвекции, теплопроводности,
теплового излучения сокращены до минимума. При дистилляции веществ с
температурой кипения до -80 °С часто бывает достаточно в целях теплоизоля-
теплоизоляции обмотать колонку асбестовым шнуром, стекло- или шлаковатой, надеть на
нее рубашку из пенопласта или поместить в стеклянную трубку, создав тем са-
самым воздушную рубашку (рис. А.73). Гораздо меньше потери тепла при ис-
использовании вакуумированной посеребренной рубашки или электрического
нагревателя, намотанного на колонку. В последнем случае удается полностью
компенсировать тепловые потери, но при этом нельзя допускать перегревания
колонки. Температуру рубашки поэтому надо поддерживать немного ниже
температуры внутри колонки.
Необходимое для разделения смеси флегмовое число можно найти графически
(разд. А,2.3.3.1). Для лабораторных целей оптимальное флегмовое число пример-
примерно равно числу теоретических тарелок, необходимому для разделения смеси. Ес-
Если колонка соответствует более высокому числу теоретических тарелок, чем это
требуется для разделения, то можно выбрать меньшее флегмовое число. Для ус-
установления определенного флегмового числа служит головка колонки. Без голов-
головки колонки можно обойтись только в случае очень легких задач по разделению
смесей (например, если разность температур кипения превышает 40 °С, а степень
чистоты дистиллята должна составлять не более 95%).
Наибольшее распространение имеют головки с полной конденсацией паров
(рис. А.73). Конденсат в таких головках с помощью крана просто и для больши-
большинства целей достаточно хорошо разделяется на дистиллят и флегму. Флегмовое
число определяется довольно точно как отношение числа капель, стекающих с
капельников / и 2. Регулировка флегмового числа облегчается, если отверстие
крана имеет насечку (ср. рис. А.22).
В технике вместо головки колонки часто используют так называемые дефлегматоры. Они
действуют как холодильники и конденсируют часть паров раньше, чем последние достигнут верх-
верхнего конца колонки. Пар, не сконденсировавшийся в
дефлегматоре, направляется в нисходящий холодиль-
холодильник. Поскольку в дефлегматоре конденсируется более
высококипящая часть паров, он обладает некоторым
разделяющим действием (порядка нескольких теоре-
теоретических тарелок). Установление определенного
флегмового числа при использовании дефлегматора
очень затруднено, и поэтому они почти не применя-
применяются в лабораториях. Однако для определенных целей
(например, при отгонке низкокипящего вещества из
реакционной смеси) известную пользу может
принести применение так называемой насадки Хана
(рис. АЛ), которая работает по принципу дефлегма-
дефлегматора. Сосуд / заполняют жидкостью, температура
кипения которой близка к температуре кипения отго-
отгоняемого вещества; в простейшем случае этот сосуд за-
Рис. А.77. Насадка Хана. полняют самим отгоняемым веществом.
2. Способы разделения веществ 73
2.3.4. ПЕРЕГОНКА С ВОДЯНЫМ ПАРОМ
Упругость паров над смесью двух взаиморастворимых жидкостей определяется
упругостями паров отдельных компонентов по закону Рауля [уравнение А.48].
Упругость паров над смесью, если только не образуется азеотропная смесь,
лежит между упругостями паров чистых компонентов, а температура кипения
смеси находится между их температурами кипения. Напротив, если два соедине-
соединения практически взаимно нерастворимы, то они и не оказывают никакого влия-
влияния на упругость паров смеси1:
/>а=/а
Ръ = Рв [А.78]
Р = Рк + Ръ
Суммарная упругость паров р над гетерогенной смесью определяется просто
суммой упругостей паров отдельных компонентов. Таким образом, суммарная
упругость паров смеси больше упругости паров каждого отдельного компонента
и температура кипения такой смеси всегда ниже температуры кипения наиболее
низкокипящего компонента.
Состав дистиллята в этом случае не зависит от абсолютных количеств компо-
компонентов. Оба вещества содержатся в дистилляте в количествах, пропорциональ-
пропорциональных упругостям их паров (при температуре кипения смеси):
Количество вещества А РА
=— [А.79]
Количество вещества В Рц
Однако уравнение [А.79] в большинстве случаев можно считать лишь приб-
приближенным, так как допущение, что жидкости совершенно взаимно нераствори-
нерастворимы, не совсем справедливо.
Наиболее важным примером такой двухфазной перегонки, встречающимся
на практике, является перегонка с водяным паром. Вещество, весьма малораство-
малорастворимое в воде, перегоняют из гетерогенной смеси с водой или пропускают в та-
такую смесь водяной пар. Таким образом можно успешно перегонять вещества,
температура кипения которых лежит значительно выше 100 °С.
Перегонку с водяным паром проводят в приборе, изображенном на рис. А.80.
Для перегонки через колбу пропускают интенсивный ток пара. Водные раство-
растворы целесообразно предварительно нагреть почти до кипения и нагревать
(особенно при длительной перегонке) содержимое колбы также и во время
пропускания водяного пара. Это позволит избежать чрезмерного увеличения
объема перегоняемой смеси.
При перегонке с водяным паром следует применять только эффективные
холодильники, так как теплота конденсации водяного пара весьма велика. Пере-
Перегонку продолжают обычно до тех пор, пока дистиллят не перестанет разделять-
разделяться на два слоя. Затем отсоединяют источник водяного пара (размыкают
соединение между пароотводящей трубкой парового котелка и трубкой ввода
пара в перегонную колбу) и только потом его выключают (почему?).
Значение символов ср. [А.48].
74
А Введение в лабораторную практику
Рис. А.80. Прибор для перегонки с паром.
1 — паровой котелок с вертикальной трубкой; 2 — делительная воронка; 3 — нагреватели;
4 — колба для перегонки.
Малые количества веществ можно перегонять с водяным паром в прибо-
приборе, изображенном на рис. А.81. При этом обычно не требуется дополнитель-
дополнительного источника пара, вполне достаточно нагреть смесь вещества с водой до
кипения.
Рис. А.81. Прибор для перегонки с водяным паром малых количеств веществ.
2. Способы разделения веществ
75
Таблица А.82. Наиболее часто
Азеотропная смесь
Вода — этанол
Вода — этилацетат
Вода — муравьиная кислота
Вода — диоксан
Вода — тетрахлоруглерод
Вода — бензол
Вода — толуол
Этанол — этилацетат
Этанол — бензол
Этанол — хлороформ
Этанол — тетрахлоруглерод
Этилацетат — тетрахлоруглерод
Метанол — тетрахлоруглерод
Метанол — бензол
Толуол — уксусная кислота
Этанол — бензол — вода
встречающиеся азеотропные
Г. кип.
компонентов, °С
100
100
100
100
100
100
100
78,3
78,3
78,3
78,3
78
64,7
64,7
110,6
78,3
78,3
78
100,7
101,3
77
80,6
110,6
78
80,6
61,2
77
77
77
80,6
118,5
80,6 100
Состав
смеси
азестропной
смеси, масс. %
4
9
23
20
4
9
20
30
32
7
16
43
21
39
72
19
96
91
77
80
96
91
80
70
68
93
84
57
79
61
28
74 7
Т. кип.
смеси, °С
78,15
70
107,3
87
66
69,2
84,1
72
68,2
59,4
64,9
75
55,7
48,3
105,4
64,9
2.3.5. ПЕРЕГОНКА АЗЕОТРОПНЫХ СМЕСЕЙ
Многие вещества образуют азеотропные смеси, т. е. при определенном соотно-
соотношении компонентов смесь имеет максимально большую или минимально ма-
малую температуру кипения (ср. табл. А.82). Азеотропную смесь невозможно раз-
разделить перегонкой на отдельные компоненты, так как жидкость и пар над ней
имеют одинаковый состав (разд. А,2.3.3 и рис. А.66). Из известных азеотропных
смесей можно назвать, например, бромоводородную кислоту (при мак-
максимальной температуре кипения смесей образуется азеотропная смесь,
т. кип. 126 °С) и 96%-ный этиловый спирт (при максимальной температуре
кипения смесей образуется азеотропная смесь, т. кип. 78,15 °С).
Образование азеотропной смеси можно использовать для извлечения одного ве-
вещества из смеси. Важное значение имеет азеотропная сушка, при которой к высу-
высушиваемому соединению добавляют по возможности малосмешивающееся с водой
на холоду вещество, образующее с водой азеотропную смесь (например, бензол). За-
Затем нагревают смесь до кипения в приборе, изображенном на рис. А.83, а. Вода об-
образует с бензолом азеотропную смесь и при охлаждении отделяется в виде капель;
выделившуюся воду сливают из градуированной трубки водоотделителя. Таким об-
образом, можно легко установить момент окончания отгонки воды и определить ее
количество. Точно так же можно наблюдать за течением химических реакций, при
которых выделяется вода. Постоянно отгоняя воду из реакционной смеси, можно
сместить равновесие химической реакции в желательном направлении.
Наиболее часто для отделения воды при азеотропной сушке применяют бен-
бензол, толуол, ксилол, хлороформ, тетрахлоруглерод. Поскольку два последних
растворителя тяжелее воды, для них следует использовать водоотделитель, изоб-
изображенный на рис. А.83, б. До начала нагревания градуированную трубку заполня-
заполняют соответствующим растворителем, засасывая его. Для отгонки больших коли-
76
А Введение в лабораторную практику
б
Рис. А.83. Приспособления, используемые при перегонке азеотропных смесей
(водоотделители).
честв воды удобнее пользоваться приспособлением, показанным на рис. А.83, в,
в котором возможен постоянный сток воды, выделяющейся при азеотропной
перегонке. Этот прибор работает безукоризненно только в том случае, если он
установлен строго вертикально и перед отгонкой был заполнен дистиллятом.
Перечисленные выше растворители можно сушить (при не слишком высо-
высоких требованиях к их влажности) также при помощи перегонки; при этом
первые мутные части дистиллята отбрасывают.
2.4. СУБЛИМАЦИЯ (ВОЗГОНКА)
Упругость паров твердых веществ также растет при повышении температуры.
Многие вещества можно перевести в газовую фазу, не расплавляя их (а при кон-
конденсации пара происходит сразу же образование твердой фазы). В этом случае
говорят о сублимации (возгонке).
2. Способы разделения веществ
77
Рис. А.84. Приборы для возгонки.
Точкой возгонки считают температуру, при которой упругость пара твердого
вещества равна внешнему давлению. При этой температуре кристаллы испаря-
испаряются не только с поверхности, но и по всему объему, что сопровождается раст-
растрескиванием и тем самым может привести к загрязнению сублимата. Поэтому
возгонку осуществляют обычно при температурах ниже точки возгонки, так что-
чтобы упругость пара оставалась ниже внешнего давления. Разделяющая
способность, достигаемая при возгонке веществ, упругости паров которых мало
отличаются, как правило, невысока.
Указание о сублимационной способности вещества можно получить, наб-
наблюдая за протеканием его плавления под микроскопом (ср. разд. А,3.1.2). На
верхнем стеклышке появляются кристаллы, еще до достижения температуры
плавления.
Простой прибор для возгонки состоит из фарфоровой чашки и опрокинутой
над ней стеклянной воронки (рис. А.84, а). Диаметр воронки должен быть нем-
немного меньше диаметра чашки. Трубку воронки неплотно закрывают ватой. Что-
Чтобы сублимат не попадал обратно в чашку, ее покрывают круглым бумажным
фильтром, в котором в нескольких местах сделаны отверстия.
Для веществ, которые не возгоняются или очень медленно возгоняются при
атмосферном давлении, процесс возгонки часто осуществляют при понижен-
пониженном давлении. Для этого используют прибор, изображенный на рис. А.84, били в.
При открывании прибора необходимо избегать встряхивания (прогреть шлиф!),
чтобы не вызвать опадения сублимата с охлаждающего «пальца».
Охлаждающая поверхность «пальца» должна быть по возможности незначи-
незначительно удалена от нагреваемого пространства, откуда происходит возгонка
(достигается более высокая скорость возгонки!). Возгонка происходит только с
поверхности вещества, и поэтому препарат надо очень тонко измельчать. При
повышении температуры можно достигнуть более высокой скорости возгонки,
но при этом образуются мелкокристаллические и в большинстве случаев менее
чистые сублиматы.
Возгонка имеет ряд преимуществ по сравнению с перекристаллизацией: как
правило, при возгонке получаются очень чистые препараты и, кроме того, этот
метод очень удобен при очистке малых количеств.
78 А Введение в лабораторную практику
2.5. ЭКСТРАКЦИЯ И РАСПРЕДЕЛЕНИЕ МЕЖДУ ДВУМЯ ФАЗАМИ
Под экстракцией понимают перевод вещества из одной жидкой фазы, в которой
оно растворено или суспендировано, в другую жидкую фазу. Этот перевод воз-
возможен, потому что распределение вещества между двумя фазами подчиняется
определенным закономерностям. Термин «распределение» в научной литерату-
литературе имеет широкий смысл: распределение между двумя фазами любой природы,
а в частном случае это распределение между двумя жидкими фазами.
Распределение растворенного вещества между двумя жидкими фазами опре-
определяется законом распределения Нернста:
— =К [А.85]
В состоянии равновесия отношение концентраций Сд и eg вещества, которое растворено в двух
несмешивающихся жидких фазах, при определенной температуре является величиной постоянной
(коэффициент распределения К). В приведенной форме закон Нернста применим только для не-
небольших концентраций (идеальные условия) и в тех случаях, когда растворенное вещество имеет в
обеих фазах одинаковую степень ассоциации. Экстракция легко осуществима, если растворимость
данного соединения в экстрагенте значительно выше, чем в фазе исходного растворителя, и коэф-
коэффициент распределения, таким образом, значительно отличается от единицы.
Для веществ с коэффициентом распределения К< 100 (если при определении А"по уравне-
уравнению [А.85] концентрация вещества в растворителе-экстрагенте обозначена сА) уже недоста-
недостаточно простой однократной экстракции. В этом случае повторяют экстракцию несколько раз,
используя свежие порции растворителя.
Два вещества (с коэффициентами распределения А", и К2) в идеальном случае распределя-
распределяются между двумя жидкими фазами независимо друг от друга.
Если различие в величинах их коэффициентов распределения достаточно велико, то такие
вещества разделить при помощи экстракции просто. Для оценки трудности разделения введен
фактор разделения р, который всегда больше или равен 1, т. е. находят отношение большего
коэффициента распределения к меньшему:
Р = — [А.86]
Ср. с относительной летучестью а при перегонке (уравнение [А.52]).
Оба вещества могут быть удовлетворительно разделены простой экстракцией только тог-
тогда, когда р>100. Для разделения смесей с р<100 следует применять методы дробной экстрак-
экстракции (разд. А,2.5.3).
Подобные соотношения нужно ожидать при распределении вещества между любыми дву-
двумя фазами. Массообмен веществ при всех способах распределения возможен только на пове-
поверхности раздела фаз. Чтобы ускорить установление равновесия, необходимо по возможности
увеличить поверхность соприкосновения фаз. Жидкости для этого встряхивают или смешива-
смешивают продавливанием через пористые фильтры. Твердые вещества перед экстракцией тонко из-
измельчают. Тем не менее во многих случаях, особенно если в распределении участвует твердая
фаза, равновесное состояние так и не достигается.
2.5.1. ЭКСТРАКЦИЯ ТВЕРДЫХ ВЕЩЕСТВ
2.5.1.1. Однократная простая экстракция
Вещество нагревают с растворителем в колбе с обратным холодильником,
фильтруют через воронку для горячего фильтрования или декантируют. С не-
небольшими количествами вещества работают в пробирке с погруженным в нее
охлаждающим «пальцем» или с воздушным холодильником.
2. Способы разделения веществ
79
2.5.1.2. Многократная простая экстракция
Для более полной экстракции следует, как правило, описанную операцию пов-
повторить несколько раз. Для этого целесообразно применять специальные авто-
автоматически работающие приборы. Эти приборы состоят из колбы, насадки-
экстрактора и обратного холодильника. Находящийся в колбе растворитель
частично испаряется; конденсат стекает на вещество, подвергаемое экстракции
и находящееся в экстракционной гильзе, проходит через него и стекает обрат-
обратно в колбу; при этом концентрация экстрагируемого компонента в растворите-
растворителе возрастает.
Насадки для экстракции. Проточный экстрактор изображен на рис. А.87. В нем
вещество непрерывно промывается стекающим из холодильника сконденсиро-
сконденсированным, но еще горячим растворителем; экстракт непрерывно стекает в колбу.
Экстрактор Сокслета (рис. А.88) отличается от проточного наличием сифо-
сифона, через который экстракт сливается в колбу периодически только тогда, когда
уровень жидкости в насадке поднимается до верхнего колена сифона. Вещество,
подвергаемое экстракции, должно быть тяжелее, чем растворитель.
Для экстракции в полумикромасштабе или для экстракции высококипящи-
ми растворителями в качестве экстракционной гильзы применяют пористый
стеклянный фильтр (рис. А.89), который закрепляют под холодильником таким
Рис. А.87. Проточный
экстрактор.
Рис. А.88. Экстрактор
Сокслета.
Рис. А.89. Прибор для
экстракции полумикроколичеств.
80 А Введение в лабораторную практику
образом, чтобы его омывали горячие пары растворителя в колбе и одновремен-
одновременно промывал стекающий из холодильника еще теплый растворитель. Для
экстракции полумикроколичеств веществ можно использовать и вышеописан-
вышеописанные экстракторы меньшего размера.
2.5.2. ЭКСТРАКЦИЯ ЖИДКОСТЕЙ
Экстракция соединений из растворов (большей частью из водных) является
очень важной и одной из обязательных операций препаративной работы в орга-
органической химии. Дискретная экстракция известна как встряхивание, непрерыв-
непрерывная экстракция — как перколяция.
2.5.2.1. Извлечение веществ из растворов или суспензий
Водный раствор (или реже суспензию) смешивают в делительной воронке
(рис. А.90) с экстрагентом-растворителем (объем которого равен l/s—'/з обще-
общего объема раствора). Если растворитель огнеопасен, следует погасить все близ-
близко расположенные горелки с открытым пламенем. Делительная воронка долж-
должна быть заполнена не более чем на 2/з- Воронку закрывают пробкой и сначала
осторожно взбалтывают, прочно удерживая пробку и кран делительной ворон-
воронки. Затем делительную воронку переворачивают краном вверх и, осторожно
открывая кран, выпускают образующиеся пары. Осторожное встряхивание с
периодическим выпусканием паров продолжают до установления равновесно-
равновесного парциального давления паров растворителя в воздушном пространстве над
жидкостью в делительной воронке (давление внутри воронки должно быть
равно внешнему); только после этого переходят к энергичному встряхиванию
в течение 1—2 мин.
При работе с сильнокислыми, щелочными или раздражающими веществами
необходимо надевать защитные очки!
После отстаивания разделяют образующиеся два слоя. Нижний слой слива-
сливают через кран делительной воронки, а верхний всегда выливают через верхнее
отверстие. В сомнительных случаях надо уточнить, какой из двух слоев предс-
представляет собой водную фазу; для этого отбирают каплю каждой из фаз и добавля-
добавляют их в пробирку с водой. При относительно
высокой растворимости экстрагируемого
вещества в воде водный слой можно насы-
насытить сернокислым аммонием или поварен-
поваренной солью. Некоторые системы склонны к
образованию эмульсий. В таком случае
смесь в делительной воронке не встряхива-
встряхивают, а только слегка взбалтывают, покачивая
воронку. Образующуюся эмульсию можно
разрушить, добавляя небольшое количество
противовспенивающего средства или ами-
Рис. А.90. Делительная воронка. лового спирта, насыщая водную фазу пова-
2. Способы разделения веществ 81
ренной солью или фильтруя раствор. Однако самое надежное средство — оста-
оставить стоять смесь для расслаивания на продолжительное время.
Ниже перечислены наиболее распространенные экстрагенты.
• Экстрагенты-растворители легче воды. Диэтиловый эфир (низкая темпе-
температура кипения, огнеопасен, склонен к образованию взрывоопасных перокси-
дов, слабо — примерно до 8% — растворим в воде), толуол (огнеопасен).
• Экстрагенты-растворители тяжелее воды. Дихлорметан (низкая темпе-
температура кипения, 41 °С), хлороформ, тетрахлоруглерод (не огнеопасный).
При простой однократной экстракции встряхиванием в особо благоприят-
благоприятных случаях можно добиться установления распределения, полностью соответ-
соответствующего равновесным условиям; при этом в экстракт в соответствии с зако-
законом Нернста переходит лишь определенное количество вещества в зависимости
от количества экстрагента и концентрации экстрагируемого вещества в раство-
растворе. Поэтому экстракцию следует повторять несколько раз. Вещества, плохо
растворимые в воде, экстрагируют тремя-четырьмя порциями растворителя; для
хорошо растворимых в воде соединений экстракцию надо повторять много раз.
В подобных случаях лучше использовать непрерывную экстракцию (перколя-
цию, ср. А.2.5.2.2).
Целесообразнее проводить экстракцию несколько раз, используя небольшие
количества растворителя, чем пытаться извлечь вещество при однократной
экстракции большим объемом растворителя. Для того чтобы определить, закон-
закончен ли процесс экстракции, небольшое количество последней порции экстрак-
экстракта помещают на часовое стекло и испаряют растворитель. Конец извлечения
окрашенных веществ обычно определяют по отсутствию окраски после получе-
получения очередной порции экстракта.
Экстракт, как правило, следует очистить от посторонних веществ, чаще все-
всего кислот или оснований. Для этого его промывают, т. е. встряхивают с несколь-
несколькими порциями разбавленных водных щелочных растворов (обычно соды или
гидрокарбоната натрия) или растворов кислот, а затем несколько раз промыва-
промывают водой.
Всегда следует иметь в виду, что при промывании экстракта содой может выде-
выделяться углекислый газ и давление в делительной воронке может повыситься,
поэтому выделяющийся газ надо осторожно выпускать через кран.
После всех операций экстракт высушивают подходящими осушителями
(см.А.1.10.2).
2.5.2.2. Непрерывная экстракция
Применяя экстракторы непрерывного действия (перколяторы; рис. А.91 иА.92),
можно извлекать вещество из жидкой фазы с помощью очень небольшого коли-
количества растворителя. Растворитель при этом испаряют в колбе и конденсируют
его пары в обратном холодильнике. Конденсат в виде капель проходит через
раствор, постепенно обогащаясь экстрагируемым веществом, и стекает через
перелив обратно в колбу. Таким образом, оказывается возможным извлекать
вещества с коэффициентом распределения А"даже меньше 1,5.
82
А Введение в лабораторную практику
I
ULn
а б
Рис. А.91. Перколяторы.
а — если экстрагент легче раствора,
из которого происходит экстракция;
б — если экстрагент тяжелее этого раствора.
Рис. А.92. Полумикроперколятор
Кутшера — Штейделя.
Необходимо обратить внимание на то, что при нагревании объем жидкости
увеличивается. Поэтому никогда нельзя заполнять до перелива холодной нижней
фазой патрон экстрактора при работе с легкими растворителями-экстрагентами
(рис. А.93, а и А.92). В экстракторы для тяжелых растворителей (рис. А.93, б) сна-
сначала надо налить немного тяжелой нижней фазы, а затем добавить раствор, под-
подлежащий экстракции (почему?).
Рис. А.93. Ротационный перколятор.
а — для легких экстрагенов; б — для тяжелых экстрагенов.
2. Способы разделения веществ
83
2.5.3. ПРОТИВОТОЧНОЕ РАСПРЕДЕЛЕНИЕ
Противоточным (дробным) распределением называют многоступенчатую экстракцию, при
которой обе жидкости (экстрагируемая и экстрагирующая) движутся в противоположном
направлении и постоянно находятся в состоянии равновесия. При этом экстракт, частично
обогащенный экстрагируемым веществом, смешивается с новыми порциями раствора, из
которого происходит извлечение, а раствор, уже частично проэкстрагированный, смешивает-
смешивается со свежим растворителем (рис. А.94, а).
Этот метод имеет практическое значение для разделения веществ с фактором разделения,
лишь несколько большим единицы.
Противоточное распределение и простую экстракцию можно сопоставить с ректификацией
и простой перегонкой. Понятие ступени разделения имеет аналогичное значение.
Получить представление о поведении вещества в условиях противоточного распределения
можно из рис. А.94, а. В первом сосуде (например, в делительной воронке) 100 частей растворен-
растворенного экстрагируемого вещества (нижняя фаза) обрабатывают равным объемом экстрагента-раст-
ворителя (верхняя фаза Sq). Фазы должны быть всегда взаимно насыщенны. Затем смесь встряхи-
встряхивают (на схеме обозначено двойной стрелкой) до установления равновесия. Если коэффициент
распределения К= 1, в верхней и нижней фазах после этого окажутся равные количества (по 50
частей) экстрагируемого вещества. Это первая ступень распределения. Верхнюю фазу переносят
в следующий сосуд и экстрагируют ею новую порцию нижней фазы (Щ, а нижнюю фазу, содер-
содержащую экстрагируемое вещество, экстрагируют новой порцией верхней фазы (S\). Этот процесс
называют первым переносом. После того как равновесие установится, вновь разделяют и перено-
переносят фазы в следующие сосуды (второй перенос) и т. д. После трех переносов в первом и четвертом
сосудах находится по 12,5 части вещества, а во втором и третьем — по 37,5 части. Таким образом,
максимальное количество вещества накапливается в средних сосудах. Для большого числа сосу-
сосудов получают колоколовидную кривую (рис. А.94, б, пунктирная кривая).
Если коэффициент распределения вещества не равен 1, то максимальное количество
вещества будет находиться в сосуде с большим (К = 3) или меньшим (К = 0,33) номером
(рис. А.94, б). К таким же результатам приходят и в том случае, когда в исходном растворе со-
Or
I
о
12
ЮО1
i
50
I
25
[25]
50
25
0
25
25
25
25
0
и2
12 3 4
Номер ячейки распределения
10 20
Число ячеек распределения
30
Рис. А.94. Способы распределения
а — схема дробного распределения; б — кривые распределения.
84 А Введение в лабораторную практику
держатся одновременно оба вещества, коэффициенты распределения которых равны соответ-
соответственно 0,33 и 3, т. е. путем экстракции эти вещества разделяются.
На практике используют автоматические приборы с несколькими сотнями распредели-
распределительных ячеек (ступеней). Более подробно с этими методами можно ознакомиться по ориги-
оригинальной литературе (см. список литературы в конце главы).
2.6. АДСОРБЦИЯ
Под адсорбцией понимают повышение содержания соединения на поверхности
твердых веществ.
Для разделения смесей органических соединений на практике имеет значе-
значение их разное сродство к твердым веществам.
Твердое вещество, на котором происходит адсорбция, называют адсорбен-
адсорбентом; вещество, которое адсорбируется, называют адсорбатом. Различают непо-
неполярные и полярные адсорбенты.
• Неполярные адсорбенты: активированный уголь, некоторые органичес-
органические смолы (например, вофатит EW), молекулярные сита.
• Полярные адсорбенты: оксид железа (Ре2Оз), оксид алюминия, силика-
гель, углеводы (крахмал, сахар, целлюлоза).
Эффективность адсорбентов падает в приведенной последовательности.
Особое значение имеют полярные адсорбенты. Их сродство к соответствую-
соответствующему адсорбируемому веществу возрастает с ростом полярности последнего.
Поэтому особенно прочно адсорбируется вода; при этом склонность активной
поверхности адсорбента к адсорбции других (менее полярных) веществ умень-
уменьшается, по мере того как большая часть поверхности покрывается молекулами
воды. Для оксида алюминия — одного из наиболее распространенных адсорбен-
адсорбентов — специальными опытами с красителями1 установлено пять условных сте-
степеней активности в зависимости от содержания воды: I (наиболее активная) —
0%; II - 3%; III - 4,5-6%; IV- 9,5%; V- 13%. Кроме того, выпускаемый оксид
алюминия может быть кислым, нейтральным или щелочным.
Адсорбируемость органических веществ обусловливается помимо полярнос-
полярности молекул также их размерами и поляризуемостью.
Органические соединения можно расположить примерно в следующем по-
порядке по возрастанию сродства к полярным адсорбентам: галогенопроизводные
углеводородов < простые эфиры < третичные амины, нитросоединения < слож-
сложные эфиры < кетоны, альдегиды < первичные амины < амиды кислот < спирты
< карбоновые кислоты.
Известны аналогичные закономерности и для растворителей. Адсорбция од-
одного и того же органического соединения из неполярного растворителя эффек-
эффективнее, чем из полярного. Напротив, уже адсорбированное соединение можно
вытеснить с адсорбента только таким растворителем, у которого сродство к ад-
адсорбенту больше, чем у адсорбированного вещества. По способности вытеснять
1 Установление активности и цветные пробы см. в работе Hesse G. и др. Angew. Chetn., 1952,
64, 103. Методику высушивания оксида алюминия до первой степени активности см. в работе
Brockmann H., SchodderH. Ber., 1941, 74, 73.
2. Способы разделения веществ
85
из адсорбента адсорбированные вещества (элюировать) растворители можно
расположить в определенном порядке (элюотропный ряд) (табл. А.95).
Таблица А.95. Элюотропный ряд растворителей
Пентан
Гексан
Петролейный эфир
Циклогексан
Дисульфид углерода
Тетрахлоруглерод
Бензол
Диэтиловый эфир
Хлорформ
Дихлорметан
Тетрагидрофуран
Этилметилкетон
Ацетон
Этилацетат
Ацетонитрил
Пиридин
Бутанол
Этанол
Метанол
Уксусная кислота
Вода
Для активированного угля — неполярного адсорбента — выполняются
обратные закономерности.
Необходимо учитывать, что с адсорбцией всегда связана поляризация моле-
молекул, которая может привести к увеличению чувствительности соединения к
свету, воздуху, влаге, действию окислителей.
2.6.1. Обесцвечивание растворов
При обесцвечивании растворов должны быть удалены загрязняющие окрашен-
окрашенные побочные продукты (главным образом высокомолекулярные соединения),
присутствие которых часто затрудняет кристаллизацию основного компонента.
Если эти примеси по своим физическим или химическим свойствам существен-
существенно отличаются от основного продукта, то, добавляя подходящий адсорбент, их
можно селективно удалить из раствора. Адсорбированные примеси отбрасыва-
отбрасывают вместе с адсорбентом.
Для того чтобы исключить потери основного продукта, следует применять
возможно меньшие количества адсорбента (максимально 1—5% от количества
очищаемого вещества). Для обеспечивания растворов в полярных растворителях
используют активированный уголь, в неполярных растворителях (от гексана до
хлороформа; см. элюотропный ряд, табл. А.95) — оксид алюминия. Вофатит EW
применяют лишь в водных растворах.
При обесцвечивании холодный раствор просто некоторое время взбалтыва-
взбалтывают (именно с активированным углем такой способ чаще всего применяется) или
кипятят.
При добавлении активированного угля к горячим растворам надо соблюдать
осторожность! Из-за неожиданного вскипания и выделения адсорбированного
углем воздуха может происходить бурное вспенивание!
Адсорбент отделяют центрифугированием или фильтрованием; иногда, если
это оказывается необходимым, добавляют средства, облегчающие фильтрова-
фильтрование (например, кизельгур). В случае необходимости обесцвечивание повторяют.
Следует обратить внимание, что при обесцвечивании активированным углем,
особенно при нагревании, чувствительные к окислению вещества под действи-
действием адсорбированного кислорода легко могут окисляться.
86 А Введение в лабораторную практику
Другой способ принят при работе с ионообменными смолами и оксидом
алюминия в качестве адсорбентов. Холодный раствор, подлежащий обесцвечи-
обесцвечиванию, фильтруют через слой адсорбента, который помещен в короткую широ-
широкую колонку, на воронку Бюхнера или воронку с пористым фильтром. Если сам
адсорбент неокрашен, насыщение его примесями можно контролировать по
передвижению окрашенной зоны.
2.7. ХРОМАТОГРАФИЯ
Хроматографические методы заключаются в разделении веществ между непод-
неподвижной (стационарной) фазой и подвижной (мобильной) фазой, протекающей
через нее. Компоненты смеси веществ, нанесенные на стационарную фазу,
удерживаются ею весьма различным образом и поэтому перемещаются с различ-
различной скоростью и могут быть таким образом разделены.
Подвижная фаза может быть жидкой или газообразной, поэтому методы
называют жидкостная хроматография (ЖХ) или газовая хроматография (ГХ). Не-
Неподвижная фаза может быть твердым веществом или жидкостью, фиксирован-
фиксированной на твердом носителе. Она находится в мелкозернистом виде в колонке (коло-
(колоночная хроматография) или в тонком слое на инертном носителе или пластинке
{тонкослойная хроматография). Специальная фильтровальная бумага может
также служить в качестве неподвижной фазы (бумажная хроматография).
Предварительно использованы две физико-химические предпосылки для
хроматографического разделения — различное распределение компонентов
из-за их отличий в растворимости в обеих фазах (см. разд. А,2.5.3) и фракцион-
фракционная адсорбция из-за различной адсорбции компонентов на неподвижной фазе
(ср. разд. А,2.6). В зависимости от преобладающего влияния говорят о распре-
распределительной хроматографии или адсорбционной хроматографии.
Оба процесса часто строго не разделимы, а действуют более или менее согласованно. По-
Пористые твердые вещества, использованные как носители для жидких неподвижных фаз при
распределительной хроматографии, могут обладать определенной адсорбционной способ-
способностью, и твердые адсорбенты для адсорбционной хроматографии, покрытые компонентами
подвижной фазы или смесью веществ, могут способствовать их распределению.
Другие хроматографические методы разделения базируются на ионном обмене (на ионо-
ионообменных смолах или гелях); при этом ионы могут быть разделены на пористых твердых веще-
веществах по величине молекулярной массы или величине биосродства (аффинная хроматография
с неподвижными фазами, модифицированными определенными лигандами — ферментами,
белками, липидами и др., способствующими селективной адсорбции). Информацию об этом
можно найти в специальной литературе.
В качестве неподвижной фазы для адсорбционной хроматографии использу-
используются главным образом силикагель и оксид алюминия (нейтральный, кислый
или основный), активность которых зависит от содержания воды. На этих по-
полярных адсорбентах разделяемые вещества адсорбируются согласно их поляр-
полярности, как это описано в разд. А,2.6. В качестве подвижных фаз используют уг-
углеводороды (пентан, гексан, гептан, изооктан), хлоруглеводороды (дихлорме-
тан, хлороформ, тетрахлоруглерод), простые эфиры (тетрагидрофуран, диоксан)
и ацетонитрил. Их элюирующая способность повышается соответственно их
элюотропному ряду, табл. А.95. Состав мобильной фазы зависит от полярности
2. Способы разделения веществ 87
разделяемых веществ. Сперва применяют неполярный растворитель, а затем
повышают, если необходимо, его элюирующую способность путем добавления
какого-либо более полярного растворителя.
Для распределительной хроматографии используют силикагель как носи-
носитель, на который наносят жидкость как стационарную фазу. Полярные и гидро-
гидрофильные неподвижные фазы — это вода, этиленгликоль, этилендиамин, эфиры
цианалканов или диметилсульфоксид. В качестве элюирующих веществ исполь-
используют менее полярные вещества, не смешивающиеся с неподвижной фазой,
прежде всего выше названные углеводороды или хлоруглеводороды. При этом,
как и в хроматографии на полярных адсорбентах, быстрее всего элюируются на-
наименее полярные вещества (ср. разд. А,2.6): алканы > галогенсодержащие сое-
соединения > простые эфиры > сложные эфиры > альдегиды > кетоны > спирты,
амины > амиды > карбоновые кислоты.
Особенно проявили себя в этом отношении так называемые «обращенные
фазы», это неполярные, гидрофобные неподвижные фазы (например, силика-
силикагель), поверхность которых модифицирована химически связанными алкиль-
ными остатками.
Такая модификация достигается обработкой поверхности силикагеля алкилхлорсилана-
ми, например:
| СНз , СНз
—Si—ОН + CI—Si—R - — Si—O-Si—R + HCI [A.96]
1 CH3 ' СНз
Чаще всего R — остаток октадецила С18Н37, но это могут быть и октил-, циклогексил- или
фенильные остатки и замещенные алкилы с полярными циано-, нитро- или аминогруппами.
Фазы, используемые для разделения энантиомеров, содержат хиральные группы, как, напри-
например, модифицированные целлюлозы, циклодекстрины (циклен из 6 или 8 глюкозных единиц)
или другие асимметричные остатки. Хроматографическое разделение с «обращенными» фаза-
фазами нельзя четко отнести к распределительной или адсорбционной хроматографии.
Обращенные фазы элюируют сильнополярными растворителями, как вода,
или смесями вода—метанол или вода—ацетонитрил. При этом полярные вещест-
вещества элюируются в обратном порядке по сравнению с хроматографией с полярны-
полярными неподвижными фазами. Они элюируются тем быстрее, чем выше их поляр-
полярность: карбоновые кислоты > спирты, фенолы > амины > сложные эфиры,
альдегиды > кетоны > галогенсодержащие соединения.
При хроматографии смесей соединений, компоненты которых сильно отли-
отличаются по своей полярности, случается, что медленно идущие соединения выхо-
выходят очень поздно или совсем не элюируются. Тогда принято в процессе хрома-
хроматографии повысить элюирующую силу подвижной фазы непрерывным добавле-
добавлением растворителя с повышенной элюирующей способностью (градиентное
элюирование). В современных хроматографах это делается автоматически в
заранее выбранных границах.
Разделяющая способность неподвижной фазы повышается при уменьшении
величины частиц носителя, но при этом повышается сопротивление потоку. Для
достижения достаточной скорости потока при величине частиц меньше 50 мкм
подвижную фазу пропускают через неподвижную под давлением. Так как разде-
разделяющая способность при использовании мелкозернистых фаз мало зависит от
88 А Введение в лабораторную практику
скорости потока подвижной фазы, можно работать при высоких скоростях
потока, а время разделения будет небольшим.
2.7.1. ТОНКОСЛОЙНАЯ ХРОМАТОГРАФИЯ
При тонкослойной хроматографии разделение веществ происходит на открытых
колонках, неподвижная фаза которых состоит из тонкого слоя адсорбента, нане-
нанесенного на инертную подложку. В качестве слоя носителя служат плоские плас-
пластинки из стекла, алюминия или полиэфирных пленок. Как адсорбент используют
силикагель или оксид алюминия, смешанные с гипсом в качестве связующего.
Для получения однородных слоев одинаковой толщины применяют специальные
приборы. Слои наносят в мокром состоянии, затем высушивают на воздухе и ак-
активируют при повышенной температуре A05—150 °С). Активность возрастает с
понижением содержания воды. Активированные пластинки хранят в эксикаторе.
Удачно используют продажные приготовленные носители. Для восходящей
хроматографии вырезают из нанесенной пластинки полоски нужной ширины
(-1,5 см на пробу) и около 10 см длиной. Для двумерной хроматографии берутся
квадраты размером 10 х 10 см.
Для подбора элюентов справедливы общие соображения, рассмотренные в
разд. А,2.6 и А,2.7. При хроматографировании смесей неизвестных веществ сна-
сначала используют циклогексан, затем толуол или хлороформ и в зависимости от
полученных результатов переходят далее к полярным растворителям. Можно
применять и смеси растворителей, например хорошие результаты дает смесь
циклогексана с возрастающими добавками этилацетата E, 10, 20% и более).
В предварительных опытах при подборе элюентов можно использовать ради-
радиальную хроматографию. При этом в центр пробы подают из микрокапилляра
растворитель и оценивают его элюирующий эффект по образующимся концент-
концентрическим зонам.
Тонкослойная хроматография может быть восходящей, нисходящей, ради-
радиальной и двумерной. Наибольшее значение имеет удобная для выполнения
восходящая ТСХ.
Вещества наносят в виде 1%-ного раствора в неполярном растворителе.
Общее количество вещества целесообразно определять при помощи предвари-
предварительного опыта. Слишком высокая концентрация разделяемых веществ приво-
приводит к появлению «хвостов» на хроматограмме и снижает эффективность разде-
разделения. Хорошо разделяемое количество вещества повышается с увеличением
активности и толщины слоя адсорбента.
Растворы испытуемых смесей наносят на пластину с помощью тонкого ка-
капилляра (например, капилляра для определения температуры плавления); при
погружении в раствор такие капилляры самопроизвольно заполняются под
действием капиллярных сил. Нанесенные пятна испытуемых образцов должны
быть расположены на расстоянии 1—2 см друг от друга, иметь небольшой размер
B—3 мм) и находиться не ближе 1,5 см от нижнего края пластины. Если раствор
образца очень разбавленный, нанесение одного пятна повторяют несколько раз,
дожидаясь после каждого нанесения испарения растворителя. Для двумерного
хроматографирования пятно располагают в одном из углов квадратной пласти-
пластины на расстоянии 1,5 см от краев.
2. Способы разделения веществ
89
Проявление хроматограммы проводят в плотно закрытой камере, атмосфера
которой насыщена парами элюента (для более полного насыщения стенки ка-
камеры оклеивают полосками фильтровальной бумаги и оставляют стоять с
элюентом в течение 30 мин). Пластину (полоску фольги) устанавливают в каме-
камере почти вертикально, погрузив конец в жидкость на ~5 мм.
Для предварительных опытов часто хроматограмму проявляют в прикрытом
часовым стеклом химическом стакане, в который вложена манжета из фильтро-
фильтровальной бумаги.
В большинстве случаев для разделения достаточно подъема фронта элюента на
высоту ~10 см. После этого пластину вынимают из камеры и сразу же с помощью
острого предмета (шпателя, карандаша) отмечают положение фронта элюента.
Проявленные пластины высушивают на воздухе. Положение пятен некото-
некоторых бесцветных веществ, которые флуоресцируют, можно установить при рас-
рассмотрении пластин в УФ-свете. Если к адсорбенту добавлен флуоресцирующий
индикатор, то все вещества, поглощающие в УФ-области, при освещении
УФ-светом проявляются на флуоресцирующем поле в виде темных пятен.
В большинстве случаев удается проявить хроматографические пятна в при-
присутствии паров иода. Для этого хроматограмму помещают в закрытый сосуд, в
котором находится кристаллик иода. Вещество проявляется в виде коричнево-
коричневого пятна или (реже) при длительном действии паров иода в виде белого пятна на
темном фоне. Для проявления хроматограммы применяют, кроме того, обра-
обработку парами брома, опрыскивание раствором перманганата калия и другими
подходящими реагентами (табл. А.97) с применением распылителя (рис. А.98).
Таблица А.97. Реагенты для опрыскивания тонкослойных хроматограмм
Определяемый класс
соединений
Реагент
Состав реагента и
некоторые замечания
Альдегиды
Высшие спирты
Амины
Амины ароматические
Аминокислоты
Эфиры, амиды и ан-
ангидриды карбоновых
кислот, лактоны
2,4-Динитрофенил-
гидразин — серная
кислота
Ванилин — серная
кислота
4-Диметиламинобен-
зальдегид — соляная
кислота
Нингидрин
Диазотированная суль-
фаниловая кислота
Нингидрин
Гидроксиламин —
хлорид железа(Ш)
1 г 2,4-динитрофенилгидразина раство-
растворяют в смеси 25 мл спирта, 8 мл воды и
5 мл концентрированной серной кислоты
0,5 г ванилина растворяют в смеси 80 мл
серной кислоты + 20 мл спирта; раствор
нагревают до 120 °С
1 г 4-диметиламинобензальдегида раст-
растворяют в смеси 25 мл конц. соляной кис-
кислоты и 75 мл метанола
0,3 г нингидрина растворяют в 100 мл бу-
танола и 3 мл ледяной уксусной кислоты
Диазотирование (см. разд. Г,8.2.1) — хро-
хроматограмму опрыскивают 1%-ным раст-
раствором, затем 1%-ным раствором соды
См. Амины
Раствор I: 2 г гидрохлорида гидроксила-
мина растворяют в 5 мл воды, прибавля-
прибавляют 15 мл спирта
90
А Введение в лабораторную практику
Таблица А.97. Продолжение
Определяемый класс
соединений
Кетоны
Метилкетоны (актив-
(активные), соединения с под-
подвижной метиленовой
группой
Углеводороды
Фенолы
Органические азотсо-
азотсодержащие соединения
Ненасыщенные соеди-
соединения, восстановители
Сахара
Реагент
о-Дианизидин
2,4-Динитрофенил-
гидразин
Нитропруссид натрия
— щелочь
Конц. серная кислота
Формальдегид —
серная кислота
Диазотированная суль-
фаниловая кислота
Хлорид железа(Ш)
Ванилин — серная
кислота
Реактив Драгендорфа
Перманганат калия
Фосфорномолибде-
новая кислота
Анисовый альдегид —
серная кислота
Перманганат калия
Состав реагента и
некоторые замечания
Раствор II: 5 г КОН растворяют в
небольшом количестве воды и добавля-
добавляют этанол до 50 мл
Реагент для опрыскивания I: смешать
растворы I и II, профильтровать
Реагент для опрыскивания II: 1 г хлорида
железа(Ш) растворяют в смеси 2 мл
конц. НС1 и 20 мл эфира
Обработка хроматограмм: опрыскива-
опрыскивание реагентом I, сушка при комнатной
температуре, опрыскивание реагентом II
Насыщенный раствор о-дианизидина
D,4 -диамино-3,3 -диметоксидифенила)
в ледяной уксусной кислоте
См. Альдегиды
1 г нитропруссида натрия растворяют в
смеси 50 мл этанола и 50 мл 2 н. NaOH и
нагревают до 150 °С
0,2 мл 37%-ного формалина растворяют
в 10 мл конц. серной кислоты
См. Амины
1-5%-ный раствор в 0,5 М НС1
См. Спирты
Раствор 1:0,085 г основного нитрата вис-
висмута растворяют в смеси 1 мл ледяной
уксусной кислоты и 4 мл воды
Раствор II: 2 г иодида калия растворяют
в 5 мл воды
Реагент для опрыскивания: 1 мл раствора I и
1 мл раствора 11 растворяют в смеси
4 мл ледяной уксусной кислоты и 20 мл воды
0,5%-ный раствор в 1 н. NaOH или в воде
10%-ный раствор в спирте. Для выявле-
выявления пятен пластину нагревают до 120 °С
до оптимального образования пятен
0,5 мл анисового альдегида растворяют в
смеси 50 мл ледяной уксусной кислоты и
1 мл серной кислоты. Пластину нагрева-
нагревают до 100 °С
См. Ненасыщенные соединения
2. Способы разделения веществ
91
Рис. А.98. Распылитель.
Рис. А.99. Тонкослойная
хроматограмма.
7 — средний фронт растворителя;
2 — вещество 2,3— вещество 1; 4 — старт.
В общем случае рекомендуется опрыскивание высушенных и освобожденных
от растворителя пластин свежеприготовленным 2%-ным аммиачно-спиртовым
раствором нитрата серебра с последующим нагреванием до 105 "С (в течение
2—5 мин). Многие вещества при этом дают черные пятна на темном фоне.
Положение пятен веществ после проявления (рис. А.99) характеризуется
значением
Расстояние между стартом и центром пятна вещества
RF = - [А. 100]
Расстояние между стартом и средним фронтом растворителя
Значение RF является характеристической величиной для каждого вещества
и может быть привлечено для его идентификации. Значения /^многих веществ
приведены в таблицах. Воспроизводимость значений RfC данным элюентом оп-
определяется прежде всего постоянством активности адсорбента, насыщения в ка-
камере (см. ниже), толщины слоя адсорбента и температуры камеры. Для иденти-
идентификации надежнее всего параллельно на той же пластине хроматографировать
определяемое вещество и известное вещество, образец сравнения, проверив
совпадение RF в двух системах растворителей. Если значения их RF отличаются
от величин, приведенных в таблицах, то и все другие значения Rf должны быть
скорректированы в одинаковых условиях.
Для дальнейшего исследования пятна окрашенных или флуоресцирующих
веществ его соскребают с пластины вместе со слоем адсорбента и подвергают за-
затем масс-спектрометрическому анализу или (после элюирования) изучению
поглощения в УФ-области.
Для разделения миллиграммовых количеств используют пластины с толстым
слоем A—3 мм). Пробу наносят на старт в виде непрерывной линии и после ис-
испарения растворителя проявляют как обычно. Для выявления зон разделенных
веществ пластину прикрывают, оставив полоску в 2 мм шириной для опрыски-
92 А Введение в лабораторную практику
вания. После этого отделяют с помощью шпателя зоны, где обнаружены веще-
вещества (не затрагивая опрысканную полосу), извлекают их подходящими раство-
растворителями. При этом следует иметь в виду, что полярные растворители (напри-
(например, спирты) могут частично переводить силикагель в коллоидный раствор.
Тонкослойная хроматография очень удобна для предварительной подготов-
подготовки к проведению колоночной хроматографии. Однако при этом надо учитывать,
что четкость разделения в «закрытой» колонке меньше, чем на пластине
(«открытой колонке»).
2.7.2. КОЛОНОЧНАЯ ЖИДКОСТНАЯ ХРОМАТОГРАФИЯ
Жидкостную хроматографию можно проводить просто в «разделительной ко-
колонке», в вертикально стоящей стеклянной трубке, которая заполнена подходя-
подходящей неподвижной фазой. Вещество, подлежащее разделению или очистке, в
растворе подают в верх заполненной колонки и вытесняют затем растворителем
через колонку. При этом в зависимости от их сродства к адсорбенту и к раство-
растворителю вещества вытесняются с разной скоростью.
В качестве хроматографических колонок используют стеклянные трубки
(рис. А.101). В зависимости от количества хроматографируемого вещества упот-
употребляются трубки различных размеров A5 х 1, 25 х 2, 40 х 3, 60 х 4 см). В ниж-
нижнюю часть трубки вставляется неплотный тампон из обычной или стеклянной
ваты, в широких трубках имеется сетчатая фарфоровая пластинка. Растворитель
добавляют из капельной воронки.
В качестве адсорбентов наиболее часто используют оксид алюминия или си-
силикагель с величиной зерна от 65 до 200 мкм. Для успешного разделения смеси
большое значение имеет равномерное наполнение хроматографической колон-
колонки. Безусловно, следует избегать пузырьков воздуха, неравномерной засыпки и
в особенности трещин в адсорбенте. При заполнении колонки лучше всего при-
приготовить суспензию тщательно высушенного адсорбента в растворителе, кото-
которым будет осуществляться элюирование, и
эту суспензию медленно перенести (при
постукивании) в хроматографическую ко-
колонку, в которую предварительно уже нали-
налито некоторое количество растворителя. Ког-
Когда заполнение колонки окончено, адсорбент
покрывают сверху крупным песком или ва-
ватой. Необходимо следить за тем, чтобы хро-
матографическая колонка ни на секунду не
оставалась «сухой» (без слоя растворителя
над адсорбентом), так как в противном слу-
случае в слое адсорбента образуются трещины.
Соотношение адсорбат : адсорбент должно
составлять примерно 1: 100.
Применяется также и сухое заполнение
колонок, при котором разделяемые вещест-
Рис. А.1О1. Хроматографическая ва в небольшом количестве растворителя
колонка. совместно с наполнителем высушивают
2. Способы разделения веществ 93
досуха в вакуумном роторном испарителе. Затем эту пропитанную неподвиж-
неподвижную фазу подают в колонку, заполненную сухим адсорбентом, и элюируют
подходящими растворителями.
В качестве носителей жидких неподвижных фаз для распределительной хро-
хроматографии в разделительных колонках используют силикагель или порошок
целлюлозы. Соотношение количеств анализируемых веществ и носителя лежит
между 1: 1000 до 1 : 3000.
При выборе подвижной фазы руководствуются соображениями, изложен-
изложенными в разд. А,2.6 и А,2.7.
Разделяемое вещество вводят в колонку в виде максимально концентриро-
концентрированного раствора, причем для растворения используют возможно менее поляр-
полярный растворитель с малой элюотропной силой, лучше всего подвижную фазу.
Как только раствор почти пройдет, в колонку начинают добавлять тот же раст-
растворитель. Скорость движения раствора в колонке не должна быть слишком
большой, чтобы достигалось адсорбционное равновесие (для колонки высотой
40 см это -3-4 мл/мин).
Если элюат стекает слишком медленно, в колонке несколько повышают дав-
давление. Для этого увеличивают столб жидкости над адсорбентом либо с помощью
насосов, либо с помощью технических газов (сжатый воздух, азот, аргон) через
склянки из дуранового стекла, проверенные на давление и закрытые винтом; это
одновременно позволяет осуществлять дополнительное дозирование элюента.
В идеальном случае каждое индивидуальное вещество в результате разделе-
разделения смеси находится внутри узкой зоны наполненной колонки. Если вещества
окрашены или, например, видимы в УФ-свете благодаря флуоресценции, то
можно осторожно вытолкнуть адсорбент с поглощенным веществом и разрезав
его на отдельные зоны, экстрагировать вещества по отдельности из каждой
зоны. Эта механическая обработка теперь мало употребима.
Предпочитают вымывать (элюировать) отдельные вещества из колонки до-
дополнительным количеством растворителя и элюат собирать фракциями по
-0,5—10 мл. Эта операция может быть автоматизирована с помощью сборника
фракций, при которой падающие капли считают в приемнике электрически
(фотоячейка) или используется объем фракций для регулировки. В собранных
фракциях с помощью подходящего аналитического метода устанавливается,
содержит ли она элюируемое вещество. Затем в случае твердых определяемых
веществ фракции упаривают в вакууме и определяют температуру плавления
остатка. Если использованный растворитель не элюирует вещество или после
вымывания одного из веществ отделяются фракции с чистым растворителем, то
надо поднять элюирующую способность растворителя. Для этого в увеличиваю-
увеличивающихся количествах — начиная с 1—2% — начинают использовать растворитель,
стоящий выше в ряду элюирующей способности, и выясняют, элюируется ли
теперь вещество из колонки. Это продолжают до тех пор, когда практически все
введенные вещества будут элюированы.
Примером применения хроматографии является извлечение кофеина из кофе
(или из чая) и хроматографическая очистка сырого кофеина на оксиде алюми-
алюминия (нейтральном) [Connor R. О. J. Chem. Educ, 1965, 42, 493].
Разделяющую способность и скорость проведения колоночной хроматогра-
хроматографии можно существенно повысить, если использовать неподвижную фазу мень-
94 А Введение в лабораторную практику
шей зернистости D0 мкм) и более плотное
наполнение и пропускать элюирующий
растворитель с помощью инертного газа
(например, азота) из газовой склянки под
давлением от 1,5 до 2 бар в колонку. Эту про-
процедуру называют флеш-хроматография. Она
объединяет небольшие затраты обычной ко-
а ~^~ лоночной хроматографии со скоростью хро-
Рис. А.1О2. Резервуар для матографии, применяемой под уменылен-
растворителя (а) и ным Давлением (ср. разд. А.2.7.3), но не дос-
регулятор потока (б) тигает ее эффективности,
для флеш-хроматографии. Флеш-хроматография обычно применя-
применяется для препаративного разделения и
очистки веществ в количествах от 0,01 до 25 г. В зависимости от величины про-
пробы используют стеклянные колонки длиной 35-60 см и внутренним диаметром
10—40 мм. Распространены короткие колонки с высотой сорбента 15 см. Ниж-
Нижняя часть колонки наполняется стеклянной ватой и прокаленным морским пес-
песком; неподвижную фазу заполняют сухим способом и покрывают подвижной
фазой из насаженного на колонку резервуара для растворителей. Применяя
инертный газ (например, азот из газовой склянки) под давлением 1,5—2 бар, уп-
уплотняют набивку колонки и освобождают ее от пузырьков воздуха. Для регули-
регулировки давления служит регулятор потока с игольчатым вентилем (рис. А. 102).
Все шлифы аппаратуры защищены клеммами или винтовыми запорами.
В качестве неподвижной фазы наряду с полярными адсорбентами силикаге-
лем и оксидом алюминия применяют и неполярные гидрофобные «обращенные»
фазы (ср. разд. А,2.7). Для хроматографии на силикагеле или оксиде алюминия в
качестве элюентов применяют и-гексан, петролейный эфир, дихлорметан, толу-
толуол, тетрагидрофуран и этилацетат, часто смеси из этилацетата и и-гексана или
петролейного эфира. Предварительные опыты по тонкослойной хроматографии
удобны для выбора системы разделения. Соотношение количеств пробы, адсор-
адсорбента и подвижной фазы должно составлять примерно 1:100:1000.
Проба, растворенная в подвижной фазе, заливается в неподвижную фазу, на-
насаживают резервуар с растворителем и с помощью регулятора скорости устанав-
устанавливают скорость протекания элюента 5 см/мин. Как и при обычной колоночной
хроматографии, собирают элюат вручную или автоматически в виде фракций
необходимой величины. Строение элюированных веществ устанавливают по
тонкослойной хроматографии, ИК- или УФ-спектроскопии, а твердые вещест-
вещества — также измерением температуры плавления остатка. Смесь веществ может
быть разделена в течение 5—10 мин.
2.7.3. ВЫСОКОЭФФЕКТИВНАЯ ЖИДКОСТНАЯ ХРОМАТОГРАФИЯ (ВЭЖХ)
Высокоэффективная жидкостная хроматография (ВЭЖХ; англ. high perfor-
performance liquid chromatography, HPLC) — это высокопроизводительная быстрая
колоночная жидкостная хроматография для разделения смесей веществ в ана-
аналитических или препаративных масштабах. Применяют неподвижные фазы с
очень маленькими частицами носителя C—10 мкм) и однородными по величи-
2. Способы разделения веществ
95
Раство- Ввод пробы
ригель Насос
Термостат
'Разделительная
колонка
Самописец
Детектор Сборник
фракций
Рис. А.103. Схема прибора для высокоэффективной жидкостной хроматографии.
не, через которые элюирующее средство продавливается под высоким давлени-
давлением E0—500 бар). Для этого имеются автоматизированные продажные приборы,
схема которых представлена на рис. А.103. Колонка состоит обычно из нержа-
нержавеющей стали и имеет при аналитических применениях диаметр от 2 до 5 мм.
Материалы, используемые для носителей, представляют собой плотную равно-
равномерную набивку с высокой разделяющей способностью, благодаря чему впол-
вполне достаточны короткие колонки длиной 5—25 см. Их число теоретических та-
тарелок (см. уравнение [А. 105]) составляет от 1000 до 100 000. ВЭЖХ может осу-
осуществляться как адсорбционная или распределительная хроматография. Чаще
всего применяют обращенные фазы. Для выбора неподвижной фазы и элюента
см. разд. А.2.7.
Смесь, подлежащую разделению, впрыскивают шприцем в начало колонки в
поток элюента. А элюируемые компоненты в конце колонки регистрируются
подходящим детектором. Для детектирования используют соответствующие
свойства веществ, которые можно измерить, например флуоресцентное излуче-
излучение, ионизацию или теплопроводность. В большинстве случаев используют
УФ-поглощение при определенной длине волны (например, 254 нм) или пока-
показатель преломления.
Разделенные вещества проявляются на хроматограмме в виде пиков опреде-
определенного расположения и величины, рис. А. 104. Временем удерживания ?r обоз-
обозначают время, которое проходит от момента ввода пробы до появления макси-
максимума пика; чистое время удерживания ?'r — это время удерживания минус время
протекания потока Ir = t'R + to. Время протекания потока, или мертвое время
to, — это время, которое необходимо подвижной фазе, чтобы пройти через не-
неподвижную фазу в колонке. При постоянной скорости потока элюента время
удерживания можно сопоставить с объемом, который обозначают какудержива-
96
А Введение в лабораторную практику
§
Сигн
*R1
¦4 >
1
Ввод прс
\
>б
Компонент
У
\
R1
1
h,
у
Компс
/
V
мент 2
(-
i
i
ь \
h2
2
Время
Рис. А. 104. Хроматограмма.
емый объем KR. Время удерживания и удерживаемый объем характеристичны
для определяемого вещества, как и значение R? (ср. уравнение А. 100). Они зави-
зависят, конечно, от природы неподвижной фазы, длины колонки, природы и ско-
скорости пропускания подвижной фазы и от температуры.
Из времени удерживания можно вычислить число теоретических тарелок п,
которое так же, как в ректификации (ср. разд. А,2.3.3.1) и противоточном расп-
распределении (разд. А,2.5.3), служит мерой эффективности колонки. При постоян-
постоянных условиях опыта
„ = 16 (—У [А. 105]
где w — ширина пика у основания.
Площадь пика пропорциональна количеству разделяемого вещества и поэто-
поэтому привлекается для его количественного анализа. Современные ВЭЖХ-хрома-
тографы интегрируют площади пиков автоматически. В приближенном виде ее
можно вычислить по методу «треугольника» путем умножения высоты пика h на
половину ширины Ь или вырезая пик и взвешивая его. Высота и площадь пика,
естественно, зависят и от чувствительности детектора к определенному вещест-
веществу, например, при УФ-детектировании — от коэффициента экстинкции при ис-
использованной длине волны. Связь между площадью пика и количеством веще-
вещества следует определять путем построения градуировочного графика, но лучше
путем добавления заведомого количества внутреннего стандарта, площадь пика
которого может быть соотнесена с площадью пиков исследуемых веществ.
С колонками большего диаметра E мм — 20 см) и при рабочих давлениях
между 70 и 80 бар высокоэффективная жидкостная хроматография может быть
использована для разделения веществ в препаративных масштабах A мг — 10 г)
(препаративная ВЭЖХ).
Более дешевым вариантом ВЭЖХ является так называемая жидкостная хро-
хроматография среднего давления (СДЖХ; англ. medium pressure chromatography,
MPLC), при которой работают с давлением 5—50 бар. Она служит главным обра-
образом для выделения и очистки веществ в количестве от 1 мг до 100 г.
2. Способы разделения веществ
97
Для СДЖХ применяют также продажные приборы. В общем используют ци-
цилиндрические стеклянные колонки с рубашкой из искусственных материалов,
диаметром от 15 до 100 мм и длиной от 25 до 50 см. Колонки с большим диамет-
диаметром менее производительны. В качестве неподвижных фаз используются такие
адсорбенты, как силикагель или оксид алюминия, а также полиамиды и обра-
обращенные фазы. Величина зерен лежит между 15 и 50 мкм. Подходящее использо-
использование подвижной фазы может быть определено предварительными опытами с
помощью детектора по теплопроводности, меньших СДЖХ-колонок или ана-
аналитической ВЭЖХ.
Раствор пробы вкалывают в резервуар для проб и, включая его между насо-
насосом и колонкой, загружают пробу в колонку. Элюат, выходящий из колонки,
протекает через УФ-детектор (не пригоден для соединений, не активных в
УФ-спектре) или детектор по лучепреломлению (не пригоден для градиентного
элюирования) и путем автоматического сборника фракций улавливается в выб-
выбранном количестве. Детекторы должны быть приспособлены для большей ско-
скорости пропускания элюата. Сигналы детектора записывает самописец в виде
хромато граммы.
2.7А. ГАЗОВАЯ ХРОМАТОГРАФИЯ
В газовой хроматографии (ГХ) применяют инертный газ в качестве подвижной
фазы, а в качестве неподвижной фазы — твердый адсорбент (газо-твердофазная,
или газоадсорбционная, хроматография) или чаще жидкость на твердом носите-
носителе (газо-жидкостная, или, точнее, газо-жидко-твердофазная, распределитель-
распределительная хроматография).
Она ограничена веществами, которые испаряются без разложения или дают
при разложении идентифицируемые газообразные продукты. Газовая хромато-
хроматография — очень производительный и быстрый способ разделения; она применя-
применяется главным образом для качественного и количественного, а также для препа-
препаративного разделения смесей веществ. Используются продажные приборы,
схематическое изображение которых приведено на рис. А. 106.
В качестве разделительной трубки или колонки применяют трубки из меди,
стали или кварца с диаметром 3—8 мм и длиной 1—6 м, в которых неподвижная
фаза находится на твердом носителе. Также используются так называемые ка-
капиллярные колонки (диаметр 0,1-0,5 мм, длина 10-300 м), которые обладают
Ввод
пробы Детектор
Сборник
фракций
Самописец
Термостат
Рис. А. 106. Схематическое изображение газового хроматографа.
98 А Введение в лабораторную практику
очень высокой разделяющей способностью. В них неподвижная фаза находится
в виде жидкости непосредственно на стенке колонки (тонкопленочные капил-
капилляры) или в тонком слое носителя (тонкослойные капилляры). С наполненны-
наполненными колонками могут быть разделены 0,5-30 мг смеси веществ, на капиллярных
колонках — количества в пределах микрограммов.
Через колонку пропускают газ (азот, гелий, аргон, водород), в верхнюю часть
колонки подается смесь разделяемых веществ, в случае жидкостей обычно вка-
вкалывается. Рабочая температура газового хроматографа (от 0 до 400 °С) должна
быть примерно на 100—200 °С ниже средней температуры кипения разделяемых
веществ. Она поддерживается с помощью термостата, и ее можно непрерывно
повышать в процессе разделения, что соответствует градиентному элюирова-
нию при жидкостной хроматографии.
В качестве неподвижной фазы в газовой распределительной хроматогра-
хроматографии используют органические жидкости с малой упругостью пара (<0,1 кПа,
или <1 мм рт. ст., при рабочей температуре), например, сквалан (разветвлен-
(разветвленный Сзо-алкан), апиезоновые масла (нефтяные фракции), силиконовые масла,
эфиры фталевой кислоты, полигликоли, полиэфиры и др. В качестве носите-
носителей используются вещества, которые при развитой поверхности имеют малую
адсорбционную активность (силикагель, глины). Эти вещества содействуют
значительному увеличению поверхности обмена жидкой фазы при разделе-
разделении, не нарушая при этом равновесного распределения между газом и жид-
жидкостью, обусловленного силами адсорбции.
Для регистрации газообразных компонентов в детекторах после разделения в
принципе можно использовать любое физическое свойство газов или паров. На-
Наиболее приемлемым является измерение теплопроводности (детектор по теплоп-
теплопроводности, катарометр), а при сгорании (или облучении) газа — измерение ио-
ионизации (пламенно-ионизационный детектор, ионизационный детектор облуче-
облучения). ИК-спектроскопия также используется для детектирования, особенно в
хроматографах, которые непосредственно связаны с масс-спектрометром. Сиг-
Сигналы детектора регистрируются с помощью чувствительного автоматического
самописца. Таким образом получают хроматограмму, которая имеет такой же
вид, как и жидкостная хроматограмма (рис. А. 104).
Площадь пиков (пологих и острых) пропорциональна количеству вещест-
вещества. Временем удерживания Ir, как описано в разд. А,2.7.3, называют время,
проходящее с момента ввода пробы до появления максимума пика вещества
(см. рис. А. 104). При постоянной скорости газа-носителя это время соответ-
соответствует объему, обозначаемому как удерживаемый объем Vr. Время удержива-
удерживания (удерживаемый объем) является для данного вещества характеристичес-
характеристической величиной. В практической газовой хроматографии вместо абсолютных
величин имеют дело с относительным удерживанием, которое определяют с
помощью соединения, прибавляемого к определяемым веществам и выступа-
выступающего в качестве стандарта:
[А. 107]
Аналогично процессам ректификации эффективность разделения колонки
для ГЖХ оценивают числом теоретических тарелок п (разд. А,2.3.3.1), которое
2. Способы разделения веществ 99
увеличивается с удлинением колонки и рассчитывается по формуле [А. 105], как
в ВЭЖХ. Кроме того, п зависит от ряда других параметров, таких, как тип и
количество неподвижной фазы, температура колонки, скорость подачи газа-но-
газа-носителя, а также от его природы и давления.
У продажных газовых хроматографов при длине колонки 2 м эффективность
разделения соответствует 2000 теоретических тарелок. При использовании ка-
капиллярных колонок длиной до 30 м эффективность возрастает в 10—20 раз. Для
прецизионных приборов она может достигать 500 000 теоретических тарелок.
Эффективность колонки в большой степени зависит от выбора подходящей
неподвижной фазы. При этом следует руководствоваться следующими прави-
правилами:
• смесь веществ с неполярными молекулами можно разделить, используя
жидкую фазу, молекулы которой также неполярны, причем последова-
последовательность разделения соответствует температуре кипения разделяемых
веществ;
• на неполярной жидкой фазе полярные молекулы перемещаются быст-
быстрее, чем неполярные;
• с увеличением полярности неподвижной жидкой фазы полярные ком-
компоненты удерживаются сильнее, чем неполярные (при одинаковой тем-
температуре кипения).
Так, для разделения углеводородов и их производных, молекулы которых об-
обладают малой полярностью (например, галогенопроизводные углеводородов),
пригодны парафиновые и силиконовые масла, трикрезилфосфат. Диалкилфта-
латы рекомендуется применять для разделения кислородсодержащих соедине-
соединений (простых и сложных эфиров, альдегидов, кетонов и др.). Смеси, содержа-
содержащие воду, хорошо разделяются на полигликолях.
Если селективность неподвижной фазы (т. е. ее способность различным образом
удерживать вещества, имеющие одинаковые температуры кипения, но отличающи-
отличающиеся своей химической структурой) является недостаточной, для разделения исполь-
используют комбинации колонок с неподвижными фазами различной полярности.
Газовая хроматография применяется для качественного и количественного
анализа смесей органических веществ. Важнейшей задачей при качественном
анализе органических соединений является идентификация и определение чис-
чистоты веществ; в последнем случае вещество хроматографируют по крайней мере
дважды на двух неподвижных фазах различной полярности. Если в обоих случа-
случаях на хроматограмме появляется только один пик, то вещество обычно можно
рассматривать как индивидуальное.
Приступая к идентификации веществ, предварительно определяют относи-
относительное удерживание этих веществ. В качестве стандарта вполне пригодны
нонан и другие углеводороды нормального строения. Сравнение эксперимен-
экспериментальных значений относительных удерживаний с табличными данными часто
позволяет сделать вывод о строении анализируемых веществ. Вероятность оши-
ошибочного вывода сводится к минимуму при повторном разделении смеси на не-
неподвижной фазе с иной полярностью. При добавлении к идентифицируемому
веществу аутентичного соединения значительно повышается надежность полу-
100 А Введение в лабораторную практику
ченных данных. В последнее время для качественного анализа фракций, получа-
получаемых с помощью газовой хроматографии, используют масс-спектрометрию
(разд. А,3.7); в этом случае на выходе газового хроматографа устанавливают
масс-спектрометр.
Для количественной оценки хроматографических данных в первом приближе-
приближении можно использовать отношение площадей пиков на хроматограмме.
Если предположить что а) число компонентов в пробе соответствует числу выде-
выделенных пиков, б) показания детектора находятся в линейной зависимости от коли-
количества пробы и в) в анализируемой смеси находятся химически родственные соеди-
соединения, то площади пиков Ft относятся друг к другу как массы компонентов
юо/;
/и,- = [А. 108]
В современных приборах площади пиков определяют измерением при помощи
интегратора. В то же время, они могут быть вычислены как площадь треугольника
путем умножения высоты h на полуширину Ь (ср. рис. А. 104) или взвешиванием
бумаги, вырезанной из хроматограммы по контурам кривой, образующей пик.
Абсолютную концентрацию одного или нескольких компонентов в анализи-
анализируемой пробе можно определить, используя либо внешнюю (построение граду-
ировочного графика), либо внутреннюю градуировку (метод внутреннего стан-
стандарта). Последний метод основан на том, что к анализируемой смеси добавляют
точно дозированное количество соединения, принятого за стандарт. Последнее
должно быть химически родственным с разделяемыми компонентами и иметь с
ними близкое время удерживания, но не перекрываться на хроматограмме ни с
одним из пиков анализируемых компонентов.
Из отношения площадей получают массовые проценты неизвестных компо-
компонентов
Mi 100 Ft
т,= [А. 109]
М„ + М, FCT
где М„ — количество вещества, принятое за стандарт.
При количественном анализе веществ, сильно отличающихся по своей
структуре, нужно ввести поправочный коэффициент, учитывающий специфи-
специфические особенности детектирования данного соединения.
Газовая хроматография применяется и для препаративного разделения смесей
веществ. Вещества, выходящие с колонки, вымораживают с помощью ловушек.
3. ОПРЕДЕЛЕНИЕ ФИЗИЧЕСКИХ СВОЙСТВ
ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Характеристику органического соединения нельзя считать исчерпывающей, если
для него приведены лишь элементный состав и молекулярная масса. Для иденти-
идентификации соединения необходимо определить также другие данные, прежде всего
физические свойства. Важнейшие из них — это температура кипения, температу-
температура плавления, плотность, показатель преломления, для некоторых соединений
вращение плоскости поляризации света, УФ-, ИК- и ЯМР-спектры, а также рас-
распад ионизированных молекул в газовой фазе (масс-спектры).
3. Определение физических свойств органических соединений 101
Полученные при идентификации данные могут одновременно служить и
для оценки чистоты вещества. Вещество можно признать чистым только тогда,
когда физические константы его не меняются после повторной очистки —
перегонки, перекристаллизации, хроматографирования и т. д.
3.1. ТЕМПЕРАТУРА ПЛАВЛЕНИЯ
Температурой плавления соединения называют температуру, при которой его
твердая фаза находится в равновесии с собственными расплавом. Чистые веще-
вещества обладают четко выраженной температурой плавления; ее точное определе-
определение (±0,01 °С) возможно путем снятия кривых плавления.
При применении обычных простых методик определения температуры
плавления, которые описаны ниже, плавление вещества наблюдается в интер-
интервале температур от нескольких десятых градуса до целого градуса. Незначитель-
Незначительные загрязнения иногда сильно понижают температуру плавления данного сое-
соединения; при загрязнении вещества более высокоплавкими примесями тоже
наблюдается, как правило, понижение температуры плавления. Кроме того,
в этих случаях наблюдается значительное увеличение интервала плавления
(более чем на 1 °С). Такое явление используется при установлении идентичнос-
идентичности двух веществ с одинаковой температурой плавления. Для этого смешивают
равные количества сравниваемых веществ (проба смешанного плавления). Если
температура плавления смеси остается неизменной, то делают заключение об
идентичности обоих веществ. Если же температура плавления пробы ниже тем-
температуры плавления исходных веществ, то, следовательно, имеются два разных
вещества. Однако для смесей изоморфных соединений даже разных по хими-
химическому составу понижение температуры плавления не обнаруживается.
Надо выработать привычку первоначально подтверждать идентичность двух
веществ, используя эти простые и быстрые методы, а уже потом привлекать
более сложные спектроскопические методы.
Многие органические соединения плавятся с разложением, что, как прави-
правило, легко обнаруживается по окрашиванию расплава или выделению газов.
В этом случае температура разложения обычно характеризуется некоторым
интервалом, сильно зависит от скорости нагревания (быстрое нагревание при-
приводит к более высокому значению температуры разложения) и поэтому очень
часто не может быть точно воспроизведена. Для некоторых веществ вообще
нельзя говорить о точке фазового перехода, так как при сильном нагревании
они обугливаются.
Между температурой плавления вещества и его молекулярным строением существует оп-
определенная зависимость. Замечено, что вещества с симметричными молекулами плавятся при
более высокой температуре, чем вещества менее симметричного строения. Так, например,
парафины нормального (неразветвленного) строения имеют более высокую температуру плав-
плавления, чем их изомеры с тем же числом атомов углерода. У стереоизомерных соединений
транс-изомер, как правило, плавится при более высокой температуре [например, для малеино-
вой кислоты (чис-форма) т. пл. 130 °С, а для фумаровой кислоты (транс-форма) т. пл. 287 °С].
Температура плавления вещества растет с увеличением степени ассоциации молекул. Так,
сложные эфиры, неспособные к образованию водородных связей, плавятся при значительно
более низких температурах, чем соответствующие карбоновые кислоты.
102
А Введение в лабораторную практику
3.1.1. ОПРЕДЕЛЕНИЕ ТЕМПЕРАТУРЫ ПЛАВЛЕНИЯ В КАПИЛЛЯРЕ
Тонкоизмельченное и хорошо высушенное вещество слоем 2—4 мм помеща-
помещают в запаянную с одного конца капиллярную трубку (диаметр ~1 мм). Для
этого открытый конец капилляра погружают в пробу вещества и набранное в
верхний конец капилляра вещество осторожно стряхивают на дно капилляра
(или бросают капилляр заплавленным концом вниз несколько раз через
длинную стеклянную трубку, вертикально установленную на жесткой под-
подставке).
Температуру плавления веществ, склонных к возгонке, определяют в капил-
капиллярах, запаянных с обоих концов, запаянный капилляр с веществом надо пол-
полностью погружать в нагревательную баню.
Капилляр для определения температуры плавления в простейшем случае
закрепляют на термометре (желательно, чтобы термометр был более точным)
при помощи резинового колечка (рис. А. 110). Проба вещества должна находить-
находиться при этом на уровне ртутного шарика термометра1.
Затем в стакане с парафиновым или силиконовым маслом (теплопередающая
среда, позволяющая проводить нагревание до -250 °С) медленно D—6 град/мин,
а вблизи точки плавления — 1—2 град/мин) повышают температуру, доводя
вещество до плавления.
Мешалка
Рис. А.110. Простейший прибор для
определения температуры плавления
низкоплавких веществ.
Рис. А. 111. Прибор Тиле
для определения
температуры плавления.
1 В наших лабораториях термометр с капилляром помещают в длинную толстостенную
пробирку, которая в верхней части пришлифована к длинногорлой круглодонной колбе. В эту
внешнюю колбу наливают подходящую жидкость (в качестве теплопередающей среды). Кол-
Колба и пробирка должны сообщаться с воздухом, иначе при нагревании возможен взрыв! —
Прим. перев.
3. Определение физических свойств органических соединений 103
Удобнее определять температуру плавления в приборе Тиле (рис. А. 111),
в котором более равномерен теплоперенос.
Ц При определении температуры плавления обязательно надевать защитные
I очки!
Температурой плавления при использовании описанной методики считается
температура в момент полного расплавления вещества (просветвления распла-
расплава). Точность определения температуры плавления при этом составляет не более
±0,5 °С. Если испытывают не совсем чистое вещество, указывается температур-
температурный интервал от момента появления первых капель жидкой фазы до образова-
образования прозрачного расплава. Определенная так температура плавления (темпера-
(температурный интервал) обычно примерно на 1 "С выше, чем установленная при помо-
помощи нагревательного столика (разд. А,3.1.2).
Для определения температуры плавления высокоплавких соединений
(т. пл. >250 °С) нагревание осуществляют в металлическом (медном или алюми-
алюминиевом) блоке. Капилляр и термометр вводят в соответствующие отверстия бло-
блока, еще одно отверстие служит для наблюдения за процессом плавления. (Это
отверстие полезно закрыть слюдяными стеклами.)
Определение температуры плавления в капилляре возможно без особенно
больших трудностей до —50 °С. В простейшем случае такие определения прово-
проводят в большом стеклянном стакане (рис. АЛЮ), наполненном охлаждающей
смесью из сухого льда с метанолом. Сначала охлаждают вещество в капилляре до
затвердевания, а затем при постоянном перемешивании дают охлаждающей
смеси медленно нагреваться.
Поскольку термометр не полностью (не на всю высоту ртутного столбика)
погружен в жидкость, необходимо ввести поправку в его показания. Если 9набл —
наблюдаемая температура плавления, истинную температуру плавления 9иет
рассчитывают по формуле
9„ст=Э„абл + иу(9„абл-Эср) [А. 112]
где 9ср — средняя температура выступающего индикаторного столбика, у —
константа, зависящая от типа термометра (для ртутного термометра из иенского
стекла у = 0,00016), п — число делений (в градусах) шкалы выступающего из
жидкости индикаторного столбика.
3.1.2. ОПРЕДЕЛЕНИЕ ТЕМПЕРАТУРЫ ПЛАВЛЕНИЯ С ПОМОЩЬЮ МИКРОСКОПА
С НАГРЕВАТЕЛЬНЫМ СТОЛИКОМ
Использование оптического микроскопа с 50—100-кратным увеличением для
изучения процесса плавления имеет ряд преимуществ по сравнению с определе-
определением температуры плавления в капилляре: количество вещества, необходимое
для определения, крайне незначительно (достаточно нескольких миллиграммов
или даже микрограммов). Все изменения, происходящие с веществом в процессе
нагревания (отщепление воды от гидратов, полиморфные превращения, возгон-
возгонку и разложение), можно отчетливо наблюдать под микроскопом (рис. А. 113).
104
А Введение в лабораторную практику
Освещение
микроскопа
Ввод вещества
Защитное кольцо
Приспособление
для слежения за
температурой
термометра
Фильтр
Ввод для термометра
Рис. А.113. Нагревательный столик с микроскопом
для определения температур плавления.
Дня этих целей микроскоп должен иметь специальный предметный столик
(конструкции Кофлера и Бётиуса), в который вмонтирован электрический нагрева-
нагреватель; скорость нагревания последнего регулируется с помощью реостата. В боковое
отверстие нагревательного столика помещают термометр, предварительно програ-
дуированный на этом же столике с использованием подходящих соединений с из-
известной температурой плавления; температуры плавления неизвестных веществ,
полученные таким методом, уже содержат поправку на выступающий столбик.
Подготовка пробы к исследованию заключается во внесении нескольких
кристалликов между специальными плоскими предметными и покровным стек-
стеклами. Большие кристаллы предварительно растирают между стеклами. Пред-
Предметное стекло вместе с направляющим кольцом помещают на покрытый стек-
стеклянной пластиной нагревательный столик; изображение вещества должно нахо-
находиться в поле зрения микроскопа.
Температуру плавления можно определять двумя различными способами.
Согласно одному из них, температуру повышают непрерывно (вблизи точки
плавления со скоростью 2—4 град/мин) до полного расплавления вещества. На-
Началом плавления считают температуру, при которой начинают округляться углы
и грани более крупных кристаллов. Точка, в которой кристаллы полностью
исчезают, считается концом плавления вещества. Согласно второму способу, с
помощью реостата устанавливают такую температуру, при которой достигается
равновесие между твердой и жидкой фазами.
3. Определение физических свойств органических соединений 105
Температуру плавления сублимирующихся веществ определяют в плоских
запаянных кюветах (кюветах Фишера).
При проведении пробы смешанного плавления на предметное стекло поме-
помещают по одному кристаллику каждого из двух испытуемых веществ и тщательно
смешивают их, слегка нажимая и передвигая покровное стекло.
3.2. ТЕМПЕРАТУРА КИПЕНИЯ
Температура кипения вещества в отличие от температуры плавления очень силь-
сильно зависит от давления (ср. [А.45]), и определить этот параметр не так просто.
В большинстве случаев за температуру кипения вещества принимается тем-
температурный интервал при перегонке вещества. При этом погрешности опреде-
определения связаны с перегревом паров и недостатками прибора (например, непра-
неправильное положение термометра, см. разд. А,2.3.2.2), что искажает истинное зна-
значение температуры кипения. Кроме того, источниками погрешностей являются
неправильное определение поправки на показания термометра (ср. [АЛ 12]) или
неправильное измерение давления (например, при ошибочных показаниях ма-
манометра на вакуумной установке). Поэтому в литературе часто для одного и того
же вещества указываются разные температуры кипения.
Влияние загрязнений на температуру кипения сильно зависит от характера
примесей. Так, значительное влияние оказывают остаточные количества легко-
легколетучего растворителя. Напротив, примесь вещества с той же самой температу-
температурой кипения вообще может не влиять (при идеальном поведении) на температу-
температуру кипения (см. закон Рауля, уравнение [А.48]). Как правило, примеси в незна-
незначительных количествах оказываются для температуры кипения гораздо менее
существенными, чем для температуры плавления.
По указанным причинам температура кипения является гораздо менее
надежным способом идентификации и критерием чистоты вещества, чем темпе-
температура плавления.
Размеры молекул и межмолекулярное взаимодействие — вот что в значительной мере
обусловливает величину температуры кипения. Так, например, температура кипения нор-
нормальных парафинов (С4-С|2) возрастает на 20-30 "С при увеличении длины углеродной цепи
на один С-атом. Соединения с разветвленными молекулами, как правило, обладают более
низкими температурами кипения, чем соответствующие соединения линейного строения.
При одинаковом числе атомов углерода температура кипения увеличивается в ряду, простые
эфиры < альдегиды < спирты, так как межмолекулярное взаимодействие (ассоциация) возрас-
возрастает в том же порядке (водородные связи в спиртах).
Температуру кипения можно точно определить с помощью эбуллиометров.
Принцип их действия основан на том, что жидкость нагревается (в приборе с об-
обратным холодильником) до кипения и измеряется температура. При соответ-
соответствующей конструкции прибора исключаются тепловые потери и перегрев пара.
Однако для работы в таких приборах обычно необходимы большие количества
вещества (не меньше нескольких миллилитров).
Если в распоряжении имеется достаточное количество жидкости (>10 мл), то
проще всего снять в приборе для перегонки кривую кипения. При этом надо об-
обратить внимание на то, чтобы шарик термометра полностью омывался парами,
106 А Введение в лабораторную практику
смачивался конденсирующейся жидкостью и не был погружен слишком глубо-
глубоко в перегретый пар (разд. А,2.3.2.2).
3.3. РЕФРАКТОМЕТРИЯ
Для идентификации жидких веществ и проверки их чистоты можно использо-
использовать также определение показателя преломления п. Если луч монохроматическо-
монохроматического света проходит через границу раздела двух сред (рис. АЛ 14), то он отклоняет-
отклоняется от первоначального направления по закону Снеллиуса
С\ sin a
— = = п
с2 sin р
где ci и с2 — скорости света в средах 7 и 2 В общем случае одной из сред служит
воздух.
Показатель преломления сильно зависит от температуры. У органических
жидкостей с ростом температуры на 1 °С он падает на ~4 • 10~4 — 5 • 10~4. Кроме
того, показатель преломления сильно меняется в зависимости от длины волны
света (дисперсия). Обычно указывается показатель преломления для спектраль-
спектральной линии желтого излучения натрия (D-линия, 589 нм). Температуру измере-
измерения и длину волны света (или спектральную линию) отмечают при значении
показателя преломления соответственно верхним и нижним индексами, напри-
например Яо25-
Показатели преломления определяют с помощью рефрактометров. В лабо-
лабораториях органической химии чаще всего это рефрактометр Аббе. Принцип из-
измерения в этом рефтактометре основан на определении предельного угла пол-
полного внутреннего отражения; кроме того, конструкция этого прибора позволяет
работать в полихроматическом свете (например, при дневном освещении), при
этом измеренное значение показателя преломления и есть значение для D-ли-
нии. Для измерения необходимо всего нес-
несколько капель жидкости, точность измере-
измерения составляет ±0,00011. Чтобы добиться та-
такой точности, во время измерения следует
поддерживать с помощью термостата посто-
постоянную температуру (±0,2 °С). Показатель
преломления целесообразно измерять при 20
или 25 "С, а у низкоплавких твердых веществ
Среда 2 несколько выше температуры плавления.
Показатель преломления растворов зави-
зависит от концентрации. Поэтому рефракто-
Рис. А.114. Преломление света метрик» используют также для определения
на границе двух сред. концентрации растворов, для проверки чис-
1 Время от времени прибор проверяют, например, измеряя показатель преломления жид-
жидкости с точно известным показателем преломления (в частности, для дистиллированной воды
при 20 °С он составляет 1,3330), и при необходимости юстируют.
3. Определение физических свойств органических соединений 107
тоты веществ и контроля за процессами разделения; например, так можно конт-
контролировать перегонку, проводимую в аналитических целях. Показатель прелом-
преломления бинарной смеси линейно зависит от объемной концентрации компонен-
компонентов, если только при их смешивании не происходит изменения объема. В других
случаях появляются отклонения от линейной зависимости и для точных опреде-
определений концентрации необходимо строить градуировочный график.
Зная показатель преломления вещества п и его плотность D, по уравнению Лорентца —
Лоренца можно рассчитать молекулярную рефракцию Mr вещества, которая является констан-
константой, не зависящей от температуры:
п2-\ М 3
MR = —-—=-nNAa [A.115]
и2+2 D 4
где М — молекулярная масса, Л^ — постоянная Авогадро; D — плотность.
Молекулярная рефракция позволяет сделать ряд преложений о строении молекулы (более
подробно с этим можно ознакомиться в учебниках). Кроме того, она прямо пропорциональна
электронной поляризуемости молекулы а (ср. [А. 115]).
3.4. ПОЛЯРИМЕТРИЯ
Определенные химические соединения обладают «оптической активностью»,
т. е. если через них пропускать плоскополяризованный свет, то они поворачи-
поворачивают плоскость колебаний света на определенный угол — угол вращения а.
Оптическая активность возникает в тех случаях, когда молекула соответствую-
соответствующего соединения хиральна. Для более подробного ознакомления с этими
вопросами следует обратиться к разд. В,7.3.1 о хиральности и энантиомерии!
Вращение плоскости поляризации может происходить вправо (обозначается
знаком «+»; для наблюдателя — по часовой стрелке) или влево (обозначается
знаком «—»). Для данного растворителя угол вращения а зависит от концентра-
концентрации с (в граммах на 100 мл раствора), толщины слоя / вещества (в дециметрах),
температуры / и длины волны X1. Для данных длины волны и температуры угол
вращения определяют по формуле
а = [а]к'Ш 1АЛ16]
где [а\х' — удельное вращение. Измерения обычно проводят для D-линии натрия
при температурах 20 или 25 °С, что обязательно указывается, например [а]о20-
Угол вращения а определяют с помощью поляриметров.
Принципиальная схема визуального поляриметра (рис. А. 117) включает ис-
источник монохроматического света 1, излучение которого поляризуется при про-
прохождении через призму Николя 2 (поляризатор); далее поляризованный луч
проходит через кювету 3 с раствором исследуемого вещества. Происходящее при
этом отклонение плоскости поляризации света определяют с помощью второй,
1 Зависимость вращения плоскости поляризации от длины волны называют дисперсией
вращения (литература по этому вопросу приведена в конце главы).
108 А Введение в лабораторную практику
Рис. А.117. Схема поляриметра.
вращающейся призмы Николя 4 (анализатора), которая жестко связана с граду-
градуированной шкалой. Наблюдаемое при этом через окуляр 5зрительное поле, раз-
разделенное на две или три части различной яркости, следует сделать равномерно
освещенным. Величину необходимого для этого поворота анализатора Усчиты-
Усчитывают со шкалы. Для проверки нулевой точки прибора проводят аналогичные из-
измерения без исследуемого раствора.
Полученный таким образом угол вращения +сс может в равной мере соотве-
соответствовать как правому вращению на угол а (или 180 °С+а), так и левому враще-
вращению на угол 180°—а (или 360°—а). Точное направление вращения определяют
поэтому при помощи повторного измерения, которое проводят либо с половин-
половинной толщиной слоя, либо с половинной концентрацией жидкости. Если при
этом получают угол вращения а/2 (или а/2+90°), то можно считать, что вещест-
вещество является правовращающим; если же новый угол вращения равен 90°—а/2
(или 180°-а/2), то имеется левовращающее вещество. Поскольку удельное вра-
вращение не очень сильно зависит от температуры, измерения обычно проводят, не
термостатируя кювету. При более точных измерениях термостатирование
необходимо.
Удельное вращение значительно зависит от растворителя, а в некоторых слу-
случаях и от концентрации вещества, так как растворенное вещество может взаимо-
взаимодействовать с растворителем. Поэтому, приводя данные по оптическому враще-
вращению, надо всегда указывать применяемый растворитель и концентрацию веще-
вещества в растворе, например [а]о25 27,3° в воде (с = 0,130 г/мл).
Поляриметрические измерения применяются не только для проверки чисто-
чистоты оптически активных веществ, но и для определения количественного содер-
содержания их в растворах. Например, этим методом можно поляриметрически опре-
определять содержание сахара в растворах (сахариметр).
3.5. ОПТИЧЕСКАЯ СПЕКТРОСКОПИЯ
Если соединение облучать электромагнитными волнами, оно может вступать во
взаимодействие с излучением и поглощать энергию, если одна из его собствен-
собственных частот соответствует частоте облучения. В зависимости от частоты погло-
поглощенного излучения различают рентгеновскую, УФ, видимую, ИК и микровол-
микроволновую спектроскопию.
Для обычных исследований в органической химии особенно интересны
ультрафиолетовая (УФ) область, видимая область (вид.) и инфракрасная область
(ИК) спектра, так как имеются недорогие спектрометры и получаемые спект-
спектральные данные могут быть относительно просто эмпирически использованы.
3. Определение физических свойств органических соединений 109
Такие приборы работают следующим образом: полихроматическое излуче-
излучение, поступающее из источника света, проходит через пробу вещества, где пог-
поглощается часть излучения, которая по частотам соответствует собственным воз-
возбужденным частотам вещества. В монохроматоре (обычно включенным вслед)
полихроматический свет разделяется на отдельные частоты, затем усиливается
и, наконец, измеряется относительная интенсивность с лучем сравнения, кото-
который непосредственно посылается через монохроматор (ИКС) или через кювету
сравнения, которая содержит только растворитель (УФ- и видимая спектроско-
спектроскопия). В последнем случае собственное поглощение растворителя и отраженные
потери компенсированы на поверхности кюветы (двухлучевой спектрометр).
Монохроматор шаг за шагом устанавливается на отдельные частоты и таким об-
образом проходят интересующую спектральную область. Интенсивность погло-
поглощения регистрируется по длине волны или частоте в виде кривой (спектр) или в
цифровой форме вычислительной машиной.
В новейших спектрометрах для УФ/вид.-области вместо монохроматора ис-
используется дисперсионный элемент, разделяющий полихроматический свет на
отдельные частоты, которые регистрируются определенным фотодиодом, уси-
усиливаются электрически и записываются. С такими приборами УФ/вид.-спектр
может быть получен за миллисекунды. И в ИК-спектрометрах имеются даль-
дальнейшие усовершенствования (ИК-спектроскопия с фурье-преобразованием,
ИКФП).
В УФ/вид.- и ИК-спектрах положение сигнала на координате энергии (дли-
(длина волны, т. е. частота) и его интенсивность имеют значение. Поглощенная
энергия Доопределяется соотношением
he
AE=hv = — [АЛ 18]
А,
где v — частота, А. — длина волны, h — постоянная Планка, с — скорость света.
Путем умножения на постоянную Авогадро получают молярную энергию из-
излучения для соответствующей длины волны.
119,6-Ю3
АЕ= кДж/моль [А. 119]
^(нм)
УФ/вид.- и ИК-спектрометры выдают график интенсивности поглощения
от длины волны и(или) от частоты. Регистрация частоты имеет то пре-
преимущество, что абсцисса представляется линейной от энергии. Частота имеет
невероятно большие числовые значения и поэтому применяют волновое число
v (см~'):
v 107
v = — = см-1 [А. 120]
Ц)
Волновое число v хорошо применять для ИК-спектров, но в случае УФ/вид .-спек-
.-спектров представляется еще очень большой величиной; в этих случаях часто
используют 103 см-1 = 1 кК (килокайзер); см. табл. А.121.
110
А Введение в лабораторную практику
Таблица А. 121
v, 103 см-1
X, нм
Д?, кДж/моль
. Частоты и области
Вакуум-УФа
>55
<180
>665
энергии
УФ
55-28
180-360
665-335
в оптической спектроскопии
Видимая область
28-13
360-750
335-160
ик
<13
>750
<160
а Вакуумная УФ-область в органической химии пока не играет роли для рутинных исследо-
исследований.
Поглощательная способность вещества характеризуется величиной — опти-
оптической плотности (А), и если вещество не склонно к ассоциации, то А следует
закону Ламберта—Бера:
А = lg —= zed
[А. 122]
где /0 — интенсивность света, поступающего в пробу, / — интенсивность про-
прошедшего света, б — молярный коэффициент поглощения (ранее коэффициент
экстинкции), с — концентрация вещества, d — толщина поглощающего слоя.
Оптическая плотность (экстинкция) пропорциональна концентрации с, ши-
ширине пленки d(cM) и молярному коэффициенту поглощения е (обычно приводит-
приводится в см2/моль); е — это константа вещества, зависящая от длины волны.
Оптическая плотность смеси веществ в случае отсутствия сильных межмоле-
межмолекулярных взаимодействий складывается аддитивно из оптических плотностей
отдельных компонентов.
Коэффициенты поглощения в УФ — видимой области могут достигать очень
больших величин вплоть до 105 см2/моль, так что в УФ/вид.-области возможно
очень чувствительное определение концентрации.
Энергия, поглощенная в оптической области, распределяется на электрон-
электронные и молекулярные колебания. Поглощения в УФ/вид.-области обусловлены в
первую очередь возбуждением электронных переходов. Поэтому УФ/вид.-спек-
роскопия является электронной спектроскопией. Колебания молекул проявля-
проявляются лишь ограниченно (так называемые тонкие структуры). ИК-спектроско-
пия, напротив, регистрирует в основном ядерные (молекулярные) колебания, в
то время как в ближней ИК-области G50—1200 нм) электронные переходы
протекают во всех случаях.
Поскольку электронные переходы не связаны так непосредственно со стро-
строением молекул, как с молекулярными колебаниями, УФ/вид.-спектроскопия
для определения структуры соединения менее значима, чем ИК-спектроскопия.
Имеются новейшие спектрометры, снабженные компьютерами, в памяти
которых хранятся библиотеки большого числа типичных спектров. Структуры
неизвестных соединений можно тогда выяснить путем сравнения их спектров с
ранее полученными.
3. Определение физических свойств органических соединений
111
3.5.1. УЛЬТРАФИОЛЕТОВАЯ И ВИДИМАЯ СПЕКТРОСКОПИЯ
Большинство органических соединений, которые содержат двойные связи
и(или) свободные электронные пары, поглощают в области выше 180 нм. Груп-
Группы атомов, ответственные за поглощение света, называются хромофорами.
При облучении светом молекула переходит из основного состояния в воз-
возбужденное состояние, которое можно рассматривать как новую молекулу с иной
структурой (длины связей, величины углов и распределение электронов). Воз-
Возбужденное состояние имеет более высокую энергию, время его жизни измеряет-
измеряется обычно наносекундами. Возбужденные состояния могут вступать в химичес-
химические реакции (фотохимия).
Совозбужденные молекулярные колебания — прежде всего из-за обмена коле-
колебаниями с растворителями — чаще всего неразрешимы, но можно наблюдать по-
полосы поглощения, перекрывающиеся иногда в неполярных растворителях с
«тонкими» структурами, которые отражают эти молекулярные колебания,
ср. рис. А.123.
Согласно простой качественной квантовохимическои модели световое воз-
возбуждение связано с переходом одного электрона из занятой молекулярной
200
А, нм 1
250 300
400 500
и -
3 -
en
2 —
1 -
1
" /
1/
-
-
-
1 1 1 1
1 1
/
/
/\
\
1 1 1 1
1 1 1 1 II
\ —
\
1
1
1
—н
\
1
\ \
\ \
\ \
\ \
\ \
\ \
\ \
\ \
\ \
\ \
\ Jc
i i i i i i i i
i i l iii
сн2=снч
вгексане
зСС^СН^
в гептане
/
/
/
, 111 ,1111
сн=о -
сн=сн2
-
-
\
S
1 1
1 1 1 1 1
55000 50000 45000 40000 35000 30000 25000 20000
•*—- v,см
Рис. А.123. УФ-спектры акролеина и пентадиена-1,3.
112
А Введение в лабораторную практику
I
п
п*
п ¦
тт
*
тг
пп - лл -
Основное возбужденное возбужденное
состояние состояние состояние
Рис. А.124. Основные и наиболее бедные энергией возбужденные состояния
акролеина (модель согласно методу МО).
орбитали основного состояния в антисвязывающую молекулярную орбиталь,
ср. рис. А. 124. Антисвязывающие орбитали обычно обозначаются звездочкой.
Можно различать переходы возбужденных состояний следующих типов:
тсл*-переходы обычно е > 104 см2/моль (рис. А. 123)
Я7г*-переходы е < 1000 см2/моль, например, в карбонильных соедине-
соединениях (рис. А. 123)
яа*-переходы е < 1000 см2/моль, например, в насыщенных спиртах,
аминах
аа*-переходы s > 104 см2/моль в насыщенных соединениях
ст*-Переходы лежат много ниже 200 нм и для рутинных исследований не играют
роли.
Различная интенсивность полос поглощения (ср. рис. А. 123) связана с тем,
что электронные переходы подчиняются условиям симметрии. Различают «раз-
«разрешенные» переходы (е > 104 см2/моль) и более или менее строго «запрещенные»
переходы (е < 104 см2/моль). ил*-Переходы карбонильных групп относительно
строго запрещены (s = 10 н- 100 см2/моль).
лл*-Состояния обычно более поляризованы, а mt'-состояния менее поляр-
ны, чем соответствующие основные состояния. Поэтому тгл'-полосы поглоще-
поглощения при повышении полярности растворителя сдвигаются в сторону более
низкой энергии (в сторону более длинных волн — батохромный, или положи-
положительный сольватохромный, сдвиг); ятс'-полосы, напротив, сдвигаются в сторону
коротких волн {гипсохромный, или отрицательный сольватохромный, сдвиг).
Это явление, связанное с различием интенсивностей разрешенных лл*-сдви-
гов и запрещенных «я'-переходов, может служить для идентификации типа
возбуждения.
Особенно большие батохромные, а также гипсохромные эффекты появляют-
появляются, когда при возбуждении светом один электрон далеко переходит от электро-
3. Определение физических свойств органических соединений
113
нодонора к электроноакцептору в той же молекуле (в солях также межмолеку-
лярно); это так называемые переходы с переносом заряда, которые наблюдают-
наблюдаются, например, в и-нитроанилине (положительная сольватохромия) или в
1-метил-4-метоксикарбонилпиридинийиодиде I или пиридинио-феноляте II
(сильная отрицательная сольватохромия). Так как сольватохромный сдвиг по-
повышается с возрастающей полярностью растворителя, то I по Косоверу (Z-зна-
(Z-значение) и II по Рейхардту и Димроту (?т-значение) могут быть использованы для
характеристики полярности растворителя (ср. разд. В.3.3).
СООСНз
[А. 124а]
Несмотря на то, что УФ/вид.-спектроскопия не очень пригодна для структур-
структурных анализов, все же можно делать некоторые общие заключения о связи между
молекулярной структурой и УФ/вид. -спектрами (ср. также литературные ссылки в
разд. А,6): ял*-переходы в УФ/вид.-области можно обнаружить. Несопряженные
двойные и тройные связи поглощают в области ниже 200 нм. Их поглощение
сдвигается батохромно путем сопряжения с другими двойными связями (С=С,
ОС, С=О, C=N, N=N). Причина этого объясняется диаграммой на рис. А.125,
где показано, как система я-электронов путем сопряжения двух изолированных
двойных связей объединяется. Некоторые примеры сопряженных ненасыщенных
соединений сопоставлены в табл. А. 126. Из нее видно, что в сопряженных полие-
нах батохромное влияние дополнительных двойных связей становится меньше.
Р-Каротин A1 сопряженных двойных связей) имеет в наиболее длинноволновой
области максимум поглощения при 494 нм (б = 150 000 см2/моль), а
при бесконечном сопряжении границу конвергенции можно ожидать при
610 нм. Молярный коэффициент поглощения повышается с увеличением
числа двойных связей. Подобным же образом, как и дополнительные двой-
двойные связи, действуют так называемые ауксохромы, группы, усилива-
с=с-с=с с=с
Рис. А. 125. Действие дополнительных сопряжений на поглощение света в олефинах
и карбонильных соединениях.
114
А Введение в лабораторную практику
Таблица А. 126. Влияние увеличивающегося сопряжения в полиенахA), цианинах (II) и
оксонолах (Ш)а
Число двойных
связей в молекуле
0
1
2
3
4
5
6
Xmax, HM (lge)
I (в гексане)
174 D,38)
227 D,38)
275 D,48)
310 D,88)
342 E,09)
380 E,17)
II (вдихлорметане)
224 D,16)
312 D,81)
416 E,08)
519 E,32)
625 E,47)
734 E,55)
III (в диметилформамиде)
190
268 D,43)
362 D,75)
455 D,88)
548 D,80)
644
сн
сн3
CH,
сн
СН-,'
ющие окраску, например, ОН, OR, !SR, NR2, если возможно сопряжение со сво-
свободной парой электронов двойных связей. Подобные группы содержатся во мно-
многих красителях и существенно ответственны за характер красителя. Два таких ти-
типа красителей (полиметиновые красители) — цианины (II) и оксонолы (III) —
приведены в табл. А. 126. В полиметинах действие дополнительных группировок
с двойными связями остается постоянным, так что возможно получение краси-
красителей, которые поглощают далеко в ИК-области (выше 1200 нм).
Изолированные С=О-группы альдегидов и кетонов показывают
тс*-поглощение в относительно узкой области 280-300 нм. Несмотря на то что
Таблица А. 127. Поглощение хромофоров с пп*
родах — растворителях)
Группа атомов
>=°
c=s
C=N—
—N=N—
—N=O
0,°
—N
О0
280
500
240
350
660
270
-связями (е
! алифатических углеводо-
lge
1,3
1,0
2,2
1Д
1,3
1,3
3. Определение физических свойств органических соединений
115
эти переходы запрещены и полосы из-за этого только слабые, УФ-спектроско-
пия может здесь внести ценный вклад в анализе функциональных групп. Это
справедливо и для других групп, ср. табл. А. 127.
В области С=1Ч-хромофоров поглощают и семикарбазоны, и тиосемикарбазо-
ны (в которых кроме того проявляется полоса поглощения при 280 нм,
б = 20 000 см2/моль). Соответствующие а,C-ненасыщенные соединения поглоща-
поглощают примерно на 20 нм в области более длинных волн. В 2,4-динитрофенилгидразо-
нах, напротив, виден переход с переносом заряда при 360 нм (е = 20 000 см2/моль).
Как следует из рис. А. 125 и А.123, пя*-поглощение в а,Р-ненасыщенных соеди-
соединениях сдвигается батохромно (обычно примерно на 20—50 нм). Гетероатомы, не-
непосредственно связанные с С=О-группой с неподеленной электронной парой,
как, например, в карбоновых кислотах, эфирах карбоновых кислот и амидов кис-
кислот, сдвигают ил* -поглощение С=О-группы гипсохромно (Xmaii ~ 200—220 нм).
о о
С?
НзС-С?
н3с-с'
Н
он
NH2
^тах = 205 НМ
ig г = 2,21
в метиловом спирте
(А. 128)
Хтах = 277нм Хтах=208нм
lge = 0,70 Ig e = 1,50
в воде в этиловом спирте
Ароматические углеводороды имеют от 2 до 3 полос средней силы в области
200—400 нм, в которых проявляется также иногда тонкая структура в неполяр-
неполярных растворителях. В более конденсированных ароматических соединениях по-
полосы сдвинуты в область более длинных волн (рис. А. 129). Подобный же бато-
хромный сдвиг проявляют и заместители, способные к сопряжению, причем как
акцепторные, так и донорные группы (рис. А. 130).
200 250 300 400 500
1 -
50000 40000 30000 20000
-•— v,cm-1
Рис. А. 129. УФ-спектры бензола,
нафталина, антрацена.
400 500
50000
40000 30000
•— V, см-1
20000
Рис.
А. 130. УФ-спектры бензальдегида,
анилина и фторбензола.
116 А Введение в лабораторную практику
Светопоглощение ароматических гетероциклов часто напоминает поглоще-
поглощение ароматических соединений, например,
[А.131]
Ig е = 2,31 Хтах = 257нм Ig E = 3,48
Хтах = 203,5 нм Ig e = 3,87 Хтах=195нм Ig e = 3,73
в воде (+ 2% метанола) в воде (+ 2% метанола)
УФ/вид.-спектры органических соединений можно получать, используя
растворы. Чтобы уловить как сильные, так и слабые полосы поглощения, нужно
измерять растворы различной концентрации. По экспериментально получен-
полученным величинам, оптической плотности рассчитывают коэффициенты е и затем
по Ig s находят А, и v (ср. рис. А. 123, А. 129, А. 130). Употребляемые растворители
должны быть высокой чистоты (спектрально чистые) и не должны проявлять
взаимодействие с растворенным веществом. По этой причине лучшую инфор-
информацию получают в гексане или циклогексане, особенно для тонких структур.
Растворитель, конечно, не должен сам поглощать в ожидаемой области. По этой
причине не следует применять продажный этанол, так как он часто содержит
еще следы бензола (поскольку его очищают азеотропной перегонкой). Спектр
органических красителей лучше снимать в ацетонитриле или этаноле, а не в
воде, так как она способствует образованию димеров. В области 180—200 нм пог-
поглощает уже кислород, поэтому оптическая часть спектрометра должна быть про-
продута аргоном или азотом. Некоторые часто используемые растворители приве-
приведены в табл. А. 132.
Таблица А. 132. Растворители для УФ/вид.-спектроскопии. Приведены границы длин волн
(нм), выше которых оптическая плотность растворов в слое 1 см может быть измерена.
Гексан
Циклогексан
Вода
Ацетонитрил
Метанол
<180
<180
190
190
203
Этанол
Диэтиловый эфир
Дихлорметан
Хлороформ
Тетрахлоруглерод
204
215
220
237
257
Вследствие часто высоких молярных коэффициентов поглощения в УФ-ви-
УФ-видимой области УФ/вид.-спектроскопия имеет особенно большое значение для
аналитических определений (при условии отсутствия сильных отклонений от
указанных закономерностей) и в связи с этим для исследования равновесий и
скоростей органических реакций.
В препаративной органической химии часто тоже целесообразно просле-
проследить за течением реакции с помощью спектров в УФ-видимой области или во-
вообще проводить реакцию в кювете спектрометра. При этом часто находят, что
все спектры пересекаются в одной (изобестической) точке одинакового погло-
поглощения. Это указывает на то, что протекает единственная реакция, при которой
исходные соединения превращаются в конечные продукты без промежуточно-
3. Определение физических свойств органических соединений 117
го образования долгоживущих продуктов, поглощающих в исследованной
области.
Часто с помощью УФ/вид.-спектров можно определить количественный
состав реакционной смеси, если известны молярные коэффициенты поглоще-
поглощения компонентов.
Перекрывающиеся полосы поглощения могут быть определены путем расче-
расчета. Но также возможно повысить степень разделения концентрационных опре-
определений тем, что первые, вторые или более высокие части кривой поглощения,
определяют путем расчета (по производным спектрам).
3.5.2. ИНФРАКРАСНАЯ СПЕКТРОСКОПИЯ
Поглощение излучения в инфракрасной области связано с возбуждением коле-
колебаний в молекуле, которые при комнатной температуре термически не возбуж-
возбуждаются. При возбуждении колебания молекулы переходят из нулевого колеба-
колебательного состояния (v = 0) преимущественно в первое возбужденное состояние
(v = 1). Такие типы колебаний называют основными, собственными или
нормальными колебаниями. Они расположены в области от 400 до 4000 см~'
B5—2,5 мкмI и соответствуют энергиям возбуждения от 4 до 40 кДж/моль
A—10 ккал/моль). (Ср. эти значения с затратами энергии, необходимыми для
возбуждения электронов!)
Число основных колебаний вытекает из числа степеней свободы молекулы.
Молекула, состоящая из п атомов, имеет Ъп степеней свободы. Из них 3 степени
свободы падают на поступательное и 3 (для линейно построенных молекул 2) —
на вращательное движение. Колебательное движение молекулы имеет Ъп — 6
(для линейных молекул Ъп — 5) степеней свободы. Такого количества нормаль-
нормальных основных колебаний и следует ожидать в спектре. Однако поглощение
ИК-излучения электромагнитного переменного поля наблюдается только в том
случае, если происходящий при этом переход на более высокий колебательный
уровень связан с изменением электрического дипольного момента молекулы.
Только такие переходы являются разрешенными. Поэтому особенно интенсив-
интенсивное поглощение обусловлено наличием в молекуле сильнополярных групп (нап-
(например, >С=О, -SO2-, -NO2 и т. д.). Напротив, неполярные группы, имеющие-
имеющиеся в симметрично построенных олефинах (R.2C=CR.2) или азосоединениях
(R—N=N—R), не проявляются в ИК-спектрах. Многие колебания, неактивные в
ИК-спектре, обнаруживаются в спектрах комбинационного рассеяния (спект-
(спектрах Рамана); последние несут особенно ценную информацию, дополняя
ИК-спектроскопическое исследование.
Помимо основных в указанной выше области могут возбуждаться также
обертонные колебания (связанные с переходом на более высокие колебательные
уровни: v = «vi, где п = 2, 3,...) и комбинационные колебания (обусловленные
наложением основных и обертонных колебаний: v = vj + V2 или v = v2 — vi).
Интенсивность этих колебаний, как правило, невелика.
, 104
v = (v — в обратных сантиметрах, X — в микрометрах).
118 А Введение в лабораторную практику
Поскольку каждый колебательный переход сопровождается изменением
вращательного состояния молекулы, инфракрасный спектр представляет со-
собой вращательно-колебательный спектр. Из-за большого количества отдель-
отдельных полос поглощения и в результате взаимодействия молекул в твердом или
жидком состояниях этот спектр проявляется не как линейный, а как спектр
полос поглощения (см. также разд. А,3.5.1).
Основные колебания подразделяются на валентные (колебания по линии
связи, при которых происходит изменение расстояния между атомами по оси
связи) и на различного рода деформационные колебания, связанные с изменени-
изменением валентных углов. Для валентных колебаний двухатомной молекулы в первом
приближении вполне применим закон Гука, выведенный для гармонического
колебания двух шаров, связанных пружиной:
1 Г т1т2
ц="^г+^г [алзз1
где ц — приведенная масса, К— силовая постоянная (мера энергии связи). Это
уравнение можно также привлекать при обсуждении валентных колебаний двух-
двухатомных структурных элементов. По закону Гука частота колебаний возрастает
с увеличением энергии связи между атомами и уменьшением приведенной
атомной массы. В соответствии с этим в ИК-спектре частота поглощения воз-
возрастает при переходе от простой связи С—С (<1200 см~!) к двойной С=С
(-1600-1700 см-1) и далее к тройной ОС (-2200 см-')-
Подобным же образом можно объяснить изменение частот поглощения в ряду групп О—Н,
N-H, С-Н, S—Н, а также различие в частотах групп О-Н и О—D или С-Н и С—D (табл. АЛ 35).
Поскольку энергия возбуждения деформационных колебаний атомов, не
слишком отличающихся по массе, гораздо меньше, чем энергия, необходимая
для смещения атомов по оси связи, то валентные колебания проявляются в
ИК-спектрах в более высокочастотной области, чем деформационные колеба-
колебания. Это положение иллюстрирует рис. А. 134, где показано отнесение трех
собственных колебаний молекулы воды (см. также табл. А. 135).
В табл. А. 135 представлены сведения о валентных и деформационных коле-
колебаниях важнейших характеристических групп и некоторых конкретных органи-
органических соединений. С положениями и формами полос можно ознакомиться,
изучив спектры, приведенные в части Д.
3756 см-1 3655 см-1 1595 см-1
антисимметричные симметричные деформационные
валентные колебания колебания
Рис. А. 134. Типы колебаний в молекуле воды.
3. Определение физических свойств органических соединений
119
Таблица А.135. Характеристические частоты колебаний некоторых структурных эле-
элементов и углерод-углеродных связей в ИК-области
Волновое число
(см-1) и качественная
оценка полосы
поглощения11
3700-3600, ср.
(узкая полоса)
3600-3200, с.
(широкая полоса)
3550-3350, ср.
3500-3100, ср.
3300-3250, ел.
3350-3150, ср.-с.
(широкая полоса)
3200-2400, ср.
(широкая полоса)
3100-3000, ср.-сл.
3000-2800, с.-ср.
2960, 2870, с.-ср.
2925, 2850, ел.
2900-2400, ср.
2830-2815, ср.
2820-2760, ср.
2820-2720, ср.
2600-2550, ел.
2300-2100, ср.-с.
2270-2000, с.
Тип колебаний и
соответствующий
структурный элемент
Валентное, —О—Н
(свободная группа)
Валентное. —О—Н
(связанная группа)
Валентное, —N—Н
(свободная группа)
Валентное, —N—Н
(связанная группа)
Валентное, эС—Н
Валентное, -NH3ffl
Валентное, —О—Н
(связанная группа)
Валентное, =С—Н
Валентное, —С—Н
Валентное, —СНз
Валентное, —СНг
Валентное, —О—D;
валентное, —N—D
Валентное, —О—СНз
Валентное, -N-CH3
Валентное, -(О)С-Н
Валентное, —S—Н
Валентное, -С=Х (X = С, N, О)
Валентное, -Y=C=X (Y = N, С;
X = О, S); валентное, —Nj
Соединения
Спирты, фенолы, кислоты, гид-
роксикетоны, эфиры гидрокси-
кислот (рис. Д.55, Д.56)
См.рис.Д.43,Д.52
Первичные B полосы) и вторич-
вторичные амины и амиды (ср. рис. Д.6.
Д-8)
Монозамещенные производные
ацетилена (рис. Д.67)
Гидрохлориды аминов и амино-
аминокислот (рис. Д. 16)
Карбоновые кислоты, хелаты
(рис.Д.36.Д.39,Д.7О)
Ароматические соединения,
олефины (рис. Д.6, Д.25, Д.39,
Д.40,Д.44, Д.55, Д.65, Д.67)
Насыщенные углеводороды,
ал килы
Насыщенные углеводороды, ал-
килы (рис. Д. 19, Д.24, Д.48, Д.52,
Д.58)
Насыщенные углеводороды,
алкилы (рис. Д.44, Д.48, Д.58)
Амины, спирты
Простые метиловые эфиры
(рис. Д.6)
N-Метиламин (рис. Д. 13)
Альдегиды
Меркаптаны, тиофенолы
(рис.Д.70)
Ацетилены, нитрилы, моноок-
монооксид углерода
Изоцианаты, изотиоцианаты,
кетены; азиды
120
А Введение в лабораторную практику
Таблица А. 135. Продолжение
Волновое число
(см~') и качественная
оценка полосы
поглощения11
2260-2190, ел.
2260, ср.
2260-2210, ср.
2185-2120, ср.
2140-2100, ср.
1850-1600, с.
1785-1700, с.
1840-1780, с.
1780-1720, с
1780-1750, с.
1760-1700, с.
1720-1690, с.
1750-1730, с.
1730-1710, с.
1745, с.
1715, с.
1705, с.
1715-1680, с.
1690-1630, с.
1690-1660, с.
1690-1650, с.
1675-1630, ср.
1650-1620, ср.
1650-1550, ср.
Тип колебаний и
соответствующий
структурный элемент
Валентное, — СзС—
Валентное, —N+sN
Валентное, —CsN
Валентное, —N=C
Валентное, — СэС
Валентное, —С=О
Валентное, —С=О
Валентное, —С=О
Валентное, —С=О
Валентное, —С=О
Валентное, —С=О
Валентное, —С=О
Валентное, —С=О
Валентное, —С=О
Валентное, —С=О
Валентное, —С=О
Валентное, —С=О
Валентное, —C=N
Валентное, —С=О
Валентное, —С=О
Валентное,—С=С
Деформационное, — Nb^
Деформационное, —N—Н
Соединения
1,2-Дизамещенные производные ацети-
ацетилена
Соли диазония
Нитрилы (рис. Д.8)
Изонитрилы
Монозамещенные ацетилены (рис. Д.67)
Карбонильные соединения
Ацилгалогениды
Ангидриды карбоновых кислот (две по-
полосы) (рис. Д.40)
Фениловые и виниловые эфиры насы-
насыщенных карбоновых кислот (рис. Д.36)
а,р-Ненасыщенные карбоновые кисло-
кислоты ароматического ряда (рис. Д.ЗО, Д.70)
Алкиловые эфиры насыщенных карбо-
карбоновых кислот
Насыщенные альдегиды и кетоны, эфи-
эфиры а,р-ненасыщенных и ароматических
карбоновых кислот (рис. Д. 19, Д.24)
Циклопентанон
Циклогексанон
Циклогептанон
а,р-Ненасыщенные и ароматические
альдегиды (рис. Д.20, Д.21)
Азометины, оксимы и др.
а,р-Ненасыщенные и ароматические
кетоны
Первичные, вторичные и третичные ами-
амиды карбоновых кислот (амидная полоса I)
Ароматические соединения, олефины
(рис.Д.39,Д.65)
Первичные амиды карбоновых кислот
(амидная полоса II)
Первичные и вторичные амины
(рис.Д6,Д.8,Д.9)
3. Определение физических свойств органических соединений
121
Таблица А. 135. Продолжение
Волновое число
(см"') и качественная
оценка полосы
поглощения11
1630-1615, ср.
1610-1590, ср.
1610-1560, о.с.
1600-1775, 1500
1570-1510, ср.
1560-1515, с.
1500-1480, ср.
1470-1400, с.-ср.
1420-1330, с.
1400-1300, с.
(широкая полоса)
1390-1370, с.
1360-1030, ср.-с.
1350-1240, с.
1200-1145, с.
1300-1020, ср.-с.
1300-1050
1275-1200, о.с.
1075-1020, с.
1150-1020, о.с.
1260-1200, с.
1200-1150, с.
1150-1100, ср.
1050-1010, с.
1070-1030, с.
970-960, с.
Тип колебаний и
соответствующий
структурный элемент
Деформационное, Н—О—Н
Углерод-углеродные связи
в ароматическом кольце
Валентное, -СО в СОО
Деформационное, —NH3+
Деформационное, —N—Н
Валентное, — NO2
Углерод-углеродные связи
в ароматическом кольце
Деформационное, —СНз и
-СН2
Валентное, —SO2
Валентное, -СО в СОО~
Деформационное, —СН3
Валентное, —С—N<^
Валентное, —NO2
Валентное, —SO2
Валентное, -С-О-С
Валентное, -С-О-С
Валентное, —С—О—С
Валентное, —С—О—С
Валентное, —С—О
Валентное, —С—О
Валентное, —С—О
Валентное, —С—О
Валентное, —S=O
Деформационное, =С—Н
Соединения
Кристаллизационная вода в гидратах
Ароматические соединения (рис. Д.8,
Д.25,Д.36,Д.55,Д.67)
Соли карбоновых кислот
Соли аммония (две полосы)
Вторичные амиды (амидная полоса 11)
Алифатаические и ароматические
нитросоединения (рис. Д.9, Д.56)
Ароматические соединения (рис. Д.8,
Д.36,Д.55,Д.67)
Насыщенные углеводороды и алкилы
(рис. Д. 16, Д.24, Д.25, Д.48, Д.52, Д.58)
Органические сульфосоединения
Соли карбоновых кислот
Насыщенные углеводороды и алкилы
(рис. Д. 16, Д.24, Д.25, Д.48, Д.52, Д.58)
Амиды,амины
Алифатические и ароматические
нитросоединения (рис. Д.9, Д.56)
Органические сульфосоединения
Простые и сложные эфиры, ангидри-
ангидриды, ацетали
Насыщенные сложные эфиры,
ангидриды (две полосы)
Ароматические и виниловые эфиры
(две полосы) (рис. Д.6)
Алифатические и алициклические
простые эфиры (рис. Д. 13, Д.43, Д.44)
Фенолы (рис. Д.55, Д.56)
Третичные спирты
Вторичные спирты (рис. Д.52)
Первичные спирты
Сульфоксиды
1,2-Дизамещенные этилены
(транс-изомеры)
122
А Введение в лабораторную практику
Таблица А. 135. Продолжение
Волновое число
(см~') и качественная
оценка полосы
поглощения3
995-985, с.
915-905, с.
920 (широкая полоса)
810-750, с.
710-690, с.
890, с.
840-810, с.
800-500, ср.-сл.
770-735, с.
770-730, с.
710-690, с.
780-720, ср.
800-600, ср.-сл.
730-680, ср.
670, с.
Тип колебаний и
соответствующий
структурный элемент
Деформационное, =С—Н
Деформационное, О—Н...О—
Деформационное, =С—Н
Деформационное, =С—Н
Деформационное, =С-Н
Валентное, —С—Hal
Деформационное, =С—Н
Деформационное, =С—Н
Деформационное, —СНг
Валентное, —С—S
Деформационное, =С—Н
Деформационное, =С—Н
Соединения
Монозамещеные этилены
(две полосы) (рис. Д.65)
Димеры карбоновых кислот
(рис.Д.36,Д.39,Д.7О)
1,3-Дизамещенные бензолы
(две полосы)
1,1-Дизамещенные этилены
1,4-Дизамещенные бензолы
(рис.Д.56,Д.62)
Ароматические и алициклические
галогенопроизводные
1,2-Дизамещенные бензолы
(рис.Д.8,Д.9,Д.7О)
Монозамещенные бензолы (две
полосы) (рис. Д.25, Д.36, Д.44,
Д.55,Д.56,Д.67)
я-Парафины, содержащие более
чытырех СН2-групп
Органические серосодержащие со-
соединения (тиолы, тиоэфиры и др.)
1,2-Дизамещенные этилены
(г<«с-изомеры)
Бензол
а Качественная оценка полосы поглощения: о.с. — очень сильная, с. — сильная,
ср. — средняя, ел. — слабая.
Исходя из изложенных закономерностей относительно взаимосвязи струк-
структуры и частоты полосы поглощения стало возможным, анализируя ПК-спект-
ПК-спектры, получать разнообразную информацию о строении химических соединений.
Соединения, молекулы которых содержат определенный структурный элемент
(например, ОН, СН, С=С, ОС, С=О, NO2 и др.), поглощают в определенной
области спектра, характерной только для этих групп. Ответственные за это пог-
поглощение нормальные колебания, в которых, вообще говоря, принимают участие
все атомы молекулы, обусловлены, следовательно, колебаниями этих структур-
структурных элементов. Отсюда возникает возможность использовать колебания в диаг-
диагностических целях. Характеристические частоты поглощения такого типа для
некоторых типов структурных элементов приведены в табл. А. 135.
3. Определение физических свойств органических соединений 123
Весьма существенным является то, что почти все структурные элементы про-
проявляют поглощение в нескольких областях ИК-спектра, так как одновременно
возбуждаются различные типы валентных и деформационных колебаний; это
означает, что наряду с интенсивными полосами поглощения с характеристичес-
характеристическими частотами в ИК-спектре всегда проявляются еще и другие полосы погло-
поглощения, соответствующие тем же структурным элементам (например, О—Н,
N—Н, С—Н, С=С, С=С, С=О, NO2 и т. д.). Наличие в молекуле определенного
структурного элемента можно считать однозначно доказанным только в том
случае, если в спектре обнаруживают все соответствующие этому элементу по-
полосы поглощения. Расшифровка структур неизвестных соединений (разд. А,3.8)
основывается прежде всего на этом требовании.
Обнаружение характеристических частот в ИК-спектре неизвестного соеди-
соединения позволяет сделать вывод не только о наличии соответствующих групп в
этом соединении, но и высказать предположения об их ближайшем окружении
(о влиянии соседних групп, наличии сопряженных и водородных связей). Так,
для валентных колебаний групп ОН и NH различают колебания «свободных»
групп, т. е. групп, не участвующих в образовании водородных связей, и колеба-
колебания «связанных» групп, т. е. групп, вовлеченных в образование водородных свя-
связей. Полосы валентных колебаний связанных групп ОН и NH обычно широкие
и нечеткие (рис. А. 137, а и б) и смещены по сравнению с полосами соответству-
соответствующих валентных колебаний свободных групп в более длинноволновую область
спектра.
В области 1400—700 см~' многие органические молекулы имеют настолько
сложные спектры, что отнесение всех полос поглощения к определенным струк-
структурным элементам сопряжено со значительными трудностями даже при наличии
обширного экспериментального материала. Однако именно эта область имеет
наибольшее значение при решении вопроса о структуре неизвестного соедине-
соединения. Как показывает опыт, два вещества (например, природное соединение и его
синтетический аналог) могут быть признаны идентичными, если их спектры в
этой области во всех деталях полностью совпадают. Поэтому эту область назы-
называют также фингерпринтовой областью или областью отпечатков пальцев.
При расшифровке ИК-спектра неизвестного вещества целесообразно внача-
вначале сделать предположения относительно углеродного скелета соединения. Для
этого служат область валентных колебаний связи С-Н C300-2800 см-'Х область
деформационных колебаний связи С—Н A540—650 см-1) и область скелетных
колебаний углерод-углеродных связей A700-600 см~')- Относя полосы в этих
областях к определенным структурным элементам в соответствии с данными
табл. А. 135, можно в большинстве случаев решить, относится ли данное соеди-
соединение к ароматическим, непредельным, алифатическим или представляет собой
смешанное (алкилароматическое) соединение.
Ароматические соединения и олефины можно различить по валентным (в об-
области 3100—3000 см~') и деформационным колебаниям (в области 950—650 см)
связи =С-Н. Для ароматических соединений, кроме того, типичны колебания
С=С-связей в ароматическом кольце (в области 1600—1500 см). Менее интен-
интенсивные колебания связи С=С олефинов расположены выше 1600 см~' (в области
1600—1660 см~')- Интенсивность полос возрастает в случае несимметрично пост-
построенных олефинов.
124 А Введение в лабораторную практику
При анализе ИК-спектра в области деформационных колебаний связи
=С—Н по числу полос и их положению в некоторых особо благоприятных слу-
случаях можно судить о взаимном расположении заместителей в бензольном коль-
кольце (табл. А. 135).
Если в ИК-спектре отсутствуют полосы поглощения, характерные для аро-
ароматических соединений и олефинов, и, напротив, имеются полосы поглощения
в области 2800—2900 см~' (валентные колебания связи С—Н), можно с уверен-
уверенностью относить неизвестное соединение к представителям алифатического ря-
ряда. Если же в ИК-спектре наблюдаются полосы поглощения, соответствующие
колебаниям обоих классов соединений, то речь в этом случае идет о смешанном
алкилароматическом (или об алифатическом непредельном) соединении.
После установления типа углеродного скелета далее путем отнесения харак-
характеристических частот, наблюдаемых в ИК-спектре, определяют, какие функци-
функциональные группы находятся в неизвестном соединении. Полезно знать качест-
качественный состав соединения, тогда как при рассмотрении его предполагаемой
структуры определенные функциональные группы можно исключить.
Группы О—Н и N—Н могут быть легко идентифицированы по наличию в
ИК-спектре довольно интенсивных полос поглощения в области 3700—3100 см~', а
системы с тройными связями —по наличию поглощения в области 2300—2100 см~'
(обычно в этой области спектра отсутствуют другие полосы поглощения); точно
так же о наличии карбонильных соединений судят по появлению интенсивного
поглощения в области 1900—1600 см~'.
Положение карбонильной полосы поглощения в ИК-спектре сильно зави-
зависит от заместителей при карбонильном углероде. В коротковолновой области
(от 1740 см и выше) расположены характеристические полосы поглощения
ацилхлоридов и ангидридов карбоновых кислот, а также а-галогензамещенных
карбонильных соединений; в средней области спектра A750—1700 см~') прояв-
проявляются, полосы поглощения, соответствующие эфирам карбоновых кислот, аль-
альдегидам и кетонам. Амиды карбоновых кислот и гетероаналоги карбонильных
соединений, как, например, азометины и оксимы (валентные колебания связи
C=N), поглощают ниже 1700 см~'.
При отнесении полос поглощения в ИК-спектрах нужно, кроме того, при-
принимать во внимание, что сопряжение карбонильной группы с ненасыщенной
группировкой приводит к сдвигу полосы поглощения карбонильной группы в
более длинноволновую область спектра (на -20 см~'), а введение галогена в ос-
положение к карбонильной группе вызывает индукционный (или индуктивный)
эффект и сдвиг полосы поглощения в более коротковолновую область. Поэто-
Поэтому отнесение волновых чисел в табл. А. 135 можно оценить, как ориентировоч-
ориентировочное, нередки отклонения от этих значений в ту или иную сторону.
ИК-спектроскопия нашла применение в препаративной химии. Таким простейшим при-
примером может служить синтез эфира акриловой кислоты из 2-хлорэтанола (этиленхлоргидри-
на); ход этого процесса на каждой стадии контролировался спектроскопически:
О
НО-СН2-СН2-С1 —- HO-CH2-CH2-CSN —- H2C=CH-C=N —- Н2С=СН-С? гд j361
ОС2Н5
I II III VI
ИК-спектр этиленхлоргидрина (рис. А. 137, а) помимо колебаний группы С—Н и основного
скелета молекулы показывает полосы, типичные для гидроксильной группы C360 см~' — вале-
3. Определение физических свойств органических соединений
125
нтные колебания связанной ОН-группы; 1080 см — валентные колебания С—О, 1393 см~' —
деформационные колебания О-Н), и связи С—С1 F63 см' — валентные колебания С-С1).
При взаимодействии 2-хлорэтанола с цианидом калия получают р-гидроксипропионит-
рил (II). В его ИК-спектре (рис. А. 137, 6) имеются все полосы, характерные для гидроксиль-
ной группы. Полоса валентных колебаний С-С1 исчезла и вместо нее наблюдается новая
полоса при 2252 см'1, соответствующая валентным колебаниям группы C=N.
3600 3400 3200 3000 2800 2600 2400 2200 2000 1800 1600 1400 1200 1000 800 700 600 5О0 400
3600 3400 3200 3000 2800 2600 2400 2200 2000 1800 1600 1400 1200 1000 В00 700 600 500 400
В 3600 3400 3200 3000 2800 2600 2400 2200 2000 1800 1600 1400 1200 1000 800 700 600 500 400
s
I
I
60
40
20
' ¦' ' I L.
Г 3600 3400 3200 3000 2800 2600 2400 2200 2000 1800 1600 1400 1200 1000 800 700 600 500 «00
Рис. А.137. ИК-спектры, измеренные на спектрометре UR 10 (VEB Carl Zeiss Jena).
а — 2-хлорэтанол; б — 2-гидроксипропионитрил; в — акрилонитрил;
г— этиловый эфир акриловой кислоты.
126
А Введение в лабораторную практику
Акрилонитрил (III), образующийся при дегидратации р-гидроксипропионитрила, облада-
обладает полностью измененным спектром. В нем нет полос, характерных для гидроксильной груп-
группы, но появляются полосы, типичные для структурного элемента СН2=СН— A620 см*1 —
валентные колебания связи С=С; 3038 и 3070 см~' — валентные колебания связи С—Н в нена-
ненасыщенных соединениях; 1420 и 980 см — деформационные колебания С—Н олефинов с
винильной группой). Валентные колебания C=N смещены B230 см-1) под влиянием сопряже-
сопряжения с двойной связью С=С.
Инфракрасный спектр этилового эфира акриловой кислоты (IV), полученного алкоголи-
зом акрилонитрила, содержит характерные для сложноэфирной группы полосы при 1735 см~'
(валентные колебания С=О) и 1205 см~' (валентные колебания С-О). В спектре больше нет
полос группы C=N, а полосы, отвечающие винильной группе, остались.
Дополнительно следует изучить ИК-спектры, приведенные в части Д!
ИК-спектры жидкостей без растворителя снимают в тонком слое между двумя дисками из
бромида калия. Из твердых веществ прессованием их с бромидом калия изготовляют таблетки,
для этого их совместно перемешивают в вибромельнице. Поскольку бромид калия, как прави-
правило, содержит немного воды, следует проявлять осторожность при отнесении полос поглоще-
поглощения воды. Для того чтобы исключить дисперсионные эффекты, величина частиц должна быть
меньше длины волны излучения, а показатели преломления пробы и смешанного с ней носи-
носителя должны совпадать. Вместо бромида калия можно использовать, например, хлорид нат-
натрия. При измерениях в растворах следует учитывать то, что все растворители имеют полосы
поглощения в ИК-области. Поэтому спектры снимают в двух растворителях, у которых
собственные полосы поглощения располагаются в разных областях. Обычно используют тет-
рахлоруглерод и дисульфид углерода. При расшифровке спектра для предотвращения ошибок
необходимо исключить область поглощения растворителя.
Закон Ламберта — Бера в общем сохраняет свою силу и в ИК-спектроскопии,
которую поэтому можно использовать для количественного определения состава
смесей, если типичные полосы компонентов достаточно удалены друг от друга.
3.6. СПЕКТРОСКОПИЯ ЯДЕРНОГО МАГНИТНОГО РЕЗОНАНСА
Спектроскопия ядерного магнитного резонанса (ЯМР-спектроскопия) предс-
представляет собой особый вид абсорбционной спектроскопии. Явление резонанса в
спектре ЯМР наступает при поглощении электромагнитного излучения пара-
парамагнитными ядрами, находящимися в постоянном внешнем магнитном поле.
Магнитным моментом обладают ядра, в состав которых входит нечетное число
нейтронов или протонов (табл. А. 138).
Таблица А. 138. Магнитные свойства некоторых атомных ядер
Ядро
'Н
2H(D)
13С
14N
15N
19F
3lp
Число
протонов
1
1
6
7
7
9
15
Число
нейтронов
0
1
7
7
8
10
16
Спин/
'/2
1
'/2
1
'/2
'/2
'/2
Магнитный
момент щ,
ядерные
магнетоны
2,79268
0,857387
0,702199
0,40347
0,28298
2,62727
1,1305
Распростра-
Распространенность
изотопа в
природе, %
99,985
0,015
1,11
99,63
0,37
100
100
3. Определение физических свойств органических соединений
127
/Л-+1/2
m—Vi
AE-hv
-m—Vi
Рис. А. 139. Парамагнитное атомное ядро в однородном магнитном поле.
а — возможные ориентации вектора магнитного момента;
б — энергия ядер с различной ориентацией.
Если парамагнитное ядро поместить в постоянное магнитное поле, то воз-
возможна различная ориентация ядерного магнитного момента по отношению к
направлению внешнего поля, что определяется магнитным квантовым числом
/п/1. При наложении дополнительного переменного электромагнитного поля,
магнитный вектор которого перпендикулярен постоянному магнитному
полю, возможна вынужденная переориентация магнитного момента ядра,
сопровождаемая поглощением энергии высокочастотного поля (ядернй резо-
резонанс, см. рис. АЛ39).
Величина этой энергии (АЕ) и соответствующая ей частота поглощенного
излучения зависят от магнитных свойств ядра (ц/ — магнитный ядерный мо-
момент, /— ядерный спин). АЕ пропорциональна внешнему магнитному полю #о
АЕ = hv =
[А. 140]
Наиболее благоприятными объектами для ЯМР-спектроскопии являются
ядра, у которых отношения ц///, а следовательно, и АЕ достаточно велики. Это
ядра со спином /= 1/2, такие, как 'Н, 13С, 15N, 19F и 31Р (табл. А. 138, уравнение
[А. 140]). Напротив, из наиболее часто встречающихся в органической химии
элементов ядра изотопов 12С, 16О и 32S имеют спин, равный 0, и не дают сигна-
сигналов в спектре ЯМР.
Для измерения ЯМР-спектров пробу исследуемого вещества (в виде жидкос-
жидкости или раствора) вносят в постоянное магнитное поле #о. Исследуемое вещест-
1 /и/может принимать значения от +/, /—!,... до —/, где /— ядерный спин.
128 А Введение в лабораторную практику
во помещают в центр индукционной катушки, создающей высокочастотное
электромагнитное поле с частотой v. Затем изменяют напряженность магнитно-
магнитного поля #0, до тех пор пока не наступит явление резонанса (см. ниже). В этот мо-
момент образец начинает поглощать энергию высокочастотного поля, и ток, про-
протекающий по катушке, возрастает. Изменение величины протекающего тока
{резонансный сигнал) может быть измерено и зарегистрировано; так получают
спектр ядерного магнитного резонанса (см., например, рис. А. 145).
Согласно уравнению [А. 140], при измерении ядерного резонанса можно
работать при постоянной напряженности магнитного поля Но, изменяя часто-
частоту электромагнитного поля. При напряженности внешнего магнитного поля
Но = 104 Гс частоты излучения, поглощаемого при явлении резонанса, нахо-
находятся в области 1—50 МГц (радиоволновая область). Максимальное разреше-
разрешение современных высокоэффективных ЯМР-спектрометров лежит между
0,1 и 0,2 Гц. Предел обнаружения метода ЯМР-спектроскопии составляет
1018 магнитных ядер.
Приведенные рассуждения относились только к ядрам, которые не имеют
электронных оболочек. Однако поскольку ядро экранировано электронной обо-
оболочкой, то внешнее магнитное поле, окружающее ядро, ослаблено (диамагнит-
(диамагнитное экранирование):
#эфф=#о-аЯо [А-141]
где а — постоянная магнитного экранирования. Таким образом, резонансный
сигнал проявляется в реальных условиях (по сравнению с неэкранированным
ядром) только при больших значениях напряженности внешнего магнитного
поля. Этот эффект называется химическим сдвигом, так как он зависит от элект-
электронного (т. е. химического) окружения ядер.
На практике химический сдвиг Д относят к резонансному сигналу эталона S
(вещества, принятого за стандарт), добавляемого к раствору исследуемого об-
образца (внутренний стандарт). Тогда в качестве меры химического сдвига можно
приводить простую разность резонансных напряженностей (или резонансных
частот) эталона и исследуемого вещества Hs—Hj (соответственно vs—v,). У совре-
современных радиочастотных генераторов с частотой, например, 100 МГц различия в
химических сдвигах неэквивалентных протонов могут составлять до 2000 Гц
(ср. уравнение [А. 143]). Эти химические сдвиги, разумеется, пропорциональны
внешнему магнитному полю (или соответственно частоте генератора). Лучшие
современные продажные генераторы, оснащенные сверхпроводящими магни-
магнитами, работают при частотах до -800 МГц.
Чтобы получить величину химического сдвига, не зависящую от напряжен-
напряженности приложенного магнитного поля или частоты генератора, делят разность
напряженностей или разность частот образца и эталона на Но (соответственно vo)
1
где 8 — безразмерная величина — имеет порядок 10~5—10~7. В качестве единицы
химического сдвига принимают 10~6 (миллионную долю, сокращенно м.д.).
При ЯМР-исследованиях на ядрах ' Н и 13С в качестве внутреннего стандарта
используют тетраметилсилан (ТМС), поскольку он дает лишь один резонанс-
3. Определение физических свойств органических соединений 129
ный сигнал, в значительной мере не зависящий от концентрации и химическо-
химического состава раствора. Кроме того, химический сдвиг ТМС столь велик, что у боль-
большинства веществ протоны (или ядра 13С) поглощают при меньших напряжен-
ностях поля. ТМС приписывают 6 = 0 м.д.; расположенные в более слабых полях
сигналы имеют 5 больше нуля1.
Если умножить значение химического сдвига 5 (в миллионных долях) на час-
частоту генератора (в мегагерцах), то получают химический сдвиг в герцах:
5 (Гц) = 5 (м. д.) v (МГц) [А. 143]
Обычно в герцах приводятся константы спин-спинового взаимодействия
(см. ниже).
3.6.1. СПЕКТРОСКОПИЯ ЯМР 1Н
В табл. А. 144 сопоставлены характеристические химические сдвиги некоторых
структурных групп. Приведенные значения показывают, что химический сдвиг
зависит от электронной плотности вблизи соответствующих протонов: элект-
роноакцепторные заместители понижают магнитное экранирование, а электро-
нодонорные — повышают. Химические сдвиги поэтому часто находятся в
линейной зависимости от электроотрицательности и а-констант Гаммета (см.
табл. В.74, с. 212).
Наряду с электронной плотностью на величину химического сдвига оказыва-
оказывают влияние также другие факторы. Это прежде всего относится к протонам, рас-
расположенным рядом с 71-связями (табл. А. 144). Приложенное магнитное поле ин-
индуцирует в л-электронных системах дополнительное поле. Наложение обоих по-
полей создает магнитноанизотропные области, влияние которых на химические
сдвиги ясно видно у альдегидов (табл. А. 144), производных ацетилена и бензола.
На рис. А. 145 приведен спектр л-ксилола. В спектре имеются два резонанс-
резонансных сигнала, созданных химически эквивалентными протонами двух металь-
метальных групп и четырьмя метановыми группами ароматического ядра. Интенсив-
Интенсивности (площади) сигналов относятся как числа соответствующих протонов, в
данном случае 3:2.
Причиной высоких значений 5 для бензольных протонов является возника-
возникающий в бензольном кольце под действием магнитного поля кольцевой ток,
обусловленный магнитной индукцией вл-электронной системе ароматического
ядра. Как видно из рис. А. 146, этот кольцевой ток повышает эффективную
напряженность магнитного поля вне кольца, что приводит к поглощению при
относительно низком внешнем поле. У ацетилена наблюдается обратная кар-
картина.
Спектры ядерного магнитного резонанса высокого разрешения становятся
более сложными, что, однако, облегчает их интерпретацию. Сигналы, появляю-
1 В употреблявшейся прежде т-шкале сигналу ТМС приписывалось значение 10 м.д. Все
сигналы, лежащие в слабом поле, получали тогда т-значения меньше 10 м.д. Для пересчета
используют соотношение т + 5 = 10.
130
А Введение в лабораторную практику
Таблица А. 144. Химические сдвиги 'Н относительно тетраметилсилана (ТМС)
Группа
(CH3LSi
/ Я
НзС-С-
\
н3с-с=с—
/
н3с-с=сч
н3с-с=о
H3C-S —
Н3С-Аг
/
H3C-N
\
НзС-О—
н3с-о-с=о
1
Н3С—1
Н3С-Вг
Н3С-С1
H3C-F
H3C-NO2
—сн2—а
—с-сн2-с=
—C-CH2-S—
\ /
— C-CH2-N
—С-СН2-Аг
\
—с-сн2-о—
Химический
сдвиг, м. д.
0
0,8-1,3
1,8-2,1
1,6-2,1
1,9-2,7
2,0-2,6
2,1-2,7
2,1-3,1
2,3-4,0
3,6
2,2
2,7
3,05
4,3
4,3
0,9-1,6
1,1-2,4
2,4-3,0
2,3-3,6
2,6-3,3
3,3-4,5
Группа
\
— С-СН2-С1
\
— C-CH2-NO2
/
Ar-CH2-O—
\
сн-а
/
\
сн-он
—СНВг2
—с=сн
R
НB)
R-CH=CH-
(цис-транс)
~~ г
Ar-SH
с6н5-он
R-OH
Аг-Н
О
R-C-H
О
R-C-OH
\
C=N-OH
\
с=с-он
/ 1
/ 1
Химический
сдвиг, м. д.
3,3-3,7
4,3-4,6
4,3-5,3
1,3-2,1
4,0
5,9
2,4
1: 4,7-5,0
2: 5,6-5,8
5 5
5,1-5,7
2,8-3,6г
4,56-г
0,7-5,5 в-г
6,9
7,27
9,7-10,1
9,7-13,0г
8,8-10,2г
15—16Г
а В насыщенных углеводородах.
6 Неассоциированная, свободная, группа.
в Положение сигнала сильно зависит от степени ассоциации.
г Сигнал пропадает при встряхивании пробы с D2O.
3. Определение физических свойств органических соединений
131
10,С
9Р
8,0 7,0 6,0 5,0 4,0 3,0 2,0 1,0
Рис. А. 145. Спектр ЯМР 1Н л-ксилола.
щиеся при различных значениях напряженности магнитного поля, в результате
так называемого спин-спинового взаимодействия часто расщепляются на дубле-
дублеты, триплеты и т. д. Подобное расщепление наступает в том случае, если рас-
матриваемый протон находится по соседству с одним или несколькими
парамагнитными ядрами. Магнитные поля, создаваемые этими ядрами, усили-
усиливают или ослабляют внешнее магнитное поле соответственно своей спиновой
ориентации (ср. рис. А. 139). Таким образом, протон, имеющий ядерный спин
/и/= + '/2 или /и/= — х/г, создает в районе другого близлежащего протона факти-
фактически два магнитных поля с большей или меньшей напряженностью; в спектре
ЯМР получают для рассматриваемого протона сигнал в виде дублета. Благодаря
этому спин-спиновому взаимодействию осуществляется в некотором роде еще
один небольшой резонансный эффект. Изменение результирующего магнитно-
магнитного поля незначительно, и расщепление сигнала в общем составляет всего нес-
несколько герц. Величина расщепления зависит от взаимного удаления этих двух
протонов во взаимодействующих молекулах, от расположения в пространстве и
от химического окружения.
Спин-спиновое взаимодействие быстро уменьшается с удалением друг от
друга взаимодействующих ядер. Как правило, в спектре ЯМР можно наблюдать
только спин-спиновое взаимодействие, проявляющиеся в расщеплении сигна-
сигналов, не далее чем на 3—4 связи. Сигналы протонов, находящихся в одинаковом
химической (магнитном) окружении, не расщепляются в спектре ЯМР.
Рис. А. 146. Кольцевые токи в молекулах бензола и ацетилена.
132
А Введение в лабораторную практику
Таблица А. 147. Константы спин-спинового взаимодействия J некоторых протонов,
связанных с атомами углеродаа
Структура
Н б
/ 'н
—С-С—
н н
н3с-сн2—
н3сх
сн—
Н3С
U
\ г
с=с
/ н
Н /
с=с
/ н
\ /
с=с
н' н
\/
\ Я"н
с=с
н ^
J, Гц
10-20
(ср. рис. Д.50)
2-9 (ср. рис. Д.5,
Д.50)
6,7-7,2 (см.
рис.Д.7,Д.42)
5,7-6,8
(см.рис.Д.53,Д.63)
0-3,5
11-18
(см.рис.Д.38,Д.42)
6-14
0,5-2 (см. рис. Д.38)
Структура
\ /
ч С-Н
/ 'н
\ J?
—с-с
н н
\
—с-с=с-н
н'
н
KJ? н
Циклогексанв
а-ав
а—е
е—е
J, Гц
4-10 (см. рис. Д.38)
1-3
2-3
орто 7—10 (рис. Д.7,
Д.10,Д.22,Д.23)
мета 2—3 (рис. Д. 10,
Д.П)
пара 1
8-10
2-3
2-3
а На рисунках в частях А и Д 1 м. д. соответствует 90 Гц.
6 При неэквивалентности обоих протонов.
в а — аксиальный атом Н, е — экваториальный атом Н. Ср. схему [В.84] на с. 218.
Мерой спин-спинового взаимодействия служит константа спин-спинового
взаимодействия (КССВ) /, которая выражает расстояние между расщепленными
линиями (в герцах). В отличие от химического сдвига ее величина не зависит от
внешнего магнитного поля. Чем выше частота радиочастотного излучения
ЯМР-спектрометра, тем легче различить оба эффекта. Исходя из величины
константы /, часто можно сделать заключение об относительном положении
взаимодействующих ядер. В табл. А. 147 приведены значения констант для неко-
некоторых структурных фрагментов.
Число сигналов в спектре ЯМР, обусловленных спин-спиновым взаимодей-
взаимодействием (мультиплетность А/), зависит от числа п\, пг,... и спинового момента /
магнитно-неэквивалентных ядер, вызывающих расщепление. Мультиплетность
сигнала определяется уравнением
[А. 148]
3. Определение физических свойств органических соединений
133
или в случае взаимодействия одинаковых ядер
= 2nl+l
или в случае протона (/= ХЦ)
[А. 149]
[А. 150]
Таким образом, группа СН в ЯМР-спектре расщепляет сигнал соседних экви-
эквивалентных протонов на дублет, СН2-группа приводит к расщеплению сигнала на
триплет и СНз-группа вызывает квадруплетное расщепление сигнала. [Самосто-
[Самостоятельно выведите расщепление сигнала протона, вступающего во взаимодей-
взаимодействие с атомами D (/= 1), 13С (/= '/г), 14N (/= 1), 15N (/= '/2) иди 19F (/= i/2).]
Интенсивности расщепленных сигналов (площади пиков или приближенно
их высота) относятся друг к другу как коэффициенты бинома с числом членов п
(отношение интенсивностей для дублета 1:1, для триплета 1:2: 1, для квадруп-
квадруплета 1:3:3:1). Спин-спиновое расщепление распознается по этим типичным
отношениям интенсивностей сигналов расщепления.
Рассмотрим конкретный пример, где проявляется спин-спиновое взаимодействие (мультип-
летность сигнала и соотношение интенсивностей), например ЯМР-спектр этанола (рис. А.151).
В молекуле этанола имеются три вида протонов (в группах ОН, СН2 и СН3); они находятся в раз-
различном химическом окружении, так что в спектре должны проявляться три основных сигнала.
На протон группы ОН, кроме того, оказывается дополнительное влияние полей двух метилено-
вых протонов. При расщеплении, обусловленном одним из этих протонов, в ЯМР-спектре воз-
возникает дублет, сигналы которого удалены друг от друга на 4,5 Гц (/[д = 4,5 Гц). Равным образом
на протон ОН-группы действует также второй протон метиленовой группы, так что в свою оче-
очередь дублет должен еще раз расщепиться и дать два дублета, что соответствует появлению четы-
четырех сигналов. Однако, поскольку константа взаимодействия в обоих случаях равна 4,5 Гц, оба
средних сигнала сливаются в один с удвоенной интенсивностью. В общем в результате взаи-
взаимодействия протона гидроксильной группы с метиленовыми протонами в спектре получают
триплет с соотношением интенсивностей 1:2:1. Появление триплета следовало также ожидать
согласно уравнению [А. 150]. Средний химический сдвиг для этого сигнала, выраженный в мил-
миллионных долях, соответствует химическому сдвигу протонов ОН-группы в отсутствие спин-спи-
спин-спинового взаимодействия. Аналогично для группы эквивалентных СНз-протонов при взаимодей-
взаимодействии с обоими метиленовыми протонами в ЯМР-спектре появляется триплет с соотношением
интенсивностей полос 1:2:1. Эквивалентные метиленовые протоны взаимодействуют с тремя
протонами СНз-группы, так что вследствие наложения отдельных сигналов вначале следует
ожидать появления квадруплета с соотношением интенсивностей 1:3:3:1. Но, поскольку на
ОН
^ Взаимодействие с СНо
щ с.
спин — спинового взаимодействия
^ Взаимодействие с СНч
^ Взаимодействие с ОН
сн,сн:он
Взаимодейсгвие с С Н 7
.Й.М.Д.
5,5
5,0
4,5
4,0
3,5
3,0
2,5
2.0
1.5
1.0
0,5
Рис. А.151. Спектр ЯМР 1Н высокого разрешения безводного этанола (в присутствии
следов кислот и воды взаимодействие протонов СН2-ОН не наблюдается).
134 А Введение в лабораторную практику
метиленовые протоны влияет, кроме того, магнитное поле протонов ОН-группы, каждый сиг-
сигнал квадруплета расщепляется еще раз на дублет (/ = 4,5 Гц). Вследствие того что величины
констант /2,з и /1,2 различны, при расщеплении сигналы не совпадают, и в спектре ЯМР прояв-
проявляется октете приведенным распределением интенсивностей сигналов (ср. уравнение [А. 148]).
Связанные сигналы можно легко распознать по равенству констант спин-спинового взаимодей-
взаимодействия /i_2 = /2,1; ^2,3 = Ji,2, как это видно из рис. А.151.
В некоторых спектрах бывает довольно трудно доказать взаимодействие
между двумя протонами, привлекая лишь константы взаимодействия или расп-
распределение интенсивностей сигналов. С помощью современных ЯМР-спектро-
метров для доказательства можно прибегнуть к так называемому двойному резо-
резонансу. При этом компенсируется взаимодействие между соответствующими
партнерами в результате помещения образца в поле с частотой, соответствую-
соответствующей резонансному сигналу одного из дающих расщепление протонов. Поглоще-
Поглощение энергии приводит к быстрой переориентации спина поглощающего прото-
протона, так что магнитное поле в районе взаимодействующего партнера усредняется
и имеющее место в первоначальном спектре ЯМР расщепление сигналов пропа-
пропадает.
В зависимости от соотношения между значениями химического сдвига Д и
константы взаимодействия / спектры подразделяют на различные типы. Если
химический сдвиг намного превышает константу взаимодействия (Д>Д то вза-
взаимодействующие ядра обозначают прописными буквами, далеко отстоящими
(в алфавите) друг от друга (А, X). Если величина химического сдвига сравнима с
константой спин-спинового взаимодействия (Д ~ J), то выбирают для обозначе-
обозначения в спектре соседние прописные буквы (А, В). Число эквивалентных протонов
указывается в индексе.
Для закрепления изложенного выше материала выведите тип спектра
протонного магнитного резонанса (ПМР) для этилиодида и укажите характер
расщепления и распределение интенсивностей в мультиплете!
Отношение Схема Тип
J/A взаимодействия спектра
0,1 I I 1_1 АХ
rf Ъ
0,2 I I LJ АВ
2,0
АВ
Рис. А. 152. Взаимодействие двух протонов в зависимости от отношения J/A.
3. Определение физических свойств органических соединений 135
Для спектров типа АХВУ картина расщепления менее наглядна, чем для
спектров типа АХХГ Например, в АВ-спектрах интенсивности сигналов не сле-
следуют коэффициентам в биноме. Наблюдают так называемый эффект крыши:
интенсивность средних сигналов мультиплета увеличивается за счет крайних
(рис. А. 152). Эффект крыши может быть полезным для распознавания связан-
связанных сигналов и для интерпретации спектра.
Если соединение содержит три неэквивалентных протона, взаимодействую-
взаимодействующих друг с другом, то для обозначения типа спектра выбирают, согласно приня-
принятому принципу, три прописные буквы. Обозначение АхМуХг относится к спект-
спектру, в котором Д>/ (в алфавите буквы отстоят далеко друг от друга).
Обратитесь к рис. А. 145 и А. 151 и укажите типы спектров «-ксилола и этанола.
Изучите спектры ЯМР 'Н в части Д и объясните сигналы протонов следую-
следующих важных групп:
СН2 CH2—
[А. 152а]
Н3С \ Н /
сн— с=с н2с=с
Н3С Н х х
3.6.2. СПЕКТРОСКОПИЯ ЯМР "С
Наряду со спектроскопией ЯМР 'Н исключительное значение для исследования
органических соединений приобрела спектроскопия ЯМР 13С, позволяющая
изучать углеродный скелет соединения и химическое окружение всех атомов
углерода.
Вследствие низкого природного содержания изотопа 13С A,108%) и его мало-
малого магнитного момента (табл. А. 138) чувствительность спектроскопии ЯМР 13С
значительно меньше и составляет примерно 1,8 • 10~4 чувствительности спект-
спектроскопии ПМР. Это требует иной техники регистрации спектров.
Обычно 13С-спектры регистрируют с помощью импульсной фурье-спектрос-
копии. (Та же техника пригодна и для снятия спектров ЯМР иных ядер, в том
числе и 1И.) При этом с помощью мощного высокочастотного импульса однов-
одновременно возбуждают все резонансные частоты ядер одного вида (например,
13С). После этого кратковременного возбуждения ядра возвращаются в равно-
равновесное состояние. Связанное с этим процессом падение индуцированной на-
намагниченности (свободный спад индукции, ССИ) измеряют перпендикулярно к
полю Но (рис. А. 139). ССИ — это сложная интерферограмма, состоящая из мно-
множества перекрывающихся колебаний. После применения математической опе-
операции, называемой фурье-преобразованием, получают обычный спектр ЯМР.
Возбуждение повторяют многократно с интервалом порядка 1 с и сигналы ССИ
накапливают в компьютере; таким путем регистрируют прекрасные спектры
(см., например, т. 2, рис. Д.31), несмотря на малую чувствительность спектрос-
спектроскопии ЯМР 13С.
136 А Введение в лабораторную практику
Время, необходимое для регистрации спектра ЯМР 13С, определяется раст-
растворимостью вещества в выбранном для работы растворителе: чем выше конце-
концентрация исследуемого раствора, тем меньше времени требуется. Ориентиро-
Ориентировочно считают, что на каждый ожидаемый сигнал в 1 мл раствора должно нахо-
находиться >5 мг изучаемого соединения. Для снятия спектров ЯМР 13С обычно
применяют дейтерированные растворители.
Спектры ЯМР 13С могут оказаться очень сложными из-за множества близких
и дальних 13С—'Н-взаимодействий. Для упрощения спектров образец дополни-
дополнительно облучают мощным импульсом, соответствующим всей области химичес-
химических сдвигов протонов (широкополосная протонная развязка). В результате муль-
типлетный сигнал каждого углеродного атома, связанного с водородом, слива-
сливается в синглет большой интенсивности. Для ядер 13С интенсивность сигналов
дополнительно повышается в результате ядерного эффекта Оверхаузера. Отно-
Отношение интенсивностей в спектрах ЯМР 13С в отличие от обычных спектров ЯМР
'Н не отвечает отношению числа соответствующих ядер (т. 2, рис. Д.31).
При использовании внерезонансной (неполной) протонной развязки образец
облучают частотами, отстоящими на 100—500 Гц от области резонанса протонов.
При этом выявляются |3С—'Н-взаимодействия с протонами, непосредственно
связанными с данным углеродным атомом. Это позволяет определить, имеются
ли группы СНз (квадруплет), СН2 (триплет), СН (дублет) или четвертичный
С-атом (синглет).
Наряду с внерезонансной существуют другие пульспрограммы как, например,
DEPT (англ. distortionless enhancement by /xMarization /ransfer), которая позволяет
быстро различать четвертичные, третичные, вторичные и первичные атомы угле-
углерода. Эти программы применяют прежде всего к спектрам природных соедине-
соединений, богатых линиями, так как иначе наличие большого числа атомов С приводит
во многих случаях к наложению мультиплетов, что затрудняет интерпретацию.
Высокая информативность спектроскопии углеродного резонанса при изуче-
изучении строения органических соединений наглядно проявляется в больших хими-
химических сдвигах, значительно превышающих сдвиги протонов. В то время как хи-
химические сдвиги протонов охватывают область около 15 м. д., эта область в ЯМР
|3С превышает 250 м. д. Поэтому даже малые изменения в химическом окруже-
окружении ядер углерода четко отражаются в спектрах углеродного резонанса. В спект-
спектрах ЯМР 13С с протонной развязкой даже сложных соединений (например, сте-
стероидов), как правило, каждому химически неэквивалентному атому углерода
отвечает один сигнал. Кроме химических сдвигов при расшифровке таких спект-
спектров может помочь и малая интенсивность сигналов четвертичных С-атомов.
Химический сдвиг и соответственно положение сигналов С-атомов зависят
от их состояния и электронного влияния окружения (индукционные и мезомер-
ные эффекты заместителей, пространственные факторы). В отличие от спектров
ЯМР 1Н магнитная анизотропия не вносит существенного вклада в химические
сдвиги 13С. Характерные химические сдвиги 13С представлены в табл. А. 153.
Для химических сдвигов 13С характерна аддитивность влияния заместителей.
При наличии достаточного материала для сравнения отнесение сигналов можно
проверить на основе расчета вклада различных заместителей.
Ниже приводятся некоторые общие правила отнесения сигналов в спектрах
ЯМР13С.
3. Определение физических свойств органических соединений
137
Таблица А. 153.
Группа
НзС-С
НзС-S
H3C-N
НзС-0
H3C-F
H3C-CI
НзС-Вг
Н3С-1
С-СН2-С
C-CH2-S
C-CH2-N
С-СН2-0
C-CH2-Hal
С2СН-С
C2CH-S
C2CH-N
С2СН-О
C2CH-Hal
Химические сдвиги 13С
Химический сдвиг
5, м.д.
0-35
10-45
25-55
40-55
75,2
24,9
10,0
-20,7
15-45
20-60
35-70
45-85
0-85
20-70
45-70
45-80
50-90
35-100
(относительно тетраметилсилана)
Группа
С3С-С
c3c-s
C3C-N
С3С-О
C3C-Hal
\ /
с=с
-ос-
С(ар.)
С (гетероар.)
\
C=N—
\
с=о
о
с=о
—и
с=о
ci
\=0
н
\ _
/ ~
Химический сдвиг
5, м.д.
30-75
45-70
50-80
50-90
35-110
90-155
70-110
95-165
100-175
145-170
105-130
165-185
165-185
165-180
180-205
185-225
В наиболее сильных полях (от 0 до 50 м. д.) располагаются сигналы я/Я-гибриди-
зованных С-атомов, за ними следуют 5р-гибридизованные F0—100 м. д.), в слабых
полях расположены сигналы л/Я-гибридизованных атомов углерода A00—200 м. д.).
Эти закономерности в общем соответствуют химическим сдвигам протонов в соот-
соответствующих СН-фрагментах. Для алифатических углеводородов величины хими-
химических сдвигов легко рассчитать по правилу Линдемана — Адамса (расчет можно
провести также по правилу Гранта — Пауля, однако расхождение с эксперименталь-
экспериментальными данными в этом случае часто оказывается более существенным):
8, = А, + ? №тапт + №уп + Ns6n [A. 154]
т=0
где п — число Н-атомов, связанных с данным углеродным атомом /; т — число
Н-атомов при ос-углеродном атоме; Nma — число групп СНт в ос-положении (т = 0,
1,2, причем а-СНз-группы не учитывают); М — число у-углеродных атомов. /Vе —
число 5-углеродных атомов; влияние (З-углеродных атомов уже учтено в других
членах. А„ — эмпирическая константа, зависящая от и: — С'Н„—СаНт—О5—О—С8—.
138
А Введение в лабораторную практику
Для закрепления изложенного рассчитайте спектр ЯМР 13С н-пентана,
используя данные, приведенные в табл. А. 155. Полученный результат сравните с
экспериментальными данными E, м. д.: С1 = 13,5; С2 = 22,4; С3 = 34,3).
Как и следовало ожидать, у замещенных углеводородов углеродный атом,
несущий заместитель, экранируется тем меньше, чем выше электроотрицатель-
электроотрицательность связанного с ним элемента (см. табл. А. 156).
Таблица А. 155. Инкременты для расчета химических сдвигов 13С по правилу Линдема-
на — Адамса
п
3
2
1
0
Ап
6,80
15,34
23,46
27,77
т
2
1
0
2
1
0
2
1
0
2
1
0
О-пт
9,56
17,83
25,48
9,75
16,70
21,43
6,60
11,14
14,70
2,26
3,96
7,35
-2,99
-2,69
-2,07
+0,86
5П
0,49
0,25
0
0
Таблица А. 156. Изменение химических сдвигов 13С в 1 -замещенных н-апканах в зави-
зависимости от электротрицательности Е центрального атома заместителя X
Заместитель X
Н
СНз
SH
NH2
Cl
ОН
F
Ex
2,1
2,5
2,5
3,0
3,0
3,5
4,0
Srch2x — §rch3 - A
0
+ 9
+ 11
+29
+31
+48
+68
В соединениях, способных к мезомерии, на химические сдвиги 13С сильно
влияют донорные и акцепторные заместители. Так, например, в сс,Р-неп-
редельных соединениях донорные группы в р-положении вызывают сдвиг в
3. Определение физических свойств органических соединений
139
сторону сильного поля, акцепторные — в сторону слабого поля. Это иллюстрирует
сравнение данных для акролеина, метилвинилового эфира и этилена:
н2с=сн-сн=о
8=136,0
Н2С=СН2
123,3
Н2С=СН-ОСН3
84,4
[А. 157]
Таблица А. 158. Инкременты для расчета химических сдвигов 13С в замещенных бен-
бензолах по уравнению 6,= 128,5 + /,,+ /г/+ ...а
Заместитель
Н
—СН3
—c2h5
-НС(СНзJ
—CH2CI
—CH2OR
-CF3
—СН=СН2
-с6н5
—сн=о
СО—СНз
—соон
—COOR
—COCI
—CONR2
—С=Ы
—ОН
—ОСН3
—ос6н5
—O-COR
— NH2
—NR2
—NHCOR
— N=N-C6H5
—NO2
—N=C=O
— SH
— SR
—SO3H
—F
—Cl
—Br
—I
1
/, M. Д.
_
9,3
15,7
20,1
9,1
13,0
2,6
7,5
13,0
7,5
9,3
2,4
2,0
4,6
5,5
-16,0
26,9
31,3
29,1
23,0
19,2
21,0
11,1
24,0
19,6
5,7
2,2
8,0
15,0
35,1
6,4
-5,4
-32,3
Д/, при различном положении заместителя
орто
_
0,6
-0,6
-2,0
0,0
-1,5
-2,6
-1,8
-1,1
0,7
0,2
1,6
1,0
2,9
-0,5
3,5
-12,6
-15,0
-9,5
-6,0
-12,4
-16,0
-9,9
-5,8
-5,3
-3,6
0,7
0,0
-2,2
-14,3
0,2
3,3
9,9
в кольце
мета
_
0,0
-од
0,0
0,2
0,0
-0,3
-1,8
0,5
-0,5
0,2
-од
0,0
0,6
-1,0
0,7
1,6
0,9
0,3
1,0
1,3
0,7
0,2
0,3
0,8
1,2
0,4
0,0
1,3
0,9
1,0
2,2
2,6
пара
_
-3,1
-2,8
-2,5
-0,2
-1,0
-3,2
-3,5
-1,0
5,4
4,2
4,8
4,5
7,0
5,0
4,3
-7,6
-8,1
-5,3
-2,0
-9,5
-12,0
-5,6
2,2
6,0
-2,8
-3,1
-2,0
3,8
-4,4
-2,0
-1,0
0,4
а Приведенные инкременты для химических сдвигов 13С в замещенных бензолах взяты из ра-
работы EwingD. F. Org. Magn. Resonance, 1979,12, 499.
140
А Введение в лабораторную практику
Аналогично на основе электронных эффектов заместителей можно объяс-
объяснить химические сдвиги в спектрах ЯМР 13С замещенных бензолов и, более то-
того, предсказать их, учитывая вклады заместителей.
Рассчитайте химические сдвиги 13С для анизола, анилина и бензонитрила,
используя данные табл. А. 158.
При расчете химических сдвигов 13С в производных бензола исходное значе-
значение для бензола E = 128,5) алгебраически суммируют с приведенными в табл.
А. 158 инкрементами заместителей, зависящими от природы и положения пос-
последних. В случае сильных электронных или пространственных взаимодействий
между заместителями (например, у ор/ио-дизамещенных бензолов) наблюдают-
наблюдаются заметные расхождения между вычисленными и экспериментальными значе-
значениями.
Таблица А. 159
Соединение
н3с-сн3
Н2С=СН2
нс=сн
NEC-CH3
ноос-сн3
сн4
CI-CH3
CI2CH2
CI3C-H
Константы спин-спинового взаимодействия 13С-1Н
'/сн, Гц
125
156
248
136
130
145
150
178
209
Соединение
Н Н
с=с
Н5С6 С6Н5
н
0=6
СНз
н
о=с
он
г
1
г
-- ^,—
у нм
н„
'/сн, Гц
155
172
222
159
Н0УСН: 1-0
Нм зусн: 7,4
Н„4Усн: -1,1
Наряду с химическими сдвигами 13С важную информацию о строении соеди-
соединения дают также 13С—'Н-взаимодействия (их сила и мультиплетность). Осо-
Особенно сильно выражены такие взаимодействия через одну связь. Константы
спин-спинового взаимодействия '/сн лежат в интервале между +120 и 320 Гц.
С увеличением вклада ^-электронов в гибридизацию (т. е. с повышением элект-
электроотрицательности заместителя) спин-спиновое взаимодействие возрастает.
В связи с этим полезно сравнить значения для этана, этилена и ацетилена, а также
для хлорзамещенных метанов (табл. А. 159). Как можно было ожидать, значения /
уменьшаются при удалении взаимодействующих ядер BJch- от —10 до +60 Гц).
О некоторых взаимосвязях между величиной /и строением соединений говорят
данные, приведенные в табл. А. 159.
В свете изложенного обсудите спектры ЯМР 13С, приведенные в части Д!
3. Определение физических свойств органических соединений
141
С новейшими высокопроизводительными ЯМР-спектрометрами могут быть разрешены
сложные структуры. Для этого используют так называемые 2О-спектры (двухмерные спект-
спектры), которые позволяют получать точные данные о взаимодействиях через несколько связей.
Применение ядерного эффекта Оверхаузера, в основе которого лежат диполь-дипольные вза-
взаимодействия между двумя ядрами с определенным пространственым расположением, позво-
позволяет делать заключения о пространственном расположении молекул, например об относи-
относительной конфигурации. Путем применения комплексов металлов с хиральными лигандами
можно также быстро и относительно просто получать сведения о соотношении энантиомеров
при стереоселективных синтезах.
Дальнейшая важная информация для решения структурных вопросов может
быть получена из взаимодействий между ядрами |3С и другими магнитными яд-
ядрами. Этот феномен, измеряемый через несколько связей, дает сведения о типе
замещения, агрегатном состоянии и др. Поскольку из-за физических величин
(гиромагнитное отношение) некоторые константы взаимодействия имеют отри-
отрицательные значения, в табл. А. 160 приведены абсолютные значения констант
гетероядерных взаимодействий.
Таблица А.160. Константы взаимодействия J A3С-Н) выбранных соединений (Гц)
(абсолютные значения)
Соединения
HaC-F
H5C6-F
HsC.-CF,
H5Ce-15NH2
H5C-PPh3
H5C-P(O)Ph2
(EtOJP(O)O-C2H5
H5C6-6Li
H5C-Si(CH3K
H5C-BPh4eNa©
['¦/ex]
162
245
272
11,4
13
104
8,0
66,5
49,4
[2/cx]
21
32
2,7
20
10
6
1,5
[Vex]
8
4
1,3
7
12
7
2,7
[Vcx]
3
1
<1
0,3
2
0,5
3.7. МАСС-СПЕКТРОМЕТРИЯ
Масс-спектрометрия (МС) представляет собой аналитический метод, позволя-
позволяющий получить данные о молекулярной массе, элементном составе и структуре
органического соединения. Для этого необходимо очень малое количество ве-
вещества, поскольку масс-спектрометр является зачастую детектором на выходе
из колонки газового (ГХ) или жидкостного (ЖХ) хроматографа. В этом случае
говорят о сочетании методов ГХ-МС или соответственно ЖХ-МС.
На рис. А. 161 приведена схема масс-спектрометра. Вещество предваритель-
предварительно переводят в газовую фазу, после чего оно ионизируется в ионизационной
камере. Изонизированая молекула и образующиеся из нее ионизированные
осколки выводятся в электрическое поле, где формируются в пучок, попадаю-
142
А Введение в лабораторную практику
Входная
щель
Электроды
Нагреватель
Выходная
щель
Коллектор
Усилитель
Регистрация
сигнала
Рис. А.161. Схема масс-спектрометра.
щий далее в магнитное поле анализатора, в котором ионы двигаются по кривым,
радиус которых зависит от отношения массы иона к его заряду (m/z), т. е. проис-
происходит разделение ионов на пучки, имеющие различное направление движения и
содержащие определенное количество ионов с одинаковой массой. Каждый из
этих пучков регистрируется на выходе из анализатора.
Ионизация молекулы осуществляется обычно в результате электронного удара
(ЭУ) в результате чего образуются молекулярные ионы (катион-радикалы):
М + е
е
[А. 162]
Поскольку энергия бомбардирующих электронов (обычно 70 эВ) значитель-
значительно выше энергии, необходимой для ионизации соединения (энергии ионизации
органических соединений составляют величины от 8 до 15 эВ), то молекулярный
ион распадается (частично) на заряженные и незаряженные осколки (фрагмен-
(фрагментация):
м© _ А© + В- [А. 163]
м© _ с© + D [A. 164]
Наряду с электронным ударом используются также и другие способы иони-
ионизации. Среди них важно отметить химическую ионизацию (XVI), при которой сна-
сначала ионизируется добавляемый в избытке так называемый газ-реагент (напри-
(например, Н2, СЬЦ, инертный газ), который далее при столкновении с анализируемой
молекулой передает ей часть своей энергии1, ионизируя ее. В этом случае боль-
большого избытка энергии сверх энергии ионизации молекула не получает. Метод
полевой ионизации — полевой десорбции (ПД): из твердой пробы под действием
мощного F—8 кВ) электрического поля вырывается электрон с образованием
1 Строго говоря, в случае метана предварительно образуется ион метония (CHs+), который
взаимодействует с молекулой вещества и протонирует ее с образованием низкоэнергетической
частицы МН. — Прим. перев.
3. Определение физических свойств органических соединений 143
положительного иона, который затем десорбируется с электрода.
В противоположность методам ЭУ и ХИ в этом случае не требуется предвари-
предварительное испарение пробы. Нечто аналогичное имеет место в методе ионизации
путем бомбардировки ускоренными атомами (БУАI, при котором проба суспен-
суспендируется в матрице (глицерин, тиоглицерин, полиэтиленгликоль) и бомбарди-
бомбардируется в ионизационной камере масс-спектрометра потоком «разогнанных»
нейтральных атомов инертных газов (аргон, криптон), имеющих высокую внут-
внутреннюю энергию и передающих свою энергию анализируемому веществу (а так-
также молекулам матрицы) с образованием соответствующих ионов.
Образующиеся на выходе из масс-спектрометра ионные токи очень малы, поэ-
поэтому их усиливают с помощью электронных умножителей и записывают самопис-
самописцами. В современных приборах аналоговые сигналы передаются в компьютер, ко-
который после обработки данных выдает масс-спектр в виде графика или таблицы.
В большинстве случаев вероятность образования в масс-спектрометрах
отрицательных ионов на порядок меньше, чем положительных, поэтому в боль-
большинстве случаев отрицательные ионы не регистрируются.
Получаемые данные о возникающих фрагментах, представленные в виде за-
зависимости интенсивности от массовых чисел (ср. рис. А. 178), называется масс-
спектром. Массовые числа приведены в значениях величин m/z, где масса т вы-
выражена в атомных единицах массы2 (а.е.м.), a z — величина заряда. Поскольку в
большинстве случаев положительный заряд равен 1, то отношение m/z числен-
численно равно массовому числу.
Под термином разрешающая способность масс-спектрометра подразумевают
величину массового числа, при котором две последующие массы могут быть чет-
четко отделены.
Поскольку большинство атомных масс не представляют собой целые числа, то
при подсчетах суммарной массы ее рассчитывают с точностью до ±1 • 10~3 а.е.м.
При этом ионы с одинаковым целочисленным массовым числом могут иметь
различный состав и, как следствие, различные величины m/z-
На масс-спектрометрах высокого разрешения, используя массы сравнения
(методом «меченых» пиков), можно определять значение массового числа от-
отдельного иона (с точностью до 10~3—10~4 а.е.м.) или зарегистрировать полный
масс-спектр высокого разрешения. Таким образом, не только для молекулярно-
молекулярного иона, но и для всех фрагментных ионов будут определены их брутто-формулы.
Поскольку масс-спектральная фрагментация зависит как от структуры, так и от
имеющихся в ней заместителей, то полученные масс-спектральные данные поз-
позволяют установить как структуру ионов, так и структуру анализируемого соеди-
соединения. Расчет брутто-формулы ионов по данным масс-спектра высокого разре-
разрешения осуществляется с помощью компьютера по специальной программе. Без
помощи ЭВМ это также возможно осуществить с помощью специальных таблиц
(см. Henneberg D., Casper К. Z. Analyt. Chem. 227 A967) 241-260; Benz W., Henne-
berg D.\ Massenspektroskopie Organischer Verbinolungen. Leipzig, 1969), однако
такая работа исключительно трудоемка и требует очень больших затрат времени.
1 Если бомбардировка осуществляется заряженными ионами, то метод называется масс-
спектрометрией вторичных ионов (ВИМС). — Прим. перев.
2 а.е.м. = Vi2 массы изотопа 12С = 1,6660 • 104 г = 931 501 кэВ.
144
А Введение в лабораторную практику
Конечно, проще всего применить масс-спектральные правила к масс-спект-
масс-спектрам, полученным методом химической ионизации или методом мягкой иониза-
ионизации, не требующим предварительного испарения образца. В этом случае наблюда-
наблюдаются только молекулярные ионы (М+-ионы) с очень небольшим количеством
фрагментных осколков. Но при этом зачастую в спектрах наблюдаются ионы с
массами выше, чем у молекулярного иона, например, за счет реакций присоедине-
присоединения (образование клатратов) при слишком низком вакууме (клатратная масс-
спектрометрия), за счет взаимодействия с газом-реагентом (ХИ), реакции с катио-
катионом (ПД), а также с протоном (например, из растворителя-матрицы в методе БУА).
В случае соединений, не содержащих азота, молекулярный ион имеет четную ве-
величину массы. Его наблюдаемая масса (или масса фрагментов) не совпадает с моле-
молекулярной массой. Это различие хорошо иллюстрирует пример с бромметаном, моле-
молекулярная масса (94,939) которого является средним значением всех комбинаций изо-
изотопов с учетом их распространенности в природе, т. е. 12С'Нз79Вг, 12С'Нз81Вг,
|3С'Нз79Вг, 13С'Нз81Вг, 12С'Н22Н79Вгит.д.1 Масс-спектрометрическая молекулярная
масса соответствует только составу 12С'Н379Вг, т. е. комбинации лишь наиболее лег-
легких изотопов (М= 94 или, точнее, 93,941 823). Другие комбинации изотопов видны
на спектре в виде ионов в соответствии с их массами (искусственно полученные или
радиоактивные изотопы здесь отсутствуют), а их интенсивность соответствует при-
природной распространенности. Таким образом, в случае бромметана в масс-спектре
наблюдаются помимо пиков молекулярных ионов с массой 94 пики изотопных ио-
ионов (М + 1) A3С'Н379Вг, пОН22Н79ВтУ; (М + 2) A2ОН381Вг, 13С'Н22Н79Вг,
12С Н2Н279Вг и т. д.). Однако вклады изотопов |3С и 2Н благодаря их низкой природ-
природной распространенности по сравнению с распространенностью изотопов брома
G9Вг: 8|Вг« 1 :1) можно не принимать во внимание2. Таким образом, в масс-спектре
бромметана регистрируются два пика с m/z94 и 96 примерно равной интенсивности.
Органические соединения, содержащие атом бора, содержат пик иона (М + 1),
интенсивность которого примерно в четыре раза выше интенсивности пика молеку-
молекулярного иона. Поясните этот факт исходя из данных табл. А. 165.
Таблица А. 165. Интенсивности / (%) наиболее интенсивных пиков ионов некоторых
элементов в соответствии с их природной распространенностью
Элемент
В
Si
Si2
S
s2
Cl
Cl2
Br
Br2
m/z
10
28
56
32
64
35
70
79
158
M
24,4
100
100
100
100
100
100
100
51,1
M+ 1
100
5,1
10,2
0,8
1,6
M + 2
3,4
7,0
4,4
8,9
32,4
64,8
98,1
100
M + 3
0,3
0,1
M + 4
0,1
0,01
0,2
10,5
48,9
1 Реально в масс-спектрах немеченых соединений пики ионов, содержащих изотоп
2Н (D), не наблюдаются, так как естественная распространенность этого изотопа составляет
0,015% от содержания изотопа 'Н. — Прим. перев.
1 Точнее, 79Вг : 81Вг = 50,54 : 49,46. — Прим. перев.
3. Определение физических свойств органических соединений 145
При большом числе атомов углерода в молекуле образца пик иона (М + 1)
имеет уже значительную интенсивность и это надо учитывать. Если соединение
содержит только С, Н, N и О, которые вносят малый вклад в интенсивность
иона (М + 1), то действует следующее приблизительное правило:
Интенсивность (М + 1) в % к М
Число атомов С = [А.166]
Если ион содержит еще и серу, то это сказывается на интенсивности пика
иона (М + 2) (см. табл. А.165). В целом, если в соединении с известным элеме-
элементным составом содержатся определенные гетероатомы (Cl, Br, S, Si, В), то их
присутствие в ионах может быть обнаружено визуально по их изотопным соот-
соотношениям.
Масс-спектры представляют или в табличной форме (величины m/z и соот-
соответствующие им интенсивности, выраженные в % от интенсивности макси-
максимального пика в спектре) или же в графическом (штриховом) виде. В этом слу-
случае также наиболее интенсивный пик (часто молекулярный) является основ-
основным, а интенсивности остальных рассчитываются от него.
В ряде случаев, в особенности в масс-спектрометрии электронного удара,
пик молекулярного иона отсутствует или же имеет низкую интенсивность.
В особенности это характерно для масс-спектров соединений, содержащих лег-
легко элиминируемые группы (например, в виде воды), так что пик с наибольшей
массой (не обязательно наиболее интенсивный) будет отвечать массе продукта
элиминирования.
Для интерпретации спектра очень важно найти «разность масс» важнейших
пиков по сравнению с пиком с наибольшей массой, поскольку эта разность с
ближайшим соседним или более дальним соседним пиком говорит об отщепле-
отщеплении определенных фрагментов, что позволяет установить структуру неизвестно-
неизвестного (отсутствующего) фрагмента. Для помощи в расчетах и интерпретации масс-
спектров служат специальные таблицы, которые можно найти в монографиях.
В случае масс-спектров высокого разрешения исследователь получает брутто-
формулу ионов, анализ которых позволяет сделать заключение о структуре
искомого соединения. Анализ изотопных пиков позволяет, как было описано
выше, обнаружить в составе соединения такие элементы, как Cl, Br, S и Si.
Затем начинают последовательно от высоких масс к более низким пытаться
приписать ионам определенную брутто- и структурную формулу. В области вы-
высоких масс следует анализировать пики всех ионов, тогда как в области низких
масс подвергают анализу лишь ионы с наибольшей интенсивностью пиков.
Для понимания и интерпретации масс-спектров необходимо знать наиболее
важные механизмы фрагментации. Первичный (молекулярный) ион, как прави-
правило, распадается с образованием термодинамически наиболее стабильного иона.
При этом расщепляются прежде всего простые связи. В ряде случаев (перегруп-
(перегруппировка Мак-Лафферти) играют роль и стерические факторы.
Чем больше молекула, тем сложнее ее масс-спектр. Многие ионы, пики ко-
которых малоинтенсивны, образованные в результате высокоэнергетических ре-
реакций различными путями, неспецифичны для конкретных структур. Гораздо
важнее специфические пути фрагментации, которые связаны с особенностями
146
А Введение в лабораторную практику
структуры соединения. Для интерпретации масс-спектра важны в особенности
пик молекулярного иона, наиболее интенсивный пик и пики ионов с характе-
характеристическими массами.
Важнейшие процессы фрагментации
а) Алканы и соединения с длинными алкильными группами расщепляют-
расщепляются следующим образом:
©
[А. 167]
При этом вероятность образования карбониевых ионов растет в ряду
первичный < вторичный < третичный
б) В алкилгалогенидах, простых эфирах, спиртах, сульфидах и т. д. (R—X)
преимущественно отщепляется X и образуется ион R+
I
/g\ + х' [А. 168]
но при X = OR, NR, SR и т. д. ионы М — X не характерны и их пики име-
имеют низкую интенсивность. Для них более типичен путь «е» (см. ниже).
в) Ионы алкильных групп могут далее распадаться под электронным уда-
ударом с отщеплением олефина:
©
©
[А. 169]
г) Олефины, имеющие не менее четырех атомов углерода, могут претерпе-
претерпевать аллильное расщепление:
[А. 170]
д) В масс-спектрах соединений, содержащих бензильную группу, как пра-
правило, присутствует интенсивный пик бензилкатиона:
/=\ ©
R-
[А.171]
Методом масс-спектроскопии активированных столкновений было по-
показано, что, в противоположность существующему мнению, этот ион
лишь частично изомеризуется в ион тропилия:
[А. 172]
3. Определение физических свойств органических соединений
147
е) В случае алифатических гетероатомных соединений преобладает расщеп-
расщепление С—С-связи, соседней с гетероатомом. Вероятность этого типа распа-
распада, называемого «ониевым» распадом (чаще р-распадом. — Пер.), возраста-
возрастает в ряду CKO<S<N.B случае анионов такой тип распада доминирует.
.©.х
[А. 173]
ж) Гетероатом со свободной парой электронов, связанный с углеродом
кратной связью, вызывает преимущественное расщепление соседних
С—С- или С—Х-связей:
R-C
/Р
©
© ,
R-C=O + X
[А. 174]
з) Системы, которые формально можно отнести к аддуктам Дильса—Аль-
дера, претерпевают масс-спектральное ретро-дильс-альдеровскоерасщеп-
ретро-дильс-альдеровскоерасщепление. Если доминируют какие-либо другие пути фрагментации, то пики
вышеупомянутых ионов имеют низкую интенсивность:
[А. 175]
и) Важным путем фрагментации является миграция атома водорода,
названная по имени автора, ее обнаружившего, перегруппировкой
Мак-Лафферти:
[А. 176]
а также
,-Н
[А. 177]
Эта перегруппировка протекает через шестичленное переходное состоя-
состояние с миграцией кратной связи A,5-сигматропный сдвиг. — Пер.).
Наряду с этими путями фрагментации известен также целый ряд других про-
процессов фрагментации и перегруппировок, с которыми можно познакомиться в
специальных монографиях.
148
А Введение в лабораторную практику
Молекулярный
(родительский)
пик
I 1 1 1 1 1 1—i 1
Рис. А. 178. ХИ-масс-спектр диоктилфталата.
В масс-спектрах могут также появляться двухзарядные ионы, которые реги-
регистрируются как ионы с кажущейся массой /и/2. Это может привести к ошибоч-
ошибочным выводам. Такие ионы могут появляться при энергиях ионизации выше
30 эВ', но исчезать при более низких ее значениях.
Если ионный ток регистрируется «напрямую» (не компьютером), то на ленте самописца
можно обнаружить расплывчатые (не острые) пики ионов с нецелочисленными массами. Они
принадлежат так называемым метастабильным ионам с кажущейся массой т'. Эти ионы обра-
образуются не в ионизационной камере, а после выхода из нее переход входом в магнитное поле.
Если при этом ион тх под действием ускоряющего напряжения получит определенную допол-
дополнительную энергию и перед входом в магнитное поле распадется с образованием нового иона
т2, то после магнитного поля он будет регистрироваться не как ион т2, а как ион с массой
т = т22/т\. Наличие иона т* является подтверждением одностадийного превращения иона т\
в т2. Для расчета метастабильных пиков используют таблицы и номограммы, имеющиеся в
специальной литературе.
Представленный на рис. А. 178 масс-спектр диоктилфталата в графической
форме получен методом ХИ, поскольку в масс-спектре электронного удара это-
этого соединения пик молекулярного иона отсутствует, хотя имеется тот же макси-
максимальный (по интенсивности) пик. Пик с максимальной массой имеет нечетную
величину (m/z — 391), хотя соединение содержит в своем составе только С, Н
1 Пики ионов с зарядом >2 особенно характерны для масс-спектров полициклических
ароматических и гетероароматических соединений, в которых они могут иметь заметную
(более 3—10%) интенсивность.
4р н
^0
/-
(,0R
П w
II
О
,R'
f
7Г
0
о-н +
0
3. Определение физических свойств органических соединений 149
и О. Сравнение с данными криоскопии (Л/= 391) позволяет утверждать, что оп-
определенно этот пик иона (М + 1). Интенсивность изотопного пика 392 составля-
составляет примерно 30% от пика 391, что согласно уравнению [А.166] соответствует 27
атомам С (действительное значение 24 для октилфталата). Наиболее интенсив-
интенсивный (максимальный) пик иона m/z 149 образуется в результате одновременного
или последовательного протекания перегруппировки Мак-Лафферти и ацилие-
вого расщепления.
+ -OR [A. 179]
Перегруппировка Мак-Лафферти с ацилиевым расщеплением приводит к
иону m/z 149, тогда как в результате двойной перегруппировки Мак-Лафферти
образуется ион m/z 167. Ионы с m/z 113, 71, 57 и 43 являются алкильными фраг-
фрагментами, образующимися из октильного фрагмента CgHn путем отщепления
алкильной группы.
Ионы m/z 149, 167 и 391, состав которых частично подтвержден масс-спект-
масс-спектрами высокого разрешения, являются экспериментальными данными, вноси-
вносимыми в таблицы. Табличные или полученные на компьютере данные имеют в
конечном случае одни и те же значения массовых чисел, соответствующие
одной и той же брутто-формуле вещества.
Пик иона m/z 149 является так называемым ключевым пиком, он присутству-
присутствует в масс-спектрах всех эфиров фталевой кислоты и является, как правило, мак-
максимальным. Если в масс-спектре какого-либо неизвестного вещества появляет-
появляется пик иона с такой массой, то следует прежде всего искать следы эфира фтале-
фталевой кислоты. Поскольку эти эфиры находят широкое применение в качестве
пластификаторов для различных синтетических материалов (в частности при
изготовлении вакуумных смазок), то они могут попасть в виде примеси в пробу
и привести к ошибочной интерпретации экспериментальных данных.
3.8. УСТАНОВЛЕНИЕ СТРУКТУРЫ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
СПЕКТРАЛЬНЫМИ МЕТОДАМИ
Спектральные методы позволяют устанавливать структуру органических соеди-
соединений с гораздо меньшими затратами времени по сравнению с чисто химичес-
химическими методами.
Однако применение одного только спектрального метода без привлечения
дополнительных сведений обычно не дает с уверенностью сделать заключение о
структуре соединений. Комбинируя несколько спектральных методов, получа-
получают всестороннее подтверждение отдельных данных о строении вещества и таким
образом надежно определяют структуру соединения (см. табл. А. 180).
При проведении синтеза установление структуры продуктов реакции облег-
облегчается еще и тем, что на основе теоретических рассуждений и опытных данных
150
А Введение в лабораторную практику
Таблица А. 180. Характеристики ИК-, УФ-, ЯМР- и масс-спектров важнейших структурных
ИК-спектры
важнейшие полосы поглощения,
см-1
другие полосы поглощения.
см-1
УФ-спектры,
*м«кс нм
(Is e)
3700—3600 п.
3600—3200 п.
О—Н (своб.)
О—Н (в Н-свя
зях); межмоле-
межмолекулярные
Н-связи разры-
разрываются при раз-
разбавлении инерт-
инертными раствори-
растворителями
1050 с. ОН (перв.)
1150—1100 с. ОН (втор.)
1200—1150 с. ОН (трет.)
1260—1200 с. фенолы
>210 спирты
не поглощают
АгОН: арома-
ароматическое погло-
поглощение
3500—3400 у.
3400—3300 у.
3300—3250 с.
3200-2400 ош.
3080 у.
2975 у.
3080 у.
3030 у.
3030 у.
3030 у.
RNH2 (своб.)
RCONH,
(своб.);
в Н-связях при-
примерно на
100 см~' ниже
R?NH
RCONHR
аС-Н
очень прочные
Н-связн, хела-
ты, карбоновые
кислоты
—СН = СН,
—сн=сн2
RjC=CH2
R2C=CHR
quc-RHC=CHR
транс-
RHC=CHR
1650—1550 с.
2160—2100 ел. (отсутствует
при высокой симметрии)
920 у., ш.
1645 у.. 990 с, 910 с.
1655 у., 890 с.
1670 ел., 840—800 с.
1660 у., 730—675 у.
(неинформативна)
1675 с. (или отсутствует)
965 с.
>220 RNH2 не
поглощают
ArNH2: арома-
ароматическое погло-
поглощение
<210
3100—3000 у. С—Н в арома
(обычно 3 по- тических соеди-
лосы) нениях
1600 п. ( + 1580 при допол-
дополнительном сопряжении)
1500 п. B полосы)
1525—1450
770—730 cv. 710—690 с.
(монозамещенные)
770—735 с. A,2-дизамещен-
ные)
810—750 с, 710—690
A,3-дизамещенные)
780—760, 745—750
A,2,3-трнзамещенные)
205—260 D)
260—300 (око-
(около 3)
3. Определение физических свойств органических соединений
151
элементов органических соединений
Спектры ЯМР 'Н. химические
сдвиги, м. д.
Спектры ЯМР 13С. хи-
химические сдвиги, м. д.
Масс-спектры, т/е важ-
важнейших осколочных нонов
(М — молекулярный ион)
R—ОН 0,5 (своб.); 5,5 (в
Н-связях); Перв., втор, и
трет. ОН различаются по
интенсивности или КССВ
Аг-ОН 4,5 (своб.); 7,0 (в
Н-связях)
Все сигналы О—Н исчезают
в D2O
RjCH-0 3,5—5,1
СНзО—
RCH2O-
RjCHO-
R3CO—
40—55
45—85
50—90
50—90
ROH: M м.; чаше
(М—18) (Н,О)
перв. ОН 31 и.
втор. ОН 45 и.
трет. ОН 59 и.
АгОН: М и.; (М—28)
(СО)
RjNH: вследствие квадру-
польного расщепления
часто широкие и едва
различимые сигналы
R2CHN 2,0—3,8
вС-Н 2,0—3,2
/(СНСвСН)=2-3 Гц
RCH==CH» 4,5—5,9 C сиг-
сигнала)
Ццис) =6-14 Гц
/(гракс).= 11—18 Гц
7A,1) -0—3,5 Гц
CHjNR2
RCH2NR2
R2CHNR2
R3CNR2
25—55
35—70
45—80
50-80
CHj 4,5-5,9 B сиг-
сигнала)
/A,1)-0—3,5 Гц
3C = CHR 5,0—5,5
CHRD«c) 5,2—5,5
B сигнала)
/(!,2)=6—14 Гц
HC = CHR(rp<2Hc) 5,2—5,5
B сигнала)
У A,2) = 11—18 Гц
RCsCR 70—110
R2C=CRj 110—150;
у алкенов обычно
6(ццс)<б (транс),
но при полярных
заместителях у
двойной связи за
висимость может
быть обратной
М нечетный при нечет-
нечетном числе атомов N;
алифатические амины —
М отсутствует; арома-
ароматические амины — М и.
М м. или отсутствует;
типичных осколочных
ионов нет
М олефинов часто ма-
малоинтенсивен, а картина
фрагментации не инфор-
информативна
Ar—H 6,5—8,5
У(орго)=7—10 Гц
J(мета) =2—3 Гц
J(napa)"\ Гц
ArCRz—H 2,2-3,1
С(Аг)
Бензол
95—165
128.5
М и.; типичные фраг-
фрагменты: СзН3 C9), С4Н5
E1), C5HS F5), СтН,
(91), CeH5N (93), C,HSO
(94), С,Н5О A05)
152
А Введение в лабораторную практику
ИК-спектрыа
важнейшие полосы поглощения,
см-1
другие полосы поглощения,
см-1
УФ-спектры,
llgEl
840—810 с. A,4-дизамещен-
ные)
810—800 A,2,3,4-тетразаме-
щенные)
870 у. A,2,3,4,5-пентазаме-
щенные)
2960—2870 у. RCH3
2925, 2850 п. R2CH2
1380 у.
1460—1420 с.
1300—1100 у.
1470—1400 у.
790—720 п.
RCH3
Алканы не по-
поглощают
>210
2830—2815 у. ROCH3
2820— R2NCH3
2760 с—у.
3000—2250 ш. R3NH
2900—2400 у. ROD
R2ND
2600—2550 ел. RSH
1150—1020 с.
250B,5)
2160—2100 ел. RCs=CR
(отсутствует
при высокой
симметрии) +
2260±20 у. RNsN
2260—2210 п. RCs*N
2270—2000 с. RN = C=
O
210
260—400 D—5)
<200
-2100 у.
R
RN3
RN = C=S
RC = C = O
RCD
1820 oc.
1760 ос.
1785—1700 ос.
1770 ос.
1770 ос.
1770 ос.
1760 ос.»'г
RCOOCOR
RCOHal
4-Лактоны
RCOOAr
RCOOC
RCOOH (мо-
(мономер)
1300—1050 с. B полосы)
1220 с, 1160 с. (дополни-
(дополнительно ароматические полосы)
1220 с, 1160 с. (дополни-
(дополнительно олефиновые полосы)
3550 п.
210
Ароматическое
поглощение
210
3. Определение физических свойств органических соединений
153
Таблица. А.180. Продолжение
Спектры ЯМР 'И. химические
сдвиги, м. д.
RCH, 0,8—1,1
R2CH2 0,9—1,6
R3CH 1,3—2,1
О—CRr-H 3,2—4,0
N—CRj—H 2,8—3,5
N—Н (триплет) /(NH)
около 50 Гц, сигналы очень
слабые или отсутствуют
RSH 1,0—2,5 (алифатиче-
(алифатические тиолы)
S—CR2—H 2,8—3,0
«ССН, 1,8—2,1
= CCH2CR3 2,2—2,8
Ns=CCR2H 2—2,5
Ne=CSCR2H 2,5-3
Ароматические сигналы, см.
выше
Олефиновые сигналы, см.
выше
RCOOH 9,5—13
HOOCCR,H 2,0—2,6
Спектры ЯМР "С. хи-
химические сдвиги, м. д.
CH3CR3
R3CCH2CR3
(RaCJCHCR:
(R3CKCCR3
RsCHOR
R2CHNR2
CH3SR
RCH2SR
R2CHSR
R3CSR
-CsCR
-CaN
—N = C|
_N=C=O
-SCsN
-OCsN
_N=C=S
RCOOCOR
RCOC1
RCOOH
0—35
15—45
20—70
30—75
50—90
45—80
10-45
20—60
45—70
45—70
70—110
105—130
130—150
115—135
110—120
105—120
120—140
150-175
105—185
160—180
Масс-спектры, m/e иж-
нейших осколочных ионов
(М — молекулярный ион)"
М обычно м.; типичные
фрагменты С9Н; D3),
С<Н9 E7)
Типичный изотопный
пик М+2; фрагментация
аналогична ROH; типич-
типичные фрагменты HS C3),
HjS C4)
М часто отсутствует,
фрагментация нехарак-
нехарактерна
Как карбоновые кислоты
М и.; типичные фрагмен-
фрагменты АгО, RCO
RCOOH: M м.
ArCOOH: M и.
154
А Введение в лабораторную практику
ИК-спектрыа
важнейшие полосы поглощения,
см-1
другие полосы поглощения.
см-1
УФ-спектры.
Л
1710 ОС.»Г
1745 осЛг
1750—1720"г
RCOOH (ди-
мер)
Циклопентано-
ны
RCOOR
1720 осЛг Альдегиды
1715 ос.вг Кетоны (линей-
(линейные, 6-и 7-член-
ные цикличе-
циклические)
1690—1650 ос. RCONH2,
RCONHR,
RCONR,
(«Амид I»)
1420
1300—) 200
920 у., ш.
1300—1100 ос. (несколько
полос)
2820 у.
2720 у.
+ 15
R2C=CRCONR2
1640-1530
1675—
1630 у., ш.
1650 ос.
1610—1560
1650—1500
1420—1330
1370—1330
1275—1200
с.
с.
с.
с.
с.
ОС
(диариловые,
виниловые
1150—1070
эфиры)
RCONH,,
RCONHR
(«Амид II»)
R2C = CRj
RC(OH) =
= CRCOR
RCOOe
RNO,
RSO2OR
RSO2NR2
:., 1075—1200 с
арилалкиловые,
эфиры)
с
(диалкиловые
3500- RCONHR
3300 (ср. N—H)
3500— RCONHR
3300 (ср. N-H)
3100—3000 (см. выше)
700—950
(ср. 3100—3000 см-')
1615 RCOR (с внутримоле-
внутримолекулярной Н-связью)
1400—1300 с, ш.
1370—1250 с.
1200—1145 с.
1180—1160 (может быть
также N—Н; см. 3500—
3400 см-1)
270—300A)
270—300A)
270—300A)
<210
<230C.4)
R2C=CRCONR2
210
Около 280A)
230C—4)
230
Ароматическое
поглощение
210
3. Определение физических свойств органических соединений
155
Таблица. А. 180. Продолжение
Спектры ЯМР 'Н, химические
сдвиги, м. д.
Спектры ЯМР "С, хи-
химические сдвиги, м. д.
Масс-спектры, т/г важ-
важнейших осколочных ионов
(М — молекулярный ион) б
ROOCCRjH
2,0—2,6
RCHO 9,4—10,0
RjCHCHO 2,0—2,6
RCH=CRCHO 5,8—6,2
RCOCRjH 2,0—2,6
RCOCR=CRH 5,8—6,2
RCONRH Слабый широкий
сигнал
R2CHCONRj 2,0—2,6
RCOOR
RCHO
155—175
175—205
RCOR 185—225
RsCCOR 160—200
RaC=CRCOR180—215
RCONR2
155—180
RCONRCRSH 2,8—3,5
ROH-O = CRi 7,0—13,0
RCH2NO2 4,2-4,5
RO2SCRjH 2,5—3,5
RCOOH: расщепление по
Мак-Лафферти; типичные
фрагменты: (M—18),
(М—17), m/e 45
ArCOOR; M и.
RCOOR: M м.
RCOOR: перегруппиров-
перегруппировка Мак-Лафферти
АгСНО: (М—1) и.
ArCOR: M и.; перегруп-
перегруппировка Мак-Лафферти
ArCONR2: M и.
RCONRj: М м. М не-
нечетный при нечетном
числе атомов N; расщеп-
расщепление по СО-группе не-
нетипично (ср. полосу при
3100—3000 см-' в
ИК-спектрах)
RNO2: M м.; ArNO2:
М и., типичные фрагмен-
фрагменты NO C0), NO» D6)
Типичны изотопный пик
(М+2) и фрагмент
(М—64)
Ароматические сигналы;
см. выше
R3COR см. выше
ROCRjH
3,3—4,0
ArOR: M и.
ROR: Мм., часто интен-
интенсивнее (М+1)
156
А Введение в лабораторную практику
ИК-спектры
важнейшие полосы поглощения,
см-1
другие полосы поглощения.
см-1
1400—1000 с. R3CF
800—500 с. R3CC1, R3CBr
ИК-спектр интерпретирует-
интерпретируется с трудом
200
250 (RI, RBr)
а Обозначения: ос. — очень сильное поглощение; с. — сильное поглощение; у. — умеренное
поглощение: ел. — слабое поглощение; п. — переменное поглощение; ош. — очень широкая
полоса; ш. — широкая полоса.
6 Обозначения: и. — интенсивный пик; м. — малоинтенсивный пик.
из нескольких предполагаемых структур можно выбрать наиболее вероятную.
Таким образом, в спектре ищут типичные сигналы ожидаемой структуры.
Квантовохимические расчеты молекул также дают предпосылки для предсказания физико-
химических параметров, таких, как химические сдвиги в ЯМР, длины волн и углы, УФ-погло-
щение, и помогают во многих случаях сузить пути синтеза. Подходящие компьютерные прог-
программы, связанные с соответствующим банком данных, могут также сэкономить время и деньги.
В табл. А.180 сопоставлены данные ИК-, УФ-, ЯМР- и масс-спектроскопии
типичных структурных элементов. В основу таблицы положены характеристи-
характеристические полосы в ИК-спектре, которые расположены в порядке уменьшения
волновых чисел. Для того чтобы избежать ошибки при определении структуры,
необходимо всегда привлекать для ее установления также характеристические
величины, получаемые с помощью других спектральных методов.
3.9. РЕНТГЕНОСТРУКТУРНЫЙ АНАЛИЗ
Спектроскопические методы аналитической химии малорезультативны для ре-
решения структурных задач в случае соединений с малым количеством атомов во-
водорода, а также содержащих много сходных атомов углерода, как, например, во
многих металлокомплексах, где из-за парамагнитных свойств атомов металла
метод ЯМР, который так информативен в других случаях, отказывает и дает зак-
заключения только для небольших частей структуры. Эти трудности преобразуют-
преобразуются рентгеноструктурным анализом, в котором из одного монокристалла путем
рассеяния рентгеновских лучей от электронных оболочек атомов соединения
могут быть определены координаты атомов в элементарной ячейке и располо-
расположение молекулы в кристалле. Соответственно волновому характеру электромаг-
электромагнитного облучения выявляются эффекты рассеяния электронов при взаимодей-
взаимодействии с рентгеновскими лучами. В уравнении Брэгга представлены условия для
3. Определение физических свойств органических соединений
157
Таблица. А.180. Продолжение
Спектры ЯМР 'Н. химические
сдвиги, м. д.
HalCR2H 2,6—3,1
J(HCF)=55 Гц
J(HC—CF)=5—20 Гц
Спектры ЯМР
13С. хн-
мнческие сдвиги, м. д.
CH3F
СН3С1
СНзВг
СН31
R3CCH2HaI
(R3CJCHHal
(R3CKCHal
75,2
24,9
10,0
-20,7
0—85
35—110
35—110
Масс-спектры
, т/е важ-
нейших осколочных ионов
(М — молекулярный нон) 6
Относительно
пиков хлор-
держащих
см. табл. 22;
каждый атом
изотопных
и бромсо-
соединений
(М+2) на
галогена
в — Для а,C-ненасыщенных соединений около -20 с\г'.
г — В случае а-галогенозамещениых соединений резонансные сигналы п зависимости от кон-
центрации смещены на 25-45 см-' выше (в расчете на 1 атом галогена).
интерференции:
п X = 2 d sin в
[А.181]
где d — межллоскостное расстояние; в — угол падения рентгеновских лучей.
Это уравнение составляет основу для оценки рентгенограммы. Все узловые
плоскости способны к отражению только тогда, когда кристалл во время облу-
облучения поворачивается. Благодаря поворотным движениям рефлексы возникают
всегда, когда узловые плоскости согласно уравнению Брэгга занимают требуе-
требуемую ориентацию относительно падающего первичного луча. Для определения
кристаллической структуры органических соединений используют четырех-
кружный дифрактометр (рис. А. 182), с монохроматическим рентгеновским из-
излучением Си-К„-линии (X = 153,9 пм) или Мо-Ка-линии (X, = 71 пм). Путем
компьютерного управления ориентацией кристалла с помощью четырех незави-
независимых друг от друга кругов можно установить нужные ориентации кристалла и
последовательно измерить все рефлексы и их интенсивности.
Рентгеновский луч и
Кристалл
^\ Детектор
Поворот + смещение
кристалла
Рис. А. 182. Схематическое изображение четырехкружного дифрактометра.
158 А Введение в лабораторную практику
Рис. А. 183. Рисунок гетероциклического хинона, полученный с помощью ORTEP-программы.
В зависимости от детектирующей системы могут быть измерены кристаллы
размером >5 мкм. С помощью компьютерной программы получают измерен-
измеренные интенсивности координат атомов и по ним вычисляют структурные пара-
параметры — длины связей, углы, торсионные углы и др.
В противоположность другим методам определения структур, рентгеност-
руктурный анализ — это прямой метод, т. е. получают не только данные о частях
структуры, таких, как хромофорная система (УФ-вид.-спектроскопия), спин-
спиновые взаимодействия магнитных ядер (ЯМР) или реакционноспособные
функциональные группы (ИК), но сразу общую структуру соединения. Значимо
также определение меж- и внутримолекулярных водородных мостиков в крис-
кристалле. Но метод не пригоден для наблюдения за динамическими процессами ти-
типа вращения, таутомерии или is/Z-изомеризации, так как они протекают в раст-
растворителях.
Для наглядного представления структуры существует много возможностей, сре-
среди них широкое распространение получило ORTEP-изображение, см. рис. А. 183.
Положения атомов рисуются в форме характеристических эллипсоидных тепловых
колебаний, которые описывают как электронная плотность при уклонении крис-
кристалла размазывается от идеальной периодической решетки.
4. ХРАНЕНИЕ ХИМИЧЕСКИХ РЕАКТИВОВ.
УНИЧТОЖЕНИЕ ВРЕДНЫХ ОСТАТКОВ1
4.1. ХРАНЕНИЕ ХИМИЧЕСКИХ РЕАКТИВОВ
Для хранения химических реактивов в большинстве случаев в лаборатории
служат склянки с пришлифованными стеклянными пробками (целесообразно
1 В разделе по приготовлению реактивов (ч. Г) для отдельных реактивов сделаны указания
относительно их опасности; об особенностях хранения, а также первой помощи при несчаст-
несчастных случаях см. части Е и Ж.
4. Хранение химических реактивов. Уничтожение вредных остатков 159
использовать сосуды с нормальными шли-
шлифами). В специальных склянках с широким
горлом (так называемых склянках для
порошков) хранятся твердые вещества или
очень вязкие жидкости. Узкогорлые
склянки предназначены главным образом
для жидкостей. Для хранения веществ,
которые взаимодействуют со стеклом (нап-
(например, для плавиковой кислоты), применя-
применяют пластмассовые или металлические
сосуды, в крайнем случае используют и
стеклянные, покрытые изнутри слоем
парафина. Щелочные металлы хранят
под керосином, а белый фосфор - под Рис. А. 184. Наполнение ампулы
водой. веществом.
Светочувствительные вещества, к которым относятся и простые эфиры, склон-
склонные, особенно под действием света, образовывать пероксиды (разд. Г, 1.5),
хранят в сосудах из темного стекла.
Вещества, выделяющие ядовитые или раздражающие пары (например, бром,
олеум, соляная или плавиковая кислоты), рекомендуется хранить на специаль-
специально отведенной полке в вытяжном шкафу.
Ни в коем случае нельзя хранить химические реактивы в сосудах, обычно пред-
предназначенных для пищевых продуктов или напитков!
Вещества в небольших количествах или вещества, чувствительные к внеш-
внешним воздействиям, часто запаивают в ампулы, которые можно изготовить из
пробирки на пламени газовой горелки (рис. А. 184). Ампулы надо заполнять не
более чем наполовину. Чтобы избежать попадания вещества на стенки запаива-
запаиваемого горла ампулы, вещество следует вводить в ампулу через маленькую ворон-
воронку с длинным тонким носиком. Во время запаивания ампул в «остром» пламени
газовой горелки ампулы с легкокипящими веществами нужно охлаждать в под-
подходящей охладительной бане.
Все без исключения сосуды с химическими реактивами должны быть отчетливо и
надежно подписаны.
На обычных бумажных этикетках целесообразно писать карандашом или
тушью (надписи чернилами и химическим карандашом довольно быстро
бледнеют под действием лабораторного воздуха и легко стираются) и для
лучшей сохранности эти этикетки надо покрывать бесцветным лаком, зак-
заклеивать сверху прозрачной липкой лентой или же наносить тонкий слой
парафина. Не следует наклеивать этикетки на старые, так как отклеивание и
потеря этикетки могут привести к путанице. Для хранения едких веществ,
легко разрушающих надпись, используют склянки с вытравленной
надписью.
160 А Введение в лабораторную практику
Ядовитые вещества (например, синильная кислота и ее соли, мышьяк и его сое-
соединения, белый фосфор, многие алкалоиды, некоторые эфиры фосфорной кис-
кислоты; см. литературу в конце этого раздела) следует хранить в сейфах. Если эти
вещества постоянно необходимы для работы, разрешается хранить небольшие
количества в лаборатории во время работы.
Правила техники безопасности, действующие в Германии, регламентируют
объемы легковоспламеняющихся жидкостей (ЛВЖ). На каждом рабочем месте
общий объем всех жидкостей классов AI и В не должен превышать
необходимого для текущей работы количества', а объем одной склянки с такой
жидкостью не должен быть больше 1 л. Эти требования распространяются так-
также на количество ЛВЖ, предназначаемое для мытья лабораторной посуды.
Общелабораторные запасы огнеопасных жидкостей и других веществ, которые
могут усилить опасность пожара, должны храниться таким образом, чтобы их
можно было быстро вынести и не допустить возгорания. Как правило, в лабора-
лаборатории должны находиться лишь небольшие количества огнеопасных веществ.
Сосуды больших размеров, наполненные химическими реактивами, нельзя пере-
переносить, держа только за горло, их следует поддерживать снизу, под дно, или ста-
ставить в специальные ящики (или корзины) для переноски реактивов.
Для переноски большого числа маленьких склянок рекомендуется использо-
использовать специальные деревянные ящики.
4.2. ОТХОДЫ И ИХ УНИЧТОЖЕНИЕ
Осколки химической посуды и другие предметы с острыми краями нельзя выбра-
выбрасывать в корзины для бумаг или другие обычные мусоросборники. Их следует со-
собирать в специальные ящики. Опасные вещества, например, такие, которые могут
выделять ядовитые газы, или самовоспламеняющиеся остатки (скелетный никель,
фосфор, щелочные металлы), нельзя выбрасывать в мусоросборники или спус-
спускать в канализационную систему. Кроме того, в канализационную систему запре-
запрещается выливать или высыпать ненужные вещества или растворы, в которых при-
присутствуют нерастворимые в воде или не смешивающиеся с ней компоненты.
Химические отходы необходимо предварительно обезвреживать путем хими-
химической обработки или сжигания в специально отведенных местах вне пределов
лаборатории (желательно на открытом воздухе). При выливании в канализацион-
канализационную сеть смешивающихся с водой огнеопасных или агрессивных жидкостей
необходимо пускать для промывания сильный поток воды.
1 ЛВЖ подразделяют на две группы по классам опасности (А и В):
А. ЛВЖ, не смешивающиеся с водой или смешивающиеся лишь частично. Они классифи-
классифицируются по температуре воспламенения:
Класс
AI
All
АШ
Температура воспламенения, °С
<21
21-55
55-100
В. Жидкости, смешивающиеся с водой в любых соотношениях и имеющие температуру
воспламенения <2ГС.
Таким образом, дисульфид углерода, эфир, бензол, петролейный эфир относятся к клас-
классу AI, а спирт, ацетон и т. п. — к классу В.
4. Хранение химических реактивов. Уничтожение вредных остатков 161
Остатки металлического натрия или калия следует уничтожать, обрабатывая
небольшие порции этих металлов в большом объеме спирта (защитные очки!)
Для уничтожения остатков калия предпочтительно применять грег-бутанол.
Необходимо всегда проверять, можно ли после определенной обработки (нап-
(например, перегонки, химической реакции) остатки снова употребить. Все ненужные
остатки надо соответственно действующим предписаниям собирать даже в
маленьких количествах или сразу обезвреживать. Об этом следует вести запись.
Чтобы лабораторные остатки веществ обезопасить, их надо помещать в
отдельные емкости с учетом химических свойств. Такие емкости должны быть
предусмотрены для следующих типов отходов:
• органические растворители, не содержащие галогены, и растворы
веществ, свободные от галогенов;
• органические растворители, содержащие галогены, и растворы веществ,
содержащие галогены;
• растворы солей, кислот, оснований, рН которых лежит в интервале 6—8;
• твердые органические лабораторные химикаты1;
• твердые неорганические вещества;
• ядовитые неорганические вещества, а также тяжелые металлы и их
растворы1;
• ртуть и неорганические ртуть-серебряные соединения1;
• ядовитые и горючие соединения1;
• цветные и благородные металлы, а также соединения, разделенные по
типу металла;
• иодсодержащие соединения;
• фильтры и массы для отсоса, хроматографические пластинки и содержи-
содержимое хроматографических колонок;
• осколки стекла.
Небольшие количества ядовитых, раздражающих и самовозгорающихся или
взрывающихся остатков веществ надо самим путем соответствующих химичес-
химических реакций превратить в безопасные соединения.
Кислоты и основания в водном растворе нейтрализуют осторожно гидрокарбона-
гидрокарбонатом натрия либо натриевой щелочью или разбавленной соляной либо серной
кислотами (контроль по рН 6-8).
Ангидриды или галогеноангидриды кислот превращают в метиловые эфиры
путем добавления по каплям избытка метилового спирта.
Фториды действием гидроксида кальция превращают в фториды кальция.
Органические перекиси восстанавливают сульфитом натрия или его раствором.
Неорганические перекиси, бром, иод восстанавливают раствором тиосуль-
тиосульфата натрия путем добавления по каплям.
Нитрилы или тиолы окисляют избытком 15%-ного раствора гипохлорита нат-
натрия путем многочасового перемешивания. Избыток гипохлорита натрия разру-
разрушают тиосульфатом натрия.
Водорастворимые альдегиды превращают в бисульфитные аддукты действи-
действием конц. водного раствора гидросульфита натрия.
1 В оригинальной упаковке производителя или в крепко закрываемой упаковке,
не бьющейся и с заметной и хорошо закрепленной этикеткой.
162 А Введение в лабораторную практику
Диазоалканы превращают в эфиры реакцией с уксусной кислотой.
Чувствительные к гидролизу элементоорганические соединения (например,
щелочнометаллические, гриньяровские), которые обычно применяют в органи-
органических растворителях, осторожно по каплям и при перемешивании добавляют в
н-бутанол. Образующиеся горючие газы отводят шлангом сразу в сброс. После
окончания выделения газа перемешивают еще 1 ч, а затем добавляют избыток
воды.
Дезактивация других опасных соединений описана в части Ж.
5. НЕОБХОДИМОЕ ЛАБОРАТОРНОЕ ОБОРУДОВАНИЕ И
ПОСУДА
Число, шт.
Холодильник Л ибиха, 2 х НШ 14,5, длина 400 мм ' 1
Воздушный холодильник, 2 х НШ 14,5, длина 400 мм 1
Холодильник Димрота, 2 х НШ 29 1
Насадка Кляйзена, 1 х НШ 29; 3 х НШ 14,5 1
Термометр, НШ 14,5, до 360 °С 1
Термометр, без шлифа, 360 "С 1
Вакуумный форштос, 2 х НШ 14,5 1
Переходные соединения, керн НШ 29, муфта НШ 14,5 1
Круглодонная или грушевидная колба, НШ 14,5, на 10 мл 1
Колба, НШ 14,5, на 25, 50 и 100 мл По 2 каждого объема
Колба, НШ 29, на 100 и 250 мл По 2 каждого объема
Колба, НШ 29, на 500 и 1000 мл По 1 каждого объема
Двугорлая колба с наклонным горлом, НШ 29; НШ 14,5, на 250 мл 1
Трехгорлая колба, 3 х НШ 29, на 1000 мл 1
Дефлегматор (колонка) Вигре, 2 х НШ 29, действующая длина 20 см 1
Дефлегматор (колонка) Вигре, 2 х НШ 14,5, действующая длина 10 см 1
Пробка, НШ 29 2
Пробка, НШ 14,5 2
Делительная воронка, на 500 мл 1
Капельная воронка, НШ 14,5 или 29, на 50 или 100 мл 1
Склянка для отсасывания, на 500 мл 1
Воронка Бюхнера, диаметр 8 см 1
Воронка Хирша, диаметр 10 мм (из пористого стекла) 1
Стеклянный фильтр, диаметр 8 см 1
Стеклянный фильтр, диаметр 4 см 1
Лабораторный стакан, на 10, 25, 50, 250 и 600 мм По 2 каждого объема
Лабораторный стакан, на 1000 мл 1
Колба Эрленмейера, на 25, 50,100 и 300 мл По 2 каждого объема
1 НШ — нормальный шлиф. — Прим. перев.
6. Литература 163
Число, шт.
Колба Эрленмейера, на 500 мл 1
Пробирка, размер 130 х 15 мм 20
Пробирка, размер 70 х 15 мм 20
Пробирка, размер 70 х 7 мм 20
Стеклянная трубка для прокаливания 10
Капилляр для определения температуры плавления 100
Часовое стекло 3
Предметное стекло (покровное стекло) 5
Хлоркальциевая трубка, НШ 29 1
Мерный цилиндр, на 10 и 100 мл По 1 каждого объема
Воздушная стеклянная баня, диаметр 16 см с двумя
керамическими крышками 1
Узкогорлая и широкогорлая склянки, на 30,50,100,250 и 500 мл По 1 каждого объема
Стеклянные трубки
Стеклянные палочки
Кроме того, необходимо также следующее оборудование: штатив для проби-
пробирок, держатель для пробирок, пинцеты, металлический шпатель, газовые, водя-
водяные и вакуумные шланги, резиновые пробки, асбестовая сетка, фильтровальная
бумага для простых и складчатых фильтров, корковое кольцо (подставка для
колб), зажимы, штативы лабораторные, горелки.
Желательно также иметь следующие приборы:
Число, шт.
Трехгорлая колба, НШ 14,5; 29; 14,5, на 500 мл 1
Двугорлая колба, НШ 14,5; 29, на 100 мл 1
Переходные соединения, муфта НШ 29, керн НШ 14,5 1
Затвор для мешалки с цилиндрическим шлифом НШ 29 1
Промывные склянки 2
Другие необходимые для работы приборы, например большие колбы и большие
лабораторные стаканы, эксикаторы, колонки для перегонки, головки колонок, мо-
моторы для перемешивания и др., должны выдаваться во временное пользование.
6. ЛИТЕРАТУРА
Исчерпывающие сведения об описанных в части А методах имеются в книгах:
Houben-Weyl: Methoden der Organischen Chemie. 4. Aufl. Bd. 3, Hrsg.: E. Muller. — Georg
Thieme Verlag, Stuttgart.
Technique of Organic Chemistry. Bd. 1-11. Hrsg.: A. Weissberger. — Interscience Publishers,
New York.
Ulmanns Encyclopadie der Technischen Chemie. 3. Aufl. — Urban & Schwarzenberg,
Munchen, Berlin, 1961. Bd. 2/1: Anwendung physikalischer und physikalisch-chemisch-
er Methoden im Laboratorium; 4. neubearb. u. erw. Aufl. — Verlag Chemie,
Weinheim/Bergstr., Deerfield Beach/Florida, Basel, 1980. Bd. 5: Analysen- und
Messverfahren, Hrsg.: H. Kelker.
164 А Введение в лабораторную практику
Analytikum. Methoden der analytischen Chemie und ihre theoretischen Grundlagen. —
Deutschen Verlag fur Grundstofflndustrie, Leipzig, 1990.
Правила безопасного обращения с химикатами
Bender H. F. Das Gefahrstoffbuch. Sicherer Umgang mit Gefahrstoffen in der Praxis. — Wiley-
VCH, Weinheim 2000.
Bernabei D. Sicherheit. Handbuch fur das Labor. — GIT Verlag, Darmstadt 1991.
Einfuhrung in die chemische Laboratoriumspraxis. Von E. Fanghanel u. a. — Deutscher
Verlag fur Grundstoffindustrie, Leipzig 1992.
Gefahrliche chemische Reaktionen, Hrsg.: L. Roth, U. Weller. — Ecomed
Verlagsgesellschaft, Landsberg/Lech 1991.
Rinze P. Gefahrstoffe an Hochschulen, Hrsg.: Gesellschafl Deutscher Chemiker. — VCH
Verlagsgesellschaft, Weinheim 1992.
Roth L, Weller U. Sicherheitsfibel Chemie. — Ecomed Verlagsgesellschaft, Landsberg/Lech 1991.
SchuferH. K, Jochum C. Sicherheit in der Chemie. — Carl Hanser Verlag, Munchen, Wien 1996.
Sicheres Arbeiten in chemischen Laboratorien. Hrsg.: Bundesverband der Unfallkassen. —
Munchen, 2002 (Schiftenreihe Theorie und Praxis der Pravention)
http://regelwerk.unfallkassen. de/daten/inform/I_8553.pdf.
Sicherheit in chemischen und verwandten Laboratorien. Hrsg.: F. Heske. — Verlag Chemie,
Weinheim 1983.
PicotA., Grenouilett P. Safety in the Chemistry and Biochemistry Laboratory. — VCH, New
York 1994.
Важные химические работы, касающиеся законов, правил и предписаний
Gesetz zum Schutz vorgefahrlichen Stoffen (Chemikaliengesetz — ChemG); BGB1 I, 2002,
S. 2090, 3082. S. 1704.
Verordnung zum Schutz vor gefa'hrlichen Stoffen (Gefahrstofrverordnung — GefStoffV);
BGB11, 1999, S. 2233; I, 2003, S. 3082.
Richtlinie 67/548/EWG zur Angleichung der Rechts — und Verwaltungsvorschriften fur die
Einstufung. Verpackung und Kennzeichnung gefahrlicher Stoffe; AB1 196, 1967, S. 1;
L225, 2001, S.I.
Anhang I der Richtlinie 67/548/EWG (Liste der gefahrlichen Stoffe); AB1 L 355, 2003, S. 1.
Betriebssicherheits verordnung (Betr Sich V); BGB1, 2002,1, S. 3777.
Kreislaufwirtschaft - und Abfallgesetz (KrW/AbfC); BGB11,1994, S. 1354; I, 2001, S. 2785.
Правила техники безопасности
Technische Regeln fur Gefahrstoffe, TRGS
TRGS 102 Technische Richtkonzentrationen (TRK) fur gefahrliche Stoffe
TRGS 201 Einstufung und Kennzeichnung von Abfallen zur Beseitigung beim Ungang
TRGS 514 Lagern sehr giftiger und giftiger Stoffe in Verpackungen und ortsbeweglichen Behaltern
TRGS 515 Lagern brandfordernder Stoffe in Verpackungen und ortsbeweglichen Bahaltern
TRGS 526 Laboratorien
TRGS 900 Grenzwerte in der Luft am Arbeitsplatz "Luftgrenzwerte"
TRGS 903 Biologische Arbeitsplatztoleranzwerte BAT-Berte
TRGS 905 Verzeichnis krebserzeugender, erbgutverandernder oder fortpflanzungsgefahr-
dender Stoffe
TRGS 907 Verzeichnis sensibilisierender Stoffe
6. Литература 165
Technische Regeln fur brennbare Fliissigkeiten (TRbF)
TRbF 20 Lager
TRbF 60 Ortsbewegliche Behalter
Technische Regeln Druckgase (TRG)
TRG 101 Gase
TRG 280 Allgemeine Anforderungen an Druckgasbehalter. Betreiben von Druckgasbehal-
tern
Работа с малыми количествами веществ
Lieb H., Schoniger W. Anleitung zur Darstellung organischer Praparate mit kleinen
Substanzmengen. — Springer-Verlag, Wien, 1961.
Ma T. S., Horak V. Microscale Manipulation in Chemistry. — John Wiley & Sons, New York,
Sidney, Toronto, 1976.
Mayo D. W., Pike R. M., Trumper P. K. Microscale Organic Laboratory. — John Wiley &
Sons, New York, 1994.
Harwood L. M., Moody C. J., Percy J. M. Experimental Organic Chemistry, Standard and
Microscale. — Blackwell Science, Oxford, 1998.
Перегонка и ректификация
Krell E. Handbuch der Laboratoriumsdestillation. — Deutscher Verlag der Wissenschaften,
Berlin, 1976. (Имеется русский перевод 1-го изд.: Крель Э. Руководство по лабора-
лабораторной ректификации. — М.: ИЛ, 1960.)
Einfuhrung in die Trennverfahren. Von E. Krell u. a. — Deutscher Verlag fur
Grundstoffindustrie, Leipzig, 1975.
Stage H., CZ-Chem. Techn. 1 A972), 263-272.
Хроматография
Gritter R. J., BobittJ. M., SchwartingA. G., Einfuhrung in die Chromatographie. — Springer-
Verlag, Berlin, Heidelberg, 1987.
Hrapia H. Einfuhrung in die Chromatographie. — Akademie-Verlag, Berlin, 1977.
Krauss G.-J., Krauss G. Experimente zur Chromatographie. — Deutscher Verlag der
Wissenschaften, Berlin, 1981.
Schwedt G. Chromatographische Trennmethoden. — Georg Thieme Verlag, Stuttgart, New
York, 1994.
Тонкослойная хроматография
Frey H. P., Zieloff K. Qualitative und quantitative Dtinnschichtchromatographie. —
VCH-Verlagsgesellschaft, Weinheim, 1992.
Dunnschicht-Chromatographie. Von H. Jork u. a. — VCH Verlagsgesellschaft, Weinheim 1989.
Kraus L., Koch A., Hoffstetter-Kuhn S. Dunnschichtchromatographie. — Springer-Verlag,
Berlin, Heidelberg, 1996.
Randerath K. Dunnschicht-Chromatographie. — Verlag Chemie, Weinheim, 1972.
StahlE. Thin-layer Chromatography. — Springer-Verlag, Berlin, Gottingen, Heidelberg, 1988.
Высокоэффективная жидкостная хроматография
Aced G., MockelH. J. Liquidchromatographie. — VCH Verlagsgesellschaft, Weinheim, 1991.
Ardrey R. E. Liquid Chromatography — Mass Spectrometry — Wiley-VCH, Weinheim, New
York, 2003.
Engelhardt #., Hochdruck-Flussigkeits-Chromatographie. — Springer-Verlag, Berlin, New
York, Heidelberg, 1977.
166 А Введение в лабораторную практику
Eppert G. J., Flilssigchromatographie. HPLC — Theorie und Praxis — Vieweg-Verlag,
Wiesbaden, 1997.
Gottwald W. RP-HPLC fur Anwender. — VCH Verlagsgesellschaft, Weinheim, 1993.
Handbuch derHPLC. Hrsg.: K. K. Unger. - GIT Verlag, Darmstadt, 1995.
Meyer V. R. Fallstricke und Fehlerquellen der HPLC in Bildern. - Wiley-VCH, Weinheim, 1999.
Meyer V. R. Praxis der Hochleistungs-Fliissigchromatographie. — Verlag Moritz Diesterweg;
Otto Salle Verlag; Verlag Sauerlander, Frankfurt/Main, 1990.
Газовая хроматография
Berezkin V. G., Zeeuw J. de: Capillary Gas Adsorption Chromatography. — Hiithig Verlag,
Heidelberg, 1996.
Ettre L. S., Hinshaw J. V., Rohrschneider L. Grundbegriffe und Gleichungen der
Gaschromatographie. — Hiithig Verlag, Heidelberg, 1996.
Gottwald W. GC fur Anwender. — VCH Verlagsgesellschaft, Weinheim, 1995.
Handbuch der Gaschromatographie, Hrsg.: E. Leibnitz, H. G. Struppe. — Akademische
Verlagsgesellschaft, Geet & Portig, Leipzig, 1984.
Jentsch D., Gaschromatographie, — Franckh'sche Verlagshandlung, Stuttgart, 1975.
KolbB. Gaschromatographie in Bildern. — Wiley-VCH, Weinheim, 1999.
Rddel W., Wolm G. Grundlagen der Gaschromatographie. — Deutscher Verlag der
Wissenschaften, Berlin, 1982.
Schomburg G. Gaschromatographie. — VCH Verlagsgesellschaft, Weinheim, 1986.
Wollrab A. Gaschromatographie. — Verlag Moritz Diesterweg; Otto Salle Verlag, Frankfurt
am Maim; Verlag Sauerlander, Aarau, 1982.
Спектроскопия (УФ, ИК, ЯМР, масс-спектрометрия)
Banwell N. С, McCash M. E. Molekiilspektroskopie — Ein Grundkurs. — В. G. Teubner,
Munchen, 1999.
Bechmann W., Schmidt J. Struktur- und Stoffanalytik mit spektroskopischen Methoden. —
B. G. Teubner, Munchen, 2000.
BorsdorfR., Scholz M. Spektroskopische Methoden in der organischen Chemie. — Akademie-
Verlag, Berlin 1982.
Cooper J. W. Spectroscopic Techniques for Organic Chemists. — John Wiley & Sons, New
York, Chichester, Brisbane, Toronto, 1980.
Derkosch J. Absorptionsspektralanalyse im ultravioletten, sichtbaren und infraroten Bereich. —
Akademie-Verlag, Leipzig, 1967.
FahrE., Mitschke M. Spektren und Strukturen organischer Verbindungen. — Verlag Chemie,
Weinheim, 1979.
Hesse M., Meier H., Zeeh B. Spektroskopische Methoden in der organischen Chemie. —
Georg Thieme Verlag, Stuttgart, 1991.
Pretsch E. Clerc J. T. Spectra Interpretation of Organic Compounds. — VCH
Verlagsgesellschaft, Weinheim, 1997.
Silverstein R. M., Webster F. X. Spectrometric Identification of Organic Compounds. — Johh
Wiley & Sons, New York, 1997.
Simon W., Clerc Th. Strukturaufklarung organischer Verbindungen mit spektroskopischen
Methoden. — Akademische Verlagsgesellschaft, Geest & Portig, Leipzig, 1967.
SternhellS., Kalman J. R. Organic Structures from Spectra. — John Wiley & Sons, New York,
1992.
Strukturanalytik. Von E. Steger u. a. — Deutscher Verlag fur Grundstoffindustrie. Leipzig,
Stuttgart, 1992.
6. Литература 167
Williams D. H, Fleming J. Strukturaufklarung in der organischen Chemie. — Georg Thieme
Verlag, Stuttgart, New York, 1991.
Спектроскопия в видимой и УФ областях
DMS-UV-Atlas. Bd. I-V. Hrsg.: H.-H. Perkampus, I. Sandemann, C. J. Timmons. —
Butterworth, London; Verlag Chemie, Weinheim, 1966-1971.
Gauglitz G. Praxis der UV-VIS-Spektroskopie. — Attempto Verlag, Tubingen, 1983.
Organic Electronic Spectral Data. Bd. 1-21. — J. Wiley & Sons, New York, 1960-1985.
Fabian J., Hartmann H. Light Absorption of Organic Colorants Theoretical Theatment and
Empirical Rules. — Springer-Verlag, Berlin, Heidelberg, New York, 1980.
Lang L. Absorptionsspektren im ultravioletten und im sichtbaren Bereich. Bd. 1-9. —
Akademiai Kiado, Budapest, 1959-1967.
Perkampus H.-H. UV-VIS-Spektroskopie und ihre Anwendungen. — Springer-Verlag,
Berlin, Heidelberg, New York, Tokyo, 1986.
Perkampus H.-H. UV-VIS-Atlas of Organic Compounds. — VCH Verlagsgesellschaft,
Weinheim, 1992.
Pestemer M. Anleitung zum Messen von Absorptionsspektren im Ultraviolett und Sicht-
Sichtbaren. — Georg Thieme Verlag, Stuttgart, 1964.
ИК-спектроскопия
Bellamy L. J. Ultrarotspektrum und chemische Konstitution. — Dr. Dietrich Steinkopff
Verlag, Darmstadt, 1974.
The IR-Spectra of Complex Molecules. — Chapman and Hall, London, 1975.
Brugel W. Einfuhrung in die Ultrarotspektroskopie. — Dr. Dietrich Steinkopff Verlag,
Darmstadt, 1969.
Faldini A., Schnepfel F. M. Schwingungsspektroskopie. — Georg Thieme Verlag, Stuttgart,
New York, 1985.
Gottwald W., Wachter G. IR-Spektroskopie fur Anwender. — VCH Verlagsgesellschaft,
Weinheim, 1997.
Gunzler H, Gremlich H.-U. IR Spectroscopy — An Introduction. — Wiley-VCH, Weinheim,
New York, 2002.
Gunzler H, HeiseH. M. IR-Spektroskopie. — VCH Verlagsgesellschaft, Weinheim, 1996.
Holly S., Sohar P. Infrarotspektroskopie. Hrsg.: G. Malewski. — Akademie-Verlag, Berlin,
1974; Absorption Spectra in the Infrared Region. — Akademiai Kiado, Budapest, 1975.
Pachler K. G., Matlok F, Gremlich H.-U. Merck FT-IR Atals. — VCH Verlagsgesellschaft,
Weinheim, 1987.
Pouchert С J. The Aldrich Library of FT-IR Spectra. Bd. 1—3. — Aldrich Chemical Company
1985-1989.
Schroder B. Raman/Infrared Atlas of Organic Compounds. — VCH Verlagsgesellschaft,
Weinheim, 1991.
Volkmann H. Handbuch der Infrarot-Spektroskopie. — Verlag Chemie, Weinheim, 1972.
Weidlein J., Mailer U., Dehnicke K. Schwingungsspektroskopie. — Georg Thieme Verlag,
Stuttgart, New York, 1988.
ЯМР-спектроскопия
BreitmaierE., VoelterW., 13-C-NMR-Spektroskopie, Verlag Chemie, Weinheim, 1989.
Breitmaier E. Vom NMR-Spektrum zur Strukturformel organischer Verbindungen. —
B. G. Teubner, Stuttgart, 1992.
Ernst L KohlenstofF-13-NMR-Spektroskopie. — Dr. Dietrich SteinkopffVerlag, Darmstadt, 1970.
Friebolin H Ein- und zweidimensionale NMR-Spektroskopie. — Wiley-VCH, Weinheim, 1999.
168 А Введение в лабораторную практику
GuntherH. NMR-Spektroskopie. — Georg Thieme Verlag, Stuttgart, 1992.
Herzog W.-D., Messerschmidt M. NMR-Spektroskopie fur Anwender. — VCH
Verlagsgesellschaft, Weinheim, 1994.
Kalinowski H.-O., Berger S., Braun S. l3C-NMR-Spektroskopie. — Georg Thieme Verlag,
Stuttgart, New York, 1984.
Kleinpeter T. NMR-Spektroskopie. Struktur, Dynamik und Chemie des Molekiils. —
J. A. Barth, Leipzig, Berlin, Heidelberg, 1992.
Kleinpeter E., BorsdorfR., 13C-NMR-Spektroskopie in der Organischen Chemie — Akademie
Verlag, Berlin, 1981.
Suhr H. Anwendung der kernmagnetischen Resonanz in der organischen Chemie. —
Springer-Verlag, Berlin, Heidelberg, New York, 1965.
Wehrli F. W., Marchand A. P., Wirtglin T. Interpretation of Carbon l3C-NMR-Spectra. —
John Wiley & Sons, New York, 1988.
Zschunke A. Kernmagnetische Resonanzspektroskopie in der organischen Chemie. —
Akademie-Verlag, Berlin, 1977.
Масс-спектрометрия
Benz W. Massenspektrometrie organischer Verbindungen. — Akademische
Verlagsgesellschaft Geest & Porting, Leipzig, 1969.
Budzikiewicz H. Massenspektrometrie. — Wiley-VCH, Weinheim, 1998.
McLafferty F. M. Interpretation of Mass Spectra. — W. A. Benjamin, New York, 1973.
Remane H., Herzschuh R. Massenspektrometrie in der organischen Chemie. — Akademie-
Verlag, Berlin, 1977.
Spiteller G. Massenspektroskopische Strukturanalyse organischer Verbindungen. —
Akademische Verlagsgesellschaft Geest & Portig, Leipzig, 1966.
Дисперсия оптического вращения
Circular Dichroism. Hrsg.: N. Berova; K. Nakanishi; R. W. Woody. — Wiley-VCH, New
York, Weinheim, 2000.
Crabbe P. ORD und CD in Chemistry and Biochemistry. — Academic Press, New York,
London, 1972.
Djemssi C. Optical Rotatory Dispersion: Application to Organic Chemistry. — McGraw Hill
Book Сотр., New York, 1960. (Русский перевод см. с. 170.)
Snatzke G. Optical Rotatory Dispersion and Circular Dichroism in Organic Chemistry. —
Heyden & Son, London, 1967.
Рентгеноструктурный анализ
BuergerM. J. Kristallographie. — Walter de Gruyter, Berlin, 1977.
Clegg W. Crystal Structure Determination. — Oxford University Press, Oxford, New York,
Tokyo, 1998.
DunitzJ. D. X-Ray Analysis and Structure of Organic Compounds. — VHCA, Basel, 1995.
Massa W. Kristallstrukturbestimmung. — B. G. Teubner, Stuttgart, 1996.
WolfelE. R. Theorie und Praxis der Rontgenstrukturanalyse. — Vieweg, Braunschweig, 1987.
Литература на русском языке1
Кейл В. Лабораторная техника органической химии. — М.: Мир, 1966.
Берлин А. Я. Техника лабораторной работы в органической химии, 2-е изд. — М.: Гос-
химиздат, 1963.
1 Литература на русском языке приведена из раннего русского издания «Органикума» (М.,
Мир, 1992. — Перевод 18-го немецкого издания 1990 года). — Прим. ред.
6. Литература 169
Вейганд К. Методы эксперимента в органической химии. — Ч. 1. — М.: ИЛ, 1951.
Воскресенский П. И. Техника лабораторных работ, 6-е изд. — М.: Госхимиздат, 1964.
Мортон Э. А. Лабораторная техника в органической химии. — М.: Госхимиздат, 1941.
Либ Г., Шенигер В. Синтез органических препаратов из малых количеств веществ. —
М.: Госхимиздат, 1957.
Линстед Р., Элвидж Дж., Вами М., Вилкинсон Дж. Современные методы исследова-
исследования в органической химии. — М.: ИЛ, 1959.
Черонис Н. Микро- и полумикрометоды органической химии. — М.: ИЛ, 1960.
Розенгардт М. И. Техника лабораторной перегонки и ректификации. — М.: Госхимиз-
Госхимиздат, 1951.
Жидкостная экстракция (теория и практика)/Ред. А. Г. Касаткин. — М.: Госхимиздат, 1958.
Фукс Н. В сб.: Реакции и методы исследования органических соединений: Т. 1. — М.:
Госхимиздат, 1951, с. 170-306.
Шемякин Ф. М., Мицеловский Э. С, Романов Д. В. Хроматографический анализ. — М.:
Госхимиздат, 1955.
Шталь Э. Хроматография в тонких слоях. — М.: Мир, 1969.
АхремА. А., Кузнецов А. И. Тонкослойная хроматография. — М.: Наука, 1965.
Шингляр М. Газовая хроматография в практике. — Химия, 1964.
Айвазов Б. В. Основы газовой хроматографии. — М.: Высшая школа, 1977.
Шай Г. Теоретические основы хроматографии газов. — М.: ИЛ, 1963.
Байер Э. Хроматография газов. — М.: ИЛ, 1961.
Руководство по газовой хроматографии. В 2-х ч.: Пер. С нем./Под ред. Э. Лейбница,
X. Штруппе. - М.: Мир, 1988.
Козицына Л. А., Куплетская Н. Б. Применение УФ-, ИК- и ЯМР-спектроскопии в
органической химии. — М.: Высшая школа. 1971.
Иоффе Б. В., Костиков Р. Р., Разин В. В. Физические методы определения строения
органических молекул. — Л.: Изд. ЛГУ, 1976.
Бранд Дж., Эглинтон Г. Применение спектроскопии в органической химии. — М.:
Мир, 1967.
Сильверстейн Р., Басслер Г., Моррил Т. Спектрометрическая идентификация органи-
органических соединений. — М.: Мир, 1977.
Рао Ч. Электронные спектры в химии. — М. Мир, 1964.
Гиллем А., Штерн Е. Электронные спектры поглощения органических соединений. —
М.: ИЛ, 1957.
Наканиси К. Инфракрасные спектры и строение органических соединений (практи-
(практическое руководство). — М.: Мир, 1965.
Кесслер И. Методы инфракрасной спектроскопии в химическом анализе. — М.: Мир,
1964.
Кросс А. Введение в инфракрасную спектроскопию. — М.: ИЛ, 1961.
Беллами Л. Инфракрасные спектры сложных молекул. — М.: ИЛ, 1963.
Жунке А. Ядерный магнитный резонанс в органической химии. — М.: Мир, 1974.
Ионин Б. И., Ершов Б. А. ЯМР-спектроскопия в органической химии. — М.: Химия,
1967.
Роберте Дж. Введение в анализ спектров ЯМР высокого разрешения. — М.: ИЛ, 1963.
Будзикевич Г., Джерасси К, Уильяме Д. Интерпретация масс-спектров органических
соединений. — М.: Мир, 1966.
БейнонДж. Масс-спектрометрия и ее применение в органической химии. — М.: Мир,
1964.
Полякова А. А., Хмельницкий Р. А. Введение в масс-спектрометрию органических сое-
соединений. — М.: Химия, 1966.
170 А Введение в лабораторную практику
Джонстон Р. Руководство по масс-спектрометрии для химиков-органиков. — М.:
Мир, 1975.
Джерасси К. Дисперсия оптического вращения. Применение в органической химии. — М.:
ИЛ, 1962.
ВеллюзЛ., Легран М., Грожан М. Оптический круговой дихроизм: Принципы измене-
изменения и применение. — М.: Мир, 1967.
ЛИТЕРАТУРА ПО ОРГАНИЧЕСКОЙ ХИМИИ
ВЕДЕНИЕ ЛАБОРАТОРНОГО ЖУРНАЛА
Данный практикум не заменяет учебника. Поэтому предваряющие лаборатор-
лабораторную методику небольшие введения надо обязательно дополнять изучением
соответствующего учебника. Кроме того, необходимо как можно раньше
познакомиться с химической литературой, которая в основном включает:
• оригинальные работы1, в которых содержатся сообщения о результатах
собственных исследований авторов; оригинальные работы публику-
публикуются в научных журналах, а также в виде патентов;
• обобщения и обзоры, чаще всего составляемые на основе критической
обработки оригинальной литературы за определенное время по опре-
определенной тематике;
• реферативную литературу, где излагается краткое содержание ориги-
оригинальных работ и содержится информация об обзорах;
• справочники табличных данных и др.
Эти публикации (в печатной форме или в режиме online) доступны через
Интернет2.
Следует выработать обязательную привычку, как можно более полно просматри-
просматривать литературу. Помните, что часто час работы с литературой может сэкономить
многие дни практической работы в лаборатории.
1. ОРИГИНАЛЬНАЯ ЛИТЕРАТУРА
1.1. СПЕЦИАЛЬНЫЕ ЖУРНАЛЫ
Оригинальные работы по определенной (обычно достаточно узко ограничен-
ограниченной) тематике публикуются, как правило, в специальных (научных) журналах.
В работах препаративного направления следует знакомиться не только с
1 Оригинальные работы в библиотечном деле называют также первичными источниками,
в то время как все монографии, справочники, реферативные издания объединяют в группу
вторичных источников.
2 Ср. Nachr. Chem. Tech. Lab. 46A998), 1188-1193.
172 Б Литература по органической химии
содержащей методики экспериментальной частью, но и изучить изложенные в
начале статьи общие положения. Важные для химика-органика журналы с их
сокращенными названиями по С. A. (Chemical Abstracts) перечислены ниже1:
Acta Chemica Scandinavica (Acta Chem. Scand.);
Analytical Chemistry (Analyt. Chem., Anal. Chem.);
Angewandte Chemie (Angew. Chem.);
Australian Journal of Chemistry (Aust. J. Chem.);
Bulletin of the Chemical Society of Japan (Bull. Chem. Soc. Japan);
Bulletin de la Societe Chimique de France (Bull. Soc. Chim. France; [Bl.]);
Canadian Journal of Chemistry (Can. J. Chem.);
Chemische Berichte (Chem. Ber.; [В.]; до 1997); продолжение журнала: Berichte der
Deutschen chemischen Gesellschaft (Ber. dt. Chem. Ges.; Ber. Deut. Chem. Ges.);
Chemical Communications (Chem. Commun.);
Chemistry A European Journal (Chem. Eur. J.);
Chemistry Letters (Chem. Letters; Chem. Lett.);
Collection of Czechoslovak Chemical Communications (Collect. Czech. Chem. Commun.);
European Journal of Organic Chemistry (Eur. J. Org. Chem.);
Helvetica Chimica Acta (Helv. Chim. Acta; [Helv.]);
Journal of the American Chemical Society (J. Amer. Chem. Soc.; J. Am. Chem. Soc.; [Am. Soc.]);
Journal of the Chemical Society, Chemical Communications (J. chem. Soc., Chem. Commun.);
Jorunal of the Chemical Society, Perkin Transactions (J. chem. Soc, [Soc] Perkin Trans.);
Journal of the Heterocyclic Chemistry (J. Heterocycl. Chem.);
Journal of Organic Chemistry (J. Org. Chem.);
Journal of Organometallic Chemistry (J. Organomet. Chem.);
Journal fur praktische Chemie (J. prakt. Chem.; [J. pr.]);
Liebigs Annalen der Chemie (LiebigsAnn. Chemie; LiebigsAnn. Chem., [А]; до 1997);
Monatshefte fur Chemie (Mh. Chemie; Monatsh. Chem.; [M]);
Organometallics (Organometallics);
Organic Letters (Org. Letters);
Synlett (Synlett);
Synthetic Communications (Synth. Commun.);
Synthesis (Synthesis);
Tetrahedron (Tetrahedron);
Tetrahedron: Asymmetry;
Tetrahedron Letters (Tetrahedron Lett.);
Доклады Академии наук СССР;
Журнал общей химии;
Журнал органической химии;
Известия Академии наук СССР, серия химическая;
Химия гетероциклических соединений.
1 Мы посчитали уместным привести здесь сокращения, соответствующие принятым в ре-
реферативном журнале «Химия», кроме того, даны сокращения, используемые в справочнике
Бейльштейна. — Прим. перев.
2. Обобщающие работы и обзоры 173
1.2. ПАТЕНТЫ
Для химика-синтетика часто представляют определенную ценность методики
получения химических соединений, содержащиеся в патентной литературе.
В большинстве стран патенты в настоящее время классифицируют по
Международной классификации изобретений (МКИ), пришедшей на смену
национальным системам. Для поиска способов получения соединений опре-
определенных классов необходимо прежде всего ознакомиться с их расположени-
расположением в последних изданиях МКИ. Например, способы получения 1,2,4-тиадиа-
золов надо было бы искать в классе МКИ С 07 D/285/08, что означает:
С раздел: химия и металлургия
07 класс: органическая химия
D подкласс: гетероциклические соединения
285 группа: гетероциклы с кольцами, содержащими атомы серы и азота
08 подгруппа: 1,2,4-тиадиазолы
При поиске пришлось бы просмотреть все патентные описания, посвящен-
посвященные 1,2,4-тиадиазолам.
В Германии с 1975 года МКИ является обязательной. Для сопоставления со
старой классификацией имеются соответствующие справочные таблицы.
В Германии описания изобретений сначала публикуются в форме «открытой
выкладки» (Offenlegungsschriften, OS), после соответствующей проверки — вто-
вторично в виде «выкладок» (Auslegeschriften, AS), и наконец, в виде патентных
описаний (Patentschriften, PS). С 1981 г. промежуточная стадия AS упразднена.
Патенты Германии примерно до 1940 г. собраны в издании Friedlander,
Fortschritte der Teerfarbenproduktion, Springer-Verlag, Berlin.
Реферативные журналы «Химия» и Chemical Abstracts реферируют хими-
химические патенты важнейших стран.
Кроме того, имеется банк данных патентов (DPCI, IFIPAT, INPADOC,
JAPIO, PATDPA, WPINDEX/WPIDS/WPIX), которые можно получить по
электронной связи через CD-ROM или также в режиме online (через сетевую
связь с центральным компьютером (см. разд. Б.6.3I.
2. ОБОБЩАЮЩИЕ РАБОТЫ И ОБЗОРЫ
Обобщающие работы и обзорные статьи дают возможность быстрого доступа к
оригинальной литературе, цитируемой в этих источниках. Однако они не дают
гарантии полноты цитирования литературы по обсуждаемой теме.
Перечисляемые ниже журналы публикуют частично наряду с оригиналь-
оригинальными работами обзоры по разным темам:
Accounts of Chemical Research (Ace. Chem. Res.);
Angewandte Chemie (Angew. Chem.)
Chemical Reviews (Chem. Rev.);
1 О бесплатной патентной информации по Интернету см. Nachr. Chem. Tech. Lab. 47
A999), 567-569.
174 Б Литература по органической химии
Chemical Society Reviews (Chem. Soc. Rev.);
Synthesis;
Tetrahedron (Tetrahedron)
Успехи химии (Усп. хим.).
Обобщающие работы и обзоры новейших достижений публикуются также в
следующих серийных изданиях:
Advances in Heterocyclic Chemistry. A. R. Katritzky (ed.). - Academic Press, New York,
London (Adv. Heterocycl. Chem.);
Advances in Organic Chemistry. R. A. Raphael, E. С Taylor, H. Wynberg (eds.). -
Interscience Publishers, New York, London (Adv. Org. Chem.);
Advances in Physical Organic Chemistry. D. Bethell (ed.). - Academic Press, New York,
London (Adv. Phys. Org. Chem.);
Advances in Organometaltic Chemistry. F. G. A. Stone, R. West (eds.). - Academic Press, San
Diego (Adv. Organomet. Chem.);
Annual Reports in Organic Synthesis. J. McMurray, R. B. Miller (eds.). - Academic Press,
New York (Ann. Rep. Org. Chem.);
Contemporary Organic Synthesis. Royal Society of Chemistry, London (Contemp. Org.
Synth.);
Neuere Methoden der organischen Chemie. W. Foerst (ed.). — Verlag Chemie,
Weinheim/Bergstrasse (Neuere Methoden);
Organic Reactions. - John Wiley & Sons, New York, London (Org. React.);
Organic Synthesis Highlights.: J. Mulzer, H. Waldmann, u. a. Wiley-VCH, Weinheim;
Progress in Organic Chemistry J. Cook, W. Carruthers (eds.). - Butterworth, London (Progr.
Org. Chem.);
Progress in Physical Organic Chemistry. S. G. Cohen, A. Streitwieser, Jr., R. W. Taft. -
Interscience Publischers, New York. London (Progr. Phys. Org. Chem.);
Реакции и методы исследования органических соединений. - М.: Химия;
Synthetic Reagents. J. S. Pizey (ed.). — Ellis Horwood Ltd., Chichester;
Topics in Current Chemistry — Fortschritte der chemischen Forschung. — Springer-Verlag,
Berlin, Gottingen, Heidelberg (Fortschr. Chem. Forsch.).
Публикации подобного рода появляются также в виде справочников и
сборников методик. Наиболее обширным сборником методик получения ве-
веществ разных классов является: Houben-Weyl, Methoden der organischen Chemie,
E. Muller, 4. Auflage. — Georg Thieme Verlag, Stuttgart (Houben-Weyl). В этом из-
издании наряду с общими химическими методами имеются также обширные
части, посвященные анализу, лабораторной технике, физическим методам1.
Широко представлены важнейшие методы препаративной органической
химии в книге: Weygand-Hilgetag, Organisch-chemische Experimentierkunst,
4. Auflage. — Johann Ambrosius Barth, Leipzig, 1970. Ценной особенностью это-
этого издания является возможность поиска по указателю не только классов сое-
соединений, но и методов (например, методы образования определенных связей,
их разрыв, перегруппировки и т. д.).
Такой принцип регистрации принят и углублен в издании: Theilheimer W.,
Synthetische Methoden der organischen Chemie (Synthetic Methods of Organic
1 Часть томов предыдущего, 3-го издания Houben-Weyl в 1930-х годах переводилась на рус-
русский язык. Из томов 4-го издания переведен материал, посвященный анализу. — Прим. перев.
3. Реферативная литература 175
Chemistry). — Verlag S. Karger, Basel, New York. T. 1—4 вышли на немецком язы-
языке; начиная с т. 5, издание выходит на английском языке. Ежегодно выходя-
выходящие дополнения освещают новые оригинальные работы, важные для препара-
препаративной органической химии. Поиск нужной реакции облегчается принятой в
книге своеобразной символикой.
Успехи в некоторых областях органической химии освещены в изданиях:
J. March, Advanced Organic Chemistry. — John Wiley & Sons, New York, 1992.
F. A. Carey, R. J. Sundberg, Organische Chemie. - VCH. Weinheim, 1995.
J. Fuhrhop, G. Penzlin, Organic Synthesis. — Concepts, Methods, Starting Materials, VCH
Weinheim, 1994.
R. О. С Norman, J. M. Coxon, Principles of Organic Synthesis. — Blackie, London, 1993.
К. С Nicolaou, E. J. Sorensen, Classics in Total Synthesis. -VCH, Weinheim, 1996.
Methodicum Chemicum.: F. Korte. — Georg Thieme Verlag, Stuttgart; Academic Press, New
York, San Fransisco, London.
Comprehensive Organic Synthesis B. M. Trost, I. Flemming (eds.). — Pergamon Press,
Oxford, 1991.
The Chemistry of Functional Groups. S. Patai (ed.). — Interescience Publishers, John Wiley &
Sons, London, New York, Sydney (Chem. Funct. Groups).
Comprehensive Organic Functional Group Transformations.: A. R. Katritzky, O. Meth-Cohn,
C. W. Rees (eds.). - Pergamon, Elsevier Science, Oxford, 1995.
The Chemistry of Heterocyclic Compounds. AS. Weissberger (ed.) — Interscience Publisherr,
New York, London.
Heterocyclic Compounds. R. C. Elderfield (ed.). — John Wiley & Sons, New York; Chapman
& Hall, London (Heterocycl. Compounds).
Очень полезным с точки зрения получения и применения важнейших реа-
реагентов является энциклопедический справочник Физер Л., Физер М. Реаген-
Реагенты для органического синтеза: Пер. с англ. Т. 1—6. — М.: Мир, 1970—1975.
Сборником тщательно проверенных новых методик с указаниями возмож-
возможностей расширения сфер их применения является серия Organic Syntheses (Org.
Synth.) - John Wiley & Sons, New York1.
Encyclopedia of Reagents for Organic Synthesis.: L. A. Paquette (ed.). — John Wiley
& Sons, Chichester. 1995.
3. РЕФЕРАТИВНАЯ ЛИТЕРАТУРА
3.1. СПРАВОЧНИК БЕЙЛЬШТЕЙНА
Важнейший источник информации для химика-органика — справочник
Бейльштейна [Beilsteins Handbuch der Organischen Chemie]. Он выходит с
1918 г. В настоящее время выходит в свет 4-е издание. В справочнике система-
систематизированы2 все опубликованные в научной литературе данные о получении
1 Некоторые тома этой серии переведены на русский язык под названием «Синтезы орга-
органических препаратов». — Прим. перев.
2 Описание системы дано в I т. основного издания, а также в работах Weissbach О. Beilstein-
Leitfaden. — Springer-Verlag, Berlin, Heidelberg, New York, 1975; Hoffmann E., Sunkel J.,
Luckenbach R. Beilsteins Handbuch der organischen Chemie - Aujbau, Inhalt, Benutzungsweise und
Bedeutung. - Chemie Labor u. Betrieb, 1982, 33, 205-214, 403,451.
176
Б Литература по органической химии
и свойствах всех соединений углерода. Приводятся данные о строении и конфи-
конфигурации, нахождении в природе и выделении из природных продуктов, получе-
получении, образовании, очистке, структурных и энергетических параметрах молекул,
физических свойствах, химическом, поведении, идентификации и анализе,
солях и продуктах присоединения. Приводимые факты критически проверены.
Внимание/При цитировании оригинальной литературы используются сок-
сокращения (разд. Б, 1.1). Информация об этом имеется также в начале каждого
тома справочника.
Справочник Бейльштейна в настоящее время состоит из шести серий
(основное издание Hauptwerk и пять дополнений Erganzungswerke) со следую-
следующим временным охватом:
Hauptwerk (основное издание)
I.
И.
III.
Erganzungswerk (дополнение)
Erganzungswerk
Erganzungswerk
III./IV. Erganzungswerk
IV.
V.
Erganzungswerk
Erganzungswerk
(на английском языке)
Сокращение
Н
EI
ЕП
ЕШ
Е Ш/Е IV (с. 17-27)
EIV
EV
Период времени (годы),
за который литература
полностью включена в
справочник
до 1909
1910-1919
1920-1929
1930-1949
1930-1959
1950-1959
1960-1979
Обычно актуальность отдельных серий за указанные годы выше, чем
полностью охваченная литература.
Справочник Бейльштейна включает 27 томов, многие из которых в новых
дополнительных сериях разрослись до нескольких книг. Общий предметный и
формульный указатели охватывают серии от Н до Е II. Кроме того, в конце
каждого тома справочника имеется предметный указатель, а начиная с серии
Е III еще и формульный. Сводные указатели меньшего объема есть к отдель-
отдельным томам серии Е III. Дополнительные серии Е II и Е IV в т. 17—27 (гетеро-
(гетероциклические соединения) объединены.
Необходимо учитывать, что начиная с серии Е Ш/Е IV и далее Е IV была
изменена номенклатура: если прежде префиксы располагались по «возрастаю-
«возрастающей сложности» (см. Е III, Bd. 5, 1. Teilband, «синие страницы»), то теперь их
записывают в алфавитном порядке (см. Е IV, общий указатель — т. 17—18,
«желтые страницы»). Другие изменения номенклатуры связаны с использова-
использованием правил ИЮПАК.
Продажные компьютерные программы {Beilstein, Key, Sandra) позволяют опре-
определить том справочника Бейльштейна, в котором должно находиться искомое со-
соединение, даже если оно еще не синтезировано и без знания системы регистров.
Информацию, содержащуюся в справочнике Бейльштейна, можно полу-
получить и электронным путем через Inhouse-Version Beilstein Crossfire (Beilstein
Informationssysteme, Frankfurt am Main).
3. Реферативная литература 177
3.2. РЕФЕРАТИВНЫЕ ЖУРНАЛЫ
Даже квалифицированный химик может регулярно просматривать лишь отно-
относительно небольшое число специальных журналов, объем и число которых пос-
постоянно возрастают. Для получения более широкой информации и поисков
используют реферативные журналы, охватывающие всю химическую литературу
и кратко (без какой-либо оценки) освещающие оригинальные работы. Материал
располагается в определенном порядке. Реферат содержит следующие данные:
автор (авторы), место публикации работы, заголовок и содержание.
Американское химическое общество издает с 1907 г. реферативный журнал
Chemical Abstracts (CA), в котором реферируется вся химическая литература.
В последние годы сервис С. A. (CAS) наряду с данными из ранних рефератов
предлагает банк данных для online-исследований по предметным указателям,
соединениям, структурам и другим вопросам, который может быть получен
через STN-банк данных (см. разд. Б,6.3). Использование этих данных требует
выхода на соответствующий главный компьютер по банку данных, наличия
соответствующего терминала и владения языком команды. Надежность и эко-
экономия времени и затрат достижимы в ручных или online-поисках только тогда,
когда профиль поиска для соединений и предметного содержания словаря
очень точно сформулирован. Для этого требуется точное знание строения и
порядка регистров в СА.
Для каждого тома СА могут быть использованы следующие регистры:
Subject Index, с 1972 г. разделен на
General Subject Index;
Chemical Substance Index (для построения и поиска см. также разд. Б,6.1.1 и Б,6.2);
Formula Index (соответствует системе НШ);
Author Index;
Numeral Patent and Patent Concordance, с 1981 г. объединен в один регистр;
Ring Index;
Index Guide.
Index Guide (издается с 1968 г.) дает в алфавитном порядке перекрестные
данные для названий соединений, которые встречаются в литературе, и систе-
систематические регистрационные названия, принятые в СА. За каждый пятилет-
пятилетний регистрационный период выпущены сводные тома Index Guide. Как в
General Subject Index, так и в Chemical Substance Index имеются указания о
когда либо действующем Index Guide.
Важные сведения о структуре и использовании СА дают четыре приложе-
приложения к Index Guide.
Приложение I поясняет порядок построения словаря для поиска по General Subject Index и
алфавитный регистр, по которому можно перепроверить выбранный поиск.
Приложение II информирует о построении и использовании выше названных регистров томов.
Приложение III объясняет методику предметного поиска в General Subject Index.
Приложение /Ксодержит правила номенклатуры для образования систематического реги-
регистрационного номера химического соединения. Они базируются в основном на правилах,
принятых в ИЮПАК (см. разд. Б,5).
178 Б Литература по органической химии
Начиная с 1969 г. все индивидуальные химические соединения получают в
СА регистрационные номера, приводимые также в рефератах и указателях.
Реферативный журнал «Химия» (РЖХим) издается с 1953 г. Всесоюзным
институтом научной и технической информации (ВИНИТИ) в Москве. Он
также реферирует всю химическую литературу. Кроме рефератов статей из пе-
периодических журналов в РЖХим приводятся краткие данные о новых книгах,
монографиях, брошюрах и других непериодических изданиях, а также о рецен-
рецензиях, диссертациях, патентах. Журнал снабжается авторским, формульным и
предметным указателями.
Старейший химический реферативный журнал Chemische Zentralblatt (С.)
на немецком языке перестал выходить в 1969 г. О построении и содержании
этого издания можно прочесть в книге Новака (см. список литературы в конце
главы), а также в раннем издании «Органикума» (пер. с нем. — М.: Мир, 1979).
3.3. СИГНАЛЬНАЯ ИНФОРМАЦИЯ
Для текущей информации по отдельным областям науки служат специальные
издания, в которых обеспечивается быстрое появление рефератов, или (что ме-
менее ценно) воспроизводятся только заголовки оригинальных работ. Такими из-
изданиями охватывается лишь ограниченное число журналов, исчерпывающий
поиск по этим источникам невозможен.
Пример подобной реферативной службы — журнал Chemische Informa-
tionsdienst (Chem. Inform.), издаваемый с 1970 г. западногерманским издатель-
издательством Verlag Chemie, Weinheim/Bergstrasse. В этом журнале обрабатываются
публикации в наиболее важных специальных журналах; рефераты для нагляд-
наглядности содержат формульные схемы. Патентная литература не включена.
Current Contents (издание Института научной информации в Филадель-
Филадельфии) содержит названия статей, опубликованных в более чем 5000 журналах.
Возможен поиск по фамилиям авторов и библиографическим данным, а также
по ключевым словам в отдельных названиях статей.
4. СПРАВОЧНИКИ ТАБЛИЧНЫХ ДАННЫХ
Самый обширный справочник табличных данных — многотомное издание
Landolt-Bsrnstein, Zahlenwerte und Funktionen aus Physik, Chemie, Astronomie,
Geophysik und Technik, 6. Aufl, а также Landolt-Bornstein, Neue Serie, Hrsg.:
K.-H. Hellwege, Springer-Verlag, Berlin, Gottingen, Heidelberg, 1961 ff.
Физические константы важнейших неорганических и органических ве-
веществ, а также табличные данные и описание функций собраны в однотомни-
однотомнике Handbook of Chemistry and Physics, CRC Press, Cleveland, Ohio. Это издание
ежегодно перерабатывается с включением новейших данных.
На немецком языке имеются справочники аналогичного содержания:
D'Ans-Lax, Taschenbuch far Chemiker und Physiker, Bd. 1/1991, Bd. 2/1983, Bd.
3/1970, Springer Verlag, Berlin, Heidelberg, New York; Chemikerkalender,
Hrsg.: C. Synowitz, K. Schafer, Springer-Verlag, Berlin, Heidelberg, New York,
1984; Tabellenbuch Chemie, Deutscher Verlag fur Grundstoffindustrie, Leipzig, 1990.
5. Указания о номенклатуре 179
5. УКАЗАНИЯ О НОМЕНКЛАТУРЕ
С течением времени химическая номенклатура сильно изменилась. Вначале
предпочитали тривиальные названия, которые и сегодня могут быть применены
во многих случаях. Первой попыткой систематического обозначения была Жене-
Женевская номенклатура, которая сегодняшним требованиям уже не удовлетворяет.
Правила, которые действуют в настоящее время, были разработаны
ИЮПАК (International Union of Pure and Applied Chemistry) на английском
языке и приняты для «Органикума» с минимальным количеством изменений.
(В русском языке традиционно использование названий бензол, толуол, наф-
нафталин, глицерин вместо benzen, toluen, naphtalen, glycerol. — Ред.)
Согласно правилам ИЮПАК название химического соединения обра-
образуется от названия родового соединения, которое в целом отражает углерод-
углеродный скелет, и названия заместителей, а также их числа и положения. Замести-
Заместители — это фрагменты молекул, так называемые остатки, алкильные, арильные
или гетероциклические, присоединенные каждый к одному из атомов С ос-
основной цепи; наличие заместителей отражено префиксами в названии соеди-
соединения; в присутствии нескольких заместителей они указываются в алфавит-
алфавитном порядке так же, как и характеристические группы (например. —ОН,
—NH2, —С1, —С=О, —СООН и др.), для которых действует шкала значимости,
т. е. «старшинства». В качестве основной группы принимают наиболее стар-
старший заместитель в этой шкале (см. табл. Б.1; заместительная номенклатура)
или дают название соединения по характеру его функционального класса
(радикало-функциональная номенклатура), что образует суффикс в названии
соединения. При нумерации атомов родовой структуры основной (старший)
заместитель получает наиболее низкую цифру. Все другие характеристические
группы указываются префиксами, которые в алфавитном порядке присоеди-
присоединяются к родовому названию.
Заместительная номенклатура и радикало-функциональная номенклатура
дают возможность обозначить соединение по правилам ИЮПАК. В первой из
них замена одного Н-атома на заместитель обозначается как префикс или
суффикс, например:
2 1
С1—СН2—СН2—ОН Родовое название: этан
Характеристические группы: С1, ОН
Основная группа: ОН; -ол
Название: 2-хлорэтанол
4 3 2 1
НО-СН2-СН2-С-СН3 Родовое название: бутан
II Характеристические группы: ОН, =0
О Основная группа: =О; -он
Название: 4-гидроксибутан-2-он
Для радикало-функционального обозначения вместо указания характерис-
характеристических групп проверяют, принадлежит ли соединение к определенному
классу (см. табл. Б.1). К названию класса (при нескольких вариантах старши-
старшинства — к наиболее старшему) добавляют в качестве префикса названия остат-
180
Б Литература по органической химии
Таблица Б. 1. Характеристические группы по убывающему старшинству в номенклатуре ИЮПАК
Характеристические
группы
Анионы, напр. —О~
Катионы, напр. —NR4"
-соон
-(С)ОО№
ОН
-SO3H
-COOR (где R - алкил)
—SO3R (где R — алкил)
-COHal
-(C)OHala
-SO2Hal
-CONH2
-SO2NH2
-CsN
-(CKNa
н>=°
п
\
\
fc)-s°
-'он
-SH
-ООН
-NH2
=NH
-PH2R (где R - алкил)
-О
—SR (где R — алкил)
—Галоген
-NO
-NO2
Обозначение
префикса
-ато-
-ониа-
карбокси-
тиокарбокси-
сульфо-
алкоксикарбонил
алкоксисульфонил
галогенформил-
галогенсульфонил-
карбамоил-
сульфамоил-
циан
формил-
оксо-
оксо-
тиоксо-
гидрокси-
сульфанил-,
меркапто-
гидроперокси-
амино-
имино-
фосфино
алк(ил)окси-
алкилсульфанил -,
алкилтио-
галоген-
нитрозо-
нитро-
Обозначение суффикса
заместитель
-(он)иум
-карбоновая кислота
-кислота
тион карбоновая кислота
-сульфоновая кислота
алкиловый эфир карбо-
карбоновой кислоты
алкиловый эфир сульфо-
сульфоновой кислоты
-галогенид карбоновой
кислоты
-галогенид кислоты
-оил галогенид
-галогенид сульфоновой
кислоты
-амид карбоновой кислоты
-амид сульфоновой кислоты
-карбонитрил
-нитрил
-карбальдегид
-ал(ь)
-тион
-ол
-тиол
-амин
-имин
радикало-
функциональная
группа
-цианид
-кетон
-тиокетон
-алкоголь (спирт]
-гидропероксид
амин
-фосфин
-алкилэфир
-алкилсульфид
-галогенид
а Атом углерода, взятый в скобки, префиксом или суффиксом не обозначается, например,
СН3СН2СООН: пропановая (пропионовая) или этанкарбоновая кислота.
6. Проведение литературного поиска 181
ков по алфавиту. Другие характеристические группы указываются в виде
префиксов в названиях остатков.
С1-СН2-СН2-ОН Характеристические группы: С1, ОН
Название класса: спирт (алкоголь)
Название остатка: этил
Название: 2-хлорэтиловый спирт
НО-СН2-СН2-С-СН3 Характеристические группы: ОН, С=О
II Название класса: кетон
О Название остатков: этил, метил
Название: метилB-гидроксиэтил)кетон
Наряду с этими вариантами номенклатуры для специфических классов со-
соединений (например, гетероциклов, природных соединений и др.) имеются
еще другие системы наименований или специальные правила (см. разд. Б,8).
С помощью компьютерных программ ACD/Name (Advanced Chemistry
Development) и AUTONOM (Beilstein) можно быстро составить названия
(в системе ИЮПАК) соединения по химической структурной формуле1.
6. ПРОВЕДЕНИЕ ЛИТЕРАТУРНОГО ПОИСКА
Способ просмотра и объем необходимой литературы существенно зависят от
конкретной задачи. Здесь мы ограничиваемся лишь несколькими наиболее
часто встречающимися случаями.
6.1. ПОИСК СВЕДЕНИЙ ОБ ОПРЕДЕЛЕННОМ
ОРГАНИЧЕСКОМ СОЕДИНЕНИИ
6.1.1. ИСЧЕРПЫВАЮЩИЙ ЛИТЕРАТУРНЫЙ ПОИСК
Прежде всего записывают молекулярную формулу соединения. Это делают на
основе известной структуры или названия, составленного в соответствии с при-
применяемыми в справочнике Бейльштейна правилами номенклатуры (Е III, том 5,
синие страницы; Е IV, общий указатель к томам 17/18, желтые страницы).
Просматривая формульный или предметный указатель справочника Бейльштей-
Бейльштейна устанавливают, было ли искомое соединение описано до 1929 г. Если было
описано, по регистрационному номеру находят дополнительные сведения в ЕIII
и ЕIV. Регистрационные номера приводятся в верхней строке страницы. Там же
на нечетных страницах дополнительных серий имеется указание, на какой стра-
странице основного издания описано данное соединение (либо было бы описано, ес-
если бы было известно до 1909 г.). Таким образом, на страницах каждого тома всех
дополнительных серий, имеющих в верхней строке указание на определенную
страницу основного издания, описываются соединения того же или близкого
строения. Если в дополнительных сериях слишком много страниц содержат ука-
указания на одну и ту же страницу основного издания, то лучше воспользоваться
формульным или предметным указателем соответствующего тома.
Если искомого соединения нет в общих указателях (т. е. оно не описано до
1929 г.), то проводят поиск в дополнительных сериях Е III, E III/IV, Е IV и EV,
1 ACD/Name: Advanced Chemistry Development Inc., Toronto: WWW:http://www.acdlabs.corn.
AUTONOM: Beilstein Informationssysteme, Frankfurt a.M., WWW:http://www.beilstein. com.
182 Б Литература по органической химии
опираясь на систему, принятую в справочнике. С системой можно ознакомиться,
например в т. 1 основного издания, благодаря чему можно определить том, в кото-
котором надлежит искать нужное соединение. После этого целесообразно продолжить
поиск, пользуясь имеющимися кданному тому формульным и предметным указа-
указателями, охватывающими основное издание и дополнения I—IV (см. разд. Б,3.1.)
Тщательный просмотр справочника Бейльштейна не требует большого тру-
труда; в то же время обращающийся к этому изданию получает для искомого соеди-
соединения всю литературу до 1949 г. (или соответственно до 1959 г.; см. с. 176). Поиск за
последующие годы выполняют с помощью СА или РЖхим. В СА начинают с
просмотра последнего имеющегося формульного указателя (указателя химичес-
химических соединений). Для того чтобы определить, под каким названием соединение
следует искать в указателе, необходимо, начиная с тома 69 A968 г.), предвари-
предварительно справиться по Index Guide. Поскольку в отдельные периоды происходи-
происходили изменения в номенклатуре, необходимо просмотреть все пятилетние сборни-
сборники Index Guide. Информацию о целях и построении Index Guide содержат
выпуски 1991 г. (т. 75) и в особенности 1977 г. Найдя нужное регистрационное наз-
название, просматривают ежегодные предметные указатели (указатели химических
соединений) или соответствующие сводные 5- и 10-летние указатели. Так находят
номера рефератов (до т. 65 за 1966 г. — номера страниц), а при ознакомлении с ни-
ними — ссылку на оригинальную работу. Если после ознакомления с рефератом не-
неясно, имеет ли данная работа отношение к интересующей проблеме, все равно
следует внимательно просмотреть оригинальную работу. Дело в том, что все
встречающиеся в оригинале соединения регистрируются в указателе, однако не
все они обязательно упоминаются в реферате; поэтому не надо удивляться тому,
что найденное по указателю соединение отсутствует в реферате.
При просмотре СА доходят (начиная с текущего года) вплоть до того года,
который является последним для литературы, включенной в справочник
Бейльштейна.
6.1.2. ПОИСК ПОДХОДЯЩЕЙ МЕТОДИКИ ПОЛУЧЕНИЯ
При поисках подходящей методики получения органического соединения, от-
отсутствующего в «Органикуме», следует прежде всего просмотреть многотом-
ник Физер Л., Физер М. Реагенты для органического синтеза. Не найдя и здесь
нужной информации, обращаются к соответствующему тому справочника
Houben-Weyl, к приведенным в разд. Б,2 сборникам методик, а затем при не-
необходимости к справочнику Бейльштейна и, наконец, СА. Можно рекомендо-
рекомендовать просмотр новейших указателей СА, как уже говорилось в разд. Б,6.3.1.
С найденными ссылками следует ознакомиться в оригинале, даже если в рефе-
реферате описываются лишь свойства искомого соединения: в оригинале часто все
же можно обнаружить и способ получения1.
1 Ценное пособие — книга Лернер И. М., Берлин А. И., Словачевская Н. М. Указатель пре-
препаративных синтезов органических соединений. — Л.: Химия, 1973, где в очень четкой форме
собраны ссылки на методы синтеза почти 20 тыс. соединений. Аналогичный справочник из-
издан и по синтезу других классов соединений: Лернер И. М., Гонор А. А. Указатель препаратив-
препаративных методов синтеза неорганических, комплексных и элементоорганических соединений. -
Л.: Химия, 1966. — Прим. перев.
6. Проведение литературного поиска 183
6.2. ПОИСК СВЕДЕНИЙ О КЛАССАХ СОЕДИНЕНИЙ
Решая задачу синтеза ранее не описанного соединения, лучше всего сначала озна-
ознакомиться с методами получения веществ соответствующего класса. Получить ис-
исчерпывающую информацию такого рода нелегко, да чаще всего в этом и нет необ-
необходимости. Начать можно с предметного указателя к «Органикуму» и с литературы,
приведенной в конце разделов. Затем можно обратиться к другим источникам в
следующей последовательности: Weigand-Hilgetag, Houben-Weyl, Theilheimer (см.
разд. Б,2). Информацию можно получить и из СА: в общепредметный указатель
включены такие понятия, как алкилирование, арилирование, реакции присоеди-
присоединения, атакже именные реакции, например реакция Канниццаро, перегруппиров-
перегруппировка Кляйзена, синтез индолов по Фишеру, реакция Гриньяра и т. д. (ср. разд. Б,3.2).
Распространение найденного метода синтеза на новые соединения, а также
поиск альтернативных методов синтеза уже известных веществ, как правило,
требует большого опыта (см. разд. В,8).
Часто возникает необходимость получить ответ на вопрос, известны ли ве-
вещества, структурно родственные искомому соединению. В справочнике
Бейльштейна такие соединения располагаются в том же томе, что и искомое,
если только наличие дополнительных функциональных групп не приведет к
регистрации их в более позднем томе (принцип наиболее позднего места).
Вследствие этого необходимо учесть систему Бейльштейна и другие издавае-
издаваемые Бейлылтейновским институтом материалы. Для поиска литературы после
1949 г. (или после 1959 г.) снова можно рекомендовать просмотр СА. В указа-
указателе химических соединений (предметном указателе) соединения объединя-
объединяются в гнезда в соответствии с их родоначальной структурой.
Например, все соединения пиридина регистрируют совместно как соеди-
соединения пиридина, далее указаны через запятую в алфавитном порядке замести-
заместители. Это дает возможность легко получить информацию о соединениях,
аналогичных искомому1.
6.3. ПОИСК С ПОМОЩЬЮ КОМПЬЮТЕРА
Поиск литературы с помощью компьютера — очень производительный метод,
тогда как выполнение поиска вручную потребовало бы много времени. Предпо-
Предпосылкой для этого является наличие персонального компьютера, который путем
телекоммуникационного устройства связан с базой данных.
В области online-исследований о химических сообщениях ведущую пози-
позицию занимает сеть Host STN (Scientific and Technical Network) International, что
позволяет высшим учебным заведениям проводить такие исследования в рам-
рамках учебных программ с выгодными условиями оплаты2.
Для информации по химическим вопросам Chemical Abstracts (CA) —
самый охватывающий источник. Online-версия Chemical Abstracts состоит из
1 Внимание! Родоначальная структура, определяющая место регистрации, может менять-
меняться. Например, вместо «индол, 3-ацетил», используют название «этанон, 1-(индол-3-ил)». В то
же время индол-3-уксусная кислота регистрируется под своим принятым названием. Всегда
следует обращаться к Index Guide.
2 STN — банк данных доступен и через Интернет: http://stnweb.fizkarlsrahe.de.
184 Б Литература по органической химии
файла СА с библиографическими сведениями и рефератами, файлом
REGISTRY со структурами и номенклатурой для всех регистрируемых
Chemical Abstracts Service (CAS) соединений и файлом CAOLD с CAS-регист-
рами и номерами Abstracts. Первые два файла охватывают литературу с 1967 г.,
а файл CAOLD - с 1957 по 1966 г.
Предшествующим дополнением к банку данных СА является банк данных
Бейльштейна (online или также Inhouse-Version Beilstein CrossFire) с базой данных
по структурам и важнейшим литературным источникам по органической химии.
Наряду с этими банками данных, ориентированными на соединения, становятся также
значимыми специальные банки данных по реакциям, такие, как CASREACT или CHEMIN-
FORMRX, см. разд. Б,6.3.3. С помощью SPECINFO при STN предоставляется также банк дан-
данных по спектрам с интегрированными программами для воспроизводства спектров. Много-
Многочисленные банки данных по патентам (INPADOC, WPIDS/WP1NDEX, PATDPA) и др. также
предоставляются online. Для постановки комплексных библиографических вопросов при STN
online имеется мультимедийный файл с функциями поиска. Эта система позволяет проводить
исследования по нескольким банкам данных (т. н. кластерам) одновременно.
Со всеми банками данных, предоставляемых STN, можно работать с помощью легко освоя-
емого языка команд Messenger.
Из примерно 20 команд для простого поиска online следующие четыре являются глав-
главными:
FILE или FIL — выбор желаемого банка данных;
EXPAND, ЕХР или Е — просмотр индекса банка данных по одному поиску;
SEARCH, SEA или S — просмотр одного файла по сообщениям;
DISPLAY, DIS или D - указания на найденные сообщения или указания на одно
сообщение со специфическим регистрационным номером.
С помощью различных операторов (логические или булевые операторы: OR, AND, NOT;
операторы относительной близости W, A, L, S, Р; арифметические операторы: =, <, >, =< или
<=, => или >=, -) возможно объединение категорий поиска. С помощью маскировочных
знаков (?,!, #) можно задавать части слов, а также учитывать незнакомые или различного типа
обозначения. Следует отметить, что часто применяемые слова и знаки, так называемые стоп-
слова (артикли, предлоги, все понятия языка команд, а также скобки, косые линии, маскировоч-
маскировочные символы и верхние запятые) по Messenger рассматриваются как элементы языка команд и
поэтому их нельзя найти. Если эти слова и знаки — элементы поиска, тогда их следует заключить
в верхние запятые — апострофы.
Искомая информация может быть введена в форме текста (название химического соеди-
соединения, ключевые слова, авторы) или графиков (структурные формулы). Корректная формули-
формулировка вопроса и точный ввод являются предпосылками успешного поиска.
Так как все банки данных STN в основном построены однотипно, перекрестные поиски
(кросс-файл-исследование, англ. crossfile searching) проводятся по тем же самым правилам
(за исключением использования отдельных специфических полей). Их результаты могут быть
использованы для поиска в других файлах.
После подключения к банку данных STN сперва попадают в File HOME. Стрел-
Стрелка в языке поиска Messenger показывает, что система ждет ввода команды. Желае-
Желаемый файл может быть затем вызван, например, файл СА — путем ввода FILE СА.
С командой SEARCH (SEA или S) и вводом категории поиска начинается
поиск1. Если вводят, например, S TOCOPHEROL, то на экране появляется:
1 Во многих случаях выгодно перед собственно поиском с помощью команды Expand
(ЕХР,Е) проверить и уточнить наличие категорий поиска в регистрах, расположенных в опре-
определенном алфавитном порядке: Basic Index (BI), Author Index (AU), Chemical Name Index
(CN). Номера, указанные в выдаваемом списке, могут быть использованы в дальнейшем как
категории поиска (например, S E2-E5).
6. Проведение литературного поиска 185
L1 12308 TOCOPHEROL
Каждый ответ от программы online получает так называемый L-номер, ко-
который может быть использован в дальнейшем в качестве категории поиска.
Цифра 12308 показывает, что термин токоферол содержится в 12308 рефератах.
Следует отметить, что в базовом индексе файла СА в качестве категорий поиска могут быть введе-
введены только регистрационный номер CAS и отдельные слова из заглавия, резюме или регистрационные
понятия, сложные слова разделяются и все знаки, не являющиеся символами алфавита, удаляются.
Большое преимущество поиска online состоит в возможности объединять
различные категории поиска путем применения операторов (AND,
OR, NOT) и таким образом искать одновременно. Если, например, вводят
S ASSYMETRIC AND SYNTESIS AND AMINES, то на экране появится
23683 ASYMMETRIC
618994 SYNTHESIS
138857 AMINES
L2 543 ASYMMETRIC AND SYNTHESIS AND AMINES
Было найдено 543 реферата с искомыми понятиями, которые могут появиться без
прямого указания одного на другой. Чтобы это исключить, можно желаемое взаимное
расположение категорий установить в контексте с соседним оператором. После
ввода, например S ASYMMETRIC(W)SYNTHESISAW)AMINES получают ответ:
L3 12 ASYMMETRIC(W)SYNTHESISAW)AMINES
т. е. только публикации, в которых слово синтез следует сразу после слова асим-
асимметричный и отделено от слова амин только одним словом. W-Оператор (nW)
позволяет поиск слов, которые отстоят друг от друга максимум на п слов. Анало-
Аналогично этому с А-оператором (пА) поиск слов возможен в любой последователь-
последовательности и максимум на расстоянии п слов. Левый или L-оператор (L), напротив,
ограничивает поиск двумя словами, которые могут быть расположены в любой
последовательности и на любом расстоянии друг от друга.
Другое полезное вспомогательное средство при формулировании понятий
поиска - маскировочные знаки. Знак вопроса (?) вслед за родовым словом (пра-
(правая маскировка; соответствующая левая маскировка более не возможна) может
заменить несколько знаков. Выражение CATALY? могло быть введено для поис-
поиска понятий CATALYST, CATALYSIS, CATALYTIC. Восклицательный знак (!) в
понятии поиска заменяет ровно один знак (например, STABILIIER для
STABILISER или STABILIZER), решетка (#) в конце слова (например, AM1N#)
заменяет один или никакой знак и может указывать на множественное число.
Число ответов в коде литературного поиска online может достигать величины,
которая уже будет неразумно большой и поэтому должна быть понижена; для это-
этого существует несколько возможностей. Так, объем исследования может быть су-
сужен. Например, вводя S LI RANGE=A990,) получают ответ L4 4715 TOCO-
TOCOPHEROL, охватывающий литературу списка 1 с 1990 г. Может быть выбран поиск
публикаций только на желаемом языке (немецком, английском, французском)
186 Б Литература по органической химии
(например, ввод S L4 AND (GEROR ENG OR FR)/LAк L5 дает 3514 ответов) или
исключены патенты (S L5 NOT P/DT понижает число ответов из L5 до 3132).
Публикации определенных авторов легко получить вводом S NAME,
VORNAME, INITIALEN/AU. И здесь принято перед поиском с помощью
Expand-приказа проверить написание в авторском регистре (/AU) фамилии,
имени, инициалов и сокращений и использовать несколько Е-номеров как
категорий поиска. Объединение различных предметных поисков или ответов с
одним или несколькими авторами часто тоже представляет интерес (например,
SL1 AND INGOLD, K?/AU или S FULLEREN? AND DIEDERICH.F7/AU).
Путем команды DISPLAY (DIS или D) и L-номера прилагаемого ответа и
форматов соответствующих полей (в СА-файле AN для номеров СА, BIB для
библиографических данных и Abstract-номеров, ABS для Abstracts и Abstract-
номеров, AU для авторов), например, D L1 1—10 BIB, ABS полученные ответы
будут выведены на экран. Online-поиск может быть затем продолжен в других
банках данных с использованием полученных L-номеров или закончен путем
команды LOGOFF или LOG Y.
6.3.1. ПОИСК ХИМИЧЕСКИХ СОЕДИНЕНИЙ И ИХ СИНТЕЗОВ
В файле CAS-Registry и Beilstein возможны поиски online по отдельным соединени-
соединениям с их точной структурой (а также регистрационным номером CAS) с применени-
применением химического названия (или фрагмента химического названия) или структурной
формулы (см. также ниже о поиске субструктур) в качестве категорий поиска. Пер-
Первый путь часто быстрее и дешевле, но менее надежен, так как он требует хорошего
знания химической номенклатуры и способов обозначения по СА, которые не
всегда совпадают с правилами ИЮПАК.
Перед online-поиском надо сформулировать название химического соеди-
соединения так, как оно должно быть введено по команде Expand или Search. В наз-
названиях со специальными знаками все выше- и нижестоящие знаки должны
быть написаны в одну строку с обычными знаками, буквы курсивом написаны
как обычные, греческие буквы выписаны и внесены в нужном месте (напри-
(например, .ALPHA.-TOCOPHEROL), квадратные скобки заменены круглыми, а
названия, которые содержат апостроф, заключены в верхние штрихи (напри-
(например, 'N,N'-DIISOPROPYL BENZIDINE' или N,N!-DIISOPROPYL BENZI-
DINE). Последнее относится и к команде Search, когда в названии содержатся
круглые скобки.
После этих приготовлений надо посмотреть с помощью бесплатной
команды Expand в поле CN, имеется ли искомое соединение в индексе (указа-
(указателеI. К примеру, ввод Е BICYCLOB.2.2)OCT-2-ENE/CN даст на экране:
=> Е BICYCLOB.2.2)OCT-2-ENE/CN
El I BICYCLOB.2.2)OCT-2-EN-2-OL,SODIUM SALT/CN
E2 1 BICYCLOB.2.2)OCT-2-EN-2-YLKETONE/CN
E3 1 -> BICYCLOB.2.2)OCT-2-ENE/CN
E4 1 BICYCLOB.2.2)OCT-2-ENE,
1,2,3,4,5,5,6,6,7,7,8,8-DODECAFLUORO-/CN
1 Указание CN-поля необходимо, так как иначе поиск будет в Basic Index (BI), который
содержит только фрагменты названий вместо полных химических названий.
6. Проведение литературного поиска 187
Если ничего не найдено (Е = 0) или если найдено названий больше чем
одно, надо проверить названия с точки зрения их написания или полноты.
Затем с помощью команды Search выполняется настоящий поиск, напри-
например, путем ввода 'BICYCLOB.2.2)-2-ENE'/CN. Вместо названия можно
ввести Е-номер, например: S E3/CN или S E1-E4/CN или S El, E3/CN.
В случае сложных химических соединений и связанной с этим неуверен-
неуверенностью в написании правильного названия вещество может быть найдено с
помощью фрагментов названия (CNS), которые отражают смысловые значения
фрагментов полного названия и могут быть маскированы. Их можно найти в реги-
регистрационном файле и в файле Бейльштейна, а также в Basic Index и в химических
названиях индекса сегментов (/CNS). Для поиска рекомендуется использовать
CNS-поле, поскольку в этом случае возможна как левая, так и правая маскировка
(для фрагментов, содержащих не менее 4 буквI. Фрагменты названий могут быть
связаны при помощи различных операторов [например, (nW), (nA), (L)
или (AND)] друг с другом или с другими категориями поиска. Ввод
S QUINOLINE(L)METHOXY(L)METHILAMINO(L)CHLORO/CNS приведет к
таким ответам, в которых описаны производные хинолина, содержащие по край-
крайней мере один атом хлора и одну метокси- и одну метиламиногруппу. Объедине-
Объединение с молекулярной формулой в случае больших молекул может значительно
уменьшить число ответов.
С помощью команды Display можно снова отобразить полученные ответы в
форматах, предоставляемых регистрационным файлом или файлом Бейль-
Бейльштейна. С помощью регистрационных номеров, полученных по команде
D RN, или L-номеров по регистрационному файлу следует затем искать новую
информацию для этих химических соединений по файлам СА2.
6.3.2. ПОИСК ПО СТРУКТУРЕ
Поиски по названиям трудоемки или практически невыполнимы, если ведется
поиск соединений, которые содержат общие структурные элементы и, кроме то-
того, еще различные заместители. Такие поиски структур возможны online в файле
CAS-регистра и Beilstein-файле (offline также по банкам данных Beilstein Cross
Fire). В течение секунд или нескольких минут можно просмотреть имеющиеся в
этих файлах миллионы соединений с данной структурой или субструктурой.
Химические структуры или субструктуры могут быть заданы online путем приме-
применения соответствующих команд (GRA, NODE, BOND, HCOUNT и т. д.) в режиме
STRUCTURE. Требуются знание этих команд и некоторый навык, особенно что-
чтобы построить сложные структуры за определенное время при применении online.
1 Использование расширения Expand-Kommandos (E Namensfragment?/CN или Е LEFT
Namensfragment/CNS) и здесь выгодно для подготовки к поиску.
2 Надежная и полная информация об отдельных химических соединениях может быть по-
получена online по банку данных СА только с регистрационным номером CAS (RN).
В случае простых соединений источником для поиска могут служить напечатанные RN-ука-
затели, как и СА Chemical Substance Index, CA Index Guides, Merck Index, справочники орга-
органических соединений и многочисленные каталоги химических соединений. Для менее
используемых соединений этот путь может быть заменен описанным online-поиском в файле
CAS-регистра или в файле Бейльштейна.
188 Б Литература по органической химии
Пакет STN EXPRESS содержит программу, с помощью которой можно
нарисовать структуры offline, затем сохранить их и в нужное время включить в
online в структурный файл и затем использовать для поиска.
Прежде чем выполнять дорогостоящие поиски субструктур, следует полу-
получить определенные навыки по поискам структур, упражняясь на учебном фай-
файле (L Registry, L Beilstein).
6.3.3. ПОИСК ХИМИЧЕСКИХ РЕАКЦИЙ
Поиск реакций может быть осуществлен наряду с Beilstein-File (здесь следует
упомянуть возможность поиска структур с помощью команды REA,
позволяющей получить информацию о химических реакциях) по специально
созданным для этого банкам данных реакций CASREACT (охватывающим
источники с 1985 г.) и CHEMINFORMRX (избранные химические реакции
из реферативной службы Cheminform с 1991 г.). Реакции соединений находят
по соответствующим указателям, регистрационным номерам или струк-
структурам.
Поскольку банк данных для реакций еще создается и охватывает только
новейшую литературу, пока часто приходится пользоваться данными, имею-
имеющимися в банке данных СА и Бейльштейна.
Банк данных Бейльштейна так же, как и Registry-File, позволяет проводить
структурные исследования при вводе различных структур или субструктур и
поиск химических соединений по химическим названиям (CN), фрагментам
названий (CNS), брутто-формулам (MF) и регистрационным номерам CAS
(RN). После нахождения химического соединения и ввода команды
отображения (D) следует указать, какой информацией вы обладаете и сколько
сообщений лежит в отдельных полях (ввод D FA). Их можно затем последова-
последовательно и по отдельности вызывать (например, с помощью команды D PRE для
способа получения, D REA для химических реакций, D МР для температур
плавления, D EAS для электронно-абсорбционных спектров и т. д.). Для хими-
химика, работающего в области синтеза, особенно интересны поля «методы получе-
получения» (PRE)- и «реакции» (REA); имеются еще субполя, как, например,
PRE.EDT (для едуктов1), PRE.RGT (для реагентов, растворителей, катализато-
катализаторов и др.), PRE.BPRO (для побочных продуктов), REA.RP (для реакционного
партнера) или REA.PRO (для продуктов реакций). Освоение многочисленных
возможностей, которые предоставляет банк данных Бейльштейна, требует
длительных упражнений.
Прекрасной альтернативой для поиска химических реакций может служить
банк данных Inhouse Cross-FireplusReactions, который содержит сведения о
10 миллионах химических реакций.
1 Едукт — это вещество, вступающее в реакцию, которое вносит свои атомы углерода в
получаемый продукт. — Прим. перев.
7. Ведение лабораторного журнала 189
7. ВЕДЕНИЕ ЛАБОРАТОРНОГО ЖУРНАЛА
Во время выполнения синтеза все численные данные и наблюдения следует
заносить в лабораторный журнал. К таким сведениям относятся, например,
количества взятых веществ, намеренные или случайные отклонения от методи-
методики, изменение окраски, повышение температуры, выход получающихся соеди-
соединений. Лабораторный журнал необходимо вести в прочной тетради с нумеро-
нумерованными страницами. В каждом случае всегда необходимо указывать дату
проведения эксперимента. В журнал записывают также методику эксперимента,
особенно если она не опубликована в руководстве к практическим занятиям
(например, методику, взятую из литературы при выполнении литературного
«синтеза»I.
По окончании опыта оформляют отчет о работе, основанный на использован-
использованной методике и собственных наблюдениях, где описывают практическое выпол-
выполнение работы. Отчет должен содержать название получаемого соединения (по но-
номенклатуре ИЮПАК, а также тривиальное); константы, взятые из литературных
источников и определенные экспериментально (температуры плавления и кипе-
кипения, плотность, показатель преломления), уравнение реакции, количества взятых
веществ (в граммах и молях), описание использованной литературы, точное опи-
описание практического проведения опыта. Если вещества очищались перегонкой, в
отчете следует привести диаграмму кипения или баланс перегонки. Кроме того, в
отчете указывают выход полученных веществ и приводят расчет выхода.
Выход вычисляется в процентном отношении к теоретическому (сокраще-
(сокращено: % от теор.). Последний определяют по уравнению реакции исходя из взя-
взятых количеств веществ. При использовании неэквимолекулярных соотноше-
соотношений расчет выхода проводят по компоненту, взятому в недостатке. В тех случа-
случаях, когда в использованной методике приводится выход продуктов реакции,
надо сравнить его с реально полученным и объяснить отклонения.
При подготовке литературных синтезов часто необходимо выбрать один
способ из нескольких описанных. Эти способы следует систематизировать и
сопоставить между собой, чтобы выбрать наилучший. В составленной для этой
цели наглядной схеме особо отмечают такие условия реакций, как давление и
температура, а также выходы продуктов реакций.
В отчете следует указать также все известные методы синтеза с соответству-
соответствующими литературными ссылками. Отчет должен содержать краткое обоснова-
обоснование выбранного метода синтеза и подробное описание его практического вы-
выполнения со всеми наблюдениями, отраженными в лабораторном журнале.
Для каждой стадии рассчитывают выход веществ; кроме того, после описания
последней стадии приводят и суммарный выход — относительно количеств
веществ, взятых для первой стадии. В заключение отчета следует обсудить про-
проведенный опыт и сравнить полученные результаты с указанными в литературе.
При приведении данных по анализу веществ (идентификации) должно быть ясно
видно, что полученное вещество охарактеризовано совершенно однозначно. В жур-
журнале надо кратко описать методы, которые привели к идентификации вещества.
1 Литературным «синтезом» обычно называют работу по сбору литературы, для того что-
чтобы выбрать методику получения данного соединения. - Прим. перев.
190 Б Литература по органической химии
8. ЛИТЕРАТУРА
Химическая информация
Macke M. Die chemische Literatur - ihre ErschlieCung und Benutzung. - Verlag Chemie,
Weinheim, 1982.
Maizell R. E. How to find chemical information. - Wiley & Sons, New York, 1998.
Loewenthal H. J. E., Zass E. Der clevere Organiker. — Johann Ambrosius Barth, Edition
Deutscher Verlag der Wissenschaften, Leipzig, Berlin, Heidelberg, 1993.
PichlerH. R. Online-Recherchen fur Chemiker. - VCH Verlagsgesellschaft, Weinheim, 1986.
Schulz, H., Georgy U. Von CAbis CAS online. - Springer-Verlag, Berlin, Heidelberg, 1994.
Номенклатура химических соединений
Номенклатурные плавила ИЮПАК по химии: Т. 1-4. - М.: ВИНИТИ, 1979-1984.
Internationale Regeln fur die chemische Nomenklatur und Terminologie. Bd. 1. Hrsg.:
Deutscher Zentralausschup fur Chemie. - Verlag Chemie, Weinheim, 1978-1990.
Nomenklatur der Organischen Chemie. G. Kruse (ed.). - VCH, Weinheim, 1997.
Handbuch zur Anwendung der Nomenklatur organisch-chemischer Verbindungen.:
W. Liebscher. - Akademie-Verlag, Berlin, 1979.
Bunzli-Trepp U. Handbuch fur die sistematische Nomenklatur der Organischen Chemie,
Metallorganischen Chemie und Koordinationschemie. Chemical-Abstracts-Richtlinien
mit IUPAC-Empfehlungen und vielen Trivialnamen. — Logos, Berlin, 2001.
Hellwich K.-H. Chemische Nomenklatur. — Govi-Verlag, Eschborn, 1998.
Reimlinger H. Nomenklatur Organisch-Chemischer Verbindungen. - Walter de Gruyter, Berlin, 1998.
Литература по банк-данным
Using CAS Online. — Chemical Abstracts Service, Columbus, Ohio, 1985.
REGISTRY. - STN International, Fachinformationszentrum Karlsruhe, 1988.
CA, CApreviews, CAOLD. — STN International, Fachinformationszentrum Karlsruhe, 1989.
BEILSTEIN, CHEMINFORMRX. - STN International, Fachinformationszentrum
Karlsruhe, 1993.
Focus on CASREACT. - STN International, Chemical Abstracts Service, Columbus, Ohio, 1992.
Arbeitsanleitungen zu BEILSTEIN CrossFire und SciFinder Scholar. —
http://www.fiz-karlsruhe.de; http://www.stn-international.de).
Детальные описания банк-данных издаются предпринимателями
(например, STN International, с/о Fachinformationszentrum Karlsruhe;
WWW: http ://www. stn- international .de).
НЕКОТОРЫЕ ОБЩИЕ ПОЛОЖЕНИЯ
1. КЛАССИФИКАЦИЯ ОРГАНИЧЕСКИХ РЕАКЦИЙ
Реакции можно классифицировать по разным признакам.
I. Весьма общий подход к классификации основан на том факте, что при
химических реакциях происходят перемещения электронов. В связи с этим раз-
различают следующие типы реакций:
1. Реакции переноса электронов, например:
R3N: + М3® ^=^ R3N® + М2® [В.1]
К такому типу относятся многие окислительно-восстановительные реакции
(разд. Г,6). Известны реакции с переносом как одного, так и двух электронов.
2. Реакции перегруппировки связей.
а) Гемолитические, протекающие с участием радикалов (радикальные реак-
реакции).
При таких реакциях связи расщепляются «симметрично» с образованием
радикалов или же связи образуются между радикалами, например:
CI—CI =?=^ CI- + CI- [В.2]
Сюда же относятся реакции гомолитического (радикального) обмена, например:
CI- + H-R > CI—Н + R- [В.З]
б) Гетеролитические (полярные, ионные) реакции.
При таких реакциях связи расщепляются (или образуются) «несимметрич-
«несимметрично», так что электронные пары не разъединяются. К этим реакциям относятся:
— гетеролитическое расщепление молекул или, наоборот, соединение
электрофильных и нуклеофильных частиц:
R-Cl -^— R® + cie [B.4]
192 В Некоторые общие положения
— гетеролитический (ионный) обмен:
Iе + r-ci ^=^ I-R + CIQ [B.5]
В| + Н-А == В-Н + IA0 [В.6]
(здесь В| — основание, НА — кислота).
При гетеролитическом образовании связи один из партнеров выступает в
роли донора пары электронов (основание Льюиса, нуклеофил), второй — в роли
акцептора пары электронов (кислота Льюиса, электрофил).
При гетеролитическом расщеплении связей уходящую с парой электронов
группу называют нуклеофугом; группу, предоставляющую электронную пару, -
электрофугом'.
в) Согласованные многоцентровые реакции.
При таких реакциях несколько связей образуются и расщепляются однов-
одновременно (согласованно, синхронно) через циклическое переходное состояние
без образования ионов или радикалов.
К этому типу относятся реакция Дильса—Альдера (разд. Г,4.4.3), перегруп-
перегруппировка Коупа (разд. Г,9.2).
[В.7]
II. С учетом структурных изменений вступающих в реакцию соединений
классификацию можно построить по-иному.
а) Реакции замещения, например:
Аг—Н + HNO3 Ar—NO2 + Н2О [В.8]
б) Реакции присоединения, например:
Н2С=СН2 + Вг2 ВгСН2-СН2Вг [В.9]
в) Реакции отщепления (элиминирования), например:
Н3С-СН2ОН Н2С=СН2 + Н2О [В. 10]
г) Перегруппировки и изомеризации, например:
Эту классификацию можно сочетать с классификацией первого типа, так j
что, например, можно говорить о гомолитическом, гетеролитическом или j
многоцентровом присоединениях. Именно такой подход главным образом и i
положен в основу дальнейшего изложения. !
1 В отечественной литературе оба термина не получили широкого распространения. -
Прим. перев.
2. Изменения энергии при химических реакциях 193
III. В классификации, основанной на молекулярно-кинетическом подходе,
отражают, какое число отдельных молекул подвергается изменениям в стадии,
определяющей скорость реакции (молекулярность реакции, см. разд. В,3).
При этом различают:
— мономолекулярные реакции;
— бимолекулярные реакции;
— тримолекулярные реакции.
2. ИЗМЕНЕНИЯ ЭНЕРГИИ ПРИ ХИМИЧЕСКИХ РЕАКЦИЯХ
Химические реакции всегда сопровождаются перераспределением энергии: из-
изменяется внутренняя энергия участников реакции, происходит обмен энергией
с окружающей средой.
При лабораторных синтезах сведения об энергетическом балансе реакции
полезны для правильного выбора условий реакции; при промышленных син-
синтезах такие сведения совершенно необходимы.
На рис. В. 12 приведена схема изменения энергии в одностадийном хими-
химическом превращении (элементарной реакции), происходящем по энергетичес-
энергетически наиболее выгодному пути («координата реакции») от исходных веществ к
продуктам реакции.
?
ИЛИ Н"
ИЛИ G"
Переходное
состояние
Исходные ^ 0 „
,. ч ^ч^ Ar" ИЛИ ArG
вещества (А) ^
Продукты (Е)
Координата реакции
Рис. В. 12. Изменение энергии во время элементарной химической реакции.
Внутренние энергии Е, энтальпии Н и свободные энергии Гиббса (G)
исходных веществ и конечных продуктов не зависят от пути реакции. Они
представляют собой величины, с которыми оперируют по законам химической
термодинамики.
Разность молярных стандартных свободных энергий образования A^G° ко-
конечных (Е) и исходных (А) продуктов равна молярной стандартной свободной
энергии реакции:
° = ДвСод° - Ав<5(А)° [В.13]
Эта величина прямо связана с константой термодинамического равновесия
реакции К:
194 В Некоторые общие положения
ArG"
Ктем больше, чем меньше ArG°:
Ak(j<0, К>\ (см.рис. B.12) [B.15]
Дк<7>0, К<\ [В.16]
Если К > 1, то равновесие в реакции сдвинуто в сторону конечных продук-
продуктов. При К < 1 равновесие реакции можно тоже иногда сместить в сторону
конечных продуктов, постоянно удаляя их из сферы реакции (ср., например,
этерификацию; разд. Г,7.1.4.1).
Разность молярной стандартной энтальпии образования Дв#° конечных (Е)
и исходных (А) продуктов равна молярной стандартной энтальпии реакции:
ДкЯ° = ЛвЯе — Дв#; [В-17]
Эта величина указывает тепловой эффект реакции и определяет (в соответ-
соответствии с изобарой реакции Вант-Гоффа) температурную зависимость констант
равновесия:
ARH° < 0 (экзотермическая реакция, рис. В. 12):
при повышении температуры А"уменьшается [В. 18]
Ar/T > 0 (эндотермическая реакция):
при повышении температуры А"растет [В. 19]
Стандартные энтальпии образования можно найти в таблицах или рассчи-
рассчитать по сумме отдельных вкладов. Это дает возможность получить представле-
представление об ожидаемом тепловом эффекте реакции и соответственно выбрать усло-
условия эксперимента.
По закону Гесса Ar/P равно разности энтальпий связей, расщепляющихся
и образующихся в реакции:
ДКЯ° = SjAotf0 — 22ДСЯ° [В.20]
где Zj — расщепленные связи; Ъг — образовавшиеся связи.
Поэтому вместо стандартных энтальпий образования для расчета ArH°
можно использовать и энтальпии диссоциации связей, участвующих в реакции
(разд. Г.1.3).
ARG°и ARH°связаны уравнением Гиббса - Гельмгольца:
ArS° — молярная стандартная энтропия реакции, равная разности станда-
стандартных энтропии конечных и исходных веществ. Энтропию можно интерпре-
интерпретировать как меру «неупорядоченности» системы (в смысле возрастания воз-
возможностей движения отдельных ее компонентов):
2. Изменение энергии при химических реакциях 195
ДяS° > 0 «неупорядоченность» возрастает при переходе от исходных веществ
к конечным продуктам, например когда число реагентов меньше числа
конечных продуктов или когда циклическое соединение превращается
в ациклическое [В.22]
ARS° < 0 возрастает «упорядоченность» (примеры, обратные предыдущим) [В.23]
В соответствии с уравнением [В.21], стандартная свободная энергия реак-
реакции (К> 1) может быть отрицательной, если энтальпия реакции отрицательна
(экзотермическая реакция) либо энтропия реакции положительна. Константа
скорости особенно возрастает, если АцН° < 0 и ArS° > 0.
Влияние энтропийного фактора в [В.21] можно рассмотреть на примере реакции гидриро-
гидрирования - дегидрирования:
\ / Катализатор \ /
С=С + Н2 . Н-С-С-Н [В.24]
/ \ Катализатор / \
Для гидрирования [реакция [В.24], направление слева направо] ARH° < 0 (экзотермическая
реакция), а также ArS°< 0 (неупорядоченность системы уменьшается, так как слева две части,
а справа только одна), т. е. энтропийный член (—TArS°) > 0. Поскольку, однако, при комнат-
комнатной температуре этот член значительно меньше, чем ArH", то для прямой реакции остается
A«G° < 0 (уравнение [В.21]), т. е. равновесие сдвинуто в сторону продуктов гидрирования. С
ростом температуры энтропийный член в конце концов может стать больше ARIT, так что ARG°
окажется положительным и гидрирование станет невозможным. При такой температуре
(-200-300 °С) происходит, наоборот, дегидрирование, т. е. реакция [В.24] идет справа налево.
Хотя дегидрирование — эндотермическая реакция, однако для него AR5" > 0, т. е. — TArS° < 0.
Свободная энергия реакции ArG°, хотя и предсказывает термодинамичес-
термодинамическую осуществимость реакции, однако ничего не говорит о том, с какой ско-
скоростью реакция проходит. Здесь могут играть роль задерживающие факторы,
которые необходимо преодолеть до начала реакции. Примером может служить
система Н2+О2 (гремучий газ): она требует поступления дополнительной энер-
энергии (поджигания), после чего реакция происходит взрывообразно.
Термодинамически возможной реакции препятствует энергетический
барьер, который необходимо преодолеть на пути от исходных веществ к конеч-
конечным продуктам (рис. В.12). Разность свободных энергий (или энергий Гиббса)
образующегося переходного состояния (активированного комплекса) и исход-
исходных веществ представляет собой энергию активации Е\ (или свободную
энергию активации AG*). Она определяет скорость реакции. Законы химичес-
химической кинетики дают соотношения
k — Ae~Eb-lRT уравнение Аррениуса [В.25]
=iSi-e e уравнение Эйринга [В.26]
h h
По этим уравнениям константа скорости к тем больше, чем меньше
АН' или ЕА[ и чем больше AS* или А.
и АН* связаны соотношением АН* = ЕА - RT.
196
В Некоторые общие положения
Предэкспоненциальный множитель А в уравнении Аррениуса или энтро-
энтропия активации AS* в уравнении Эйринга отражают пространственные требо-
требования к переходному состоянию. А или AS* тем меньше, чем более упорядо-
упорядочение переходное состояние по сравнению с исходными веществами (см. вли-
влияние энтропийного фактора [В.22] и [В.23]).
На рис. В. 12 видно, что энергия активации обратной реакции определяется
суммой энергии активации прямой реакции и энтальпии активации. Тем
самым обеспечивается связь термодинамики и кинетики, выражающаяся
уравнением
прямой реакции
>братной реакции
[В.27]
Энергия активации в принципе не может быть меньше энтальпии реакции.
В общем же она часто тем меньше, чем более экзотермична реакция, т. е. чем
больше запас энергии в исходных веществах и чем менее энергоемки конечные
продукты (рис. В.28).
По Эвансу и Поляни, между обеими величинами может выполняться соот-
соотношение
?A=aAR//:4-P [B.29]
Часто аналогичные соотношения существуют между энергией реакции и
энергией активации (линейное соотношение свободных энергий; разд. В,5.2):
AG+ = aAnG° + b [В. 30]
Эти эмпирические соотношения удовлетворительно соблюдаются лишь в
пределах рядов сходных реакций.
Из рис. В.28 видно, что имеется связь между тепловым эффектом реакции
(т. е. ArG°) и положением переходного состояния на координате реакции (пос-
(постулат Хэммонда): активированный комплекс экзотермической реакции распо-
располагается «раньше» на координате реакции, по энергии и структуре он сходен с
исходными веществами. Для эндотермической реакции вероятнее более «позд-
«позднее» расположение этого комплекса на координате реакции, причем он более
ARH°(ARG°2)-
Активи-
Исходные рованный
вещества комплекс Продукты
Координата реакции
Рис. В.28. Схема энергетического профиля реакций с различной свободной
энергией (соответственно и энтальпией реакции).
3.0 протекании органических реакций во времени 197
близок к конечным продуктам. Для оценки в этом случае можно принять
конечные продукты за модель переходного состояния, а также считать
AG* « ArG\ так что скорость образования продуктов реакции тем меньше, чем
больше запас энергии в них (разд. ГД.З, Г.2.1.1, Г,5.1.2 и др.).
Постулат Хэммонда может служить основанием для установления связи
между селективностью и реакционной способностью (разд. ГД.З).
3. О ПРОТЕКАНИИ ОРГАНИЧЕСКИХ РЕАКЦИЙ ВО ВРЕМЕНИ
Скорость г химической реакции, например:
А+В->С [В.31]
зависит от природы и концентрации реагирующих веществ. В простых случаях
г=к[А]"[В]ь [В.32]
где к — константа скорости реакции, величина которой зависит от AG* (урав-
(уравнение [В.26]); она является мерой реакционной способности веществ А и В.
В соответствии с уравнениями [В.25] и [В.26] к зависит от температуры.
В грубом приближении скорость многих органических реакций при повышении
температуры на 10 °С возрастает в 2—Зраза.
Сумма показателей степени а + Ь + ... при концентрациях [А], [В] и т. д.
в уравнении [В.32] называется порядком реакции. Какие именно вещества
участвуют в стадии, определяющей скорость, зависит от механизма реакции.
Таким образом, экспериментальные данные о протекании реакции во време-
времени могут служить основой для выводов о ее механизме.
У протекающих в одну стадию элементарных реакций картина проста.
Здесь в кинетическом уравнении представлены все участники реакции; поря-
порядок реакции совпадает с ее молекулярностью, т. е. числом участвующих в реак-
реакции молекул. В этих случаях скорость зависит от концентрации всех исходных
веществ, возрастая с ростом концентрации.
3.1. ПОСЛЕДОВАТЕЛЬНЫЕ РЕАКЦИИ
Большинство органических реакций осуществляется не одностадийно, а в виде
последовательности элементарных реакций. Механизм реакции определяется
при этом типом и числом элементарных реакций, их временной последователь-
последовательностью и возникающими промежуточными продуктами.
Реакция, отвечающая стехиометрическому уравнению [В.31], может проте-
протекать, например, в две стадии через образование промежуточного продукта Z
А ? г [В.ЗЗ]
Z + B X С [В.34]
198
В Некоторые общие положения
а Координата реакции б Координата реакции
Рис. В.35. Энергетические профили двухстадийной реакции типа [В.33], [В.34].
Как показано на рис. В.35, промежуточный продукт Z часто представляет
собой высокоэнергетическую частицу (например, ион или радикал), поэтому
он чрезвычайно реакционноспособен (ДС*, Дб^ << AG*, т. е. jfc_i, kj »k\).
Реакционноспособная частица по мере своего образования немедленно реаги-
реагирует дальше, так что ее концентрация очень мала (принцип стационарности
Боденштейна, см. учебники по кинетике). Уравнение скорости суммарной
реакции приобретает вид:
[А] [В]
+ МВ] ^
r_
Это сложное кинетическое уравнение для двух практически важных случаев упрощается:
а) Если &_i << к2 [В] (рис. В.35, а), то уравнение [В.36] переходит в [В.37]:
r=k1[A]
[В.37]
Это кинетическое уравнение первого порядка, совпадающее с таковым для первой стадии
[В.33]. Таким образом, стадией, определяющей скорость реакции [В.31], в данном случае ока-
оказывается образование промежуточного продукта Z. В кинетическое уравнение суммарной ре-
реакции входит лишь концентрация участвующего в стадии [В.33] партнера, т. е. концентрация
вещества А, но не концентрация участвующего в следующей стадии вещества В. Это значит,
что скорость реакции нельзя увеличить, повышая концентрацию вещества В.
Характерные примеры реакций такого типа - мономолекулярное нуклеофильное замеще-
замещение (разд. Г,2.1.1), атакже мономолекулярное элиминирование (разд. Г,3.1.1.1).
б) Если к-х »кг [В] (рис. В.35, б), то членом к2 [В] в знаменателе уравнения [В.36] можно пре-
пренебречь. В результате выражение [В.36] превращается в кинетическое уравнение второго порядка
[В]
[В.38]
где константа скорости к — произведение константы равновесия второй стадии кг и констан-
константы равновесия предшествующей первой стадии К\. Большая скорость реакции в этом случае
может быть как следствием высокой реакционной способности (кг), так и следствием высокой
тенденции к образованию промежуточного продукта Z, т. е. его высокой концентрации
([Z] ~ Ki). Обыкновенно промежуточные продукты, концентрация которых высока, имеют
малую реакционную способность: большим величинам К\ соответствуют малые кг и наоборот.
Часто высокие к реализуются при средней реакционноспособности промежуточных продуктов.
Примером реакций, описываемых кинетическим уравнением [В.38], являются многие прев-
превращения, катализируемые кислотами и основаниями, в которых участвуют промежуточные про-
продукты Z, возникающие из субстрата А при протонировании или депротонировании в предвари-
предварительной стадии кислотно-основного равновесия (см., например, разд. Г,4.1.3, Г.7.1.4 и др.).
3. О протекании органических реакциях во времени 199
3.2. КОНКУРИРУЮЩИЕ (ПАРАЛЛЕЛЬНЫЕ) РЕАКЦИИ
Взаимодействие органических веществ довольно часто сопровождается побоч-
побочными процессами (параллельные, конкурирующие или одновременные реак-
реакции). Например, соединение А вступает в реакцию одновременно с двумя веще-
веществами В и С с образованием продуктов D и Е.
А + В -*¦ D
А + С X Е |В-39'
При необратимых конкурирующих реакциях одного порядка количественное
соотношение продуктов D и Е, образующихся в результате суммарной реак-
реакции, постоянно; это соотношение характеризует относительную реакционную
способность соединений В и С по отношению к А (см. ряды реакционной
способности, разд. Г, 1.3 и Г,2.2):
[D] MB]
При этом под В и С не обязательно подразумевают два разных соединения,
это могут быть две различные части одной и той же молекулы (разд. Г, 1.3);
вещества с подобными свойствами называют амбифункциональными или амби-
дентными (см. рис. В.80, разд. Г,2.3, Г.5.1.2, Г,7.2.1.10, Г.7.4.2.1).
Если одна или несколько конкурирующих реакций обратимы, то это зна-
значительно усложняет общую картину протекания реакции. Допустим, вещест-
вещество А обратимо реагирует (константа скорости к\) с образованием вещества В
(константа скорости обратной реакции к-\). Одновременно вещество А может
необратимо превращаться в вещество С (константа скорости кг). Допустим.
ЧтоА:! >k-i »к2.
С il A5= В [В.41]
Пусть вещество С среди данных веществ термодинамически наиболее ста-
стабильно. Тогда вследствие высокого значения к\ или же благоприятного поло-
положения равновесия реакции А <-» В (К = к\/к-\) вскоре после начала реакции об-
образуется относительно большое количество вещества В и лишь небольшое —
вещества С (константа скорости мала). Если в этот момент прервать реакцию,
то соединение В можно выделить в качестве главного продукта. В этом случае
говорят о кинетическом контроле реакции.
Если, наоборот, реакция продолжается, то вещество А выводится из равновес-
равновесной системы в результате медленно протекающей конкурирующей реакции с реа-
реагентом С. Поэтому, поскольку система равновесна, еще некоторое количество В
должно перейти в А; последнее превращается в С. Таким образом, в конце концов
соединение В полностью переходит в термодинамически более стабильное С.
Отсюда очевидно, что если реакция продолжается достаточно долго, можно выде-
выделить вещество С в качестве главного продукта реакции. В этом случае говорят о
термодинамическом контроле реакции. [Примеры — сульфирование нафталина
(см. [Г.5.21], разд. Г,5.1.4), ацилирование по Фриделю — Крафтсу (разд. Г,5.1.7).]
200 В Некоторые общие положения
Конкурирующие реакции в большинстве случаев по-разному реагируют на
изменение температуры: в соответствии с уравнением [В.25] различные энер-
энергии активации обусловливают различную температурную зависимость скорос-
скорости реакции. Реакции с высокой энергией активации при повышении темпера-
температуры ускоряются больше, чем реакции с низкой энергией активации.
Изменения температуры могут также влиять на положения равновесий
конкурирующих реакций.
3.3. ВЛИЯНИЕ РАСТВОРИТЕЛЕЙ
НА РЕАКЦИОННУЮ СПОСОБНОСТЬ
В основе сольватации лежат кулоновские, дисперсионные и дипольные взаимодей-
взаимодействия, а также специфические химические процессы (например, образование водо-
водородных связей, электронодонорная и электроноакцепторная способности). Природа
и сила таких взаимодействий зависят от свойств растворенных частиц и растворителя.
Сольватация понижает (иногда очень сильно) энергию активации соедине-
соединений и переходных состояний (рис. В. 12) в растворах по сравнению с газообраз-
газообразным состоянием. Таким образом, сольватирующая способность растворителей
может существенно влиять на скорость реакций, а также на положение равно-
равновесия. Поэтому сольватирующую способность растворителей надо обязатель-
обязательно учитывать при выборе реакционной среды.
Для грубой оценки сольватирующих свойств растворителей можно вос-
воспользоваться, например, величинами диэлектрической проницаемости е, ко-
которые в основном определяются электростатическими взаимодействиями
между ионами и полярными веществами. Это макроскопический параметр, и
поэтому на молекулярном уровне он недостаточно точно описывает специфи-
специфические взаимодействия между растворителем и растворенным веществом.
Именно это обстоятельство стимулировало попытку описать влияние раство-
растворителей на реакции с помощью одно- и многопараметрических уравнений с
эмпирическими параметрами. Здесь полезно использование линейного соот-
соотношения энергий (ср. уравнение [В.30] и разд. В,5.2).
Различают следующие типы растворителей.
Неполярные и малополярные растворители; к ним относят углеводороды
(s = 2) и простые эфиры, например диоксан (е = 2,2), диэтиловый эфир
(е = 4,2), тетрагидрофуран (е = 7,4). Эфиры проявляют нуклеофильные свойства.
Полярные протонные растворители; эта группа включает такие важные раство-
растворители, как вода (е = 78), спирты, карбоновые кислоты, аммиак и формамид
(е = 109). Вследствие большой диэлектрической проницаемости эти растворители
способствуют диссоциации ионных пар и солей. Кроме того, за счет своих свобод-
свободных электронных пар они могут путем нуклеофильной сольватации стабилизиро-
стабилизировать центры с недостатком электронов (например, катионы), а за счет кислотных
Н-атомов — электрофильно стабилизировать электроноизбыточные центры (нап-
(например, анионы). Эти особенности проявляются также в том, что данные раство-
растворители обычно существуют в ассоциированном состоянии. Тенденция к образо-
образованию водородных связей возрастает с ростом кислотности растворителя, поэто-
поэтому она особенно сильно выражена, например, у муравьиной кислоты.
3. О протекании органических реакциях во времени 201
Полярные апротонные растворители; к этой группе нуклеофильных раство-
растворителей относятся ацетон (е = 20), ацетонитрил (е = 37), нитрометан (е = 37),
диметилформамид (б = 37), гексаметилфосфортриамид (е = 30), диметилсуль-
фоксид (е = 47), тетрагидротиофендиоксид-1,1 (сульфолан, е = 44), диэфиры
этиленгликоля, этилен- и пропиленкарбонаты (е = 65), М,Ы'-диметилпропи-
ленмочевина, диэтиловый эфир этиленгликоля и др.
Поскольку у этих растворителей нет достаточно кислых Н-атомов, они
сольватируют анионы не за счет образования водородных связей, а за счет зна-
значительно более слабых дисперсионных взаимодействий. При s > 30 эти раство-
растворители способствуют диссоциации ионных пар и солей.
3.4. КАТАЛИЗ
Многие реакции ускоряются при добавлении катализатора. Катализатор реаги-
реагирует с одним из исходных веществ, образуя реакционноспособный промежуточ-
промежуточный продукт, дальнейшее превращение которого в конечные продукты сопро-
сопровождается регенерацией катализатора. Таким образом, создается новый путь к
конечным продуктам, который также требует энергии активации, но уже гораз-
гораздо меньшей, по сравнению с некатализируемой реакцией.
Энергетические отношения между исходными веществами и конечными
продуктами при этом не изменяются, так что катализатор не влияет на положе-
положение равновесия: прямую и обратную реакции он ускоряет одинаково.
Рассмотрим катализируемую протонами енолизацию метилкетона:
О и ОН ОН
С + Н0 =Д= Н3С-@С -*С Н2С=С + Н® [В.42]
R k R R
Вспомнив принцип Боденштейна, запишем кинетическое уравнение [В.43, а]
г = *'** [^"'[Н@] - ПН®] [кетой] = к' [кетон] [в 43]
б в
Поскольку катализатор Не во время реакции не расходуется, его концентрация остается
постоянной, и при экспериментальном определении кинетики получают уравнение [В.43, в] с
константой скорости к'= к[Нф]. Если концентрация Не возрастает, то растет и к, а тем самым
и скорость реакции. Деление к на концентрацию Не дает константу к в полном кинетическом
уравнении (б).
Таким образом, катализатор является веществом, которое входит в кинети-
кинетическое, но не входит в стехиометрическое уравнение реакции.
Примеры кислотно-основного катализа можно найти в разд. Г,7, примеры
катализа кислотами Льюиса — в разд. Г,2.2.1, Г,5.1.5, Г,5.1.7 и др., примеры
катализа комплексами переходных металлов — в разд. Г,4.5.
При гетерогенном катализе, когда в качестве катализатора действует твер-
твердое вещество, промежуточные продукты образуются при хемосорбции реаги-
реагирующих веществ на поверхности катализатора. Здесь к химико-кинетическим
процессам прибавляются транспортные: диффузия исходных веществ к пове-
поверхности катализатора и продуктов реакции от катализатора. Это нередко при-
приводит к очень сложным соотношениям. Примеры гетерогенных каталитичес-
202 В Некоторые общие положения
ких реакций можно найти в разд. Г.4.5.2 и Г,6.3.2. Процессы переноса играют
роль и в межфазном катализе, описанном в разд. Г,2.4.2.
4. КИСЛОТНО-ОСНОВНЫЕ РЕАКЦИИ
Кислотно-основные реакции представляют собой типичные равновесные про-
процессы. Очень многие органические превращения проходят гетеролитически,
т. е. как кислотно-основные реакции (уравнение [В.1]). Поэтому количествен-
количественное рассмотрение этих процессов имеет большое значение для проблемы реак-
реакционной способности в органической химии.
По Бренстеду кислотами являются вещества, способные отдавать протоны
(т. е. доноры протонов), а основаниями — вещества, способные принимать
протоны (акцепторы протонов):
кислота основание протон 1 '
Протонированное основание А—Н часто называют сопряженной кислотой ос-
основания А~.
Кислотно-основный характер не связан с каким-то определенным зарядом молекулы
(достаточно сравнить кислоты Н-С1, Н—NH^, HSO.i~).
Реакция между кислотой Бренстеда и основанием Бренстеда состоит в
переносе протона от кислоты к основанию
А—Н + В ^ Ае + Н-В©
кислота 1 основание 2 основание 1 кислота 2
[В.446]
Из этого уравнения следует, что кислотность и основность взаимосвязаны,
т. е. они должны рассматриваться только совместно. Кислотность и основ-
основность — величины относительные, зависящие от реакционных партнеров и
реакционной среды.
В водных растворах кислот в роли основания выступает вода
Л—Н + Н2О 5? Ае + Н3О® [В.44в]
Константу равновесия этой реакции, умноженную на концентрацию воды
(постоянную в разбавленных растворах), принято называть константой кис-
кислотности Ка
R _ [Н®] [А®] ,В451
До (НА] lB'45J
Эта константа характеризует кислотность кислоты Бренстеда в водном
растворе. Взятый с обратным знаком десятичный логарифм (аналогично рН)
обозначают рКа
-\gKa = VKa [B.46]
рКа тем меньше, чем больше кислотность соединения А—Н.
4. Кислотно-основные реакции
203
В водных растворах оснований вода выступает в роли кислоты
Н2О 5?
[B.47]
Мерой основности аналогично рКа служит взятый с обратным знаком деся-
десятичный логарифм константы основности (рАв).
Кислотность НВе, сопряженной кислоты основания В, можно выразить
через рКа или рАцв®
НВ© + Н2О ^ В + Н3О©
[В.48]
Поскольку в водных растворах кислотность и основность сопряженной
кислотно-основной пары связаны друг с другом через ионное произведение
воды (Aw = 10~14 л2 • моль~2 при 25 °С), то
ИЛИ
[В.49]
Для более наглядного сравнения основность часто выражают в шкале рАд, т. е.
используют рАцв® сопряженной кислоты для характеристики основания В.
Таблица В.50. Величины рКа в
Кислота
НзС-Н
СН3СН2—Н
QHsCHs—H
Н2С=СНСН2-Н
Н2С=СН—Н
нс=с-н
NsCCH2—H
СН3СОСН2—Н
N = C-H
НО—Н
СНзО—Н
СвН5О—Н
СНзСОО—Н
С6Н5СОО—Н
Н2О-Н
СНзОН-Н
(СН3)гО-Н
СН3С(О—Н)?
(СНзJС=О-Н
Вода
~50
~50
~40
~40
~40
~40
~25
~25
20
9,2
15,74
16
10,0
4,76
4,21
-1,74
-2,2
-3,8
-6
-7
воде и диметилсульфоксиде (ДМСО) при 25 °С
дмсо
56
43
44
31,3
26,5
12,9
31,2
29,0
18,0
12,3
11,1
Кислота
H2N—H
CH3NH—H
C6H5NH—H
СНзСОЫН—Н
C2HNH2-H
(CsHsbN-H
е
H3N-H
QHsNHj— H
CH3CONH2—H
CH3C^N—Н
HS-H
C2HSS—Н
СбНбЗ-жгН
(CH3JS-H
CH3SH—Н
F-H
С1—Н
Вг—Н
I—Н
Вода
~35
—35
~25
17
10,6
9,76
9,24
4,6
-10
7,00
10,6
6,5
-5
-7
3,2
-7
-9
-10
ДМСО
41
30,6
25,5
11,0
9,0
10,5
3,6
10,3
15
1,8
0,9
204 В Некоторые общие положения
Сопряженная кислота какого-либо основания тем слабее, а рАнв© тем
больше, чем сильнее основание.
рКа некоторых веществ приведены в табл. В.50. Видно, что кислотность и
основность сильно зависят от природы заместителей в молекуле, поэтому
величины рКа используются также для количественной оценки влияния замес-
заместителей (разд. В,5.2).
Энергия ионов в кислотно-основных реакциях во многом определяется их
сольватацией, и, таким образом, кислотность и основность ионов зависит от
растворителя. Так, в апротонных средах анионы сольватированы гораздо сла-
слабее, чем в протонных растворителях, молекулы которых могут связываться во-
водородными мостиковыми связями с анионами (разд. В,3.3). Этим объясняется
необычное увеличение основности (и, нуклеофильности) анионов при добав-
добавлении воды в апротонный растворитель.
Сравните большие различия значений рКа галогеноводородов, карбоновых кислот, фено-
фенолов и спиртов в воде и диметилсульфоксиде. Например, у карбоксилат-ионов в воде основ-
основность гораздо меньше (на 6 рА"-единиц!), чем у алифатических аминов, а в диметилсульфокси-
диметилсульфоксиде, наоборот, они гораздо основнее алифатических аминов.
Для многих органических реакций важно, что амфотерные соединения
(как-то НО—Н, RO—H, RCOO—Н) в зависимости от партнера в реакции могут
выступать в роли как кислот, так и оснований. Их характеризуют двумя значе-
значениями рКа (рАна и рАнв+), например:
©
СН3СН2ОН2 н
сн3сн2он •
ь Н2О
+¦ н2о
^^ СН3СН2ОН +
:^= СН3СН2О|е •*
н3о0
¦ н3о0
р/Снв® = 2,2
рКНА = 18
[В.51]
[В.52]
Протонированная форма спиртов [В.51] важна, в частности, в катализируемых
минеральными кислотами реакциях образования простых эфиров (разд. Г,2.5.2),
при этерификации неорганическими кислотами (Г,2.5.1) и катализируемом кис-
кислотами образовании олефинов (разд. Г,3.1.4). Протонированная форма карбоно-
карбоновых кислот R—С (ОНJФ важна при образовании сложных эфиров со спиртами
(уравнение [Г.7.39]).
Обратите внимание на то, какие сильные кислоты возникают при протони-
ровании карбонильных и карбоксильных групп (табл. В.50)!
Кислотами Льюиса называют соединения, которые из-за незаполненной
внешней электронной оболочки выступают в качестве акцепторов электрон-
электронных пар. Основаниями Льюиса считают соединения с п- или я-электронами,
выступающие в роли доноров электронных пар.
Кислоты Льюиса:
Н®, R3C®, BF3, AICI3, R2CI (карбены), R-Hal, R2C=O (при С=) [В.53]
Основания Льюиса:
анионы, I NR3, R—O-R, R2C=CR2, арены, R2C=O (при =0) [В.54]
Определения кислот и оснований Бренстеда и Льюиса не перекрываются.
Рассмотрите с этой точки зрения приведенные примеры [В.53] и [В.54].
5. Влияние заместителей 205
При реакциях, в основе которых лежат диполь-дипольные взаимодействия,
электрофильные участки реакции играют роль кислот Льюиса, нуклеофиль-
ные — роль оснований Льюиса. Их реакционноспособность часто изменяется
параллельно кислотности и основности по Льюису. В качестве характеристики
нуклеофильной реакционной способности (нуклеофильности) реагентов
используют константы скорости их реакций с определенным электрофильным
субстратом. Аналогично для характеристики электрофильности служат
константы скорости реакций электрофильных реагентов с определенным нук-
леофилом. Таким образом, в основе определения нуклеофильности и электро-
электрофильности лежит кинетика в отличие от определения кислотности и основ-
основности. При этом речь идет об относительных величинах, зависящих от партне-
партнера и реакционной среды. Поэтому по отношению к разным электрофильным
(нуклеофильным) партнерам, а также разным растворителям обычно устанав-
устанавливаются различные ряды нуклеофильности (электрофильности) реагентов.
Об этом см. также в разд. Г,2.2.2 (нуклеофильное замещение).
Сила и реакционноспособность кислот Льюиса (электрофилов) и основа-
оснований Льюиса (нуклеофилов) существенно зависят от того, являются ли они
«жесткими» или «мягкими».
Жесткие кислоты Льюиса предпочтительно реагируют с жесткими основа-
основаниями, а мягкие кислоты — предпочтительно с мягкими основаниями [принцип
жестких и мягких кислот и оснований (ЖМКО) Пирсона; см. также разд. В,6].
Жесткость кислоты или основания Льюиса возрастает вместе с ростом
плотности заряда и уменьшением поляризуемости. Наоборот, мягкость воз-
возрастает с уменьшением плотности заряда и ростом поляризуемости.
ф Таким образом, жесткие кислоты — это, например, Н+, BF3, А1СЬ, SO3,
RC=O; жесткие основания — это, например, Н2О, HO~, F~, СНзСОО", ROH,
NH3.
К числу мягких кислот Льюиса относятся ионы переходных металлов в низ-
низких степенях окисления, радикалы (например, С1-, Вг, R-, RO), карбены; кмяг-
ким основаниям — R2S, RSH, R3P, I", SCN~, CN~, олефины, арены, Н~ и R~.
5. ВЛИЯНИЕ ЗАМЕСТИТЕЛЕЙ НА РАСПРЕДЕЛЕНИЕ
ЭЛЕКТРОННОЙ ПЛОТНОСТИ И РЕАКЦИОННУЮ
СПОСОБНОСТЬ ОРГАНИЧЕСКИХ МОЛЕКУЛ
5.1. ПОЛЯРНЫЕ ЭФФЕКТЫ ЗАМЕСТИТЕЛЕЙ
Полярные заместители за счет своей электронодонорной или электроноакцеп-
торной способности изменяют распределение электронной плотности (распре-
(распределение зарядов) в органических молекулах. Тем самым они оказывают влияние
на реакционную способность.
Электроноакцепторные заместители X понижают (по сравнению с X = Н)
электронную плотность у реакционного центра Z. Тем самым они способ-
способствуют атаке нуклеофильного реагента |Nu и затрудняют атаку электрофиль-
ного Е:
206 В Некоторые общие положения
X-R-Z +NINu X-R-Zr + E
увеличение уменьшение [В.55]
реакционной реакционной
способности способности
Электронодонорные заместители Y, напротив, повышают электронную
плотность на реакционном центре (по Сравнению с Y = Н). Они понижают ре-
реакционную способность по отношению к нуклеофильным реагентам, повы-
повышают к электрофильным:
уменьшение увеличение [В.56]
реакционной реакционной
способности способности
Таким образом, представления о влиянии полярных заместителей позволя-
позволяют оценить реакционную способность органических соединений.
Индукционный эффект. Индукционный эффект (или эффект поля) основан
главным образом на электростатическом притяжении или отталкивании, воз-
возникающем между заместителем и остальной частью молекулы. Это взаимодей-
взаимодействие подчиняется закону Кулона и поэтому быстро убывает с расстоянием,
исчезая в цепи более трех С-атомов. Заместителю приписывают —/-эффект,
если он, образуя связь с углеродом, притягивает электроны сильнее, чем водо-
водород. При +/-эффекте, наоборот, заместитель притягивает электроны слабее,
чем водород.
Индукционный эффект возрастает с ростом электроотрицательности и за-
заряда заместителей. —/-Эффект тем больше, чем правее и выше в периодичес-
периодической системе находится центральный атом заместителя. Все без исключения
ненасыщенные группы обладают —/-эффектом, который возрастает с преоб-
преобладанием 5-характера гибридных орбиталей (двойная связь < тройная связь).
Относительная величина и направление индукционного эффекта различ-
различных заместителей указаны ниже:
-/: -NR2 < -OR < -F -I < -Br < -Cl < -F
-NR3 < -OR2 -NR2 < =NR < =N
-CR=CR2 < -К У < -C=CR
+ /: _Q|° < -N-R
Алкильным группам длительное время приписывали +/-эффект. Послед-
Последний, однако, очень мал, близок к нулю. Но все же из-за их поляризуемости
алкильные группы способны стабилизировать соседние положительные, а так-
также отрицательные заряды. Этот эффект поляризуемости повышается с увели-
увеличением размера заместителя в следующем ряду.
/ - F
-СН3 < -СН2-СН3 < -СН < -С-СНз [В.58]
СНз СНз
5. Влияние заместителей 207
Для количественной оценки силы и направления индукционного эффекта
можно использовать индукционные константы заместителей а/(табл. В.74).
Индукционный эффект проявляется как в насыщенных, так и в ненасы-
ненасыщенных соединениях; в насыщенных молекулах его проявление — единствен-
единственный эффект заместителя.
Мезомерный эффект. Кроме индукционного эффекта заместители могут
проявлять еще и мезомерные эффекты, если заместитель связан с ненасыщен-
ненасыщенной системой или с атомом, имеющим свободную электронную пару, и может
вступать с ним в сопряжение.
В согласии с квантовохимической моделью сопряжением называется перек-
перекрывание р-орбитали (на которой могут находиться два, один или ни одного
электрона) заместителя с молекулярной л-орбиталью соседней двойной связи.
Таким образом, становится возможной делокализация п-электронов, что приво-
приводит к характерным отличиям от несопряженных соединений: уменьшению
энергии (стабилизации), уменьшению длины формально одинарных связей,
перераспределению зарядов. В результате распределение электронов в сопря-
сопряженных соединениях уже не удается выразить с помощью единственной струк-
структурной формулы. Реальное состояние передают с помощью нескольких
«граничных» формул1, запись которых основана на классических структурных
формулах, например:
Н3С-СН=СН-СН = О Н3С-СН-СН=СН-О|е = НзС-СН-СН-СН-0 [В-59]
H2N-CH=O H2N=CH-QI = H2N -CH-0 [й.ЬО]
Н2С=СН-СН > H2C-CH=Ci® = H2C -CH-CI [B.61]
Граничные формулы - это только способ условной записи, физической реальности за ни-
ними нет. Поэтому двустороннюю стрелку (символ мезомерии) ни в коем случае не следует пу-
путать с символом равновесия^. Реальное распределение электронов примерно соответствует
наложению граничных формул: его можно передать написанными справа формулами (схемы
[В.59]—[В.61]). Недостаток таких формул проявляется в том, что в них непосредственно не
видно числа делокализованных электронов.
Явление, когда действительная структура молекулы находится между ис-
использованными для их описания (фиктивными) граничными формулами, на-
называют мезомерией («между частицами»). Мезомерный эффект заместителя
обозначают знаком заряда, который приобретает рассматриваемая группа2.
Например, в схемах [В.59] и [В,60] группа С=О проявляет отрицательный
мезомерный эффект (—Л/), а в [В.61] С1-атом — +М-эффект.
1 Или используя одну формулу с символикой зарядов и пунктирных связей, как это, в
частности, показано в правой части схем (В.59)—(В.61). — Прим. перев.
2 Знак мезомерного эффекта обратный по сравнению со знаком константы заместителя а
(разд. В.5.2).
208 В Некоторые общие положения
+Л/-эффект тем больше, чем левее стоит заместитель в периоде периоди-
периодической системы и чем больше его отрицательный заряд. Для —М-эффекта со-
соотношение обратное. В общем случае мезомерный эффект подчиняется следу-
следующим закономерностям:
+ М: -?| < -OR < -NR2
-II < -Brl < -СП < -Fl* [В.62]
-OR < -01°
_ ©
-М: = NR < =NR2
= CR2 < =NR < =0 [B.63]
Этот ряд основан на том, что необходимые для участия в сопряжении электроны нахо-
находятся у С-атома на 2^-орбитали (или соответствующей гибридной орбитали); так обстоит дело
и у атома фтора. Соответствующие орбитали у хлора, брома и иода — это 3/ь, 4/>- и 5/>-орбита-
ли, что делает их пространственно менее выгодными для перекрывания с 2/>-орбиталью атома
углерода.
В меньшей степени ст-электроны насыщенных остатков также в состоянии
вступать в сверхсопряжение (гиперконъюгацию) с л-электронной системой. На
этом основан незначительный +А/-эффект алкильных групп.
Группы, способные к мезомерии, одновременно всегда проявляют и ин-
индукционный эффект. Знак обоих эффектов может либо совпадать, либо быть
противоположным. У атома хлора —/-эффект больше, чем +М-эффект, так
что хлор, связанный с ненасыщенным остатком, ведет себя как акцептор
электронов. Для -NR.2 картина обратная: -/ < +М, поэтому группа -NR2
действует как донор электронов. Величина мезомерного эффекта сильно за-
зависит от остальной части молекул (в связи с этим см. разд. Г,5.1.2 о втором
электрофильном замещении в аренах), поэтому суммарный эффект оценить
не всегда легко. В этом плане значительную ценность имеют константы за-
заместителей Гаммета ст (табл. В.74), в которых отражается суммарное влияние
индукционного и мезомерного эффектов заместителя, связанного с бензоль-
бензольным ядром.
Разлагая на компоненты суммарные константы заместителей о, можно полу-
получить количественную меру мезомерного эффекта в виде констант or (табл. В.74).
Схема [В.59] показывает, что в результате мезомерии положительный заряд
5+ с карбонильного углерода ^С=0 частично переносится на атом углерода
С-3. В результате метильная группа приобретает свойства, сходные с теми,
которые она имеет при непосредственной связи с карбонильной группой в
структуре СН3-СН=О. Подобный перенос влияния заместителя винильной
группой называют принципом винилогии.
В согласии со сказанным аминогруппа р-аминокротонового эфира [В.64]
имеет свойства не амина, а амида. То же относится и к аминогруппе в 4-ами-
5. Влияние заместителей
209
ноацетофеноне [В.65]; этот последний пример иллюстрирует принцип фени-
логии:
с=сн-с
H2NI
[B.64]
H2N
[B.65]
СН3
Рассмотрите также распределение зарядов в енамине [В.66] с учетом прин-
принципа винилогии, а также химический сдвиг винилогичного протона кротоно-
вой кислоты (см. рис. Д.38) в спектре ЯМР 'Н:
Н
R2N-CH=C
[В.66]
Аг
5.2.
КОЛИЧЕСТВЕННЫЙ ПОДХОД К
ПОЛЯРНЫМ ЭФФЕКТАМ ЗАМЕСТИТЕЛЕЙ.
УРАВНЕНИЕ ГАММЕТА
Эмпирически установлено, что полярные заместители по своему влиянию на кислотность и
основность располагаются в том же порядке, что и по влиянию на реакционную способность
органических соединений. Для реакций мета- и «о/>а-замещенных производных бензольного
ряда подобную корреляцию можно описать количественно. Логарифмы скоростей реакций
замещенных бензолов находятся в линейной зависимости от рКа (логарифмов констант диссо-
диссоциации Ка) замещенных бензойных кислот (рис. В.68).
= pig K0
[В.67]
где р - константа реакции.
Уравнение [В.67] представляет собой одну из форм обсуждавшегося ранее [В.30] линейно-
линейного соотношения свободных энергий ArCh ДО*. Это уравнение получается из [В.30] при подс-
подстановке [В. 14] и[В.26].
Рис. В.68. Зависимость скорости гидролиза этиловых эфиров мета- и
лара-замещенных бензойных кислот от кислотности соответствующих кислот.
210
В Некоторые общие положения
Если из уравнения [(В.67] вычесть соответствующее выражение для незамещенного соеди-
соединения (X = Н, к = къ, К= Ко), то получим уравнение Гаммета:
]g — = pa [B.69]
К
где a—\g—rr~ (а— константа заместителя).
К
[В.70]
Уравнение Гаммета справедливо и для многих других реакций, в которых принимают участие
мета- и ла/кг-замещенные производные бензола; наряду с применением его для расчета констант
скоростей реакций оно может быть также использовано для расчета констант равновесия.
Константы заместителей а характеризуют электронные эффекты заместителей в мета- и
«ара-положениях. Они дают сумму индукционных и мезомерных эффектов заместителей.
Некоторые значения приведены в табл. В.74.
Константа реакции р характеризует чувствительность реакции к влиянию полярных замести-
заместителей. Реакции, протекающие быстрее (или более полно), по мере того, как реакционный центр
становится более положительным (или отрицательным), характеризуются положительным значе-
значением р.
Нуклеофильныереакции: р положительна; ускорение реакции в результате электроноакцеп-
торного действия заместителей.
Электрофильные реакции: р отрицательна; ускорение реакции в результате электро-
нодонорного действия заместителей.
Уравнение Гаммета используют обычно в графической форме: наносят по оси абсцисс
значения о отдельных заместителей, а по оси ординат — экспериментальные значения lg к/ко
(рис. В.71). Тангенс угла наклона прямой соответствует константе реакции р, по знаку которой
можно сделать вывод относительно механизма данной реакции (см. выше).
Рис. В.71. Зависимость скоростей реакции от величин сг-констант Гаммета.
7 — щелочной гидролиз этилбензоатов B5 "С; р = +2,54);
2 — взаимодействие замещенных анилинов с бензоилхлоридом B5 *С; р = -2,78);
3 — бромирование замещенных толуидинов (8О'С;р = -1,39); относительно а+-констант см. текст.
5. Влияние заместителей 211
Уравнение Гаммета неприменимо в том случае, когда кроме электронных эффектов замес-
заместителей на реакционный центр оказывают влияние другие факторы, например пространствен-
пространственные, как это имеет место в случае реакций с участием opmo-замещенных производных бензола и
у алифатических соединений1.
Отклонения от уравнения Гаммета встречаются также и в том с случае, когда возникает
прямое сопряжение (мезомерия) между заместителем X и реакционным центром Y. Так,
заместители в пара-положении, обладающие —М-эффектом (NO2, CN и др.), увеличивают
кислотность фенолов или ионов анилиния сильнее, чем это следует из их о-констант, так как
образующиеся фенолят-ион и ион анилиния дополнительно стабилизируются в результате
сопряжения
[В.72]
©
NH2
©4
В этих случаях необходимо применять особые значения констант заместителей, обозначае-
обозначаемые а~.
Напротив, электронодонорные заместители (группы, обладающие +М-эффектом) повы-
повышают скорость реакций, протекающих по положительному (карбений-подобному) реакцион-
реакционному центру, гораздо сильнее, чем это следует из их а-констант. В таких случаях для этих за-
заместителей необходимо использовать ст+-константы, которые учитывают дополнительную
стабилизацию положительного реакционного центра, вызываемую данными заместителями.
Например, это необходимо в случае SNl-реакций (см. разд. Г,2.1) замещенных а,а-диметил-
бензилгалогенидов (кумилгалогенидов), когда в качестве промежуточного продукта образует-
образуется соответствующий катион:
НзСО-Чч />-С-С1 —q- НзСО—(ч />-С -—- Н3Сд=( )=С [В.73]
^—^ СН3 -С1 ^—^ СН3 ^^ СН3
а+-Константы Гаммета играют особую роль, так как они могут применяться в случае реакций
электрофильного замещения в ароматическом ядре (разд. Г,5.1.2). Некоторые значения этих
констант приведены в табл. В.74. В случаях лета-заместителей константы а, а~и а+, как и
ожидалось, практически идентичны, так как в этом положении невозможно сколько-нибудь
заметное электронное сопряжение (мезомерия). Поэтому, по-видимому, а„ отражает один ин-
индукционный эффект заместителя; а так как о„ - сумма его индукционного и мезомерного
эффектов, то разность а„ — ом характеризует мезомерный эффект2. Однако оказалось, что эти
соотношения выполняются не строго, поскольку при л<ета-замещении возможно также неп-
непрямое проявление сопряжения с реакционным центром молекулы. Если ст-константу рассмат-
рассматривать частично как а/ (обусловлена индукционным эффектом) и частично как ctr (обусловле-
(обусловлена мезомерным эффектом), то тогда получим
Оп = а, + aR [B.75]
Ом := О г -I- — 0р г т> nri
'~ 3 IB.76J
Константы а/ проще всего получить из величин рКа замещенных уксусных кислот
Х-СНгСООН; наряду с стя-константами они представлены в табл. В.74. Видно, что эти конс-
константы правильно характеризуют обсуждавшиеся в разд. В,5.1 индукционные и мезомерные
эффекты заместителей.
1 В соотношениях свободных энергий формальные термодинамические величины комби-
комбинируют с величинами, характеризующими процессы на молекулярном уровне. Поэтому они
не имеют строгости макроскопических термодинамических законов.
2 CTj, - константа для ле/иа-замещенных производных, о„ — то же для лара-производных. -
Прим. перев.
212
В Некоторые общие положения
Таблица
№
1
2
3
4
5
6
7
8
9
10
И
12
13
14
15
16
17
18
1 В.74. Константы Гаммета для некоторых заместителей
Заместитель
N(CH3b
NH2
ОН
ОСНз
СН3
С(СН3)з
с6н5
н
F
С1
Вг
I
COOQHs
СОСНз
CN
SO2CH3
NO2
N(CH,),«
ам
-0,21
-0,16
0,12
0,12
-0,07
-0,10
0,06
0
0,34
0,37
0,39
0,35
0,37
0,38
0,56
0,60
0,71
0,88
-0,83
-0,66
-0,37
-0,27
-0,17
-0,20
-0,01
0
0,06
0,23
0,23
0,18
0,45
0,50
0,66
0,72
0,78
0,82
-1,7
-1,3
-0,92
-0,78
-0,31
-0,26
-0,18
0
-0,07
0,11
0,15
0,14
0,48
0,66
0,79
0,41
0,10
0,10
0,25
0,25
-0,05
-0,07
0,10
0
0,52
0,47
0,45
0,39
0,30
0,28
0,58
0,59
0,63
0,86
-0,76
-0,61
-0,50
-0,13
-0,13
-0,09
0
-0,44
-0,24
-0,22
-0,10
0,20
0,25
0,07
0,14
0,15
0,00
(~±М-эффект)
0,62
0,50
0,49
0,39
0,10
0.10
0,07
0
0,28
0,14
0,16
0,17
-0,08
-0,12
-0,10
-0,12
-0,07
0,06
5.3. ПРОСТРАНСТВЕННЫЕ ЭФФЕКТЫ
Помимо электронных (полярных) эффектов на реакционную способность вли-
влияют и пространственные эффекты.
Объемистые заместители затрудняют подход реагента («пространственные
препятствия»); для преодоления этого влияния необходима повышенная энер-
энергия активации. Кроме того, при наличии пространственных препятствий энт-
энтропия активации AS* становится более отрицательной. На реакционную спо-
способность влияют также конформационные (стереохимические) эффекты.
Соответствующие примеры рассмотрены в разд. Г.3.1.3.
Напряженные циклические системы (например, циклопропильная, цикло-
бутильная, бицикло[2.2.1]гептильная) из-за деформации валентных углов не-
несут в себе дополнительный запас энергии. Такие системы особенно реакцион-
носпособны в тех случаях, когда в результате реакции снимается напряжение.
6. РАССМОТРЕНИЕ РЕАКЦИОННОЙ СПОСОБНОСТИ
С ПОЗИЦИИ ТЕОРИИ ВОЗМУЩЕНИЙ
Известны попытки подойти к решению проблемы химической реакционноспо-
собности средствами теории возмущения молекулярных орбиталей. Таким пу-
путем можно описать начальную стадию реакций, т. е. начальный энергетический
подъем кривой к переходному состоянию (рис. В. 12), рассматривая разносто-
разносторонние возможности сближения реагирующих веществ и возникающие при
этом изменения энергии. Правильные представления о переходном состоянии и
о реакции в целом будут следовать из такого рассмотрения лишь в тех случаях,
если начальная стадия реакции определяет дальнейший ход реакции (рис. В.28).
6. Рассмотрение реакционной способности
213
Е к
-0,8 +0,6
+0,6 +0,8
Энергетические Коэффициенты
уровни
НСМО
ВЗМО
Рис. В.77. Энергии и населенности ВЗМО и НСМО я-связи в группе СО.
Квадраты коэффициентов соответствуют электронной плотности на атомах.
В качестве ВЗМО здесь выступает высшая занятая л-орбиталь, хотя в действительности
высшей занятой орбиталью группы С=О является л-орбиталь.
Основой при рассмотрении реакционной способности с позиций теории возмущений слу-
служат энергии Е молекулярных орбиталей (МО), рассчитанные квантовохимическими метода-
методами, а также коэффициенты, относящиеся к ним (их величина и знак), как это представлено на
рис. В.77 для 71-связи в карбонильной группе С=О. Эти данные получают путем изучения
линейных комбинаций атомных орбиталей (ЛКАО). Взаимодействие электронов при этом в
общем не учитывают.
Как правило, достаточно иметь такую информацию для высшей занятой молекулярной
орбитали (ВЗМО) и низшей свободной молекулярной орбитали (НСМО) реагирующих
веществ. ВЗМО и НСМО называют граничными орбиталями. Предполагается, что читатель
знаком с употребляемыми здесь квантовохимическими величинами и их значением.
Энергии граничных орбиталей можно выразить через экспериментально доступные вели-
величины. Мерой энергии ВЗМО (?взмо) является энергия ионизации соответствующего соедине-
соединения (отнесенная к энергии электрона в вакууме, ?взмо= энергия ионизации) или полуволна
потенциала окисления.
Мерой энергии НСМО (?нсмо) для данного соединения является сродство к электрону
(отнесенное опять же к энергии электрона в вакууме, ?нсмо = сродство к электрону) или полу-
полуволна потенциала восстановления. Энергии ионизации и сродства к электрону можно
оценить, например, по спектрам поглощения комплексов с переносом заряда, а энергии иони-
ионизации - также по фотоэлектронным спектрам.
Крутизна энергетической кривой и величина энергии активации определя-
определяются несколькими противоположно действующими факторами. Взаимодей-
Взаимодействия между занятыми орбиталями реагирующих веществ, а также увеличение
межатомных расстояний в реагирующих молекулах особенно повышают энер-
энергию, необходимую для реакции. Факторы, снижающие энергию, в дальней-
дальнейшем рассмотрены подробнее. Хотя они существенно слабее, однако именно
они характерно влияют на ход энергетической кривой и, как правило, на вели-
величину энергии активации.
При рассмотрении с позиций теории возмущения учитывают следующие
факторы, понижающие энергию:
а) кулоновское взаимодействие в случае заряженных или полярных
реагирующих веществ (уравнение [В.78]);
214 В Некоторые общие положения
б) взаимодействие между занятыми орбиталями одного участника реакции
и свободными орбиталями другого (уравнение [В.78]).
Чаще всего рассматривают только ВЗМО и НСМО реагирующих веществ,
поскольку взаимодействия других занятых и незанятых орбиталей обычно не
приводят к заметному выигрышу энергии.
-?взмо характеризует нуклеофил, который выступает в качестве донора
электронов, вносящего свою электронную пару в образующуюся ковалентную
связь. ?нсмо аналогично описывает электрофил.
Выигрыш энергии Доопределяется по уравнению
eR ?взмО(»г-?нсм0(е) [В.78]
кулоновское граничные
взаимодействие орбитали
где Q — общий заряд нуклеофила (и) и электрофила (е);е— локальная диэлект-
диэлектрическая проницаемость; R — расстояние между нуклеофилом и электрофи-
лом; с — коэффициенты орбиталей ВЗМО и НСМО нуклеофила и электрофила
(и и е)\ р - резонансный интеграл (энергия взаимодействия двух атомных орби-
орбиталей в нуклеофиле и электрофиле); Е— энергия ВЗМО и НСМО.
Если преобладающим в [В.78] является кулоновское взаимодействие, то
говорят, что реакция контролируется зарядом. Если преобладает гранично-
орбитальный член уравнения, то говорят об орбитальном контроле. На боль-
большинство реакций в различной степени влияют оба фактора.
В дальнейшем мы подробнее рассмотрим лишь реакции, управляемые в
основном орбитальными факторами. Анализ граничноорбитального вклада в
уравнении [В.78] позволяет установить, что выигрыш энергии возрастает по
мере того, как
а) растут вклады, характеризующие участие во взаимодействии центров;
б) уменьшается различие между энергией ВЗМО одного партнера и
НСМО другого.
Это означает, что предпочтительны ВЗМО — НСМО-взаимодействия с
малой затратой энергии (рис. В.79), что соответствует отнесению участников
реакции к числу нуклеофилов и электрофилов (см. схемы [В.1] и [В.4]).
Энергии граничных орбиталей позволяют теоретически обосновать каче-
качественную характеристику нуклеофилов и электрофилов как жестких или мяг-
мягких кислот и оснований (разд. В,4):
• Жесткие кислоты (электрофилы) имеют относительно богатую энер-
энергию НСМО (относительно низкое сродство к электрону), относитель-
относительно высокую плотность положительного заряда и малую поляризуе-
поляризуемость.
• Мягкие кислоты (электрофилы) имеют, напротив, НСМО относительно
небольшой энергии (относительно большое сродство к электрону) и
высокую поляризуемость.
• Жесткие основания (нуклеофилы) имеют ВЗМО с относительно низкой
энергией (относительно высокая энергия ионизации), относительно
высокую плотность отрицательного заряда и малую поляризуемость.
6. Рассмотрение реакционной способности
215
НСМО
\нсмо
ж
Рис. В.79. Разности энергии ВЗМО (НСМО) и НСМО (ВЗМО) двух участников реакции
А и В (/) и достигаемый при их взаимодействиях выгрыш энергии (// и ///).
ВЗМО - НСМО-взаимодействия и в том и в другом случае (// и ///) относятся к одной и
той же точке в начале энергетической кривой (рис. В. 12).
• Мягкие основания (нуклеофилы) имеют ВЗМО с несколько большей
энергией (относительно низкая энергия ионизации) и высокую поля-
поляризуемость.
Поскольку мягкие основания имеют высоколежащую ВЗМО, а мягкие кис-
кислоты — низколежащую НСМО, разность энергий орбиталей в этом случае
меньше, чем при комбинации мягкого основания (высоколежащая ВЗМО) с
жесткой кислотой (высоколежащая НСМО). Таким образом, предпочтитель-
предпочтительнее реакция мягкого с мягким.
Между жесткими основаниями (низколежащая ВЗМО) и жесткими кислота-
кислотами (высоколежащая НСМО) различия в энергии относительно велики, так что
эта реакция протекает преимущественно как контролируемая зарядом, т. е.
в уравнении [В.78] определяющим становится кулоновский вклад.
Можно обосновать и влияние заместителей на нуклеофильность или электро-
фильность реагирующих веществ. Если, например, сравнить энергии ионизации
(как меру -Евзмо) различных замещенных бензолов, то можно заметить падение
нуклеофильности в ряду: диметиламинобензол (—7,51 эВ), бензол (—9,40 эВ); нит-
нитробензол (—10,26 эВ). Качественно тот же самый ряд получают при рассмотрении
мезомерного влияния заместителей на молекулу бензола (разд. В,5), а также из
величины и знака констант заместителей в уравнении Гаммета (табл. В.74).
О влиянии донорных и акцепторных заместителей на ?взмо и ?нсмо У
олефинов см. разд. Г,4.4.
Орбитально-контролируемые превращения реагентов с несколькими
электрофильными и нуклеофильными центрами (т. е. амбифункциональных
или амбидентных систем) можно описать через энергии орбиталей с учетом
коэффициентов:
— у амбидентных электрофилов — наиболее электрофилен центр с наи-
наибольшим вкладом в НСМО;
— у амбидентных нуклеофилов — наиболее нуклеофилен центр с наиболь-
наибольшим вкладом в ВЗМО.
216 В Некоторые общие положения
Ср. С/О-алкилирование
(разд.Г,2иГ,7.4)
Ср. О-алкилирование(разд. Г, 2)
0 }*^ ®Ф—Я и реакции фенолятов
О и | Л (разд. Г, 5)
О nWn Ср. образование алифатических
О' О O-vVo нитросоединений или эфиров
азотистой кислоты (разд. Г,2)
®CEN гОь—J <~'р' обРазование цианидов
иизоцианидов(разд. Г, 2)
Рис. В.80. Амбидентные (амбифункционапьные) нуклеофилы: отрицательный заряд
делокализован; изображены также предельные структуры и населенности ВЗМО.
Таковы следствия анализа граничноорбитального члена в уравнении [В.78].
На рис. В.80 даны примеры амбидентных нуклеофилов (ср. также разд. Г,2.3).
Для взаимодействия с электрофилами у цианид-аниона имеются две орбитали с
относительно небольшой разностью энергий: высшая занятая а-МО и высшая
занятая л-МО (на рис. В.80 характеристики обеих МО объединены). При обсуж-
обсуждении региоселективной реакции циклоприсоединения (разд. Г,4.4) учитывают
также амбивалентность электрофильного партнера реакции.
Граничноорбитальный подход к реакциям, протекающим преимуществен-
преимущественно при орбитальном контроле, ограничивается рядом факторов, особенно
если речь идет о предсказаниях течения реакций. Проблемы возникают, нап-
например, в связи с доступностью данных об орбитальных энергиях. Так, кванто-
вохимически рассчитанные энергии для ВЗМО и НСМО можно сравнивать
лишь тогда, когда они получены с одинаковыми приближениями. Точные
экспериментальные данные о величинах ?нсмо получить трудно.
Квантовохимически рассчитанные орбитальные энергии, а также энергии
ионизации и сродства к электрону относятся лишь к газовому состоянию.
Сольватационные эффекты учесть здесь трудно. К тому же доля зарядового и
орбитального контроля в разных реакциях различна.
По всем этим причинам численные данные о реакционноспособности
молекул и относительной активности различных реагентов (разд. В,3.2) не
являются абсолютными, всегда постоянными величинами; они зависят от
типа реакции, от партнера, от среды.
7. СТЕРЕОИЗОМЕРИЯ
Соединения одинакового строения (топология, связывание атомов), которые
отличаются только пространственным расположением атомов, называют
стереоизомерами. Они могут иметь различные физические свойства и отличать-
отличаться реакционной способностью.
7. Стереоизомерия
217
7.1. КОНФОРМАЦИЯ
Стереоизомеры, которые образуются путем поворота вокруг простой связи,
называют конформерами, соответствующее пространственное расположение
атомов — конформацией.
Некоторые конформации бутана, которые получены при повороте вокруг
С—С-связи, например центральной С—С-связи, изображены на схеме [В.81].
Так как заместители у обоих атомов С влияют по-разному в зависимости от уг-
угла поворота вокруг С—С-связи, энергии отдельных конформеров различаются,
ср. рис. В.82. Как правило, конформации, такие, как1и II [В.81], в которых за-
заместители у соседних атомов С скошены {англ. staggered), из-за небольшого
стерического взаимодействия беднее энергией, чем в заслоненных {англ.
eclipsed) конформациях III и IV. Самой стабильной обычно является пол-
полностью скошенная конформация — трансоидная (антиперипланарная), в
которой самые большие заместители находятся в анти- {транс) положениях.
Наибольшее стерическое взаимодействие проявляется в полностью заслоненной,
синперипланарной конформации IV, в которой большие заместители расположе-
расположены в син (цмс)-ориентации. Она поэтому энергетически самая высокая.
Перспективные
проекции
(«КОЗЛЫ»)
Проекции
Ньюмена
("*?*"
1 Н^Н СНз
( хНз
1 HYTY
1 -<>.
1 н^~~г^н
СНз
1
трансоидная
антиперипланарная
заторможенная
анти, гране
н5>СНз
нЖнн
СН3
Ну+уСНз
Н ПГ Н
н
II
скошенная
синклинальная
ГОШ
Н
НзС J\
UH СНз
н-^н
п п
НСН3
/-К
1^ Л^>^СНз
н'^'^н
III
частично заслоненная
антиклинальная
СНз
н-^н
П П
Н3ССН3
S) 1
н^сЫ-н
н н
IV
полностью заслоненная
синперипланарная
син, цис
[В.81]
Для изображения конформеров на бумаге применяются различные проек-
проекции. В так называемом способе изображения «козлы» или «перспективная
проекция»1 связь С—С-рисуют диагонально и немного удлиненной и проециру-
Е0ТН, кДж/моль
0° 60° 120° 180° 240° 300° 360°
Рис. В. 82. Энергия бутана в зависимости от поворота вокруг внутренней С-С-связи.
1 См. Потапов В. М. Стереохимия. — М: 1988. — Прим. перев.
218 В Некоторые общие положения
ют заместители у обоих атомов углерода на плоскость бумаги (см. [В.81] навер-
наверху). По проекции Ньюмена С—С-связь изображается наклонно и между С-ато-
мами рисуют кружок ([В.81] внизу).
Если, однако, между заместителями у обоих атомов углерода существует при-
притяжение, как, например, между двумя гидроксильными группами, которые
связаны водородным мостиком, то синклинальная и синперипланарная кон-
формации более стабильны, чем антиперипланарная (I) и антиклинальная (III).
В соединениях с открытой цепью различие в энергиях между отдельными конформерами в
целом невелико, например, в бутане [В.81] между конформациями I и II оно едва составляет
4 кДж/моль, между I и III — около 16 кДж/моль, ср. рис. В.82. При нормальных условиях поэто-
поэтому поворот вокруг С-С-связи мало ограничен и различные конформеры не удается выделить.
Различие в энергии Гиббса в 4 кДж/моль соответствует согласно уравнению [В. 14] константе
равновесия К= 5,0 при 25 °С, а в случае различия в энергии Гиббса в 16 кДж/моль - константе
скорости к = 1,0 ¦ 10" с~' (по уравнению [В.26]), т. е. термодинамическое равновесие для кон-
формеров I и II устанавливается при их соотношении 5:1, но они взаимопревращаются друг в
друга с такой скоростью, что их выделить по отдельности невозможно.
Если же заместители у соседних атомов С такие большие, что невозможно
свободное вращение вокруг С-С-связи, то конформеры могут быть разделе-
разделены, как, например, в случае 1,1,2,2-тетра-/и/>е/и-бутилэтана или в о-замещен-
ных дифениленах. В этих так называемых атропоизомерах оба бензольных
кольца повернуты почти на 90 "С.
R
[В.83]
При различных о-заместителях оба атропоизомера соотносятся как картинка и ее зеркаль-
зеркальное изображение, которые не могут быть совмещены и поэтому являются энантиомерами, см.
разд. В,7.3.1.
В алициклических соединениях число возможных конформаций ограниче-
ограничено, так как полный поворот вокруг одной С—С-связи в цикле невозможен.
У циклогексана имеются две конформаций, в которых угол между С—С-связями
тетраэдрический и поэтому свободен от напряжения, это формы кресла и ванны.
На На н н Н Н
Мл I II II
На на нн нн н н [В84]
форма кресла форма ванны твист-форма
(а — аксиальный,
е — экваториальный)
Форма ванны, которая содержит заслоненные конформаций, примерно на
27 кДж/моль богаче энергией, чем форма кресла, в которой только трансоид-
ные конформаций. Несколько менее напряжена твист-форма, но все же на
21 кДж/моль энергетически богаче, чем форма кресла. Циклогексан и больши-
большинство его производных существуют поэтому предпочтительно в форме кресла.
7. Стереоизомерия 219
Отдельные конформеры могут путем поворота вокруг С—С-связи и «вывора-
«выворачивания» кольца через твист-форму переходить друг в друга. Энергия актива-
активации этого перехода составляет 45 кДж/моль и константа скорости при 25 °С
В замещенных циклогексанах положение заместителя у С-атома кольца
может быть либо аксиальным (а), или экваториальным (е от англ. equatorial),
т. е. ближе к плоскости цикла (см. [В.84]). Аксиальные и экваториальные кон-
конформеры находятся в равновесии, так как существует возможность легкого вы-
выворачивания кольца. Равновесие сдвинуто в сторону обладающего меньшей
энергией экваториального конформера и тем больше, чем больше заместитель.
-СНз(е)
[В.85]
СНз (а) дкG = -7,5 кДж/моль, К= 21
Так, например, экваториальная форма металциклогексана на 7,5 кДж/моль беднее энер-
энергией, чем аксиальная, и в равновесии она составляет 95,4% (К= 21). Для mpem-бутилциклогек-
сана соответствующие значения составляют 20 кДж/моль, или 99,97% {К= 3200).
В химических реакциях циклогексанов, например реакциях элиминирова-
элиминирования (ср. разд. Г,3.1.3), всегда следует считаться с превращениями экваториаль-
экваториальных и аксиальных конформаций1.
7.2. ЦИС, ГРАНС-ИЗОМЕРИЯ
В соединениях с двумя заместителями у атомов, связанных связью неспособ-
неспособной к вращению, возникают цис, транс -изомеры (ранее обозначались как гео-
геометрические изомеры). Это происходит в соединениях с двойными связями и в
циклических соединениях. В замещенных алкенах ^нс-изомеры содержат за-
заместители с одной стороны двойной связи, в транс -изомерах — на противопо-
противоположных сторонах, например:
ноосч роон ноосч н
соон [В86]
с
соон
цис- или (Z)-3TeH-1,2-дикарбоновая кислота гране- или (Е)-этен-1,2-дикарбоновая кислота
(малеиновая кислота) (фумаровая кислота)
Так как в соединениях с двумя различными заместителями цис-транс-соот-
несение неоднозначно, их в настоящее время называют по системе E/Z. Если,
оба заместителя с наивысшим значением в КИП-системе (см. с. 222) — с одной
стороны двойной связи, то это B)-изомер (от нем. zusammen — вместе),
другой — (?)-изомер (от нем. entgegen — напротив).
1 Если особенности строения делают молекулу жесткой, такое превращение невозможно,
например, в конденсированных циклических системах, как в транс-декалине [В.90].
220
В Некоторые общие положения
Н3С
соон
с=с
н" сн3
Н3С СНз
с=с
н' соон
(Z)
[В.87]
(?)- и B)-алкены - различные химические соединения, различающиеся физическими и
химическими свойствами.
Температура плавления малеиновой и фумаровой кислот [В.86] составляет 130 и 286 °С, а
их константа кислотности рК„ равна 1,8 и 3,0 соответственно.
(?)- и (/?)-алкены являются диастереомерами. Обозначения (?) и (Z) используются и для
других связей, например C=N, N=N.
Ph
ОН
Ph
\
\
/
C = N
Н
[В.88]
Н ОН
(Z)- или сш-оксим бензальдегида (Е)- или анга-оксим бензальдегида
Более старые син, анти-обозначения применимы и сейчас.
Дизамещенные циклические соединения также обозначаются цис,транс-
символикой. В цис-изомере заместители расположены с одной стороны от плос-
плоскости кольца, в m/ншс-изомере — по разные стороны.
Н3С I СН3
н н н
цис-1,2-диметилциклопропан
Н,С
[В.89]
гранс-1,2-диметилциклопропан
Эти соединения также являются диастереомерами (см. разд. В,7.3.2). В них замещенные
С-атомы цикла — хиральные центры с четырьмя различными заместителями, так что каждый
из г<«с,/и^анс-изомеров может существовать в двух оптически активных энантиомерных фор-
формах, которые обычно оставляют рацемат (см. В,7.3.3.1). Если оба заместителя у атомов углеро-
углерода кольца, как в [В.90], одинаковые, то «wc-изомер не хирален и существует только в одной,
оптически неактивной, жезо-форме.
Обозначения цис/транс применяются и в высших циклах, а также для характеристики
сочленения в конденсированных циклах, например
цис-декалин
Н Н
гранс-декалин
[В.90]
В [В.90], как и обычно принято (см. разд. В,7.3.1), заместители, расположенные выше
плоскости кольца, представляют клинообразно, а те, что ниже плоскости - заштрихованной
связью.
7. Стереоизомерия 221
7.3. ХИРАЛЬНОСТЬ И СТЕРЕОИЗОМЕРИЯ
7.3.1. ЭНАНТИОМЕРИЯ
Соединение с одним атомом углерода, который тетраэдрически окружен четырьмя
различными заместителями, например, глицериновый альдегид [В.91], может суще-
существовать в виде двух стереоизомеров, которые представляют собой зеркальные
отображения, не совместимые друг с другом. Такие объекты, которые не совмести-
совместимы со своим зеркальным изображением (как левая рука и правая рука), называют хи-
ральными (от греческого слова, означающего «рука»), а оба оптических стереоизомера—
энантиомерами (ранее их также называли оптическими изомерами или антиподами)
Н он
но н
НОН2С СНО
(+)-глицериновый зеркальная (-)-глицериновый I '
альдегид плоскость альдегид
Центральный атом С при этом называется асимметрическим, а расположе-
расположение заместителей вокруг него — конфигурацией1. Асимметрический С-атом —
стереогенный, или хиральный, центр2.
Оба энантиомера имеют одинаковое строение и различаются между собой
только конфигурацией (топографией). В ахиральном окружении они обладают
одинаковыми физическими и химическими свойствами, например, одинако-
одинаковой энергией, температурой плавления, температурой кипения, раствори-
растворимостью, спектрами и одинаковой реакционной способностью. В хиральном
окружении, напротив, они ведут себя по-разному, например по отношению к
поляризованному свету или хиральным реагентам.
Раствор одного энантиомера вращает плоскость линейно поляризованного
света направо, раствор другого энантиомера — налево, что обозначается как
(+) и (—). Это свойство называется оптической активностью. На основании
различной оптической активности оба энантиомера могут быть количественно
определены, см. разд. А,3.4 «Поляриметрия». В смеси из одинаковых коли-
количеств обоих энантиомеров (+)- и (—)-формы компенсируют вращение и такая
рацемическая смесь {рацемат) оптически неактивна.
Чтобы пространственную конфигурацию хирального соединения с асимметри-
асимметрическим центральным атомом представить на плоскости бумаги, две из четырех свя-
связей «размещают» на плоскости бумаги, как в [В.91], и соединяют с центральным
атомом заместители, лежащие спереди и сзади плоскости, соответственно черной и
штриховой клинообразными линиями. Более длинные цепочки углеродных атомов
рисуют в виде зигзагообразной линии без обозначений атомов С и Н, как в [В.93].
1 Соединения с другими асимметрическими атомами, например, Si, N, Р, также хиральны.
2 Кроме хирального центра хиральность может быть обусловлена также осью хиральнос-
ти, как в атропоизомерах [В.83] и алленах, или плоскостью хиральности, как в циклоалкенах.
R., / А Н,, ,СН2
"с=с=с • I *'с=с;
Н-< V,. / СН/' Н .-
п
Аллен Циклоалкен
222 В Некоторые общие положения
Согласно более старому (по Фишеру) написанию формул проецируют тет-
тетраэдр на плоскость рисунка, при этом два из заместителей изображают гори-
горизонтально, а два остальных заместителя — выше и ниже. Наиболее длинная
углеродная цепь при этом изображается вертикально с наиболее окисленной
группой наверху (см. [В.92]). ОН-группа в (+)-энантиомере находится справа
(лат. dexter) от асимметрического С-атома, а в (-)-энантиомере - слева (laeus),
и такие конфигурации обозначают как D или L соответственно.
сно
н«4-он
СН2ОН
сно
н—|— он
СН2ОН
сно у он
но«4— н = нон2с-4—сно = онс«-4-сн2он
СН2ОН ОН Н
СНО Н ОН [В.92]
но—]—н = нон2с—[—сно = онс—|— сн2он
СН2ОН ОН Н
О-(+)-глицериновый И-)-глицериновый
альдегид альдегид
Следует учесть, что проекционную форму Фишера на плоскости бумаги можно повернуть
только на 180° без изменения конфигурации. При повороте на 90° в плоскости бумаги или на
180° вдоль длины оси молекулы два заместителя, лежащие напротив друг друга, следует поме-
поменять местами, как это видно из стереоформул L-(—)-глицеринового альдегида, ср. [В.92].
Способность соответствующего энантиомера вращать поляризованный луч
в поляриметре вправо (+) или влево (—) не простым образом зависит от его
D- или L-конфигурации, бывает, что и D-соединения могут вращать влево, а
L-соединения - вправо, как например в тетрозах [В.93].
Поскольку DL-система обозначений в случае сложных соединений ведет к
проблемам, Кан, Ингольд и Прелог ввели однозначную систему для описания
конфигурации (КИП-система). Она устанавливает порядок расположения
четырех заместителей у асимметрического центра соответственно их старшин-
старшинству. Старшинство падает в следующем порядке:
— с понижением порядкового номера связанного атома;
— при одном и том же атоме в первой сфере связей > во второй сфере
связей > в третьей сфере связей;
— тройная связь > двойная связь > одинарная связь между атомами.
Этим путем получен следующий ряд старшинства:
I > Вг > С1 > SO3H > SR > F > OR > ОН > NR2 > NHR > NH2 > COOR > COOH >
CHO > CH2OH > CR3 > CHR2 > CH2R > CH3 > H
Если теперь посмотреть на тетраэдр со стороны, противоположной к
«самому младшему» заместителю, т. е. к Н (в случае глицеринового альдегида
[В.91] и [В.92; 3-я и 4-я формулы] Н расположен сзади), то соответствующий
энантиомер обозначается как R (от лат. rectus — правый), когда переход от
7. Стереоизомерия
223
заместителя наивысшего по старшинству к заместителю наименьшему по
старшинству происходит по часовой стрелке, или S (от лат. sinister — левый),
когда этот переход совершается против часовой стрелки. Так, (+)-глицериновый
альдегид — это (Л)-глицериновый альдегид, а (—)-глицериновый альдегид — это
E)-глицериновый альдегид.
7.3.2. ДИАСТЕРЕОМЕРИЯ
Соединения с двумя (в общем случае п) асимметрическими С-атомами выступа-
выступают в четырех B") оптически активных формах, как, например, простейшие саха-
сахара со структурой НОСН2-СНОН-СНОН-СНО (тетрозы) [В.93]. Из них (+)- и
(—)-эритрозы и (+)- и (—)-треозы являются зеркальными изображениями, т. е.
парами энантиомеров. Напротив, ни одна из эритроз не является зеркальным
изображением треоз. Стереоизомеры, которые не являются зеркальными изоб-
изображениями, называют дшстереомерами1. В противоположность энантиомерам,
они различаются по своим физическим и химическим свойствам.
ОН
ОН
СНО
"СНО
он
он
но y сно
он
СН2ОН
Хпн
СН2ОН
4Н
СН2ОН
jH
СН2ОН
1oh
[В.93]
сно
(Я,Я)-(-)-эритроза
сно
(S,S)-(+)-3pnTpo3a
СНО
BS,3R)-(-)-Tpeo3a
СНО
BR,3S)-(+)-Tpeo3a
В [В.93] представлены четыре тетрозы также в виде ньюменовских проекций
их трансоидных конформаций. Их конфигурация обозначена по КИП-системе,
числа дают номера соответствующих С-атомов (начиная с СНО-группы).
По DL-системе, если положить в основу фишеровские проекции, получа-
получаются обозначения, приведенные в [В.94]. Для соотнесения D- или L-конфигу-
рации исходят из положения ОН-группы при асимметрическом С-атоме,
наиболее удаленном от СНО-группы.
сно
Н—j—OH = Н
h—Uoh ~ н—
^ "ОН
СН2ОН
сно
он
он
СН2ОН
)-эритроза
СНО
но—|— н
но—|— н
СН2ОН
1_-(+)-эритроза
СНО
СНО
но-
н-
-н
-он
СН2ОН
D-(—)-треоза
Н-
но-
-ОН
-Н
СН2ОН
[В.94]
L-(+)-Tpeo3a
Руководствуясь конфигурациями тетроз, обозначения эритро/трео используют так-
также для наименования других диастереомеров с двумя соседними асимметрическими
С-атомами, см., например, схему [Г.7.124]. В трансоидных конформациях при
изображении в перспективных проекциях или проекциях Ньюмена (см. с. 217) эритро-
1 В этом плане ^/Z-изомеры олефинов — диастереомеры (ср. разд. В,7.2), но они не хиральны.
224 В Некоторые общие положения
формы заместители соответствующего старшинства расположены в ян/ии-положении, в
трео-форме — в «/«-положении. В полностью заслоненных проекциях Фишера наобо-
наоборот, эти заместители в эритро-форме стоят в сын-, а в /ирео-форме — в анти-положени-
анти-положениях, ср. [В.93] и [В.94].
Соединения с двумя асимметрическими С-атомами представлены только
тремя стереоизомерными формами, как например, винные кислоты
ОН ОН ОН
НООС. А НООС. А, НООС. Д^
^{ СООН Y СООН ^Y СООН
он он он
СООН СООН СООН
нч— он но-
но—^н н-
-н н-
-он н-
-он
-он
СООН СООН СООН
(Я,Д)-(+) или L-(+) (S,S)-(-) или D-(-) (R,S)- или мезо-винная кислота
Два из них энантиомеры и оптически активны, в то время как диастерео-
мерная .мезо-винная кислота оптически неактивна. Она содержит плоскость
симметрии (ее след обозначен штриховой линией в [В.95], плоскость располо-
расположена перпендикулярно плоскости чертежа), так что зеркальные изображения
могут быть совмещены. Оптическая активность обоих асимметрических
С-атомов внутренне компенсируется.
7.3.3. СИНТЕЗ ХИРАЛЬНЫХ СОЕДИНЕНИЙ
Предпосылкой для синтеза хирального соединения из ахирального является
прохиральное исходное соединение. Ахиральная молекула или часть молекулы
называется прохиральной, если при введении в нее нового ахирального замести-
заместителя она становится хиральной. Прохиральными являются молекулы с атомами
С, которые содержат три различных заместителя. Это могут быть тригональные
плоские С-соединения с двумя различными остатками и одной двойной связью
[В.96] или тетраэдрические С-соединения с четырьмя остатками, из которых два
одинаковые [В.97]. Если в эту молекулу вводят еще заместитель путем присоеди-
присоединения по двойной связи (как в [В.96]) или путем замещения одного из двух
одинаковых остатков (как в [В.97]), то возникает асимметрический атом С и
продукты становятся хиральными.
Прохиральное соединение обладает плоскостью симметрии, которая делит
молекулу на две половины, которые являются зеркальными изображениями.
Обе стороны обозначают тогда как энантиотопы. В зависимости от того, с
какой стороны вводится четвертый заместитель, образуется один или другой
энантиомер.
7. Стереоизомерия
225
ОХ [В.96]
R y\ „, старшинство ОХ > R" '> R" > R'
R" R
R»y\ старшинство R'" > R" > R' > R [B.97]
R"' R'"
(З)-энантиомер
R"
(Я)-энантиомер
Если субстрат кроме прохиральности содержит и центр хиральности, то
плоскость, соответственно проведенная через молекулу, делит ее на два так
называемых диастереотопа. При введении нового заместителя с разных
сторон возникают тогда диастереомеры, ср. [В.99].
7.3.3.1. Разделение рацематов
В ахиральном окружении обе энантиотопные стороны прохирального соедине-
соединения идентичны. Присоединение по карбонильной группе [В.96] или замещение
С-атома [В.97] приводят тогда к рацемической смеси (рацемату), так как оба
энантиомера образуются с одинаковой скоростью реакции через переходное
состояние одинаковой энергии и с одинаковой энергией активации. Из рацема-
рацемата можно получить чистые энантиомеры различными способами.
Можно, например, рацемическую смесь обработать хиральным соединени-
соединением, при котором образуются диастереомеры. Они в противоположность энан-
тиомерам имеют различные физические свойства, например растворимость, и
могут тогда быть разделены фракционной кристаллизацией или хроматогра-
фически. Из разделенных диастереомеров затем отщепляют оптически актив-
активное вспомогательное соединение и так получают чистые энантиомеры. Этот
метод особенно просто применим, если разделяемые энантиомеры содержат
кислотные или основные группы, которые с оптически активными основани-
основаниями (например, хинином, бруцином, ос-фенилэтиламином) или кислотами
(например, винной) дают диастереомерные соли.
(R)-B + (S)-ha
(S)-B + (S)-HA
рацемат
(R)-BH®(S)-Ae
(S)-BH®(S)-A®
диастереомеры
[B.98]
Подобный обмен, как при солеобразовании, лежит в основе комплексообразо-
вания энантиомеров с одним хиральным партнером (ср. разделение О,Ь-<х-фенил-
этиламина, разд. Г,7.3.2) и при адсорбции на одном твердом носителе (ср. разд.
А,2.7, хроматографическое разделение рацематов, например, фермента, когда
превращается только один из двух энантиомеров, а другой остается неизмен-
неизменным).
226 В Некоторые общие положения
Наконец, рацемические смеси могут быть быстро и экономно разделены
путем кинетического разделения рацематов, при котором в стереоселективной
реакции с одним хиральным реагентом, например, ферментом, превращается
только один из двух энантиомеров, а другой остается (см. ниже).
7.3.3.2. Стереоселективные синтезы
В хиральном окружении обе стороны или оба остатка прохирального соедине-
соединения неравноценны. В преращениях они реагируют с различной скоростью
([В.96] или [В.97]), и один из энантиомеров образуется предпочтительно (стере-
оселективный или асимметрический синтез).
Хиральность, которая обусловливает стереоразличия у реакционного цент-
центра и необходима для стереоселективного синтеза, обозначается как асиммет-
асимметрическая индукция. Она может быть достигнута несколькими путями.
Если исходное соединение уже содержит хиральный структурный элемент
(субстратная индукция), например, субстрат содержит хиральную группу (как
карбонильное соединение с асимметрическим С-атомом в [В.99]), то реакция
протекает через диастереомерные переходные состояния, которые отличаются
по своей энергии. Тогда один из диастереомерных продуктов образуется пред-
предпочтительно (диастереоселективный синтез).
Эти отношения хорошо исследованы для реакций нуклеофильного присо-
присоединения с хиральными карбонильными соединениями (ср., например, разд.
Г.7.3.1.1)
О 1.Nue Nu ОН НО, .Nu
2. Н® М^ X. М
R X^R +
К*° K'G [В 991
основной продукт побочный продукт Lu--7-'J
К — маленький, М — средней величины, G — большой остаток;
Nu"— гидрид или С-нуклеофил.
Если остатки у асимметрического С-атома различимо большие (К < М< G),
то исходят из того, что в переходном состоянии наибольший остаток располо-
расположен наклонно относительно карбонильной группы и что нуклеофильный ре-
реагент Nu~ вступает с противоположной стороны в углу, который соответствует
углу образующейся связи Nu—С—О в реакционном продукте (модель Фель-
кина — Ана [В. 100]). Из двух возможных конформаций I и II беднее энергией
та, в которой нуклеофил соседствует с маленьким остатком К (II), чем та, в ко-
котором он находится рядом с большим остатком М (I). Следовательно, предпоч-
предпочтительно образуется диастереомер (III), в котором нуклеофил оказывается меж-
между маленьким остатком (К) и средним (М) и ОН-группа - между средним и
наибольшим остатком.
0 к м о он
1 II III
7. Стереоизомерия 227
Об ауксилиарной индукции говорят, если прохиральный субстрат путем прев-
превращения с хиральным вспомогательным соединением (ауксилиар) переводится в
хиральное промежуточное соединение, с которым затем осуществляется стерео-
селективный синтез. Из продукта реакции хиральное вспомогательное соедине-
соединение затем отщепляют (оно может вновь выделено и использовано). Например,
если превратить карбонильные соединения с хиральными гидразинами в хираль-
ные гидразоны, реакция последних предпочтительно приведет к одному
из энантиомеров, ср. разд. Г,7.4.2.1, уравнение [Г. 7.305] (энантиоселективный
синтез).
Реакционная индукция имеется тогда, когда прохиральный субстрат непосре-
непосредственно превращается в нужный хиральныи продукт при реакции с хиральным
реагентом. Стереохимическая информация может быть тогда передана или со
стехиометрическим количеством введенного реагента или предпочтительно с
хиральным катализатором (асимметрический катализ). Так как при реакцион-
реакционной индукции, в противоположность к ауксилиарной индукции, не образуются
асимметрические промежуточные соединения, такие реакции должны проте-
протекать высокоэнантиоселективно. Как пример стехиометрического варианта сле-
следует упомянуть асимметрическое гидроборирование (ср. разд. Г,4.1.8). Синтети-
Синтетически значимыми примерами для асимметрического катализа являются гидри-
гидрирование в присутствии металлокомплексов с хиральными лигандами (см. разд.
Г,4.5.1), асимметрическое эпоксидирование и дигидроксилирование (см.
Г,4.1.6), а также реакции, катализируемые ферментами (см. разд. Г,7.3.1.5).
Если хиральныи субстрат реагирует с хиральным реагентом, то получается
двойное диастереоразличие. Соответственно возникающим тогда различным
влияниям двух хиральных партнеров можно различать два случая. Если индук-
индукции субстрата и реагента дополняют друг друга положительно, то говорят о
равных парах (англ. matched pair). Если оба они действуют по-разному, т. е.
имеют противоположную стереохимическую направленность, то речь идет о
непохожих, неравных парах (mismatched pair).
Поясняется это на примере эпоксидирования по Шарплессу — Кацуки (разд. Г,4.1.6)
одного из аллиловых спиртов, полученных из D-глицеринового альдегида (/-Ви — трет-
бутил, z-Pr — изопроприл)г
frBu00H
Ti@i-PrL
[В. 101]
Без эфира винной кислоты 2,3 : 1
(+)-диэтилтартрат 1 : 2
(-)-диэтилтартрат 90 : 1
В то время как без добавления эфира винной кислоты оба диастереомера образуются в со-
соотношении 2,3:1, при добавлении второго хирального реакционного партнера (здесь один из
двух энантиомерных эфиров винной кислоты, которые участвуют в каталитически влияющем
комплексе) происходит изменение соотношения диастереомерного состава. При добавлении
природного (+)-Я,Л-диэтилового эфира винной кислоты преимущественно образуется другой
диастереомер, однако селективность при этом невелика. Напротив, (-)-51,5-диэтиловый эфир
228 В Некоторые общие положения
винной кислоты вызывает резкое различие и приводит с большим избытком к диастереомеру,
который образуется преимущественно и при отсутствии эфира винной кислоты. Соответ-
Соответственно этому в первом случае имеют место противоположные (неравные) пары, а во втором —
равные пары. Так как хиральность эфира винной кислоты, который в комбинации с алкоголятом
титана и /и/>етя-бутилпероксидом образует стерически затрудненный комплекс, в обоих случаях
преобладает, здесь имеет место (хотя и в незначительной степени) слабый контроль реагентов.
Резко выраженный контроль реагентом стехиометрии или катализа позво-
позволяет осуществлять целенаправленный синтез только желаемого стереоизомера
путем выбора энантиомера, вводимого в реакцию, независимо от элементов
хиральности, имеющихся в субстрате, что предпочтительно.
Контроль реагентом может быть использован и для кинетического разделе-
разделения рацематов, если скорости реакции обоих энантиомеров субстрата сущест-
существенно различаются. С помощью ферментов, а также с применением новых
(современных) хиральных реагентов многочисленные смеси рацематов могут
быть разделены быстро и экономично путем селективного превращения толь-
только одного из энантиомеров.
Степень предпочтительного образования одного энантиомера при энантиоселективном
синтезе обычно указывается в форме процентного содержания энантиомерного избытка (епап-
tiomericexcess, ее):
F - F Е+ — масса энантиомера, образовавшегося в избытке
ее% = + р ¦ 100 [В.102]
?+ + ?¦_ Е- — масса энантиомера, образовавшегося в недостатке
Из равенства [В.26] следует, что стереоселективность тем выше, чем больше различие в
энергии Гиббса (свободной энергии) между переходными состояниями, которые приводят к
энантиомерам, и чем ниже температура.
^± = ^±* [В. 103]
?+ _ ДДС*
|пё:-~лг
Если различие в энергии составляет, например 10 кДж/моль, тогда при 25 "С достигается
соотношение Е+ : ?_ = 56,5 : 1, что соответствует избытку энантиомера 96,5%. При —78 °С эти
значения Е+/Е- = 475 и ее = 99,6%. Стереоселективные синтезы поэтому проводят при
возможно более низких температурах.
8. ПЛАНИРОВАНИЕ СИНТЕЗОВ
Как в лаборатории, так и в промышленности целью органических синтезов яв-
являются материалы определенной структуры со специальными свойствами — это
фармацевтические препараты, растительные защитные средства, душистые ве-
вещества, красители и т. д. Обычно конечные продукты не удается получить в одну
стадию из доступных исходных веществ, а приходится получать их в несколько
стадий, при этом часто возможно использование различных путей последующих
реакций. В случае таких сложных молекул, как многие натуральные вещества и
медикаменты, число возможных реакционных путей и требуемых стадий может
быть очень большим и бывает затруднительно найти оптимальный реакцион-
реакционный путь. Из известных возможностей, естественно, стремятся к тем, в которых
наименьшее число стадий и наибольшие суммарные выходы, приводящие к
цели. Играют также роль экономические и экологические факторы для выбора,
8. Планирование синтезов 229
т. е. цена, токсичность исходных, промежуточных и побочных продуктов, а так-
также возможность вторичного использования нецелевых продуктов и растворите-
растворителей.
8.1. РЕТРОСИНТЕЗЫ
Планировать путь синтеза помогает ретросинтетический анализ {англ. discon-
disconnection approach). Пробуют различные варианты, по которым целевая молеку-
молекула может быть построена из частичных структур путем знакомых и целесооб-
целесообразных реакций. Для этого на бумаге «разлагают» ее путем «разрыва» соответ-
соответствующих связей на отдельные части— синтоны (синтетические эквиваленты).
Этим синтонам затем подбирают реальные реагенты, взаимодействие с кото-
которыми приводит к исходной молекуле. Полученные таким образом предшест-
предшественники целевой молекулы теперь рассматриваются как новые целевые струк-
структуры и они снова могут быть «расщеплены» на части. Эту процедуру повторяют
до тех пор, пока не приходят к простым и доступным или легко синтезируемым
соединениям как к исходным. Целевая молекула таким образом ступенчато
разбивается на промежуточные соединения, из которых она может быть син-
синтезирована.
Отдельные ретросинтетические шаги обычно символизируют стрелкой (->).
Во многих случаях во время синтеза вводятся функциональные группы,
которые не присутствуют в целевой молекуле и которые еще должны быть уда-
удалены. Эту операцию обозначают как превращение функциональных групп
(англ. functional group interconversion).
При ретросинтетическом анализе обычно учитывают, что в большинстве
методов синтеза используются полярные реакции, в которых электрофильный
и нуклеофильный реагент взаимодействуют друг с другом. Поэтому при разло-
разложении молекулы связи разрывают гетеролитически, так чтобы образовались
синтоны с электрофильными электроноакцепторными центрами и нуклео-
фильными донорными центрами. Функциональные группы, присутствующие
в молекуле, благодаря своим полярным эффектам заместителей влияют на то,
какой партнер принимает электронную пару, какой заряд получают части мо-
молекулы.
Может случиться, что при разложении молекулы образуется синтон с неес-
неестественным зарядом, которому никакой реагент с соответствующей поляр-
полярностью реакционного центра не может быть применен. В этом случае необхо-
необходима переполюсовка: акцепторный центр (электрофил) следует перевести
путем подходящей реакции в донорный центр (нуклеофил) или наоборот. Воз-
Возможности такой реакционной инверсии иллюстрированы на примере карбо-
карбонильной функции в разд. Г,7.2.1.6. Простой ретросинтетический анализ ди-
этиламида 3-бензоилпропионовой кислоты приведен на рис. В. 104. Сразу вид-
видно, что имеется несколько возможностей разделить молекулу путем разрыва
одной из связей. Они ведут к различным синтонам, а затем к различным путям
синтеза и реагентам.
Разрыв A) расщепляет связь С—С между фенильным остатком и кетогруп-
пой. Для образовавшегося синтона могут быть использованы бензол и ангид-
230
В Некоторые общие положения
Превращение функциональной группы
Рис. В. 104. Ретросинтетический анализ диэтиламида 3-бензоилпропионовой кислоты.
рид янтарной кислоты как реагенты, которые по реакции Фриделя — Крафтса
дадут 3-бензоилпропионовую кислоту. Путем последующей синтетической
стадии карбоксильная группа должна быть переведена в диэтиламид.
Другой путь разложения B) включает разрыв в ациланион и эфир акрило-
акриловой кислоты. В то время как в эфирной части из-за а,р-ненасыщенной карбо-
карбонильной системы присутствует акцептор Михаэля, второй компонент должен
быть создан из бензальдегида путем изменения заряда. Представленный здесь
металлированный 1,3-дитиан в принципе синтетически доступен, но более
склонен к нуклеофильной атаке карбонильной группы A,2-присоединение
как конкуренция 1,4-присоединению).
При третьем пути распада C) происходит алкилирование енолята ацетофе-
нона этиловым эфиром уксусной кислоты. Здесь происходит сперва перепрото-
нирование, обусловленное более высокой С—Н-кислотностью эфира хлорук-
сусной кислоты. Альдольная реакция, протекающая затем, связана с внутримо-
внутримолекулярным отщеплением от хлорида и приводит вместо желаемого продукта к
глицидному эфиру (реакция Дарзана — Кляйзена, разд. Г,7.2.1.3).
В заключение приведено разложение фенилвинилкетона (разд. Г,7.4.1.3) на
акцептор Михаэля и цианид-ион как нуклеофил.
Несмотря на то, что для этого примера существуют 4 различных варианта
синтеза (наряду с другими, которые здесь не приведены), только первый и чет-
четвертый годятся для построения целевой молекулы.
8. Планирование синтезов
231
При многоступенчатых синтезах по экономическим и экологическим
причинам надо выбирать самый короткий. При этом различают линейные и
конвергентные синтезы. По первому типу целевой продукт синтезируют шаг за
шагом из исходного продукта. При пятистадийном линейном синтезе, каждая
стадия из которых протекает с высокими выходами в 90%, общий выход на
конечной стадии составит 59%. Поэтому при синтезе прежде всего нату-
натуральных веществ придерживаются в основном конвергентной стратегии, при
которой сперва получают два или три частичных продукта, соединение
которых затем путем соответствующей реакции сочетания в финале дает целе-
целевой продукт. Весьма элегантны и экономичны каскады нескольких следующих
друг за другом реакций (так называемые тандем- или домино-реакции), благо-
благодаря чему возможно быстрое построение сложных молекул.
8.2. ЗАЩИТНЫЕ ГРУППЫ
В случае многих синтезов необходима дезактивация присутствующих в молеку-
молекуле функциональных групп, чтобы препятствовать их участию в реакции. Для
Таблица В. 105. Защитные группы
Защищаемая
группа
Окси НО-
Амино H2N-
Защитная группа,
защищенная группа
трет-Алкил- (эфир),
R3CO-
Бензил- (бензиновый
эфир),
PhCH2O-
Триалкилсилил-
(силиловый эфир),
R3SiO-
Ацеталь,
ROCHRO-
mpem-Бутилоксикар-
бонил- (ВОС)
/-BuOCONH-
Бензилоксикарбонил,
PhCH2OCONH-
Фталимид, ср. [2.78],
Бензил-,
(PhCH2JN-
Триалкилсилил-,
(R3SiJN-
Введение путем
превращения
Изобуген, трифенилметил-
хлорид, ср. разд. Г,4.1.3 и
Г.2.6.2
Бензилгалогенид, ср. разд.
В.2.6.2
Триметилсилилхлорид и
т.п., ср. разд. Г,2.7
Карбонильные соединения
(ср. разд. Г,7.1.2), винило-
виниловые эфиры (ср. [Г,9.21])
или алкоксиалкилхлориды
Ди-треот-бутилдикарбонат
и др., ср. разд. Г,7.1.4.2
Бензиловый эфир хлор-
угольной кислоты, ср. разд.
Г,7.1.4.2
Фталимид, ср. разд. Г,2.6.4
Бензилбромид, ср. разд. Г,2.6.4
Триметилсилилхлорид,
ср.разд.Г,2.7
Путь расщепления
Кислотный гидро-
гидролиз
Гидрогенолиз, кис-
кислотами Льюиса
Кислотный гидро-
гидролиз
Кислотный гидро-
гидролиз, кислотами
Льюиса
Трифторуксусная
кислота, л-толуол-
сульфокислота
Гидрогенолиз с
Н2 Pd/C
Гидразинолиз,
ср. [Г.2.80]
Гидрогенолиз
Гидролиз
232
В Некоторые общие положения
Таблица В. 105. Продолжение
Защищаемая
группа
Карбонил,
-СО-
Карбокси-,
-соон
Защитная группа,
защищенная группа
0,0-, S,S-h
0,5-ацетали, ср.
[Г.7.28], [Г.7.32]
Алкил- (алкиловый
эфир),
-COOR
Бензил- (бензиловый
эфир),
-COOCH2Ph
1,3-Оксазолин, ф.
[Г.7.48]
Введение путем
превращения
Эгандиол, этандитиол,
2-меркаптоэтанол, ср.
разд. Г,7.1.2иГ,7.1.3
Переэтерефикация,
ср. разд. Г,7.1.4.1 и Г,2.6.3
Бензилбромид, ср. разд.
Г.2.6.4
2-Аминоалканолы или ази-
ридины, ср. разд. Г,7.1.4.2
Путь расщепления
Кислотный гидро-
гидролиз
Основный гидролиз
Гидрогенолиз
Кислотный гидролиз
этого применяют так называемые защитные группы, которые затем удаляют.
Употребляемые защитные группы, которые в этой книге упоминаются, приве-
приведены в табл. В. 105.
Важные критерии для выбора защитной группы:
• Введение и распад должны осуществляться по возможности в мягких
условиях и с выходами близкими к количественным без изменения
остатка молекулы.
• Реагенты, необходимые для введения защитной группы, должны быть
легко доступны и обладать минимальной токсичностью.
• Защищенная функциональная группа должна быть устойчивой в усло-
условиях дальнейших реакций синтеза.
Наряду с чисто химическими методами для удаления защитных групп при-
применяют и такие методы, как фотохимические, электрохимические или
ферментативные. В пептидных или нуклеотидных синтезах используют преи-
преимущественно полимерно связанные защитные группы.
9. ЛИТЕРАТУРА
Для более полного охвата и обучения по разделам, рассмотренным в этой главе,
могут служить книги по физической органической и физической химии, а
также:
9. Литература 233
Термодинамика органических химических соединений
Barin I. Thermochemical Data of Pure Substances. — VCH Verlagsgesellschaft, Weinheim,
1993.
Benson S. W. Thermichemical Kinetics. - John Wiley & Sons, New York, 1968.
Benson S. W., Cruickshank F. R., Golden D. M., Haugen G. R., O'Neal H. E., Rodgers A. S.,
ShawR., Walsh R., Chem. Rev. 69 A969), 279-324.
Cox J. D., Pilcher G. Thermochemistry of Organic and Organometallic Compounds. -
Academic Press, London, 1970.
Stull D. R., Westrum E. F., Sinke G. С The Chemical Thermodynamics of Organic
Compounds. — John Wiley & Sons, New York, 1969.
Кинетика
Frost A. A., Pearson R. G. Kinetik und Mechanismen homogener chemischer Reaktionen. —
Verlag Chemie, Weinheim, 1964.
Homann К. Н. Reaktionskinetik. - Dr. Dietrich Steinkopff Verlag, Darmstadt, 1975.
Huisgen R., in: Houben-Weyl. Bd. 3/1 A955), S. 99-162.
Laidler K. J. Reaktionskinetik. — Bibliographisches Institut, Mannheim, 1970.
Logan S. R. Grundlagen der chemischen Kinetik. - VCH Verlagsgesellschaft, Weinheim,
1997.
Schwetlick K. Kinetische Methoden zur Untersuchung von Reaktionsmechanismen. —
Deutscher Verlag der Wissenschaften, Berlin, 1971.
Techniques of Chemistry, Hrsg.: A. Weissberger. Bd. 6/1-2. - John Wiley & Sons, New York,
1974.
Эффекты растворителей
Parker A. J, Chem. Rev. 69 A969), 1-32.
Reichardt C, Angew. Chem. 91 A979), 119.
Reichardt С Solvernts and Solvent Effects in Organic Chemistry. - VCH Verlagsgesellschaft,
Weinheim, 1988.
Waddington Т. C, Nicht-waprige Losungsmittel. - Huthig-Verlag, Heidelberg, 1972.
Кислоты и основания
Bell К. Р. The Proton in Chemistry. - Chapman and Hall, London, 1973.
BordwellF. G, Ace. Chem. Res. 21 A988), 456-463.
Chalbna Ju. L, Usp. Khim. 49 A980), 1174.
Дюмаев К. М., Королева Б. А. Усп. хим. 49 A980), 2065-2085.
Ebel H. F. Die Aciditat der CH-Sauren. - Georg Thieme Verlag, Stuttgart, 1969.
Izutzu K. Acid Base Dissociation Constants in Dipolar Aprotic Solvents. — Blackwell
Scientific Publications, Oxford, 1990.
Jensen W. В., Chem. Rev. 78 A978), 1-22.
Jensen W. B. The Lewis Acid-Base Concepts. - John Wiley & Sons, New York, 1980.
Jones J. R. The Ionisation of Carbon Acids. — Academic Press, London, 1973.
Perrin D. D. Dissociation Constants of Organic Bases in Aqueous Solution. - Butterworths,
London, 1965.
Reutov 0. A., Beletskaya I. P., Butin K. P. CH-Acids. - Pergamon Press, New York,
1979.
234 В Некоторые общие положения
Serjeant E. P., Dempsey В. Ionisation Constants of Organic Acids in Aqueous Solution. -
Pergamon Press, New York, 1979.
Уравнение Гаммета
Correlation Analysis in Chemistry. Hrsg.: N. B. Chapman, J. Shorter. — Plenum Press, New
York, London, 1978.
JaffeH. #., Chem. Rev. 53 A953), 191.
Johnson С D. The Hammett Equation. - Cambridge University Press, Cambridge,
1973.
Пальм И. А. Основы количественной теории органических реакций. Пер. с англ. - Л.:
Химия, 1977.
Ritchie С. D., Sager W. К, Progr. Phys. Org. Chem. 2 A964), 323.
Shorter J., Qquart. Rev. 24 A970), 433.
Wells P. R. Chem. Rev. 63 A963), 171.
Теория помех в органических реакциях
Chemical Reactivity and Reaction Paths. Hrsg.: G. Klopman. - John Wiley & Sons, New
York, 1974.
Fleming I. Grenzorbitale und Reaktionen organischer Verbindungen. - Verlag Chemie,
Weinheim, 1979.
Hudson R. F, Angew. Chem. 85 A973), 63-84.
Стереоизомерия
IUPAC. Rules for the Nomenclature of Organic Chemistry. Section E: Stereochemistry. Pure
Appl. Chem. 54 A976), 13-30.
Bahr W., Theobald H. Organische Stereochemie - Begriffe und Definitionen. - Springer-
Verlag, Berlin, Heidelberg, New York, 1973.
ElielE. L, Wilen S. H. Organische Stereochemie. - Wiley-VCH. Weinheim, 1998.
Eliel E. L, Wilen S. H., Doyle M. P., Basic Organic Stereochemistry. - Wiley-VCH,
Weinheim, New York, 2001.
Hauptmann S., Mann G. Stereochemie. - Spektrum Akademischer Verlag, Heidelberg,
Berlin, Oxford, 1996.
Kagan H. B. Organische Stereochemie. — Georg Thieme Verlag, Stuttgart, 1977.
Стереоселективный синтез
Chiral Reagents for Asymmetric Synthesis. Hrsg.: L. H. Paquette. — Wiley-VCH, Weinheim,
2003.
Stereoselective Synthesis. Hrsg.: G. Helmchen, R. V. Hofmann, J. Mulzer, E. Schaumann.
Houben-Weyl, Vol. E 21. - Georg Thieme Verlag, Stuttgart, New York, 1995.
Asymmetric Synthesis. Hrsg.: J. D. Morrison. —Academic Press, New York, 1983—1985.
Ager D. J., East M. B. Asymmetric Synthetic Methodology. - CRC Press, Boca Raton,
1996.
Aitken R. A., Kilenyi S. N. Asymmetric Synthesis. - Blackie Academic & Professional,
1994.
9. Литература 235
Atkinson R. S. Stereoselective Synthesis. - John Wiley & Sons, New York, 1995.
Cerwinka O. Enantioselective Reactions in Oragnic Chemistry. — Harwood, London,
1994.
Gawley R. E., Aube J. Principles of Asymmetric Synthesis. - Pergamon, Oxford,
1996.
Izumi Y., Tai A. Stereodifferentiating Reactions. - John Wiley & Sons, New York,
1995.
ManderL. N. Stereoselektive Synthese. - Wiley-VCH, Weinheim, 1998.
МоррисонДж., Мошер Г. Асимметрические органические реакции: Пер. с англ. — М.:
Мир, 1973.
Nogradi M. Stereoselective Synthesis. — VCH Verlagsgesellschaft, Weinheim, New York,
1995. (Есть русский перевод раннего издания: Ногради М. Стереоселективный
синтез. — М.: Мир, 1989.)
Otto E., Schollikopf К., Schulz B.-G. Stereoselective Synthesis. - Springer-Verlag, Berlin,
Heidelberg, New York, 1993.
Rahman A.-U., Sha Z. Stereoselective Synthesis in Organic Chemistry. — Springer-Verlag,
New York, 1993.
Stephenson G. R. Advanced Asymmetric Synthesis. — Blackie Academic & Professional,
1996.
Winterfeld E. Prinzipien und Methoden der stereoselektiven Synthese. — Vieweg Verlag,
Braunschweig, Wiesbaden, 1988.
Catalytic Asymmetric Synthesis. Hrsg.: I. Ohma. - VCH Publishers, New York,
1993.
Comprehensive Asymmetric Catalysis. Hrsg.: E. N. Jacobsen. A. Pfaltz, H. Yamamoto. —
Springer Verlag, Berlin, Heidelberg, New York, 1999.
Brunner H., Zettlmeyer W. Handbook of Enantioselective Catalysis. - VCH
Verlagsgesellschaft, Weinheim, 1993.
Noyori R. Asymmetric Catalysis in Organic Synthesis. — John Wiley & Sons, New York,
1994.
Планирование синтеза
Willis С. L., Wills M. Syntheseplanung in der Organischen Chemie. - VCH, Weinheim,
1997.
Warren S. Organische Retrosynthese. - B. G. Teubner, Stuttgart, 1997.
Warren S. The Disconnection Approach. — John Wiley & Sons, Chichester,
1982.
Corey E. J., Cheng X.-M.: The Logic of Chemical Synthesis. - J. Wiley & Sons, New York,
1999.
Fuhrhop J., Li G. Organic Synthesis — Concepts and Methods. — Wiley-VCH, Weinheim,
1994.
Larock R. C. Comprehensive Organic Transformations. — VCH. Weinheim, 1989.
Seebach D. Methoden der Reaktivitatsumpolung. - Angew. Chem. 91 A979), 259-278.
Umpoled Synthons. A Survey of Sources and Uses in Synthesis. Hrsg.: T. P. Hase. — J. Wiley
& Sons, New York, 1987.
Tse-Lok Ho: Tandem Organic Reactions. — John Wiley & Sons, New York, 1992.
Tietze J. F., Beifuss U. Sequentielle Tranformationen in der Organischen Chemie - eine
Synthesestrategie mit Zukunft. - Angew. Chem. 105 A993), 137-170.
Tietze L. F. Domino Reactions in Organic Synthesis. - Chem. Rev. 96 A996), 115-136.
236 В Некоторые общие положения
Защитные группы
Greene Т. W., Wuts P. G. M. Protecting Groups in Organic Synthesis. - John Wiley & Sons,
New York, 1999.
Kocienski P. J. Protecting Groups. - Georg Thieme Verlag, Stuttgart, 1994.
Schelhaas M., Waldmann H., Angew. Chem. 108 A996), 2192-2219.
г
ПРЕПАРАТИВНАЯ ЧАСТЬ
О ПОЛЬЗОВАНИИ МЕТОДИКАМИ И ТАБЛИЦАМИ
Приведенные в данной книге методики для синтеза отдельных препаратов в большинстве слу-
случаев являются общими и могут служить руководством для соответствующего метода синтеза.
Область их возможного применения выходит за рамки указанных в таблицах примеров. При
распространении этих методик на другие соединения или типы соединений надо учитывать
химические особенности последних, в первую очередь это касается разделения продуктов ре-
реакций. Хотя приведенные общие методики и передают сущность соответствующих методов,
применение их для конкретного синтеза не всегда дает возможность достичь наилучшего вы-
выхода. Для этого необходимо соблюдать специальные условия, что вообще является особен-
особенностью органической химии.
I Перед началом эксперимента необходимо получить сведения о реагентах (часть
Е) и о возможных опасностях при их применении (часть Ж).
В приложении (часть Г, см. т. 2) представлена табл. П.1, которая включает
принятую в Германии систематизацию опасных свойств веществ с помощью
R-символов опасности и S-символов по мерам безопасности (см. внутреннюю
сторону обложки). В табл. П.1 тем не менее приведены только те вещества, данные
об опасных свойствах которых имеются в Приложении I Правил по систематике
опасных веществ (Anhang I der Richtlinie 67/548/EWG; TRGS 900,905). Указания о
веществах, которые не представлены в табл. П.1, можно получить из каталогов и
книг по безопасным способам получения реагентов и по технике безопасности,
которые указаны в части Ж (т. 2, с. 412).
I Вещества, для которых имеются данные по их опасным свойствам, следует рас-
рассматривать как опасные и обращаться с ними в соответствии с S-индексами 22,
23, 24 и 25.
В методиках эксперимента, приведенных в Препаративной части (часть Г;
см. т. 1 и т. 2), на опасные свойства применяющихся реагентов указывает рядом
стоящий символ Q).
I Указания об опасных свойствах и о технике безопасности являются обязатель-
обязательными элементами рабочей инструкции.
Методику эксперимента рекомендуется оформлять в форме инструкции по
эксплуатации, в которой указываются потенциальная опасность реагентов,
238
Г Препаративная часть
реакции и продуктов, меры предосторожности, средства первой помощи и ука-
указание в целесообразности уничтожения отходов.
Если особо не оговаривается, методики могут применяться для получения
веществ как в макро-, так и в полумикромасштабе.
Под макроколичествами следует понимать количества порядка 0,1-1 моль,
под полумикроколичествами — количества от 1 до 10 ммоль (примерно 0,1-2 г);
микроколичества — от 15 до 150 мг. Сведения о необходимых различиях в техни-
технике работы имеются в части А.
Некоторые методики предназначены для аналитических целей, в связи с чем
они могут отличаться некоторыми особенностями. Так, основное внимание в ме-
методиках для качественного функционального анализа направлено не на получе-
получение высоких выходов, а на возможность более широкого применения данной
методики. Поэтому аналитические методики в книге специально отмечены.
Приведенные температуры плавления и кипения взяты из литературы или оп-
определены экспериментально для определенного образца. В соответствии с указа-
указаниями, сделанными в разд. А,2.3.2.2, студент должен уметь сам определять интер-
интервал температуры кипения. Давление, приводимое для температуры кипения,
должно одновременно служить указанием, надо ли перегонять вещество при ат-
атмосферном давлении, в вакууме водоструйного насоса или в высоком вакууме.
Давление, как правило, указывается в скобках в миллиметрах ртутного столба.
Указание растворителя в скобках за точкой плавления означает, что этот раст-
растворитель пригоден для перекристаллизации данного вещества.
Наряду с методиками во многих местах даны ссылки на литературные источ-
источники, в которых описаны другие возможные пути препаративного получения ве-
веществ. При этом рекомендуются руководства на русском, английском или фран-
французском языках, чтобы дать студентам возможность уже во время подготовки к
практическим занятиям улучшить свою языковую подготовку. Практически
обычно указывается литература, касающаяся препаратов, которые иллюстрируют
приведенную вначале общую методику или какой-либо другой вариант метода
для получения данного класса веществ. В некоторых случаях литературные ссыл-
ссылки относятся к получению исходных веществ, необходимых для приготовления
препаратов, приведенных в таблицах части Г.
Ниже представлены некоторые важные сокращения:
Ас
Ас2О
АсОН
Ви
ВиОН
i-BuOH
t-BuOH
Chlf.
DMF
Et
EtOH
EtjO
Hal
ГМФА
ацетил
ацетангидрид
уксусная кислота
бутил
бутанол
изобутанол
m/je/и-бутанол
хлороформ
диметилформамид (ДМФА)
этил
этанол
диэтиловый эфир
галоген
гексаметилфосфортриамид
Me
МеОН
Ме2СО
Ph
PhH
PhMe
Pr
PrOH
i-PrOH
ТГФ
W
метил
метанол
ацетон
фенил
бензол
толуол
пропил
пропанол
изопропанол
тетрагидрофуран
вода
показатель преломления
D-линии натрия при
20 "С
1. Радикальное замещение
239
[cx]du удельное вращение (для D-линии натрия при 20 °С)
D42o плотность (при 20 "С,
по отношению к НгО при 4 °С)
е диэлектрическая проницаемость или молярный коэффициент поглощения
т. кип. температура кипения
т. пл. температура плавления
1. РАДИКАЛЬНОЕ ЗАМЕЩЕНИЕ
При реакции замещения в субстрате RX заместитель X замещается заместите-
заместителем Y:
R-X + Y-Z
R-Y + X—Z
[Г.1.1]
Такие реакции могут протекать по различным механизмам (ср., например,
разд. Г,2; Г,5; Г,7.1.4). По радикальному (гемолитическому) механизму прохо-
проходит замещение при насыщенном атоме углерода.
Важные реакции радикального замещения приведены в табл. Г. 1.2
Реакции радикального замещения [Г.1.1], в которых в качестве короткожи-
вущих промежуточных частиц (продуктов) выступают «свободные» радикалы
(R- и Y*), протекают по цепному механизму (см. ниже разд. ГД.2). Радикалы —
Таблица Г.1.2. Радикальное замещение
R-H + Y-Y -
Y = F, CI, Вг
R-H + CI-Z -
R-Y + H-Y
R-CI + Н- Z
Z = -NRJ, -SO2CI, -РСЦ, -COCI,
-ОС(СН3)з, -CCI3
R-H
Br-Z
О
Z= -N
R-Br + H-Z
, -ОС(СН3)з, -СС13
R-H + О2 R-O-O-H
R-H + SO2 + Cl2 R-SO2CI + H-CI
2 R-H + 2 SO2 + О2 2 R-SO3H
R-H + NO2-Z -
Z = -OH, -NO2
R-X + H-MR3 -
R-NO2 + H-Z
R-H + X-MR3
X = -Hal, -OSOjR1, -OCS2R'
M = Sn, Si
Галогенирование молекулярным галогеном
Хлорирование N-хлораминами, N-хлор-
сукцинимидом, сульфурилхлоридом, пен-
тахлоридом фосфора, фосгеном, трет-бу-
тилгипохлоритом, тетрахлорметаном
Бромирование N-бромсукцинимидом,
mpem-бутилгипобромитом, бромтрихлорме-
таном
Пероксигенирование
Сульфохлорирование
Сульфоксидирование
Нитрование
Восстановление галогенсодержащих соеди-
соединений, эфиров сульфокислот, эфиров ди-
тиокарбоновых кислот триалкилстаннана-
ми и триалкилсиланами
240
Г Препаративная часть
это молекулы или атомы с одним или несколькими неспаренными электрона-
электронами1, основой которых является валентная ненасыщенность, крайняя неустой-
неустойчивость и высокая реакционная способность.
Для инициирования радикальной реакции замещения прежде всего необходимо
образование из исходных веществ RX или YZ одного из двух радикалов R • или Y •.
1.1. ОБРАЗОВАНИЕ РАДИКАЛОВ И ИХ СТАБИЛЬНОСТЬ
Самым важным способом образования радикала(ов) является гомолиз — «сим-
«симметричное» расщепление гомеополярной связи, сопровождающееся разрывом
связывающей электронной пары.
Y-Z Y- + -Z
[Г.1.3]
С1—CI - CI- + -CI
При таком гомолитическом расщеплении связи всегда должна затрачиваться
некоторая энергия на диссоциацию (табл. Г. 1.4). Эта энергия может быть приоб-
приобретена различными способами, в соответствии с чем имеются разные возмож-
возможные пути образования радикалов.
Таблица Г.1.4. Стандартные молярные энтальпии диссоциации связей (при 25 °С)
Связь
Н-Н
F—F
Cl-Cl
Вг—Вг
I—I
НзС-СН3
H2N—NH2
но-он
СНз-Н
сн3сн2-н
(СН3JСН-Н
C6Hs—H
СН2=СНСН2-Н
С6Н5—СН2-Н
кДж/моль
432
155
239
190
149
370
253
214
435
4П
396
458
371
356
Связь
H-F
H-Cl
Н-Вг
Н—I
НО—Н
ноо-н
(СНз)зСО-Н
(СНзЬСО-ОС(СНз)з
С6Н5СОО-ОСОСбН5
CH3-rN=NfCH3
сн3 сн,
I • 1
NC— C-fN=N-f С—CN
сн3 сн3
CH3Hg-CH3
До"".
кДж/моль
566
428
363
295
499
375
435
157
126
210
131
218
Не следует путать радикалы с остатками R в химических формулах.
1. Радикальное замещение 241
Расщепление связи под действием тепловой энергии (термолиз). Равновесие
диссоциации связей, для расщепления которых необходима небольшая энергия,
уже при низких температурах заметно смещено в сторону продуктов диссоциа-
диссоциации. Так, например, в 0,1 М бензольном растворе 6-дифенилметилен-З-трифе-
нилметилциклогексадиена-1,4 (Дп#°с-с = 46 кДж/моль) при комнатной темпе-
температуре до 2% вещества диссоциировано на трифенилметильные радикалы1
р\ _/=\,н PtK-_Ph
р^ \=ЛС ~ t I1"-1-5]
Ph Ph Kn
Соединения, у которых энтальпия диссоциации составляет от 120 до
170 кДж/моль, как это имеет место у пероксидов и алифатических азосоеди-
нений, уже при невысоких температурах (от 70 до 150 °С) претерпевают гомо-
литический распад. Такие соединения могут быть использованы в качестве
инициаторов радикальных реакций, например для инициирования радикаль-
радикальных цепных реакций (см. ниже).
При температурах выше 800 °С даже устойчивые молекулы углеводородов
претерпевают радикальный распад. При этой температуре большинство реак-
реакций с органическими веществами протекает по радикальному механизму (пиро-
(пиролиз, крекинг-процесс).
Расщепление связи под действием излучения (фотолиз, радиолизJ. Энергия Е
светового кванта, согласно уравнению Планка, равна hv. Отсюда следует, что,
например, ультрафиолетовый свет с длиной волны X = 300 нм эквивалентен
энергии 400 кДж/моль. При сопоставлении данных табл. Г. 1.4 становится по-
понятным, что под действием облучения коротковолновым (ультрафиолетовым)
светом можно расщепить большинство связей.
Подсчитайте энергию желтого (X = 600 нм) и фиолетового (X = 400 нм) света
и подумайте, может ли красный свет (А. = 700 нм) расщепить молекулу хлора!
Фотохимическое действие оказывает только поглощенное излучение. При этом не всегда
требуется, чтобы поглощение осуществлялось реагирующими компонентами; непосредствен-
непосредственно не участвующий в реакции сенсибилизатор также может поглощать свет и переносить по-
полученную энергию на реагирующие вещества (например, сенсибилизаторами могут служить
органические красители, кетоны и др.).
Образование радикалов при окислительно-восстановительных процессах (хими-
(химическая энергия). Многие окислительно-восстановительные процессы связаны с
переходом электронов и образованием радикалов, например:
R-O-Q-H + Fe2© R-O- + еЮН + Fe3© [Г.1.6]
1 Изображенные здесь сопряженные структуры были предложены в 1905 г. Якобсоном;
ранее долгое время димеру трифенилметилрадикала приписывалась формула гексафенилэта-
на. Благодаря более поздним исследованиям сопряженные структуры были подтверждены.
2 Фотолиз — расщепление связи под действием видимого или ультрафиолетового света.
Радиолиз — расщепление связи под действием излучения большой энергии.
242 Г Препаративная часть
(см. об этом в разд. Г, 1.5). Аналогичным процессом является синтез углеводоро-
углеводородов путем электролиза солей карбоновых кислот (метод Кольбе):
-557 R'
[Г. 1.7]
2R- R-R
Расщепление связи под действием механической энергии. Ультразвук, очень
быстрое перемешивание или перемалывание веществ на вибромельнице могут
вести к разрывам связи («механохимия»).
Из табл. Г. 1.4 видно, что энтальпии диссоциации отдельных связей весьма
различны. Даже для одной связи, например С—Н, эта величина сильно зависит
от структуры остальной части молекулы. Вообще энтальпия диссоциации связи
тем ниже, чем беднее энергией (стабильнее) радикалы, которые образуются при
расщеплении связи. Термодинамическая стабильность радикалов зависит от
того, насколько сильно неспаренный электрон может быть делокализован в
остальной части молекулы. Этому содействуют способные к мезомерии замес-
заместители, например фенильные и аллильные группы:
н2с-сн-сн2 = н2с-сн=сн2 ^^ н2с=сн-сн2
Вследствие этого энтальпия диссоциации связи С—Н в бензильном (аллиль-
ном) радикале ниже C56 кДж/моль), чем для таких же связей в других соедине-
соединениях.
По этой же причине особенно мала энтальпия диссоциации такой углерод-
углеродной связи, при расщеплении которой образуются трифенилметильные
радикалы (см. схему [Г. 1.5])!
Пространственные эффекты заместителей также оказывают большое вли-
влияние на энтальпию диссоциации связей. Алкильные радикалы имеют плоское
строение, поэтому при гемолитическом расщеплении связи С—X атом угле-
углерода из тетраэдрической конфигурации переходит в тригональную плоскую.
Находящиеся при этом С-атоме заместители отодвигаются друг от друга, что
уменьшает испытываемые ими пространственные препятствия. Вследствие
этого энтальпия диссоциации связи с увеличением объема заместителей
уменьшается. Подобные пространственные эффекты являются причиной
уменьшения энтальпии диссоциации при переходе от первичных к вторич-
вторичным и третичным С—Н-связям (ср. С—Н-связи в метане, этане, пропане и
изобутане).
1. Радикальное замещение 243
1.2. РЕАКЦИИ И ВРЕМЯ ЖИЗНИ СВОБОДНЫХ РАДИКАЛОВ.
ЦЕПНЫЕ РАДИКАЛЬНЫЕ РЕАКЦИИ
Радикалы могут реагировать:
а) с потерей радикальных свойств
— рекомбинация двух радикалов1, например:
2<^ V- СН2 - / V- СН2-СН2—^ \ [ГЛ.9]
— диспропорционирование двух радикалов, например:
2 НзС-Ь - Н2С=С + Н3С-СН [Г. 1.10]
СН СНз СНз
б) с переносом радикальных свойств
— расщепление или изомеризация радикала, например:
С\л — С")-+ со* [глл1]
о- ^—J
— присоединение радикалов по кратным связям, например:
Вг- + Н2С=СН2 Вг-СН2-СН2 [ГЛ.12]
Реакции этого типа обсуждаются в разд. Г,4.3.
— отщепление атомов или групп под действием радикалов, например:
R-H + о R- + H-CI [ГЛ.13]
Реакции этого вида являются существенной частью радикального замещения
(см. ниже).
Большинство радикалов отличается высокой реакционноспособностью
(разд. Г, 1.3), с подходящим субстратом они легко реагируют по схемам [ГЛ.12] и
[Г. 1.13]. Поэтому радикалы присутствуют, как правило, в очень низких концен-
концентрациях и имеют очень короткое время жизни (менее 10~3 с).
В присутствии подходящих реакционных партнеров, которыми могут быть
также растворители, радикалы быстро реагируют по реакции присоединения
[Г. 1.12] или замещения [Г.1.13]. Меньшее значение имеют собственные реакции
радикалов при их рекомбинации [Г. 1.9] или диспропорционировании [ГЛ. 10].
(Вероятность того, что два радикала столкнутся и рекомбинируются в неради-
нерадикальное соединение, чрезвычайно мала).
1 При рекомбинации двух радикалов освобождается энергия диссоциации вновь образу-
образующейся связи. Многоатомные молекулы в состоянии сохранить эту энергию, в то время как
при рекомбинации атомов энергия должна быть отведена при столкновении с третьим парт-
партнером (иная молекула, стенка сосуда).
244 Г Препаративная часть
Для радикалов с меньшей реакционноспособностью, которые медленно
вступают во взаимодействие с данным субстратом или растворителем, процессы
рекомбинации и диспропорционирования часто служат единственной возмож-
возможностью осуществления реакции. Кроме того, этим процессам благоприятствует
более высокая в данном случае концентрация радикала.
В предельном случае реакционная способность радикала может оказаться
столь низкой, что он будет уже неспособен к полной димеризации. Подобные
радикалы могут в отсутствие кислорода существовать в высоких концентрациях
и иметь длительное время жизни (например, РпзС, схема [Г.1.5]).
Малореакционноспособные радикалы, имеющие время жизни от несколь-
нескольких секунд до нескольких лет, называют долгоживущими (персистентными).
Если время жизни столь велико, а реакционная способность так мала, что ради-
радикалы можно изолировать в качестве индивидуальных веществ и манипулировать
с ними, говорят о стабильных радикалах (например, дифенилпикрилгидразил,
ДФПГ; 2,2,6,6-тетраметилпиперидин-1-оксил, ТЕМПО):
O2N
Ме"Ам^~ме [Г.1.1
Me i Me
O2N
ДФПГ ТЕМПО
На реакцдрнноспособность и время жизни свободных радикалов существен-
существенно влияю<пространственные эффекты заместителей. Большие заместители
(фенил, трет-бутил) по соседству с радикальным электроном затрудняют под-
подход реагента и снижают реакционную способность радикала, как, например, в
случае трифенилметила или ДФПГ. Пространственные факторы могут стабили-
стабилизировать даже термодинамически нестабильные радикалы, например радикалы
ТЕМПО или (трет-ВиJСИ', который имеет в бескислородном растворе при
25 °С среднее время жизни около 1 мин.
Радикальные цепные реакции. Перенос радикальных свойств на другие моле-
молекулы очень часто повторяется в определенной последовательности; так протека-
протекают цепные радикальные реакции. Например, такой цепной реакцией является
радикальное галогенирование органических соединений, при котором по
С—Н-связи происходит замещение водорода галогеном (X — галоген):
Y-Y
Y- +
R- +
и т. д.
2 Y-
R-H
Y-Y -
R- + Н-
R-Y +
-Y
Y-
Реакция зарождения цепи
Реакция переноса цепи
[Г.1
[Г.1
[Г.1
.15а]
.156]
.15в]
Этот цикл повторяется до тех пор, пока цепь не оборвется. Важнейшими
реакциями обрыва цепи являются процессы рекомбинации и диспропорциони-
диспропорционирования переносчиков цепи (R-, X*):
Y- + Y- Y—Y
R* + Y* •¦ R—Y Реакция обрыва цепи \Г \ 15г1
R- + R- R-R
1. Радикальное замещение 245
Обрыв цепи может быть следствием взаимодействия переносчика цепи с мо-
молекулами растворителя или специально добавленными веществами, так называ-
называемыми ингибиторами. Ингибиторами могут быть или сами радикалы (кислород
[Г. 1.42], оксид азота, стабильные радикалы), которые вступают в реакцию с
переносчиками цепи, или соединения (ариламин, фенол, хинон), слишком бед-
бедные энергией, чтобы продолжать цепную реакцию (ср., например, с реакцией
[Г.1.19]).
В реакции зарождения цепи образуются реакционноспособные переносчики
цепи. Для этого пригодны все перечисленные в разд. Г, 1.1 реакции образования
радикалов. Так, например, фотолиз молекулы хлора дает два атома хлора, кото-
которые являются переносчиками цепи при радикальном хлорировании (уравнения
[ГЛ.15]). Часто для начала цепи добавляют инициатор, т. е. соединение, которое
уже при небольшой затрате энергии распадается на радикалы (пероксиды, азо-
соединения; см. табл. Г. 1.4). Возникающие при этом радикалы при дальнейшем
взаимодействии образуют переносчик цепи, например:
(Н3С)зСО-ОС(СН3)з 2(Н3СKСО-
[Г. 1.16]
(Н3СKСО- + R-H - (Н3СKСО-Н + R-
Напишите уравнение цепной реакции, протекающей при действии брома
на толуол с азодиизобутиронитрилом или пероксидом бензоила в качестве
инициатора!
Число реакционных циклов на один возникший радикал называют длиной
цепи. При фотохимическом инициировании число реакционных циклов, при-
приходящихся на один поглощенный квант света, называют квантовым выходом.
1.3. РЕАКЦИОННАЯ СПОСОБНОСТЬ И СЕЛЕКТИВНОСТЬ
ПРИ РЕАКЦИЯХ РАДИКАЛЬНОГО ЗАМЕЩЕНИЯ
В общем случае радикал способен вступать в реакцию замещения тогда, когда с
этим связано выделение энергии, т. е. если реакция протекает экзотермически1.
При подобных цепных реакциях сумма энтальпий всех стадий должна оставать-
оставаться отрицательной, хотя отдельные стадии могут быть эндотермическими.
Молярная стандартная энтальпия реакции Ar//° может быть рассчитана по
уравнению [В.20] из энтальпий диссоциации связей, расщепляющихся и обра-
образующихся во время реакции. ArH° тем меньше (т. е. реакция тем экзотермичнее),
чем слабее расщепляющиеся связи и чем сильнее образующиеся. Так, для пер-
первой стадии переноса цепи [Г. 1.156] реакции хлорирования этана имеем
CI- + Н-СН2СН3 - CI-H + гз
[ГЛ.17]
ДиН?э8= AdHch3ch2-h-AdHh-ci= D11-428)=-17кДж/моль
1 Это утверждение справедливо не для всех химических реакций (см. уравнение [В.21]).
Большой экспериментальный опыт показывает, что оно справедливо лишь для радикальных
реакций.
246 Г Препаративная часть
Атом хлора разрывает в этане стабильную связь С—Н; при этом образуется еще
более стабильная связь Н—С1. Поскольку при хлорировании этана вторая стадия
передачи цепи (уравнение [Г.1.15в]) экзотермична, весь процесс протекает как
цепная реакция, если она была инициирована атомом хлора, который образует-
образуется при гомолитическом расщеплении молекулы хлора при относительно
небольшой затрате энергии.
Напротив, атом иода не в состоянии реагировать с этаном, поэтому прямое
иодирование углеводородов обычными способами не удается. Хотя для расщеп-
расщепления молекулы иода требуется меньшая энергия, чем для расщепления молеку-
молекулы хлора, однако при образовании связи Н—I выделяется всего 295 кДж/моль1,
поэтому реакция атомарного иода с этаном была бы эндотермична:
I- + Н-СН2СН3 1-Н + -СН2СНз
ARH2°98= ДвН°НзСН2_н -Д0Н,°.н = D11-295) = 116 кДж/моль
Поэтому иод действует как ингибитор радикальных процессов, так как, при-
принимая радикальные свойства, он не может передать их субстрату:
R- + I-I R-I + I-
[ГЛ.19]
I- + R-H —#— Н-1 + R-
Хотя с помощью термодинамического подхода можно только установить,
возможна ли радикальная реакция замещения или нет, при этом ничего не гово-
говорится о скорости реакции (уравнение [В.2]). Однако в общем известно, что более
экзотермические реакции проходят быстрее, чем менее экзотермические (см.
рис. В.28). Поэтому с помощью приведенных выше термодинамических рассуж-
рассуждений можно оценивать реакционноспособность радикалов и связей.
Реакционная способность радикала относительно данного субстрата, следо-
следовательно, тем выше, чем больше выигрыш энергии при образовании вновь воз-
возникающей связи, т. е. чем больше энергия диссоциации этой связи.
Напротив, если рассмотреть реакцию определенного радикала с различ-
различными С—Н-связями, то окажется, что взаимодействие протекает тем более
экзотермично, чем ниже энергии диссоциации этих связей. По этой причине
реакционная способность связей С—Н третичного2 углерода выше, чем реак-
реакционная способность «вторичных» и «первичных» С—Н-связей. Особенно
легко подвергаются воздействию связи С—Н в бензильном и аллильном поло-
положениях (например, в пропилене и толуоле). Объясните это, пользуясь данны-
данными табл. Г. 1.4.
Мерой реакционной способности радикала служит константа равновесия
реакции, в которой он атакует данную связь. Для того чтобы определить относи-
1 Очевидно, что способность атаковать определенную связь нельзя непосредственно
отождествлять со стабильностью (легкостью образования) радикала. Ср., например, низкую
энергию диссоциации молекулы фтора с чрезвычайно высокой реакционной способностью
радикала фтора.
2 Иногда встречается сокращение (Г)-, B°)-, C°)-С-Н-связи для первичных, вторичных
и третичных С—Н-связей.
1. Радикальное замещение
247
тельную реакционную способность (ср. разд. В,3.2) разных радикалов, исследу-
исследуют их взаимодействие с одним и тем же субстратом (например, толуолом) и от-
относят полученные константы скоростей реаакций к одному из радикалов. В ре-
результате таких исследований стало возможным расположить радикалы по их
реакционной способности в следующий ряд:
F- > НО- > CI- > СН3- > Br
ROO-
[Г. 1.20]
Для определения относительной реакционной способности (Г)-, B°)- и
C")-связей С—Н оставляют постоянным второй реагент, т. е. проводят реакцию
с одним и тем же радикалом, и относят полученные данные, например, к пер-
первичной связи С—Н, для чего можно использовать даже связи в одной и той же
молекуле. В табл. Г. 1.21 приведены результаты исследования такого рода. Срав-
Сравнимыми являются только значения, стоящие в одной горизонтальной строке
таблицы. Очевидно, что наиболее реакционноспособной при атаке любым из
трех указанных в таблице радикалов выступает третичная связь, труднее подвер-
подвергается атаке вторичная связь и наиболее трудно — первичная связь С—Н.
Из данных табл. Г. 1.21 можно сделать еще один важный вывод, а именно от-
относительные реакционные способности трех типов связей С—Н не остаются
постоянными для всех реакций. Например, при фторировании они отличаются
очень мало, тогда как при бромировании различия достигают нескольких
порядков.
Таблица Г.1.21. Относительная реакционная способность связей С-Н в бутане или
изобутане при атаке радикалами галогенов (газовая фаза, 27 °С)
Радикал
F-
С1-
Вг-а
Первичная связь
С-Н
1
1
1
Вторичная связь
С-Н
1,2
3,9
32
Третичная связь
С-Н
1.4
5,1
1600
' При 127 °С.
Объяснение можно дать с помощью постулата Хэммонда (разд. В,2; с. 196). Ре-
акционноспособный атом фтора взаимодействует с R-H-связью экзотермично и
переходное состояние напоминает исходные вещества (кривая 3на рис. В.28). Реак-
Реакции менее активного брома напротив чаще всего эндотермичны, активированный
комплекс похож на продукты реакции (кривая / на рис. В.28). В переходном состо-
состоянии R • • • Н • • • F связь R—Н разрыхлена лишь незначительно; в R • • • Н • ¦ • Вг она,
напротив, в значительной степени расщеплена. Поэтому различные по силе R—Н-свя-
зи лишь в малой степени влияют на скорость реакции с высокоактивным F-, т. е.
селективность мала. С менее реакционноспособным радикалом брома Вг разные свя-
связи R—Н реагируют с сильно различающимися скоростями (большая селективность).
Часто действует следующее правило: высокая реакционная способность обусловли-
обусловливает низкую селективность и наоборот. В соответствии с этим избирательность
248
Г Препаративная часть
радикальных реакций с повышением температуры падает, так как реакционная
способность радикалов возрастает с температурой. Впрочем, это влияние невелико.
Например, для относительных реакционных способностей связей С—Н (Г: 2°: 3°) в
насыщенных углеводородах при взаимодействии с атомарным хлором в жидкой фа-
фазе при —50 "С найдено соотношение 1:7,2:11,8, тогда как при +50 "С = 1:2,9 :4,5.
В промышленности этот процесс применяется в широких масштабах (пиролиз, крекинг-про-
крекинг-процесс, галогенирование, окисление), так как там часто имеют дело со смесями изомеров.
Приведенные до сих пор рассуждения часто недостаточны, чтобы в полной
мере объяснить ход радикальных реакций. Прежде всего должны учитываться
полярные влияния как на реакционную способность радикала, так и на относи-
относительные реакционные способности подвергаемых воздействию связей С—Н.
В соответствии с положением атомов в периодической системе радикалы об-
обладают различным сродством к электрону. Так, например, атомы галогенов и
кислородсодержащие радикалы (НО-, НОО, RO, ROO) обладают ярко выра-
выраженными электрофильными свойствами. Поэтому они атакуют предпочтитель-
предпочтительно места с более высокой электронной плотностью. По этой причине заместите-
заместители, обладающие +/- и +М-эффектами, повышают реакционную способность
соседних связей С—Н относительно таких радикалов, а группы, проявляющие
—/- и — М-эффекты, понижают ее. Несколько примеров приведено в табл. Г. 1.22.
Таблица Г.1.22. Относительная реакционная способность и изомерный состав при
хлорировании С-Н-связей
Изомерный состав, %
Относительная реакционная способность
31
1
31
1
21
64
3,1
69
3,3
47
5
Q
0,24
0
0
22
—С
сопи*
9
1
3,4
1,6 0,7
С1
а Хлорирование карбонильных соединений может происходить также в присутствии избыт-
избытка галогена по полярному механизму; в таком случае образуются побочные а-замешенные
продукты (разд. Г.7.4.2.2).
чзмо
R-
%
S
—
немо
взмо
2
чзмо
R.
/
ж.
S
\—немо
J
-%-взмо
а б
Рис. Г.1.23. Взаимодействие ВЗМО-ЧЗМО и НСМО-ЧЗМО в ходе радикальных реакций.
а — электрофильный радикал; б — нуклеофильный радикал.
1. Радикальное замещение 249
При расчетах изомерного состава продуктов реакции из относительных реакционных спо-
способностей надо учитывать, кроме того, также число соответствующих связей С-Н. Например,
при хлорировании бутиронитрила имеются три первичные С-Н-связи с реакционной способ-
способностью 1, две 3-С—Н-связи с реакционной способностью 3,3. Отсюда получим изомерный сос-
состав C • 1): B ¦ 3,3) = 31% : 69%. Рассчитайте изомерный состав при хлорировании н-бутана и
изобутана, пользуясь данными табл. ГЛ.21!
Полярные свойства радикалов можно хорошо понять на основе теории воз-
возмущений молекулярных орбиталей (разд. В,6). Граничная орбиталь радикала R-
занята одним электроном, ее можно обозначить «частично занятая молекуляр-
молекулярная орбиталь» (ЧЗМО). Она может взаимодействовать как с НСМО-, так и с
ВЗМО-орбиталями партнера S. В обоих случаях имеется выигрыш в энергии,
как это ясно видно на рис. Г. 1.23. На рис. Г. 1.23, а выигрыш энергии составляет
2АЕ2—АЕ1, поскольку вновь образующаяся орбиталь с более низкой энергией
занята двумя электронами, а более богатая энергией орбиталь — только
одним.
Радикалы с низкорасположенной ЧЗМО, например радикал хлора (ечзмо = —13 эВ), в ко-
котором радикальный электрон находится при электроотрицательном атомном ядре, называют
электрофильными. Подобные радикалы вступают во взаимодействие преимущественно с
ВЗМО участника реакции S (рис. Г. 1.23, а). Если же энергия ЧЗМО высока, как, например, в
/яре/и-бутилрадикале (ечзмо = —6,9 эВ), то более благоприятно взаимодействие с НСМО парт-
партнера S, т. е. этот радикал реагирует предпочтительно как нуклеофильный (рис. Г. 1.23,6). В сог-
согласии с этим радикалы хлора и метильные радикалы отщепляют водород из а- и р-положений
пропионовой кислоты в следующих соотношениях:
,С1 ,СН3.
50( I < M
Н Н Н Н [Г.1.24]
н2с-сн-соон н2с-сн-соон
Ра Ра
а-С—Н-орбитали, как правило, имеют низкую энергию. Карбоксильная группа своим индук-
индукционным эффектом понижает энергию а-С—Н-орбитали сильнее, чем C-С—Н-орбитали.
В согласии с рис. Г. 1.23, а ЧЗМО радикала хлора оказывается ближе по энергии к ВЗМО р-С—Н-свя-
р-С—Н-связи, чем а-С—Н-связи: это значит, что а-С—Н-связь имеет большую реакционную способность.
Для реакции метильных радикалов, напротив, решающее значение имеет ЧЗМО — НСМО-взаи-
модействие. Энергии НСМО С-Н-орбиталей (а*), как правило, расположены высоко. Электро-
ноакцепторная карбоксильная группа уменьшает энергию НСМО а-С—Н-связи сильнее, чем
р-С—Н-связи. Тем самым а-С-Н-орбиталь придвигается ближе к ЧЗМО метильного радикала и
отрыв а-водорода становится предпочтительной реакцией.
1.4. РАДИКАЛЬНОЕ ГАЛОГЕНИРОВАНИЕ
Замена водорода галогеном1 является препаративно важной реакцией радикаль-
радикального замещения, которая протекает как типичная цепная реакция. Отдельные
стадии этой реакции были описаны выше [уравнения (Г. 1.15)].
Замещение водорода С—Н-связи на галоген — это окисление (разд. Г.6.1).
250 Г Препаративная часть
Галогены очень сильно различаются по своей реакционной способности
(разд. Г, 1.3). При действии элементного фтора на большинство органических
соединений происходит взаимодействие со взрывом, ведущее к высокофтори-
рованным соединениям и сопровождающееся частичным разложением молеку-
молекулы (образование углерода, тетрафторуглерода). Поэтому, чтобы получить опре-
определенные фторсодержащие соединения, необходимо идти обходным путем
(разд. Г,2.6.7иГ,8.3.1).
В противоположность фтору иод не способен к радикальной атаке связей
С-Н, приводящей к замещению (разд. Г, 1.3). Здесь часто реакция идет в обрат-
обратном направлении: иодалканы, которые могут быть легко получены, например из
соответствующих спиртов (разд. Г,2.5.1), восстанавливаются иодоводородом до
углеводородов:
RI + HI RH + 12 [Г. 1.25]
Поэтому практическое значение имеют только хлорирование и бромиро-
вание.
1.4.1. ХЛОРИРОВАНИЕ
Хотя хлорирование элементным хлором протекает гладко, селективность этой
реакции мала. Поэтому она имеет препаративное значение главным образом для
хлорирования алкилароматических соединений в боковую цепь, так как реакци-
реакционная способность а-связей С—Н гораздо выше, чем связей С—Н в других поло-
положениях. Кроме того, различие в относительной реакционной способности
а-связей С—Н (например, в толуоле, бензилхлориде, бензилидендихлориде) так
велико, что своевременное прекращение хлорирования позволяет получить
любой из трех возможных продуктов хлорирования.
Бензилидендихлорид и трихлорметилбензол нашли применение благодаря
их способности гидролизоваться до бензальдегида и бензойной кислоты.
При проведении таких реакций хлорирования надо заботиться о том, чтобы
в реакционной смеси отсутствовали катализаторы Фриделя — Крафтса (они
являются кислотами Льюиса; разд. Г,5.1.5 и Г,5.1.7), которые ускоряют ионное
замещение в ядре. По этой причине нельзя проводить, например, хлорирование
в железных сосудах.
Для инициирования реакции хлорирования применяется главным образом
световое излучение с большой энергией. Квантовый выход при фотохлорирова-
фотохлорировании может составить до 40 000. Однако в присутствии ничтожных количеств
кислорода, который действует как ингибитор, квантовый выход в большинстве
случаев не превышает 2000.
(ff\ Общая методика фотохлорирования ароматических соединений в боковую цепь (табл. Г. 1.27)
^^ Из-за трудности дозировки хлора методика подходит для работы только с макроколичества-
макроколичествами веществ.
¦ Внимание*. Защищать глаза при работе с погружной лампой опасного УФ-излуче-
ния, а также соблюдать безопасность соседних рабочих мест! Хлор очень ядовит
и работать следует под хорошо действующей тягой.
1. Радикальное замещение
251
Учтите, что хлор очень ядовит (тяга!) (см. часть
Е), а бензилгалогениды оказывают сильное
раздражающее действие на кожу (см. также
разд. Г,1.4.2). По окончании синтеза все при-
приборы обработать метанольным раствором
КОН. Работать в резиновых перчатках!
Хлорирование лучше всего проводить в трехгор-
лой колбе с погруженной в нее ртутной лампой,
трубкой для ввода газа и эффективным обратным
холодильником (рис. Г. 1.26). Если в распоряжении
нет погружной ртутной лампы, можно освещать
колбу снаружи 500-ваттной фотолампой или вести
хлорирование на прямом солнечном свету. В этом
случае реакция протекает несколько медленнее и
выходы обычно уменьшаются. Хлор берут из балло-
баллона и пропускают для высушивания через промывную склянку с концентрированной серной
кислотой. С обеих сторон к этой склянке присоединяют по одной пустой склянке в качестве
предохранительных сосудов1.
Углеводород в описанном выше приборе доводят до кипения на бане, соблюдая при этом
соответствующие меры предосторожности, и пропускают энергичный ток хлора. Высококи-
пящие углеводороды хлорируют при 180 °С. Из холодильника не должен выходить хлор (наб-
(наблюдать по окраске паров на выходе из холодильника!). Хлорирование продолжают до достиже-
достижения колбой рассчитанной массы или до тех пор, пока энергично кипящая реакционная смесь
не достигнет определенной температуры (табл. Г. 1.27).
Таблица Г.1.27. Фотохлорирование алкиларенов
Рис. Г.1.26. Прибор для
проведения фотореакций.
Продукт реакции
Бензилхлорид
Бензилидендихлорид
Трихлорметилбензол
о-Мет ил бензилхлорид
1-Фенил-1-х лорэтанв
о-Хлорбензилхлорид
о-Хлорбензилиденди-
у ПППМТ1
AJ1U |Л1Д
я-Хлорбензилхлорид
л-Хлорбензилиден-
ТТНУЛППИ П
дпллирид
п-Нитробензилиденди-
хлорид
Исходное
вещество
Толуол
Толуол
Толуол
о-Ксилол
Этилбензол
о-Хлортолуол
о-Хлортолуол
о-Хлортолуол
я-Хлортолуол
гс-Нитротолуол
Конечная
температура",
•с
157
187
175
205
Т. кип., "С
(мм рт. ст.)е
69 A5)
86 A4)
111 B3)
91 A8)
77 A5)
92 A2)
100 A0)
92 A0);
Т. пл. 28
129 B2)
Т. пл. 46
(этанол —
гексан)
1,5390
1,5509
1,5581
1,5427
1,5273
1,5621
1,5633
1.565Г
—
Выход,
%
80
80
90
70
60
85
75
85
85
80
а При внешнем освещении хлорируют до тех пор, пока внутри колбы не будет достигнута ука-
указанная температура. При использовании погружной лампы хлорирование контролируют по уве-
увеличению массы колбы.
6 Физические константы (т. кип., т. пл., nD20) относятся здесь и в следующих таблицах, если нет
других указаний, к продуктам.
в Наряду с этим образуется 15—20% 1-фенил-2-хлорэтана.
г /id25 ДЛЯ переохлажденного расплава.
1 Об этом см. также разд. А, 1.6 и А, 1.10.1.
252 Г Препаративная часть
Выпадающие при охлаждении продукты хлорирования можно отфильтровать и очистить пе-
перекристаллизацией. Жидкие продукты реакции фракционируют перегонкой в вакууме на
20-сантиметровой колонке Вигре, предварительно добавив небольшое количество (на кончике
шпателя) гидрокарбоната натрия. Если из продукта хлорирования надо получить далее спирт,
альдегид или карбоновую кислоту, то вполне достаточно собрать фракцию с интервалом кипе-
кипения в 10 °С. Для получения более чистого вещества полученную основную фракцию ректифици-
ректифицируют повторно, отбирая фракции в более узких пределах. Необходимо подводить баланс
перегонки и для отдельных фракций определять их физические константы (разд. А,2.3.3.2).
Легко и избирательно также хлорируются ароматические альдегиды и их
N-производные (оксимы, гидразоны, азины; разд. Г,7.1), причем образуются
хлорангидриды карбоновых кислот (или их N-производные):
X X
R-c' + С12 R-c" + HCl
Н Ъ [Г. 1.28]
=Х: =0, =N-OH, =N-NR'2 и др.
о-Хаорбензоилхаорид из бензальдегида: Clarke Л. Т., Taylor E. R. Org. Syntheses,
Coll. Vol. 1A956I55.
Кроме элементного хлора для хлорирования применяют также другие реа-
реагенты, например, тетрахлорметан (тетрахлоруглерод) ССЦ, фосген С12С=0,
N-хлорсукцинимид (NCS, [Г. 1.32]), пентахлорид фосфора РС15, трет-бутт-
гипохлорит (СНз)зСОС1 или сульфурилхлорид SO2C12 (ср. табл. Г. 1.2). Они ре-
реагируют по радикальному цепному механизму, как это описано для элемент-
элементного галогена [Г. 1.15], и активируются для начала цепи пероксидом (напри-
(например, [Г. 1.16]) или УФ-светом.
Простым и важным препаративным методом хлорирования углеводородов
является действие сульфурилхлорида в присутсвтии инициатора цепной реакции:
RH + SO2CI2 RQI + SO2 + HCl [Г. 1.29]
Хлорирование сульфурилхлоридом протекает более избирательно, чем хлорирование эле-
элементным хлором; например, получить трихлорметилбензол из толуола с сульфурилхлоридом
уже не удается. Это указывает на то, что действительным переносчиком цепи является не ради-
радикал хлора, а менее активный 8О2С1-радикал:
Инициатор - 2R'-
Зарождение цепи
R'- + SO2CI2 R'CI + SC^CI
[Г. 1.30]
RH + SO2CI R- + HCl + SO2
Рост цепи
R- + SO2CI2 ^ RCI + SO2CI
В случае алкилароматических соединений, содержащих бром в ароматическом ядре, про-
происходит замена брома в ядре и водорода в боковой цепи, в результате чего образуется смесь
продуктов реакции.
Из-за удобного дозирования сульфохлорида эта реакция подходит для рабо-
работы с полумикроколичествами и поэтому часто заменяет хлорирование молеку-
молекулярным хлором.
1. Радикальное замещение
а Ир5 для переохлажденного расплава.
6 Не вполне чистый препарат, содержит примесь 1-фенил-2-хлорэтана.
253
Таблица Г.1.31. Хлорирование углеводородов сульфурилхлоридом
Продукт реакции
Хлорциклогексан
Бензилхлорид
о-Хлорбензилхлорид
п-Хлорбензилхлорид
1 -Фенил-1 -хлорэтан
Бензилидендихлорид
Исходное
вещество
Циклогексан
Толуол
о-Хлортолуол
«-Хлортолуол
Этилбензол
Толуол
Т. кип., "С
(мм рт. ст.)
67 F2)
61 A0)
92 A2)
92 A0)
28 (т. пл.)
77 A5)
86 A4)
V
1,4626
1,5390
1,5621
1.56513
1,5278
1,5503
Выход,
%
60
80
75
70
85б
75
) Общая методика хлорирования углеводородов сульфурилхлоридом (табл. Г Л .31)
Внимание*. Для охраны труда ср. предыдущую методику. Во время реакции обра-
образуются диоксид серы и хлороводород. Тяга! Конические шлифы во время реак-
реакции заедает, поэтому между керном и муфтой надо проложить тефлоновую плен-
пленку. Можно использовать также винтовые зажимы с тефлоновым уплотнением
(рис. А. 1, д) или шлифы из тефлона.
Для получения монохлорпроизводного наиболее подходит молярное отношение углеводо-
углеводорода к сульфурилхлориду 1,2 : 1, для получения бензилидендихлоридов — молярное отноше-
отношение 1 : 2.
К углеводороду и SO2CI2 добавляют 0,002 моль бензоилпероксида или еще лучше азо-бис-
изобутиронитрила (на 1 моль SO2CI2) и нагревают смесь до кипения в круглодонной колбе с
хорошим эффективным обратным холодильником (почему?), снабженным хлоркальциевой труб-
трубкой. Примерно через 1 ч добавляют снова такое же количество инициатора. Реакция считается
законченной, когда не наблюдается больше выделения газов (через 8—10 ч). Дают реакционной
смеси охладиться, промывают водой (см. разд. А,2.5.2.1), сушат сульфатом магния и фракциони-
фракционируют на 20-сантиметровой колонке Вигре. В табл. Г. 1.21 даны выходы продуктов в расчете на
сульфурилхлорид.
Для получения небольших количеств хлоридов арилгидроксимовых кислот,
которые могут применяться в качестве предшественников для нитрилоксидов (см.
[Г.4.91]), подходит хлорирование аральдоксимов N-хлорсукцинимидом (NCS):
о о
NOH )L NOH }L
+ Cl-N ] Ai—( + H-N ] [Г.1.32]
н у а у
О О
I Общая методика хлорирования замещенных бензальдоксимов N-хлорсукцинимидом1 (табл.
Г.1.33)
Внимание*. Бензилдроксимоилхлорид действует разъедающе на кожу и
слизистые. Тяга! Защитные перчатки!
По Kou-Chang Liu, Shelton, B.R., Howe, R.K. J.Org.Chem. 45 A980), 3916.
254
Г Препаративная часть
Таблица Г. 1.33. Получение бензгидроксимоилхлорида хлорированием бензаль-
доксимов N-хлорсукцинимидом
Замещ. бензгидроксимоил-
хлорид (продукт)
З-Хлор-
4-Хлор-
2-Метокси-
4-Трифторметил-
2,4,6-Триметил-
Замещ. бензалвдоксим
(исходное соед.)
З-Хлор-
4-Хлор-
2-Метокси-
4-Трифторметил-
2,4,6-Триметил-
Т. пл., °С
58-61
87-89
105-108
89-91
62-69
Выход сырого
продукта, %
86
89
85
80
80
В трехгорлой колбе на 500 мл , снабженной мешалкой и термометром, растворяют в 250 мл
диметилформамида 0,3 моль замещенного бензальдоксима1, нагревают до 25—30 °С и прибав-
прибавляют при перемешивании от 1/10 до 1/5 части 0,3 моль N-хлорсукцинимида. В течение 10 мин
происходит повышение температуры минимум на 3 °С. Если реакция не начинается, можно
ввести прибл. 20 мл газообразного НС1. Как только реакция начнется, температуру удержива-
удерживают не выше 35 °С и прибавляют остаток N-хлорсукцинимида, время от времени охлаждая ре-
реакционную смесь смесью лед/соль. После охлаждения определяют окончание реакции по сла-
слабой или отрицательной реакции капли раствора с влажной иодкрахмальной бумажкой. Затем
прибавляют 4-кратный объем ледяной воды и дважды встряхивают с 250 мл эфира. Эфирный
экстракт трижды промывают водой, сушат Na2SO4 и эфир упаривают при 30—40 °С в вакууме.
Образующийся продукт может быть использован после контроля температуры плавления для
нитрилоксидных реакций.
С необыкновенно высокой селективностью хлорирование можно провести
при помощи N-хлораминов и серной кислоты в присутствии солей металлов
или при освещении:
RH +
RCI + R-2NH
[Г. 1.34]
Реакция происходит по цепному механизму. Переносчиками цепи являются
катион-радикалы аминов, образующиеся в результате переноса электронов:
j©
©
RbNHCI
©¦
Fe2©+ RjNHCI Fe3©+ R2NH + CIG
[Г.1.35]
©¦
R2NH + RH
©
R- + R2NHCI
©
R2NH2 + R •
©•
RCI + R2NH
Алкилгалогениды, спирты, жирные кислоты и их эфиры при этом хлориру-
хлорируются преимущественно по положению (ю—1):
сн3—сн2—сн2—сн2-сн2-сн2-он
СН3-СН-СН2-СН2-СН2-СН2-ОН [Г. 1.36]
С1
Получение см. разд. Г.7.1.1.
1. Радикальное замещение
255
Таблица Г.1.37. Технически наиболее важные хлорпроизводные углеводородов
Продукт хлорирования
Метилхлорид
Метилендихлорид
Хлороформ
Тетрахлоруглерод
Этилхлорид
Монохлорпентаны
(амилхлориды)
Полихлор циклопентаны
Высшие монохлорпара-
фины
Аллилхлорид
Бензилхлорид
Бензилидендихлорид
Бензотрихлорид
Применение11
Хладагент
—*• Силиконы
—>¦ МетилцеллюлоЗа
Растворитель и экстрагент масел, жиров, синте-
синтетических материалов (ацетилцеллюлозы, поливи-
нилхлорида) и лаков
Растворитель жиров, масел, смол, пенициллина
и др.
—>- CHF2CI —> C2F4 —> политетрафторэти-
политетрафторэтилен (тефлон)
Растворитель жиров, масел, смол, лаков.
Средство для чистки текстильных материалов и
металлов
—>• Этилцеллюлоза
Хладагент
Средство для наркоза и анестезии
Растворитель и экстрагент
—>¦ Амиловые спирты —»¦ амиловые эфиры (рас-
(растворители и пластификаторы)
—> Гексахлорциклопентадиен —>- (средства для
борьбы с вредителями сельскогохозяйства)
—>¦ Алкилбензолсульфонаты (моющие средства)
—> Алкилнафталины (присадки к смазочным
маслам)
—> Аллиловый спирт—
аллиловый эфир
(пластификатор)-»полимеры
— акролеин
—> Эпихлоргидрин рч. глицерин
Aхлор-2,3-эпокси-Ч
пропан) Ц. эпоксидные смолы
—<¦ Аллиламин
—> Бензилцеллюлоза
—»• Бензиловый спирт
—>¦ Бензилцианид и др.
—> Бензальдегид
—>• Бензоилхлорид
—>¦ Трифторметилбензол (средство для защиты
растений)-> лекарства, краски
а Стрелка показывает продукт, для получения которого может использоваться данное хлорпро-
изводное.
В промышленности хлорирование в больших объемах метана, этана, пентана, высших
парафинов, пропилена и толуола производится элементным хлором. Применение получа-
получающихся при этом продуктов см. табл. ГЛ.37.
256 Г Препаративная часть
1.4.2. БРОМИРОВАНИЕ
Бромирование элементным бромом в лабораторных целях часто удобнее, чем
хлорирование, поскольку дозировка галогенирующего средства осуществляется
легче и процесс обладает большей избирательностью. Однако для технического
использования бром слишком дорог.
По тем же причинам, о которых шла речь при хлорировании, бромирование
применяется главным образом для препаративного получения бензилбромида и
бензилидендибромида. Алкиларены, имеющие заместители в ядре, бромируют-
ся довольно гладко, как и незамещенные производные. Скорость вступления
второго атома брома в боковую цепь (скорость образования бензилидендибро-
бензилидендибромида) гораздо ниже, чем скорость образования соответствующего бензилброми-
бензилбромида. Бензотрибромид уже не образуется.
Кинетическая длина цепи радикального бромирования мала, так как цеп-
цепная реакция лишь слабоэкзотермична. Так, квантовый выход при бромирова-
нии циклогексана при комнатной температуре равен ~2. Для инициирования
пригоден видимый свет.
^р) Общая методика фотобромирования алкиларенов в боковую цепь (табл. Г. 1.38)
Внимание] Бензилбромид и аналогичные бромпроизводные алкиларенов в боль-
большинстве случаев вызывают сильное слезотечение и раздражающе действуют на
кожу. Поэтому работать с ними следует только под тягой, а при экстрагировании
и других подобных видах работы следует надевать резиновые перчатки и защит-
защитные очки (разд. Г, 1.4, фотохлорирование). При ожогах сразу промойте кожу спир-
спиртом, но не водой! Пока с пораженных мест полностью не удалены остатки вещест-
вещества, нельзя применять никакую мазь, так как она только способствует впитыванию
вещества в кожу. При попадании в глаза их надо промыть слабо подщелоченной
водой (разбавленным раствором гидрокарбоната натрия). Меры предосторож-
предосторожности и другие особенности при работе с бромом см. в части Е.
Растворяют 0,2 моль алкиларена в пятикратном количестве сухого тетрахлоруглерода (см.
часть Е) и помещают в двугорлую колбу с обратным холодильником и хорошо укрепленной ка-
капельной воронкой1. Кончик капельной воронки надо погрузить в жидкость, чтобы уменьшить
потери брома. Колбу нагревают до кипения и добавляют по каплям 0,205 моль брома (предва-
(предварительно высушенного встряхиванием с конц. серной кислотой). Во время прибавления бро-
брома колбу освещают 500-ваттной фотолампой. Скорость прибавления брома надо регулировать
так, чтобы скапывающий из обратного холодильника тетрахлоруглерод все время был почти
бесцветным. Для получения монобромсоединений требуется от 30 мин до 2 ч, для получения
дибромсоединений — от 2 до 10 ч.
[Выделяющийся бромоводород отводят через обратный холодильник в наполненную до
половины колбу Эрленмейера, которая соединяется с холодильником с помощью керна с на-
надетым на него резиновым (лучше поливинилхлоридным) шлангом. Газоотводная трубка не
должна быть погружена в воду, а должна оканчиваться на расстоянии примерно 1 см от пове-
поверхности воды (почему?). Образующуюся разбавленную бромоводородную кислоту очищают,
перегоняя на короткой колонке; собирают азеотропно кипящую при 126 °С G60 мм рт. ст.)
48%-ную бромоводородную кислоту (последняя используется для реакций этерификации и
расщепления простых эфиров, см. разд. Г,2.5.2)].
По окончании реакции освещение прекращают. Если должен получиться твердый про-
продукт, то горячий раствор сразу выливают в колбу Эрленмейера (Осторожно1. Тяга, резиновые
1 Плотность брома 3,14 г/см3! Капельную воронку и колбу надо монтировать на одном
штативе.
1. Радикальное замещение
257
Таблица Г.1.38. Фотобромирование алкиларенов
Продукт реакции
Бензилбромид
о-Метилбензилбромид
о-Хлор бензилбромид
JH-Хлор бензилбромид
л-Хлор бензилбромид
о-Бромбензилбромид
л-Бромбензилбромид
п-Бромбензилбромид
я-Нитробензилбромид
Бензилидендибромид
л-Хлорбензилидендибро-
мид
ж-Ацетоксибензилиденди-
оромид
л-Нитробензилиденди-
?\ V\ fl \Я 1Ж П
иромид
2,4-Дихлорбензилиден-
дибромид
1,2-Бис(дибромметил)-
бензол
1,3-Бис(дибром метил)-
бензол
1,4-Бис(дибромметил)-
бензол
Исходное
вещество
Толуол
о-Ксилол
о-Хлортолуол
л(-Хлортолуол
п-Хлортолуол
о-Бромтолуол
ж-Бромтолуол
л-Бромтолуол
л-Нитротолуол
Толуол
л-Хлортолуол
л-Крезилацетат
л-Нитротолуол
2,4-Дихлорто-
луол
о-Ксилол
ж-Ксилол
/г-Ксилол
Т. ПА., °С
21
17,5
50 (этанол или
петэ эсЬио^
11 Ч* а yj ¦ w ЯГ Г f
31 (этанол или
лигроин)
41 (этанол)
61 (этанол)
99 (этанол)
78 (этанол)
116 (хлоро-
(хлороформ)
107 (хлоро-
(хлороформ)
170 (хлоро-
(хлороформ)
Т. кип., °С
(мм рт. ст.)
78 A5)
104 A4)
104 A2)
109 A0)
124 B0)
130 A2)
126 A2)
120 A5)
145 A2)
167 A1)
90 @,8)
Выход,
%
70
80
80
60
70
80
75
65
70
80
50
70
75
65
50
50
80
перчатки, защитные очки!), дают веществу закристаллизоваться (если нужно, ставят в холо-
холодильный шкаф) и очищают перекристаллизацией.
Для твердых продуктов, которые не кристаллизуются из реакционной смеси или кристал-
кристаллизуются в незначительных количествах, надо дать смеси лучше охладиться, быстро промыть
ледяной водой, затем охлажденным на льду водным раствором гидрокарбоната натрия, еще
раз ледяной водой, высушить сульфатом магния и упарить тетрахлоруглерод на водяной бане
в слабом вакууме; остаток перекристаллизовывают или перегоняют в вакууме, используя наг-
нагревательную баню (перед перегонкой добавляют небольшое количество — на кончике шпате-
шпателя — гидрокарбоната натрия).
Для получения оптимальных выходов, применяют чистые исходные вещества и перераба-
перерабатывают маточные растворы. Для этого их упаривают в вакууме на водяной бане, а остаток
перекристаллизовывают.
Бензилбромиды при температурах выше 150 "С склонны к разложению. При
стоянии они окрашиваются в красный цвет, и лучше всего их сразу использовать
для дальнейшей работы. Они малоустойчивы к гидролизу.
Радикальным бромированием в промышленности получают большие количества метил-
бромида. Его применяют для обеззараживания почвы. Получают также бромированные выс-
высшие углеводороды, используемые в огнетушителях для электрических установок и автомашин.
Вместо элементного брома для радикального бромирования можно также при-
применять другие бромирующие соединения, например бромтрихлорметан ВгССЬ,
/я/?ет-бутилгипобромит ВгОС(СН3)з или N-бромсукцинимид, ср. табл. Г. 1.2.
Широко применяемым бромирующим агентом является N-бромсукцини-
N-бромсукцинимид. Его исключительное значение состоит в том, что он способен к замести-
258 Г Препаративная часть
\ /
с=с
/ Ън
/ ¦
0
к
+ Br-N
Y
i/
о
1 \
/
—- р=с +
/
СН-Вг
/
О
\\
>
H-N
>
И
о
тельному бромированию олефинов в аллильное положение, причем двойная
связь сохраняется:
[Г. 1.39]
При этом речь идет о радикальной цепной реакции, в которой бромирующим агентом яв-
является молекулярный бром, образующийся в незначительной концентрации из N-бромсукци-
нимида.
Подобно аллильным С—Н-связям при бромировании N-бромсукциними-
дом ведут себя и С-Н-связи, находящиеся по соседству с ароматическим
ядром. Таким образом, при бромировании N-бромсукцинимидом можно полу-
получить ос-бромалкиларены. В качестве реакционной среды используют тетрахлор-
углерод, в котором N-бромсукцинимид нерастворим. Растворы реагента в
полярных растворителях приводят к другим реакциям, например происходит
присоединение Вг и замещение в ядре, что характерно для полярных примесей
(солей, кислот).
«Ту) Общая методика бромирования N-бромсукцинимидом в аллильное положение (табл. Г. 1.40)
I Внимание'. Бензилбромид и подобные соединения обладают слезоточивым
действием и раздражают кожу (см. выше).
Растворяют 0,1 моль исходного вещества в 100 мл тетрахлоруглерода, высушенного над
фосфорным ангидридом (см. часть Е), добавляют 0,1 моль высушенного неперекристаллизо-
ванного N-бромсукцинимида и затем 0,2 г азодиизобутиронитрила. Эту смесь осторожно наг-
нагревают в круглодонной колбе с обратным холодильником до тех пор, пока не начнется реак-
реакция, что можно обнаружить по выделению тепла (сильное вскипание!). В случае необходимос-
необходимости надо немного охладить колбу, следя, однако, за тем, чтобы реакция не прекратилась.
Конец реакции можно заметить по тому, что N-бромсукцинимид, имеющий большую
плотность, растворяется и переходит в сукцинимид, который всплывает на поверхность. Для
полноты реакции нагревание до кипения продолжают еще 10 мин. Реакция с олефинами за-
заканчивается примерно за 1 ч; алкиларены требуют большего времени. После охлаждения отса-
отсасывают сукцинимид1, промывают его небольшим количеством тетрахлоруглерода, из объеди-
объединенных фильтратов отгоняют растворитель в слабом вакууме на водяной бане. Остаток, если
ожидают твердый продукт, оставляют стоять в холодильном шкафу или в охладительной
смеси, образовавшиеся кристаллы отсасывают и перекристаллизовывают. Жидкие продукты
очищают, перегоняя в вакууме, для нагревания используют баню.
Методика пригодна для работы с полумикроколичествами.
1.5. ПЕРОКСИГЕНИРОВАНИЕ
Молекула кислорода представляет собой бирадикал О—О. Поэтому она может
реагировать с некоторыми органическими соединениями по радикальному
механизму, причем сначала образуются пероксиды:
R-H + О2 R-O-O-H [Г. 1.41]
Сукцинимид собирают и снова используют для получения N-бромсукцинимида (см. часть Е).
1. Радикальное замещение
259
Таблица Г.1.40. Бромирование N-бромсукцинимидом
Продукт реакции
З-Бромциклогексен
I -Бромметилнафта лин
2-Бромметилнафталин
о-Х лорбензил бромид
Исходное вещество
Циклогексена
I -Метилнафталин
2-Метил нафталин
о-Хлортолуол
Физические константы
Т. кип. 75 °С
A5 мм рт. ст.):
По20 1,5285
Т. кип. 175 °С
A0 мм рт. ст.);
Т. пл. 53 °С (этанол)
Т. кип. 1504-170 °С
A6 мм рт. ст.);
Т. пл. 56 °С (этанол)
Т. кип. 104 °С
A2 мм рт. ст.)
Выход, %
40
60
60
80
а Кипятят 1 ч над фосфорным ангидридом и перегоняют.
Такие реакции часто медленно протекают уже в мягких условиях, например
при комнатной температуре. Их называют реакциями аутоокисления. Речь идет
о цепных реакциях со следующими стадиями:
R-H + -О-О- R- + НОО-
R-O-O-
R-O-O-H + R'
зарождение цепи
рост цепи
[Г. 1.42]
Обрыв цепи происходит преимущественно в результате взаимодействия
переносчиков цепи ROO друг с другом.
Окисление ускоряется в присутствии генераторов радикалов (пероксидов,
азосоединений), освещения и следов ионов тяжелых металлов. Поскольку пе-
роксиды образуются в процессе реакции, то эта реакция протекает аутокатали-
тически. Каталитическое действие ионов тяжелых металлов основывается на
образовании радикалов из пероксидов, например (см. также [Г.1.6]):
ROOH + М2® ROO- + Н© + М© [Г. 1.43]
ROOH + М© RO- + ОН© + М2© [ГЛ.44]
RO- + H-R ROH + R- [Г. 1.45]
Радикал пероксида малоактивен (Ad#°hoo-h = 375 кДж/моль) и поэтому до-
довольно селективен. Он атакует преимущественно связи С—Н, имеющие высо-
высокую реакционную способность (расположенные по соседству с ароматическим
ядром, в алл ильном положении, третичные, а также связи С—Н, находящиеся по
соседству с кислородом, например, в альдегидах, простых эфирах).
Промышленное значение имеет окисление изобутана до m/je/и-бутилгидроперксида и
изопропилбензола (кумола) до а,а-диметилбензилгидропероксида. Напишите уравнение этих
реакций по стадиям!
Гидропероксид mpe/n-бутила служит окислителем при получении пропиленоксида из про-
пропилена (ср. разд. Г,4.1.6). При кислотной обработке гидропероксида кумола образуются фенол
и ацетон (ср. Г,9.1.3). Кроме того, оба пероксида применяются в качестве инициаторов реак-
реакции полимеризации и как реагенты для сшивания полимеров.
260
Г Препаративная часть
При температурах выше 100 °С в присутствии пероксидов и солей тяжелых металлов раз-
разрываются также вторичные С-Н-связи. На этом основано технически важное окисление
парафинов (разд. Г,6.5).
Также затвердение, называемое высыханием, некоторых сльноненасыщенных масел в
присутствии солей тяжелых металлов («сиккативов») — процесс аутоокисления, который на-
начинается прежде всего с реакционноспособного аллильного положения. Подобные реакции
протекают при некоторых нежелательных процессах, например при прогоркании жиров,
масел, при старении каучука и других полимеров.
Имеет значение аутоокисление альдегидов, которое ведет сначала, согласно описанной
выше цепной реакции, к образованию пероксикислоты, которая вступает затем в катализиру-
катализируемую кислотой реакцию с поляризованной карбонильной группой следующей молекулы
альдегида, образуя карбоновую кислоту:
Р
.0
R-C
О2
R-C
R-CHO(H®)
ООН
но.
R'
Н(О
С) С
[Г.1.46]
О
.;C-R
2 R-C
\* I—/
о-о
он
Эта реакция используется в промышленности для получения уксусной кислоты из ацеталь-
дегида. Кроме того, она часто оказывается нежелательным процессом при хранении альдегидов,
особенно в присутствии следов солей тяжелых металлов и на свету. Ароматические амино- и
гидроксисоединения (например, гидрохинон) ингибируют цепные реакции (разд. Г, 1.2) и
поэтому добавляются в качестве антиоксидантов.
Большинство пероксидных соединений богато энергией и поэтому имеет
склонность разлагаться со взрывом. Особенно опасны пероксиды простых
эфиров, которые на воздухе и на свету легко образуются прежде всего из диэ-
тилового эфира, диизопропилового эфира, тетрагидрофурана и диоксана'.
Образующиеся пероксиды менее летучи, чем исходные простые эфиры, поэто-
поэтому при отгонке растворителей накапливаются в перегонной колбе.
Поэтому эфир перед использованием всегда проверяют на содержание пе-
пероксидов; для этого его встряхивают с водным подкисленным серной кислотой
раствором сульфата титанаA\/) или уксуснокислым раствором иодида калия.
В присутствии пероксидов появляется желтое окрашивание.
Как всякие соединения кислого характера, гидропероксиды образуют со щело-
щелочами нерастворимые в эфире соли. Поэтому указанные растворители всегда
хранят над гидроксидом калия в темных склянках.
^П) Общая методика получения гидропероксидов из углеводородов (табл. Г. 1.47)
Очистка углеводородов. Окисляемые углеводороды не должны содержать олефинов. Угле-
Углеводороды встряхивают с концентрированной серной кислотой: приблизительно на 1 объем уг-
углеводорода берут 0,1 объема кислоты. (Осторожно! Происходит небольшое разогревание!).
Эту процедуру повторяют до тех пор, пока серная кислота не перестанет окрашиваться в
коричневый или желтый цвет. Затем два раза промывают водой, сушат над твердым гидрокси-
гидроксидом калия и перегоняют над натрием.
Проведение окисления. В колбу с эффективным обратным холодильником, трубкой для вво-
ввода газа, соединенной с предохранительной склянкой (разд. А, 1.6), помещают 0,2 моль очи-
очищенного углеводорода и 0,1 г азодиизобутиронитрила. Нагревают колбу на водяной или гли-
глицериновой бане при указанной температуре и в течение 8—10 ч через углеводород медленно
пропускают ток кислорода.
1 К образованию пероксидов склонны также ненасыщенные углеводороды, кетоны,
тетралин.
1. Радикальное замещение
261
Таблица Г.1.47. Гидропероксиды из углеводородов
Продукт реакции
а,а-Диметилбензилгидроперок-
сид (кумилгидропероксид)
а-Этил-а-метилбензилгидро-
1,2,3,4-Тетрагидронафтил-1 -гид-
ропероксид (тетралингидропе-
Декалин-9-гидропероксид
1 -Метилциклогексилгидропер-
оксид
Исходное вещество
Кумол
вгор-Бутилбензол
1,2,3,4-Тетрагидро-
нафталин (тетралин)
Декалин
Метилциклогексан
Температура
реакции,
•с
80
120
80
80
80
Содержание
гидропероксида
в растворе,
%
20
12а
20
20
3,5
20% через 20 ч.
Определение содержания гидропероксида. В колбе Эрленмейера на 200 мл, снабженной
пришлифованной пробкой, точно взвешивают 0,2—0,5 г раствора, отобранного из реакцион-
реакционного сосуда. Добавляют 1 г иодида калия, 10 мл уксусного ангидрида и несколько раз встряхи-
встряхивают до тех пор, пока весь иодид не растворится. Через 10 мин реакционную смесь разлагают,
добавляя 50 мл воды, далее энергично встряхивают в течение 30 с; выделившийся иод титруют
0,1 н. раствором тиосульфата натрия в присутствии крахмала в качестве индикатора.
Содержание Объем Na2S2O3 (мл) • Молекулярная масса пероксида
гидропероксида =
в растворе (%) Навеска (г) • 200
Определение гидропероксида повторяют через каждые 2 ч.
1.6. ДРУГИЕ РЕАКЦИИ РАДИКАЛЬНОГО ЗАМЕЩЕНИЯ
Совместное действие хлора и диоксида серы (так называемое сульфохлорирование) на высшие
парафины (с длиной углеродной цепи C^-Cis) находит широкое техническое применение.
Эта реакция также является цепной, протекающей с участием радикалов:
[Г. 1.48]
Cl2 ^^ 2 CI ¦
R-H + О ¦ R- + HCI
R- + S02 R-S;
Образующиеся при омылении алкилсульфонилхлоридов соли сульфоновых кислот
являются хорошими моющими средствами:
RSO2CI + 2 NaOH RSO3Na + NaCI + H2O
Сульфонилхлориды находят применение также в качестве дубильных средств.
[Г. 1.49]
262 Г Препаративная часть
В результате цепной реакции окисления парафинов кислородом в присутствии диоксида
серы (сульфоокисление) сначала образуются пероксосульфоновые кислоты:
R-H + SO2 + О2 > R-SO2-O-O-H [Г.1.50]
которые в последующих реакциях с водой и диоксидом серы переходят в алкилсульфоновые
кислоты и серную кислоту.
При подходящих условиях удается также провести нитрование алифатических углеводоро-
углеводородов1. В промышленности проводят нитрование низких (газообразных) углеводородов парами
азотной кислоты при -450 °С; для высших углеводородов этот метод непригоден, поскольку в
этом случае происходит глубокий крекинг (разложение) молекулы. Высшие углеводороды
нитруют, например при температурах 170— 180 °С, в жидкой фазе (в случае необходимости под
давлением) азотной кислотой или диоксидом азота. Широкое распространение в промышлен-
промышленности получило нитрование пропана, продуктами которого являются нитрометан, нитроэтан
и нитропропан — важные растворители и промежуточные продукты. Нитроциклогексан
используют в качестве исходного продукта для получения s-капролактама; для этого получае-
получаемый нитрованием циклогексана нитроциклогексан каталитически гидрируют до циклогекса-
ноноксима, из которого затем получают е-капролактам (разд. Г,7.1.4.2 и Г,9.1.2.4).
Можно проводить радикальное нитрозирование диоксидом азота алканов и алкиларенов
при повышенной температуре или под действием излучения высокой энергии. Нитрозирова-
Нитрозирование удается также провести действием смеси N0 с хлором или действием нитрозилхлорида
NOC1 при освещении УФ-светом. Как побочный продукт образуется хлороводород. В присут-
присутствии хлороводорода первоначально образующиеся первичные и вторичные нитрозосоедине-
ния перегруппировываются в оксимы. Так в промышленности получают циклогексаноноксим
(полупродукт в производстве е-капролактама; см. выше) фотонитрозированием циклогексана.
Напишите соответствующие уравнения реакций!
Рассмотренное ранее радикальное замещение относилось исключительно к за-
замене атома водорода различными функциональными группами. Можно также нао-
наоборот замещать водородом функциональные группы в органических соединениях.
Такое «радикальное восстановление» удается с помощью некоторых гидридов ме-
металлов, таких как станнаны R3SnH, германы R3GeH и силаны R3SiH, например:
R-X + Bu3Sn-H R-H + Bu3Sn-CI [Г.1.51]
Реакции протекают по радикальному цепному механизму и начинаются с
помощью обычных инициаторов (азосоединения, пероксиды, УФ-свет):
Инициатор 2R'- Начало цепи
R'- + Bu3SnH R'H + Bu3Sn- Рост цепи rj. j ~i
RX + Bu3Sn- - R- + Bu3SnX
R- + Bu3SnH RH + Bu3Sn-
С помощью наиболее часто применяемого гидрида трибутилцинка таким
методом можно восстановить до углеводородов нитросоединения (R—NCb), га-
логениды (R-Cl, R-Br, R-I) и сложные эфиры других неорганических и орга-
органических кислот, таких как я-толуолсульфонаты (R—OSO2C6H4CH3) и эфиры
дитиоугольной кислоты (R-OCS2CH3J. Обе вышеуказанные реакции подходят
также для превращения спиртов в углеводороды:
R-OH R-X - R-H [ГЛ.53]
1 Реакция Коновалова. — Прим. перев.
2 Для восстановления перечисленных соединений используют также другие гидриды
металлов, например, LiAlH4, NaBH4 (ср. разд. Г,7.3.1.1). Однако реагируют они по ионным
механизмам (нуклеофильное замещение) и поэтому с иной селективностью.
1. Радикальное замещение 263
Как описано в разд. Г,3.2 и Г,8.5, сначала из спиртов получают перечислен-
перечисленные выше сложные эфиры, а затем последние восстанавливают гидридом трибу-
тилцинка.
Во всех случаях обнаружена зависимость реакционной способности от при-
природы R в следующем ряду:
Аллил, бензил > трет-алкил > втор-апкт > первичный алкил » арил [Г. 1.54]
(почему?). Можно легко восстановить стерически затрудненные соединения
RX, которые трудно или совсем не реагируют по ионному механизму. Кроме то-
того возможна реакция с винил-Х и при определенных условиях также с арил-Х.
1-Хлор-2-метил-2-фенш1циклопропан из 1,1-дихлор-2-метил-2-фенилцикло-
пропана: McKinney M.A., Nagarajan S.C. J. Org. Chem. 44 A979), 2233.
1-Хпорбицикло[4.1.0]гептан из 7,7-дихлорбицикло[4.1.0]гептана: Seyferth D.,
Ymazaki H., Alleston D.L. J. Org. Chem. 28 A963), 703.
З-Дезокси-1,2:5,6-ди-О-изопропилиден-а-В-рибогексофураноза из изопропи-
лиденглюкофуранозы: lacono S., Rasmussen J. R. Org. Synth. 64 A986), 57.
1.7. ЛИТЕРАТУРА
Общие сведения о радикальных реакциях
Radicals in Organic Synthesis. Vol. 1: Basic Principles; Vol. 2: Applications. Hrsg.:
P. Renaud, V. P. Sibi. - Wiley-VCH, Weinheim, 2001.
C-Radikale, Hrsg.: M. Regitz, B. Giese, in: Houben-Weyl. Bd. E19aA989), S. 1-1567.
Curran D. P., Synthesis 1988, 417-439; 489-513.
Curran D. P., Porter N. A., Giese B. Stereochemistry of Radical Reactions. — VCH
Verlagsgesellschaft, Weinheim, 1996.
Davies D. I., Parrott M. J. Free Radicals in Organic Synthesis. — Springer-Verlag, Berlin,
Heidelberg, 1978.
Fossey J., Lefort D., Sorba J. Free Radicals in Organic Chemistry. — John Wiley & Sons, New
York, 1995.
Free Radicals. J. K. Kochi (ed.) Bd. 1-2. - John Wiley & Sons, New York, 1973.
Linker Т., Schmittel M. Radikale und Radikaleonen in der Organischen Synthese. — Wiley-
VCH, Weinheim, 1998.
Motherwell W. В., Crich D. Free Radical Chain Reactions in Organic Synthesis. — Academic
Press, London, 1992.
Nonhebel D. C, Tedder J. M., Walton J. С Radicals. — Cambridge University Press,
Cambridge, 1979.
Perkins M. J. Radical Chemistry. — Ellis Horwood, New York, 1994.
Pryor W. A. Einfuhrung in die Radikalchemie. — Verlag Chemie, Weinhein, 1974.
Ramaiah M. Tetrahedron. 1987,43, 3541-3676.
Riichardt Ch. Steric Effects in Free Radical Chemistry. — In: Topics in Current Chemistry,
Bd. 88. — Springer-Verlag, Berlin, Heidelberg, New York, 1980, p. 1-32.
Tedder J. M. Angew Chem., 1982,94,433-442.
Семенов Н. Н. О некоторых проблемах химической кинетики и реакционной способ-
способности. - М.: Изд. АН СССР, 1958.
264 Г Препаративная часть
Бучаченко А. Л., Вассерман А. М. Стабильные радикалы. Электронное строение, реак-
реакционная способность и применение. — М.: Химия, 1973.
Ингольд К., Роберте Б. Реакции свободнорадикального замещения. — М: Мир, 1974.
Галогенирование N-галогенаминаш (и другие гемолитические замещения)
Deno N. С, Methods Free Radical Chem. 1972, 3, 135-154.
MinisciF. Synthesis, 1973, 1-36.
Sosnovsky G., Rawlinson D. J. Adv. Free-Radical Chem. 1972, 4, 703.
Хлорирование
Poutsma M. L. Methods Free Radical Chemistry, 1969, 1, 79.
Stroh H. In: Houben-Weyl, Bd. 5/3, 1962. S. 511-528, 564-650, 735-748.
Смолин С. С, Пырялова П. С, Курдюмова Н. А. Усп. хим., 1960, 29, 23-54.
Бромирование
RoedigA. In: Houben-Weyl, Bd. 5/4, I960, S. 153-162, 331-347.
Thaler W. A. Methodes in Free Radical Chemistry, 1969, 2, 121.
N-Бромсукцинимид как бромирующий агент
Homer L, Winckelmann E. H. In: Neuere Methoden. Bd. 3., 1961, 98-135; Angew. Chem.,
1959, 71, 349-365.
Мачшская И. В., Бархаш В. А. В сб.: Реакции и методы исследования органических
соединений. Вып. 9, с. 287 — М.: Госхимиздат, 1959.
Терентьев А. П., Яновская Л. А. В сб.: Реакции и методы исследования органических
соединений. Вып. 6, с. 7-342. — М.: Госхимиздат, 1957.
Ивановский Э. Э. В сб.: Реакции и методы исследования органических соединений.
Вып. 2, с. 209-2147. — М.: Госхимиздат, 1952.
Восстановление гидридом трибутилцинка
Kuivila H. G. Synthesis, 1970, 499-509.
Neumann W. P. Synthesis, 1987, 665-683.
Окисление молекулярным кислородом
Autoxidation von Kohlenwasserstoffen, von W. Pritzkow u.a. — Deutscher Verlag fur
Grundstofflndustrie, Leipzig, 1981.
CriegeeR. In: Houben-Weyl, Bd. 8, 1952, S. 9-27.
Эмануэль Н. М. Теория и практика жидкофазного окисления. — М.: Наука, 1974.
Эмануэль Н. М., Денисов Е. Т., Майзус Е. К. Цепные реакции окисления углеводородов
в жидкой фазе. — М.: Наука, 1965.
Hiatt R. In: Organic Peroxides, vol. 2, D. Swern (ed.). — Wiley-Interscience, New York,
1971.
KropfH., Mailer W., WeickmannA., in:Hoben-Weyl, Bd. 4/la A981), S. 77-87.
KropfH., MunkeS., in: Hoben-Weyl, Bd. E13/1 A988), S. 59-126.
Шенберг А. Препаративная органическая фотохимия. — M.: ИЛ, 1963.
Pritzkow W. u.a Autooxidation von Kohlenwasserstoffen. — Deutscher Verlag fur
Grundstoffindustrie, Leipzig, 1981.
2. Нуклеофильное замещение при насыщенном атоме углерода 265
2. НУКЛЕОФИЛЬНОЕ ЗАМЕЩЕНИЕ ПРИ
НАСЫЩЕННОМ АТОМЕ УГЛЕРОДА
2.1. ХОД РЕАКЦИИ И ЕЕ МЕХАНИЗМ
В реакциях нуклеофильного замещения при насыщенном атоме углерода нук-
леофильный реагент Nu замещает связанный с углеродом атом или группу
атомов X; при этом Nu предоставляет пару электронов для возникающей связи,
а X уходит с парой электронов субстрата:
Nul + R-X - Nu-R + XI [Г.2.1]
Реакционную частицу Nu называют нуклеофильнымреагентом; важнейшими
представителями нуклеофильных реагентов являются нейтральные вещества со
свободной электронной парой и анионы, например:
Nul = ICII® IBrl0 III® HOI0, ROI0 HSI0, RSI0 INSCI0, H-O-H, R-O-H, NH3, RNH2, R2NH [Г.2.2]
О реакциях карбанионов, которые получаются из С—Н-кислотных соедине-
соединений (кетонов, сложных эфиров и др.), речь пойдет в разд. Г',7.4.2.1.
Ненасыщенные углеводороды и арены также могут выступать в роли нук-
нуклеофильных агентов, например при алкилировании по Фриделю—Крафтсу
(табл. Г.2.4). Этот тип реакций обсуждается в разд. Г,5 как электрофильное
замещение в ароматических соединениях.
Замещаемый заместитель представляет собой, как правило, электроноак-
цепторную группировку, которая благодаря индукционному эффекту поляри-
поляризует связь С—X, например:
-X= -Cl, —Br,—I, -O-SO2-OH,-O-SO2-OR, -O-Su2-\\ ir
[Г.2.3]
© ,H © ,R © ©
-O , -O , -NR3, -N=N идр.
(Непосредственно ОН-, R0- и R2N-rpynnbi заместить нельзя,, замена
удается после протонирования или алкилирования (при R2N), ср. разд. Г,2.2.1.)
Нуклеофильное замещение у насыщенного атома углерода — очень распро-
распространенный тип реакций, как представлено в табл. Г.2.4.
Реакции нуклеофильного замещения включают два процесса — нуклео-
фильную атаку реагентов Nu на RX, приводящую к образованию связи Nu—С, и
нуклеофугное удаление заместителя X при расщеплении связи С—X:
Nul^C^X [Г.2.5]
Оба этих процесса могут происходить одновременно в одну стадию или друг за
другом в две стадии. Поэтому существуют три возможности протекания реакции:
а) Разрыв связи С—X предшествует образованию связи С—Nu. На первой стадии,
определяющей скорость, прежде всего в качестве промежуточного продукта,
266
Г Препаративная часть
Таблица Г.2.4. Нуклеофильное замещение у насыщенного атома углерода и у ато-
атома кремния
Реакция
Процесс
R—ОН+НХ ** R—Х+Н2О
R—OH + R'OH =** R-OR'+H2O
R—X' + OH9 —* R—С
R—X+OR'e —¦ R_OR' + Xs
R—X+R'COOe —* ROCOR' + XQ
R-X + S№ -
R—X+SR'e
R—S
R—SR'+ Xе
R-X+SCNe^ R-SCN +
R-X+SR2' — R—SR2'+
R—X+NHR2' — R—NR2'
R—X+NR3' —¦ R—iW+Xe
R—X+CNe —* R—CN+Xe
R—X+NO2e
R-
( + R-NC)
( + R—O—NO)
R—X'+Xe
e /COR'
R-X + ICHC —
NCOR'
R—x + H-Ar
—Si—X + NHR2
+ CNe
N3S
,COR'
+ Xe
4COR'
+ HX
—Si— NR2 + HX
—Si—CN +¦ Xе
I
—Si-N3 + Xе
Этерификация спиртов га-
логеноводородными кисло-
кислотами и другими неоргани-
неорганическими кислотами; кислот-
кислотный гидролиз алкилгалоге-
нидов, алкилсульфатов и т. д.
Образование простых эфи-
ров в кислых условиях, рас-
расщепление простых эфиров
Щелочное омыление
Синтез простых эфиров по
Вильямсону
Синтез эфиров карбоновых
кислот
Синтез тиолов
Синтез тиоэфиров
Синтез алкилтиоцианатов
Образование сульфониевых
соединений
Алкилирование аминов
Кватернизация аминов
Синтез нитрилов по Кольбе
(Синтез изонитрилов)
Синтез нитроалканов
(Синтез эфиров азотистой
кислоты)
Реакция Финкельштейна
Алкилирование СН-кислот-
ных соединений (см. разд.
Г.7.4.2.1)
Алкилирование по
лю — Крафтсу (см.
разд. Г.5.1.7)
Фриде-
Си нтез
нов
Синтез
нидов
Синтез
дов
триалкилсилилами-
триалкилсилилциа-
триалкилсилилази-
—Х-—С1, —Вг. —I, —О—SO2OH (моноалкилсульфаты). —О—SOi—OR (диалкилсульфаты),
г\_/ 3 [толуолсульфонаты (тозилаты)].
который на второй стадии быстро реагирует с нуклеофилом Nu-, возникает кар-
бениевый ион. В качестве нового богатого энгергией соединения он быстро ре-
реагирует с нуклеофилом Nu. Энергия при этом изменяется, как представлено на
рис. В.35, а. В стадии, определяющей скорость, участвует только R-X, поэтому
такой механизм называют монамалекулярным нуклеофшьным замещением (Sn 1).
2. Нуклеофильное замещение при насыщенном атоме углерода 267
б) Образование связи Nu—С и расщепление связи С-Х происходит однов-
одновременно и согласованно в одну стадию (на рис. В. 12 показан соответству-
соответствующий энергетический профиль). В этой элементарной стадии участвуют
оба партнера реакции, такой механизм называют бимолекулярным нуклео-
фильным замещением (S]sj2).
в) Образование связи Nu—С предшествует расщеплению связи С—X. Подоб-
Подобный случай несогласованного нуклеофильного замещения у насыщенного
углеродного атома невозможен: он сопровождался бы расширением октет-
ной оболочки атома углерода, что потребовало бы очень большой затраты
энергии. Однако такой механизм встречается например, у соединений
кремния (разд. Г, 2.7), а также при нуклеофильном замещении в аренах
(разд. Г,5.2.1) и карбоксильных соединениях (разд. Г,7.1.4).
2.1.1. МОНОМОЛЕКУЛЯРНОЕ НУКЛЕОФИЛЬНОЕ ЗАМЕЩЕНИЕ (SN1)
В соответствии с указанным выше, этот механизм следует изобразить следую-
следующим образом:
\ медленно \ ©
С-Х . X'
+ хг
•о-"' ^ ,м.._и быстро
С + INu-H
X-Nu + Н®
рование ©
перегруп-
перегруппировка
олефин + H2NU
(разд. Г, 3.1.1)
продукты перегруппировки
(разд. Г, 9)
Аналогично реагируют и заряженные нуклеофилы Nu~: конечный продукт нук-
нуклеофильного замещения образуется тогда непосредственно из карбениевого иона.
Карбениевый ион1 — высокоэнергетический промежуточнй продукт (см.
рис. В.35, а) со своей собственной судьбой; он быстро и неселективно превраща-
превращается в продукт замещения и дальнейшие продукты (олефины, продукты пере-
перегруппировки).
В общем случае SnI -реакцию можно обнаружить по следующим типичным
характеристикам.
а) В начальном периоде2 она протекает как реакция первого порядка и ее
скорость определяется уравнением
[Г.2.7]
At
1 Переходное состояние, которое предшествует образованию карбениевого иона, похоже
на него, так как поздно расположено на реакционной координате; см. постулат Хэммонда
(разд. В,2, с. 196).
2 В дальнейшем все большую роль начинает играть обратная реакция с участием Х~ и
уравнение скорости реакции несколько усложняется; см. разд. В.З.1.
268 Г Препаративная часть
Поскольку партнер реакции Nu не принимает участия в стадии, определяющей
скорость, то повышение его концентрации не приводит к ускорению реакции,
б) В SN1-реакции центральный атом углерода субстрата RX в стадии, опре-
определяющей скорость реакции, переходит из четырехсвязанной тетраэдри-
ческой формы (состояние ^/^-гибридизации) в трехсвязанный карбени-
евый ион, имеющий форму треугольника с углеродным атомом посере-
посередине. В этом промежуточном состоянии субстрата реагент Nu, участвую-
участвующий в быстрой второй стадии, может приблизительно с одинаковой
вероятностью подойти с каждой из двух сторон, причем образуются два
новых тетраэдрических продукта реакции RNu [Г.2.8]. Поэтому оптичес-
оптически активные соединения в ходе SN1-реакции рацемизуются.
Nu® Nu
-f
[Г.2.8]
NuG L
1 -Реакции в малополярных растворителях с участием реакционноспособ-
ных карбениевых ионов часто проходят преимущественно с обращением кон-
конфигурации [как 8к2-реакции; уравнение (Г.2.9)]. В этих случаях во время реаак-
ции уходящая группа Х~ остается связанной с карбениевым ионом в виде
ионной пары, так что реагент Nu может подойти к нему только со стороны, не
прикрытой Х~\
в) При Sn 1 -реакциях, как правило, в качестве побочных продуктов образу-
образуются олефины или продукты перегруппировки. Олефины нередко
составляют даже основную часть продуктов реакции (о соотношении
элиминирования и замещения см. разд. Г.3.1.1).
г) Поскольку при БгЛ-реакциях ионы, образующиеся на стадии, определя-
определяющей скорость, должны быть сольватированы, большое влияние на ско-
скорость реакции оказывает сольватирующая способность растворителя
(разд.Г,2.2.1).
д) Энтропии активации Б^-реакций, как правило, близки к нулю, так как
образование промежуточного карбениевого катиона происходит без
существенных пространственных препятствий (см. разд. В,2).
2.1.2. БИМОЛЕКУЛЯРНОЕ НУКЛЕОФИЛЬНОЕ ЗАМЕЩЕНИЕ (SN2)
При этом типе реакций разрыв и образование связи протекают одновременно
(согласованно). Реагент Nu приближается к поляризованной молекуле R—X со
стороны, противоположной заместителю X, и вступает во взаимодействие с R.
Синхронно с процессом образования связи R—Nu происходит разрыхление связи
между R и X. При этом молекула проходит через переходное состояние, в котором
Nu связан еще не очень прочно, а X еще не полностью освободился от остатка:
Nul + ,..С-Х
t
Nu—С—X
С
XI [Г.2.9]
2. Нуклеофильное замещение при насыщенном атоме углерода 269
Это переходное состояние обладает наибольшей энергией на координате
реакции (рис. В. 12).
Ниже перечислены характерные признаки для 8^-реакций.
а) Так как оба реакционных партнера Nu и RX на одной стадии превраща-
превращаются в продукт RNu и X, Б^-реакция описывается уравнением скорос-
скорости 2-го порядка:
dj^xj [Г210]
Повышение концентрации Nu увеличивает скорость реакции.
б) Во всех стадиях реакции центральный атом углерода сохраняет все свои
связи с другими группами; поэтому оптическая активность асимметри-
асимметрического атома углерода полностью сохраняется. При этом образуется оп-
оптический изомер, относящийся к исходному соединению как предмет к
своему зеркальному изображению (обращение конфигурации, инвер-
инверсия). Весь процесс можно наглядно представить, если сравнить его с
выворачиванием зонтика. Его называют вальденовским обращением.
в) При Б^-реакциях, в противоположность SNl-реакциям, можно избе-
избежать образования как олефинов, так и продуктов перегруппировки,
подобрав подходящие условия.
г) Энтропия активации 8н2-реакций в большинстве случаев сильноотри-
сильноотрицательна, так как переходное состояние представляет собой структуру с
высокой степенью упорядоченности, пространственные требования при
образовании которой высоки.
2.2. ФАКТОРЫ, ВЛИЯЮЩИЕ НА ХОД НУКЛЕОФИЛЬНОГО ЗАМЕЩЕНИЯ
Реакции, протекающие исключительно как SnI или как Sn2, представляют со-
собой граничные случаи, между которыми располагается целый «спектр» проме-
промежуточных. В какой степени осуществляется SnI- или Б^-механизм нуклео-
фильного замещения, зависит в первую очередь от структуры субстрата RX и
условий реакции (прежде всего от растворителя и катализатора) и в определен-
определенной степени можно прогнозировать (разд. Г,2.2.1).
Реакции, протекающие в пограничной области между SN1 и SN2, можно со-
соответствующим выбором условий сдвинуть в сторону SnI- или Б^-типа. Это
важно с препаративной точки зрения, так как продукты реакции зависят от ее
механизма. При SN2-peaKu™x выбором соответствующих условий часто можно
избежать образования продуктов элиминирования и перегруппировки, в то вре-
время как при SNl-реакциях это обычно невозможно (разд. Г,3.1.1).
Разные продукты реакции могут возникать и в самом процессе замещения. Это
наблюдается при конкуренции различных нуклеофилов, а также при реакциях би-
бифункциональных нуклеофилов. Подробнее об этом будет идти речь в разд. Г,2.3.
2.2.1. РЕАКЦИОННАЯ СПОСОБНОСТЬ СУБСТРАТА RX
На реакционную способность субстрата RX как в SnI-, так и в SN2-peaiojHHX су-
существенно влияют заместители у центрального атома углерода. Так, SNl-реак-
270
Г Препаративная часть
ционная способность RX возрастает, если положительный заряд карбениевого
иона R+, образующегося в скоростьопределяющей стадии реакции, может ста-
стабилизироваться (делокализоваться) +/- или +М-эффектом заместителей. По
этой причине склонность к SnI -реакциям повышается от метальных к трет-бу-
тильным соединениям:
Н3С X < НзС~СН2~Х
н3с
Н3С
н3с
СН-Х « НзС-С-Х
с н-,с'
[Г.2.11]
В том же направлении действует и пространственный (стерический) эффект
алкильных групп. При переходе тетраэдрического исходного соединения RX в
тригональный плоский карбениевый ион заместители отодвигаются друг от дру-
друга, что снижает пространственное напряжение. У объемистых /я/?е/я-алкильных
остатков этот эффект проявляется сильнее, чем у вторичных, у первичных же
менее всего. В результате тенденция к гетеролитическому расщеплению связи
R—X в стадии, определяющей скорость реакции SnI, возрастает от первичных к
третичным RX (табл. Г.2.12).
Напротив, в реакциях Sn2 на реакционную способность реагента RX прост-
пространственные эффекты алкильных групп оказывают противоположное влияние.
В ряду этилгалогенид<изопропилгалогенид</я/>е/и-бутилгалогенид возрастает
затруднение атаки нуклеофила на центральный атом углерода «с обратной
стороны» связи С—X [схема (Г.2.9)] и потому замедляется скорость реакции
(см. табл. Г.2.12).
В свете сказанного действует более общее правило: третичные алкильные со-
соединения реагируют при нуклеофильном замещении обычно мономолекулярно
(SnI), первичные — напротив, бимолекулярно (SN2). Вторичные алкильные со-
соединения часто реагируют в «пограничной области», где проявляются признаки
как SN2-, так и SNl-реакций. В этих случаях можно с помощью подходящего
растворителя (разд. Г,2.2.2) направить реакцию либо по SN1-, либо по SN2-nyra.
Аналогично алкильным системам, переход от SN2- к SNl-THny осуществля-
осуществляется и в ряду бензил-, дифенилметил- и трифенилметилгалогенидов или в ря-
ряду бензил-, 2-метоксибензил-, 2,4-диметоксибензилгалогенидов. В этих слу-
случаях карбениевый ион стабилизируется +Л/-эффектами арильных или меток-
сигрупп. Напишите соответствующие схемы!
SN2-nepexoflHoe состояние может стабилизироваться также мезомерными
эффектами. По этой причине бензильные и аллильные системы реагируют тоже
Таблица Г
Механизм
SnI
Sn2
.2.12. Сольволиз а апкилбромидов в 80%-ном этаноле при
Константа
скорости реакций
105 Ai.c
105 k2, Л'МОЛЬ 1-С
СН3Вг
0,35
2040
5840
СН3СН2Вг
0,14
171
1230
(СНзЬСНВг
0,24
5,0
21
55 °С
(СН,),СВг
1010
очень мала
0
а Сольволизом называют реакции, в которых растворитель одновременно является
нуклеофильным реагентом.
2. Нуклеофильное замещение при насыщенном атоме углерода
271
по механизму SN2, однако в 100-200 раз быстрее, чем соответствующие алкиль-
ные соединения. Как схематически показано ниже, основой такой стабилиза-
стабилизации является перекрывание я-орбитали с квази-^-орбиталями, возникающими
в переходном состоянии SN2-pearaniH:
е
Nu|
|G
/\
\ /
!!' I '"
/\
+ xi
е
[Г.2.13]
Для таких соединений характерны очень высокие скорости реакций. Приме-
Примером может служить взаимодействие с KI в ацетоне при 60°С.
[Г.2.14]
Высокую реакционную способность используют при селективном превра-
превращении эфиров хлоруксусной кислоты в эфиры аминоуксусной кислоты, для по-
получения «активированных эфиров» из соли карбоновой кислоты и, например,
хлорацетонитрила и при синтезе фенациловых эфиров (см. разд. Г,2.6.3).
Помимо прямого сопряжения заместитель реакционного центра, находя-
находящийся в пространственно выгодном положении, может влиять и путем внутри-
внутримолекулярной нуклеофильной атаки:
НзС-СН2-СН2-С1
"отн 1
NEC-CH2-CI
2800
H2C=CH-CH2-CI
90
Ph-CO-CH2-CI
32 000 G5 'С)
Ph-CH2-CI
250
EtOOC-CH2-CI
1600
¦Ни
.АСИ2)п
'Vz,
,Nu
[Г.2.15]
Благодаря этому влиянию соседних нуклеофильных групп Z (R2N—, RS—,
RO-, галоген, особенно иод, арил) снижается энергия образующегося карбени-
евого иона и повышается скорость SnI-реакции. В подобных реакциях осущес-
осуществляется двукратное вальденовское обращение, так что в итоге конфиругация у
реакционного центра сохраняется. Ознакомьтесь с этим вопросом по учебнику.
Снижение скорости реакций SN2 под влиянием пространственных факторов
может быть обусловлено наличием объемистых заместителей не только в а-, но
и в C-положении. Так, неопентильные соединения (трет-ВиСНгХ) реагируют в
106 раз менденнее, чем соединения СН3СН2Х.
Нуклеофильное замещение галогена в винил- и арилгалогенидах требует го-
гораздо более жестких условий по сравнению с реакциями алкилгалогенидов.
Энергии диссоциации связей в случае фенильных соединений повышены по
сравнению с приведенными в табл. Г.2.16 примерно на 60 кДж/моль; нуклео-
нуклеофильное замещение протекает здесь обычно по другим механизмам (разд. Г,5.2).
В соответствии со схемой [Г.2.5] заместительХв ходе нуклеофильного заме-
замещения уходит из субстрата R—X со связывающей парой электронов. При этом
272
Г Препаративная часть
Таблица Г. 2
газовой фазе
Me-NH2
1240
16. Энергии
Me—ОН
1150
гетеролитического расщепления
Ме-ОСНз
1150
Me—F
1070
В растворах эта энергия сильно понижается
(разд. Г,2.2.2).
Me—Cl
950
связей (кДж/моль) в
Me—Br
920
из-за сольпатационныд
Me—I
890
эффектов
должна быть затрачена энергия на гетеролитическое расщепление связи R—X,
которая слагается из энергии гемолитического расщепления связи, энергии ио-
ионизации R- и сродства к электрону Х\
В табл. Г.2.16 приведены энергии гетеролитического расщепления некото-
некоторых связей. В соответствии с данными таблицы реакционная способность R—X
(в диметилформамиде) возрастает в ряду
R-NH2 « R-OH « R-F « R-CI < R-Br < R—I
[Г.2.17]
Этот ряд действителен как для SnI-, так и для Б^-реакций. Ряд одновремен-
одновременно отвечает уменьшению основности аниона Х~, т. е. в первом приближении
склонность к отщеплению группы X тем больше, чем меньше ее основность. По
этой причине алкиламины, спирты и алкилфториды в качестве субстрата RX в
реакциях нуклеофильного замещения имеют чрезвычайно низкую реакционную
способность, несмотря на высокую электроотрицательность их гетероатома.
Основность уходящей группы можно изменить введением заместителей.
Низкую способность ОН-группы к отщеплению можно сильно увеличить прев-
превращением ее в в значительно более слабоосновную и-толуолсульфонатную груп-
группу («тозилатную» группу, TsO~). Еще более высокую реакционную способность
имеют трифторметансульфонаты («трифлат», R—OSO2CF3), которые по реакци-
реакционной способности активнее тозилатов в 10 000 раз.
В соответствии с основностью способность к отщеплению я-толуолсульфо-
натной группы попадает между хлоридом и бромидом. В действительности, од-
однако, алкилтозилаты гораздо реакционноспособнее, чем алкилбромиды и
алкилиодиды. Это доказывает, что основность не является единственным кри-
критерием для отщепления группы. Кроме того, склонность к отщеплению зависит
как от природы остатков R и нуклеофильных реагентов Nu, так и от природы
растворителя, так что данный ряд по склонности к отщеплению не имеет обще-
общего характера.
Так, при Бы2-реакциях установлено, что субстраты, в которых уходящая
группа X легко поляризуема («мягкие»; например ,1~), быстрее реагируют с мяг-
мягкими реагентами, тогда как группы со слабо поляризующимся гетероатомом
(«жесткий»; например, TsO~) быстрее вытесняются жестким нуклеофилом. От-
Относительная реакционная способность метилтозилата и метилиодида по отно-
отношению к различным нуклеофильным реагентам Nu~ в метаноле при 25 "С сос-
составляет, например:
[Г.2.18]
Nu©
^MeOTs / ^Mel
N?
6,6
CH3O©
4,6
CIS
2,8
Br©
0,72
SCN©
0,28
I©
0,13
2. Нуклеофильное замещение при насыщенном атоме углерода 273
При SiMl-реакциях в протонных растворителях алкилсульфонаты также
обычно более реакционноспособны, чем алкилиодиды.
Тенденцию НО- или RO-группы к отщеплению можно сильно повысить
также протонированием, таким образом, становится возможным нуклеофиль-
нуклеофильное замещение, например при образовании эфиров неорганических кислот или
расщеплении простых эфиров (разд. Г,2.5.1 и Г,2.5.2). Аналогично аминогруппы
легко отщепляются после кватернизации, что препаративно используют при
Е2-реакциях (элиминирование по Гофману, разд. Г,3).
Первичные аминогруппы диазотированием можно превращать в диазоние-
вые группы (разд. Г,8). Алифатические диазоний-катионы неустойчивы и
самопроизвольно распадаются с образованием карбениевых ионов и элемент-
элементного азота. Диазониевая группа — наиболее легко отщепляющийся заместитель,
она всегда замещается по механизму SnI.
Нуклеофугное отщепление (см. с. 192) заместителей X из RX можно ускорить
также протонными растворителями. Такие растворители особенно хорошо
сольватируют анионы благодаря образованию водородных связей (разд. В,3.3),
это повышает стабильность образующихся анионов Х~ и облегчает их отщепле-
отщепление. Подобное влияние растворителей тем более заметно, чем выше Н-кислот-
ность растворителя и чем жестче уходящая группа X.
Кроме того, различия между отдельными уходящими группами в данном
случае существенно меньше, чем в полярных апротонных растворителях, т. е.
протонные растворители в определенной степени выравнивают тенденцию к
отщеплению.
С другой стороны, образование водородных связей в протонных раствори-
растворителях приводит к снижению нуклеофильности реагента (разд. Г,2.2.2). В ре-
результате переходное состояние сдвигается в SN1 -область тем больше, чем боль-
больше кислотность растворителя. Насколько велико влияние растворителя на
SnI-реакцию, показывают относительные скорости сольволиза трет-бутш-
хлорида (образование т/?еяг-бутилкатиона):
[Г.2.19]
Подобно протонным растворителям влияют также кислоты Льюиса, которые
выполняют роль электрофильных катализаторов, вступающих во взаимодей-
взаимодействие со свободными электронными парами заместителя X. В качестве кислот
Льюиса следует прежде всего упомянуть Ag+, SnCl4, BF3, A1C13, FeCl3, ZnCl2,
SbCb; они особенно сильно взаимодействуют с соединениями галогенов. Это
обстоятельство используют при алкилировании по Фриделю—Крафтсу (разд.
Г.5.1.7).
2.2.2. НУКЛЕОФИЛЬНОСТЬ РЕАГЕНТОВ
В процессе нуклеофильного замещения между нуклеофильными реагентами Nu
и электрофильным субстратом RX (или карбениевым ионом R+ при Зг
Растворитель
*отн B5 °С)
Этиловый спирт
1
Метанол
9
Муравьиная
кислота
12 200
Вода
335000
274 Г Препаративная часть
циях) образуется новая связь С—Nu, при этом выделяется энергия гетеролиза
связи. Из-за потери обоих связывающих электронов электронная плотность
реагента уменьшается; анионный нуклеофил Nu~ превращается в нейтраль-
нейтральный продукт RNu, нейтральный нуклеофил (например, в спирт или амин), в ка-
катион RNu+:
NuG+R-X [NU--R--xj* Nu-R +X® [Г.2.20]
Nu + R-X [NU--R-—X] - Nu-R +XU [Г.2.21]
При реакции в полярных растворителях изменения зарядов приводят к соль-
ватационным и энергетическим изменениям. В случае [Г.2.20] нуклеофил Nu~
при переходе в активированный комплекс частично десольватируется, что вы-
вызывает выделение энергии; в случае [Г.2.21] переходное состояние и реакцион-
реакционный продукт сольватированы сильнее, чем исходные реагенты, что отражается
на уменьшении энергии.
Оба этих фактора — выделение энергии вследствие образования связи
R—Nu и изменения энергии из-за изменения состояния сольватации — опреде-
определяют реакционную способность нуклеофильных реагентов — их нуклеофиль-
ность.
Из-за различного влияния этих факторов для различных растворителей и
различных реакционных партнеров ряд нуклеофильности реагентов во всех ре-
реакциях не одинаков, а зависит от природы элекрофильных партнеров и раство-
растворителей. В качестве меры нуклеофильности используют относительные скорос-
скорости по отношению к определенным электрофильным субстратам.
Наиболее просты отношения в полярных апротонных растворителях, таких
как диметилформамид (ДМФА), диметилсульфоксид (ДМСО) или гексаметил-
фосфортриамид (ГМФТ).
В них нуклеофилы сольватированы не так сильно, как в протонных раст-
растворителях (разд. В,3.3) и выгрыш энергии при образовании связи Nu—R, т. е.
склонность нуклеофильного реагента к электрофильному партнеру R, становит-
становится решающим фактором его реакционной способности. Показано, что в первом
приближении нуклеофильность реагента тем больше, чем выше его бренстедо-
вская основность. Это особенно очевидно в ряду похожих нуклеофилов, напри-
например с одинаковым нуклеофильныым атомом:
RO© > PhOQ > RCOO© > ROH > PhOH > RCOOH [Г.2.22]
Таким образом, в апротонных растворителях нуклеофильность галогенид-
ионов изменяется параллельно их основности (см. [Г.2.25]).
Становится понятной связь между нуклеофильностью и основностью, так
как и в реакциях нуклеофильного замещения, и в бренстедовских кислотно-ос-
кислотно-основных реакциях нуклеофильные реагенты Nu действуют как доноры электро-
электронов (основания Льюиса). Однако бренстедовская основность характеризуется
термодинамическим равновесием донора электронов с Н+-ионом, тогда как
нуклеофильность отражает скорость реакции с одним более или менее положи-
положительно заряженным атомом углерода.
2. Нуклеофильное замещение при насыщенном атоме углерода
275
Так как согласно ЖМКО-концепции (разд. В,4) алкилгалогениды и алкил-
сульфонаты являются мягкими электрофилами (в противоположность жестко-
жесткому Н+), они легче реагируют с мягкими основаниями. В соответствии с этим в
8к2-реакциях мягкие, высокополяризованные нуклеофилы часто оказываются
более реакционноспособными, чем жесткие нуклеофилы, имеющие с ними
одинаковую основность, например:
RSee > Rse > roo1 [Г.2.23]
Наряду с основностью поэтому по поляризуемости определяют нуклеофиль-
ность реагентов. В отдельных случаях трудно оценить, какой фактор преобладает.
Таблица Г.2.24. Константы скорости 5м2-реакции метилиодида с различными
нуклеофилами Nit в диметилформамиде и метаноле (при О °С)
Nu©
CN©
СН^СОО©
4-NO2C6H4S©
Nf
F©
CIS
Br©
I©
SeCN©
SCN©
4-NO2C6H4O©
к, л - моль-1
ДМФА
30
2,0
1,36
0,31
од
0,24
0,12
9,2 • 10-2
6,9 • 10-3
' 1,4-10-3
•c-i
MeOH
3,3 • 10-5
4,5 • 10-8
5,7 ¦ 10-з
3,0 • 10-6
6,3 • 10-8
1,0-10-'
1,8 ¦ 10-6
1,6 -1(H
4,0 • 10-»
3,0 • 10-5
9,6 ¦ 10-8
&MeOH
^ДМФА
1-10-6
2-10-8
4-10-3
1 10-5
6-1O-7
4-10-7
1 • 10-5
4-10-3
4-10-3
7-10-5
Экспериментально по Б^-реакции с метилиодидом в ДМФА (табл. Г.2.24)
установлен следующий ряд увеличения нуклеофильности:
SCN6 < ie < Br©
F9 < N6 < СН3СОО© < CN©
[Г.2.25]
Этот ряд может изменяться при реакциях с другими субстратами, что можно
использовать для увеличения селективности амбидентных нуклеофилов (разд.
Г,2.3). В апротонных средах также значительное влияние на нуклеофил оказы-
оказывает природа растворителя. Так как анионный нуклеофил при переходе в акти-
активированный комплекс Б^-реакции частично десольватируется, его реакцион-
реакционная способность становится сразу тем больше, чем слабее сольватация. При
повышенной льюисовой кислотности растворителя (способность к сольватации
анионов) нуклеофильность анионного реагента падает в апротонных раство-
растворителях приблизительно в ряду гексаметилфосфотриамид > ацетон > диметил-
формамид > пропиленкарбонат « ацетонитрил « диметилсульфоксид > нитро-
метан.
1 Так как в газовой фазе для этих ионов наблюдается обратный порядок нуклеофильнос-
нуклеофильности, соответствующий их основности, в растворе степень реакционной способности можно
объяснить эффектом сольватации. Малые ионы сильнее сольватированы в апротонных раст-
растворителях и поэтому менее реакционноспособны, чем более крупные.
276 Г Препаративная часть
При реакции нейтральных нуклеофилов, таких как спирт или амин, переход-
переходное состояние, напротив, сольватировано сильнее, чем исходное вещество, ско-
скорость реакции возрастает в апротонных растворителях с повышенной
льюисовой кислотностью в обратном порядке.
В протонных растворителях нуклеофильнък реагенты сольватированы гораз-
гораздо сильнее, чем в апротонных, поскольку между Nu~ и растворителем образуют-
образуются водородные связи. Это повышает расход энергии на дссольватацию Nu~ при
переходе к активированному комплексу 8н2-реакции (схема [Г.2.9]); соответ-
соответственно снижается нуклеофильность. Поскольку кислые Н-атомы протонных
растворителей с точки зрения концепции ЖМКО считаются жесткими, особен-
особенно прочные водородные связи образуются с жесткими, т. е. сильноосновными и
слабополяризуемыми нуклеофилами, реакционная способность которых пони-
понижается вследствие этого особенно сильно. Напротив, мягкие (т. е. слабоосновные
и сильнополяризуемые) нуклеофилы образуют лишь слабые водородные связи,
поэтому их реакционная способность снижается протонными растворителями
не столь сильно. В результате ряд нуклеофильности при переходе от апротонных
полярных растворителей к протонным почти обращается:
СНзСОО© = F© < С1© < ОН© < Вгв = \э < CN© < SCNe ^ S2O32© [Г.2.26]
Таким образом, в протонных растворителях нуклеофильность определяется
уже не основностью ионов, а главным образом их поляризуемостью.
Падение реакционной способности при переходе от полярных апротонных к
протонным растворителям очень велико у жестких нуклеофилов. Так, для реак-
реакции RBr с Х~ в ацетоне и воде отношение скоростей составляет 8 • 106 для F~,
1-104дляСг-и1-102для1-.
Соотношения становятся еще более сложными, из-за того что ионные нуклеофилы вво-
вводятся в реакцию в виде солей, в то время как полную активность они приобретают только в ви-
виде свободных ионов, а ионные пары (например, Li+Bt~) практические не реагируют. Экспери-
Экспериментально наблюдаемые суммарные константы скорости 8м2-реакций определяются помимо
прочего константами диссоциации соответствующих солей в данном растворителе. Диссоци-
Диссоциация, а тем самым и реакционная способность соли в первом приближении возрастают с
полярностью (диэлектрической проницаемостью) растворителя и с размером катиона и анио-
аниона, например,
LiCl<NaCl<KCI или LiF < LiCl < LiBr < Lil [Г.2.27]
В менее полярных растворителях с диэлектрической проницаемостью менее 30 (напри-
(например, в ацетоне) соли с малыми по размеру ионами диссоциированы не полностью: в этих слу-
случаях наблюдают зависимость нуклеофильности от размера ионов в соответствии с рядом
[Г.2.27]. Для галогенидов этот ряд противоположен наблюдаемому в апротонных растворите-
растворителях [Г.2.25]; это обусловлено неполной диссоциацией ионных пар. Следовательно, для полно-
полного использования реакционной способности анионных нуклеофилов лучше применять соли
калия, а не лития. Подходящими оказываются также тетраалкиламмониевые ионы; соли пос-
последних применяются в межфазном катализе, обсуждаемом в разд. Г,2.4.2.
Диссоциацию ионной пары, а тем самым и реакционную способность соли можно повы-
повысить, используя более полярный растворитель с хорошей способностью сольватировать кати-
катионы. Апротонные растворители по этим признакам располагаются примерно в следующий
ряд:
Et2O < ТГФ < МеОСН2СН2ОМе < ДМФА< ДМСО [Г.2.28]
2. Нуклеофильное замещение при насыщенном атоме углерода 277
Аналогичный, но более сильный эффект достигается при добавлении в каче-
качестве комплексообразователей криптандов, например краун-эфиров (разд.
Г.2.4.1) или полигликолей; при этом стабилизируются катионы (но не анионы).
Их действие может быть усилено тем, что, например, KF в подобной комплекс-
комплексной форме хорошо растворяется в ацетонитриле или бензоле. Тогда из-за высо-
высокой основности фторид-ионов (ряд [Г.2.25]) становится возможным нуклео-
нуклеофильное замещение, которое не удается в протонных растворителях:
R-OSO2Ar +KT" ~ R-F+ArSO3 K~ [Г 2 291
криптанд в ацетонитриле l •^••^•^j
R-X + CH3COO®Na® CH3COOR + X®Na® [Г.2.30]
криптанд в ацетонитриле
Кроме рассмотренных электронных и сольватационных факторов на реак-
реакционную способность нуклеофилов оказывают влияние также стерические эф-
эффекты. Большие объемистые группы реагента затрудняют или даже препятству-
препятствуют приближению к электрофильному С-атому субстрата RX и снижают нуклео-
фильность. Эти эффекты следует использовать для того, чтобы предотвратить
протекание нежелательных реакций замещения со стерически затрудненными
основаниями, как, например, при индуцированном основаниями элиминиро-
элиминировании, см. разд. Г,3.1.1.2.
2.3. РЕГИОСЕЛЕКТИВНОСТЬ РЕАКЦИЙ
С АМБИФУНКЦИОНАЛЬНЫМИ НУКЛЕОФИЛАМИ
Некоторые нуклеофилы обладают не одним, а двумя или несколькими реакци-
реакционными центрами. Их называют амбифункциональными или амбидентными
реагентами. В зависимости от условий реакции и структуры субстрата RX они
образуют разные продукты реакций, например:
[Г.2.31]
[Г.2.32]
[Г.2.33]
[Г.2.34]
Соотношение образующихся изомеров зависит от растворителя и типа реак-
реакции; в частности, от того, протекает ли она при зарядовом или орбитальном
контроле (разд. В,6).
Х®+ R-O-N=0 -
эфир азотистой
кислоты
е © Q
X + R-N=CI
O=N-OIQ + R-X
е
IC=NI + R-X -
изоцианид
Х®+ R-N=C=S
изотиоцианат
(горчичное масло)
\ OR
С) \ /
X + С=С
продукт
О-апкилирования
(эфир енола)
INEC-SI® + R-X
C=c' + R-X -
R-N4 + Xu
Ъ
нитросоединение
R-C=N| + X®
нитрил
R-S-СЭМ + X®
тиоцианат
(роданид)
продукт
С-алкилирования
(разд. Г, 7.4.2.1)
278
Г Препаративная часть
а) Влияние растворителей на региоселективность SN2-peaKUHft амбидентных реагентов ос-
основано на том, что сольватация и потому нуклеофильность обоих центров может быть раз-
различной в разных растворителях. Так, в апротонных растворителях центр с большей плот-
плотностью заряда оказывается более основным и предпочтительнее реагирует с RX.
В протонных растворителях по мере роста их Н-кислотности этот центр все сильнее соль-
ватируется за счет образования водородных связей, его нуклеофильность снижается. Таким
образом, в сильнокислотных растворителях (фенол, трифторэтанол) реакция происходит
по меннее сольватированному и тем самым более нуклеофильному мягкому центру.
Типичный пример — реакция фенолята (сходного с енолятом в схеме [Г.2.34]) с аллил-
бромидом. В апротонном диметиловом эфире этиленгликоля и в слабокислом протонном
растворителе метаноле (рАнл 16) и в воде (рАнд 15,7) образуется 100% аллилфенилового
эфира (продукт О-алкилирования), в более кислом феноле (рАнд 10) — только 23% аллил-
аллилфенилового эфира и 77% смеси о- и л-аллилфенолов (продукты С-алкилирования).
б) В данном апротонном растворителе региоселективность, кроме того, зависит от
субстрата R-X. Так, при алкилировании енолята пропиофенона действием R—X в гек-
саметилфосфортриамиде продукты О-алкилирования составляют: R—115%; R-Br40%;
R-C1 67%; R-OTs 85%. Реакция во всех случаях идет по механизму SN2. Такой ряд
реакционной способности можно объяснить в рамках концепции ЖМКО: у амбидент-
ного енолята кислородный атом является жестким, р-углеродный атом — мягким
центром. С другой стороны, жесткость а-углеродного атома в субстрате R-X зависит от
заместителя X. Алкилиодиды и алкилбромиды имеют мягкий реакционный центр,
алкилхлориды и тозилаты — жесткий. Взаимодействия «жесткий — жесткий» и
«мягкий — мягкий» всегда предпочтительнее комбинации «жесткий — мягкий», что
позволяет легко объяснить наблюдаемую региоселективность:
[Г.2.35]
в) Жесткость центрального углеродного атома в субстрате R—X увеличивается при пере-
переходе реакции от механизма SN2 к SN1, поскольку в ходе SNl-реакции возникает карбе-
ниевый ион, а с ростом положительного заряда жесткость возрастает. По этой причи-
причине, например, первичные алкилгалогениды в SN2-peaKUHH с нитритом серебра дают
70—80% нитроалканов, а третичные алкилгалогениды нитроалканов вовсе не образуют,
но образуют -60% т/>стя-алкилнитрита наряду со значительными количествами олефи-
нов. Это указывает на S»l -механизм реакции.
г) У хелатов металлов с р-дикарбонильными соединениями существуют свои особеннос-
особенности: вследствие квазиароматического характера они не диссоциируют даже в сильно-
сильнополярных апротонных растворителях. В этих случаях обычно образуются почти исклю-
исключительно продукты С-алкилирования; лишь при использовании самых жестких
алкилирующих агентов (диметилсульфат, R-O-SO2CF3) образуются значительные
количества О-алкилированных продуктов.
Изложенные выше рассуждения на основе концепции ЖМКО соответствуют теории
граничных орбиталей (разд. В,6): реакции жестких центров проходят преимущественно при
зарядовом контроле, тогда как реакции мягких центров — при орбитальном контроле.
В новейших работах приходят к выводу, что региоселективность при алкилировании амбидент-
амбидентных анионов может быть вызвана также тем, что реакции у (жесткого) заряженного центра проте-
протекают по SnI- или 5н2-механизму, а реакции мягких центров сопровождаются одноэлектронным
переносом, т. е. являются реакциями радикалов и радикал-анионов. Таким образом, особенности
региоселективности являются результатом конкуренции двух коренным образом различных меха-
механизмов реакции. С литературой по этому вопросу вы ознакомитесь в разд. Г,2.8.
2. Нуклеофильное замещение при насыщенном атоме углерода 279
2.4. УСЛОВИЯ РЕАКЦИЙ НУКЛЕОФИЛЬНОГО ЗАМЕЩЕНИЯ
С АНИОННЫМИ НУКЛЕОФИЛАМИ
2.4.1. ВОЗМОЖНОСТИ ОСУЩЕСТВЛЕНИЯ РЕАКЦИЙ
При реакциях с незаряженными нуклеофилами (например, аминами) выбор ус-
условий, в которых нуклеофильные замещения протекают с достаточными ско-
скоростями, обычно не составляет труда. По-иному обстоит дело с используемыми
в виде щелочных солей анионными нуклеофилами. Они хорошо растворяются в
воде, в большинстве же органических растворителей — плохо или вовсе нераст-
нерастворимы. В то же время субстраты часто нерастворимы в воде, но хорошо раство-
растворимы в обычных органических растворителях.
Экспериментально оправдывает себя ряд возможностей проведения реак-
реакций, которые протекают в гомогенной среде.
Реакции щелочных солей проводят в смешивающихся с водой растворителях
(например, спирт, ацетон), добавляя столько воды, чтобы соль растворилась,
а субстрат не выпал в осадок. Частыми препятствиями оказываются выпадение
субстрата уже при небольшом добавлении воды или очень малая концентрация
соли в растворе.
Поскольку щелочные соли ограниченно растворимы в полярных раствори-
растворителях (протонных — этиленгликоль и апротонных — ДМФА, ДМСО), они при-
пригодны в качестве реакционной среды при нуклеофильном замещении (разд.
Г,2.2.2). Недостатком является высокая стоимость таких растворителей, а иног-
иногда и трудность их удаления при обработке реакционной смеси.
Интересную возможность создают краун-эфиры, с помощью которых ще-
щелочные соли можно растворить даже в неполярных растворителях. При этом
катионы щелочных металлов комплексуются с краун-эфирами и частично с
эфирами полигликоля за счет взаимодействия со свободными электронными
парами атомов кислорода таких эфиров (которые сами по себе обладают липо-
фильными свойствами). На схеме [Г.2.36] изображен комплекс соли калия с
циклическим полиэфиром 18-краун-б1:
О
О'
[ .,*:... ] nu° [г.2.36]
о
о
Относительно реакционной способности нуклеофильных анионов в таких
комплексах см. разд. Г,2.2.2. Препятствиями для широкого использования
краун-эфиров являются их высокая цена, а также заметная токсичность.
Анионные нуклеофилы хорошо растворяются также в виде их четвертичных
аммониевых солей, в особенности если алкильная группа содержит достаточно
много С-атомов (в сумме 16 и более), что придает ей липофильные свойства.
1 Циклические эфиры такого типа называют краун-эфирами. В начале названия указыва-
указывают число звеньев в цикле, в конце — число эфирных атомов кислорода.
280 Г Препаративная часть
Многие анионы с помощью четвертичных аммониевых ионов в виде ионных
пар количественно переводятся из водной среды в органическую. Поэтому нет
необходимости специально получать соответствующие соли. Чем больше поля-
поляризуемость анионов, тем лучше они «экстрагируются» определенным аммоние-
аммониевым остатком. Представлен ряд анионных остатков по их способности экстраги-
экстрагироваться в виде солей тетрабутиламмония из воды в хлороформ:
СНзСООе < а© < С6Н5СООе < Вгв < NOf> < ie [Г.2.37]
Методически удобной формой использования подобной экстракции ионов
является проведение реакций нуклеофильного замещения с помощью межфаз-
межфазного катализа.
2.4.2. МЕЖФАЗНЫЙ КАТАЛИЗ
Межфазным катализом называют ускорение реакций в двухфазных системах
(например, вода — органический растворитель или соль щелочного металла —
органический растворитель), причем реагент находится в водной или твердой
фазе, и его перенос к субстрату через границу фаз осуществляется с помощью
так называемых катализаторов фазового переноса. В качестве таковых для
систем жидкость-жидкость используют липофильные четвертичные аммоние-
аммониевые соли с возможно более жестким анионом, таким как гидросульфат или хло-
хлорид, например гидросульфат тетрабутиламмония, аликват 336, адоген 464. Пос-
Последние два названия относятся к техническим продуктам, состоящим главным
образом из метилтридециламмонийхлорида. Для жидкостно-твердых систем
особенно пригодны краун-эфиры (разд. Г,2.2.2). Реакции нуклеофильного
замещения в условиях межфазного катализа проводятся чаще всего в системах
вода — дихлорметан, вода — хлороформ или вода — дихлорэтан.
Липофильная четвертичная аммониевая соль переносит анионный нуклео-
фил в виде ионной пары из водной среды в органическую, где и происходит ре-
реакция с RX. После этого ион аммония связывается с отщепившимся Х~, пере-
переносит его в водную фазу; цикл повторяется, так что достаточно каталитических
количеств соли аммония. Катализу описанного типа благоприятствует то, что
более жесткий Х~ из R-X по сравнению с нуклеофилом Nu~ сильнее гидрати-
руется. Поэтому в реакциях с переносом фаз алкилхлориды и алкилбромиды
применять выгоднее, чем алкилиодиды: иодид-анион, освобождающийся в
соответствии со схемой [Г.2.37], сильно уменьшает возврат катализатора в
водную фазу.
На схеме [Г.2.38] показана реакция азид-иона с алкилгалогенидом в услови-
условиях межфазного катализа. Ионная пара Q+N3~ вместе со своей гидратной оболоч-
оболочкой переносится в органическую фазу, так что нуклеофильность аниона
примерно отвечает таковой в водном растворе:
м ©м Sn®vS м ©w0 м ©v©
водная фаза Na , N3 , Q , X Na , X Na , X
' ^ J~~^ , ?-, [Г.2.38]
органическая фаза R-X R-X + [q®N30] R-N3, [q®X0]
2. Нуклеофильное замещение при насыщенном атоме углерода 281
Проведение реакций нуклеофильного замещения в двухфазной системе
жидкость—жидкость дает следующие преимущества по сравнению с реакциями
в гомогенных условиях:
— высокие скорости при низких температурах, т. е. в мягких условиях;
— удобство обработки реакционной смеси (продукт реакции находится
только в органической фазе;
— использование дешевых растворителей, которые к тому же не должны
быть безводными (см. также ниже).
Применение межфазных катализаторов типа четвертичных солей аммония
оказывается полезным и для промежуточного получения таких препаративно
важных нуклеофильных реагентов, как алкоголят-, тиолат- или карбанионы,
которые в форме солей не очень устойчивы и поэтому не предлагаются в качест-
качестве продажных препаратов. Обычно их приходится получать в безводных раство-
растворителях из соответствующих слабых кислот и сильных оснований (алкоголяты,
амид натрия и др.).
В двухфазной же системе, используя четвертичные аммониевые соли, можно
работать с водной щелочью. Четвертичные аммониевые соли влияют на равно-
равновесие депротонирования слабой кислоты благодаря тому, что после депротони-
рования уносят образующиеся липофильные анионы с поверхности раздела в
органическую фазу, где они и реагируют с электрофильным субстратом. На схе-
схеме [Г.2.39] подобный процесс показан на примере депротонирования хлорофор-
хлороформа 40%-ным раствором гидроксида калия с образованием трихлорметанид-ани-
она С13С-:
водная фаза К , ОН © НгО К , CI , НгО
С,зС-н' "* еСС,3 г 'el "" [Г.2.39]
органическая фаза [q©c| e] [Q©| [Q©CCI3e] — =CCI2 + [q@CIQ]
На поверхности раздела молекулы хлороформа отдают протон в водную фа-
фазу с образованием молекулы воды. Липофильный трихлорметанид-анион зак-
закрепляется в органической фазе на поверхности раздела, поскольку гидратиро-
ванный противоион калия К+ не может перейти из водной фазы в органическую.
Находящийся в органической фазе липофильный аммониевый ион (ионная па-
пара Q+X~), отдавая Х~ в водную фазу, может удалить трихлорметанид-анион с
границы раздела и в виде ионной пары перенести его в органическую фазу, где
он далее превращается в дихлоркарбен (см. также разд. Г,3 и Г,4).
В подобных простых и удобных условиях даже относительно слабокислые
соединения могут быть превращены в их нуклеофильные анионы и введены в
дальнейшие реакции (ср. также, например, табл. Г.7.306).
2.5. НУКЛЕОФИЛЬНОЕ ЗАМЕЩЕНИЕ В СПИРТАХ И ПРОСТЫХ ЭФИРАХ
Как было уже обосновано в разд. Г,2.2.1, нуклеофугный уход и нуклеофиль-
ная замена гидроксильной или алкоксильной группы возможны только после
предварительной активации введением электроноакцепторной группы. Легче
282 Г Препаративная часть
всего активацию можно осуществить присоединением протона к ОН группе1,
но и можно при этом использовать замещение атома Н в ОН-группе на
сульфонильную или фосфониевую группы (как при реакции Мицунобу, см.
[2.58]). По этой причине все реакции нуклеофильного замещения в спиртах
или простых эфирах катализируются кислотами.
Для спиртов важнейшей реакцией замещения является этерификация
сильными неорганическими кислотами:
,©
©
R-OH + Н~ ^=^ R-OH2
© @ [Г.2.40]
R-OH2 + НХ =^= R-X + Н2О + НГ
где НХ — галогеноводородная, серная, азотная или борная кислота.
Кислотный гидролиз алкилгалогенидов, алкилсульфатов и других эфиров опи-
описывается теми же уравнениями реакций в обратном направлении. Эти процессы
обсуждаются в разд. Г,2.6.1. Важнейшими побочными продуктами являются оле-
фины (вследствие элиминирования, см. разд. Г,3.1.4) и простые эфиры. Простой
эфир может образоваться в результате взаимодействия с находящимся в реакцион-
реакционной смеси спиртом, который также может выступать как нуклеофильный агент:
R-OH2 + R-OH т^р R-6; — R-O-R [Г.2.41]
Повышение температуры благоприятствует и образованию эфира, и элими-
элиминированию. Избыток спирта способствует образованию эфира, избыток кисло-
кислоты — этерификации. Степень эфирообразования зависит, кроме того, от струк-
структуры спирта: тенденция к образованию симметричного простого эфира у
третичных спиртов меньше из-за пространственных затруднений.
Реакция образования простых эфиров также обратима, и в общем очень
инертные простые эфиры можно расщепить в присутствии лишь сильных
кислот. При этом эфир сначала протонируется, а во второй стадии происходит
нуклеофильное замещение анионом кислоты:
R-6' + НХ ===== R-X + ROH + Н® (Sn1iuimSn2) [Г.2.42]
н
Этот процесс совершенно аналогичен этерификации спиртов неорганичес-
неорганическими кислотами (уравнение [Г.2.40]).
2.5.1. ЗАМЕЩЕНИЕ ГИДРОКСИЛЬНОЙ ГРУППЫ В СПИРТАХ
НА ОСТАТОК НЕОРГАНИЧЕСКОЙ КИСЛОТЫ
Наиболее простой метод образования алкилгалогенидов из спиртов — взаимо-
взаимодействие с галогеноводородными кислотами:
ROH + НХ -У— R-X + Н2О [Г.2.43]
Ониевые соли могут образовываться также с кислотами Льюиса (ZnCl2; BF3), например:
R4_ R4© e
О + BF3 - O-BF3
Н Н
2. Нуклеофильное замещение при насыщенном атоме углерода 283
Реакционная способность галогеноводородных кислот понижается в ряду
HI > НВг > НС1 > HF (уменьшаются сила кислоты и нуклеофильность анио-
анионов; ср. ряды [Г.2.26]).
Реакции с иодоводородной и бромоводородной кислотами осуществляются
в большинстве случаев легко, тогда как хлороводородная кислота уже настоль-
настолько малоактивна, что ею без трудностей этерифицируются только наиболее
реакционноспособные спирты (третичные и бензиловые). В других случаях
необходимо создать по возможности более высокую концентрацию хлороводо-
рода путем насыщения спирта газообразным НС1 и даже проводить реакцию в
запаянной ампуле при повышенной температуре. Прибавление безводного
хлорида цинка повышает реакционную способность как спирта, так и соляной
кислоты, однако одновременно в значительной степени усиливает и побочные
реакции (перегруппировку, изомеризацию, в особенности при реакциях по
SnI-механизму).
При замещении ОН-группы спиртов на фтор хорошие результаты получены
с растворами фтороводорода в пиридине. При этом катионные полимеризация
и перегруппировки протекают лишь в незначительной степени.
Реакционная способность спиртов понижается с увеличением длины угле-
углеродной цепи. Скорость этерификации возрастает при переходе от первичных
спиртов к третичным. Первичные спирты, как правило, реагируют с галогеново-
дородными кислотами по бимолекулярному механизму, давая алкилгалогени-
ды, третичные — по мономолекулярному механизму, вторичные — по погра-
пограничному.
Поскольку при этерификации спиртов неорганическими кислотами речь
идет о типично равновесной реакции, то из закона действующих масс вытекают
следующие возможности оптимизации выхода: а) увеличение концентрации
одного из двух реагентов; б) удаление продуктов реакции.
Образующуюся при этерификации воду можно удалять из реакционной
смеси с помощью водоотнимающих средств (например, добавляя конце-
концентрированную серную кислоту) или иногда отгонкой в виде азеотропной смеси
(разд. А,2.3.5). Не рекомендуется применять серную кислоту в виде водоот-
нимающего средства при этерификации вторичных и третичных спиртов, так
как при этом могут легко образоваться олефины. По той же причине при
этерификации этих спиртов работают при возможно более низкой темпера-
температуре.
В случае низших алкилгалогенидов часто можно отгонять образующийся
эфир, поскольку он обладает более низкой температурой кипения, чем спирт
(почему?). Иногда эфир может быть удален из реакционной среды экстракцией
(экстрактивная этерификация, см. разд. Г,7.1.4.1).
Иодоводород может действовать на образовавшийся алкилиодид как восста-
восстановитель, причем в результате получают углеводород (уравнение [Г. 1.25]). Эта
реакция особенно легко протекает для третичных алкилиодидов, поэтому для их
получения лучше исходить из спирта, иода и красного фосфора (табл. Г.2.52)
или же использовать замещение по Финкельштейну (разд. Г,2.6.7).
284
Г Препаративная часть
Если структура спирта обусловливает протекание реакции главным образом по Sn 1 -механизму,
то наряду с образованием олефина особенно следует ожидать перегруппировок в качестве побоч-
побочных реакций. Даже при этерификации вторичных спиртов существует такая опасность. Так, спирт,
содержащий ОН-гругшу у 2-го атома углерода, превращается в 2-галогенид. Для разветвленных в
а-положении первичных и вторичных спиртов перегруппировка становится даже основной реак-
реакцией, так что образуются третичные алкилгалогениды (см. схему [Г.2.6] и разд. Г,9), например:
Н3С-С-СН2ОН
H3c'
+ н@, -н2о_
НзС~~С~~СН2
н3с/
с-сн2-сн3
Н3С
[Г.2.44]
Вг-С-СН2-СН3
СН3
В этих случаях галогениды лучше всего получать, используя РХз в пиридине или SOCb в
пиридине или же через соответствующие тозилаты (разд. Г,2.6.7).
Ф Общая методика этерификации спиртов бромоводородной кислотой (табл. Г.2.45). К 1 моль
соответствующего первичного спирта добавляют при охлаждении 0,5 моль концентриро-
концентрированной серной кислоты, а затем 1,5 моль 48%-ной свежеперегнанной бромоводородной
кислоты и доводят смесь до кипения. Этерификацию вторичных и третичных спиртов осущест-
осуществляют без прибавления серной кислоты, чтобы ограничить образование олефинов.
Способ А. Низкокипящие алкилбромиды отгоняют непосредственно из реакционной смеси
B0-см колонка Вигре, нисходящий холодильник, скорость перегонки 2—3 капли/с).
Способ Б. Для получения труднолетучих алкилбромидов реакционную смесь кипятят 6 ч с
обратным холодильником. Затем перегоняют с водяным паром и отделяют алкилбромид с
помощью делительной воронки.
Таблица Г.2.45. Этерификация спиртов бромоводородом
Продукт реакции
1-Бромэтан
1-Бромпропан
2-Бромпропан
1-Бромпропен-2
(аллилбромид)
1-Бромбутан
2-Бромбутан
1 -Бром-2-метилпро-
пан (изобутилбромид)
трег-Бутилбромид
1 -Бромпентан
1-Бромгексан
Бромциклогексан
1 -Бромгептан
1 -Бромоктан
1-Бромдекан
1-Бромдодекан
2-Фенил-1-бромэтан
1,3-Дибромпропан
1,4-Дибромбутан
Т. кип., 'С
(мм рт. ст.)
38
71
59
70
100
91
92
73
129
154
164
59 A0)
93 B2)
118 A6)
148 A6)
98 A4)
167
98 A2)
"D26
1,4239
1,4341
1,4251
1,4689
1,4398
1,435
1,437
1,4283
1,4446
1,4478
1,4956
1,4506
1,4526
1,4559
1,4581
1,556
1,5233
1,5175
1
1
1
1
dtto
,4586
,3539
,425
,432
,2829
,2556
,256
,2220
,219
,175
,140
,П2
1,0683
1,0382
1,359
1,9822
1,8080
Выход,
%
90
80
80
80
80
80
80
60
80
80
65
80
80
90
90
70
80
80
Способ
обработки
А
А
А
А
Б
А
А
А
Б
Б
Б
Б
Б
Б
Б
Б
Б
Б
Примечания
Приемник ох-
охлаждают ледя-
ледяной Н2О
Без H2SO4
Без H2SO4
Без H2SO4
Без H2SO4
Без H2SO4
2. Нуклеофильное замещение при насыщенном атоме углерода 285
Очистка продуктов реакции, полученных способами Аи Б. Сырой продукт два раза осторожно
встряхивают (опасность образования эмульсий!) с концентрированной серной кислотой (объем
кислоты должен быть в 5 раз меньше объема продукта реакции) или с равным объемом концент-
концентрированной соляной кислоты, чтобы удалить образовавшийся в качестве побочного продукта
простой эфир. Промывают сырой бромид водой, а если очищаемый алкилбромид кипит при
температуре выше 100 °С, то двумя порциями по 75 мл 40%-ного водного метанола. Затем про-
продолжают промывать раствором гидрокарбоната натрия для нейтрализации остатков кислоты и
еще несколько раз водой, сушат хлоридом кальция и фракционируют на 20-см колонке Вигре.
Щ Внимание! Проводя очистку с помощью делительной воронки, всегда следует
Ц проверить, в каком слое находится алкилбромид (разд. А.2.5.2.1).
Методика пригодна для синтеза в полумикроколичествах.
Алкшшориды могут быть получены принципиально таким же путем (при этом
на 1 моль спирта добавляют 2 моль концентрированной соляной кислоты и
2 моль безводного хлорида цинка) [Vogel A. I. J. Chem. Soc. [London], 1943, 636].
Получение трет-бутилхлорида см. Норрис Дж., Олмстед А. В сб.: Синтезы
органических препататов. Сб. 1. — М.: ИЛ, 1949, с. 482.
В промышленности метил- и этилхлориды получают этерификацией из соответствующих
спиртов (метанола и этанола) действием хлороводорода. Другие важнейшие методики получе-
получения и данные о применении этих веществ см. разд. Г,1.4.1,табл. ГЛ.37.
Алкилгалогениды, кроме того, можно получать из спиртов и неорганических
галогенидов, таких как три- или пентахлорид фосфора и тионилхлорид:
3 ROH + РХ3 3 RX + Н3РО3 [Г.2.46]
ROH + РХ5 RX + НХ + РОХ3 [Г.2.47]
ROH + SOCI2 RCI + HCI + SO2 [Г.2.48]
Реакции протекают по сложным механизмам. В качестве промежуточного
продукта всегда образуются сложные эфиры соответствующей неорганической
кислоты, например, РС13 превращается в эфир фосфористой кислоты:
ROH + РС13 -^р ROPCI2 4^Г (R0JPCI ^if* (П0)зР [Г149]
Сложные эфиры далее реагируют с образовавшимся галогеноводородом:
H-CI + R-OPCI2 R-CI + НОРС12 [Г.2.50]
Если такое замещение протекает по механизму Sn2, а реакционный центр
хирален, то наблюдается инверсия конфигурации (вальденовское обращение).
При реакциях спиртов с тригалогенидом фосфора можно для связывания
кислоты прибавлять такие вещества, как пиридин; однако их не следует приме-
применять в эквимолярных количествах, так как при этом образуются эфиры фосфо-
фосфористой кислоты (схема [Г.2.49]), а не алкилгалогениды.
При реакциях спиртов с тионилхлоридом первоначально образуются эфиры
сернистой кислоты (схема [Г.2.51]), которые далее могут реагировать двумя
286
Г Препаративная часть
путями. В присутствии пиридина такой эфир атакуется хлорид-ионом по
SN2-MexaHH3My, причем с обращением конфигурации образуется RC1. В отсут-
отсутствие пиридина образующийся сложный эфир вступает в реакцию внутреннего
нуклеофильного замещения (SnI), давая алкилхлорид с конфиругацией исходного
спирта (сохранение конфигурации):
CI
I
-HCI
[Г.2.51]
При взаимодействии тионилхлорида со спиртами обычно для связывания кислоты добавля-
добавляют пиридин или другие третичные амины. Таким образом реакцию удается проводить при низ-
низких температурах. Оксихлорид фосфора дает, как правило, только соответствующие эфиры фос-
фосфорной кислоты и поэтому этот реактив малопригоден для синтеза алкилгалогенидов. В РСЬ
можно использовать лишь один атом хлора.
Получение алкилгалогенидов с помощью указанных хлорангидридов неор-
неорганических кислот в случае сильноразветвленных первичных, а также вторич-
вторичных и третичных спиртов предпочтительнее, чем непосредственная этерифика-
ция галогеноводородными кислотами. Прежде всего при низких температурах
образуется меньше олефинов и продуктов перегруппировок.
Галогенангидриды неорганических кислот обычно применяют в избытке.
Необходимо обращать внимание на то, чтобы продукт реакции можно было вы-
выделить перегонкой.
Трибромид и трииодид фосфора можно получать во время реакции взаимо-
взаимодействием красного фосфора с соответствующими галогенами. Этот метод осо-
особенно удобно применять при получении алкилиодидов, поскольку исключается
образование иодоводорода, который может действовать на алкилиодиды как
восстановитель (уравнение [ГЛ.25]).
Ф Общая методика получения алкилиодидов из спирта, иода и красного фосфора (табл.
Г.2.52). Прибор для реакции (рис. А.87) состоит из круглодонной колбы, снабженной
проточным экстрактором и обратным холодильником. В насадку помещают пористый
вкладыш, в котором находится 0,5 моль иода. В круглодонную колбу прибавляют 1 моль соот-
соответствующего спирта (абсолютного!) и 0,33 моль красного фосфора. Затем нагревают до кипе-
кипения. Возвращаемый холодильником спирт растворяет иод; температуру бани регулируют так,
чтобы держать реакцию под контролем. Выделяющегося при реакции тепла иногда бывает
достаточно для циркуляции спирта. По окончании реакции смесь подвергают обработке.
Таблица Г.2.52. Алкилиодиды из спирта, иода и красного фосфора
Продукт реакции
Метилиодид
Этилиодид
1 -Иодпропан
2-Иодпропан
1 -Иодбутан
1-Иодгексан
Иодциклогексан
2-Иодоктан
Т. кип., 'С
(мм рт. ст.)
42,5
72
102
89
130
60 A3)
82 B0)
92 A2)
,5320
,5140
,5050
,4996
,5006
,4926
,5475
1,4888
Выход, %
80
80
80
80
80
80
80
90
Способ
обработки
А
В или А
В
А
В или Б
Б или В
В или Б
Б или В
2. Нуклеофильное замещение при насыщенном атоме углерода 287
Способ А. Если продукт реакции кипит при температуре ниже 100 °С, то его прямо отгоняют,
промывают небольшим количеством воды, сушат сульфатом магния и перегоняют.
Способ Б. В случае высших алкилиодидов охлажденную реакционную смесь разбавляют водой,
отделяют органический слой, а водную фазу экстрагируют эфиром. Органический слой и эфирные
вытяжки объединяют, сушат сульфатом натрия, отгоняют эфир, а остаток фракционируют.
Способ В. В большинстве случаев рекомендуется перегонка продукта реакции с водяным
паром. Полученный отгон извлекают эфиром, сушат и фракцирнируют.
Для проведения синтеза с полумикроколичествами реакцию проводят в круглодонной колбе
с обратным холодильником.
Получение пентилхлорида из пентилового спирта и тионилхлорида в присут-
присутствии пиридина: Whitemore F. С, Karnatz F. A., Popkin A. H. J. Am. Chem. Soc,
1938, 60, 2540.
В лабораториях этерификация других неорганических кислот проводится реже. Однако в
промышленности большое значение имеют эфиры серной и азотной кислот:
R-OH + HO-SO2-OH - RO-SO2-OH + Н2О [Г.2.53]
Натриевые соли высших алкилгидросульфатов (часто неправильно называемые сульфоната-
ми жирных спиртов) являются важными моющими средствами.
Из этилового спирта через этилгидросульфат в зависимости от условий реакции можно
получить диэтиловый эфир или этилен (разд. Г,2.5.2 и Г,3.1.4).
Из метилгидросульфата («метилсерной кислоты») при нагревании получают важный мети-
метилирующий агент диметилсульфат:
2CH3OSO2OH > (CH3OJSO2 + H2SO4 [Г.2.54]
Согласно другому методу, диметилсульфат получают из диметилового эфира
(табл. Г.2.61) и серного ангидрида:
СН3ОСН3 + SO3 (CH3OJSO2 [Г.2.55]
Сложные эфиры азотной кислоты с полигидроксисоединениями являются важными
взрывчатыми веществами: тринитрат глицерина (нитроглицерин), динитрат гликоля (нит-
рогликоль), динитрат дигликоля, динитрат целлюлозы (коллодий), тринитрат целлюлозы
(пироксилин) и тетранитрат пентаэритрита (нитропента). Динитрат целлюлозы применяет-
применяется, кроме того, как пластмасса (целлулоид) и как сырье для получения лака (нитролак).
Эфиры борной кислоты также можно получить непосредственной этерификацией бор-
борной кислоты или борного ангидрида. Такие эфиры представляют собой кислоты Льюиса, и
поэтому они взаимодействуют со следующей молекулой спирта, давая комплексное соедине-
соединение. Раствор образующейся при этом одноосновной кислоты проводит электрический ток
лучше, чем сама борная кислота. Этот факт используется для установления относительного
положения ОН-групп в циклических диолах-1,2 (например, в сахарах), а именно для иденти-
идентификации цис- и транс-изомеров, поскольку образование эфира стерически вожможно лишь
в первом случае:
V .ОН но НО /
-с. +НП'В"ОН+ .с-
/ "он но но' \
\
—с
1
—с
-Оч
ЧЕ
-о"
О
1
О
-с—
i -
-С —
\
С \§/
| В
-с—
1
©
н
[Г.2.56]
Другая возможность увеличения способности спирта для взаимодействия с
кислотами НХ состоит в его предварительном превращении в алкоксифосфони-
евую соль, которая может образовываться при реакции с трифенилфосфином и
азодикарбоновым эфиром (реакция Мицунобу):
+ EtOOC-N=N-COOEt + Ph3P rncTi
R0H + HX -EtOOC-NHNH-COOEt-Ph3P=O RX [Г2-5?]
288 Г Препаративная часть
Сначала из трифенилфосфина, диэтилового эфира азодикарбоновой
кислоты и НХ образуется гидразофосфониевая соль, которая взаимодействует
со спиртом с отщеплением диэтилового эфира гидразодикарбоновой кислоты и
образованием алкоксифосфониевой соли. Последняя затем реагирует с нуклео-
филом Х~ с образованием целевого RX и отщеплением трифенилфосфиноксида
как хорошо уходящей группы:
COOEt Ph3R® COOEt
PhsPI^ ~^N^M + H-^X N-n' + IXG
ЕЮОС' "-- -^ ЕЮОС H
+ ROH
-EtOOC-NHNH-COOEt
Ph3P = O + R-X
По атому углерода, который содержит гидроксильную фуппу, происходит
инверсия (инверсия Мицунобу). Нуклеофил атакует, как при вальденовском
обращении, уходящую группу с противоположной стороны (ср. [Г.2.9]).
По реакции Мицунобу можно также с помощью спирта алкилировать такие
слабокислые соединения НХ, как азотистоводородную и карбоновые кислоты,
фенолы, имиды, тиолы, тиоамиды и р-дикарбонильные соединения. Напишите
эти реакции!
цис-1-Азидо-2-хлорциклогексан из /и/>анс-2-хлорциклогексанола: Leubner H.;
ZbiralE., Helv. Chim. Acta, 1976, 59, 2100.
B5)-(+)-Октантиол из тиогликолевой кислоты: Volante R. P.; Tetrahedron
Lett. 1981,22,3119.
Реакцию Мицунобу применяли к редокс-конденсации по Мукайама. В этом случае полу-
получают фосфониевую соль (Ph3PY+ ; X") из трифенилфосфина и соединения, в котором присут-
присутствует связь гетероатом-гетероатом или гетероатом-углерод (X—Y = Hal-Hal, Hal-CHal3,
PhS—SPh, RSe-CN и т. д.). Фосфониевый ион реагирует со спиртом с выбросом нуклеофила Х~,
который затем взаимодействует с алкоксифосфониевым ионом с образованием RX и отщепле-
отщеплением трифенилфосфиноксида:
© О +ROH © о гг 1 «П1
Ph3P + Y—X Ph3P-Y + X —ткг~ Ph3P-OR + Xй - Ph3PO + RX [Г.2.59]
— HY
Напишите реакции приведенных соединений X—Yco спиртами R—ОН!
2.5.2. ОБРАЗОВАНИЕ ПРОСТЫХ ЭФИРОВ ИЗ СПИРТОВ С УЧАСТИЕМ КИСЛОТ.
РАСЩЕПЛЕНИЕ ПРОСТЫХ ЭФИРОВ
Получение простых эфиров по схеме [Г.2.41] из спиртов в присутствии сильных
кислот редко применяется в лаборатории; это главным образом нежелательная
побочная реакция. В промышленности, однако, этот метод используется в
широких масштабах, в частности для получения диэтилового эфира из этанола,
тетрагидрофурана из бутандиола-1,4 и диоксана из этиленгликоля.
Одним из вариантов этого метода является получение простых эфиров в
газовой фазе (каталитическая дегидратация на оксиде алюминия, сульфате
алюминия).
Получение простых эфиров из спиртов с участием кислот можно проводить
в две стадии; при этом сначала из спирта и серной кислоты получают алкилсер-
2. Нуклеофильное замещение при насыщенном атоме углерода
289
ную кислоту (Г.2.53), которая затем с новой порцией спирта при повышенной
температуре превращается в простой эфир:
RO-SO2-OH + HOR
ROR + H2SO4
[Г.2.60]
Поскольку алкилсерная кислота может быть получена и путем присоедине-
присоединения серной кислоты к олефину (уравнение [Г.4.16а]), простые эфиры можно по-
получать из олефинов и серной кислоты.
По той же причине в результате любой катализируемой кислотами реакции
присоединения воды к олефинам в качестве побочного продукта образуется
простой эфир. Некоторые технически важные простые эфиры перечислены в
табл. Г.2.61.
В отличие от редкого в лабораторной практике получения простых эфиров из
спиртов в присутствии сильных кислот расщепление этих эфиров сильными
кислотами нашло широкое применение прежде всего в аналитических целях.
Алифатические простые эфиры лучше всего расщеплять иодоводородной
кислотой свежеперегнанной (высокая реакционная способность иодоводорода,
более легкое выделение низших алкилиодидов по сравнению с бромидами).
Алкилароматические простые эфиры также можно расщеплять иодоводо-
иодоводородной кислотой. Однако при этом происходят побочные процессы (например,
иодирование ароматического ядра). Диариловые эфиры обычно не расщепля-
расщепляются иодоводородной кислотой; для их идентификации используют замещение
в ароматическое ядро (сульфохлорирование; см. разд. Г,5.1.4).
Вместо дорогой иодоводородной кислоты для расщепления простых эфиров
можно применять раствор A:1) 48%-ной бромоводородной кислоты в ледяной
уксусной кислоте. Поскольку низшие алкилбромиды легколетучи, этот способ
Таблица Г.2.61. Технически важные простые эфиры и их применение
Простой эфир
Диметиловый эфир3
Диэтиловый эфир
Диизопропиловый эфир6
Метил-трет-бутиловый эфир (МТБЭ)
6ис-2-Хлорэтиловый эфир
Тетрагидрофуран
1,4-Диоксан
Применение
Метилирующий агент;
-> диметилсульфат
Растворитель; например, смесь со спиртом — раст-
растворитель коллодия (целлулоида); очень распрост-
распространенный растворитель в лаборатории
Высокооктановое моторное топливо; растворитель
Тоже
Растворитель для этилцеллюлозы, смол и жиров;
-> Тиопласт
Растворитель для полимеров;
-> политетраметиленгликоль -> полиуретан;
-> 1,4-дихлорбутан (уравнение [Г.2.62])
Растворитель
а Побочный продукт при синтезе метанола из монооксида углерода.
6 Побочный продукт при синтезе изопропанола из пропилена и серной кислоты (табл. Г.4.20).
290 Г Препаративная часть
пригоден только для высших простых эфиров, а также для простых эфиров
фенола с низшими алкильными остатками, если нет необходимости определять
строение алкильного остатка спирта.
Расщепление простых эфиров (общая методика для качественного анализа).
Способ А. Симметричный1 алифатический эфир кипятят 3-4 ч с обратным холодильником
приблизительно с пятикратным по объему количеством свежеперегнанной иодоводородной
кислоты. Затем прибавляют четырехкратный объем воды и отгоняют алкилиодид с водяным
паром; органическую фазу извлекают небольшими порциями эфира, сушат и идентифициру-
идентифицируют алкилиодид в виде S-алкилтиоурониевой соли (разд. Г,2.6.6).
Способ Б. Кипятят с обратным холодильником 1 ч 0,5 г простого эфира фенола с 5 мл сме-
смеси A : 1) ледяной уксусной кислоты и 48%-ной бромоводородной кислоты. Затем выливают
реакционную смесь в 20 мл воды, доводя до слабощелочной реакции при помощи едкого нат-
натра и извлекают эфиром непрореагировавший эфир фенола и, возможно, присутствующий еще
алкилбромид. После подкисления разбавленной серной кислотой извлекают эфиром фенол и
идентифицируют его в виде подходящего производного (разд. Д,2.5.3).
Примеры расщепления простых эфиров в препаративных целях:
Получение 1,4-дихлорбутана из тетрагидрофурана: Fried S., Kleene R. D.
J. Am. Chem. Soc, 1941, 63, 2691; Reppe W. Lieb. Ann., 1955, 596,90, 118.
Получение 1,4-дибромбутана из тетрагидрофурана: Fried S., Kleene R. D.
J. Am. Chem. Soc, 1940, 62, 3258.
Расщепление простых эфиров находит применение в количественном анализе для опреде-
определения метоксигрупп. Образующийся при действии иодоводородной кислоты метилиодид от-
отгоняется и затем определяется титрованием.
В промышленности расщепление простых эфиров применяют, например, для получения
1,4-дихлорбутана из тетрагидрофурана и хлороводорода:
[Г.2.62]
1,4-Дихлорбутан служит исходным продуктом для получения найлона (уравнение
[Г.2.111]).
2.6. НУКЛЕОФИЛЬНОЕ ЗАМЕЩЕНИЕ В АЛКИЛГАЛОГЕНИДАХ,
АЛКИЛСУЛЬФАТАХ И АЛКИЛСУЛЬФОНАТАХ
2.6.1. ГИДРОЛИЗ
При взаимодействии с водой алкилгалогениды дают спирты и галогеноводород-
ные кислоты (реакция, обратная образованию алкилгалогенидов):
RX + НОН - ROH + НХ [Г.2.63]
Однако вода представляет собой слабонуклеофильный реагент. Поэтому
только очень реакционноспособные алкилгалогениды легко могут быть гидро-
лизованы водой (см. ниже получение тритилового спирта).
1 Аналогично можно идентифицировать несимметричные простые эфиры, если смесь об-
образующихся алкилгалогенидов может быть разделена перегонкой или идентифицирована с
помощью ГЖХ.
2. Нуклеофильное замещение при насыщенном атоме углерода 291
Недостаточные электронодонорные свойства воды можно компенсировать
путем поляризации связи С—Hal, например, при помощи кислот Льюиса (хлор-
(хлорное железо и т. д.):
FeCI3 - ^Q-R + X-FeCI30 HOR + HX + FeCI3 [Г.2.64]
H
Гидролиз алкилгалогенидов можно также ускорить, добавляя щелочь. Нук-
леофильность или основность гидроксид-иона гораздо выше, чем молекулы
воды. Кроме того, в щелочной среде одновременно смещается положение рав-
равновесия в сторону продуктов гидролиза, так как обратная реакция невозможна.
Алкилгалогениды нерастворимы в воде. Поэтому гидролиз может происхо-
происходить только на поверхности раздела фаз. Чтобы получить гомогенную смесь,
часто в качестве растворителя добавляют спирт или работают в двухфазной
системе в условиях межфазного катализа.
Спирт, образующийся при гидролизе, а также прибавляемый для гомогени-
гомогенизации, делает возможным протекание некоторых побочных реакций. Он нахо-
находится в равновесии (которое, впрочем, сильно сдвинуто влево) с гидроксид-ио-
нами, так что образуются небольшие количества алкоголята, который реагирует
с алкилгалогенидом, образуя простой эфир (эту реакцию можно превратить в
главную; см. синтез Вильямсона, разд. Г,2.6.2):
R-O-H + еОН ^=^ R-OG + Н2О [Г.2.65а]
R-OQ + R-X R-O-R + X0 [Г.2.656]
Кроме образования простых эфиров — нежелательной побочной реакции —
сильные основания часто приводят к элиминированию галогеноводородов, в
результате чего образуются олефины или ацетилены (разд. Г,3).
Объясните образование простых эфиров при кислотном гидролизе алкилга-
алкилгалогенидов, алкилсульфатов и т. д.
Указанных побочных реакций можно избежать, если проводить омыление
алкилгалогенидов водой в присутствии влажного оксида («гидроксида») сереб-
серебра. Реакция протекает на поверхности твердого «гидроксида» серебра.
Ф Получение трифенилкарбинола (тритилового спирта). Трифенилхлорметан нагревают в
водной суспензии в течение 10 мин. После охлаждения отсасывают образовавшийся
трифенилкарбинол и перекристаллизовывают (из тетрахлоруглерода или спирта). Выход
;т. пл. 162 °С.
Геминальные ди- и тригалогениды также можно гидролизовать в кислой или
щелочной среде. Альдегиды образуются при гидролизе 1,1-дигалогенидов, кото-
которые получают действием эфиров галогеноводородных кислот на гидраты альде-
альдегидов:
ТР5Г R-c^ [Г.2.66]
Тригалогениды в качестве продуктов гидролиза дают карбоновые кислоты.
В случае ароматических соединений, содержащих трихлорметильную группу,
292
Г Препаративная часть
реакцию можно остановить на стадии хлорангидрида кислоты:
CI он о
+ н2о
CI
CI
[Г.2.67]
Для гидролиза геминальныхдигалогенидов нельзя применять сильные осно-
основания, так как образующиеся альдегиды чувствительны к щелочам. Поэтому
гидролиз проводят в присутствии карбоната кальция, формиата натрия или ок-
салата калия. Для двух таких случаев ниже приведены описанные в литературе
методики.
Бензилидендихлориды и бензилидендибромиды во многих случаях очень
гладко гидролизуются при обработке концентрированной серной кислотой в со-
соответствующие бензальдегиды. Электронодонорные группы в ядре (например,
гидроксильные группы) облегчают гидролиз, электроноакцепторные — затруд-
затрудняют его (почему?). В последнем случае надо повысить температуру реакции
(однако не выше 110 "С, так как при температуре более 90 °С образующиеся
альдегиды частично уже заметно окисляются серной кислотой).
Ф Общая методика гидролиза бензилидендигалогенидов в концентрированной серной кислоте
(табл. Г.2.68)
S Внимание] Бензилиденгалогениды оказывают сильное раздражающее действие
на слизистую оболочку и кожу. Работать под тягой в защитных перчатках!
Таблица Г.2.68. Получение альдегидов путем гидролиза бензилидендигалогени-
бензилидендигалогенидов концентрированной серной кислотой
Альдегид
Бензальдегид
я-Хлорбензальде-
гид
о-Хлорбензальде-
гид
2,4-Дихлорбен-
зальдегид
л-Нитробензальде-
гид
Терефталевый аль-
альдегид
Бензилидендигалогенид
Бензилидендихлорид
или бензилидендибро-
мид
л-Хлорбензилиден-
дихлорид или л-хлор-
бензилидендибро-
мид
о-Хлорбензилиден-
дихлорид
2,4-Дихлорбензили-
дендихлорид или
2,4-дихлорбензили-
дендибромид
л-Нитробензилиденди-
бромид
1,4-Бис(дибромме-
тил)бензол
Темпе-
Температура
реак-
реакции, "С
0
20
20
90а
90а
90а
Физические константы
Т. кип. 64 °С
A3 мм рт. ст);
лп20 1,5446
Т. кип. 111 °С
B0 мм рт. ст.);
т. пл. 48 "С (лигроин)
Т. кип. 84 °С
A0 мм рт. ст.);
лвм 1,5670
Т. пл. 71 °С (лигроин)
Т. пл. 196 "С
(эфир+петр. эфирN
Т. пл. 115вС
(вода + метанол; 9 : 1)
Выход,
%
65
70
70
80
85
80
Реакцию можно проводить также при 110°С; в этом случае она заканчивается в
течение нескольких минут.
" Для очистки можно также использовать перегонку с водяным паром.
2. Нуклеофильное замещение при насыщенном атоме углерода 293
Соответствующий бензилидендихлорид или бензилидендибромид обрабатывают при пе-
перемешивании восьмикратным (по массе) количеством концентрированной серной кислоты.
Реакцию проводят в трехгорлой колбе, снабженной эффективной мешалкой, обратным холо-
холодильником и длинным капилляром, который служит газовводной трубкой. Через капилляр
пропускают поток азота, и одновременно при помощи водоструйного насоса, соединенного с
верхним концом холодильника, создают пониженное давление. В случае реакционноспособ-
ных бензилидендигалогенидов уже при О °С начинается сильное выделение галогеноводорода.
Более инертные бензилиденгалогениды нагревают на водяной или гликолевой бане до указан-
указанных в таблице температур. Во всех случаях реакционная смесь интенсивно окрашена в красно-
коричневый цвет.
Когда выделение галогеноводорода прекратится (в указанных условиях протекание реак-
реакции требует от 45 мин до 2 ч), реакционную смесь выливают на лед и образовавшийся альде-
альдегид три раза извлекают эфиром. Эфирный экстракт обрабатывают раствором гидрокарбоната
натрия, затем промывают водой и сушат сульфатом магния. После испарения эфира альдегид
перегоняют в вакууме; если же альдегид твердоплавок, то его перекристаллизовывают. При
подкислении раствора гидрокарбоната можно получить кислоту (побочный продукт). Она
появляется из бензотригалогенида, который присутствует в виде примеси в недостаточно
очищенном бензилидендигалогениде или в результате окисления образующегося альдегида
концентрированной серной кислотой либо воздухом.
С полумикроколичествами работают при атмосферном давлении в открытой колбе Эрлен-
мейера. От мешалки можно отказаться, так как реакционная смесь достаточно энергично
перемешивается потоком газа.
м-Фталевый и о-фталевый альдегиды получают гидролизом бис(дибромме-
тил)бензола в водно-спиртовом растворе в присутствии оксалата калия: Thiele J.,
Gtinther О. Lieb. Ann., 1906, 347, 106.
м-Гидроксибензальдегид получают гидролизом .м-ацетоксибензилидендибро-
мида [3-(дибромметил)фенилацетата] в водно-спиртовом растворе в присут-
присутствии формиата натрия: Eliel E. L., Nelson К. W. J. Chem. Soc, 1955, 1628.
В некоторых случаях для гидролиза алкилгалогенидов целесообразно
использовать обходной путь через сложные эфиры карбоновых кислот (разд.
Г.2.6.3):
R-X + eO-CO-R' R-O-CO-R1 + XQ
[Г.2.69]
R'COOR + H2O - R'COOH + ROH
При взаимодействии алкилгалогенида с анионом кислоты не происходит,
как правило, образования олефина, так как, хотя анион кислоты и обладает
высокой нуклеофильностью (реакционной способностью к углероду в алкилга-
логениде), он имеет сравнительно низкую основность (реакционную способ-
способность к протону).
Подобно алкилгалогенидам можно гидролизовать и эфиры других неоргани-
неорганических кислот.
Гидролиз алкилхлоридов и алкилсульфатов является важным промышленным методом
синтеза спиртов. Исходные алкилхлориды получают либо путем хлорирования углеводородов
(разд. Г,1), либо путем присоединения хлора или хлорноватистой кислоты к олефинам (разд.
Г,4). Алкилгидросульфаты получают путем присоединения серной кислоты к олефинам (разд.
Г,4). Таким путем в промышленности в больших масштабах получают амиловый и аллиловый
спирты (табл. Г. 1.37), этиленгликоль, глицерин (табл. Г.4.26), этиловый, изопропиловый и
бутиловые спирты (табл. Г.4.20).
294 Г Препаративная часть
2.6.2. СИНТЕЗ ПРОСТЫХ ЭФИРОВ ИЗ АЛКОГОЛЯТОВ ИЛИ ФЕНОЛЯТОВ
Простые эфиры образуются при взаимодействии алкилгалогенидов, диалкил-
сульфатов, эфиров толуолсульфоновых кислот и т. д. с алкоголятами или фено-
фенолятами:
R-O® + R'-X R-O-R1 + Xе [Г.2.70]
Эта реакция была уже описана как побочный процесс при щелочном гидро-
гидролизе алкилгалогенидов в присутствии спирта (реакция [Г.2.65]).
Поскольку фенолы обладают относительно высокой кислотностью, образо-
образование их натриевых солей происходит уже в водном растворе едкого натра, тог-
тогда как равновесие образования алкоголята в этом случае сильно сдвинуты в сто-
сторону свободного спирта (Г.2.65а). (Почему фенолы гораздо более сильные
кислоты, чем спирты?)
При получении метиловых эфиров обычно используют более реакционнос-
пособный и дешевый диметилсульфат и лишь редко легко летучий и дорогой ме-
тилиодид. В обычных условиях реакции (водный раствор, низкая температура)
используется только одна алкильная группа диметилсульфата для алкилирова-
ния, например:
0ю
f~ ]hO-R + WIQ-SO2-OR [Г.2.71]
101
Для получения простых этиловых эфиров можно аналогично использовать
диэтилсульфат. Для получения простых эфиров высших спиртов наиболее удоб-
удобными реагентами являются алкилбромиды и алкилиодиды.
Ф Общая методика получения простых эфиров фенола при метилировании диметилсульфатом
(табл. Г.2.72).
Щ Внимание] Диметилсульфат — сильный яд! Он способствует заболеванию
Щ раком. Работать под тягой только в резиновых перчатках!
Прибор состоит из трехгорлой колбы с обратным холодильником, мешалкой, термомет-
термометром и капельной воронкой. Соответствующий фенол быстро обрабатывают щелочью, добав-
добавляя при энергичном помешивании 1,25 моль 10%-ного раствора едкого натра на каждую кис-
кислотную группу. В случае полифенолов содержимое колбы тотчас приобретает темную окраску
в результате окисления кислородом воздуха. В этом случае прибор изолируют от кислорода с
помощью клапана Бунзена1. Затем из капельной воронки добавляют по 1 моль диметилсуль-
диметилсульфата на каждую фенольную гидроксильную группу2, подлежащую метилированию; реакцион-
реакционную смесь при этом хорошо перемешивают. Необходимо следить, чтобы температура не под-
1 Обратный холодильник закрывают пробкой, через которую проходит стеклянная труб-
трубка с коротким куском резинового шланга. Вдоль шланга лезвием бритвы делают короткий
разрез, а с другого конца шланг закрывают пробкой.
2 Карбоксильные группы из-за своей незначительной нуклеофильности реагируют труд-
труднее, чем фенольные гидроксилы. Поэтому и удается получить ароматические алкоксикарбо-
новые кислоты.
2. Нуклеофильное замещение при насыщенном атоме углерода
295
нималась выше 40 °С (охлаждение водой). Чтобы реакция прошла полностью, а также для того,
чтобы разложился непрореагировавший диметилсульфат, нагревание на кипящей водяной
бане продолжают еще 30 мин. Если продукты реакции — жидкости, то для их выделения
после охлаждения реакционной смеси отделяют органический слой, а водный экстрагируют
эфиром. Объединенные органические слои промывают разбавленным едким натром,
затем водой, сушат хлоридом кальция и фракционируют. Твердые продукты реакции
отделяют фильтрованием, промывают водой и перекристаллизовывают. Из водных растворов
после подкисления и извлечения эфиром можно выделить не вступивший в реакцию
фенол.
В тех случаях, когда должны получиться частично этерифицированные фенолы или когда
они образуются в качестве побочных продуктов (в каком случае это происходит?), реакцион-
реакционную смесь сначала подщелачивают и извлекают эфиром нейтральные эфиры фенола. После
подкисления раствора концентрированной соляной кислотой выделяют частично этерифици-
этерифицированные фенолы, которые обрабатывают описанным выше способом, однако эфирный
экстракт в этом случае не промывают водной щелочью (почему?).
Таблица Г.2.72. Простые эфиры фенолов, получаемые при метилировании диме-
тил сульфатом
Простой эфир
Метилфениловый эфир
(анизол)
о-Крезилметиловый эфир
ж-Крезилметиловый эфир
л-Крезилметиловый эфир
Метил-р-нафтиловый
эфир (неролин)
Монометиловый эфир
гидрохинона8
Диметиловый эфир гид-
гидрохинона"
Монометиловый эфир
рсоОрЦИпа
Диметиловый эфир
резорцина
л-Метоксибензойная
(анисовая) кислота
3,4,5-Триметоксибензой-
ная кислота (тримети-
ловый эфир галловой
кислоты)
3,4-Диметоксибензальде-
гид (верартовый альде-
альдегидI1
о-Нитроанизол
Исходное вещество
Фенол
о-Крезол
ж-Крезол
п-Крезол
Р-Нафтол
Гидрохинон
Гидрохинон
Резорцин
Резорцин
я-Гидроксибензой-
ная кислота
3,4,5-Тригидрокси-
бензойная (галло-
(галловая) кислота
З-Метокси-4-гидр-
оксибензальдегид
(ванилинI"
о-Нитрофенол
Т.кип.'.'С
154
64 A4)
65 A4)
65 A4)
Т. пл. 72
(этанол)
128 A2);
т. пл. 56 (петр.
эфир)
109 B0);
т. пл. 56
(этанол)
144 B5)
ПО B0)
Т. пл. 184
(водн. уксусная
кислота)
Т. пл. 170
(водн. этанол)
153 (8);
т.пл. 46 (лигроин)
133 A1)
1,5173
1,5179
1,5130
1,512
1,5223
1,5620
Выход,
%
85
80
80
80
73
60
95
50
85
75
70
70
50
а Цифры в скобках указывают давление (мм рт. ст.), которому соответствует
данная температура кипения. Для твердых веществ также приведена температура
плавления.
6 Не перегоняется с водяным паром, содержит примесь дниетнлового эфира.
в Перегоняется с водяным паром.
г Ванилин и верартовый альдегид устойчивы к щелочам. Верартовый альдегид сле-
следует хранить в закрытых склянках.
Таблица Г.2.73. Получение простых эфиров по Вильямсону
Продукт реакции
я-Бутилметиловый эфир
я-Бутилэтиловый эфир
я-Пентилметиловый
эфир6
к-Гексилметнловыйб
к-Гексилэтиловый эфир6
Этокснбензол (фенетол)
к-Пропокси бензол
к-Бутокси бензол
Бензилфениловый эфир
л-Нитрофенетол
Т. кип., "С
(ммрт. ст.);
"d
71; 1,3736
92; 1,3818
99; 1,3873
126; 1,3972
142; 1,4008
57A2);
1,5080
81A2);
1,5014
87(9);
1,5049
Т. пл. 40 °С
(этанол)
Т. пл. 60 °С
(водн. эта-
этанол)
Исходное
соединерние
н-Бутанол
Метанол
н-Бутанол
Этанол
н-Пентанол
я-Гексанол
я-Гексанол
Фенол
Фенол
Фенол
Фенол
п-Нитрофенол
Т. кип
(ммрт.
Vе
117;
117;
138;
156;
156;
1,
1.
1.
1,
1
,"С
ст.);
3993
3993
4099
4179
4179
Алкирующий агент
СН31, СНзОТз* диметил-
сульфат
C4H9Br, C4H,OTs
C2H5Br, C2H5OTs
С4Н9Вг, C4H9OTs
CH3I, CHjOTs, диметил-
сульфат
CH3I, CHjOTs, диметилсульфат
С2Н5Вг, C2H5OTs
CjH5Br, C2H5I, C2H5OTs
C3H7Br, QH7I, C3HrOTs
C4HsBr, C«H,OTs
Бензилхлорид
С2НБВг, C3H5I, C2H6OTs
Вариант
В
A
В
A
В
В
В
Б
Б
Б
Б
Б
Выход,
80
80
80
80
80
80
80
80
80
80
80
60
а R-OTs: алкил-л-толуолсульфонат.
6 Хорошо получается также при обратной комбинации, т. е. из метилового (или этилового) спирта и алкилирующего агента С5 (по вариан-
варианту А).
2. Нуклеофильное замещение при насыщенном атоме углерода 297
Таким же путем выделяют простые эфиры фенолкаброновых кислот. При работе с полу-
микроколичествами компоненты встряхивают в круглодонной колбочке, закрытой пробкой,
нагревают, как описано выше, с обратным холодильником на водяной бане и затем обрабаты-
обрабатывают, как обычно. Можно обойтись без контроля температуры реакционной смеси.
Получение арилалкиловых эфиров алкилированием фенолов в условиях меж-
межфазного катализа: McKillopA., FiaudJ.-C, HugR. R. Tetrahedron, 1974, 30, 1379.
Ф Общая методика получения простых эфиров, спиртов и фенолов с помощью алкилгалогени-
дов, толуолсульфонатов или диметилсульфата (синтез Вильямсона) (табл. Г.2.73).
§ Внимание*. Диметилсульфат — сильный яд! Он оказывает канцерогенное
действие. Работать под тягой в резиновых перчатках!
Для получения алифатических простых эфиров вначале в трехгорлой колбе с мешалкой и
обратным холодильником готовят раствор алкоголята из 0,25 моль натрия и 1,2 моль соответ-
соответствующего абсолютного спирта1. Затем в колбу добавляют либо 0,2 моль алкилиодида (алкил-
бромида) или алкил-л-толуолсульфоната, либо 0,14 моль диметилсульфата2 (если применяют
малоактивные алкилбромиды, то вносят немного безводного иодида калия) и нагревают при
перемешивании3 без доступа влаги 5 ч.
Для получения простых эфиров фенолов также сначала получают раствор этилата натрия
из 0,25 моль натрия и 300 мл абсолютного спирта. Затем к раствору добавляют 0,2 моль фено-
фенола в небольшом количестве абсолютного этилового спирта. После добавления алкилирующе-
го средства процесс ведут так же, как описано выше. Поскольку фенолят имеет высокую
нуклеофильность, он реагирует с алкилирующим средством легче, чем спирт.
Обработка
Способ А. Реакционную смесь по охлаждении перемешивают с пятикратным количеством
воды, отделяют эфир, промывают еще раз водой, сушат хлоридом кальция и перегоняют.
Способ Б. Из реакционной смеси при перемешивании тщательно отгоняют спирт на 20-см
колонке Вигре, выливают охлажденный остаток от перегонки в 100 мл 5%-ного едкого натра,
извлекают органическую фазу диэтиловым эфиром, промывают водой, высушивают хлоридом
кальция, отгоняют растворитель, а остаток перекристаллизовывают или фракционируют.
Способ В. Реакционный продукт при интенсивном перемешивании отгоняют непосред-
непосредственно из реакционной среды до тех пор, когда будет достигнута температура кипения
используемого спирта. Дистиллят, состоящий из эфира и исходного спирта, затем фракциони-
фракционируют в узких границах с помощью 30-см колонки Вигре. Для близкокипящих фракций опре-
определяют показатель преломления. Фракции, которые содержат целевой эфир, объединяют и
повторно перегоняют над 5% натрия до достижения нужного значения показателя преломле-
преломления.
Для полумикро- и микроколичеств подходит такая комбинация, для которой можно приме-
применять методы А или Б, при этом можно не использовать перемешивание. Продукт следует подвер-
подвергать тщательному фракционированию, для микроколичеств можно использовать прибор,
показанный на рис. А.34, в.
1 Получение см. часть Е. В случае низкомолекулярных спиртов (Ci-C3) можно брать
трехкратный избыток спирта, чтобы реакционную смесь легче было перемешивать.
2 В этих условиях используются обе метильные группы диметилсульфата.
3 При использовании легкокипящих алкилирующих средств нужно применять эффектив-
эффективный холодильник.
298 Г Препаративная часть
Примеры методик синтеза простых эфиров фенолов по Вильямсону без
предварительного получения этилата натрия (реакция в присутствии карбоната
калия в ацетоне): Аллен Ч., Гейтс-мл. Дж. В сб.: Синтезы органических препара-
препаратов. Сб. 3. — М.: ИЛ, 1952, с. 134. Описано получение ряда простых эфиров
о-нитрофенола.
Получение аллилфенилового эфира: Tarbell D. S. Org. Reactions, 1944, 2, 26.
Путем образования простых эфиров можно «блокировать» гидроксильные группы. Если
надо, например, окислить соединение с сохранением гидроксильной функции, то последнюю
можно защитить перед реакцией алкилированием и после проведения окисления снова рас-
расщепить простую эфирную связь. Особенно удобен для блокирования первичных гидроксиль-
ных групп трифенилметилхлорид (тритилхлорид), который легко реагирует с первичными
спиртами в пиридине. Простой трифенилметиловый эфир можно уже на холоду гидролизо-
вать в кислой среде. Эта реакция, которую называют тритилированием, прежде всего находит
применение в химии Сахаров.
Образование простых эфиров при реакции с диметилсульфатом (и особенно с хлоруксус-
ной кислотой) используется для идентификации фенолов:
С \— ОН + CI—СН2-СООН + 2NaOH {. \—O-CH2-COONa + NaCI + 2 Н2О [Г.2.74]
Таким же путем можно получить 2,4-дихлор- и 2,4,5-трихлорфеноксиуксусную кислоты,
которые находят применение в качестве ускорителей роста растений и средств борьбы с сор-
сорняками.
По принципу Вильямсона из щелочной целлюлозы и алкилхлоридов или хлоруксусной
кислоты в промышленности в крупных масштабах получают простые эфиры целлюлозы.
Метил- и карбоксиметилцеллюлозы растворимы в воде и играют важную роль в производстве
клеев, красящих веществ, вспомогательных текстильных материалов и моющих средств. Нера-
Нерастворимые в воде этил- и бензилцеллюлозы имеют важное значение как сырье в производстве
лаков, клеев, пластмасс.
По внутримолекулярному варианту синтеза Вильямсона (из хлоргидринов, см. разд. Г,4.1.4)
получают эпоксисоединения, которые находят широкое применение в промышленности.
2.6.3. СИНТЕЗ ЭФИРОВ КАРБОНОВЫХ КИСЛОТ
Реакции алкилгалогенидов, эфиров серной или сульфоновых кислот с аниона-
анионами карбоновых кислот аналогичны их взаимодействию с анионами спиртов или
фенолов с образованием эфирной связи, которая находится в этом случае по со-
соседству с карбонильной группой, так что образуется сложный эфир. По своему
механизму эта реакция принципиально отлична от нормальной (катализируе-
(катализируемой кислотами) этерификации карбоновой кислоты спиртом (разд. Г.7.1.4.1),
при которой в эфирную группу включается атом кислорода из спирта.
Лишь в особых случаях получение сложных эфиров алкилированием анио-
анионов карбоновых кислот имеет препаративное значение. Однако эта реакция
используется для идентификации карбоновых кислот. Для этого применяют па-
/?а-замещенные фенацилбромиды и я-нитробензилбромид, которые очень реак-
ционноспособны и образующиеся сложные эфиры хорошо кристаллизуются:
—- Br—(. V-CO-CH2-O-CO-R + Вг® [Г.2.75]
Поскольку названные галогениды чрезвычайно легко омыляются щелочью, ¦
реакцию проводят в очень слабокислом растворе или в смеси триэтиламина с
2. Нуклеофильное замещение при насыщенном атоме углерода
299
Таблица Г.2.76
Продукт реакции
Бутилбензоат
Аллилбензоат
Гексилбензоат
Бензилбензоат
а Добавить
. Эфиры бензойной кислоты
Исходное вещество
Бутилбромид
Аллилбромид
Гексилбромид
Бензилхлорида
2 г иодида натрия.
в условиях межфазного катализа
Т. кип., 'С (мм рт. ст.)
250; ПО A0)
230; 106 A2)
138^-140 (8)
172-173 A0)
Выход,
%
70
60
80
70
ацетоном. Триэтиламин способен связывать образующийся галогеноводород,
смещая равновесие в сторону образования сложного эфира.
Ф Общая методика получения эфиров бензойной кислоты в условиях межфазного катализа
(табл. Г.2.76)
«Внимание*. Фенацилбромиды оказывают сильное раздражающее действие на
слизистую оболочку и кожу! Работать под тягой в резиновых перчатках!
В многогорлой колбе на 500 мл с мешалкой и обратным холодильником 4 ч при энергич-
энергичном перемешивании кипятят смесь 0,2 моль бензоата натрия, 0,2 моль алкилбромида,
0,01 моль аликвата 336 (разд. Г.2.4.2) и 100 мл воды. По охлаждении органический слой отде-
отделяют, водный дважды извлекают эфиром (по 60 мл). Объединенные органические фазы про-
промывают 20 мл воды, сушат сульфатом магния и перегоняют.
Получение фенациловых и л-нитробензиловых эфиров (общая методика для качественного анализа)
Способ А (для чистых кислот). Раствор 1 ммольтриэтиламина в 2 мл сухого ацетона (см. часть
Е) нейтрализуют соответствующей кислотой и затем прибавляют к нему раствор 0,5 ммоль соот-
соответствующего фенацилбромида (фенацилбромида, и-бромфенацилбромида, л-фенилфенацил-
бромида) в 3 мл сухого ацетона. Вскоре выпадает осадок бромида триэтиламмония. Выдержива-
Выдерживают смесь 3 ч при комнатной температуре, разбавляют 10 мл воды, отсасывают выпавший эфир и
тщательно промывают 5%-ным водным раствором гидрокарбоната натрия и затем водой. После
этого перекристаллизовывают из водного этанола.
и-Нитробензиловые эфиры можно получать по этой же методике, однако вследствие более
низкой реакционной способности л-нитробензилхлорида надо добавить -10 мг иодида натрия и
реакционную смесь кипятить 2 ч с обратным холодильником. Наряду с ацетоном в качестве
растворителей можно использовать спирты.
Способ Б (для водного раствора кислот). 2 мл слабосолянокислого водного раствора, в котором
содержится —0,1 г карбоновой кислоты, смешивают с 2 мл раствора 0,2 г фенацилбромида в спирте
и нагревают с обратным холодильником (продолжительность нагревания монокарбоновых кислот
1 ч, дикарбоновых 2 ч, трикарбоновых 3 ч). Иногда в процессе нагревания выпадают кристаллы, ко-
которые растворяют, добавляя небольшое количество спирта. После завершения реакции реакцион-
реакционную смесь охлаждают, образовавшиеся кристаллы отсасывают и перекристаллизовывают.
2.6.4. АЛКИЛИРОВАНИЕ АММИАКА И АМИНОВ
Алкилгалогениды, алкилсульфаты и другие алкилирующие агенты взаимодей-
взаимодействуют с аммиаком:
[Г.2.77]
R-X -
R-X ¦
<- NH3
t- R-NH2 -
©
- R-NH3 +
©
R2NH2
X0-
+ xe
^=^ R-NH2 -
^= R2NH н
t- HX
- HX
300
Г Препаративная часть
Образующийся в первой стадии первичный амин, будучи сильным основанием
конкурирует с аммиаком за следующую молекулу алкилгалогенида. Поэтому обра
зуются не только первичные, но и вторичные и далее третичные амины, а такж
соли четвертичных аммониевых оснований. Напишите уравнения этих реакций!
Применяя большой избыток аммиака либо добавляя карбонат или хлорщ
аммония, можно повысить выход первичных аминов.
Однако, из-за образования также высших продуктов алкилирования для полу
чения чистых первичных или вторичных аминов часто приходится выбирать об
ходные пути. При этом во всех случаях в реакцию с галогенидом вводят блокиро
ванное производное аммиака, которое содержит лишь один свободный атом водо-
водорода. Блокирующую группу отщепляют после реакции алкилирования. Для такогс
обратимого блокирования используют, например, фталимид (синтез Габриэля)
Аминогруппа фталимида или сульфамида вследствие оттягивания электронов обе-
обеими карбонильными группами (или сульфонильной группой) не обладает доста-
достаточно выраженным основным характером, что необходимо для реакции с алкилга-
логенидами. Наоборот, она проявляет кислотные свойства, поэтому со щелочам»
образуются соли, которые и могут вступать в реакцию:
О О
R-X
-КХ
N-R
[Г.2.78]
О О
Образующийся N-алкилфталимид как амид кислоты может быть гидролизован до фтале-
вой кислоты и чистого первичного амина:
О
,СООН
N-R
+ 2Н2О, Н
©
+ H2N-R
[Г.2.79]
соон
Поскольку гидролиз приходится проводить, как правило, при повышенных температурах
под давлением, удобнее использовать гидразинолиз:
О
N-R
+ NH2NH2
+ H2N-R
[Г.2.80]
Для синтеза вторичных аминов с разными алкильными остатками можно использовать
взаимодействие алкилгалогенидов с сульфонамидами первичных аминов, например:
©
-SO2-N-CH3 + С2Н51 -^7Г
®
К
СН3
-SO3H + HN
С2Н5
Для этой же цели могут применяться также азометины1:
С2Н5
Н2О, Н^
[Г.2.81]
-CH=NR' + RX
©Л
-CH=N
R
0 + Н2О_
-нх
NH +
-сн=о [Г.2.82]
R'
О получении и способности к гидролизу азометинов и уротропина см. разд. Г,7.1.1
2. Нуклеофильное замещение при насыщенном атоме углерода 301
Реакция уротропина с алкилгалогенидами также приводит к солям четвертичных аммони-
аммониевых оснований, которые могут быть гидролизованы разбавленными кислотами до первичных
аминов (реакция Делепина).
© Я Н О®
RX + (CH2NN4 > N3(CH2NNRX ——- RNH2 [Г.2.83]
Легко протекающую реакцию первичных аминов с бензилгалогенидом до
двукратно аралкилированных (третичных) аминов можно использовать для бло-
блокирования аминогруппы при реакциях аминосоединений. Если, например,
проводят восстановление литийалюминийгидридом аминокислоты до аминос-
пирта (разд. Г,7.3.1.1), то прежде всего превращают аминокислоту действием
бензилбромида в 1Ч,К-дибензильное производное. При этом и карбоксильная
группа превращается в бензиловый эфир. Оба бензильных остатка на аминог-
аминогруппе действуют как защитные группы и после проведения восстановления
сложноэфирной группы их снова удаляют. Например, это возможно просто с
помощью гидрогенолиза над палладием на угле в качестве катализатора, при
этом бензильные остатки восстанавливаются до толуола.
R-CH-COOH + 3 PhCH2Br ——- R-CH-COOCH2Ph
I — 3 НВг I
NH2 N(CH2PhJ
[Г.2.84]
CH2OH
N(CH2PhJ N(CH2PhJ " NH2
Ф Получение бензилового эфира (ф-Р^ГЧ-дибензилаланина1. Суспендируют 0,2 моль E)-ала-
нина, 50 мл 2 н. NaOH и 36 г карбоната калия в 100 мл этанола и нагревают приблизитель-
приблизительно до 40 "С. Затем в течение 15 мин при хорошем перемешивании прибавляют по каплям
0,65 моль свежеперегнанного бензилбромида и нагревают с обратным холодильником при даль-
дальнейшем перемешивании в течение 12 ч (за ночь). Отделенный от водной фазы органический
слой еще дважды по 75 мл экстрагируют эфиром, объединенные органические фазы тщательно
промывают насыщенным раствором поваренной соли и сушат сульфатом магния. После удале-
удаления растворителя и избытка исходного соединения (при температуре бани до 110 °С и при
1,3 кПа, т. е. 10 мм рт. ст.) перегоняют на роторном испарителе при температуре бани 40-50 °С
@,002 кПа, т. е. 0,015 мм рт. ст.). Выход 65%. Продукт используют далее без дополнительной
очистки.
Ф Получение дициклогексилэтиламина. 2 моль М,1Ч-дициклогексиламина нагревают 2 ч на
кипящей водяной бане с 2 моль диэтилсульфата без доступа влаги. Реакцию проводят в
трехгорлой колбе емкостью 1 л, снабженной обратным холодильником, мешалкой и ка-
капельной воронкой. Перемешивание продолжают еще 15 ч, поддерживая заданную температу-
температуру; затем в охлажденную смесь добавляют 2,5 моль 50%-ного раствора едкого кали, выделив-
выделившийся амин отделяют, а водную фазу четырежды экстрагируют эфиром. Амин и эфирные
вытяжки объединяют и сушат в течение ночи над едким кали, затем упаривают эфир и фрак-
фракционируют амин в вакууме на 30-сантиметровой колонке Вигре. Т. кип. 138 °С A4 мм рт. ст.).
Выход 337 г (94% на вошедший в реакцию амин). Первая фракция представляет собой непро-
реагировавший дициклогексиламин (выход -15%); т. кип. 125 °С A6 мм рт. ст.). Степень чис-
чистоты полученного продукта контролируют газохроматографически2.
R-CH-COOCH2Ph ^ R-CH-CH2OH n'u - R-CH-CH2OH
1 I - ? ГПОП3 I
1 Частное сообщение H.-U. Reissig.
2 Для отделения дициклогексилэтиламина от дициклогексиламина методом ГЖХнеобхо1
димы: колонка длиной 1 м; неподвижная фаза — простой эфир гексаманнита и пропионитри-
ла A0%) на носителе — кизельгуре; разделение проводят при температуре 195 "С и скорости
подачи водорода (газ-носитель) 4 л/ч.
302
Г Препаративная часть
Дициклогексилэтиламин представляет собой третичный амин, атом азота
которого сильно экранирован объемистыми циклогексильными группами. Он
является важным реагентом при получении олефинов (разд. Г,3.1.5).
При аммонолизе сс-галогенкарбоновых кислот, протекающем аналогично
аммонилизу алкилгалогенидов, образуются а-аминокислоты. Для получения
высших жирных аминокислот лучше применять ct-бромпроизводные, так как
реакция с соответствующими хлоридами очень длительна.
Ф Общая методика получения а-аминокислот из а-галогенкарбоновых кислот (табл. Г.2.85).
В круглодонной колбе с обратным холодильником нагревают 8 моль карбоната аммония
в 140 мл воды до 55 °С; встряхивая, охлаждают до 40 °С и, поддерживая эту температуру,
добавляют 6 моль концентрированного водного раствора аммиака. Смесь оставляют на
30 мин. Затем порциями прибавляют 1 моль соответствующей а-галогенкарбоновой кислоты
и выдерживают при температуре 40-50 °С (в случае бромзамещенных кислот оставляют на
24 ч, а в случае хлорзамещенных — на 40 ч). Аммиак и углекислоту удаляют, упаривая раствор
в фарфоровой чашке на голом пламени, доводят температуру раствора до 110 °С. Содержимое
чашки охлаждают до 60 °С и переносят в 3 л метилового спирта. Выпавший за ночь в холодиль-
холодильнике осадок отфильтровывают и промывают метиловым спиртом. Получаемая аминокислота
имеет высокую степень чистоты.
Таблица Г.2.85. а-Аминокислоты из а-галогенкарбоновых кислот
Продукт реакции
а-Аминоуксусная кисло-
кислота (глицин)
а-Аминопропионовая кис-
кислота (аланин)
а-Аыиномасляная кис -
лота
сс-Аминовалериановая
кислота (норвалин)
а-Аминоизокапроновая
кислота (лейцин)
а-Амннокапроновая кис-
кислота (норлейцин)
Исходное вещество
С1СН2СООН
СНзСНВгСООН
СНзСНаСНВгСООН
СН3СН2СН2СНВгСООН
(СНзЬСНСН2СНВгСООН
СН3(СН2)зСНВгСООН
Т. пл., *С
232
295
Разл.
303 (в запаян-
запаянном капилляре)
292 (в запаян-
запаянном капилляре)
275
Выход,
%
70
60
60
60
50
65
Третичные амины можно перевести при помощи алкилирующих агентов в
соли четвертичных аммониевых оснований и таким образом идентифицировать.
ФКватернизация третичных аминов (общая методика для качественного анализа). 0,5 г соот-
соответствующего третичного амина и 1 г кватернизующего реагента (метилиодид, метило-
метиловый эфир л-толуолсульфоновой кислоты и др.) растворяют отдельно в двойных объемах
нитрометана, ацетонитрила или спирта (растворители перечислены по убывающей их приго-
пригодности для кватернизации). Растворы объединяют, оставляют на 1 ч и затем еще нагревают
30 мин на водяной бане. Иногда соли четвертичных аммониевых оснований сразу выпадают в
осадок, в других случаях раствор упаривают в вакууме и перекристаллизовывают соль из сме-
смеси сухого этилацетата и этилового спирта.
Соли четвертичных аммониевых оснований с более длинным алкильным остатком
(С,2—Cis) обладают поверхностно-активными и бактерицидными свойствами. Они использу-
используются как текстильные вспомогательные материалы, флотационные и дезинфицирующие сред-
средства. Четвертичная соль дипиридила, например паракват, применяется в качестве гербицида.
2. Нуклеофильное замещение при насыщенном атоме углерода
303
Экономически важным гербицидом является глифосат, Г>[-(фосфонометил)глицин, кото-
который можно получить алкилированием глицина хлорметилфосфоновой кислотой.
Me-N
Паракват
N-Me 2CI (HOJP-CH2-NH-CH2-COOH
Глифосат
NH N(CH2CH2CIJ
Циклофосфамид
[Г.2.85а]
Алкилирующее действие бис(р-хлорэтил)аминов (выделение азота) на дезоксирибонуклеи-
новую кислоту используют в химиотерапии рака. К лекарствам такого рода принадлежит цикло-
циклофосфамид.
Низшие алифатические амины (первичные, вторичные и третичные) получают в промыш-
промышленности из аммиака и спиртов каталитическим отщеплением воды на оксиде алюминия.
Образующиеся смеси разделяют перегонкой.
2.6.5. АЛКИЛИРОВАНИЕ СОЕДИНЕНИЙ ФОСФОРА
2.6.5.1. Алкилирование третичных фосфинов
Подобно третичным аминам можно кватернизовать также соответствующие
фосфины, используя подходящий алкилирующий агент:
R-X + PR3
[R-PR3] X
е
[Г.2.86]
Ф Общая методика получения алкилтрифенилфосфониевых солей (табл. Г.2.87). К 0,1 моль
трифенилфосфина прибавляют хорошо охлажденный раствор 0,1 моль алкилгалогени-
да в 150 мл абсолютного толуола, смесь нагревают 20 ч в автоклаве при 130 "С. Осадок
отсасывают и тщательно промывают горячим толуолом.
При небольших загрузках используют запаянную трубку, а алкилгалогениды, кипящие
выше 80 °С, можно нагревать с фосфином в толуольном растворе 48 ч (с обратным холодиль-
холодильником).
Получаемые из алкилтрифенилфосфониевых солей илиды используют для
синтеза олефинов через стадию присоединения к карбонильным соединениям
(реакция Виттига; разд. Г,7.2.1.7.2).
Таблица Г.2.87. Алкилтрифенилфосфонийгалогениды
Продукт реакции
Метилтрифенилфосфонийбромид
Метилтрифенилфосфонийиодид
Этилтрифенилфосфонийбромид
Изопропилтрифенилфосфонийбромид
Бутилтрифенилфосфонийбромид
Бензилтрифенилфосфонийбромид
Алкилгалогенид
Метилбромид
Метилиодид
Этилбромид
Изопропилбромид
Бутилбромид
Бензилбромид
Т. пл., 'С
228
185
209
238
243
280
Выход,
%
90
90
90
80
80
80
304 Г Препаративная часть
2.6.5.2. Реакция Михаэлиса — Арбузова
В реакции Михаэлиса — Арбузова алкилируется атом фосфора в эфирах кислот
трехвалентного фосфора. При этом алкилирующий агент сначала кватернизует
нуклеофильный трехвалентный фосфор в соединении I с образованием II (схе-
(схема [Г.2.88]). Этот неустойчивый промежуточный продукт в условиях реакции
переходит в соединение пятивалентного фосфора III:
Y eY Y
Ry _j_ d - /"\r"}' fc q . гэ - ^^DT -4- V ^^^ ъ. О ' D — ^^ Д- D' .V
Л ~ Г\ \J П П r, \J П ~ Л П г\— \J ~ П Л
Z Z Z [Г.2.88]
i и in
Таким путем из эфиров фосфористой кислоты (Y = Z = О-алкил) получают
диэфиры алкилфосфоновых кислот, из диэфиров алкилфосфоновых кислот
(Y = алкил; Z = О-алкил) — эфиры диалкилфосфиновых кислот, а из эфиров
диалкилфосфиновых кислот (Y = Z = алкил) — триалкилфосфиноксиды.
GТ)) Общая методика получения диэтиловых эфиров алкилфосфоновых кислот (табл. Г.2.89)
И Внимание1. Избегать попадания на кожу фосфорорганических соединений!
Нагревают 0,5 моль триэтил фосфита и 0,55 моль алкил галогенида до 150-155 °С (темпера-
(температура бани 160 °С) и выдерживают при этой температуре 1,5—2 ч для реакционноспособных ал-
килгалогенидов и 3—5 ч для менее реакционноспособных. При работе с алкилхлоридами при-
применяют колбу с обратным холодильником и выделяющийся при этом этилхлорид отгоняют
или собирают в охлаждаемую ловушку. Для алкилбромидов и алкилиодидов используют аппа-
аппарат для отгонки, снабженный длинной колонкой Вигре, быстро отгоняют выделяющийся
этилгалогенид и одновременно следят за протеканием реакции. После завершения отгонки
этилгалогенида продукт перегоняют.
Получение диэтилового эфира бензилфосфоновой кислоты из диэтилфосфита
и бензилбромида: Fedorynski F, Wojciechowski К., Matacz Z., Makosza M. J. Org.
Chem. 1978, 43, 4682.
Получение диизопропилового эфира метилфосфоновой кислоты из триизопропил-
фосфита и метилиодида: Ford-Moore A. H., Репу В. J. Org. Syntheses, СоБ. Vol. IV A963),
325.
Получение диэтилового эфира этилфосфоновой кислоты из триэтил фосфита и
этилиодида: KosolapoffG. M. Org. Reactions, 1951, 6, 286.
Получение З-бромпроп-1-илфосфоновой кислоты из триэтилфосфита и
1,3-дибромпропана: KosolapoffG. M. Ogr. Reactions, 1951, 6, 287.
Фосфоновые эфиры являются исходными соединениями для промышленно
важной реакции Хорнера—Вадсворта—Эммонса, ср. разд. Г,7.2.1.7.1.
Фосфорорганические соединения в настоящее время находят применение в качестве ин-
инсектицидов. Таковы, например, паратион, Е 605 [D-нитрофенил)диэтилтионфосфат], хлорти-
он [(З-хлор-4-нитрофенил) диметилтионфосфат].
Чрезвычайно токсичные боевые отравляющие вещества (DFP — диизопропилфторфосфат,
зоман — пинаконовый эфир метилфторфосфорной кислоты, зарин — изопропиловый эфир той
же кислоты, эфир Таммелина — холиновый эфир той же кислоты и др.) являются фторангидрида-
ми фосфорной или фосфоновой кислоты. Они действуют как ингибиторы холинэстеразы.
Последняя ответственна за расщепление образующегося при раздражении вегетативной нервной
2. Нуклеофильное замещение при насыщенном атоме углерода
305
Таблица Г.2.89. Получение диэтиловых эфиров алкилфосфоновых кислот по реак-
реакции Михаэлиса - Арбузова
Продукт
Диэтиловый эфир
цианометилфосфоновой
кислоты
Диэтиловый эфир бен-
зилфосфоновой кислоты
Диэтиловый эфир
нафт-1 -илметилфосфо-
новой кислоты
Диэтиловый эфир
B-бромэтил)фосфоно-
вой кислоты
Диэтиловый эфир
бутил фосфоновой
кислоты
Диэтиловый эфир
этилфосфоновой кислоты
Исходное
соединение
RXB[r.2.88]
Хлорацетонит-
рила
Бензилхлорид
Бензилбромид
1-Хлорметил-
нафталин
1,2-Диромэтан
Бутилбромид"
Прибавляют 0,5 г
NaP
Т. кип., °С
(мм. рт. ст.)
148-150A0)
169-171 B5)
205-206 E)
86-87 BN
74A)
80-83A1)
Выход, %
80
90
80
65
85
90
Время
реакции, ч
1,5
5
4
3
2
3
а Прикапывать при 150 °С.
6 Не привышать указанную температуру.
в Начинать при температуре 110 °С, температуру повышать так, чтобы отгонялся только
С2Н5Вг.
г Из триэтилфосфита промежуточно образуется этилиодид.
системы ацетилхолина до холина и уксусной кислоты. Эти яды блокируют действие холинэстера-
зы путем ее фосфорилирования. При концентрации этих соединений в воздухе 5 • 10~7 г/л у людей
уже через 1-2 мин проявляются симптомы сильного отравления. Ультраяды могут применяться в
качестве боевых отравляющих веществ. Однако по международной конвенции запрещено получе-
получение, хранение и применение этих веществ.
2.6.6. АЛКИЛИРОВАНИЕ СЕРОСОДЕРЖАЩИХ СОЕДИНЕНИЙ
По аналогии с щелочным гидролизом протекает взаимодействие алкилгалогени-
дов, алкилсульфатов и т. д. с гидросульфидом натрия; при этом образуются тиолы:
[Г.2.90]
R-X
®
s-н
R-S-H + X"
В качестве побочных продуктов получаются симметричные тиоэфиры, так
как образующийся из тиолов при щелочном гидролизе тиолат-ион реагирует с
306
Г Препаративная часть
еще не вступившим в реакцию алкилгалогенидом:
R-X + eS-R - R-S-R + Xе [Г.2.91]
Этот процесс можно сделать основным, если вводить в реакцию с сульфидом
натрия два моля алкилгалогенида:
2R-X + Na2S R-S-R + 2NaX [Г.2.92]
или по аналогии с синтезом Вильямсона в реакцию с алкилгалогенидами вво-
вводить тиолат. В результате можно получить также несимметричные тиоэфиры.
(ГП) Общая методика получения симметричных диалкилсульфидов (табл. Г.2.93)
Внимание1. Многие тиолы и некоторые сульфиды даже в очень малых концент-
концентрациях обладают чрезвычайно неприятным запахом! Работать под тягой и в за-
защитных перчатках! Для уменьшения запаха необходимо после завершения экс-
эксперимента обрабатывать посуду раствором перманганата калия!
В трехгорлой колбе с эффективным обратным холодильником, капельной воронкой и ме-
мешалкой растворяют 1,2 моль сульфида натрия Na2S • 9Н2О в 250 мл воды и 100 мл метилового
спирта. При перемешивании добавляют 2 моль соответствующего алкилбромида и кипятят 5 ч
с обратным холодильником при очень интенсивном перемешивании. По охлаждении отделяют
органический слой, а водный встряхивают с двумя эфирными порциями по 75 мл. Объединен-
Объединенные органические слои промывают несколько раз 10%-ным раствором едкого натра и водой,
сушат хлоридом кальция и перегоняют. Если получены твердые вещества, их отфильтровывают,
промывают водой и перекристаллизовывают.
Синтезы с полумикроколичествами проводят без мешалки в круглодонной колбе с обрат-
обратным холодильником.
Таблица Г.2.93. Тиоэфиры, полученные алкилированием сульфида натрия
Тиоэфир
Диэтилсульфид
Ди-н-пропилсульфид
Ди-«-бутилсульфид
Дибензилсульфид
Алкилирующий
агент
Этилбромид
«-Пропилбромид
к-Бутилбромид
Бензилхлорид
Физические константы
Т. кип. 91 °С; яп20 1,4426
Т. кип. 142 eC; nDM 1,4473
Т. кип. 75°С (Юммрт. ст.);
лс20 1,4529
Т. пл. 49 "С (метанол)
Выход,
Ж
65
70
70
85
Получение 2,2'-дигидроксидиэтилсульфида (тиодигликоля) из 2-хлорэтанола
(этиленхлоргидрина): Фабер Е., Миллер Дж. Е. В сб.: Синтезы органических
препаратов. Вып. 2. — М.: ИЛ, 1949, с. 453.
Алкилтиоцианаты (алкилроданиды) являются ценными промежуточными
продуктами для дальнейших превращений, как, например, для синтеза алкилтио-
лов, тиоэфиров и диалкилсульфидов, которые легко получаются при взаимодей-
взаимодействии первичных и вторичных алкилгалогенидов и щелочных солей тиоцианатов:
- R-S-C=N + Xе [Г.2.94]
R-X +
Э
S-C=N
При этом амбидентные нуклеофилы предпочтительнее реагируют с субстра-
субстратом, в котором центральный атом, например сера, обладает высокой поляризу-
поляризуемостью. С алкилгалогенидами, которые реагируют предпочтительно по SnI-
2. Нуклеофильное замещение при насыщенном атоме углерода 307
механизму, как, например, трет-бутилгалогениды, образуют, напротив, смесь
алкилтиоцианата и алкилизотиоцианата (о реакционном поведении амбифу-
нкциональных нуклеофильных реагентов см. разд. В.6 и Г.2.3). Так как алкили-
зотиоцианаты являются термодинамически стабильными продуктами, тиоциа-
наты в случае легкого образования алкил-, бензгидрил- или аллильного катио-
катионов (при добавлении кислоты Льюиса) можно термически превращать в
соответствующие изотиоцианаты. См. литературу, по получению трет-бутип-
изотиоцианата, а также указанную в конце главы.
Реакционную способность алкилтиоцианата определяют благодаря легкой
способности к разрыву S—CN-связи под действием оснований. Так, с помощью
щелочного расщепления эти соединения легко могут быть переведены в алкилти-
олаты [Г.2.95] или использоваться при замещении С-нуклеофилов в виде замас-
замаскированных алкилсульфенильных соединений, таких как ацетилиден [Г.2.96]:
RS-CN + ОН + Н2О RSG + NH3 + СО2 [Г.2.95]
RS-CN + ICHC-R' RS-C=C-R' + CN0 [Г.2.96]
В следующих методиках представлен твердофазный способ получения ал-
килтиоцианатов, при котором алкилтиоцианат наносят на силикагель и в таком
виде смешивают с чистым алкилгалогенидом (без прибавления растворителя).
Реакция протекает гладко и с хорошим выходом.
Ф Общая методика получения алкилтиоцианатов на силикагеле, активированном тиоциана-
том калия (табл. Г.2.97I
I Внимание] Арилметилгалогениды сильно раздражают кожу и слизистую
оболочку. Тяга! Работать в перчатках!
В круглодонной колбе растворяют 7,5 г @,075 моль) тиоцианата калия в 25 мл
дистиллированной воды и в раствор быстро вносят 7,5 г силикагеля 60, отгоняют на роторном
испарителе воду B0 мбар) при температуре бани 50 °С и высушивают силикагель в этих услови-
условиях в течение 4 ч. К охлажденным реагентам прибавляют 0,025 моль алкилгалогенида. Если веще-
вещества при комнатной температуре твердые, то смесь нагревают на водяной бане несколько выше
температуры плавления алкилгалогенида, колбу закрывают и несколько раз встряхивают. Снова
повторно встряхивают и оставляют на 48 ч при комнатной температуре. Затем силикагель пере-
переносят на стеклянный фильтр и при небольшом вакууме продукт экстрагируют пятью порциями
по 20 мл дихлорметана. После удаления растворителя продукт очищают вакуумной перегонкой
или перекристаллизацией.
Смесь mpem-бутилтиоцианата и трет-бутилизотиоцианата из трет-бугкл-
хлорида и щелочной соли тиоцианата, а также превращение этой смеси в трет-
бутилизотиоцианат: Schmidt E., Striewsky W., Seefelder M., Hitzler F. Liebigs Ann.
Chem. 1950, 568, 193.
Применение метода см. Kodamari M., Kuzuoka Т., Yoshitomi S. Synthesis, 1983, 141.
308
Г Препаративная часть
Таблица Г.2.97. Получение алкилтиоцианатов из алкилгалогенидов
Продукт
Бутилтиоцианат
Гексилтиоцианат
Бензилтиоцианат
4-Метилбензилтиоциа-
нат
2- Метилбензилтиоциа-
нат
1,4-Бис(тиоцианатоме-
тил)бензол
4- Метоксибензилтиоци-
анат
2-Бромбензилтиоцианат
1 -Тиоцианатометил-
нафталин
2-Тиоцианатометил-
нафталин
Алкилгалогенид
Бутилиодид
Гексилодид
Бензилхлорид
4-Метилбензил-
хлорид
4-Метилбензил-
бромид
2-Метилбензил-
хлорид
2-Метилбензил-
бромид
1,4-Бис(дибромме-
тил) бензол
4-Метоксибензил-
хлорид
2-Бромбензилбро-
мид
1-Хлорметил наф-
нафталин
2-Бромметилнаф-
талин
Физические константы
Т. кип. 64 °С A2 мм рт. ст.)
Т. кип. 93 °С A0 мм рт. ст.)
Т. пл. 39 °С (ЕЮН)
Т. кип. 151 °С B0 мм. рт. ст.)
Т. пл. 18 °С (этанол)
Т. пл. 33 °С (этанол)
Т. кип. 134 °С A мм рт. ст.)
Т. кип. 113 °С@,13 мм рт. ст.)
Т. пл. 91 °С (этанол)
Т. пл. 101 °С (этанол)
Выход в %
85
87
95
93
90
92
90
96
98
95
Сульфиды, подобно третичным аминам и фосфинам, реагируют с алкилга-
логенидами, образуя третичные сульфониевые соли:
R-X + SR2 [r-sr2] Xе [Г.2.98]
Сера в тиомочевине также обладает высокой нуклеофильностью, так что это
соединение легко алкилируется; поэтому при добавлении алкилгалогенидов
легко образуются S-алкилтиоурониевые соли:
NH2
R-X + S =С
NH2
-
©
R-S=C
NH2
——¦
NH2
©,NH2
R-^-C -
\lH2
©
NH2
-» R-S-C ~~ Я
4NH2
-
NH2
NH2
©
xe-
R-S-C
V
[г.:
-
NH2
NH2
1.99}
xe
2. Нуклеофильное замещение при насыщенном атоме углерода 309
Эти соли применяются для идентификации алкилгалогенидов. Обычно для
таких целей из солей получают хорошо кристаллизующиеся пикраты (табл. Д.45).
Получение пикратов S-алкилтиоурония (общая методика для качественного анализа). К раствору
0,2 г тиомочевины в 0,6 мл воды и 0,4 мл этилового спирта прибавляют 0,2 г алкилгалогенида.
Нагревают смесь на водяной бане в колбе с обратным холодильником, пока не исчезнет слой
алкилгалогенида. Нагревание продолжают еще 15 мин. Затем еще горячий раствор прибавля-
прибавляют к 40 мл кипящего 1 %-ного водного раствора пикриновой кислоты. После охлаждения отса-
отсасывают выпавшие кристаллы, промывают водой и перекристаллизовывают из водного спирта.
Определение эквивалентной массы пикратов алкилтаоурония. Точную навеску пикрата алкилтиоуро-
ния @,3—0,35 г) растворяют в 25—50 мл ледяной уксусной кислоты и титруют 0,1 н. хлорной кисло-
кислотой в ледяной уксусной кислоте (см. часть Е) по кристаллическому фиолетовому как индикатору:
Расчет:
-. Навеска (г) 1000
Эквивалентная масса пикрата = ¦—¦
Объем НС1О4(мл)-Нормальность НС1О4(гэкв.)
Эквивалентная масса спирта = Эквивалентная масса пикрата—288,2
Хлорид S-бензилтиоурония образует с сульфоновыми кислотами и многими
карбоновыми кислотами труднорастворимые, хорошо кристаллизующиеся
S-бензилтиоурониевые соли, которые можно использовать для идентификации
этих кислот.
S-Алкилтиоурониевые соли легко могут гидролизоваться щелочами, при
этом образуются тиолы:
-10
NH2
R-S-C.
NH
,е
ОН°+Н2О R-S-H + СО2 + 2NH3 [Г.2.100]
Общая методика получения тиолов через S-алкилтиоурониевые соли (табл. Г.2.101)
Внимание*. Из-за чрезвычайного неприятного запаха с тиолами надо работать в
специальной комнате под очень хорошей тягой и при любых возможных контактах
(мытье посуды и т. д.) надевать резиновые перчатки. Для очистки использованной
посуды следует обработать ее концентрированной азотной кислотой или раство-
раствором перманганата калия. Тиолы при этом окисляются и теряют навязчивый запах.
К 1,1 моль тиомочевины в 50 мл 95%-ного спирта в круглодонной колбе добавляют либо
1 моль алкилбромида или алкилхлорида, либо 0,5 моль диалкилсульфата и кипятят 6 ч с обрат-
обратным холодильником. Для получения дитиолов берут двойное количество тиомочевины и спир-
спирта. При охлаждении кристаллизуется S-алкилтиоурониевая соль. Ее отсасывают и без дальней-
дальнейшей очистки омыляют в тиол. Для омыления в двугорлую колбу помещают 1 моль тиоурониевой
соли @,5 моль в случае сульфата тиоурония), 600 мл 0,5 н. раствора едкого натра и нагревают с
обратным холодильником 2 ч, пропуская слабый поток азота. Чтобы предотвратить распростра-
распространение неприятного запаха, азот, выходящий из реакционного сосуда и содержащий тиол, про-
пропускают через раствор перманганата калия. Охлажденную реакционную смесь подкисляют 2 н.
соляной кислотой, отделяют слой тиола, сушат сульфатом магния (при получении высококипя-
щих тиолов с т. кип. > 130 "С осушитель промывают эфиром) и фракционируют на колонке Виг-
ре. Перегонку в вакууме надо проводить в атмосфере азота1.
При получении высших тиолов нельзя полностью избежать образования дисульфидов,
при фракционировании они остаются в перегонной колбе.
Методика пригодна для работы с полумикроколичествами.
1 См. разд. А, 2.3.2.2.
310
Г Препаративная часть
Таблица Г.2.101. Тиолы, полученные через S-алкилтиоурониевые соли
Тиол
Бутантиол-1
2-Метилпроп а нтио л-1
Бутантиол-2
Гексантиол-1
Додекаитиол-1
Фенилметантиол
2-Фенилэтантиол-1
Пропандитиол-1,3
Гександитиол-1,6
Т. кип?, "С
98
88
85
151
154 B0)
73 A0)
105 B3)
57 A2)
119 A5)
Vs
,4401
,4358
,4338
,4473
,4575е
,5730б
,5642"
,5403е
Выход, %
90
55
60
70
70
70
70
70
60
Цифры в скобках указывают давление (мм рт. ст.), которому соответствует данная
температура кипения.
6 При 20 °С.
в При 18 "С.
Получение дитиогликоля {этандитиола-1,2) из дибромэтана и тиомочевины:
Специале А. В сб.: Синтезы органических препаратов. Сб. 4. — М.: ИЛ, 1953,
с. 569.
Некоторые меркаптосоединения (тиолы) имеют техническое значение, например, как ус-
ускорители вулканизации, антиоксиданты и т. д. Тиогликолевая кислота, которую получают из
натриевой соли хлоруксусной кислоты и гидросульфида или тиосульфата натрия, является
активным компонентом средств для холодной завивки волос.
Додецилтиол (из додецилхлорида и гидросульфида натрия) применяется как инициатор
полимеризации бутадиена.
2.6.7. СИНТЕЗ АЛКИЛГАЛОГЕНИДОВ ПО РЕАКЦИИ ФИНКЕЛЬШТЕЙНА
Галоген в алкилгалогенидах можно в равновесной реакции заменить другим
галогеном (реакция Финкелыптейна)
R-X + Y"
R-Y + X
©
[Г.2.102]
Реакцию чаще всего используют для синтеза алкилфторидов и первичных
алкилиодидов, поскольку последние часто не удается получить из соответствую-
соответствующего спирта и иодоводородной кислоты.
Первичные алкилиодиды получают из соответствующих хлоридов и бромидов.
Растворителем служит ацетон, в котором иодид натрия растворим, а хлорид и бро-
бромид натрия нерастворимы. Образующиеся в ходе реакции хлорид или бромид нат-
натрия выпадают в осадок, что смещает равновесие в сторону образования алкилиоди-
да. В случае вторичных и особенно третичных галогенидов реакция не идет.
Таким путем можно ввести также фторид-ион. Поскольку фторид — плохая
уходящая группа (разд. Г,2.2.1), то равновесие [Г.2.102] сдвигается в сторону об-
образования алкилфторида. Для этого лучше всего реакционноспособные алкили-
алкилиодиды и алкилтозилаты вводить в реакцию с фторидом калия в полярных апро-
2. Нуклеофильное замещение при насыщенном атоме углерода
311
Таблица Г.2.103. Алкилфториды, полученные через алкилтозилаты
Алкил фторид
н-Бутилфторид
м-Пентилфторид
м-Гексилфторид
н-Гептилфторид
н-Октилфторид
Т. кип., 'С
33
64
93
120
142
Vs
1,3398
1,3600
1,3750
1,3872
1,3960
Выход, %
50
50
50
60
60
тонных растворителях (например, диметилформамид, тетраметиленсульфон).
Выходы можно существенно повысить, работая в ацетонитриле или бензоле в
присутствии краун-эфира (разд. Г,2.4.1) или в условиях межфазного катализа
(разд. Г,2.4.2). Реакцию можно проводить также в присутствии кислот Льюиса, в
частности, AgF, HgF2, HF/SbF3 (ср. также схему [Г.2.104.]).
Замещение остатка тозилата на бром (взаимодействие с бромидом лития в
ацетоне, бромидом натрия в диметилсульфоксиде или бромидом кальция в
спирте), иод (взаимодействие с иодидом калия в ацетоне) имеет большое значе-
значение для получения таких алкилгалогенидов, которые в кислой среде склонны к
перегруппировкам (разд. Г,2.5.1).
Получение вторичных алкилбромидов из тозилатов и бромида натрия в диме-
диметилсульфоксиде: CasonJ., CorreiaJ. S.J. Org. Chem., 1961, 26, 3645.
Получение алкил- и бензилиодидов, а также эфиров иодуксусной кислоты из соот-
соответствующих бромидов с безводным иодидом натрия в ацетоне: Finkelstein H. Вег.,
1910, 43, 1528. Для удаления иода можно вместо ртути использовать встряхивание
с разбавленным раствором тиосульфата натрия или гидросульфита натрия.
(ГП) Общая методика получения алкилфторидов из алкилтозилатов (табл. Г.2.103)
Л Внимание! Алкилфториды — сильные яды!1 Работать под тягой!
В приборе для перегонки с погруженным в жидкость термометром (колба с боковым нисхо-
нисходящим отводом, см. рис. А.4) растворяют 1,5 моль растертого в тонкий порошок сухого фторида
калия в 8—10-кратном (по массе) количестве диэтиленгликоля при температуре около 50 °С. За-
Затем прибавляют 1 моль соответствующего эфира я-толуолсульфоновой кислоты и нагревают
примерно 1 ч при температуре 110-120 "С. При этом низшие алкилфториды (приблизительно до
С5) частично отгоняются. Затем перегоняют остаток при температуре -200 °С (для алкилфтори-
алкилфторидов с длиной цепи более С7 перегонку заканчивают в слабом вакууме). Дистиллят промывают
водой, сушат сульфатом натрия и фракционируют на 20-сантиметровой колонке Вигре.
В промышленности фторхлоралканы получают из полихлоралканов и безводной плавиковой кис-
кислоты в присугсвтии кислот Льюиса, чаще всего SbCb- Катализатор изменяет механизм реакции на Sn 1:
R - Cl + SbCI
H-F+ R
Piw + SbCI6s
R-F+ Н®
[Г.2.104]
Хлордифторметан является промышленно важным продуктом, пиролизом которого при
700 °С получают тетрафторэтилен — исходное соединение для получения химически инертного
политетрафторэтилена (тефлон).
Ср. Pattison F. L. M., Norman J. J. J. Am Chem. Soc. 79 A959), 2311.
312 Г Препаративная часть
Применявшиеся ранее в качестве хладагентов и газоносителей фреоны
CF2C12, CFCI3, CF2C1—CFCI2 способствуют сокращению стратосферного озоно-
озонового слоя, поэтому с 1995 г. применение этих продуктов в Германии запрещено.
2.6.8. ПОЛУЧЕНИЕ НИТРОАЛКАНОВ^
Алкилиодиды и алкилбромиды реагируют с нитритами металлов, давая смеси
нитроалканов и эфиров азотистой кислоты (так называемые изонитроалканы).
О реакциях амбифункциональных нуклеофильных анионных реагентов см.
разд. В,6, Г,2.3 и уравнение [Г.2.31].
Из первичных галогенидов образуются преимущественно нитроалканы.
В этом случае даже при применении нитрита серебра не образуется больших
количеств алкилнитрита, если работают в неполярных растворителях (эфир).
В реакциях же с участием вторичных иодидов и бромидов с нитритом сереб-
серебра в эфире нитросоединения образуются с выходом около 15%. Из третичных
галогенидов практически образуются не нитроалканы, а эфиры азотистой
кислоты и главным образом олефины путем элиминирования.
При взаимодействии вторичных галогенидов с нитритом натрия, например
в диметилформамиде, образуются преимущественно нитроалканы. Реакции
с третичными галогенидами и в этих условиях дают главным образом
олефины.
Нитрит серебра (благодаря своей высокой реакционной способности и обра-
образованию нерастворимого галогенида серебра) дает довольно хорошие выходы
первичных нитроалканов. Нитрит натрия, однако, гораздо дешевле, так что при-
приходится мириться с несколько заниженными выходами.
Для реакций с участием вторичных алкилгалогенидов следует предпочесть
нитрит натрия в диметилформамиде. Последний очень удобен благодаря
довольно хорошей способности растворять оба реакционых компонента и
незначительной способности сольватировать анионы.
Ф Общая методика получения нитроалканов (табл. Г.2.105). К смеси 0,5 моль нитрита нат-
натрия и 0,5 моль мочевины (мочевина добавляется для повышения растворимости нит-
нитрита в диметилформамиде) в 600 мл сухого диметилформамида (см. часть Е) быстро до-
добавляют 0,3 моль соответствующего алкилгалогенида и перемешивают 1—6 ч (время переме-
перемешивания зависит от реакционной способности галогенида) при комнатной температуре. Затем
выливают в 1,5 л смеси воды со льдом, несколько раз извлекают эфиром, сушат хлоридом
кальция и перегоняют на 30-сантиметровой колонке Вигре. Из первых фракций можно выде-
выделить кипящий при более низкой температуре эфир азотистой кислоты, который образуется
как побочный продукт.
Получение первичных нитроалканов при взаимодействии алкилбромидов или
алкилиодидов с нитритом серебра в эфире: Kornblum N., Taub В., Ungnade H. Е.
J. Am. Chem. Soc, 1954, 76, 3209; Корнблюм Н., УнгнадеХ. В сб.: Синтезы органи-
органических препаратов, Сб. 10. — М.: ИЛ, 1960, с. 58.
В лаборатории нитрометан лучше всего получать действием натриевой соли
хлоруксусной кислоты на нитрит натрия в водном растворе. (Почему нельзя
1 О получении нитроалканов прямым нитрованием алифатических углеводородов см.
разд. Г, 1.6.
Таблица Г.2.105. Получение нитроалканов и эфиров азотистой
Продукт реакции
2-Нитропропан
1-Ннтрогексан
1-Нитрооктаи
2-Нитрооктан
Фенилнитрометан
Исходное вещество
2-Иодпропан6
1-Бромгексан
1-Иодгексан
1-Бромоктан
2-Иодоктан
Бензилбромид
Продолжительность
реакции, ч
4
4
1
4
8
5Г
кислоты иа
Т. кип.,
•с
120
82 A5)
111 A5)
98 A4)
93 C)
алкилгалогенидов
л 20
"d
1,3971
1,4235
1,4323
1,4279
1,5323
Выход,
%
26
52
55
50
52
Алкилнитрит
Т. кип.,
48В
32 A5)
85 A5)
60 A4)
66 C)
V0
1,3990
1,4301
1,4082
1,5010
Выход,
%
23
27
28
25
Цифры в скобках указывают давление (мм рт. ст.). которому соответствует данная температура кипения.
2-Иодпропан необходимо сначала освободить от следов иодоводорода. Для этого его встряхивают с охлажденным во льду раствором
соды, промывают ледяной водой, высушивают несколько раз сульфатом магния; для реакции можно использовать неперегнанное вещество.
в Перегоняется с эфиром.
г Работают при температуре от —20 до —15 °С.
314 Г Препаративная часть
вводить в реакцию свободную хлоруксусную кислоту, а надо сначала нейтрализо-
нейтрализовать ее?) Получаемая нитроуксусная кислота декарбоксилируется при нагрева-
нагревании (напишите уравнение реакции). Образующееся в некоторых случаях изонит-
росоединение нельзя выделить, так как оно гидролизуется в реакционном раст-
растворе.
Ф Получение нитрометана1. В большом стакане готовят раствор 1,05 моль хлороуксусной кис-
кислоты в 200 мл воды, нейтрализуют его содой и прибавляют раствор 1 моль нитрита натрия
в 120 мл воды. В приборе для перегонки (емкость перегонной колбы 500 мл) нагревают
100 мл этого раствора; нагревание проводят с помощью газовой горелки через проволочную сет-
сетку. При этом происходит выделение ССЬ и вместе с водой отгоняется нитрометан. Через капель-
капельную воронку, укрепленную на отводе для термометра, можно добавлять остаток реакционного
раствора к горячему раствору в перегонной колбе так, чтобы скорость перегонки можно было хо-
хорошо контролировать. Перегонку продолжают до тех пор, пока в приемник поступают масля-
маслянистые капли, затем заменяют приемник и отгоняют еще 100 мл воды. Нитрометан отделяют из
первой фракции, оба водных раствора объединяют, насыщают хлоридом натрия и вновь отгон-
отгоняют около 1/4 этого раствора. При этом получают еще некоторое количество нитрометана, кото-
который также отделяют. После высушивания хлоридом кальция нитрометан перегоняют еще раз.
Т. кип. 101 °С; «D251,3827. Выход 20-24 г C3-3
Приблизительно с тем же выходом можно получать нитрометан из диметил-
сульфата с нитритом натрия: Decombe M. J. Bull. Soc. Chim. France. 1953, 1038.
Перевод алкилгалогенидов в нитросоединения можно использовать для
идентификации первичных, вторичных и третичных алкилгалогенидов (или
соответствующих спиртов). Образующиеся при этом первичные и вторичные
нитросоединения дают с азотистой кислотой вещества, которые можно легко
различать (ср. разд. Г,8.2.3), тогда как третичные нитросоединения вообще не
получаются (см. выше).
2.6.9. ПОЛУЧЕНИЕ АЛКИЛЦИАНИДОВ (СИНТЕЗ НИТРИЛОВ ПО КОЛЬБЕ)
При взаимодействии алкилгалогенидов с цианидами металлов, так же, как и при
реакции с нитритами, имеются две возможности для атаки амбифункциональ-
ного цианид-иона и обычно образуется смесь нитрилов и изонитрилов:
R~X + IC=N * [Г.2.106]
Соотношение нитрилов и изонитрилов зависит от типа и условий реакции.
О реакциях амбидентных нуклеофилов см. разд. В,6 и Г,2.3.
В случае алифатических первичных алкилгалогенидов и бензилгалогенидов
реакция с алкилцианидами даже в протонных растворителях (спиртах, водно-
спиртовых смесях) протекает главным образом с образованием нитрилов и неже-
нежелательное образование изонитрилов, которые могут быть легко обнаружены по
чрезвычайно неприятному запаху, протекает лишь в очень небольшой степени.
IC=N (
R-CSN +
' нитрил
\ 0 е
R-N=CI +
изонитрил
хе
хе
SteinkopfW., KirchhoffG. Ber., 1909,42, 3438.
2. Нуклеофильное замещение при насыщенном атоме углерода 315
В случае замещенных бензилгалогенидов (например, при наличии +/- и +Л/-за-
местителей типа алкил- и алкоксигрупп; см. разд. Г,2.2.1) лучше работать в апро-
тонном растворителе. Таким образом затрудняется также возможный для этих
реакционноспособных галогенидов сольволиз до бензиловых спиртоб и простых
бензиловых эфиров.
Вторичные бромиды или хлориды могут вступать в реакцию, но выходы
очень низкие, тогда как третичные галогениды совсем не реагируют в желатель-
желательном направлении.
Галогенопроизводные спиртов, простых эфиров и карбоновых кислот (пос-
(после нейтрализации карбоксильной группы) реагируют гладко. Вместо алкилгало-
генидов можно часто использовать соответствующие сульфаты или сульфонаты.
Взаимодействие с цианидом серебра приводит, как и следует ожидать, в
основном к изонитрилам.
иТи Общая методика получения нитрилов (табл. Г.2.107)
Внимание] Цианиды щелочных металлов — сильные яды! Особенно опасна об-
образующаяся при подкислении синильная кислота. Необходимо работать в вы-
вытяжном шкафу с мощной вентиляцией! При уничтожении остатков надо соблю-
соблюдать максимальную осторожность. Галогенметилсодержащие ароматические
соединения сильно раздражают кожу и слизистую оболочку. Работать в
защитных перчатках! См. также часть Е.
Способ А (для реакционноспособных алкилгалогенидов). В двугорлой колбе на 2 л, снабжен-
снабженной обратным холодильником, кипятят 20 ч без доступа влаги 1 моль соответствующего гало-
генида с 1,5 моль растертого в тонкий порошок и высушенного при 105 °С цианида натрия,
0,05 моль иодида натрия и 500 мл сухого ацетона. Затем реакционную смесь охлаждают, соль
отделяют фильтрованием и промывают 200 мл ацетона. Отфильтрованный осадок уничтожа-
уничтожают с соблюдением всех мер предосторожности (в нем содержится цианид натрия). Из объеди-
объединенных фильтратов отгоняют ацетон и остаток фракционируют в вакууме.
При работе с полумикроколичествами реакцию проводят в круглодонной колбочке без
перемешивания.
Способ Б (для инертных галогенидов).
Вариант Б1. При получении цианидов вместо ацетона используют 90%-ный этанол. При
отгонке растворителя часто выпадает немного соли, которую отфильтровывают перед пере-
перегонкой продукта.
Вариант Б2. В трехгорлой колбе на 1 л, снабженной мешалкой, обратным холодильником и
термометром (который опущен в реакционную смесь), при хорошем перемешивании осторож-
осторожно нагревают 250 мл триэтиленгликоля, 1,25 моль хорошо растертого сухого цианида натрия и
1 моль алкилбромида или алкилхлорида. Начало сильноэкзотермической реакции в случае низ-
низших алкилгалогенидов можно обнаружить по сильному вскипанию раствора. Медленно повы-
повышают температуру до 140 °С (в случае бензилгалогенидов только до 100 °С) и продолжают пере-
перемешивание при этой температуре еще 30 мин.
Дальнейшую обработку ведут в зависимости от температуры кипения образовавшегося
нитрила и его растворимости в воде.
2.1. Низшие, легкорастворимые в воде и легколетучие нитрилы (длина цепи менее Cs) от-
отгоняют прямо из реакционной смеси, иногда в слабом вакууме, промывают насыщенным
раствором поваренной соли, сушат хлоридом кальция и вновь перегоняют на 30-сантиметро-
30-сантиметровой колонке Вигре.
2.2. При получении высших нитрилов реакционную смесь выливают в воду (~ 1 л) и извле-
извлекают четырьмя порциями хлороформа по 150 мл. Из объединенных хлороформных экстрактов
удаляют изоцианид путем встряхивания в течение ~5 мин с 100 мл 5%-ной серной кислоты,
затем нейтрализуют хлороформный слой, встряхивая с разбавленным раствором гидрокарбо-
гидрокарбоната натрия, промывают водой, сушат над хлоридом кальция, очищают перегонкой.
Таблица Г.2.107. Нитрилы, полученные из алкилгалогенидов
Нитрил
Бензилцианид
4-Метоксибензилцианид
3,4-Диметоксибензилцианид
2,5-Диметоксибензилциаяид
2,4-Диметилбензилцианид
2,5-Диметилбензилцианид
2,4,6-Триметил бензилцианид
2-Хлорбензилцианид
3 -Хлорбензилцианид
4-Хлорбензилцианид
2-Бромбензилцианид
З-Бромбензилцианид
Исходное вещество
Бензилхлорид
4-Метоксибензилхлорид
3,4-Диметоксибензилхлорид
2,5-Диметоксибензилхлорид
2,4-Диметилбензилхлорид
2,5-Диметилбензилхлорид
2,4,6-Триметилбензилхлорид
2-Хлорбензилхлорид
(или бромид)
3 -Хлорбензилбромид
4 -Хлорбензилхлорид
(или бромид)
2-Бромбензилбромид
З-Бромбензилбромид
Способ
А
А
А
А
А
А
А
Б B.2)
Б B.2)
Б B.2)
Б B.2)
Б B.2)
Т. кип., "С*
109 A3)
94 @,3)
150A,5); т. пл. 68 (этанол)
162A2); т. пл. 55 (этанол)
138A1)
102 A); т. пл. 28 (этанол)
163B2);
т. пл. 80 (петрол. эфир)
120A1);
т. пл. 24
136 A6)
139A2);
т. пл. 32
146 A3)
147A0)
V°
1,5211
1,5288
Выход,
%
80
80
80
70
70
70
90
80
80
80
80
80
Таблица Г.2.107. Продолжение
я-Бромбензил цианид
се-Нафтилацетонитрил
Ацетонитрил
Пропионитрил
н-Бутиронитрил
к-Валеронитрил
Пентилцианид
Гексилцианид
к-Октилцианид
м-Децилцианид
к-Ундецилцианид
к-Додецилцианид
к-Тридевдлцианид
Сукцинонитрил
Глутаронитрил
Адипонитрил
л-Бромбензилбромид
1-Хлорметилнафталин
Диметилсульфат6
Диэтилсульфат6
1-Бромпропан
1-Бромбутан; 1-хлорбутан
1-Бромпентан; 1-хлорпентан
1-Бромгексан; 1-хлоргексан
1-Бромоктан
1-Бромдекан
1-Бромундекан
1-Бромдодекан; 1-хлордоде-
как
1-Бромтридекан
1,2-Дибромэтан
1,3-Дибромпропан; 1,3-ди-
хлорпропан
1,4-Дибромбутан; 1,4-дихлор-
бутан
Б B.2)
А
Б B.1)
Б B.1)
Б B.1)
Б B.1)
Б B.2); Б A)
Б B.2); БA)
Б B.2)
Б B.2)
Б B.2)
Б B.2); БA)
Б B.2)
БA)
БA)
БA)
156A2)
176A1)
81
97
118
139
80 E0)
96 E0)
98 A0)
131 A2)
142A2)
160 A8)
167 A0); т. пл. 19
114 B); т. пл. 53
101 A,5)
115A)
1,6173
1,3418
1,3656
1,3815
1,3939
1,4050
1,4125
1,4235
1,4312
1,4341
1,4389
1,4392
1,4339
1,4369
80
80
75
90
60
80
80
80
75
80
90
80
85
50
60
60
а Цифра в скобках указывает давление (мм рт. ст.), которому соответствует данная температура кипения.
6 Сульфат применяют из-за его более высокой температуры кипения. В этих условиях в реакцию вступают обе алкильные группы. Продукт
реакции не следует промывать раствором поваренной соли.
318 Г Препаративная часть
При полумикроколичествах можно работать без перемешивания и внутреннего термомет-
термометра, температуру контролируют температурой нагревательного прибора.
Алифатические нитрилы можно получать с прекрасными выходами при ис-
использовании в качестве растворителя диметилсульфоксида: Smiley R. A., Arnold С.
J. Org. Chem., I960, 25, 257; Friedman L., Shechter H. J. Org. Chem., I960, 25, 877
(цитируемые работы содержат многочисленные примеры).
В препаративном отношении нитрилы являются очень важными соединения-
соединениями, так как они легко доступны и обладают способностью вступать в разнообраз-
разнообразные реакции. Так, при гидролизе они превращаются в амиды и далее в карбоно-
вые кислоты (разд. Г,7.1.5), а при восстановлении в алкиламины (разд. Г,7.1.7 и
Г.7.3.1.1):
R-CEN + Н2О - R-C 2— NH3+R-C [Г.2.108]
NH2 ОН
R-CEN + 2H2 R-CH2-NH2 [Г.2.109]
Реакции цианидов щелочных металлов с галогенсодержащими соединениями нашли при-
применение также в промышленности, например:
Сложные эфиры:
Ph-CH2-COOH (— "ахучие Bei«eCTBa<
rii on2 owwn 1 фармацевтические
/ Фенилуксусная препараты)
Ph-CH2-CI Ph-CH2-CN ( кислота
бензилцианид \ [Г.2.110] 11 -2.110J
Ph-CH2-CH2-NH2
^-фенилэтиламин
С1-СН2-СООН NC-CHj-COOH НООС-СН2-СООН (-^фармацевтические
циануксусная малоновая препараты) [Г.2.111]
кислота кислота
D-изобутилфенил)ацетонитрил [Г.2.112]
i-Bu—f ^>—CH-CN i-Bu—(/ ~%— СН-СООН
СН3
ибупрофен
(противовоспалительное,
противоревматическое
средство)
2.7. НУКЛЕОФИЛЬНОЕ ЗАМЕЩЕНИЕ
В КРЕМНИЙОРГАНИЧЕСКИХ СОЕДИНЕНИЯХ
Определенные кремнийорганические соединения способны реагировать так же,
как их углеродные аналоги. Так, силаны с соответствующей уходящей группой у
атома кремния можно подвергнуть нуклеофильному замещению. Триметилси-
лилхлорид реагирует с аминами, амидами, спиртами и другими нуклеофилами,
2. Нуклеофильное замещение при насыщенном атоме углерода
319
образуя триметилсилиламины, триметилсилиламиды, алкокситриметилсиланы
и другие соединения с триметилсилильной группой, например:
Н3с
Н3С—Si—Cl
Н3С
2HNR2
Н3С
H3C-Si—NR2
Н3С
©
R2NH2CI
0
[Г.2.113]
Реакция идет по механизму SN2. В особых случаях в качестве промежуточных
продуктов обсуждалась возможность образования соединений пятикоордина-
ционного кремния с участием d-электронов.
Многие триметилсилильные соединения получают не прямым действием
триметилсилилхлорида; целесообразнее осуществить реакцию обмена с предва-
предварительно синтезированным силилированным амином или амидом, например:
Me3Si—NEt2 + H2N-R
-HNEt2
Me3Si—NH-R
[Г.2.114]
Реакция катализируется кислотами, а иногда также сульфатом аммония.
(ГП) Общая методика триметилсилилирования амино- и гидроксильных соединений (табл. Г.2.1 IS)
I Внимание! Все используемые приборы должны быть сухими, а реагенты безводными!
В трехгорлую колбу на 250 мл, снабженную мешалкой, капельной воронкой и обратным
холодильником с хлоркалыщевой трубкой, помещают 100 мл абсолютного эфира, 0,2 моль ис-
исходного соединения и 0,2 моль триэтиламина (в случае гидроксильных производных —
0,2 моль пиридина). При реакциях с амидами в качестве растворителя используют 100 мл то-
толуола. Добавляют в течение 20—30 мин раствор 0,2 моль свежеперегнанного триметилхлорси-
лана в 50 мл применяемого растворителя. При реакциях с аминосоединениями реакционную
смесь нагревают 2 ч на водяной бане.
Затем реакционную смесь тщательно закрывают и оставляют на 12 ч, после этого отсасы-
отсасывают, осадок промывают 30 мл растворителя, который затем отгоняют в вакууме, продукт ре-
реакции перегоняют.
Таблица Г.2.115. Триметилсилилирование амино- и гидроксильных соединений
Продукт реакции
Исходное вещество
Т. кип.а, -С
Выход,
%
N-Триметилсилилднэтиламин
N-Триметилсилилпиперндин
N-Триметилсилилацетамид11
N-Метил-М-триметилсилил-
формамид
Бутокситриметилсцлан
Циклогексилокситрнмстил-
силан
п-Бромфенокснтриметилси-
лан
Диэтиламин6
Пиперидин
Ацетамид
N-Метилформамид
Бутанол
Циклогексанол
п-Бромфенол
126; 40 B0)
161; 66 B5)
84 A3);
78 (8)
64A2)
123ч-124
169-М70;
53 A0)
113 A4)
1,4112
1,4423
1,4179
1,4408
1,3930
1,4315
1,5145
75
60
65
70
50
65
55
а В скобках указаны цифры, относящиеся к давлению (мм рт. ст.), для которого
приведена температура кипения.
Для реакции взять 0,4 моль; добавлять триэтиламин не нужно.
в Т. пл. 29^-33 °С. Если происходит кристаллизация в холодильнике, перекрывают
подачу воды и дают холодильнику немного прогреться.
320 Г Препаративная часть
Триметилхлорсилан — важное вспомогательное вещество в органическом
синтезе. Триметилсилильный остаток используется как защитная группа для
карбоксильной группы, легко отщепляемая гидролизом, например при синтезе
пептидов (см., например, [Г.7.54]). Силильные эфиры енолов — важные проме-
промежуточные продукты при замещении карбонильных соединений в р-положении
(разд. Г,7.4.2). Триметилсилильный остаток можно направленно заместить
электрофилами, например в реакции с алкилгалогенидами. В аналитических це-
целях смеси термически неустойчивых амино- и гидроксилсодержащих соедине-
соединений превращают в триметилсилильные производные и затем разделяют методом
газо-жидкостной хроматографии. Триметилхлорсилан служит также для маски-
маскирования известных нуклеофильных агентов:
Me3Si-CN -К™ Me3Si-CI N°N?, - Me3Si—N=N=N1 [Г.2.116]
Триметилсилилцианид для некоторых реакций предпочтительнее свободной
синильной кислоты (уравнение [Г.7.117]). Триметилсилилазид использовать
удобнее, чем взрывчатую азотоводородную кислоту.
Триметилсилилазид: BirkhoferL., Wegener P. Org. Syntheses. 1970, 50, 107.
Триметилсилилхлорид в промышленности получают из метилхлорида и кремния (синтез
Мюллера — Рохова). Он легко гидролизуется до триметилсиланола, который далее быстро
межмолекулярно конденсируется с образованием гексаметилдисилоксана. Эту особенность
хлорсиланов используют в технике при синтезе силиконов. При этом диалкил- или диарил-
дихлорсиланы гидролизуют до дизамещенных силандиолов, которые далее вступают в реак-
реакцию поликонденсации.
Силиконы представляют собой высокомолекулярные полисилоксаны, они производятся в
виде силиконовых масел, смол или каучуков.
При персилилировании целлюлозы с помощью триметилхлорсилана получают раствори-
растворимые производные целлюлозы, благодаря чему расширяются возможности применения этих
биополимеров.
О фториндуцированном расщеплении простых силиловых эфиров, включающих цикли-
циклические пероксиды, которое сопровождается хемилюминисценцией, и о значении этой реак-
реакции для медицинского и биохимического анализа см.: Albrecht S., Brand! H., Adam W.
Chem.unserer Zeit, 1990, 24, 227-238.
2.8. ЛИТЕРАТУРА
Механизм нуклеофильного замещения у насыщеного атома
Bentley Т. W., Schleyer P. R. Adv. Phys. Org. Chem. 1977, 14, 1.
Bunton С. A. Nucleophilic Substitution at a Saturated Carbon Atom. Elsevier, Amsterdam,
London, New York, 1963.
GompperR. Angew. Chem., 1964, 76, 412-424.
Harris J. M. Progr. Phys. Org. Chem., 1974, 11, 89-173.
Hartshorn S. R. Aliphatic Nucleophilic Substitution. Cambridge University Press, Cambridge, 1973.
De la Mare P. B. D., Swedlund B. E. In: Chemistry of the Carbon-Halogen Bond. S. Patai
(ed.). — John Wiley & Sons, London, New York, Sidney, Toronto, 1973, p. 407-490.
Parker A. I. Quart. Rev. (Chem. Soc., London), 1962,16,163-187; Adv. Org. Chem., 1965,
5, 1; Adv. Phys. Org. Chem., 1967, 5, 173.
Рудаков Е. С Кожевников И. В., Самашиков В. В. Усп. хим., 1974, 43, 707-726.
2. Нуклеофильное замещение при насыщенном атоме углерода 321
Streitwieser jun. A. Solvolytic displacement reactions. — McGraw-Hill Book Сотр.,
New York, 1962; Chem. Rev., 1956, 56, 571-752.
Thornton E. R. Solvolysis Mechanisms. — Ronald Press Сотр., New York, 1964.
Нуклеофильное замещение с участием анион-радикалов
Белецкая И. П., Дрозд В. Н. Усп. хим., 1979, 48, 793-828.
Chanon M., Tobe M. L. Angew. Chem., 1982, 94, 27.
Kornblum N. Angew. Chem., 1975, 87, 797-808.
Нуклеофильное замещение в условиях межфазного катализа
DehmlowE. V. Angew. Chem., 1977, 89, 521; 1974, 86, 187.
Демлов Э., Демлов С. Межфазный катализ. — М.: Мир, 1987.
Starks С. М., Liotta С. Phase Transfer Catalysis. — Academic Press, New York, 1978.
Вебер В., Гокель Г. Межфазный катализ в органическом синтезе. — М.: Мир, 1980.
Получение алкилгалогенидов из спирта
RoedigA. In: Houben-Weyl, Bd. 5/4, 1960, S. 361-411, 610-628.
Stroh R. In: Houben-Weyl, Bd. 5/3, 1962, S. 830-838, 862-870.
РеакцияМицунобу
Mitsunobu О. Synthesis, 1981, 1-28.
Hughes D. L, Org. React. 1992, 42, 335-656.
Гидролиз геминальных дигалогенидов в альдегиды
Bayer О. In: Houben-Weyl, Bd. 7/1, 1954, S. 211-220.
Получение простых эфиров
Meerwein H. In: Houben-Weyl, Bd. 6/3, 1965, S. 10-40.
Расщепление простых эфиров
Bhatt M. V., Kalkqmi S. U. Synthesis 1983, 249-282.
BurwellJr. R. L. Chem. Rev., 1954, 54, 615-685.
Meerwein H. In: Houben-Weyl, Bd. 6/3, 1965, S. 143-171.
Roth H., Meerwein H. In: Houben-Weyl, Bd. 2, 1953, S. 423-425.
Press J. В., Synth. Commun. 1979, 9, 407.
Получение тиолов, тиоэфиров и тиоцианатов
Field L. Syntheses, 1972, 101; 1978, 713.
Gundermann K.-D., Humke K. In: Houben-Weil. Bd. Ell/1, 1985, S. 32-63, 158-187.
SchoberlA., Wagner A. In: Houben-Weyl, Bd. 9, 1955, S. 7-19, 97-113.
Bdgemann M., Petersen S., Schultz O.-E., SoilH. In: Houben-Weil. Bd. 9, 1955, S. 856-867.
HartmannA. In: Houben-Weil. Bd. E4, 1983, S. 834-883, 940-970.
Получение эфиров карбоновых кислот алкилированием солей карбоновых кислот
Henecka H. In: Houben-Weyl, Bd. 8, 1952, S. 514-543.
PielartzikH., Irmisch-PielartzikB., Eicher T. In: Houben-Weil. Bd. E 5/1, 1985, S. 684-690.
322 Г Препаративная часть
Реакция Финкельштейна
Miller J. A., Nunn M. J. J. Chem. Soc, Perkin Tr. 1,1976,416.
RoedigA. In: Houben-Weyl, Bd. 5/4, 1960, S. 595-605.
Получение фторидов
Бокемюллер В. В кн.: Новые методы препаративной органической химии. — М.: ИЛ,
1950, с. 7-31.
Forche W. E. In: Houben-Weyl, Bd. 5/3, 1962, S. 1-397.
Хеше А. В сб.: Органические реакции. Сб. 2. — М.: ИЛ, 1950, с. 51-105.
Получение алифатических нитросоединений
Корнблюм Н. В сб.: Органические реакции Сб. 12. — М.: Мир, 1965, с. 117.
Padeken И. G. In: Houben-Weyl, Bd. 10/1, 1971, S. 46-60.
Получение нитрилов из алкилгалогенидов, алкилсульфатов и
других алкилпроизводных
Gundermann С. In: Houben-Weil. Bd. E 5/2, 1985, S. 1447-1485.
KurtzP. In: Houben-Weyl, Bd. 8, 1952, S. 290-311.
MowryD. T. Chem. Rev., 1948, 42,189-284.
Получение аминов из галогенопроизводных
Gibson M. S., Bradshaw R. W. Ang. Chem., 1968, 80, 986.
Spielberger G. In: Houben-Weyl, Bd. 11/2, 1975, S. 24-108.
Получение четвертичных аммониевых соединений
GoerdelerJ. In: Houben-Weyl, Bd. 11/2, 1958, S. 591-630.
Получение четвертичных фосфониевых соединений
Josden К. In: Houben-Weil. Bd. E 1, 1982, S. 491-572.
Sasse K. In: Houben-Weil. Bd. 12/1, 1963, S. 79-104.
Реакция Михаэлиса - Арбузова
Bhattacharva А. К., Thyagarajan G. Chem. Rev., 1981, 81, 415-130.
HeydtH., RegitzM. In: Houben-Weil. Bd. E2, 1982, S. 19-23, 198-204, 366-377.
Sasse K. In: Houben-Weil. Bd. 12/1, 1963, S. 150-152, 251-257, 433-446.
Реакции нуклеофильного замещения в триалкилсилилгалогенидах
Burkhofer L., Stuhl O. In: Topics in Current Chemistry, Bd. 88, — Springer-Verlag, Berlin,
Heidelberg, New York, 1980, p. 33-88.
Klebe J. F. In: Advances in Organic Chemicstry, Bd. 8, — Wiley-Interscience, New York,
London, 1972, p. 97-178.
3. Элиминирование с образованием кратных углерод-углеродных связей
323
3. ЭЛИМИНИРОВАНИЕ С ОБРАЗОВАНИЕМ
КРАТНЫХ УГЛЕРОД-УГЛЕРОДНЫХ СВЯЗЕЙ
В реакциях элиминирования из одной молекулы отщепляются два атома или
две группы (например, Y и X). Взаимное расположение отщепляющихся групп
определяет строение продукта реакции и может служить основой для классифи-
классификации. Наиболее важные случаи:
а) а, р (или 1,2)-элиминирование протекает с образованием кратных связей:
Y-C-C-X
I I
\ /
С=С
/ \
[Г.3.1]
б) ос,а (или 1,1)-элиминирование приводит к образованию структур с
секстетом электронов, например, карбенов:
Y-C-X
1С
[Г.3.2]
Элиминирование может протекать по ионному, радикальному или перицик-
лическому механизму реакции.
3.1. РЕАКЦИИ ИОННОГО а,р-ЭЛИМИНИРОВАНИЯ
Наиболее важные реакции ионного а,р-элиминирования приведены в табл.
Г.3.3. С помощью а,р-элиминирования наряду с олефинами и ацетиленами
Таблица Г.З
44- _
н он
44- -
HOR
44- -
Н Hal
н' XHal
1 ^0
Н Cl
JU- -
®NR3 ОН9
3. Важнейшие реакции ионного элиминирования
Схема связи
- X
- X
- >=<
-^ -с^с- +
- >=с=о
+ н-он
+ H-OR
+ H-Hal
H-Hal
+ H-Cl
+ H-NR3 ОН9
* H20 + NR3
Название процесса3
Дегидратация
(а,Р-гидрогидроксиэлимини-
рование)
а,Р-Гидроалкоксиэлиминиро-
вание
а,Р-Дегидрогалогенирование
Синтез алкина
Синтез кетенов
Расщепление по Гофману
четвертичных аммониевых
солей
(а,Р-Гидротриалкиламино-
элиминирование)
В скобках по номенклатуре ИЮПАК.
324
Г Препаративная часть
препаративно доступными становятся и гетероаналоги непредельных соедине-
соединений (например, >C=N-, -C=N).
3.1.1. ЗАМЕЩЕНИЕ И ЭЛИМИНИРОВАНИЕ КАК КОНКУРИРУЮЩИЕ РЕАКЦИИ.
МЕХАНИЗМ РЕАКЦИЙ ИОННОГО ЭЛИМИНИРОВАНИЯ
Нуклеофильное замещение (разд. Г.2) и ионное элиминирование являются про-
процессами, которые протекают по приведенным механизмам. В обоих случаях
речь идет о реакции одного основания Льюиса В| с субстратом RX, причем из RX
вытесняется нуклеофуг X. При нуклеофильном замещении основание В| реаги-
реагирует как нуклеофил и образует с R связь R—В [Г.2.5]; в случае элиминирования
В уже как основание отщепляет протон от р-углеродного атома остатка R с об-
образованием олефина:
н-с-схх
нв
\ /
С=С + XI
/ \
0
[Г.3.4]
И нуклеофильный агент, и уходящая группа могут быть также незаряженными основани-
основаниями Льюиса.
Кроме протона от подходящего субстрата можно отщеплять также другие положительно
заряженные молекулярные фрагменты, например:
0
0 I I
н2о-с-с-с-он
Н2О
V
+ С-ОН
-нг
[Г.3.5]
но-с-с-с-он
с=о
Реакции такого типа называют реакциями фрагментации [Grob С. A., Schiess P. W.
Angew. Chem., 1967, 79, 1; Grob С. A. Angew. Chem., 1969, 81, 543]. Формально эли-
элиминирование можно рассматривать как особый случай этого типа реакций.
При ионном а,р-элиминировании в зависимости от соотношения скоростей
расщепления С—Х-связи и расщепления С—Н-связи можно различить несколь-
несколько крайних случаев.
Мономолекулярное элиминирование Е1. Расщепление С—Х-связи предшеству-
предшествует расщеплению связи С—Н. Подобно SnI-реакциям, прежде всего образуется
карбениевый ион, из которого во второй стадии отщепляется протон и образу-
образуется олефин:
BI
0
I I
н-с-с-х
I /
н-с-с©
I \
I /
н-с-с©
I \
нв +
\ /
/с=с\
[Г.3.6]
3. Элиминирование с образованием кратных углерод-углеродных связей 325
Скорость реакции определяет первая стадия; в уравнение скорости Е1-реакции
входит только концентрация субстрата RX, а концентрация основания В на ско-
скорость реакции не влияет.
У соединений, дейтерированных по р-углеродному атому, наблюдается вто-
вторичный изотопный эффект: &hAd~ 1,1—1,2. Электронодонорные заместители у
а-углеродного атома, как и можно ожидать, повышают скорость элиминирова-
элиминирования. В случае этих субстратов в уравнении Гаммета константа реакции оказыва-
оказывается отрицательной (разд. В,5.2).
Бимолекулярное элиминирование Е2. Перегруппировка связей проходит в
одну стадию через переходное состояние, в котором участвуют субстрат и
реагент:
+ н-с-с-х
I I
8" I I 8-Г \
е
B—H — C-C—X HB + C=C + ХГ [Г.3.7]
\
Субстрат и основной агент фигурируют в уравнении скорости реакции. У соеди-
соединений, дейтерированных по р-углероду, наблюдается большой первичный
изотопный эффект (&hAd = 3—7). В случае элиминирования соответствующих
субстратов константа реакции Гаммета положительна и имеет величину 2—3.
Мономолекулярное элиминирование ElcB. Расщепление С—Н-связи предшест-
предшествует расщеплению связи С-Х. В первой стадии реакции образуется карбанион
(сопряженное основание «сВ» субстрата), который теряет X на второй мономо-
мономолекулярной стадии:
„0
\е
вг + н-с-с-х ^^ нв + ю-с-х
II / I
\е I \ / G
ic-c-x —- с=с + хи
/ I / \
У соединений, дейтерированных по р-С-атому, не обнаруживается кинети-
кинетического изотопного эффекта. Константа реакции для этого типа реакции имеет
большое положительное численное значение1.
В действительности наблюдаемые механизмы лежат между описанными
крайними случаями, т. е., как правило, наблюдается согласованное, но не синх-
синхронное отщепление X и Н.
На соотношение между элиминированием и замещением влияют следующие
факторы:
— электронные и пространственные эффекты в субстрате и реагенте;
— эффекты растворителя;
1 Этот ElcB-механизм реализуется только в случае очень устойчивых промежуточных
карбанионов.
326 Г Препаративная часть
- температура (повышение температуры в общем благоприятствует эли-
элиминированию):
HpS04 180'С Г Г Т 01
С2Н5ОН —-— - Н2С=СН2 + Н2О (Элиминирование) l J
HpSOj 130'С
2С2Н5ОН —— С2Н5-О-С2Н5 + Н2О (Замещение) [Г.3.10]
3.1.1.1. Мономолекулярное элиминирование
Для препаративных целей Е1-реакции в общем нежелательны, поскольку про-
промежуточно образующийся катион может далее реагировать не только в направ-
направлении элиминирования, но также вступать в побочные реакции замещения и
перегруппировки. К тому же Е1-реакции часто оказываются обратимыми
(электрофильное присоединение; разд. Г,4). При этом, если конкурирующая
SnI-реакция, наоборот, необратима, равновесие в условиях термодинамическо-
термодинамического контроля смещается в сторону продукта замещения (разд. В,3.2).
В условиях кинетического контроля названные выше факторы влияют на
соотношение замещения и элиминирования по-разному.
Влияние электронных и пространственных эффектов в субстрате и реагенте.
Как и при SnI-реакциях, субстраты, карбениевый ион которых стабилизирован
+/- или +Л/-эффектами, склонны к El-реакциям. Решающим для образования
олефина является строение алкильного остатка в субстрате. Так, при катализи-
катализируемой кислотами дегидратации спиртов доля олефинов (по отношению к
продуктам замещения — простым эфирам) возрастает в ряду первичный
спирт < вторичный спирт < третичный спирт. Особенно благоприятны условия
образования карбениевых ионов и протекания реакции по механизму элимини-
элиминирования Е1 при каталитической дегидратации вторичных и третичных спиртов,
например:
НзС © я, СН3 СН3
н3с-с-он2 медленн? н3с-с + н2о —- н2с=с + н3о® [Г.3.11]
Н3С СНз СН3
В быстрой стадии протежирования, предшествующей скоростьопределяющей стадии, об-
образуется ион оксония I, который мономолекулярно распадается с образованием карбениевого
иона II и энергетически обедненной молекулы воды.
Как указывалось в разд. Г,2.2, в промежуточном карбениевом ионе и олефи-
не уменьшается пространственное напряжение, создаваемое объемистыми за-
заместителями. Поэтому элиминирование является тем более выгодным, чем
больший объем занимают окружающие карбениевый углерод остатки. Так, нап-
например, при сольволизе /л/?ет-пентилхлорида образуется 34% олефина, а при
сольволизе 4-хлор-2,2,4-триметилпентана и 4-хлор-2,2,4,6,6-пентаметилгепта-
на выходы олефинов составляют соответственно 65 и даже 100%. (Напишите
уравнения этих реакций!)
3. Элиминирование с образованием кратных углерод-углеродных связей 327
При строго мономолекулярном протекании реакции природа уходящей
группы не влияет на соотношение продуктов замещения и элиминирования,
поскольку этот фактор существен лишь для образования карбениевого иона.
Легко уходящие группы X (F<<CKBr<KN2+) способствуют элиминирова-
элиминированию. При катализируемой кислотами дегидратации спиртов типичной уходя-
уходящей группой X является Н2О, при образовании олефинов из эфиров серной
кислоты или сульфоновых кислот — группы ROSC>3~ и RSC>3~.
Поскольку отщепление протона происходит из очень энергетически обога-
обогащенного катиона, эта стадия обычно протекает чрезвычайно быстро. Поэтому
на ходе реакции обычно не сказывается основность реагентов В.
Влияние реакционной среды. Сильно сольватирующие полярные протонные
растворители (вода, первичные спирты, муравьиная кислота) стабилизируют
анионы и карбениевые ионы, благоприятствуя тем самььм протеканию мономо-
мономолекулярных реакций. Например, по El-механизму проходит сольволитическое
дегидрогалогенирование вторичных и третичных алкилгалогенидов:
/)— СН-СНз В НС00Н. <{ \_Сн-сНз рг- <л \— СН = СН2 [Г.3.121
// I J кипячение \ // и® \ // ц-->.1.ь|
/ С| / -н \—/
и сольволиз эфиров серной кислоты или сульфоновых кислот:
\ / кипячение \ / _н© \ // [Г.3.13]
Образования какого побочного продукта можно ожидать в реакции (Г.3.13)?
3.1.1.2. Бимолекулярное элиминирование
Механизмы Е2 и SN2 различаются между собой значительно сильнее, чем Е1 и Sn 1 •
При Е2-реакции атака В направлена на атом Н при р-углероде субстрата, т. е. на пе-
периферию молекулы; при 8н2-реакции, напротив, атакуется а-углеродный атом.
Поэтому соотношение продуктов элиминирования и замещения в бимолекуляр-
бимолекулярных реакциях легче регулируется. Для этих целей удобно использовать действие
электронных и пространственных факторов, а также эффекты растворителей.
Е2-механизм предпочтительнее в препаративном отношении также потому, что
при этих реакциях меньше опасность побочных перегруппировок. Реакции элими-
элиминирования во вторичных алкильных соединениях, которые часто протекают по
промежуточному между Е1 и Е2 механизму, можно сдвинуть в сторону механизма
Е2, более предпочтительного для препаративных целей. Этого можно достичь даже
в случае третичных алкильных соединений, которые в обычных условиях склонны
к образованию карбениевых ионов, т. е. к Е1-реакциям.
Влияние электронных эффектов. Атака основания на протон при р-углероде
субстрата облегчается, если а-углерод—это (по-возможности) мягкий электрофиль-
ный центр, т. е. если уходящая группа X не создала на нем слишком большого поло-
положительного заряда. Низкая тенденция X к отщеплению благоприятствует синхрон-
синхронному протеканию трех стадий элиминирования. К бимолекулярному элиминирова-
элиминированию особенно склонны соединения с заместителями— NR/, -PR3+ и -SR2+
(гофмановское расщепление гидроксидов аммония, фосфония и сульфония).
328 Г Препаративная часть
При Е2-реакции В выступает в роли партнера протона. Поэтому реакцион-
реакционная способность реагентов в реакциях элиминирования определяется его основ-
основностью (разд. В,4). Таким образом, для гидроксид—и алкоголят-анионов наблю-
наблюдается следующий ряд реакционной способности: OH~<MeO-<EtO"<
изо-РЮ~<трет-ВиО~. Наряду с гидроксидами и алкоголятами в качестве реа-
реагентов часто применяют также R3N, Nfb", RCOO. Существенно также то, что
основность реагентов не ослабляется протонными растворителями.
Применяя сильные и концентрированные основания (концентрация В вхо-
входит в уравнение скорости Е2-реакции), часто оказывается возможным сдвинуть
элиминирование в область механизма Е2 или ElcB. Так, с помощью сильных ос-
оснований удается бимолекулярно отщеплять кроме упомянутых выше и другие
нуклеофугные группы, например С1~, Вг~ и I" при дегидрогалогенировании
алкилгалогенидов, —OSO2R и —OSO2OR при образовании олефинов из эфиров
сульфоновых кислот или серной кислоты. (Напишите уравнения этих реакций!)
Влияние пространственных эффектов. Экранирование а-углерода субстрата
сильно разветвленными алкильными остатками затрудняет реакцию замещения
по сравнению с элиминированием, при котором В атакует периферию молеку-
молекулы. В таком же направлении действуют объемистые основания, которые могут
приблизиться к протону у р-, но не у а-углерода.
Поэтому при действии объемистых сильных оснований на третичные алкил-
галогениды идет исключительно отщепление галогеноводорода, а не замещение
у а-углерода. Последнее с подобными субстратами можно осуществить только в
условиях сольволиза.
К очень объемистым основаниям относятся, например, трет-бугтаты щелоч-
щелочных металлов, дициклогексилэтиламин, а также некоторые амидины:
1,5-диазабицикло[4.3.0]нонен-5 (ДБН) и 1,8-диазабицикло[5.4.0]ундецен-7 (ДБУ):
[Г.3.14]
ДБН ДБУ
Например, из октилбромида при элиминировании действием дициколгек-
силэтиламина получают 99% октена-1. Замещение (кватернизация амина) прак-
практически не наблюдается. Названные амидины1 позволяют осуществлять реак-
реакции элиминирования при низких температурах, как, например, дегалогенирова-
ние чувствительных субстратов.
Влияние реакционной среды. Апротонные растворители, например диметил-
формамид и диметилсульфоксид, слабо стабилизируют анионы и поэтому не
способствуют отщеплению X из RX. Поскольку такие растворители не ослабля-
ослабляют основность реагента В из-за образования водородных связей, они представ-
представляют собой хорошую среду для бимолекулярного элиминирования.
Для того чтобы сдвинуть реакции элиминирования в препаративно выгод-
выгодную Е2-область, выбирают уходящие группы X с низкой тенденцией к отщепле-
1 Такие амидины препаративно легкодоступны (разд. Г,7.1.5). Об их применении при
реакциях элиминирования см. OedigerH., MollerF., EiterK., Synthesis, 1972, 591.
3. Элиминирование с образованием кратных углерод-углеродных связей 329
нию, используют объемистые, сильные основания в большой концентрации и
работают с полярными апротонными растворителями.
3.7.2. ВЛИЯНИЕ ПОРЯДКА РЕАКЦИИ И ОБЩИХ ПРОСТРАНСТВЕННЫХ
СООТНОШЕНИЙ НА НАПРАВЛЕНИЕ ЭЛИМИНИРОВАНИЯ
Для вторичных и третичных исходных соединений элиминирование может идти
в двух направлениях и приводить к олефинам с различным положением двой-
двойных связей:
/3 а /3
4 13 12 И
—с-с-с-с
I I -НХ
Н X Н
По Зайцеву
(Д-олефин)
n n ~-n По Гофману
I I (А-олефин)
[Г.3.15]
Если образуется олефин с наибольшим числом алкильных групп при двой-
двойной связи, то говорят об элиминировании по Зайцеву. Протон отщепляется
здесь от р-углерода — атома, который несет наибольшее число алкильных групп.
Элиминирование по Гофману имеет место тогда, когда образуется олефин с
меньшим числом алкильных групп. Протон отщепляется в данном случае от
C-углеродного атома, несущего небольшое количество алкильных групп. Как
правило, олефины, образующиеся по Зайцеву, термодинамически более
стабильны, чем получающиеся по Гофману.
В большинстве случаев мономолекулярное элиминирование дает преимуще-
преимущественно соединения по Зайцеву, например сольволитическое дегидрогалогени-
рование вторичных и третичных алкилгалогенидов и тозилатов, а также дегидра-
дегидратация вторичных и третичных спиртов.
сн3
— Н3С-СН=С-СН3 87,5% (поЗайцеву)
н3с-сн2-с-сн3
СН3
i 15%-HanH2SO4
I J -Н2О СН3
ОН '
— Н3С-СН2-С=СН2 12,5% (поГофману)
[Г.3.16]
Надо отметить, что на результат элиминирования [Г.3.16] влияет еще и вероятностный
фактор: для элиминирования в гофмановский олефин (двойная связь при а-С) в распоряже-
распоряжении имеются шесть атомов водорода по сравнению с всего двумя атомами водорода при эли-
элиминировании по Зайцеву. Таким образом, гофмановская реакция должна быть в три раза
более вероятной, чем образование р-олефина. Обратите внимание на этот фактор и в других
случаях при объяснении направления элиминирования.
При бимолекулярном элиминировании часто предпочтительным оказывает-
оказывается образование термодинамически менее устойчивого олефина.
Элиминированию по Гофману способствуют:
1) уменьшение тенденции к отщеплению нуклеофугной группы, что выра-
выражается рядом
330
Г Препаративная часть
N2©>
R2S®>
R3N®
[Г.3.17]
Данные табл. Г.3.18 подтверждают общее правило: легко уходящие группы благоприят-
благоприятствуют протеканию элиминирования по Зайцеву. Субстраты с положительно заряженными
группами (например, триалкиламмониевые) обычно дают продукты элиминирования по Гоф-
Гофману. Термическое разложение триалкиламмонийгидроксидов называют элиминированием
по Гофману в узком (историческом) смысле.
Таблица Г.3.18. Влияние легкости отщепления заместителя на направление эле-
минирования
СН3-СН2-СН2-СН-СНз -^
X
X
Выход олефина по Зайцеву, %
по Гофману, %
CjHjl
F
17
83
CH3-CH2-CH2-CH=CH2
сн3—сн2—сн=сн—сн3
Cl
64
36
по Гофману
по Зайцеву
Br
75
25
I
80
20
2) увеличение объема и силы основания, а также степени экранирования в
субстрате (табл. Г.3.19).
Таблица Г.3.19. Влияние пространственного фактора на выход олефина при дегид-
робромировании 2-бром-2-метилбутана различными алкоголятами калия
сн3
н3с-сн2-с-сн3
Вг
+ ROK
- ROH, - КВг
СН3
Н 3С С Н —~ С
СН3
н3с-сн2-с=сн2
&2-Олефин (по Зайцеву)
А^-Олефин (по Гофману)
Основание
Выход Д1-олефина, %
Выход Д2-олефина,%
Н3С-СН2-ОК
38
62
сн3
н3с-с-ок
СН3
73
27
СН3
н3с-с-ок
н3с-сн2
78
22
Н3С CH2
н3с-сн2-с-ок
Н3С СНз
89
11
Некоторые примеры влияния свойств и природы оснований приведены в табл. Г.3.19.
С небольшим по объему молекулы метилатом калия наблюдается преимущественное об-
образование продукта по Зайцеву, тогда как более объемистые основания приводят к гофмано-
вскому продукту. Следует ожидать, что элиминирование по Гофману становится более пред-
предпочтительным, по мере того как затрудняется подход отщепляющего протон основания к
«внутреннему» водороду, уходящему при элиминировании по Зайцеву. Влияние этого прост-
пространственного фактора может быть обусловлено строением как реагента, так и субстрата. Так,
при дегидратации 2,4,4-триметилпентанола-2 образуется преимущественно олефин по
Гофману (путь А):
3. Элиминирование с образованием кратных углерод-углеродных связей
331
Me Me
Me Me
Me Me
Me,' ^l Me
Me ОН
н н
Me'
Me
Me Me
по Гофману
(80 %)
[Г.3.20]
по Зайцеву
Me B0%)
Причиной этого является тот факт, что основание, необходимое для отщепления протона
(здесь вода), лишь с трудом может подойти к водороду при С-3 (путь В), так как он экраниро-
экранирован объемистыми метальными группами. Этот пример относится к довольно редкому случаю,
когда Е1-элиминирование идет с образованием олефина по Гофману, тогда как обычно осно-
основание, отщепляющее протон, без затруднения может подойти к «внутреннему» водороду.
3.7.3. СТЕРЕОЭЛЕКТРОННЫЕ ЭФФЕКТЫ И НАПРАВЛЕНИЕ ЭЛИМИНИРОВАНИЯ.
ПРОСТРАНСТВЕННЫЙ ХОД ЭЛИМИНИРОВАНИЯ
Параллельное расположение /?-атомных орбиталей, которое возникает при элиминировании
по а- и р-С-атомам, способствует их перекрыванию и может привести к образованию тс-связи.
Этот стереоэлектронный эффект при реакциях элиминирования необходимо дополнительно
учитывать наряду с рассмотренными общими эффектами других пространственных факторов.
Согласно правилу Ингольда, бимолекулярное элиминирование протекает особенно глад-
гладко, когда отщепляющиеся заместители находятся относительно друг друга в антиперипланар-
ной конформации (ср. [В.81], I). При этом четыре центра X, С, С и Y, принимающие участие в
реакции, расположены в одной плоскости. Поэтому также говорят об аноти-элиминировании.
Условия копланарности выполняются также в случае синперипланарной конформации
[В.81, IV]. Поэтому в особых случаях возможно также протекание Е2-элиминирования из этой
конформации (так называемое смн-элиминирование). Однако, в силу того что данная конфор-
мация энергетически невыгодна (см. выше), Е2-реакции протекают в общем стереоспецифич-
но как шдам-элиминирование (табл. Г.3.21).
Стереоэлектронные эффекты имеют значение для ациклического ряда соединений при
бимолекулярном элиминировании в тех случаях, если образующийся олефин может существо-
существовать в виде (Z)-, (?)-изомеров'. Покажем это на примере отщепления НВг или DBr из эритро-
2-бром-1-дейтеро-1,2-дифенилэтана. При действии алкоголята образуется (?)-стильбен,
Таблица Г.3.21. Пространственные эффекты при бимолекулярном элиминирова-
элиминировании НХ
1 Для однозначного отнесения (<ис-т/>анс-изомеров по (.^^-номенклатуре см. разд. В,7.2.
При этом заместители в олефине учитываются по правилу [Cahn R. S., Ingold С. К., Prelog V.,
Angew. Chem., 1966, 78,413]: если два старших заместителя стоят по одну сторону двойной свя-
связи, изомер получает обозначение (Z) (zusammen), в противоположном случае — (Е) (entgegen).
332 Г Препаративная часть
причем молекула теряет 91% своего дейтерия. Элиминирование происходит он/яи-копланарно
из конформации [Г.3.23, IV]. Она энергетически выгодна, так как оба объемистых фенильных
остатках пространственно не мешают друг другу:
D H Ph H
h .. )=( [Г.3.22]
Вг "°ВГ Н Ph
Ph H Ph „ D
H+D. ^оЛг H>cVBr
H-^Br' Sk PrAKD
I PhPh Ph DPh Ph HPh Ph
I II III IV V VI
Из конформации II и V невозможно ни син-, ни он/яи-копланарное элиминирование. Из
конформации I и III возможно син-копланарное, а из конформации IV и VI — онти-копланар-
ное элиминирование. Рассмотрите, отщепляется при этом НВг или DBr, а также образуется
(?)- или (ZJ-олефин.
Влияние конформации приобретает большое значение прежде всего для алициклических
соединений, так как здесь относительное положение заместителей закрепляется в результате
образования цикла. Поясним это на примере системы циклогексана. В системе циклогексана
плоское расположение атомов углерода не представляется возможным из-за возникающих нап-
напряжений и поэтому молекула может существовать в форме кресла или ванны, см. разд. В,7.1.
Приложение правила Ингольда к таким алициклическим системам приводит, согласно
Бартону, к следующей закономерности: бимолекулярное элиминирование в ряду циклогекса-
циклогексана протекает гладко лишь в том случае, когда оба уходящих заместителя занимают аксиальное
(транс) положение (заторможенная конформация). Бимолекулярное отщепление двух замес-
заместителей издиэкваториального (транс) положения, как правило, невозможно, цис-Соединения
(один заместитель в положении е, другой в положении а) реагируют с большим трудом или
вообще не реагируют.
Впрочем, ее-конформация подлежащах элиминированию заместителей может, согласно вы-
вышесказанному, легко переходить в необходимую для бимолекулярного элиминирования аа-кон-
формацию (схема [Г.3.24]). Следует, однако, отчетливо представлять себе, что путем инверсии
цикла цис-заместители в конформациях ае или еа никогда не могут занять /и/гднс-положения!
Поясним вытекающие из этих закономерностей следствия для направления бимолекуляр-
бимолекулярного элиминирования на конкретном примере — отщеплении л-толуолсульфоновой кислоты
от ментилтозилата с помощью алкоголята:
Tfi> TSO Дсн,
В л-ментилтолизате I тозильная группа расположена экваториально. Поэтому она может
вступать в реакцию бимолекулярного элиминирования только после инверсии цикла A-»П).
В этой конформации имеется только один аксиальный водород, способный к элиминированию,
и образуется исключительно я-ментен-2 (IIII. Другие реакции бимолекулярного элиминирова-
1 Для скелета углеводорода ментана общепринята следующая нумерация атомов уг-
углерода. (Связи, изображенные пунктиром, лежат за плоскостью рисунка.) В 2-замещен-
ных л-ментанах и я-неоментанах метальная и изопропильная группы ориентированы
диаксиально (или диэкваториально). У производных я-ментана метальная группа и
заместитель в положении 3 занимают диэкваториальное положение (при инверсии
переход в диаксиальное); в производных и-неоментана их взаимное расположение эква-
экваториально-аксиальное (при инверсии — аксиально-экваториальное).
3. Элиминирование с образованием кратных углерод-углеродных связей
333
ния, например превращения 1,2-дибромсоединений в олефины при взаимодействии с иодидом
калия в ацетоне, в случае алициклических соединений также предполагают наличие диаксиаль-
ного (анти) положения атомов брома, например:
Вг(а)
111® Вг(а)
Вг
Br(a)
вме) s CXBr
[Г.3.25]
Хотя Е2-реакции протекают предпочтительно при он/тш-элиминировании, может проис-
происходить смн-элиминирование, особенно в том случае, если структурные факторы затрудняют
или делают невозможным о«тм-элиминирование. Например, в дейтерированном норбор-
нильном производном [Г.3.26] из-за жесткости бициклической системы оказывается невоз-
невозможным ан/ям-копланарное расположение остатка X и р-Н-атома. Реакция поэтому протека-
протекает как «/«-элиминирование DX с образованием не содержащего дейтерий норборнена.
D
X=Br, OTs, NMe3 [Г.3.26]
Также в других циклических системах, в которых в отличие от циклогексана невозможна инвер-
инверсия цикла, для реализации он/яи-конформации элиминирующейся группы предпочтительным яв-
является а/к-элиминирование, как показано в табл. Г.3.27 на примере циклоалканов разного размера.
Таблица Г.3.27. Доля син-элиминирования при Е2-реакции дейтероциклоалкил-
триметиламмонийгидроксидов
Размер цикла
Циклобутил
Циклогексил
Циклопентил
Циклогептил
син-Элиминирование, %
90
46
4
37
Вообще тетраалкиламмониевые соединения проявляют высокую тенденцию к образова-
образованию продуктов «/«-элиминирования. Предполагают, что субстрат образует с основанием цик-
циклический комплекс, в котором фиксируется син-расположение отщепляющегося заместителя:
\©
H.©.NR3
О'
Н
-H2O,-NR3
[Г.3.28]
Другие, ранее рассмотренные факторы — влияние природы заместителей, строения и ос-
основности основания и т. д. — также надо учитывать, но в случае циклических соединений они
имеют второстепенное значение по сравнению со стереоэлектронными. Однако если по кон-
формационным причинам нельзя ожидать предпочтительного направления элиминирования,
то эти факторы могут стать решающими и для циклических соединений. Это видно из реакции
элиминирования хлороводорода из неоментилхлорида с помощью этилата натрия. Здесь по
отношению к хлору в т/?анс-положении находятся (аксиальные) атомы водорода при С-2 и
С-4 атомах. Поэтому в соответствии с термодинамическими соотношениями образуется смесь
75% ментена-3 и 25% ментена-2 (предпочтительное образование изомера по Зайцеву):
334 Г Препаративная часть
Н(а) Н(а)
75 % 25 %
Мономолекулярное элиминирование протекает через плоский карбениевый ион или карба-
нион в качестве промежуточного продукта. Это возможно из любой конформации (син или
анти) и обычно нестереоспецифично. Иное положение наблюдается у непредельных соедине-
соединений, где заместители фиксированы двойной связью, не допускающей свободного вращения:
CIWCI ?<L н-сс-с, _s?- %J [Г.з.30,
' \. быстро медленно ' \
Н Н Н CI
(Предполагают, что (?)-изомер в [Г.З.ЗО] вообще реагирует лишь после перегруппировки в
(ZJ-изомер.)
3.1.4. ОТЩЕПЛЕНИЕ ВОДЫ ОТ СПИРТОВ (ДЕГИДРАТАЦИЯ) И
ОТЩЕПЛЕНИЕ СПИРТОВ ОТ ПРОСТЫХ ЭФИРОВ
В присутствии сильных кислот часто можно очень гладко отщепить молекулу
воды от спиртов в жидкой фазе. Легкость элиминирования возрастает при пере-
переходе от первичного спирта к третичному, так как дегидратация происходит глав-
главным образом по Е1-механизму. Эта дегидратация приводит к олефину по Зайце-
Зайцеву. (Напишите уравнение реакции!)
Чтобы соотношение между продуктами замещения (образование сложных
эфиров спиртов, образование простых эфиров) и элиминирования существенно
сдвинуть в сторону элиминирования, надо использовать для первичных спиртов
высокие температуры A80—200 °С) (разд. Г,3.1.1), а для того, чтобы повысить
скорость реакции, следует создать высокую концентрацию сильной кислоты
(серной, фосфорной).
При таких жестких условиях образуются значительные количества побочных
продуктов (см. ниже), и поэтому катализируемая кислотами дегидратация пер-
первичных спиртов применяется реже, чем каталитическая дегидратация на оксиде
алюминия.
Вторичные спирты, напротив, реагируют уже очень гладко при температуре
-140 °С в присутствии фосфорной кислоты. В случае третичных спиртов жела-
желательное элиминирование воды осуществляется уже при помощи щавелевой или
фосфорной кислоты при температуре -100 °С. Для этой цели можно с успехом
применять также каталитические количества и-толуолсульфоновой кислоты.
Очень легко реагируют также р-гидроксикарбонильные соединения (про-
(продукты альдольной конденсации, см. табл. Г.7.123), так как элиминирование
воды приводит в данном случае к энергетически выгодным ос,р-ненасыщенным
карбонильным соединениям. В этом случае для реакции пригодны условия
дегидратации третичных спиртов. Отщепление воды особенно удобно осущес-
осуществлять в присутствии -1% иода, причем собственно катализатором здесь, веро-
вероятно, является образующийся иодоводород.
3. Элиминирование с образованием кратных углерод-углеродных связей 335
Поскольку реакция имеет Е1 -характер, то обычно происходят такие перег-
перегруппировки промежуточного иона карбения, которые могут приводить к
образованию энергетически более выгодного иона (см. схемы [Г.2.44] и [Г.4.17]).
Они обусловливают прежде всего образование изомерных олефинов с
различным положением двойной связи1. Поэтому в этих случаях не удается
при помощи кислотной дегидратации спиртов получить какой-либо один
олефин:
+ н® ©
НзС-СН2-СН2-СН2-ОН ——— Н3С-СН2-СН2-СН2 — Н3С-СН2-СН=СН2
— Н2О _ и^
Образование
низкоэнергетического иона [Г.3.31]
©
Н3С-СН2-СН-СН3 тг» Н3С-СН=СН-СН3
-н®
но:
©
Н3С~СН2 — СН2~ СН2
Н3С-СН2-СН-СН3 -^ Н3С-СН2-!н-СН3 ^ (вь-сокознергетический) [Г.3.32]
I - Н 2О \
I Н 2О
он -н® н3с-сн=сн-сн3
Иногда (см. разд. Г.9.1.1.2, перегруппировка Вагнера — Меервейна) при де-
дегидратации образуются преимущественно продукты перегруппировки углерод-
углеродного скелета. Так, при дегидратации 3,3-диметилбутанола-2 образуется
2,3-диметилбутен-2 (а не 3,3-диметилбутен-2, как это можно было ожидать):
© Н3С СН3
Н3С-С-СН-СН3 -—- С=С [Г.3.33]
н3с он н3с сн3
При дегидратации спиртов в присутствии кислот в качестве дальнейшей по-
побочной реакции может легко протекать полимеризация олефина (разд. Г,4.1.9).
Чтобы сместить равновесие дегидратации в желательном направлении, если
возможно, сразу отгоняют образовавшийся олефин (и таким образом одновре-
одновременно предотвращают его дальнейшие превращения — изомеризацию, полиме-
полимеризацию) или в случае высококипящих олефинов удаляют из реакционной
смеси воду путем азеотропной отгонки с толуолом.
Дегидратацию спиртов удается также гладко осуществить в газовой фазе при
температурах 300—400 "С на оксиде алюминия, фосфате алюминия, оксиде то-
тория, диоксиде титана и т. д. При этом (как и в случае первичных спиртов) обра-
образуется меньше побочных продуктов. При дегидратации на оксиде алюминия
можно почти совершенно подавить перегруппировки, если частично отравить
кислотные центры катализатора пиперидином или другими основаниями.
1 Известно, что кислоты могут вызывать изомеризацию олефинов. Поэтому можно пред-
предположить, что далее происходит изомеризация образовавшегося олефина в соответствии со
схемой [Г.4.17].
Таблица Г.3.34. Кислотная дегидратация спиртов
Продукт реакции
Пентен-2
2-Метилбутен-2
1,1 -Дифенилэтилен
2,3-Диметилбутен-2
Циклогексен
Циклопентен
З-Метилбутен-З-он-2
4-Метилпентен-3-он-2 (мези-
тилоксид)
З-Метилпентен-З-он-2 д
Бутен-2-алье (кротоновый
альдегид)
Исходное соединение
Пентанол-2
2-Метилбутанол-2
1,1 -Д ифенилэтанол
2,3-Диметилбутанол-2
Циклогексанол
Циклопентанол
4-Гидрокси-З-метилбута-
нон-2
4-Гидрокси-4-метилпента-
нон-2 (диацетоновый спирт)
4-Гидрокси-З-метилпента-
нон-2
З-Гидроксибутаналь(ацет-
альдоль)
Т. кип.", "С
37
38
134 A0)
73Г
83
45
36 A00)
131
63 E0)
102
V0
1,3830
1,3859
1,6085
1,4115
1,4464
1,4223
1,4432
1,4425
1,4489
1,4366
Выход, %
70
80
70
80
80
80
85
90
80
80
Примечания
б
в
Содержит ~10% соответ-
соответствующего производного бу-
тена-1
То же
а Цифры в скобках указывают давление (мм рт. ст.), которому соответствует данная температура кипения.
6 Изучите ИК-спектр олефина и после отнесения полос установите, образуется ли наряду с пентеном-2 также пентен-1 и находится пентен-2 в B)-
или (?)-форме. (Пентен-1: 912, 994, 1642, 3083 см; (?)-пентен-2:964, 1670, 3027 см-'; B)-пентен-2: 933, 964, 1406, 1658, 3018 смЧ)
в Исследуйте смесь изомеров B-метилбутен-1,2-метилбутен-2 и 3-метилбутен-1) с помощью ГЖХ и оцените их количественное соотношение. Для
разделения изомеров необходимы следующие условия: длина колонки 1 м, носитель—кизельгур, фаза — парафиновое масло, температура 20 °С, газ-
носитель — водород E л/ч). Относительные времена удерживания (по эфиру): 2-метилбутена-1 1,0; 2-метилбутена-2 1,45; 3-метилбутена-1 0,55.
г В качестве предгона получают немного 2,3-диметилбутена-1. Т. кип. 55 °С.
д Перегонять в вакууме, используя в качестве стабилизатора 0,5% СНзСООН (ледян.) и гидрохинона.
с При повторной перегонке стабилизировать 0,5% СНзСООН и 0,5% гидрохинона.
3. Элиминирование с образованием кратных углерод-углеродных связей 337
Ф Общая методика дегидратации вторичных и третичных спиртов и продуктов альдольной
конденсации в присутствии кислот (табл. Г.3.34). Вторичные спирты смешивают с
85%-ной фосфорной кислотой, взятой в количестве 50% от массы спирта, третичные
спирты — с безводной щавелевой кислотой, взятой в количестве 20% от массы спирта,
или с 85%-ной фосфорной кислотой, взятой в количестве 5% от массы спирта; к Р-гидрокси-
кетонам или Р-гидроксиальдегидам прибавляют 1% иода.
Эту смесь нагревают в приборе для перегонки на масляной или металлической бане до тем-
температуры 120—160 °С, чтобы образующийся олефин постоянно отгонялся. Необходимо следить
за тем, чтобы перегонялся только олефин. В случае низкокипящих олефинов надо применять
20-сантиметровую колонку Вигре, а приемник дополнительно охлаждать ледяной водой.
Дистиллят отделяют от водной фазы в делительной воронке, сушат сульфатом натрия и
перегоняют; к малоустойчивым соединениям (диены, <х,Р-ненасыщенные карбонильные
соединения) бывает полезно добавить ингибитор полимеризации (например, гидрохинон) и,
кроме того, перегонять при возможно более низкой температуре.
Методику можно использовать для работы с полумикроколичествами.
Получение некоторых стиролов дегидратацией а-гидроксиэтилбензолов:
Overberger С. G., Sounders J. И. Org. Synth., Coll. Voll. Ill, 1955, 204; имеется пере-
перевод 1-го издания: Овербергер К., СаундерсДж. В сб.: Синтезы органических пре-
препаратов, Сб. 4. — М.: ИЛ, 1953, с. 540.
Получение амида метакриловой кислоты из ацетонциангидрина: Wiley R. И.,
Waddey W. E., Org. Syntheses, Coll. Vol. Ill, 1955, 560.
Принцип элиминирования спиртов из простых эфиров аналогичен принци-
принципу отщепления воды из спиртов. Как правило, этот метод не обладает по сравне-
сравнению с дегидратацией никакими преимуществами. Однако из ацеталей можно
получить таким образом простые эфиры енолов:
н®
R-CH2-CH(OR'J > R-CH = CH-OR' [Г.3.35]
— R ОН
Эта реакция катализируется уже -0,5%-ными добавками фосфорной кисло-
кислоты, поскольку в качестве промежуточного продукта образуется относительно
бедный энергией ион карбения (почему?). Отщепившийся спирт отгоняют из
реакционной смеси. Это связано с трудностями, когда отщепляемый спирт и об-
образующийся простой эфир енола обладают близкими температурами кипения
(низшие члены гомологических рядов). В таких случаях приходится работать с
эффективной ректификационной колонкой.
Надо отметить, что ацетали также можно расщеплять на спирты и простые
эфиры енолов в газовой фазе на оксиде алюминия.
Ф Общая методика получения простых эфиров енолов из ацеталей элиминированием спир-
спиртов (табл. Г.3.36). Соответствующий ацеталь смешивают с 85%-ной фосфорной кис-
кислотой @,5%) и пиридином A,2%), после чего смесь нагревают до кипения в кругло-
донной колбе с насадкой Хана (рис. А.77) и нисходящим холодильником. В качестве
охлаждающей жидкости для насадки Хана используется тот же спирт, который отщепляет-
отщепляется от ацеталя. Медленно образующийся во время реакции спирт отгоняется и собирается в
мерный цилиндр, поэтому легко можно следить за ходом реакции.
По окончании отщепления спирта содержимое колбы перегоняют.
Аналогичным путем можно получить 2-этоксибутадиен-1,3 из винилметил-
кетона через 1,3,3-триэтоксибутан (напишите уравнения реакций): Dykstra H. В.
J. Am. Chem. Soc, 1935, 57, 2255.
338
Г Препаративная часть
Таблица Г.3.36. Простые эфиры енолов из ацеталей
Простой эфир енола
2-Этоксигексен-1
а-Этоксистирол
Р-Метоксистирол
1 -Этоксициклогексен
Исходное соединение
Диэтилацеталь м-бутилме-
тилкетона
Диэтилкеталь ацетофе-
нона
Диметилацеталь фенил-
ацетальдегида
Диэтилацеталь циклогек-
санона
т.кип.а
135
89 A1)
99 A3)
160
V
1,4180
1,5292
1,5620
1,4580
Выход,
%
90
95
90
95
Цифры в скобках указывают давление (им рт. ст.), которому соответствует данная
температура кинения.
Другие примеры: Назаров И. Н., Маркин С. М., Крупное Б. К. ЖОХ, 1959,
29, 3692.
Дегидратация гидроксисоединений имеет промышленное значение для получения олефи-
нов и винилпроизводных, которые являются важными промежуточными продуктами и моно-
мономерами для производства синтетических материалов и искусственных волокон (табл. Г.3.37).
Однако основное количество промышленно важных олефинов получают при дегидрировании
и пиролизе углеводородов нефти (см. разд. Г,6.6).
Примером аналитического применения кислотной дегидратации может слу-
служить количественное определение третичных спиртов в смеси с первичными и
вторичными. При этом смесь кипятят с ксилолом в присутствии небольшого ко-
количества хлорида цинка или иода. Прибор снабжен обратным холодильником и
водоотделителем. В этих условиях дегидратируется только третичный спирт. Его
содержание в смеси рассчитывают по количеству выделившейся воды.
Таблица Г.3.37. Технически важные ненасыщенные соединения, получаемые
путем дегидратации, и их применение
Исходное соединение
Ацетальдоль
(см. разд. Г,7.2.1.3)
Ацетонциангидрин
Ускусный ангидрид
Конечный продукт
Кротоновый альдегид
Метиловый эфир ме-
такриловой кислоты
Кетен
Применение
-> Кротоновая кислота -» Сополи-
Сополимер
-» Сорбиновая кислота (консер-
(консерванты)
-» З-Метоксибутанол (гидравличес-
(гидравлическая жидкость) -> З-Метоксибутила-
цетат (растворитель для лаков)
-> Полиметилметакрилат (плекси-
(плексиглас)
-> Ацетилирующий агент
3. Элиминирование с образованием кратных углерод-углеродных связей 339
3.1.5. ОТЩЕПЛЕНИЕ ГАЛОГЕНОВОДОРОДОВ ОТ АЛКИЛГАЛОГЕНИДОВ
(ГИДРОГАЛОЭЛИМИНИРОВАНИЕ; ДЕГИДРОГАЛОГЕНИРОВАНИЕ)
Хотя для получения олефинов из спиртов простейшим способом является пря-
прямая дегидратация, часто выбирают обходной путь — дегидрогалогенирование
соответствующих алкилгалогенидов или детозилирование эфиров я-толуол-
сульфоновой кислоты. Прежде всего этот путь наиболее приемлем, когда дегид-
дегидратация, протекающая по El-механизму, может привести к смеси олефинов или
продуктам перегруппировки. При использовании высоких концентраций силь-
сильных оснований названная реакция почти всегда протекает по бимолекулярному
механизму.
В качестве оснований прежде всего применяют гидроксиды щелочных ме-
металлов в спиртах или в полярных апротонных растворителях, алкоголяты в соот-
соответствующих спиртах или диметилсульфоксиде, амиды щелочных металлов в
инертном растворителе и третичные органические основания, такие, как пири-
пиридин, хинолин, диметиланилин, этилдициклогексиламин, а также амидины ДБН
иДБУ[Г.3.14].
Природа основания и растворителя, а также температура реакции должны
быть подобраны соответственно субстрату и строению получаемого олефина. Так,
первичные алкилгалогениды склонны к реакциям замещения и при действии гид-
роксидов щелочных металлов или алкоголятов образуют главным образом соотве-
соответствующие простые эфиры (разд. Г,2.6.2). Поэтому для получения олефинов при-
применяют «пространственно-затрудненные» основания, например этилдицикло-
этилдициклогексиламин, или основания, обладающие высоким значением рЛв, например
т/>ет-бугилат калия (отщепление проводят при высокой температуре и в присут-
присутствии подходящих растворителей). Дегидрогалогенирование вторичных и третич-
третичных алкилгалогенидов протекает в более мягких условиях.
Дегидрогалогенирование удается также проводить действием едкого натра в
двухфазных системах с использованием межфазных катализаторов (разд. Г,2.4.2
и препараты из разд. Г,3.1.6).
Электроноакцепторные группы, находящиеся рядом с отщепляемым атомом
водорода, ускоряют элиминирование, так как они повышают кислотность связи
С—Н и тем самым облегчают отщепление протона. Конкурирующая реакция
нуклеофильного замещения при этом подавляется. р-Хлорпропионовая кислота
реагирует уже с разбавленной щелочью, давая акриловую кислоту, а из р-хлор-
этилметилкетона и диэтиланилина образуется винилметилкетон (разд. Г,3.1.6).
(Напишите уравнения этих реакций!)
Подвергаются элиминированию заместители, в общем не являющиеся ти-
типичными уходящими группами, если они находятся в р-положении к электро-
ноакцепторным группам. (В качестве примера напишите уравнение реакции
расщепления р-пиперидинпропиофенона, катализируемой основаниями и при-
приводящей к винилфенилкетону и пиперидину.) Ацилгалогениды, имеющие
соседнюю связь С-Н, уже при температуре ниже О °С реагируют с триэтилами-
ном с образованием кетенов:
n r
[Г.3.38]
340
Г Препаративная часть
Большинство образующихся кетенов неустойчиво в условиях проведения ре-
реакции, так как амин катализирует их димеризацию. Однако образование кете-
кетенов подтверждается вторичными реакциями (см. уравнение [Г.7.314]). Из хло-
рангидрида дифенилуксусной кислоты и триэтиламина получают устойчивый
дифенилкетен. (Напишите уравнение реакции!)
Аналогично из хлорангидридов алифатических сульфоновых кислот образу-
образуются сульфены (R2C=SO2), которые нельзя выделить в свободном виде.
Дегидрогалогенирование находит применение в промышленности прежде всего для полу-
получения галогенированных олефинов:
1,2-Дихлорэтан —> винилхлорид (-> поливинилхлорид)
1,1,2,2-Тетрахлорэтан -> трихлорэтилен (растворитель) -» хлоруксусная кислота
2,4-Дихлор-2-метилбутан —> изопрен (-> полимеры)
Хлорированные циклопентаны -> гексахлорциклопентадиен (-> инсектициды)
Гексахлорциклогексан -> 1,2,4-трихлорбензол (-> 2,4-дихлорфенол -> 2,4-дихлорфенок-
сиуксусная кислота; см. разд. Г,2.6.2).
Аналогично олефинамдегидрогалогенированием 1,1-или 1,2-дигалогенидов
могут быть получены ацетилены (алкины):
Н н
i i
-с-с—
I I
X X
Н X
I I
-с-с—
I I
Н X
—с=с—
-2НВ, -2Х
[Г.3.39]
Отщепление двух молекул галогеноводородов требует в общем жестких усло-
условий проведения реакций; в большинстве случаев применяют суспензии амидов
щелочных металлов в неполярных растворителях или их растворы в жидком
аммиаке, а также спиртовой раствор едкого кали или алкоголятов щелочных
металлов.
Под действием сильных щелочей и при повышенных температурах тройная
углерод-углеродная связь обычно перемещается в середину молекулы или про-
происходит изомеризация в алленовую группировку С=С=С— . Поэтому для полу-
Таблица Г.3.40. Дегидрогалогенирование алкилбромидов этилдициклогексил-
амином
Олефин
Гексен-1
Гептен-1
Октен-1
Децен-1
Додецен-1
Алкилбромид (т. кип., *С)
1-Бромгексан A56)
1-Бромгептан A78)
1-Бромоктан B00)
1-Бромдекан
1-Бромдодекан
Методика
А
А
А
Б
Б
Т.иш.
олефина, 'С
(мм рт. ст.)
63
93
122
52 A1)
96 A5)
1,3877
1,3998
1,4091
1,4215
1,4308
Выход,
%
80
90
95
80
90
3. Элиминирование с образованием кратных углерод-углеродных связей 341
чения алкинов с концевой тройной связью рекомендуется применять амид нат-
натрия в жидком аммиаке (температура таких реакций, как правило, ниже —30 °С)
или неполярные растворители, как, например, лигроин. В этой среде натриевые
соли алкинов-1 нерастворимы, и таким образом удается избежать дальнейших
побочных реакций1. Более простым препаративным методом является дегидро-
галогенирование с едким кали в триэтиленгликоле (тригликолеJ, которое,
правда, не может быть применено для соединений, содержащих чувствительные
к щелочам группы. В этом случае опасность изомеризации связи —С=С— сведе-
сведена к минимуму, так как образующийся алкин тотчас же отгоняют из реакцион-
реакционной среды.
Ф Общая методика дегидрогалогенирования алкилгалогенидов с этилдициклогексилами-
ном3 (табл. Г.3.40). В трехгорлой колбе на 250 мл, снабженной либо насадкой для пе-
перегонки и нисходящим холодильником (методика А), либо термометром, мешалкой и
обратным холодильником с хлоркальциевой трубкой (методика Б), нагревают 0,1 моль
алкилгалогенида и 0,15 моль этилдициклогексиламина до температуры 180 °С при сильном
перемешивании. В случае низкокипящих алкилгалогенидов температуру поддерживают на
20 "С выше температуры кипения алкилгалогенида. Поскольку алкилбромиды имеют более
высокую температуру кипения, они удобнее, чем соответствующие алкилхлориды. (Какие
преимущества дает более высокая температура кипения?)
Олефины, кипящие при температуры ниже 130 °С, отгоняются непосредственно из реак-
реакционной смеси (методика А). К концу реакции, когда в перегонной колбе остается уже
небольшое количество олефина, температуру повышают до 230 °С. По окончании реакции,
которая продолжается около 15-20 ч, отогнанный олефин сушат хлоридом кальция и ректи-
ректифицируют.
В случае олефинов, кипящих при температуре выше 130 "С (методика Б), реакционную
смесь нагревают (с обратным холодильником) при перемешивании 20 ч, дают охладиться и от-
отсасывают выпавшую этилдициклогексиламмониевую соль. Осадок на фильтре промывают
петролейным эфиром. Из объединенных фильтратов отгоняют петролейный эфир и остаток
фракционируют на 20-сантиметровой колонке Вигре в вакууме.
Этилдициклогексиламин надо регенерировать. В методике Б к остатку после фракциони-
фракционирования олефинов, содержащему в основном амин, прибавляют разбавленную соляную кис-
кислоту до кислой реакции. Водный раствор обрабатывают эфиром для удаления непрореагиро-
вавшего алкилбромида и остатков олефина. Объединяют с отфильтрованным бромидом этил-
дициклогексиламмония и выделяют свободный амин действием избытка 50%-ного едкого
кали. Дальнейшую обработку см. разд. Г,2.6.4.
В методике А с остатком в колбе поступают аналогично. Остаток растворяют в соляной
кислоте (контроль на кислую реакцию!), обрабатывают эфиром. Подщелачивают едким кали,
отделяют амин и т. д.
Ф Общая методика дегидрогалогенирования (детозилирования) с едким кали в тригликоле
(табл. Г.3.41). Реакцию проводят в приборе, состоящем из двугорлой колбы с мешалкой,
насадкой для перегонки и нисходящим холодильником. При нагревании на металличес-
металлической бане до температуры ~ 100 °С растворяют 0,25 моль едкого кали на каждые 0,1 моль отщеп-
1 Напротив, амид натрия вызывает изомеризацию алкинов, имеющих неконцевую трой-
тройную связь, в алкин-1.
2 НО-СН2-СН2-О-СН2-СН2-О-СН2-СН2-ОН — а-гидро-ш-гидрокситри(оксиэти-
лен). Более употребительны тривиальные названия. Они используются в данной книге, хотя и
не отвечают правилам номенклатуры.
3 Можно также применять метилдициклогексиламин, ДБН или ДБУ [Г.3.14] (с. 328).
342
Г Препаративная часть
Таблица Г.3.41. Дегидрогалогенирование (детозилирование) гидроксидом калия в
тригликоле
Продукт реакции
Гексин-1
Октин-1
Децин-1
Додецин-1
Фенилацетилен
Циклогексен"
Циклогексадиен-1 ,За'6
(+) -я-Ментен-2"
Исходное соединение
1,2-Дибромгексан
1,2-Дибромоктан
1,2-Дибромдекан
1,2-Дибромдодекан
1,2-Дибромэтилбензол
Бромциклогексан
1,2-Д ибромциклогексан
(—) -я-Ментил-3-тозилат
Т. кип., *С
(мм рт. ст.)
71
127
69 A0)
95 A5)
143
83
80
56 A5)
V5
1,3960
,4134
,4242
,4351
.5460
,4438
,4730
,4506
Выход,
%
60
60
80
80
90
90
65
85
Не экстрагировать эфироы! Органическую фазу отделить, высушить, перегнать.
б Можно прибавить к реакционной смеси только черверть исходного соединения,
затем нагреть до начала реакции и прибавить по каплям остаток дибромида. Продукт содер-
содержит 10—15% циклогексадиена-1,4.
в Перегнать над натрием. Определите поляриметрически (см. разд. А.3.4) содержа-
содержание л-ментена-2: для л-ментена-2 [a) Dx +132°, для л-ментена-3 [a]D20 +110° (без раство-
растворителя).
ляющегося галогеноводорода в 60 мл тригликоля (коричневое окрашиваниеI. Раствор немного
охлаждают, добавляют соответствующий алкилгалогенид или алкилтозилат и затем медленно
поднимают температуру бани до 200 °С; при этом отгоняется продукт элиминирования. Приме-
Применять внутренний термометр не следует, так как стекло термометра очень легко разъедается горя-
горячим раствором едкого кали! Реакция может начинаться неожиданно (со вспениванием), поэто-
поэтому нагревание надо проводить осторожно. В большинстве случаев реакция заканчивается за
-30 мин.
Продукт реакции отделяют от воды (которая имелась в растворителе и образовалась за
счет побочных процессов), водный слой извлекают эфиром и объединенные органические фазы
сушат сульфатом натрия. После отгонки эфира продукт элиминирования фракционируют.
Методики можно использовать и для работы с полумикроколичествами веществ. В этом слу-
случае обходятся без мешалки; синтез проводят в простом приборе для перегонки.
\ЦУ) Получение дикетена
8 Осторожно1. Дикетен ядовит и раздражает органы дыхания, а также кожу! Тяга! Работать
в защитных перчатках! Посуду мыть разбавленной щелочью!
1 моль ацетилхлорида растворяют в 400 мл диэтилового эфира, при хорошем перемешивании
прибавляют по каплям смесь 1 моль триэтиламина и 400 мл эфира. Прибавление регулируют
так, чтобы реакция шла не слишком бурно. Эфирный раствор отсасывают от образующегося
гидрохлорида триэтиламина, вводя в реакционную колбу трубку с пористой перегородкой
(рис. Г.3.42). Продукт перегоняют в небольшом вакууме с использованием 20-сантиметровой
колонки ВиГре. Т. кип. 72 °С A00 мм рт. ст.); выход 55%.
Для дальнейшего использования кетен обычно перегонять не надо. Поскольку кетен с тру-
трудом отделяется от эфира, последующие реакции лучше всего проводить непосредственно в
полученном эфирном растворе.
Объем колбы должен быть по крайней мере в два раза больше объема реакционной смеси.
3. Элиминирование с образованием кратных углерод-углеродных связей
343
Склянка Вульфа
водоструйный насос
Фильтр
Рис. Г.3.42. Схема обратного фильтрования.
Получение диэтилацеталя кетена из диэтилацеталя бромацетальдегида:
McElvain S. M., Kundiger D. Org. Syntheses, Coll. Vol. Ill, 1955, 506; имеется пере-
перевод раннего издания: Мак-Эльвен С, Кундигер Д. В сб.: Синтезы органических
препаратов. Сб. 3. — М: ИЛ, 1952, с. 244.
Получение диэтилацеталя акролеина из ацеталя р-хлорпропионового альде-
альдегида: Witzemann E. J., Evans W. L., Hass H., SchroederE. F. Org. Syntheses, Coll. Vol.
II, 1943, 17; имеется перевод: Витцеман Е., Эванс В., Хасс Г., Шредер Е. В сб.:
Синтезы органических препаратов. Сб. 2. — М.: ИЛ, 1949, с. 64.
Получение 2-винилтиофена из 2-A-хлорэтил)тиофена: Emerson W. S.,
Patrik Т. М. Org. Syntheses, Coll. Vol. IV, 1963, 980; имеется перевод раннего
издания: Эмерсон У., Патрик Т.-мл. В сб.: Синтезы органических препаратов.
Сб. 10. - М.: ИЛ, 1960, с. 16.
Получение дмфенгм/се/иешиздифенилацетилхлорида: Taylor Т. С, МсКШорА.,
Hawks G. H. Org. Synth., 1972, 52, 36.
Приведем несколько примеров синтезов алкинов дегидробромированием в
спиртовых растворах едкого кали:
Получение ацетилендикарбоновой кислоты из мезодибромянтарной кислоты:
Аббот Т., Арно Р., Томпсон Р. В сб.: Синтезы органических препаратов. Сб. 2. — М.:
ИЛ, 1949, с. 71.
Получение фенилпропиоловой кислоты из этилового эфира а,р-дибромкорич-
ной кислоты: Аббот Т. В сб.: Синтезы органических препаратов. Сб. 2. — М.:
ИЛ, 1949, с. 520.
Получение толана из 1,2-дибромстильбена: Smith L. J., Falkoff M. M. Org.
Syntheses, Coll. Vol. Ill, 1955, 350.
Дегидробромирование с использованием межфазных катализаторов: получение
диэтилацеталя пропионового альдегида из диэтилацеталя 2,3-дибромпропионового
альдегида, а также другие примеры: Le CoqA., GorguesA. Org. Syntheses, 1979, 59, 10.
3.1.6. ЭЛИМИНИРОВАНИЕ ТРИАЛКИЛАМИНА ИЗ
ЧЕТВЕРТИЧНЫХ АММОНИЕВЫХ ОСНОВАНИЙ
(ГОФМАНОВСКОЕ РАСЩЕПЛЕНИЕ)'
При алкилировании аминов избытком алкилирующего агента (чаще всего при-
применяют метилиодид или диметилсульфат) получают соли четвертичных аммо-
аммониевых оснований, которые при помощи гидроксида серебра или ионообмен-
Не надо путать с гофмановской перегруппировкой амидов кислот (разд. Г,9).
344 Г Препаративная часть
ников легко перевести в четвертичные аммониевые основания. Последние рас-
распадаются при простом нагревании или упаривании водного раствора, давая
олефин, третичный амин и воду:
I I © е \ /
H-C-C-N(CH3K + ОН С=С + N(CH3K + Н2О [Г.3.43]
При наличии в молекуле нескольких р-водородных атомов это бимолекулярное
элиминирование протекает во многих случаях по правилу Гофмана, например:
в ,
н3с-сн-сн2-сн3 + он —
©N(CH3K
Н2С=СН-СН2-СН3 95%
Н3С-СН=СН-СН3 5%
[Г. 3.44]
Если в р-положении не имеется ни одного атома водорода и поэтому реакция
элиминирования невозможна, то происходит замещение, например:
CH3N(CH3K + ОН СН3ОН + N(CH3K [Г.3.45]
Гофмановское расщепление в свое время имело большое значение для установления стро-
строения азотсодержащих природных соединений (алкалоидов). Сущность метода заключается в
том, что соответствующий амин кватернизуют избытком метилиодида (исчерпывающее мети-
метилирование) и исследуют образующийся при пиролизе олефин. Таким путем Гофман установил
строение пиперидина:
^ Mel, + 72Ад2О, + У2Н2О
-Agl I © И ©
МеТме°Н
Me
[Г.3.46]
. |Г| ill - /-^ /<^
- Н2О, - NMe3
пиперилен (пентадиен-1,3)
С классическими работами Вильштетера (расщепление псевдопельтьерина до циклоокта-
тетраена) можно ознакомиться в учебниках по органической химии.
В настоящее время гофмановское расщепление иногда применяют в лабора-
лабораториях, если надо получить определенный олефин (например, с концевой
двойной связью) в относительно мягких условиях. Во многих случаях при этом не
обязательно переводить четвертичную аммониевую соль в свободное аммоние-
аммониевое основание. Так, соли оснований Манниха строения [Г.3.47] (см. разд.
Г,7.2.1.5) можно легко перевести в соответствующие винилкетоны при нагрева-
нагревании их до температуры 100-150 °С.
©,R
Ft-CO-CH2-CH2-N-R
Н
Xе [Г.3.47]
Часто удается также осуществить расщепление при помощи перегонки с во-
водяным паром. Легкость элиминирования объясняется тем, что может образо-
образоваться энергетически выгодная сопряженная система электронов. (Напишите
уравнение реакции для приведенного ниже примера.)
3. Элиминирование с образованием кратных углерод-углеродных связей 345
«Г)) Получение метилвинилкетона
Л Осторожно*. Метилвинилкетон ядовит и обладает в безводном состоянии силь-
I ным слезоточивым действием. Тяга!
Растворяют 1 моль гидрохлорида 4-диметиламинобутанона-2 или 4-диэтиламинобутанона-21 в
минимальном количестве воды и добавляют 1 г гидрохинона и 1 мл ледяной уксусной кислоты.
Этот раствор прибавляют по каплям при перемешивании в течение 1—2 ч к 250 мл диэтилфталата
(служит в качестве «внутреннего» теплоносителя); последний находится втрехгорлой колбе на 1 л,
снабженной эффективной мешалкой, капельной юронкой, внутренним термометром и насадкой
для перегонки с нисходящим холодильником, и нагрет до 160 °С. Образующийся метилвинилкетон
отгоняется вместе с реакционной водой. Приемник, присоединенный к холодильнику через ваку-
вакуумный форштос, дополнительно охлаждают в ледяной воде. Для повышения устойчивости метил-
метилвинилкетона в него добавляют 0,5 г гидрохинона и 0,5 мл ледяной уксусной кислоты.
По окончании реакции дистиллят насыщают поташом, метилвинилкетон отделяют, сушат
сульфатом натрия и перегоняют в слабом вакууме2, причем и в перегонную колбу, и в прием-
приемник добавляют 0,5 г гидрохинона и 0,5 мл ледяной уксусной кислоты. Приемники охлаждают
смесью льда с солью. Т. кип. 33 °С A00 мм рт. ст.); выход 80%.
Получение гексена-1 гофмановским расщеплением к-гексиламина: Коуп А.,
Трумбулл Э. В. В сб.: Органические реакции. Сб. 11. — М.: Мир, 1965, с. 327.
Алкены путем элиминирования по Гофману с использованием ионообмен-
ников для получения четвертичных аммониевых оснований (гидроксидов):
дифенилметилвиниловый эфир: Kaiser С, WeinstockJ. Org. Synth. 1976, 55, 3.
3.2. ТЕРМИЧЕСКОЕ СИН-ЭЛИМИНИРОВАНИЕ
Принцип такого элиминирования лучше всего разобрать на примере классической
реакции Чугаева. Прежде всего из алкоголята калия готовят соответствующий
ксантогенат калия путем обработки дисульфидом углерода [Г.3.48а]. Алкилирова-
нием ксантогената (чаще всего с помощью метилиодида) получают О,8-диалкило-
вый эфир (эфир ксантогеновой кислоты [Г.3.486], который далее в результате пи-
пиролиза распадается на олефин, алкилтиол и оксид-сульфид углерода [Г.3.48в]:
м I I /Я ©
н-с-с-ок + cs2 —- н-с-с-о-с Q к° [г.3.48а]
1 I о
I I /Я +R| I I /Я
н-с-с-о-с —^ н-с-с-о-с [Г.3.486]
I I \ р) I© \ L J
II SU -' II SR
Н-С-С-О-С' -^^- ЧС=С7 + RSH + COS [Г.3.48В]
II SR / \
1 Гидрохлорид, полученный по реакции Манниха, можно применять непосредственно
без очистки. При использовании свободного основания Манниха его нейтрализуют при
охлаждении льдом эквимолярным количеством концентрированной НС1.
2 Перегонку надо выполнять при возможно более низкой температуре. Однако при пол-
полном вакууме, создаваемом водоструйным насосом, температура кипения метилвинилкетона
уже ниже комнатной температуры.
346 Г Препаративная часть
Пиролиз протекает как мономолекулярная реакция, однако в отличие от
Е1-реакций не через свободные ионы, а через циклическое переходное состоя-
состояние, в котором разрыв и образование связей происходят одновременно. Пос-
Поскольку переходное состояние имеет циклическую структуру, стерическое тече-
течение реакции приобретает характер син-элиминирования:
/-и и + i (—- COS + R-SH) [Г.3.49]
Так, при элиминировании по Чугаеву из (—)-ментола образуется 66% и-мен-
тена-3 (и 34% и-ментена-2), из чего можно сделать вывод о сня-элиминирова-
нии, поскольку при С-4 нет водородного атома в ан/ии-положении:
! I I —х= —OCSMe
и
S
[Г.3.50]
(ср. полученные результаты с бимолекулярным элиминированием, например,
метилтозилата; разд. Г,3.1.3). Подобным образом при пиролизе распадаются и
другие эфиры, например уретаны, эфиры угольной и карбоновых кислот, селен-
и сульфоксиды, а также аминоксиды (элиминирование по Коупу), причем
склонность соединений к пиролизу уменьшается в последовательности:
Р V^ ^ > V-^ ^П > |-ч /II > 1-4 /"И > |-ч /
7^> ~Я(* -/CV° 7CV° -CV°
Так, для пиролиза эфиров уксусной кислоты необходима температура
400—500 "С, тогда как ксантогенаты претерпевают пиролитический распад уже
при нагревании до 120-200 °С, а аминоксиды — при нагревании до 80-160 °С.
Напротив, селеноксид уже частично разлагается при его получении при ком-
комнатной температуре. (Напишите уравнения пиролиза каждого упомянутого
здесь соединения.)
Направление отщепления при таком термическом элиминировании, как
правило, трудно указать однозначно. В случае соединений с открытой цепью
влияние пространственных (конформационных) и термодинамических факто-
факторов часто компенсируется, так что направление элиминирования определяется
главным образом статистически (см. примечание в разд. Г.3.1.2). Например, при
пиролизе 2-бутилацетата получают 51% бутена-1 и 49% бутена-2.
Для циклических соединений конформация может иметь решающее значение. Если из
конформации возможно элиминирование в двух направлениях, то преимущественно образу-
образуется термодинамически более устойчивый продукт. Например, элиминирование 1-метилцик-
логексилацетата приводит к получению 75% 1-метилциклогексена-1 и 25% метиленциклогек-
сана.
3. Элиминирование с образованием кратных углерод-углеродных связей 347
Хотя для превращения эфира уксусной кислоты в олефин надо проводить
пиролиз в довольно жестких условиях, тем не менее ацетаты часто используются в
качестве исходных соединений из-за своей доступности. Кроме того, пиролиз
ацетатов не сопровождается значительными побочными реакциями или изомери-
изомеризацией двойной связи, несмотря на то что во многих случаях необходимы высокие
температуры. Так, из н-алкилацетатов получают довольно чистые алкены-1. Даже
ацетат 2,2-диметилпентанола-З дает наряду с 77% нормального продукта элими-
элиминирования D,4-диметилпентена-2) всего -7% изомерного олефина, тогда как при
кислотной дегидратации происходит более глубокая изомеризация углеродного
скелета (схема [Г.3.33]). Нитрильные группы, метокси- или нитрогруппы или
различные сложноэфирные группы в общем случае не препятствуют реакции,
поэтому становится возможным получение а,C-ненасыщенных нитрилов, нитро-
соединений и эфиров из а- или р-ацилоксисоединений. Гладко можно получить
и сопряженные диены. Ацетаты вторичных и третичных спиртов реагируют при
температурах от 400 до 500 °С практически полностью, тогда как ацетаты первич-
первичных спиртов при таком температурном режиме часто не вступают в реакцию.
Получение ненасыщенных соединений при пиролизе ацетатов: Organikum, 20.
Aufl., Wiley-VCH, Weinheim, 1999, S. 274.
Акрилонитрил и эфиры акриловой кислоты: Bums Н., Jones D. Т., Rorchie P. D. J.
Chem. Soc. (London), 1935, 400.
Бута-1,3-диен-1-карбонитрил: Gudgeon H., Hill R., Isaacs E. J. Chem. Soc.
(London), 1951, 1926.
a,p-Ненасыщенные кетоны из a-ацетоксикетонов: ColongeJ., Dubin J.-C. Bull.
Soc. Chim. France, 1960, 1180.
Примеры элиминирования по Чугаеву эфиров ксантогеновой кислоты в слу-
случае сильноразветвленных вторичных спиртов типа 3,3-диметилбутанола-2:
Нэс Г. Р. В сб.: Органические реакции. Сб. 12. — М.: Мир, 1965.
Получение метиленциклогексана элиминированием по Коупу: Коуп А., Сиге-
нек Е. В сб.: Синтезы органических препаратов. Сб. 11. — М.: ИЛ, 1961, с. 36.
Через образование циклического переходного состояния по аналогии с меха-
механизмом пиролиза сложных эфиров протекают и некоторые другие реакции, из
которых здесь следует упомянуть декарбоксилирование малоновых кислот и
Р-оксокислот. При этом также речь идет в основном о пиролитическом образо-
образовании олефина, который, однако, в данном случае представляет собой енол и
сразу же переходит в более бедную энергией кетоформу. По этой причине р-ок-
сокислоты совершенно стабильны лишь в том случае, если они не могут давать
енол, как, например, камфоркарбоновая кислота; в этом случае правило Бредта1
не допускает образования енольной связи (схемы реакций [Г.3.52]—[Г.3.54]).
Поскольку диоксид углерода, отщепляющийся при декарбоксилировании,
представляет собой очень энергетически бедное соединение, эти реакции элими-
элиминирования протекают уже при низких температурах (малоновые кислоты декар-
боксилируются при температурах 140-160 °С, р-оксокарбоновые кислоты — даже
ниже 100 °С). О практическом проведении этих реакций см. разд. Г,7.1.4.3.
1 Правило Бредта: В мостиковых бициклических соединениях двойная связь не может
находиться в голове моста, так как это привело бы к очень большому напряжению.
Бицикло[х,у,г]алкены-1 устойчивы лишь, если х + у + z > 7.
348
Г Препаративная часть
\/
°v°
нагревание
-С02
замещенная малоновая
кислота
\/
р-оксокарбоновая
кислота
-СО2
С
ОН
\/
о
замещенная уксусная
кислота
\/
rSrR
II
О
кетон
[Г.3.52]
[Г.3.53]
ноос -о 'он
Аналогично протекает пиролиз уксусного ангидрида до кетена:
А
700'С
СН3
сн2
с
II
о
он
I
О"" "СН3
Кетен можно получить также пиролизом ацетона:
Н3С-СО-СН3 - Н2С=С=О + СН4
[Г.3.54]
[Г.3.55]
[Г.3.56]
Оба способа применяются в промышленности. О некоторых реакциях кете-
нов см. разд.Т,7.1.6 и Г, 7.4.2.3.
3.3. а,а-ЭЛИМИНИРОВАНИЕ
При а,а-элиминировании геминальных заместителей образуются карбены
R2C: — короткоживущие, очень реакционноспособные промежуточные соеди-
соединения.
Так, при дегидрогалогенировании ди- и тригалогенметанов, а также ди- и
тригалогенускусных кислот (с последующим декарбоксилированием) образуют-
образуются короткоживущие галогенкарбены На1(Н)С| или На12С|, например:
[Г.3.57]
[Г.3.58]
Cl Q
CI
CI
ci-c-coo® —-
н
я
1С
CI
Я1
1С
н
+ н0 +
+ со2
cie
+ CI
3. Элиминирование с образованием кратных углерод-углеродных связей 349
Напротив, метилены R2CI (R = алкил, арил или Н) образуются из алифати-
алифатических диазосоединений (см. разд. Г,8.4) при пиролизе, УФ-облучении или в
присутствии катализаторов (см. также разложение а-диазокетонов; схема
[Г.9.22]).
Аналогично образованию карбенов из диазоалканов при термолизе или фотолизе азидов
возникают другие короткоживущие промежуточные соединения — нитрены:
0 © _ __
R-N-N=N R-N + N2 [Г.3.59]
Нитрены вступают в те же реакции, что и карбены.
В силу наличия на карбеновом атоме углерода секстета электронов карбены в общем
случае являются электрофильными агентами. Два несвязывающих электрона карбенового
атома углерода могут иметь противоположные спины; тогда они образуют электронную пару.
Такое состояние называют синглетным. В случае неспаренных электронов (бирадикал)
говорят о триплетном состоянии. Как правило, карбены образуются в синглетном состоянии
и затем переходят в энергетически более устойчивое триплетное состояние, последующие
реакции которого осуществляются по радикальному механизму. От спинового состояния
карбена в существенной мере зависит как механизм, так и стереохимия его реакций.
Ознакомьтесь подробнее с этой проблемой в приведенной в конце раздела литера-
литературе.
Укажем важнейшие реакции карбенов.
а) Включение в ковалентные связи, например в связь С-Н. Так, при фотолизе диазоме-
тана в диэтиловом эфире наряду с небольшими количествами этилена образуется
смесь этилпропилового и этилизопропилового эфиров:
C2H5-O-CH2-CH3
—- C2H5-O-CH2-CH2-CH3
+ ICH2
с2н5-о-сн-сн3
Путем внутримолекулярного включения в связь С—Н из алкилкарбенов легко образуются цик-
циклопропаны. (Напишите схему реакции!) Примером давно известной реакции включения
дихлоркарбена (образующегося из хлороформа в присутствии гидроксида натрия) может
служить карбшшминная реакция, применяющаяся как для препаративного получения изоци-
анидов, так и для аналитического обнаружения первичных амидов (см. разд. Д, 1.2.8.1), а также
синтез о-фенолкарбальдегидов по Реймеру-Тиману. (Прочтите об этой реакции в учеб-
учебнике!)
б) Электрофильное присоединение карбенов к кратным связям; см. разд. Г.4.4.1.
в) Внутри- и межмолекулярное отщепление атома водорода и последующие превраще-
превращения образующихся радикалов:
R-CH + H-R' R-CH2 + R1 [Г.3.61]
Рекомбинация образующихся радикалов может, например, привести к продуктам включения.
В противоположность высокой реакционной способности описанных короткоживущих
карбенов при депротонировании подходящих N- или 8,М-гетероциклических солей получают
350 Г Препаративная часть
стабильные карбены, которые можно выделить и охарактеризовать. Стабильность этих карбе-
нов объясняется мезомерией между электронной парой атома углерода и соседнего гетероато-
ма, например,
R R R
>1- [Г.3.62]
R R R
Данные об устойчивых карбенах представлены в литературе, указанной в разд. Г,3.4.
3.4. ЛИТЕРАТУРА
О механизмах реакций элиминирования
Алескеров М. А., Юфит С. С, Кучеров В. Ф. Усп. хим., 1978, 47, 233-259.
Banthorpe D. V. Elimination reactions. Elsevier, Amsterdam, London, New York,
1963.
BunnetJ. F. Angew. Chem. 1962, 74, 731-741.
Cockerill A. F, Harrison R. G. In: The Chemistry of Doublebonded Functional Groups.
S.Patai (ed.). — John Wiley & Sons, London, 1977.
Ingold С Proc.Chem. Soc. 1962, 265-274.
Sounders Jr., W. H. In: The Chemistry Alkenes. S. Patai (ed.). — Interscience Publishers,
London, New York, Sydney, 1964.
Sounders Jr., W. H, Cockerill A. F: Mechanisms of elimination reactions. — John Wiley &
Sons, New York, 1973.
SicherJ. Angew. Chem. 1972, 84, 177-191.
Синтезы олефинов
Houben-Weyl, Bd. 5/lb. — Georg Thieme Verlag, Stuttgart, 1972.
Hubert A. J, ReimlingerH. Synthesis, 1969, 97; 1970, 405-430.
KndzingerH. Angew. Chem., 1968, 80, 778-792.
Левша Р. Я., Скварченко В. Р. Усп. хим., 1949,18, 515-545.
Элиминирование по Гофману и Коупу
Коуп А., Тримбум Э. В сб.: Органические реакции, Сб. 11. — М.: Мир, 1965, с. 327.
Элиминирование по Чугаеву
Нэс Г. В сб.: Органические реакции, Сб. 12. — М.: Мир, 1965, с. 71.
Синтезы ацетиленов
Craig J. С, Bergenthal M. D., Fleming I, Harley-Mason J. Angew. Chem., 1969, 81,
437-446.
4. Присоединение к неактивированным кратным углерод-углеродным связям 351
Franke W. u. a. Angew. Chem., I960, 72, 391-400; Neuere Methoden, 1961, 3,
261-279.
Джекобе Т. В сб.: Органические реакции. Сб. 5. — М.: ИЛ, 1951, с. 7—92.
Kdbrich G. Angew. Chem., 1965, 77, 75-94.
Левина Р. Я., Викторова Е. А. В сб. Реакции и методы исследования органических
соединений. Сб. 7. — М.: Госхимиздат, 1951, с. 7—98.
Получение кетенов
Хенфорд В., Зауер Дж. В сб.: Органические реакции. Сб. 3. — М.: ИЛ, 1951,
с. 110-139.
Schaumann К, Scheiblich S. In: Houben-Weyl, Bd. E15/2 A993), S. 2353-2530.
Tidwell Т. Т. Ketenes. — John Wiley & Sons, New York, 1994.
Карбены (метилены), нитрены
Burke St. D., Griego P. A. Org. React., 1979, 26, 361-475.
Carbene (Carbenoide). Hrsg, M. Regitz. In: Houben-Weyl., Bd. E19b A989),
S. 1-2214.
Chinoporos E. Chem. Rev. 1963, 63, 235-255.
Dave V., WarnhoffE. W. Org. React., 1970,18, 217-401.
Gilchrist T. L., Rees C. W. Carbenes, Nitrenes, Arynes. — Th. Nelson & Sons, London,
1969.
Jones M., Jr., Moss R. A. Carbenes, Bd. 1—2. — John Wiley & Sons, New York,
1973-1975.
Kirmse W. Angew. Chem. 1959, 71, 537-541; 1961, 73, 161-166; 1965, 77, 1-10; Prog.
Org.Chem. 1964, 6, 164-216; Carbene Chemistry. -Academic Press, New York,1964;
Carbene, Carbenoide und Carbeneanalyse. — Verlag Chemie, Weinheim/Bergstr.
1969.
Lwowski L. Angew. Chem. 1967, 79, 922-931.
Розанцев Г. Г., Файнзильберг А. А., Новиков С. С. — Усп. хим. 1965, 34, 177—218.
Wentrup С. In: Topic in Current Chemistry. Bd.62. — Springer-Verlag, Berlin, Heidelberg,
New York, 1976, S. 173-251.
Получение дигалогенокарбенов
с помощью межфазных реакций
Weber W. P., Gokel G. W. In: Phase Transfer Catalysis in Organic Synthesis. — Springer-
Verlag, Berlin, Heidelberg, New York, 1977, p. 18-72.
Стабильные карбены из гетероциклов
ArduengoA. J., Krafcyk R. Chem. Unserer Zeit, 1998, 32, 6-XX.
Herrmann W. A., Kocher С Angew. Chem. 1997,109, 2256-2282.
352 Г Препаративная часть
4. ПРИСОЕДИНЕНИЕ К НЕАКТИВИРОВАННЫМ
КРАТНЫМ УГЛЕРОД-УГЛЕРОДНЫМ СВЯЗЯМ
Для двойных и тройных связей типичны следующие реакции присоединения:
С=С + X-Y =^^= X-C-C-Y [Г.4.1а]
/ \ I I
\ /
—СНС— + X-Y ^=^ С=С [Г.4.16]
7 V
С формальной точки зрения речь идет о реакциях, обратных рассмотренным
в разд. Г,3 процессам элиминирования.
Важнейшие реакции присоединения суммированы в табл. Г.4.2. я-Связи
алкенов легко поляризуются. Нуклеофильность алкенов отражена и в относи-
относительно высокой энергии их я-ВЗМО1 (см. разд. В,6). Поэтому несопряженные
олефиновые двойные связи преимущественно подвергаются электрофильной
атаке.
Энергия я-ВЗМО алкинов ниже, чем у олефинов2. По этой причине они
реагируют преимущественно с нуклеофильными реагентами.
Кроме того, и двойные, и тройные связи способны вступать в реакции,
протекающие по радикальному механизму.
В связи с этим различают следующие реакции присоединения:
а) электрофильное присоединение (символ Ас1е),
б) нуклеофильное присоединение (символ AcIn),
в) радикальное присоединение (символ AcIr).
Полярность кратных углерод-углеродных связей особенно повышают замес-
заместители, способные к сопряжению. +Л/-Заместители (NR2, OR, ОН) повышают
электронную плотность соседней двойной связи1 и облегчают взаимодействие с
электрофильными агентами (см. разд. Г,7.4.2). —М-Заместители (карбонильная,
нитрильная и нитрогруппы) понижают электронную плотность на соседних
двойных связях1 и таким путем повышают их реакционноспособность по отно-
отношению к нуклеофильным реагентам (см. разд. Г,7.4.1).
У особенно «богатых электронами» олефинов (например, у N-алкилированных тетраами-
ноэтенов) тенденция к отдаче электронов настолько велика, что они являются восстановите-
восстановителями, растворимыми в органических растворителях. Напротив, олефины с пониженной
электронной плотностью, например, тетрацианэтилен, являются окислителями.
1 Энергия тс-ВЗМО неактивированного олефина равна ~ -10,5 эВ, а энергия НСМО равна
~ —1,5 эВ. Для олефинов эти параметры составляют около -9,0 эВ и +3,0 эВ (в случае
электродонорных заместителей) и -10,9 эВ и 0 эВ (в случае электроноакцепторных
заместителей) соответственно.
2 Энергии ВЗМО и НСМО алкинов изменяются в диапазонах от —10,9 до -11,4 эВ и от
+ 1,1 до +0,6 эВ соответственно.
4. Присоединение к неактивированным кратным углерод-углеродным связям
353
Таблица Г.4.2. Наиболее важные реакции присоединения к алкенам и алкинам
Схема реакции
Примечания
+ H-Hal
+ H-OSO3H
+ H-OH
+ H-OR
+ Hat-Hal
+ HO-Cl
+ (HgOAc)ffl/OHe
+ )b-h
1/2 02
:CR2
03
H«
H-C-C-Hal
I I
H-C-C-OSO3H
H-C-C-OH
I I
H-C-C-OR
Hal
-U-
Hat
I I
HO-C-C-Ct
AcOHg-C-C-OH
I I
HO
1 I
.1 I
c-c—
V
ho-c-c-oh
c
R R
Присоединение
HC1, HBr, HI с обра-
образованием алкилгало-
генидов
Присоединение
H2SO4 с образовани-
образованием гидросульфатов
Кислотная гидрата-
гидратация с образованием
спиртов
Присоединение
спиртов с образова-
образованием простых эфиров
Присоединение гало-
галогенов и межгалогенных
соединений с образова-
образованием вицинальных ди-
галогеналканов
Присоединение кисло-
кислородсодержащих кис-
кислот галогенов в низ-
низших степенях окисле-
окисления (типа хлорнова-
хлорноватистой) с образовани-
образованием галогенгидринов
Оксимеркурирование
Восстановление про-
продуктов меркуриро-
вания до спиртов
Гидроборирование
Окисление
нических
до спиртов
борорга-
Эпоксидирование и
гидроксилирование до
эпоксидов и 1,2-ди-
гидр оксисоединени й
Циклоприсоединение
карбенов с образова-
образованием циклопропанов
Озонирование
Восстановление озо-
нидов до альдегидов
и кетонов
354
Г Препаративная часть
Таблица Г.4.2. Продолжение
Схема реакции
Примечания
+ -C=N-Oe
О
+ со
Катали-
затор
Катали^
затор
Катали-
Катализатор*
I
н-сн-с
н-i-c
¦ н-сн-С-сн2он
i i
I V/
-C=C- + H-OH
+ H-OR
+ )C-R
=r/'
:=c
он
\
,c-cx
,c-cx
\
OR
/C=cN
OCOR
+ Hal-Hal
Hal
\c_c/
Hal
Циклоприсоединение
1,3-диполей с образова-
образованием пятичленных гете-
роциклов (например,
Д2-1,2-оксазолинов)
Циклоприсоединение
диенов с образовани-
образованием циклогексенов (ре-
(реакция Дильса — Аль-
дер а)
Гидрирование
Реакция с СО/Н2 с
образованием альде-
альдегидов (гидроформи-
лирование) и далее
спиртов
Олигомеризация, цик-
циклизация и полимери-
полимеризация
Гидратация ацетиле-
ацетиленов с образованием
альдегидов (кетонов)
Присоединение спир-
спиртов к ацетиленам с
образованием про-
простых эфиров енолов
Присоединение кис-
кислот к ацетиленам с
образованием слож-
сложных эфиров енолов
Присоединение ами-
аминов к ацетиленам с
образованием енами-
нов
Присоединение гало-
галогенов к ацетиленам
с бразованием ви-
цинальных дигалоген-
алкенов
4. Присоединение к неактивированным кратным углерод-углеродным связям 355
4.1. ЭЛЕКТРОФИЛЬНОЕ ПРИСОЕДИНЕНИЕ
К ОЛЕФИНАМ И АЦЕТИЛЕНАМ
4.1.1. МЕХАНИЗМ ЭЛЕКТРОФИЛЬНОГО ПРИСОЕДИНЕНИЯ
Электрофильное присоединение к олефинам можно рассматривать как кислот-
кислотно-основное взаимодействие. Реакции этого типа протекают тем легче, чем бо-
более основен (точнее, более нуклеофилен) олефин и чем кислотнее (точнее, более
электрофилен) реагент.
Упоминавшееся выше влияние +М- и +/- либо —М- и -/-заместителей отра-
отражает следующий экспериментально найденный ряд повышения основности
олефинов:
CI-CH=CH2 = НООС-СН=СН2 < Н2С=СН2 < Alk-CH=CH2
[ГАЗ]
< (AlkJC=CH2 < Alk-CH=CH-Alk < (AlkJC=C(AlkJ
В качестве электрофильных агентов к олефинам присоединяются протонные
кислоты и кислоты Льюиса, в том числе галогеноводородные кислоты, серная
кислота, азотная кислота, НзО+, галогены, межгалогенные соединения (IC1,
ВгС1 и т. д.), гипогалогенные кислоты и другие соединения.
Свободные галогены являются потенциальными кислотами Льюиса, пос-
поскольку они могут поляризоваться под влиянием электрофильного агента (раст-
(растворителя или катализатора, например А1С13, ZnCl2, BF3 и т. д.). Реакционная
способность присоединяющихся реагентов возрастает с повышением их кис-
кислотности (например, у галогеноводородных кислот в ряду HF « НСК НВг < HI,
у галогенов в ряду h < Вг2 < С1г) и в присутствии электрофильных катализа-
катализаторов.
Электрофильный агент сначала взаимодействует с я-связью; при этом может
образовываться я-комплекс (комплекс с переносом заряда).
Ароматические соединения также образуют л-комплексы, обнаруживаемые, например,
по характеристическим полосам поглощения в УФ-диапазоне. Визуально л-комплексы мож-
можно обнаружить по интенсивной окраске растворов иода или А1С13 в ароматических углево-
углеводородах.
На первой стадии присоединения (схема [Г.4.4]) образуется карбениевый
ион I. На следующей быстрой стадии нуклеофильный агент присоединяется к
карбениевому иону, образуя конечный продукт реакции II. На схеме [Г.4.4] хлор
присоединяется под воздействием электрофильного агента Е (растворителя или
катализатора), поляризующего молекулу хлора:
356
Г Препаративная часть
\© I Q
C-C-CI + C1---E
I
[Г.4.4]
CI-C-C© + CI
I \
ci-c-c-ci
I I
До стадии образования карбениевого иона I аналогично протекает и электрофильное хло-
хлорирование ароматических соединений (см. разд. Г,5.1.5).
Двухстадийный механизм электрофильного присоединения доказывается
тем, что во второй стадии реакции (взаимодействие с карбениевым ионом)
могут конкурировать другие нуклеофильные агенты, например при взаи-
взаимодействии хлора с олефинами в присутствии бромида натрия, воды или
спирта1:
I ©/
а—с-с
I \
+ СГ
+ Вг
е
CI-C-C-CI
Cl-C-C-Br
+ Н2О I ©
——- а-с-с-он2
-н
©
CI-C-C-OH
CI-C-C-OR
[Г.4.5а]
[Г.4.56]
[Г.4.5в]
[Г.4.5г]
н
Степень протекания отдельных конкурирующих реакций определяется
реакционной способностью и концентрацией каждого из нуклеофильных
агентов и может быть изменена подбором условий (например, концентра-
концентраций).
При реакциях электрофильных агентов с сопряженными диенами перво-
первоначально также образуется карбениевый ион. Однако его положительный
заряд делокализован по всей сопряженной системе. Поэтому нуклеофильный
реагент может присоединяться как к атому С-2, так и к атому С-4, например:
1 Схема [Г.4.5в] соответствует присоединению кислородсодержащих кислот галогенов в
низших степенях окисления.
ci—сн2-сн^сн-^сн2 + cie
4. Присоединение к неактивированным кратным углерод-углеродным связям 357
н2с=сн-сн=сн2 + ci-ci —- а-сн2-сн-сн=сн2 — а-сн2-сн=сн-сн2 + ае [Г.4.6а]
С1-СН2-СН=СН-СН2-С1 A,4-аддукт)
" f
[Г.4.66]
CI—СН2-СН-СН=СН2 A,2-аддукт)
CI
Поэтому в общем случае образуются смеси 1,2- и 1,4-аддуктов. Термодина-
Термодинамически устойчивее 1,4-аддукт, так что он часто образуется в большем количе-
количестве (в приведенном примере 1,4-аддукта образуется в четыре раза больше, чем
1,2-аддукта).
4.1.2. НАПРАВЛЕНИЕ НУКЛЕОФИЛЬНОЙ АТАКИ И ВЛИЯНИЕ
ПРОСТРАНСТВЕННЫХ ЭФФЕКТОВ ПРИ РЕАКЦИЯХ
ЭЛЕКТРОФИЛЬНОГО ПРИСОЕДИНЕНИЯ
Большинство электрофильных агентов, присоединяющихся к олефинам в пер-
первой стадии (см. схему [Г.4.4]), может затем внутримолекулярно взаимодейство-
взаимодействовать с карбениевым атомом углерода, выступая уже в роли «нуклеофилов». При
этом карбениевый ион временно стабилизируется за счет образования
«мостика».
При этом можно представить себе следующие граничные случаи:
Xе
С
^с-с^ «,.с-с- ^с#с< Н^ [ГА7]
* ' ^ © '* ^ ^ е ^ э Y "*
I II III IV
С учетом этих граничных случаев можно объяснить направление нуклеофильной атаки
при присоединении к несимметричным олефинам и стерические эффекты при нуклеофиль-
ном присоединении.
На схеме [Г.4.7] I — классический карбениевый ион. На второй стадии нуклеофильный
агент Х| может атаковать треугольный плоский карбениевый ион с обеих сторон. Такое присо-
присоединение нестереоселективно. Карбениевый ион II имеет несимметричную мостиковую
структуру и поэтому допускает только стереоселективное ак/ш-присоединение Х|. Симмет-
Симметричный мостиковый катион III можно представить также как делокализованный л-комплекс.
Он также допускает лишь он/им-присоединение. Направление присоединения X и Y к несим-
несимметричным олефинам в этом случае неоднозначно. Если расщепление агента X—Y затруднено,
то смн-присоединение может происходить через четырехцентровое переходное состояние IV
(см., например, реакцию гидроборирования).
Если несимметричный реагент (например, HHal, H2O) присоединяется к
несимметрично замещенному олефину, то образуется практически исключи-
исключительно один из двух возможных аддуктов:
358 Г Препаративная часть
R-CH-СНз
R-CH=CH2 + НХ
Ч* Л [Г.4.8]
R-CH2-CH2-X
Присоединившийся на первой стадии протон не способен создать мостик в
карбениевом ионе. Поэтому образуется лишь тот из двух классических карбе-
ниевых ионов, который выгоднее энергетически. Так, из пропена и
НС1 возникает карбениевый ион I (схема [Г.4.9]), в котором положительный
заряд стабилизирован +/-эффектом и поляризационным эффектом двух
метильных групп. В качестве конечного продукта образуется изопропилхло-
рид:
0 е
н3с-сн-сн3 + с\
Н3С-СН=СН2 + HCI <\ ' [Г 4 91
^Ч © е
н3с-сн2-сн2 + а
Этот результат можно обобщить следующим образом: электрофильное при-
присоединение протонных кислот к олефинам протекает через образование наибо-
наиболее стабильного катиона. Иными словами, при электрофильном присоединении
протонных кислот к несимметрично замещенным олефинам атом водорода присое-
присоединяется к углеродному атому двойной связи, уже имеющему наибольшее число
водородных атомов (правило Марковникова).
Это правило можно распространить и на гипогалогенные кислоты, а также
межгалогенные соединения. Однако здесь возможны отклонения от правила
Морковникова, которые связаны с возможностью образования симметричной
мостиковой структуры типа III на схеме [Г.4.7].
[Г.4.10]
Вообще правило Марковникова также выполняется, если может возникать
четырехцентровое переходное состояние IV на схеме [Г.4.7] (ср. разд. Г,4.1.8,
присоединение бороводорода к олефинам).
Для реакций нуклеофильного и радикального присоединения правило
Марковникова теряет силу!
Стереохимия реакций присоединения имеет значение лишь в тех случаях, когда возника-
возникают два асимметрических атома углерода, а также у циклических соединений, в которых затруд-
затруднено вращение вокруг простой связи С-С. Закрепляющий конфигурацию эффект электро-
фильного агента, присоединившегося к олефину на первой стадии реакции, возрастает при
повышении его поляризуемости в ряду С1< Вг < I.
Присоединение брома осуществляется через симметричный мостиковый карбениевый
ион, поэтому на второй стадии бромид-ион может присоединяться только в снтм-положение:
R
i
СН
II
СНо
X
+ 1
н
он
1
н
CI
1
1
он
1
X
4. Присоединение к неактивированным кратным углерод-углеродным связям 359
+ Вг—Вг
или
Вг
• d-c-"iv
-Вг
Вг
Вг
Вг
Вг
[Г.4.11]
Малеиновая кислота присоединяет бром, образуя с 80%-ным выходом рацемическую ди-
бромянтарную кислоту, а фумаровая в аналогичной реакции дает мезодибромянтарную кислоту:
+ВГ2
соон
-СООН
<r Вг<Э
Н
Вг
]/
СООН
Вг
СООН
Вг
H-yiCOOH
Вг
СООН
Вг——Н D-дибром-
и пг янтарная
1~ кислота
СООН
СООН
н-
Вг-
-Вг L-дибром-
-Н
СООН
янтарная
кислота
[Г.4.12а]
НООС-%
соон
ноос н
ноос н
соон
У циклогексена из двух возможных транс-форм получают обычно только й/с-аксиальную
(см. [Г.3.25]):
Вг
Br2
-Вг
©
Вг2
-Вг
е
Вг
Вг
Вг Вг
Вг
©
[Г.4.13]
4.1.3. ПРИСОЕДИНЕНИЕ ПРОТОННЫХ КИСЛОТ И ВОДЫ
К ОЛЕФИНАМ И АЦЕТИЛЕНАМ
Присоединение сильных кислот (галогеноводородных, серной и т. д.) к олефи-
нам протекает в соответствии с механизмом, описанным в разд. Г,4.1.1 как
двухстадийный процесс, на первой стадии которого присоединяется протон, в
то время как анион реагирует лишь на второй стадии:
с=с
н-х
I /
н-с-с©
I \
+ X
©
н-с-с-х
[Г.4.14]
360
Г Препаративная часть
При присоединении галогеноводородных кислот по этому механизму полу-
получаются алкилгалогениды, а взаимодействие с серной кислотой приводит к
моноал кил сульфатам.
Вода не может непосредственно присоединяться к олефинам, поскольку ее
кислотные свойства очень слабо выражены (концентрация НзО+ очень мала).
Но это присоединение легко идет в присутствии таких сильных кислот, как
серная, азотная и др. Оно представляет собой прямой процесс и протекает без
промежуточного образования сложного эфира:
\ /
/С=С\
I /
н-с-с© +
Н2О
I I ©
н-с-с-он2
н-с-с-он + н [Г.4.15]
Аналогично с промежуточным карбениевым ионом могут реагировать и
другие имеющиеся в реакционной смеси нуклеофильные реагенты, например
анион кислоты, применяемой в качестве катализатора (см. схему [Г.4.14]), уже
образовавшийся спирт и оставшийся олефин. Так, при взаимодействии олефи-
на с водной H2SC>4 протекают конкурирующие реакции [Г.4.16]:
+ ho-so2-o
0
I /
н-с-с©
I \
I I
+H-C-C-OSO2O
е
I I
H-C-C-OSO2OH
моноал килсульфат
h-c-c-o-so2-c—с-с-н
диалкилсульфат
-Н2О
I I
+н-с-с-он
I I
I I ©
Н-С-С-ОН2
н-с-с-о-с-с-н
Mill
III/
н-с-с-с-с© —%г
II I \ -н®
I I
н-с-с-он
I I
спирт
[Г.4.1ба]
[Г.4.166]
[Г.4.1бв]
н-с-с-о-с-с-н [Г.4.16г]
М II
эфир
I I /
Н-С-С-С=С и т. д.
I I I \
димеры,полимеры
[Г.4.16Д]
С безводными или очень концентрированными кислотами практически возмож-
возможно только образование сложных эфиров и полимеризация, причем большие количе-
количества кислоты способствуют образованию сложного эфира, а высокая основность оле-
фина — полимеризации. В присутствии разбавленных кислот большую роль играет
прямая гидратация, при которой в качестве побочных продуктов всегда образуются
простые эфиры.
Реакционная способность олефинов и кислот обсуждалась в разд. Г,4.1.1. Чем
инертнее олефины, тем сильнее или концентрированнее должна быть применяемая
кислота. Например, этилен не взаимодействует с концентрированной соляной кис-
кислотой, но реагирует с бромоводородной или иодоводородной кислотами. Напротив,
изобутен легко реагирует с хлороводородом, который взаимодействует с этиленом
4. Присоединение к неактивированным кратным углерод-углеродным связям 361
только в присутствии кислотных катализаторов, например хлорида алюминия.
Изобутен и другие третичные олефины легко реагируют с серной кислотой уже при
О 'Си концентрации кислоты 65%. Для пропена и н-бутена нужно применять
85%-ную кислоту, а этилен быстро взаимодействует лишь с 98%-ной серной
кислотой при нагревании. Поэтому можно, например, из С4-фракции крекинг-газа
легко «вымыть» изобутилен 60—65%-ной серной кислотой.
При высоких концентрациях серной кислоты, которые применяются при крупномасштаб-
крупномасштабных промышленных синтезах, основной реакцией становится протекающее наряду с гидрата-
гидратацией образование моноалкилсульфатов. Кроме того, в реакционной смеси, обнаруживают
также диалкилсульфат (см. разд. Г,2.5.2 и табл. Г.4.20), который путем кислотного гидролиза
или алкоголиза перерабатывают в спирт или простой эфир.
При присоединении кислот и воды к олефинам получают не только однород-
однородные и ожидаемые по правилу Марковникова продукты, но и смеси. Карбениевый
ион II (схема [Г.4.17]), возникший после присоединения протона к двойной связи
олефина I, может перегруппироваться в обедненный энергией ион III. При пов-
повторном отщеплении протона образуется олефин IV (см. также схему [Г.3.31]):
+ н® ©
н3с-сн2-сн2-сн=сн2 - д* н3с-сн2-сн2-сн-сн3 и
[Г.4.17]
+ н ©
Н3С-СН2-СН=СН-СН3 - ' Н3С-СН2-СН-СН2-СН3
IV *
Так, концентрированная серная кислота при —10 °С присоединяется к доде-
цену-1, давая смесь изомерных додецилсульфатов. По этим же причинам под
действием кислот двойная связь в олефинах легко мигрирует. Если реакция про-
протекает достаточно долго, получаются различные олефины в количествах, соотве-
соответствующих их термодинамической устойчивости.
Электрофильное присоединение воды, спиртов и кислот к ацетиленам про-
протекает только в присутствии специальных катализаторов (ртутных и медных
солей), так как ацетиленовые соединения недостаточно реакционноспособны
по отношению к электрофильным агентам (см. разд. Г,4, введение). Механизм
этого присоединения в деталях еще не выяснен.
Енолы, образующиеся при присоединении воды к ацетиленам, тотчас же
перегруппировываются в карбонильные соединения:
-СНС- + Н2О Нг5О"Н95°*. \-Г — -L/ [г.4.18]
н \ н х
Из самого ацетилена образуется ацетальдегид, а из замещенных ацетиленов —
кетоны. Из соответствующих ацетиленов можно получить сс,р-ненасыщенные
кетоны и а-гидроксикетоны. Олефиновые двойные связи в условиях гидратации
ацетиленов, как правило, не реагируют.
Ф Общая методика гидратации ацетиленовых соединений (табл. Г.4.19). В трехгорлую колбу на
500 мл, снабженную мешалкой, обратным холодильником и капельной воронкой, помещают
8 мл концентрированной серной кислоты, 5 г сульфата ртути(Н) и 200 мл воды, нагревают до 60 °С и в
течение 1 ч при хорошем перемешивании добавляют по каплям 0,5 моль соответствующего алкина.
Перемешивание продолжают еще 3 ч при 60 °С, затем охлаждают в бане со льдом и извлекают пятью
порциями эфира (по 40 мл). Объединенные эфирные вытяжки промывают насыщенным раствором
хлорида натрия до нейтральной реакции и сушат сульфатом натрия. После удаления эфира кетон пе-
перегоняют. Для кетона с большой молекулярной массой можно повысить температуру реакции до 80 °С.
362
Г Препаративная часть
Таблица Г.4.19. Кетоны, полученные гидратацией ацетиленовых производных
Продукт реакции
Гексанон-2
Гептанон-2
Октанон-2
A -Гвдроксициклогексил)метилкетон
A -Гидроксицихлопентил)метилкетон
З-Гидрокси-3-метилпента-
нон-2
Алкин
Гексин-1
Гептин-1
Октин-1
1 -Этинилциклогексанол
1 -Этинилциклопентанол
З-Гидрокси-3-метилпен-
тин-1
Т. кип., 'С
(мм рт. ст.)
126
148
168
93 A5)
77 A0)
72 E0)
V5
1,3985
1,4066
1,4134
1,4670
1,4619
1,4200
Выход,
Ж
78
85
90
65
65
60
При работе с полумикроколичествами можно использовать видоизмененную методику.
Предварительно из 100 мг красного оксида ртути, 10 мгтрихлоруксусной кислоты, 0,25 мл ме-
метанола и 0,15 мл эфирата трифторида бора получают катализатор, для чего смесь в пробирке
нагревают в течение 1 мин при 50-60 °С. Растворяют 1 г соответствующего алкина в 3 мл мета-
Таблица Г.4.20. Применение продуктов присоединения неорганических кислот,
воды и спиртов к алкенам и алкинам
Продукт
Этилхлорид (см. табл. Г. 1.37)
Этанол
Изопропанол
вгор-Бутанол
грег-Бутанол
Моноалкилсульфаты Си—Qe
грег-Бутилметиловый эфир
Винилхлорид»
Винилацетата
Ацетальдегид*
2-Хлорбутадиен-1,3
Применение
—*¦ Этилцеллюлоза (см. разд. Г.2.6.2)
Растворитель
—»¦ Ацетальдегид —>• уксусная кислота
—>¦ Диэтиловый эфир (см. табл. Г. 2.61)
—>• Сложные эфиры (растворители)
—> Хлораль —>¦ ДДТ (см. разд. Г,5.1.8.5)
Растворитель
—>¦ Сложные эфиры (растворители)
—> Метилэтилкетон (растворитель)
Алкилирующее средство (-* грег-бутилфенол
и т.д.)
Моющие средства
—> Перспективный антидетонатор; понижа-
понижает содержание СО в выхлопных газах
—>¦ Поливинилхлорид (см. табл. Г 3.37)
—»¦ Поливинилацетат (—*¦ поливини-
поливиниловый спирт)
—> Сополимеры (например, с винилхлори-
дом)
—> Уксусная кислота, ацетангидрид
—>• Этилацетат (реакция Кляйзена — Ти-
щенко, см. разд. Г,7.3.1.3)
—«-Ацетальдоль-»-кротоновый альдегид (см.
табл. Г. 3.37)
—>¦ Пентаэритрит
—>¦ Хлораль —>¦ ДДТ (см. разд. Г,5.1.8.5)
—>• Надуксусная кислота
—>• Глиоксаль
—> Пиридин и алкилпиридины
—>• Хлоропрен
Эти продукты получают не из ацетилена, а из более дешевого этилена.
4. Присоединение к неактивированным кратным углерод-углеродным связям 363
нола, смешивают с раствором катализатора и нагревают 30 мин при 50—60 °С. За счет присое-
присоединения двух молей спирта получается ацеталь; при этом выпадает серый осадок. Добавлени-
Добавлением 2—3 мл воды ацеталь омыляют до кетона; для связывания кислоты прибавляют 10% карбо-
карбоната калия. Извлекают эфиром и дальнейшую обработку проводят по описанной выше мето-
методике.
Этот вариант пригоден для аналитической идентификации ацетиленов,
тогда как образующиеся кетоны (сырой продукт) следует охарактеризовывать
подходящими производными.
Примеры гидратации ацетиленовых производных: Купин Б. С, Петров А. А.
ЖОХ, 1961,31,2963.
Гидратация 1,1-дизамещенных пропаргиловых спиртов при одновременной
дегидратации третичных спиртов до а,/5-ненасыщенных кетонов в присутствии
ионообменников: Newman M. S. J. Am. Chem. Soc, 1953, 75, 4740.
Присоединение неорганических кислот и воды к олефинам не имеет препара-
препаративного значения. Обычно простые галогениды легко получить другим путем,
например из спиртов, и часто, напротив, сами галогениды служат для получения
олефинов.
В промышленности же присоединение хлороводорода, серной кислоты и воды к олефи-
олефинам, а также присоединение воды, хлороводорода, синильной и уксусной кислот к ацетиле-
ацетиленам1 нашло очень широкое применение. Важнейшие продукты перечислены в табл.
Г.4.20.
Для присоединения воды к олефинам наряду с уже описанным прямым процес-
процессом с применением серной кислоты (схема [Г.4.15]) используют еще газофазный
процесс, при котором олефин в смеси с водяным паром при повышенной
температуре и давлении (примерно 300 °С и 7 МПа) пропускают над кислотными
катализаторами (например, фосфорной кислотой на кизельгуре,
TiO2/Fe2O3).
4.1.4. ПРИСОЕДИНЕНИЕ ГАЛОГЕНОВ И
ГИПОГАЛОГЕННЫХ КИСЛОТ К ОЛЕФИНАМ И АЦЕТИЛЕНАМ
Присоединение хлора и брома является характерной реакцией на двойные связи
С=С; реакция протекает легко и во многих случаях количественно. Влияние ре-
реакционной способности (основности) олефинов на скорость присоединения
брома видно из ряда [Г.4.3]. Отдельные олефины либо из-за малой плотности
электронов у двойной углерод-углеродной связи, либо из-за пространственных
затруднений совсем не присоединяют бром или присоединяют его с трудом
(например, тетрацианэтилен, тетрафенилэтилен, а,р-ненасыщенные кислоты и
кетоны).
Присоединение иода, как правило, не удается осуществить из-за понижен-
пониженной активности этого галогена (см. разд. Г,4.1.1). Оно протекает удовлетвори-
Другие технически важные продукты винилирования указаны в разд. Г.4.2.2.
364 Г Препаративная часть
тельно только в случае очень реакционноспособных олефинов (например,
стирола, аллилового спирта и др.)- Напротив, взаимодействие фтора с двойны-
двойными углерод-углеродными связями происходит так энергично, что олефин распа-
распадается на осколки с меньшим числом углеродных атомов.
У олефинов, имеющих разветвление при двойной углерод-углеродной связи
(изобутилен, триметилэтилен), при действии хлора происходит замещение
актома водорода с сохранением двойной связи:
Н3С Н2С
С=СН2 + С12 С-СН2-С1 + HCI [Г.4.21]
Н3С H3c'
металлилхлорид
При этом также вначале присоединяется катион хлора, но образовавшийся
карбениевый ион отщепляет протон с образованием олефингалогенида.
При более высоких температурах D00—500 "С; «горячее» хлорирование)
w-олефины также хлорируются с замещением по радикальному механизму в
аллильное положение (см. также разд. Г, 1.4.1):
Н2С=СН-СН3 + С12 5°° °- Н2С=СН-СН2С1 + HCI [Г.4.22]
аллилхлорид
В реакциях с галогенами тройная связь менее реакционноспособна, чем двой-
двойная (см. разд. Г,4, введение). Это подтверждает, например, следующая реакция:
Н2С=СН-СН2-СНСН -^- Н2С-СН-СН2-СЕСН [Г.4.23]
Вг Вг
Если присоединение галогенов к олефинам проводят в водном растворе, то по-
получают р-гидроксигалогениды (см. схему [Г.4.5в]). Чтобы поддерживать малую
концентрацию хлорид-иона и тем самым подавлять побочную реакцию присоеди-
присоединения галогена, процесс останавливают, после того как взаимодействие пройдет
лишь на несколько процентов. Поэтому более выгодно препаративное получение
р-гидроксигалогенидов (галогенгидринов) прямым присоединением гипо-
галогенной кислоты (например, из хлорамина Т, mpem-бутилгипохлорита).
Присоединение галогенов является основным препаративным методом получе-
получения вицинальных дигалогенидов, которые имеют большое значение при синтезе
ацетиленов и диенов (см. схему [Г.3.39]). Присоединение брома можно также ис-
использовать для очистки олефинов, отщепляя галоген из легче очищаемых диброми-
дов под действием цинковой пыли или иодида калия в ацетоне (см. схему [Г.3.28]).
(ГП) Общая методика присоединения брома к олефинам и ацетиленам (табл. Г.4.24)
11 При работе с бромом необходимо соблюдать осторожность (часть Е)! Работать
| под тягой в резиновых перчатках!
В трехгорлой колбе, снабженной мешалкой, капельной воронкой и внутренним термомет-
термометром, раствор непредельного соединения в двух- или трехкратном количестве тетрахлоруглерода
или хлороформа охлаждают до 0 °С. При температуре 0-5 °С и хорошем перемешивании прибав-
прибавляют по каплям раствор эквимолярного количества брома (примерно в двойном объеме того же
растворителя). Температура должна поддерживаться в указанном диапазоне. Концентрация
непрореагировавшего брома в реакционной смеси не должна быть высокой (контролируется по
Таблица Г.4.24. Присоединение брома к кратным СС-связям
Продукт
присоединения
1,2-Дибромгексан
1,2-Дибром гептан
1,2-Дибром октан
1,2-Дибромдекан
1,2-Дибромдодекан
транс-1,2-Дибромцикло-
гексана
1,2-Дибром-1 -фенилэтан
Мезодибромянтарная
кислота
1,2,3-Трибром пропан
(?) -1,2-Дибромстиль-
бен
1,2-Дибром стирол
DL-Дибромянтарная
кислота
Исходное
соединение
Гексен-1
Гептен-1
Октен-1
Деиен-1
Додецен-1
Циклогексен
Стирол
Фумаровая
кислота
Аллилбромид
Дифенилацетилен
Фенилацетилен
Малеиновый
ангидрид
Т. кип., "С
(мм рт. ст.)
90 A8)
103 A2)
117 A4)
160A8)
174 B5)
96A1)
133 A9); 74 (т. пл.)
256 (т. пл.)
100A8)
211 (т.пл.; этанол)
133A5); 74 (т.пл.;
аТаНОЛ)
166—167 (т. пл.;
вода)
«в20
1,5010
1,5015
1,4956
1,5010
1,5540
1,5868
Выход, %
90
90
90
90
90
95
95
80
90
60
78
80
Примечания
Для работы берут бром в недоста-
недостаточном количестве (на ~10%), так
как в противном случае заметно про-
происходит реакция замещения, что
сильно понижает выход
Применять свежеперегнанный стирол.
Продукт разъедает кожу (резиновые
перчатки!)
Готовят суспензию фумаровой кисло-
кислоты в двукратном объеме кипящей
воды. При кипении смеси по каплям
добавляют бром (без растворителя).
Продукт присоединения выпадает
при охлаждении до —10 °С и отмы-
отмывается от брома водой
Реакцию проводят в эфире, который
легко растворяет одновременно полу-
получающийся (Z)-изомер (т. пл. 64 °С)
Реакцию проводят при 50 °С. Выпав-
Выпавший после охлаждения продукт от-
отделяют и перекристаллизовывают из
горячей воды
а Для дополнительной очистки продукт реакции встряхивают 5 мин с 20%-ным спиртовым раствором гидроксида калия (в количестве
1/3 объема очищаемого продукта), разбавляют равным объемом воды, отмывают от щелочи, сушат сульфатом натрия и перегоняют. При очист-
очистке теряется 10% продукта.
366
Г Препаративная часть
окраске). Выпадающий в осадок продукт присоединения отсасывают либо отгоняют раствори-
растворитель и остаток очищают перегонкой или перекристаллизацией.
Реакция хорошо проходит с полумикроколичеегвами веществ. В этом случае отказываются от ме-
мешалки и внутреннего термометра, а при добавлении брома реакционную смесь хорошо встряхивают.
Реакция присоединения брома служит для качественного определения двой-
двойной связи С=С.
/jn^j) Качественная реакция на С=С-двойные связи: «бромная проба». Разбавленный раствор бро-
^-^ ма в хлороформе или ССЦ медленно по каплям прибавляют к раствору олефина в том же
растворителе. Если бром тотчас обесцвечивается, то очень возможно присутствие соедине-
соединения с двойной связью. Если обесцвечивание происходит медленно или если одновременно выделя-
выделяется бромоводород в виде тумана, то эта проба для данного соединения является малопригодной, так
как насыщенные соединения (некоторые кетоны, спирты, амины и ароматические соединения) так
реагируют с бромом путем замещения или окисления. Кроме того, как уже упоминалось, некоторые
олефины не реагируют с бромом вообще или реагируют медленно.
Для количественного определения двойных связей С=С обычно используют присоедине-
присоединение брома (бромное число), реже иода (йодное число). Имея дело со смесями веществ, всегда
надо считаться с возможностью побочных реакций, которые могут завысить результат опреде-
определения. Кроме того, некоторые алкены присоединяют галоген в неэквивалентных количествах
(например, в результате 1,4-присоединения к сопряженным диенам), поэтому этот метод дает
лишь приближенную оценку степени ненасыщенности исследуемой пробы.
В промышленных масштабах широко используют присоединение хлора к алкенам и алки-
нам, а также получение гидроксихлоралканов (хлоргидринов) (табл. Г.4.26).
В результате присоединения S2Cl2 к двойным связям С=С при холодной вулканизации
происходит сшивание ненасыщенных полимерных цепей S—S-мостиками:
2 С=С + S2CI2 Cl—C-C-S-S-C-C-CI [Г.4.25]
/ \ II I I
Таблица Г.4.26. Важные продукты, получаемые присоединением галогенов и кис-
кислот типа хлорноватистой к алкенам и алкинам
Продукт присоединения
Этиленхлорид
A,2-дихлорэтан)
2,3-Дихлорбутен-1
1,4-Дихлорбутен-1
1,1,2,2-Тетрахлорэтан
2,3-Дихлорпррпанол-1
A,2-дихлоргидрин)
З-Хлорпропандиол-1,2
A-хлоргидрин)
Исходные
вещества
Этилен
Бутадиен
Бутадиен
Ацетилен
Аллилхло-
рид
Аллиловый
спирт
Применение
Растворитель для смол, каучука
—>• Винилхлорид (см. табл. Г.4.20 и
разд. Г.3.1.5)
—>¦ Трихлорэтан —>¦ винилиден-
дихлорид
—> Тетрахлорэтан —* трихлор-
этилен
—>¦ Этилендиамин
2-Хлорбутадиен-1,3 —>• хлоропрен
—»¦ 1,4-Дицианобутен-2 —> адипо-
нитрил (см. табл. Г.2.107)
-¦Гексаметилендиамин —>• полиимиды,
полиуретановые лаки
—>¦ Трихлорэтилен (растворитель)
—>• Монохлоруксусная кислота
—>¦ Эпихлоргидрин A-хлор-2,3-эпок-
сипропан) —>¦ эпоксидные смолы
—>¦ Глицерин (см. табл. Г.4.34)
—> Глицерин (см. разд. Г.4.1.6)
4. Присоединение к неактивированным кратным углерод-углеродным связям
367
4.1.5. ОКСИМЕРКУРИРОВАНИЕ
Ионы двухвалентной ртути, обладая большой электрофильностью, легко присо-
присоединяются к олефинам. При действии ацетата ртути(П) в водной среде в прису-
присутствии солюбилизаторов сначала образуется карбениевый ион I (схема [Г.4.27];
см. также II на схеме [Г.4.7]), который в результате взаимодействия с водой дает
продукт оксимеркурирования II (схема [Г.4.27]). Обычно его сразу восстанавли-
восстанавливают до соответствующего спирта III. Метод представляет собой ценный вари-
вариант гидратации алкенов (в особенности малореакционноспособных). Как пра-
правило, спирты образуются в соответствии с правилом Марковникова:
R-CH=CH2
HgOAc
©.-•' I +H2O NaBH4,OHu
R-CH-CH2 - - R-CH-CH2-HgOAc »- R-CH-CH3 [Г.4.27]
-H* 0H -Hg,-HOAc ^
(СП) Общая методика получения спиртов оксимеркурированием (табл. Г.4.28)
8 Внимание*. Ацетат ртути ядовит! Образующуюся при реакции ртуть нельзя сли-
сливать в канализацию, ее необходимо собирать для последующей переработки!
Таблица Г.4.28. Получение спиртов оксимеркурированием алкенов
Продукт реакции
Октанол-2
Деканол-2
1-Фенилэтанол
2-эяЗо-Гидроксинор-
борнан
Исходное
вещество
Октен-1
Децен-1
Стирол
Норборнен
Т. кип., 'С
(мм рт. ст.)
87 B0)
105-
106A3)
94 A2);
Т. пл. 20 СС
Т. пл.
149—150 °С
1,4260
1,4306
1,5275
Выход, %
85
90
95
90
В колбе Эрленмейера на 250 мл с магнитной мешалкой готовят раствор 0,05 моль ацетата
ртути(П) в 50 мл воды. К этому раствору прибавляют 50 мл тетрагидрофурана, свободного от пе-
роксидов (см. часть Е), и затем 0,05 моль олефина. Температура реакционной смеси при этом
не должна подниматься выше 30 °С. Перемешивание продолжают до тех пор, пока в отобранных
пробах при добавлении гидроксида натрия не перестанет выпадать оксид ртути. После этого к
реакционной смеси добавляют 50 мл 3 н. NaOH, затем по каплям раствор 0,025 моль борогидри-
да натрия в 50 мл 3 н. NaOH. По окончании добавления перемешивают еще 1 ч. Выделившуюся
металлическую ртуть отделяют в делительной воронке, водный слой насыщают хлоридом нат-
натрия, дважды извлекают порциями по 50 мл эфира. Эфирный экстракт сушат сульфатом натрия,
эфир отгоняют, продукт реакции очищают перекристаллизацией или перегонкой в вакууме.
4.1.6. ЭПОКСИДИРОВАНИЕ И ДИГИДРОКСИЛИРОВАНИЕ
Кислород присоединяется к олефинам, давая оксираны (эпоксиды). В качестве
электрофильного агента может служить молекулярный или химически связан-
связанный кислород в пероксикислотах (например, в пероксибензойной, л*-хлор-
пероксибензойной, монопероксифталевой, надмуравьиной, надуксусной,
пероксовольфрамовой), гидропероксидах и в пероксиде водорода:
368 Г Препаративная часть
V
II
,н-о
1-0 "
О'С
- гид - > * >
\ ° R \ °
Во многих случаях эпоксиды можно вьщелить, если работать в инертных
растворителях, например с пероксибензойной кислотой в эфире или хлорофор-
хлороформе (реакция Прилежаева). В иных случаях эпоксид в реакционном растворе
подвергается гидролизу или сольволизу до соответствующих диолов-1,2 или их
сложных эфиров.
В очень мягких условиях в нейтральной среде олефины можно эпоксидиро-
вать диоксираном, циклическим пероксидом:
Чс/ о R ~-c R
Д + OXR' ~ ^9° + /"*' [Г.4.29а]
Диоксиран можно легко получить из кетона и гидропероксисульфата калия и применять in
situ (без выделения вещества). В качестве окислителя можно использовать также каталитичес-
каталитические количества кетона и гидропероксисульфата калия. Применяют диоксиран из простейших
кетонов, таких как ацетон или трифторацетон, и из хиральных кетонов, например производ-
производных фруктозы, с которыми протекает энантиоселективное эпоксидирование.
Раскрытие эпоксидного кольца в разбавленных кислотах или щелочах явля-
является 8^-реакцией, приводящей в случае г<ис-эпоксидных соединений к транс-
диолам-1,2:
?\^ Л © © I I © I I
Н20 + "рС-С"" + Н Н2О-С-С-ОН =р= Н + НО-С-С-ОН [Г.4.30]
(Напишите схему щелочного гидролиза эпоксидов!)
Дигидроксилирование Z-олефинов пероксидом водорода в присутствии ка-
каталитических количеств оксидов металлов протекает через стадию образования
эпоксисоединений, что приводит к /и/ишс-диолам. При проведении реакций в
муравьиной или уксусной кислотах в качестве промежуточных соединений
образуются надмуравьиная или надуксусная кислота; продуктами этих реакций
могут быть ацетаты или формиаты /ту?а«с-диолов.
Для эпоксидирования в инертном растворителе или дигидроксилирования в
кислой среде необходимо использовать олефин со сравнительно сильно выра-
выраженными основными свойствами (этилен, замещенный алкильными или ариль-
ными группами), а, р-Ненасыщенные кетоны или альдегиды в этих условиях не
реагируют, но могут быть эпоксидированы пероксидом водорода в слабощелоч-
слабощелочной среде.
Первичные аллиловые спирты могут быть превращены в эпоксид энантиосе-
лективно по Шарплессу и Кацуки с помощью т/7е/я-бутилгидропероксида в
присутствии (Л,Л)-или (^6)-диэтилового эфира винной кислоты и изопропила-
та титана(ГУ).
Эти «АЕ»-реакции (асимметрическое эпоксидирование) протекают через
комплекс, в котором координационно связаны как эфир винной кислоты,
так и аллиловый спирт и т/>е/я-бутилгидропероксид. При этом абсолютная
4. Присоединение к неактивированным кратным углерод-углеродным связям
369
Таблица Г.4.31. Получение эпоксидов из олефинов
Эпоксид
1,2-Эпоксициклогек-
сан
1,2-Эпоксициклопен-
тан
1,2-Эпоксиэтилбензол
Исходное вещество
Циклогексен
Циклопентен
Стирол
Т. кип., *С
(мм рт. ст.)
132
100
77 A1)
1,4519
1,4341
1,5361
Выход, %
80
30
70
конфигурация применяющегося эфира винной кислоты управляет конфигу-
конфигурацией образующегося эпоксида, ср. разд. С.7.3.3.2. Химический выход в реак-
реакции находится в пределах 70—90%; образующийся эпоксид, энантиомерный
избыток которого составляет чаще всего >90%, является дорогим хиральным
исходным веществом для синтеза углеводов, терпенов, антибиотиков и т. д.
Получение B8,35)-3-пропилоксиранэтанола из ?-гексен-2-ола-1: Hill J. G.,
Sharpless К. В., Ехопе С. М., Regenje R. Org. Synth. 1985, 63, 66.
((Jj) Общая методика эпоксидирования олефинов (табл. Г.4.31)
Внимание1. Эпоксидирование и гидроксилирование могут протекать очень
энергично, поэтому реакция должна проводиться всегда за защитным экраном.
р; При работе с неизвестными веществами надо ставить предварительные опыты
¦J с малыми количествами. Продукты реакции можно подвергать перегонке толь-
только тогда, когда они уже не дают реакцию на пероксиды (см. ниже)!
К раствору 0,30 моль пероксибензойной кислоты в 500 мл эфира осторожно при 0 °С при-
прибавляют 0,29 моль соответствующего олефина. Раствор при эффективном перемешивании ос-
оставляют на 24 ч при 0 "С. Можно следить за ходом реакции, отбирая время от времени по 2 мл
раствора, прибавляемого к смеси 15 мл хлороформа, 10 мл ледяной уксусной кислоты и 2 мл
насыщенного водного раствора иодида калия, и оставляя смесь на 5 мин. После добавления
75 мл воды выделившийся иод титруют 0,1 н. тиосульфатом. По окончании эпоксидирования
реакционный раствор несколько раз промывают 10%-ным раствором гидроксида натрия,
затем водой, сушат сульфатом магния и фракционируют.
Ф Получение т/шмс-циклогександиола-1,2 (транс-дигидроксилирование смесью муравьиной
кислоты и пероксида водорода). Прибор состоит из трехгорлой колбы на 250 мл, мешал-
мешалки, обратного холодильника и капельной воронки. К смеси 100 мл 90%-ной муравьи-
муравьиной кислоты1 и 0,12 моль 30%-ного пероксида водорода (пергидроля) в течение 5 мин прибав-
прибавляют по каплям 0,1 моль циклогексена. При этом реакционная смесь нагревается до 65-70 "С
и становится гомогенной2. Затем смесь оставляют на 2 ч при этой температуре, которую под-
поддерживают с помощью водяной бани: взятая после этого проба не должна выделять иод из
раствора иодида калия, в противном случае нагревание надо продолжить. Ббльшую часть
муравьиной кислоты и воды отгоняют в вакууме, для гидролиза формиата остаток нагревают
1 Также можно применять соответствующие количества 88%-ной кислоты.
2 При больших загрузках, как при описанном режиме работы, может нарушиться указан-
указанный температурный режим и реакция может выйти из под контроля. В этом случае следует
сделать необходимые изменения методики, ср. Roebuck A., Adkins H. Org. Synth., Coll. Voll. Ill,
1955, 217.
370 Г Препаративная часть
45 мин на паровой бане с 50 мл 20%-ного гидроксида натрия. Реакционную смесь охлаждают,
нейтрализуют разбавленной соляной кислотой и отгоняют растворитель на водяной бане в
вакууме. Остаток несколько раз извлекают горячим этилацетатом, отгоняют растворитель;
продукт перекристализовывают или перегоняют. Т. кип. 123 °С A4 мм рт. ст.); т. пл. 103 °С
(этанол). Выход 70%.
Реакцию можно проводить также с полумикроколичествами. Смесь при этом встряхива-
встряхивают, пока она не станет гомогенной, а затем обрабатывают, как описано выше.
Аналогично из стирола можно получить фенилэтандиол-1,2, т. пл. 67 °С (лигроин), выход 40%.
Ф Получение глицерина из аллилового спирта (гидроксилирование смесью пероксовольф-
рамовой кислоты и пероксида водорода). В трехгорлой колбе, снабженной мешалкой,
обратным холодильником и капельной воронкой, нагревают до 70 °С 9%-ный водный
раствор аллилового спирта и при хорошем перемешивании добавляют по каплям пергидроль
A0%-ный избыток), в котором предварительно растворяют триоксид вольфрама
C%-ный избыток относительно аллилового спирта). Нагревание при 70 °С продолжают до тех
пор, пока проба на пероксид с подксиленным раствором иодида калия не станет отрицатель-
отрицательной (около 3 ч). Затем смесь перегоняют в вакууме. Т. кип. 180 °С A3 мм рт. ст.). Выход 90%.
Приведенная выше методика повторяет в лабораторных условиях современный промыш-
промышленный метод получения глицерина. Относительно других методов см. табл. ГЛ.37 и Г.4.26.
Глицерин является важнейшим продуктом химической промышленности (см. табл. Г.4.34).
Традиционные методы получения эпоксидов в промышленности (через гидроксихлориды
или путем окисления пероксикислотами) в настоящее время применяются главным образом для
промышленного синтеза пропиленоксида, поскольку при прямом окислении молекулярным
кислородом или гидропероксидами затрагивается метальная группа пропилена и образуется
преимущественно акролеин. Особое значение приобрело окисление этилена до этиленоксида
кислородом на серебряном контакте и реакция пропилена с гидропероксидами в присутствии
металлов V и VI побочных групп. Другие олефины, особенно в присутствии молибденовых и ва-
ванадиевых соединений в качестве катализаторов, также можно окислять гидропероксидами
(/я/ити-бутилгидропероксидом, /и/>е/и-пентилгидропероксидом, <х,а-диметилбензилгидроперок-
сидом) с образованием эпоксидов. В качестве побочных продуктов образуются спирты.
Эпоксиды очень реакционноспособны; кроме воды и кислот (см. схему
[Г.4.30]) они присоединяют в присутствии кислотных и щелочных катализаторов
также и другие нуклеофильные реагенты, например спирты, тиолы, амины, маг-
нийорганические соединения. В промышленности такие реакции с этиленокси-
дом проводят в больших масштабах. Образующиеся таким путем соединения мо-
могут сами присоединяться к этиленоксиду, в результате чего при взаимодействии с
водой кроме этиленгликоля получают ди-, три- и полиэтиленгликоли1, при
присоединении спиртов — простые эфиры гликоля и полигликолей, а при взаи-
взаимодействии с аммиаком — моно-, ди- и триэтиламины:
+ ROH
н2с-сн2
- но-сн2-сн2-он —- но-сн2-сн2-о-сн2-сн2-он и т.д. [Г.4.32а]
-HO-CH2-CH2-OR —-HO-CH2-CH2-O-CH2-CH2-OR и т.д. [Г.4.326]
- HO-CH2-CH2-NH2 —- (HO-CH2-CH2JNH —- (HO-CH2-CH2KN [Г.4.32в]
При избытке этиленоксида получают в основном высокомолекулярные про-
продукты присоединения.
а-Гидро-со-гидроксиди-, -три- или -поли(оксиэтилен).
4. Присоединение к неактивированным кратным углерод-углеродным связям
371
Таблица Г.4.34. Применение продуктов, получаемых из эпоксидов
Продукт
Применение
Из этиленоксида (эпоксиэтана, оксирана):
Этиленгликоль
Диэтиленгликоль
Триэтиленгликоль
Полиэтиленгликоли
Моноалкиловые эфиры низших
спиртов (Ci—С4) и моно- или
диэтиленгликоля
Полипропиленгликольалкиловые и
полипропиленгликольмоноариловые
эфиры высших спиртов (Ci2—Си)
и алкилфенолов (алкил: Cg—C12)
Этаноламины
Антифриз (глизантин)
—>¦ Полиэфиры с терефталевой кислотой
(искусственные волокна)
—* Динитрат (взрывчатое вещество, см.
разд. Г.2.5.1)
Пластификатор для целлофана
—> Динитрат (взрывчатое вещество)
—> Полиэфиры с малеиновой кислотой
(твердые полиэфирные смолы)
Тормозные жидкости
—>¦ Полиуретаны (реакция с диизоциана-
тами; см. разд. Г.7.1.6)
—»• Полиэфирные смолы (спиртовой компо-
компонент)
Растворители для лаков идр.(целлозольвы или
карбитолы)
Неионогенные биодеструктируемые моющие
средства, работающие в жесткой воде;
вспомогательные материалы для текстиль-
текстильной промышленности; эмульгаторы (дис-
персал)
Абсорбционные жидкости для очистки га-
газов от H2S, CO2 и др.
—> Производные жирных кислот (эмуль-
(эмульгаторы для минеральных масел)
Из пропиленоксида:
Сополимеры с этиленом
Эпихлоргидрин
1-Хлор-2,3-эпоксипропан
2,3-Эпоксипропанол
Неионные пенообразующие ПАВ
—> Глицерин
—>¦ Эпоксидные смолы (эпилокс) — про-
продукты взаимодействия с бисфенолом А
—> Алкидные смолы (глифтали) —продук-
—продукты взаимодействия с фталевой и другими
поликарбоновыми кислотами
—> Глицерин — Увлажнитель табачных из-
изделий, антифриз, пластификатор (для цел-
целлофана), косметическое средство
—»¦ Продукты конденсации с этилен- и про-
пиленоксидами (триполиэфиры глицерина)
—¦* Поверхностно-активные вещества
—>¦ Полиуретаны и алкидные смолы (спир-
(спиртовой компонент)
—> Тринитрат (взрывчатое вещество)
372 Г Препаративная часть
Реакции типа [Г.4.32в] используют в процессах затвердевания эпоксидных
смол, причем реагентом служит амин, например бисB-аминоэтил)амин.
Присоединение к этиленоксиду синильной кислоты дает C-гидроксипропи-
онитрил (этиленциангидрин), а магнийорганических соединений (см. разд.
Г,7.3.1.5) — первичные спирты, причем алкильный остаток реактива Гриньяра
удлиняется на два атома углерода:
о
RMgX + H2C-CH2—- R-CH2-CH2-OMgX _'"'° • R-CH2-CH2-OH [Г.4.33]
В табл. Г.4.34 перечислены важнейшие продукты, получаемые из этиленок-
сида и пропиленоксида в промышленности.
Эпоксидирование встречается и в природе, например при деструкции кон-
конденсированных ароматических углеводородов в организме животных. Так, с
помощью фермента цитохрома Р 450 в печени из бензопирена образуется ди-
эпоксид, которому приписывают сильное канцерогенное действие:
О.
[Г.4.35]
При действии кислот Льюиса (например, трифторида бора) эпоксиды перег-
перегруппировываются в альдегиды или кетоны (см. схему [Г.9.17]). Эту реакцию
можно применять для идентификации олефинов.
Метод эпоксидирования пригоден для количественного определения двойной
связи С=С. Реактивом является пероксибензойная или монопероксифталевая
кислота в безводном растворителе. Число двойных связей устанавливают иодо-
метрическим титрованием избытка пероксикислоты, не вступившей в реакцию.
В отличие от гидроксилирования Z-олефинов через эпоксиды, которое при-
приводит к транс-толам (схема [Г.4.30]), при гидроксилировании перманганатом
калия на холоду в нейтральной или щелочной среде или тетраоксидом осмия по-
получают г<ис-диолы:
V oso4 -с-Ч н2о А-он н2о -V°N e мпо? V [ГА361
и *- i OsO2 „ -- i -—^тт ' МпО2 и
/Сч -с~0' -озоз _с_он _Мп0© _-с.о- хсч
При омылении промежуточно образующихся циклических эфиров атаке
подвергается атом металла, поэтому г<ыс-диольная структура сохраняется.
Тетраоксид осмия дает очень хорошие выходы, однако реагент токсичен и
очень дорог.
Этот недостаток можно обойти (уменьшить) при использовании каталитичес-
каталитических количеств тетраоксида осмия. В качестве стехиометрического количества
окислителя здесь можно применять аминоксиды, такие как N-метилморфолин-
N-оксид (NMO), тре/я-бутилгидропероксид, хлорат бария и пероксид водорода,
которые повторно окисляют образующийся триоксид осмия в соответствующий
тетраоксид.
4. Присоединение к неактивированным кратным углерод-углеродным связям 373
Большое число различных замещенных прохиральных олефинов можно селективно дигид-
роксилировать по Шарплессу с помощью стехиометрического окислителя и каталитических коли-
количеств осмата калия и гексацианоферрата(Ш) калия в присутствии фталазинового производного
энантиомерно чистого алкалоида дигидрохинина или дигидрохинидина соответственно. С по-
помощью «АД-реакций» (асимметрическое дигидроксилирование) в зависимости от выбора реакци-
реакционной смеси — «АД-смесь-а « (содержит производное дигидрохинина) или «АД-смесь-р « (содер-
(содержит производное дигидрохинидина) — можно синтезировать один или другой энантиомер.
Дигридроксилирование перманганатом калия легко приводит к разрыву свя-
связей С—С и дальнейшему окислению продуктов расщепления до альдегидов и
кислот (см. разд. Г,6.5). Дигидроксилирование циклогексена может служить
примером реакций алкенов с перманганатом в нейтральной среде.
Ф Получение цис-циклогександиола-1,2. о, 1 моль циклогексена, растворенного в 200 мл этанола,
помещают в охлаждаемую смесью льда с солью трехгорлую колбу на 750 мл с внутренним тер-
термометром, капельной воронкой и мешалкой. При энергичном перемешивании прибавляют
раствор 0,09 моль перманганата калия и 10 г сульфата магния в 250 мл воды. Скорость прибавления
регулируют так, чтобы температура реакционной смеси поддерживалась между 0 и 5 °С. По оконча-
окончании прибавления перемешивают еще два часа, осадок диоксида марганца отфильтровывают и про-
промывают 3 раза порциями по 50 мл ацетона. Соединенные фильтраты упаривают до 120 мл в вакууме
водоструйного насоса, остаток насыщают хлоридом натрия и извлекают 4-5 раз порциями по 50 мл
хлороформа. После сушки сульфатом натрия отгоняют хлороформ, остаток перекристаллизовывают
из бензола. Т. пл. 99—102 °С; т. кип. 118 °С A4 мм рт. ст.); выход 65%.
цис-Циклогександиол-1,2 из циклогексена и N-метилморфолин-М-оксида в
присутствии OsO4 (катализатор): van Rheenen V., Cha D. Y, Hartley W. H. Org.
Syntheses, 1978, 58, 43.
2-Метилпропандиол-1,2 из изобутилена с тетраоксидом осмия: Milas N. А.,
Sussman S. J. Am. Chem. Soc, 1936, 58, 1302.
цис-Диолы-1,2 реакцией олефинов с перманганатом калия в условиях меж-
межфазного катализа: Weber W. P., ShepardJ. P. Tetrahedron Lett., 1972, 4907.
(R,R)-1,2-Дифенилэтандиол-1,2 (стильбендиол) из стильбена асимметричес-
асимметрическим дигидроксилированием: МсКее В. Н., Gilheany D. G., Sharpless К. В. Org.
Synth. 1992, 70,47-53.
4.1.7. ОЗОНИРОВАНИЕ
Присоединение озона к олефинам и аренам (озонирование) в настоящее время
имеет препаративное значение лишь для получения карбонильных соединений,
трудно доступных иными путями.
Первая стадия реакции является перициклическим 1,3-диполярным присое-
присоединении озона по двойной связи (ср. циклоприсоединение, разд. Г.4.4):
V ->с-Ч
и + Оз • О [Г.4.37]
/°\ /О
Циклоаддукт в дальнейшем распадается и его осколки соединяются по типу
диполярного 1,3-циклоприсоединения, образуя собственно озонид:
-Vov Я° W [ГА38]
1 ~~ \
374 Г Препаративная часть
При восстановительном расщеплении озонидов (диметилсульфидом, цинко-
цинковой пылью в уксусной кислоте, каталитическим гидрированием над палладием на
карбонате кальция, дитионитом натрия и др.) образуются альдегиды или кетоны:
\°уС -^- -VV- ^~ 2 )<0Н 2>=О +2Н2О [Г.4.39]
0-0 ОН ОН х ОН '
При низких температурах с озоном взаимодействуют лишь двойные связи С=С в алкенах,
в то время как арены и алкины не реагируют; не окисляются и другие группы. Поэтому озони-
озонирование используют для установления строения непредельных соединений, в особенности
эластомеров и природных соединений. Озон также используется в промышленности для прев-
превращения олеиновой кислоты в азелаиновую, для получения додекандикарбоновой кислоты из
циклододекатриена через соответствующий моноциклоалкен, для получения ванилина из изо-
эвгенола и гелиотропина из изосафрола.
Аппаратура для озонирования, общая методика озонирования и конкретные
методики: Органикум. — М.: Мир, 1979, т. 1, с. 351.
Дифенил-2,2'-дикарбальдегид (дифеновый альдегид) и 2'-формилдифенилкарбо-
новая-2-кислота (дифеновая альдегидокислота): Bailey P. S., Erickson R. E. Org.
Syntheses, 1961,41,41,46.
4.1.8. ГИДРОБОРИРОВАНИЕ
Легко и количественно протекает сын-присоединение бороводорода к неактиви-
неактивированным двойным связям С=С, приводящее к алкилборанам. К олефинам с
концевой двойной связью бораны присоединяются против правила Марковни-
кова1, причем атом бора выступает в качестве электрофила (см. также схему
[Г.4.9]), при этом возникает четырехцентровое переходное состояние.
Гидроборирование проводят, как правило, действием диборана2, который
обычно образуется in situ, например из NaBH4 и эфирата BF3 в диглиме или в
кислом растворе из NaBH4 (см. общую методику). При реакции с алкеном заме-
замещаются все атомы Н диборана и образуется соответствующий триалкиддиборан:
X
н-в
/в-
8-H--BS
\
-с-с
Н В— [Г.4.40]
\
3 RCH=CH2 + ВН3 (RCH2CH2KB
Гидроборирование — важный, имеющий широкую сферу применения метод
препаративной органической химии. Образующиеся борорганические соедине-
соединения обычно без выделения подвергают дальнейшим превращениям. В схеме
[Г.4.41] представлены важнейшие продукты таких превращений:
1 Присоединение борана к двойной связи происходит региоселективно против правила
Марковникова таким образом, что атом бора оказывается связанным с наименее замещенным
атомом углерода при двойной связи, т. е. стереоспецифично как cww-присоединение. — Прим. перев.
2 Точнее, диборана F); число атомов водорода в боранах указывают арабской цифрой в
скобках после названия.
4. Присоединение к неактивированным кратным углерод-углеродным связям
375
(RCH2CH2KB
Н2О2, ОН
,©
H2NOSO3H
Вг2, ОН
,е
R'-COOH
R'-COOD
AgNO3, ОН
,в
СО
3 RCH2CH2OH
3 RCH2CH2NH2
3RCH2CH2Br
3RCH2CH3
3RCH2CH2D
Уг RCH2CH2CH2CH2R
(RCH2CH2KC-B=O
[Г.4.41]
H2O2, OH
(RCH2CH2KC-OH
Наибольшее значение имеет окисление борорганических соединений перок-
сидом водорода в щелочной среде, приводящее к образованию спиртов1. Итогом
реакции оказывается присоединение воды к исходному олефину, формально
протекающее против правила Марковникова.
Общая методика получения спиртов гидроборированием (табл. Г.4.42) [Hach V. Synthesis,
1974, 340]
Внимание*. Образующийся в ходе реакции газообразный диборан очень ядо-
ядовит! Работать с хорошо действующей тягой. Приборы по окончании реакции
оставить на несколько часов с раствором щелочи или водного аммиака.
Растворы, содержащие боран, не хранить, так как они склонны к разложе-
разложению со взрывом.
Аппаратура состоит из трехгорлой колбы объемом 500 мл, снабженной капельной ворон-
воронкой, внутренним термометром, трубкой для подвода азота, обратным холодильником с хлор-
кальциевой трубкой, магнитной мешалкой. В колбу помещают 0,1 моль безводного (высушен-
(высушенного молекулярным ситом 4А) алкена и 0,037 моля раствора борогидрида натрия в 250 мл без-
безводного тетрагидрофурана (см. часть Е). Охлаждая льдом, осторожно прибавляют по каплям
раствор 0,37 моль безводной уксусной кислоты (высушенной молекулярным ситом 4А) в 50 мл
безводного тетрагидрофурана. Температуру реакционной смеси поддерживают в пределах
10—20 °С. По окончании прибавления перемешивание продолжают еще 2 ч, затем осторожно
добавляют по каплям раствор 4 г гидроксида натрия в 20 мл воды и после этого 14 мл 30%-но-
го пероксида водорода. Перемешивают еще 2 ч, отделяют органическую фазу, водную извле-
извлекают трижды 10 мл эфира. Соединенные вытяжки сушат сульфатом магния, отгоняют раство-
растворитель, остаток перегоняют в вакууме или перекристаллизовывают.
Дилкилбораны с объемистыми заместителями являются ценными селектив-
селективными гидроборирующими агентами. Таков, например, дисиамилборан, получа-
получаемый присоединением диборана к 2-метилбутену-2. При присоединении дисиа-
милборана к B)-4-метилпентену-2 (табл. Г.4.43) связь С-В образуется премуще-
ственно у пространственно менее затрудненного атома С-2. Напротив, при
гидроборировании дибораном образуется смесь двух изомерных спиртов. Чистые
Здесь имеет место 1,2-перегруппировка у атома кислорода; см. разд. Г,9.1.3.
376
Г Препаративная часть
первичные спирты в этих случаях можно получить, используя способность бора-
нов изомеризоваться примерно при 160 °С. По механизму элиминирования —
присоединения бор при этом мигрирует к концевому атому углерода.
Таблица Г.4.42. Получение спиртов реакцией гидроборирования
Продукт реакции
Октанол-1
Деканол-1
2-Фенилэтанола
зкзо- (+) -Норборна-
нол-26
(—) -Изомиртанол"
[(IS, 2#)-пинанол-10]
(—) -Изопинохамфеол
[ (\R, 2R, 3R) -пинанол-3]
Исходное
вещество
Октен-1
Децен-1
Стирол
Норборнен
(-)-Р-Пи-
нен
( + )-сс-Пи-
нен
Т. кип., °С
(мм рт. ст.)
100B0)
111—113A1)
98—110A2)
128 (т. пл.;
петр. эфир)
118A5)
72C);
т. пл. 56
1,4925
1,4368
1,5240
1,4910
Мл20
-32,8е
A0%;
бензол)
Выход, %
80
80
70
70
70
75
0 В реакционной сиесн содержится также 20% 1-феннлэтанола, т. кип. 94 °С
A2 мм рт. ст.).
После перекристаллизации продукт реакции возгоняют в ва-
вакууме водоструйного насоса.
НО
в [<x]D*> —20,9° D% в CHCls); если работать в диглиме и реакционную смесь нагреть
до 140 "С до прибавления раствора пероксида водорода, образуется (—)-миртанол
[A5,25-пинанол-10), [а]„ш —28,5° D% в СНСЦ).
НзС^/^j/ н3с/«:сн2он h3cyCH3
сн2он ^Нз
(-)-изомиртанол (-)-миртанол
(-)-изопинокамфеол
Таблица Г.4.43. Влияние стерических эффектов при присоединении гидробориру-
ющих реагентов к B)-4-метилпентену-2
Исходное соединение
(Z) -4-Метилпентен-2
Продукт реакции
4-Метилпентанол-2
2-Метилпентанол-З
Выход продукта (%) при
использовании
BjH,
57
43
днснамилборана
97
3
4. Присоединение к неактивированным кратным углерод-углеродным связям
377
Гидроборирование (—)-а-пинена приводит к (+)-диизопинокамфеилборану
(+)-AрсJВН, который можно использовать в качестве хирального гидробориру-
ющего реагента. Так, гидроборирование прохирального Z-алкена с помощью
(+)-AрсJВН с последующим окислением спирта протекает с высоким выходом
одного из энантиомеров:
норборнен
(бицикло[2.2.1]-
гептен-2)
НО
экзо-норбонеол
(бицикло[2.2.1]-
гептанол-2)
оптическая
чистота 83%
[Г.4.44]
4.1.9. КАТИОННАЯ ОЛИГОМЕРИЗАЦИЯ И ПОЛИМЕРИЗАЦИЯ
Олефины могут полимеризоваться при каталитическом действии кислот. Кар-
бениевый ион, образовавшийся после присоединения протона к олефину, явля-
является (как кислота Льюиса) электрофильным реагентом, который, подобно про-
протону, катиону хлора и др., может присоединиться к следующей молекуле олефи-
на. В результате после отщепления протона имеет место димеризация или (при
повторении этого процесса) полимеризация олефина (см. схему [Г.4.16д]). Ход
реакции можно иллюстрировать на примере изобутена:
н®
сн.
СНз
нгс=с
СН3
н3с-с©
-Н2С=С(СН3J
СНз
сн.
Н3С-С-СН2-С© и т.д. [Г.4.45]
СН3
СН3
СНЯ
Присоединение протекает здесь в соответствии с правилом Марковникова.
Обрыв реакционной цепи большей частью происходит путем отщепления
протона, причем одновременно образуются алкены-1 и алкены-2 (см. схему
[Г.3.20]):
СНз Ch
Н3С-С-СН2-С©
СН3
-н
снз СНз
н3с-с-сн=сч
сн3
B0%)
сн3
[Г.4.46]
.сн2
? „
Н3С-С-СН2-С
СНз Ч
(80%)
Как упоминалось выше, полимеризация является побочной реакцией при ка-
катализируемой кислотами гидратации олефинов (см. схему [Г.4.16]). Олефины ти-
типа изобутена особенно легко полимеризуются по катионному типу, так как,
с одной стороны, промежуточно образующиеся третичные алкильные катионы
энергетически более выгодны (см. разд. Г,2.2.1) и, с другой стороны, изобутен об-
обладает высокой основностью и поэтому может быстро реагировать с катионом.
378 Г Препаративная часть
Степень полимеризации в зависимости от катализатора и температуры бывает
различной. Цепь удлиняется с понижением температуры и с повышением чисто-
чистоты применяемого олефина. Средняя молекулярная масса может достигать 105.
Ф Получение изооктена (смеси изомеров в соответствии со схемой [Г.4.46]). Смесь 170 г
холодной 60%-ной серной кислоты и 50 г трет-бутанола нагревают 20 ч на водяной ба-
бане. Получающийся димер отделяется во время нагревания в виде маслянистого слоя.
Димер промывают водой и раствором соды, сушат сульфатом магния и кипятят 5 ч с обратным
холодильником над металлическим натрием. После фракционирования на эффективной ко-
колонке (почему?) получают 25 г смеси изомерных углеводородов с т. кип. 100—128 "С (при пов-
повторной перегонке собирают фракцию с т. кип. 100—105 °С).
Карбениевые ионы, полученные другим путем, могут реагировать с алкенами также в со-
соответствии со схемой [Г.4.45]. Так, например, можно легко провести катионную полимериза-
полимеризацию стирола (см. ниже).
^П) Катионная полимеризация стирола
Приготовление катализатора: В трехгорлой круглодонной колбе на 100 мл, снабженной
обратным холодильником, мешалкой и капельной воронкой и помещенной на водяную баню,
нагревают 20 г безводного хлорида алюминия в 30 г толуола приблизительно до 100 °С и затем
медленно прибавляют по каплям 0,6 мл воды. В результате бурной реакции (выделение НС1)
образуется красно-коричневый каталитический комплекс. После завершения прибавления
воды реакционную смесь перемешивают 5 мин при 90 °С.
Полимеризация: В колбу (из сульфированного стекла) на 250 мл, снабженную мешалкой, тер-
термометром, обратным холодильником и капельной воронкой и охлажденную приблизительно до
15 °С на водяной бане, помещают 20 г стирола в 80 мл толуола. При сильном перемешивании и
охлаждении прибавляют по каплям 0,5 мл раствора приготовленного катализатора. При этом
температура реакционной смеси не должна подниматься выше 25 °С. После окончания прибав-
прибавления перемешивают дополнительно 15 мин и затем оставляют стоять 30 мин. Отделяют деканта-
декантацией полимеризационный раствор, промывают 4 мл воды и затем 4 мл 15% -ного раствора едко-
едкого натра и сушат сульфатом магния. При промывании окраска полимеризационного раствора
должна измениться от красно-коричневой до желтой. После вакуумной отгонки растворителя от-
отделяют порошок полимера. Светло-желтый осадок имеет вид смолы. Выход 80—85%, температу-
температура размягчения 50-100 °С, молекулярная масса 500—1000 г/моль.
Также карбокатионы можно получать из алкена (пропена, бутена) действием
безводной фтороводородной кислоты; они реагируют с изобутаном, причем в
результате переноса атома водорода образуется т/гет-бутил-катион. Последний
затем присоединяется по механизму электрофильного присоединения (AdE) к
алкену. Таким путем из изобутана и смесей пропена, бутена и изобутена образу-
образуются так называемые алкилатбензшы (HF-алкилирование).
Другие примеры применения катионной олиго- и полимеризации алкенов
приведены в табл. Г.4.47.
4.2. НУКЛЕОФИЛЬНОЕ ПРИСОЕДИНЕНИЕ
4.2.1. АНИОННАЯ ПОЛИМЕРИЗАЦИЯ ОЛЕФИНОВ
Вследствие их основного характера олефины обычно не способны присоединять
нуклеофильные агенты (см. разд. Г,4.1.1). Однако нуклеофильность некоторых
металлоорганических соединений настолько высока, что нуклеофильное присо-
присоединение становится возможным. Важнейшей реакцией этого типа является
анионная полимеризация моноолефинов и сопряженных диенов.
4. Присоединение к неактивированным кратным углерод-углеродным связям
379
Таблица Г.4.47. Промышленное применение катионной димеризации и полимери-
полимеризации алкенов
Алкен
Сз/СгАлкены
Изобутен
Пропен
Этилен
Смесь стирола, индена и их
метильных производных
Применение
Алкилатбензин
—>¦ Изооктен —> изооктан (стандарт мо-
моторного топлива, октановое число 100)
—h Полиизобутен
А1С1з
—•¦ Бутилкаучук (к изобутену добавляют
2% изопрена)
—> «Тетрапропилен> (—»¦ алкилбензолсуль-
фонаты)
—>• Синтетические смазочные масла
—> Углеводородные смолы (инден-кумаро-
новые смолы), компоненты для клеев, ла-
лаков, красок, изолирующих материалов
Диены благодаря более сильной делокализации я-электронов и связанной с этим большей
поляризуемости двойных связей несколько реакционноспособнее моноолефинов и могут
присоединять натрий.
В соответствии со схемой [Г.4.48] инициаторами реакций полимеризации в растворах
являются металлоорганические соединения, например фениллитий, нафтилнатрий, а также
амид натрия:
Старт:
Рост цепи:
Ph
Ph
Ph
Ph
Ph Ph
[Г.4.48]
U и т. д.
Ph Ph Ph
При анионной полимеризации невозможен обрыв цепи за счет рекомбинации. Если при-
принять меры, предотвращающие обрыв цепи загрязнениями, то полимерные анионы оказывают-
оказываются как бы «живыми», сохраняя способность к росту при новых добавках мономера. Это обстоя-
обстоятельство используют при блок-сополимеризации, например, из стирола и бутадиена получают
термопластичные эластомеры; в них чередуются относительно длинные последовательности
стирольных и бутадиеновых звеньев. В живые полимеры могут быть введены и реакционноспо-
собные концевые группы, например, при взаимодействии с этиленоксидом. За счет поликон-
поликонденсации этих групп также могут быть созданы блок-сополимеры.
Напишите схему полимеризации бутадиена и этиленоксида, аналогичную схеме [Г.4.48].
Полимеризация бутадиена натрием ранее использовалась для получения каучуков типа
«буна».
4.2.2. НУКЛЕОФИЛЬНОЕ ПРИСОЕДИНЕНИЕ К АЦЕТИЛЕНАМ
Вследствие большей склонности ацетиленов к нуклеофильному присоедине-
присоединению (ср. разд. Г.4, введение) сильноосновные анионы спиртов легко присоеди-
380 Г Препаративная часть
няются к тройной связи, например:
е OR H OR
• \ / \
Образующийся первоначально сильноосновный карбанион I отрывает от
молекулы спирта протон, причем получается простой виниловый эфир II. Пос-
Поскольку в результате опять возникает алкоголят-ион, то последний может быть
взят в каталитических количествах. Алкоголят-ион является также активным
агентом при присоединении спиртов в присутствии гидроксида натрия (см. схе-
схему [Г.2.65а]).
Таким же образом может происходить нуклеофильное присоединение к аце-
ацетилену карбоновых кислот, фенолов, тиолов, а также некоторых амидов, вто-
вторичных аминов и др. Часть таких реакций винилирования1 протекает также в
присутствии солей тяжелых металлов по электрофильному механизму (см. схе-
схему [Г.4.18]).
Синтез простых виниловых эфиров идет легко и с высокими выходами.
Однако, чтобы достигнуть необходимой температуры и требуемой скорости ре-
реакции, нужно, как правило, работать под давлением. При этом должны строго
соблюдаться соответствующие меры предосторожности (см. ниже).
(СП) Общая методика винилирования спиртов (табл. Г.4.50)
Внимание1. Автоклав, применяемый при этих реакциях, должен выдерживать по
крайней мере 10-кратное давление по сравнению с ожидаемым при нормаль-
нормальном течении реакции (на практике берут автоклав, испытанный на 35 МПа,
350 атм). Так как с серебром и медью ацетилен образует взрывчатые соедине-
соединения, то автоклав и приборы к нему (манометр!) не должны иметь деталей из этих
металлов, которые могут соприкасаться с ацетиленом. Автоклав не должен про-
пропускать газ, чтобы в рабочем помещении не могла образоваться взрывчатая
смесь ацетилена с воздухом. По той же причине при промывании автоклава и
при снятии давления ацетилен должен отводиться из рабочего помещения (см.
также разд, А, 1.8). Поскольку работа с ацетиленом под давлением связана с
непредвиденными опасностями, проведение этого опыта допускается только в
том случае, если будут приняты соответствующие меры предосторожности.
См. также Reppe W- Neue Entwicklungen auf dem Gebiet der Chemie des
I Acetylens und Kohlenoxids. — Springer-Verlag, Berlin. 1949; Reppe W. Chemie und
I TechnikderAcetylen-Drick-Reaktion. — Verlag Chemie, Weinheim/Bergstr., 1951.
В автоклав из легированной стали с мешалкой или во встряхиваемый автоклав, общий
объем которого должен быть равен по меньшей мере 5-кратному (лучше 10-кратному) объему
реакционной смеси, помещают 1 моль соответствующего спирта или фенола, а также около
10% (по массе) тонкоразмолотого гидроксида калия. Чтобы уменьшить смолообразование,
при винилировании фенолов прибавляют, кроме того, немного воды A5-17 мл на 1 моль фе-
фенола). Затем автоклав закрывают, тщательно вытесняют воздух ацетиленом или азотом, пода-
подают ацетилен под давлением 0,8—1,6 МПа (8—16 атм) и медленно нагревают до 180 °С (винили-
рование фенолов) или до 140 °С (винилирование спиртов). Как только ацетилен перестанет
1 Присоединение ацетилена к соединениям, содержащим группы ОН, SH или NH, назы-
называют реакцией винилирования.
Таблица Г.4.50. Винилирование
Продукт реакции
Этилвиниловый эфир
н-Пропилвиниловый эфир
м-Бутилвиниловый эфир
Изобутилвиниловый эфир
Циклогексилвиниловый эфир
Бензилвиниловый эфир
Фенилвиниловый эфир
Виниловый эфир гваякола
Дивиниловый эфир этиленгли-
коля A,2-дивинилоксиэтен)
Этиленацеталь ацетальдегида
зпиртов и фенолов
Исходное вещество
Этанол
к-Пропанол
и-Бутанол
Изобутанол
Циклогексанол
Бензиловый спирт
Фенол
Монометиловый эфир пиро-
пирокатехина (гваякол)
Этиленгликоль
Этиленгликоль
Т. кип., °С
(мм рт. ст.)
36
65
94
83
53 B3)
47 A5)
156
112 C0)
127
82
"d
1,3790
1,3913
1,4017
1,3981
1,4547
1,5185
1,5224
1,5356
1,4338
1,3972
Выход,
%
80
90
75
80
68
63
70
60
60
68
Примечания
Добавить 1 моль воды
Добавить 1 моль воды
Ацетилен подают до насыще-
насыщения; побочно образуется 14%
этиленацеталя ацетальдегида
Работают при 190 °С; после
окончания поглощения ацети-
ацетилена выдерживают еще 3 ч
при 190°С; побочно образует-
образуется 8% 1,2-дивинилоксиэтена
382 Г Препаративная часть
поглощаться и давление станет постоянным, автоклав охлаждают, снова подают ацетилен до
давления 0,8—1,6 МПа и опять нагревают до указанной выше температуры. Эту операцию пов-
повторяют, пока не поглотится рассчитанное количество ацетилена1 и реакция не закончится.
Присоединение алифатических спиртов к ацетилену протекает очень быстро, поэтому обычно
после достижения температуры реакции автоклав сразу охлаждают и снова подают ацетилен.
Когда винилирование закончится, автоклав охлаждают и давление снижают.
Фенилвиниловые эфиры выделяют из реакционной смеси перегонкой с водяным паром,
алифатические виниловые эфиры перегоняют в вакууме непосредственно из реакционного
раствЬра на 20-сантиметровой колонке Вигре.
Виниловые эфиры сушат карбонатом калия и еще раз перегоняют. При этом целесообраз-
целесообразно прибавить небольшое количество карбоната калия, чтобы исключить полимеризацию, ко-
которая катализируется кислотами.
Винилирование н-бутанола при атмосферном давлении в присутствии большого
количества гидроксида калия (чтобы достичь приблизительно 140 "С — необходи-
необходимой температуры винилирования): DuhamelA. Bull. Soc. Chim. France, 1956, 156.
Из-за высокой реакционной способности двойной связи виниловые эфи-
эфиры широко применяются в различных синтезах, например, их можно исполь-
использовать в качестве диенофила при циклоприсоединении (ср. разд. Г,4.4).
Виниловые эфиры в кислой среде легко присоединяют воду, образуя полуацеталь ацеталь-
дегида, который распадается на ацетальдегид и спирт (см. разд. Г,7.1.2):
OR u _ ,_,© OR О
I + Н9О1 Н / // ,r- . -, ,
Н2С=С — - Н3С-С-ОН ^=^ Н3С-С + ROH [Г.4.51]
Н Н ЧН
Виниловые соединения имеют большое значение в технике для получения полимеров (см.
разд. Г,4.3), имеющих различные свойства и области применения. Отдельные виниловые сое-
соединения используют также при сополимеризации.
Препаративно просто осуществить присоединение аминов к замещенным
ацетиленам. Например, легко и с высоким выходом удается провести реакцию
аммиака, первичных и вторичных аминов с эфирами ацетиленмоно- и дикарбо-
новых кислот. Продукты реакций являются енаминами, у которых двойная
связь сопряжена с карбонильной группой; они достаточно устойчивы. Если у
азота имеются атомы водорода, способные к хелатированию, то предпочтитель-
предпочтительной оказывается B)-форма:
R"NH2 + R-CEC-COOR' R"—N C-OR1 [Г 4 52]
;с=с
R Н
Подобные енамины представляют особый интерес в синтезах гетероцикли-
гетероциклических соединений (см. схему [Г.7.288]).
1 Поглощенное количество ацетилена можно рассчитать с достаточной точностью по об-
общему газовому уравнению из уменьшения давления. Регистрацию давления производят каж-
каждый раз при охлажденном автоклаве, так как при этом можно пренебречь давлением паров
жидких компонентов. Для полного винилирования спирта применяют несколько большее ко-
количество ацетилена, чем требуется по расчету (принимая во внимание побочные реакции, а
также растворимость ацетилена в виниловом эфире).
4. Присоединение к неактивированным кратным углерод-углеродным связям
383
Таблица Г.4.53. Присоединение аминов к ацетилендикарбоновым эфирам
Продукт реакции
Диметиловый эфир амино-
фумаровой кислоты6
Диэтиловый эфир аминофу-
маровой кислоты
Диэтиловый эфир N-метил-
аминофумаровой кислоты
Диэтиловый эфир N-бензил-
аминофумаровой кислоты
Диэтиловый эфир анилино-
фумаровой кислоты
Диметиловый эфир N-rper-
бутиламинофумаровой кис-
кислоты
Диметиловый эфир N.N-ди-
метиламиномалеиновой кис-
кислоты»
Исходное
веществоа
Аммиак, М
Аммиак, Э
Метиламин, Э
Бензиламии, Э
Анилин, М
трег-Бутиламин, М
Диметиламин, М
Т. кип. 'С
(мм рт. ст.)
62@,7);
т. пл. 30
136 A6)
140 A8)
154 @,7)
132 @,2)
85 @,3)
Т. пл. 83
(метанол)
"r,so
D
1,5106
1,4928
1,5130
1,5568
1,5820
1,4880
Выход,
%
80
75
90
80
90
85
70
а М — диметиловый эфир ацетилендикарбоновой кислоты, Э — диэтиловый эфир аце-
тилендикарбоновой кислоты.
" Сначала образуется преимущественно диметиловый эфир аминомалеиновой кисло-
кислоты (?-форма), т. пл. 92°С(эфир); при перегонке он переходит в (ZJ-форму.
в После отгонки эфира продукт очищают перекристаллизацией.
(ГП) Общая методика присоединения аминов к эфирам ацетилендикарбоновых кислот (табл. Г.4.53)
I Внимание*. Эфиры ацетилендикарбоновых кислот обладают слезоточивым
действием! Работать под тягой!
Реакцию проводят в 250-мл трехгорлой колбе, снабженной мешалкой, внутренним термо-
термометром, капельной воронкой и обратным холодильником с хлор кальциевой трубкой. В колбу
помещают раствор 0,11 моль безводного амина (высушенного над гидроксидом калия) в
100 мл сухого эфира и при энергичном перемешивании по каплям прибавляют раствор
0,1 моль ацетилендикарбонового эфира в 50 мл сухого эфира. Скорость прибавления регули-
регулируют так, чтобы температура не поднималась выше 15-20 °С (в нужных случаях применяют на-
наружное охлаждение ледяной водой). Газообразные амины после сушки (см. часть Е, аммиак) в
небольшом избытке медленно вводят в эфирный раствор ацетилендикарбонового эфира при
охлаждении (температура реакционной смеси 15-20 °С). Смесь оставляют на 5 ч при комнат-
комнатной температуре, эфир отгоняют, остаток фракционируют в вакууме.
4.3. РЕАКЦИИ РАДИКАЛЬНОГО ПРИСОЕДИНЕНИЯ И ПОЛИМЕРИЗАЦИИ
Кратные углерод-углеродные связи могут присоединять также и радикалы.
Подобно радикальному замещению (см. разд. Г,1), радикальное присоединение
имеет цепной характер. Инициирующий радикал R1*, возникший в результате
фотолиза или термолиза, реагирует прежде всего с аддендом X—Y:
R'- + Y-X
R-Y + Х-
[Г.4.54]
384
Г Препаративная часть
Оразовавшийся Х-радикал присоединяется к олефину. При этом возникает
Р-замещенный алкильный радикал, который реагирует на второй стадии роста
цепи с X—Y с переносом Y-радикала (проверить!)
х-
I /
\ /
с=с
/ \
I /
х-с-с-
I \
Х-С-С• + Y-X
X-C-C-Y + Х-
[Г.4.55а]
[Г.4.556]
Обрыв цепи происходит при рекомбинации Х-р-алкильного радикала.
На схемах [Г.4.56] показано присоединение брома к этилену. Электрофильный атом
брома, частично занятая молекулярная орбиталь (ЧЗМО) которого имеет сравнительно
низкую энергию (см. рис. Г. 1.23), вступает во взаимодействие с ВЗМО олефина:
Вг- + Н2С=СН2
Вг-СН2-СН2
= -21кДж/моль [Г.4.56а]
Образовавшийся углеродный радикал стабилизируется реакцией с молекулой
брома, одновременно возникает новый бром-радикал:
Вг-СН2-СН2 + Вг2
Вг-СН2-СН2-Вг + Вг-
° = -60кДж/моль [Г.4.566]
Обе стадии цепной реакции экзотермичны. Реакция протекает самопроизволь-
самопроизвольно, длина цепей велика.
Хлор, у которого энергия ЧЗМО ниже, чем у брома, реагирует с алкенами
легче. Кроме того, энергия диссоциации связи С—С1 примерно на 50 кДж/моль
выше энергии диссоциации связи С—Вг; следовательно первая стадия, анало-
аналогичная схеме [Г.4.56а], экзотермична (см. также уравнение [В.17]). Радикал
иода, напротив, недостаточно реакционноспособен, для того чтобы атаковать
двойную связь, поскольку первая стадия эндотермична (около 50 кДж/моль).
Радикальное присоединение к олефинам характерно также для бромоводо-
рода, альдегидов, спиртов, сложных эфиров, полигалогеноалканов (хлорофор-
(хлороформа, тетрахлоруглерода), сероводорода, тиолов тиокислот, гидросульфита и мно-
многих других реагентов:
-НВг
R-CH=CH2
- R'CH2CHO
+ ССЦ
+ CHBr3
+ СНС13
R-CH2-CH2-Br
R-CH2-CH2-CO-СНг-R1
R-CHCI—CH2-CCI3
R-CHBr-CH2-CHBr2
R-CH2-CH2-CCI3
[Г.4.57]
(Напишите схемы цепных реакций с этими соединениями.)
4. Присоединение к неактивированным кратным углерод-углеродным связям 385
В отличие от бромоводорода, иодо- и хлороводород не присоединяются к
олефинам по радикальному механизму. В случае иодоводорода первая стадия
цепной реакции (иод-радикал+олефин) невозможна, так как радикал иода не-
недостаточно реакционноспособен, а у хлороводорода гемолитический разрыв
связи Н—С1 требует слишком большого расхода энергии, отчего вторая стадия
цепной реакции эндотермична и не реализуется.
Органические радикалы с пониженной под влиянием электроноакцептор-
ных заместителей спиновой плотностью у радикального центра (например, ра-
радикалы полигалогенометанов) легко реагируют с более основными олефинами
(например, виниловыми эфирами), а также с обычными олефинами. Напротив,
в случае этилендикарбоновых эфиров радикал, образующийся в первой стадии
аналогично схеме [Г.4.55а], обладает столь малой энергией, что он уже не спосо-
способен инициировать цепную реакцию путем стадии переноса по схеме [Г.4.556].
Алифатические альдегиды и спирты иногда с высоким выходом присоединя-
присоединяются по радикальному механизму к перфторолефинам и а,р-ненасыщенным
карбонильным соединениям, особенно к эфирам малеиновой кислоты.
Из альдегидов, разветвленных в ос-положении, только с небольшим выходом
можно получить кетоны, так как образующийся вначале ацильный радикал в
первую очередь подвергается декарбонилированию (как это можно объяснить?):
R3C-CHO R3C-CO R3C + СО [Г.4.58]
В гемолитических условиях, обусловливающих образование радикалов, даже
хлор присоединяется к бензолу, причем возникает смесь стереоизомерных гек-
сахлорциклогексанов (в каких условиях можно подавить всегда заметно протека-
протекающую при этом конкурентную реакцию замещения; см. разд. Г,5.1.5?); в смеси
содержится около 15% у-изомера — важного инсектицида (гаммексан, у-ГХЦГ,
гексахлоран).
Присоединение галогенов к ацетиленам также, вероятно, протекает по ради-
радикальному механизму. На основании соображений, приведенных во введении к
разд. Г,4, электрофильное присоединение в данном случае затруднено.
Необходимо отметить, что описанное выше радикальное присоединение
протекает против правила Марковникова. Это и понятно, так как один из двух
радикалов, которые могут образоваться на первой стадии, например радикал I в
реакции
^^ R-CH-CH2-Br (I)
R-CH=CH2 + Br-
^^ R-CH-CH2 (II) [Г.4.59]
Br
обладает меньшей энергией, чем радикал II, по причинам, указанным в
разд. Г, 1.
Также возможно, что радикальное присоединение бромоводорода к олефи-
олефинам в присутствии пероксидов протекает против правила Марковникова
(пероксидный эффект).
Если реагент может присоединяться и по ионному, и по радикальному механиз-
механизму, то направление реакции зависит от условий. Так, в присутствии кислоты Льюи-
Льюиса, например АШгз, пероксидный эффект при присоединении НВг подавляется и
реакция идет по ионному механизму в соответствии с правилом Марковникова.
386 Г Препаративная часть
(ГП) Общая методика радикального присоединения к олефинам (табл. Г.4.60)
I Внимание1. Защищать глаза при работе с погружной лампой опасного УФ-излу-
чения. Соблюдать правила безопасности для соседнего рабочего места!
С целью предотвращения образования теломеров (см. [Г.4.72]) олефин берут в недостатке.
Выход рассчитывают на вступивший в реакцию олефин.
Прибор состоит из трехгорлой колбы, снабженной газоподводящей трубкой, по которой
газ поступает непосредственно в реакционную смесь, эффективным обратным холодильни-
холодильником и внутренним термометром. При реакциях, инициируемых фотохимически, необходимо
также иметь охлаждаемую (!) погружную ртутную лампу1. Вступающие в реакцию олефины в
ходе реакции находятся в газообразном состоянии, присоединение проводят в атмосфере азо-
азота (в прибор медленно пропускают ток свободного от кислорода азотаJ.
Для достижения высоких выходов совершенно необходимо точно поддерживать указан-
указанную температуру. Если заданный температурный интервал узок, пользуются металлической
баней с реле и контактным термометром (см. разд. А, 1.7.1) или еще лучше термостатом.
Реакционную смесь перегоняют в вакууме, используя 40-сантиметровую колонку Вигре.
После фракционирования получают фракции исходных соединений и продукта присоедине-
присоединения (остаток после перегонки обычно состоит из полимеров и теломеров; см. [Г.4.72]).
Если продукт перегоняется в широком интервале температур, то перегонку проводят на
60-сантиметровой колонке Вигре. Если же продукт во время проведения реакции выпадает в
осадок, реакционную смесь оставляют на ночь в холодильнике, образовавшийся продукт отса-
отсасывают и перекристаллизовывают.
Вариант А. Присоединение СС14.1 моль олефина, 4 моль СС14, 0,06 моль бензоилпероксида
нагревают 5 ч с обратным холодильником.
Вариант Б. Присоединение альдегидов. 1 моль олефина, 4 моль альдегида и 0,03 моль бензоил-
бензоилпероксида нагревают 8 ч при указанной в методике температуре. После этого (т. е. через 8 ч
нагревания) добавляют еще 0,03 моль пероксида и далее продолжают нагревание еще 16 ч.
Вариант В. Присоединение тиолов. 1 моль олефина и 1 моль тиола облучают 5 ч при ком-
комнатной температуре.
Вариант Г. Присоединение газообразных соединений. При облучении в реакционную среду мед-
медленно вводят газ до насыщения или до начала кристаллизации продукта. В последнем случае
(с целью предотвращения закупоривания) используют газоподводящую трубку с колоколо-
образным расширением на конце или применяют приспособление, показанное на рис. А. 13.
Методика малопригодна для полумикроколичеств.
Получение диэтилового эфира B-ацетоксиэтил)малоновой кислоты присое-
присоединением диэтилового эфира малоновой кислоты к винилацетату Гриттер Р.
В сб.: Органические реакции. Сб. 13. — М.: Мир, 1966, с. 157.
Обе участвующие при радикальном сдваивании части X и Y при образовании
олефина могут принадлежать двум исходным продуктам.
Синтетически важным присоединением этого типа являются реакции олефи-
нов с алкилгалогенидами (RX) в присутствии металлоорганических гидридов,
например гидрида трибутилолова (QHgbSn—H:
1 Внешнее УФ-облучение даже при удвоении времени реакции приводит к малым выходам
(это относится и к случаям, когда реакцию проводят в колбе из кварца или увиолевого стекла).
Для присоединения хлора достаточно облучения обычной лампой накаливания B00 В), распо-
расположенной рядом с колбой.
2 Ср. часть Е.
Таблица Г.4.60. Радикальное присоединение
Продукт
1,1,1,3-Тетрахлороктан
1,1,1,3-Тетрахлорнонан
Диэтиловый эфир бути-
рилянтарной кислоты
Диэтиловый эфир гепта-
ноилянтарной кислоты
1 -Бензилтио-2-фенилэта н
1 -Фенил-2-фенилтиоэтан
Р-Фенилтиопропионитрил
о-Гексахлорциклогексан
1,3-Днбромпропан
1 -Хлор -3-бромпропан
Исходное вещество
Тетрахлоруглерод
Тетрахлоруглерод
Масляный альде-
альдегид
Энантовый альде-
альдегид (гептаналь)
Бензилтиол
Тиофенол
Тиофенол
Хлор
Бромоводород
Бромоводород
к алкенам
Олефин
Гептен-1
Октен-1
Диэтиловый эфир
малеиновой кис-
кислоты
То же
Стирол
Стирол
Акрилонитрил
Бензол
Аллилбромид
Аллилхлорид
Вари-
Вариант
А
А
Б
Б
Б
В
В
Г
Г
Г
Т. кип., °С
(мм рт. ст.)
130 A9)
78 @,1)
112A)
113@,5)
156 C)
188A5)
154 (8)
Т. пл. 158
(бензол)
167
142
"D
1,4772
1,4770
1,4376
1,4392
(пРи25°С)
1,5894
1,6042
1,5735
1,5232
1,4950
Выход,
%
50
40
75
55
80
70
80
70
95
95
Примечания
Кипятят с обратным
холодильником
Нагревают до 82—
85 °С
Нагревают на водя-
водяной бане
Сухой хлор пропускают
в нагретый до кипения
сухой, свободный от
тиофена бензол
Охлаждают льдом
Охлаждают льдом
388 Г Препаративная часть
\ /
R-X + С=С + H-Sn(C4H9K - R-C-C-H + X-Sn(C4H9K [Г.4.61]
Радикальная цепная реакция включает после инициирования (например, с по-
помощью азобисизобутиронитрила):
R'—N=N-Rf 2R'- + N2
[Г.4.62]
R'- + H-SnBu3 - R'—H + -SnBu3
три следующие стадии развития цепи:
R-X + -SnBu3 R- + X-SnBu3
R- + H2C=CH-Z - R-CH2-CH-Z [Г.4.63]
R-CH2-CH-Z + H-SnBu3 R-CH2-CH2-Z + -SnBu3
В результате реакция [Г. 4.61] сводится к присоединению R-H к олефину.
Таким образом можно в мягких условиях реакции осуществить радикальное
образование С—С-связи, что делает доступным большое число соединений
(Z = CN, COR, COOR, OR, арил и т. д.).
1-Дезокси-2,Ъ,А,6-тетра-О-ацетил-\-(\-цианэтил)-а-Т)-глюкопираноза:
Giese В., DupuisJ., NixM. Org. Synth. 1987, 65, 236-242.
Аналогичное присоединение протекает и с ртутьорганическими гидридами:
R-Hg-H + С=С - R-C-C-H + Нд [Г.4.64]
Ртутьорганический гидрид RHgH можно получать in situ при действии гидридов металлов
на ртутьорганические соединения, и реакция проходит спонтанно, так как связь R—Hg очень
слабая (см. табл. Г. 1.4):
RHgX ^- RHgH C-=^^ R. [Г.4.65]
Далее R- реагируют с олефинами по следующему радикально-цепному механизму:
R- + H2C=CH-Z R-CH2-CH— Z
R-CH2-CH-Z + H-HgR - R-CH2-CH2-Z + -HgR ' ' ' '
R-Hg- - R- + Hg
Ртутьорганические соединения RHgX можно получить по реакции Гриньяра из RX и гало-
генида ртути:
RX J*L RMgX -^gj- RHgX [Г-4.67]
или из олефина и органоборана (см. гидроборирование, разд. Г.4.1.8) и ацетата ртути:
\
\ / + в~н / I I
Vc/ -!— H-C-cV +Нд(ОСОСНзJ Н-С-С-НдОСОСН3 [Г.4.68]
/ \ II \ II
Присоединенный остаток R возникает из алкилгалогенида и олефина, которые таким образом
могут быть соединены с субстратным олефином НгС=СН2. Поэтому реакция является мно-
многоплановой для синтеза с образованием С-С-связи.
Названные реакции можно также использовать для внутримолекулярного присоединения
и затем циклизации:
4. Присоединение к неактивированным кратным углерод-углеродным связям
389
[Г.4.69]
Таким образом получают циклопентильные и циклогексильные соединения на основе гексен-
5-ил-1- и гептен-6-ил-1-соединений RX.
Напишите эти реакции!
Радикальная полимеризация. Углеводородный радикал, образовавшийся на
первой стадии радикального присоединения согласно схеме [Г.4.70а], может ре-
реагировать со следующей молекулой олефина (б). Таким образом реакция может
продолжаться дальше (реакция роста) и приводить к цепочечным полимерам (в):
[Г.4.70а]
(б)
и т.д.
[Г.4.70б,в]
При радикальном присоединении полимеризация является нежелательным
побочным процессом, тогда как целенаправленная радикальная полимеризация
приводит к ценным промышленным полимерам.
С понижением температуры, уменьшением концентрации исходных радика-
радикалов и повышением концентрации мономера растет величина соответствующей
макромолекулы. Средняя молекулярная масса достигает 107.
Не все олефины одинаково склонны к радикальной полимеризации. Наиболее
легко этот процесс происходит с соединениями, заместителями в которых стаби-
стабилизируют радикалы, возникающие во время роста, например винильные, ариль-
ные и карбонильные группы (см. разд. Г, 1.1). Обрыв цепи происходит путем дис-
пропорционирования (а) или рекомбинации (димеризации) (б) макрорадикалов:
2 X
(а)
(б)
[Г.4.71]
R R R
Поэтому радикальная цепь вообще обрывается. Обрыв цепи может также проис-
происходить в результате переноса радикальной функции на другую молекулу (раство-
(растворитель; образовавшуюся макромолекулу; другие добавленные вещества, способ-
способные к образованию радикалов, например, регуляторы полимеризации). При
этом растущий полимер становится «мертвым», а вновь образовавшийся ради-
радикал может служить началом новой цепи.
Подобные реакции переноса цепи специально используют при теломериза-
ции. При направленной полимеризации, подбирая подходящие условия реак-
реакции (например, концентрацию), можно получить теломеры, т. е. полимеры с
390 Г Препаративная часть
небольшой степенью полимеризации, несущие определенные концевые груп-
группы. Из-за сравнительно малой молекулярной массы концевые группы опреде-
определяют свойства теломеров в большей степени, чем в случае высокомолекулярных
соединений. В качестве соединений, образующих концевые группы, использу-
используют тетрахлоруглерод, хлороформ, причем указанная выше промежуточная
степень теломеризации обусловливается подходящей (сравнительно высокой)
концентрацией этих веществ, например:
Иницирование цепи: R'OOR' - 2 R'O
Рост цепи:
R'O- + H2C=CHR - R'OCH2CHR
R'OCH2CHR + n H2C=CHR RlOCH2CH(CH2CH)n.1CH2CH [Г.4.72]
R R R
Пререносцепи:
R'OCH2CHCH2CH + ССЦ R'OCH2CHCH2CHCI + -CCI3
R R R R
Рост цепи: CI3C- + H2C=CHR CI3CCH2CHR
Участвующие в теломеризации соединения называют таксогеном (мономер)
и телогеном (например, ССЦ, HSiCl3, ROH, HC1).
Ф Полимеризация полистирола (полимеризация в растворе). В пробирке с холодильником
растворяют 2 г стирола (свежеперегнанного над небольшим количеством серы) в 10 мл
ксилола, добавляют 50 мг бензоилпероксида и нагревают 2 ч на водяной бане примерно при
80 °С. При перемешивании выливают раствор в 100 мл метанола, налитого в ступку. Выпавший
продукт хорошо растирают в ступке для удаления мономера и ксилола. Через 2 ч полимер
отфильтровывают, промывают метанолом и сушат в вакуум-эксикаторе.
Полимеризация стирола в суспензии и эмульсионная сополимеризация стирола и
изопрена: Sorenson W. R. J. Chem. Educ, 1965, 42, 8.
Сополимеризация диметилфумарата и винилацетата: Abell P. J. In: KochiJ. К.,
Free Radicals. — John Wiley & Sons, New York, 1973, Vol. 2, p. 96.
Радикальная полимеризация винильных соединений является важнейшим
методом получения пластмасс, синтетических волокон и искусственного каучука.
Большую часть применяемых в настоящее время полимеров получают из смесей мономе-
мономеров. Причем последние могут располагаться в полимерной молекуле в порядке регулярного
или нерегулярного чередования или блоками (см. табл. Г.4.73).
Сополимеризацией различных винильных мономеров можно получать полимеры с зара-
заранее заданными свойствами.
Различают процессы полимеризации в массе, в растворе и в эмульсии. Представляет опре-
определенный интерес и полимеризация в суспензии. В качестве инициаторов применяют перок-
сиды, например бензоилпероксид, персульфат калия и а,а-диметилбензилгидропероксид в
присутствии сульфата железа(П).
Теломеры также приобретают все большее значение. Например, взаимодействием
а,а,а-трихлор-со-хлорпарафинов с аммиаком с последующим омылением можно получать
ш-аминокарбоновые кислоты, лактамы которых полимеризуются до полиамидов.
4. Присоединение к неактивированным кратным углерод-углеродным связям
391
Таблица Г.4.73. Технически важные полимеры, получаемые из мономеров, и их
применение
Мономеры
Применение полимеров
Этилен
Винилхлорид
Стирол
Тетрафторэтилен
Акрилонитрил
Бутадиен
Винилацетат
Эфиры метакриловой
кислоты
Бутадиен+стирол
Бутадиен+стирол+
акрилонитрил
Бутадиен+акрилонитрил
Стирол+акрилонитрил
Этилен+винилацетат
Винилхлорид+винили-
дендихлорид
Стирол+ненасыщенные
полиэфиры
Полиэтилен высокого давления: пленки, упако-
упаковочные материалы, емкости, трубы, изоляционные
материалы
Поливинилхлорид (ПВХ): пленки, кислотоустой-
кислотоустойчивые трубы, сосуды, пластины, электроизоля-
электроизоляционные материалы
Полистирол: электропромышленность, пленки, пе-
нополистирол (упаковочный материал)
Политетрафторэтилен (тефлон): высокая стой-
стойкость к химикатам, футеровка, аппаратуры, шлан-
шланги, уплотнения
Полиакрилонитрил (ПАН): волокна
Полибутадиен (буна): эластомеры
Поливинилацетат: латекс, жевательная резинка
Полиметакрилат (плексиглас), органическое
стекло
Буна S: эластичный или ударостойкий полисти-
полистирол
ABS-полимеры: прочный на разрыв полимер
Буна N (пербунан)
Ударостойкий полистирол
EVA-полимеры: термопластичный, мягкий
материал. Олигомеры: добавки к дизельному
топливу, улучшающие текучесть
Волокна, щетки
Полиэфирные смолы, армированные стекловолок-
стекловолокнами, ламинаты
4.4. РЕАКЦИИ ЦИКЛОПРИСОЕДИНЕНИЯ
Реакции присоединения к олефинам, ведущие к циклическим продуктам, назы-
называют циклоприсоединением. По виду и числу атомов двух компонентов, вносимых
ими в создаваемый цикл, образование карбо- и гетероциклов разделяют на сле-
следующие типы:
[1 + 2]-Циклоприсоединение
\
X:
с
II
\
х5
[Г.4.74]
Сюда относятся реакции присоединения карбенов (X: = R2C:) и нитренов
(X: = R-N:) к олефинам, а также эпоксидирование олефинов (разд. Г,4.1.6), и
образование тииранов.
[2 + 2]-Циклоприсоединение
X
II
Y
V
II
/к
Х-С
I i
Y-C-
\
[Г.4.75]
392 Г Препаративная часть
К этой группе относятся циклоприсоединения олефинов к олефинам с обра-
образованием циклобутанов (термически оно осуществляется_редко, но легко фото-
фотохимически); присоединение синглетного кислорода (О=О) к олефинам с обра-
образованием циклических пероксидов (диоксетанов) и фотохимическое присоеди-
присоединение альдегидов или кетонов к олефинам с образованием оксетанов (реакция
Патерно — Бюхи).
[3 + 2]-Циклоприсоединение A,3-биполярное циклоприсоединение)
Y +п Y i гг л 7fti
zi° /\ VV [ГА761
Эти термически легко протекающие реакции циклоприсоединения имеют осо-
особое значение для синтеза гетероциклов. В качестве 1,3-биполярных реагентов
в 0
(Х+ —Y—Z~) используются диазосоединения N=N—CH—R (см. разд. Г,8.4), ази-
ф В. ф О ® 9
ды N=N—N—R, нитрилоксиды R—C=N—О|, нитрилимины R—C=N—N—R и
нитрилилиды R— C=N—CR2, которые часто образуются непосредственно в
реакционной смеси.
[4 + 2]-Циклоприсоединение (диеновый синтез, реакция Дильса — Альдера)
'„W \ / yj^ /
? + ? ~ fi ?~ [Г.4.77]
Y-z /c\ Y-z'V
Важнейшая из реакций указанного типа — легко осуществляющаяся термичес-
термически реакция Дильса — Альдера. Диенами-1,3 (W=X—Y=Z) могут быть как линей-
линейные соединения (например, бутадиен-1,3), так и циклические (циклопентадиен,
циклогесадиен-1,3). С гетероаналогами диенов (а,р-ненасыщенными альдеги-
альдегидами, а,C-ненасыщенными кетонами, азинами и др.) получают шестичленные
гетероциклы. Вместо олефина (диенофила) может участвовать, например, синг-
летный кислород; тогда в результате реакции образуются шестичленные цикли-
циклические пероксиды.
Реакции циклоприсоединения могут протекать по перициклическому меха-
механизму. При этом участвующие электроны в циклическом переходном состоянии
подвергаются согласованному сдвигу; замыкание цикла стереоспецифично, а
скорость реакции почти не зависит от растворителя. Нестереоспецифичное
циклоприсоединение обычно осуществляется в две стадии через радикалы или
ионы. При ионном механизме скорость реакции зависит от природы раствори-
растворителя.
На основе концепции граничных орбиталей1 можно объяснить пространственную направ-
направленность перициклических реакций циклоприсоединения, а также определить, разрешена
термически или фотохимически индуцированная реакция. Это объяснение базируется на зна-
знаках коэффициентов ВЗМО, НСМО и ЧЗМО обоих реагирующих соединений. Как известно,
образования ковалентной связи можно ожидать только в том случае, если у реагирующих мо-
молекул перекрываются орбитальные области одинакового знака. Если это условие выполнено
1 Данная здесь трактовка является применением правил Вудворда — Хоффмана о сохра-
сохранении орбитальной симметрии.
4. Присоединение к неактивированным кратным углерод-углеродным связям
393
немо
взмо
немо
супрафасное
ВЗМО
немо
чзмо
[2+1 ] -Циклоприсоединение
термически индуцированное, разрешено,
конфигурация олефина
сохраняется
[2+2]-Циклоприсоединение
а) термически индуцированное, б) фотохимически
запрещено, в других случаях индуцированное,
возможно как разрешено
супра-антарафасное (супра-супрафасное
присоединение присоединение)
немо
ВЗМО
немо
ВЗМО
немо
ВЗМО
[3+2]-Циклоприсоединение,
термически индуцированное,
разрешено, супрафасное присоединение
[4+2]-Циклоприсоединение,
термически индуцированное,
разрешено, супрафасное присоединение
Рис. Г.4.78. Механизм циклоприсоединения в рамках теории граничных орбиталей.
хотя бы для одного из двух возможных взаимодействий ВЗМО-НСМО реагирующих молекул,
то оказывается возможным термически согласованное циклоприсоединение. При этом однов-
одновременно предопределяется и стереохимический ход реакции.
Для термически индуцированных реакций характерно, что обе новые ст-связи образуются
с одной стороны л-связи. Подобный механизм реакции называют супрафасным (супраповерх-
ностным). При антарафасном (антараповерхностном) механизме реакции взаимодействуют
имеющие одинаковый знак области, расположенные на противоположных сторонах двух
л-систем (рис. Г.4.78).
В соответствии с рис. Г.4.78 возможны термически индуцированные перициклическое
[1 + 2]-циклоприсоединение синглетных карбенов к олефинам, а также [3 + 2]- и [4 + 2]-цикло-
присоединения. Напротив, термически согласованное [2 + 2]-циклоприсоединение, как правило,
невозможно. Образование связи между ВЗМО одной молекулы олефина и НСМО второй
могло бы осуществиться по супрафасному механизму только между двумя атомами. Образование
второй связи требует антарафасного взаимодействия. Если же одна из олефиновых молекул реаги-
реагирует в фотохимически возбужденном состоянии, то между ее ЧЗМО и НСМО второй молекулы
олефина возможно перекрывание орбитальных областей одинакового знака по супрафасному
типу. Фотохимическое перициклическое [2 + 2]-циклоприсоединение разрешено. Однако реаги-
реагирующая система не обязательно будет следовать механизму, соответствующему концепции
граничных орбиталей: она может найти выход в двухступенчатом механизме.
4.4.1. РЕАКЦИИ [1 + 2]-ЦИКЛОПРИСОЕДИНЕНИЯ.
ПРИСОЕДИНЕНИЕ КАРБЕНОВ И КАРБЕНОИДОВ
Карбены реагируют с олефинами по схеме [Г.4.74] с образованием циклопропа-
циклопропанов. Однако стереоспецифичное синхронное присоединение следует ожидать
лишь для синглетного карбена [Г.4.79], в то время как для образующегося в раст-
394
Г Препаративная часть
воре триплетного карбена оказывается невозможной синхронная реакция и
поэтому не происходит стереоспецифичной реакции [Г.4.80]:
\
R
\
С::
/ -
c
\
C
/
[Г.4.79]
\ R
/ •• 'A*
7 Я.-R1
\ Lr
p.'?
/ c.
[Г.4.80]
Арены также присоединяют карбены, причем сначала образуются бицик-
ло[4.1.0]гептадиены (норкарадиены), которые являются валентными таутомера-
ми циклогептатриенов и легко в них изомеризуются. Тем самым открывается
простой препаративный путь (посредством расширения цикла) к циклогепта-
триенам, труднодоступным иными путями:
+ :СН2
[Г.4.81]
Ф Общая методика присоединения дихлоркарбена к олефинам (табл. Г.4.82). 0,1 моль оле-
фина и 1 ммоль бензилтриэтиламмонийхлорида растворяют в 0,4 моль хлороформа,
содержащего 1 мл этанола. После добавления охлажденного льдом свежеприготовлен-
свежеприготовленного 50%-ного раствора 0,4 моль гидроксида натрия смесь энергично перемешивают
Таблица Г.4.82. Получение 1,1-дихлорциклопропанов присоединением дихлор-
дихлоркарбена
Продукт реакции
7,7-Дихлорбицикло [4.1.0] -
гептан
1,1 - Дихлор-2-фенилцикло-
1,1 - Д ихлор -2-метил-2-фе-
нилциклопропан
1,1-Дихлор-2,2-дифенилцик-
лопропан
1,1 - Д ихлор -2-гексилцикло-
1,1 - Дихлор -2,2-диметилцик-
лопропан
Исходное соединение
Циклогексен
Стирол
ос-Метилстирол
1,1 - Д ифенилэтилен
Октен-1
Изобутен а
Т. кип., *С
(мм рт. ст.)
78—79 A5)
114 A3)
66 @,6)
Т. пл. 114°С
50—52 @,5)
118—120
V
1,5028
1,5515
1,5410
1,4555
1,4499
Выход,
%
78
84
75
70
60
60
Изобутен вводят сначала при —10 "С, а затем при постепенной повышении тем-
температуры до комнатной.
4. Присоединение к неактивированным кратным углерод-углеродным связям 395
1 ч при комнатной температуре и еще 5 ч при 50 °С. Охлажденную смесь выливают в 500 мл во-
воды, органический слой отделяют, водный встряхивают со 100 мл хлороформа. Соединенные
органические фазы сушат сульфатом натрия, растворитель упаривают в вакууме, остаток пере-
перегоняют, а твердые продукты реакции перекристаллизовывают.
1,1-Дихлор-транс-2,3-дифенилциклопропан из транс-стльбена и дихлоркар-
бена в условиях межфазного катализа: Dehmlow E. V., Schdnfeld J. Lieb. Ann.
Chem., 1971,744,42.
В качестве источника для генерации карбена следует также учитывать а-эли-
минирование галогеналканов (разд. Г,3.3), а также другие реакции образования
карбенов, такие как разложение алифатических диазосоединений (разд. Г,8.4.2
и Г,9.1.1.3). Если эти соединения разлагаются в присутствии катализаторов на
основе переходных металлов (оптически активные Си-, Rh-соли) промежуточно
возникают не свободные карбены, а металл-карбеновые комплексы [4.120],
которые реагируют с олефинами с образованием циклопропанов.
Из 2,5-диметилгексадиена-2,4 и диазоуксусного эфира таким путем можно также и в про-
промышленности получить этиловый эфир хризантемовой кислоты:
Cu-кат \ AV [Г.4.83]
N2CHCOOEt \ / COOEt + N2
Эфиры хризантемовой кислоты и структурно подобных кислот выделены из пиретрума,
одного из природных веществ, из которого получены различные производные хризантемовой
кислоты с инсектицидным действием. Получены и синтетические продукты, так называемые
пиретроиды, которые применяются в качестве инсектицидов.
Этиловый эфир экзо-трицикло(З.ЗЛ,О2-4)октан-3-анти-карбоновой кислоты из
норборнена и диазоуксусного эфира в присутствии CuCN: Sauers R. Я, Sonnett P. E.
Tetrahedron, 1964, 20, 1029.
Этиловый эфир 2,3-диметилциклопропанкарбоновой кислоты из бутена-2 и ди-
диазоуксусного эфира в присутствии C11OSO2CF3: Salomon R. G., Kochi J. К. J. Am.
Chem. Soc. 1973, 95, 3300.
Этиловый эфир 2-ацетоксициклопропанкарбоновой кислоты из винилацетата и
диазоуксусного эфира в присутствии Ш^ОСОСНз)-»: Anciaux A. J., Hubert A. J.,
Noels А. ?., PetiniotN., Teyssie P. J. Org. Chem. 1980, 45, 695.
Соединения, которые реагируют с олефинами с образованием циклопропа-
циклопропанов без генерации промежуточных свободных карбенов, называются карбенои-
дами. К ним принадлежат также так называемые реагенты Симмонса — Смита,
например дииодметан и цинк-медная пара (из цинковой пыли и Cu2Cl2, C11SO4
или Cu(OAch), которые при сын-присоединении олефинов стереоспецифичес-
ки образуют соответствующие циклопропаны. Активно реагирующими агента-
агентами являются цинкорганические соединения, которые входят в уравнение равно-
равновесия Шленка [Г.7.195]:
2ICH2Znl =^= (ICH2JZn + Znl2 [Г.4.84]
396 Г Препаративная часть
V •** 7 1 ¦ -?
и + нс/ i:;:ch/ jn i
;:ch2/ j i ^сн2 + zni2 [Г.4.85]
/ ¦¦',, I ^1 *- .
По другому варианту можно использовать взаимодействие диэтилцинка с
или другими дигалогенидами.
Бицикло[4.1.0]гептан (норкаран) из циклогексена и дииодметана в присут-
присутствии Zn/CuCl: Rawson R. J., Harrison I. T. J. Org. Chem. 1970, 35, 2057.
4.4.2. [2+2]-ЦИКЛОПРИСОЕДИНЕНИЕ
Реакции [Г.4.75], которые приводят к карбо- или гетероциклическим четырех-
четырехчленным циклам, как можно видеть на рис. Г.4.78, представляют перицикличес-
кий процесс, разрешенный фотохимически. Напротив, перициклический про-
процесс для олефинов запрещен термически, так как геометрически невозможно
осуществить супра-антарафасное взаимодействие орбиталей.
Это ограничение снимается в том случае, если в присоединении может участвовать другая,
перпендикулярная к ^-тг-орбиталям /э-орбиталь ip-гибридизованного атома углерода, как в
гетерокумуленах (например, в кетенах и изоцианатах).
взмо -w v/.- vssa sro [Г.4.86]
Алкен
(См. ниже.)
Фотохимически с помощью [2+2]-циклоприсоединения можно осуществить
превращение олефинов в циклобутан. Если при этом возникает синглетное сос-
состояние, реакция протекает как смн-присоединение. При внутримолекулярном
варианте такого фотоциклоприсоединения открываются важные аспекты
использования полициклических соединений с циклобутановым кольцом.
[Г.4.87]
Норборнадиен Квадрициклан
Однако часто присоединение протекает не как перициклический процесс, а
как многостадийный процесс, что установлено по присутствию открытоцепо-
чечных интермедиатов в форме 1,4-бирадикалов или 1,4-диполей. цис-Селек-
тивность исчезает в том случае, если вращение вокруг связей в этих интермеди-
атах происходит быстрее, чем замыкание цикла.
Другим вариантом фотохимически инициируемого [2+2]-циклоприсоедине-
ния является реакция Патерно — Бюхи. При циклизации альдегидо- или кетокар-
бонильной группы с двойной связью образуются синтетически важные оксетаны.
Получение 3-метил-6[5-пропионилоксиметил-2,7-диоксабицикло[3.2.0]гепте-
на-3 и 1р-метил-бр-пропионилоксиметил-2,7-диоксабицикло[3.2.0]гептена-3 по
реакции Патерно — Бюхи: Just G. In: Photochemical Key Steps in Ogranic Synthesis.
J. Mattay, A. G. Griesbeck (eds). VCH, Weinheim 1994, 43-44.
4. Присоединение к неактивированным кратным углерод-углеродным связям 397
При облучении триплетного кислорода в присутствии сенсибилизаторов об-
образующийся синглетный кислород при наличиии структурно подходящего
олефина присоединяется с образованием 1,2-диоксетана. Возникающий энер-
энергетически обогащенный четырехчленный циклический пероксид при химичес-
химической или термохимической активации может распадаться на два карбонильных
соединения, причем может возникнуть возбужденное состояние. Избыток энер-
энергии может быть выделен в форме света (хемилюминесценция).
Кетен, у которого НСМО является низкоэнергетической, тоже может реаги-
реагировать с двойной связью С=С. Благодаря супра- и антарафасному взаимодей-
взаимодействию с ВЗМО двойной связи реакционных партнеров оказывается разрешен-
разрешенным по симметрии термическое [2+2]-циклоприсоединение (см. выше). Синте-
Синтетическое применение находят особенно кетен-олефин-циклоприсоединение,
при котором кетен, например, может in situ легко возникать из хлорангидридов
карбоновых кислот (разд. Г,3.1.5).
о
н о
Et3N /°-4-/ [Г.4.88]
Получение 5-метил-2-оксабицикло[3.2.0]гептанона-7 по реакции [Г.4.88]:
SneiderB. В., Hui R. A. H. F. J. Org. Chem. 1985, 50, 5167-5176.
В отсутствие реагента (партнера реакции) образуются многочисленные цик-
лодимеры кетена. Из самого кетена при этом возникает дикетен.
4.4.3. [3 + 2]-ЦИКЛ0ПРИС0ЕДИНЕНИЕ A,3-БИПОЛЯРНОЕ ПРИСОЕДИНЕНИЕ)
Разнообразие структур 1,3-биполярных соединений открывает доступ к пя-
тичленным карбо- и особенно гетероциклам различного строения путем
[3 + 2]-циклоприсоединения. Например, диазоалканы (см. разд. Г,8.4) с биполя-
рофилами даютД'-пиразолины (см. схемы [Г.4.90] и [Г.4.76]):
Н2С|Э Н2С1°
N© _> N|
N N©
[Г.4.90]
CN
Региоселективность реакции обычно удается предвидеть, учитывая полярность
реагентов (см. схему [Г.4.90]).
Наряду с устойчивыми 1,3-биполярными соединениями, например озоном
(см. разд. Г,4.1.7), оксидом азотаA) N2O, азидами (напишите их 1,3-биполярную
структуру!), часто применяются и другие нестабильные интермедиа™, возника-
возникающие лишь в реакционной смеси, например в результате отщепления НС1 (нит-
рилимины, нитрилоксиды и т. д.). Необходимые для синтеза Д2-1,2-оксазолинов
398
Г Препаративная часть
(III на схеме [Г.4.91]) и -оксазолов (IV на схеме [Г.4.91]) нитрилоксиды II полу-
получают аналогично из гидроксимоилхлоридов I в безводной среде. Промежуточно
образующийся нитрил оксид реагирует с биполярофилами, образуя гетероцик-
гетероциклическое соединение:
NOH
Ph-C
CI
-на
© 0"
Ph-C=N-0
© е
Ph-C=N-0
\
— с=с—
Ph
Ph
и-
\ /
с=с
/ \
(III)
(IV)
[Г.4.91]
Напишите схемы синтеза других пятичленных гетероциклов из указанных
1,3-биполярных соединений.
Ф Общая методика синтеза 3-D-хлорфенил)-Д2-1,2-оксазолинов и 3-D-хлорфенил)-1,2-
оксазолов с помощью 1,3-биполярного циклоприсоединения (табл. Г.4.92). В трехгорлой колбе на
250 мл с мешалкой и капельной воронкой смешивают раствор 0,02 моль 4-хлорбензгидрокси-
моилхлорида (см. разд. Г, 1.4.1) в 50 мл CH2Cl2 и 0,06 моль алкена. К зтому раствору в течение
45 мин прибавляют по каплям раствор 0,023 мольтриэтиламина в 30 мл СН2С12. По окончании при-
прибавления перемешивают еще 1 ч, добавлением 10 мл воды растворяют выделившийся триэтиламмо-
нийхлорид. Органическую фазу отделяют, дважды промывают небольшим количеством воды, сушат
сульфатом натрия. После отгонки растворителя и избытка алкена в вакууме остаток перегоняют в
высоком вакууме или перекристаллизовывают из абсолютного этанола.
Таблица Г.4.92. Получение 3-D-хлорфенил)-Д2-1,2-оксазолинов и 3-D-хлорфе-
нил)-1,2-оксазолов 1,3-биполярным циклоприсоединением 4-хлорбензонитрилок-
сида к алкенам и алкинам
Продукт реакции
3-D-Хлорфенил)-5-фенил-
Д2-1,2-оксазолин
3-D-Хлорфенил)-5-циано-
Д2-1,2-оксазолин
5-Ацетокси-3-D-хлорфенил-
Д2-1,2-оксазолин
3-D-Хлорфенил)-5-фенил-
1,2-оксазол
Исходное
соединение
Стирол
Акрилонитрил
Винилацетат
Фенилацетилен
Т. пл., *С
132-133
122-123
81-83
177-179
Выход,
%
80
65
87
72
4.4.4. [4+2]-ЦИКЛОПРИСОЕДИНЕНИЕ (ДИЕНОВЫЙ СИНТЕЗ, РЕАКЦИЯ ДИЛЬСА-АЛЬДЕРА)
Важнейшей из реакций [4 + 2]-циклоприсоединения является реакция Дильса —
Альдера (см. рис. Г.4.78 и схему [Г.4.77]). Реакция Дильса — Альдера протекает осо-
особенно гладко, если диен обогащен электронами, а «ен» (диенофил) обеднен ими
или наоборот. В последнем случае говорят о «диеновом синтезе с обращенными
электронными требованиями».
400
Г Препаративная часть
молекул циклопентадиена такова, что становится возможной дополнительная стабилизация
за счет вторичных орбитальных взаимодействий (на схеме [Г.4.95] они обозначены пунк-
пунктиром).
Реагируя одновременно как диен и как диенофил, могут аналогично димери-
зоваться а,р-ненасыщенные карбонильные соединения, в том числе акролеин,
метил винил кетон и др., например: •
О
О
[Г.4.96]
Подобную региоселективность, весьма часто наблюдаемую в реакции Дильса — Альдера,
можно объяснить на основе концепции граничных орбиталей (см. разд. В,6). При этом необ-
необходимо учитывать энергии, а также величины и знаки коэффициентов для ВЗМО и НСМО
двух реагентов. При реакции Дильса — Альдера оба возможных взаимодействия (ВЗМОдиен —
НСМОолефин и НСМОдИен — ВЗМОолефи„) в принципе относятся к связывающим.
Чтобы определить структуру основного продукта реакции, поступают следующим образом:
- ВЗМО—НСМО-пары реагентов формируют так, чтобы атомы с наибольшими коэффи-
коэффициентами орбиталей могли взаимодействовать друг с другом;
- Если в обоих случаях получается одно и то же соединение, то оно и явится главным
продуктом реакции;
- Если при сравнении коэффициентов два сочетания ВЗМО—НСМО приводят к разным
продуктам, то следует установить, для какой пары ВЗМО-НСМО разность энергий
(са/сьJ/(.Евзмо@)-?нсмоD)) (ср. [В.78]) меньше; такое сочетание приведет к главному
продукту. Значения квадратов коэффициентов аналогичны как для малых, так и для
больших различий в энергии (&Е) ВЗМО—НСМО.
Для реакции 2-фенилбутадиена-1,3 со стиролом (схема [Г.4.97а]) сравнение коэффициен-
коэффициентов позволяет предположить, что реакция будет осуществляться в основном по пути «а».
Действительно, 1,4-дифенилциклогексен-1 и 2,4-дифенилциклогексен-1 образуются в соот-
соотношении 20: 1:
Ph
Ph
Ph
20
немо взмо
+0,7 эВ -9,1 эВ
АЕ - 9,8 эВ
[Г.4.97а]
Ph
Ph
ВЗМО НСМО
-8,5 эВ +1,0эВ
Д? = 9,5 эВ
[Г.4.976]
Взаимодействие 1-этоксибутадиена-1,3 с акролеином приводит почти исключительно к
2-этоксициклогексен-3-карбальдегиду-1. Сравнение коэффициентов показывает, что хотя
4. Присоединение к неактивированным кратным углерод-углеродным связям
401
реакция [Г.4.98] в принципе может идти как по пути «а», так и по пути «б», однако АЕзначи-
АЕзначительно меньше на пути «а» (см. также уравнение [В.78]):
OEt
OEt
О
СНО
ВЗМО НСМО
-8,5 эВ ОэВ
Д? = 8,5эВ
[Г.4.98а]
OEt
-Н-
сно
нсмо взмо
+2,5эВ -10,9
ДЕ = 13,4 эВ
[Г.4.986]
Поскольку многие реакции Дильса — Альдера при повышении температуры
становятся обратимыми, работать следует при возможно более низких темпера-
температурах. Катализаторы обычно не требуются, хотя в некоторых случаях кислоты
Льюиса (ССЬСООН, А1С13) повышают скорость и региоселективность1. В случае
исходных веществ, склонных к полимеризации, прибавляют подходящие инги-
ингибиторы полимеризации (например, гидрохинон).
Взаимно растворимые вещества обычно вводят в реакцию без растворителя.
В других случаях, а также при бурно протекающих реакциях можно работать в
инертном растворителе (толуоле, ксилоле).
Ф Общая методика диенового синтеза (табл. Г.4.99). 0,1 моль диенофила растворяют в ми-
минимальном количестве растворителя и смешивают с растворенным в том же раствори-
растворителе диеном. В случае жидких диенов их избыток может служить растворителем.
Газообразные исходные вещества предварительно растворяют в указанном в табл. Г.4.99 раст-
растворителе или конденсируют в сосуде под давлением. После того как закончилось самопроиз-
самопроизвольное выделение тепла, реакцию продолжают при указанных в таблице условиях. При обра-
обработке реакционной смеси жидкие продукты (после удаления растворителя в вакууме) перего-
перегоняют, твердые отсасывают и затем перекристаллизовывают.
Методика пригодна для синтезов в полумикромасштабе.
Фуран и аналогичные пятичленные гетероциклы гладко реагируют в качест-
качестве диенов. В бициклических аддуктах под действием кислоты размыкается кис-
кислородный мостик и образуются ароматические соединения. Аддукты на основе
ацетиленового диенофила изомеризуются в фенолы, например:
О
COOEt
COOEt
[Г.4.100]
1 Протонирование понижает энергии ВЗМО и НСМО а,р-непредельных карбонильных
соединений, так что ВЗМО диена и НСМО карбонильного соединения сближаются и их вза-
взаимодействие оказывается доминирующим.
Таблица Г.4.99. Реакции по Дильсу — Альдеру
Продукт
Диен: диенофил
(молярное отношение)
Растворитель
Условия синтеза
Физические
константы
Выход,
Ангидрид чмс-циклогексен-4-ди-
карбоновой-1,2 кислоты
Циклогексен-3-карбальдегид-1
Д5-Норборненкарбоновая-2 кис-
кислота (бицикло [2.2.1 ]гептен-5-
карбоновая-2 кислота)а
Ангидрид цис-бицикло [2,2,2] -
октен-5-дикарбоновой-2,3 кис-
кислоты
1,1а,4,4а,8,8а,9а,10а-Октагидро-
1,4; 5,8-диэтаноантрахинон
Ангидрид Ч"с-9,10-дигидро-
9,10-этаноантрацендикарооно-
вой-11,12 кислоты
2 -Этокси-2,3-дигидро-2Н-пиран
Бутадиен-1,3: малеи-
новый ангидрид A:1)
Бутадиен-1,3 :акролеин
A:1)
Циклопентадиен^: ак-
акриловая кислота
A:2)
Циклогексадиен": ма-
леиновый ангидрид
A:1)
Циклогексадиенв:
: л-бензохинон D:1)
Антрацен : малеино-
вый ангидрид A:1)
Акролеин1": этилвини-
ловый эфир D : 5)
Бензол
Без растворителя
Эфир
Бензол
Без растворителя
Ксилол
Без растворителя
5 ч, 100 °С, толсто-
толстостенная запаянная ам-
ампула или автоклав
1 ч, 100 °С, запаянная
ампула или автоклав
6 ч, кипячение с об-
обратным холодильни-
холодильником
30 мин, кипячение с
обратным холодиль-
холодильником
24 ч, кипячение с об-
обратным холодильни-
холодильником
10 мин, кипячение с
обратным холодиль-
холодильником
2 ч, 185 °С, автоклав
или запаянная
ампула
Т. пл. 103 °С (лиг-
(лигроин)
Т. кип. 51 °С
A3 мм рт. ст.)
Т. кип. 132 °С
B2 мм рт. ст.)
Т. пл. 147 °С (лиг-
(лигроин)
Т. пл. 196 °С (эта-
(этанол)
Т. пл. 262 °С (кси-
(ксилол)
Т. кип. 109 °С
A00 мм рт. ст.);
по20 1,4420
90
90
80
90
80
90
85
Для рационального обозначения бициклических систем, часто образующихся при диеновых синтезах, после приставки «бицикло» в
квадратных скобках указывают число углеродных атомов для всех трех мостиков между узловыми атомами. Затем следует название угле-
углеводорода, включающее все атомы бицикла. Счет начинают с головы мостика. Затем нумеруют самый длинный мостик, возвращаются к
ближайшему более короткому и, наконец, нумеруют последний мостик.
Бицикло D,4,01 декан
Вицикло [3,2,1] октан
Бицикло [2,2,2] октен-2
° Получают, перегоняя при нормальном давлении «дициклопентадиен».
в Перегоняют над небольшим количеством натрия.
После прибавления небольшого количества гидрохинона перегоняют, добавив в приемник также около 1% гидрохинона, который
предотвращает полимеризацию.
4. Присоединение к неактивированным кратным углерод-углеродным связям
403
Адцукты на основе олефиновых диенофилов могут быть ароматизованы при
отщеплении воды. Из оксазолов таким же путем можно получить пиридины.
Из оксазола и циклическиго ацеталя бутен-2-диола-1,4 такие реакции при-
применяют в техническом синтезе пиридоксина (витамин Вб), например [Г.7.536]:
н3с
OEt
о +
О
О
к
OEt
HsCXfc =
-Н2О
OEt
он
[Г.4.101]
+2Нг0 НзСyVCH20H
V
СН2ОН
4.5. ПРИСОЕДИНЕНИЕ, КАТАЛИЗИРУЕМОЕ МЕТАЛЛАМИ И
КОМПЛЕКСАМИ МЕТАЛЛОВ
4.5.1. РЕАКЦИИ ОЛЕФИНОВ И АЛКИНОВ В УСЛОВИЯХ ГОМОГЕННОГО КАТАЛИЗА
Катализ металлоорганическими комплексами позволяет осуществлять с оле-
финами и алкинами особые реакции создания связей С—С и С—Н, которые из-
за больших энергий активации без катализаторов реализовать, как правило, не
удается.
Существенной предпосылкой для катализа металлоорганическими комплек-
комплексами является активация ненасыщенного субстрата за счет образования коорди-
координационной связи с центральным атомом комплекса переходного металла. При
этом прежде всего образуется я-комплекс. Олефины образуют подобные
л-комплексы с солями и комплексами многих переходных металлов [например,
Fe, Co, Ni, Pd(II), Pt(II), Cu(I), Ag(I)].
По модели ЛКАО-МО координационная связь состоит из я-акцепторной
доли взаимодействия незанятой атомной орбитали металла с занятой
я-ВЗМО олефина, а также я-донорной доли взаимодействия между занятой
атомной орбиталью центрального атома и НСМО (я*-орбиталью) олефина
(рис. Г.4.102). Если преобладает я-акцепторная доля связи, то становится воз-
возможной нуклеофшьная атака, в случае сво-
свободного олефина неосуществимая.
Реакционную способность олефина и
направление реакции определяет распреде-
распределение электронной плотности в я-комплек-
се. Это распределение зависит от типа и
заряда переходного металла и от природы
лигандов. Часто собственно катализатор
возникает первым во время так называемой рис f 4 102 ОбразованИе
«стадии формирования» в реакционной сме- связей в олефиновых комплексах
си (см. схему [Г.4.115]). переходных металлов.
С =
404 Г Препаративная часть
В результате важнейших реакций олефинов, катализируемых комплексами
переходных металлов, получаются замещенные олефины или продукты присое-
присоединения к олефинам. Эти процессы осуществляются в виде сложной последова-
последовательности реакций, для которой характерен ряд типичных элементарных актов.
При синтезе замещенных олефинов связанная в л-комплексе молекула
олефина прежде всего формально внедряется между металлом и одним из лиган-
дов; при этом лиганд нуклеофильно присоединяется к р-положению олефина.
Подобное нуклеофильное присоединение лиганда {реакция внедрения) — важный
элементарный акт при катализе комплексами металлов:
\ нуклеофильное
у, присоединение i , Ц Р-элимини- ¦
L-/C-H лиганда ¦¦ м-С-Св- -В^^^ м-н + W [Г.4.103]
М \ [_/ I" v гтдрид-иона ^ / чх
Отрыв продукта замещения от центрального атома сопровождается элими-
элиминированием гидрид-иона из р-положения; р-элиминирование гидрид-иона
представляет собой следующий важный элементарный акт. Гидрид-ион взаимо-
взаимодействует с металлом.
По этой схеме осуществляется, например, промышленный синтез ацетальде-
гида из этилена и кислорода в присутствии каталитических количеств PdCl2 и
(вакер-процесс):
(H2C=CH2)PdCI2 *"'"• HOCH2CH2-PdCI
Н2С=СН2
-н2с=сн-он—»н3с-сн=о [Г.4.104]
+ 2CuCI2
- Pd@)
На первой стадии реакции происходит присоединение воды к активирован-
активированному палладием алкену. Образующийся гидроксипалладиевый комплекс пре-
претерпевает характерное р-гидридное элиминирование с образованием енола
ацетальдегида и Н—Pd—С1-частиц, которые распадаются на НС1 и Pd(O).
Pd(O) окисляется СиСЬ до Pd(II) и при этом образующаяся Cu(I) под действи-
действием кислорода снова превращается в Cu(II):
2CuCI + \ог + 2HCI - 2CuCI2 + Н2О [Г.4.105]
Вакер-окисление прежде всего можно применять к многочисленным терми-
терминальным алкенам, которые региоселективно приводят к метилкетонам. Метод
также может быть распространен на другие нуклеофил ы и применен для получе-
получения, например, винилацетата, акрилонитрила и винилхлорида.
Ф Общая методика вакер-окисления терминальных олефинов (табл. Г.4.106) [Tsuji J.,
Nagashima H., Nemoto H. Org.Synth. 1984, 62, 9.]
В трехгорлую колбу на 250 мл, снабженную капельной воронкой с компенсацией давле-
давления, помещают 10 ммоль PdCl2, 0,1 моль CuCl и 80 мл смеси диметилформамида и воды
4. Присоединение к неактивированным кратным углерод-углеродным связям
405
Таблица Г.4.106. Метилкетоны из терминальных олефинова
Продукт
Деканон-2
Додеканон-2
Октанон-2
Исходное
соединение
Децен-16
Додецен-1
Октен-1
Т. кип., °С
43—50 Aмм рт. ст.)
105-108 E мм рт. ст.)
Т.пл. 17-20
Т.пл. 168
«о25
1,4253
1,4348
1,4134
Выход, %
65
60
60
а Рекомендованный в методике хлорид медиA) CuCl реагирует селективнее, чем СиС12 (так
как продукт может хлорироваться).
6 Использовать свежеперегнанный.
(ДМФА: Н2О = 7:1).' В капельную воронку помещают 0,1 моль соответствующего олефина и
ко второму горлу колбы присоединяют ввод от баллона с кислородом (все пробки и шлифы
должны быть надежными!). Реакционную смесь для поглощения кислорода перемешивают
1 ч, затем медленно (~ 20 мин) при интенсивном перемешивании прибавляют из капельной
воронки соответствующий олефин, раствор в течение 15 мин из зеленого становится черным
и затем постепенно снова зеленым. После 24-часового выдерживания реакционной смеси на
холоду с 3 н. НС1 C00 мл) экстрагируют пять раз по 100 мл эфира. Объединенные эфирные вы-
вытяжки тщательно последовательно промывают 150 мл насыщенного раствора NaHCO3 и
150 мл насыщенного раствора NaCl и сушат над MgSC>4. После отфильтровывания осушителя
и удаления растворителя на роторном испарителе остаток фракционируют на колонке Вигре.
Неоднократное повторение стадии внедрения без отщепления продукта от
центрального атома приводит к олигомерам или полимерам.
Важнейшим примером является реакция полимеризации этилена при низ-
низком давлении (открыта Циглером и Наттой). В качестве каталитически актив-
активных систем применяют соединения переходных металлов, например трихлорид
титана, и металлоорганические соединения главных подгрупп I—III групп пери-
периодической системы, например (С2Нз)зА1. Сначала в результате алкилирования
образуется алкилтитановый комплекс, который затем присоединяет олефин на
свободное место, образуя я-комплекс:
TiCI,
RTiCI2
+ Н2С=СН2
RTi(H2C=CH2)CI2
[Г.4.107]
Поскольку титан (центральный атом) беден d-электронами, то внедрение
олефина по связи R—Ti осуществляется быстрее, чем р-элиминирование гидрид-
иона. Освободившееся место у центрального атома занимает новая молекула оле-
олефина. Данная реакция повторяется многократно с образованием полимера:
RTi(H2C=CH2)CI2 -
- R-CH2-CH2—TiCI2 +H2C~CHg. RCH2CH2—Ti(H2C-CH2)CI2 и т.д. [Г.4.108]
Диметилформамид (ДМФА) перед употреблением перегнать.
406 Г Препаративная часть
Полимеризация такого типа протекает стереоселективно (например, в
полипропилене каждый второй атом углерода является асимметрическим!):
_6н-сн2-сн-сн2-сн-
СН3 СН3 СНз l J
(В этой связи ознакомьтесь с атактической, синдиотактической и изотактической полиме-
полимеризацией и зависимостью свойств полимера от типа полимеризации!)
Если центральный атом богат (/-электронами (например, никель), то р-элиминирование
гидрид-иона облегчается и реакция приводит к димеризации олефина:
НгС> \ н2с=сн2 , Н2<"
^ /СН2 _ Nj_CH2_CH3 _1—!i_2_ L^
X
Н2С=СН2
L
1~^.,. -н2с=сн-сн2-сн3 \
NK - Ni-(CH2K-CH3
X X
Ф Получение полиэтилена. Описанный ниже катализатор (амиллитий — тетрахлорид ти-
титана) удобнее для полимеризации этилена при атмосферном давлении в лабораторных
условиях, чем самовоспламеняющийся катализатор триалкилалюминий — тетрахлорид
титана. Эксперимент удается только при условии тщательного удаления воды и кислорода из
реакционной массы.
Сначала из 400 мл петролейного эфира (т. кип. 60-80 °С), высушенного над КОН, удаляют
кислород, для чего его несколько минут нагревают с обратным холодильником до кипения,
пропуская ток свободного от кислорода азота (см. часть Е). После охлаждения 50 мл приготов-
приготовленного таким образом растворителя переносят в трехгорлую колбу на 250 мл, из которой воздух
также вытеснен свободным от кислорода азотом. Колба снабжена мешалкой, обратным холо-
холодильником, газоподводящей трубкой и капельной воронкой. В ходе всех операций медленно
пропускают ток азота, не содержащего Ог- В колбу помещают 3 г мелконарезанного лития и по
каплям при эффективном перемешивании добавляют 2 мл раствора, полученного из 18 г амил-
хлорида в 25 мл петролейного эфира (освобожденного от воздуха). После начала реакции, о чем
свидетельствует осаждение хлорида лития, добавляют по каплям в течение 20 мин (при эффек-
эффективном перемешивании и охлаждении смесью льда с солью) остаток раствора амилхлорида.
Перемешивание продолжают еще 2,5 ч, дают хлориду лития осесть и отбирают сухой пипеткой
со шприцем (ее предварительно промывают азотом) 30 мл находящегося над осадком раствора1,
который в токе азота разбавляют оставшимся свободным от воздуха петролейным эфиром в
трехгорлой колбе емкостью 500 мл (снабженной мешалкой, обратным холодильником, газо-
газоподводящей трубкой). Прибавляют 2 мл тетрахлорида титана; содержимое колбы после переме-
перемешивания в течение 20 мин представляет собой готовый катализатор. Теперь вместо азота пропус-
пропускают этилен2. Реакцию прекращают, когда выпавший полиэтилен начинает затруднять переме-
перемешивание; катализатор разрушают, добавляя 40 мл бутанола. Полиэтилен отфильтровывают и
промывают смесью концентрированной НС1 и метанола A:1) до обесцвечивания, а затем водой,
сушат в сушильном шкафу при 80 °С. Для очистки продукт растворяют в горячем тетралине или
декалине и оставляют на холоду. Выпадает полиэтилен с т. пл. 120—130 °С.
Катализируемое комплексами присоединение к олефинам в общем случае
включает следующие элементарные стадии:
1 Небольшие количества захваченного хлорида лития не мешают. Остаток реакционной
смеси и нерастворившийся литий осторожно разлагают этанолом.
2 Этилен, как и азот, очищают от кислорода (см. часть Е).
4. Присоединение к неактивированным кратным углерод-углеродным связям 407
Окислительное
присоединение
+ XY
Восстановительное
элиминирование
\ _ /
^ L-M-L
v_i-iv ГГ41111
'х 9"9 Y L1.4.111J
t
L—M-L
Реагент X—Y окислительно присоединяется к центральному атому, причем его окислитель-
окислительное и координационное числа увеличиваются вдвое. Это взаимодействие, являющееся реак-
реакцией син-присоединения, либо приводит к комплексам, условием образования которых явля-
является разрыв связи между X и Y (например, Н—Н, H-Hal, H-OR, H-SR, Hal-Hal), либо X-Y
без разрыва связи занимает два соседних координационных центра (например, -О—О—,
—S-S-, O=NR, RN=NR и др.). Тенденция к окислительному присоединению уменьшается в
ряду Hal-Hal>H-H»R3C-H.
После реакции с координационно связанным субстратом конечный продукт восстанови-
восстановительно элиминируется из комплекса. По этой схеме протекают, например, реакции гомогенно-
гомогенного гидрирования:
\ / Катализатор _\ ' _
с=с + н2 - ^-с-с— [г.4.112]
' х н н
В качестве промежуточных продуктов при этом образуются гидридные комплексы (X = Y = Н),
возникающие при окислительном присоединении Н2, например к комплексам родия или
иридия.
Катализатор Уилкинсона — трис(трифенилфосфин)родийхлорид, получение которого пред-
представлено реакцией [Г.4.113], широко применяется как в промышленности, так и в лаборатории.
PMVR<C, ^± ™3Р><С, =^ Ph3P-R^I 5 = растворители
Если в качестве катализатора применяют оптически активные фосфины, то таким путем
удается осуществлять асимметрическое гидрирование, например, получение (Zl-a-ацетил-
амидо-3,4-дигидроксикоричной кислоты с оптической чистотой более 90% в ходе синтеза
ДОФА [Ь-р-C,4-дигидроксифенил)-а-аланина], применяющегося при лечении болезни
Паркинсона.
Большое промышленное значение имеют реакции карбонилирования. Так, при гидрофор-
милировании (оксосинтез) при взаимодействии олефинов со смесью Нг/СО на кобальтовом
или родиевом катализаторе получают альдегиды или спирты:
* R-CH2-CH2-CHO -^- R-CH2-CH2-CH2OH
R-CH=CH2 + CO + Н2 (^ [Г.4.114]
> R-CH-CHO ^~ R-CH-CH2OH
СН3 СН3
Реакция протекает по общему механизму (схема [Г. 111]). После формирования катализа-
катализатора и образования л-комплекса происходят две реакции внедрения; при этом сначала образу-
образуется связь металл-алкил, а в нее внедряется СО. После окислительного присоединения Н2 к
комплексу ацилметалл происходит восстановительное элиминирование альдегида и регенера-
регенерация катализатора:
408 Г Препаративная часть
Со2(СО)8 + Н 2 ===== 2 НСо(СОL ===== 2 НСо(СОK + 2 СО
RHC=CH2 + НСо(СОK ===== HCo(RCH=CH2)(COK ===== RCH2-CH2-Co(COK
RCH2-CH2-Co(COK + СО ===== RCH2-CH2-Co(COL ====== RCH2-CH2-CO-Co(COK
RCH2-CH2-CO-Co(COK + H2 RCH2-CH2-CHO + HCo(COK ГГ 4 1151
В ходе последующей реакции альдегид может гидрироваться.
Смеси жирных спиртов, получаемые таким путем в промышленности, можно затем пере-
переработать, например, в сульфаты жирных спиртов (синтетические моющие средства); см. схему
[Г.2.53] и разд. Г,7.1.7.1. Карбонилирование метанола на родиевом катализаторе приводит к
уксусной кислоте с почти количественным выходом.
Циклоолигомеризацию олефинов и ацетилена, имеющую большое значение для
синтеза сложных олефинов, средних циклоолефинов и циклопарафинов, мож-
можно рассмотреть на примере бутадиена. Окислительное присоединение бутадие-
бутадиена к №°-комплексу приводит при образовании связи С—С к системе бис-тг-ал-
лил-никель:
+ NiL _
-NIL" =^" [Г.4.116]
В зависимости от пространственных и электронных свойств лигандов (L,L')
промежуточный продукт I (схема [Г.4.116]) реагирует по-разному. Восстанови-
Восстановительное элиминирование с разрушением №°-комплекса приводит к циклоокта-
диену-1,5 из которого каталитическим дегидрированием получают циклоокта-
тетраен. В комплекс I может внедриться еще одна молекула бутадиена; тогда по-
получают (в том числе и в промышленном масштабе) циклододекатриен-1,5,9.
[ГА117]
В приведенной ниже методике образуется транс,транс,цис-юомер циклодо-
декатриена.
Дальнейшая обработка по известным методикам приводит к циклододекано-
ну (исходному веществу в синтезе найлона-12), к додекандикарбоновой-1,12
кислоте (использующейся как кислотный компонент при синтезе полиэфиров,
полиамидов и др.).
Ацетилен на никелевом катализаторе можно тетрамеризовать в циклоокта-
тетраен:
4 нснсн ' 2-
В промышленности таким путем из азелаиновой кислоты получают циклододекадиен-1,5,
который применяется как промежуточный продукт при получении алкидных смол, полиамидов,
ударопрочных полиэфиров и в качестве размягчителя.
4. Присоединение к неактивированным кратным углерод-углеродным связям 409
(Пл Получение транс,/я/>анс,г<иоциклододекатриена \BreilH. et al. Makromol. Chem., 1963,69, 18]
Внимание'. Все используемые реактивы не должны содержать даже следов вла-
влаги (см. часть Е). Реакцию проводят в атмосфере сухого инертного газа (чистого
азота или аргона).
Прибор состоит из однолитровой четырехгорлой колбы, снабженной термометром, ме-
мешалкой, газоподводящей трубкой и обратным холодильником, который присоединен к счет-
счетчику пузырьков, заполненному вазелиновым маслом. В 400 мл безводного бензола суспенди-
суспендируют тонкую взвесь 160 ммоль гидрида кальция и 40 ммоль безводного А1С13. Перемешивание
реакционной смеси в атмосфере инертного газа продолжают 3 ч, затем прибавляют
20 ммоль тетрахлорида титана. После получасового интенсивного перемешивания доводят
температуру до 40 °С, продолжая перемешивание при сильном охлаждении (баня — лед с
солью), подают ток сухого бутадиена таким образом, чтобы счетчик пузырьков не регистриро-
регистрировал выделение газа. Через 40 мин прекращают подачу газа в реакционную смесь и для заверше-
завершения реакции перемешивают еще 1 ч. Затем добавляют 50 мл ацетона и отфильтровывают вы-
выпавший полибутадиен. Фильтрат фракционируют при 20 мм рт. ст., собирая основной продукт
при 110-120 °С.
Чистоту определяют газохроматографически и в случае необходимости перегоняют еще
раз на 20-сантиметровой колонке Вигре. Т. кип. 115 "С B9 мм рт. ст.); nD20 1,5078; выход
80-85%.
(ГП) Получение циклооктатетраена из ацетилена [Reppe W. et al. LiebigsAnn. Chem., 1948,560, 1]
Щ Внимание*. Обратите внимание на указания по работе с ацетиленом (см. разд.
й Г,4.2.2). Реакцию можно проводить только в том случае, если соблюдаются все
щ меры предосторожности, на которые там обращалось внимание (в особеннос-
|| ти необходим предохранительный вентиль между автоклавом и баллоном).
В двухлитровый автоклав с магнитной мешалкой (или во стряхиваемый автоклав, выдер-
выдерживающий большое давление) помещают смесь 20 г ацетилацетоната никеля (см. часть Е),
60 гтонкоизмельченного карбида кальция и 500 мл безводного тетрагидрофурана (см. часть Е).
После вытеснения кислорода воздуха путем многократного промывания автоклава чистым
азотом автоклав нагревают до 70 °С (датчик температуры введен внутрь автоклава) при началь-
начальном давлении азота 5 атм. После достижения заданной температуры начинают подачу ацети-
ацетилена в автоклав (до давления 15-25 атм). Подачу ацетилена возобновляют несколько раз по
мере снижения давления, пока ацетилен не перестанет поглощаться. После окончания реак-
реакции C0—60 ч) автоклав охлаждают и давление спускают (выпускают газ через крышку), содер-
содержимое фильтруют и фракционируют при 60 мм рт. ст. на 20-сантиметровой колонке Вигре.
После отгонки растворителя и небольшого количества бензола циклооктатетраен перегоняет-
перегоняется в виде желто-золотистой жидкости при 64—65 °С. Перегонку в вакууме повторяют.
Т. кип. 64-65 °С F0 мм рт. ст.) или 42-45,5 °С A7 мм рт. ст.); nD201,5390; выход 200 г.
Через комплексы олефинов с переходными металлами протекает также промышленно важный
синтез олефинов (метатезис, или диспропорционированиё), например, катализируемое вольфрамом
или молибденом диспропорционированиё двух молекул пропена на этилен и бутен-2:
2 Н3С-СН=СН2 ^=^ Н3С-СН=СН-СН3 + Н2С = СН2 [Г.4.119]
Промежуточными продуктами являются, вероятно, комплексы металл-кар-
бен LXM=CH-R:
R
\=МЦ R\ R.
+ ^=^ | | =^= J + II [Г.4.120]
/=\ ?¦ о1 R' R1
410 Г Препаративная часть
Каталитический метатезис замыкания цикла а,со-диолефинов приводит к
циклическим продуктам, при этом могут быть получены почти все большие
циклы. Движущей силой реакции является отщепление летучего этилена. В ка-
качестве катализатора используют преимущественно рутениевые или молибдено-
молибденовые комплексы, которые in situ до стадии инициирования могут превращаться в
реакционоспособные частицы [М]=СНг, совместимые с большим числом
других функциональных групп.
^ П + II [Г-4.121]
4.5.2. ГЕТЕРОГЕННОЕ КАТАЛИТИЧЕСКОЕ ГИДРИРОВАНИЕ
Присоединение водорода к кратным углерод-углеродным связям С—С протекает
легко и используется очень часто. Обычная неактивированная кратная связь не
восстанавливается реагентами, пригодными для восстановления активирован-
активированных двойных связей (например, цинком в соляной кислоте, амальгамой натрия,
натрием в спирте) (см. разд. Г,7.1.7). Напротив, и активированные, и неактиви-
неактивированные двойные связи, а также ацетилены можно гидрировать газообразным
водородом в присутствии катализаторов. В качестве катализаторов гидрирования
применяют металлы побочных подгрупп, их оксиды и сульфиды. В лабораторных
условиях используют в основном металлы.
Катализатор должен быть тонко измельчен. Для этого существуют различные способы, и
получаемые вещества известны под следующими названиями:
а) черни; металл осаждают из раствора его соли восстановлением; такой катализатор
должен быть свежеприготовленным;
б) катализаторы Адамса; платину и палладий получают тонкоизмельченными из све-
свежеприготовленных оксидов, восстанавливая их водородом непосредственно в реак-
реакционном сосуде;
в) скелетные катализаторы (катализаторы Ренея); активный катализатор получают в виде
«губчатого металла» из двойных сплавов (никеля, железа, меди или кобальта с алюминием
или кремнием) путем растворения одного компонента сплава в кислоте или щелочи; не-
небольшая примесь растворяемого металла в катализаторе часто повышает его активность;
г) катализаторы на носителях; металл в виде черни может быть осажден на поверхности но-
носителя; в этом случае достаточны лишь небольшие количества дорогостоящего благород-
благородного металла, поэтому такие катализаторы нашли широкое применение в промышлен-
промышленности; носитель, не обладающий сам по себе каталитическими свойствами, часто оказы-
оказывает синергетический эффект (носителями служат, например, уголь, диоксид кремния,
оксид алюминия, сульфаты и карбонаты щелочноземельных металлов);
д) оксидные и сульфидные катализаторы; эти катализаторы из-за устойчивости к катали-
заторным ядам и дешевизны находят применение главным образом в промышлен-
промышленности (например, хромиты меди, цинка, сульфиды молибдена и вольфрама и др.).
Механизм гетерогенного гидрирования родствен механизму соответствую-
соответствующего гомогенного процесса. На поверхности металлического катализатора
адсорбируется как водород, так и олефин с образованием гидридных или
олефиновых комплексов соответственно (хемосорбция). При внедрении комп-
комплексно-связанного олефина по связи М—Н и восстановлении алкил-М-комп-
лекса образуется насыщенный углеводород:
4. Присоединение к неактивированным кратным углерод-углеродным связям 411
ч \/ \' '/
Н /C|CV н Н -С Н н Н [Г.4.122]
-м—м—м— ^=^ —м—м—м— =^=^ —м—м—м—
Реакции каталитического гидрирования, особенно в нейтральных или кислых растворах,
как правило, протекают по схеме сын-присоединения. Так, из салициловой кислоты и ее эфи-
ров на платине или скелетном никеле образуются преимущественно ^мс-гексагидросалицило-
вая кислота и ее эфиры.
Гидрирование с благородными металлами в качестве катализаторов протека-
протекает в кислой среде быстрее, чем в щелочной, а в полярном растворителе быстрее,
чем в неполярном.
Реакционная способность олефинов различается мало. Особенно легко гид-
гидрируется ацетиленовая связь, причем, если после поглощения рассчитанного
количества водорода реакцию прекратить, можно добиться селективного гидри-
гидрирования до олефинов. В промышленности целесообразно применять с этой
целью «частично отравленный» солями тяжелых металлов или хинолином пал-
ладиевый катализатор {катализатор Лшдлара). Вследствие высокой устойчи-
устойчивости ароматических систем для гидрирования ароматических и гетероцикли-
гетероциклических соединений необходимы более жесткие условия по сравнению с гидри-
гидрированием простых олефинов. Конденсированные ароматические соединения
гидрируются несколько легче, а именно вначале одно кольцо, затем при более
жестких условиях другие кольца. У ароматических соединений с ненасыщенны-
ненасыщенными боковыми цепями последние легко гидрируются.
О гидрировании других ненасыщенных систем (нитрозо-, нитро-, карбо-
карбонильные соединения, азометины, нитрилы) и его значении см. разд. Г,7 и Г,8.
О гидрировании двойных углерод-углеродных связей в присутствии карбо-
карбонильных групп см. разд. Г.7.1.7 и табл. Г.4.126.
Практическое выполнение каталитического гидрирования
а) Катализаторы. В лабораториях для гидрирования кратных углерод-угле-
углерод-углеродных связей применяют чаще всего платину1, палладий и скелетный никель.
Самый активный из этих катализаторов — платина; с ее помощью можно гидри-
гидрировать при комнатной температуре без давления даже устойчивые ароматичес-
ароматические связи. Скелетный никель и палладий (в виде оксида палладия или осажден-
осажденный на активированном угле, сульфате бария, стронция или карбонате кальция)
менее активны, однако дают возможность гидрировать при комнатной темпера-
температуре неароматические кратные связи. Благодаря этому становится возможным
селективное гидрирование, например, стирола до этилбензола. Гидрирование
ароматических структур на менее активном катализаторе, например на скелет-
скелетном никеле, требует повышенных температур (порядка -150 °С) и высокого
давления водорода A50—200 атм).
В известных границах активность катализаторов зависит от условий получе-
получения. Выбор катализатора определяется устойчивостью гидрируемого вещества в
В виде оксида платины (катализатор Адамса, см. выше).
412
Г Препаративная часть
Утка» для гидрирования
Магнит
в стеклянной
оболочке
а От баллона б
Рис. Г.4.123. Схема прибора для каталитического гидрирования.
а — прибор для гидрирования со встряхиванием;
б — прибор для гидрирования с магнитной мешалкой.
данных условиях (термическая стабильность, устойчивость в щелочной или кис-
кислой среде), конструкцией прибора и ценой.
Металлические катализаторы очень чувствительны1 к контактным ядам,
особенно к серо- и галогенсодержащим веществам. Следовательно, должны
применяться максимально чистые вещества и растворители2.
б) Растворители. В качестве растворителей при каталитическом гидрирова-
гидрировании применяют чаще всего воду, этиловый и метиловый спирты, этилацетат, ди-
оксан3, ледяную уксусную кислоту и смеси этих растворителей. Жидкие вещест-
вещества можно гидрировать без добавления растворителя. Гидрирование на оксиде
платины надо проводить в нейтральной, еще лучше кислой, среде (в уксусной
кислоте или в присутствии минеральной кислоты). При работе со скелетным
никелем предпочтительна нейтральная или щелочная среда.
в) Аппаратура. Вещество, растворитель, катализатор хорошо встряхивают
или перемешивают в атмосфере водорода, чтобы осуществить максимальный
контакт катализатора и водорода. По этой же причине аппаратуру нужно запол-
заполнять не полностью. Гидрирование проводят в аппарате для встряхивания либо в
автоклаве.
Аппарат для встряхивания присоединяют к резервуару с водородом (газомет-
(газометру) под небольшим избыточным давлением так, что понижение давления водо-'
1 Пример использования преднамеренного (частичного) отравления: восстановление по
Розенмунду (см. разд. Г,7.1.7.1), а также вышеупомянутое частичное гидридование ацетилена.
2 Кипячением вещества и растворителя со скелетным никелем перед гидрированием
освобождаются от незначительных количеств катализаторного яда. Поэтому для гидрирова-
гидрирования необходимо использовать только свежеприготовленный катализатор.
3 При температуре не выше 150 °С; в противном случае диоксан взрывоопасен.
4. Присоединение к неактивированным кратным углерод-углеродным связям 413
рода в результате поглощения постоянно компенсируется (рис. Г.4.123, а). Если
позволяет конструкция прибора, можно работать под давлением 1—2 атм при
нагревании или на холоду.
Для многих целей подходящим является сосуд для гидрирования с магнит-
магнитной мешалкой (рис. Г.4.123, б). Он представляет собой колбу Эрленмейера на
300 мл с круглым дном и шлифом НШ 29. Такой сосуд можно безопасно эвакуи-
эвакуировать (до 100 мм рт. ст.) и вводить до 200 мл исходного раствора для гидрирова-
гидрирования C0—50 г вещества).
Если необходимы большие загрузки, гидрирование проводят главным обра-
образом в автоклаве (см. разд. А, 1.8.2). С повышением рабочего давления увеличива-
увеличивается скорость гидрирования. При этом со значительно меньшими количествами
катализатора достигают такой же скорости гидрирования, как при работе без
давления. Повышение температуры для увеличения скорости, напротив, не ре-
рекомендуется, так как иногда это приводит к изменению направления гидриро-
гидрирования (например, к гидрированию ароматического кольца), а следовательно, к
другим продуктам.
(у^) Общая методика каталитического гидрирования (табл. Г.4.124)
Необходимо соблюдать правила техники безопасности (разд. А.1.8.2). Перед
нагреванием автоклава надо оценить ожидаемое максимальное давление
(газовые законы!). Водород нельзя выпускать в рабочее помещение, а нужно
выводить в атмосферу через стальную капиллярную трубку. В присутствии ме-
металлических катализаторов смеси водорода с воздухом взрывоопасны, поэто-
поэтому катализатор должен быть покрыт жидкостью, пока воздух не будет полностью
удален из сосуда для гидрирования. По этой же причине его вносят в аппарат под
слоем жидкости. Остатки катализатора нельзя выбрасывать в ящик для бумаг, так
как, например, скелетный никель легко самовоспламеняется!
Приготовление катализатора см. часть Е.
В сосуд для гидрирования помещают гидрируемое соединение или его раствор и добавля-
добавляют катализатор в следующих количествах (относительно массы гидрируемого соединения):
оксид платины 0,5-1%, более дешевый скелетный никель 5-10%, палладий на угле 5%'. Сосуд
для гидрирования трижды эвакуируют для удаления кислорода воздуха, после каждого раза его
заполняют водородом или пропускают через прибор 10 мин инертный газ (азот). При гидри-
гидрировании в автоклаве его дважды заполняют водородом и затем спускают давление.
После этого включают мешалку или аппарат для встряхивания и сразу же записывают объ-
объем (при работе с аппаратом для встряхивания) или давление (при работе с автоклавом) водо-
водорода и время. Часто водород потребляется вначале быстро, так как им насыщается раствор.
При применении оксида платины идет восстановление катализатора, что сопровождается не-
небольшим индукционным периодом, который, однако, не должен превышать 5—10 мин, иначе
это говорит о том, что оксид платины плохого качества. Записывают потребление водорода во
времени и строят соответствующий график. Перемешивают (или встряхивают) до тех пор, по-
пока гидрирование не закончится (т. е. когда кривая гидрирования станет параллельной оси вре-
времени). Если кривая насыщения не достигается, то это говорит о негерметичности прибора или
о гидрировании других частей молекулы.
1 Скелетный никель и другие активные катализаторы следует всегда хранить под слоем
растворителя. Так как катализаторы лучше всего применять свежеприготовленными, реко-
рекомендуется готовить нужное количество непосредственно перед опытом, например путем
выщелачивания никель-алюминиевого сплава с известным содержанием никеля.
Таблица Г.4.124. Каталитическое гидрирование кратных углерод-углеродных связей
Продукт реакции
Этилбензол
З-Фенилпропионо-
вая кислота
З-Фенилпропионо-
вая кислота
Янтарная кислота
Бутандиол-1,4
4-Фенилбутанон-2
1,5-Дифенилпента-
нон-3
1,3-Дифенилпропа-
нон
Метилизобутилке-
тон
Исходное вещество
Стирол
Коричная кислота
Калиевая соль ко-
коричной кислоты
Малеиновая кис-
кислота
Бутин-2-диол-1,4
Бензилиденацетон
Дибензилиденаце-
тон
Бензилиденацето-
фенон (халкон)
Мезитилоксид
(изопропилиден-
ацетон)
Катализа-
Катализатор
Ni
PtO2
Ni
PtO2
Pd/C, Ni
PtO2> Ni,
Pd/C
Тоже
Тоже
Тоже
Давление,
температура
а
а •
а
а
а
а
а
а
а
Растворитель
(содержание,
мл/моль)
Без раствори -
теля
Метанол (800)
Вода (900)
Этанол A500)
Метанол D00)
Этанол A50)
Этанол B500)
(суспензия)
Этилацетат
A500)
Без раствори -
теля
Физические
константы
Т. кип. 136 "С;
па* 1,4959
Т. пл. 47 °С
(разб. НС1)
Т. пл. 47 °С
(разб. НС1)
Т. пл. 185°С
(вода)
Т. кип. 129 °С
B3 мм рт. ст.)
Т. кип. 115 "С
A2 мм рт. ст.);
по20 1,5124
Т. кип. 209 °С
A0 мм рт. ст.);
т пл 13 °С-
по* 1 5586
Т. пл. 73 °С
(этанол)
Т. кип. 115°С;
по20 1,3959
Выход
80
90
90
95
92
95
80
90
95
Примечания
Побочно образуется
немного метилового
эфира фенилпропио-
новой кислоты
Коричную кислоту Aмоль)
растворяют в экви-
эквивалентном количестве
едкого кали+900 мл
воды. После отделе-
отделения катализатора
подкисляют НС1 и
отфильтровывают
Нейтральный никель
Ренея инактивируют
0,1%-ной уксусной
кислотой, чтобы
предупредить вос-
восстановление карбо-
карбонильной группы
(разд. Г.7.1.7.1). Ка-
Катализатор, получен-
полученный из 1 г сплава,
промывают 2 раза
водой порциями по
15 мл и 4 раза 1%-ной
уксусной кислотой
2-Метилциклогек-
санол
Дигидрорезорцин
(циклогександион-1,3)
Метиловый эфир
Ч«с-гексагидроса-
лициловой
кислоты
ЧНС-Декалинол-2
(декагидронафтали-
нол-2)
о-Крезол
Мононатриевая
соль резорцина
Метиловый эфир
салициловой кис-
кислоты
Р-Нафтол
Ni
Ni
Ni (нейт-
(нейтральный)
Ni
10—20 МПа,
комнатная
температура
20 МПа,
135 °С
6 МПа, 90 "С
Без раствори-
растворителя
Вода B00)
Метанол F00)
Этанол
Т. кип. 68 вС
A2 мм рт. ст.);
лпм 1,4636
Т. пл. 104 °С
(этилацетат)
Т. кип. 97 °С
(8 мм рт. ст.);
Ло20 1,4665
Т. кип. 115—
128 °С
A4 мм рт. ст.);
лс20 1,5120
90
95
80
50
Образуется смесь изо-
изомеров с преоблада-
преобладанием G0%) DL-транс-
соединения. О катализаторе
см. предыдущее примечание
Резорцин растворить
в водном растворе
NaOH A,2 моль
NaOH на 1 моль ре-
резорцина). Обязательно
гидрировать до конца,
так как в противном
случае конечный
продукт не кристал-
кристаллизуется. После
отфильтровывания
катализатора фильт-
фильтрат подкисляют НС1,
охлаждают до 0 °С
и отсасывают
Таблица Г.4.124. Продолжение
Продукт реакции
Диметилацеталь
циклогексилаце-
тальдегида
Пиперидин8
Метилциклогексан
1,2-Диметилцикло-
гексан
1,3-Диметилцикло-
гексан
1,4-Диметилцикло-
гексан
Исходное вещество
Диметилацеталь
фенилацетальде-
гида
Пиридинг
Толуол
о-Ксилол
ж-Ксилол
л-Ксилол
Катали-
Катализатор
N1
Ni
д
д
д
д
Давление,
температура
а
20 МПа,
150°С
4 МПа,
250 °С
То же
> »
Растворитель
(содержание,
мл/моль)
Без раство-
растворителя
То же
» >
» »
» »
> »
Физические
константы
Т. кип. 80—82 "С
A2 мм рт. ст.);
/id20 1,4458
Т. кип. 106 °С;
/to20 1,4530
Т. кип. 101 °С;
/id20 1,4230
123—130 °С
120—125 °С
118—125 "С
Выход
95
80
99
95
95
95
Примечания
Смесь изомеров с
преобладанием транс-
трансизомера
Смесь изомеров с
преобладанием цис-
изомера
Смесь изомеров с
преобладанием транс-
трансизомера
а Комнатная температура и небольшое избыточное давление (работать в приборах, представленных на рис. Г.4.123 и Г.4.125).
6 цис относительно связей в кольце; смесь изомеров из цис-цис- и транс-цис-соединений.
а Перед перегонкой на 30-сантиметровой колонке Вигре еще раз высушивают над КОН. По показателю преломления рассчитывают содержание
пиридина (т. кип. 115 °С; nD201,5100) и сравнивают с результатами газохроматографического анализа.
г Пиридиккне должен содержать катализаторных ядов; см. часть Е.
д В качестве катализатора применяют Pt-катализатор риформинга.
4. Присоединение к неактивированным кратным углерод-углеродным связям
417
При проведении частичного гидрирования процесс прекращают, как только появляется
отчетливый перелом на кривой гидрирования.
По окончании гидрирования прибор, в котором происходило перемешивание, снова эваку-
эвакуируют или промывают инертным газом (если использовался автоклав, из него спускают остаточ-
остаточное давление), и катализатор отфильтровывают через пористую пластинку или через хороший
плотный фильтр. Надо обратить внимание на то, что тонкоизмельченный, не смоченный раст-
растворителем металл пирофорен, поэтому катализатор должен всегда содержаться влажным. Его
можно использовать повторно для тех же целей. Если это дорогой катализатор из благородного
металла, то его надо собирать столь же тщательно, как и в количественном анализе.
После упаривания растворителя остаток перегоняют или соответственно перекристалли-
зовывают.
Если в табл. Г.4.124 отсутствуют особые указания, то гидрирование проводят при нормаль-
нормальном давлении и комнатной температуре. При больших загрузках и в этих случаях целесообраз-
целесообразно работать в автоклаве при повышенном давлении.
Полумикрогидрирование удобно проводить в приборе, изображенном на рис. Г.4.123, б.
Получение $-холестанола гидрированием холестерина на платине: Брюс В.,
РоллеДж. В. В сб.: Синтезы органических препаратов. Сб. 2. М.: ИЛ, 1949, с. 195.
Каталитическое гидрирование имеет также значение как аналитический ме-
метод количественного определения кратных связей. Прибор, описание которого
приведено ниже, позволяет в большинстве случаев получать достаточно точные
данные (рис. Г.4.125).
Вакуумный
кран 4
Колба
с веществом
Сосуд для
регулирования
уровня
Кран 2 Кран 1
[Г ТГ К вакуумному
y ° насосу
К источнику
водорода
Магнитная
мешалка
КранЗ
Рис. Г.4.125. Схема простой установки для количественного гидрирования.
418
Г Препаративная часть
Таблица Г.4.126. Примеры промышленно важных процессов каталитического гид-
гидрирования кратных углерод-углеродных связей
Продукт гидрирования
Жиры
Бутандиол-1,4
Масляный алльдегид
Циклогексанол
Циклогексан
Циклогексанкарбоновая
кислота
Циклододекан
Тетрагидронафталин
Декагидронафталин (декалин)
Пергидроаценафтен
Исходное вещество
Растительные масла
Бутин-2-диол-1,4
Кротоновый альдегид
Фенол
Бензол
Бензойная кислота
Циклододекатриен
Нафталин
Нафталин
Аценафтен
Применение
Маргарин
-> Полиэфир, полиуретан
-> Тетрагидрофуран (раствори-
(растворитель) -> политетраметиленгли-
коль N-метилпирролидон (экст-
рагент)
-> у-Бутиролактон -> N-винил-
пирролидон
-> и-Бутанол
-> 2-Этилгексанол
-> Циклогексанон
Растворитель
-> Нитроциклогексан -> цикло-
гексаноноксим -> найлон 6
->Аципиновая кислота -» найлон 66
Е-Капролактам -> найлон 6
-> Смесь циклододеканола и
циклододеканона -> циклододе-
каноноксим -> полиамиды (най-
(найлон 12)
-> Додекан-1,12-дикислота
-> Полиэфиры
Растворитель, переносчик водо-
водорода
-> а-Тетралон -> а-Нафтол
Растворитель
-> Адамантан -> термостойкие
полиэфиры
Ф Количественное гидрирование. В толстостенную колбу на 70 мл помещают 15 мл чистой
ледяной уксусной кислоты и примерно 0,1 г оксида платины или 2 мл спиртовой суспен-
суспензии выпавшего при получении скелетного никеля. Точную навеску (около 20 мг) неиз-
неизвестного вещества помещают в специальную колбу (взвешивание производится непосред-
непосредственно в этой колбе), вещество растворяют в 3-5 мл ледяной уксусной кислоты или спирта и
присоединяют к сосуду для гидрирования. Затем при открытом кране 3 опускают грушу, под-
поддерживающую необходимый уровень (кран /должен соединять прибор с атмосферой и закры-
закрывать выход к вакуумному насосу, кран 2 должен соединять прибор с источником водорода и
быть открытым на атмосферу); при этом из бюретки и восходящей трубки уходит вода. Затем
закрывают кран 3 и возвращают грушу в самое высокое положение. Шлифы должны быть со-
совершенно светлыми и прозрачными (смазка Рамзая, силиконовая или аналогичная по вязкос-
вязкости). Резиновое соединение с водородным баллоном имеет клапан Бунзена (см. разд. А,1.6).
Прибор трижды откачивают, каждый раз заполняя его водородом (откачивание: кранами 1 и 2
соединяют прибор с вакуумным насосом и закрывают соединения с источником водорода и с
атмосферой; заполнение водородом: открывают баллон, водород выходит через вентиль Бунзе-
Бунзена). Затем кран / поворачивают так, чтобы вакуумный насос был открыт на атмосферу, а через
4. Присоединение к неактивированным кратным углерод-углеродным связям 419
кран 2 медленно впускают в прибор водород. Открывая кран 3, несколько поднимают уровень
воды в восходящей трубке; затем закрывают кран 2 (соединив источник водорода с простран-
пространством между кранами 1 и 2), а кран 4 на короткое время открывают, пока жидкость в бюретке
не поднимается настолько высоко, чтобы мениск лежал в градуированной части бюретки. При
отчетах объема высота уровней в трубке и бюретке должна быть одинаковой. Включают магнит-
магнитную мешалку и гидрируют сначала катализатор, поддерживая давление при помощи столба во-
воды. Гидрирование продолжают, пока водород не перестанет потребляться. Перемешивают еще
10 мин, при этом объем должен быть постоянным. Его точно отсчитывают и подают анализи-
анализируемый раствор в суспензию катализатора, переворачивая грушевидную колбу с веществом на
180 °С. Остаток на стенках груши не дает заметной ошибки. Затем снова гидрируют до постоян-
постоянного объема и снимают показания уровня в бюретке. Все отсчеты надо проводить при одинако-
одинаковых условиях (температура и давление). Обычно достаточно проводить измерения при комнат-
комнатной температуре, которая остается практически постоянной. При расчете поглощенного водо-
водорода надо из показаний давления (барометр) вычесть давление насыщенных водяных паров при
соответствующей температуре.
Каталитическое гидрирование кратных углерод-углеродных связей имеет большое значе-
значение в промышленности. Его проводят обычно в газовой фазе, чтобы процесс был непрерыв-
непрерывным. Важнейшие примеры приведены в табл. Г.4.126.
(Ознакомьтесь с промышленно важными методами гидрирования угля, нефти, смол и
монооксида углерода.)
4.6. ЛИТЕРАТУРА
Обзор реакций олефинов и ацетиленов
Азингер Ф. Химия и технология моноолефинов. — М.: Гостоптехиздат, 1960.
Bergmann E. D. The Chemistry of Acetylene and Related Compounds. — Interscience, New
York, 1948.
Houben-Weyl, Bd. 5/lb, 1972, S. 946-1117.
Raphael R. A. Acetylenic Compound in Organic Syntheses. — Butterworth, London, 1955.
The Chemistry of Alkenes. S. Patai (ed.). — Intersciense Publischers, London, New York,
Sydney, 1964.
The Chemistry of Double-Bonded Functional Groups. S. Patai (ed.). — Wiley, Chichester, 1977.
The Chemistry of Triple-Bonded Functional Groups. S. Patai (ed.). — Wiley, Chichester, 1994.
The Chemistry of Carbon-Carbon Triple Bond. S.Patai (ed.). — Wiley, Chichester, 1978.
Теоретическая трактовка реакций присоединения и циклоприсоединения
DewarM. J. S. Angew. Chem., 1971, 83, 859-875.
EpiotisN. D. Angew. Chem., 1974, 86, 825-855.
Halevi E. A. Angew. Chem., 1976, 88, 664-679.
Houk K. N. In: Topics in Current Chemistry. Bd. 79. — Springer-Verlag, Berlin, Heidelberg,
New York, 1979, pp. 1-40.
Fleming I. Grenzorbitale und Reaktionen organischer Verbindungen. — Verlag Chemie,
Weinheim/Bergstr., New York, 1979.
Gilhirst T. L., Storr R. С Organic Reactions and Orbital Symmetry. — Cambridge University
Press, Cambridge, 1979.
Halivi E. A. Angew. Chem. 1976, 88, 664-679.
Hemdon W. С Chem. Rew. 1972, 72, 157-179.
Trong Anh N. Die Woodward-Hoffmann-Regeln und ihre Anwendung. — Verlag Chemie,
Weinheim, 1972.
420 Г Препаративная часть
Вудворд Р., Хоффман Р. Сохранение орбитальной симметрии. — М.: Мир, 1971; Angew.
Chem., 1969, 81, 797-869.
Электрофильное присоединение
De la Маге Р. В. D., Bolton R. Electrophilic Additions to Unsaturated Systems. — Elsevier,
Amsterdam, 1982.
Присоединение воды и протонных кислот к олефинам и ацетиленам
Gilbert E. Е. Sulfonation and Related Reactions. — Interscience, New York, 1965.
Кренцель Б. А. Усп. хим., 1951, 20, 759-775.
RoedigA. In: Houben-Weyl, Bd. 5/4, 1960, S. 102-132, 535-539.
Stroh R. In: Houben-Weyl, Bd. 5/3, 1962, S. 813-825.
Присоединение синильной кислоты к олефинам и ацетиленам
KurtsP. In: Houben-Weyl, Bd. 8, 1952, S. 265-274.
Присоединение галогенов, хлорноватистой, бромноватистой и
иодноватистой кислот и их сложных эфиров к олефинам и ацетиленам
De la Mare Р. В. D. Electropfilic Halogenation. — Cambridge University Press, London,
1976.
RoedigA. In: Houben-Weyl, Bd. 5/4, 1960, S. 38-100, 133-151, 530-535, 540-547.
Stroh R. In: Houben-Weyl, Bd. 5/3, 1962, S. 529-556, 768-780.
Вьюнов К. А., ГинакА. И. Усп. хим., 1981, 50, 273-295.
Оксимеркурирование
ChattJ. Chem. Rev. 1951, 48, 7-43.
Larock R. С. Solvomercuration/Demercuration Reaction in Organic Synthesis. — Springer-
Verlag, Berlin, Heidelberg, New York, 1986.
Straub H., ZellerK. P., Leditschke H. In: Houben-Weyl, Bd. 13/2b, 1974, S. 130-153.
Получение и реакции эпоксидов, дигидроксилирование
Dims G. In: Houben-Weyl, Bd. 6/3, 1965, S. 371-487.
Gunstone F. D. Advances Org. Chem., 1960, 1, 103-147.
Малиновский М. С. Усп. хим., 1957, 26, 801-823.
Parker R. E., Isaacs N. S. Chem. Rev., 1959, 59, 737-799.
SchroederM. Chem. Rev., 1980, 80, 187-213.
СвернД. В сб.: Органические реакции. Сб. 7. — М.: ИЛ, 1956, с. 476.
Cha J. К., Kim N.-S. Chem. Rev. 1995, 95, 1761-1795 (дигидроксилирование).
Эпоксидирование по Шарплессу - Кацуки
Irgensen К. A. Chem. Rev. 1989, 89, 431-458.
PfenningerA. Synthesis, 1986, 89-116.
Jonhson R. A., Sharpless К. В. In: Catalytic Asymmetric Synthesis. I. Ojma-VCH (ed.),
New York 1993, 103-158.
Katsuki Т., Martin V. S. Org. React. 1996, 48, 1-299.
4. Присоединение к неактивированным кратным углерод-углеродным связям 421
Дигидроксилирование по Шарплессу
Becker H., Soler M. A., Sharpless К. В. Tetrahedron 1995, 51, 1345-376.
Kolb H. С, VanNieuwenhze M. S., Sharpless К. В. Chem. Rev. 1994, 94, 2483-2547.
Kolb Н. С, Sharpless К. В. In: Transition Met. Org. Synth. Beller M., Bolm С (eds.).
Bd. 2 - Wiley-VCH, Weinheim, 1998, S. 2219-242.
Озонирование
Bailey P. S:. Ozonation in Organic Chemistry. Bd. 1-2. — Academic Press, New York,
1978-1982.
Bailey P. S. Chem. Rev., 1958, 58, 925-1010.
Bayer 0. In: Houben-Weyl, Bd. 7/1, 1954, S. 333-345.
Меняйло А. Т., Поспелов М. В. Усп. хим., 1967, 36, 662—685.
Criegee R. Angew. Chem., 1975, 87, 765-771.
Одинокое В. Н., Толстиков Г. А. Усп. хим. 1981, 50, 1207-1251.
Разумовский С. Д., ЗаиковГ. Е. Усп. хим., 1980, 49, 2345-2376.
Zvilichovsky G., Zvilichovsky В. In: The Chemistry of Hydroxyl, Ether Pepoxide Groups.
S. Patai (ed.). - Wiley, Chichester, 1993, 687-784.
Гидроборирование
Brown H. С Organic Synthesis via Boranes. — John Wiley & Sons. New York, 1975.
Brown H. С Angew. Chem. 1980, 92, 675-683.
Cragg G. M. L. Organoboranes in Organic Synthesis. — Marcel Derrer, New York, 1973.
KropfH., Schroder R. In: Houben-Weyl, Bd. 6/la, 1979, T. 1. S. 494-554.
Onak T. Organoborane Chemistry. — Academic Press, New York, 1975.
SchenkerE. In: Neuere Methoden. Bd. 4, 1966, S. 173-293.
Zzuzuki A., Dhillon A. In: Topics Current Chemistry, Bd. 130, — Springer-Verlag, Berlin,
Heidelberg, New York, 1985.
Цвейфель Г., Браун X. В сб.: Органические реакции. Сб. 13. — М.: Мир, 1966, с. 5-65.
PelterA., Smith К., Brown H. С. Borane Reagents. — Academic Press, San Diego, 1988.
Greene A. E., Luche M.-J., Sena A. A. J. Org. Chem. 1985, 50, 3957-3962.
Винилирование
Фаворский А. Е., Шостаковский М. Ф. ЖОХ, 1943, 13, 1-20.
Reppe W. u. a. Lieb. Ann. Chem., 1956, 601, 81-138.
Шостаковский М. Ф. Усп. хим., 1964, 33, 129-150.
Радикальное присоединение к двойным углерод-углеродным связям
(См. также литературу к разд. ГЛ.)
Стэси Ф., Гаррис Дж. В сб.: Органические реакции. Сб. 13. — М.: Мир, 1966,
с. 170-487.
VogelH. H. Synthesis, 1970, 99-140.
Уоллинг Ч., Хойзер Э. Веб.: Органические реакции. Сб. 13. — М.: Мир, 1966, с. 103-169.
422 Г Препаративная часть
Радикальное присоединение при С-С-связывании
Curran D. P. Synthesis 1988, 417-439; 489-513.
Giese В. Radicals in Organic Synthesis: Formation of Carbon-Carbon Bonds. — Pergamon
Press, Oxford, 1986.
Ramaiah M. Tetrahedron 1987, 43, 3541-3676.
Jasperse С P., Curran D. P., Fevig T. L. Chem. Rev. 1991, 91, 1237-1286.
Полимеризация ненасыщенных соединений
EliasH. G. Makromolekule. — Dr. Alfred Hiithig Verlag, Heidelberg, 1971.
Freidlina R. Kh., Chukovskaja E. C. Synthesis, 1974, 477—488 (теломеризация).
Henciri-Olive G., Olive S. Polymerisation. Katalyse, Kinetik, Mechanismen. — Verlag
Chemie, Weinheim, 1976.
Houben-Weil. Hrsg.: Bartl H., Falbe J. Bd.E 20/1-2, 1987.
Houwink R. Chemie und Technologie der Kunststoffe. Bd. 1—2. — Akadem. Verlagsges.
Geest & Portig, Leipzig, 1962/1963.
Kennedy J. P. Cationic Polymerization of Olefins. — Interscience, New York, 1975.
Kern W., Schulz R. С u. a. In: Houben-Weyl, Bd. 14, 1961, S. 24-1182.
Polymer Syntheses, Dandier S.R., Kara W. (eds.) Bd . 1-3. 1974-1980.
Rempp P., MerrillE. W. Polymer Synthesis. — Hiithig & Wepf Verlag, Basel, Heidelberg, New
York, 1986.
Vollmert В.: Grundriss der Macromolekularen Chemie. Bd. 1-5. — E. Vollmert Verlag,
Karlsruhe, 1982.
Циклоприсоединение
Oppolzer W. Angew. Chem. 1977, 89, 10-24.
Ulrich H. Organic Chemistry. Vol. 9. — Academic Press, New York, 1967. (Циклоприсое-
(Циклоприсоединение с участием гетерокумуленов.)
Carruthers W. Cycloaddition Reactions in Organic Synthesis. — Pergamon Press, Oxford, 1990.
11 +2]-Циклоприсоединение
Parham W. E., Schweizer E. E. Ogr. Reactions, 1963, 13, 55-90.
Brookhart M., Studabaker W. B. Chem. Rev., 1987, 87, 411—432 (циклопропанирование с
металлокарбеновыми комплексами).
Charette А. В. In: Organozinc Reagents. Hrsg. P. Knochel, P. Jones. — Oxford University
Press, Oxford, 1999, 263-285 (циклопропанирование с цинкорганическими
соединениями).
[2+2]-Циклоприсоединение
Bach T. Synthesis, 1998, 683-703 (стереоселективное внутримолекулярное
[2+2]-фотоциклоприсоединение и его применение в синтезе).
Braun M. In: Organic Synthesis Highlights. Hrsg. J. Mulzer. — VCH, Weinheim, 1991,
105-110 (применение реакции Патерно — Бюхи).
Jones G., II. Org. Photochem. 1981, 5,1—122 (реакция Патерно — Бюхи).
Oppolzer W. Ace. Chem. Res. 1982, 15, 135-141 (внутримолекулярное
[2+2]-фотоциклоприсоединение в органическом синтезе).
Bauslaugh P. G. Synthesis, 1970, 287-300 (синтез с применением фотохимического
циклоприсоединения енонов и алкенов).
4. Присоединение к неактивированным кратным углерод-углеродным связям 423
[3+2]-Циклоприсоединение
Black D. St. С, Crozier R. R., Davis V. СИ. Synthesis, 1975, 205-221 (присоединение к
нитронам).
Huisgen R. Angew. Chem., 1963, 75, 604-637, 742-754; Helv. Chim. Acta, 1967, 50,
2421-2439.
PadwaA. Angew. Chem., 1976, 88, 131 — 144 (внутримолекулярное присоединение).
Stuckwisch С. G. Synthesis 1973, 469-483.
Tsuge O., Hatta Т., Hisano T. In: The Chemistry of Double-Bonded Functional Groups. S.
Patai (ed.). - Wiley, Chichester, 1989, 345-475.
1,3-Dipolar Cycloaddition Chemistry. Bd. 1-2. Padwa A. (ed.). — J. Wiley & Sons, New
York, 1984.
[4+2]-Циклоприсоединение (реакция Дильса — Альдера)
Арбузов Ю. А. Усп. хим., 1962, 31, 529-558.
Brieger G., BennetJ. N. Chem. Rev. 1980, 80, 63-67.
CiganekE. Org. React. 1984, 32, 1-374.
Холмс Г. Л. В сб.: Органические реакции. Сб. 4. — М.: ИЛ, 1951, с. 86.
Клетцель М. С. В сб.: Органические реакции. Сб. 4. — М.: ИЛ, 1951, с. 7-85.
SauerJ. Angew. Chem., 1966, 78, 233; 1967, 79, 76.
SauerJ., Sustmann R. Angew. Chem., 1980, 92, 773-801.
TaberD. F. Intramolecular Diels—Alder and Alder Ene Reactions. — Springer-Verlag, Berlin,
Heidelberg, New York, 1984.
TatacchiA., Fringuelli F. Diels-Alder-Reaction. — Wiley-VCH, Weinheim, 2002.
Corey E. J. Angew. Chem. 2002, 114, 1724-1741 (каталитические энантиоселективные
реакции Дильса — Альдера).
Craig D. Chem. Soc. Rev. 1987, 16, 187—238 (внутримолекулярная реакция Дильса —
Альдера).
FallisA. G. Ace. Chem. Res. 1999, 32, 464—474 (внутримолекулярная реакция Дильса —
Альдера).
Титов Ю. А. Усп. хим., 1962, 31, 529-558
Wagner-Jauregg T. Synthesis, 1980, 3, 165-214; 1980, 10, 769-798.
WollweberH. In: Houben-Weyl, Bd. 5/lc, 1970, S. 911-1139.
WollweberH. Diels-Alder-Reaktionen. — Georg. Thieme Verlag, Stuttgart, 1972.
Металлокомплексный катализ и гомогенное гидрирование
Applied Homogeneous Catalysis with Organometallic Compounds. B. Cornils,
W. A. Hermann (eds.). Bd. 1-3. - Wiley-VCH, Weinheim, 2002. .
Aspects of Homogeneous Catalys. R. Ugo (ed.). Bd. 1—4. — D. Reidel Publ. Сотр.,
Dordrecht, Boston, London, 1970-1981.
Birch A. J., Williamson D. H. Org. React. 1976, 24, 1-186 (гомогенное гидрирование).
Bird С. W. Transition Metal Intermediates in Organic Synthesis. — Academic Press,
New York, 1967.
CassarL., Chiusoli G. P., Guerrieri F. Synthesis 1973, 509—523 (карбонилирование олефи-
нов и ацетиленов).
Handbook of Metathesis. R. H. Grubbs (ed.). Bd. 1-3. - Wiley-VCH, Weinheim, 2002.
Heimbach P., Schenkluhn. In: Topics in Current Chemistry. Bd. 92 — Springer-Verlag,
Berlin, Heidelberg, New York 1980, S. 45-108.
424 Г Препаративная часть
Henrici-Olive G, Olive S. In: Topics in Current Chemistry. Bd. 67 — Springer-Verlag, Berlin,
Heidelberg, с S. 107-127.
Houben-Weil. Hrsg.: Falbe E. Bd. E 18/1-2, 1986.
Джеймс Б. Гомогенное гидрирование. — М.: Мир, 1976.
Nakamura A., Tsutsui M. Principles and Applications of Homogeneous Catalysis. — John
Wiley & Sons, New York, London, 1980.
Pracejus H. Koordinationschemische Katalyse ogranischer Reaktionen. — Verlag Theodor
Steinkopff, Dresden, 1977.
Schuster M., Blechert S. Angew. Chem. 1997, 109, 2124—2144 (каталитическая
перегруппировка с замыканием кольца).
SemmelhackM. F. Org. React. 1972, 19, 115-198.
Takaya H., Ohta Т., Noyori R. In: Catalytic Asymmetric Synthesis. Hrsg.: I. Ojima — VCH,
New York, 1993, 1-39 (энантиоселективное гомогенное гидрирование).
Taube R. Homogene Katalyse. — Academie-Verlag, Berlin, 1988.
Transition Metals in Homogeneous Catalysis. Hrsg. Schrauzer G. N. — Marcel Dekker,
New York, 1971.
Transition Metals for Organic Synthesis. Beller M., Bolm С (eds.) Bd. 1+2 — Wiley-VCH,
Weinheim, 1998.
TsujiJ. Fortschr. Chem. Forsch. 1972, 28, 41-84; Adv. Org. Chem. 1969, 6, 109-255.
Гидрирование олефинов, ацетиленов и ароматических соединений.
Общие сведения о катализаторах и аппаратуре
Bogoslowski В. М., Kasakova S. S. Skelettkatalysatoren in organischen Chemie. — Deutscher
Verlag derWissenschaften, Berlin, 1960.
Caine D. Org. React. 1976, 23, 1—258 (восстановление С=С-связей в а,р-ненасыщенных
карбонильных соединениях).
Grundmann С. In: Neuere Methoden. Bd. 1, 1944, S. 117-136.
Komarewsky V. I., Riesz C. H., Morritz F. L. In: Technique of Organic Chemistry.
A. Weissberger (ed.). - Interscience, New York, 1965. Bd. 4/2, 1965, S. 1-164.
Rylander P. N. Catalytic Hydrogenation over Platinum Metals. — Academic Press,
New York, 1967.
Rylander P. N. Hydrogenation Methods. —Academic Press, London 1985.
Schiller G. In: Houben-Weyl. Bd. 4/2, 1955, S. 248-303.
SchrdterR. In: Neure Methoden. Bd. 1. 1944, S. 75-116.
WimmerK. In: Houben-Weyl. Bd. 4/2, 1955, S. 143-152, 163-192.
5. ЗАМЕЩЕНИЕ В АРОМАТИЧЕСКОМ КОЛЬЦЕ
Ароматические соединения (например, бензол, нафталин) имеют плоскую или
почти плоскую систему сопряженных двойных связей и, подобно олефинам, об-
обладают основными свойствами (см. разд. Г,4.1.1). В связи с этим они реагируют,
как и олефины, прежде всего с электрофильными соединениями, но в отличие
от олефинов в данном случае происходит замещение атома водорода с сохране-
сохранением в конечном продукте ароматической системы.
Если основность ароматических соединений из-за влияния групп с сильны-
сильными -/- и -Л/-эффектами сильно понижена, то возможен также нуклеофильный
обмен заместителей. Этот тип реакций, однако, менее распространен.
5. Замещение в ароматическом кольце
425
2л
6л
6л
6л
С
/©\
\ /
с-с
с© с
с
II
^ v; © i/
I II III IV
Рис. Г.5.1. Углеродные кольца с ароматическими свойствами.
Такие заместители, как галогены, в особенности иод, а также другие легко
уходящие группы, например диазогруппа, легко замещаются в присутствии
катализаторов — переходных металлов.
Ранее реакции замещения считались характерным критерием ароматичности структуры со-
соединения. Однако позже оказалось, что чем стабильнее система кратных сопряженных связей
соединения (включая ионогенные структуры), тем менее вероятно, что реакции замещения для
них будут доминировать, но могут и вообще не иметь места (см., например, разд. Г,5.1 — реак-
реакции 9,10-присоединения у антрацена).
В настоящее время для характеристики ароматичности структуры используют прежде все-
всего физические свойства, например энергию стабилизации, определяемую термохимическим
способом, выравнивание длин связей в сопряженных системах и появление кольцевых токов,
регистрируемых с помощью спектроскопии ЯМР (см. разд. А,3.5.3).
Что касается моноциклических соединений, то на основании простого квантовохимичес-
кого анализа критерием ароматичности можно считать наличие (An + 2) 71-электронов (п = О,
1, 2, ...) (правило Хюккеля), которые делокализованы по всему кольцу. Такая делокализация
возможна только тогда, когда соединение имеет плоское строение. Судя по этому критерию,
циклопропенил-катион I, циклопентадиенил-анион II и циклогептатриенил-катион IV (ион
тропилия) являются ароматическими системами (см. рис. Г.5.1).
Бензол как незаряженную конъюгированную систему с шестью л-электронами
рассматривают как прототип ароматических соединений. Группа бициклических сис-
систем и циклов с гетероатомами (кислорода, азота, серы и др.) также обладает типичны-
типичными «ароматическими» свойствами. Примерами могут служить фуран, тиофен, пиррол,
пиридин, катион пирилия, соответствующие бензопроизводные, нафталин и азулен.
5.1. ЭЛЕКТРОФИЛЬНОЕ АРОМАТИЧЕСКОЕ ЗАМЕЩЕНИЕ
Электрофильное замещение в ароматическом кольце состоит, как правило, в
замене ароматически связанного атома водорода электрофильным агентом.
Важнейшие реакции этого вида указаны в табл. Г.5.2.
5.1.1. МЕХАНИЗМ ЭЛЕКТРОФИЛЬНОГО АРОМАТИЧЕСКОГО ЗАМЕЩЕНИЯ
Как правило, электрофильные агенты, перечисленные в табл. Г.5.2, предварительно
при помощи подходящего реагента или катализатора (кислоты или кислоты Льюиса)
переводят в положительно заряженную реакционноспособную форму, например:
426
Г Препаративная часть
Таблица Г.5.2. Электрофильное замещение в ароматическом кольце
Схема
Тип реакции
АгН + HNO3 —> ArNO2 + H20
АгН + H2SO4 =^ ArSO3H + Н20
АгН + С12 —> ArCl + HCl
АгН + (SCNJ + ArSCN + HSCN
ArR + HCl
АгН + RCN + HCl
АгН + /N-Cf
СН3 Н
ArC=NH2Cl
R
> ArCOR
9
сн3
АгН + Н2С=О
ArCH2OH
АгН + Н2С=О + HNR2 —> Ar-CH2-NR2 + Н2О
Агн + н2с=о + на —> Агсн2а + н2о
он
АгН + R-C=O > Ar-CH-R
Н
АгН + СО2 1- АгСООН
АгН + HNO2 > ArNO + Н2О
АгН + Ar'-NsN Cle + Ar-N=N-Ar' + HCl
АгН + HgX2 *• ArHgX + НХ
Х- остаток органической или неорганической
кислоты
Нитрование
Сульфирование
Галогенирование
(хлорирование)
Роданирование
Алкилирование по
Фриделю—Крафтсу
Ацилирование по
Фриделю—Крафтсу
Синтез Гаттерма-
на — Коха
Синтез Гаттермана
Синтез Губена —
Геша
Синтез Вильсмейера
Гидроксиметилиро-
вание
Аминометилирова-
ние (см. реакция
Манниха;
разд. Г.7.2.1.5)
Хлорметилирова-
ние (реакция
Блана)
Реакции с альде-
альдегидами или кето-
нами
Синтез Кольбе —
Шмитта
Нитрозирование
Азосочетание (см.
разд. Г.8.3.3)
Металлирование
(меркурирование)
5. Замещение в ароматическом кольце 427
Н®+ НО -Vf _ == Н2О + Nof
0 [Г.5.3]
8* 6"
CI-CI + AICI3 =^=^ CI--CI-—А1С13
Для электрофильного ароматического замещения доказан следующий ме-
механизм:
[Г.5.4а]
о-комплекс
+ нв® [Г.5.46]
В ходе кислотно-основной реакции арен присоединяет электрофильный
агент (катион Y® на схеме [Г.5.4а]), образуя о-комплекс (бензениевый ионI.
В некоторых случаях его можно обнаружить с помощью ИК-спектроскопии или
лучше ЯМР.
Изменение потенциальной энергии вдоль координаты реакции имеет харак-
характер типа представленного на рис. В.35. ст-Комплекс находится в энергетической
впадине. Он сходен с карбениевым ионом, образующимся при электрофильном
присоединении к олефинам, однако стабилизируется не путем присоединения
основания, а посредством отщепления протона. Роль основания в схеме (Г.5.46)
играет противоион, возникший при образовании Y®, сам арен или растворитель.
Какая из стадий схемы (Г.5.4) окажется скоростьопределяющей, зависит от конкретной
системы. Кинетический анализ показывает, что в большинстве случаев скорость электрофиль-
электрофильного ароматического замещения определяет стадия образования с-комплекса. Тогда реакция
имеет второй порядок, ее скорость зависит от концентрации арена и реагента. Здесь налицо
случай, представленный на рис. В.35, а, когда переходное состояние X] расположено выше, чем
Х2. В редких случаях скорость реакции определяет стадия отщепления протона; это наблюдает-
наблюдается чаще всего в очень реакционноспособных системах. В этой ситуации при использовании
дейтерированных аренов наблюдается первичный кинетический изотопный эффект (кц/ко),
переходное состояние Хг расположено выше, чем Xi (см. рис. В.35, б), а концентрация основа-
основания влияет на скороЪть реакции.
5.1.2. ВЛИЯНИЕ ЗАМЕСТИТЕЛЕЙ НА РЕАКЦИОННУЮ СПОСОБНОСТЬ АРЕНОВ
И НАПРАВЛЕННОСТЬ ПОСЛЕДУЮЩИХ РЕАКЦИЙ ЗАМЕЩЕНИЯ
Реакция между нуклеофильным ядром и электрофильным агентом протекает
тем легче, чем сильнее основные свойства ароматической системы и чем силь-
сильнее кислотные свойства этого агента (см. разд. Г,4.1.1). Основность ядра повы-
1 Переходное состояние при образовании а-комплекса структурно аналогично последне-
последнему, так как на координате реакции лежит позже. См. постулат Хэммонда, разд. В,2.
428 Г Препаративная часть
шается при наличии заместителей, которые увеличивают электронную плот-
плотность благодаря индукционным или мезомерным эффектам:
Алкил +/
—OH<-NH2<— NHR<— NR2 +M>— I [T 5 5]
—01е +М; +1
Реакционная способность ядра понижается при наличии следующих групп:
-COR; —СООН; -COOR; -CN; —N02 —M; —I
Галогены -j-Ai < —/ [Г.5.6]
-NR3© -/
Это влияние заместителей на реакционную способность ароматического
ядра становится особенно заметным при вторичном электрофильном замеще-
замещении у различных производных нафталина. Так, в 1-нитронафталине второй
заместитель направляется в еще не замещенное, а в 1-метилнафталине — в уже
замещенное кольцо.
В л-электроноизбыточных гетероциклах, таких как тиофен или пиррол,
активирующим действием обладает сам гетероатом (соответственно, сера или
NH). Напротив, в электрононедостаточных гетероциклах, например пиридине,
гетероатом (азот) дезактивирует молекулу. (С теоретическими объяснениями
такого влияния гетероатомов следует ознакомиться в учебниках.)
Селективность, с которой агент с определенными электрофильными свой-
свойствами реагирует с аренами разной основности (субстратная специфичность),
бывает иногда очень значительной. Эти различия имеют большое значение для
практического проведения реакций замещения в ароматическом кольце.
Если в уже замещенное бензольное кольцо должен быть введен второй элект-
рофильный заместитель, то в общем случае возможны три различных варианта
замещения1:
орго-продукт
[Г.5.7]
Уже имеющиеся в кольце заместители оказывают определенное направляю-
направляющее действие; в результате преимущественно образуются те или иные изомеры
1 В принципе реагент может атаковать также уже замещенное положение арена («ипсо-за-
мещение»). Примером может служить замещение групп SO3H нитрогруппами при получении
пикриновой кислоты, желтого Марциуса и нафтолового желтого S (см. разд Г.5.1.4).
5. Замещение в ароматическом кольце 429
(региоселективность). Для определения положения вступления второго замести-
заместителя используют эмпирически найденные приближенные правила, сформули-
сформулированные ниже.
а) Заместители «первого рода» направляют второй заместитель преимуще-
преимущественно в орто- и пара-положения. К заместителям первого рода относят-
относятся группы или атомы, которые усиливают основные свойства ядра (см.
заместители в рядах [Г.5.5]), и галогены.
б) Заместители «второго рода» направляют второй заместитель преимуще-
преимущественно в мета-положение. К заместителям второго рода относятся за-
заместители, которые уменьшают реакционную способность бензольного
кольца (см. заместители в рядах [Г.5.6]), за исключением галогенов.
Повышение или понижение реакционной способности ароматических соединений (влия-
(влияние на легкость замещения), вызванное уже имеющимся в ядре заместителем, ничего не гово-
говорит о его влиянии на направление замещения. Объяснение правил ориентации основано на ана-
анализе мезомерных граничных структур монозамещенных ароматических соединений. При этом
предполагается, что заместители не только влияют на общую основность ядер в основном состо-
состоянии, но и у отдельных ядер углеродных атомов создают различную плотность электронов.
В случае заместителей — эффективных доноров электронов (ОН, OR, NH2, NHR, NR2) —
квадраты коэффициентов, относящихся к атомам углерода в о- и и-положениях, указывают на
повышенную электронную плотность уже в основном состоянии. Это подтверждается также
данными спектроскопии ЯМР 13С. Таким образом, для аренов с заместителями указанного ти-
типа на основе анализа мезомерных структур можно в принципе предсказать направление вто-
вторичного замещения. В случае аренов с акцепторными заместителями (галогены, NO2, CN и
др.) результаты изучения спектров ЯМР 13С не во всех случаях совпадают с обусловленной ме-
зомерным эффектом поляризацией. Следовательно, одной только электронной плотностью в
основном состоянии нельзя объяснить ориентацию повторного замещения. Единое объясне-
объяснение направляющего эффекта донорных и акцепторных заместителей на вторичное замещение
в аренах можно дать, рассмотрев энергетическое состояние ст-комплекса.
Образующийся ст-комплекс определяет, в какое положение (орто-, пара- или мета-) всту-
вступает новый заместитель, так как имеющиеся в кольце заместители оказывают различное вли-
влияние на энергию активации трех возможных переходных состояний (см. ниже). Различие в
энергиях активации обусловливает, согласно уравнению Аррениуса (В.25), различие в скорос-
скоростях конкурирующих стадий. Поскольку энергии переходных состояний, приводящих к трем
возможным ст-комплексам, неизвестны, вместо них рассматривают энергии ст-комплексов.
Предполагается, что неточность, связанная с этим упрощением, невелика. Это позволяет
представить себе энергию ст-комплекса и энергию переходного состояния (которое приводит
к ст-комплексу) для орто-, пара- и жетяа-заместителей (рис. В.35).
ст-Комплекс можно изобразить следующим образом:
Н © Н Н
I II III IV
Из этих формул видно, что в орто- и пара-положениях относительно атакующего заместителя
Y появляются частичные положительные заряды; суммарно это изображено формулой IV.
Вследствие наличия положительного заряда ст-комплекс представляет собой богатую энерги-
энергией систему. Чем более делокализован заряд внутри мезомерной системы, тем стабильнее, т. е.
тем беднее энергией данная система. Для оценки энергии ст-комплекса нужно, таким образом,
исследовать, в какой степени уже имеющийся заместитель может способствовать дальнейшей
делокализации положительного заряда в комплексе. Если в ядре уже имеется электронодонор-
ный +/-заместитель X, то он может компенсировать частичный положительный заряд тем
больше, чем ближе к заряду он расположен. Следовательно, заместитель X, находясь в орто- и
иа/>а-положениях к группе Y, компенсирует заряд сильнее, чем в л*ета-положении:
430
Г Препаративная часть
[Г.5.9]
Такое же действие оказывают и +Л/-заместители, например:
Н Н
МеО
[Г.5.10]
Поэтому в случае +/- или +Л/-заместителей энергия активации образования ст-комплекса при
орто- и ла/>а-замещениях меньше, чем при .«ema-замещении; орто- и ла/>а-производные обра-
образуются быстрее (орто- и ло/?а-ориентация заместителей).
Напротив, —/- или — М-заместитель увеличивает положительный частичный заряд тем
сильнее, чем ближе он к нему находится, т. е. в орто- и лора-положениях (по сравнению с
жета-положением):
/5-
8* Н
[Г.5.11]
Следовательно, в этом случае энергия активации наименьшая для л<ета-замешения, и эта
реакция протекает быстрее (.ме/яа-ориентация заместителей).
Если заместители проявляют —/и +Л/-эффекты, то в положительно заряженном, а следо-
следовательно, сильноэлектрофильном ст-комплексе преобладает донорное влияние, вытекающее
из наличия свободной электронной пары. Таким образом, подобные заместители понижают
энергию активации, необходимую для образования орто- и пара-продуктов замещения. Это
относится и к галогенам, хотя последние принадлежат к заместителям, затрудняющим заме-
замещение (понижение общей основности ядра, так как в основном состоянии +М< -/; см. ряды
[Г.5.6])!
Эти закономерности справедливы только для кинетически контролируемых реакций
(см. разд. В,3.2). Если условия реакции способствуют образованию термодинамически конт-
контролируемых продуктов, то необходимо принимать в расчет изомеризацию, которая приво-
приводит к значительному изменению соотношения между орто- и пара-продуктами (см., напри-
например, сульфирование и алкилирование по Фриделю — Крафтсу).
Вот несколько примеров электрофильного вторичного замещения в аренах.
Электрофильное замещение нитробензола происходит преимущественно в
мета-положение; оно протекает быстрее, чем с бензолом. Для анилина и фено-
фенола электрофильное замещение происходит преимущественно в орто- и пара-по-
пара-положения; оно протекает легче, чем для бензола. Для хлорбензола наблюдают
главным образом орто- и ла/?а-замещение, однако по сравнению с бензолом оно
затруднено.
(Как влияет солеобразование фенола и анилина на легкость и положение
замещения при введении в эти соединения второго заместителя?)
У незамещенного нафталина электрофильное замещение протекает преиму-
преимущественно в а-положение (см., однако, схему [Г,5.21]).
Обратите внимание на легкость и направленность электрофильного замеще-
замещения у гетероциклов: теофена, пиррола (по аналогии с фенолом), пиридина
(по аналогии с нитробензолом), индола, N-оксида пиридина и др.
5. Замещение в ароматическом кольце 431
Кроме рассмотренных электронных факторов на характер вторичного заме-
замещения могут также влиять пространственные эффекты. Последние, очевидно, в
первую очередь препятствуют замещению в o/wio-положение. При этом соотно-
соотношение образующихся орто- и иа/ю-изомеров оказывается гораздо меньшим, чем
это можно было бы ожидать B:1), исходя только из статистических соображе-
соображений. С увеличением размера уже имеющихся или вновь вступающих в аромати-
ароматическое кольцо заместителей доля о/?/яо-продуктов уменьшается. Так, при хлори-
хлорировании толуола молекулярным хлором в уксусной кислоте соотношение между
орто- и яара-изомерами составляет 1,5; тот же процесс для тре/я-бутилбензола
приводит к отношению 0,28. При введении изопропильной группы в трет-бу-
тилбензол o/wzo-изомер вообще не образуется.
Если же электрофильные агенты имеют возможность вступать во взаимодей-
взаимодействие с уже имеющимися в ядре основными заместителями (—ОН, —OR) преж-
прежде, чем произойдет замещение в opmo-положение, то это, напротив, способству-
способствует орто-замещению (см., например, синтез салициловой кислоты, разд. Г,5.1.8.6
и список литературы в разд. Г,5.4).
Региоселективного замещения в аренах можно достичь также с помощью об-
обратимого блокирования. Так, в яяра-положение к первому заместителю можно
ввести /я/>е/я-бутильную группу. При последующем (третьем) электрофильном
замещении реакция будет осуществляться исключительно в opmo-положение к
первому заместителю. После этого т/?е/и-бутильную группу можно отщепить в
виде изобутилена или перенести на другой арен (переалкилирование). В качест-
качестве защитных групп в определенных случаях пригодны также галогены (хлор и
бром). Их можно удалить восстановлением; хлор в этих условиях сохраняется
как заместитель в ядре.
Продукты реакции, ожидаемые при замещении в ароматическом ядре, мож-
можно рассчитать с хорошим приближением с помощью уравнения Гаммета (разд.
В,5.2), причем следует использовать и+-константы заместителей.
5.1.3. НИТРОВАНИЕ
Действующим электрофильным агентом при нитровании является нитроний-ка-
тион (NO24"), источником которого могут служить многие соединения, например1:
1 Ацетилнитрат CH3COONO2 — взрывчатое соединение, о чем ни в коем случае нельзя за-
забывать при его выделении. Чистый ацетилнитрат обычно не применяют. Можно растворить
нитруемое соединение в смеси ледяной уксусной кислоты и уксусного ангидрида и при хоро-
хорошем охлаждении медленно добавить 100%-ную азотную кислоту, контролируя температуру.
Образующийся ацетилнитрат тут же расходуется. И в этом случае реакцию надо проводить
очень осторожно (обязателен контроль температуры!). Следует помнить о мерах предосто-
предосторожности и при выделении продуктов. Аналогичные замечания относятся к реже применяе-
применяемому бензоилнитрату.
Несчастные случаи при нитровании ацетилнитратом: Chem. Tech., 1955, 7, 121; Angew.
Chem., 1955, 67, 157; Nachr. Chem. Techn., 1963, 299; Chem. Labor. Betrieb, 1966, 346.
Применение тетрафторбората нитрония описано в работах: Olah G., Kuhn S., Mlinko A. J.
Chem. Soc, 1956, 4257; Kuhn S. J., Olah G. A. J. Am. Chem. Soc, 1961, 83, 4564; Olah G. A.,
Lin H. С Synthesis, 1974, 444; Физер Л., Физер М. Реагенты для органического синтеза. — М.
Мир, 1970, Т. II, С. 463; 1971, Т. V, С. 336; 1978, Т. VII, С. 390.
432 Г Препаративная часть
HO-NO2, O2N-O-NO2, RCO-O-NO21 , Nof BF® и др. [Г.5.12]
Склонность к образованию нитроний-катиона увеличивается с повышением
элеткроотрицательности заместителя, связанного с нитрофуппой.
Гидроксильная группа как таковая отщепляться не может (см. схему [Г.2.3]),
поэтому нитроний-катион из азотной кислоты образуется только в кислой среде:
© ©Я © ©Я m /?
Н +HO-NN ===== H2O-Nv ==== Н2О+ Nv [Г.5.13]
Ъ Ъ Ъ
В простейшем случае азотная кислота может протонироваться сама (авто-
протолиз):
HONO2 + HONO2 == H2O-NO2 + O-NO2 [F.5.14J
Однако равновесие сильно смещено влево, так что одна азотная кислота —
слабый нитрующий агент. При добавлении концентрированной серной кисло-
кислоты концентрация катиона N02® сильно возрастает:
HNO3 + 2H2SO4 ===== N02®+ H30®+ 2HSO4G [Г.5.15]
Нитрующее действие смеси азотной и серной кислот (нитрующей смеси) по-
поэтому намного сильнее, чем чистой азотной кислоты. Еще большего повышения
реакционной способности можно достигнуть, применяя дымящую азотную
кислоту и олеум. Другие известные нитрующие средства не имеют столь общего
значения. (Напишите в общем виде уравнение реакции нитрования!)
На практике нужно согласовывать активность нитрующего средства с реакци-
реакционной способностью ароматического соединения. Фенолы и простые эфиры фе-
фенолов нитруются уже разбавленной азотной кислотой, в то время как нитрование
бензальдегида, бензойной кислоты, нитробензола и т. д. требует смеси дымящей
азотной и серной кислот (почему?). л*-Динитробензол с трудом нитруется даже
смесью дымящей азотной и серной кислот E сут, ПО °С, выход 45%). Напротив,
нитрование jw-динитробензола тетрафторборатом нитрония N02+ BF4~ в среде
фторсульфоновой кислоты через 3 ч дает 61% 1,3,5-тринитробензола.
При нитровании наиболее распространенной побочной реакцией является
окисление. Ему благоприятствует повышение температуры реакции. Процесс
окисления обнаруживается по выделению оксидов азота. Например, амины
вследствие их легкой окисляемости можно нитровать либо в виде их ацетильных
производных, либо в очень концентрированном сернокислом растворе. В пос-
последнем случае получают главным образом jwe/яа-изомер (почему?). Альдегиды,
алкиларилкетоны и в меньшей степени алкилбензолы при нитровании также в
известной степени подвергаются окислению. По этой причине фенолы можно
сравнительно гладко нитровать только в разбавленной азотной кислоте. Причем
продуктами являются мононитросоединения. Прямое нитрование фенолов до
полинитросоединений таким путем невозможно. В этом случае применяют об-
обходной путь: сначала соединение сульфонируют, а затем замещают сульфогруп-
1 См. примечание на с. 431.
5. Замещение в ароматическом кольце 433
пы нитрогруппами (например, так поступают при получении пикриновой кис-
кислоты и нафтолового желтого S, см. разд. Г,5.1.4).
Нитрогруппа сильно понижает реакционную способность ароматического
кольца по отношению к электрофильным заместителям, поэтому опасность вто-
вторичного нитрования существует только в случае очень реакционноспособных
веществ.
Самой трудной частью препаративной работы обычно является разделение
изомерной смеси, особенно орто- и яа/>а-изомеров, которые часто получаются в
почти равных количествах. Для разделения используют вымораживание, перек-
перекристаллизацию, фракционную перегонку, перегонку с водяным паром (напри-
(например, о-нитрофенолы в противоположность я-нитрофенолам перегоняют с водя-
водяным паром). Часто эти методы приходится комбинировать.
В общей методике приведены только такие примеры, когда получается толь-
только один изомер или легкоразделяемые смеси изомеров.
Общая методика нитрования ароматических соединений (табл. Г.5.16)
Внимание! При работе с азотной и серной кислотами необходимо соблюдать
меры предосторожности (защитные очки, тяга!) (см. также часть Е). Ди- и поли-
нитросоединения перегонять нельзя, так как при этом возможны взрывы.
Для приготовления нитрующей смеси к азотной кислоте медленно при охлаждении ледя-
ледяной водой и перемешивании или встряхивании приливают серную кислоту.
Состав нитрующей смеси определяется реакционной способностью нитруемого вещества.
На 0,1 моль ароматического соединения берут:
Вариант А (для инертных соединений): 10 мл @,23 моль) 100%-ной азотной кислоты (d= 1,5)
и 14 мл концентрированной серной кислоты.
Вариант Б (для соединений средней реакционной способности): 10 мл @,15 моль) концентрирован-
концентрированной азотной кислоты F8%-ной, d= 1,41) и 12 мл концентрированной серной кислоты.
Вариант В (дляреакционноспособных аренов): 33 мл @,3 моль) 40%-ной азотной кислоты.
В трехгорлую колбу на 250 мл, снабженную мешалкой, капельной воронкой и внутренним тер-
термометром (прибор не должен быть герметичным!) помещают 0,1 моль ароматического соедине-
соединения. Затем медленно при хорошем перемешивании и охлаждении (баня со льдом) из капельной
воронки добавляют нитрующую смесь, предварительно охлажденную по меньшей мере до 10 °С;
температура должна поддерживаться в интервале 5—10 °С. При нитровании реакционноспособных
соединений (вариант В) по окончании добавления азотной кислоты перемешивание при комнат-
комнатной температуре продолжают еще 30 мин, в других случаях (варианты А и Б) 2—3 ч.
После этого реакционную смесь осторожно выливают примерно в 300 мл ледяной воды и
хорошо перемешивают. Твердые продукты нитрования отфильтровывают, тщательно промы-
промывают водой и очищают (обычно перекристаллизацией). Жидкие нитросоединения отделяют
при помощи делительной воронки, водный раствор экстрагируют один раз эфиром, объеди-
объединенные органические фракции промывают водой, раствором гидрокарбоната натрия до нейт-
нейтральной реакции и еще раз водой, сушат хлоридом кальция и перегоняют.
Методика пригодна для синтеза нитросоединений в полумикроколичествах. Мешалкой,
капельной воронкой и контролем за температурой смеси в этом случае можно пренебречь.
Нитрующую смесь медленно прибавляют при хорошем охлаждении и встряхивании.
З-Нитроацетофенон: Керсон Б., Хазен Р. В сб.: Синтезы органических препа-
препаратов. Сб. 2. - М.: ИЛ, 1949, с. 361.
З-Нитрофталевая кислота: Moser С. М., GompfTh. J. Org. Chem., 1950,15, 583.
4-HumponupuduH-N-oKCud, 4-нитрохинолин-Ъ)'-оксид, 4-нитропиридин: OchiaE.
J. Org. Chem., 1953, 18, 534.
Таблица Г.5.16. Нитрование ароматических соединений
Продукт реакции
•«-Динитробензол
2,4-Динитротолуол
Метиловый эфир .м-нит-
робензойной кислоты
.и-Нитробензальдегид
л-Бромнитробензол
л-Нитробензилцианид
Нитробензол
1-Нитронафталин
о-Нитротолуол
л-Нитротолуол
4-Нитровератрол
о-Нитрофенол
л-Нитрофенол
Исходное вещество
Нитробензол
я-Нитротолуол
Метилбензоат
Бенз альдегид
Бромбензол
Бензилцианид
Бензол
Нафталин
Толуол
Толуол
Вератрол
Фенол
Фенол
Вариант
А
А
А
А
А
А
Б
Б
Б
Б
В
В
В
Т. пл., "С
90 (этанол)
71 (метанол)
78 (метанол)
58 (водн. этанол)
126 (этанол)
117 (80%-ный этанол)
Т. кип. 99 B0 мм рт. ст.);
яо20 1,5532
57 (этанол)
Т. кип. 94 A0 мм рт. ст.);
«D20 1,5472
Т. кип. 101 A0 мм рт. ст.);
55 (этанол)
98 (этанол)
46 (этанол)
114 (вода)
Выход, %
80
80
80а
40
80
60
80
606
40в
20"
70г
зол
10*
Примечания
Нитрующую смесь при-
прибавляют по каплям при
60 °С и 30 мин нагрева-
нагревают при 80"С
Бензальдегид прибавля-
прибавляют по каплям к нитрую-
нитрующей смеси
Работают при —5 °С
-и-Нитробензойную кислоту целесообразно получать гидролизом соответствующего метилового эфира (Камм О. и др. В сб.: Синтезы
органических препаратов. Сб. 1—М.: ИЛ, 1956, с. 295), так как при прямом нитровании бензойной кислоты образуется трудноразделимая
смесь изомеров.
В нитрующую смесь при 45—50 "С вносят тонкоизмельченный нафталин, перемешивают 45 мин при 60 "С. Из сырого продукта пере-
перегонкой с водяным паром выделяют непрореагировавший нафталин.
в л-Изомер вымораживают смесью льда с хлоридом натрия, быстро отфильтровывают, промывают небольшим количеством холодного пет-
ролейного эфира. Из фильтрата перегонкой в вакууме на 30-сантиметровой колонке Вигре с электрическим обогревом выделяют о-изомер; из
остатка после перегонки вымораживанием можно выделить еще немного л-изомера.
г Вератрол предварительно промывают 10%-ным раствором гидроксида натрия и водой, затем перегоняют (для очистки от гваякола);
продукт очищают перекристаллизацией с активированным углем.
д К разбавленной азотной кислоте по каплям добавляют фенол, содержащий небольшое количество воды. Нитрующую смесь декан-
декантируют с полутвердой смеси нитрофенолов. Последнюю дважды промывают водой. о-Изомер отгоняют с водяным паром, остаток охлажда-
охлаждают, отфильтровывают л-нитрофенол и перекристаллнзовывают его из 3%-ной НС1 с добавлением активированного угля.
5. Замещение в ароматическом кольце 435
3,5-Динитробензойная кислота: Брюстер Р., Уильяме Б., Филлипс Р. В сб.:
Синтезы органических препаратов. Сб. 3. — М.: ИЛ, 1952, с. 214.
Примеры нитрования аминов
2-Ацетиламино-1-нитронафталин\ Хартман В., Смит Л. В сб.: Синтезы орга-
органических препаратов. Сб. 2. — М.: ИЛ, 1949, с. 359.
4-Метокси-2-нитроанилин через соответствующий ацетанилид: Фанта П.,
ТарбеллД. В сб.: Синтезы органических препаратов. Сб. 3. — М.: ИЛ, 1952, с. 358.
M-Humpo-N,N-duMemunaHwiuH (нитрование в концентрированном растворе
серной кислоты): Фитч Г. В сб.: Синтезы органических препаратов. Сб. 3. — М.:
ИЛ, 1953, с. 374.
В лаборатории нитросоединения применяют главным образом в качестве
исходных веществ при получении аминов и ряда других продуктов восстановле-
восстановления (см. разд. Г,8.1).
В промышленности нитрование является одним из важнейших процессов. Нитросоединения
применяются в качестве взрывчатых веществ (тринитротолуол, тринитробензол, смесь нитробен-
нитробензола с N2O4 под названием «панкласит»). Особенно большое значение имеет восстановление нит-
росоединенпй до аминов, которые используются как промежуточные продукты при получении
красителей и лекарственных веществ (см. разд. Г,8.1). Из 2,4-динитротолуола, его смеси с 2,6-ди-
нитротолуолом и из других динитросоединений получают диизоцианаты, перерабатываемые
затем в полиуретаны. л<-Фенилендиамин, получаемый из л<-динитробензола, используется для
получения термостойких полиамидных волокон. Производные л-фенилендиамина, дифенила-
дифениламина, аминофенолов, получаемые восстановлением соответствующих нитросоединений,
используются как антиоксиданты и средства, защищающие материалы от старения.
Значение нитроаренов как промежуточных продуктов в получении других соединений можно
проиллюстрировать на двух следующих примерах (напишите соответствующие схемы превращений):
- л-нитротолуол: л-толуидин, 4-нитро-2-хлортолуол, л-нитробензойная кислота, 2,4-ди-
нитротолуол;
— /1-нитрохлорбензол: л-нитрофенол, л-хлоранилин, л-нитродифениламин, л-нитроани-
зол, л-нитрофенетол, л-фенилендиамин, л-нитроанилин, 4-нитро-1,2-дихлорбензол.
Нитрование можно использовать для идентификации ароматических соеди-
соединений. Последующее восстановление образующихся продуктов нитрования до
аминов делает возможным получение характерных производных (см. разд. Д,2.6).
Такие нитросоединения, как пикриновая и стифниновая кислоты, 1,3,5-три-
нитробензол, 2,4-динитрофенилгидразин, 3,5-динитробензойная кислота и др.,
являются важными реактивами для идентификации органических соединений
(см.разд.Д,2.1.1.3,Д,2.2.7.2иД,2.5).
5.1.4. СУЛЬФИРОВАНИЕ
Самыми распространенными сульфирующими средствами являются 70— 100%-ные
растворы серной кислоты и олеум с различным содержанием SO3.
Считают, что собственно сульфирующим агентом является серный ангидрид
или катионы HSO3®:
11
S°3H [Г.5.17]
436 Г Препаративная часть
или соответственно
+ Н® ©/Я
H2SO4 + SO3 ^=^ H2S2O7 === H2SO4 + HO-S4
H(j) \\
[Г.5.18]
1 С © — w
O^ VOH + H©
Сульфирование в противоположность нитрованию и большинству других
реакций электрофильного замещения в ряду ароматических соединений предс-
представляет собой обратимую реакцию:
ArH + H2SO4 == ArSO3H + Н2О [Г.5.19]
Гидролиз сульфоновых кислот в зависимости от их устойчивости происходит
под действием серной кислоты различной концентрации или даже воды, осо-
особенно при повышенной температуре.
Концентрированная азотная кислота также может вытеснять сульфогруппу
{«unco-замещение»). Таким путем можно получать нитросоединения. Метод
имеет практическое значение в том случае, если соответствующее ароматичес-
ароматическое соединение неустойчиво по отношению к азотной кислоте. Так, пикрино-
пикриновую кислоту B,4,6-тринитрофенол) можно получить через устойчивую к окис-
окислению фенолдисульфоновую-2,4 кислоту:
ОН
X. /SO3H
__ Г | +3HNO3
-2Н2О k^/J -2H2SO4, - Н2О
SO3H NO2
В принципе этим же методом можно получать 2,4-динитронафтол-1 (желтый
Марциуса)>и 2,4-динитронафт-1-ол-7-сульфоновую кислоту (нафтоловый жел-
желтый S).
Поскольку реакция сульфирования обратима, положение, в которое
сульфогруппа вступает в ароматическое ядро, может зависеть от условий реак-
реакции. Так, при сульфировании нафталина при низких етмпературах (<80 °С,
кинетически контролируемый процесс, см. разд. В,3.2) получают главным об-
образом ос-нафталинсульфоновую кислоту. Однако при более высокой темпера-
температуре A80 °С, термодинамически контролируемый процесс) равновесие [Г.5.21]
так сильно сдвигается в сторону исходных веществ, что а-кислота снова рас-
распадается на исходные компоненты. Поэтому здесь получается р-нафталин-
сульфоновая кислота1, процесс образования которой в этих условиях необ-
необратим:
1 Последние данные указывают, что процесс идет не путем перегруппировки а-кислоты в
р-изомер.
5. Замещение в ароматическом кольце
437
SO3H
-so3
180'С
[Г.5.21]
SO3H
Обратимость реакции сульфирования можно использовать для защиты реак-
ционноспособных положений ароматического кольца.
При сульфировании нужно подбирать активность сульфирующего агента в
соответствии с реакционной способностью ароматического ядра. Серная кисло-
кислота как наименее активное из распространенных сульфирующих средств может
применяться только для сульфирования реакционноспособных соединений.
В ходе сульфирования скорость реакции уменьшается в результате разбавления
водой серной кислоты, образующейся в реакции, благодаря чему реакция при-
приостанавливается. Чтобы сместить равновесие сульфирования как можно больше
вправо, следует либо применять избыток серной кислоты (что, однако, может
затруднить выделение продукта), либо удалять воду. В простейшем случае этого
можно достигнуть азеотропной перегонкой (см. разд. А,2.3.5), для чего добавля-
добавляют подходящий растворитель (хлороформ, лигроин) или избыток сульфируемо-
сульфируемого соединения. Ароматические амины сульфируют, нагревая сухие гидросуль-
гидросульфаты (метод спекания) или длительно нагревая с серной кислотой:
NH3® HSO4G
HO3S
NH2 + Н2О
[Г.5.22]
Для сульфирования менее реакционноспособных соединений чаще всего
применяют олеум. Реакции проводят с 5—30%-ными растворами олеума при
различных температурах, что зависит от реакционной способности данного со-
соединения и желательной степени сульфирования. Так, 10%-ный олеум сульфи-
сульфирует бензол при комнатной температуре до монокислоты, а при 200—250 °С — до
jw-дисульфоновой кислоты.
Для сульфирования в мягких условиях подходят растворы серной кислоты
или триоксида серы в различных растворителях (хлороформе, жидком диоксиде
серы).
Адцукты SO3 с третичными основаниями (например, пиридином) или циклическими
эфирами (например, диоксаном) представляют собой селективные сульфирующие агенты,
особенно пригодные для сульфирования ароматических соединений с повышенной
я-электронной плотностью. Их реакционная способность снижается при уменьшении
нуклеофильности растворителя. Обсудите различную реакционную способность SO3 в
нитробензоле, хлороформе и жидком SO2.
В промышленности все большее значение приобретает сульфирование в газовой фазе
смесью триоксида серы и воздуха.
438 Г Препаративная часть
Образование сульфонов — обычная побочная реакция при сульфировании,
когда уже образовавшаяся сульфоновая кислота действует как сульфирующий
агент (напишите схему реакции!). Этот побочный процесс можно исключить,
если применять большой избыток серной кислоты (или олеума, или хлорсуль-
фоновой кислоты). Высокие температуры способствуют этой реакции.
Серная кислота (в еще большей степени олеум) при повышенных температу-
температурах может окислять органические соединения (выделение SO2!), приводя к обуг-
обугливанию.
Сульфоновые кислоты, за исключением аминосульфоновых кислот (внут-
(внутреннее солеобразование), хорошо растворимы в воде и представляют собой
сильные кислоты. Они, как правило, растворимы и в избытке сульфирующего
агента, поэтому выделение сульфоновых кислот часто встречает трудности. Во
многих случаях натриевую соль сульфоновой кислоты высаливают из водных
растворов хлоридом или сульфатом натрия:
ArSO3H + NaCI ^=^ ArSO3Na + HCI [Г.5.23]
Натриевые соли непосредственно используются для последующих реакций.
Бариевые и кальциевые соли сульфоновых кислот в противоположность
сульфатам, как правило, растворимы в воде. Поэтому избыточную серную кис-
кислоту можно удалить в виде сульфата щелочноземельного металла. Из солей ще-
щелочноземельных металлов сульфоновую кислоту можно выделить в чистом виде
с помощью ионообменных смол.
Проблему выделения можно также обойти, если в качестве сульфирующего
агента применять хлорсульфоновую кислоту. При этом образуются сульфонил-
хлориды, которые труднорастворимы и в воде гидролизуются медленнее, чем
большинство ацилхлоридов:
ArH + CISO3H ArSO3H + HCI
[Г.5.24]
ArSO3H + CISO3H - ArSO2CI + H2SO4
Свободную сульфоновую кислоту легко получают из сульфонилхлорида гид-
гидролизом. Во многих случаях сульфонилхлориды даже удобнее, чем сульфоновые
кислоты или их соли. Поэтому в лаборатории сульфохлорирование часто пред-
предпочитают сульфированию. Как видно из схемы [Г.5.24], на каждый моль арена
необходимо не менее двух молей хлорсульфоновой кислоты. При сульфирова-
сульфировании менее реакционноспособных соединений хлорсульфоновая кислота часто
берется с большим избытком, чтобы подавить образование сульфонов. Иногда
выходы зависят в значительной степени от чистоты хлорсульфоновой кислоты
(см. часть Е). В отличие от большинства сульфоновых кислот, многие сульфо-
хлориды можно перегонять.
^у) Общая методика сульфохлорирования аренов (табл. Г.5.25)
JJ Внимание1. При работе с хлорсульфоновой кислотой соблюдайте меры предос-
Щ торожности (тяга, защитные очки, защитные перчатки!) (см. также часть Е).
При загрузках 0,5 моль ароматического соединения реакцию ведут в литровой трехгорлой
колбе, снабженной мешалкой, обратным холодильником с газоотводной трубкой, внутренним
термометром и при необходимости капельной воронкой.
Таблица Г.5.25. Сульфохлорирование ароматических соединений
Продукт реакции
л-Нитробензолсульфонил-
хлорид
Бензолсульфонилхлорид
л-Толуолсульфонилхлорид
о-Толуолсульфонилхлорид
л-Этилбензолсульфонил -
хлорид
л-Пропилбензолсульфонил-
хлорид
Исходное
соединение
Нитробензол
Бензол
Толуол
Толуол
Этилбензол
Пропилбензол
Вариант
А
Б
Б
Б
Б
Б
Физические константы
Т.пл. 62 °С (эфир)
Т. кип. 114°С
A0 мм рт. ст.);
т. пл. 14,5 "С; по20 1.5521
Т. пл. 69 °С (петролей-
ный эфир)
Т. кип. 126 °С
A0 мм рт. ст.);
по20 1,5565
Т. кип. 168 °С
A5 мм рт. ст.);
по* 1,5469
Т. кип. 170 °С
A5 мм рт. ст.)
Выход, %
75
75
30
25
60
60
Примечания
Остаток после перегонки
представляет собой дифе-
нилсульфон; т. кип. 225 СС
A0 мм рт.ст.);т. пл. 128 °С
(метанол)
Работают при температуре <5°G
после упаривания хлоро-
хлороформа лара-изомер вымора-
вымораживают и отфильтровыва-
отфильтровывают (см. рис. А.40).
Если добавлять хлорсуль-
фоновую кислоту к толуо-
толуолу, то образуется только
л-изомер с выходом 65%
орто-Изомер выделяют из
фильтрата перегонкой с ко-
колонкой Вигре
Таблица Г.5.25. Продолжение
Продукт реакции
л-Изопропилбензолсульфо-
нилхлорид
л-Бутилбензолсульфонил-
хлорид
л-Ацетамидобензолсульфо-
нилхлорид
Л-Хлорбензолсульфонил-
хлорид
л-Метоксибензолсульфонил-
хлорид
Исходное
соединение
Изопропил бензол
Бутилбензол
Ацетанилид
Хлорбензол
Анизол
Вариант
Б
Б
Б
Б
В
Физические константы
Т. кип. 142 "С
A2 мм рт. ст.)
Т. кип. 182°С
A5 мм рт. ст.)
Т. пл. 149 °С (ацетон)
Т. кип. 147 °С
A5 мм рт. ст.);
т. пл. 53 °С (эфир)
Т. кип. 105 °С
@,3 мм рт. ст.);
т. пл. 42 °С
(петролейный эфир)
Выход, %
60
60
80
80
55
Примечания
Ацетанилид добавляют при
15°С, реакцию проводят
при 60°С; для очистки про-
продукт растворяют в неболь-
небольшом количестве ацетона при
35 °С, охлаждают до
—10 °С, отфильтровывают
и промывают охлажденным
льдом толуолом
Хлорбензол добавляют по
каплям к раствору хлор-
сульфоновой кислоты в
хлороформе B50 мл на
1 моль кислоты); смесь пе-
перемешивают 1 ч при 25°С
5. Замещение в ароматическом кольце 441
A. Малореакционноспособные ароматические соединения. К арену сразу прибавляют трех-
трехкратный мольный избыток чистой хлорсульфоновой кислоты и при перемешивании медлен-
медленно нагревают смесь до 110—120 °С так, чтобы выделение хлороводорода происходило непре-
непрерывно. В заключение температуру повышают на 10 "С. Процесс заканчивают, когда хлорово-
дород перестает выделяться. Дальнейшую обработку см. ниже.
Б. Ароматические соединения средней реакционной способности. Арен добавляют по каплям
при перемешивании и охлаждении до 0—5 °С к трехкратному мольному избытку хлорсульфо-
хлорсульфоновой кислоты. Перемешивают при комнатной температуре, пока не прекратится выделение
хлороводорода.
B. Реакционноспособные ароматические соединения (моносульфохлорированиё). Исходное со-
соединение растворяют в сухом хлороформе B50 мл на каждый моль) и к нему по каплям при-
прибавляют 2 моль хлорсульфоновой кислоты при хорошем перемешивании и охлаждении до
приблизительно -10 °С. Перемешивание при этой температуре продолжают до прекращения
выделения хлороводорода. Затем нагревают реакционную смесь до комнатной температуры и
перемешивают до прекращения выделения хлороводорода.
Обработка. Реакционную смесь очень осторожно при хорошем перемешивании выливают
на измельченный лед (тяга!) Сульфонилхлорид отфильтровывают (если он твердый) или
экстрагируют хлороформом, тетрахлоруглеродом или бензолом (если он жидкий). Твердые
сульфонилхлориды тщательно промывают водой, а экстракты жидких промывают водой, раст-
раствором гидрокарбоната натрия, а затем снова водой. Наконец, либо перегоняют (при этом вода
удаляется в виде азеотропной смеси), либо перекристаллизовывают предварительно высушен-
высушенное на воздухе вещество.
Реакция может быть проведена с полумикроколичествами. В таком виде она имеет значе-
значение для качественного анализа ароматических соединений.
Получение нафтол-2-сульфоновой-1 кислоты сульфохлорированием р-наф-
тола и ее дальнейшая переработка в 2-аминонафталинсулъфоновую-1 кислоту
{кислоту Тобиаса): Fuerz-David H. E., Blangey L. Grundlegende Operationen der
Farbenchemie, 8. Aufl. — Springer-Verlag, Wien, 1952, S. 189.
Сульфохлорирование (общая методика для качественного анализа). В пробирке растворяют 0,5 г
реакционноспособного арена в 3 мл хлороформа и добавляют по каплям при охлаждении
льдом 3 мл хлорсульфоновой кислоты. Оставляют на 20 мин при комнатной температуре, за-
затем осторожно выливают на примерно 30 г размолотого льда, отделяют хлороформный слой и
промывают его водой. Хлороформ удаляют выпариванием и сырой продукт перекристаллизо-
перекристаллизовывают или переводят в сульфонамид (см. разд. Г,8.5).
Для аренов с малой и средней реакционной способностью аналитическую
методику видоизменяют в соответствии с условиями общей методики сульфо-
хлорирования.
Ф Получение пиридинсульфоновой-3 кислоты [McElvain S. M., Goese M. A. J. Am. Chem. Soc,
1943, 65, 2233]
Н Осторожно при обращении с олеумом! Защитные очки! Работать в вытяжном
Л шкафу (SO3)!
В колбу на 500 мл помещают 400 г 20—22%-ного олеума и осторожно, медленно, по каплям
при хорошем перемешивании и охлаждении ледяной водой добавляют 1 моль пиридина. Затем
прибавляют 2,5 г сульфата ртути @,8 мол. %); колбу снабжают насадкой Кляйзена, к которой
присоединяют воздушный холодильник, а через вакуумный форштос — сосуд с небольшим
количеством концентрированной серной кислоты. Реакционную смесь нагревают на металли-
металлической бане 20 ч при 200-230 °С. Затем отгоняют в вакууме 230—240 г серной кислоты
[т. кип. 180 °С B мм рт. ст.)].
442 Г Препаративная часть
К темно-коричневому маслянистому остатку добавляют при охлаждении 200 мл абсолют-
абсолютного этанола и оставляют раствор при 0 °С на несколько часов для кристаллизации продукта.
После отсасывания сульфоновую кислоту растворяют в 500 мл воды, пропускают через раст-
раствор сероводород для осаждения еще содержащихся в ней солей ртути, нагревают суспензию до
80 °С и отфильтровывают выпавший сульфид ртути. Фильтрат упаривают до начала кристал-
кристаллизации, добавляют 150 мл этанола, охлаждают и сульфоновую кислоту отфильтровывают.
Т. шт. 352-356 °С; выход 40%.
Ф Получение л-толуосульфоновой кислоты. В трехгорлой колбе на 500 мл, снабженной во-
водоотделителем (см. рис. А.83а) и мешалкой, кипятят с обратным холодильником на ме-
металлической бане смесь 2 моль чистого толуола и 0,5 моль концентрированной серной
кислоты до прекращения выделения воды (около 5 ч). Поскольку применяемые реагенты так-
также содержат влагу, количество выделившейся воды бывает несколько больше рассчитанного.
После охлаждения к реакционной смеси прибавляют 0,5 моль воды; при этом сульфокис-
лота выкристаллизовывается в виде гидрата. Для удаления избытка толуола и побочно образо-
образовавшейся о-толуолсульфоновой кислоты полученную массу фильтруют на воронке со стек-
стеклянным фильтром, а затем прессуют на глине. Для окончательной очистки гидрат л-толуол-
сульфоновой кислоты растворяют в небольшом количестве горячей воды, кипятят некоторое
время с активированным углем, горячий раствор фильтруют, охлаждают; охлажденный раст-
раствор насыщают хлороводородом. Выпавшие кристаллы быстро отсасывают на воронке со стек-
стеклянным фильтром и промывают холодной концентрированной соляной кислотой. Такую
очистку повторяют дважды: гидрат кислоты сушат затем в эксикаторе над КОН и концентри-
концентрированной серной кислотой (см. разд. А,1.10.3), пока проба на хлороводород не будет отрица-
отрицательной. Кристаллы имеют вид бесцветных призм. Т. пл. 105 °С (в запаянном капилляре);
выход 40%. Вещество очень гигроскопично.
Гидрат л-толуолсульфоновой кислоты можно перекристаллизовать из большого количест-
количества хлороформа или дихлорэтана [Perron R. Bull. Soc. Chim. France, 1952, 966].
(f|^ Получение пикриновой кислоты1.
Внимание! При синтезе выделяются оксиды азота. Работать под тягой! Осто-
Осторожно при обращении с концентрированными кислотами! Защитные очки!
Пикриновая кислота — взрывчатое вещество. Большие количества должны
храниться только во влажном состоянии (около 10% воды).
В колбе Эрленмейера на 500 мл к 0,5 моль фенола добавляют 1,5 моль концентрированной
серной кислоты и нагревают 1 ч на кипящей водяной бане; при этом образуется дисульфоновая
кислота. Колбу охлаждают смесью поваренной соли со льдом до 0 °С и при этой температуре
медленно по каплям при перемешивании прибавляют смесь 2 моль азотной кислоты (d = 1,5) и
равного (по массе) количества концентрированной серной кислоты. Реакционную смесь остав-
оставляют на ночь при комнатной температуре, затем нагревают 1 ч при 30 °С, после чего медленно
повышают температуру до 45 °С. Для завершения реакции часть реакционной смеси (около
50 мл) нагревают на кипящей водяной бане; к подогретому раствору при перемешивании до-
добавляют остальное количество реакционного раствора, следя, чтобы раствор не очень бурно
вспенивался и не было сильного выделения нитрозных газов2. После этого нагревают еще 2 ч на
кипящей водяной бане, осторожно добавляют 50 мл воды и охлаждают на ледяной бане. Выпав-
Выпавшие кристаллы отсасывают, хорошо промывают холодной водой и перекристаллизовывают из
разбавленного этанола A объем спирта +2 объема воды). Т. пл. 122 °С; выход 90%.
Метод пригоден для полумикроколичеств.
1 Описанная методика аналогична проведению реакции в промышленности.
2 При таких условиях можно безопасно работать с бо'льшими загрузками. При меньших
загрузках @,2 моль и меньше) можно осторожно нагревать на водяной бане сразу всю реакци-
реакционную смесь.
5. Замещение в ароматическом кольце 443
Ряд ароматических сульфоновых кислот имеет промышленное значение. Высшие алкил-
бензолсульфонаты с алкильным остатком из 12—15 углеродных атомов все шире применяются
в качестве моющих средств и средств для химической чистки (табл, Г.5.43). Низшие алкилнаф-
талинсульфонаты (прежде всего соединения с бутильным остатком) используются как смачи-
смачивающие, эмульгирующие и флотационные средства («некали»). Поскольку сульфогруппа мо-
может быть заменена на гидроксильную сплавлением с гидроксидом натрия (см. разд. Г,5.2.2), то
из соответствующих сульфоновых кислот получают, например, фенол, резорцин, нафтолы,
ализарин. Сульфаниловая кислота и большое число сульфированных нафтолов и нафтилами-
нов представляют собой важные полупродукты при получении растворимых в воде азокрасите-
лей. Сульфированием сшитого полистирола получают сильнокислую ионообменную смолу.
Процессы сульфохлорирования используются в промышленности для получения хлор-
сульфоновых кислот для последующего синтеза лекарственных препаратов и средств защиты
растений (сульфонамиды, сульфонилмочевины, см. разд. Г,8.5), а также о-толуолсульфонил-
хлорида (синтез сахарина, см. разд. Г,6.2.1).
В аналитической химии сульфохлорирование применяется для идентифика-
идентификации алкилированных и галогенированных ароматических соединений. В лабо-
лабораториях сульфонилхлориды применяются, кроме того, как исходные соедине-
соединения при синтезе сульфиновых кислот, тиофенолов и др., а также при идентифи-
идентификации гидрокси- и аминосоединений (см. разд. Г,8.5 и Д,2.1.1.2).
5.7.5. ГАЛОГЕНИРОВАНИЕ
Галогенирующими средствами служат в первую очередь сами галогены. Правда,
фтор очень агрессивен к углерод-углеродным связям и разрушает ароматичес-
ароматические соединения. Поэтому нельзя получить при прямом фторировании вполне
определенные фторароматические соединения.
Хлор, бром и иод в неполярных растворителях реагируют очень медленно.
Под действием сильнополярных растворителей, или так называемых переносчи-
переносчиков галогенов (кислот Льюиса, например, А1С13, FeCb, а также металлического
железа), молекула галогена поляризуется и приобретает свойства кислоты
Льюиса (см. разд. Г,4.1.1). Тем самым электрофильное замещение значительно
облегчается:
н
+ CI-CI + FeCI3 > К © I "' + CI®--FeCI3 > Г \\ + HCI + FeCI3 [Г.5.26]
Низкие энтропии активации таких реакций галогенирования указывают на
то, что катализатор специфически связывается в переходном комплексе, как это
показано на схеме [Г.5.26]. Реакционная способность галогенов возрастает от
иода к хлору.
Галогенирование протекает очень энергично, если использовать агенты, в
которых галоген в результате поляризации имеет сильный положительный заряд
или даже существует как катион. Так, очень инертный л*-динитробензол удается
быстро при комнатной температуре бромировать, например, действием дибром-
изоциануровой кислоты в концентрированной серной кислоте1. Считают, что
бромирующим агентом в этом случае является протонированная форма
Gottardi W. Monatsh. Chemie, 1968, 99, 815.
444 Г Препаративная часть
дибромизоциануровой кислоты:
о о
н 1 н
+ сАгАо [Г52?1
NO2 H
Сравните условия реакции с другими препаративными методиками. Ту же
реакцию с выходом 52% можно провести за 11 ч при 100 °С действием брома в
концентрированной серной кислоте в присутствии сульфата серебра1. Полага-
Полагают, что в этом случае промежуточно образуется бром-катион:
2Br2 + Ag2SO4 2Br® + 2AgBr + SO|® [Г.5.28]
Кислоты типа хлорноватистой в кислой среде также дают галоген-катионы:
Н®+Н0-Вг — Н2О-Вг ^ Н2О + Вг® [Г.5.29]
Такие кислоты образуются, например, при реакции галогена с водой (напи-
(напишите схему реакции!). Источником хлорноватистой кислоты может служить и
хорошо дозируемый хлорамин Т в кислом растворе:
н3с—<f у—so2-n-ci + на + н2о —- н3с—<f V-so2-nh2 + ноа + Naci [Г5 30]
W Na® W
Реакционная способность элементного иода в реакциях замещения в арома-
ароматическом ядре незначительна, так что прямое иодирование возможно только в
случае фенола и ароматических аминов. Добавление окислителей (концентри-
(концентрированная серная кислота, азотная кислота или оксид ртути), которые необходи-
необходимы для образования иод-катионов и связывания образующегося свободного HI,
способствует прямому иодированию инертных аренов..
Практическое значение реаций галогенирования ограничивается тем, что
большинство ароматических соединений дает смеси различных изомернозаме-
щенных продуктов галогенирования, которые, как правило, трудно разделить.
При галогенировании алкилароматических соединений приходится, кроме
того, считаться с конкурирующей реакцией радикального замещения в боковой
цепи (см. разд. Г, 1.4). Установлено, что нагревание до температуры кипения, сол-
солнечный свет способствуют галогенированию боковой цепи. Напротив, охлаждение,
катализатор благоприятствуют галогенированию ядра.
При отсутствии переносчика галогенов в условиях, которые способствуют
радикальным реакциям, галогенирование протекает преимущественно в боко-
боковую цепь.
В лаборатории проще всего осуществлять бромирование. Бромирование
является наиболее простым процессом для осуществления в лабораторных усло-
условиях. Каким образом необходимо учитывать различие в реакционной способ-
DerbushireD. H., Waters W.A.J. Chem. Soc. [London], 1950, 573.
5. Замещение в ароматическом кольце 445
ности ароматических соединений1 при выборе условий галогенирования, видно
на примере приведенной ниже общей методики бромирования. Так, бромиро-
вание реакционноспособных ароматических соединений (фенолов, их простых
эфиров, аминов) нужно проводить в разбавленном растворе при низкой темпе-
температуре, если необходимо получить монозамещенные производные. В этих слу-
случаях удобно вводить бром в реакционную смесь, распыляя его при помощи тока
воздуха, проходящего через промывную склянку с бромом.
Ф Общая методика бромирования ароматических соединений элементным бромом (табл.
Г.5.31)
Ц Внимание! Осторожно при работе с бромом (см. часть Е). Капельная воронка
в должна быть хорошо укреплена (плотность брома 3,14 г/см3!!
Бромирование 0,5 моль ароматического соединения проводят в трехгорлой колбе на 250 мл,
снабженной мешалкой, обратным холодильником, внутренним термометром и капельной
воронкой. Выделяющийся бромоводород растворяют в воде и затем перерабатывают в бромо-
водородную кислоту (см. разд. Г, 1.4.2).
Целесообразно перед синтезом высушить бром, встряхивая его с концентрированной
серной кислотой.
A. Инертные соединения. 0,6 моль ароматического соединения нагревают с 4 г порошка желе-
железа (лучше всего восстановленного железа) при перемешивании до 100-150 "С (см. табл. Г.5.31) и
при этой температуре как можно быстрее добавляют 0,35 моль брома так, чтобы из холодильни-
холодильника улетучивалось минимальное количество брома. Для сокращения потерь брома трубку капель-
капельной воронки опускают почти до поверхности жидкости. По окончании добавления реакцион-
реакционную смесь перемешивают еще 1 ч при указанной температуре и затем вводят еще 4 г восстанов-
восстановленного железа и 0,35 г моль брома. После перемешивания в течение 2 ч при 150 °С продукт
реакции отгоняют с водяным паром (должно быть не менее 2 л дистиллята), извлекают дихлор-
метаном или хлороформом, тщательно промывают 10%-ным раствором гидроксида натрия и
водой и отгоняют растворитель. Остаток перегоняют или перекристаллизовывают.
Б. Соединения средней реакционной способности. К 0,5 моль ароматического соединения и
1 г порошка железа добавляют по каплям при комнатной температуре и хорошем перемешива-
перемешивании 0,5 моль брома. Если после добавления небольшого количества брома через некоторое
время, необходимое для индукционного периода, бромоводород не выделяется, то реакцион-
реакционную смесь можно осторожно подогреть до 30-40 °С. После того как реакция начинается, про-
продолжают работать при комнатной температуре. Реакционную смесь оставляют на ночь, затем
промывают водой, к которой добавлено немного гидросульфита натрия, 10%-ным раствором
гидроксида натрия, снова водой и перегоняют в вакууме.
B. Реакциожоспособные соединения. Растворяют 0,5 моль ароматического соединения в
200 мл тетрахлоруглерода, раствор охлаждают до 0 °С и при хорошем перемешивании медлен-
медленно, по каплям прибавляют 0,4 моль брома (или соответственно больше, если должно быть
введено большее число атомов брома) в 50 мл тетрахлоруглерода; температура должна под-
поддерживаться в пределах 0—5 °С (смесь хлорида натрия со льдом). После того как бром добав-
добавлен, перемешивание продолжают еще 2 ч при той же температуре, доводя до конца. Обработ-
Обработка, как в методике Б.
Реакцию можно проводить в полумикромасштабе, особенно если не образуются трудно-
разделяемые изомеры и продукт реакции твердый.
Бромирование ацетофена в присутствии хлорида алюминия, который предо-
предотвращает бромирование боковой цепи: Пирсон Д. X, Поп X., Харгров У. В сб.:
Синтезы органических препаратов. Сб. 12. — М.: 1964, с. 11.
Например, скорости хлорирования фторбензола и анизола различаются в 107 раз.
Таблица Г.5.31. Бромирование ароматических соединений элементным бромом
Продукт реакции
я-Бромнитробензол
2-Бром-4-нитротолуол
м-Бромбензойная кис-
кислота
Бромбензол(+л-дибром-
бензол)
л-Бром-грег-бутилбен-
зол
Броммезитилен
1-Бром-2-метилнафта-
лин
n-Бром анизол
п-Бром фенол8
2,4-Дибромфенолв
Исходное
соединение
Нитробензол
4-Нитротолуол
Бензойная кислота
Бензол
грег-Бутилбензол
1,3,5-Триметилбен-
зол (мезитилен)
2-Метилнафталин
Анизол
Фенол
Фенол
Вари-
Вариант
А
А
А
Б
Б
Б
Б
В
В
В
Т. кип., 'С
(мм рт. ст.)
138 A8)
156; 54 B0)
105 A4)
105 A6)
155 A4)
108 B0)
122 A5)
154 A1)
Т. пл., "С
56 (разб.
этанолj
77 (разб.
этанол)
155 (вода)
63 (хлоро-
(хлороформ)
40
1,5598
1,5309
(при 25 °С)
1,5527
1,6487
1,5605
Выход,
%
60
80
70а
65
75
40б
80
75
60
70
Примечания
Работать при 145—
1 ?,Г\ аГ
Работать при 120—
loU *-.
Перегонять на 30-санти-
30-сантиметровой колонке Вигре.
Остаток содержит л-ди-
бромбензол, т. пл. 89 °С
(этанол)
Часто кристаллизуется
лишь при охлаждении
твердым СО2 в метаноле
Работают при 140—150 °С; перемешивают 2 ч при 150 "С и затем еще 3 ч при 260 "С. С паром не перегоняют, продукты реакции раст-
растворяют в растворе карбоната натрия, фильтруют и л-бромбензойную кислоту осаждают разбавленной соляной кислотой.
Берут 0,6 моль брома, реакцию проводят в темноте. Сырой продукт содержит гидролизуемый бром (в боковых цепях). После про-
промывания кипятят 3 ч с обратным холодильником с 100 мл 10%-ного спиртового раствора КОН, выливают в 400 мл воды, отделяют, про-
промывают до нейтральной реакции и перегоняют.
в Осторожно! При обработке не промывать реакционную смесь щелочью! Соединение обладает отвратительным и очень навяз-
навязчивым запахом.
5. Замещение в ароматическом кольце
447
Таблица Г.5.32. Бромирование малоактивных аренов дибромизоциануровой
кислотой
Продукт
л-Бромнитробензол
6-Бром-2,4-динитрото-
луол
1 -Бром-3,5-динитробен-
зол
З-Бромбензойная кис-
кислота
3-Б ромацетофенон
Исходное
вещество
Нитробензол
2,4-Динитротолуол
ж-Динитробензол
Бензойная кислота
Ацетофенон
Т.пл., "С
51 (этанол)
55 (этанол)
72 (этанол)
150 (водн. мета-
метанол)
Т. кип. 131 °С
A6 мм рт. ст.);
/id20 1,5755;
т. пл. 8°С
Выход, %
70
80
80
75
50
Бромирование фенолов (методика для качественного анализа). 7,5 г бромида калия растворяют в
50 мл водь1 и добавляют 5 г брома. Этот раствор добавляют по каплям при встряхивании к 0,5 г
фенола в воде, диоксане или этаноле, пока не появится устойчивая светло-желтая окраска.
После добавления 20 мл воды отсасывают бромированный продукт, промывают разбавленным
раствором гидросульфита натрия и перекристаллизовывают из этанола или водного этанола.
Ф Общая методика бромирования малоактивных аренов дибромизоциануровой кислотой
(табл. Г.5.32)
Получение дибромизоциануровой кислоты [Gottardi W. Monatsh. Chem., 1967, 98, 507]
11 Внимание! Соблюдайте осторожность при работе с бромом (см. часть Е).
| Защитные очки!
К раствору 0,2 моль гидроксида лития и 0,1 моль тщательно измельченной в ступке циану-
ровой кислоты в 1 л воды при 20 °С в один прием добавляют 0,4 моль B0 мл) брома1. Энергич-
Энергичным встряхиванием бром переводят в раствор и оставляют смесь медленно охлаждаться в холо-
холодильном шкафу в течение 24 ч, периодически встряхивая. Продукт отфильтровывают, промы-
промывают небольшим количеством охлажденной (на ледяной бане) бромной воды, воду удаляют
тщательным прессованием на необожженной глиняной тарелке, сушат в вакуум-эксикаторе
целый день над КОН, затем 24 ч над Р2О5. Т. пл. 307-309 °С (с разложением); выход 90%.
Продукт без дальнейшей очистки применяют для бромирования.
Бромирование дибромизоциоануровой кислотой [Gottardi W. Monatsh. Chem., 1968, 99, 815]
Внимание! Осторожно обращайтесь с
Защитные очки!
концентрированной серной кислотой!
15 ммоль ароматического соединения (в случае ацетофенона 30 ммолей) растворяют при
комнатной температуре в стаканчике в 16 мл концентрированной серной кислоты. Включив
магнитную мешалку, по каплям прибавляют на каждые 15 ммоль арена 7,5 ммоль раствора
дибромизоциануровой кислоты в 24 мл концентрированной серной кислоты. Бромирующий
агент растворяют в серной кислоте при слабом нагревании. Реакционную смесь оставляют на
45 мин при комнатной температуре, затем выливают на лед. Твердые продукты реакции, нера-
1 Для ускорения растворения циануровой кислоты можно применить нагревание, однако
перед прибавлением брома раствор необходимо охладить до 20 °С.
448 Г Препаративная часть
створимые в щелочах, для отделения от циануровой кислоты кипятят с разбавленным гидрок-
сидом натрия, отсасывают, промывают водой, перекристаллизовывают. Если продукт реакции
растворим в щелочах или представляет собой масло, то после выливания на лед его экстраги-
экстрагируют дихлорметаном (циануровая кислота не растворяется в дихлорметане), экстракт промы-
промывают водой, сушат сульфатом натрия. После отгонки растворителя продукт перекристаллизо-
перекристаллизовывают или перегоняют в вакууме.
2,6-Дибром-4-нитроаншшн: Meyer R., Meyer W., Taeger K. Ber. Deut. Chem.
Ges., 1920, 53, 2034.
В промышленности в качестве галогенирующего агента применяется главным образом хлор,
причем в первую очередь в больших количествах получают хлорбензол и некоторые хлорфенолы.
Ранее хлорбензол перерабатывали преимущественно в ДЦТ (см. разд. Г,5.1.8.5). Распростра-
Распространенный прежде синтез фенола из хлорбензола ныне в значительной мере вытеснен технически
более выгодным получением фенола по Хоку (см. разд. Г,9.1.3). Образующийся при хлорирова-
хлорировании бензола и-дихлорбензол применяется для борьбы с насекомыми (в частности, как средство
против моли). Хлорфенолы и хлоркрезолы используются как дезинфицирующие средства.
2,4-Дихлор- и 2,4,5-трихлорфенолы — исходные вещества для получения соответствую-
соответствующих хлорфеноксиуксусных кислот (см. разд. Г,2.6.2), находящих применение как селективные
средства для борьбы с сорняками. Пентахлорфенол — важный препарат для защиты древеси-
древесины. Кроме того, моно- и полихлорбензолы являются промежуточными продуктами при полу-
получении красителей и лекарственных препаратов.
Многократно или полностью бромированные арены (политрибромстирол, пентабромто-
луол, бромированный дифениловый эфир и др.) применяют в качестве пламягасителей, а по-
лииодзамещенные ароматические соединения используются в качестве контрастных веществ
в рентгенографии.
5.1.6. ТИОЦИАНИРОВАНИЕ (РОДАНИРОВАНИЕ)
С помощью псевдогалогена диродана (дициандисульфана) (SCNJ удается ввес-
ввести тиоцианогруппу (—SCN) в активные арены. Реакция проходит при комнат-
комнатной температуре или ниже ее. Поскольку диродан склонен к полимеризации,
целесообразно получать его непосредственно в реакционной смеси из тиоциа-
ната аммония (или тиоцианатов щелочных металлов) и брома или хлора:
2SCN0+Br2 ~ (SCNJ + 2BrG [Г.5.33]
В реакцию роданирования вступают фенолы, ароматические амины, кон-
конденсированные ароматические углеводороды (например, антрацен), некоторые
гетероциклы и С—Н-кислотные соединения. Такие дополнительные заместите-
заместители, как —NO2, —Hal, —СООН, —COOR, не мешают реакции с активированны-
активированными ароматическими соединениями, если свободны орто- или «ара-положения
по отношению к донорному заместителю. Тиоцианатная группа вступает преи-
преимущественно в иора-положение, а если оно занято, то в op/no-положение. При
этом иара-замещенные анилины легко претерпевают замыкание цикла с образо-
образованием 2-аминобензотиазолов:
+ (SCNJ + HSCN Г j| ' [ I VNH2 [Г.5.34]
В качестве растворителей пригодны ледяная уксусная кислота, муравьиная
кислота или насыщенные хлоридом (бромидом) натрия метанол или метилаце-
5. Замещение в ароматическом кольце
449
Таблица Г.5.35. Введение тиоцианатной группы
Продукт реакции
л-Аминофеиилтиоцианат
я- (N.N-Диметиламино) фенил-
тиоцианат
л-Гидроксифенилтиоцианат
2-Амино-6-этоксибензотиазол
М-B-Аминобензотиазол -6-ил)-
ацетамид
2-Амино-6-хлорбензотиазол
2-Амино-6-метоксибензотиазол
2-Амино-6-метилбензотиазол
Исходное вещество
Анилин
N.N-Диметилаии-
лин
Фенол
л-Фенетидин
4-Аминоацета-
нилид
4 Хлоранилин
п-Анизидин
га-Толуидин
Т. пл., "С
58 (этанол — вода,
циклогексан)
74 (лигроин с
т. кип. 90—
100°С)
58 (метанол —
вода)
174 (этанол—вода)
192 (этанол—вода)
198 (этанол —
вода)
165 (метанол)
130 (этанол—вода)
Выход,
%
60
65
80
90
75
85
тат. Гидролизом сильными щелочами (щелочное плавление) как из тиоциано-
аренов, так и из 2-аминобензотиазола можно получить соответствующие тиофе-
нолы (см. также разд. Г,8.5).
(yj) Общая методика введения тиоцианатной группы (табл. Г.5.35)
I] Из неокисленной тиоциановой кислоты в ходе побочной реакции образуется
И синильная кислота. Работать под тягой!
0,1 моль ароматического соединения, 0,22 моль щелочной или аммониевой соли тиоциа-
тиоциановой кислоты в 75 мл ледяной уксусной кислоты в стаканчике охлаждают до 10—20 °С. При
механическом перемешивании прибавляют по каплям раствор 0,1 моль брома в 20 мл ледяной
уксусной кислоты, поддерживая температуру ниже 20 °С. Реакционную смесь оставляют на
3 ч при комнатной температуре, выливают в шести-восьмикратное количество воды, при
охлаждении нейтрализуют концентрированным аммиаком, оставляют для кристаллизации,
продукт отсасывают и очищают перекристаллизацией.
5.1.7. АЛКИЛИРОВАНИЕ ПО ФРИДЕЛЮ - КРАФТСУ
Подобно галогенам, алкилгалогениды могут быть так сильно поляризованы
кислотами Льюиса (хлоридами алюминия и цинка, трифторидом бора и др.),
что они становятся способными к электрофильному замещению в ароматичес-
ароматическом ядре:
R-CI + AICI
R —CI---AICI3
I
R®AICI4°
[Г.5.36]
(Напишите схему реакции алкилирования!)
Поляризация связи R—X, обусловленная образованием комплекса (схема
[Г.5.36]), возрастает от первичных к третичным алкилгалогенидам (почему?),
следовательно, электрофильная активность алкилгалогенидов увеличивается в
450 Г Препаративная часть
этой же последовательности1. Реакционная способность уменьшается от алкил-
фторидов к алкилиодидам (ср., однако, ацилирование по Фриделю — Крафтсу,
разд. Г,5.1.8.1), так как образование комплекса с катализатором затрудняется с
_ ростом радиуса галогена. Наряду с алкилгалогенидами в качестве алкилирую-
щих средств применяют алкилтозилаты, спирты и некоторые олефины:
R-OH + Н-Х =^= R-O--H--X =^ R®+ Н2О + X® [Г.5.37]
н
© 0
R-CH=CH2 + H2SO4 == R-CH-СНз + HSO4 [Г.5.38]
Реакция с олефинами протекает в соответствии с правилом Марковникова.
При алкилировании олефинами или спиртами в качестве катализаторов
обычно применяют протонные кислоты. Их активность падает в ряду
H2F2 > H2SO4 > (Р4О10) > Н3РО4 [Г.5.39]
Каталитическое действие кислот Льюиса тоже различно:
AICI3 > FeCI3 > SbCI5 > SnCI4 > BF3 > TiCI4 > ZnCI2 [Г.5.40]
Кислоты Льюиса, вероятно, действуют тоже только в виде протонных кис-
кислот; последние образуются в присутствии воды, спиртов или галогеноводоро-
дов, например:
НХ + BF3 н® + XBF3® [Г.5.41]
Указанные последовательности активностей нельзя считать абсолютными,
так как активность катализатора зависит также от условий реакции и строения
взаимодействующих соединений.
Спирты требуют в качестве катализаторов по меньшей мере молярных коли-
количеств кислот Льюиса (так как вода, образующаяся при реакции, инактивирует
эквимолярное количество катализатора), в то время как в реакциях с участием
алкилгалогенидов и олефинов достаточно добавлять незначительные количест-
количества катализатора.
В лаборатории алкилирование по Фриделю — Крафтсу имеет ограниченное
значение, так как обычно при этой реакции образуются неоднородные продук-
продукты, что обусловлено следующими причинами.
а) Образующийся продукт алкилирования более основен, чем исходное
соединение, поэтому дальше преимущественно алкилируется продукт.
Если хотят получить продукты моноалкилирования, то берут большой
избыток ароматического соединения.
б) Как и сульфонирование, реакция алкилирования по Фриделю — Крафт-
Крафтсу обратима.
1 Комплексы алкилгалогенидов с активными катализаторами реакции Фриделя — Крафт-
са обычно существуют в виде ионных пар типа II в схеме [Г.5.29], что следует из образования
перегруппированных продуктов алкилирования.
5. Замещение в ароматическом кольце 451
Выше описанные правила замещения действуют только до тех пор, пока алкилирование
протекает в условиях кинетического контроля (см. разд. В,3.2). Поэтому процесс должен быть
вовремя остановлен, что удается сделать при низких скоростях реакции, т. е. при мягких усло-
условиях ее проведения (при пониженной температуре и недостатке катализатора; см. описание
методики). В условиях же термодинамического контроля, т. е. при более высоких температу-
температурах, большем времени реакции и больших количествах катализатора, при алкилировании за-
замещенных ароматических соединений чаще всего образуются .м-замещенные продукты. Так,
например, при метилировании толуола метилхлоридом в присутствии хлорида алюминия при
О °С образуется 27% л«-ксилола, при 55 °С — уже 87%, а при 90 °С — даже 98% этого изомера.
Кроме того, при использовании высокоактивных катализаторов легко протекают процессы
дезалкилирования и переалкилирования. Например, в присутствии хлорида алюминия наряду
с л«-ксилолом образуются бензол, толуол, триметилбензол и т. д., тогда как при использовании
в качестве катализаторов серной кислоты, фтороводорода, фторида бора или других среднеак-
тивных катализаторов такие побочные продукты образуются в минимальных количествах.
в) даже в мягких условиях первичные и вторичные алкилгалогениды дают
преимущественно вторичные или третичные алкиларены соответственно.
Это становится понятным для условий, приближающихся к SnI -реакции
(см. схему [Г.2.6]I. Перегруппировки можно избежать, если работать при
низких температурах.
Алкилирование н-олефинами также ведет к смеси вторичных алкиларенов,
так как промежуточно образующийся карбениевый ион изомеризуется согласно
схеме [Г.4.17].
Перегруппировки могут претерпевать также и алкильные группы, уже нахо-
находящиеся в ароматическом кольце в качестве заместителей. Однако такие превра-
превращения протекают только в более жестких условиях.
Поскольку синтез связан с перечисленными выше осложнениями, далее
приведена лишь методика алкилирования бензола. Относительно гладко реаги-
реагируют также фенолы и их простые эфиры, в то время как алкилирование менее
основных ароматических соединений (например, нитробензола и пиридина) в
таких условиях не идет.
Ф Общая методика алкилирования бензола по Фриделю — Крафтсу (табл. Г.5.42). Реакцию про-
проводят в трехгорлой колбе на 1 л, снабженной мешалкой, внутренним термометром, капель-
капельной воронкой и обратным холодильником с хлоркальциевой трубкой, выход из которой
шлангом соединен с колбой Эрленмейера, наполовину наполненной водой. При этом конец
шланга не должен быть опущен в воду, но должен заканчиваться над ее поверхностью (Тяга!).
В реакционную колбу помещают:
A) при действии алкилгалогенидов: 5 моль свободного от тиофена сухого бензола (см. часть
Е), 0,1 моль безводного хлорида алюминия;
Б) при действии спиртов: 5 моль свободного от тиофена бензола, 1 моль безводного хло-
хлорида алюминия;
B) при действии олефинов: 5 моль сухого бензола, 1 моль концентрированной серной
кислоты.
К содержимому колбы прибавляют по каплям при перемешивании 1 моль алкилирующе-
го средства. При этом приливают сначала несколько миллилитров без охлаждения и ждут, по-
пока реакция не начнется. Затем при охлаждении ледяной водой прибавляют по каплям остаток
таким образом, чтобы температура в колбе не превышала 20 °С. Часто реакционная смесь об-
образует два слоя. Перемешивание продолжают в течение ночи до окончания выделения хлоро-
1 Электрофильное ароматическое замещение при действии алкилгалогенидов можно так-
также рассматривать как нуклеофильное замещение алкилгалогенида аренами, выступающими в
роли оснований (см. табл. Г.2.4).
452
Г Препаративная часть
Таблица Г.5.42. Алкилирование бензола по Фриделю -
Продукт реакции
Изопропилбензол
(кумол)
грег-Бутилбензол
втор-Ъ утил бенз ола
Циклогексил бензол
Алкилирующее средство
к-Пропилхлорид
н- Пропилбромид
Изопропилхлорид
Изопропилбромид
Изопропиловый спирт
Пропилен6
грег-Бутилхлорид
грег-Бутилбромид
грег-Бутанол
Изобутен8
н-Бутилхлорид
н-Бутилбромид
2-Хлор бутан
2-Бромбутан
Бутанол-2
Циклогексен
Вариант
А
Б
В
А
Б
В
А
Б
В
Крафтсу
Физические
константы
Т. кип.
152 °С;
лв20 1,4915
Т. кип.
169 °С;
/id20 1,4926
Т. кип.
173 °С;
ло20 1,4901
Т. кип.
ПО "С
A0 мм
рт. ст.);
т. пл. 8°С;
лп20 1,5260
Выход, %
80
50
75
60
80
60
60
60
65
Наряду с данным соединением при алкилировании с помощью к-алкилгалогенидов
получают немного к-алкилбензола.
Газообразный олефин. Поэтому сосуд для реакции должен быть снабжен газо-
подводящей трубкой вместо капельной воронки. О дозировании газов см. разд. А. 1.6.
водорода, затем содержимое колбы выливают на лед. Органическую фазу промывают водой,
раствором карбоната натрия и снова водой до нейтральной реакции и сушат сульфатом маг-
магния. Растворитель отгоняют и остаток либо фракционируют на 20-сантиметровой колонке
Вигре, либо перекристаллизовывают.
Тетрахлоруглеродом бензол алкилируется в зависимости от стехиометричес-
кого соотношения до трифенилхлорметана (трифенилметилхлорида, тритил-
хлорида) или дифенилдихлорметана. Если продукт реакции быстро обрабаты-
обрабатывать при низких температурах, то можно выделить эти галогениды; в противном
случае они гидролизуются (см. разд. Г,2.6.1) и образуется трифенилкарбинол
или бензофенон соответственно.
Ф Получение трифенилметилхлорида (тритилхлорида). Прибор состоит из трехгорлой кол-
колбы на 1 л, мешалки, капельной воронки и холодильника с хлоркальциевой трубкой. К
суспензии 0,6 моль хлорида алюминия хорошего качества (см. часть Е) в 6 молях сухо-
сухого, свободного оттиофена бензола (см. часть Е) прибавляют по каплям 0,4 моль хорошо
высушенного тетрахлоруглерода. Перемешивают до тех пор, пока не прекратится выделение
хлороводорода. Затем содержимое колбы выливают при перемешивании на смесь 300 г льда с
300 мл концентрированной соляной кислоты, причем температура не должна быть выше 0 °С.
Органический слой отделяют, трижды промывают охлажденной на льду разбавленной соля-
соляной кислотой, и, наконец, ледяной водой. Пока тритилхлорид находится в соприкосновении
с водой, надо работать по возможности быстро, чтобы предотвратить образование трифенил-
5. Замещение в ароматическом кольце 453
Таблица Г.5.43. Промышленно важные продукты алкилирования по Фрццелю - Крафтсу
Продукт
Применение
Этилбензол
Кумол
Алкилбензолы (Сю—
2,4-Ди-т/>ет-бутилфенол и
2,6-ди-/и/)ет-бутил-4-метил-
фенол
Алкилфенолы (С4—С8)
Алкилфенолы (C
Бутилнафталин
а
Стирол —> полистирол, Буна S
Гидропероксид кумола —»¦ фенол (см. разд. Г, 1.5)
Алкилбензолсульфонаты (ПАВ)
Антиоксиданты
Бактерициды, антиоксиданты
Фенолформальдегидные смолы
Алкилфенилполигликолевые эфиры (см. табл. Г,4.34)
Бутилнафталинсульфонат (см. разд. Г.5.1.4)
Алкилбензолсульфонаты с линейной или малоразветвленной боковой цепью экологически
безопаснее, так как быстрее подвергаются биодеградации.
карбинола. После сушки хлоридом кальция растворитель отгоняют и остаток перекристалли-
зовывают из лигроина с т. кип. 90-100 °С с добавкой небольшого количества ацетилхлорида
или тионилхлорида. Чистый препарат получают перегонкой в высоком вакууме в сабельной
колбе. Т. кип. 170 °С @,4 мм рт. ст.); т. пл. 114 °С; выход 75%.
Ф Получение бензофенона. В прибор, описанный в методике синтеза тритилхлорида, по-
помещают 1,5 моль сухого тетрахлоруглерода (см. часть Е) и 0,3 моль хлорида алюминия
хорошего качества (см. часть Е). Колбу охлаждают до 10-15 °С и добавляют сразу 2 мл
(при больших загрузках также не более 2 мл!) из общего количества 0,7 моль бензола. Когда ре-
реакция начнется, поддерживают температуру 5-10 °С и прибавляют по каплям остаток бензола
(строгий контроль температуры!). После прибавления всего бензола продолжают перемешива-
перемешивание еще 3 ч при 10 °С, а затем оставляют на ночь при комнатной температуре.
Обратный холодильник заменяют нисходящим. Через капельную воронку осторожно при-
прибавляют 250 мл воды (охлаждать не нужно, так как галогенопроизводное должно быть гидро-
лизовано). Избыток тетрахлоруглерода отгоняют. Наконец, для гидролиза дигалогенида про-
пропускают через раствор в течение 30 мин водяной пар. После охлаждения органический слой
отделяют, водный слой извлекают бензолом, объединенные органические слои промывают
водой и сушат сульфатом магния. После отгонки растворителя продукт фракционируют в
вакууме. Т. кип. 190 "С A5 мм рт. ст.); т. пл. 48 °С; выход 65%.
С помощью алкилирования по Фриделю — Крафтсу возможна обратимая
защита различных положений в ароматическом ядре при электрофильном заме-
замещении. Для этого вводят трет-бутлъный остаток, который будучи объемным,
защищает, кроме того, два соседних с ним ор/яо-положения и может быть снова
отщеплен в виде изобутилена либо удален путем переалкилирования.
Изомеризацию ксилолов используют в технике для получения я-ксилола. При этом в ка-
качестве катализатора применяют платинированную кремневую кислоту на глине. Выход ксило-
ксилолов можно повысить за счет реакций переалкилирования в системе толуол — три- или тетра-
метилбензол. В промышленности реакция Фриделя — Крафтса имеет очень большое значение,
особенно если в качестве алкилирующих средств используются олефины. Важнейшие
промышленные продукты приведены в табл. Г.5.43.
454
Г Препаративная часть
Промышленный полный синтез токоферола (витамин Е) включает стадию алкилирования
по Фриделю — Крафтсу 2,3,5-триметилгидрохинона изофитолом:
[Г.5.43а]
5.1.8. ЭЛЕКТРОФИЛЬНОЕ АРОМАТИЧЕСКОЕ ЗАМЕЩЕНИЕ ПРИ
ВЗАИМОДЕЙСТВИИ С КАРБОНИЛЬНЫМИ СОЕДИНЕНИЯМИ
Такие карбонильные соединения, как альдегиды, кетоны, карбоновые кислоты
и их производные, а также родственные им соединения (например, имидохло-
риды карбоновых кислот) представляют собой кислоты Льюиса, так как имеют
полярную карбонильную группу (см. разд. Г,7) и поэтому в принципе способны
к электрофильному замещению в ароматических соединениях:
О5"
R-SC° [Г.5.44]
X
Электрофильная активность этих соединений, однако, относительно неве-
невелика и должна быть повышена действием кислот Льюиса или протонных кислот.
При этом кислотный катализатор Е (схема [Г.45]) атакует атом кислорода карбо-
карбонильного соединения (или атом азота соединения, аналогичного карбонильно-
карбонильному) и, смещая электронную плотность, повышает положительный заряд сосед-
соседнего атома углерода;
8"
R_5^"E v-u R. [Г.5.45]
П чу Л — П. П
\
X
В случае хлорангидридов кислот катализатор может атаковать как атом
кислорода, так и атом галогена (см. схему [Г.5.47]). •
В качестве катализаторов применяют соединения, используемые для синте-
синтезов по Фриделю — Крафтсу и перечисленные в предыдущем разделе; там же
обсуждалась и их эффективность; см. ряды [Г.5.39] и [Г.5.40].
Реакционная способность карбонильных соединений возрастает в ряду (под-
(подробнее см. ряд [Г.7.3]):
Р
со2 < —с
NR2
о
OAr1
о
—с
—С
—С X = галоген, остаток кислоты [Г.5.46]
Сложные эфиры алифатических спиртов взаимодействуют как алкилирующие агенты.
5. Замещение в ароматическом кольце 455
Применение электрофильного ароматического замещения с помощью кар-
карбонильных соединений ограничено: ароматические ядра с сильноинактивирую-
щими заместителями (-NO2, -COR, -CN) не реагируют вообще, если только
эффект этих групп не компенсируется дополнительными гидрокси-, алкил-,
амино- и другими группами.
Самые реакционноспособные карбонильные соединения — ацилхлориды —
могут еще взаимодействовать по Фриделю — Крафтсу в присутствии очень эф-
эффективного хлорида алюминия с относительно инертными галогенобензолами,
в то время как хлорметилирование формальдегидом в присутствии хлороводоро-
да и хлорида цинка происходит лишь с ароматическими соединениями, имею-
имеющими реакционную способность, близкую к реакционной способности бензола.
Формилирование амидами кислот в присутствии РОСЬ по Вильсмейеру удается
гладко только для полициклических углеводородов, фенолов и их эфиров, ами-
аминов. Наконец, очень инертный диоксид углерода реагирует без добавления
электрофильного катализатора только с самыми реакционноспособными аро-
ароматическими соединениями — фенолятами.
5.1.8.1. Ацилирование по Фриделю - Крафтсу
Ацилирование ароматических соединений по Фриделю — Крафтсу является
важнейшим методом синтеза жирноароматических кетонов. В качестве ацили-
рующего средства применяют ацилгалогениды (чаще всего ацилхлориды), ан-
ангидриды кислот, а иногда также карбоновые кислоты.
При взаимодействии амбидентного ацилхлорида с катализатором реакции
Фриделя — Крафтса могут образовываться собственно электрофильные агенты
I, II и карбениевый ион III:
5? 8+//О ^
R-C = R-C 8. =- R-C = O+AICI4°
CI С1-АЮ13 11 .ЭЛ/]
I II III
Положение равновесия зависит от природы реагентов и от растворителя: вы-
высокая диэлектрическая проницаемость благоприятствует образованию III.
(Напишите общие схемы реакций ацилирования арена электрофильными
агентами I и II!)
Поскольку в большинстве случаев промежуточно образующийся комплекс
(Г.5.47,1 или II) имеет значительный объем, то при реакции Фриделя — Крафт-
Крафтса с монозамещенными бензолами, как правило, образуются исключительно
и-изомерные соединения. Благодаря такой региоселективности процесс ацили-
ацилирования по Фриделю - Крафтсу имеет важное препаративное значение.
Выбор катализатора определяется реакционной способностью ароматичес-
ароматического соединения. Чаще всего применяют хлорид алюминия и только для очень
реакционноспособных систем (например, для тиофена) используют хлорид
цинка, серную кислоту и др.
Тригалогениды алюминия образуют комплексы и с ацилирующим сред-
средством, и с образующимся карбонильным соединением; комплекс с последним в
условиях реакции устойчив. Для синтезов по Фриделю — Крафтсу с ацилгалоге-
456 Г Препаративная часть
нидами необходимы поэтому по меньшей мере мольные количества катализато-
катализатора. При взаимодействии с ангидридами кислот получающаяся кислота связыва-
связывает еще один моль катализатора, поэтому в целом необходимы по крайней мере
два моля. В каждом случае по окончании реакции образовавшийся комплекс
кетона с хлоридом алюминия должен быть гидролитически разрушен (соляной
кислотой со льдом).
Ацилирование по Фриделю - Крафтсу более реакционноспособных, чем
бензол, аренов и гетероаренов удается и с каталитическими количествами ката-
катализаторов, в качестве которых используют хлорид железа(Ш), иод, хлорид цин-
цинка или железо. Считается, что при необходимых для этих реакций высоких тем-
температурах комплекс катализатора с карбонильным соединением диссоциирует;
после этого катализатор вновь вступает в реакцию.
Трифторметансульфоновая кислота может катализировать ацилироваиие по Фриделю —
Крафтсу действием ацилхлоридов. По-видимому, промежуточно при этом образуется ангид-
ангидрид карбоновой и сульфоновой кислот:
x=/ CI
О
CI
82%
Реакцию ацилирования по Фриделю — Крафтсу удается распространить на
ароматические углеводороды (в том числе полициклические), галогенопроиз-
водные и реакционноспособные гетероциклы (например, тиофен, фуран). Аро-
Ароматические амины образуют с катализатором неацилирующийся комплекс.
Если же аминогруппа защищена ацетилированием, то реакция удается.
Фенолы реагируют неоднозначно, и поэтому для получения ароматических
гидроксикетонов предпочитают внутримолекулярную перегруппировку сложных
эфиров фенолов в присутствии хлорида алюминия {перегруппировка Фриса):
СОСН3
Д1П~ I I
ОСОСНз
[Г.5.49]
А\ ]) _ ОН
Н3С
А1С13
20'С
н3с
СОСНз
Ароматические соединения с сильноинактивирующими заместителями,
например с нитро-, циано- и карбонильными группами, не ацилируются по
Фриделю — Крафтсу. Поэтому при ацилировании можно не опасаться вторич-
вторичного и полизамещения.
Наряду с взаимодействием по Фриделю — Крафтсу с простыми ацилхлори-
дами и ангидридами кислот препаративный интерес представляет взаимодей-
5. Замещение в ароматическом кольце 457
ствие с ангидридами дикарбоновых кислот. При этом получают оксокислоты,
которые в дальнейшем можно перевести в хиноны, например:
о 9 °
с
О
о 6
Антрахинон
Ароилбензойные кислоты применяются для идентификации алкил- и гало-
генопроизводных (см. часть Д). [Напишите схему получения 1,4-дигидрокси-
антрахинона (хинизарина)!].
Растворителем при ацилировании по Фриделю — Крафтсу может служить
избыток ацилируемого углеводорода. Очень часто применяют сероуглерод, так
как он практически не влияет на реакционную способность хлорида алюминия.
Комплекс образовавшегося ароматического кетона с хлоридом алюминия оста-
остается при этом чаще всего в твердой фазе, поэтому при больших загрузках реак-
реакционная смесь с трудом перемешивается и обрабатывается. Кроме того, сероуг-
сероуглерод ядовит и очень легко воспламеняется (уже нагретые до 100 °С предметы
вызывают опасность воспламенения, см. также разд. А, 1.7.2). В нитробензоле
или галогеноуглеводородах (дихлорэтане или трихлорэтилене) активность ката-
катализатора несколько понижена из-за комплексообразования, но ацилирование
по Фриделю — Крафтсу в них можно вести в значительной степени гомогенно.
Галогеноуглеводороды можно применять только при температурах ниже 50 °С,
так как в противном случае они сами вступают в реакцию.
В менее полярном 1,2-дихлорэтане (этиленхлориде) из нафталина получают
а-кетон, а в сильнополярной среде (нитробензоле) — C-кетон (см. ниже).
(ГП) Общая методика ацилирования ацилхлоридами по Фриделю — Крафтсу (табл. Г.5.51)
fj Внимание! Выделяется хлороводород. Тяга!
В трехгорлой колбе на 1 л, снабженной мешалкой и капельной воронкой с хлоркальциевой
трубкой, смешивают 400 мл 1,2-дихлорэтана с 1,2 моль тонкорастертого хлорида алюминия и
добавляют по каплям при перемешивании и охлаждении ледяной водой 1,05 моль ацилхлори-
да. Затем из капельной воронки при охлаждении водой добавляют 1 моль ароматического
соединения так, чтобы температура смеси поддерживалась около 20 "С. Реакционную смесь
перемешивают еще 1 ч и оставляют на ночь. При ацилировании галогенобензолов нагревают
5 ч при 50 °С, причем ацилируемое соединение используется и в качестве растворителя (все
количество галогенобензола сразу помещают в колбу).
Для разложения комплекса кетона с хлоридом алюминия содержимое колбы осторожно
выливают на 500 мл льда, выпадающий гидроксид алюминия переводят в раствор, добавляя
небольшое количество концентрированной соляной кислоты. Затем в делительной воронке
отделяют органический слой, а водный дважды экстрагируют дихлорэтаном. Объединенные
вытяжки тщательно промывают водой, 2%-ным раствором гидроксида натрия и снова водой.
После сушки карбонатом калия растворитель отгоняют, а кетон перегоняют в вакууме.
Приведенная методика пригодна также для полумикросинтезов.
Получение $-нафггшлметилкетона ацилированием нафталина в нитробензо-
нитробензоле: Bassilios H. F., MakarS. M., Salem A. Y. Bull. Soc. Chim. France. 1958, 1430.
З-Нитробензофенон из бензола и л-нитробензоилхлорида: Oelschldger H. Arch.
Pharmaz. Ber. Deut. Pharmaz. Ges., 1957, 290, 587.
Таблица Г.5.51. Получение кетонов ацилированием по Фриделю - Крафтсу
Продукт реакции
Ацетофенон
Пропиофенон
Бутирофенон
4-Фенилацетофенон
4-Метилацетофенон
2,4-Диметилацетофенон
Метил-а-нафтилкетон"
4-Метоксиацетофенон
3,4-Диметоксиацетофенон
4-Хлорацетофенон
4-Бромацетофенон
Исходное вещество
Бензол
Бензол
Бензол
Дифенил
Толуол
л-Ксилол
Нафталин
Анизол
Вератрол
Хлорбензол
Бромбензол
Ацилирующее
средство
Ацетилхлорид
Пропионилхлорид
Бутирилхлорид
Ацетилхлорид
»
>
»
Т. кип., °С
(мм рт. ст.)
94 B0)
92 (И)
105 (II)
195—210 A8)
ПО A4)
93 E)
166 (I2N
139 A5)
155 (9)
118 B0)
130 A5)
Т. пл., °С
20
21
12
120 (этанол)
39
21
50
V
1,5340
1,5270
1,5202
1,5340
Выход,
%
70
70
70
60
70
75
60
60
60
80
80
а Нафталин прикапывают в растворителе, добавляют 1,2 моль ацетилхлорида; работают в интервале температур 20—30 °С.
6 Содержит около 5% метил-р-нафтилкетона. Для чистого соединения лр20 1,6285.
5. Замещение в ароматическом кольце 459
$-Бензоилпропионовая кислота из бензола и янтарного ангидрида: Сомер-
Сомервилл Л., Аллен К. В сб.: Синтезы органических препаратов. Сб. 2. — М.: ИЛ,
1949, с. 95.
$-Бензоилакриловая кислота из бензола и малеинового ангидрида: Груммит О.,
Беккер X., Миссе К. В сб.: Синтезы органических препаратов. Сб. 4. — М.: ИЛ,
1953, с. 87.
а-Тетралон из бензола и у-бутиролактона: Олсон Ц., БэдэрА. В сб.: Синтезы
органических препаратов. Сб. 6. — М.: ИЛ, 1956, с. 49.
а-7е/и/?а/гонизу-фенилмасляной кислоты: Мартин X., ФизерЛ. В сб.: Синте-
Синтезы органических препаратов. Сб. 2. — М.: ИЛ, 1949, с. 448.
2-Ацетил- и 2-бензоилтиофены: Hartough H. D., KosakA. I. J. Am. Chem. Soc,
1946, 68, 2639.
Ацилирование аренов карбоновыми кислотами по Фриделю — Крафтсу в прису-
присутствии полифосфорной кислоты: Klemm L. H., Bower G. M. J. Org. Chem., 1958, 23,
344.
4,4'-Диметоксибензофенон и 2-бензоилтиофен ацилированием по Фриделю —
Крафтсу с использованием каталитических количеств кислот Льюиса: Pearson D. Е.,
Buhler С. A. Synthesis, 1972, 533.
Ацилирование аренов по Фриделю — Крафтсу ацилхлоридами, катализируемое
трифторметансульфоновой кислотой: EffenbergerF., Epple G. Angew. Chem., 1972,
84, 295.
Ацилирование ароматических углеводородов фталевым ангидридом (общая методика для качественно-
качественного анализа). К смеси 0,5 г углеводорода, 0,6 г фталевого ангидрида в 2-3 мл сухого дихлорметана до-
добавляют при охлаждении льдом 2,5 г тонкоизмельченного хлорида алюминия. В зависимости от
интенсивности реакции оставляют после этого при комнатной температуре или нагревают с обрат-
обратным холодильником, пока не перестанет выделяться хлороводород (около 30 мин). Охлажденный
продукт реакции разлагают, добавляя 5 мл смеси концентрированной соляной кислоты и льда,
твердый осадок отсасывают и промывают водой. Для очистки его растворяют при нагревании в
5 мл концентрированного раствора карбоната натрия, кипятят 5 мин с небольшим количеством
активированного угля, фильтруют горячим и после охлаждения подкисляют разбавленной A:1)
соляной кислотой по конго красному. Выпавшую ароилбензойную кислоту перекристаллизовы-
вают из водного спирта или из смеси толуола с петролейным эфиром.
Многие получаемые по реакции Фриделя — Крафтса кетоны являются важными проме-
промежуточными продуктами при промышленном синтезе лекарственных препаратов. Например,
пропиофенон используется в синтезе эфедрина, а безофенон — в синтезе метадона (анальге-
(анальгетик, более сильный, чем морфин, используется в качестве замены героина в программе борь-
борьбы с наркоманией). О структуре этих соединений прочтите в учебнике. Замещенные бензофе-
ноны (например, кетон Михлера) служат исходными веществами для трифенилметановых
красителей. Бензофеноны, содержащие гидроксильные группы, используют как поглотители
УФ-излучения, применяющиеся для придания устойчивости к свету искусственным материа-
материалам, а также в составе солнцезащитных средств.
5.1.8.2. Синтез Гатгермана
Ацилированием по Фриделю — Крафтсу формилгалогенидами можно было бы
получать ароматические альдегиды, что и удается при использовании формил-
фторида (напишите схему реакции!). Вместо неустойчивого формилхлорида
можно применять смесь хлороводорода и монооксида углерода в присутствии
хлорида алюминия и хлорида медиA) (синтез Гатгермана — Коха).
460 Г Препаративная часть
Каталитический эффект хлорида медиA), вероятно, обусловлен его способностью связы-
связывать монооксид углерода в комплекс, устойчивый при высоком давлении.
Получение п-толуилового альдегида: Колеман Г. X, Крейг Д. В сб.: Синтезы
органических препаратов. Сб. 2. — М.: ИЛ, 1949, с. 464.
Альдегиды, которые не удается получить методом Гаттермана — Коха из фе-
фенолов и их простых эфиров, часто можно синтезировать с помощью синильной
кислоты и хлороводорода в присутствии хлорида алюминия или хлорида цинка
(синтез Гаттермана):
/NH +AICI3 /JH-AICIa
H-C=N + HCI ^=^ Н-С' - Н-С' [Г.5.52]
Cl CI
Электрофильным агентом здесь является комплекс катализатора с форм-
имидохлоридом. Вследствие этого при синтезе образуется вначале гидрохлорид
альдимина, который при нагревании с разбавленными кислотами или основа-
основаниями легко гидролизуется до альдегида:
АгН + HCN + HCI АЮ'3 - ArCH=NH2CIG Гидр0Лиз. ДгСНО [Г.5.53]
Совершенно аналогично протекает синтез кетонов по Губену — Гешу, где
вместо синильной кислоты применяются нитрилы.
Модификация синтеза Гаттермана по Адамсу позволяет исключить приме-
применение свободной синильной кислоты. Последнюю получают непосредственно в
ходе реакции действием хлороводорода на цианид цинка. Одновременно при
этом получается хлорид цинка, активность которого как катализатора бывает
достаточна для реакции с реакционноспособными фенолами. В других случаях
надо дополнительно добавлять хлорид алюминия.
Синтез Гаттермана осуществим не только для фенолов и их эфиров, но так-
также и для некоторых углеводородов и гетероциклических соединений (производ-
(производных фурана, пиррола, индола; незамещенные соединения не реагируют) или ти-
офена. При наличии заместителей, инактивирующих ядро, реакция не идет.
Синтез неприменим для ароматических аминов (почему?).
Альдегидная группа вступает (с хорошей избирательностью) обычно в пара-
положение к активирующей группе и лишь затем — в ор/ио-положение, если
пара-положенж занято.
Общую методику и другие примеры применения синтеза Гаттермана —
Адамса см.: Органикум. — М.: Мир, 1979, т. I, с. 425.
5.1.8.3. Синтез Вильсмейера
При синтезе Вильсмейера в качестве формилирующего агента применяют ами-
амиды муравьиной кислоты. Особенно часто применяют диметилформамид и
N-метилформанилид. N-Метилформанилид несколько реакционноспособнее,
чем диметилформамид, хотя последний дешевле. Катализатором обычно служит
оксихорид фосфора, образующий с амидом комплекс, который в случае N-ме-
тилформанилида был выделен:
5. Замещение в ароматическом кольце 461
ое о
i CI ii о
\ /Р \JL,,°~P^ \_®_Я~Р~С| Ci
N-C + POCI3 > N-C ci Cl ^=±: N-C ci
' н / Vi 7 Vi
[Г.5.54]
a k© Cl л—ъ. N— r—j. О
\\ + /М^орос!2 _ R^-Q-foroci, H20. ^-F\-$
Вместо оксихлорида фосфора применяют также фосген (особенно в про-
промышленности).
Синтез Вильсмейера применим к реакционноспособным ароматическим со-
соединениям, прежде всего к полициклическим соединениям, фенолам, их прос-
простым эфирам, а также к реакционноспособным гетероциклам, содержащим
кислород, серу и азот. В отличие от синтезов Гаттермана, Гаттермана — Коха и
Гаттермана — Адамса в эту реакцию также вступают вторичные и третичные
ароматические амины.
Область применения синтеза Вильсмейера в последнее время значительно
расширилась за счет применения винилогов формамидов1, причем образуются
ненасыщенные альдегиды, например:
О г л О
+ R'2N-CH=CH-C/ 3. ¦ R2N—^ у— СН=СН-с' [Г.5.55]
н 2 \=/ Vi
Растворителями в синтезе Вильсмейера обычно служат бензол, хлорбензол,
о-дихлорбензол или избыток диметилформамида.
При применении N-метилформанилида температура реакции не должна
превышать 70 "С, так как в противном случае он перегруппировывается в л-ме-
тиламинобензальдегид.
^у) Общая методика формилирования по Вильсмейеру (табл. Г.5.56)
JJ Внимание*. Оксихлорид фосфора обладает раздражающим действием! Тяга!
И Защитные очки!
Реакцию проводят в трехгорлой колбе на 250 мл, снабженной мешалкой, обратным холо-
холодильником с хлоркальциевой трубкой, внутренним термометром и капельной воронкой.
К смеси арена и N-метилформанилида или диметилформамида при хорошем перемешива-
перемешивании и охлаждении ледяной водой прибавляют по каплям оксихлорид фосфора так, чтобы темпе-
температура в колбе не поднималась выше 20 °С. Перемешивание продолжают еще 1 ч при этой же
температуре, а затем температуру повышают (указания см. в последней графе табл. Г.5.56).
В зависимости от реакционной способности ароматического соединения применяют
различные количества формилирующего комллекса.
Вариант А. 0,2 моль ароматического соединения, 0,3 моль N-метилформанилида и
0,3 моль оксихлорида фосфора нагревают 3 ч (температура указана в табл. Г.5.56).
Вариант Б. 0,2 моль ароматического соединения, 0,2 моль N-метилформанилида и
0,2 моль оксихлорида фосфора нагревают 2 ч при 60 "С.
О принципе винилогии см. разд. В,5.1 и Г,7.4.
Таблица Г.5.56. Получение альдегидов методом
Продукт реакции
Анисовый альдегид
4-Этоксибензальдегид
2-Метоксинафтальдегид-1
4-Метоксинафтальдегид-1
2,4-Диметоксибензальде-
гид
3,4-Диметоксибензальде-
гид
4-Диметиламинобензаль-
дегид
4-Диэтиламинобензаль-
дегид
2,4-Дигидроксибензаль-
дегнд
Тиофенкарбальдегид-2
Индолкарбальдегид-3
Коричный альдегид
Исходное вещество
Анизол
Фенетол
2-Метоксинафталин
1 -Метоксинафталин
Диметиловый эфир ре-
резорцина
Вератрол
N.N-Диметиланилин
N.N-Диэтиланилин
Резорцин
Тиофен
Индол
Стирол
Вильсмеиера
Вари-
Вариант
А
А
А
А
Б
Б
В
В
В
В
В
В
Т. кип., *С
(мм рт. ст.)
135 A6)
140 B0)
205 A8)
210 A0)
ПО @,1)
169 B1)
166 A7)
124 B)
198
129 B0)
Т. пл., °С,
39
84 (этанол)
34 (этанол)
70 (води, этанол
или лигроин)
45 (циклогексан)
73 (води, этанол)
41 (води, этанол)
136 (вода)
1,5888
192 (этанол)
1,6195
Выход,
Ж
30
30
65а
80
85
40
80
80
40б
75
90
30
Примечания
При 60 °С; очищать че-
через аддукт с гидросуль-
гидросульфитом
То же
При 80 "С
Т. кип. 165 °С
A0 мм рт. ст.)
Очищать через аддукт
с гидросульфитом
1,5 ч при 35 °С
1 ч при 80°С
Добавляют 50 мл толуола, температура реакции 80 °С. Сырой продукт растворяют в этаноле, кипятят 5 мин с активированным углем,
фильтруют; добавлением воды к маточному раствору можно выделить дополнительное количество альдегида.
Берут только 0,2 моль диметилформамида, так как в противной случае сравнительно легкорастворимый в воде альдегид может
быть выделен лишь с трудом. Не нагревать!
5. Замещение в ароматическом кольце 463
Вариант В. 0,2 моль ароматического соединения, 0,6 моль диметилформамида (из них
только 0,2 моль необходимо для реакции, остальное служит растворителем), 0,2 моль оксихло-
рида фосфора нагревают 3 ч на водяной бане (это не относится к очень реакционноспособным
или чувствительным к нагреванию ароматическим соединениям; см. табл. Г.5.56).
Для разложения комплекса, получающегося в процессе реакции, к реакционной смеси до-
добавляют при охлаждении 200 г льда и нейтрализуют кислоту до рН 6, добавляя 5 н. раствор гид-
роксида натрия. Твердые выпавшие продукты затем извлекают эфиром или отфильтровывают.
Объединенные эфирные вытяжки нейтрализуют водным раствором гидрокарбоната и сушат
сульфатом натрия. Эфир отгоняют, а остаток очищают перегонкой или перекристаллизацией.
Для некоторых альдегидов простых эфиров фенолов, получающихся лишь с посредствен-
посредственными выходами, рекомендуется очистка через аддукт с гидросульфитом (см. табл. Г.5.56). Для
этого эфирную вытяжку встряхивают с 40%-ным раствором гидросульфита натрия, выпадаю-
выпадающий аддукт отфильтровывают и промывают эфиром. Затем аддукт разлагают нагреванием с
2 н. серной кислотой до прекращения выделения диоксида серы, альдегид извлекают эфиром,
нейтрализуют, сушат и перегоняют.
Если вещества получаются с хорошим выходом, то эту методику можно использовать для
полу ми кросинтезов.
Получение антраценкарбалъдегида-9. Campaigne E., Archer W. L. J. Am. Chem.
Soc, 1953, 75, 989.
Формилирование ароматических соединений диметилформамидом и дибром-
трифенилфосфораном: Bestmann H. J. et al. Lieb Ann. Chem.. 1968, 718, 24.
В качестве формилирующих средств наряду с формамидами могут быть
использованы также «скрытые» производные муравьиной кислоты, такие как
дихлорметилалкиловые эфиры, которые при взаимодействии с "ПСЦ или SnCU
образуют очень реакционноспособные формилирующие реагенты.
Формилирование ароматических и гетероароматических соединений
дихлорметилметиловым эфиром: Gross H., RiecheA., Hoft E., Beyer E. Org. Synth.,
Coll.V. 5A973), 365.
5.1.8.4. Электрофильное замещение при реакциях с формальдегидом
Благодаря высокой реакционной способности, а также по пространственным
причинам формальдегид весьма склонен к участию в электрофильном арома-
ароматическом замещении. С самыми активными аренами (фенолятами) он реаги-
реагирует даже в отсутствие кислотного катализатора, причем гидроксиметилено-
вые группы вступают в орто- и пара-положения (гидроксиметилирование):
н ю*~^ о (о;
Реакцию трудно остановить на этой стадии: как правило, получают многократно
гидроксиметилированные, а в некоторых случаях — высококонденсированные
продукты. Поэтому препаративное использование этих реакций незначительно.
В промышленности, однако, эти процессы широко используются (резолы, см.
[Г.5.61]).
Однако в некоторых случаях благодаря пространственным препятствиям конденсация мо-
может быть направлена на образование с высокими выходами циклических продуктов, так назы-
называемых каликсаренов. В общем случае я-трет-бутилфенолы реагируют с формальдегидом с об-
образованием л-т/>е/л-бутилкаликс[л]аренов — циклических продуктов конденсации с 4, 6 или 8
ароматическими фрагментами в зависимости от условий реакции (температуры, растворителя,
свойств и концентрации основания).
464
Г Препаративная часть
(СН2О)»/ОН
,Э
R = грег-Алкил
[Г.5.57а]
л-3 л=4, 6, 8
Получение п-трет-бутилкаликс[п]аренов: л = 4: Gutsche С. D., Iqbal M.,
Synth., Coll., A993), Vol. VIII, 75-77; п = 6: Gutsche С. D., Dhawan В., Leonis M.,
Stewart D., Oig. Synth., Coll., A993), Vol. VIII, 77-79; n = 8: Munch S. H.,
Gutsche CD., Onj. Synth., Coll., A993), Vol. VIII, 80-81.
Такие реакционноспособные арены, как фенолы и некоторые гетероциклы,
могут вступать в реакцию аминометилирования (при взаимодействии с формаль-
формальдегидом и вторичным амином; см. реакция Манниха, разд. Г,7.2.1.5).
Аминометилирование фенолов 1,3,5-триалкилгексагидро-1,3,5-триазинами:
Reynolds D. D., CosarB. С. J. Heterocycl. Chem., 1971, 8, 597, 605.
Амидометилирование ароматических соединений: Zaugg H. E. Synthesis, 1970,49.
В присутствии кислотных катализаторов формальдегид взаимодействует и с
менее реакционноспособными аренами, например с бензолом. Реакции с арил-
галогенидами требуют уже особо жестких условий и дают неудовлетворительные
выходы. При этом реакция протекает по обычному механизму:
н
сн2он
СН2ОН
[Г.5.58]
-Н'
Однако в условиях реакции (присутствие кислотных катализаторов) взаимо-
взаимодействие не останавливается обычно на стадии бензилового спирта, а путем алки-
лирования по Фриделю — Крафтсу еще не замещенного углеводорода образуются
диарилметаны I (схема [Г.5.59]). Если же замещение у ароматического ядра фор-
формальдегидом проводят при высоких концентрациях хлороводорода, то из проме-
промежуточно образующегося бензилового спирта путем нуклеофильного замещения
получают соответствующий бензилхлорид II (схема [Г.5.59]) (хлорметилирование,
реакция Блана):
+ нш © ©
Аг-СН2ОН ===== Аг-СН2-ОН2 ====== Аг-СН2 + Н20
-н®
@
Аг-СН2
Аг-СН2-Аг + Н® (I)
Ar-CH2-CI (ID
[Г.5.59]
5. Замещение в ароматическом кольце
465
Образование диарилметанов I по схеме [Г.5.59] не всегда можно подавить в
условиях хлорметилирования, особенно если применяемый арен очень реакци-
онноспособен. Поэтому фенолы и их простые эфиры должны вводиться в реак-
реакцию при соблюдении особых мер предосторожности (разбавление инертным
растворителем).
Для более реакционноспособных аренов бывает иногда достаточно катали-
каталитического действия хлороводорода; для реакций с более инертными соединени-
соединениями, чтобы ускорить процесс, необходимо дополнительно вводить катализатор
(серную или фосфорную кислоту, хлорид цинка). Хлорметилирующим агентом
может быть также хлордиметиловый эфир1.
В присутствии следов кислоты хлорметильные соединения легко переходят в
производные диарилметана. (Как это можно объяснить?) Поэтому при перегон-
перегонке целесообразно добавлять немного твердого гидрокарбоната натрия.
VyJS) Общая методика хлорметилирования ароматических соединений (табл. Г.5.60)
Внимание! Промежуточно образующийся гидроксиметилкатион обладает кан-
канцерогенным действием. Тщательно избегайте контактов с реакционной
смесью! Многие галогенметильные ароматические соединения сильно раздра-
раздражают кожу и глаза. Тяга! Защитные очки! Резиновые перчатки!
При ожогах соответствующие места промыть спиртом. До этого никакие мази не
применять, так как они способствуют всасыванию (см. также разд. Г, 1.4.2).
А. Бензол и моноалкиларены. В трехгорлой колбе, снабженной мешалкой, газоподводящей
трубкой и обратным холодильником с хлоркальциевой трубкой, нагревают до 60 °С 4 моль со-
соответствующего соединения (избыток в 3 моль служит в качестве растворителя), 1 моль пара-
формальдегида, 60 г свежеприготовленного плавленого, тонкорастертого порошка хлорида
цинка и одновременно с перемешиванием пропускают энергичный ток сухого хлороводорода
Таблица Г.5.60. Хлорметилирование аренов
Продукт реакции
Бензилхлорид
4-Метилбензилхлоридв
2,4-Диметил бензилхлорид
2,5-Диметилбензилхлорид
2,4,6-Триметилбензил-
хлорид
3,4-Диметоксибензил-
хлорид (вератрилхлорид)
Исходное
вещество
Бензол
Толуол
ж-Ксилол
л-Ксилол
Мезитилен
Вератрол
Используют 30 г хлорида цинка.
6 При 20 "С.
в Содержит 35% 2-метилбензилхлорида;
Вари-
Вариант
А"
А
Б
Б
Б
В
Т. кип., "С
(мм рт. ст.)
70A5)
90B0)
103П2)
103A2)
115A0);
37(т. пл.)
103A);
55 (т. пл.,
лигроин)
для чистого вещества г
л и
"D
1,5390*5
1,5371
1,5368
сю 1,5342.
Выход,
%
75
80
65
60
55
65
1 Внимание'. Имеются данные, что образующийся промежуточный протонированный
формальдегид канцерогенен (ср. также Org. React. 1972, 19, 482).
466 Г Препаративная часть
(см. часть Е). Нагревают до прекращения поглощения хлороводорода (около 20 мин) и израс-
израсходования основного количества параформальдегида. После охлаждения органический слой
тщательно промывают ледяной водой и охлажденным на льду водным раствором гидрокарбо-
гидрокарбоната натрия, тщательно сушат карбонатом калия и перегоняют в вакууме над небольшим коли-
количеством гидрокарбоната натрия. Часть исходного вещества, взятого в качестве растворителя,
перегоняется в виде головной фракции.
Б. Ди- и полиалкиларены. Смешивают 1 моль углеводорода, пятикратное (по массе) коли-
количество концентрированной соляной кислоты с 1,3 моль параформальдегида или с соответству-
соответствующим количеством 40%-ного раствора формалина и нагревают 7 ч при 60—70 °С в трехгорлой
колбе, снабженной мешалкой, обратным холодильником и газоподводящей трубкой, однов-
одновременно пропуская сильный ток хлороводорода. Отделившееся масло растворяют в толуоле и
обрабатывают, как описано в варианте А.
В. Простые эфиры фенола. В трехгорлой колбе, снабженной мешалкой, внутренним термо-
термометром, газоподводящей трубкой и хлоркальциевой трубкой, растворяют 1 моль простого
эфира фенола в 600 мл хлорбензола и при перемешивании и охлаждении (ледяная баня) насы-
насыщают сухим хлороводородом, поддерживая температуру 5—10 °С. Продолжая интенсивно
перемешивать и пропускать хлороводород, вносят 1,3 моль параформальдегида. При этом тем-
температура не должна подниматься выше 20 °С. После перемешивания в течение 60 мин и про-
пропускания хлороводорода сливают жидкость с небольшого осадка на дне колбы; хлорбензоль-
ный раствор промывают и сушат, как описано в варианте А, и перегоняют в вакууме, добавив
гидрокарбонат натрия (на кончике шпателя).
Если продукт хлорметилирования далее используют для получения соответствующего
нитрила (см. разд. Г,2.6.9), то применяют неочищенный продукт, который остается после
отгонки хлорбензола в вакууме.
4-Метоксибензилхлорид {анизилхлорид): MtillerA., Meszaros M., Lempert-Sreter M.,
Srdra I. J. Org. Chem., 1951,16, 1013.
1-Хлорметилнафталин: Груммит О., БэкА. В сб.: Синтезы органических пре-
препаратов. Сб. 3. — М.: ИЛ, 1952, с. 481.
2-Хлорметилтиофень. Виберг К., Мак-Шейн Г. В сб.: Синтезы органических
препаратов. Сб. 4. — М.: ИЛ, 1953, с. 537.
2-Хлорметил-4-нитрофенол (применение хлордиметилового эфира в качест-
качестве источника формальдегидаJ: Бюхлер К., Кирхнер Ф., ДибелДж. В сб.: Синтезы
органических препаратов. Сб. 3. — М.: ИЛ, 1952, с. 478.
Бензилгалогениды, полученные хлорметилированием, очень реакционно-
способны (см. разд. Г,2.6.1); их легко можно перевести в соответствующие спир-
спирты, простые эфиры, нитрилы, кислоты и их производные, амины и альдегиды
(реакция Соммле). Кроме того, группа CH2CI может быть восстановлена до
метильной группы. Метилирование ароматических соединений этим путем дает
меньше побочных продуктов, чем метилирование по Фриделю — Крафтсу
(поскольку реакция хлорметилирования более селективна).
Аналогично хлорметилированию проходит реакция бромметшшрования (ток
бромоводорода). С гомологами формальдегида (уксусным, пропионовым, мас-
масляным альдегидами) реакции галогеналкилирования большей частью проходят
медленнее, поскольку они менее реакционноспособны.
Конденсацией формальдегида с анилином в присутствии кислот по схеме [Г.5.59,1] в про-
промышленности получают в больших количествах л,л'-метилендианилин и далее обработкой
1 Даже при хранении 2-хлорметилтиофена на холоду и в темноте он способен разлагаться
со взрывом с выделением хлороводорода. Не хранить!
2 Внимание] а-Галогенэфиры сильно канцерогенны! См. Org. Reactions, 1972, 19, 422.
5. Замещение в ароматическом кольце 467
фосгеном — п,п '-метиленбис(фенилизоцианат) — исходный продукт для получения полиуре-
полиуретанов (см. разд. Г,7.1.6).
Конденсация формальдегида с фенолами проводится в промышленности в больших
масштабах для получения синтетических материалов (фенолформальдегидных смол). При вза-
взаимодействии в щелочной среде (сода, аммиак, едкий натр) с избытком формальдегида снача-
сначала получают полигидроксиметилированные фенолы (см. схему [Г.5.57]) и далее линейные
поликонденсаты (резолы) со свободными метилольными группами:
ОН
/СН2 -^/^-СН2 -^^/СНг ~^Л^
[Г.5.61]
Н ^^"
СН2ОН СН2ОН
При нагревании резолы претерпевают дальнейшую конденсацию, приводящую к сшиванию
цепей (отвердению). Такие вещества {дуропласты) не растворяются ни в каких известных раст-
растворителях и не плавятся.
При взаимодействии фенола с недостатком формальдегида в кислой среде получаются по-
поликонденсаты (так называемые новолаки), которые не содержат свободных метилольных
групп, при этом плавятся и не отвердевают (являются термопластичными). Однако при нагре-
нагревании с гексаметилентетрамином, который при этом распадается на формальдегид и аммиак,
они также могут отвердевать.
Фенолформальдегидные смолы (бакелиты) принадлежат к самым первым пластмассам,
полученным в промышленности; их производство и в настоящее время является одним из ос-
основных в промышленности пластмасс. Они широко используются в качестве прессовочных
масс (с такими наполнителями, как древесная мука, текстиль, бумага), литьевых смол, а также
в качестве исходных веществ в производстве лаков и клеев.
5.1.8.5. Катализируемые кислотами реакции ароматических соединений
с другими альдегидами или кетонами
Также, как формальдегид, при действии кислотных катализаторов могут взаимо-
взаимодействовать с реагентами и другие альдегиды и кетоны. При этом сначала полу-
получаются соответствующие замещенные бензиловые спирты, которые в условиях,
аналогичных реакции (Г.5.59,1), превращаются в производные дифенилметана.
Примером может служить промышленно важный синтез контактного инсек-
инсектицида ди(и-хлорфенил)-C-трихлорэтана (ДЦТ) из трихлорацетальдегида (хло-
раля) и хлорбензола:
f/ ^\ H2SO4 f/ ^\ f/ ^\
С13С-СНО + 2 (' у— CI ——=«• CI—(' х>— СН—(' у— CI + Н2О гг s fi?l
\ / \ / I \ / L1 -J-u^J
^^ N ' CCI3 Ч~^
ДЦТ является одним из самых эффективных инсектицидов, применение которого, однако,
в Европе запрещено из-за его высокой устойчивости к разложению и способности накапли-
накапливаться в жировых тканях человека и животных. Однако в других странах он до сих пор произво-
производится и используется для борьбы с комарами Anofeles, переносчиками малярии. Его аналог —
2,2-бисD-метоксифенил)-1,1,1-трихлорэтан (метоксихлор), получаемый взаимодействием
хлораля с метоксибензолом (анизолом), обладает несколько меньшей инсектицидной актив-
активностью, однако в отличие от ДЦТ не аккумулируется в организме и является одним из важней-
важнейших безопасных инсектицидов.
Аналогично из ацетона и фенола в присутствии серной кислоты образуется
2,2-бис(л-гидроксифенил)пропан (диан, бисфенол А), играющий важную роль
468 Г Препаративная часть
при получении синтетических материалов (эпоксидных смол, поликарбонатов,
модифицированных фенолформальдегидных смол).
При взаимодействии бензальдегида с ароматическими соединениями обра-
образуются производные трифенилметана, например:
NMe2 + Н2О
[Г.5.63]
Аналогично должны были бы реагировать диарилкетоны типа кетона Михлера (в качестве
карбонильного компонента), образуя замещенные тетрафенилметаны. Однако образующийся
вначале ион карбения I вполне устойчив как в кислом или нейтральном растворе, так и в крис-
кристаллическом виде:
MezN—<ч /X ,—, MezN—<;ч Дон
РОС13
[Г.5.64]
I
Такая высокая стабильность карбениевого иона объясняется тем, что положительный заряд
делокализован по всей молекуле (карбений-иммониевый ион; I и II в схеме [Г.5.64]). Вслед-
Вследствие делокализации я-электронов такие ионы окрашены (цветные соли; основные трифе-
нилметановые красители; см. также разд. А,3.5.1).
Из кетона Михлера и диметиланилина при действии кислотных катализаторов образуется
кристаллический фиблетовый (схема [Г.5.64]), используемый в качестве красителя для копи-
копировальной бумаги и «химического» карандаша.
Ознакомьтесь с другими методами получения красителей трифенилметанового ряда.
Ф Синтез 2,2-ди(л-хлорфенил)-1,1,1-трихлорэтана (ДДТ). В трехгорлой колбе, снабженной
мешалкой, внутренним термометром, капельной воронкой и обратным холодильни-
холодильником, к смеси 0,3 моль безводного хлораля1, 0,5 моль хлорбензола и 70 мл концентриро-
концентрированной серной кислоты прибавляют по каплям при 20-25 °С в течение 30 мин 50 мл 30%-ного
олеума (защитные очки!). Перемешивание продолжают 4 ч при 30 °С, затем осторожно выли-
выливают на 500 г льда. При этом сначала отделяется вязкое маслянистое вещество, которое затвер-
затвердевает достаточно быстро при стоянии. Вещество отсасывают, хорошо промывают водой,
переносят в фарфоровую чашку, где кипящей водой отмывают от кислоты, тщательно раз-
разминая кусочки. Воду декантируют, продукт перекристаллизовывают из спирта.
Т. пл. 108 °С; выход 65%.
Ф Получение кристаллического фиолетового. Смесь 0,02 моль диметиланилина, 0,004 моль
4,4'-бисAЧ,1Ч-диметиламино)бензофенона (кетон Михлера) и 0,01 моль оксихлорида
фосфора нагревают в пробирке 3 ч на кипящей водяной бане. Синий расплав растворя-
растворяют в 50 мл воды, подщелачивают 2 н. раствором гидроксида натрия и избыточный диметил-
1 Соответствующее количество хлоральгидрата встряхивают в делительной воронке с че-
четырехкратным (по массе) количеством теплой концентрированной серной кислоты; отделяю-
отделяющийся хлораль используется для данного синтеза.
5. Замещение в ароматическом кольце 469
анилин отгоняют с водяным паром. После охлаждения «карбинольное основание» отсасыва-
отсасывают, промывают водой, тщательно растирают в ступке и кипятят с 50 мл 0,4%-ной НС1. Горя-
Горячий раствор фильтруют, краситель высаливают, добавляя тонкорастертый хлорид натрия, и
перекристаллизовывают из воды. Получаются крупные призмы с бронзовым блеском. Выход
количественный.
5.1.8.6. Карбоксилирование
Феноляты взаимодействуют даже со слабоэлектрофильным диоксидом углерода,
образуя соответствующие фенолкарбоновые кислоты. Правда, для этого в случае
монофенолов необходимо нагревание, а для получения хороших выходов —
повышенное давление.
Ниже приведена схема реакции для промышленно важного синтеза салици-
салициловой кислоты:
Я нЯ
Na
I
xNa [Г.5.65]
Ph"- ' " """ PhONa +
ONa "^ "OH
Реакция происходит по хелатному механизму, где ион натрия играет роль
электрофильного катализатора, повышающего полярность связи С=О (I ->• II).
Фенолят-анион отщепляет протон от соединения II, давая хелат III. Так образу-
образуется динатриевая соль салициловой кислоты — конечный продукт синтеза
Кольбе без давления. При работе под давлением (синтез Кольбе — Шмитта)
реакция, как показано на схеме, идет до мононатриевой соли. (Какого макси-
максимального выхода можно достигнуть без давления?)
Положение ароматического кольца, в которое вступает карбоксильная группа,
зависит от щелочного металла, применяемого для образования фенолята, и темпе-
температуры. Тенденция к образованию хелата уменьшается отлития через натрий к ка-
калию, т. е. с увеличением ионного радиуса. Преимущественная o/wio-ориентация в
случае фенолята натрия объясняется значительным выигрышем энергии, который
получается при образовании хелата. Если ионный радиус металла слишком велик,
что затрудняет образование хелата, то карбоксилируется сильно поляризующееся и
вследствие этого более реакционноспособное пора-положение.
Вторая гидроксильная группа в орто- или jwema-положении делает фенол та-
таким реакционноспособным, что карбоксилирование возможно уже в водно-ще-
водно-щелочном растворе. Напротив, .мета-аминогруппа или /ш/>а-гидроксигруппа вы-
вызывает лишь незначительное повышение реакционной способности кольца.
Карбоксилировать удается и гетероциклические соединения: пиррол (аналогич-
Таблица Г.5.66. Карбоксилирование фенолов (синтез Кольбе - Шмитта)
Продукт реакции
2,4-Дигидрокси бензойная
(р-резорциловая кис-
кислота)
2,4,6-Тригидроксибензой-
ная кислота
2,5-Дигидрокситерефта-
левая кислота
4-Аминосалициловая кис-
кислота
Салициловая кислота
4-Гидроксибензойная кис-
кислота
3- Гидроксинафталнн-
- 2-карбоновая кислота3
Исходное вещество
Резорцин
Флороглюцин
Гидрохинон
*-Аминофенол
Фенол
Фенол
р-Нафтол
Вариант
А
А
Б
Б
В
В
В
Т. пл., 'С
213 (разл.)
Разл. при 60 °С [декарб-
оксилируется, поэтому в
расплаве находится уже
флороглюцин, т. пл.
219°С(возг.)]
197
151 (т. пл. гидрохлорида
222 "С)
159
214
216
Выход, %
50
30
50
70
70
70
60
Примечания
Кислота теряет СОг уже при
кипячении с водой, поэтому не
перекристаллизовывать, а рас-
растворить в растворе гидрокар-
гидрокарбоната калия и осаждать НС1.
После этого отмывать водой
до нейтральной реакции
Щелочной раствор подкислить
конц. НС1 до изменения окрас-
окраски конго красного. (При под-
кислении до рН 1 выкристал-
выкристаллизовывается гидрохлорид.)
Для очистки осаждать из рас-
раствора в гидрокарбонате нат-
натрия
Применять натриевую соль;
нагревать 24 ч
Применять калиевую соль; на-
нагревать 12 ч
Применять натриевую соль;
нагревать 24 ч
а Термодинамический контроль
5. Замещение в ароматическом кольце 471
но фенолу) дает пирролкарбоновую-2-кислоту, карбазол превращается в карба-
золкарбоновую-1 -кислоту.
При карбоксилировании менее реакционноспособных фенолов необходимо
применять тщательно высушенные феноляты: вода дает с фенолом более проч-
прочные хелаты, чем диоксид углерода, вода проявляет также более сильные кислот-
кислотные свойства, а поэтому в присутствии воды из фенолята вьщеляется свободный
фенол. Кроме того, под действием влаги фенолят комкуется, что также затруд-
затрудняет его взаимодействие с диоксидом углерода.
(Qj) Общая методика карбоксилирования фенолов (табл. Г.5.66)
A. Очень реакционноспособные фенолы. Нагревают 2 ч с обратным холодильником
1 моль фенола с 5 моль гидрокарбоната калия в 1 л воды. После охлаждения образовавшуюся
кислоту осаждают концентрированной соляной кислотой, охлаждают до О °С, отсасывают и
перекристаллизовывают из воды с добавлением активированного угля.
Б. Фенолы средней реакционной способности. Тщательно смешивают 1 моль соответствую-
соответствующего фенола с 2,5 моль свежепрокаленного карбоната калия. Смесь переносят в автоклав, по-
подают диоксид углерода до давления 25—40 атм и нагревают 6 ч при 130 °С. После охлаждения и
снятия давления расплав растворяют в воде и обрабатывают, как описано выше.
B. Малореакционноспособные фенолы. Если должна быть получена о-гидроксикарбоновая
кислота, то 1 моль соответствующего фенола смешивают с раствором 1,05 моль гидроксида нат-
натрия в 100 мл воды. Для лора-карбоксилирования берут такое же количество гидроксида калия.
Упаривают в вакууме досуха и вьщерживают еще 4 ч на масляной или металлической бане при
150 °С. Твердый остаток растирают в порошок, переносят в автоклав и подают диоксид углерода
до давления 5 атм @,5 МПа). Затем нагревают 24 ч (или 12 ч, см. табл. Г.5.66) при 190 °С, причем
время от времени возобновляют подачу углекислоты, поддерживая давление примерно постоян-
постоянным. После охлаждения автоклава давление снимают и далее обрабатывают, как описано выше.
Наряду с салициловой кислотой, которую в значительной степени перерабатывают в лекар-
лекарственные препараты (салол, аспирин) и красители, в промышленности путем карбоксилирова-
карбоксилирования соответствующих фенолов получают также 4-амино-2-гидроксибензойную кислоту
(и-аминосалициловую кислоту, ПАСК, — средство против туберкулеза), 2-гидроксинафталин-
карбоновую-3-кислоту (для получения красителей типа нафтола AS) и другие кислоты.
5.7.9. НИТРОЗИРОВАНИЕ
Нитрозирование представляет собой электрофильное ароматическое заме-
замещение атома водорода на нитрозогруппу при взаимодействии с азотистой
кислотой.
Реакция аналогична нитрованию азотной кислотой. Подобно иону нитро-
ния при нитровании, в процессе нитрозирования в качестве электрофильного
агента выступает, по-видимому, нитрозил-катион NO:
га © ® гг <; ?71
Н + HO-N=O ^=^ H2O-N=O =^^ H2O + N=O 11.5.O/J
Поскольку ион NO+ менее реакционноспособен по сравнению с NCV",
нитрозирование возможно только для реакционноспособных аренов (фенолы,
третичные ароматические амины). Оно дает преимущественно пара-заме-
щенные продукты. и-Нитрозофенол является таутомером хинонмоно-
оксима:
472 Г Препаративная часть
Н°~СЗ H°-^3-N=0 ~ 0=<^bN0H [Г.5.68]
Первичные и вторичные ароматические амины с азотистой кислотой реаги-
реагируют по атому азота (образование солей диазония из первичных и N-нитрозами-
нов из вторичных аминов; см. разд. Г,8.2.1). При действии минеральных кислот
N-нитрозамины перегруппировываются в С-нитрозосоединения.
Ф Получение 1Ч,1Ч-диметил-/1-нитрозоанилина. В химический стакан на 25 мл помещают
4 мл концентрированной НС1 и 10 г льда; к этой смеси добавляют 10 ммоль диметила-
нилина. Затем медленно при охлаждении на ледяной бане и перемешивании прибавля-
прибавляют раствор 12 ммоль нитрита натрия в 3 мл воды, причем температура не должна быть выше
5 °С. Не следует допускать выделения оксидов азота. После охлаждения в течение 15 мин на ле-
ледяной бане желтый осадок гидрохлорида1 отсасывают или центрифугируют и промывают ох-
охлажденной на льду разбавленной НС1, а затем спиртом. Т. пл. 177 °С (разл.).
Для получения свободного основания влажный гидрохлорид медленно при перемешива-
перемешивании вносят в разбавленный раствор карбоната натрия, на который налит слой эфира. После
растворения гидрохлорида отделяют эфирный слой, водный еще раз экстрагируют эфиром,
соединенные эфирные вытяжки сушат сульфатом натрия, эфир отгоняют. Изумрудно-зеле-
Изумрудно-зеленые кристаллы с т. пл. 88 °С (петролейный эфир); выход 95%.
Нитрозирование диалкиланилинов используется для синтеза чистых диалкиламинов
(нуклеофильное ароматическое замещение, см. ниже):
ON—/ V-N + ОН® ON^' ^0®+HN^ [Г.5.69]
Уясните себе, что речь идет о гидролизе фенилога амида азотистой кислоты. (О принципе
фенилогии и винилогии см. разд. В,5.1 и Г,7.4.)
Нитрозогруппа является сильным хромофором, поэтому нитрозосоединения в свободном
мономерном состоянии окрашены в синий или зеленый цвет (см. разд. А,3.5.1).
5.2. НУКЛЕОФИЛЬНОЕ АРОМАТИЧЕСКОЕ ЗАМЕЩЕНИЕ
Ароматические соединения, имеющие систему сопряженных двойных связей,
являются основаниями Льюиса. Поэтому обмен заместителей под действием
нуклеофильных агентов (например, введение гидрокси- или аминогруппы)
удается в целом значительно труднее, чем электрофильное замещение:
INuG ~ Г Т + IXе [Г.5.70]
При нуклеофильной реакции заместитель X отщепляется вместе со связыва-
связывающей парой электронов. Поэтому имеет весьма существенное значение, какую
1 и-Нитрозосоединения вторичных и третичных ароматических аминов дают с минераль-
минеральными кислотами нейтральные соли, имеющие хиноидную структуру:
NOH
CI
0
5. Замещение в ароматическом кольце 473
частицу он может образовать: энергетически бедный анион или незаряженную
молекулу. Это важно при замене галогена (-> галоген-анион), сульфогруппы
(-» сульфит-ион), диазониевой группы (-> молекулярный азот) и в других анало-
аналогичных реакциях, которые сравнительно легко протекают по нуклеофильному
механизму. Напротив, нуклеофильное замещение атома водорода протекает с
трудом и удается, как правило, только в том случае, если сильноосновный и
реакционноспособный гидрид-анион, образующийся в результате реакции,
может быть переведен (например, окислением) в нейтральную частицу.
Нуклеофильное замещение диазогруппы рассматривается в разд. Г,8.3.
5.2.1. НУКЛЕОФИЛЬНОЕ ЗАМЕЩЕНИЕ В
АКТИВИРОВАННЫХ АРОМАТИЧЕСКИХ СОЕДИНЕНИЯХ
—/ и —М-заместители понижают основность ароматического ядра, а потому зат-
затрудняют электрофильное замещение (см. разд. Г,5.1.2), но благоприятствуют
нуклеофильному замещению. Нуклеофильное замещение у активированных
аренов происходит по механизму присоединения—элиминирования. Ниже этот
механизм иллюстрируется на примере применяемого в промышленности гидро-
гидролиза л-хлорнитробензола:
С
Нуклеофильное замещение в активированных ароматических соединениях
имеет определенное сходство с бимолекулярным нуклеофильным замещением
(SN2) в алифатическом ряду. Оно, как правило, также протекает бимолекулярно,
причем самой медленной стадией оказывается образование аниона II (схема
[Г.5.71]). В отличие от реакций Sn2, но аналогично а-комплексу, образующему-
образующемуся при электрофильном ароматическом замещении, анион II представляет
собой не переходное состояние, а реальное промежуточное соединение.
Реализация подобного механизма предполагает, что реакционный центр
позитивирован, а отрицательный заряд в анионе II (схема [Г.5.71]) может быть
особенно эффективно делокализован за счет сопряжения. Эти условия создают-
создаются —^-заместителями в орто- и ла/иг-положениях к реакционному центру. Их
активирующее действие в соответствии с электроноакцепторными свойствами
уменьшается в ряду:
©
— NHNI > —NO > —NO2 > —CN > —СНО > —СОСН3 > —N=N-C6H5 > —Cl
По аналогичной причине легко осуществляется нуклеофильное замещение в
гетероциклах с пониженной я-электронной плотностью, например в пиридине
(аналогичном нитробензолу), хинолине.
Замещению особенно способствует дополнительное позитивирование
реакционного центра уходящей группой (хлором в схеме [Г.5.71]). По этой
причине в активированных аренах в общем случае замещение облегчается в ря-
ряду К Вг < С1 « F. Этот ряд коренным образом отличается от ряда, найденного
474 Г Препаративная часть
для реакций Sn2: I > Вг > Cl » F, в которых в отличие от рассматриваемых ре-
реакций отщепление происходит синхронно с присоединением нуклеофильного
реагента. Однако скорость нуклеофильного замещения в активированных аре-
аренах зависит не только от уменьшения электронной плотности в реакционном
центре, но и от активности нуклеофильного реагента, которая возрастает при
повышении его основности.
В активированных аренах таким путем галоген, водород и другие заместители
в мягких условиях могут быть замещены на гидроксильную, амино-, меркапто- и
другие группы (см. препаративные примеры). Если в качестве нуклеофильного
агента использовать анион диметилсульфоксида (-|СНг—SO—CH3) или диметил-
оксосульфонийметилид (~|СН2—+8О(СНз)г), то удается метилировать, например,
хинолин, изохинолин, акридин, нитробензол. Роль уходящей группы играет
анион метилсульфеновой кислоты или диметилсульфоксид соответственно.
(Напишите схему метилирования хинолина в положение 4!) В то время как, нап-
например, хлорбензол удается гидролизовать до фенола только в очень жестких усло-
условиях (см. разд. Г,5.2.2), замена галогена в орто- или /шра-хлорнитробензоле про-
происходит уже при действии карбоната натрия при 130 °С, а пикрилхлорид A-хлор-
2,4,6-тринитробензол) по реакционной способности напоминает ацилхлорид.
В лаборатории и промышленности особенно важны реакции нуклеофильно-
нуклеофильного замещения с участием активированных арилгалогенидов. Напишите схемы
следующих реакций:
2,4-Динитрохлорбензол —>¦ 2,4-динитрофенол —?- пикриновая кислота
2,4-Динитрохлорбензол ^> 2,4-динитрофенилгидразин
л-Нитрозо-Н,Ы-диалкиланилины -> диалкиламины + л-нитрозофенол (см. схему [Г.5.69])
Синтез антибиотика ципрофлоксацша (противовоспалительное средство для
лечения пневмонии) включает на конечных стадиях как раз два процесса нукле-
нуклеофильного ароматического замещения:
[Г.5.72]
Взаимодействие 2,4-динитрофтор- и 2,4-динитрохлорбензола с аминами,
спиртами и тиолами можно использовать для идентификации аминов, спиртов
и тиолов. Особенно большое значение эта реакция имеет для определения кон-
концевых аминокислот в пептидах. Для этого пептид арилируют 2,4-динитрофтор-
бензолом и гидролизуют. Концевая аминокислота в виде 2,4-динитрофенильно-
5. Замещение в ароматическом кольце 475
го производного (ДНФ-производного) может быть легко отделена от других
аминокислот и идентифицирована (Ф. Сенгер):
H2N-CH-CO-nenTMfl—- O2N—<f ч—NH-CH-CO-пептид—- O2N—<f ^—NH-CH-COOH
R 4=\ R "=\ R
NO2 NO2
-•-аминокислоты [I-j./jJ
Из нуклеофильных замещений у активированных аренов, которые протека-
протекают путем замены атома водорода, особое значение имеет синтез по Чичибабину
2- или 4-аминопиридинов и 2- или 4-аминохинолинов с применением амида
натрия. Образующийся при этом гидрид натрия реагирует с активным водоро-
водородом аминопиридина:
Na®
Аналогично протекает алкилирование или арилирование пиридинов или хи-
нолинов под действием литийалкилов и литийарилов. В этом случае при низких
температурах даже может быть выделен промежуточный продукт I:
20>с ¦¦ ¦ - ¦¦ I + ин
SN R [Г.5.75]
i ' к \Г"
Li *-'
I
Гидрид лития, образующийся при нагревании, выпадает в осадок и выводит-
выводится таким образом из равновесия. Промежуточное соединение I можно разру-
разрушить также водой, при этом образуется 1,2-дигидросоединение, которое следу-
следует затем окислить.
Легко происходит и гидроксилирование солей N-алкилпиридиния. Перво-
Первоначально образующееся дигидросоединение при действии окислителей превра-
превращается в N-алкилпиридон II:
G fi^l K3[Fe(CNN]
^ + он —- (I 1 н -
R R R
I II
Получение 1-метилпиридона-2\ Прим Е. А., Мак Элвейн С. М. В сб.: Синтезы
органических препаратов. Сб. 2. — М.: ИЛ, 1949, с. 333.
Легко гидроксилируются ароматические нитросоединения. Так, нитробен-
нитробензол уже при стоянии над твердым гидроксидом калия дает о-нитрофенол, а воз-
возникающий гидрид-ион восстанавливает избыточный нитробензол, в том числе
до азосоединений (красное окрашивание) (см. схему [Г.8.9]). Поэтому нитросо-
нитросоединения не следует сушить гидроксидом калия.
Промышленное значение имеют реакции нуклеофильного замещения в антрахинонах для
получения красителей и полупродуктов. Так, из 2-аминоантрахинона при сплавлении со
476 Г Препаративная часть
смесью гидроксида калия или натрия в присутствии окислителей (нитрата натрия или хлората
калия) при 220 °С получают важнейший кубовый краситель индантрон (индантреновый синий
RS):
О О
[Г.5.77]
О О
В этом случае образующийся гидрид-ион разрушается окислителем.
Ф Получение арил- и алкил-2,4-динитрофенилсульфидов (общая методика для качественно-
качественного анализа). Растворяют 5 ммоль соответствующего тиола или тиофенола и 5 ммоль
2,4-динитрохлорбензола в 15 мл этанола; к этому раствору приливают раствор 5 ммоль
гидроксида натрия в 2 мл этанола. Реакционную смесь нагревают 10 мин с обратным холо-
холодильником. Горячий раствор отфильтровывают от выпавшей соли. При охлаждении раствора
кристаллизуется тиоэфир, который можно перекристаллизовать из этанола.
Ф Получение 2,4-динитрофенилгидразина. В трехгорлой колбе на 500 мл, снабженной внут-
внутренним термометром, мешалкой и обратным холодильником, растворяют 0,25 моль чис-
чистого 2,4-динитрохлорбензола (т. пл. 51-52 °С) в 125 мл теплого диэтилегнликоля. При
15—20 °С при перемешивании и охлаждении добавляют по каплям 0,3 моль гидразингидрата
F0—65%-ный водный раствор). После окончания экзотермической реакции реакционную
смесь нагревают при перемешивании 20 мин на кипящей водяной бане с 50 мл метанола, что-
чтобы перевести в раствор непрореагировавший 2,4-динитрохлорбензол. После охлаждения
выпавший 2,4-динитрофенилгидразин отсасывают, промывают небольшим количеством мета-
метанола и перекристаллизовывают. Т. пл. 200 "С (бутанол или диоксан); выход 80%.
(^р) Получение 2-аминопиридина
Осторожно! Амид натрия разлагается при добавлении воды со взрывом! В при-
присутствии воздуха, диоксида углерода и влаги он образует взрывчатые вещест-
вещества, которые можно опознать по их желтой окраске. Подобные окрашенные пре-
препараты нельзя применять для синтезов. Защитные очки! Защитные перчатки!
Чтобы синтез прошел удачно, амид натрия должен быть безупречного качества.
В трехгорлой колбе на 500 мл, снабженной эффективной мешалкой, капельной воронкой и
обратным холодильником с осушительной трубкой, наполненной натронной известью, готовят
суспензию, состоящую из 0,5 моль тонкоразмолотого амида натрия (см. часть Е) в 75 мл диметил-
анилина, тщательно высушенного гидроксидом калия и перегнанного. При перемешивании до-
добавляют по каплям 0,4 моль тщательно высушенного (порошкообразным гидроксидом калия или
гидроксидом бария) и перегнанного пиридина, заменяют капельную воронку на внутренний тер-
термометр и нагревают 10 ч при 105-110 °С (до прекращения выделения водорода). Реакционная
смесь окрашивается при этом в коричневый (до черного) цвет и через некоторое время отвердева-
отвердевает (прекратить перемешивание). После охлаждения реакционную смесь разлагают, медленно
добавляя 80 мл разбавленного раствора гидроксида натрия, и выливают в 300 мл воды; для завер-
завершения гидролиза натриевой соли раствор насыщают твердым гидроксидом натрия и отделяют ор-
органическую фазу. Затем сушат гидроксидом калия и перегоняют в вакууме на 40-сантиметровой
колонке Вигре. Вначале отгоняется диметиланилин, т. кип. 81—82 °С A3 мм рт. ст.), затем 2-ами-
нопиридин, т. кип. 95—96 °С A3 мм рт. ст.), т. пл. 56 °С (лигроин). Из промежуточной фракции
с т. кип. 82-95 °С A3 мм рт. ст.), добавляя петролейный эфир, можно выделить еще немного
2-аминопиридина. Общий выход 60%.
5. Замещение в ароматическом кольце 477
5.2.2. НУКЛЕОФИЛЬНОЕ ЗАМЕЩЕНИЕ В
НЕАКТИВИРОВАННЫХ АРОМАТИЧЕСКИХ СОЕДИНЕНИЯХ
Ароматически связанный галоген, который не активирован —/- или — Л/-замес-
тителем, как правило, не может быть замещен на гидроксильную, амино- или
цианогруппу в мягких условиях, которые были описаны для реакций SN2
(см. разд. Г,2). Для гидролиза хлорбензола в присутствии 10—15%-ного раствора
гидроксида натрия необходима температура около 350 °С.
Если метить связанный с хлором углеродный атом хлорбензола изотопом
14С, то в конечном продукте гидроксильную группу находят не только у этого
E8%), но и у соседнего с ним углеродного атома D2%). По-видимому, при эли-
элиминировании хлороводорода вначале образуется производное бензола с фор-
формальной тройной связью («арин», «дегидробензол»), после чего нуклеофильно
присоединяется вода (механизм элиминирования — присоединения):
(^^\ +НОН;ОН°
ИГ|-
Н ^ Ч)Н Ч
При нуклеофильном замещении у галогенобензолов, имеющих другой
заместитель, такой механизм приводит к образованию изомеров. Так, при взаи-
взаимодействии и-хлортолуола с амидом натрия в жидком аммиаке получают смесь
м- и я-толуидинов F2 : 38).
Однако во многих случаях нуклеофильное замещение в неактивированных
ароматических соединениях протекает как через арины, так и по механизму,
соответствующему схеме [Г.5.71]. Тогда перегруппировки наблюдаются в
незначительной мере или вообще не наблюдаются, как, например, при щелоч-
щелочном плавлении а- или р-нафталинсульфоновых кислот, которые дают исклю-
исключительно а- или C-нафтолы соответственно. Из ароматических сульфоновых
кислот при взаимодействии с цианидами металлов также обычно без перегруп-
перегруппировок получаются соответствующие нитрилы.
(^) Получение В-нафтола [May С. Е. J. Am. Chem. Soc, 1922, 44, 650]
jf Осторожно1. Защитные перчатки! Защитные очки!
Нагревают 0,75 моль гидроксида натрия и 3 мл воды в никелевом тигле (емкостью около 75 мл)
до 270 °С, медленно вносят 0,044 моль тонкоразмолотой натриевой соли р-нафталинсульфо-
новой кислоты1. При этом раствор перемешивают термометром, который вставляют в никеле-
никелевую гильзу, наполненную высококипящим парафином. Затем постепенно повышают темпе-
температуру внутри тигля в течение 20 мин до 315 °С и выдерживают при этой температуре 3 мин.
Расплав выливают на кафель, измельчают, переносят в стакан, растворяют в воде и при охлаж-
охлаждении сильно подкисляют концентрированной соляной кислотой. Оставляют на ночь,
отфильтровывают, промывают водой, сушат и перекристаллизовывают из воды. Т. пл. 122—
123 °С; выход 80%.
Известны также реакции замещения в неактивированных аренах, связанные
с переносом одного электрона, т. е. радикальные процессы, а часто даже цепные
1 Целесообразно применять свободный от карбонатов, без остатка растворяющийся в воде
р-нафталинсульфонат натрия, в противном случае реакционная смесь сильно вспенивается.
478 Г Препаративная часть
реакции. Подобные реакции SnrI приводят к тем же результатам, что и ионное
нуклеофильное замещение.
При реакциях Snr на первой стадии на субстрат переносится электрон от
подходящего донора электронов с низким окислительным потенциалом (им мо-
может быть и сам нуклеофил). Перенос может быть осуществлен и электрохими-
электрохимически с катода на субстрат Аг—X.
Образующийся по схеме [Г.5.79 (а)] анион-радикал I субстрата диссоцииру-
диссоциирует, активный радикал вступает во взаимодействие с нуклеофилом, давая анион-
радикал II, который в результате переноса электрона снова образует I:
Ar—X
Ar-X?
Ar- + INuQ
Ar—Nu? + Ar—X
в© -Y
есольв > '
• Ar—X-
1
- Ar • + X
* Ar-Nu?
II
- Ar-Nu + Ar—X®
(а) Стартовая реакция
(б) Реакция роста цепи
(в) Реакция роста цепи
(г) Реакция переноса
[Г.5.79]
По механизму SnrI в аренах и гетероаренах нуклеофильно замещаются га-
галогены, группы —SC6H5, —N+(CH3K, —OPO(OEtJ, —N2+ (о замещении -N2+
анионом I~ по Зандмейеру см. разд. Г,8.3.2). В качестве нуклеофилов при этом
используют сильноосновные амины, алкоголяты, тиолаты, еноляты, сульфина-
ты, соли аци-нитросоединений, анионы ацетонитрила. Таким образом, при
этом типе реакций возможно и образование связей С—С между аренами или ге-
тероциклами и карбанионом. (Напишите схемы указанных ниже препаратив-
препаративных реакций!)
Для реакций Snr с участием аренов типично образование (в присутствии
протонных растворителей) продуктов восстановления, например восстановлен-
восстановленных аренов:
Аг- + H-R Аг-Н + R- [Г.5.80]
В качестве реакционной среды пригодны апротонные растворители, напри-
например диметилсульфоксид, диметилформамид, а также система жидкий аммиак —
щелочной металл. Поскольку при фотовозбуждении как окислительные, так и
восстановительные потенциалы веществ возрастают (на величину энергии
фотовозбуждения), реакции SnrI часто удается инициировать фотохими-
фотохимически.
Получение фенилацетона: RossiR. A., BunnettJ. F. J. Org. Chem., 1973, 38,1407.
Диэтилфенилфосфонат: BunnettJ. F., Weiss R. H. Org. Syntheses, 1978, 58, 134.
Как гидролиз хлорбензола, так и щелочное плавление ароматических сульфоновых кислот
широко используется в промышленности для получения фенолов. Важнейшими продуктами та-
таких реакций являются резорцин (из бензоддисульфоновой-1,3-кислоты), jn-аминофенол (из
л-аминобензолсульфоновой кислоты; применение см. в разд. Г,5.1.8.6), р- и а-нафтолы и их
производные (из соответствующих сульфоновых кислот, см. разд. Г,5.1.4 и табл. Г.8.35), пирока-
пирокатехин (катехол) и 2,4,5-трихлорфенол (из о-хлорфенола и 1,2,4,5-тетрахлорбензола соответ-
соответственно, см. разд. Г.5.1.5).
5. Замещение в ароматическом кольце 479
Различные гидрокси- и аминоантрахиноны получают из хлорантрахинонов и антрахи-
нонсульфоновых кислот. Они представляют собой важные красители и промежуточные
продукты в их производстве.
5.3. АРОМАТИЧЕСКОЕ ЗАМЕЩЕНИЕ ПРИ
СОДЕЙСТВИИ МЕТАЛЛОВ
Ароматические соединения, для которых нуклеофильное замещение протекает
с трудом или вообще не идет, например, неактивированные арилгалогениды
АгХ (см. разд. 5.2), могут зачастую легко реагировать с рядом металлов, их комп-
комплексами или металлоорганическими соединениями, замещая атом галогена X.
Арилгалогениды в таких реакциях ведут себя как обычные галогенорганические
соединения (см. разд. Г,7.2.2).
С неблагородными металлами, такими, как литий или магний, образуются'
соответствующие металлированные арены:
Аг—X + 2 Li Ar—Li + LiX [Г.5.81а]
Ar-X + Mg Ar-Mg-X [Г.5.816]
Процесс обмена галогена на металл может протекать и с металлоорганичес-
металлоорганическими соединениями, содержащими сильноэлектроположительные металлы,
например, алкиллитием:
Аг—X + Li—R > Ar—Li + R-X [Г.5.82]
Алкиллитий может металлировать даже ароматическую связь С—Н:
Аг-Н + Li—R > Ar—Li + R-H [Г.5.83]
Благодаря наличию полярной металлуглеродной связи образующиеся метал-
лоорганические соединения обладают высокой нуклеофильностью и активно
реагируют с большинством электрофильных агентов. Важные превращения
такого типа будут обсуждены позднее в разд. Г,5.3.1 и Г,7.2.2.
С комплексами низковалентных переходных металлов, особенно с паллади-
ем@) или с никелем(О), арилгалогениды, арилсульфонаты и другие замещенные
арены, содержащие легко уходящие группы, взаимодействуют по механизму
окислительного присоединения (см. разд. Г,4.5.1) по атому металла:
Агч
Аг—X + Pd°L2 „Pd" L2 Цлиганд) = РЯз идр. [Г.5.84]
При этом образуется металлоорганическое соединение, способное претерпе-
претерпевать далее самые различные превращения. И прежде всего — это реакции арома-
ароматического замещения, катализируемые переходными металлами (см. разд.
Г,5.3.2идалее).
Наконец, арилгалогениды, сульфонаты и другие арены АгХ при взаимо-
взаимодействии непосредственно с металлоорганическими соединениями или же при
480 Г Препаративная часть
каталитическом содействии переходных металлов могут образовывать новые
связи С—С.
Аг—X + M-R Ar—R + MX M = Cu, MgX, ZnR, BR2, SnRj и др. [Г.5.85]
Препаративное значение имеют прежде всего реакции с медь-, магний-,
цинк-, бор- и оловоорганическими соединениями, обсуждаемые в разд. Г,5.3.2 и
Г.7.2.2.
Все упомянутые выше превращения могут протекать как с аренами, так и с
соответствующими алкенами, что существенно увеличивает их синтетический
потенциал.
5.3.1. МЕТАЛЛИРОВАНИЕ АРОМАТИЧЕСКИХ СОЕДИНЕНИЙ
Металлирование аренов может быть осуществлено, исходя из арилгалогенидов
путем обмена галогена (см. схемы [Г.5.81] и [Г.5.82]) или исходя из Аг—Н путем
замены атома водорода (схема [Г.5.83]).
Реакции арилгалогенидов (по схеме [Г.5.81]) с участием неблагородных ме-
металлов, среди которых наиболее важное препаративное значение имеют литий и
магний, подробно рассматриваются в разд. Г.7.2.2.
Литийароматические соединения в большинстве случаев получают путем об-
обмена галогена на металл при взаимодействии арилгалогенидов с алкиллитием
согласно схеме [Г.5.82]. Такой обмен осуществим, поскольку алкилы металлов
являются более сильными основаниями, чем арилметаллы (благодаря большей
кислотности аренов по сравнению с алканами, см. табл. В.50).
С увеличением энергии диссоциации связи арил-галоген реакционная
способность арилгалогенидов падает в ряду арилиодиды > арилбромиды >
арилхлориды. Арилфториды вообще не реагируют. Иод- и бромарены реаги-
реагируют с бутил- или /и/?е/и-бутиллитием в эфирном растворе (диэтиловый эфир
или тетрагидрофуран) даже при температуре от -60 до -100 "С с очень высо-
высокой скоростью. При таких низких температурах не затрагиваются даже такие
заместители в ароматическом ядре, которые при комнатной температуре сами
реагируют с алкиллитием, например, амидные-, эпокси-, циано- и нитро-
группы.
Ограничивающими факторами таких реакций являются трудности получения в свое рас-
распоряжение чистых арилгалогенидов, поскольку при прямом галогенировании аренов зачастую
образуется смесь изомеров, разделение которых затруднено (см. разд. Г,5.1.5) или же для их
получения необходимо использовать селективные методы синтеза (см., например, разд.
Г.8.3.2, реакция Зандмейера).
Такие препятствия можно обойти, если использовать реакции прямого ме-
таллирования ароматической связи С—Н алкиллитием по схеме [Г.5.83]. Этот
процесс протекает также по упомянутому выше направлению благодаря боль-
большей, чем у алканов, кислотности ароматической связи С—Н. По сравнению с
обменом металл—галоген процесс требует более высоких температур (от —30 до
—40 °С), однако добавление в реакционную смесь диаминов (например,
1Ч,>1,1Ч',1ч['-тетраметилэтилендиамин, ТМДА), или/и /ирет-бутилата калия при-
приводит к резкому увеличению скорости реакции.
5. Замещение в ароматическом кольце 481
В таких условиях возможно осуществить металл ирование самого бензола (и даже этилена):
диамин образует хелатированные комплексы с литием, разрушая ассоциаты алкиллития в
растворе, повышая таким образом его реакционноспособность.
Использование смеси алкиллития с т/>ет-бутилатом калия, называемой суперосновани-
супероснованием, позволяет за счет промежуточного образования алкилкалия, более реакционноспособно-
го, чем алкиллитий, получить в конечном счете арилкалий:
R-Li + f-BuOK == R-K + Г-BuOLi
[Г.5.86]
Ar-H + R-K - Ar-K + R-H
Заместители, имеющие свободную пару электронов, способную координа-
координационно взаимодействовать с металлом, оказывают сильное влияние на скорость
и региоселективность металл ирования. Такие группы (группы направленного ме-
таллирования, в английской литературе, обозначаемые DMG — directing metalla-
tion groups) повышают реакционноспособность аренов и направляют металл в
o/wjo-положение по отношению к заместителю (dirigierte orthometallierung,
«DoM»):
,0.87,
rVf
+ Li—R ?|| | + R-H
Y = OR', SR', NR 2, CH2OR', CONRj, OCONRj, —^ ~V_, SOR\ SO2R'
N"\
По той же причине гетероциклические ароматические соединения в общем
случае металлируются в соседнее с гетероатомом положение.
Используя метод направленного ор/ио-металлирования с последующей реакцией с электро-
фильными агентами [Г.5.88], возможно в мягких условиях региоселективно ввести заместитель в
соседнее с активирующей группой положение. Это позволяет получать и другие региоселектив-
ные соединения, которые при обычном электрофильном замещении или образуются в виде сме-
смеси орто-пара-изомеров или же вообще дают только мета-изомерный продукт (см. разд. Г,5.1.2).
Гетероароматические соединения, например пиридин и индол, замещаются при этом в положе-
положение 2, что невозможно осуществить обычным электрофильным замещением.
Получаемые по реакции металлирования литий- и магнийорганические сое-
соединения содержат сильнополярную связь металл—углерод, причем отрицатель-
отрицательный заряд сосредоточен преимущественно на атоме углерода, что повышает его
основность и нуклеофильность. Благодаря этому такие соединения легко реаги-
реагируют с электрофильными агентами с замещением атома металла:
8+ 5~ 8* 6" ггсоо1
М-Аг + Е-Х - Аг—Е + MX [Г.5.88]
Некоторые важные реакции с участием соединений ариллития и арилмагния
представлены в табл. Г.5.89. Аналогично реагируют алкил- и алкенилметаллоор-
ганические соединения. Взаимодействие металлоорганических соединений с
карбонилсодержащими соединениями обсуждены в разд. Г,7.2.2.
Диметил-1,3-оксазолинильная группа используется для защиты СООН-груплы, ср. [Г.7.48].
482
Г Препаративная часть
Таблица Г.5.89. Реакции арилметаллических соединений
Ar-M
Ar—M
Ar—M
Ar-M
Ar-M
Ar-M
Ar-M
Ar-M
Ar-M
+ H-X - Ar—H + MX
+ R-X - Ar-R + MX
+ { O2 Ar—OM - Ar—OH + MX
, +RX
+ 1 S8 Ar-SM Ar-SR + MX
-^~ Ar-SH + MX
+ R-S-S-R Ar-SR + RSM
+ MeO-NH2 Ar—NH2 + MeOM
+ l2 -—- Ar—1 + Ml
+ HX
+ R2C=O Ar-CR2-OM —j^- Ar-CR2-OH
+ HX
+ CO2 Ar-CO-OM -щ- Ar-COOH
Реакция с ОН-, NH-, CH- и
другими Н-кислотными сое-
соединениями; см. разд. Г.7.2.2
Реакция с алкилгалогенидами
и алкилсульфонатами с обра-
образованием алкиларенов
Реакция с кислородом с обра-
образованием фенолов
Реакция с серой с образовани-
образованием тиоэфиров и тиолов
Реакция с дисульфидами,
приводящая ктиоэфирам
Реакция с метоксиламином с
образованием аминов
Реакция с иодом с образова-
образованием арилиодидов
Реакция с карбонилсодержа-
щими соединениями с обра-
образованием спиртов; см. разд.
Г.7.2.2
Реакция с диоксидом углерода
с образованием карбоновых
кислот; см. разд. Г.7.2.2
Реакции с кислородом и водой упомянуты специально, чтобы подчеркнуть
необходимость защиты металлоорганических соединений от влаги и воздуха.
В качестве примера алкилирования с направленным металлированием и об-
образованием соответствующего литииарена ниже приводится схема и методика
синтеза 3-децил-1,2-диметоксибензола C-децилвератрола).
ОМе ОМе ОМе
[Г.5.90]
Обратите внимание на то, какой измерный алкилвератрол должен получать-
получаться при ацилировании вератрола по Фриделю — Крафтсу (разд. Г,5.1.8.1) с после-
последующим восстановлением кетона по Вольфу — Кижнеру (разд. Г.7.3.1.6).
Получение 3-децилвератрола1
Внимание! н-Бутиллитий исключительно легко гидролизуется при наличии вла-
влаги, а на воздухе самовоспламеняется. Все используемые вещества должны
быть абсолютно обезвожены, а все стеклянное оборудование — тщательно
высушено. Реакция проводится в токе сухого аргона.
По Ng G. В., Damon С. R. J. Oig. Chem., 1978, 43, 3205.
5. Замещение в ароматическом кольце 483
Процесс проводят при перемешивании магнитной мешалкой и охлаждении на бане. Для
реакции используют трехгорлую колбу емкостью 500 мл, снабженную обратным холодильни-
холодильником с хлоркальциевой трубкой, трубкой для пропускания газа и затвора с септой.
К продутому током аргона раствору 0,25 моль C4,5 г) вератролав 150 мл сухого тетрагидро-
фурана (см. часть Е) в атмосфере аргона при температуре 0 °С (ледяная баня!) при перемешива-
перемешивании, с помощью шприца в течение 10 мин впрыскивают через септу 0,17 моль
н-бутиллития A23 мл 1,36 М раствора и-бутиллития в диэтиловом эфире). Перемешивают еще
1,5 ч при 0 °С, после чего впрыскивают шприцем продутый аргоном раствор 0,083 моль A8,5 г)
1-бромдекана в 20 мл ТГФ. После трехчасового нагревания с обратным холодильником смесь
охлаждают до комнатной температуры. Для гидролиза осторожно прикапывают 100 мл
10%-ной соляной кислоты, разделяют фазы, водный слой дважды встряхивают со 150 мл эфи-
эфира, объединенную органическую фазу промывают 20 мл 10%-ного гидроксида натрия, водой,
сушат безводным сульфатом магния и удаляют растворитель на роторном испарителе. Остаток
перегоняют на короткой колонке Вигре в вакууме и после отгонки вератрола получают
17 г G3% от стехиометрии) продукта в виде бесцветной жидкости, т. кип. 139—140 °С при
0,5 мм рт. ст.
2-Тиофентиол получают литиированием тиофена с последующим взаимо-
взаимодействием с серой: Jones E., Moodie I. M., Org. Synth., 1970, 50, 104.
а,а-Дифенил-2-фурилметанол получают литиированием фурана с последую-
последующим взаимодействием с бензофеноном: Gschwend H. W., Rodriguez W. R., Org.
React. 1979, 26, 97.
B-Диметиламино-5-метилфенш1)дифенилметанол получают направленным
металлированием литием Ы^-диметаламино-л-толуидина с последующим вза-
взаимодействием с бензофеноном: HayJ. К, Harris Т. М., Org. Synth., Coll., 1988, Vol.
VI, 478.
С препаративной точки зрения очень важными являются реакции литий- и
магнийорганических соединений с галогенидами других элементов с образова-
образованием соответствующих элементоорганических соединений:
Аг—М + М'—X Аг—М' + MX M' = R2P, R2P(O), R2B, R3Si, R3Sn и др. [Г.5.91]
Таким образом могут быть получены фосфор-, бор-, кремний-, олово- и дру-
другие металлоорганические соединения {трансметаллирование, см., например,
уравнения [Г.7.201, Г.7.217]). Получаемые при этом элементоорганические
соединения сами могут вступать в разнообразные реакции замещения (см. сле-
следующий раздел и разд. Г,7.2.2).
У атома бора с помощью литий- и магнийорганических соединений можно заместить
алкоксигруппу в триалкоксиборе, например:
Аг-М + В(ОМеK Аг-В(ОМеJ + МОМе [Г.5.92]
Получаемые из триалкилборатов эфиры арилборных кислот используют в других реакци-
реакциях (см., например, схему [Г.5.104]). Так, взаимодействием с пероксидом водорода в уксусной
кислоте можно окислить их до арилборатов, гидролиз которых дает фенолы:
Аг_в(омеJ ±^ Ar-O-B(OMeJ _ „^еь Ar-OH [Г.5.93]
Последовательность реакций с участием арилборных соединений открывает возмож-
возможность получения фенолов, исходя из Аг—Н или АгХ (по схемам [Г.5.83] и [Г.5.81]). Эти реак-
реакции протекают с гораздо лучшими выходами, чем прямое окисление арилметаллов кислоро-
кислородом (см. табл. Г.5.89).
484 Г Препаративная часть
5.3.2. СОЧЕТАНИЕ АРОМАТИЧЕСКИХ СОЕДИНЕНИИ С
МЕТАЛЛООРГАНИЧЕСКИМИ
Арилгалогениды, сульфонаты и др. арены Аг—X, прежде всего арилиодиды,
арилбромиды и арилтрифторметилсульфонаты (трифлаты) реагируют по схеме
[Г.5.85] с металлоорганическими соединениями непосредственно или при ката-
литизе переходными металлами, замещая X органическим остатком. Алкенил-
галогениды и алкилтрифлаты реагируют аналогично. При таких реакциях, на-
называемых перекрестным сочетанием (или кросс-сочетанием), образуются новые
С—С-связи, что имеет огромное значение в синтезе органических соединений.
5.3.2.2. Сочетание металлоорганических соединений
щелочных металлов с медьорганическими
С сильнополярными и сильноосновными металлоорганическими соединениями щелочных
металлов арилгалогениды реагируют даже в отсутствие катализаторов. Например, они сочета-
сочетаются с алкиллитием в тетрагидрофуране с образованием алкиларенов (возможно, через проме-
промежуточный обмен галогена на металл; схема [Г.5.85]):
Аг—X + R-Li ^=^ Ar—Li + R-X Ar—R + LiX [Г.5.94]
При этом побочные реакции, такие как симметричное сочетание и элиминирование с об-
образованием олефинов, заметно снижают выход продуктов.
Алкилметаллы могут быть получены также in situ из галогенсодержащих соединений и ще-
щелочных металлов. Так реагируют смеси арилгалогенида и алкилгалогенида с натрием в эфире
с образованием алкиларена (реакция Вюрца — Фиттига):
Аг—X + R-X + 2Na Ar-R + 2 NaX [Г.5.95]
Такой процесс является вариантом реакции Вюрца, при которой органические галогениды
реагируют с натрием с образованием симметричных продуктов сочетания; так, из арилгалоге-
нидов образуются биарилы (правда, с малыми выходами).
Аналогичные реакции сочетания могут быть осуществлены и при взаимодействии с други-
другими щелочными металлами, например с литием.
В любом случае промежуточно образуются металлоарены щелочных металлов, реагирую-
реагирующие далее с галогенидами с образованием продуктов сочетания:
Аг~х тщ~ Аг~м ^ш* Аг~Аг [г-5-96!
Поскольку металлоорганические соединения щелочных металлов «несовместимы» со мно-
многими функциональными группами (—COR, —CN, —NO:, —SO2R, —ОН, —NH2 и др.; см. табл.
Г.5.89 и разд. Г,7.2.2), то такие производные использовать в реакциях сочетания невозможно.
Медьорганические соединения также могут реагировать с алкилгалогенидами
в отсутствие катализаторов, но поскольку они не взаимодействуют со многими
полярными группами, то их использование в реакциях сочетания имеет важ-
важное препаративное значение.
Литийдиалкилкупраты R^CuLi, легко образующиеся при взаимодействии
алкиллития с солями одновалентной меди (см. [Г.7.217]), при реакции с арили-
одидами в эфире или тетрагидрофуране образуют с хорошими выходами алкила-
рены (см. схему [Г.2.19]):
Ar—I + R2CuLi - Ar-R + RCu + Lil [Г.5.97]
5. Замещение в ароматическом кольце 485
Ацетилениды меди при взаимодействии с арил-(а также с алкенил-)-галоге-
нидами образуют арил-(соответственно алкенил-)-ацетилены {сочетание по
Стефенсу — Кастро):
Ar—I + CuC=CR > Ar—C=C-R + Cul [Г.5.98]
Дифенилацетилен и замещенные дифенилацетилены из арилиодидов и фенил-
ацетиленидов медиA): Stefens R. D., Castro С. Е., J. Org. Chem., 1963, 28, 3313.
2-Фенилфуро[3,2-Ь]пиридин из З-гидрокси-2-иодпиридина и фенилацетиле-
нида медиA): Onsley D. С, Castro С. Е., Org. Synth., 1972, 52, 128.
При температуре выше 200 °С арилгалогениды с цианидом меди образуют бензонитрилы
(реакция Розенмунда — фон-Брауна):
Ar—Br + CuCEN Ar-C=N + CuBr [Г.5.99]
1-Цианонафталин из 1-бромнафталина и CuCN: Newman M. S., Org. Synth., Coll. 1955, Vol.
111,631.
9-Цианофенантрен из 9-бромфенантрена и CuCN: Callen J. E., Domfeld C. A., Org. Synth.,
Coll., 1955, Vol. Ill, 212.
Медьорганические соединения являются промежуточными продуктами реакции Ульмана —
димеризации арилгалогенидовдо диарилов при взаимодействии с порошком меди при высоких
(выше 200 °С) температурах:
2Ar—X + 2CU Ar—Ar + 2CuX [Г.5.100]
Возможно, эта реакция, как и [Г.5.96], протекает через стадию промежуточного образова-
образования арилмеди.
Чаще всего применяют арилиодиды, однако арилбромиды и арилхлориды также могут
быть использованы. Активации субстрата способствуют электроноакцепторные заместители в
o/wm-положении, например o-NO2 или o-CN. Так, из смеси арилиодида с арилбромидом или
арилхлоридом получают несимметричные диарилы Ar—Ar1 (перекрестнаяреакция Ульмана).
При значительно более низких температурах протекает сочетание арилгалогенидов в диа-
диарилы в гомогенной фазе с растворимыми солями медиA) особенно с трифлатом. Аналогичное
содействие оказывают также комплексы никеля(О).
2,2'-Динитродифенил из 2-нитрохлорбензола с медной бронзой: Fuson R. С,
Cleveland E. A., Org. Synth, Coll, 1955, Vol. Ill, 339.
4,4'-Дицианодифенил из 4-бромбензонитрила и бисA,5-циклооктадиен)никеля
Ni(codJ: Semmelhack M. К, HelqistP. M., Jones L. D., J. Am. Chem. Soc, 1971,93,5908.
5.3.3.2. Перекрестное сочетание (кросс-сочетание),
катализируемое переходными металлами
Большинство металлоорганических соединений сочетается с арил (и алкенил-)-гало-
генидами только в присутствии каталитических количеств переходных металлов, в
качестве которых чаще всего используются комплексы палладия(О) и никеля(О).
Их каталитическое действие основывается на способности к окислительному присоедине-
присоединению [Г.5.85], трансметаллированию [Г.5.88] и восстановительному элиминированию (см. разд.
Г,4.5.1, схема [Г.4.111]). Эти три стадии, охватывающие каталитический цикл, схематически
приведены на схеме [Г.5.101] для комплекса палладия без указания лиганда:
Pd°
+Аг-Х / N. -Ar-R
/ \ [Г.5.101]
Ar-Pd"-X +"~M ¦ Ar-Pd"-R
- MX
486 Г Препаративная часть
Каталитическая реакция начинается с окислительного присоединения арилгалогенида
АгХ к Pd@) с образованием комплекса Ar-Pd(II)-X, который далее реагирует с металлооргани-
ческим реагентом RM, и в результате трансметаллирования образуется комплекс Ar-Pd(II)-R.
Последний в процессе восстановительного элиминирования образует продукт перекрестного
сочетания Ar—R, при этом высвобождается катализатор Pd@).
Все три реакции цикла в соответствии с характером реагентов, лиганда и растворителя
протекают по своим собственным механизмам. Восстановительное элиминирование, напри-
например, может осуществляться непосредственно из четырехкоординированного плоского квад-
квадратного комплекса Ar-Pd(II)L2-R, что возможно только в случае ч«с-конфиругации, в которую
должен изомеризоваться первично образующийся комплекс, имеющий т/>аис-конфигурацию
(см. [Г.5.102а]). Такой путь реакции наиболее вероятен в случае арил- и алкенилпалладиевых
комплексов. В то же время элиминирование может протекать в несколько стадий с первона-
первоначальным отщеплением лиганда с образованием соответствующего трехкоординированного
комплекса [Г.5.1026].
Ar.
Arx ,
Pd
r" ч
L
L
Ar
1
R
[Г.5.102а]
R 4
+ l> Аг\„.> Ar
Pd ^=^ R + PdL [Г.5.1026]
Кроме арилгалогенидов в реакции перекрестного сочетания, катализируе-
катализируемой комплексами переходных металлов, в качестве субстратов могут быть ис-
использованы также арилсульфонаты ArOSO2R', арилдиазониевые соли ArN2+X~ и
другие Аг—X с легко уходящими группами. Особенно эффективно использова-
использование трифторметилсульфонатов (трифлатов) Ar—SO2CF3, получаемых из фено-
фенолов. Реакционная способность таких соединений изменяется в ряду Ar—I >
Ar—OTf > Ar—Br > Ar—Cl. Реакционная способность электрофильных субстра-
субстратов может быть повышена наличием в структуре электроноакцепторных замес-
заместителей или понижена электронодонорными заместителями. Обратная зависи-
зависимость имеет место в случае, если металлоорганический субстрат является нукле-
офильным агентом.
Важной с препаративной точки зрения является катализируемая палладие-
выми комплексами реакция арил- (и алкенил-) -галогенидов и трифлатов с аце-
тиленидами меди [Г.5.95]. Поскольку ацетилениды меди могут быть получены in
situ из соответствующего ацетилена и соли медиA) в основной среде, арилирова-
ние (и винилирование) терминальных ацетиленов проводят в присутствии осно-
оснований с каталитическими количествами солей медиA) и палладиевого комплек-
комплекса (реакция Соногашира):
Аг_х + н-CEC-R PdUCU''^NH. Ar-CEEC-R + НХ [Г.5.1ОЗ]
В качестве катализаторов обычно используют иодид медиA) и PdCl2(PPri3J,
Pd(PPh3L, Pd(OAcJ или PdCl2, а в качестве основания — алкиламины, такие как
диизопропиламин или триэтиламин.
Даже если используются комплексы Pd(II), например PdC^PPlbh, все равно каталити-
каталитическое действие оказывает комплекс Pd@), образующийся из комплекса Pd(II) при его восста-
восстановлении в реакционной среде, например, аминами.
5. Замещение в ароматическом кольце
487
Таблица Г.5.104. Диарилацетилены, получаемые арилированием фенилацетилена
с палладиевым катализатором
Конечный продукт
4-Метокситолан
4-Хлортолан
4-Метилтолан
4-Нитротолан
1-A-Нафтил)-2-фенилацетилен
1-B-Нафтил)-2-фенилацетиленв
Исходный продукт
4-Иоданизол
4-Хлориодбензол
4-Иодтолуол
4-Иоднитробензол
1-Иоднафталин
2-Бромнафталин
Т. пл., °С
58-59
80-81
78-79
119-120 (ЕЮН)
54—55 (гексан)
117 (ЕЮН)
Выход, %
74 [12]а
88
63
65 [2]*
706
65
а Выход по отношению к выделенному 1,4-дифенилбутадиину, т. пл. 88-89 °С (95% МеОН).
6 Если твердый продукт не выпадает, то водную фазу дважды экстрагируют порциями по
50 мл диэтилового эфира, объединенные экстракты промывают разбавленной соляной кислотой
и водой до нейтральной реакции, сушат и упаривают растворитель на роторном испарителе.
Остаток хроматографируют на силикагеле (высота слоя 20 см, диаметр 3 см).
в Берут 1,25 ммоль фенилацетилена, к реакционной смеси добавляют 20 мг Nal. Температура
реакции 130 °С, время реакции 8 ч. Хроматографируют по методике, указанной выше (см. снос-
сноску «б»), первая фракция содержит 2-бромнафталин.
Реакция Соногашира протекает в мягких условиях при комнатной температуре и при на-
наличии в структурах обоих партнеров по сочетанию таких заместителей, как гидрокси-, амино-,
карбонильная, сложноэфирная или амидная группы.
Если ацетиленид меди не достаточно быстро взаимодействует в каталитическом цикле, то
протекает побочный процесс редокс-димеризации с образованием диинов R—C=C—C=C—R
(см. табл. Г.5.104).
(ГП) Общая препаративная методика проведения реакции Соногашира (табл. Г.5.104)
Используют трехгорлую колбу на 50 мл с мешалкой, трубкой для ввода газа, хлоркальцие-
вой трубкой и затвором с септой.
К раствору 1 ммоль арилиодида, 1 мол.% тетракис(трифенилфосфин)палладия и 2 мол.%
иодида медиA) в 20 мл диметилформамида и 5 мл диизопропиламина, через который предва-
предварительно в течение 30 мин пропускали ток аргона. При перемешивании через септу впрыски-
впрыскивают 1,05 ммоль фенилацетилена. Перемешивают 2 ч при комнатной температуре, сохраняя
слабый ток аргона (использовать счетчик пузырьков). Затем реакционную массу выливают на
смесь примерно 100 г льда и 200 мл 0,1 н. соляной кислоты, оставляют стоять под тягой при
охлаждении льдом на 2—3 ч. Выпавший осадок отделяют, промывают водой до нейтральной
реакции, сушат и хроматографируют на короткой колонке с силикагелем с циклогексаном в
качестве элюента для отделения бутадиинов от продукта. Качество разделения контролируют
методом ТСХ на алюминиевых пластинках с силикагелем (с флуоресцентным индикатором),
элюент — циклогексан. Фракции, содержащие чистый продукт, объединяют и остаток после
испарения растворителя перекристаллизовывают из смеси метанол-вода 19 : 1 (об.).
Арил- и алкенилгалогениды и -трифлаты могут сочетаться при каталитичес-
каталитическом действии комплексов палладия в присутствии оснований также и с борорга-
борорганическими соединениями (органоборанами, органоборными кислотами и их
эфирами — сочетание по Сузуки), например:
Аг—X + R-B(OHJ
PdL2 +NaOH_
-NaX
Ar—R + B(OHK
apiui, алкенил, алкил [Г.5.105]
488 Г Препаративная часть
Борорганические соединения содержат в значительной степени гомеополярную С—В-связь,
благодаря чему являются более слабыми нуклеофилами по сравнению с полярными металлоор-
ганическими соединениями. В каталитическом цикле [Г.5.101] они реагируют с органопалладий-
галогенидами достаточно быстро только в присутствии оснований. В то же время они инертны по
отношению к большому числу функциональных групп, таких как ОН, NH, CO, NO2, CN, а так-
также по отношению к кислороду и воде, благодаря чему работа с ними не требует предосторожнос-
предосторожностей, принимаемых при работе с обычными металлоорганическими соединениями, и реакции с их
участием проводятся даже в воде. Другими преимуществами их использования является неток-
нетоксичность, доступность и высокая селективность в реакциях кросс-сочетания.
В качестве калализаторов применяют прежде всего палладийфосфиновые
комплексы и соли палладия(И) в комбинации с третичными фосфинами или
без них. Обычно в качестве основания используют карбонаты, фосфаты, гид-
роксиды или алкоксиды щелочных металлов, фторид цезия или тетрабутилам-
мония. Растворителем могут служить водорастворимые спирты, кетоны, прос-
простые эфиры и амиды кислот, можно также работать в суспензиях в диоксане,
диметилформамиде или в ароматических углеводородах.
Реакция Сузуки успешно применяется для получения производных диари-
лов (R = Аг). Необходимые для этого арилборные кислоты получают из арил-
магнийгалогенидов или ариллития и эфиров борной кислоты по схеме [Г.5.92] с
последующим гидролизом образующихся эфиров арилборных кислот.
Сочетание по Сузуки позволяет получать замещенные диарилы с высокой региоселектив-
ностью. В этом случае, в противоположность классическому синтезу из арилгалогенидов с
промежуточным образованием медьорганических соединений (реакция Ульмана [Г.5.97]), для
образования С—С-связи достаточно более мягких условий (около 100 °С), что позволяет иметь
в ароматических ядрах разнообразные заместители.
Несимметрично замещенные диарилы из арилборных кислот и арилиодидов
или -бромидов с использованием ацетата палладия(П) и трифенилфосфина:
Huff В. Е., Koenig Th. M., Mitchell D., StaszakM. A., Org. Synth., 1998, 75, 53-56.
Несимметрично замещенные дифенилы из арилборных кислот и арилиодидов с
использованием ацетата палладия(П) или соответственно смеси трис(дибензи-
лиденацетон)дипалладия и три(о-толил)фосфина: Goodson F. Е., Wallow Th. I.,
Novak В. M., Org. Synth., 1998, 75, 61-68.
Несимметрично замещенные дифенилы из мезитиленборной кислоты и арил-
арилгалогенидов, тетракис(трифенилфосфин)палладия и гидроксида таллия:
Anderson J. С, NamliH, Roberts С. A., Tetrahedron, 1997, 53, 15123-15134.
Катализируемое палладием кросс-сочетание органических галогенидов и
трифлатов с оловоорганическими соединениями (реакция Стилле)
R._R + xSnRa I™. 106]
R'-Х
[где R, R' = арил-, алкенил-, алкинил-, бензил-, аллил-, ацил-; R" = алкил (Me,
Bu)] представляет собой реакцию с самыми различными компонентами. Ее
можно использовать также и для синтеза замещенных аренов.
Необходимые для кросс-сочетания по Стилле ароматические оловоорганические соедине-
соединения могут быть легко получены из арилгалогенидов с электроноакцепторными заместителями
путем нуклеофильного ароматического замещения (см. разд. Г,5.2) с натрийтриалкилстанна-
том [Г.5.107] или путем катализируемого палладием взаимодействия с гексаалкилдистаннатом
5. Замещение в ароматическом кольце 489
[Г.5.108], или же, наконец, в случае неактивированных арилгалогенидов через литийоргани-
ческие соединения и хлорид триалкилолова [Г.5.91].
Аг—X + Me3SnNa > Ar —SnMe3 + NaX [Г.5.107]
PdLp
Ar—X + Me3SnSnMe3 -~ Ar—SnMe3 + XSnMe3 [Г.5.108]
Если реакцию Стилле проводить в присутствии СО A бар), то протекает про-
процесс карбонилирования с образованием ароматических альдегидов и кетонов:
О
Arl + RSnMe3 + СО PdL - Аг—с' + ISnMe3 [Г.5.109а]
4R
Arl + HSnBu3 + СО Pdl~2- Аг-с' + ISnBu3 [Г.5.1096]
Существенной особенностью реакции Стилле является токсичность исполюуемых олово-
органических соединений.
Условия и характер протекания реакции Стилле соответствуют типам палладийкатализи-
руемых реакций. Приведенные ниже примеры являются иллюстрацией некоторых аспектов
таких реакций.
Палладийкатализируемое получение карбальдегидов из арилгалогенидов,
гидрида трибутилолова и монооксида углерода: Baillargeon V. P., Stille J. К., J. Am.
Chem. Soc, 1986, 108, 452-461.
Палладийкатализируемое получение толуолов из солей диазония и тетраме-
тилстаннана: Kikukawa К., Копо К., Wada К, Matsuda T. J. Org. Chem., 1983, 48,
1333-1336.
Для осуществления кросс-сочетания арил- и алкенилгалогенидов и -сульфо-
натов с магнийорганическими реагентами {реактивами Гриньяра)
Аг—X + R-MgX - Ar-R + MgX2 [Г.5.110]
в качестве катализаторов преимущественно используются комплексы никеля,
такие как дихлор[1,2-бис(дифенилфосфино)этан]никель(П) NiCl2(dppe) и
дихлор[1,3-бис(дифенилфосфино)пропан]никель(П) NiCl2(dppp), хотя могут
применяться также палладий- и другие металлсодержащие катализаторы.
Но и в этом случае каталитически активной частицей процесса являются всегда комплек-
комплексы Ni@), образующиеся в процессе реакции в результате восстановления комплекса Ni(II) ре-
реактивом Гриньяра. Реакция протекает по тому же механизму, что и сочетание, катализируемое
палладием [Г.5.101].
С помощью комплексов никеля можно осуществлять сочетание алкильных ре-
реактивов Гриньяра, имеющих в р-положении атом водорода, что невозможно в слу-
случае использования Pd-комплексов, поскольку соединения алкилпалладия претер-
претерпевают легкое р-элиминирование с образованием олефинов (см. [Г.4.103]).
Одной из особенностей магнийорганических соединений является их склон-
склонность к симметризации, а также несовместимость с большим числом функцио-
функциональных групп (—ОН, —SH, —NH2, >С=О, —СООН,—NO2, —CN и т. д.), в то
же время привлекает их доступность (см. разд. Г,7.2.2).
490
Г Препаративная часть
1,2-Дибутилбензол из бутилмагнийбромида, а также другие примеры
кросс-сочетания арил- и алкенилгалогенидов с реагентами Гриньяра в присут-
присутствии NiCh(dppp): Kumada M., Татао К., Sumitani К., Org. Synth. 1978, 58,
127-133.
5.3.3. РЕАКЦИЯ ХЕКА
Арилгалогениды, арилтрифлаты и соли арилдиазония в присутствии оснований
и при каталитическом действии комплексов палладия реагируют с алкенами с
образованием алкениларенов {реакция Хека)
[Г.5.111]
Аг
Наряду с ароматическими соединениями в реакции Pd-катализируемого винилирования
могут принимать участие также и олефины (см. разд. Г,4.5).
В качестве катализатора используются прежде всего комплексы палладия(О)
с моно- и бидентантными третичными фосфиновыми лигандами, а также соли
Pd(II) плюс фосфин. В последнем случае используемые реагенты (олефин,
амин) восстанавливают Pd(II) до необходимой частицы Pd(O).
Механизм реакции Хека представляет собой комбинацию стадий палладийкатализируе-
мых реакций арилгалогенидов [Г.5.101] и описанных в разд. Г.4.5.1 каталитических превраще-
превращений олефинов. Весь каталитический цикл представлен на схеме [Г.5.112]. На стадии 1 проис-
происходит окислительное присоединение к комплексу Pd(O) с образованием частицы ст-арил-Pd,
НВХ
Аг—X
IB
H-PdL2X
Pd°U
Аг—PdL2X
©
Ar4
PdL2X
Ar PdLaX
H R
Рис. Г.5.112. Каталитический цикл реакции Хека.
1 — окислительное присоединение; 2 — образование я-комплекса; 3 — цис-внедрение;
4 — внутримолекулярный поворот; 5 — син-элиминирование;
6 — восстановительное элиминирование НХ при содействии основания.
5. Замещение в ароматическом кольце 491
которая обратимо образует с олефином я-комплекс (стадия 2). Далее происходит синхронное
внедрение олефина в связь Ar—Pd (м,ис-внедрение) (стадия 3) с образованием на решающей
стадии реакции новой С—С-связи. Поворот вокруг этой связи D) приводит к син-конформа-
ции палладия и атома водорода, способствующей их элиминированию с образованием конеч-
конечного продукта E) с транс-конфигурацией. Отщепившаяся частица Pd(II) при взаимодействии
с основанием в результате восстановительного элиминирования НХ снова образует исходный
катализатор F).
Как и в других Pd-катализируемых реакциях, в общем случае реакционная
способность субстрата увеличивается в ряду арилхлорид < арилбромид < арил-
иодид. Арилфториды вообще не реагируют.
Варьируя фосфановые лиганды, можно изменять реакционную способность реагента. Для
большинства реакций катализатором служит тeтpaкиc(тpифeнилфocфин)Pd@). В растворе он
диссоциирует, отщепляя лиганд и образуя трис(трифенилфосфин)Рс1@), из которого образу-
образуется каталитически активный двухкоординированный комплекс палладия(О) PdLj. Часто так-
также используют смесь ацетата Pd(II) с трифенилфосфином. В противоположность последнему,
стерически более чувствительные лиганды, например три(о-толил)фосфин, способны предот-
предотвратить протекание побочных реакций.
Для винилирования арилхлоридов необходимо использовать комплексы Pd@) с более ос-
основными фосфинами, такими как, например, три(/я/>е/и-бутил)фосфин. Подбирая катализа-
катализатор возможно в полигалагензамещенных аренах заместить только один наиболее реакцион-
носпособный атом галогена. С другой стороны, возможно в одну стадию заместить одно-
одновременно несколько заместителей с одинаковой реакционной способностью.
В реакции Хека могут принимать участие субстраты с разнообразными
заместителями, такими как алкил-, арил-, алкокси-, амино- и цианогруппы.
Особенно плодотворно использование в реакции Хека в качестве реагентов
терминальных олефинов, которые арилируются региоспецифично по стеричес-
стерически незатрудненному C-положению, тогда как в случае 1,2-дизамещенных алке-
нов можно ожидать образования смеси изомерных аддуктов:
100% 100% 100% 93 7
I I I н| |н
Н2С = СН-СООСН3 H2C=CH-Ph H2C=CH-CN C=C l ' ' J
НзС' Vh
Направленность реакции Хека с циклическими олефинами имеет свои особенности, кото-
которые иллюстрирует приводимый ниже пример арилирования циклогексена:
Ar-X
о
[Г.5.114]
Аг
Поскольку в промежуточно образующемся продукте арилирования имеется только один
атом водорода, находящийся в сын-положении к палладиевому фрагменту, то образуется иск-
исключительно несопряженный 3-арилциклогексен с хиральным атомом углерода в положении 3.
Поэтому использование в реакции хирального фосфинового лиганда даст возможность осуще-
осуществить энантиоселективное протекание реакции Хека.
В качестве растворителей в реакции Хека используют прежде всего диметил-
формамид, диметилацетамид, ацетонитрил, тетрагидрофуран, диоксан, а также
492
Г Препаративная часть
Таблица Г.5.115. Арилирование акриламида (реакция Хека)
Продукт
(?)-Амид коричной кислоты
(?)-Амид 4-метоксикоричной кислоты
(?)-Амид 4-метилкоричной кислоты
(?)-3-A-нафтил)акриламид
Исходное
соединение
Иодбензол
и-Иоданизол
л-Иодтолуол
1-Иоднафталин
Т. пл., °С
142-144
193-195
189-190
176
Выход, %
42
45
63
65
ароматические углеводороды. Особенно пригодны апротонные, диполярные
растворители, способные комплексоваться с палладием. Температура реакции
зависит от реакционной способности субстрата и колеблется, как правило, в ин-
интервале от комнатной до 120 °С. Для нейтрализации образующихся в процессе
реакции кислот обычно применяют как третичные амины, так и неорганические
основания, например карбонаты щелочных металлов. Для защиты каталитичес-
каталитически активных частиц палладия(О) реакцию проводят в токе инертного газа (азот,
аргон) и при полном отсутствии влаги.
(ГП) Арилирование акриламида (реакция Хека). Общая методика (табл. Г.5.115)
В трехгорлую колбу с обратным холодильником, трубкой для пропускания газа и магнит-
магнитной мешалкой с нагревом (прибор погружен в силиконовую баню) помещают 15 мл сухого ди-
метилформамида (см. часть Е), добавляют 0,4 ммоль иодарена, 0,41 ммоль акриламида и 1,5 мл
триэтиламина. Через полученный раствор в течение 30 мин пропускают ток аргона, после чего
добавляют 50 мг тетракис(трифенилфосфин)палладия. Сохраняя слабый ток аргона (применя-
(применяют счетчик пузырьков), смесь нагревают в течение 6 ч при 120 °С. После охлаждения до комнат-
комнатной температуры отделяют основное количество твердых частиц на стеклянном пористом
фильтре, фильтрат выливают на 150 мл смеси воды со льдом и всю массу помещают на ночь в
холодильник. Продукт отфильтровывают и перекристаллизовывают из водного этанола A:1).
Стереохимическую конфигурацию продуктов подтверждают методом ПМР-спектроскопии
(см. разд. А, 3.6).
2-Метил-З-фенилпропаналь из иодбензола и металлилового спирта в присут-
присутствии триэтиламина и ацетата палладия(П): Buntin S. A., Heck R. F., Org. Synth.,
1983, 61, 82-84.
5.3.4. СОЧЕТАНИЕ АРИЛ-ГЕТЕРОАТОМ
Переходные металлы могут эффективно катализировать процессы образования
не только С—С-связи, но и связи углерод—гетероатом. При этом реагентами мо-
могут служить первичные и вторичные амины, спирты, фосфины и тиолы.
Аг—X + H-YR
Ar— YR Y = NH,NR, О, S, PR, P(O)R
[Г.5.116]
Напишите соответствующие уравнения реакций и назовите получающиеся
продукты!
Реакции протекают по аналогичному механизму, что и в случае взаимодействия арилгало-
генидов с металлоорганическими соединениями [Г.5.101] (М—R = Н—YR). В качестве катали-
катализатора используются преимущественно те же комплексы никеля и палладия.
5. Замещение в ароматическом кольце 493
Из всех Pd-катализируемых реакций наиболее важными являются синтезы
ариламинов из арилгалогенидов, арилтрифлатов или арилдиазониевых солей и
первичных или вторичных аминов, поскольку селективное образование С—N-свя-
зи, приводящее к вторичным или третичным аминам, другими методами можно
осуществить лишь путем многостадийного синтеза (ср., например, разд. Г,2).
Кроме того, реакции внутри- и межмолекулярного арилирования аминов и ами-
амидов имеют большое значение как для синтеза природных соединений, так и при
создании новых материалов.
В реакциях Pd-катализируемого аминирования амины являются нуклеофилами, а арома-
ароматические соединения — электрофилами. Поэтому в аминах желательно иметь электронодо-
норные группы, а в субстрате — электроноакцепторные. Нуклеофильность аминов может быть
повышена станнилированием М,М-диметиламинотрибутилоловом. Напишите эти реакции!
В качестве лигандов для катализаторов используют прежде всего стерически объемные
фосфины, такие как, например, три(о-толил)- или три(тре/я-бутил)фосфин, в комбинации с
ацетатом Pd(II) или трис(дибензилиденацетон)дипалладием Pd2(dbaK. При получении вто-
вторичных аминов зачастую используют бидентатные хелатизированные лиганды, например
1,Г-ди(дифенилфосфино)ферроцен (ddpf) или 2,2'-бис(дифенилфосфино)-1,Г-динафтил
(BINAP). Напишите структуры этих соединений!
Для проведения процессов аминоарилирования необходимо иметь достаточно сильные
основания, например трет-бутилат натрия, карбонаты калия или кальция. Растворителями
служат те же соединения, что и в других Pd-катализируемых реакциях (реакция Хека).
Третичные амины из арилиодидов и вторичных аминов с использованием
Рд2(с1Ьа)з/(о-толил)зР и трет -бутил ата натрия: Wolfe J. P., Buchwald S. L., J. Org.
Chem., 1996,61, 1113-1135.
Получение бензоконденсированных от 5- до 7-членных N-гетероциклов внутри-
внутримолекулярным арилированием М,1Ч-арилалкиламиносоединений: Wolfe J. P.,
Rennets R. A., BuchwaldS. L, Tetrahedron, 1996, 52, 7525-7546.
Синтез N-apuA-N-алкиламинов арилированием алкиламинов с использова-
использованием в качестве катализатора Pd(dppf)Cl2: Driver M. S., HartwigJ. К, J. Am. Chem.
Soc, 1996,118,7217-7218.
5.4. ЛИТЕРАТУРА
Общие вопросы ароматического замещения
BaciocchiE., Illuminati G, in: Prog. Phys. Org. Chem. 1967, 5, 1-79.
Berliner E., in; Prog. Phys. Org. Chem. 1964, 2, 253-321.
Effenberger F., Angew. Chem. 1980, 92, 147-168.
Minkin V. I., Glaknovtsev M. N.,SimkinB. Y. Aromaticity and Antiaromaticity. — John Wiley
& Sons, New York, 1994.
Norman R. O. C, Taylor R.: Electrophilic Substitution in Benzenoid Compounds. — Elsevier,
Amsterdam, 1965.
Pearson D. E., Buehler С A., Synthesis, 1971, 455-477.
Sainsbury M. Aromatenchemie. — VCH Verlagsgesellschaft, Weinheim, 1995.
Taylor R. Electrophilic Aromatic Substitution. — John Wiley & Sons, Chichester, 1990.
Yakobson G. G, Furin G. G, Synthesis, 1980, 345-364.
494 Г Препаративная часть
Замещение в ряду гетероароматических соединений
Акселърод 3. С, Березовский В. М. Усп. хим., 1970, 39, 1337-1368.
Cook M. J., KatritzkyA. R., in: Adv. Heterocycl. Chem. 1974,17, 255-356.
Goldfarb Ya. L, Volkenshteiin Yu. В., Belenkii L. I., Angew. Chem. 1968, 80, 547-557.
KatritzkyA. K, Johnson С D., Angew. Chem. 1967, 79, 629-656.
Marino G, in: Adv. Heterocycl. Chem. 1971,13, 235-314.
Обратимое блокирование при ароматическом замещении
Tashiro M., Synthesis 1979, 921-936.
Нитрование
Hartshorn S. R., SchofieldK., in: Prog. Phys. Org. Chem. 1973, 8, 278.
Nitration and Aromatic Reactivity. Von J. G. Hoggett et al — Cambridge University Press,
Cambridge, 1971.
Olah G. A., Malhotra R., NarangS. C: Nitration. — VCH Publishers, New York, 1989.
Schofleld K.: Aromatic Nitration. — Cambridge University Press, Cambridge, 1980.
Seidenfaden W., PawellekD., in: Houben-Weyl. Bd. 10/1 A971), S. 479-818.
Stock L. M., in: Prog. Phys. Org. Chem. 1976,12, 21-47.
TumoeA. И. Усп. хим., 1958, 27, 845-890.
Сульфирование
Cerfontain H:. Mechanistic Aspects in Aromatic Sulfonation and Desulfonation. —
Wiley-Interscience, New York, 1968.
Gilbert E. E.: Sulfonation and Related Reactions. — Interscience, New York, 1965.
Muth E, in: Houben-Weyl. Bd. 9 A955), S. 429-535.
Сьютер С. М., ВестонА. В. В сб.: Органические реакции. Сб. 3. — М.: ИЛ, 1951,
с. 140-189.
Сульфохлорирование
Muth E, in: Houben-Weyl. Bd. 9 A955), S. 572-579.
Хлорирование, Хромирование, иодирование
Dela Mare Р. В. D:. Electrophilic Halogenation. — Cambridge University Press, Cambridge, 1976.
RoedigA., in: Houben-Weyl. Bd. 5/4 A960), S. 233-331, 557-594.
Stroh R., in: Houben-Weyl. Bd. 5/3 A962), S. 651-725.
Роданирование
Bdgemann M., Petersen S., Schultz O.-E., Soli #., in: Houben-Weyl. Bd. 9 A955), S. 859-863.
Алкилирование по Фриделю — Крафтсу
AsingerE, VogelH. #., in: Houben-Weyl. Bd. 5/la A970), S. 501-539.
Olah G. A.: Friedel-Crafts and Related Reactions. Bd. 2. — Interscience, New York, 1964.
Olah G. A.: Friedel-Crafts Chemistry. — John Wiley & Sons, New York, 1973.
5. Замещение в ароматическом кольце 495
Прайс Ч. В сб.: Органические реакции. Сб. 3. — М.: ИЛ, 1951, с. 7-87.
Roberts R. M., Khalaf A. A.: Friedel-Crafts Alkylation Chemistry. — Marcel Dekker,
New York, Basel, 1984.
Ацилирование по Фриделю — Крафтсу
Берлинер Э. В сб.: Органические реакции. Сб. 5. — М.: ИЛ, 1951, с. 195-270.
ChevierB., Weiss R., Angew. Chem. 1974, 86, 12-21.
Gore P. #., Chem. Rev. 1955, 55, 229-281.
Olah G. A.: Friedel Crafts and Related Reactions. Bd. 3. — Interscience, New York,
1965.
Pearson D. E., Buehler С A., Synthesis 1972, 533-542.
Schellhammer C.-fV., in: Houben-Weyl. Bd. 7/2a A973), S. 15-378.
РеакцияФриса
БлапгтА. В сб.: Органические реакции. Сб. 1. — М.: ИЛ, 1948, с. 455-491.
Непеска #., in: Houben-Weyl. Bd. 7/2a A973), S. 379-389.
Реакция Гаттермана — Коха
Bayer О., in: Houben-Weyl. Bd. 7/1 A954), S. 16-20.
Мачинская И. В. В сб.: Реакции и методы исследования органических соединений.
Сб. 7. — М.: Госхимиздат, 1958, с. 277-306.
Краунз Н. В сб.: Органические реакции. Сб. 5. — М.: ИЛ, 1951, с. 271-283.
Синтез Гаттермана
Bayer О., in: Houben-Weyl. Bd. 7/1 A954), S. 20-29.
Мачинская И. В. В сб.: Реакции и методы исследования органических соединений.
Сб. 7. - М.: Госхимиздат, 1958, с. 307-365.
Трюс У. В сб.: Органические реакции. Сб. 9. — М.: ИЛ, 1959, с. 45-81.
Синтез Геша
Schellhammer C.-W., in: Houben-Weyl. Bd. 7/2a A973), S. 389-421.
Cneppu П., Дюбуа А. В сб.: Органические реакции. Сб. 9. — М.: ИЛ, 1959, с. 45-81.
Зильберман Е. В., Усп. хим. 1962, 31, 1309-1347.
Синтез Вильсмейера
Bayer О., in: Houben-Weyl. Bd. 7/1 A954), S. 29-36.
Jutz С, in: Adv. Org. Chem. 1976, 9, 1, 225-342.
Marson С. М., Tetrahedron 1992, 48, 3659-3726.
Marson C. M., Giles P. R.: Synthesis Using Vilsmeier Reagents. — CRC Press, Boc Raton,
1994.
Muhkuh В. Я., Дорофеенко Г. Н. Усп. хим., 1960, 29, 1301-1335.
Хлорметилирование (реакция Блана)
Беленький Л. И., Волькеншнейн Ю. Б., Карманова И. Б. Усп. хим., 1977, 46, 1998.
496 Г Препаративная часть
Фьюзон Р., Мак-Кивер И. В сб.: Органические реакции. Сб. 1. — М.: ИЛ, 1948,
с. 84-114.
Stroh R., in: Houben-Weyl. Bd. 5/3 A962), S. 1001-1007.
Карбоксилирование
Henecka H., Ott E., in: Houben-Weyl. Bd. 8 A952), S. 372-384.
LindseyA. S., Jeskey H., Chem. Rev. 1957, 57, 583-620.
Нуклеофильное ароматическое замещение
BunnettJ. F, ZahlerR. E., Chem. Rev. 1951, 49, 273-412.
Illuminati G, in: Adv. Heterocycl. Chem. 1964, 3, 285-371.
RossS. D., in: Prog. Org. Chem. 1963, 1, 31-74.
SauerJ., Huisgen R., Angew. Chem. 1960, 72, 91-108; 294-315.
... через арины
Heaney #., Chem. Rev. 1962, 62, 81-97.
Hoffmann R. W:. Dehydrobenzene and Cycloalkynes. — Verlag Chemie, Weinheim;
Academic Press, New York, London, 1967.
Kauffmann Th., Angew. Chem. 1965, 77, 557-571.
Miller J.: Aromatic Nucleophilic Substitution. — Elsevier, Amsterdam, 1968.
Wittig G, Angew. Chem. 1965, 77, 752-759.
Zoltewicz J. A., in: Topics in Current Chemistry. Bd. 59. — Springer-Verlag, Berlin,
Heidelberg, New York, 1975. S. 33-64.
Реакции SRN1
Белецкая И. П., Дрозд В. Н. Усп. хим., 1979, 48, 793-828.
BunnettJ. F., Асе. Chem. Res. 1978,11, 413.
Chanon M., Tobe M. L, Angew. Chem. 1982, 94, 27-49.
Rossi M., Tobe R. A., Ace. Chem. Res. 1982, 15, 164-170.
Rossi R. A., De Rossi R. H.\ Aromatic Substitution by the SrnI Mechanism. — American
Chemical Society, Washington, 1983.
Rossi R. A., PieriniA. N., Org. React. 1999, 54, 1-271.
Аминирование гетероциклических оснований амидами щелочных металлов
Леффлер М. В сб.: Органические реакции. Сб. 1. — М.: ИЛ, 1948, с. 115—132.
MdllerF., in: Houben-Weyl. Bd. 11/1 A957), S. 9-17.
Пожарский А. Ф., Симонов A. M., Доронкин В. H. Усп. хим., 1978, 47, 1933-1969.
Направленное металлирование ароматических соединений
GschwendH. W., Rodriguez H. R-, Org. React. 1979, 26, 1-360.
Narasimhan N. S., Mali R. S., Synthesis 1983, 957-986.
Snieckus V., Chem. Rev. 1990, 90, 879-933.
5. Замещение в ароматическом кольце 497
Перекрестное сочетание, катализируемое переходными металлами
Bolm С, HildebrandJ. P., Muniz К, Hermanns N., Angew. Chem., 2001, 113, 3382-3407
(каталитическое асимметричное арилирование).
Handbook of Organopalladium Chemistry for Organic Synthesis. E. Negishi,
A. de Meijere (eds.), Bd. 1-2. - Wiley-VCH, Weinheim, 2002.
Metal-catalyzed Cross-coupling Reactions. F. Diederich, P. J. Stang (eds.). — Wiley-VCH,
Weinheim, 1998.
Transition Metals for Organic Synthesis, Vol. 1. M. Beller, С Bolm (eds.). — Wiley-VCH,
Weinheim, 1998.
Tsuji J.: Palladium Reagents and Catalysts. — John Wiley & Sons, Chichester, 1995.
Heck R. F.: Palladium Reagents in Organic Synthesis. — Academic Press, New York,
1985.
Kumada M., Pure Appl. Chem. 1980, 52, 669-679.
Felkin H., Swierczewski G., Tetrahedron 1975, 31, 2735—2748 (с реагентами
Гриньяра).
Kalinin V. N., Synthesis 1992, 413-432 (гетероциклы).
Реакция Сузуки
Miyaura N., Suzuki A., Chem. Rev. 1995, 95, 2457-2483.
Реакция Стилле
Farina V., Krishnamurthy V., Scott W. /., Org. React. 1997, 50, 1-652.
Farina V., Krishnamurthy V., Scott W. J:. The Stille Reaction. — John Wiley & Sons,
New York, 1998.
Mitchell T. N., Synthesis 1992, 803-815.
Stille J. K, Angew. Chem. 1986, 98, 504-519.
Duncton M. A. J., Pattenden G. J. Chem. Soc, Perkin Trans. 1,1999, 1235-1246 (внутримо-
(внутримолекулярная реакция Стилле)
Реакция Хека
ShibasakiM., Boden С. D. J., KojimaA., Tetrahedron 1997, 53, 7371-7395.
De Meijere A., Meyer E., Angew. Chem. 1994, 106, 2473-2506.
Jeffery Т., Adv. Met.-Org. Chem. 1996, 5, 153-260.
SchmalzH.-G., Nachr. Chem. Tech. Lab. 1994, 42, 270-276.
Altenbach H.-J., Nachr. Chem. Tech. Lab. 1988, 36, 1324-1327.
ReibigH.-U., Nachr. Chem. Tech. Lab. 1986, 34, 1066-1073.
HeckR. F, Org. React. 1982, 27, 345-390.
Heck R. F: Palladium Reagents in Organic Synthesis. — Academic Press, New York, 1985,
p. 179-321.
498 Г Препаративная часть
Синтезы ариламинов и арилэфиров
HartwigJ. E, Angew. Chem. 1998, ПО, 2154-2177.
BarananoD., Mann G, HartwigJ. F., Curr. Org. Chem. 1997, 1, 287-305.
Modern Animation Methods. A. Ricei (ed.). — Wiley-VCH, Weinheim, 2000.
Реакция Ульмана
LeyS. V., Thomas A. W., Angew. Chem. 2003, 115, 5558-5607.
Hassan J., Sevignon M., Gozzi C., Shulz G., Lemaire M. Chem. Rev. 2002, 102,
1359-1469.
ОГЛАВЛЕНИЕ
Or переводчиков двадцать второго издания 5
Предисловие к двадцать второму изданию 6
Предисловие к двадцать первому изданию 6
z?\Q введение в лабораторную практику 7
1. Оборудование и приемы, используемые при проведении реакций в
органической химии 7
1.1. Сорта стекла, способы соединения стеклянных деталей 7
1.2. Химическая посуда 10
1.3. Холодильники 10
1.4. Стандартное оборудование для проведения реакций в органической
химии 13
1.5. Перемешивание и встряхивание 16
1.5.1. Типы мешалок 16
1.5.2. Крепление мешалок в приборе и применяемые уплотнения 18
1.5.3. Моторы 18
1.5.4. Встряхивание 19
1.6. Введение газов в реакцию и их дозирование 19
1.7. Нагревание и охлаждение 22
1.7.1. Нагреватели, теплопередача, нагревательные бани 22
1.7.2. Нагревание огнеопасных жидкостей 24
1.7.3. Охлаждающие средства 25
1.8. Работа поддавлением 26
1.8.1. Толстостенные ампулы для работы под давлением 27
1.8.2. Автоклавы 27
1.8.3. Баллоны для газов 29
1.9. Работа под уменьшенным давлением 31
1.9.1. Создание вакуума ; 31
1.9.2. Измерение давления в вакуумных системах 33
1.9.3. Работа под вакуумом 34
1.10. Высушивание 35
1.10.1. Сушка газов 36
1.10.2. Обезвоживание жидкостей 37
1.10.3. Сушка твердых веществ 38
1.10.4. Наиболее распространенные осушители 39
1.11. Работы в микромасштабе 39
2. Способы разделения веществ 46
2.1. Фильтрование и центрифугирование 46
2.2. Перекристаллизация 49
2.2.1. Выбор растворителя 49
2.2.2. Проведение перекристаллизации 50
2.2.3. Кристаллизация из расплава 52
2.3. Перегонка и ректификация 53
2.3.1. Зависимость температуры кипения от давления 53
2.3.2. Простая перегонка 55
2.3.2.1. Физические основы процесса разделения 55
2.3.2.2. Проведение простой перегонки 56
500 Оглавление
2.3.2.3. Отгонка растворителей 61
2.3.2.4. «Короткая» перегонка. Перегонка с шарообразными трубками 62
2.3.3. Ректификация 63
2.3.3.1. Физические основы ректификации 63
2.3.3.2. Проведение ректификации 67
2.3.4. Перегонка с водяным паром 73
2.3.5. Перегонка азеотропных смесей 75
2.4. Сублимация (возгонка) 76
2.5. Экстракция и распределение между двумя фазами 78
2.5.1. Экстракция твердых веществ 78
2.5.1.1. Однократная простая экстракция 78
2.5.1.2. Многократная простая экстракция 79
2.5.2. Экстракция жидкостей 80
2.5.2.1. Извлечение веществ из растворов или суспензий 80
2.5.2.2. Непрерывная экстракция 81
2.5.3. Противоточное распределение 83
2.6. Дцсорбция 84
2.6.1. Обесцвечивание растворов 85
2.7. Хроматография 86
2.7.1. Тонкослойная хроматография 88
2.7.2. Колоночная жидкостная хроматография 92
2.7.3. Высокоэффективная жидкостная хроматография (ВЭЖХ) 94
2.7.4. Газовая хроматография 97
3. Определение физических свойств органических соединений 100
3.1. Температура плавления 101
3.1.1. Определение температуры плавления в капилляре 102
3.1.2. Определение температуры плавления с помощью микроскопа
с нагревательным столиком 103
3.2. Температура кипения 105
3.3. Рефрактометрия 106
3.4. Поляриметрия 107
3.5. Оптическая спектроскопия 108
3.5.1. Ультрафиолетовая и видимая спектроскопия 111
3.5.2. Инфракрасная спектроскопия 117
3.6. Спектроскопия ядерного магнитного резонанса 126
3.6.1. Спектроскопия ЯМР 'Н 129
3.6.2. Спектроскопия ЯМР 13С 135
3.7. Масс-спектроскопия 141
3.8. Установление структуры органических соединений спектральными
методами 149
3.9. Рентгеноструктурный анализ 156
4. Хранение химических реактивов. Уничтожение вредных остатков 158
4.1. Хранение химических реактивов 158
4.2. Отходы и их уничтожение 160
5. Необходимое лабораторное оборудование и посуда 162
6. Литература 163
)а ЛИТЕРАТУРА ПО ОРГАНИЧЕСКОЙ ХИМИИ.
ВЕДЕНИЕ ЛАБОРАТОРНОГО ЖУРНАЛА 171
1. Оригинальная литература 171
1.1. Специальные журналы 171
1.2. Патенты 173
Оглавление 501
2. Обобщающие работы и обзоры 173
3. Реферативная литература 175
3.1. Справочник Бейльштейна 175
3.2. Реферативные журналы 177
3.3. Сигнальная информация 178
4. Справочник табличных данных 178
5. Указания о номенклатуре 179
6. Проведение литературного поиска 181
6.1. Поиск сведений об определенном органическом соединении 181
6.1.1. Исчерпывающий литературный поиск 181
6.1.2. Поиск подходящей методики получения •. 182
6.2. Поиск сведений о классах соединений 183
6.3. Поиск с помощью компьютера 183
6.3.1. Поиск химических соединений и их синтезов 186
6.3.2. Поиск по структуре 187
6.3.3. Поиск химических реакций 188
7. Ведение лабораторного журнала 189
8. Литература 190
)Q НЕКОТОРЫЕ ОБЩИЕ ПОЛОЖЕНИЯ 191
1. Классификация органических реакций 191
2. Изменения энергии при химических реакциях 193
3. О протекании органических реакций во времени 197
3.1. Последовательные реакции 197
3.2. Конкурирующие (параллельные) реакции 199
3.3. Влияние растворителей на реакционную способность 200
3.4. Катализ 201
4. Кислотно-основные реакции 202
5. Влияние заместителей на распределение электронной плотности и
реакционную способность органических молекул 205
5.1. Полярные эффекты заместителей 205
5.2. Количественный подход к полярным эффектам заместителей.
Уравнение Гаммета 209
5.3. Пространственные эффекты 212
6. Рассмотрение реакционной способности с позиции теории возмущений ...212
7. Стереоизомерия 216
7.1. Конформация 217
7.2. цис, транс-Изомерия 219
7.3. Хиральность и стереоизомерия 221
7.3.1. Энантиомерия 221
7.3.2. Диастереомерия 223
7.3.3. Синтез хиральных соединений 224
7.3.3.1. Разделение рацематов 225
7.3.3.2. Стереоселективные синтезы 226
8. Планирование синтезов 228
8.1. Ретросинтезы 229
8.2. Защитные группы 231
9. Литература 232
(Г,
ПРЕПАРАТИВНАЯ ЧАСТЬ.
О ПОЛЬЗОВАНИИ МЕТОДИКАМИ И ТАБЛИЦАМИ 237
Радикальное замещение 239
1.1. Образование радикалов и их стабильность 240
502 Оглавление
1.2. Реакции и время жизни свободных радикалов. Цепные радикальные
реакции 243
1.3. Реакционная способность и селективность при реакциях радикального
замещения 245
1.4. Радикальное галогенирование .- 249
1.4.1. Хлорирование 250
1.4.2. Бромирование 256
1.5. Пероксигенирование 258
1.6. Другие реакции радикального замещения 261
1.7. Литература 263
2. Нуклеофильное замещение при насыщенном атоме углерода 265
2.1. Ход реакции и ее механизм 265
2.1.1. Мономолекулярное нуклеофильное замещение (SnI) 267
2.1.2. Бимолекулярное нуклеофильное замещение (SN2) 268
2.2. Факторы, влияющие на ход нуклеофильного замещения 269
2.2.1. Реакционная способность субстрата RX 269
2.2.2. Нуклеофильность реагентов 273
2.3. Региоселективность реакций с амбифункциональными
нуклеофилами 277
2.4. Условия реакций нуклеофильного замещения с анионными
нуклеофилами 279
2.4.1. Возможности осуществления реакцийё 279
2.4.2. Межфазный катализ 280
2.5. Нуклеофильное замещение в спиртах и простых эфирах 281
2.5.1. Замещение гидроксильной группы в спиртах на остаток
неорганической кислоты 282
2.5.2. Образование простых эфиров из спиртов с участием кислот.
Расщепление простых эфиров 288
2.6. Нуклеофильное замещение в алкилгалогенидах, алкилсульфатах и
алкилсульфонатах 290
2.6.1. Гидролиз 290
2.6.2. Синтез простых эфиров из алкоголятов или фенолятов 294
2.6.3. Синтез эфиров карбоновых кислот 298
2.6.4. Алкилирование аммиака и аминов 299
2.6.5. Алкилирование соединений фосфора 303
2.6.5.1. Алкилирование третичных фосфинов 303
2.6.5.2. Реакция Михаэлиса — Арбузова 304
2.6.6. Алкилирование серосодержащих соединений 305
3.6.7. Синтез алкилгалогенидов по реакции Финкельштейна 310
3.6.8. Получение нитроалканов 312
3.6.9. Получение алкилцианидов (синтез нитрилов по Кольбе) 314
2.7. Нуклеофильное замещение в кремнийорганических соединениях 318
2.8. Литература 320
3. Элиминирование с образованием кратных углерод-углеродных связей 323
3.1. Реакции ионного а,Р-элиминирования 323
3.1.1. Замещение и элиминирование как конкурирующие реакции.
Механизм реакций ионного элиминирования 324
3.1.1.1. Мономолекулярное элиминирование 326
3.1.1.2. Бимолекулярное элиминирование 327
3.1.2. Влияние порядка реакции и общих пространственных соотношений
на направление элиминирования 329
3.1.3. Стереоэлектронные эффекты и направление элиминирования.
Пространственный ход элиминирования 331
Оглавление 503
3.1.4. Отщепление воды от спиртов (дегидратация) и отщепление
спиртов от простых эфиров 334
3.1.5. Отщепление галогеноводородов от алкилгалогенидов
(гидрогалоэлиминирование; дегидрогалогенирование) 339
3.1.6. Элиминирование триалкиламина из четвертичных аммониевых
оснований (гофмановское расщепление) 343
3.2. Термическое смн-элиминирование ,. 345
3.3. а,ос-Элиминирование 348
3.4. Литература 350
4. Присоединение к неактивированным кратным углерод-углеродным
связям 352
4.1. Электрофильное присоединение к олефинам и ацетиленам 355
4.1.1. Механизм электрофильного присоединения 355
4.1.2. Направление нуклеофильной атаки и влияние пространственных
эффектов при реакциях электрофильного присоединения 357
4.1.3. Присоединение протонных кислот и воды к олефинам и
ацетиленам 359
4.1.4. Присоединение галогенов и гипогалогенных кислот
к олефинам и ацетиленам 363
4.1.5. Оксимеркурирование 367
4.1.6. Эпоксидирование и дигидроксилирование 367
4.1.7. Озонирование 373
4.1.8. Гидроборирование 374
4.1.9. Катионная олигомеризация и полимеризация 377
4.2. Нуклеофильное присоединение 378
4.2.1. Анионная полимеризация олефинов 378
4.2.2. Нуклеофильное присоединение к ацетиленам 379
4.3. Реакции радикального присоединения и полимеризации 383
4.4. Реакции циклоприсоединения 391
4.4.1. Реакции [1 + 2]-циклоприсоединения. Присоединение карбенов
и карбеноидов 393
4.4.2. [2 + 2]-циклоприсоединение 396
4.4.3. [3 + 2]-циклоприсоединение A,3-биполярное присоединение) 397
4.4.4. [4 + 2]-циклоприсоединение (диеновый синтез, реакция
Дильса - Альдера) 398
4.5. Присоединение, катализируемое металлами и комплексами
металлов 403
4.5.1. Реакции олефинов и алкинов в условиях гомогенного катализа 403
4.5.2. Гетерогенное каталитическое гидрирование 410
4.6. Литература 419
5. Замещение в ароматическом кольце 424
5.1. Электрофильное ароматическое замещение 425
5.1.1. Механизм электрофильного ароматического замещения 425
5.1.2. Влияние заместителей на реакционную способность аренов
и направленность последующих реакций замещения 427
5.1.3. Нитрование 431
5.1.4. Сульфирование 435
5.1.5. Галогенирование 443
5.1.6. Тиоцианирование (роданирование) 448
5.1.7. Алкилирование по Фриделю — Крафтсу 449
5.1.8. Электрофильное ароматическое замещение при взаимодействии
с карбонильными соединениями 454
5.1.8.1. Ацилирование по Фриделю - Крафтсу 455
504 Оглавление
5.1.8.2. Синтез Гаттермана 459
5.1.8.3. Синтез Вильсмейера 460
5.1.8.4. Электрофильное замещение при реакциях с
формальдегидом 463
5.1.8.5. Катализируемые кислотами реакции ароматических
соединений с другими альдегидами или кетонами 467
5.1.8.6 Карбоксилирование 469
5.1.9. Нитрозирование 471
5.2. Нуклеофильное ароматическое замещение 472
5.2.1. Нуклеофильное замещение в активированных ароматических
соединениях 473
5.2.2. Нуклеофильное замещение в неактивированных ароматических
соединениях 477
5.3. Ароматическое замещение при содействии металлов 479
5.3.1. Металлирование ароматических соединений 480
5.3.2. Сочетание ароматических соединений с металлоорганическими 484
5.3.2.2. Сочетание металлоорганических соединений щелочных
металлов с медьорганическими 484
5.3.3.2. Перекрестное сочетание (кросс-сочетание),
катализируемое переходными металлами 485
5.3.3. Реакция Хека 490
5.3.4. Сочетание арил—гетероатом
5.4. Литература 493
лучший 1
зарубежный
УЧЕБНИК
Учебное издание
Беккер Хейнц, Беккерт Райнер, Бергер Вернер и др.
ОРГАНИКУМ
В двух томах
1
Зав. редакцией канд. хим. наук Т. И. Почкаева. Ведущий редактор И. С. Беленькая
Художник М. М. Иванов. Технический редактор Е. В. Денюкова
Оригинал-макет подготовлен А. А. Пудовым, Е. В. Денюковой
Подписано к печати 01.10.07. Формат 70 * 100/16. Бумага офсетная. Печать офсетная.
Бум. л. 15,75. Печ. л. 31,50. Изд. № 3/9915. Тираж 2000 экз. Зак. 12233.
Издательство «МИР»
Министерства культуры и массовых коммуникаций РФ.
107996, ГСП-6, Москва, 1-й Рижский пер., 2.
Диапозитивы изготовлены в издательстве «Мир»
Отпечатано с готовых диапозитивов в ОАО «ИПК «Южный Урал»,
460000, г. Оренбург, пер. Сиободппа, 4.