






















































































































Похожие




Текст
Практикум
по органической химии
Practical Organic
Chemistry
A student handbook of techniques
J.T. Sharp, I. Gosney
University of Edtnburg
A. G. Rowley
Consultant in analytical chemistry
London New York
CHAPMAN AND HALL
ДЖ. ШАРП, И. ГОСНИ, А. РОУЛИ
Практикум
по органической химии
Перевод с английского
канд. хим. наук В. А. Павлова
под редакцией
д-ра хим. наук, проф. В. В. Москвы
МОСКВА "МИР1*
1993
ББК 24.2
Ш25
УДК 547
Шарп Дж Госни И., Роули А.
Ш25 Практикум по органической химии: Пер. с англ. -М:
Мир, 1993. - с. 240, ил.
ISBN 5-03-002126-4
Кинга авторов из Великобритании может служить руководством по
технике эксперимента в органической химии. Она подскажет, как
провести ту или иную Операцию, расскажет о маленьких хитростях,
которые помогут достичь наилучшего результата, обратит внимание на
меры безопасности непосредственно по ходу эксперимента. В нее
включены последние достижения и разработки в области
эксперимента в органической химии (например, цМаш-вакуумный пиролиз) и
практические ценные советы по выделению и очистке веществ,
особенно методом преп ра вно хроматографии.
Для студентов, преподавателей и аспирантов химических и нехими-
ческик вузов, изучающих органическую химию.
1705000000 - 066
Редакция литературы по химии
Федеральна! целевая программа книгоиздания России
Учебное издание
Джон Шарп. Иэн Госни, Алан Роули
ПРАКТИКУМ НО ОРГАНИЧЕСКОЙ ХИМИИ
Заведующий редакцией вка^еник О. А Реутов
дожеегкн реяи . <-.<р В. И Шаповалов
Технический М. яебона.Коррех И. Киселей
Иб№ T7WI
Подписано к пеит* 26.03ЭЗ. Формат бОаЗД IJU. Букага офоспм
Гарнитуре таИнс Шьем 7.50 6,™ я Уел геч л. 15. Усл. чр.-с
Иэд NSsyT7S7 Тираж 1MOUSC1 Зш»э№9 СМИ.
"Мир" Мнинстдх™ леч« нифорнаиин РосснвсиоЯ Ф<и
Филиал МИ ПЕрви обримовая типография 113114. Мнхва, м
ISDN 5-ОЗ-О02126-4 <русск.» ©1989 J Т. Sharp. I Gosney,
ISBN 0-412-28230-5 (англ.) A G Rowley
© перевод не русский t
Павлов В.Л.. 1993
ПРЕДИСЛОВИЕ АВТОРОВ
Самым замечательным и увлекательным в органической
химии представляется экспериментальная работа за
лабораторным столом". Овладение этим искусством необходимо не
только профессиональным исследователям, но и ст>.ентам
на самых ранних стадиях об ени П «вл кательность
самого предмета органической химии хвязана с бесконечным
множеством различных типов реакций и почти неисчерпаемым
ообразием химических соединений. Каждая реакция в
каждое новое соединение ставят свои собственные особые
проблемы, бросая вызов мастерству и изобретательности
химика независимо от того, работает ли он в лаборатории на
первом году обучения или же находится на переднем крае
научных исследований.
Назначение этой книги - дать основное руководство по
технике экспериментальны к работ, используемой в типичном
студенческом к рс Она в сжатой форме охватывает весь
набор требуемых практических навыков как для начинающих
студентов, еще ие имеющих опыта лабораторных работ, так
и для студентов, заканчивающих курс обучения, когда они
обычно привлекаются к более сложной и ответственной
экспериментальной исследовательской работе под руковдством
научного руководителя.
Нашей целью было создание руководства по технике экс-
римента, которое может быть использовано в различных
практических курсах во время всего периода обучения
студентов. Преподаватели, ведущие практические занятия,
обычно очень заинтересованы в подборе частных экспериментов
или упражнений для своих студентов, и поэтому редко
бывает так, чтобы книга, описывающая эксперименты для одного
раздела химии был бы пригодной для другого. Однако
существуют экспериментальные приемы общие для всех
разделов, и мы надеемся, что эта книга будет источником
полезной информации по ряду методов, влякнци я неотъемлемой
частью современной химической практики. Мы включили не
только классические и вечные методы, как, например,
очистка соединений кристаллизацией и перегонкой, но также и
более современные приемы работы с реагентами,
чувствительными к влаге и воздуху, без которых успех современной
органической химии был бы невозможен. Кроме того, включены
ременны методы репара ной хрома графин например
"флэш"-хроматография и хроматография "среднего давления",
6 Предисловие авторов
а также аппаратурное оформление хроматографии, газо-жид-
костная хроматографии (ГЖХ) и высокоэффективная
жидкостная хроматография (ВЭЖХ), играющие важнейшую роль в
контроле и анализе реакций.
Книга такого объема не может претендовать на
исчерпывающий характер; кроме тога, как и при венком
практическом обучении, некоторые приборы и методы лучше
демонстрировать, чем описывать. В этом случае прн выборе метода
синтеза каких-либо соединений или проведении изучаемых
реакции весьма существенны руководство и совет опытного
преподавателя.
Так как книга рассчитана на неодинаковую подготовку
студентов, разные главы ее построены таким образом, что в
начале каждого раздела основное внимание уделяется тому,
как обращаться с прибором, и важнейшим аспектам метода.
В дальнейшем материал дается в более развернутом виде,
например рассматривается оптимизация текущих условий и
параметров. Основная теория, например хроматографии,
приводится лишь на таком уровне, который необходим для
эффективной практической работы
Хотя эта книга предназначена в основном для студентов,
мы надеемся, что она окажется ценной и для аспирантов и
стажеров в качестве руководства по основным
экспериментальным методам, которые они могут усовершенствовать
модифицировать и распространить на отдельные области
исследования.
Мы признательны нашим многочисленным коллегам
(бывшим и настоящим), дипломникам и студентам за их ценные
советы относительно того, что в настоящее время считается
хорошей, эффективной и безопасной лабораторной практикой.
Особенно мы благодарны доктору Д. Рейду (ЯМР служба
Эдинбургского университета) за его советы, касающиеся
подготовки образцов для спектроскопии ядерного магнитного
резонанса (ЯМР). Наконец, мы выражаем надежду, что многие
из тех, кто впервые подойдет вплотную к проблемам
практической органической химии, получат такое же большое
удовольствие и удовлетворение от нее, какое имеем мы сами.
Эдинбург, Дж. Шарп, К. Гост и А. Роули
октябрь 1988 г.
ПРЕДИСЛОВИЕ РЕЦЕНЗЕНТА
Существовало два аспекта в преподавании органической
химии, которые вызвали мое пристальное внимание к этому
предмету.
Во-первых, использование представлений о механизме
реакции для разумного объяснения экспериментальных
наблюдений и, во-вторых, наиболее важное - это радость и
наслаждение, испытываемые при выполнении синтезов, приводящих
к чистым продуктам.
Хотя в настоящее время для анализа этих продуктов
используются главным образом инструментальные методы, для
химика-органика основной все же остается экспериментальная
работа по синтезу соединений и выделению их в чистом
виде. Чтобы получить удовлетворение от эксперимента, химик
должен овладеть искусством практической органической
химии. К сожалению, с сокращением практического содержания
многих студенческих курсов трудно выработать необходимые
навыки. Настоящая книга дает прекрасную основу для
обучения экспериментальным методам, необходимым в
практической органической химии, начиная с первого года обучения
вплоть до аспирантуры и дальше, и делает это с учетам
наиболее существенных мер безопасности, принятых в
современной практике.
Имеется несколько блестящих книг, в которых особое
внимание уделяется ряду интересных экспериментов и реакций.
Однако в них отсутствует глубокая проработка основных
экспериментальных требований, необходимых для успешного
проведения реакций и выделения чистых веществ - двух
решающих условий, которыми должны овладеть начинающие
химики. Авторы написали книгу, которая найдет широкое
применение, поскольку ее можно использовать в сочетании с
другими источниками, где описываются стандартные методики
синтезов. Хотя разрабатываются сложные компьютерные
программы для поиска оптимальных путей синтеза, следует
помнить, что огромное количество продуктов, продаваемых
химической и фармацевтической промы енностью является
чистыми соединениями, а не компьютерными распечатками.
Производство этих соединений требует от npnpcw»K*4.iM«cici
химика высокого мастерства.
Таким образом, данная книга, обучающая молодых
химиков правильному проведению практических работ, играет
I
S Предисловие рецензента
важную роль в овладении чстг^пктллммм методами и
поэтому займет достойное место на лабораторном столе.
Октябрь 1988 г. р_ Рамадж
Профессор химии
Эдинбургского университета
БЛАГОДАРНОСТИ
Мы благодарны фирме "J.Bibby Scieace Products Ltd" за
разрешение приводить в Схемах ваших приборов рисунки
екоторой части их продукции: стандартной конической
стеклянной посуды "Quicxfit", пластиковых хомутикоя ("Bibby")
и запорных краников Rotaflo.
Мы также признательны фирмам "American Chemical
Society", "Marcel Dekker Inc.", "Aldrich Chemical Company
Lid*' и "John Wiley and Sons Inc." за разрешение поместить
некоторые охраняемые авторским правом выдержки из текста,
диаграммы или таблицы, как указано в тексте.
МЕРЫ БЕЗОПАСНОСТИ И КОНТРОЛЬ
ЗА РАБОТОЙ В ЛАБОРАТОРИИ
ОСНОВНЫЕ МЕРЫ ПРЕДОСТОРОЖНОСТИ
1. Работайте в лаборатории только в отведенное для этого
время, под контролем преподавателя или других
сотрудников.
2. ВСЕГДА над вайте защитные очки (или защитную
маску). (Те, кто носит контактные линзы, читайте разд.
1.3.1.)
3. Не принимайте пищу, не пейте и не курите в
лаборатории.
4. Если вы испытываете какие-либо сомнения в методике
эксперимента или в технике безопасности, прежде чем
продолжить работу проконс льтир йтесь вашим
преподавателем.
Более детальные меры предосторожности даны в разд. 1.3;
их необходимо прочесть до начала эксперимента.
КОНТРОЛЬ В ЛАБОРАТОРИИ
Методы, описанные в настоящей книге, представляют
собой общепринятую экспериментальную практику. Однако
необходимо подчеркнуть, что это - общие правила. При
проведении конкретной химической реакции или при работе с
определенным химическим соединением может потребоваться их
модификация, чтобы сделать их эффективными для данного
частного случая или по причинам безопасности. В связи с
этим важно, чтобы студенты и другие неопытные
исследователи проводили практическую работу ОЛЬКО под контролем
квалифицированного персонала с должным вниманием к
технике безопасности (см. руководство "Ouide to Safe Practices
in Chemical Laboratories", опубликованное "Royal Society of
Chemistry', London) и законодательству.
ССБТ, "Организация (Сучения безопасности труда. Общие положения"
ГОСТ 12 О О04-90 Москва. ССБТ. "Работы учебные лабораторные. Общие
требования безопасности", ГОСТ 12.4.113-82 Москва - Прим перев.
1. ВВЕДЕНИЕ
1 1. ЭКСПЕРИМЕНТАЛЬНЫЕ МЕТОДИКИ
Многое из практической органической химии относится к
тому, что обычно называют "препаративной" или
"синтетической" работой, целью которой является проведение
химической реакции или серии реакций для получения
определенного химического соединения в чистом виде с максимально
возможным выходом. Такие процессы лежат в основе
высокоэффективной химической промышленности, производящей
огромное число химических продуктов, начиная от простых
веществ, используемых при производстве пластмасс и других
полимеров, и до сложнейших соединений медицинского
назначения.
Лабораторный синтез обычно СОСТОЯТ ИЗ трех
последовательных операций: а) химической реакции, т. е. превращения
реагирующих соединений в продукты реакции; б) отделения
нужного продукта или продуктов от растворителей, побочных
веществ или неорганических материалов и, наконец, в)
очистки и идентификации продукта. В общем, методы
проведения реакций (см. пункт "а") достаточно просты и включают
в себ« смешение реагентов в соответствующих количествах и
стимулирование реакции путем нагревания или облучения
(гл. 2). Однако в последнее время эта область стала более
ежной, так как прогресс в синтезе определяется все возра-
ающим использованием реакционно-способных,
чувствительных к воздуху и влаге реагентов для работы с которыми
необходимы низкие температуры или инертная атмосфера.
Методы, относящиеся к группе "б", при помощи которых
требуемый продукт выделяют из реакционной смеси, часто
называют методами "обработки". В некоторых случаях это
сделать очень легко, но обычно это наиболее сложная часть
работы, в которой экспериментатору приходится использошль
вес свои знания и умение, чтобы грамотно комбинировать
различные методы, например экстракцию, хроматографию или
кристаллизацию, дли достижения нужного результата. В
простых студенческих лабораторных работах методы выделения
продукта обычно четко изложены в методических
руководствах но в более сложной исследовательской работе именно
здесь требуется принятие решений (гл. 3 и 4). Поэтому
много внимания в книге уделено методам выделения и очистки
веществ, включая необходимую теоретическую подготовку.
12
Глава 1
для того чтобы оценить их возможности и недостатки.
Кроме этих методов, напрямую связанных с
препаративной работой, химику часто необходимо наблюдать за
протеканием реакции, т. е. проверять, насколько она прошла, или
определять точный выход либо соотношение продуктов. Для
этого обычно пользуются одним из инструментальных хрома-
тографических методов, например газо-жидкостной
хроматографией (ГЖХ) (разд. 4.1.2) или высокоэффективной
жидкостной хроматографией (ВЭЖХ) {разд. 4.1.3), которые
позволяют проводить анализ всего лишь на микрограммовых или
миллиграммовых образцах реакционной смеси.
Когда в результате химической реакции получено какое-
либо соединение, возникает необходимость его
идентификации. Идентификацию известных соединений (полученных
ранее, чьи свойства уже описаны в химической литературе)
традиционно проводили по их физическим свойствам,
например по температуре плавления, температуре кипения и
показателю преломления. Эти константы и сейчас играют очень
важную роль и как критерии идентификации, и как
критерии чистоты, но помимо них имеется целый ряд
спектральных методов, являющихся необычайно мощным инструментом
идентификации известных, а также новых соединений.
Наиболее распространенные среди них - инфракрасная (ИК) и
ультрафиолетовая (УФ), спектроскопия, масс-спектрометрия, а
также ядерный магнитный резонанс (ЯМР). Данная книга не
включает интерпретацию спектров (по этим вопросам имеется
много блестящих учебников), но все-таки содержит
информацию по подготовке образцов для спектроскопии (гл. 5).
1.2. ХОРОШАЯ ЛАБОРАТОРНАЯ ПРАКТИКА*
'Ключом к успеху в практической работе является
заповедь: "думайте прежде, чем делать". I Практическую работу
никогда нельзя сводить к простому упражнению с
"поваренной книгой" в руках, т. с. к ней не следует подходить со
слепой верой в инструкции и в блаженном неведении xhmhhj
При выполнении работы, на которую даны подробные
инструкции, вам следует сначала прочесть их полностью, лучше
до прихода в лабораторию, а) убедиться в том, что вы пони-
Под "хорошей лабораторной практикой" (Good Laboratory Practice, CLP)
подразумевается набор стандартных требований, предъявляемых к условиям
проведения эксперимента. GL.P - является критерием доверии к надежности
экспериментальных данных и позволяет считать результаты эксперимента
безусловно достоверными. - Прим. перев.
введение
13
маете химизм процесса и цель работы, б) удостовериться,
что вам ясны смысл эксперимента и приемы работы, и если
нет, внимательно изучить их прежде, чем приступать к
работе, и в) обдумать заранее каждое действие и постараться
мысленно представить себе, что вам предстоит проделать как
с точки зрения химизма процесса, так и приемов работы и,
таким образом, предвидеть возможные затруднения или
степень риска. Думать - означает, что ваше путешествие в мир
синтеза будет более многообещающим, прибывать к цели вы
будете гораздо чаще и по пути даже немного изучите
химию.
Вы также убедитесь, что хорошую химию делать намного
легче, если вы организованны и работаете в чистых и
опрятных условиях. Часто приходится проводить несколько
экспериментов одновременно, например, некоторые реакционные
смеси необходимо кипятить с обратным холодильником в
течение нескольких часов, и за это время вы могли бы
заняться идентификацией "неизвестного" соединения или
работать с другой реакционной смесью. По мере приобретения
опыта проводить параллельную работу становится легче, но
при этом важно не увлекаться, чтобы не делать
эксперименты поспешно и небрежно, толком не завершив их.
Это относится, в частности, к исследовательской работе,
где крайне легко увлечься и унестись на волне энтузиазма -
поэтому помните, (что проведение реакций - это относительно
легкая часть работы; ее завершение, выделение и индентифи-
кация продукта, занимают наибольшую часть времени.!
Однако, несмотря на самые благие намерения и хорошие
методики, иногда что-то не будет получаться или вам
покажется, что все идет неправильно. Обычно, это случается из-
за неправильного применения какого-нибудь
экспериментального метода, вследствие отсутствия опыта или недостаточно
внимательного наблюдения за протеканием реакции без
использования тонкослойной хроматографии (ТСХ) или
газожидкостной хроматографии (ГЖХ) (разд. 4.1.1 и 4.1.2). В
исследовательской работе может даже оказаться ошибочной
теория.
1.3. МЕРЫ БЕЗОПАСНОСТИ В ЛАБОРАТОРИИ
Меры безопасности. Ключевые моменты и основные
меры предосторожности даны перед введением (с. J0).
Практическая органическая химия при надлежащей работе
вполне безопасное занятие, но чтобы сделать ее таковой, не-
14 Глава 1
обходимы внимание и осторожность. Многие из веществ,
используемых в органической химии, являются в той или иной
мере воспламеняющимися или токсичными или теми и
другими одновременно. Разработка правильных рабочих методик
гарантирует, что с такими веществами можно работать
безопасно.
Меры безопасности. Все лаборатории в высших учебных
заведениях разрабатывают собственные меры безопасности
при работе, и <туде ы должны неукоснительно их
соблюдать. Ниже даны некоторые общие советы, касающиеся
обычных мер предосторожности, но они не являются ис^
черпывающими и должны быть дополнены специальными
мерами, разработанными для конкретных экспериментов.
1.3.1- Меры безопасности при работе с химическими
веществами
В общем, органические реагенты, предназначенные для
работы в учебных лабораториях, должны быть низкой
токсичности. Но даже если это и так, нелишне руководствоваться
следующим:
1. Держите ВСЕ соединения и растворители подальше от
рта, кожи, глаз и одежды.
2. Избегайте вдыхания паров или пыли.
3. Никогда ничего не пробуйте в лаборатории на вкус.
Особая осторожность необходима при работе с сильными
кислотами, едкими и летучими реагентами и
воспламеняющимися растворителями. При выполнении исследовательской
работы помните, что вы можете получить, преднамеренно или
случайно, новые химические соединения с неизвестными
биологическими свойствамии.
а) Средства индивидуальной защиты
I) Глаза
Меры безопасности. Защитные очки (или маску, если
требуется полная защита лица) необходимо надевать
всегда, когда находишься в лаборатории.
2) Контактные линзы
Студент, который носит контактные линзы, должен
принимать самые строгие меры, чтобы не допустить попадания в
Введение 15
глаза любого вещества. Едкие или токсичные вещества могут
быстро проникнуть под контактную линзу, и тогда
промывание становится почти невозможным.
3) Руки
При осторожной и аккуратной работе химикаты, как
правило, не должны попадать на ваши руки. Однако при
работе с вредными, едкими или токсичными материалами
разумно надевать защитные перчатки, но помните, что ваши
движения станут более неуклюжими.
4) Одежда
В лаборатории необходимо находиться в застегнутом
лабораторном халате. Это обеспечивает некоторую
индивидуальную защиту и позволяет избежать загрязнения одежды.
Лабораторные халаты следует регулярно стирать (соблюдая
соответствующие меры предосторожности, если они загрязнены).
б) Общие меры предосторожности
1. Не нагревайте, не смешивайте, не лейте и не
взбалтывайте реактивы вблизи от лица. Всегда направляйте горло
сосуда от лица и тела.
2. Никогда не засасывайте жидкость в пипетку ртом, всегда
пользуйтесь грушей или специальным приспособлением для
наполнения пипеток.
3. Будьте осторожны с сильными кислотами или щелочами,
особенно при нагревании. Никогда не добавляйте воду к
концентрированным кислотам или щелочам.
4. С материалами, выделяющими вредные пары, следует
работать только в вытяжном шкафу, надевая защитные
перчатки. К таким материалам относятся галогениды
фосфора, бром, все хлорангидриды кислот, уксусный ангидк
рид, дымящая азотная кислота, концентрированный
раствор аммиака, жидкий аммиак, диоксид серы и другие.
Если у вас возникли какие-то сомнения,
ПОСОВЕТУЙТЕСЬ с преподавателем.
в) Удаление отработанных химикатов
Меры безопасности. Не сливайте органические
растворители или другие органические вещества в раковину.
Отработанные растворители следует выливать в
специальные приемники; другие остатки удаляют согласно
инструкции.
1.3.2. Пожароопасность
Большинство органических растворителей и многие другие!
органические жидкости летучи и легко воспламеняются.
Некоторые из них при контакте с воздухом образуют
взрывчатые пероксиды (см. пункт 5 ниже) Ниже приведены общие
меры предосторожности, чтобы избежать пожара.
1. Не нагревайте органические жидкости, даже в малых
количествах, на открытом огне или рядом с ним. Для этого!
пользуйтесь водяной баней (разд. 2.1.4), масляной баней
(разд. 2.1.4) или электронагревателем (разд. 2.1.4). Особая]
осторожность необходима при работе с эфиром, петролей-
ным эфиром и сероуглеродом, которые очень летучи и i
имеют низкие температуры вспышки.
2. Не нагревайте органические жидкости в открытом сосуде.
Обязательно установите холодильник - либо обратный
(разд. 2.1.4), либо прямой для отгонки (разд. 3.4.2). В
некоторых методиках синтезов требуется отделить
растворитель от продукта реакции "выпариванием". Для этого I
используют либо роторный испаритель (разд. 3.1.2), либо]
отгонку (разд. 3.4). НЕЛЬЗЯ проводить выпаривание рас-]
творителя непосредственно в атмосферу .
3. Никогда не нагревайте закрытую систему любого типа.
4 Перед использованием эфира (или любого другого летуче-1
го легковоспламеняющегося растворителя), например для |
экстракции (разд. 3.1.3), удостоверьтесь, что поблизости]
нет открытого огня или других источников возгорания
(ваших или вашего соседа). Часто безопаснее работать в]
вытяжном шкафу, чем за лабораторным столом.
5. Некоторые растворители, особенно эфиры и углеводороды. |
прн хран нии самопрои вольно разуют взрывчатые пе-]
роксиды. Перегонка содержащих пероксиды растворителей
очень опасна, так как остатки, в которых накапливаются]
пероксиды, прн нагревании могут взорваться. Поэтому та-|
кие растворители НИКОГДА не следует выпаривать или I
перегонять до тех пор, пока тест не покажет отсутствии]
пероксидов (проверить с вашим преподавателем). Ряд тес-1
тов для обнаружения пероксидов и методы их удаления!
прййедены в работах [1 - 31 (гл. 3).
1.3.3. Работа с вакуумом и давлением
I. Вакуумный эксикатор, находящийся под вакуумом, необ-1
Это допускается только fina воды. Прим. переа
jt ведение П
ходимо помещать в специальную безопасную оплетку .
2. Не вакуумируйте плоскодонные колбы за исключением
колб Бунзена.
3 Все приборы под давлением или вакуумом должны
находиться за защитным экраном. Не пользуйтесь
поцарапанной посудой.
1.4. ВЕДЕНИЕ ЗАПИСЕЙ
Ведение соответствующих записей - жизненно важная
часть всей экспериментальной работы. Окончательный отчет
должен быть точным, ясным и кратким и содержать такое
количество информации, чтобы любой профессиональный
химик смог точно повторить работу Ниже изложены
общепринятые положения и установившийся порядок ведения рабочего
журнала.
1.4.1 Запись экспериментальных данных
Ведите все записи в прочном лабораторном журнале.
Каждый эксперимент должен иметь номер, заглавие и дату его
проведения. По ходу эксперимента все наблюдения,
взвешивания, температуры и другие данные заносите
непосредственно в журнал (не пишите их на клочках бумаги, которые
легко потерять).
1.4.2. Окончательный отчет
После того как эксперимент завершен, необходимо
написать окончательный отчет (как проиллюстрировано ниже),
который должен включать:
1 Краткую формулировку цели эксперимента.
2. Написанный своими словами сжатый отчет о
непосредственно проведенном эксперименте, а ке простую копию
данной методики. Количества веществ помешают в скобки
после их названия. Приведем такой пример*. "Сухие
магниевые стружки (0,45 г, 0,018 моль) поместили в
высушенную в печи 25-мл трехгорлую колбу, снабженную
капельной воронкой, обратным холодильником (и то, и
другое с хлоркальциевыми трубками) и магнитной мешалкой.
В капельную воронку залкли раствор бромбензола
(2,65 г, 0,017 моль) в сухом эфире (9 мл) и прикапыва-
Можно просто обмотать его полотенцем - Прим. черт.
г. ПРОВЕДЕНИЕ РЕАКЦИИ
18 Глава I
ли его и течение примерно 5 мин с перемешиванием
После прикапывания первых нескольких капель, раствор
помутнел и стал нагреваться. Прикапывание было
продолжено с такой скоростью, чтобы эфир спокойно кипел".
Детальные описания стандартных экспериментальных методик,
например перегонки или кристаллизации, обычно не
требуются (за исключением экспериментов, специально
разработанных для обучения этим методам), но они должнь
включать сведения о любых изменениях, которые важны
для данного конкретного эксперимента.
3. Массу каждого продукта и его выход в процентах:
4. Температуру плавления или кипения каждого продукта, i
также литературные данные для сравнения [последние
можно получить из справочной литературы, имеющейся i
лаборатории или библиотеке (см. гл. б) ].
5. Спектроскопические данные о продуктах, если требуети
определить их подлинность или чистоту. Известные сое
аинения обычно идентифицируют по их инфракрасныг
спектрам (ИК) или спектрам ядерного магнитного резо
нанса (ЯМР); в последнем случае также следует приве
сти литературные данные (гл. б). Для ИК-спектров обыч
но достаточно сослаться только на поглощение существен
ных характеристических групп, но для спектров ЯМ1
следует привести полный спектр (как химические сдвиги
так и константы расщепления).
6. Заключительную часть, суммирующую результаты и ком
ментирующую их.
[.5. ОБРАЗЦЫ И СПЕКТРЫ
Сохраните в небольшом количестве образцы всех продук
тов, интермедиатов и производных и навесите на ампулу (
образцом ваше имя, номер эксперимента, дату, название сое
динения и его температуру плавления (т. пл.) или кипени)
(т. кип.). Спектры должны иметь аналогичные пометки, и
кроме того, на них следует указать условия и параметры
при которых они были записаны.
Для проведения препаративной реакции (А+В -» С)
прежде всего необходимо подобрать такие условия, при которых
исходные вещества А и В будут реагировать с подходящей
скоростью с минимальным образованием побочных продуктов
и при которых продукт С устойчив. Условия проведения
реакции определяются в значительной степени ее природой,
еобходимо контролировать главным образам температуру
реакции и порядок смешения реагентов; в некоторых случаях
требуется изолировать реакционную смесь от воздействия на
нее кислорода воздуха и влаги.
В экспериментальной работе, выполняемой в учебной
лаборатории, особенно на начальных стадиях обучения, условия
реакции обычно подробно описаны в инструкции к
проведению эксперимента. Следовательно, отпадает необходимость
подбирать температуру реакции, растворитель и
продолжительность реакции. Однако на более позднем этапе обучения,
и особенно в исследовательской работе, этот выбор
приобретает решающее значение и требует тщательного анализа
природы химической реакции, с которой вы имеете дело, а
также применения опыта и профессиональных навыков,
приобретенных в предшествующей работе.
В исследовательской работе трудно переоценить значение
контроля за ходом реакции. Для этого обычно используют
один из аналитических хроматографических методов (разд.
4.1), позволяющий следить за исчезновением реагентов
и(или) образованием продукта реакции. Время, потраченное
на отработку метода контроля, обычно полностью
компенсируется временем, сэкономленным на том, что не приходится
повторять реакции и что, когда цело доходит до выбора
наилучшего метода извлечения полученного продукта, состав
реакционной смеси известен.
2 1. ОСНОВНЫЕ МЕТОДЫ
2.1.1. Оборудование и приборы
В лаборатории химические реакции обычно проводят в
борах из боросиликатного стекла (пирекс), снабженных
ифами - коническими соединениями (муфта - керн) из
матового стекла (рис. 2.1). Реакции обычно проводят в круг-
ладонных (КД) колбах (рис. 2.2) с одним или несколькими
Керн
Муфта
Ж
Пластиковый
хомутик
t"Bibby")
э Переходная
Т муфта с
I БОЛЬШЕ ГС
) шлифа на
' меньший
Г Переходная
I муфта с
а меньшего
|шлифа на
I больший
Рис. 2.1 Конические шлифы стандартного размера (муфта
Рис 2.2. Круглодонные реакционные колбы.
горлами, в которые вставляют обратный холодильник,
капельные воронки, мешалки и т. д., как показано, например, №
рис. 2.3. Для одногорлых колб можно использовать двух- i
трехгорлые насадки (рис. 2.4).
Для соединения частей прибора со шлифами^ разных рая
меров существует целый набор переходных муфт (ряс. 2.1).I
На лабораторном жаргоне переходные муфты часто
1ми". - Прим. перев.
прояснение реакции 21
В большинстве случаев шлифы можно соединять "сухими"
(без смазки). Легкий толчок и поворот при сборке
обеспечивают слабый скрепляющий эффект за счет трения. Здравый
смысл подсказывает, что различные части собранного прибора
необходимо укрепить на штативах при помощи лапок. Для
закрепления двух частей прибора пои необходимости можно
использовать пластиковые хомутики (рис. 2.1), как,
например, для присоединения колбы к переходной муфте в
роторном испарителе (рис. 3.1).
Сухие шлифы обеспечивают хорошее уплотнение, тем не
менее жидкость проникает на поверхность матового стекла, а
некоторые растворы (особенно водный раствор гидроксида на-
Трехгорлая
круглоданная
келва
Реакционная
смесь
-Лопасть мешалки
"jc 2.3 Типичный прибор (в собранном виде) для проведения (
Пластиковые хомутики можно заменить резниками, нарезанными :
старой велоситушой камеры. - Прим. перев.
4f
Pml- 2.4. Двух- и трехгорлые насадки.
трия) вызывают необратимое разъедание обеих частей шлифа
Этого можно избежать, если поверх керна надеть тонкую
тефлоновую муфту или нанести тонкий слой смазки *углево-
ородной ма ки апиезон или силиконовой ). Шлифы следует
слегка смазать, чтобы они не пропускали воздуха или чтобы.
их части могли поворачиваться друг относительно друга.
СПОЛН
случаях
2.1-2. Смешение реагентов
Реакции можно проводить несколькими различными
бами в зависимости от их природы: I) в некоторых случая:
требуемые навески реагентов сразу смешивают в реакционном
сосуде перед началом реакции; 2) чаще один из реагентов
помешают в реакционный сосуд, а другой постепеннс добаИ
л«ют в ходе реакции в течение определенного промежутка
времени и 3} в редких случаях оба реагента добавляют
постепенно в ходе реакции.
а) Взвешивание и загрузка
В некоторых случаях (в мелкомасштабных реакциях) рея
генты можно взвесить непосредствектш в реак, н ом сосуде
но обычно лучше взвешивать их в отдельных емкостях и
затем переносить в реакционный сосуд. Твердые вещества лН
ще взвешивать в химическом стакане, покрытом часовые
стеклом, и засыпать в реакционный сосуд через воронку дл|
сыпучих веществ (рис. 2S), чтобы избежать загрязнение
шлифа.
более досту1|ный и дешевы!
описано в разд. 2 21
материалов, чувствите.-л
: действию воздуха и
Рис 2 5 Воронки для сыпучих веществ.
Если предстоит использовать растворитель, то часто
твердое вещество удобнее растворять в нем перед загрузкой.
Жийкеет'ь можно взвешивать в закрытых пробками
конических -олбак или (что более удобно) отмеривать
определенный ее объем (если известна плотность) с помощью мерного
цилинд реноса жидкости в реакционный сосуд
следует пользоваться обычной или капельной воронкой.
6) Постепенное добавление реагентов
и крупномасштабных
прикапывать из ка-
!енн'
■1
1. Жидкости и растворы. В средне-
реакциях жидкости я растворы ^можно
пельной воронки (рис. 2.3 и 2.6) ,
Чтобы легко было контролировать скорость прикапывания,
стеклянные краны необходимо слегка смазать, но не следует
наносить много смазки, ибо она может полностью или
частично закупорить отверстие в кране, а затем раствориться,
что приведет к изменению скорости прикапывания. В кранах
типа ротафло ("Rotaflo") с жидкостью контактируют только
тефлоновые части, которые не требуют смазки.
2. Твердые вещества. Постепенное добавление твердых
веществ - нелегкое дело, поэтому лучше всего вводить их в
раство■;-:', если это возможна Если ж такси возможности
1 то твердое ■ ществ следу вводить порциями через во-
Р°нку для Сыпучих веществ. Однако в большинстве случаев
Ри ведении реакции в кипящем растворителе необходим
^^ обавлением порции вещества дать
реакционному сосуду слегка остыть.
Р"Чы для правления небольших объемов
Рис. 2 6. Капельные воронки со стеклянньсм краном <а) и краником i
ротафяо ("Roiano") {©-
2.1.3. Перемешивание реакционных смесей
Перемешивание реакционной смеси обеспечивает хорошее
смешение по мере введения реагентов, удерживает твердьк
вещества и масла в виде суспензии или эмульсии и
способствует равномерному кипению при проведении реакции с
обратным холодильником (разд. 2.1.4). Существуют два
основных приема: а) при помощи лопастной мешалки, установлен]
ной на оси, непосредственно соединенной с моторчиком (рия
2.3) и б) посредством магнитного бруска, приводимого в
движение вращающимся магнитом, установленным под
реакционным сосудом (рис. 2.7).
В препаративной работе редко пользуются вибраторами,
которых встряхиваются весь сосуд и его содержимое. Однак<
иногда они находят применение, как, например, при интен
сивном перемешивании двухфазной смеси в реакциях фаэовс
го переноса.
Установить лопастные мешалки сложнее, чем магнитны*
(см. ниже), ни ини необхцдимы для перемешивании болышп
количеств реагентов, а также вязких растворов или раство -'
пддццв ие реакции
— Мешалка,
комбинированная
с электроплиткой
Рис 2 7. Использование комбинированной магнитной мешрлки с эле«1ро-
илиткой для перемешивания реакционной смеси и иа!ревания на масляной
ров. содержащих много взвешенного твердого вещества, когда
гнитные мешалки неэффективны. Выбирая тип мешалки,
необходимо помнить, что в некоторых реакциях образуются
твердые вещества или комплексные соединения и по мере
протекания реакции смесь все труднее перемешивать. Если у
вас возникли сомнения, используйте лопастную мешалку.
Втулка
(вращающаяся
с осью-)
Направляющий
стакан (непо -
ДБИЖНЫЙ ) _
Затвор из матового
стекла (смазанный)
Тефл ежовая
прокладка
Конусообразный -
шлиф для
подсоединения КОЛБЫ
Тефлоновая
лопасть.
Рис. 2.8. Лопзстая
к с завинчивающимся
Переходник
с эавмкчивающиис
колпачком
i с затвором в собранном виде (о) и перехо t
i в качестве простейшего затвора для ме
а) Лопастные мешалки
Наиболее эффективные мешалки этого типа снабж<
тефлоновой (политетрафторэтиленовой) лопастью, по форм
соответствующей колбе и Hai. ..жатой (так, что ее можн
снять) на вращающуюся стеклянную ось (рис. 2.8,а). (ТЩ
лон инертен ко всем реагентам, за исключением фторидов
расплавов щелочных металлов; его можно использовать пр
температурах вплоть до 250°С.) В горло колбы вставлен
правляющин стакан (рис* 2.8,й), в котором имеется затвор и
матового стекла, смазанный вязким маслом (силиконовым Щ
медицинским жидким парафином) для обеспечения герм
ности; это предотвращает утечку паров кипящих расл
лей или попадание в колбу воздуха и водяных паров. Вту
ГТропедепие реакции
(вращающаяся часть затвора) прикреплена к оси мешалки с
помощью уплотнительного кольца из силиконовой резины
расположенного в завинчивающемся колпачке . При медлен'
ном непродолжительном перемешивании в качестве за™а
можно использовать непосредственно завинчивающийся
колпачок (рис. 2.8,6), смазав кольцо из силиконовой резине на"о-
сГзкой. ВНУФИ НСГ°- ЖИДК"М ""Р**™* «™ силикон
Для облегчения установки мешалки и плавности ее поа-
щения необходимо гибкое соединение моторчика с
веющейся осью мешалки. Для этого в качестве соедините^™
элемента используют короткий кусок толстостенной S™°
труоки, который закрепляют двумя скобами или скр1™й
медной проволокой (рис. 2.3). врученной
Меры безопасности. Моторчики, приводящие е движение
мешалки, при вращении искрят, поэтому их нельзя
устанавливать там, где есть воспламеняющиеся пары. Они
обычно имеют встроенный регулятор скорости, но в целом
по мере разогрева их скорость возрастает- Следовательно,
прежде нем оставлять такое устройство без присмотра,
необходимо выждать определенное время для достижения
равномерного перемешивания. Особое внимание следует уде-
чя/пь также надежному соединению частей прибора, так
как вибрация мешалки может привести к расшатыванию
зажимов и шлифовых соединений.
б) Магнитные мешал:
Как отмечалось выше, они очень удобны и эффективны
для легкоперсмешиваемых систем. Перемешивающие бруски
сейчас сочти всегда имеют тефлоновос покрытие
(ограничения иа использование тефлона см. выше, в п. "а"), но
остерегайтесь использования более старых и более дешевых их
разновидностей, покрытых другими пластиками, которые
могут быть неустойчивыми к действию ряда растворителей или
высоких температур В реакциях с участием расплавов
щеточных металлов, вызывающих потемнеине тефлона, следует
применять перемешивающие бруски с изоляцией из стекла.
Комбинированные магнитные мешалки с нагревательным
лементом (см., например, рис. 2.7) можно использовать ъ
I it,. "мес™ заиинчивающегосн колпачка
резиновой прокладкой i
ma - Прим персе.
Часто в качестве смазки используют глицерин. - Прим. переа.
28
качестве обычных мешалок для реакций при комнатной тем
пературе. В то же время они очень удобны для проведения
реакций, требующих одновременного перемешиваиня и
нагревания (см. также следующий раздел).
в) Вибраторы
Наиболее распространенный тип вибратора имеет приводи-'
мую в движение электродвигателем лапку для крепления реч
акционного сосуда, которая движется вверх и вниз, осуществи
ляя перемешивание.
пдлирдигае реакции
Меры безопасности. Здесь важны четыре момента: Я
пробку реакционного сосуда следует зажать или замотать
проволокой; 2) вибратор следует уравновесить, поместив
груз на противоположную сторону лапки; 3) скорость
вибраторов по мере разогрева электродвигателя возрастает,
поэтому их не следует оставлять без присмотра, пока н£
установилась постоянная требуемая скорость; 4) вибратор,
разумно размещать за защитным экраном в вытяжном
шкафу на случай, если колба разобьется.
2.1.4. Контроль за температурой
а) Нагревание реакционных смесей
Почти все препаративные реакции проводят в жидкой i
зе в каком-либо растворителе, даже если сами реагенты
жидкие. Особенно важно знать температуру кипения растворите-1
ля, так как на этом основан самый удобный способ контроля
за температурой реакции (см. п. 1 ниже). В учебных
синтезах растворитель указан в описании методики, а для иссле-|
довательской работы некоторую полезную информацию
свойствах растворителей и методах вх очистки можно i
в справочниках [З-б].
I) Кипячение с обратным холодильником
При проведении реакции в кипящих растворителях (как,
например, на рис. 2.3 и 2.1) для возвращения сконденсирс
ванного растворителя в реакционную колбу используют уста-!
новленныи вертикально холодильник. Этот способ обычно
называют "кипячение с обратным холодильником". Для равнг'
мерного кипения в раствор помещают кипятильнички или
перемешивают его.
СПОСОБЫ НАГРЕВАНИЯ. Электрические колбонагрезате-
ли (рис. 2.9) обеспечивают самый безопасный и наиболее
эффективный способ нагревания круглодонных колб с обратным
холодильником. Для повышения эффективности и
безопасности нагревания нужен колбонагреватель подходящего размера
(размер колб всегда указан на кожухе колбонагревателя).
Подачу энергии к чехлу следует отрегулировать так, чтобы
обеспечить медленное возвращение растворителя в реакцией
ную колбу.
:- 2 9 Электрический колбонагрева
качестве кинягнльничкоя можно использовать кусочки предварительно
Рокаденшго фарфора (например, от разбитой посуды) или дробленого крас-
га кир,шча, а также стекловату - Прим перев.
rJE2fSg£i
Для проведения реакций с обратным холодильником п-
температурс кипения растворителя инже 80°С можно таи
использовать нагреваемые на электроплитке водяные бани.
некоторых случаях предпочтительнее именно водяные бани,
не колбонагреватели; так, например, если реагенты или п .
дукты реакции очень чувствительны к действию температур!
то на поверхности колбы температура должна быть ка
можно ниже. Недостатки водяных бань очевидны: 1) й
того, что вся вода может выкипеть, они не подходят Д
проведения реакций без присмотра или в течение ночи;
водяной пар и конденсат, покрывающие прибор, затрудня
проведение реакций в безводной среде; 3) они нспригод •
для проведения реакций с участием натрия и других вэ
ществ, энергично реагирующих с водой, поскольку во ври
опыта колба может треснуть или произойдет разгерметизаци
прибора.
ОБРАТНЫЕ ХОЛОДИЛЬНИКИ. Для охлаждения жид .
стей с температурой кипения выше 50° С пригоден обыч
водяной холодильник Либиха (рис. 2.10), но для жидкостей
более низкой температурой кипения, например дизтилчж!
эфира (т. кип 350С)У требуется более эффективный обра
ный холодильник с дьойной рубащкой (рис. 2.10).
Воздушный
ХОЛОДИЛЬНИК
Рис 2.10 Холодильники.
Надевая резиновую трубку на холодильник, оберните его
полотенцем, чтобы не порезать руку при поломке, и смочите
трубку водой, этанолом или глицерином либо нанесите
немного вакуумной смазки Эти трудности не возникают при
использоваини современных холодильников с пластмассовыми
отводами для воды с нарезкой
Меры безопасности. Если вы хотите оставить реакцию
ни ночь, водяной шланг следует закрепить на холодильнике
и водопроводном кране медной проволокой (или зажимами).
(Будьте осторожны: старая, протершаяся резиновая
трубка при намотке проволоки может порваться.) Необходимо
также поставить автоматический выключатель,
реагирующий на подачу воды, который отключает подачу энергии к
колбонагревателю, если поступление воды в холодильник
прекратится.
Таких потенциальных трудностей иногда можно избежать,
используя холодильник типа "Airflux" (фирмы "Jeneons
Scientific Ltd," Leighton Buzzard, Bedfordshire), который
охлаждается водой и в то же время не нуждается в ее подаче.
Он представляет собой модифицированный холодильник
Либиха, в котором охлаждающая вода охлаждается сама путем
циркуляции через алюминиеаый поглотитель тепла.
Теплоемкость его, однако, ограничена, и такие холодильники
подходят только для жидкостей с температурой кипения в
пределах 60 - 150°С.
Для охлаждения высококипящих жидкостей (т. кип.
150°С) подходят воздушные холодильники (рис. 2.10).
2) Проведение реакций без обратного холодильника
В некоторых случаях для проведения реакции требуется
нагревание при достаточно высоких температурах, для
которых трудно подобрать соответствующий растворитель, чтобы
осуществить кипячение с обратным холодильником. В таких
^Тучаях реакционную колбу нагревают на масляной бане или
(если температуры очень высокие) на бане из расплавов
металлов (металлические бани).
МАСЛЯНЫЕ БАНИ. Для проведения реакции в интервале
Температур от комнатной до 250° С используют прибор (рис.
1п. в котором колба нагревается на масляной бане с
помощью электроплитки. Обычно применяется комбинированная
"^тка со встроенной магнитной мешалкой, и перемешивание
^уществляется как в реакционной колбе, так и в масляной
32 Гдавд j
бане. Реакционную смесь при необходимости можно также
перемешивать лопастной мешалкой (разд. 2.1.3). Температур;
реакционной смеем контролируется с помощью термометра,
погруженного в реакционную смесь (рис. 2.7), другой
термометр контролирует температуру бани.
Легкость контроля за температурой в значительной
степени определяется конструктивными особенностями плитки
Лучшие типы плиток имеют термореле с
термочувствительным элементом, погруженным в масло. Они обеспечивают
точное соблюдение заданного температурного режима
независимо от окружающих условий. Более дешевые плитки имею-
простой регулятор подачи энергии, и при пользовании ивд
необходимо начинать с низкого уровня, постепенно
увеличивая подачу энергии, пока не будет достигнута необходима!
температура бани. При каждом изменении положения
переключателя энергии системе требуется некоторое время ДЛ1
достижения равновесия, причем в силу инерционности воа
можны перегревы. Контроль температуры в последнем случа<
менее точен, но вполне достаточен для проведения
большинства препаративных реакций. При этом изменения наружно:
температуры и скорости перемешивания, а также сквозняки :
вытяжных шкафах могут вызвать значительные колебани
температуры бани.
"Масло", находящееся в масляных банях, - это обычны]
медицинский жидкий парафин (или же минеральное масло>
который дешев и обеспечивает нагрев до 200°С . Выше это:
температуры он парит, быстро темнеет и может воспламе
ниться. При температурах выше 150°С с масляной баней не
обходимо работать в вытяжном шкафу из-за появления не
приятного запаха "горячего масла" и выделения паров. Сили
коновые масла обеспечивают более широкий диапазон темпе
ратур, например, силиконовая жидкость марки "Dow Согшц
550", применяется в интервале температур от комнатной д
250 ° С, однако они очень дороги и обычно используютс
только при высоких температурах.
В качестве бань обычно служат прозрачные сосуды - крв
сталлизаторы из пирексного стекла такого размера, чтобы
них помещались колбы емкостью до 250 мл. Это довольв
мелкие емкости с плоским дном для хорошей теплопередг
чи от плитки. Колбы большего размера (500 мл и более
Во многих случаях более удобны глицериновые или гликолевые (пол
этиленгликоль, триэтиленгликоль, диэтиленгликоль. этиленглнколь) бани. В
можно использовать дли проведения реакций, протекающих при температур!
до 200° С. - Прим. перев.
ддр-^риис реакции , 33
лучше всего помещать в алюминиевые кастрюли (но не в
эмалированные железные, поскольку железо блокирует
действие магнитных мешалок).
Меры безопасности. Если в масляную баню попадает
вода, то при температуре выше 100°С она становится
опасной из-за вспенивания и разбрызгивания. Такой баней
не следует пользоваться или нужно удалить из нее воду.
МЕТАЛЛИЧЕСКИЕ БАНИ. Для получения более высоких
температур следует пользоваться банями из легкоплавких
сплавов. Наибольшее распространение получил сплав Вуда
(сплав свинца, висмута, олова и кадмия), который плавится
при 70°С и может применяться до 350°С. Его обычно
помещают в эмалированную железную кружку или кастрюлю и
нагревают на горелке Бунзена. Так как сплав Вуда
расширяется при затвердевании, любые стеклянные предметы
(например, термометры) во избежание их разрушения необходимо
вынуть из еще жидкого сплава.
б) Реакции при температурах ниже комнатной
Эти редкции обычно проводят в приборах, аналогичных
изображенному на рис. 2.7, но колбу погружают в
охлаждающую до требуемой температуры баню (см., например, рис.
2.16). Для охлаждения до температуры -20СС в качестве
сосуда для бани подходит кристаллизатор, а для получения
более низких температур требуется неглубокий широкий сосуд
Дыоара. Для того чтобы обеспечить хороший контроль
реакции, особенно при низких температурах, важно регулировать
температуру как реакционной смеси, так и охлаждающей
бани. Обыкновенные ртутные термометры обычно градуированы
до -10, -20°С; при более низких температурах нужен
спиртовой термометр (с диапазоном от -120 до +30°С). Ниже
рассмотрены наиболее часто встречающиеся охлаждающие
агенты.
■') Лед
Кашица из дробленого льда и воды имеет постоянную
температуру 0°С. Более низкие температуры дают хорошо
перемешанные смеси дробленого льда и неорганических солейт
*ак. например, смесь льда и хлорида натрия в соотношении
л 1 обеспечивает температуру -20° С .
" Г~
В аналогичных целях вместо льда можно успешно использовать снег. -
**«•* перев.
Таблица 2.1 Охлаждающие бани из смесей льда и с
Соль
КС1
NH/3
ttaCI
NaBr
МйС12
CaClj
Ced2
бн2о
6HzO
Количество,
г соли/100 г льда
30
25
33
66
85
[23
из
Температура. ° С
я Взято вз более обширного набора данных, приведенных в роботе [31 Пе
Iwiieuai'UHU с разрешении издательства "Jut™ Wiley and Sana lrc".
2) Твердый диоксид углерода (сухой лед)
С помощью сухого льда температуру охлаждающих 6ai
можно понизить до -78°С. Сухой лед обычно лоставля*
большими кусками, от которых молотком или киркой
льда откалывают более мелкие куски, заворачивают из
плотную ткань и дробят деревянным молотком или бруско!
Чтобы ие обморозить руки, с сухим льдом надо работать ■
стро, в плотных резиновых перчатках.
Меры безопасности. Бани с сухим льдом следует нагих
пять и держать в вытяжном шкафу, так как при это
может выделяться .много диоксида углерода и паров pai
творителя, особенно при ее наполнении
СУХОЙ ЛЕД/АЦЕТОН (-78"С). Сухой лед чаще всего
комбинации с ацетоном дает бани с постоянной температур*
-78 "С. Баню приготавливают следующим образом: сначала
сосуд Дыоара помещают ацетон, а затем медленно добавляй
измельченный сухой лед до образования избытка последИ
(остерегайтесь сильного вспенивания в начальный момент)
СУХОЙ ЛЕД/ДРУГИЕ РАСТВОРИТЕЛИ. Бани с отнЛ
тельно постоянной температурой выше -78 е С можно пригот
вить добавлением небольшого избытка твердых кусочков с;
кого льда к органическим растворителям (или смеси раств<
рителей) с температурой замерзания выше -78°С [11. ЭтД
вариант описанных ниже вязких бань. Применяют следуюпш
pjnBPiTFHHe реакции
Объемная доля о-ксипола в смеси
1'ис 2-1! Температура бонн, охлпиущемой смесью о- и л ксилола с
сухим ЛЬДОМ.
раст ригели- четыреххлористый углерод (-23 ° С), гептанон-3
(38°С), циклогексанон (-46°С) и хлороформ (-61 °С). При
использовании смеси о- и л-ксилола получают бани с
промежуточными температурами (рис. 2.П).
3) Жидкий азот
Жидкий азот сам по себе дает охлаждение до -1%°С и
часто применяется в комбинации с органическими
растворителями для приготовлении "вязких бань" с более высокими
температурами.
ВЯЗКИЕ БАНИ Их готовят следующим образом: к соот-
ветствующему растворителю (см. примеры в тзбл. 2.2) [21,
помещенному в сосуд Дьюара, очень медленно при
интенсивном перемешивания добавляют жидкий азот (в вытяжном
ШкаФу> до тех пор, пока значительная часть растворителя не
затвердеет. Пока в смеси будет присутствовать твердый рас-
творитель, температура оставшейся части растворителя буди!
близка к температуре замерзания.
Меры безопасности. Никогда не смешивайте жидкий i
дух или жидкий кислород с органическими растворителями
- это может привести к сильному взрыву.
2.1.5. Реакции в безводной среде и инертной атмосфере
Многие органические реагенты, промежуточные соединен]
и растворители легко взаимодействуют с водой и (или) кислД
родом воздуха или углекислым газом. Это часто требует прс
ведения ряда препаративных синтезов в абсолютно безводны
условиях без доступа воздуха. Ниже рассмотрены основны
методы проведения реакций, протекающих только в том i
чае, если реагенты, растворитель и аппаратура будут сухи!
(например, реакции Гриньяра, синтезы на основе малоновога
эфира и т. д.), а сама реакция будет осуществлена без
доступа воздуха и влаги. (Более сложные методы работы с
такими чувст тельными к воздуху и влаге реагентами,
растворы литийорганических соединений, обсуждаются в |
2 2.1.)
а) Реакции в безводных условиях
I) Методы сушки реагентов и растворителей
В учебном лабораторном практикуме реагенты и раствори]
тели для экономии времени обычно очищают заранее. Eon
же этого не сделано, тогда необходимо ознакомиться со cfl
циальными методами их очистки. Ниже приведены основны
методы сушки жидкостей и твердых веществ.
ЖИДКОСТИ, Методы сушки органических жидкостей Л
агентов и растворителей) достаточно специфичны и
от природы соединения Если не приведены детали, то npd
де, чем приступить к работе, необходимо проконсультировав
ся у преподавателя.
В справочной литературе [3 - 6] можно найти много иД
формации по сушке и очистке широкого круга растворителей
и реагентов.
Метод сушки обычно включает в себя добавление к жил
кости небольшого количества неорганического осушающег
агента, который либо поглощает воду (как, например, мол
кулярные сита или безводные соли типа сульфата мапп
Та6дниа 2.3. Вязкие бани с жидким i
Растворитель Температура, °С
™,-
Растворитель
37
ратура, °С
циклогексаи
циьлогептая
0-Дик.чорбензол
л-дихлорбензол
о-Ксилол
Еронбенмл
Хлорбензол
„-Октан
Этилацетат
8 Переплат*
Chemical Society
б
-12
-18
-25
-29
-30
-45
56
-84
но с разрешения i
(1966)
Гептан
Цихлочентан
Гексан
Толуол
Метилацетат
Циклогексен
Этиловый спирт
н-Пешан
2-Метилбутаи
13 работы |2]. Авторе»
(вязкий)
ое право
-91
-94
-95
-98
-104
-116
-131
-160
American
или кальция), либо необратимо реагирует с водой (как,
например, металлический натрий, гидрид кальция или литий -
алюм нийгидрид) Ясно, что осушающий агент не должен
реагировать с самим соединением. В некоторых случаях смесь
осушающего агента и органической жидкости просто
перемешивают при комнатной температуре и затем, чтобы удалить
осушающий агент, фильтруют через сухую воронку Шота в
приемник (стр. 95). После этого жидкость перегоняют, как
обычно, в сухом приборе. В других случаях органическую
жидкость кипятят с обратным холодильником над осушающим
агентом, а затем отгоняют. Кипячение с обратным
холодильником и последующую отгонку можно проводить по
стандартной методике, но прибор, изображенный на рис. 2.12,
позволяет одноврем нно кипятить и еобирать жидкость без
разборки аппарата. Кран X во время кипячения держат открытым,
а затем закрывают, чтобы собрать сухой отогнанный
растворитель в емкость. Этот прибор особенно полезен, когда
требуется регулярная подача свежелерегяанного растворителя
типа тетрагидрофурана или диметоксиэтана.
Меры безопасности. При работе этого прибора и
перечне любого растворителя совершенно необходимо, чтобы
1 е колбе оставался достаточный запас растворителя, т. е.
, Ч1Т!обы перегонка не велась досуха или почти досуха. При-
I ^°Р НЕ СЛЕДУЕТ оставлять без присмотра во время от-
г°чки. Общие рекомендации по мерам безопасности при пе-
\tetoiiKe растворителя см. в разд. 1.3.2 и 34.1.
Рис. 2.12 Прибор, используемый для сутки и отгонки нсболыш
честв растворителя.
ТВЕРДЫЕ ВЕЩЕСТВА. Методы высушивания твердых J
ществ менее специфичны: для сушки при комнатной темпе!
туре обычно используют эксикатор (рис. 2.13), а для суш
ддл»ие реакции
Орогези
Подсоединение
вакуума
Смазанная шлифованная поверхность
иди '"О'-кольцевой затвор (изолирую
^ щая лента}
Высушиваемо;
вещество
Осушитель
Рис 2 13 Вакуумный эксикатор.
при повышенных температурах - сушильный пистолет (рис.
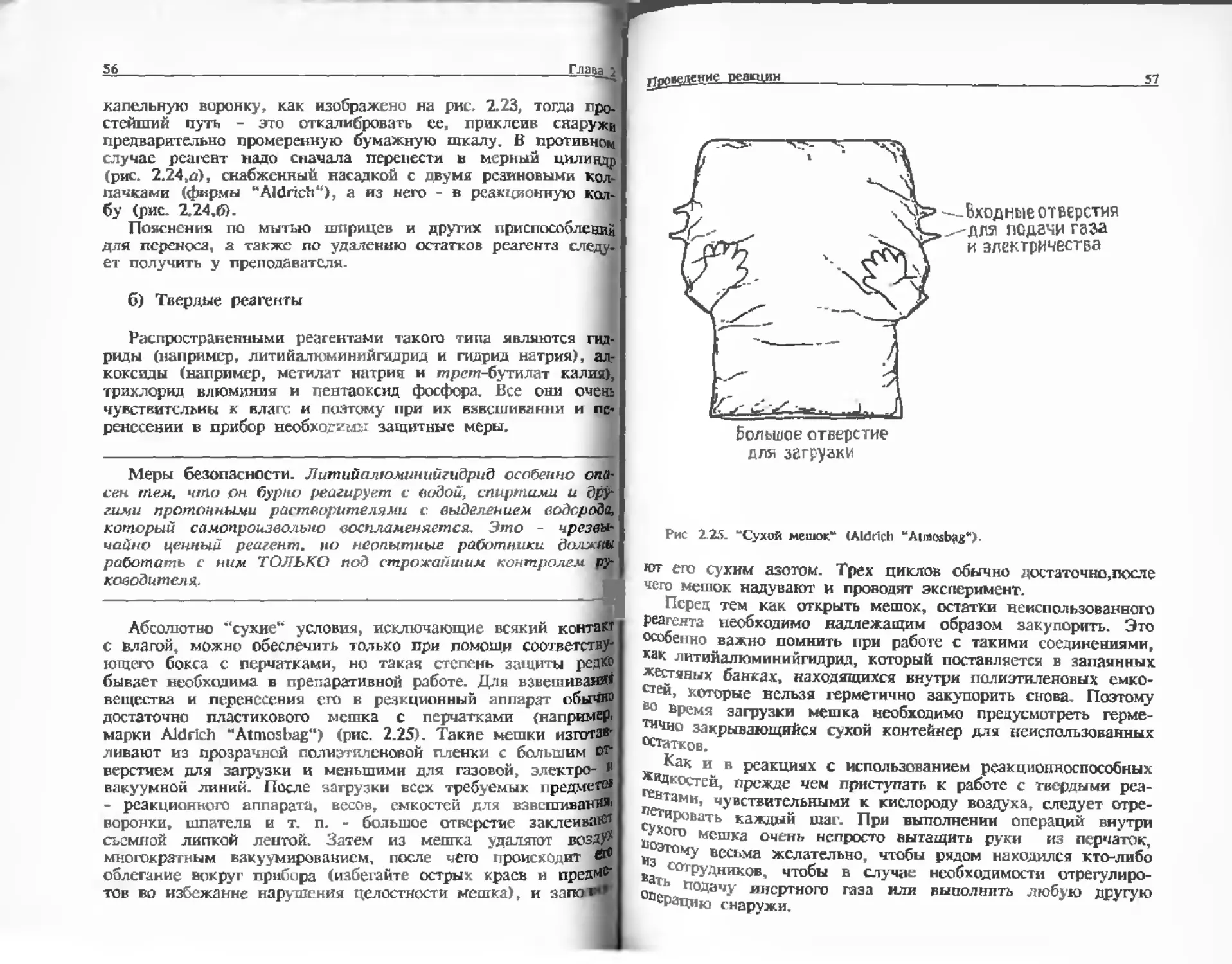
3.14, с. 99), или вакуумную печь. Органические твердые
вещества НИКОГДА не следует сушить в обычной
лабораторной печи.
В эксикаторе осушитель хранят в отделычой чашке под
металлической сеткой . В качестве Осушителя обычно
используют силикагель, который меняет окраску в зависимости от
увлажнения (будучи сухим, он окрашен в голубой цвет;
розовая окраска указывает на необходимость регенерации -
нагревания в печи при 125°С). В некоторых случаях требуется
более эффективный осушитель типа пентаоксида фосфора.
Меры безопасности. Осторожно! Пеитаоксид фосфо
энергично реагирует с eodoii.
При сушке твердое вещество должно быть в виде порошка
"ли в мелкокристаллической форме. Его следует насыпать в
i ш лабораториях используют керамическую сетку. - Прим.
40 Eassaj
стеклянную чашку тонким слоем (при сушке в вакууме при.
крыть часовым стеклом). Сушка происходит быстрее и эф.
фективнее в вакууме; эксикатор можно вакуумировать с
помощью водоструйного насоса до давления 10 - 12 мм рт. ст.
(разд. 3.4.8, а) или с помощью ротационного масленого насоса
до давления 0,1 мм рт. ст. (разд. 3.4.8,6).
Меры безопасности. Иногда при работе вакуумные
эксикаторы взрываются. Поэтому перед включением насоса и
в процессе вакуумирования эксикатор необходимо покрывать
металлической сеткой .
При стравливании вакуума, перед запуском воздуха обрат».
но в эксикатор, закройте конец трубки небольшим кусочком
фильтровальной бумаги и затем осторожно откройте кран.
Фильтровальная бумага сдерживает струю^воздуха, предотвра
щая распыление кристаллов и осушителя .
В некоторых случаях - для нелетучих твердых веществ -
может потребоваться нагревание в вакууме. Этот метод чаще
используется для сушки продуктов реакции, чем реагентов
(гл. 3).
2) Высушивание приборов
Стеклянную посуду легче всего высушить в лабораторной
печи. Нагревания в течение 1 - 2 ч при 120° С вполне до
статочно, чтобы удалить воду с поверхности посуды и обес
печить необходимую для учебных лабораторных целей сте
пень высушивания. Однако, чтобы получить более высокий
выход, или если требуется абсолютно сухая посуда, ее следу
ет прокалить для удаления адсорбированной воды при 125 °£
в течение ночи или при 140°С в течение 4 ч. Предметы,
содержащие притертые части (шприцы или затворы
мешалок), перед прокаливанием необходимо разобрать. Не
забудьте высушить вспомогательную посуду: мерные цилиндры,
капельные пипетки, колбы, химические стаканы и стеклянные
чашки для измерения или взвешивания реагентов.
Пластиковые детали, включая тефлоновые, прокаливать не следует, вс
их надо высушить в эксикаторе.
Металлическую сетку можно заменить, обмотав эксикатор пшюте11'
цем. - Прим. перед.
Лучше заполнять эксикатор сухим воздухом, пропуская его через &У
шитель - Прим. перев.
ggnrgyKHe реакции
41
Трувка.
заполненная
осушителем
Моторчик
для
мешалки
трувка,
заполненная
осушителем
Осушитель
Реакционная
смесь
Рис. 2.14. Прибор для работы в безводных условиях, снабженный
трубками с осушителем.
После просушки прибор следует собрать, пока он горячий
(наденьте перчатки), немного смазав все шлифы, чтобы
предотвратить заклинивание при разборке и обеспечить
герметизацию. Собранному прибору дайте затем остыть, используя
при этом для защиты от атмосферной влаги (см. следующий
Раздел) трубки, заполненные осушителем (например,
хлоридом кальция), на холодильниках, капельных воронках и т. д.
<Рис. 2.14) или, что более предпочтительно, продувая сухой
аз°т (см. подраздел "б" ниже). Небольшие по размеру пред-
Меты, такие, как мерные цилиндры, шприцы и т. д., следует
охлаждать в эксикаторе.
42 Гла!
3) Приборы для проведения реакций без доступа влаги
Обычную препаративную аппаратуру (как, например, I
рис. 2.14) можно легко модифицировать для проведения pes
ций в безводной среде а) установкой хлоркальциеьых труб
(см. ниже) на обратных холодильниках и капельных воя
ках, б) нанесением смазки на шлифы и в) использован!
магнитной мешалки или герметичного затвора на лопастн
мешалке (разд. 2.1.3).
ТРУБКИ ДЛЯ ЗАЩИТЫ ОТ АТМОСФЕРНОЙ ВЛАШ
Трубки для защиты от влаги (рис. 2.14) обычно наполнен
гранулированным силикагелем, изменяющим окраску от roj
бой в суком состоянии до розовой, когда необходима его!
генерация, или гр^. -лированяым хлоридом > зльция, пол
щенным между двумя пробками из стекловаты
Меры безопасности. При работе со стекловатой I
девайте резиновые перчатки или берите пинцет.
Трубки посте заполнения осушителем следует прока!
при 125°С и перед использованием охладить в эксикаторе!
Когда требуется абсолютно сухая среда, лучше вт
применять как обезвоживающие средства, так и инертн.
среду, создаваемую продувкой в систему сухого ииертт
газа (обычно азота), как описано в следующем разделе. 1
6) Реакции в инертной атмосфере
I) Подача инертного газа
Азот - наиболее широко распространенный инертный I
так как он дешев и поставляется с высокой степенью чис1
ты. Не содержащий кислорода азот производства фирн
"British Oxygen" марки "Wi Spot на самом деле содерж
только 0.0002 кисл рода и 0 0003% воды). Газ такой cti
пени чистоты можно r.i подвергать дальнейшей очистке и
сушке. Для дальнейшей осушки или удаления кислорода i
часто пропускают через различные абсорбционные погло!
тельные склянки, но обычно это приводит к противоположи
му эффекту.)
Применяют также аргон или гелий, но эти газы нам!
дороже.
—^пдрнне реакции
Меры безопасности. Не следует пользоваться дрессельны
ми склянками, содержащими концентрированную серную
кислоту, так как они неэффективны и представляют
большую опасность ш-за возможного разбрызгивания кислоты
при неожиданном увеличении давления.
Газовый баллон должен быть оборудован понижающим
давление редуктором <до 1,75 кг/см3}, соединенным с
игольчатым клапаном, контролирующим скорость подачи газа. В
случае отсутствия игольчатого клапана его можно заменить
винтовым зажимом на газовой трубке рядом с редуктором.
Меры безопасности. Диафрагменпые редукторы при их
открывании имеют склонность к "заеданию", что может
привести к резкому скачку давления. Лучше поступать
следующим образом: закрыть игольчатый клапан (или
винтовой зажим), таким образом изолируя редуктор от
реакционного аппарата, установить редуктором требуемое
давление газа (0,21 - 0,35 кг/см*) и затем открыть клапан
(или винтовой зажим), чтобы установить требуемую
скорость подачи газа.
Если баллонов с азотом не хватает или их нельзя
доставить к месту проведения эксперимента, то в некоторых
случаях можно воспользоваться резиновыми камерами (см.,
например, рис. 2.17). Однако азот в них недолго сохраняет
высокую степень чистоты из-за диффузии кислорода и влаги
через стенки вшеры.
2) Приборы для проведения реакций в инертной
атмосфере
Как и при проведении реакций в безводной среде, в этом
случае также необходимо слегка смазать стеклянные шлифы
" установить на лопастные мешалки герметичный затвор.
Д-чя заполнения системы (продувки) азотом (или другим
инертным газом) и поддержания в ней на протяжении всей
реакции слегка повышенного давления требуется лишь немно-
113 усовершенствовать стандартное препаративное оборудова-
КЯе Для этого обычно используют разные методы барботиро-
ва"ия (как, например, на рис. 2.15).
Ба 5отер обеспечивает направленный выход азота и
предка ает попадание воздуха в прибор. Существуют барботе-
Барботер
\_ ^ Выбор
Подача
азота _
Переходник с
завинчивающимся
колпачком
Рис 2 15 Прибор для проведения реакций н инертной (азот) ат юсфе]
ры нескольких типов (рис. 2.15 и 2.16), обычно запо
ных медицинским жидким парафином (менее опасным,
ртуть). Место подачи азота в прибор можно расположит!
нескольких местах. Если оно удалено от барботера Я
2.15), то облегчается предварительная продувка системы
разборки отдельных ее частей. При работе с небольшими
личествами веществ азот удобно вводить через иглу
впрыскивания, которой прокалывают резиновую перегорел
{разд. 2.2.1).
Азот можно вводить и через трубку, непосредственно Щ
диненную с барботером {как, например, изображено на 1
2.16), но при продувании таких систем необходимо уб|
у или резиновую перегородку в капельной воронке, что-
fy обеспечить хороший ток азота череэ прибор. В таких
(Яучаяк удобен комбинированный ввод газа и установка
барботера, как показано па рис. 2.16. Для реакций, проводимых
с обратным холодильником, барботер такого типа следует
установить на верхнем конце холодильника.
Возможно множество вариантов на данную тему в
зависимости от требований эксперимента, что предоставляет
большой простор для изобретательности при конструировании
эффективных систем
Азот можно продуть либо до, либо после загрузки колбы
растворителем и реагентом в зависимости от типа реак-
Пробка или
^^- резиновый
JlT колпачок
/?г\ Капельная воронка
с отводом для
выравнивания
давления
Термометры
Рис 216. Прибор для проведения реакций при i
и»Чтюй атмосфере.
ции. В простейших опытах это указано в руководстве,
исследовательской работе вам придется это продумать. Поел]
продувки ток азота убавляют, чтобы он очень медленно npoj
булькивал через барботер. Однако во время охлаждения
прибора после реакции не забудьте увеличить скорость подащ
газа, чтобы избежать затягивания через барботер воздуха ]
систему, поскольку газы и пары в ней сжимаются при охла-
3) Прибавление реагентов в инертной атмосфере
Здесь предполагается, что сами реагенты не слитком I
акционнеспособны по отношению к кислороду воздуха и в]
re {работа с такими реагентами описана в разд 2.2.1).
ЖИДКОСТИ И РАСТВОРЫ. При работе с умереннв!
или большими количествами веществ необходимый о&Я
жидкости можно быстро отмерить мерным цилиндром и пр
капать через капельную воронку с уравновешенным давле!
см (как, например, изображено на рис. 2.16). При малз
объемах {до 10 мл) намного удобнее пользоваться шприи
и впрыскивать реагент в реакционный сосуд через резиной
перегородку {использование шприцев и перегородок подроЯ
обсуждается в разд. 2.2.1).
ТВЕРДЫЕ ВЕЩЕСТВА. Часто твердые реагенты {наЛ
мер, магний в резкциях Гриньяра) помещают в реакционна
колбу до продувки ее азотом. Постепенное прибавление тш
дых реагентов в процессе реакции в инертной атмосфере э
труднительно. Поэтому, когда это возможно, их следует I
бавлять в виде растворов. Для крупно- и среди емасштаби
процессов эффективны шпековые системы подачи, приводив
в движение электродвигателем, однако они мало пригсИ
вие промышленных лабораторий. Самый распространенный i
бораторный метод представляет собой некий вариант систем
изображенной на рис. 2.17, в которой твердое вещество В
гружают в изогнутую трубку, соединенную с боковым I
клонным горлом реакционной колбы. Вращением трубке
легким постукиванием добиваются постепенного, контролищ
мого присыпания твердого вещества. Кран на трубке не (Г
зателен, но он нужен при заполнении системы азотом. Вг!
не очевидно, что эту систему в том виде, в котором 1
изображена на рис. 2.17, нельзя использовать при проведен
реакции, требующей кипячения с обратным холодильником;
данном случае между колбой и трубкой нужно вставить!
роткий холодильник с широким внутренним каналом.
-«рвение резкцот
Все шлифы
закреплены
Резиновая камера
Запорный краник
Рис 2.17 Прибор дли медленного добавления твердого реагента,
иллюстрирующий использование резиновой камеры дли подачи азота.
2.2. ТЕХНИКА СПЕЦИАЛЬНЫХ РАБОТ
2.2.1. Работа с реагентами, чувствительными
к влаге и кислороду воздуха
В последние годы в связи с бурным развитием и
усовершенствованием синтетических методов очень большое
распространение получили реагентм с высокой реакционной
способностью, чувствительные к влаге и (или) кислороду воздуха.
Поэтому важно, чтобы все химики были обучены методам
безопасного обрищения с такими соединениями В этом
разделе приведены основные правила по перенесению реакционно-
способных жидкостей и твердых веществ. Дополнительную
информацию по технике таких работ можно почерпнуть на
Рядя великолепных статей и технических бюллетеней [7 -
И].
Мы благодарны фирме "Aldrlch Chemical Co. Lid"
материал из работ 17 91
Меры безопасности- Хорошо разработанные правила
ращения с реагентами, чувствительными к влаге и кисЩ
роду воздуха, делают эти работы легко выполнимымиТ
безопасными. Однако экспериментаторы должны всегда Щ
мнить о том, что многие из этих реагентов очень опас
ны, так как они чрезвычайно энергично реагируют с вддш
а некоторые из них являются пирофорными (самопрои:
вольно воспламеняются при контакте с воздухом). Исаи
дователи должны убедиться в том, что они приняли соот
ветсщвующие меры предосторожности (разд. 1.3), илеГ
виду, что некоторые из этих операций могут проводитЩ
при повышенном давлении.
Операции, описанные, в частности, в этом разд ч.
студенты или другие неопытные работники должны прЖ
дить ТОЛЬКО под наблюдением опытного персонала в щ
чение всего эксперимента Хорошей тренировкой перед
Сотой с реакционным материалом является проведение ее.
го эксперимента с использованием безвредной жидкости М
твердого вещество.
а) Жидкие реагенты и ристворы
Неорганические жидкости типа тстрахлорида титана и тег
рахлорида олова, а также металдоорганические соединения |
типа литийорганических, борорганических и реактивов Грияь-
ярз легко приготовить в виде растворов в эфире или
углеводородах. Многие такие реагенты в настоящее время : родают-
ся в виде стандартных растворов в герметичной упакош
(например, в фирменных бутылях "Aldrich Sure/Seal"),
реагенты не должны соприкасаться с воздухом ни при
приготовлении, ни при транспортировке или использовании]
К приемам работы б безводной среде и инертной атмос!
ре (разд. 2.1.5) прибегают только тогда, когда необходим
перенести эти жидкости из одного сосуда в другой, напригЯ
из колбы, в которой реагент был приготовлен, в капельку
воронку другого прибора. Такие же условия необходимо с<
блюдать при переливании продажных реагентов (напримС!
растворов литийорганических реагентов) из упаковоч]
склянки в реакционную колбу или капельную воронку.
Ниже описан легкий метод переливания таких жидкое
прокалыванием резиновой перегородки при помощи иглы. П]
переливании небольших количеств веществ применя1
шприц, для больших объемов - двустороннюю иглу. В обоих
случаях необходимо, чтобы на бутылях для храни ■ (рас-
2.21), реакционных колбах (рис. 2.22) и капельных воронка)
es£^H e реакции
Игла
ifta
Мед|
I провс
- Стеклянная трувка I Медная
или мугрта ишифг | проволока
Рис. 2.18. Использование ьолпачка "Siiba - Seal" (в) н его закрепление
тонкой медной проволокой (б)
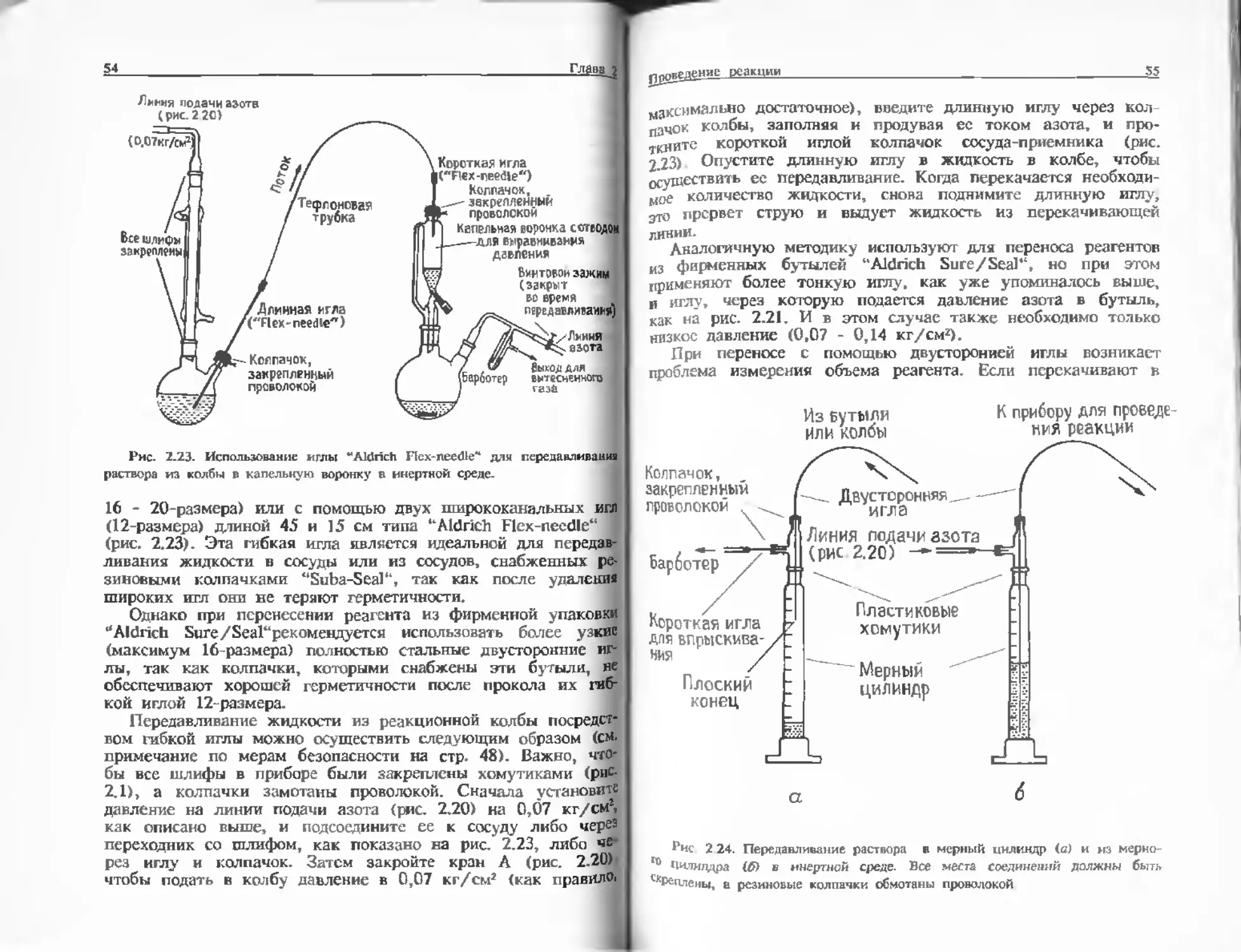
{рис. 2.23) были надеты резиновые колпачки (рис. 2.18). Эти
колпачки легко прокалываются медицинской иглой, а после
удаления иглы снова становятся герметичными. Колпачки
(например, резиновые колпачки фирмы "Suba-Seal") подходят
к большинству размеров муфт и трубок. При использовании
повышенного давления (см. ниже) или при воздействии на
Рис 2 19. Переходник для >
Азот,
Манометр
±
АЗОТ, / .
поступающий Игольчатый
из регулятора кпэпам,регули-
даЕия Р "рующии поток
Резиновый
s колпачок
Пластиковая
...трубка
Барботе
е I
Короткая игл
для впрыски»
Рис. 2.20. Линия падачи язога.
колпачек паров растворителя его следует обмотать прав
кой (рис. 2.16,6), чтобы предотвратить набухание и вы
колпачка из муфты.
При проведении реакций с обратным холодильником I
ше свести к минимуму площадь контакта с парами, иоГ
зуа переходник (рис. 2.19) и маленький (около 6 мм) ко.
чок, чем надевать на муфту большой колпачок. ITponf
тельность жизни колпачка можно увеличить, если ел
смазать его сверху силиконовой смазкой, чтобы об лея
прокалывание иглой.
Для перенесения жидкостей с помощью шприца или I
сторонней иглы нужно через гибкую линию (пластни
трубку), заканчивающуюся короткой медицинской игло^
или 20-размера, подать сухой азот под небольшим давля
(0,7 - 0,21 кг/см2) {разд. 2 1.5).
Необходим также барботер с резиновым колпачком, ч;
продуть шприцы путем заполнения их азотом и опорожне
Прибор, изображенный на рис. 2.20, служит обеим це
Очень важно контролировать давление азота в пределах!
0,35 кг/смг), а для этого необходимы хороший клапан и
нометр.
I) Техника работы со шприцами
Обычно применяют стеклянные шприцы емкостью до
мл. Иглы должны быть длинными (30 - 60 см) и гиб!
Наиболее употребляемые размеры 18 или 20, однако!
вязких жидкостей может понадобиться 16 размер. Работа
шприцем легче и безопаснее, если между ним и иглой и
■ гтить запорный краник (марки *'Luer-Lock*') (рис. 2.21), но
■ Хто не обязательно. Все части высушивают н охлаждают по
■ сдельности (разд. 2.1.5); при их сборке на соединение
Линия подачи азота
(рис. 2.20)
Кольцо, фиксирующее
бутыль б целях
безопасности
Бутыль (Aldrich
—^ Sure/ Seal)
"ic 2.21 Заполнение шприца реагентом из бутыли (Aldrich Sure/Seal)
* Удаление газовых пузырьков к возвращение избыточного реагента б бу-
Luer" (и запорный краник, если им снабжен шприц) hJ
сят немного силиконовой смазки для обеспечения гззоиеп]
ницаемости.
Шприцы используют для отмеривания и быстрого пера
сения небольших количеств реагента (до 50 мл). Их об J
не применяют для медленного прибавления реагента н -Я
ние длительного времени, поскольку вокруг поршня немиьг
мо происходит гидролиз что может привести к заеданию,
ли поршень долго остается в статическом состоянии. Заедай
можно предотвратить, смазав поршень одной или двумя
каплями силиконовой жидкости или минерального масла (бели
такое загрязнение приемлемо). Однако, если требуется ыЖ
ленное прибавление жидкости, то лучше поместить реаген^И
Линия подачи азота
_Jpnc. 2.20)
Рис 2.22. За юляение шприца из реакционной колб;
капельную воронку с отводом для выравнивания давления.
реактив можно перенести из бутыли для хранения,
снабженной резиновым колпачком (например, бутыли фирмы
"Aldnch Sure/Seal"), следующим образом {см. меры
безопасности на стр. 48).
Бутыль необходимо жестко закрепить
яа штативе с помощью кольца (рис. 2.21). Установите на ли-
рай подачи азота {рис. 2.20) начальное давление 0,07 - 0,]4
кг/см2 и при помощи игольчатого клапана отрегулируйте
медленный ток газа через барботер (при открытом кране А).
Затем через резиновый колпачок бутыли введите короткую
иглу В, подождите немного и закройте кран А. Эта
процедура необходима для того, чтобы стравить сначала все
избыточное давление в бутыли, возникшее во время хранения.
После этого подайте в бутыль небольшое давление до 0,07 -
0,14 кг/см2 (0,14 кг/см2 - это максимально необходимое для
этой операции давление - большего давления НЕ подавайте).
Закройте запорный краник ("Luer-lock") на шприце и
введите ипу шприца через колпачок в жидкость (рис. 2.21 ,с).
Откройте запорный краник; тогда повышенное давление
направит жидкость в шприц; при этом вы должны внимательно
наблюдать за движением поршня назад.
Если поршень застрянет, слегка потяните его; не тяните
поршень сильно, так как это приводит к утечкам жидкости и
образованию в шприце пузырьков газа
Наберите немного больше реагента, чем требуется, и
затем надавите на поршень в направлении, указанном на рис.
1.11,6, так чтобы все пузырьки газа оказались у конца иглы
Затем, открыв кран А (рис. 2.20), стравите давление из
бутыли и выпустите весь газ и избыток реактива из шприца
обратно в бутыль.
После того кзк удален избыток реактива, наберите в
шприц немного азота, чтобы отвести реагент от иглы, и
перед извлечением иглы из бутыли закройте запорный краник
на шприце. Быстро перенесите шприц и через резиновый
колпачок введите реагент в реакционный аппарат.
Аналогичным способом можно переносить растворы из
снабженных колпачками реакционных колб. В таких
приборах (рис. 2.22) колпачок следует замотать проволокой, а все
Шлифы скрепить пластиковыми хомутиками (рис. 2.1).
2) Перенесение при помощи двусторонней иглы
Этим методом удобно переносить растворы объемом бо-
^ SO мл. Перекачка осуществляется через длинную
двустороннюю иглу из нержавеющей стали {длиной 60 или 90 см,
Короткая игла
I ("Flex-needle")
Колпачок,
- закрепленный
проволокой
капельная воронка с отер
-для выравнивания
давления
бинтовой зам!
(закрыт
во время
передавливай!
-о >> Линия
- ааота
Рис. 2.23. Использование иглы "Aldrich Flex-needle" для передавливаи
раствора из колбы в капельную воронку в инертной среде-
16 - 20-размера) или с помощью двух ширококанальных и
(12-размера) длиной 45 и 15 см типа "Aldrich Flex-needl^
(рис. 2.23). Эта гибкая игла является идеальной для перед;
ливания жидкости в сосуды или из сосудов, снабженных^
зиновыми колпачками "Suba-Seal", так как после удале
широких игл они не теряют герметичности.
Однако при перенесении реагента из фирменной упаковки
"Aldrich Sure/Seal"pi мендуется использовать более узкие
(максимум 16-размера) полностью стальные двусторонние
иглы, так как колпачки, которыми снабжены эти бутыли,
обеспечивают хорошей герметичности после прокола их
кой иглой 12-размера.
Передавливание жидкости из реакционной колбы посред!
вом гибкой иглы можно осуществить следующим образом (с*^
примечание по мерам безопасности на стр. 48). Важно,
чтобы все шлифы в приборе были закреплены хомутиками (рис
2.1), а колпачки замотаны проволокой. Сначала установи^"
давление на линии подачи азота {рис. 2.20) на 0,07 кг^Г
как описано выше, и подсоедините ее к сосуду либо чер'
переходник со шлифом, как показано на рис. 2.23, либо^
рез иглу и колпачок. Затем закройте кран А (рис. 2.21
чтобы подать в колбу давление в 0,07 кг/см2 (как npaaF
д^дрние реакции
максимально достаточное), введите длинную иглу через
колпачок колбы, заполняя и продувая ее током азота, и
прочите короткой иглой колпачок сосуда-приемника (рис.
2.23) Опустите длинную иглу в жидкость в колбе, чтобы
осуществить ее передавливание. Когда перекачается необходи-
мое количество жидкости, снова поднимите длинную иглу,
зто прервет струю и выдует жидкость из перекачивающей
линии-
Аналогичную методику используют для переноса реагентов
из фирменных бутылей "Aldrich Sure/Seal", но при этом
применяют более тонкую иглу, как уже упоминалось выше,
в иглу, через которую подается давление азота в бутыль,
как на рис. 2.21. И в этом случае также необходимо только
низкое давление (0,07 - 0,14 кг/см2).
При переносе с помощью двусторонней иглы возникает
проблема измерения объема реагента. Если перекачивают в
К прибору для
проведения реакции
Линия подачи азота }\
Рис 2 24. Передавливание раствора в мерный цилиндр (а) и из мерно-
^ Чмлипдра 16) в инертной среде. Все места соединений должны быть
обмотаны проволокой
капельную воронку, как изображено на рис. 2.23, тогда щ _
стейший путь - это откалибровать ее, приклеив снаружи
предварительно промеренную бумажную шкалу. Б противном}
случае реагент надо сначала перенести в м рный цилиндр
(рис. 2.24,о), снабженный насадкой с двумя резиновыми кг
пачками (фирмы "Aldrich"), а из него - в реакционную ■
бу (рис. 2.24,6).
Пояснения по мытью шприцев и других приспособле
для nepeHocai а также по удалению остатков реагента сл|
ет получить у преподавателя.
б) Твердые реагенты
Распространенными реагентами такого типа являются
риды (например, литийалюминийгидрид и гидрид натрия) ,1
кокезды (например, метилат натрия и трсти-бутилат кал:
трихлорид влюмшшя и пентаоксид фосфора. Все они о!
чувствительны к влаге и поэтому при их взвешивании иг
ренссении в прибор необходимы защитные меры.
Меры безопасности. Литийалюминийгидрид особенно о\
сен тем, что он бурно реагирует с водой, спиртами w
гимн протонными растворителями с выделением водоШ
который самопроизвольно воспламеняется. Это - чрМ
чайно ценный реагент, но неопытные работники <?<
работать с ним ТОЛЬКО под строжайшим контролем
ководителя-
Абсолютно "сухие" условия, исключающие всякий контаИ
с влагой, можно обеспечить только при помощи
соответствующего бокса с перчатками, но такая степень защиты редй*
бывает необходима в препаративной работе. Для взвешиванй*
вещества и перенесения его в резщионный пп рз обычно
достаточно пластикового мешка с п рчатками {напримерг
марки Aldrich "Atmosbag") (рис. 2.25). Такие мешки
изготавливают из прозрачной полиэтиленовой пленки с болы им
отверстием для загрузки и меньшими для газовой, э.-ектро- "
вакуумной линий. После загрузки всех требуемых предмеЯ
- реакционного аппарата, весов, емкостей для взвешиванй*
воронки, шпателя и т. п. - большое отверстие заклеивав
съемной липкой лентой. Затем из мешка >даляют воздУ*
многократным вакуумированисм, поелг- его роисходит ^Ш
облегание вокруг при ра и егайт остры краев пред*16'
тов во избежание нарушения целостности мешка), и запо^»
ч М£> --Входные отверстия
-для подачи газа
и электричества
Рис 2.25. "Сухой мешок" (Aldrich "АижкЬзд").
юг его сухим азотом. Трех циклов обычно достаточно.после
чего мешок надувают и проводят эксперимент.
Перед тем как открыть мешок, остатки неиспользованного
рьэ необходимо надлежащим образом закупорить. Это
особе важно помнить при работе с такими соединениями,
как итийалюминийгидрид, который поставляется в запаянных
жестяных банках, находящихся внутри полиэтиленовых
емкостей, которые нельзя герметично закупорить снова Поэтому
80 время загрузки мешка необходимо предусмотреть герме-
ТИЧн крываклцийся сухой контейнер для неиспользованных
Как и в реакциях с использованием реакционноспгхобных
ид TS&j прежде чем приступать к работе с твердыми реа-
петТа увствительными к кислороду воздуха следует отре-
Су н '"""''-■ каждый шаг. При выполнении операций внутри
йоч^ ешка очень непросто вытащить руки из перчаток,
ИзЭг°Му весьма желательно, чтобы рядом находился кто-либо
'""•'Днихов, чтобы в случае необходимости отрегулиро-
Лвеведе-
Меры безопасности. Особое внимание необходимо уделят
индивидуальным средствам защиты на случай, если меш
порвется или будет проколот. Рекомендуется надеван*
двойные перчатки (тонкие хирургические перчатки даю
дополнительную защиту, при этом почти не теряет
ловкость) и устанавливать сухой мешок в вытяжном гит
фу или под вытяжным колпаком.
2,2.2. Реакции в жидком аммиаке
Жидкий аммиак (т. кип. -33°С) - распространенный |
творитель, особенно в реакциях с участием амидов щелочных I
металлов (например, амида натрия NaNH2), где он служит ъ
как растворитель, и как реагент для получения основания.
Меры безопасности. Нет ничего сложного в обращении t
жидким аммиаком в лабораторных условиях, но так как с
чрезвычайно вреден и токсичен, важно, чтобы студенты i
другие неопытные работники работали с ним ТОЛЬКО^
строжайшим контролем квалифицированного персонала,
жидким аммиаком следует работать только в выт51^Ж
шкафу с очень хорошей вентиляцией, причем в таком^^
те, откуда весь персонал можно быстро эвакуировать^
несчастном случае или при отключении вытяжной систем
в шкафу. Следует также надевать индивидуальные средст
защиты (разд. 1.3).
Жидкий аммиак поставляют в лаборатории в баллон!
разных размеров. Для лабораторных целей наиболее удобны
по размеру и форме небольшие баллоны (б кг). Аммиак s
жидком виде отбирают из перевернутого баллона,
закрепленного в специальной подставке, с вентилем внизу (рис. 2.26)
Каким бы ни был размер баллона, его необходимо надежно
укрепить и сделать безопасным. Перед использованием
баллон следует снабдить соответствующим контрольным
клапаном (в дополнение к основному вентилю на баллоне) и гиб"
кой подводящей трубкой, закрепив ее на вентиле проволокой.
а) Реакционные аппараты
Реакции можно проводить в обычных трехгорлых кЛ
(рис. 2.27). Если необходимо провести реакцию при темпс|
туре кипения аммиака (-33°С), то колбу следует homi
в большой полиэтиленовый таз или кастрюлю, установив
Вентиль
Рис 2 26 Баллон
при отборе жидкого аммиака.
Перевернутый баллон
с аммиаком (надежно
закрепленный)
Гибкая пластиковая
трубка
Контрольный клапан
i опрокинутом
на корковое кольцо. Кастрюлю свободно заполняют
изоляционным материалом типа вермикулита . Это обеспечивает
также сбор содержимого колбы, если она разобьется. Для
проведения реакций при более низких температурах колбу следует
поместить в обычную охлаждающую баню (разд. 2.1.4).
Чтобы свести к минимуму потери аммиака, целесообразно
установит ратный холодильник, заполненный сухим льдом
(рис 27). Колбу можно снабдить лопастной или магнитной
меш -: ;й? капельной воронкой с выравнивающей давление
Щ'{' или другими обычными приспособлениями. Для пред-
2твРа нии доступа влаги используют предохранительные
РУок заполненные гранулированной натронной известью
чо не хлоридом кальция), или защитную колонку {рис.
-"). которая имеет меньшее сопротивление потоку газа.
Б) Загрузка реакционного сосуда
бал загРУзке реакционной колбы жидким аммиаком как
л°н, так и колба должны находиться в вытяжном шкафу.
г представляет собой разновидность слюды. - Прим черев.
Закрепление
трубки
проволокой
Стеклянная
труокэ г
Переходник
с завинчивающимся
колпачком
Рис. 2.27 Прибор для проведения реакций ь жидком аммиаке.
Пластиковую трубку для подачи аммиака необходимо
крутить проволокой с обоих концов.
Соберите прибор (рис. 2.27) и поместите толченый ■
лед (разд. 2.14) в углубление охлаждающего пальца обрап
го холодильника. Убедитесь, что дверцы вытяжного шка
опущены и что имеется хорошая тяга. Проверьте, чтобы ю
трольный вентиль на баллоне был закрыт. После этогг
кройте основной вентиль, а затем плавно - контрольный »->•
тиль. Жидкий аммиак s начальный момент будет быстро
улетучиваться до тех пор, пока подающая трубка и колба ч*
охладятся, поэтому все операцни надо проводить осторс
По заполнении колбы закройте контрольный и основной
тили, выждите некоторое время, пока жидкость не улст^
ся из подающей линии, и затем отсоедините ее от код'
измените насадку с завинчивающимся колпачком на пробку
иди капельную воронку в зависимости от потребности.
Пр» добавлении реагентов имейте в виду, что многие
реакции являются экзотермическими и, следовательно, быстрое
приба ление реагентов может вызвать сильное вскипание
аммиака Поэгоьгу разумнее осторожно продолжать процесс,
слепя за тем, чтобы обратный холодильник был заполнен
доверху сухим льдом. Вы обнаружите, что снаружи колба
покрошен слоем льда, затрудняя наблюдение за тем, что
происходят циутри. При необходимости вы можете очистить "окно",
соскоблив лед или смыв его струей ацетона или этанола из
промывной бутыли, а затем удалив растворитель мягкой
бумагой или тряпочкой.
Меры безопасности. Помните, что эти растворители
являются легковоспламеняющимися и летучими, и примите
соответствующие меры предосторожности.
После проведения реакции реакционную смесь обычно
оставляют на ночь или до тех пор, пока сухой лсд и жидкий
аммиак полностью не испарятся. Нежелательно выпускать
через вытяжной шкаф большое количество вредного материала,
поэтому следует подсоединить к выходному отверстию
защитной колонки соответствующее устройство для абсорбции газа
(прок хультируйтесь у вашего преподавателя). Проводите эти
работы в вытяжном шкафу.
2-2.3. Каталитическое гидрирование
Этот процесс включает* в себя взаимодействие органическо-
*о Реагента с газообразным водородом в присутствии
катализатора - металла или оксида металла. Реакцию проводят при
энергичном перемешивании или встряхивании раствора
резита, содержащего суспендированный порошкообразный
катализатор, в атмосфере водорода (рис. 2.28) до тех пор, пока
ке прекратится поглощение водорода. Давление водорода и
^^кратуру варьируют в зависимости от природы восстанав-
"ЯЧемой функциональной группы. Катализаторы и условия
!ВИро': обсуждаются в работах 112 - 14J. Эти условия мож-
Разделить на три категории и проводить реакцию соответ-
bc*iho в аппаратах различных типов: а) гидрирование при
^йсферном давлении и при комнатной темп ратуре; б) гнд-
" Звание при слабом давлении (до 4,2 кг/см1) и в) гидри-
ше при повышенном давлении (свыше 4,2 кг/см2). Ме-
и» гидрирования, упомянутая в п. "а", описана ниже, а
выходит за рамки данной книги.
rjjfiS^
|рние реакции
для подачи газа
Толстостенный Д
гибкий
шланг
Рис 2.28. Прибор я гидрирования при атмосферном давлении.
Меры безопасности. При гидрировании могут возникну*
следующие опасные моменты: I) водород легко воспламеп
ется, а смесь водорода с воздухом взрывав я. 2) в не
торых случаях используется высокое давление; 3) ката,
затор сильно реакционноспособен и может вызвать Щ
произвольное воспламенение паров растворителя как
так а после его использованиям Поэтому студенты аИ
пышные исследователи должны проводить реакции гиЬри%
вания ТОЛЬКО под строжайшим контролем квалифицЩ
ванного персонала.
а) Гмдри.*
t при атмосферном давлении
Прибор (рис. 2.28) состоит из гребенки, от которой отя
дят отводы к 1) баллону с водородом и ртутном барботерУ
2} нескольким прокалиброванным газовым бюреткам разди4'
ных размеров оказана только одна), из которых во
реакция отбирается водород, 3) реакционной колбе, г
диненной гибким шлангам, и 4) вакуумной системе, св«
ной с водоструйным насосом.
jVlepu безопасности. С этим прибором необходимо рабо-
I тать в вытяжном шкафу с хорошей вентиляцией (венти-
I яямор е искрозащитном исполнении), причем совершенно
I очевидно, что поблизости не должно быть никакого пламе-
| нн> нагревательных приборов, печей инфракрасных спектра-
[ метров или других источников воспламенения. Отводи ю
I трубку от ртутного барботера следует поместить вбишзи
1 вентиляционного канала.
I Перед проведением операции из системы первоначально
| удаляют весь воздух путем многократиого вакуумирования
гребенки и реакционного сосуда и заполнении их водородом
I Затем заполняют водородом газовую бюретку подходящего
I размера и интенсивно перемешивают реакционный раствор до
I прекращения поглощения водорода. Подробное описание
методики следует ниже. (Приборы для гидрирования слегка
различаются по конструкции, но из приведенного здесь описания
I можно легко понять принцип работы на каждом из них.)
I) Приготовление реакционной смеси
I Отвесьте нужное количество катализатора, поместите его в
колбу для гидрирования и залейте раствор реагента в инерт-
I иом растворителе (например, в уксусной кислоте, этаноле,
I метаноле, циклогексане или гексане) и опустите в колбу
брусок магнитной мешалки.
Меры безопасности. Катализатор нужно поместить в
I каЩ/ ДО прибавления растворителя; изменение порядка
загрузки может оказаться причиной воспламенения раство-
ХРчтеля.
ел ^ол^у не следует заполнять более чем наполовину. Подсо-
чините колбу к гидрогенизатору, нанеся немного силиконо-
I ми Сма3ки в месте присоединения, и укрепите ее пружинка-
защИли пластиковыми хомутиками (рие. 2.1). Оградите колбу
| Щвтвым экраном. Закройте кран Д.
«„ ' Удаление воздуха из системы и заполнение Zt овой
у"°Реткц
^ ' %и закрытых кранах А и Д откройте краны Б, В и Г
п°Днимите уравнивающий сосуд, чтобы вытеснить весь
газ из газовой бюретки, затем закройте кран Г. Onyci
уравнивающий сосуд в самое низкое положение.
2. Включите водоструйный насос, откройте кран Д и
кройте кран В, чтобы начать вакуумирование; при ;
уровень ртути в манометре изменится, так как из сШ
мы откачивается воздух.
3. Пока происходит вакуумирование, попросите вашего пр(
подавателя помочь установить контролируемый поток во
дорода из баллона и пропустите интенсивную струю этом
газа через ртутный барботер и отверстие выходной труО
ки.
4. Когда уровень ртути в манометре установится, закрой
кран Б и очень осторожно откройте кран А, чтобы
полнить гребенку и реакционный сосуд водородом. Ш
5. Закройте кран А и откройте кран Б, чтобы снова вак
мировать систему.
6. Повторите операции 4 и 5 и затем операцию 4 с
чтобы гребенка и реакционная колба снова запо
водородом; при этом кран Б закрыт, а кран А открыт.
7. Откройте кран Г и заполните водородом газовую бюр
ку. Когда она заполнится, закройте кран А и прекрат
подачу водорода.
8. Откройте кран В и отключите водоструйный насос!
3) Гидрирование
Выровняйте уровни жидкости в газовой бюретке и зам
шите полученное показание. Включите магнитную мешали
чтобы обеспечить интенсивное перемешивание реакциовИ
смеси, и следите за абсорбцией водорода регулируя ее времи
от времени уравнивающим сосудом. Когда поглощение вода-
рода прекратится, выровняйте уровни и запишите
окончательное показание бюретки. Произведите расчет, чтобы убедитьЯ
в том, что поглощенный объем соответствует теоретический
Иногда реакция преждевременно прекращается из-за отравлг
ния катализатора, и тогда необходимо добавить еще катал**
затора.
Меры безопасности. Катализатор СЛЕДУЕТ Ъобав)
в виде суспензии в используемом растворителе, но РЧ
КОЕМ случае в виде сухого порошка (см. выше разд. !/■
4} Разборка прибора и обработка реакционной смеси
1. Закройте кран Г и отключите магнитную мешалку!
■ lugBCS^ реакции оз
Г бы дать катализатору осесть. Затем круговым движением
смойте прилипший к стенкам колбы катализатор, чтобы
0н также оказался на дне, но НЕ снимайте при этом
колбу
2 Включите водоструйный насос, закройте кран В и
откройте кран Б, чтобы удалить газ из гребенки и реакционной
колбы
3, Когда манометр установится, откройте кран В, а затем
осторожно кран Б, чтобы впустить в систему воздух.
4. Снимите реакционную колбу и обработайте смесь, как
описано в п. 5.
Меры безопасности. Неопытным сотрудникам следует
выполнять эту операцию под контролем, так как катали
заторы, подвергшиеся действию водорода, могут
самопроизвольно загореться, если им дать высохнуть.
5. Чтобы удалить катализатор, отфильтруйте при слабом
отсасывании реакционную смесь через воронку Шота, в
которой пористая поверхность покрыта тонким слоем цсли-
та , но ни в косм случае НЕ давайте катализатору
высохнуть. Промойте катализатор небольшой порцией
растворителя и снова не давайте ему высохнуть. Отделите
фильтрат для последующей обработки и сразу
тщательно промойте катализатор водой. Проконсультируйтесь у
препецавателя относительно дальнейшего обезвреживания
катализатора. Тщательно промойте водой реакционную
колбу и магнитную мешалку для удаления следов катали
»■ Проведите операцию, описанную в п. 1 разд. 2, чтобы
подготовить прибор для последующей работы, и
отключите водоструйный насос.
2.2.4. Фотохимия (фотолиз)
а) Прибор для проведения реакций
Препаративные фотохимические реакции обычно проводят
реакторах типа изображенного на рис. 2.29 . В этих при-
Рах ультрафиолетовая (УФ) лампа помешена в снабженный
„Мелит (Celn> - это броуимиллерит AICaFeOs Прим. черев.
"ИКтпры такого типа можно заказать в фирме "Applied i'holopliys'cs
* 2°3 205 Klnesion Road. Leatherhead, KT22 7РВ.
Ввод
охлаждающего
агента
ТОЛЬ
Подача инертного
газа
Рис. 2.29 Типичный фотохимический реактор с погруженным в ието р
зернуаром и УФ-ламшй.
охлаждающей рубашкой центральный резервуар, который i
гружен в облучаемый раствор. Реакционный сосуд
иметь различные размеры, но в любом случае лампа долж
быть погружена полностью, чтобы обеспечить максимально
поглощение излучения. Погружаемый резервуар может быть
сделан из кварца (очень дорогой материал), пропускают^1*1
излучение приблизительно до 200 нм, или из более дешевоГ" I
дпрдрдрние реакции 67
рексного или другого боросиликатного стекла, прозрачного
^льк 300 нм Реакционные сосуды изготавливают из пи-
оекса Ри использовании популярных ртутных ламп среднего
igjyi is (см. ниже), работающих при высокой температуре,
необходима охлаждающая рубашка для поддержания
комнатной ратуры реакционного раствора (или для проведения
пеак при любых необходимых температурах путем погру-
женм епосредственно самого реакционного сосуда в
термостат и пропускания через рубашку воды той же самой
температуры) Во время облучения раствор обычно
перемешивают магнитной мешалкой или пропуская через раствор ток
азота или другого инертного газа.
Меры безопасности. УФ-излучение весьма вредно для глаз
и кожи. Поэтому во время работы реакторы должны быть
полностью экранированы. Если необходимо работать на
приборе при светящейся лампе, то даже на короткое
время необходимо надевать специальные темные очки
(поглощающие излучение ниже 400 нм), а также защищать кожу
(Яеждой, перчатками и защитным кремом от загара.
б) Источники УФ-излучения
В качестве источников УФ-излучения чаще всего
используют ртутные лампы (подробные данные по другим типам
я^мп даны в монографии [I5J>.
I) Ртутные лампы низкого давления
Это лампы низкой мощности (5 - 20 Вт), дающие
примерно 90% излучения в диапазоне 254 нм (другое основное
^злученае в области 185 нм через кварц не передается).
Даль :i.L подробности приведены в книге Калверта и Пит-
*® [161. При работе эти лампы охлаждаются и при
погружении r реактор требуют минимальной подачи охлаждающего
агента в рубашку (рис. 2.29).
2) Pt
тутные лампы среднего давления
Тп к Я Этих значительно более мощных (100 - 400 Вт) ламп
V о гея около 15 мин для достижения высокой рабочей
^«чц ,чтуры и полной мощности. Они выделяют много тепла,
г "0ЭТ0МУ их обычно используют в реакторах, в которых по-
(пи 1ЫИ резервуар с лампой имеет рубашку охлаждения
2-29), чтобы избежать нагревания реакционного раство-
Таблица 2.3. Ч> ьтрующие растворы*
Отфильтрованные длины
Ниже 250
Ниже 305
Ниже 330
Ниже 355
Ниже 400
Ниже 460
Выше 360
Выпи 450
Na2W0,
SnClj в водн. НО (0.1 М раствор в 2:3
НС1/Нг0)
г м мвлуо4
В1СЦ в НС)
КН-фталат ♦ KN02 (в гликоле при I
рНП)
KjCrO^ <0 I М раствор в N1I„0H/NH^
при рШО)
I M NiSO, +■ I M CuSO,, tu 5%-Д
H^S04)
CoSO, + CuSO„
'
" Для получения более детальной информации по этим и другим
фильтрующим растворам см книгу: Zimmerman И Е. Mol. Pholochem., v 3, р. 2SI
U«7I)
pa. Такие лампы испускают УФ-излучение в более широко^
диапазоне с очень сильной полосой при 365 - 366 им, бол£{'
слабыми полосами при 334, 313, 303, 297, 265 и 254 нмЖ
также значительное излучение в видимой области при 4^Г
408, 436, 546 и 577 - 579 нм (более подробно см. ссылку
П6]>.
Этот широкий диапазон излучения делает лампы средне*
давления высокоэффективными при проведении
фотохимических реакций с целым рядом органических соединений,
ющнх различные спектры УФ-поглошения.
Если требуется облучение при определенной длине »w..-^
тогда излучение с другими длинами волн может быть ^Ж
фильтровано посредством различных стекол [17] или ра^Н
ров неорганических солей, сведения о которых прсдстаьленИ
в табл. 2 3 и ссылке, приведенной в той же таблице.
1ХИМИЧС-
1Й, 4*1
ВОЛЯб'Г
в) Условия реакций и применяемые растворители
,
Фотохимические реакции обычно проводят в весьма
бавленных растворах (0,01 - 0,05 М) при интенсивном ■
мешивании. Сильное разбавление способствует хорошему "|
никновению излучения в реакционный раствор и сводиТ
1
^еаеаегиедемщии и
минимуму осадок полимеров и смол на стенках погруженного
-пдгйтного резервуара лампы, которые могут загородить свет
я остановить реакцию.
В качестве растворителя можно использовать различные по
полярности вещества от углеводородов до спиртов [171. Ясно,
что растворитель не должен заметно пог щать излучение на
щстоте возбуждения реагента 118] и подвергаться
фоторазложению В некоторых случаях весьма важно, чтобы раствори
тель был инертен к интермедиатам, генерируемым при
фотолизе. Все растворители необходимо тщательно очистить от
любых пероксидов и (или) примесей, поглощающих в УФ-об-
ласти (см. сылки [3 - 6] к гл. 2 и [4] к гл. 3), и затем
проконтролировать с помощью УФ-спектра. Весьма нелишне
записывать УФ-спектры всех новых партий растворителя,
чтобы проверить Уф-поглощение примесей.
Растворенный кислород может оказывать вредное влияние
ва многие фотохимические процессы, и поэтому перед облу
чением его следует удалить из реакционного раствора
непосредственно в реакционном сосуде. Проще всего это сделать,
барботируя через него ток очищенного от кислорода азота,
гелия или аргона в течение примерно 30 мин. Ясно, что
реакцию затем надо проводить в инертной атмосфере (разд.
2.1.5).
12.5. Флаш-вакуумный пиролиз
Реакции пиролиза - это такие реакции, в которых
вещество при нагревании разлагается. Однако при пиролизе в
раство вследствие бимолекулярных реакций субстрата (или
°бра ся реакцнонноспособных интермедиатов) с самим
собой . (или) растворителем часто образуется целый набор
ПР°ДУ ов, полимеров и смол. Этого можно избежать при
флат-вакуумном пиролизе (ФВП) путем разложения молекул
субстрата в паровой фазе при высоком вакууме и очень
короткой выдержке при высокой температуре. В данных услови-
"х вещества в газовой фазе хорошо отделены друг от друга,
4x0 сводит к минимум бимолекулярные процессы; кроме то-
. отсутствие растворителя исключает реакцию с горячими
»№пк или О0"раз ташлмися при термолизе резкционнос-
°е°бйыми интермеднатами.
т fi рис 2.30 изображен аппарат для ФВП. Пиролизную
2 г КУ из кварца длиной примерно 30 см и диаметром
в сч нагревают в печи с точным температурным контрол м
•■s гРеделах 200 - 1000°С (например, в трубчатой печи
(jJ^ton-Redcroft LM" 8Ю0>. Вещество, испаряясь на входе
KiTert Е всего входную трубку нагревать в печи "ВисЫ
^ Гопг">, попадает в пиролизную трубку, продукты со-
Ловушки 1 азота Ловушна 2
(див уламива- \Вануумныи\
■■-продукта) \мннометр \ ц доеной |
жидкий азот
Рис. 2.30 Прибор для флат-вакуумного пиролиза
бирают в ловушки, охлаждаемые жидким азотом. Во время
пиролиза систему вакуумируют до 1СИ мм рт. ст. или выдк
Большинство экспериментов ФВП проводят при давл^
Ю-1 - 10"2 мм рт. ст., легко получаемого с помощью рогат
онного масляного насоса. Однако иногда возникает необхо;
мость в более низком давлении, и тогда используют масл:
ный диффузионный насос, но такие случаи встречаютс!^^
дко.
Эксперименты по пиролизу легко выполнимы. Перед на<
лом самого эксперимента очистите пиролизную трубку, наг]
вая ее на воздуке примерно до 800°С, отсоединив вход]
трубку и ловушки. После ее охлаждения поместите peai
во входную трубку и соберите ловушки, как показано
рис. 2.30 Заполните ловушку 2 жидким азотом, вклго»
насос и вакуумируйте систему до необходимого давления I
ниже). Включите печь и дождитесь, пока установится треб;
мая температура (см. ниже). Летучие загрязнения, содШ
щиеся в системе или субстрате, конденсируются в локушн
Затем залейте жидкий азот в ловушку 1, предназначу
для сбора продукта, и установите иа входной печи ■nta
температуру, при которой субстрат будет испаряться в пир'
лиэную трубку со скоростью примерно 0,5 - 1,0 г/ч. КсгдЗ
все вещество пройдет через трубку, отключите печь и да***
ей остыть, включите вакуумный насос, впустите в системУ
сухой азот и доведите температуру ловушек до комнатной
Продукт можно затем извлечь из ловушки растворителе^
(например, дихлорметаном) и обработать обычным способом.
Меры безопасности. Соблюдайте обычные меры пМ
сторожности при работе с вакуумом (разд. 13 3). Лре'<
S
Иачать пиролиз, тщательно проанализируйте химизм
и продумайте, что вы будете делать с токсичны-
летучими материалами (например, HCN, гаяогеноводора-
'япми, SOv NH3), образующимися при пиролизе и конденси-
„ющимися в охлаждаемой ловушке вместе с требуемым
клиническим продуктом. Возможно, что будет достаточно
подсоединять ловушки по мере их нагревания к
соответствующей химической поглотительной склянке. Другой путь
удаления летучих веществ - их постепенное испарение, т.
е в то время, когда система все еще находится под
вакуумом, ловушку 2 оставляют погруженной в жидкий азот, а
ловушке I дают постепенно нагреться, что приводит к
отгонке летучих ееществ (см. разд. 2.1.4 об охлаждающих
банях)
В настоящее время имеется много обзоров по ФВП и его
использованию в препаративной работе [19 - 211, которые
дают информацию об области применения метода и
температурах и давлении, необходимых при пиролизе соединений
различных классов. При использовании очень высоких
температур в печи (400 - 1000° С) может на первый взгляд
создаться впечатление, что это - "очень грубый метод"
разрушающего воздействия на молекулу, однако это не так
Сочетание высокой температуры с очень коротким временем
контакта (обычно 1 - 20 мс> придает ФВП неожиданную
тонкость и избирательность при разрыве связи. Даже
незначительные изменения температуры (на 50 е С) могут измените
направление реакции, поэтому большинство реакций ФВП
протекает очень селективно с высоким или количественным
выходом.
Приведенное выше описание касается только основы
препаративного использования ФВП. В более сложных случаях
^от метод пригоден для получения крайне нестабильных и
коротко живущих веществ, а также для работы по изучению
"еханизма реакции, когда продукты пиролиза улавливаются
пРи низкой температуре и непосредственно исследуются
методами инфракрасной спектроскопии и другими спектральными
методами.
ВЫД£Щ
продуктов реакции
3. ВЫДЕЛЕНИЕ И ОЧИСТКА ПРОДУКТ01
РЕАКЦИИ
3.1. ОСНОВНЫЕ ОПЕРАЦИИ НА
ЗАВЕРШАЮЩЕЙ СТАДИИ ЭКСПЕРИМЕНТ!
3.1.1. Общие положения
Препаративные органические реакции редко приводят В
полному превращению исходных соединений в конечный прс-
дукт. В хорошей в синтетическом отношении реакции можно
ожидать 70 - 80%-ныЙ выход продукта и более, но в то же
самое время в реакционной смеси 64 присут гвовать в
небольших количествах другие органические вещества,
представляющие собой побочные продукты В некоторых случат;
в ходе реакций образуется полимерный маг риал,
окрашенный в желтый или коричневый цвет Помимо органически*
веществ, многие реакции приводят к образованию эквимоляр-
них количеств неорганических продуктов, например галогени-
дов металлов, выделяющихся в типовом синтезе эфира:
.
HCIljBr + NaOCfl, -* RCH2OCH2 + NuBr
Таким образом, смесь, полученная в результате резкц]
протекающей даже с хорошим выходом, может быть довольно
сложной. Она состоит из растворителя, используемого в
реакции, основного продукта, побочных продуктов, непрореагиро-
вавших исходных веществ, возможно также из полиме[^И
материала и неорганического продукта. Очевидно, что ifl
этом целью эксперимента является выделение основного ПИ
дукта с максимально высоким выходом и как можно в бал#
чистом состоянии Имеются реакции (к сожалению, их очей*
мало), в которых основной продукт выпадает в осадок в вйР
кристаллов при охлаждении реакционной смеси до комнатное
температуры Однако как правило, для выделения продув
требуются более сложные операции, включающие последов^
тельное применение различных методов "-"':;- процесс обычй"
называют "обработкой'- pea ционно см< и Потребность *
тех или иных методах определяется природой смеси а так*6
химическими и физическими свойствами выделяемых соедия^
ний. В необратимых реакциях с одним основным продукт^
последовательность операций обычно такова, что сначала У^3' I
лают растворитель и неорганические продукты, а затем отйу
лют основной продукт от других органических веществ,
присутствующих в смеси, посредством кристаллизации (для твер-
пых веществ) или перегонкой (для жидкостей). В более
сложи реакциях, где возможно образование нескольких
основ ых" продуктов, может возникнуть необходимость раз-
пелен их посредством одного из методов хроматографирова-
"ИЯ *?■ 4К « е
В большинстве учебных препаративных методик химич
ского интеэа и в книгах по синтетической химии процесс
обра .. ■ смеси обычно разобран в деталях, и даже в описа-
вяи экспериментальной части какой-либо новой работы в хи-
мичес х журналах обычно находишь подробности обработки
реакционной смеси и выделения продуктов. Поэтому студенту
редко приходится самом, решать как выделить продукт, но
на всех этапах работы важно быть уверенным в том, что вы
понимаете, почему выполняется именно эта операция и к
чему на приведет. Такое понимание подготовит вас к
самостоятельной иссл'. ов:-тельской работе, .-...да для к ждой
реакции придется отрабатывать наилучший способ выделения и
очистки продуктов. В этом вам поможет широкий набор
практически навыков, описанный в последующи- разделах
и, они -сь на свой опыт, вы сможете решить, какие из них
окажутся самыми эффективными для конкретной реакционной
смеси
3.1.2. Удаление растворителя с помощью роторного
испарителя
На какой-то стадии в процессе выделения продуктов
реакции возникает необходимость в удалении растворителя из
реакционной массы или растворителя, использованного в
процессе экстракции (разд. 3.1.3). В методике может быть про-
с*0 сказано, что растворитель "выпаривают". Это НЕ
означает, что из испарительной емкости его следует выпарить в ат-
Мосфе Растворители удаляют либо отгонкой (разд. 3.4),
либо
вляющем большинстве случаев на роторном
испарителе (рис. 3.1).
Роторный испаритель - это по существу устройство для
ыстро удаления растворителей отгонкой их в вакууме. При
10 тьзовании соблюдают следующую последовательность
Помещают раствор, подлежащий выпариванию, в
tiriyto колбу соответствующего размера (заполненную
^■"^е чем наполовину). Подсоединяют ее к каналу, по
> екР0М^ °УДУТ удзляться пары растворителя, используя при
вым 1лимости переходник, и скрепляют это место пластико-
>омутиком или резинкой, чтобы колба не сорвалась в
водяную баню. В том случае, если вакуум создается с Ш
мощью водоструйного насоса, нет необходимости смазывай
шлифы Убедитесь, что приемная колба пуста и тоже
закреплена. На Этой стадии водяная баня должна быть холод но&
Опускают прибор таким образом, чтобы колба на одну
треть погрузилась в воду, подключают воду к холодильнику
и затем включают водоструйный насос (разд. 3 4.8.) По*3
набирается вакуум включают моторчик, чтобы колба в №
торой проводят выпаривание, вращалась. Когда вакуум стаб!"
лизируется, медленно нагревают водяную баню до тех o°ff
пока не начнет отгоняться растворитель. Во избежание в»*"
нивания и выброса раствора не перегревайте баню.
> соблюдать перечисленную выше
последовательность. НИКОГДА не погружайт колбу испарителя в
ГОРЯЧУЮ водяную баню, поскольку это, как правила,
приводит к бурному кипению, вспениванию и выбросу раствора в
приемник.
50
с вило, проще бывае ню Прим-
д.-
» * I
I Че.
Подключение испарителя к водоструйному насосу создает
fVM. позволяющий легко удалить "летучие" растворители
^мдературой кипения до 100°С при температуре бани до
- 60°С (При этом необходим хороший вакуум. Если вы
можете достичь вакуума 20 - 30 мм рт. ст., проконсуль-
уитесь с вашим преподавателем, как это лучше сделать,
людайте технику безопасности при работе с вакуумом.)
™£ч Удаления растворителей с более высокой температурой
яения предпочтительнее использовать высоковакуумную мо-
испаритсля (рис. 3.2).
а) Высоковакуумный роторный испаритель
В этой разновидности испарителя водяной холодильник за.
менен ловушкой, охлаждаемой жидким азотом или смесь»
сухого льда с ацетоном {разд. Z1.4). Вакуум создается с во.
мощью ротационного масляного насоса (разд. 3.4.8). Этот ме-
■год аналогичен описанному выше; следует только удостове-
риться что жидкость, которую необходимо выпарить, не
содержит никаких "летучих*' растворителей. В противном
случае она может сильно вспениться при подаче вакуума даяц
при комнатной температуре. Поэтому безопаснее сначала ра-
ботать на обычном роторном испарителе, нагревая баню
примерно до 60°С, чтобы удалить летучие вещества, а затем,
перед тем как использовать высоковакуумный испаритель,
охладить жидкость снова до комнатной температуры.
3.1.3. Экстракция
а) Использование делительной воронки
Большинство простых экстракций выполняют в делите-И
ной воронке (рис. 3.3). Сама процедура включает в себя
смешение органического раствора (в растворителе, не
смешивающемся с водой) с водой (водной кислотой или щелочью!,
встряхивание воронки, чтобы полностью перемешать ^~
слоя, отстаивание смеси, пока оба слоя не разделятся снова,
и затем отделение нижнего слоя в другой сосуд. Смешение
позволяет веществу перейти из одного слоя в другой в !
симости от его относительной растворимости в каждом ИЗ
них.
Существуют два основных вида экстракции: 1) экстракод
водой для удаления из смеси водорастворимого вещества
(обычно неорганического) и 2) экстракция водным раствором
кислоты или щелочи, чтобы удалить из органического ело*
соответственно органические основания [реакция (3.1) ] вЛ"
органические кислоты [реакция (3 2) J:
RNH2 + HC1 (водн.) -* rSHjCI"
(водонерастворимое) (водорастворимое)
RC02H + ИаОШводн.) -* RC02"\a+
(ограничено (очень хорошо
растворимое в воде) растворимое в воде)
ПРОЦЕСС ЭКСТРАКЦИИ. Выберите делительную воро!
соответствующего размера, которую можно было бы зап#
нять не более чем на две трети объема. Проверьте кра!
iililL-" пчистка "РОДУ"10" Реакции
Поддерживайте!
в этом месте
Поддерживайте Т
в этом месте i
6
[ воронка; б положение для стравливания i
и убедитесь, что он свободно вращается (нанесите немного
смазки на наружные края стеклянных кранов, краны из
тефлона не смазывают). Закройте кран и залейте органический
раствор (через воронку). Добавьте воды (или водной кислоты
или основания) и заткните пробкой.
Меры безопасности. При работе с кислотами или осно-
°иниямц нелишне надавать резиновые перчатки.
Придерживая пробку одной рукой и закрытый кран - дру-
ГОи1 переверните вороту и плавными рутовыми движениями
вращайте се в течение нескольких секунд, чтобы образовался
Еодоворот (но НЕ трясите). При перевернутой воронке
откройте кран, чтобы стравить давление (рис. 3 3,6) (давление
^ожет значительно возрасти при использовании летучих
растлителей, например эфира). Закройте кран, повращайте бо
с& энергично и стравите давление снова Проделайте это не-
колько раз до прекращения увеличения давления. После
^ого делительную воронку можно потрясти, чтобы переме-
грян.
5
шать ее содержимое, но ве трясите слитком энергично ц^
долго, поскольку может образоваться эмульсия, которую бу-
дет трудно разрушить.
Меры безопасности. Не трясите воронку вблизи от саое-
го лица.
Затем зажмите воронку лапкой или вставьте в кольцо н
дайте состоять, чтобы произошло разделение фаз и между
ними установилась резкая граница. Для этого может
потребоваться много времени, если взбалтывание было слишком
сильным. Иногда коагуляцию капель можно ускорить
плавным вращением. Образовавшаяся эмульсия иногда
разрушается при добавлении нескольких капель спирта типа i нтанола
или октанола или небольшого количества нейтрального
электролита^ например хлорида натрия (который растворится в
водном слое). Однако здесь необходимо терпение, иначе
разделение двух слоев будет затруднено и продукт будет потер!
После разделения слоев пробку удаляют и нижний сло^™
вают в другой сосуд. Нижний слой может быть органичи
или водным в зависимости от плотности органического
творителя (если вы не уверены, наберите в пипетку
несколько капель жидкости и выясните, смешивается ли она с
водой). Процесс экстракции обычно повторяют несколько раз,
объединяя порции экстракта для дальнейшей обработки.
Важно во всех случаях хранить оба слоя до завершения работы.
ВЫСАЛИВАНИЕ. В некоторых случаях экстракция води
усложняется тем, что требуемое органическое соединение
слегка растворяется в воде, и поэтому некоторая его часть
остается в обоих слоях, тогда как в идеальном случае оио
должно полностью перейти в органический слой. В таких
случаях растворимость органического соединения в воде
заметно снижается в присутствии растворенных солей, обычно
хлорида натрия для нейтральных или основных веществ или
сульфата аммония - для кислых веществ. Этот эффект мож*
но использовать в различных вариантах экстракции, и в
экспериментальных методиках обычно указывается, требуется J*
высаливание.
б) Непрерывная экстракция
Экстракция с использованием делительной воронки ДЭ*Т
удовлетворительные результаты только тогда, когда выдеЛ^И
мое соединение имеет такой высокий коэффициент распреД6"
■дри-ииа и очистка продумтов реакции
^олбонагреватеяь
рис 3.4. Непреры
01 растворите
зритель Е
-: ^створитель t
-5^Г Коябонагреватель |r_f
£ния, что оно переходит почти полностью в тот или ниой
слой. Однако некоторые органические соединения (к счастью,
°гнссительно небольшое количество) типа карбоновых или
Ч'льфокислот легк0 растворяются как в органических
растворителя так и в воде. Такие вещества можно экстрагировать
^ного раствора только непрерывной экстракцией, напри
Ч> в аппарате, показанном на рис. 3.4,с. Он применяется в
Учае растворителей, которые тяжелее воды (например.
Экстрагируемое
твердое
вещество
Растворитель
Колбонагреватель
Рис. 3.5. Экстрактор Сокслста >
ществ.
i непрерывной экстракции твердым
дихлорметана). Растворитель непрерывно отгоняется в холО"
дильник, где он конденсируется и затем проходит через
водный раствор в виде мелких капелек собирается на дне *
возвращается в колбу. При прохождении через водный СД°В
растворитель экстрагирует немного извлекаемого соединен**
которое затем по мере экстракции и. капливается в кол"
Несколько видоизмененный способ применяется в том слл^Н
когда растворители легче воды (рис. 3.4,6).
,рннс очистка продуктов реакции 81
' v экстракция твердых веществ
I Иногда необходимо извлечь твердый органический продукт
I тверД0И смеси, содержащей другие вещества (обычно неор-
нЯЧ • и|'■'* котоРЬ1е ме растворяются нн в органических
растворителях, ни в воде. Это легче сделать, если смесь сначала
'змель в ступк Если органическое твердое вещество
пегко растворяется в органическом растворителе, то его легко
-«делит*' например поместив смесь в воронку Шота (рис-
311). затем добавляют растворитель, перемешивают и отса-
I емваюг раствор. Однако, если органическое твердое вещество
труднорзстворимо, используют метод непрерывкой экстракции
в экстракторе Сокслета (рис. 3.5). Смесь помещают в
пористую гильзу. Растворитель перегоняют в экстракционную
камеру; когда она заполнится, растворитель через сифон
поступает обратно в колбу. В результате многократного
повторенная этого цикла твердое вещество переносится в колбу.
3.1.4. Высушивание органических растворов
I По окончании водной экстракции органический слой содер-
I жнт некоторое количество воды как в виде небольших
капелек, так и в растворенном виде [многие растворители
способны растворять значительные количества воды (табл. 3.2) ].
Воду можно удалить добавлением небольшого количества по-
I рошкообраэного неорганического осушителя, который затем
| удаляют фильтрованием. Для этой цели использовали многие
осушители, но недавно было показано [1], что для быстрой
(15 - 30 мин) сушки сильно увлажненных эфирных
растворов особенно эффективны безводный хлорид кальция,
безводный су 1ьфат магния и порошкообразные молекулярные ента.
I Однако хлорид кальция реагирует с соединениями многих
классов, и поэтому его нельзя применять для сушки
органически" г, :пиртов фенолов минов, амидов, альдегидов,
кегонов эфиров Наибол эффе ти ропгкообразны
молекулярные сита (тип 4А) (см. ниже) можно использовать
I Для сушки соединений почти всех классов, но они имеют вы-
I ^Уо стоимость. Поэтому в качестве осушителя общецелево-
иазпачения предпочтение отдают безводному сульфату
(ешев, эффективен и нейтрален по отррщению к
функциовачьных групп.
Процедура сушки заключается в следующем, органический
помещают в коническую колбу и добавляют
примерив 1 г с льфата магния (или другого осушителя) на 10 мл
„. ™ора Смесь оставляют на 15 - 30 мин, время от време-
помешивая или встряхивая се, а затем фильтруют под
магния
бальи
Раство
82
вакуумом (разд- 3.2.3) через сухую воронку Шота. Так
вслед за сушкой обычно следует выпаривание на рот^Н
вание непосредственно в круглодонную колбу подходяЛ
размера (см. рис. 3.11,в).
Если необходима более тщательная сушка, то лучше все*
провести первичную сушку, как описано выше, и затемЯ
сушить раствор при помощи безводного сульфата кальца,
(драйерита) или молекулярных сит (типа 4А или 5А). Моле,
кулярные сита действуют быстро в порошкообразной фор«.
(например, содержание воды в насыщенном водой эфире
15 мин снижается до 0,092 мг на грамм), тогда как
гранулированная форма намного менее эффективна (чтобы сниз|
содержание воды в эфире до 0,29 мг на грамм, треЯ
6 ч).
3.1.5. Выделение целевого продукта (или продуктов) 1
После первичной обработки, в результате которой уда
ются все неорганические вещества и органические раствор^И
ли, остается сырой органический продукт (или продукты).
а) Реакции, приводящие к одному основному продукту i
Если реакция простая, протекающая в одном направленна,
тогда наверняка целевое соединение можно выделить
кристаллизацией или перегонкой, как будет указано в методике
эксперимента Однако иногда возникают проблемы с выделением
продуктов, затвердевающих при окончательном удалении
растворителя без кристаллизации. В таких довольно часто встрг-
чающихся случаях выделяется вязкое масло которое не к^*'
сталлизуется даже после тщательного удаления растворитйМ
на роторном испарителе. Если оставить масло на ночь nj"
комнатной температуре, то это часто приводит к
кристаллизации, но если кристаллизации не произошло, то существуй
несколько приемов, с помощью которых можно ее вызвав
Обычно зги приемы лучше опробовать па небольшой порй*й
продукта (в маленькой пробирке) и в случае успеха испол*-
зовать полученное твердое вещество в качестве "затраы*"
для основной массы материала. Наиболее распространена!''
прием - потереть стеклянной палочкой о внутреннюю стей*
сосуда у поверхности масла. Это должно привести к образе
ванию центров кристаллизации, которые могут стимулировЭ^
рост кристаллов. Если этот прием при комнатной темпера*?'
ре не эффективен, то иногда едует i хладить образец ^
-Я0°С или даже в жидком азоте и затем дать ему мсдл^И
ipgas м очистка продуктов реакция
^петься. Для сиропообразных смесей особенно эффективным
„J&«0 '• является растирание или "втирание" масла с петро-
ейя эфиром или гексаном. Масло покрывают небольшим
11 дч твом растворителя и растирают стеклянной палочкой
raw сосуда. В благоприятных случаях растворитель раство-
- ,, ^nw^oii vrvmnup unmiT uPmsiTk кппгтя лпнзянки
рвет
следы примесей, которые могут мешать кристаллизации,
£,raa 1к трение вызывает образование центров кристаллиза-
Ёсли описанные выше приемы не дали результата,
необходимо взять небольшое количество смеси и разделить ее
хрома графически (см следующую главу), чтобы получить
иалень ий образец чистого соединения, которое легко
кристаллизуется. Его можно затем использовать в качестве "за-
травк для основной массы образца. Это часто быстрее и
дешевл чем хроматографирование всего продукта.
Если нн один из перечисленных выше методов не
подойдет, необходимо сделать вывод, что "твердое вещество"
слишко загрязнено, чтобы закристаллизоваться, и его
следует выделять хроматографическя.
б) Реакции, дающие смеси
Смеси жидкостей обычно можно разделить фракционной
перегонкой (разд. 3.4.3), но в некоторых случаях перегонка
неасущ жгвима либо из-за слишком малого количества
вещества, либо слишком низкой летучести жидкости или слишком
высок -- i м- i к нагреванию. Небольшие
количества (менее 1 мл) "летучих" жидкостей можно разделить
лрепа ивной газо-жидкостной хроматографией (разд 4.1 .2).
^гим методом удается проводить разделения, которые
невозможно осуществить перетонкой, ■■ ■■ он представляет
неоценимое значение при получении urti .u < ■ количеств
образцов чистого вещества для спектрального анализа Однако
зт - г существу техника малых количеств, обычно
неприятная для препаративных разделений. Нелетучие жидкости,
которые не поддаются перегонке, можно выделить методами
Хроматографии, используемыми для твердых веществ.
Смеси твердых веществ обычно разделяют жидкостно-ад-
^рбционной хроматографией. Такие смеси следует сначала
исслед ть методом тонкослойной хроматографии (ТСХ)
(разд -; j ])( чтобы выяснить, сколько в них компонентов и
*** >о легко они разделяются. Затем можно провести пре-
^Рати^ое разделение посредством флаш-хроматографии.
Флат- роматографии на сухой колонке или хроматографии
i ^Днего давления (разд. 4.2.2).
3.2. КРИСТАЛЛИЗАЦИЯ
Кристаллизация - наиболее общий метод очистки органа,
ческих твердых веществ, не слишком загрязненных другим^
веществами , чаще всего применяемый в практической ори.
нической химии. Как правило, это трудный метод* в для то.
го чтобы выполнить его хорошо, требуется большая
практика, особенно при работе с малыми количествами вещества.
3.2.1. Общие принципы
Метод основывается на том, что растворимость тверды);
соединений в горячих растворителях намного выше, чем в
холодных. Таким образом, если вы приготовите насыщенны!
горячий раствор соединения А и дадите ему остыть, он
станет пересыщенным и соединение А выделится в виде
кристаллов Если соединение А не очищено, скажем содержит
несколько процентов другого соединения В, тогда примесь
также растворится в горячем растворителе, но при
охлаждении раствор не будет пересыщен соединением В потому что
оно присутствует в малой концентрации), которое останется в
растворе, в то время как основной компонент, соединение А,
закристаллизуется. Таким образом, чистое кристаллическое
соединение А можно отфильтровать, тогда как примесь
соединения В останется в фильтрате (обычно известном как
маточный раствор).
На практике метод кристаллизации состоит из следую^И
пяти стадий: 1) растворение твердого вещества в
минимальном объеме кипящего растворителя; 2) фильтрование горячего
раствора для удаления нерастворимых примесей (если они
присутствуют); 3) охлаждение раствора и после кристаллов
ции части твердого вещества выдерживание его до полнот
кристаллизации; 4) отделение кристаллов от маточного рас*
твора фильтрованием и 5) высушивание кристаллов. №
третьей стадии второстепенные компоненты (растворимые при*
меси) остаются в растворе, тогда как основной компо^И
выкристаллизовывается. Для того чтобы достичь абсолютно*
чистоты, может потребоваться многократная перекристаллиз*
Дня.
Ключ к успешной кристаллизации лежит в использоваЯЯ*
наилучшего растворителя, который легко растворяет вещг^^
Щац ц!ЗёЭ£И
В rex случаях, ког присутствуют заметные количества других
нений <примерно 5 - 10% и более), обычно необходимо разделать
хроматографически (гл 41.
продуктов реахцю
I и нагревании, но в котором основной компонент почти не
растворим при охлаждении, что позволяет ему почти
полночи выкристаллизоваться. На начальной стадии обучения в
^ебн заданиях растворитель для кристаллизации обычно
ргова ется, н° R последующей работе выбор подходящего
«аство теля (разд. 3.2.4) является существенным моментом,
требую[цим тщательной подготовки и здравого суждения.
3.2.2. Температура плавления как критерий чистоты
Температура плавления дает ценную информацию о
чистоте твердого вещества (разд. 3.3), так как присутствие
примесей понижает температуру плавления (7*гл) и расширяет
интервал температур плавления. Ее следует измерять как до,
так и после кристаллизации вещества. По достижении
максимального значения Твя , которая не изменяется при
дальнейшей пере ристаллизации, можно судить о чистоте соединения.
Этот прием известен как "кристаллизация до постоянной
Меры безопасности. Большинство растворителей,
используемых при кристаллизации, являются летучими и
воспламеняющимися, а некоторые - токсичными. Примите
соответствующие меры (разд. 1.3), чтобы предотвратить
их вдыхание или возгорание.
3.2.3. Методы кристаллизации
Выбор метода кристаллизации зависит от количества
образца. Метод, приведенный ниже, в разд. "а", пригоден для
кристаллизации любого количества вещества, вплоть до 100
нг или даже менее. Однако при нижнем пределе, скажем в
МО мг или менее, легче использовать метод, описанный в
РЗЭД- б (стр. 100), особенно если требуется горячее
фильтрована,. Этот метод можно применять при наличии до 10 мг
а) Кристаллизация с использованием методики колба -
•оронка
I Ча^ Вди растворитель для кристаллизации не указан, то сна-
4Ла просмотрите разд. 3.2.4.
(""ел* "асгпворение твердого вещества в кипящем раствори-
Ч>Дое вещество необходимо растворять в минимальном
Вывод воды
Ввод воды
Твердое вещество и
растворитель для
перекристаллизации
Электрически
нагреваемая
" водяная баня
Рис. 3.6- Прибор для кристаллизации
объеме кипящего растворителя в конической колбе . Коли
должна иметь шлиф для того, чтобы к ней можно было^В
соединять обратный холодильник (рис. 3.6). Можно
использовать колбы размером до 5 мл- Размер колбы должен бй®
таким, чтобы во время кристаллизации она была заполяеяз
твердым веществом и растворителем не более чем напол^Н
ну. Требуемый размер можно определить, проделав прспг
тельный опыт по выбору растворителя или руководств;
собственным опытом; например, для I г твердого вешг
потребовалась бы колба на 25 или 50 мл.
МЕТОДИКА. Взвесьте вещество, которое необходимо ^Н
ся***
Кристаллизацию никогда не следует проводить в химическом
или любом другом открытом сосуде Из конической колбы легко *^Ш
1 крумюдопной 5
; кристаллизации, тогда i
:0л^
I ^едряис и очистка продуктов реакции gl
дргаллизовать, и через воронку для сыпучих тел (см, рис.
25) перенесите его в колбу. Добавьте ровно столько раство-
-ятелн, чтобы покрыть им твердое вещество, но недостаточ-
{фе для его полного растворения при кипении Добавьте
несколько гранул кипятильничков. Подсоедините обратный
холодильник (разд. 2.1.4), если необходимо, через переходник.
Меры безопасности. Независимо от объема обратный хо-
доЗцлЫ! * необходим, поскольку опасно нагревать любой
растворитель в открытом сосуде.
Установите колбу и холодильник на водяную баню, если
растворители имеют Т^ до 80° С (рис. 3.6), или на
электрическую плитку в случае более высококипящих растворителей
Нагрев йте смесь до тех пор, пока не начнется равномерное
кипение растворителя с конденсацией в обратном
холодильнике. Спустя несколько минут часть твердого вещества должна
раствориться. Продолжайте добавлять из пипетки через
обратный холодильник небольшие порции растворителя до тех
пор, пока твердое вещество не растворится полностью. Это
нужно делать медленно, давая растворителю покипеть в
течение нескольких минут после каждого добавления, чтобы
твердое вещество успело раствориться. Помните, что
необходимо использовать минимальный объем растворителя.
Запишите объем израсходованного растворителя.
Если раствор прозрачен (нет взвешенных твердых
веществ) в сла^о окрашен смолистыми примесями, то его сле-
^^«тзвить для кристаллизации (см. ниже разд. 3).
Однако в двух случаях необходимы дальнейшие действия
Первый - когда раствор содержит нерастворимый материал,
Z*!?m*eP пыль или следы неорганического вещества. Все это
J-ofaoflHMo удалить фильтрованием горячего раствора (см.
0 Дующий раздел). Второй - когда образец загрязнен сильно
. ™Пенн|Ыми смолистыми примесями (как, например, в том
Пхт6* ССли твеРД°е вещество - это сырой продукт реакции).
^Римеси можно удалить следующим образом. Раствор немно-
(1 ^?жа10т> добавляют порошкообразный древесный уголь
щ~ *Уо от массы органического твердого вещества), затем
к!™*18'1' раствор до кипения и кипятят с обратным холодиль-
ом в течение нескольких минут.
*****
Гранулы
о другую коническую I
Древесный уголь абсорбирует примеси (в большинстве слу.
чаев), и затем его удаляют горячим фильтрованием (следущ.
щий раздел).
Меры безопасности. Не добавляйте древесный уголь efr
посредственно в кипящий раствор, так как это может ви-
звать сильное вспенивание.
2) Фильтрование горячего раствора
Эта операция необходима ТОЛЬКО в том случае, еслив
растворе присутствуют нерастворимые примеси. Она вызывает
множество проблем у неопытных работников (а иногда также
и у опытных). Трудности возникают в том случае, если
раствор остывает во время фильтрования; тогда твердое
вещество может качать кристаллизоваться на воронке, затрудняя
дальнейшее фильтрование. Вероятность кристаллизации ш
воронке меньше в том случае, если раствор не является
абсолютно насыщенным в горячем состоянии, поэтому при
горячем фильтровании в заключ ние следует добавить еще
примерно 5% растворителя (разд. I).
Здесь можно использовать несколько приемов. Первый
(описанный ниже) - вакуумное фильтрование через
предварительно нагретую воронку, которое применяют для образца
небольшого и среднего объема (скажем, до 50 - 100 мл)
Второй прием - простое фильтрование, используемое, как
правило, для крупномасштабных операций, обычно через
нагретую воронку для горячего фильтрования.
ВАКУУМНОЕ ФИЛЬТРОВАНИЕ ЧЕРЕЗ
ПРЕДВАРИТЕЛЬНО НАГРЕТУЮ ВОРОНКУ Эта быстрая и эффекв
ная методика удобна в случае не очень летучих растворит*;
лей [эфир, 40/60 бензин и дихлорметан (разд. 3.2.W
Фильтрование можно проводить либе через стеклянную &
ронку Шота (рис. 3.7,с), либо через воронку Бюхнера ^Н
Хирша, снабженную фильтровальной бумагой (рис. 3.7,6 и й
Воронка последнего типа необходима при фильтровании с я*'
тивированным утлем (см последнюю часть этого разд^'
Основное требование к воронкам Шота заключается в тя*
чтобы пористый материал был чистым и свободно пропу^И
раствор; это позволяет проводить фильтрование быстро *•
минимальным отсосом. Частично забитый пористый фиЛ"*
замедляет фильтрование, что приводит к охлаждению раС^
ра и делает всю операцию неудачной. Поэтому перед Ф*1^,
ровани м нужно проверить пропускную способность ПОриСТС
продуктов решщин
Фильтровальная
бумага
7
Перегородка
с отверстиями
Y
Рис 3.7 Воронки для фильтровании, а - воронки Шота с пористой стек-
жадной перегородкой; 6 - воронка Бюхнера. в - воронка Хирша.
^льт11а, пропустив через него немного чистого растворителя
["аналогичных условиях. Чтобы избежать возникновения этой
Чюолемы, многие предпочитьют фильтровать через воронку
^Ра или Хирша с фильтровальной бумагой.
80ЛИЛЬтР°вать следует через воронку подходящего размера.
Ф^т"3 С боковым отводом (рис. 3.10) особенно удобна для
ЕгшитьР0Ванаа неболыпих объемов. Фильтрование можно про-
З.й Г1 Ли(ю непосредственно в коническую колбу (рис.
6biL., ли6° <* случае небольших объемов) в маленькую про-
Ч1«У (рис. 3.8,©.
«ой ^Дики с использованием воронки Шота и фильтроваль-
i ЧУмаги слегка различаются, о чем будет сказано ниже.
г&*ът2>дика с использованием воронки Шота. Предвари-
йагрейте воронку в термостате до температуры, не-
Пробирка
Пробирка для
фильтрования с 1
боковым отводом J
Спираль us медной,
проволоки
ШЕ )
Гис 3.8 Прибор для фильтрования больших la) и мнкро (б) количеств.
много превышающей температуру кипения растворителя.
Быстро подсоедините се к колбе или пробирке для фильтрования
{рис. 3-8), залейте горячий раствор и подсоедините к колбе
очень слабый вакуум (используйте водоструйный насос с
частично открытым входным воздушным краном, стр. 137). ■
Подсасывание должно обеспечивать быстрое просачиванш
растворителя через воронку, не вызывая его быстрого испаре:
нья и охлаждения. Продолжайте добавлять горячий раствор)
так чтобы пористое дно воронки было все время покрыто
слоем жидкости. Фильтрат следует нагреть для растворения
всех кристаллов, которые могли бы в нем появиться, и затей
дать ему медленно остыть.
Методика с использованием воронки Бюхнера или XupW0
и фильтровальной бумаги. Этот метод незначительно отлич»'
ется от описанного выше. Нагрейте воронку, как описано вм'
ше, установите ее на колбу для фильтрата (рис. 3-8), поло-
жите фильтровальную бумагу и увлажните ее горячим чвс-
тым растворителем .нагретым отдельно), чтобы она пршн^И
к воронке. Затем подключите слабый вакуум (используй1^
водоструйный насос с частично открытым входным воздУ"
ным краном) и сразу же вылейте горячий раствор в и-е'У^
коронки. Важно подать ncyicoc до чаливки жидкости, чт°^0
предотвратить всплывакие фильтровальной бумаги с сетчато
дудение и очистка продудев реакции 91
«снования воронки. Затем продолжайте поступать так, как
одисано выше, во втором абзаце предыдущего раздела.
фильтрование растворов, содержащих древесный уголь
или д угие коллоидные включения. Древесный уголь содержит
очень мелкие частицы, которые могут необратимо засорить
стеклянный пористый фильтр воронки Шота Поэтому всегда
Аильт через ; с : ш бумагу, как описано выше.
Однако и сама ;. . ч ■ , ■ бумага может быстро
забиться рсвесным углем. Чтобы избежать этого, используйте
порошок для фильтрования, например целит. Этот мелкий
порош следует добавлять к раствору (а количестве до
0,5% растворенного вещества) непосредственно перед
фильтрованием или насыпать его слоем (толщиной 3 мм) на
увлажненную фильтровальную бумагу, а затем увлажнять
горячим растворителем.
Тот же самый прием можно использовать, чтобы быстро
отфильтровать горячий или холодный раствор, загрязненный
другими очень мелкими или коллоидными примесями.
ПРОСТОЕ ФИЛЬТРОВАНИЕ. Его обычно используют для
фильтрования больших объемов раствора. При этом
применяют стеклянную воронку с коротким концом (рис. 3.9),
снабженную складчатым бумажным фильтром (попросите вашего
преподавателя показать, как приготовить складчатый фильтр).
Фильтрование через складчатый фильтр намного быстрее, чем
обычным методом. Существенна, конечно, поддерживать
уровень жидкости ниже края фильтровальной бумаги. Поскольку
в этом методе площадь фильтровальной бумаги больше, чем
при использовании воронок Бюхнера м Хирша, и поскольку
бумага впитывает достаточно много раствора, он непригоден
Для работы с малыми объемами жидкости. При работе с
относительно небольшими количествами раствора воронку мож-
чо просто предварительно нагреть в печи до температуры
'"Уть выше температуры кипения растворителя. Но при
фильтровании большого объема раствора воронка и ее содержимое
Должны оставаться горячими (рис. 3.9). Имеются различные
"W* нагревателей: некоторые из них заполняют горячей во-
Дои, другае _ нагревают с помощью электричества.
Меры безопасности. Эту операцию необходимо выпол-
ы £ вытяжном шкафу, и при фильтровании поблизости
е должна быть источников возгорания, открытого пламе-
tiJlu электрических приборов.
Складчатый
бумажный
фильтр
льтрование горячего раствора через складчатый I
Рис. 3 9- Простое i
мажиый фильтр.
3) Образование кристаллов
Насыщенному (или почти насыщенному) горячему раст.0-
из него обычно выпадают кристаллы. Этот процесс может
быть медленным, и поэтому раствору следует дать „остсЯ
Обы^Т™" на6люлая- пои « завершите, кристаллизации
Обычно при медленном охлаждении образуются кр,..ные цт-
fwSl', ™°Р,! 6ыСТ,юм " <ме"ь мелкие- порошкообразные.
t^hS . ДИ"е""!' " Р3"»^"™» ■« оказывают „а-га-
*^- ™„™""е ? Ч^тадлмафпо. Быстрое охлалщение
может привести к образованию масел (см. ниже) I
выв,«5"ег3аЛеР,Ш:""* кРгсталл»ииии кристаллы отфильтро-
™^Г;^У " ЮМШВМ1ЭТ следующий раздел). Низки*
S ™ Щ °ГО в™?™ называет на то, что использу!
с^иптк™ мТТ'т,™ 6ЫЛ "Я™™™ <™ от, было взято
слишком много). В таких случает из фильтрата (маточного
^
| ццдгуие и очистка продугтон реакции 93
„детвора) можно дополнительно выделить кристаллы
охлаждением его ниже комнатной температуры в смеси воды со
льдом или даже до более низкой температуры в
морозильнике или охлаждающей бане (табл. 2.1). Если соединение
представляет особую ценность, маточную жидкость можно
сконцентрировать на роторном испарителе (разд. 3.1.2) н по
охлаждении получить еще одну порцию вешества. Как
правило, эти последующие порции менее чистые, чем основная; их
следует хранить отдельно до тех пор, пока их чистота не
будет определена по температуре плавления или каким-либо
другим способом.
ПРОБЛЕМЫ: КРИСТАЛЛЫ НЕ ОБРАЗУЮТСЯ. В
некоторых случаях кристаллы при охлаждении раствора не
образуются, даже если раствор пересыщен. Это обычно
происходит из-за отсутствия подходящих центров, инициирующих
обра аиие кристалла, и чаще всего случается с растворами,
очищенными от пыли горячим фильтрованием. Вызвать
кристаллизацию можно несколькими способами. Лучшим из них
явл ется добавка "затравочного" кристалла ("затравки") того
же самого вещества (для этой цели полезно оставить
небольшое количество неочищенного материала). Другой способ -
это потереть острой стеклянной палочкой по внутренней
стенке колбы на уровне жидкости, что приводит к образова
кию неровностей на стеклянной поверхности, которые служат
центрами роста кристаллов. Иногда помогает дальнейшее
охлаждение раствора, но некоторые растворители при сильном
охлаждении, дают вязкие растворы, которые никогда не
кристаллизуются. Если эти методы не приводит к успеху, то
иногда полезно просто заткнуть колбу, подписать ее и
оставить в лабораторном шкафу или холодильнике на несколько
дней.
ПРОБЛЕМЫ: ОБРАЗУЮТСЯ МАСЛА. Низкоплавкие
вещества могут выделяться не в виде кристаллов, а в виде
часта особенно при быстром охлаждении раствора. В
образовавшемся масле могут растворяться примеси или некоторое
количество растворителя, которые снижают температуру
плавления, поэтому масло может не закристаллизоваться даже
прн температуре значительно ниже ожидаемой температуры
члавлеиия соединения. Даже если оно все-таки затвердеет
пРи стоянии, вероятнее всего, оно будет сильно
загрязненным.
Для того чтобы прежде всего предотвратить обра зова ни
часел, необходимо экспериментировать с разбаалением, введе-
ем затравки и очень медленным охлаждением. Таким обра-
-JTjffia
зом, когда при охлаждении раствора начинает выделять^!
масло, следует снова нагреть раствор до растворения масла *
затем дать раствору очень медленно остыть, одновременно оЯ
торожно помешивая его стеклянной палочкой. Если масло оя
разуется снова, нагрейте раствор как прежде, дайте немного
остыть и затем добавьте несколько кристалликов затравки"
или потрите колбу стеклянной палочкой (см выше) пд
дальнейшем охлаждении. Беля кристаллики "затравки"
растворились раньше, чем появилось масло или твердое
вещество, добавьте их еще немного. Если и это не даст ж аемого
эффекта, снова нагрейте раствор, добавьте еще немного
растворителя и повторите процедуру. Для того чтобы
образовались кристаллы, часто требуется много терпения и упорства,
но даже тогда полученный выход может быть не очень
высоким. Кристаллизация низкоплавких веществ, содержащих
значительные количества примесей, намного более трудоемка; в
таких случаях, прежде чем приступать к кристаллизации, их
лучше очистить хроматографически (гл. 4).
4) Отделение кристаллов
После завершения кристаллизации кристаллы отделяют!
холодного маточного раствора вакуумным фильтровани
Обычно используют два типа фильтровальных воронок: а)
стеклянной пористой пластинкой (см., например, рис. 3,7,с),
в которых не нужна фильтровальная бумага Гспекшлеся
стеклянные фильтры имеют различную степень пористости - от О
(крупнозернистые) до 5 (мелкие); пористость 3 является
наиболее распространенной jj и б) воронки типа Бюхнера и Хир-
ша (рис. 3.7,6 и о) с сетчатой пластинкой, на которую ухла-
продуктов реакции
■- ОТСОС
(вакуум)
fh-
Рис 3 10 Воронка Шота для фильтрования микроколичеств.
Рис 3 11 Приборы дли отфильтровывания кристаллов
дывают фильтровальную бумагу Оба типа воронок имеют
широкий набор размеров. Лучше использовать воронки со
спекшимся пористым стеклянным фильтром, известные просто
как "воронки Шота", так как после фильтрования твердое
вещество можно аккуратно соскоблить, не рискуя при этом
порвать фильтровальную бумагу и загрязнить кристаллы.
Очень небольшие количества твердого вещества (20 мг и
менее) можно отфильтровать на самых маленьких воронках
Шота (рис. 3.10) с фильтрующим диском с нижним
диаметром примерно 7 мм (другие методы фильтрования малых
количеств обсуждаются на стр. 100). Важно, чтобы
фильтрующая часть была чистой и свободно пропускала жидкость
(проконсультируйтесь у преподавателя или лаборанта
относительно очистки фильтров, имеющих плохую пропускную
способность).
Фильтровальная воронка соединяется с фильтровальной
колбой (Бунзена) (рис. 3.11 ,а) или фильтровальной
пробиркой (рис. 3.11,6) либо посредством шлифа (рис. 3.11), либо
при помощи эластичной резиновой круглой пластинки (рис.
3-8), которая играет роль соответствующего вакуумного
уплотнения, когда воронка слегка продавливается вниз. Исполь
зуют также переходник с боковым отводом (рис. 3.11,е) либо
с конической, либо с круглодонной колбой. Последняя более
предпочтительна, если фильтрат затем концентрируют на
ровном испарителе.
Отсасывание обеспечивается вакуумным водоструйн]
насосом (разд. 3.4.8).
ПРОЦЕСС ФИЛЬТРОВАНИЯ (Фильтрование pacrnJ
охлажденных до температуры ниже 0°С, см. в с едую
подразделе.) При фильтровании через воронку Бюхнера |
Хнрша фильтровальная бумага на дне воронки должна!
крывать всю сетчатую пластинку, не загибаясь у стенок
Фильтровальную бумагу необходимо слегка смочить чистым
растворителем, чтобы она прилипл, к перфо рованному дну.
Подайте к системе слабый вакуум. Плавно вращая
кристаллизационную колбу с кристаллами и маточным г «твором
аккуратно вылейте смесь в воронк;- Подсос долж. быть
таким, чтобы через фильтр проходила ровная струя фильтрата.
Когд фильтрование закончится, стравите вакуум и, если
необходимо, снова залейте часть маточного раствора в
кристаллизационную колбу, чтобы смыть кристаллы, прилипшие к
стенкам.
После того как все кристаллы будут перенесены на
воронку, их промывают небольшой порцией холодною чистого рЯ
творителя. Для этого надо убрать подсос, - Г.вить
растворитель, чтобы он только покрыл кристаллы, слегка nej-емешать
шпателем и подключить снова слабый вакуум. Промывание
кристаллов следует делать с большой осторожностью, так как
это может привести к значительным потерям, если кристаллы
заметно растворимы в холодном растворителе
Наконец, покройте воронку часовым стеклом для защи-
кристаллов от пыли и подсоедините вакуум, чтобы удзя
как можно больше растворителя. При работе со средним^
большими количествами кристаллов во время этой onepai
кристаллы можно на фильтре отжать от растворителя ши
кой стеклянной пробкой, но при работе с небольшими ко,
чествами этого делать не рекомендуется, так как может
алечь за собой потерю вещества.
ФИЛЬТРОВАНИЕ ОХЛАЖДЕННЫХ РАСТВОРОВ Ино
при кристаллизации, чтобы получить хороший выход
вещества, необходимо охлаждать смесь до температуры ниже окрл
жающей. В таких случаях целесообразно перед
использованием охлаждать фильтрующую bopohkv во избежали агрева-
ния раствора при фильтровании. Охлаждение воронки легко
провести, поместив ее (предпочтительно в полиэтиленовом
пакете, чтобы она оставалась чистой и сухой) в холодильнИ!
или морозильник. При фильтровании охлажденных растворе
возникают трудности в результате конденсации и влаги
дик
1
Азот
Рис. 3 12. Фильтрование под азотом
оронке, растворе и кристаллах, избежать которую полностью
почти невозможно. Однако если соединение не чувствительно
к влаге, то это практически не имеет значения, так как
кристаллы можно высушить (см. ниже) после фильтрования.
Самое лучшее, что можно пре ринять без тщательных
приготовлений, - это быстро установить воронку под струю
сухого азота (рис. 3.12) и покрыть ее на время фильтрования
Часовым стеклом.
Пробирки Крейга (Craig) (см. рис. 3,16) обладают
значительными преимуществами при низкотемпературной крист
зацян небольших количеств раствора, так как при их
использовании не нужна операциз по перемещению кристаллов.
5) Сушка кристаллов
ПРИ КОМНАТНОЙ ТЕМПЕРАТУРЕ. Большинство образ-
цов можно эффективно высушить при комнатной температу.
ре, используя вакуум для удаления следов оставшегося рас-
творителя. После удаления максимально возможного
количества растворителя отегсыван на фильтровальной в. ронке
перенесите кристаллы во взвешенный кристаллнза <р (рис!
3.13) и покройте его часовым стеклом Подберите ч «сталли-
затор такого размера, чтобы кристаллы покр< ■:■ ■-.■■ его лео
сравнительно тонким слоем Очень маленькие образцы можно
высушивать в пробирке, покрытой кусочком алюминиевой
фольги с проколотыми в ней отверстиями. Поместите
кристаллизатор (или пробирку) в вакуумный ксикатор (рис.
2.13). Для устойчивости поставьте пробирку в маленький
химический стаканчик Установите предохранительное покрытие
над эксикатором (или обмотайте его полотенцем) и вакууми-
руйте его Ък ьшинство ра ворител й с Т^т до 80°С легко
улетучивается в вакууме водоструйного насоса (разд. 3.4.8,
п."а"); для менее летучих растворителей типа толуола, про-
панола, бута ноля и т. п. требуется более высокий вакуум,
шла Баемый роторным масляным насосом (разд. 3.4 8, п."б").
После сушки в течение 0,5 -1ч стравите вакуум (стр. 40),
взвесьте образец и определите его температуру плавление
Поскольку небольшие следы растворителя могут понизить Щ
стоит повторить процедуру сушки еще в течение 0,5
снова определить Тяя.
ПРИ ПОВЫШЕННОЙ ТЕМПЕРАТУРЕ, Иногда нсобход!
мо высушить кристаллы при повышенной температуре
например, при испол ии для к и аллизации очень нел чего
растворителя или при подютивке образца для аналитических
целей (разд 3 2.5, п. 'а") И снова эту процедуру ocyinecij
Ю).
ия.
ди-
Часовое стекло
Рис. 3 13 Сушка кристаллов в кристаллизаторе
■gin-.ние и очистки продуктов реакции
к вак\У MH0MV часосу
Кристаллы
Лодочка
Колбонагремтель
Рис. 3 14 Сушильный
пне комнатной.
длв сушки в вакууме при i
!
Ляют в вакууме при температуре значительно ниже Тпг_
(или температуры сублимации) образца. Большие количества
щества можно высушить в кристаллизаторе, помешенном в
уумный сушильный шкаф. Меньшие количества высушива
*зт в лодочке в сушильном пистолете (рис. 3.14).
Очень маленькие образцы высушивают в пробирке, не-
отно прикрытой алюминиевой фольгой с проколотыми в
*е* отверстиями и помещенной в лодочку.
Камеру сушильного пистолета нагревают либо парами рас-
рителя 'рис. 3.14, а также табл. 3.2 с температурами
кипения), либо электрически Во всех случаях образец
помещают в холодный вакуумный сушильный шкаф или пистолет,
одключают требуемый вакуум (обычно вакуум масляного
насоса (разд. 3.4.8, п. "б")) и затем начинают нагревать.
НИКОГДА не проводите эту процедуру в нагретом лабораторном
сушильном шкафу при атмосферном давлении по многим причинам" плавления
рмического разложении, токсичности, загрязнения.
Хаавя
6) Кристаллизация микроколичеств
Часто препаративную работу в органической химии
выполняют на образцах малого объема, получая при этом ■
500 мг (0,5 г) продукта. Кристаллизацию образцов массой ■
100 мг можно выполнить методами колба - воронка,
описанными в предыдущем разделе, в очень маленьких прибора
Если не нужно проводить горячего фильтрования, можно
закристаллизовать даже еще меньшие количества в крошечных
конических колбочках или микропообирках. Однако при
работе с очень маленькими объемами , например при
подготовке образцов к элементному анализу или спектроскопии,
необходима абсолютная чистота.
При этом настоятельно рекомендуется удалять пыль
горячим фильтрованием. Для фильтрования образцов массой до
100 мг и менее удобнее всего работать на приборе,
изображенном на рис. 3.15, в котором скомбинированы колба и
фильтр. Этот прибор сводит до минимума потери при рабоЯ
и переносе вещества, которые могут быть значительными,
если применять традиционные методы работы с маленькими
образцами; он удобен для работы с образцами до 500 мг, ио
самые оптимальные размеры - 10, 5 и 2 мл. К боковому
отводу подсоединяют воздушный насос с резиновой камерон
снабженной подходящим по размеру шлифом.
1) Растворение образца и горячее фильтрование
Меры безопасности. Эту операцию следует проводить в\
вытяжном шкафу вдали от источников возгорания.
Используя микрошпатель**, засыпьте образец в баллончик]
Миллиграммовые количества часто легче очистить сублимацией (раЗД-
3.4.7), но она подходит не для всех соединений.
Работа с маленькими количествами мелкого порошка или кристаллов
часто становится невозможной из-за статического электричества, под
воздействием которого частицы твердого вещества либо прилипают к стеклянным
поверхностям, либо разлетаются с лопатки шпателя во все стороны. Эту
проблему можно преодолеть, используя антистатический пистолет (например!
фирмы Aldrich "Zerostai") непосредственно перед загрузкой.
ц^рние и очистка продуктов реакции
Лапка для
крепления
Воздушный насос с
резиновой камерой
Пористая
стеклянная
пластинка
Рис. 3.15. Приборы для кристаллизации малых количеств веществ с
горячим фильтрованием.
через боковой отвод. Добавьте одну или две гранулы кипя-
тильничков и затем вводите пипеткой немного растворителя
для кристаллизации, чтобы смыть весь образец, прилипший к
боковому отводу. Укрепите прибор в указанном месте и
осторожно нагрейте баллончик на маленькой водяной бане [или
лучше на маленькой масляной бане, подогреваемой плиткой
(рис. 2.7)]. При нагревании на водяной бане "носик" можно
защитить от конденсации, неплотно обернув его кусочком
фольги или надев чистый колпачок от капельной пипетки.
Отрегулируйте нагревание таким образом, чтобы
растворитель конденсировался на уровне пористой пластинки (но не
выше) и плавно стекал вниз. Длинный боковой отвод будет
служить в качестве обратного холодильника. Продолжайте
постепенно заливать растворитель через боковой отвод до
полного растворения соединения. Не прекращайте осторожного
Кипячения до тех пор, пока пары растворителя не нагреют
Гористой пластинки. Выньте приборчик из баии, быстро
оботрите масло или воду с внешней стороны баллончика,
наденьте на носик маленькую коническую колбочку или пробирку
Крейга (рис. 3.16), переверните приборчик (рис. 3.15) и при
Помощи резиновой камеры слегка повысьте давление, чтобы
вЬ1давить горячюю жидкость через фильтр в приемник. Про-
водить горячее фильтрование маленьких объемов раствора под
давлением намного легче, чем под вакуумом, так как
устраняется проблема с испарением и охлаждением.
Резкое охлаждение раствора при его попадании в приема
ник может вызвать быструю кристаллизацию. В этом случае
раствор следует снова нагреть до растворения кристаллов и
затем дать ему медленно остыть
2) Выделение и сушка кристаллов
При работе с 50 - 500 мг горячего раствора его можно
отфильтровать в маленькую коническую колбочку или
микропробирку, где кристаллизация произойдет обычным способом
(разд. 3.2.3). Кристаллы затем отфильтровывают на
маленькой воронке Шота, изображенной на рис. 310- Однако для
фильтрования 100 мг раствора н менее лучше пользоваться
пробиркой Крейга <рис 3.16), как описано ниже, что
значительно снижает потери при работе с кристаллами и их
переносе.
ИСПОЛЬЗОВАНИЕ ПРОБИРКИ КРЕЙГА Отфильтруй^
горячий раствор в пробирку Крейга подходящего размера, к!
это описано выше, закройте ее неплотно пробкой и оставь]
пробки все
кристаллов маточную и-ид,^.,.. __
жидкость и переверните пробирку Крейга в hcxi
;. Постучите по пробирке, чтобы удалить - ..г. _
Т"" необходимости приподнимите пробку, смойте
—-" иягтпгапнтеля и
НИК. ииы;-....
кристаллы. При необходимости приподнят.,,., ._
кристаллы несколькими каплями охлажденного растворителя
отц риф ги уй При cvunce кристаллов снимите
пробку, ншлотн. рикройте . робирку алюминиевой фольгой
* поместите ее в сушильный пистолет (рис, 314). Если трс-
ся дальнейшая перекристаллизация, то се выполняют в
iwpKe Крейга, добавив отфильтрованный растворитель и
.ирожно нагревав основание пробирки на водяной или мас-
ной бане (в вытяжном шкафу), чтобы снова растворить
исталлы Пробирка сама по себе будет действовать как об-
•Чтпт иикл можно повторять столько
бу тся
пробирке Крейга, добавив
_ "агре-
(в
тныи холидп.1*,,1„... Этот цикл
раз, сколько потребуется, без каких-;
кристаллов.
3.2.4. Выбор растворителя для кристаллизации
Успех кристаллизации зависит в значительной степени от
правильного выбора растворителя. Идеальный растворитель -
тот, в котором очищаемое вещество легко растворяется при
нагревании, но лишь умеренно на холоду и в котором очень
хорошо растворимы примеси.
ш_
Таблица 3.2. Раствори;
Растворитель8
1. обычно используемые при кристаллизации
Диэлектрическая
проницаемость
Растворимость в
воде, г/100 г^
Пентан
Гексан
Петролей! эфир
Шгелогексан
Толуол
Днэтиловый эфир
Этилацетат
Уксусная кис
Дихлорметан
Пропанол-2*
Ацетон
Этанол
Метанол
иметилформамнд
иметнлеульфоксид
гл
1.9
0,03
Нераств.
Нераств.
Сл раств
Сл. раств.
7.5
9.0
Раств.
2.0
Раств.
Раств.
Раств
• Растворители, помеченные звездочкой, прежде всего рекомендуются
пробной кристаллизации (см текст).
а) Общие закономерности
На практике растворитель подбирают путем серии
пробных кристаллизации (см. следующий раздел), но некоторьи
указания можно получить из общих положений, изложенных'
ниже. Наиболее известен принцип "подобное растворяется в\
подобном", т. е. полярные соединения более растворимы в
полярных растворителях, чем в нсполярных и наоборот. Не-'
которые общие характеристики растворимости и полярности
растворителей даны в табл. 3.1, а некоторые значения
диэлектрических проницаемостей растворителей (указывающие
на полярность) приведены в табл. 3.2. Например, полярное!
соединение, содержащ гидроксильную (ОН) группу,
вероятно, растворимо в метаноле (CrLOH), но хуже растворяется
последовательно в этаноле (С2Н^ОН) и в высших спиртах,
полярность которых убывает по мере удлинения
углеводородной цепи. Сильнополярные соединения, например, с
несколькими гидроксильными (ОН) группами либо с карбоксильной I
!
рйДЕ.]|е"ие " очистка продуктов ркв^ц—.
(С0,Н) или сульфокислотиой (SO^H) группой, как правило,
до крайней мерс частично растворимы в воде, но это,
очевидно, зависит от того, что еще присутствует в молекуле,
ра римость в воде ухудшается при наличии таких
гидрофобных групп, как углеводородные цепи или кольца. Обычно
большинство органических соединений, за исключением ис-
$ол щого их числа, содержащих группы ОН, COjH или
SO нерастворимы в воде, поэтому вода редко бывает
хорошим растворителем для кристаллизации. Неполярные
соединения например углеводороды или алкилгалогениды, почти
нерастворимы в воде, но легко раствог^ются в неполярных
растворителях типа петролейного эфира или тоуола.
ВЫБОР ТЕМПЕРАТУРЫ КИПЕНИЯ РАСТВОРИТЕЛЯ.
Обычно лучше всего подбирать растворитель с широким
температурным интервалом, так чтобы различие в растворимости
между холодным и горячим было как можно больше. По
этой причине следует по возможности избегать использования
таких ннзкокипятцих растворителей, как эфир (диалоговый
эфир), 40/60 бензин и дихлорметан. При работе с высоко-
кипящими растворителями также имеются трудности: они
намного менее летучи, и поэтому их трудно удалить с
кристаллов после фильтрования. Следует избегать растворителей
с температурой кипения выше температуры плавления
вещества, которое необходимо перекристаллизовать. Такое
соединение расплавится, перед тем как растворится, и при
охлаждении выпадет скорее в виде масла, чем в виде кристаллов.
Наиболее популярны растворители с температурой
кипения в интервале 60 - 90°С: 60/80 бензин, гексан, цикло-
гексан, этилацетат и низшие спирты (до Съ или С,); см.
табл. 3.2. Если соединение не растворяется ни в одном из
Петролейный эфир - это смесь углеводородов, а НЕ эфир; он имеет
несколько интервалов температур кипения, например 40 60°С, 60 - 80°С.
80 - 100*C. Эти фракции обычно называют "40У60 бензин". "60/80 бен-
зни" м т д Все они неполярны и легко воспламеняются.
Такие популярные растворители, как четырехкдорнстый углерод, бензол
хлороформ, валяются токсичными или канцерогенными веществами, и их
не следует использовать для кристаллизации
. LM
них, следует попробовать более высококипящие растворителе,
например уксусную кислоту (ледяную), пиридни высшие
спирты, диметилформамид или диметилсульфоксид.
б) Пробные кристаллизации
Растворители, отмеченные звездочкой в табл. 3.2, прежде
всего рекомендуются для пообной кристаллизации. Немного
твердого вещества (-"50 мг) помещают в чистую пробирку
(7,5x10 мл) я при перемешивании по каплям добавляют
0,25 - 0,5 мл растворителя. Бели твердое вещество растворяв
ется на колоду, то растворитель, очевидно, подходит только
в качестве "хорошего" растворителя в смеси двух
растворителей (см. ниже). Если же оно не растворяется, нагрейте
пробирку на водяной бане, встряхивая ее при атом до тех пор,
пока растворитель не закипит. Если твердое вещество
растворилось не полностью, добавьте еще несколько небольших
порций растворителя до общего объема примерно 1,5 мл.
Если часть твердого вещества все же не растворится, то
следует попробовать другой растворитель. При получении
прозрачного раствора дайте ему остыть. Если после того, как
раствор постоял при комнатной температуре в течение
нескольких минут, кристаллы не появились, добавьте затравочный"
кристаллик или потрите стенку пробирки стеклянной
палочкой (разд. 3.2.3).
В хорошем растворителе выход кристаллов хороший.
Небольшой выход кристаллов означает, что данный растворитель
не подходит из-за высокой растворимости в нем на холоду.
Повторите процесс с несколькими растворителями и выбеоите
тот из них, который дает наибольший выход кристаллов . ■
СМЕШАННЫЕ РАСТВОРИТЕЛИ. В некоторых случаях
может оказаться так, что ни один растворитель не подойдет.
Тогда необходимо использовать смесь двух растворителей, в
одном из которых вещество растворяется легко, а в другом
лишь умеренно. Оба растворители, конечно, должны
полностью смешиваться друг с другом и по возможности иметь
близкие температуры кипения. Далее можно следовать двум
методикам: 1) суспендировать твердое вещество в небольшом,
количестве "слабого" растворителя, изгреть до кипения и
затем добавить по каплям "хороший" растворитель до растворе-
При работе с малыми количествами вещества згу цифру можно
зтъ до 10 мг и пропорционально снизить количество растворителя.
Если соединение фактически нерастворимо во всех распространенных
растворителях см. раза 3.2.5
|Дрцрнмд и очистка продуктов реакции
i
■J
для твердого вещества или 2) растворить твердое вещество в
деболыном количестве "хорошего" растворителя при кипяче-
ляи с обратным холодильником и вводить по каплям ела
бый растворитель, до тех пор пока не появится слабая опа-
десценция, а затем добавить еще кашпо или две "хорошего"
рзстворителя, чтобы устранить опалесценцию. В каждом слу-
ча когда раствор становится опалесцирующим при
охлаждении и соединение начинает выделяться в виде масла,
следует добавить еще несколько капель "хорошего" растворителя и
снова нагреть раствор. После того как найден эффективный
метод его следует использовать для кристаллизации основной
кассы вещества
3,2.5. Специальные темы
а) Подготовка кристаллических образцов для анализа
сжиганием
сследовательская работа часто приводит к с_,
новых соединений, которые раньше не были получены и
описаны в химической литературе. Такие соединения обычно
ифицируют путем элементного анализа, а также с
помощью информации, полученной различными
спектроскопическими методами (гл. 5) Эти данные и такие физические ха-
ра ристики, как температура плавления (или показатель
преломления для жидкостей), затем приводятся в литературе,
чтобы то же самое соединение могли легко опознать другие
иссл ватели. Эти данные надежны и воспроизводимы только
для абсолютно чистого соединения. Присутствие примесей
может дать неправильную информацию, что сильно затруднит
ктурное отнесение, а в худшем случае приведет к тому,
что соединению припишут неправильную структуру.
Термин "элементный анализ" обычно относят к
определению эмпирической формулы соединения (например, C^HN и
т д > путем его сжигания. Он жизненно важен не только
Для определения элементного состава (который можно также
выполнить с помощью масс-спектрометрии), но и как
критерий чистоты. По причинам, отмеченным выше, данные о
новом соединении нельзя опубликовать в авторитетных
химических журналах до тех пор, пока их чистота не будет под-
ждена точными результатами анализа на сжигание. Это
важная гарантия дл^я серьезной науки и высоких
профессиональных стандартов .
В вцде исключения для очень нестабильных соединений, которые нель-
* проанализировать сжиганием, допустимы некоторые другие доказательства
108.
При анализе сжиганием очень маленькую навеску соедн..
нения (3-5 мг) сжигают в избытке кислорода я определяю*
количество СО,, ИД) и N2, а затем находят процентное Ж
держание С, Н и N. Расчет покажет, соответствуют ли эти
экспериментальные данные предложенной эмпирической фМ
муле (допустимая погрешность ±0,3%),
Так как на анализ берут только очень малое количество
образца, последний должен быть не только химически
чистым, но и не содержать пыли, волокон фильтровальной
бумаги и любого другого постороннего материала Поэтому поя
готовка образцов для анализа требует большой тщательности.
Все приборы необходимо тщательно вымыть, прополоскать
дистиллированной водой и высушить в сушильном шкафу
(лучше всего в большом кристаллизаторе, покрытом часовым
стеклом для предохранения от пыли). Для удаления пыли во
время кристаллизации необходимо горячее фильтрование либо
под вакуумом (разд. 3.2 3.), либо методом, описанным на
стр. 100, который является более легким при работе с
маленькими образцами и требует использования минимального
числа единиц прибора. Выделение и сушка кристаллов в
пробирке Крейга (рис. 3.16) позволяют свести к минимуму
число операций и действие пыли. Кристаллы можно также
отфильтровать на маленькой воронке Шота (рис. 3.10) или
даже на воронке Хирша со специальной твердой
фильтровальной бумагой. Однако в последнем методе кристаллы следует
быстро высушить отсасыванием на фильтре, чтобы избежать
попадания пыли на образец. И, наконец, для удаления
окклюдированного растворителя кристаллы надо высушить в
сушильном пистолете (рис. 3.14). Перед тем как отдать
образец на анализ, его следует внимательно осмотреть через
лупу, чтобы убедиться в отсутствии пыли и других включений.
6) Кристаллизация соединений с очень низкой
растворимостью
При пробной кристаллизации может оказаться, что соедиИ
нение очень слабо растворяется во всех растворителях,
используемых для кристаллизации обычным методом. Такие
соединения можно удовлетворительно закристаллизовать
продолжительной экстракцией в аппарате Сокслета, как опневно я
разд. 3.1,3. Твердое вещество для кристаллизации помещают
в пористую гильзу (рис. 3,5), а круглодонную колбу
заполняют примерно на дае трети объема соответствующим
растворителем, желательно не высококипящим, потому что
длительное кипячение с обратным холодильником при высокой
температуре может привести к термическому разложению соеди-j
Iff
яеция. Если либо растворитель (разд. 3.2.4), либо соединение
^вствительны х действию воздуха, процесс следует проводить
s инертной среде (например, в азоте) (разд. 2.1.5). При
длительной экстракции соединение постепенно переносится в
колбу, где оно кристаллизуется.
3,3- ТЕМПЕРАТУРА ПЛАВЛЕНИЯ
3.3.1. Общие принципы
Температура плавления {Твл) - важный параметр,
характеризующий твердое органическое вещество. Нахождение этого
параметра служит способом идентификации и определения
меры чистоты соединения. Ее использование для
идентификации основано на том факте, что все чистые соединения
имеют определенную температуру плавления или, чтобы быть
более точными, узкий температурный интервал перехода из
твердого состояния в жидкое. Этот температурный интервал
для чистых соединений составляет максимум 1 - 2°С. Таким
образом, Т чистого образца, например бензойной кислоты,
следовало бы записать как 120 - 122° С. Использование Тв:
как меры чистоты основывается на том факте, что
присутствие примесей, во-первых, понижает Трл и, во-вторых,
расширяет интервал температур плавления. Например, слегка
загрязненный образец бензойной кислоты плавится при 114 -
119°С.
Использование Твл для идентификации, очевидно,
отличается большой неопределенностью, так как существует
несколько миллионов органических соединений и вследствие
совпадений многие из них имеют одинаковую температуру
плавления. Однако этот метод можно сделать намного более
определенным, следуя методике, известной как определение
"температуры плавления смешанной пробы". Эту методику
можно осуществить, только если у вас имеется "подлинный"
образец соединения, о котором идет речь. Она заключается в
плательном измельчении равных количеств "неизвестного*1 и
одлннного" образцов и определении температуры плавления
смеси. Если Тпя приготовленной смеси фактически та же, что
и у каждого из двух образцов, весьма вероятно, что они
представляют собой одно и то же вещество, если же Тол
смеси ниже, велика вероятность того, что это разные
соединения.
3.3.2. Определение температуры плавления
В большинстве случаев при рутинном определении Тпл не-
I ^_---термсметр
Отверстия для трех
капилляров
большой образец вещества помещают в тонкостенный
капилляр. Капилляр (рис. 3.17) затем медленно изтровают в
приборе опред ения ш и определяют интервал
В наиболее распространенно приборе я опред ения 7„л
(ряс. 3.17) имеется электрически обогреваемый металлический
блок i отверстием для термометра тремя рстиями дли
капилляров в отверстием для наблюдения. Для защиты от
сквозняков блок вмонтирован в футляр, который бжен
увеличительным стеклом и систем й ос щеяия для наблюден
ния за поведением образцов во время нагревают Наличие
трех отверстий для капилляров позволяет одновременно
исследовать оба образца и их смесь. Скорость и гревания блока
регулируют реостатом. В некоторых более дорогих приборах
вместо металлического блока для нагревания используют
жидкую баню (для лучшего теллопереноса) или вместо
обычного термометра применяют термометр с сопротивлением из —
11
ли
детины с цифровым температурным табло.
Капилляры для определения Тпа обычно поставляются в
виде трубочек с открытыми концами длиной около 10 см.
Каждый капилляр легко превратить в два капилляра, нагрев
его в центре на небольшом горячем пламени микрогорелки и
затем растянув (получаются два запаянных конца). Это
требует практических навыков, так как концы должны быть
достаточно симметричными и неизогнутыми, иначе капилляр
нельзя будет вставить в прибор для определения температуры
плавления. Капилляры для определения Гпд следует хранить
в пробирке, закрытой пробкой, чтобы они оставались
чистыми и не пылились.
Перед заполнением капилляра слегка разотрите образе
шпателем на чистом часовом стекле, а затем вдавите
открытый конец капилляра в порошок, чтобы получилась
маленькая "пробка" из твердого вещества, которая при осторожном
постукивании запаянного конца по твердой поверхности или
при проведении напильничком по наружной стенке капилляра
у его верхнего конца (что создает вибрацию) переместится
вниз. Наконец, следует еще раз постучать капилляром о
твердую поверхность (или бросить в стеклянную трубку дли-
но 60 см) и, таким образом, утрамбовать столбик твердого
вещества высотой 2-3 мм.
Если приблизительная ТПЛ соединения неизвестна, то,
чтобы сэкономить время и сделать процедуру не такой
утомительной, сначала проводят грубое определение, используя
довольно быстрое нагревание. Затем прибору дают остыть на
30°С, вставляют новый образец и, контролируя нагревание,
медленно повышают температуру - примерно на 1°С в
минуту вблизи Tu.
Слишком быстрое нагревание приведет к заниженному
результату, потому что температура термометра будет
отставать от температуры блока.
Фиксируйте интервал Тп с точностью до 0,5°С, начиная
с появления первого признака плавления (начала появления
жидкости) до полного исчезновения твердого вещества.
3.3-3. Другие методы
а) Предметный столик Кофлера
Предметный столик Кофлера (рис. 3.18,о) предназначен
Для быстрого (1 мин) рутинного определения Тпг в интервале
Отполированная
металлическая
г _ пластинка.
Горячий Кристалл.
конец
Круглый стеклянный
колпачок с
загрунтованной кромкой
Мостик из стекла над
предметным стеклом
v подставкой
i для определения температуры г
Рис- 3.18. Предметный столик Кирлера.
Рис. 3 19. Нагреваемая i
ЛЕ11ИЯ П(Щ МИКрОСКОПОМ.
на пластинку помещают несколько кристалликов
вещества в передвигают указатель до тех пор, пока он не
сравняется с границей между твердым и расплавленным
веществом (рис. 3.18,6). Затем по температурной шкале определяют
Тся Прибор имеет точность примерно ±1°С, но для
получения более правильных результатов его необходимо проградуи
ровать вблизи ожидаемой 7"пч, используя подготовленные для
этого одно или два калибровочные соединения с точно
известной Тпя.
6) Столик с микроскопом для определения Тпя
Это намного более совершенный прибор, где для
определения Г требуется всего несколько кристалликов. Такие
приборы обычно используют для точного определения Гпл неиз-
вестного соединения. Кристаллы помещают на обогрева мыи: J
освещаемый предметный столик микроскопа (рис. 3.19) и ещ
блюдают при большом увеличении за их плавлением.
Процедура определения Тпд заключается в следующем.
Несколько кристалликов образца помещают на предметное стен
ло (рис. 3.19), покрывают стеклянной пластинкой, затея
стеклом в виде мостика и сверку круглым стеклянным
колпачком Далее микроскоп фокусируют на кристаллах и
довольно быстро настраивают реостат на температуру, лежащую
из 10°С ниже 7*^ образца, а затем повышают ее на 1 *С 1
минуту. В микроскоп видно, что кристаллы как раз перед
плавлением теряют острые грани, объединяясь в маленькие
капельки. Зарегистрируйте температурный интервал между
появлением мелких капелек вокруг каждого кристалла щ
полного плавления.
3.4. ПЕРЕГОНКА
3.4.1. Общие положения _
Перегонка является наиболее важным и широко
используемым методом очистки органических жидкостей и разделения
жидких смесей Этот метод заключается в кипячении и
выпаривании жидкости с последующей конденсацией паров в
дистиллят. Разделение двух жидкостей с разницей температур
кипения 50 - 70° С или более можно осуществить простой
перегонкой: (разд. 34.2), но если эта разница меньше
необходима фракционная перегонка на более сложном приборе
(разд 3.4.3). Некоторые жидкости с высокими температурами
кипения в процессе п регонки при атмосферном давлении
разлагаются.
При снижении давления температура кипения понижается)
что позволяет перегонять высококипящие жидкости и масла
легко и безопасно. Этот метод называется вакуумной
перегонкой (разд. 3.4.5).
Меры безопасности. Так как большинство органических
жидкостей легко воспламеняется, перегонять их следует t
большой осторожностью. Иаибольш ю опасность
представляют легко летучие вещества (например, эфир с Тг
35"С). При переганке таких соединений нельзя допускаШ
чтобы их пары контактировали с открытым пламенШ
искрящими моторчиками, биметаллическими силовыми
регуляторами, источниками инфракрасного излучения или kokw
ми-либо другими источниками возгорания. Опасны также
пероксиды, образующиеся при контакте с воздухом иекот
т с
лав-
иль,
чем,
■гуси-
же
го-
деление и очистка продуктов реакции И^
рых соединений, особенно эфиров и углеводородов (стр. 16).
Ж кости, содержащие пероксиды, НЕЛЬЗЯ перегонять без
предварительного их удаления. Вещества, используемые в
учебных лабораториях, не должны содержать пероксиды.
Однако в исследовательской работе эту опасность всегда
следует предусматривать. Ряд тестов на определение пе-
роксидов и методы их удаления приведены в работах [2 -
4j В работе (4] обсуждаются и сравниваются доступные
методы и даются рекомендации по использованию
индикаторных молекулярных cum (4A) в качестве эффективного
pea. <нта для удаления гидропероксидов из обычных эфирных
растворителей типа диэтилового эфира и тетрагидро-
фурана.
3.4 2. Простая перегонка
а) При атмосферном давлении
Обычный прибор для простой перегонки в собранном виде
гок зан на рис. 3.20,а. Такие приборы малоэффективны и
пригодны лишь для разделения жидкостей с температурами
кипения, различающимися не менее чем на 50 - 70° С. При
пцкгонке на этом приборе смесей с более близкими темпера-
гурами кипения более летучий компонент, хотя и отгоняется
первым, уже на ранних стадиях перегонки загрязнен более
высококнпящнм компонентом.
ПРИБОРЫ Растворы большого объема обычно перегоняют
в круглодонных колбах, а образцы объемом до 100 мл - в
евидных. Необходимо подобрать колбу такого размера,
чтобы в начале перегонки она была заполнена не более чем
на две трети Колбу подсоединяют к холодильнику при
помощи простой насадки (насадки Вюрца) (рис. 3 20,о) Растворы
объемом 10-100 мл перегоняют в специальных перегонных
колбах (рис. 3 21,(2) с боковым отводом. Не следует
использовать для перегонки конические колбы. Иногда при перегон-
жидкостей с высокой температурой кипения полезно обер-
куть насадку лентой из стекловолокна (или асбестовым
шнуром). Это предотвратят излишнюю конденсацию, и, таким
°бразом, пары смогут попасть в боковой отвод.
Термометр для измерения температуры перегонки вставля-
107 в насадку. Для правильного измерения температуры
шарик термометра необходимо полностью погрузить в пары чуть
**иже уровня бокового отвода.
Выбор типа холодильника в каждом конкретном случае
определяется температурой кипения перегоняемой жидкости
Кипятильнмчки
Лабораторная подставка,, J
плитки или деревянные йрусш
Осушительная трубка
Рис. 3-20. Перегонка при атмосферном давле ни
Простой холодильник Либиха (см, рис. 2.10) применяется для
перегонки жидкостей с температурами кипения выше 50°С,
но для переганки легколетучих жидкостей, на рим р эфира
№жкп 35°С), необходим холодильник с двойной поверхностью
охлаждения (холодильник Дэвиса) (рис, 2.10). Вода подается
в нижний отвод холодильника и выходит в верхний. Обычно
вода подается под слабым напором; излишнее давление
вызовет перемещение отводного резинового шланга под действия
реактивной силы струи. При температуре кипения 150°С~
выше вполне достаточно воздушного холодильника.
1 - грушевидная,
Арбузова
Переходник, соединяющий холодильник с приемной колбой
(н ываемый "алонжем"), должен сообщаться с атмосферой
Б противном случае давление в системе может возрасти и
ра шить прибор. Если необходимо провести перегонку без
доступа влаги, к отводному отверстию на алонже можно при
Шмощи короткого резинового шланга присоединить неплотно
набитую осушительную трубку или использовать специальный
а/юнж, изображенный на рис. 3.20,6.
Перегонную колбу можно нагревать на открытом пламени
горелки Бунзена ТОЛЬКО в редких случаях при перегон к
водного раствора. Для перегонки органических жидкостей
перегонную колбу нужно погрузить в какую-либо
нагревательную баню (рис. 3.20,а). Тип бани и природа источника
тепла в значительной степени зависят от того, что перегоняется.
На водяной бане с электрическим обогревом можно
перегонять жидкости с температурой кипения до 80°С. При перс-
гонке эфира или других летучих легковоспламеняющихся
Жидкостей необходимо, чтобы вблизи не было открытого
пламени или другого источника возгорания. (При работе с
растворами небольших объемов химический стакан с водой мож-
ко нагреть на электрической плитке.)
\
Меры безопасности. Летучие легковоспламеняющиеся
^идкжти типа диотиловаго или петролейнога эфира всегда
следует перегонять в вытяжном шкафу, а не просто на
Лабораторном столе, где один из ваших соседей неожиданно
Решит зажечь бунзеловскую горелку.
чг
Для перегонки жидкостей с температурой кипения свыше
80°С требуется масляная баня. Различные типы масел и
температурные пределы, при которых их можно использовать
рассмотрены в разд. 2,1,4, Верхний температурный предел
для масляных бань составляет примерно 250° С (с силиково-
вой жидкостью). Идеальной баней является специально
разработанная малоинерционная элсктрообогреваемая масляная
баня с точным термостатическим контролем температуры,
однако такие бани дороги и редко используются в учебных
лабораториях. При проведении крупномасштабных работ масло
обычно наливают в кастрюлю, а в случае мелких работ - в
алюминиевую кружку и нагревают на горелке Бунзсна (или
микрогорелке) либо на электроплитке. Последняя обычно
очень инерционна и не обеспечивает хорошего контроля^"
температурой бани. Поэтому чаше всего для нагревания ис
пользуют горелку Бунзена или микрогорелку с зажимом^
резиновой трубке, чтобы точно контролировать размер плав
Меры безопасности. Нагреваемые на открытом пламеы
бани НЕЛЬЗЯ использовать для перегонки летучих
легковоспламеняющихся жидкостей. Колбонагре.ватели пе
подходят для нагревания перегонных колб из-за высокой
температуры на поверхшкти, которая может подниматься по
мере падения уровня жидкости.
Масляная баня, работающая при температуре примерней
50°С выше ее верхнего предела, немного дымит и неприятно
пахнет, поэтому лучше всего устанавливать ее в вытяжио)
шкафу. Если требуется нагревание выше 250°С, то приме!
ют баню из металлического сплава В уда (стр. 33) ■
350°С). Однако лучше и безопаснее перегонять высококил:
■дне жидкости при пониженном давлении (см. следуюпн
раздел "б").
При сборке прибора понадобится несколько лапок для ■
поддержки. Их число можно свести к минимуму
использованием пластиковых хомутиков (см рис 2.1), чтобы
предотвратить падение алонжа и приемника Когда прибор уже
собран, его перемещение вверх или вниз затруднено, поэтому *
самом начале сборки прибор следует устанавливать на
правильной высоте. Она определяется высотой, на которой ycj
новлена масляная (или водяная) баня, поэтому нужно нач
нать сборку именно с этого конца с установки перегони
колбы ка правильной высоте.
J3-
рдение и очирты продуктов реакции
ПРОЦЕСС ПЕРЕГОНКИ. Соберите установку, принимая
^ внимание изложенные выше замечания. Взвесьте приемные
колбы. В перегонную колбу через воронку влейте жидкость.
Это лучше делать до установки бани чтобы и случае пере
ива жидкость не попала в баню. В начале перегонки колбу
следует заполнить не более чем на две трети. Добавьте
несколько гранул (кусочков) "кипятильничков", чтобы кипение
было спокойным. Если перегонку необходимо прервать, перед
ее возобновлением следует добавить еще "кипятнльничков**.
НИКОГДА не добавляйте нх в кипящую или перегретую
жидкость (так как это может вызвать неожиданное бурное
вскипание и вспенивание). Удостоверьтесь, что нее
соединения герметичны, а приемник надежно закреплен. Проверьте,
чтобы прибор через отверстие в алонже соединялся с
атмосферой. Если жидкость легко воспламеняется, к отводу в
алонже следует подсоединить кусок резиновой трубки и
вывести ее в безопасное место с хорошей вытяжкой, подальше от
источника возгорания (см. примечания по мерам безопасности
на стр. 114). Проверьте, чтобы к холодильнику была
подключена вода. Если вы собираетесь работать на масляной
бане, удостоверьтесь, что в ней нет воды (стр. 33),
Убедившись, что все нормально, начинайте ОСТОРОЖНО нагревать
баню, чтобы температура поднималась медленно. За это
время происходит перенос тепла к жидкости в колбе. Когда
жидкость закипит, уменьшите скорость нагревания и,
контролируя температуру бани, добейтесь медленной и стабильной
перегонки. Вы обнаружите, что для обеспечения необходимой
скорости перегонки температура в бане должна быть из
30°С выше температуры перегонки. Наилучшее разделение
достигается при очень медленной перегонке (10 капель в
минуту). Запишите интервал температур кипения каждой
фракции, например Твмп 71 - 73°С. НЕ перегоняйте досуха так
как некоторые остатки иногда содержат взрывчатые перокси-
Ды (стр. 16 и 114). После перегонки взвесьте каждую
фракцию
б) Перегонка при пониженном давлении (вакуумная
перегонка)
Многие органические соединения нельзя перегонять при
втмосферном давлении, поскольку оин частично или
полночью разлагаются при нормальной температуре ^сипения.
Понижение давления до менее чем 30 мм рт ст. значительно
ТО,™, м„ю»«,р»х « и»»еряш,ьиь„ прибор» Шк-Леода (760 »м рт с.
■ I >тм, I мм рт с- - I "РР " 133-Э ""А
снижает емпера ру кипения, что позволяет проводить пере,
гонку без разложения. Обычно используются два типа ваку
умных насосов. Во-первых, водяной насос (известный как лд
бораторный водоструйный насос или аспиратор), понижаюг^Н
давление до 10 - 20 мм рт. ст., что позволяет снизить тем
пературу кипения на 100 - 125°С. Действие вакуумных сие.
тем с водоструйным насосом обсуждается в разд. 3.4.8,а. Во-
вторых, ротационный масляный насос, дающий давление ад
0,01 мм рт. ст. Ниже 30 мм рт. ст. при каждом понижении
давления в 2 раза температура кипения снижается примерно
на 10°С Работа с масляными вакуумными насосами описана
в разд. 3.4.8,6.
При очень низких давлениях «0,1 мм рт. ст.) плотность
паров слишком низка, чтобы фракционная перегонка была
эффективна и иногда целесообразней использовать процесс
известный как молекулярная перегонка (разд. 3.4.5) Молеку
лярная перегонка применяется для очистки термически
нестабильных или высокомолекулярных жидкостей
Меры безопасности, при всех вакуумных пррегонках
соблюдайте все меры предосторожности по работе с вакуум
ными системами (разд. 1.3.3).
ум-
ПРИБОРЫ. Типичная установка для перегонки при noi
женном давлении изображена на рис. 3.22. Она
сколько важных отличий от установки для перегонки при
атмосферном давлении. В данном случае используют те же
перегонные колбы, но снабженные двугорлой насадкой Кляйзе
на, одно горло которой предназначено для термометра, а
другое для капилляра (см. ниже). Небольшие объемы
жидкостей (100 мл или менее) удобнее перегонять в колбе Кчяй-
зена (перегонная колба вместе с насадкой) (рис. 3.21,6),
нежели в отдельной колбе с насадкой. Колбы Кляйзена
выпускаются объемом от Ю до 100 мл. Присоединение короткой
колонки Вигре (рис. 3.21 ,й) дает небольшое, но важное
увеличение эффективности. В начале перегонки колбы должны
быть заполнены наполовину или на две трети.
В колбу опускают тонко вытянутый капилляр, так чтобы
он не доходил до ее дна примерно на 2-3 мм. Когда система
находится под вакуумом, через капилляр пропускают воЗДУ*
(или азот); при этом образуется поток очень мелких пузыр*'
ков, которые способствуют спокойному кипению ("кипятиль-
нички" не работают под вакуумом). В тех случаях, когда
перегоняемая жидкость чувствительна к окислению, в калиЛ'
ляр при низком давлении (0,07 кг/смг) подастся азот
'
■дрпр не н очистка продуктов реакц и
Воздух п
ИГИ83ПТ Г
TepMoweip
ит взот !| Переходник с
завинчивающимся
, колпачком
Капилляр
К вакуум-насосу
Дистиллят
Рис 3.22. Перегонка при
Для каждой перегонки необходимо вытягивать новый
капилляр. Он должен быть как можно тоньше, но тем не менее
пропускать поток мелких пузырьков без потери вакуума.
Технику вытягивания капилляра легче продемонстрировать,
чем описать, но основные приемы даны в разд. 3.4.8,в.
Холодильник Либиха можно подсоединить к приемнику
посредством простого алонжа, изображенного на рис. 3.22, но
при этом главное неудобство заключается в том, что при
замене колбы происходит потеря вакуума и нарушение
равновесия и непрерывности перегонки. Этого можно избежать,
используя более сложные системы сбора дистиллята. Наиболее
Ра ространенная из них - это "элонж с несколькими
отводами (рис. 3.23,о), обычно известный как "паук",
позволяющий поворачивать каждую колбу в положение для приема
Дистиллята. Шлиф, по которому происходит вращение, дол-
*е быть смазан Существует множество аналогичных конст-
Р>кцнй вращающихся сборников фракций, причем некоторые
чз них содержат до 12 накопительных трубок. Альтернативой
является треугольник Перкина, изображенный на рис. 3.23,6.
XSSSlj
Холодильник
Рис. 3 23. Типы алоижей- а - иращлемый иноголвпчатый "гтаук".^И
форштос Перкина.
Используя это приспособление, путем переключения
кранов в правильной последовательности можно ваменять
приемник, для того чтобы изолировать колбу, которую необходиИ
удалить, подать к ней воздух, заменить ее и затем вак уми-
ровать новую колбу перед подключением ее к системе. I
Перегонную колбу следует нагревать на масляной бане,
как это описано для перегонки при атмосферном пиления
Фазд. 3.4.2).
При сборке приборов удобно пользоваться пластиковый»
утиками рис. 2.1) для скрепления колб с алонжеы
("пауком') и паука с холодил ком В проти ном случа они
гут п съ еще подключения вакъл-ыа. Для систем,
работающих при давления водостр иного насоса (10 20 мм
рт. ст.), вакуумную смазку обычно не наносят (за исклнтче-
ни м места »ращения "паука"), но для перегонки при
высоком вакууме, когда применяется масляный насос, необходЯ
умеренная вакуумная смазка.
ВАКУУМНАЯ ПЕРЕГОНКА
Жидкость, предназначенная для перегонки, не должна Ш
держать летучих растворителей типа эфира, в против*
случае при подаче вакуума неожиданный спад давления
I
дцтт! TieiiHC и очистка продуктов реакции 123
дйЗй/л к неконтролируемому вспениванию содержимого колбы
и его выбросу через насадку в холодильник.
Установите прибор, принимая- во внимание приведенные
выше замечания; залейте жидкость в колбу (не более чем на
две трети объема), но не вставляйте на этой стадии
капилляр (иначе он заполнится жидкостью) и не нагревайте колбу
до подачи вакуума. Проверьте весь прибор. Проверьте
вакуумную систему, перед тем как подсоединить ее к
перегонному аппарату, чтобы убедится в том, что она дает
необходимый вакуум. Если все в порядке, вставьте "капилляр,
подсоедините вакуумную систему и медленно вакуумируйте
перегонный аппарат После того как будет достигнуто требуемое
дашк-ние, можно приступать к нагреванию масляной бани.
При сборке дистиллята записывайте давление и температур-
ны интервал для каждой фракции. По завершении
перегонки уберите источник нагревании и дайте прибору остыть,
перед тем как стравить вакуум.
Вакуум стравливают не путчем отключения насоса, а
открыванием, крана в системе для впуска воздуха (разд.
3.4 8. стр. 137).
Примите все необходимые меры, чтобы приемные колбы
«с упали при стравливании вакуума.
3.4 3 Фракционная перегонка
Эффективность разделения можно заметно улучшить,
используя колонку для фракционной перегонки В лучших
случаях это позволяет разделить жидкости, температуры
кипения которых различаются только на несколько градусов
Цельсия. Дифлегматор (см , например, рис. 3 24) состоит из
- ановленной над перегонной колбой вертикальной трубки,
заполненной материалом с большой поверхностью, на которой
происходит частичная конденсация пара. Необходимо, чтобы
Установился самый тесный контакт между поднимающимся
"аром и спускающимся конденсатом, который непрерывно
стекает через нижнюю часть колонки в перегонную колбу. В
•Тих условиях в дифлегматоре устанавливается равновеси
при котором пар в верхней части состоит главным образом
из более летучего компонента Описываемый процесс известен
Как контактная ректификация; его эффективность зависит от
поверхностной площади стекающей внутри дифлсгматора
жидкости
ХОЛОДИльщ,
Термометр
Колонка
Вигрв
Капилляр для
подачи воздуха
или азота (при
перегонке под
вакуумом)
\
Термометр
— Холодильник
Вакуумная
Конструкции дифлсгматоров и работа с ними - это
сложная и интересная область, которая рассмотрена здесь только
вкратце.
а) Типы колонок
Дифлсгматоры бывают нескольких общих типов. К
первому типу можно отнести те из них, в которых высокая плв»
iodine и очистка иродуктов реакции
Моторчик
Вакуумная
—-рубашка
Скрученная
вращающаяся
лента
Рис. 3 25 фракционные колонки;
си скрученной вращающейся лентой
К мешалке в
перегонной колбе
i виде концентрических -
пшдь поверхности достигается путем заполнения
цилиндрической колонки (рис. 3.24,6) неплотным материалом, например,
кольцами Рашига (короткие отрезки стеклянных трубок),
спиралями Фенке (маленькие одно- или двухвитковые
спиральки из стекла или металла) или сетками из нержавеющей
стали. Их иногда называют "насадками", поэтому колонки
этого типа получили название "насадочные" колонки.
В колонках второго типа имеются прокладки, сделанные
из заранее отлитых сеток из нержавеющей стали.
Третий тип представлен тонкопленочными колонками с
относительно открытым внутренним пространством. Они имеют
такое устройство, что пары проходят близко к поверхности
стекла, по которому стекает конденсат. К ним относятся ко-
аонкн Вигре, представляющие собой простую стеклянную
трубку с направленными вниз вырезами (рис. 3.24,с). .Наибо-
126 Глдва
лее эффективными из них являются колонки из концентру.
ческих трубок (рис. 3.25,о); в таких колонках пар
поднимается через узкое кольцевое пространство между двумя
стенками, по которым стекает конденсат, направляемый
спиральными канавками. В продаже имеется целый ряд таких коло-
нок (колонка "Spallrahr") -
Наконец, существуют колонки с вращающейся лент
(рис. 3.25,6), в которых конденсат и пары смешиваются и
помощи быстро вращающейся скрученной ленты
6) Характеристики колонок
Представляют интерес три характеристики колонок.
1) Эффективность
Эффективность колонки измеряется в теоретических тара
ках (одна теоретическая тарелка - это эффективность н
каждой элементарной "ступени" перегонки). Для сравнении
колонок различных типов пользуются понятием "высота,
эквивалентная теоретической тарелке" <ВЭТТ) Колонки с
насадками (спирали и кольца), колонки в виде
концентрических трубок и колонки с вращающейся лентой имеют
высокий КПД (ВЭТТ в пределах 0,5 - 2 См). Эффективность
колонки Вигрс намного ниже (в лучшем случае ВЭТТ
достигнет 15 см).
2) Удерживающая способность
Это объем жидкости, необходимый для того, чтобы
покрыть внутреннюю поверхность колонки и ее насадку. Тая
как эту часть жидкости нельзя извлечь, се считают
"утерянной" при перегонке. Колонка с насадкой имеет высокуй
удерживающую способность и поэтому не подходит для перс-
гонки малых объемов вещества.
Колонка Вигре, колонки с концентрическими трубкам
или вращающейся лентой имеют низкую удерживающую
способность.
3) Перепад давлений
Этот важный при вакуумной перегонке показатель харЯ
теризует давление в колбе, необходимое, чтобы прогнать гн
Перегонная система "Fisher Spaltrohr", выпускаеман фирмой "ОЛ
Scientific" Midtilelon в г Манчестере, Великобритании.
I ддрление и очистка продуктов реакции 127
-и через колонку в головку. Очевидно, что чем меньше
перепад давлений, тем глубже вакуум в колбе и тем ниже
температура кипения; это важно при перегонке неустойчивых
при нагревании соединений. Насадочные колонки имеют
высокий перепад давлений, тогда как колонки Вигрс, колонки
из концентрических трубок и с вращающейся лентой -
низкий
Для уменьшения потерь тепла при работе фракционной
колонки используют либо вакуумную рубашку (иногда
посеребренную) (см., например, рис. 3.24,а), либо рубашку с
электрическим обогревом (рис, 3.24,6).
4) Головки
Значительного улучшения простой перегонки можно
добиться, если просто поместить между колбой и обычной иа-
I садкой Вюрца (являющейся головкой колонки) колонку Вигре
или короткую вдеадочную колонку (рис. 3.24,0); это устрой-
I ство пригодно для перегонки при атмосферном давлении или
в вакууме. В такой установке наилучшее разделение
обеспечивается при медленной перегонке.
Когда требуется очень высокая эффективность разделения,
обычную головку заменяют устройством, сочетающим
обратный холодильник с частичным отбором (рис. 3.24,6), которое
I лозволяет контролировать флегмОвое число (отношение
количества жидкости, отбираемой в качестве дистиллята, к тому
ее количеству, которое возвращается в колонку). Обычно,
I чем медленнее отбор, тем выше чистота отбираемого
вещества Для вакуумной перегонки требуется более сложная
головка, облегчающая многократный фракционнный отбор.
ИСПОЛНЕНИЕ При перегонке на колонке с
обогреваемой рубашкой в верхней части колонки обычно
поддерживается температура примерно на 5°С ниже температуры
перегонки Первоначально система работает под общим обратным
холодильником, чтобы сначала полностью увлажнился наса-
дочный материал, а затем установилось начальное равновесие.
Это может занять несколько часов; на равповесие указывает
температура в верхней части колонки, достигающая
минимального значения. Более точное показание получают с
помощью ГЖХ (разд 4 1.2), причем некоторые головки имеют
Разделенные перегородкой отверстия для ввода проб, чтобы
постоянно контролировать состав конденсата в головке. Затем
инают отбор, используя при этом флегаовое число, по
рому судят о необходимой полноте разделения (и снова
ПКХ чрезвычайно облегчает установление соответствующей
скорости отбора)
IBBjJ
3.4.4. Перегонка мккроколичеств
При очистке небольших количеств жидкости (1 - 10 мл)
перегонкой важно правильно выбрать прибор, в противно^
случае возможны большие потери вещества, образующего
пленку на поверхности колбы, холодильника и других
стеклянных предметах. Эти потери большого количества вещества
в приборе, являющиеся безвозвратными, представляют
главную проблему. Их можно Свести к минимуму, сечи
использовать специально сконструированный м нькнй прибор с
минимальной поверхностью. Все маленькие перегонные колбы
имеют грушевидную форму, что позволяет свести к
минимуму термическое разложение.
В приборе типа изображенного на рис. 3.26, в котором
пальчиковый холодильник соединен с приемным переходни-
^ Термометр
Капилляр
"Ч Вращающийся 1
четырехлапчатыи |
приемник
Рис. 3.26. Перегонка микроколичеств
продуктов реакции
^
Рис. 3 27 Перетопка микроколичеств
ком, можно перегнать от 10 до 1 мл раствора. При работе в
верхней части приведенного интервала можно использовать
колбу типа показанной на рис. 3.2б,а с короткой елочной
насадкой для проведения фракционирования и с капилляром
Однако для перегонки меньших количеств более
предпочтительна простая грушевидная колба (размером до 2 мл, рис.
3 26,6). В таких маленьких колбах нерационально
использовать капилляр; для предотвращения вспенивания в нижнюю
часть колбы пометают стекловату или порошок пемзы.
Очень маленькие объемы (до 1 мл> можно перегонять на
иборе. изображенном на рис. 327. Перегон мую жидкость
ещают на дно наружной трубки и нагревают на
маленькой масляной бане. Дистиллят конденсируется в охлаждаемом
■• ьчнкьиОМ холодильнике и собирается в стеклянном ковши-
к Чтобы предотвратить вспенивание, используют стекловату,
Как обсуждалось выше. В альтернативном методе при
перегонке используют трубку с коротким боковым отводом,
который действует как приемник (см. рис. 3.32,(5). Жидкость
помещают в трубку с помощью длинной пипетки Пастера или
лучше в ампулку для образца, как в случае сублимации
(разд. 3.4.7). После перегонки боковой отвод отрезают.
Рис. 3.2В. Перегони, «трети.™ . „о»*»»»* прием™».
Другой метод умгаьшитъ потери - убрать холодильник и
перепоить непосредствен™ в охлаждаемый прие„Г„к ф„"
А28). Этот метод дает хорошие результаты, но его недоста
Дробленый
сухои лед
Моторчик
(необязательно)
-.. ■ Р^кцумнЯ
Перегоняемое
вещество
/ Вторая
/ фракция
Диафрагменная
перегородка
Рис 3.29 Перегонка в шарообразной трубке (Кюге.и.рора)
i очистка продуктов реакции
J
состоит б том, что при замене приемника нарушается
вакуум. Развитие и усовершенствование этого принципа на-
ило отражение в создании популярной установки для пере-
joh Кюгельрора (рис. 3.29). В ней можно перегонять от
20 мт до 20 г вещества. Так как она позволяет осуществлять
перегонку быстро при минимальном нагреве, это делает ее
особенно полезной для перегонки термически неустойчивы*
соединений и высококипящих масел. Однако на такой
установке нельзя проводить эффективное фракционирование; луч-
pie всего применять ее для перегонки смесей веществ с
температурами кипения, различающимися по крайней мере на
20 - 30° С. Перегонка ьедется в наборе шарообразных трубок
■рис 3.29), соединенных с помощью шлифовых соединений.
Этот прибор устанавливают на скользящую подпорку, так
чтобы в печь можно было поместить любое количество
шаров В более дорогих установках трубки вращаются
моторчиком (подобно колбе в роторном испарителе). Печь имеет
точный i-смпературный контроль, в более простом исполнении
она сделаш иг металла, а в более дорогом - из стекла
(покрытого металлической нагревающей пленкой). Входное
отверстие печи снабжено перегородкой типа диафрагмы, которую
можно закрывать в любом месте соединения шарообразных
трубок.
Перегоняемую жидкость наливают в крайнюю трубку (или
при перегонке больших объемов в стандартную 25- или
50-мл круглодонную колбу). Очень мвленькис количества
нелетучих жидкостей растворяют в эфире или дихлорметане и
затем удаляют растворитель на роторном испарителе. В
шарик можно поместить стекловату, чтобы предотвратить
вспенивание, но это значительно снижает скорость перегонки и
не рекомендуется для термически нестабильных веществ. При
работе с "роторным" прибором Кюгельрора в этом нет
необходимости.
При простой перегонке, когда ожидается только одна
кция, в печь вставляют только один крайний баллон,
после чего закрывают диафрагму, которая защищает наружные
пловы от нагревания Затем осторожно, во избежание
вспенивания, подают вакуум и начинают вращение (если это
можно). После этого поднимают температуру печи до тех
пор, пока не начнется перегонка Для жидкостей с низкой и
средней температурой кипения необходимо охлаждать баллон,
в котором происходит конденсация. Для этого
непосредственно над баллоном (рис. 3.29) укрепляют широкую трубку и
олняют ее дробленым сухим льдом (разд. 2.1,4). При вы-
полнйши фракционной перегонки в печь помещают все
баллоны, ва исключением одного крайнего, который служит при-
133 Гляадд
емннком первой фракции. Затем по мере необходимости бал-
лоны выдвигают для сбора последующих фракций.
Фракционная перегонка малых объемов из-за потерь
остается неэффективной, что исключает использование насадоч-
ных колонок (разд. 3.4.3). Однако существенные улучшения
могут быть достигауты при использовании тонкопленочных
колонок (разд. 3.4.3), для которых эта проблема менее остра.
Некоторое увеличение эффективности наблюдается при работе
с васадкой Вигре (рис. 3.26), желательно снабженной
вакуумной рубашкой для уменьшения потерь тепла Очень высокой
эффективности удалось достичь (но намного большей ценой)
при помощи микросистемы Шпалтроура Spallrohr (разд.
3.4.3, стр. 126), в которой перегоняют 1 - 10 мл вещества;
эффективность этой системы 15 тарелок, потери составляют
0,1 мл. Очень маленькие количества (<1 мл) нельзя
эффективно фракционировать; их надо разделять препаративной га-
зо-жидкостной хроматографией (если вещество достаточно
летуче) или препаративной тонкослойной хроматографией, либо
какой-либо из форм колоночной хроматографии.
3.4.5. Молекулярная перегонка
Некоторые жидкости нельзя подвергать обычной вакуумной
перегонке при давлениях до 10 ' мм рт. ст. либо из-за
высокой температуры кипения, либо из-за термической
нестабильности. В большинстве случаев такие вещества можно
очистить молекулярной перегонкой при очень низком
давлении (10"э - 10"* мм рт. ст.) в специально сконструированном
приборе, где пространство между поверхностью жидкости и
холодильником меньше, чем средний свободный пробег
молекулы. Самая простая конструкция такого прибора изображена
на рис. 3.30,й. При молекулярной перегонке молекулы
переходят прямо с поверхности жидкости на холодильник без
прохождения воздушного барьера, и поэтому только
незначительное их количество возвращается в жидкость. Обычное
понятие "температура кипения" здесь неприменимо, так как не
существует больше равновесия между жидкостью и паром.
На простом приборе (рис. 3.30,о) или его модификации с
"ковшиком" (рис. 3.27) можно перегонять только очень
маленькие образцы. Большие образцы обрабатывают на
приборах для молекулярной возгонки (один из них схематнчве
изображен на рис. 3.30,0, в которых перегоняемое вещество
растекается тонкой пленкой по внутренней стенке полого на
гревателя и охлаждается на трубке в центре. В высоком
вакууме осуществима также перегонка из баллона в баллон,
как в установке Кюгельрора (рис 3.29); в установке с "вра
Непсрегнанный
" остаток
Рис. 3.30. а - молекулярная перегонка или сублимация микроколичеств,
6 - молекулярная перегонка по типу "падающей пленки".
вдающимся баллоном" возможность достижения высокого
вакуума ограничена из-за вакуумного затвора.
Необходимый высокий вакуум можно полупить с помощью
диффузионного насоса, соединенного с роторным масляным
асосом. При перегонке микроколичеств вещества вакуум
Нужно подавать очень плавно во избежание вспенивания, а
мпературу бани поднимать очень медленно, чтобы избежать
толчков при кипении.
3.4.6. Перегонка с водяным паром
Некоторые органические соединения, почти не
смешивающиеся с водой, можно отделить от нелетучих вагрязнений,
включая неорганические примеси, перегонкой с водяным па-
Пративоразбрызги
вандцая насадка^
Пар —.
Рис. 3.31 Перетопка
ром. Этот процесс, по существу, являющийся совместной
перегонкой с водой, обычно заключается в пропускании струи
пара через горячую смесь перегоняемого вещества и воды
Если вещество характеризуется высоким давлением паров (5
мм рт. ст. или более при 100°С), оно переносится с паром
и, будучи несмешивающимся с водой, легко отделяется от
дистиллята. Одним из преимуществ перегонки с паром
является то, что она ведется при температуре кипения воды. Это
позволяет очищать высококипящне вещества, чувствительные
к нагреванию, не выдерживающие обычной перегонки. Перс-
гонка с водяным паром имеет также важное значение при
отделении летучих продуктов от смолистого вещества, которое
часто образуется в ходе органической реакции и не
удаляется перегонкой или кристаллизацией.
Прибор, обычно используемый для перегонки с паром
больших количеств вещества, показан на рис. 3.31.
Перегоняемое вещество помещают в 500-мл круглодонную колбу,
снабженную предохранительной насадкой. Такав форма
насадки сводит к минимуму возможность переноса нелетучего
вещества в холодильник в результате вспенивания или
разбрызгивания. Выходное отверстие для паров соединено с эф- \
фективным холодильником; дистиллят собирается в большой,
колбе подходящего размера.
Пар можно получать из генератора (БУДЬТЕ ВНИМЛ
1
i р^л целен не и очистка продуктов редкими. 135
ТЕЛЬНЫ) или из паропровода. В последнем случае обычно
необходим сифон для удаления конденсата перед подачей
пара в перегонную колбу. Обычно перегонку стремятся
провести как можно быстрее, но не превышая при этом емкости
системы конденсации. Так как в перегонной колбе всегда
конденсируется некоторое количество водяного пара, во время
перегонки ее следует нагревать на сетке, заполнив примерно
наполовину. Если перегоняемый образец является
низкоплавким таердым веществом, следует внимательно следить за тем,
чтобы холодильник не забился. Любое вещество,
скапливающееся в холодильнике, можно быстро удалить из него путем
отвода воды из охлаждающей рубашки на несколько минут;
в результате этого пар перенесет расплавленное твердое
вещество в приемник.
Для перегонки с паром микроколичеств вещества часто
удобно генерировать пар in situ кипячением водного раствора
или суспензии органического соединения. Во время перегонки
иногда приходится добавлять горячую воду, чтобы раствор не
становился слишком концентрированным. Это делают, не
разбирая прибора, установив на боковом отводе перегонной
колбы капельную воронку.
3.4.7- Сублимация твердых веществ
Сублимация - один из самых удобных методов очистки
небольших количеств органических твердых веществ В этом
процессе вещество испаряют при нагревании до температуры
ниже его температуры плавления {Твя) и пары
конденсируются непосредственно в твердое состояние иа холодном
приемнике. Только органические соединения с относительно
высоким давлением паров могут возгоняться при атмосферном
влении при температурах ниже их Т. Их относительно
немного, и подавляющее большинство сублимируется только
при сильно пониженном давлении. Этот метод лучше всего
подходит в случае неполяриых соединений, которые обычно
более летучи, чем полярные с аналогичной молекулярной
массой; он особенно ценен для соединений, которые
гигроскопичны или расплываются. Его преимущества заключаются в
простоте, легкости исполнения и минимальных потерях веще-
ва. Главный его недостаток в том, что, как и при перегон-
е, процесс разделения зависит от разницы в давлении
паров, и поэтому соединения со сходной летучестью будут воз-
няться вместе. Типичный, полностью стеклянный
сублиматор показан на рис. 3.30,« (его можно также использовать
Для молекулярной перегонки (разд. 3.45)). Для работы с
количествами от нескольких миллиграммов до I г применяют
приборы различных размеров
Сщ^
ПРОЦЕСС СУБЛИМАЦИИ. Сублимируемое вещество аз-
мельчают в порошок и помещают на дно трубки, в которую
вставлен пальчиковый холодильник Расстояние между днои
сублиматора и кончиком холодильника должпо быть
небольшим во достаточным, чтобы сублимат не загрязнился ил
разбрызгивании таердого вещества, С этой проблемой часто
сталкиваются, если перед сублимацией не удалены
растворители и другие летучие вещества. В большинстве случаев
достаточно расстояние около 1 см.
После вакуумнрования на водном или масляном насосе
сублиматор погружают в мелкую масляную баню и
постепенно нагревают до тех пор, пока на поверхности пальчикового
холодильника не образуется пленка сублимата. Температура
не должна подниматься слишком быстро, иначе вещество
может разбрызгаться По завершении сублимации осторожно
отключают вакуум и вынимают пальчиковый холодильник.
Затем сублимированное вещество соскребают микрошпателсм на
часовое стеклышко или плотную фильтровальную бумагу
либо смывают его растворителем (хотя это и приводит к потере
того преимущества, которое дает непосредственное получения
чистого твердого вещества).
Очень малые количества лучше возгонять в длинной
стеклянной трубке (диаметром около 9 мм), нагреваемой в
наклонном, электрически обогреваемом металлическом блок!
(рис. 3 32 а) Неочищенное вещество переносят в маленьку
Термометр
Образец в
ампулке
( мнкроколичеств с использованием нагрева
- труби для перегонки мнкроколичеств.
-
1шд2"'|"|е " очистм "раду"0' «дани
137
ампулку, которую помещают на дно трубки Маленькая про
(ючка из ваты в верхней части трубки предотвращает
попадание загрязняющих органических веществ из резинового
шлчига Трубку устанавливают под упом 45° и затем
медленно вакуумируют, чтобы частички образца не засосало в
вс нкж> часть трубки Очень летучие твердые вещества кон-
де ируются на значительном отрезке трубки, поэтому иногда
тр бку необходимо обмотать охлаждающим змеевиком или
влажной ватой, чтобы обеспечить локальную конденсацию
При удалении сублимата очень осторожно стравливают
вакуум трубку с обеих сторон от сублимата обрезают, затем
удаляют сублимат, соскабливая его микрошпателсм.
3 4.8 Приложения
а) Водоструйные вакуумные системы
Водоструйный насос (аспиратор) позволяет легко получить
средний вакуум (10 - 25 мм рт ст.). Нижний предел
зависит от давления водяных паров и, следовательно от их
температуры, но, как правило, при использовании обычной
одной воды достигает 10 - 12 мм рт ст Обычная
вакуумная система изображена на рис. 3 33 Все резиновые
шланги должны быть толстостенными, выдерживающими давление.
При перегонке необходим манометр, который информирует о
работе системы. Давление измеряется по разнице между
двумя уровнями ртути. При работе водопроводный кран всегда
полностью открывают, невозможно контролировать вакуум,
регулируя скорость потока воды. При работе таких насосов
существует опасность того, что вода может быть "затянута"
в вакуумированный прибор, если давление воды падает
Этого можно избежать, установив водяную ловушку, как
показано на рис. 3 33. По этой же причине кран не следует вы
почать до тех пор, пока не будет спущен вакуум после
открытия краиа для впуска воздуха
б) Ротационные масляные насосы
Ротационные масляные насосы, часто используемые в виде
рсносных насосов в учебных лабораториях, обеспечивают
ы окий вакуум (до 0,01 мм рт. ст.) при перегонке и сушке
уумная система (рис 3.34) должна включать
охлаждаемую ловушку, чтобы масло в насосе не загрязнялось парами
растворителя. Перед подсоединением вакуумной системы к
ибору ее следует испытать. Эта процедура заключается в
следующем Удостоксрьтссь, что охлаждаемая ловушка пуста
Давление *
Рис. 3.33. Вакуумная система с водоструйным насосом.
И се шлиф слегка смазан силиконовой смазкой. Медленно по- I
грузите ловушку в сосуд Дьюара с жидким азотом, вакройте I
краны А и Б, чтобы изолировать систему, и затем сразу же I
включите насос.
Меры безопасности. Не оставляйте ловушку в жидком
азоте, если система не под вакуумом, иначе в ловушке I
сконденсируется жидкий кислород. Смеси легко еоспламеня-1
ющихся растворителей и жидкого кислорода могут вызвать I
сильный взрыв.
Проверьте вакуум. Если он не достигает 1 мм рт. ст.,
есть подозрение на подсос. Наиболее распространенной
причиной подсоса является образование каналов в смазке кранов i
или испорченные резиновые шланги. Если подсоса нет, то
.
црцяме и пчиелса продуктов реакции
Рис. Э 34. Вакуумная система с ротационным масляным насосом.
проблема может заключаться в присутствии паров
растворителя в масле насоса. Их можно удалить, открыв на некоторое
время воздушный балластный клапан, чтобы выдуть
растворители. При этом вакуум будет слабым, но он должен ваметно
улучшиться, когда клапан снова закроют.
Когда будет получен удовлетворительный вакуум,
подсоедините систему и медленно провакуумируйте ее. Чтобы снять
вакуум, сначала отключите вакуумированную систему, затем
ройте кран А, впустите воздух, отключите насос и выньте
ушку из жидкого азота. Никогда не кончайте работу
простым выключением насоса, оставив ловушку под вакуумом,
иначе масло из изеоса засосет в ловушку
> Воздушные капилляры
Воздушный капилляр для вакуумной перегонки вытягивают
из куска трубки, как показано на рис. 3-35. Чтобы
размягчить стекло трубки, газовое пламя должно быть обогащено
кислородом и иметь достаточно высокую температуру, не вы-
ывая, однако, слишком сильного размягчения стекла, что де-
ет непригодным его для работы. Описываем, я методика
известна как метод "двойной вытяжки". Сначала медленно
вращайте трубку в пламени, пока она не размягчится;
продолжайте вращение, пока стенки не станут толще, и ватем
медленно растяните за оба конца до образования равномерного
nzt
Место нагрева
:
Рис 3.35 Вытягивание воздушного капилляра для вакуумной перегя
а - капиллярная трубка, б первое сужение, в после второго вытяпшаюн
сужения (рис. 3.35,6). Дайте стеклу слегка охладиться и за-*
тем нагрейте сужение в кончнхе пламени, пока стекло не
размягчится, и снова осторожно потяните за концы, до
образования второго очень тонкого сужения (рис. 3.35,в).
Отрежьте капилляр до необходимой длины, дайте ему остыть и
испытайте его, погрузив один конец в растворитель (только не
возле пламени) и аккуратно продув. Хороший воздушный
капилляр даст только струйку очень мелких пузырьков
Нагревайте трубку не ближе, чем на 2 см к шлифу,
иначе он деформируется; в этом случае капилляр следует
отдать стеклодуву, чтобы напаять еще кусочек трубки.
4 ХРОМАТОГРАФИЧБСКОБ РАЗДЕЛЕНИЕ
' СМЕСЕЙ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
"Хроматография" охватывает ряд очень эффективных
экспериментальных методов разделения смесей органических
соединений. В этой главе хроматографические методы разделены
на две группы: а) маломасштабные "аналитические" методы
качественного н количественного анализа смесей и б) более
ма табные "препаративные" методы разделения в
количествах от миллиграмма до нескольких грамм.
Профессиональные навыки, необходимые для
использования этих методов, обычно накапливаются в учебных
лабораториях годами. На ранних стадиях обучения основное внима-
ни уделяют тому, как обращаться с оборудованием, поэтому
в методиках будут указаны точные рабочие параметры
(природа сорбентов и элюирующих растворителей, насадки
колонок в газо-жидкостной хроматографии и т п.), которые
предстоит использовать. По мере приобретения опыта
экспериментатор начинает все лучше понимать, как эти параметры али-
яют на процесс разделения, к учится оптимизировать их для
различных смесей и продуктов реакций. Построение каждого
из приведенных ниже разделов ставит своей целью облегчить
этот процесс познания. Сначала рассматриваются простые
операции, а затем оптимизация параметров разделения Для
последнего важно понимать общие принципы, лежащие в
основе процессов разделения; они описаны в общих чертах в
отдельном разделе (разд. 4.3.1).
4.1. АНАЛИТИЧЕСКИЕ МЕТОДЫ
4 1. Тонкослойная хроматография
Тонкослойная хроматография (ТСХ) - один из наиболее
широко используемых хроматографических методов - имеет
громное значение для быстрого качественного анализа
смесей, контроля реакций и определения рабочих параметров,
которые следует использовать в препаративной колоночной
роматографии. Препаративная ТСХ рассматривается в разд.
а) Общее описание
Разделение проводят на плоской пластинке, покрытой тон-
Рис Л 1 Тонкослойная хроматография. _
ким слоем сорбента - силикагелем или оксидом алюминия
(pHi. 4.1,о). Разделяемую смесь, растворенную в
соответствующем растворителе, наносят в виде капель на пластинку
(рис, 4.1,6) и после испарения растворителя помещают пла-И
стинку в проявителыгую камеру (рис 4.1,в), в которую на-
лито немного растворителя. Растворитель поднимается по
слою сорбента под действием капиллярных сил. При этом
различные соединения, находящиеся в смеси, поднимаются с
разными скоростями в зависимости от их сродства к
сорбенту. По достижении растворителем верхнего слоя сорбента
соединения в идеальном случае должны полностью разделиться
(рис. 4.1,г). Процесс разделения - это одна из форм жидко-
стно-адсорбционной хроматографии, которая более подробно
обсуждается в разд. 4 3 1
6) ТСХ-пластинки и сорбенты
атастинки для ТСХ состоят
„_.„,.. «о ичдлижки ооычно стекла,
а иногда пластмассы или толстой алюминиевой фольги, на.
1пиаюграФическое разделение смесей 143
которую нанесен тонкий слой сорбента толщиной примерно
0,25 мм (рис. 4.1,о) Сорбентом обычно служит либо силика-
гель, либо оксид алюминия с размером частиц до 10 -
30 мкм, к которым добавлено связующее (до 10% гипса или
крахмала), обеспечивающее прочность слоя. Пластинки
бывают двух основных типов: многоразового использования и
одноразовые.
Пластинки многоразового использования изготавливают
из толстого стекла, на которое при помощи специального
оборудования наносят сорбент, а затем после использования
пластинки счищают его для повторного покрытия. Пластинки
бывают разных размеров от 20 х 5 см до дешевых
пластинок, сделанных из микроскопных предметных стекол (75 х
25 мм), пригодных для контроля реакций. В учебных
лабораториях обычно используют пластинки с заранее нанесенным
слоем; для научно-исследовательской работы детальное
описание процесса покрытия приведено в разд. 4,3.3.
Одноразоаые пластинки поставляются различными
изготовителями с готовым слоем сорбента, нанесенным на основу
из тонкого стекла, пластмассовую пластинку или
алюминиевую фольгу. Особенно удобны пластинки последних двух
типов, так как большие листы легко разрезать ножницами на
полоски нужного размера-
Одноразовые пластинки дают лучшее разрешение, чем
пластинки многоразового использования, причем для
большинства разделений вполне достаточна пластинка высотой 5
см При работе с ними отрезают полоску размером 5 х 20
см, проводят карандашом слабую линию на расстоянии 5 мм
от длинной нижней кромки, чтобы указать, куда наносить
капли образца, и затем нарезают куски соответствующетс
размера (см., например, рис. 4.19).
Готовые пластинки имеют определенную активность (разд
4 3 2), обычно от 11 до 111 степени, их следует хранить над
силикагелем
в) Нанесение образца
Образец обычно наносят с помощью капилляра в виде 1 -
2%-ного раствора в летучем растворителе типа дихлормстана
или эфира (избегайте полярных растворителей, например
этанола); раствор удобнее всего готовить в маленьком пузырьке.
Капилляр для нанесения капель легко сделать из капилляра
Для определения температуры плавления; для этого среднюю
часть капилляра помещают в пламя микрогорелки, а затем
осторожно растягивают и ломают в месте растяжения.
Место нанесения капель в зависимости от размера пла-
144
ава
стинки должно быть на расстоянии 1 см от нижнего края на
больших (5 х 20 см) пластинках и около 5 мм на маленЯ
ких одноразовых пластинках. Важно также наносить капЯ
достаточно далеко от нижнего края (рис. 4.1,6 и в), чтобЯ
они не погружались в проявляющий растворитель.
Перед нанесением образца заполните капилляр, погрузив
его конец в раствор (капилляр заполняется раствором под
действием капиллярных сил), затем слегка прикоснитесь
заполненным кончиком к сорбенту, стараясь при этом не
повредить его поверхности, чтобы не вызвать некоторого
искажения капли при проявлении хроматограммы. Важно
наносить как можно более маленькую каплю, а это требует
тонкого капилляра. Перед проявлением хроматограммы дайте
капле полностью высохнуть.
г) Проявление хроматограммы
Меры безопасности. Б этой операции обычно
используются летучие легковоспламеняющиеся растворители,
поэтому ее нужно проводить вдали от источников возгорания
предпочтительно в вытяжном шкафу.
Хроматограмму проявляют погружением нижнего края
пластинки в проявляющий растворитель в сосуде соответствуй
щего размера (рис. 4.1 ,в). Большие пластинки проявляют в
специальных камерах (20 х 5 см), а маленькие - в широкЯ
горлых бутылях с завинчивающимися крышками. РекомендЯ
ции по выбору проявляющего растворителя приведены в разя
"ж" ниже. Камеру изнутри следует частично выложить
фильтровальной бумагой, которая погружалась бы в раствори-
тель, чтобы создать атмосферу, насыщенную парами раствЯ
рителя, и свести к минимуму испарение с пластинки. При
проявлении хроматограммы сначала залейте в банку стольЯ
растворителя, чтобы пятно образца оказалось над его поверЯ
ностью, а затем опустите в банку пластинку, стараясь
расположить ее вертикально.
Это легко проделать с большими пластинками, но с ма-|
ленькими легкими одноразовыми пластинками необходимо
обращаться с большой осторожностью, используя щипцы или
пинцет, для опускания и вынимания их из камеры. На
время проявления камера должна оставаться закрытой. Когда
фронт растворителя поднимется почти до верха пластинкЯ
выньте ее из камеры и сразу же карандашом или шпате лед
отметьте положение фронта растворителя. Высушите пластиЯ
ку в вытяжном шкафу.
уроматографическое разделение смесей |45
д) Просмотр хроматограммы
Если соединения в образце окрашены, то после
проявления их легко различить визуально, однако для бесцветных
соединений требуется какой-либо метод визуализации.
Наиболее употребительный метод - введение в слой
сорбента неорганического флуоресцентного агента ( 0,5%). В
продаже имеются такие одноразовые пластинки. При
освещении такой пластинки УФ-лампой (254 нм) сорбент начинает
светиться бледно-зеленым или голубым светом, а
органические соединения, которые гасят флуоресценцию, выделяются
в виде темных пятен.
Меры безопасности, Хотя специальные УФ-лампы для
ТСХ имеют низкую мощность, их все же следует
вмонтировать в смотровой бокс или колпак, чтобы защитить
глаза наблюдателя от УФ-излучения и чтобы дневной свет
не попадал в камеру.
Еще один^ распространенный метод состоит в
использовании склянки с иодом. Она представляет собой емкость
такого же размера, как и камера для проявления, куда помещено
несколько кристалликов иода.
Если сухую проявленную пластинку поместить в камеру
на несколько минут, пары иода растворяются в органических
"пятнах", окрашивая их в коричневый цвет, до тех пор пока
вся пластинка не потемнеет.
Какой бы метод ни использовался для визуализации,
положение пятен для проведения дальнейших измерений
необходимо пометить карандашом или шпателем (рис. 4.1,г).
Необходимо отметить, что, хотя описанные выше методы
в целом довольно эффективны, одни соединения
"проявляются" сильнее, другие слабее, а некоторые иногда не видны
совсем. Поэтому не принимайте относительную яркость пятен
даже в качестве грубой характеристики относительных
концентраций в смеси.
Имеется целый ряд реагентов, нанесение которых из
пульверизатора на пластинку после проявления хроматограммы,
окрашивает соединения определенных классов в различные
цвета [1, 2].
В качестве склянки можно использовать эксикатор или другую емкость
с плотно притертой крышкой. - Прим. перев.
К)6729
146
Хйавал
ВНИМАНИЕ! Идентичность хроматографического
поведения даже на разных сорбентах с различными растворШ
телями нельзя рассматривать как абсолютное
доказательство структурной идентичности. Изучаемое соединение
необходимо выделить методом препаративной хроматографиш
и затем идентифицировать, например, методом ИКС или
ЯМР.
ж) Выбор проявляющего растворителя (подвижной фазы)
Высота, на которую поднимается по пластинке "пятно"!
соединения, зависит от сродства последнего к сорбенту и
силы (полярности) проявляющего растворителя (см. табл. 4.5).
Полярные соединения (спирты, кетоны и т. п.) сорбируют-1
ся сильно и поэтому плохо продвигаются при использован!^
слабых проявляющих растворителей типа гексана, тогда как
неполярный углеводород типа нафталина хорошо поднимаете
по пластинке. Наилучший растворитель приходится находитИ
методом проб и ошибок. Силу растворителя легче всего регИ
лировать, используя смеси сильного и слабого растворителе^И
Обычно начинают с 1:1-смеси эфира (сильный растворител^И
с гексаном (или 60/80 петролейным эфиром) (слабый
растворитель) и затем соответственно меняют соотношение.
Очевидно, что, если чистый эфир не способен поднять "пятна"
вверх по пластинке, необходимо перейти к более "сильному"
растворителю (табл. 4.5).
В случаях когда разделению препятствует сильное переД
крыванне двух пятен, даже если они хорошо поднимаются по 1
ург1матографическое разделение смесей 147
пластинке (/?, 0,6 - 0,9), можно попытаться улучшить
разделение двумя путями: 1) изменением химической природы
проявляющего растворителя при сохранении его силы (это
изменит значение к' и, следовательно, а; см. стр. 204) или
2) изменением природы неподвижной фазы, например
заменой оксида алюминия на силикагель и наоборот. На
практике при наличии разных пластинок последний путь часто
является более эффективным и быстрым.
В случаях когда смесь содержит несколько полярных и
неполярных соединений, может потребоваться двукратное
проявление - сначала в слабом растворителе для отделения
неполярных соединений (полярные остаются на старте), а затем
в более сильном растворителе для отделения полярных
соединений (неполярные при этом сходятся вместе у фронта
растворителя) .
4.1.2. Газо-жидкостная хроматография
Газо-жидкостная хроматография (ГЖХ) используется для
разделения смесей "летучих" соединений, т. е. практически
соединений с молекулярной массой до 500. Она имеет
огромное значение как для качественного, так и для
количественного анализа и благодаря очень высокой чувствительности
позволяет обнаруживать микрограммовые (до 10~6 г)
количества соединений.
а) Общее описание
Принципиальная схема газового хроматографа показана на
рис. 4.2,й. Само разделение смеси происходит на колонке с
неподвижной фазой - термически стабильной нелетучей
жидкостью, нанесенной на инертное гранулированное твердое
вещество,
В качестве неподвижной фазы обычно используют
силиконовые масла, углеводородные смазки и высокомолекулярные
полиэфиры. Через колонку непрерывно пропускают
подвижную фазу, представляющую собой газ (обычно азот),
называемый газом-носителем. Колонку помещают в термостат,
который можно нагревать до 300 ° С. На одном конце колонки
устанавливают ввод для газа-носителя и устройство для
впрыскивания пробы (инжектор) с резиновой мембраной,
через которую шприцем вводят образец.
В колонку впрыскивается маленький объем смеси, где она
испаряется иногда с помощью флаш-нагревателя. Компоненты
смеси сами распределяются между неподвижной жидкой
фазой и подвижной фазой, с которой они передвигаются вдоль
°YKU Детектор г-
{мембрана "v-pL.^
Газ-носитель гзсг^ -J cz
_аЛ
Выход газа-но-i
»ситеяя и про-:
дукгое сгорания
Хромэтограмма
Газовая струя,
выходящая из
колонки
Рис. 4.2. Газо-жидкостная хроматографи:
колонки (в идеальном случае с различными скоростями),
разделяются на отдельные группы и элюируются один за другим
через детектор. Это устройство реагирует на присутствие
органического соединения в потоке газа-носителя и посылает
электрический сигнал к самописцу или дисплею.
Таким образом, каждое соединение смеси дает пик,
записываемый на ленте самописца (см. рис. 4.2,й и 4.5). Обычно
более летучие соединения быстрее проходят через колонку.
Процесс разделения более детально обсуждается в разд. 4.3.1.
б) Прибор
На ранних этапах обучения методу ГЖХ прибор для
конкретного эксперимента в учебной лаборатории обычно
подготовлен заранее с необходимой колонкой, температурой
термостата и скоростью потока газа-носителя. Выбор этих
параметров рассматривается в последующих разделах.
Большинство газо-жидкостных хроматографов снабжено
пламенно-ионизационным детектором (ПВД) (рис 4.2,6).
В нем струя газа-носителя из колонки смешивается с
водородом в пропорции 1:1, и получаемая при этом смесь подается
в изолированную форсунку в корпусе детектора, где и
сжигается. Туда же подается воздух, чтобы поддержать пламя
и удалить продукты сгорания. Конец форсунки служит одним
Хроматографическое разделение смесей
149
из пары электродов; другим электродом является цилиндр,
окружающий пламя. При сгорании очередного из элюируемых
органических соединений оно ионизируется. Образующиеся
заряженные частицы увеличивают удельную
электропроводность между электродами, и через зазор протекает слабый
электрический ток (10-J2 - 10"7 А). После усиления он дает
сигнал, который подается на самописец.
Пламенно-ионизационные детекторы популярны благодаря
своей высокой чувствительности (до 10"1г r/мл); кроме того,
они дают линейную зависимость между высотой пика и
количеством сжигаемого соединения.
Некоторые газовые хроматографы снабжены детекторами
по удельной теплопроводности (катарометрами), которые
контролируют изменения удельной теплопроводности потока газа-
носителя, когда в нем присутствует органическое соединение.
Их чувствительность намного ниже, чем у
пламенно-ионизационных детекторов, и поэтому они реже устанавливаются в
современных приборах.
Усилитель снабжен аттенюаторным (ослабляющим)
переключателем, который контролирует величину выходного
сигнала подобно регулятору громкости на радио. Это позволяет
регулировать высоту пиков или приводить их в соответствие
со "шкалой", если они очень большие, либо увеличивать
высоту пика побочных компонентов, так чтобы они были
достаточно большими для точного измерения.
Аттенюатор градуируют в интервале от xl (наиболее
чувствительное положение) до х500 000 (наименее
чувствительное), причем каждый пик на диаграмме должен быть
помечен в соответствии с ослаблением, использованным при его
записи,
в) Ввод образца
Образцы обычно впрыскиваются в виде растворов в
летучем растворителе. Обычно удобно работать с концентрацией
1%, но можно использовать и намного более разбавленные
растворы.
Меры безопасности. При работе с горячим
хроматографом невоспламеняющиеся растворители типа дихлормета-
на безопаснее, например, чем эфир, как при растворении
образца, так и при промывании шприца.
НАСАДОЧНЫЕ КОЛОНКИ. Впрыскивание производится
мнкролитровым шприцем (обычно емкостью 10 мкл). Эти
150
Глава 4
Рис. 4.3. Головка для ввода пробы в газовом хроматографе.
шприцы хрупкие и очень дорогие; обращайтесь с ними с
большой осторожностью и не оставляйте их на движущейся
диаграммной ленте. Длина иглы меняется от прибора к
прибору, но она должна быть достаточно большой, чтобы
образец попадал в насадку колонки, а не в пустое пространство
над ней (рис. 4.3).
Перед вводом сначала удостоверьтесь, что шприц чистый;
для этого погрузите иглу в какой-либо растворитель, налитый
в коническую колбу (но не в бутыль, где хранится
растворитель), и несколько раз осторожно наполните и опорожните
его. Если движение поршня затруднено или забита игла,
обратитесь к своему преподавателю, так как может
понадобиться более основательная промывка.
Затем погрузите иглу в раствор образца, несколько раз
подвигайте поршень вверх и вниз, чтобы удалить воздух из
цилиндра, и затем лабернте необходимое количество (обычно
1-5 мкл) раствора.
Вытрите иглу бумажной салфеткой, проверьте, чтобы в
жидкости не было каких-либо значительных воздушных
пузырьков (достаточно трудно и необязательно удалять
последний пузырек) и произведите впрыскивание.
ХроматограФическое разделение смесей 151.
При прокалывании мембраны обязательно левой рукой
придерживайте иглу, чтобы придать ей устойчивость, а затем
введите ее на полную длину. На некоторых приборах
имеется маркировочная кнопка, которая делает отметку на
диаграмме, указывая время впрыскивания (см. рис. 4.5), Если
этого нет, сделайте на диаграмме пометку карандашом.
КАПИЛЛЯРНЫЕ КОЛОНКИ. Эти колонки могут
выдерживать только 1% нагрузки насадочной колонки. Устройство
ввода пробы (инжектор) обычно снабжено делителем потока,
который направляет несколько процентов от введенной пробы
в колонку и выпускает остальную часть. В более
современных приборах инжекторы не имеют делителя потока (см,
руководство к прибору).
г) Запись хроматограммы •
Прежде чем будет получена удовлетворительная хромато-
грамма со всеми пиками на шкале, может понадобиться одна
или более пробных попыток. Для первого раза установите
аттенюатор в среднюю позицию на шкале (если в вашем
руководстве по эксплуатации не указано что-либо другое) и
затем наблюдайте за пиками по мере их появления.
Если вы увидите, что пик уходит за край диаграммной
ленты, то по мере приближения пера к верхнему краю
бумаги постепенно регулируйте аттенюатор до тех пор, пока не
будет достигнута вершина пика, и отметьте на диаграмме
используемое положение аттенюатора; сделайте это для каждого
пика. Когда запись хроматограммы закончится, просмотрите
все представляющие интерес маленькие пики на хроматограм-
ме и решите, какое требуется усиление, чтобы получить их
приемлемую высоту (выше половины шкалы).
Затем снова впрысните тот же самый объем образца и
перед появлением каждого пика настраивайте аттенюатор на
соответствующее значение. В некоторых отдельных случаях
все пики можно записать при одинаковом усилении. Если
прибор отрегулирован должным образом (см. разд. 1, стр.
163), изменение усиления при нахождении пера самописца
на базовой линии не должно приводить к какому-либо
заметному сдвигу пера.
Такой способ регулировки интенсивности пика с помощью аттенюатора
не подходит при использовании электронного интегратора для измерения
площади пика; см. инструкции по применению интегратора.
(а)
J
Рис. 4.4. формы пиков в ГЖХ.
ФОРМА ПИКА. При записи хроматограммы понаблюдайте
за формой пиков. Искажение (рис. 4.4,6) обычной
симметричной формы (рис. 4.4,а) может быть обусловлено двумя
обстоятельствами. Более просто исправить лишнюю загрузку
образца, которая приводит к тому, что перо медленно
поднимается в начале пика и после прохождения вершины круто
падает вниз к базовой линии. Для устранения этого
необходимо снизить количество образца и соответственно увеличить
усиление.
Труднее исправить "хвостатые" пики, фронтальная сторона
которых выглядит нормально, но которые имеют длинный
вытянутый "хвост" перед возвращением пера к базовой линии.
Это обычно является симптомом несовместимости между
образцом и неподвижной фазой (см. разд. "з" ниже). Однако
иногда они обусловлены неправильным впрыскиванием
образца: либо низкой температурой зоны впрыскивания (проверьте
флаш-нагреватель), либо слишком короткой иглой шприца.
Хроматограмма трехкомпонентной смеси (все пики
получены при одинаковом усилении) показана на рис. 4.5. В
последующих двух разделах она будет использована для
иллюстрации применения ГЖХ в качественном и количественном
анализе.
д) Идентификация методом ГЖХ
Для данного набора условий (тип колонки, температура,
расход газа-носителя) каждое соединение характеризуется
j; ромаго» рафич? ;кое разделение смесей
Площадь пика
М — Ю./р X Ь
Рис. 4.5. Хроматограмма трехкомпонентной смеси, полученная на
газовом хроматографе.
временем удерживания (Г, - Т3, рис. 4.5) . Поэтому, чтобы
идентифицировать какое-либо соединение в смеси, сравнивают
Если быть более точными, то следует рассчитать удерживаемый объем,
т. е. объем газа-носителя, необходимый для вымывания соединения
(удерживаемый объем равен времени удерживания, умноженному на расход
газа-носителя). Однако в учебной лаборатории не всегда можно измерять расход
газа-носителя.
154
Глава 4
его время удерживания с временем удерживания
"подлинного" образца соединения (вещества сравнения), записанного
отдельно. Метод усиления пика позволяет удостовериться в
том, что время удерживания одинаково. Для этого добавьте к
образцу немного вещества сравнения и еще раз снимите хро-
матограмму. Если рассматриваемый пик увеличивается в
размере, но форма его не искажается, это подтверждает, что
время удерживания не изменилось.
На занятиях в учебной лаборатории одинаковое время
удерживания считается достаточным основанием для
идентификации соединения, ио в исследовательской и
профессиональной работе этого недостаточно. Сомнения происходят из
того факта, что два совершенно различных соединения могут
иметь одно и то же время удерживания. Если вещество
сравнения и "неизвестное" соединение имеют одинаковое время
удерживания на двух различных неподвижных фазах (разд.
"з", см. ниже), то это служит ,более убедительным
доказательством, ио даже и тогда необходимы другие
доказательства идентичности, например получение масс-спектра на масс-
спектрометре, спаренном с газовым хроматографом. Иногда
возникает необходимость в проведении препаративной ГЖХ и
получении спектра ЯМР.
е) Количественный анализ
ГЖХ широко используется для количественного анализа
смесей. Это относительно простая процедура, но она занимает
некоторое время, так как требует градуировки прибора на
образцах чистых соединений, которые предстоит
идентифицировать.
Первое, что необходимо сделать, - это измерить площади
пиков на хроматограмме. Для этого лучше всего использовать
электронный интегратор, но он не всегда имеется, особенно в
учебных лабораториях. Однако, если пик достаточно
симметричен, хорошие результаты дает и ручное измерение. Пример
измерения площади пика М(АМ) показан на рис. 4.5.
Площадь пика получают в результате умножения его высоты (Л)
на ширину основания на половине высоты (н*1/2)- Для
получения высокой точности измерения скорость диаграммы должна
быть достаточно высокой во избежание получения очень
узких пиков. Диаграммный измеритель с подсветкой облегчает
измерение ширины и делает это намного точнее, чем при
измерении линейкой.
Площадь пика прямо пропорциональна массе вещества,
проходящего через детектор. Таким образом, из рассмотрения
пика X на рис. 4.5 следует, что Wx пропорциональна Ах, где
Хроматографическое разделение смесей 155
Wx - масса вещества, соответствующего пику с площадью Ах.
Константа пропорциональности между этими двумя
величинами кх является коэффициентом отклика соединения X. Таким
образом:
wx = кхАх
В принципе кх можно было бы найти путем впрыскивания
известного количества Wx и измерения площади пика Ах.
Однако впрыскивать точные количества очень трудно,
поэтому данный метод практически неосуществим. Обойти эту
проблему позволяют два метода: 1) метод внутреннего
стандарта, пригодный для анализа всех смесей, и 2) метод
внутренней нормализации, обычно используемый только тогда,
когда все компоненты имеют одинаковый коэффициент
отклика (константу пропорциональности).
1) Метод внутреннего стандарта
В этом методе к анализируемой смеси добавляют
известное количество внутреннего стандарта, чтобы получить пик,
отделяемый от пиков неизвестных соединений, например пик
М на рис. 4.5. Содержание X и Y в смеси можно вычислить
измерением отношений площадей пиков Ах/Ам и Ау/А^,
Эта процедура выглядит следующим образом:
1. Методом проб и ошибок найдите подходящий
внутренний стандарт. Он должен быть чистым, стабильным и нере-
акциоииоспособным по отношению к компонентам смеси. Для
этого часто используют углеводороды типа бифенила, дибеи-
зила или нафталина или различные эфиры.
2. Добавьте к смеси внутренний стандарт в количестве,
достаточном для получения пика такого же размера, как и
пик неизвестного соединения.
3. Сделайте не менее трех впрыскиваний смеси, чтобы
получить воспроизводимые значения (±3 %) Ах/Ам и Ау / Ам.
(Если воспроизводимые значения, не получаются, возможно,
имеются какие-либо трудности с прибором (см. разд. 1 ниже)
или вы вводите слишком концентрированные растворы.)
4. Соотношение пиков X и М:
wx = кхАх и wM = кмАм
таким образом:
WX/WM = (kx/kuHAx/AM)
Отношение кх/кц известно как градуировочный
коэффициент Кх соединения X.
Следовательно:
156
Глава 4
В этом уравнении массовое соотношение во введенном
образце (Wy/Wji/) то же самое, что и во всей смеси
независимо от дозы впрыска. Таким образом, если JVM - это масса
внутреннего стандарта, добавленного на второй стадии (см.
выше), тогда Wx - это количество соединения X в смеси.
Следовательно, Wx можно вычислить по уравнению (4.1),
если известно Кх.
Процесс нахождения Кх прост. Готовят градуировочную
смесь, содержащую известные количества соединений, и после
ее впрыска измеряют отношение площадей пиков. Затем по
уравнению (4.1) вычисляют Кх. На практике для
определения Кх обычно готовят несколько градуировочных смесей и
вычерчивают график зависимости WX/WM от Ах/Ам. Если
необходимо определить несколько неизвестных соединений, то
все их следует ввести в градуировочную смесь и вычертить
градуировочный график для каждого. Градуировочные смеси
должны быть приблизительно такого же разбавления, что и
неизвестная смесь, т. е. пики необходимо получать при
одном и том же положении аттенюатора хроматографа. Чтобы
получить согласующиеся значения отношения площадей
пиков, нужно сделать ие менее трех вводов проб.
2) Метод внутренней нормализации
Этот метод количественного ГЖХ-анализа применяется
реже в случае, когда элюируются все соединения смеси, давая
пики, которые можно измерить. В отличие от метода
внутреннего стандарта его нельзя использовать в тех случаях,
когда реакционные смеси содержат высокомолекулярные, смо-
лсобразные или полимерные продукты, нелетучие в ГЖХ.
Самый простой случай применения метода внутренней
нормализации - это когда все компоненты смеси имеют один
и тот же коэффициент отклика. Здесь можно упомянуть
гомологические ряды соединений или смеси изомеров. В данном
случае концентрации компонентов можно определить по
формуле
Х(%) = iAx/Aj}100
где Ах - площадь пика компонента X, а АТ - общая
площадь пиков всех компонентов (за исключением пика
растворителя). Перед тем как использовать этот метод, важно
показать, что все компоненты имеют одинаковый коэффициент
отклика. Этот метод можно модифицировать с целью учета
при расчете различных коэффициентов отклика, но в общем
метод внутреннего стандарта намного более удобен.
смесей
ill
ж) Запись данных ГЖХ
На каждой хроматограмме должна быть сделана
следующая запись:
1) номер эксперимента или номер образца;
2) количество введенной пробы;
з) используемая колонка, т. е. неподвижная фаза и
процент ее нанесения, материал носителя, длина;
4) расход газа-носителя;
5) рабочая температура;
6) усиление, используемое для каждого пика;
7) скорость диаграммы.
В качестве примера можно привести такую запись:
Опыт 1, образец 1/4,
1 мкл;
200 см 10% смазки апиезои APL на хромосорбе W;
150°С; N2, 60 мл/мин;
все пики при 50*102;
диаграмма, 60 см/ч.
3) Выбор неподвижной фазы и рабочих параметров
Для получения хорошего разделения неизвестной смеси
необходимо методом проб и ошибок подобрать оптимальные
колонку и рабочую температуру, -используя общие замечания,
приведенные ниже. По мере накопления опыта это
становится делать легче и быстрее. В целом, процесс осуществляется
в следующих направлениях: подберите колонку с
определенным количеством нанесенной неподвижной фазы (разд. 1 и
2) из доступного материала. Установите ее в прибор с
соответствующим расходом газа-носителя (разд. 4) и прикиньте
рабочую температуру (разд. 2 и 3). Введите образец,
посмотрите на результаты и затем, варьируя рабочую температуру,
попытайтесь добиться достаточного разрешения пиков за
минимальное время анализа. Если пару перекрывающихся пиков
нельзя разделить варьированием температуры, необходимо
поменять неподвижную фазу и начать все сначала. Если смесь
содержит известное число соединений, тогда ясно, когда
будет достигнуто требуемое разделение. Однако совершенно
новая смесь, полученная в исследовательской работе, может
содержать неизвестное число компонентов и тогда важно
проделать процедуру, описанную в разд. 2, чтобы ие упустить
какого-либо компонента.
1) Выбор неподвижной фазы
ТРАДИЦИОННЫЕ НАСАДКИ КОЛОНОК. Общие прин-
Таблица 4.1. Основные типы неподвижных фаз для ГЖХ
Полярность
Пример
Неполярные
Промежуточной
полярности
Полярные
Апиезонные смазки
Силиконовые каучуки
Силиконовые жидкости
Силиконы с полярными
группами
Полифениловые эфиры
Полизтиленгликоли
(карбоваксы) в широком
диапазоне молекулярных
масс
Полиамиды
Эфиры и полиэфиры
APL
SE30/E301, OV1 (диметил)!
OV17 и ОУ25(фенилметил)|
ХЕ60, (цианоэтилметил), I
QF1 (фтор)
Versamide 900
Неопентил гл ико льсу к -
цинат (NPGS), диэти-
ленгликольсукцинат
(DEGS), полиэтилен-
гл икол ьад и 1 нтат
(PEGA)
ципы разделения ГЖХ обсуждаются в разд. 4.3.1,6, который]
следует внимательно прочесть, прежде чем пользоваться реко-1
мендациями этого раздела. В основном следует ожидать, что!
более летучие вещества пройдут через колонку быстрее, чем|
менее летучие, поэтому впрыснутые соединения будут элюи- ]
роваться в порядке возрастания их температуры кипения. Од- I
нако этот порядок сильно нарушается специфическими взаи- 1
модеиствиями между соединениями и неподвижной жидкой]
фазой. Они зависят от природы соединения
(полярное/неполярное) и природы неподвижной фазы, чьи свойства также
можно трактовать с точки зрения "полярности" (основные
типы перечислены в табл. 4.1). В продаже имеется несколько
сотен неподвижных фаз, но многие из них имеют сходные
свойства. В большинстве лабораторий хранятся только
некоторые основные типы неподвижных фаз, и этого хватает для
проведения большинства разделений. Некоторые
специфические примеры приведены в табл. 4.2, где неподвижные фазы
расположены в порядке возрастания их полярности.
Каждая конкретная жидкая фаза имеет рекомендуемую
максимальную рабочую температуру (табл. 4.2), выше
которой колонка начинает течь (происходит потеря неподвижной
фазы) или возникает термическое разложение.
Правильный выбор неподвижной фазы для конкретного
.^^графическое разделение смесей
Таблица. 4.2. Некоторые распространенные неподвижные фазы
Неподвижная фаза Максимальная рабочая
температура, ° С
Сквален
Апиезон L
Силиконовая смола (метил) SE30
Силикон (метил) OV1
Силиконовая смола (фенил) SE52
Силикон (фенил) OV17
Силикон (фенил) OV25
Силикон (фтор) QF1
Силиконовая смола (нитрил) ХЕ60
Поли-л!-фениловый эфир (6 колец)
Неопентилгликольсукцинат (NPGS)
Карбовакс 20М
Карбовакс 1000
Этиленгликольфталат
Диэтиленгликольсукцинат (DEGS)
Цнаноэтилсахароза
150
250
250
300
250
300
300
225
225
250
200
220
150
200
190
125
разделения не прост и может состоять из многих проб и
ошибок. Данные по удерживающей способности приводятся во
многих литературных источниках, из которых их можно
почерпнуть при работе с известными соединениями или
смесями, но при анализе новых соединений хроматографист
должен руководствоваться некоторыми основными положениями и
константами Рохршнейдера Мак-Рейнольдса [За, б], дающими
количественную оценку полярности и сведения по
селективности. Обычно "подобное удерживает подобное", например,
на "неполярных" колонках неполярные соединения элюируют-
ся более медленно, чем полярные соединения с той же
температурой кипения. На "полярных" колонках полярные
соединения удерживаются сильнее по сравнению с неполярными
с такой же температурой кипения.
При анализе ГЖХ полярных соединений типа спиртов,
аминов и карбоновых кислот могут возникнуть трудности,
связанные с тем, что эти соединения имеют тенденцию
давать пики "с хвостами" (рис. 4.4,6) в результате сильной
адсорбции на активных центрах поверхности насадки. Этого
можно избежать, по крайней мере частично, использованием
насадки, дезактивированной промывкой кислотой и силаниза-
цией (такие насадки часто маркируются AW/DMCS). Это
особенно важно для колонок, содержащих менее чем 5% не-
Глава 4j
Г
подвижной фазы. Наиболее подходящими для полярных соеЯ
динений являются специальные насадки, обсуждаемые ниже. 1
ПОРИСТЫЕ ПОЛИМЕРНЫЕ ГРАНУЛЫ. Эти материалы,!
используемые как в качестве насадки, так и в качестве
неподвижной фазы, выпускают различных типов (например, по-
рапак и хромосорб). Они обеспечивают прекрасное разделение
полярных соединений, сводя к минимуму "хвосты" пиков, а
также пригодны для анализа газов.
СВЯЗАННЫЕ НЕПОДВИЖНЫЕ ФАЗЫ. Неподвижны*
жидкие фазы (НЖФ) могут быть связаны с поверхностыбИ
насадки. В некоторых случаях, когда количество связанном
НЖФ очень низко (0,2%) (см. ниже), поверхность почтИ
полностью дезактивирована, так что полярные соединения не
дают "хвостов". Связывание, однако, может изменить
характеристики разделения неподвижной фазы.
2) Процент нанесения неподвижной фазы и рабочая I
температура
Количество неподвижной фазы, нанесенной на насадку, I
для обычных аналитических колонок обычно составляет от 1
до 10% по массе. Наносить можно до 25% неподвижной фаЛ
зы, но выше этого насадка становится липкой. При данном
температуре объем удерживания соединения пропорционален
количеству неподвижной фазы; таким образом, например, со-Я
единение пройдет через колонку намного быстрее при нанесет*
нии 1 или 2% неподвижной фазы, чем 10%. Колонки с не-J
большим количеством НЖФ (1 - 5%), следовательно, имеютШ
огромное значение при анализе соединений с высокой моле-J
кулярнои массой и низкой летучестью; для их анализа наЯ
колонке с 10% неподвижной фазы потребовалось бы очень J
много времени.
На скорость прохождения соединения через колонку
оказывает также влияние температура колонки - наиболее легкви
контролируемый рабочий параметр газо-жидкостной хромато-J
графии. Чем выше температура, тем меньше время
удерживания данного соединения. Однако при повышении температур
ры происходит сближение соседних пиков, поэтому на
практике оптимизация температуры - это компромисс между соШ
крашением времени анализа и сохранением соответствующего*
разрешения между представляющими интерес соединениями. 4
Оптимальные температуру и процент нанесения неподвиж-Л
иой фазы для конкретного разделения можно подобрать толь-Л
ко методом проб и ошибок, но все же при этом советуемЯ
УР2^ятлграФическое разделение
Пии Площадь
8772
6362
4237
Изотермическая,
60°С
Колонка: 2,5% 0V1 на
хромосорбе G с размером
частиц 80-100 меш
Программирование
температуры: 2мин при бо°С,
затем повышение на гес
в 1 мин до 100°С
и
Рис. 4.6. ГЖ-хроматограмма без программирования температуры (а) и с
программированием температуры (6).
Руководствоваться следующим:
1. Рабочая температура колонки с 10% неподвижной
фазы должна быть на 30 - 50° С ниже температуры кипения
(760 мм рт. ст.) разделяемых соединений. При разделении на
колонках с более низкими количествами неподвижной жидкой
•разы для получения аналогичных объемов удерживания
потребуются, конечно, более низкие температуры.
2. Термически нестабильные соединения лучше всего
разделяются на колонках с небольшим содержанием НЖФ, что
162
Глава 4\
позволяет сохранять по возможности низкую рабочую темпЛ
ратуру.
3. Высокомолекулярные соединения с низкой летучестьЛ
разделяют на слегка загруженных колонках с небольшим
содержанием НЖФ, что позволяет работать при температураИ
ниже максимально допустимых для жидкой фазы; например,
тетрафенилиафталин (мол. масса 432) можно хроматографиро-
вать только на колонке с 1% НЖФ при 220° С.
4. Напротив, для разделения очень летучих соединений]
может потребоваться колонка с большими количествами
НЖФ (10 - 25%), обеспечивающая приемлемые временя
удерживания при минимальной рабочей температуре колонка
(обычно комнатной температуре).
5. Возможно использование программирования температу-J
ры. Для смесей, содержащих компоненты с широким
интервалом температур кипения, термостат колонки можно запроя
граммировать сначала на низкую температуру и разделите
летучие компоненты, а затем в процессе анализа повышатш
температуру, чтобы за приемлемое время элюировались менее
летучие компоненты (рис. 4.6). Программирование температуи
ры особенно полезно при анализе неизвестных смесей,
полученных в новых реакциях. В этих случаях наиболее важным
является то, что температура колонки программируется п«
всему доступному интервалу температур вплоть до предельЯ
ной температуры колонки. Смесь также необходимо
пропустить - через колонки как с высоким, так и с низким
содержанием неподвижной фазы. В противном случае легко "потш
рять" либо очень летучие продукты, выходящие вместе с
растворителем, либо малолетучие компоненты с большим
временем удерживания, которые могут остаться в колонке.
3) Температуры ввода пробы и детектирования
В большинстве газовых хроматографов конец колонки, ку-|
да вводят образец, можно нагреть выше температуры
термостата чтобы произошло мгновенное "флаш" испарение образ!
ца (рис. 4.2,о). Обычно температура "флаш"-нагревателя доя
жиа быть иа 50° С выше температуры термостата колонки,
ио при этом необходимо принять меры, чтобы избежать
термолиза лабильных соединений. В последнем случае темпераи
тура инжектора при впрыске должна быть такой же, каш
температура термостата колонки, а образец следует впрыски!
вать непосредственно в насадку колонки шприцем с длинной
иглой. В некоторых приборах у детекторов имеется отдельные
термостат; его температура также должна быть на 50° С
выше температуры термостата колонки.
УррцдгпграФическое разделение смесей 163
4) Скорость потока (расход) газа-носителя
Природа газа-иосителя ие оказывает большого влияния на
разрешение, ио его расход очень важен, так как линейная
скорость потока газа влияет на эффективность колонки. При
низких скоростях потока :*ффс ми юность низка, но с
возрастанием скорости потока достигает максимума и затем снова
снижается при высоких скоростях потока. Колонки диаметром
4 мм обычно работают при скорости 40 - 60 мл/мин,
диаметром 2 мм - при скорости 15 - 20 мл/мин, а
капиллярные колонки - при- скорости 1-2 мл/мин.
и) Установка прибора. Некоторые общие положения
Каждый прибор необходимо устанавливать в соответствии
с инструкцией по его эксплуатации. Однако существует
несколько общих положений, которые следует выполнить, чтобы
гарантировать хорошую работу. При количественном анализе,
в частности, необходимо подбирать правильные скорости
газового потока как для колонки, так и для пламенио-иоиизаци-
онного детектора (см. инструкцию). При неправильной подаче
воздуха отклик детектора будет непостоянным,
непоследовательным и нелинейным, а при неправильной подаче водорода
пламя ие будет стабильным. В системе не должно быть
никаких утечек; даже маленькие утечки вокруг мембраны или
в стыках колонки приведут к невоспроизводимым
результатам, поэтому, перед тем как включать нагреватель
термостата, стыки всегда следует проверять на наличие утечек
мыльным раствором (10%-ный водный раствор). Необходимо
также соответствующим образом настроить электрическую
систему. У каждого прибора своя методика настройки, но по
существу она состоит в установке усилителя и записывающего
устройства иа нулевые уровни и последующего поддержания
уровня фона регулировкой тока детектора. После правильной
настройки прибора перо самописца не должно значительно
отклоняться от базовой линии при изменении положения
аттенюатора.
После работы ГЖХ-колонок в течение определенного
времени они загрязняются небольшими количествами
малолетучих соединений, которые при нормальных условиях очень
медленно элюируются и дают нестабильную волнистую
базовую ^линию на самописце. Такие загрязнения удаляют
"очисткой" колонки путем ее нагревания (обычно в течение
ночи) при максимальной рабочей температуре (табл. 4.2.) и
малом расходе газа-носителя с отсоединенным от колонки
детектором.
Глава ^I
Емкость для
злюента
I I
^Ч p«-j Предохра-
\s) нательная
.. ^—^ |—I колонка
, Манометр Клапан,
регулирующий
давление
[—j Фильтр
Регулятор,
_| I выравнивающий
1 —> пульсацию при
подаче злюента
J Насос
Детектор
Хроматограчма
Рис. 4-7. Схема прибора для ВЭЖХ.
4.1.3. Высокоэффективная жидкостная хроматография
Высокоэффективная жидкостная хроматография (ВЭЖХ) н
"инструментальная" форма колоночной хроматографии для
малых количеств вещества. Этот универсальный метод можно
использовать для качествениого и количественного анализИ
смесей соединений всех классов - летучих и нелетучих,
термически стабильных и нестабильных, полярных и иеполяд
ных.
а) Общее описание метода
Основная схема используемого оборудования показана на
рис. 4.7. Колонка содержит неподвижную фазу - обычно сорГ
бент (например, силикагель или оксид алюминия) или
связанную^ жидкую фазу (см. стр. 209) - в виде очень мелкиЯ
частиц . Жидкая подвижная фаза (элюент) подается насосо»
Используются также другие виды хроматографии - ионообменная, сито-1
вая, - которые здесь ие рассматриваются.
^графическое разделение смесей
ысокого давления. Раствор смеси впрыскивается в верхнюю
Бясть насадки колонки микролитровым шприцем.
Содержащиеся в смеси соединения переносятся затем в нижнюю часть
колонки элюентом, причем различные соединения
перемещаются с разными скоростями в зависимости от их
относительного сродства к неподвижной фазе и движущемуся элюенту.
В идеале они разделяются полностью, давая отдельные
полосы (как показано на примере препаративной колонки на рис.
4.16), которые последовательно обнаруживаются детектором.
Более детально процессы разделения описаны в разд. 4.3.1.
Детектор реагирует на каждое проходящее через него
соединение и регистрирует пик с помощью самописца или в
самых современных приборах на дисплее. Результаты в
основном представлены в таком же виде, как в ГЖХ. Типичная
хроматограмма показана на рис. 4.13. Метод, описанный в
этом разделе, применяется для аналитических разделений,
при которых впрыскиваются субмиллиграмовые количества
образца. Такие разделения редко занимают больше 30 мин, а
часто длятся намного меньше.
б) Оборудование
Типичная установка для проведения ВЭЖХ (рис. 4.7)
состоит из следующих частей.
1) Колонка
ВЭЖХ колонка представляет собой трубку из
нержавеющей стали (диаметром 0,2 - 0,5 см и длиной 8-25 см). В
полости трубки имеются два металлических пористых
фильтра из нержавеющей стали, пространство между которыми
заполнено неподвижной фазой.
Трубка подсоединена к насосу, подающему растворитель, и
к детектору при помощи различных приспособлений. После
набивки колонки могут использоваться продолжительное
время при аккуратном обращении с ними. Обычно к входу
колонки подсоединяют короткую предохранительную колонку,
которая защищает дорогую аналитическую колонку от пыли,
смол и других примесей, присутствующих в образце.
Предохранительную колонку набивают тем же материалом, что и
основную.
2) Система подачи растворителя
Растворитель прокачивается через колонку обычно
поршневым насосом под давлением до 210 - 420 атм, но чаще
всего требуется давление всего в несколько десятков атмос-
Подача смешанного
растворителя на
колонку
Рис 4 8 Двухнасоснаы i
я градиентного элюированнв i
фер. В насосах этого типа установлена задвижка для вырав
нявания пульсации потока растворителя между ходам
поршня.
В простейшем приборе имеется один насос (рис. 4.7) да
изократического элюирования (постояншя сила растворителя]
В более современных приборах предусмотрено градиентное
элюированис, контролирующее изменение силы растворителя
во время разделения. В этом случае устанавливают два
насоса (рис, 4.8), один из которых подает сильный, а другой - 1
слабый растворитель сначала к камере смешения, а оттуда -
к колонке. Соотношение двух растворителей контролируется I
микропроцессором или компьютером, который в соответствии 1
с особенностями разделения запрограммирован таким образом, I
чтобы регулировать изменение соотношения между раствори- I
телями (градиент) либо ступенъчзто, либо непрерывно
Во всех случаях растворители перед использованием
необходимо дегазировать, чтобы свести к минимуму образование I
воздушных пузырьков в детекторной камере, и отфнльтро- I
вать, чтобы удалить пыль (фильтр обычно устанавливается I
на линии, подающей растворитель к насосу).
3) Детектор L
Детекторы реагируют на свойства раствора, подавая элект-^|
рический сигнал, когда соединение появляется в потоке рас-
«ческое разделение с
йтеля Обычно применяют два типа детекторов -
ультрафиолетовый (уф) и рефрактометрический Первый является
амного более распространенным и при 254 нм фиксирует
Яочти все классы соединений, кроме полностью насыщенных.
Очевидно, элюирующий растворитель должен быть прозрачен
„лл У ета. Рефрактометрические детекторы менее
чувствительны, но незаменимы при обнаружении простых и сложных
насьппгнных соединений. Они работают по
дифференциальному принципу, сравнивая показатель преломления раствора,
выходящего из колонки, с показателем преломления чистого
растворителя.
4) Система ввода пробы
Исследуемый образец в виде раствора (проба) вводят либо
непосредственно в верхнюю часть колонки шприцем (рис,
4 9), либо в поток элюента через кран (рис. 4.10).
ВВОД ПРОБЫ ЧЕРЕЗ МЕМБРАНУ. Этот метод
используют только при давлениях растворителя до 100 кг/см2.
Колонка снабжена головкой для ввода пробы (рис. 4.9) с
силиконовой резиновой мембраной, покрытой снаружи ПТФЭ Иг-
Элгсирующий
растворитель
Сетка из
нержавеющей стали
Шприц
Колпачок
направляющий иглу
рис 4 9 Головка для вводя пробы в ВЭЖХ.
От насоса
К колонке
Шприц
Место для ввода иглы
Дозировочная петля
От насоса
К колонке
Рис. 4.10. Кран для ввода пробы в ВЭЖХ (схема): а в положении
"загрузка"; б - в положении "ввод".
ла шприца должна быть такой длины, чтобы проба
впрыскивалась непосредственно в центр над верхней металлическоЯ
сеткой (фильтром) - обычно в слой мелких стеклянных
шариков над насадкой колонки. Шарики периодически заменяют
новыми, чтобы удалить кусочки резины, отламывающиеся
при прокалывании мембраны. Этот метод обеспечивает
высокое разрешение, ио имеет и свои недостатки, главный из ков
торых - частое забивание иглы шприца резиной мембраныш
Забивание иглы можно свести к минимуму, если заранее
проделать отверстие в мембране и использовать тупую иглу с
выпуклым наконечником, но все же это может вызыва-Я
трудности, особенно у неопытных операторов.
PJuSLcn/™bi ™РЕЗ КРЛН- КРЖЫ W --ода проб!
различаются по конструкции, но общий принцип их действи!
ддгряфическое разделение смесей 169
оказан на рис. 4.10. При загрузке растворенный образец
помещается при атмосферном давлении в специальную
дозировочную петлю, в то время как элюент подается иасосом
непосредственно в колонку. Когда кран установлен в положение
"ввод", элюент поступает через дозировочную петлю и
смывает образец в колонку. Дозировочные петли для
аналитической работы имеют объем от 5 до 20 мкл, ио бывают и
петли больших размеров, что позволяет использовать край для
препаративной работы на больших колонках. Кран может
работать в двух режимах: либо дозировочная петля заполнена
раствором полиостью, либо в большую петлю (20 мкл) из
микрошприца впрыснут небольшой объем (несколько
микролитров) и использован специальный затвор для иглы,
расположенный на одном из боковых отверстий. Последний метод
удобен для исследовательской работы, так как позволяет
варьировать объем образца, ио зато первый дает совершенно
надежный способ ввода абсолютно воспроизводимых по
объему проб.
в) Работа на приборе
1) Подготовка
После настройки и оптимизации ВЭЖ-хроматографа для
конкретного разделения работа иа нем очень проста. В
простейшем варианте с одним насосом (рис. 4.7)
последовательность операций следующая:
Меры безопасности. Обычно в хроматографии
применяются летучие и легковоспламеняющиеся растворители,
поэтому примите соответствующие меры предосторожности
(разд. 1.3) для предотвращения их возгорания и вдыхания.
Все операции по смешению следует проводить в вытяжном
шкафу.
** Дегазируйте элюирующий растворитель (или смесь
растворителей) перед тем, как поместить его в соответствующий
резервуар. Простейший способ - подсоединить колбу с
растворителем к вакуумной линии водоструйного иасоса с
одновременным перемешиванием (магнитной мешалкой) или
взбалтыванием ее содержимого. Это - быстрый метод, ио
°и может привести к нарушению соотношения в смеси
растворителей в результате испарения более летучего из
иих. Другой, более предпочтительный способ заключается в
том, что растворитель кипятят с обратным холодильником,
дают ему остыть и сразу же используют.
170
2
Доведите скорость потока растворителя до указанного 3HaJ
чения (оно зависит от диаметра колонки, например,
скорость 1-3 мл/мин подходит для колонки диаметром 5 мм!
и включите нягпг
4
и включите насос.
3. Пропускайте растворитель через колонку и детектор до]
тех пор, пока в элюате не исчезнут пузырьки воздуха. 1
4. В это время включите детектор и установите переключая
тель диапазонов в положение наименьшей чувствительно-!
сти.
5. Включите самописец.
Через несколько минут базовая линия должна стабилизи- I
роваться. Когда она установится, проверьте ее на всех
диапазонах переключателя чувствительности. Если иа этой стадиЯ
возникает какая-либо проблема, проконсультируйтесь с препон
давателем, так как она может быть обусловлена
загрязнением колонки или просто попаданием пузырька воздуха в
ячейку детектора.
2) Запись хроматограммы
Концентрацию образца в растворе следует отрегулировать!
таким образом, чтобы количество вводимого вещества
находилось в пределах от нескольких микрограммов до 0,5 мг.
Образец необходимо растворить в растворителе, который ие дает
пика (или один пик минимального размера) на хроматограм-
ме, т. е. прозрачный для УФ-света при использовании УФ-
детектора, а при обнаружении йа детекторе по показатели
преломления доба*ликте по возможности немного элюирующе-
го растворителя. Наилучшее разрешение получают при иен
пользовании небольших образцов и малых объемов пробыл
При вводе пробы шприцем непосредственно в колонку либо в
дозировочную петлю (через кран) ее объем обычно находится
в пределах 1-5 мкл, а при вводе через кран при
полностью заполненной дозировочной петле - 5 или 10 мкл.
ВВОД ПРОБЫ ЧЕРЕЗ МЕМБРАНУ. Промывание и за-1
полнение шприца описано в разделе, посвященном ГЖХ
(разд. 4.1.2). Прн вводе пробы игла должна проколоть плотно
прижатую мембрану (рис. 4.9), а затем при введении
раствора необходимо преодолеть высокое обратное сопротивление
(до 100 атм). Для облегчения введения иглы во время
сборки прибора мембрану можно проткнуть иглой большего
размера или использовать мембрану с заранее сделанным
проколом. При вводе пробы нужно крепко держать поршень,
чтобы он не выталкивался из цилиндра под давлением раствори-
чрекое разделение смесей
а игла должна полностью войти в прибор. Ввод пробы
через кран намного легче.
А ВВОД ПРОБЫ ЧЕРЕЗ КРАН. Общие правила введения
раствора через кран (рис. 4.10) приведены в п. "б" на стр.
168 ио при этом имеются некоторые второстепенные
различия' в зависимости от особенностей операции и типа прибора.
Сначала из большого шприца промойте дозировочную петлю.
Проверьте ее чистоту, заполнив петлю чистым растворителем.
Если на хроматограмме не появилось никаких других пиков,
кроме маленького пика растворителя (который может быть
положительным или отрицательным), тогда вводите пробу.
ОПТИМИЗАЦИЯ ОБЪЕМА ПРОБЫ И УСТАНОВКА
ДИАПАЗОНА. Введите первую пробу при среднем положении
регулятора диапазонов (аттенюатора) на детекторе. По мере
появления пиков настройте аттенюатор так, чтобы на
диаграмме прописался каждый пик, и сделайте рядом с ним
пометку. Иногда при высокой концентрации образца
появляются нежелательные нелинейные эффекты, поэтому если для
какого-либо из пиков требуется минимальное (наименее
чувствительное) положение регулятора, тогда либо разбавьте
образец, либо в следующий раз введите порцию меньшего
объема. Понаблюдайте также за формой пиков: несимметричная
форма свидетельствует о возможной перегрузке колонки (или
детектора), а также еще раз напоминает о том, что
необходимо уменьшить объем пробы (или концентрацию). Помните
также, что разрешение становится лучше, когда количество
образца уменьшается. Однако при очень низкой
концентрации образца наступает тот предел, когда шумы базовой
линии будут мешать при установке очень чувствительных
диапазонов, необходимых для очень низких концентраций
образца. Для оптимизации объема образца и установки диапазонов
может потребоваться введение нескольких проб. В конечной
пробе перед появлением каждого пика приведите диапазон к
соответствующему значению (см. ^примечание на стр. 151).
г) Качественный и количественный анализ
1) Идентификация соединений
Как и в ГЖХ, в ВЭЖХ каждое соединение при данном
наборе рабочих условий имеет определенное время
удерживания (или объем удерживания) (рис. 4.11). Эта особенность
ожег быть использована для предварительной
идентификации путем сравнения с "эталонным" образцом. Методы, опи-
Ш Главам
санные для ГЖХ (разд. 4.1.2), в равной степени относятся (|1
к ЮЖХ, а результаты имеют одну и ту же погрешность
Однако в ВЭЖХ легче получить дополнительную
информацию по идентификации вещества путем накопления образца
и снятия его масс-спектра.
2) Количественный анализ
Метод внутреннего стандарта (разд. 4.1.2) в равной степе- I
ни применим к ВЭЖХ. При использовании УФ-детектора его
следует установить на длину волны, при которой все исслеИ
дуемые соединения проявляют достаточно сильное
поглощение
Важно понять, что грубые оценки относительных коли-1
честв соединений в смеси, сделанные просто на основаниЯ
относительных площадей пиков, могут быть весьма
ошибочными, особенно при УФ-индикации. Это объясняется тем, чтЛ
при использовании детектора, фиксирующего длину волны,
коэффициенты экстинкции соединений могут очень сильнЯ
различаться.
Разрешение R
соединений А и Б
Эффективность
колонки
*=16©'
Неудерживаемое
растворенное веществ|
Рис. 4.11. Параметры удерживания в ВЭЖХ.
ijiirif^^ разделение смесей
173
д) Основы удерживания и разрешающей способности
W этот раздел содержит определения некоторых из терминов,
используемых в ВЭЖХ, и дает представление о том, как
влияют на разрешающую способность те переменные, которые
находятся под контролем оператора. В дальнейшем
предполагается, что читатель ознакомился с разд. 4.3.1 (стр. 204), в
котором приводятся общие принципы разделения.
разделение двух компонентов А и В изображено на рис.
4.11. Каждый пик имеет характеристическое время
удерживания Т при данном наборе условий, ио его обычно переводят
в объем удерживания V, т. е.
V = Т х (скорость потока элюента)
"Неудерживаемый" пик соответствует неполярному
веществу (растворителю для образца), которое прошло через
колонку с той же скоростью, что и элюирующий растворитель. Его
объем удерживания V0 известен как объем пустой части
колонки, иногда просто называемый "объемом колонки".
Разделение в ВЭЖХ обычно обсуждается в терминах к' для
каждого соединения (см. разд. 4.3.1, п. "а")). Значение к' легко
получить из данных по удерживанию, например:
* А = (VA " V/V0 "™ * А = <ГА " WT0
Таким образом, к'А представляет собой меру удерживания
соединения А (в объемах колонки) и известно как фактор
емкости соединения А. Очевидно, чем больше различие
между к' и к'в, тем лучше разделение между соединениями А и
В. Мера разделения определяется соотношением к'А/к'в,
которое известно как коэффициент разделения а. Для того
чтобы получить хорошее разделение, а должно быть как
можно больше.
Разрешение R двух пиков, т. е. мера того, наскольхо
хорошо они разделены, зависит не только от 7\ - Тв
(рис. 4. П), ио также от ширины этих пиков. Последнее
св«зано с эффективностью колонки, которую обычно
выражают через число теоретических тарелок N. Разрешение можно
выразить в значениях трех варьируемых переменных а> N к
R = \iCL - \)N1'2 [Jf/(1 + Jfc>] (4.2)
гДе k - среднее между k\ и к', а = *А/&В и N - число
еоретических тарелок колонки. Уравнение (4,2) дает
понимание того, как оператор может оптимизировать разделение.
—Едава t
*д = 4,9
*в =4,3
а = 1,13
■М
Рис. 4,12. Неполное разрешение двух компонентов » ВЭЖХ.
е) Оптимизация разделения
Рассмотрим ситуацию, когда первоначальная попытка раз!
деления соединений А и В не дала полного разрешения (рис.
4.1Z). Ниже мы рассматриваем, что может сделать оператор
для улучшения ситуации посредством изменения N> к' и а.
1) Число теоретических тарелок N
Эффективность колонки существенно зависит от размера!
частиц насадки и от плотности ее упаковки. Однако для
дайной конкретной колонки она также зависит от скорости
потока, а этот параметр легко регулируется оператором.
Оптимальные значения зависят от внутреннего диаметра (в. д.)
колонки: 0,1-1 мл/мин для в. д. колонки 2 - 3 мм и 1 -
J мл/мин для в. д. 3 - 5 мм. Однако R зависит только от
W/ поэтому улучшение разрешения может быть
недостаточно эффективным.
2) Фактор емкости к'
Объемы удерживания соединений (и, следовательно,
значения к) зависят от силы элюирующего растворителя (табл.
4 5). Используя более слабые растворители, можно увеличить
объемы удерживания, ио ключевым вопросом по-прежнему J
огсэ3^ЧРСк0е разделение смесей
ется: улучшит ли это разрешение? Ответ положительный,
оста некоторыми ограничениями. Разрешение R пропорцио-
!1°льно [к/(Х + JOL поэтому по мере увеличения к' разре-
"'Гние сначала быстро возрастает, а затем увеличивается
более медленно и после к' = 5 совсем незначительно. Таким
образом, нет смысла в чрезмерном ослаблении силы
растворителя (£°) и удлинении времени удерживания, так как в
дальнейшем выигрыш в разрешении будет небольшим.
Оптимальный интервал к' - между 1 и 10, когда образцы будут
Впвдэлюированы 2-11 колоночными объемами растворителя.
3) Коэффициент разделения а
* Рассмотрим ситуацию, когда подобран растворитель такой
силы, чтобы к' находился в оптимальном интервале, но два
соединения все же еще не разделились. По определению а =
I к'А/к'в. Индивидуальные значения к' для соединений
зависят от их коэффициентов распределения между неподвижной
и подвижной фазами (разд. 4.3.1, п. "а"). Эти
коэффициенты (и, следовательно, ее) можно изменять путем изменения
состава подвижной фазы вследствие селективных
взаимодействий между растворенным веществом и растворителем.
Поэтому необходимо поддерживать полярность (силу) растворителя
более или менее постоянной, сохраняя оптимальный к', но
варьировать состав подвижной фазы, чтобы изменить се.
Такая возможность обсуждается в последующих разделах
применительно к различным хроматографическим методам.
ж) Выбор вида хроматографии
Методика ВЭЖХ может быть использована для различных
видов хроматографии (ионообменная и ситовая хроматография
здесь не рассматриваются). Распространенными видами
являются жидкостно-адсорбционная хроматография (жидкость -
твердая фаза) (ЖТХ) и распределительная хроматография
(жидкостно-жидкостная) (ЖЖХ) с использованием связанных
^идких неподвижных фаз. Адсорбционная хроматография
(ЖТХ) применима для разделения широкого круга неионных
соединений, от умеренно неполярных до умеренно полярных,
но недостаточно эффективна для разделения либо совсем
неполярных (углеводороды), либо сильнополярных соединений,
™ких, как амины, спирты или кислоты. Связаннофазовая
ЖЖХ в своей обычной и обращеннофазовой форме примени-
Ма для разделения почти всех типов соединений, особенно
полярных соединений, которые не разделяются ЖТХ. Это
обстоятельство, а также тот факт, что ЖЖХ в некоторых ас-
пектах легче в экспериментальном исполнении, чем ЖТХ
объясняет значительный рост ее популярности за последние
пять лет.
з) Жидкостно-адсорбционная хроматография [жидкость-
твердая фаза (ЖТХ)]
В общих чертах принципы разделения рассмотрены ниже
в разд. 4.3.1, п."а".
1) Сорбенты
В качестве сорбента почти всегда используют силикагель
или оксид алюминия, специально подготовленные в виде псе
ристых сферических частиц размером от 3 до 10 мкм. И<Л
пользование этих очень маленьких частиц - ключ к
высокому разрешению, получаемому в ВЭЖХ. Типичная "широкаяш
колонка (10 см в длину и 4 мм в диаметре) # имеет эффеЛ
тивность примерно 6000 теоретических тарелок при заполнЛ
нии материалом с размером частиц 5 мкм и 10000 тарелок!
частицами размером 3 мкм. Колонки обычно покупают готЛ
выми, заполненными для конкретного применения.
Как силикагель, так и оксид алюминия пригодны для
разделения соединений различных типов. Следует избегать очеЛ
сильных взаимодействий между соединением и сорбентом, таи
как они могут привести к появлению нежелательных хвосте™
у пиков. Поэтому амины лучше анализировать на оксиди
алюминия, чем на силикагеле (который является кислотным)!
а фенолы - лучше на силикагеле, чем на оксиде алюминия
Разделение сильнополярных соединений не дает хороших рЛ
зультатов, и их лучше анализировать распределительной
ЖЖХ (см. п. 1 в следующем разделе на стр. 181).
Предварительная проверка с помощью ТСХ может
показать, какой из сорбентов лучше всего подходит для конкре'в
ного образца, но в тех случаях, где с помощью ТСХ нельзя
добиться разрешения, следует опробовать оба типа колоноИ
для ВЭЖХ.
2) Контроль активности сорбента
На поверхности сорбента имеются участки, на которых действ
вуют различные адсорбционные силы. Для получения
u«J лЭФФеК™ВН0СТЬ любой "Роматографической колонки, как и ректифик!
ционнои колонки (Разд. 3.4-3). измеряется в теоретических тарелках: «Л
выше их число, тем лучше разделение (рис. 4-II).
^Pf-rr*. разделение смесей
тих по форме пиков и воспроизводимых времен удержи-
*о1Х>я необходимо, чтобы наиболее активные участки были
ва5и доованы каким-либо сильно адсорбированным соедине-
м: например водой или спиртом. Активность поверхности
^вис'ит от того, насколько заблокированы центры адсорбции.
Обычно для дезактивации используют воду, так как такая
цеДУра в целом надежна, если .выполнена должным обра-
ом Однако оиа отнимает много времени, поскольку требует
Тщательного контроля за содержанием воды в элюирующем
растворителе. Вместо воды можно использовать другие
полярные "модификаторы" поверхности (см. ниже).
ДЕЗАКТИВАЦИЯ ВОДОЙ. Обычно при этом около
половины поверхности сорбента покрыто водным монослоем.
Колонку "доводят" до этого уровня активации пропусканием
через иее около 10 - 20 колоночных объемов эфира,
содержащего необходимое количество воды. Для силикагеля
требуется эфир, примерно на 50% насыщенный водой, а для
оксида алюминия необходим 25%-ный водный эфир [4]. Для
приготовления влажного эфира сухой эфир смешивают с
эфиром, насыщенным водой, в соответствующих пропорциях. По
мере прохождения влажного эфира вниз по колонке он
приходит в равновесие с сорбентом, либо поглощая, либо
отдавая воду до тех пор, пока ие будет достигнуто равновесие .
Для достижения иужиой активности можно варьировать
процентное содержание воды в эфире. Например, разделение
четырех изомерных углеводородов (рис. 4.13) было получено
после насыщения оксида алюминия 15%-ным водным эфиром,
тогда как на менее активной колонке, полученной
насыщением чаще используемым 25%-ным водным эфиром, не было
получено никакого разделения. Таким образом, колонку
довольно быстро и легко довести до требуемой активности.
Проблема заключается в том, чтобы активность оставалась
постоянной в течение всего времени анализа. Все элюирую-
Щие растворители могут поглощать воду из сорбента или
отдавать ее в зависимости от его насыщенности водой. Чтобы
активность была постоянной, элюент должен содержать то же
самое количество воды, что и исходный растворитель. Для
этого необходимо приготавливать элюент как в сухом, так и
в насыщенном водой состояниях и смешивать нх в соответст-
вУЮщих пропорциях.
"Насыщение" колонки эфиром служит также для того, чтобы смыть
е Загрязнения, оставшиеся на колонке от предыдущих анализов. Полезно
Р°Д0лжать промывание до тех пор, пока не будет получена стабильная
базовая линия.
Глава 4
Рнс. 4.13. Разделение смеси углеводородов с помощью ВЭЖХ на оксиде
алюминия.
ДРУГИЕ ПОВЕРХНОСТНЫЕ МОДИФИКАТОРЫ. Вместо
воды в качестве поверхностных модификаторов можно
использовать органические соединения типа метанола, изопропа-
нола или ацетонитрила. Их добавляют к элюирующему
растворителю в очень малых количествах (0,01 - 0,5%),
необходимых для установления и поддержания требуемой
активности.
Количество добавляемого модификатора зависит от его
природы и природы элюента: так, например, добавление
0,15% метанола или 0,3% изопропанола к дихлорметаиу
дает такую же активность, как и 50%-ное насыщение водой.
Хроматографическое разделение смесей 179
Сообщалось, что в некоторых случаях использование иевод-
ных модификаторов приводит к пикам невыразительной
формы.
3) Подвижная фаза
Элюеиты выбирают из более или менее того же самого
набора органических растворителей, которые применяются для
ТС и препаративной колоночной хроматографии (табл. 4.5).
Очевидно, что при использовании УФ-детектора не подходят
растворители, поглощающие в УФ-области, в таких случаях
применяют специально очищенные и высушенные
растворители для ВЭЖХ. Редко бывает так, что один растворитель
обладает оптимальной силой (см. разд. 4.3.1, п. "а") для
конкретного анализа; обычно применяют смеси гексаиа как
слабого растворителя с подходящим сильным растворителем,
например эфиром, чтобы достичь необходимого значения е °
(рис. 4.23). Для каждого нового анализа в ВЭЖХ самым
ответственным этапом является выбор состава растворителя
подходящей силы. Подход к этой проблеме обсуждается в
следующем разделе.
4) Сила и состав элюирующего растворителя
Состав подвижной фазы подбирают с целью достижения
разделения или его улучшения и сведения к минимуму
продолжительности анализа.
Первоначальную силу растворителя, который предстоит
опробовать, можно оценить предварительно с помощью ТСХ.
Необходимо найти смесь растворителей (обычно гексаиа и
сильного растворителя; см. табл. 4.5), которая дала бы R, =
= 0,2 - 0,3 для разделяемых соединений. Если смесь
содержит соединения, сильно различающиеся по полярности, то
потребуется ступенчатое или градиентное элюирование (см.
последний параграф этого раздела).
Обычно лучше начинать элюирование достаточно сильной
смесью растворителей, позволяющей довольно быстро вымыть
все компоненты, а затем ослаблять ее (увеличением
количества слабого растворителя), чтобы оптимизировать разделение,
придерживаясь рекомендаций, приведенных в разд. 4.1.3,
п. "е". Если к' для пары компонентов уже оптимизирован,
но разделение еще ие достигнуто, то в этом случае
необходимо поддерживать более или меиее постоянную полярность
растворителя (е°), сохраняя оптимальный к\ но варьируя
состав подвижной фазы, чтобы изменить а. Например, два
компонента могут иметь значение о* = 1,05 при использова-
180
Глава 4
нии подвижной фазы с 50 об. % эфира в гексане и, таким
образом, могут разделиться неполностью, и в то же самое
время использование 6 об. % диоксана в гексане может
дать а, равное 2, и полное разделение, хотя обе смеси
растворителей имеют одинаковую полярность (е° - 0,3).
Варьирование а, руководствуясь данными табл. 4.5 и рис.
4,23, означает привнесение метода проб и ошибок, так как
не существует надежного способа предсказания того, как
изменяется а с изменением состава элюента. Однако Снайдер
[5] показал, что смеси растворителей, содержащие либо
очень низкие, либо очень высокие концентрации сильного
растворителя, дадут высокое значение а с большей
вероятностью, нежели смеси с промежуточной концентрацией сильного
растворителя. Например, при разделении 1-ацетилнафталина
и 1,5-динитронафталина смесь растворителей из 5 об. %
пиридина в пентане дала а = 2,4, тогда как использование
25 об. % эфира в пентане дало а *= 1,16, хотя обе смеси
имеют е° - 0,25.
ГРАДИЕНТНОЕ ЭЛЮИРОВАНИЕ. В некоторых случаях
смеси содержат как быстро продвигающиеся слабополярные
соединения, так и более сильно удерживаемые полярные
вещества. В этих случаях требуется либо ступенчатое, либо
градиентное элюирование. Хорошие устройства для
градиентного элюирования (рис. 4.8) могут быть запрограммированы
на увеличение концентрации более сильного растворителя
либо ступенчато, либо градиентно или комбинацией и того и
другого. Однако при наличии даже простых иасосов состав
растворителей можно изменять ступеньчато вручную, что
позволяет проводить элюирование полярных компонентов.
Наиболее важным моментом при градиентном элюировании
является чистота растворителей; например, при использовании
УФ-детектора любой поглощающий в УФ-области
загрязнитель, который присутствует в более сильном растворителе,
вызывает смещение базовой линии по мере увеличения его
концентрации. Любые полярные примеси в слабом
растворителе можно сорбировать на колонке при низких
значениях е° растворителя, но позже, при увеличении его
полярности они проявляются как пики или бугры на базовой линии.
н) Жидкостно-жидкостная хроматография (ЖЖХ) на
связанных фазах
Общие принципы ЖЖХ приведены в разд. 4.3.1, п. "в".
Этот эффективный метод пригоден для разделения широкого
круга соединений различной природы, включая те, которые с
ХроматограФическое разделение смесей 181
трудом разделяются адсорбционной хроматографией (ЖТХ),
например, углеводороды (которые слабо удерживаются
сорбентами) и полярные соединения (которые не дают пики
хорошей формы из-за сильной здеорбции). Другое главное
преимущество ЖЖХ заключается в том, что отпадает
необходимость доводить колонку "до кондиции", а затем
контролировать ее активность, как в ЖТХ (см. предыдущий подраздел
"з").
1) Неподвижная фаза
Неподвижная фаза состоит из монослоя органической
жидкости, химически связанной с поверхностью частиц силикаге-
ля, как показано на рис. 4.25. Размеры частиц такие же,
как в ЖТХ (3 - 10 мкм). Связанные фазы бывают двух
типов: простые длинноцепочечные углеводороды (например,
С8Н17 или С№Н37), создающие липофильный слой на
поверхности частиц, и их аналоги с более короткой цепью,
содержащие полярную группу типа циано-, амино- или диольной
группы. В соответствии с этим существуют два
различающихся метода разделения: обращеннофазовый в первом случае и
прямофазовый - в последнем.
2) Обращеннофазовая ЖЖХ
В этом методе неподвижная фаза (липофильный
углеводородный монослой на поверхности частиц) неполярна, а элю-
ент (обычно смесь воды с полярным органическим
растворителем типа метанола, ацетонитрила, диоксана или тетрагид-
рофурана) сильнополярен. Это противоположно ситуации,
наблюдаемой в адсорбционной хроматографии, отсюда и ее
название. Когда полярные соединения вводятся в колонку, они
имеют большее сродство к полярному элюенту, чем к
неполярной неподвижной фазе, и поэтому быстро проходят через
нее, тогда как неполярные соединения избирательно
переходят в неподвижную фазу и удерживаются ею. Таким
образом, в ЖЖХ порядок элюирования обратный по сравнению с
ЖТХ, т. е. полярные соединения элюируются раньше
неполярных.
ПОДГОТОВИТЕЛЬНАЯ РАБОТА И ПРОВЕДЕНИЕ
РАЗДЕЛЕНИЙ. Первой задачей при разделении является
нахождение соотношения между количеством органического
растворителя и водой в элюенте, которое бы дало приемлемое
время удерживания для разделяемых соединений. Ее решают
методом проб и ошибок, причем намного быстрее при наличии
182
Глава 4
двухнасосной системы Срис. 4.8). Информацию о наиболее
подходящем органическом компоненте и процентном составе
элюента для соединений различных классов можно получить
из литературы (см., например, работу [6]). Однако в
отсутствие других сведений в качестве органического компонента
при выполнении исследовательской работы обычно используют
метанол. Помните, что это обращеннофазовый метод, т. е.
полярные соединения элюируются быстрее всего слабыми
элюентами с высоким содержанием воды, а для вымывания
неполярных соединений требуется более сильный элюент,
обогащенный органическим компонентом.
Если пики имеют плохое разрешение, то, снизив силу
растворителя, можно увеличить время удерживания (и
значение к')> но, как и в ЖТХ, не имеет смысла делать к'
намного большим 10 (см. предыдущий подраздел, п. "е" на
стр. 173). Если же разрешение пиков не улучшится, следует
заменить органический компонент и тем самым попытаться
изменить коэффициент разделения а, оставив с ° более или
менее постоянной.
В связаннофазовой ЖЖХ неожиданно возникает новый
фактор, влияющий на разделение, - насадка колонки. На
фактически одной и той же неподвижной фазе, изготовленной
разными производителями (и даже на разных партиях из
одного и того же места), можно получить различающиеся
характеристики, что вызвано присутствием остаточных сила-
нольных групп на поверхности силикагеля
Границы описанного здесь обращеииофаэового метода
можно расширить добавлением к подвижной фазе буферных
смесей, кислот, оснований или детергентов [2, 6].
3) Прямофазовая ЖЖХ
В этой форме связаннофазовой ЖЖХ неподвижная фаза
имеет полярные группы (некоторые полярные связанные
фазы, например циаиопропильную можно использовать как в
прямофазовом так и обращеннофазовом вариантах), а элюен-
том, как и в адсорбционной хроматографии (ЖТХ), служит
углеводород. Колонки для прямофазовой ЖЖХ могут
заменять колонки, заполненные силикагелем в ЖТХ, с тем
преимуществом, что связанные фазы дают широкую область
избирательности. Они лучше всего подходят для полярных
соединений и дают лучшее разделение и форму пиков, чем
ЖТХ. Элюент обычно состоит из гексана или 2,2,4-триметил-
пентана в смеси с сильным растворителем (табл. 4.5),
который добавляют для достижения необходимого значения е °,
как и в ЖТХ. Оптимальную силу растворителя н его состав
ХроматограФическое разделение смесей
183
Фронт
""растворителя
\
Полосы
компонентов
/
Стартовая
полоса
Рис. 4.14. Препаративная ТСХ.
снова приходится подбирать методом проб и ошибок, поэтому
следует еще раз обратиться к рекомендациям по применению
этого метода, приведенным на стр. 179.
4.2. ПРЕПАРАТИВНЫЕ МЕТОДЫ
4.2.1. Препаративная тонкослойная хроматография (ТСХ)
Препаративная ТСХ представляет собой пропорционально
увеличенный вид аналитической ТСХ (разд. 4.1.1),
позволяющий проводить разделение в препаративном масштабе.
Используемые в ней пластинки (см. например, рис. 4.14)
больше, чем аналитические (обычно 20 х 20 см), и покрыты
более толстым слоем сорбента (толщиной 0,5 или 1 мм).
Пластинка такого размера пригодна для разделения до 100 - 150
мг смеси в зависимости от трудности разделения.
Раствор образца наносится в виде полосы вдоль нижней
кромки пластинки. Его можно наносить в виде
перекрывающихся капель (с помощью пипетки или шприца) или в виде
непрерывной линии (с помощью усеченной пастеровской
пипетки с ватным тампоном) (рис. 4.15). В обоих методах
нанесения требуется направляющее приспособление, приподнятое
над поверхностью сорбента. Для нанесения узкой однородной
полосы необходима особая тщательность. Если предстоит
большой объем работы по препаративной ТСХ, то желательно
пользоваться механическим приспособлением для нанесения
полос. Оно имеет каретку для перемещения шприца вдоль
184
Глава 4
Ватный тампон
^тсх-пластинка
Рис. 4.15. Нанесение раствора образца на пластинку для препаративной
тех.
пластинки, которая автоматически распределяет раствор по
ходу передвижения. Чтобы получить узкую полосу, лучше
всего нанести несколько тонких полосок одна поверх другой,
давая каждой высохнуть перед нанесением последующей.
После проявления (разд. 4.1.1) пластинки компоненты
обычно обнаруживают УФ-методом (разд. 4.1.1). Реагенты
для опрыскивания и иод разрушили бы образец, поэтому
отмечают положения полос, затем осторожно соскребают
сорбент с пластинки.
Меры безопасности. Эту операцию следует выполнять в
вытяжном шкафу.
Затем соединения извлекают, помещая соскобленный
сорбент в стеклянную воронку Шота и добавляя
соответствующий растворитель, обычно дихлорметан или этилацетат в
зависимости от полярности соединения.
4.2.2. Колоночная хроматография
Колоночная хроматография - это единственный наиболее
важный метод в распоряжении химика-органика для разделения
смесей в препаративном масштабе (от нескольких
миллиграммов до десятков граммов). Обычно разделение проводят жид-
костно-адсорбционной хроматографией, которая эффективна
для большинства неионных соединений. Общие принципы
разделения описаны в разд. 4.3.1. Та же экспериментальная
методика лежит и в основе ионообменной и ситовой хрома-
Хроматографическое разделение смесей
185
графии, которые используются для разделения ионных
"биоорганических" соединений и высокомолекулярных веществ
[7].
Элюирующий
растворитель
Рис 4.16. Колоночная хроматография (с "гравитационным** элюированием
т. е. под дейстпием собственной силы тяжести).
186
Глава 4
а) Общее описание
Разделение выполняют на колонке, представляющей собой
стеклянную трубку, заполненную сорбентом (рис. 4.1б,о),
выполняющим роль пористого слоя через который протекает
подвижная фаза. Подвижной фазой, обычно называемой "элюи-
рующий растворитель" или "элюент", является органический
растворитель типа гексана.
Разделяемая смесь подается в верхнюю часть колонки, где
она сорбируется неподвижной фазой, а затем через колонку
непрерывно пропускают элюент (рис. 4.16,6). Каждый
компонент смеси переносится вниз по колонке подвижной фазой со
скоростью, которая зависит от его сродства к сорбенту. В
идеальном случае смесь разделяется на ряд отдельных
компонентов (полос) (рис. 4.1б,б), которые медленно опускаются
вдоль колонки и в конечном итоге собираются в приемник.
Сильно адсорбирующиеся полярные соединения, например
спирты (ROH), амины (RNH2) или карбоновые кислоты
(RC02H), продвигаются медленнее, чем менее полярные
соединения, такие, как альдегиды и кетоны (RCOR'), эфиры
(R20> и углеводороды, которые адсорбируются менее сильно.
Скорость, с которой полосы движутся вниз по колонке,
можно контролировать, регулируя силу (полярность) элюиру-
ющего растворителя (разд. 4.3.1).
Обычно элюат собирают порциями (фракциями) и каждую
проверяют с помощью ТСХ (или ГЖХ, если возможно),
чтобы определить, какой в ней присутствует компонент (если он
вообще там присутствует). Затем соответствующие фракции
объединяют, удаляют растворитель на роторном испарителе и
ващеляют соединение.
При использовании любой формы колоночной
хроматографии (см. следующий раздел) непременным условием
"хорошей" экспериментальной практики является взвешивание
смеси перед помещением ее в колонку и взвешивание каждого
компонента после разделения. Если этого не сделать, то
можно "потерять" какое-либо соединение из сложной смеси,
оставив его необнаруженным иа колонке.
б) Выбор метода
Существует много разновидностей препаративной
колоночной хроматографии, которые различаются по типам колонок
и особенно по методам пропускания элюирующего
растворителя через колонку. Исторически сложилось так, что в
течение многих лет стандартной практикой была колоночная хро-
Хроматографическое разделение смесей 187
матография с "гравитационным элюированием" (рис. 4.16),
но за последние примерно десять лет ее значительно
потеснили такие более быстрые и эффективные методы, как
флаш-хроматография, хроматография среднего давления и
флаш-хроматография на сухой колонке.
В начальный период обучения при выполнении заданий
обычно указываются метод разделения, сорбент и элюирую-
щий растворитель. Однако в дальнейшем необходимо
проводить предварительную проверку смеси с помощью ТСХ. Это
позволяет установить число соединений в смеси, легко ли
или трудно они разделяются, а также подобрать наилучший
сорбент и растворитель (илн смесь растворителей). Пластинки
для ТСХ не обязательно имеют ту же активность, что и
колонка (см. разд. 4.3.1 и 4.3.2), но все же дают полезную
информацию, которой можно руководствоваться прн
разделении.
В общем случае соединения, которые разрешены на ТСХ-
пластинках с разницей в R, более чем 0,10 - 0,15, можно
разделить либо флаш-хроматографией (см. ниже), либо
флаш-хроматографией на сухой колонке (разд. 4.2.2, п. "г").
Метод "среднего давления" (разд. 4.2.2, п. "д") обеспечивает
более высокую разрешающую способность и приводит к
хорошему разделению, даже когда на ТСХ нет четкого
разделения пятен (в этом случае для получения ясной картины
следует использовать ВЭЖХ). Традиционная хроматография с
"гравитационным элюированием" также бывает эффективной,
но имеет тенденцию быть утомительной из-за низкой
скорости потока элюента, особенно в длинных колонках.
Большинство разделений может быть выполнено более эффективно и
более быстро с помощью одного из вышеупомянутых методов.
Меры безопасности. Пыль от хроматографических
сорбентов вредна при вдыхании. Работающий должен носить
маску от пыли, и все операции, связанные со свободным
сорбентом, должны выполняться в вытяжном шкафу.
Используемые растворители обычно летучи, легко воспламеняются
и до некоторой степени токсичны. Работающие должны
принимать надлежащие меры, предотвращающие загорание
и вдыхание растворителей (разд. 1.3). Операции, при
которых растворители остаются в открытых емкостях,
следует выполнять в вытяжном шкафу.
Прохождение элюента через колонку под действием собственной силы
тяжести. - Прим. перев.
188
Глава 4
в) Флаш-хроматограф и я
Флаш-хроматография была разработана профессором К.
Стиллом [8] в конце 1970-х гт. в качестве эффект и ного и
очень быстрого метода для рутинного разделения смесей со
средним разрешением. Он позволяет проводить разделение
соединений, которые различаются по Rf (в ТСХ) на 0,15 и
более. ^
Прибор , используемый для проведения флаш-хроматогра-
фии, изображен на рис. 4.17,о. Для разделения разных по
объему образцов требуется набор колонок различного
диаметра (но одинаковой длины) (табл. 4.3). Сорбентом служит си-
ликагель 60 тонкого зернения (с размером частиц 40 -
63 мкм), например типа мерк 9385 (Merck type 9385). Во
время работы элюирующий растворитель прогоняется вниз по
колонке давлением воздуха или азота. Для достижения
хорошего разделения весьма важна скорость потока элюента,
причем наилучшие результаты получаются, когда уровень
растворителя в колонке над сорбентом падает со скоростью
примерно 5 см/мин. При соблюдении рекомендуемых ниже
условий для элюирования требуется незначительное время
(обычно примерно 15 мин).
1) Выбор элюирующего растворителя
Для общего использования в качестве растворителя
рекомендуется смесь петролейного эфира (фракция с т. кип. 40 -
60° С) и этилацетата или (для более полярных соединений)
смесь петролейного эфира и ацетона или дихлорметана и
ацетона. Процедура заключается в следующем. Проведите
предварительную проверку с помощью ТСХ, чтобы подобрать
такое соотношение двух растворителей, при котором Rf
искомого соединения будет 0,35. Если несколько соединений на
ТСХ поднимаются рядом, подберите такой растворитель,
чтобы среднее пятно имело Rt 0,35. Если соединения сильно
разделены, подберите такой элюент, при котором наименее
подвижный компонент будет иметь R, 0,35. Правильный
подбор силы растворителя является решающим условием
успешного проведения разделения, и стоит потратить на это время.
Мы благодарны Американскому химическому обществу за разрешение
использовать материалы из этой статьи, особенно табл. 4.3 и рис. 4-19.
Авторское право Американского химического_ общества (1978).
Необходимые приборы изготовляются фирмой "Aldrich Chemical Co.
Ltd.".
ХроматограФическое разделение смесей
Отверстие для
стравливания
давления С
Места стыков,
закрепленные
зажимами и(или)(
прочными
резиновыми лентами
Стекловата
Клапан,регулирую-
щи и лоток Б
$.
Манометр
Линия подачи
азота или
воздуха
От редуктора,
регулирующего
давление
Элюент
Стекловата
6
Рис. 4.17. Прибор для флаш-хроматографии {а); способ установки пробки
из стекловаты <б).
Если требуется очень маленькое процентное содержание
сильного растворителя «2%), лучше уменьшить его еще
наполовину, что облегчает управление колонкой.
2) Набивка колонки
Выберите колонку подходящего размера (табл. 4.3). В ос-
Таблица 4.3. Размеры колонок в флаш хроматограф и
Диаметр
колонки
мм
10
20
30
40
50
а Типичный
Объем
элюента".
мл
100
200
400
600
1000
объем, необходим*
Типичная загрузка
образца,
Ад>0,2
100
400
900
1600
мг
Дя^о.1
40
160
360
600
25О0 1000
1Й при заполнении и элюир
Типичный объем
фракции, мл
5
10
20
30
50
звании.
нование стеклянной трубки при помощи стеклянной палочки
вставьте маленькую пробку из стекловаты (рнс. 4.17,6).
Насыпьте немного крупного песка, чтобы получился ровный
слой толщиной 2 мм, и затем в один прием засыпьте сухой
силикагель 60 (40 - 63 мкм), чтобы получилась колонка
длиной 15 см (меры безопасности см. на стр. 187). Для
уплотнения силикагеля осторожно постукайте колонкой о стол
(в вертикальном положении) при открытом запорном кране,
а затем добавьте крупный песок слоем толщиной 2 мм .
Осторожно заполните колонку элюирующим растворителем.
Установите регулятор потока и закрепите для безопасности все
места соединений соединительными зажимами и прочными
эластичными лентами (рис. 4 17,о)
Меры безопасности. Предусмотрите меры
предосторожности, необходимые при работе с емкостями под давлени-
Откройте кран В регулятора потока, подсоедините линию
подачи азота и установите на редукторе газового баллона
давление примерно 0,5 кг/см2. Азот будет энергично
выходить через отверстие С. Закройте кран и дождитесь, пока
давление на манометре не достигнет 0,5 кг/см2. Тогда
растворитель быстро пройдет через силикагель. вытесняя воз-
Колонки наилучшего качества получаются при быстром заполнении
одной порцией сухого силикагеля и последующем уплотнении слоя сорбента
многократным вакуумированием и резким девакуумироваинем колонки. -
Прим. перев.
Хроматографическое разделение смесей 191.
дух из колонки. Важно, чтобы поток растворителя был
непрерывным до тех пор, пока не будет удален весь воздух и
не охладится силикагель [в противном случае произойдет
разделение на отдельные зоны (стр. 202) 1. После этого
установите регулятор потока (кран В) так, чтобы уровень
растворителя в колонке падал со скоростью 5 см/мин, и запишите
необходимое для этого давление. Продолжайте снижение
уровня растворителя до тех пор, пока он не опустится до
верхнего слоя неподвижной фазы, а затем остановите его,
полностью открыв кран В и закрыв запорный кран на
колонке. Не давайте сорбенту высохнуть, иначе колонка будет
выведена из рабочего состояния. Растворитель,
применявшийся при набивке колонки, следует использовать вновь во
время элюирования (см. п. 4 ниже).
3) Ввод образца
Подачу смеси предпочтительно осуществлять в виде
довольно концентрированного [25% (масса /об.) ] раствора в
элюирующем растворителе, как детально обсуждается ниже.
Однако во многих случаях образец недостаточно растворим
в исходном элюенте. Во всех формах колоночной
хроматографии важно избегать нанесения образца в виде разбавленного
раствора в элюенте или в виде раствора в более сильном
(более полярном) растворителе, поскольку это ведет к уши-
рению полос и плохому разрешению. Если образец плохо
растворяется в элюенте, лучше предварительно адсорбировать
его на сорбенте, как описано ниже.
НАНЕСЕНИЕ ОБРАЗЦА В ВИДЕ РАСТВОРА,
Приготовьте раствор образца в элюирующем растворителе [25%
(масса/об.) 1. Осторожно закапайте его из пипетки на верхний
слой сорбента и затем подайте на колонку небольшое
давление, чтобы продавить раствор в силикагель (постарайтесь не
продавить воздух через слой сорбента). Промойте стенки
небольшим количеством элюеита и продавите смывной элюент
через силикагель.
ПРЕДВАРИТЕЛЬНАЯ АДСОРБЦИЯ ОБРАЗЦА. В этом
методе образец предварительно адсорбируется на пятикратном
по массе количестве сорбента и все это помещается в
верхнюю часть колонки. Процедура заключается в следующем.
Растворите навеску образца в легколетучем растворителе
(например, в дихлорметане) в колбе роторного испарителя.
Добавьте сорбент (в пятикратном избытке по массе) и затем
удалите растворитель на роторном испарителе при слабом на-
192
Глава 4
г— 1Л
1
Крупнопористый стеклянный фильтр
Рис. 4.18. Переходник с пористым стеклянным фильтром, используемый
для предварительной адсорбции.
гревании. Присутствие твердого вещества обычно служит
причиной сильного толчкообразного кипения смеси (смесь кипит
"толчками"), и поэтому необходимо подсоединить переходник
(рис. 4.18) с крупнопористым стеклянным фильтром, чтобы
предотвратить потерю твердого вещества в переходном канале
испарителя. В конечном итоге будет получен свободнотекучий
порошок. При уровне элюента в колонке в несколько
сантиметров над слоем сорбента добавьте через воронку очень
тонкой струйкой адсорбированный образец, осторожно
постукивая по колонке, чтобы он равномерно распределился по
верхнему слою неподвижной фазы. Присыпание должно
производиться медленно с добавлением при необходимости
растворителя, чтобы избежать захвата воздуха. Имеется
альтернативный метод, при котором предварительно
адсорбированный образец суспендируют в минимальном объеме
растворителя и заливают суспензию в колонку, где она затем
отстаивается (стр. 202). После загрузки понижают уровень
растворителя и обмывают стенкн, как описано выше.
4) Элюирование
Заполните колонку и резервуар элюнрующим
растворителем. Подсоедините и закрепите регулятор потока н
установите давление, необходимое для обеспечения нужной скорости
потока (как описано выше в п. 2). Соберите фракции
соответствующего объема (табл. 4.3), используя коллектор
фракций.
Разделенные компоненты можно определить с помощью
ТСХ, как показано, например, на рис. 4.19, используя для
этого небольшую стеклянную ТСХ-пластинку или для
большего удобства полоску пластиковой одноразовой пластинки.
Хроматографическое разделение смесей . 193
/ 123456789 10 11
Смесь Фракции
Рис. 4.19. ТС-хроматограмма фракций, полученных в флаш-хроматогра-
фии.
г) Флаш-хроматография на сухой колонке
Этот метод разработан д-ром Л. Харвудом из
Манчестерского университета в начале 80-х гт. [91 . Он не требует
специального оборудования, его разделяющая способность по
крайней мере не хуже, чем у флаш-хроматографии, а
разрешение сравнимо с разрешением, получаемым на пластинке
для аналитической ТСХ.
В этом методе имеется несколько главных отступлений от
традиционной колоночной хроматографии. Разделение
проводят на силикагеле марки "для ТС хроматографии"
[например, на кизельгуре марки мерк 60 ("Merck Kieselgel 60") ] с
частицами немного меньшего размера (примерно 15 мкм),
чем в обычной колоночной хроматографии. Силикагель
помещают в стандартную стеклянную воронку Шота (рис. 4.20) и
прогоняют через него элюирующий растворитель всасыванием
в вакууме водоструйного насоса. Самое необычное в этом
методе, однако, то, что при элюировании добавляются
предварительно отмеренные порции растворителя и после
добавления каждой порции колонке дают "высохнуть". Это очень
существенное различие, но описываемый метод необыкновенно
эффективен.
Меры безопасности. Всю операцию следует проводить в
вытяжном шкафу, вдали от источников возгорания.
9 Описанный здесь метод опирается на это сообщение и нашу
собственную экспериментальную методику. (Мы благодарим компанию "AJdrich
Chemicai Co. Inc.** за разрешение использовать материал из этой статьи )
13-6729
194
Глава 4
Сорбент
Водоструйный
насос
Рис. 4.20. Прибор для флаш-хроматографии на сухой колонке.
1) Прибор
Собирают такой же прибор, как для фильтрования (рис.
4.20), используя цилиндрическую воронку Шота, размер
которой соответствует количеству образца (табл. 4.4).
2) Набивка колонки
Заполните воронку до краев сухим силикагелем для ТСХ
(см. примечание по мерам безопасности на стр. 187) и
осторожно постучите по ней, чтобы уплотнить порошок и
удалить все пустоты. Затем подсоедините вакуум водоструйного
насоса, сначала слабый, а затем полный и тщательно
утрамбуйте силикагель плоской пробкой. !!>го надо делать сначала
по окружности, а затем по направлению к центру, чтобы
получился горизонтальный, хорошо утрамбованный слой, над
которым появляется свободное пространство для добавления
смеси и элюента.
3) Выбор исходного растворителя
Проведите предварительные эксперименты по ТСХ, чтобы
подобрать смесь растворителей, которая быстрее всего подни-
Хроматографическое разделение смесей 195
Таблица 4.4. Размеры воронок в флаш-хроматографии на сухой колонке
Размер воронки. Масса силикагеля Масса образца". Объем фракции
мм (прибл.), г мг растворителя,
мл
диаметр длина
30 45 15 200 1(М5
40 50 30 500 15-30
70 55 100 1000-2000 20-50
а Эти количества приблизительны; для "трудных" смесей может подойти
меньшая загрузка, а для "легких** - большая.
мет интересующий вас компонент на высоту R. = 0,2 , и
используйте ее как исходный растворитель. Для соединений
средней полярности в качестве слабого растворителя обычно
применяют гексан (или петролейный эфир) с добавлением
эфира или этилацетата для достижения требуемой силы (см.
разд. 4.3.1, п. "а"). Всегда можно подобрать подходящий
растворитель, вполне удовлетворяющий широкому интервалу
полярности образцов, комбинацией гексана (или пентана),
эфира, этилацетата или метанола.
4) Предварительное элюирование
Предварительно проэлюируйте колонку исходным
растворителем под вакуумом, добавляя его очень осторожпо во
избежание нарушения поверхности силикагеля. Если набивка
была сделана правильно, то будет видно, как опускается
горизонтальная линия фронта растворителя. Если же произойдет
образование каналов, колонку необходимо досуха
отфильтровать и снова набить. Во время предварительного элюирования
поверхность должна быть покрыта растворителем, пока он не
прошел в приемник. Затем дайте силикагелю "высохнуть"
под всасыванием, пока объем вытекающего из колонки
растворителя не уменьшится до нескольких капель.
5) Загрузка образца
В идеальном случае смесь следует вводить в колонку в
Эта величина зависит от активности ТСХ-пластин; иногда необходимо
провести несколько предварительных экспериментов, чтобы установить
наилучшую корреляцию для вашей системы
виде концентрированного [25% (масса/об.) ] раствора в
исходном растворителе. (Если она недостаточно растворима,
следуйте методике "предварительного адсорбирования",
приведенной в следующем абзаце. При открытом кране А (рис.
4.20) равномерно залейте раствор на поверхность силикагеля,
а затем закройте кран А. Когда после отсасывания колонка
станет сухой, начинайте элюирование (следующий подраздел).
Бели смесь плохо растворяется в исходном растворителе,
ее следует предварительно адсорбировать силикагелем для
ТСХ, как описано на стр. 191. Равномерно насыпьте порошок
на утрамбованный сорбент (после его предварительного элюи-
рования) и спрессуйте его под вакуумом (тщательно и
равномерно), как и при обычной подготовке колонки.
6) Элюирование
Используя коллектор фракций, приготовьте в отдельных
пробирках элюирующий растворитель, чтобы суммарный
объем соответствовал рекомендованному в табл. 4.4. Наилучшие
результаты разделения достигаются при увеличении силы
растворителя. Приготовление элюента начинайте с двух или
трех пробирок с исходным растворителем (см. выше, разд. 3,
"Выбор исходного растворителя"), а затем в каждой
последующей пробирке увеличивайте количество сильного
растворителя примерно на 5 - 10%.
Четырехходовой
кран Г
Элнент
Манометр
<£>Фильтр <]—|
Клапаи,
регулирующий
давление
А
В"..
4
В'- 3
?. -'
Раствор
образца
Колонка
предварительной
очистки
Шприц
Основная
колонка
Коллектор
Фракции
Рис. 4.21. Схема прибора для хроматографии среднего давления.
роматографическое разделение смесей 197
После того как пробирки с элюирующим растворителем
подготовлены, содержимое первой из них равномерно
разлейте по поверхности силикагеля, на которую предварительно
уже нанесена разделяемая смесь; при этом вакуум должен
быть подсоединен. Подождите, пока колонка не станет
"сухой", и затем слегка приоткройте кран А снимите колбу и
вылейте элюат обратно в ту же- пробирку. Сделайте то же
самое с каждой последующей порцией элюирующего
растворителя, "насухо отсасывая" колонку после каждого
добавления. Компоненты определяют тестированием фракций с
помощью ТСХ, как описано для флаш-хроматографии. С
приобретением опыта легко осуществимы разделения с такой же
эффективностью, как и в ТСХ. Эта методика не подходит
для легко окисляющихся соединений.
д) Жидкостная хроматография среднего давления
Описанная ниже методика базируется на методе,
разработанном профессором Мэйерсом [101 . В жидкостной
хроматографии среднего давления (ЖХСД) при помощи насоса для
подачи растворителя решаются многие проблемы элюирова-
ния, возникающие при нормальном давлении. Это позволяет
применять длинные колонки с более мелкими частицами
сорбента, что приводит к более высокой разделяющей
способности без увеличения продолжительности анализа. ЖХСД -
намного более эффективный метод, чем флаш-хроматография и
флаш-хроматография на сухой колонке; она позволяет
разделять смеси, не дающие четкого разделения пятен в
аналитической ТСХ. Его можно успешно сочетать с любым из
"флаш"-методов разделения сложных смесей, используя
"флэш"-методику для быстрого предварительного разделения
и ЖХСД для последующего разделения "трудных"
соединений. Он экономит сорбент, так как основную колонку можно
использовать повторно после очистки посредством промывки
между разделениями.
1) Прибор
Прибор для ЖХСД изображен на рис. 4.21. Колонки
изготавливают из стекла со стойкими к действию растворителей
соединительными трубками и заполняют силикагелем 60
(40 - 63 мкм), например типа мерк 9385, или оксидом алю-
Мы благодарны Американскому химическому обществу за разрешение
миния использовать материал этой статьи, [авторское право Американского
химического общества (1979)].
198
Глава 4
аналогичного качества. Процедура набивкн описана ниже.
Размер главной разделительной колонки зависнт от масштаба
операции и легкости разделения, которое предстонт провести.
Колонки размером 1000 х 15 мм применяют для небольших
разделений (до 2 - 3 г), тогда как на колонках размером
1000 х 25 мм можно разделять вплоть до 10 г образца. Эти
цифры являются только ориентировочными: в случае легких
разделений они выше, тогда как трудные смеси разделяются
только при очень небольших загрузках колонки. Для того
чтобы запдитить главную колонку от загрязнения
сильнополярными "смолистыми" веществами, присутствующими во
многих реакционных смесях, в результате чего ее довольно
часто приходится перенабивать, используют предварительную
очистку на маленькой (200 х 15 мм) колонке.
Насос поршневого или диафрагменного (с покрытой
тефлоном диафрагмой) типа должен подавать растворитель со
скоростью 5-50 мл/мин при давлениях до 7 атм, причем
клапан, регулирующий давление, должен срабатывать при 7
атм.
Четырехходовой кран имеет два положения: в положении
А насос соединен с колонкой, а шприц - с емкостью для
образца; -в положении В образец вводится в колонку при
помощи шприца (5 - 20 мл) обычно "газонепроницаемого" типа.
Необходимо использование механического коллектора фракций
любого типа, который способен отбирать фракции объемом 25
или 50 мл (см. примечания по мерам безопасности на стр.
187 и 190).
2) Выбор элюирующего растворителя
Для того чтобы приготовить элюирующий растворитель
требуемой силы, нужно взять в качестве базового слабого
растворителя петролейный эфир (т. кип. 40 - 60° С) и
добавлять к нему необходимое количество сильного растворителя
типа эфира или этилацетата (см. стр. 204 - 206) Перед
разделением на колонке смесь необходимо исследовать с
помощью ТСХ, чтобы подобрать такое соотношение двух
растворителей, которое бы дало для разделяемых соединений
Rf = 0,2 - 0,3 (для трудных разделений - более низкое
значение). Нужен некоторый опыт, чтобы на его основе выбрать
растворитель оптимальной силы, так как последняя зависит
от относительной активности колонки и пластинки. Для
смесей, содержащих соединения с широким диапазоном
полярности, сила растворителя, конечно, будет увеличиваться по
мере разделения, также как это обычно делается при
нормальном элюнровании под действием силы тяжести.
"' »' СЦ-_^_
199
После подбора исходного растворителя промойте им
систему, подключив колонку предварительной очистки, если
только НЕ предстоит использовать метод нанесения образца с
предварительным его адсорбированием (см. следующий
подраздел).
3) Загрузка образца
Образец обычно вводят в колонку в виде
концентрированного 25%-ного (маса/об.) раствора в исходном элюирующем
растворителе. Как и в любой колоночной хроматографии,
следует избегать растворения образца в очень полярном
растворителе, так как это препятствует первоначальной
адсорбции образца верхним слоем сорбента и приводит к ушире-
нию полос и плохому разрешению. Если образец растворим
только в сильнополярных растворителях, лучше
предварительно адсорбировать его на силикагеле (стр. 190) и в сухом
виде перенести в верхнюю часть колонки предварительной
очистки (о методе набивки см. ниже).
Образец в виде раствора вводят через двухходовой кран
(рис. 4.21) следующим образом. Установите кран в
положение А, наберите раствор в шприц, затем остановите насос,
переведите кран в положение В и введите образец (введение
осуществляется легче, если во время этой операции колонка
предварительной очистки отсоединена от основной колонки).
При использовании метода предварительной очистки
загруженную колонку предварительной адсорбции (сухоупакован-
ную) соединяют с системой после промывки основной
колонки исходным элюентом. Воздух из колонки предварительной
очистки быстро удаляется по мере прокачивания растворителя
под давлением.
4) Элюирование
Скорость потока при элюировании зависит от трудности
разделения (от величины ДЮ; она должна быть
оптимальной, обеспечивая разделение без ухудшения его качества.
Минимальная скорость составляет 5 мл/мин для узких (15
мм) и 20 мл/мин для широких (25 мм) колонок.
Определение разделенных компонентов, находящихся в коллекторе
фракций, производится с помощью ТСХ, как описано для
флаш-хроматографии [см. п. "в" выше].
5) Промывание колонок обратной струей
Колонки можно использовать многократно, если их очи-
200
Колонки
Насос
Бутыль для сбора
отходов
растворителя
Рис, 4.22. Промывание колонок в ЖХСД обратной струей.
щать, удаляя все оставшееся вещество промыванием обратной
струей между анализами. Колонку предварительной очистки
необходимо менять чаще в зависимости от ее цвета.
По завершении разделения прибор следует подсоединить,
как показано на рис 4.22, отверстием 4 к выходу главной
колонки, а отверстием 3 к бутыли для отходов. Установив
кран в положение В, промойте систему этилацетатом и
прокачайте через нее еще около 300 мл этого растворителя.
Затем систему нужно промыть очередным элюирующим
растворителем, используемым прн следующем разделении, или
неполярным растворителем, если колонку предстоит
законсервировать.
6) Набивка колонки
Колонки набивают сухим сорбентом методом "заполнения
и постукивания*', при котором сорбент добавляют
небольшими порциями (на 3 мм по высоте колонки) н уплотняют
многократным постукиванием колонки в вертикальном
положении о твердую поверхность. При набивке некоторых
колонок необходимо постукивание о деревянный брусок во
избежание повреждения нижней части колонки. Метод
"заполнения-постукивания" позволяет довольно легко получить эффек-
■М емкф
ж
тивность, равную 900 - 1000 тарелок для коротких (250 мм)
и 3000 - 4000 для длинных (1000 мм) колонок. Эти
измерения были сделаны скорее на аналитических, чем
препаративных колонках, при введении небольших количеств образца и
низких скоростях потока.
В обычном препаративном методе эти показатели не
удается достичь из-за перегрузки колонки и последующего уши-
рения полос, но такие измерения нужны для проверки
качества операции набивкн и легко выполнимы при наличии
детекторной системы ВЭЖХ (см. рис. 4.11). В лаборатории
авторов метод "заполнения-постукивания" оказался гораздо
успешнее, чем другой метод с утрамбовкой сухого сорбента.
По окончании набивки колонки соединяют и промывают
обратной струей, как показано на рис, 4.22, а; затем
прокачивают через них растворитель (обычно петролейный эфир)
до тех пор, пока не будет вытеснен весь воздух, После этого
колонка готова к употреблению.
При хранении колонку увлажняют неполярным
растворителем и закрывают для предотвращения доступа воздуха.
е) Хроматография с элюированнем самотеком (под
действием силы тяжести)
В этом методе элюирующий растворитель подается нз^
емкости (обычно капельной воронки), подсоединенной к верхней
части колонки (рис 4.16). Его преимуществом является
простота, т. е. в смысле механики мало что может оказаться
неправильным, а главным недостатком - то, что скорость
потока элюеита сильно замедляется в длинных колонках или
при маленьких размерах частиц сорбента. Однако этот метод
применим для относительно легких разделений, которые
можно быстро выполнить на коротких широких колонках.
1) Колонка
Колонка представляет собой стеклянную трубку с краном
на одном конце и шлифом на другом, в который вставляют
емкость с растворителем. Сорбент удерживается либо диском
из пористого стекла (как в воронке Шота), впаянным в
колонку (рнс 4.16,а), либо при его отсутствии - пробкой из
стекловаты.
См. замечания по мерам безопасности на стр. 187.
202 Глава 4
2) Сорбент
В качестве сорбента используют как оксид алюминия, так
и силикагель с размером частиц 50 - 200 мкм. Обычно
предпочитают оксид алюминия не потому, что силикагель
менее эффективен при разделении смесей, а потому, что он
имеет некоторые специфические особенности, затрудняющие
его использование в гравитационных (обычных) колонках.
Основная трудность состоит в том, что колонки, заполненные
силикагелем, склонны к "растрескиванию" - образованию
пустот в слое сорбента. Это, как правило, происходит при
изменении силы растворителя, особенно если оно слишком
резкое, или в результате выделения тепла при взаимодействии
растворителя с сорбентом. Если же предстоит использовать
силикагель, важно применять изократическое элюнрование
(растворитель постоянной силы) или изменять силу
растворителя очень медленно. Оксид алюминия бывает нейтральной,
основной и кислой разновидностей, которые активированы до
степени I по шкале Брокмана (см. разд. 4.3.2).
"Нейтральный" оксид подходит для большинства соединений, тогда как
"основной" н "кислый" очень активно сорбируют
органические кислоты и основания соответственно. Однако все
разновидности оксида алюминия при сильной активации могут
катализировать некоторые замечательные "внутриколоночные"
реакции. Поэтому важно всегда контролировать, чтобы то,
что выходит из колонки, соответствовало тому, что было в
нее подано (МСХ, 5ШР). Сорбент необходимо
дезактивировать до требуемого уровня (стр. 210) перед тем, как
наполнять колонку. Чтобы достичь сравнимого с ТСХ разделения,
активность сорбента должна быть выше активности ТСХ-пла-
стины (обычно степени II или III), используемой для
выяснения условий разделения.
Необходимое количество сорбента в некоторой степени
зависит от трудности разделения; для легких смесей массовое
соотношение между сорбентом и образцом должно составлять
примерно 50:1 в случае оксида алюминия и 75:1 в случае
силикагеля, в то время как для трудных смесей оно может
достигать 300:1.
3) Набивка колонки
Для заполнения колонки используют подвижную
суспензию сорбента в исходном элюенте [см. подраздел 5 (ниже) ].
Это проделывают следующим образом. Частично заполните
колонку исходным растворителем и затем через воронку
влейте суспензию непрерывной струей. Дайте растворителю
т
Хроматографическое разделение смесей 203
слиться через кран, чтобы сорбент осел, и постучите
легонько по колонке куском толстостенного резинового шланга,
чтобы уплотнить сорбент. Не допускайте падения уровня
растворителя ниже уровня слоя сорбента.
4) Загрузка образца
В ВИДЕ РАСТВОРА. Самый простой метод - это ввести
образец в колонку в виде концентрированного раствора в
небольшом количестве исходного элюирующего растворителя (но
не более сильного растворителя). При этом часто
сталкиваются с тем, что разделяемая смесь либо нерастворима, либо
только слабо растворима в растворителе. В этом случае
необходимо использовать метод предварительной адсорбции,
рассмотренный на стр. 191.
Если же разделяемая смесь растворима в исходном элюи-
рующем растворителе, то процедура загрузки следущая.
Доведите уровень элюента в колонке чуть выше слоя сорбента.
Из пипетки осторожно прикапайте раствор образца и затем
I слейте большую часть растворителя, чтобы его уровень опять
стал чуть выше слоя сорбента. Тщательно смойте стенки
колонки небольшим количеством исходного растворителя и
снова слейте его до уровня чуть выше слоя сорбента. Осторожно
заполните колонку исходным элюентом, стараясь не нарушить
верхний слой набивки, и начинайте элюированне.
Обычно на верхний слой сорбента помещают тонкий слой
крупного песка, чтобы предотвратить помутнение элюента
при заполнении им колонки.
5) Элюирование колонки
Колонку элюируют органическими растворителями или
смесью растворителей строго контролируемой силы (см. стр.
205, 206). Обычно используют петролейный эфир (т. кип.
40 - 60° С) в качестве слабого растворителя, смешанный с
эфиром или дихлорметаном или каким-либо другим сильным
растворителем, чтобы получить требуемую силу. Исходным
растворителем для заполнения колонки служит обычно
петролейный эфир или смесь растворителей, подобранная с
помощью ТСХ (на пластинках, активированных до того же
самого уровня, что и сорбент в колонке), которая дает R, =
0,1 - 0,2 для наиболее быстро движущегося компонента
смеси. По мере хроматографирования сила элюента может быть
постепенно увеличена путем соответствующего увеличения
доли сильного растворителя. Когда в конечном итоге доля силь-
II ного растворителя (например, эфира) достигнет 100%, силу
204
Глава 4
растворителя можно увеличивать постепенным приливаннем
еще более сильного растворителя (например, этилацетата) н
т. д. Некоторую информацию относительно того, что нужно
для каждого компонента, можно получить с помощью
предварительных ТСХ-экспериментов.
6) Сбор фракций
Если соединения не окрашены, то не видно, когда какой
компонент появляется в элюате. Обычно собирают "фракции"
элюата равного объема по ходу хроматографирования (см.
табл. 4.3 для руководства по объему фракций) и проверяют
их с помощью ТСХ (см., например, рис 4.19) или ГЖХ,
чтобы определить соединения.
4.3. ПРИЛОЖЕНИЯ
4.3.1. Общие принципы хроматографического разделения
Все виды хроматографии основаны на использовании
двухфазной системы, состоящей из неподвижной и подвижной
фаз. Проиллюстируем это на примерах.
а) Жидкостно-адсорбционная хроматография (жидкость-
твердое вещество)
Она известна еще как адсорбционная хроматография,
потому что неподвижной фазой являются мелкие частицы
твердого сорбента (обычно силикагель или оксид алюминия).
Подвижной фазой (элюентом) служит органический растворитель
типа гексана или эфира. Сорбент может быть упакован в
колонку (рис 4.16,й) или нанесен в виде тонкого слоя на
пластинку (рис. 4.1,а). Во время хроматографирования элюент
проходит через сорбент, стекает вниз по колонке или
поднимается вверх по пластинке. Соединение Ау помещенное на
сорбент, в результате установления динамичесокго равновесия
распределяется между сорбентом н элюентом. Коэффициент
распределения k'A этого соединения зависит от его
относительного сродства к сорбенту и элюенту:
к А = «SA/«MA
где nsA - общее число молекул соединения А,
адсорбированных твердым веществом, а пмА - общее число молекул
соединения А, растворенных в элюенте.
По мере продвижения элюента через сорбент он переме-
Хроматографическое разделение смесей 205
щает образец вдоль слоя в виде медленно движущейся
полосы. Чем больше содержание фракции А в элюенте, тем
быстрее она пройдет через сорбент, и наоборот, чем сильнее
фракция А сорбируется твердым веществом, тем медленнее
она передвигается. Таким образом, скорость движения
фракции А зависит от к'А.
Если в верхнюю часть колонки подается смесь двух
различных соединений (А н В), то имеется большая вероятность
того, что онн будут иметь разное сродство к подвижной фазе
и сорбенту, т. е. к'А будет отличаться от А'в, и,
следовательно, две полосы будут двигаться с разными скоростями.
Очевидно, чем больше разница между к' и А'в, тем лучше
разделение между соединениями А н В. Отношение к'А и к'в
известно как коэффициент разделения а.
Скорость, с которой полосы проходят вниз по колонке или
вверх по пластинке, можно контролировать варьированием
свойств сорбента (об активности сорбента см. ниже) и (или)
свойств элюирующего растворителя (о силе растворителя см.
ниже).
АКТИВНОСТЬ СОРБЕНТА. В жидкостной хроматографии
в качестве неподвижной фазы, как правило, используют
только два сорбента: силикагель (Si02-xH20) и гидратирован-
ный оксид алюминия (А1203). Хроматографические свойства
этих веществ зависят от присутствия активных
адсорбционных центров на их поверхности, взаимодейстующих с
органическим молекулами. Вообще говоря, чем более полярна
органическая молекула, тем сильнее она сорбируется.
(Функциональные группы биполярного характера, например С=0, C=N,
ОН, NH2, C02H и т. п., придают органической молекуле
полярный характер.) Однако активные центры могут также
адсорбировать молекулы воды н связывать их так сильно, что
центр эффективно блокируется, а поверхность
дезактивируется. Активность (адсорбирующую способность), следовательно,
можно контролировать, регулируя содержание воды в
сорбенте. Активность определяют количественно по шкале Брокмана
1-V, где степень I соответствует максимальной активности.
Практические методы контроля и измерения активности
обсуждаются в разд. 4.3.2.
СИЛА РАСТВОРИТЕЛЯ. Другим главным фактором,
контролирующим скорость прохождения соединения вниз по
колонке, является сила, или полярность, элюирующего
растворителя. Чем сильнее (полярнее) растворитель, тем больше
его сродство к соединению в колонке и тем быстрее это
соединение проходит через колонку. Количественно сила растворите-
206 Глава 4
Таблица 4.5. Сила (значения С °) некоторых растворителей для
хроматографии в<6
Растворитель С° Растворитель €°
Пентан (CSH12) 0,00 Тетрагидрофуран <C4Hg0) 0,45
Гексан (СбН14) 0,01 1,2-Дихлорэтан (СН2С1СН2С1) 0,49
Циклогексан (С6Н12) 0,04 Диоксан <C4Hg02) 0,56
Тетрахлорид углерода (СС14) 0,18 Этилацетат (CHgCOjCgHg) 0,58
Толуол (С6Н5СНз> 0,29 Диметилсульфоксид (CHgSOCH^ 0,62
2-Хлорпропан (CH^CHClCiy 0,29 Ацетонитрил (CHgCN) 0,65
Диэтиловый эфир (С2Н5ОС2Н5) 0,38 1- и 2-Пропанол (C3HjOH) 0,82
Хлороформ (СНС13) 0,40 Этанол (СН^СН^Н) 0,88
Дихлорметан (CP^Cl) 0,42 Метанол (СН3ОН) 0,95
а Приведены значения для хроматографирования на оксиде алюминия, но
аналогичный порядок наблюдается и для силикагеля.
6 Более обширные данные и обсуждение свойств хроматографических
растворителей можно найти в работах [II - 13]
ля определяется как его значение е ° (табл. 4.5), которое
находится в пределах от 0 для неполярных углеводородов
типа пентана до 0,95 для сильнополярного метанола.
При хроматографировании необходимо очень внимательно
контролировать силу растворителя, для чего обычно
используют различные смеси сильного растворителя типа эфира
(€°=0,38) со слабым растворителем типа гексана (е°«0,01).
На рис 4.23, например, показано, как изменяется сила
растворителя в зависимости от его состава для бинарных смесей
гексана с различными сильными растворителями.
б) Газо-жидкостная хроматография
Принцип ее действия такой же, как в жидкостно-адсорб-
ционной хроматографии, но при этом неподвижной фазой
является жидкость, а подвижной фазой - газ. Селективность
обусловлена скорее распределением, чем поглощением.
Жидкая неподвижная фаза представляет собой
высокомолекулярную нелетучую жидкость - обычно тяжелое масло, смолу или
полимер (более детально см. стр. 158), и поэтому основной
проблемой является сохранение ее неподвижности. Это
достигается двумя способами. Согласно одному нз них, жидкость
наносится в виде тонкой пленки на поверхность
гранулированного инертного твердого вещества - носителя (рис. 4.24,а)
(обычно одна из форм целита - днатомитовой глины с боль-
Хроматографическое разделение смесей
207
Рис. 4.23. Сила растворителя (С°) для смесей сильных растворителей с
гексаном.
шой площадью поверхности). Носитель с неподвижной
жидкой фазой, называемый "насадка колонки", набивают в
свернутую в спираль стеклянную или металлическую трубку
(внутренним диаметром 2-4 мм н длиной от 1 да 5 м),
известную как ГЖХ-колонка. Таким образом, большая
площадь поверхности жидкой фазы доступна для подвижной
газообразной фазы, обычно азота, который непрерывно
проходит через колонку при ее работе (рнс 4.2,а). Насадочные
колонки этого типа достаточно дешевы, имеют
продолжительное время жизни и находят широкое применение.
В качестве альтернативы неподвижная жидкая фаза может
наноситься в виде тонкой пленки на внутреннюю стенку
208 Глава 4
Стандартная
насадочная
колонка
Гранулм носителя
с нанесенной на
них неподвижной
фазой
Капиллярная
■ колонка
Стенка капил-.
лярной трубим,
покрытая тонком
"пленкой
неподвижной фазы
Рис. 4.24. Газо-жидкостная хроматография на насадочной колонке (а) и
капиллярной колонке (б).
капиллярной трубки очень узкого диаметра (0,2 - 0,5 мм)
(рис. 4.24,а). Капиллярные трубки первоначально делали из
металла, но сейчас в основном используют кварцевые
капиллярные колонки. Такие колонки имеют очень низкое
сопротивление газовому потоку, и поэтому они могут быть очень
длинными (до 100 м). Большая длина делает их очень
эффективными, что позволяет проводить разделения, которые
невозможны на насадочных колонках. Однако кварцевые
капиллярные колонки очень дороги. Эффективность колонки
измеряется в теоретических тарелках (подобно ректификацион-
Хроматографическое разделение смесей 209
ным колонкам); насадочная колонка обычно имеет примерно
20000 тарелок, в то время как капиллярная колонка -
примерно 250000.
После введения соединения в колонку оно распределяется
в соответствии с динамическим равновесием между
неподвижной и подвижной фазами в количествах, которые зависят от
его летучести и сродства к неподвижной жидкой фазе.
Каждое соединение при определенной рабочей температуре и
конкретной жидкой фазе имеет свой собственный коэффициент
распределения (к\ стр. 204). Так как подвижная фаза
проходит через колонку непрерывно, введенное соединение в виде
полосы перемещается вдоль колонки со скоростью, которая
зависит от к' (см. стр. 204). При введении смесн двух
соединений с различными значениями к' более летучее нз них
распределится в газовой фазе в более высокой концентрации
н, таким образом, пройдет вдоль колонки быстрее, чем менее
летучее. В идеальном случае произойдет полное разделение
двух полос, и каждое соединение отдельно элюнруется в
конце колонки в детектор, в котором самописец зафиксирует
пик на диаграммной ленте (см., например, рис. 4.2,а).
в) Жидкостно-жидкостная (распределительная)
хроматография
Она основана на том же принципе, что и ГЖХ, но
подвижная и неподвижная фазы в ней являются жидкими. Это,
казалось бы, ставит значительные экспериментальные
трудности в связывании неподвижной жидкой фазы и
предотвращении ее смывания подвижной фазой. Однако за последние
несколько лет эта трудность была преодолена путем разработки
связанных жидких фаз. В них неподвижная жидкая фаза,
как и в ГЖХ, наносится тонкой пленкой на поверхность
гранулированного инертного твердого вещества, но различие
состоит в том, что молекулы жидкой фазы химически связаны
с поверхностью частицы и поэтому не могут быть смыты.
Чаще всего используют сферические частицы силикагеля (рис.
4.25), к которым в качестве неподвижной жидкости
прикреплен одним концом длинноцепочечный углеводород (например
октадекан (С^Н^)). Природу и полярность связанной
жидкости можно менять путем включения в углеводородную цепь
полярных (например, циано-, амино- или диольной) групп.
Эти связанные вещества помещают в колонки (они очень
важны для ВЭЖХ) или наносят на ТСХ-пластинки. Методы
проведения разделения во многом те же, что и в жидкостно-
адсорбционной хроматографии, но с использованием других
систем растворителей в качестве элюентов.
14-6729
CioH,
CioH4-
Si(OH)2
1
0 OH
1 1
Si Si
Si(oh)
/ \
0 0
1 1
Si Si
ч\
WW4
Частичка силикагеля
Xy jfe^"^ Связанные углеводородные
Рис. 4.25. Связанная неподвижная фаза в распределительной
хроматографии.
4.3.2. Контроль активности сорбента
Активность сорбента контролируют путем регулирования
водного содержания (стр. 205); ее количественно определяют
по шкале Брокмана (степени I-V) |14]. Наилучший способ
получить нужную активность - сначала удалить всю воду на-
Таблица 4.6. Дезактивация силикагеля и оксида алюминия водой
Содержание воды"
в силикагеле, %
Степень активности
0
5
15
25
38
j
II
III
IV
V
Содержание воды а
в оксиде алюминия, %
0
3
6
10
15
а По массе: X воды + (100-Х) сорбента.
X ■ i > ич . - . ие смесей 211
Таблица 4.7. Определение степени активности8 по Брокману
Краситель Степень активности
II Ш IV V
Азобензол 0,59 0,74 0,85 0,95
п-У . « 0,16 0,49 0,69 0,89
п-Аминоазобензол 0,00 0,03 0,08 6,19
а Значения R, на силикагеле или оксиде алюминия при использовании в
качестве элюента 20% толуола + 80% петролейного эфира.
греванием сорбента в муфельной печи. Для того чтобы
достичь степени активности I, оксид алюминия нагревают в
течение 3 ч прн 500°С, а силнкагель 16 ч при 400°С. Затем
материал дезактивируют до нужного уровня добавлением
воды, как показано в табл. 4.6.
Дезактивацию производят, помещая сорбент и требуемое
количество воды в плотно закрытые емкости и тщательно
перемешивая в течение 5 ч. С большими количествами
сорбента это можно проделать, вращая электродвигателем
стеклянную или металлическую емкость, посаженную на вал, а с
малыми количествами - в закрытом роторном испарителе.
После приготовления сорбенты следует хранить в плотно
закупоренных емкостях, чтобы предотвратить поглощение воды
нз атмосферы.
Степень активности по Брокману можно определить
нахождением значений R, различных азокрасителей (табл. 4.7).
4.3.3. Приготовление ТСХ-пластин
БОЛЬШИЕ ПЛАСТИНЫ. Для нанесения покрытий
используют специальные приспособления. Хорошие результаты
дает тщательное обезжиривание пластины детергентом перед
нанесением покрытия. Приспособление для нанесения
покрытия требует использования суспензии сорбента (размер частиц
10-30 мкм) в воде (2 мл воды иа 1 г сорбента).
Ориентировочно, чтобы покрыть пластинку площадью 100 х 20 см
слоем тощиной 0,25 см, требуется 50 г сорбента. После
нанесения покрытия пластины оставляют сохнуть до тех пор,
пока нанесенный слой не застынет, затем нх переносят на
специальную подставку и нагревают в термостате при 120 -
130° С в течение 4 ч. Такой способ дает пластины с
активностью по Брокману степени IMII. Пластины должны
храниться в ящике над силикагелем.
Глава 4
по-
ПРЕДМЕТНЫЕ СТЕКЛА. Их покрывают, погружая п
парно прижатыми тыльной стороной друг к другу в
суспензию сорбента в метаноле 1:1; затем стекла б£3рт выним^г
LrtTT ДЛНННЫС КРМ б0ЛЬШИМ и Удельным пальце
„»^ /еЗИН°ВЫе пеРтатки>- Покрытые пластинки
разъединяют, кладут в горизонтальном положении на подходящую
Ж""* " ак™виР>"от. нагР»ая 30 мин в термостате 7р"
v„ С-,Толщина "О» зависит от состава суспензии.
Пластинки удобно хранить в небольшой широкогорлой банке с
завинчивающейся крышкой и нспользоватГпо мере необходимей
ш
5. ПОДГОТОВКА ОБРАЗЦОВ ДЛЯ
СПЕКТРОСКОПИИ
Содержание этой главы ограничено ее заглавием и не
затрагивает теорию спектроскопии илн интерпретацию спектров.
Предполагается, что читатель знаком с теоретическими
основами каждого метода и принципами работы на приборах.
Здесь не приводятся инструкции по работе на инфракрасных
(ИК) и ультрафиолетовых (УФ) спектрометрах, так как онн
разнообразны и лучше всего усваиваются во время
демонстрации приборов.
5.1. ИНФРАКРАСНАЯ СПЕКТРОСКОПИЯ
Существует несколько рутинных методов подготовки
образцов. Наиболее популярные в лабораториях органического
синтеза "жидкие пленки" и "суспензии в вазелиновом масле"
обсуждаются в подразделе "а", а краткое описание
использования таблеток КВг и растворов - в подразделах "б" и "в"
соответственно.
а) Жидкие пленки и суспензии в вазелиновом масле
Самый простой метод заключается в использовании двух
пластин поваренной соли (NaCl) - обычно диаметром 25 мм
н толщиной 4 мм, отшлифованных до получения оптически
плоских поверхностей. Анализируемое вещество просто
наносится в виде тонкой пленки между двумя пластинками (рис.
5.1). Жидкие образцы анализируют в виде тонкой пленки
чистой жидкости, твердые же вещества необходимо сначала
растереть с инертной жидкостью до пастообразной тонкой
суспензии. Приготовление образца и нанесение его на
пластинки более детально обсуждается ниже. Из-за хрупкости и
дороговизны с пластинками необходимо обращаться очень
осторожно. Совершенно очевидно, что поскольку они сделаны
нз соли, то являются водорастворимыми и нх поверхности
быстро выходят из строя прн контакте с влажными
образцами или кожей пальцев. С ними следует работать в
резиновых перчатках (брать только за края), используя щипцы илн
пинцет, и хранить в маленьком эксикаторе над
самоиндикаторным силнкагелем. При нанесении образца на пластинки
положите их на лист чистой фильтровальной бумаги или
бумажное полотенце (а не просто на стол).
214
Глава 5
Пластинки
поваренной
соли
/\
ГИЛ
Жидкая пленка или пленка
в вазелиновом масле
Рис. 5.1. Пластинки, используемые в ИК-спектроскопии, для нанесения
жидких пленок или суспензий в вазелиновом масле.
ЖИДКОСТИ. Поместите каплю чистого жидкого образца
на одну нз пластинок, а затем накройте второй пластинкой,
чтобы размазать жидкость в пленку.
ТВЕРДЫЕ ВЕЩЕСТВА. Поместите немного твердого
вещества (5 мг) в маленькую агатовую ступку и разотрите его
в мелкий порошок. Затем добавьте капельку нуйола (илн
другой суспендирующей жидкости) н разотрите смесь до
получения однородной пастообразной массы. Для того чтобы
взять необходимое количество вазелина, нужна практика;
большинство людей в первое время берут его слишком много,
тогда как необходимо самое минимальное количество,
позволяющее получить пастообразную массу. После тщательного
растирания маленьким шпателем соскоблите пасту со ступки
и нанесите ее тонким слоем в центре одной из пластинок.
Сверху положите вторую пластинку н слегка придавите ее,
чтобы размазать суспензию в тонкую пленку.
Качество суспензии оказывает существенное влияние на
качество спектра. Слишком маленькое количество вазелина
дает грубую суспензию, которую трудно размазать, что
приводит к широким, плохо разрешенным пикам в спектре,
слишком слабое растирание также приводит к плохому
разрешению, а слишком большое количество вазелина дает
Нуйол (вазелин) - это вязкая смесь насыщенных углеводородов,
дающая ИК-поглощение С-Н-связей при 2950, 1450, 1375 см"1, которое,
конечно, перекрывает поглощение образца при этих частотах. Альтернативным
веществом, исключающим поглощение С-Н связей, является гексахлорбутадиен.
Подготовка образцов для спектроскопии 215
спектр, в котором доминируют три пика вазелина со
слабыми пиками поглощения образца.
После нанесения образца на пластинки вставьте их в со-
ответствющий держатель и поместите держатель в один из
лучей (обычно ближайший) прибора. Большинство держателей
пластинок приводит к некоторому ослаблению луча, поэтому
необходимо уравновесить его влияние, поместив аналогичный
держатель в другой луч.
После записи спектра пластинки следует сразу же
вымыть. Отделите их щипцами или пинцетом и промойте
каждую струей дихлорметана нз промьгаалки.
Меры безопасности. Промывайте пластинки над
склянкой для отходов, предпочтительно в вытяжном шкафу.
Вытрите пластинки насухо бумажной салфеткой и храните
над силикагелем.
б) Таблетки КВг
Второй, наиболее популярный метод, используемый для
твердых образцов, - это растереть их с бромидом калия до
порошкообразного состояния и затем спрессовать смесь при
высоком давлении (700 - 1050 атм) до получения твердой
прозрачной таблетки. Обычно для этого требуется 1 - 2 мг
образца и примерно 100 мг бромида калия (общее
необходимое количество вещества зависит от размера формочки
пресса, но содержание его должно составлять около 1% от
массы образца).
Вещество следует растереть как можно тоньше в агатовой
ступке или лучше в маленькой шаровой мельнице. Это
следует делать как можно быстрее, чтобы свести к минимуму
^поглощение атмосферной влаги бромидом калия. Смесь затем
переносят в формочку гидравлического пресса и прессуют.
Приготовленная таким образом таблетка должна быть
идеально прозрачной для видимого света. Однако хорошие спектры
могут иногда давать полупрозрачные или даже совсем
светонепроницаемые таблеткн. Непрозрачность таблетки обычно
вызывается поглощенной влагой или использованием слишком
большого количества образца.
Специального качества для ИК измерений, высушенный при 110° С и
хранящийся в эксикаторе.
216
Глава 5
Рис. 5.2. Кювета для записи ИК-спектров
Совершенно очевидно, что на изготовление таблеток
уходит больше времени, чем на приготовление пленки в
вазелиновом масле, и для этого требуется больше единиц
оборудования, но преимущество таблеток состоит в том, что в
спектрах отсутствуют какие бы то нн было пнки поглощения
вазелинового масла (или растворителя), закрывающие пики
поглощения образца.
в) Спектры растворов
Как твердые, так и жидкие образцы можно исследовать в
виде растворов. Главное практическое достоинство этого
метода заключается в том, что он не деструктивен и ценные
образцы могут быть восстановлены Раствор помещают в
кювету (рис 5.2) с окошками из поваренной соли [см. в
подразделе "а** меры предосторожности прн работе с ними],
толщину которой можно регулировать при помощи прокладок в
пределах 0,015 - 1,0 мм. Самые тонкие прокладки (0,015 -
0,05 мм) обеспечивают толщину, позволяющую снимать
спектры чистых жидкостей, тогда как спектры растворов обычно
снимают при толщине в пределах 0,1 - 1,0 мм. Обычно
кювета толщиной 0,5 мм имеет объем 0,3 мл; для сильнопогло-
щающнх соединений требуется 0,03 М концентрация
раствора. Кювету заполняют шприцем, слегка приподнимая один
конец во время заполнения, для того чтобы весь воздух был
вытеснен. После использования кювету следует сразу же
вымыть сухим растворителем (обычно дихлорметаном), продуть
чистым воздухом или азотом н хранить в эксикаторе над си-
лнкагелем.
Подготовка образцов для спектроскопии
217
РАСТВОРИТЕЛИ. ИК-поглощение применяемого
растворителя должно быть как можно меньше, особенно в той
области, которая представляет интерес при исследовании образца.
Не существует растворителей, полностью прозрачных по
всему диапазону (660 - 4000 см-1) ИК-поглощения, но четырех-
хлористый углерод и дисульфид углерода имеют узкую
область поглощения и поэтому наиболее часто используются в
качестве растворителей.
Меры безопасности. Оба этих растворителя токсичны
при вдыхании, а дисульфид углерода имеет очень низкую
температуру воспламенения.
Свободные от поглощения области ("окна") для этих
растворителей (рнс. 5.3) перекрываются, поэтому их совместное
использование покроет всю область. Чтобы компенсировать
поглощение растворителей, в контрольный луч спектрометра
помещают ячейку с чистым растворителем. Следует отметить,
однако, что, хотя это и предотвращает появление пиков
растворителя в ИК-спектре образца, любые появившиеся в
данном месте пикн поглощения образца будут маскироваться и
либо не проявятся совсем, либо пропишутся с очень малой
интенсивностью.
5.2. ЯДЕРНЫЙ МАГНИТНЫЙ РЕЗОНАНС
Спектры ЯМР на ядрах 1Н или UC или других магнитных
ядрах получают, используя растворы образца в подходящем
растворителе (не перекрывающем нужную область). При
работе иа большинстве спектрометров в ампулу диаметром
5 мм (рис. 5 4,а) заливают 0,4 мл раствора образца на глу
cs2
CCU
ww<
вфф*
^//f.irp^p:
1666 1250 1000
Волновое число, см-"1
ЪШШТА
Рис. 5,3. "Окна" в ИК-области для дисульфида углерода и четыреххло-
ристого углерода.
о5 мм
Ы Фильтр
\й^-из ваты
Воздух
6
Рис. 5.4- Приготовление образца для спектроскопии ЯМР.
бнну примерно 35 мм. Необходима тщательная подготовка
раствора, так как качество полученного спектра зависит не
только от чистоты образца, но также от чистоты ампулы и
растворителя н, в частности, от полноты удаления
взвешенных частичек при фильтровании.
а) Ампула для образца в ЯМР
Ампула - это точное изделие, изготовленное из
тонкостенной трубки, открытый конец которой легко откалывается или
отламывается. Обращайтесь с ней осторожно, так как
стоимость ампул очень высока. Сразу же после использования
ампулу следует тщательно промыть ацетоном или дихлорме-
таном, высушить, продув через нее чистый воздух или азот
при помощи тонкой металлической трубки (рис. 5.4,6), и
затем покрыть колпачком, чтобы не допустить попадания пыли
при хранении. Сушка ампул в сушильном шкафу не
рекомендуется, так как они захватывают пыль, если их оставить
на длительное время в открытом состоянии; кроме того, ечн-
"^■™""j OfrflMQi Ml ТТГШНШММ
219
тается, что горячая сушка может привести к деформации.
Весьма важным является немедленное промывание, так как
старые растворы могут разлагаться и полимеризоваться, что
приводит к образованию трудиоудаляемых остатков. В таких
случаях отрезок трубки, используемый для чистки (рис.
5.4,6), можно применять в качестве "шомпола*1, на который
наматывают ватку, смоченную с*от.ч - -г ующим органическим
растворителем. Если это не помогает, тогда
проконсультируйтесь у преподавателя; не используйте абразивные материалы
или хромовую кислоту.
б) Приготовление раствора
В большинстве случаев при проведении лабораторной
работы вам укажут, сколько образца и какой растворитель
использовать, - если нет, тогда смотрите подразделы "в" и "г**
ниже. Раствор удобнее всего готовить в небольшой пробирке
(3 мл), предварительно вымытой и высушенной в термостате.
Взвестьте образец, добавьте из градуированной пипетки
объемом 1 мл 0,4 мл растворителя и слегка постучите по
пробирке, чтобы перемешать содержимое и растворить образец.
Весьма важно профильтровать раствор для удаления пыли
или других взвешенных частиц. Это можно быстро и легко
сделать прн помощи пастеровской пипетки с ватным
фильтром (рис 5.4,0). Чтобы приготовить фильтр, возьмите
маленький кусочек чистой сухой ваты, сожмите его и вставьте
в узкий конец пипетки, а затем при помощи другой пипетки
довольно плотно утрамбуйте его.' Для получения наилучших
результатов пропустите через фильтр немного чистого
растворителя, чтобы смыть отдельные волокна. Затем поместите эту
пипетку в ампулу для ЯМР (рис. 5.4,в) и закапайте в нее
раствор при помощи другой пастеровской пипетки, которая
предназначена только для отбора чистого растворителя, чтобы
избежать его загрязнения.
По завершении фильтрования накройте ампулы крышкой
н держите их в вертикальном положении вдали от света и
тепла до тех пор, пока не запишите спектр.
в) Растворители для ЯМР
В ЯМР применяют различные предварительно дейтериро-
Для некоторых спектрометров требуется наличие внутреннего стандарта,
обычно тетраметилсилана [(CHp.Si]. Fcj™ он необходим для вашего
персонального прибора, то его, вероятно, уже добавили в приготовленную бутыль
с растворителем для ЯМР (0,5%). Узнайте о наличии стандарта у вашего
преподавателя и при необходимости добавьте его на этой стадии.
220
лава 5
ванные растворители, например дейтерохлороформ (CDC13)
или дейтеробеизол (C6D6). Совершенно очевидно, что
использование дейтерированных растворителей необходимо в
спектроскопии ЯМР *Н, чтобы поглощение *Н растворителя не
закрывало спектр. Однако дейтерированные растворители
необходимы также (хотя это и менее очевидно) при записи
спектров ЯМР на других ядрах (например, UC), так как в
большинстве приборов ядро дейтерия используется в качестве
стандарта. Для *Н спектроскопии требуемое содержание
дейтерия составляет 99,5% или более. Наиболее широко
распространенный растворитель - дейтерохлороформ (CDCL,: А* ОН)
7,25; 8(иС) 76,9); он обладает хорошей растворяющей
способностью, нереакционноспособен по отношению к
большинству функциональных групп (реагирует с некоторыми аминами)
и довольно дешев.
Меры безопасности. Хлороформ токсичен, и с ним
следует работать в вытяжном шкафу.
Другие используемые растворители (но намного более
дорогие, чем CDCL,) приведены в табл. 5.1.
Проверку растворимости, конечно, не следует проводить в
дейтерированных растворителях.
г) Количество образца
Здесь очень трудно давать определенные рекомендации,
так как количество образца зависит от ядра, спектр которого
предстоит изучить, и природы спектрометра. Однако
некоторые советы относительно 'Н и иС следует высказать.
ЯМР 'Н. Многие рутинные спектры для учебных целей и
исследовательской работы записывают при рабочей частоте
60 МГц; для этих целей спектр с хорошим соотношением
сигнал/шум можно получить при использовании около
0,1 ммоль образца (скажем, 30 - 50 мг для соединений с
молекулярной массой до 500). На более сложном фурье-спек-
трометре удается получить спектры при наличии значительно
меньшего количества образца (1 мг и менее), но, чтобы
сократить время записи на приборе, следует брать примерно
20 мг образца.
Полезно знать химический сдвиг 'Н, так как это поглощение можно
наблюдать в спектрах, записанных с использованием очень разбавленных
растворов, когда концентрация остаточных 0,5% протонов, содержащихся в
растворителе, становится сравнимой с концентрацией растворенного вещества.
Таблица 5.1. Растворители для ЯМР
Дейтерированный растворитель
s его- fi(uo
Хлороформ (CDCy
Бензол (C6D6)
Толуол (CD3C6D5)
Ацетон (CDjCOCDj)
Диметилсульфоксид <CD3SOCD3)
Трифторацетоуксусная кислота (CF,C02D)
Вода (D20)
а Смотри сноску на стр. 220.
7,25
7,16
1,99 и
6,98 - 7,09
2,05
2,50
11,30
4,8 (при рН 7;
зависит от рН)
76,9
128,0
19,2 и
128,9; 137,5
29,8; 206,0
39,5
115,7; 163,8
ЯМР 13С. Из-за низкого содержания в природе и более
слабого магнитного момента ЯМР UC примерно в 6000 раз
менее чувствителен, чем ЯМР 1Н. Поэтому растворы должны
быть как можно более концентрированными. При работе на
обычном высокочастотном фурье-спектрометре (90 МГц для
ВС) с количеством образца 0,1 ммоль для получения полного
спектра с протонной развязкой требуется накопление
примерно в течение I ч и еще в течение I ч для получения
двумерного спектра по методу DEPT (неселективная передача
поляризации) с целью определения протонной мультиплетно-
сти углеродов (т. с. СН3, СН2, СН или четвертичного).
Между временем накопления и концентрацией имеется следующее
важное соотношение: если концентрацию уменьшить
наполовину, то необходимое время накопления возрастает в четыре
раза. Таким образом, для образца с концентрацией 0,025
ммоль потребовалась бы запись на спектрометре в течение
всей иочи.
5.3. МАСС-СПЕКТРОМЕТРИЯ
Это высокочувствительный метод, позволяющий получать
спектры при использовании всего лишь нескольких
микрограммов вещества, однако работать удобнее с большими
количествами образца (в размере нескольких миллиграммов,
если вы ими располагаете). Не требуется никакой
предварительной подготовки образца, так как при необходимости эту
работу выполняет оператор. Оператор должен знать
приблизительную молекулярную массу соединения и его Тм или
222
Глава 5
Рис. 5.5. Кювета для образца в УФ-спектроскопии.
71кнп; кроме того, ему полезно дать некоторую информацию о
термической стабильности, если она известна.
Важно, чтобы образец был чистым. Масс-спектры
содержащих примеси неочищенных соединений могут ввести вас в
заблуждение, так как пики слегка более летучих примесей,
присутствующих даже в мизерных количествах, могут быть
записаны с более высокой интенсивностью, чем пики
основного компонента.
5.4. УЛЬТРАФИОЛЕТОВАЯ СПЕКТРОСКОПИЯ
Запись УФ-спектра требует приготовления очень
разбавленного раствора образца (известной концентрации) в
растворителе, который прозрачен к УФ-излучению. Метод
приготовления раствора описан ниже.
а) Кювета для образца
Обычная кювета для образца изображена на рис. 5.5.
Длина пути оптического луча в кювете, как правило,
составляет 10 мм (объем 3 мл), но широко используются и 1-мм,
и 40-мм кюветы. Эти весьма точные и очень дорогие
изделия изготавливают из кварца. Подобные кюветы, сделанные
из стекла, применяют для записи спектров в видимой
области света. Важно избегать появления малейших царапин или
повреждений прозрачных стенок кюветы. После использования
ШИЯШНИ ра****'1"* Л" *МЕ*ТШЯЯ№
223
кювету следует тщательно промыть растворителем и продуть
чистым воздухом или азотом. Для более тщательной
промывки пользуйтесь одним из детергентов для стеклянной посуды
с последующим промыванием дисгнлли.оиаяной водой и
затем этанолом.
Никогда не пользуйтесь абразивными порошками или
ершиками для пробирок.
При записи спектра кювету с раствором образца
помещают в один луч спектрометра, а в другой луч устанавливают
такую же кювету, содержащую только растворитель.
б) Растворители
В качестве растворителей обычно используют насыщенные
углеводороды (например, гексаи) или простые спирты
(например, этанол). Они должны быть специального
"спектроскопического" качества и не содержать примесей, поглощающих в
УФ-области, наличие которых возможно в растворителях,
предназначенных для проведения обычных лабораторных
работ. Поскольку обычно требуются разбавленные растворы
(5 - 50 мг/л), растворимость редко является проблемой.
в) Концентрация
Требуемая концентрация связана с молярным
коэффициентом экстинкции образца (см. уравнение ниже). Полученный
вами спектр отражает зависимость поглощательной
способности А (или оптической плотности) от длины волны.
Оптическая плотность зависит от концентрации раствора с и длины
пути оптического луча в кювете (толщины кюветы) с
образцом /, а е - константа пропорциональности, которая
связывает Леси/ при определенной длине волны, т. е.
А = ed
(с обычно выражают в молях на литр, а / - в сантиметрах;
в таком случае е - это молярный коэффициент экстинкции,
который обычно приводится без размерностей, хотя он не
является безразмерным). Таким образом, для получения
значения оптической плотности бензойной кислоты С6Н5С02Н
(ЛЕ'(он)юм = 228 им; е^ = 10660), равного 1,9 (при
использовании почти полной высоты диаграммы), в кювете
толщиной 1 см потребуется раствор с концентрацией 1,8-10"4
моль/л (0,022 г/л).
224
Глава 5
г) Приготовление раствора
Непосредственное приготовление необходимых для УФ-
слектроскопин очень разбавленных растворов потребовало бы
высокой точности взвешивания крошечных количеств образца
или использования большого количества дорогого растворителя
"спектроскопического" качества. Чтобы избежать этого,
обычно предварительно готовят раствор, концентрация которого в
10 - 100 раз превышает необходимую концентрацию, а
затем, используя пипетки и мерные колбочки, разбавляют его
до требуемой концентрации. Если УФ-спектр соединения
известен, ознакомьтесь с ним (см. ссылки иа стр. 227) и по
уравнению, приведенному в предыдущем подразделе,
вычислите требуемую концентрацию. -Например, чтобы получить
УФ-спектр бензойной кислоты [см. подраздел "в"],
предварительно приготовьте этанольиый раствор бензойной кислоты
(55 мг) в мерной колбочке на 25 мл и после ее полного
растворения разведите I мл этого раствора до 100 мл в
другой мерной колбе. Если соединение новое, тогда вам
придется экспериментировать с различными последовательными
разведениями ранее приготовленного раствора, записывая спектр
каждого, пока не будет получена требуемая концентрация.
Приготовление основного раствора и последующие
разбавления необходимо производить в соответствии с обычными
правилами, используемыми в количественной работе: все
пипетки и колбы должны быть абсолютно чистыми и не
содержать органических загрязнений и растворителей, которые
могли бы дать ложные поглощения.
6. ПОИСК ХИМИЧЕСКОЙ ИНФОРМАЦИИ
Использование литературы по органической химии -
необходимое дополнение к экспериментальной работе на всех
уровнях. (Для более подробного ознакомления отсылаем
читателя к книге: Maizell R. Е. How to Find Chemical
Information, 2nd Edn., Wiley lnterscience, New York, 1987).
Под "литературой" мы подразумеваем огромный объем
информации, хранящейся в библиотеках, по химическим
реакциям и многим миллионам соединений, которые были
синтезированы со времени зарождения органической химии в
середине прошлого столетия.
Основной источник информации - это первичная
литература (первоисточники). Новые химические сведения сначала
сообщаются их первооткрывателем в "оригинальной" статье в
одном из сотен химических журналов, выходящих по всему
свету на многих языках. В большинстве химических
библиотек хранятся полные комплекты наиболее важных из этих
журналов, от первых номеров вплоть до настоящего дня.
Менее известные журналы можно обычно получить по
требованию во временное пользование из центральных библиотек.
Кратчайший доступ к этому обширнейшему кладезю знаний
открывает ознакомление с информацией в журнале Chemical
Abstracts, в котором содержится краткое изложение (реферат)
каждой статьи, патента и т. п, причем вся информация
сгруппирована в большие разделы, имеющие заголовок и
индекс (см. разд. 6.4 ниже). Многое из этой информации
впоследствии собирается, перерабатывается и обсуждается в целом
ряде обзоров и монографий, посвященных конкретным темам,
например определенным типам реакций или определенным
классам соединений. Публикации такого рода составляют
"вторичную" литературу; они бесценны для получения
информации об отдельных областях химии при написании
очерков или подготовке к научному докладу. В дополнение к
этому много физических и спектроскопических данных,
особенно касающихся простых соединений, собрано в целом ряде
разнообразных справочников и атласов.
В будущем большая часть работы при поиске литературы
будет выполняться путем компьютерного просмотра баз
данных и рефератов. На пути к этой цели уже достигнуты
большие успехи, но по причинам скорее цены, нежели
практической возможности, мы все еще в значительной мере
полагаемся на ручные методы.
15 6729
326
Глава б
Давайте сначала остановимся иа различных видах
информации, которая требуется при выполнении практической
работы. Во-первых, к ней относятся физические свойства
отдельных соединений, их температуры плавления или кипения
и детали ИК-, УФ-, ЯМР- или масс-спектров; такая
информация требуется при идентификации неизвестных соединений.
Часто окончательное подтверждение структуры зависит от
сравнения физических и (или) спектроскопических свойств
неизвестного соединения со свойствами соединения аналогичной
структуры, описанными в литературе. В прошлом методы
идентификации состояли главным образом в получении
одного или нескольких кристаллических производных неизвестного
соединения и сравнении их температур плавления с данными,
опубликованными в литературе. Сейчас этот подход стал
применяться реже, так как спектроскопические методы
идентификации стали более эффективными и получили более
широкое распространение при обучении студентов, но тем ие
менее им все еще пользуются в некоторых случаях.
Физические и спектроскопические данные, необходимые для
идентификации неизвестных соединений, часто получают из
различного рода справочников (см. ниже, разд. 6.1 и 6.2), но в
случае идентификации менее распространенных соединений
обычно необходимо обратиться к справочнику Бейльштейна
(Beihteins Handbuch) (разд. 6.3) или к оригинальной
литературе через реферативный химический журнал Chemical
Abstracts (разд. 6.4) или РЖХим.
Второй вид информации, необходимой при практической
работе, касается методов получения конкретных соединений и
наиболее общих аспектов их реакционной способности.
Например, вам может понадобиться просмотреть литературу,
чтобы найти наилучший способ получения какого-либо
соединения для использования его в качестве исходного вещества.
Вы можете также заинтересоваться, является ли соединение,
которое вам необходимо приготовить или которое, возможно,
только что получили, "новым" в том смысле, что оно
никогда не было синтезировано ранее. Такую информацию можно
почерпнуть из справочника Бейльштейна, в котором собраны
обширные сведения по методам "получения и свойствам
огромного числа соединений с ссылками на оригинальную
литературу (см. табл. 6.1); пояснение, как им пользоваться,
обсуждается в разд. 6.3. Однако в большинстве случаев
необходимо обращаться к журналу Chemical Abstracts или РЖХим
и тщательно просматривать первоисточники.
6.1. ФИЗИЧЕСКИЕ СВОЙСТВА
Данные по температурам плавления и кипения распростра-
По*П1, «и«ичесали ин;-';>м ии 227
иенных органических соединений и температурам плавления
их кристаллических производных можно почерпнуть из
многих книг, касающихся идентификации органических
соединений, например из "Практикума по органической химии"
Фогеля (Vogel. Practical Organic Chemistry), в котором
соединения классифицируются по их функциональной группе и
перечислены в таблицах в порядке увеличения их температур
кипения или плавления. Наиболее исчерпывающий набор
данных содержится в CRC Handbook of Tables for Organic
Compound Identification. Информацию по еще более
широкому перечню соединений можно получить из "Словаря
органических соединений*' (Dictionary of Organic Compounds). В
этом многотомном справочнике соединения приведены в
алфавитном порядке и, что очень полезно, имеются ссылки иа
методы получения, а также данные по соответствующим
производным.
Полезные физические данные (7,пл, Тжт, плотность,
показатель преломления, растворимость) для широкого набора
распространенных органических соединений можно почерпнуть
из CRC Handbook of Chemistry and Physics.
Самое большое собрание данных по органическим
соединениям представлено в справочнике Бейльштейна (Beilstein's
Handbuch) (см. разд. 6.3), в котором собрана информация по
некоторым физическим свойствам (7^, Ткт и растворимости),
а также по методам синтеза и реакциям.
6.2. СПЕКТРОСКОПИЧЕСКИЕ ДАННЫЕ
Общее руководство по определению структуры совместным
применением УФ-, ИК-, ЯМР- и масс-спектроскопии
(например, блестящий учебник "Спектроскопические методы в
органической химии" Вильямса и Флеминга) является настольной
книгой для студентов всех лабораторных курсов. В книгах
этого типа описаны принципы, лежащие в основе каждого
спектроскопического метода, и приведены примеры
расшифровки структуры, а также таблицы данных по корреляции
для интерпретации спектров неизвестных соединений.
Часто бывает необходимо в целях сравнения найти
спектры конкретных соединений. Самые доступные справочники
перечислены ниже. Однако необходимо подчеркнуть, что оии
ие являются исчерпывающими, и поэтому за необходимой
информацией приходится обращаться к оригинальной
литературе (см. разд. 6.3. и 6.4).
УФС: 1) The Sadtler Handbook of Ultraviolet Spectra содержит
спектры 46 000 соединений.
2) Organic Electronic Spectral Data, в котором соединения раз-
Глава б
мещены согласно их эмпирической формуле; приведены
максимумы поглощения и литературные ссылки.
3) UV Atlas of Organic Compounds содержит спектры почти
1000 соединений, занесенных в указатель по хромофорной
группе.
4) Ultraviolet Spectra of Aromatic Compounds приводит
спектры 597 соединений.
ИКС: 1) The Aldrich Library of Infrared Spectra содержит
свыше 12000 спектров, расположенных в порядке
возрастания сложности молекулы. Представлен 51 класс
химических соединений, расположенных в алфавитном порядке и
по молекулярной формуле. Это прекрасный справочник для
студентов.
2) Documentation of Molecular Spectroscopy содержит наиболее
обширный набор данных, занесенных в тщательно состав-
- ленный перекрестный указатель на закодированных
карточках.
3) The Sadtler Handbook of Infrared Spectra содержит 55000
спектров (призма и дифракционная решетка) и ежегодно
пополняется.
ЯМР: I) The Aldrich Library of NMR Spectra содержит около
8500 протонных спектров, записанных при частоте
60 МГц. Это прекрасный атлас для студентов.
2) Atlas of Carbon-13 NMR Data приводит химические сдвиги
и отнесения более чем для 3000 соединений. Может также
использоваться в качестве базы данных для компьютерного
поиска.
3) Carbon-13 NMR Spectra содержит примерно 500 спектров.
Масс-спектры
ЗАМЕЧАНИЕ. К результатам идентификации соединений,
полученным путем сравнения снятых масс-спектров с
библиотечными данными, следует подходить с осторожностью,
поскольку состав осколочных иоиов может в значительной
степени зависеть от типа прибора.
1) The Eight Peak Index of Mass Spectra - каталоги восьми
наиболее распространенных ионов для более 30000
соединений.
2) American National Bureau of Standards Data Base -
каталог из более чем 42 000 спектров, который часто
используют в качестве базы данных в компьютерах современных
масс-спектрометров для сравнения с полученными
спектрами.
3) Lederberg's Computation of Molecular Formulas for Mass
Spectrometry - справочное пособие по масс-спектрометрии
Поиск химической информации 229
высокого разрешения, где приведены все возможные
молекулярные формулы, удовлетворяющие данной молекулярной
массе, величина которой известна до 5 или 6 десятичного
знака.
6.3. СПРАВОЧНИК БЕЙЛЬШТЕЙНА
(Beilstein's Handbuch)
В это уникальное справочное пособие (иа немецком
языке) вошли все органические соединения, описанные в
литературе вплоть до 1959 г.; в настоящее время оио постоянно
расширяется, с тем чтобы охватить публикации вплоть до
1979 г. (табл. 6.1). В нем дана информация по методам
синтеза, реакциям и физическим свойствам. Этим справочником
легко пользоваться, если усвоить принцип, по которому
построен материал.
Полная серия состоит из основного издания (Hauptwerk,
Н) и ряда дополнительных изданий (Ergansungbande, EI, Ell
и т. д.), часть из которых еще не завершена и которые
охватывают периоды, перечисленные в табл. 6.1. Основное
издание состоит из глав, посвященных ациклическим (тома 1 -
4), карбоциклическим (тома 5 - 16) и гетероциклическим
(тома 17 - 27) соединениям. Каждое дополнительное издание
также включает 27 томов (или групп томов) и имеет такое
Таблица 6.1. Серия "Справочник Бейлынтейна" (4-е издание)
Серия Сокращен- Период, за Цвет этикетки
ное обоз- который ох- на корешке
начение вачена вся
литература
Основное издание
Дополнительное издание 0)
Дополнительное издание (11)
Дополнительное издание (Ш)
н
Е I
Е II
Е III
Дополнительное издание (I1I/IV) Е HI/IVa
Дополнительное издание (IV)
Дополнительное издание (V)
Е IV
Е V
До 1909 г. Зеленый
1910
1920
1930
1930
1950
1960
1919 Красный
1929 Белый
1949 Синий
1959 Синий/черный
черный
1959 Будет публико-
1979 ваться, начи-ная
с 1984 - 1985 гг.
и далее
а Тома 17-27 дополнительных серий Ш и IV, охватывающие
гетероциклические соединения, объединены в один выпуск.
230
Глава б
же расположение материала, как и основное издание.
Основное и первые два дополнительных издания, охватывающие
литературу вплоть до 1929 г., снабжены сводным предметным
указателем (указателем названий соединений, приведенных в
справочнике) и сводным формульным указателем (указателем
эмпирических формул). Впоследствии каждое дополнительное
издание (начиная с ЕШ и далее) снабжается отдельным
указателем. Чрезвычайно полезной особенностью справочника
Бейльштейна является нумерация каждого соединения (и
соединений с близкими структурами), которая позволяет быстро
находить его в последующих дополнительных изданиях под
тем же самым номером (дающих относительно новые
сведения, опубликованные в литературе). Кроме того, номер
страницы, иа которой впервые описано соединение, повторяется
жирным шрифтом в верхней части страниц различных
дополнительных изданий, что позволяет легко находить любую
дополнительную информацию.
Если вы хотите найти соединение, то проще всего
обратиться к указателю. У начинающего химика часто возникают
проблемы, связанные с нюансами номенклатуры, и поэтому
проще и быстрее отыскать его по сводному формульному
указателю (2-е издание) по молекулярной формуле
соединения. В формульном указателе первыми указаны С и Н, а за
ними в алфавитном порядке другие элементы. Давайте
предположим, что мы хотим идентифицировать соединение общей
формулы С10Н10О (Тпд 41 - 42°С), которое, как известно из
его спектроскопических и химических свойств, является а,В-
ненасыщенным кетоном. К соединениям этого класса можно
отнести два названия из перечисленных под формулой
Бензилиденацетон 7,364, 1 192, II 287
о>-Этилиденацетофенон 7,368, I 194, II 290
Это означает, что оба соединения встречаются в томе 7;
номера трех страниц, которые идут следом за номером тома,
относятся к основному (Н) и первым двум дополнительным
изданиям (EI и ЕН). Обратите внимание на то, что номер
тома всегда остается одним и тем же.
В отдельных статьях методы синтеза даются первыми, затем
идут физические свойства (Г^, 71(.ип, растворимость и
т. д.), и, наконец, описываются химические свойства,
начиная с действия нагревания, окислительных и
восстановительных агентов. Если соединение образует соли, они приводятся
в конце статьи.
В статьях о двух упомянутых выше кетонах указывается,
что бензилиденацетон - это действительно твердое вещество,
Поиск химической информации
231
плавящееся при 41 - 42° С, а этилиденацетофенон - при
обычных температурах жидкость и поэтому исключается как
возможное соединение.
Третье дополнительное издание (ЕШ), охватывающее
литературу вплоть до 1949 г., завершено, но пока не имеет
сводных указателей. Для нахождения конкретных соединений
в третьем дополнительном издании либо посмотрите в
предметный или формульный указатель, приведенный в конце
каждого тома (том 7 для двух примеров выше), либо
отметьте номер страницы, на которой соединение приведено в
основном издании (Hauptwerk). Этот номер приводится в
середине верхней части соответствующей страницы в каждом
дополнительном издании. Так, бензилиденацетон можно найти
на странице 1399 в томе 7 третьего дополнительного издания.
6.4. РЕФЕРАТИВНЫЙ ЖУРНАЛ CHEMICAL
ABSTRACTS
Этот журнал, издаваемый Американским химическим
обществом начиная с 1907 г., охватывает всю литературу в
области химии. Журнал выходит еженедельно и состоит из
коротких рефератов на английском языке, излагающих суть
оригинальных статей или патентов, опубликованных где-либо
в мире. В журнале реферируются до стандартных размеров
свыше 10000 различных журналов на многих языках, причем
в каждом реферате указывается:
1) номер реферата,
2) заглавие статьи,
3) имена авторов,
4) адрес авторов,
5) сокращенное название журнала, в котором
опубликована статья,
6) год, том журнала и номер страницы,
7) язык статьи.
Ежегодные предметные, авторские, формульные и
патентные указатели составлены для каждого тома до 1962 г.,
когда выпускалось два тома в год, каждый с отдельным
указателем. С 1972 г. предметные указатели разделены на
подразделы "Химические соединения" и "Общий предметный
указатель". С интервалом в 10 лет (а с недавних пор в 5 лет)
указатели к каждому тому собираются вместе и публикуются
как "Сводные указатели". Опубликованные к настоящему
времени указатели, перечисленные в табл. 6.2, охватывают
литературу вплоть до 1986 г. По более поздним публикациям
необходимо обращаться к двухгодичным указателям.
Поиск конкретного соединения должен начинаться с самых
232
Таблица 6.2. Указатели для
Сводные
указатели
1
2
3
4
5
6
7
8
9
10
11
Номера Предмет
томов
1-20
11-20
21-30
31-40
41-50
51-55
56-65
66-75
76-85
86-95
96-105
1907-1916
1917-1926
1927-1936
1937-1946
1947-1956
1957-1961
1962-1966
1967-1971
1972-1976
1977-1981
1982-1986
Chemical Abstracts
Химические
соединения
1937-1946
1972-1976
1977-1981
1982-1986
Автор
1907-1916
1917-1926
1927-1936
1937-1946
1947-1956
1957-1961
1962-1966
1967-1971
1972-1976
1977-1981
1982-1986
Формула
1920-1946
1947-1956
1957-1961
1962-1966
1967-1971
1972-1976
1977-1981
1982-1986
Глава 6
Номер
патента
1907-1936
1937-1946
1947-1956
1957-1961
1962-1966
1967-1971
1972-1976
1977-1981
1982-1986
последних указателей и продолжаться в обратном
направлении по крайней мере до 1949 г., начиная с которого можно
обращаться к Beilstein's Handbuch. Неискушенный в поиске
исследователь обнаружит, что легче начинать с формульного
указателя, так как тогда удается избежать неприятностей,
связанных с неправильным названием соединения. Такие
проблемы, однако, можно намного упростить использованием
Index Guide ("Путеводитель к указателям"). Он выпускается
с каждым "Сводным указателем" (с восьмого) н дает
указания по индексации, альтернативные названия соединений,
перекрестные ссылки и типичные структурные схемы. Другим
бесценным помощником, призванным помочь исследователю
быстро найти соединение, является "Указатель циклических
структур" (Ring Index), в котором перечислены
систематические названия циклических соединений в соответствии с
числом колец, а также их размером и атомным составом.
Например, соединение
классифицированное как 5, 6, 6, 6 и C.N-C5N-C6-C6,
окажется в "Указателе циклических структур в разделе тетрацик-
лических под названием 1Ш-индоло[3,2-с]хинолин.
ЛИТЕРАТУРА
Литература к главе 2
1. Phipps А М., Hume D. N. J. Chem. Educ, 1968, v. 45., p. 664.
2. Rondeau R.E. J. Chem. Eng. Data, 1966, v. 11, p. 124.
3. Гордон А, Форд О. Спутник химика. - М.: Мир, 1976.
4. Физер Л., Физер М. Реагенты для органического синтеза, т. 1-VIL- М.:
Мир, 1970-1978. Fiezer L. Е., Fiezer M. Reagenls for Organic Synthesis,
vols. 1-13, Wiley-Interscience, New York, 1967-88.
5. Riddick J. A, Burger W. B. Organic Solvents: Physical Properties and
Methods of Purification, in Techniques of Chemistry, 3rd edn., voi. 2, Wiley-
Interscience, New York, 1970.
6. Penin D. D., Armarago W. L, F., Perrin D. R. Purification of Laboratory
Chemicals, 2nd edn., Perganion Press, New York, 1980.
7. Handiing Air-Sensitive Reagents, Aidrich Technical Buiietm, AL-134.
8. Equipment for Handling Air-Sensitive Reagents, Aidrich Technical Billetin, AL-
135.
9. Gill G. В., Whiting D. A Guidelines for handling air-sensitive compounds,
Aidrichlmica Acta, v. 19(2), p. 31, 1986.
10. Shriver D. F. The Manipulation of Air-Sensitive Compounds, McGraw-Hill,
Hew York, 1981.
11. Kramer G. W.t Levy А. В., Midland M. M., Brown II. С Organic
Synthesis via Boranes, Wiiey, New York, ch. 9, 1975.
12. Augustine Я L. Catalytic Hydrogenation, Edward Arnoid, London, Marcel
Dekker, New York. 1965.
13. Freifelder M. Practical Catalytic Hydrogenation, Wiiey-Interscience, New
York, 1971.
14. Rylander P. N. Catalytic Hydrogenation in Organic Synthesis, Academic
Press, New York, 1979.
15. Schenk G. O. Preparative Organic Photochemistry (ed. G. Schonberg),
Springcr-Verlag, Berlin, 1968, Ch. 46.
16. Калверт Дж., Питтс Дж. Фотохимия. - М.:Мир, 1968.
17. Synthetic Organic Photochemistry, ed. Horspool W. M., Plenum Press, New
York, 1984.
18. UV Atlas of Organic Compounds, Butterworths, London, 1965.
19. Wiersum U. E. Flash Vacuum thermolysis, a versatile method in organic
chemistry, part 1, General aspects and techniques. Reel. Trav. Chim. Pays-
234 Литература
Bas, 1982, v. 101, p. 317-332-
20. Wiersutn U. E. Preparative flash-vacuum thermolysis. The revival of pyroiytic
synthesis, Aidrichimlca Acta, 1984, v. 17, p. 31-40.
21. Brown R. F. С Pyroiytic Methods In Organic Chemistry, Academic Press,
New York, 1980.
Литература к главе 3
1. Burfield D. R., Smithers R. H. J. Chem. Educ, 1982, v. 59, p. 703.
2. Riddick J. A., Burger W. B. Organic Solvents: Physical Properties and
Methods of Purification, in Techniques of Chemistry, Wiley Intersclence, New
York, 1970, 3rd edn, v 2.
3. Гордон А, Форд Р. Спутник химика. - M.: Мир, 1976.
4. Burfield D. R. J. Org. Chem., 1982, v. 47, p. 3821.
Литература к главе 4
1. TLC Visualisation Reagents and Chromatographic Solvents, Eastman Organic
Chemicals, Rochester, New York (Kodak Publication No. 335).
2. Dyeing Reagents for Thin-Layer and Paper Chromatography, E. Merck AG.
Darmstadt, Germany.
3a. Supina W. R., Rose L. P. J. Chromatogr, Sci., 1970, v. 8, p. 214.
3b. McReynolds W. O. J. Chomatogr. Sci., 1970, v. 8, p. 685.
4. Snyder L. R„ Kirkland J. 3. An Introduction to Modern Liquid
Chromatography, 1979, 2nd edn, Wiley-Intersclence, New York.
Я Snyder L. R. J. Chromatogr., 1971, v. 63, p. 15.
6. Pryde Af Gilbert M. T. Appilcation of High Performance Liquid
Chromatography, Chapman and Hall, London, 1979.
7. Morris С J. O. R., Morris P. Separation Methods in Biochemistry, 1976, 2nd
edn, Interscience, New York.
8. 5/(7/ W. C, Kahn M., Mitra A J. Org. Chem., 1978, v. 43, No 14, p.
2923-2925.
9. Harwood L. M. Aldrichimica Acta, 1985, v. 18, p. 25.
10. Meyers A. I., Slade J., Smith R. K., Mihelich E D., Hershenson F. M.,
Liang С D. J. Org. Chem., 1979, v. 44, No 13, p. 2247-2249.
11. Snyder L. R. Principles of Adsorption Chromatography, Marcel Dekker, New
York, 1968-
12- Snyder L. R., Kirkland J. J. Modern Liqnld Chromatography, 2nd edn.,
VViley-Interscience» New York, 1979.
13. Knox J. H. High Performance Liquid Chromatography, Edinburgh University
Press, Edinburgh.
Литература 235
14. Вгосктапп H., Schodder H. Chem. Ber., 1941, Bd 74B, S. 73.
Литература к главе 6
Beiistein's Handbuch der Organischen Chemle', 4th edn., Springer-Verlag, Berlin,
1918-present. Multi-volume.
Breilmaier E., Hess C, Voelter W. Atlas of Carbon-13 NMR Data, Vols. 1 and
2, Heyden, London, 1979.
Chemical Abstracts. American Chemical Society. Chemical Abstracts Service,
Columbus, Ohio, 1907-present. 2 vols, per year with volume indexes.
Реферативный журнал Химия (РЖХим), с 1953 г. по настоящее время.
CRC Handbook of Tables for Organic Compound lndentification, 3rd edn.
compiled by Z. Rappoport. CRC Press Inc., Cleveland, Ohio, 1967.
CRC Handbook of Chemistry and Physics, ed. R.C. West, CRC Press Inc.,
Cleveland. Ohio, Annual.
Documentation of Molecular Spectroscopy, Butlerworths, London, A coiiection of
spectra on coded cards, elaborately cross Indexed.
Eight Peak Index of Mass Spectra, 2nd edn., UKCIS, The University,
Nottingham, U. K. 1974.
Dictionary of Organic Compounds, 5th edn., Chapman and Hall, London, 1982.
5 vols, plus annual supplements and 2 vols, of indexes.
Johnson L. F., Jankowski W. C, Carbon-13 NMR Spectra, a Collection of
Assigned, Coded and Indexed Spectra, Wiley, New York, 1972.
Lederberg J., Computation of Molecular Formulas for Mass Spectrometry, Holden-
Day, San Francisco, 1964.
Pouchert C. J. The Aldrich Library of Infrared Spectra, 3rd edn. Aidrich
Chemical Co., Inc., Milwaukee, Wl, 1981.
Pouchert С Л The Aidrich Library of NMR Spectra, 2nd edn., Aidrich
Chemical Co., Inc., Milwaukee, Wl, 1983
Organic Electronic Spectral Data, Wiiey, New York, 1960-present. Annual since
1972 (covers literature back to 1946).
The Sadtler Handbook of Infrared Spectra, ed. W. W. Simons, Heyden, London,
1978.
UV Atlas of Organic Compounds. Butterworths, London, 1965.
Vogel A. Textbook of Practical Organic Chemistry, 4th edn., Longman, London,
1978.
Williams D. H., Fleming I., Spectroscopic Methods in Organic Chemistry, 3rd
edn.. McGraw-Hill (U.K.), 1980.
предметный указатель
Активность сорбента 205, " тРУ6ки А"я защиты от влаги
210 - 211 воздуха 42
Арбузова колба перегонная 117 Выход процентный 18
Ъейлыитейна справочник
229 - 231
Бюхнера воронка 89, 90 - 91
Величина Rj, расчет 142
Вигре колонка 124, 125
Водяные бани 29 - 30
Воронки
- Бюхнера 89, 90 - 91
- делительные 76 - 78
- для горячего фильтрования
91 - 92
- - твердых сыпучих веществ 23
- капельные 23 - 24
- Хирша 89, 90 - 91
- Шота 88 - 90, 94
Вуда сплав 33
Высокоэффективная жидкостная
хроматография (ВЭЖХ)
- - - выбор элюирующего
растворителя 179 - 180, 181 - 182
- - - градиентное элюирование 180
- - - запись хроматограммы 170
- - - качественный анализ 171 - 172
- - - количественный анализ 172
- - - методы ввода пробы 167 - 169,
170 - 171
- - - на связанных фазах 180 - 181
- - - неподвижные фазы 176 - 179,
181
- - - оборудование 164 - 169
- - - обращеннофазовая 181
- - - объем пробы 171
- - - оптимизация разделения 173 -
175
- - - принцип разделения 173
- - - прчмофазовая 182 - 183
Высушивание
- кристаллов 98 - 100, 102 - 103
- пистолет сушильный 39 и ел., 99
- приборов 40-41
- растворителей 36 - 37
- растворов 81-82
- твердых веществ 38-40
Газо-жидкостная хроматография
(ГЖХ)
- - детекторы 148 - 149
- - запись хроматограммы 151 - 152
- - качественный анализ 152 - 154
- - количественный анализ 154 - 156
- - колонки 149 - 151
- - неподвижные фазы 157 - 162
- - общие положения прн работе на
хроматографе 163
- - принципы разделения 206 - 209
- - расход газа-носителя 163
- - схема хроматографа 148 - 149
- - температурный режим 160 - 162
- - форма пика 151 - 152
Гидрирование каталитическое 61 65
- - при атмосферном давлении 62 -
65
Дефлегматоры 123 - 126
Дэвиса холодильник см.
Холодильники
Жидкостная хроматография; см.
также Хроматография с элюиро-
ваннем самотеком, Флаш-хрома-
тография. Высокоэффективная
жидкостная хроматография
- - активность сорбента 205, 210 -
211
- - сила растворителя 202 - 206
- - среднего давления 197
- - - - аппаратурное оформление
197 - 198
- - - - выбор элюирующего
растворителя 198 - 199
- - - - загрузка образца 199
- - - - набивка колонки 200 - 201
- - - - промывание колонок обратной
струей 199 - 200
- - - - скорость потока при элюиро-
ванин 199
Запись результатов эксперимента
17 - 18
Предметный указатель
.
Инертная атмосфера
- - перенос и загрузка реагентов и
реакционных смесей 48 - 58
- - проведение реакций 42 - 58
- - - приборы 43 - 46
- - - техника 46 - 58
Инфракрасная спектроскопия
- - выбор растворителей 217
- - жидкие пленки 213 - 214
- - растворы 216 - 217
- - суспензии в вазелиновом масле
214 - 215
- - таблетки бромида калия 215 -
216
Капилляры воздушные для вакуумной
перегонки 139 - 140
"Кипятильнички" 28, 119
Кляйзена колба 117, 120
Колбонагреватели 29
Кофлера столик для определения
температуры плавления 112 - 113
Кристаллизация 84 и ел.
- выбор растворителя 103 - 107
- затруднения 93 - 94
- микроколичеств 100 - 103
- подготовка образцов для анализа
сжиганием 107 - 108
- соединений с низкой
растворимостью 108 - 109
Кюгельрора прибор для вакуумной
перегонки 130 - 131
Лгибиха холодильник см.
Холодильники
Масляные бани 31-33
Масс-спектрометрия 221 - 222
Меры безопасной работы 13-17;
см. также Меры безопасности
при описании конкретных работ
Металлические бани 33
Мешалки
- лопастные 26 - 27
- магнитные 27 - 28
Нагревание с обратным холодиль
ником 28 - 31
Насосы вакуумные
- - водоструйный 111 - 112
- - масляный ротационный 112 - 113
Охлаждающие агенты 33 - 36
Очистка веществ
- - кристаллизацией 84 - 109
- - перегонкой 114 - 135
- - сублимацией 135 - 137
- - хроматографическая
см. Хроматография
Пальчиковый холодильник
см. Холодильники
Перегонка 114 - 140
- вакуумная 119 - 123
- микроколичеств 128 - 132
- молекулярная 132 - 133
- при атмосферном давлении 115 -
119
- с водяным паром 133 - 135
- фракционная 123 - 127
Перекристаллизация см.
Кристаллизация
Перемешивание 24-26
Перкина форштос 121 - 122
"Пистолет" сушильный 39 и ел,, 99
Приборы для реакций; см. также
Конкретные методы
- - - вибраторы 28
- - - воронка для сыпучих веществ
23
- - - капельные воронки 23, 24
колбы 19 - 22
- - - мешалки 24 - 28
насадки 20 - 21
- - - холодильники 30 - 31
- - - шлифы 19-22
Растворители
- осушка см. Высушивание
растворителей
- удаление 73 - 76
Рашига кольца 125
Реакции в жидком аммиаке 58 - 61
Реферативный журнал Chemical
Abstracts 231 - 233
Роторный испаритель 73 - 76
Сокслета экстрактор 80-81
Спектроскопические данные,
справочная литература 227 - 229
Спектроскопия
- инфракрасная 213 - 217
- масс-спектромегрия 221 - 222
- подготовка образцов и
растворителей 213
- ультрафиолетовая 222 - 224
- ядерного магнитного резонанса
217 - 221
238
Справочная литература по
органической химии 225 и ел.
Сублимация твердых веществ 135 -
137
"Сухой лед" 34
Температура плавления
- - как критерий чистоты 85
- - методы определения 109 - 114
- - - - в капилляре 109 - 112
- - - - на столике Кофлера 112 -
113
----- столике с микроскопом
ИЗ - 114
- - общие принципы 109
Техника проведения реакций
- - - в жидком аммиаке 58
- - - каталитического гидрирования
61
- - - с реагентами, чувствительными
к влаге и кислороду воздуха 47
- - - флаш-вакуумного пиролиза 69
- - - фотохимических 65
Тонкослойная хроматография
- - аналитическая 141 - 147
- - величина R, 142, 146
- - выбор растворителя 146 - 147
- - идентификация компонентов
смеси 144 - 146, 184
- - качественный анализ 145 - 146
- - нанесение образца 143 - 144,
183 - 184
предварительный анализ для
колоночной хроматографии
186 - 187
- - препаративная 183 - 184
- - приготовление пластин 211 - 212
- - проявление хроматограммы 144
- - сорбенты 142 - 143
Ультрафиолетовая спектроскопия 224
- - концентрация растворов для 223
- - кювета для образца 222 - 223
- - растворители для 223
Фенке спирали 125
Физические свойства веществ,
справочные данные 226 - 227
Фильтрование
- горячих растворов 88 - 92
- кристаллов 94 - 96
- микроколичеств 100 - 103
- охлажденных растворов 96 - 97
- под азотом 97
Флаш-вакуумный пиролиз 69-71
Флаш-хроматография
- выбор элюента 188 - 189
- набивка колонок 189 - 191
- нанесение образца 191 - 192
- на сухой колонке 193 - 197
- размеры колонок 190
Фотохимические реакции (фотолиз)
- - источники УФ-излучения 67 -
68
- - прибор 65 - 67
- - условия и растворители 68 - 69
Хирша воронка 89, 90 - 91
Холодильники
- воздушный 30, 31
- Дэвиса с двойной рубашкой
охлаждения 30
- Либиха 30
- пальчиковый 128 - 129
- типа "AIRFLUX" 31
Хроматография; см. также
Конкретные методы
- аналитическая 141 - 183
- препаративная 183 - 204
- принципы 204 - 212
- с элюированием самотеком
- - - - загрузка образца 203
- - - - колонки 201
- - - - набивка колонки 202 - 203
- - - - сбор фракций 204
сорбенты 201 - 202
- - - элюирование 203 - 204
Шлифы 19-20
Шота воронка 88 - 90, 94
Эксикаторы 38 - 39
Экстракция
- жидкостей 76 - 78
- непрерывная 78-81
- твердых веществ 81
Ядерный магнитный резонанс 217
- - - ампулы для образцов 218 - 219
- - - количество образца 220 - 221
- - - приготовление раствора образца
219
растворители 219 - 220, 221.
ОГЛАВЛЕНИЕ
Предисловие авторов 5
Предисловие рецензента 7
Меры безопасности и контроль за работой в лаборатории 10
1. Введение 11
1.1. Экспериментальные методики 11
1.2 Хорошая лабораторная практика 12
1.3. Меры безопасности в лаборатории 13
1.3.1. Меры безопасности при работе с химическими веществами.. 14
1.3.2. Пожароопасность 16
1.3.3. Работа с вакуумом и давлением 16
1.4. Ведение записей 17
1.4.1. Запись экспериментальных данных 17
1.4.2. Окончательный отчет 17
1.5. Образцы и спектры 18
2. Проведение реакции 19
2.1. Основные методы 19
2.1.1. Оборудование и приборы 19
2.1.2. Смешение реагентов 22
2.1.3. Перемешивание реакционных смесей 24
2.1.4. Контроль за температурой 28
2.1.5. Реакции в безводной среде и инертной атмосфере 36
2.2. Техника специальных работ 47
2.2.1 Работа с реагентами, чувствительными к влаге и кислороду
воздуха 47
2.2.2. Реакции в жидком аммиаке 58
2-2 3. Каталитическое гидрирование 61
2.2 4 Фотохимия (фотолиз) 65
2.2.5. Флаш-вакуумный пиролиз 69
3. Выделение и очистка продуктов реакции - 72
3.1. Основные операции на завершающей стадии эксперимента 72
3.1.1. Общие положения 72
3 1.2. Удаление растворителя с помощью роторного испарителя 73
3 13. Экстракция 76
3.1.4. Высушивание органических растворов 81
3.1.5. Выделение целевого продукта (или продуктов) 82
3-2 Кристаллизация 84
3.2.1. Общие принципы , 84
3 2.2. Температура плавления как критерий чистоты 85
240 Оглавление
3.2.3. Методы кристаллизации 85
3-2.4 Выбор растворителя для кристаллизации... 103
3.2.5. Специальные темы 107
3.3. Температура плавления ... 109
З.ЗЛ. Общие принципы 109
3-3-2. Определение температуры плавления 109
3.3-3- Другие методы 111
3 4. Перегонка... 114
3.4.1. Общие положения 114
3.4-2. Простая перегонка •, _ 115
3.4.3. Фракционная перегонка 123
3.4.4. Перегонка микроколичеств.. 128
3.4.5. Молекулярная перегонка 132
3.4.6. Перегонка с водяным паром 1331
3.4.7. Сублимация твердых веществ 135
3.4.8. Приложения 137
4. Хроматографическое разделение смесей органических соединений 1411
4.1. Аналитические методы 141
4-1.1. Тонкослойная хроматография 141
4-1.2. Газо-жидкостная хроматография 147
4-1.3. Высокоэффективная жидкостная хроматография 164
4-2. Препаративные методы 183
4.2.1 Препаративная тонкослойная хроматография (ТСХ) 183
4.2.2. Колоночная хроматография 184
4-3. Приложения 2041
4.3.1. Общие принципы хроматографического разделения 204
4.3.2. Контроль активности сорбента 210
4.3.3. Приготовление ТСХ-пластин 211
5. Подготовка образцов для спектроскопии 2131
5.1. Инфракрасная спектроскопия 213|
5.2 Ядерный магнитный резонанс - ^1
5.3. Масс-спектрометрия 221
5.4. Ультрафилетовая спектроскопия 222 '
6. Поиск химической информации 225
6.1. Физические свойства 226
6.2. Спектроскопические данные 227 '
6-3. Справочник Бейлыитейна (Beilstein's Handbuch) 229
6.4- Реферативный журнал Chemical Abstracts 231
Литература 233
Предметный указатель 236