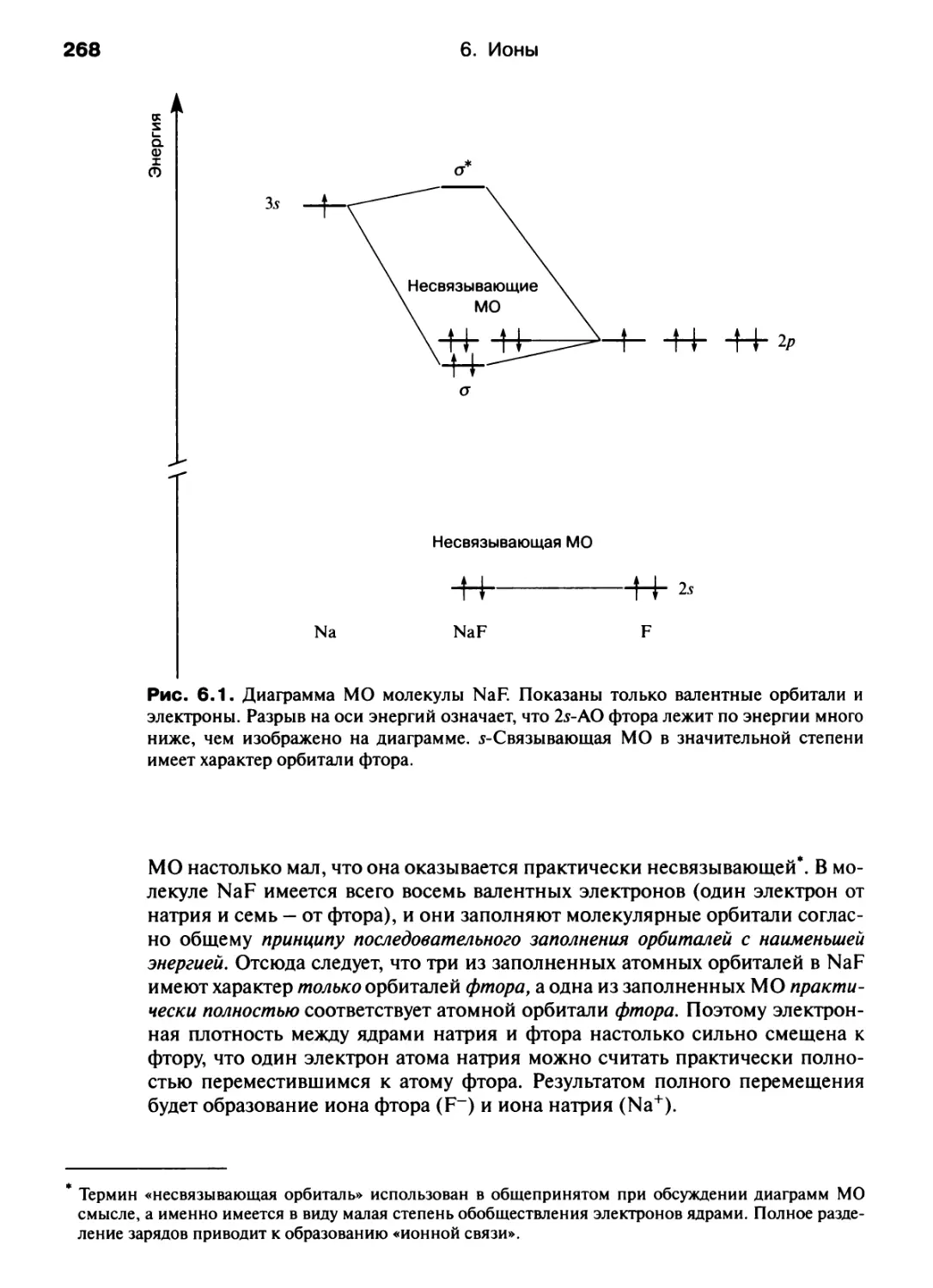
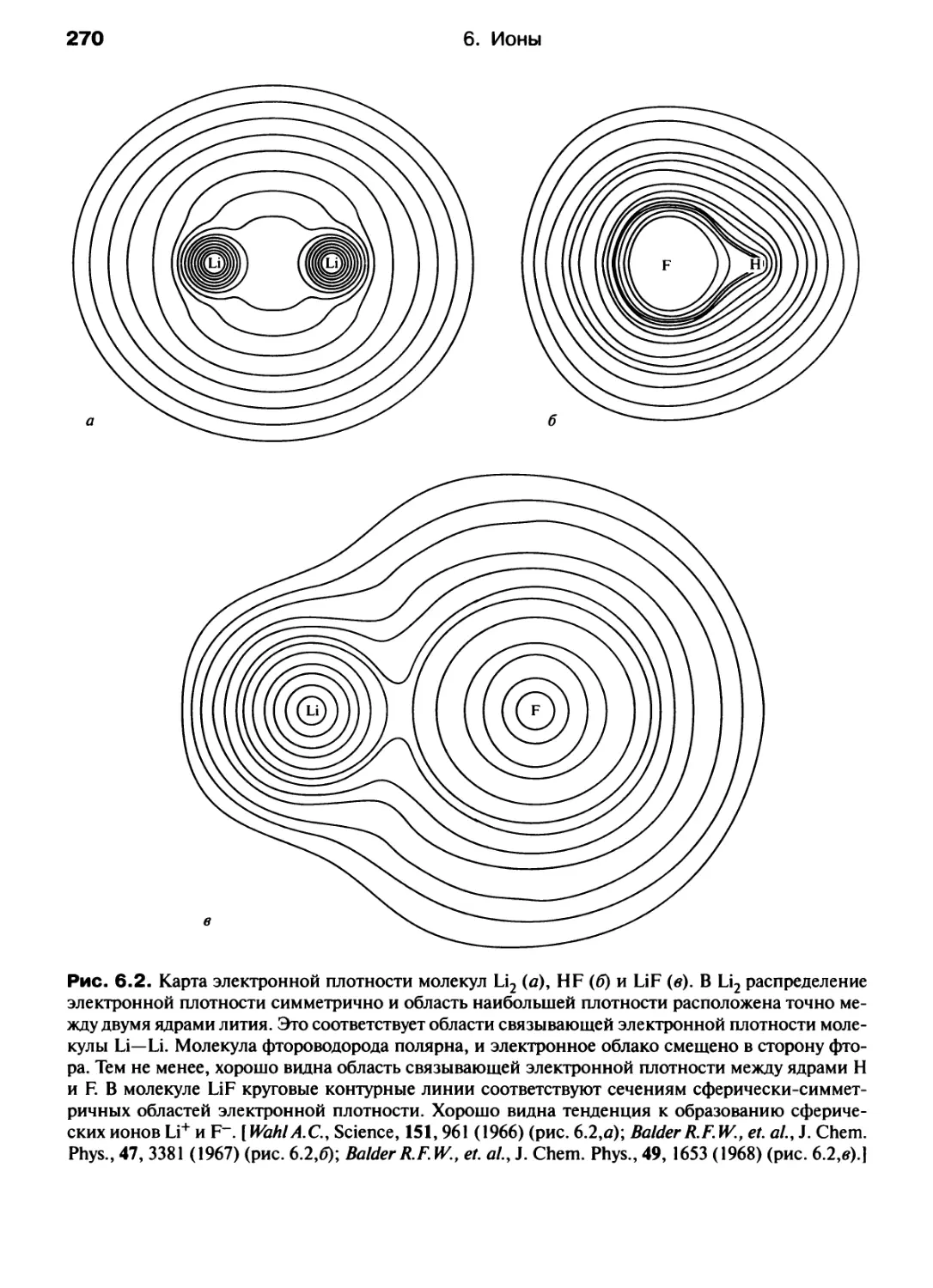
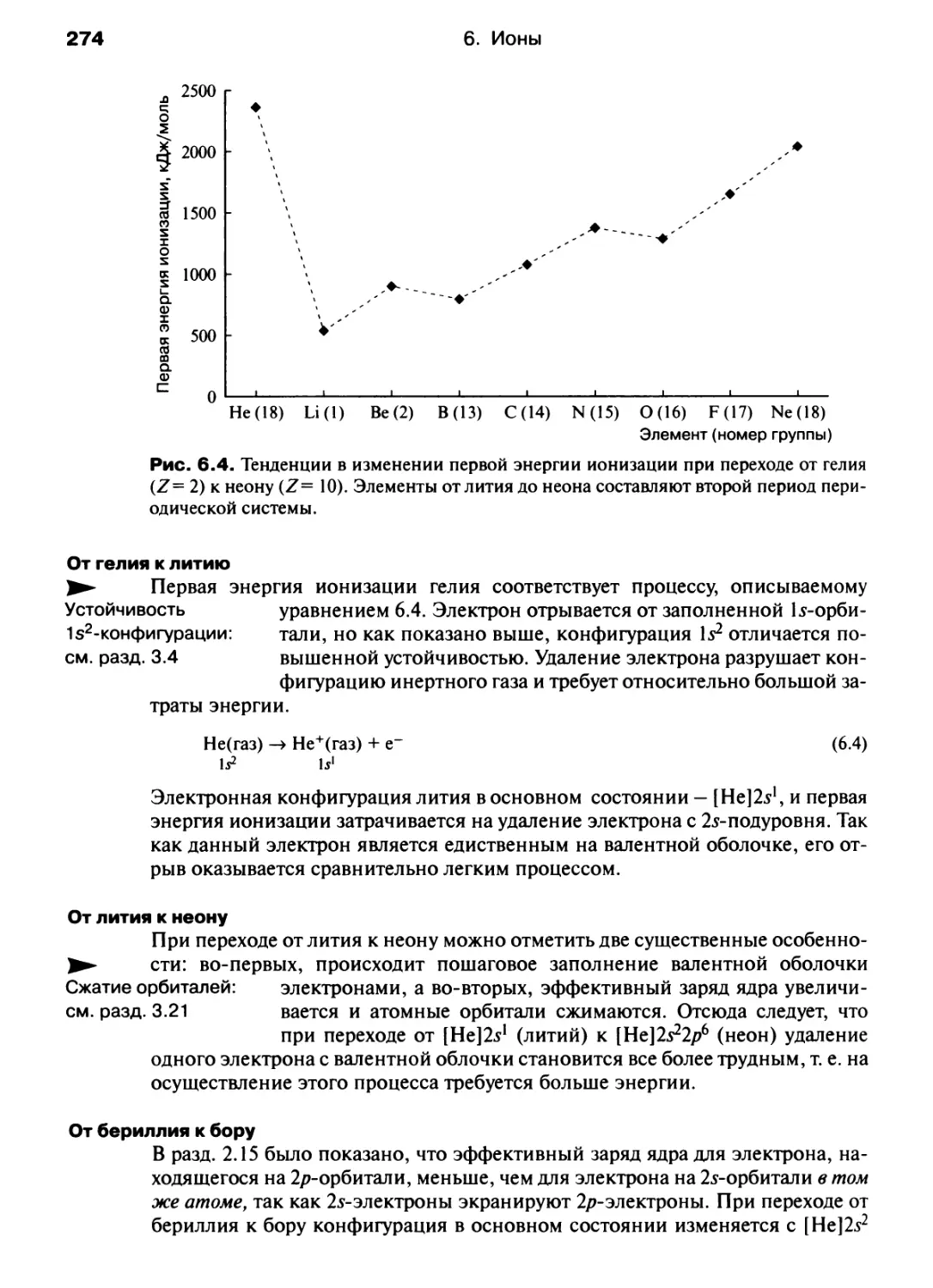
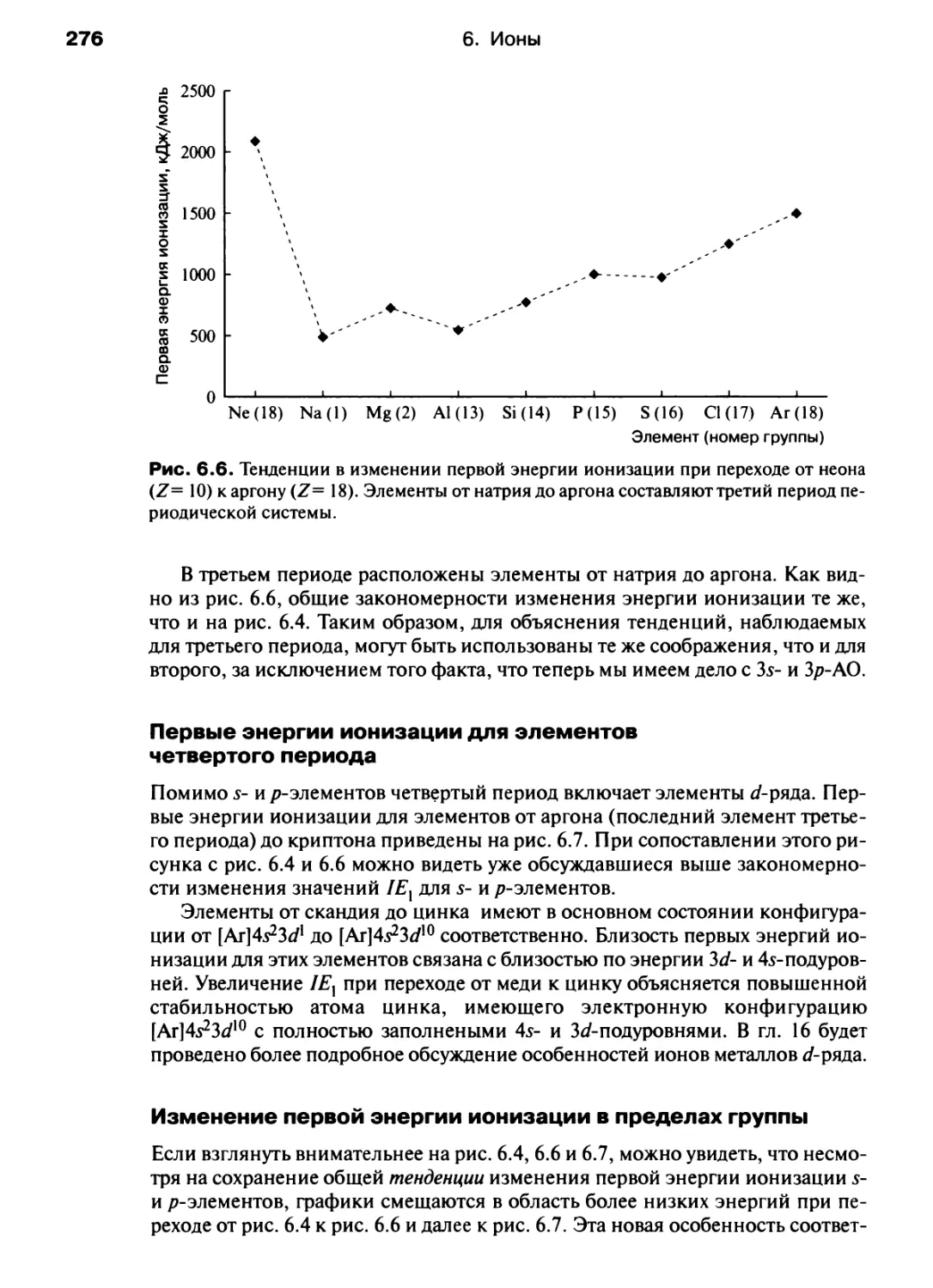
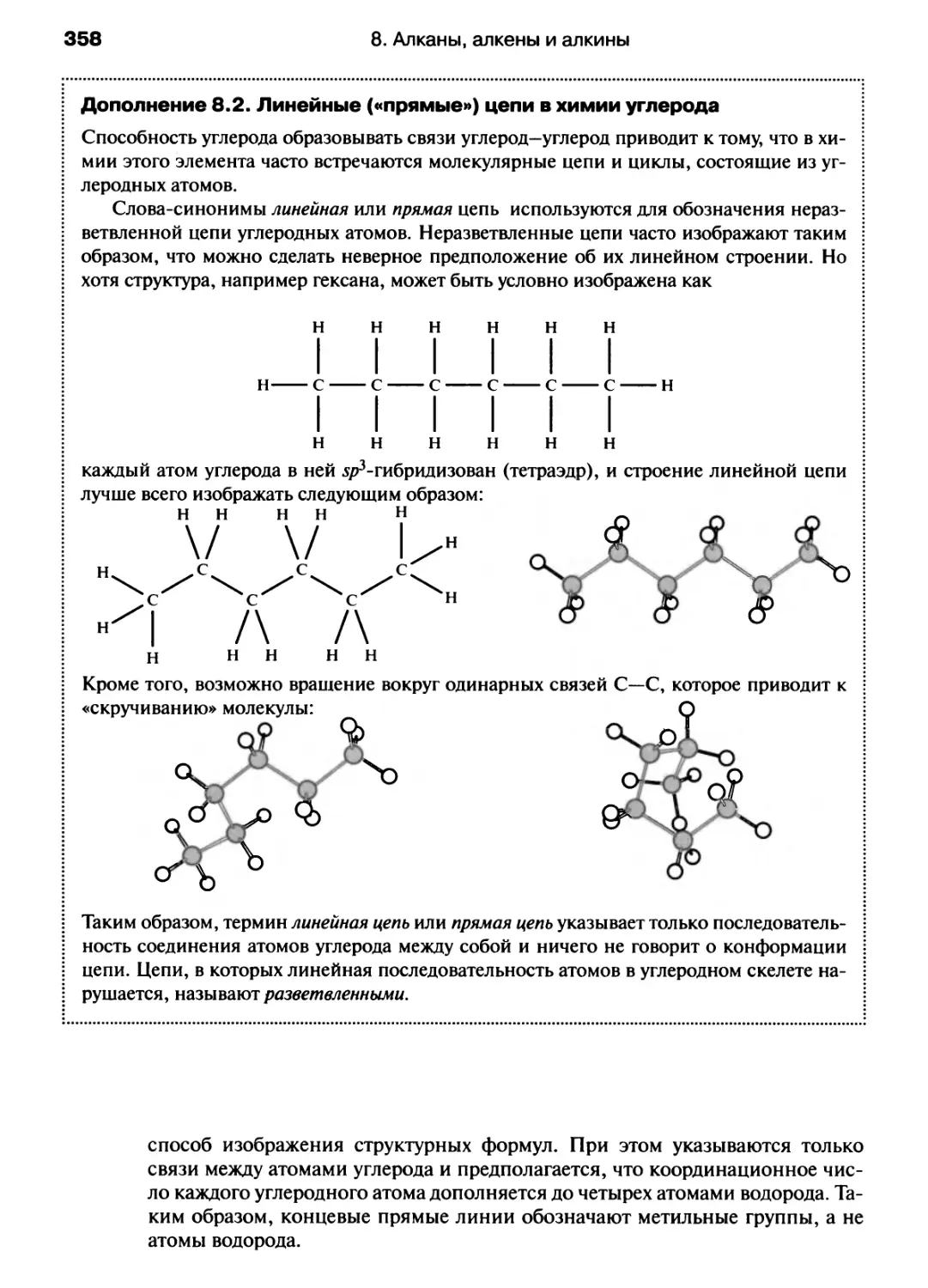
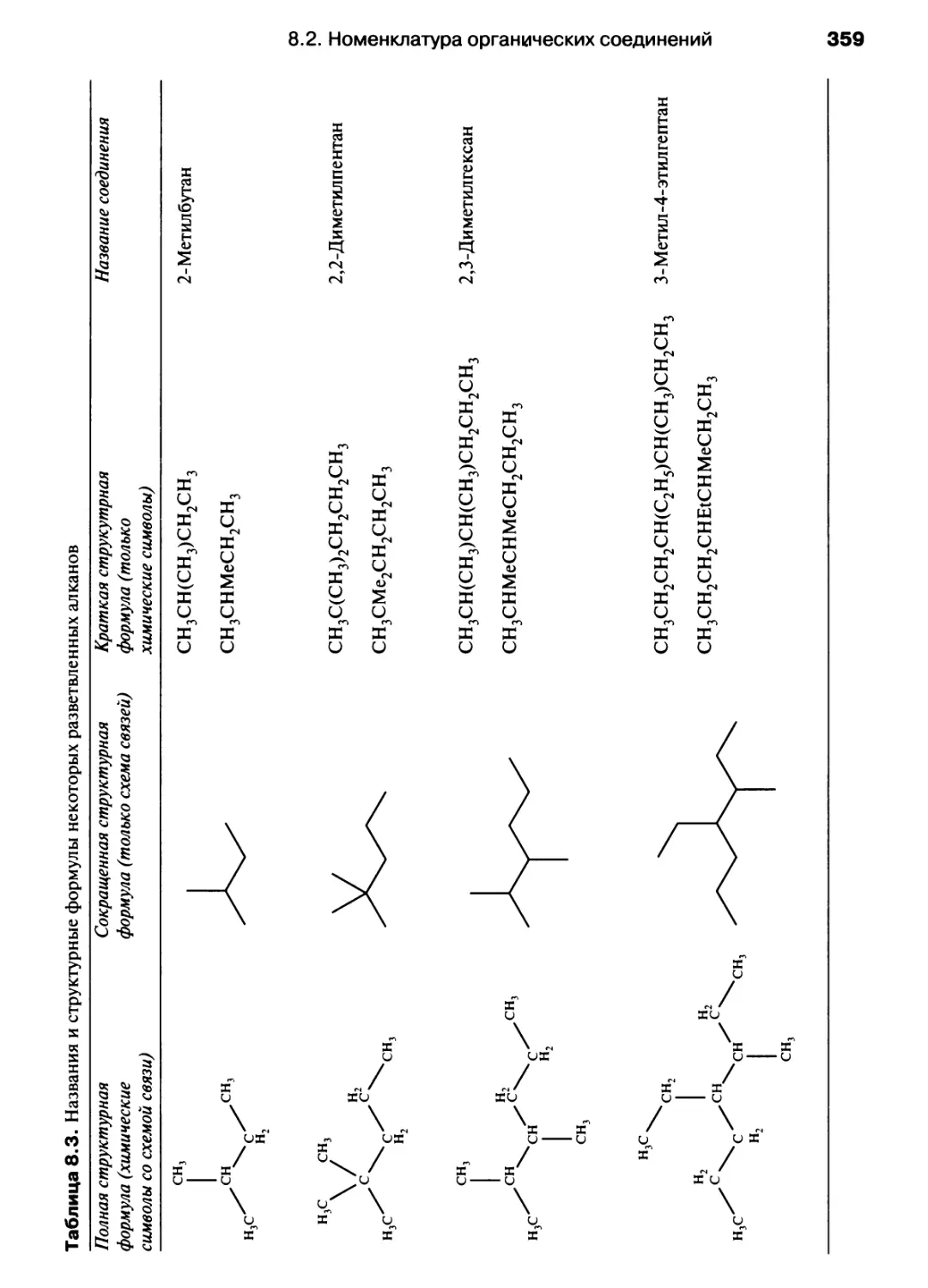
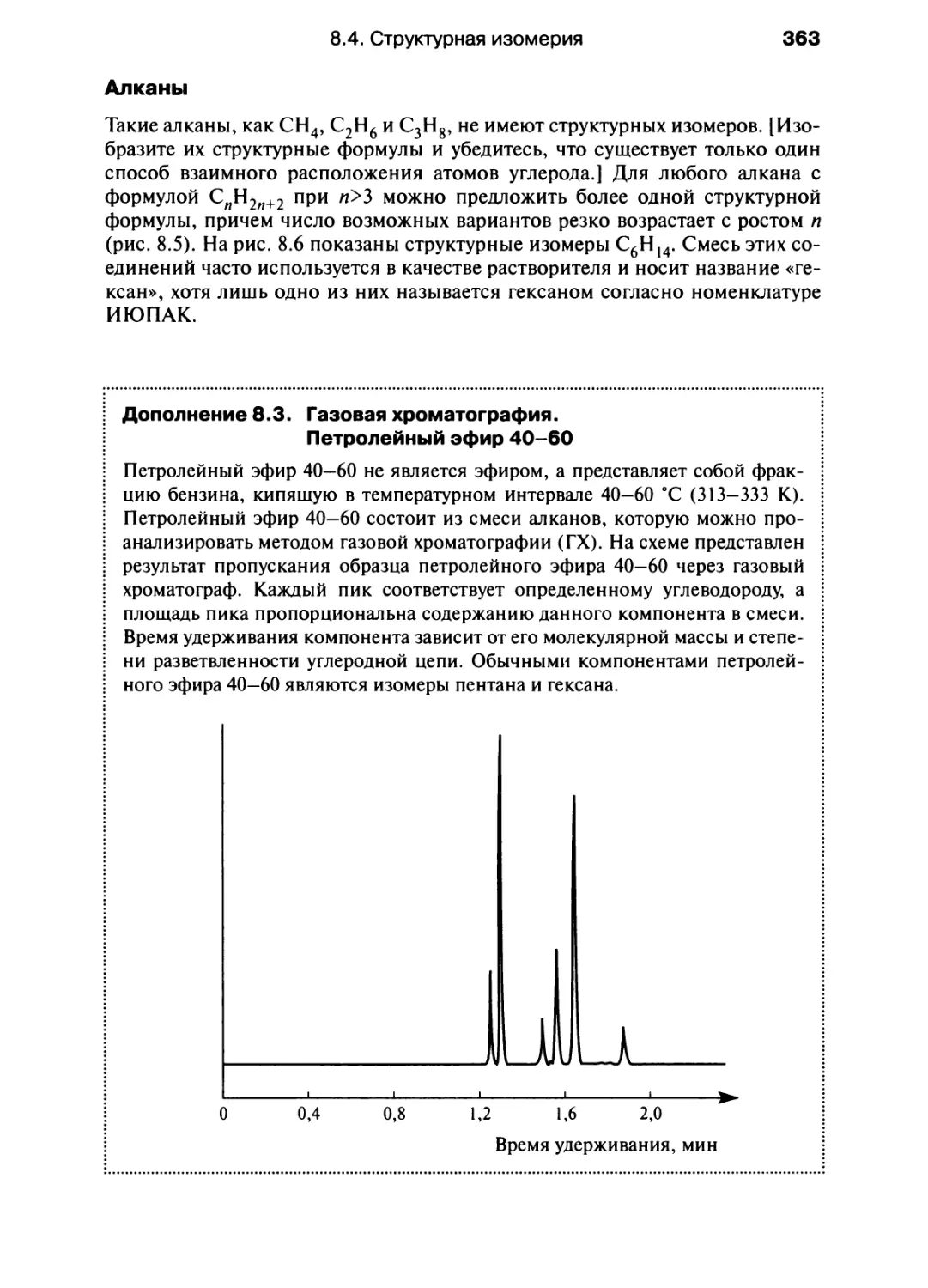
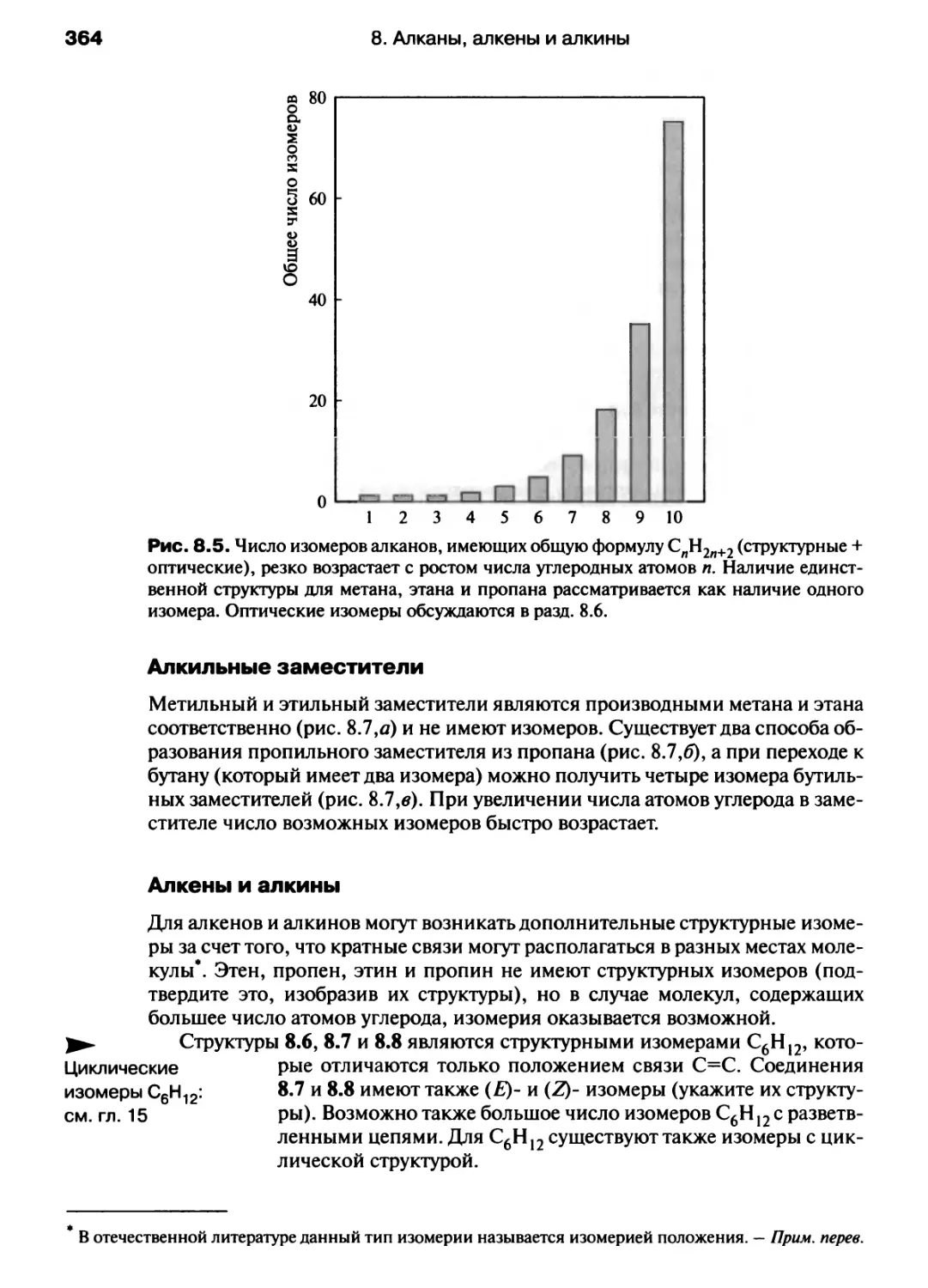
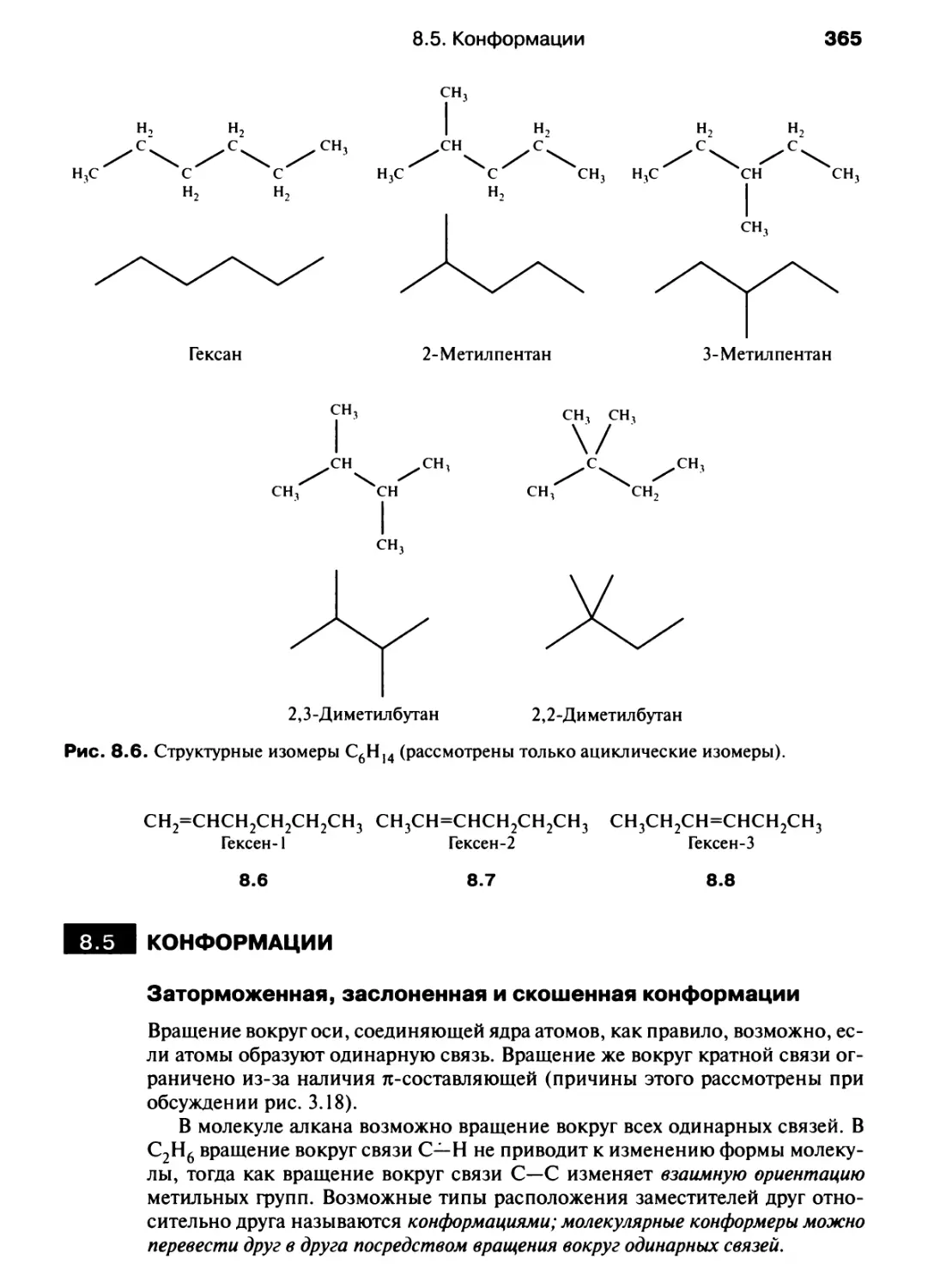
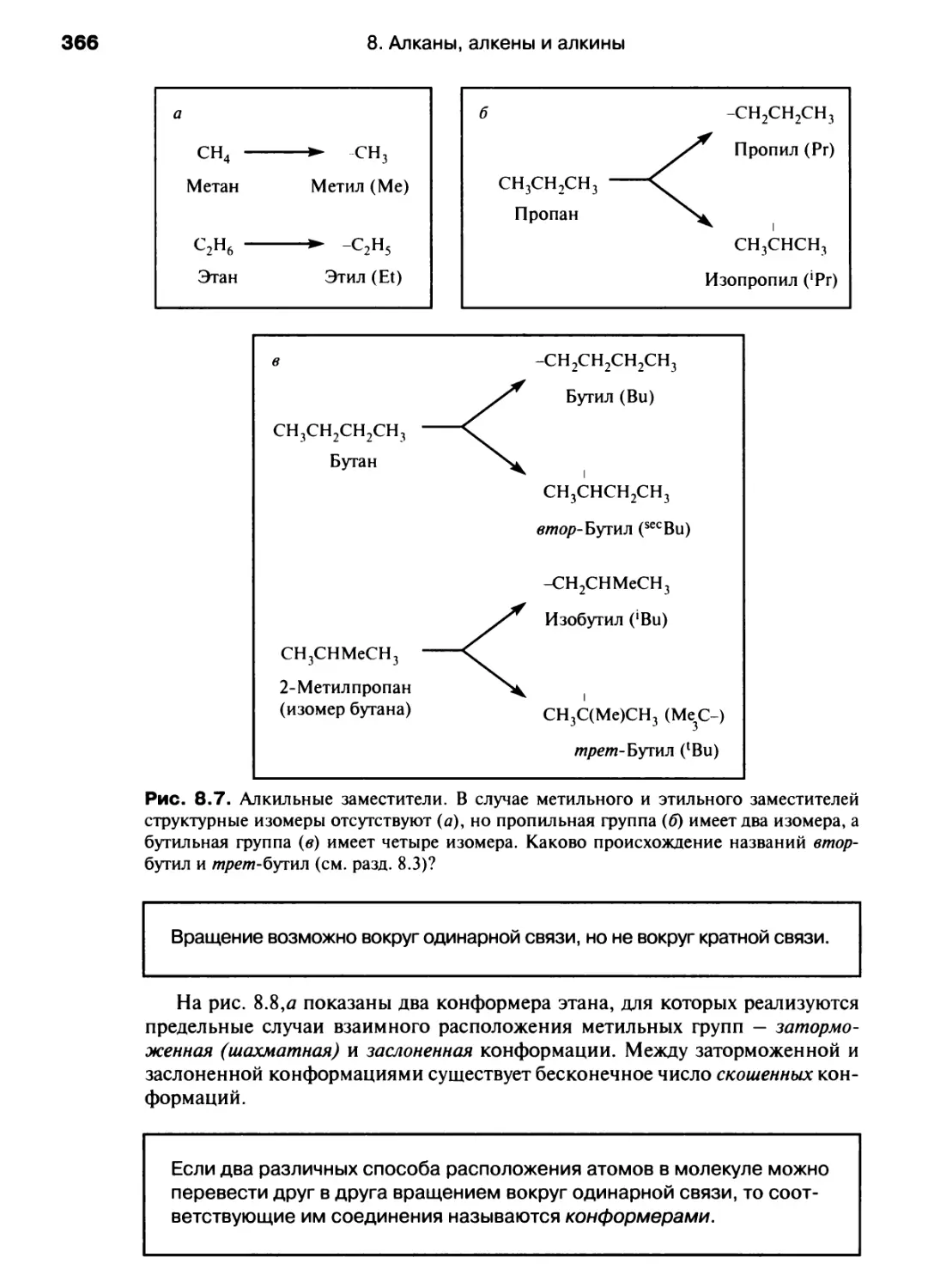
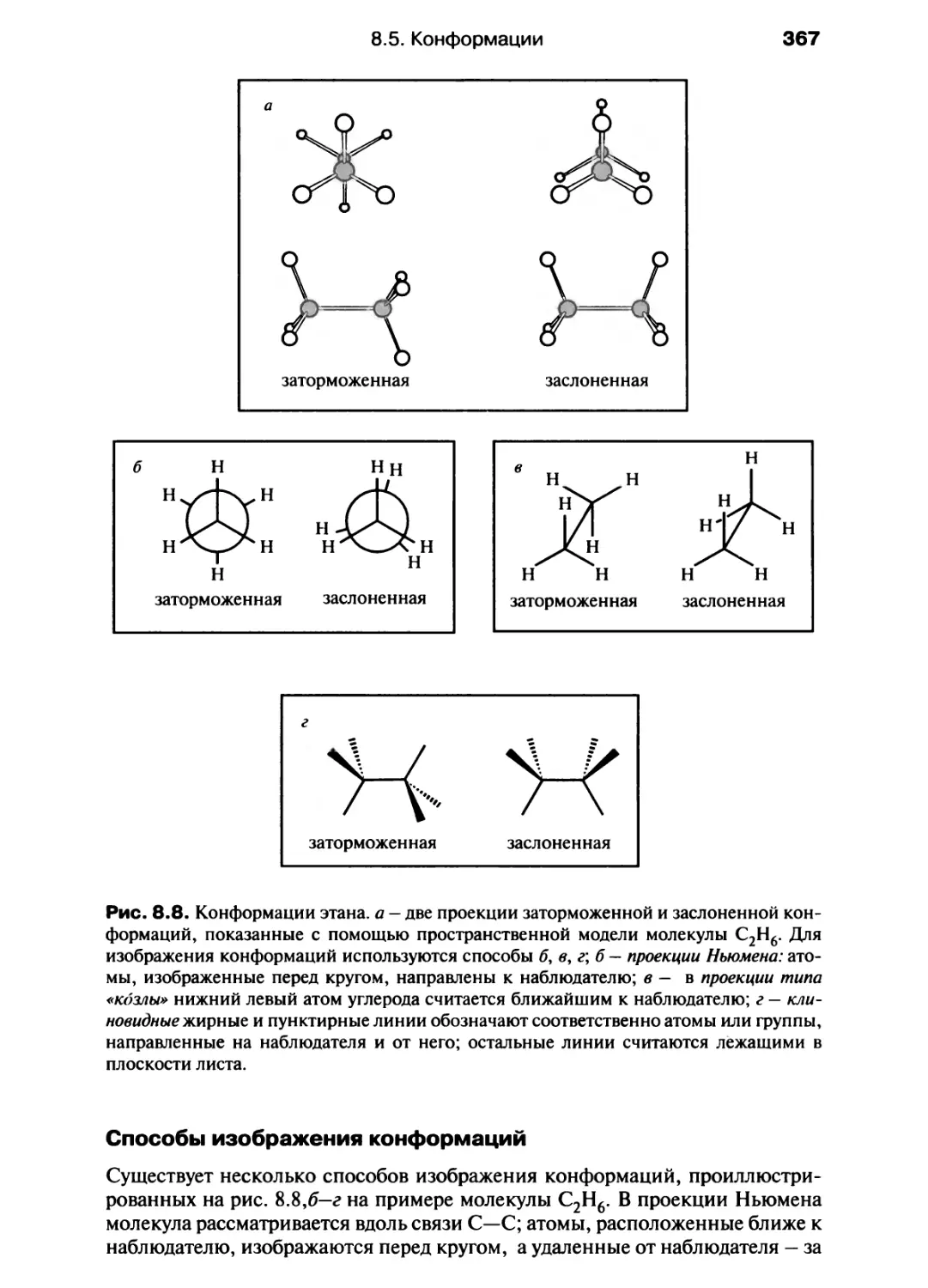
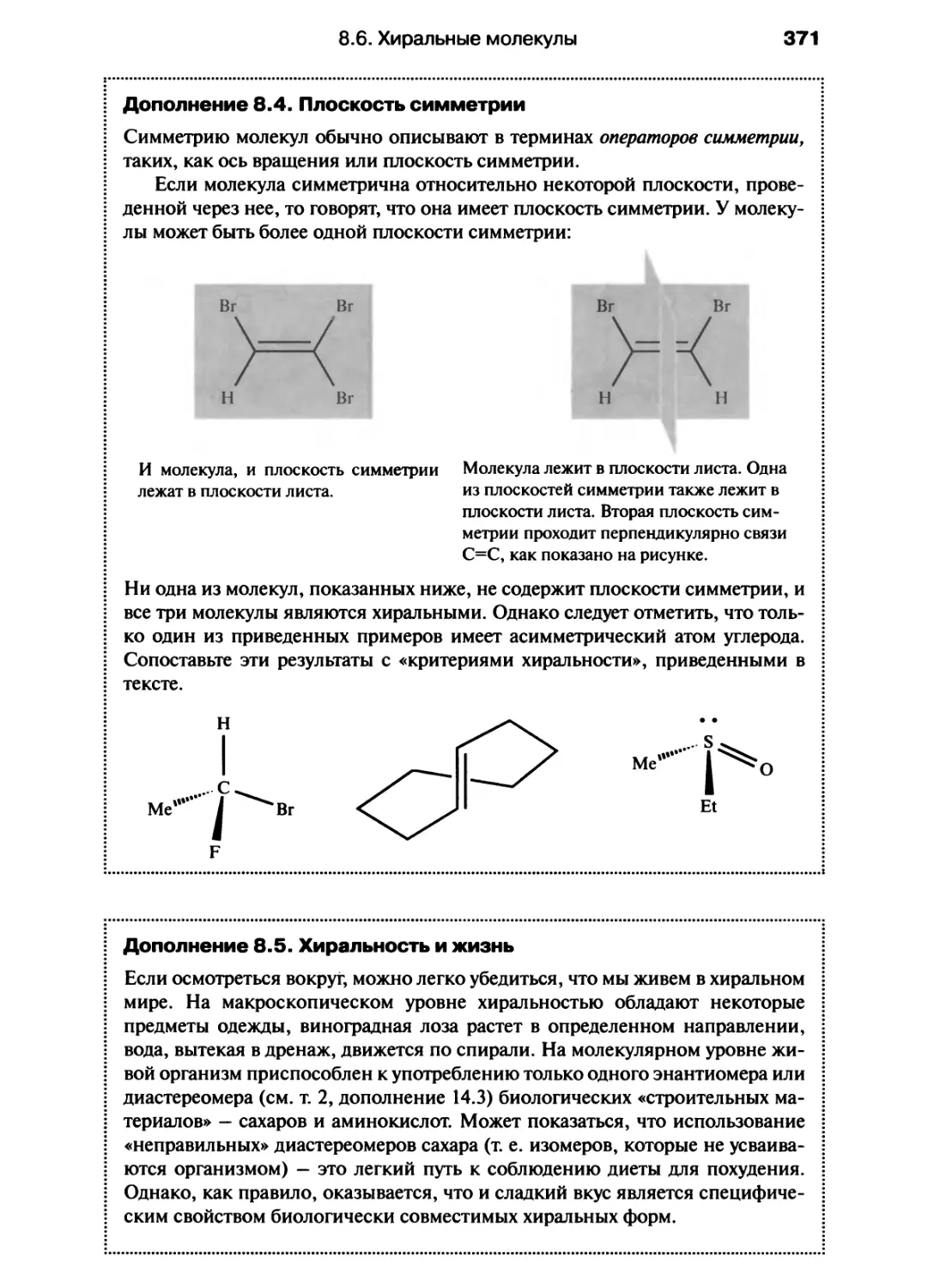
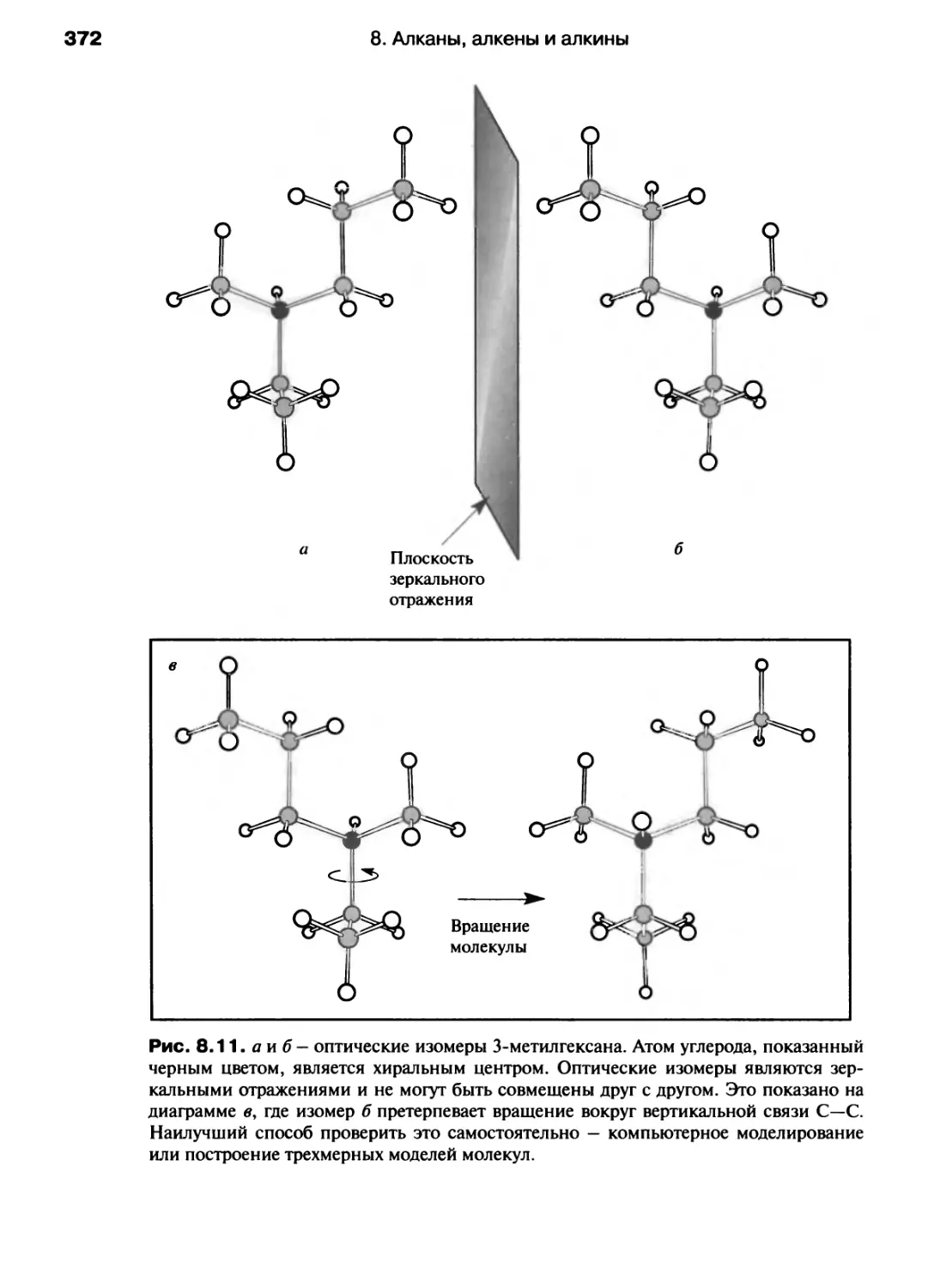
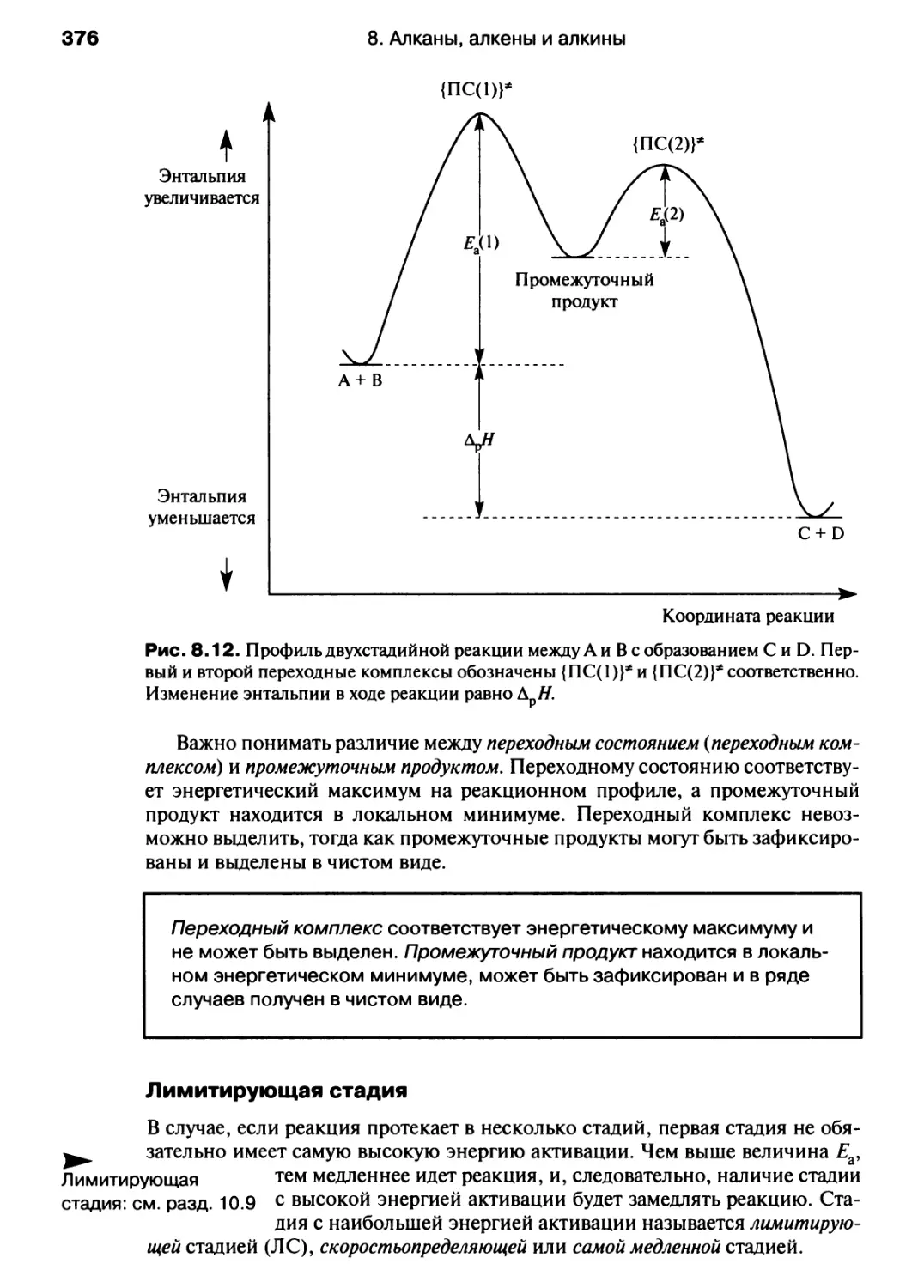
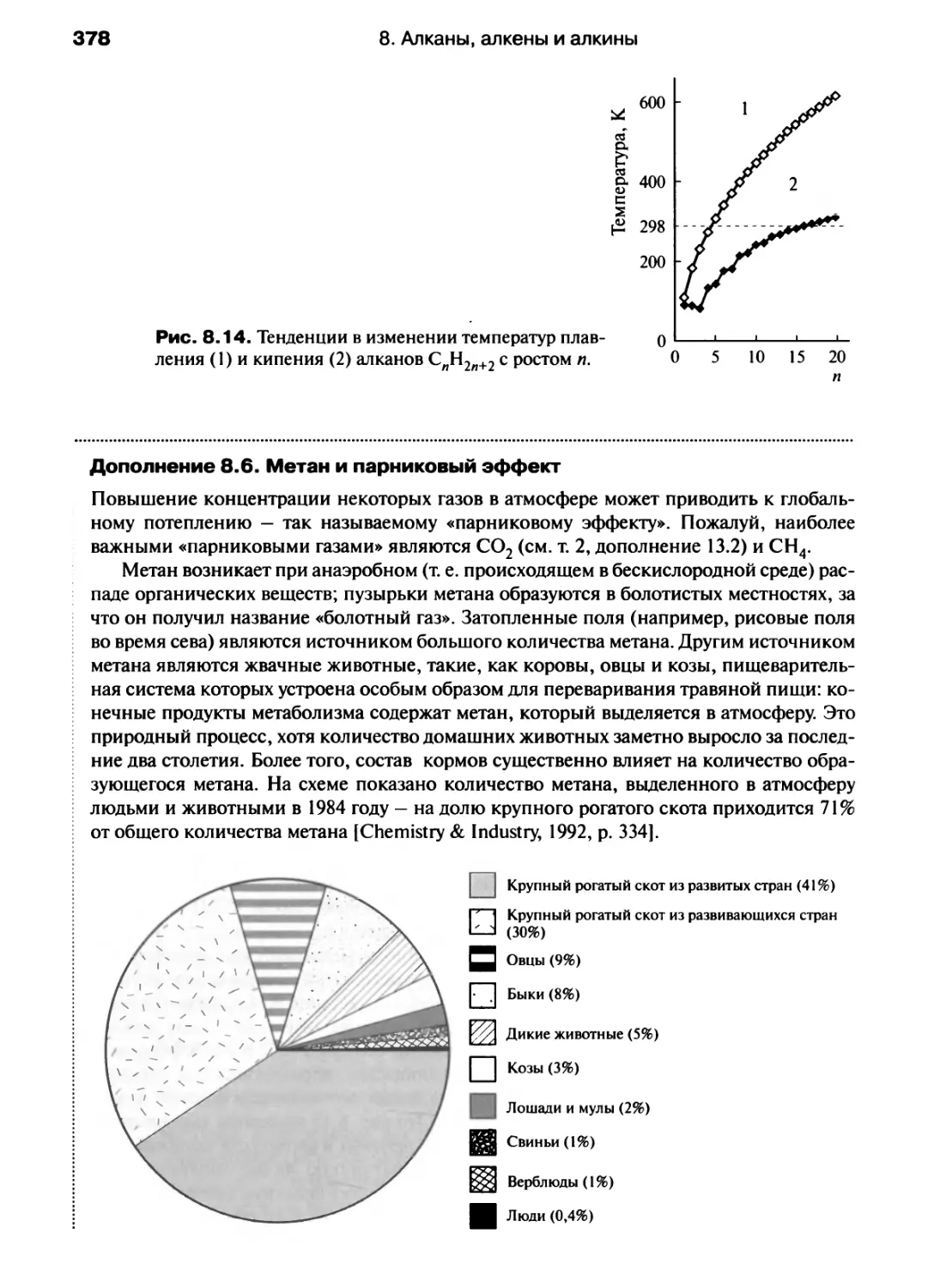
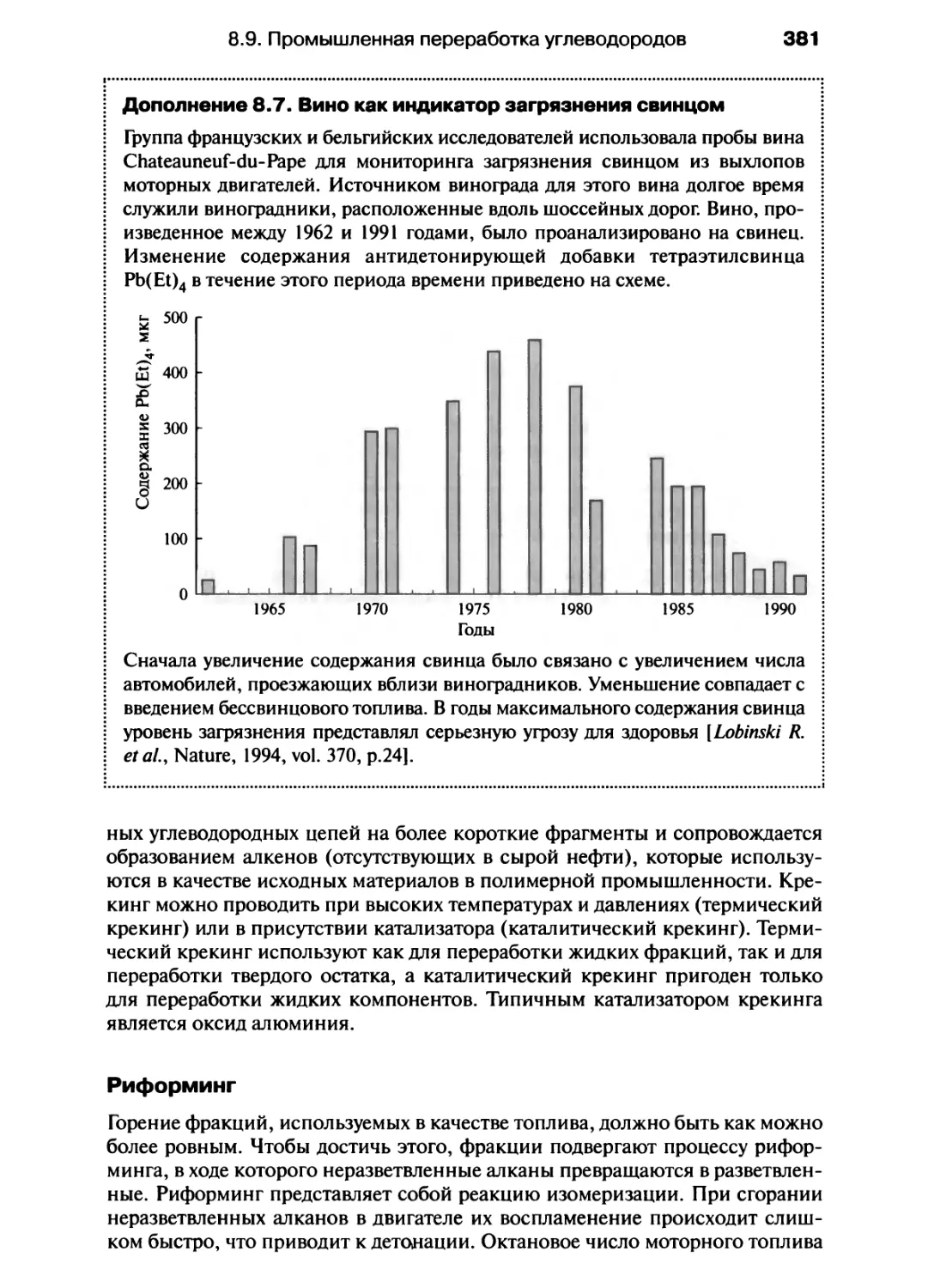
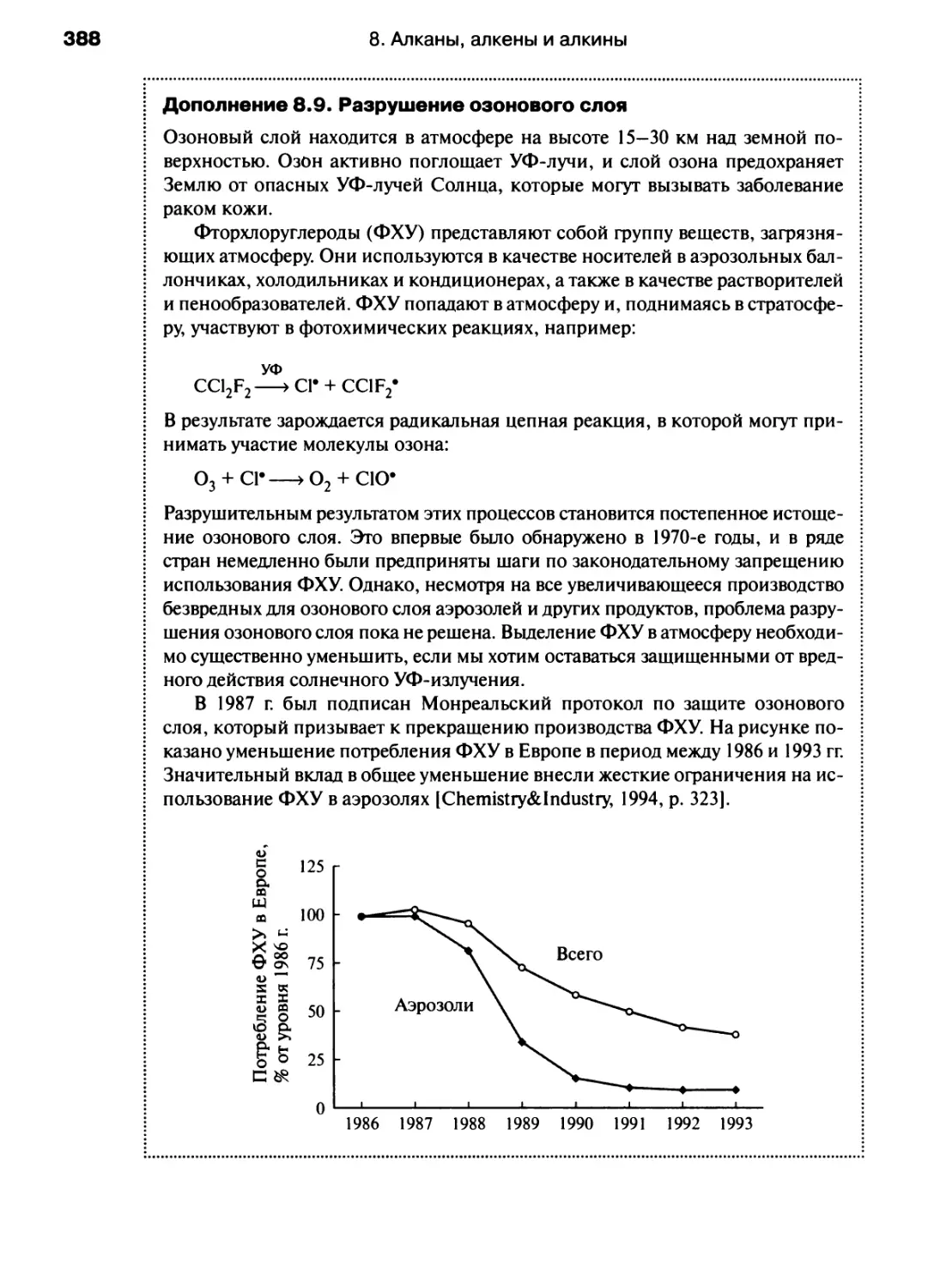
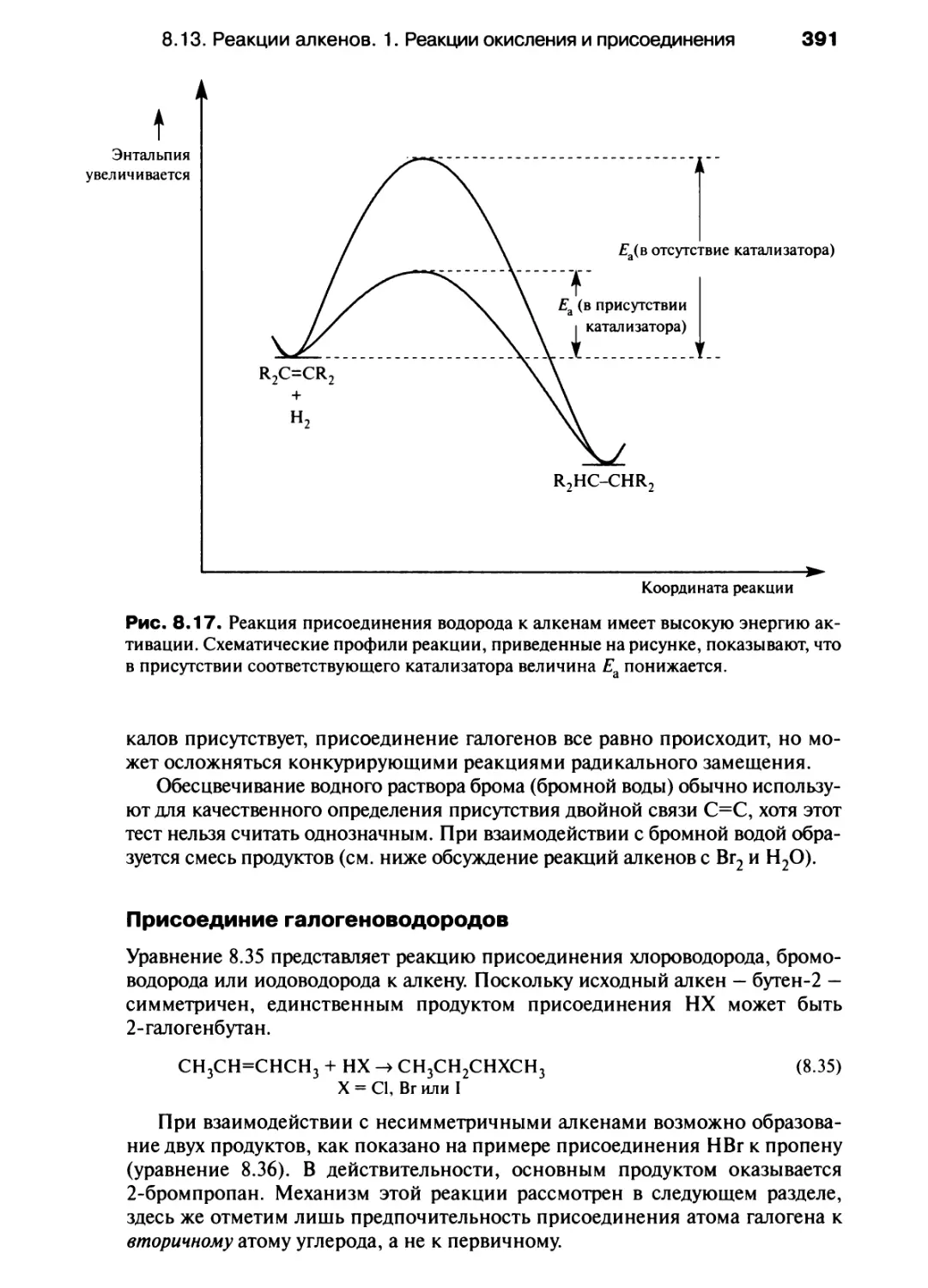
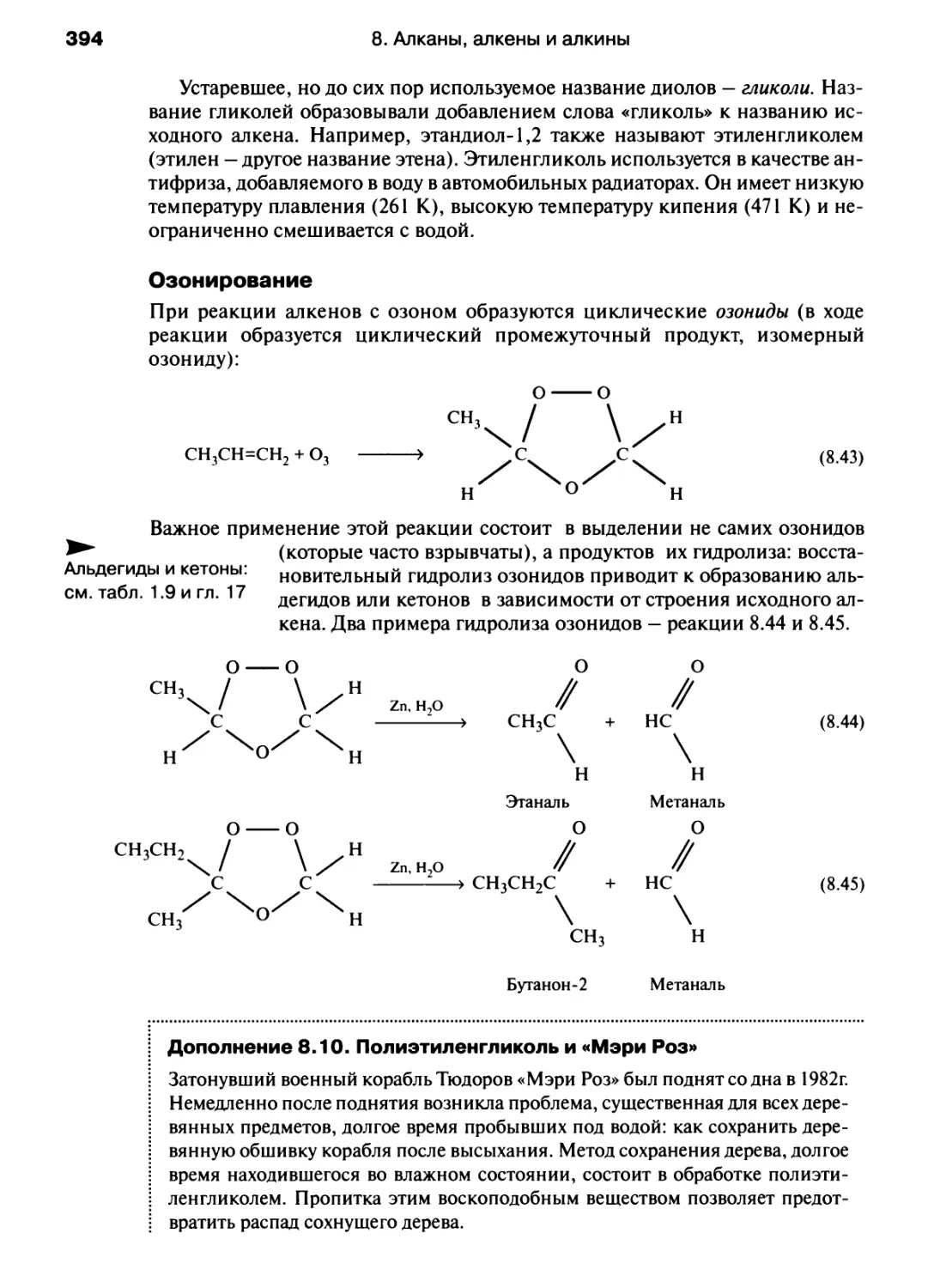
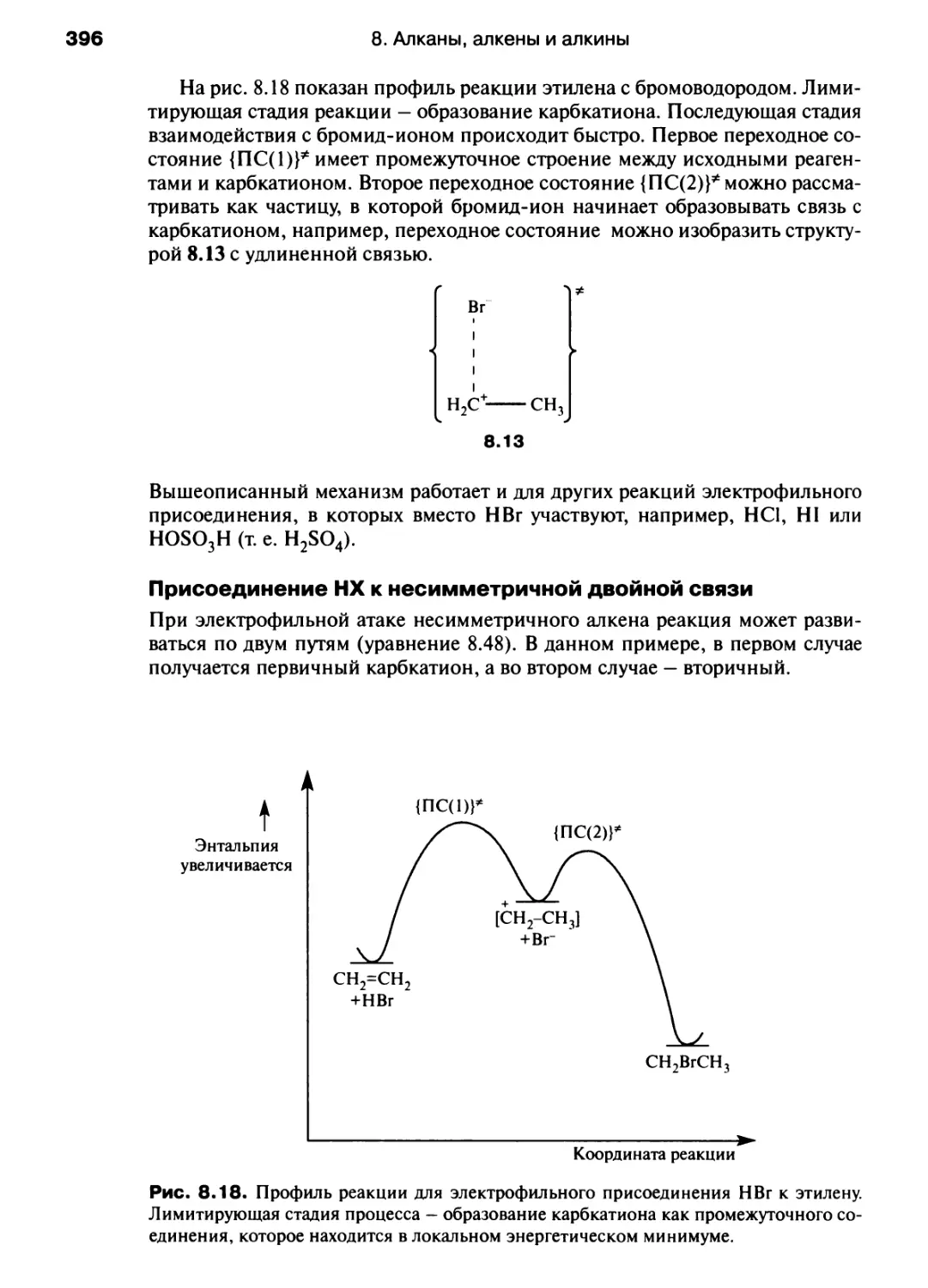
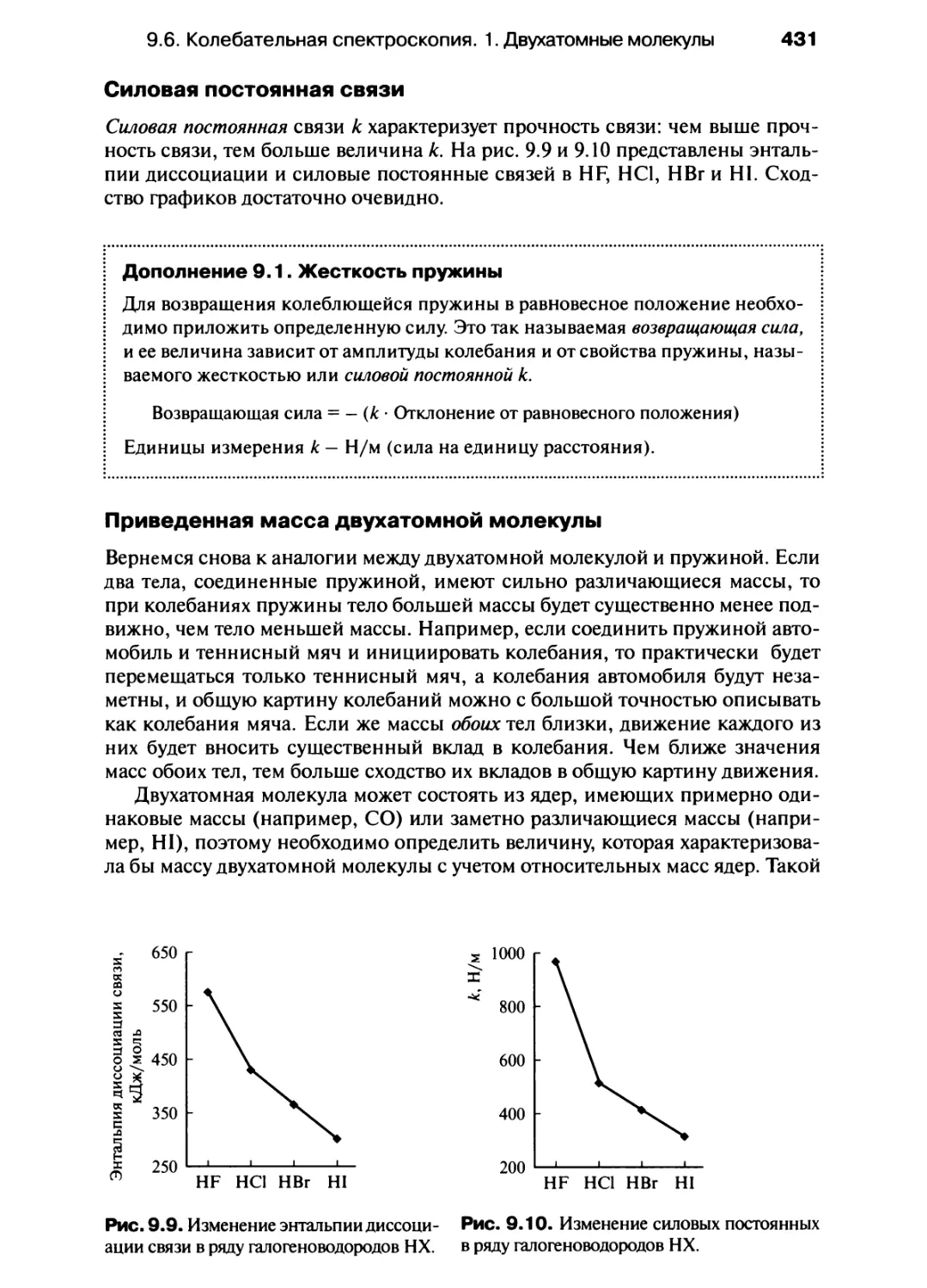
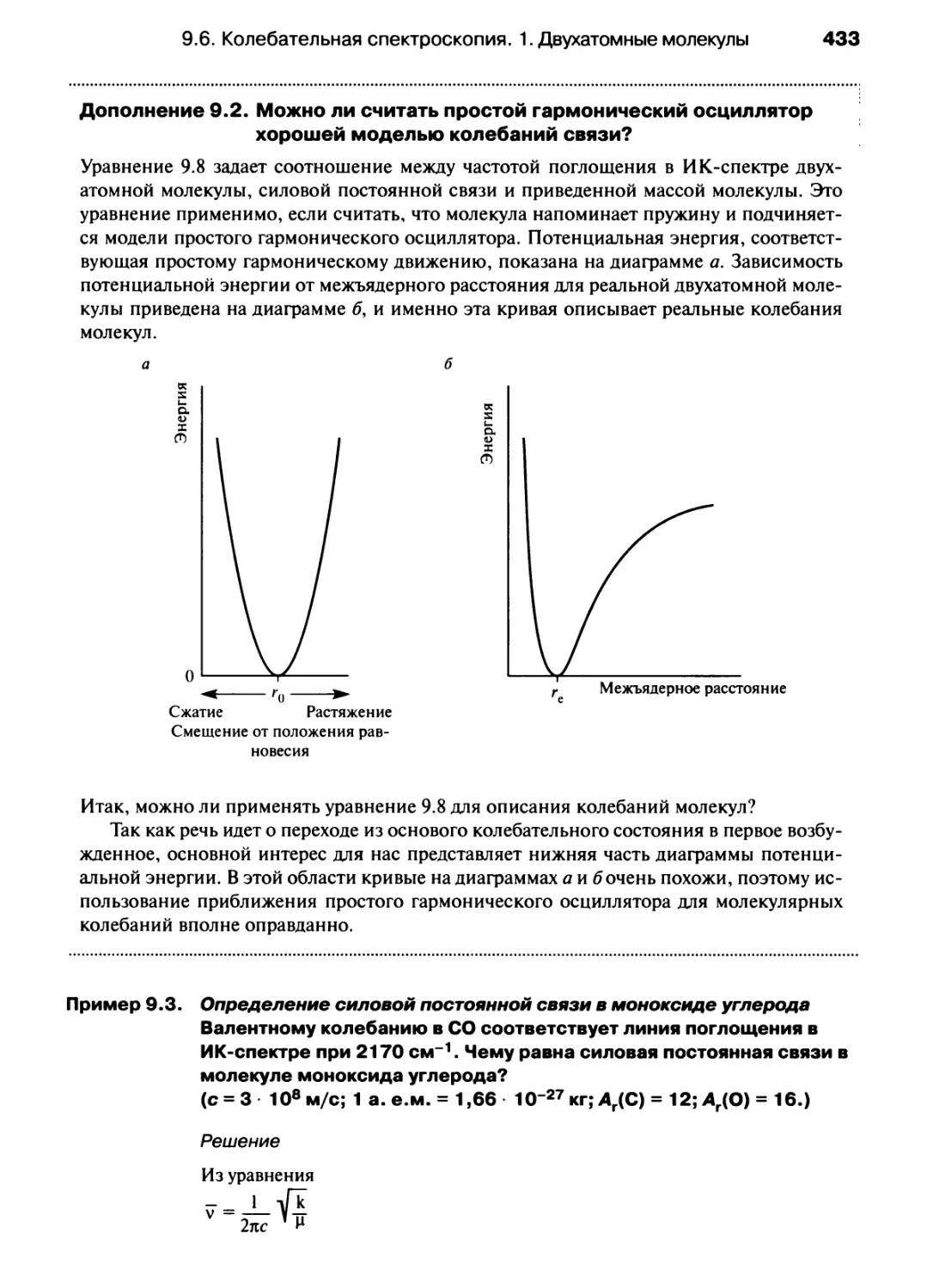
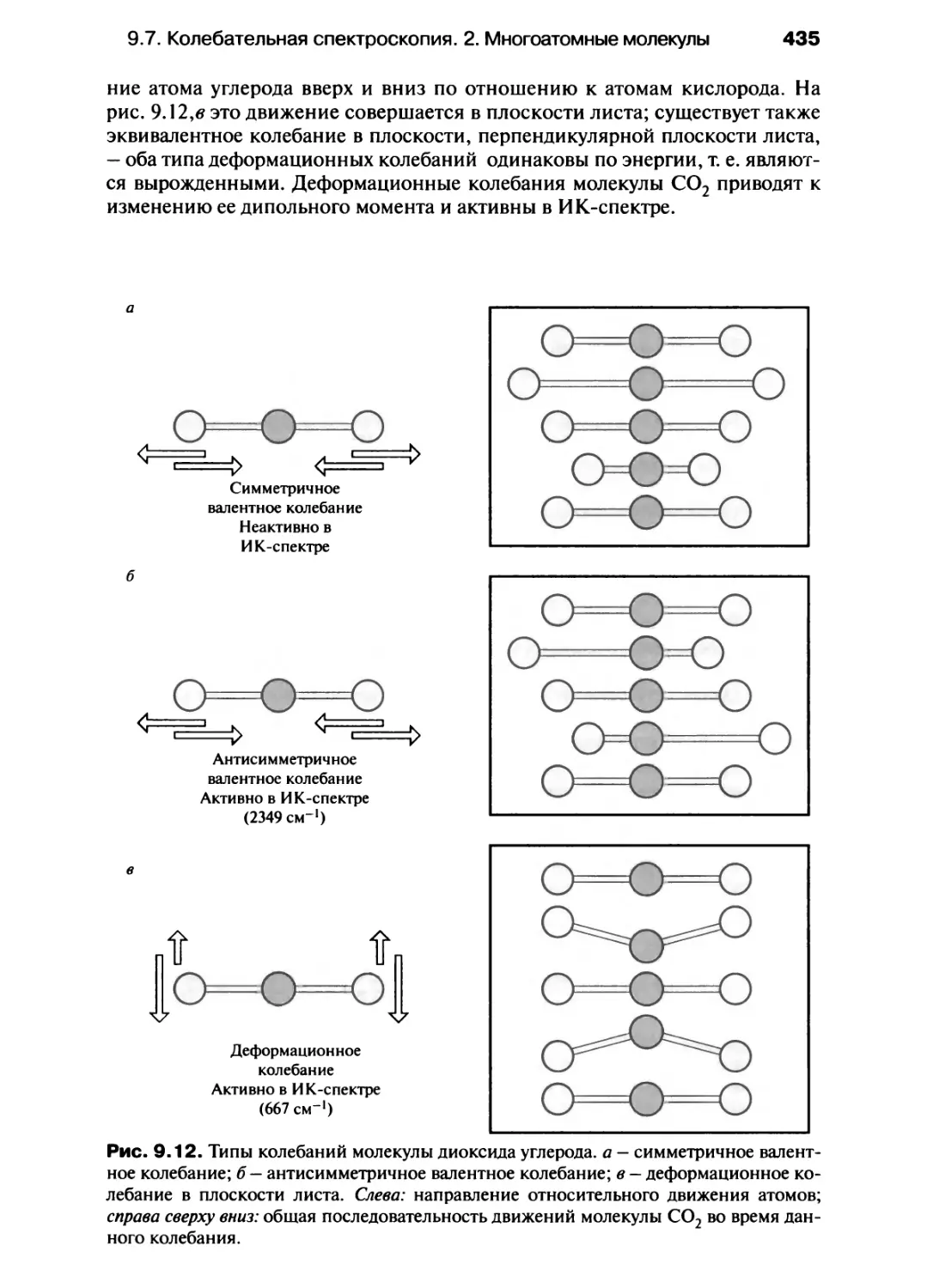
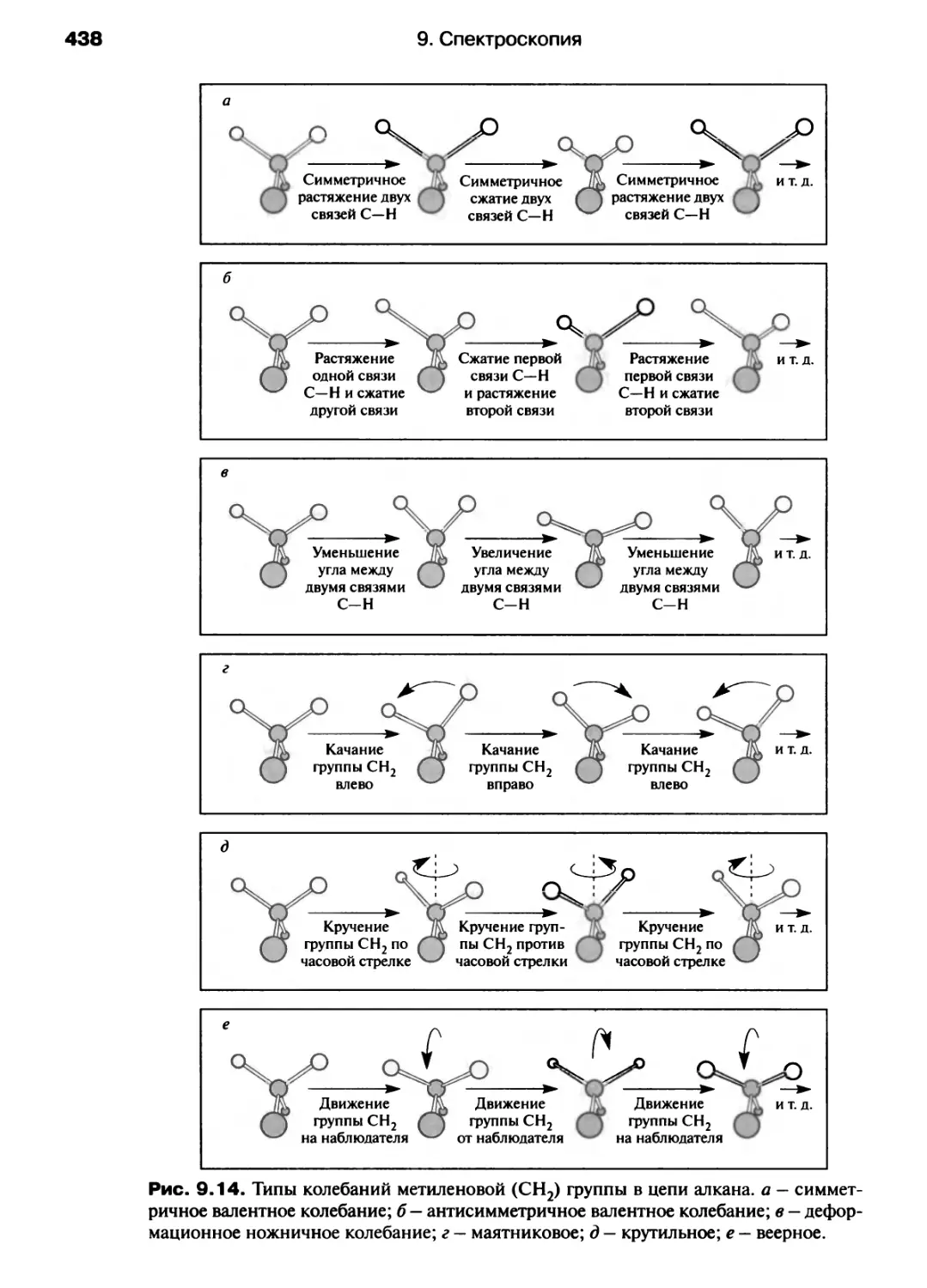
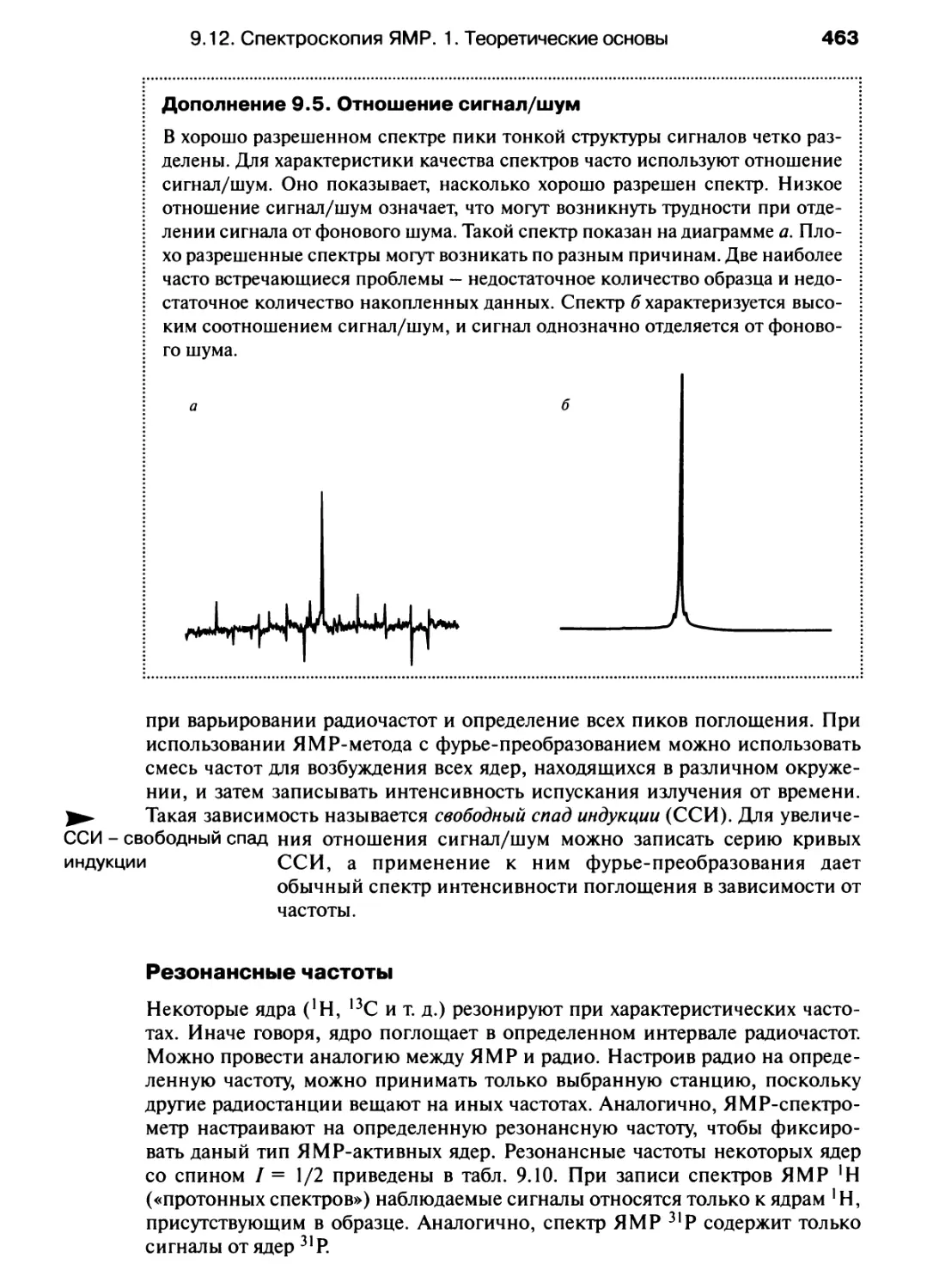
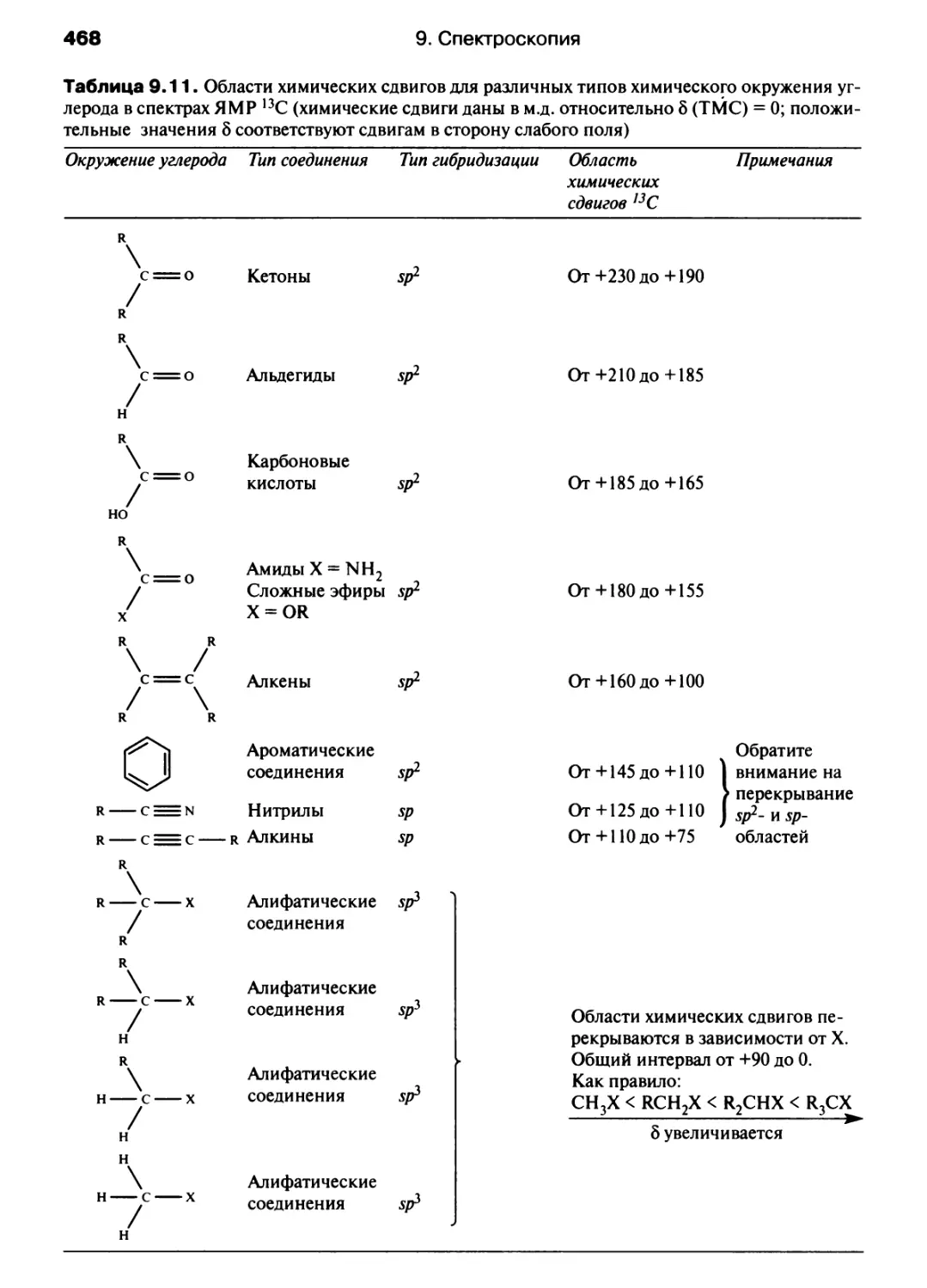
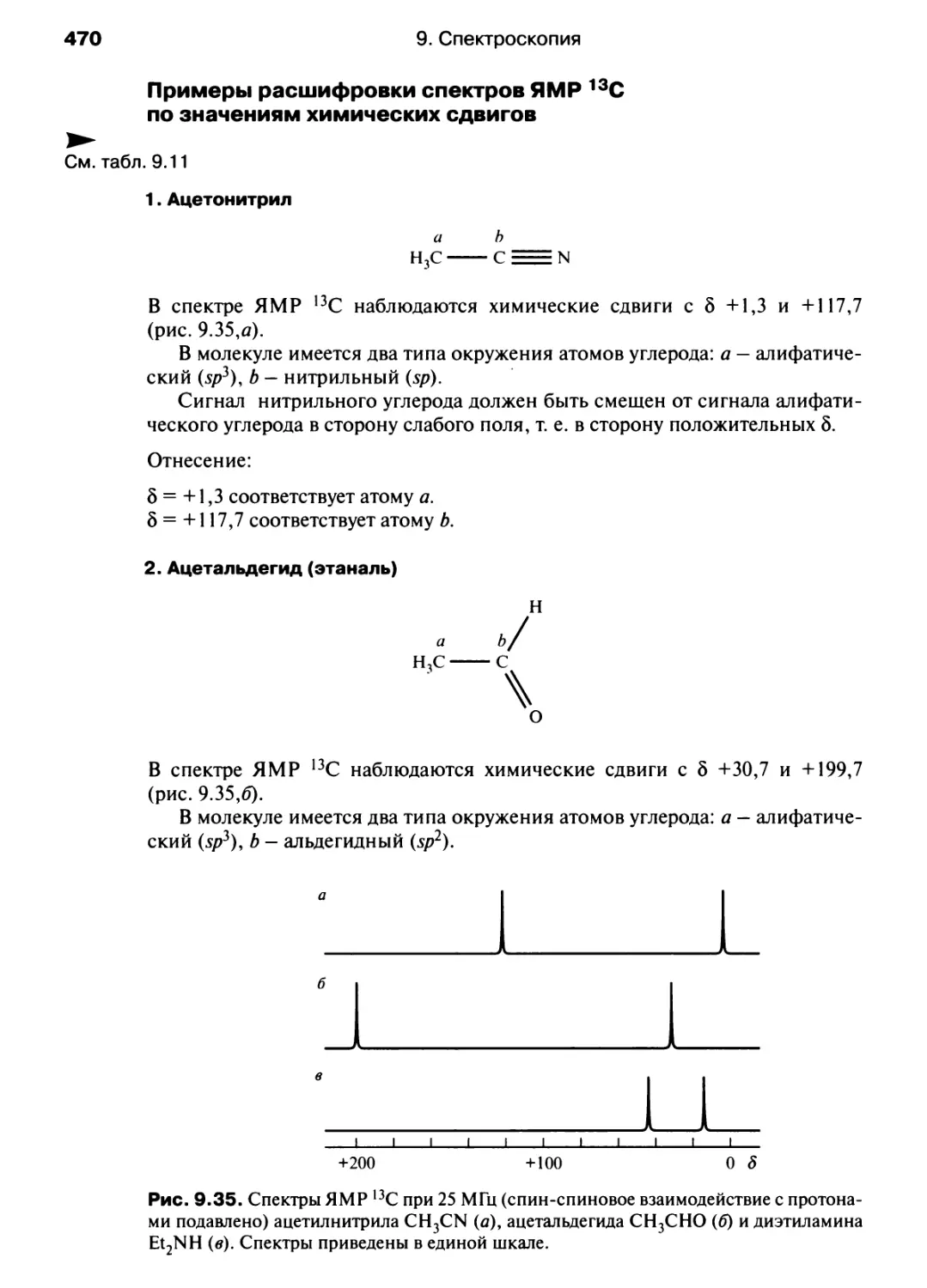
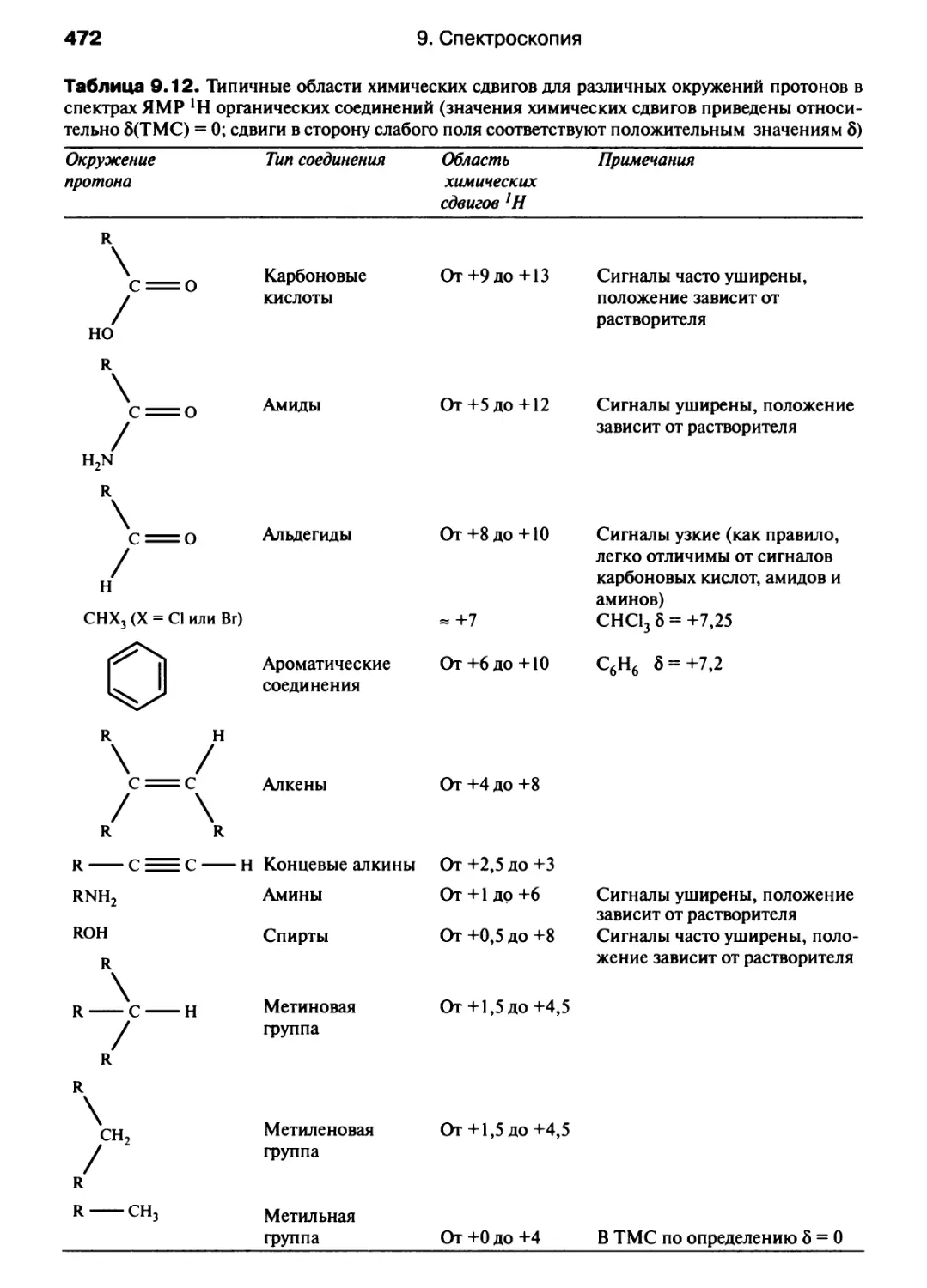
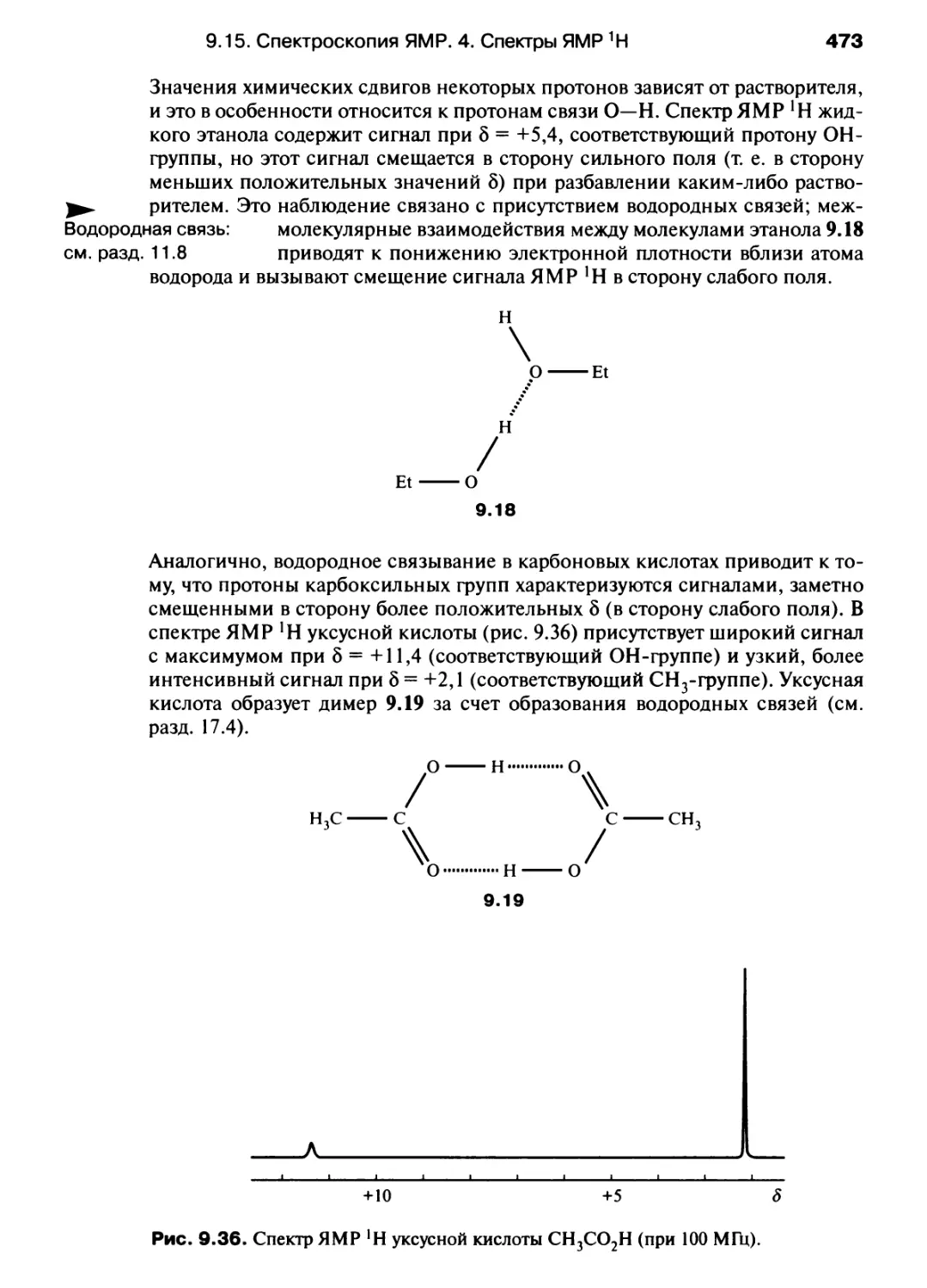
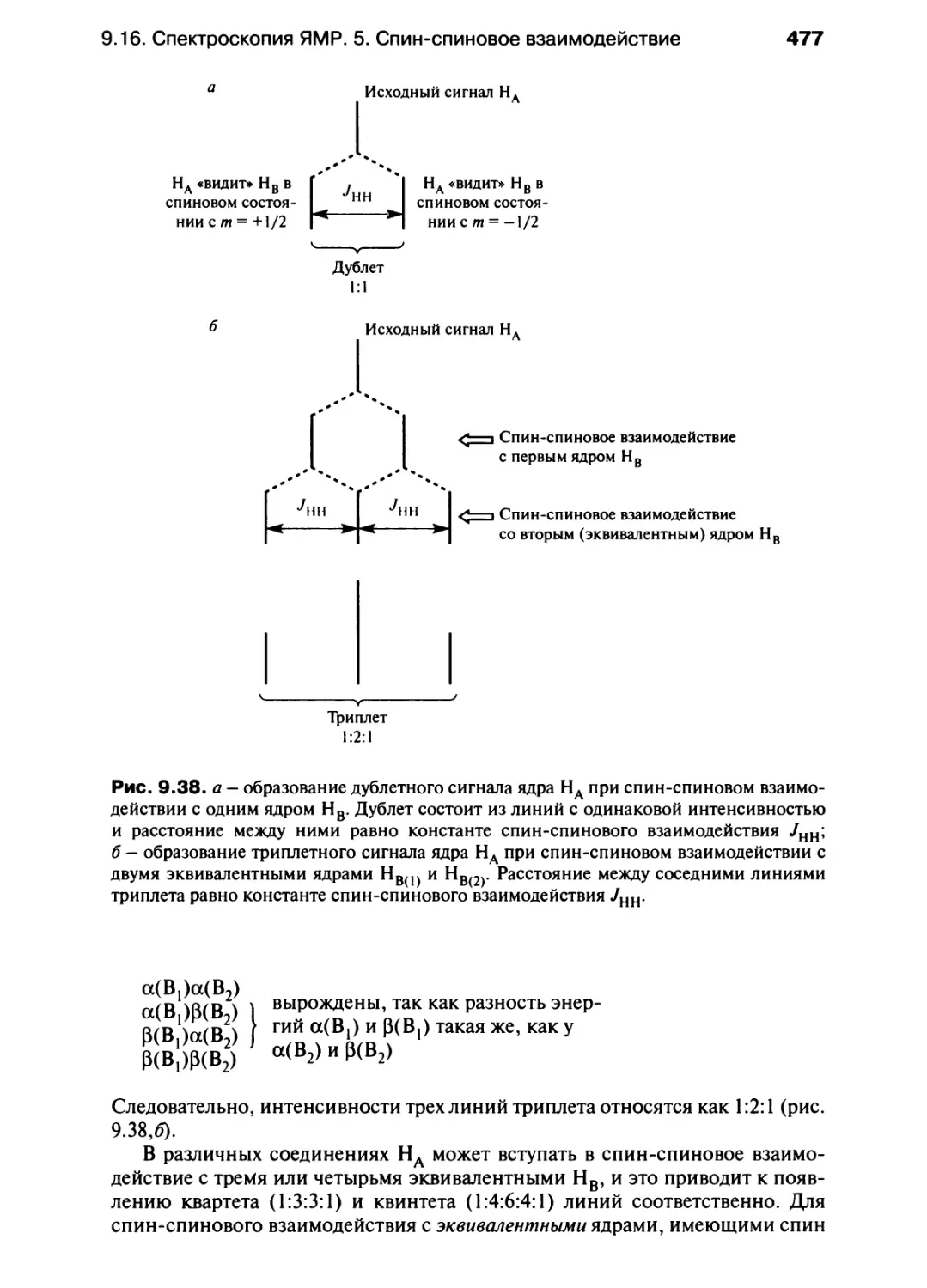
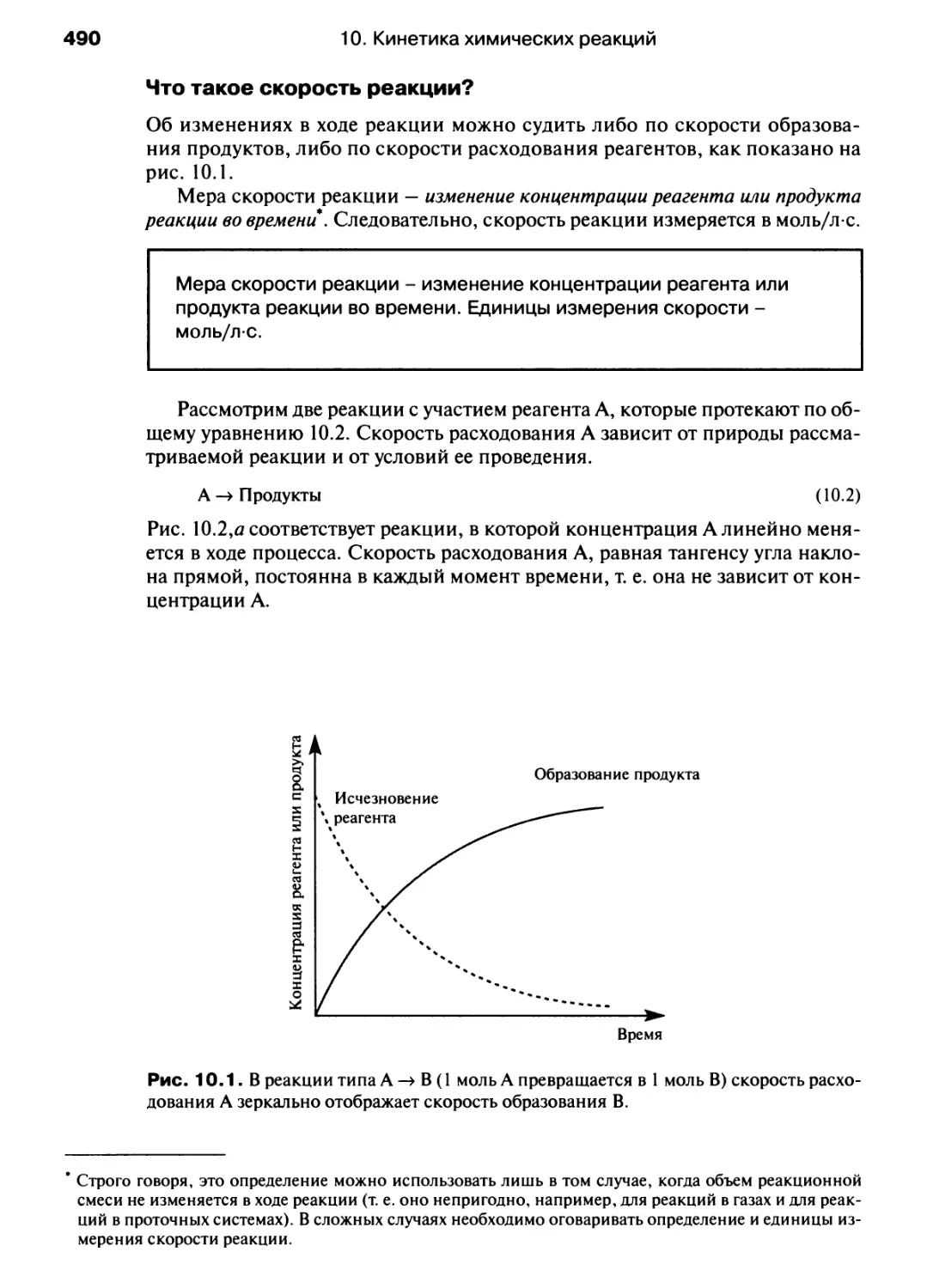
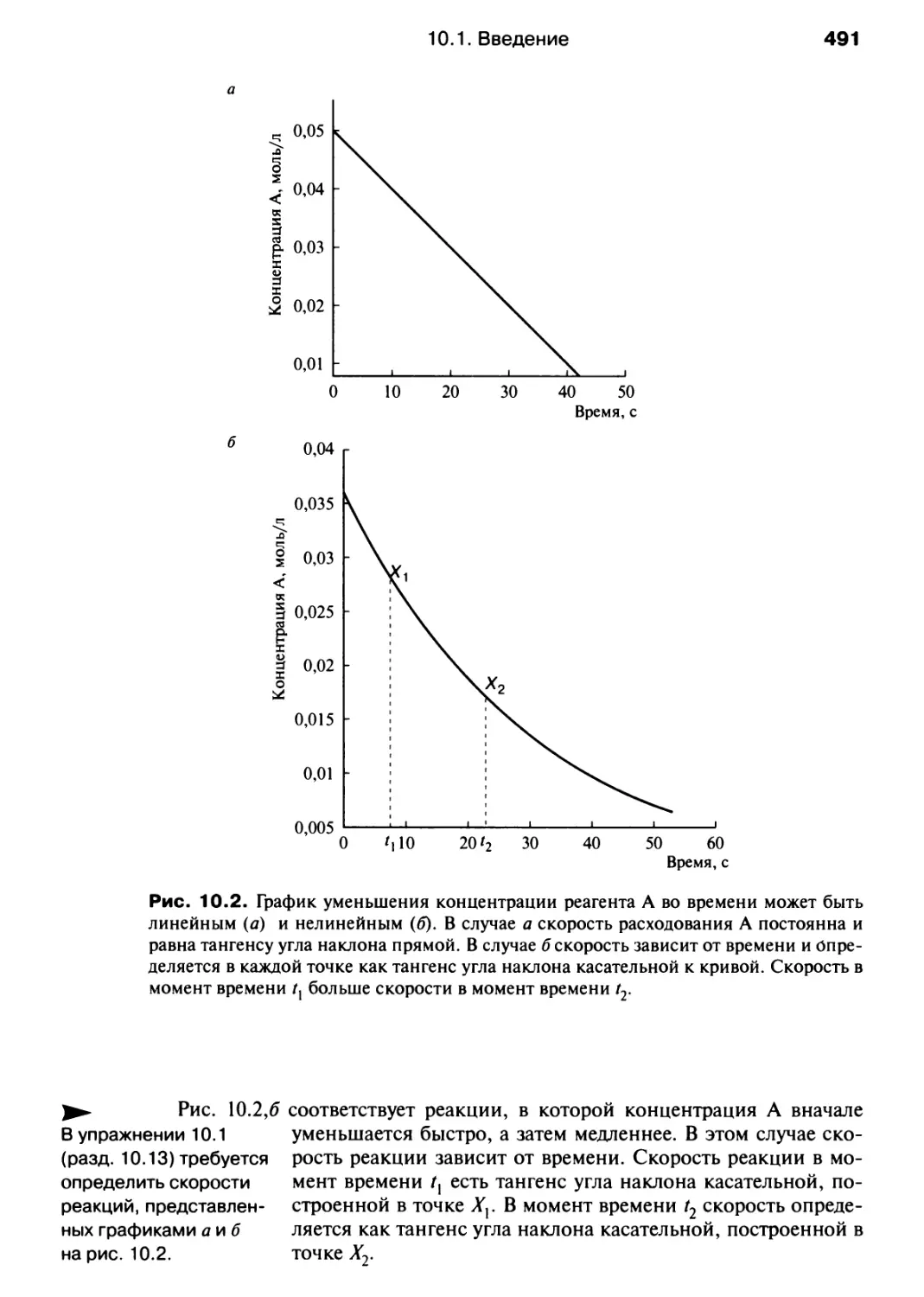
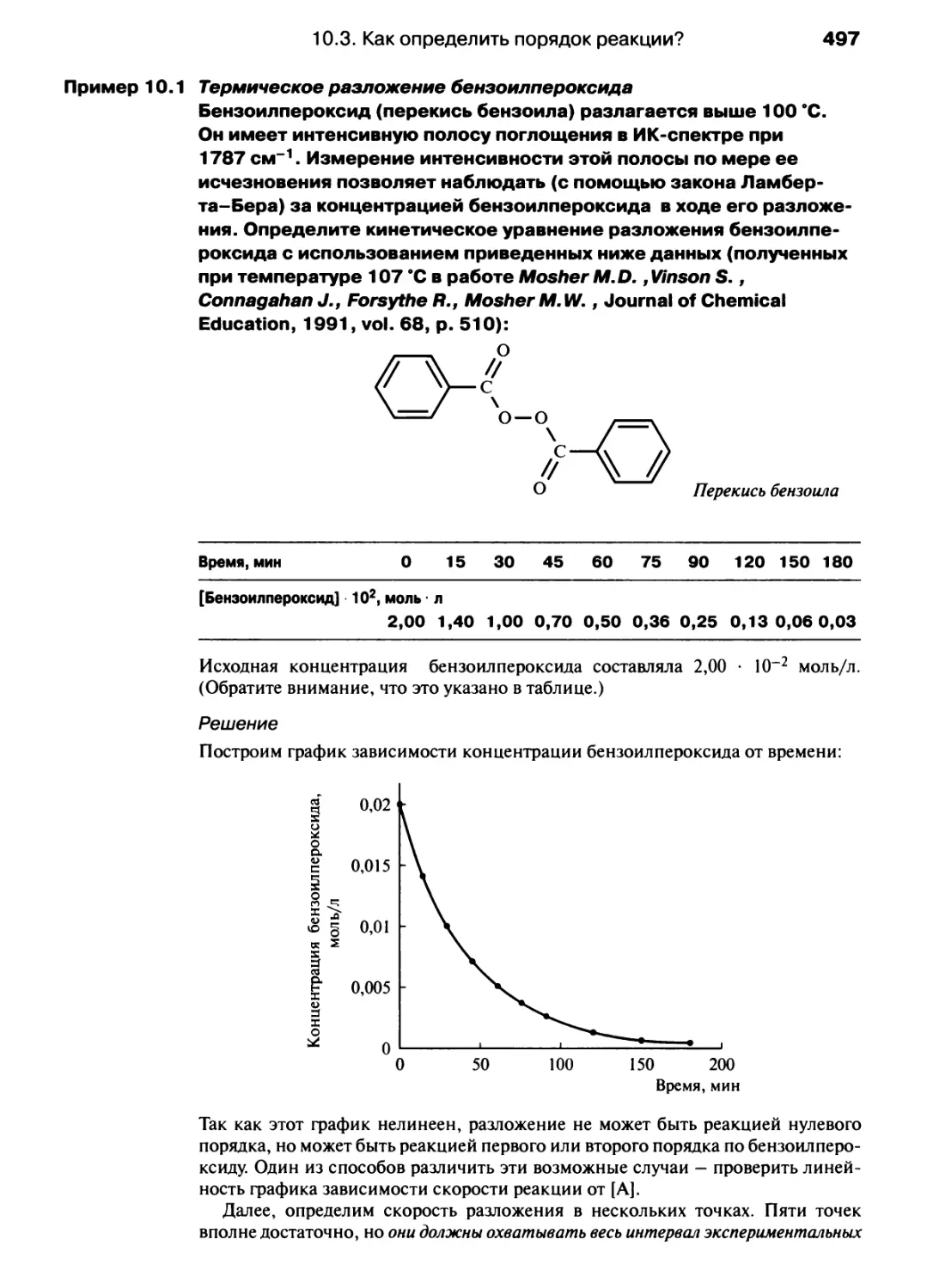
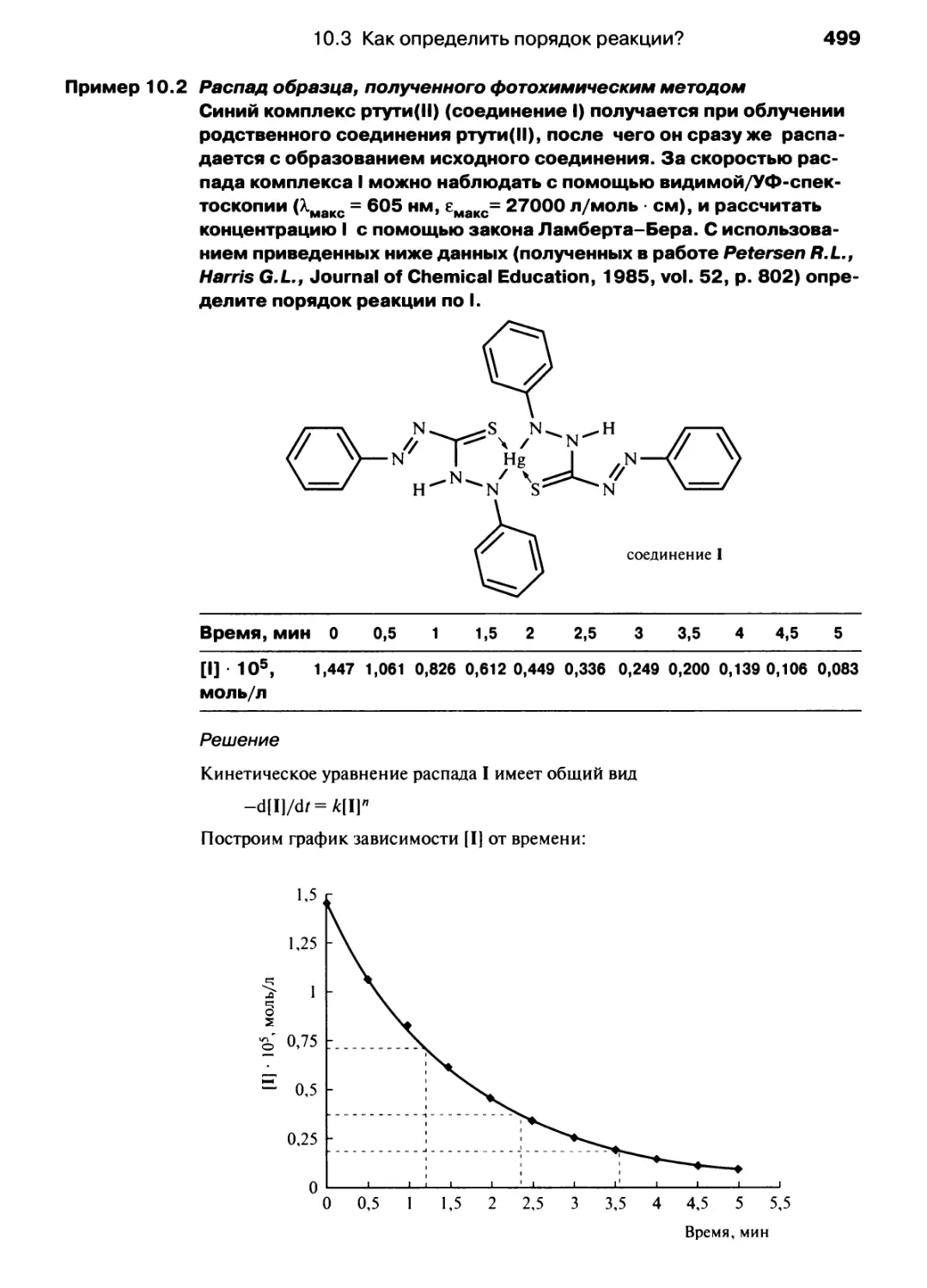
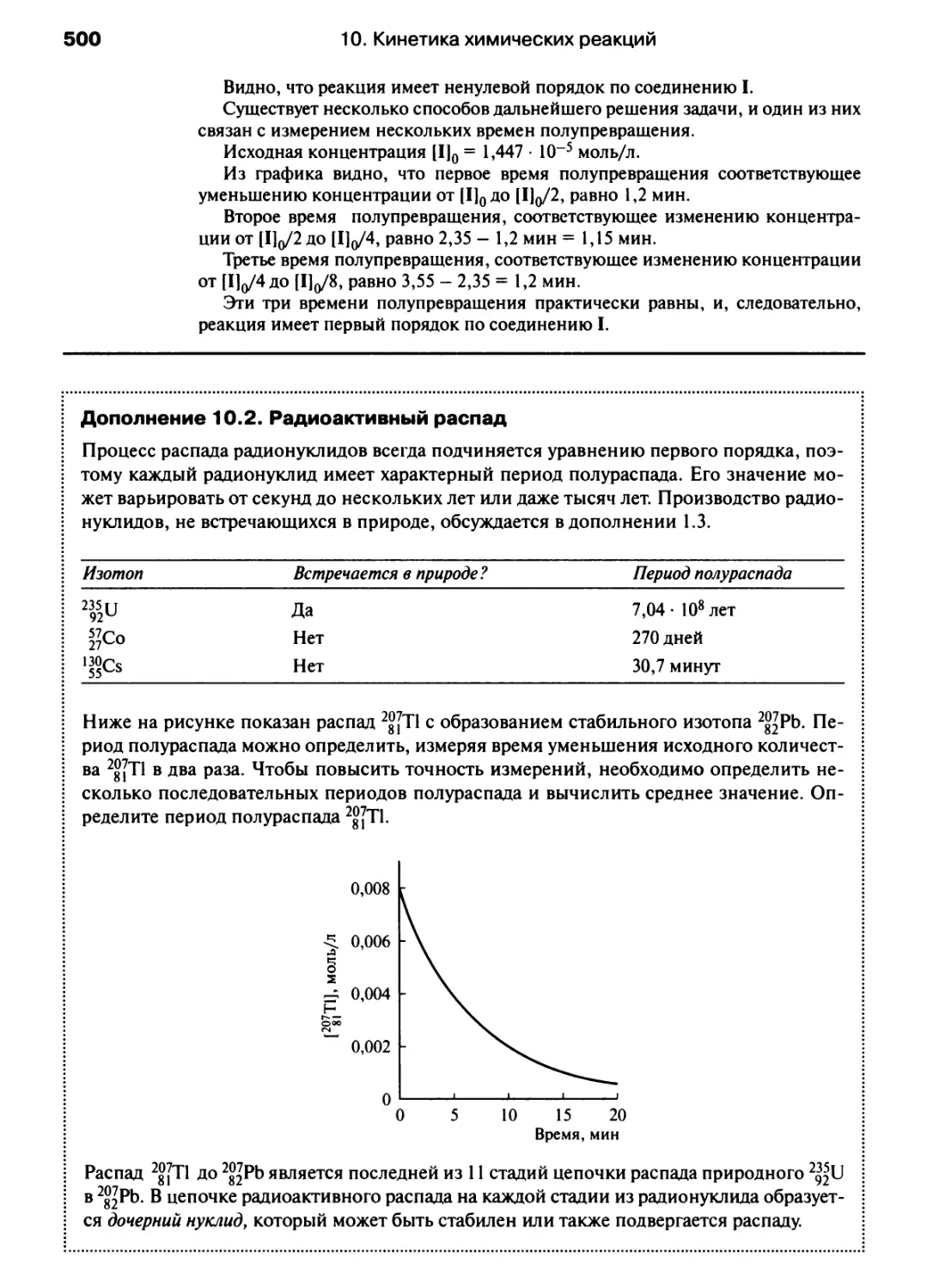
/
























































































































































































































































































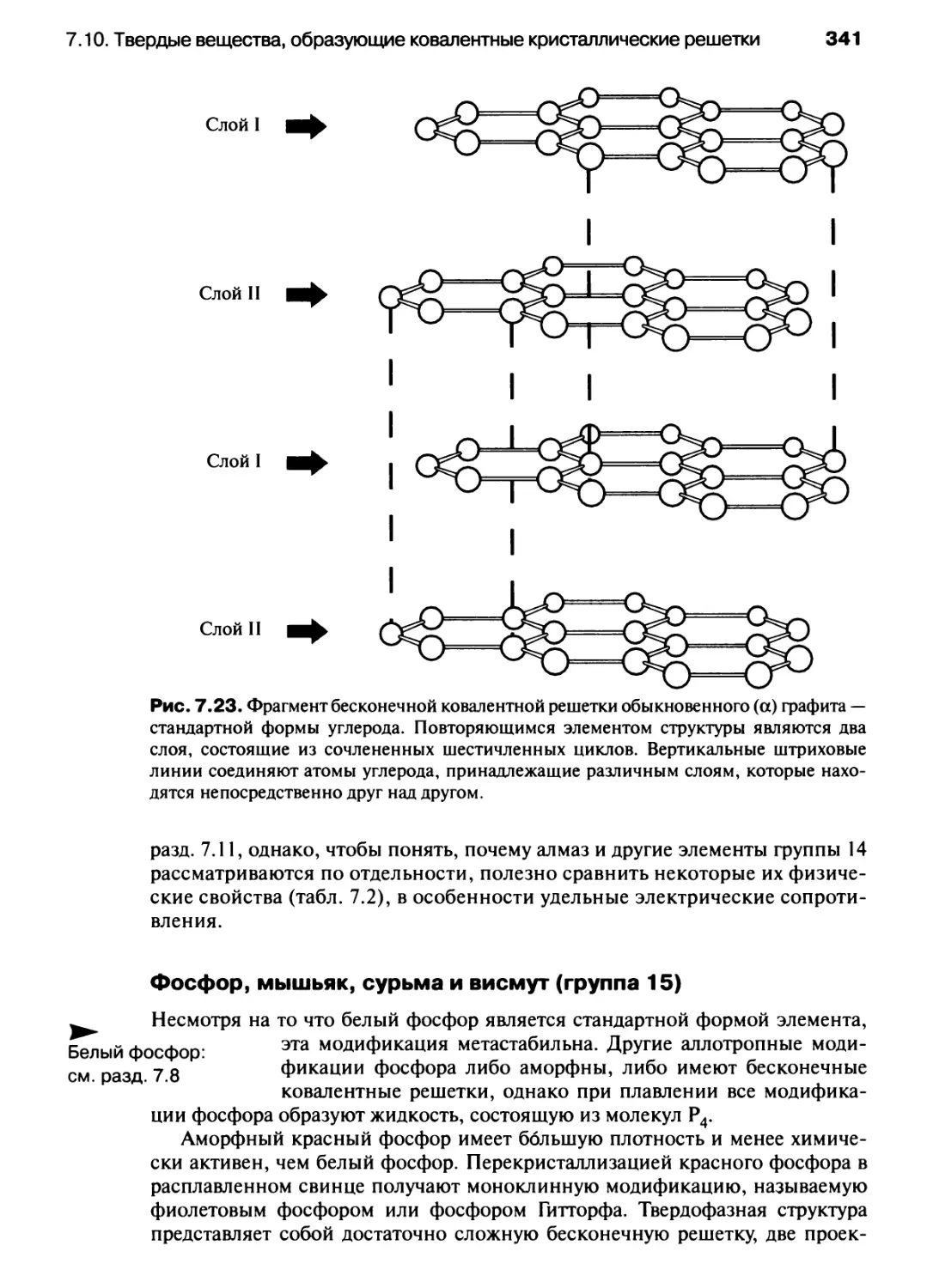
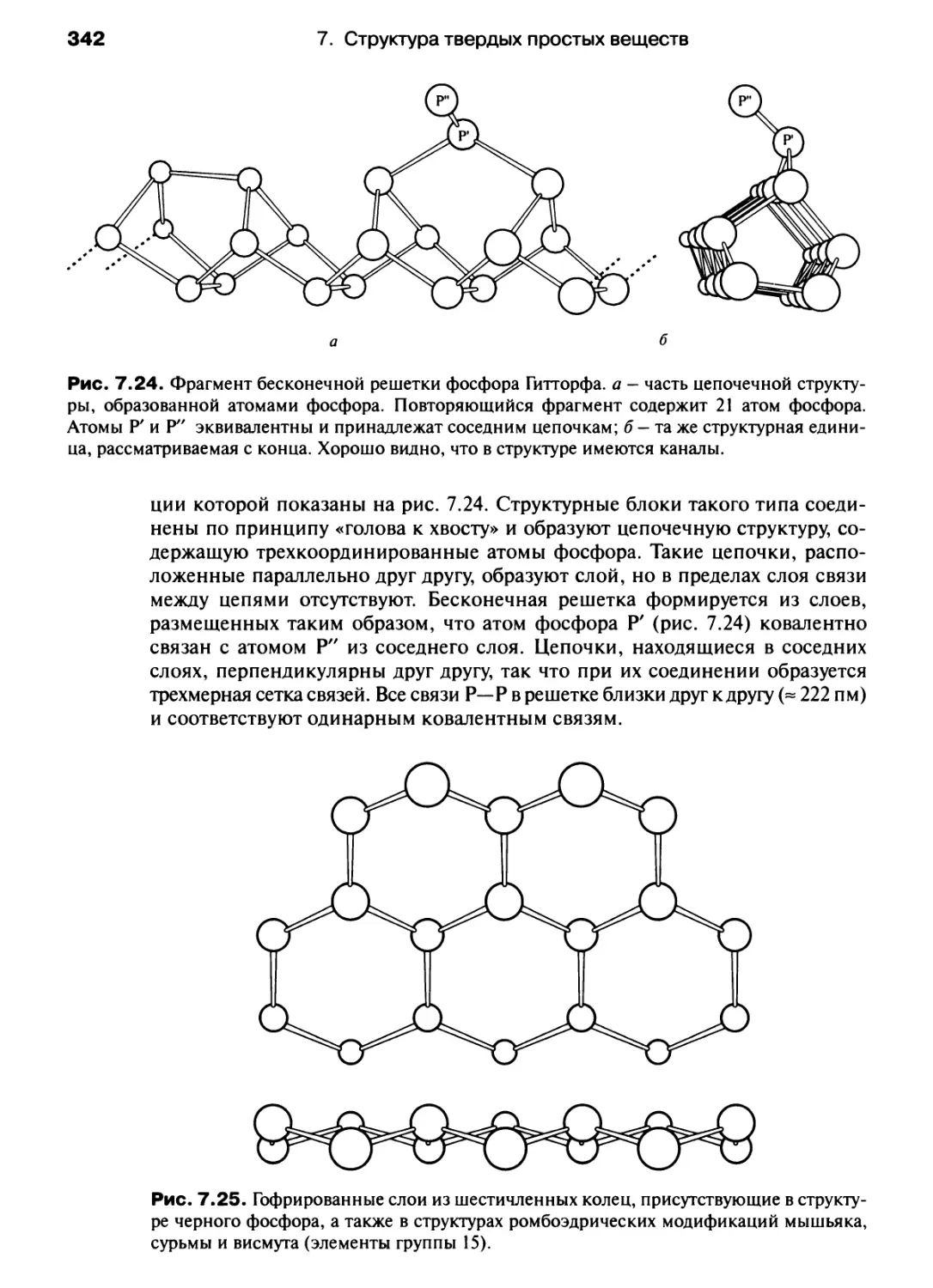







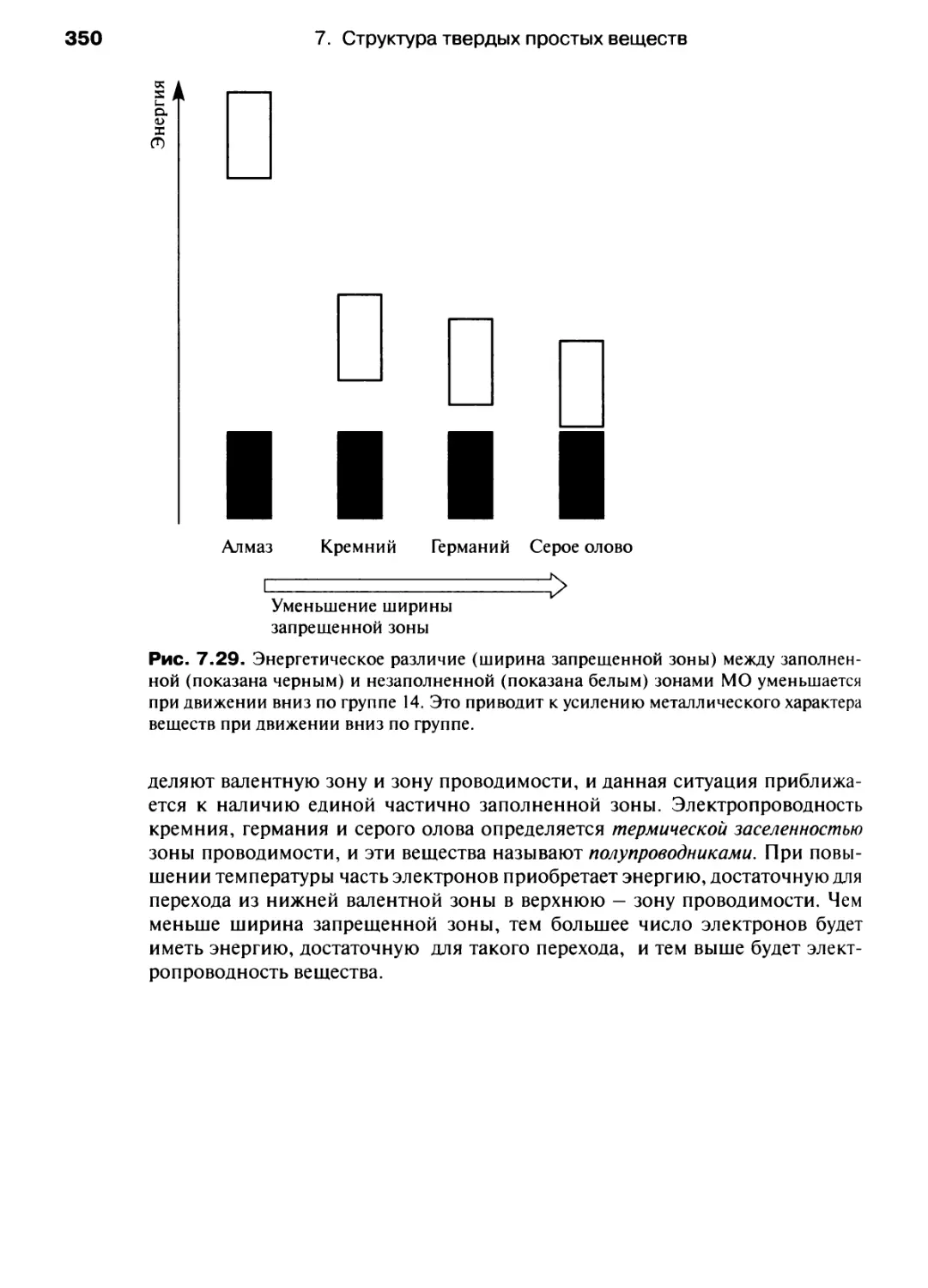




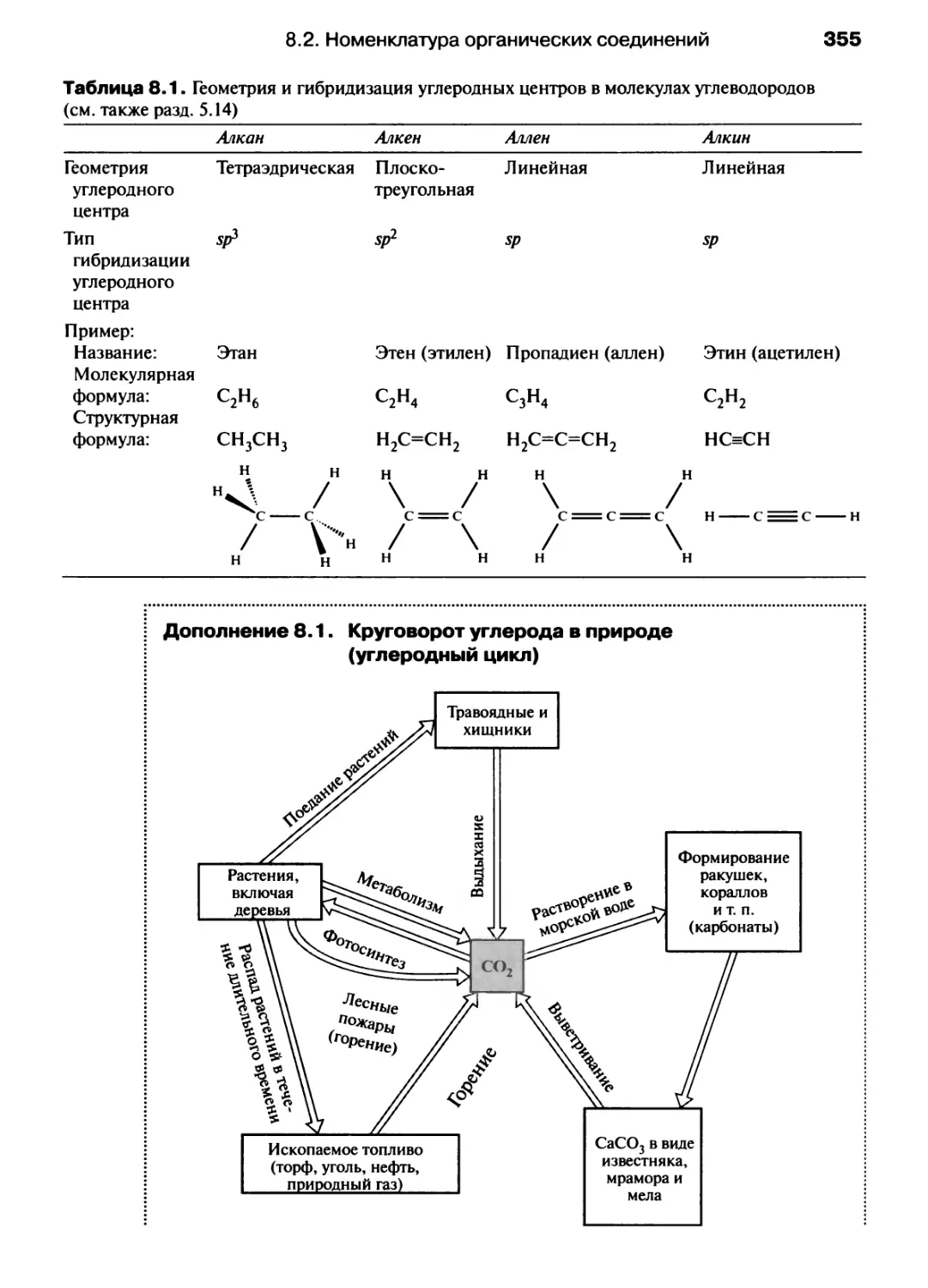























































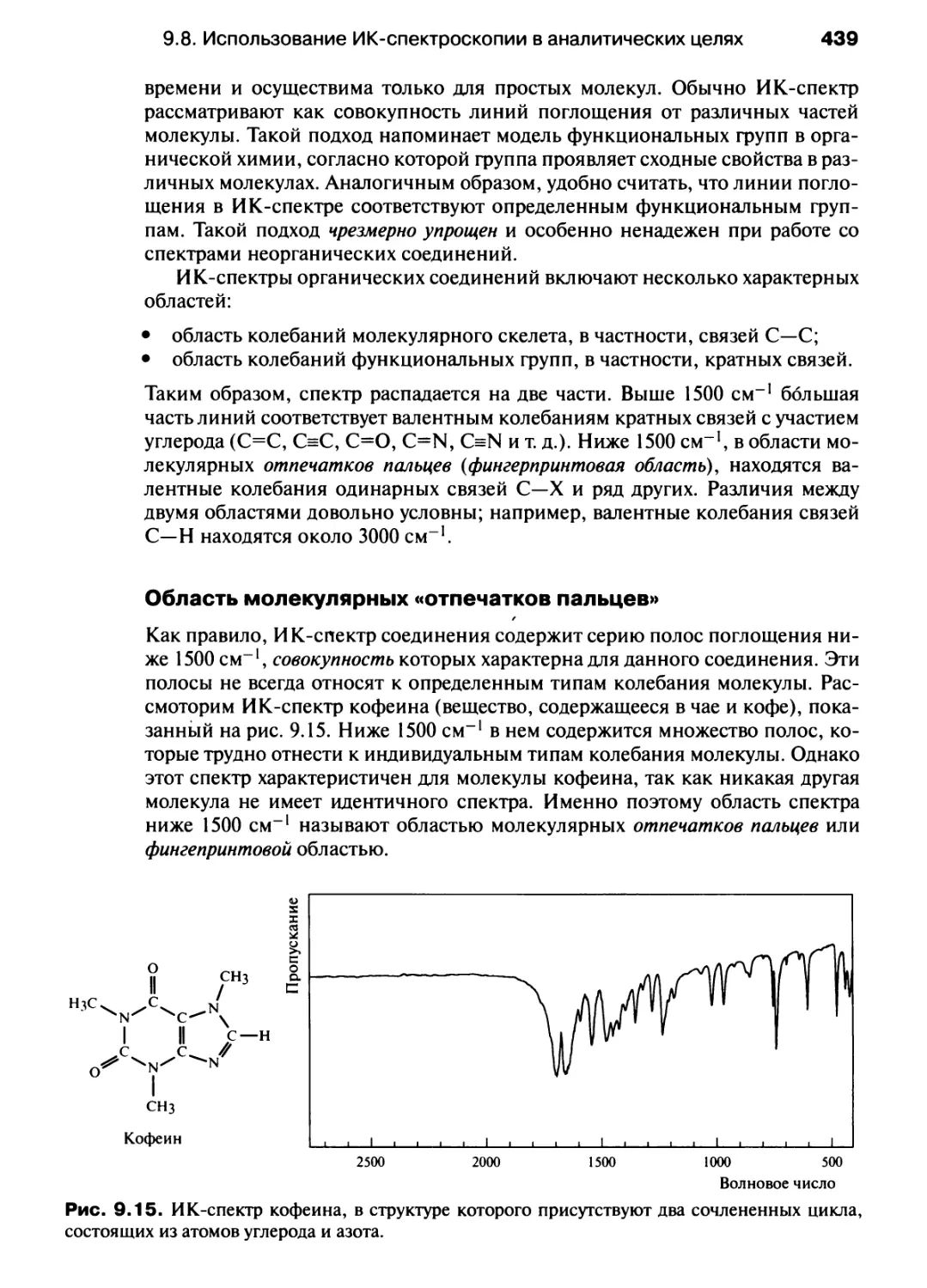

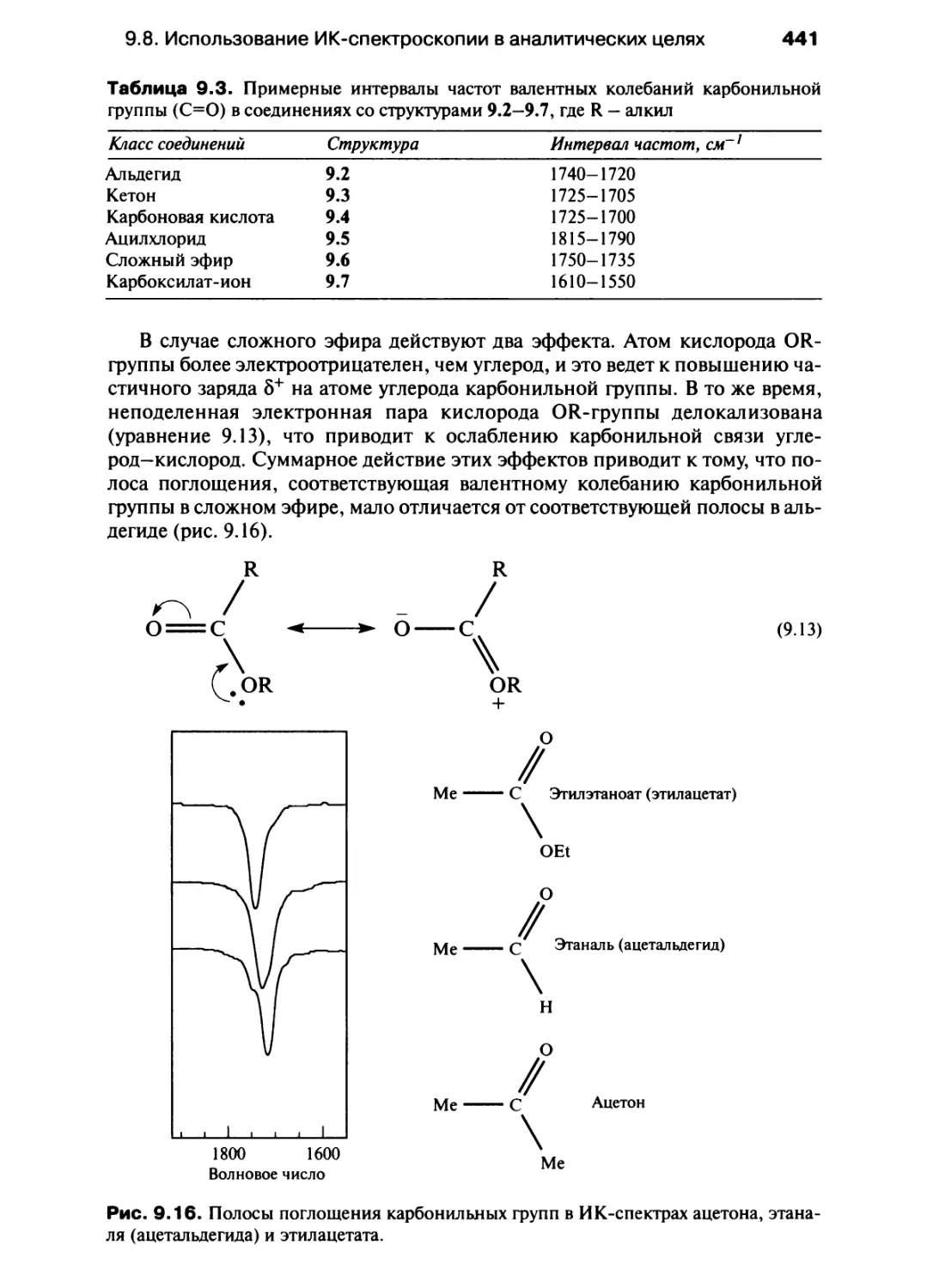
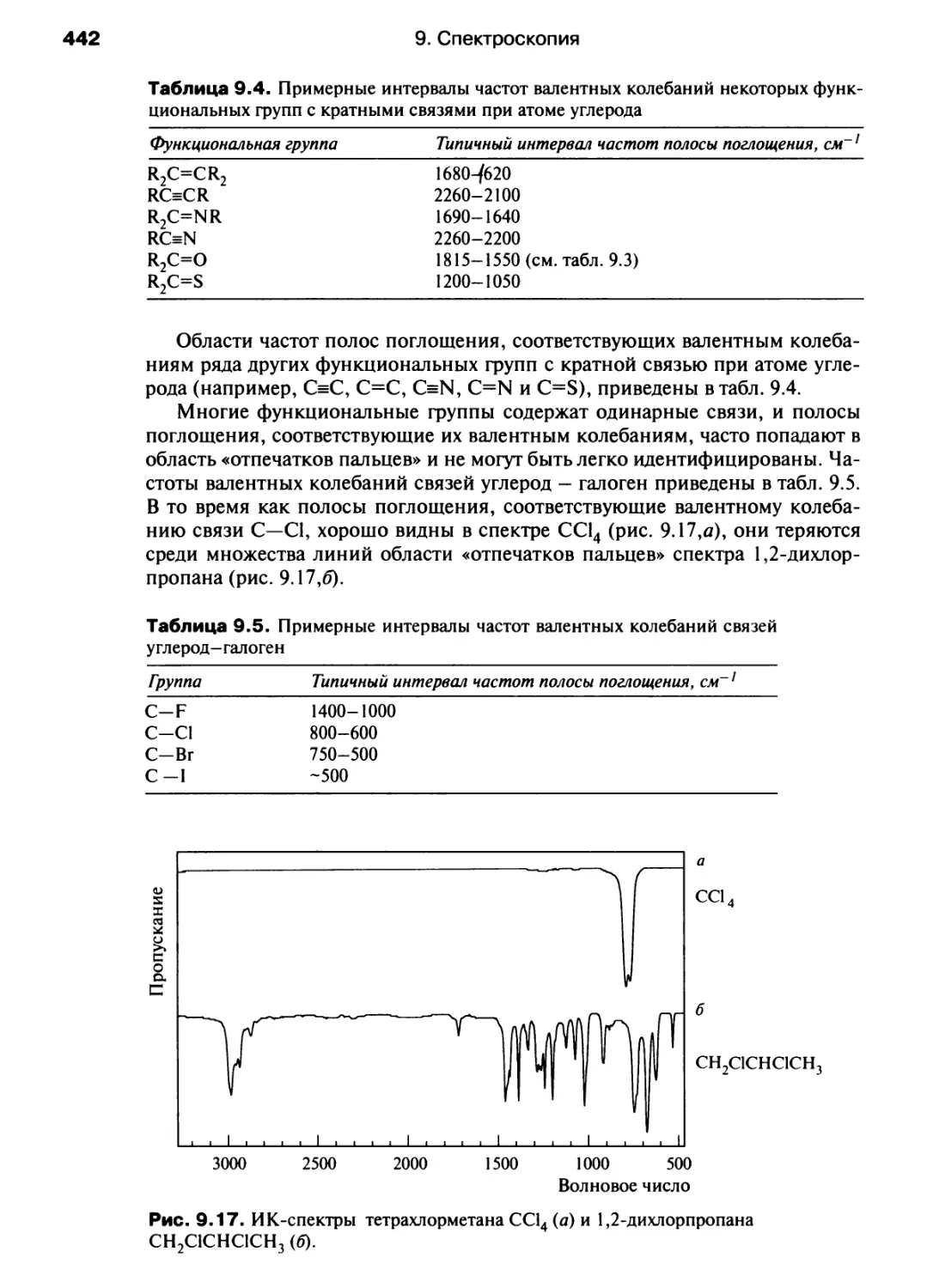
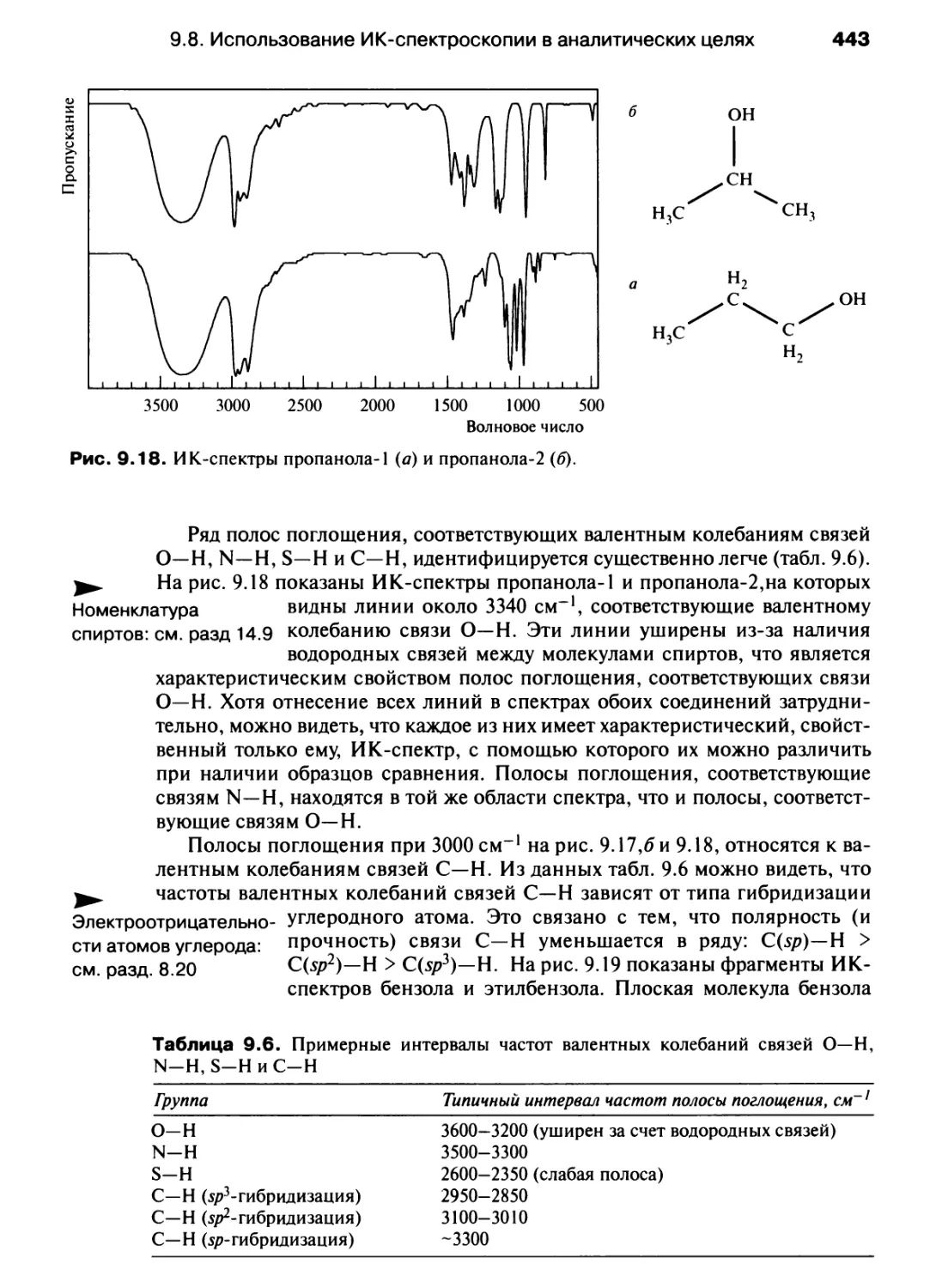
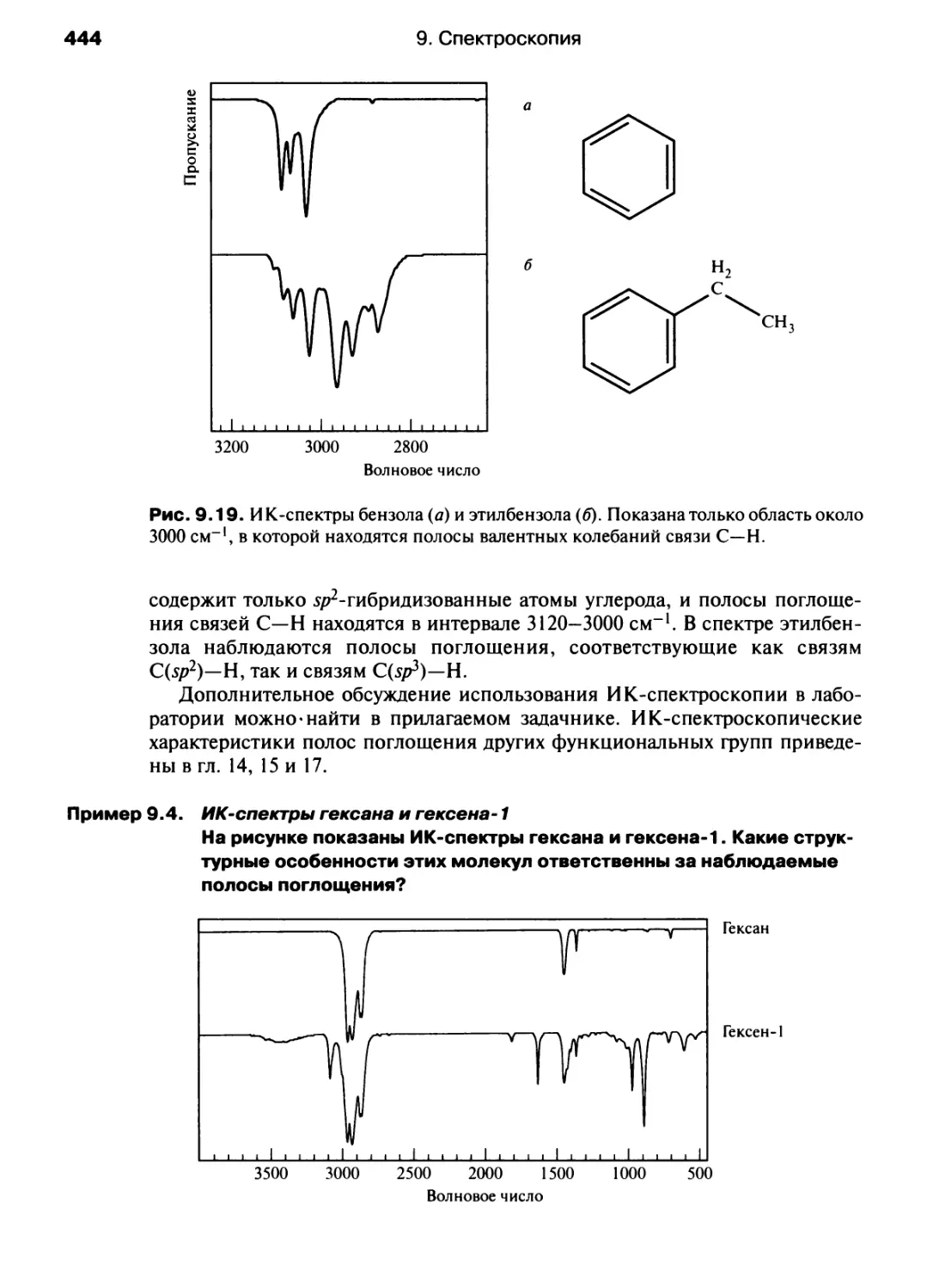
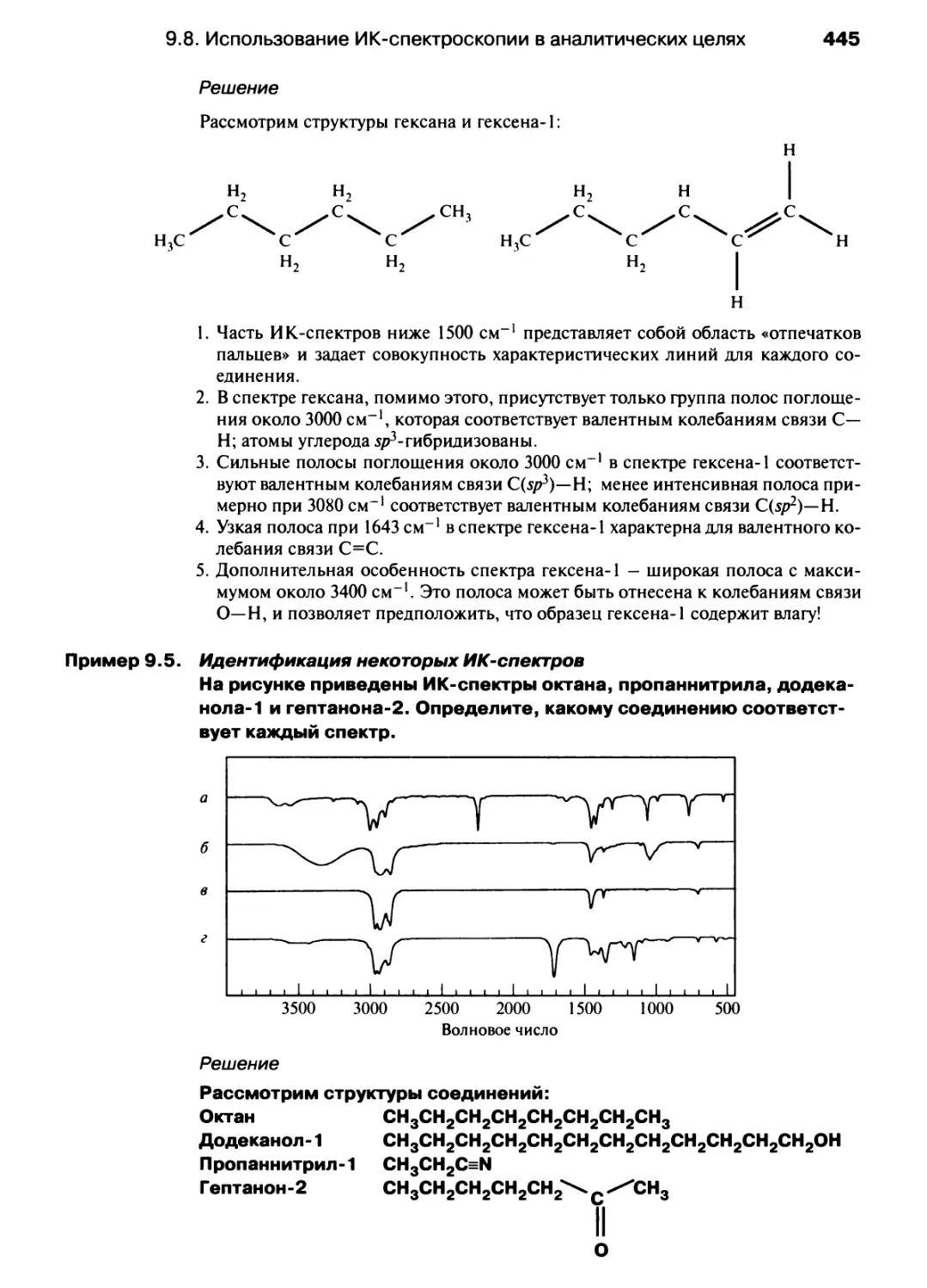

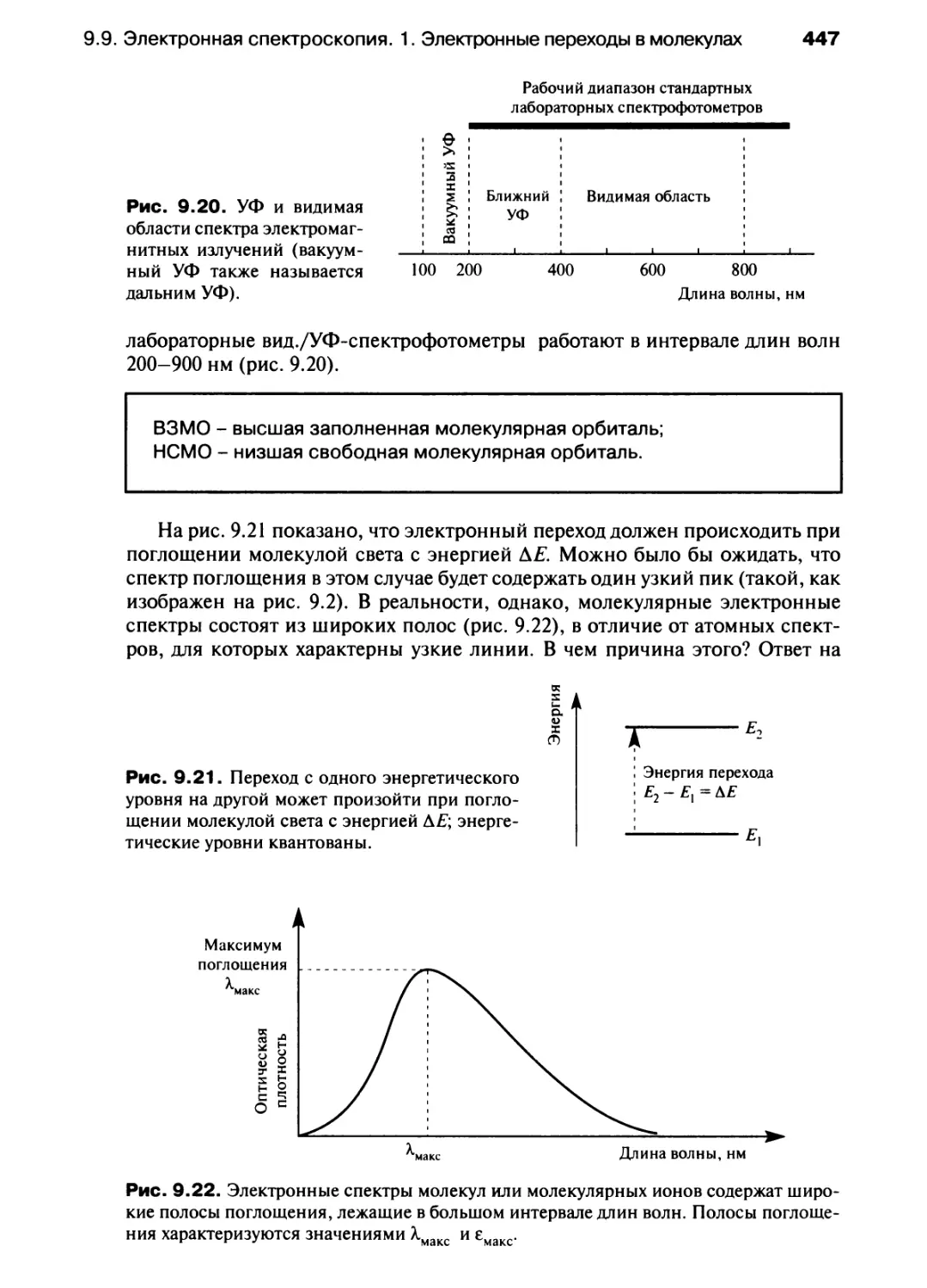

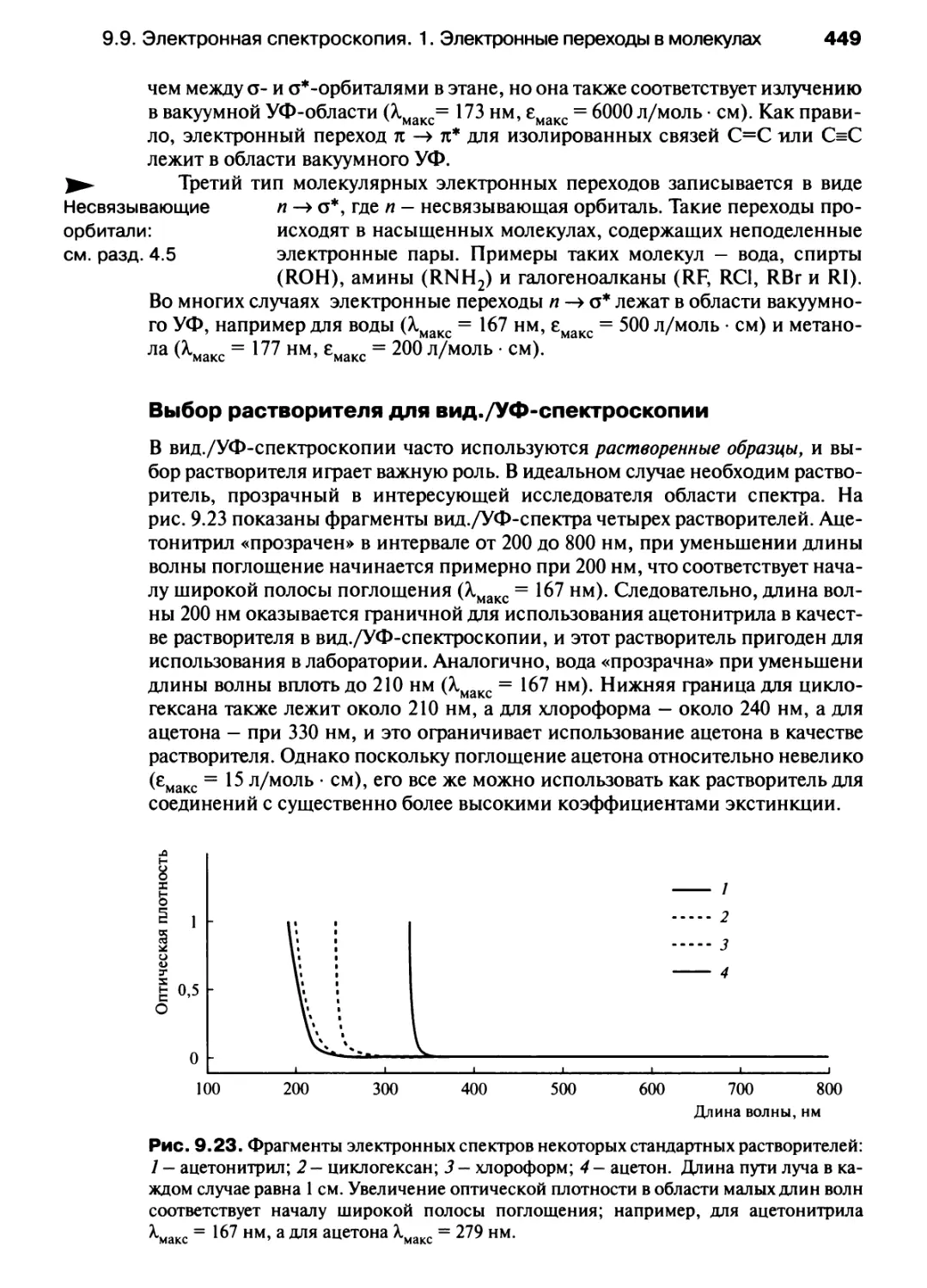












































































Автор: Хаускрофт К. Констебл Э.
Теги: химия общая и неорганическая химия задачи по химии общая химия
ISBN: 5-03-003310-6
Год: 2002




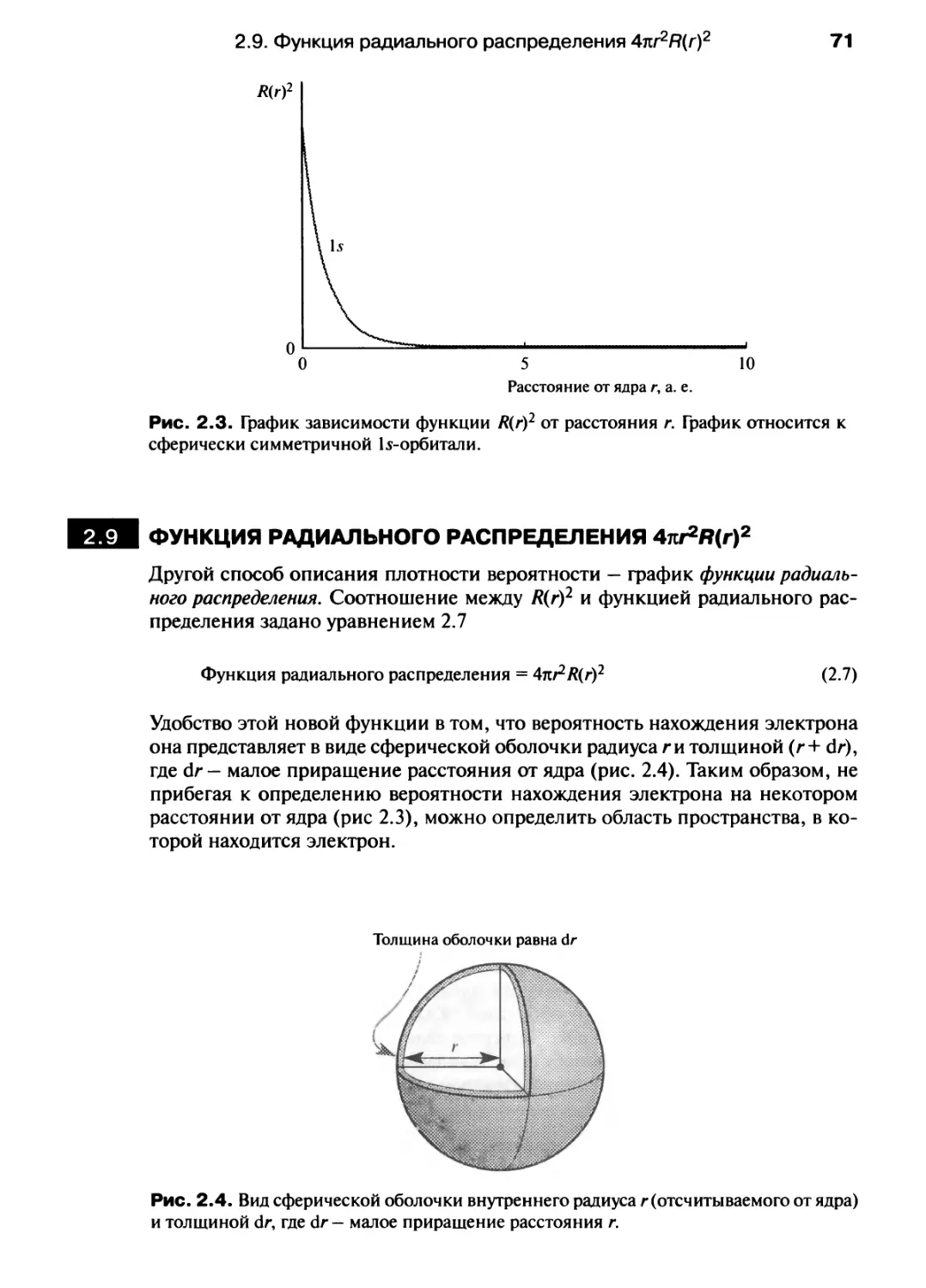
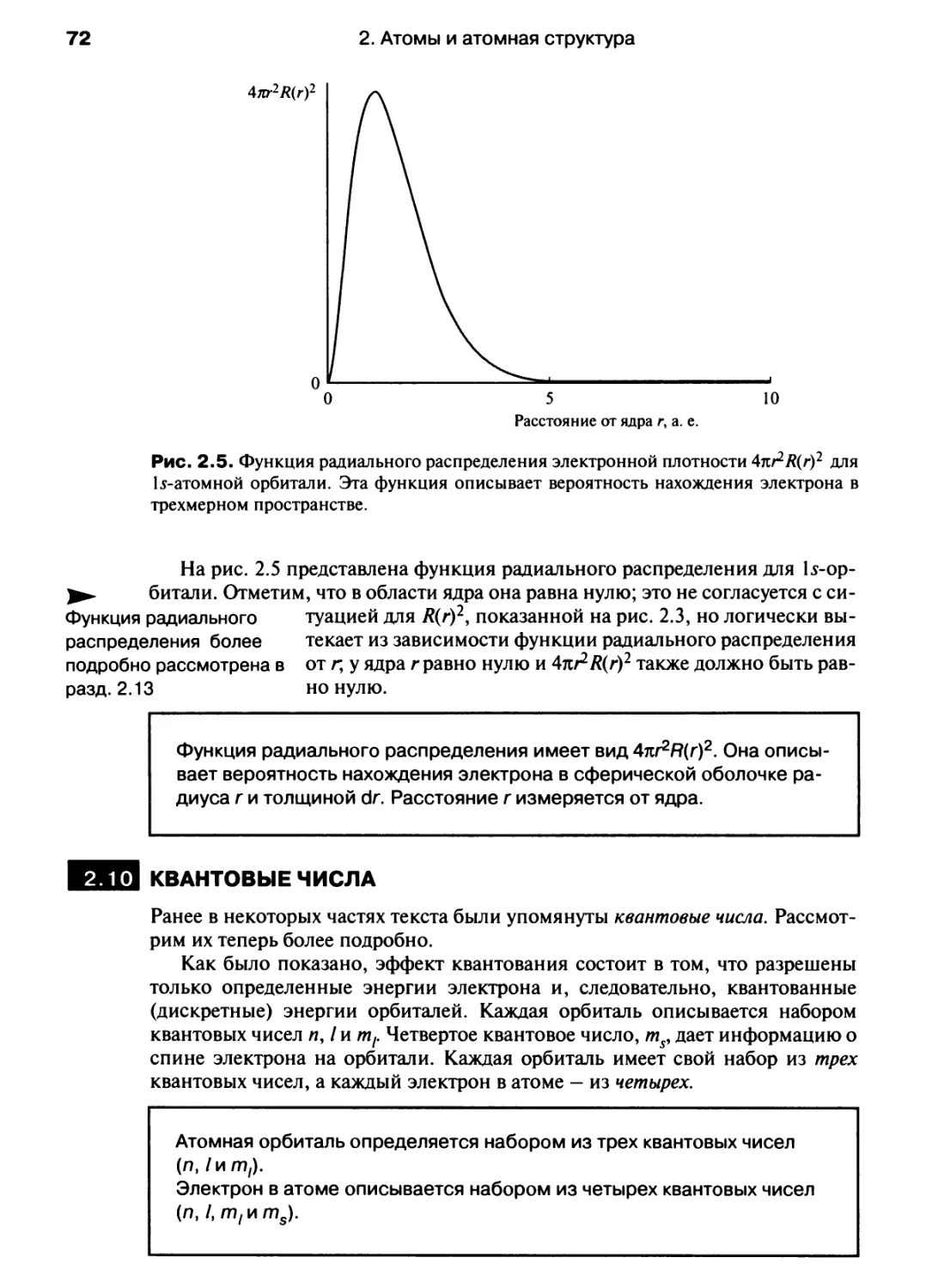
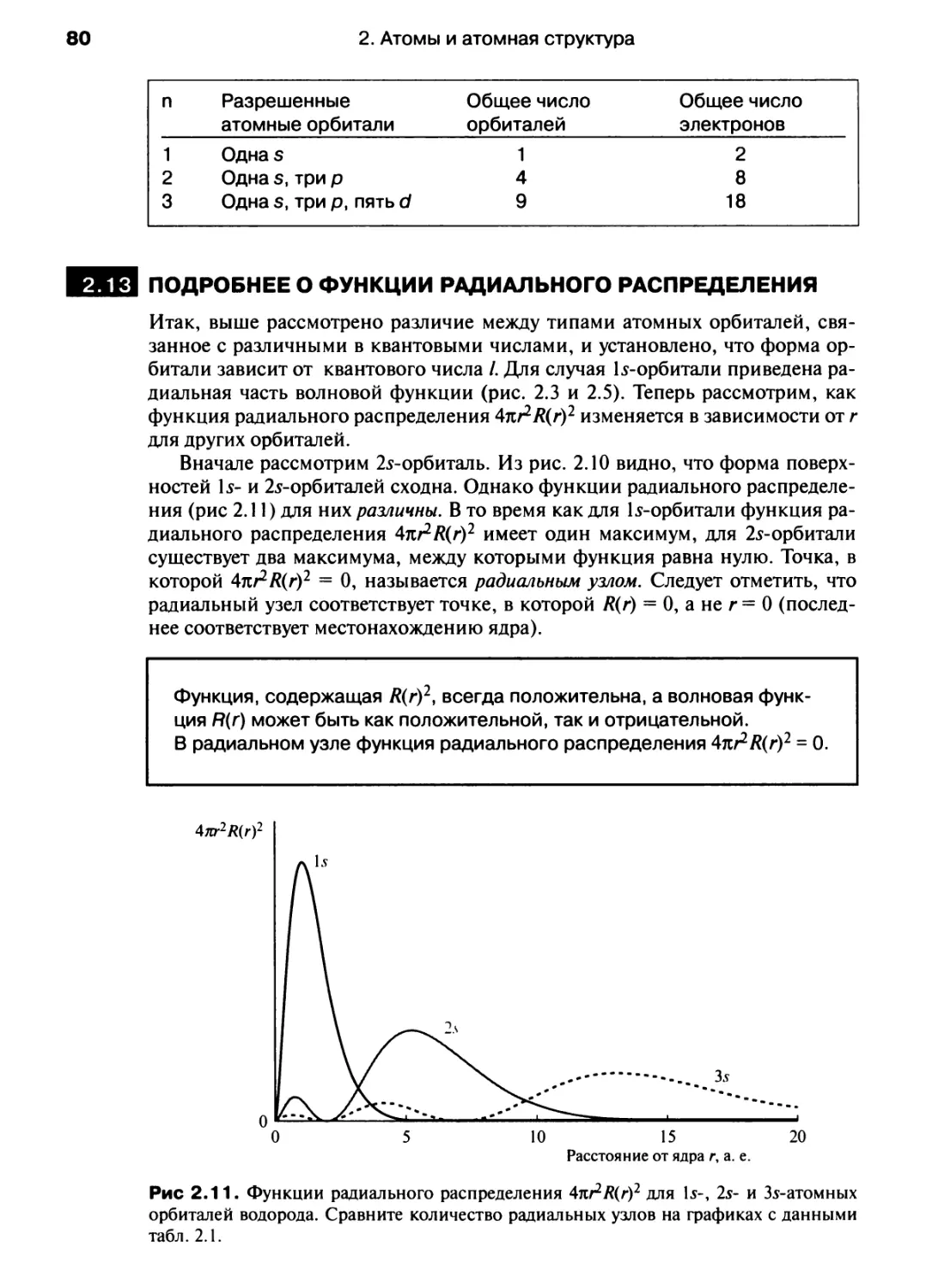
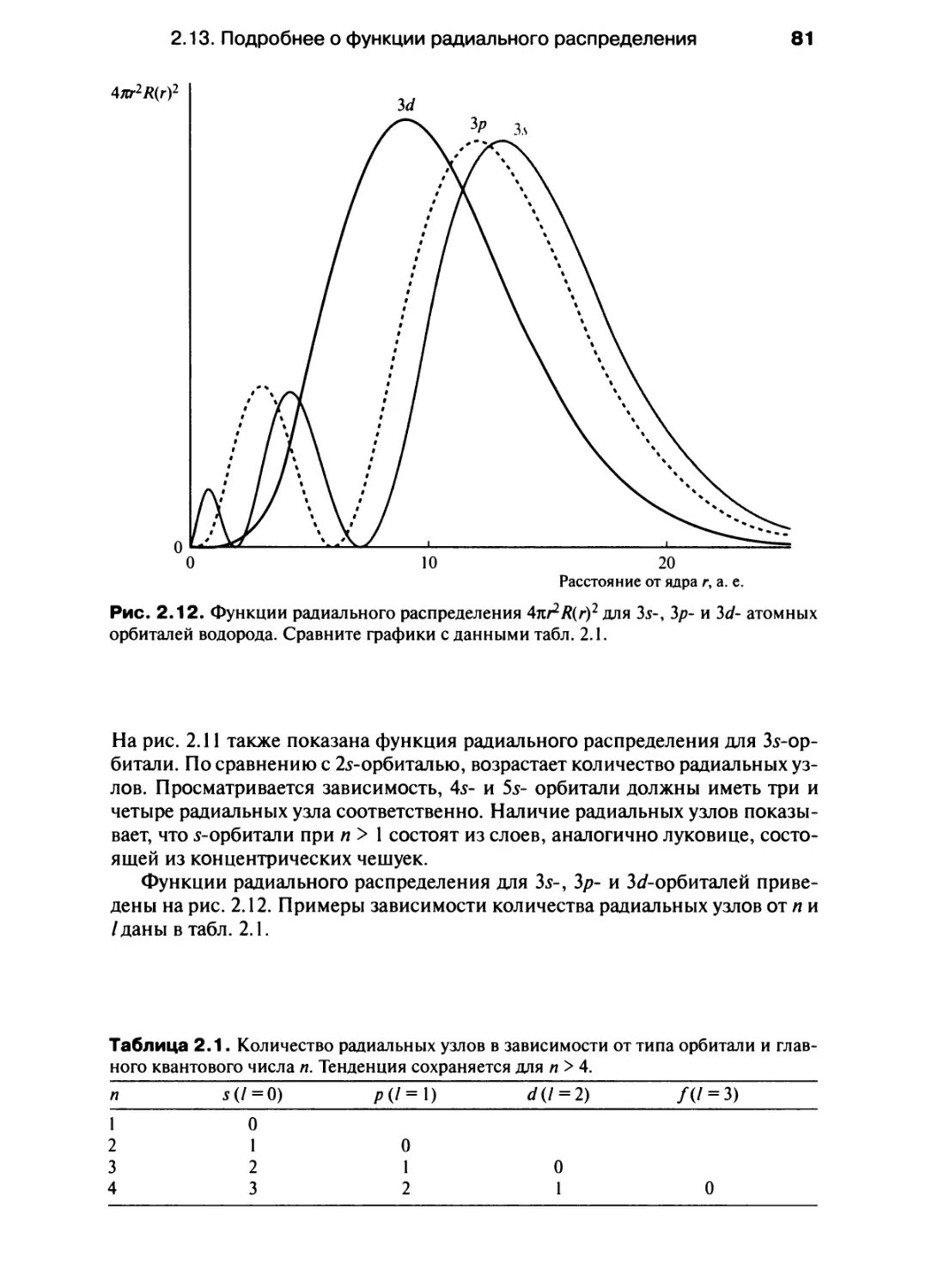
Текст
ЛУЧШИЙ
| ЗАРУБЕЖНЫЙ |
УЧЕБНИК
К. Хаускрофт, Э. Констебл
СОВРЕМЕННЫЙ
КУРС
ОБЩЕЙ ХИМИИ
L_V"
Издательство «МИР»
CHEMISTRY
An Integrated Approach
Catherine E. Hausecroft
Edwin C. Constable
Longman
ЛУЧШИЙ w
зарубежный
УЧЕБНИК
К. Хаускрофт, Э. Констебл
СОВРЕМЕННЫЙ
КУРС
ОБЩЕЙ ХИМИИ
В двух томах
1
Перевод с английского
канд. хим. наук Я. А. Ребане,
М. А. Дикусар
и канд. хим. наук А. А. Вертегела
под редакцией
профессора, д-ра хим. наук В. П. Зломанова
Москва «Мир» 2002
УДК 54
ББК24.1
Х26
Хаускрофт К., Констебл Э.
Х26 Современный курс общей химии. В 2-х т. Т. 1: Пер. с англ. —
М.: Мир, 2002. — 540 с, ил. - (Лучший зарубежный учебник)
ISBN 5-03-003310-6
В учебном издании, написанном английскими авторами, химия
рассматривается как единая наука без традиционного разделения на
органическую, неорганическую, физическую и т. п. химию, с современных
позиций излагаются основные концепции и законы общей химии. Для
лучшего усвоения материала каждая глава сопровождается
упражнениями, примерами решения типовых задач, а также дополнениями,
расширяющими кругозор студентов. Книга хорошо иллюстрирована.
В русском издании выходит в двух томах. Том 1 включает первые 10
глав.
Для студентов университетов и химических вузов.
ББК24.1
Федеральная программа книгоиздания России
Редакция литературы по химии
© Addison Wesley Longman, 1997.
This translation of Chemistry: An Integrated Approach,
ISBN 5-03-003310-6 (русск.) First Edition is published by arrangement with Addison
ISBN 5-03-003412-9 Wesley Longman Limited, London
ISBN 0 582 25342 X (англ.) О перевод на русский язык, оформление, «Мир», 2002
ПРЕДИСЛОВИЕ РЕДАКТОРА
ПЕРЕВОДА
Предлагаемая читателю книга «Современный курс общей химии» является
фундаментальным учебным изданием. Новизна, оригинальность и принципиальное
отличие этого учебника от ранее издававшихся на русском языке заключается в
интегральном подходе к преподаванию химии как единой науки, а не в виде
исторически сложившихся разделов: неорганической, органической и физической
химии. О единстве химии как науки о превращениях вещества свидетельствуют
реальные процессы, протекающие в живых организмах, в природе вокруг нас.
Отсутствие существенных различий между неорганической и органической химией
особенно заметно в химии соединений неметаллов, образующих протяженные
цепи, циклы, клетки, кластеры и т. д.
Композиция книги и подбор материала соответствуют основной идее -
неделимости химии. Серьезное внимание уделено строению атома, сопоставлению и
использованию метода молекулярных орбиталей (МО) с классической
концепцией валентных связей (ВС) для объяснения строения и свойств двух- и
многоатомных молекул. Теоретические положения термодинамики, кинетики и
строения вещества сопровождаются конкретными примерами из различных
разделов химии. Особенность учебника — анализ основных закономерностей
строения и свойств химических соединений с использованием представлений
об изоэлектронных аналогах.
Учебник написан на основе курса лекций, который авторы читали в различных
университетах и колледжах Великобритании, США, Канады, Швейцарии и т. д.
Необходимо отметить ясность и четкость изложения и прекрасные
информативные иллюстрации, что способствует углубленной проработке приводимых
практических данных. Учебник построен так, что материал не обязательно изучать
последовательно, отдельные главы можно изучать выборочно, следуя традиционной
схеме — неорганическая, органическая и физическая химия. Для лучшего
понимания и усвоения материала в учебнике имеются примеры решения типовых задач и
упражнения. К основному тексту учебного курса, выделенного в этот двухтомный
учебник, прилагается как дополнение «Задачник» (полное название:
«Современный курс общей химии. Пособие для самостоятельной работы»), в котором
рассмотрены такие важные темы, как теория кислот и оснований, буферные системы,
многостадийный органический синтез и т. д., а также конкретные задачи для
самостоятельного решения.
Перевод учебника осуществлен коллективом авторов — Я.А. Ребане и
М.А. Дикусар (гл. 1-5), А.А. Вертегелом (гл. 6-10), Р.А. Ничипорук (гл. 11-13) и
А.А. Молодык (гл. 14—17).
Можно надеяться, что появление этого учебного издания на русском языке
будет полезным для преподавателей и студентов химических и нехимических
высших учебных заведений, а также для учителей и учащихся средних школ с
углубленным изучением химии.
В. П. Зломанов
ПРЕДИСЛОВИЕ АВТОРОВ
К РУССКОМУ ИЗДАНИЮ
По мере развития междисциплинарных исследований границы между
органической, неорганической и физической химией стираются. Поэтому в настоящем
учебнике (Chemistry: An Integrated Approach) мы постарались показать
студентам первого курса химию как единый предмет, а не совокупность трех разных
дисциплин. Чтобы понять применение основ химии в науке и технике на
современном этапе развития нашего общества, нужно отдавать себе отчет, что
объединение органической, неорганической и физической химии при обучении в одну
целостную науку сейчас диктуется потребностями жизни. В университетах и
колледжах многих стран программы обучения изменяются именно в этом
направлении. Мы надеемся, что предлагаемый учебник соответствует требованиям
интегрального подхода к изучению химии. В то же время некоторые новые
химические проблемы и достижения специально выделены в виде дополнений. Об
основных задачах, которые стояли перед нами при написании учебника, мы
рассказали в предисловии.
Мы выражаем благодарность профессору В.П. Зломанову за организацию
перевода нашей книги и редактирование русского издания, что сделало наш труд
доступным для русской аудитории.
Катрин Е. Хаускрофт
Эдвин К. Констебл
БЛАГОДАРНОСТИ
Мы выражаем благодарность за разрешение на использование в нашей книге
следующих материалов:
рис. 6.2, а: воспроизведен с разрешения из статьи Wahl А. С. Science, 151,
р. 961 (1966). Copyright 1966 American Association for the Advancement of
Science;
рис. 6.2, а и б: Journal of Chemical Physics, American Institute of Physics and
Professor Richard F. W. Bader;
рис. 6.22: Zeitschrift fur Naturforschung, Section A: Physical Sciences, Verlag
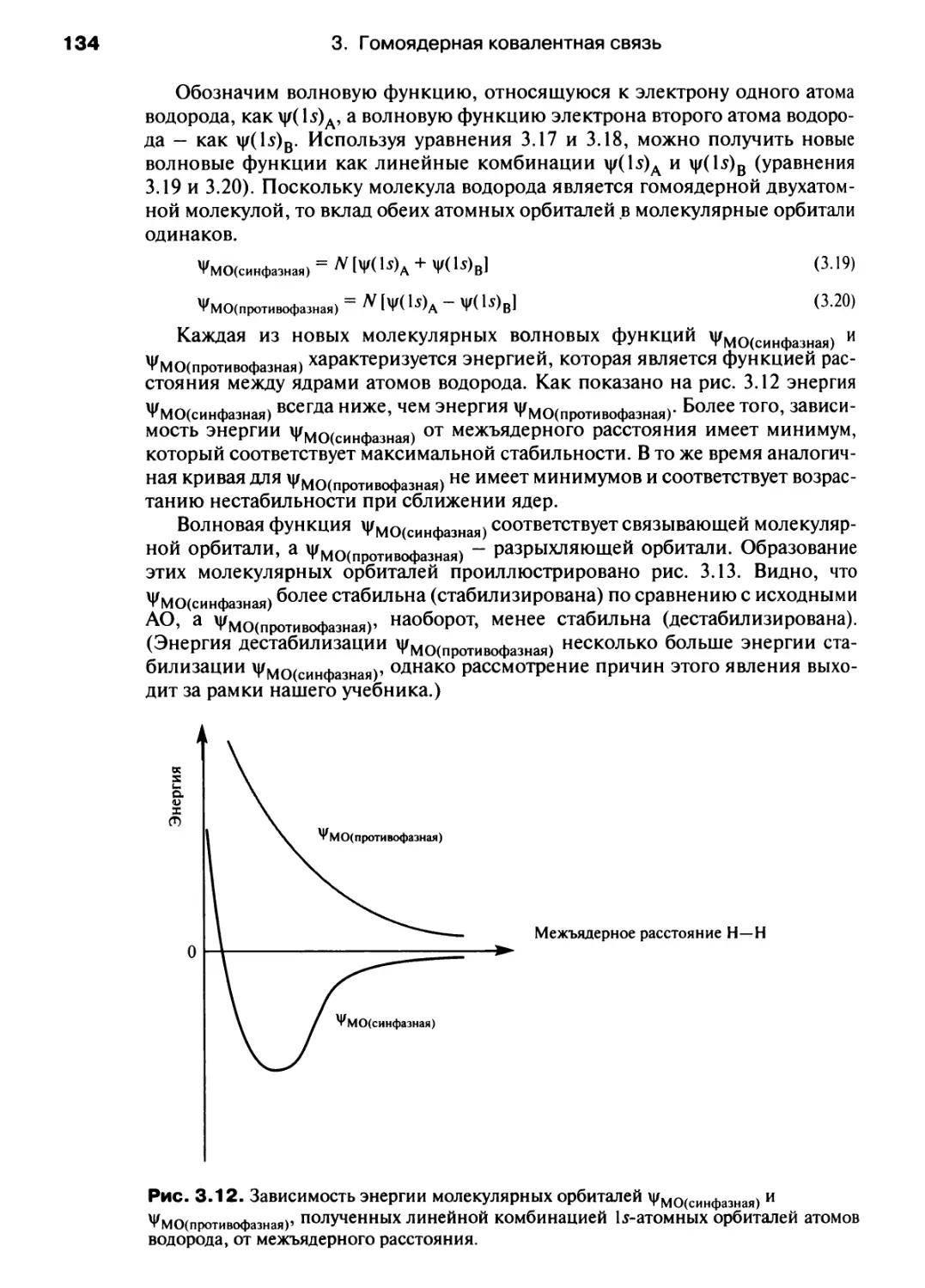
der Zeitschrift fur Naturforschung;
рис. 7.14: Chemical Reviews, Gordon and Breach Publishers.
Несмотря на наши усилия найти владельцев авторского права мы
пользуемся этой возможностью принести наши извинения всем владельцам
авторского права, чьи права мы неумышленно нарушили.
Посвящается Филби и Изис,
которые по-своему помогали нам
в написании этого учебника
ПРЕДИСЛОВИЕ
Итак, перед вами учебник под названием «Современный курс общей химии»
("Chemistry: An Integrated Approach"). Что означает такое название и каковы
наши цели? Почему читателю следует предпочесть эту книгу многим другим
учебникам по химии?
Всем нам, преподавателям и студентам, часто приходится сталкиваться с тем,
что один и тот же материал преподносится с использованием различных
подходов и смысловых оттенков. Например, на первом курсе университетов при
изучении химии приходится по крайней мере четыре раза обращаться к
термодинамике. Другая общая проблема заключается в том, что студенту слишком рано
приходится иметь дело со «строгим» изложением материала. Ведь совсем не так
уж необходимо излагать математическое решение уравнения Шредингера, чтобы
оценить характер и полезность модели атомных и молекулярных орбиталей для
понимания природы химической связи.
В настоящем учебнике различные концепции вводятся на том уровне, который
соответствует университетской программе первого курса. Однако излагаются они
таким образом, что при дальнейшем изучении их не придется изучать снова. Мы не
использовали слишком упрощенных и поэтому неверных приближений.
Химия — это единая наука, что и отражено в композиции учебника: в нем нет
глав, посвященных исключительно органической, неорганической или
физической химии, хотя в содержании отдельной главы может преобладать
соответствующая тематика. Разграничение между органической и неорганической,
структурной и любой другой химией является искусственным. Этот учебник
предназначен для того, чтобы поощрять студентов не раскладывать получаемые
«химические знания» по «закрытым ящикам», которые открываются только на
экзаменах. В учебник включены различные примеры из реальной жизни, в том
числе из окружающей нас природы, рассказывается о применении химии в
биологии и промышленности. Это помогает понять, что неорганическая,
органическая и физическая химия являются взаимопроникающими (а вовсе не
взаимоисключающими) разделами единой науки.
В настоящем учебнике различные разделы химии тематически связаны
между собой. Это удобно для преподавания химии на первом курсе. Так, гл. 8, 14,
15 и 17, посвященные органической химии, могут быть изучены как отдельный
раздел. Физические методы исследования, применяемые в химии, а именно
инфракрасная спектроскопия, спектроскопия видимого и ультрафиолетового
излучения, ядерный магнитный резонанс, изложены в гл. 9; эта глава может быть
полезна при изучении строения вещества.
Следует сказать несколько слов о математическом аппарате учебника. После
тщательных раздумий мы приняли простой подход к математическим выводам:
не предлагая детальных математических выкладок, мы просто формулируем
концепцию (это особенно видно на примере гл. 10, посвященной кинетике),
поскольку на ранней стадии изучения химии гораздо важнее понимать следствия из
закона или формулы, чем уметь воспроизводить различные математические вы-
Предисловие
воды. Наиболее четко это использовано в гл. 12, где термодинамика изложена с
привлечением экспериментальных данных, а не математических выражений.
Как основа для изложения в учебнике принят уровень А английской
программы по химии, существующий с 1994 г. Однако в гл. 1 представлены обновленные
сведения о фундаментальных концепциях и законах, которые потребуются при
изучении последующих глав. Теория кислот и оснований Брёнстеда в первой главе
не рассматривается, но ее основные положения, а также задачи с расчетами
представлены в гл. 11, где обсуждается химия водорода.
Дополнением к основному тексту учебника служит «Задачник», в который
включены дополнительные примеры, а также некоторые новые темы (масс-спек-
трометрия, кинетическая теория газов, индикаторы, буферные системы,
многостадийный органический синтез и т. д.). Это расширяет содержание основного
курса, изложенного в учебнике. Разделы «Задачника» соответствуют разделам
данного учебника, так что сведения по предложенному набору вопросов
нетрудно найти, чтобы усвоить предлагаемый курс общей химии или получать
образование, следуя модульной, или традиционной, системе изучения химии.
Если это не оговорено особо, то физические данные цитируются по
следующим источникам: hide D.R., Editor in chief, Handbook of Chemistry and Physics, 74th
ed. CRC Press, Boca Raton, FL; Gordon A.J., Ford R.A., The Chemist's Companion: A
Handbook of Practical Data, Techniques and References, Wiley, New York (1972) (есть
перевод: Гордон А., Форд Р. Спутник химика. - М.: Мир, 1976). Трехмерные
рисунки структур взяты из базы данных Кембриджского кристаллографического
общества; графические работы выполнены в Техническом университете Цюриха.
Номенклатура соответствует рекомендациям ИЮПАК, но многие тривиальные
названия сохранены, так как они традиционно используются в лабораториях; в
приложении 10 даны названия наиболее широко используемых реактивов,
которые обычно известны по их традиционным, а не систематическим названиям.
Мы хотели бы поблагодарить Малкольма Герлоха, Мартина Мейса, Джона
Майера, Даяну Смит и рецензентов, предложенных издательской группой «Эд-
дисон Уэсли Лонгман», комментарии и предложения которых мы не могли не
оценить; Зою Левин за консультации по вопросам номенклатуры; Шерил Кук и
Монику Буркхард за помощь в получении некоторых опубликованных данных.
Конечно, мы искренне признательны своим коллегам из «Эддисон Уэсли
Лонгман» - Кате Хикк, Пауле Тернер, Паулине Гиллерт, Кевину Ансиенту, Мартину
Клоштоку, Нилу Пателю, Роз Эмери и, особенно, Крис Лидинг, Алексу Сибруку
и Джейн Гленденинг - за их поддержку в период написания книги, когда и
«перья» (или, точнее, принтеры) готовы были сломаться.
Процесс написания этой книги проходил в компании наших двух крошек
Филби и Изис, которые часами могли наблюдать за экраном компьютера,
засыпая, играя и прыгая на его клавиатуре и лишь иногда давая понять нам, что
соблюдение времени их кормления более важно, чем лишний час работы над
книгой. Их здравомыслие помогало и нам оставаться в здравом уме, когда процесс
написания книги становился невыносимым.
Нам доставило большую радость писать этот учебник. Мы надеемся, что вы
также получите удовольствие, читая его, и найдете рассмотренный подход
полезным. Просим студентов и преподавателей сообщить свое мнение о книге по
адресу: housecroft@ubaclu.unibas.ch или constable@ubaclu.unibas.ch.
Базель, 1996
Катрин Хаускрофт
Эдвин Констебл
СПИСОК ОБОЗНАЧЕНИЙ
(Обозначения лигандов: см. табл. 16.2)
А
\
Аг
ах
В
Ви
с
D
d
Е
е
е~
Е1
Е2
Еп
ЕЛ
eq
Et
F
G
H
Oi
h
/TV
/
/
IE
Tr
J
поглощение, оптическая плотность
относительная атомная масса
угловая волновая функция
арильная группа
аксиальный
напряженность магнитного поля
бутильная группа
коэффициент (в волновой функции)
теплоемкость (при постоянном
давлении)
теплоемкость (при постоянном
объеме)
энтальпия разрыва (диссоциации)
химической связи
средняя энтальпия разрыва
(диссоциации) химической связи
расстояние
энергия
заряд электрона
электрон
одномолекулярное отщепление
бимолекулярное отщепление
энергия активации
кинетическая энергия
потенциал электрохимической
ячейки
стандартный электродный
потенциал
сродство к электрону
экваториальный
этил ьная группа
число Фарадея
энергия Гиббса
энтальпия
гамильтониан (оператор
Гамильтона)
постоянная Планка
высоэнергетическое излучение (для
реакции фотолиза)
сила электрического тока
интенсивность прошедшего
излучения
спиновое квантовое число
интенсивность падающего
излучения
энергия (потенциал) ионизации
изопропильная группа
константа спин-спинового
взаимодействия (ССВ)
к
к
К
К*
*ь
*с
*р
**
*w
/
т
т,
ms
К
Me
N
"а
п
п
п
р
г*
Ph
Рг
Я
R
R
г
R(r)
г
ион
'ков
мет
rv
S
S
V
SN2
Т
t
lBu
и
V
константа скорости реакции
силовая постоянная химической
связи
константа равновесия
константа диссоциации кислоты
константа диссоциации основания
константа равновесия реакции в
растворе
константа равновесия газовой
реакции
произведение растворимости (ПР)
константа ионизации воды
орбитальное квантовое число
масса
магнитное квантовое число
магнитное спиновое квантовое число
относительная молекулярная масса
метильная группа
нормировочный множитель
число Авогадро
показатель степени в уравнении Борна
число молей
главное квантовое число
давление
парциальное давление
компонента X
фенильная группа
пропильная группа
заряд
алкильная группа
универсальная газовая постоянная
межъядерное или межатомное
расстояние
радиальная волновая функция
ионный радиус
ковалентный радиус
металлический радиус
радиус Ван-дер-Ваальса
энтропия
спиновое квантовое число
мономолекулярное нуклеофильное
замещение
бимолекулярное нуклеофильное
замещение
температура
время
mpem-бутлъная группа
внутренняя энергия
объем
10
Список обозначений
V
W
[X]
Z
^эфф
z_
а
р
8
6"
6+
Д
Дат"
\ор"
Добр"
Дгидр"
Дреш"
ДраствЯ
Асолы>Я
Дисп"
ДобрС
АР^
е
М
^эфф
Р
разность потенциалов
совершенная работа
концентрация компонента X
число молей электронов,
перемещенных в
электрохимической ячейке
атомный номер химического
элемента
эффективный заряд ядра
модуль отрицательного заряда
модуль положительного заряда
угол
константа устойчивости (комплекса)
химический сдвиг (в ЯМР-спектре)
частичный отрицательный заряд
частичный положительный заряд
нагревание (на стрелке в уравнениях
реакций)
энергия расщепления в
октаэдрическом поле
энтальпия атомизации
энтальпия горения
энтальпия образования
энтальпия плавления
энтальпия гидратации
энтальпия образования ионной
кристаллической решетки
энтальпия реакции
энтальпия растворения
энтальпиия сольватации
энтальпия испарения
энергия Гиббса образования
энергия Гиббса реакции
молярный коэффициент
экстинкции
молярный коэффициент
экстинкции, соответствующий
максимуму полосы поглощения
(в электронных спектрах)
диэлектрическая проницаемость в
вакууме
угол
длина волны
длина волны, соответствующая
максимуму полосы поглощения
(в электронных спектрах)
электрический дипольный момент
эффективный дипольный момент
мостиковый лиганд
плотность
vOP
2с-2е
Зс-2е
частота
волновое число
магнитная восприимчивость
электроотрицательность по шкале
Оллреда — Рохова
электроотрицательность по шкале
Малликена
электроотрицательность по шкале
Полинга
волновая функция
двухцентровая двухэлектронная
связь
трехцентровая двухэлектронная
связь
(символ в верхнем индексе)
активированный комплекс;
переходное состояние
а.е.м.
АО
ВЗ
ВЗМО
ВС
вид./УФ
ДМФ
ик
кч
Л К АО
ЛС
МО
немо
ПМР
ПР
атомная единица массы
атомная орбиталь
валентная зона
(англ. HOMO) высшая
заполненная молекулярная орбиталь
валентная связь
1 видимая — ультрафиолетовая
область спектра
диметилформамид
инфракрасная область спектра
координационное число
линейная комбинация атомных
орбиталей
лимитирующая стадия реакции
молекулярная орбиталь
(англ. LUMO) низшая свободная
молекулярная орбиталь
протонный магнитный резонанс
произведение растворимости
(англ. KSD)
{ПС}*
ПФ
РЧ
сси
ТГФ
тмс
УФ
ФЭС
ЭПР
ЯМР
ЯМР'Н
переходное состояние
преобразование Фурье
радиочастота
свободный спад индукции
тетрагидрофуран
тетраметилсилан
ультрафиолетовая область спектра
фотоэлектронная спектроскопия
электронный парамагнитный
резонанс
ядерный магнитный резонанс
протонный магнитный резонанс
ОСНОВНЫЕ ПОНЯТИЯ
ЧТО ТАКОЕ ХИМИЯ И ПОЧЕМУ ОНА ВАЖНА
Любая форма материи в мире животных, растений или минералов состоит из
элементов или их соединений. Известно более сотни химических элементов;
далеко не все из них широко распространены. Подавляющее большинство
элементов встречается в природе, но некоторые, такие, как технеций и
кюрий, были получены искусственно.
Задача химии — изучить свойства элементов, т. е. узнать, как они
взаимодействуют друг с другом и какие химические превращения претерпевают их
соединения.
Живые (биологические) системы состоят в основном из углерода,
водорода, азота и кислорода. Правильное понимание биологии, и в частности
молекулярной биологии, должно быть основано на детальном знании структур,
свойств и реакционной способности биомолекул. Эти фундаментальные
знания приобретаются при изучении химии.
Грань, разделяющая химические и физические науки, часто оказывается
трудно определяемой, условной, как, например, между физикой и быстро
^ развивающейся химией сверхпроводников — соединений, обладающих ни-
j; чтожно малым электрическим сопротивлением. Сверхпрово-
Сверхпроводники: см. -
димость проявляется обычно при очень низких температурах,
дополнение 7.4 ^
поэтому для широкого использования сверхпроводимости
необходимо синтезировать новые сверхпроводники, которые
работали бы при обычных температурах. Таким образом, хотя определение
сверхпроводимости выполняет физик, основной прогресс в этой области
достигается при помощи химического синтеза и изучения химического состава
этих материалов.
Химия играет ключевую роль среди естественных наук. Она дает
фундаментальные знания, необходимые для прикладных наук, таких, как
астрономия, материаловедение, химическая технология, медицина и фармакология.
Вот почему надо изучать химию на первом курсе высших учебных заведений.
В любой области науки необходимо понимание основных химических
концепций.
Традиционно химия делится на неорганическую, органическую и
физическую (теоретическая химия может рассматриваться как раздел физической
► химии). Однако в действительности эти области очень сильно перекрывают-
Окисление: ся. Рассмотрим один пример — вакер-процесс, в ходе которого
см. разд. 1.16 этилен окисляется до ацетальдегида (уравнение 1.1).
1. Основные понятия
С2Н4 + [PdCl4]2- + Н20 -> СН3СНО + 2НС1 + 2С1~ + Pd (1.1)
Этилен Этаналь
(ацетальдегид, уксусный альдегид)
До недавнего времени на этой реакции был основан важный метод
промышленного получения уксусного альдегида (ацетальдегида). Реакция протекает
в водном растворе в присутствии комплексного аниона тетрахлоропаллада-
та(Н) [PdCl4]2~ (1.1). Палладий(О), получающийся по реакции 1.1, снова
переходит в ион [PdCl4]2_ при реакции с медью(Н) и хлорид-ионами
(уравнение 1.2); образующаяся при этом медь(1), которая присутствует в растворе в
виде иона [СиС12]~ , окисляется кислородом по реакции 1.3. В этом
10.7 случае [PdCl4]2~ и Си2+ играют роль катализатора, так как они в
целом не подвергаются химическому превращению при
протекании реакций описанного цикла.
2Cu2+ + Pd + 8Cl- -> [PdCl4]2" + 2[СиС12Г (1.2)
4Cu+ + 4H+ + 02 -> 4Cu2+ + 2H20 (1.3)
Основная реакция (превращение этилена в ацетальдегид) относится к
органической химии, превращение катализатора - к неорганической, а для
интерпретации кинетики и понимания механизма реакции, а также для того,
чтобы определить оптимальные условия промышленного проведения
процесса, нужна физическая химия. Таким образом, все три области химии
используются для понимания вакер-процесса. Это один из многочисленных
примеров, когда рассмотрение химического процесса или концепции
требует одновременного применения двух или нескольких разделов химии.
В книге «Современный курс общей химии» темы расположены таким
образом, чтобы обеспечить наиболее естественное усвоение студентами
первого курса необходимого химического материала. В книге нет никаких
искусственных барьеров между главами, которые мы намеренно не стали разделять
на неорганическую, органическую или физическую химию. Мы надеемся,
что таким способом вы научитесь применять пройденный материал в
нестандартных условиях.
Цель остальной части данной главы — напомнить основные понятия и
определения.
ЧТО ТАКОЕ ИЮПАК?
Поскольку химия как наука постоянно развивается и число известных
соединений продолжает расти с фантастической быстротой, совершенно
необходимо принять набор правил, в соответствии с которым можно было бы назы-
1.3. Единицы СИ
13
вать эти вещества, т. е. договориться о номенклатуре, и, что более важно, о
том, чтобы эти правила использовались всеми химиками.
ИЮПАК (от англ. The /nternational i/hion of Aire and Applied Chemistry-
Международный союз теоретической и прикладной химии ) кроме решения
разнообразных задач занимается разработкой рекомендаций по
номенклатуре органических и неорганических соединений, а также по нумерации и
названиям групп в периодической системе элементов.
В этой книге мы будем по возможности придерживаться номенклатуры,
рекомендованной ИЮПАК, но следует учитывать, что в словаре химика су-
^ шествует большое число тривиальных названий, применение которых оказы-
Тривиальные названия: вается часто более рациональным, чем использование гро-
см. приложение 10 моздкой систематической номенклатуры. Некоторые основ-
в т 2 ные правила и общая система номенклатуры органических и
неорганических соединений приведены в разд. 1.24. Мы
детально разбираем системные методы названий новых классов соединений по
мере их обсуждения в тексте.
ЕДИНИЦЫ СИ
Система стандартизованных и общепризнанных единиц измерения не менее
важна, чем система названий химических соединений. Международная
система единиц (СИ - русская транскипция SI, System /nternational) дает нам
общепринятую систему мер.
Основные величины СИ
Существует семь основных величин СИ (табл. 1.1) и две дополнительные
единицы измерения: радиан и стерадиан. Радиан (рад) - единица измерения
плоских углов; стерадиан (ср) — единица телесного угла (в частности, в
стереометрии). Важной характеристикой единиц СИ является их
самодостаточность. Из семи основных единиц мы можем получить все остальные
единицы измерения (см. примеры 1.1, 1.2 и 1.5). Некоторые из наиболее
употребительных производных единиц имеют свои собственные названия. Величины
такого типа, применяемые в химии, перечислены в табл. 1.2.
Таблица 1.1. Основные единицы СИ
Физическая величина
Масса
Длина
Время
Электрический ток
Температура
(термодинамическая)
Количество вещества
Сила света
Обозначение
т
1
t
I
Т
п
к
Единица
наименование
килограмм
метр
секунда
ампер
кельвин а
моль
кандела
измерения
русские обозначения
кг
м
с
А
К
моль
кд
а Градус Цельсия °С — более старая, чем кельвин, единица измерения температуры. (Обратите
внимание на обозначение — правильно писать °С и К, а не °К.)
14
1. Основные понятия
Таблица 1.2. Некоторые производные единиц СИ
Физическая
величина
Давление
Сила
Частота
Электрическая емкость
Электрический заряд
Электрическое
сопротивление
Электродвижущая сила
Энергия
Единица
измерения
паскаль
ньютон
герц
фарада
кулон
ом
вольт
джоуль
Русское
обозначение
Па
Н
Гц
Ф
Кл
Ом
В
Дж
Связь с основными
единицами СИ
кг • м-1 • с-2
кг•м • с-2
С"1
А2 • с4 • кг-1 • м-2
А-с
кг • м2 • с-3 • А-2
кг • м2 • с-3 • А-1
кг • м2 • с-2
Производные единиц СИ
Если для расчета физической величины используется ее
физико-математическое выражение, то ее единица измерения может быть определена
непосредственно из этого выражения. Рассмотрим объем параллелепипеда со
сторонами а, Ь, с (уравнение 1.4). Если единицей длины в СИ является метр
(табл. 1.1), то единица измерения объема в СИ может быть определена по
уравнению 1.5. Это очень простой пример, но он показывает, что исходя из
основных величин СИ можно легко определить единицу измерения любой
необходимой физической величины.
Объем параллелепипеда = Длина Ширина Высота
= а • b • с = abc (1.4)
СИ-единица объема = СИ-единица длины • СИ-единица ширины
• СИ-единица высоты
= м • м • м = м3 (1.5)
Некоторые из старых единиц измерения до сих пор находят свое
применение, например, атмосфера (атм) используется вместо паскаля, а калория
(кал) вместо джоуля, однако единицы СИ являются общепринятыми и
используются во всем мире.
Пример 1.1 Единицы измерения
Какая единица СИ используется для измерения плотности?
Решение
Плотность равна массе, деленной на объем.
Отсюда
СИ-единица массы
СИ-единица плотности
СИ-единица объема
= кг/м3
1.3. Единицы СИ
15
Большие и малые числа
Очень часто числа, которыми оперирует та или иная наука, либо очень малы,
либо чрезвычайно велики. Например, длина типичной связи С—С,
выраженная в стандартных единицах длины (м) равна 0,000 000 000 154 м. Писать
такой ряд нулей очень неудобно и, кроме того, это увеличивает вероятность
ошибок. Полезно записывать эту величину как 1,54 Ю-10 м. Это гораздо
более удобный и наглядный способ записи информации о длине связи. Было
бы еще более удобно вовсе не писать множитель Ю-10. Так как обычно
длины связей одинаковы по порядку величины, было бы полезно для длины
связи С—С писать просто 1,54. Это приводит к возникновению единицы
измерения 10~10 м, которая получила название «ангстрем» (А). Длина С—С связи
равна, таким образом, 1,54 А. К сожалению, ангстрем не принадлежит к
числу единиц СИ, хотя широко используется. Система СИ требует применения
для кратных величин одной из перечисленных в табл. 1.3 приставок.
Расстояние 1,54 • 10~10 м равно 154 • 10~12 м или 0,154 • 10~9 м. Из табл. 1.3 видно,
что эти величины равны соответственно 154 пикометрам (пм) или 0,154
нанометрам (нм). Оба варианта могут использоваться в системе СИ. (В этой
книге в качестве единицы длины химической связи используются пикомет-
ры и нанометры.)
Совместимость единиц измерения
Небольшое предупреждение относительно применения приставок,
перечисленных в табл. 1.3: при вычислениях необходимо согласовать размерности
единиц. Например, если плотность выражена в г/см3, а объем — в м3, то
нельзя использовать эти размерности в одной формуле; скажем, при вычислении
массы (уравнение 1.6):
Масса = Объем • Плотность (1.6)
Прежде всего, входящие в формулу величины необходимо перевести в
одинаковый масштаб размерностей, как это показано в примере 1.2.
Наиболее надежный способ избежать ошибок при расчетах - перевести в шкалу,
соответствующую основным единицам СИ (табл. 1.1), все величины до
подстановки их в формулу. В книге довольно много подобных примеров.
Таблица 1.3. Масштабные приставки, применяемые в СИ для обозначения кратных
единиц
Масштабный Название Символ Масштабный Название Символ
коэффициент коэффициент
1012 тера Т Ю-2 санти с
109 гига Г 10~3 милли м
106 мега М 10~6 микро мк
103 кило к Ю-9 нано н
102 гекто г Ю-12 пико п
10 дека да Ю-15 фемто ф
Ю-1 деци д 10~18 атто а
16
1. Основные понятия
Пример 1.2. Размерности физических величин при расчетах
Какой объем занимаеют 10 г ртути, если плотность ртути при 298 К
равна 1,36 104 кг/м3?
Решение
Уравнение, которое связывает массу т, объем Vи плотность р, может быть
записано как
m m
о- — или V - —
V р
Перед подстановкой необходимо привести размерности данных величин к
единому масштабу. Плотность дана в кг/м3, а масса - в граммах, поэтому ее
следует выразить в кг:
10 г = 10- 10"3кг= 1 • Ю-2
кг
Теперь можно рассчитать объем ртути:
K=^=i-^1- =7,35- НЯмЗ
р 1,36 104
Проверьте соответствие единиц:
к=^-=^!1
КГ • М~
ПРОТОНЫ, ЭЛЕКТРОНЫ И НЕЙТРОНЫ
Атом состоит в основном из частиц трех типов: протонов, электронов и
нейтронов*. Некоторые важные свойства этих частиц приведены в табл. 1.4.
Нейтрон и протон имеют примерно одинаковую массу покоя, по сравнению с
которой масса электрона пренебрежимо мала. Заряд протона равен по
абсолютной величине, но противоположен по знаку заряду электрона. Таким
образом, комбинация равного числа протонов и электронов обеспечивает атому
электронейтральность. Нейтрон, как следует из его названия, нейтрален, т. е.
не имеет заряда. Расположение и энергия электронов в атомах и ионах
обсуждаются в гл. 2.
Таблица 1.4. Свойства протона, электрона и нейтрона
Протон Электрон Нейтрон
Заряд, Кл
Относительный заряд
Масса покоя, кг 1,673 • Ю"27 9,109 • 10"31 1,675 • 10"
Относительная масса
+ 1,602- 10"19
1
1,673- Ю-27
1837
-1,602- 10"19
1
9,109- 10"31
1
0
0
1,675
1839
* Химики считают их фундаментальными, однако физики доказали, что эти частицы состоят из еще
более мелких.
1.5. Химические элементы 17
Н9 ХИМИЧЕСКИЕ ЭЛЕМЕНТЫ
ИЮПАК рекомендует следующее определение химического элемента:
Химический элемент - это совокупность атомов с одинаковым
положительным зарядом ядра.
Химическим элементам соответствуют общепринятые символы, однако
названия некоторых элементов являются предметом лингвистических
дискуссий.
Металлы, неметаллы и полуметаллы
Элементы могут быть разделены на металлы, неметаллы и полуметаллы*.
Названия большинства металлов заканчивается на -ww, например, литий,
натрий, магний, кальций, алюминий, скандий, ванадий, гафний, рутений,
родий, иридий, осмий, палладий. Исключения составляют хром, титан, олово,
свинец, железо, кобальт, медь, цинк, вольфрам, платина, серебро и золото.
По рекомендации ИЮПАК, термин «полуметаллы» более предпочтителен,
чем «металлоиды» (см. также гл.7).
Аллотропные модификации
Некоторые элементы существуют в нескольких структурных формах, и это их
свойство называется аллотропией. Рассмотрим углерод - его широко
распространенными аллотропными модификациями являются графит и алмаз
(рис 1.1), оба имеющие бесконечно протяженную кристаллическую решетку.
Обе аллотропные модификации состоят только из атомов углерода, сгорают
в избытке кислорода с образованием диоксида углерода С02. Однако
внешний вид этих двух аллотропных модификаций резко различается. Алмаз
термодинамически неустойчив при комнатных температуре и давлении, однако,
к счастью, его переход в графит - процесс, чрезвычайно медленный, и алмаз
является метастабильным. В 1980-х годах была открыта еще одна
аллотропная модификация углерода - фуллерены (например, С60; см. рис. 1.1),
которые присутствуют в саже в количестве нескольких процентов
разд. 7.9 по Macce> причем наиболее распространены - отдельные
молекулы С60. Олово, фосфор, мышьяк, кислород, сера и селен
также имеют аллоторопные модификации.
Аллоторопными модификациями называют различные структурные
формы одного элемента.
Понятию «полуметаллы» в русской научной литературе ранее соответствовал термин «металлоиды».
В настоящее время этот термин считается устаревшим, и все элементы подразделяют на металлы и
неметаллы. К неметаллам относят 22 элемента: Н, В, С, Si, N, P, As, О, S, Se, Те, галогены и
инертные газы, к металлам — все остальные элементы. — Прим. ред.
С60: см.
18
••-8П»
Графит Алмаз Фуллерен С^
Рис. 1.1. Структуры трех аллотропных модификаций углерода; фуллерены представлены
формой С60.
Дополнение 1.1. Названия элементов
Происхождение названий и символов химических элементов часто бывает связано с
историей их открытия или использования и отражает различия между языками. Три
примера это наглядно иллюстрируют.
Ртуть (Ид)
Серебристо-белый, жидкий металл ртуть был известен еще древним грекам, которые
называли его «жидким серебром». В латыни это название превратилось в hydrargyrum,
от которого произошел современный символ Hg. Англичане называли ртуть quicksilver
(живое серебро), в Германии элемент был известен как Quecksilber. Слово mercury
(ртуть) произошло от имени посланника римских богов Меркурия.
Кобальт (Со)
Этот ^-элемент в Германии получил название Kobalt. Никель и кобальт в различных
количествах присутствуют в железных рудах. Когда в ранние времена их перерабатывали,
то из руды, содержащей большое количество кобальта, получали железо низкого
качества. Эти руды были известны как ложные руды и их называли Kobold (по имени
злобных существ, якобы живших в рудниках).
Вольфрам (W)
Символ вольфрама W происходит от названия металла Wolfram, которое он получил в
Германии по названию его руды вольфрамита. Другая руда вольфрама называлась
Scheelite, по имении шведского химика Шееле первооткрывателя вольфрама, но
прежде чем руду назвали шеелитом, она была известна как «тангстен», что означает
«тяжелый камень», отсюда — английское название вольфрама tungsten.
1. Основные понятия
1.6. Состояние вещества
19
СОСТОЯНИЕ ВЕЩЕСТВА
Твердые вещества, жидкости и газы
При определенной температуре вещество находится в одном из трех
физических состояний — твердом, жидком или газообразном (рис 1.2). Газом
называют пар при температуре выше критической.
В твердом состоянии атомы всегда расположены регулярно; твердое
вещество имеет фиксированный объем (при данных давлении и температуре) и
форму. Жидкость также при данных условиях имеет определенный объем, но
не имеет формы. Она принимает форму сосуда, в котором находится.
Частицы (атомы или молекулы) газа хаотично движутся и занимают большой
объем. Газу не присуща форма.
На рис. 1.2 показано, что поверхность твердого тела не зависит от формы
сосуда. Газ, если он не ограничен контейнером, не имеет четкой
фиксированной поверхности, что позволяет двум (или нескольким) газам смешиваться, и
этот процесс называется диффузией (рис. 1.3). Жидкость повторяет форму
сосуда, но на ее границе действует поверхностное натяжение, которое и
определяет ее поверхность. Если жидкости способны смешиваться, они
называются смешивающимися (например, гексан и октан или этанол и вода). Если
жидкости не смешиваются (например, вода и масло или вода и гексан), то
они называются несмешивающимися.
Если жидкости способны смешиваться, они называются
смешивающимися. Несмешивающиеся жидкости расслаиваются.
В химических уравнениях всегда указывается состояние вещества, для чего
используются следующие стандартные обозначения:
Твердое вещество (тв.)
Жидкость (ж.)
Газ (газ)
Водный раствор (водн.)
Го
о
-\
о
Твердое вещество Жидкость Газ
Рис. 1.2. Организация частиц (атомов или молекул) в твердом теле, жидкости и газе.
Твердое тело имеет вполне определенную форму. Жидкость повторяет форму сосуда.
Атомы или молекулы газа свободно перемещаются в сосуде.
Ч*5
О ^О
20
1. Основные понятия
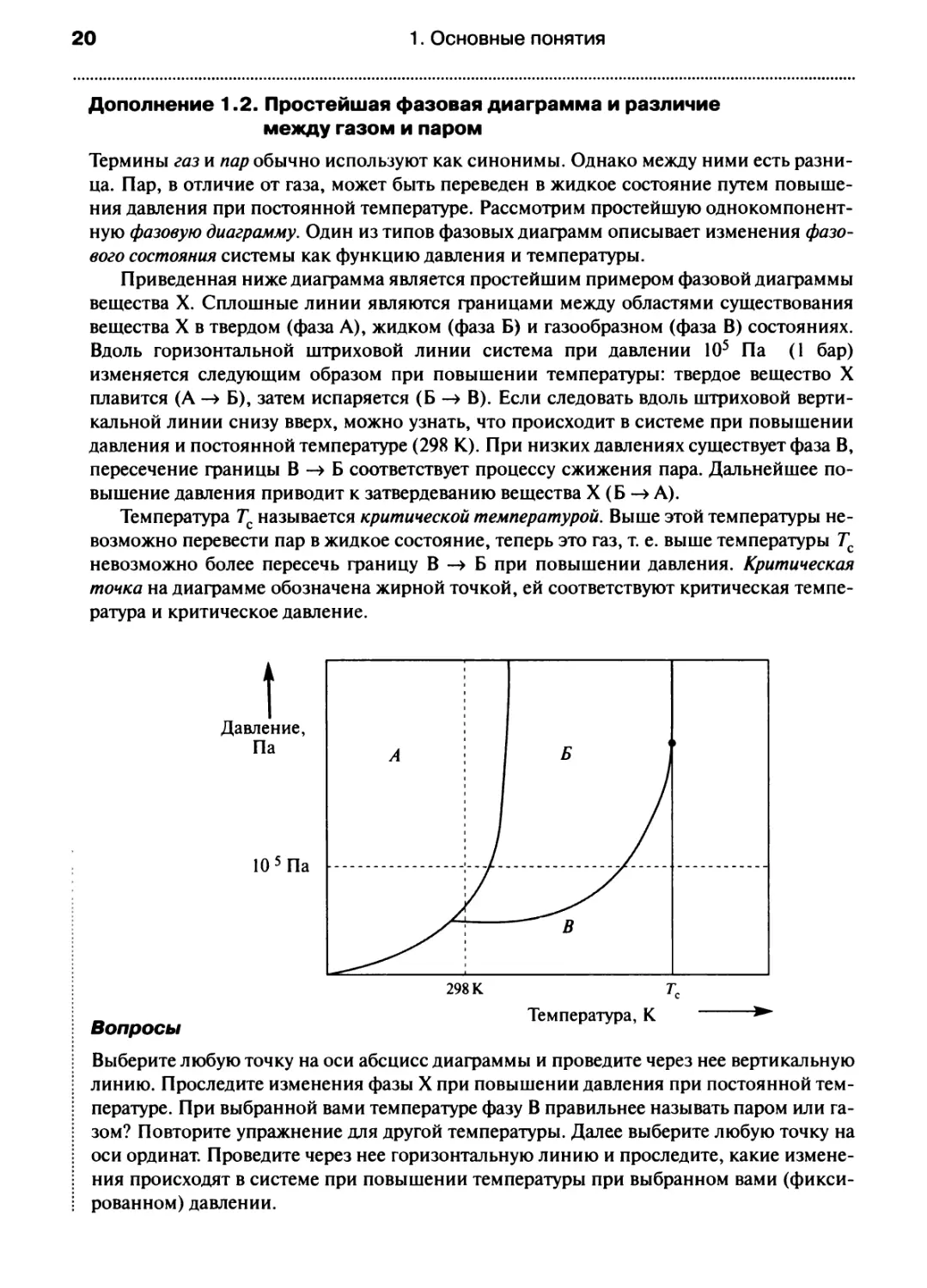
Дополнение 1.2. Простейшая фазовая диаграмма и различие
между газом и паром
Термины газ и пар обычно используют как синонимы. Однако между ними есть
разница. Пар, в отличие от газа, может быть переведен в жидкое состояние путем
повышения давления при постоянной температуре. Рассмотрим простейшую однокомпонент-
ную фазовую диаграмму. Один из типов фазовых диаграмм описывает изменения
фазового состояния системы как функцию давления и температуры.
Приведенная ниже диаграмма является простейшим примером фазовой диаграммы
вещества X. Сплошные линии являются границами между областями существования
вещества X в твердом (фаза А), жидком (фаза Б) и газообразном (фаза В) состояниях.
Вдоль горизонтальной штриховой линии система при давлении 105 Па (I бар)
изменяется следующим образом при повышении температуры: твердое вещество X
плавится (А -> Б), затем испаряется (Б -> В). Если следовать вдоль штриховой
вертикальной линии снизу вверх, можно узнать, что происходит в системе при повышении
давления и постоянной температуре (298 К). При низких давлениях существует фаза В,
пересечение границы В -> Б соответствует процессу сжижения пара. Дальнейшее
повышение давления приводит к затвердеванию вещества X (Б -> А).
Температура Тс называется критической температурой. Выше этой температуры
невозможно перевести пар в жидкое состояние, теперь это газ, т. е. выше температуры Тс
невозможно более пересечь фаницу В -» Б при повышении давления. Критическая
точка на диаграмме обозначена жирной точкой, ей соответствуют критическая
температура и критическое давление.
Давление,
Па
105Па
А
z
\^^s
)
Б
L^y
в
►
298 К
Вопросы
Температура, К
Выберите любую точку на оси абсцисс диаграммы и проведите через нее вертикальную
линию. Проследите изменения фазы X при повышении давления при постоянной
температуре. При выбранной вами температуре фазу В правильнее называть паром или
газом? Повторите упражнение для другой температуры. Далее выберите любую точку на
оси ординат. Проведите через нее горизонтальную линию и проследите, какие
изменения происходят в системе при повышении температуры при выбранном вами
(фиксированном) давлении.
1.7. Атомы и изотопы
21
Го
о
о
о I
о
о о|
о
о
о
о
Удаление
перегородки
Рис. 1.3. Объем газа ограничен сосудом, в котором он находится. Два газа могут
смешиваться (диффундировать), если убрать перегородку между ними.
Обозначение «водн.» относится к водным растворам, это не состояние
вещества.
Фазы
Три состояния вещества являются фазами, но и одно состояние может
включать несколько фаз. Например, каждая аллотропная модификация углерода
является отдельной фазой, но все они находятся в твердом состоянии.
Каждая фаза существует при определенных условиях (давлении и температуре),
эта информация представляется в форме фазовых диаграмм. Использование
простейшей фазовой диаграммы рассмотрено в дополнении 1.2.
Фазовые переходы
При данном давлении (или данной температуре) переход одной фазы в
другую происходит при одной температуре (или при одном давлении). Обычно
имеют дело с переходами при атмосферном давлении. Химический элемент
(или конкретная аллотропная модификация, если элемент обладает
аллотропией) переходит из твердого состояния в жидкое при температуре плавления
(Гпл), а из жидкости в пар - при температуре кипения (Ткип).
Третье состояние следует называть паром, а не газом до тех пор,
пока не будет достигнута критическая температура.
Температуры плавления и кипения некоторых элементов приведены в
табл. 1.5. Углерод и сера не включены в таблицу, так как их
фазовые переходы более сложны. При нагревании при
определенном давлении некоторые элементы (например, иод) сразу переходят
из твердого состояния в пар. Этот процесс называется сублимацией или
возгонкой.
Изменения
энтальпии при
плавлении и
испарении: см. разд. 1.20
АТОМЫ И ИЗОТОПЫ
Атомы и атомный номер
Атом состоит из положительно заряженного ядра и отрицательно
заряженных электронов. Простейшим атомом является водород, состоящий из
протона и электрона. Протон в атоме водорода является ядром, но ядро любого
другого атома состоит из протонов и нейтронов.
22
1. Основные понятия
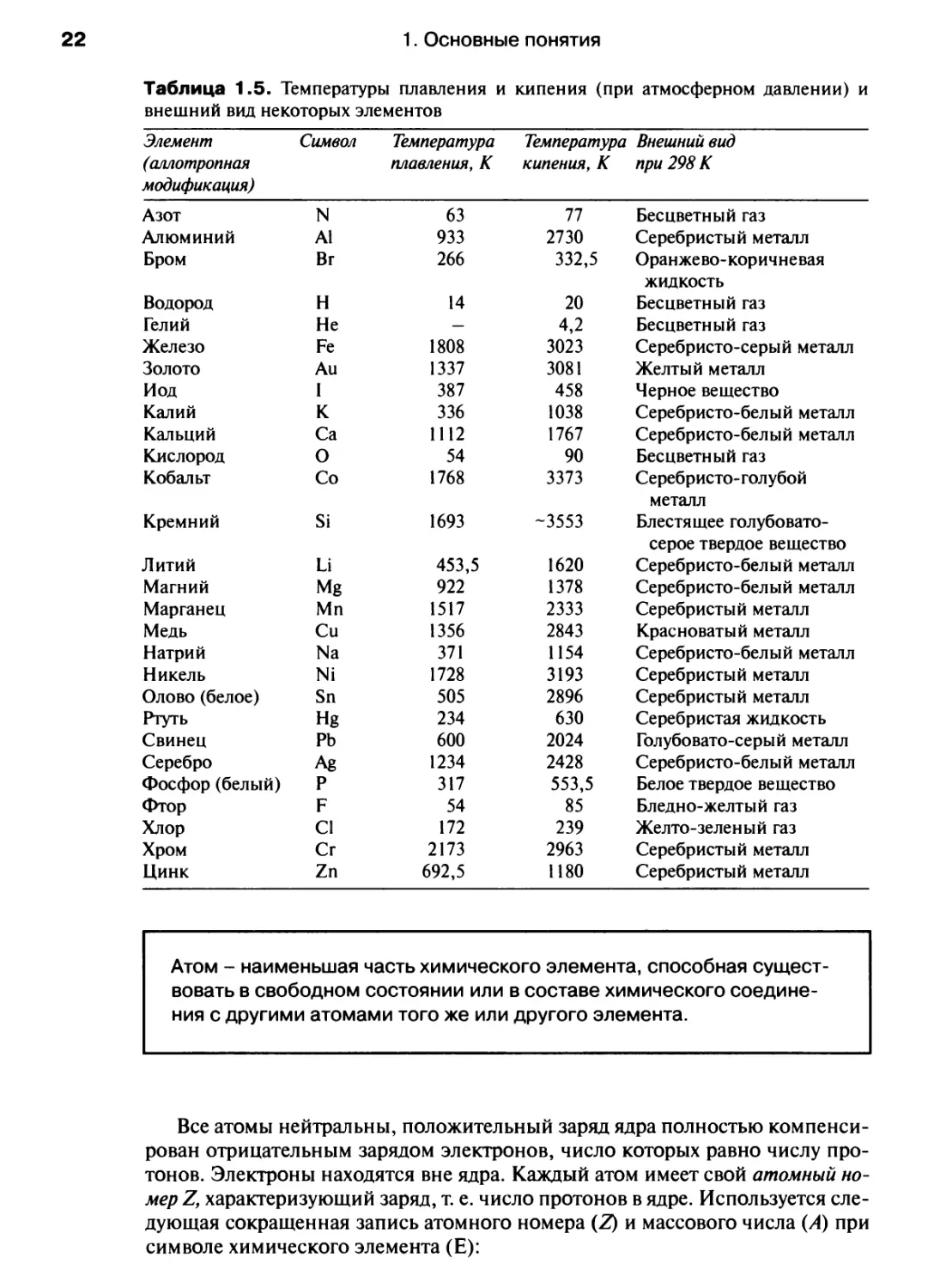
Таблица 1.5. Температуры плавления и кипения (при атмосферном давлении) и
внешний вид некоторых элементов
Элемент Символ Температура Температура Внешний вид
(аллотропная плавления, К кипения, К при 298 К
модификация)
Азот
Алюминий
Бром
Водород
Гелий
Железо
Золото
Иод
Калий
Кальций
Кислород
Кобальт
Кремний
Литий
Магний
Марганец
Медь
Натрий
Никель
Олово (белое)
Ртуть
Свинец
Серебро
Фосфор (белый)
Фтор
Хлор
Хром
Цинк
N
Al
Вг
Н
Не
Fe
Au
I
К
Са
О
Со
Si
Li
Mg
Mn
Си
Na
Ni
Sn
Hg
Pb
Ag
P
F
CI
Cr
Zn
63
933
266
14
-
1808
1337
387
336
1112
54
1768
1693
453,5
922
1517
1356
371
1728
505
234
600
1234
317
54
172
2173
692,5
77
2730
332,5
20
4,2
3023
3081
458
1038
1767
90
3373
-3553
1620
1378
2333
2843
1154
3193
2896
630
2024
2428
553,5
85
239
2963
1180
Бесцветный газ
Серебристый металл
Оранжево-коричневая
жидкость
Бесцветный газ
Бесцветный газ
Серебристо-серый металл
Желтый металл
Черное вещество
Серебристо-белый металл
Серебристо-белый металл
Бесцветный газ
Серебристо-голубой
металл
Блестящее голубовато-
серое твердое вещество
Серебристо-белый металл
Серебристо-белый металл
Серебристый металл
Красноватый металл
Серебристо-белый металл
Серебристый металл
Серебристый металл
Серебристая жидкость
Голубовато-серый металл
Серебристо-белый металл
Белое твердое вещество
Бледно-желтый газ
Желто-зеленый газ
Серебристый металл
Серебристый металл
Атом - наименьшая часть химического элемента, способная
существовать в свободном состоянии или в составе химического
соединения с другими атомами того же или другого элемента.
Все атомы нейтральны, положительный заряд ядра полностью
компенсирован отрицательным зарядом электронов, число которых равно числу
протонов. Электроны находятся вне ядра. Каждый атом имеет свой атомный
номер Z, характеризующий заряд, т. е. число протонов в ядре. Используется
следующая сокращенная запись атомного номера (2) и массового числа (А) при
символе химического элемента (Е):
1.7. Атомы и изотопы
23
Массовое число —> л
Символ элемента —> |н ii^°
Атомный номер —> Z
Атомный номер Z = Число протонов в ядре = Число электронов.
Массовое число Л = Число протонов + Число нейтронов.
Относительная атомная масса
Масса атома сконцентрирована в ядре, где находятся протоны и нейтроны.
Если сложить реальные массы протонов и нейтронов, получатся очень
малые, нецелочисленные значения, и для удобства используют относительные
атомные массы, выраженные в атомных единицах массы. Атомная единица
массы (а.е.м.) приблизительно равна 1,660 • 10~27 кг, и это примерно
соответствует массе протона или нейтрона (см. табл. 1.4). В расчетах массу протона
или нейтрона принимают равной 1 а.е.м. Шкала относительных атомных
масс (АТ) строится относительно массы атома углерода 12С, принятой равной
12,0000.
Изотопы
Для данного химического элемента может существовать более одного типа
атомов. Они называются изотопами. Не следует путать изотопы и
аллотропные модификации. Аллотропы - это разные структурные формы элемента,
возникающие при различном пространственном расположении атомов (см.
рис. 1.1). Изотопы - это атомы одного и того же элемента, но с различным
числом нейтронов в ядре. Некоторые изотопы, не существующие в природе,
могут быть получены искусственно.
Элемент характеризуется числом протонов, которое при сохранении
электронейтральности должно быть равно числу электронов. Однако число
нейтронов в элементе может различаться. Например, водород имеет три
изотопа. Наиболее распространенным является протий ]Н (99,984%), у которого
есть протон и электрон, но нет нейтронов. Второй изотоп - дейтерий или
«тяжелый водород» (0,0156%), обозначаемый символами 2Н или D, — имеет
протон, нейтрон и электрон. Один из 1017 атомов в образце природного
водорода - тритий (3Н или Т); он радиоактивен. Атомная масса природного
водорода отражает наличие всех трех изотопов и равна средневзвешенному
значению массы присутствующих изотопов. Относительная атомная масса
водорода равна 1,0080; это количество близко к единице, так как изотоп !Н с
атомным номером 1 составляет 99,984% природной смеси изотопов.
Другими примерами элементов, существующих в природе в виде смеси
изотопов, являются литий (^Li и ^Li), хлор (\jC\ и ^С1) и медь (^Си и ^Си).
Элементы, существующие в виде единственного природного изотопа,
называют монотопами, например фосфор (]\Р) и кобальт (27С0). Изотопы могут
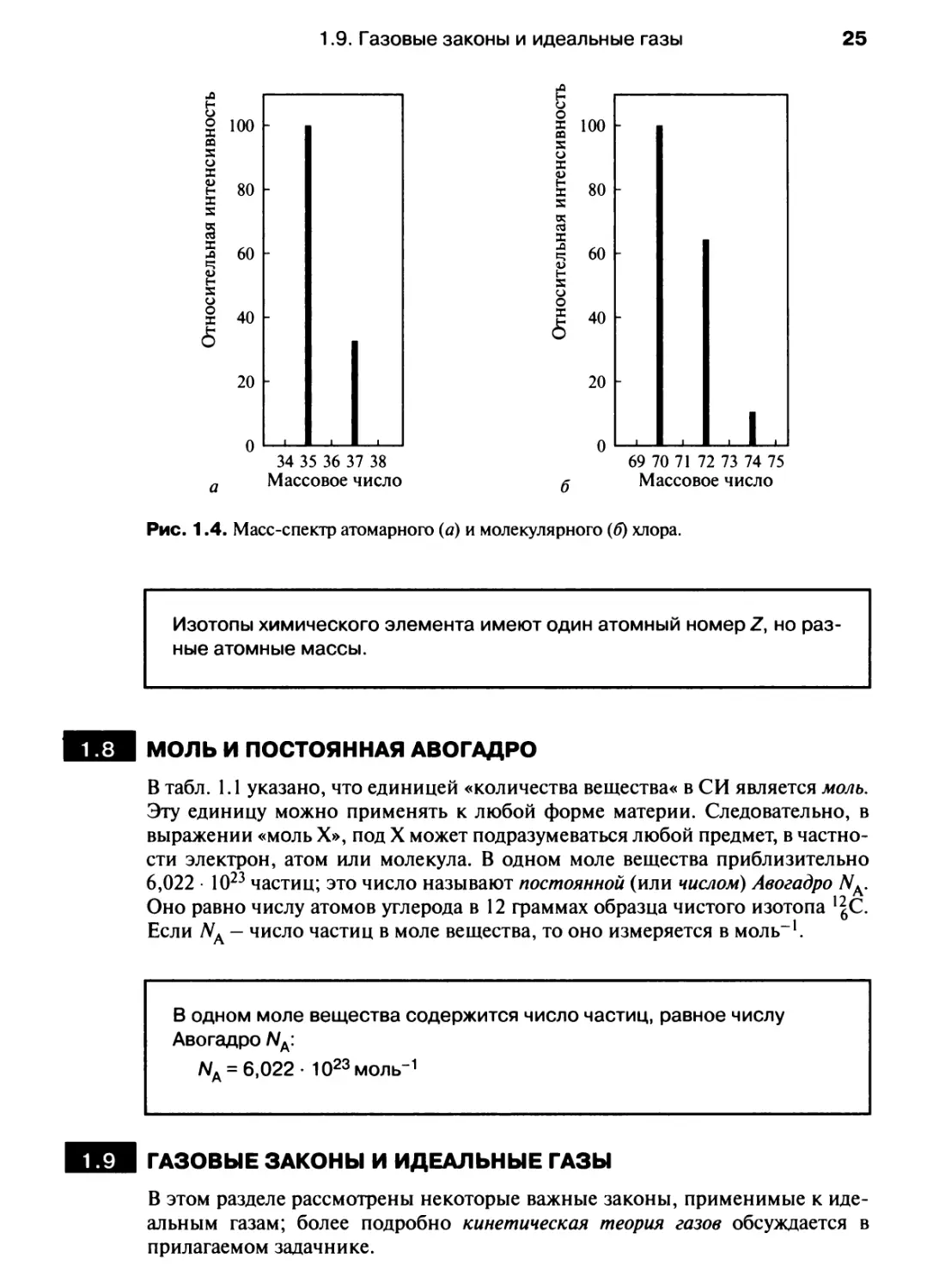
быть разделены при масс-спектрометрии. На рис 1.4,д представлено
распределение изотопов в атомарном хлоре. В масс-спектре С12 (рис 1.4,6)
присутствуют три пика, соответствующие различным комбинациям двух изотопов
24
1. Основные понятия
Дополнение 1.3. Искусственные изотопы и р-распад
Некоторые изотопы, в особенности наиболее тяжелых элементов, получают,
бомбардируя ядра элементов другими частицами, обычно нейтронами.
Примером искусственного изотопа может служить изотоп плутония 2^Ри.
Его получают в результате ряда ядерных реакций из изотопа урана ^U. При
бомбардировке 2||U нейтронами относительная атомная масса продукта
реакции на одну единицу больше, чем исходного изотопа, так как
присоединился нейтрон:
««U + Jn
•^U
Изотоп 2Ц\] спонтанно теряет Р-частицу (т.е. электрон) и образует изотоп
нептуния 29з^р, который также претерпевает Р-распад и превращается в
2$Ри:
^U-^39Np + p-
2$Np -> 2$Pu + р-
Р-Распад
Эмиссия Р-частиц (Р-распад) имеет место, когда ядро сложным путем теряет
электрон, что приводит к «превращению нейтрона в протон»*. Масса ядра
при Р-распаде не меняется, так как Р-частица (электрон) имеет ничтожную
массу. Атомный номер ядра, претерпевающего р-распад, возрастает на
единицу, так как ядро приобретает протон:
о11
1р + Р"
* Это не совсем точно. Распад ядра, сопровождающийся потерей р-частицы, -
сложный процесс, изучением которого занимаются физики-ядерщики. Для нас интересен
лишь тот факт, что при р-распаде атомная масса остается неизменной, а атомный
номер возрастает на единицу.
хлора. [Упражнение: Используя данные рис. 1.4,а, вычислите массовые числа
и относительные интенсивности пиков на рис 1.4,6.]
Пример 1.3. Относительная атомная масса
Вычислите атомную массу природного магния, если он имеет
следующий изотопный состав: 78,7% ^Мд, 10,1% ^|Мд и 11,2% ^Мд.
Решение
Относительная атомная масса магния - это средневзвешенное относительных
атомных масс трех изотопов:
rV ' \ 100 / \ 100 / \ 100 /
= 24,325
1.9. Газовые законы и идеальные газы
25
100
80
60
40
20
п
l_J
1 1 1
1 1
о
о
£ 100
CD
S
CJ
X
о>
х 80
s
S
X
€ 60
<и
н
S
о
о
| 40
20
~
1 1
1
1
34 35 36 37 38
Массовое число
69 70 71 72 73 74 75
Массовое число
Рис. 1.4. Масс-спектр атомарного (а) и молекулярного (б) хлора.
Изотопы химического элемента имеют один атомный номер Z, но
разные атомные массы.
МОЛЬ И ПОСТОЯННАЯ АВОГАДРО
В табл. 1.1 указано, что единицей «количества вещества« в СИ является моль.
Эту единицу можно применять к любой форме материи. Следовательно, в
выражении «моль X», под X может подразумеваться любой предмет, в
частности электрон, атом или молекула. В одном моле вещества приблизительно
6,022 1023 частиц; это число называют постоянной (или числом) Лвогадро NA.
Оно равно числу атомов углерода в 12 граммах образца чистого изотопа Х\С.
Если NA — число частиц в моле вещества, то оно измеряется в моль-1.
В одном моле вещества содержится число частиц, равное числу
Авогадро Л/А:
Л/А = 6,022- 1023 моль"1
ГАЗОВЫЕ ЗАКОНЫ И ИДЕАЛЬНЫЕ ГАЗЫ
В этом разделе рассмотрены некоторые важные законы, применимые к
идеальным газам; более подробно кинетическая теория газов обсуждается в
прилагаемом задачнике.
26
1. Основные понятия
Давление и закон Бойля-Мариотта
Давление определяется как сила, действующая на единицу поверхности:
Давление = Сила : Площадь (1.7)
и единицей для давления в СИ является паскаль (Па) (см. табл. 1.2).
Хотя обычно рабочее давление в лаборатории равно атмосферному
(исключая случаи использования пониженного или высокого давления), И ЮПАК
рекомендовал в качестве стандартного давления 1 бар (105 Па)*. Конечно,
условия работы могут не совпадать со стандартными; в этом случае необходима
корректировка (см. ниже).
Когда фиксированная масса газа сжимается (давление на газ увеличивается)
при постоянной температуре, объем газа уменьшается по закону Бойля-
Мариотта (уравнение 1.8), откуда видно, что увеличение давления вдвое
уменьшает объем газа в два раза, а уменьшение давления вдвое увеличивает
объем газа в два раза.
Давление ос 1/Объем
Рос 1/^при постоянной температуре (закон Бойля-Мариотта) (1.8)
Закон Гей-Люссака
Закон Бойля—Мариотта применим только при постоянной температуре, так
как объем газа зависит от температуры и давления одновременно. Объем и
температура фиксированной массы газа связаны законом Гей-Люссака
(уравнение 1.9); из прямой пропорциональности между объемом и температурой
следует, что при увеличении температуры вдвое объем газа возрастает во
столько же раз.
Объем ос Температура
Кос Гпри постоянном давлении (1.9)
Комбинация законов Бойля-Мариотта и Гей-Люссака дает соотношение
(1.10) между давлением, температурой и объемом фиксированной массы :
РосТ/У (1.10)
PVI T= const (уравнение состояния идеального газа) (1.11)
Определение объема газа при каких-либо значениях давления и температуры
по известному объему при других значениях давления и температуры, может
быть выполнено по уравнению 1.12, поскольку обе части уравнения равны
одному и тому же постоянному числу (const).
p\v\ pivi
* До 1982 г. за стандартное принималось давление 1 атм (1 атм = 101300 Па), и это значение
по-прежнему используется во многих учебниках и физических таблицах. Бар - это «нестандартная» единица,
так же как и ангстрем.
1.9. Газовые законы и идеальные газы
27
Пример 1.4. Зависимость объема газа от давления и температуры
При 273 К и 105 Па образец гелия занимает объем 0,0227 м3.
Какой объем занимает тот же образец при 293 К и 1,04 105 Па?
Решение
Необходимо использовать соотношение (1.12)
_P1V^_ = _P2V2_
Tt Т2
Сначала проверьте соответствие единиц: У в м3, Т в К, Р в Па. (В данном случае
несовместимость единиц, в которых указаны Р и V, компенсирована. Объясните,
почему)
/>, = Ю5Па К, = 0,0227 м3 Г, = 273 К
Р2= 1,04- 105Па У2 = 1 72 = 293 К
105. 0,0227 _ 1,04. 105. У2
273 293
103-0,0227-293 , 3
2 273-1,04-105
Идеальные газы
В действительности мы имеем дело с «реальными» газами, но удобно считать,
что большинство газов ведут себя как идеальные, т. е. без взаимодействия
частиц. Такой газ подчиняется закону идеального газа (уравнение 1.13), в
котором константа из уравнения 1.11 - это универсальная газовая постоянная R,
а количество газа составляет 1 моль.
PV
—-=/?=8,314Дж моль-'К"1 (1.13)
Для п молей газа этот закон можно переписать в виде уравнения 1.14
РУ
—— = nR или РУ= nRT (уравнение состояния идеального газа)* (114)
Универсальная газовая постоянная одинакова для всех газов независимо от
того, являются ли частицы газа атомами (Ne, He), молекулами, состоящими
из одинаковых атомов (02, N2) или химическими соединениями разных
атомов (С02, H2S, NO).
* В отечественной научной литературе уравнение состояния идеального газа в виде 1.14 называется
также уравнением Клапейрона—Менделеева. — Прим. ред.
28
1. Основные понятия
По рекомендации ИЮПАК стандартными считаются давление 105 Па
(1 бар) и температура 273,15 К. Для удобства обычно принимают в качестве
стандартной температуры 273 К*. Объем одного моля газа в стандартных
условиях (1 бар; 273 К) равен 0,0227 м3 или 22,7 дм3 (уравнение 1.15).
Совместимость единиц в этом уравнении рассмотрена в примере 1.5.
nRT
Объем одного моля идеального газа =
Р
1 8,314 273 ЛЛ^, , (115)
-= 0,0227 м3
105
Необходимо заметить, что этот объем отличается от 22,4 дм3 - молярного
объема при стандартном давлении в 1 атм (101300 Па); молярный объем
22,7 дм3 относится к давлению 1 бар (100 000 Па).
Молярный объем идеального газа при стандартном давлении (105 Па)
и температуре (273 К) равен 22,7 дм3.
Пример 1.5. Производные единицы СИ
Определите единицу СИ для универсальной газовой постоянной Я.
Решение
Из закона идеального газа
PV=nRT
где Р — давление, V— объем, Т- температура, п — число молей газа, следует
выражение для R:
R- РУ
пТ
СИ-единица _ (СИ-единица давления) • (СИ-единица объема)
Для ** (СИ-единица количества вещества) • (СИ-единица температуры)
[СИ-единицей давления является Па, но это производная единица, в основных
же единицах Па - кг • м-1 ■ с~2 (табл. 1.1 и 1.2)]
= (кг м-1 с-2)(м3)
(моль) К
= кг • м2 • с-2 • моль-1 • К"1
Эта единица корректна в отношении соответствия системе СИ, но ее можно
упростить, так как джоуль (Дж) равен кгм2с~2 (табл. 1.2):
СИ-единица для R = Дж • Кг'моль-1 или Дж/(К • моль).
* Стандартная температура 273 К не равна температуре стандартного состояния 298 К, используемой в
термодинамике, см. разд. 1.17 и гл.12.
1.9. Газовые законы и идеальные газы
29
Упражнение: Исходя из уравнения 1.14 и используя основные единицы СИ, покажите, что
единицы объема м3 совместимы с единицами давления Па, температуры К и
универсальной газовой постоянной R — Дж/(К ■ моль).
Пример 1.6. Объем газа
Определите объем, занимаемый 1 молем диоксида углерода при
300 К и давлении 1 бар. [Я = 8,314 Дж/(К • моль).]
Решение
Если принять, что газ идеальный, то можно применить закон идеального газа:
PV=nRT
Сначала необходимо проверить совместимость единиц — давление должно быть
в Па:
I бар= Ю5Па
Для определения объема выполним преобразование уравнения состояния
идеального газа:
V=_"RT
v=
р
nRT _l 8,314 300
Р 105
= 0,0249 м3
= 24,9 дм3
Замечание: У вас должно войти в привычку оценивать, насколько реальным
является числовое решение задачи. В данном случае известно, что молярный
объем идеального газа при 105 Па и 273 К равен 22,7 дм3, а в задаче получен ответ
24,9 дм3 при 105 Па и 300 К. Такое возрастание объема согласуется со
сравнительно малым изменением температуры.
Закон парциальных давлений Дальтона
Суммарное давление смеси газов равно сумме парциальных давлений
отдельных компонентов этой смеси. Это закон парциальных давлений Дальтона:
Р= РА+ />в + Рс + ... (1.16)
где Р— суммарное давление, РА - парциальное давление компонента А и т. д.
Парциальное давление каждого газа пропорционально числу его молей.
Уравнение 1.17 представляет собой соотношение между парциальным
давлением компонента газовой смеси (Рх) и суммарным давлением смеси Р.
Парциальное давление компонента X =
Число молей X
Число молей смеси
Суммарное давление смеси (117)
30
1. Основные понятия
Пример 1.7. Парциальные давления
При 290 К и 105 Па 25 дм3 образца газа содержат 0,35 моль аргона
и 0,61 моль неона, а) Это единственные компоненты газовой
смеси? б) Каковы парциальные давления этих двух газов?
[Я = 8,314 Дж/(К моль.)]
Решение
а) Закон идеального газа (PV= nRT) позволяет найти суммарное количество
молей (п) газа в образце. Объем должен быть переведен в м3: 25 дм3 = 25 Ю-3 м3.
PV 105-25-КН
-= 1,04
RT 8,314 290
Так как имеется всего 0,35 моль аргона и 0,61 моль неона, то должно быть еще
0,08 моль одного (или нескольких) других компонентов смеси.
б) Зная суммарное количество молей газовой смеси, можно определить
парциальные давления аргона (РАт) и неона (/^е).
Парциальное давление компонента X
Число молей X
Число молей смеси
Суммарное давление смеси
0 35
Для аргона: />= ' 105 = 33654 Па
Аг 1,04
0,61
Для неона: PNp =—-^— • 105 = 58654 Па
™е 1,04
Парциальные давления используются при определении констант
равновесия А_ для газообразных систем, это в дальнейшем будет рассмотрено в
разд. 1.22 и гл.12.
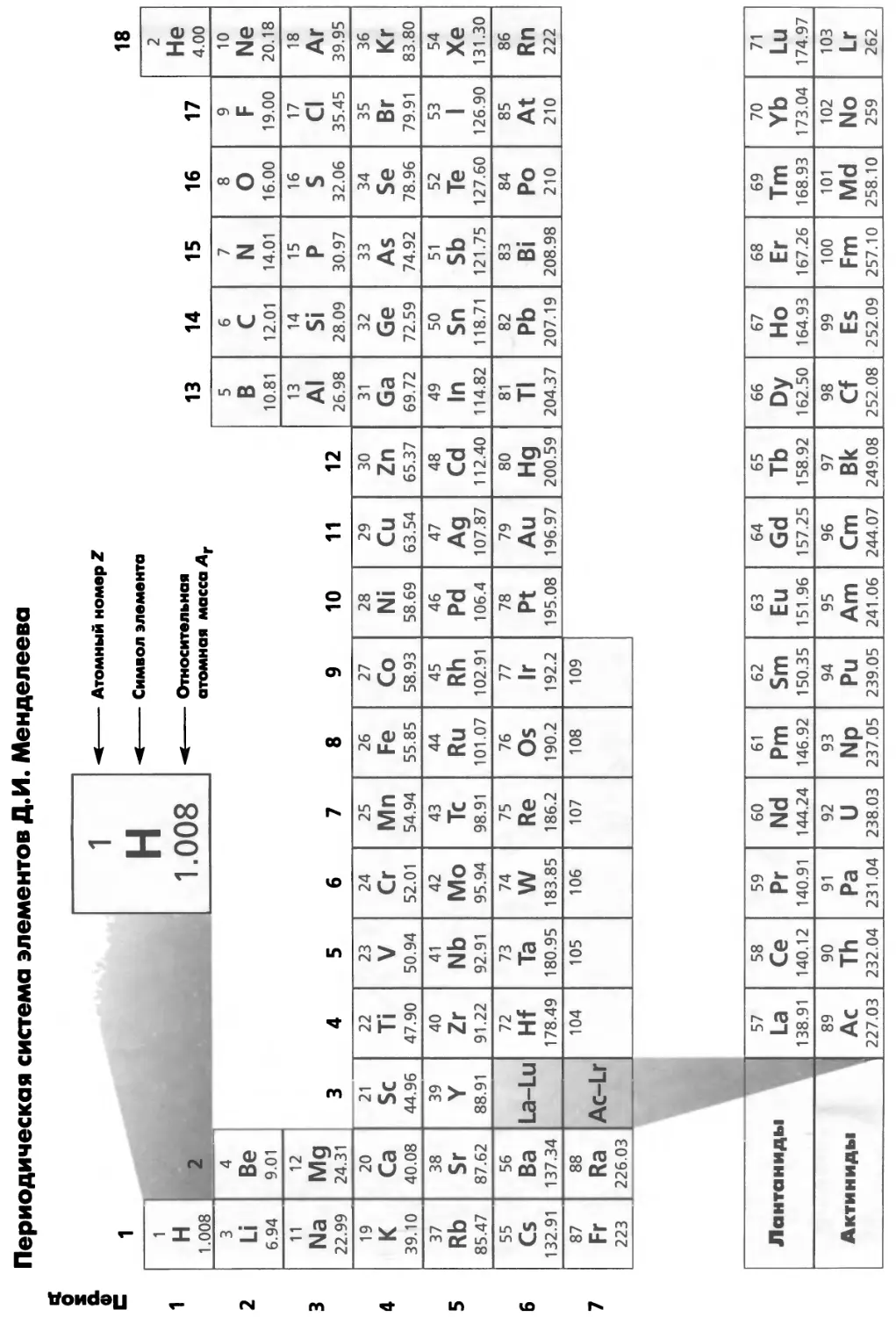
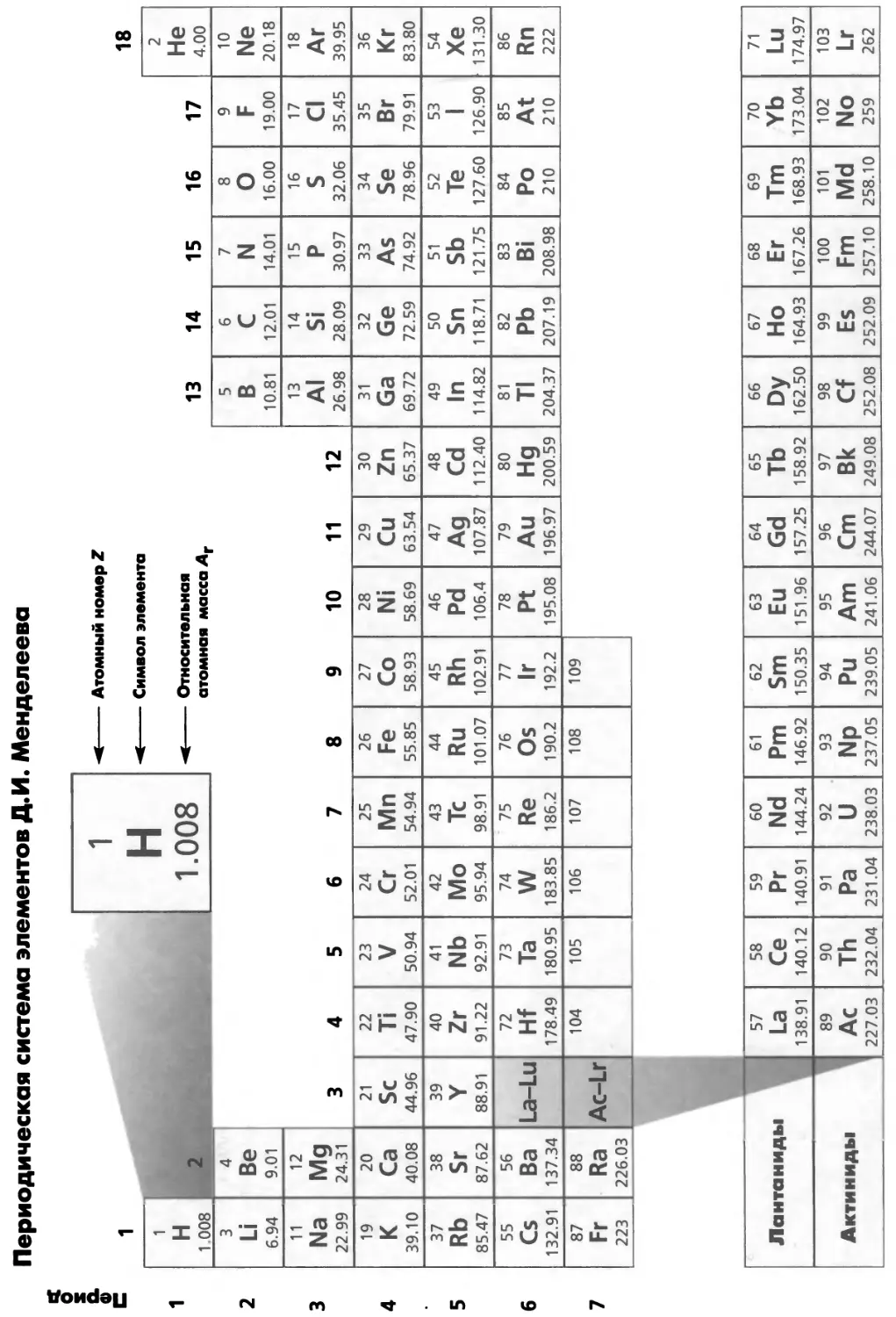
ПЕРИОДИЧЕСКАЯ СИСТЕМА ХИМИЧЕСКИХ ЭЛЕМЕНТОВ
Расположение химических элементов
в периодической системе
В периодической системе (рис. 1.5) элементы расположены в порядке
возрастания заряда ядра, т. е. числа протонов. Распределение по группам (вертикаль-
w^ ные колонки) производится в соответствии с количеством валентных элект-
Валентные электроны: ронов. По рекомендации ИЮПАК группы нумеруются
арабом, разд. 2.19 скими цифрами от 1 до 18 (рис. 1.5).
d-Элементы называют также переходными элементами, хотя
цинк, кадмий и ртуть (группа 12) по решению ИЮПАК не относят к переход-
► ным, так как они не имеют частично заполненных </-орбиталей. «Лантанои-
Номенклатура неорга- дЫ» и «актиноиды» - корректные термины, и они предпочти-
нических соединений: тельнее терминов «лантаниды» и «актиниды»*. Рекомендован-
см. разд. 1.24 ные ИЮПАК названия некоторых групп даны в табл. 1.6.
* Название «лантаноид» по-гречески означает «подобный лантану». Однако свойства элементов,
следующих за лантаном, - Се, Рг и т. д., отличаются от свойств лантана и подобными назвать их нельзя;
то же справедливо и для актиноидов, поэтому в переводе мы предпочли термины «лантаниды» и
«актиниды». — Прим. ред.
1.10. Периодическая система химических элементов
31
Период
Номер группы
7 8 9 10 11 12 13 14 15 16 17 18
Благородные газы
Гн
Li
Na
К
Rb
Cs
Fr
Be
Mg
Ca
Sr
Ba
Ra
J-Элементы
t \
Sc
Y
La|
Ac|
Ti
Zr
Hf
V
Nb
Та
Cr
Mo
W
Mn
Tc
Re
Fe
Ru
Os
Co
Rh
Ir
Ni
Pd
Pt
Cu
Ag
Au
Zn
Cd
Hg
t
В
Al
Ga
In
Tl
^-Элементы
л
С
Si
Ge
Sn
Pb
N
P
As
Sb
Bi
О
s
Se
Те
Po
N
F
CI
Br
I
At
He]
Ne
Ar
Kr
Xe
Rn
Лантаниды
Актиниды
(/'-элементы)
|ce
JTh
Pr
Pa
Nd
U
Pm
Np
Sm
Pu
Eu
Am
Gd
Cm
Tb
Bk
Dy
Cf
Ho
Es
Er
Fm
Tm
Md
Yb
No
Luj
Lr
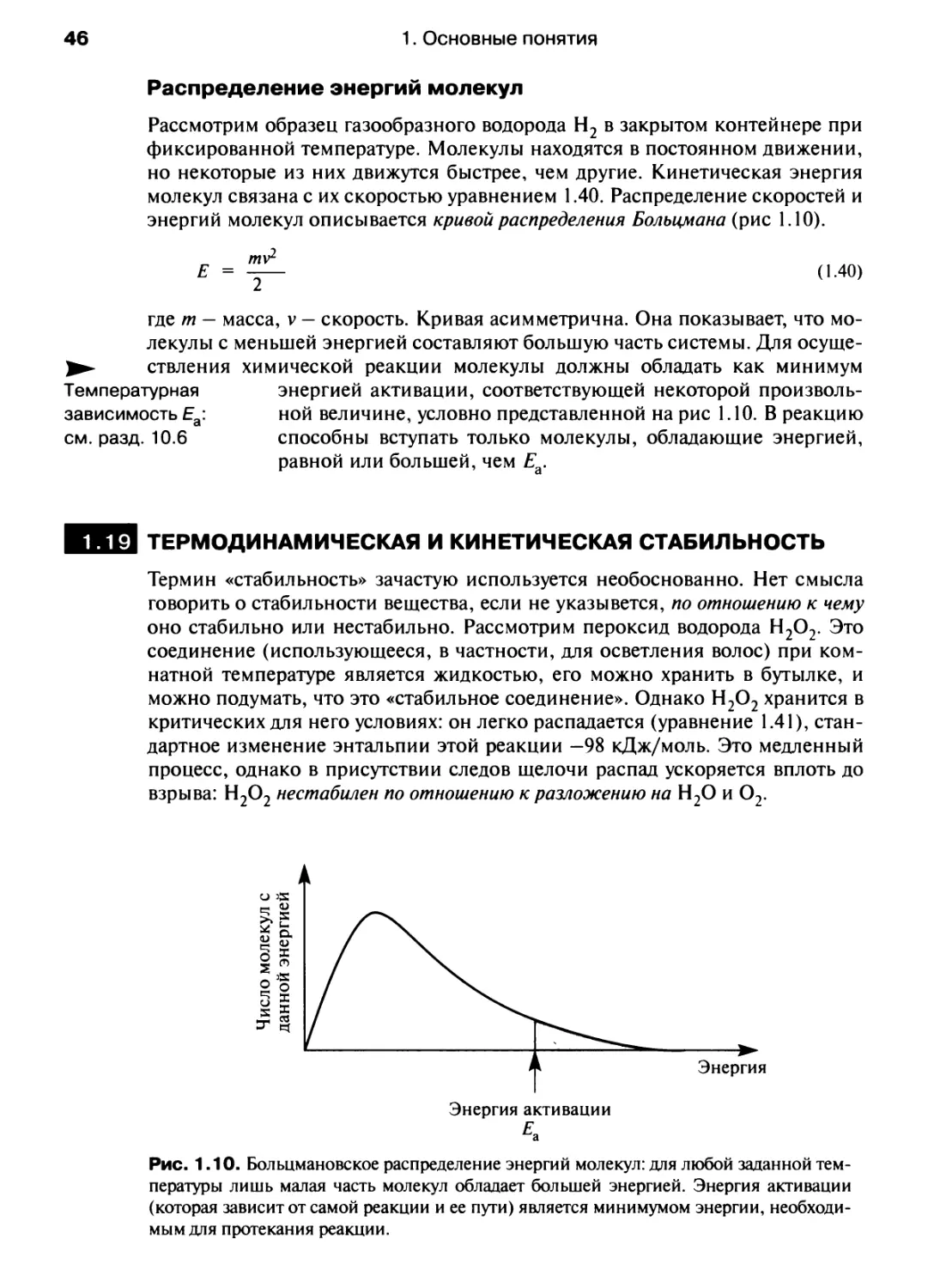
Рис. 1.5. Периодическая система. Показаны s-, /?-, </-элементы, а также семейства лан-
танидов и актинидов.
Таблица. 1.6. Названия некоторых групп периодической системы,
рекомендованные ИЮПАК
Номер группы
Рекомендованное название
I (кроме Н)
2
15
16
17
18
1 (кроме Н), 2, 13, 14, 15, 16, 17, 18
Щелочные металлы
Щелочноземельные металлы
Пникогены а
Халькогены
Галогены
Благородные газы
Главные группы элементов
а Название пникогены было предложено, но окончательно не принято ИЮПАК: Leigh G. J. (ed.)
IU РАС Nomenclature of Inorganic Chemistry: Recomendations 1990. Blackwell Scientific Publications,
Oxford, 1990.
Валентные электроны
Очень важно знать расположение элемента в периодической системе, так как
оно закономерно связано с его свойствами. Фундаментальным свойством
элемента является электронная конфигурация основного состояния (см. гл. 2),
которая может быть определена по расположению элемента в периодической
системе. Например, элементы групп 1 и 2 имеют один или два электрона на
внешней (валентной) оболочке соответственно. После блока ^-элементов (в
котором 10 групп) число валентных электронов может быть определено вы-
1. Основные понятия
читанием десяти из номера группы. Азот (N) находится в группе 15 и имеет
5 валентных электронов, теллур (Те) - в группе 16 и имеет 6 валентных
электронов. Структура таблицы отражает периодическое изменение свойств
элементов {периодичность).
Нет необходимости заучивать таблицу наизусть, но весьма полезно
понять общие закономерности изменения свойств элементов внутри групп и
периодов. Знание того, что селен находится в одной группе с кислородом
немедленно укажет вам на наличие сходства в химическом поведении этих
элементов. Однако необходима осторожность — при движении по группе вниз
свойства могут сильно меняться. Сравните элементы 14-й группы: углерод -
это неметалл; он обычно образует соединения с валентностью 4, тогда как
свинец (РЬ) — металл, имеющий две степени окисления, +2 и +4. Подробнее
эти закономерности рассмотрены в гл. 7 и 13.
РАДИКАЛЫ И ИОНЫ
Радикалы, катионы и анионы
Наличие одного или нескольких неспаренных электронов в атоме или
молекуле наделяет их свойством радикала. Для обозначения радикальной
частицы, имеющей неспаренный электрон, используется химический символ с
точкой. Нейтральный атом ^F с одним неспаренным электроном является
радикалом (рис 1.6).
У радикала есть по меньшей мере один неспаренный электрон.
Атом фтора легко принимает один электрон (рис. 1.6 и уравнение 1.18),
октета: чтобы образовать ион с конфигурацией благородного газа.
.2.20
F*(ra3) + e-->F-(ra3) (1.18)
В ионе F~ появляется дисбаланс между положительным зарядом ядра,
содержащим девять протонов, и отрицательным зарядом десяти электронов,
окружающих ядро: при добавлении одного электрона нейтральный радикал
Добавление
электрона
Атом фтора Ион фтора
(радикал) (не радикал)
Рис. 1.6. Атом фтора (радикал) при добавлении электрона становится отрицательно
заряженным ионом фтора. Показаны только валентные электроны. Ион фтора имеет
электронную конфигурацию благородного газа.
1.11. Радикалы и ионы
33
фтора становится отрицательно заряженным ионом фтора. Для обозначения
отрицательно заряженного иона используется окончание -ид: фторид-ион.
Отрицательно заряженный ион называют анионом.
Потеря одного электрона нейтральным атомом приводит к образованию
положительно заряженного иона — катиона. Атом натрия может отдать один
электрон и образовать катион натрия (уравнение 1.19). Положительный
заряд возникает из-за разницы между числом протонов в ядре и числом
электронов вокруг ядра.
Na'(ra3) -> Na+(ra3) + e~ (1.19)
Анион - это отрицательно заряженный ион, а катион - положительно
заряженный ион.
Хотя в уравнениях 1.18 и 1.19 нейтральные атомы обозначены как радикалы,
обычно в химических уравнениях для атомов химических элементов не
указывается, являются ли они радикалами; например, уравнение 1.20
обозначает то же, что и уравнение 1.19:
Ыа(газ) -> Ыа+(газ) + е" (1.20)
Термины двухзарядный, трехзарядный анион (или катион) используются для
обозначения заряда иона. Двухзарядный катион имеет заряд +2 (например,
Mg2+, Са2+), двухзарядный анион — заряд -2 (О2-, Se2~), трехзарядный
катион — заряд +3 (Al3+, Fe3+), трехзарядный анион - заряд —3 (N3~, PO^~).
Пример 1.8. Образование ионов
На основании периодической системы (рис. 1.5) назовите наиболее
вероятные ионы элементов Na, Ca, Br.
Решение
Сначала определите положение каждого элемента в периодической системе.
Помните, что элементы с числом валентных электронов до 4 склонны отдавать,
а с большим числом — принимать электроны, чтобы в каждом из этих случаев
получалась устойчивая электронная конфигурация благородного газа.
Na находится в группе 1.
У него 1 валентный электрон.
Na легко отдает один электрон с образованием электронной конфигурации [Ne].
Na образует ион Na+.
Са находится в группе 2.
У него 2 валентных электрона.
Са легко отдает два электрона с образованием электронной конфигурации [Аг].
Са образует ион Са2+.
Вг находится в группе 17.
У него 7 валентных электронов.
Вг принимает один электрон с образованием электронной конфигурации [Кг].
Вг образует ион Вг~.
34
1. Основные понятия
МОЛЕКУЛЫ И ХИМИЧЕСКИЕ СОЕДИНЕНИЯ:
ОБРАЗОВАНИЕ СВЯЗИ
Образование ковалентной связи
Молекула является отдельной нейтральной частицей, которая
получается при образовании ковалентной связи между одним или
несколькими атомами.
Когда два радикала объединяют свои электроны, образуется молекула.
Уравнение 1.21 представляет реакцию образования молекулы водорода из
двух атомов (радикалов).
2Н'->Н2 (1.21)
Гомоядерные и гетероядерные молекулы
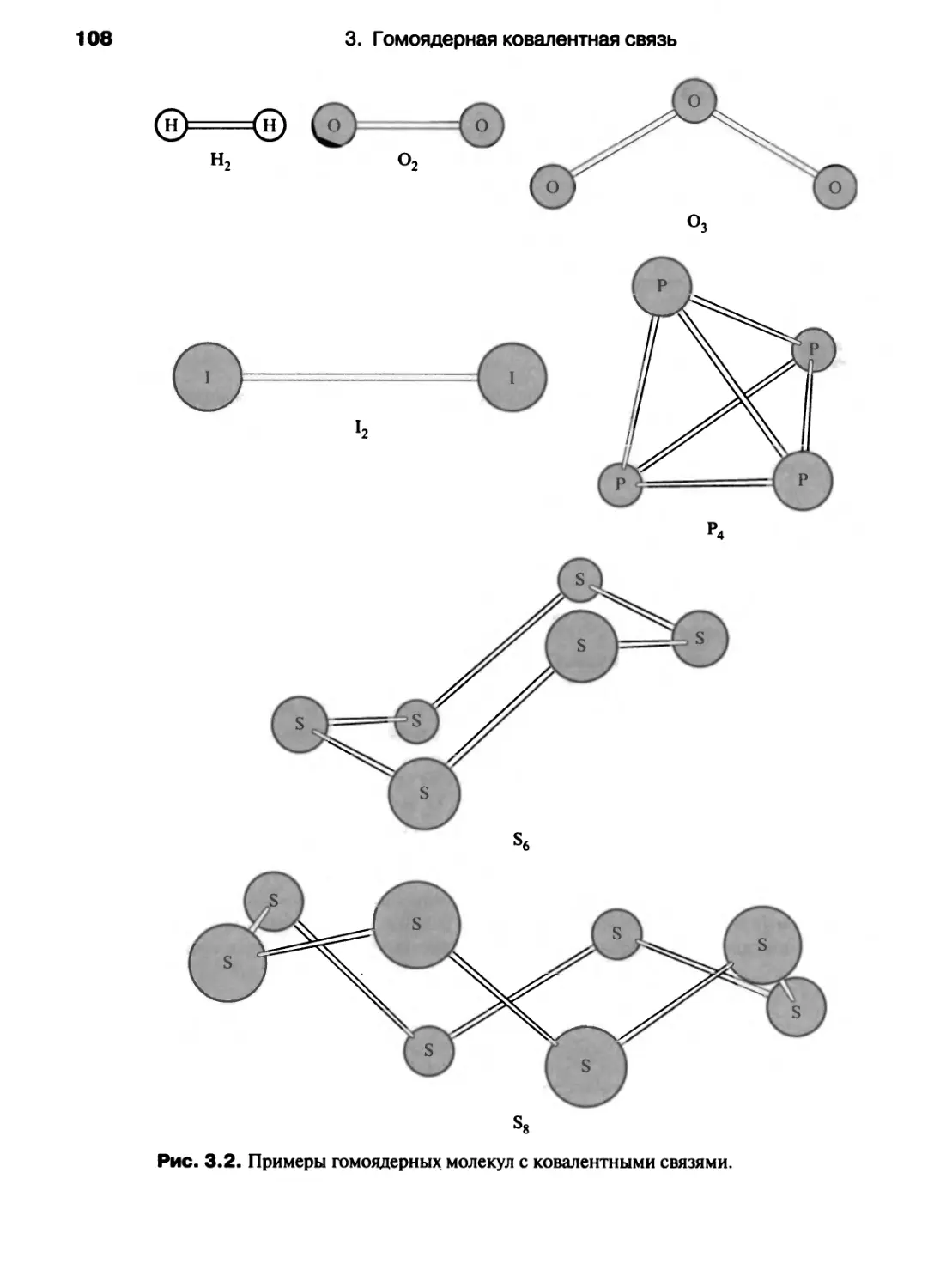
Молекула Н2 является гомоядерной двухатомной молекулой. Термин «двух-
w, атомный» означает, что Н2 состоит из двух атомов, а «гомоядерный» - что эти
Гомоядерные двух- атомы одинаковые. Н2 — это молекулярный водород, в отличие
атомные молекулы: от атомарного водорода Н. Примерами других гомоядерных
см гл з молекул могут служить молекулярные кислород (02), азот (N2),
фтор (F2), озон (03), молекулярная сера (S8).
Гетероядерные молекулы содержат атомы более чем одного химического
элемента. Моноксид углерода СО — гетероядерная двухатомная молекула.
Когда молекула содержит три или более атомов, она называется многоатомной;
диоксид углерода С02, метан СН4, этанол С2Н5ОН - многоатомные молекулы.
Ковалентные и ионные связи
Важное различие между ковалентной и ионной связью - распределение
связывающих электронов между ядрами. Электроны, образующие ковалентную
связь, поровну распределены между ядрами, как показано на рис. 1.7,а для
молекулы С12, имеющей одинарную связь CI—C1. Так как два атома
идентичны, то два связывающих электрона расположены симметрично между
ядрами хлора.
си: ■
ZJ
Рис. 1.7. Образование ковалентной связи (а) в молекуле Cl2, где каждый атом
предоставляет по одному электрону, и ионной связи (б) между положительно заряженным ионом
натрия и отрицательно заряженным ионом хлора. Молекулы С12 существуют в виде
дискретных частиц, тогда как ионные пары NaCl составляют ионную решетку
кристаллического NaCl.
1.13. Молекулы и химические соединения
35
Полное перемещение пары связывающих электронов к одному из ядер
приводит к образованию ионной связи. На рис. 1.7,5 схематически показано
► положение пары электронов в решетке хлорида натрия; между
Трехмерная ионная ионами существует область, в которой электронная плотность
решетка: см. гл. 6 почти нулевая. На рис 1.7,5 показана изолированная пара
ионов Na+ и С1~, но в действительности это не так: положительные и
отрицательные ионы притягиваются, и эта цепь взаимодействий приводит к
образованию трехмерной ионной решетки.
В случае ковалентной связи обобществленные электроны связи
поровну распределены между атомами. При образовании ионной связи
происходит перенос одного или более электронов от одного атома к
другому.
Молекулы и химические соединения
Следует уточнить понятия молекулы и химического соединения. Соединение
нейтрально; оно включает частицы с ковалентной или ионной связью,
например NaF (состоит из ионов Na+ и F~), СО (ковалентная двухатомная
молекула), SF6 (ковалентная многоатомная молекула) и т. д. Молекула -
дискретная частица, и из этих трех химических соединений только СО и SF6
состоят из молекул.
ВВ МОЛЕКУЛЫ И ХИМИЧЕСКИЕ СОЕДИНЕНИЯ:
ОТНОСИТЕЛЬНАЯ МОЛЕКУЛЯРНАЯ МАССА И ЧИСЛО МОЛЕЙ
Относительная молекулярная масса
Относительная молекулярная масса МТ химического соединения является
суммой относительных атомных масс составляющих его атомов. Например,
моноксид углерода СО имеет относительную молекулярную массу 28 (так как
относительные атомные массы С и О равны 12 и 16), а диоксид углерода С02 —
44. (Ранее относительную молекулярную массу Мт неправильно называли
молекулярным весом химического соединения.)
Число молей
Относительная молекулярная масса численно равна массе (в фаммах)
одного моля вещества. Уравнение 1.22 определяет связь между массой (в фаммах)
и количеством молей.
Масса в граммах
Число молей = ——
Относительная молекулярная масса
(1.22)
36
1. Основные понятия
Пример 1.9. Относительная молекулярная масса и число молей
Сколько молей молекул содержат 3,48 г ацетона? [АГ(С) = 12,
ЛГ(0) = 16,АГ(Н) = 1.]
Решение
Запишем формулу ацетона:
СН3С(0)СН3
А/г = (3- 12)+ (1 • 16)+ (6- 1) = 58
Масса (г) 3,48
Число молей = — = ——— = 0,06
М 58
KKQ КОНЦЕНТРАЦИЯ РАСТВОРОВ
Молярность
Химические реакции часто проходят в растворах, и концентрация растворов
указывает, какое количество химического соединения или иона содержится в
единице объема раствора. Единицы СИ для концентрации — моль/м3,
однако на практике используют единицы моль/дм3, или моль/л. Раствор,
содержащий 1 моль растворенного вещества в объеме 1 дм3, называется
одномолярными М) раствором.
Концентрация и объем раствора
В действительности не всегда имеют дело с одномолярными растворами, и
количество молей растворенного вещества может быть найдено по
уравнению 1.23, а для раствора объемом 1 см3 (что чаще используется в
лабораторной практике) — по уравнению 1.24.
Число молей = Объем (дм3) • Концентрация (моль/дм3) (123)
Число молей = Объем (см3) • Концентрация (моль/дм3) / 1000 (124)
Пример 1.10. Концентрация раствора
Какова концентрация раствора, если 1,17 г хлорида натрия
растворены в 100 см3 воды? [Ar(Na) = 23,4r(CI) = 35,5.]
Решение
Сначала необходимо определить число молей NaCl в 1,17 г:
Л/г = 23+ 35,5 = 58,5
1,17
Число молей NaCl = —r^-=— — 0,02
1.15. Стехиометрия реакции
37
Для дальнейших вычислений необходимо убедиться в том, что единицы
совместимы. Объем выражен в см3; следовательно, для нахождения концентрации
необходимо использовать уравнение 1.24.
Концентрация = Число молей • 1000 / Объем (см3)
Концентрация = 0,02 • 1000/100 = 0,2 моль/дм3
Пример 1.11. Концентрация раствора
Сколько иодида калия (KI) надо растворить в 50 см3 воды, чтобы
получить раствор с концентрацией 0,05 М [4Г(К) = 39, Аг(\) = 127.]
Решение
Определим количество молей К1 в 50 см3 0,05 моль/дм3 раствора:
Число молей = Объем (см3) • Концентрация (моль/дм3) / 1000
Число молей KI = 50 • 0,05/1000 = 2,5 • 10"3
Переведем число молей в массу:
Число молей = Масса (г) / Л/г или Масса = Число молей • Л/г
Для KI Л/г = 39 + 127 = 166. Следовательно,
Масса KI = (2,5 • 10"3) • 166 = 0,415 г
СТЕХИОМЕТРИЯ РЕАКЦИИ
Стехиометрическое уравнение реакции показывает, в каких молярных
соотношениях находятся реагенты и продукты реакции. В реакции 1.25 одна
молекула пентана реагирует с восьмью молекулами кислорода, в результате
чего получается пять молекул диоксида углерода и шесть молекул воды.
С5Н12(газ) + 802(газ) -> 5С02(газ) + 6Н20(ж.) (1.25)
Пентан
Пример 1.12. Количество исходных веществ, необходимое для полного
протекания реакции
Сколько цинка необходимо для полного протекания реакции с
30 см3 1М хлороводородной (соляной) кислоты? [Ar(Zn) = 65.]
Решение
В результате реакции металла с кислотой получается соль и Н2. Запишем
стехиометрическое уравнение реакции:
Zn(TB.) + 2НС1(водн.) -> 2пС12(водн.) + Н2(газ)
Определим число молей НС1 в растворе:
Число молей = Объем (см3) • Концентрация (моль/дм3)/ 1000.
Число молей НС1 = 30 1/1000 = 3 • 10"2
38
1. Основные понятия
Рассмотрим уравнение реакции - один моль Zn реагирует с двумя молями НО
и, значит, 3 • 10~2 моль НС1 реагируют с 1,5 • 10~2 моль Zn.
Теперь можно определить, сколько цинка потребуется для данной реакции:
Масса (г) = Число молей • Л/г = 1,5 • 10~2 • 65 = 0,975 г
Пример 1.13. Количество продукта реакции
Сколько С02 образуется при сгорании 0,3 г углерода в стандартных
условиях? [АГ(С) = 12, 1 моль газа при 105Па и 273 К занимает
объем 22,7 дм3.]
Решение
Запишем уравнение реакции:
С(тв.) + 02(газ) -> С02(газ)
Из одного моля углерода образуется один моль С02 (при условии полного
сгорания).
Определим число молей углерода:
Число молей = Масса (г) / Ат = 0,3/12 = 0,025
(В уравнении используется Аг так как речь идет об атомах химического элемента.)
При стандартных температуре (273 К) и давлении (105Па) один моль С02
занимает 22,7 дм3.
Объем 0,025 моль С02 = 0,025 • 22,7 = 0,5675 дм3
[Упражнение: Какой объем занимает газ, если продукт сгорания углерода — мо-
ноксид углерода?]
ОКИСЛЕНИЕ, ВОССТАНОВЛЕНИЕ И СТЕПЕНЬ ОКИСЛЕНИЯ
Окисление и восстановление
Когда металл сгорает в атмосфере кислорода с образованием оксида, он
окисляется (уравнение 1.26).
2Mg(TB.) + 02(газ) -> 2MgO(TB.) (1.26)
При обратной реакции, когда оксид металла реагирует с водородом и
превращается снова в металл, элемент восстанавливается (уравнение 1.27).
Нагревание
СиО(тв.) + Н2(газ) > Си(тв.) + Н20(газ) (1.27)
В реакции 1.26 02 — окислитель, а в реакции 1.27 Н2 — восстановитель.
Хотя обычно окисление воспринимается как присоединение кислорода, а
восстановление — как потеря кислорода, необходимо дать и другие
определения этих процессов. Восстановление может означать присоединение водорода; в
уравнении 1.28 хлор восстанавливается, а водород окисляется.
С12(газ) + Н2(газ) -> 2НС1(газ) (1.28)
1.16. Окисление, восстановление и степень окисления 39
Потеря водорода может быть окислением; например, хлор окисляется, когда
переходит из НС1 в С12.
Окисление и восстановление также могут быть определены в рамках
представления о переносе электронов — электроны присоединяются при
восстановлении и теряются при окислении (уравнения 1.29 и 1.30).
S+ 2е~ —> S2- Восстановление (129)
Zn -> Zn2++ 2e~ Окисление (1.30)
Однако порой бывает трудно применить эти простые определения к
конкретной реакции. Например, что происходит с водой в реакции натрия с водой
(уравнение 1.31)?
2Na(TB.) + 2Н20(ж.) -> 2№ОН(водн.) + Н2(газ) (1.31)
Степень окисления
Понятие степени окисления позволяет различить процессы окисления и
восстановления. Степени окисления приписываются каждому атому в составе
соединения и являются формальными понятиями, хотя для таких ионов, как
Na+, заряд +1 можно считать его степенью окисления +1.
Степень окисления химического элемента в простом веществе принимается
равной нулю. Это относится как к атомам (например, Не), так и к гомоядер-
ным молекулам (Н2, Р4, S8).
Чтобы определить степень окисления элемента в соединении,
необходимо следовать ряду общих правил (но осторожно):
• Сумма степеней окисления атомов в нейтральном соединении равна нулю.
• Сумма степеней окисления атомов в составе иона равна заряду иона
(например, в сульфат-ионе SO|~ сумма степеней окисления S и О должна
быть -2).
• В соединении с неметаллическим элементом степень окисления водорода
равна +1, а в соединении с металлическим элементом она равна —1.
• Степень окисления фтора всегда -1.
• Степень окисления хлора, брома и иода обычно -1 (исключая случаи
соединений с другими галогенами и кислородом; см. гл. 13).
• Степень окисления кислорода в соединении обычно -2.
• Степень окисления металлов группы 1 в соединениях равна +1.
• Степень окисления металлов группы 2 в соединениях равна +2.
• d-Металлы обычно имеют положительные степени окисления (за
исключением некоторых соединений с низкими степенями окисления; см.
разд. 16.14).
К этим правилам следует добавить замечание о том, что элементы групп 13,
14, 15 и 16 могут проявлять различные степени окисления, см. например,
дополнение 8.8. Обычно необходимо иметь полную картину структуры
соединения, прежде чем определять степени окисления.
40
1. Основные понятия
Пример 1.14. Определение степеней окисления
Какие степени окисления имеет каждый атом в следующих
частицах: Kl, FeCI3, Na2S04, NO3?
Решение
KI: Металл группы 1 имеет степень окисления +1. Это согласуется с тем, что иод
имеет степень окисления -1 и их сумма равна 0.
FeCl3: Хлор обычно имеет степень окисления —1 и, так как имеются три атома О,
для сохранения нейтральности соединения степень окисления железа
должна быть +3.
Na2S04: Из трех элементов S имеет переменную степень окисления и она будет
рассмотрена последней. Na - элемент группы 1, он имеет степень окисления
+ 1. Кислород обычно имеет степень окисления —2. Степень окисления
серы определяется, исходя из суммарной степени окисления, равной нулю:
(2 • Степень окисления №)+(Степень окисления S)
+(4 • Степень окисления О) = 0.
(+2) + (Степень окисления S) + (-8) = О
Степень окисления S =0 + 8 — 2 = + 6
NOj: Кислород имеет обычно степень окисления -2. Общий заряд равен -1,
значит:
(Степень окисления N) + (3 • Степень окисления О) = — 1
Степень окисления N = -1 + 6 = +5
Изменение степени окисления
При окислении степень окисления возрастает (становится более
положительной), а при восстановлении - уменьшается (становится более
отрицательной). В уравнении 1.29 изменение степени окисления серы от 0 до —2
соответствует восстановлению, а в реакции 1.30 изменение степени окисления
цинка от 0 до +2 соответствует окислению.
В окислительно-восстановительных («редокс») процессах изменения
степеней окисления при восстановлении и окислении должны быть равны. В
реакции 1.32 железо окисляется до железа(Н), а хлор восстанавливается до
хлорид-иона. Общее увеличение степени окисления железа в стехыометриче-
ской реакции должно быть компенсировано общим уменьшением степени
окисления хлора.
Fe(TB.) + С12(газ) -> FeCl2(TB.)
Степень окисления:
Изменение =
Окисление
%
Изменение = 2 (-1) =-2
Восстановление (1.32)
Пример 1.15. Изменение степени окисления в процессе реакции
Какие элементы окисляются и восстанавливаются в процессе
реакции 2К(тв.) + 2Н20(ж.) -> 2КОН(водн.) + Н2(газ)?
Решение
Рассмотрим структуры Н20 и КОН:
1.17. Термохимия
41
Н
Н-0
К+ [-0-Н]
и определим степени окисления:
Ожидаемые степени окисления К = +1 и О = -2; с учетом
нейтральности соединения, степень окисления Н = +1 (так как Н
связан с кислородом, неметаллом)
2К +
2Н20
2КОН
Элементное
состояние, степень
окисления равна О
Водород связан с неметаллом, степень окисления
равна +1. Это находится в соответствии с
ожидаемой степенью окисления кислорода О —2
Элементное
состояние, степень
окисления равна О
Теперь рассмотрим изменения степеней окисления.
К Оба атома К изменяют свою степень окисления от 0 до +1; окисление.
Н Два атома Н сохраняют степень окисления +1.
Два атома Н восстанавливаются от +1 до 0.
О Нет изменения (было и осталось -2).
Вывод: Общее возрастание степеней окисления сбалансировано общим их
уменьшением, окисление сбалансировано восстановлением.
ТЕРМОХИМИЯ
Протекание химической реакции регулируется термодинамическими и
кинетическими факторами. Первые связаны с энергией системы, тогда как вторые -
со скоростью процесса. Исследование кинетики реакции зависит от
предполагаемого механизма реакции, т. е. от пути, по которому по нашему мнению
взаимодействуют атомы и молекулы в процессе реакции.
Изменение энтальпии реакции
Термохимия имеет дело с измерениями изменения энтальпии Д# — теплового
эффекта реакции. Изменение энтальпии, сопровождающее реакцию,
соответствует количеству поглощенного или выделившегося в результате реакции
тепла при температуре Т. Стандартная энтальпия реакции есть изменение
энтальпии в результате реакции, исходные вещества и продукты которой
находятся в стандартных состояниях. Эта величина обозначается Ар//°(Т), где А
означает «изменение», индекс р — «реакции», Н— символ энтальпии, кружок
42
1. Основные понятия
в верхнем индексе означает «стандартное состояние», (7) - «при
температуре 7». Такая запись используется для широкого круга термохимических (и
термодинамических) функций, которые будут рассмотрены далее.
Стандартное состояние вещества — состояние, наиболее стабильное при
давлении 1 бар (105 Па) и некоторой температуре Т. Обычно Г принимается
равной 298,15 К, а обозначение стандартного изменения энтальпии реакции
при 298,15 К принимает вид АрЯ>(298,15К). Обычно достаточно писать
А #°(298 К). Не путайте стандартную термодинамическую температуру
(298 К) со стандартной температурой (273 К), применяемой при
определении объемов газов в стандартных условиях (разд. 1.9).
Изменение энтальпии может быть определено экспериментально калори-
метинескими методами или вычислено при помощи табличных величин
стандартных энтальпий образования (см. ниже).
По определению, отрицательная величина АЯ соответствует выделению
тепла в результате реакции (экзотермическаяреакция, АЖ 0), а
положительная — поглощению тепла из окружающей среды (эндотермическая реакция,
ДЯ>0).
В результате экзотермической реакции тепло выделяется
(отрицательная величина АН).
При эндотермической реакции тепло поглощается (положительная
величина АН).
Стандартная энтальпия образования
Стандартная энтальпия образования соединения До6 /Г(298 К) представляет
собой изменение энтальпии при образовании соединения в его стандартном
состоянии из составляющих элементов, каждый из которых также находится
в стандартном состоянии.
Стандартное состояние элемента — термодинамически наиболее
стабильная форма элемента при давлении 1 бар (105 Па) и температуре 298 К.
Исключением является фосфор, для которого стандартным состоянием
считается белый фосфор, тогда как термодинамически более стабильны красная и
черная формы*. Стандартная энтальпия образования элемента в стандартном
состоянии принимается равной нулю.
Ао6 Н°(298 К) элемента в стандартном состоянии равно нулю.
Образование метана из углерода и водорода приведено в уравнении 1.33.
Стандартным состоянием водорода является двухатомная молекулярная
Определение стандартного состояния элемента и исключение для фосфора были приняты
Национальным Бюро Стандартов (США).
1.17. Термохимия
43
форма в газовой фазе, а для углерода — графит. Изменение энтальпии,
сопровождающее эту реакцию при 298 К, — это стандартная энтальпия
образования метана.
2Н2(газ) + С (графит) -> СН4 (газ)
Аобр//°(298 К) СН4 (газ) = -75 кДж /(моль метана)
Отрицательное значение соответствует экзотермической реакции.
(1.33)
Вычисление стандартной энтальпии реакции
В общем случае для реакции веществ А и В с образованием продуктов С и D
стандартная энтальпия реакции А //°(298 К) является разностью между
суммами стандартных энтальпий образования продуктов и реагирующих
веществ (уравнение 1.35).
А + В -> С + D, Ар//°(298 К) - стандартная энтальпия реакции (134)
Др/У<298 К) = XAo6pW<298 К)продукты - 1А0бр/Г(298 К)реагенть1 (1.35)
Если теплосодержание (энтальпия) продуктов больше, чем реагентов,
реакция является эндотермической (Ар//° > 0), в противном случае — реакция
экзотермическая (А Н° < 0). Это схематически показано на рис. 1.8 в виде
диаграммы, представляющей профиль реакции.
Распространенным примером экзотермической реакции является
горение топлива, например бутана С4Н10 (уравнение 1.36)
С4Н,0(газ) + ,3/202(газ) -> 4С02(газ) + 5Н20(ж.) (1.36)
Значение А /Г(298 К) для реакции 1.36 можно определить, исходя из
стандартных энтальпий образования продуктов и реагентов. Величины
Аоб //°(298 К) для С4Н10(газ), 02(газ), С02(газ), Н20(ж.) составляют -126, 0,
—393,5 и —286 кДж/моль соответственно. В уравнении 1.37 показано, как
вычислить А //°(298К) для реакции 1.36.
Положи- ,
тельная i
энтальпия
t
Отрицательная
энтальпия
Экза
Ь /Л
А + В ♦
АН
C + D
Координата реакции
термическая реакция
Положи-i
тельная ^
энтальпия
t
Отрицательная
энтальпия
Эндот
^ /
А + В
t
АН
\
C + D
Координата реакции
ермическая реакция
Рис. 1.8. Профиль реакции для экзотермической (а) и эндотермической (б) реакций.
44 1. Основные понятия
Др /Г(298 К) = ХД^ Я°(298 К)продукты - ZA^ Я°(298 К)реагенты
= [4 • (-393,5)] + [5 • (-286)] - (-126)
=-2878 кДж/моль (1.37)
Сжигание вещества в кислороде относится к процессам горения (окисления)
и изменение энтальпии является энтальпией горения А Я.
Пример 1.16. Стандартная энтальпия реакции
Какова энтальпия реакции газообразного Cl2 с этиленом с
образованием 1,2-дихлорэтана:
С2Н4(газ) + С12(газ) ->С2Н4С12(ж.)
если величины АобрН°(298 К) для С2Н4 (газ) и 1,2-C2H4CI2 (ж.) равны
+52,5 и -167 кДж/моль соответственно?
Решение
При 298К С12(газ) является стандартным состоянием элемента и для него
ДобЯ'(298К) = 0.
Стехиометрическое уравнение реакции показывает, что из одного моля
этилена образуется один моль 1,2-дихлорэтана.
При 298 К: Д^/Г = [АобрЯ° С2Н4С12(ж.)] - [Аобр/Г С2Н4(газ)]
= -167-52,5
= -219 кДж/моль реакции
Запомните: Выражение «на моль реакции» означает, что мы вычисляем
изменение энтальпии по отношению к реакции в том виде, в котором она записана.
Если бы в реакции участвовало два моля этилена:
2С2Н4(газ) + 2С12(газ) -> 2С2Н4С12(ж.)
изменение энтальпии было бы вдвое большим (-439 кДж).
Закон Гесса постоянства сумм теплот реакции
Проделанные выше вычисления основаны на законе Гесса постоянства сумм
теплот реакции, согласно которому тепловой эффект реакции не зависит от
пути реакции, т.е. изменение энтальпии зависит только от начального и
конечного состояний системы. В уравнении 1.37 мы предполагали, что бутан и
кислород, а также вода и диоксид углерода образовались прямо из
составляющих их элементов. Эти величины изменения энтальпии являются частью
термохимического цикла, который заканчивается уравнением 1.36. Схема
уравнений 1.38 описывает полный цикл.
Др//° (298 К)
С4Н10(газ) + ,3/202(газ) ► 4С02(газ) + 5Н20(ж.)
Д/Л (298 К) \ \ / Д/Л(298К)
4С(графит) + 5Н2(газ) + ,3/202(газ)
(1.38)
1.18. Болыдмановское распределение энергий молекул 45
На схеме показан путь от элементов в стандартном состоянии до диоксида
углерода и воды. Закон Гесса связывает три величины изменения энтальпии
в термохимический цикл (уравнение 1.39).
Др/Г + Д/Г, = АН°2
(при 298 К)
(1.39)
Преобразуя это уравнение, можно выразить Аоб Я° как разность между
суммами стандартных энтальпий образования продуктов (АЯ°2) и реагентов
(А#° j), как это было сделано в уравнении 1.37. Закон Гесса особенно важен в
более сложных случаях, таких, как определение энергии решетки (разд. 6.12)
или изменения энтальпии при растворении солей (разд. 12.11).
Закон Гесса постоянства сумм теплот реакции утверждает, что
изменение энтальпии при образовании продуктов из реагентов не зависит
от пути реакции.
БОЛЬЦМАНОВСКОЕ РАСПРЕДЕЛЕНИЕ ЭНЕРГИЙ МОЛЕКУЛ
Энергия активации
На рис. 1.9 показано, что для образования продуктов С и D из реагентов А и
В необходимо преодолеть энтальпийный барьер. Этот начальный барьер
называют энергией активации. Он является мерой кинетического барьера реак-
^ ции (кинетика реакции: см. гл. 10). Чем выше этот барьер, тем менее вероят-
Энергия активации: но протекание реакции. Предпочтительнее использовать
терем, разд. 10.6 мин «энергия активации» Е'.
Энергия активации реакции Еа представляет собой избыток энергии
(по отношению к основному состоянию), необходимый для
протекания реакции.
Рис. 1.9. Профиль
экзотермической реакции,
показывающий актива-
ционный барьер или
энергию активации £а
(см. также гл. 10).
Положительная
энтальпия
Отрицательная
энтальпия
C + D
Координата реакции
46
1. Основные понятия
Распределение энергий молекул
Рассмотрим образец газообразного водорода Н2 в закрытом контейнере при
фиксированной температуре. Молекулы находятся в постоянном движении,
но некоторые из них движутся быстрее, чем другие. Кинетическая энергия
молекул связана с их скоростью уравнением 1.40. Распределение скоростей и
энергий молекул описывается кривой распределения Больцмана (рис 1.10).
/wv2
(1.40)
где т — масса, v — скорость. Кривая асимметрична. Она показывает, что
молекулы с меньшей энергией составляют большую часть системы. Для осуще-
^~ ствления химической реакции молекулы должны обладать как минимум
Температурная энергией активации, соответствующей некоторой произвол ь-
зависимость Еа: ной величине, условно представленной на рис 1.10. В реакцию
см. разд. 10.6 способны вступать только молекулы, обладающие энергией,
равной или большей, чем Е .
ТЕРМОДИНАМИЧЕСКАЯ И КИНЕТИЧЕСКАЯ СТАБИЛЬНОСТЬ
Термин «стабильность» зачастую используется необоснованно. Нет смысла
говорить о стабильности вещества, если не указывется, по отношению к чему
оно стабильно или нестабильно. Рассмотрим пероксид водорода Н202. Это
соединение (использующееся, в частности, для осветления волос) при
комнатной температуре является жидкостью, его можно хранить в бутылке, и
можно подумать, что это «стабильное соединение». Однако Н202 хранится в
критических для него условиях: он легко распадается (уравнение 1.41),
стандартное изменение энтальпии этой реакции -98 кДж/моль. Это медленный
процесс, однако в присутствии следов щелочи распад ускоряется вплоть до
взрыва: Н202 нестабилен по отношению к разложению на Н20 и 02.
Энергия
Энергия активации
Рис. 1.10. Больцмановское распределение энергий молекул: для любой заданной
температуры лишь малая часть молекул обладает большей энергией. Энергия активации
(которая зависит от самой реакции и ее пути) является минимумом энергии,
необходимым для протекания реакции.
1.21. Межмолекулярные взаимодействия
47
2Н202(ж.) -> 2Н20(ж.) + 02(газ) (1.41)
Выше уже отмечалось, что алмаз неустойчив по отношению к переходу в
графит, но этот переход происходит очень медленно.
3 ЭНТАЛЬПИИ ПЛАВЛЕНИЯ И ИСПАРЕНИЯ
Энтальпия плавления
При плавлении твердого вещества происходит фазовый переход из твердого
состояния в жидкое, для которого необходим подвод теплоты. Например,
при плавлении металла твердая решетка (которая является жесткой
структурой) разрушается, хотя атомы и не отделяются друг от друга полностью
(рис. 1.2). Энтальпия такого фазового перехода называется энтальпией
плавления Лпл#. Когда жидкость затвердевает, тепловая энергия высвобождается,
так как образуются связи, и изменение энтальпии равно -Апл#. Если
плавление — эндотермический процесс, то кристаллизация — экзотермический. В
уравнении 1.42 представлены фазовые переходы меди.
+ 13 кДж/моль
Си(тв.) ( -» Си(ж.)
-13 кДж/моль
Энтальпия испарения
(1.42)
Изменение энтальпии, сопровождающее переход жидкости в пар,
называется энтальпией испарения Аисп#. Когда жидкий металл испаряется,
необходима энергия для разделения атомов — плавление является эндотермическим
процессом. При обратном процессе, когда пары металла конденсируются,
тепловая энергия выделяется, так как образуются связи. Фазовый переход для
свинца приведен в уравнении 1.43.
— 180 кДж/моль
РЬ(ж.) ( > РЬ(газ)
+ 180кДж/моль W-^J
МЕЖМОЛЕКУЛЯРНЫЕ ВЗАИМОДЕЙСТВИЯ
Межмолекулярные взаимодействия и ковалентные связи
В предыдущем разделе рассмотрены фазовые переходы металла на основе
взаимодействий атомов. Например, в молекулах метана 1.2 атомы связаны
друг с другом ковалентными связями.
Н
h^""*'"h
н
1.2
1. Основные понятия
Таблица 1.7. Межмолекулярные силы
Взаимодействие
Дисперсионные
силы Лондона
Диполь—д и пол ьные
взаимодействия
И он-ди пол ьные
взаимодействия
Водородные связи
Действует между
большинством
молекул
полярными
молекулами
ионами и
полярными
молекулами
атомом Н и
электроотрицательным
атомом (N,0, F)
Типичная энергия,
кДж/моль
<2
2
15
25
Дальнейшее
обсуждение
Разд. 2.21
(наведенные диполи;
межатомные силы)
Разд. 11.8 и 12.11
Разд. 11.8
В газовой фазе эти молекулы разделены в пространстве и мало влияют
друг на друга. Но в реальных газах, в отличие от идеального газа, молекулы
все же взаимодействуют. Когда метан сжижается, молекулы сближаются, а
когда жидкость затвердевает, образуется упорядоченная структура, в которой
между молекулами СН4 возникают межмолекулярные взаимодействия.
Переходы пар —> жидкость и жидкость -> твердое вещество являются
эндотермическими, но межмолекулярные взаимодействия очень слабы: Аисп# =
8 кДж/моль и Апл#= 1 кДж/моль. При фазовых переходах ковалентные связи
в молекуле СН4 не затрагиваются.
Типы межмолекулярных взаимодействий
Сила межмолекулярных (вандерваальсовых) взаимодействий зависит от их
конкретной природы (табл 1.7). Наиболее слабые взаимодействия возникают
между электронными облаками соседних молекул и называются
дисперсионными силами Лондона. Именно эти взаимодействия проявляются между
молекулами метана в твердом состоянии.
Межмолекулярные взаимодействия описаны в нескольких разделах
данной книги, но на данный момент важно запомнить, что энтальпии плавления
и испарения (а так же температуры плавления и кипения) молекулярных
частиц отражают степень межмолекулярных взаимодействий. При переходе к
ионным твердым веществам, в которых между ионами действуют
электростатические силы, энергия, необходимая для разделения ионов, всегда гораздо
больше, чем для разделения молекул. Энтальпии плавления твердых веществ
у ионных соединений гораздо выше, чем у молекулярных.
КОНСТАНТЫ РАВНОВЕСИЯ И ПРИНЦИП ЛЕ ШАТЕЛЬЕ
Принцип Ле Шателье
Рассмотрим реакцию дихромат-иона и гидроксид-иона (уравнение 1.44).
12-гпппы ъ + H.rvw ^ (1.44)
[Сг207]2"(водн.) + 20Н-(водн.)^
оранжевый
>2[СЮ4]2"(водн.) + Н20(ж.)
желтый
1.22. Константы равновесия и принцип Ле Шателье 49
При реакции с ионом ОН~ дихромат-ион переходит в хромат-ион желтого
цвета, но в присутствии кислоты процесс идет в обратную сторону. Варьируя
концентрацию кислоты и щелочи, можно сдвинуть равновесие в одну или
другую сторону.
Это пример принципа Ле Шателье, который утверждает, что при внешнем
воздействии на систему, находящуюся в равновесии, состояние системы
изменяется так, чтобы компенсировать это воздействие. Если в уравнении 1.44
в равновесную систему добавить гидроксид-ион, будет идти та реакция, при
которой расходуются ионы ОН~, т. е. слева направо. Если же добавить
кислоту, будет идти та реакция, при которой расходуется кислота, т. е. справа
налево. [Вопрос: Как используется кислота?]
Согласно принципу Ле Шателье, при внешнем воздействии на
систему в равновесии состояние системы изменяется так, чтобы
компенсировать это воздействие.
Положение равновесия
Положение равновесия дает информацию о том, насколько реагенты (левая
часть уравнения) превалируют над продуктами (правая часть) или наоборот.
Эта информация выражается константой равновесия, причем необходимо
отдельно рассматривать равновесия в растворах и газовой фазе.
Константа равновесия в растворе Кс
Если в равновесии участвуют частицы в растворе, константа равновесия
может быть выражена в концентрациях компонентов равновесной смеси. Для
реакции 1.45 константа равновесия Кс записывается в виде 1.46, где в
квадратных скобках стоят концентрации частиц.
aA + bB^cC + dD (1.45)
где компоненты - жидкости или растворенные вещества.
К = L JL J, (1.46)
Пример 1.17. Константа равновесия
Уксусная кислота и этанол реагируют следующим образом:
сн3со2н + сн3сн2он <=► сн3со2с2н5 + н2о
Уксусная Этанол
кислота
При реакции одного моля уксусной кислоты с одним молем этанола
в реакционной смеси в равновесии при 298 К присутствует
0,67 моль СН3С02С2Н5. Чему равна константа равновесия?
50 1. Основные понятия
Решение
Сначала необходимо найти число молей каждого компонента в равновесной
смеси, а затем определить их концентрации. Предположим, что общий объем
смеси равен К дм3.
сн3со2н + сн3сн2он £» сн3со2с2н5 + н2о
Начальное число молей: 1 1 0 0
Число молей в равновесии: 0,33
Концентрации в состоянии 0*33
равновесия: у
Константа равновесия определяется выражением
/0,67 \/0,67\
к Jv И V > - (0,67)2_1 12
С АЗЗ ч/0,33 \ (0,33)2
В данном случае объем ^сокращается в уравнении, но так будет не всегда.
Реакции в газовой фазе: константа равновесия Кр
►- Для реакций в газовой фазе константа равновесия выражается в парциальных
Парциальные давле- давлениях компонентов. Для реакции 1.47 константа равнове-
ния: см. разд. 1.9 сия записывается в виде выражения 1.48
дА(газ) + №(газ) <=> сС(газ) + сЮ(газ)
*п=— ^-т
Р (РаГ(Ръ)ь
Пример 1.18. Константа равновесия в газовой фазе
Водород и иод реагируют следующим образом:
Н2(газ) + 12(газ) <=> 2Н1(газ)
При 400 К /(р = 6,3. Если при давлении 105 Па смешать два моля Н2 и
два моля l2, каков будет состав равновесной смеси?
Решение
Необходимо определить число молей каждого компонента в равновесной
смеси, а затем найти их парциальные давления.
Парциальное давление компонента X =
= (Число молей X / Общее число молей газа) Общее давление
0,33 0,67 0,67
0,33 0,67 0,67
"~v v V
(1.47)
(1.48)
1.22. Константы равновесия и принцип Ле Шателье 51
Допустим, число молей HI в равновесии равно 2х (следует помнить о
стехиометрии реакции при введении данного неизвестного).
х моль Н2 и дс моль I2 дают 2х моль HI.
Н2(газ) + 12(газ) <=> 2Н1(газ)
Начальное число молей: 2 2 2
Число молей в равновесии: (2-х) (2-х) 2х
Общее число молей газа в равновесии = (2-х) + (2-х) + 2х = 4.
(2 — jc)
Парциальные давления в состоянии равновесия: /> = р. = — 105 Па
н2 i2 4
л«=-т" 105Па
Константа равновесия определяется следующим образом:
К= =6,3
Р <V(/V
\2
(^ю5) (2fio5)
= 6,3
(2_^.105)(2_,.105) (2_,.ш5)
(2л)2
= 6,3
(2-х)2
4х2 = 6,3 • (4 - Ах + л-2) = 25,2 - 25,2jc + 6,3jc2
О = 2,3jc2 - 25,2* + 25,2
Это квадратное уравнение вида
ах1 + Ьх + с = О
и оно может быть решено следующим образом:
-Ъ ± <bz - Лас
Х = Та
25,2 ± V25,22 - (4 • 2,3 • 25,2)
х =
2-2,3
У этого уравнения два решения, но только одно имеет смысл (почему?).
х= 1,1
Состав равновесной смеси: 2,2 моль HI, 0,9 моль Н2 и 0,9 моль 12.
52
1. Основные понятия
ПЕЕ1 ЭМПИРИЧЕСКИЕ, МОЛЕКУЛЯРНЫЕ И СТРУКТУРНЫЕ ФОРМУЛЫ
Эмпирические и молекулярные формулы
Эмпирическая формула соединения показывает соотношение атомов
элементов в данном соединении. Однако она не всегда совпадает с молекулярной
формулой, показывающей число атомов данного элемента с учетом
молекулярной массы соединения. Разницу между эмпирической и молекулярной
формулой соединения можно пояснить на примере этана, в котором
соотношение углерод : водород равно 1:3. Это означает, что эмпирическая формула
этана СН3. Относительная молекулярная масса этана — 30, что соответствует
двум единицам СН3 в молекуле, т. е. молекулярная формула этана - С2Н6.
Однако в метане эмпирическая формула СН4 совпадает с молекулярной формулой.
Пример 1.19. Эмпирическая и молекулярная формулы
Алканы имеют общую формулу СлН2л+2. Определите, какой алкан
содержит 83,7% углерода по массе. [АГ(С) = 12, АГ{И) = 1.]
Решение
Пусть формула соединения будет СХН . Процентный состав алкана 83,7% С и
16,3% Н.
% углерода по массе = (Масса углерода /Масса соединения) 100.
% водорода по массе = (Масса водорода /Масса соединения) • 100.
Масса 1 моль алкана = Относительная молекулярная масса Мт.
%С = 83,7 Jl2'x) . ЮО
Мт
О -у)
%Н= 16,3 = У • 100
мт
Значение Мх неизвестно, однако можно определить эмпирическую формулу, т.е.
соотношение молей атомов С : Н в соединении:
у _ 100 16,3- 12 _233
jc 100 • 83,7
Эмпирическая формула алкана: СН2 33 или С3Н7.
Соединение должно подходить к семейству алканов с общей формулой
СлН2л+2, откуда следует, что молекулярная формула соединения С6Н ,4.
На этом можно закончить решение данной задачи; на практике
эмпирическую формулу можно определить следующим образом:
%С = 83,7 ЛГ(С)=12
%Н=16,3 АТ(Н)=\
Соотношение С:Н = 83,7/12 : 16,3/1 * 7 : 16,3 = 1 : 2,33.
1.24. Номенклатура химических соединений
53
Структурная формула
^ Ни структурная, ни молекулярная формулы не дают информацию о том, как
Структура и образование связаны атомы в молекуле. Молекулярная формула H2S не
связи: см. гл. 3-5 отражает расположения трех атомов в молекуле сульфида
водорода, а структурная формула 1.3 такую информацию
дает. Ее также можно получить, исходя из количества валентных электронов,
способных образовывать связь.
► Для некоторых молекулярных формул возможно несколько путей соеди-
Изомеры: см. также разд. нения атомов в молекуле, и такие молекулы называют изо-
5.11, 8.4, 8.6 и 16.5 мерами. Примером может служить С4Н10, для которого
можно привести две структурные формулы, 1.4 и 1.5.
1.3 1>4
НОМЕНКЛАТУРА ХИМИЧЕСКИХ СОЕДИНЕНИЙ
В этом разделе рассмотрены некоторые аспекты номенклатуры и изложены
наиболее важные правила ИЮПАК для органических и неорганических
соединений. Суммированы некоторые широко используемые «тривиальные»
названия соединений с целью ознакомить вас с ними так же хорошо, как и с
их систематическими названиями в соответствии с рекомендациями
ИЮПАК. Более детальные правила и специфика нумерации атомов углерода в
органических молекулах изложены в гл. 8, 14, 15 и 17.
Классификация органических цепей:
алканы с неразветвленной цепью
Общая формула алканов с неразветвленной цепью СяН2л+2; это насыщенные
молекулы, которые содержат только связи С-С и С-Н. Простейшим
представителем этого семейства является метан СН4 (1.6). Название метан
содержит в себе следующую информацию: мет- означает, что углеродная цепь
состоит из одного атома; -ан указывает на то, что соединение является алканом.
1.6
Названия органических соединений имеют основу, содержащую
информацию о количестве атомов углерода в основной цепи молекулы. Они
перечислены в средней колонке табл 1.8. Для алканов с неразветвленной цепью к
основам, перечисленным в таблице, добавляется окончание -ан. Соединение
1.7 — это пропан, а 1.8 — гептан.
1. Основные понятия
1.7
1.8
Таблица 1.8. Описание числа атомов в углеродной цепи и числа заместителей в
названиях соединений
Число Основа, обозначающая число
атомов С в углеродной цепи
Приставка, используемая для
описания числа заместителей
I
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
Мет-
Эт-
Проп-
Бут-
Пент-
Гекс-
Гепт-
Окт-
Нон-
Дек-
Ундек-
Додек-
Триде-
Тетраде-
Пентадек-
Гексадек-
Гептадек-
Октадек-
Нонадек-
Икос-
Моно-
Ди-
Три-
Тетра-
Пента-
Гекса-
Гепта-
Окта-
Нона-
Дека-
Ундека-
Додека-
Тридека-
Тетрадека-
Пентадека-
Гексадека-
Гептадека-
Октадека-
Нонадека-
икоса-
Классификация органических соединений:
функциональные группы
Функциональная группа молекулы обусловливает ее характеристическую
реакционную способность. Функциональная группа алкена - двойная связь
С=С, а спирта - группа -ОН. Функциональные группы, описываемые в этой
книге, перечислены в табл. 1.9. Наличие большинства этих групп можно
определить такими инструментальными методами, как инфракрасная или
электронная спектроскопия, а также спектроскопия ядерного магнитного
резонанса (см. гл. 9).
Номенклатура неорганических соединений
Цель номенклатуры ИЮПАК - дать соединению или иону однозначное
название. При работе с неорганическими элементами часто возникает
проблема возможности существования разных степеней окисления. Простейшим
примером является различие между двумя оксидами углерода — моноксидом
1.24. Номенклатура химических соединений 55
Таблица 1.9. Некоторые функциональные группы органических молекул
Название функциональ- Функциональная
ной группы группа
Пример (в скобках приведено
тривиальное название, если оно
широко используется)
Спирт
— ОН
Этанол СН3СН2ОН
Альдегид
О
У/
— С
\
ЭтанальСН3СНО
(ацетальдегид)
Кетон
/
О
— С
\
R R*H
Диметилкетон СН3С(0)СН3
(ацетон)
Карбоновая кислота
У/
—с
\
о—н
Этановая кислота СН3СООН
(уксусная кислота)
Сложный эфир
Амид
\
О—R
Этиловый эфир уксусной кислоты
сн3со2с2н5
(этил ацетат)
Простой эфир
Амин
(где R — алкильный
радикал, например)
R R=R'
/ mjimR*R'
R'—О
— NH2
Диэтиловый эфир С2Н5ОС2Н5
(эфир, этиловый эфир)
Этиламин CH3CH2NH2
О
//
—с
\
NH,
Этанамид CH3CONH2
(ацетамид)
Галогеноалкан
(алкилгалогенид)
—X
X = F, CI, Br, I
Бромэтан СН3СН2Вг
Хлорангидрид карбоновой
кислоты (ацетилхлорид)
—С
/
\
CI
Этаноилхлорид СН3СОС1
(карбон илхлорид)
Нитрил
-C=N Этанонитрил CH3CN
(ацетонитрил)
Нитросоединение ™и2 Нитрометан CH3NO;
56
1. Основные понятия
и диоксидом, т. е. СО и С02 соответственно. Используемые численные
приставки приведены в табл. 1.8. Отметим, что предпочтительно использовать
ди- вместо би-.
Бинарные соединения
Бинарные соединения состоят только из атомов двух разных элементов. Для
начала рассмотрим такие примеры, где нет неясности относительно степени
окисления составляющих элементов. Примерами таких бинарных
соединений могут служить NaCl, CaO, HC1, Na2S и MgBr2. В формуле более
электроположительный элемент (обычно это металл) должен быть записан первым.
Соединения называются следующим образом:
NaCl хлорид натрия
СаО оксид кальция
НС1 хлороводород
Na2S сульфид натрия
MgBr2 бромид магния
Нет необходимости называть «сульфид динатрия» или «дибромид магния»,
так как для металлических s-элементов нет неопределенности относительно
степени окисления.
Бинарные соединения состоят из атомов двух разных элементов.
Анионы
Окончания -ид, -ит и -am обычно обозначают анионные частицы (см. табл.
1.10). Окончания -ит и -am обычно указывают на наличие кислорода в
составе аниона (т.е. на оксоанион) и используются для анионов, образованных от
оксокислот; например, оксоанион, образованный от серной кислоты, - это
сульфат-ион.
Существует несколько приемлемых способов назвать оксоанионы таких
элементов, как сера, азот или фосфор (табл. 1.10). Старые названия, такие,
как сульфат, сульфит, нитрат и нитрит, по-прежнему используются ИЮПАК.
Однако более информативно определять степени окисления связанного с
кислородом элемента, в данном случае серы, и использовать название «тет-
раоксосульфат(1У)» вместо «сульфат». Это указывает не только на степень
окисления атома серы, но и на количество атомов кислорода. Третье
приемлемое название — тетраоксосульфат(2—). В этой книге мы придерживаемся в
основном номенклатуры ИЮПАК, вместе с тем оставлены наиболее часто
используемые традиционные названия, например сульфат.
Степени окисления
Обычно степени окисления указываются на основе системы Штока с
римской нумерацией*. Числа всегда целые и проставляются после элемента, к
которому относятся; в табл. 1.10 это показано в применении к некоторым
* Нулевая степень окисления обозначается 0, хотя это не римская цифра.
1.24. Номенклатура химических соединений
57
Таблица 1.10. Названия распространенных анионов. В некоторых случаях ИЮПАК
рекомендует более одного названия
Формула аниона
н-
он-
F"
ci-
Вг"
I-
о2-
s2-
Se2"
N3"
N3"
рЗ-
CN"
NH2"
OCN-
SCN"
so42-
S032"
NO3-
N02"
ро43-
Р033"
сю4-
со32-
Название аниона
Гидрид
Гидроксид
Фторид
Хлорид
Бромид
Иодид
Оксид
Сульфид
Селенид
Нитрид
Азид
Фосфид
Цианид
Амид
Цианат
Тиоцианат
Сульфат или тетраоксосульфат(1У)
Сульфит или триоксосульфат(1У)
Нитрат или триоксонитрат(У)
Нитрит или диоксонитрат(Ш)
Фосфат или тетраоксофосфат(У)
Фосфит или триоксофосфат(Ш)
Перхлорат или тетраоксохлорат(УП)
Карбонат или триоксокарбонат(1У)
анионам. Степень окисления может быть нулевой, отрицательной или
положительной*. Степень окисления принимается положительной, если перед
цифрой не стоит отрицательного знака. Так, (III) равносильно (+III); но для
отрицательной степени окисления необходимо писать (-III).
В формуле степень окисления записывается в виде верхнего индекса
(например, Mnv,,04~), но в названиях - в строке (например, бромид железа(Н)).
Это важно в том случае, если название может быть неоднозначным (см. ниже).
Теперь рассмотрим бинарные соединения, для которых возможна
неоднозначность, обусловленная степенью окисления электроположительного
элемента (обычно металла). Примерами таких соединений могут быть FeCl3,
S02, S03, C1F, C1F3, NiS04 и SnCl2. Просто название «хлорид железа» может
Степень окисления атома какого-либо элемента в любом химическом соединении есть заряд,
которым обладал бы этот элемент, если бы электроны в каждой связи этого атома были смещены к более
электроотрицательному атому. - Прим. ред.
58
1. Основные понятия
относиться и к хлориду железа(П) и к хлориду железа(Ш). Для FeCl3
необходимо указывать степень окисления: «хлорид железа(Ш)». Другое возможное
название — «трихлорид железа».
Степень окисления серы в S02 может быть определена уже из названия
«оксид серы(1У)», но другим приемлемым названием может быть «диоксид
серы». Аналогично, S03 может быть назван оксид серы(У1) или триоксид серы.
Далее приведены названия для OF, C1F3, NiS04 и SnCl2:
CIF фторид хлора(1) или монофторид хлора
CI F3 фторид хл ора( 111) ил и трифторид хлора
NiS04 сульфат никеля(II)
SnCl2 хлорид олова(П) или дихлорид олова
Катионы
Для обозначения катионов металлов, имеющих обычно одну степень
окисления (т. е. для 5-элементов), могут использоваться названия самих металлов
(например, ион натрия, ион бария), хотя можно также указывать заряд
(например, ион натрия(1) или ион натрия(1+), ион бария(Н) или ион ба-
рия(2+))/
Там, где возможна неопределенность, заряд надо указывать обязательно
(например, ион железа(Н) или ион железа(2+), ион меди(П) или ион ме-
ди(2+), ион таллия(1) или ион таллия(1+)).
Названия многоатомных катионов вводятся в книге по мере изложения
материала, но в табл. 1.11 перечислены наиболее распространенные
неорганические неметаллические катионы, с которыми вы уже, вероятно, знакомы.
Отметим, что окончание -ий обычно соответствует катиону, хотя это также и
окончание для названий металлов (см. разд. 1.15). Многие ионы металлов,
особенно {/-элементов, могут образовывать комплексные ионы. Они
описаны в гл. 16.
Таблица 1.11. Названия некоторых распространенных неметаллических катионов
Формула катиона Название катиона
Н+ Ион водорода
Н30+ Гидроксоний-ион
NH4+ Аммоний-ион
NO+ Нитрозил-ион
NO^ Нитрил-ион
N2H5+ Гидразиний-ион
В отличие от системы Штока, по системе Эвенса-Бассета заряд иона указывается арабскими
цифрами со знаком заряда в круглых скобках сразу после названия элемента. - Прим. ред.
1.26. Упражнения
59
K^S ЗАКЛЮЧЕНИЕ
Первая глава напомнила вам об основных химических понятиях, которые
необходимо знать прежде чем приступать к изучению университетского курса
химии. Если в дальнейшем понятие или какая-либо концепция будут
названы известными, то вам необходимо пересмотреть соответствующий
материал гл.1 и задачи. В разд 1.24 уже введены основные понятия номенклатуры
органических и неорганических соединений, а более детальное рассмотрение
будет проведено по ходу повествования.
Мы намеренно не стали называть главу 1 «Введение». Обычно существует
тенденция пролистывать названные так главы, не обращая на них особого
внимания. В данном случае гл. 1 направлена на то, чтобы помочь вам
вспомнить основные понятия химии.
ЙЕН упражнения
1.1. Сколько а) мм, б) пм, в) см, г) нм
содержится в 0,0006 м?
1.2. Типичная длина двойной связи С=0
в органическом альдегиде равна 122
пм. Какова она в нм?
1.3. Относительная молекулярная масса
NaCl равна 58,4 г/моль, а его
плотность — 2,16 г/см3. Чему равен объем
одного моля NaCl в м3?
1.4. Уравнение Е = h\ связывает
постоянную Планка h с энергией и
частотой. Определите единицы СИ для
постоянной Планка.
1.5. Кинетическая энергия задается
уравнением Е= \ mv2. Используя
основные единицы СИ, покажите, что
правая и левая части данного
уравнения сопоставимы.
1.6. Вычислите относительную атомную
массу образца природного бора,
содержащего 19,9% ^В и 80,1% У В.
1.7. Масс-спектр молекулярного брома
содержит три линии
соответствующего иона BrJ. Изотопы брома: 35ВГ
(50%) и ^Вг(50%). Объясните
наличие трех линий в спектре и
предскажите их интенсивности и
соответствующие им атомные массы. Как
должен выглядеть масс-спектр НВг
(данные об изотопах водорода
приведены в разд. 1.7)?
1.8. Приведите нижеследующие объемы
к стандартным температуре (273 К) и
давлению (1 бар=105 Па) и выразите
ваш ответ в м3 в каждом случае.
а) 30,0 см3 С02 при 290 К и 101325 Па
(1 атм);
б) 5,30 дм3 Н2 при 298 К и 100 кПа
(1бар);
в) 0,300 м3 N2 при 263 К и 102 кПа;
г) 222 м3 СН4 при 298 К и 200 000 Па
(2 бар).
1.9. Парциальное давление гелия в
50 дм3 газовой смеси при 285 К и
105 Па равно 4 • 105 Па. Сколько
имеется молей гелия? (Объем одного
моля идеального газа при 273 К и
105 Па (1 бар) составляет 22,7 дм3.)
1.10. 20 дм3 образца газа при 273 К и
давлении 2 бар содержат 0,5 моль N2 и
0,7 моль Аг. Каково парциальное
давление каждого газа и есть ли в
смеси другие газы? (Объем одного моля
идеального газа при 273 К и 105 Па
(1 бар) составляет 22,7 дм3.)
1.11. Определите число молей для
каждого случая: а) 0,44 г PF3, б) 1 дм3 газо-
60
1. Основные понятия
образного PF3 при 293 К и 2 • 105 Па,
в) 3,48 г Мп02, г) 0,042 г MgC03.
ИГ(Р) = 31, Лг(Мп) = 55, AT(Mg) = 24,
у4г(0) = 16, АТ(С) = 12; объем одного
моля идеального газа при 273 К и
105 Па (1 бар) составляет 22,7 дм3.]
1.12. Какую массу твердого вещества
необходимо растворить, чтобы
получить следующие концентрации
растворов: а) 0,01 моль/л KI; б) 0,2
моль/л NaCl; в) 0,05 моль/л Na2S04?
ИГ(К) = 39, Аг(\) = 127, AT(Na) = 23,
/(r(Cl) = 35,5,y(r(S) = 32,>(r(0)=16;
1 л = 1 дм3.]
1.13. Напишите в соответствии с
периодической системой наиболее
вероятные формулы соединений между
а) натрием и иодом, б) магнием и
хлором, в) магнием и кислородом,
г) кальцием и фтором, д) литием и
азотом, е) кальцием и фосфором,
ж) натрием и серой и з) водородом и
серой.
1.14. Используя информацию
периодической системы, предскажите
наиболее вероятные формулы оксида,
хлорида, фторида и гидрида алюминия.
1.15. Запишите стехиометрические
уравнения образования соединений,
указанных в задаче 1.13, из
составляющих элементов.
1.16. Что вы понимаете под следующими
терминами: протон, электрон,
нейтрон, ядро, атом, радикал, ион,
катион, анион, молекула, ковалентная
связь, соединение, изотоп,
аллотропная модификация?
1.17. Предположите, какой тип связи
(ковалентная или ионная связь) будет
характерен для следующих
соединений? Какие частицы являются
молекулами? a) NaCl; б) N2; в) S03; г) К1;
д) N02; e) Na2S04; ж) МпО^-;
з) СН3ОН; и) С02; к) С2Н6; л) НС1;
м) S042".
1.18. Определите степень окисления азота
в каждом из следующих оксидов:
a) N20; б) NO; в) N02; г) N203;
д) N204; e) N205. Какие можно
сделать предположения? Посмотрите на
рис. 13.18 и структуру 13.30 (т. 2, гл.
13) — повлияло ли знание структуры
на ваши выводы?
1.19. Для каждой из следующих реакций
определите окислительную и
восстановительную части, а для (б)-(ж)
покажите, что изменения степеней
окисления окислителя и
восстановителя сбалансированы:
а) Си2+(водн.) + 2е~ -» Си(тв.)
б) Mg(TB.) + Н2804(водн.) ->
->MgS04^H.) + Н2(газ)
в) 2Са(тв.) + 02(газ) -> 2СаО(тв.)
г) 2Fe(TB.) + ЗС12(газ) ->
->2FeCl3(TB.)
д) Си(тв.) + 2AgN03(Bon,H.) -»
->Cu(N03)2(BOflH.) + 2Ag(TB.)
е) CuO(tb.) + Н2(газ) -> Си(тв.) +
Н20(газ)
ж) МпО^"(водн.) + 5Ре2+(водн.) +
+ 8Н+(водн.) ->
—»Мп2+(водн.) + 5Ре3+(водн.) +
+ 4Н20(ж.)
1.20. а) В соединении кислород обычно
имеет степень окисления -2.
Каковы его степени окисления в
аллотропных модификациях 02 и
Оэ, а также в соединении Н202 и
ионах 0\~ и 02? б) Каковы
окислительная и восстановительная
части в реакции распада пероксида
водорода (уравнение 1.41)?
1.21. Что означают термины изменение
энтальпии, стандартная энтальпия
образования, энтальпия плавления!
Что понимают под стандартным
состоянием вещества? Опишите, что
значат символы Д^ /Т(298 К), Апл#,
Дисп^Агор"'
1.22. Используя данные табл. 1.12,
рассчитайте стандартные энтальпии
реакций при 298 К: а) горения
1.26 Упражнения
61
этилена; б) дегидратации (потери
Н20) этанола с образованием
этилена; в) реакции Вг2 с этиленом с
образованием 1,2-дибромэтана.
Предположите, что все соединения
находятся в своих стандартных
состояниях при 298 К.
Таблица 1.12. Данные, необходимые для
решения упражнения 1.22
Соединение,
Диоксид углерода
Вода
Этилен
Этанол
1,2-Дибромэтан
1.23. При реакции одного моля N2 и трех
молей Н2
N2(ra3) + ЗН2(газ)^2ЫН3(газ)
равновесная смесь при 500 К и
давлении 1 бар содержит 0,48 моль
NH3. Каково значение К' ?
Формула и
стандартное
состояние
С02(газ)
Н20(ж.)
С2Н4(газ)
С2Н5ОН(ж.)
С2Н4Вг2(ж.)
(298 К),
кДж/моль
-393,5
-286
+52
-278
-81
1.24. Для равновесия
Н2(газ) + С12(газ)^2НС1(газ)
величина К при 1500 К равна
4,1 • 103. Сколько молей НС1
присутствует в равновесной смеси,
если изначально взято по 0,5 Н2 и
С12 и общее давление равно 105 Па?
1.25. Дайте систематическое название
каждому следующему соединению:
а) Na2C03; б) FeBr3; в) CoS04;
г) ВаС12; д) Fe203; e) Fe(OH)2; ж) Lil;
з) KCN; и) KSCN; к) Са3Р2.
1.26. Напишите формулы следующих
соединений: а) иодид никеля(П);
б) нитрат аммония; в) гидроксид
бария; г) сульфат железа(Ш);
д) сульфат железа(П); е) гидрид
алюминия; ж) оксид свинца(1У);
з) сульфид олова(Н).
1.27. Из скольких атомов состоит
углеродная цепь в а) октане,
б) гексане, в) пропане, г) декане,
д) бутане?
Q АТОМЫ И АТОМНАЯ СТРУКТУРА
131 ЗНАЧЕНИЕ ЭЛЕКТРОНОВ
В гл.1 указывалось, что химия изучает атомы и частицы, полученные
соединением атомов. В гл. 2 рассмотрены некоторые аспекты строения атома,
дано описание атомной структуры, принятое в настоящее время в химии и
использованное в книге при рассмотрении структуры многоатомных ионов и
молекул.
Атом состоит из ядра и окружающих его электронов; ядро состоит из про-
►- тонов и нейтронов. Нейтронов нет только у протия (]И). Химические свойст-
Нейтроны, протоны, ва в основном определяются поведением электронов; свойства
электроны: ЯДра важны главным образом для ядерной физики. Ионы обра-
см. разд. 1.4 зуются при потере или присоединении электрона; ковалентная
связь возникает при обобществлении электронов атомами,
образующими связь. Число электронов в атоме химического элемента
обусловливает его химические свойства.
Элементы со сходными свойствами эмпирически собраны в группы
периодической системы элементов.
Возникает вопрос: если химические свойства атомов одного элемента
зависят от числа электронов, то почему элементы одной группы
периодической системы, имеющие разное число электронов, ведут себя одинаково? Для
ответа на этот вопрос необходимо рассмотреть электронную структуру и
способ организации электронов в атоме.
ЕВ КЛАССИЧЕСКОЕ ОПИСАНИЕ СТРОЕНИЯ АТОМА
В своем развитии современная атомная теория прошла путь от классической
до квантовой механики. Переход был связан с несоответствием между
теорией и экспериментальными данными, а также с фундаментальными
изменениями в науке в конце XIX — начале XX веков.
Простейшие модели строения атома не включали детального описания
положительно заряженного ядра и отрицательно заряженных электронов.
Структура основывалась на притяжении частиц, несущих различный заряд.
После ряда экспериментов было установлено, что положительно заряженное
ядро окружено отрицательно заряженными электронами. Электроны
притягиваются к протону. Единственная сила, которая могла бы удерживать их на
расстоянии от ядра, — электростатическое отталкивание между одинаково
заряженными электронами. Модель не предполагала, что электроны
находятся на некотором расстоянии от ядра.
2.3. Классическая модель строения атома - модель Бора 63
Дополнение 2.1. Законы Ньютона
Первый закон
Тело находится в состоянии покоя или равномерного движения по прямой,
если на него не действуют внешние силы.
Второй закон
Изменение момента количества движения тела пропорционально
приложенной силе и происходит в направлении действия силы.
Третий закон
Сила действия равна силе противодействия.
Это привело к попытке описать строение атома на основе представления
о движении электронов вокруг ядра. В 1911 г. Эрнест Резерфорд предположил,
что атом состоит из положительно заряженного ядра и отрицательно
заряженных электронов, которые двигаются вокруг него по кольцевым орбитам.
В соответствии с классической моделью такого атома, электроны в нем
подчиняются законам Ньютона. Модель Резерфорда была ошибочна, так как
электроны должны были бы упасть на ядро. Кроме того, классические пред-
^ ставления описывали относительное расположение и движение ядра и элек-
Атомный спектр водо- тронов только в атоме водорода. Для атомов с большим числом
рода: см. разд. 2.16 электронов невозможно было алгебраически решить
уравнения, описывающие их движение. Однако даже для водорода
модель Резерфорда не могла объяснить некоторые наблюдаемые
спектроскопические свойства.
Е9 КЛАССИЧЕСКАЯ МОДЕЛЬ СТРОЕНИЯ АТОМА - МОДЕЛЬ БОРА
В 1913 г. Нильс Бор предложил квантовую модель строения атома. В атоме
Бора электроны вращаются вокруг ядра по кольцевым, планетарным орбитам.
Основное предположение, отличавшее модель Бора от предыдущих моделей,
в том, что энергия электрона на орбите остается постоянной и единственно
возможной для данной орбиты и каждой орбите соответствует своя энергия.
Квантованность означает, что величина может иметь только
дискретные значения.
В рамках представлений классической механики, электрон не может
двигаться по кольцевой орбите, если нет силы, удерживающей его на ней; если
нет силы притяжения, действующей на электрон, он сойдет с орбиты (то же
самое происходит, если вы вращаете привязанный к веревке мячик, а потом
2. Атомы и атомная структура
А Вектор скорости v
Направление
кругового у ф Электрон
движения
электрона
Рис 2.1. Модель Бора. Отрицательно заряженный электрон движется по кольцевой
орбите вокруг положительно заряженного ядра. Между ними есть сила притяжения,
равная центробежной силе, воздействующей на электрон и побуждающей его сойти с
орбиты.
Дополнение 2.2. История развития атомной теории
1801 Юнг демонстрирует волновые свойства света.
1888 Герц обнаруживает возникновение радиоволн при ускорении
электрического заряда, что свидетельствует о электромагнитной природе света.
1900 Рэлей и Джине пробуют вычислить распределение энергии излучения
абсолютно черного тела, но приходят к выводу об «ультрафиолетовой
катастрофе» (см. дополнение 2.3).
1900 Планк устанавливает, что электромагнитное излучение квантовано,
т. е. поглощается и излучается порциями (£= Av).
1905 Эйнштейн предполагает, что световые волны ведут себя как частицы,
энергия которых равна hv. Позднее (1926) они были названы фотонами.
1909 Резерфорд, Гейгер и Марсден показывают, что когда ос-частицы
ударяются о золотую фольгу, некоторые из них отражаются (а-частица — это
ион Не2+). Резерфорд предполагает, что атом содержит положительно
заряженное ядро и отрицательно заряженные электроны.
1911 Резерфорд предлагает модель стороения атома из положительно
заряженного ядра, вокруг которого по кольцевым орбитам движутся
электроны; модель неверна, так как такое движение будет нарушаться
вследствие притяжения электрона к ядру (см. разд. 2.2).
1913 Бор предлагает модель атома водорода, где электрон движется вокруг
ядра по орбите с определенной энергией. Возможно также
существование других орбит с дискретными энергиями (см. разд. 2.3).
1924 Де Бройль предполагает, что все частицы, включая электрон,
проявляют одновременно свойства волны и частицы (корпускулярно-волновой
дуализм (см. разд. 2.5).
1926 Открыто волновое уравнение Шредингера (см. разд. 2.7).
1927 Дэвиссон и Джермер экспериментально подтверждают теорию де
Бройля.
1927 Гейзенберг утверждает, что существует принцип неопределенности,
согласно которому из-за корпускулярно-волнового дуализма невозможно
одновременно определить координату и импульс (момент количества
движения) любой микрочастицы, включая электрон (см. разд. 2.6).
2.5. Корпускулярно-волновой дуализм
65
отпускаете веревку). На рис. 2.1 показан электрон, движущийся со скоростью
v по кольцевой орбите вокруг неподвижного ядра. Скорость - это векторная
величина, направленная по касательной в каждой точке орбиты.
Скалярная величина, например масса, имеет только размер.
Векторная величина, например скорость, имеет размер и направление.
В атоме Бора центробежная сила, пытающаяся свести электрон с орбиты,
точно равна силе притяжения между отрицательно заряженным электроном
и положительно заряженным ядром. Таким образом, электрон остается на
некотором расстоянии от ядра. В модели атома Бора возможны несколько
орбит со строго определенными энергиями. Это было важным достижением в
развитии атомной теории. Допустив, что электроны могут двигаться только
по конкретным орбитам, Бор создал квантовую модель строения атома. Эта
модель подтверждалась спектроскопическими данными, но все же была не в
состоянии объяснить результаты некоторых экспериментов.
ВЯ КВАНТ
В предыдущих разделах обсуждалось поведение электрона в атоме с точки
зрения классической механики или «классической» квантовой механики. В
конце XIX столетия был обнаружен существенный (можно сказать,
исторический) недостаток классической теории — так называемая
«ультрафиолетовая катастрофа» (см. дополнение 2.3). Это привело Макса Планка к идее
дискретных, квантованных, энергий электромагнитного излучения.
Уравнение 2.1 устанавливает соотношение между энергией Ей частотой v.
электромагнитного излучения. Коэффициент пропорциональности h — это
постоянная Планка (И = 6,626 ■ 10~34 Дж • с).
E=hv (2.1)
E=hv
Единица энергии - джоуль (Дж), частоты - с-1 или герц (Гц), а
постоянной Планка - Дж • с.
ЕЯ1 КОРПУСКУЛЯРНО-ВОЛНОВОЙ ДУАЛИЗМ
Модель Планка предполагала, что свет- это электромагнитные волны. В
1905 г. Альберт Эйнштейн предположил, что электромагнитное излучение
ведет себя как частица (корпускула). Однако было очевидно, что, хотя
электромагнитное излучение обладает свойствами, полностью объяснимыми в
рамках классических представлений о его корпускулярной природе, другие
свойства могут быть объяснены только с учетом его волновой природы. Частица
электромагнитного излучения была названа фотоном, и из уравнения 2.1
видно, что каждый фотон света с частотой v имеет энергию hv.
2. Атомы и атомная структура
Дополнение 2.3. Излучение абсолютно черного тела и
«ультрафиолетовая катастрофа»
Абсолютно черное тело поглощает все излучение любой длины волны,
падающее на его поверхность. При нагревании черное тело излучает.
Если попытаться применить законы классической физики для описания
этого процесса (что сделали Рэлей и Джине), то получится удивительный
результат: при комнатной температуре осцилляторы с короткой длиной волны
X должны иметь сильное излучение, интенсивность которого будет
постоянно возрастать и никогда не достигнет максимума. Этот результат не
согласовался с экспериментальными данными, показывающими наличие максимума
при Хмакс и падение интенсивности излучения при большей или меньшей
длине волны. Очень короткие длины волн соответствуют ультрафиолетовому,
рентгеновскому или у-излучению. Отсюда возник термин «ультрафиолетовая
катастрофа».
Ситуацию можно исправить, только отойдя от классических идей. Этот
подход был реализован Максом Планком. Он предположил, что
электромагнитное излучение может иметь только дискретные значения энергий.
Отсюда вытекало важное уравнение
Е=Ьм
где Е— энергия, v — частота, а И — постоянная Планка. Это означает, что
энергия излучаемого или поглощаемого электромагнитного излучения должна
быть кратна hv. Напомним, что частота v и длина волны X связаны
уравнением с = Xv, где с - скорость света.
Теперь вернемся к электрону в атоме. В 1927 г. Луи де Бройль доказал, что
если излучение может одновременно проявлять свойства волны и частицы
(что противоречило законам классической механики), то же относится и к
электрону или любой другой движущейся частице. Это явление известно под
названием корпускулярно-волнового дуализма.
Соотношение де Бройля (уравнение 2.2), показывает, что частица
(электрон) с моментом mv (т — масса, a v — скорость частицы) является также
волной с длиной X. Уравнение 2.2 связывает концепцию классического
механического момента с волновыми свойствами.
где h — постоянная Планка. Таким образом, происходит переход от
классической к квантовой (или волновой) механике. Посмотрим, что это означает для
электрона в атоме.
ПРИНЦИП НЕОПРЕДЕЛЕННОСТИ
Для частицы относительно большой массы волновые свойства
несущественны, и ее координаты и направление движения могут быть определены и
измерены достаточно точно. Однако для электрона с крошечной массой это
2.7. Уравнение Шредингера
67
не так. С классической точки зрения, электрон может двигаться по
определенной траектории, как брошенный мячик. Однако если мы наделяем
электрон волновыми свойствами, становится невозможным определить его
координаты и момент количества движения одновременно с одинаковой
точностью. Это принцип неопределенности Гейзенберга.
Теперь необходимо рассматривать электрон с новых позиций и говорить
не об определенных координатах и моменте, а о вероятности нахождения
электрона в данной области пространства.
Вероятность нахождения электрона в данной точке пространства
определяется величиной \|/2, где \|/ - это волновая функция. Волновая
функция - математическое выражение, определяющее поведение
электрона-волны.
Определить волновую функцию (у) и вероятность нахождения электрона
в данной точке (\|/2) можно исходя из волнового уравнения Шредингера.
УРАВНЕНИЕ ШРЕДИНГЕРА
Уравнение
Выражение 2.3 представляет собой одну из форм уравнения Шредингера.
M\f = Еу (2.3)
При первом взгляде на уравнение 2.3 напрашивается вопрос: «Можно ли
сократить \\г и сказать, что 9(= £?». Это разумный вопрос, и для того, чтобы
обосновать отрицательный ответ на него, необходимо понять смысл
величин, составляющих данное уравнение.
В правой части уравнения 2.3 \|/ представляет собой волновую функцию, а
Е— общую энергию, соответствующую этой функции. Левая часть
представляет собой математический оператор над \|/. ^называется оператором
Гамильтона или гамильтонианом.
Собственные значения и собственные векторы
Уравнение 2.3 в более обобщенной форме можно представлять в виде
уравнения 2.4, согласно которому применение оператора к функции (в нашем
случае, у) дает произведение скалярной величины (£) на эту функцию (\|/). В
таком соотношении функция называется собственной функцией, а скалярная
величина — собственным значением. В уравнении 2.3 \|/ — собственная
функция, Е- собственное значение, 0<— оператор Гамильтона.
Оператор, примененный
к функции
= (Скалярная величина) • (Функция)
(2.4)
68
2. Атомы и атомная структура
#ty = E\\f - одна из форм волнового уравнения Шредингера. Я-
оператор Гамильтона (гамильтониан), волновая функция \|/ - собственная
функция, энергия Е - собственное значение.
Какую информацию можно извлечь
из уравнения Шредингера?
Очень трудно понять физический смысл уравнения Шредингера, но целью
данного обсуждения является не детальное его изучение, а простая
демонстрация того, как из этого уравнения могут быть получены значения \|/ и у2
(точные или приблизительные). Из него могут быть определены энергии,
соответствующие конкретным волновым функциям. Квантованность энергии
вытекает как следствие из уравнения Шредингера.
Необходимо четко запомнить, что уравнение Шредингера имеет решение
только для задачи двух тел, т. е. для частиц, состоящих из ядра и одного
электрона (например, Н, Не+, Li2+) - это водородоподобные частицы.
Волновая функция V|/ является решением уравнения Шредингера и
описывает поведение электрона в области пространства, называемой атомной
орбиталью. Результат может быть использован для расчета молекулярных орби-
талей в более сложных частицах.
►- Атомная орбиталь обычно описывается тремя целыми квантовыми числа-
Квантовые числа: мы. Главное квантовое число п - положительное целое число в
см. разд. 2.10 интервале от 1 до <». Два других квантовых числа, / и т{, также
могут быть определены, и комбинация всех этих трех чисел
описывает единственную орбиталь.
Дополнение 2.4. Математические операторы
Как можно предположить из названия, математический оператор производит
действие (операцию) над функцией.
Дифференциал d/dx — пример математического оператора.
Он означает, что надо продифференцировать переменную по jc.
Пусть переменной будет у, причем у = 8х Найдем dy/dx
EZ= ffi =8
dx dx
Оператор Гамильтона Я— сложный дифференциальный оператор, он
записывается в виде:
V(x,y,z)
где дудх2, д2/ду* и d2/dz2 — частные производные (см. дополнение 12.1), а
V(x, у, z) — потенциальная энергия. Сложная структура !Н необходима для
описания положения электрона в трехмерном декартовом пространстве.
В уравнении 2.3 гамильтониан применен к волновой функции \|/.
2.7. Уравнение Шредингера
69
Волновая функция у - математическое выражение, содержащее всю
необходимую информацию о поведении электрона (как волны).
Описываемая этой функцией область пространства называется
атомной орбиталью.
Каждая атомная орбиталь может быть однозначно определена тремя
квантовыми числами: п, /, тг
Радиальная и угловая составляющие волновой функции
До сих пор положение электрона задавалось в прямоугольной, декартовой,
системе координат (х, у, z). Часто удобно использовать другую систему
координат - радиальную координату (г) и две угловые компоненты (6 и ф,
рис. 2.2). Она называется сферической полярной системой координат. Нет
необходимости здесь останавливаться на математической связи между двумя
системами. Однако полезно уметь разделять радиальную (г) и угловую (0, ф)
компоненты функции \|/. Это делается в соответствии с уравнением 2.5, где
R(r) и Л(0,ф) - новые радиальная и угловая волновые функции соответственно.
. (x,y,z) = \|/
» Ууглов^Ф) = *(гЦ(е,ф)
(2.5)
тпрямоуг v '■" ' ~радиальну ' туглову
В уравнении 2.5 радиальная компонента зависит от квантовых чисел ли/,
тогда как угловая — от / и тг Таким образом компоненты записываются в виде
9 измеряется по этой дуге
Точка 9 = 0
Точка Qc полярными
координатами (г, 9, ф)
ф измеряется по этой дуге
Рис. 2.2. Определение полярных координат (г, 6, ф) для точки Q\ r- радиальная
координата, 6 и ф — угловые координаты. Показаны также оси прямоугольной системы
координат (х ,у, z). В центре рисунка находится точка отсчета, соответствующая
началу координат.
70 2. Атомы и атомная структура
Атомная волновая функция у может быть разделена на радиальную
R(r) и угловую Л(0, ф) компоненты.
Rnl(r), и А/т(0, ф). Однако в данной книге обозначение упрощено. R(r)
обозначает радиальную, а Л(0, ф) — угловую компоненты волновой функции.
ПЛОТНОСТЬ ВЕРОЯТНОСТИ
Функции у2, R{r)2 и Д(6, ф)2
Функция \|/2 пропорциональна плотности вероятности нахождения
электрона в данной точке пространства*. Зная у2 в каждой точке пространства
вокруг ядра, можно определить область, в которой электрон проводит,
например, 95% времени. Сделав это, можно изобразить поверхности атомных ор-
биталей. Отметим, что у2 может быть представлена в виде радиальной и
угловой компонент, R(r)2 и Л(6, ф)2.
1s-Атомная орбиталь
Для атома водорода решением уравнения Шредингера с наиболее низкой
энергией является ls-орбиталь. Она имеет форму сферы, т. е. сферически
симметрична относительно ядра. Если рассматривать вероятность нахождения
электрона только в зависимости от расстояния до ядра, можно получить
радиальную составляющую R(r). Величина R(r)2 наиболее велика вблизи ядра и
быстро уменьшается при движении от центра вдоль радиуса. Если
вероятность обнаружить электрон далеко от ядра очень мала, то поверхность орби-
тали будет такова, чтобы включать в себя 95% плотности вероятности полной
волновой функции \|/2. На рис 2.3 показана зависимость R(r)2 от расстояния г.
Кривая для R(r)2 достигает нуля при бесконечном радиусе.
Нормирование
Волновая функция обычно нормируется на единицу, т. е. вероятность
нахождения электрона где-то в пространстве принимается равной I. Другими
словами, электрон должен где-то быть!
Математически нормирование описано уравнением 2.6, согласно
которому полный интеграл по всему пространству dx от функции \|/2 (а это
плотность вероятности) должен быть равен единице.**
Jv^dT=l (2.6)
Хотя здесь употребляется \|f, правильнее было бы написать 1|Л}/*, где \|/* — комлексно сопряженная
величина функции у. В одном направлении (например, х) вероятность нахождения электрона в
некоторых пределах от хдо (jc+dx), где dx — бесконечно малое изменение х, пропорциональна функции
\\f(x)\\f*(x)dx. Для трех направлений используется \|A|/*dx, что позволяет определить вероятность
нахождения электрона в элементе объема dx. Если рассматривать только радиальную составляющую
волновой функции, выражение будет иметь вид R(r)R*(r).
Более строго уравнение 2.6 записывается в виде J\|Aj/*dx = 1, где \|/* - комплексно сопряженное
функции у.
2.9. Функция радиального распределения 4яг2Я(г)2
R(r)2
71
10
Расстояние от ядра г, а. е.
Рис. 2.3. График зависимости функции R(r)2 от расстояния г. График относится к
сферически симметричной ls-орбитали.
ФУНКЦИЯ РАДИАЛЬНОГО РАСПРЕДЕЛЕНИЯ 4яг2Я(г)2
Другой способ описания плотности вероятности — график функции
радиального распределения. Соотношение между R(r)2 и функцией радиального
распределения задано уравнением 2.7
Функция радиального распределения = 4кг2R(r)2
(2.7)
Удобство этой новой функции в том, что вероятность нахождения электрона
она представляет в виде сферической оболочки радиуса г и толщиной (г + dr),
где dr- малое приращение расстояния от ядра (рис. 2.4). Таким образом, не
прибегая к определению вероятности нахождения электрона на некотором
расстоянии от ядра (рис 2.3), можно определить область пространства, в
которой находится электрон.
Толщина оболочки равна dr
Рис. 2.4. Вид сферической оболочки внутреннего радиуса г (отсчитываемого от ядра)
и толщиной dr, где dr- малое приращение расстояния г.
72
2. Атомы и атомная структура
4лг2Л(г):
10
Расстояние от ядра г, а. е.
Рис. 2.5. Функция радиального распределения электронной плотности 4лг2/?(г)2 для
Ь-атомной орбитали. Эта функция описывает вероятность нахождения электрона в
трехмерном пространстве.
На рис. 2.5 представлена функция радиального распределения для ls-op-
битали. Отметим, что в области ядра она равна нулю; это не согласуется с си-
Функция радиального
распределения более
подробно рассмотрена в
разд. 2.13
туацией для R(r)2, показанной на рис. 2.3, но логически
вытекает из зависимости функции радиального распределения
от г, у ядра г равно нулю и 4кг2R(r)2 также должно быть
равно нулю.
Функция радиального распределения имеет вид Лк^Щг)2. Она
описывает вероятность нахождения электрона в сферической оболочке
радиуса г и толщиной dr. Расстояние г измеряется от ядра.
3 КВАНТОВЫЕ ЧИСЛА
Ранее в некоторых частях текста были упомянуты квантовые числа.
Рассмотрим их теперь более подробно.
Как было показано, эффект квантования состоит в том, что разрешены
только определенные энергии электрона и, следовательно, квантованные
(дискретные) энергии орбиталей. Каждая орбиталь описывается набором
квантовых чисел л, / и т{. Четвертое квантовое число, ms, дает информацию о
спине электрона на орбитали. Каждая орбиталь имеет свой набор из трех
квантовых чисел, а каждый электрон в атоме - из четырех.
Атомная орбиталь определяется набором из трех квантовых чисел
(п,/ит,).
Электрон в атоме описывается набором из четырех квантовых чисел
(п,/, т,ит5).
2.10. Квантовые числа
73
Главное квантовое число п
Главное квантовое число п может принимать любые положительные целые
значения от 1 до °°. Число п характеризует энергетический уровень электрона
или «оболочку». Для конкретного энергетического уровня существуют
подуровни или «подоболочки», определяемые квантовым числом /.
Главное квантовое число: п = 1, 2, 3, 4, 5...°°.
Орбитальное квантовое число /
^ Квантовое число / называют орбитальным (или реже азимутальным) кванто-
Квантовоечисло/называютор- вым числом. Для определенного значения главного
битальным (или реже азиму- квантового числа п орбитальное квантовое число /
мотальным) квантовым числом жет принимать целые значения отО до (я-1). Так, при
п = 3 для / разрешены значения 0, 1 и 2. Каждое
значение /соответствует определенному типу атомной орбитали.
Орбитальное квантовое число: /= 0, 1, 2, 3, 4,.
При п = 1
л = 2
л = 3
п = 4
/=0;
/=0,1;
/=0, 1,2;
/=0, 1,2,3.
-,(п-1)
Дополнение 2.5. Угловой момент
Угловой момент = Масса Угловая скорость
Электрон на атомной орбитали имеет одновременно орбитальный и
спиновый угловые моменты. Оба типа моментов квантованы.
Угловая скорость - это угол а, на который поворачивается движущаяся
по кругу точка за секунду. Единицы СИ для угловой скорости — радианы в
секунду.
Положение частицы
\ момент времени / = 1с
Направление
кругового
вращения
частицы
Положение частицы
в момент времени t = 0 с
Угловая скорость = а рад/с
74
2. Атомы и атомная структура
Величина /дает детальную информацию об области пространства, в
которой может двигаться электрон — в частности /характеризует форму орбитали.
Число /также определяет угловой момент электрона на орбитали, т. е.
орбитальный угловой момент.
Магнитное квантовое число т1
Магнитное квантовое число т{ характеризует пространственное
расположение орбиталей и может принимать целые значения в интервале от —/до /.
Если /= 1, разрешенные значения для т{ равны —1, 0 и 1.
Магнитное квантовое число: mt= -/, (-/+1),...0,...(/-1), /.
Пример 2.1. Значение квантовых чисел и их физический смысл
Определите набор возможных квантовых чисел для п = 2 и
объясните, что они означают.
Решение
Пусть л = 2.
Отметим, что значение п определяет энергетический уровень.
Возможные значения /лежат в интервале от 0 до (л—1).
Следовательно, при п = 2 /= 0 и 1.
Это означает, что уровень п = 2 имеет два подуровня, один с / = 0, а другой с / = 1.
Теперь определим возможные значения квантового числа т[. величины /улежат
в интервале от —1 до +1.
Для подуровня / = 0 есть одно значение mf
m, = 0
Для подуровня / = 1 есть три значения ml
т,= -1,0, 1
Возможен следующий набор квантовых чисел для п = 2:
Итак,
п
2
2
2
2
1
0
1
1
1
ml
0
-1
0
+ 1
Физический смысл этого набора состоит в том, что для уровня с п = 2
возможны два типа орбиталей (так как возможны два значения /)• Для /= 1 будут
существовать три орбитали одного типа, но разнонаправленные. (Подробнее орбитали
описаны в разд. 2.11.)
Спиновое квантовое число s и
магнитное спиновое квантовое число ms
В соответствии с классической моделью, электрон может вращаться вокруг
своей оси и таким образом обладать спиновым угловым моментом в допол-
2.11. Атомные орбитали
75
нение к орбитальному угловому моменту, описанному выше. Спиновое
квантовое число s определяет величину спинового углового момента электрона и
может быть равным только 1/2. Магнитное спиновое квантовое число ms
определяет направление спинового углового момента электрона и может быть
равно + 1/2 или —1/2.
Магнитное спиновое квантовое число т = ±1/2.
^- Одна атомная или молекулярная орбиталь может нести максимум 2 элек-
Квантовые числа: трона. Два значения ms соответствуют двум электронам, зани-
см. также разд. 2.18 мающим одну орбиталь. При этом один имеет величину ms =
+ 1/2, а другой — ms = —1/2, т. е. оба электрона имеют
противоположные спины.
Пример 2.2. Набор квантовых чисел, описывающих конкретный электрон
Определите набор квантовых чисел, описывающий электрон на
атомной орбитали с л = 1.
Решение
Сначала определим, сколько существует орбиталей с п = I. Для n = l /лежит в
пределах от 0 до (n— l). Следовательно, для п = I возможно только значение / = 0.
Теперь определим возможные значения для /и,. Они лежат в пределах
-L.0...+1. Значит, для /= 0 возможно одно значение т,= 0.
Итак, данная орбиталь определяется следующим набором квантовых чисел:
n= l,/=0,w/=0
Эта орбиталь может содержать два электрона. Отсюда, каждый электрон может
быть определен следующим набором квантовых чисел:
n= l,/=0,w/ = 0,mv=+l/2
или
n= 1,/=0, т, = 0, ет5 = -1/2
АТОМНЫЕ ОРБИТАЛИ
Типы атомных орбиталей
В предыдущем разделе было сказано, что квантовое число / определяет тип
атомной орбитали. Наиболее часто встречаются четыре типа атомных
орбиталей: 5, /?, d и/ Буквы 5, р9 d и /используются только как символы. Число
/ = 0 соответствует 5-орбитали, / = 1 — /ьорбитали, 1=2 — ^/-орбитали, а
/= 3 —/-орбитали.
Различие между этими четырьмя типами орбиталей состоит в их форме и
симметрии (рис. 2.6), а их форма определяется квантовым числом /.
Для s-орбитали / = 0, для /ьорбитали / = 1, для d-орбитали / = 2, для
/-орбитали /= 3.
76
2. Атомы и атомная структура
5-Орбиталь сферически симметрична относительно ядра, и ее граничная
поверхность (т. е. волновая функция электрона) имеет постоянный знак. Это
означает, что волновая функция s-орбитали всегда либо положительна, либо
отрицательна (рис. 2.6,я).
Для /?-орбитали существует одна смена знака, и это происходит при
пересечении орбитали узловой плоскостью (рис 2.7). Каждая часть орбитали
называется долей. Знак волновой функции определяется как положительный или
отрицательный (что соответствует наличию или отсутствию штриховки на
рис. 2.6,6). Узловая плоскость соответствует узлу поперечной волны (рис. 2.8).
a zk
I \s
Рис. 2.6. Два способа представления фазы волновой функции. В качестве примеров
приведены ls-орбиталь, для которой не существует изменения знака волновой
функции (а) и 2/?-орбиталь (в данном случае 2pz), для которой существует одно изменение
знака (б).
Дополнение 2.6. Символы орбиталей
Символы 5, р, d и /берут свое начало от слов sharp (острый), principal
(главный), diffuse (диффузный) и fundamental (основной). Это соответствует
характеристикам спектральных линий водорода (см. разд. 2.16). В настоящий
момент вам следует относиться к ним просто как к символам.
Известны также обозначения других атомных орбиталей: g, h и т. д.
Значение /
Символ атомной орбитали
0 12 3 4 5
s p d f g h
*
2.11. Атомные орбитали
77
z А
Рис 2.7. Смена знака у атомной /ьорби-
тали. Здесь приведена /у орбиталь и
узловая плоскость соответствует плоскости xz.
Узловая плоскость в данном
случае соответствует
плоскости xz
Амплитуда волновой функции, соответствующая s-орбитали, либо
положительна, либо отрицательна. Граничная поверхность s-орбитали
не меняет знак и не имеет узловой плоскости.
Граничная поверхность р-орбитали меняет знак один раз и имеет
одну узловую плоскость.
Рис. 2.8. Поперечная волна.
Число и форма орбиталей данного типа
Для s-орбитали орбитальное квантовое число / равно 0. Это соответствует
единственной величине m{. I = 0, ml = 0. Для данного квантового числа п есть
только одна s-орбиталь, и говорят, что она однократно вырождена. s-Орбиталь
сферически симметрична (рис. 2.9).
Для /ьорбитали орбитальное квантовое число / равно 1. Это приводит к
трем возможным величинам 1^= +1, 0, — 1. Физический смысл этого
утверждения состоит в том, что уравнение Шредингера дает три решения для р-ор-
биталей при данном значении л, где п >2 (почему п должно быть больше
единицы?). Эти три решения обычно изображают, как показано на рис 2.9. По-
78
2. Атомы и атомная структура
Р, Ру Р=
Рис. 2.9. Для данного значения главного квантового числа п существует одна s-орби-
таль. Для данного п при п > 2 возможно существование трех/?-орбиталей.
верхности трех л/?-орбиталей идентичны, но орбитали направлены под
прямым углом друг к другу, и говорят, что /ьорбиталь трехкратно (или трижды)
вырождена. Обычно р-орбиталь, направленную по оси х, называют рх-ор6и-
талью, тогда как р - и /^-орбитали направлены по осям у и z соответственно.
Это и определяет направление осей х, у и z\
► Для ^/-орбитали орбитальное квантовое число / = 2. Это приводит к нали-
ФормасУ-орбиталей: Чию пяти возможных значений т(. (Для /=0^=2, 1, 0, -1,
см. разд. 16.1 _2.) Уравнение Шредингера дает пять действительных
решений для </-орбитали при данном значении п > 2 (почему п
должно быть больше 2?), и говорят, что ^-орбитали пятикратно вырождены.
Вырожденные орбитали имеют одинаковую энергию.
Размер орбиталей
Для данного атома орбитали одной симметрии (т. е. одинаковые числа / и /я7),
но с разным значением главного квантового числа (например, 15, 25, 35, 45)
отличаются по своему относительному размеру (объему внутреннего
пространства). Чем больше величина п, тем больше атомная орбиталь. Это
показано на рис 2.10 для я5-атомных орбиталей, хотя следует отметить, что эта
зависимость не линейна. Увеличение размера также соответствует тому, что
орбиталь становится более рыхлой, диффузной.
2.12. Типы орбиталей и главное квантовое число
79
\s 2s 3s 4s
Рис 2.10. Увеличение размера «5-атомных орбиталей для п= 1, 2, 3 и 4. 45-Орбиталь
более диффузна, чем Зз-орбиталь, которая в свою очередь более диффузна, чем 25- или
15-орбиталь.
ЕХН ТИПЫ ОРБИТАЛЕЙ И ГЛАВНОЕ КВАНТОВОЕ ЧИСЛО
Для данного главного квантового числа п можно определить (см. разд. 2.10)
серию чисел / и /я,. Из этого следует, что для данного квантового числа п
разрешен набор орбиталей определенных типов, и он также может быть
определен. Мы обозначаем орбитали одного типа, относящиеся к разным главным
квантовым числам, как Я5-, np-, nd-... - орбитали. Рассмотрим случаи, когда
п= 1,2,3.
п=1
Если п = I, то / = 0 и т1 = 0.
Величина /= 0 соответствует однократно вырожденной 5-орбитали.
Существует только одно разрешенное значение для т1 (т1 = 0).
Тогда, для п = 1 разрешена 15-атомная орбиталь.
п = 2
Если п = 2, то / = 0 или 1.
Рассмотрим отдельно каждое значение /.
Для / = 0 существует одна s-орбиталь, это 25-орбиталь.
Для / = 1 разрешенные значения для т{ 1, 0, —1 соответствуют трем /?-орби-
талям.
Итак, для п = 2 разрешенными орбиталями являются 25, 2рх, 2р и 2pz.
п = 3
Если п = 3, то / = 2, 1 или 0.
Рассмотрим отдельно каждое значение /.
Для / = 0 существует одна 5-орбиталь, обозначаемая 35.
Для /= 1 разрешенные значения для т1 1, 0, -1 соответствуют трем /?-орби-
талям.
^ Для 1=2 т1 может быть равно 2, 1, 0, —1 и —2, это соответству-
Символы Зс/-орбита- ет набору из пяти ^/-орбиталей.
лей: см. разд. 16.1 Следовательно, для п = 3 разрешены 35-, Ърх-, Зру-, 3pz- и пять
3^-орбиталей.
2. Атомы и атомная структура
Разрешенные
атомные орбитали
Общее число
орбиталей
Общее число
электронов
1 Одна s
2 Одна s, три р
3 Одна s, три р, пять d
1
4
9
2
8
18
ПОДРОБНЕЕ О ФУНКЦИИ РАДИАЛЬНОГО РАСПРЕДЕЛЕНИЯ
Итак, выше рассмотрено различие между типами атомных орбиталей,
связанное с различными в квантовыми числами, и установлено, что форма
орбитали зависит от квантового числа /. Для случая ls-орбитали приведена
радиальная часть волновой функции (рис. 2.3 и 2.5). Теперь рассмотрим, как
функция радиального распределения 4кг2R(r)2 изменяется в зависимости от г
для других орбиталей.
Вначале рассмотрим 2^-орбиталь. Из рис. 2.10 видно, что форма
поверхностей 15- и 2^-орбиталей сходна. Однако функции радиального
распределения (рис 2.11) для них различны. В то время как для ls-орбитали функция
радиального распределения 4nr2R(r)2 имеет один максимум, для 2$-орбитали
существует два максимума, между которыми функция равна нулю. Точка, в
которой 4nr2R(r)2 = 0, называется радиальным узлом. Следует отметить, что
радиальный узел соответствует точке, в которой R(r) = 0, а не г = 0
(последнее соответствует местонахождению ядра).
Функция, содержащая /?(г)2, всегда положительна, а волновая
функция R(r) может быть как положительной, так и отрицательной.
В радиальном узле функция радиального распределения 4nr2R(r)2 = 0.
4Kr2R(r)2
oEW-
10 15
Расстояние от ядра г, а. е.
20
Рис 2.11. Функции радиального распределения 4кг2R(r)2 для Is-, 2s- и 35-атомных
орбиталей водорода. Сравните количество радиальных узлов на графиках с данными
табл. 2.1.
2.13. Подробнее о функции радиального распределения 81
О 10 20
Расстояние от ядра г, а. е.
Рис. 2.12. Функции радиального распределения 4кг2R(r)2 для 35-, Зр- и 3d- атомных
орбиталей водорода. Сравните графики с данными табл. 2.1.
На рис. 2.11 также показана функция радиального распределения для 35-ор-
битали. По сравнению с 25-орбиталью, возрастает количество радиальных
узлов. Просматривается зависимость, 45- и 55- орбитали должны иметь три и
четыре радиальных узла соответственно. Наличие радиальных узлов
показывает, что 5-орбитали при п > 1 состоят из слоев, аналогично луковице,
состоящей из концентрических чешуек.
Функции радиального распределения для 35-, Зр- и 3^-орбиталей
приведены на рис. 2.12. Примеры зависимости количества радиальных узлов от л и
/даны в табл. 2.1.
Таблица 2.1. Количество радиальных узлов в зависимости от типа орбитали и
главного квантового числа п. Тенденция сохраняется для п > 4.
п 5(/ = 0) /»(/ = !) d(l = 2) /(/ = 3)
1 0
2 I 0
3 2 I 0
4 3 2 I 0
82
2. Атомы и атомная структура
BQ РЕШЕНИЕ УРАВНЕНИЯ ШРЕДИНГЕРА ДЛЯ АТОМА ВОДОРОДА
► В этом разделе рассмотрено решение уравнения Шредингера
Уравнение Шредингера: ддЯ атома водорода, т. е. для одной из немногих систем, для
см. разд. 2.3 которых данное уравнение может быть решено почти точно.
Можно подумать, что это очень мало, учитывая, что
существует еще более ста элементов, не считая молекул и ионов. Однако решение
можно аппроксимировать к системам большим, чем водородный атом.
Здесь опущены все математические преобразования, и решение
уравнения Шредингера просто постулировано. В конце концов, химики
используют именно результат решения уравнения.
Волновая функция
Решение уравнения Шредингера относительно \|/ позволяет получить
информацию, на основании которой можно построить орбитали атома водорода.
Каждое решение для \|/ представляет собой комплексное математическое
выражение, содержащее описание радиальной R(r) и угловой Л(6, ф) частей.
Решение соответствует конкретному набору квантовых чисел л, / и тг
В табл. 2.2 приведены некоторые решения уравнения Шредингера для \|/,
что дает возможность представить, какова их математическая форма.
Энергия
Значения энергии для атома водорода, получаемые из уравнения
Шредингера, соответствуют энергиям орбиталей (энергетическим уровням) и могут
быть записаны в виде уравнения 2.8.
Е= - -L- (2.8)
Таблица 2.2. Решения уравнения Шредингера для атома водорода, определяющие
Is-, 25- и 2/ьорбитали
Атомная п I т1 Радиальная часть Угловая часть
орбиталь волновой функции волновой функции
R(r) A(Q$)
и
Is
Ъх
2Р=
2Ру
1
2
2
2
2
0
0
1
1
1
0
0
+1
0
-1
2е~Г
2^С--2Ит)
лг**
лг"*
лг"*
1
1
V5(sin Э cos ф)
V3(cos 6)
2*с
V5(sin 6 sin ф)
2^г
2.14. Решение уравнения Шредингера для атома водорода 83
где Е — энергия, п — главное квантовое число, к — константа. Константа к
равна 1,312 • 103 кДж/моль. Итак, по уравнению 2.8 мы можем определить
энергию ls-орбитали, подставив туда значение /7=1. Аналогично можно
поступить и для других значений п.
Отметим, что из уравнения 2.8 может быть получено только одно
значение энергии для каждого п. Это означает, что в атоме водорода электрон
имеет одинаковую энергию вне зависимости от того, орбиталь какого типа для
данного главного квантового числа он занимает. Такие орбитали называют
вырожденными. Например, энергия электрона одинакова, занимает он 2s-
или 2/ьорбитали. Аналогично, одинаковую энергию имеет электрон на 35-,
3/7- и 3*/-орбиталях атома водорода. (Следует отметить, что электрон в атоме
водорода может занимать другие, кроме 15, орбитали, когда находится в
возбужденном состоянии; см. ниже).
На рис. 2.13 представлены энергетические решения уравнения
Шредингера для атома водорода. Эти решения соответствуют исключительно атому
водорода. Уровни энергии сближаются при возрастании числа л, и этот
результат является общим для всех атомов.
Где находится электрон в атоме водорода?
Для атома водорода были определены орбитали и их энергии. Электрон
занимает орбиталь с наиболее низкой энергией. Это соответствует основному
(наиболее стабильному) состоянию атома.
<ъ
Рис. 2.13. Схематическое (не соответствующее по
масштабу) представление энергетических решений
уравнения Шредингера для атома водорода.
(Энергетические уровни между п = 6 и бесконечностью не
показаны. В этой области при п -> «> они
сближаются и переходят в континуум.)
\7
I
Энергетические
уровни
сближаются
л = 6
и = 5
л = 3
/7 = 2
84
2. Атомы и атомная структура
Следовательно, в атоме водорода электрон занимает ls-орбиталь, и
электронная конфигурация основного состояния атома водорода записывается, как
\sx или 15.
Другие «водородоподобные» системы — Не+ и Li2+. Их электронная
конфигурация такая же, как у атома Н, хотя заряды ядер другие.
►► Когда электрон в атоме водорода по тем или иным причинам получает до-
Заполнение атомных полнительную энергию, он переходит на более высокую атом-
орбиталей электрона- ную орбиталь, чем 15. Например, он может занять 2/?-орбиталь.
ми: см. разд. 2.18 Такое состояние называется возбужденным.
Тип атомной орбитали и количество электронов на ней обозначаются
следующим образом:
ns* или пр*, или лсР и т. д.
где п - главное квантовое число, ах- число электронов на орбитали
или группе орбиталей.
Обозначение 1s1 соответствует одному электрону на ls-атомной
орбитали.
BBS ПРОНИКНОВЕНИЕ ОРБИТАЛИ К ЯДРУ И ЭКРАНИРОВАНИЕ
Как электроны в атоме влияют друг на друга?
Атом или ион с одним электроном называют водородоподобной
частицей.
Атом или ион с большим числом электронов называют
многоэлектронной частицей.
Итак, было рассмотрено взаимодействие одного электрона с ядром. Перед
переходом от атома водорода к атомам с большим числом электронов
следует обсудить, как влияют электроны друг на друга.
В разд. 2.13 были рассмотрены радиальные свойства различных атомных
орбиталей. При заполнении этих орбиталей электронами возникают два
типа электростатических взаимодействий:
• притяжение между ядром и электроном;
• отталкивание между электронами.
В водородоподобных атомах 2s- и 2/?-орбитали вырождены, как и все
орбитали с одинаковым главным квантовым числом. Электрон может занимать
только одну орбиталь в данный конкретный момент. Свободная орбиталь
виртуальна, она не имеет физического смысла.
Теперь рассмотрим атом, содержащий два электрона, один на 25-, а другой
на 2/ьорбитали. Функции радиального распределения этих двух орбиталей
изображены на рис 2.14. Наличие радиального узла у 25-орбитали означает,
что вблизи от ядра существует область, где может быть обнаружен
25-электрон. Вследствие этого электрон на 25-орбитали будет проводить больше вре-
2.15. Проникновение орбитали к ядру и экранирование 85
Расстояние от ядра г, а. е.
Рис. 2.14. Функции радиального распределения 4кг2R(r)2 для 25- и 2/7-атомных ор-
биталей.
мени ближе к ядру, чем электрон на 2/?-орбитали. В таком случае говорят, что
25-орбиталь проникает (к ядру) больше, чем 2/?-орбиталь. В атоме водорода,
где есть только один электрон, энергии электрона на 2s- и 2/>-орбиталях
одинаковы. Большее среднее расстояние от ядра до 25-электрона, чем до 2р-
электрона, компенсируется наличием радиального узла на 25-орбитали.
Что произойдет при наличии электронов на обеих орбиталях? Наличие
радиального узла на 25-орбитали означает, что существует область
отрицательного заряда (из-за наличия там 25-электрона) между ядром и областью
расположения электрона на 2/>-орбитали. Потенциальная энергия электрона,
заданная уравнением 2.9, относится к электростатическому притяжению
между положительно заряженным ядром и отрицательно заряженным
электроном.
Eoc L (2.9)
где Е- кулоновская (потенциальная) энергия, а г- расстояние между двумя
заряженными точками.
Несмотря на это, положительный заряд (ядра), действующий на электрон
на 2/ьорбитали, «слабеет» из-за наличия 25-электронной плотности рядом с
ядром. 25-Электрон экранирует 2/ьэлектрон. Заряд ядра, действующий на
электрон на 2/?-орбитали, меньше действующего на 25-электрон. Принимая
во внимание этот эффект, для многоэлектронного атома необходимо
заменить заряд ядра Z эффективным ядерным зарядом Z^. Величина Z3^ для
данного атома варьирует для различных орбиталей и зависит от числа и
расположения электронов. Величины Z^ могут быть определены эмпирически
на основе правил Слейтера*.
Правила Слейтера здесь детально не обсуждаются; при необходимости см. Хьюи Дж.
Неорганическая химия. Строение вещества и реакционная способность. М., Химия, 1987.
86
2. Атомы и атомная структура
Эффективный ядерный заряд Z^^ - величина положительного
заряда, действующего на электрон, с учетом экранирования другими
электронами.
25-Орбиталь проникает ближе к ядру и экранирует 2/?-орбиталь.
Электроны на 2р-орбитали испытывают меньшее электростатическое притяжение и
поэтому имеют более высокую энергию (они менее стабилизированы) по
сравнению с электронами на 2$-орбитали. Аналогичным образом, 3^-орбита-
ли имеют более высокую энергию, чем Зр-, а те в свою очередь больше, чем
35- (рис. 2.15).
Изменения в энергии атомных орбиталей
в зависимости от изменения атомного номера
В этом разделе кратко рассмотрено, как энергия конкретной атомной орби-
тали изменяется с возрастанием атомного номера; например, энергия 2s-op-
битали атома лития отличается от энергии той же орбитали в других атомах.
С возрастанием атомного номера возрастает заряд ядра, и по отношению
к электрону на данной ns- или л/ьорбитали также возрастает эффективный
ядерный заряд. Следствием этого является уменьшение (плавное, хотя и нели-
Энергия Е
/\
/1 = 3 <
3d
IP
3s
Рис. 2.15. Схематическое (без соблюдения масштаба) представление энергетических
решений (для п = I, 2 и 3) уравнения Шредингера для многоэлектронного атома.
2.16. Атомный спектр водорода и правила отбора
87
нейноё) энергии атомной орбитали. Итак энергии nd- и «/-атомных орбиталей
обычно уменьшаются с увеличением атомного номера, но эта взаимосвязь
достаточно сложна, и мы не будем ее здесь рассматривать*.
g атомный спектр водорода и правила отбора
Если единственный электрон на ls-орбитали водорода возбудить (т. е.
сообщить ему энергию), он может быть переведен в более высокое
энергетическое состояние. Новое состояние является неустойчивым, и электрон
возвращается на более низкий энергетический уровень, излучая при этом энергию.
Доказательством дискретной природы орбиталей является наличие
спектральных линий в спектре испускания (эмиссионном спектре) водорода
(рис. 2.16). Особенно важно здесь то, что наблюдаются отдельные линии;
эмиссия не постоянна во всем интервале частот. Подобные же отдельные
частоты наблюдаются и в спектре поглощения.
Некоторые электронные переходы, дающие начало определенным
частотам, наблюдаемым в эмиссионном спектре атомарного водорода,
представлены на рис. 2.17. Отметим, что некоторые переходы запрещены — должны
Правила отбора показывают,
какие спектроскопические
переходы разрешены: см. также
разд. 9.6.
соблюдаться правила отбора, записанные в виде
уравнений 2.10 и 2.11.
Величина Ал в уравнении 2.10 — изменение
величины главного квантового числа я, а согласно правилам
отборам, переходы с изменением главного квантового
числа не запрещены. В уравнении 2.11 А/- изменение орбитального
квантового числа /, и оно накладывает следующее ограничение на переход: /должно
изменяться только на единицу. Это уравнение также известно как правило
отбора Лапорта и соответствует изменению углового момента на единицу.
Дл = 0, 1,2,3,4...
А/ = +1 или — 1
(правило отбора Лапорта)
(2.10)
Серия Балыиера
Серия Лаймана
ВИДИМАЯ ОБЛАСТЬ
УФ-ОБЛАСТЬ
Увеличение энергии
Рис. 2.16. Часть эмиссионного спектра (спектра испускания) атомарного водорода.
Группы линий имеют определенные названия, например, серии Бальмера и Лаймана.
Для дополнительной информации см. Саито К.у Химия и периодическая таблица. М., Мир, 1982.
88
2. Атомы и атомная структура
Дополнение 2.7. Спектры поглощения и испускания
Основное состояние атома (или частицы) соответствует электронному
окружению с наиболее низкой энергией.
Если атом (или другая частица) поглощает электромагнитное излучение,
происходят дискретные электронные переходы и наблюдается спектр
поглощения (абсорбционный спектр).
Когда энергия (тепло или свет) сообщается одному или нескольким
электронам в атоме или частице, то они могут перейти из своего основного
энергетического уровня в более высокоэнергетическое состояние.
Возбужденное состояние неустойчиво и электрон возвращается в основное
состояние. Так возникает спектр испускания (эмиссионный спектр).
Абсорбционные и эмиссионные переходы могут быть разделены
следующим образом:
Испускание: (высокоэнергетическое состояние) -
Поглощение: (высокоэнергетическое состояние) ■
(низкоэнергетическое состояние)
»(низкоэнергетическое состояние)
Линии в спектрах поглощения и испускания могут быть определены при
помощи частоты v:
Переход
Переход
Поглощение Испускание
Полезно использовать следующие соотношения:
Е= h\
где Е- энергия, v - частота, /- длина волны, с — скорость света, h -
постоянная Планка.
Основные правила отбора для электронного спектра:
Дл = 0, 1,2,3,4...
Д/= +1 или -1 (правило отбора Лапорта)
Например, переход с уровня п = 3 на один из уровней п = 2 разрешен толь-
^» ко в случае, если / изменяется на ±1, что на примере конкретных атомных
Вырожденные энерге- орбиталей соответствует переходам Зр -» 2s или 3d -> 2р, но не
тические уровни в ато- 35 -> 2s или 3d -» 2s. Последние запрещены в соответствии с
ме Н: см. разд. 2.14 уравнением 2.11. (Чему равно А/для каждого из этих перехо-
2.16. Атомный спектр водорода и правила отбора
89
Соотношение между
энергией и частотой:
E=/7V
дов?) Конечно, в атоме водорода атомные уровни с одним значением п
вырождены и переход с An = О (скажем, 2р -> 25) не приводит к изменению
энергии и не может давать начало наблюдаемой спектральной линии.
Линии в спектре испускания атомарного водорода разбиваются на
несколько серий; две из этих серий показаны (частично) на рис. 2.17. Эти
линии определены в табл. 2.3. Относительные энергии переходов
различаются, и поэтому каждая группа спектральных линий
появляется в определенной части электромагнитного спектра
(см. т. 2, приложение 1). Серия Лаймана линий типа п' —» 1
(2 —> 1, 3 —> 1, 4 —> 1...) отвечает энергетическим переходам,
соответствующим частотам в ультрафиолетовой области. Серия Бальмера
линий типа п' -> 2 (3 -> 2, 4 -> 2, 5 -» 2...) отвечает энергетическим переходам,
соответствующим частотам в видимой области. Конечно, в атоме водорода
nd-, пр- и /ю-орбитали имеют одинаковую энергию и переходы пр —> (п— \)s и
nd —> (п— \)р происходят при одинаковой частоте для данного значения п.
Серия Лаймана имеет особое значение, так как может быть использована
для определения потенциала ионизации атома водорода, т. е. энергии,
требуемой для полного удаления 1 s-электрона из атома. Частоты линий серии
/ = 0
/=1
1=2
Серия Бальмера
п = ©о (континуум)
Серия Лаймана
п= 1
Рис. 2.17. Некоторые разрешенные переходы, приводящие к образованию серий
Лаймана и Бальмера в спектре испускания атомарного водорода. Отметим, что
должны соблюдаться правила отбора (уравнения 2.10 и 2.11)
2. Атомы и атомная структура
Таблица 2.3. Серии линий, наблюдаемые в спектре испускания атома водорода
Название серии ri->n Область, в которой наблюдаются переходы
Лаймана 2->1, 3->1, 4->1, и т. д.
Бальмера 3—>2, 4—>2, 5-»2, и т. д.
Пашена 4->3, 5->3, 6-»3, и т. д.
Брэкетта 5->4, 6—>4, 7—>4, и т. д.
Пфунда 6->5, 7->5, 8->5, и т. д.
Ультрафиолетовая
Видимая
Инфракрасная
Дальняя инфракрасная
Дальняя инфракрасная
Лаймана позволяют определить частоты, соответствующие спектральным
переходам п' -> л = 2 -> 1,3-> 1,4-> 1. Значения могут быть
экстраполированы для определения частоты, соответствующей переходу «> —> 1. Этот метод
разобран в примере 2.3.
Первый потенциал ионизации атома - энергия, требуемая для
полного удаления наиболее просто отделяемого электрона из атома в
газовой фазе. Для атома X это соответствует процессу
Х(газ) -> Х+(газ) + е~
2.3. Потенциал ионизации водорода
Частоты некоторых спектральных линий серии Лаймана для
атомарного водорода равны 2,466, 2,923, 3,083, 3,157, 3,197, 3,221 и
3,237 (х 1015) Гц. Используя эти данные, определите потенциал
ионизации атомарного водорода.
Решение
Данные представляют собой частоты v спектральных переходов.
Задача заключается в определении потенциала ионизации атомарного
водорода, который соответствует величине v для перехода п' —> п = <» —»l.
Уровень /i' = ©о (континуум) соответствует полному удалению электрона из
атома. По мере приближения к этому уровню разница между двумя соседними
уровнями становится все меньше, пока (на бесконечности) не окажется равной
нулю. В этой точке ядро не влияет на энергию электрона и она перестает быть
квантованной. Спектральные переходы на уровень п = I с уровней близких к п
= °о, имеют практически одинаковую энергию.
Для определения первого потенциала ионизации атома водорода необходимо
найти точку, в которой разность энергий для двух последовательных переходов
на соседние уровни приближается к нулю. Это соответствует сходимости
предела по частоте.
Сначала вычислим разности Av между последовательными значениями
известных V.
(2,923 - 2,466)
(3,083 - 2,923)
(3,157-3,083)
(3,197-3,157)
(3,221-3,197)
(3,237-3,221)
10,5 = 0,457
10,5 = 0,160
1015 = 0,074
1015 = 0,040
1015 = 0,024
1015 = 0,016
1015Гц
1015Гц
1015 Гц
1015 Гц
1015Гц
10,5Гц
2.17. Многоэлектронные атомы
Теперь построим график зависимости Av от v.
0,5 г
91
Кривая достигает нуля при v = 3,275 • 1015 Гц. Тогда соответствующая этому
значению v энергия будет определяться как
Е = Mr = (6,626 10-34Джс)(3,275- 1015 с"1)
Но это величина в расчете на атом.
Потенциал ионизации должен быть выражен в единицах энергии на моль, что
означает необходимость умножения на число Авогадро (т. е. на число атомов в
моле).
Потенциал ионизации
водорода = (6,626 • 10-34)(3,275 • 10,5)(6,022 • 1023Дж/моль)
= 1,310- 106Дж/моль
= 1310 кДж/моль
МНОГОЭЛЕКТРОННЫЕ АТОМЫ
Ранее было сказано, что нейтральный атом, содержащий более одного
электрона, называется многоэлектронным, и понятно, что под эту категорию
попадают все атомы, кроме водорода. Было показано, что должно быть учтено
электростатическое отталкивание между электронами в многоэлектронном
атоме, и решение уравнения Шредингера для таких атомов невозможно. При
попытке получить решения относительно Е и х\г необходимо использовать
92
2. Атомы и атомная структура
различные приближения, и, по крайней мере для атомов, можно получить
численные решения достаточно высокой точности.
Нет смысла далее углубляться в эту проблему, следует лишь еще раз
отметить важные результаты разд. 2.15.Следствием эффектов проникновения орби-
талей к ядру и экранирования является то, что во всех атомах, в которых
число электронов более одного, орбитали с одним значением я, но разными
значениями / имеют различную энергию (рис. 2.15).
ПРИНЦИП ПОСЛЕДОВАТЕЛЬНОГО ЗАПОЛНЕНИЯ
НАИНИЗШИХ ОРБИТАЛЕЙ
Этот принцип включает несколько правил для определения электронной
структуры основного состояния атомов. Рассмотрим их на примере атомных
(а не молекулярных) орбиталей.
Принцип последовательного заполнения наинизших орбиталей используется
в сочетании с двумя другими — правилом Хунда и принципом Паули. Они
могут быть суммированы следующим образом:
• Орбитали заполняются, начиная с имеющих самую низкую энергию.
w • Если имеется набор вырожденных орбиталей, спаривание электронов на
о w орбитали невозможно до тех пор, пока каждая орбиталь не
«Вырожденный» ,_к КД v
будет содержать один электрон. Одиночные электроны, за-
значит «имеющий ту-7 к ,_ ^ „ г >
нимающие набор вырожденных орбиталей, имеют
одинаковые (параллельные) спины. Это правило Хунда.
• Согласно принципу Паули, два электрона в атоме не могут иметь
одинаковый набор значений квантовых чисел я, /, mt и ms. Это означает, что
каждая орбиталь может нести максимум два электрона с различными
значениями ms (различными спинами).
Первое из приведенных выше утверждений не требует дальнейшего
разъяснения. Второе утверждение может быть проиллюстрировано на примере двух
электронов, занимающих несколько вырожденных /ьорбиталей. Электроны
могут быть обозначены стрелочками, ориентация которых показывает
направление спина (ms = +1/2 или —1/2 ). На рис. 2.18,а показано правильное
расположение двух электронов в соответствии с правилом Хунда. Два других
возможных варианта (рис. 2.18,5 и 2.18,в) противоречат этому правилу (хотя
и соответствуют возможным возбужденным состояниям атома).
Принцип Паули запрещает двум электронам с одинаковыми спинами
занимать одну орбиталь, ведь тогда они имели бы одинаковый набор из
четырех квантовых чисел. На рис. 2.19 приведены разрешенное и запрещенное
расположение электронов на ls-орбитали.
Разрешено Запрещено Запрещено
а б в
Рис. 2.18. Правило Хунда: два электрона на вырожденных /7-орбиталях в основном
состоянии должны располагаться на различных орбиталях и иметь одинаковые
(параллельные) спины.
2.18. Принцип последовательного заполнения наинизших орбиталей
93
Рис. 2.19. Принцип
Паули: два электрона,
занимающих
15-атомную орбиталь, имеют
различные наборы из
четырех квантовых
чисел л, /, /и, и ms только в
том случае, если их
спины различны.
Запрещено
А А
1 V ' '
is
Электрон 1 Электрон 2
\ п=\ п=\
1 = 0 1 = 0
\ mi = 0 w/ = 0
ms = +1Л ms = +lA
Разрешено
А
15
T
Электрон 1 Электрон 2
п=\ п=\
1 = 0 1 = 0
nij = 0 mt = 0
ms = +'/2 ms = ~ХЛ
Принцип Паули: никакие два электрона в атоме не могут иметь
одинаковый набор квантовых чисел п, /, mt и ms.
Это означает, что каждый электрон в атоме имеет уникальный набор
из четырех квантовых чисел.
Правило Хунда: при заполнении вырожденных орбиталей спаривание
электронов невозможно до тех пор, пока каждая орбиталь не будет
нести по одному электрону. Одиночные электроны, занимающие
вырожденные орбитали, имеют параллельные спины.
Принцип последовательного заполнения орбиталей с наинизшей
энергией - группа правил, которые должны выполняться при
заполнении электронами атомных или молекулярных орбиталей. Он сочетает в
себе правило Хунда и принцип Паули со следующими дополнениями:
• орбитали заполняются последовательно, начиная с самой низшей
по энергии;
• орбиталь, содержащая два электрона, полностью занята.
Пример 2.4. Использование принципа последовательного
заполнения наинизших орбиталей
Определите расположение электронов в атоме азота в основном
состоянии.
Решение
Атомный номер азота 7; следовательно, у него 7 электронов.
Самую низкую энергию имеет 15-орбиталь (п = 1, /= 0, т{ = 0).
Максимальное число электронов на ls-орбитали - два.
Следующая низшая по энергии орбиталь - 25 (я = 2, / = 0, т{ = 0).
94
2. Атомы и атомная структура
Максимальное число электронов на 2$-орбитали — два.
Далее следует набор из трех вырожденных 2/ьорбиталей (п = 2, /= 1, т1 = + 1,
0,-1).
На 2р-орбитали можно расположить максимально 6 электронов, но осталось
только 3 (из 7 электронов азота по два уже расположено на 15- и 2$-орбиталях).
Оставшиеся электроны займут 2р-орбиталь так, чтобы было соблюдено
правило Хунда. Каждый электрон займет отдельную орбиталь и все они будут иметь
параллельные спины.
Расположение электронов в атоме азота может быть представлено
следующим образом:
Энергия
2?
2s
\s
ЭЛЕКТРОННАЯ КОНФИГУРАЦИЯ
Электронная конфигурация основного состояния
Принцип последовательного заполнения наинизших орбиталей является
методом, позволяющим предсказать порядок заполнения атомных орбиталей
электронами и определить расположение электронов в основном состоянии
^~ (т. е. электронную конфигурацию основного состояния атома). Результат может
Z- атомный номер быть представлен в виде диаграммы, как это сделано для
основного состояния атома углерода (Z= 6) на рис. 2.20.
Электронная конфигурация основного состояния атома соответствует
наиболее низкоэнергетическому состоянию.
Ниже приведен другой способ представления электронной
конфигурации. Для атома водорода (разд. 2.14) наличие одного электрона на ls-орбита-
ли может быть отображено символом Is1. Такие символы могут быть
использованы для обозначения электронной конфигурации любого атома. Так, для
углерода (рис. 2.20) электронная конфигурация может быть записана
следующим образом: Xp-l^lp1. Аналогичным образом, электронные
конфигурации гелия и лития (рис. 2.21): Is2 и \s22sx соответственно.
2.19. Электронная конфигурация
95
Энергия
Рис. 2.20. Расположение электронов
на орбиталях в основном состоянии
атома углерода.
2р
2s
\s
С (Z = 6)
Обычно порядок заполнения атомных орбиталей в основном состоянии
атома следующий (начиная с низкоэнергетических):
15 < 25 < 2р < 35 < Ър < 45 < 3d< 4р < 55 < 4d< 5р < 6s < 5d~ 4/< 6p<7s< 6d= 5/
Следует особо отметить, что эта последовательность может меняться от
атома к атому, потому что на энергию электронов на орбиталях влияет заряд
ядра, присутствие других электронов на той же орбитали (или том же
подуровне) и суммарный заряд. Этот факт будет упомянут при рассмотрении
некоторых аспектов химии ^-элементов (гл. 16).
Обычный порядок заполнения (начиная с низкоэнергетических)
атомных орбиталей:
1s<2s<2p<3s<3p<4s<3d<4p<5s<4p<5p<6s<5rf«4f<6p<7s<6d«5f
Энергия
15
Энергия
2s
\s
Не (Z = 2) Li (Z = 3)
Рис. 2.21. Расположение электронов на атомных орбиталях гелия и лития в
основном состоянии.
96 2. Атомы и атомная структура
Таблица 2.4. Электронные конфигурации основного состояния для первых 20
элементов.
Атомный
номер
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
Символ
элемента
Н
Не
Li
Be
В
С
N
О
F
Ne
Na
Mg
Al
Si
P
S
CI
Ne
К
Ca
Электронная
конфигурация
\?
h2
\s22s]
\s22s2
\s22s22px
\s22s22p2
\s22s22p3
\s22s22p4
\s22s22p5
\s22s22p6
\s22s22p63s]
\s22s22p63s2
\s22s22p63s23p]
\s22s22p62s23p2
\s22s22pe3s23p3
\s22s22p6ls2lp4
\s22s22pe3s23p5
b22^2/763.V23/76
\s22s22pb?>s2lp4sx
lj22s22/763s23/?64s2
Сокращенная запись
электронной
конфигурации
\?
Ь2 = [Не]
[Не]2лл
[He]2s2
[He]2s22p]
[He]2s22p2
[He]2s22p3
[He]2s22p4
[He]2s22p5
[He]2s22p6 = [Ne]
[Nep*1
[Ne]3s2
[Ne]3523/71
[Ne]3s23/?2
[Ne]3s23/?3
[Ne]3s23/?4
[Ne]3*23/?5
[Ne]3s23/>6 = [Ar]
[Аг]4^
[Ar)4s2
Зная последовательность орбиталей и правила их заполнения, можно
записать электронные конфигурации основного состояния большинства
атомов. Для элементов с атомными номерами от 1 до 20 они приведены в табл.
2.4. При движении вниз по таблице электронная конфигурация низших
заполненных квантовых уровней не меняется. Благодаря этому может быть
использована сокращенная запись. Так, выделяются только электроны,
занимающие новый квантовый уровень (с новым значением п), а внутренние
уровни («остов») обозначаются предыдущим элементом из группы 18 (Не, Ne
или Аг).
Электронные конфигурации основного состояния элементов с атомным
номером >20 мы не будем пока рассматривать, так как приведенная выше
закономерность для них не так надежна.
Валентные электроны и электроны внутренних оболочек
Теперь будут введены понятия валентной электронной конфигурации и
валентных электронов, имеющие глубокий смысл. Валентные электроны в атоме -
это такие электроны, которые расположены на внешних
(высокоэнергетических) квантовых уровнях; именно они прежде всего определяют химические
свойства элемента. Рассмотрим натрий (Z= 11) — его основная электронная
^» конфигурация 1522522р6351. Главные квантовые уровни с п = 1 и 2 полностью
Валентные электроны заняты и содержат остовные электроны (электроны внутрен-
и периодичность: них оболочек). Натрий имеет один валентный (3s) электрон, и
см. разд. 2.22 это согласуется с его химическими свойствами.
2.20. Правило октета
97
Пример 2.5. Электронная конфигурация основного состояния
Определите электронную конфигурацию основного состояния
натрия (Z= 11).
Решение
Имеется 11 электронов.
Последовательность заполнения орбиталей \s < 2s < 2р < 3s.
Максимальная заселенность s-уровня - два электрона.
Максимальная заселенность/^-уровня - шесть электронов.
Значит, электронная конфигурация основного состояния натрия
1522522р6351,или^е]351.
ЕВЗ ПРАВИЛО ОКТЕТА
При внимательном рассмотрении электронных конфигураций основного
состояния (табл. 2.4) может быть выявлена ясная тенденция, отраженная в
правой колонке таблицы. Заполненные главные квантовые уровни
обозначаются как «строительные блоки» внутри электронных конфигураций
последующих элементов. Данная тенденция - один из примеров периодичности —
повторяемости конфигурации внешних электронов для различных значений
главного квантового числа.
Видна повторяемость последовательностей ns(\ + 2) и nstnpil + 6). Общее
число электронов, которые могут располагаться на орбиталях с п = 1, — два,
но для п = 2 общее число электронов вырастает до восьми (2s22p6). Число
восемь также соответствует числу электронов, необходимых для заполнения 3s-
и 3/>-орбиталей. Это основа правила октета.
Как предполагает название*, правило октета основывается на
наблюдении, что атомы 5- и р-элементов имеют тенденцию отдавать, принимать или
объединять электроны так, чтобы заполнить восьмью электронами внешнюю
оболочку.
Электронная конфигурация основного состояния фтора записывается как
[Не]2522/?5. До полной конфигурации [Не]2522/?6, т. е. до образования октета
внешних электронов, не хватает одного электрона. Фтор легко принимает
один электрон и образует ион F~ с электронной конфигурацией [He]2s22/?6.
Атому азота ([Не]2522/?3) требуется три электрона для получения конфигурации
[He]2s22/?6, и легко может быть получен ион N3-. Атому углерода ([He]2s22/?2)
► необходимо для достижения конфигурации [Не]2522/?6 образовать ион С4-, что
Образование ионов: энергетически не выгодно. Атому углерода удобнее обобщест-
см. гл. 6 вить (объединить) четыре электрона с другим атомом, завершив
формирование октета без образования иона.
Атом удовлетворяет правилу октета, когда он, путем присоединения,
потери или обобществления электронов с другими атомами получает
конфигурацию ns2np6, т.е. имеет 8 электронов на внешней
электронной оболочке.
Слово «октет» происходит от латинского octo, что значит восемь.
98
2. Атомы и атомная структура
Несмотря на все достоинства, концепция октета имеет некоторые
ограничения. Она может быть удовлетворительно применена к главному квантовому
уровню п = 2. Уровень п = 2 полностью занят, если он содержит восемь
электронов. Но исключения есть даже здесь: элементы литий и бериллий,
содержащие внешние 25-электроны (табл. 2.4), с большей легкостью теряют
электроны и приобретают устойчивую электронную конфигурацию гелия (Ь2);
молекулы типа BF3 с шестью валентными электронами тоже стабильны.
А как работает правило октета на квантовом уровне п = 3? Здесь атомы
натрия и магния (табл. 2.4) легче теряют электроны, образуя конфигурацию
неона. Алюминий с конфигурацией основного электронного состояния
[Ne^s^/?1 может отдать три электрона и образовать А13+ (т. е. «неоновую»
частицу) или может участвовать в образовании связи, чтобы достигнуть октета
путем объединения электронов. Но алюминий также может образовывать
соединения, не отвечающие формально правилу октета, например А1С13. Атомы
элементов с основными электронными состояниями между [Ne^s^/?2 и
^ [Ne^s^/?6 могут также объединять или принимать электроны для образова-
Галогениды алюминия: ния октета, но здесь есть много сложностей. Для п = 3 сущест-
см. разд. 13.4 вует другой подуровень — 3*/-орбитали, — и это приводит к
возможности расширения октета.
Итак, правило октета — удобный инструмент для предсказания возмож-
^~ ности образования ионов или ковалентной связи, но оно ограничено сравни-
Расширение октета: тельно малым числом элементов. Однако принцип правила ок-
см. разд. 5.16 тета очень важен и может быть расширен до правила 18
электронов, которое учитывает заполнение ns-, np-, и я^-подуров-
ней.
Пример 2.6. Правило октета
Предположите, почему фтор образует молекулу F2, а кислород
может образовывать дифторид OF2 (для F Z = 9, для О Z = 8).
Решение
Запишем электронные конфигурации основного состояния F и О.
F 1522522/75
О 1522522/
Фтор может получить октет электронов, объединив электрон с другим атомом
фтора. Таким образом, квантовый уровень /7 = 2 оказывается завершенным.
Кислород может достигнуть октета внешних электронов, объединив
электроны с двумя атомами фтора в молекуле OF2. При этом оказывается достигнутым
октет для каждого атома F, так как каждый атом получает электрон за счет
спаривания с электронами кислорода.
|Я1 ОДНОАТОМНЫЕ ГАЗЫ
Благородные газы
При внимательном рассмотрении электронных конфигураций основного
состояния видно, что заполненные электронные оболочки являются «строи-
2.21. Одноатомные газы 99
Таблица 2.5. Электронная конфигурация основного состояния благородных газов
Благородный
газ
Гелий
Неон
Аргон
Криптон
Ксенон
Радон
Символ
Не
Ne
Аг
Кг
Хе
Rn
Атомный
номер
2
10
18
36
54
86
Электронная конфигурация
основного состояния
\s2
\s22s22p6
1л22л-22/76Зл23/76
1s22s22p63s23p4s23d]04pb
\s22s22pb3s23pb4s23dw4p65s24dw5pe
\s22s22pb3s23p4s23dl04p65s4dl05pb6s24f45dw6p6
тельными блоками» для конфигураций более тяжелых элементов. Элементы,
имеющие эти целиком заполненные оболочки, относятся к одной группе
периодической таблицы - группе 18. Это так называемые благородные газы*
Электронные конфигурации основного состояния благородных газов
приведены в табл. 2.5. Отметим, что у каждого из этих элементов (исключая
гелий) электронная конфигурация содержит блок nsPnp6 для наибольшего
значения п\ например, конфигурация аргона оканчивается на 3s23pe. Эта
конфигурация обычно называется «конфигурацией инертного
(благородного) газа». Наличие заполненных d- и/-оболочек в дополнение к s- и
^-оболочкам у более тяжелых элементов не имеет значения в данном случае.
Тот факт, что каждый благородный газ имеет заполненную внешнюю яр-
оболочку объясняет тот факт, что элементы группы 18 существуют в
элементном состоянии как моноатомные частицы — нет движущей силы для
образования связи между атомами.
Силы, действующие между атомами
Благородные газы (за исключением гелия) затвердевают только при
исключительно низких температурах. Температуры плавления неона и аргона со-
^ ставляют 24,5 К и 84 К соответственно. Столь малые величины показывают,
Межатомные что силы межатомного взаимодействия в твердом состоянии
взаимодействия: очень слабы, и легко преодолеваются при переходе в жидкое
см. разд. 1.21 состояние. Удивителен узкий интервал температур, в котором
элементы 18 группы представляют собой жидкости. Сеть
межатомных взаимодействий в жидкости очень слаба и процесс
испарения проходит очень легко из-за низких значений
энтальпии (табл. 2.6).
Какова природа и форма сил взаимодействия между атомами?
Рассмотрим два атома аргона. Каждый представляет собой сферу с положительно
заряженным ядром в центре, окруженным отрицательно заряженными
электронами. На большом расстоянии есть только слабое взаимодействие между
атомами. Однако при сближении атомов их электронные облака начинают
отталкиваться. На очень малых межъядерных расстояниях сила
отталкивания, обусловленная взаимодействием электронов, будет очень велика. Хоро-
ДиспН:см. разд. 1.21
Термин «благородные газы» рекомендован ИЮПАК, хотя чаще их называют «инертными газами»;
см. разд. 1.10.
100
2. Атомы и атомная структура
Таблица 2.6. Некоторые физические свойства благородных газов
Элемент Т„, К
Гелий -а
Неон 24,5
Аргон 84
Криптон 116
Ксенон 161
Радон 202
а Гелий не может быть
Ткип> К
4,2
27
87
120
165
211
переведен в
\fl> '
кДж/моль
-
0,3
1,1
1,4
1,8
-
4,сД
кДж/моль
0,1
2
6,5
9
13
18
Вандер-
ваальсов
радиус (гу),
пм
99
160
191
197
214
-
Первый
потенциал
ионизации,
кДж/моль
2372
2080
1520
1351
1170
1037
твердое состояние ни при каких давлении и температуре.
шее математическое приближение показывает, что сила отталкивания
зависит от межъядерного расстояния в соответствии с уравнением 2.12.
Потенциальная энергия, связанная с этой силой, изменяется по уравнению 2.13. Этот
потенциал отталкивания (который принято считать положительной
энергией) между двумя атомами аргона, представлен верхней кривой на рис. 2.22.
Сила отталкивания между атомами « \/d]3 (2.12)
Потенциальная энергия отталкивания ос \/dn (2.13)
Если бы в системе существовала только сила отталкивания, то элементы
благородного газа никогда не образовали бы кристаллическую решетку. Надо
признать, что также существует сила притяжения между атомами, которая
превосходит электронное отталкивание.
Отталкивание
Рис. 2.22. Потенциалы отталкивания
и притяжения между двумя атомами
аргона как функция межъядерного
расстояния d.
200 400 600 800
Межъядерное расстояние d, пм
2.21. Одноатомные газы
101
Атом 1 Атом 2 Атом 3 Атом 4
Рис. 2.23. Диполь, образующийся в атоме 1, индуцирует диполь в атоме 2, который
индуцирует диполь в атоме 3 и т. д. Расстояние rv - это вандерваальсов радиус, равный
половине межъядерного расстояния между двумя максимально сближенными
несвязанными атомами.
Мы можем следующим образом обрисовать возникновение силы
притяжения. Внутри атома аргона позиции электронов и ядра не фиксированы. В
^ любой выбранный момент центр электронной плотности может не совпадать
Дипольный момент: с центром положительного заряда, т. е. с ядром. Этот диполь
см. разд. 4.11 временный, но он может индуцировать диполь в соседнем
атоме (рис. 2.23) и в результате между атомами появится
электростатическое диполь-дипольное взаимодействие. Так как
диполь временный, то и каждое отдельное взаимодействие является
временным, но их сеть образует силу притяжения между атомами.
Асимметричность заряда в атоме или молекуле создает диполь: в
одной части атома или молекулы образуется повышенная плотность
отрицательного, а в другой - положительного заряда.
Диполь может быть временным (как в атоме аргона) или постоянным
(как в молекуле хлороводорода).
Сила притяжения, обусловленная диполь-дипольными взаимодействиями,
может быть представлена в виде уравнения 2.14. Это приводит к наличию
энергетического потенциала притяжения, который изменяется с
расстоянием (уравнение 2.15), и этот потенциал притяжения (который принято считать
отрицательной энергией) между атомами аргона показан на рис. 2.22 в виде
нижней кривой.
Сила притяжения между атомами «= \/сР (2.14)
Потенциальная энергия притяжения « \/(f> (2.15)
Наконец, на рис. 2.24 показан суммарный потенциал (притяжение плюс
отталкивание) между атомами аргона как функция межъядерного расстояния d.
На больших расстояниях доминирует притяжение. При сближении двух
атомов потенциал притяжения возрастает (по абсолютной величине) до
некоторой точки максимальной стабилизации (наинизшей энергии). Однако, если
атомы продолжают сближаться, возрастает отталкивание между
электронами. На очень коротких расстояниях электрон-электронное отталкивание
доминирует и это проявляется в очень нестабильном расположении атомов,
обладающем положительным потенциалом.
102
2. Атомы и атомная структура
ё О
О)
200
2
</ ^г400 600 800
Межъядерное расстояние */, пм
Рис. 2.24. Суммарный потенциал
взаимодействия двух атомов аргона как
функция межъядерного расстояния d.
Величина d в минимуме энергии (de)
соответствует удвоенному вандерваальсову радиусу
атома аргона.
Не Ne Аг Кг Хе
Рис. 2.25. Объемы атомов
благородных газов, определенные исходя
из вандерваальсовых радиусов.
Объем возрастает при движении вниз по
группе 18.
Вандерваальсов радиус
Величина d в точке на рис. 2.24, где потенциал достигает минимума,
соответствует оптимальному расстоянию dt между двумя атомами аргона. Это
несвязанное состояние. Отметим, что de — это не расстояние, на котором силы
притяжения и отталкивания равны и потенциал равен нулю.
Удобно определить dt в рамках атомных свойств - величина dJ2
определяется как вандерваальсов радиус rv атома аргона. Величина rv - это
половина наименьшего межъядерного расстояния между соседними атомами
аргона. Величины радиусов Ван-дер-Ваальса для благородных газов приведены в
табл. 2.6. В случае моноатомного благородного газа эти радиусы могут быть
использованы для определения размеров атома. При помощи
вандерваальсовых радиусов может быть определен объем атома: объем сферического атома
равен 4/зя/у3- Возрастание атомного объема при движении вниз по группе 18
представлено на рис. 2.25.
Вандерваальсов радиус rv атома X определяется как половина
минимального расстояния между двумя несвязанными атомами X.
2.22. Периодичность
103
ШЕЯа периодичность
В конце этой главы мы кратко рассмотрим определяющую роль электронных
конфигураций атомов в химии элементов и примеры, иллюстрирующие
некоторые важные выводы.
Благородные газы
Ранее уже были отмечены некоторое сходство между благородными газами и
принадлежность к одной группе периодической таблицы. Все эти элементы
обладают полностью заполненными атомными орбиталями на внешнем
квантовом уровне. То, что каждый благородный газ имеет внешнюю п£прь-
конфигурацию, показывает, что они подчиняются правилу октета.
Физические и химические свойства благородных газов отражают тот
факт, что это стабильные частицы. Однако для ксенона и в меньшей степени
для криптона наличие завершенного электронного октета еще не означает
полной потери химической активности. Некоторые реакции с участием
ксенона приведены в уравнениях 2.16—2.18, а причины, по которым ксенон и
криптон могут вступать в эти реакции, рассмотрены в разд. 13.13.
Xe + F2 -> XeF2 (2.16)
3XeF4 + 6Н20 -> Xe03 + 2Хе + \02 + 12HF (2.17)
XeF6+H20 -> XeOF4 + 2HF (2.18)
Группа 1: Щелочные металлы
^ Тенденции в химическом поведении элементов определяются числом ва-
s-Элементьг см гл 11 лентных электронов и порядком их расположения на орбита-
о-Элементьг см гл 13 лях* Периодическая таблица содержит 18 групп элементов,
d-Элементьг см гл 13 скомпонованных в "фи блока (исключая лантаниды и актини-
' iC ды): 5-, р- и ^-элементы.
И ID _ г
Для 5-элементов характерна конфигурация валентных
электронов ns1 (группа 1) или ns2 (фуппа 2). Отдельные данные по
химическим и физическим свойствам элементов группы 1 (щелочные металлы)
приведены в табл. 2.7. При 298 К все элементы данной группы - твердые
вещества, но их температуры плавления относительно низки, потому что легко
преодолеть их межатомные взаимодействия в твердом состоянии, например,
в некоторых частях света цезий жидкий при температуре окружающей среды.
Интервал температур, в котором элементы первой группы остаются
жидкостями, значительно выше, чем у благородных газов (табл. 2.6).
Элементы первой группы ведут себя одинаково, как и элементы группы
^ 18. Величины первого потенциала ионизации элементов груп-
Потенциал ионизации: . , - « -ч /
„ rt пы 1 (табл. 2.7) относительно низки (по сравнению с элемента-
СМ. раЗД. 6.3 ч t <> im
ми других групп), ионизация (уравнение 2.19) проходит легко.
Это согласуется с конфигурацией валентных электронов ос-
^ новного состояния данных элементов - ns1. На рис 2.26 про-
Тенденции в изменении водится сравнение первых потенциалов ионизации элементов
потенциала^онизации: групп i и ]g . Для атома благородного газа образование одно-
см. упражнения 2.11 и зарядного иона (реакция 2.19) является невыгодным - должна
2.12 в разд. 2.24 быть разрушена целиком заполненная электронная оболочка.
104
2. Атомы и атомная структура
Таблица 2.7. Физические и химические свойства элементов группы 1 (щелочные
металлы)
Элемент
Литий
Натрий
Калий
Рубидий
Цезий
Символ
Li
Na
К
Rb
Cs
' Ш.Л* *»
453,5
371
336
312
301,5
Т К
1кип* л
1615
1154
1032
961
978
Электронная Первый
конфигурация потенциал
[He]2s*
[Nep*1
[Ar]4sl
[Kr]5sl
[Xe]6sl
ионизации,
кДж/моль
520
496
419
403
376
ОбразуеХимическая
мые ионы формула
Li+
Na+
К+
Rb+
Cs+
хлорида
LiCl
NaCl
КС1
RbCl
CsCl
М(газ) -» М+(газ) + е~
(2.19)
Образование иона М+ элементами первой группы накладывает отпечаток на
все их химические свойства, уравнения 2.20—2.23 показывают лишь
несколько реакций с их участием.
(2.20)
(2.21)
(2.22)
2Na + С12 -)
2NaCl
2К + 2Н20 -> 2КОН + Н2
4Na + 02 ->
Li2C03——
2Na20
-^ Li20 + C02
(2.23)
Более детальное обсуждение периодичности проведено в гл. 11 и 13.
II
S с
X
250U
2000
1500
1000
500
л
• Не
N.Ne
\ Аг
•
Li Na v
- j -
Кг
"•••
Rb
Хе
Cs
i
Группа 1
•
Группа 18
о
25
50
75 100
Атомный номер
Рис. 2.26. Тенденции в изменении первых потенциалов ионизации элементов в
группах 1 и 18.
2.23. Заключение
105
^Е1 ЗАКЛЮЧЕНИЕ
В этой главе были рассмотрены некоторые
сложные концепции, однако они
необходимы для понимания химии элементов. При
рассмотрении других разделов данной кни-
Теперь вы должны уметь:
• кратко объяснить, что такое корпускулярно-
волновой дуализм;
• кратко объяснить, что такое принцип
неопределенности;
• кратко объяснить, что такое решение
волнового уравнения Шредингера;
• описать значение радиальной и волновой
частей волновой функции;
• определить, что такое главное квантовое
число;
• определить, что такое орбитальное и
спиновое квантовые числа и соотнести их
разрешенные значения с главным квантовым
числом;
• описать атомную орбиталь и электрон(ы) с
точки зрения величин квантовых чисел;
• кратко объяснить, почему используют
нормированные волновые функции;
• графически изобразить функции
радиального распределения для 1s-, 2s-, 3s-, 2p-,
Зр-, Зс/-орбиталей (что они означают?);
• обрисовать экранирующий эффект
электронов;
• нарисовать форму, показывающую знак,
для ns- и лр-атомных орбиталей;
• характеризовать лрх-, лру-, лр2-атомные ор-
битали;
• объяснить, почему в водородном атоме ор-
битали с одним главным квантовым числом
вырождены;
ги представится возможность много раз
использовать эти концепции и будет ясно,
почему они введены в самом начале.
• объяснить, почему в многоэлектронном
атоме заполненные орбитали с одинаковым
главным, но разными орбитальными
квантовыми числами не вырождены;
• определить первый потенциал ионизации
атома водорода исходя из набора частот
серии Лаймана спектральных линий;
• описать, как использовать принцип
последовательного заполнения наинизших
орбиталей для определения конфигурации
основного электронного состояния атома;
• написать порядок следования по энергии
орбиталей от 1s до 4р. Указать, какая из них
имеет низшую энергию и что влияет на эту
последовательность;
• для данного атомного номера (до Z = 20)
записать символьную форму электронной
конфигурации основного состояния;
• для данного атомного номера (до Z = 20)
нарисовать диаграмму энергетических
уровней и показать электронную конфигурацию
основного состояния;
• объяснить, как возникает кривая
энергетического потенциала (рис. 2.24) и каково
значение энергетического минимума;
• объяснить значение периодичности на
примере элементов групп 1 и 18.
Что означают след
• Вектор
• Скаляр
• Квантованность
• Фотон
• Волновая функция
• Собственная функция
• Собственное значение
• Квантовые числа л, /, mf
и ms
• Функция радиального
распределения
• Атомная орбиталь
• Узловая плоскость
• Доля (орбитали)
• Знак орбитали
• Граничная поверхность
орбитали
1ие термины?
• Вырожденные орбитали
• Невырожденные орбитали
• Диффузность (по
отношению к атомной орбитали)
• Проникновение орбитали
к ядру
• Экранирование
• Эффективный ядерный
заряд
• Водородоподобный атом
или ион
• Многоэлектронный атом
• Первый потенциал
ионизации
• Принцип
последовательного заполнения
наинизших орбиталей
• Правило Хунда
• Принцип Паули
• Электронная
конфигурация основного состояния
• Правило октета
• Электронная
конфигурация благородного
(инертного)газа
• Электроны внутренних
оболочек (остовные
электроны)
• Валентные электроны
• Вандерваальсов радиус
• Наведенный дипольный
момент
106
2. Атомы и атомная структура
Ю9 УПРАЖНЕНИЯ
2.1. Объясните разницу между векторной
и скалярной величинами, приведите
примеры.
2.2. Определите длину волны
электромагнитного излучения с частотой
а) 2- 1013 Гц, б) 4,5- 1015 Гц,
в) 2,1 • 1017 Гц. По спектру в
приложении 1 (см. т. 2) определите для
каждой волны тип излучения
(например, рентгеновское).
2.3. Определите возможные значения /и
/и, для п = 4. Исходя из этого,
определите возможное число 4/-орбиталей.
2.4. Определите набор из четырех
квантовых чисел, определяющих каждый
электрон в атомах а) ^Не и б) ^Вв
их основных состояниях.
2.5. Определите энергии уровней
водорода для п = 1, 2 и 3. {Указание: см.
уравнение 2.8.)
2.6. Используя данные примера 2.3,
нарисуйте зависимость Дл от большего
значения частоты и определите
первый потенциал ионизации водорода.
2.7. Объясните следующие понятия:
а) волновая функция, б) радиальная и
угловая части волновой функции, в)
атомная орбиталь, г) радиальный узел,
е) проникновение орбиталей к ядру и
экранирование, ж) радиальное
распределение электронной плотности.
2.8. Определите электронную
конфигурацию основного состояния для Be
(Z=4), F(Z=9), P(Z= 15),
К (Z= 19), записав ответ а) в
символьной форме и б) в форме
диаграммы энергетических уровней.
2.9. Как вы понимаете «правило октета»?
2.10. Фосфор образует хлориды РС13 и
РС15. Подчиняются ли эти
соединения правилу октета?
2.11. Используя данные табл. 2.6, 2.7 и
рис. 2.26, предскажите первый
потенциал ионизации франция Fr.
2.12. Посмотрите на рис. 2.26.
Предположите, почему убывает первый
потенциал ионизации для а) элементов
первой группы и б) благородных газов.
ВГОМОЯДЕРНАЯ КОВАЛЕНТНАЯ
СВЯЗЬ
ШЯШ ВВЕДЕНИЕ
В разд. 1.12 было введено понятие «молекула». Также утверждалось, что кова-
лентная связь образуется при обобществлении электронов атомами.
Молекула — дискретная нейтральная частица, образующаяся за счет
ковалентных связей между двумя или несколькими атомами.
В этой главе более детально разбирается представление о ковалентном
связывании.
Рассмотрим образование связи между одинаковыми атомами — гомоядер-
ные связи, например, связь С—С в этане С2Н6, связь N—N в гидразине N2H4,
связь О—О в пероксиде водорода Н202 (рис. 3.1). На рис. 3.2 приведены
примеры гомоядерных молекул, т. е. молекул, построенных из одинаковых атомов.
Каждая из этих молекул — молекулярная форма элемента. Для некоторых
элементов известно несколько аллотропных модифика-
Аллотропные модификации: ций (например, 02 и 03).
см. разд. 1.5 Мы попытаемся сначала «увидеть» ковалентные
связи глазами экспериментатора, а затем обсудим их с
использованием теоретических моделей.
Согласно представлению об образовании связей в молекулах
наблюдаемые в них межатомные расстояния значительно короче тех, которые можно
ожидать из вандерваальсовых радиусов атомов. (В гл. 2 были приведены ван-
Рис. 3.1. Молекулы с одной гомоядерной связью: этан С2Н6, гидразин N2H4, пероксид
водорода Н202.
108 3. Гомоядерная ковал ентная связь
©=© ©Н=®
н2 о2
Оз
Рис. 3.2. Примеры гомоядерных молекул с ковалентными связями.
(ИЧ)
3.2. Измерение межъядерных связей
109
дерваальсовы радиусы элементов, образующих только одноатомные частицы
простого вещества, но эти радиусы могут быть определены и для
большинства других элементов. На практике их определяют, исходя из кратчайших
межмолекулярных расстояний.)
Из этого следует, что межатомное расстояние является очень важным
параметром при описании строения вещества. Что касается химических связей,
то их длина (или, что более правильно, усредненное во времени межатомное
расстояние) является их фундаментальной характеристикой.
В следующем разделе представлено несколько экспериментальных
методов определения межатомных и межмолекулярных расстояний. Если вы не
хотите прерывать обсуждение химического связеобразования, то переходите
к разд. 3.3, а разд. 3.2 может быть проработан позже.
EBI ИЗМЕРЕНИЕ МЕЖЪЯДЕРНЫХ РАССТОЯНИЙ
►- Существует ряд методов определения важных структурных параметров. Все
Колебательная спект- эти методы могут быть разделены на две большие группы: спек-
роскопия в приложе- тральные и дифракционные. Детальное рассмотрение экспери-
нии к двухатомным мо- ментальных процедур выходит за рамки этой книги. В этом
лекулам: см. разд. 9.6 разделе приведены лишь основные характеристики методов и
получаемых данных.
Обзор дифракционных методов
Для определения структуры твердых веществ чаще всего используются два
метода: дифракция рентгеновских лучей и дифракция нейтронов. Для изучения
строения молекул в газовой фазе больше всего подходит дифракция
(рассеяние) электронов ( хотя этот метод имеет более широкое применение):
электроны имеют отрицательный заряд, а кванты рентгеновского излучения и
нейтроны электронейтральны. Поэтому рентгеновское излучение и нейтроны
могут глубже внедряться в молекулы, так как не испытывают
электростатического отталкивания от валентных (внешних) и остовных (внутренних)
электронов. Говорят, что рентгеновские лучи и нейтроны «обладают большей
проникающей способностью» по сравнения с электронами. Некоторые
сравнительные характеристики трех дифракционных методов приведены в табл. 3.1.
При выборе дифракционного метода исследования следует иметь в виду,
что типичное расстояние между связанными атомами равно 100-300 пм
(1 пм = Ю-12 м). Длина волны излучения, которое планируется использовать,
должна иметь величину, близкую к длине исследуемых связей. Детали
приведены в дополнении 3.1.
Необходимо подчеркнуть некоторые детали, влияющие на тип
информации, которая может быть получена каким-либо дифракционным методом:
• Электроны несут отрицательный заряд, и их дифракция определяется
суммарным электростатическим полем, создаваемым электронами и
положительно заряженными ядрами изучаемого вещества.
• Рентгеновские лучи - электромагнитное излучение. Они рассеиваются
главным образом электронами, независимо от того, имеет вещество
110
3. Гомоядерная ковалентная связь
Таблица 3.1. Сравнение электронной, нейтронной и рентгеновской дифракции
Дифракция
электронов
Дифракция
рентгеновских лучей
Дифракция нейтронов
Примерная длина
волны, м
Тип образцов для
исследования
Положение чего
определяется
Преимущества
Недостатки
ю-"
Обычно газы, редко
жидкости и
поверхности твердых тел
Электронная
плотность
Получение
структурных данных о
молекулах в газовой фазе.
Другие методы не
применимы для таких
исследований
Уточнение данных для
больших молекул
затруднительно, неточное
определение
положения атомов водорода
,0-ю
Обычно
монокристаллы и порошки, редко
растворы
Электронная
плотность
Метод широко
распространен и
сравнительно прост, возможно
изучение
молекулярных и ионных
кристаллов, возможно
получение точных параметров
связи для
неводородных атомов
Положение атомов
водорода не всегда
определяется точно
,0-ю
Обычно монокристаллы
и порошки
Ядра
Возможно определение
положение атомов
водорода, очень точное
определение структурных
параметров, возможно
изучение молекулярных и
ионных кристаллов
Метод дорог, уточнение
данных для больших
молекул затруднительно
Дополнение 3.1. Дифракционные методы
Дифракционные методы могут быть использованы для определения
межатомных расстояний, но длина волны применяемого излучения должна быть
того же порядка величины, что и межатомные расстояния.
Обычно длины химических связей лежат в интервале 100-300 пм.
Поскольку длина волны рентгеновского излучения равна примерно 100 пм, то
оно может быть использовано для определения межъядерных расстояний в
твердых веществах.
Длина волны пучка электронов варьирует при изменении ускоряющего
напряжения, величина порядка 10 пм является вполне обычной и
используется в дифракционных экспериментах.
Существует возможность замедления быстрых нейтронов, генерируемых в
ядерных реакторах, вплоть до длин волн порядка 100 пм. Кроме того, для
дифракционных экспериментов существуют и специально разработанные
источники медленных нейтронов.
Для более детального знакомства с дифракционными методами
исследования вещества см. Ebsworth E.A.V., Rankin D.W.H., Cradock S., Structural
Methods in Inorganic Chemistry, 2nd edn., Blachwell Scientific Publications,
Oxford (1991), а также Порай-Кошиц МЛ. Основы структурного анализа
химических соединений, М., Высшая школа, 1989.
3.2. Измерение межъядерных связей
111
Дополнение 3.2. Сложность определения положения атомов
водорода методами рентгеновской и электронной дифракции
Рассмотрим связь С—Н. Электронная конфигурация основного состояния
атома углерода l522522/?2, а атома водорода Is1. Углерод имеет четыре
валентных электрона и два электрона на внутреннем уровне. У атома водорода
внутренних электронов нет вообще.
Водород использует свой единственный электрон на образование связи
С—Н, что приводит к дальнейшему снижению на нем электронной
плотности.
Связывающие электроны
С
\7
Н
—•
ZV
/V
15-
электроны
(внутренние)
вокруг
ядра
Нет
внутренних
электронов
вокруг
ядра
При попытках определения длины связи С—Н методами рентгеновской или
электронной дифракции возникают серьезные трудности. Оба метода
определяют положение областей с высокой электронной плотностью. Положение
ядер устанавливается по положению максимумов плотности отрицательного
заряда, т. е. по положению электронов.
Положение атома углерода может быть определено по положению
электронной плотности, отвечающей внутренним 1 s-электронам. Атом же
водорода не может быть локализован таким простым методом. Его положение может
быть приблизительно определено по положению электронной плотности,
отвечающей связывающим электронам, но такие оценки приводят к
заниженной величине длины связи С—Н.
Экспериментально определяется длина связи d\ тогда как действительная
величина межатомного расстояния равна d.
112
3. Гомоядерная ковалентная связь
молекулярную или ионную структуру. Положение ядер может быть
приблизительно определено благодаря тому, что рентгеновское
излучение в основном рассеивается электронами внутренних уровней, которые
концентрируются вокруг ядер. Следствием этого является тот факт, что
определение положения ковалентно связанных атомов водорода, не
имеющих внутренних электронов, представляет серьезную проблему как для
рентгеновской, так и для электронной дифракции (см. дополнение 3.2).
• Нейтроны рассеиваются ядрами, вот почему именно этот метод
используется для точного определения координат атомов.
Напомним, что целью является определение межатомных расстояний.
Дифракция нейтронов кажется наиболее подходящим и универсальным
методом; однако эксперименты по дифракции нейтронов очень дороги и
требуют очень сложного оборудования. Таким образом, рентгеновская дифракция
остается наиболее распространенным методом получения структурной
информации.
Дифракция электронов
Газовая фаза отличается от твердого тела тем, что молекулы находятся в
состоянии постоянного хаотического движения, а межмолекулярные
взаимодействия носят случайный характер. Поэтому дифракционные
эксперименты на газах дают информацию главным образом о межатомных расстояниях
внутри молекул. Исходные экспериментальные данные представляют собой
зависимость интенсивности от угла рассеяния пучка электронов. Она может
быть пересчитана в зависимость интенсивности от расстояния. Пики на
такой кривой соответствуют межатомным расстояниям в молекуле.
Рассмотрим результаты экспериментов по дифракции электронов при
изучении строения трихлорида бора в газовой фазе. При обработке данных
были получены расстояния В—CI и CI—CI (рис. 3.3). Все связи В—CI имеют
одинаковую длину, как и расстояния CI—CI между несвязанными атомами
CI. Путем несложных тригонометрических вычислений было установлено,
что все углы CI—В—CI равны 120°. Молекула имеет плоское треугольное
строение.
Рентгеновская дифракция
Рассеяние рентгеновских лучей электронами внутренних уровней является
источником наиболее ценной информации о расположении атомов в
молекуле. Чем тяжелее элемент, тем больше электронов располагается на его вну-
Рис. 3.3. Расстояния между атомами в молекуле
трихлорида бора определенные из экспериментов по
электронной дифракции. Величины валентных углов (углов
между связями) рассчитаны, исходя из этих
межатомных расстояний.
3.3. Ковалентный радиус атома 113
Таблица 3.2. Расстояния S—S между соседними атомами в некоторых аллотропных
модификациях серы
Аллотропная модификация Конфигурация молекулы Расстояние S—S, нм
S8 (а-форма)
S8 (Р-форма)
sio
s„
s
\\2
s
Sl8
20
Кольцо
Кольцо
Кольцо
Кольцо
Кольцо
Кольцо
Кольцо
Кольцо
Спиральная цепь
0,206
0,206
0,205
0,206
0,206
0,205
0,206
0,205
0,207
тренних уровнях и тем выше его способность рассеивать рентгеновское
излучение. В результате положение тяжелых элементов может быть определено
гораздо легче и точнее, чем для легких элементов. Выше уже отмечалось, что
особенно большие трудности возникают при определении положения атомов
водорода.
В твердом состоянии движение молекул затруднено. При кристаллизации
под действием сил межмолекулярного взаимодействия происходит образова-
► ние упорядоченной структуры. Эксперименты по рентгеновской дифракции
Аллотропные являются источником информации как о расположении ато-
модификации серы: мов внутри молекулы, так и о взаимном расположении молекул
см. разд. 7.8 в кристалле. Для иллюстрации рассмотрим кристаллическую
структуру различных аллотропных модификаций серы.
Рентгеновские дифракционные эксперименты показывают, что
кристаллы ромбической серы (а-модификация, обычное состояние серы) состоят из
восьмичленных колец S8 (рис. 3.2).
Все расстояния S—S в кольце одинаковы (206 пм). В табл. 3.2
перечислены некоторые аллотропные модификации серы и расстояния между
соседними атомами в них. Во всех случаях молекулы образуются за счет
одинарных ковалентных связей, длины которых практически одинаковы.
Кратчайшие расстояния между атомами серы соседних колец все же больше
расстояний S—S между атомами, образующими кольцо, и равны 337 пм для
Sn и 323 пм для Sl0. Это означает, что ковалентного связывания между
кольцами не происходит.
КОВАЛЕНТНЫЙ РАДИУС АТОМА
Рассмотрим двухатомную гомоядерную молекулу, такую, как С12 в
газообразном состоянии. Эта молекула образуется за счет одинарной связи длиной
199 пм. Было бы удобно иметь какой-либо параметр для описания размера
атома, участвующего в образовании ковалентной связи. За ковалентный
радиус гков атома X, связанного одинарной связью, примем половину длины
типичной простой гомоядерной связи X—X. Тогда ковалентный радиус атома
хлора равен 99 пм.
114 3. Гомоядерная ковалентная связь
За ковалентный радиус гков атома X принимается половина длины
простой (одинарной) гомоядерной связи X—X.
Ковалентные радиусы различаются для одинарной, двойной и
тройной связей.
Часто пользуются усредненными величинами ковалентных радиусов, так как
длины одинарных связей X—X несколько различаются для разных молекул.
Ковалентный радиус серы можно оценить, находя среднюю величину
половины длины одинарной связи S—S для различных аллотропных модификаций.
Из табл. 3.2 видно, что среднее расстояние между атомами серы равно 0,206 нм
(или 206 пм; 1 нм = 10~9 м = 1000 пм), что соответствует гков = 0,103 нм.
^ В табл. 3.3 сопоставляются ковалентные и вандерваальсовы радиусы водо-
р-Элементы: см. гл.13 рода и р-элементов. Так как вандерваальсовы радиусы
вычисляются для атомов, не образующих друг с другом химической
связи, то они больше соответствующих ковалентных радиусов, которые
имеют смысл только для связанных атомов.
Как правило, при движении вниз по группе и вандерваальсовы, и
ковалентные радиусы увеличиваются (рис. 3.4).
Таблица 3.3. Вандерваальсовы и ковалентные радиусы водорода и р-элементов
(Приводимые величины являются обобщением результатов, полученных для
широкого круга соединений.)
Элемент Вандерваальсов радиус, нм Ковалентный радиус, нм
Н 0,120 0,037а
Группа 13 В 0,208 0,088
А1 - 0,130
Ga - 0,122
In - 0,150
Tl - 0,155
Группам С 0,185 0,077
Si 0,210 0,118
Ge - 0,122
Sn - 0,140
Pb - 0,154
Группа 15 N 0,154 0,075
P 0,190 0,110
As 0,200 0,122
Sb 0,220 0,143
Bi 0,240 0,152
Группа 16 О 0,140 0,073
S 0,185 0,103
Se 0,200 0,117
Те 0,220 0,135
Группа 17 F 0,135 0,071
CI 0,180 0,099
Br 0,195 0,114
I 0,215 0,133
a В некоторых случаях для органических соединений лучше использовать величину 0,030 нм.
3.4. Энергия связи. Образование двухатомной молекулы Н2 115
5
С
Радиус
250
200
150
100
50
0
N Р As Sb Bi О S Se Те F CI Br I
Группа 15 Группа 16 Группа 17
□ ™ГмШНЫе Р3" □ вандерваальсовы радиусы
Рис. 3.4. Изменение ковалентных и вандерваальсовых радиусов элементов в группах
15, 16, 17.
ЭНЕРГИЯ СВЯЗИ.
ОБРАЗОВАНИЕ ДВУХАТОМНОЙ МОЛЕКУЛЫ Н2
Перейдем к рассмотрению энергий связи. Если в предыдущем разделе
сравнивались длины ковалентных и вандерваальсовых связей, то начало этого
раздела будет посвящено сравнению энергии, необходимой для сближения
двух атомов водорода и двух атомов аргона (см. разд. 2.21). Для атомов
аргона следует рассматривать два фактора: взаимное притяжение атомов за счет
диполь-дипольного взаимодействия и электростатическое отталкивание их
электронных оболочек. Можно ли использовать тот же подход для описания
взаимодействия двух атомов водорода?
Существует фундаментальное различие между этими двумя случаями.
При любом расстоянии между атомами аргона наиболее сильным
притягивающим взаимодействием будет оставаться взаимодействие между
наведенными диполями и образования ковалентной связи, основанной на
обобществлении электронов, происходить не будет. При сближении же атомов
водорода может произойти образование ковалентной связи.
Это можно понять при рассмотрении числа электронов в валентной
оболочке каждого атома. Каждый атом аргона обладает полностью заполненной
оболочкой [NelS^/j6, так что для него нет движущей силы для изменения
электронной конфигурации.
В отличие от этого, атом водорода имеет конфигурацию Is1. Так же, как и
электронные конфигурации других благородных газов, заполненная ls-обо-
лочка (атом гелия) обладает повышенной стабильностью. Для атома
водорода существуют две возможности для достижения стабильной заполненной
октета: s-оболочки:
. 2.20 1) атом может захватить электрон и стать анионом Н~ с
электронной конфиругацией Is2 или
2) атом водорода может обобществить электроны с каким-
либо другим атомом, например со вторым атомом водорода, что приведет к
образованию молекулы Н2 (рис. 3.5).
116
3. Гомоядерная ковалентная связь
Рис. 3.5. Атом водорода имеет электронную
конфигурацию \sK Образование молекулы Н2
происходит при обобществлении электронов
двумя атомами. При этом каждый атом
водорода приобретает конфигурацию
благородного газа (гелия) - Is2.
н*
ь1
Не
Ь2
•н
ы
=>
н: н
Дополнение 3.3. Основное электронное состояние атома гелия
Электронная конфигурация атома гелия [Не] соответствует одному из общих
правил периодической системы. Каждый элемент группы 1 легко теряет один
электрон и приобретает электронную конфигурацию благородного газа:
К
[ArHs1
Na
[Ne^s1
Li
[He]25!
-> K+ + e"
[Аг]
-> Na+ + e"
[Ne]
-> Li+ + e"
[He]
Гелий не подчиняется правилу октета, как таковому, но имеет полностью
заполненную оболочку с главным квантовым числом п = 1, на которой может
разместиться только два электрона.
При сближении атомов водорода происходит образование ковалентнои
связи, так как обобществление электронов становится выгодным. На рис. 3.6
приведена зависимость потенциальной энергии системы от расстояния
между двумя атомами водорода. Хотя эта кривая внешне очень похожа на
зависимость, приведенную на рис. 2.24, она является результатом суммирования
принципиально других слагаемых. Можно выделить четыре основных
взаимодействия:
1) взаимное отталкивание электронов;
2) притяжение между электроном и протоном одного атома;
3) притяжение между электроном и протоном другого атома;
4) отталкивание протонов.
Эти взаимодействия представлены на рис. 3.7.
На самом деле взаимодействия 2 и 3 вносят заметный вклад только при
больших межъядерных расстояниях. При сближении атомов происходит
образование ковалентнои связи, и нет смысла рассматривать взаимодействие
одного электрона с одним протоном.
3.4. Энергия связи. Образование двухатомной молекулы Н2
117
250
о
С
-250
-500
0,3 0,4
Расстояние, нм
Рис. 3.6. Зависимость потенциальной энергии системы от расстояния между двумя
атомами водорода. Минимум на кривой соответствует равновесию.
Межъядерное расстояние г
Рис. 3.7. Сближение двух атомов водорода, каждый из которых имеет один протон и
один электрон. Наблюдаются четыре типа взаимодействий: I) электрон-электрон
(отталкивание); 2) электрон-протон внутри одного атома (притяжение); 3)
электрон-протон между разными атомами (притяжение); 4) протон—протон (отталкивание).
Какую информацию можно получить из рис. 3.6? Известно, что
нормальным состоянием водорода является молекулярный, а не атомарный водород.
Минимум на кривой потенциальной энергии соответствует равновесному
расстоянию между ядрами в молекуле Н2. Равновесие - состояние системы,
118
3. Гомоядерная ковапентная связь
при котором ее энергия минимальна, а межъядерное расстояние
соответствует длине простой ковалентной связи Н—Н и должно быть равно 0,074 нм. (В
действительности из-за асимметрии кривой потенциальной энергии
экспериментально измеренная длина связи несколько отличается от
межъядерного расстояния, соответствующего минимуму энергии.)
ВЯ ЭНЕРГИЯ СВЯЗИ
В предыдущем разделе было показано, что образование молекулы Н2
соответствует минимуму на кривой потенциальной энергии системы, состоящей
из двух атомов водорода. Кривую, подобную приведенной на рис. 3.6, можно
получить для любой гетероядерной (X—Y) или гомоядерной (Y—Y)
ковалентной связи. До сих пор все сводилось к рассмотрению геометрического
фактора — на каком расстоянии друг от друга находятся взаимодействующие
атомы. Теперь необходимо выяснить, сколько энергии выделяется при
образовании и сколько энергии необходимо затратить для разрыва (диссоциации)
связи?
Внутренняя энергия и энтальпия
Минимуму на кривой, приведенной на рис. 3.6, соответствует энергия,
равная 458 кДж/моль. К сожалению, здесь возникает проблема, так как энергия,
выделяющаяся при образовании связи Н—Н, или, наоборот, необходимая
для ее разрыва, не равна точно 458 кДж/моль.
Кривая на рис. 3.6 соответствует внутренней энергии системы (AU) при
температуре 0 К. Однако часто более удобно работать с энтальпиями (А//),
измеренными при температуре 298 К. На практике разница между АЯи Д£/
равна приблизительно 3 кДж/моль. (Смысл понятий «энтальпия» и
«внутренняя энергия» будет подробно разобран в гл.12; сейчас необходимо
запомнить, что разница между этими величинами для данной системы невелика.)
Более серьезная проблема возникает по причине того, что даже при 0 К
атомы водорода сохраняют некоторую подвижность и колеблются около по-
►- ложения равновесия. Необходимо помнить, что низшее колебательное со-
Колебательные стояние молекулы водорода лежит на 26 кДж/моль выше дна
состояния: см. гл. 9 ямы на кривой на рис. 3.6. Эта остаточная энергия называется
нулевой энергией. Любые экспериментальные измерения
связаны с реальными молекулами и самым низшим энергетическим состоянием, в
котором они могут находиться. Таким образом, экспериментальные
измерения «достигают» не минимума потенциальной энергии, а лишь низшего
колебательного уровня.
Ш и АН (см
AL/ =
где Р -
= АН-
. разд
12.2) связаны уравнением
РДУ при постоянном давлении
- давление,
V- объем.
3.5. Энергия связи
119
Диссоциация гомоядерных связей в двухатомных молекулах
Наиболее удобным параметром для описания энергии связи Н—Н в
молекуле водорода является энергия, необходимая для разрыва этой связи
(уравнение 3.1) в молекуле, находящейся на низшем колебательном уровне. В
табл. 3.4 приведены величины изменения внутренней энергии Л(/и
энтальпии Д#, которые могут быть использованы для описания реакции
диссоциации молекулы водорода.
Н2(газ) -> 2Н(газ) (3.1)
Так как величины АН и А £/близки, в дальнейшем их различие
упоминаться не будет, а для характеристики прочности связи будет использоваться
энтальпия разрыва. Таким образом, энергию связи Н—Н мы принимаем равной
436 кДж/моль.
Таблица 3.4. Изменение внутренней энергии и энтальпии при диссоциации
молекулы Н2. Величины А£/и АЯ связаны уравнением AU = АЯ— PAV, где Р— давление,
V — объем.
Процесс Величина
Н2(газ) -» 2Н(газ) (О К) А£/(0 К) = 432 кДж/моль
Н2(газ) -> 2Н(газ) (298 К) Д£/(298 К) = 433 кДж/моль
АЯ(298 К) = 436 кДж/моль
Изменение внутренней энергии (AL/) и изменение энтальпии (АН) при
диссоциации двухатомной молекулы X—X относится к процессу
Н2(газ) ->2Н(газ)
протекающему в газовой фазе при фиксированной температуре
(обычно или О К, или 298 К).
Разрыв гомоядерных связей в многоатомных молекулах
В многоатомных молекулах энергия диссоциации связи является энергией,
необходимой для разрыва одной вполне определенной связи. Уравнения
3.2-3.4 соответствуют разрыву гомоядерных связей в молекулах,
представленных на рис. 3.1. Рис. 3.8 иллюстрирует разрыв связи С—С в этане С2Н6.
С2Н6(газ) -> 2СН3" (газ) (3.2)
1Ч2Н4(газ) -* 2NH2"(ra3) (3.3)
Н202(газ) -> 2HO * (газ) (3.4)
На рис. 3.8 также показан результат разрыва гомоядерной связи в
циклической молекуле. При разрыве одной из связей S—S в циклической молекуле S6
происходит раскрытие кольца. При этом концевые атомы получающейся
цепи имеют по одному неспаренному электрону. Разрыв следующей связи
приведет к образованию фрагментов S . Величины изменения энтальпии в
120
3. Гомоядерная ковалентная связь
2СН,
S6 -S-S-S-S-S-S*
Рис. 3.8. Разрыв связи в молекуле этана приводит к образованию двух радикалов, а в
циклической молекуле S6 — к раскрытию кольца.
Дополнение 3.4. Средние и индивидуальные энтальпии
разрыва связей
Молекула метана — тетраэдрическая молекула с четырьмя химически
эквивалентными связями С—Н одинаковой длины. Это доказано
экспериментально.
СН4(газ) -> С(газ) + 4Н(газ)
При полной диссоциации метана на атомы (т. е. при атомизации) можно
определить среднюю энергию разрыва связи С—Н, т. е. D(C—H). Если энергия
распада СН4 на атомы равна ДатЯ° — 1664 кДж/моль, то D(C—H) =
1664 кДж/моль/4 = 416 кДж/моль.
Однако если проводить процесс ступенчато, то энергия каждой стадии не
будет равна 416 кДж/моль:
СН4(газ) -> СН3(газ) + Н(газ), Д#= 435 кДж/моль
СН3(газ) -» СН2(газ) + Н(газ), ЛЯ = 460 кДж/моль
СН2(газ) -> СН(газ) + Н(газ), ДЯ = 427 кДж/моль
СН(газ) -► С(газ) + Н(газ), ДЯ = 338 кДж/моль
Из этих величин можно сделать вывод, что, хотя все четыре связи С—Н в
молекуле метана эквивалентны, их прочность будет отличаться от прочности
трех связей С—Н в СН3, от прочности двух связей С—Н в СН2 и от
прочности связи в радикале СН.
3.6. Стандартная энтальпия атомизации элемента 121
этих реакциях будут несколько различаться. Это различие в энергии
диссоциации одной и той же связи в разных молекулах обсуждается в дополнении
3.4. Чаще всего, однако, используются усредненные величины энергий
связей.
В дальнейшем для обозначения энтальпии разрыва связи в какой-либо
Н определенной молекуле будет использоваться обозначение D:
D = ЛДИССЯ, а усредненная для разных молекул энергия
разрыва какой-либо связи будет обозначаться как D. Например, энтальпия
разрыва связи С—С в этане (уравнение 3.2) равна 376 кДж/моль. Однако
после усреднения энтальпий разрыва той же связи в различных молекулах
или нескольких связей С—С с разным окружением в одной молекуле
получается величинаD, равная приблизительно 346 кДж/моль. Величины/)
для одной и той же связи несколько варьируют от одного литературного
источника к другому в зависимости от того, сколько и каких веществ было
использовано для усреднения. Следует помнить, что энтальпия разрыва
связи зависит от ее кратности: величина D для простой связи С—С равна
346 кДж/моль, D для двойной связи С=С равна 598 кДж/моль, а для
тройной связи D(C=C) = 813 кДж/моль.
Энтальпия диссоциации связи D для связи X—X является
усредненной величиной. Для одинарной (X—X), двойной (Х=Х) и тройной (Х=Х)
связей эта величина должна определяться отдельно.
СТАНДАРТНАЯ ЭНТАЛЬПИЯ АТОМИЗАЦИИ ЭЛЕМЕНТА
Стандартная энтальпия атомизации
Рассмотрим процесс разрыва химической связи, представленный
уравнением 3.1. Разрыв связи Н—Н приводит к образованию двух атомов водорода, а
энергия, необходимая для проведения этой реакции, называется энтальпией
атомизации. Стандартной энтальпией атомизации элемента (Дат#°)
называется изменение энтальпии системы, необходимое для перевода одного моля
атомов элемента из стандартного состояния в газообразное атомарное
состояние. (Стандартное состояние: см. разд. 1.17). Для Н2 это соответствует
уравнению 3.5, а для ртути и никеля уравнениям 3.6 и 3.7 соответственно.
0,5Н2(газ)-> Н(газ), ДатЯ° = 218 кДж/моль (3.5)
Щ(ж.) -> Hg(ra3), АатЯ° = 61 кДж/моль (3.6)
Ni(TB.) -> Ni(ra3), Дат#° = 430 кДж/моль (3.7)
В стандартном состоянии одни элементы твердые, другие — жидкие или
газообразные. Если стандартным состоянием вещества является твердая фаза, то
в стандартную энтальпию атомизации (при 298 К) входят:
1) энтальпия, необходимая для увеличения температуры элемента от
298 К до точки плавления;
122
3. Гомоядерная ковалентная связь
2) энтальпия плавления элемента (перехода из твердой фазы и жидкую);
3) энтальпия, необходимая для увеличения температуры элемента от
точки плавления до точки кипения (Т К);
4) энтальпия испарения элемента (перехода из жидкой фазы в
газообразную);
5) энтальпия разрыва связей в многоатомных молекулах газовой фазы;
6) энтальпия охлаждения моноатомного газа элемента от Г К до 298 К.
Стандартной энтальпией атомизации элемента АатН° называется
энтальпия образования одного моля одноатомного газа (при 298 К) из
стандартного состояния этого элемента. Единицы измерения в
СИ - кДж на моль атомов в газовой фазе.
Таблица 3.5. Энтальпии атомизации некоторых элементов
Элемент
As
Br
С
CI
Си
F
Н
Hg
I
К
N
Na
О
Р
S
Sn
Стандартное состояние элеменг
As(tb.)
Вг2(ж.)
С(графит)
С12(газ)
Си(тв.)
F2(ra3)
Н2(газ)
Нё(ж.)
12(тв.)
К(тв.)
N2(ra3)
Na(TB.)
02(газ)
Р4(белый)
58(ромбическая сера)
БпСбелое олово)
А.ЛТН°(298К), кДж/моль
302
112
717
121
338
79
218
61
107
90
473
108
249
315
277
302
Более полный список: В табл. 3.5 приведены энтальпии ионизации некоторых эле-
см. приложение 7 ментов.
Пример 3.1. Слагаемые теплоты атомизации
Определите теплоту атомизации ртути, используя закон Гесса.
Необходимые данные приведены ниже.
Решение
Температура кипения: Т"кип = 655 К.
Теплоемкость жидкой ртути: С = 27,4 Дж/(моль • К).
Теплоемкость газообразной ртути: Ср = 20,8 Дж/(моль • К).
Энтальпия испарения: ДИСПН = 59,1 кДж/моль.
3.6. Стандартная энтальпия атомизации элемента 123
Стандартной энтальпии атомизации ртути соответствует теплота,
необходимая для перевода одного моля атомов ртути из жидкого состояния
(стандартного состояния этого элемента) в одноатомный газ.
Молярная теплоемкость (С ) равна теплоте, которую необходимо затратить
для нагревания одного моля вещества на один градус при постоянном давлении
(см. разд. 12.3).
Ниже дана схема цикла, состоящего из отдельных стадий перевода ртути из
жидкого состояния в состояние одноатомного газа при 298 К:
Слагаемые процесса атомизации:
Лат"°
Щ(ж.) при 298 К ► Hg(ra3) при 298 К
АЯ,
А
щ
АЯ.
НЕ(ж.) при 655 К (Г) - ► Нё(газ) при 655 К (Г)
АЯ, (теплота, необходимая для нагревания 1 моль жидкой ртути от
298 до 655 К ) =
= (Теплоемкость жидкости) • (Изменение температуры)
= 27,4 • (655-298)
= 9782 Дж/моль
=9,8 кДж/моль
АЯ2 (теплота испарения) =
= 59,1 кД ж/моль
АЯ3 (теплота, выделяющаяся при охлаждении 1 моль паров ртути
от 655 до 298 К) =
= —(Теплоемкость газа) • (Изменение температуры)
= -20,8 • (655-298)
= -7426 Дж/моль
= —7,4 кДж/моль
По закону Гесса:
Д„Я° = АЯ, + АЯ2 + АЯ3
= 9,8 + 59,1-7,4
= 61,5 кДж/моль
Энтальпия атомизации и энтальпия разрыва связи
для двухатомных газов
Взаимосвязь между энтальпией атомизации и энтальпией разрыва связи для
двухатомных газов имеет важное значение.
Энтальпия разрыва связи (D) определяется как энергия, необходимая для
разрыва 1 моль этих связей (уравнение 3.8). Для водорода D = 436 кДж/моль.
В то же время, энтальпия атомизации определяется в расчете на 1 моль
атомов элемента (уравнение 3.9). Это различие часто приводит к ошибкам в
расчетах.
Н2(газ) -> 2 Н(газ), D = 436 кДжДмоль связей) (3.8)
0,5Н2(газ) -» Н(газ), \ТН° = 218 кДжДмоль атомов в газовой фазе) (3.9)
124 3. Гомоядерная ковалентная связь
Для двухатомных газов:
энтальпия разрыва связи равна удвоенной энтальпии атомизации.
0 = 2ДатН-
ОПРЕДЕЛЕНИЕ ЭНТАЛЬПИИ СВЯЗЕЙ
ПО СТАНДАРТНЫМ ТЕПЛОТАМ ОБРАЗОВАНИЯ
„ ~ Энтальпия разрыва связи в двухатомных молекулах может быть
Применение колеба- J „
определена путем термохимических измерении или из спект-
тельнои спектроско-
ральных данных,
пии для изучения двух- ~ _
Однако для больших молекул прямые методы измерения не
атомных молекул: п
Q ft всегда применимы. В таких случаях можно проводить расчеты,
см. разд. У.о _ г^
основанные на законе Гесса. Экспериментально возможно из-
jr r- мерять энтальпии сгорания как элементов, так и сложных ве-
Закон Гесса: ,-
л ,_ ществ, а затем использовать эти данные для расчета теплот об-
см. разд. 1.17 /А иоч
к разования (Д^ /Г) химических соединении, которые в свою
очередь являются исходным материалом для определения
энтальпий разрыва химических связей. При этом следует помнить, что
получаемые таким образом величины D зависят от точности определения многих
других параметров (см. ниже).
Пусть необходимо определить энергию разрыва одинарной связи N—N.
Эта величина не может быть получена прямыми методами, так как в
молекуле азота связь является тройной. Следовательно, необходимо обратиться к
какому-нибудь соединению со связью N—N, например к гидразину N2H4
(рис. 3.1). Эта молекула имеет одну одинарную связь между атомами азота и
четыре связи N—Н. Для того чтобы разрушить эту молекулу до атомарного
состояния, необходимо разорвать все связи. Соответствующее изменение
энтальпии будет определяться уравнением 3.10. Необходимо отметить, что в
данном случае используются величины Z)(N—N) и Z)(N—H), так как для
расчета энтальпии разрыва связи N—N во вполне конкретном соединении
используется усредненная энтальпия разрыва связи N—Н.
N2H4(ra3) -> 2N(raa) + 4Н(газ), ДатД° = AN-N) + 4Z)(N-H) (3.10)
Таким образом, для определения энтальпии разрыва связи N—N вводится
еще одна неизвестная величина: D(N—H). Молекула аммиака образуется
только за счет связей N—Н, следовательно, зная ее энергию атомизации,
можно рассчитать среднюю энергию такой связи (уравнение 3.11). Величина
Аат/У° для процесса, которому соответствует уравнение 3.11, может быть
определена из стандартной энтальпии образования аммиака (уравнение 3.12) по
закону Гесса (уравнение 3.13). Стандартные энтальпии атомизации азота и
водорода равны 473 и 218 кДж/моль соответственно.
NH3(ra3) -> N(raa) + ЗН(газ), \ТН° = 3AN-H) (3.11)
0,5 1Ч2(газ) + 1,5 Н2(газ) -> NH3(ra3), A^H0 = -46 кДж/моль (3.12)
3.7. Определение энтальпии связей
125
NH3(ra3)
3Z>(N-H)
ДобрЯ°
N(ra3) + ЗН(газ)
A/f(N)+3A /Г(Н)
(3.13)
0,5N2(ra3)+ 1,5Н2(газ)
Из уравнений 3.13 получаем
3fl(N-H)= AaT/f(N) + ЗАат/Г(Н) - Д^/Г (ЫН3,газ)
35(N-H) = 473 + (3 • 218) - (-46) = 1173
5(N-H) = 391 кДж/моль
Этот результат можно использовать для расчета энергии связи N—N,
основываясь на уравнении 3.10.
N2H4(ra3)
Z)(N-N) + 4AN-H)
2N(ra3) + 4Н(газ)
2Дат/Г (N) + 4Д/Г (Н)
(3.14)
N2(ra3) + 2Н2(газ)
Стандартная энтальпия образования газообразного гидразина равна
95 кДж/моль.
Из уравнений 3.14 получаем:
«N-N) + 4ДТМ-Н) = 2AaT/T(N) + 4ДатДв(Н) - А0брЯ,(Ы2Н4, газ)
Z)(N-N) + 4AN-H) = (2 • 473) + (4 • 218) - 95 = 1723
Z>(N-N) = 1723 - 4Z)(N-H)
Так как Z)(N—H) = 391 кДж/моль (см. выше)
AN-N) = 1723 - (4 • 391) = 159 кДж/моль
Чтобы выполнить все приведенные выше расчеты, необходимо было
принять, что энергия разрыва связи N—Н одинакова в аммиакае и гидразине.
Это предположение выглядит разумным, так как оба вещества близки по
своим свойствам. Однако подобные действия с молекулами разных типов
должны производиться с крайней осторожностью. Естественно, величина Д
определенная для простой связи, не может быть использована для связи
кратной, так как оба типа связей имеют принципиально различные энтальпии
разрыва.
Некоторые типичные энтальпии разрыва простых ковалентных гомоядер-
ных связей приведены в табл. 3.6. Часть этих величин получена путем прямых
измерений тепловых эффектов диссоциации соответствующих двухатомных
молекул (например, ДН—Н), D(F— F) и ДО—О)). Другие были получены
126
3. Гомоядерная ковалентная связь
Таблица 3.6. Энтальпии разрыва некоторых простых гомоядерных ковалентных связей
Часть приводимых величин получена путем прямых измерений (например, для связи
Н—Н), некоторые величины получены при усреднении данных для многих молекул и
являются /)-величинами (например, для связи С—С), а некоторые энтальпии (например, для
связи N—N) зависят от точности определения энтальпий разрыва других связей.
Связь Х—Х
Н-Н
С-С
Si-Si
Ge-Ge
Sn-Sn
N-N
P-P
As—As
D(X-
436
346
196
163
152
159
200
177
-X), кДж/моль
Связь Х—Х
0-0
S-S
Se-Se
F-F
Cl-Cl
Br-Br
I-I
D(X—X), кДж/моль
146
266
193
159
242
193
151
при изучении распада небольших молекул; например, величина />(S—S)
была измерена при диссоциации S8 (см. рис. 3.2), а некоторые энтальпии
(например, связи N—N) зависят от точности определения энтальпий разрыва
других связей. Такие величины, как Z)(N—N), рассчитываются в
предположении о возможности перенесения значения энтальпии разрыва связей от
молекулы одного вещества к молекуле другого вещества.
ЕЯ ПРИРОДА КОВАЛЕНТНОЙ СВЯЗИ В Н2
В предыдущих разделах обсуждались два экспериментально наблюдаемых
параметра, с помощью которых можно описывать химические связи типа
Н—Н: межъядерное расстояние, из которого можно определить ковалентный
радиус атома, и энтальпия разрыва связи. Эксперименты показывают, что
при сближении двух атомов водорода, они могут образовывать связь длиной
0,074 нм с энтальпией диссоциации 436 кДж/моль.
Причиной образования связи является тенденция каждого атома
водорода к приобретению электронной конфигурации гелия, что достигается путем
обобществления электронов.
Ковалентная связь в молекуле водорода может быть представлена с
помощью структуры Льюиса. Но для более полного описания необходимо
использовать другие методы, из которых наиболее известны теория валентных
связей (ВС) и метод молекулярных орбиталей (МО). Следующие разделы
посвящены использованию этих теоретических подходов.
ИЕИ СТРУКТУРЫ ЛЬЮИСА ДЛЯ МОЛЕКУЛЫ ВОДОРОДА
Структуры 3.1 и 3.2 схематически показывают образование связи в молекуле
водорода и называются структурами Льюиса. В структуре 3.1 электроны
показаны точками; связь между атомами может быть представлена линией
(структура 3.2).
Хотя структуры Льюиса показывают образование связи между атомами
водорода, они не содержат информации о характере этой связи и
пространственном распределении электронной плотности.
3.9. Структуры Льюиса для молекулы водорода 127
Н : Н Н Н
3.1 3.2
Дополнение 3.5. Структуры Льюиса
В 1916 году Дж. Н. Льюис предложил простой, но информативный способ
представления распределения валентных электронов в молекулах. В соответствии с этим методом
электроны изображаются точками или точками и крестами, а ядра — символами
элементов. (Точки и кресты используются для того, чтобы различать электроны,
принадлежащие разным атомам, однако следует помнить, что в действительности электроны
неразличимы.)
По возможности, все электроны должны образовывать пары. При наличии неспа-
ренного электрона частица является радикалом.
Пример: С\2
Электронная конфигурация основного состояния атома хлора — [Ne^s^/?5. При
образовании молекулы С12 происходит обобществление неспаренных электронов. Атомы
хлора приобретают устойчивую электронную конфигурация типа [Аг].
Структура Льюиса для молекулы С12 приведена ниже:
: а : ci :
Эта структура может быть упрощена путем выделения связывающих электронов и не-
поделенных пар электронов:
: ci — а :
Структуры Льюиса очень удобны как способ изображения связывания атомов в
молекулах. Ниже приведено несколько примеров:
Н Н Н
• • •• •• ••
:f:h :s:h h:n: \t н : с : н
н
HF H2S NH3 CH4
н н н
•••• •• ••
н:с:о: h:n:n:h
•••• •• ••
н н
: ci :
: ci : рь : ci :
: ci :
СН3ОН N2H4 PbCl4
128
3. Гомоядерная ковалентная связь
И ЗАДАЧА ОПИСАНИЯ ЭЛЕКТРОНОВ В МОЛЕКУЛАХ
Задача
В разд. 2.7 обсуждалось уравнение Шредингера и его применение для
описания динамического поведения электронов в атоме. Также отмечалось, что
точное математическое решение этого уравнения может быть получено
только для водородоподобных частиц. При описании поведения электронов в
молекулах проблема остается - невозможно получить точное решение
волнового уравнения, если система состоит более чем из одного ядра и одного
электрона. Ситуацию можно упростить, если пренебречь движением ядра, но это
не решит проблему в целом.
Задача состоит в том, чтобы получить молекулярные волновые функции
(\|/мол), которые описывали бы поведение электронов в молекулах. Что же для
этого необходимо сделать?
Два способа решения
Существует два способа приближенного нахождения молекулярных
волновых функций - теория валентных связей и метод молекулярных орбиталей,
которые основываются на различных исходных предположениях, но, в
принципе, должны приводить к одним и тем же результатам. Прежде чем
детально ознакомиться с этими методами, необходимо отметить некоторые
проблемы, возникающие при рассмотрении молекул, содержащих более
двух электронов. Хорошим примером может служить молекула воды.
Ниже представлены структуры Льюиса (3.3 и 3.4) для молекулы воды.
Показаны две связывающие (О—Н) и две неподеленные пары электронов.
Основой структур Льюиса является представление о химической связи как
о паре электронов, обобществленной двумя атомами. Это же представление
является основой и для теории валентных связей (ВС).
н
• • I
: о : н : о —
• • • •
3.3 3.4
w Теория валентных связей базируется на положении, что любая молекула об-
Теория валентных разуется за счет ряда дискретных химических связей. Все связи
связей: см. разд. 3.11 являются полностью изолированными, и каждая из них может
быть описана как взаимодействие двух электронов с двумя
ядрами. Эта модель связывания называется два центра — два электрона
(2с-2е). Необходимо еще раз отметить, что в рамках этой теории все связи
считаются строго локализованными. В теории валентных связей делается
попытка записать волновые функции для каждой отдельной пары электронов,
а затем скомбинировать из них волновые функции молекулы в целом.
3.11. Теория валентных связей
129
► Метод молекулярных орбиталей (МО), напротив, не предпо-
Метод молекулярных лагает наличия двухцентровых двухэлектронных связей. Моле-
орбиталей: см. кулярные орбитали создаются таким образом, что они окружа-
разд. 3.12 ют любое число атомов или все атомы молекулы.
Взаимодействие, которое захватывает более двух ядер, называется
многоцентровым или делокализованным.
Связывающее взаимодействие называется локализованным, если в
него вовлечено два атома, и делокализованным, если в него
вовлечено три или более атомов.
Метод молекулярных орбиталей гораздо сложнее с математической точки
зрения и менее интуитивно понятен, чем теория валентных связей. В случае
молекулы воды значительно легче представить образование двух
двухцентровых связей О—Н, чем рассуждать о многоцетровых взаимодействиях типа
Н—О—Н. Однако в случае многих молекул метод молекулярных орбиталей
дает адекватное описание связывания. Эта ситуация во многом напоминает
ситуацию с энтальпиями разрыва химических связей. Известно, что все
четыре связи С—Н в метане эквивалентны, но, как было показано в
дополнении 3.4, при последовательном отрыве атомов водорода тепловые эффекты
оказываются различными.
И теория валентных связей, и метод молекулярных орбиталей являются при-
ближенными способами построения молекулярных волновых функций умол. Оба
метода должны в конце концов приводить к одинаковому набору этих функций.
В теории валентных связей электроны распределяются по двухцент-
ровым связывающим орбиталям или оказываются локализованными
на отдельных атомах.
В методе молекулярных орбиталей электроны делокализованы по
всей молекуле.
ТЕОРИЯ ВАЛЕНТНЫХ СВЯЗЕЙ
Общие замечания
В теории валентных связей образование двухатомной молекулы является
следствием взаимного проникновения электронных облаков
взаимодействующих атомов. На практике это означает, что делается попытка
аппроксимировать «реальную» волновую функцию молекулы путем комбинирования
волновых функций отдельных двухэлектронных взаимодействий.
Математические детали выходят за рамки этой книги. Для более глубокого изучения
данного вопроса можно рекомендовать книгу: McWeeny R., Coulson's Valence,
3rd edn., Oxford University Press, Oxford (1979). [См. также Ч. Коулсон,
Валентность, М., Мир, 1965. - Ред.]
130 3. Гомоядерная ковалентная связь
Образование связи в молекуле Н2. Исходное приближение
Рассмотрим образование молекулы Н2. Каждый атом водорода состоит из
одного протона и одного электрона. На рис.3.9 ядра обозначены как НА и Нв, а
электроны - как I и 2. Если атомы находятся на значительном удалении друг
от друга, то электрон I полностью принадлежит ядру НА, а электрон 2 — ядру
Нв. Пусть такое состояние системы описывается волновой функцией ц/,.
Хотя электроны и имеют разные обозначения, на самом деле они
совершенно неразличимы. Поэтому при сближении атомов становится
невозможно определить, какой из электронов принадлежит атому НА, а какой - атому
Нв. Пусть это новое состояние системы описывается волновой функцией \|/2.
Обобщенное описание системы, состоящей из двух атомов водорода
(\|/ков), может быть представлено уравнением
Уков = Ч>1 + У2 (115)
Волновая функция \|/ков определяет энергию системы. На кривой
зависимости энергии от межъядерного расстояния (Н—Н) обнаруживается
минимум при 0,087 нм. Энергия системы в минимуме равна 303 кДж/моль. Таким
образом, применение уравнения 3.15 не дает величин, хорошо
согласующихся с экспериментальной длиной связи 0,074 нм и энергией диссоциации
436 кДж/моль. Требуется дальнейшее уточнение модели.
Образование связи в молекуле Н2. Уточнение модели
До сих пор к рассмотрению принимались случаи, когда атомы НА и Нв были
связаны с одним из электронов 1 или 2. Теперь необходимо учесть, что оба
электрона одновременно могут находиться вблизи одного из атомов.
Возможны четыре варианта:
1) ядро НА связано с электроном 1, а ядро Нв - с электроном 2;
2) ядро НА связано с электроном 2, а ядро Нв - с электроном 1;
3) ядро НА связано с электронами 1 и 2, а ядро Нв не связано с электронами;
4) ядро HA не связано с электронами, а ядро Нв связано с электронами 1 и 2.
Тогда как в двух первых случаях электронейтральность сохраняется, два
последних варианта соответствуют переносу электрона от одного ядра к
другому. Следствием этого является возникновение двух ионов — Н~ и Н+. Если
с ядром НА не связано ни одного электрона, то образуются ионы НА+ и Нв~.
Наоборот, если с ядром НА связаны оба электрона, то возникают ионы НА~ и
Нв+. Так как атомы водорода одинаковы, то и образование [НА+НВ~] и
[Нд~Нв+] равновероятно.
Допущение возможности существования [НА+НВ~] и [НА~НВ+] приводит
к тому, что волновая функция, описывающая распределение электронов в
Рис. 3.9. Обозначения, используемые
для ядер и электронов в при описании
образования связи в молекуле Н2
методом валентных связей.
3.12 . Метод молекулярных орбиталей
131
пространстве между ядрами может иметь не только введенную выше кова-
лентную составляющую, но и ионную составляющую. Эта ситуация
выражается уравнением 3.16.
Умол = Уков + С • Уион <ЗЛ6>
Коэффициент с соответствует относительному вкладу волновой функции
ц/ион в общую волновую функцию \|/мол.
В случае молекулы водорода уравнение 3.16 означает, что связывание в
молекуле водорода имеет ковалентную и ионную составляющие и должно
быть представлено суммой двух структур 3.5а и 3.56. Структуры 3.5а и 3.56
^ называются резонансными, что подчеркивается знаком обратимости.
Более подробно о
резонансных Н Н -*—► Н+ Н
структурах:
см. разд. 5.17 3.5а 3.56
Необходимо помнить, что чисто ионной и чисто ковалентной форм
молекулы водорода не существует. Все изложенное выше является только
попыткой описания связывания в этой молекуле с помощью волновой функции
\|/мол как суммы вкладов двух предельных моделей, которые выражаются
через \|/ков и 1|/ион.
► Варьируя параметр с в уравнении 3.16, можно подобрать такое межъядер-
Нормирование: Ное расстояние, которому будет, с одной стороны, соответство-
см. разд. 2.8 вать минимум энергии, и который, с другой стороны, будет
близок к экспериментально наблюдаемой длине связи Н—Н. Для
нормированных волновых функций при с = 0,25 рассчитанная длина связи
Н—Н равна 0,075 нм, что соответствует экспериментальным данным.
Предсказываемая энтальпия разрыва связи оказывается равной 398 кДж/моль.
Дальнейшее математическое уточнение модели может существенно улучшить
согласие этой величины с экспериментом, однако это происходит за счет
сильного усложнения первоначально простой физической картины.
МЕТОД МОЛЕКУЛЯРНЫХ ОРБИТАЛЕЙ
Общие замечания
Метод молекулярных орбиталей отличается от теории валентных связей тем,
что не предполагает образования локализованных двухцентровых связей в
молекулах, а напротив направлен на нахождение волновых функций для всей
молекулы в целом. Конечной целью является нахождение молекулярных
орбиталей - областей с высокой электронной плотностью. В некоторых
случаях электрон может быть делокализован по всей молекуле, в других -
ситуация может свестись к двухцентровым двухэлектронным орбиталям.
Обычно исходной точкой метода молекулярных орбиталей является
рассмотрение атомных орбиталей, которые могут принимать участие в
образовании связей в молекуле. Это приближение широко известно как МО ЛКАО
(молекулярные орбитали — линейная комбинация атомных орбиталей). Оно
основывается на нахождении приближенных молекулярных волновых
132
3. Гомоядерная ковалентная связь
функций (умо) как линейных комбинаций волновых функций отдельных
атомов молекулы (\|/А0). Необходимо напомнить, что атомные волновые
функции также являются приближенными и рассчитываются из волновых
функций атома водорода.
Взаимодействие между двумя атомными орбиталями будет:
• разрешено, если орбитали совместимы по симметрии;
• значительным, если область перекрывания будет значительной;
• значительным, если атомные орбитали близки по энергии.
Смысл первого пункта станет вполне понятным в конце этой главы. Что
касается второго пункта, то необходимо прояснить значение термина
«перекрывание». Это слово широко используется в методе молекулярных орбита-
лей, а мерой самого перекрывания является величина интеграла
перекрывания S. На рис. 3.10 показаны две 15-атомные орбитали. На рис. 3.10,а они
расположены близко друг к другу, но не имеют общей области пространства, т. е.
вероятность пребывания электронов обеих орбиталей в одной и той же точке
пространства практически равна нулю. Нулю же приблизительно равен и
интеграл перекрывания этих двух орбиталей. (Интеграл перекрывания не
может быть точно равен нулю, границы орбиталей не заключают в себе
область пространства, в которой вероятность нахождения электрона равна
100%, см. гл. 2). На рис. 3.10,5 видно, что орбитали имеют небольшую
область перекрывания, однако интеграл перекрывания по-прежнему
недостаточен для образования молекулярной орбитали. На рис. 3.10,в показана
ситуация значительного перекрывания орбиталей, интеграл перекрывания лежит
в интервале 0<5<1. Необходимо заметить, что S никогда не достигает
значения 1, так как в силу взаимного отталкивания ядра не могут очень близко
подойти друг к другу.
Чтобы понять важность ситуации, изображенной на рис. 3.10,в,
вспомните рис. 2.10 и подумайте, что произойдет при попытке комбинирования 15- и
45-атомных орбиталей; сравните эту ситуацию с комбинацией двух 15-орби-
талей. 15- и 45-АО имеют разную энергию и разную протяженность в
пространстве. Помните, что для эффективного взаимодействия орбитали
должны иметь значительное перекрывание.
Взаимодействие атомных орбиталей может быть выражено через
линейные комбинации атомных волновых функций. Рис. 3.11 показывает, что
происходит при синфазной и противофазной комбинации поперечных волн.
Синфазная комбинация приводит к интерференции с усилением: если ис-
OOGOGD
а б в
Рис. 3.10. Приводимые схемы отражают различные варианты перекрывания орбита-
лей и величины интеграла перекрывания S. а — две ls-орбитали не перекрываются,
S~ 0; б — перекрывание незначительно, величина Sочень мала; в — эффективное
перекрывание атомных орбиталей, величина Sзначительна. Ядра показаны точками в
центрах орбиталей.
3.12. Метод молекулярных орбиталей
133
Амплитуда
к/
X
1
Синфазная
комбинация
>
Амплитуда
1 : ►
)
V
1 Ч^
S
1
Противофазная
комбина-
l
Рис. 3.11. Интерференция двух поперечных волн одинаковой амплитуды и длины может
происходить либо с усилением, либо с взаимным гашением колебаний.
ходные волны одинаковы, то амплитуда волны после интерференции
удваивается. Противофазная комбинация приводит к взаимному гашению волн:
если исходные волны одинаковы по амплитуде, то гашение колебаний будет
полным.
Теперь перейдем от поперечных волн к атомным орбиталям, которые,
будучи «электронными волнами», могут находится в фазе или противофазе друг
к другу. Уравнения 3.17 и 3.18 соответствуют син- и противофазной
комбинации двух волновых функций, \|/j и ц/2, которые соответствуют двум
атомным орбиталям.
УмО(синфазная) = N' ^1 + ^ <ЗЛ7>
УмО(противофазная) = N' IVi " V21 <318)
N— нормировочный множитель, который необходим, чтобы вероятность
нахождения электрона в пространстве вблизи ядра была равна единице.
Общее правило заключается в том, что число создаваемых молекулярных
орбиталей должно быть равно числу исходных атомных орбиталей. Оно
становится особенно важным при построении диаграмм в следующих разделах.
При комбинировании атомных орбиталей следует помнить, что
Число получаемых МО = Число исходных МО
Молекулярные орбитали водорода
Рассмотрим образование связи в молекуле водорода с точки зрения теории
молекулярных орбиталей.
Каждый атом водорода имеет одну ls-атомную орбиталь. Описание
связывания в молекуле Н2 с позиции метода молекулярных орбиталей
основывается на предположении о возможности перекрывания этих атомных
орбиталей при сближении атомов. ls-Орбитали могут перекрываться, так как они
имеют одинаковую симметрию. (Вопрос о роли симметрии более подробно
рассмотрен при обсуждении связывания в других двухатомных молекулах.)
134
3. Гомоядерная ковалентная связь
Обозначим волновую функцию, относящуюся к электрону одного атома
водорода, как \|/( ls)A, а волновую функцию электрона второго атома
водорода - как \|/(ls)B. Используя уравнения 3.17 и 3.18, можно получить новые
волновые функции как линейные комбинации \|/(Ь)А и \|/(ls)B (уравнения
3.19 и 3.20). Поскольку молекула водорода является гомоядерной
двухатомной молекулой, то вклад обеих атомных орбиталей в молекулярные орбитали
одинаков.
^МО(синфазная) = "М^д + VO*)Bl 0-19)
УмО(противофазная) = N №0*)А ~ ^U^ (32°)
Каждая ИЗ НОВЫХ МОЛекуЛЯрНЫХ ВОЛНОВЫХ ФУНКЦИЙ Ч'мО(синфазная) И
^моспротивофазная) характеризуется энергией, которая является функцией
расстояния между ядрами атомов водорода. Как показано на рис. 3.12 энергия
^мо(синфазная) все™а ниже> чем энергия У МО( противофазная г Более того,
зависимость энергии Умо(синфазная) от межъядерного расстояния имеет минимум,
который соответствует максимальной стабильности. В то же время
аналогичная кривая для Умо(противофазная)не имеет минимумов и соответствует
возрастанию нестабильности при сближении ядер.
Волновая функция Ч'мо(синфазная) соответствует связывающей
молекулярной орбитали, а Ч>М0(пРотивофазная) ~ разрыхляющей орбитали. Образование
этих молекулярных орбиталей проиллюстрировано рис. 3.13. Видно, что
Умо(синфазная) более стабильна (стабилизирована) по сравнению с исходными
АО, а Умо(противофазная)> наоборот, менее стабильна (дестабилизирована).
(Энергия дестабилизации Умо(противофазная) несколько больше энергии
стабилизации Умо(синфазная)> однако рассмотрение причин этого явления
выходит за рамки нашего учебника.)
тМО(п роти вофазная)
Межъядерное расстояние Н—Н
Рис. 3.12. Зависимость энергии молекулярных орбиталей Умо(синфазная) и
Умо(противофазная)> полученных линейной комбинацией Ь-атомных орбиталей атомов
водорода, от межъядерного расстояния.
3.12. Метод молекулярных орбиталей
135
(Г)
УмО(противофаэная) УмО(раэрыхляюшая) MV( 1*)А ~ У( ls)B]
¥(\S)
)=mos)A + ws)B]
^МО(синфазная) т МО(связываюшая
Рис. 3.13. Синфазная и противофазная комбинации двух ls-атомных орбиталей
приводят к двум молекулярным орбиталям, одна из которых стабилизирована (имеет
меньшую энергию), а вторая дестабилизирована (имеет большую энергию) по
сравнению с исходными АО.
Для комбинации 15-атомных орбиталей нормировочный множитель
равен 1/VT. Теперь можно переписать уравнения 3.19 и 3.20 в их окончательной
форме:
^(связывающая) = 0/^ )[У(1*)А + у(15)в] (3.21)
МЧ10(разры™яющая)= O/Vl )[l|/(l5)A - y(\s)B] (3.22)
Размещение электронов по орбиталям в молекуле водорода
► До сих пор при обсуждении метода молекулярных орбиталей совершенно не
Правила заполнения упоминались электроны! Но поскольку волновые функции уже
орбиталей: рассчитаны, то можно разместить на них электроны путем
заем, разд. 2.18 пол нения сначала нижних, а затем и более высоко лежащих
орбиталей.
В молекуле водорода всего два электрона, которые заполняют низшую по
энергии орбиталь и имеют противоположные спины. Энергетическая
диаграмма молекулярных орбиталей для молекулы Н2 показана на рис. 3.14.
Важно, что оба электрона оказываются на связывающей орбитали, а
разрыхляющая орбиталь остается пустой.
Как выглядят молекулярные орбитали водорода?
Связывающая и разрыхляющая орбитали молекулы Н2 схематически
показаны на рис. 3.15. Комбинирование двух ls-орбиталей может быть нагляднее
всего представлено перекрыванием двух окружностей либо в фазе, либо в
противофазе. Это показано слева на рис. 3.15,а и б. Такого типа изображения
будут использованы во всей книге.
Справа на рис. 3.15,а и б молекулярные орбитали показаны более
подробно. На рисунке связывающей орбитали показана область наиболее вероятно-
136
3. Гомоядерная ковалентная связь
тМО(разрыхляющая)
т МО(связывающая)
н н2 н
Рис. 3.14. Связывающая и разрыхляющая орбитали молекулы Н2, полученные
линейной комбинацией атомных ls-орбиталей. Два электрона заполняют низшую по
энергии связывающую орбиталь. (Значение символов a(ls) и a*(\s) объясняется ниже
в разд. 3.12 и 3.15.)
го пребывания электронов. Эта ситуация соответствует образованию связи
Н—Н в теории валентных связей. Отрицательный заряд сконцентрирован
преимущественно в области между двумя ядрами, что приводит к снижению
энергии их электростатического отталкивания.
Разрыхляющая орбиталь молекулы водорода показана на рис. 3.15,5.
Наиболее важная ее черта — наличие узловой плоскости, проходящей между
ядрами: при заполнении этой орбитали вероятность обнаружения электрона
в любой точке узловой плоскости равна нулю. Следствием этого является тот
факт, что вероятность обнаружить электрон, находящийся на этой
разрыхляющей орбитали, в пространстве между ядрами меньше, чем для случая
просто двух не взаимодействующих ядер, и гораздо меньше, чем для электрона,
находящегося на связывающей орбитали. Таким образом, при заполнении
этой орбитали происходит увеличение отталкивания между ядрами.
Обозначение молекулярных орбиталей в молекуле Н2
На рис. 3.14 появились некоторые новые обозначения. Связывающая МО
обозначается, как a(ls), а разрыхляющая - как o*(ls). Эти символы содержат
информацию о симметрии орбиталей.
^~ Молекулярная орбиталь имеет a-симметрию, если она симметрична отно-
Более детально о сительно линии связи, т. е. не происходит изменения знака ор-
a и а*-орбиталях: битали при ее вращении вокруг линии, соединяющей атомы,
см. разд. 3.15 Молекулярная а*-орбиталь должна отвечать двум
требованиям:
1) быть a-орбиталью, т. е. не менять знак при повороте вокруг линии
связи;
2) иметь узловую плоскость между ядрами, что отражено наличием
звездочки в обозначении этой орбитали.
х
3.13. Свойства молекулы Н2
137
эквивалентна
Ч'МО(связывающая)
• I эквивалентна
т МО(разрыхляюшая)
Узловая
плоскость
Рис. 3.15. Схематическое изображение связывающей (а) и разрыхляющей (б)
орбиталей молекулы Н2. Ядра показаны черными точками.
По отношению к паре ядер:
а-орбиталь симметрична относительно линии связи;
а*-орбиталь симметрична относительно линии связи и имеет узловую
плоскость между ядрами.
Используя эти новые обозначения, можно записать электронную
конфигурацию основного состояния молекулы водорода, как o(ls)2, что соответствует
образованию а-связывающей орбитали из двух 15-атомных орбиталей и ее
заполнению двумя электронами.
КАКИЕ СВОЙСТВА МОЛЕКУЛЫ Н2 МОГУТ БЫТЬ
ПРЕДСКАЗАНЫ ТЕОРИЕЙ ВАЛЕНТНЫХ СВЯЗЕЙ И
МЕТОДОМ МОЛЕКУЛЯРНЫХ ОРБИТАЛЕЙ?
Теперь можно сравнить описания связывания в молекуле водорода в рамках
метода молекулярных орбиталей и теории валентных связей.
1. Структура Льюиса показывает, что молекула водорода имеет одну
одинарную ковалентную связь Н—Н и все валентные электроны в ней спарены.
3. Гомоядерная ковалентная связь
2. Теория валентных связей показывает, что связь в молекуле Н2 может
быть описана волновой функцией, имеющей как ковалентную, так и ионную
составляющие. Основной является ковалентная составляющая. Два
валентных электрона спарены.
3. В соответствии с методом молекулярных орбиталей связь в молекуле
водорода является главным образом ковалентной и осуществляется за счет
локализованной связывающей орбитали. Два валентных электрона спарены.
Таким образом, во всех случаях получаются очень близкие результаты.
ГОМОЯДЕРНЫЕ ДВУХАТОМНЫЕ МОЛЕКУЛЫ s-ЭЛЕМЕНТОВ -
ЭЛЕМЕНТОВ ВТОРОГО ПЕРИОДА
В этом и следующих разделах данной главы для двухатомных молекул Х2 (X -
элемент второго периода) построены энергетические диаграммы,
аналогичные приведенной на рис. 3.14. Для каждой молекулы сделана попытка с
помощью метода молекулярных орбиталей предсказать некоторые ее свойства,
а затем сравнить их с экспериментальными данными. Прежде всего
рассмотрено связывание в двухатомных молекулах Li2 и Ве2. Первая из них
действительно существует в газовой фазе, тогда как о существовании второй ничего
не известно. Можно ли объяснить нестабильность Ве2?
Li2
Электронная конфигурация основного состояния атома лития: [He^s1. На
рис. 3.16 показано, какие молекулярные орбитали получаются при
комбинировании 25-АО двух атомов лития. Совершенно очевидно сходство с
образованием МО в молекуле водорода (рис. 3.14), ходя главное квантовое число п
в данном случае равно не 1, а 2.
Из рис. 3.16 видно, что электронная конфигурация молекулы Li2 — a(2s)2,
или в более полной форме - a(ls)2a*(ls)2o(2s)2. Однако из шести электронов
молекулы Li2 четыре, находящиеся на a(ls) и a*(ls) МО, являются
электронами внутренних уровней и их суммарный вклад в связывание равен нулю.
Главную роль в связеобразовании играют валентные электроны,
находящиеся на а(2$)-орбитали.
В соответствии с методом молекулярных орбиталей два валентных
электрона занимают связывающую МО, т. е. молекула Li2 устойчива вследствие
образования простой ковалентной связи. Такой же результат получается и при
рассмотрении структур Льюиса 3.6 и 3.7. В теории валентных связей этому
соответствует образование локализованной 5-связи Li—Li.
Li I Li Li Li
3.6 3.7
Be2
Электронная конфигурация основного состояния атома Be: [He^s2. На
рис. 3.17 показано, какие молекулярные орбитали получаются при
комбинировании 25-АО двух атомов бериллия. Сами по себе орбитали совершенно
аналогичны орбиталям Li2. Снова возможно пренебречь вкладом внутренних
15-электронов, однако на этот раз на МО, полученных из 25-атомных орбита-
лей, необходимо разместить четыре электрона. Из рис. 3.17 видно, что кон-
3.14. Гомоядерные двухатомные молекулы s-элементов
139
а
X
о {2s)
2s
2s
G{2s)
Li,
Li
Рис. 3.16. Связывающая и разрыхляющая орбитали молекулы Li2, полученные
линейной комбинацией 25-атомных орбиталей. Ь-Орбитали с внутренними
электронами не показаны.
фигурация основного состояния молекулы Ве2 должна быть g(2s)2g*(2s)2. И
на связывающей, и на разрыхляющей МО находится по два электрона. Т. е.
образования связи в молекуле Ве2 не должно происходить, что вполне
соответствует ее нестабильности.
Прежде чем перейти к другим элементам второго периода, необходимо
рассмотреть перекрывание /ьорбиталей и ввести понятие «порядок связи».
о
5
<ъ
2s
Be
<ИЗД
°(2s)
Be.
2s
Be
Рис. 3.17. Энергетическая диаграмма молекулярных орбиталей для молекулы Ве2.
Ь-Орбитали с внутренними электронами не показаны.
140
3. Гомоядерная ковалентная связь
ШКШ ПЕРЕКРЫВАНИЕ р-ОРБИТАЛЕЙ
В разд. 3.12 было показано, что перекрывание двух s-орбиталей приводит к
образованию молекулярных орбиталей симметрии о и а*. Так как s-орбитали
имеют сферическую форму, то результат их перекрывания никак не зависит
от их взаимной ориентации. Чтобы убедиться в этом, посмотрите на рис. 3.15
и мысленно поверните одну из орбиталей вокруг оси, соединяющей ядра.
Изменит ли это характер перекрывания орбиталей? Повторите тот же
эксперимент, повернув орбиталь в другую сторону.
Ситуация с атомными /7-орбиталями совершенно другая. Поместим два
ядра вдоль оси z прямоугольной системы координат на расстоянии,
соответствующем длине химической связи. (Выбор оси z в данном случае является
произвольным, но общепринятым.) 2pz-AO направлены друг на друга, их
перекрывание может происходить или в фазе, или в противофазе, так, как это
показано на рис. 3.18,я и б соответственно. Образующаяся при этом связыва-
^~ ющая молекулярная орбиталь показана в правой части рис. 3.18,а. Эта
Напомним, чтоа-орби- орбиталь является а-орбиталью. Соответствующая ей разрых-
тали симметричны от- ляющая молекулярная орбиталь имеет узловую плоскость, про-
носительно вращения ходящую между ядрами (рис. 3.18,6). Эта плоскость возникает в
вокруг оси, соединяю- дополнение к узловым плоскостям, которые имеет каждая из
щей ядра атомов. двух участвующих в ее образовании р-АО. Эта разрыхляющая
орбиталь является а*-орбиталью.
Рассмотрим перекрывание 2/^-атомных орбиталей (рис. 3.18,в и г). Так
как эти орбитали направлены перпендикулярно оси z, на которой
расположены ядра, то и взаимодействовать они могут только за счет бокового
перекрывания. Это перекрывание менее эффективно, чем прямое перекрывание,
которое возможно для 2/у-АО, т.е. интеграл перекрывания S имеет меньшее
значение. Результатом синфазной комбинации 2/^-атомных орбиталей
является связывающая МО, узловая плоскость которой совпадает с узловыми
плоскостями исходных АО (рис. 3.18,в). Эта молекулярная орбиталь
называется л-орбиталью. 71-МО является асимметричной по отношению к
вращению вокруг оси, соединяющей ядра. При повороте на 180° знак орбитали
меняется на противоположный.
Результатом противофазной комбинации 2рх-АО является образование
разрыхляющей молекулярной орбитали, которая, сохраняя узловые
плоскости исходных 2/^-атомных орбиталей, имеет и дополнительную,
проходящую между взаимодействующими ядрами (рис. 3.18,г). Эта МО
обозначается как к*.
Молекулярная орбиталь имеет симметрию л*, если она удовлетворяет
двум критериям:
1) меняет знак (фазу) при повороте вокруг оси, соединяющей
взаимодействующие ядра, т.е. является я-орбиталью;
2) имеет узловую плоскость между взаимодействующими ядрами, что
отражено наличием звездочки в символе орбитали.
Перекрывание двух 2/уатомных орбиталей происходит так же, как и
перекрывание 2рх-АО, но возникающие при этом связывающая и
разрыхляющая МО будут повернуты на 90° относительно МО, сформированных орбита-
лями 2рх. Чтобы проиллюстрировать образование к(2р )- и л*(2/> )-МО,
нарисуйте диаграммы, аналогичные приведенным на рис. 3.18.
3.15. Перекрывание р-орбиталей
141
-ОЭОЭ-? ■=> -О
2Р.-
2PZ
*(%»,)
I '-,, %_ k k
2/>-
2/>z
o*(2p2)
0>
У У '
IP*
1PX
<=$
t=c>
n{2Px)
0 ,
z
x*Vpx)
Y
Рис. 3.18. Перекрывание двух 2/ьатомных орбиталей. о - прямое перекрывание
вдоль оси z дает а-связывающую МО; б - образование а*-МО (разрыхляющей);
в — перекрывание боковых поверхностей 2/уатомных орбиталей приводит к
формированию связывающей я-МО'; г — образование разрыхляющей я*-МО.
По отношению к паре ядер:
я-орбиталь является асимметричной, т.е. меняет знак при повороте
вокруг оси, соединяющей взаимодействующие ядра;
я*-орбиталь также является асимметричной, но кроме того имеет
узловую плоскость, проходящую между ядрами.
142
3. Гомоядерная ковалентная связь
(Г)
о*(2р:)
2р
2р
°(2Р:)
Рис. 3.19. Энергетическая диаграмма с- и я-МО, возникающих из двух наборов
вырожденных 2рх- , 2р■ -, 2pz- атомных орбиталей. Ядра атомов лежат вдоль оси z.
В гомоядерных двухатомных молекулах комбинация двух наборов
вырожденных 2/7-АО (по три орбитали на атом) приводит к образованию одной а- и
двух я-связывающих и одной о*- и двух к*- разрыхляющих молекулярных
орбиталей. (Необходимо напомнить, что шесть атомных орбиталей дают шесть
молекулярных орбиталей.) Единственная разница между двумя я-орбиталя-
ми заключается в их различной ориентации; следовательно, они являются
энергетически вырожденными. То же утверждение относится и к
разрыхляющим я*-МО. На рис. 3.19 показана диаграмма энергетических уровней,
возникающих из двух наборов 2/?-атомных орбиталей. Как будет показано в
дальнейшем, относительная энергия образующихся МО может меняться.
И порядок связи
Порядок ковалентной связи является мерой взаимодействия между атомами.
Три наиболее распространенных типа связи: простая (одинарная) связь (по-
►- рядок связи 1), двойная (порядок 2) и тройная (порядок 3). Необходимо от-
Нецелочисленные метить, что порядок связи не ограничивается только целыми
порядки связей: числами.
см. разд. 3.22 Вполне понятно, что подразумевается, когда говорят об
образовании одинарной связи в молекуле водорода, но при
рассмотрении порядка связи в других молекулах следует соблюдать осторож-
^~ ность. Порядок ковалентной связи может быть определен по уравнению 3.23.
Несвязывающие МО: Электроны, расположенные на несвязывающих орбиталях, в
см. разд. 4.5 расчет не принимаются.
Порядок связи
=.,г
исло связывающих
электронов
Число разрыхляющих \
электронов / ^ '
Для молекулы водорода порядок связи равен 1, так как на связывающей
орбитали находится два электрона, а разрыхляющая МО пуста (см. рис. 3.14).
Аналогично получается порядок связи 1 и для Li2 (рис. 3.16 и уравнение 3.24).
Необходимо принимать во внимание только валентные электроны. Исполь-
3.18. Гомоядерные двухатомные молекулы р-элементов: F2 и 02 143
зуя уравнение 3.23, можно понять, почему в разд. 3.14 говорилось, что
электроны внутренних уровней компенсируют влияние друг друга.
Порядок связи в Li2 = 0,5(2 - 0) = I (3.24)
Для молекулы Ве2 порядок связи равен 0, так как на связывающей
орбитали находится два электрона, но и на разрыхляющей МО — также два
(рис. 3.17). Т. е. связь Be—Be не образуется, что вполне соответствует
экспериментальным данным.
Порядок ковалентной связи может быть рассчитан при рассмотрении
энергетических диаграмм молекулярных орбиталей.
Порядок связи = 0.5^^^^^ . Чис^даь^яющих )
ВЗАИМОСВЯЗЬ МЕЖДУ КРАТНОСТЬЮ СВЯЗИ,
ЕЕ ДЛИНОЙ И ЭНЕРГИЕЙ РАЗРЫВА
В разд. 3.5 при обсуждении энтальпии разрыва химических связей было
отмечено, что эта величина для тройной связи углерод—углерод С=С больше,
чем для двойной С=С, которая в свою очередь больше, чем энтальпия
разрыва простой связи С—С. На рис. 3.20 приведены зависимости энтальпии и
длины химической связи от ее порядка, а также зависимость энергии разрыва
(диссоциации) связи от длины этой связи. На основании этих данных можно
сделать следующие выводы:
• энтальпия диссоциации химической связи возрастает при увеличении
кратности связи;
• длина связи уменьшается при увеличении ее кратности;
• энтальпия диссоциации химической связи уменьшается при увеличении
ее длины.
Рис. 3.20 показывает, что эти закономерности имеют общий характер для
всех р-элементов.
ГОМОЯДЕРНЫЕ ДВУХАТОМНЫЕ МОЛЕКУЛЫ
р-ЭЛЕМЕНТОВ ВТОРОГО ПЕРИОДА: F2 и 02
В этом разделе и разд. 3.20 образование молекул В2, С2, N2, 02 и F2
рассмотрено с точки зрения метода молекулярных орбиталей и теории валентных
связей. Получаемые результаты сопоставлены с данными, которые могут
быть извлечены из структур Льюиса. Образование молекул Li2 и Ве2
происходит за счет перекрывания 2s-AO, в случае других гомоядерных двухатомных
молекул элементов второго периода в образовании связи участвуют и 2/7-
атомные орбитали.
В ходе предстоящих рассуждений наличие внутренних b-электронов
игнорируется (по причинам, обсуждаемым выше).
144
3. Гомоядерная ковалентная связь
1000 г
750
500
5 9 250
х
1000 г
160
150
^ 140
го
5 по
ее
I 12°
поЬ
100
750
500
g S 250
2 3
Порядок связи
100 ПО 120 130 140 150 160
Длина связи, пм
связи С—С
-о связи N—N
♦ связи О—О
Порядок связи
Рис. 3.20. Взаимозависимость энтальпии, длины и кратности связей С—С, N—N и
О-О.
Экспериментальный факт: стандартное состояние фтора - диамагнитный газ F2.
Электронная конфигурация основного состояния атома фтора: [Не]2522/75.
Строение молекулы F2 может быть представлено двумя структурами Льюиса
(3.8 и 3.9). Каждый атом фтора имеет завершенный октет электронов,
происходит образование простой ковалентной связи F—F. Теория валентных
связей также объясняет устойчивость этой молекулы за счет образования
простой связи.
: F
3.8
3.9
На рис. 3.21 приводится диаграмма молекулярных орбиталей,
молекулярные орбитали получены путем линейной комбинации атомных орбиталей. В
молекуле 14 валентных электронов, которые заполняют МО снизу вверх
согласно принципу последовательного заполнения наинизших орбиталей. Все
электроны спарены, что соответствует диамагнетизму F2.
3.18. Гомоядерные двухатомные молекулы р-элементов: F2 и 02 145
о(2рг)
-н-
G{2s)
Рис. 3.21. Энергетическая диаграмма молекулярных орбиталей для F2. Is-АО не
показаны. Ядра атомов фтора лежат на оси z.
Частица диамагнитна, если у нее нет неспаренных электронов.
Частица парамагнитна, если у нее есть неспаренные электроны.
Электронная конфигурация молекулы фтора может быть записана как
► c(2s)2a42s)M2pz)M2px)M2py)2n42px)2n*(2py)2
Как изменится эта п с -> "»-> <-\
Порядок связи может быть определен по уравнению 3.23. Он
конфигурация, если оказывается равным единице (уравнение 3.25), что соответству-
принять в расчет ет величине полученной при рассмотрении структур Льюиса.
электроны внутренних
уровней?
Порядок связи в F2 = 0,5(8 - 6) = 1 (3.25)
Таким образом, и структуры Льюиса, и теория валентных связей, и метод
молекулярных орбиталей приводят к одному результату.
146
3. Гомоядерная ковалентная связь
Экспериментальные факты: При 298 К кислород - газ, при 90 К он
конденсируется в голубую жидкость. Кислород парамагнитен, имеет два неспаренных
электрона.
Электронная конфигурация основного состояния атома кислорода:
[\\ъ\2!?-2рА. Диаграмма молекулярных орбиталей для 02 показана на рис. 3.22.
В молекуле 12 валентных электронов. В соответствии с правилом Хунда два
последних электрона должны занять две вырожденные по энергии к*(2рх)- и
к*(2рJ-МО. Этот вывод совпадает с экспериментальными данными:
молекула кислорода парамагнитна и имеет два неспаренных электрона.
Электронная конфигурация молекулы кислорода может быть записана
как
а(2*)2 о*(25)2 с(2р)2 к(2рх)2 к(2р)2 к*(2рхУ п*(2р/
(3.26)
Порядок связи в 02 = 0,5 (8 - 4) = 2
Рассмотрим структуру Льюиса для этой молекулы. Электронная
конфигурация основного состояния атома кислорода: [Не]2522/?4, и, как показано в
структуре ЗЛО, атом кислорода имеет два неспаренных электрона. Эти элек-
(Т)
-н-
2р
О
o*(2pz)
<т(2Р:)
<j(2s)
О,
2р
О
Рис. 3.22. Энергетическая диаграмма молекулярных орбиталей для 02. \s-AO не
показаны. Ядра атомов кислорода лежат на оси z.
3.19. Смешивание орбиталей и а-л-взаимодействие 147
троны и используются для образования двойной (в соответствии с
уравнением 3.26) связи. Каждый атом кислорода в структурах 3.11 и 3.12 подчиняется
правилу октета.
О
о : о
О:
:0
3.10
3.11
3.12
Однако проблема заключается в том, что структуры Льюиса не
предсказывают наличия в молекуле 02 двух неспаренных электронов; простая теория
валентных связей также приходит к этому результату. В то же время метод
молекулярных орбиталей дает результаты, наилучшим образом совпадающие с
экспериментальными данными (двойная связь 0=0 и наличие двух
неспаренных электронов в молекуле). Объяснение парамагнетизма 02 является
классическим примером успехов метода молекулярных орбиталей.
СМЕШИВАНИЕ ОРБИТАЛЕЙ И а я-ВЗАИМОДЕЙСТВИЕ
Прежде чем приступить к описанию образования связей в молекулах В2, С2 и
N2, необходимо рассмотреть две новые концепции: смешивание орбиталей и
а-л-взаимодействие, приводящее к обращению с-л-последовательности
орбиталей. Однако понимание причин возникновения данных явлений не обя-
сг*(2/7г)
2р
2р
о (2s)
Рис. 3.23. Диаграмма молекулярных орбиталей, показывающая примерный порядок
орбиталей, образующихся при комбинировании 2s- и 2/?-атомных орбиталей. Эта
диаграмма применима к формированию гомоядерных двухатомных молекул
из/?-элементов конца второго периода (О и F), ядра которых лежат на z-оси молекул.
148
3. Гомоядерная ковалентная связь
зательно для качественного описания молекулярных орбиталей этих
двухатомных молекул, и можно вернуться к этой теме после обсуждения разд. 3.20.
При построении молекулярных орбиталей из линейных комбинаций
атомных орбиталей первоначальный подход предполагал возможность
перекрывания только сходных орбиталей. На рис. 3.23 показано перекрывание
между 2s-АО двух идентичных атомов с образованием g(2s)- и a*(2s)-MO и
перекрывание 2/ьатомных орбиталей с образованием о(2р)-, к(2р)-, к*(2р)- и
а*(2/?)-МО. Порядок следования орбиталей приблизителен. Отметим, что
с(2р) имеет более низкую энергию, чем л(2/?)-уровни.
Теперь можно ввести новую концепцию: если возможно смешивание АО,
имеющих одинаковую симметрию и энергию, с образованием МО, то, вероятно,
также могут смешиваться и МО, имеющие одинаковую симметрию и энергию.
(Т)
о\2р)
<**(2р)
Л2р)
Л2р)
Л2р)
о(2р)
7Г(2/7)
*(2р)
o\2s)
c{2s) ^^
o{2s)
Смешивание
о-орбиталей
^>
а(2р)
к(2р)
<f(2s)
а(Ъ)
Смешивание
а*-орбиталей
:>
Рис. 3.24. Влияние смешивания молекулярных орбиталей на их взаимное
расположение для двухатомных молекул элементов второго периода. Смешивание о- и
^-орбиталей рассмотрено отдельно.
3.19. Смешивание орбиталей и о-л-взаимодействие 149
На рис. 3.23 показаны две а-орбитали [o(2s) и с(2р)] и две а*-орбитали
[o*(2s) и а*(2/?)]. Для р-элементов начала второго периода 2s- и 2р-АО отно-
^ сительно близки по энергии (550 кДж/моль для бора), тогда как для элемен-
Различие энергий тов конца второго периода они сильно разнятся (2100 кДж/моль
s-ир-орбиталей: ДЛЯ фтора). Таким образом, молекулярные орбитали, получен-
см. разд. 2.15 ные из 2s- и 2/?-атомных орбиталей легких р-элементов, близки
друг к другу. Если орбитали имеют близкую энергию и
симметрию, то они могут смешиваться. Результаты смешивания o(2s) и а(2р), а так
же c*(2s) и с*(2р) показаны на рис. 3.24.
Таким образом, если 2s- и 2р-АО близки по энергии, то смешивание o(2s)-
и а(2/?)-молекулярных орбиталей даст а(2/?)-орбиталь, лежащую выше по
энергии, чем тс(2/?)-уровни. Это верно для р-элементов начала второго
периода (В, С и N). Конечно, смешивание орбиталей происходит и в случае более
тяжелых элементов, однако энергетическое различие делает этот процесс
менее значительным, и о(2/?)-орбиталь по-прежнему лежит ниже к(2р).
Наконец, на рис. 3.25 показана полная (с учетом смешивания) схема
молекулярных орбиталей, образованных из 25- и 2р-АО. Обратите внимание,
что отдельные атомные орбитали принимают участие в образовании
нескольких молекулярных орбиталей. Эта тема будет повторно обсуждаться при
рассмотрении связывания в молекуле СО в разд. 4.14. Необходимо отметить, что
несмотря на эффекты смешивания общее число МО равно числу исходных
атомных орбиталей.
Теперь можно перейти к рассмотрению молекул В2, С2 и N2. При этом
будет использована схема МО, приведенная на рис. 3.25.
Рис. 3.25. Энергетическая диаграмма МО, образованных из 2s- и 2/7-атомных
орбиталей. Учитываются эффекты смешивания молекулярных орбиталей. Эта диаграмма
может быть использована при описании связывания в двухатомных гомоядерных
молекулах р-элементов начала второго периода (В, С, N).
3. Гомоядерная ковалентная связь
ГОМОЯДЕРНЫЕ ДВУХАТОМНЫЕ МОЛЕКУЛЫ р-ЭЛЕМЕНТОВ
ВТОРОГО ПЕРИОДА: В2, С2 и N2
В2
Экспериментальный факт: Пары бора содержат парамагнитные молекулы В2.
Электронная конфигурация основного состояния атома бора: [Не]2522/?1.
Диаграмма молекулярных орбиталей молекулы В2 показана на рис. 3.26.
Каждый атом бора имеет три валентных электрона. Четыре электрона
заполняют g(2s)- и g*(2s)-MO. Чтобы молекула была парамагнитной, оставшиеся два
электрона должны расположиться по одному на двух вырожденных к(2рх)- и
71(2^)-орбиталях. Таким образом, парамагнетизм В2 является
экспериментальным доказательством а-я-взаимодействия (более детальное обсуждение
приведено ниже в разделе, посвященном С2).
Из рис. 3.26 видно, что электронная конфигурация молекулы В2 может
быть записана как
a(2s)2o*(2s)2п(2рх)] к(2р/
Порядок связи в молекуле, в соответствии с уравнением 3.27, равен 1.
Порядок связи в В2 = 0,5 (4 — 2) = 1 (3.27)
Рассмотрим структуру Льюиса для этой молекулы. Можно ожидать
спаривания шести электронов (по три от каждого атома бора) с образованием
тройной связи (структуры 3.13 и 3.14). Правило октета не выполняется.
Такой порядок связи не соответствует ни предсказаниям метода молекулярных
орбиталей, ни экспериментальным данным. Существует альтернативный
вариант структуры Льюиса (структуры 3.15 и 3.16): порядок связи равен
единице, а у каждого атома бора остается неподеленная пара электронов. Хотя этот
вариант кажется более приемлемым с точки зрения самой связи, он не
объясняет парамагнетизма молекулы. Необходимо помнить, что в соответствии
с методом Льюиса два электрона всегда будут давать пару.
в : в в^^в : в : в : : в — в :
3.13 3.14 3.15 3.16
Таким образом, метод структур Льюиса не дает удовлетворительного
объяснения свойств В2. В то же время метод молекулярных орбиталей
предсказывает и порядок связи, и магнитные свойства этой молекулы,
соответствующие экспериментальным данным.
с2
Экспериментальный факт: Молекула С2 диамагнитна.
Электронная конфигурация основного состояния атома углерода: [Не]2522/?2.
Структура Льюиса молекулы С2 соответствует образованию двойной связи
3.20. Гомоядерные двухатомные молекулы р-элементов: В2, С2 и N2 151
с?)
0*(2/О
п*(2рх) я*(2ру)
о(2р.)
я(2рх) я(2р})
o*(2.v)
-н-
-и-
ст(2л)
в2
| А 0*(2,О
X
я*(2рх) я*(2ру)
о{2р:)
-Н- -Н-
лг(2/7х) я(2/»,.)
<т*(2л)
-н-
-н-
ст(2л)
с2
о. -
/r*(2/>t) **(2^.)
о(2р.)
-н- -н-
л(2/>л) тг(2/7,)
о*(2л)
-Н-
-Н-
CF(2l)
N,
Рис. 3.26. Энергетическая
диаграмма молекулярных ор-
биталей молекулы В2. Орби-
тали o(\s) и o*(\s) с
внутренними электронами не
показаны.
Рис. 3.27. Энергетическая
диаграмма молекулярных ор-
биталей молекулы С2. Орбита-
ли o(\s) и g*(\s) с
внутренними электронами не показаны.
Рис. 3.28. Энергетическая
диаграмма молекулярных ор-
биталей молекулы N2. Орби-
тали o(\s) и a*(ls) с
внутренними электронами не
показаны.
С=С и наличию у каждого атома неподеленной пары электронов (структуры
3.17 и 3.18). Все электроны спарены.
С : С :
3.17
3.18
Диаграмма молекулярных орбиталей приведена на рис. 3.27. Восемь
валентных электронов располагаются на МО, возникающих в результате
комбинирования 2s- и 2/7-атомных орбиталей. Четыре электрона занимают o(2s)-
и o*(2s)-MO. Если к(2рх) и к{2р) лежат ниже по энергии, чем о(2р^, то
оставшиеся четыре электрона полностью займут эти две вырожденные орбитали.
Т. е. эта молекула должна быть диамагнитна, что соответствует
экспериментальным данным. Это подтверждает правильность предположения о том, что
в данном случае а(2/?г)-орбиталь имеет большую энергию, чем к(2рх)- и
к(2р)-МО. Если бы это было не так, то электронная конфигурация
молекулы С2 была бы o(2s)2 g*(2s)2 с(2р^)2 к(2рх){ к(2рУ, что соответствовало бы
образованию парамагнитной частицы и противоречило бы эксперименту.
Исходя из рис. 3.27 можно записать электронную конфигурацию
молекулы как g(2s)2 o*(2s)2 к(2р)2 к(2р)2.
152
3. Гомоядерная ковалентная связь
Порядок связи может быть рассчитан, исходя из уравнения 3.28 и
рис. 3.27.
Порядок связи в С2 = 0,5 (6 - 2) = 2 (3.28)
В заключение следует отметить, что метод структур Льюиса, теория
валентных связей и метод молекулярных орбиталей одинаково успешно объясняют
свойства молекулы С2. В данном случае в отношении метода молекулярных
орбиталей именно лучше использовать выражение «объясняет», чем
«предсказывает», так как путем простых вычислений практически невозможно
ответить на вопрос, будут ли к(2рх)- и к(2ру)-МО лежать выше или ниже орби-
тали o(2pz). Однако этот метод позволяет объяснить магнитные свойства С2.
щ
Экспериментальные факты: Молекула N2 обладает сравнительно низкой
реакционной способностью. Этот газ часто используется для создания инертной
атмосферы, если исходные реагенты или продукты реакции легко
взаимодействуют с кислородом воздуха или парами воды. Энтальпия диссоциации
молекулы N2 очень высока, а длина связи напротив мала (табл. 3.7). Молекула
азота диамагнитна.
Структура Льюиса для молекулы N^ Электронная конфигурация
основного состояния атома азота: [Не]2522/?3. Три 2р-электрона не спарены
(структура 3.19). Таким образом для завершения октетов требуется образование
тройной связи N=N. Это показано на структурах 3.20 и 3.21. Теория валентных
связей также объясняет устойчивость молекулы азота образованием тройной
связи, кроме того у каждого из атомов остается еще и неподеленная пара
электронов.
* N *
3.19
: n
n :
3.20
3.21
Таблица 3.7. Результаты экспериментальных исследований, выполненных для
двухатомных гомоядерных молекул элементов второго периода
Молекула
йг
Ве2
(не наблюдалась
экспериментально)
в2
с2
N2
о2
F2
Длина
связи, нм
0,267
-
0,159
0,124
0,110
0,121
0,141
Энтальпия диссоциации
связи,
кДж/людь
ПО
-
297
607
945
498
159
Магнитные свойства
Диамагнитна
-
Парамагнитна
Диамагнитна
Диамагнитна
Парамагнитна
Диамагнитна
3.20. Гомоядерные двухатомные молекулы р-элементов: В2, С2 и N2 153
Диаграмма молекулярных орбиталей для N2 приведена на рис. 3.28.
Заполнение МО десятью валентными электронами приводит к их полному
спариванию, что соответствует экспериментальным данным о диамагнетизме
молекулы азота.
^ Для N2 а-тс-взаимодействия к(2р)- и а(2/?)-орбиталей не влияет ни на по-
Фотоэлектронная рядок связи, ни на количество неспаренных электронов. Но
спектроскопия: см. предположение о том, что к(2рх)- и к(2р)-МО лежат ниже по
дополнение 3.6 энергии, чем о(2/^)-орбиталь, подтверждается результатами
исследований методом фотоэлектронной спектроскопии.
Исходя из рис. 3.28 можно записать электронную конфигурацию
молекулы азота как
а(25)2 o*(2s)2 к(2рх)2 к(2р/ а(2р/
Из уравнения 3.29 следует, что порядок связи равен трем, что соответствует
короткому межъядерному расстоянию и большой энтальпии диссоциации
связи (табл. 3.7).
Порядок связи в N2 = 0,5 (8 - 2) = 3 (3.29)
В заключение следует еще раз указать на тот факт, что применение структур
Льюиса, теории валентных связей и метода молекулярных орбиталей
приводит к одному и тому же результату: молекула азота диамагнитна и устойчива
за счет образования тройной связи.
Дополнение 3.6. Фотоэлектронная спектроскопия (ФЭС)
Фотоэлектронная спектроскопия — метод измерения энергии заполненных
атомных и молекулярных орбиталей.
Облучение молекул или атомов электромагнитным излучением энергии Е
приводит к эмиссии электронов из изучаемого образца. Каждый электрон
характеризуется так называемой энергией связи, которая соответствует
минимальной энергии, необходимой для отрыва этого электрона от атома или
молекулы.
Энергия, которой обладает электрон после эмиссии, определяется
уравнением
Энергия электрона = Е — Энергия связи электрона
где £ должна быть больше энергии связи. В этом заключается суть метода
измерения энергии связи.
Теорема Купманса устанавливает соответствие между энергией связи
электрона и орбиталью, которую он занимает. То есть энергия связи может
служить мерой энергии орбитали.
Фотоэлектронная спектроскопия является важным экспериментальным
методом измерения энергий связи как валентных, так и внутренних
электронов. Зная эти энергии, можно судить о взаимном расположении орбиталей в
атомарных или молекулярных системах. О фотоэлектронной спектроскопии
см. также разд. 5.22.
154
3. Гомоядерная ковалентная связь
о*<2/>)
п*{2р) ..^ ^ " аЧ1р)
а(2р) .. ^ ЧХ
^ * """+- Н— -и- -Н- лтр)
""" -+- ■+■ -++■ -н- ++■ ++'::>,.. ^ ^
ЧГН--Н- -И--И- «<ад
о*(2«) -Н- "Н" ст(2р)
+^ _^ -••"++--...
ст(2л)
"И" *<*)
°2 F2
Рис. 3.29. Конфигурации основного электронного состояния и изменение энергии
молекулярных орбиталей для двухатомных гомоядерных молекул ^-элементов второго периода.
р-Элементы второго периода. Заключение
Итак, за счет чего происходит связывание в двухатомных молекулах Х2, где X =
В, С, N, О или F? На рис. 3.29 показано, как изменяется энергия и взаимное
расположение молекулярных орбиталей o(2s), c*(2s), g(2/?), a*(2p), к(2р) и
к*(2р) при переходе от В2 к F2.
Отметим следующие главные моменты:
1. Наблюдается эффект смешивания молекулярных орбиталей, который
приводит к а-я-взаимодействию, т. е. к обращению взаимного расположения
о(2р)- и к(2р)-ЫО.
2. Молекулярные орбитали, так же как и атомные, заполняются снизу
вверх, т. е. в соответствии с принципом последовательного заполнения
наинизших орбиталей.
3. Энергетические диаграммы молекулярных орбиталей могут быть с
успехом применены для определения порядка связи в молекулах.
4. Энергетические диаграммы молекулярных орбиталей могут быть
использованы для предсказания и объяснения диа- и парамагнитных свойств
молекул.
3.21. Периодические закономерности в свойствах элементов второго периода 155
ПЕРИОДИЧЕСКИЕ ЗАКОНОМЕРНОСТИ В СВОЙСТВАХ
ДВУХАТОМНЫХ ГОМОЯДЕРНЫХ МОЛЕКУЛ
ЭЛЕМЕНТОВ ВТОРОГО ПЕРИОДА
В этом разделе систематизированы данные о двухатомных гомоядерных
молекулах элементов второго периода — от Li2 до F2, за исключением «Ве2».
На рис. 3.30 показано, как изменяется длина и энтальпия диссоциации
(разрыва) связи в молекулах типа Х2 при переходе от Li к F. В ряду от В2 до N2
происходит уменьшение длины связи и одновременное увеличение
прочности связи. В ряду от N2 до F2 длина связи, наоборот, увеличивается, но
одновременно происходит и уменьшение энтальпии разрыва этой связи. Эти
изменения совпадают с изменением порядка связи в молекулах:
В2(одинарная) <С2(двойная) <М2(тройная) > 02(двойная) >Р2(одинарная)
Необходимо отметить, что установить точное количественное соответствие
между длиной связи и энтальпией разрыва связи невозможно. Такие факторы,
как межъядерное и межэлектронное отталкивание, сильно влияют на
реально наблюдаемые значения этих параметров. Например, Li2, как и В2 и F2,
имеет простую связь между атомами. Однако длина связи в Li2 равна 0,267
нм, что значительно больше, чем в двух остальных молекулах.
Соответственно энтальпия диссоциации связи Li2 очень мала. Это различие определяется
протяженностью орбиталей, вовлеченных в образование связи. 25- и 2/?-ор-
битали атома бора находятся под влиянием большего эффективного заряда
ядра, чем 2^-орбитали лития, и, следовательно, имеют меньшую
пространственную протяженность. Для достижения эффективного перекрывания
орбиталей атомы бора должны сблизиться гораздо сильнее, чем атомы лития.
о
аз
X
I
X
0,3
0,2
0,2
0.15
1000 г
800
3 600
2
:3
400
200
Li Be В
б
N О
Рис. 3.30. Изменение длины связи X—X (а) и энтальпии разрыва молекул Х2 (б).
(X - элементы второго периода от Li до F).
156
3. Гомоядерная ковалентная связь
ЕЕ9 ДВУХАТОМНЫЕ ЧАСТИЦЫ 02, [02]+, [02]~ и [02]2~
Энергетические диаграммы молекулярных орбиталей, построенные для
двухатомных гомоядерных молекул, могут быть использованы для
предсказания свойств или объяснения экспериментальных данных, полученных для
частиц, являющихся производными этих молекул. В качестве примера
рассмотрим 02, [02]\ [02]" и [02]2" (см. табл. 3.8).
Диаграмма молекулярных орбиталей для 02 показана на рис. 3.22.
Электронная конфигурация
c(2s)2 а*(2*)2 c(2pz)2 п(2рх)2 к(2ру)2 к*(2рхУ к*(2РуУ
соответствует образованию двойной связи и объясняет парамагнетизм
молекулы кислорода.
Что произойдет при окислении или восстановлении 02? Добавление
электронов соответствует восстановлению, а удаление электронов —
окислению. Используя диаграммы МО, можно проследить за изменением длины,
порядка и энтальпии диссоциации связи, за изменением количества неспа-
ренных электронов при окислении или восстановлении этой молекулы.
Правила заполнения орбиталей электронами остаются неизменными: при
добавлении электрона он занимает низшую по энергии свободную орбиталь, при
удалении электрона освобождается высшая по энергии занятая орбиталь.
Первой стадии окисления молекулы кислорода (уравнение 3.30)
соответствует удаление электрона с я*-орбитали (рис. 3.22). Диаграмма
молекулярных орбиталей показывает, что частица [02]+ имеет один неспаренный
электрон, т. е. является парамагнитной: [02]+ — это катион-радикал.
Окисление и восстановление - процессы, связанные с потерей или
соответственно приобретением электронов.
Окисление - процесс потери одного или более электронов.
Восстановление - процесс приобретения одного или более
электронов.
°2 -> [02]+ + е" (3.30)
Порядок связи в [02]+ = 0,5 (8 - 3) = 2,5 (3.31)
Таблица 3.8. Экспериментальные данные, полученные для молекулы 02 и ее
производных
Частица Длина связи, им Энтальпия разрыва Магнитные свойства
связи, кДж/моль
02 0,121 498 Парамагнитна
[02]+ 0,112 644 Парамагнитна
[02]~ 0,132 360 Парамагнитна
[02]2" 0,149 149 Диамагнитна
3.23. Двухатомные гомоядерные молекулы
157
При переходе от 02 к [02]+ порядок связи увеличивается от 2 до 2,5
(уравнения 3.26 и 3.31). Это отражается в увеличении энергии диссоциации связи и
уменьшении ее длины (табл. 3.8).
Одно- и двухэлектронное восстановление молекулы кислорода
происходит по уравнениям 3.32 и 3.33. Из рассмотрения диаграммы молекулярных
орбиталей (рис. 3.22) видно, что супероксид-ион [02]~ образуется при
добавлении одного электрона на одну из л*-орбиталей. Ион [02]~ парамагнитен
(это анион-радикал), порядок связи в нем равен 1,5. Данные табл. 3.8
подтверждают ослабление и удлинение связи кислород-кислород при переходе
от 02 к [02]".
Катион-радикал - это катион, имеющий неспаренный электрон.
Анион-радикал это анион, имеющий неспаренный электрон.
02 + е" -> [02]" (3.32)
02 + 2е~ -> [02]2" (3.33)
^ Добавление двух электронов молекуле 02 с образованием пероксид-иона
Оксиды, пероксиды и [°2l2~ приводит к формированию диамагнитных частиц, и по-
супероксидьг см рядок связи становится равным 1. Это также согласуется с экс-
разд 11 12 и 13 10 периментальными данными. Дианион [02]2~ имеет более
длинную и слабую связь, чем 02 и [02]~.
ДВУХАТОМНЫЕ ГОМОЯДЕРНЫЕ МОЛЕКУЛЫ.
ИЗМЕНЕНИЕ СВОЙСТВ ПО ГРУППАМ
В этом разделе рассмотрены изменения в свойствах двухатомных гомоядер-
ных молекул при движении по группам элементов, а не по периодам.
Прежде всего внимание уделено молекулам щелочных металлов и галогенов, затем
рассмотрены элементы группы 15, для которых изменение энтальпии
разрыва связи X—X сильно влияет на строение.
Группа 1: щелочные металлы
(из обсуждения исключен водород)
Все элементы группы I имеют электронную конфигурацию основного
состояния /is1. Гомоядерные двухатомные молекулы щелочных металлов могут
быть обнаружены в газовой фазе.
Образование молекулы Li2 из двух атомов лития показано на рис. 3.16.
Аналогичные диаграммы могут быть нарисованы и для образования Na2, K2,
Rb2, Cs2 и Fr2, хотя экспериментальные данные становятся все более
разрозненными при движении сверху вниз по группе (табл. 3.9).
Метод молекулярных орбиталей предсказывает образование простых
связей для всех этих молекул. Такой же результат получается и при
использовании структур Льюиса и теории валентных связей. Каждый атом щелочного
158
3. Гомоядерная ковалентная связь
Таблица 3.9. Экспериментальные данные, полученные для гомоядерных
двухатомных молекул элементов группы I
Молекула Электронная конфигурация Длина связи, нм Энтальпия
Х2 основного состояния диссоциации
элемента X связи, кДж/моль
U2 [He]2s! 0^267 ПО
Na2 [NeP*1 0,308 74
К2 [АГ1451 0,390 55
Rb2 [Кф*1 - 49
Cs2 [Xe]6sl - 44
металла имеет один валентный электрон, который и используется на
образование связи с другим атомом элемента первой группы.
Изменение энтальпии диссоциации связей и длин связей при переходе от
Li2 к Na2 и К2 (табл. 3.9) показывает, что при движении по группе вниз
происходит ослабление и удлинение связи металл—металл. Такая же зависимость
прослеживается и для энтальпий связей Rb—Rb и Cs—Cs. Эти изменения
являются следствием изменения значения главного квантового числа (п)
валентных электронов и пространственной протяженности соответствующих
орбиталей (см. рис. 2.10). Необходимо вспомнить, как изменяются
радиальные функции распределения 5-атомных орбиталей при увеличении п.
Радиальные функции распределения задают вероятность обнаружения электрона
в сферической оболочке радиуса г и толщины (г + dr). На рис. 2.11 эти
функции показаны для 15-, 25- и 35-АО. Валентными орбиталями Li и Na
являются 25- и 35-АО соответственно. ф
Группа 17: галогены
Каждый элемент этой группы имеет конфигурацию валентной оболочки
пР-пр5. Структуры Льюиса для С12 и Вг2 напоминают структуры для F2 (3.8 и
3.9). Все эти молекулы устойчивы за счет образования простых связей.
Диаграммы молекулярных орбиталей для С12, Вг2 и 12 аналогичны диаграмме,
приведенной на рис. 3.21, с той лишь разницей, что изменение главного
квантового числа валентных орбиталей приводит к изменению их
относительной энергии. В любом случае, порядок связи равен единице, а молекулы
должны быть диамагнитны.
В табл. 3.10 приведены некоторые данные о молекулах галогенов.
Относительные межъядерные расстояния и энтальпии диссоциации вполне
соответствуют образованию простых связей. Однако вместо монотонного
уменьшения прочности и удлинения связей при движении вниз по группе, в
молекуле F2 связывание оказывается значительно более слабым, чем можно было бы
ожидать. Это показано на рис. 3.31. Ослабление связи обычно объясняется
отталкиванием неподеленных электронных пар двух атомов фтора. Хотя все
атомы галогенов имеют по три неподеленные пары, в случае фтора их
отталкивание выражено наиболее сильно.
3.23. Двухатомные гомоядерные молекулы
159
250
§1
о s
X
200
150
F CI Br I
As Sb
Рис. 3.31. Изменение энтальпии
диссоциации двухатомных гомоя-
дерных молекул галогенов.
Рис. 3.32. Изменение энтальпии
диссоциации двухатомных гомоядерных
молекул элементов группы 15.
Таблица 3.10. Экспериментальные данные, полученные для гомоядерных
двухатомных молекул галогенов
Молекула
С12
Вг,
Электронная конфигурация
основного состояния
атома X
[Не]2522/т5
[Ne]3523/?5
[Аг]4524р5
[Kr^Sp5
Длина связи, нм
0,141
0,199
0,228
0,267
Энтальпия
диссоциации
связи, кДж/моль
159
242
193
151
Группа 15
Энтальпии диссоциации молекул N2, P2, As2, Sb2 и Bi2 приведены на
рис. 3.32. При переходе от N2 к Р2 наблюдается очень резкое уменьшение
прочности связи, которое при дальнейшем движении вниз по группе
сменяется более плавным.
Каким образом атомы элементов группы 15 могут объединяться в
молекулы? Наличие трех неспаренных электронов дает возможность для
образования либо двухатомной молекулы с тройной связью, либо четырехатомной
молекулы с шестью простыми связями (рис. 3.33).
Энтальпия разрыва связи является одним из факторов, определяющим,
каким образом будет происходить образование молекулы. Четыре атома
азота, находящихся в газовой фазе, могут дать две молекулы N2 (уравнение 3.34)
или одну молекулу N4 (уравнение 3.35).
160
3. Гомоядерная ковалентная связь
Рис. 3.33. Атомы группы 15 теоретически могут образовывать либо двухатомные
молекулы с тройной связью, либо четырехатомные молекулы с шестью одинарными
связями. Условия образования молекул Р4, As4, Sb4 и Bi4 резко отличаются от условий
образования молекулы азота.
4N(ra3) -» 2N2(ra3)
(3.34)
4N(ra3) -> N4(ra3) (3.35)
В первом случае изменение энтальпии, связанное с образованием двух
тройных связей N=N равно:
A#°[4N(ra3) -> 2N2(ra3)] = -{2 • D(N=N)}
= -(2 • 945)
= -1890 кДж/(моль реакции)
Во втором случае происходит образование шести одинарных связей:
ДЯ°[4Ы(газ) -> N4(ra3)] = -{6 • 0(N-N)}
= -(6 • 159)
^» = -954 кДж/(моль реакции)
Величина D(N—N) уаким образом, образование двух молекул N2 энергетически бо-
взята из табл. 3.6 лее ВЫГОдНо, чем одной молекулы N4, которую вообще не удается
наблюдать экспериментально. Результатом высокой энтальпии
диссоциации тройной связи N=N является исключительная стабильность
молекулы N2, которая была принята за стандартное состояние азота.
Ниже приведены аналогичные вычисления для фосфора
4Р(газ) -> 2Р2(газ) (3.36)
4Р(газ) -> Р4(газ) (3.37)
Тепловой эффект образования двух молекул Р2 равен:
ДД°[4Р(газ) -> 2Р2(газ)] = -{2 • ДР^Р)}
= -(2-490)
= -980 кДж/(моль реакции)
Тепловой эффект образования молекулы Р4 равен:
Д//°[4Р(газ) -> Р4(газ)] = -{6 • ДР-Р)}
= -(6-200)
^ = -1200 кДж/(моль реакции)
Величина D(P—P) В данном случае более выгодно образование молекулы Р4, хотя
взята из табл. 3.6 разница в энтальпиях между двумя возможными молекулярными
3.23. Двухатомные гомоядерные молекулы
161
формами фосфора значительно меньше, чем в случае азота. Стандартным
состоянием фосфора является белый фосфор - твердое вещество,
образованное молекулами Р4.
Выше 827 К молекулы Р4 распадаются на молекулы Р2 (уравнение 3.38).
Исходя из приведенных выше данных, изменение энтальпии при этом
переходе можно оценить как Д#° = 220 кДж/моль. Экспериментально найденная
величина равна 217 кДж/моль.
Р4(газ) -> 2Р2(газ) (3.38)
В газовой фазе мышьяк присутствует в виде молекул As4. При относительно
низких температурах сурьма также образует молекулы Sb4. Пары сурьмы и
висмута, разбавленные инертными газами, содержат молекулы Sb4, Sb2, Bi4 и
Bi2. Однако все эти частицы не существуют при 298 К. В стандартном
состоянии мышьяк, сурьма и висмут имеют кристаллическую структуру,
напоминающую структуру черного фосфора. Этот вопрос рассматривается в гл. 7.
Пример 3.2. Аллотропные формы кислорода и серы
Попытайтесь объяснить, почему кислород образует двухатомные, а
не циклические молекулы Ов, в то время как при 298 К и
атмосферном давлении сера построена именно из шестичленных циклов, а не
из молекул S2.
Энтальпии диссоциации связей:
D(0—О) = 146 кДж/моль
D(S—S) = 266 кДж/моль
0(0=0) = 498 кДж/моль
D(S=S) = 425 кДж/моль
Решение
Рассмотрим образование молекул 02 и 06 из атомов кислорода. Чтобы расчеты
имели смысл, число исходных атомов кислорода должно быть одинаковым:
60(газ) -»302(газ)
60(газ) -» 06(газ)
При образовании 3 моль молекул 02(газ)
А/Г = -{3D(0=0)}
= -(3 • 498)
= —1494 кДж/(моль реакции)
При образовании 1 моль молекул 06(газ)
А/Г =-{6D(0-0)}
= -(6 • 146)
= -876 кДжДмоль реакции)
Таким образом, образование двухатомных молекул кислорода более выгодно,
чем образование циклов. (Необходимо помнить, что изменение энтальпии не
единственный фактор, определяющий направление протекания химической
реакции. См. гл.12.)
В случае серы также необходимо рассчитать тепловые эффекты приведенных
ниже реакций:
162
3. Гомоядерная ковалентная связь
6S(ra3) -> 3S2(ra3)
6S(ra3) -» S6(ra3)
При образовании 3 моль молекул S2(ra3)
АЯ°= -{3^S=S)} = -(3 • 425) = -1275 кДж/(моль реакции)
При образовании 1 моль молекул S6(ra3)
Д#° = -{6 Z)(S-S)} = -(6 • 266) = -1596 кДж/(моль реакции)
Следовательно, образование кольцевых молекул S6 является более выгодным,
чем S2. (Существуют и другие молекулы типа Sn; см. табл. 3.2.)
ЗАКЛЮЧЕНИЕ
В этой главе рассматривалось образование связей в гомоядерных молекулах.
Что означают следующие термины?
Ковалентная связь
Гомоядерный
Ковапентный радиус
Энтальпия (энергия)
разрыва (диссоциации)
связи
Энтальпия атомизации
Структура Льюиса
Теория валентных связей
Метод молекулярных орби-
талей
Линейная комбинация
атомных орбиталей
Локализованная связь
Делокализованная связь
о- и я-связи
Связывающие и
разрыхляющие
орбитали
Порядок связи
Диамагнитный
Парамагнитный
Катион-радикал
Анион-ради кал
Теперь вы должны уметь:
кратко изложить суть методов, применяемых
для определения длин связей;
связать энтальпии разрыва связей с
энтальпиями атомизации;
различать средние энтальпии разрыва связей
и энтальпии разрыва отдельных, вполне
определенных химических связей;
используя закон Гесса, рассчитать энтальпии
разрыва связей, исходя из энтальпий
образования соединений и энтальпий атомизации;
нарисовать структуры Льюиса для простых ко-
валентных молекул;
изложить принципы метода молекулярных
орбиталей и теории валентных связей;
объяснить образование молекулы водорода с
точки зрения метода молекулярных орбиталей
и теории валентных связей, сравнить
результаты обоих методов;
нарисовать диаграммы молекулярных
орбиталей для гомоядерных двухатомных молекул
s- и р-элементов, использовать
экспериментальные данные для доказательства
правильности предложенных вами диаграмм;
знать зависимость между порядком гомоя-
дерной связи и ее длиной и энтальпией
диссоциации;
рассказать, как меняются длины и энтальпии
разрыва гомоядерных связей в молекулах в
ряду элементов второго периода или в
группах s- и р-элементов.
3.25. Упражнения
163
НЗЭ УПРАЖНЕНИЯ
3.1. Дайте определения следующих
терминов: а) ковалентный радиус; б)
энтальпия разрыва связи; в)
стандартная энтальпия атомизации элемента.
3.2. а) Что вызывает дифракцию
рентгеновских лучей? б)Объясните, почему
по данным рентгеновской
дифракции длина двухцентровой двухэлек-
тронной связи В—Н в ионе [В2Н7]~
равна 0,103 нм, а дифракция
нейтронов дает для той же связи величину
0,118нм.
3.3. Можно ли использовать одну и ту же
величину энтальпии разрыва а) для
связей углерод-углерод в этане С2Н4
и этилене С2Н4; б) для связей
азот—азот в гидразине N2H4h N2F4;
в) для связей водород-кислород в
молекулах воды и пероксида
водорода Н202. Ответ объясните.
3.4. Используя данные табл. 3.6, оцените
величины энтальпий атомизации
следующих молекул в газовой фазе:
а) Р4; б) S8; в) As4; г) 12. Структуры
этих молекул приведены в тексте
главы.
3.5. Используя ответ к задаче 3.4 и
стандартную энтальпию атомизации
белого фосфора (табл. 3.5),
рассчитайте стандартную энтальпию
образования (при 298 К) газообразного Р4.
3.6. Стандартные энтальпии образования
метана СН4 и этана С2Н6 при 298 К
равны —75 и —85 кДж/моль
соответственно. Определите величины
энтальпий диссоциации связей С—С и
С—Н при 298 К, если стандартные
энтальпии атомизации водорода и
углерода равны 218 кДж/моль и
717 кДж/моль соответственно.
Сравните свой ответ с величинами,
приведенными в табл. 3.6. Если
табличные данные не будут совпадать с
расчетом, объясните причину различий.
3.7. Принимая во внимание только
валентные электроны, нарисуйте
структуры Льюиса для следующих
молекул: а) С12; б) Na2; в) S2; г) NF3;
д) НС1; е) ВН3; ж) SF2.
3.8. Принимая во внимание только
валентные электроны, нарисуйте
структуры Льюиса для следующих
молекул: а) метан СН4; б) бром метан
СН3Вг; в) этан С2Н6; г) этанол
С2Н5ОН;д) этилен С2Н4.
3.9. Сколько структур Льюиса вы можете
нарисовать для молекулы С2? В тексте
для этой молекулы приводилась
только одна структура. Объясните, почему
остальные, предложенные вами,
варианты не имеют смысла. (Подсказка:
подумайте, как распределены в
пространстве связывающие электроны.)
3.10. Используя метод молекулярных ор-
биталей, объясните, почему не
происходит образования молекулы Не2.
3.11. Постройте полную диаграмму
молекулярных орбиталей для Li2,
используйте для этого и валентные
электроны, и электроны внутренних
уровней. Определите порядок связи в
этой молекуле. Изменится ли
порядок связи, если исключить из
рассмотрения внутренние электроны?
3.12. Какая частица получится, если
путем окисления из молекулы Li2
удалить один электрон? Используя
диаграмму, построенную в предыдущей
задаче, и данные табл. 3.9, ответьте,
как при этом изменится порядок,
длина и энтальпия разрыва связи
Li—Li, а также магнитные свойства
этой молекулы.
3.13. Используя метод молекулярных
орбиталей, объясните закономерности,
вытекающие из данных табл. 3.11.
Диамагнитны или парамагнитны
частицы [N2]~ и [N2]+ ?
164
3 Гомоядерная ковалентная связь
Таблица 3.11. Данные к упражнению 3.13
Частица Длина связи, Энтальпия разрыва
нм связи, кДж/моль
N2 0,110 945
[N2]" 0,119 765
[N2]+ 0Л_12 84]
3.14. В табл.3.12 приведены энтальпии
разрыва связей в двухатомных
молекулах элементов группы 16.
Предложите объяснение наблюдаемых зако-
Таблица 3.12. Данные к упражнению 3.14
Молекула Длина связи,
нм
02 0,121
S2 0,189
Se2 0,217
Те2 0,256
Энтальпия разрыва
связи,
, кДж/моль
498
425
333
260
номерностей. Соответствуют ли
значения длин связей в молекулах 02
и S2 энтальпиям разрыва этих связей?
ГЕТЕРОЯДЕРНЫЕ ДВУХАТОМНЫЕ
МОЛЕКУЛЫ
ВВЕДЕНИЕ
В гл. 3 обсуждались гомоядерные связи, например в гомоядерных
двухатомных молекулах Н2 или N2, многоатомных молекулах Р8 или S8 или даже в ге-
тероядерных молекулах С2Н6 и Н202, в которых имеются гомоядерные связи
С—С и О—О соответственно. В этой главе рассмотрены гетероядерные связи
на примере двухатомных частиц.
СИА
NH3
Рис. 4.1. Молекулы с гетероатомными связями: метан СН4, этан С2Н6, трифторид
бора BF3, аммиак NH3.
4. Гетероядерные двухатомные молекулы
В гомоядерной двухатомной молекуле Х2 оба атома X вносят в связь
одинаковый вклад: диаграммы молекулярных орбиталей, приведенные в гл. 3,
выглядят симметричными, и каждая молекулярная орбиталь содержит
равные вклады от соответствующих атомных орбиталей.
Гетероядерная связь образуется между различными атомами, например
связь С—Н в метане и этане, связь В— Рвтрифторидебораисвязь1Ч1—Н в
аммиаке (рис. 4.1). Химическая наука в большинстве случаев имеет дело с
молекулами и ионами, содержащими более одного типа атомов, и необходимо
знать, какие взаимодействия могут определять образование связи между
различными типами атомов. Существует бесчисленное множество гетероядер-
ных многоатомных молекул, но здесь будет рассмотрено образование связи на
примере двухатомных молекул HF, LiF и УН.
СТРУКТУРЫ ЛЬЮИСА ДЛЯ HF, LiF И LiH
Структуры Льюиса для атомов водорода, лития и фтора и электронные
конфигурации их основных состояний приведены на рис. 4.2,а.
Атомы водорода и лития имеют по одному валентному электрону, тогда
как у фтора их семь. По аналогии с образованием молекул Н2 и F2, в которых
ковалентная связь образуется путем обобществления электронов двумя
атомами, можно точно так же объединить в пару атомы водорода и фтора, чтобы
получить молекулу HF, и такой же подход распространить на описание
связывания в молекулах LiF и LiH (рис. 4.2,б); при этом предполагается, что в
каждой молекуле образуется одинарная связь.
В газовой фазе HF, LiF и LiH могут существовать как молекулярные
частицы. Однако, как будет показано позднее, при комнатной температуре и
атмосферном давлении, связывание не всегда такое простое, как предполагает
подход Льюиса.
ПРИМЕНЕНИЕ ТЕОРИИ ВАЛЕНТНЫХ СВЯЗЕЙ
ДЛЯ ОБЪЯСНЕНИЯ ОБРАЗОВАНИЯ МОЛЕКУЛ HF, UF И UH
Различие между гомоядерной двухатомной молекулой Н2
и гетероядерной молекулой XY
В гл. 3 (разд. 3.11) показано, как применить метод валентных связей (ВС) к
молекуле Н2 и описать образование связи с помощью резонансных структур
(4.1—4.3). Структуры 4.2 и 4.3 эквивалентны по энергии.
а
•
Н
Ы
•
Li
[Hejli-1
• •
• f :
[He]2s22p5
Рис. 4.2. a — структуры Льюиса и электронные конфигурации основных состояний
атомов водорода, лития и фтора; б - структуры Льюиса для фторо водорода, фторида
лития и гидрида лития.
б
• •
н : f :
• •
..
н—f :
••
• •
Li : f :
• •
• •
Li—f :
• •
Li Г H
Li H
4.3. Применение теории ВС к HF, LiF и LiH
167
н — н -«—*- нА+нв -«—► нА нв+
4.1 4.2 4.3
Аналогичный подход можно использовать при описании образования связей
в гетероядерных двухатомных молекулах. Однако следует отметить важное
различие между гомо- и гетероядерными частицами. В случае двухатомных
молекул XY наряду с резонансной структурой 4.4, которая описывает кова-
лентную составляющую суммарного связывания, существуют еще две
(различающиеся по энергии) структуры (4.5 и 4.6), которые соответствуют ионной
составляющей.
X Y -«—► X+Y -«—► X-Y+
4.4 4.5 4.6
Вклад этих трех резонансных структур (4.4—4.6) зависит от их относительной
энергии. Применим это подход для HF, LiF и LiH.
Фтороводород
Для молекулы фтороводорода метод валентных связей предполагает
существование трех резонансных форм - 4.7, 4.8 и 4.9.
Ковалентная форма 4.7 подразумевает, что атомы Н и F обобществляют два
Н F -«—► H+F- «*—-► H-F+
4.7 4.8 4.9
электрона так, что каждый атом имеет конфигурацию благородного газа;
образование простой (одинарной) ковалентной связи между атомами в
молекуле HF позволяет атому водорода получить такую же внешнюю электронную
оболочку, как у атома гелия, а фтору — как у атома неона.
Чтобы оценить относительные энергии ионных форм, рассмотрим
электронную конфигурацию основного состояния атома фтора. Резонансная
структура 4.8 показывает, что электрон переходит от атома Н к атому F
(уравнения 4.1 и 4.2).
Н->Н+ + е" (4.1)
F + е" -> F" (4.2)
Электронная конфигурация основного состояния атома фтора:
[Не]2522/?5, и добавление одного электрона дает конфигурацию неона:
[Не]2522/?6 = [Ne]. Ион F~ имеет электронный октет на внешней оболочке.
Ионная форма 4.8 является выгодной альтернативой ковалентной структуре
4.7.
Резонансная структура 4.9 показывает, что электрон переходит от фтора к
водороду (уравнения 4.3 и 4.4).
F -> F+ + е" (4.3)
Н + е" -> Н-
(4.4)
168
4. Гетероядерные двухатомные молекулы
Хотя образование иона Н~ и дает конфигурацию гелия (Is2 = [He]),
энергетический выигрыш на этой стадии недостаточен, чтобы компенсировать
энергетический проигрыш при образовании F+. Потеря одного электрона атомом
фтора, приводящая к катиону F+, означает образование частицы с шестью
валентными электронами. Такая структура будет, по-видимому, менее
стабильна, чем анион F~ с октетом электронов. Преимущества присоединения
электрона к атому F намного больше, чем при потере электрона; поэтому
реакция 4.2 предпочтительнее реакции 4.3. (В упражнении 6.6 в разд. 6.20
предлагается определить энергии реакций 4.1-4.4.)
С учетом вышесказанного нет смысла предполагать, что резонансная
структура, включающая F+, может иметь существенное значение при
описании образования связи во фтороводороде. Итак, связь во фтороводороде
может быть эффективно описана в рамках ковалентной структуры и ионной
формы, в которую входят ионы Н+ и F-.
Фторид лития и гидрид лития
Теорию валентных связей можно аналогично применить и для описания
образования связей в молекулах LiF и LiH. Резонансные структуры для этих
молекул приведены на рис. 4.3.
Для LiF можно вновь исключить резонансную структуру, включающую
ион F+, тогда связь во фториде лития можно быть описать в терминах
ковалентной и ионной резонансных структур.
Для LiH можно предположить три резонансные структуры, все они
вносят вклад в суммарное связывание в соединении. Атомы лития и водорода
(см. рис. 4.2) имеют электронную конфигурацию основного состояния nsx и
могут свободно отдать электрон для образования ионов Li+ и Н+ с
конфигурацией ns°. Но водород может, кроме того, получить электрон и образовать
ион Н~ — частицу с электронной конфигурацией атома гелия.
Теория ВС: выводы
Теория валентных связей объясняет образование молекул HF, LiF и LiH с
использованием представлений о ковалентной и ионной резонансных
структурах. Однако в данном наборе резонансных структур некоторые формы
играют более важную роль, чем другие. Итак, качественная интерпретация в
терминах стабильности возможных ионов вполне удачна, и метод дает хорошее
интуитивное описание образования связи в простых молекулах. Конечно,
можно более строго подойти к проблеме и установить вклады в общую связь
каждой резонансной структуры. Однако удобно рассматривать
относительные вклады каждой резонансной структуры, как это сделано выше.
Рис. 4.3. Резонансные структуры молекул LiF (а) и LiH (б). Для LiF вклад правой
структуры в общее связывание пренебрежимо мал.
4.4. Применение метода молекулярных орбиталей 169
ЕЕВ ПРИМЕНЕНИЕ МЕТОДА МОЛЕКУЛЯРНЫХ ОРБИТАЛЕЙ
ДЛЯ ОПИСАНИЯ СВЯЗЫВАНИЯ В ГЕТЕРОЯДЕРНЫХ
ДВУХТОМНЫХ МОЛЕКУЛАХ
В гл. 3 метод молекулярных орбиталей (МО) был использован для описания
образования связи в гомоядерных двухатомных молекулах: МО-диаграммы
были симметричными; атомные орбитали одного типа, принадлежащие
разным атомам, имели одинаковую энергию (например, для F2; см. рис. 3.21).
Каждая МО молекулы Х2 содержит равные атомные вклады от каждого
атома X.
Теперь рассмотрим гетероядерную молекулу XY. В этой главе
рассматривается случай, когда атомы X и Y — разные атомы, с атомным номером <10.
Существенное отличие между X и Y в том, что у них разные эффективные
ядерные заряды Z^. Если Y находится в группе периодической таблицы с
^ большим номером, чем X, то электрон на данной атомной орбитали испыты-
Эффективный ядерный вает воздействие Z^(Y)> Z^X). Так как гэфф возрастает
заряд- см разд 2 15 ВД°ЛЬ строки периодической таблицы, энергия конкретной АО
уменьшается (больший отрицательный заряд). Это
схематически проиллюстрировано на рис. 4.4 для комбинации 25-АО
атомов X и Y при образовании c(2s)- и а*(2$)-МО в молекуле XY.
Важной чертой МО-диаграммы на рис. 4.4 является то, что энергия
связывающей МО ближе к 25-АО атома Y, чем к 25-АО атома X, а энергия
разрыхляющей МО ближе к 2s-АО атома X, чем к 25-АО атома Y. Это влияет на
природу молекулярных орбиталей так, как показано на рис. 4.5. 25-АО атома
Y дает больший вклад в связывающую о(2$)-орбиталь, чем 25-АО атома X.
Для разрыхляющей МО ситуация обратная, и 25-АО атома Y дает меньший
вклад в о*(25)-МО, чем АО атома X, так как а(25)-МО ближе по энергии к
о,
<?(2s)
Рис. 4.4. Схематическое представление МО-диаграмм для комбинации 2s-AO
атомов X и Y в гетероядерной двухатомной молекуле; Z3(bd)(Y)> Z3dKb(X).
4. Гетероядерные двухатомные молекулы
X Y X Y
х |y
Рис. 4.5. Схематическое представление o(2s)-MO (а) и a*(2s)-MO (б) в гетероядер-
ной двухатомной молекуле XY. Ядра показаны черными точками.
25-орбитали атома Y, а a*(2s)-MO — к 2^-орбитали атома X (т. е. a(2s)-MO
обладает ^-характером от Y, а a*(2s)-MO - 2s-xapaKmepoM от X). На рис. 4.5,а и
4.5,5 наглядно представлены больший и меньший 2$-вклады; заметьте, как
это отражается на окончательной форме молекулярных орбиталей и делает их
асимметричными.
Волновая функция для МО в гетероядерной двухатомной молекуле
должна показывать, какой вклад дает каждая составляющая ее АО. Это делается
при помощи коэффициентов сп. В уравнении 4.5 коэффициенты с{ и с2
отражают соотношение составляющих молекулярную орбиталь атомных орбита-
лей \|/х и \|/Y. Относительные величины с, и с2 выбираются в соответствии с
эффективными ядерными зарядами атомов X и Y.
УМО = С\ ' Ух + С2 ■ VY» ГДе С1 > С2 (45)
Уравнение 4.6 дает выражение для волновой функции, которая
описывает связывающую орбиталь, показанную на рис. 4.4 и 4.5.
Тот факт, что коэффициент с2 больше, чем ср соответствует тому, что
результирующая МО по энергии ближе к 2^-орбитали Y, чем к 2$-орбитали X,
что приводит к расположению МО в основном у атома Y (рис. 4.5,я).
В гл. 3 была отмечена важность энергетического совпадения между
атомными орбиталями как критерия для их эффективного взаимодействия. В случае
гомоядерных двухтомных молекул, когда перекрывание ограничивалось
аналогичными орбиталями, это не вызвало проблем, так как энергии двух ls-AO
водорода были одинаковыми. В гетероядерных двухтомных молекулах всегда
будет разница между энергиями орбиталей двух различных атомов, которая
иногда оказывается слишком большой для достижения эффективного
взаимодействия. Это положение будет проиллюстрировано позднее при
обсуждении образования связи в LiF и LiH.
4.5. Применение метода МО к LiH, LiF и HF
171
ПРИМЕНЕНИЕ МЕТОДА МОЛЕКУЛЯРНЫХ ОРБИТАЛЕЙ
ДЛЯ ОПИСАНИЯ ОБРАЗОВАНИЯ СВЯЗИ В LiH, UF И HF
Два важных предварительных замечания
• При использовании метода молекулярных орбиталей для описания
сравнительно простых молекул во внимание принимается только
перекрывание валентных атомных орбиталей.
• Метод молекулярных орбиталей описывает ковалентное связывание. Он не
рассматривает детально ионные вклады, как это делается в методе
валентных связей.
Гидрид лития
Электронная конфигурация основного состояния водорода и лития - \sx и
[HePs1 соответственно. ls-Орбиталь водорода имеет меньшую энергию, чем
2s-AO лития. Симметрии 15-АО водорода и 2s-АО лития совместимы и
перекрывание возможно. Комбинация этих АО, дающая две молекулярные орби-
тали, показана на рис. 4.6.
Два валентных электрона займут низшую по энергии свободную а-связы-
вающую МО и будут иметь противоположные спины. Порядок связи в
молекуле LiH равен 1 (уравнение 4.7).
Порядок связи:
см. разд. 3.16
Порядок связи в LiH = 0,5 • (2 - 0) = 1
(4.7)
Рис. 4.6 показывает, что ls-AO водорода дает больший (74%) вклад в а-свя-
зывающую МО LiH, чем 2s-АО лития (26%). Это означает, что а-связываю-
щая МО ближе по энергии к орбитали водорода, чем лития (рис. 4.7.) В
результате электроны больше времени «ассоциированы» с ядром водорода,
чем лития.
Рис. 4.6. Диаграмма
МО молекулы LiH.
Показаны только валентные
атомные орбитали и
электроны.
172
4. Гетероядерные двухатомные молекулы
Рис. 4.7. Представление а-связывающей
МО LiH. Li H
Волновая функция, представленная в уравнении 4.8, описывает о-связы-
вающую МО LiH.
\|/(LiH, a-MO) = с, • \|/Li(2y) + с2 • \|/H(lv), где с, < с2 (4.8)
Из рис. 4.6 следует, что а*-МО ближе по энергии к орбитали лития, чем
водорода. [Сделайте схематический рисунок а*-МО LiH.]
Фтороводород
Электронная конфигурация основного состояния водорода и фтора - Is1 и
[Не]2522/?5 соответственно. Прежде чем начать обсуждение перекрывания
атомных орбиталей, необходимо оценить их относительные энергии. При
движении от водорода к фтору эффективный ядерный заряд значительно
возрастает, и это существенно понижает энергии атомных орбиталей фтора
по отношению к Is-АО водорода. Так, несмотря на разницу в главном
квантовом числе, 25- и 2р-орбитали фтора лежат в области более низких энергий,
чем ls-AO водорода. Это проиллюстрировано на рис. 4.8; ось энергии на
рисунке рассечена, так что 2$-орбиталь фтора реально даже ниже по энергии,
чем показано.
Вспомните, что для эффективного перекрывания разница между
энергиями двух атомных орбиталей не должна быть слишком велика. Можно было
бы допустить взаимодействие Н Is- и F 25-орбиталей друг с другом, как в
случае LiH, но, хотя взаимодействие разрешено по симметрии, разница в энергии
слишком велика. Итак, перекрывание неблагоприятно, и это оставляет 2s-
АО фтора неиспользованной - она становится несвязывающей орбиталью в
молекуле HF.
Несвязывающая молекулярная орбиталь не является ни
связывающей, ни разрыхляющей.
Ядра водорода и фтора расположены на оси z\ следовательно, Is-АО
водорода и 2pz-AO фтора могут перекрываться, как показано на рис. 4.9,а. На
рис. 4.8 видно, что энергии этих орбиталей достаточно близки, чтобы при их
перекрывании возникли о- и а*-МО. Вклад 2pz-AO фтора в а-связывающую
МО больше, чем вклад ls-орбитали водорода, и связывающая орбиталь
оказывается ближе по энергии к 2/?-орбитали атома фтора, чем к ls-орбитали
водорода.
Две оставшиеся 2р-АО фтора лежат под углом к оси межъядерного
взаимодействия Н—F и не перекрываются с ls-орбиталью водорода; рис. 4.9,6 ил-
О-
4.5. Применение метода МО к LiH, LiF и HF
173
i*H-
b+f
2р
Несвязывающая МО
-Н
-4-2S
И
HF
Рис. 4.8. Диаграмма МО молекулы HF. Показаны только валентные атомные орби-
тали и электроны. Разрыв на вертикальной оси (энергии), отражает то, что 2s-AO
фтора лежит гораздо ниже, чем это показано.
Разрыхляющее
взаимодействие
15
Н
F
Связывающее вза
имодействие
Рис. 4.9. а - разрешенное по симметрии перекрывание \s-AO водорода и 2pz-AO
фтора (отметим, что ядра F и Н лежат на оси межъядерного взаимодействия г); б -
комбинация ls-AO водорода и 2рх-АО фтора приводит к равным по величине
связывающему и разрыхляющему взаимодействиям, и суммарное взаимодействие является
несвязывающим; аналогичная ситуация возникает для 2/у-орбитали.
174
4. Гетероядерные двухатомные молекулы
люстрирует это для 2рх-АО. Так 2рх- и 2/? -АО становятся несвязывающими
орбиталями в молекуле HF.
В молекуле HF восемь валентных электронов, которые согласно принципу
последовательного заполнения орбиталей занимают наинизшие по энергии
свободные МО так, как это показано на рис. 4.8. Только два электрона
занимают связывающую орбиталь в Н—F, остальные шесть находятся на
несвязывающих МО и локализованы на атоме фтора. Эти три несвязывающие пары
электронов аналогичны неподеленным парам электронов в структуре
Льюиса для молекулы HF на рис. 4.2.
Порядок связи во фтороводороде (уравнение 4.9) может быть определен
обычным путем, так как электроны на несвязывающих МО не оказывают
влияния на связь.
Порядок связи в HF = 0,5 (2 - 0) = I (4.9)
Электрон на несвязывающей МО не влияет на порядок связи.
Можно следующим образом просуммировать применение метода МО к
молекуле HF:
• Перекрывание между Is-АО водорода и 2pz-AO фтора дает одну о-связы-
вающую МО и одну а*-разрыхляющую МО.
• 2рх- и 2/7 -АО фтора не перекрываются с Is-АО водорода и являются
несвязывающими.
• 25-Орбиталь фтора может перекрываться с Is-АО водорода по условиям
симметрии, но несовпадение в энергиях атомных орбиталей делает
взаимодействие слабым - 25-орбиталь фтора в основном несвязывающая.
• Из восьми валентных электронов в молекуле HF два занимают
связывающую МО, а шесть — несвязывающие орбитали, локализованные на атоме
фтора.
• а-Связывающая МО в HF смещена к атому фтора.
Фторид лития
Подход метода МО к связыванию в LiF полностью соответствует
описанному для HF, только с той разницей, что электронная конфигурация основного
состояния лития — [Не]2s1, а не Is1, как у водорода.
На рис. 4.10 приведена диаграмма МО фторида лития. Ядра лития и
фтора лежат на оси z (структура 4.10). Как и в HF, 2s-АО фтора имеет низкую
энергию и образует несвязывающую орбиталь в молекуле LiF 2px- и 2л-АО
фтора так же, как и в HF, являются несвязывающими.
• • >z
Li F
4.10
Перекрывание возможно между 2s-AO лития и 2pz-AO фтора. Однако
разница в энергиях между этими орбиталями значительна, и взаимодействие
4.5. Применение метода МО к LiH, LiF и HF
175
Ь-++^ЬИ/>
Несвязываюшая МО
-И
-Чг
Is
Li
LiF
Рис. 4.10. Диаграмма МО для молекулы LiF. Показаны только валентные атомные
орбитали и электроны. Разрыв на вертикальной оси (энергии), отражает тот факт, что
2s-AO фтора лежит гораздо ниже, чем это показано.
не очень эффективно. Из рис. 4.10 видно, что а-связывающая орбиталь лишь
немного стабильнее, чем 2pz-AO фтора. Сравнение рис. 4.8 и 4.10 показывает,
что стабилизация а-связывающей орбитали в LiF меньше, чем в HF Что это
означает для молекулы LiF?
Волновая функция, описывающая связывающую МО на рис. 4.10, задана
уравнением 4.10.
\|/(LiF, а-МО) = сх • \|/Li(2,) + с2 • у¥{2рт]
где с, « с2
(4.10)
Если коэффициент с} очень мал, то волновая функция описывает орбиталь,
сильно сдвинутую в сторону атома фтора: в уравнении 4.10 произведение
с2 • V|/F(2n) гораздо больше, чем сх • V|/Li(25)- Это означает, что электрон на
связывающей *мО чаще находится в окрестности атома фтора, чем лития.
В заключение отметим, что картина связывания в LiF предполагает одну
связывающую МО; она занята двумя электронами и порядок связи в
молекуле равен единице. Есть также три заполненные несвязывающие орбитали,
локализованные на атоме фтора; они являются аналогами отдельных пар
электронов, изображенных в структуре Льюиса LiF (рис. 4.2). Наибольший
вклад в связывающую МО в двухатомной молекуле LiF дает 2/?-орбиталь
фтора (~97%). Итак, хотя метод МО построен на ковалентной модели, результат
показывает нам, что связывание в LiF может быть описано как близкое к
ионному. Мы вернемся к LiF в разд. 6.2.
176
4. Гетероядерные двухатомные молекулы
Метод МО: выводы
• Применение метода молекулярных орбиталей к описанию связывания в
молекулах LiH, HF и LiF показывает, что для всех них порядок связи
равен единице. В каждой молекуле связывающей является а-орбиталь.
• Связывающая МО в LiH сформирована путем перекрывания 2s-AO Li и
ls-AO H. Связывающая МО в HF сформирована из ls-AO H и 2/ьАО F.
Связывающая МО в LiF формируется за счет вкладов 2s-AO Li и одной из
2/7-орбиталей F.
• Относительные вклады атомных орбиталей различных атомов в
связывающую молекулярную орбиталь сильно зависят от природы этих атомов. В
LiH связывающая МО ближе по энергии к орбитали водорода, а не лития.
В HF связывающая МО ближе по энергии к орбитали фтора. В LiF
связывающая МО почти полностью формируется за счет 2/ьорбитали фтора.
Если известна относительная энергия связывающей МО в молекулах LiH,
HF и LiF, то возможно сделать выводы о распределении электронной
плотности между атомами в этих молекулах. В отличие от симметричного
распределения электронной плотности между ядрами в гомоядерных двухатомных
молекулах, в LiH, HF и LiF она сдвинута в сторону одного из атомов. В LiH
максимум электронной плотности сдвинут ближе к ядру Н, чем к Li, тогда
как в HF он ближе к ядру F. Попытки рационализировать эти результаты
приводят нас к концепции электроотрицательности и полярности связи, но
предварительно мы рассмотрим энтальпии диссоциации гетероядерных связей.
ЭНТАЛЬПИИ ДИССОЦИАЦИИ ГЕТЕРОЯДЕРНОЙ СВЯЗИ
В разд. 3.7 был описан способ применения закона Гесса для установления
величины энтальпии диссоциации связи. Прежде всего необходимо установить
энтальпии диссоциации связей в гомоядерных молекулах, но чтобы сделать
это, необходимо привлечь энтальпии диссоциации гетероядерных связей -
величина D(N—N) в N2H4 зависела от значения величины Z)(N—H) в NH3 и
N2H4.
^nQnuan -итап.П|._ Итак, известно, как величины средней энтальпии диссоци-
диссоциации связи ации связи могут быть получены из стандартных энтальпий
атомизации. Это показано для СС14 (4.11) и 1,2-С2Н4Вг2 (4.12)
в уравнениях 4.11 и 4.12.
ДатЯ°СС14(газ) = 4 Д С-С1) (4.11)
Дат/ГС2Н4Вг2(газ) = 4ДС-Н) + ДС-С) +2ДС-Вг) (4.12)
Вг Н
С\
\
С1
.С... Hi
L '""a
\
a » Вг
4.11 4.12
4.6. Энтальпии диссоциации гетероядерной связи 177
В разд. 4.6 была особо отмечена связь между энальпией диссоциации связи D
и стандартной энтальпией атомизации молекул гомоядерного двухатомного
газа. Это показано для фтора в уравнении 4.13.
AaT//°F = 0,5Z)(F-F) (4.13)
Можно ли использовать аналогичную стратегию для установления энтальпии
диссоциации гетероядерной связи? Можно ли для одинарной связи X—Y
установить величины, которые характеризовали бы вклады атомов X и Y в
величину ДХ—Y)? Логично было бы взять половину ДХ—X) и сделать ее
мерой вклада атома X в величину энтальпии связи ДХ—Y). Аналогично для
Y использовать половину D(Y—Y) как меру вклада атома Y в величину
ДХ—Y). Тогда величины ДХ—Y) могут быть установлены в соответствии с
уравнением 4.14.
ДХ-У)=0,5ДХ-Х) + 0,5 Z)(Y-Y)
или (4.14)
AX-Y)=0,5 [ДХ-Х) + ДY-Y)]
Но обоснованно ли такое правило аддитивности? Ответ на этот вопрос можно
дать, сравнив некоторые величины, полученные из уравнения 4.14, с
экспериментально определенными величинами энтальпий диссоциации связи
(табл. 4.1). Для некоторых молекул (BrI, BrCl и HI) эта модель дает хорошие
величины энтальпий диссоциации связи, но для других (HF и НС1)
расхождения велики, так что модель не всегда применима.
Невозможность использования этой простой модели аддитивности
вкладов в энтальпию диссоциации гетероядерной связи заставила Лайнуса По-
линга ввести понятие электроотрицательности.
Таблица 4.1. Энтальпии диссоциации связи D для гомоядерных и гетероядерных
двухатомных молекул
Для определения величины D(X—Y) (уравнение 4.14) используются экспериментальные
величины ДХ—X) и D(Y—Y). Затем делается сравнение с экспериментальными данными.
Величина AZ) в последней колонке это разность между экспериментально полученной
(колонка 6) и вычисленной (колонка 5) величинами D(X—Y) (см. уравнение 4.15).
X
н
н
н
н
F
С1
С1
Вг
Y
F
С1
Вг
I
С1
Вг
I
I
D(X-X)
вХ2,
кДж/моль
436
436
436
436
159
242
242
193
D(Y-Y)
eY2,
кДж/моль
159
242
193
151
242
193
151
151
0,5[D(X-X)
+D(Y-Y)],
кДж/моль
298
339
315
294
201
218
197
172
3kch.D(X-Y)
eXY,
кДж/моль
570
432
366
298
256
218
211
179
AD (и-
4.15)
iуравнения
272
93
51
4
55
0
14
7
178 4. Гетероядерные двухатомные молекулы
Дополнение 4.1. Лайнус Полинг (1901-1994)
Возможно, имя Полинга ассоциируется у вас с понятием электроотрицательности, но
его вклад в химию и науку вообще намного шире. Его воображение и гений оставили
долговременный след в основополагающих теориях современной химии.
Лайнус Карл Полинг родился в Орегоне в 1901 г. Он получил ученую степень в
области прикладной химии в 1922 г. В то время его интерес был сфокусирован на методах
рентгеновской дифракции и исследовании кристаллических структур. В 1926 г. Полинг
работал со Шредингером и Бором, развивая квантовую теорию и теорию строения
атома. В 1920-х годах начали расшифровывать структуры силикатных минералов, но
сложность их строения не согласовывалась со структурными воззрениями. В 1929 г.
Полинг вывел правила, основанные на ионных радиусах и зарядах, которые помогли
дать рациональное объяснение строения силикатов. Его внимание к химической связи
в то время привело к пониманию многих аспектов связывания, не имевших ранее
объяснения. Он скомбинировал квантовую теорию с другими теориями, например с
теорией Г. Н. Льюиса. Полинг также выступал защитником концепции резонансных
структур, понимая, что химические свойства могут быть лучше поняты при использовании
комбинации различных структурных форм. Разработка теории валентных связей
Полинга предоставила химикам новую основу для построения химических моделей, и в
конце 1930-х годов книга Полинга «Природа химической связи» многим открыла
глаза на мир химической связи и структуры. Эта книга по-прежнему остается одним из
шедевров химической литературы.
Биохимия также испытала на себе благотворное влияние идей Полинга. Он был
первым, кто предположил, что ot-спираль является мотивом структуры белка, и его идеи
были подкреплены экспериментами по рентгеновской дифракции в Кембридже
(Великобритания) в конце 1940-х годов. В 1954 г. за вклад в химию Полинг был удостоен
первой из двух его Нобелевских премий; вторую он получил в 1963 г. за усилия в деле
защиты мира. Он был несгибаемым борцом против ядерного вооружения и одно время был в
натянутых отношениях с правительством США. С 1966 г. внимание Полинга было
направлено на витамин С, исследованию которого он посвятил много лет. Он был
убежден, что витамин С является лекарством от всех болезней, включая рак, и принимал
большие дозы его каждый день.
4.7. Электроотрицательность по Полингу 179
EBI ЭЛЕКТРООТРИЦАТЕЛЬНОСТЬ ПО ПОЛИНГУ
Определение величин электроотрицательности
При сравнении расчетных и экспериментальных величин D(X—Y),
перечисленных в табл. 4.1, видно, что расчетная величина всегда намного меньше. В
1932 г. Полинг предположил, что эта разница (уравнение 4.15) может быть
объяснена в рамках теории валентных связей.
AD=D(X-Y)3Kcn - 0,5 [ДХ-Х) + «Y-Y)] (4.15)
Полинг высказал предположение о том, что AD является мерой ионного
характера связи X—Y. В терминах теории валентной связи, это соответствует
вкладу резонансных структур X+Y~ и X~Y+ в связывание в молекуле XY и
тому, насколько этот вклад больше, чем вклад структур Х+Х~ и Y~Y+ для Х2 и
Yj соответственно. Далее Полинг предположил, что резонансные формы
X^Y~ и X~Y+ неравноценны. (Это предположение было обосновано для HF в
разд. 4.3.)
Если доминирует форма X+Y~, это не означает, что молекула XY
действительно существует в виде пары ионов Х+ и Y~. Утверждается, что электроны
связи X—Y сдвинуты более в сторону атома Y, чем X, т. е. в молекуле XY атом
Y имеет большую способность оттягивать на себя электроны (т. е. электроно-
акцепторную силу), чем атом X.
Полинг ввел термин электроотрицательность (х11)* и определил его как
«способность атома в молекуле оттягивать электроны на себя». Этот подход
рассматривает полинговскую электроотрицательность как свойство атома, а
не связи. Важно запомнить, что электроотрицательность это свойство атома
в молекуле, а не фундаментальное свойство отдельного атома.
Хотя к представлениям об электроотрицательности мы пришли при
рассмотрении энергии образования двухатомной молекулы XY, это понятие
также пригодно при рассмотрении связей X—Y в более сложных молекулах.
Электроотрицательность хп была определена Полингом как
способность атома в молекуле оттягивать электроны на себя.
Величины электроотрицательности Полинга (табл. 4.2) вычисляются
следующим образом. Разность AZ) (уравнение 4.15) обычно измеряется в
единицах кДж/моль. Величина %п связана с термохимическими величинами,
измеряемыми во внесистемных единицах электрон-вольт (эВ). Величина А/)оце-
96,5 кДж/моль нивается как разность электроотрицательностей атомов X и Y,
где величина Л/) находится из уравнения 4.15 и измеряется в
эВ.
Xn(Y)-Xn(X) = VAD (4.16)
Решая уравнение 4.16 для различных комбинаций атомов X и Y, Полинг
определил самосогласованный набор величин электроотрицательностей.
Верхний индекс в символе хп означает полинговскую электроотрицательность и отличает ее от
величин электроотрицательности, используемых в соответствии с другими шкалами.
1эВ =
180 4. Гетероядерные двухатомные молекулы
Таблица 4.2. Величины электроотрицательности Полинга (х11) для некоторых
элементов периодической системы
Для элементов, которые могут проявлять несколько степеней окисления, %п даны для
указанных степеней окисления. %п — величина безразмерная.
Группа Группа Группа Группа Группа Группа Группа
12 13 14 15 16 17
н
2,2
Li
1,0
Na
0,9
К
0,8
Rb
0,8
Cs
0,8
Be
1,6
Mg
1,3
Ca
1,0
Sr
0,9
Ba
0,9
(J-Эле-
менты)
В
2,0
Al(III)
1,6
Ga(III)
1,8
In(III)
1,8
T1(I)
1,6
Tl(III)
2,0
С
2,6
Si
1,9
Ge(IV)
2,0
Sn(II)
1,8
Sn(IV)
2,0
Pb(II)
1,9
Pb(IV)
2,3
N
3,0
p
2,2
As(III)
2,2
Sb
2,1
Bi
2,0
О
3,4
S
2,6
Se
2,6
Те
2,1
Po
2,0
F
4,0
CI
3,2
Br
3,0
I
2,7
At
2,2
По Полингу хП(Н) ~ 2 и x (F) = 4. Чем больше величина % , тем выше
способность атома оттягивать на себя электроны; в табл. 4.2 фтор является
самым электроотрицательным атомом.
Спустя годы после того, как Полинг ввел данную концепцию, стали
доступны более точные термохимические данные и величины хп были
скорректированы. В табл. 4.2 перечислены используемые в настоящее время
величины хп- Тем же путем, используя термохимические данные для связей X—X и
Y—Y в молекулах Х2 и Y2 (например, связь С—С в С2Н6 или О—О в Н202),
можно получить величины %п. Это дает возможность определять величины
электроотрицательности для элементов в различных степенях окисления.
Пример 4.1. Определение энтальпии диссоциации
гетероядерной ковалентной связи
Определите энтальпию диссоциации связи в молекуле хлорида
иода, если известно, что
D(CI-CI) = 242 кДж/моль
£>(1—I) = 151 кДж/моль
ХП(С1) = 3,2
Хп(!) = 2,7
4.8. Зависимость электроотрицательности от степени окисления 181
Решение
Необходимо использовать уравнения 4.15 и 4.16:
AD = Z)(X-Y) - 0,5 [D(X - X) + D(Y - Y)]
Xn(Y)-Xn(X) = VAD
В этих уравнениях величины D используйте в эВ. Сначала определите Л/).
VAD = хП(С1) - ХП(1)
= 3,2-2,7
= 0,5
AD = (0,5)2 = 0,25 эВ
Выразите эту величину в единицах кДж/моль.
AZ) = 0,25 • 96,5 = 24,1 кДж/моль
Теперь определите Щ\—О):
AD = D(X-Y) - 0,5 [ДХ-Х) + D(Y-Y)]
24,1 = D(I-C1) - 0,5 [D(l-l) + Z)(C1-C1)]
D(I-Cl) =24,1+0,5(151 +242)
= 220,6 кДж/моль
ЗАВИСИМОСТЬ ЭЛЕКТРООТРИЦАТЕЛЬНОСТИ ОТ
СТЕПЕНИ ОКИСЛЕНИЯ И ПОРЯДКА СВЯЗИ
Степень окисления
Некоторые элементы существуют в нескольких степенях окисления, и от
этого зависят величины электроотрицательности; так, в табл. 4.2 некоторые
величины хп даны не просто для элемента, а для элемента в определенной сте-
► пени окисления, например %п Sn(II) = 1,8 и %u Sn(IV)=2,0.
Степень окисления: Рассмотрим свинец - элемент группы 14. Это металл, и он
см. разд. 1.16. может использовать все свои валентные электроны для
образования соединений свинца(1У), например РЬС14. С другой стороны, он может
использовать только два валентных электрона и образовывать соединения
свинца(П) - РЬС12 и РЫ2. Способность ядра свинца(1У) оттягивать на себя
электроны выше, чем у свинца(Н), и это отражается на величинах
электроотрицательности.
В общем случае величина электроотрицательности данного элемента
возрастает с увеличением положительной степени окисления.
Порядок связи
Электроотрицательность атома углерода, участвующего в одинарной связи,
не равна электроотрицательности углеродного атома, образующего кратную
связь.
В углеводородах С2Н2 (4.13), С2Н4 (4.14), С2Н6 (4.15) прочность связи
углерод-углерод меняется, и можно ожидать различных величин %п, но в
4. Гетероядерные двухатомные молекулы
н н н н
_ = _ \=/ _\ _ /_
/ \ н / \ н
н н н н
4.13 4.14 4.15
табл. 4.2 дана только одна величина для xn(Q. Часто различие между %п
бывает слишком мало, однако оно может быть достаточно для того, чтобы
получить неправильные результаты при определении распределения электронной
плотности между двумя атомами. Эта проблема будет рассмотрена в
разд. 4.13.
ОБЗОР СВЯЗЫВАНИЯ В HF: ВЫВОДЫ
В этом разделе объединены идеи методов структур Льюиса, валентной связи
и молекулярных орбиталей, а так же энтальпии диссоциации связи и элект-
роотри цател ьности.
• Резонансная структура H+F~ гораздо лучше описывает связь, чем H~F+.
• Метод молекулярных связей показывает, что а-связывающая МО ближе
по энергии к орбитали атома фтора.
• В методах валентных связей и молекулярных орбиталей большая
способность атома фтора оттягивать на себя электроны приводит к тому, что
распределение электронной плотности вдоль вектора связи HF сдвинуто в
сторону атома фтора.
• Использование принципа аддитивности при вычислении энтальпии
диссоциации связи Н—F дает неудовлетворительные результаты.
• Теория электроотрицательности Полинга дает возможность
скорректировать искомую величину ДН—F) с учетом ионного характера связи.
Разность электроотрицательностей Н и F составляет 1,8 (см. табл. 4.2).
Используя уравнения 4.15 и 4.16, можно получить скорректированную
величину для ДН—F) = 610 кДж/моль. Экспериментальная величина при
этом составляет 570 кДж/моль. Обычно вычисляемые этим путем
энтальпии диссоциации связи отличаются от экспериментальных на ±10%.
ДРУГИЕ ШКАЛЫ ИЗМЕРЕНИЯ ЭЛЕКТРООРИЦАТЕЛЬНОСТИ
Хотя метод Полинга работает хорошо, он имеет ряд недостатков. Величины
Xй вычисляются эмпирическим путем. Почему мы считаем, что разность
электроотрицательностей двух атомов равна квадратному корню от разности
в энтальпиях диссоциации связи? Если величина хп является свойством
атома, то от чего она зависит? Как величины электроотрицательности Полинга
соотносятся с распределением электронной плотности в молекулах?
Для ответа на эти вопросы были разработаны другие методы, основанные
на различных свойствах атома. В данном разделе обсуждаются два из них.
4.10. Другие шкалы измерения электроорицательности 183
Величины электроотрицательности Малликена (хм)
Рассмотрим двухатомную молекулу XY. Среднее положение максимума
электронной плотности зависит от относительных величин способности атомов X
и Y оттягивать на себя электронную плотность. Для каждого атома
существует предпочтение иметь отрицательный или положительный заряд. Эта идея
лежит в основе метода Малликена определения величин
электроорицательности. Малликен предположил, что величина электроотроотрицательности
атома может быть вычислена, исходя из свойств атома, связанных с
образованием катионов и анионов. Это первый потенциал ионизации и сродство к
электрону. Эти величины будут рассмотрены в гл. 6, но определения
приведены также и здесь.
Первый потенциал ионизации (1Е{) атома — это энергия, необходимая для
отрыва первого валентного электрона (уравнение 4.17); общее изменение
энергии вычисляется для процесса в газовой фазе.
Х(газ) -> Х+(газ) + е" (4.17)
Сродство элемента к электрону (ЕЛХ) — это общая энергия со знаком минус,
необходимая для присоединения электрона (уравнение 4.18); эта величина
также измеряется для процесса в газовой фазе.
У(газ) + е" -> Х-(газ) (4.18)
Метод Малликена вытекает из полинговского определения
электроотрицательности как способности атома в молекуле оттягивать электроны на себя и
согласуется с концепцией теории валентных связей о том, что ионные
резонансные формы X+Y~ и X~Y+ неодинаково важны для связывания в гетеро-
ядерной двухатомной молекуле XY
Величины электроотрицательности Малликена вычисляются по
уравнению 4.19, однако полученные значения несопоставимы с величинами По-
линга, хотя они и построены в соответствии с его принципами.
м 1ЕХ + ЕАХ
Xм = 2 (4.19)
где 1Е] и ЕЛХ выражены в эВ (1эВ = 96,5 кДж/моль).
Величины электроотрицательности Оллреда-Рохова (хОР)
Оллред и Рохов разработали другую шкалу электроотрицатель-
Правила Слейтера: ностей, основанную на эффективном ядерном заряде атома
см. разд. 2.15 ^эфф* Эффективный ядерный заряд может быть вычислен при
помощи правил Слейтера.
Величины электроотрицательности Оллреда-Рохова (хор) определяют по
уравнению 4.20, которое дает величины, напрямую сравнимые с хп.
Х°? = 3590 • эфф + 0,744 (4.20)
г2
ков
где /■„__ даются в пм.
184
4. Гетероядерные двухатомные молекулы
ЕКП ПОЛЯРНЫЕ ДВУХАТОМНЫЕ МОЛЕКУЛЫ
Гомоядерные молекулы, такие, как Н2 или N2, имеют симметричное
распределение электрического зарада, и два конца молекулы неразличимы. Такие
молекулы называют неполярными.
Гетероядерная двухатомная молекула состоит из двух различных атомов.
Если эти атомы обладают разной способностью притягивать электроны, то
электронная плотность будет сдвигаться по направлению к более электроот-
^ рицательному атому. Такие молекулы называют полярными, они обладают
Магнитный момент: электрическим дипольным моментом и. Термин «электриче-
см. разд. 16.13 ский» вводится для того, чтобы отличать эту величину от
магнитного дипольного момента, но он не всегда употребляется.
В полярной двухатомной молекуле максимум распределения
электрического заряда сдвинут к одному из концов молекулы. Такие
молекулы обладают электрическим дипольным моментом ц. Дипольный
момент - векторная величина.
В единицах СИ ц измеряется в кулон-метрах (Кл • м); однако
для удобства часто используют внесистемную единицу дебай
(1Д = 3,336-10-30Клм).
У полярной двухатомной молекулы один конец заряжен отрицательно по
отношению к другому. Это показано на рис. 4.11 для НВг. Обозначение 5+ и 5~
показывает, что в молекуле существует некоторое частичное разделение
заряда; это обозначение не указывает на количественную степень такого
разделения. Для указания направления дипольного момента может быть
использована перечеркнутая стрелка (рис. 4.11,в). В табл. 4.3 перечислены величины \х
для некоторых двухатомных молекул с одинарной связью.
Для того чтобы определить, будет ли молекула полярной, а также найти
величину дипольного момента, можно использовать величины
электроотрицательности. Рассмотрим бромоводород. Электроотрицательность %и для Н
и Вг равна 2,2 и 3,0 соответственно. Из этого следует, что атом брома
притягивает электрон с большей силой, чем атом водорода, и можно предсказать,
что дипольный момент будет направлен, как показано на рис. 4.11.
Рис. 4.11. а - полярность молекулы НВг обусловлена асимметричным
распределением электронной плотности; б — это делает связь полярной; в — диполь может быть
обозначен перечеркнутой стрелкой, направленной в сторону отрицательно
заряженного конца молекулы.
4.11. Полярные двухатомные молекулы 185
Дополнение 4.2. Дипольные моменты
Дипольные моменты возникают при наличии электрического заряда.
Предположим, что в двухатомной молекуле XY, атомы которой
по-разному притягивают электроны, частичные заряды равны +# и —q.
+q -q
Молекула в целом нейтральна, поэтому величины заряда (выражаемые
числом электронов) равны по модулю и обратны по знаку (за единицу принят
заряд электрона).
Пусть расстояние между двумя заряженными центрами равно г.
Дипольный момент ц определяется выражением
д = (Электрический заряд) • (Расстояние между зарядами)
Величина заряда, выраженная числом электронов, показанная на верхней
диаграмме, соответствует электрическому заряду, равному (q • е), где е — заряд
электрона. (Заряд электрона равен 1,602 • Ю-19 Кл.)
\x = q- е- г
Заряд измеряется в кулонах, расстояние - в метрах.
В единицах СИ \i измеряется в кулон-метрах (Кл • м), но для удобства ц
обычно задается в дебаях, 1Д = 3,336 ■ Ю-30 Кл м.
Пример
Дипольный момент в HF равен 1,83 Д. Длина связи Н—F составляет 92 пм.
Определите, как распределен заряд в молекуле HF.
+q -q
Дипольный момент определяется выражением
» = q-e-r
где q — заряд атома, определяемый числом электронов (заряд, равный
единице, соответствует заряду одного электрона).
Если предположить, что центры зарядов совпадают с ядрами Н и F, то г
равно длине связи.
Длина связи Н—F равна 92 пм = 9,2 Ю-1 [м.
1,83 3,336 10 30 _
1,602- 1019 9,2- Ю-11
Данная величина показывает, что заряд в HF распределен так, что атом
фтора получил, а атом водорода отдал 0,4 электрона по сравнению с
нейтральными атомами.
4. Гетероядерные двухатомные молекулы
1,83
1,11
0,83
0,45
0,89
0,52
1,42
1,24
0,73
5,88
6,33
H(6+)F(8-)
Н(8+)С1(6-)
Н(6+)Вг(6")
Н(6+)1(8-)
C1(5+)F(5")
Вг(5+)С1(5-)
Br(6+)F(8-)
1(8+)С1(5")
1(5+)Вг(5-)
Li(5+)H(5")
Li(5+)F(6")
Таблица 4.3. Дипольные моменты ц для некоторых двухатомных молекул в газовой
фазе.
Значения даны в дебаях (Д); это внесистемная единица, которая используется для удобства
(1Д = 3,336-10-30Клм).
Молекула XY //, Д Полярность (S* и 8~)
HF
на
НВг
HI
C1F
BrCl
BrF
IC1
IBr
LiH
LiF
Величина дипольного момента может быть оценена по разности электро-
отрицательностей атомов в гетероядерной молекуле. В серии галогеноводо-
родов хп уменьшается в ряду F> С1> Вг> I. В каждом случае %п (галоген)>
ХП(Н). Соответственно можно предсказать, что наиболее полярной
молекулой в этой группе будет HF, а наименее полярной — HI, но в каждом галоге-
новодороде НХ дипольный момент будет направлен, как показано в
формуле 4.16.
^ 6
Н X
4.16
Необходимо с осторожностью использовать величины электроотрица-
тельностей для молекул с кратными связями, следует учитывать степень
окисления участвующих в связи атомов.
ИЗОЭЛЕКТРОННЫЕ ЧАСТИЦЫ
В этом разделе сделано некоторое отступление с целью ввести важную
концепцию изоэлектронных частиц.
Количество валентных электронов в молекуле N2 равно 10 — по пять от
каждого атома азота. В молекуле СО также десять валентных электронов -
четыре от углерода и шесть от кислорода. Молекулы N2 и СО имеют
одинаковое общее количество электронов (валентные + электроны внутренних ор-
биталей) и называются изоэлектронными. Термин «изоэлектронный»
обозначает «имеющий одинаковое число электронов».
Концепция изоэлектронных частиц широко используется и упоминается
во многих местах этой книги.
Две частицы называют изоэлектронными, если они имеют
одинаковое общее число электронов (валентные + электроны внутренних
орбиталей).
4.12. Изоэлектронные частицы
187
Дополнение 4.3. Изоэлектронные частицы
При помощи периодической системы легко определить, являются ли две
частицы изоэлектронными.
Ниже показаны некоторые/?-элементы.
Группа 13
nsLnpy
В
Z = 5
Al
Z= 13
Ga
Z=31
Группа 14
ns2np2
С
Z = 6
Si
Z= 14
Ge
Z = 32
Группа 15
ns2np3
N
Z-l
P
Z= 15
As
Z = 33
Группа 16
ns2np4
О
Z = 8
S
Z= 16
Se
Z = 34
Группа 17
ns2np5
F
Z = 9
CI
Z= 17
Br
Z = 35
Перемещение на одну ячейку вправо по строке прибавляет один
валентный электрон: у фтора на один электрон больше, чем у кислорода.
Перемещение на одну ячейку влево по строке в таблице означает, что
число электронов уменьшается на один: у фосфора на один электрон меньше,
чем у серы.
Для элементов данной группы число валентных электронов остается
постоянным, но число электронов на внутренних оболочках меняется: азот имеет
электронную конфигурацию основного состояния Xs^lP-lp*, а фосфор -
1522522р63523/?3. Это означает, что эти элементы не изоэлектронны между
собой. Они имеют разное число электронов на внутренних оболочках, но
одинаковое число валентных электронов. Однако из-за их близкого родства их с
оговоркой можно называть «изоэлектронными по отношению к валентным
электронам».
Пример 4.2. Изоэлектронные атомы и ионы
Покажите, что атом фтора и ион О" изоэлектронны (Z(F)=9; Z(0)=8).
Решение
Электронная конфигурация основного состояния атома фтора: [Не]2^22/75.
Электронная конфигурация основного состояния атома кислорода: [Не]2522/?4.
Электронная конфигурация основного состояния иона 0~: [He]2s22/?5.
Поэтому ион О" изоэлектронен атому фтора.
188
4. Гетероядерные двухатомные молекулы
Пример 4.3. Изоэлектронные молекулы и ионы
Покажите, что частицы СО, CN" и N2 изоэлектронны (Z(C)=6; Z(N)=7;
Z(0)=8).
Решение
Атом кислорода и ион N- имеют по 8 электронов.
Молекула СО имеет (6 + 8) = 14 электронов.
Анион CN~ имеет (6 + 8) = 14 электронов.
Молекула N2 имеет (7 + 7) = 14 электронов.
Следовательно, частицы СО, CN~ и N2 изоэлектронны.
ОПИСАНИЕ ОБРАЗОВАНИЯ СВЯЗИ В СО ПРИ ПОМОЩИ
СТРУКТУР ЛЬЮИСА И ТЕОРИИ ВАЛЕНТНЫХ СВЯЗЕЙ
Структуры 4.17 и 4.18 — это структуры Льюиса для гомоядерных двухатомных
^ молекул С2 и 02. То что для них характерно образование двойных связей, по-
Образование связи в зволяет предположить существование аналогичной молекулы
С2 и 02: из атомов углерода и кислорода — структура 4.19 изображает
см. разд. 3.18 и 3.20 СО (моноксид углерода).
:с = с:
4.17
:о=о:
• •
4.18
:с = о[
4.19
Структура Льюиса 4.19 неудовлетворительно описывает молекулу СО.
Образование двойной связи С=0 позволяет кислороду получить восемь
электронов на внешней электронной оболочке (октет), углероду — только
шесть. Изоэлектронность молекул СО и N2 — важный ключ к пониманию
того, как решается эта проблема в молекуле СО.
Атом азота имеет пять валентных электронов. В соответствии со
структурой 4.20 в молекуле N2 два атома азота образуют тройную связь, что дает
каждому атому октет валентных электронов.
:n^n:
4.20
Атом азота изоэлектронен С~ и 0+, и, сравнивая СО и N2, можно
изобразить диаграмму Льюиса для молекулы СО, комбинируя С" и 0+. Это
показано на рис. 4.12 (ср. со структурой 4.20).
Структуры 4.19 и 4.21 показывают две резонансные формы моноксида
углерода. Заметим, что структура 4.19 предполагает наличие двойной связи
С=0, а структура 4.21 — тройной. В соответствии со структурой 4.21 каждый
атом имеет октет электронов.
4.13. Описание СО по теории ВС
189
-+» : с
о : -<-
о :
Атом С
о+
Атом О
: с-
+
ю:
Рис. 4.12. Используя изоэлектронность СО и N2, можно описать образование связи
в СО как комбинацию ионов С~ и 0+.
Если структура 4.21 дает существенный вклад в образование связи в СО,
молекула должна быть полярной и иметь заряд 8~ на углероде. (Это не
соответствует величинам электроотрицательностеи для углерода и кислорода из
табл. 4.2, особенно если учесть зависимость % от порядка связи.) Наблюдае-
^ мый дипольный момент СО равен 0,11 Д. Это сравнительно небольшая ве-
Химия СО: см. разд. личина, так что молекула СО почти неполярна - углеродный
13.6 и 16.14 конец молекулы несет малый отрицательный заряд по
отношению к кислородному концу. Для химии СО это важное
заключение.
Если же значимой является структура 4.19, то энтальпия диссоциации
связи и расстояние углерод-кислород должны соответствовать двойной
связи. Структура 4.21 предполагает более прочную тройную связь. В табл. 4.4 пе-
Дополнение 4.4. Моноксид углерода - токсичный газ
Моноксид углерода очень ядовит, потому что он связывается с атомами
железа в гемоглобине сильнее, чем 02. Это прекращает поглощение и транспорт
кислорода в крови. На рис. а показана структура одной из цепей кислород-
связывающего белка гемоглобина млекопитающего. Для простоты атомы
водорода не показаны. Атом железа связан с гетероциклической молекулой —
гемом (эта часть структуры выделена на рисунке). На рис. б изображено
окружение железа, где этот атом связан с пятью атомами азота. Когда 02 или СО
связываются с железом, они располагаются так, что этот атом получает окта-
эдрическое окружение.
113
1076
0,П
Диамагнитен
ПО
945
0
Диамагнитен
190 4. Гетероядерные двухатомные молекулы
Таблица 4.4. Сравнение некоторых свойств СО и N2
Свойство СО N2
Длина связи, пм
Энтальпия диссоциации связи, кДж/моль
Дипольный момент, Д
Магнитные свойства
речислены некоторые важные данные для СО и N2; отметим, что N2 имеет
тройную связь. Длина связи в СО близка к длине связи в N2, однако связь еще
прочнее. Это свидетельствует о наличии тройной связи, т. е. подтверждает
структуру 4.21.
В заключение следует отметить, что теория валентных связей описывает
моноксид углерода в терминах резонансных структур с двойной или тройной
связью. Все электроны спарены, что подтверждается диамагнитностью со-
^- единения. Наблюдаемые свойства СО (сильная короткая связь, слабая по-
Диамагнетизм: лярность молекулы С5~05+) могут быть объяснены одновре-
см. разд. 3.18 менным существованием резонансных структур 4.19 и 4.21. В
дополнение следует отметить, что дипольный момент,
определяемый электроотрицательностями Полинга, предполагает разделение
заряда, обратное тому, которое показано для структуры 4.21.
ОПИСАНИЕ ОБРАЗОВАНИЯ СВЯЗИ В СО
МЕТОДОМ МОЛЕКУЛЯРНЫХ ОРБИТАЛЕЙ
Исходные данные
Рассмотрим образование связи в моноксиде углерода с использованием
метода молекулярных орбиталей и принципов, изложенных в гл. 3. При этом
мы опираемся на следующие начальные данные:
• эффективный ядерный заряд атома кислорода больше, чем атома
углерода;
• 2s-AO кислорода лежит по энергии ниже, чем 2s-AO углерода;
• 2/7-АО кислорода лежат по энергии ниже, чем 2р-АО углерода;
• разница в энергиях между 2р- и 2я-орбиталями кислорода больше, чем у
углерода.
Линейная комбинация атомных орбиталей углерода и кислорода дает МО-
диаграмму, показанную на рис. 4.13. Связывающие МО [a(2s), o(2/?) и тс(2/?)],
ближе по энергии к атому кислорода, чем углерода. В частности, из рис. 4.13
видно, что a(2s)-MO по энергии почти совпадает с 25-орбиталью кислорода.
В первом приближении эта диаграмма удовлетворительна - она дает
верный порядок связи СО (три) и предсказывает диамагнитность соединения, что
согласуется с экспериментальными данными. Однако эта простая картина
дает неправильное распределение молекулярных орбиталей по энергиям и
предполагает, что G-MO являются либо связывающими, либо разрыхляющими.
На этом можно завершить обсуждение связи в СО; остальная часть
раздела посвящена важности смешивания орбиталей.
4.14. Описание СО методом МО
191
О)
С?(2р)
2р
Рис. 4.13. Диаграмма МО молекулы СО, учитывающая перекрывание орбиталей
25(C) и 25(0), а также 2р(С) и 2р(0).
Смешивание орбиталей
В разд. 3.19 обсуждалась важность смешивания орбиталей. Если 2s- и 2р-
^ атомные орбитали в гомоядерной двухатомной молекуле достаточно близки
а-л-Взаимодействие: по энергии, то могут взаимодействовать a(2s)- и а(2/?)-молеку-
см. разд. 3.19 лярные орбитали. Аналогично, возможно смешивание a*(2s)-
и а*(2/?)-М0. Это приводит к изменению энергий
молекулярных орбиталей.
Относительные энергии атомных орбиталей в моноксиде углерода
таковы, что возможно смешивание молекулярных орбиталей. Это показано на
рис. 4.14. Смешивание g(2s)- и а(2/>)-орбиталей понижает энергию a(2s)-M0
(обозначенной как la) и повышает энергию а(2р) (обозначенной как За).
Энергия За-орбитали выше, чем я(2/?)-орбиталей. Аналогично, смешивание
o*(2s)- и а*(2/?)-орбиталей понижает энергию a*(2.s) (обозначенной как 2а) и
повышает энергию а*(2/?) (обозначенной как 4а). Как следствие, 2s- и 2/?-ор-
битали атомов кислорода и углерода дают вклад во все четыре а-МО
молекулы СО. Наиболее замечательным результатом является то (ср. рис. 4.13 и
4.14), что наивысшей заполненной МО является орбиталь a-типа, а не л-ти-
па. Степень смешивания контролирует точный порядок следования по
энергии la- и 2а-орбиталей.
К сожалению, даже рис. 4.14 не полностью информативен, потому что не
показывает индивидуальные вклады АО углерода и кислорода в каждую из
четырех a-орбиталей. Эти вклады не равны, и следствием этой запутанности
является то, что характер a-орбиталей не понятен — являются они
связывающими (а) или разрыхляющими (а*). Именно по этой причине на рис. 4.14
они обозначены как la, 2a, За и 4a.
192
4. Гетероядерные двухатомные молекулы
X
(Г)
2р
Рис. 4.14 Диафамма МО молекулы СО, предполагающая смешивание орбиталей
o(2s) и а(2/?), а также o*(2s) и о*(2р). По сравнению с рис 4.13 изменяется
последовательность а- и я-орбиталей. Это приводит к тому, что наивысшая занятая МО имеет
а-симметрию.
Более детальное рассмотрение МО в молекуле СО приводит к следующим
выводам:
• наиболее близкая по энергии к 2^-орбитали кислорода la-МО является
несвязывающей по отношению к взаимодействию углерод-кислород;
• поскольку вклад 2s- и 2/ьорбиталей кислорода в За-МО невелик, Зо-МО
расположена у атома углерода и также может считаться несвязывающей.
Эти результаты дают возможность изобразить измененную МО-диаграмму
моноксида углерода (рис. 4.15). Следует отметить, что хотя эта диаграмма
является упрощенной, она все же представляет собой удобную модель
образования связи в молекуле.
Выше был сделан вывод о том, что За-МО является в основном
несвязывающей, что явно показано на рис. 4.15 — За-МО не соединена с орбиталями
атома кислорода. Это несколько меняет наше представление о
несвязывающей орбитали. Ранее было показано, что несвязывающая орбиталь должна
совпадать по энергии с атомной орбиталью; в качестве примера можно
привести несвязывающие орбитали, построенные из 2/7-орбиталей фтора на
рис. 4.8. Однако в молекуле СО За-МО не совпадает по энергии с атомной
орбиталью углерода. Ее энергия оказывается средневзвешенной энергией 2s-
и 2/7-АО углерода.
4.14. Описание СО методом МО
193
X
■+-
2р
-н-
ler
СО
2р
-И" 2.
О
Рис. 4.15. Диафамма МО для СО, допускающая эффект смешивания орбиталей, но
также и учитывающая, что не все АО дают одинаковый вклад в а-МО. Основное
изменение, по сравнению с рис. 4.14, заключается в том, что 1о- и Зо-МО становятся не-
связывающими. Заметим, что это упрощенная схема.
Применение метода МО для описания образования связи
в СО: выводы
На основании МО-диаграммы образования связи в молекуле СО,
представленной на рис. 4.15, можно сделать следующие выводы:
• 2а- и л(2/?)-связывающие МО заняты шестью электронами, остальные
четыре электрона занимают несвязывающие орбитали (1а и За). 4а- и
к*(2р)-МО являются разрыхляющими.
• Порядок связи в СО равен трем (уравнение 4.21).
Порядок связи в СО = 0,5 (6 - 0) = 3 (4.21)
• Молекула СО диамагнитна — это совпадает с экспериментальными
данными.
• Наиболее высокоэнергетическая занятая МО в СО локализована
преимущественно на атоме углерода и содержит вклады Is- и 2/ьАО, что
отражено на рис. 4.16. В разд. 16.14 мы обсудим некоторые химические свойства
СО, являющиеся, в частности, следствием природы этой орбитали и того
факта, что низшая по энергии свободная МО имеет л-симметрию.
• На простом качественном уровне рассмотрения метода МО трудно
сделать какие-либо выводы о полярности молекулы СО. Распределение
заряда в молекуле следует из суммарной картины занятых молекулярных орби-
4. Гетероядерные двухатомные молекулы
Рис. 4.16. Схематическое, упрощенное ^ИР"^^
изображение самой высокоэнергетиче- -wk ^СЗ * ^
ской занятой МО в моноксиде углерода. z
Черными точками обозначены ядра С и О. с О
талей; свободные орбитали не влияют на свойства молекулы. Электронная
плотность занятой 1а-МО локализована на атоме кислорода (О5-), тогда
как электроны 2а- и За-МО расположены ближе к атому углерода (Cs~).
Обе 7с(2р)-МО ближе по энергии к атому кислорода (О5-), чем углерода .
ИОНЫ CN~ И NO+, ИЗОЭЛЕКТРОННЫЕ СО
Цианид-ион CN- и нитрозил-катион NO+ являются хорошо известными
химическими частицами.
У атома углерода четыре валентных электрона, а у атома азота - пять.
Таким образом, в цианид-ионе 10 валентных электронов [4 + 5 + 1 (из-за
наличия отрицательного заряда)].
У атома азота пять валентных электронов, а у атома кислорода — шесть. В
нитрозил-катионе также 10 валентных электронов [5 + 6—1 (благодаря
положительному заряду)].
Углерод, кислород и азот имеют одинаковое число электронов на
внутренних оболочках. Следовательно, ионы CN~ и NO+ изоэлектронны. Они
также изоэлектронны молекулам СО и N2.
Ранее было отмечено, что существует некоторое сходство и различие
между описаниями образования связи в гомоядерной двухатомной молекуле N2
и гетероядерной молекуле СО. Входят ли ионы CN~ и NO+ в эту серию?
Одинаков ли порядок связи у этих частиц? Совпадают ли магнитные свойства?
Какие изменения в процесс образования связи вносит наличие
дополнительного заряда? Можно ли предсказать, на каком атоме в частицах CN" и NO+
локализован заряд?
В течение всего обсуждения следует помнить, что С и N+ изоэлектронны по
отношению друг к другу; N и 0+, а также N~ и О тоже составляют пары изо-
электронных частиц.
Теория валентных связей
Резонансные структуры для CN~ можно изобразить, используя СО в
качестве основы. Атом С может быть замещен на изоэлектронный N+, а атом О -
на N-, как это показано на рис. 4.17. Структура II для каждой частицы может
быть сопоставлена со структурой Льюиса для N2; следует помнить, что атом
азота изоэлектронен одновременно С~ и 0+.
Ранее было показано, что наличие сильной короткой связи в СО
предполагает, что резонансная структура II дает существенный вклад в связь в этой
молекуле, и акцентирует внимание на аналогии в образовании связи в СО и
N2. Длина связи в анионе CN~ равна 114 пм, а в катионе NO+ она составляет
106 пм. Эти величины сравнимы с таковыми для СО и N2 (113 и 110 пм соот-
4.15. Ионы CN" и NO+, изоэлектронные СО
195
ветственно) и согласуются с предположением о сильном влиянии
резонансной структуры II на образование связи.
Малый дипольный момент СО может быть объяснен, если допустить, что
резонансные структуры I и II частично компенсируют друг друга, несмотря
на различие в электроотрицательностях двух атомов. Однако трудно
использовать теорию валентных связей, чтобы предсказать полярность связи
углерод-кислород. Обе резонансные формы для NO+ предполагают разную
полярность иона (рис. 4.17): в I азот заряжен положительно, а в II
положительный заряд несет кислород. Аналогичная проблема возникает, когда мы
пытаемся предсказать, какой из атомов будет нести отрицательный заряд в
ионе CN~.
Метод молекулярных орбиталей
Несмотря на различие в МО-диаграммах для N2 (рис. 3.28) и СО (рис. 4.15),
результаты рассмотрения согласуются с наблюдаемым диамагнетизмом,
порядок связи равен трем в обоих случаях. Распределения заряда в молекулах
следует из энергий занятых молекулярных орбиталей. В N2 МО содержат
одинаковые вклады от каждого атома и связь неполярна (|д = О Д). В монокси-
де углерода разница в энергиях атомных орбиталей углерода и кислорода
вызывает изменение энергий МО, и в результате связь углерод—кислород
оказывается полярной (ji = 0,11 Д).
Для гетероядерных ионов CN~ и NO+ при описании образования связи
методом молекулярных орбиталей в качестве точки отсчета лучше
использовать СО, чем N2. Во многих отношениях такой подход правильный, но
невозможно дать абсолютно точный ответ, используя лишь качественную картину.
Для описания связи в гетероатомных частицах, изоэлектронных СО, в
качестве первого приближения возможно использовать самую простую
МО-диаграмму (рис. 4.13). Ответ может оказаться не совсем корректным из-за
несовпадения некоторых результатов с экспериментальными данными. Однако
при наличии достаточного опыта можно научиться предсказывать, как
энергии атомных орбиталей могут влиять на состав молекулярных орбиталей и,
следовательно, на молекулярные свойства.
:n==n :
+
:с = о; ** ► :с==о:
+ . +
:n^=o; *+ ► :n==o:
:c = n; ** ► :c==n:
i и
Рис. 4.17. Резонансные структуры для CN~ и NO+ могут быть получены исходя из
структур СО и N2 с учетом изоэлектронности ChN+,NhO+,3 также N~ и О.
196
4. Гетероядерные двухатомные молекулы
Для начала можно использовать диаграмму молекулярных орбиталей на
рис. 4.13, учитывая, что уменьшение электроотрицательностей ряду О > N > С
(табл. 4.2) означает, что АО кислорода будут ниже по энергии, чем АО азота,
а те в свою очередь ниже углеродных. Теперь можно предположить, что:
• порядок связи в СО, CN~ и NO+ равен трем;
• эти частицы диамагнитны.
Такой же результат можно получить, используя более сложную
МО-диаграмму (рис. 4.15). Однако более сложные расчеты выявляют более тонкие
различия. Молекулярная орбиталь 1а, которая была преимущественно не-
связывающей в СО, становится связывающей в ионах CN~ и NO+.
Молекулярная орбиталь 2а в СО связывающая, а в CN~ и NO+ разрыхляющая.
Орбиталь За, локализованная на атоме углерода в СО, в ионах CN~ и NO"1"
имеет характер связывающей орбитали, но так же асимметрична (локализована
на углероде в CN~ и на азоте в NO+).
ЧАСТИЦЫ NO+, NO, NO"
Рассмотрим очень близкие частицы NO+, NO и NO~. Отметим, что частица
N0 изоэлектронна О^, тогда как N0" изоэлектронна 02; полезно сравнить
некоторые новые данные, приведенные в этом разделе, с деталями,
изложенными в разд. 3.22 для дикислородных частиц. Некоторые свойства N0 и 02+
представлены в табл. 4.5.
Как и в разд 4.15 при рассмотрении иона N0+, здесь при обсуждении
образования связи в N0 и N0" может быть использована диаграмма рис. 4.13.
Моноксид азота N0 имеет на один валентный электрон больше по
сравнению с N0+, и, согласно принципу последовательного заполнения наинизших
молекулярных орбиталей, он займет одну из л*(2/>)-орбиталей; см. рис. 4.13.
Таким образом, молекула N0 имеет один неспаренный электрон и должна
быть парамагнитной. Это согласуется с экспериментальными данными:
N0 - радикал.
Порядок связи в N0 может быть определен в соответствии с уравнением
4.22. Увеличение длины связи в N0 (115 пм) по сравнению с N0+ (106 пм)
связано с уменьшением порядка связи от 3 до 2,5.
Порядок связи в N0 = 0,5 (6 - 1) = 2,5 (4.22)
Нечетный электрон занимает разрыхляющую МО, которая ближе по энергии
к атому азота, чем кислорода, так, что нечетный электрон будет
располагаться ближе к атому N, чем к О. Конечно, то, насколько это положение верно,
зависит от относительных энергий атомных орбиталей N и О.
Таблица 4.5 Некоторые свойства моноксида азота N0 и изоэлектронного
катиона Oj
Свойство NO Oy+
Длина связи, пм
Энтальпия диссоциации связи, кДж/моль
Магнитные свойства
Дипольный момент, Д
115 112
631 644
Парамагнитен Парамагнитен
0,16 О
4.17. Заключение
197
Дополнение 4.5. Роль молекулы NO в биологии
N
О
Известно, что моноксид азота играет важную роль в биологии и в настоящее
время является предметом тщательного изучения. Это малая молекула,
которая легко диффундирует сквозь клеточную стенку. Она играет активную роль
в таких процессах у млекопитающих, как регулирование кровяного давления,
мускульная релаксация и передача нервных импульсов.
Одним из наиболее замечательных свойств N0 является его цитотоксич-
ность (способность разрушать индивидуальные клетки), которая влияет на
способность иммунной системы убивать опухолевые клетки.
Более подробно об этом можно прочитать в статье Батлера Г. [Butler A.R.
The biological role of nitric oxid, Chem.a.Ind., p. 828 (1995).] [См. также:
Зеленина В.К. Оксид азота(Н): Новые возможности давно известной
молекулы. Соросовский Образовательный Журнал, 1997, №10, с. 105. - Ред.]
Анион N0" имеет по сравнению с NO+ два дополнительных валентных
электрона. Они занимают наинизшие по энергии две свободные я*(2р)-орби-
тали (см. рис. 4.13). Ион NO~ по аналогии с молекулой 02 парамагнитен.
Порядок связи равен двум (уравнение 4.23).
Порядок связи в NO- = 0,5 (6 — 2) = 2
(4.23)
ЗАКЛЮЧЕНИЕ
В этой главе рассматривался вопрос образования связей в гетероядерных двухатомных
молекулах.
Что означают следующие термины?
Гетероядерный
Резонансная структура
Характер орбитали
Коэффициент в выражении
волновой функции
Близость орбиталей по
энергиям
Несвязывающая
молекулярная орбиталь
Электроотрицательность
Полярная молекула
Электрический дипольный
момент
Изоэлектронность
Теперь вы должны уметь:
• суммировать наиболее важные различия
между гомоядерными и гетероядерными
двухатомными молекулами;
• описать образование связи в HF, LiF, LiH, CO,
NO+ , CN~ , NO и NO" с точки зрения теории
валентных связей;
• описать образование связи в HF, LiF, LiH, CO,
NO+ , CN", NO и NO" с точки зрения метода
молекулярных орбиталей;
• схематично изображать диаграммы
молекулярных орбиталей для гетероатомных
двухатомных молекул и уметь их интерпретировать;
• использовать энтальпии диссоциации связи
для определения энтальпий атомизации;
кратко изложить концепцию
электроотрицательности;
определить понятия (электрического) диполь-
ного момента и полярной молекулы;
определять, будет ли молекула полярной, с
использованием величин электроотрицатель-
ностей Полинга;
написать ряд изоэлектронных молекул и
ионов;
использовать картину образования связи в
одной молекуле для описания связывания в
изоэлектронных ей молекуле или ионе.
198
4. Гетероядерные двухатомные молекулы
ЕКВ УПРАЖНЕНИЯ
4.1. Волновая функция связывающей
МО в гетероядерной двухатомной
молекуле АВ задана уравнением
\|/(МО) = 0,9\|/А + 0,3\|/в
Как соотносятся величины Z3(M)
атомов А и В?
4.2. Нарисуйте резонансную структуру
(структуры) для гидроксид-иона
ОН~. Как можно описать
образование связи в этом ионе в рамках
теории валентных связей?
4.3. Учитывая, что ион ОН- изоэлектро-
нен HF, изобразите МО-диаграмму
для этого иона. Каков порядок связи
в ОН-? Отличается ли МО-картина
связеобразования, полученная этим
методом, от полученной ранее по
теории валентных связей?
4.4. Вычислите энтальпию диссоциации
связи для фторида хлора C1F, если
энтальпии диссоциации связи для F2
и Cl2 равны 159 и 242 кДж/моль
соответственно. %U(F) = 4,0 и хп(С1) =
3,2 (1эВ = 96,5 кДж/моль).
Сравните ваш ответ с эксперименальными
данными.
4.5. Уравнения 4.15 и 4.16 могут быть
применены к многоатомным
молекулам. Используя данные табл. 3.6 и
4.2, вычислите энтальпии
диссоциации связей а) О—Н, б) С—О, в) S—F.
4.6. Используя полученные в
предыдущем упражнении энтальпии
диссоциации связей, определите
энтальпии атомизации а) Н20, б) СС14,
в) SF4.
4.7. Используйте величины первого
потенциала ионизации и сродства к
электрону, указанные в приложениях
5 и 6, для определения величин
электроотрицательности Малликена
Xм атомов N, О, F, О, Вг, I.
Нанесите эти величины на график и
покажите тенденции в изменении хм
вдоль периода /^-элементов и вниз по
группе 17. Как эти изменения
согласуются с тенденциями для х11 (см.
табл. 4.2)?
4.8. Укажите пять ионов (анионов или
катионов), изоэлектронных неону.
4.9. Объясните, будут ли пары данных
частиц изоэлектронны: а) Не и Li+,
б) Se^- и Вг2, в) N0 и 0+, г) F2 и C1F,
д) N2 и Р2, е) Р3" и С1-.
4.10. Это упражнение относится к
материалу дополнения 4.2. Определите
распределение заряда в молекуле
СО, если дипольный момент равен
0,11 Д, а длина связи С=0 равна
113 пм. Атом углерода несет
заряд 5~.
Н МНОГОАТОМНЫЕ МОЛЕКУЛЫ:
ФОРМА И ХИМИЧЕСКАЯ СВЯЗЬ
ЕП ВВЕДЕНИЕ
Геометрия молекул
В гл. 3 и 4 основное внимание уделялось двухатомным молекулам, однако
форма этих молекул не рассматривалась, потому что двухатомные молекулы
всегда линейны\ Однако двухатомные молекулы составляют лишь малую часть
от общего числа молекул, и в этой главе рассмотрены многоатомные частицы,
т. е. состоящие из трех и более атомов.
Многоатомные молекула или ион содержат три или более атомов.
Некоторые многоатомные молекулы и ионы линейны (например, С02,
HC^N, 1^~ , НС=СС=СН), но большинство таких частиц имеют нелинейное
строение.
В качестве основного признака для описания трехмерной структуры
многоатомной молекулы удобно использовать геометрию окружения
центрального атома. В табл. 5.1 перечислены некоторые геометрические типы
молекул общей формулы XYn. Ниже мы опишем их подробнее.
Углы между связями
Длина связи и угол между связями (валентный угол) чрезвычайно важны при
описании строения молекул. Угол между связями хорошо описывает
молекулярную геометрию, он определяется для трех соединенных вместе атомов*.
Например, для плоскотреугольной молекулы XY3 каждый угол Y—X—Y
равен 120° (рис. 5.1,я).
Форма молекул всегда описывается в терминах идеальной геометрии,
соответствующие идеальные углы для основных структурных типов указаны в
правой колонке табл. 5.1. Отметим, что реальная геометрия молекул далеко не
всегда отвечает идеальной. На практике большинство молекул описывается
Мы не рассматриваем торсионные углы, определяемые для четырех атомов как степень поворота
вокруг центральной связи и, таким образом, как степень выхода из плоскости.
200 5. Многоатомные молекулы: форма и химическая связь
Таблица 5.1. Молекулярная геометрия многоатомных молекул ХУЛ
Формула Координацией- Геометрия
XYn ное число
атома X
XY,
XY,
XY,
XY,
XY,
Пространственная Идеальный угол
форма Y—X— Y
Линейная
Угловая (изогнутая)
Плоскотреугольная
Тригонально-
пирамидальная
Т-образная
У X У
Y^ ^У
У
/
У X
\
У
Y^i'^Y
У
Ya
Yb X
I
180°
Различный (*180°)
120°
Различный (< 120е)
Ya-X-Yb = 90°
Ya-X-Ya= 180°
(атомы лежат в
одной плоскости)
Y
XY4
Тетраэдрическая
.х
Y^i"""Y
109,5°
XY4
Плоскоквадратная
У Y
\ /
X
Y/\Y
90°
XY.
Дисфеноидная
(искаженный
тетраэдр)
хС
..»»»Ye(
Y.q-X-Y.,-120-
XY,
XY,
XYt
Тригонально-
бипирамидальная
j -^ Yax-X-Yeq = 90'
t^Y, Yeq-X-Yeq=120-
Квадратно-
пирамидальная
Октаэдрическая
Yax
Y
1
1
x
Y | Y Y
Y
Y *»».. 1 ..♦♦» Y
^>X>1
Y^ | ^*Y2
Y
Различный
Y,-X-Y2 = 90'
1 Хотя угол определяется между атомами Y1 и У2 , все атомы Y эквивалентны.
5.1. Введение
201
а
Y
Для плоскотреугольной молекулы XY3
в1 = 02 = 03 = 120°
Рис. 5.1. а- угол между связями определяется как угол, образованный тремя
связанными атомами. В треугольной молекуле XY3 идеальный угол Y—X—Y равен 120°; б -
углы между связями равны 120°, если все заместители одинаковы, но если это не так,
углы могут различаться; тогда геометрия считается приблизительно трнгоналъной.
перечисленными в табл. 5.1 структурами без учета степени идеальности углов.
Отклонения от идеальности обусловлены неравноценностью заместителей.
Молекулы BF3 и ВС13 имеют форму равностороннего треугольника, a BC12F
и BC1F2 — такие лишь приблизительно. Атомы хлора больше атомов фтора, и
этим объясняется отклонение углов между связями от 120° (рис. 5.1 ,б). Длина
связи dтакже различается: d(B—CI) > d(B—F).
В табл. 5.1 перечислены молекулы общей формулой XYW, имеющие в центре
атом X. Большое количество многоатомных молекул имеют более одного
центрального атома; этан 5.1 содержит два атома углерода, имеющих тетра-
эдрическое окружение, а циклогексен 5.2 — шесть атомов углерода, из кото-
v ,
II i
н с ч
А н
5.2
^ /
/ \'""н
5.1
202
5. Многоатомные молекулы: форма и химическая связь
рых четыре имеют тетраэдрическое окружение, а два - треугольную
геометрию. Форма больших молекул определяется совокупностью окружений всех
атомных центров (разд. 5.3).
Координационное число
Координационным числом (КЧ) атома называют число присоединенных к нему
атомов или групп атомов. Оно не зависит от типа связи (одинарная, двойная
или тройная), а также от того, является связь локализованной или делокали-
зованной.
Координационным числом атома называют число присоединенных к
нему атомов или групп атомов.
В табл. 5.1 приведены примеры координационных чисел центрального
атома X в молекуле XYW, изменяющиеся от двух до шести. В частицах BF3 (5.3) и
COj (5.4) координационное число центрального атома равно трем, хотя типы
связей не одинаковы. Аналогично, в СН4 (5.5) и S02C12 (5.6)
координационное число центрального атома равно четырем.
F
i
F F
5.3
В этой главе описаны формы молекул и ионов XYn с одним центральным
атомом X, причем Y может быть как атомом, так и группой атомов. Мы
проследим за тем, как усложнение структур молекул по сравнению с
двухатомными частицами влияет на дипольный момент, и рассмотрим подходы к
описанию образования связи в многоатомных молекулах. Сначала проведем
краткий обзор р-элементов и суммируем наблюдаемое разнообразие
молекулярных структур. Обсуждение начинается с трехатомных молекул.
^£Ш ГЕОМЕТРИЯ ТРЕХАТОМНЫХ МОЛЕКУЛ
Трехатомные молекулы могут иметь линейное или угловое (изогнутое)*
строение. Примерами линейных молекул могут служить диоксид углерода (5.7) и
циановодород (5.8), а угловых - вода (5.9) и диоксид серы (5.10).
* Угловые (изогнутые ) молекулы иначе называют нелинейными или V-образными.
Н
О
о-
^
О О
5.4
h^V"" о^Ч""4
С1
5.5
5.6
5.2. Геометрия трехатомных молекул
203
О:
= С =
5.7
О
-СЕ
5.8
О
5.9
5.10
Линейные трехатомные молекулы и ионы
На рис. 5.2 представлены структуры молекулы диоксида углерода,
нитрил-катиона* NOJ, азид-аниона Nj и цианат-иона NCO~. Эти частицы изоэлек-
тронны и все имеют линейную структуру - они изоструктурны. Замена атома
кислорода в NCO- на атом серы дает тиоцианат-анион NCS-. Если учитывать
только валентные электроны, то NCO~ и NCS~ изоэлектронны и имеют
аналогичные структуры (рис. 5.2). Другим примером изоэлектронных по
валентным электронам и изоструктурных частиц могут служить линейные полига-
логенид-анионы ICl2~ (5.11) и 13~ (5.12).
^ Можно ожидать, что изоэлектронные частицы будут также
Изоэлектронные изоструктурны. В основном это действительно так у много-
частицы. см. разд. 4.12 ат0МНых частиц, и трехатомные молекулы не являются
исключением. Поскольку радиусы атомов могут существенно отли-
С1-
■о]'
[ ]
5.11
5.12
Термин «изоструктурные» означает «имеющие одинаковую структуру».
®=©=®
со,
[NOJ+
[ © # =® ]
[NJ-
=©
[NCO]-
®=&
[NCS]-
<D
Рис. 5.2. Изоэлектронные частицы С02, N0^, Njh NCO~ имеют линейную
структуру. Цианат-ион NCO" и тиоцианат-ион NCS" имеют одинаковое число валентных
электронов и поэтому могут рассматриваться как изоэлектронные.
* Старое название иона NOJ - ион нитрония - до сих пор часто встречается в литературе.
204
5. Многоатомные молекулы: форма и химическая связь
чаться, то и длины связей в однотипных молекулах или ионах могут быть
неодинаковыми, но геометрия окружения центрального атома, которая
определяется углами между связями, остается фактически неизменной. Конечно,
если принимать в расчет только валентные электроны, то формально изо-
электронные молекулы могут иметь принципиально различную структуру.
Классический пример — С02 и Si02: при комнатной температуре С02 -
линейная молекула, a Si02 образует гигантскую решетку, в которой атомы
кремния окружены четырьмя атомами кислорода, образующими тетраэдр. В то же
время, некоторые молекулы действительно подчиняются изложенному выше
правилу.
Изоэлектронные молекулы и ионы обычно изоструктурны.
Но изоструктурные частицы не обязательно изоэлектронны.
Угловые молекулы и ионы
На рис. 5.3 показаны некоторые угловые молекулы. Обратите внимание на
то, что валентный угол, обозначенный как а в молекуле 5.13, не имеет какой-
то определенной величины. Точная геометрия угловой молекулы становится
известной только после экспериментальных измерений угла а, но термин
«угловая» может быть применен к любой трехатомной молекуле XY2 с
0<а<180°.
.<^>.
5.13
н2о
so2
Рис 5.3. Некоторые угловые молекулы, иллюстрирующие набор углов между
связями: Н20 (ZH-0-H = 104,5°), F20 (ZF-O-F = 103°), SF2 (ZF-S-F = 98°) и S02
(ZO-S-0=119e).
5.2. Геометрия трехатомных молекул
205
Рис. 5.4. Структура N02 (ZO—N—О =
134°). Сравните со структурой NOj на
рис. 5.2.
NO,
HOC1
C1NO
FSN
Рис. 5.5. Некоторые асимметричные угловые трехатомные молекулы:
НОС1 (ZH-0-C1 = 102,5е), C1NO (ZO-N-C1 = 113°) и NSF (ZN-S-F =
117°).
I
О
I
2
I
X
i
117
114
111
108
С1
Вг
Размер атома X
Рис. 5.6. В молекулах FNO, C1NO и BrNO угол X—N—О возрастает с увеличением
размера атома галогена X.
Из приведенных на рис. 5.3 можно выделить три пары родственных
молекул: OF2 и SF2 изоэлектронны (имеют одинаковые наборы валентных
электронов); Н20 и OF2 сходны в том, что в обоих случаях центральным является
атом кислорода, образующий две одинарные связи. Кроме того, в молекулах
SF2 и S02 атом серы имеет координационное число 2. Однако в SF2 связи
одинарные, а в S02 двойные.
Ранее отмечалось, что полигалогенид-анионы 1С12~ и 13~ линейны. В то
же время, полигалогенид-катионы 1С12+ и 1^" имеют угловое строение.
Изменение структуры, которое в данном случае сопровождает изменение заряда,
5. Многоатомные молекулы: форма и химическая связь
не является уникальным. Например, катион N0^ (рис. 5.2) линеен, тогда
как и N02, и N02~ изогнуты, хотя валентные углы в них разные [134° у N02
(рис. 5.4) и 115е у NOf].
Некоторые асимметричные угловые трехатомные частицы показаны на
рис. 5.5. Структура хлорноватистой кислоты НОС1 аналогична Н20, а С120
аналогичен OF2 (рис. 5.3). Замена атома хлора в C1NO другим элементом
группы 17 дает аналогичные молекулы FNO и BrNO, hoc другими углами
между связями (рис. 5.6). Тенденции в изменении углов между связями корел-
лируют с размером и/или электроотрицательностью атомов.
МОЛЕКУЛЫ С ЧИСЛОМ АТОМОВ БОЛЕЕ ТРЕХ
ЛИНЕЙНОЙ ИЛИ УГЛОВОЙ ГЕОМЕТРИИ
Хотя линейная или угловая геометрия обычно ассоциируется с
трехатомными молекулами или ионами, это понятия могут быть с успехом применены и
к большим молекулам.
Молекула или ион, содержащие скелет из трех атомов, могут быть
сопоставлены с трехатомными частицами. На рис. 5.7,а показаны родственные
молекулы — циановодород (трехатомная линейная молекула) и ацетонитрил
MeCN, где Me - метильная группа СН3, которая может рассматриваться как
&
<Е)
HCN
о—®
MeCN
Рис. 5.7. Структурное родство между циановодородом HCN и ацетонитрилом
MeCN (а) и H2S и Me2S (б). Если рассматривать метильную группу как псевдоатом, то
MeCN можно рассматривать как молекулу с линейным CCN-скелетом, a Me2S - как
угловую молекулу.
,5.4. Геометрия молекул, содержащих р-элементы второго периода
(н>=Цс)==0==^
207
нс=сн
=©=&
МеС = СМе
Рис. 5.8. Структурное родство между алкинами НС=СН и МеС^СМе. Молекула
НС^СН действительно линейна, а МеС^СМе имеет линейный СССС-скелет.
«псевдоатом». Удобно рассматривать MeCN как имеющий молекулярный
скелет CCN, это позволяет легко описать геометрию молекулы: ацетонитрил —
линейная молекула.
Аналогично можно провести параллель между диметилсульфидом Me2S и
H2S (рис. 5.7,6). Me2S сохраняет угловую геометрию при атоме серы, только
угол между связями меняется от 92° в H2S до 99° в Me2S. Соединение MeSH
имеет аналогичную структуру, но ZC—S—Н = 96,5°. В родственных
соединениях Н20, МеОН и Ме20 центр угловой структуры находится на атоме
кислорода. (Обратите внимание на данную формулировку: речь идет о
конфигурации относительно центрального кислородного атома. Описание ничего не
говорит о геометрии углеродных центров.)
Алкины RC=CR (где R — органическая группа) имеют Линейный
четырехатомный скелет; в него включены первые атомы каждой R-группы. На
рис. 5.8 представлены структуры НС=СН и МеС^СМе. Замена водорода на
метильные группы не влияет на общую линейную геометрию центральной
части молекулы.
Следует запомнить, что при описании МеС=СМе как линейной
молекулы имеется ввиду только геометрия четырех центральных атомов углерода. С
другой стороны, молекула НС=СН действительно линейна.
Примеры этой главы подтверждают важное утверждение — геометрия
больших молекул может быть адекватно описана в рамках локальной
геометрии отдельных атомных центров.
ГЕОМЕТРИЯ МОЛЕКУЛ,
СОДЕРЖАЩИХ р-ЭЛЕМЕНТЫ ВТОРОГО ПЕРИОДА
1-2
S-
элементы
3-12
6-
элементы
13-18
Р-
элементы
В этом разделе рассмотрены молекулы, содержащие в
качестве центрального атома р-элементы второго
периода (см. периодическую систему на с. 31).
Рассмотрение ограничено молекулами типа XYn с одним
центральным атомом и одинаковыми заместителями. На
рис. 5.9 показаны пять типов геометрии таких
молекул, центральным атомом которых может быть бор,
5. Многоатомные молекулы: форма и химическая связь
о
Линейная
Угловая
Плоскотреугольная
Три тонально-пирамидальная
Тетраэдрическая
Рис. 5.9. Пять типов геометрии молекул XYn с р-элементом второго периода в
качестве центрального атома.
углерод, азот или кислород. В табл. 5.2 перечислены некоторые реальные
молекулы, имеющие геометрию одного из этих типов. Фтор, являющийся
элементом второго периода, редко является центральным атомом, чаще он
занимает концевую позицию, например в BF3, CF4, NF3 и OF2.
s-Элементы расположены в группах 1 и 2 периодической системы
элементов, of-элементы - в группах 3-12, р-элементы - в группах
3-18.
Бор (группа 13)
Молекулы и ионы, в которых бор проявляет координационное число три,
имеют форму равностороннего треугольника, например, BF3, BCl3, BMe3 и
В033~. Тетраэдрически координированный бор существует в анионах типа
ВН4" и BF4".
Таблица 5.2. Примеры молекул и ионов общей формулы XY„, XY™+ или XY^_, где
X — р-элемент второго периода (от бора до фтора)
Форма молекулы
(см. табл. 5.1 и рис. 5.9)
Примеры (Me — метил CHj)
Линейная С02, N02+, NCO", N3" = NNN"
Угловая Н20, OF2, HOF, N02, N02", NH2"
Плоскотреугольная BF3, BC13, BMe3, B033", C032", N03"
Тригонально-пирамидальная NH3, NF3, NMe3, H30+
Тетраэдрическая ВН4", BF4~, CH4, CMe4, CF4, NH4+, NMe4+
5.4. Геометрия молекул, содержащих р-элементы второго периода 209
Углерод (группа 14)
Для углеродсодержащих молекул чаще всего наблюдаются три типа
геометрии - линейная, плоскотреугольная и тетраэдрическая. Трехатомные
частицы с центральным атомом углерода (С02, NCO~, HCN) линейны.
Примерами молекул, имеющих плоскотреугольную форму, могут быть Н2С=0,
С12С=0, НС02Н (5.14) и С032-.
Тетраэдрически координированный углерод встречается очень часто,
например в насыщенных углеводородах (алканах) типа СН4. Изоэлектронные
молекулы СН4 и ВН4~ имеют и одинаковую геометрию (рис. 5.10).
®
[bhj-
©
®^А^®
CHd
®
[NHJ+
NH,
[НэО]+
Рис. 5.10. Частицы ВН4~, СН4, NH4+ изоэлектронны и изоструктурны. NH3 и
Н30+ также изоэлектронны и изоструктурны.
О
/
\
Н
ОН
5.14
N ...
C'""R
5.15
Азот (группа 15)
Геометрия окружения атома азота может быть линейной (рис. 5.2), угловой
(рис. 5.4), плоскотреугольной, тригонально-пирамидальной и тетраэдриче-
ской.
210
5. Многоатомные молекулы: форма и химическая связь
Нейтральные молекулы с центральным атомом азота, имеющим
координационное число 3, обычно имеют форму тригональной пирамиды,
например NH3 (рис. 5.10) и NF3. В эту же группу могут быть включены
органические амины общей формулы RNH2, R2NH или R3N (5.15), где R -
органический радикал.
Ион аммония NH4+ имеет форму тетраэдра и изоэлектронен ВН4~ и СН4
(рис. 5.10).
Атом азота в нитрат-ионе N03~ имеет плоскотреугольную геометрию
окружения. По принципу изоэлектронности такую же геометрию должны
иметь и ионы С032- и В033~.
Кислород (группа 16)
Простые молекулы с центральным атомом кислорода имеют угловое
строение (рис. 5.3). В соединениях с координационным числом 3 геометрия
окружения кислорода - тригональная пирамида. Простейшим примером
является ион гидроксония Н30+, изоэлектронный молекуле аммиака (рис. 5.Ю).
Обзор р-элементов второго периода
• Соединения ^-элементов второго периода обычно имеют одну из
геометрических форм, приведенных на рис. 5.9.
• Форма молекул, содержащих бор, углерод и азот, более разнообразна, по
сравнению с кислород- и фторсодержащими молекулами.
• Изменение заряда молекулы или иона часто приводит и к изменению
геометрии; ср., например, N02 и N02~.
р-ЭЛЕМЕНТЫ ТРЕТЬЕГО, ЧЕТВЕРТОГО И ПЯТОГО ПЕРИОДОВ
Элементы, которые обсуждаются в этом разделе, перечислены на рис. 5.И,
а формы соответствующих молекул показаны на рис. 5.9 и
рис. 5.12. В табл. 5.3 приводятся примеры для каждого типа
структуры.
Химия р-элементов:
см. гл. 13
Группа 13
Группа 14 Группа 15
Группа 16 Группа 17
3-й период
4-й период
5-й период
6-й период
А1
Алюминий
Ga
Галлий
In
Индий
Т1
Таллий
Si
Кремний
Ge
Германий
Sn
Олово
РЬ
Свинец
Р
Фосфор
As
Мышьяк
Sb
Сурьма
Bi
Висмут
S
Сера
Se
Селен
Те
Теллур
Ро
Полоний
С1 1
Хлор
Вг
Бром
Иод
At
Астат
Рис. 5.11. р-Элементы третьего, четвертого, пятого и шестого периодов (исключая
инертные газы).
5.5. р-Элементы третьего, четвертого и пятого периодов
211
Таблица 5.3. Примеры молекул XYW, где X - ^-элемент
(см. также рис. 5.9 и рис. 5.12)
Форма
Примеры
Линейная
Угловая
Плоскотреугольная
Тригонально-пирамидальная
Т-образная
Тетраэдрическая
Дисфеноидная
(искаженно-тетраэдрическая)
Плоскоквадратная
Тригонально-бипирамидальная
Квадратно-пирамидальная
Октаэдрическая
1С12", 13- = I-I-I-
SnCl2, H2S, SF2, S02, H2Se, SeCl2, TeCl2
A1C13, AlBr3, S03
PF3, PCI3, AsF3, SbCl3, PMe3, AsMe3, P033",
S032", Se032", CI03-
C1F3, BrF3
SiH4, GeH4, SiF4, SiCl4, GeF4, SnCl4, PbCl4,
AIH4", GaBr4", InCl4~, T1I4", PBr4+, P043",
so42-, cio4-
SF4, РВг4", C1F4+, IF4+
C1F4", BrF4", IC14"
PC15, AsF5, SbF5, SOF4, SnCl5"
CIF5, BrF5, IF5, SF5", TeF5", SbF52"
SF6, SeF6, Te(OH)6, IF6+, PF6", AsCl6", SbF6",
SnF62-,AIF63"
O
Т-образная
О
¥
A
Дисфеноидная
(искаженный тетраэдр)
Плоскоквадратная
Тригонально-
бипирамидальная
Квадратно-пирамидальная
О
Октаэдрическая
Рис. 5.12. Формы молекул (в дополнение к приведенным на рис. 5.9) XYn, где X
р-элемент третьего, четвертого, пятого или шестого периода.
212
5. Многоатомные молекулы: форма и химическая связь
Группа 13
^~ При высоких температурах тригалогениды А1С13, А1Вг3 и А113 существуют в
Зависимость строения виде изолированных молекул, имеющих плоскотреугольную
хлорида алюминия структуру. Однако для Al, Ga и In более обычна тетраэдриче-
от температуры: екая координация, которая реализуется в анионах типа А1Н4",
см. разд. 13.4 А1С14", GaBr4" и 1пС14".
^ги два типа геометрии координационного окружения
наблюдаются и для соединений бора, но более тяжелые элементы группы 13
имеют тенденцию к увеличению координационного числа. Например, ионы
A1F63~ (5.16) и Ga(H20)63+ имеют октаэдрическое строение.
"|з-
F
5.16
С\
CI-
-Sn.
kCl
rCl
CI
5.17
Группа 14
В газовой фазе SnCl2 имеет угловую геометрию, однако другие соединения
двухвалентного олова имеют более сложное строение. Многие соединения
элементов группы 14 по своему составу напоминают соединения углерода, но
при проведении таких аналогий следует соблюдать определенную осторож-
^- ность: по составу Si02 действительно напоминает С02, но их
Диоксиды углерода и строение принципиально различно.
кремния: см. разд. 13.6 Силан SiH4 так же, как и СН4, имеет тетраэдрическое
строение. Аналогично, изоструктурными и изоэлектронными
являются пары SiH4 и А1Н4~; СН4 и ВН4~; GeH4 и GaH4~.
Молекулы SiF4, GeF4, SnCl4 и РЬС14 также имеют тетраэдрическое строение.
Для элементов группы 14 координационные числа более 4 реализуются в
анионах SnCl5~ (5.17) и Me2SnCl3~, которые имеют тригонально-бипирами-
дальную геометрию, и в октаэдрическом двухзарядном анионе SnF62".
Группа 15
Разнообразие структур оказывается еще более богатым при рассмотрении
элементов пятнадцатой группы.
Тригональная пирамида — самая обычная геометрия молекул,
содержащих фосфор, мышьяк и сурьму, например PF3, AsF3, SbCl3, PMe3 (5.18) и
Me
V*""'Me
Me
Br
>Br
rBr
Br
5.18
5.19
5.5. р-Элементы третьего, четвертого и пятого периодов
213
Рис. 5.13. В SbF52~ атомы фтора
образуют квадратную пирамиду
вокруг центрального атома сурьмы.
/Л
F # F F
F
2-
Квадратная
пирамида
Р033~. Хотя наше рассмотрение ограничивается молекулами с одним
центральным атомом — элементом группы 15, следует помнить, что в молекуле
Р4 каждый атом фосфора находится в вершине тригональной пирамиды
(рис. 3.2).
Координационное число 4 чаще всего связано для элементов группы 15 с
тетраэдрической координацией, хотя иногда наблюдается и дисфеноидная
геометрия (рис. 5.12). Ионы РС14+, РВг4+, Р043" имеют форму тетраэдра.
Однако изменение знака заряда с РВг4+ на РВг4~ приводит к изменению
геометрии на дисфеноидную (5.19).
Число молекул и ионов с КЧ 5 центрального атома довольно велико. В
газовой фазе пентагалогениды PF5, AsF5 и SbCl5 имеют форму тригональной
бипирамиды, но изменение заряда снова приводит к изменению геометрии -
в твердой фазе ион SbF52~ имеет структуру квадратной пирамиды (рис. 5.13).
Октаэдрическая геометрия также обычна для элементов группы 15,
например, PF6~, AsF6~ и SbF6~.
Группа 16
Для кислорода характерна только угловая и тригонально-пирамидальная
форма молекул и ионов, но для его более тяжелых аналогов геометрия может
быть более разнообразной.
Примерами угловых молекул могут быть H2S, SF2, S02, SeCl2 и TeCl2,
плоскотреугольную форму имеет S03 (5.20), тогда как SOCl2, SeOCl2, S032~ и
Те032~ имеют структуру тригональной пирамиды. И опять изменение заряда
приводит к изменению геометрии, как это происходит при переходе от S03 к
S032~. Координационное число 4 реализуется для серы, селена и теллура в
виде тетраэдра и дисфеноида. Ион S042~ - тетраэдр, тогда как SF4, SeF4 и
Me2TeI2 имеют геометрию дисфеноида. Для КЧ 5 центрального атома
возможны геометрии тригональной бипирамиды (SOF4, 5.21) и квадратной
пирамиды (SbF5~). В число октаэдрических молекул входят SF6, SeF6 (5.22) и
TeF6.
О
4? ^
5.20
О:
F
5.21
F^^ I ^^F
F
5.22
5. Многоатомные молекулы: форма и химическая связь
Группа 17
В отличие от фтора более тяжелые галогены образуют молекулы с
разнообразной геометрией. Иод является центральным атомом в линейном анионе
13~ и угловом катионе 13+. Для центральных атомов с КЧ 3 известны
молекулы Т-образной и тригонально-пирамидальной формы (рис. 5.12). C1F3
является примером Т-образной молекулы, хотя два угла между связями F-C1-F,
которые должны быть равны 90° в идеальном случае, на самом деле чуть
меньше (87°). Анион СЮ3~ имеет форму тригональной пирамиды.
Элементы группы 17 в своих соединениях реализуют КЧ 4 тремя
способами. Тетраэдрическое строение имеют, например, ионы С104~ и Ю4~, тогда
как катионы типа C1F4+ и IF4+ имеют форму дисфеноида, т. е. искаженного
тетраэдра (C1F4+ изоэлектронен SF4). При переходе от C1F4+ к C1F4~
происходит трансформация геометрии от дисфеноидной к плоскоквадратной.
Шестиатомные молекулы с центральным атомом галогена (КЧ 5) имеют
форму либо тригональной бипирамиды (C102F3), либо квадратной пирамиды
(C1F5 и IF5). В то же время катион IF6+ является примером октаэдрической
частицы с КЧ 6 атома иода.
Общие координационные свойства р-элементов
3-6-го периодов
• р-Элементы 3-6 периодов проявляют в своих соединениях большее
разнообразие геометрии координационного окружения по сравнению с их
аналогами из 2-го периода.
• При переходе от элементов группы 13 к элементам группы 17 возможная
геометрия молекул становится более разнообразной.
• Изменение заряда частицы при сохранении ее элементного состава, как
правило, приводит к изменению геометрии.
МОДЕЛЬ ОТТАЛКИВАНИЯ ЭЛЕКТРОННЫХ ПАР
ВАЛЕНТНОЙ ОБОЛОЧКИ (ТЕОРИЯ ГИЛЛЕСПИ)
В предыдущем разделе была рассмотрена геометрия молекул и молекулярных
ионов без объяснения приводимых фактов. Отмечены некоторые
закономерности в изменении формы координационного окружения при увеличении
номера группы или периода. В этой главе мы рассмотрим метод предсказания
и объяснения строения молекул и ионов.
Модель отталкивания электронных пар валентной оболочки (Valence-Shell
Electron-Pair Repulsion, VSEPR) была первоначально предложена Н.В. Сид-
жвиком и Дж. Пауэллом в 1940 г. и развита в работах Р. Найхолма и Р. Гиллес-
пи в 1957—1961 гг. Теория Гиллеспи основывается на рассмотрении
взаимодействия пар валентных электронов и исходит из предположения, согласно
которому только электроны валентной оболочки определяют геометрию
молекулы или иона.
Минимизация межэлектронного отталкивания
По теории Гиллеспи, для достижения стабильности пары валентных
электронов должны находиться на максимальном расстоянии друг от друга, что
соответствует минимуму межэлектронного отталкивания.
5.6. Модель отталкивания электронных пар валентной оболочки 215
Предположим, что в молекуле XYn все атомы Y связаны с центральным
атомом одинарными связями и в образовании этих связей принимают участие
все валентные электроны. Таким образом, каждой паре электронов
соответствует одна связь X—Y. Относительное положение п электронных пар,
которому соответствует минимум энергии системы, может быть определено
следующим образом: каждая пара электронов представляется в виде точечного
отрицательного заряда, который помещается на поверхность некоторой сферы.
Для п = 2 отталкивание этих электронных пар будет минимальным, если
они расположены на противоположных сторонах сферы (рис. 5.14,д). Такой
ситуации соответствует линейная геометрия молекул типа XY2.
Для /2 = 3 суммарная энергия электростатического отталкивания для трех
точечных зарядов на поверхности сферы будет минимальной, если эти
заряды расположены в вершинах равностороннего треугольника (рис. 5.14,6). Это
соответствует плоскотреугольной геометрии молекул XY3.
Аналогично, для п = 4, 5 и 6 оптимальным с точки зрения минимума
энергии расположением точечных зарядов на поверхности сферы является распо-
^ ложение по вершинам тетраэдра, тригональной бипирамиды и октаэдра со-
Энергетические раз- ответственно. На рис. 5.15 показаны эти полиэдры (а) и соот-
личия этих структур: ветствующие тетрагональная, тригонально-бипирамидальная и
см. разд. 5.12 октаэдрическая формы молекул XY4, XY5 и XY6 (б).
Хотя в табл. 5.1 для п = 2, 3 и 4 представлено несколько
возможных типов геометрии молекул, в каждом случае лишь одна из них
соответствует минимуму энергии.
а б
Отталкивание Отталкивание
Отталкивание
~ Отталкивание
Отталкивание
Y
Рис. 5.14. Пары валентных электронов могут рассматриваться как точечные заряды.
Минимуму энергии такой системы будет соответствовать положение, когда эти
заряды максимально удалены друг от друга, а — два точечных отрицательных заряда
занимают положения на противоположных сторонах сферы. Таким образом, молекула XY2
должна быть линейной при условии, что все валентные электроны использованы на
образование связей; б— взаимное отталкивание трех точечных зарядов приводит к их
расположению в вершинах равностороннего треугольника. Это соответствует
плоскотреугольной геометрии молекул XY3, если все валентные электроны использованы на
образование связей (ср. рис. 5.16).
216 5. Многоатомные молекулы: форма и химическая связь
Рис. 5.15. Для четырех, пяти и шести одноименных точечных зарядов,
расположенных на поверхности сферы, суммарная энергия отталкивания будет минимальной,
если эти заряды расположены по вершинам тетраэдра, тригональной бипирамиды и
октаэдра соответственно. Для молекул типа ХУЯ, в которых все валентные электроны
принимают участие в образовании связей, это отвечает образованию тетраэдрических,
тригонально-бипирамидальных и октаэдрических молекул типа XY4, XY5 и XY6.
Неподеленные электронные пары
Не все валентные электроны принимают участие в образовании связей.
Структура Льюиса для молекулы воды (5.23) показывает, что атому
кислорода принадлежат две неподеленные пары валентных электронов. В молекуле
СН4 (5.24) все валентные электроны участвуют в образовании связей.
н
н : ° : н : с : н
н н
5.23 5.24
В межэлектронном отталкивании участвуют как связывающие, так и
неподеленные электронные пары валентной оболочки центрального атома,
поэтому наличие одной или нескольких неподеленных пар также влияет на геометрию
молекул. Слева на рис. 5.16,д и б приведены структуры Льюиса для ВН3 и
NH3. Атом бора использует все три свои валентных электрона на образова-
5.6. Модель отталкивания электронных пар валентной оболочки
217
а
н : в : н
• •
н
Л\\
\Л
"I
Рис. 5.16. Применение модели Гиллеспи для предсказания формы молекул ВН3 и
NH3. Обратите внимание на роль неподеленной пары электронов в молекуле аммиака.
ние одинарных связей В—Н, тогда как у атома азота, имеющего пять
валентных электронов, остается одна неподеленная пара. В молекуле ВН3 атом
бора окружен тремя парами электронов, что в соответствии с моделью
Гиллеспи соответствует плоскотреугольной геометрии (рис. 5.16,я). В молекуле
аммиака в валентной оболочке атома азота присутствуют четыре электронные
пары и теория предсказывает, что эта молекула должна иметь форму
тетраэдра, одна из вершин которого занята неподеленной парой электронов (рис.
5.16,6). Таким образом, форма молекулы NH3 может быть описана как тетра-
эдрическая, хотя на самом деле атом азота и три атома водорода образуют
тригональную пирамиду (рис. 5.9).
Относительные величины межэлектронного отталкивания
Для молекулы ХУЛ, содержащей только связывающие пары электронов,
можно предположить, что все они отталкиваются друг от друга одинаковым
образом, если все эти связи эквивалентны.
• Кратные связи характеризуются большей плотностью электронов между
ядрами по сравнению с одинарными, поэтому и межэлектронное
отталкивание с участием кратных связей сильнее, чем отталкивание между
простыми связями (рис. 5.17).
• Если в молекуле несколько неподеленных электронных пар, то
отталкивание электронных пар уменьшается в ряду:
неподеленная —
неподеленная»
)>(
неподеленная
связывающая
/ связывающая -
1 связывающая
Zy>Zp>Za
Рис. 5.17. Кратные связи характеризуются большей плотностью электронов между
ядрами по сравнению с одинарными, поэтому и межэлектронное отталкивание с
участием кратных связей сильнее, чем отталкивание между простыми связями.
218 5. Многоатомные молекулы: форма и химическая связь
V \ V \ V \
н н н
0=109.5° 0=107.5° 0=104.5°
Рис. 5.18. а — для тетраэрического расположения четырех пар электронов
наблюдается шесть парных отталкивающих взаимодействий между ними; б— геометрия
молекулы метана СН4 определяется шестью эквивалентными взаимодействиями типа
«связывающая пара — связывающая пара»; как следствие, все четыре валентных угла
Н—С—Н равны. Геометрия молекулы аммиака NH3 определяется тремя
взаимодействиями типа «связывающая пара - связывающая пара» и тремя типа «неподеленная
пара — связывающая пара», поэтому валентный угол Н—N—Н меньше угла Н—С—Н в
молекуле метана.
В молекуле воды существует одно взаимодействие «неподеленная пара —
неподеленная пара», одно — типа «связывающая пара — связывающая пара» и четыре - типа
«неподеленная пара — связывающая пара», и валентный угол Н—О—Н меньше, чем
угол Н—N—Н в молекуле аммиака, и тем более меньше, чем угол Н—С—Н в
молекуле метана.
Эта разница в величинах взаимного отталкивания влияет на геометрию
молекул, что может быть проиллюстрировано на примере молекул СН4, NH3 и
Н20. Общее число отталкивающих межэлектронных взаимодействий в этих
молекулах одинаково и равно шести (рис. 5.18,а). В случае СН4 это
отталкивание только между связывающими электронными парами, тогда как геоме-
Модель отталкивания электронных пар, предложенная Гиллеспи,
используется для предсказания или объяснения геометрии молекул.
Эта модель рассматривает только отталкивание электронных пар
валентной оболочки центрального атома молекулы или иона.
• Пары электронов располагаются в пространстве таким образом,
чтобы минимизировать энергию взаимного отталкивания.
• Величина межэлектронного отталкивания уменьшается в ряду:
«неподеленная пара - неподеленная пара» > «неподеленная пара -
связывающая пара» > «связывающая пара - связывающая пара».
• Для молекул с различным числом п электронных пар в валентной
оболочке центрального атома характерны следующие типы
геометрии:
п = 2 линейная
п = 3 плоскотреугольная
п = 4 тетраэдрическая
п = 5 тригонально-бипирамидальная
п = 6 октаэдрическая
5.6. Модель отталкивания электронных пар валентной оболочки 219
трия молекул аммиака и воды определяется также взаимодействиями типа
«неподеленная пара - связывающая пара» и «неподеленная пара - неподе-
ленная пара». Увеличение отталкивания между электронными парами
приводит к уменьшению валентного угла в этих молекулах (рис. 5.18,6).
Применение теории Гиллеспи для молекул или ионов,
содержащих р-элементы
На основании модели отталкивания электронных пар можно предсказать
геометрию молекулы по следующему алгоритму:
1. Нарисуйте структуру Льюиса молекулы или иона, геометрию которого
необходимо предсказать, и определите число связывающих и неподеленных
пар электронов в валентной оболочке центрального атома.
2. «Базовая» геометрия (табл. 5.4) определяется числом «точечных»
отрицательных зарядов, которые соответствуют простым (одинарным), кратным
связям или неподеленным электронным парам.
3. Если все связи в молекуле — простые, то степень отклонения реальной
структуры (см. табл. 5.1) от идеальной определяется относительной
энергией возможных парных взаимодействий. Отталкивание максимально
между двумя неподеленными парами, несколько меньше при взаимодействии
связывающей и неподеленной электронной пары и минимально при
взаимодействии двух связывающих пар электронов.
4. Если в молекуле или ионе есть кратные связи, необходимо принять во
внимание, что энергия отталкивания возрастает с увеличением кратности
связи.
5. В тригональной бипирамиде неподеленные пары занимают
экваториальные позиции, а не аксиальные. Кратные связи также располагаются в
экваториальной плоскости.
6. В октаэдре две неподеленные пары занимают транс-положения, т. е.
оказываются максимально удаленными друг от друга .
Пример 1. H2S
Сера - элемент группы 16 с шестью валентными электронами, атом
водорода имеет один валентный электрон. Молекула сероводорода имеет
приведенную ниже структуру Льюиса:
н : s :
н
Таблица 5.4. Геометрия распределения точечных зарядов (неподеленных или
связывающих электронных пар). См. также табл. 5.1.
Число точечных отрицательных зарядов Геометрия
2 Линейная
3 Плоскотреугольная
4 Тетраэдрическая
5 Тригонально-бипирамидальная
6 Октаэдрическая
220
5. Многоатомные молекулы: форма и химическая связь
Эту молекулу можно представить как совокупность четырех точечных
отрицательных зарядов (две неподеленные и две связывающие пары
электронов), расположенных вокруг положительно заряженного центра.
«Базовая» геометрия молекулы — тетраэдр; следовательно, молекула
сероводорода - угловая (5.25). Угол Н—S—Н должен быть меньше идеального те-
траэдрического угла, равного 109,5°. Действительно, по экспериментальным
данным этот угол равен 92,1°.
или
г
5.25
Пример 2. NH4+
Азот расположен в группе 15 и имеет пять валентных электронов. Если
приписать положительный заряд атому азота, то N+ будет иметь четыре
валентных электрона. Атом водорода имеет один валентный электрон. Иону NH4+
соответствует приводимая ниже структура Льюиса:
Н
• •
н : n : н
н
В ионе NH4+ можно выделить четыре точечных отрицательных заряда
(четыре пары связывающих электронов), неподеленных пар нет и,
следовательно, структура иона аммония (5.26) — неискаженный тетраэдр (см.
рис. 5.10).
N ..
V"'"H
5.26
Пример 3. С02
Углерод имеет четыре, а кислород — шесть валентных электронов. Структура
Льюиса молекулы углекислого газа показана ниже.
О
О
Атом углерода принимает участие в образовании двух двойных связей,
которые соответствуют двум точечным отрицательным зарядам; таким образом,
5.6. Модель отталкивания электронных пар валентной оболочки 221
молекула С02 должна быть линейной (5.27). Сравните этот результат с рис.
5.2. (Почему неподеленные пары электронов атомов кислорода не
оказывают влияния на геометрию молекулы?)
5.27
Пример 4. S02
И сера, и кислород имеют по шесть валентных электронов. Структура
Льюиса приведена ниже.
•• •
о '
: s
о .
• • •
Можно выделить три точечных отрицательных заряда, два из которых
соответствуют двойным связям сера-кислород, а один - неподеленной
электронной паре атома серы. Таким образом, структура молекулы диоксида
серы, является производной от треугольной геометрии (5.28). Сама молекула -
угловая с углом О—S—О 119°, что лишь немногим меньше идеального (120°).
Это отклонение от идеального случая объясняется неэквивалентностью
взаимодействий типа «неподеленная пара - двойная связь» и «двойная связь -
двойная связь».
О О
. / /
S или S
\ ч
О О
5.28
Пример 5. SF4
У серы шесть валентных электронов, а у фтора - семь. Структура Льюиса SF4
приведена ниже.
: f : s : f :
• • • •
• • • •
*F F*
• • • •
• • • •
Можно выделить пять точечных зарядов — четыре связывающие пары и
одну неподеленную пару электронов серы. «Базовая» структура - тригональ-
ная бипирамида, причем неподеленная пара для минимизации энергии
отталкивания занимает одну из экваториальных позиций. Следовательно,
молекула SF4 (5.29) имеет форму дисфеноида (искаженного тетраэдра). Атомы
фтора оказываются неэквивалентными (два экваториальных F и два
аксиальных Fax). (Влияют ли неподеленные электронные пары атомов фтора на
геометрию молекулы SF4?)
222
5. Многоатомные молекулы: форма и химическая связь
или
5.29
Угол F —S—F больше угла Fax—S—F . Принимая во внимание
относительную величину отталкивания «неподеленная пара - неподеленная пара» >
«неподеленная пара - связывающая пара» > «связывающая пара -
связывающая пара», можно предположить, что угол Fax—S—Fax меньше или равен
-F больше 90°, но меньше 120°. Эксперимент показыва-
180°, а угол Feq-S-
ет, что угол F — S—F
равен 173°, а угол F —S—F равен 102°
Пример 6.1С14
Иод и хлор относятся к группе 17 и имеют по семь валентных электронов.
Отрицательный заряд может быть отнесен к центральному атому иода. 1~ имеет
восемь валентных электронов.
Структура Льюиса 1С14~ приведена ниже.
с\ #..# а
• • • 4
С1 *••' С1
Центральный атом иода окружен шестью точечными зарядами (две непо-
деленные пары и четыре связывающие пары электронов).
Геометрия иона 1С14~ (5.30) является производной от октаэдра. Неподе-
ленные пары располагаются в противоположных вершинах (находятся в
транс-положении друг относительно друга), чтобы минимизировать взаимное
отталкивание. Таким образом, ион 1С14~ имеет плоскоквадратное строение.
ci ci
С1^
с\^
_
• 1 •
^!С
*|*
^С1
*^С1
_
или
С1 С1
5.30
НЕДОСТАТКИ ТЕОРИИ ГИЛЛЕСПИ
Главным достоинством теории отталкивания электронных пар валентной
оболочки является ее простота. Эта удачная модель получила очень широкое
распространение. Однако существуют молекулы, геометрия которых не
может быть надежно предсказана на основании теории Гиллеспи. В этом
разделе рассмотрены некоторые такие случаи.
5.7. Недостатки теории Гиллеспи
223
Трифторид хлора
Структура Льюиса для C1F3 приведена ниже:
С1
Атом хлора окружен пятью точечными отрицательными зарядами (две
неподеленные и три связывающие электронные пары). Следовательно,
структура C1F3 является производной от тригональной бипирамиды. Вопрос
заключается лишь в том, какие позиции, экваториальные или аксиальные,
будут занимать неподеленные пары? Ниже приведены возможные варианты, а
проблема заключается в оценке преимуществ каждой из них.
■С1С . F C1J*
..»*
F
С\.
•I* F F
5.31 5.32 5.33
1. Неподеленные пары занимают аксиальные положения, что
минимизирует энергию отталкивания типа «неподеленная пара - неподеленная пара»
(5.31), но в то же время возрастает число и суммарная энергия
взаимодействий типа «неподеленная пара - связывающая пара».
2. Неподеленные пары могут быть расположены в экваториальной
плоскости (5.32); при такой конфигурации они оказываются под углом 120°. При
этом остаются еще четыре взаимодействия типа «неподеленная пара —
связывающая пара» между электронными парами, расположенными под
углом 90°, и еще два взаимодействия того же типа, но между парами,
находящимися под углом 120°. Возникают еще два 90-градусных
взаимодействия типа «связывающая пара — связывающая пара».
3. Существует еще один вариант, когда неподеленные пары располагаются
под углом 90° (5.33). Эта конфигурация отвечает наивысшей энергии
взаимодействия между неподеленными парами и может быть исключена из
рассмотрения.
Обе структуры, 5.31 и 5.32, кажутся вполне разумными, и сделать
обоснованный выбор в пользу одной из них на основании чисто качественных
рассуждений невозможно. Экспериментальные измерения показывают, что
молекула C1F3 имеет Т-образную геометрию с углом F^—CI—F 87°. Эти
результаты соответствуют структуре 5.32, в которой обе неподеленные пары
находятся в экваториальной плоскости тригональной бипирамиды.
Уменьшение угла Fax—CI—Feq по сравнению с идеальным (90°) может быть
объяснено взаимодействием типа «неподеленная пара - связывающая пара».
224
5. Многоатомные молекулы: форма и химическая связь
Анионы SeCl|~ и ТеС1§~
Se и Те имеют по шесть валентных электронов. В ионах SeCl|~ и ТеС1|~ в
валентной оболочке центрального атома находится 14 электронов (6 от
центрального атома, по одному от каждого атома хлора и еще два возникают за
счет заряда). Теория Гиллеспи предсказывает, что геометрия этих ионов будет
производной от полиэдра с семью вершинами, одна из которых будет занята
неподеленной парой. Однако экспериментальные измерения показывают,
что реальная форма этих частиц - правильный октаэдр (5.34), т. е. наличие
неподеленной пары не влияет на геометрию этих ионов, как того следовало бы
ожидать из теории Гиллеспи. Такая неподеленная пара называется
стереохимически неактивной.
С1^^ I ^^С1
С1
5.34
Предсказать строение ионов SeCl62~ и ТеС162~ невозможно, можно только
объяснить их геометрию, вводя понятие стереохимически неактивной пары
электронов. Стереохимически неактивные электронные пары чаще всего
наблюдаются для молекул сравнительно тяжелых элементов. Склонность s-
электронов валентной оболочки играть роль несвязывающих электронов
называется эффектом инертной пары.
Если присутствие неподеленной электронной пары влияет на
геометрию молекулы или иона, то такая пара называется стереохимически
активной.
Если присутствие неподеленной электронной пары не влияет на
геометрию молекулы или иона, то такая пара называется
стереохимически неактивной.
Склонность s-электронов валентной оболочки играть роль
несвязывающих электронов называется эффектом инертной пары.
НЯ МОДЕЛЬ КЕПЕРТА
1-2 3-12 13-18 Модель Гиллеспи обычно используется для предсказания и
объяснения геометрии молекул или ионов, содержащих в
качестве центрального атома р-элементы. Кеперт развил
эту модель в применении к ^-элементам. Рассмотрим
молекулы с общей формулой МЬЛ, где М - d-элемент, a L -
лиганд, т. е. группа, связанная с центральным атомом
металла.
S-
элементы
d-
элементы
Р-
элементы
5.9. Применение модели Кеперта
225
w. В модели Кеперта атом металла лежит в центре сферы, а лиганды могут
Лиганды: см. разд. 16.3 свободно двигаться по поверхности этой сферы.
Считается, что лиганды отталкиваются друг от друга, как и
точечные отрицательные заряды в модели Гиллеспи. Модель Кеперта
предсказывает взаимное расположение лигандов, как и модель Гиллеспи
предсказывает геометрию молекул XYW, однако заряд иона (МЦ/"+ или МЦ/"-) не
принимается во внимание. Полиэдры, перечисленные в табл.5.4, могут быть
применены к молекулам, в которых центральным атомом является как/?-, так
и d-элемент.
Предсказываемая по модели Кеперта форма молекулы не зависит от
электронной конфигурации основного состояния атома металла. Главное
отличие этой модели от модели Гиллеспи заключается в том, что полностью
игнорируется наличие неподеленных электронных пар в валентной оболочке
центрального атома.
Модель Кеперта предсказывает геометрию соединений d-элементов
типа М1_л, учитывая только взаимное отталкивание лигандов. Модель
игнорирует наличие неподеленных электронных пар.
ЕЕЯ ПРИМЕНЕНИЕ МОДЕЛИ КЕПЕРТА
Линейная геометрия
В анионе Au(CN)^~ две цианидные группы связаны с центральным атомом
золота. Их взаимное отталкивание минимально, если они расположены под
углом 180°. Таким образом, ион должен быть линейным (5.35), что
подтверждается экспериментальными данными.
Анионы HgCl2~ и АиС12~ имеют ту же структуру, что и Au(CN)^~.
|NC Au CNI ~
5.35
Плоскотреугольная геометрия
Если центральный атом окружен тремя лигандами, как в анионе Cu(CN)|~,
то для минимизации энергии отталкивания, они должны располагаться по
вершинам равностороннего треугольника. Это соответствует реально
наблюдаемой структуре, приведенной на рис.5.19.
Тетраэдрическая геометрия
Тетраэдрические комплексные соединения d-элементов являются
достаточно распространенными. Примером может служить ион СоС142~, в котором
четыре атома хлора окружают центральный атом кобальта. При тетраэдриче-
ской геометрии отталкивание между атомами хлора минимально (5.36).
Аналогично была предсказана и подтверждена экспериментально тетраэдриче-
226
5. Многоатомные молекулы: форма и химическая связь
Рис. 5.19. Плоскотреугольная структура иона
одновалентной меди Cu(CN)32~.
6
екая структура следующих ионов и нейтральных молекул: Мп04~, СЮ42~,
NiCl42", VCl4-, ZrCl4 и Os04.
Cl
CI
.Co
V"
Cl
"Cl
2-
N
С
NC CdC
*CN
rCN
5.36
С
N
5.37
ОС-
о
с
Fe.
*СО
rCO
С
О
5.38
Структура с КЧ 5
Для комплекса [Cd(CN)5]3~ с центральным ионом Cd2+ устойчивой будет
тригонально-бипирамидальная геометрия, которая отвечает минимуму
энергии отталкивания между цианидными группами (5.37). Аналогичное
строение может быть предсказано и для карбонила железа [Fe(CO)5] (5.38).
В теории Гиллеспи неподеленные пары электронов занимают в триго-
нальной бипирамиде преимущественно экваториальные позиции, так как
энергия взаимодействий с их участием сравнительно велика. Аналогично и в
теории Кеперта наиболее объемистый лиганд стремится занять именно
экваториальную позицию.
►- Структура 5.39 соответствует предсказанной геометрии комплекса двух-
Ме-метил СН3 валентного никеля [NiCl2(PMe2Ph)3], где PMe2Ph - объеми-
Ph - фенил С6Н5 стые органические фосфины, которые занимают три
экваториальные позиции.
Предсказанная геометрия соединения [NiBr3(PMe2Ph)2]
представлена структурой 5.40, а на рис. 5.20 проводятся результаты,
полученные методом рентгеновской дифракции. Отталкивание между пятью лиган-
дами минимизируется, если две диметилфенилфосфиновые группы
максимально удалены друг от друга. Бромид-ионы также являются крупными ли-
гандами, и их отталкивание минимально, если они располагаются под углом
120°, а не 90°. (См. упражнение 5.7 в разд. 5.24.)
Ci PMe2Ph
PhMe2P ■
■Ni^
*PMe2Ph
Br
Br Ni,
r PMe2Ph
rBr
Cl
PMe2Ph
5.39
5.40
5.10. Исключение из модели Кеперта
227
Рис. 5.20. Структура комплекса
трехвалентного никеля [NiBr3(PMe2Ph)2],
определенная поданным
рентгеновской дифракции.
Октаэдрическая структура
Среди молекул и ионов, содержащих в качестве центрального атом
3d-элемента, октаэдрическая геометрия встречается очень часто; эта же геометрия
предсказывается моделью Кеперта, например для иона [Со(Н20)6]2+ (5.41).
Аналогичное строение предсказано и определено экспериментально для
ионов [Ni(H20)6]2+, [Fe(CN)6]3", [Fe(CN)6]4" и [NiF6]2".
"12-
О
н2о„„.
.Со,
*он9
гон,
о
Н2
5.41
ИСКЛЮЧЕНИЕ ИЗ МОДЕЛИ КЕПЕРТА:
ПЛОСКОКВАДРАТНАЯ ГЕОМЕТРИЯ
Хотя модель Кеперта с успехом применяется для предсказания геометрии
большого числа комплексных соединений 3*/-элементов, ее простая форма,
изложенная выше, принципиально не может предсказать образование
плоскоквадратных частиц.
В теории Гиллеспи плоскоквадратная геометрия ICl4~ (5.30) является
производной от октаэдра, две вершины которого заняты неподеленными
парами. Однако в модели Кеперта основное внимание уделяется отталкиванию
между лигандами и игнорируется влияние неподеленных пар. Для КЧ 4 такое
рассмотрение неизбежно приводит к тетраэдрической геометрии.
Для тетраэдрической геометрии иона [СоС14]2~ (5.36), предсказываемой
теорией Кеперта, угол CI—Co—CI должен быть равен 109,5°. Эта же величи-
228
5. Многоатомные молекулы: форма и химическая связь
на получается из экспериментальных данных по дифракции. В то же время
ион [PtCl4]2~ имеет плоскоквадратное строение (5.42) с углом CI—Pt—C1 90°.
Ионы [Ni(CN)4]2~, [PdCl4]2~ и [AuCl4]~ также имеют плоскую геометрию.
Известно много плоскоквадратных молекул и ионов с d-элементами в
качестве центральных атомов, геометрия которых не контролируется чисто сте-
рическими (размерными) факторами*.
а П2-
а-
■pt-
■а
а
5.42
ГЕОМЕТРИЧЕСКАЯ ИЗОМЕРИЯ
Геометрическая изомерия будет проиллюстрирована на примере частиц с
тригонально-бипирамидальной, квадратной и октаэдрической структурой, а
также соединений с двойными связями.
Если два соединения имеют одинаковую молекулярную формулу, но
отличаются расположением атомов или групп атомов относительно
центрального атома или двойной связи, то эти соединения
называются геометрическими изомерами.
Тригонально-бипирамидальная структура
В тригональной бипирамиде существует два типа позиций - экваториальные
(eq) и аксиальные (ах), как, например, в [Fe(CO)5] (5.43).
CO..
аОС-
Fe^
coax
5.43
*СС>
rCCL
При замещении карбонильной группы, например на триметилфосфин
(РМе3), могут возникнуть два геометрических изомера [Fe(CO)4PMe3], 5.44 и
5.45:
* Детальное рассмотрение электронных эффектов, приводящих к стабилизации плоскоквадратной
геометрии, приводится в книге Gerloch Л/., Constable £.CTransition Metal Chemistry: The Valence Shell
in d-Block Chemistry, VCH, Weinheim (1994). [См. также Картмел Э., Фоулс Г. Валентность и строение
молекул, M.-J1., Химия, 1979. — Ред.]
5.11. Геометрическая изомерия
229
О
РМе3 С
.и* СО I tlt*PMe3
ОС Fe ^Г^ ОС Fe ,^^
^^СО I ^^СО
С С
О О
5.44 5.45
Тригонально-бипирамидальная молекула PC12F3 имеет три
геометрических изомера, отличающихся числом атомов хлора, занимающих аксиальные
позиции: 1) оба атома хлора в аксиальных позициях (5.46), 2) оба атома
хлора в экваториальных позициях (5.47), 3) один атом хлора в аксиальной,
другой - в экваториальной позиции (5.48).
CI F C1
^ С1 Р ,^ С1 Р ^^
^Cl I ^F
CI F F
5.46 5.47 5.48
Плоскоквадратная геометрия
В анионе [PtCl4]2~ (5.42) все четыре атома хлора эквивалентны. При
замещении только одного атома хлора получается единственный продукт, не
имеющий геометрических изомеров [PtCl3(NH3)]~ (5.49). Однако при замещении
двух групп могут возникать изомеры [PtCl2(NH3)2] со структурами 5.50 и
5.51. Изомер 5.50 называется цис-изомером, а 5.51 — транс-изомером. Эти
структурные различия решающим образом сказываются на изменении
свойств вещества. Так, ^wc-изомер [PtCl2(NH3)2] является эффективным
препаратом для лечения некоторых форм рака, тогда как транс-изомер в
десятки раз менее активен.
Плоскоквадратные частицы общей формулы XY2Z2 имеют два
геометрических изомера. В цис-изомере группы Y (а следовательно, и
группы Z) занимают соседние положения. В гране-изомере группы Y (и
группы Z) занимают противоположные положения.
Z Y
цис транс
230
5. Многоатомные молекулы: форма и химическая связь
Структуры *<wc-[PtCl2(PMePh2)2] и /w/?0«c-[NiBr2(PMe2Ph)2] были
определены рентгенодифракционным методом и представлены на рис. 5.21.
С1-
С1
■Pt-
NH,
С1
5.49
С1-
NH,
■Pt-
■NH,
H3N
CI
■Pt-
■NFL
CI
5.50
CI
5.51
Октаэдрическая структура
Во всех частицах типа SF6, [Co(H20)6]2+ и [МоС16]3~ центральный атом
окружен шестью одинаковыми группами. Если одна из этих групп заменяется на
какую-либо другую, то образуется один-единственный продукт, например
[МоС15(Н20)]2~ (5.52), который не имеет геометрических изомеров.
Если замещаются две группы, то становится возможным образование
двух изомерных форм. В ионе [CoCl2(NH3)4]+ атомы хлора могут находится в
цис- (5.53) или транс- (5.54) положении относительно центрального атома.
С1
Cli
.мо,:
>ci
rci
a
5.52
NFL
H3N„
Co.
H3N1
^Cl
NH3
5.53
H3N,
CI
Co С
NH
rNH
CI
5.54
Если октаэдрическая частица имеет общую формулу типа XY3Z3, то также
возможно образование двух изомеров, представленных на рис. 5.22. Пристав-
Рис. 5.21. Геометрические изомеры, экспериментально определенные рентгеностру-
ктурным методом для октаэдрических структур: а — *<wc-[PtCl2(PMePh2)2]; б— транс-
[NiBr2(PMe2Ph)2].
5.11. Геометрическая изомерия
231
Октаэдрические частицы общей формулы XY2Z4 имеют два
геометрических изомера. В цис-изомере группы Y занимают соседние
положения. В транс-изомере группы Y занимают противоположные,
максимально удаленные друг от друга положения.
k z z "•..
z
цис
Z
транс
Рис. 5.22. У октаэдрических частиц
общей формулы XY3Z3 могут
образовываться два геометрических изомера: гран- и ос-
изомеры.
гран-изомер
ос-изомер
ка гран- (граневый) означает, что все три группы Y (или Z) образуют одну из
граней октаэдра. Приставка ос- (осевой) указывает на то, что все три группы
Y (или Z) лежат в одной плоскости с центральным атомом.
Экспериментально определенные структуры г/?д«-[Мп(СО)3(ЫН3)3]+ и
ос-[RhCl3(py)3] представлены на рис. 5.23. Необходимо отметить, что гране-
вое расположение СО-групп в [Mn(CO)3(NH3)3]+ означает, что и группы NH3
располагаются аналогичным образом. Аналогично, если атомы хлора в
^ [RhCl3(py)3] лежат в одной плоскости с центральным атомом
Пиридин (ру): (ос-изомер), то и молекулы пиридина также лежат в одной пло-
см. разд. 15.12 скости с центральным атомом.
Октаэдрические частицы общей формулы XY3Z3 имеют два
геометрических изомера. В гран-изомере группы Y образуют одну из граней
октаэдра. В ос-изомере группы Y лежат в одной плоскости с
центральным атомом.
Y
Y/"#.. I ...»*z
Y^^ I ^^Z
I
Z
гран
Z
1
Y"# I ...n*»xY
Y^^ I ^^Z
I
Z
oc
232
5. Многоатомные молекулы: форма и химическая связь
Рис. 5.23. Экспериментально определенные гран- и ос-структуры, а - гран-
[Mn(CO)3(NH3)3]+; б- oc-[RhCl3(py)3], где ру - пиридин C5H5N.
Двойные связи
Каждый атом углерода в этилене (этене) (5.55) имеет плоскотреугольное
окружение, сама молекула также плоская, вращение вокруг двойной связи С=С невоз-
^» можно. В 1,2-дихлорэтене к каждому атому углерода присоединено по одному
Номенклатура атому хлора. Однако у этого соединения существуют два геометри-
алкенов: см. разд. 8.2 ческих изомера. В ^ис-изомере (Z-изомере, 5.56) оба атома хлора
лежат по одну сторону относительно двойной связи. В
транс-изомере (^-изомере, 5.57) атомы хлора лежат по разные стороны двойной связи.
н н ci а а н
\w/ \ = / \ = /
/ \ / \ / \
н н н н н ci
5.55 5.56 5.57
Этот тип изомерии встречается не только у органических алкенов.
Неорганическое соединение N2F2 проявляет ту же изомерию. Наличие у азота непо-
деленной электронной пары приводит к тому, что угол N—N—F меньше 180°,
а следовательно, возможно образование геометрических изомеров 5.58 и 5.59.
F
\
F F N = N
\ / \
N = N F
5.58
5.59
5.12. Тригональная бипирамида и квадратная пирамида 233
ESQ ДВЕ БЛИЗКИЕ ПО ЭНЕРГИИ СТРУКТУРЫ:
ТРИГОНАЛЬНАЯ БИПИРАМИДА И КВАДРАТНАЯ ПИРАМИДА
Твердая фаза
В твердой фазе форма аниона [Ni(CN)5]3~ зависит оттого, какой катион
входит в состав рассматриваемого соединения. В некоторых солях анион
[Ni(CN)5]3~ по форме ближе к квадратной пирамиде 5.60, чем тригональной
^» бипирамиде 5.61. Это показывает, что обе геометрии чрезвычайно близки по
en - этилендиамин энергии. Более того, в соединении [Cr(en)3][Ni(CN)5] • 1,5Н20,
H2N-CH2-CH2-NH2 где катионом является [Сг(еп)3]3+, анион [Ni(CN)5]3~ присут-
см. гл.16, табл. 16.2 ствует в двух формах . На рис. 5.24 показаны четыре аниона в
тех формах, в которых они присутствуют в монокристаллах
[Cr(en)3][Ni(CN)5] • 1,5Н20. Внимательное рассмотрение углов между
связями показывает, насколько на самом деле близки структуры, названные как
«тригональная бипирамида» и «квадратная пирамида».
3-
N
N
С
NC
Ni
I
CN
С
N
С
N
NC
■NiC
xCN
rCN
5.60
С
N
5.61
159
Тригональная
бипирамида
о-от
Квадратная
пирамида
Тригональная
бипирамида
Квадратная
пирамида
Рис. 5.24. Анионы [Ni(CN)5]J- в монокристаллах [Cr(en)3][Ni(CN)5] • 1,5Н20. (Для
простоты на рисунке не показаны катионы и молекулы кристаллизационной воды.)
5. Многоатомные молекулы: форма и химическая связь
Тригональная Квадратная
бипирамида I ^> пирамида
Рис. 5.25. Превращение тригонально-бипирамидальной молекулы в
квадратно-пирамидальную происходит за счет небольшого изменения углов при минимальных
затратах энергии. На рисунке квадратная пирамида показана в необычном ракурсе
(ср. рис. 5.12).
Хотя межлигандное отталкивание минимально в тригональной бипирами-
де, достаточно небольшого изменения энергии молекулы, чтобы превратить
эту структуру в квадратную пирамиду. В тригональной бипирамиде.угол
между двумя аксиальными лигандами равен 180°, а углы между экваториальными
лигандами равны 120°. На рис. 5.25 показано, как тригональная бипирамида
превращается в квадратную пирамиду при уменьшении угла между
аксиальными лигандами и при увеличении угла между парой экваториальных.
Малая разница в энергиях между этими двумя геометриями означает, что
в твердом состоянии реальная форма молекулы или иона с КЧ 5 будет
определяться взаимодействием между частицами, образующими кристалл.
Существует большое число примеров, когда реальная структура молекулы или
иона является промежуточной между тригональной бипирамидой и квадратной
пирамидой.
Растворы: псевдовращение Берри
В растворах малая разница в энергии между тригонально-бипирамидальной
и квадратно-пирамидальной геометрией имеет особенно важное значение. В
растворах PF5 или [Fe(CO)5] движение молекул гораздо менее затруднено по
сравнению с твердым состоянием, поэтому процесс, изображенный на
рис. 5.25, становится динамическим, т. е. протекающим в реальном времени.
Более того, последующие искажения углов могут приводить к
восстановлению исходной тригонально-бипирамидальной геометрии. Эти
низкоэнергетические переходы называются псевдовращением Берри.
Псевдовращение Берри - низкоэнергетический процесс
взаимопревращения тригонально-бипирамидальной и
квадратно-пирамидальной структур частиц, следствием которого является взаимное
замещение экваториальных и аксиальных лигандов.
5.12. Тригональная бипирамида и квадратная пирамида
235
Тригональная бипирамида
(атомы 2 и 3 расположены
аксиально)
2/
Квадратная пирамида
Тригональная бипирамида
(атомы 4 и 5 расположены
аксиально)
Рис. 5.26. Механизм псевдовращения Берри. Происходит превращение одной три-
гональной бипирамиды в другую через стадию образование квадратной пирамиды.
Нумерация лигандов показывает, что при этом процессе экваториальные заместители
становятся аксиальными и наоборот. Первая стадия этого превращения показана
более детально на рис. 5.25.
Важным следствием псевдовращения Берри является постоянное
взаимозамещение аксиальных и экваториальных групп в тригонально-бипирами-
дальной структуре (рис. 5.26). Т. е. все пять групп могут меняться местами
друг с другом. Это делает молекулы стереохимически нежесткими.
Как можно наблюдать такое поведение? Рассмотрим молекулу XY5. Если
время обмена между экваториальными и аксиальными положениями групп Y
меньше характеристического времени эксперимента (см. разд. 9.3), то все
группы Y окажутся совершенно неразличимыми (одинаковы). Если же это время
обмена позициями больше характеристического времени эксперимента, то
становится возможным экспериментально различить экваториальные и
аксиальные группы Y.
Молекулы или молекулярные ионы называются стереохимически
нежесткими, если происходит динамический процесс обмена
позициями между экваториальными и аксиальными лигандами.
Часто для изучения стереохимически нежестких систем используют метод
ядерного магнитного резонанса (ЯМР). Здесь будут рассмотрены только
результаты этих экспериментов. Более детальное рассмотрение самого метода
ЯМР дана в гл. 9.
Рассмотрим молекулу [Fe(CO)5] (5.38). Атом углерода 13С ЯМР-активен,
т. е. может исследоваться этим методом. Два типа окружения (атомы
углерода в двух аксиальных и трех экваториальных группах СО) должны давать два
различных сигнала в спектре, однако при комнатной температуре тепловой
энергии молекул оказывается достаточно для того, чтобы происходило
псевдовращение Берри, и все СО-группы оказываются эквивалентными.
Следствием этого является наличие лишь одного пика в ЯМР-спектре карбонила
железа при комнатной температуре.
5. Многоатомные молекулы: форма и химическая связь
ЛА
■3
Ю
Тригональная би-
пирамида
Тригональная би-
пирамида
Рис. 5.27. Схематическое представление энтальпии активации перехода между три-
гональной бипирамидой и квадратной пирамидой и далее снова в тригональную бипи-
рамиду. Энергии двух тригональных бипирамид одинаковы. Квадратная пирамида
располагается выше по энергии.
Каждый процесс взаимопревращения имеет характеристическую энтальпию
активации (рис. 5.27). Чем ниже температура, тем меньше молекул обладают
энергией, достаточной для преодоления активационного барьера. Путем
понижения температуры можно попытаться «заморозить» псевдовращение. Однако в
случае [Fe(CO)5] этого не удается сделать даже при 103 К, обмен позициями
между экваториальными и аксиальными лигандами продолжается и при этой
температуре, а в ЯМР-спектре по-прежнему присутствует лишь один пик.
ФОРМА И ДИПОЛЬНЫЕ МОМЕНТЫ МОЛЕКУЛ
В гл. 4 обсуждались полярные молекулы и электрический дипольный момент.
У двухатомных молекул один конец оказывается отрицательно заряженным
относительно второго. В случае многоатомных молекул ситуация более
сложная и требует тщательного рассмотрения.
Линейные молекулы
Рассмотрим молекулу С02. Величины электроотрицательности по Полингу
Хп равны 2,6 для углерода и 3,4 для кислорода. Таким образом, каждая связь
С—О полярна (Сб+05~). Однако дипольный момент молекулы равен нулю,
так как молекула линейна, а дипольный момент — векторная величина (т. е.
характеризуется абсолютной величиной и направлением). На рис. 5.28,а
показано, что, хотя каждая связь и полярна, но дипольные моменты равны по
величине и направлены в противоположные стороны, поэтому суммарный
дипольный момент молекулы равен нулю (ц = 0).
5.13. Форма и дипольные моменты молекул
237
Рис. 5.28. а - молекула диоксида
углерода: каждая связь С—О полярна, но
одинаковые по абсолютной величине и
разнонаправленные дипольные моменты
компенсируют друг друга; б — молекула
диоксида серы: каждая связь S—О
полярна, а изогнутая (угловая) структура
молекулы приводит к взаимному усилению ди-
польных моментов связей. Направление
дипольного момента молекулы показано
стрелкой.
8-
О-
5+
-С-
<5-
Дипольный
момент
молекулы
Молекулы С02 (рис. 5.28,а) и OCS (рис. 5.29,а) обе линейны, однако
первая из них симметрична относительно атома углерода, а вторая нет.
Величины электроотрицательности (рис. 5.29,а) показывают, что молекула OCS
должна иметь дипольный момент, направленный к атому кислорода,
несущему некоторый отрицательный заряд. Экспериментально определенный
дипольный момент молекулы OCS равен 0,72 Д (табл. 5.5).
Угловые молекулы
Дипольные моменты некоторых угловых молекул приведены в табл. 5.5.
Сравним дипольные моменты С02 и S02. Хотя сера и углерод имеют
одинаковую электроотрицательность по Полингу %и (2,6), дипольные моменты
молекул различны и равны 0 для С02 и 1,63 Д для S02. Такая разница
определяется различием структур молекул. На рис. 5.28 показано, что дипольные
моменты связей компенсируют друг друга в случае С02 и, наоборот, усиливают
друг друга в случае S02, приводя к значительной величине дипольного
момента молекулы в целом. Этот пример показывает, как важно бывает различать
дипольные моменты молекул и отдельных связей.
3,4
l
2,6 2,6 3,4 /^N
S=C^O Н Н
П ^ Z,Z
а
3,2
CJ
r2,0
С1 С\
3,2 3,2
ц = 0 (неполярная)
б
к
z.,z. ^r
3,0
N.
2.2 н
2,2
2,(
' ♦
/S\ I
н н
2,2 2,2 4-
в
i
Н 2,2
к
Рис. 5.29. Полярные и неполярные многоатомные молекулы. Числами показаны
величины электроотрицательности отдельных атомов. Стрелками показано
направление дипольных моментов (ц) молекул.
238 5. Многоатомные молекулы: форма и химическая связь
Таблица 5.5. Дипольные моменты некоторых многоатомных молекул
Молекула
ВС13
NC13
NF3
NH3
РС13
PF3
РН3
н2о
H2S
OF2
SF2
SF4
CF4
co2
ocs
so2
N02
N20
Форма молекулы
Плоскотреугольная
Тригонально-пирамидальная
"
"
"
"
u
Угловая
**
"
"
Дисфеноидная (искаженный тетраэдр)
Тетраэдрическая
Линейная
"
Угловая
"
Линейная
Дипольный момент /v, Д
0
0,39
0,24
1,47
0,56
1,03
0,57
1,85
0,97
0,3
1,05
0,63
0
0
0,72
1,63
0,32
0,16
Способность воды быть растворителем отчасти объяснятся наличием ди-
польного момента у молекулы Н20. Величины, приведенные в табл. 5.5,
показывают, что дипольный момент молекулы воды достаточно высок и равен
1,85 Д, тогда как у H2S он значительно меньше (р = 0,97 Д). Хотя угол между
связями в Н20 и H2S остается неизменным, уменьшение полярности связей
приводит к снижению общего дипольного момента молекулы (рис. 5.29).
Плоскотреугольные и тригонально-пирамидальные молекулы
Плоскотреугольные молекулы типа ВС13 (рис. 5.29,г) неполярны, дипольные
моменты связей компенсируют друг друга.
Молекулы типа NC13, NF3, NH3, PC13, PF3 и РН3 все имеют тригонально-
пирамидальную структуру и все полярны (табл. 5.5). Однако величина и
направление дипольных моментов этих молекул зависит от полярности связей
и от присутствия неподеленных электронных пар. Рассмотрим молекулу
аммиака рис.5.29,д (р = 1,47 Д). Каждая связь N5- — Н5+ полярна, а у атома
азота имеется еще и неподеленная пара. Общий дипольный момент молекулы
таков, что азот несет небольшой отрицательный заряд. Молекула NF3 имеет
ту же геометрию, что и молекула NH3, но полярность связей имеет другое
направление N5+—F5-. Суммарный дипольный момент молекулы NF3
(р = 0,24 Д) меньше, чем дипольный момент молекулы NH3, так как
неподеленная пара компенсирует положительный заряд на атоме азота,
возникающий за счет большей электроотрицательности фтора.
Молекулы с КЧ 4
Тетраэдрические молекулы XY4 с четырьмя одинаковыми группами Y
неполярны. Например, в молекуле CF4 дипольные моменты отдельных связей
компенсируют друг друга. То же относится и к плоскоквадратным молекулам XY4.
5.13. Форма и дипольные моменты молекул
239
Дополнение 5.1. Расчет дипольного момента BCI3
Молекула имеет плоскотреугольную форму. Угол CI—В—CI равен 120°.
Электроотрицательности по Полингу равны 2,0 для бора и 3,2 для хлора.
3,2
С\
СГ 2,0 ^а
3,2 3,2
Каждая связь В—С1 полярна, а дипольный момент связи - векторная
величина. Все три диполя отдельных связей имеют одинаковую абсолютную
величину V, но различные направления, как показано на схеме ниже.
У
к
>Кб0о;60°>^
v ^-^r^ v
Спроектируем векторы дипольных моментов связей на вертикальную ось:
Сумма векторов, направленных вверх, равна ^(одна связь)
Сумма векторов, направленных вниз, равна 2 (Kcos 60°) = 2 (V/2) = V
Таким образом, дипольный момент молекулы трихлорида бора равен
сумме двух равных по абсолютной величине, но разноправленных векторов.
Следовательно, молекула ВС13 не имеет дипольного момента (ц = 0).
Молекулы XY4 с геометрией дисфеноида имеют дипольный момент.
Дипольный момент SF4 (5.29) равен 0,63 Д. Каждая связь S—F полярна. Два
диполя, находящиеся в аксиальных позициях, компенсируют друг друга, однако
в экваториальной плоскости компенсации дипольных моментов связей нет.
Молекулы с КЧ 5 и 6
Молекулы XY6, в которых все группы Y одинаковы, не имеют дипольного
момента (SF6). Связи X—Y могут быть полярны, но их дипольные моменты
компенсируют друг друга. То же утверждение относится и к молекулам с три-
гонально-бипирамидальной геометрией. Например, в молекуле PF5
дипольные моменты аксиальных связей Р—F имеют одинаковую абсолютную
величину, но противоположное направление. Дипольные моменты
экваториальных связей компенсируют друг друга точно так же, как это
происходит в молекуле ВС13.
Труднее предсказать, какой дипольный момент будет иметь молекула,
имеющая квадратно-пирамидальную геометрию, так как очень многое
зависит от точной формы молекулы. Дипольный момент IF5 равен 2,18 Д. В этом
5. Многоатомные молекулы: форма и химическая связь
случае суммирование проекций векторов дипольных моментов отдельных
связей на вертикальную ось не приводит в двум одинаковым по абсолютной
величине и противоположно направленным векторам.
В многоатомных молекулах наличие дипольного момента
определяется как дипольными моментами отдельных связей, так и точной
геометрией молекулы.
Выводы
• Геометрия молекулы самым непосредственным образом влияет на
величину дипольного момента.
• Наличие полярных связей является необходимым, но не достаточным
условием наличия дипольного момента у молекулы.
• Простого рассмотрения электроотрицательностей атомов, образующих
данную молекулу, недостаточно для предсказания величины и
направления ее дипольного момента.
УГЛЕРОД ИМЕЕТ ЛИШЬ ТРИ ТИПА
КООРДИНАЦИОННОГО ОКРУЖЕНИЯ
Выше мы рассмотрели геометрию частиц с координационным числом
центрального атома 2, 3, 4, 5 и 6. Тяжелые элементы групп 15, 16 и 17, а также
металлы J-ряда проявляют большее разнообразие форм координационного
окружения по сравнению с р-элементами второго периода. В этом разделе
рассмотрены формы координационного окружения углерода. В последующих
главах показано, как они влияют на его химию.
Линейная координация
Помимо двух- и трехатомных молекул, таких, как СО, CN~, C02, CS2 и HCN,
углерод с КЧ 2 чаще встречается в алкинах 5.62 и алленах 5.63 (R — любой
органический заместитель). (Аллены - это диены, в которых один из атомов
углерода принимает участие в образовании сразу двух двойных связей.)
Наличие тройной или сопряженных двойных связей приводит к линейной
координации атома углерода и жесткости молекулы.
R R
\_ =/
/ с \
R R
5.63
5.62
5.14. Углерод имеет лишь три типа координационного окружения 241
Плоскотреугольное окружение
Оба атома углерода в структуре 5.63 имеют плоскотреугольное окружение.
Это является характерной чертой алкенов 5.64.
Координационное число атома углерода, равное трем, чаще всего
является следствием наличия одной двойной связи при этом атоме. Эта двойная
связь может быть С=С-связью, как в алкенах, или С=0-связью, как в
карбонильных соединениях, которые рассмотрены в гл. 17. Одним из
фундаментальных строительных блоков ароматических соединений является
бензольное кольцо 5.65, состоящее из шести атомов углерода, каждый из которых
имеет координационное число три. Плоские кольца встречаются во многих
молекулах, родственных бензолу: пиридин 5.66, нафталин 5.67.
Две аллотропные модификации углерода тоже имеют в своем составе
^ атомы углерода с КЧ 3. Структура графита состоит из бесконечных слоев
Графит и фуллерены: шестичленных колец. В фуллеренах наблюдаются некоторые
см. разд. 7.9 и 7.10 отклонения от строго планарной геометрии
координационного окружения, так как в молекулах Сп (например, п = 60)
шестичленные кольца связаны с пятичленными, что необходимо для
достижения приблизительно сферической формы этих молекул.
Н
сг с
\ / н\с^с/н н
с = с I II
5.64 I 5.66
Н
5.65
Н
Q С С
н/ ^с/ \с^ \н
н н
5.67
5.68
242
5. Многоатомные молекулы: форма и химическая связь
Тетраэдрическое окружение
Насыщенные углеводороды (алканы) и их производные содержат тетраэдри-
чески координированные атомы углерода. В так называемых линейных алка-
.7* ^ нах это приводит к зигзагообразной, форме углеродного скеле-
Конформации алкенов: „ ^ u ,- ,оч ^
^ к п _ та, такой, как у нонана CQH9n (5.68). Такие органические цепи
см. разд. 8.5 ~ g. wu
к гораздо более гибки по сравнению с цепями, содержащими
углеродные атомы с координационным числом два или три. Это приводит к
существенно большему числу возможных конформаций цепи.
ФОРМА МОЛЕКУЛ И ПРАВИЛО ОКТЕТА
В разд. 5.4 и 5.5 было показано, что:
• среди /^-элементов 2-го периода бор, углерод и азот проявляют большее
разнообразие типов координационного окружения, чем кислород и фтор;
• более тяжелые р-элементы групп 13-17 проявляют большее разнообразие
типов координационного окружения, чем ^-элементы второго периода;
• для тяжелых/?-элементов (3-й,4-й,5-й периоды) разнообразие типов
координационного окружения увеличивается при переходе от группы 13 к
группе 17.
В этом и последующих разделах первое утверждение объясняется с точки
зрения правила октета и показано, как это правило должно быть уточнено при
рассмотрении остальных двух правил.
Фтор
Электронная конфигурация основного состояния фтора — [Не]2.у22/75. Для
завершения октета фтору необходимо присоединить лишь один электрон.
Фтор образует только одну одинарную связь, а следствием этого являются все
связанные с этим и обсужденные выше структурные ограничения.
Кислород
Электронная конфигурация основного состояния кислорода — [He]2.s22/74.
Октет будет завершен, если кислород присоединит к своей валентной
оболочке два электрона. Это требование может быть выполнено путем
образования
1) двух одинарных связей (например, Н20, OF2);
2) одной двойной связи (например, 0=0, 0=С=0).
Те типы координационного окружения, которые реализуются для атома
кислорода, могут быть объяснены с использованием правила октета.
Образование двух одинарных связей приводит к молекулам угловой формы (Н20).
Если кислород образует двойную связь, то это неизбежно приводит к тому,
что этот атом оказывается концевым (С02).
На первый взгляд может показаться, что атом кислорода не может
образовывать три простые связи. Однако это не так. Примером может служить три-
5.15. Форма молекул и правило октета
243
о:
О-центр
Для заполнения октета
образуются две
одинарные связи
^>
.О-
н \
н
Угловая молекула Н20
Для заполнения октета
образуются три
одинарные связи
о- i :>
0+-центр
н \
Н
Тригонально-пирамидальный ион Н30+
Рис. 5.30. а - нейтральный атом кислорода, образуя две одинарные связи,
подчиняется правилу октета; б— в катионах атом кислорода подчиняется правилу октета, образуя
три одинарные связи. Локализация положительного заряда на атоме кислорода
является формальным приемом, облегчающим объяснение образования связей в Н30+.
гонально-пирамидальный ион Н30+ (рис. 5.10). Все частицы с центральным
атомом кислорода и с КЧ 3 сходны в одном — все они катионы.
Если локализовать положительный заряд иона Н30+ на центральном
атоме кислорода, то образование трех одинарных связей может быть объяснено
тем, что заполнение октета в данном случае происходит не на атоме
кислорода, а на ионе 0+ (рис. 5.30). Нейтральный атом кислорода (рис. 5.30,я)
подчиняется правилу октета, образуя две одинарные связи, а теория Гиллеспи
предсказывает, что молекула воды должна быть угловой. В катионах атом
кислорода подчиняется правилу октета, образуя три одинарные связи (рис. 5.30,6),
а в соответствии с теорией Гиллеспи катион Н30+ должен иметь форму три-
гональной пирамиды.
Для завершения октета атом кислорода образует две одинарные
связи, а положительно заряженный центр 0+ - три одинарные связи.
Азот
Азот так же, как фтор и кислород, подчиняется правилу октета. Структуры
азотсодержащих частиц можно объяснить на основании этого правила.
Атом азота имеет пять валентных электронов и может завершить октет,
образуя:
244 5. Многоатомные молекулы: форма и химическая связь
Дополнение 5.2. Формальная локализация заряда на
центральном атоме
В разд. 5.14 и 5.15 обсуждаются факторы, определяющие количество связей,
которое могут образовывать атомы р-элементов. В некоторых случаях
реально наблюдаемое количество связей оказывается больше ожидаемого. В этих
случаях следует применять прием, основанный на формальной локализации
всего заряда иона на центральном атоме.
В ионе Н30+ атом кислорода образует три одинарные связи О—Н . Такое
поведение становится вполне понятным, если предположить, что связи
образует не атом кислорода, а ион кислорода 0+.
В ионе ВН4~ бор образует четыре связи В—Н. Это становится понятным,
если предположить, что завершить октет валентных электронов стремится не
атом бора, а ион В~.
Образование шести связей Si—F в SiF62~ становится понятным, если
предположить, что центральный атом кремния несет заряд -2, т. е. имеет
шесть валентных электронов и, следовательно, может образовывать шесть
одинарных связей. Заметим, что в SiF62~ не выполняется правило октета для
атома кремния. (Сколько электронов оказывается в валентной оболочке
атома кремния после образования иона SiF62-?)
Хотя этот метод очень прост и хорошо работает, следует помнить, что это
лишь формальный прием. Было бы необоснованным предполагать, что
центральный атом в действительности несет весь заряд иона. Совсем не
обязательно, чтобы в Н30+ атом кислорода нес заряд +1.
Также следует помнить, что величина заряда, приписываемого
центральному атому, каким-то образом связана с его степенью окисления.
В Н30+ степень окисления атома кислорода —2, а атомов водорода +1.
В ВН4~ степень окисления бора +3, а атомов фтора —1.
В SiF62~ степени окисления кремния и фтора равны +4 и — 1
соответственно.
1) три одинарные связи (NH3, NF3, NMe3);
2) одну одинарную и одну двойную связи (CI—N=0, F—N=0);
3) одну тройную связь (Н—ON, ON~).
Отсюда следуют и возможные формы координационного окружения
атома азота. Образование трех одинарных связей и наличие неподеленной пары
электронов у атома азота приводит к молекулам тригонально-пирамидаль-
ной формы (рис. 5.31,д), молекулы с одной двойной и одной одинарной
связями имеют угловое строение, а одна тройная связь дает линейные молекулы
или ионы.
Существует большое число частиц, в которых атом азота имеет
координационное число 4. Насколько важен этот тип координационного окружения в
химии азота, становится понятным уже после рассмотрения большого
количества солей, образуемых катионами NH4+ и NMe4+. Учитывая правило
октета, образование четырех простых связей становится понятным, если заряд
+ 1 приписать центральному атому (рис. 5.31,6). Теория Гиллеспи
предсказывает, что ион NH4+ должен иметь тетраэдрическое строение.
5.15. Форма молекул и правило октета
245
Для завершения октета атом азота должен образовать три одинарные
связи, а положительно заряженный центр N+ - четыре одинарные связи.
Для заполнения октета
образуются три
одинарные связи
•N-
N-центр
Тригонально-пирамидальная
молекула NH3
Для заполнения октета
образуются четыре
одинарные связи
•N*
1М+-центр
^
- \
Н
Тетраэдрический ион NH4+
Рис. 5.31. а — нейтральный атом азота достраивает свою валентную оболочку до
октета при образовании трех одинарных связей; б — центр N+ получает восемь
валентных электронов, образуя четыре одинарные связи.
Углерод
Для завершения октета атому углерода ([Не]2522/?2) требуется четыре
электрона, которые могут быть приобретены одним из следующих способов: путем
образования
1) четырех одинарных связей (СН4);
2) двух одинарных и одной двойной связи (0=СС12);
3) одной одинарной и одной тройной связи (Н—C=N);
4) двух двойных связей (0=С=0)*
Эти варианты соответствуют трем обсужденным выше предпочтительным
формам координационного окружения углерода — тетраэдрической,
плоскотреугольной и линейной. Офаниченное число форм координационного
окружения является следствием подчинения углерода правилу октета.
* Октет может быть завершен и при образовании четверной связи, но это лишь теоретическая
возможность.
5. Многоатомные молекулы: форма и химическая связь
Бор
Большинство нейтральных молекул, содержащих один атом бора, имеют
плоскотреугольную форму, что является прямым следствием электронной
конфигурации его основного состояния ([Не]2522/?). Три валентных
электрона принимают участие в образовании трех одинарных связей, однако при
этом бор не получает завершенного октета.
Частицы типа ВН4"и BF4~, содержащие бор с КЧ 4, имеют одно общее
свойство — все они анионы. Ион В~ изоэлектронен атому углерода и имеет
четыре валентных электрона. Он может образовывать четыре одинарные связи
и достигать таким образом полностью завершенного октета. Тетраэдрическая
форма частиц ВН4~ и BF4~ прямо следует из теории Гиллеспи.
РАСШИРЕНИЕ ОКТЕТА
Полное заполнение восьмиэлектронной валентной оболочки эквивалентно
образованию четырех одинарных связей. Однако, как было показано в в
предыдущих разделах этой главы, тяжелые/?-элементы (/?-элементы 3-го,4-го
и т. д. периодов) способны образовывать соединения, в которых р-элемент
имеет
1) в валентной оболочке больше восьми электронов (C1F3, SF4) и
2) координационное число >4 (SF6, PC15).
(Почему тяжелые р-элементы проявляют свойства, не наблюдающиеся у
элементов 2-го периода?)
Расширенный октет с точки зрения теории валентных связей
р-Элементы второго периода имеют электронную конфигурацию основного
состояния 2s22pm, где т = 1-И>. Правило октета возникает как следствие
определенной стабильности состояния, в котором орбитали с главным квантовым
числом п = 2 полностью заполнены.
р-Элементы третьего периода имеют электронную конфигурацию
основного состояния 3s23/?"1, где т = 1-И). Для частиц, подчиняющихся правилу
октета, Зр-орбитали полностью заполнены. Однако в этом случае существуют и
другие орбитали с тем же значением главного квантового числа - 3^-орбита-
ли, на которых могут разместиться 10 электронов. Таким образом,
максимальное заполнение отвечает конфигурации З^З/^З*/10 (18 электронов).
Наличие J-орбиталей дает возможность тяжелым р-элементам (рис. 5.11)
образовывать больше четырех связей и делает их химические свойства
существенно отличными от свойств р-элементов второго периода.
Хотя максимально устойчивой была бы конфигурация 2s22pe или
З^З/^З*/10, на практике валентная оболочка редко заполняется 8 или 18
электронами, соответствующими ее максимальной емкости. Чаще всего число
электронов оказывается в интервале между 8 и 18. Число образующихся
одинарных связей отражает число электронов в валентной оболочке. Иод
образует следующие фториды: IF (нестабилен), IF3,IF5 и IF7, а также ионы IF2~,
IF2+, IF4+ и IF6+. В гл. 13 рассмотрена химия /ьэлементов и приведены
примеры, когда валентная оболочка содержит больше 8 электронов.
5.17. Теория валентных связей: резонансные структуры 247
р-Элементы второго периода подчиняются правилу октета, за
исключением бора, который во многих своих соединениях имеет лишь 6
электронов в валентной оболочке.
Более тяжелые р-элементы могут в своих соединениях подчиняться
правилу октета, но могут и увеличивать емкость валентной оболочки
максимум до 18 электронов.
ТЕОРИЯ ВАЛЕНТНЫХ СВЯЗЕЙ: РЕЗОНАНСНЫЕ СТРУКТУРЫ
В гл. 3 и 4 строение двухатомных молекул было разобрано с точки зрения
теории валентных связей и метода молекулярных орбиталей. В этом и
следующих разделах те же подходы будут использованы для описания
многоатомных молекул и ионов. Вначале рассмотрим Н20, BF3 и S042~ с позиций
теории валентных связей.
Эквивалентность связей
В молекуле воды длины связей О—Н одинаковы, и любая теоретическая
модель должна это учитывать. Аналогично, экспериментальные данные
показывают, что все три связи В—F в трифториде бора одинаковы так же, как
одинаковы и все связи S—О в сульфат-ионе.
Резонансные структуры
Резонансные структу- Резонансные структуры для молекулы воды приведены на рис.
рьг см разд 3 11 и 4 3 5.32,я. Резонансные формы, обозначенные как «ковалентная»
и «ионная», соответствуют симметричной форме молекулы
Н20 (длины обеих связей Н—О одинаковы). Две «частично
ионные» структуры должны рассматриваться совместно для сохранения
эквивалентности взаимодействий О—Н в молекуле.
Даже для трехатомной молекулы (очень малой и простой ) рисование
резонансных структур может оказаться утомительным занятием. Все ли эти
структуры необходимы? Все ли они одинаково важны? Чтобы ответить на эти
вопросы надо использовать интуицию. Чрезвычайно важно учитывать
следующие моменты:
1) разделение зарядов должно отражать реальные
электроотрицательности элементов;
2) не следует приписывать большие отрицательные или положительные
заряды какому-либо одному атому;
3) заряды соседних атомов должны иметь противоположные знаки (два
близких положительных или отрицательных заряда дестабилизируют
молекулу).
Рассмотрим снова структуры, приведенные на рис. 5.32,а, учитывая, что
электроотрицательности по Полингу для кислорода и водорода равны 3,4 и
2,2 соответственно. Вклад ионной резонансной структуры пренебрежимо
248
5. Многоатомные молекулы: форма и химическая связь
н н
ковалентная
Н+ Н
частично ионная
/
О
н н+
частично ионная
О2
н+ н+
ионная
Рис. 5.32. а — резонансные структуры молекулы воды; б— трех структур достаточно
для адекватного описания связывания.
мал, так как в данном случае трудно ожидать столь полного разделения
зарядов в молекуле. Таким образом, структуры, приведенные на рис. 5.32 Д дают
приближенное, но адекватное описание связывания в молекуле воды.
(Вопрос: Почему среди структур, показанных на рис. 5.32,я, отсутствуют
структуры с ионами 0+ и Н~?)
Аналогично связывание в BF3 и СН4 может быть также представлено
наборами резонансных структур. Рис. 5.33,д показывает, насколько трудоемкой
F F
В2+
F F
/ВЧ
F F
В3+
F- F
ВЧ
F F
/в\
F F
" в+ **—*> В2+ ^—^ о2+
F F F F F F
+F F F F+
F F F F F+ F
I ** „ I « I « И „ I
/в\ /ВЧ ВЧ /B+ /B\ ^B\
F FF FF FF FF F+F F
/4
F F+
Рис. 5.33. a - резонансные структуры BF3; б - только приведенные на рис. б резонансные
формы вносят существенный вклад в связывание.
5.17. Теория валентных связей: резонансные структуры 249
о о
_ I ** I _ _ „
о о о о
Рис. 5.34. Резонансные формы сульфат-иона.
о^|Хо о^||Хо о^|Ч o^fl^o -o'f|4 о^Ч
становится эта работа при увеличении сложности молекул. Трифторид бора -
очень малая молекула, но для нее можно выписать 11 резонансных форм.
Можно сгруппировать эти структуры в соответствии с зарядом центрального
атома и исключить некоторые из них как наименее существенные для
описания связывания в структуре. Например, можно не рассматривать структуры с
зарядом центрального атома больше единицы. На рис. 5.33,5 показаны резо-
^» нансные формы, вносящие наибольший вклад в связывание в BF3.
BF3: см. разд. 5.20 Такое описание соответствует действительности. Однако
необходимо рассматривать все три частично ионные структуры
одновременно, чтобы сохранить эквивалентность связей В—F.
Прежде чем рисовать резонансные структуры для иона, необходимо
решить, на каком атоме должен быть локализован его заряд. На рис. 5.34
показан набор шести резонансных форм для сульфат-иона с разумным
распределением зарядов и сохранением эквивалентности всех четырех связей
сера-кислород. В принципе, можно привести и другие варианты, но их вклад в
связывание будет ничтожен.
Пример 5.1. Резонансные структуры нитрат-иона
Нарисуйте резонансные структуры нитрат-иона и предскажите его
форму.
Решение
Нитрат-ион имеет формулу N03~.
Азот находится в группе 15 и имеет пять валентных электронов.
Кислород находится в группе 16 и имеет шесть валентных электронов.
Оба элемента находятся во втором периоде и подчиняются правилу
октета.
Резонансные структуры, вносящие максимальный вклад в связывание:
о- о о
О О О О О О
Ион имеет плоскотреугольную геометрию.
Было бы неправильно рисовать на резонансных структурах две двойные
связи и одну одинарную. Почему?
250 5. Многоатомные молекулы: форма и химическая связь
^ХВ ТЕОРИЯ ВАЛЕНТНЫХ СВЯЗЕЙ И ГИБРИДИЗАЦИЯ
Ранее в этой главе для определения числа валентных электронов и числа
связей, которые может образовывать данный атом, использовалась электронная
конфигурация основного состояния. Также было показано, что для
^-элементов второго периода подчинение правилу октета является следствием
определенной стабильности полностью заполненных 2s- и 2/ьорбиталей.
В этом разделе с точки зрения теории валентных связей будет
рассмотрено участие различных атомных орбиталей в связывании в многоатомных
молекулах.
Направленность атомных орбиталей
► За исключением сферически симметричных s-орбиталей остальные атомные
Форма атомных орби- орбитали вытянуты вдоль определенных направлений. Оказы-
талей: см. разд. 2.11 вает ли влияние направленность орбиталей на взаимное
расположение атомов в молекулах?
Рассмотрим образование плоскотреугольной молекулы,
например BF3. Валентная оболочка атома бора состоит из 2s- и трех 2/>-атомных
орбиталей, которые показаны на рис. 5.35. 2$-Орбиталь имеет сферическую
симметрию, каждая из 2р-орбиталей вытянута вдоль одной из осей
прямоугольной системы координат. На рис. 5.35 также приведено расположение
атомов молекулы трифторида бора в той же координатной системе. Атом
бора помещен в начало координат, а один из атомов фтора лежит на оси —у. Вся
молекула лежит в плоскости ху. Хотя одна из связей В—F совпадает по
направлению с 2л-орбиталью, для остальных двух связей такого соответствия не
существует. Аналогичные трудности возникают при рассмотрении молекул
других типов геометрии.
Теперь можно сформулировать задачу. Как объяснить образование связей
в молекуле, если направление атомных орбиталей не совпадает с
наблюдаемой формой этой молекулы?
Гибридизация атомных орбиталей
В этом разделе в рамках теории валентных связей предложен метод, который
позволит моделировать пространственное направление орбиталей,
необходимое для образования локализованных связей. Эти орбитали называются
гибридными и могут быть использованы для построения связывающих орбиталей
в молекулах точно так же, как и обычные атомные орбитали. Следует
отметить, что гибридизация (смешивание орбиталей) является хорошим приемом для
описания связеобразования в рамках теории ВС, хотя нет оснований
предполагать, что такой процесс происходит в действительности.
Гибридизация возможна, если исходные атомные орбитали близки по
энергии. Характер гибридной орбитали зависит от того, какими атомными
орбиталями она образована и каков вклад каждой из них. Количество
образуемых гибридных орбиталей равно общему числу использованных атомных,
что совпадает с правилом, существующим в теории молекулярных орбиталей
(число образуемых молекулярных орбиталей равно суммарному числу
исходных атомных).
5.18. Теория валентных связей и гибридизация 251
** у
► >'
2Р.
ху-плоскость
Рис. 5.35. Для рассмотрения связывания в плоскотреугольной молекуле, такой, как
BF3, необходимо рассмотреть ориентацию валентных атомных орбиталей атома бора и
удобным образом расположить атомы фтора в прямоугольной системе координат. Все
три связи BF не могут совпадать по направлению с 25-атомными орбиталиями
центрального атома.
Самым простым случаем является случай смешивания атомных одной s-
и одной р-орбиталей с одинаковым значением главного квантового числа (2s
и 2р\ 3s и Зр). На рис. 5.36 показано, что, если s- и /7-орбитали
перекрываются в фазе (т. е. s-орбиталь и перекрывающаяся с ней доля />-орбитали имеют
одинаковый знак), то происходит их взаимное усиление. Если же орбитали
перекрываются в противофазе, то происходит уменьшение размера
получающейся гибридной орбитали. Таким образом получаются две гибридные sp-op-
битали, каждая их которых имеет 50% /^-характера и 50% s-характера и
сохраняет направленность исходной /ьорбитали.
Гибридизация одной s- и двух р-АО. Так как исходных орбиталей три, то и
гибридных орбиталей тоже должно быть три (рис. 5.37). Получающие
орбитали называются з/^-гибридными и лежат в одной плоскости. В данном
случае, в плоскости ху, так как для смешивания были выбраны s-, рх- и /уАО,
что соответствует плоскотреугольной геометрии.
252
5. Многоатомные молекулы: форма и химическая связь
5/>-гибридная
орбиталь
СХЭ
или
оэ * о
О
sp- гибридная
орбиталь
Рис. 5.36. Образование гибридных s/ьорбиталей из комбинации атомных s- и р-ор-
биталей. Влияние знака s-AO на получающуюся гибридную орбиталь показано на
нижней части рисунка.
Четыре одинаковых, направленных к вершинам тетраэдра 5/?3-орбитали
образуются при смешивании 5-, рх-, р - и pz-AO так, как это показано на
рис. 5.38. Каждая их них на 25% ^-характера и на 75% /^-характера.
Для орбиталей с главным квантовым числом п = 2 могут образовываться
только 5/7-, sp1-, 5/?3-гибридные орбитали, так как доступными являются
только 25- и 2/ьАО. Но при увеличении главного квантового числа в
гибридизации могут принимать участие и ^/-орбитали.
Смешивание 5-, рх-, р -, pz- и dz2-AO дает пять 5/>3</-гибридных орбиталей,
расположение которых в пространстве соответствует тригональной бипира-
2
Рх
О^
Т
Рис. 5.37. Комбинация одной 25- и двух 2/ьАО дает три 5/?2-гибридные орбитали.
5.18. Теория валентных связей и гибридизация 253
О
Pz
Ру
Рх
т
sp3- гибридная
орбиталь
^-гибридная
орбиталь
.^-гибридная
орбиталь
V
s/?3-гибридная
орбиталь
Рис. 5.38. Комбинация одной 25- и трех 2/7-орбиталей дает четыре sp3-гибридные
орбитали.
миде (рис. 5.39,я). Этот набор орбиталей необычен тем, что состоит из двух
аксиальных и трех экваториальных орбиталей.
Смешивание s-, рх-, ру-, р-, dz2- и d^i^-AO дает шесть .у/^-гибридных
орбиталей, направленных к вершинам октаэдра (рис. 5.39,6).
Атомные орбитали могут смешиваться, давая гибридные орбитали.
Каждому набору гибридных орбиталей соответствует определенный
способ их пространственной ориентации:
sp линейный
sp2 к вершинам треугольника
sp3 к вершинам тетраэдра
sp3d к вершинам тригональной бипирамиды
sp3cfi к вершинам октаэдра
254
5. Многоатомные молекулы: форма и химическая связь
Дополнение 5.3. Направленность 5р2-гибридных орбиталей
На рис. 5.37 показано, что комбинация s-, рх- и р -АО приводит к
образованию трех одинаковых $р2-гибридных орбиталей, отличающихся лишь своей
направленностью. Откуда возникает эта разнонаправленность?
Во-первых, каждая р-орбиталь вытянута вдоль определенного
направления (рх — вдоль оси х, р — вдоль оси у). Три гибридные орбитали лежат в
плоскости ху. Так как р^-орбиталь не участвует в гибридизации, то нет и
компонента, направленной вдоль оси z.
Процесс смешивания s-, рх-и р -орбиталей может быть разбит на
следующие стадии:
1. Вклад 5-орбитали должен быть равным образом распределен между тремя
гибридными орбиталями.
2. Каждая из гибридных орбиталей должна иметь один и тот же вклад от р-
орбиталей, т. е. одинаковый процент /^-характера.
3. Пусть направление одной из гибридных орбиталей совпадает с
направлением, например, /уорбитали. Тогда первая из sp2-гибридных орбиталей
будет на одну треть иметь 5-характер, а на две трети р -характер:
+ 3
sp1- гибридная
орбиталь
4. Остающиеся компоненты: одна треть р -орбитали, целая рх-орбиталь и
две трети 5-орбитали. Эти фрагменты должны быть скомбинированы в две
одинаковые гибридные орбитали, направленность которых определяется
участием ру- и рх-орбиталей. Гибридные орбитали получаются после
прибавления к каждой из них по одной трети 5-орбитали.
Ч_^^Р
СХ)
+ 3 ( J
sp1- гибрид
sp1- гибрид
(Изменение знака р-орбитали приводит к изменению ее направления.)
Заметим, что расположение гибридных орбиталей в плоскости ху
является совершенно произвольным. Эти же орбитали могут лежать и в плоскости
yz, если для смешивания взять р- и р7 -АО, или в плоскости xz в случае р- и
P.-AO.
5.19. Гибридизация и форма молекул, содержащих р-элементы 255
а
= t
У?
X
•
¥••""
г
аксиальная
|
аксиальная
экваториальная
sp^d
1 г
экваториальная
экваториальная
Рис. 5.39. а — пять 5/?3^-гибридных орбиталей; 5— шесть 5/73^/2-гибридных орбиталей,
показанных относительно прямоугольной системы координат.
ГИБРИДИЗАЦИЯ И ФОРМА МОЛЕКУЛ,
СОДЕРЖАЩИХ р-ЭЛ ЕМЕНТЫ
В предыдущем разделе показано, что каждый набор гибридных орбиталей
вполне определенным образом ориентирован в пространстве. В данном
разделе обсуждается применение представлений о гибридных орбиталях для
описания связывания в некоторых молекулах, содержащих р-элементы.
Сравнивая взаимное расположение гибридных орбиталей и форму молекул,
можно прийти к заключению, что в случае линейных трехатомных молекул
(типа С02) центральный атом находится в состоянии sp-гибридизации, в
случае плоскотреугольных молекул (типа BF3) — в состоянии sp1-гибридизации,
а в случае тетраэдрических молекул (типа СН4) — в состоянии
^-гибридизации.
Важно отметить, что молекула принимает определенную геометрию не из-за
того, что происходит та или иная гибридизация центрального атома;
представление о гибридизации является лишь удобным инструментом описания
реальной формы молекулы в рамках теории валентных связей.
Гибридная орбиталь может рассматриваться как домен, внутри которого
располагается пара электронов. При этом проявляется четкая связь между
представлениями о гибридизации и теорией Гиллеспи. Для линейных (13~),
плоскотреугольных (ВВг3), тетраэдрических (SiCl4), тригонально-бипирами-
256 5. Многоатомные молекулы: форма и химическая связь
— sp3-Гибридная орбиталь
t с^
Н
Рис. 5.40. Теория Гиллеспи предсказывает, что геометрия молекулы аммиака
определяется тетраэдрическим расположением четырех пар электронов, при этом четыре
атома располагаются в вершинах треугольной пирамиды. Атом азота находится в
состоянии ^-гибридизации, а неподеленная пара занимает одну из этих орбиталей.
дальных (РС15) и октаэдрических (SiF6) молекул можно предположить, что
центральный атом находится в состоянии sp-, sp2-, sp3-, sp^d- и sp3 сР-гибриди-
зации соответственно. Однако в предыдущих разделах описывались
молекулы и ионы, которые имели другую геометрическую форму - угловую,
Т-образную, дисфеноидную (искаженного тетраэдра), плоскоквадратную и
квадратно-пирамидальную. В теории Гиллеспи существование таких молекул
объясняется наличием в валентной оболочке центрального атома неподелен-
ной пары электронов. При использовании представлений о гибридизации
предполагается, что неподеленная пара занимает в валентной оболочке один
домен точно так же, как и любая связывающая пара.
Рассмотрим молекулу NH3. Теория Гиллеспи предсказывает, что
геометрия молекулы аммиака определяется тетраэдрическим расположением
четырех пар электронов (рис. 5.16,6). Можно предположить, что атом азота
находится в состоянии 5р3-гибридизации, а неподеленная пара занимает одну из
этих орбиталей (рис. 5.40).
Аналогично, теория Гиллеспи предсказывает, что геометрия молекулы SF4
определяется пятью парами электронов, расположенными по вершинам три-
гональной бипирамиды. Это соответствует 5/т3^/-гибридизации атома серы и
неподеленной паре, занимающей одну из орбиталей в экваториальной
плоскости (рис. 5.41).
F
. I. F
i F
F
Рис. 5.41. Теория Гиллеспи предсказывает, что геометрия молекулы SF4
определяется пятью парами электронов, расположенными по вершинам тригональной
бипирамиды. Атом серы находится в состоянии sp3d-гибридизации, а неподеленная пара
занимает одну из орбиталей в экваториальной плоскости.
4
Аксиальная sp3J-гибридная орбиталь
, Экваториальная $/?3</-гибридная орбиталь
5.20. Гибридизация: роль негибридных орбитапей 257
Д ГИБРИДИЗАЦИЯ: РОЛЬ НЕГИБРИДНЫХ ОРБИТАЛЕИ
Негибридные орбитали
Набор из одной s- и трех /ьорбиталей (рх, ру и pz) может принимать участие в
трех типах гибридизации. Если центральный атом находится в состоянии sp3-
гибридизации, то у него оказываются задействованными все орбитали
валентной оболочки. Однако в состоянии 5/?2-гибридизации у атома остается
одна негибридная /?-орбиталь, а в случае sp-гибридизации — две /ьорбитали.
Аналогично, при sp3d- и з/т^-гибридизации без изменения остаются
соответственно четыре или три ^/-орбитали.
В этом разделе рассмотрена роль, которую играют эти негибридные
орбитали при образовании связей в молекулах.
Скелет из о-связей
Молекула метана образуется за счет четырех одинарных связей С—Н.
Молекула СН4 имеет форму тетраэдра, что соответствует sp3-гибридизации
центрального атома. Взаимное расположение гибридных орбиталей совпадает с
расположением С—Н-связей (рис. 5.42). Каждая гибридная орбиталь
углерода перекрывается с 15-АО атома водорода, а два валентных электрона (по
одному от атома углерода и атома водорода) занимают образующуюся при этом
локализованную связывающую орбиталь. Таким образом, каждая С—Н-связь -
это локализованная (5-связь; четыре такие связи образуют с-скелет молекулы,
что полностью описывает связывание в СН4 в рамках теории валентных
связей. Аналогично можно объяснить строение молекулы аммиака,
геометрия которой определяется тремя а-связями и одной неподеленной парой
электронов.
Плоскотреугольная молекула ВН3
Атом бора в молекуле ВН3 находится в состоянии 5р2-гибридизации. В
соответствии с теорией валентных связей образование этой молекулы происходит
за счет перекрывания 5/?2-гибридных орбиталей бора с 15-АО атомов
водорода (рис. 5.43). Каждой связывающей орбитали соответствует одна пара
электронов и одна локализованная а-связь В—Н. У атома бора остается также и
не принимающая участия в гибридизации пустая 2р-орбиталь.
.С .
\
'Н
Атом углерода в состоянии
5/?3-гибридизации
Н
Рис. 5.42. Применение теории валентных сййзей для описания образования
молекулы метана. Взаимное расположение 5р3-гибридных орбиталей совпадает с
расположением С—Н-связей. Каждая гибридная орбиталь перекрывается с Is-АО атома
водорода, образуя четыре локализованные а-связи С—Н.
258 5. Многоатомные молекулы: форма и химическая связь
^^_ Негибридизованная 2/уАО
Атом бора
в состоянии
5/72-гибридизации
Рис. 5.43. Применение теории валентных связей для описания образования
молекулы ВН3. Взаимное расположение трех $/>2-гибридных орбиталей совпадает с
расположением В—Н-связей. Каждая гибридная орбиталь перекрывается с Ь-АО атома
водорода, образуя три локализованные а-евязи В—Н. Если принять, что молекула лежит в
плоскости ху, то у центрального атома остается еще неиспользованная 2рг-орбиталь.
Плоскотреугольная молекула BF3
У BF3 набор а-связей такой же, как и у ВН3. а-Связи В—F образуются за счет
перекрывания 5/?2-гибридных орбиталей центрального атома с орбиталями
атомов фтора. Образующаяся при этом а-связывающая орбиталь занята
парой электронов.
Как и в случае ВН3, после образования трех а-связей у атома бора в
молекуле BF3 остается негибридизованная пустая (незанятая) 2/?-орбиталь.
Однако, в отличие от случая ВН3, каждый атом фтора имеет три неподеленные
пары электронов, одна из которых имеет ту же ориентацию, что и пустая 2/ьор-
биталь (например, 2/7^-АО) центрального атома бора. На рис. 5.44 показано,
что атом бора может образовывать л-связь с любым из атомов бора за счет
перекрывания /ьорбиталей. Напомним, что некоторые из резонансных
структур, приведенных для трифторида бора на рис. 5.33, характеризуются
наличием двойной связи B=F.
Случай BF3 является иллюстрацией общего правила: негибридизованные
р-орбитали могут принимать участие в образовании к-связей. В качестве
дополнительных примеров можно привести ВС13, В033~, С032~, N03~. Все эти
частицы обладают плоскотреугольной геометрией, при которой центральный
атом находится в состоянии 5/^-гибридизации, сохраняя неизменной одну из
/ьорбиталей, которая может перекрываться с /ьорбиталями соседних атомов
с образованием я-связи. (Какие атомные орбитали вовлечены в образование
л-связей в ВС13 и С032"?)
Линейная молекула С02
Атом углерода в линейной молекуле С02 находится в состоянии
^-гибридизации и сохраняет без изменения две 2р-АО (рис. 5.45). Молекулярный
скелет из двух а-связей образуется за счет перекрывания s/ьгибридных орбита-
лей центрального атома углерода с орбиталями двух атомов кислорода.
Каждая локализованная а-связывающая орбиталь содержит пару электронов
(один - от углерода, один - от кислорода).
Две оставшиеся 2р-АО углерода перпендикулярны друг другу и каждая
перекрывается с 2/ьАО одного из атомов кислорода. Это приводит к образова-
5.20. Гибридизация: роль негибридных орбиталей 259
Заполненная
'2р-АО фтора
...** F
F в^^
Заполненная | Заполненная
2р-АО фтора Пустая 2р-АО фтора
2р-АО бора
Рис. 5.44. Применение теории валентных связей для описания образования
молекулы трифторида бора. Прежде всего происходит образование а-связей с
использованием 5/?2-гибридных орбиталей центрального атома. Оставшаяся 2р-АО бора по своей
симметрии может перекрываться с заполненными /ьорбиталями атомов фтора с
образованием я-связи.
нию двух л-связей кислород-углерод (рис. 5.45). На каждой ^-связывающей
орбитали находится пара электронов (формально, один - от углерода, один -
от кислорода).
Общим результатом является образование двух двойных связей С=0,
каждая из которых имеет а- и л-компоненты.
я-Связывание с использованием d-орбиталей
Для тяжелых р-элементов характерны частицы с КЧ 5 или 6. При этом
центральный атом находится в состоянии sp3d- или ^/^-гибридизации
соответственно. В обоих случаях несколько ^/-орбиталей не принимают участия в
гибридизации и могут быть использованы для образования тс-связей.
Образование
я-связи
0=С=0 виЖСЬ, О
sp- Гибридная sp- Гибридная
орбиталь орбиталь
О-
t
Образование
л-связи
Рис. 5.45. Применение теории валентных связей для описания стороения молекулы
углекислого газа. Молекулярный скелет из двух а-связей С—О образуется за счет
sp-гибридных орбиталей атома углерода. Оставшиеся 2р-АО углерода и
соответствующие /ьорбитали кислорода могут быть использованы для образования я-связей
углерод—кислород.
260
5. Многоатомные молекулы: форма и химическая связь
Например, в случае SOF4 образование молекулярного а-скелета
происходит за счет перекрывания $/>3*/-гибридных орбиталей атома серы с орбиталя-
ми кислорода и фтора (рис. 5.46). На каждой из локализованных а-связыва-
ющих орбиталей находится по два электрона (один - от атома серы, один -
от кислорода или фтора). Допустим, молекула SOF4 ориентирована таким
образом, что связь S—О лежит вдоль оси у, а аксиальные связи S—F - вдоль оси
z. Из четырех не участвующих в гибридизации d-AO серы </ -орбиталь
ориентирована так, что может перекрываться с 2pz-AO кислорода, что приводит
к образованию л-связи сера—кислород. Таким образом, в молекуле SOF4 12
связывающих электронов, распределенных между пятью о- и одной л-связы-
вающими орбиталями.
Гибридизация: выводы
Используя представления о гибридизации, для всех приводимых ниже
молекул нарисуйте схему образования связей. (Напомним, что а-связь - это
одинарная связь.)
р-Элементы второго периода
• sp3- Гибридизация соответствует образованию четырех а-связей и тетраэд-
рической форме молекулы (например, CF4).
• 5/?2-Гибридизация соответствует образованию трех а-связей и
плоскотреугольной форме молекулы; возможно образование л-связи за счет
использования 2р-АО центрального атома (например, СО|~).
• sp-Гибридизация соответствует образованию двух а-связей и линейной
геометрии молекулы; возможно образование л-связей при использовании
двух оставшихся 2р-АО центрального атома (например, NOp.
Тяжелые р-элементы
• sp3-Гибридизация соответствует образованию четырех а-связей и тетраэд-
рической форме молекулы; л-связывание возможно при использовании
d-AO (например, С104~).
zk 2p. АО 3Jv.AO
о= s^^ —►
^F У
F Пять
5/>3</-ГИбрИДНЫХ
орбиталей
Рис. 5.46. Образование молекулярного скелета из а-связей в молекуле SOF4 может
быть описано, исходя из предположения о sp3tf-гибридизации центрального атома
серы. Одна из оставшихся 3d-AO серы может перекрываться с 2р-АО кислорода, давая
л-связь сера-кислород.
5.21. Теория молекулярных орбиталей и многоатомные молекулы 261
• sp1- Гибридизация соответствует образованию трех о-связей и
плоскотреугольной форме молекулы; возможно образование л-связи за счет
использования 2/7-АО центрального атома (например, S03); d-AO также могут
принимать участие в образовании связей.
• sp-Гибридизация соответствует образованию двух а-связей и линейной
геометрии молекулы (например, 1С12~); возможно образование я-связей
при использовании оставшихся р- и d-AO центрального атома.
• sp3d-Гибридизация соответствует образованию пяти о-связей и тригональ-
но-бипирамидальной форме молекулы (например, PF5); ^-связывание
возможно при использовании d-AO (например, SOF4).
• sp3cfl- Гибридизация соответствует образованию шести а-связей и октаэд-
рической форме молекулы (например, SF6); я-связывание возможно при
использовании d-AO (например, IOF5).
Я ТЕОРИЯ МОЛЕКУЛЯРНЫХ ОРБИТАЛЕЙ
И МНОГОАТОМНЫЕ МОЛЕКУЛЫ
Проблема описания многоатомных молекул
В этой книге применение метода молекулярных орбиталей для описания
многоатомных молекул не будет рассматриваться подробно. Основная
проблема, которая при этом возникает, заключается в выборе способа
представления. Каждая сторона на диаграммах молекулярных орбиталей, например,
для Н2 (рис. 3.14) или HF (рис. 4.8), соответствует набору атомных орбиталей
лишь одного из атомов двухатомной молекулы. А как же поступить в случае
многоатомной молекулы? Если попытаться сохранить прежний способ
представления данных, то диаграммы получатся либо очень сложными, либо
просто недоступными для понимания.
Общий подход к этой проблеме заключается в попытке свести задачу к
задаче с двумя неизвестными. В случае СН4, вместо того, чтобы рассматривать
взаимодействие 2s- и 2/ьорбиталей атома углерода с отдельными ls-орбита-
лями каждого атома водорода (это стало бы задачей «с пятью
неизвестными»), моделируется взаимодействие орбиталей центрального атома со всеми
четырьмя орбиталями водорода сразу. Это так называемый метод групповых
орбиталей лигандов.
Групповые орбитали лигандов
Молекула метана имеет тетраэдрическую форму. Для упрощения описания
связывания в этой молекуле по методу молекулярных орбиталей полезно
представить связь между тетраэдром и кубом так, как это показано на
рис. 5.47, что приводит к удобному расположению атомов водорода в
прямоугольной системе координат. Атом углерода находится в центре куба.
Атом углерода имеет четыре валентные орбитали: 25, 2рх, 2р и 2pz
(рис. 5.48,а); 2^-орбиталь имеет сферическую симметрию, а 2/?-орбитали
направлены вдоль осей координат (рис. 5.47 и 5.48).
262
5. Многоатомные молекулы: форма и химическая связь
Рис. 5.47. Связь между тетраэдрической
формой молекулы метана и кубом.
Каждое ребро куба параллельно одной из осей
прямоугольной системы координат.
Каждая из четырех Is-АО атомов водорода может иметь как
положительный, так и отрицательный знак. Следовательно, можно предложить
различные комбинации знаков этих четырех ls-орбиталей. Исходя из четырех
атомных орбиталей, можно получить четыре групповые орбитали лигандов (ГОЛ)
(рис. 5.48,6), так как число образуемых ГОЛ равно общему числу исходных
атомных орбиталей. Синфазная комбинация АО (знаки всех АО одинаковы)
обозначена как ГОЛ 1.
Теперь используем связь между тетраэдром и кубом (рис. 5.47). Каждая из
плоскостей ху9 xz и yz делит куб в различных направлениях. Например, на
рис. 5.47 два атома водорода лежат выше плоскости yz, а два - ниже.
Аналогичная ситуация наблюдается и для плоскостей ху и xz. В случае ГОЛ2 четыре
исходные атомные орбитали разделены плоскостью yz на две пары; орбитали
каждой пары синфазны друг другу и находятся в противофазе по отношению к
орбиталям второй пары. Аналогичные рассуждения могут быть проведены и
для ГОЛЗ и ГОЛ4.
Рис. 5.48. а — валентные орбитали углерода: 2s, 2рх, 2р и 2р ; б— комбинация четырех ls-op-
х' г у
биталей атомов водорода дает четыре групповые орбитали лигандов (ГОЛ).
5.21. Теория молекулярных орбиталей и многоатомные молекулы 263
Число образуемых групповых орбиталей лигандов равно общему
числу исходных атомных орбиталей.
Комбинирование орбиталей центрального атома
с групповыми орбиталями лигандов
Теперь необходимо подобрать соответствующие комбинации валентных
орбиталей атома углерода с групповыми орбиталями лигандов (ГОЛ).
Критерий соответствия — симметрия. Необходимо для каждой атомной орбитали
атома углерода найти соответствующую ГОЛ.
2^-Орбиталь центрального атома имеет ту же симметрию, что и ГОЛ 1.
Если поместить 25-орбиталь углерода в центр ГОЛ 1, то она будет
перекрываться с каждой из четырех ls-орбиталей атомов водорода. Это взаимодействие
приводит к образованию одной связывающей МО: o(2s). Это наиболее низко
лежащая МО (рис. 5.49) делокализована по всем четырем С—Н-связям.
2/уОрбиталь углерода имеет ту же симметрию, что и ГОЛ2. Если
поместить 2/уорбиталь в центр ГОЛ2, то она будет перекрываться с каждой из
четырех ls-орбиталей атомов водорода. Это взаимодействие приводит к
образованию второй связывающей МО в молекуле метана: с(2рх). Эта МО имеет
узловую плоскость, совпадающую с плоскостью yz. Аналогично 2/? -орбиталь
соответствует ГОЛЗ, а 2/?г-орбиталь - ГОЛ4. Эти комбинации приводят к
формированию еще двух связывающих МО. Три молекулярные орбитали, об-
о*ЙР)
2р
+- "И -+
ГОЛ четырех
ls-орбиталей
атомов водорода
СН4
4Н
Рис. 5.49. Схема молекулярных орбиталей молекулы СН4.
5. Многоатомные молекулы: форма и химическая связь
разованные комбинациями 2/ьорбиталей углерода и ГОЛ2, ГОЛЗ и ГОЛ4,
отличаются только ориентацией, вырождены по энергии и обозначены на
рис. 5.49 как с(2р). Все эти МО делокализованы по всем четырем С—Н-связям.
Таким образом, молекула метана существует за счет образования четырех
связывающих (и, следовательно, четырех разрыхляющих) молекулярных ор-
биталей. Эти орбитали делокализованы по всем четырем С—Н-связям.
Заполнение орбиталей электронами
В завершение необходимо определить общее число валентных электронов и
распределить их по орбиталям в соответствии с общими правилами.
В молекуле метана восемь валентных электронов, которые заполняют
молекулярные орбитали так, как показано на рис. 5.49. Все электроны спарены,
и молекула должна быть диамагнитной, что соответствует
экспериментальным данным.
СРАВНЕНИЕ РЕЗУЛЬТАТОВ ТЕОРИИ ВС И МЕТОДА МО
ПРИ ОПИСАНИИ СВЯЗЫВАНИЯ В МОЛЕКУЛЕ МЕТАНА
Строение молекулы метана по теории ВС и в методе МО
В соответствии с теорией валентных связей (ВС) молекула метана образуется
в результате перекрывания четырех 5/?3-гибридных орбиталей центрального
атома и ls-орбиталей атомов водорода, которое приводит к возникновению
четырех эквивалентных и локализованных а-связывающих орбиталей.
Метод молекулярных орбиталей (МО) также описывает образование
молекулы СН4 за счет четырех МО, однако в этом случае одна из орбиталей
имеет энергию ниже энергии трех остальных (рис. 5.49). Кроме того, эти МО
оказываются делокализованными по всем четырем связям С—И.
Насколько различны два этих описания? Какое из них неверно?
Насколько они соответствуют действительности?
Фотоэлектронная спектроскопия
Одно из основных различий в описании связывания в молекуле метана
между теорией ВС и методом МО заключается в том, что теория ВС
предсказывает вырожденность всех четырех связывающих молекулярных орбиталей,
тогда как метод МО указывает на несколько более низкую энергию одной из
них по сравнению с энергией трех оставшихся.
В дополнении 3.6 было кратко описано, как с помощью фотоэлектронной
спектроскопии можно измерять энергию молекулярных орбиталей.
Экспериментальные данные, полученные для молекулы метана, показывают, что
метод МО более правильно описывает образование связей в СН4. Значит ли
это, что теория ВС дает неправильный ответ?
Теория ВС и метод МО
В соответствии с методом МО молекула метана образуется за счет
размещения электронов на четырех связывающих МО. o(2s)-MO имеет сферическую
симметрию, и вносит одинаковый вклад во все четыре связи С—Н.
5.23. Заключение
265
а(2рх)-у а(2р)- и c(2pz)-M0 связаны друг с другом через операцию поворота
на 90°. Поскольку они вырождены, их необходимо рассматривать не по
отдельности, а все вместе как единый набор орбиталей. Вместе взятые они
вносят одинаковый вклад во все четыре связи С—Н. Таким образом, все связи в
молекуле метана оказываются одинаковыми, несмотря на то, что не все
связывающие орбитали эквивалентны.
Теория ВС вводит представление о $/?3-гибридизации центрального атома
и об образовании четырех эквивалентных по энергии и локализованных
молекулярных с-орбиталей, которые соответствуют четырем связям С—Н.
Оба описанных выше метода могут быть использованы для моделирования
связывания в молекуле метана. Оба метода показывают эквивалентность всех
связей С—Н. Хотя метод молекулярных орбиталей дает более реалистичное
объяснение наблюдаемому расположению энергетических уровней в
молекуле, описание, даваемое теорией валентных связей, значительно проще. Хотя
метод молекулярных орбиталей теоретически может быть применен к любым
молекулам, расчеты даже для сравнительно простых объектов оказываются
весьма трудоемкими.
ЗАКЛЮЧЕНИЕ
В этой главе рассмотрены геометрия многоатомных молекул и образование связей в них.
Что означают следующие термины?
Многоатомный
Координационное число
Изоструктурный
Плоскотреугольный
Тригонально-пирамидаль-
ный
Тетраэдрический
Тригонально-бипирами-
дальный
Квадратно-пирамидальный
Октаэдрический
Плоскоквадратный
Дисфеноидный
Модель Гиллеспи
Модель Кеперта
Стереохимически
неактивная неподеленная пара
Эффект инертной пары
Геометрическая изомерия
Стереохимически нежесткий
Псевдовращение Берри
Дипольный момент
молекулы
Расширение октета
Гибридная орбиталь
Теперь вы должны уметь:
приводить примеры молекул и ионов с
координационным числом центрального атома
два, три, четыре, пять и шесть;
объяснить, почему типы координационного
окружения у бора, углерода и азота более
разнообразны, чем у кислорода и фтора;
объяснить, почему типы координационного
окружения у тяжелых р-элементов более
разнообразны, чем у первых р-элементов той же
группы в периодической системе;
ответить, когда р-элементы подчиняются
правилу октета и когда и как валентная оболочка
может быть расширена;
нарисовать геометрические изомеры для
молекул или ионов, имеющих форму
плоскоквадратную, тригонально-бипирамидальную и ок-
таэдрическую, а также для молекул с двойной
связью;
объяснить, почему у углерода существует
весьма ограниченное число типов
координационного окружения;
описать механизм, который приводит к
обмену позициями между экваториальными и
аксиальными лигандами в молекулах с
координационным числом пять;
определить, имеет ли многоатомная молекула
дипольный момент;
нарисовать резонансные структуры для
простой многоатомной молекулы или иона;
объяснить, что обозначает понятие
«гибридизация орбиталей»;
рассказать, как связаны различные типы
гибридизации с геометрией молекул;
используя представления о гибридизации,
объяснить образование связей в простых
многоатомных молекулах с одинарными и
двойными связями.
266 5. Многоатомные молекулы: форма и химическая связь
ЕЕ! упражнения
5.1. Нарисуйте структуры Льюиса для
С02, N02+, N3", N20.
5.2. Используя теорию Гиллеспи,
предскажите форму аниона 13~.
Предскажите и объясните форму анионов
1С12~, 1Вг2-, C1F2~ и BrF2~. (Обратите
внимание, что в этих анионах
центральный атом тяжелее лигандов.)
5.3. Используя в качестве исходных
линейную молекулу С02 и угловую
молекулу S02, предскажите,
основываясь на принципе изоэлектронности,
геометрию следующих молекул и
ионов: a) N02+; б) N02"; в) Оэ; г) CS2;
д) N20; e) NCS".
5.4. Использую теорию Гиллеспи,
предскажите геометрию следующих
молекул и ионов (Me = CH3): а) ВВг3;
б) NMe3; в) SC12; г) 13+; д) ВгС13;
е) PF5; ж) PC1F4; з) NMe4+; и) XeF4;
к) XeF2.
5.5. Использую модель Кеперта,
предскажите геометрию следующих
молекул и ионов, содержащих
^-элементы: а) АиВг2"; б) [FeF6]3~;
в) [Ag(CN)2]-; г) [Ni(NH3)6]2+;
д) TiCl4; e) [MoS4]2-; ж) [Сг(СО)6];
3)Ni(CO)4;M)VCl3(NMe3)2].
5.6. Нарисуйте геометрические изомеры
(Et = этил С2Н5, MeCN (рис. 5.7)
связывается с металлом через атом
азота): а) СН3СН=СНСН3;
б) PdCl2(PPh3)2; в) [FeH(CO)4]-;
г) W(CO)4(NCMe)2;
д) CrCl3(PEt3)3;
е) IrBr2(Et)(PMe3)3.
5.7. Какую структуру предсказывает
модель Кеперта для частицы типа МЦ?
Основываясь на этой геометрии,
ответьте, сколько изомеров может
иметь соединение NiBr3(PMe2Ph)2.
Какой из этих изомеров будет
характеризоваться наименьшим
отталкиванием между лигандами? (Ответ
объясните.)
5.8. Предскажите форму следующих
молекул: a) BF3; б) PF3; в) PF5;
г) BBrF2; д) IF5; e) C1F3; ж) H2Se;
з) S03. Какие из этих молекул могут
обладать дипольным моментом ?
(Электроотрицательности по Полин-
гу приведены в табл. 4.2.)
5.9. В табл. 5.6 приведены величины ди-
польных моментов некоторых
родственных органических молекул.
Объясните наблюдаемые зависимости,
учитывая, что
электроотрицательности по Полингу для С, Н, С1 и F
равны 2,6, 2,2, 3,2 и 4,0 соответственно.
Таблица 5.6. Данные для задачи 5.9
Молекула Дипольный момент, Д
сн4
CF4
СС14
CH3F
CH2F2
CHF3
CH3CI
СН2С12
СНС13
0
0
0
1,86
1,98
1,65
1,89
1,60
1,04
5.10. Объясните, каким образом
комбинирование 25- и 2/ьорбиталей
углерода может дать гибридные орбита-
ли, необходимые для образования
связей в карбонат-ионе.
5.11. Какова гибридизация центрального
атома в молекулах: a) SiH4; б) NF3;
B)PF5;r)BF3^)[CoF6]3-;e)IF3;
ж) TiCl4?
5.12. Рассмотрим молекулу С02.
а) Предскажите ее форму, используя
теорию Гиллеспи.
б) Нарисуйте резонансные
структуры для этой молекулы.
в) Объясните образование связей в
этой молекуле, используя
представления о гибридизации. (Помните,
что все возможные варианты
должны давать один и тот же результат.)
ионы
ВВЕДЕНИЕ
В трех предыдущих главах рассмотрены ковалентные связи. Электронная
плотность в гомоядерных молекулах сконцентрирована точно между двумя
атомами, образующими связь, однако в случае гетероядерной ковалентной
связи электронная плотность смещена к более электроотрицательному атому.
Снова о молекуле HF
В рамках теории валентных связей (ВС) резонансные молекулярные
структуры учитывают вклады различных типов связи. В молекуле HF различие
между электроотрицательностями водорода (хп = 2,2) и фтора (х11 = 4,0) означа-
►" ет, что резонансная структура, соответствующая ионной форме
Резонансные структу- h+F~, вносит существенный вклад в суммарную модель обра-
ры для молекулы HF: зования связи. При использовании метода молекулярных
орем, разд. 4.3 биталей (МО) для описания связи в молекуле HF можно
видеть, что в силу различия по энергии между атомными орбиталями водорода
^ и фтора а-связывающая молекулярная орбиталь принадлежит в большей сте-
Диаграмма МО для HF: пени фтору, чем водороду. Электронная плотность в молекуле
см. разд. 4.5 HF смещена к атому фтора, и эта связь полярна. В обоих
рассмотренных моделях атом фтора в HF имеет частичный
отрицательный заряд (8~), а атом водорода — частичный положительный
заряд (8+).
Связеобразование во фториде натрия
согласно моделям ВС и МО
В газовой фазе фторид натрия состоит из молекул NaF (или, выражаясь
точнее, из формульных единиц NaF), хотя в водном растворе, в расплаве или в
твердой фазе присутствуют ионы Na+ и F~.
Различие между электроотрицательностями натрия (хп = 0,9) и фтора
(ХП = 4,0) больше, чем эти различия для водорода и фтора. Поэтому в резо-
насных структурах фторида натрия преобладает вклад ионной формы (Na+F~).
Диаграмма МО для NaF приведена на рис. 6.1. 35-Валентная АО натрия
^~ находится выше по энергии, чем 2s- и 2/7-валентные АО фтора. Поэтому сме-
Энергетическое соот- шивание указанных орбиталей незначительно и вклад в связы-
ветствие орбиталей: вающую МО вносят в основном орбитали фтора, а не натрия.
см. разд. 3.12 и 4.4 Приближенно можно считать, что вклад орбитали натрия в эту
268
6. Ионы
а
ш
х
СО
3*
Ь -Н- -НИ,
Na
Несвязывающая МО
-Н- -Н-*
NaF F
Рис. 6.1. Диаграмма МО молекулы NaF Показаны только валентные орбитали и
электроны. Разрыв на оси энергий означает, что 2s-AO фтора лежит по энергии много
ниже, чем изображено на диаграмме. s-Связывающая МО в значительной степени
имеет характер орбитали фтора.
МО настолько мал, что она оказывается практически несвязывающей*. В
молекуле NaF имеется всего восемь валентных электронов (один электрон от
натрия и семь — от фтора), и они заполняют молекулярные орбитали
согласно общему принципу последовательного заполнения орбиталей с наименьшей
энергией. Отсюда следует, что три из заполненных атомных орбиталей в NaF
имеют характер только орбиталей фтора, а одна из заполненных МО
практически полностью соответствует атомной орбитали фтора. Поэтому
электронная плотность между ядрами натрия и фтора настолько сильно смещена к
фтору, что один электрон атома натрия можно считать практически
полностью переместившимся к атому фтора. Результатом полного перемещения
будет образование иона фтора (F~) и иона натрия (Na+).
Термин «несвязывающая орбиталь» использован в общепринятом при обсуждении диаграмм МО
смысле, а именно имеется в виду малая степень обобществления электронов ядрами. Полное
разделение зарядов приводит к образованию «ионной связи».
6.2. Карты электронной плотности
269
Ионы
В разд. 1.11 был рассмотрен процесс образования ионов. Атом фтора
(электронная конфигурация в основном, невозбужденном, состоянии: [Не]2522/?5)
может принять один электрон с образованием аниона фтора F~ с
конфигурацией инертного газа ([Не]2^22/?6, или [Ne]). Потеря одного электрона
нейтральным атомом натрия (электронная конфигурация в основном состоянии
[Ne]3s') приводит к образованию положительно заряженного катиона (Na+),
также имеющего конфигурацию инертного газа [Ne].
Между атомами или молекулами, имеющими дипольный момент
(постоянный либо наведенный), действуют вандерваальсовы силы или силы ди-
поль-дипольного взаимодействия. Между сферически симметричными
ионами, несущими заряды противоположного знака, существуют также
сильные электростатические взаимодействия. Во фториде натрия действуют силы
притяжения между катионами Na+ и всеми близлежащими фторид-ионами.
Аналогичные силы действуют между анионами F~ и всеми близлежащими
катионами Na+. Силы электростатического взаимодействия зависят только от
расстояния, но не от направления в пространстве. В конечном итоге, это
приводит к образованию ионной решетки.
Анион - отрицательно заряженный ион. Катион - положительно
заряженный ион.
Настоящая глава посвящена ионам и изменениям энтальпии, которые
сопровождают процессы образования катионов и анионов. Какие силы
действуют между противоположно заряженными ионами? Насколько велика
энергия этих взаимодействий? Каково расположение ионов в твердом теле?
И модель ковалентной связи, и ионная модель являются предельными
моделями образования связи. Тем не менее, ниже будет показано, что точно так
же, как теория валентных связей может быть улучшена введением ионных
резонансных структур, модель ионной связи можно изменить таким образом,
чтобы принимать в расчет ковалентный характер связывания. В самом деле,
лишь очень ограниченное число систем могут рассматриваться как чисто
ионные или чисто ковалентные. В большинстве случаев более правильно
рассматривать связывание как носящее промежуточный характер между
этими двумя предельными случаями.
Е^| КАРТЫ ЭЛЕКТРОННОЙ ПЛОТНОСТИ
Различие между полностью ковалентной связью X—Y и ионным
взаимодействием X+Y~ можно видеть при рассмотрении областей электронной плотности
вокруг ядер X и Y. Выше ковалентная связь уже рассматривалась как наличие
^ «общей» электронной плотности. Распределение электронной плотности меж-
Уравнение ду центрами атомов в молекуле может быть исследовано с помо-
Шредингера: щью соответствующих вычислений при решении уравнения
см. разд. 2.7 Шредингера. Такие результаты могут быть представлены в виде
270
6. Ионы
Рис. 6.2. Карта электронной плотности молекул Li2 (я), HF (б) и LiF (в). В Li2 распределение
электронной плотности симметрично и область наибольшей плотности расположена точно
между двумя ядрами лития. Это соответствует области связывающей электронной плотности
молекулы Li—Li. Молекула фтороводорода полярна, и электронное облако смещено в сторону
фтора. Тем не менее, хорошо видна область связывающей электронной плотности между ядрами Н
и F. В молекуле LiF круговые контурные линии соответствуют сечениям
сферически-симметричных областей электронной плотности. Хорошо видна тенденция к образованию
сферических ионов Li+ и F". [WahlA.C, Science, 151, 961 (1966) (рис. 6.2,я); BalderR.F.W., et. al.9 J. Chem.
Phys., 47, 3381 (1967) (рис. 6.2,6); Balder R.F.W., et. al.y J. Chem. Phys., 49, 1653 (1968) (рис. 6.2,*).]
6.2. Карты электронной плотности
271
карты изолиний. Карта электронной плотности состоит из изолиний,
которые соединяют точки с одинаковой электронной плотностью. Изолинии,
проведенные вблизи ядер, в основном соответствуют электронной плотности
внутренних уровней.
На рис. 6.2 показаны три карты электронной плотности. Каждая из них
представлена как сечение молекулы некоторой плоскостью и характеризует
распределение электронной плотности в этой плоскости. На рис. 6.2,д
представлена электронная плотность гомоядерной молекулы Li2. Карта
симметрична, так что распределение электронной плотности около одного из
атомов Li является зеркальным отражением плотности вблизи другого. Область,
в которой электронное облако симметрично расположено между двумя
ядрами, представляет собой связывающую электронную плотность; связь Li—Li
носит в основном ковалентный характер.
Карта электронной плотности для молекулы HF показана на рис 6.2,6.
Изолинии электронной плотности на этой диаграмме типичны для молекулы
с полярной ковалентной связью. Карта является асимметричной и
распределение электронной плотности имеет форму яйца. Большая электронная
плотность сосредоточена вблизи ядра атома фтора. Тем не менее, часть
электронного облака распределена между атомами водорода и фтора, и это
соответствует электронной плотности ковалентной связи.
Рис. 6.2,в иллюстрирует распределение электронной плотности в
молекуле LiF. Теперь электронная плотность может быть наилучшим образом
описана как сконцентрированная в двух круговых областях (которые
соответствуют сферическим при переходе к трехмерной картине). Каждая окружность
имеет в центре ядро одного из двух атомов. Удаленные от ядер изолинии
соединяют точки с малой электронной плотностью. Связь в молекуле LiF
может быть охарактеризована как приближающаяся к ионной.
Карта электронной плотности на рис. 6.2,в иллюстрирует ситуацию, уже
обсуждавшуюся в разд. 6.1 на примере фторида натрия. С уменьшением
степени ковалентного взаимодействия между двумя атомами проявляется
тенденция к образованию двух сферически-симметричных ионов. В предельном
случае карта электронной плотности должна выглядеть так, как показано на
рис. 6.3. В первом приближении можно полагать, что ионы имеют
сферическую форму, хотя ниже в данной главе мы обсудим, до какой степени такое
описание является корректным.
272 6. Ионы
ЕВ ЭНЕРГИЯ ИОНИЗАЦИИ
В разд. 4.10 уже упоминалось об энергии (или потенциале) ионизации в
связи с вопросом о шкале электроотрицательностей по Малликену. Первая
энергия ионизации атома (1Е}) была определена как изменение внутренней
энергии при температуре 0 К (А£/(0 К)), связанннное с удалением первого
валентного электрона (см. уравнение 6.1). Изменение энергии определяется
96,5 кДж/моль для процесса в газовой фазе. Единицы измерения энергии
ионизации — кДж/моль или эВ.
Х(газ) -> Х+(газ) + е" (6.1)
Первой энергией ионизации атома, находящегося в газовой фазе,
называется изменение внутренней энергии при О К (Д1/(0 К)),
происходящее при удалении первого валентного электрона по уравнению
Х(газ) -> Х+(газ) + е"
Для термохимических расчетов используют соответствующее
изменение энтальпии:
ДН(298 К) * Ш(0 К)
Часто бывает необходимо включать значения энергии ионизации в расчеты
Цикл Гесса: по термохимическим циклам Гесса; в циклах Гесса удобнее ис-
см. разд. 1.17 пользовать изменения энтальпии А#(298 К), а не внутренней
энергии А£/(0 К). Этот вопрос уже обсуждался в разд. 3.5; более
подробное рассмотрение различий между А£/и А# проводится в гл. 12.
Значения энергии ионизации (IE) мало отличаются от значений
соответствующей энтальпии и поэтому могут быть использованы для термохимических
расчетов, если требования к точности расчетов не слишком высоки.
Вторая энергия ионизации атома (1Е2) соответствует процессу,
определяемому уравнением 6.2; отметим также, что она является первой энергией
ионизации катиона Х+. Может также происходить последующий отрыв
электронов, при этом третья энергия ионизации (1Е3) соответствует потере
третьего электрона (уравнение 6.3) и т. д. Как правило, нас интересует удаление
электронов только с валентной облочки атома. Удаление внутренних
электронов требует существенно большей энергии. Например, для элементов
группы 2 периодической системы первая и вторая энергии ионизации,
соответствующие удалению двух валентных электронов, относительно малы,
тогда как энергии отрыва IE внутренних электронов существенно более велики.
Х+(газ) -> Х2+(газ) + е" (6.2)
Х2+(газ) -> Х3+(газ) + е" (6.3)
Значения энергий ионизации для некоторых элементов приведены в
приложении 5 в т. 2.
1 эВ =
6.4. Тенденции в изменении энергий ионизации 273
Дополнение 6.1. Энергии ионизации и соответствующие
значения энтальпии
Первой энергией ионизации атома 1ЕХ называется изменение внутренней
энергии при О К (Д£/(0 К)), происходящее при удалении первого валентного
электрона; эта величина определена для процесса, происходящего в газовой фазе:
Х(газ) -> Х+(газ) + е"
Для расчета термохимических циклов необходимо знание изменения
энтальпии при 298 К (Д#(298 К)).
В предположении, что и X, и Х+ в газовой фазе представляют собой
идеальные газы, величины Д£/(0 К) и Д#(298 К) соотносятся следующим образом:
Д#(298 К) = Д£/(0 К) + (| • R • AT)
= Д£/(0 К) + (f ■ 8,314 ■ 10-3 • 298)
= Д£/(0 К) + 6,2 кДж/моль
где R — универсальная газовая постоянная (R = 8,314 Дж/мольК), а Д Т—
разность между двумя температурами (О К и 298 К).
Как правило, энергии ионизации, соответствующие удалению валентных
электронов, имеют величину порядка 1000 кДж/моль, и добавлением
величины 6,2 кДж/моль можно пренебречь. Таким образом, в большинстве
термодинамических расчетов возможно использование табулированных значений
энергий ионизации А (7(0 К) вместо требуемых значений энтальпии
ионизации ДЯ(298 К).
БЕЯ ТЕНДЕНЦИИ В ИЗМЕНЕНИИ ЭНЕРГИЙ ИОНИЗАЦИИ
Первые энергии ионизации элементов второго периода
Во втором периоде находятся элементы отлития (группа 1) до неона (группа
18). Электронная конфигурация двух s-элементов (Li и Be) в основном
состоянии - [He]25! и [He]2^ соответственно, а для шести р-элементов (от бора до
неона) - [Ht]2s22p\ [He]2s22p2, [He]2s22/>3, [He]2s22p\ [He]2^2/?5 и
[Не]2522/?6.
На рис. 6.4 показано изменение первых энергий ионизации (/£,) по мере
увеличения атомного номера элементов во втором периоде; для сравнения на
рисунке также приведено значение /Е{ для гелия (инертный газ,
предшествующий литию в периодической системе). Следует отметить три особенности
графика:
• 1ЕХ резко уменьшается при переходе от Не к Li;
• 1ЕХ в целом увеличивается с ростом порядкового номера элемента в
пределах периода;
• Существует два очевидных разрыва при переходе от Be к В и от N к О.
Данные наблюдения находят объяснение в рамках представлений о
заполнении электронных уровней валентными электронами.
274
6. Ионы
л 2500 г
§
Д 2000 г
я 1500
к 1000 h
i_
О.
0
l 500 h
CD
a
a)
C 0
He (18) Li(l) Be (2) В (13) С (14) N(15) О (16) F(17) Ne(18)
Элемент (номер группы)
Рис. 6.4. Тенденции в изменении первой энергии ионизации при переходе от гелия
(Z= 2) к неону (Z= 10). Элементы отлития до неона составляют второй период
периодической системы.
От гелия к литию
► Первая энергия ионизации гелия соответствует процессу, описываемому
♦
Устойчивость
"^-конфигурации:
см. разд. 3.4
уравнением 6.4. Электрон отрывается от заполненной ls-орби-
тали, но как показано выше, конфигурация Is2 отличается
повышенной устойчивостью. Удаление электрона разрушает
конфигурацию инертного газа и требует относительно большой
затраты энергии.
Не(газ)
Is2
Не+(газ) + е"
(6.4)
Электронная конфигурация лития в основном состоянии - [He]2s', и первая
энергия ионизации затрачивается на удаление электрона с 25-подуровня. Так
как данный электрон является едиственным на валентной оболочке, его
отрыв оказывается сравнительно легким процессом.
От лития к неону
При переходе отлития к неону можно отметить две существенные особенно-
► сти: во-первых, происходит пошаговое заполнение валентной оболочки
Сжатие орбиталей: электронами, а во-вторых, эффективный заряд ядра увеличи-
см. разд. 3.21 вается и атомные орбитали сжимаются. Отсюда следует, что
при переходе от [He^s1 (литий) к [Wt\ls22pb (неон) удаление
одного электрона с валентной облочки становится все более трудным, т. е. на
осуществление этого процесса требуется больше энергии.
От бериллия к бору
В разд. 2.15 было показано, что эффективный заряд ядра для электрона,
находящегося на 2/ьорбитали, меньше, чем для электрона на 2$-орбитали в том
же атоме, так как 2s-электроны экранируют 2/>-электроны. При переходе от
бериллия к бору конфигурация в основном состоянии изменяется с [Не]2s2
6.4. Тенденции в изменении энергий ионизации 275
на [Не]2522р1. Количество протонов в ядре также возрастает на единицу. Тем
не менее, реальное увеличение заряда ядра не перевешивает эффекта
экранирования и в конечном итоге удаление 2р-электрона с атома бора оказывается
более энергетически выгодным, чем отрыв электрона с 25-орбитали бериллия.
От азота к кислороду
Тенденция к увеличению энергии ионизации в ряду ^-элементов
прерывается при переходе от азота к кислороду. Небольшое уменьшение значений
1ЕХ при этом связано с особенностями электронной конфигурации азота в
основном состоянии: [He]2s22/?3 (рис. 6.5). 2р-Подуровень азота является
полузаполненным, и это приводит к повышенной стабильности такой
электронной конфигурации*. Таким образом, осуществить отрыв электрона с
валентной оболочки атома азота оказывается несколько труднее, чем можно
было бы ожидать**.
Энергия
2р
2s
у Валентные электроны
И
Внутренние электроны = [Не]
Рис. 6.5. Электронная конфигурация азота в основном состоянии. Следует отметить
наличие полузаполненного 2р-подуровня.
Первые энергии ионизации элементов третьего периода
На рис. 6.6 показано изменение первых энергий ионизации при переходе от
неона к аргону. Резкое уменьшение 1ЕХ при переходе от неона к натрию
соответствует разнице между энергозатратами на удаление электрона с
полностью заполненной валентной оболочки неона и на отрыв единственного
валентного электрона с 3^-орбитали натрия.
Причины такой стабильности весьма сложны. В данном случае мы просто отмечаем особую
устойчивость полузаполненных конфигураций, таких, как/?3 и d5.
"Альтернативным объяснением, не требующим введения малообоснованного с квантовомеханиче-
ской точки зрения понятия о повышенной устойчивости полузаполненных уровней, является
повышение энергии 2^-подуровня кислорода за счет энергии спаривания при появлении первого
спаренного электрона. - Прим. перев.
276
6. Ионы
л 2500
о
5
2000 h
1500
| 1000 к
а
О)
z
о
(0
ш
а
а>
♦^.
500 г
Ne(18) Na(l) Mg(2) Al(13) Si (14)
P(15) S(16) CI (17) Ar(18)
Элемент (номер группы)
Рис. 6.6. Тенденции в изменении первой энергии ионизации при переходе от неона
(Z= 10) к аргону (Z= 18). Элементы от натрия до аргона составляют третий период
периодической системы.
В третьем периоде расположены элементы от натрия до аргона. Как
видно из рис. 6.6, общие закономерности изменения энергии ионизации те же,
что и на рис. 6.4. Таким образом, для объяснения тенденций, наблюдаемых
для третьего периода, могут быть использованы те же соображения, что и для
второго, за исключением того факта, что теперь мы имеем дело с 35- и 3/ьАО.
Первые энергии ионизации для элементов
четвертого периода
Помимо s- и р-элементов четвертый период включает элементы d-ряда.
Первые энергии ионизации для элементов от аргона (последний элемент
третьего периода) до криптона приведены на рис. 6.7. При сопоставлении этого
рисунка с рис. 6.4 и 6.6 можно видеть уже обсуждавшиеся выше
закономерности изменения значений 1ЕХ для s- и р-элементов.
Элементы от скандия до цинка имеют в основном состоянии
конфигурации от [Аг^^З*/1 до [Ar]4523rf10 соответственно. Близость первых энергий
ионизации для этих элементов связана с близостью по энергии 3d- и 4л-подуров-
ней. Увеличение 1ЕХ при переходе от меди к цинку объясняется повышенной
стабильностью атома цинка, имеющего электронную конфигурацию
[Ar]4^23t/10 с полностью заполнеными 45- и 3^-подуровнями. В гл. 16 будет
проведено более подробное обсуждение особенностей ионов металлов </-ряда.
Изменение первой энергии ионизации в пределах группы
Если взглянуть внимательнее на рис. 6.4, 6.6 и 6.7, можно увидеть, что
несмотря на сохранение общей тенденции изменения первой энергии ионизации s-
и р-элементов, графики смещаются в область более низких энергий при
переходе от рис. 6.4 к рис. 6.6 и далее к рис. 6.7. Эта новая особенность соответ-
6.4. Тенденции в изменении энергий ионизации
277
20UU
1500
1000
500
п
- ♦
\
4''
—i—
*-
--♦--
-♦-
------
-♦--
—i—
.-♦--
--♦--
--♦--
--♦--
--♦''
.'4
V
,♦''
,♦--
--V
ж'
,♦
i
о.
с Аг К Са Sc Ti V Cr Mn Fe Co Ni Cu Zn Ga Ge As Se Br Кг
(18) (1) (2) (3) (4) (5) (6) (7) (8) (9) (10) (11) (12) (13) (14) (15) (16) (17) (18)
Элемент (номер группы)
Рис. 6.7. Тенденции в изменении первой энергии ионизации при переходе от аргона (Z= 18) к
криптону (Z= 36). Элементы от калия до криптона составляют четвертый период
периодической системы. В группах 1 и 2 находятся 5-элементы, в группах от 3-й до 12-й — d-элементы, в
группах от 13-й до 18-й - р-элементы.
ствует общему правилу: первая энергия ионизации понижается при смещении по
группе сверху вниз.
Рассмотрим элементы группы 1. Значения 1ЕХ для лития, натрия, калия,
рубидия и цезия приведены в левой части рис. 6.8. Наблюдается постепенное
уменьшение значений 1ЕХ в пределах группы. Первая энергия ионизации
лития соответствует отрыву одного электрона с 2^-АО; в случае натрия теряется
Зя-электрон, тогда как для калия, рубидия и цезия удаляются 45-, 5s- и 6s-
электроны соответственно. Уменьшение энергии ионизации происходит из-
за роста расстояния между ядром и валентным электроном с увеличением
главного квантового числа.
а
CD
а
с:
2000
1500 Ь
1000
500 h
Группа 1
Г *♦-♦-■♦
I 1 1 1 1 1
Группа 2
i i i i i
Группа 15
i i i i i
Группа 17
Ч
iiii
Li Na К Rb Cs Be Mg Ca Sr Ba N P As Sb Bi F CI Br I
Рис. 6.8. Первая энергия ионизации уменьшается сверху вниз в пределах группы
s- или р-элементов.
278
6. Ионы
Те же соображения могут быть использованы для объяснения тенденций
изменения первой энергии ионизации в пределах групп 2, 15 и 17 (рис. 6.8).
Последовательные энергии ионизации атома
До сих пор рассматривалось лишь образование однозарядных катионов
(уравнение 6.1). Для щелочного металла с электронной конфигурацией
валентного уровня nsx потеря более одного электрона требует значительных
энергетических затрат. Первая и вторая ионизации атома натрия протекают
по уравнениям 6.5 и 6.6.
Первая ионизация: Na(ra3) —> Na+(ra3) + e~ (6.5)
Вторая ионизация: Na+(ra3) -» Na2+(ra3) + e~ (6.6)
Значение 1ЕХ для натрия составляет 496 кДж/моль, а 1Е2 — 4563 кДж/моль.
Это почти десятикратное увеличение происходит потому, что второй
электрон удаляется:
1) от положительно заряженного иона;
2) с целиком заполненного 2р-подуровня.
Рассмотренное выше объяснение высокой энергии ионизации неона в
полной мере относится к п. 2: Ne и Na+ являются изоэлектронными частицами.
П. 1 отражает то обстоятельство, что внешние электроны в Na+
подвергаются действию большего эффективного заряда ядра, чем внешние электроны в
нейтральном атоме натрия.
Первые пять энергий ионизации атома натрия представлены на рис. 6.9.
Можно видеть резкое увеличение энергий ионизации по мере
последовательного отрыва внутренних электронов.
Рассмотрим теперь последовательную ионизацию атома магния с
электронной конфигурацией в основном состоянии [Ne^s2. Первые пять энер-
15000
. 10000
Q.
ш
i
(О
5000
3 4 5 6
Число удаленных электронов
(1 - первый электрон и т.д.)
Рис. 6.9. Изменение пяти последовательных потенциалов ионизации атомов натрия
([Ne^s1), магния ([Ne^s2) и алюминия ([Ne^A/?1).
6.5. Сродство к электрону
279
гий ионизации приведены на рис. 6.9; из них первые три соответствуют
процессам, описываемым уравнениями 6.7-6.9:
Первая ионизация: Mg(ra3) -> Mg+(ra3) + e~ (6.7)
Вторая ионизация: Mg+(ra3) —> Mg2+(ra3) + e~ (6.8)
Третья ионизация: Mg2+(ra3) —> Mg3+(ra3) + e~ (6.9)
Анализ последовательных энергий ионизации для магния на рис. 6.9
показывает, что после относительно небольшого начального увеличения следует
резкий скачок, который наблюдается после того, как был удален второй
электрон (уравнение 6.8). Такая картина объясняется тем, что первые два
электрона удаляются с валентной Зя-оболочки, а дальнейшая ионизация включает
отрыв внутренних электронов.
Аналогично, на графике первых пяти энергий ионизации алюминия
(электронаая конфигурация в основном состоянии [Ne]3523/?1) можно видеть
постепенное увеличение энергии, соответствующей потере трех валентных
электронов, с последующим гораздо более быстрым ростом IE при отрыве
внутренних электронов.
Как у магния, так и у алюминия второй валентный электрон удаляется
труднее, чем первый, поскольку отрыв второго электрона происходит от
положительно заряженного иона. Подобным же образом, в энергию отрыва
третьего валентного электрона от алюминия, включается дополнительная
работа, необходимая для преодоления притяжения между эффективным
зарядом ядра иона А12+ и удаляемым электроном:
А12+(газ) -> А13+(газ) + е" (6.10)
Хотя попытка объяснить наблюдаемые степени окисления натрия, магния и
алюминия (а также и других элементов) только с точки зрения
последовательных энергий ионизации на первый взгляд представляется разумной, такой
подход весьма опасен. В качестве упражнения, иллюстрирующего это,
попытайтесь рассчитать (используя значения IE, приведенные в приложении 5,
см. т. 2) сумму первых двух энергий ионизации атома натрия (т. е. общую
энергию, необходимую для образования иона Na2+ из газообразного натрия) и
сумму первых трех энергий ионизации алюминия (т. е. общую энергию,
требуемую для образования иона А13+ из газообразного алюминия). Очевидно, что
ионы А13+ могут присутствовать в соединениях элемента, а ионы Na2+ — нет.
Но, с другой стороны, остается неясным, почему, например, соединения,
содержащие А12+, очень редки, хотя затраты энергии, необходимые для
образования этого иона, существенно меньше, чем для образования А13+. Очевидно,
энергии ионизации являются не единственным фактором, определяющим
стабильность той или иной степени окисления. Несколько позже мы
вернемся к этому вопросу.
СРОДСТВО К ЭЛЕКТРОНУ
Определение
Следует иметь в виду, что согласно принятым обозначениям знаки энергий
сродства к электрону противоположны знакам, обычно используемым в
термодинамике, и в ряде случаев это приводит к путанице. Первая энергия срод-
280
6. Ионы
ства к электрону (ЕАХ) равна изменению внутренней энергии при
присоединении одного электрона газообразным атомом, взятому с обратным знаком
(ЕА = -А£/(0К), см. уравнение 6.11). Вторая энергия сродства к электрону
соответствует уравнению 6.12.
У(газ) + е" -> У-(газ) (6.11)
У-(газ) + е" -> У2~(газ) (6.12)
В цикле Гесса необходимо использовать не изменение внутренней энергии
(-Д£/(0К)), а изменение энтальпии (-Д#(298К)) в реакциях 6.11 и 6.12.
Различие между соответствующими значениями (-Д//(298К)) и (-А£/(0К))
можно считать малым, и им можно пренебречь в расчетах, хотя при
необходимости возможно введение поправки. Таким образом, значения энергии
сродства к электрону могут быть напрямую использованы в
термохимических расчетах, разумеется при условии, что знак выбран правильно:
экзотермическому процессу соответствует отрицательная энтальпия (АЯ), но
положительная энергия сродства к электрону (ЕА)*.
Значения АЯ = — ЕА, соответствующие присоединению электрона к ато-
^ мам и отрицательно заряженным ионам, приведены в прило-
1 эВ = 96,5 кДж/моль жении 6 в т. 2. Единицы измерения сродства к электрону -
кДж/моль или эВ.
Первая энергия сродства к электрону (ЕА^) равна изменению
внутренней энергии (AL/(0 К)), происходящему при присоединении одного
электрона газообразным атомом, взятому с обратным знаком:
У(газ) + е~->У-(газ)
Для термохимических расчетов используют соответствующее
изменение энтальпии:
АН(298 К) * AU(0 К) = -ЕА
Пример 6.1. Энтальпия образования аниона
Энтальпия диссоциации хлора на атомы при 298 К равна 242
кДж/моль, а первая энергия сродства к электрону равна 3,61 эВ.
Определите стандартную энтальпию образования хлорид-иона
(1 эВ = 96,5 кДж/моль).
Решение
Сначала необходимо использовать одинаковые единицы измерения и
правильно выбрать знаки. Имеющиеся данные соответствуют следующим процессам:
* В ряде справочников приводятся значения сродства к электрону ЕА, тогда как другие содержат
информацию непосредственно об энтальпии присоединения электрона. Перед использованием этих
данных в расчетах необходимо обращать особое внимание на то, какая именно величина приведена
в справочной литературе.
6.5. Сродство к электрону
281
1) Разрыв связи:
С12 (газ) -» 2С1 (газ)
D = 242 кДж/моль
Это значение энтальпии, его знак выбран правильно, так как процесс разрыва
связи CI—C1 является эндотермическим.
2) Присоединение электрона:
СЦгаз) + е" -> С1"(газ)
ЕЛ = 3,61 эВ = (3,61 • 96,5) кДж/моль = 348 кДж/моль
Заметим, что, во-первых, величина ЕЛ имеет знак, противоположный обычно
используемому в термодинамике. Процесс присоединения электрона к атому
хлора является экзотермическим, следовательно, его энтальпия должна быть
отрицательной.
Во-вторых, величина ЕЛ соответствует не изменению энтальпии, а
изменению внутренней энергии, а именно:
-ЕЛ = Д£/(0 К) * ДЯ(298 К)
АЯ(298 К) * -ЕЛ * -348 кДж/моль
Цель задачи — найти стандартную энтальпию образования хлорид-иона
(Д^ Я°298). Она соответствует процессу:
^С12(газ) + е" -> С1~(газ)
стандартное
состояние
Построение цикла Гесса:
Лобр/Г(298 К)
£С12(газ)
С1"(газ)
С1(газ)
Ло6рЯ(298 К)
= АЯ, + АЯ2
= \D+ (-ЕЛ)
= 121 -348
= —227 кДж/моль.
Изменения энтальпии, сопровождающие присоединение
первого и второго электронов
Изменение энтальпии при присоединении одного электрона к атомам
некоторых s- и р- элементов, приведены в табл. 6.1. За исключением азота, для
которого изменение энтальпии практически равно нулю, значения ЛЯ
отрицательны, что указывает на экзотермичность данного процесса. Также
приведены АЯ присоединения электрона к анионам 0~ и S~; оба процесса являются
эндотермическими.
282
6. Ионы
Таблица 6.1. Приближенные значения изменения энтальпии (Л//(298 К)) при
присоединении одного электрона к газообразному атому или аниону. (В
действительности, приведенные в таблице численные значения, соответствуют изменению
внутренней энергии AU(0 К), см. пояснения в тексте.)
Реакция -АН, кДж/моль
Н(газ) + е" -> Н-(газ) -73
1л(газ) + е~ -> У~(газ) -60
Na(ra3) + е~ -> Na~(ra3) -53
К(газ) + е" -> К-(газ) -48
1М(газ) + е~ -> N"(ra3) ~0
Р(газ + е~ -> Р-(газ) -72
О(газ) + е" -> О-(газ) -141
О-(газ) + е~ -> 02~(газ) +798
S(ra3) + е" -> S"(ra3) -201
S"(ra3) + e" -> S2"(ra3) +640
F(ra3) + е" -> F"(ra3) -328
С1(газ) + е~ -> С1~(газ) -349
Вг(газ) + е" -> Вг"(газ) -325
1(газ) + е~ -> Г(газ) -295
Когда электрон присоединяется к атому, на него действуют кулоновские
силы отталкивания от отрицательно заряженной внешней электронной
оболочки и силы притяжения к положительно заряженному ядру. В общем
случае, полное изменение энтальпии для процесса, описываемого уравнением
6.11, оказывается отрицательным.
При присоединении электрона к аниону силы отталкивания существенно
возрастают, и для преодоления отталкивания необходим дополнительный
подвод энергии. В результате изменение энтальпии оказывается
положительным.
Рассмотрим теперь образование газообразного оксид-иона О2- из атома
кислорода (уравнение 6.13). Общее изменение энтальпии процесса равно
сумме значений АЯ последовательных присоединений двух электронов
(уравнение 6.14).
О(газ) + 2е" -> 02"(газ) (6.13)
Д//[0(газ) -> 02"(газ)] = А//[0(газ) -> О" (газ)]
+ Д//[0-(газ) -> 02-(газ)]
= -141 + 798 = 657 кДж/моль (6.14)
Таким образом, образование оксид-иона из атомов кислорода в газовой фазе
оказывается сильно эндотермическим процессом и может показаться
невыгодным. В разд. 6.16 (при рассмотрении процесса образования КО) будет
показано, что эндотермические эффекты такого рода могут быть
компенсированы за счет формирования ионной решетки твердофазного оксида.
ЭЛЕКТРОСТАТИЧЕСКИЕ ВЗАИМОДЕЙСТВИЯ МЕЖДУ ИОНАМИ
Рассмотрим образование ионной пары X+Y~ в газовой фазе. Необходимо
помнить, что в любом изолированном химическом соединении катионы не могут
6.6. Электростатические взаимодействия между ионами 283
существовать в отсутствие анионов*. В простейшем случае электрон,
теряемый при образовании однозарядного катиона Х+, переносится ко второму
атому с образованием аниона Y~. Суммарная реакция описывается
уравнением 6.15:
Х(газ) -> Х+(газ) + е"
У(газ) + е- -> У-(газ)
ХУ(газ)
(6.15)
Термин «ионная пара» относится к единичной паре ионов, имеющих
заряды противоположного знака. Например, ионная пара в хлориде
натрия - пара ионов Na+CI".
Электростатическое (кулоновское) притяжение
Сферический ион можно рассматривать как точечный заряд. Между
точечными зарядами действуют силы электростатического (кулоновского)
взаимодействия; ионы, имеющие заряды противоположного знака, притягиваются друг
к другу, а одноименно заряженные ионы отталкиваются. Хотя
изолированные ионные пары обычно не встречаются, в качестве упрощенной модели
рассмотрим притяжение между парой ионов Na+ и С1~. Уравнение 6.16
описывает образование ионов Na+ и О" и последующее образование из них
изолированной ионной пары в газовой фазе.
Na(ra3) -> Na+(ra3) + e~
С1(газ) + е" -> С1"(газ)
NaCl(ra3)
(6.16)
Сферически-симметричные ионы можно рассматривать как точечные
заряды.
Между ионами действуют силы электростатического (кулоновского)
взаимодействия. Разноименно заряженные ионы притягиваются, а
одноименно заряженные ионы отталкиваются.
Изменение внутренней энергии AU(0 К), которое связано с
электростатическим притяжением между двумя изолированными ионами Na+ и С1~,
определяется уравнением 6.17 (единицы измерения А (/(О К) - джоули):
1К\ ■ И ■ е2 \
Для изолированной ионной пары: Д£/(0 К)= — I— ) (6.17)
М- ТС- £п- г/
В ряде особых случаев в роли аниона выступает не химическая частица, а электрон. Примером
является раствор натрия в жидком аммиаке — см. разд. 11.12.
284
6. Ионы
где \z+\ — модуль положительного заряда (для Na+ \z+\ = 1, для Са2+ \z+\ = 2)*;
|z_| - модуль отрицательного заряда (для С1~ \z_\ = 1, для О2" \z_\ = 2); е -
заряд электрона = 1,602 • 10~19 Кл; е0 — диэлектрическая проницаемость
вакуума = 8,854 • 10~12 Ф/м; г— расстояние между ядрами ионов (единицы
измерения - метры)
^» Если имеется один моль частиц хлорида натрия, в которых каждый ион
Число Авогадро: Na+ взаимодействует только с одним ионом С1~ и каждый ион
см. разд. 1.8 С1~ взаимодействует только с одним ионом Na+, уравнение 6.17
домножается на число Авогадро (УУА = 6,022 • 1023 моль-1), что
дает уравнение 6.18 (единицы измерения AU(Q К) — Дж/моль):
/jVa.|z+|.|z|.£>2\
Для моля ионных пар: AU(0 К) = — I— 1 (6.18)
W • к • е0 ■ г i
Пример 6.2. Кулоновское взаимодействие между ионами
Рассчитайте изменение внутренней энергии AU(0 К) за счет сил
притяжения между катионами и анионами в одном моле ионных пар
хлорида натрия, если расстояние между ядрами ионов Na+ и С1~
равно 236 пм.
Необходимые данные: NA = 6,022 • 1023 моль-1, е = 1,602 • 10-19 Кл,
е0 = 8,854 Ю-12 Ф/м.
Решение
I NA-\z+\'\z\e2\
Для моля ионных пар: Д£/(0 К) = —I —л 1 Дж/моль
V 4 ■ к- £q • г /
Но в этом уравнении расстояние г измеряется в метрах:
г= 236 пм = 2,36- 10-10м
/6,022- 1023- 1 • 1 • (1,602 1019)2\
МАРК) - ~U 3,142 8,854 1012 • 2,36 Ю10/
= -588502 Дж/моль
= -588,5 кДж/моль.
Кулоновское отталкивание
Если ионные пары расположены близко друг к другу, то катион каждой
ионной пары будет отталкиваться от катионов других ионных пар; точно так же
силы отталкивания будут действовать между анионами.
Внутренняя энергия кулоновского отталкивания может быть вычислена с
использованием уравнения 6.17 (или уравнения 6.18 при расчете на один
моль вещества). Однако следует помнить, что если внутренняя энергия,
соответствующая силам притяжения, имеет отрицательный знак (т. е. система
стабилизируется), то внутренняя энергия, соответствующая отталкиванию,
Модуль действительного числа всегда положителен, поэтому значения и \z+\ и \z_\ положительны,
несмотря на то что значения z+ и z_ имеют разные знаки.
6.7. Ионные решетки
285
имеет положительный знак (т. е. система дестабилизируется). Если мы имеем
дело только с силами отталкивания, следует использовать уравнение 6.19.
(\zA-\z2\-e2\
Для двух одноименно заряженных ионов: А £/(0 К) = -I 1 (6.19)
Ч • /г- е0- г'
где 1^1 и |z2| - модули зарядов соответствующих ионов.
Силы отталкивания между ионами, принадлежащими различным
ионным парам, становятся существенными только тогда, когда ионные пары
расположены достаточно близко друг к другу. В силу этого, для ионных пар в
газовой фазе, которые мы рассматривали до сих пор, силами кулоновского
отталкивания можно пренебречь. Однако в твердофазных соединениях вклад
этих сил весьма существен.
WSM ИОННЫЕ РЕШЕТКИ
Ионы в твердой фазе
В газообразном хлориде натрия каждая ионная пара состоит из одного
катиона Na+ и одного аниона С1~. Попробуем представить себе, что происходит
при конденсации газовой фазы с образованием твердого хлорида натрия.
Катионы и анионы теперь располагаются гораздо ближе друг к другу, и если
придерживаться чисто электростатической модели, считая ионы точечными
зарядами, то каждый катион Na+ притягивается ко всем близлежащим
анионам С1~. Аналогично, каждый анион С1~ притягивается ко всем
близлежащим катионам Na+. В то же время все хлорид-ионы, расположенные близко
друг к другу, будут отталкиваться, то же можно сказать о близко расположе-
ных ионах Na+.
Результатом сложения всех сил электростатического притяжения и оттал-
► кивания будет образование упорядоченной структуры, называ-
Структуры ионных емой ионной решеткой, в которой внутренняя энергия
электрорешеток можно статических взаимодействий принимает минимальное значе-
описывать в терминах ние Зто достигается за счет такого взаимного расположения
плотнейших шаровых ионов, при котором каждый ион в решетке окружен ионами с
упаковок: см. гл. 7 зарядом другого знака.
Во многих соединениях, содержащих простые ионы, расположение ионов
соответствует одному из небольшого количества структурных типов ионных
решеток. Каждый структурный тип носит имя родоначального соединения
(например, тип хлорида натрия или тип хлорида цезия) или минерала (типы
флюорита, рутила, цинковой обманки или вюрцита). В данной главе мы
рассмотрим несколько наиболее часто встречающихся структурных типов,
чтобы проиллюстрировать различные способы расположения ионов в твердой
фазе*. Например, структурный тип NaCl описывает не только расположение
ионов Na+ и С1~ в хлориде натрия, но и расположение ионов Са2+ и О2- в
решетке СаО.
Более детальную информацию о разнообразных типах ионных решеток можно найти в книге:
Уэллс Л. Структурная неорганическая химия, М., «Мир», 1987.
286
6. Ионы
В твердом состоянии соединение, состоящее из ионов, образует
упорядоченную структуру, называемую ионной решеткой. Структура
большинства соединений, состоящих из простых ионов, принадлежит
одному из небольшого числа общих структурных типов решеток.
Элементарная ячейка
Все ионные решетки могут распространяться сколь угодно далеко во всех
направлениях в трехмерном пространстве, при этом они составлены из
бесконечно большого числа повторяющихся фрагментов. Такие фрагменты
называются элементарными ячейками. Они должны быть достаточно велики,
чтобы содержать всю информацию, необходимую для построения единственной
бесконечной решетки (см. упражнение 6.10 в разд. 6.20). В ряде последующих
разделов мы проиллюстрируем понятие элементарной ячейки на примере
ряда наиболее часто встречающихся структурных типов.
Минимальный повторяющийся фрагмент ионной решетки называется
элементарной ячейкой.
Координационное число
Координационным числом (КЧ) иона в ионной решетке называется число
наиболее близких к нему ионов с противоположным зарядом.
В последующих разделах мы рассмотрим структурные типы NaCl, CsCl,
CaF2, Ti02 и ZnS. В решетках этих соединений находятся ионы, имеющие КЧ
3,4, 6 и 8. На рис. 6.10,я показано октаэдрическое окружение иона с КЧ 6. На
рис. 6.10,6 центральный ион имеет КЧ 4; это означает, что он находится в
центре тетраэдра из четырех ионов с зарядом противоположного знака.
Координационным числом (КЧ) иона в решетке называется число
наиболее близких к нему ионов с противоположным зарядом.
Рис. 6.10. a — шестикоординированный
ион в октаэдрическом окружении; б — че-
тырехкоординированный ион втетраэдри-
ческом окружении.
6.8. Структурный тип хлорида натрия (каменной соли) 287
СТРУКТУРНЫЙ ТИП ХЛОРИДА НАТРИЯ (КАМЕННОЙ СОЛИ)
На рис. 6.1 \,а показана элементарная ячейка решетки хлорида натрия.
Решетка является кубической и содержит катионы Na+ и анионы С1~.
В центре элементарной ячейки находится хлорид-ион*. Его
непосредственными соседями являются шесть ионов Na+, расположенные по вершинам
октаэдра; таким образом, ион С1" имеет КЧ 6 (рис. 6.1 \,б).
Рассмотрим теперь ион Na+ в центре квадратной грани куба (рис. 6.1 \,а).
Чтобы завершить его координационное окружение, необходимо достроить
решетку, присоединив еще одну элементарную ячейку к рассматриваемой
грани (рис. 6.11,в). Можно видеть, что КЧ иона натрия также равно шести, а
его окружение также является октаэдрическим (рис. 6.11,г).
^» Некоторые соединения, ионная решетка которых относится к тому же
Стехиометрия: структурному типу, что и решетка NaCl, приведены в табл. 6.2.
см. разд. 1.15 Заметим, что все они они имеют стехиометрию 1:1, т. е.
отношение числа катионов к числу анионов равно единице.
Na+
тт^^^адт ^Й^>
Рис. 6.11. а — элементарная ячейка решетки хлорида натрия. В центре элементарной
ячейки находится хлорид-ион; аналогично может быть построена элементарная
ячейка, в центре которой находится катион Na+; б- КЧ каждого хлорид-иона равно
шести; в — две элементарные ячейки хлорида натрия, имеющие общую грань; г — КЧ
каждого иона Na+ также равно шести.
* Поскольку все позиции ионов натрия и хлора в структуре NaCl эквивалентны, альтернативная
элементарная ячейка может быть изображена с ионом натрия в центре.
6. Ионы
Таблица 6.2. Некоторые соединения, имеющие структуру типа NaCl (рис. 6.11)
твердом состоянии при комнатной температуре
Соединение Формула Катион Анион
Хлорид натрия
Фторид натрия
Гидрид натрия
Хлорид лития
Бромид калия
И од ид калия
Фторид серебра
Хлорид серебра
Оксид магния
Оксид кальция
Оксид бария
Оксид железа( II)
Сульфид магния
Сульфид свинца(Н)
NaCl
NaF
NaH
LiCl
КВг
KI
AgF
AgCl
MgO
CaO
BaO
FeO
MgS
PbS
Na+
Na+
Na+
Li+
K+
K+
Ag+
Ag+
Mg2+
Ca2+
Ba2+
Fe2+
Mg2+
Pb2+
ci-
F"
H-
ci-
Br-
I-
F"
ci-
o2-
o2-
o2-
o2-
s2-
s2-
ОПРЕДЕЛЕНИЕ СТЕХИОМЕТРИИ СОЕДИНЕНИЯ ПО СТРОЕНИЮ
ЭЛЕМЕНТАРНОЙ ЯЧЕЙКИ НА ПРИМЕРЕ NaCl
В разд. 6.7 было сформулировано утверждение, что вся информация,
необходимая для построения бесконечной ионной решетки, должна содержаться в
ее элементарной ячейке. Отсюда следует, что стехиометрия ионного
соединения также определяется структурой элементарной ячейки.
Можно видеть, что на рис. 6.11,а число ионов Na+ не равно числу ионов
О". Тем не менее, поскольку на рисунке изображена элементарная ячейка
хлорида натрия, она должна указывать на соотношение ионов Na+:C1" =1:1.
Рассмотрим структуру более подробно. Важным обстоятельством является
то, что в решетке NaCl элементарная ячейка, показанная на рис. 6.11 ,д,
окружена шестью другими ячейками, непосредственно прилегающими к граням
куба. Одна из таких ячеек показана на рис. 6.11 ,в.
Ионы, находящиеся на границе соприкасающихся элементарных ячеек,
являются общими более чем для одной элементарной ячейки. Существует три
типа позиций ионов, в которых они обобществляются различным
количеством элементарных ячеек. Эти позиции проиллюстрированы на рис. 6.12 для
кубической элементарной ячейки.
В левой части рис. 6.12,а показаны две элементарные ячейки, каждая из
которых содержит ион в центре грани. Если эти блоки соединены, ион
обобществляется двумя элементарными ячейками. На рис. 6.12,5 показано, что
ион, расположенный на середине ребра, обобществляется четырьмя
элементарными ячейками. Наконец, если ион расположен в вершине куба, можно
видеть, что он принадлежит восьми элементарным ячейкам одновременно
(рис. 6.12,в).
Вернемся к элементарной ячейке NaCl (рис. 6.13). Во-первых,
необходимо определить различные типы позиций (узлов):
• единственный центральный узел;
• 6 узлов в центре граней;
• 12 узлов в центре ребер;
• 8 узлов в вершинах куба.
6.9. Определение стехиометрии соединения
289
а
А /
i—Н
А Л
-' \А
) V
1 ^
А А у
А
щ
[/
71
б
/
t^
ш
у. дэ
ZLj
,i—
i
А
w—
7
1^
^^ )
^1 ,
/; —
/
1^
/: /:
>
А
■г
/
/
в
/: /1
ZZT]
[—т а ,
>
/
Р
A~W\
)-\)
1 [/
-» L'"\a
А 9—v
У
Л ,
ЩЛ—\ (
1 >}—г
> V
L v
)
w
71 *
)
А\
А Л
/\А : А II
! >•
\ >•
- - г -у\ -А--Л
- ' 'к
±Щ
Рис. 6.12. Обобществление ионов в кубической решетке, а — ион в центре фани
обобществляется двумя элементарными ячейками; б — ион в центре ребра
одновременно принадлежит четырем элементарным ячейкам; в — ион в вершине принадлежит
восьми элементарным ячейкам.
Теперь рассчитаем, какая часть каждого из ионов действительно
«принадлежит» рассматриваемой элементарной ячейке, т. е. каким образом слияние
ячеек влияет на ионы в различных позициях.
Единственный центральный узел
Ион в этом узле принадлежит только элементарной ячейке, показанной на
рис. 6.13, и не обобществляется соседними ячейками.
Узлы в центре граней
Каждый ион в центре фани обобществляется двумя элементарными
ячейками. На рис. 6.13 присутствует шесть таких ионов (все они являются
катионами Na+) и каждый из них лишь наполовину принадлежит ячейке,
изображенной на рисунке.
290
6. Ионы
Ион на ребре
принадлежит
четырем
элементарным
ячейкам
Ион в вершине
принадлежит
восьми
элементарным
ячейкам
Ион в центре ячейки принадлежит
одной элементарной ячейке
Ион в центре грани принадлежит
двум элементарным ячейкам
Рис. 6.13. В элементарной ячейке хлорида натрия имеется четыре типа позиций, в
которых находятся ионы. Единственная позиция в центре элементарной ячейки, в
которой находится ион Cl~, полностью принадлежит только одной элементарной
ячейке; кроме того, имеется 6 позиций на гранях куба, 12 позиций в середине ребер и 8
позиций в вершинах куба.
Узлы в центре ребер
При образовании бесконечной решетки каждый ион, находящийся в центре
ребра, принадлежит одновременно четырем элементарным ячейкам. На рис.
6.13 можно найти 12 таких ионов, все они являются анионами С1~. Каждый
из таких ионов лишь на четверть принадлежит данной ячейке.
Узлы в вершинах куба
При образовании решетки каждый из ионов, находящихся в таких узлах,
обобществляется восемью элементарными ячейками. На рис. 6.13
присутствует восемь таких ионов Na+; каждый из них лишь на одну восьмую
принадлежит ячейке, изображенной на рисунке.
Стехиометрия хлорида натрия определяется суммированием вкладов от
каждого из ионов, принадлежащих некоторой элементарной ячейке. В табл.
6.3 показано, каким образом проводится процесс суммирования. Общее
число ионов Na+, принадлежащих элементарной ячейке, равно четырем; таково
же число ионов С1~. Отношение Na+:C1~ равно 4:4 или 1:1, откуда следует, что
эмпирическая формула хлорида натрия может быть записана как NaCl.
Таблица 6.3. Определение стехиометрии хлорида натрия из рассмотрения
структуры элементарной ячейки
Положение (см. рис. 6.13) Количество ионов Na+
Количество ионов С1~
Центральный ион
Центр грани
Ребро
Вершина
Всего
0
(6
0
(8
4
1/2) = 3
1/8 )= 1
1
О
(12- 1/4) = 3
О
4
6.11. Структуный тип фторида кальция (флюорита) 291
Е£П СТРУКТУРНЫЙ ТИП ХЛОРИДА ЦЕЗИЯ
Элементарная ячейка хлорида цезия CsCl показана на рис. 6.14,а\ такая ре-
► шетка называется кубической объемноцентрированной. Центральный ион Cs+
Кубическая объемно- имеет координационное число, равное восьми,
центрированная На рис. 6.14,5 дополнительно показаны некоторые ионы,
решетка: см. также принадлежащие соседним элементарным ячейкам. Можно ви-
разд. 7.3 деть, что хлорид-ионы являются также восьмикоординирован-
ными и располагаются в центре куба из ионов цезия. Таким
образом, элементарная ячейка CsCl может быть изображена как с ионом Cs+,
так и с ионом С1~ в центре куба.
Стехиометрия хлорида цезия может быть определена исходя из
расположения ионов в элементарной ячейке. Из рис. 6.14,а видно, что ион Cs+
находится в центре куба и целиком принадлежит единственной элементарной
ячейке. Каждый из восьми ионов СГ~, лежащих в вершинах куба,
обобществляется сразу восемью элементарными ячейками, так что количество хлорид-
ионов на одну ячейку равно (8 • 1/8 ) = 1. Значит, отношение Cs+:C1~ =1:1.
Хлорид таллия(1) в твердом состоянии также имеет решетку типа CsCl, в
которой ионы таллия располагаются в позициях цезия.
ЕЛ СТРУКТУНЫЙ ТИП ФТОРИДА КАЛЬЦИЯ (ФЛЮОРИТА)
Элементарная ячейка минерала флюорита, представляющего собой фторид
кальция CaF2, показана на рис. 6.15,а. Заметим, что расположение ионов
Са2+ (черные кружки) такое же, как расположение ионов Na+ в решетке NaCl
(ср. рис. 6.1 \,а). Однако взаимное расположение ионов Са2+ и F~ в решетке
флюорита отличается от расположения ионов Na+ и С1~ в NaCl.
Рис. 6.14. а — элементарная ячейка хлорида цезия, центральный атом цезия имеет
КЧ 8; б- дополнение элементарной ячейки соседними атомами цезия иллюстрирует,
что хлорид-ионы также имеют КЧ 8.
292 6. Ионы
Рис. 6.15. а — элементарная ячейка фторида кальция; б— каждый ион F~ имеет КЧ
4; в — каждый ион Са2+ имеет КЧ 8.
Каждый фторид-ион во флюорите находится в тетраэдрической позиции
и имеет КЧ 4 (рис. 6.15,6). Координационное окружение иона кальция
можно определить, если рассмотреть прилегающие элементарные ячейки. Проще
всего это сделать для иона кальция, находящегося в центре грани
элементарной ячейки. Можно видеть, что ион кальция имеет КЧ 8 и располагается
внутри куба из ионов фтора (рис. 6.15,в).
Число ионов Са2+ и F~ в элементарной ячейке рассчитывается по
уравнениям 6.20 и 6.21 (см. упражнение 6.11 в разд. 6.20). Отношение Ca2+:F~ = 4:8 =
1 : 2 соответствует формуле фторида кальция CaF2.
Число ионов Са2+
в элементарной ячейке флюорита = (6 • 1/2 ) + (8 • 1/8) = 4 (6.20)
Число ионов F~
в элементарной ячейке флюорита = 8 • 1 = 8 (6.21)
Некоторые соединения, имеющие в твердом состоянии структуру
флюорита, приведены в табл. 6.4.
Таблица 6.4. Некоторые соединения, имеющие флюоритоподобную структуру (т. е.
решетку CaF2) в твердом состоянии при комнатной температуре
Соединение Формула
Фторид кальция CaF2
Фторид бария BaF2
Хлорид бария ВаС12
Фторид ртути(Н) HgF2
Соединение Формула
Фторид свинца(Н) p-PbF2
Оксид циркония(1У) ZK)2
Оксид гафния(1У) НГО2
Оксид урана(1У) U02
6.12. Структурный тип рутила
293
СТРУКТУРНЫЙ ТИП РУТИЛА
Тетрагональная ячейка:
см. т. 2, приложение 12
Три структурных типа, рассмотреных выше, имели кубические элементарные
ячейки. В отличие от них, в структуре минерала рутила (оксид титана(1У)
Ti02) элементарная ячейка тетрагональна (рис. 6.16,а).
Восемь атомов титана расположены в вершинах
элементарной ячейки и один находится в центре. Каждый из
центральных атомов октаэдрически окружен шестью атомами
кислорода и имеет КЧ 6 (рис. 6.16,6). Если рассмотреть
прилегающие элементарные ячейки, можно видеть, что все атомы титана,
расположенные в вершинах тетрагональной ячейки, также имеют октаэдри-
ческое окружение. Каждый атом кислорода трехкоординирован и находится
в центре треугольника из атомов титана (рис. 6.16,в).
Стехиометрия рутила может быть рассчитана исходя из структуры
элементарной ячейки. Расчет количества атомов титана(1У) и кислорода в
элементарной ячейке проводится по уравнениям 6.22 и 6.23. Согласно этим
расчетам, в рутиле отношение титан : кислород равно 2:4 или соответственно
1 : 2. Это подтверждает формулу оксида титана(1У) ТЮ2.
Число атомов титана
в элементарной ячейке рутила = (1 • 1) + (8 • 1/8 ) = 2
(6.22)
Число атомов кислорода
в элементарной ячейке рутила = (4 • 1/2 ) + (2 • 1) = 4 (6.23)
В табл. 6.5 перечислены соединения, имеющие в твердом состоянии
структуру рутила.
°*t
о2-
Рис. 6.16. а - элементарная ячейка рутила (ТЮ2); б - каждый атом титана имеет
КЧ 6; в - каждый атом кислорода имеет КЧ 3.
294
6. Ионы
Таблица 6.5. Некоторые содинения, имеющие структуру типа рутила (рис. 6.16) в
твердом состоянии при комнатной температуре
Соединение Формула
Оксид титана( IV) ТЮ2
Оксид марганца(IV) |3-Мп02
Оксид олова(1У) Sn02
Соединение Формула
Оксид свинца( IV) РЬ02
Фторид магния MgF2
Фторид железа( II) Fe F2
I Дополнение 6.2. Оксид титана(М) - белее белого
! Оксид титана(1У) — белое, кристаллическое, нетоксичное соединение, харак-
| теризующееся высокой термической стабильностью. Он широко использует-
! ся в качестве белого красителя; в частности, ТЮ2 применяют для придания
j ярко-белого оттенка высококачественной бумаге для рисования. В настоящее
I время диоксид титана практически вытеснил применявшиеся ранее в качест-
I ве белых красителей токсичные соединения свинца, сурьмы и цинка. К опти-
; ческим свойствам ТЮ2, которые делают его столь ценным материалом, отно-
; сятся отсутствие поглощения видимого света (см. разд. 9.5) и весьма высокий
j коэффициент преломления.
ESQ СТРУКТУРНЫЕ ТИПЫ СУЛЬФИДА ЦИНКА(М)
В твердом состоянии сульфид цинка ZnS может существовать в виде двух
структурных модификаций - цинковой обманки* и вюрцита.
Решетка цинковой обманки
Элементарная ячейка цинковой обманки представлена на рис. 6.17,а.
Сравнение с рис. 6.11 ,а и 6.15,я показывает, что ионы Zn2+ в структуре цинковой
обманки занимают те же позиции, что ионы Na+ в структуре NaCl и ионы
Са2+ в структуре CaF2. Однако расположение ионов S2- отличается от
расположения анионов в структурах каменной соли и флюорита. Каждый сульфид-
ион в цинковой обманке четырехкоординирован и находится в тетраэдриче-
ском окружении (рис. 6.17,6).
Так как ни один из ионов цинка не располагается в центре элементарной
ячейки, координационное окружение цинка можно найти только при
построении нескольких прилегающих элементарных ячеек. Оказывается, что
каждый катион Zn2+ также находится в тетраэдрическом окружении
(рис. 6.17,в).
Стехиометрию цинковой обманки можно рассчитать исходя из структуры
элементарной ячейки. Согласно уравнениям 6.24 и 6.25, отношение
Zn2+ : S2~ в цинковой обманке равно 4 : 4, или соответственно 1:1, что дает
эмпирическую формулу ZnS.
Другое широко распространенное название цинковой обманки — сфалерит. — Прим. перев.
6.13. Структурные типы сульфида цинка(И)
295
Zn2+
Рис. 6.17. а — элементарная ячейка цинковой обманки; б — каждый сульфид-ион
имеет КЧ 4; в — каждый ион Zn2+ также имеет КЧ 4. В обоих случаях окружение
является тетраэдрическим.
Число ионов Zn2+ в элементарной ячейке
цинковой обманки = (6 • 1/2 ) + (8 • 1/8 ) = 4
Число ионов S2- в элементарной ячейке
цинковой обманки = (4 • I) = 4
(6.24)
(6.25)
Структурный тип вюрцита
^ Элементарная ячейка минерала вюрцита показана на рис. 6.18,а. Это соеди-
Гексагональная
нение является примером соединении с гексагональной эле-
плотнеишая упаковка:
см. разд. 7.2
. ментарной ячейкой. И катионы Zn2+, и анионы S2- в вюрците
находятся в тетраэдрическом окружении и имеют КЧ 4
(рис. 6.18,6и 6.18,*).
Стехиометрию цинковой обманки можно подтвердить исходя из
структуры элементарной ячейки с использованием уравнений 6.26 и 6.27. Следует
отметить, что в бесконечной решетке, состоящей из гексагональных
структурных единиц, ионы в центре граней обобществляются двумя
элементарными ячейками (рис. 6.19,я), ионы на вертикальных ребрах принадлежат
одновременно трем элементарным ячейкам (рис. 6.19,6), а ионы в вершинах
гексагональной призмы - шести элементарным ячейкам (рис. 6.19,в).
Число ионов Zn2+ в ячейке вюрцита = (6-1/3 ) + (4-1) = 6
(6.26)
Число ионов S2" в ячейке вюрцита = (2-1/2 ) + (12-1/6 ) + (3-1) = 6 (6.27)
В вюрците отношение числа ионов Zn2+ : S2~ равно 4 : 4, или 1:1, что дает
эмпирическую формулу ZnS.
296
6. Ионы
?Фя^>
о
Zn2+
s2-
Рис. 6.18. а - элементарная ячейка вюрцита*; б— каждый сульфид-ион имеет КЧ 4;
в — каждый ион Zn2+ также имеет КЧ 4. В обоих случаях окружение является тетраэд-
рическим.
РАЗМЕРЫ ИОНОВ
Определение межъядерных расстояний в ионной решетке
Для определения структуры ионных соединений можно использовать метод
дифракции рентгеновских лучей (см. разд. 3.2), который позволяет
рассчитать межъядерные расстояния. Так, дифракционные эксперименты
показывают, что расстояние между ядрами Na и CI в твердом NaCl равно 281 пм, а
расстояние между ядрами Na и F в твердом NaF равно 231 пм. Приведенные
цифры отвечают равновесному расстоянию между ближайшими ионами,
несущими заряды противоположного знака, в соответствующих
кристаллических решетках.
В рамках электростатической модели ионы рассматривают как сферы
конечного размера. Также принимают, что соседние ионы в кристаллической
решетке касаются друг друга, как это показано на рис. 6.20 для ионных пар
Na+F~ и Na+Cl~. На предыдущих рисунках, где были представлены
структуры NaCl, CsCl, CaF2, ТЮ2 и ZnS, ионы изображались на заметном
расстоянии друг от друга; однако такое представление было использовано
исключительно в целях повышения наглядности. На самом деле ионы занимают
значительную часть пространства внутри кристаллической решетки, как
показано на рис. 6.21 на примере элементарной ячейки CsCl.
Строго говоря, фрагмент структуры вюрцита, изображенный на рис. 6.18,а, не является
элементарной ячейкой, поскольку он не является минимальным повторяющимся фрагментом структуры.
Действительно, можно видеть, что бесконечная решетка может быть однозначно построена исходя из
параллелограмма, имеющего в основании ромб их четырех атомов серы. Таким образом, фрагмент
структуры, показанный на рис. 6.18,д, содержит три элементарные ячейки вюрцита. - Прим. перев.
6.14. Размеры ионов
297
а
^
I
\
s
<Г^
N
-•,
^ь
^
N
Щ-1
б
<
TFjh
si
Ы f
к
fn
к I
1
LJ
i
>
^ , гяо 1
^^ 1 (
\| 1
й
41 ^
<ЗИ>
М
<! ">
ъ
Рис. 6.19. Обобществление ионов между гексагональными структурными
единицами, а — ион в центре грани принадлежит одновременно двум элементарным ячейкам;
б- ион на вертикальном ребре принадлежит одновременно трем элементарным
ячейкам; в — ион в вершине гексагональной призмы принадлежит одновременно шести
элементарным ячейкам.
Ионный радиус
Ранее были рассмотрены вандерваальсовы и ковалентные радиусы атомов.
Для ионов появляется новый параметр — ионный радиус (гион), который
можно получить из данных рентгеновской дифракции. Однако, как видно из
298
6. Ионы
231 пм
281 пм
Рис. 6.20. Экспериментальные данные по дифракции рентгеновских лучей
позволяют определить межъядерные расстояния. Во фториде натрия расстояние между
ближайшими ядрами Na и F равно 231 пм. В хлориде натрия это расстояние равно 281 пм.
Если предположить, что ионы - это сферы, которые в решетке касаются друг друга, то
межъядерное расстояние можно считать равным сумме соответствующих ионных
радиусов.
Рис. 6.21. Заполнение пространства ионами в элементарной ячейке CsCl (а).
Сравните этот рисунок с использованным ранее представлением той же ячейки (б).
Рисунок а более реалистичен, но структура элементарной ячейки более удобна для
понимания при использовании схем типа б.
рис. 6.20, эта задача не столь проста, поскольку экспериментальные данные
позволяют рассчитать лишь расстояние между ядрами, которое равно сумме
ионных радиусов катиона и аниона (уравнение 6.28):
Межъядерное расстояние между _
катионом и ближайшим анионом в решетке катион анион
(6.28)
Для некоторых соединений были получены карты электронной
плотности. Карта, представленная на рис. 6.22, показывает наличие двух типов
ионов в решетке NaCl, это ионы Na+ и С1~. Ядра ионов расположены в центрах
сферических* областей электронной плотности. Расстояние между ядрами
* Карты электронной плотности являются сечениями трехмерной решетки, и окружности на таких
картах соответствуют сечениям сферически-симметричных областей электронной плотности.
6.14. Размеры ионов
299
двух соседних ионов Na+ и С1~ равно 281 пм. Как можно видеть из рис. 6.22,
электронная плотность принимает минимальное значение в некоторой
точке, расположенной между двумя ядрами. В предельном случае, когда
связывание является чисто ионным, электронная плотность в минимуме должна
обращаться в нуль. Эта точка соответствует «границе» между ионами Na+ и
С1~. Таким образом, расстояние между ядрами можно разбить на две
составляющие: радиус катиона (гкатион) и радиус аниона (/*анион). В случае NaCl
ионные радиусы определяются уравнением 6.29. Если электронную плотность
между ядрами удается рассчитать с достаточной точностью, то положение
«границы» можно использовать для определения обоих ионных радусов.
281 nM = rNa+ + rQ_ (6.29)
Метод, рассмотренный выше, относительно редко применяют для оценки
величин ионных радиусов. Альтернативой является расчет
самосогласованного набора ионных радиусов при наличии достаточно большого количества
данных по межъядерным расстояниям по системе уравнений, задаваемых
выражением 6.28. Предполагается, что ионный радиус иона,
присутствующего в двух различных соединениях в сходном окружении (например, в окта-
эдрической координации), остается неизменным. Другое допущение состоит
Рис. 6.22. Фрагмент карты электронной плотности решетки хлорида натрия, на
котором показаны два иона Na+ и два иона Cl~. [Schoknecht (?., Z. Naturforsh., Teil. A, 12,
983(1957)].
300
6. Ионы
Таблица 6.6. Ионные радиусы некоторых ионов. Приведенные значения
соответствуют ионам с КЧ 6. Ионные радиусы меняются при изменении координационного числа
иона (см. разд. 6.14). Более полные данные приведены в приложении 3, см. т. 2.
Анионы
Анион
F"
С1-
Вг"
I"
о2-
s2-
Se2"
N2"
133
181
196
220
140
184
198
171
Катионы
Катион
Гион> "М
Li+
Na+
К+
Rb+
Cs+
Mg2+
Ca2+
Al3+
Ti3+
Cr2+
Cr3+
Mn2+
Fe2+
Fe3+
Co2+
Co3+
Ni2+
Cu2+
Zn2+
76
102
138
149
170
72
100
54
67
73
62
67
61
55
65
55
69
73
74
Ионный радиус (гион) является характеристикой размера
сферического иона.
Межъядерное расстояние между
катионом и ближайшим анионом в решетке катион анион
Величина гион может изменяться в зависимости от координационного
числа иона.
в том, что как минимум для одного соединения из набора должна быть
возможность экспериментального измерения межъядерного расстояния между
двумя соседними анионами. Необходимость этого допущения поясняется в
дополнении 6.3.
Значения ионных радиусов для некоторых ионов приведены в табл. 6.6.
Ионные радиусы катионов и анионов зависят от их координационного
окружения. Так, например, для шестикоординированного катиона ^ион(Са2+)
равен 100 пм, а для восьмикоординированного - 112 пм. Значения,
приведенные в табл. 6.6, относятся к шестикоординированным ионам.
Если для элемента характерно несколько устойчивых степеней окисления,
он образует различные типы ионов. Как правило, это характерно для
d-элементов; например, известен и Fe2+, и Fe3+. Учет эффективных зарядов ядер дает
ответ на вопрос, почему радиус иона Fe3+ (гион = 55 пм) меньше радиуса Fe2+
(гион = 61 пм). Некоторые другие аналогичные пары приведены в табл. 6.6.
6.14. Размеры ионов 301
Дополнение 6.3. Расчет самосогласованного набора ионных радиусов
Приведенные ниже соединения образуют
решетки, относящиеся к структурному
типу NaCl. Для каждого из них
расстояние между катионом и ближайшим
анионом можно определить с помощью рент-
геноструктурного анализа.
Соединение
Межъядерное
Формула расстояние,
Хлорид лития LiCl 257
Хлорид натрия NaCl 281
Фторид лития LiF 201
Фторид натрия NaF 231
Используя уравнение 6.28, можно
записать следующую систему уравнений:
ги+ + tr = 257
rNa+ + rn- = 281
'С1-
л+ + гр_ = 201
'Na
+ + rF_ = 231
но для ее решения и определения всех
четырех ионных радиусов не хватает
данных. Введение дополнительных
соединений приведет к появлению новых
переменных, и не может улучшить ситуацию.
Чтобы решить уравнения, необходимо
оценить величину одного из ионных ра-
дусов независимым способом. Если
рассмотреть решетку, в которой присутствует
большой анион и маленький катион,
можно предположить, что в ней
соприкасаются не только ближайшие катион и
анион, но и два ближайших аниона. На
схеме показано, как эта ситуация
реализуется в случае LiCl, для которого
указанное предположение действительно верно.
Оба межъядерных расстояния а и Ь могут'
быть определены экспериментально:
а = 1i+ + га-
= 257 пм
(D
(Н)
b = 2 ra-= a VT= 363 пм
Из уравнения (II):
гс,- = 181,5 пм
а подстановка этой величины в уравнение
(I) дает
rLi+ = 257-181,5 = 75,5 пм
В качестве упражнения попробуйте
использовать эти данные для расчета
ионных радиусов Na+ и F~ по системе
четырех уравнений, приведенной выше. Затем
перепроверьте значения радиусов Li+ и
С\~ с использованием полученных
результатов. Вы увидите, что эти величины
несколько отличаются от полученных
ранее, и, следовательно, для получения
надежных усредненных величин
необходимо рассматривать большее количество
соединений.
302
6. Ионы
250
I
« 200
о
S
,
250
200 h
JQ
X
X
о
S
150
100
50
Li+ Na+ K+ Rb+ Cs+
Рис. 6.23. Увеличение ионных радиусов
щелочных металлов в пределах группы 1.
150 Ь
100
50 h
F C1 Вг
Группа 17
О2" S2- Se2
Группа 16
Рис. 6.24. Увеличение радиусов галогенид-
ионов в пределах группы 17 и двузарядных
анионов в пределах группы 16.
Тенденции изменения ионных радиусов в пределах групп
В разд. 2.21 отмечалось, что вандерваальсовы радиусы инертных газов
увеличиваются при движении вниз по группе 18, а рис. 3.4 иллюстрирует
увеличение ковалентных радиусов атомов с ростом их порядкового номера в группах
15, 16 и 17. Аналогичные тенденции наблюдаются и для ионных радиусов (см.
табл. 6.6) при условии сравнения радиусов ионов, имеющих одинаковый
заряд, например ионов М2+. Как видно из рис. 6.23, ионные радиусы ионов М+
возрастают по мере движения вниз по группе 1. Аналогичная тенденция
наблюдается для галогенид-анионов (Х~) в группе 17 и для двузарядных
анионов элементов группы 16 (рис. 6.24).
ЭНЕРГИЯ РЕШЕТКИ: ЧИСТО ИОННАЯ МОДЕЛЬ
Энергией решетки ионного соединения Д£/(0 К) называется изменение
внутренней энергии, сопровождающее образование одного моля твердой фазы из
составляющих газообразных ионов при температуре 0 К. Эту величину
можно оценить в предположении, что твердофазная решетка состоит из
сферических ионов. Оценка границ применимости такого приближения проведена в
разд. 6.17 и 6.18. Для того чтобы рассчитать энергию решетки, необходимо
принимать во внимание все силы притяжения и отталкивания, действующие
между ионами.
6.15. Энергия решетки: чисто ионная модель
303
Кулоновские силы в ионной решетке
В разд. 6.6 были рассмотрены электростатические взаимодействия между
двумя ионами, имеющими заряды противоположного знака. Изменение
внутренней энергии при образовании одного моля ионных пар за счет кулонов-
ского взаимодействия в пределах ионной пары можно рассчитать по уравнению
6.18. При этом принимается, что ионные пары являются изолированными и
силы взаимодействия между ними отсутствуют.
Ионы, расположенные в твердофазной решетке, испытывают действие
кулоновских сил притяжения и отталкивания от окружающих ионов. Это
становится очевидным, если рассмотреть некоторый ион в любой из
решеток, показанных на рис. 6.11, 6.14, 6.15, 6.16, 6.17 или 6.18.
Электростатические взаимодействия можно подразделить на следующие подгруппы:
• притяжение между соседними катионами и анионами;
• более слабые силы притяжения между удаленными катионами и анионами;
• отталкивание между ближайшими одноименно заряженными ионами;
• более слабое отталкивание между удаленными одноименно заряженными
ионами.
Количество и величина этих сил зависит от геометрии решетки и
межъядерных расстояний между ионами.
Изменение внутренней энергии
за счет кулоновского взаимодействия
Чтобы определить изменение внутренней энергии AU(0 К), связанное с
образованием ионной решетки, удобно рассматривать образование решетки из
газообразных ионов согласно уравнению
Х+(газ) + У-(газ) = XY(tb.) (6.30)
В величину Д£/(0 К), связанную с образованием ионной решетки, вносят
вклад силы кулоновского притяжения (см. уравнение 6.18) и отталкивания
(см. уравнение 6.19). Уравнения 6.18 и 6.19 имеют одинаковый вид и могут
быть легко просуммированы. Количество и относительные вклады
кулоновских взаимодействий учитываются с помощью коэффициента А,
называемого константой Маделунга. В табл. 6.7 приведены величины А для различных
типов кристаллических решеток, описанных выше.
Таблица 6.7. Константы Маделунга для некоторых часто встречающихся типов
решеток. Приведенные значения рассчитаны геометрическим методом, описанным в
дополнении 6.4. А - безразмерная величина
Тип решетки Решетка показана на рис. А
Хлорид натрия NaCl 6.11 1,7476
Хлорид цезия CsCl 6.14 1,7627
Флюорит CaF2 6.15 2,5194
Рутил ТЮ2 6.16 2,408
Цинковая обманка P-ZnS 6.17 1,6381
Вюрцита-ZnS 6.18 1,6413
304
6. Ионы
Теперь можно записать уравнение 6.31, в котором величина кулоновско-
го взаимодействия Л U(0 К) соответствует понижению внутренней энергии за
счет кулоновских сил, возникающих в одном моле ионного твердофазного
XY, образовавшегося из 1 моль газообразных ионов Х+ и 1 моль газообразных
ионов Y~. Так как при образовании решетки энергия высвобождается,
величина A U (О К) является отрицательной. Единицы измерения Л (/(О К) в
уравнении 6.31 - Дж/моль. Для ионной решетки:
(N - А • \z I • \z I • е2\
—^ ' +' ' "' (6.31)
4 • к • е0 • г /
где \z+\ — абсолютная величина положительного заряда;
\z_\ — абсолютная величина отрицательного заряда;
е — заряд электрона = 1,602 • 10~19 Кл;
е0 — диэлектрическая проницаемость вакуума = 8,854 • 10~12 Ф/м;
г- межъядерное расстояние между ионами (единицы измерения - м);
А — константа Маделунга (см. табл. 6.7).
Силы Борна
Ионы в реальной ионной решетке не являются точечными зарядами, как это
принимается в электростатической модели, а имеют конечный размер.
Наряду с электростатическими силами, рассмотренными выше, существуют также
борновские силы отталкивания между соседними ионами. Эти силы
возникают из-за электрон-электронного отталкивания и отталкивания между
ядрами.
Зависимость внутренней энергии, связанной с силами Борна, от
межъядерного расстояния определяется уравнением
Д£/(0 К) (Борн) ос _L (6.32)
где г— межъядерное расстояние, п — показатель Борна.
^- Величина п зависит от электронной конфигурации ионов в решетке; эти
К+, СГ изоэлектронны Аг; значения перечислены в табл. 6.8. Например, для хлорида
Na+ изоэлектронен Ne; калия, состоящего из ионов, являющихся электронными
СГ изоэлектронен Аг. аналогами аргона, показатель Борна равен 9. Для хлорида
натрия, который содержит ионы, являющиеся
электронными аналогами неона и аргона, п = (| + §) = 8.
Уравнение Борна-Ланде
Уравнение 6.31 можно уточнить путем учета энергии борновского
отталкивания:
Энергия решетки = А 1/(0 К) = Ina'А ' \z+\\z-\e \ Л М (6 33)
V 4 • я • £q • г /V п 1
Уравнение 6.33 можно использовать, если известны формула соединения,
межъядерное расстояние и структурный тип решетки. Величины г и п можно
получить из экспериментов по дифракции рентгеновских лучей. Константа
6.15. Энергия решетки: чисто ионная модель
305
Дополнение 6.4. Определение величины константы Маделунга
Константа Маделунга учитывает кулоновские силы притяжения и отталкивания,
действующие на некоторый ион в кристаллической решетке.
Рассмотрим решетку NaCl, показанную на рисунке.
В разд. 6.8 показано, что каждый ион Na+ и каждый ион
С1~ имеют КЧ 6. Эта величина указывает число
ближайших ионов, несущих заряды противоположного знака.
Как видно из схемы, центральный ион С1~ окружен
шестью ионами Na+, каждый из которых находится на
расстоянии г, соответствующем межъядерному расстоянию,
измеренному с помощью метода дифракции
рентгеновских лучей (281 пм). Это верно для всех хлорид-ионов в
решетке, поскольку все они занимают эквивалентные
позиции. Аналогично, каждый ион Na+ окружен шестью
ионами С1~, каждый из которых находится на
расстоянии г = 281 пм от него.
Рассмотрим теперь, какие еще ионы находятся в
непосредственной близости от центрального хлорид-иона. Во-первых, это 12 ионов СГ,
расположенных на расстоянии а от центрального иона; между ними и центральным ионом
действуют силы отталкивания. Все 12 ионов С1~ показаны на рисунке, хотя расстояние а
отмечено только для трех из них. Величина а зависит от г следующим образом:
а2 = р. + ft = 2,2
а = ЫТ
Далее, имеется восемь ионов Na+, каждый из которых находится на расстоянии Ь от
центрального хлорид-иона; между ними и центральным ионом действуют силы притяжения.
Величина b связана с г по уравнению:
Ь2 = г2 + а2
b2 = i2 + (пП)2
b = ЫТ
Имеется также взаимодействие (как притяжение, так и отталкивание) с более удаленными
ионами, однако кулоновские взаимодействия уменьшаются с ростом соответствующих
межъядерных расстояний. Следует помнить, что энергия кулоновского взаимодействия
обратно пропорциональна расстоянию между точечными зарядами (см. уравнение 6.17).
Константа Маделунга А учитывает все силы притяжения и отталкивания, действующие
на центральный ион. Для решетки NaCl значение константы Маделунга определяется
выражением
1 ч м /о . 1
/4 = 6-(12
) + (8
)-•
где ряд может быть продолжен добавлением членов, соответствующих взаимодействиям на
больших расстояниях. Следует отметить, что значение г не входит в это выражение. Таким
образом, величина Л, рассчитанная по данному уравнению, будет верна для всех ионных
решеток со структурным типом NaCl, в то время как г меняется в зависимости от природы
присутствующих ионов (см. табл. 6.2). Это связано с тем, что отношение расстояний г: a: b
всегда равно 1 : VT: VT. Величины, обратные этим, возникают в выражении для константы
Маделунга потому, что энергия кулоновского взаимодействия обратно пропорциональна
расстоянию между точечными зарядами, как это уже указывалось выше.
Учет всех членов ряда дает величину А = 1,7476 для решетки типа NaCl.
Задание: Выше приведены только три первых члена ряда для расчета константы
Маделунга решетки типа NaCl. Каковы будут два следующих члена этого ряда?
306
6. Ионы
Таблица 6.8. Значения показателя Борна п ионных соединений XY для различных
электронных конфигураций [X+][Y~]. Величины п являются безразмерными.
Значения п для ионных соединений, содержащих ионы с различными электронными
конфигурациями, равны усредненному значению п для компонентов. Например, п = 7 для
NaF, но для LiF п = § + \ =6.
Электронная конфигурация
ионов в ионном соединении XY
Примеры ионов
[Не][Не]
[Ne][Ne]
[Аг][Аг] или [3<У10][Аг]
[Кг][Кг] или [4rf10][Kr]
[Хе][Хе] или [5</10][Хе]
Н", Li+
F", О2", Na+, Mg2+
CI", S2-, K+, Ca2+, Cu+
Br-, Rb+, Ba2+, Ag+
I", Cs\ Au+
5
7
9
10
12
Маделунга Л также определяется структурным типом решетки (см. табл. 6.7).
Величину г можно рассчитать по сумме радиусов катиона и аниона
(уравнение 6.28). Вопросы расчета величины г затронуты в упражнении 6.13 в
разд. 6.20.
Энергия решетки ионного соединения есть изменение внутренней
энергии, происходящее при образовании одного моля твердой фазы
из составляющих газообразных ионов при температуре 0 К.
В рамках модели ионной решетки, состоящей из
сферически-симметричных ионов, величину энергии ионной решетки можно оценить с
помощью уравнения Борна-Ланде:
k+||z_|-e2
(N
—
4 • п ■ еп
К'-Чг)
Пример 6.3. Энергия решетки
Оцените изменение внутренней энергии при образовании 1 моль
CsCI из газообразных ионов при выполнении электростатической
модели твердофазных решеток.
Решение
Необходимые данные: Л/А = 6,022 • 1023 моль"1, А = 1,7627,
е = 1,602 • 10~19 Кл, е0 = 8,854 • Ю-12 Ф/м, показатель Борна для
CsCI п = 10,5, межъядерное расстояние г = 356 пм.
Изменение внутренней энергии (энергии решетки) задается уравнением
Борна-Ланде:
дадк) = (^_Ш^)(,__и
\ 4/г-£0г / \ п /
Величина г должна измеряться в метрах: 356 пм = 3,56 ■ 10~10 м.
6.16. Энергия решетки: экспериментальные данные 307
Д£/(0 К)
6,022 • 1023 • 1,7627 • 1 • 1 • (1,602 • 10"19)2
4 -3,142 -8,854- 1012 • 3,56 • КГ10
2,724- 10 14
V3,961 10 20
= - 622 373 Дж/моль
= — 622 кДж/моль.
-) • 0,905
10,5 J
ЭНЕРГИЯ РЕШЕТКИ: ЭКСПЕРИМЕНТАЛЬНЫЕ ДАННЫЕ
Как можно видеть, чисто ионная модель может быть использована для
оценки энергии решетки. В данном разделе показано, каким образом можно
экспериментально определить эту величину.
Использование закона Гесса: цикл Борна-Габера
^ Энергии решетки не могут быть измерены напрямую: это следует из опреде-
Закон Гесса: ления, данного в разд. 6.15. Поэтому для их определения ис-
см. разд. 1.17 пользуют закон Гесса. Например, хотя энергия решетки
хлорида калия есть изменение внутренней энергии в реакции 6.34
при 0 К, можно определить энтальпию решетки (А /Г) как изменение
энтальпии этой реакции в стандартных условиях. Согласно закону Гесса, Ареш//°
определяется уравнением 6.35.
К+(газ) + СГ(газ) = КС1(тв.)
(6.34)
Ареш^(КС1' ТВ) = АобрЯ°(КС1' ТВ.) " Аобр"°(К+' газ> " ДобрЯЧСГ, газ> <6-35)
В правой части уравнения 6.35 присутствует стандартная энтальпия образо-
^ вания КО, которую можно найти в справочной литературе (как
Значения ДобрН°: и цдя большинства других неорганических соединений).
Станем, т. 2, приложение 8 дарТНые энтальпии образования газообразных ионов можно
определить по закону Гесса (уравнения 6.36 и 6.37).
К(тв.)
Добр"°
*К+(газ)
Ао.Я(
АН * /£,
К(газ)
ДобрЯ° <К+' Га3> = Дат^0 (К>газ> + ,Е\ (К'га3>
*С12(газ);
Добр"°
■С1-(газ)
(6.36)
АН=0
С1(газ)
АобрЯ° (С1~* Га3) = iD <С12' Га3> + АЕАЯ (С1' Га3>
(6.37)
308
6. Ионы
Рис. 6.25. Цикл Борна-
Габера для хлорида калия.
ДатЯ°(К) + ±/)(С12)
К(тв.) + £С12(газ) ► к(газ) + С1(газ)
АобР^°
КС1(тв.)
Дреш"°
/£,
AvaH
К+(газ) + С1 (газ)
где ДЕА#- изменение энтальпии, связанное с присоединением электрона.
^ Значения стандартных энтальпий атомизации Аат//\ энергии диссоциа-
Д НГ и энтальпии дис- ции связей и энергии ионизации хорошо известны для боль-
социации связей:
см. разд. 3.6
шинства элементов, однако величины сродства к электрону
измерены с достаточной точностью лишь для небольшого числа
случаев.
Теперь уравнения 6.35—6.37 можно использовать для построения особого
цикла Гесса, известного как цикл Борна-Габера (рис. 6.25), который
позволяет определить величину А /Г (уравнения 6.38 и 6.39). Значение А /У°,
определенное таким образом, представляет собой экспериментальную
величину, поскольку ее вычисляют исходя из экспериментальных данных.
Добр7У°(КС1, тв.) = Дат#°(К, газ) + £ДС12, газ) + /Я,(К, газ)
+ ДедЯСС!, газ) + Д Д°(КС1, тв.)
(6.38)
Уравнение 6.38 можно переписать, чтобы получить выражение для энтальпии
решетки:
Дреш/Г(КС1, тв.) = +Добр/Г(КС1, тв.) - Дат/Г(К, газ) - £«С12, газ)
- /^(К, газ) - ЬЕАЩС1 газ) (6.39)
Как уже было показано ранее, значения энергий ионизации и энергий
сродства к электрону являются изменениями внутренней энергии, но могут
быть заменены изменениями энтальпии, поскольку возникающая при этом
ошибка относительно мала. Аналогично, А /У° ~ А6/(0 К).
Таким образом, энергию решетки КС1 можно определить по уравнению
6.39 с использованием экспериментальных значений Аоб //°(КС1, тв.) =
-437 кДж/моль, Аат/Г(К, газ) = 90 кДж/моль, ДС12, газ) = 242 кДж/моль,
/ЕХ(К, газ) = 419 кДж/моль, ДЕА#(С1, газ) = -349 кДж/моль следующим
образом:
Д Щ0 К) = Др^ЖКС!, тв.) = -437 ■
90 - 121 - 419 + 349 = -718 кДж/моль (6.40)
СРАВНЕНИЕ ЗНАЧЕНИЙ ЭНЕРГИИ РЕШЕТКИ, ОПРЕДЕЛЕННЫХ
ПО УРАВНЕНИЮ БОРНА-ЛАНДЕ И ПО ЦИКЛУ БОРНА-ГАБЕРА
В табл. 6.9 приведены значения энергии решетки некоторых ионных
соединений, рассчитанных по уравнению Борна-Ланде (Д£/(0 К) (Борна-Ланде))
и определенных с помощью цикла Борна-Габера (~Д£/(0 К) (Борна-Габера)).
Соединения сгруппированы в таблице по периодическому принципу и вклю-
6.17. Сравнение значений энергии решетки
309
Таблица 6.9. Значения энергии решетки для некоторых соединений, определенные
по уравнению Борна-Ланде (6.33) и по циклу Борна-Габера (рис. 6.25 и уравнение
6.39). Расчеты по циклу Борна-Габера дают величину А //°, которая приближенно
равна Д£/(0 К); величины в данной таблице рассчитаны из термохимических данных,
приведенных в соответствующих таблицах в этой книге
Соединение
NaF
NaCl
NaBr
Nal
KF
KC1
KBr
KI
AgF
AgCl
AgBr
Agl
Определяете
AU(0 К) ^A^Ji AU(OK) no
no циклу
Борна-Габера,
кДж/моль
-931
-786
-736
-671
-827
-718
-675
-617
-972
-915
-888
-858
О I^QV ATUAIIIPULIA -
и K.dK uiношение
уравнению
Борна—Ланде,
кДж/моль
-901
-756
-719
-672
-798
-687
-660
-622
-871
-784
-758
-737
ДЦО Ю(Борна-Габера) -
[АЩОК)
(Борна-Габера)
-АЩОК)
(Борна—Ланде)],
кДж/моль
^30
-30
-17
+ 1
-29
-31
-15
+5
-101
-131
-130
-121
Различие между
значениями
АЩОК), %а
й
3,8
2,3
«0
3,5
4,3
2,2
0,8
10,4
14,3
14,6
14,1
- Д£/(0 К) (Борна-Ланде)
Л6/(0 К) (Борна-Габера)
чают галогениды двух металлов группы l(Na и К) и одного металла группы 11
(Ag). За исключением иодида серебра(1), все галогениды, приведенные в
таблице, имеют структуру типа NaCl; Agl имеет при комнатной температуре и
давлении 1 атм структуру вюрцита.
Различие между значениями Д£/(0 К) (Борна-Габера) и Д£/(0 К)
(Борна-Ланде) для каждого из соединений приведено в процентах относительно
экспериментального значения (см. последнюю колонку табл. 6.9). Для галоге-
нидов натрия и калия экспериментальные и теоретически предсказанные
величины энергии решетки хорошо согласуются друг с другом, и это
свидетельствует о том, что модель дискретных ионов, используемая в уравнении
Борна-Ланде, является хорошим приближением для указанных соединений*.
Для всех галогенидов серебра расчеты по уравнению Борна-Ланде дают
величину энергии решетки, заниженную более чем на 10%. Это означает, что
модель дискретных ионов не является хорошим приближением для
галогенидов серебра(1).
В разд. 6.1 уже упоминалось, что ионная модель может быть улучшена
введением представлений о частичном ковалентном связывании, аналогично
тому, как модель ковалентных связей можно уточнить, принимая в расчет
Использование уравнения Борна-Ланде не единственный метод расчета энергии решетки.
Возможно использование более сложных уравнений, которые позволяют получать более точные результаты.
Однако общие тенденции, отмеченные в табл. 6.9, остаются неизменными. Более детальное
обсуждение можно найти в книге Dasset W.E. , Inorganic Energetics, 2nd edn, Cambridge University Press,
Cambridge (1984). [См. также Джонсон Д. Термодинамические аспекты неорганической химии, М.:
Мир, 1985. - Ред.]
310
6. Ионы
вклад от ионного взаимодействия. Именно с этим связаны неточности,
возникающие при расчете энергии решетки для галогенидов серебра(1).
ЁВШ поляризация ионов
Заканчивая данную главу, вернемся к обсуждению предположения о
сферичности ионов. Всегда ли оно верно?
Рассмотрим ион малого размера, такой, как Li+, и предположим, что он
является сферическим. Так как площадь поверхности иона Li+ мала,
поверхностная плотность заряда оказывается относительно большой:
Заряд иона
Поверхностная плотность заряда иона = — (6.41)
Площадь поверхности иона
Теперь рассмотрим большой анион, такой, как 1~, имеющий малую
поверхностную плотность заряда. Если иодид-ион находится вблизи катиона с
высокой поверхностной плотностью заряда, распределение зарядов может
искажаться и уже не может рассматриваться как сферически-симметричное.
Искажение означает, что «центр тяжести» электронной плотности в иодид-
анионе не совпадает с положением ядра, в результате чего возникает д и пол ь-
^ ный момент, индуцированный соседним малым катионом. Электронная
Индуцированный плотность оказывается смещена в направлении катиона. Ио-
(наведенный) диполь: дид-анион является легко поляризуемым. Катион, вызывающий
см. разд. 2.21 поляризацию, называют поляризующим катионом.
„ Заряд иона
Плотность заряда иона =
Площадь поверхности иона
Малые катионы, такие, как Li+, Mg2+, Al3+, Fe3+ и Ti4+, обладают высокой
поверхностной плотностью заряда и оказывают сильное поляризующее
воздействие.
Анионы, такие, как I~, Н~, N3~, Se2~ и Р3~, легко поляризуемы. При
движении вниз по группе периодической системы, например от фтора к иоду,
заряд анионов остается постоянным, а размер ионов увеличивается, что
приводит к увеличению поляризуемости ионов.
Когда ион (как правило, катион) индуцирует диполь в соседнем ионе (как
правило, анионе), возникают ион-дипольные взаимодействия.
ЗАКЛЮЧЕНИЕ
В этой главе рассмотрены образование катионов и анионов из газообразных атомов и
взаимодействие между ионами в твердой фазе.
Что означают следующие термины?
Карта электронной
плотности
Энергия ионизации
Сродство к электрону
Ионная пара
Кулоновское
взаимодействие
Ионная решетка
Элементарная ячейка
Координационное число
(в ионном соединении)
Ионный радиус
Энергия решетки
Константа Маделунга
Показатель Борна
Уравнение Борна-Ланде
Цикл Борна-Габера
Поверхностная плотность
заряда иона
Теперь вы должны уметь:
• обсудить взаимосвязь между электронным
строением атома и ходом изменения его
последовательных потенциалов
ионизации;
• описать структуры хлорида натрия, хлорида
цезия, флюорита, рутила и сульфида
цинка(П);
• определять стехиометрический состав
ионного соединения по строению
элементарной ячейки его твердофазной решетки;
• записать уравнение Борна-Ланде и знать,
как его использовать для оценки энергий
решеток;
оценить верность допущении, использова-
ных в модели Борна-Ланде;
составить цикл Борна-Габера и определять
с его помощью энергию решетки
соединения, а также использовать цикл
Борна-Габера для расчета других
термодинамических величин, таких, как энергия сродства к
электрону, при известном значении
энергии решетки;
обсудить вопрос, почему величины энергии
ионных решеток, рассчитанные с помощью
уравнения Борна-Ланде, согласуются или
не согласуются с экспериментальными
значениями.
ЗИЛ УПРАЖНЕНИЯ
6.1. На примере атома кислорода
объясните, что вы понимаете под а)
первой энергией ионизации; б) второй
энергией ионизации; в) первой
энергией сродства к электрону;
г) второй энергией сродства к
электрону; д) изменением энтальпии при
присоединении электрона.
6.2. Первые пять энергий ионизации
атома X равны 589, 1148, 4911, 6494 и
8153 кДж/моль. В какой группе
периодической системы находится
этот элемент?
6.3. Первые энергии ионизации
кислорода (Z= 8), фтора (Z= 9), неона
(Z= 10) и натрия (Z= 11) равны
1312, 1681, 2080 и 495 кДж/моль
соответственно. Объясните ход
изменения энергии ионизации в этом
ряду на основании электронного
строения атомов.
6.4. Рассчитайте изменение энтальпии,
происходящее в ходе процесса
^Li2(ra3) -» 1л+(газ) + е~
если энтальпия диссоциации дили-
тия равна 110 кДж/моль, а величина
первой энергии ионизации 1ЕХ для
атома лития равна 520 кДж/моль.
6.5. Рассчитайте изменение энтальпии,
происходящее в ходе процесса
^Li2(ra3) + е~ -> Ы~(газ)
если энтальпия диссоциации дили-
тия равна 110 кДж/моль, а величина
ЕАХ для атома лития равна
60 кДж/моль.
312
6. Ионы
6.6. Рассчитайте изменение энтальпии
для реакций (в газовой фазе)
H* + F#->H+ + F- (1)
H*+F*->H- + F+ (2)
и объясните результаты с
использованием представлений о
резонансных структурах в разд. 4.3.
(Необходимые данные см. в табл. 6.1 и
приложениях 5 и 6 в т. 2.)
6.7. С использованием данных табл. 6.1
определите изменение энтальпии,
происходящее при образовании
газообразного иона S2- из
газообразного атома серы. Объясните знак и
величину полученного результата
исходя из электронного строения
атома серы, а также ионов S~ и S2-.
6.8. Укажите различие между терминами
вандерваальсов радиус, ковалентный
радиус и ионный радиус. Как можно
оценить ионный радиус катиона
натрия? Укажите, какая
дополнительная информация вам потребуется и
какие допущения необходимо
сделать.
6.9. Докажите, что уравнение 6.19
корректно с точки зрения размерности
входящих в него величин.
(Подсказка: Си. табл. 1.2.)
6.10. На рис. 6.11 показана элементарная
ячейка решетки хлорида натрия.
Почему его элементарная ячейка не
может быть меньшего размера,
например, куб, содержащий только четыре
ионные пары?
6.11. Выведите уравнения 6.20 и 6.21
исходя из позиций, занимаемых
ионами в элементарной ячейке фторида
кальция.
6.12. Почему межъядерное расстояние
между соседними ионами Na+ и С1~
в газообразном NaCl (236 пм)
короче, чем в твердофазном соединении
(281 пм)?
6.13. а) Изобразите строение
сульфат-иона S042~ и тетрафтороборат-иона
BF4~ и объясните, почему эти
частицы можно приближенно считать
сферическими, б) Предложите
способ расчета радиусов ионов S042~ и
BF4~, исходя из энергий
кристаллических решеток сульфата и тетраф-
торбората натрия. Если для решения
этой задачи потребуется введение
каких-либо приближений, укажите
их. Какая дополнительная
информация вам потребуется?
6.14. Определите показатели Борна для
следующих соединений с
использованием данных табл. 6.8: a) NaCl;
б) LiCl; в) AgF; г) MgO.
6.15. В примере 6.3 (см. разд. 6.15) энергия
решетки CsCl рассчитана с
использованием экспериментально
измеренного межъядерного расстояния
356 пм. Рассчитайте энергию
решетки хлорида цезия с использованием
значений ионных радиусов гион
Cs+ (170 пм) и С1" (181 пм).
Объясните различие между двумя
значениями Д£/(0 К).
6.16. Рассчитайте энергию решетки оксида
магния двумя способами: с
использованием уравнения Борна—Ланде и
по циклу Борна-Габера. Решетка
MgO относится к структурному типу
NaCl. Все необходимые данные для
расчета по методу Борна—Ланде
имеются в гл. 6; термохимические
данные см. в приложениях 5-8 в т. 2.
С использованием полученных
данных прокомментируйте адекватность
приближения сферических ионов
для решетки MgO.
СТРУКТУРА ТВЕРДЫХ ПРОСТЫХ
ВЕЩЕСТВ
ВВЕДЕНИЕ
В разд. 2.21 рассмотрены вандерваальсовы силы, действующие между
атомами инертных газов, и показано, что слабость этих взаимодействий
проявляется в некоторых физических свойствах инертных газов (табл. 2.6). Также
отмечалось, что эти элементы переходят в твердое состояние только при очень
низких температурах, а твердый гелий может быть получен лишь при
повышенных давлениях. Когда элементы группы 18 переходят в твердое
состояние, их атомы образуют упорядоченную решетку с плотнейшей упаковкой.
Перед тем как рассмотреть особенности строения инертых газов в твердом
состоянии, необходимо обсудить значение понятия плотнейшая шаровая
упаковка. Оно, вероятно, уже знакомо некоторым студентам; краткий обзор этой
темы дан в разд. 7.2. В разд. 7.3 рассмотрены простая и объемноцентрирован-
ная кубические упаковки.
В данной главе обсуждаются не только твердые простые вещества с плот-
нейшими упаковками атомов, но и простые вещества, состоящие из молекул
различного размера или имеющие в твердом состоянии
бесконечно-протяженные ковалентные решетки.
ПЛОТНЕЙШИЕ ШАРОВЫЕ УПАКОВКИ
Гексагональная и кубическая плотнейшие упаковки
Предположим, что имеется прямоугольный ящик, в котором необходимо
разместить шары одинакового размера. При условии упорядоченного
расположения наиболее эффективным способом размещения первого слоя шаров на
дне ящика будет способ, показанный на рис. 7.1. Это часть плотнейшей
упаковки, в которой каждый шар, находящийся в окружении других шаров,
контактирует с шестью шарами в пределах данного слоя. На рис. 7.1 особо
отмечен гексагональный мотив упаковки.
Добавление следующего слоя плотноупакованных шаров проводится
таким образом, чтобы шары второго слоя попадали в пустоты между шарами
первого слоя, при этом может быть заполнена лишь половина пустот
(рис. 7.2). Когда добавляется третий слой, шары снова заполняют только
половину пустот второго слоя, но теперь для них имеется два различающихся
набора пустот. В одном из них пустоты расположены непосредственно над
шарами первого слоя; в верхней части рис. 7.3 они обозначены буквой А. Пус-
314
7. Структура твердых простых веществ
Рис. 7.1. Слой одинаковых шаров
размещается упорядочение на дне прямоугольного
ящика. Наиболее эффективным способом
размещения является плотнейшая упаковка.
Линиями на рисунке показан
повторяющийся гексагональный мотив упаковки.
тоты, обозначенные буквой С, располагаются над пустотами первого слоя,
которые не были заняты шарами второго слоя. В зависимости от того, какой
из двух наборов пустот будет занят третьим слоем шаров, реализуется одна из
двух плотнейших шаровых упаковок.
В плотнейшей шаровой упаковке реализуется наиболее эффективное
заполнение пространства.
f
Рис. 7.2. Если три шара расположены в вершинах треугольника и касаются друг
друга, в центре треугольника возникает пустота. Слой плотноупакованных шаров
образует упорядоченную структуру таких пустот, а — между семью плотноупакованными
шарами имеется шесть пустот. Если шары второго слоя располагаются над пустотами
первого слоя, то окажется, что по стерическим причинам возможно заполнение
только половины пустот; сферы второго слоя помещаются в каждую из пустот,
помеченных на рисунке точкой; б- вид сбоку иллюстрирует, каким образом производится
размещение второго слоя над первым.
7.2. Плотнейшие шаровые упаковки
315
V
щ
1 Первый слой
==л Второй слой
Шары 3-го слоя помещаются в
лунки над пустотами типа А
Шары 3-го слоя помещаются в
лунки над пустотами типа С
1 Слой А Слой С
> Слой В Слой В
<=> Слой А Слой А
Рис. 7.3. Второй слой плотнейшей шаровой упаковки имеет пустоты двух типов: А -
непосредственно над шарами первого слоя и С - над пустотами первого слоя. Таким образом, имеется две
возможности размещения шаров третьего слоя. Возможные последовательности заполнения
показаны на рисунке в проекции сверху (а, б) и сбоку (в, г): ABA (я), ABC (б), ABA (в), ABC (г).
Последовательность слоев ABC, изображенная на рис. г, образует плоскость с квадратичным
мотивом расположения шаров.
На рис. 7.3,д шары третьего слоя располагаются непосредственно над
шарами первого слоя; это дополнительно иллюстрируется видом сбоку,
показанным на рис. 7.3,в. Слои разного типа обозначаются буквами А и В;
размещение последующих слоев описанным выше способом можно выразить
записью АВАВАВ...
316
7. Структура твердых простых веществ
б
Рис. 7.4. Каждый шар в плотнейшей упаковке имеет 12 ближайших соседей. Это выполняется
для обоих способов расположения слоев: ABA (а) и ABC (б).
На рис. 7.3,6и 7.3,г показан случай, когда шары третьего шарового слоя не
располагаются непосредственно над шарами первого. Эти слои
обозначаются А, В и С, и дальнейшее послойное заполнение можно выразить записью
АВСАВС...
В обоих рассмотренных плотнейших упаковках каждый шар находится в
контакте с шестью шарами в пределах собственного слоя, а также с тремя
шарами нижнего и тремя шарами верхнего соседних слоев. Таким образом,
каждый шар имеет 12 ближайших соседей, т. е. КЧ 12. На рис. 7А,а и 7.4,5
показано ближайшее окружение шаров для упаковок типа ABA и ABC
соответственно. Отметим, что различие между двумя типами упаковок заключается
лишь во взаимной ориентации треугольников, образованных шарами
верхнего и нижнего слоев, показанных на рисунке.
Расположение слоев типа АВАВАВ... называют гексагональной плотней-
^ шей упаковкой шаров (ГПУ), а расположение типа АВСАВС... известно как
кубическая плотнейшая упаковка шаров (КПУ).
Соответствующие элементарные ячейки показаны на рис. 7.5.
Элементарная ячейка, показанная на рис. 7.5,д, имеет
форму куба, откуда и происходит название данного типа
плотнейшей шаровой упаковки. Она содержит восемь шаров в вершинах куба и шесть
шаров в центре граней. Другое название кубической плотнейшей упаковки -
гранецентрированная кубическая (ГЦК) структура.
Легко видеть, как элементарная ячейка, показанная на рис. 7.5,5,
возникает при размещении слоев типа АВАВАВ..., изображеных на рис. 7.3.
Гораздо сложнее представить себе соответствие между элементарной ячейкой на
рис. 7.5,д и расположением слоев типа АВСАВС... На рис. 7.6,а показаны три
соседние ГЦК-ячейки. Если эту структуру повернуть на 45°, расположение
слоев видно более наглядно.
В правой части рис. 7.6,я шары слоя одного типа окрашены в одинаковый
цвет. Та же структура изображена на рис. 7.6,6 под другим углом зрения,
чтобы было видно взаимное расположение слоев А, В и С. Для наглядности,
рекомендуется использовать трехмерные модели или графические
компьютерные программы.
Элементарная ячейка:
см. разд. 6.7
7.2. Плотнейшие шаровые упаковки 317
Рис. 7.5. Плотнейшие шаровые упаковки типа АВСАВС... и АВАВАВ... называются
кубической (а) и гексагональной (б) плотнейшими упаковками соответственно. Если «уменьшить»
размеры шаров, кубическая (в) и гексагональная (г) элементарные ячейки видны более
наглядно. Кубическая плотнейшая упаковка также называется кубической гранецентрированной
решеткой. Можно видеть, что в центре каждой фани кубической ячейки, показанной на рис. а и
в, располагается шар.
Шары одинакового размера можно разместить в пространстве с
образованием плотнейшей упаковки как минимум двумя способами.
В гексагональной плотнейшей упаковке (ГПУ) шаровые слои
располагаются по типу АВАВАВ...
В кубической плотнейшей упаковке (КПУ), которую также называют
гранецентрированной кубической структурой (ГЦК), шаровые слои
располагаются по типу АВСАВС...
Как в ГПУ, так и в КПУ каждый шар имеет 12 ближайших соседей.
318
7. Структура твердых простых веществ
Поворот на 45°
эквивалентно
о о
ХУ
Слой С
Слой В
Слой А
Слой С
Слой В
о о
Рис. 7.6. а - связь между ГЦК-структурой (на рисунке показаны три элементарные
ячейки) и последовательностью АВСАВС... слоев плотноупакованных шаров можно
легко увидеть, повернув кубические ячейки на 45°; б — последняя структура на рис. а
перерисована таким образом, чтобы последовательность слоев АВСАВС... была видна
более наглядно.
Внутренние пустоты
Если шар разместить над центром треугольника, образованного тремя
другими шарами, образуется тетраэдр (рис. 7.7,а), внутри которого имеется
свободное пространство — тетраэдрическая пустота.
В плотноупакованной структуре появляется еще один вид пустот - окта-
эдрические (рис. 7.7,6). Оба типа внутренних пустот присутствуют в
элементарной ячейке КПУ, показанной на рис. 7.5,а. Дальнейшее обсуждение
внутренних пустот приводится в дополнении 7.1.
Не вполне очевидно, почему октаэдрические пустоты присутствуют
только в плотноупакованной структуре, однако из рис. 7.3 и 7.6 можно
заметить, что трехслойная последовательность в КПУ-структуре порождает
плоскости с квадратичным мотивом расположения шаров. Например, левая
боковая сторона фигуры, изображенной на рис. 7.3,г, образована именно
такой плоскостью, такие же плоскости образуют передние стороны двух
структур, показанных в правой части рис. 7.6,а. Размещение шаров сверху и
снизу над центром квадрата приводит к формированию октаэдрической
пустоты (рис. 7.7,6). Для того чтобы проверить и наглядно представить
вышеупомянутые утверждения, рекомендуется использовать трехмерные модели
КПУ-структуры.
7.2. Плотнейшие шаровые упаковки
319
Дополнение 7.1. Альтернативный подход к описанию
ионных решеток
Ниже показано гранецентрированное кубическое (ГЦК) расположение шаров (кубическая
плотнейшая упаковка, КПУ). На левом рисунке показана октаэдрическая пустота,
расположенная в центре ГЦК-элементарной ячейки. На правом рисунке изображена одна из тетра-
эдрических пустот; в одной ГЦК-элементарной ячейке имеется восемь таких пустот.
Для упаковки, составленной из сфер одинакового размера, внутренние пустоты
остаются незаполненными.
Ионная решетка состоит из сфер (ионов), которые имеют различные размеры. В
большинстве случаев (например, для NaCl) размеры анионов превышают размеры катионов,
поэтому удобно рассматривать решетку как плотнейшую упаковку анионов, содержащую
катионы во внутренних пустотах.
Рассмотрим кубическую плотнейшую упаковку сульфид-ионов. Сама по себе такая
упаковка была бы неустойчива, поскольку между анионами действуют силы отталкивания, но
если в некоторых внутренних пустотах разместить катионы цинка, можно получить ионную
решетку, стабилизированную за счет сил притяжения между катионами и анионами, как в
элементарной ячейке цинковой обманки (ZnS):
Ионы S2- образуют ГЦК-упаковку. Половина тетраэдринеских пустот занята ионами
Zn2+. Катионы располагаются таким образом, что заполненные тетраэдрические пустоты
чередуются с незаполненными.
Такое описание структуры цинковой обманки предполагает, что катионы достаточно
малы, чтобы разместиться во внутренних пустотах между большими анионами,
образующими плотнейшую упаковку. Заметим, что при таком описании в вершинах элементароной
ячейки располагаются анионы; сравните это с изображением элементарной ячейки ZnS на
рис. 6.17, где в вершинах элементарной ячейки расположены ионы цинка. Позволяют ли
эти два способа изображения элементарной ячейки получать эквивалентную информацию
о структуре решетки в целом?
Аналогичным образом можно описать структуры NaCl и CaF2 в терминах катионов,
занимающих пустоты в плотнейшей упаковке анионов; эти случаи рассмотрены в конце главы в разд. 7.16.
7. Структура твердых простых веществ
Рис. 7.7. а — тетраэдрическая пустота, присутствующая как в ГПУ, так и в КПУ,
образуется при помещении шара в выемку над тремя плотноупакованными шарами;
б - октаэдрическая пустота образуется, когда два шара располагаются сверху и снизу
квадрата, образованного четырьмя шарами; такое взаимное расположение шаров
встречается в КПУ и ГПУ.
В плотноупакованных шаровых структурах присутствуют как октаэд-
рические, так и тетраэдрические внутренние пустоты.
ПРОСТАЯ И ОБЪЕМНОЦЕНТРИРОВАННАЯ
КУБИЧЕСКИЕ ШАРОВЫЕ УПАКОВКИ
В кубической и гексагональной плотнейших упаковках каждый шар имеет 12
ближайших соседей, причем шары размещены наиболее эффективным
образом, так что объем «пустого пространства» минимален. Существует, однако,
возможность создания упорядоченного размещения одинаковых шаров, при
котором они не будут упакованы плотнейшим образом. Примером такой
структуры является простая кубическая упаковка, показанная на рис. 7.8.
Элементарная ячейка содержит восемь шаров, размещенных в вершинах
куба, при таком расположении каждый шар в бесконечной решетке имеет
шесть ближайших соседей в октаэдрической координации.
7.3. Простая и объемноцентрированная кубические шаровые упаковки 321
Рис. 7.8. а — расположение шаров однакового размера в простой кубической
упаковке; б— элементарная ячейка простой кубической упаковки: шары находятся в
вершинах куба.
Простая кубическая упаковка шаров приводит к упорядоченному
расположению, но при такой упаковке остается значительное количество
неиспользованного пространства. Внутренние («кубические») пустоты слишком
малы, чтобы вместить еще один шар того же размера. Поэтому размещение
еще одного такого же шара в указанной пустоте приводит к тому, что
исходные шары слегка разойдутся в стороны. Результатом будет образование объ-
емноцентрированной кубической (ОЦК) упаковки. Различие между простой
и объемноцентрированной кубическими упаковками видно из сравнения
рис. 7.8,а и 7.9,а. Хотя в случае ОЦК-упаковки пространство используется
более эффективно, чем в случае простой кубической упаковки, данный
способ размещения шаров все равно менее эффективен, чем в кубической или
гексагональной плотнейших упаковках.
Элементарная ячейка ОЦК-решетки показана на рис. 7.9,6;
координационное число для упаковки данного типа равно восьми (рис. 1.9,в).
Повторение кубических элементарных ячеек задает простую
кубическую упаковку, в которой каждый шар имеет КЧ 6.
В объемноцентрированной кубической (ОЦК) упаковке элементарная
ячейка представляет собой куб из восьми не соприкасающихся
шаров, в центре которого находится еще один шар. Каждый шар имеет
КЧ8.
Простая и объемноцентрированная упаковки не являются плотней-
шими упаковками.
322 7. Структура твердых простых веществ
Рис. 7.9. а — объемноцентрированная кубическая (ОЦК) упаковка шаров одинакового
размера; б — элементарная ячейка ОЦК-решетки шаров представляет собой куб, в центре которого
расположен шар; центральный шар касается каждого из шаров в вершинах куба, но шары в
вершинах куба не касаются друг друга; в — каждый из шаров в ОЦК-упаковке имеет восемь
ближайших соседей.
7.5. Кристаллические и аморфные твердые тела 323
БЕЯ СХОДСТВО И РАЗЛИЧИЕ МЕЖДУ ПЛОТНЕЙШИМИ И
НЕПЛОТНЕЙШИМИ УПАКОВКАМИ
Гексагональная плотнейшая упаковка (рис. 7.5,а и 7.5,в)
• Структура может быть представлена как чередование слоев плотноупако-
ванных атомов типа АВАВАВ...
• Каждая сфера имеет 12 ближайших соседей.
• Упаковка содержит тетраэдрические и октаэдрические внутренние пустоты.
Кубическая плотнейшая упаковка (КПУ),
или гранецентрированная кубическая (ГЦК) упаковка
(рис. 7.5,6 и 7.5,г)
• Структура может быть представлена как чередование слоев плотноупако-
ванных атомов типа АВСАВС...
• Каждая сфера имеет 12 ближайших соседей.
• Упаковка содержит тетраэдрические и октаэдрические внутренние пустоты.
Простая кубическая упаковка (рис. 7.8)
• Сферы не являются плотноупакованными.
• Каждая сфера имеет 6 ближайших соседей.
Объемноцентрированная кубическая упаковка
• Сферы не являются плотноупакованными.
• Каждая сфера имеет 8 ближайших соседей.
Относительная эффективность упаковки
Относительная эффективность упаковки сфер одинакового размера
меняется в зависимости от типа упаковки следующим образом:
ГПУ = КПУ (ГЦК) > (ОЦК) > Простая кубическая упаковка
ЕЕЯ КРИСТАЛЛИЧЕСКИЕ И АМОРФНЫЕ ТВЕРДЫЕ ТЕЛА
В кристаллических твердых телах атомы, молекулы или ионы располагаются
упорядоченным образом, так что некоторая элементарная ячейка
повторяется во всей кристаллической решетке. Для проведения рентгеноструктурного
эксперимента (см. разд. 3.2), как правило, требуется монокристалл.
Разрушение монокристалла происходит вдоль определенных плоскостей скола. Это
приводит к тому, что кристаллы определенного вещества обладают
характерной формой*.
В подавляющем числе случаев характеристическая форма монокристаллов задается не в процессе их
разрушения, а в процессе их роста из жидкой или газовой фазы, и определяется в первую очередь
различиями в скоростях роста в разных кристаллографических направлениях. Например, в случае
кристаллов, имеющих кубическую элементарную ячейку, наибольшая скорость роста наблюдается в
направлениях, перпендикулярных граням куба, тогда как скорость роста в диагональных
направлениях понижена. В результате монокристаллы таких соединений обычно имеют октаэдрическую
форму.— Прим. перев.
7. Структура твердых простых веществ
В аморфных твердых телах частицы располагаются беспорядочно.
Разрушение аморфных тел приводит к образованию порошков, тогда как при
разрушении кристаллических тел образуются микрокристаллические материалы.
Впрочем, для невооруженного глаза микрокристаллы могут быть
неотличимы от порошков.
В кристаллическом твердом теле атомы, молекулы или ионы
образуют упорядоченную решетку с характерной элементарной ячейкой.
ТВЕРДЫЕ ПРОСТЫЕ ВЕЩЕСТВА,
ОБРАЗОВАННЫЕ ЭЛЕМЕНТАМИ ГРУППЫ 18
Элементы группы 18 называют также благородными газами, и кажется не
вполне обычным их рассмотрение в каком-либо ином агрегатном состоянии,
кроме газообразного. Благородные газы затвердевают при низких
температурах (см. табл. 2.6), причем их молярная энтальпия плавления весьма мала.
Это свидетельствует о слабых вандерваальсовых взаимодействиях между
атомами в твердом состоянии. В кристаллических веществах атомы элементов
группы 18 образуют плотнейшие упаковки. Твердые неон, криптон и ксенон
образуют кубические плотнейшие упаковки, тогда как в случае гелия
реализуется гексагональная плотнейшая упаковка.
Дополнение 7.2. Жидкий гелий - важный хладагент
Хотя при обычных условиях элементы группы 18 встречаются в газообразном
состоянии, другие агрегатные состояния имеют большое практическое
значение. В частности, весьма важным является применение жидкого гелия в
качестве хладагента.
В промышленности и лабораторных исследованиях наиболее часто в
качестве хладагента применяют жидкий азот (т.кип. 77 К). Однако
использование жидкого азота не позволяет достигать сверхнизких температур,
поскольку при сжижении газообразного азота под давлением образуется жидкость с
температурой лишь немногим ниже точки кипения. Чтобы достичь более
низких температур, требуется жидкость с еще более низкой, чем у жидкого
азота, температурой кипения. Для этой цели, как правило, используется
жидкий гелий. Как известно, гелий кипит при 4,2 К, поэтому он дает
возможность достигать температур, близких к абсолютному нулю. Ниже 2,2 К
изотопно-чистый 4Не претерпевает превращение в так называемый гелий-Н. Эта
необычная жидкость обладает почти нулевой вязкостью и
теплопроводностью, которая намного превосходит теплопроводность меди.
До недавнего времени использование жидкого гелия было необходимо
при охлаждении сверхпроводников для перевода их в сверхпроводящее
состояние. Сейчас известны высокотемпературные сверхпроводники (см.
дополнение 7.4).
7.7. Твердые простые вещества, состоящие из двухатомных молекул 325
ШЕШ ТВЕРДЫЕ ПРОСТЫЕ ВЕЩЕСТВА,
СОСТОЯЩИЕ ИЗ ДВУХАТОМНЫХ МОЛЕКУЛ
На рис. 3.2 показаны некоторые гомоядерные молекулы (Н2, 02, 03,I2, Р4> S6
и S8). Каждая из них является молекулярной формой простого вещества (т. е.
вещества, состоящего из атомов одного элемента). В газовой фазе молекулы
отделены друг от друга, но в твердом состоянии они взаимодействуют между
собой за счет вандерваальсовых сил. В этом и двух последующих разделах мы
рассмотрим строение твердых водорода и простых веществ, образуемых
элементами групп 16 и 17, одну аллотропную модификацию фосфора и
аллотропные модификации углерода. Все перечисленные вещества являются
неметаллами, имеющими в твердом состоянии молекулярную структуру.
Водород Н2 и фтор F2
Газообразный Н2 сжижается при охлаждении до 20,4 К*. Дальнейшее
охлаждение до 14,0 К приводит к образованию твердого водорода. Даже при
приближении к температуре абсолютного нуля (0 К) молекулы Н2 имеют
достаточную энергию, чтобы свободно вращаться вокруг некоторой точки в
твердофазной решетке. Как видно из рис. 7.10, из-за вращения каждую
молекулу водорода в решетке можно рассматривать как сферу, центр которой
совпадает с серединой связи Н—Н. В твердом водороде реализуется ГПУ
таких сфер, каждая из которых соответствует молекуле водорода.
Использование модели плотноупакованных сфер возможно только потому, что
молекулы Н2 свободно вращаются при температуре, при которой производилось
определение структуры.
Молекулярный фтор F2 затвердевает при 53 К. Ниже 45 К молекулы F2
претерпевают свободное вращение и структура может быть описана как
слегка искаженная плотнейшая упаковка сфер, в которой каждая сфера
представляет собой молекулу F2**. Энтальпия плавления F2 равна 0,5 кДж/моль, и
столь низкое значение позволяет предположить, что для плавления фтора
необходимо преодолеть только вандерваальсовы силы.
Ситуация, с которой мы встретились в случае кристаллических Н2 и F2,
довольно необычна. В большинстве твердых тел молекулы не могут свободно
вращаться. В действительности наблюдаются некоторые виды теплового
движения, например колебания связей, но координаты атомов в молекулах
могут быть определены с достаточной степенью точности. Это означает, что
модель плотноупакованных сфер не может быть использована для большинства
молекулярных твердых тел, поскольку молекулы, образующие решетку, не
► являются сферическими. Данная модель применима для кри-
Кристаллические сталлов, образованных элементами группы 18 (поскольку они
решетки металлов: моноатомны), для Н2 и F2 (поскольку эти молекулы могут
своем, разд. 7.11 бодно вращаться) и для металлов.
Здесь и далее в этой главе рассматриваются фазовые переходы при атмосферном давлении, если это
не оговорено особо.
** Выше 45 К устойчивой является твердая фаза с более сложной структурой.
326
7. Структура твердых простых веществ
Рис. 7.10. Молекулы Н2 свободно вращаются в твердом состоянии, а - некоторые
возможные ориентации молекулы Н2 относительно середины связи Н—Н; б—
различные ориентации наложены друг на друга, чтобы проиллюстрировать, почему каждая
вращающаяся молекула может рассматриваться как сфера; центр сферы совпадает с
серединой связи Н—Н.
Хлор Cl2, бром Вг2 и иод l2 (группа 17)
Некоторые физические и структурные свойства Cl2, Вг2 и 12 приведены в
табл. 7.1. При 298 К (и давлении 1 атм) 12 — твердое вещество, Вг2 - жидкость,
а С12 - газ. В твердом состоянии их структуры похожи, но отличаются от
структуры твердого F2.
В твердом состоянии молекулы С12 (а также Вг2 или 12) образуют слои
зигзагообразных цепочек (рис. 7.11), расположенные друг над другом. В
структуре имеется три типа характерных расстояний, которые особенно
информативны. Рассмотрим структуру твердого С12. Расстояние между
атомами в молекуле С12 (а на рис. 7.11) равно 198 пм. Эта величина равна
удвоенному ковалентному радиусу хлора (см. табл. 7.1). В пределах слоя можно
также определить межмолекулярные расстояния С1...С1, кратчайшее из
которых равно 332 пм (Ь на рис. 7.11). Это расстояние короче, чем удвоенный
вандерваальсов радус атома хлора; следовательно, имеется химическое
взаимодействие между молекулами хлора внутри слоя. Кратчайшее расстояние
между слоями равно 374 пм. Различие между межмолекулярными
расстояниями в пределах слоев и межслоевыми расстояниями становится еще более
существенным при переходе от С12 к Вг2 и от Вг2 к 12 (см. табл. 7.1). Отметим
7.8. Молекулярные твердые простые вещества, групп 15 и 16 327
Таблица 7.1. Некоторые физические и структурные свойства хлора, брома и иода. (Более
детальное описание строения этих веществ в твердой фазе см. в тексте и на рис. 7.11.)
Элемент ТЛ1, К \^> Ковалент- Вандерва- Межатом- Межмоле- Расстояние Межатом-
кДж/моль ный радиус альсов ра- ное рассто- кулярное между ное рассто-
гков> пм диус гv, пм яние а расстояние слоями, пм яние в газо-
(рис. 7.11), в пределах образной
пм слоя Ь молекуле,
(рис. 7.11), пм
пм
Хл^ i7U5 M 99 180 198 332 374 199
Бром 265,8 10,6 114 195 227 331 399 228
Иод 386,7 15,5 133 215 272 350 427 267
Рис. 7.11. В твердом состоянии молекулы Cl2 (а также Вг2 или 12) образуют слои
зигзагообразных цепочек молекул Х2 (X = С1, Вг или I). Часть одного из слоев показана
на рисунке. Значения межатомных (а) расстояний X—X и межмолекулярных (Ь)
расстояний Х-Х, приведены в табл. 7.1.
также, что длина связи I—I в твердом 12 больше, чем в газообразной
молекуле (табл. 7.1), а длины связей CI—C1 и Br—Вг практически не изменяются при
переходе от газообразных С12 и Вг2 к твердому состоянию. В твердом иоде
межмолекулярное взаимодействие приводит к увеличению расстояния I—I
внутри молекулы 12.
В9 МОЛЕКУЛЯРНЫЕ ТВЕРДЫЕ ПРОСТЫЕ ВЕЩЕСТВА,
ОБРАЗОВАННЫЕ ЭЛЕМЕНТАМИ ГРУПП 15 И 16
Сера (группа 16)
Сера образует ряд циклических и цепочечных структур со связями S—S. В
табл. 3.2 приведены некоторые аллотропные модификации серы. Одна из
модификаций образована молекулами S6, имеющими циклическое строение и
328
7. Структура твердых простых веществ
►- присутствующими в конформации кресла (рис. 7.12). При кри-
Конформации сталлизации кольца S6 образуют весьма эффективную упаков-
«кресло» и «ванна»: ^ так что данная модификация обладает наибольшей плотно-
см. разд. 15.2 стью (2,2 г/см3) среди других аллотропных модификаций серы.
Только вандерваальсовы силы действуют между кольцами.
Кристаллическая ромбическая сера (ос-модификация,
являющаяся стандартной формой* элементной серы) состоит из
циклов S8 (рис. 7.13), связь между которыми осуществляется за
► счет вандерваальсовых взаимодействий. Расстояние S—S в цик-
rKOB(S) = 103nM
ле равно 206 пм, что соответствует одинарным связям. Взаимное
расположение циклов S8 в кристаллическом состоянии показано
на рис. 7.14. Циклы в структуре не располагаются строго один под другим.
Моноклинная сера ф-форма) также состоит из колец S8, но они
расположены менее плотно (1,94 г/см3), чем в ромбической сере (2,07 г/см3). При
нагревании ромбической серы до 368 К происходит реорганизация циклов в
решетке и образуется моноклинная модификация. Однако при быстром нагре-
Конформация молекулы описывает относительное расположение
атомов в пространстве. Две наиболее часто наблюдаемые
конформации шестичленных циклов - кресло и ванна.
кресло ванна
Рис. 7.12. Вид молекулы S6 при рассмотрении с двух различных точек. Одна из
проекций подчеркивает, что молекула имеет конформацию кресла.
* Стандартная аллотропная форма отвечает стандартному состоянию (см. разд. 1.17) вещества, так как
обычно это наиболее термодинамически устойчивая форма, хотя встречаются исключения из этого
правила (например, для фосфора: см. ниже). - Прим. ред.
7.8. Молекулярные твердые простые вещества, групп 15 и 16
329
Рис. 7.13. Вид молекулы S8 при рассмотрении с двух различных точек. Такую форму
цикла часто называют «короной». Такое же строение имеет молекула Se8.
Рис. 7.14. Расположение циклов S8 в кристаллической ромбической сере [Meyer /?.,
Chem. Rev., 76, 367(1976)].
7. Структура твердых простых веществ
вании монокристаллов ромбической серы можно избежать перехода в
моноклинную форму и достичь точки плавления (385 К). Если кристаллизацию
жидкости проводить при 373 К, образуется моноклинная сера, но
полученные кристаллы необходимо быстро охладить до комнатной темературы,
чтобы избежать перехода в ромбическую серу. При комнатной температуре
моноклинная сера превращается в ромбическую модификацию в течение
нескольких недель.
Большая часть аллотропных модификаций серы состоит из циклических
структурных единиц (табл. 3.2), но в ряде случаев в структуре присутствуют
цепи Sx, имеющие различную длину. В каждой цепи атомы серы связаны
одинарными связями S—S и образуют геликоидальную (спиральную) структуру
(рис. 7.15). Важной особенностью геликоида является закрученность. Закру-
ченность может быть право- или левосторонней, и эти две формы не могут
быть совмещены друг с другом при наложении.
Существуют различные формы цепочечной серы (или катена-серы*),
молекулы которой состоят из смеси циклов и цепей, в их число входит и
пластическая сера. Нити пластической серы можно вытянуть из перегретой
расплавленной серы; их строение меняется во времени и при 298 К
пластическая сера постепенно переходит в ромбическую. Существует два хорошо
охарактеризованных примера аллотропных модификаций серы,
содержащих спиральные цепи: нитевидная и ламинарная сера. В структуре
нитевидной серы цепи расположены параллельно друг другу, причем право- и
левосторонние спирали пристутствуют в равном количестве. В ламинарной
сере геликоидальные цепи частично перекрещиваются.
а
otPozPo^fxyi^fP
б
оЯ2р%ЯосРосРоь
Рис. 7.15. Цепочка геликоидальной серы (S^), закрученной в правую (а) или левую
(б) стороны. Обе цепи (а и б) не могут быть совмещены при наложении.
Префикс катена- используется в номенклатуре ИЮПАК для обозначения цепочечных структур.
7.9. Молекулярные аллотропные модификации углерода: С60 331
Селен и теллур (группа 16)
Элементные селен и теллур образуют как циклы, так и геликоидальные цепи.
У селена имеется несколько аллотропных модификаций. Кристаллический
моноклинный красный селен содержит циклы Se8, которые, как и молекулы
S8, имеют форму короны (рис. 7.13). Термодинамически устойчивой формой
элемента является серый (или металлический) селен, кристаллическая стру-
^ ктура которого состоит из геликоидальных цепей Se^. У теллура имеется
Se^ и Те^ - цепи только одна кристаллическая модификация, которая также по-
бесконечной длины строена из геликоидальных цепей Те^. Как в структуре
теллура, так и в структуре серого селена оси цепей располагаются
параллельно друг другу. В плоскости, перпендикулярной этим
осям, атомы образуют гексагональные слои.
Фосфор (группа 15)
Белый фосфор принят в качестве стандартной формы фосфора. Это
уникальная стандартная форма элемента, так как белый фосфор не является
термодинамически стабильным*, а наиболее стабильная форма фосфора —
черный фосфор. О черном и красном фосфоре см. разд. 7.10.
Кристаллический белый фосфор состоит из тетраэдрических молекул Р4
(рис. 7.16). Расстояния между атомами фосфора в молекуле Р4 составляют
221 пм, что соответствует одинарной связи Р-Р (г = 110 пм).
МОЛЕКУЛЯРНЫЕ АЛЛОТРОПНЫЕ
МОДИФИКАЦИИ УГЛЕРОДА: С60
► До недавнего времени, как правило, рассматривались две основные алло-
Алмаз и графит: тропные модификации углерода: алмаз и графит. В середине
см. разд. 7.10 1980-х гг. были открыты новые аллотропные модификации
углерода — фуллерены.
Фуллерены состоят из дискретных молекул. Наиболее широко изучен
фуллерен, состоящий из молекул С60 (рис. 7.17,а). Сферическая оболочка из
60 атомов образована пяти- и шестичленными циклами; все атомы углерода
в молекуле идентичны. Каждый пятичленный цикл соединен с пятью шести-
Рис. 7.16. Белый фосфор состоит из
тетраэдрических молекул Р4. Все расстояния Р-Р в молекуле
одинаковы.
Использование белого фосфора в качестве стандартной формы связано с тем, что более
термодинамически стабильные красная и черная модификации не имеют, в силу особенностей своего
строения, строго определенных физических констант (например, Гкип, 7^ и т. п.). — Прим. перев.
332 7. Структура твердых простых веществ
Дополнение 7.3. Происхождение названия «фуллерены»
Архитерктор Роберт Бакминстер Фуллер разработал необычные сферические
здания, одно из которых было построено на международной выставке ЕХРО-
67 в Монреале. Основным мотивом постройки были повторяющиеся
гексагональные фрагменты, однако их сочленение позволяет получать только пла-
нарные фигуры. Для того чтобы геометрическое тело приобрело кривизну,
необходимую для построения объемной конструкции, необходимо в
определенных местах ввести пятиугольные фрагменты. Структура С60 также
напоминает футбольный мяч, сшитый из пятиугольных (обычно окрашенных в
черный цвет) и шестиугольных (обычно белых) фрагментов. С60 также
называют бакминстерфуллереном или баки-боллом*.
Название «фуллерены» было дано классу аллотропных модификаций
углерода с практически сферическими молекулами, такими, как С60, С70 и С84.
Более подробно об увлекательных событиях, завершившихся открытием
С60, можно прочитать в статьях Харольда В. Крото: [С60: Buckminsterfulleren,
The Celestial Sphere that Fell to Earth. Angewante Chemie, International Edition in
English, 1992, vol. 31, p. Ill] или И.В. Золотухина [Фуллерит — новая форма
углерода. Соросовский образовательный журнал, 1996, JSfe 2, с. 51-56] и В.Ф.
Мастерова [Физические свойства фуллеренов, там же, 1997, № 1, с. 92-94].
В октябре 1996 г., сэр Харольд В. Крото из университета Суссекса
(Великобритания) и профессоры Ричард Смолли и Роберт Керл из университета
Райе (США) были удостоены Нобелевской премии за открытие новой формы
углерода - С60.
* Игра слов: в сочетании bucky-ball, которое можно перевести как «футбольный мяч»,
первое слово имеет тот же корень, что и во втором имени Фуллера—Buckminster. —
Прим. перев.
Рис. 7.17. Молекула одного из фуллеренов - С60. а - молекула С60 состоит из
соединенных пяти- и шестичленных циклов, образованных углеродными атомами, из
которых формируется практически сферическая молекула; б— вид молекулы С60, на
котором отсутствует изображение атомов углерода, иллюстрирует расположение
локализованных одинарных и двойных связей углерод — углерод (ориентация молекулы такая
же, как в случае а).
7.9. Молекулярные аллотропные модификации углерода: С^ 333
Дополнение 7.4. Сверхпроводимость: фуллериды
щелочных металлов М3С60
Щелочные металлы М способны восстанавливать фуллерены с образованием солеоб-
разных фуллеридов [М+]3[С60]3_. При низких температурах эти соединения
становятся сверхпроводниками. Сверхпроводники способны проводить электрический ток без
сопротивления. Температура, при которой материал становится сверхпроводником,
называется его критической температурой (Тс). До 1987 г. не было известно соединений,
которые бы находились в сверхпроводящем состоянии при температуре выше 20 К.
В 1987 г. были открыты высокотемпературные сверхпроводники. Большинство
высокотемпературных сверхпроводников представляет собой смешанные оксиды металлов,
например YBa2Cu408 (ТС = 80 К), YBa2Cu307 (ТС = 95 К), Ва2СаСи2Т1208 (ТС = 110 К),
Ва2Са2Си3Т12О10 (Тс = 128 К)*.
Сверхпроводящие фуллериды М3С60 имеют более простое строение, чем
смешанные оксиды металлов. В их структуре атомы щелочных металлов занимают внутренние
пустоты в плотнейшей упаковке, образованной частицами С60. Поскольку каждый
анион [С60]3~ имеет почти сферическую форму, приближение плотнейшей шаровой
упаковки является вполне обоснованным. В К.3С60 и Rb3C60 частицы [С60]3~ образуют
гранецентрированную кубическую (ГЦК) упаковку.
Если вернуться к дополнению 7.1, можно видеть, что в элементарной ячейке ГЦК-
упаковки имеются одна октаэдрическая пустота и восемь тетраэдрических пустот.
Также присутствуют 12 октаэдрических пустот, каждая из которых принадлежит
одновременно двум соседним элементарным ячейкам. Катионы щелочных металлов в К3С60 и
Rb3C60 полностью занимают все октаэдрические (на схеме черные кружки) и тетраэд-
рические (светлые кружки) пустоты.
Значения Тс для К3С60 и Rb3C60 равны 18 К и 28 К соответственно, а для соединения
Cs3C60, в котором частицы С60 образуют объемноцентрированную кубическую
решетку, Тс = 40 К. Среди известных сегодня представителей семейства фуллеридов
(См. продолжение)
334
7. Структура твердых простых веществ
(Продолжение)
щелочных металлов Cs3C60 имеет наибольшую критическую температуру. [Какой тип
пустот могут занимать ионы Cs+ в ОЦК-решетке?] Na3C60 имеет структуру, сходную со
структурами калиевого и рубидиевого аналогов, но не является сверхпроводником. В
данной области химии фуллеренов ведутся активные исследования с целью
дальнейшего повышения значения Тс.
Серию статей, посвященных различным аспектам сверхпроводимости, можно
найти в журнале Chemistry in Britain, 1994, vol. 30, pp. 722—748. [См. также Успехи химии,
2000, вып. 1,с. 3-40. -Ред.]
* Рекордсменами среди сверхпроводников, обладающими самыми высокими значениями Тс,
являются ртутьсодержащие сверхпроводники, например, HgBa2Ca2Cu308+5 (ГС = 153 К), открытые
сотрудниками Химического факультета МГУ Е.В. Антиповым и С.Н. Путилиным. — Прим. перев.
Рис. 7.18. Часть кристаллической структуры С60 • 4С6Н6. Молекулы С60 образуют
упорядоченную структуру, а молекулы бензола занимают пустоты между ними.
Формула С60 • 4С6Н6 показывает, что один моль С60 кристаллизуется с четырьмя молями
бензола; это соотношение очевидным образом отражено на рисунке.
7.10. Твердые вещества, образующие ковалентные кристаллические решетки 335
членными. В молекуле отсутствуют пятичленные циклы, соединенные друг с
другом.
Расположение соседей вокруг углеродного атома может быть либо
линейным, либо плоскотреугольным, либотетраэдрическим. Несмотря на
кажущуюся сложность, структура С60 соответствует этим ограничениям. Каждый
атом углерода в С60 ковалентно связан с тремя соседними, которые образуют
приближенное плоскотреугольное окружение. Поскольку поверхность С60
достаточно велика, отклонение от планарности в окружении каждого
углеродного центра сравнительно невелико. Связи С—С в молекуле С60 можно
подразделить на две группы: связи при сочленении двух шестичленных
циклов (139 пм) и связи при сочленении пяти- и шестичленного циклов (145 пм).
На рис. 7.17,5— традиционное представление молекулы С60 с
локализованными одинарными и двойными углерод-углеродными связями.
В твердом состоянии при 298 К сферические молекулы С60 образуют
плотноупакованную структуру. Однако большая часть рентгеноструктурных
исследований С60 была проведена не для чистых кристаллических фуллере-
нов, а для сольватированных образцов. Например, С60 растворим в бензоле
(С6Н6), и монокристаллы, выращенные при упаривании растворителя из
бензольного раствора, имеют состав С60- 4С6Н6. На рис. 7.18 показана часть
кристаллической структуры С60 • 4С6Н6. Молекулы С60 расположены
упорядоченным образом, а молекулы бензола занимают пустоты между ними.
Кристаллы, выращенные из раствора, могут содержать кристаллосоль-
ваты, состав которых указывается при записи формулы соединения.
EKQ ТВЕРДЫЕ ВЕЩЕСТВА, ОБРАЗУЮЩИЕ
БЕСКОНЕЧНЫЕ КОВАЛЕНТНЫЕ КРИСТАЛЛИЧЕСКИЕ РЕШЕТКИ
Некоторые неметаллические ^-элементы кристаллизуются с образованием
бесконечных ковалентных решеток. Ниже рассмотрены хорошо известные
примеры таких веществ — алмаз и графит, а также некоторые аллотропные
модификации бора, кремния, фосфора, мышьяка и сурьмы. При плавлении
этих веществ происходит разрыв ковалентных связей.
Бор (группа 13)
Стандартная форма бора — (3-ромбоэдрический бор. Структура этой
аллотропной модификации достаточна сложна, поэтому мы начнем
рассмотрение со строения ос-ромбоэдрического бора. Основным структурным блоком,
из которого построены как а-, так и ^-модификации, является икосаэдр В,2
(рис. 7.19). Каждый атом бора ковалентно связан с пятью атомами бора в
икосаэдре, несмотря на то, что каждый атом бора имеет всего три валентных
электрона. Связи в частице В,2 делокализованы, поэтому важно помнить, что
связи В—В, показанные на рис. 7.19, нельзя рассматривать как двухцентро-
вые двухэлектронные.
Структура сс-ромбоэдрического бора состоит из блоков В12, образующих
решетку, близкую к кубической плотнейшей упаковке. Атомы бора в икоса-
336
7. Структура твердых простых веществ
эдрических блоках располагаются на поверхности сферы, поэтому модель
плотнейшей шаровой упаковки применима. Однако в отличие от случаев,
рассмотренных выше, «сферы» в решетке ос-ромбоэдрического бора
соединены друг с другом ковалентными связями. Часть структуры (фрагмент слоя
бесконечной решетки), приведена на рис. 7.20. В трехмерной структуре такие
слои расположены по типу АВСАВС... (см. рис. 7.3 и 7.5). Наличие ковалент-
ных связей между икосаэдрами В12 отличает данную бесконечную ковалент-
ную решетку от рассмотренных ранее истинных плотнейших упаковок.
Структура (J-ромбоэдрического бора состоит из блоков В84, которые
связаны между собой блоками В10. Частицы В84 удобно рассматривать как
суперпозицию трех субчастиц: (В12)(В12)(В60). В центре частицы В84 находится
икосаэдр В12 (рис. 7.21,д), в котором каждый атом бора радиально соединен
с другим атомом бора (рис. 7.21,6). Термин «радиальный» использован здесь,
чтобы подчеркнуть, что линии, соединяющие внешние 12 атомов бора с
атомами внутреннего блока В12, проходят через центр частицы. Построенный
таким образом блок (В12)(В12) располагается внутри «ячейки» В60, имеющей
такую же геометрию, как молекула С60 (ср. рис. 7.21,в и 7.17,а). Вся частица
В84 показана на рис. 7.21,г. Заметим, что каждый атом из внешнего блока В12
связан с пятиугольником блока В60. В бесконечной решетке Р-ромбоэдриче-
ского бора блоки В84, показанные на рис. 7.21 ,г, связаны между собой
блоками В10. Как и в случае сс-ромбоэдрической модификации, решетка в целом
оказывается весьма прочной, в результате чего кристаллический бор имеет
очень высокую твердость и характеризуется крайне высокой температурой
плавления (2453 К для ^-ромбоэдрического бора).
Углерод (группа 14)
Алмаз и графит — аллотропные модификации углерода, имеющие
бесконечные ковалентные решетки в твердой фазе. Они сильно отличаются по
физическим свойствам. Кристаллы алмаза прозрачны, обладают высокой
твердостью и высоко ценятся в ювелирном деле. Кристаллы графита скользкие на
ощупь и окрашены в черный цвет.
На рис. 7.22 показан фрагмент кристаллической решетки алмаза. Каждый
атом углерода образует одинарные связи С—С с четырьмя соседними
атомами, расположенными по вершинам тетраэдра; в результате решетка в целом
оказывается очень прочной.
Рис. 7.19. Икосаэдр В,2 - основная
структурная единица в а- и Р-ромбоэдри-
ческом боре. Обе алолотропные
модификации бора имеют в твердом состоянии
бесконечные ковалентные решетки.
7.10. Твердые вещества, образующие ковалентные кристаллические решетки 337
Рис. 7.20. Фрагмент слоя в бесконечной решетке ос-ромбоэдрического бора. Слой
построен из икосаэдров В,2. Трехмерную структуру можно приближенно
рассматривать как кубическую плотнейшую упаковку сфер. Делокализованные ковалентные
связи между блоками В12 приводят к образованию прочной бесконечной решетки.
Стандартной формой углерода является графит, точнее, сс-графит,
поскольку существует еще одна модификация - (3-графит. Структуры а- и р-
графита представляют собой бесконечные ковалентные решетки, состоящие
из параллельных слоев сочлененных шестичленных циклов. В ос-графите
слои чередуются через один, тогда как в Р-графите повторяющийся элемент
струкутры содержит три слоя. Нагревание Р-графита до 1298 К вызывает его
переход в сс-графит.
^- Структура ct-графита (обыкновенный графит) показана рис. 7.23. Все рас-
гК0В(С) = 77 пм стояния С—С внутри слоев эквивалентны (142 пм), а расстоя-
rv(C) = 185 пм ние между соседними слоями равно 335 пм. Это говорит о
слабом взаимодействии между слоями. Об этом же
свидетельствуют физические свойства графита: он легко раскалывается вдоль плоскостей
шестичленных циклов. Это свойство позволяет использовать графит в
качестве смазочного материала. Упаковка атомов углерода в обыкновенном
фафите менее эффективна, чем в алмазе; плотности графита и алмаза равны 2,3
и 3,5 г/см3 соответственно.
Электрическая проводимость - важное свойство графита; ее
возникновение объясняется особенностями структуры и связывания. Электронная
конфигурация валентной оболочки углерода — [Не]2522/?2, и поскольку каждый
атом углерода в фафите образует ковалентные связи с тремя углеродными
атомами в слое, один валентный электрон остается неиспользованным. Лиш-
338 7. Структура твердых простых веществ
Рис. 7.21. Строение частицы В84, являющейся основным структурным блоком в
решетке р-ромбоэдрического бора, а — в центре частицы расположен икосаэдр В,2;
б — над каждым атомом бора в первичном икосаэдре находится еще один атом бора;
в - внешней оболочкой частицы В84 служит «ячейка» В60; г - итоговая частица В84
состоит из трех ковалентно-связанных субчастиц (Bl2)(Bl2)(B60).
ний электрон может перемещаться в плоскости шестичленных циклов.
Удельное электрическое сопротивление графита в направлении,
параллельном этим плоскостям, равно 1,3 • 10~5 Омм при 293 К, тогда как в
направлении, перпендикулярном плоскостям, оно равно 1 Омм. Удельное
электрическое сопротивление алмаза равно 1 • 10й Омм, поэтому алмаз является
превосходным изолятором. Все валентные электроны в алмазе участвуют в
образовании локализованных одинарных связей С—С.
Удельное электрическое сопротивление р материала - мера его
сопротивления R протеканию электрического тока. Другими словами,
для проволоки с единичной площадью сечения S = 1 м2 удельное
сопротивление (р) имеет размерность Ом • м, где сопротивление R
измеряется в омах, а длина / проволоки - в метрах; Я = p(//S). Хорошие
проводники имеют очень низкое удельное электрическое
сопротивление, а изоляторы - высокое удельное электрическое сопротивление.
7.10. Твердые вещества, образующие ковалентные кристаллические решетки 339
Дополнение 7.5. Связь между структурами алмаза и цинковой
обманки
Структура цинковой обманки (ZnS) обсуждалась в разд. 6.13; ее элементарная
ячейка приведена на рис. 6.17. Если и атомы цинка, и атомы серы заменить на
атомы углерода, получится следующая структура:
Если посмотреть на элементарную ячейку под другим углом, можно легко
видеть соответствие со структурой алмаза, показанной на рис. 7.22.
Таким образом, структуры алмаза и цинковой обманки родственны.
Таблица 7.2. Некоторые физические и структурные свойства алмаза, кремния,
германия и серого олова. (Все эти вещества имеют одинаковый тип решетки: см. рис. 7.22.)
Элемент Внешний вид Т„я. Ка Энталь- Плот- Межатомное Удельное
вещества пия плав- ность, расстояние сопротив-,
ления, г/см3 вкристал- ление Омм
кДж/моль лической
решетке, пм
Углерод
(алмаз)
Кремний
Германий
Олово
Прозрачен 3820
Сине-серый, 1683
блестящий
Серовато- 1211
белый,
блестящий
Тускло-серый -
(серая
модификация)
105
40
35
-
3,5
2,3
5,3
5,75
154
235
244
280
1- 10й (293 К)
1-Ю-3 (273 К)
0,46 (295 К)
11-10"8 (273 К)
а Серое олово является низкотемпературной модификацией. Выше 286 К устойчиво белое олово,
которое плавится при 505 К.
340
7. Структура твердых простых веществ
Кремний, германий и олово (группа 14)
В твердом состоянии при 298 К кремний образует алмазоподобную
кристаллическую структуру (рис. 7.22). Германий и серая модификация олова также
имеют аналогичные структуры, но по своим свойствам ближе к
полуметаллам, чем к неметаллам. Их аллотропные модификации рассмотрены ниже в
Рис. 7.22. Фрагмент бесконечной ковале нтной решетки одной из аллотропных
модификаций углерода - алмаза.
Дополнение 7.6. Углеродные нанотрубки
Мы привыкли считать, что слои углеродных атомов в графите являются
плоскими (рис. 7.23), но исследования последних лет показали возможность создания
структур, представляющих собой графитовые слои, свернутые в трубки
диаметром около 10 нм. Эти структуры называют нанотрубками. Углеродные
нанотрубки можно получать в относительно больших количествах — они образуются на
поверхности графитового катода в электрической дуге между графитовыми
электродами в атмосфере гелия. Совместные работы исследовательских групп
из Швейцарии и Бразилии привели недавно к получению монослоев
углеродных нанотрубок, вертикально прикрепленных к поверхности полимера.
Отдельные нанотрубки в таком монослое могут при определенных условиях испускать
электроны, которые можно фиксировать на аноде. Потенциальное применение
таких приборов связано с созданием плоских дисплеев, подобных тем, что
используются сейчас в портативных компьютерах. Предполагается, что
использование нанотрубок позволит существенно улучшить качество таких экранов.
О красоте трехмерных структур нанотрубок и других углеродсодержащих
систем Сп можно прочитать в статье "Beyond C60: Graphite Struvtures for the
Future", Chemical Society Reviews, 1995, vol. 24, p. 341. [См. также статью
«Углеродные нанотрубки» в Соросовском общеобразовательном журнале,
1999, №3, с. \\\.-Ред.]
7.10. Твердые вещества, образующие ковалентные кристаллические решетки 341
Слой I
Слой II
Слой!
Слой II
Рис. 7.23. Фрагмент бесконечной ковалентной решетки обыкновенного (а) графита —
стандартной формы углерода. Повторяющимся элементом структуры являются два
слоя, состоящие из сочлененных шестичленных циклов. Вертикальные штриховые
линии соединяют атомы углерода, принадлежащие различным слоям, которые
находятся непосредственно друг над другом.
разд. 7.11, однако, чтобы понять, почему алмаз и другие элементы группы 14
рассматриваются по отдельности, полезно сравнить некоторые их
физические свойства (табл. 7.2), в особенности удельные электрические
сопротивления.
Фосфор, мышьяк, сурьма и висмут (группа 15)
^ Несмотря на то что белый фосфор является стандартной формой элемента,
Белый фосфор- эта м°ДиФикаиия метастабильна. Другие аллотропные моди-
см оазд 7 8 фикации фосфора либо аморфны, либо имеют бесконечные
ковалентные решетки, однако при плавлении все
модификации фосфора образуют жидкость, состоящую из молекул Р4.
Аморфный красный фосфор имеет большую плотность и менее
химически активен, чем белый фосфор. Перекристаллизацией красного фосфора в
расплавленном свинце получают моноклинную модификацию, называемую
фиолетовым фосфором или фосфором Гитторфа. Твердофазная структура
представляет собой достаточно сложную бесконечную решетку, две проек-
342 7. Структура твердых простых веществ
Рис. 7.24. Фрагмент бесконечной решетки фосфора Гитторфа. а - часть цепочечной
структуры, образованной атомами фосфора. Повторяющийся фрагмент содержит 21 атом фосфора.
Атомы Р' и Р" эквивалентны и принадлежат соседним цепочкам; б— та же структурная
единица, рассматриваемая с конца. Хорошо видно, что в структуре имеются каналы.
ции которой показаны на рис. 7.24. Структурные блоки такого типа
соединены по принципу «голова к хвосту» и образуют цепочечную структуру,
содержащую трехкоординированные атомы фосфора. Такие цепочки,
расположенные параллельно друг другу, образуют слой, но в пределах слоя связи
между цепями отсутствуют. Бесконечная решетка формируется из слоев,
размещенных таким образом, что атом фосфора Р' (рис. 7.24) ковалентно
связан с атомом Р" из соседнего слоя. Цепочки, находящиеся в соседних
слоях, перпендикулярны друг другу, так что при их соединении образуется
трехмерная сетка связей. Все связи Р—Р в решетке близки друг к другу (= 222 пм)
и соответствуют одинарным ковалентным связям.
Рис. 7.25. Гофрированные слои из шестичленных колец, присутствующие в
структуре черного фосфора, а также в структурах ромбоэдрических модификаций мышьяка,
сурьмы и висмута (элементы группы 15).
7.11 Структуры металлов при 298 К
343
Черный фосфор является наиболее термодинамически стабильной
формой элемента. Структура ромбоэдрического черного фосфора состоит из ше-
стичленных колец Р6 (рис. 7.25). Кольца Р6 присутствуют также в структуре
ромбического черного фосфора.
В твердой фазе ромбоэдрические модификации мышьяка, сурьмы и
висмута изоструктурны ромбоэдрическому черному фосфору. При движении
вниз по группе 15 координационное число элемента возрастает с трех
(соседние атомы в пределах слоя) до шести (три связи с атомами в слое и три - с
атомами соседнего слоя). Эти аллотропные модификации мышьяка, сурьмы
и висмута получили название металлических; их металлический характер
возрастает при движении вниз по группе.
СТРУКТУРЫ МЕТАЛЛОВ ПРИ 298 К
Простые вещества, образованные элементами групп 1 и 2 (^-элементами), а
также </-элементами, являются металлами. В случае /^-элементов металлы
можно приблизительно отделить от неметаллов диагональной линией
(рис. 7.26), хотя переход от неметаллов к металлам происходит плавно.
Структуры металлов в твердой фазе хорошо описываются в рамках моделей плот-
нейших шаровых упаковок. В табл. 7.3 приведены типы кристаллических
решеток s- и ^-металлов, а также указаны их температуры плавления.
Металлы - s-элементы
В твердой фазе при 298 К атомы всех щелочных металлов (группа 1)
образуют объемноцентрированную кубическую упаковку (рис. 7.9). Все щелочные
металлы мягкие, для них характерны относительно низкие температуры
плавления (табл. 7.3). В соответствии с этими данными, энтальпии плавления
также невелики и уменьшаются при движении вниз по группе от
3,0 кДж/моль для лития до 2,1 кДж/моль для цезия.
За исключением бария, имеющего объемноцентрированную кубическую
решетку, щелочноземельные металлы (группа 2) кристаллизуются с
образованием гексагональной плотнейшей упаковки. Их температуры плавления
выше, чем температуры плавления соответствующих щелочных металлов.
13 14 15 16 17 18
В
А1
Ga
In
TI
С
Si
N
P
Ge As
Sn
Pb
О
s
Se
Sb Те
Bi
Po
F
CI
Br
I
At
Ne
Ar
Kr
Xe
Rn
Рис. 7.26. «Диагональная линия», которую часто проводят для приблизительного
разделения положения неметаллов (справа от жирной линии) и металлов (слева от
жирной линии) в периодической системе элементов. Переход не является резким, и
элементы, примыкающие к границе, часто проявляют как металлические, так и
неметаллические свойства. Из-за этого их часто относят к полуметаллам.
344 7. Структура твердых простых веществ
Таблица 7.3. Структуры (при 298 К) и температуры плавления металлов
+ гексагональная плотнейшая упаковка
О кубическая плотнейшая упаковка
• объемноцентрированная кубическая решетка
1 2 3 4 5 6 7 8 9 10 11 12
Li
•
454
Na
•
371
К
•
337
Rb
•
312
Cs
•
301
Be
1560
Mg
♦
923
Ca
1115
Sr
♦
1050
Ba
•
1000
Sc
♦
1814
Y
♦
1799
La
1193
Ti
♦
1941
Zr
♦
2128
Hf
2506
V
•
2183
Nb
•
2750
Та
•
3290
Cr
•
2180
Mo
•
2896
W
•
3695
Mn
см. текст
1519
Тс
♦
2430
Re
♦
3459
Fe
•
1811
Ru
♦
2607
Os
♦
3306
Co
1768
Rh
a
2237
Ir
О
2719
Ni
a
1728
Pd
a
1828
Pt
О
2041
Cu
n
1358
Ag
О
1235
Au
О
1337
Zn
♦
693
Cd
♦
594
Hg
см. текст
234
Металлы - d-элементы
При 298 К металлы d-ряда. имеют структуры ГПУ, КПУ (ГЦК) либо ОЦК;
исключение составляют марганец и ртуть, о которых речь пойдет ниже. Из
табл. 7.3 можно видеть, что тип структуры зависит от положения металла в
периодической системе элементов. Большинство металлов d-ряла образуют
одну из плотнейших упаковок при 298 К, в то время как при повышенных
температурах стабильной является ОЦК-структура. Исключением является
железо, которое имеет ОЦК-структуру при 298 К, но выше 1179 К
происходит переход в ГЦК-структуру, а выше 1674 К ОЦК-решетка снова становится
наиболее устойчивой.
Температуры плавления ^/-металлов групп 3-11 намного выше, чем у
5-металлов, соответственно выше и энтальпии плавления. Два элемента
^-ряда заслуживают особого обсуждения. Первый из них — ртуть. Все три
элемента группы 12 выделяются среди других ^/-металлов относительно низкими
температурами плавления, а ртуть занимает особое место, поскольку при 298
К находится в жидком состоянии. Величина теплоты плавления ртути
(2,3 кДж/моль) нетипична для металлов J-ряда и близка к теплотам плавления
щелочных металлов. В кристаллическом состоянии ртуть имеет искаженную
простую кубическую решетку (см. рис. 7.8). Второй металл, выпадающий из
общих тенденций, - марганец. В твердом состоянии этот металл имеет
сложную кубическую решетку, в которой присутствует четыре типа атомов Мп с
координационными числами 12, 13 и 16. Причины такого отклонения от
обычных структурных типов не вполне понятны.
7.11. Металлический радиус
345
Металлы и полуметаллы в ряду р-элементов
По своим физическим и химическим свойствам тяжелые элементы групп 13,
14 и 15 относятся к металлам. Такие элементы, как германий, обладают
свойствами, промежуточными между металлами и неметаллами, поэтому их
иногда называют полуметаллами. Ниже рассмотрены структуры этих элементов,
устойчивые при 298 К.
Алюминий образует кубическую плотнейшую упаковку атомов, что
типично для металлов. Температура плавления (933 К) лишь немного
превышает температуру плавления магния, предшествующего алюминию в
периодической системе, и существенно ниже температуры плавления бора (2453 К).
^» Атомы таллия образуют ГПУ-структуру, также типичную для металла, а ре-
Элементный бор: шетка индия представляет собой слегка искаженную плотней-
см. разд. 7.10 шую упаковку атомов. Структура твердого галлия достаточно
сложна; каждый атом галлия имеет одного ближайшего соседа
на расстоянии 247 пм и шесть атомов галлия на расстояниях от 270 до 279 пм.
Галлий имеет низкую температуру плавления (303 К), и это означает, что в
некоторых районах земного шара галлий будет находиться в жидком состоянии,
а в других местах - в твердом. Кристаллическую структуру галлия можно
рассматривать как промежуточную между металлической решеткой'и
молекулярным твердым телом, состоящим из молекул Ga2.
«Диагональная» линия на рис. 7.26 проходит через группу 14 между
кремнием и германием, из чего можно было бы сделать вывод, что кремний
относится к неметаллам, а германий — к металлам. Однако это различие
выражено нечетко. В твердой фазе оба элемента образуют одинаковые бесконечные
ковалентные решетки алмазоподобного типа, но их удельные электрические
сопротивления существенно ниже, чем у алмаза (табл. 7.2), что указывает на
металлические свойства. Самый тяжелый элемент подгруппы, свинец, имеет
КПУ-решетку. Промежуточным элементом является олово. Белое (р) олово
стабильно при стандартных условиях (298 К), а при температурах ниже 286 К
оно переходит в серую (а) модификацию, имеющую алмазоподобную
структуру (табл. 7.2). Структура белого олова получается искажением структуры
серого олова таким образом, что координационное число олова возрастает с
четырех до шести. Плотность белого олова (7,31 г/см3) выше, чем плотность
серой модификации (5,75 г/см3); такое увеличение плотности при переходе
от низкотемпературной модификации к высокотемпературной является
весьма необычным. Переход из белого олова в серое протекает крайне
медленно, но может иметь драматические последствия. В XIX веке во время
особенно холодных зим происходило разрушение оловянных пуговиц на
солдатских мундирах. В 1851 году жители Зейца обнаружили, что оловянные
органные трубы в их церкви превратились в порошок!
Структура висмута описана в разд. 7.10.
^ЯВ МЕТАЛЛИЧЕСКИЙ РАДИУС
Металлическим радусом называется половина расстояния между
ближайшими соседними атомами металла в кристаллической решетке металла. В
табл. 7.4 приведены металлические радиусы для s- и ^-элементов. В группах 1
и 2 размеры атомов увеличиваются при движении вниз по группе. В триадах,
346 7. Структура твердых простых веществ
Таблица 7.4. Металлические радиусы (пм) s- и d-элементов
1 2 3 4 5 6 7 8 9 10 11 12
Li
157
Na
191
К
235
Rb
250
Cs
272
Be
112
Mg
160
Ca
197
Sr
215
Ba
224
Sc
164
Y
182
La
187
Ti
147
Zr
160
Hf
159
V
135
Nb
147
Та
147
Cr
129
Mo
140
W
141
Mn
137
Tc
135
Re
137
Fe
126
Ru
134
Os
135
Co
125
Rh
134
lr
136
Ni
125
Pd
137
Pt
139
Cu
128
Ag
144
Au
144
Zn
137
Cd
152
Hg
4-й период
5-й период
6-й период
составляющих каждую группу </-элементов, как правило, наблюдается суще-
Вандерваальсов
радиус: см. разд. 2.21
Ковалентный радиус:
см.разд. 3.3
Ионный радиус:
см. разд. 6.14
ственное увеличение радиуса при переходе от элемента 4-го
периода к элементу 5-го периода, но лишь очень небольшое
изменение радиуса происходит при переходе от металла 5-го
периода к металлу 6-го периода. Последнее связано с
присутствием у элементов 6-го периода заполненного 4/-подуровня,
наличие которого приводит к так называемому лантаныдиому
сжатию (лантаниды располагаются между лантаном La и гафнием
НО- Плохо экранирующие /-электроны находятся достаточно
близко к ядру и их влияние на радиус элемента невелико.
Металлическим радиусом называется половина расстояния между
ближайшими соседними атомами металла в кристаллической
решетке металла.
МЕТАЛЛИЧЕСКАЯ СВЯЗЬ
Металлы - проводники электрического тока
Важнейшим физическим свойством металлов является их низкое удельное
электрическое сопротивление, иначе говоря, все металлы прекрасно
проводят электрический ток. За исключением ртути, все металлы при 298 К
находятся в твердом состоянии. Несмотря на то что модели шаровых упаковок
хорошо описывают структуры металлов в твердом состоянии, они не дают
информации о способе связывания атомов между собой. Между тем, такая
информация необходима для объяснения высокой электрической
проводимости, поскольку электроны в металлах должны легко перемещаться по
7.13 Металлическая связь
347
структуре в целом. Для того чтобы объяснить высокую электрическую
проводимость металлов, необходимо понять природу связи между атомами в них.
Электрический проводник имеет низкое сопротивление (измеряемое
в омах, Ом) протеканию электрического тока (измеряемого в
амперах, А). Изолятор имеет высокое сопротивление.
Электронный газ
Одна из ранних моделей металлической связи (теория Друде-Лоренца)
основана на предположении, что валентные электроны каждого атома металла
могут свободно двигаться по кристаллической решетке. Таким образом,
предполагается, что кристаллическая решетка металла состоит не из
нейтральных атомов, а из положительно заряженных ионов (ядер, окруженных
внутренними электронами) и электронов (валентных электронов). При
приложении к куску металла разности потенциалов* валентные электроны
движутся от высокого потенциала к низкому и возникает электрический ток.
Согласно данной модели, положительно заряженные ионы металлов,
образующие, например, плотнейшую упаковку, погружены в «электронный
газ». Такое представление дает удовлетворительное качественное объяснение
высокой электрической проводимости металлов, однако не может объяснить
более детальные соотношения между электрической проводимостью
различных металлов. Существует ряд других теорий, из которых наиболее общей
является зонная теория.
Зонная теория
Зонная теория следует из рассмотрения энергии молекулярных орбиталей
совокупности множества атомов металла. Для определения основного
(невозбужденного) состояния молекулы ранее мы использовали принцип
последовательного заполнения электронами наинизших орбиталей и
располагали электроны таким образом, чтобы добиться минимальной энергии
состояния. В присутствии вырожденных орбиталей это может привести к наличию
наивысших молекулярных орбиталей, занятых одним электроном (как,
например, в случае молекул В2 и 02).
^ Для МО-диаграмм, описывающих связывание в металлическом твердом
ЛКАО: см. разд. 3.12 теле, характерно присутствие большого числа орбиталей,
очень близких по энергии. Эти орбитали образуют
непрерывную полосу разрешенных по энергии состояний, называемую зоной
(полосой) разрешенных энергий. Возникновение зоны происходит следующим
образом. Рассмотрим взаимодействие между АО одного типа (например, 2s с 2s
или 2р с 2р) в рамках подхода ЛКАО. На рис. 7.27 показаны результаты
перекрывания 2s-АО лития в зависимости от количества атомов лития,
участвующих во взаимодействии. Все исходные 2s-AO имеют одинаковые энергии. При
Примеры использования уравнения V = IR (разность потенциалов = сила тока • сопротивление)
приведены в прилагаемом задачнике.
7. Структура твердых простых веществ
взаимодействии двух атомов лития перкрывание двух 2s-AO приводит к
возникновению двух МО. В случае трех атомов лития, возникают три МО, в
случае четырех — четыре МО и т. д. Если имеется п атомов лития, то образуется
п молекулярных орбиталей, но поскольку все исходные 25-АО лития имели
одинаковую энергию, результирующие МО также очень близки по энергии и
их можно рассматривать как одну энергетическую зону. Степень заполнения
зоны определяется количеством доступных валентных электронов. Каждый
атом лития имеет один валентный электрон, и зона, показанная на рис. 7.27,
оказывается полузаполненной. Это ведет к тому, что связывающие электроны
в металле делокализованы и металлическая связь является ненаправленной.
Когда атомы металла имеют более одного типа АО, то возникает
соответствующее число энергетических зон. Если две зоны близки по энергии, они
могут перекрываться с образованием единой зоны смешанного типа
(например, s- и /?-типа). Таким образом разрешенные энергетические состояния
атомов расщепляются в энергетические зоны близко расположенных
уровней, которые могут быть отделены друг от друга запрещенными участками,
или запрещенными зонами. В зависимости от числа валентных электронов
разрешенные зоны могут быть пустыми (верхняя часть рис. 7.28) или частично
(средняя часть рис. 7.28) либо полностью (нижняя часть рис. 7.28)
заполненными. Поскольку орбитали, составляющие разрешенную зону, близки по
энергии, то в пределах частично заполненной зоны электроны могут
перемещаться из одного энергетического состояния в другое . В металлах зоны
оказываются заполненными электронами лишь частично. Поэтому при
наложении электрического поля электроны передвигаются в направлении от
высокого потенциала к низкому и возникает электрический ток. Таким образом,
Li2 Li3 Li4 Li5 Li„
Рис. 7.27. Перекрывание двух 25-АО в молекуле Li2 приводит к образованию двух
МО. В случае трех атомов лития образуются три МО и т. д. В \лп имеется п
молекулярных орбиталей, но поскольку все 25-АО имеют одинаковую энергию, энергии
получающихся МО близки друг к другу и их можно рассматривать как зону.
7.13 Металлическая связь
349
х
О)
Рис. 7.28. Молекулярные орбитали,
описывающие связывание в металле,
очень близки по энергии и представляют
собой зоны. Зоны могут быть полностью
заполнены электронами (показано
черным), незаполнены (показано белым) или
частично заполнены. На рисунке
схематически показано расположение зон в
металле.
<^ I
Незаполненная зона
<^ | Частично
заполненная зона
Полностью
заполненная зона
характерной чертой металла является наличие частично заполненной зоны, в
то время как запрещенные участки между различными зонами могут быть
достаточно велики.
Зона - группа молекулярных орбиталей, очень близких по энергии.
Энергетические различия настолько малы, что в пределах зоны
изменение энергии можно считать непрерывным, неквантованным. Между
зонами может возникать запрещенный участок (запрещенная зона).
Полупроводники
При движении вниз по группам 13—16 происходит переход от неметалла к
металлу, при этом для ряда элементов реализуется промежутное состояние —
полуметалл. Как уже отмечалось для элементов группы 14, нельзя провести
четкую границу между металлами и неметаллами.
Рис. 7.29 иллюстрирует изменение ширины запрещенной зоны при
переходе от неметалла к металлу. Зонная модель для алмаза может быть
представлена как совокупность полностью заполненной и незаполненной зон*. У
алмаза большая ширина запрещенной зоны (520 кДж/моль), поэтому он
является изолятором. Сходная картина наблюдается для кремния и германия, но
в этих случаях ширина запрещенной зоны существенно меньше (106 и
64 кДж/моль соответственно). В случае серого олова только 8 кДж/моль раз-
Наивысшая из разрешенных зон, заполненная электронами, называется валентной зоной.
Наинизшую из незаполненных разрешенных зон называют зоной проводимости, а расстояние между этими
зонами — запрещенной зоной. — Прим. ред.
350
7. Структура твердых простых веществ
1111
Алмаз
Кремний Германий Серое олово
^
Уменьшение ширины
запрещенной зоны
Рис. 7.29. Энергетическое различие (ширина запрещенной зоны) между
заполненной (показана черным) и незаполненной (показана белым) зонами МО уменьшается
при движении вниз по группе 14. Это приводит к усилению металлического характера
веществ при движении вниз по группе.
деляют валентную зону и зону проводимости, и данная ситуация
приближается к наличию единой частично заполненной зоны. Электропроводность
кремния, германия и серого олова определяется термической заселенностью
зоны проводимости, и эти вещества называют полупроводниками. При
повышении температуры часть электронов приобретает энергию, достаточную для
перехода из нижней валентной зоны в верхнюю — зону проводимости. Чем
меньше ширина запрещенной зоны, тем большее число электронов будет
иметь энергию, достаточную для такого перехода, и тем выше будет
электропроводность вещества.
7.15. Упражнения
351
ЗАКЛЮЧЕНИЕ
В данной главе были рассмотрены структуры элементов в твердой фазе.
Что означают следующие термины?
• Плотнейшая шаровая упа- •
ковка •
• Кубическая плотнейшая
упаковка •
• Гексагональная плотнейшая •
упаковка
• Простая кубическая решетка
• Объемноцентрированная ку- •
бическая решетка •
Теперь вы должны уметь:
Внутренние пустоты
Кристаллическое твердое
тело
Аморфное твердое тело
«Кресло» и «ванна» - кон-
формации шестичленного
цикла
Геликоидальная структура
Ка тена - структуры
Удельное электрическое
сопротивление
Изолятор
Металлический радиус
Металлическая связь
Запрещенная зона
Полупроводник
обсудить причины возникновения по
меньшей мере двух типов плотнейших шаровых
упаковок;
обсудить связь между простой кубической
и объемноцентрированной кубической
шаровыми упаковками;
определить число ближайших соседей в
КПУ-, ГПУ-, простой кубической и ОЦК-ре-
шетках;
сравнить эффективность упаковки в КПУ-,
ГПУ-, простой кубической и ОЦК-решетках;
определить, почему модели шаровых
упаковок применимы для некоторых, но не для
всех кристаллических решеток;
различать внутри- и межмолекулярные
связи в твердофазных структурах (всегда ли
присутствуют оба типа связей?);
описать структурное различие между
аллотропными модификациями бора, углерода,
фосфора и серы;
описать сходство и различие между
структурами твердых простых веществ,
образованных элементами группы 17;
привести примеры металлов с различными
типами решеток;
объяснить с использованием простейшей
зонной теории, почему металлы проводят
электрический ток.
УПРАЖНЕНИЯ
7.1. Укажите основные черты,
характеризующие гексагональную и
кубическую плотнейшие шаровые
упаковки. Изобразите элементарные
ячейки для каждой из структур.
7.2. Что такое внутренние пустоты?
Какие типы пустот присутствуют в 7.5.
ГПУ- и КПУ-структурах?
7.3. Как расположены сферы в объемно-
центрированной кубической
решетке? Чем отличается
объемноцентрированная кубическая решетка от
простой кубической?
7.4. Что означает понятие «ближайший 7.6.
сосед» в шаровой упаковке? Сколько
ближайших соседей имеет каждая
сфера в: а) кубической плотнейшей
упаковке; б) гексагональной
плотнейшей упаковке; в) простой
кубической решетке; г)
объемноцентрированной кубической решетке?
Укажите электронные конфигурации
атомов хлора, серы и фосфора в
основном состоянии и используйте
эти данные для описания
связывания в молекулах, присутствующих в
а) твердом хлоре С12(тв.); б)
ромбической сере; в) нитевидной сере;
г) белом фосфоре.
Что такое удельное электрическое
сопротивление? Подтвердите выска-
352
7. Структура твердых простых веществ
зывание «Все металлы являются
хорошими проводниками» с помощью
данных табл. 7.5.
Таблица 7.5. Данные к упражнению 7.6. Все
удельные сопротивления измерены при
температуре 273 К, если иное не оговорено
Элемент
Удельное электрическое
сопротивление, Ом • м
Медь
Серебро
Алюминий
Железо
Галлий
Олово
Ртуть
Висмут
Марганец
Кремний
Бор
Фосфор
1,5
1,5
2,4
8,6
1,4
3,9
9,4
1,1
1,4
1,0
1,8
1,0
ю-8
ю-8
ю-8
ю-8
ю-7
ю-7
ю-7
10"6
10"6
ю-3
104
109(293 К)
7.7. Кратко обсудите аллотропию
углерода. Какая модификация
соответствует стандартному состоянию
элемента? Объясните причины сильного
различия электрических
сопротивлений алмаза и а-графита. Почему
электрическое сопротивление
графитового стержня зависит от
направления?
7.8. Что означает понятие кристаллосоль-
ват? При кристаллизации С60 из
раствора в бензоле и дииодметане
образуются сольватированные
кристаллы состава С60 *С6Н6 >>СН212.
Определите возможную
стехиометрию этого соединения, если при
удалении растворителей молярная
масса уменьшается на 32,4%.
7.9. Какая структура решетки типична
для щелочноземельных металлов?
В табл. 7.6 приведены значения
энтальпий плавления и атомизации
щелочных металлов. Опишите
каждый из процессов в терминах
межатомных взаимодействий. С
использованием данных табл. 7.6 постройте
. * графики изменения температур
плавления, энтальпий плавления и
энтальпий испарения элементов
группы 1. Какова связь между этими
величинами? Как объяснить
наблюдаемые тенденции изменения
указанных величин?
Таблца 7.6. Данные к упражнению 7.9
Щелочной Темпера- Энтальпия Энтальпия
металл тура плав- плавления, атомиза-
ления, К кДж/моль ции,
кДж/моль
Литий 454
Натрий 371
Калий 337
Рубидий 312
Цезий 301
3,0
2,6
2,3
2,2
2,1
162
108
90
82
78
7.10. Прокомментируйте соотношение
между металлическим (197 пм) и
ионным (100 пм) радиусами кальция.
7.11. Локализованная ковалентная s-связь
является направленной, тогда как
металлическая связь ненаправленна.
Обсудите свойства ковалентной и
металлической связи, которые
приводят к возникновению такого
различия.
7.12. С использованием простейшей
зонной теории опишите различие между
электрической проводимостью в
металле (например,литии)и
полупроводнике (например, германии).
Q АЛКАНЫ, АЛКЕНЫ И АЛКИНЫ
ЕП ВВЕДЕНИЕ
Органическая химия изучает соединения углерода. Как правило,
элементный углерод, оксид углерода(Н), оксид углерода(1У) и карбонаты относят к
неорганическим соединениям. Но такое разделение является искусственным
и зачастую бесполезным, поскольку химия «органических» и
«неорганических» соединений основывается на одних и тех же подходах. В настоящее
время многочисленные междисциплинарные исследования показали, что
разграничение органической химии, неорганической химии, биохимии и
биологии нельзя считать строго определенным, и эти дисциплины
неразрывно связаны между собой. Их нельзя рассматривать по отдельности;
настоящий прогресс в науке невозможен без обмена идеями и знаниями между
исследователями различных областей науки.
Что такое углеводороды?
Углеводороды состоят только из атомов углерода и водорода. Углеводороды
подразделяются на алифатические и ароматические, а алифатические углево-
^ дороды — на насыщенные (предельные) и ненасыщенные (непредельные)
Ароматические и цикли- (рис. 8.1). Термин «алифатический» ранее использовался по
ческие соединения: отношению к жирам* и родственным им соединениям, но те-
см. т. 2 гл. 15 перь употребляется для названия углеводородов, содержащих
только открытые углеродные цепи (ациклические
углеводороды), а также для циклических соединений, которые имеют сходные свойства
с ациклическими углеводородами и называются алициклическими или цик-
лоалифатическими углеводородами**. В данной главе обсуждаются только
В ациклических углеводородах атомы углерода образуют открытые
цепи. Если в соединении имеется замкнутая цепь углеродных атомов,
то оно называется циклическим.
Отсюда устаревшее, но все еще распространенное другое название алифатических углеводородов -
углеводороды жирного ряда. — Прим. ред.
** В отечественной литературе к алифатическим углеводородам относят только ациклические
углеводороды, а ал и циклические (т. е. циклоалифатические) углеводороды, которые также
подразделяются на насыщенные (циклопарафины) и ненасыщенные, объединяют вместе с ароматическими в
большую группу карбо- и гетероциклических углеводородов. — Прим. ред.
354
8. Алканы, алкены и алкины
Углеводороды
содержат только атомы углерода и водорода
/
\
Алифатические углеводороды Ароматические углеводороды (см. гл. 15)
Насыщенные углеводороды Ненасыщенные углеводороды
содержат одинарные связи содержат как минимум одну
С—С и С—Н. Эти кратную связь углерод-углерод,
соединения называются а также одинарные связи С—Н
алканами
/
\
Алкены содержат Алкины содержат
связь С=С связь С=С
Рис. 8.1. Классификация углеводородов.
ациклические алифатические углеводороды; их открытые углеродные цепи
могут быть неразветвленными (их называют также линейными или прямыми) и
разветвленными (см. дополнение 8.2).
Соединения углерода обычно классифицируют по функциональным
группам, которые они содержат, так как эти группы вступают в характерные
химические реакции. Алкены и алкины содержат функциональные группы
С=С и С=С соответственно. Насыщенные алифатические углеводороды не
содержат никаких функциональных групп. Их старое название «парафины»
имеет латинские корни и означает «малое сродство». Общая формула
ациклического алкана: CwH2w+2; каждый атом углерода в них образует четыре
одинарные связи.
Функциональная группа - группа атомов в молекуле, вступающая в
характерные химические реакции.
Геометрия углеродных связей и гибридизация
Углеводороды в зависимости от присутствия или отсутствия кратных связей
углерод-углерод могут содержать атомы углерода в тетраэдрическом (sp3),
►- плоскотреугольном (sp1) или линейном (sp) окружении связанных с ними
Геометрия углерод- других атомов. Некоторые примеры приведены в табл. 8.1. Гиб-
ных центров: ридизация в углеводородах рассматривается в упражнении 8.8 в
см. разд. 5.14 разд. 8.23.
НОМЕНКЛАТУРА ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
В разд. 1.23 и 1.24 рассмотрены различия между эмпирическими,
молекулярными и структурными формулами, а также основы номенклатуры
органических соединений. Хотя в данной книге используется номенклатура ИЮПАК,
некоторые распространенные тривиальные названия даны в приложении 10.
8.2. Номенклатура органических соединений
355
Таблица 8.1. Геометрия и гибридизация углеродных центров в молекулах углеводородов
(см. также разд. 5.14)
Алкан
Алкен
Аллен
Алкин
Геометрия
углеродного
центра
Тип
гибридизации
углеродного
центра
Пример:
Название:
Молекулярная
формула:
Структурная
формула:
Тетраэдрическая
Плоскотреугольная
SP3
Этан
с2н6
сн3сн3
s/r
Линейная
sp
Линейная
sp
Этен (этилен) Пропадиен (аллен) Этин (ацетилен)
с2н4
Н2С—CH2
с3н4
Н2С=С=СН2
с2н2
носн
/
/ \
с с
\'"""н /
/
с
\
\
с
/
/
:С H-
\
Дополнение 8.1. Круговорот углерода в природе
(углеродный цикл)
Травоядные и
хищники
Ископаемое топливо
(торф, уголь, нефть,
природный газ)
Формирование
ракушек,
кораллов
и т. п.
(карбонаты)
известняка,
мрамора и
мела
356
8. Алканы, алкены и апкины
Названия углеводородов оканчиваются на -ан (насыщенный
углеводород); -ен (ненасыщенный углеводород с двойной связью С=С); -ин
(ненасыщенный углеводород с тройной связью С=С).
Неразветвленные алканы
Названия неразветвленных алканов даются по числу углеродных атомов в
цепи. В табл. 1.8 приведены названия, соответствующие углеродным цепям с
различным числом атомов. В табл. 8.2 перечислены названия некоторых
неразветвленных алканов, их молекулярные и структурные формулы. Эти
углеводороды образуют гомологический ряд, в котором соседние члены
отличаются на одну СН2 (метиленовую) группу.
Разветвленные алканы
Корень названия разветвленного алкана определяют по числу атомов
углерода в самой длинной неразветвленной цепи. Следует обратить особое внимание
на то, что самая длинная неразветвленная цепь не обязательно изображается
прямой. Например, на рис. 8.2 показана структура разветвленного алкана,
для которого можно выделить три возможных варианта неразветвленных
цепей. Неразветвленные цепи, выделенные цифрами-локантами на рис. 8.2,д и
8.2,в, содержат по шесть атомов углерода, а цепь, выделенная на рис. 8.2,5,
содержит семь атомов углерода, — следовательно, название данного алкана
производится от слова гептан.
После выбора самой длинной неразветвленной цепи атомы углерода в ней
нумеруются таким образом, чтобы номера атомов углерода, соединенных с
боковыми цепями, были минимальны. Для алкана, показанного на рис. 8.2,5,
боковая цепь присоединена к атому С(4) независимо от того, с какого конца
начинается нумерация главной цепи, содержащей семь углеродных атомов.
Название боковой цепи (заместителя) производится от названия
соответствующего ей алкана. Общее название таких заместителей — алкильная группа;
заместитель СН3 называется «метил» (Me), С2Н5 - «этил» (Et) и т. д. (см.
также рис. 8.7 в разд. 8.4). Таким образом, правильное название углеводорода,
показанного на рис. 8.2,5, — 4-этилгептан. Отметим две особенности
данного названия:
• между номером, указывающим положение заместителя, и названием
заместителя ставится дефис;
• название заместителя и корень пишутся слитно.
Таблица 8.2. Названия некоторых неразветвленных алканов
Название Молекулярная формула Структурная формула
Метан СН4 СН4
Этан С2Н6 СН3СН3
Пропан С3Н8 СН3СН2СН3
Бутан С4Н10 СН3СН2СН2СН3
Пентан С5Н,2 СН3СН2СН2СН2СН3
Октан С8Н18 СН3СН2СН2СН2СН2СН2СН2СН3
Декан С10Н22 СН3СН2СН2СН2СН2СН2СН2СН2СН2СН3
8.2. Номенклатура органических соединений
357
Н2 б Н2 Н2 7
с^ ^с^ ^сн3 .cw ^cw ^сн,
сн с н3с сн с
I н2 I н2
СН2 ^СН-)
н2с2
н2 н2
Н3С" з СН С
1
н
Рис. 8.2. Согласно номенклатуре ИЮПАК, название органического соединения
производится от самой длинной неразветвленной цепи в молекуле. На рисунке
показан разветвленный алкан, содержащий девять атомов углерода. Неразветвленные
цепи могут быть выделены тремя способами (д, б, в), и содержать: а) шесть атомов С;
б) семь атомов С; в) шесть атомов С.
Если присутствует более одного заместителя, их названия приводятся в
алфавитном порядке, перед каждым заместителем указывается его положение в
основной цепи. Для обозначения нескольких одинаковых заместителей
используют префиксы ди-9 три- и тетра-, например, «диметил» означает
наличие двух метильных групп, «тетраэтил» — четырех этильных групп. При
наличии различных заместителей, их названия располагаются в алфавитном
порядке независимо от количества групп одного типа (например, правильным
названием будет 4-метил-3,3-диэтил октан, а не 3,3-диэтил-4-метилоктан). В
табл. 8.3 приведен ряд примеров, а также проиллюстрирован упрощенный
Основные правила номенклатуры разветвленных алканов:
1. Найдите самую длинную неразветвленную цепь в молекуле, она
определяет корень в названии углеводорода.
2. Эта цепь нумеруется таким образом, чтобы заместители получили
наименьшие номера.
3. Перед корнем ставится название заместителя; если присутствует
несколько заместителей, их названия ставятся в алфавитном
порядке.
358
8. Алканы, апкены и алкины
Дополнение 8.2. Линейные («прямые») цепи в химии углерода
Способность углерода образовывать связи углерод—углерод приводит к тому, что в
химии этого элемента часто встречаются молекулярные цепи и циклы, состоящие из
углеродных атомов.
Слова-синонимы линейная или прямая цепь используются для обозначения нераз-
ветвленной цепи углеродных атомов. Неразветвленные цепи часто изображают таким
образом, что можно сделать неверное предположение об их линейном строении. Но
хотя структура, например гексана, может быть условно изображена как
н
н
н
н
н
н
н н н н н н
каждый атом углерода в ней 5р3-гибридизован (тетраэдр), и строение линейной цепи
лучше всего изображать следующим образом:
н н н н
н
\/ \/
н
с с с
\ / \
с с с
н
н
"- I /\ /\
н н н н н
Кроме того, возможно вращение вокруг одинарных связей С—С, которое приводит к
«скручиванию» молекулы:
Таким образом, термин линейная цепь или прямая цепь указывает только
последовательность соединения атомов углерода между собой и ничего не говорит о конформации
цепи. Цепи, в которых линейная последовательность атомов в углеродном скелете
нарушается, называют разветвленными.
способ изображения структурных формул. При этом указываются только
связи между атомами углерода и предполагается, что координационное
число каждого углеродного атома дополняется до четырех атомами водорода.
Таким образом, концевые прямые линии обозначают метильные группы, а не
атомы водорода.
Таблица 8.3. Названия и структурные формулы некоторых разветвленных алканов
Полная структурная
формула (химические
символы со схемой связи)
Сокращенная структурная
формула (только схема связей)
Краткая струкутрная
формула (только
химические символы)
Название соединения
сн,
хн
нх'
хн.
СН3СН(СНз)СН2СНз
СН3СНМеСН2СН3
2-Метил бутан
н,с сн,
\/
н,сг
сн.
.сн
н,<г сн
сн,
н,с>
^с/СН^снх
гн,
СН3С(СНз)2СН2СН2СНз
СН3СМе2СН2СН2СНз
СН3СН(СНз)СН(СНз)СН2СН2СНз
СН3СНМеСНМеСН2СН2СНз
2,2-Диметилпентан
2,3-Диметилгексан
СН3СН2СН2СН(С2Н5)СН(СНз)СН2СНз З-Метил-4-этилгептан
СНзСН2СН2СНЕХНМеСН2СН3
со
!°
X
о
CD
X
ш
■о
ш
о
■о
3
X
S
X
(D
О
*
X
о
о
2
S
X
(D
X
S
СО
(Л
<0
360
8. Алканы, алкены и алкины
Неразветвленные алкены
Корень названию алкена дает самая длинная неразветвленная цепь,
содержащая двойную связь, поэтому при названии алкена эту цепь необходимо
определить в первую очередь. При введении двойной связи в углеводородную
цепь в названии надо указать:
• число атомов углерода в цепи и
• положение двойной связи.
Углеводородная цепь нумеруется таким образом, чтобы положение двойной
связи имело наименьший номер. В структуре 8.1 имеется цепь из пяти атомов
углерода и название должно содержать корень пент-. Цепь нумеруется
справа налево, поскольку двойная связь находится ближе к правому концу.
Правильное название углеводорода - пентен-2, а не пентен-3. В структуре 8.2
имеется восемь углеродных атомов и название должно содержать корень
олти-, а нумерация атомов ведется слева направо (почему?). Навание
соединения: октен-3.
СН3(
сн2
\
с =
/
н
н
/
= с
\
сн3
н
\
с =
/
CH-jCH^
н
/
= с
\
сн3
У\
(£)-Пентен-2 (Z)-T\qhtqh-2
Н Н
\ / \ /
с = с с = с
/ \ / \
(£)-Октен-3 (2)-Октен-3
Рис. 8.3. Название геометрических изомеров алкенов.
8.2. Номенклатура органических соединений
361
сн3сн2сн=снсн3
сн3сн2сн=снсн2сн2сн2сн3
8.1
8.2
► Алкены 8.1 и 8.2 могут иметь геометрические изомеры, но из
приведенных формул нельзя определить, о каких именно геометриче-
(Е)- и (Z)- изомеры: ских изомерах идет речь. На рис. 8.3 показаны структуры гео-
см. разд. 5.11 метрических изомеров; для их названия используются
префиксы (£)- и (Z).
Диены и триены
Если в структуре присутствуют две двойные связи, углеводород называется
диеном, диены являются частным случаем алкенов. Правила названия
диенов те же, что и для алкенов, но в название включаются два номера,
указывающие положения двух двойных связей, и префикс ди-. Аналогично, триены
содержат три двойные связи. Примеры приведены на рис. 8.4, там же
показано использование префиксов (£)- и (Z)- для обозначения геометрических
изомеров гептадиена-2,4.
Диены содержат две двойные связи ОС.
1 2 3 4 5 6 7
СН3СН=СНСН=СНСН2СН3
Гептадиен-2,4
1 2 3 4 5 6 7 8 9 10
СН3СН=СНСН2СН=СНСН2СН=СНСН3
Декатриен-2,5,8
(2Д4£)-Гептадиен-2,4
(2£,4£)-Гептадиен-2,4
г
у~\
(2£,42)-Гептадиен-2,4
(2Д42)-Гептадиен-2,4
Рис. 8.4. а — номенклатура диена и триена; б — геометрические изомеры
гептадиена-2,4; чтобы избежать неясности, в скобках указывают как цифровое обозначение
положения связи, так и префиксы (£)- и (Z)-.
362
8. Алканы, алкены и алкины
Особым случаем диенов являются аллены, в них один из атомов углерода
образует две двойные связи. Простейший пример аллена - пропадиен
(табл. 8.1), который носит тривиальное название «аллен».
В алленах один из атомов углерода образует две двойные связи
(-С=С=С-).
Алкины
Правила названия алкинов такие же, как для алкенов, хотя в этом случае
невозможно образование геометрических изомеров. Таким образом, соединение
СН3С=ССН3 называется бутин-2, а соединение СН3СН2С=СН - бутин-1
(а не бутин-3).
ПЕРВИЧНЫЕ, ВТОРИЧНЫЕ, ТРЕТИЧНЫЕ И ЧЕТВЕРТИЧНЫЕ
АТОМЫ УГЛЕРОДА
Атом углерода называют первичным, вторичным, третичным или
четвертичным в зависимости от числа углеродных атомов, с которыми он связан:
• первичный атом углерода связан только с одним атомом углерода;
• вторичный атом углерода связан с двумя атомами углерода;
• третичный атом углерода связан с тремя атомами углерода;
• четвертичный атом углерода связан с четырьмя атомами углерода.
Например, этан содержит два первичных атома углерода. Пропан 8.3
содержит один вторичный и два первичных атома углерода; в 2-метилпропане
8.4 присутствуют один третичный и три первичных атома углерода; в 2,2-ди-
метилпропане 8.5 — один четвертичный и четыре первичных атома углерода.
Вторич- Н Н Пер- Н СН3 Третич- Н3С СН3
ный \ f s- винный \ ш ный \ ш Четвер-
J\^_у J / тичный
Н3С
8.5
Различие между разными типами углеродных центров становится
существенным при обсуждении реакционной способности алифатических соединений.
СТРУКТУРНАЯ ИЗОМЕРИЯ
Как было показано выше, алкены (но не алканы и не алкины) могут иметь
геометрические изомеры. С другой стороны, все указанные классы
соединений проявляют структурную изомерию, поскольку атомы углерода могут
соединяться в различной последовательности.
8.4. Структурная изомерия
363
Алканы
Такие алканы, как СН4, С2Н6 и С3Н8, не имеют структурных изомеров.
[Изобразите их структурные формулы и убедитесь, что существует только один
способ взаимного расположения атомов углерода.] Для любого алкана с
формулой CwH2w+2 ПРИ п>3 можно предложить более одной структурной
формулы, причем число возможных вариантов резко возрастает с ростом п
(рис. 8.5). На рис. 8.6 показаны структурные изомеры С6Н14. Смесь этих
соединений часто используется в качестве растворителя и носит название «ге-
ксан», хотя лишь одно из них называется гексаном согласно номенклатуре
ИЮПАК.
Дополнение 8.3. Газовая хроматография.
Петролейный эфир 40-60
Петролейный эфир 40—60 не является эфиром, а представляет собой
фракцию бензина, кипящую в температурном интервале 40—60 °С (313—333 К).
Петролейный эфир 40-60 состоит из смеси алканов, которую можно
проанализировать методом газовой хроматографии (ГХ). На схеме представлен
результат пропускания образца петролейного эфира 40—60 через газовый
хроматограф. Каждый пик соответствует определенному углеводороду, а
площадь пика пропорциональна содержанию данного компонента в смеси.
Время удерживания компонента зависит от его молекулярной массы и
степени разветвленности углеродной цепи. Обычными компонентами
петролейного эфира 40—60 являются изомеры пентана и гексана.
U
I L
0,4 0,8 1,2 1,6 2,0
Время удерживания, мин
364
8. Алканы, алкены и алкины
гнпЩН III! I I
123456789 10
Рис. 8.5. Число изомеров алканов, имеющих общую формулу СлН2л+2 (структурные +
оптические), резко возрастает с ростом числа углеродных атомов п. Наличие
единственной структуры для метана, этана и пропана рассматривается как наличие одного
изомера. Оптические изомеры обсуждаются в разд. 8.6.
Алкильные заместители
Метильный и этильный заместители являются производными метана и этана
соответственно (рис. 8.7,я) и не имеют изомеров. Существует два способа
образования пропильного заместителя из пропана (рис. 8.7,6), а при переходе к
бутану (который имеет два изомера) можно получить четыре изомера бутиль-
ных заместителей (рис. 8.7,в). При увеличении числа атомов углерода в
заместителе число возможных изомеров быстро возрастает.
Алкены и алкины
Для алкенов и алкинов могут возникать дополнительные структурные
изомеры за счет того, что кратные связи могут располагаться в разных местах
молекулы*. Этен, пропен, этин и пропин не имеют структурных изомеров
(подтвердите это, изобразив их структуры), но в случае молекул, содержащих
большее число атомов углерода, изомерия оказывается возможной.
^» Структуры 8.6, 8.7 и 8.8 являются структурными изомерами С6Н12, кото-
Циклические рые отличаются только положением связи С=С. Соединения
изомеры С6Н12: 8.7 и 8.8 имеют также (£)- и (Z)- изомеры (укажите их структу-
см. гл. 15 ры). Возможно также большое число изомеров С6Н12 с
разветвленными цепями. Для С6Н12 существуют также изомеры с
циклической структурой.
В отечественной литературе данный тип изомерии называется изомерией положения. - Прим. перев.
OKJ
60
В
ю
О
40
20
8.5. Конформации
365
сн,
н3с
н,
с
н,
н2
с\
с
н
I н2
сн с
н3с с сн3 н3с'
н,
н2
сн
сн3
сн3
Гексан
2-Метилпентан
3-Метил пентан
сн,
сн хн
сн, сн
сн,
сн3 сн3
сн,
сн3
2,3-Д и метил бутан 2,2-Диметилбутан
Рис. 8.6. Структурные изомеры С6Н]4 (рассмотрены только ациклические изомеры).
сн2=снсн2сн2сн2сн3 сн3сн=снсн2сн2сн3
Гексен-1
8.6
Гексен-2
8.7
сн3сн2сн=снсн2сн3
Гексен-3
8.8
КОНФОРМАЦИИ
Заторможенная, заслоненная и скошенная конформации
Вращение вокруг оси, соединяющей ядра атомов, как правило, возможно,
если атомы образуют одинарную связь. Вращение же вокруг кратной связи
ограничено из-за наличия л-составляющей (причины этого рассмотрены при
обсуждении рис. 3.18).
В молекуле алкана возможно вращение вокруг всех одинарных связей. В
С2Н6 вращение вокруг связи С—Н не приводит к изменению формы
молекулы, тогда как вращение вокруг связи С—С изменяет взаимную ориентацию
метильных групп. Возможные типы расположения заместителей друг
относительно друга называются конформациями; молекулярные конформеры можно
перевести друг в друга посредством вращения вокруг одинарных связей.
366
8. Алканы, алкены и алкины
СН4 ■
Метан
с2н6 ■
Этан
СН,
Метил (Me)
- -с2н5
Этил (Et)
б
сн3сн-)Сн3
Пропан
—СН2СН2СН3
уЛ Пропил (Рг)
<
^ •
СН3СНСН3
Изопропил (*Рг)
в
сн3сн2сн2сн3 <
Бутан
СН3СНМеСН3 <
2-Метил пропан
(изомер бутана)
-сн2сн2сн2сн3
У^ Бутил (Ви)
X ,
СН3СНСН2СН3
втор- Бутил (secBu)
-СН2СНМеСН3
уг Изобутил ('Ви)
Х •
СН3С(Ме)СН3 (МеС-)
трет- Бутил (lBu)
Рис. 8.7. Алкильные заместители. В случае метильного и этильного заместителей
структурные изомеры отсутствуют (я), но пропил ьная группа (б) имеет два изомера, а
бутильная группа (в) имеет четыре изомера. Каково происхождение названий втор-
бутил и трет-бугил (см. разд. 8.3)?
Вращение возможно вокруг одинарной связи, но не вокруг кратной связи.
На рис. 8.8,а показаны два конформера этана, для которых реализуются
предельные случаи взаимного расположения метильных групп -
заторможенная (шахматная) и заслоненная конформации. Между заторможенной и
заслоненной конформациями существует бесконечное число скошенных кон-
формаций.
Если два различных способа расположения атомов в молекуле можно
перевести друг в друга вращением вокруг одинарной связи, то
соответствующие им соединения называются конформерами.
8.5. Конформации 367
а
\ л
заторможенная
s
Я П
заслоненная
* v^
V
Л^
н н
,н
заторможенная
н 1
1
нрн
Л^
н н
заслоненная
б Н
н >jk н
н
заторможенная
нн
А\
н
заслоненная
г
Ч-/
/>
заторможенная
Ч-И
ГЛ
заслоненная
Рис. 8.8. Конформации этана, а - две проекции заторможенной и заслоненной кон-
формаций, показанные с помощью пространственной модели молекулы С2Н6. Для
изображения конформации используются способы 5, в, г; б — проекции Ньюмена:
атомы, изображенные перед кругом, направлены к наблюдателю; в — в проекции типа
«козлы» нижний левый атом углерода считается ближайшим к наблюдателю; г —
клиновидные жирные и пунктирные линии обозначают соответственно атомы или группы,
направленные на наблюдателя и от него; остальные линии считаются лежащими в
плоскости листа.
Способы изображения конформации
Существует несколько способов изображения конформации,
проиллюстрированных на рис. 8.8,5-г на примере молекулы С2Н6. В проекции Ньюмена
молекула рассматривается вдоль связи С—С; атомы, расположенные ближе к
наблюдателю, изображаются перед кругом, а удаленные от наблюдателя — за
368
8. Алканы, алкены и алкины
кругом. В случае рисунка типа «козлы» связь С—С рассматривается под
углом; согласно принятой договоренности, считается, что ближайший к
наблюдателю атом располагается в нижнем левом углу. Третий метод
изображения — использование клиновидных линий. Связь С—С лежит в плоскости
листа. Жирные линии изображают связи, выступающие над плоскостью
листа, а пунктирные линии соответствуют связям, удаленным от наблюдателя.
Использование таких схем позволяет однозначно представлять стереохимию
сложных молекул достаточно простыми способами.
Стереохимия молекулы описывает пространственное расположение
атомов в ней.
Изменение энергии стерических взаимодействий
►■ Когда молекула этана находится в заторможенной конформации, стериче-
Стерические ские взаимодействия между атомами водорода, принадлежа-
взаимодействия: щими соседним углеродным атомам, минимальны - это озна-
см. разд. 5.9 чает, что молекула имеет минимальную стерическую энергию.
По мере вращения вокруг связи С—С стерические
взаимодействия усиливаются. Ход изменения стерической энергии показан на рис. 8.9.
В заслоненной конформации энергия достигает максимума. При полном
обороте вокруг связи С—С на 360° молекула проходит через три одинаковые
по энергии заторможенные конформации и через три одинаковые по
энергии заслоненные конформации.
Разность энергий заторможенной и заслоненной конформации этана
составляет около 12,5 кДж/моль. Поскольку эта величина относительно
невелика, можно считать, что при 298 К молекула этана может свободно
вращаться вокруг связи С—С.
240 300 360
Угол вращения вокруг связи С—С, град.
Рис. 8.9. Стерическая энергия этана меняется по мере вращения одной метильной группы
относительно другой вокруг связи С—С. Два из шести одинаковых атомов водорода закрашены
черным, чтобы было понятно их взаимное расположение в процессе вращения.
8.6. Хиральные молекулы
369
Если на рис. 8.9 заменить атомы водорода алкильными группами, затор-
^- моженная конформация останется наиболее энергетически выгодной, а
См. также упражнение барьер вращения увеличится. Важно сознавать, что благодаря
8.9 в разд. 8.23 способности молекул алканов претерпевать вращение вокруг
связей С—С, так называемые «линейные» цепи (вытянутые
^ конформации) в действительности могут сворачиваться в шаро-
См. дополнение 8.2 образные глобулы. Подвижность цепей в алканах лучше всего
иллюстрируется молекулярными или компьютерными
моделями. Структура 8.9 представляет собой конформер гептана, в
котором углеродный скелет не является вытянутым.
ВЦ ХИРАЛЬНЫЕ МОЛЕКУЛЫ
Молекулы, которые нельзя совместить с их зеркальным отражением,
называют хиральными. Многие хиральные молекулы содержат асимметрический
атом углерода, т. е. атом углерода, который связан с четырьмя различными
группами (рис. 8.10). Объект и его зеркальное отображение называются
оптическими изомерами или энантиомерами, которые представляют собой
^ частный случай стереоизомеров. Оптические изомеры различаются только
Спиральные молекулы взаимодействием с другими хиральными объектами. Приме-
S : см. разд. 7.8 Ры хиральных объектов — руки перчатки; на правую руку
°° можно надеть только правую перчатку, но не левую.
Спиральные молекулярные цепи также хиральны.
Оптические изомеры или энантиомеры представляют собой стерео-
изомеры, структуры которых несовместимы с их зеркальными
отражениями.
Проверка на хиральность - несовместимость зеркального отражения с
исходным объектом. Существует несколько быстрых тестов, которые
позволяют проверить возможность того, что молекула является хиральной, но все
они неоднозначны:
370
8. Алканы, алкены и алкины
Зеркальная плоскость (плоскость отражения)
Рис. 8.10. Асимметрический атом углерода соединен с четырьмя различными
заместителями. Молекулы, изображенные в левой и правой частях рисунка, являются
зеркальными отображениями, несовместимыми друг с другом; они являются
оптическими изомерами.
• Наличие асимметрического атома углерода часто использовалось в
прошлом. Однако известно много хиральных молекул, которые не содержат
асимметрического атома углерода, и, что более существенно, многие
молекулы, содержащие два или более асимметрических атома углерода,
не являются хиральными.
• Наличие плоскости симметрии. Молекулы, имеющие плоскость
симметрии, не являются хиральными. Однако и в данном случае имеются
исключения - некоторые типы молекул, не обладающие плоскостью симметрии,
являются, тем не менее, ахиральными*.
В 3-метилгексане один из атомов углерода (показан черным цветом на
рис. 8.1 \,а) соединен с атомом водорода, метильной группой, этильной
группой и пропильной группой. Два оптических изомера 3-метилгексана
показаны на рис. 8.11 ,а и 8.11,б. На рис. 8.11 ,в показано, что изомер б не может быть
переведен в изомер а за счет простого вращения, и, следовательно, эти
изомеры не совпадают друг с другом (обратите особое внимание на этот
рисунок).
Если положение и природа заместителя таковы, что при его введении в
молекулу у нее исчезает плоскость симметрии, в результате такого замещения
обычно получается хиральная молекула. Для алканов, которые имеют
большое число структурных изомеров, это приводит к значительному увеличению
общего числа изомеров. В общее число изомеров, учтенных на рис. 8.5,
включены все возможные энантиомеры.
* Однозначным критерием, позволяющим утверждать, что молекула хиральна, является
одновременное отсутствие у нее центра и плоскостей симметрии.- Прим. перев.
8.6. Хиральные молекулы 371
Дополнение 8.4. Плоскость симметрии
Симметрию молекул обычно описывают в терминах операторов симметрии,
таких, как ось вращения или плоскость симметрии.
Если молекула симметрична относительно некоторой плоскости,
проведенной через нее, то говорят, что она имеет плоскость симметрии. У
молекулы может быть более одной плоскости симметрии:
И молекула, и плоскость симметрии Молекула лежит в плоскости листа. Одна
лежат в плоскости листа. из плоскостей симметрии также лежит в
плоскости листа. Вторая плоскость
симметрии проходит перпендикулярно связи
С=С, как показано на рисунке.
Ни одна из молекул, показанных ниже, не содержит плоскости симметрии, и
все три молекулы являются хиральными. Однако следует отметить, что
только один из приведенных примеров имеет асимметрический атом углерода.
Сопоставьте эти результаты с «критериями хиральности», приведенными в
тексте.
Н
Ме°
Et
Me- Г -о
Дополнение 8.5. Хиральность и жизнь
Если осмотреться вокруг, можно легко убедиться, что мы живем в хиральном
мире. На макроскопическом уровне хиральностью обладают некоторые
предметы одежды, виноградная лоза растет в определенном направлении,
вода, вытекая в дренаж, движется по спирали. На молекулярном уровне
живой организм приспособлен к употреблению только одного энантиомера или
диастереомера (см. т. 2, дополнение 14.3) биологических «строительных
материалов» — Сахаров и аминокислот. Может показаться, что использование
«неправильных» диастереомеров сахара (т. е. изомеров, которые не
усваиваются организмом) — это легкий путь к соблюдению диеты для похудения.
Однако, как правило, оказывается, что и сладкий вкус является
специфическим свойством биологически совместимых хиральных форм.
372 8. Алканы, алкены и алкины
Рис. 8.11. а и б- оптические изомеры 3-метилгексана. Атом углерода, показанный
черным цветом, является хиральным центром. Оптические изомеры являются
зеркальными отражениями и не могут быть совмещены друг с другом. Это показано на
диаграмме в, где изомер б претерпевает вращение вокруг вертикальной связи С—С.
Наилучший способ проверить это самостоятельно - компьютерное моделирование
или построение трехмерных моделей молекул.
8.7. Основные особенности механизмов реакций 373
Пример 8.1. Хиральные и ахиральные алканы
Объясните, почему гептан и 4-метилгептан не являются хиральными,
а 3-метил гептан хирален.
Молекула гептана имеет плоскость симметрии, проходящую через атомы
центральной метиленовой группы, в том числе через атом С(4). Если ввести метиль-
ный заместитель в это положение, то плоскость симметрии сохранится (следует
помнить, что вокруг связи С(4)—С(Ме) возможно вращение). Поэтому ни гептан,
ни 4-метилгептан не являются хиральными. Если же ввести метильную группу в
положение 3, плоскость симметрии исчезает, так как один из атомов углерода в
этой хиральной молекуле связан с четырьмя различными заместителями.
И
Замещение в положение С(4)
н,
\ ^Мс
н9
с с
с чсх "сн.
н7
н7
Плоскость симметрии сохраняется
Замещение в положение С(3)
С С С
\ / \ /
Н3С С С "СН,
/ V щ
Me
Плоскость симметрии исчезает
ОСНОВНЫЕ ОСОБЕННОСТИ МЕХАНИЗМОВ РЕАКЦИЙ
Прежде чем перейти к обсуждению реакций углеводородов, необходимо
пояснить, что подразумевается под механизмом реакции. Это понятие широко
используется в химии для описания пути, по которому реализуется
превращение молекул исходных реагентов в молекулы конечных продуктов.
В данном разделе определены основные понятия, используемые при
обсуждении механизмов реакций. Эти понятия являются наиболее
фундаментальными, хотя в последующих разделах будет введен ряд других. Более
подробно вопросы кинетики химических реакций рассмотрены в гл. 10.
Следует особо подчеркнуть, что механизм реакции является не более чем
удобной моделью реальных процессов, сопровождающих химическое
превращение. Механизм невозможно доказать, можно доказать лишь его
непротиворечие с экспериментом.
Энергия активации
► Энергия активации представляет собой энергетический барьер, который не-
Энергия активации: обходимо преодолеть для осуществления реакции. Рассмотрим
см. разд. 1.18 и 10.6 реакцию 8.1.
А+ В->С + D
(8.1)
374
8. Алканы, алкены и алкины
Чтобы молекулы А и В прореагировали друг с другом с образованием С и D,
необходимо разорвать химические связи и/или образовать новые. Точная
последовательность разрыва и образования связей называется путем реакции.
Как правило, существует множество путей реакции между А и В с
образованием С и D, и каждый из них характеризуется своей величиной энергии
активации. Между скоростью реакции и величиной Еа существует тесная
зависимость. Обычно реакция протекает по пути, для которого энергия
активации минимальна*.
Разрыв связей
Разрыв связей может происходить как гомолитически, так и гетеролитически.
В случае гомолитического разрыва связи X—Y на каждый из двух атомов
переносится по одному электрону. Перенос каждого из электронов
обозначается «полустрелкой». Следующее уравнение показывает пример
гомолитического разрыва связи с образованием двух радикалов:
Х-рГ ► X# + Y# (8.2)
При гетеролитическом разрыве оба электрона связи X—Y переносятся на
один из атомов. Такой переход обозначается «полной» стрелкой (уравнение
8.3). Двухэлектронный перенос, как правило, приводит к возникновению
ионов (уравнение 8.4), хотя это не всегда так, - в случае уравнения 8.5 общее
число ионов в ходе процесса остается неизменным.
X-^Y ► Х+ + Y" (8.3)
Ме3С С\ ► Ме3С+ + СГ (8.4)
н н
\ \
0+ Н ► О Н + Н+ (8.5)
Р
н
Гомолитический разрыв одинарной связи X—Y сопровождается
переходом одного электрона на каждый из атомов с образованием
радикалов X* и Y".
Гетеролитический разрыв связи X—Y сопровождается переносом
пары электронов на один из атомов X или Y и, как правило, приводит к
образованию ионов.
* Это утверждение не вполне корректно. В реальности, некоторая доля молекул может преодолевать
практически сколь угодно большие барьеры. Однако основное количество продукта образуется при
преодолении минимального энергетического барьера.
8.7. Основные особенности механизмов реакций 375
Одноэлектронный перенос изображается полустрелкой:
Двухэлектронный перенос изображается полной стрелкой:
При изображении переноса электронов стрелками, их начало и конец
должны быть четко показаны. Не допускается неясное положение стрелок.
Электрофилы и нуклеофилы
Гетеролитический разрыв связи является прямым следствием реакции с
участием электрофила или нуклеофила.
Электрофил является акцептором электронов. Он притягивается к
центрам с повышенной электронной плотностью, имеющим отрицательный
заряд (5—). Нуклеофил является донором электронов и притягивается к
областям, имеющим положительный заряд (5+).
Примеры некоторых наиболее часто встречаемых электрофилов и нук-
леофилов приведены в табл. 8.4.
Электрофил - акцептор электронов; нуклеофил - донор электронов.
Переходное состояние и промежуточные продукты
Для реакции, протекающей по одностадийному механизму (путь которой
показан, например, на рис. 1.9), существует только одна энергия активации.
Однако реакция может идти в несколько стадий, каждая из которых
характеризуется собственной величиной £а. Профиль реакции на рис. 8.12
показывает двухстадийную реакцию между веществами А и В. Энергия активации
первой стадии обозначена £а(1). По окончании первой стадии образуется
промежуточный продукт (интермедиат), который может вступать в
дальнейшую реакцию образования конечных продуктов С и D с энергией активации
Еа(2). Каждый из максимумов энергии соответствует переходному
состоянию. На рис. 8.12 {ПС(1)}* обозначает переходное состояние первой стадии,
а переходное состояние {ПС(2)}* соответствует второй стадии реакции. Для
обозначения переходного состояния обычно используют фигурные скобки и
значок * в верхнем индексе.
Таблица 8.4. Наиболее часто встречаемые электрофилы и нуклеофилы
Название электрофила
Протон
Хлор-катион
Бром-катион
Иод-катион
Метил-катион
Нитрил-катион3
Формула
Н+
С1+
Вг+
1+
Ме+
N02+
Название нуклеофила
Гидрид-анион
Хлорид-анион
Бромид-анион
Метил-анион
Гидроксид-анион
Вода
Формула
н-
С1-
Вг"
Ме"
он-
Н20
а Устаревшее название, которое до сих пор широко используется, — ион нитрония.
376
8. Алканы, алкены и алкины
t
Энтальпия
увеличивается
Энтальпия
уменьшается
♦
{ПС(2)Г
C + D
Координата реакции
Рис. 8.12. Профиль двухстадийной реакции между А и В с образованием С и D.
Первый и второй переходные комплексы обозначены {ПС(1)}* и {ПС(2)}* соответственно.
Изменение энтальпии в ходе реакции равно А Я.
Важно понимать различие между переходным состоянием (переходным
комплексом) и промежуточным продуктом. Переходному состоянию
соответствует энергетический максимум на реакционном профиле, а промежуточный
продукт находится в локальном минимуме. Переходный комплекс
невозможно выделить, тогда как промежуточные продукты могут быть
зафиксированы и выделены в чистом виде.
Переходный комплекс соответствует энергетическому максимуму и
не может быть выделен. Промежуточный продукт находится в
локальном энергетическом минимуме, может быть зафиксирован и в ряде
случаев получен в чистом виде.
Лимитирующая стадия
В случае, если реакция протекает в несколько стадий, первая стадия не обя-
^ зательно имеет самую высокую энергию активации. Чем выше величина £а,
Лимитирующая тем медленнее идет реакция, и, следовательно, наличие стадии
стадия: см. разд. 10.9 с высокой энергией активации будет замедлять реакцию.
Стадия с наибольшей энергией активации называется
лимитирующей стадией (ЛС), скоростьопределяющей или самой медленной стадией.
8.8. Физические свойства алканов
377
АН
Реагенты
Продукты
Реагенты
Продукты^
Координата реакций
Координата реакций
Рис. 8.13. Профили двух трехстадийных реакций, а — стадия 1 является лимитирующей, так
как имеет наибольшую энергию активации; стадии 2 и 3 протекают быстро; б— стадия 2
является медленной.
Лимитирующая стадия (ЛС) - самая медленная стадия реакции.
На рис. 8.13 показаны профили двух трехстадийных реакций. На рис. 8.13,а
максимальную энергию активации имеет стадия 1, которая и является
лимитирующей стадией. После преодоления этого барьера, стадии 2 и 3
протекают быстрее стадии 1. На рис. 8.13,5 лимитирующей стадией является стадия
2, а стадии 1 и 3 протекают быстро.
ФИЗИЧЕСКИЕ СВОЙСТВА АЛКАНОВ
Физические свойства алканов можно рассматривать в разных аспектах.
Например, как меняются физические свойства у нескольких структурных
изомеров? Однако наиболее разумный путь для определения закономерностей
изменения физических свойств - анализ гомологических рядов.
Гомологический ряд алканов
Электроотрицательности углерода и водорода по Полингу равны 2,6 и 2,2
соответственно. Следовательно, дипольный момент связи С—Н достаточно
мал, и связь является практически неполярной.
Алканы представляют собой почти неполярные молекулы, поэтому
межмолекулярные взаимодействия как в жидкости, так и в твердом теле
осуществляются за счет слабых вандерваальсовых сил. На рис. 8.14 показано
изменение температур плавления и кипения в гомологическом ряду алканов
С/7Н2п+2 для п от 1 до 20. Следует отметить, что при 298 К и атмосферном
давлении метан, этан, пропан и бутан газообразны, неразветвленные алканы от
пентана до гептадекана находятся в жидком состоянии, а высшие алканы
представляют собой твердые вещества. На рис. 8.15 показаны шаростержне-
вая и полусферическая модели молекулы пентана в вытянутой конформации.
Шаростержневая модель подчеркивает вытянутую форму молекулы. И в
жидкости, и в твердой фазе силы Ван-дер-Ваал ьса действуют между атомами,
расположенными на поверхности молекул. На рис. 8.16 показано взаимное
расположение молекул икозана в кристаллической решетке. Общее повыше-
378
8. Алканы, алкены и алкины
л 600
о.
& 400
С
£ 298
200
Рис. 8.14. Тенденции в изменении температур плав- о
ления (I) и кипения (2) алканов СлН2л+2 с ростом п. 0 5 10 15 20
п
Дополнение 8.6. Метан и парниковый эффект
Повышение концентрации некоторых газов в атмосфере может приводить к
глобальному потеплению - так называемому «парниковому эффекту». Пожалуй, наиболее
важными «парниковыми газами» являются С02 (см. т. 2, дополнение 13.2) и СН4.
Метан возникает при анаэробном (т. е. происходящем в бескислородной среде)
распаде органических веществ; пузырьки метана образуются в болотистых местностях, за
что он получил название «болотный газ». Затопленные поля (например, рисовые поля
во время сева) являются источником большого количества метана. Другим источником
метана являются жвачные животные, такие, как коровы, овцы и козы,
пищеварительная система которых устроена особым образом для переваривания травяной пищи:
конечные продукты метаболизма содержат метан, который выделяется в атмосферу. Это
природный процесс, хотя количество домашних животных заметно выросло за
последние два столетия. Более того, состав кормов существенно влияет на количество
образующегося метана. На схеме показано количество метана, вьщеленного в атмосферу
людьми и животными в 1984 году - на долю крупного рогатого скота приходится 71%
от общего количества метана [Chemistry & Industry, 1992, p. 334].
Крупный рогатый скот из развитых стран (41%)
Крупный рогатый скот из развивающихся стран
(30%)
Овцы (9%)
Быки (8%)
Дикие животные (5%)
Козы (3%)
Лошади и мулы (2%)
Свиньи (1%)
Верблюды (1%)
Люди (0,4%)
i i i i_
8.8. Физические свойства алканов 379
Рис. 8.15. Модели молекулы пентана в вытянутой конформации. а — шаростержне-
вая модель; б— полусферическая модель. В случае полусферической модели, размеры
шаров-атомов показывают область пространства, в котором электроны проводят
95—99% времени. Для построения шаровых моделей можно использовать значения
ковалентных и вандерваальсовых радиусов атомов. Шаростержневая модель является
удобным способом изображения пространственного строения молекулы.
Рис. 8.16. Молекулы икозана в кристаллической решетке имеют вытянутую кон-
формацию и располагаются параллельно друг другу.
ние температур плавления и кипения с ростом длины цепи отражает тот факт,
что межмолекулярные взаимодействия усиливаются с увеличением
поверхности молекулы.
Плотности неразветвленных алканов вначале увеличиваются с ростом
длины углеродной цепи, а затем выходят на постоянную величину порядка
^ 0,8 г/см3; следовательно, все алканы легче воды. Алканы не смешиваются с
Плотность воды = водой, поскольку они неполярны, а вода полярна. При попыт-
1,00 г/см3 ке смешивания жидкого алкана с чистой водой алкан образует
верхний слой.
Если две жидкости смешиваются, они образуют раствор. Если
жидкости не смешиваются, они образуют два слоя, при этом более
плотная жидкость находится в нижнем слое.
Влияние разветвления цепи на температуру кипения
В табл. 8.5 приведены температуры кипения изомеров С6Н14. Как правило,
изомеры с разветвленной цепью имеют более низкие температуры кипения,
8. Алканы, алкены и алкины
Таблица 8.5. Температуры кипения гексана и его структурных изомеров,
измеренные при атмосферном давлении
Изомер Углеродный скелет Температура кипения, К
Гексан /"N/^\/ 342
2-Метилпентан /\/\ 333
З-Метилпентан -^^Y^^ 336
2,3-Диметилбутан у^^^ 331
2,2-Диметилбутан W 323
чем изомеры с неразветвленной цепью, так как молекулы алканов при
увеличении степени разветвления принимают форму, более близкую к
сферической и силы межмолекулярного взаимодействия уменьшаются по мере
уменьшения площади контакта между молекулами.
ПРОМЫШЛЕННАЯ ПЕРЕРАБОТКА УГЛЕВОДОРОДОВ
Переработка нефти
После выделения из природных источников сырую нефть разделяют на
основные фракции (газ, моторные топлива, керосин, дизельное топливо,
парафин, смазочные масла и битум) путем ректификации (фракционной
перегонки) и отделения газов. Для перегонки используются ректификационные
колонны, каждая из которых содержит около 40 тарелок; сырая нефть
вводится в колонну при температуре порядка 600 К. Пары поднимаются вверх по
колонне, температура которой в верхней части ниже, чем внизу колонны.
Только наиболее низкокипящие вещества (имеющие самые низкие
молекулярные массы) достигают тарелок, расположенных в верхней части колонны.
На каждой тарелке собирается фракция углеводородов, кипящая в
определенном интервале температур. Самые высококипящие углеводороды
остаются внизу колонны и в дальнейшем подвергаются перегонке при пониженном
давлении. Вторичная перегонка фракции приводит к ее дальнейшему
разделению на фракции, кипящие в более узком интервале температур.
Крекинг
Не все фракции, полученные после ректификации, состоят из
углеводородов, пригодных для коммерческого использования, — в них содержится
слишком много примесей углеводородов с высокой молекулярной массой.
Такие фракции подвергают крекингу. Этот процесс приводит к разрыву длин-
8.9. Промышленная переработка углеводородов 381
Дополнение 8.7. Вино как индикатор загрязнения свинцом
Группа французских и бельгийских исследователей использовала пробы вина
Chateauneuf-du-Pape для мониторинга загрязнения свинцом из выхлопов
моторных двигателей. Источником винограда для этого вина долгое время
служили виноградники, расположенные вдоль шоссейных дорог. Вино,
произведенное между 1962 и 1991 годами, было проанализировано на свинец.
Изменение содержания антидетонирующей добавки тетраэтилсвинца
Pb(Et)4 в течение этого периода времени приведено на схеме.
500 г
ш 400 |-
I
s
х
3
а
§
U
300 h
200
100
EL
nu
1965
1970
1975
Годы
1980
1985
1990
Сначала увеличение содержания свинца было связано с увеличением числа
автомобилей, проезжающих вблизи виноградников. Уменьшение совпадает с
введением бессвинцового топлива. В годы максимального содержания свинца
уровень загрязнения представлял серьезную угрозу для здоровья [Lobinski R.
etai, Nature, 1994, vol. 370, p.24].
ных углеводородных цепей на более короткие фрагменты и сопровождается
образованием алкенов (отсутствующих в сырой нефти), которые
используются в качестве исходных материалов в полимерной промышленности.
Крекинг можно проводить при высоких температурах и давлениях (термический
крекинг) или в присутствии катализатора (каталитический крекинг).
Термический крекинг используют как для переработки жидких фракций, так и для
переработки твердого остатка, а каталитический крекинг пригоден только
для переработки жидких компонентов. Типичным катализатором крекинга
является оксид алюминия.
Риформинг
Горение фракций, используемых в качестве топлива, должно быть как можно
более ровным. Чтобы достичь этого, фракции подвергают процессу рифор-
минга, в ходе которого неразветвленные алканы превращаются в
разветвленные. Риформинг представляет собой реакцию изомеризации. При сгорании
неразветвленных алканов в двигателе их воспламенение происходит
слишком быстро, что приводит к детонации. Октановое число моторного топлива
382
8. Алканы, алкены и алкины
характеризует устойчивость топлива к детонации; октановое число может
изменяться от 0 до 100, причем 100 соответствует топливу, полностью
устойчивому к детонации. В топливо можно добавлять антидетонационные добавки,
такие, как тетраэтилсвинец Pb(Et)4, но соединения свинца загрязняют
окружающую среду, поэтому современные двигатели разрабатываются для
топлива, не содержащего свинца.
ESQ РЕАКЦИИ АЛКАНОВ
Для алканов характерны следующие типы реакций:
• горение;
• крекинг (см. разд. 8.9);
• радикальное замещение.
Горение
^~ Алканы горят в кислороде с образованием диоксида углерода и воды (уравне-
См. разд. 1.17 ния 8.6 и 8.7); горение алканов является экзотермическим
процессом.
СН4 + 202 -> С02 + 2Н20 (8.6)
СИН24+ 1702-> 11С02+ 12Н20 (8.7)
На практике наблюдается неполное сгорание алканов, при котором наряду с
С02 образуются различные количества элементного углерода и других
углеродных соединений. Присутствие элементного углерода и больших
ароматических молекул приводит к образованию значительного количества сажи при
горении высших углеводородов. Если количество 02 ограничено (например,
при горении топлива в закрытом пространстве), горение может приводить к
образованию моноксида углерода СО. Образование СО при неполном
сгорании углеводородов в неисправном домашнем обогревательном обрудовании
может приводить к смертельным случаям.
Пример 8.2. Определение формулы углеводорода
При полном сгорании ациклического алкана X образуется 211,2 г
С02 и 97,2 г Н20. Определите углеводород. [Относительные
атомные массы: АГ(С) = 12,4Г(0) =16, АГ(И) = 1.]
Решение
Молекулярная формула углеводорода задает соотношение молей атомов
С : Н; его можно найти по отношению молей С02 и Н20, образовавшихся при
сгорании.
Количество молей = Масса/Относительная молекулярная масса
Количество молей С02 = 211,2/44 = 4,8
8.10. Реакции алканов
383
Дополнение 8.8. Степени окисления в соединениях углерода
Как правило, степени окисления углерода не рассматриваются, но поскольку
некоторые органические реакции относятся к реакциям окисления или
восстановления, полезно кратко рассмотреть возможные формальные степени
окисления углерода.
Окисление соответствует: Восстановление соответствует:
• отщеплению атомов водорода или • присоединению атомов водорода или
• присоединению атомов кислорода или • отщеплению атомов кислорода или
• потере электронов • присоединению электронов.
Следует выбирать наиболее удобное из указанных определений окисления и
восстановления в зависимоости от ситуации:
Восстановление: С2Н4 -I- Н2 —> С2Н6
Окисление: С3Н8 —> С3Н6 + Н2
Окисление: СН4 + 202 -> С02 + 2Н20
Элементы группы 14 - кремний, германий и олово - образуют гидриды
общего состава ЕН4 (Е = Si, Ge или Sn). В этих соединениях элементу Е
приписывается формальная степень окисления +4, а водороду — степень окисления
-1, и эти значения находятся в соответствии с шкалой
электроотрицательности Полинга (табл. 4.2). СН4 является членом этого ряда соединений, но
значения хп(С) = 2,6 и хп(Н) = 2,2 показывают, что атом углерода имеет
небольшой отрицательный заряд 6-. Поэтому в СН4 углероду приписывается
формальная степень окисления -4, а водороду +1.
Если атомы водорода в алкане замещаются на атомы галогена, ситуация
меняется на противоположную [хп(С) = 2,6; xn(F) = 4,0; хП(С1) = 3,2;
Хп(Вг) = 3,0; хп0) = 2,7] и атом углерода имеет степень окисления +4 (CF4,
CF2C12, CC14). В СН3С1, как и в метаноле СН3ОН, степень окисления
углерода равна —2. При формальном расчете степени окисления можно не
учитывать связи С—С (вспомните, какова степень окисления фтора в F2). Степень
окисления углерода в С2Н6 равна —3. В молекуле СО степени окисления О и
С равны —2 и +2 соответственно.
Вопрос: Какова степень окисления углерода в следующих соединениях: CH3F,
СН2С12, СН3СН2ОН, С12С=0, С02, С032?
Количество молей Н20 = 97,2/18 = 5,4
Количество молей С в сгоревшем соединении X равно количеству молей С02,
т. е. 4,8.
Количество молей Н в сгоревшем соединении X равно удвоенному количеству
молей Н20, т. е. 10,8.
Отношение С : Н = 4,8 : 10,8 = 1 : 2,25
Следовательно, формула X должна быть СЯН2 25я, но мы знаем, что X —
ациклический алкан, имеющий формулу СяН2я+2.
2л + 2 = 2,25л
384
8. Алканы, алкены и алкины
0,25л = 2
п = 2/0,25 = 8
Таким образом, X имеет формулу С8Н18 (октан или один из его изомеров).
Галогенирование
Если смесь СН4 и С12 поставить на солнечный свет, произойдет реакция, в
результате которой образуются хлороводород и ряд хлоропроизводных метана
►- СН4_ЛС1Л, где п = 1, 2, 3 или 4. Уравнения 8.8—8.11 показывают
Спектр электромагнит- образование хлорметана и последующие реакции с С12; символ
ных излучений: см. /^ означает «облучение» и указывает на то, что для иницииро-
приложение 1 в т. 2 вания реакции реагенты необходимо облучить светом. Это фо-
толитическая реакция, для ее инициирования обычно
используют излучение с длиной волны 200-800 нм .
Фотолитические реакции происходят при облучении (обозначаемом /tv)
СН4 + С12 ^> СН3С1 + НС1 (8.8)
СН3С1 + С12 Л СН2С12 + НС1 (8.9)
СН2С12 + С12 Л СНС13 + НС1 (8.10)
СНС13+ С12 -> СС14 + НС1 (8.11)
Указанные реакции являются реакциями замещения. Атомы хлора замещают
атомы водорода в СН4 — этот частный случай замещения называется
реакцией хлорирования. Бромирование метана происходит при фотолизе смеси Вг2 и
СН4, однако аналогичная реакция с иодом не происходит. Реакция с F2
протекает со взрывом даже в отсутствие света. Ряд изменения реакционной
способности галогенов по отношению к метану имеет следующий вид:
F2»Cl2>Br2»I2
Хотя иод не реагирует с метаном, он включен в этот ряд, поскольку такая
закономерность справедлива для многих реакций галогенов.
Фторирование, хлорирование и бромирование высших алканов приводит
к образованию сложных смесей продуктов замещения. По мере увеличения
числа углеродных атомов в молекуле растет число возможных позиций для
замещения. В качестве упражнения напишите возможные продукты
монохлорирования октана.
В реакции замещения один атом или группа атомов обмениваются на
другие.
8.11. Хлорирование метана. Радикальная цепная реакция 385
ЕЖП ХЛОРИРОВАНИЕ МЕТАНА.
РАДИКАЛЬНАЯ ЦЕПНАЯ РЕАКЦИЯ
В данном разделе обсуждается механизм реакции хлорирования СН4, но он
имеет общий характер для реакций хлорирования и бромирования алканов, а
взаимодействие с F2 может протекать по другому механизму. Реакция между
СН4 и С12 — пример радикальной цепной реакции.
Основные особенности радикальных цепных реакций
► Радикальная цепная реакция включает следующие стадии:
Различие между
линейными и разветв- • зарождение (инициирование) цепи;
ленными цепными т рост цепи;
реакциями: . обрыв цепи
см. разд. 10.11
На стадии зарождения цепи происходит образование свободных
радикалов из одного или нескольких реагентов. Радикалы могут возникать под
действием нагревания, при облучении реакционной смеси, а также при действии
химического инициатора. В частности, в качестве инициатора может быть
использован «стабильный» свободный радикал. Уравнение 8.12 показывает
типичную стадию зарождения цепи, в которой радикалы X* образуются из
молекулы Х2 за счет гомолитического разрыва связи при облучении светом с
определенной длиной волны.
Av
Х2 -> 2Х* Зарождение цепи (8.12)
Органические радикалы часто (хотя и не всегда) имеют короткое время
жизни. При их столкновении с другими частицами могут происходить реакции,
результатом которых становится либо рост, либо обрыв цепи. При
столкновении радикала с нерадикалом может произойти рост цепи, если при этом
образуется новый активный радикал (уравнение 8.13). Новый радикал может
далее вступать в реакцию с очередной частицей с образованием третьего
радикала (уравнение 8.14). Хотя нерадикалы в уравнениях 8.13 и 8.14
обозначены одинаково (RY), в действительности это не обязательно одно и то же
соединение (см. ниже):
X* +RY -»RX + V Рост цепи (8.13)
V + RY -> Y2 + R* Рост цепи (8.14)
При обрыве цепи происходит исчезновение радикалов из реакционной смеси.
Стадия обрыва цепи иллюстрируется уравнением 8.15. В данном случае
обрыв цепи происходит по реакции, обратной зарождению цепи. Радикал X*
может также соединяться с любыми другими радикалами, присутствующими
в реагирующей системе.
2Х# -> Х2 Обрыв цепи (8.15)
Общая последовательность указанных стадий и представляет собой цепную
реакцию, которая поддерживается за счет стадии роста цепи. Поскольку кон-
386
8. Алканы, алкены и алкины
центрация радикалов в реагирующей системе в каждый момент времени
мала, столкновение радикалов с нерадикалами {рост цепи) гораздо более
вероятно, чем столкновение радикалов между собой (обрыв цепи). Между
реакциями зарождения и обрыва цепи успевает произойти большое число реакций
роста. Как правило, образование одного свободного радикала приводит к
получению нескольких тысяч молекул продукта, прежде чем произойдет обрыв
цепи. Горение алканов и других органических соединений также относится к
классу радикальных цепных реакций.
Радикальная
цепная
реакция
I
1. Зарождение цепи (образование радикалов)
2. Рост цепи (радикалы вступают в реакцию
и образуются новые радикалы)
3. Обрыв цепи (удаление радикалов из реакции)
Механизм реакции хлорирования метана
При поглощении света молекулой С12 происходит гомолитический разрыв
связи CI—C1 (уравнение 8.16):
С12->2С1* (8.16)
Радикалы хлора могут сталкиваться с молекулами метана, в результате чего
происходит рост цепи, включающий стадии 8.17—8.20. На каждой из этих
стадий один радикал исчезает, а другой появляется. В уравнениях 8.17 и 8.19
радикал О* отбирает атом водорода от молекулы реагента с образованием
НС1.
СГ + СН4 -> НС1 + СН3* (8.17)
СН3* + С12 -> СН3С1 + СГ (8.18)
СГ + СН3С1 -* НС1 + СН2СГ (8.19)
СН2С1* + С12 -> СН2С12 + СГ (8.20)
Обрыв цепи происходит при взаимодействии двух радикалов. Уравнения
8.21—8.23 показывают три возможных способа обрыва цепи, но существует и
ряд других, например с участием хлоралкильных радикалов (типа СН2С1').
2СГ->С12 (8.21)
СГ + СН3# -» СН3С1 (8.22)
2СН3# -» С2Н6 (8.23)
Реакция 8.21 обратна реакции зарождения цепи (уравнение 8.13), в то время
как реакция 8.22 приводит к образованию конечного продукта. Реакции типа
8.23 усложняют процесс, поскольку приводят к росту числа атомов углерода
в цепи. Молекула этана также может участвовать в реакциях, аналогичных
8.12. Хлорирование пропана и 2-метилпропана 387
реакции 8.17, а это в свою очередь может приводить к дальнейшему росту
углеродного скелета и образованию высших алканов. Таким образом,
радикальные цепные реакции неспецифичны.
ХЛОРИРОВАНИЕ ПРОПАНА И 2-МЕТИЛПРОПАНА
Для алканов, содержащих более двух углеродных атомов в цепи, возникает
дополнительное осложнение: радикал хлора может атаковать различные
углеродные центры. Данная проблема проиллюстрирована на примере
хлорирования пропана и 2-метилпропана.
Реакция пропана с хлором
Зарождение цепи при реакции пропана с хлором происходит по реакции 8.16.
Дальнейший рост цепи может идти двумя способами, поскольку в молекуле
пропана присутствуют два типа атомов водорода, находящиеся
соответственно на первичном и вторичном углеродных центрах (уравнения 8.24—8.26).
вторичный
,НС1 + СН3СН2СН2
СН3СН2СН3-^< (8.24)
НС1 + СН3СНСН3
первичный первичный
СН3СН2СН2* + С12 -> СН3СН2СН2С1 + СГ (8.25)
1-Хлорпропан
СН3СНСН3 + С12 -> СН3СНС1СН3 + СГ (8.26)
2-Хлорпропан
В смеси конечных продуктов преобладает 2-хлорпропан, несмотря на то что
вероятность столкновения хлоридного радикала с атомом водорода при
первичном атоме углерода выше, чем вероятность его соударения с атомом
водорода при вторичном атоме углерода. Таким образом, удаление атома
водорода с образованием вторичного алкильного радикала предпочтительнее, чем
удаление атома водорода с образованием первичного алкильного радикала.
Реакция хлора с 2-метилпропаном
После зарождения цепи (реакция 8.16) реакция радикалов хлора с
2-метилпропаном (уравнение 8.27) и последующие стадии роста цепи могут
приводить к двум изомерным продуктам (уравнения 8.28 и 8.29).
СН3
| * НС1 + СН3СН(СН3)СН2#
СИ -^< (8-27)
нзС сн3 ^ на + сн3с(сн3)сн3
8. Алканы, алкены и алкины
Дополнение 8.9. Разрушение озонового слоя
Озоновый слой находится в атмосфере на высоте 15-30 км над земной
поверхностью. Озон активно поглощает УФ-лучи, и слой озона предохраняет
Землю от опасных УФ-лучей Солнца, которые могут вызывать заболевание
раком кожи.
Фторхлоруглероды (ФХУ) представляют собой группу веществ,
загрязняющих атмосферу. Они используются в качестве носителей в аэрозольных
баллончиках, холодильниках и кондиционерах, а также в качестве растворителей
и пенообразователей. ФХУ попадают в атмосферу и, поднимаясь в
стратосферу, участвуют в фотохимических реакциях, например:
УФ
CC12F2 >C1# + CC1F2#
В результате зарождается радикальная цепная реакция, в которой могут
принимать участие молекулы озона:
03 + С1*—>02 + С10*
Разрушительным результатом этих процессов становится постепенное
истощение озонового слоя. Это впервые было обнаружено в 1970-е годы, и в ряде
стран немедленно были предприняты шаги по законодательному запрещению
использования ФХУ. Однако, несмотря на все увеличивающееся производство
безвредных для озонового слоя аэрозолей и других продуктов, проблема
разрушения озонового слоя пока не решена. Выделение ФХУ в атмосферу
необходимо существенно уменьшить, если мы хотим оставаться защищенными от
вредного действия солнечного УФ-излучения.
В 1987 г. был подписан Монреальский протокол по защите озонового
слоя, который призывает к прекращению производства ФХУ. На рисунке
показано уменьшение потребления ФХУ в Европе в период между 1986 и 1993 гг.
Значительный вклад в общее уменьшение внесли жесткие ограничения на
использование ФХУ в аэрозолях [Chemistry&Industry, 1994, р. 323].
1986 1987 1988 1989 1990 1991 1992 1993
8.12. Хлорирование пропана и 2-метилпропана 389
СН3СН(СН3)СН2# + С12 -> СН3СН(СН3)СН2С1 + СГ (8.28)
1 -Хлоп-2-метилпгюпан
1 -Хлор-2-метилпропан
СН3С(СН3)СН3 + С12 -> СН3СС1(СН3)СН3 + СГ (8.29)
2-Хлор-2-метилпропан
Эксперименты показывают, что преобладающим продуктом является 2-хлор-
2-метилпропан, так что удаление атома водорода с образованием третичного
алкильного радикала предпочтительнее, чем удаление атома водорода с
образованием первичного алкильного радикала.
Первичные, вторичные и третичные радикалы
Примеры, приведенные выше, позволяют сделать важное обобщающее
заключение. В радикальной реакции третичные радикалы образуются
предпочтительнее, чем вторичные, а вторичные — предпочтительнее, чем первичные. Это
влияет на состав продуктов цепной реакции, а также на ее скорость.
Экспериментальные данные показывают, что устойчивость радикалов
изменяется в следующем порядке:
третичный (R3C#) > вторичный (R2CH#) > первичный (RCH2#) > метальный (СН3*)
где R — алкильный заместитель.
Атом углерода в радикале имеет только семь валентных электронов и
является электронодефицитным. Наблюдаемый ряд изменения устойчивости
радикалов показывает, что алкильные группы лучше, чем атомы водорода,
стабилизируют электронодефицитные центры. Можно сказать, что
алкильные группы обладают электронодонорными свойствами, поскольку в связи
С—R электроны смещаются к атому углерода: С5- — R5+.
Алкильные группы R обладают элетронодонорными свойствами, и
стабильность радикалов изменяется в следующем ряду:
R3C# > R2CH# > RCH2# > СН3#
РЕАКЦИИ АЛКЕНОВ
1. РЕАКЦИИ ОКИСЛЕНИЯ И ПРИСОЕДИНЕНИЯ
Аналогично алканам, алкены горят в кислороде с образованием воды и
углекислого газа:
СН3СН2СН=СН2 + 602 -> 4С02 + 4Н20 (8.30)
В то время как для алканов характерны реакции замещения, в алкенах связь
С=С вступает в реакции присоединения:
А В
+ АВ > 4 L (8.31)
390 8. Алканы, алкены и алкины
Для алкенов характерны реакции:
• электрофильного присоединения;
• радикального присоединения;
• полимеризации.
В данном разделе приведены примеры некоторых реакций окисления и
присоединения по двойной связи. Механизмы этих реакций обсуждаются в
разд. 8.14. Другие реакции рассмотрены в последующих разделах.
Гидрирование
При гидрировании алкенов происходит присоединение водорода, при этом
ненасыщенный углеводород превращается в насыщенный; это важный спо-
^ соб синтеза алканов:
Катализаторы:
см. разд. 10.7 СН3СН=СН2 + Н2 атализатор )СН3СН2СН3 (8.32)
Присутствие катализаторов, в качестве которых используются
высокодисперсные металлические никель, палладий или платина, необходимо,
поскольку энергия активации реакции водорода с алкеном достаточно высока.
На рис. 8.17 схематически показано действие катализатора, которое состоит
в понижении энергии активации при переходе от алкена (R2C=CR2) к
продукту гидрирования (R2CH—CHR2); в результате скорость реакции
увеличивается. Использование металлов в качестве катализаторов реакции между
газами или между газом и жидкостью является примером гетерогенного катализа.
Гидрирование - реакция присоединения водорода.
Присоединение галогенов
Алкены присоединяют С12 и Вг2 с образованием дихлор- и дибромпроизвод-
ных соответственно (уравнения 8.33 и 8.34). Атомы галогенов в продуктах
реакции соединены с соседними атомами углерода - такие соединения
называют также вицинальными дигапогенидами.
СН3СН2СН=СНСН3 + С12 -> СН3СН2СНС1СНС1СН3 (8.33)
2,3-Дихлорпентан
СН2=СНСН2СН3 + Вг2 -> СН2ВгСНВгСН2СН3 (8.34)
1,2-Дибромбутан
Взаимодействие со фтором протекает весьма энергично, а реакции с иодом,
напротив, идут очень медленно или не идут совсем. Хлорирование или бро-
мирование протекает при температуре 298 К или ниже, для инициирования
реакции не требуется облучение. Это позволяет предположить, что в данной
реакции не участвуют свободные радикалы. Если источник свободных ради-
8.13. Реакции алкенов. 1. Реакции окисления и присоединения 391
Энтальпия
увеличивается
Еа(в отсутствие катализатора)
£а (в присутствии
i катализатора)
R2HC-CHR2
Координата реакции
Рис. 8.17. Реакция присоединения водорода к алкенам имеет высокую энергию
активации. Схематические профили реакции, приведенные на рисунке, показывают, что
в присутствии соответствующего катализатора величина Е понижается.
калов присутствует, присоединение галогенов все равно происходит, но
может осложняться конкурирующими реакциями радикального замещения.
Обесцвечивание водного раствора брома (бромной воды) обычно
используют для качественного определения присутствия двойной связи С=С, хотя этот
тест нельзя считать однозначным. При взаимодействии с бромной водой
образуется смесь продуктов (см. ниже обсуждение реакций алкенов с Вг2 и Н20).
Присоединие галогеноводородов
Уравнение 8.35 представляет реакцию присоединения хлороводорода, бромо-
водорода или иодоводорода к алкену. Поскольку исходный алкен — бутен-2 —
симметричен, единственным продуктом присоединения НХ может быть
2-галогенбутан.
СН3СН=СНСН3 + НХ -
> сн3сн2снхсн3
(8.35)
X = CI, Br или I
При взаимодействии с несимметричными алкенами возможно
образование двух продуктов, как показано на примере присоединения НВг к пропену
(уравнение 8.36). В действительности, основным продуктом оказывается
2-бромпропан. Механизм этой реакции рассмотрен в следующем разделе,
здесь же отметим лишь предпочительность присоединения атома галогена к
вторичному атому углерода, а не к первичному.
392
8. Алканы, алкены и алкины
СН,СН=СН9 + НВг
СН3СН2СН2Вг
1-Бромпропан
СН3СНВгСН3
2-Бромпропан
Побочный продукт
Основной продукт
(8.36)
Аналогично, при реакции НС1 с 2-метилпропеном основным продуктом
является 2-хлор-2-метилпропан, т. е. атом галогена предпочительнее
присоединяется к третичному (а не к первичному) атому углерода.
Присоединение серной кислоты и воды
Алкены реагируют с холодной концентрированной серной кислотой
(уравнение 8.37). В результате реакции образуется алкилгидросульфат, который
имеет структуру 8.10.
СН3СН=СН2 + H2S04 -> CH3CH(OS03H)CH3 (8.37)
Реакция заключается в присоединении О—Н-группы, имеющейся в
молекуле серной кислоты, по двойной связи С=С. Реакция аналогична
присоединению галогеноводородов, рассмотренному выше. В продукте реакции 8.37
атом кислорода также предпочтительнее присоединяется к вторичному
атому углерода, как и атом брома в примерах, обсуждавшихся выше.
ч
ОН
СИ
8.10
сн.
о
сн
8.11
н
сн,
►* Соли алкилгидросульфаты, как правило, не выделяют, но при добавлении во-
Нуклеофил: см. разд. 8.7 ды и нафевании происходит гидролиз (уравнение 8.38) и об-
Детальный механизм разуется спирт. Гидролиз соединения 8.10 в 8.11 протекает по
нуклеофильного механизму нуклеофильного замещения, при этом нуклеофилом
замещения: см. гл. 14 является молекула воды (а не гидроксид-ион).
Процесс гидролиза состоит в замещении некоторой группы X на
ОН-группу в результате реакции с Н20.
CH3CH(OS03H)CH3 + Н20
> СН3СН(ОН)СН3 + H2S04
Пропанол-2
(8.38)
8.13. Реакции алкенов. 1. Реакции окисления и присоединения 393
Реакции 8.37 и 8.38 позволяют синтезировать спирты из алкенов, а серная
кислота выступает в качестве катализатора, поскольку H2S04 хотя и
реагирует в первой реакции, но регенерируется во второй. Если целевым продуктом
является спирт, обе реакции можно проводить одновременно с
использованием водного раствора кислоты (уравнение 8.39).
Катализатор Н+
СН3СН=СН2 + Н20 > СН3СН(ОН)СН3 (8.39)
^~ Реакцию 8.39 можно проводить в обоих направлениях — при нагревании
Дегидратация спир- спиртов с концентрированными кислотами происходит обра-
тов: см. разд. 14.10 зование алкенов наряду с другими продуктами.
Реакции с галогенами и водой
Хлорная или бромная вода реагирует с алкенами по уравнениям
С12, Н20
СН3СН=СН2—-—^->СН3СН(ОН)СН2С1 (8.40)
1-Хлорпропанол-2
Вг9, Н90
СН3СМе=СН2 ——2-> СН3СМе(ОН)СН2Вг (8.41)
1-Бром-2-метилпропанол-2
Гидроксильная группа предпочтительнее присоединяется к вторичному или
третичному углеродному атому. Образование вицинальных дигалогенидов
может конкурировать с образованием продуктов реакций 8.40 и 8.41, как это
наблюдается при проведении качественной реакции на алкены с бромной водой.
Образование диолов
Окисление алкенов холодным щелочными раствором перманганата калия*
приводит к образованию вицинальных диолов:
Холодный щелочной
раствор КМпО>.
СН3СН2СН=СН2 - >СН3СН2СН(ОН)СН2ОН (8.42)
Бутандиол-1,2
Предполагается, что реакция протекает с образованием промежуточного
продукта, имеющего структуру 8.12; в ходе реакции алкен окисляется, а
марганец восстанавливается.
О О
\/
MnV
\ /
— С С —
/ \
8.12
И ЮПАК допускает использование названия перманганат для иона МпО"^, и оно широко
используется. Систематическое название этого аниона - тетраоксоманганат(УН).
394
8. Алканы, апкены и алкины
Устаревшее, но до сих пор используемое название диолов — гликоли.
Название гликолей образовывали добавлением слова «гликоль» к названию
исходного алкена. Например, этандиол-1,2 также называют этиленгликолем
(этилен - другое название этена). Этиленгликоль используется в качестве
антифриза, добавляемого в воду в автомобильных радиаторах. Он имеет низкую
температуру плавления (261 К), высокую температуру кипения (471 К) и
неограниченно смешивается с водой.
Озонирование
При реакции алкенов с озоном образуются циклические озониды (в ходе
реакции образуется циклический промежуточный продукт, изомерный
озон иду):
О-
■О
СН3
сн3сн=сн2 + о3
\
/ \/н
/V/ \
н ° н
(8.43)
Важное применение этой реакции состоит в выделении не самих озонидов
^* (которые часто взрывчаты), а продуктов их гидролиза: восста-
Альдегиды и кетоны: новительный гидролиз озонидов приводит к образованию
альдегидов или кетонов в зависимости от строения исходного
алкена. Два примера гидролиза озонидов - реакции 8.44 и 8.45.
см. табл. 1.9 и гл. 17
СН
О О
с с
о — о
сн3сн2 / \ н
с с
о
о
Zn, H,0
СН3С
/
\
/
н
Этаналь
О
НС
\
Н
Метаналь
О
(8.44)
Zn, H20
-> СН3СН2С
/
\
НС
/
\
(8.45)
CHi
н
Бутанон-2
Метаналь
Дополнение 8.10. Полиэтиленгликоль и «Мэри Роз»
Затонувший военный корабль Тюдоров «Мэри Роз» был поднят со дна в 1982г.
Немедленно после поднятия возникла проблема, существенная для всех
деревянных предметов, долгое время пробывших под водой: как сохранить
деревянную обшивку корабля после высыхания. Метод сохранения дерева, долгое
время находившегося во влажном состоянии, состоит в обработке полиэти-
ленгликолем. Пропитка этим воскоподобным веществом позволяет
предотвратить распад сохнущего дерева.
8.13. Реакции алкенов. 1. Реакции окисления и присоединения 395
МЕХАНИЗМ ЭЛЕКТРОФИЛЬНОГО ПРИСОЕДИНЕНИЯ
Присоединение может идти либо по радикальному, либо по электрофил ьно-
му механизму. Протеканию реакции по электрофильному механизму
способствует использование полярных растворителей, тогда как для радикального
механизма необходим инициатор.
Обсуждение деталей механизма позволит понять многие
экспериментальные результаты, описанные в предыдущем разделе.
Присоединение НХ к симметричной двойной связи
Электрофил (например, Н+) притягивается к области повышенной
электронной плотности двойной связи С=С, и на первой стадии электрофильно-
го присоединения два электрона я-связи переходят к электрофилу с
образованием новой а-связи (уравение 8.46).
Н
НпС=±=СНч > НоС СНч
Карбкатион
(8.46)
Электрофил является акцептором электронов.
Карбкатион (карбений-ион) имеет общую формулу R3C+, где R - атом
водорода или органическая группа.
Атом углерода, который не образует новую с-связь, является электронодефи-
цитным и имеет только шесть электронов на валентной оболочке. Он несет
формальный положительный заряд и называется карбкатионом или карбени-
евым ионом (устаревшее название, которое все еще используется, - карбоние-
вый ион.) Карбкатион является промежуточным соединением в данной
реакции. Карбкатион восприимчив к атаке нуклеофила, например, Вг~.
Н Вг Н
ВГЛ I I I
Н2С СН2 > Н2С СН2 (8.47)
Бромэтан
Нуклеофил - донор электронов.
Нуклеофил и электрофил не обязательно добавляются по отдельности. Часто
нуклеофил образуется в результате образования карбкатиона. Например,
молекула НВг полярна (Н5+—Вг5-), и водородный конец молекулы
притягивается к двойной связи С=С. Это приводит к дальнейшей поляризации
молекулы НВг и гетеролитическому разрыву связи Н—Вг. В результате появляется
ион Н+ и происходит реакция 8.46.
396
8. Алканы, алкены и алкины
На рис. 8.18 показан профиль реакции этилена с бромоводородом.
Лимитирующая стадия реакции - образование карбкатиона. Последующая стадия
взаимодействия с бромид-ионом происходит быстро. Первое переходное
состояние {ПС(1)}* имеет промежуточное строение между исходными
реагентами и карбкатионом. Второе переходное состояние {ПС(2)}* можно
рассматривать как частицу, в которой бромид-ион начинает образовывать связь с
карбкатионом, например, переходное состояние можно изобразить
структурой 8.13 с удлиненной связью.
Вг
I
I
Н2С+-
•сн,
8.13
Вышеописанный механизм работает и для других реакций электрофильного
присоединения, в которых вместо НВг участвуют, например, HCl, HI или
HOS03H (т. е. H2S04).
Присоединение НХ к несимметричной двойной связи
При электрофильной атаке несимметричного алкена реакция может
развиваться по двум путям (уравнение 8.48). В данном примере, в первом случае
получается первичный карбкатион, а во втором случае — вторичный.
Энтальпия
увеличивается
{ПС(0}*
{ПС(2)Г
+НВг
СН2ВгСН3
Координата реакции
Рис. 8.18. Профиль реакции для электрофильного присоединения НВг к этилену.
Лимитирующая стадия процесса - образование карбкатиона как промежуточного
соединения, которое находится в локальном энергетическом минимуме.
8.14. Механизм электрофильного присоединения 397
+
[СНзСН2СН2] Побочный продукт
Первичный карбкатион
СН3СН=СН2 + Н+ <( (8.48)
[сн3£нсн3]
I Вторичный карбкатион
Основной продукт
Как уже говорилось, радикалы стабилизируются электронодонорными
алкильными фуппами. Точно так же электронодефицитные карбкатионы
стабилизируются алкильными группами, соединенными с положительно
заряженным атомом углерода. Таким образом, третичный карбкатион
устойчивее вторичного карбкатиона, а вторичный карбкатион устойчивее первичного
карбкатиона. Ряд изменения устойчивости выглядит следующим образом:
R3C+ > R2CH+ > RCH2+ > СН3+
Ион СН3СНСН3 оказывается наиболее устойчивым промежуточным
соединением в реакции 8.48.
Относительная устойчивость карбкатионов меняется следующим
образом:
R3C+ > R2CH+ > RCH2+ > СН3+
где R - алкильная группа.
Основным продуктом реакции пропена с НВг является 2-бромпропан (а не
1-бромпропан, уравнение 8.36), и это следует из предпочтительности
образования вторичного карбкатиона (уравнение 8.49). Эта реакция называется ре-
гиоселективной, поскольку предпочтительно образуется один из возможных
продуктов, хотя в нуклеофильном алкене имеется два положения, которые
могут реагировать с электрофилом (Н+).
Вг
СН3СН=СН2 + Н+ > [СН3СНСН3] > СН3СНВгСН3 (8.49)
Большинство молекул НХ поляризовано следующим образом: Н5+—X5-.
Предпочтительное присоединение атома водорода молекул НХ к углеродному
атому, соединенному с максимальным числом атомов водорода, называют
правилом Марковникова. Это правило выполняется для присоединения многих
молекул НХ (HI, HC1, HOS03H, т. е. H2S04) к несимметричным алкенам.
Если в результате реакции возможно образование более одного
продукта, но в действительности преобладает один из продуктов, такая
реакция называется региоселективной.
398
8. Алканы, алкены и алкины
Кислотно-катализируемое присоединение воды
Уравнение 8.39 показывает кислотно-катализируемое присоединение Н20 к
алкену. Теоретически, атом водорода из полярной связи О—Н может
выступать в роли электрофила, но на практике, чтобы получить промежуточный
карбкатион, приходится дополнительно использовать кислоту.
Молекула воды полярна и обладает нуклеофильными свойствами,
поэтому она реагирует с карбкатионом (уравнение 8.50). При этом положительный
заряд переносится вначале на атом кислорода, а затем на атом водорода.
Перенос заряда происходит, когда кислород отдает пару электронов центру С+,
а затем при переходе пары электронов со связи О—Н на центр 0+, что
приводит к потере Н+ и регенерации катализатора.
н
\
+
СН3СНСН3
— н
н н
сн
-> Н3С ^СН3
(8.50)
СН3СН(ОН)СН3 + Н+
Кислотно-катализируемое присоединение воды подчиняется правилу Мар-
ковникова.
Присоединение хлора и брома
^» Молекулы хлора и брома неполярны. Однако электроны двойной связи С=С
Индуцированный ди- могут индуцировать диполь в молекуле С12 (рис. 8.19) или Вг2.
поль: см. разд. 2.21 При этом облегчается гетеролитический разрыв связи на
лимитирующей стадии в реакции алкена с галогеном. Из уравнения
8.51 видно, что гетеролитический разрыв связи
сопровождается присоединением электрофила С1+ (с образованием соответствующего
карбкатиона) и присоединением нуклеофила С1~.
(i
С1
(
Н2С = СН2
сь
J
-> н9с-
С1
■сн9
-> СН2С1СН2С1
1,2-Дихлорэтан
(8.51)
8.14. Механизм электрофильного присоединения 399
С\
i
С1
V
н2с = сн2 > н2с = сн2
Рис. 8.19. Молекула хлора неполярна, но при приближении Cl2 к двойной связи
С=С электроны двойной связи индуцируют диполь в молекуле Cl2.
Экспериментально доказано, что при реакции алкена CR2=CR2 с Вг2
образуется промежуточный ион бромония 8.14, который очень сходен с
промежуточным продуктом в реакции 8.51.
Вг+
ч / \ ,-**
с с
У Ч
R^ ^R
8.14
Син- и анги-присоединение
До сих пор не был затронут вопрос о стереохимии реакций присоединения.
Присоединение НХ к двум атомам углерода при исходной двойной связи
С=С изменяет тип гибридизации с sp1 на sp3. На рис. 8.20 показано, что при
присоединении атомы Н и X из НХ могут подходить к молекуле алкена либо
с одной стороны (сш*-присоединение), либо с разных сторон (яилш-присое-
динение). Если все заместители в алкене одинаковы (А = В на рис. 8.20), нет
разницы между продуктами син- и анти-присоединения.
Если на рис. 8.20 А ф В, то при сын-присоединении НХ все заместители А
и В оказываются по одну сторону двойной связи. Разумеется, может
происходить вращение вокруг одинарной связи С—С, и один из конформеров
(заторможенный) конечного продукта показан на рис. 8.20,д. Если же
происходит aH/ww-присоединение НХ, одна группа заместителей оказывается ниже
двойной связи, а другая - выше. При этом расположение заместителей в
продукте я«/ш/-присоединения (рис. 8.20,6) отличается от расположения
заместителей в продукте сын-присоединения.
В большинстве случаев нуклеофил присоединяется к алкену после
электрофила. В промежуточном соединении типа иона бромония 8.14 электрофил
блокирует связь углерод—углерод с одной стороны, так что нуклуеофил
может подойти только с противоположной: бром вступает в реакцию анти-при-
соединения к алкенам. В случае промежуточного карбкатиона со структурой
8.15 нуклеофил может атаковать с любой стороны плоского (sp2) карбениево-
го центра, что сопровождается син- или анти-присоединением. Если карбка-
тион имеет достаточно большое время жизни, может происходить вращение
а*-
I
С1*
400
8. Алканы, алкены и алкины
А
\
С =
/
В
А
/
= С
\
В
эквивалентно
А, .„и А
В*""* ^В
v „.•»•«• А
сш/-Присоеди- \
нение \
► С-
7
Свободное
вращение
X вокруг связи Н
/ с-с \
■с ► с-
\'"**А А**"/
В В
IS
б н
1
i
X
а«/им-При-
г соединение
Н А
\ И
С С
»*7 \
В X
Рис. 8.20. Молекула алкена АВС=САВ является плоской (5/т2-гибридизованные атомы
углерода). При присоединении НХ к двойной связи С=С атомы Н и X могут подходить к молекуле
алкена а) с одной стороны (сми-присоединение) или б) с разных сторон (анти-присоединение).
В случае, если в алкене имеются два заместителя типа А и два заместителя типа В, стереохимия
продуктов сын- и анти-присоединения будет различаться.
вокруг одинарной связи С—С и присоединение будет неспецифичным, хотя
такие факторы, как стерические затруднения, могут влиять на соотношение
продуктов син- и ди/ии-присоединения.
8.15
Другие примеры син- и ди/им-присоединения приводятся в прилагаемом
задачнике; там же рассмотрены следствия появления асимметрического атома
углерода в продуктах присоединения к алкенам.
8.15. Реакции апкенов. 2. Радикальное замещение 401
РЕАКЦИИ АЛКЕНОВ. 2. РАДИКАЛЬНОЕ ЗАМЕЩЕНИЕ
Большинство алкенов содержит не только двойную связь С=С, но и
алкильные заместители, которые могут вступать в реакции радикального
замещения.
Атомы водорода, соединенные с углеродными атомами при двойной
связи С=С, называются винильными (рис. 8.21); они не активны в реакциях
замещения. Однако аллильные атомы водорода, т. е. соединенные с ближайшим к
двойной связи углеродным атомом, отличаются повышенной активностью.
В радикальной реакции пропена с бромом стадии роста цепи 8.52 и 8.53
приводят к образованию бромзамещенного продукта.
СН3СН=СН2 + Вгф
НВг + СН2СН=СН2
(8.52)
сн2сн
=СН2 + Вг2
> СН2ВгСН=СН2 + Вг*
(8.53)
►- Радикал СН2СН=СН2 называется аллильнымрадикалом. Его от-
Резонансные носительно высокая стабильность связана с вкладом резонанс-
структуры: ных структур 8.16. В общем случае, устойчивость молекулы,
раем, разд. 5.17 дикала или иона тем выше, чем больше резонансных структур
можно записать для этой частицы.
Н2С
*ч
сн,
~ н,с^ ^
сн,
8.16
При особых условиях можно наблюдать радикальное присоединение к ал-
кенам, которое приводит к антимарковниковским продуктам. Это означает,
что НХ присоединяется к двойной связи С=С против правила Марковни-
кова.
Вин ильные атомы
водорода
Аллильные атомы
водорода
R*
Рис. 8.21. Атомы водорода, связанные с углеродными атомами двойной связи С=С
называются винильными, а атомы водорода, соединенные с ближайшим к двойной
связи углеродным атомом, называются аллильными. (В данном случае R и R' - алкильные
группы.)
8. Алканы, алкены и алкины
Винильные водородные атомы соединены с углеродными атомами
связи С=С.
Аллильные водородные атомы соединены с зр3-гибридизованным
атомом углерода, соседним со связью С=С.
РЕАКЦИИ АЛКЕНОВ. 3. РАДИКАЛЬНАЯ ПОЛИМЕРИЗАЦИЯ
Полимерами называются макромолекулы, содержащие повторяющиеся
фрагменты. Алкены вступают в реакцию присоединительной (или аддиционной)
полимеризации, которая является частным случаем реакций присоединения, и
поэтому другое ее название — полиприсоединение. В этом процессе молекулы
алкена (мономеры) присоединяются друг к другу с образованием полимера.
Полимеры постоянно встречаются нам в повседневной жизни. Это
иллюстрирует рис. 8.22, на котором показаны различные области применения поли-
винилхлорида (ПВХ), мономером которого является хлорэтен (более
известный под названием винилхлорид).
Полимером называется макромолекула, содержащая повторяющиеся
фрагменты - мономерные единицы.
Полимеризация алкенов, например этилена, может протекать по
радикальному механизму. В этом случае необходим инициатор, например пероксид.
Реакция начинается с образования радикалов из некоторого обобщенного
инициатора Y2*
/TV
Y2 > 2Y* Зарождение цепи (8.54)
а при столкновении радикалов с молекулами алкена (мономерами)
начинается стадия роста радикальной цепной реакции. Уравнение 8.55 показывает,
как атака радикала на атом углерода в этилене приводит к гомолитическому
разрыву я-связи углерод-углерод. Отметим, что стадия роста не включает
отщепление группы и ее присоединение к атакующему радикалу, в отличие от
стадии роста в уравнении 8.18 для реакции метана с радикалом хлора,
которая включает отщепление Н".
Y* + H2C = CH2 > YCH2CH2* Рост цепи (8.55)
Дальнейший рост цепи происходит по реакциям 8.56 и 8.57; далее
полимерная цепь растет по аналогичным реакциям.
YCH2CH2* + СН2=СН2 -> YCH2CH2CH2CH2* Рост цепи (8.56)
8.16. Реакции алкенов. 3. Радикальная полимеризация 403
Трубы и соединения (27%)
J Элементы конструкций (18%)
щ Жесткие пленки и покрытия (10%)
□
Кабели (9%)
VZ\ Бутылки и другие формованные
"^ изделия (9%)
| | Мягкие пленки (7%)
| Линолеум (6%)
| Покрытия (3%)
Pffjj Гибкие трубки (3%)
| Пены (2%)
■ИЗ Материалы для обуви (2%)
Прочее (4%)
Рис. 8.22. Использование ПВХ в Западной Европе в 1995 г. [Chemistry & Industry,
1996, p. 211.]
ych2ch2ch2ch2* + сн2=сн2 -> ych2ch2ch2ch2ch2ch2#
Рост цепи
(8.57)
Нерадикальные продукты могут получаться на стадиях роста или обрыва
цепи (уравнения 8.58 и 8.59).
YCH2CH2CH2CH2CH2CH2* + Y2 -» YCH2CH2CH2CH2CH2CH2Y + Y#
Рост цепи
(8.58)
2YCH2CH2CH2CH2* -> YCH2CH2CH2CH2CH2CH2CH2CH2Y
Обрыв цепи (8.59)
Суммарный процесс записывается уравнением 8.60, где п — любое целое
число между 1 и ©о. Длина полимерной цепи зависит от момента, когда
происходит обрыв цепи.
Н Н
\ /
п С = С
/ \
н н
Мономер
Н Н
Х-/-\
/ \
н
(8.60)
Полимер
Интересные осложнения возникают, если мономер является диеном.
Рассмотрим атаку радикала Y* на двойную связь в октадиене-1,7 (уравнение 8.61).
8. Алканы, алкены и алкины
Y*
(8.61)
Радикал, полученный в такой реакции, может либо реагировать с другой
молекулой мономера, либо вступать во внутримолекулярную реакцию
(уравнение 8.62). Циклические молекулы, полученные таким образом, могут
вступать в дальнейшие реакции полимеризации или в реакцию обрыва цепи с
инициатором.
СН,
(8.62)
Нетрудно видеть, сколько проблем возникает при попытке вести
контролируемую свободнорадикальную полимеризацию алкенов или диенов. Тем не
менее, полимеризация, протекающая по механизму радикального
присоединения, остается важным промышленным способом производства полимеров.
Другой способ полимеризации алкенов — катионная полимеризация,
например, полимеризация децена-1 для производства смазочных материалов.
Дополнение 8.11. Натуральный каучук: природные источники
Натуральный каучук - хорошо известный полимер, находящий широкое
применение. Его источник — каучуковые деревья, произрастающие в
Бразилии. Однако природный каучук содержит белки, вызывающие аллергическую
реакцию у некоторых людей. Недавно стал использоваться новый природный
источник каучука - пустынный вечнозеленый полукустарник гваюла.
В (Z)-1,4-полиизопреновом каучуке гваюлы содержится гораздо меньшее
количество аллергенных белков. Однако имеется ряд недостатков: гваюла
выделяет латекс лишь три месяца в году и ее каучук нельзя добывать простым
надрезанием коры, как это делается в случае каучуковых деревьев.
Н
Me
Н
Me
Изопрен
м
(Z)-1,4-Полиизопрен
8.17. Реакции алкенов. 4. Миграция двойной связи 405
Дополнение 8.12. Полимеры для контактных линз
Полимерные материалы для мягких контактных линз должны быть оптически
прозрачны, химически инертны, устойчивы к постоянному действию влаги,
должны обладать определенными механическими свойствами и быть
проницаемы для 02, так как роговица глаза получает необходимый для
жизнедеятельности 02 непосредственно из атмосферы, а не с потоком крови. Такой
набор требований заставил химиков-синтетиков затратить немалые усилия.
Соединение 1 — мономер, используемый при производстве полимеров для
контактных линз. Его радикальная полимеризация приводит к образованию
гидрогеля, так как полимер адсорбирует воду, но не растворяется в ней.
Наличие небольшого количества воды в контактных линзах необходимо, потому
что содержание воды непосредственно связано с проницаемостью для 02.
Относительно недавно перед производителями контактных линз возникла
проблема разработки линз, которые можно было бы не снимать долгое время.
Полимеры для изготовления таких линз должны обладать повышенной
проницаемостью для 02. Эта задача может быть решена либо созданием более
тонких линз, либо повышением содержания воды в гидрогеле. Последнее
предпочтительнее, поскольку уменьшение толщины линз приводит к
уменьшению их прочности и делает их склонными к разрыву. Сополимеризация
мономера 1 с гидрофильными мономерами 2 и 3 оказалась успешным
способом повышения содержания воды.
°Ч ^6 "2-
1 2 3
ESQ РЕАКЦИИ АЛКЕНОВ. 4. МИГРАЦИЯ ДВОЙНОЙ СВЯЗИ И
ИЗОМЕРИЗАЦИЯ АЛКЕНОВ
При обработке кислотой алкенов, содержащих четыре или более атомов
углерода, может происходить изомеризация, сопровождающаяся миграцией
двойной связи вдоль углеродной цепи. Обработка сильным основанием,
таким, как амид калия KNH2, также может приводить к изомеризации.
Кислота или основание действуют как катализаторы; такие превращения
называются кислотно-катализируемыми или основно-катализируемыми. Кислотно-
катализируемая изомеризация бутена-1 идет по уравнению
Катализатор Н+
СН3СН2СН=СН2 >СН3СН=СНСН3 (8.63)
i-
406
8. Алканы, алкены и алкины
Дополнение 8.13. Еще о полимерах
Ниже приведены некоторые промышленные полимеры, а также мономеры, из которых
их производят.
Коммерческое навание Название мономера
полимера (русск./лат.)
Структура мономера
Полиэтилен (ПЭТ/PET) Этен (этилен)
Поливинил хлорид
(ПВХ/PVC)
Хлорэтен (винилхлорид)
Полипропилен (ПП/РР) Пропен (пропилен)
Тефлон (PTFE)
Полистирол
Тетрафторэтен
Фенилэтен (стирол)
Полиметилметакрилат Метил 2-метилпропионат
(ПММА/Perspex) (метил метакрилат)
н н
\ /
с = с
/ \
н н
Н CI
\ /
с = с
/ \
Н Н
Н СН,
\ /
с = с
/ \
н н
F F
\ /
с = с
/ \
F F
», Р
с = с
/ \
H H
н сн,
\ /
с = с
/ \
н с — о
// \
о снч
Сополимеры
Вышеприведенные соединения представляют собой гомополимеры, т. е. полимеры, для
получения которых используется только один мономер. Если использовать более
одного мономера, образуется сополимер. Сополимеризация является одним из методов
«подгонки» свойств полимера к необходимым для коммерческого использования.
Существует три типа сополимеров (мономерные блоки обозначены А и В):
чередующиеся сополимеры, в котором мономеры соединены в последовательности
АВАВАВАВАВ;
блок-сополимеры, например, АААВВВАААВВВ;
статистические сополимеры, в которых расположение мономеров хаотично:
АВВАВААВА.
(См. продолжение)
8.17. Реакции алкенов. 4. Миграция двойной связи 407
(Продолжение)
Изотактические, синдиотактические и атактические полимеры
В таких полимерах, как полипропилен, заместители могут располагаться различными
способами относительно углеродного скелета полимерной цепи. Это расположение
играет важную роль, так как влияет на упаковку полимерных цепей в твердом состоянии.
В ызотактинеском полипропилене все метильные группы располагаются по одну
сторону полимерной цепи:
В синдиотактическом полипропилене метильные группы регулярно чередуются вдоль
углеродной цепи:
В атактическом полипропилене метильные группы располагаются случайным
образом относительно углеродной цепи:
В твердом состоянии молекулы изотактического и синдиотактического
полипропилена упакованы достаточно эффективно и материал оказывается кристаллическим, а
атактический полипропилен мягок и эластичен. Поэтому изотактический и синдиотакти-
ческий полипропилен можно использовать, например, в качестве материала для
пластиковых труб и покрытий, а атактический полипропилен не может быть использован
для этих целей.
Вопрос: Почему невозможно превратить синдиотактический или атактический
полимеры в изотактический путем вращения вокруг одинарных связей С—С углеродного
скелета полимера?
(См. продолжение)
408 8. Алканы, алкены и алкины
(Продолжение)
Вторичная переработка пластмассе
Как показано на рис. 8.22 на примере ПВХ, полимеры имеют очень широкие области
применения. Жизнь в современном мире нельзя представить себе без полимеров и
пластмасс, но как избавиться от многих тонн синтетических материалов после их
использования? Вторичная переработка пластиковых отходов является одним из важных аспектов
защиты окружающей среды, и европейские страны имеют ряд программ, посвященных
вторичной переработке. Как показывает нижеприведенная схема, в 1990 г. Западная
Европа перерабатывала только 7% пластиковых отходов. В настоящее время отношение
меняется, но до полного решения проблемы еще далеко. Однако количество коммерчески
используемых пластмасс, которые можно подвергать биодеградации, фотодеградации,
или пластмасс, способных к автодеградации, постоянно растет. [Chemistry & Industry,
1992, p. 401.]
| | Попадает в почву (74%)
I Сжигается без регенерации энергии (4%)
I Сжигается с регенерацией энергии (15%)
$Щ Механическая вторичная переработка
(7%)
Миграция двойной связи происходит таким образом, чтобы повысилось
число алкильных групп при двойной связи и соответственно уменьшилось
► число атомов водорода, присоединенных к ней. Данная тенденция непосред-
Электроотрицатель- ственно следует из большей электроотрицательности я/^-гиб-
ности различных ридизованного атома углерода по сравнению с 5/?3-гибридизо-
углеродных центров: ванным; электронодонорная алкильная группа предпочитает
см. разд. 8.20 присоединяться к 5/?2-гибридизованным центрам.
Чем длиннее углеродная цепь, тем больше изомеров можно получить при
миграции двойной связи С=С. На практике, как правило, получаются смеси
изомеров, в которых пробладает наиболее термодинамически стабильный
изомер.
В реакциях миграции атом или функциональная группа
«перемещается» по молекуле из одного положения в другое.
8.17. Реакции алкенов. 4. Миграция двойной связи 409
Механизм миграции двойной связи, катализируемой кислотой
Как было показано, добавление кислоты к алкенам приводит к образованию
карбкатионов; при реакции пентена-1 с Н+ образование вторичного карбка-
тиона предпочтительнее, чем образование первичного (уравнение 8.64). В
отсутствие нуклеофила, который бы завершил реакцию присоединения
(например, реакцию 8.47), карбкатион может терять протон и образовывать
либо пентен-1 (уравнение 8.65), либо пентен-2 (уравнение 8.66). Чем больше
алкильных групп соединено с двойной связью, тем более стабилен алкен,
поэтому пентен-2 является преобладающим продуктом. Суммарная реакция
изомеризации с миграцией двойной связи представляет собой совокупность
реакций 8.64 и 8.66.
СН3СН2СН2СН=СН2 + Н+ -> СН3СН2СН2СНСН3 (8.64)
Н
♦ Л
СН3СН2СН2СН СН2 > СНзСН2СН2СН=СН2 + Н+
Пентен-1 (8.65)
Н
СНзСНзСН-1- СНСН3 > СН3СН2СН=СНСН3 + Н+
Пентен-2 (8.66)
Механизм миграции двойной связи,
катализируемой основанием
Аллильные атомы водорода могут отщепляться не только в радикальных
реакциях, но и при атаке основанием, поскольку аллильные атомы водорода
относительно кислотны. В реакции 8.67 основание В~ отщепляет ион водорода
от углеродного атома, соседнего с двойной связью С=С. Образующийся
промежуточный продукт называется карбанионом.
(8.67)
■> RCHCH=CH2 + ВН
Карбанион имеет общую формулу R3C~, где R - атом водорода или
органическая группа.
d
снсн=сн2
410
8. Алканы, алкены и алкины
► Полученный аллильный карбанион стабилизирован резонанс-
Резонансные ным вкладом структур 8.17, который приводит к делокализации
структуры: л-связи по трем углеродным центрам. Альтернативный способ
см. разд.5. 17 изображения резонансной пары 8.17 - структура 8.18.
Н Н
^ *ч **—** ^
RHC СН, RHC
1
8.17
RHO' " ^СН2
8.18
Протонирование аллильного карбаниона может идти либо по атому С(1),
либо по атому С(3), и преобладающим продуктом будет алкен с
максимальным количеством алкильных групп, соединеных с двойной связью С=С. Это
непосредственно следует из различия в электроотрицательности между sp2- и
5/?3-гибридизованными атомами углерода. 5/?2-Гибридизованный атом
углерода имеет большую электроотрицательность, чем 5/?3-гибридизованный, и
наиболее стабильным продуктом будет тот, в котором максимальное число
электронодонорных алкильных групп соединено с з/?2-гибридизованными
углеродными атомами. В результате двойная связь С=С мигрирует к центру
углеродной цепи (уравнение 8.68).
Н
sl?£^ + ВН > RCH=CHCH3 + В (8.68)
RHC-' - ^СН2
Источником протонов является протонированное основание ВН,
полученное в реакции 8.67. Катализатор В~ регенерируется в реакции 8.68.
РЕАКЦИИ АЛКЕНОВ. 5. ГИДРОБОРИРОВАНИЕ
Гидроборирование с ВН3
►" Реакция алкенов с ВН3 приводит к присоединению свя-
«ВН3» означает здесь либо Зи В-Н к двойной связи С=С (уравнение 8.69). Эта
В2Н6, либо аддукт с реакция называется реакцией гидроборирования.
основанием Льюиса,
см. разд. 11.5
8.19. Реакции алкинов. 1. Присоединение
411
Н3С
н сн2
в +зн2С = СН2 > /В\ /Сн3
I
сн3
Триэтилборан (8.69)
Если алкен стерически затруднен из-за присутствия крупных органических
заместителей, только одна или, быть может, две связи В—Н вступают в
реакцию гидроборирования; примером может служить реакция 8.70.
СНз СНз СНз СНз
ВНз + С = С > Н С С ВН2 (8.70)
/ \ / \
СНз СНз СНз Сг*з
Использование таких реакций позволяет получать борорганические
соединения типа RBH2 или R2BH (R - алкил). Дальнейшие реакции с другими алке-
нами приводят к получению борорганических соединений, содержащих
различные алкильные заместители. Присоединение борана к несимметричному
алкену региоселективно; при этом атом бора образует связь с атомом
углерода, который соединен с наибольшим числом водородных атомов. Это
присоединение идет против правила Марковникова, поскольку водород более
электроотрицателен, чем бор. Связь бор-водород поляризована
следующим образом: В5+—Н6-, и в данном случае атом водорода не является электро-
филом. Таким образом, гидроборирование не нарушает правила, и называется
антимарковниковским только из-за положения, в которое присоединяется
атом водорода.
^* Борорганические соединения можно окислить пероксидом
Гидроборирование водорода в присутствии гидроксида натрия, при этом образу-
при синтезе спиртов: ются соответствующие спирты (уравнение 8.71). Эта реакция
см. разд. 14.9 является важным примером использования борорганических
соединений.
он-
(СН3СН2)3В + ЗН202 >ЗСН3СН2ОН + В(ОН)3 (8.71)
Триэтилборан Борная кислота
РЕАКЦИИ АЛКИНОВ. 1. ПРИСОЕДИНЕНИЕ
Подобно алканам и алкенам, алкины горят в кислороде с образованием
диоксида углерода и воды (уравнение 8.72).
412
8. Алканы, алкены и алкины
2СН3ОСН3 + 1Ю2 -> 8С02 + 6Н20 (8.72)
Алкины вступают в реакции присоединения с образованием производных
алкенов, а затем — производных алканов. На первой стадии реакции
образуется смесь изомерных продуктов (уравнение 8.73).
Н Н Н X
\ / \ /
н—с^с—н > с = с + с = с
/ \ / \
X XX Н
(2)-Изомер (£)-Изомер (8.73)
Присоединение водорода
Присоединение Н2 к алкинам с образованием алкенов легко происходит в
^» присутствии катализаторов - переходных металлов, таких, как никель, пал-
(Z)- и (Е)-изомеры: ладий или платина. Селективное восстановление с
образованием, разд. 5.11 ем только (£)- или только (Z)- изомеров может быть
достигнуто разными способами. Например, реакция с водородом в
присутствии катализатора Pd/BaS04 приводит к образованию (2)-изомера, а
использование натрия в жидком аммиаке в качестве восстановителя дает
преимущественно (£)-изомеры. Однако натрий в жидком аммиаке можно
использовать только для восстановления группы —OCR (но не —С=СН; см.
разд. 8.20).
При восстановлении алкинов до алкенов могут возникнуть осложнения,
связанные с возможностью последовательного восстановления
образующегося алкена до алкана. Полезным примером катализатора селективного
восстановления тройной связи С=С до двойной связи С=С может служить
катализатор Линдлара, который представляет собой металлический палладий,
нанесенный на поверхность СаС03/РЬО.
Присоединение галогенов и галогеноводородов
Механизм присоединения галогенов и галогеноводородов к алкинам сходен
с механизмом аналогичной реакции алкенов. Пропин (метилацетилен)
реагирует с Вг2 по уравнению 8.74, при этом преимущественно образуется (Z)-
изомер алкена.
СН3ОСН —i+ (Z)-CH3CBr=CHBr —-> СН3СВг2СНВг2 (8.74)
Пропин (2)-1,2-Дибромпропен 1,1,2,2-Тетрабромпропан
(метилацетилен)
В условиях электрофильного присоединения алкины присоединяют галоге-
новодороды согласно правилу Марковникова. Второе присоединение по
правилу Марковникова приводит к образованию соединения, в котором два
галогенидных заместителя соединены с одним атом углерода (уравнения 8.75 и
► 8.76). Такие соединения иногда называют гем-дигалоге-
гем означает геминальные н идам и.
8.20. Реакции алкинов. 2. Алкины как кислоты
413
сн3осн
НС!
СН3СС1=СН2
на
—> ch3cci2ch3
2,2-Дихлорпропан
(8.75)
сн3с=сн
НС1
СН3СС1=СН2
НВг
СН3СС1ВгСН3
2- Бром-2-хлорпропан
(8.76)
аИЛ РЕАКЦИИ АЛКИНОВ. 2. АЛКИНЫ КАК КИСЛОТЫ
Зависимость электроотрицательности
от типа гибридизации углеродного атома
В табл. 4.2 приведены электроотрицательности углерода и водорода по
шкале Полинга, равные 2,6 и 2,2 соответственно. Однако при этом была сделана
существенная оговорка, что электроотрицательность атома зависит от
степени окисления и порядка связи. При изменении типа гибридизации атома
углерода с sp3 к sp2 и sp, электроотрицательность изменяется (рис. 8.23).
Существенное увеличение электроотрицательности означает, что связь С—Н в ал-
кинах более полярна, чем в алкенах, а связь в алкенах в свою очередь более
полярна, чем в алканах.
Значения электроотрицательности позволяют предсказать, что анион
RC=C~ будет более устойчив, чем анион R2C=C(R)~, который в свою очередь
^ окажется стабильнее, чем анион R3C~. Это подтверждается
р/<а = -lg Ka экспериментальными наблюдениями, и приближенные
значения рКа приведены в табл. 8.6. Следует помнить, что кислотный
характер аллильного водородного атома на sp3-углеродном
центре в R2C=CRCH2R связан с особой устойчивостью аллильного карбани-
она 8.18, стабилизировнного резонансом.
^ Некоторые реакции алкинов доказывают кислотность группы —С=С—Н.
Кислоты и основания: Однако необходимо подчеркнуть, что алкины - очень слабые
см. разд. 11.9 кислоты, и этин (или ацетилен; уравнение 8.77) является
гораздо более слабой кислотой, чем вода.
Рис. 8.23. График зависимости
электроотрицательности атома углерода от типа его
гибридизации. Приведенные значения
электроотрицательности вычислены из потенциалов
ионизации и сродства к электрону и соответствуют
шкале электроотрицательности по Маллике-
ну—Джаффе; значения отнормированы таким
образом, чтобы их можно было сопоставлять с
электроотрицательностью по шкале Полинга.
414 8. Алканы, алкены и алкины
Таблица 8.6. Кислотность связей С—Н
Соединение
рК
Тип гибридизации атома углерода
СН4
н2с=сн2
нсьсн
48
44
25
spJ
sp2
sp
H-GeC-H ^ H-OC" + H+ (8.77)
Только алкины с концевой тройной связью (терминальные алкины) проявляют
кислотные свойства, поскольку кислотность характерна только для группы
-ОСН.
Солеобразные ацетилениды
Терминальные алкины реагируют с натрием (уравнение 8.78) и с амидом
калия (уравнение 8.79) с образованием ацетиленидов щелочных металлов.
2СН3СН2С=СН + 2Na -> 2[CH3CH2OC]-Na+ + H2 (8.78)
СН3ОеСН + K[NH2] -> [СН3С=С]-К+ + NH3 (8.79)
Также возможна реакция с ионами серебра(1) (уравнение 8.80), хотя в
сухом виде ацетилениды серебра взрывчаты. Аналогичные реакции могут быть
использованы для получения солей меди(1).
СН3С=СН + [Ag(NH3)2]+ -> CH3C=CAg + NH3 + NH4+ (8.80)
Ацетилениды щелочных металлов используют в важной реакции синтеза
алкинов с более длинной углеродной цепью. Движущая сила этой реакции -
образование галогенида щелочного металла (уравнение 8.81).
[CH3CH2OC]-Na+ + СН3СН2С1 -> СН3СН2С=ССН2СН3 + NaCl (8.81)
РЕАКЦИИ АЛКИНОВ. 3. ДИМЕРИЗАЦИЯ
При нагревании терминальных алкинов с растворами солей меди(Н) в
пиридине 8.19 происходит реакция димеризации, в результате которой
получаются диины (уравнение 8.82). Это достаточно общая реакция; так, уравнение
8.83 - пример использования ее для синтеза циклического гексина из
терминального диина.
2СН3СН2С=СН
Си2+ в пиридине
-> сн3сн2с=с-а=ссн2сн3
(8.82)
8.22. Заключение
415
Cu2+ в пиридине
/ %
(8.83)
Диины содержат две тройные связи С=С.
ЗАКЛЮЧЕНИЕ
Эта глава посвящена химии ациклических алканов, алкенов и алкинов. Помимо химических
свойств этих соединений, в ней рассматриваются некоторые важные подходы, используемые в
органической химии.
Что означают следующие термины?
Смешивающиеся и несме-
шивающиеся жидкости
Первичный, вторичный,
третичный и четвертичный
атомы углерода
Конформер
Асимметрический атом
углерода
Хиральное соединение
Оптический изомер (энанти-
омер)
Стереоизомер
Гомологический ряд алканов
Шаростержневая и
полусферическая диаграммы
Гомолитический и гетероли-
тический разрыв связи
Промежуточный продукт
Переходное состояние
Электрофил
Нуклеофил
Фотолиз
Гидролиз
Радикальное замещение
Радикальная полимеризация
Электрофильное
присоединение
Региоселективность
Теперь вы должны уметь:
обсудить структурную и геометрическую
изомерию алифатических углеводородных
молекул;
описать различие между заторможенными,
заслоненными и скошенными конформаци-
ями, а также изменение стерической
энергии, связанное с вращением вокруг связей
С-С;
рисовать проекции Ньюмена и диаграммы
типа «кбзлы», а также использовать
клиновидные линии для изображения конформа-
ций молекул;
обсудить, что в реальности означает
понятие «углеводород с линейной (прямой)
цепью»;
обсудить вопросы, должно ли хиральное
соединение обязательно иметь
асимметрический атом углерода и что такое хираль-
ность спиральной цепи;
указать два «теста на хиральность»
(являются ли они однозначными?);
определять изменение
окислительно-восстановительного состояния углерода в ходе
реакции;
уметь отличать промежуточный продукт
реакции от переходного состояния и указать
их положение на профиле реакции;
обсудить реакции, характерные для
алканов, и указать основные черты механизма
реакций радикального замещения;
416
8. Алканы, апкены и алкины
• обсудить типичные реакции алкенов и
указать основные черты механизма реакций
электрофильного присоединения;
• различать присоединение по правилу Мар-
ковникова и против правила Марковникова,
приводить примеры реакций обоих типов;
объяснить, почему в том или ином случае
реализуется определенный тип
присоединения;
ЕЕЭ УПРАЖНЕНИЯ
8.1. Выпишите названия неразветвлен-
ных алканов, имеющих следующие
формулы: а) С3Н8; б) С7Н16;
в) С12Н26; г) С20Н42.
8.2. Выпишите молекулярные формулы
следующих неразветвленных
алканов: а) гексан; б) нонан; в) декан;
г) тетрадекан.
8.3. Изобразите структурные формулы
следующих алканов: а) 2-метилпен-
тан; б) 3-этилоктан; в) 2,2-диметил-
бутан; г) 2,2,4,4-тетраметилгексан.
Сколько типов углеродных атомов
имеет каждое соединение?
8.4. Почему название 1,1-ди метил пропан
нельзя использовать для соединения
Ме2СНСН2СН3? Каково
систематическое название этого соединения?
8.5. Каково систематическое название
соединения с формулой
СН3СН2СНМеСНМеСН2СН2СН2СНМеСН3?
8.6. Укажите названия следующих групп
или заместителей: а) СН3; б) С2Н5;
в) С4Н9; г) Ме3С; д) Ме2СН.
8.7. Рассмотрим изомеры гексана на
рис. 8.6. а) Почему на рисунке
отсутствует изомер с названием 4-метил-
пентан? б) Почему отсутствует изомер
с названием 3,3-диметилбутан? в)
Почему отсутствуют изомеры, название
которых происходит от корня
«пропан»? г) Почему циклогексан не
является структурным изомером гексана?
• объяснить, когда и почему происходит
миграция двойной связи С=С;
• объяснить, что такое син- и
анти-присоединение к алкенам, и обсудить, в каких
случаях реализуются эти два типа
присоединения;
• обсудить типичные реакции алкинов и
прокомментировать слабые кислотные
свойства терминальных алкинов.
8.8. В каждой из приведенных молекул
укажите тип гибридизации каждого
углеродного центра:
12 3 4 5
а) СН3СН=СНСН2СН3;
1 2 34
б) CH3GeCCH3;
12 3 4 5 6 7 8
в) СН3СН=С=СНСН2СН2СН=СН2;
г) СН3СМе=СМеСН2СН2СМе2СН3.
8.9. На рис. 8.24 показано изменение
стерической энергии при вращении
вокруг связи С(2)—С(3) в молекуле
бутана. За нуль принята энергия
заторможенной конформации
(угол = 0°). а) Изобразите проекции
Ньюмена для конформации бутана в
точках А, В, С и D. [Подсказка:
Рассмотрите проекции молекулы вдоль
оси связи С(2)—С(3).] б) Объясните
вид графика на рис. 8.24.
8.10. Какие вещества окисляются, а какие
восстанавливаются в следующих
реакциях?
а) 2СН3СН2С1 + 2Na -> С4Н10 + 2NaCl
б) С2Н6 + С12 -» СН3СН2С1 + НС1
в) 2NO + 02 -> 2N02
г) СН3ОСН + Н2 -> СН3СН=СН2
д) 2СН3СН2СееСН + 2К ->
2[СН3СН2С=С]К + Н2
[Подсказка: См. дополнение 8.8.]
8.23. Упражнения
417
360
Угол вращения, град.
Рис. 8.24. К заданию 8.9.
8.11. Что означают следующие термины:
а) промежуточный продукт; б)
переходное состояние; в) электрофил;
г) гетеролитический разрыв связи;
д) гемолитический разрыв связи;
е) стереохимия; ж) региоселектив-
ность?
8.12. На рис. 8.25 изображена зависимость
-АгорЯ алканов с формулой СпН2п+2
от значения п. Почему этот график
линеен?
8.13. Почему добавление 02 существенно
влияет на ход многих радикальных
реакций? [Подсказка: Вернитесь к
гл. 3.]
8000
< 4000 г
12
п
Рис. 8.25. К упражнению 8.12.
8.14. Реакции ядерного распада часто
называют цепными реакциями. В чем их
отличие от реакции между СН4 и С12?
8.15. Уравнения 8.17—8.20 представляют
собой четыре возможные стадии
роста цепи в реакции хлорирования
метана. Они показывают
образование только двух членов семейства
алкилхлоридов (хлоралканов)
СН4_ЛС1Л. Допишите стадии роста
цепи, которые приводят к
получению остальных членов семейства.
Какие еще хлоралканы могут
получаться при хлорировании метана в
качестве побочных продуктов?
8.16. Средние значения энтальпии
диссоциации связей С=С, С—С, Н—Н и
С—Н составляют 598, 346, 436 и
412 кДж/моль соответственно.
Определите изменение энтальпии при
гидрировании этилена. Сопоставьте
ваш ответ с полученным при
использовании стандартных энтальпий
образования этилена (+52,5 кДж/моль)
и этана (-83,8 кДж/моль).
8.17. Предложите механизм
взаимодействия 2-метилбутена-1 с НС1 в
отсутствие облучения. Предскажите
соотношение между продуктами реакции и
обоснуйте свой ответ.
418
8. Алканы, алкены и алкины
8.18. Предскажите, какие продукты
образуются из пентена-2 при: а) реакции
с Вг2; б) горении в кислороде; в)
реакции с озоном и с последующей
обработкой продуктов этого
взаимодействия водой в присутствии
цинка; г) реакции с иодоводородом;
д) смешивании с хлором и
облучении светом.
8.19. Уравнение 8.40 показывает
взаимодействие пропена с С12/Н20. В тексте
отмечается, что спиртовая группа
предпочтительнее присоединяется к
вторичному атому углерода.
Предложите механизм, который согласуется
с экспериментальными результатами.
8.20. 9-Борабицикло[3.3.1]нонан (обычно
обозначаемый 9-BBN) является
селективным гидроборирующим
агентом.
9-BBN
Предложите способ синтеза 9-BBN
исходя из подходящего циклического
диена. Какие еще изомеры могут
получаться в этой реакции?
8.21. Почему использование Pd/BaS04 в
качестве катализатора реакции
гидрирования алкинов приводит к
преимущественному образованию
(2)-изомеров? Объясните, почему
натрий в жидком аммиаке, как
правило, нельзя использовать для
гидрирования терминальных алкинов.
Ш СПЕКТРОСКОПИЯ
ЧТО ТАКОЕ СПЕКТРОСКОПИЯ?
Спектроскопические методы анализа стали неотъемлемой частью совре-
► менной химии. Выше уже упоминались атомный спектр водорода и фото-
Атомный спектр водорода: электронная спектроскопия Существует множество
см. разд. 2.16 спектроскопических методов (табл. 9.1), и использова-
Фотоэлектронная ние различных типов спектроскопии позволяет исследо-
спектроскопия: вать разнообразные аспекты строения атомов и молекул,
см. дополнение 3.6 В данной главе рассмотрены инфракрасная (ИК) и
электронная (в ультрафиолетовой — видимой области) спектроскопия, а
также спектроскопия ядерного магнитного резонанса (ЯMP) — методы, которые
наиболее часто встречаются в повседневной работе в лаборатории.
Цель настоящей главы - предоставить практически полезную
информацию, не углубляясь в теорию спектроскопии.
Спектры поглощения и спектры испускания
Понятия абсорбция (поглощение) и эмиссия (испускание) фундаментальны для
► спектроскоспии. Ранее уже обсуждалось различие между атомными спектра-
Спектры поглощения и ми поглощения и испускания В общем случае, поглощение
испускания: электромагнитного излучения атомом, молекулой или ио-
см. дополнение 2.7 ном вызывает переход с низшего энергетического уровня
на высший. (Ниже в данной главе понятие «уровень» обсуждается более
поднобно.) Следует помнить (см. гл. 2), что только определенные переходы
являются разрешенными. Энергия поглощенного излучения соответствует
энергетической разности между двумя уровнями. На рис. 9.1 показана
обобщенная схема абсорбционного спектрометра, который используется для
измерения поглощения электромагнитного излучения. «Источник» создает
электромагнитное излучение, частоту которого можно варьировать в
некотором интервале. Когда излучение проходит через образец, часть его поглоща-
^~ ется, а часть пропускается, детектируется и записывается в виде спектра
E=hv (рис. 9.2). Спектр представляет собой график зависимости
поглощения или пропускания излучения от его частоты.
Интенсивность поглощения (интерпретацию терминов «поглощение» и
«пропускание» см. на рис. 9.2) зависит от нескольких факторов:
• вероятности наблюдаемого перехода;
• заселенности уровней;
• количества (концентрации) образца.
420
9. Спектроскопия
Таблица 9.1. Некоторые важные спектроскопические методы. [Обратите внимание
на то, что электронная, рентгеновская и нейтронная дифракция (гл. 3), равно как и
масс-спектрометрия не являются спектроскопическими методами.]
Название метода
Примечания
Атомно-абсорбционная
спектроскопия
Спектроскопия
электронного парамагнитного
(спинового) резонанса
(ЭПР)
Электронная
спектроскопия, или вид./УФ-
спектроскопия, т. е.
спектроскопия видимой и
ультрафиолетовой (УФ)
областей
Дальняя инфракрасная
спектроскопия
Флуоресцентная
спектроскопия
Инфракрасная (ИК)
спектроскопия
Микроволновая
спектроскопия
Мессбауэровская
(у-резонансная)
спектроскопия
Спектроскопия ядерного
магнитного резонанса
(ЯМР)
Фотоэлектронная
спектроскопия (ФЭС)
Рамановская
(комбинационного рассеяния, КР)
спектроскопия
Используется для элементного анализа, позволяет
наблюдать спектры поглощения атомов в газовой фазе
Используется для исследования веществ с одним или
несколькими неспаренными электронами
Спектроскопия поглощения [100-200 нм (вакуумный
УФ), 200—800 нм (ближний УФ и видимая область)]
используется для исследования переходов между
электронными энергетическими уровнями в атомах и
молекулах (см. разд. 9.9—9.11)
Инфракрасная спектроскопия в области волновых
чисел менее 200 см-1
Используется для исследования флуоресцирующих,
фосфоресцирующих и люминесцирующих веществ.
Флуоресценция — испускание света, которое может
происходить после поглощения УФ или видимого
излучения. Это свойство проявляется лишь некоторыми
веществами. Длина волны испускаемого света обычно
превышает длину волны поглощенного излучения
Вариант колебательной спектроскопии; величина
поглощения обычно записывается в интервале
200—4000 см-1. Используется для идентификации
веществ, поскольку ИК-спектр вещества, как правило,
уникален (см. разд. 9.6—9.8)
Спектроскопия поглощения используемая для
исследования вращательных спектров молекул
Поглощения у-излучения некоторыми ядрами (например,
57Fe и 197Аи). Используется для определения
химического окружения атомов, в том числе степени
окисления
Поглощение или испускание радиочастотного излучения,
позволяет наблюдать состояния ядерных спинов (см. разд.
9.12-9.16). Мощный аналитический инструмент,
применяемый для выявления молекулярных структур и
исследования динамического поведения веществ в
растворах и твердой фазе
Спектроскопия поглощения, используемая для изучения
энергетических состояний заполненных атомных и
молекулярных орбиталей (см. дополнение 3.6)
Вариант клебательной спектороскопии, имеющий, в
отличие от ИК-спектроскопии, другие правила отбора.
Некоторые колебания, неактивные в ИК-спектрах,
активны в рамановских спектрах
9.1. Что такое спектроскопия?
421
Источник
электромагнитного
излучения
>J Детектор
Самописец
Образец
Рис. 9.1. Обобщенная схема абсорбционного спектрометра. Электромагнитное
излучение создается в источнике и направляется на образец, в котором часть излучения
поглощается, а часть пропускается. Излучение, прошедшее через образец, попадает на
детектор и полученная информация выводится в виде спектра.
В спектрах поглощения измеряется частота и количество поглощенного
излучения, т. е. удаленного из исходного излучения, в то время как в
спектрах испускания измеряется излучение, испускаемое возбужденным
состоянием частиц образца. В эмиссионном спектрофотометре образец подвергают
возбуждению (термически, электрически или действием электромагнитного
излучения) в короткоживущее высокоэнергетическое состояние. При
обратном переходе на уровень с низкой энергией происходит испускание
излучения. Спектр записывается как график зависимости интенсивности
испускаемого излучения от частоты. Энергетическая разность между высоко- и
низкоэнергетическим уровнями соответствует энергии испускаемого излучения.
Частота
поглощения
Поглощение
Частота v
Частота
поглощения
А
Частота v
Т
п—г
100%
А
Пропускание
Рис. 9.2. Схематическое изображение спектра поглощения, в котором наблюдается
одна полоса. Из спектра можно получить информацию об интенсивности и частоте
поглощения. Интенсивность можно измерять в единицах поглощения (а) или
пропускания (б). Частоту можно пересчитать в величины энергии или длины волны.
9. Спектроскопия
Для проведения эмиссионно-спектроскопического эксперимента важно
определить достаточное время между исходным возбуждением и записью
испускания. Если период между этими событиями слишком велик, то переход с
высшего уровня на низший успеет закончиться, а если он слишком мал,
эмиссия не успеет произойти.
СПЕКТР ЭЛЕКТРОМАГНИТНОГО ИЗЛУЧЕНИЯ И
СПЕКТРОСКОПИЧЕСКИЕ МЕТОДЫ
Шкала электромагнитных излучений приведена в приложении 1 (см. т. 2).
Электромагнитное излучение можно характеризовать с помощью частоты
(v), длины волны (X) или энергии (£). Однако существует еще один удобный
способ, связанный с использованием волновых чисел. Волновое число -
величина, обратная длине волны (уравнение 9.1); его единица измерения —
обратный сантиметр. Эту величину удобно использовать, поскольку она
пропорциональна энергии.
Волновое число V = 1/ (Длина волны X) (9.1)
Различные спектроскопические методы связаны с различными частями
электромагнитного спектра, поскольку излучение с определенной энергией
возбуждает определенный вид переходов в атоме или молекуле. Например,
Волновое
число, см
На 1 моль:
-I
1 • 106
I
1 • 104
I
100
1
Частота,
Гц
3- 1016
I
3- 1014
I
3- 1012
3- 1010
. Энергия,
~~г кДж/моль
1,2- Ю-2
Ультрафиолетовое и видимое из-
у лучение; электронная
спектроскопия
Инфракрасное излучение;
колебательная спектроскопия
(ИК-спектроскопия)
Радиочастоты; изменение
ядерных спинов (ЯМР-спектроско-
пия)
1 • Ю-4
3 106
1,2- 10-6 J
А = 6,26 • Ю-34 Дж/моль, с = 3 • 108 м/с, N. — число Авогадро
Рис. 9.3. Часть электромагнитного спектра. Излучение можно характеризовать
энергией, частотой или волновым числом. Связь с длиной волны показана в
приложении 1 вт. 2. Разные области спектра соответствуют различным спектроскопическим
методам.
9.3. Шкала времени
423
► спектральные линии серии Лаймана в спектре атомарного во-
Серии Лаймана: дорода лежат между 2,466 • 1015 Гц и 3,237 • 1015 Гц. Это соот-
см. пример 2.3 ветствует ультрафиолетовой области спектра
электромагнитных излучений. На рис. 9.3 показаны области спектра
электромагнитных излучений, которым соответствуют методы Я MP, И К и
электронной спектроскопии.
^ В спектроскопии ядерного магнитного резонанса рассматриваются перехо-
ЯМР-спектроскопия: ды между ядерными спиновыми состояниями. Для таких
переем, разд. 9.12-9.16 ходов требуются небольшие затраты энергии (<0,01 кДж/моль),
и они могут быть вызваны излучением в радиочастной области
электромагнитного спектра.
Колебательная спектроскопия имеет дело с переходами между
колебательными состояниями молекул. Энергия, необходимая для того, чтобы происхо-
^ дили такие переходы, обычно находится в интервале от 1 до 100 кДж/моль и
ИК-спектроскопия: соответствует инфракрасной области электромагнитного
см. разд. 9.6-9.8 спектра. Эта область лежит в интервале волновых чисел от 100
до 10 000 см-1. Обычный лабораторный ИК-спектрометр
работает в интервале 200-4000 см-1.
^ Электронная спектроскопия изучает переходы между электронными
Электронная энергетическими уровнями молекул. Энергетическая разность
спектроскопия: между этими уровнями соответствует излучению в видимой и
см. разд. 9.9-9.11 УФ-областях спектра электромагнитных излучений, откуда и
возникло название вид./УФ-спектроскопия. Интервал длин
волн лабораторного вид./УФ-спектрофотометра — от 200 до 900 нм;
измерения, как правило, проводят в единицах длин волн.
ШКАЛА ВРЕМЕНИ
Теперь необходимо сказать о спектроскопической шкале времени. Это
сложная тема, и мы коснемся лишь одного из двух ее аспектов. Главный вопрос,
который мы будем обсуждать: позволяет ли данный спектроскопический
метод сделать «фотоснимок» неподвижной молекулы в некоторый момент
времени или мы имеем дело с усредненными результатами?
Традиционное «статическое» представление о молекуле неверно —
молекулы постоянно колеблются и вращаются со скоростью от 1012 до 1014 раз в
секунду (рис. 9.3). Если спектроскопический метод работает быстрее этого
движения, то получается «мгновенный снимок» молекулы, а если медленнее,
мы видим результат, усредненный по многим движениям. Электронная
спектроскопия позволяет получать «мгновенный снимок» молекулы в
определенном колебательном и вращательном состоянии, а спектроскопия ЯМР часто
дает усредненный результат, так как характеристическое время данного
метода больше, чем время колебательного или вращательного движения.
Вторая особенность состоит в том, что исследователь обычно имеет дело
не с единственной молекулой, а с ансамблем молекул. Хотя электронная
спектроскопия дает «мгновенный снимок» каждой отдельной молекулы в
определенном колебательном и вращательном состоянии, типичный образец
содержит порядка 1018 молекул, и не все они находятся в одинаковом
колебательном и вращательном состоянии.
424
9. Спектроскопия
Наконец, в молекуле может происходить динамический процесс типа
^- псевдовращения Берри, и это создает дополнительные проблемы с вре-
Псевдовращение менной шкалой. Понижение температуры замедляет динами-
Берри: см. разд. 5.12 ческий процесс, и он может стать медленнее, чем
спектроскопическое измерение. Однако, как упоминалось выше для
Fe(CO)5, даже при 103 К аксиальные и экваториальные лиганды СО быстро
меняются местами и спектроскопия ЯМР 13С показывает присутствие
единственного (усредненого) типа атома 13С.
ЗАКОН ЛАМБЕРТА-БЕРА
Интенсивность поглощения излучения зависит в основном от количества
образца. Если молекулы поглощают излучение с определенной частотой, то,
чем больше присутствует молекул, тем больше излучения данной частоты
поглощается и соотвественно меньше пропускается. Поглощение, или
оптическая плотность, образца связано с пропусканием уравнением 9.2; эта
зависимость графически представлена на рис. 9.4.
Оптическая плотность = -lg (Пропускание), т. е. Л = - lg T (9.2)
Пропускание ^принимает значения от 0 до 1, но экспериментально
измеренные величины часто выражают в процентах. Согласно уравнению 9.2,
пропускание 100% соответствует нулевой оптической плотности, а при Г—> 0
кривая на рис. 9.4 стремится к бесконечному значению оптической плотности;
для Т = 0,01 величина оптической плотности равна 2.
Пропускание равно отношению интенсивности прошедшего излучения
(/) к интенсивности падающего излучения (/0). Учитывая это, из уравнения
9.2 можно получить уравнение 9.3. Рис. 9.5 иллюстрирует связь между /и /0 и
показывает, как определяют оптическую плотность образца.
A = -\gI/I0 (9.3)
В спектрофотометре образец помещается в специальную кювету с точно
известными размерами. Величина оптической плотности связана с длиной
S
X
^
>>
с
С
X
се
*
&
с
8.
(%п
(100%)
0,75
(75%)
0,5
(50%)
0,25
(25%)
(0%) 0 0,5 1 1,5 2
Оптическая плотность
Рис. 9.4. Связь между пропусканием Г и
оптической плотностью А (см. уравнение
9.2). Г->0приЛ->оо.
9.4. Закон Ламберта-Бера
425
Источник
Падающее
излучение
•И-
Прошедшее излучение
Детектор
и система
Рис. 9.5. В кювете А содержится раствор образца, а в кювете В — чистый
растворитель, тот же, что в кювете А; обе кюветы имеют одинаковую толщину (т.е. длину пути
луча). Если растворитель не поглощает излучение, то интенсивность луча,
прошедшего через кювету В, равна интенсивности падающего излучения (/0); образец в кювете
А поглощает часть излучения и интенсивность луча, прошедшего через кювету А,
равна интенсивности прошедшего излучения (/). Пропускание образца равно отношению
/: /0, а оптическую плотность можно определить по уравнению 9.3.
пути луча / и концентрацией раствора законом Ламберта—Вера (уравнение
9.4), где е — молярный коэффициент экстинкции (или коэффициент
поглощения) растворенного соединения.
А = -lg ///0= e • с • / (9.4)
Концентрация с измеряется в моль/л, а длина пути луча - в см, поэтому
размерность е —л/(моль • см). Коэффициент экстинкции не зависит от
концентрации и является свойством соединения.
Закон Ламберта-Бера связывает оптическую плотность раствора
образца с молярным коэффициентом экстинкции, концентрацией
раствора и длиной пути луча в кювете:
А = е•с • /
Пример 9.1. Использование закона Ламберта-Бера
Нафталин - ароматическое соединение со следующей структурой:
Растворы нафталина поглощают свет с длиной волны 321 нм, а
коэффициент экстинкции для этого перехода равен 288 л/моль см.
Раствор нафталина в этаноле в кювете толщиной 1 см имеет
оптическую плотность 1,2. Определите концентрацию нафталина.
Решение
Согласно закону Ламберта-Бера:
А = гс1
Отсюда:
с = А/(г ■ I)
= 1,2/(288- 1) = 4,2 Ю-3 моль/л
426
9. Спектроскопия
ШШШ КОЛОРИМЕТРИЯ
Из прямой зависимости между оптической плотностью образца и его
концентрацией (уравнение 9.4) непосредственно следует возможность применения
закона Ламберта-Бера в аналитических целях. Для большинства соединений
закон Ламберта—Бера выполняется с хорошей точностью в разбавленных рас-
►- творах. Метод колориметрии используется для определения концентрации
Типы окрашенных окрашенных соединений в растворах. При этом измерение
соединений: оптической плотности позволяет не только определять
конем, разд. 9.11, центрации индивидуальных растворов, но и исследовать
а также т. 2, гл. 16 изменения концентрации окрашенного компонента в ходе
реакции.
Цвета
Когда окрашенное вещество растворяют в некотором растворителе,
интенсивность окраски зависит от концентрации раствора, а сам цвет раствора — от
длины волны видимого света, поглощаемого образцом.
Белый свет содержит непрерывный спектр электромагнитных излучений с
длиной волны от 400 до 750 нм. Когда белый свет попадает на раствор соеди-
Цвет раствора зависит от длины волны видимого света, поглощаемого
образцом, а интенсивность окраски зависит от концентрации раствора.
нения, поглощающего в видимой области, пропускаемый свет окрашен в
цвета, дополнительные к цвету поглощенного излучения. Сульфат меди(И)
поглощает оранжевый свет, и в результате раствор кажется синим, поскольку
синий цвет является дополнительным к оранжевому. В табл. 9.2 указаны
цвета, соответствующие длины волн и дополнительные цвета видимого спектра.
Таблица 9.2. Видимая область спектра электромагнитных излучений
Цвет
поглощенного
света
Красный
Оранжевый
Желтый
Зеленый
Синий
Фиолетовый
Приблизительный интервал
длин волн, нм
750-620
620-580
580-560
560-490
490-430
430-380
Цвет пропускаемого света
(дополнительный к цвету
поглощенного)
Зеленый
Синий
Фиолетовый
Красный
Оранжевый
Желтый
Пример 9.2. Зависимость оптической плотности от концентрации
0,1 М раствор сульфата меди(И) имеет оптическую плотность 0,55.
Как изменится оптическая плотность при удвоении концентрации
раствора? Считайте, что для измерений используется одна и та же
кювета.
9.5. Колориметрия
427
Решение
Связь между оптической плотностью и концентрацией задается законом
Ламберта-Бера:
А = е • с • I
При постоянных значениях длины путргяуцд_ (одна и та же кювета) и константы
е можно записать:
Ах/А2 = сх/с2
где Ах - поглощение при концентрации с,, а А2 — поглощение при
концентрации с2.
Ах = с, -А2/с2 = 0,2 0,55/0,1= 1,1
Согласно закону Ламберта-Бера, для заданного соединения величина А
линейно зависит от с при постоянной длине пути луча /.
Колориметр
На рис. 9.5 схематически показан способ измерения оптической плотности
растворов. В колориметре источник излучения настраивается на
определенную длину волны. Этот выбор делается на основе знания длины волны, при
которой происходит поглощение излучения исследуемым соединением.
Целью колориметрических исследований может быть либо определение
концентрации раствора, либо исследование реакции (см. ниже). Еще один
пример применения колориметрии — определение стехиометрического
состава комплексного соединения (см. разд. 9.11).
1. Анализ растворов железа(Н)
Для определения ионов железа(Н) используется 4,7-дифенил-1,10-фенант-
ролин (9.1).
При добавлении 9.1 к раствору, содержащему ионы железа(П), образуется
интенсивно окрашенное красное соединение, которое поглощает свет с
длиной волны 533 нм и имеет молярный коэффициент экстинкции 22 000. Если
колориметр настроен на длину волны 533 нм, концентрацию красного
соединения можно определить по величине оптической плотности. В одном моле
красного соединения содержится один моль ионов Fe2+, и, если считать, что
все железо(Н), присутствующее в растворе, вступило в реакцию с
соединением 9.1, можно рассчитать концентрацию Fe2+. На рис. 9.6 показано
изменение оптической плотности раствора (при 533 нм) в зависимости от
концентрации ионов железа(П). Такой градуировочный график можно использовать
428
9. Спектроскопия
250
Н
8
X
| 200
с
О
100
50
0
0 0,002 0,004 0,006 0,008 0,01
Концентрация, моль/л
Рис. 9.6. Зависимость оптической плотности (при 533 нм) от концентрации
раствора соединения, образующегося при взаимодействии ионов железа(Н) с 4,7-дифенил-
1,10-фенантролином. Длина пути луча в кювете равна 1 см. График иллюстрирует
закон Ламберта- Бера.
для определения концентрации серии растворов (см. упражнение 9.3 в разд.
9.18). Поскольку величина г очень большая, то даже очень низкие
концентрации ионов Fe2+ можно определять с высокой точностью.
2. Исследование реакции между металлической медью и
дихроматом(VI) калия
Метод колориметрии можно использовать для исследования реакций с
участием окрашенных образцов; при этом можно наблюдать образование или
исчезновение окрашенных реагентов или продуктов. Такое исследование
дает ценную информацию о скорости реакции (см. гл. 10).
Металлическая медь реагирует с дихроматом калия в кислой среде
(уравнение 9.5). В ходе реакции оранжевый цвет раствора дихромата калия
исчезает и раствор окрашивается в сине-зеленый цвет.
ЗСи(тв.) + Сг2072-(водн.) + 14H+ -> ЗСи2+(водн.) + 2Сг3+(водн.) + 7Н20 (9.5)
Ион Сг20^~ поглощает примерно при 500 нм, а продукты реакции
поглощают при ббльших длинах волн. Если колориметр настроить на 500 нм, можно
наблюдать за ходом реакции, измеряя изменение оптической плотности во
времени, как показано на рис. 9.7.
ЕЯ КОЛЕБАТЕЛЬНАЯ СПЕКТРОСКОПИЯ
1. ДВУХАТОМНЫЕ МОЛЕКУЛЫ
Колебательная спектроскопия занимается изучением молекулярных
колебаний. Существует два варианта колебательной спектроскопии — инфракрас-
9.6. Колебательная спектроскопия. 1. Двухатомные молекулы 429
Рис. 9.7. Изменение
оптической плотности (при 500 нм)
во времени. Можно видеть,
как ионы Сг2072~ исчезают в
ходе реакции с металлической
медью в кислой среде.
3000
Время, с
ная (ИК) и рамановская (комбинационного рассеяния, КР) спектроскопия,
и первый из этих методов широко используется в лабораторных
исследованиях. ИК-спектр фиксирует поглощение инфракрасного излучения,
связанное с колебательными состояниями молекул.
Колебания двухатомной молекулы
Двухатомная молекула состоит из двух связанных атомов и может быть
описана в рамках классической модели двух тел (атомов), соединенных
пружиной (электронами связи). Колебания пружины соответствуют растяжению и
сжатию, т. е. валентным колебаниям молекулы (рис. 9.8). Даже при 0 К
молекула Н2 совершает колебания и обладает внутренней энергией, называемой
нулевой энергией. Эта энергия соответствует энергии наинизшего
колебательного состояния, т. е. основного колебательного состояния молекулы, но если
молекуле сообщить соответствующую энергию, могут происходить переходы
на высшие колебательные уровни, на которых молекула колеблется более
энергично, чем на наинизшем колебательном уровне. Если молекуле сообщить
достаточное количество энергии, она диссоциирует.
В основном колебательном состоянии молекула обладает нулевой
энергией.
Аналогия между пружиной и молекулой используется часто, однако
следует помнить о существенном различии. Пружина в состоянии равновесия
неподвижна, в то время как молекула никогда не бывает неподвижна - она
всегда колеблется, даже в основном колебательном состоянии. Поэтому при
обсуждении колебательных состояний молекул на самом деле имеются в
виду переходы из одного колебательного состояния (обычно основного) в другое
(возбужденное) колебательное состояние и обратно. Другое важное различие
между колебаниями пружины и молекулы состоит в том, что колебания
молекулы квантованы и между ними разрешены только определенные переходы.
430
9. Спектроскопия
^^АААААЛААААдГ^)
^fcaAAAAAAAAAAAAAAAAAj^)
Растяжение Растяжение
Рис. 9.8. Колебания гетероя-
дерной двухатомной молекулы.
а — равновесное положение;
б — растяжение связи; в — ежа- ■ > < »
тие связи. Сжатие Сжатие
^ЬлАААЛЛ/^)
В общем случае энергетическая разность между основным колебательным
состоянием и первым возбужденным такова, что при 298 К большая часть
молекул находится в основном колебательном состоянии; доля молекул,
находящихся в некотором состоянии, называется заселенностью этого состояния.
При 298 К заселенность основного колебательного состояния велика, а
высшие состояния характеризуются низкой заселенностью. Заселенность
колебательных уровней описывается больцмановским распределением. В
данной главе принимается, что единственным существенным переходом
является переход из основного состояния в первое возбужденное состояние, и при
обсуждении молекулярных колебаний подразумевается именно этот переход.
► Вернемся теперь к двухатомной молекуле и рассмотрим ее взаимодейст-
Больцмановское вие с И К-излучением. Колебательный переход будет происхо-
распределение: дить только в том случае, если в молекуле имеется колебание
см. разд. 1.18 определенной частоты, и эта частота зависит от прочности ко-
валентной связи в двухатомной молекуле. В ИК-спектре
фиксируется частота излучения, при которой происходит колебательный
переход, но далеко не все типы колебаний дают полосы в ИК-спектре.
Выполняется правило отбора: в ИК-спектре проявляются только те колебания,
которые связаны с изменением дипольного момента молекулы.
Чтобы колебание было ИК-активно, оно должно приводить к
изменению дипольного момента молекулы.
Рассмотрим молекулу Н2. Она неполярна, и растяжение либо сжатие
связи Н—Н не приводит к возникновению дипольного момента. То же верно для
любой гомоядерной двухатомной молекулы. Следовательно, колебания
молекулы Н2 неактивны в ИК-спектре. Молекула СО имеет дипольный момент
(|i = 0,11 Д), который изменяется при изменении расстояния между атомами
углерода и кислорода (см. дополнение 4.2 в гл. 4):
ц = (Электрический заряд) • (Расстояние между зарядами) (9.6)
При колебаниях молекулы СО происходит изменение ее дипольного
момента, и колебания становятся ИК-активны.
9.6. Колебательная спектроскопия. 1. Двухатомные молекулы 431
Силовая постоянная связи
Силовая постоянная связи к характеризует прочность связи: чем выше
прочность связи, тем больше величина к. На рис. 9.9 и 9.10 представлены
энтальпии диссоциации и силовые постоянные связей в HF, HC1, НВг и HI.
Сходство графиков достаточно очевидно.
j Дополнение 9.1. Жесткость пружины
I Для возвращения колеблющейся пружины в равновесное положение необхо- I
j димо приложить определенную силу. Это так называемая возвращающая сила, I
! и ее величина зависит от амплитуды колебания и от свойства пружины, назы- ;
j ваемого жесткостью или силовой постоянной к. \
Возвращающая сила = - (к- Отклонение от равновесного положения)
| Единицы измерения к — Н/м (сила на единицу расстояния).
Приведенная масса двухатомной молекулы
Вернемся снова к аналогии между двухатомной молекулой и пружиной. Если
два тела, соединенные пружиной, имеют сильно различающиеся массы, то
при колебаниях пружины тело большей массы будет существенно менее
подвижно, чем тело меньшей массы. Например, если соединить пружиной
автомобиль и теннисный мяч и инициировать колебания, то практически будет
перемещаться только теннисный мяч, а колебания автомобиля будут
незаметны, и общую картину колебаний можно с большой точностью описывать
как колебания мяча. Если же массы обоих тел близки, движение каждого из
них будет вносить существенный вклад в колебания. Чем ближе значения
масс обоих тел, тем больше сходство их вкладов в общую картину движения.
Двухатомная молекула может состоять из ядер, имеющих примерно
одинаковые массы (например, СО) или заметно различающиеся массы
(например, HI), поэтому необходимо определить величину, которая
характеризовала бы массу двухатомной молекулы с учетом относительных масс ядер. Такой
650
550
о J 450
350
250
s 1000
"* 800 Ь
600
hf на нвг hi
400
200
hf на нвг hi
Рис. 9.9. Изменение энтальпии диссоци- Рис. 9.10. Изменение силовых постоянных
ации связи в ряду галогеноводородов НХ. в ряду галогеноводородов НХ.
432
9. Спектроскопия
величиной является приведенная масса \х , которая определяется для двух
ядер с массами тх и т2:
1/М=1/т,+1/т2 (9.7)
Частота колебаний двухатомной молекулы
По аналогии с движением пружины, двухатомную молекулу можно считать
простым гармоническим осциллятором с учетом относительного движения
обоих ядер в молекуле.
Переход из основого колебательного состояния в первое возбужденное
приводит к появлению в ИК-спектре двухатомной молекулы
фундаментального поглощения — при условии, что колебание ИК-активно. Частота этого
поглощения (в Гц) связана с силовой постоянной связи (в Н/м) и приведенной
массой двухатомной молекулы (в кг):
=1VI
2я
(9.8)
Единицы измерения v в уравнении 9.8 - Гц, а шкала И К-спектрометра
обычно градуирована в волновых числах (см-1). Уравнение 9.9 дает связь между
^~ волновым числом и частотой валентных колебаний; комбинация уравнений
Волновые числа: 9.8 и 9.9 дает уравнение 9.10. Пример 9.3 иллюстрирует приме-
см. разд. 9.2 нение уравнения 9.10.
v = v/c
r=-i-VI
(9.9)
(9.10)
Отсюда следует, что растяжение связи (т. е. валентное колебание) требует
меньших затрат энергии, если связь образована тяжелыми, а не легкими
элементами. На рис. 9.11 показано изменение частот фундаментальных
валентных колебаний в ряду галогеноводородов. Сопоставление с рис. 9.9 и 9.10
подчеркивает связь между частотой валентных колебаний, энтальпией
диссоциации и силовой постоянной связи.
т 4500
£ 4000
I
Б 3500
>
3000 г
2500
2000
HF HC1 НВг HI
Рис. 9.11. Изменение частоты
фундаментального валентного колебания
в ряду галогеноводородов.
Символ [х используется для обозначения как приведенной массы, так и дипольного момента (а
также для ряда других величин), но по контексту всегда можно определить, о какой именно величине
идет речь.
9.6. Колебательная спектроскопия. 1. Двухатомные молекулы 433
Дополнение 9.2. Можно ли считать простой гармонический осциллятор
хорошей моделью колебаний связи?
Уравнение 9.8 задает соотношение между частотой поглощения в ИК-спектре
двухатомной молекулы, силовой постоянной связи и приведенной массой молекулы. Это
уравнение применимо, если считать, что молекула напоминает пружину и
подчиняется модели простого гармонического осциллятора. Потенциальная энергия,
соответствующая простому гармоническому движению, показана на диаграмме а. Зависимость
потенциальной энергии от межъядерного расстояния для реальной двухатомной
молекулы приведена на диаграмме б, и именно эта кривая описывает реальные колебания
молекул.
Межъядерное расстояние
Сжатие Растяжение
Смещение от положения
равновесия
Итак, можно ли применять уравнение 9.8 для описания колебаний молекул?
Так как речь идет о переходе из основого колебательного состояния в первое
возбужденное, основной интерес для нас представляет нижняя часть диаграммы
потенциальной энергии. В этой области кривые на диаграммах а и кочень похожи, поэтому
использование приближения простого гармонического осциллятора для молекулярных
колебаний вполне оправданно.
Пример 9.3. Определение силовой постоянной связи в моноксиде углерода
Валентному колебанию в СО соответствует линия поглощения в
ИК-спектре при 2170 см-1. Чему равна силовая постоянная связи в
молекуле моноксида углерода?
(с = 3 10е м/с; 1 а. е.м. = 1,66 10"27кг; АГ{С) = 12; Аг(0) = 16.)
Решение
Из уравнения
v
_lVa
2кс !й
434
9. Спектроскопия
определяем
к = 4я2 • с2 • v2 • ц
Удостоверимся, что единицы измерения всех величин соответствуют друг другу.
Волновое число поглощения равно 2170 см-1
Для приведения к одинаковым единицам: с = 3 • 108 м/с = 3 • 1010 см/с.
Приведенная масса ц рассчитывается по уравнению:
_!_ = _!+ -1 = ! + 1 - 9 70 1Г)25
|i тх т2 2- 1,66- Ю-27 16- 1,66- Ю-27 ' *
1
Ц" 8,79-1025 КГ
Таким образом, силовая постоянная равна:
А: = 4я2 • с2 • v"2- м
= 4-3,1422-(3- 1010)2 - 21702
8,79 • 1025
= 1904 Н/м
Е^1 КОЛЕБАТЕЛЬНАЯ СПЕКТРОСКОПИЯ
2. МНОГОАТОМНЫЕ МОЛЕКУЛЫ
Колебательные степени свободы
В то время как для двухатомной молекулы существует единственное
валентное колебание (рис. 9.8), типы колебательных движений многоатомных
молекул более сложны.
С02: линейная трехатомная молекула
Молекула диоксида углерода линейна и содержит две одинаковые связи
С=0. У этой молекулы существует два типа колебаний. На рис. 9.12,а
показано симметричное валентное колебание, при котором атомы кислорода вначале
удаляются друг от друга, а затем сближаются. На рис. 9.12,5
проиллюстрировано антисимметричное валентное колебание. В этом случае одна из связей С=0
► удлинняется, а другая одновременно сжимается. Молекула диоксида углерода
ХП(С) = 2,6; хп(0) = 3,4 не имеет дипольного момента, хотя каждая из связей С=0 по-
лярна. В ходе симметричного валентного колебания дипольный
момент остается равен нулю, и это колебание ИК-инактивно. В ходе
антисимметричного валентного колебания происходит изменение дипольного
момента; в этом можно убедиться, рассмотрев правую часть рис. 9.12,5.
Следовательно, антисимметричное валентное колебание ИК-активно.
Если линейная трехатомная молекула имеет дипольный момент (например,
HCN), то и симметричное, и антисимметричное валентные колебания
оказываются активными в ИК-спектре.
У линейной трехатомной молекулы имеется также деформационное
колебание (рис. 9.12,в); в случае С02 это колебание можно представить как движе-
9.7. Колебательная спектроскопия. 2. Многоатомные молекулы 435
ние атома углерода вверх и вниз по отношению к атомам кислорода. На
рис. 9.12,в это движение совершается в плоскости листа; существует также
эквивалентное колебание в плоскости, перпендикулярной плоскости листа,
- оба типа деформационных колебаний одинаковы по энергии, т. е.
являются вырожденными. Деформационные колебания молекулы С02 приводят к
изменению ее дипольного момента и активны в ИК-спектре.
о
о
Симметричное
валентное колебание
Неактивно в
ИК-спектре
о=о=о
Антисимметричное
валентное колебание
Активно в ИК-спектре
(2349 см"1)
„t t
(МИ)
\7
V
Деформационное
колебание
Активно в ИК-спектре
(667 см"1)
0=0=0
0=0=0
0=0=0
0=00
0=0=0
0=0=0
0=00
0=0=0
0=0=0
о===о=о
0=0=0
0=0=0
0=0=0
Рис. 9.12. Типы колебаний молекулы диоксида углерода, а - симметричное
валентное колебание; б— антисимметричное валентное колебание; в — деформационное
колебание в плоскости листа. Слева: направление относительного движения атомов;
справа сверху вниз: общая последовательность движений молекулы С02 во время
данного колебания.
436
9. Спектроскопия
Термин «вырожденные» означает «имеющие одинаковую энергию».
Частоты антисимметричного валентного и деформационного колебаний
молекулы С02 равны 2349 и 667 см-1 соответственно. Это говорит о том, что
для антисимметричного валентного колебания требуется большая затрата
энергии, чем для деформационного.
Н20: угловая трехатомная молекула
Три типа колебаний молекулы воды показаны на рис. 9.13. Молекула воды
обладает дипольным моментом (ц = 1,85 Д), который изменяется во время
симметричного валентного колебания. Это верно и для других угловых
молекул, для которых симметричное валентное колебание ИК-активно.
Антисимметричное валентное колебание также ИК-активно. Движение атомов
водорода по направлению друг к другу, а затем в обратном направлении приводит
к симметричному деформационному колебанию типа «ножницы». При этом
также происходит изменение дипольного момента и наблюдается полоса
поглощения в ИК-спектре.
Колебательные степени свободы
В вышеприведенном обсуждении число колебаний молекул С02 и Н20 легко
определимо; очевидно, что при рассмотрении более сложных молекул
определение всех возможных типов колебаний может оказаться трудной задачей.
Однако существуют простые правила, которые позволяют определить число
разрешенных колебаний.
Если молекула состоит из п атомов, она имеет Ъп степеней свободы.
Молекула в целом может перемещаться в пространстве, т. е. совершать
поступательное движение. Смещение - векторная величина, поэтому на поступательное
движение приходится три степени свободы, соответствующие смещению
относительно трех осей координат (например, jc, у и zY молекула имеет три
поступательные степени свободы.
Симметричное
валентное колебание
Активно в ИК-спектре
(3657 см"1)
Антисимметричное
валентное колебание
Активно в ИК-спектре
(3756 см"1)
Ч
/
Симметричное деформационное
колебание типа «ножницы»
Активно в ИК-спектре
(1595 см"1)
Рис. 9.13. Типы колебаний в молекуле воды, а - в ходе симметричного валентного колебания
оба атома водорода сначала одновременно удаляются от атома кислорода, а затем
одновременно приближаются к нему; б — в ходе антисимметричного валентного колебания один из атомов
водорода удаляется от атома кислорода, а другой приближается к нему; в - в ходе
симметричного деформационного колебания типа «ножницы» угол связей Н—О—Н периодически
увеличивается и уменьшается; это колебание происходит в плоскости листа.
9.8. Использование ИК-спектроскопии в аналитических целях 437
Молекула, состоящая из п атомов, имеет Зп степеней свободы.
Так как на поступательное движение молекулы приходится три степени
свободы, на все остальные типы движения остается (Зп - 3) степеней
свободы, которые подразделяются на вращательные и колебательные. Подобно
поступательному движению, вращение можно описывать как изменение
координат молекулы. Принимается*, что любая нелинейная молекула имеет три
вращательные степени свободы, а линейная - только две вращательные
степени свободы. Отсюда следует, что число колебательных степеней свободы
зависит от числа атомов в молекуле и от того, является молекула линейной
или нелинейной. Уравнения 9.11 и 9.12 суммируют эти выводы.
Число колебательных степеней свободы в нелинейной молекуле = Зл — 6 (9.11)
Число колебательных степеней свободы в линейной молекуле = Зп — 5 (9.12)
Теория Гиллеспи: Применим уравнения 9.11 и 9.12 для некоторых простых моле-
см. разд. 5.6 кул, геометрия которых предсказывается теорией Гиллеспи.
1) S02 — угловая молекула: п = 3.
Число колебательных степеней свободы = Зп — 6 = 3.
2) С02 — линейная молекула: п = 3.
Число колебательных степеней свободы = Зп — 5 = 4.
3) СН4 - тетраэдрическая молекула: п = 5.
Число колебательных степеней свободы = Зп - 6 = 9.
Дополнительно к валентным и деформационным ножничным
колебаниям, описанным для молекул С02 и Н20, более крупные молекулы могут
испытывать также деформационные маятниковые, крутильные и веерные
колебания. Детальное обсуждение этих типов колебаний выходит за рамки
данной книги, но на рис. 9.14 показаны примеры сложных колебаний мети-
леновой группы в цепи алкана.
ЕЕЯ ИСПОЛЬЗОВАНИЕ ИК-СПЕКТРОСКОПИИ
В АНАЛИТИЧЕСКИХ ЦЕЛЯХ
Рассмотрим применение ИК-спектроскопии в лаборатории. Метод
позволяет исследовать вещества в твердом и газообразном состояниях, а также в
растворе. Существует два подхода к расшифровке ИК-спектров. Расшифровка
спектра как свойства молекулы в целом всегда корректна, но требует много
Для более подробного обсуждения см. Atkins P W., Physical Chemie, 5th ed., Oxford Univ. Press, Oxford,
1994. [Есть русский перевод раннего издания: Эткинс П., Физическая химия, М, Мир, 1980.
Готовится к выпуску перевод 7-го издания: Эткинс П., де Паула Дж., Физическая химия: В 3-х т. Пер. с
англ. - М.: Мир, 2003.]
438
9. Спектроскопия
Симметричное
растяжение двух
связей С—Н
Симметричное
сжатие двух
связей С—Н
Симметричное
растяжение двух
связей С—Н
Растяжение
одной связи
С—Н и сжатие
другой связи
Сжатие первой
связи С—Н
и растяжение
второй связи
Растяжение
первой связи
С—Н и сжатие
второй связи
Уменьшение
угла между
двумя связями
С-Н
Увеличение
угла между
двумя связями
С-Н
Уменьшение
угла между
двумя связями
С-Н
Кручение jj& Кручение груп
группы СН2 по (л пы СН2 против
часовой стрелке ^-^ часовой стрелки
Кручение
группы СН2 по
часовой стрелке
Движение
группы СН2
на наблюдателя
Движение
группы СН2
от наблюдателя
Движение
группы СН2
на наблюдателя
Рис. 9.14. Типы колебаний метиленовой (СН2) группы в цепи алкана. а -
симметричное валентное колебание; б - антисимметричное валентное колебание; в -
деформационное ножничное колебание; г — маятниковое; д — крутильное; е — веерное.
9.8. Использование ИК-спектроскопии в аналитических целях 439
времени и осуществима только для простых молекул. Обычно ИК-спектр
рассматривают как совокупность линий поглощения от различных частей
молекулы. Такой подход напоминает модель функциональных групп в
органической химии, согласно которой группа проявляет сходные свойства в
различных молекулах. Аналогичным образом, удобно считать, что линии
поглощения в ИК-спектре соответствуют определенным функциональным
группам. Такой подход чрезмерно упрощен и особенно ненадежен при работе со
спектрами неорганических соединений.
ИК-спектры органических соединений включают несколько характерных
областей:
• область колебаний молекулярного скелета, в частности, связей С—С;
• область колебаний функциональных групп, в частности, кратных связей.
Таким образом, спектр распадается на две части. Выше 1500 см-1 большая
часть линий соответствует валентным колебаниям кратных связей с участием
углерода (С=С, С=С, С=0, C=N, C=N и т. д.). Ниже 1500 см-1, в области
молекулярных отпечатков пальцев (фингерпринтовая область), находятся
валентные колебания одинарных связей С—X и ряд других. Различия между
двумя областями довольно условны; например, валентные колебания связей
С—Н находятся около 3000 см-1.
Область молекулярных «отпечатков пальцев»
Как правило, ИК-спектр соединения содержит серию полос поглощения
ниже 1500 см-1, совокупность которых характерна для данного соединения. Эти
полосы не всегда относят к определенным типам колебания молекулы. Рас-
смоторим ИК-спектр кофеина (вещество, содержащееся в чае и кофе),
показанный на рис. 9.15. Ниже 1500 см-1 в нем содержится множество полос,
которые трудно отнести к индивидуальным типам колебания молекулы. Однако
этот спектр характеристичен для молекулы кофеина, так как никакая другая
молекула не имеет идентичного спектра. Именно поэтому область спектра
ниже 1500 см-1 называют областью молекулярных отпечатков пальцев или
фингепринтовой областью.
н3с>
Кофеин
2500
2000
1500
1000 500
Волновое число
Рис. 9.15. ИК-спектр кофеина, в структуре которого присутствуют два сочлененных цикла,
состоящих из атомов углерода и азота.
440
9. Спектроскопия
Наиболее эффективный способ использования области «отпечатков
пальцев» в ИК-спектрах — сравнение частот и относительных интенсивно-
стей полос поглощения со справочными данными.
Функциональные группы и расшифровка ИК-спектров
Функциональные группы в молекулах могут давать полосы поглощения при
определенных частотах от 400 до 4000 см-1. Проще всего идентифицировать
функциональную группу по полосам, которые не попадают в область
«отпечатков пальцев».
► Рассмотрим в качестве примера карбонильную группу
Карбонильные С=0, которая содержится в ряде соединений, включая альде-
соединения: см. гл. 17 гиды 9.2, кетоны 9.3, карбоновые кислоты 9.4, ацилхлориды
9.5, сложные эфиры 9.6 и карбоксилат-анионы 9.7.
R R R R
/
= с
\
н
9.2
/
R
9.3
R
/
о = с
\
OR
9.6
О:
О:
/
= С
\
ОН
9.4
R
/
= С
\
о-
9.7
/
о = с
\
а
9.5
Валентное колебание связи С=0 обычно дает сильную (интенсивную)
полосу поглощения около 1700 см-1 (см. рис. 17.6), но точное значение частоты
зависит от прочности связи, на которую в свою очередь влияют другие
группы молекулы или иона. Возьмем за точку отсчета альдегид RCHO. При
переходе от альдегида к кетону атом Н заменяется на электронодонорную ал-
^» кильную группу и атом углерода в группе С=0 кетона имеет меньший час-
Электронодонорные тичный заряд 5+. Следовательно, полярность карбониль-
свойства: см. разд. 8.12 ной связи (С5+—О5-) в кетоне меньше, чем в альдегиде, а
значит, в кетоне растяжение связи С=0 происходит легче
и валентное колебание С=0 поглощает свет на меньшей частоте, чем в
альдегиде. На рис. 9.16 ясно видно уменьшение частоты ИК-поглощения
карбонильной группы при переходе от этилацетата к ацетону.
Теперь сравним альдегид RCHO с соответствующим ацилхлоридом
RCOC1. Хлор является акцептором электронов, и атом углерода в С=0-груп-
► пе ацилхлорида имеет больший частичный заряд 8+, чем в альдегиде. Это оз-
ХП(С1) = 3,2; хп(Н) = 2,2 начает, что связь С=0 в ацилхлориде имеет более ионный
характер, и ее растяжение происходит труднее, соответственно
повышается частота валентного колебания (табл. 9.3).
9.8. Использование ИК-спектроскопии в аналитических целях 441
Таблица 9.3. Примерные интервалы частот валентных колебаний карбонильной
группы (С=0) в соединениях со структурами 9.2-9.7, где R - алкил
Класс соединений
Альдегид
Кетон
Карбоновая кислота
Ацилхлорид
Сложный эфир
Карбоксилат-ион
Структура
9.2
9.3
9.4
9.5
9.6
9.7
Интервал ц
1740-1720
1725-1705
1725-1700
1815-1790
1750-1735
1610-1550
В случае сложного эфира действуют два эффекта. Атом кислорода OR-
группы более электроотрицателен, чем углерод, и это ведет к повышению
частичного заряда 8+ на атоме углерода карбонильной группы. В то же время,
неподеленная электронная пара кислорода OR-группы делокализована
(уравнение 9.13), что приводит к ослаблению карбонильной связи
углерод-кислород. Суммарное действие этих эффектов приводит к тому, что
полоса поглощения, соответствующая валентному колебанию карбонильной
группы в сложном эфире, мало отличается от соответствующей полосы в
альдегиде (рис. 9.16).
R
О-
/
(9.13)
OR
Me-
О
/
• С Этилэтаноат (этилацетат)
\
OEt
О
Ме«
//
. q Этаналь (ацетальдегид)
\
Н
О
Me-
1800 1600
Волновое число
/
С
\
■ с Ацетон
Me
Рис. 9.16. Полосы поглощения карбонильных групп в ИК-спектрах ацетона, этана-
ля (ацетальдегида) и этилацетата.
442
9. Спектроскопия
Таблица 9.4. Примерные интервалы частот валентных колебаний некоторых
функциональных групп с кратными связями при атоме углерода
Функциональная группа
Типичный интервал частот полосы поглощения, см~1
R2C=CR2
RGeCR
R2C=NR
RON
R2C=0
R2C=S
1680^620
2260-2100
1690-1640
2260-2200
1815-1550 (см. табл. 9.3)
1200-1050
Области частот полос поглощения, соответствующих валентным
колебаниям ряда других функциональных групп с кратной связью при атоме
углерода (например, С=С, С=С, ON, C=N и C=S), приведены в табл. 9.4.
Многие функциональные группы содержат одинарные связи, и полосы
поглощения, соответствующие их валентным колебаниям, часто попадают в
область «отпечатков пальцев» и не могут быть легко идентифицированы.
Частоты валентных колебаний связей углерод — галоген приведены в табл. 9.5.
В то время как полосы поглощения, соответствующие валентному
колебанию связи С—С1, хорошо видны в спектре СС14 (рис. 9.17,а), они теряются
среди множества линий области «отпечатков пальцев» спектра 1,2-дихлор-
пропана (рис. 9.17,6).
Таблица 9.5. Примерные интервалы частот валентных колебаний связей
углерод—галоген
Группа
Типичный интервал частот полосы поглощения, см
-I
C-F
С-С1
С-Вг
C-I
1400-1000
800-600
750-500
-500
CCL
СН2С1СНС1СН3
3000 2500 2000 1500 1000 500
Волновое число
Рис. 9.17. ИК-спектры тетрахлорметана СС14 (а) и 1,2-дихлорпропана
СН2С!СНС1СН3 (б).
9.8. Использование ИК-спектроскопии в аналитических целях 443
(Г—1
i i i i i i i i I i i i i
ОН
сн
'сн3
он
3500
3000
2500
2000
1500 1000 500
Волновое число
Рис. 9.18. ИК-спектры пропанола-1 (а) и пропанола-2 (б).
Ряд полос поглощения, соответствующих валентным колебаниям связей
О—Н, N—H, S—Н и С—Н, идентифицируется существенно легче (табл. 9.6).
^ На рис. 9.18 показаны ИК-спектры пропанола-1 и пропанола-2,на которых
Номенклатура видны линии около 3340 см-1, соответствующие валентному
спиртов: см. разд 14.9 колебанию связи О—Н. Эти линии уширены из-за наличия
водородных связей между молекулами спиртов, что является
характеристическим свойством полос поглощения, соответствующих связи
О—Н. Хотя отнесение всех линий в спектрах обоих соединений
затруднительно, можно видеть, что каждое из них имеет характеристический,
свойственный только ему, ИК-спектр, с помощью которого их можно различить
при наличии образцов сравнения. Полосы поглощения, соответствующие
связям N—Н, находятся в той же области спектра, что и полосы,
соответствующие связям О—Н.
Полосы поглощения при 3000 см-1 на рис. 9.17,6 и 9.18, относятся к
валентным колебаниям связей С—Н. Из данных табл. 9.6 можно видеть, что
^. частоты валентных колебаний связей С—Н зависят от типа гибридизации
Электроотрицательно- углеродного атома. Это связано с тем, что полярность (и
сти атомов углерода: прочность) связи С—Н уменьшается в ряду: C(sp)—H >
см. разд. 8.20 C(sp2)—H > C(sp3)—Н. На рис. 9.19 показаны фрагменты ИК-
спектров бензола и этилбензола. Плоская молекула бензола
Таблица 9.6. Примерные интервалы частот валентных колебаний связей О—Н,
N-H, S-НиС-Н
Группа
Типичный интервал частот полосы поглощения, см
0-U
N-H
S-H
С—Н (5/?3-гибридизация)
С—Н (sp2-гибридизация)
С—Н (sp-гибридизация)
3600—3200 (уширен за счет водородных связей)
3500-3300
2600-2350 (слабая полоса)
2950-2850
3100-3010
-3300
9. Спектроскопия
сн.
^^
3200
3000
2800
Волновое число
Рис. 9.19. ИК-спектры бензола (а) и этилбензола (б). Показана только область около
3000 см-1, в которой находятся полосы валентных колебаний связи С—Н.
содержит только з/?2-гибридизованные атомы углерода, и полосы
поглощения связей С—Н находятся в интервале 3120-3000 см-1. В спектре
этилбензола наблюдаются полосы поглощения, соответствующие как связям
C(sp2)—H, так и связям Cisp*)—Н.
Дополнительное обсуждение использования ИК-спектроскопии в
лаборатории можно»найти в прилагаемом задачнике. ИК-спектроскопические
характеристики полос поглощения других функциональных групп
приведены в гл. 14, 15 и 17.
Пример 9.4. ИК-спектры гексана и гексена-1
На рисунке показаны ИК-спектры гексана и гексена-1. Какие
структурные особенности этих молекул ответственны за наблюдаемые
полосы поглощения?
J
ТЩ
rvYH
Гексан
Гексен-1
3500 3000 2500 2000 1500
Волновое число
1000
500
9.8. Использование ИК-спектроскопии в аналитических целях 445
Решение
Рассмотрим структуры гексана и гексена-1:
Н
Н2 Н2 Н2 Н |
н3с с с н3с с с^ н
2 2 2 I
н
1. Часть ИК-спектров ниже 1500 см-1 представляет собой область «отпечатков
пальцев» и задает совокупность характеристических линий для каждого
соединения.
2. В спектре гексана, помимо этого, присутствует только группа полос
поглощения около 3000 см-1, которая соответствует валентным колебаниям связи С—
Н; атомы углерода $/?3-гибридизованы.
3. Сильные полосы поглощения около 3000 см-1 в спектре гексена-1
соответствуют валентным колебаниям связи С(5/?3)—Н; менее интенсивная полоса
примерно при 3080 см-1 соответствует валентным колебаниям связи Cisp1)—H.
4. Узкая полоса при 1643 см-1 в спектре гексена-1 характерна для валентного
колебания связи С=С.
5. Дополнительная особенность спектра гексена-1 - широкая полоса с
максимумом около 3400 см-1. Это полоса может быть отнесена к колебаниям связи
О—Н, и позволяет предположить, что образец гексена-1 содержит влагу!
Пример 9.5. Идентификация некоторых ИК-спектров
На рисунке приведены И К-спектры октана, пропаннитрила, додека-
нола-1 и гептанона-2. Определите, какому соединению
соответствует каждый спектр.
, , ■ , I , , ■ , I , ■ ■ ■ I , ■ , , I , , , ■ I , , , ■ I ■ , ■ , И
3500 3000 2500 2000 1500 1000 500
Волновое число
Решение
Рассмотрим структуры соединений:
Октан СН3СН2СН2СН2СН2СН2СН2СН3
Додеканол-1 СН3СН2СН2СН2СН2СН2СН2СН2СН2СН2СН2СН2ОН
Пропаннитрил-1 CH3CH2C=N
Гептанон-2 СН3СН2СН2СН2СН2\ /СН3
II
О
446 9. Спектроскопия
Идентификацию спектров следует начать с поиска характеристических полос
поглощения присутствующих функциональных групп.
Спектр а содержит сильную полосу поглощения около 2250 см-1. Ее можно
отнести к группе C=N в пропаннитриле. Спектр также содержит полосы около
3000 см-1, характерные для валентных колебаний алифатических связей С—Н.
В спектре б имеется сильная полоса с максимумом около 3200 см-1,
характерная для спиртовой группы. Этот спектр соответствует додеканолу-1. Как можно
ожидать для этого соединения, в спектре присутствуют линии,
соответствующие валентным колебаниям С—Н.
В спектре г имеется сильная полоса поглощения примерно 1700 см-1,
свидетельствующая о наличии карбонильной группы. Этот спектр соответствует гек-
санону-2. Кроме области «отпечатков пальцев» в спектре имеются линии, со-
тветствующие валентным колебаниям алифатических связей С—Н, которые и
должны присутствовать в этом спектре.
В спектре в выше области «отпечатков пальцев» наблюдаются только полосы,
соответствующие валентным колебаниям связей С—Н; по-видимому, в этом
соединении отсутствуют функциональные группы. Этот спектр соответствует
октану
Выводы:
Спектр Отнесение
а Пропаннитрил
б Додеканол-1
в Октан
г Гептанон-2
ЭЛЕКТРОННАЯ СПЕКТРОСКОПИЯ
1. ЭЛЕКТРОННЫЕ ПЕРЕХОДЫ В МОЛЕКУЛАХ,
ВАКУУМНЫЙ УФ И ВЫБОР РАСТВОРИТЕЛЯ
^» Электронная спектроскопия рассматривает переходы между электронными
Атомный спектр водо- энергетическими уровнями, примером которых может служить
рода: см. разд. 2.16 атомный спектр водорода. В данной главе обсуждаются
электронные спектры молекул и особое внимание уделяется
использованию электронной спектроскопии в лаборатории.
Электронные спектры принято приводить в единицах длин волн (нм).
Поглощение ультрафиолетового и видимого света
Молекулярные орбитали подразделяются на связывающие, несвязывающие
и разрыхляющие. Связывающие и разрыхляющие орбитали могут иметь
симметрию а- и л-типа. При поглощении молекулой соответствущего количества
энергии электрон с заполненной орбитали может переходить на частично
заполненную или пустую орбиталь. Так как энергии орбиталей квантованы,
каждому переходу соответствует строго определенное количество энергии.
Энергия перехода электрона с высшей заполненной молекулярной орбитали
(ВЗМО) на низшую свободную молекулярную орбиталь (НСМО), как
правило, соответствует ультрафиолетовой (УФ) или видимой области спектра
электромагнитных излучений, в интервале длин волн 100-800 нм. Стандартные
9.9. Электронная спектроскопия. 1. Электронные переходы в молекулах 447
£
Рабочий диапазон стандартных
лабораторных спектрофотометров
Рис. 9.20. УФ и видимая
области спектра
электромагнитных излучений
(вакуумный УФ также называется
дальним УФ).
2
£
Ближний
УФ
Видимая область
100 200
400
600
800
Длина волны, нм
лабораторные вид./УФ-спектрофотометры работают в интервале длин волн
200-900 нм (рис. 9.20).
ВЗМО - высшая заполненная молекулярная орбиталь;
НСМО - низшая свободная молекулярная орбиталь.
На рис. 9.21 показано, что электронный переход должен происходить при
поглощении молекулой света с энергией АЕ. Можно было бы ожидать, что
спектр поглощения в этом случае будет содержать один узкий пик (такой, как
изображен на рис. 9.2). В реальности, однако, молекулярные электронные
спектры состоят из широких полос (рис. 9.22), в отличие от атомных
спектров, для которых характерны узкие линии. В чем причина этого? Ответ на
Е А
Рис. 9.21. Переход с одного энергетического
уровня на другой может произойти при
поглощении молекулой света с энергией А£;
энергетические уровни квантованы.
Энергия перехода
Длина волны, нм
Рис. 9.22. Электронные спектры молекул или молекулярных ионов содержат
широкие полосы поглощения, лежащие в большом интервале длин волн. Полосы
поглощения характеризуются значениями X
448
9. Спектроскопия
этот вопрос связан с различием временных шкал. Дело в том, что
поглощение фотона света — очень быстрый процесс (~10~18 с), протекающий
существенно быстрее молекулярных колебаний и вращений. Соответственно
электронный переход застает молекулу в определенном колебательном и
вращательном состоянии в некоторый момент времени. Так как энергия
молекулярных орбиталей зависит от геометрии молекулы, то реально наблюдается
широкий интервал значений А£ (соответствующий различным
колебательным и вращательным состояниям). Колебательные и вращательные уровни
расположены гораздо ближе друг к другу, чем электронные, поэтому в
электронном спектре наблюдается широкая полоса. Если колебательные и
вращательные переходы ограничены (например, при охлаждении), полосы
становятся более узкими. Аналогично, если переходы в существенной степени
локализованы на отдельных атомных центрах, спектр также состоит из узких
полос, как это наблюдается в случае соединений лантанидов.
Хотя полоса поглощения занимает широкий интервал длин волн, ее
положение можно описать длиной волны, отвечающей положению максимума.
Эта величина обозначается А,макс и определяется, как показано на рис. 9.22.
^» Полосы поглощения в электронных спектрах имеют характеристические
Закон Ламберта-Бера: интенсивности, значения которых зависят от молярных коэф-
см. разд. 9.4 фициентов экстинкции (е), определямых по закону
Ламберта-Бера. Уравнение 9.14 задает связь между емакс и оптической
плотностью в максимуме Лмакс; сравните это уравнение с уравнением 9.4.
^макс =Ашкс/(с'0 (9-14)
Величины емакс варьируют от близких к нулю (для очень слабого
поглощения) до > 10000 л/моль • см (интенсивное поглощение). Для полного
описания полос поглощения в электронных спектрах необходимо использовать и
макс » Емакс*
Электронные спектры молекул, как правило, состоят из широких
полос, для описания которых необходимо использовать величины
^макс(нм) и емакс (л/мОЛЬ-СМ).
Электроные переходы в вакуумном УФ
Исходная заполненная молекулярная орбиталь при электронном переходе
может быть как связывающей (а или л), так и несвязывающей. Обычно,
электронный переход с аМО на о*МО соответствует излучению в вакуумной
УФ-области электромагнитного спектра*. Например, этан С2Н6 поглощает
излучение с длиной волны 135 нм, и этот переход (обозначаемый о -> а*), не
попадает в рабочий интервал обычного спектрофотометра. Энергетическая
разность между я- и л*-орбиталями в этине (или ацетилене НС=СН) меньше,
Область ниже 200 нм называется вакуумным или дальним УФ-спектром. Для спектроскопических
исследований в этой области необходимо вакуумировать рабочие кюветы, так как 02 поглощает в
этой области.
9.9. Электронная спектроскопия. 1. Электронные переходы в молекулах 449
чем между о- и а*-орбиталями в этане, но она также соответствует излучению
в вакуумной УФ-области (Хмакс= 173 нм, емакс = 6000 л/моль • см). Как
правило, электронный переход к —> к* для изолированных связей С=С шш С=С
лежит в области вакуумного УФ.
^- Третий тип молекулярных электронных переходов записывается в виде
Несвязывающие п —> а*, где п — несвязывающая орбиталь. Такие переходы про-
орбитали: исходят в насыщенных молекулах, содержащих неподеленные
см. разд. 4.5 электронные пары. Примеры таких молекул - вода, спирты
(ROH), амины (RNH2) и галогеноалканы (RF, RC1, RBr и RI).
Во многих случаях электронные переходы п -»а* лежат в области
вакуумного УФ, например для воды (X = 167 нм, е = 500 л/моль • см) и
метанола а
177 нм, е = 200 л/моль • см).
Выбор растворителя для вид./УФ-спектроскопии
В вид./УФ-спектроскопии часто используются растворенные образцы, и
выбор растворителя играет важную роль. В идеальном случае необходим
растворитель, прозрачный в интересующей исследователя области спектра. На
рис. 9.23 показаны фрагменты вид./УФ-спектра четырех растворителей. Аце-
тонитрил «прозрачен» в интервале от 200 до 800 нм, при уменьшении длины
волны поглощение начинается примерно при 200 нм, что соответствует
началу широкой полосы поглощения (Я,макс = 167 нм). Следовательно, длина
волны 200 нм оказывается граничной для использования ацетонитрила в
качестве растворителя в вид./УФ-спектроскопии, и этот растворитель пригоден для
использования в лаборатории. Аналогично, вода «прозрачна» при уменыиени
длины волны вплоть до 210 нм (А.макс = 167 нм). Нижняя граница для цикло-
гексана также лежит около 210 нм, а для хлороформа - около 240 нм, а для
ацетона - при 330 нм, и это ограничивает использование ацетона в качестве
растворителя. Однако поскольку поглощение ацетона относительно невелико
(емакс = 15 л/моль • см), его все же можно использовать как растворитель для
соединений с существенно более высокими коэффициентами экстинкции.
0,5
100
Г Г
1!
\i
V
i i —i
/
2
3
-1 i i i
200
300 400 500 600 700 800
Длина волны, нм
Рис. 9.23. Фрагменты электронных спектров некоторых стандартных растворителей:
/ - ацетонитрил; 2 — циклогексан; 3 — хлороформ; 4 - ацетон. Длина пути луча в
каждом случае равна 1 см. Увеличение оптической плотности в области малых длин волн
соответствует началу широкой полосы поглощения; например, для ацетонитрила
^макс = 167 нм, а для ацетона Хмакс = 279 нм.
9. Спектроскопия
ЭЛЕКТРОННАЯ СПЕКТРОСКОПИЯ. 2. я-СОПРЯЖЕНИЕ
Зависимость я -> я*-перехода от количества и
взаимного расположения я-связей
На рис. 9.24 показан сдвиг ^макс для я-> я*-переходов в ряду этилен,
бутадиен-1,3, гексатриен-1,3,5. Соответствующие величины емакс равны 15000,
21 000 и 34 600 л/моль ■ см. Связь между этими молекулами состоит в том, что
увеличение длины углеродной цепи происходит за счет добавления
чередующихся одинарной и двойной углерод-углеродных связей. Сдвиг в область более
длинных волн с увеличением длины цепи — общее явление, указывающее на
уменьшение энергетической разности между уровнями я и я*. Почему это
происходит? Чтобы ответить на этот вопрос, рассмотрим, каким образом
увеличение длины цепи и взаимное расположение одинарных и двойных связей
влияет на я-связывание между атомами углерода. Для решения этой задачи
будем последовательно добавлять атомы углерода к цепи С2.
Локализованные и делокализованные я-связи
я-Связь в этилене образуется при положительном перекрывании двух 2/?-АО,
лежащих перпендикулярно плоскости молекулы, а я*-МО образуется при
отрицательном перекрывании двух 2р-АО (рис. 9.25,а). я-Связь углерод -
углерод локализована.
Ранее уже обсуждалось связывание в аллильном анионе в рамках теории
ВС (структуры 8.17 и 8.18). Рассмотрим теперь модель МО. Анион является
плоским, и после формирования «скелета» из а-связей каждый атом
углерода имеет одну 2р-АО, расположенную перпендикулярно плоскости частицы.
я-Связывание в аллильном анионе возникает за счет перекрывания этих
атомных орбиталей, причем из трех АО должно образоваться три МО
(рис. 9.25,6). я-Связывающая орбиталь возникает при положительном
перекрывании всех трех углеродных 2/ьАО, и эта связь «размазана» по всем трем
углеродным центрам: говорят, что я-связь делокализована, и этот результат
находится в соответствии со структурой 9.8, полученной по модели ВС.
300 г
X
и I
** 250 г
200 г
150 ' ' ■ '
1 2 3
Число связей С=С
Рис. 9.24. В ряду этилен - бутадиен-1,3 - гексатриен-1,3,5 Хмакс п -» л*-перехода
сдвигается в сторону более длинных волн. Это означает, что при наложении трех
спектров полоса поглощения заметно смещается.
9.10. Электронная спектроскопия. 2. л-Сопряжение
451
Этилен
A s
Алл ильный
анион
Две
узла узловые
плоскости
Один Одна
узел узловая
плоскость
Нет Узловые
узлов плоскости
отсутствуют
Несвязы-
вающая
МО
Стоячая волна между
двумя точками
Образование я-МО
из трех 2р-АО
Рис. 9.25. а — я-связывающая и тс*-разрыхляющая МО этилена; б — возникновение л-МО в ал-
лильном анионе можно объяснить в терминах стоячей волны, закрепленной между двумя
точками и имеющей 0, I и 2 узла.
Резонансная пара 9.9 эквивалентна 9.8. я*-Разрыхляющая орбиталь легко
получается при отрицательном перекрывании трех 2р-АО и оказывается
разрыхляющей для каждой пары соседних атомов углерода.
\А/
I I
н н
9.8
н\с^с^г/-н _
i i
н н
9.9
н\ё/сх^н
9. Спектроскопия
Возникновение третьей МО несколько сложнее для понимания. На
рис. 9.25 показано, что в этой МО отсутствует вклад от среднего атома
углерода, и эта МО помечена как несвязывающая. Чтобы понять этот результат,
рассмотрим стоячую волну между двумя фиксированными точками -
концевыми атомам углерода в аллильном анионе. Если амплитуда стоячей волны
не изменяет знак между двумя точками (отсутствие узлов), то волна
соответствует я-связывающей МО на рис. 9.25,5. Амплитуда стоячей волны может
менять знак между фиксированными точками один или два раза с
образованием одного или двух узлов, и эта ситуация соответствует двум высшим по
энергии орбиталям на рис. 9.25,6. Единственная узловая плоскость второй
МО проходит через центральный атом углерода, и это означает, что 2р-АО
этого атома не вносит вклад в я*-связывание. Следовательно, эта орбиталь
оказывается несвязывающей для цепи С—С—С. Четыре электрона в
аллильном анионе полностью занимают две низшие по энергии МО.*
я-Сопряжение в бутадиене-1,3
Увеличим теперь углеродную цепь до четырех атомов и рассмотрим я-связы-
вание в бутадиене-1,3 (структура 9.10). После образования всех о-связей в
бутадиене-1,3 у каждого атома углерода остается по одной 2/?-АО, из которых
образуется четыре я-связи (рис. 9.26). Как и в случае аллильного аниона, ор-
битали характеризуются разным числом узловых плоскостей. Низшая по
энергии МО не имеет узловых плоскостей, и я-связь делокализована по всем
четырем углеродным центрам. Следующая МО имеет одну узловую
плоскость, и две я-связи в этом случае локализованы, как это изображено на
структуре 9.10. Высшие МО имеют две и три узловые плоскости соответственно.
Две низшие по энергии я*-МО (целиком заполненные электронами)
являются я-связывающими, а две высшие я-МО (незаполненные) — я-разрыхляю-
щими. Из анализа связывания следует, что, хотя название «бутадиен-1,3»
предполагает наличие локализованных я-связей, в действительности система
я-связей распределена по всему углеродному скелету. Такие связи
называются сопряженными.
н н
\ /
н с = с
\ / \
с = с н
/ \
н н
9.10
Если углеродная цепь полиена содержит более одной двойной связи
С=С, которые чередуются с одинарными связями С—С, говорят, что
молекула имеет систему сопряженных я-связей.
9.10. Электронная спектроскопия. 2. я-Сопряжение 453
Рис. 9.26. 7Е-МО в
бутадиене-1,3.
Три узла
Два узла
НСМО
л*
Один узел
B3MO
к
Нет узлов
Влияние сопряжения на переход я -> я*
В результате сопряжения энергетическая разность между высшей я-МО и
низшей я*-МО уменьшается, и соответствующий я -» я*-переход сдвигается
в сторону больших длин волн. В бутадиене-1,3 я -> я*-переход происходит с
ВЗМО на НСМО (рис. 9.26) и наблюдается в ближней УФ-области спектра
(^макс = 217нМ)'
Группа —С=С—С=С— называется хромофорной. Эта группа атомов
ответственна за поглощение света бутадиеном-1,3 в вид./УФ-спектрах. Появление
дополнительных связей С=С усиливает я-сопряжение и вызывает дальнейший
сдвиг я —> я*-перехода в область ббльших длин волн (рис. 9.24). Это явление
называется красным сдвигом (поскольку сдвиг наблюдается в сторону красного
конца спектра, см. табл. 9.2) или батохромным сдвигом. Приблизительный
сдвиг максимума поглощения при добавлении каждой дополнительной связи
С=С к цепи чередующихся связей —С =С—С = С— составляет 30 нм. Помимо
этого, поглощение становится более интенсивным (увеличение емакс).
Хромофорная группа - группа атомов в молекуле, ответственная за
поглощение электромагнитного излучения.
454
9. Спектроскопия
Таблица 9.7. Наблюдаемые значения А,макс для некоторых сопряженных полиенов.
Сопоставьте эти значения с предсказываемыми (см. текст), используя величину А.макс
в бутадиене-1,3 в качестве исходного значения
Соединение КаксJ
^^ 217 21000
227 23000
263 30000
352 147000
Добавление алкильных заместителей к атому углерода в хромофорной
группе также приводит к усилению сопряжения, однако поскольку этот
эффект связан с взаимодействием а-электронов алкильной группы с я-электро-
нами полиеновой цепи, его величина относительно мала. Красный сдвиг в
этом случае составляет около 5 нм на каждый алкильный заместитель.
Данные табл. 9.7 иллюстрируют эффекты сопряжения в некоторых полиенах.
я-Сопряжение и, следовательно, хромофорные свойства в полиенах
могут быть усилены введением в цепь дополнительной связи С=С или
алкильного заместителя.
Другие заместители также могут оказывать влияние на положение
максимума поглощения в электронных спектрах. Например, группа OR (где R - ал-
кил) вызывает красный сдвиг около 6 нм, группа SR — красный сдвиг
порядка 30 нм, а группа NR2 - красный сдвиг порядка 60 нм. Каждая из указанных
групп может служить донором электронной плотности в углеродную систему
я-связей за счет неподеленной электронной пары, что приводит к
увеличению сопряжения в молекуле и усилению хромофорных свойств. Это
иллюстрируется резонансными структурами 9.11.
9.11
Сопряжение также имеет место в алкине с чередующимися связями С—С и
С=С. С увеличением длины хромофорной цепи происходит красный сдвиг и
увеличение интенсивности пика поглощения в электронном спектре.
Сопряжение я-связей возможно не только в случае кратных связей
углерод-углерод. Примером других соединений, поглощающих в ближней УФ-
области, могут служить ос,р-ненасыщенные кетоны и альдегиды (см. разд.
17.13). Структура 9.12 показывает группу атомов, которая присутствует в этих
соединениях; атомы углерода двойной связи С=С на схеме помечены аир.
а ^
9.12
9.10. Электронная спектроскопия. 2. я-Сопряжение 455
Дополнение 9.3. Полиены как природные красители
Каротиноиды — группа полиенов, являющихся природными красителями и
придающих желтые, оранжевые и красные оттенки большому числу растений и некоторым
животным тканям. Ниже приведен ряд примеров. Обратите внимание на элементы
структуры, общие для этих молекул. Каротиноиды подразделяются на две группы:
каротины и ксантофилы. Каротины относятся к классу углеводородов, тогда как ксанто-
филы являются производными каротинов, включающими гидроксильные или другие
кислородсодержащие функциональные группы.
Помидоры окрашены в красный цвет в основном из-за присутствия ликопена
р-Каротин приводит к появлению оранжевой окраски моркови и манго (А,макс = 452 нм);
зеаксантин присутствует также в манго и придает желтый цвет яичным желткам.
а-Каротин и виолаксантин присутствуют в апельсинах.
Розовая окраска лосося и омаров связана с присутствием астаксантина.
456 9. Спектроскопия
Дополнение 9.4. Спектроскопия и контроль чистоты воздуха
Наблюдение за чистотой воздуха — важный метод контроля загрязнений
окружающей среды. В Великобритании в настоящее время действует несколько
автоматических станций наблюдения. Спектроскопические методы
используются для контроля содержания СО, 03, N02 и S02 в атмосфере.
Моноксид углерода поглощает в ИК-спектре при 2174 см-1, а озон
можно определять методом УФ-спектроскопии (Х,макс = 254 нм). Контроль уровня
N02 осуществляется более сложным образом: вначале N02 термически
разлагается до N0, а затем проводится хемилюминесцентная реакция между N0 и
03, и испускаемый свет детектируется и используется для измерения
концентрации N0. Для определения количества S02 в атмосфере применяется
флуоресцентная спектроскопия.
Другие загрязняющие вещества определяются с помощью ряда
неспектроскопических методов, например методом газовой хроматографии (см.
дополнение 8.3).
Заметный вклад в загрязнение атомсферы вносят частицы пыли. Ниже
приведены данные по составу образцов, отобранных в Лидсе
(Великобритания) в 1982-1983 гг.
(5,3%)
7С-Электроны в сс,р-ненасыщенных кетонах и альдегидах делокализованы,
как и в случае чередующихся С=С/С—С-связей, и это приводит к к ->
^-переходу, которому соответствует интенсивный пик поглощения около 220 нм.
Присутствие заместителей при а- и р-атомах углерода может приводить к
существенным сдвигам максимумов поглощения; например, для структуры
9.13 А,макс к -> тс*-перехода составляет 219 нм (емакс = 3600 л/моль • см), а для
структуры 9.14 - 235 нм (емакс = 14000 л/моль ■ см).
9.13
9.14
9.11. Электронная спектроскопия. 3. Видимая область спектра 457
Таблица 9.8. Некоторые данные по электронным спектрам соединений,
проявляющих п -> я*-переход
Класс соединений (R — алкил) ^макс нм емакс л/моль *см
R2C=0 (кетоны) 270-290 10-20
RHC=0 (альдегиды) 290 15
RCOC1 (ацилхлориды) 280 10-15
RC(0)OR' (сложные эфиры) <200—210 40-100
или RC02H (карбоновые кислоты)
1^Ч[=М1(азосоединения) 350-370 10-15
Помимо пика, отвечающего п -» л*-переходу, электронные спектры ct,(i-
ненасыщенных кетонов и альдегидов содержат менее интенсивный пик
поглощения, соответствующий электронному переходу, в котором участвует не-
поделенная электронная пара кислорода, т. е. п —> л*-переходу. Переход п —>
тс* наблюдается для альдегидов, кетонов, ацилхлоридов, карбоновых кислот,
сложных эфиров и азосоединений; некоторые спектроскопические данные
приведены в табл. 9.8.
ЭЛЕКТРОННАЯ СПЕКТРОСКОПИЯ
3. ВИДИМАЯ ОБЛАСТЬ СПЕКТРА
Хромофорная группа —N=N—
Электронные спектры многих органических соединений имеют максимумы
только в области ближнего УФ, и эти соединения не имеют окраски. Однако,
как видно из табл. 9.8, азосоединения поглощают свет, длина волны
которого расположена на границе УФ- и видимой областей. Подобно тому, как
максимум поглощения полиенов может быть сдвинут введением заместителей в
углеродную цепь, так и ^макс хромофоров —N=N— можно сместить в
видимую область. Из этого вытекает важное следствие: большинство
азосоединений окрашено, и многие из них используются в качестве коммерческих
красителей. Три примера показаны на рис. 9.27, а вид./УФ-спектр метилоранжа
приведен на рис. 9.28.
Ионы металлов d-ряда
Соединения металлов d-ряда (см. гл. 16) часто бывают окрашены, и во
многих случаях их окраска малоинтенсивна, но типична для данного
соединения. Водные растворы солей титана(Ш) содержат аквакатионы [Ti(H20)6]3+
(9.15). Электронные спектры водных растворов титана(Ш) содержат полосу
с А.макс = 510 нм, соответствующую поглощению октаэдрического катиона
[Ti(H20)6]3+, и водные растворы титана(Ш) окрашены в сине-фиолетовый
цвет. Другие металлы также образуют характерно окрашенные ионы
[М(Н20)6]"+ (табл. 9.9).
458
9. Спектроскопия
НО
Пара-краситель
.«.-А-/"^
S03-Na+
Метилоранж
S03"Na+
Конго красный
K.Wr = 497 нм
Рис. 9.27. Примеры азокрасителей, каждый из которых содержит хромофорную
группу —N=N—. Пара-краситель и конго красный используются как биологические
красители, а метилоранж - в качестве кислотно-основного индикатора. Конго
красный также используют при определении свободных минеральных кислот.
**акс = 464 нм
200 250 300 350 400 450
500 550 600 650
Длина волны, нм
Рис. 9.28. Спектр водного раствора метилоранжа в вид./УФ-области.
9.11. Электронная спектроскопия. 3. Видимая область спектра
459
Таблица 9.9. Окраска некоторых гидратированных ионов элементов
d-ряда (см. также гл. 16)
Гидратированный ион металла
Окраска
[Ti(H20)6]3+
[V(H20)6]3+
[Сг(Н20)6]3+
[Сг(Н20)6]2+
[Fe(H20)6]2+
[Со(Н20)6]2+
[Ni(H20)6l2+
[Cu(H20)6]2+
Сине-фиолетовая
Зеленая
Фиолетовая
Голубая
Бледно-зеленая
Розовая
Зеленая
Голубая
Н2
О
Н2(Х |
Ti
Н2° \
ОН,
о
9.15
3+
Если молекулы воды замещаются на другие электронодонорные группы,
новые содинения могут поглощать свет при другой длине волны. Например,
добавление аммиака к раствору голубого [Cu(H20)6]2+ приводит к
образованию темно-синего раствора.
Происходящие электронные переходы называют d— ^-переходами; более
детально они обсуждаются в разд. 16.10. Эти переходы связаны с тем, что
ионы металлов d-ряда имеют частично заполненные d-орбитали, поэтому
между flf-орбиталями могут происходить электронные переходы. Цинк(П) имеет
полностью заполненную 3^-оболочку, в пределах которой электронные
переходы невозможны, поэтому растворы соединений цинка(Н) бесцветны.
Колориметрическое определение состава
комплексных ионов: метод Жоба
Колориметрия может быть использована для определения стехиометрии
реакций, приводящих к образованию окрашенных соединений, по методу
Жоба. Рассмотрим реакцию между ионами железа(Н) и 2,2'-бипиридином
9.16, в результате которой образуется красный продукт. Цель эксперимента -
определить соотношение Fe(II): 2,2'-бипиридин в окрашенном продукте.
9.16
460
9. Спектроскопия
10
3456789 10
Объем 0,001 М раствора (NH4)2Fe(S04)2, мл
7 6 5 4 3 2 10
Объем 0,001 М раствора 2,2'-би пиридина, мл
Рис. 9.29. Использование метода Жоба для определения стехиометрии реакции
между ионами железа(Н) и 2,2'-бипиридином; источник ионов железа(Н) - двойной
сульфат железа-аммония.
Вначале определяют ^макс комплекса из его спектра поглощения, и
колориметр настраивают на эту длину волны. Затем в серию кювет (с одинаковой
длиной пути луча) помещают растворы с различным молярным
соотношением железа(Н) и 2,2'-бипиридина, имеющие одинаковый суммарный объем,
как показано на горизонтальной оси рис. 9.29. Полученные значения
оптической плотности каждого из растворов в зависимости от состава раствора
наносят на график. Максимальная оптическая плотность соответствует макси-
^ мальной концентрации окрашенного продукта в растворе. В данном случае
Структура (1 : 3)-комплекса: максимальную величину оптической плотности опреде-
см. рис. 16.7 ляли интерполяцией, она соответствует соотношению
железо(Н): 2,2'-бипиридин =1:3.
СПЕКТРОСКОПИЯ ЯДЕРНОГО МАГНИТНОГО РЕЗОНАНСА
1. ТЕОРЕТИЧЕСКИЕ ОСНОВЫ
Ядерные спиновые состояния
Ядра атомов обладают спином. Наличие спина следует из квантовой
механики. С точки зрения классической механики, физический смысл спина можно
представить как вращение ядра вокруг оси. Ядерные спины (угловые
моменты ядер) квантованы, и их значения характеризуют спиновым квантовым
числом /. Спиновое квантовое число может принимать значения /= 0, 1/2, I,
3/2, 2, 5/2 и т. д. В данной главе рассматриваются только элементы, у которых
/= 1/2, например !Н, 13С, 19F и 31Р.
Ядра имеют положительный заряд, и (если продолжать аналогию с
классической механикой) вращающийся заряд порождает магнитное поле,
которое может взаимодействовать с внешним магнитным полем. В классическом
случае магнитное спиновое квантовое число т учитывает направление
вращения; т принимает значения +/, +(/-1), ..., -/; для ядер с /= 1/2 т = +1/2
9.12. Спектроскопия ЯМР. 1. Теоретические основы
461
т = -I -т Е2 (магнитное поле, порождаемое вращением ядер,
^ противоположно внешнему магнитному полю)
| ЛЕ = Е2 - £,
_ ' £", (магнитное поле, порождаемое вращением ядер,
т ~ * совпадает по направлению с внешним магнитным полем)
Рис. 9.30. Расщепления ядерных спиновых состояний при наложении внешнего
магнитного поля.
или -1/2. Само по себе направление вращения ядра не влияет на его
энергию. Но направление вращения влияет на направление порождаемого
магнитного поля. При наложении внешнего магнитного поля его направление
может совпадать или быть противоположным направлению поля,
возникающего в результате вращения ядер. Наложение внешнего поля приводит к
появлению энергетической разности между спиновыми состояниями с т =
+ 1/2 и -1/2 (рис. 9.30). Поскольку при наложении внешнего магнитного
поля происходит расщепление спиновых состояний по энергии, оказывается
возможным использовать спектроскопический метод для определения этих
энергетических состояний.
Такой спектроскопический эксперимент приводит к ядерному
магнитному резонансу (ЯМР). Энергетическая разность между спиновыми
состояниями с т = +1/2 и —1/2 зависит от напряженности приложенного магнитного
поля, но эта разность относительно мала при всех достижимых полях. Как
правило, энергия расщепления не превышает 0,01 кДж/моль, и поглощаемое
при этом электромагнитное излучение относится к радиочастотной (РЧ) об-
^. ласти спектра. Поглощение соответствующей РЧ-энергии приводит к возбу-
РЧ - радиочастоты ждению ядер из низшего энергетического состояния в высшее,
т. е. из состояния с т = +1/2 в состояние с т = —1/2. После
этого происходит релаксация ядер, т. е. переход их из высшего состояния в низшее.
Только некоторые ядра активны в ЯМР-спектре, например 1Н, 13С, 19F
и31Р.
Запись ЯМР-спектров
Спектры ЯМР представляют собой зависимость интенсивности поглощения
от частоты. Простейший способ записи таких спектров - поместить образец
в магнитное поле и варьировать радиочастоты до тех пор, пока не будет
наблюдаться поглощение. Хотя этот способ широко используется*, у него
имеется один существенный недостаток. Энергетическая разность между
спиновыми состоянимя с т = +1/2 и -1/2 настолько мала, что если рассмотреть
* В большинстве ЯМР-спектрометров варьируется напряженность внешнего магнитного поля, а
радиочастота поддерживается постоянной. — Прим. перев. •
сг>
462
9. Спектроскопия
► распределение Больцмана, окажется, что различие в заселенности низшего и
Больцмановское высшего уровней составляет около одного ядра на миллион!
распределение: Это означает, что методу ЯМР-спектроскопии присуща низкая
см. разд. 1.18 чувствительность. Экспериментально чувствительность метода
можно оценить с помощью отношения сигнал/шум в
записываемом спектре.
Одним из способов увеличения отношения сигнал/шум является
многократная запись одного и того же спектра с последующем сложением
спектров. При сложении п спектров интенсивность сигнала растет
пропорционально я, а интенсивность шума — пропорционально я1/2; соответственно,
отношение сигнал /шум возрастает пропрционально л1/2. Однако
многократное сканирование одних и тех же частот требует значительных затрат
времени.
Выше уже отмечалось, что энергетическая разность между низшим и
высшим энергетическими уровнями очень мала. В то же время, и равновесная
заселенность состояний, и время восстановления равновесной заселенности
состояний после возбуждения, т. е. время релаксации, зависят от
энергетического различия между уровнями. Чем меньше энергетическая разность, тем
больше время восстановления равновесной заселенности. В случае ЯМР-
спектроскопии время релаксации может варьировать от нескольких секунд
до нескольких часов. Это накладывает еще одно ограничение на сложение
спектров, поскольку каждый раз перед накоплением спектров требуется
ждать восстановления равновесной заселенности.
В современном способе записи ЯМР-спектров эффекты, обсуждаемые
выше, используются для частичного решения указанных проблем.
Необходимая нам зависимость интенсивности поглощения от частоты эквивалентна
зависимости интенсивности испускаемого образцом излучения от частоты,
и, поскольку релаксационные процессы относительно медленны, можно
легко записывать излучение образца после возбуждения. Само по себе это не
дает никаких преимуществ — в действительности, это даже менее эффективно,
так как интенсивность излучения в каждый момент времени будет ниже, чем
интенсивность поглощения, в связи с тем, что поглощение происходит
практически мгновенно, а излучение занимает определенное время (см. рис. 9.31).
Однако зависимость интенсивности излучения образца от времени содержит
абсолютно ту же информацию, что и зависимость интенсивности поглощения от
частоты, и эти данные можно перевести друг в друга с помощью математи-
ческогой операции, называемой преобразованием Фурье (ПФ).
Рассмотрим теперь важный случай. Точная частота поглощения ядра
зависит от его химического окружения, и если ядра в соединении имеют
несколько типов окружения, обычный эксперимент включает запись спектра
Рис. 9.31. Зависимость потери энергии
возбуждения ядерным спиновым состоянием от
времени. Данная кривая называется кривой
свободного спада индукции (ССИ).
Время
9.12. Спектроскопия ЯМР. 1. Теоретические основы
463
Дополнение 9.5. Отношение сигнал/шум
В хорошо разрешенном спектре пики тонкой структуры сигналов четко
разделены. Для характеристики качества спектров часто используют отношение
сигнал/шум. Оно показывает, насколько хорошо разрешен спектр. Низкое
отношение сигнал/шум означает, что могут возникнуть трудности при
отделении сигнала от фонового шума. Такой спектр показан на диаграмме а.
Плохо разрешенные спектры могут возникать по разным причинам. Две наиболее
часто встречающиеся проблемы — недостаточное количество образца и
недостаточное количество накопленных данных. Спектр Охарактеризуется
высоким соотношением сигнал/шум, и сигнал однозначно отделяется от
фонового шума.
Дг44р»-и*и~
J
V.
при варьировании радиочастот и определение всех пиков поглощения. При
использовании ЯМР-метода с фурье-преобразованием можно использовать
смесь частот для возбуждения всех ядер, находящихся в различном
окружении, и затем записывать интенсивность испускания излучения от времени.
^- Такая зависимость называется свободный спад индукции (ССИ). Для увеличе-
ССИ - свободный спад ния отношения сигнал/шум можно записать серию кривых
индукции ССИ, а применение к ним фурье-преобразования дает
обычный спектр интенсивности поглощения в зависимости от
частоты.
Резонансные частоты
Некоторые ядра ('Н, 13С и т. д.) резонируют при характеристических
частотах. Иначе говоря, ядро поглощает в определенном интервале радиочастот.
Можно провести аналогию между ЯМР и радио. Настроив радио на
определенную частоту, можно принимать только выбранную станцию, поскольку
другие радиостанции вещают на иных частотах. Аналогично, ЯМР-спектро-
метр настраивают на определенную резонансную частоту, чтобы
фиксировать даный тип ЯМР-активных ядер. Резонансные частоты некоторых ядер
со спином / = 1/2 приведены в табл. 9.10. При записи спектров ЯМР 'Н
(«протонных спектров») наблюдаемые сигналы относятся только к ядрам ] Н,
присутствующим в образце. Аналогично, спектр ЯМР 31Р содержит только
сигналы от ядер 31Р.
464
9. Спектроскопия
Распространенность в природе
Ранее уже упоминалось, что в ЯМР-спектре активны только ядра,
обладающие ненулевым спином. Другим важным свойством является естественное
содержание ЯМР-активных ядер, их распространенность в природе (табл.
9.10). Фтор имеет один изотоп, и при записи спектров ЯМР 19F все ядра
фтора вносят вклад в спектр. Сходная ситуатция наблюдается для водорода, так
как 99,9% природного водорода приходится на долю ядер 'Н. В спектрах
ЯМР 13С присутствует отклик только от 1,1 % присутствующих в образце ядер
углерода, поскольку остальные 98,9% состоят из ядер 12С, которые
неактивны в ЯМР-спектре (/ = 0).
Хотя низкое естественное содержание может привести к возникновению
проблем, оно не означает, что ядро нельзя исследовать методом ЯМР-спект-
роскопии. Для повышения отношения сигнал/шум можно проводить
большее число ССИ или использовать изотопно-обогащенные образцы.
Химические сдвиги и стандарты в ЯМР-спектроскопии
Резонансные частоты для ядер *Н сильно зависят от химического окружения,
и аналогичные эффекты наблюдаются для других ЯМР-активных ядер. С чем
это связано?
Как было сказано выше, энергетическая разность между состояниями со
спинами т = + 1/2 и т = -1/2 возникает в результате взаимодействия между
магнитным полем вращающегося ядра и внешним магнитным полем.
Однако локальное поле В, действующее на ядро, не совпадает с приложенным
магнитным полем. Это связано с тем, что связывающие электронные пары в
молекуле также представляют собой движущиеся заряды и также порождают
небольшие локальные магнитные поля. Поэтому результирующее поле Яесть
сумма приложенного поля В0 и малых магнитных полей. Локальное поле,
создаваемое электронами, меняется в зависимости от химического окружения
ядра. Как правило, различия между полями очень малы, и наблюдаемое раз-
Таблица 9.10. Резонансные частоты (100 МГц,1!-!)3 для некоторых ядер с /= 1/2
Ядро
Резонансная
частота, МГц
Естественное
содержание, %
•н
19р
31р
ll9Sn
,3С
193ft
29Si
lO'/Ag
103Rh
100
94,0
40,48
37,29
25,00
21,46
19,87
4,05
3,19
-99,9
100
100
8,6
1,1
33,8
4,7
51,8
100
Стандартное вещество для
определения химического сдвига
Me4Si
CFC13
Н3Р04 (85% водный раствор)
Me4Sn
Me4Si
Na2[PtCl6]
Me4Si
Водный раствор Ag+
Rh (металл)
a Я MP-спектрометры работают при различных магнитных полях," поэтому их удобно
классифицировать по частотам, при которых поглощают протоны. Спектрометр ЯМР на 100-МГц записывает
спектр ЯМР 'Н при 100 МГц, спектрометр на 250 МГц записывает спектр ЯМР 'Н при 250 МГц.
Магнитное поле в спектрометре на 100 МГц составляет 2,35 Тл, а в спектрометре на 250 МГц-
5,875 Тл.
9.12. Спектроскопия ЯМР. 1. Теоретические основы
465
личие частот составляет величину порядка миллионных долей (м.д.). Эти
различия и приводят к возникновению различных сигналов в спектре.
Положение сигнала характеризуют величиной так называемого химического сдвига
8 наблюдаемого сигнала от сигнала некоторого стандартного соединения. В
качестве стандартного соединения для спектроскопии ЯМР !Н и 13С
используется тетраметилсилан (ТМС; структура 9.17). 12 атомов водорода в ТМС
эквивалентны и единственный тип химического окружения протонов
приводит к появлению единственного сигнала в спектре ЯМР ХН этого
соединения. Величина химического сдвига этого сигнала принимается за нуль (6 = 0).
Аналогично, в ТМС присутствуют атомы углерода с окружением только
одного типа, и химический сдвиг в 13С-ЯМР-спектре ТМС принимается за нуль.
сн3
ТМС
9.17
Сигналы в ЯМР-спектрах всегда приводятся относительно стандартного
образца (табл. 9.10). Следует обращать внимание на то, что иногда в качестве
стандартов могут быть использованы разные соединения. Хотя обычно в
качестве стандартного соединения для спектроскопии ЯМР 31Р используется
85% водный раствор фосфорной кислоты, иногда применяется Р(ОМе)3
(триметилфосфит), поэтому при ссылке на ЯМР-спектр важно указывать
стандартное соединение.
Сигналы от разных ядер соединения, наблюдаемые в ЯМР-спектрах,
сдвинуты относительно сигнала стандартного соединения. Положительные
значения 8 называются химическим сдвигом в сторону слабого поля, а
отрицательные 8 — химическим сдвигом в сторону сильного поля.
Величина химического сдвига 8 в ЯМР-спектрах измеряется
относительно сигнала стандартного соединения, для которого принято
значение 8 = 0. Положительному сдвигу соответствует меньшая, а
отрицательному - ббльшая величина поля.
Что означает шкала 8? Проблемой ЯМР-спектроскопии является то, что
энергетическая разность между спиновыми состояниями с т = +1/2 и
т = -1/2 зависит от внешнего магнитного поля. Следовательно, разница
между частотами поглощения ядер в различном химическом окружении также
зависит от приложенного поля. Каким образом сравнивать данные,
полученные при различных внешних полях? Решением проблемы оказалось введение
параметра, не зависящего от внешнего поля, — химического сдвига 8.
Величина 8 получается делением разности частот (Av) в Гц между
интересующим экспериментатора сигналом и сигналом от стандартного образца
(v0) на абсолютную частоту сигнала от стандартного образца. Для удобства
9. Спектроскопия
пользования величиной 5, это отношение умножают на 106 (уравнение 9.15)
и получают величину химического сдвига 6 в миллионных долях (м.д.).
(v-vn)-106 AvlO6
8 = - °- = (9.15)
Интервалы химических сдвигов
Интервал химических сдвигов (т. е. интервал сдвигов, в котором могут
присутствовать сигналы в спектре) зависит от рассматриваемого ядра. Например,
сигналы ЯМР !Н обычно лежат в области 8 от +15 до -35; сигналы спектров
ЯМР 13С обычно фиксируются в области 8 от +250 до —50, а сигналы
спектров ЯМР 31Р попадают в интервал 8 между +300 и —300. Однако химические
исследования могут привести к открытию ядер в необычном химическом
окружении, так что экспериментатор не должен считать «спектральные окна»
чем-то неизменным.
Выбор растворителя
Хотя ЯМР-спектры можно снимать и в твердом состоянии, в большинстве
случаев исследованию подвергаются жидкие или растворенные образцы.
Молекулы большинства растворителей содержат атомы углерода и водорода,
поэтому в спектрах ЯМР 'Н и 13С растворенных образцов спектр
растворителя накладывается на спектр образца. Так как растворитель, как правило,
присутствует в большом избытке по сравнению с образцом, сигналы от
растворителя часто оказываются самыми заметными на спектрах.
Спектры ЯМР !Н легко «засоряются» из-за влияния растворителя, что
мешает определению сигналов образца. Эта проблема решается при
использовании дейтерированных растворителей, в которых ядра 'Н замещены на ядра
2Н (D). Например, водород в одном из стандартных растворителей -
хлороформе — можно заместить на дейтерий с образованием CDC13. Хотя ядра 2Н
активны в ЯМР-спектре (/=1), их резонансная частота существенно
отличается от частоты 'Н, и, следовательно, дейтерированные растворители
«невидимы» в спектрах ЯМР !Н. Такие растворители имеются в продаже, и степень
изотопного обмена в них обычно превышает 99,5%. Остаточные молекулы,
содержащие ядра !Н, хотя и присутствуют, но дают относительно
малоинтенсивные сигналы в спектрах ЯМР !H.
СПЕКТРОСКОПИЯ ЯДЕРНОГО МАГНИТНОГО РЕЗОНАНСА
2. МОЛЕКУЛЫ С ЕДИНСТВЕННЫМ ТИПОМ
ХИМИЧЕСКОГО ОКРУЖЕНИЯ
Спектроскопия ядерного магнитного резонанса может быть использована для
определения числа ядер определенного типа, имеющих в данной молекуле
разное химическое окружение. Спектроскопия ЯМР 'Н позволяет наблюдать
окружение протонов, а спектроскопия ЯМР 13С может быть использована
для определения различных типов атомов углерода в молекуле. Число
сигналов в спектре соответствует числу различных типов химического окружения.
9.14. Спектроскопия ЯМР. 3. Разные типы химического окружения 467
Л_ Я
i i i i i i i i i i i i
+200 +100 0 8 +200 +100 0 S
Рис. 9.32. Спектры ЯМР 31Р соединений фосфора РМе3 (а) и РС13 (б).
Молекулы с единственным типом химического окружения
На первый взгляд может показаться, что наличие единственного сигнала
позволяет констатировать лишь присутствие единственного типа химического
окружения. В данном разделе показано, что из ЯМР-спектра можно извлечь
больше информации. Величина химического сдвига позволяет сделать
важные выводы о типе окружения, например о возможной геометрии центра или
о связанных с ним функциональных группах.
На рис. 9.32 показаны 31Р-ЯМР-спектры РМе3 и РС13. В каждом спектре
имеется один сигнал, показывающий, что в молекуле присутствуют атомы
фосфора только одного типа. Однако следует отметить, что природа
заместителя оказывает существенное влияние на химический сдвиг сигнала:
различие между химическими сдвигами для этих двух тригонально-пирамидаль-
ных молекул PR3 особенно велико. Для PF3, РВг3 и Р13 (все молекулы имеют
геометрию тригональной пирамиды) химические сдвиги 8 в спектрах ЯМР
31Р равны +96, +228 и +177 м.д. соответственно. Каждая из молекул СН4,
СН3С1, СН2С12 и СНС13 имеет тетраэдрическое строение, однако
присутствие хлоридных заместителей влияет на химический сдвиг 8 (м. д.) в спектрах
ЯМР 13С: СН4 (+2), СН3С1 (+22), СН2С12 (+54) и СНС13 (+77).
ESQ СПЕКТРОСКОПИЯ ЯДЕРНОГО МАГНИТНОГО РЕЗОНАНСА
3. МОЛЕКУЛЫ С РАЗЛИЧНЫМИ ТИПАМИ
ХИМИЧЕСКОГО ОКРУЖЕНИЯ
Химические сдвиги в спектрах ЯМР 13С
Химический сдвиг сигнала в спектрах ЯМР 13С связан с типом гибридизации
углеродного центра. 5/?2-Гибридизованные атомы углерода имеют больший
положительный химический сдвиг, чем 5/ьгибридизованные углеродные
центры, которые в свою очередь имеют больший положительный химический
сдвиг, чем 5/73-гибридизованные атомы углерода. Табл. 9.11 дает примерные
области химических сдвигов атомов углерода в различных окружениях в
органических молекулах. Соединения с карбонильной группой (С=0) характери-
► зуются положительными химическими сдвигами в спектрах ЯМР 13С. Число
Спектры ЯМР 13С кар- соединений, содержащих з/?3-гибридизованные атомы угле-
бонильных соедине- рода, очень велико, и включает циклические и ациклические
ний: см. разд. 17.4 алканы, спирты (X = ОН), алкилгалогениды (X = F, C1,
468
9. Спектроскопия
Таблица 9.11. Области химических сдвигов для различных типов химического окружения
углерода в спектрах ЯМР 13С (химические сдвиги даны в м.д. относительно 8 (ТМС) = 0;
положительные значения 6 соответствуют сдвигам в сторону слабого поля)
Окружение углерода Тип соединения Тип гибридизации Область
химических
сдвигов 13С
Примечания
\
с = о
/
R
R
\
с = о
/
н
R
\
/
R
\
С = 0
/
X
R R
\ /
с = с
/ \
R R
Кетоны
Альдегиды
Карбоновые
кислоты
Амиды X = NH2
Сложные эфиры
X = OR
Алкены
V2
V2
SP2
sp2
sp2
О
Ароматические
соединения sp1
R C^=N
R
\
R С X
/
R
R
\
R С X
/
Н
R
\
Н С X
/
Н
Н
\
Н С X
/
Нитрилы
-r Алкины
Алифатические
соединения
Алифатические
соединения
Алифатические
соединения
Алифатические
соединения
sp
sp
s? 1
sp3
sp3
SP>
От +230 до +190
От+210 до+185
От+185 до+165
От+180 до+155
От+160 до+100
Обратите
От+145 до+110 I внимание на
> перекрывание
От+125 до+110 J 5р2.И5р.
От +110 до +75 областей
Области химических сдвигов
перекрываются в зависимости от X.
Общий интервал от +90 до 0.
Как правило:
СН3Х < RCH2X < R2CHX < R3CX^
8 увеличивается
9.14. Спектроскопия ЯМР. 3. Разные типы химического окружения
469
Алкены
Алкины
Алканы
140
120
100
80
60
40
20
0 8
Рис. 9.33. Типичные значения химических сдвигов в спектрах ЯМР 13С алканов, ал-
кенов и алкинов.
Вг и I), амины (X = NH2), тиолы (X = SH) и простые эфиры (X = OR).
Различие между химическими сдвигами в спектрах ЯМР 13С алканов, алкенов и
алкинов проиллюстрировано на рис. 9.33.
Интенсивность сигналов
Относительная интенсивность ЯМР-сигналов позволяет облегчить
отнесение пиков, поскольку отношение интенсивностей дает информацию о
соотношении числа атомов, находящихся в различном окружении.
Спектр ЯМР 13С ацетона содержит два сигнала (рис. 9.34), которые легко
^~ отнести, поскольку отношение их интенсивностей равно 1 : 2. Однако следу-
Релаксация: ет проявлять осторожность. Интенсивности сигналов в спект-
см. разд. 9.12 pax ЯМР 13С могут привести к ошибочным выводам ввиду
различных времен релаксации разных типов ядер 13С. Проблема
возникает из особенностей ЯМР-метода с фурье-преобразованием. Поскольку
данные по испусканию излучения записываются как функция времени, то при
записи последовательных ССИ необходимо каждый раз дожидаться
восстановления больцмановского распределения для ядер во всех окружениях. Если
этого не делать, то быстро релаксирующие ядра будут давать больший вклад в
спектр, чем медленно релаксирующие. На практике, в силу ограниченной
продолжительности эксперимента, иногда сокращают время между
последовательными возбужениями. В результате этого, отношение между интенсивно-
стями сигналов не всегда соответствует отношениям между количеством
атомов в различном окружении. В частности, такая ситуация встречается для
спектров ЯМР 13С *. К счастью, для отнесения сигналов можно использовать
численные значения химических сдвигов (см. примеры, рассмотренные в
следующем разделе).
О:
/
снч
сн.
+200
+ 100
Рис. 9.34. Спектр ЯМР 13С ацетона (спин-спиновое взаимодействие с протонами
подавлено). Спектр записан при 25 МГц.
* Более детальное обсуждение см. в кн. Hunter B.K., Sanders J.К.М., Modem NMR Spectroscopy: A Guide
for Chemists, 2nd edn., Oxford University Press, Oxford (1993).
470 9. Спектроскопия
Примеры расшифровки спектров ЯМР 13С
по значениям химических сдвигов
См. табл. 9.11
1. Ацетонитрил
а Ъ
Н3С С = N
В спектре ЯМР 13С наблюдаются химические сдвиги с 5 +1,3 и +117,7
(рис. 9.35,я).
В молекуле имеется два типа окружения атомов углерода: а —
алифатический (sp3), b — нитрильный (sp).
Сигнал нитрильного углерода должен быть смещен от сигнала
алифатического углерода в сторону слабого поля, т. е. в сторону положительных 5.
Отнесение:
8 = +1,3 соответствует атому а.
5 = +117,7 соответствует атому Ь.
2. Ацетальдегид (этаналь)
а Ъ/
н3с с
ч
О
В спектре ЯМР 13С наблюдаются химические сдвиги с 8 +30,7 и +199,7
(рис. 9.35,6).
В молекуле имеется два типа окружения атомов углерода: а —
алифатический (sp3), Ъ — альдегидный (sp2).
+200 +100 0 8
Рис. 9.35. Спектры ЯМР 13С при 25 МГц (спин-спиновое взаимодействие с
протонами подавлено) ацетилнитрила CH3CN (а), ацетальдегида СН3СНО (б) и диэтиламина
Et2NH (в). Спектры приведены в единой шкале.
9.15. Спектроскопия ЯМР. 4. Спектры ЯМР1Н
471
Сигнал альдегидного углерода должен быть смещен от сигнала
алифатического углерода в сторону положительных 8.
Отнесение:
5 = +30,7 соответствует атому а.
5 = +199,7 соответствует атому Ь.
3. Диэтиламин
С С
В спектре ЯМР 13С наблюдаются химические сдвиги с 8 +15,4 и +44,1
(рис. 9.35,*).
В молекуле имеется два типа окружения $/?3-гибридизованных атомов
углерода: а — концевая группа СН3, Ь — группа RCH2X.
Как правило, сигнал от группы RCH2X смещен от сигнала концевой
группы СН3 в сторону положительных 8.
Возможное отнесение:
8 = +15,4 соответствует атому а.
8 = +44,1 соответствует атому Ь.
СПЕКТРОСКОПИЯ ЯДЕРНОГО МАГНИТНОГО РЕЗОНАНСА
4. СПЕКТРЫ ЯМР1 Н
Химическое окружение в спектрах ЯМР 1Н
Поскольку область химических сдвигов для ядер {Н относительно мала,
области, характеные для различных типов окружения протонов, существенно
перекрываются. В табл. 9.12 приведены примерные значения сигналов в
спектрах ЯМР 'Н для некоторых групп органических соединений*.
Из табл. 9.12 видно, что протоны в группах ОН и NH2 могут давать
широкие сигналы. Это явление возникает из-за того, что протоны связей О—Н и
^ N—Н могут быстро обмениваться, причем характеристическое время обмена
Временная шкала: сравнимо с характеристическим временем ЯМР-спектроско-
см. разд. 9.3 пии. Уравнение 9.16 иллюстрирует обмен водородного атома
этанола с протоном воды. Скорость обмена можно уменьшить
при понижении температуры, и это приводит к уменьшению ширины и
сдвигу сигналов.
СН3СН2ОН + /ГОН ^ СН3СН20Я + НОН (9.16)
Более детальное обсуждение см. в книге Williams D.H., Fleming /., Spectroscopic Methods in Organic
Chemistry, 4th edn., McGraw-Hill, London (1989).
472
9. Спектроскопия
Таблица 9.12. Типичные области химических сдвигов для различных окружений протонов в
спектрах Я MP !H органических соединений (значения химических сдвигов приведены
относительно 8(ТМС) = 0; сдвиги в сторону слабого поля соответствуют положительным значениям 8)
Окружение
протона
Тип соединения
Область
химических
сдвигов ]Н
Примечания
НО
с — о
/
Карбоновые
кислоты
От+9 до+13
Сигналы часто уширены,
положение зависит от
растворителя
H.N
\
/
Амиды
От +5 до +12 Сигналы уширены, положение
зависит от растворителя
\
С
/
с==о
Альдегиды
СНХ3 (X = С1 или Вг)
Ароматические
соединения
R Н
\ /
С = С Алкены
/ \
R R
R С ^= С Н Концевые алкины
RNH,
ROH
\
R С Н
/
R
R
\
СН2
/
R
R СН,
Амины
Спирты
Метиновая
группа
Метиленовая
группа
Метальная
группа
От +8 до +10
«+7
От+6 до+10
От +4 до +8
От+2,5 до+3
От+1 до+6
От +0,5 до +8
От+1,5 до+4,5
От+1,5 до+4,5
От +0 до +4
Сигналы узкие (как правило,
легко отличимы от сигналов
карбоновых кислот, амидов и
аминов)
СНС13 8 = +7,25
С6Н6 8 = +7,2
Сигналы уширены, положение
зависит от растворителя
Сигналы часто уширены,
положение зависит от растворителя
В ТМС по определению 8 = 0
9.15. Спектроскопия ЯМР. 4. Спектры ЯМР 1Н
473
Значения химических сдвигов некоторых протонов зависят от растворителя,
и это в особенности относится к протонам связи О—Н. Спектр ЯМР !Н
жидкого этанола содержит сигнал при 5 = +5,4, соответствующий протону ОН-
группы, но этот сигнал смещается в сторону сильного поля (т. е. в сторону
меньших положительных значений 8) при разбавлении каким-либо раство-
^» рителем. Это наблюдение связано с присутствием водородных связей; меж-
Водородная связь: молекулярные взаимодействия между молекулами этанола 9.18
см. разд. 11.8 приводят к понижению электронной плотности вблизи атома
водорода и вызывают смещение сигнала ЯМР 1Н в сторону слабого поля.
\
О-
■Et
Et-
/
■О
9.18
Аналогично, водородное связывание в карбоновых кислотах приводит к
тому, что протоны карбоксильных групп характеризуются сигналами, заметно
смещенными в сторону более положительных 8 (в сторону слабого поля). В
спектре ЯМР !Н уксусной кислоты (рис. 9.36) присутствует широкий сигнал
с максимумом при 8 = +11,4 (соответствующий ОН-группе) и узкий, более
интенсивный сигнал при 8 = +2,1 (соответствующий СН3-группе). Уксусная
кислота образует димер 9.19 за счет образования водородных связей (см.
разд. 17.4).
н3С-
/
С
\
.о-
н-
н -
9.19
С
/
■сн.
-I I I I I I I I I I I
+10 +5 8
Рис. 9.36. Спектр ЯМР !Н уксусной кислоты СН3С02Н (при 100 МГц).
9. Спектроскопия
Перекрывание характеристических областей спектров ЯМР ]И может
затруднить отнесение пиков. В пропине 9.20 сигналы от протона группы
Сапкин—Н и от метильной группы совпадают (8 = +1,8).
СН2СН3
/
9.20 \
сн3
9.21
В спектре ЯМР !Н бутанона-2 (9.21) присутствуют три сигнала при
8 = +1,1, +2,1 и +2,5. Хотя относительные интенсивности позволяют
отличить сигнал СН2-группы от сигналов СН3-групп, остается неясным, как
различить сигналы, соответствующие двум метильным группам. (После чтения
разд. 9.16 см. упражнение 9.24 в разд. 9.18.)
В этом случае на помощь приходит особенность Я MP-спектроскопии,
которая ранее не рассматривалась: спины магнитно-неэквивалентных ядер могут
взаимодействовать друг с другом, что приводит к характеристическому
расщеплению сигналов ЯМР.
СПЕКТРОСКОПИЯ ЯДЕРНОГО МАГНИТНОГО РЕЗОНАНСА
5. СПИН-СПИНОВОЕ ВЗАИМОДЕЙСТВИЕ
ЯДЕРНЫХ СПИНОВ ЯДЕР С / = 1 /2
Взаимодействие между двумя неэквивалентными ядрами 1Н
Ядра водорода могут находиться в одном из двух возможных спиновых
состояний (т = +1/2 или т = -1/2) и, как упоминалось выше, энергетическая
разность между этими состояниями зависит от внешнего магнитного поля.
Указывалось также, что более тонкие различия в энергии (приводящие к
возникновению различающихся химических сдвигов) появляются при движении
электронов в химических связях. Что произойдет, если два
магнитно-неэквивалентных (с разным магнитным окружением) водородных ядра окажутся в
молекуле рядом?
Рассмотрим систему из двух водородных атомов НА и Нв, находящихся в
различном магнитном окружении. Введем краткие обозначения:
НА с т = +1/2 обозначим а(А)
НА с т = —1/2 обозначим Р(А)
Нв с т = +1/2 обозначим а(В)
Нв с т = -1/2 обозначим (3(B)
В простейшем случае магнитное поле, действующее на НА (или Нв), не
зависит от спинового состояния Нв (или НА). Другими словами, одно ядро не
«чувствует» локальное магнитное поле, порождаемое вторым ядром. Спектр
будет состоять из двух пиков поглощения, соответствующих переходам
ос(А) -> Р(А) и сс(В) -> (3(B) (рис. 9.37), т. е. спектр состоит из двух синглетов и
спин-спиновое взаимодействие между НА и Нв отсутствует.
9.16. Спектроскопия ЯМР. 5. Спин-спиновое взаимодействие 475
Дополнение 9.6. Более строгое объяснение возникновения
двух синглетов для двух не взаимодействующих ядер
со спинами / = 1/2
При обсуждении в тексте опущено следующее правило отбора: разрешены
переходы с Am = ± I.
Альтернативный способ описания системы ядерных спинов НА и Нв —
введение общего спинового состояния для обоих ядер. При этом возникают
четыре энергетических уровня, показанные на диаграмме. Отметим, что
диаграмма относится к энергии системы в целом, без выделения взаимодействия
между НАи Нв.
0(A) /3(B)
/3(A) а(В)
т
2
т
4
{
i
L
3
1
а(А) /3(B)
а(А) а(В)
С учетом правил отбора Ат(НА) = ±1 и Д/и(Нв) = ±1 в данной системе
разрешены четыре перехода:
1. а(А)а(В)-,а(А)Р(В)1 н
2. Р(А)а(В) -> Р(А)Р(В) j F B
3. а(А)а(В)-,р(А)а(В) „
4. а(А)Р(В) -» Р(А)р(В) } р д а
Если взаимодействие между ядрами отсутствует, энергетическая разность
между состояниями а(А)а(В) и а(А)р(В) равна энергетической разности между
состояниями Р(А)а(В) и Р(А)Р(В), поэтому энергия переходов 1 и 2 должна
быть одинакова. Аналогично, переходы 3 и 4 происходят при одинаковой
энергии, хотя и отличной от энергии переходов 1 и 2. Другими словами, в
ЯМР-спектре видны два пика, соответствующих НА и Нв, каждый из которых
представляет собой синглет.
Допустим теперь, что ядро НА подвергается действию магнитного поля
ядра Нв. Следствием этого будет расщепление сигнала НА на два одинаковых по
интенсивности пика, которые соответствуют присутствию соседнего Нв в
состоянии а или р. Сигнал Нв также расщепляется на два пика совершенно
аналогичным образом. Возникает спин-спиновое взаимодействие между ядрами НА
и Нв, и спектр состоит из двух дублетов.
Спин-спиновое взаимодействие возможно только между
неэквивалентными ядрами. Величина взаимодействия определяется константой
спин-спинового взаимодействия J> измеряемой в герцах (Гц). Константа взаимодействия не
476
9. Спектроскопия
Нд
Рис. 9.37. а — два синглетных
резонансных сигнала наблюдаются для протонов НА
и Нв в случае отсутствия спин-спинового
взаимодействия; б - при спин-спиновом
взаимодействии синглетные сигналы
расщепляются в дублеты с одинаковым
расщеплением.
зависит от приложенного поля в ЯМР-спектрометре. Существенно, что
константа взаимодействия измеряется в герцах, так как если бы она измерялась в
единицах 8, ее значение зависело бы от внешнего поля. Например, константа
спин-спинового взаимодействия, равная 25 Гц, соответствует 0,18 для
спектрометра на 250 МГц и 0,258 для спектрометра на 100 МГц (уравнение 9.17).
У( Гц) = Л8 • (Напряженность магнитного поля спектрометра, МГц) (9.17)
где А8 = (8 пика 1) — (8 пика 2).
Взаимодействующие ядра следует указывать подстрочными символами
после обозначения константы J. Например, если константа спин-спинового
взаимодействия между НА и Нв равна 10 Гц, ее обозначают как Ун н = 10 Гц
(или /нн = 10 Гц).
■а^в
Константа спин-спинового взаимодействия измеряется в герцах (Гц):
Л(Гц) = Л8 • (Напряженность магнитного поля спектрометра, МГц)
Схема расщепления пика, соответствующего НА, при его спин-спиновом
взаимодействии с Нв показана на рис. 9.38,а\ там же показано измерение
величины У,
нн-
Спин-спиновое взаимодействие
между несколькими неэквивалентными ядрами 1Н
Рассмотрим случай, когда НА может взаимодействовать с двумя
эквивалентными ядрами Нв, обозначенными НВ(1) и НВ(2). Ядро НА может
взаимодействовать и с НВф, и с НВ(2). Этот процесс, разбитый на две стадии,
показан на рис. 9.38,5. В результате сигнал НА в спектре ЯМР 1Н
расщепляется на три линии (триплет). Каждая линия соответствует одной из возможных
комбинаций спиновых состояний НВ(1) и НВ(2), которые «детектируются»
протоном НА:
9.16. Спектроскопия ЯМР. 5. Спин-спиновое взаимодействие 477
НА «видит» Нв в
спиновом
состоянии с т = +1/2
Исходный сигнал Нд
НА «видит» Нв в
спиновом
состоянии с т = — 1/2
Дублет
1:1
Исходный сигнал НА
Лш
Am
I Спин-спиновое взаимодействие
с первым ядром Нв
i Спин-спиновое взаимодействие
со вторым (эквивалентным) ядром Нв
Триплет
1:2:1
Рис. 9.38. а — образование дублетного сигнала ядра НА при спин-спиновом
взаимодействии с одним ядром Нв. Дублет состоит из линий с одинаковой интенсивностью
и расстояние между ними равно константе спин-спинового взаимодействия Унн;
б - образование триплетного сигнала ядра НА при спин-спиновом взаимодействии с
двумя эквивалентными ядрами НВ(1) и НВ(2). Расстояние между соседними линиями
триплета равно константе спин-спинового взаимодействия Унн.
а(В,)сс(В2)
а(В,)Р(В2) 1
Р(В,)а(В2) }
Р(В,)Р(В2)
вырождены, так как разность
энергий a(Bj) и p(Bj) такая же, как у
сс(В2) и Р(В2)
Следовательно, интенсивности трех линий триплета относятся как 1:2:1 (рис.
9.38,6).
В различных соединениях НА может вступать в спин-спиновое
взаимодействие с тремя или четырьмя эквивалентными Нв, и это приводит к
появлению квартета (1:3:3:1) и квинтета (1:4:6:4:1) линий соответственно. Для
спин-спинового взаимодействия с эквивалентными ядрами, имеющими спин
478 9. Спектроскопия
Си н глет 1
Дублет 1 1
Триплет 1 2 1
Квартет 13 3 1
Квинтет 14 6 4 1
Секстет 1 5 10 10 5 1
Септет 1 6 15 20 15 6 1
Октет 1 7 21 35 35 21 7 1
Рис. 9.39. Треугольник Паскаля позволяет определить относительные
интенсивности компонент мультиплетных сигналов при спин-спиновом взаимодействии между
ядрами со спином /= 1/2. Обратите внимание, что каждая строка начинается и
заканчивается единицей. Остальные элементы каждой строки можно определить
суммированием пар чисел, расположенных надданным элементом в предыдущей строке.
Триплет имеет интенсивности 1 : (1 + 1) : 1, а квартет - 1 : (1+2) : (1+2) : 1. Треугольник
можно продолжить вниз аналогичным образом (см. упражнение 9.23 в разд. 9.18).
/= 1/2, число линий, на которые расщепляется простой сигнал (мультиплет-
ность сигнала), определяется по уравнению 9.18, а отношение интенсивно-
стей компонент можно определить с помощью треугольника Паскаля (рис.
9.39). Общее название дублета, триплета, квартета и т.д. - мулыпиплет, и это
понятие используется в тех случаях, когда нельзя установить истинный
характер сигнала.
Мультиплетность = п +1 (9.18)
где п — число эквивалентных ядер со спином / = 1/2, участвующих в спин-
спиновом взаимодействии с данным ядром.
Весьма важно запомнить, что магнитно-эквивалентные ядра не участвуют
в спин-спиновом взаимодействии друг с другом.
к-^
JL
+ 10 +5 0 8
Рис. 9.40. Спектр ЯМР !Н этанола (при 100 МГц).
9.16. Спектроскопия ЯМР. 5. Спин-спиновое взаимодействие 479
Спин-спиновое взаимодействие происходит между
неэквивалентными ядрами. Если некоторое ядро взаимодействует с п другими
ядрами со спином /= 1/2, которые эквивалентны между собой, то мульти-
плетность сигнала равна п + 1.
Рассмотрим спектр ЯМР 'Н этанола 9.22. В спектре (рис. 9.40)
присутствуют три сигнала: широкий пик при 5 = +2,6, триплет при 5 = +1,2 и квартет
при 8 = +3,7. Широкий сигнал относится к протону группы ОН. Обратите
внимание, что спин-спиновое взаимодействие с участием протона
ОН-группы в спиртах, как правило, не наблюдается, хотя его можно было бы ожидать.
Протоны типа а участвуют в спин-спиновом взаимодействии с двумя
эквивалентными протонами типа Ь, и сигнал от протонов а представляет собой
триплет. Протоны типа b взаимодействуют с тремя эквивалентными протонами
типа д, и сигнал, соответствующий протонам Ъ, представляет собой квартет.
Отнесение спектра на основании отношения интегральных интенсивностей
обоих сигналов (я : b = 2 : 3) подтверждает полученный результат. Обратите
внимание на термин «интегральная интенсивность». Поскольку сигнал
расщепляется из-за спин-спинового взаимодействия, необходимо измерять
суммарную интенсивность компонент сигнала. Наилучший способ сделать
это — определить площади под каждым пиком и сложить их; это и
называется интегральной интенсивностью сигнала.
О
н
1
н
/V,
Н Н
9.22
Возникает вопрос: «Насколько далеко может распространяться спин-
спиновое взаимодействие вдоль углеродной цепи?» На этот вопрос трудно
дать определенный ответ, но, как правило, считают, что спин-спиновое
взаимодействие 'Н—'Н возможно через одну связь С—С (как в этаноле). В
реальности можно наблюдать спин-спиновое взаимодействие и на больших
расстояниях, и эту возможность следует иметь в виду при расшифровке
спектров. Ряд примеров приведен в прилагаемом задачнике.
Если вернуться к рис. 9.40, и триплет, и квартет характеризуются
значением Унн = 7 Гц. Это не случайное совпадение, так как и триплет, и квартет
возникают из-за спин-спинового взаимодействия одного и того же набора ядер.
Значения констант спин-спинового взаимодействия зависят от многих
факторов, и в данной книге не приводится детальное обсуждение этой
проблемы. Рассмотрим, как зависимость значения константы спин-спинового
взаимодействия от геометрии алкена может быть использована в
аналитических целях.
480
9. Спектроскопия
\
с =
/
/
= с
\
\
с
/
/
\
\
с
/
/
\
9.23
9.24
9.25
Во всех алкенах 9.23—9.25 заместители X и Y неодинаковы, поэтому в
каждой из структур протоны НА и Нв магнитно-неэквивалентны и участвуют в
спин-спиновом взаимодействии. Как правило, для структур типа 9.23 Унн =
0-2 Гц, для 9.24 Унн = 12-18 Гц, а для 9.25 Унн = 6-12 Гц. Это позволяет
использовать спектры ЯМР 'Н для того, чтобы различать изомеры алкенов.
Расшифровка некоторых спектров
со спин-спиновым взаимодействием 1Н-1Н
1. 2-Хлорпропановая кислота
ь н ci
\/
Н3С
с ^°
ОН с
В спектре ЯМР !Н присутствуют три сигнала: при 8 = +Н,2 (широкий),
8 = +4,5 (квартет) и 5 = +1,7 (дублет) (рис. 9.41).
Сигнал с наибольшим положительным 8 (в слабом поле) соответствует
протону ОН-группы.
Протон b взаимодействует с тремя метильными протонами я, поэтому
сигнал протона Ь представляет собой квартет.
Три метильных протона а взаимодействуют с протоном Ь, и сигнал от
протонов а представляет собой дублет. Таким образом, отнесение сигналов с
8 = +4,5 и 8 = +1,7 однозначно.
А
1
_| L.
_| i_
_| 1_
+ 10
+5
Рис. 9.41. Спектр ЯМР ]Н 2-хлорпропановой кислоты СН3СНС1С02Н (при
100 МГц).
9.16. Спектроскопия ЯМР. 5. Спин-спиновое взаимодействие 481
Рис. 9.42. Сигнал при 5 = +1,6 в спектре про-
панола-1 представляет собой секстет из-за того,
что значения констант спин-спинового
взаимодействия /н н и JH H примерно равны.
+2,0
+ 1,6
2. Пропанол-1
Н3С
он
В спектре ЯМР !Н присутствуют четыре сигнала: при 8 = +3,6 (триплет),
5 = +2,3 (широкий), 8 = +1,6 (секстет) и 8 = +0,9 (триплет).
Широкий сигнал соответствует протону ОН-группы.
Сигнал концевой метильной группы должен расщепляться на триплет из-
за спин-спинового взаимодействия с с двумя эквивалентными протонами Ь.
Поскольку сигналы протонов концевой метильной группы, как правило,
расположены при меньших 8, чем сигналы протонов метиленовой группы,
сигнал при 8 = +0,9 можно отнести к протонам а .
Осталось произвести отнесение секстета при 8 = +1,6 и триплета при
8 = +3,6. Наилучший способ расшифровки сигналов — попытаться предсказать
ожидаемые сигналы и сравнить предсказания с реальными наблюдениями.
Метиленовые протоны с должны взаимодействовать с двумя
эквивалентными протонами b с образованием триплета. Спин-спиновое
взаимодействие с протоном ОН-группы в принципе возможно, но, как было показано
ранее (рис. 9.40), такое взаимодействие не наблюдается в этаноле, и это
оказывается общей тенденцией. Поэтому триплет при 8 = +3,6 можно отнести к
протонам метиленовой группы с.
Протоны Ъ участвуют в спин-спиновом взаимодействии и с тремя
эквивалентными протонами а и с двумя эквивалентными протонами с, но
значения констант /н н и JH H примерно равны, поэтому в спектре
присутствует секстет, соответствующий спин-спиновому взаимодействию с пятью
протонами (рис. 9.42). (Вопрос: Как выглядел бы сигнал на рис. 9.42, если бы
выполнялось одно из следующих условий: Ун н > Ун н или Ун н < Ун н ?)
3. 2-Иодпропан
Н3С
а
сн3
н ъ
482
9. Спектроскопия
Спектр ЯМР 1Н 2-иодпропана содержит дублет при 8 = +1,9 и септет при
8 = +4,3.
В молекуле существует два типа протонов: а (шесть протонов) и b (один
протон).
Протон b взаимодействует с протонами я, и сигнал b представляет собой
септет.
Протоны а взаимодействуют с протоном Ь, и сигнал а представляет собой
дублет.
Таким образом, сигнал при 8 = +1,9 относится к протонам метильной
группы а, а сигнал при 8 = +4,3 - к протону Ь.
Химическая и магнитная эквивалентность
В примерах, рассмотренных выше, проводилось однозначное соответствие
между магнитно-неэквивалентными и химически-неэквивалентными
протонами. Рассмотрим теперь химически-эквивалентные, но
магнитно-неэквивалентные ядра.
Н
Н
\
II
/
С1
\\
ь ъ%
9.26
Фуран 9.26 содержит два типа протонов (aw b). Как и можно было
ожидать для системы с двумя типами химического окружения протонов, в
спектре ЯМР !Н фурана присутствуют два сигнала, но оба сигнала (8 = +6,6 и 8 =
+7,4) представляют собой мультиплеты вместо ожидаемых дублетов. Это
объясняется следующим образом.
По отношению к протону а протон Ь, соединенный с соседним углеродным
центром, отличается от другого протона Ъ'. Хотя протоны b и Ь' химически
эквивалентны, они оказываются магнитно-неэквивалентными (по отношению к
протону а). Поэтому возникает два типа спин-спинового взаимодействия: 1)
между а и b с образованием дублета и 2) между а и Ь' с образованием дублета. В
результате сигнал представляет собой дублет дублетов.
'н„н,,
Рис. 9.43. В спектре фурана 9.26
протон а взаимодействует с протонами
Ь и Ь' с образованием дублета дублетов.
j\
НаН//
7Н(/Н/,
Протон b
взаимодействует с
протоном а с
образованием дублета
Протон Ь'
взаимодействует с
протоном а с
образованием дублета
9.16. Спектроскопия ЯМР. 5. Спин-спиновое взаимодействие 483
Дополнение 9.7. Сателлитные пики в ЯМР-спектрах
Как было показано выше, спин-спиновое взаимодействие между ядрами ,3С
и !Н пренебрежимо мало в спектрах ЯМР !Н, но должно приниматься во
внимание в спектрах ЯМР ,3С. Это различие следует из разного природного
содержания изотопов.
Теперь рассмотрим спектр ЯМР !Н SnMe4, изображенный ниже:
1
Олово имеет десять изотопов, два из которых ЯМР-активны: 117Sn (/ =
1/2, 7,6%) и 119Sn (/= 1/2, 8,6%). В образце SnMe4 статистически
распределены все природные изотопы олова.
83,8% молекул SnMe4 содержат ядра Sn с /= 0.
7,6% молекул SnMe4 содержат ядра 117Sn.
8,6% молекул SnMe4 содержат ядра 119Sn.
Это означает, что в спектре ЯМР 'Н SnMe4 сигнал от 83,8% ядер 'Н
представляет собой синглет (спин-спиновое взаимодействие с оловом отсутствует), а
7,6% протонов взаимодействуют с ' 17Sn (с образованием дублета) и 8,6%
протонов взаимодействуют с ,19Sn (также с образованием дублета). Константы
спин-спинового взаимодействия У119 . и У117 , равны 54 Гц и 52 Гц
соответственно, и, следовательно, два дублета отделены друг от друга. Эти
дублеты называются сателлитными пиками.
Упражнение: В вышеприведенном спектре укажите
• синглет;
• два дублета;
• как определить значения У119 , и У117 ,
Аналогично, протоны а и а' имеют различное по отношению к протону b
магнитное окружение, и сигнал протона b также представляет собой дублет
дублетов (рис. 9.43).
Спин-спиновое взаимодействие между ядрами 13С
При обсуждении спектров ЯМР 13С возможность спин-спинового
взаимодействия между ядрами 13С не учитывалась. С чем это связано? Для ответа на
этот вопрос рассмотрим вероятность спин-спинового взаимодействия. Пос-
484
9. Спектроскопия
Рис. 9.44. Спектр ЯМР ,3С
ацетона (при 25 МГц );
величина Усн для квартета
составляет 172 Гц.
ДА1_
_i i i i i i i i i i i
+200 +100 8
кольку природное содержание изотопа 13С составляет только 1,1%,
вероятность присутствия в молекуле двух атомов 13С, соединенных друг с другом
оказывается крайне мала. Таким образом, пренебрежение спин-спиновым
взаимодействием Усс действительно вполне оправданно, если только
образец не обогащен изотопом 13С.
Взаимодействие между ядрами 1Н и 13С:
гетероядерное спин-спиноеое взаимодействие
Спин-спиновое взаимодействие не обязательно происходит между
одинаковыми ядрами (например, 1Н—!Н). Любые неэквивалентные ядра с /> 0 могут
взаимодействовать друг с другом. Взаимодействие между разными ядрами
называется гетероядерным спин-дпиновым взаимодействием. В качестве
примера рассмотрим взаимодействие между ядрами 1Н и 13С.
В спектрах ЯМР !Н спин-спйновое взаимодействие 1Н-13С не
наблюдается по причинам, изложенным выше для взаимодействия ,3С-13С в
спектрах ЯМР ,3С.
Рассмотрим ацетон 9.27. Вероятность соединения ядра 1Н с ядром 13С
очень мала, и возможностью JCH можно пренебречь — в спектре ЯМР 'Н
присутствует синглет (5 = +2,1). В то же время, поскольку природное
содержание !Н составляет ~99,9%, практически каждое ядро 13С в метильных
группах ацетона соединено с ядром 1Н. Поэтому в спектре ЯМР 13С
действительно наблюдается спин-спиновое взаимодействие ,3С— *Н (рис. 9.44).
СН,
О
9.27
Гетероядерное спин-спиновое взаимодействие может быть подавлено с
помощью специального инструментального метода, поэтому в лабораторной
практике вам могут встретиться спектры ЯМР 13С с «подавлением
протонов». Сравнение рис. 9.34 и 9.44 показывает различие между спектрами ЯМР
13С ацетона с «подавлением протонов» и без него.
9.17. Заключение
485
ESQ ЗАКЛЮЧЕНИЕ
В этой главе рассмотрены некоторые вопросы спектроскопии; особое внимание уделено
использованию трех спектроскопических методов в лаборатории.
Что означают следующие термины?
Спектроскопическая шкала
времени
Спектр поглощения
Спектр испускания
Волновое число
Закон Ламберта-Бера
Оптическая плотность
Пропускание
Молярный коэффициент
экстинкции
ИК-спектроскопия
Основное колебательное
состояние
Энергия нулевого
колебательного уровня
Типы колебаний
Приведенная масса
Область «отпечатков
пальцев»
Видимая/УФ-спектроско-
пия
°макс
Сопряженные полиены
Хромофорная группа
Спектроскопия ЯМР 1Н
Спектроскопия ЯМР 13С
Химический сдвиг
Интенсивность сигнала
Интегральная
интенсивность сигнала
Спин-спиновое
взаимодействие
Константа спин-спинового
взаимодействия
Теперь вы должны уметь:
• сопоставить относительные энергии,
частоты и длины волн ИК-, УФ-, видимого и
РЧ-излучения;
• знать соотношение между длинами волн
поглощенного и пропускаемого
излучения и понимать, как они связаны с
наблюдаемой окраской растворов;
• использовать закон Ламберта-Бера для
определения концентраций, исходя из
данных по оптической плотности и
пропусканию;
• использовать закон Ламберта-Бера для
определения стехиометрии некоторых
реакций;
• обсудить использование модели
колеблющейся пружины для описания
колебаний двухатомной молекулы;
• определить число независимых
колебательных степеней свободы для двух- и
трехатомных молекул и указать, какие из
них (и почему) активны в ИК-спектре;
• идентифицировать ИК-спектры простых
соединений с использованием области
«отпечатков пальцев»; определять
присутствующие в соединении
функциональные группы;
• понимать, как возникают некоторые
электронные спектры, например спектры
полиенов;
объяснить, почему пики поглощения в
электронных спектрах молекулярных
соединений, как правило, представляют
собой широкие линии;
объяснить причины сдвига значений >.макс
в рядах родственных соединений;
привести примеры содинений,
поглощающих в ближней УФ и видимой областях;
описать, как используется метод Жоба;
перечислить свойства, которые
ответственны за активность ядер в ЯМР-спект-
рах;
кратко обсудить, почему использование
фурье-преобразования в ЯМР-спектро-
скопии привело к существенному
улучшению данного метода;
идентифицировать типы химического
окружения в спектрах ЯМР 13С;
понимать, почему в спектроскопии ЯМР
1Н обычно используют дейтерированные
растворители;
идентифицировать спектры ЯМР 1Н на
основе понятий различных типов
химического окружения протонов и простого
спин-спинового взаимодействия.
486
9. Спектроскопия
ESQ УПРАЖНЕНИЯ
9.1. Что такое относительные энергии
переходов, наблюдаемых в
колебательной, электронной и ЯМР-спект-
роскопии?
9.2. Водные растворы, содержащие ионы
никеля(П), имеют зеленую окраску.
Оцените длину волны света,
поглощаемого гидратированными ионами
никеля(Н).
9.3. Обратитесь к рис. 9.6. Что означает
тангенс угла наклона прямой?
Определите концентрацию ионов
железа(П) в растворе, имеющем
оптическую плотность а) 20; б) 300.
9.4. Частота валентного колебания в СО
составляет 2170 см-1, а силовая
постоянная связи равна 1905 Н/м.
Определите частоту валентного
колебания молекулы NO, если силовая
постоянная связи равна 1595 Н/м.
9.5. Определите приведенную массу
следующих двухатомных молекул: Н2,
HD (D = 2Н), Н35С1 и Н37С1.
Определите отношение приведенных масс
для пар молекул, содержащих
разные изотопы. Будут ли наблюдаться
различия в химическом поведении
а) Н2 и HD; б) Н35С1 и Н37С1?
9.6. Обратитесь к рис. 9.12,6 и 9.12,в.
Каково направление дипольного
момента при а) антисимметричном
валентном колебании молекулы
С02; б) деформационном
колебании? Возникает ли в каждом из этих
случаев постоянный дипольный
момент?
9.7. Рассмотрите симметричное
валентное, антисимметричное валентное и
деформационное колебания в
следующих молекулах: a) HCN; б) CS2;
в) OCS; г) XeF2. Какие из этих
колебаний активны, а какие — неактивны
в ИК-спектре?
9.8. В ИК-спектре молекулы Н20
присутствуют три полосы поглощения,
соответствующие антисимметричному
валентному колебанию (3756 см-1),
симметричному валентному
колебанию (3657 см-1) и деформационному
колебанию типа «ножницы»
(1595 см"1). В ИК-спектре D20 эти
полосы находятся при 2788, 2671 и
1178 см-1 соответственно. Объясните
это наблюдение.
9.9. В табл. 9.3 перечислены частоты
валентных колебаний карбонильной
группы. Поясните, почему для кар-
боновых кислот и
карбоксилат-ионов эти величины, как правило,
меньше, чем для соответствующих
альдегидов.
9.10. Для каждого из следующих
соединений укажите природу
функциональной группы и область частот в ИК-
спектре, при которых можно
ожидать появления соответствующих
полос поглощения: а) ацетонитрил;
б) бутанол-2; в) этиламин; г) пропа-
наль; д) пентен-1.
9.11. На рис 9.45 показаны ИК-спектры
полиэтилена высокой плотности, до-
декана, гексана и гексена-1. а)
Почему спектры полиэтилена, додекана и
гексана так похожи друг на друга?
б) Какие две особенности спектра
гексена-1 отличают этот спектр от
спектра соответствующего алкана?
9.12. На рис. 9.46 приведены четыре ИК-
спектра а—г. Определите, какой из
спектров соответствует каждому из
следующих соединений: циклогек-
сан, бензол, толуол (метилбензол) и
фенол.
9.13. Как с помощью ИК-спектроскопии
различить следующие пары
соединений:
9.18. Упражнения
487
"Г
г
у—-
Полиэтилен высокой
плотности
Додекан
Гексан
Гексен-1
■ ' ' ■ ' ' ■ ■ i_J i i i i_J_i i i i 1_1 1_1 i 1—1 i i i Lj i 1—1—
3500 3000 2500 2000 1500 1000 500
Волновое число, см-1
Рис. 9.45. К упражнению 9.11.
2500 2000 1500
Волновое число, см-1
Рис. 9.46. К упражнению 9.12.
а) СН3С(0)СН3 и СН3СН(ОН)СН3;
б) СН3СН2СН=СН2 и СНзСН2СН2СНз;
в) СН3СН2СН2С1 и CH3CH2CH2NH2?
В каких из указанных случаев
использование спектроскопии ЯМР
,3С может дать столь же
однозначный ответ?
9.14. Запишите три уравнения,
описывающие три я-МО алл ильного аниона
с использованием обозначений,
введенных в уравнении 4.5.
9.15. Укажите порядок я-связи в
а) аллильном катионе; б) аллильном
анионе.
488
9. Спектроскопия
9.16. Комплексное соединение X
поглощает при 466 нм (емакс = 75 л/моль см)
и при 338 нм (емакс = 68 л/моль см).
Каково отношение оптических
плотностей двух линий в спектре 0,1 М
раствора X? Зависит ли ваш ответ от
длины пути луча в кювете
спектрофотометра?
9.17. Что вы понимаете под терминами
а) поглощение; б) оптическая
плотность; в) пропускание?
9.18. Какому типу перехода (о -> а*, я ->
л*, п -> а* или п -> л*)
соответствуют самые интенсивные пики
поглощения в электронных спектрах
следующих соединений: а) пентана;
б) пентена-1; в) октатриена-2,4,6;
г) этанола?
9.19. С использованием данных табл. 9.10,
определите резонансную частоту
ядер 31Р на 400-МГц-спектрометре и
резонансную частоту ядер 13С на
250-МГц-спектрометре.
9.20. В спектре ЯМР 13С уксусной
кислоты присутствуют сигналы при 6 =
+20,6 и 6 = +178,1. Проведите
отнесение спектра.
9.21. В спектре ЯМР 13С 2-метилпропано-
ла-2 присутствуют сигналы при 6 =
+31,2 и 6 = +68,9. Предложите
отнесение этих сигналов. Какая
особенность спектра позволит подтвердить
ваши отнесения?
9.22. В спектре ЯМР 13С следующего
соединения содержатся четыре сигнала
с примерно одинаковой
интенсивностью: 5 = +20,5; 8 = +52,0; 5 = +80,8
и 6 = +170,0. Предложите отнесение
этих сигналов.
9.23. Предскажите отношение интенсив-
ностей и мультиплетность сигналов
из-за спин-спинового
взаимодействия в спектре ЯМР {Н
2-метилпропана.
9.24. Отнесите каждый из следующих
спектров ЯМР !Н. Укажите
отношение интегральных интенсивностей
сигналов.
Соединение
а) Ме2СНС!
EN
б) СН3СН2С
н,с
/
\
NH,
сн2сн3
d, м.д.
+ 1,3 (дублет)
+2,7 (септет)
+ 1,1 (триплет)
+2,2 (квартет)
+6,4 (очень
широкий)
+ 1,1 (триплет)
+2,1 (синглет)
+2,5 (квартет)
О
г) СН3СН2-0-СН2С02Н +1,3 (триплет)
+3,7 (квартет)
+4,1 (синглет)
+ 10,9 (уширенный)
д) СН3СНВг2
е) СН2С1СН2СН2С1
+2,5 (дублет)
+5,9 (квартет)
+2,2 (квинтет)
+3,7 (триплет)
9.25. Объясните, почему в спектре ЯМР
!Н CH2C1CF2C1 присутствует
триплет при 5 = +4,0. [Подсказка:
обратитесь к табл. 9.10.] Предскажите
мультиплетность сигналов в спектре
ЯМР^СРзСНзОН.
\
с
/
/
\
сн.
/
с
\
КИНЕТИКА
ХИМИЧЕСКИХ РЕАКЦИЙ
ВВЕДЕНИЕ
В разд. 1.18 были рассмотрены активационные барьеры, а в гл. 8 обсуждались
механизмы и пути реакций. Информацию о механизме реакций получают, в
частности, из изучения скоростей реакций. Такая информация связана с
закономерностями кинетики химических реакций, и данная глава посвящена
детальному рассмотрению этого вопроса. В вводном разделе суммированы
некоторые данные о скоростях реакций, с которыми вы, вероятно, уже знакомы.
Термодинамика и кинетика
При обсуждении термодинамики реакций можно оперировать различными
функциями энергии (см. гл. 12), но в данном разделе рассматривается только
энтальпия.
То обстоятельство, что реакция термодинамически выгодна, вовсе не
означает, что она обязательно должна протекать быстро. Например, реакция
алканов с кислородом (уравнение 10.1) экзотермична, но она не протекает
самопроизвольно - ее необходимо инициировать искрой или пламенем.
Смеси алканов с кислородом могут оставаться без изменения в течение
долгого времени.
С5Н12(ж.) + 802(газ) -> 5С02(газ) + 6Н20(ж.) (10.1)
Др#= -3509 кДж/моль С5Н12
С другой стороны, многие реакции, протекающие самопроизвольно,
являются эндотермическими (например, растворение нитрата аммония в воде).
Очевидно, что изменение энтальпии нельзя считать решающим фактором,
от которого зависит самопроизвольность реакции. Более важную роль играет
другое понятие — изменение свободной энергии А (7 (гл. 12). Пока же общий
вывод состоит в том, что знание об изменении термодинамических
параметров в ходе реакции не позволяет сделать однозначных выводов о скорости
превращения исходных продуктов в реагенты.
В гл. 8 обсуждалась лимитирующая стадия реакции. На рис. 8.13 показаны
профили двух трехстадийных реакций. Можно видеть, что хотя каждая из
трех стадий является экзотермической, одна из них имеет наибольшую
энтальпию активации и контролирует скорость протекания реакции в целом.
490
10. Кинетика химических реакций
Что такое скорость реакции?
Об изменениях в ходе реакции можно судить либо по скорости
образования продуктов, либо по скорости расходования реагентов, как показано на
рис. 10.1.
Мера скорости реакции — изменение концентрации реагента или продукта
реакции во времени*. Следовательно, скорость реакции измеряется в моль/лс.
Мера скорости реакции - изменение концентрации реагента или
продукта реакции во времени. Единицы измерения скорости -
моль/лс.
Рассмотрим две реакции с участием реагента А, которые протекают по
общему уравнению 10.2. Скорость расходования А зависит от природы
рассматриваемой реакции и от условий ее проведения.
А -»Продукты
(Ю.2)
Рис. 10.2,а соответствует реакции, в которой концентрация А линейно
меняется в ходе процесса. Скорость расходования А, равная тангенсу угла
наклона прямой, постоянна в каждый момент времени, т. е. она не зависит от
концентрации А.
кта
>>
прод
X
g
£
н
X
<и
СО
&
0?
S
я
ев
е-
X
и
Я
X
о
^
^
Образование прод
; Исчезновение
~
\ реагента ^
» ^-^^
\ /
/ "Ч
/ %%
/ **»^
L ^
Время
Рис. 10.1. В реакции типа А -» В (1 моль А превращается в 1 моль В) скорость
расходования А зеркально отображает скорость образования В.
' Строго говоря, это определение можно использовать лишь в том случае, когда объем реакционной
смеси не изменяется в ходе реакции (т. е. оно непригодно, например, для реакций в газах и для
реакций в проточных системах). В сложных случаях необходимо оговаривать определение и единицы
измерения скорости реакции.
10.1. Введение
491
0,04 г
0,005
40 50
Время, с
50 60
Время, с
Рис. 10.2. График уменьшения концентрации реагента А во времени может быть
линейным (а) и нелинейным (б). В случае а скорость расходования А постоянна и
равна тангенсу угла наклона прямой. В случае б скорость зависит от времени и
Определяется в каждой точке как тангенс угла наклона касательной к кривой. Скорость в
момент времени tx больше скорости в момент времени /2.
Рис. 10.2,6 соответствует реакции, в которой концентрация А вначале
В упражнении 10.1
(разд. 10.13) требуется
определить скорости
реакций,
представленных графиками а и б
на рис. 10.2.
уменьшается быстро, а затем медленнее. В этом случае
скорость реакции зависит от времени. Скорость реакции в
момент времени /, есть тангенс угла наклона касательной,
построенной в точке Xv В момент времени t2 скорость
определяется как тангенс угла наклона касательной, построенной в
точке Х2.
492 10. Кинетика химических реакций
Факторы, влияющие на скорость реакций
Ниже перечислены некоторые факторы, влияющие на скорость реакций:
• концентрация (или давление для реакций с участием газов);
• температура;
• площадь поверхности (для реакций с участием твердофазных реагентов);
• давление.
На рис. 10.3 показана скорость выделения диоксида углерода из двух
реакционных сосудов при взаимодействии карбоната кальция с разбавленной
соляной кислотой (уравнение 10.3).
СаС03(тв.) + 2НС1(водн.) -> СаС12(водн.) + Н20(ж.) + С02(газ) (10.3)
Мерой скорости реакции является скорость выделения С02. Две реакции
протекают с различными скоростями: можно видеть, что кривая I возрастает
примерно вдвое быстрее кривой II. Очевидно, что условия проведения
реакции в разных сосудах не одинаковы — скорость может зависеть от
концентрации соляной кислоты, от площади поверхности карбоната кальция
(например, порошок или гранулы) или от температуры.
Как выбрать экспериментальный метод
для наблюдения за ходом реакции?
При выборе метода исследования реакции необходимо найти свойства,
измерение которых позволит количественно зафиксировать разницу между
исходными реагентами и конечными продуктами. Удобнее всего использовать
методы, не оказывающие влияния на ход реакции. Можно использовать
обычные спектроскопические методы или методы, основанные на
измерении физических свойств, таких, как показатель преломления, вязкость или
объем. Некоторые реакции протекают слишком быстро, чтобы за их ходом
можно было следить с помощью обычных спектроскопических методов.
В данной главе не ставится задача рассмотреть специфические методы, но
ряд экспериментальных методик проиллюстрирован ниже при обсуждении
некоторых реакций. Уравнение 10.3 - пример реакции, за ходом которой
можно наблюдать по скорости газовыделения; в разд. 9.5 была рассмотрена
реакция, сопровождаемая изменением окраски.
Рис. 10.3. Скорость реакции между
твердофазным карбонатом кальция и
соляной кислотой можно исследовать,
измеряя объем выделяющегося С02.
Скорость реакции I приблизительно
вдвое выше скорости реакции II.
Каковы возможные различия между
условиями проведения этих реакций?
0 Время
10.2. Кинетические уравнения. Зависимость скорости от концентрации 493
КЕБ КИНЕТИЧЕСКИЕ УРАВНЕНИЯ.
ЗАВИСИМОСТЬ СКОРОСТИ ОТ КОНЦЕНТРАЦИИ
Запись скорости реакции в форме
дифференциального уравнения
В данном разделе обсуждается обобщенная реакция:
А -> Продукты
Скорость этой реакции определяется уравнением 10.4, где производная
—d[A]/d/ означает скорость изменения концентрации А в момент времени г,
концентрация А обозначается [А].
Скорость реакции = - d[A]/d/ (10.4)
Концентрация компонента А обозначается [А].
Общее кинетическое уравнение
В начальный момент реакции концентрация А имеет максимальное
значение; концентрация уменьшается в ходе реакции. Уравнение 10.5 дает общий
вид зависимости скорости реакции от концентрации А.
Скорость реакции = -d[A]/d/« [А]Л (10.5)
Показатель степени п в уравнении 10.5 называется порядком реакции по Aw
может принимать целочисленные (включая нуль) или дробные значения.
Порядок реакции характеризует зависимость скорости реакции от [А]. В данной
главе основное внимание уделено реакциям со значениями п = 0, 1 или 2.
Уравнение 10.5 часто записывают в форме 10.6, где к — константа
скорости реакции. Единицы измерения константы скорости меняются в
зависимости от порядка реакции (см. ниже). Константа скорости реакции, в
отличие от скорости реакции, не зависит от концентрации. Важно уметь различать
эти два понятия. Чем больше величина к при фиксированной концентрации,
тем быстрее протекает реакция.
Скорость реакции = -d[A]/d/ = k[A]n (10.6)
Общее кинетическое уравнение для реакции А
записывается в виде:
Скорость реакции =
где [А] - концентрация
и п - порядок реакции
-d[A]/dfoc
к[А]п
А в момент времени
по А.
f,/c
-> Продукты
- константа скорости
494
10. Кинетика химических реакций
Дополнение 10.1. Скорость как производная -d[A]/df
Рассмотрим реакцию:
А —> Продукты
в которой концентрация А, т. е. [А], линейно уменьшается во времени как показано на
диаграмме.
[А]2 0,02
0,01
0 30 60 90 120 150
'i h Время, с
Скорость расходования А постоянна. Из диаграммы видно, что за время от /, до /2
концентрация А уменьшается от [A]j до [А]2.
Скорость расходования А = 0,02/60 = 3,33 • Ю-4 моль/л с
Скорость реакции можно выразить через скорость расходования А. Математически эта
величина представляет собой производную [А] по времени, взятую с обратным знаком.
Скорость реакции = -d[A]/d/
В реакции:
А->В + С
скорость расходования А равна скорости образования В и С:
Скорость реакции = -d[A]/d/ = d[B]/d/ = d[C]/d/
Обратите внимание, что [А] или [А], означает концентрацию А в момент времени t.
Исходная концентрация А обозначается [А]0.
Реакции нулевого порядка по А
Рис. 10.2,0 соответствует реакции, скорость которой не зависит от
концентрации А. В этом случае показатель степени п в уравнении 10.6 равен нулю
(напомним, что возведение любого числа в нулевую степень дает единицу;
так что [А]0 = 1). Таким образом, скорость реакции равна константе скорости
(уравнение 10.7)
Скорость реакции = -d[A]/d/ = А:[А]° = к (10.7)
и реакция имеет нулевой порядок по А. В этом случае единицы измерения
константы скорости такие же, как и у самой скорости: моль/лс Скорость
реакции не зависит от времени.
10.3. Как определить порядок реакции?
495
Если реакция имеет нулевой порядок по А, ее скорость
концентрации А, а кинетическое уравнение имеет вид
Скорость реакции =
Единицы измерения к
-d[A]/df = fr
- моль/лс.
не зависит от
Реакции первого порядка по А
Если скорость реакции прямо пропорциональна концентрации реагента А,
реакция имеет первый порядок по А. Соответствующее кинетическое
уравнение имеет вид
Скорость реакции = —d[A]/d/ = k[A\ (10.8)
Это означает, что по мере уменьшения [А] в ходе реакции скорость реакции
также уменьшается, и зависимость [А] от времени имеет нелинейный характер.
Единицы измерения константы скорости в уравнении 10.8 - с-1. См.
упражнение 10.3 в разд. 10.13.
Если реакция имеет первый порядок по А, зависимость ее скорости
от концентрации А определяется следующим кинетическим
уравнением:
Скорость реакции = -d[A]/df = /c[A]
Единицы измерения к - с-1.
Реакции второго порядка по А
Если скорость реакции пропорциональна квадрату концентрации А, реакция
имеет второй порядок по А, а закон изменения скорости задается уравнением
Скорость реакции = -d[A]/d/= k[A\2 (10.9)
Зависимость [А] от времени снова оказывается нелинейной.
Единицы измерения константы скорости к в уравнении 10.9 — л/мол ьс.
Если реакция имеет второй порядок по реагенту А, ее кинетическое
уравнение имеет следующий вид:
Скорость реакции = -d[A]/df = k[A]2
Единицы измерения к - л/мольс.
КАК ОПРЕДЕЛИТЬ ПОРЯДОК РЕАКЦИИ?
В данном разделе снова рассматривается обобщенная реакция
А -> Продукты (10.10)
496 10. Кинетика химических реакций
А-> Продукты
Постройте зависимость
[А] от времени.
Является ли она линейной?
I Нет
Т
Да
Реакция имеет нулевой
порядок по А
Реакция имеет ненулевой порядок по А
Определите скорость реакции (по тангенсам) в
нескольких точках кинетической кривой.
Постройте зависимость скорости от [А].
Является ли она линейной?
I Нет
Т
Реакция имеет порядок,
отличный от нулевого и первого
Определите скорость реакции (по тангенсам) в
нескольких точках кинетической кривой.
Постройте зависимость скорости от [А]2.
Является ли она линейной?
У
Нет
Реакция имеет порядок,
отличный от нулевого,
первого и второго
Да
Реакция имеет первый
порядок по А
Да
Реакция имеет второй
порядок по А
Рис. 10.4. Схема графического метода, позволяющего определить, является ли
реакция А —> Продукты реакцией нулевого, первого или второго порядка.
Графические методы
Если график зависимости [А] от времени представляет собой прямую,
можно сделать вывод, что реакция имеет нулевой порядок по А. С другой
стороны, если этот график нелинеен (как на рис. 10.2,6), точное значение порядка
нельзя определить немедленно и можно лишь сказать, что порядок ненулевой.
Можно ограничиться лишь теми случаями, когда порядок реакции близок к
первому или второму. Как различить их?
Существует несколько способов обработки экспериментальных
результатов. Схема одного из них показана на рис. 10.4*. Выбор координат для
построения графиков непосредственно следует из уравнений 10.8 и 10.9. Как
уже указывалось, скорость реакции в данный момент времени можно
определить по тангенсу угла наклона касательной к кинетической кривой.
* Более простой способ - построить фафик зависимости в координатах lg d[A]/d/ - lg [А]; тангенс
угла наклона этой прямой равен порядку реакции п согласно уравнению 10.6.- Прим перев.
10.3. Как определить порядок реакции?
497
Пример 10.1 Термическое разложение бензоилпероксида
Бензоилпероксид (перекись бензоила) разлагается выше 100 °С.
Он имеет интенсивную полосу поглощения в ИК-спектре при
1787 см-1. Измерение интенсивности этой полосы по мере ее
исчезновения позволяет наблюдать (с помощью закона Ламбер-
та-Бера) за концентрацией бензоилпероксида в ходе его
разложения. Определите кинетическое уравнение разложения
бензоилпероксида с использованием приведенных ниже данных (полученных
при температуре 107 °С в работе Mosher M.D., Vinson S.,
Connagahan J., Forsythe R., Mosher M.W., Journal of Chemical
Education, 1991, vol. 68, p. 510):
o-
О
//
С
\
О —О
к>
Перекись бензоила
Время, мин
[Бензоилпероксид]
ю2,
0
моль
2,00
15
л
1,40
30
1,00
45
0,70
60
0,50
75
0,36
90
0,25
120
150
180
0,13 0,06 0,03
Исходная концентрация бензоилпероксида составляла 2,00 • Ю-2 моль/л.
(Обратите внимание, что это указано в таблице.)
Решение
Построим график зависимости концентрации бензоилпероксида от времени:
150 200
Время, мин
Так как этот фафик нелинеен, разложение не может быть реакцией нулевого
порядка, но может быть реакцией первого или второго порядка по бензоилперо-
ксиду. Один из способов различить эти возможные случаи - проверить
линейность графика зависимости скорости реакции от [А].
Далее, определим скорость разложения в нескольких точках. Пяти точек
вполне достаточно, но они должны охватывать весь интервал экспериментальных
498
10. Кинетика химических реакций
значений. Скорость реакции в момент времени /определяется построением
касательной к кинетической кривой в точке /. Тангенсы углов наклона касательных
имеют отрицательные значения, поскольку для построения графика
использовано изменение концентрации бензоилпероксида. Так как бензоилпероксид
расходуется в процессе реакции, отрицательные значения тангенсов соответствуют
положительной скорости реакции.
Построим таблицу с новыми данными; вы можете использовать
вышеприведенный график для того, чтобы проверить данные, приведенные в таблице.
[Бензоилпероксид] • 102, моль/л
Скорость реакции • 104, моль/лмин
2,00
4,44
1,40
3,27
1,00
2,23
0,50
1,17
0,13
0,25
Теперь построим зависимость скорости реакции от концентрации
бензоилпероксида:
i
ъ
8
О.
О
и
0,005
0,01 0,015 0,02 0,025
[Бензоилпероксид], моль/л
Прямая линия означает, что реакция имеет первый порядок по бензоилперок-
сиду, и кинетическое уравнение можно записать в виде:
Скорость реакции = -d [Бензоилпероксид]/ск = к [Бензоилпероксид]
Константу скорости реакции к можно найти из второго графика: она равна
тангенсу угла наклона прямой к = 2,25 • 10~2 мин-1, или 3,75 • Ю-4 с-1.
Построение касательных к кривой - недостаточно точный метод получения
ответа. Ниже в данной главе рассматривается альтернативный способ, который
можно использовать для решения примера 10.1.
Метод времени полупревращения
Время, необходимое для уменьшения концентрации реагента А от величины
[А], до величины [А]/2 (где [А], - концентрация А в определенный момент
времени /), называется временем полупревращения.
Для реакции первого порядка времена полупревращения, измеренные на
стадиях:
[А],->[А]/2, [А]/2 -> [А]/4, [А]/4 -> [А]/8, [А]/8-> [А]/16ит. д.
равны между собой. Это не выполняется для реакций второго порядка.
10.3 Как определить порядок реакции?
499
Пример 10.2 Распад образца, полученного фотохимическим методом
Синий комплекс ртути(Н) (соединение I) получается при облучении
родственного соединения ртути(М), после чего он сразу же
распадается с образованием исходного соединения. За скоростью
распада комплекса I можно наблюдать с помощью видимой/УФ-спек-
тоскопии (^макс = 605 нм, емакс= 27000 л/моль • см), и рассчитать
концентрацию I с помощью закона Ламберта-Бера. С
использованием приведенных ниже данных (полученных в работе Petersen R.L.,
Harris G.L., Journal of Chemical Education, 1985, vol. 52, p. 802)
определите порядок реакции по I.
£>-'"ГкХЧ>
соединение I
Время,
[I] Ю5,
моль/л
мин
0
1,447
0,5
1,061
1
0,826
1,5 2
0,612 0,449
2,5
0,336
3
0,249
3,5
0,200
4 4,5
0,139 0,106
5
0,083
Решение
Кинетическое уравнение распада I имеет общий вид
-d[l]/dt=k[l]n
Построим график зависимости [I] от времени:
1,5
1,25
Ъ 0,75
— 0,5
0,25
0
0 0,5 1 1,5 2 2,5 3 3,5 4 4,5 5 5,5
6
Время, мин
10. Кинетика химических реакций
Видно, что реакция имеет ненулевой порядок по соединению I.
Существует несколько способов дальнейшего решения задачи, и один из них
связан с измерением нескольких времен полупревращения.
Исходная концентрация [1]0 = 1,447 • 10~5 моль/л.
Из графика видно, что первое время полупревращения соответствующее
уменьшению концентрации от [1]0 до [IJq/2, равно 1,2 мин.
Второе время полупревращения, соответствующее изменению
концентрации от [IJq/2 до [1]()/4, равно 2,35 - 1,2 мин =1,15 мин.
Третье время полупревращения, соответствующее изменению концентрации
от [1]о/4 до [I]q/8, равно 3,55 - 2,35 = 1,2 мин.
Эти три времени полупревращения практически равны, и, следовательно,
реакция имеет первый порядок по соединению I.
Дополнение 10.2. Радиоактивный распад
Процесс распада радионуклидов всегда подчиняется уравнению первого порядка,
поэтому каждый радионуклид имеет характерный период полураспада. Его значение
может варьировать от секунд до нескольких лет или даже тысяч лет. Производство
радионуклидов, не встречающихся в природе, обсуждается в дополнении 1.3.
Изотоп
2i52U
gCo
'3«Cs
Встречается в природе?
Да
Нет
Нет
Период полураспада
7,04- 108лет
270 дней
30,7 минут
Ниже на рисунке показан распад 2^Т1 с образованием стабильного изотопа ^РЬ.
Период полураспада можно определить, измеряя время уменьшения исходного
количества 2^Т1 в два раза. Чтобы повысить точность измерений, необходимо определить
несколько последовательных периодов полураспада и вычислить среднее значение.
Определите период полураспада 2%]Т\.
0,008
^ 0,006
§
— 0,004
ь_
©оо
0,002
0
0 5 10 15 20
Время, мин
Распад 2^Т1 до 2g]Pb является последней из 11 стадий цепочки распада природного ^U
в ^РЬ. В цепочке радиоактивного распада на каждой стадии из радионуклида
образуется дочерний нуклид, который может быть стабилен или также подвергается распаду.
10.4. Кинетические уравнения для реакций с участием нескольких реагентов 501
КИНЕТИЧЕСКИЕ УРАВНЕНИЯ ДЛЯ РЕАКЦИЙ
С УЧАСТИЕМ НЕСКОЛЬКИХ РЕАГЕНТОВ
Простая реакция реагента А, рассмотренная выше, редко встречается в
реальных химических системах. Гораздо чаще в реакции участвуют два или
несколько реагентов, и скорость реакции зависит от концентрации нескольких
соединений. Рассмотрим влияние этих факторов на кинетическое уравнение.
В данном разделе обобщенная реакция задается уравнением
А+ В -> Продукты (Ю.Н)
Общее кинетическое уравнение и порядки реакции
Скорость реакции I0.ll может зависеть:
• только от [А];
• только от [В];
• йот [А], йот [В];
• ни от [А], ни от [В].
Скорость реакции можно выразить через скорость расходования А или В или
через скорость образования продуктов. Так как А и В реагируют в
соотношении 1:1, они расходуются с одинаковой скоростью. Общее кинетическое
уравнение записывается в виде уравнения
Скорость реакции = -d[A]/d/= -d[B]/d/= А:[А]"[ВГ (10.12)
Реакция имеет порядок п по А, порядок т по В, а общий порядок реакции
равен (т + п). Сравните этот результат с реакцией 10.10, для которой общий
порядок совпадает с порядком реакции по А. Константа скорости реакции
относится ко всему уравнению в целом. Например, кинетическое уравнение
гидролиза сахарозы (реакция 10.13 ирис. 10.5) задается уравнением 10.14.
Реакция имеет первый порядок по сахарозе и первый порядок по воде;
соответственно общий порядок реакции равен двум.
С12Н22Ои + Н20 -> С6Н1206 + С6Н1206 (10.13)
Сахароза Глюкоза Фруктоза
Скорость реакции = ^[С12Н22Оп][Н20] (10.14)
Важно помнить, что стехиометрия реакции в общем случае никак не связана с
кинетическим уравнением реакции. В реакции 10.13 концентрации обоих
исходных реагентов присутствуют в кинетическом уравнении, но это происхо-
► дит далеко не всегда. Например, кинетическое уравнение реак-
Обсуждение молеку- цИИ 10.15 «г соответствует ее стехиометрии (уравнение 10.16).
лярности реакций:
см. разд. 10.9. Мп04" + 8Н+ + 5Fe2+ -» Mn2+ + 4Н20 + 5Fe3+ (10.15)
Скорость реакции * A:[Mn04-][H+]8[Fe2+]5 (10.16)
502
10. Кинетика химических реакций
НО
НО
Сахароза
н сн7он
н н
но
Рис. 10.5. Структуры сахарозы, глюкозы и фруктозы.
ОН
Исследование скоростей реакции
с участием нескольких реагентов
В реакциях с участием более чем одного реагента концентрации всех
реагентов уменьшаются во времени. В то же время, чтобы определить порядок
реакции, необходимо наблюдать зависимость скорости реакции от
концентрации каждого из реагентов по отдельности. Этого можно достичь при условии,
что все реагенты, кроме одного, находятся в большом избытке, так что в ходе
реакции не происходит существенного изменения концентраций этих
реагентов.
Рассмотрим реакцию между А и В (уравнение Ю.11). Пусть в I л
исходного раствора присутствует Ы0~4 моль А и ЬЮ-2 моль В. К моменту, когда
прореагирует половина исходного количества А, 9,95-Ю-3 моль В останутся
неизрасходованными. Следовательно, концентрация В практически не
изменилась, тогда как концентрация А изменилась существенно. (В качестве
упражнения определите концентрации А и В в момент начала реакции и в
момент, когда прореагирует половина вещества А. На сколько процентов
изменится каждая из концентраций?) Изменение скорости реакции будет
происходить в основном за счет изменения концентрации А. Чтобы определить,
как скорость зависит от [В], небходимо повторить реакцию в условиях либо
большого избытка А, либо при других концентрациях В (но всегда в
условиях его большого избытка), как показано в примере 10.3.
Рассмотрим случай, когда общее кинетическое уравнение реакции
задается уравнением 10.17. Если В присутствует в большом избытке, величину [В]
10.5. Интегральная форма кинетических уравнений 503
можно считать постоянной (известной), и кинетическое уравнение
принимает вид 10.18.
См. примеры
10.3-10.5
Скорость реакции = -d[A]/d/ = &[А][В]
Скорость реакции = —d[A]/d/ = ^[A]
(10.17)
(10.18)
где к* = к[В]. В этом случае реакция имеет кинетику
псевдопервого порядка. В действительности, она имеет первый порядок
по А, а условие постоянства концентрации В приводит к тому,
что общий порядок реакции кажется первым.
ИНТЕГРАЛЬНАЯ ФОРМА КИНЕТИЧЕСКИХ УРАВНЕНИЙ
До сих пор скорости реакций записывались в форме дифференциальных
уравнений, и рис. 10.4 иллюстрирует графический метод нахождения
простых кинетических уравнений. Однако определение скорости реакции по
касательным к кинетическим кривым не всегда позволяет получать достаточно
точные результаты.
Экспериментальные данные часто получают в форме зависимости [А] от
времени /, поэтому полезно знать, как [А] зависит от /для разных
кинетических уравнений, вместо того, чтобы обрабатывать данные, как показано на
рис. 10.4. Таким образом, возникает вопрос: какой вид имеют зависимости
[А] от /для случаев реакций нулевого, первого и второго порядков по А?
Эти соотношения получают при записи кинетического уравнения в
интегральной форме. Детали интегрирования приводятся в дополнении 10.3; в
основном тексте эти уравнения приводятся без вывода. Все выражения
относятся или к реакции 10.2, или к случаям, когда все реагенты, кроме А,
присутствуют в большом избытке (см. разд. 10.4).
Дополнение 10.3. Вывод интегральных кинетических уравнений
Уравнение прямой
Общее уравнение прямой имеет вид
у= ах+ b
где х и у — переменные, a — тангенс угла наклона прямой и b — свободный
член.
Нулевой порядок по А
Дифференциальная форма кинетического уравнения для реакции нулевого
порядка имеет следующий вид:
Скорость реакции = —d[A]/d/ = k
Однако экспериментальные результаты, как правило, получают в виде
значений [А] при определенных временах /. График зависимости [А] от /
представляет собой прямую, и уравнение этой прямой имеет вид
(См. продолжение)
504
10. ^нетика химических реакций
(Продолжение)
у = ах+ Ь
где у — это [А], а — тангенс угла наклона прямой, х — это /, Ь — свободный член
(офезок, отсекаемый на оси у). Каков физический смысл тангенса угла
наклона и свободного члена?
Полное уравнение можно найти, проинтегрировав дифференциальную
форму кинетического уравнения. Символ J означает интефал некоторой
функции; помимо этой функции необходимо указывать переменную, по
которой ведется интефирование. Так, Jdjc означает, что интегрирование ведется
по переменной х.
И [А], и /являются переменными, и перед тем, как приступить к интефи-
рованию дифференциальной формы кинетического уравнения, необходимо
провести разделение переменных, т. е. поместить их в разные части
уравнения. Таким образом, имеем
-d[A] = kdt
При интефировании обеих частей этого уравнения получаем
-Jd[A] = \kdt
b-[A] = kt
где b — постоянная интефирования.
Чтобы найти Ъ, воспользуемся начальным условием: при / = О
концентрация [А] равна начальной концентрации [А]0. Подставляя эти значения в
предыдущее уравнение, получаем
[А]0 = *
и, следовательно:
[А]-[А]0 = А:/,или[А] = [А]0-А:/
Это выражение представляет собой уравнение прямой, полученной при по-
сфоении фафика зависимости [А] от /для реакции нулевого порядка по А.
у= ах + b
t
|A1
\
t I
t
r |
-к [А]0 и с
Следовательно, константу скорости реакции можно найти непосредственно
из тангенса угла наклона прямой. ,„
(См. продолжение )
10.5. Интегральная форма кинетических уравнений 505
(Продолжение)
Аналогичным образом могут быть получены интегральные формы
кинетических уравнений первого и второго порядков.
Реакция первого порядка no А
Дифференциальная форма кинетического уравнения имеет вид
-d[A]/d/=A:[A]
После разделения переменных можно записать:
d[A] _
ТАГ
kdt
При интегрировании получаем:
d[A] _
\kdt
[А]
Ь - In [A] = kt
где Ь — постоянная интегрирования.
При / = О [А] = [А]0. При подстановке этих значений в предыдущее
уравнение получаем
1п[А]0 = />
и, следовательно,
1п[А]0 - ln[A] = kt
или
ln[A] = ln[A]0 - kt ln[A]
1п[А]0
у— ax + b
1п[А]|/ |
-k ln[A]0
Таким образом, если график зависимости In [А] от времени представляет
собой прямую, реакция имеет первый порядок по А, а константа скорости
определяется по тангенсу угла наклона прямой.
Реакция второго порядка по А
Дифференциальная форма кинетического уравнения имеет вид
-d[A]/dr=A:[A]2
(См. продолжение)
506
10. Кинетика химических реакций
(Продолжение)
После разделения переменных можно записать:
[А]г
При интегрировании получаем
1/[А] = b + kt
где Ь — постоянная интегрирования.
При / = О [А] = [А]0. При подстановке этих значений в предыдущее
уравнение получаем
1/[А]0 = *
и, следовательно,
1/[А] = 1/[А]0 + */
у = ах + b
'[All' I
1/IA]
А
1
[Alo
tg a = -к /
Лц
1 1 1 iw
-к 1/[А]0
Л с
Таким образом, если фафик зависимости 1/[А] от времени представляет
собой прямую, реакция имеет второй порядок по А, а константа скорости
определяется по тангенсу угла наклона прямой.
Некоторые неопределенные интегралы, которые необходимо
запомнить:
Jdx = x\ \adx= ах(а- константа);
idx/x=\nx; idx/x2 = -\/x
Необходимо также помнить общее уравнение прямой:
у = ах+ b
где х и у - переменные, а - тангенс угла наклона прямой и b —
свободный член.
10.5 Интегральная форма кинетических уравнений 507
Нулевой порядок по А
Интегральная форма кинетического уравнения для реакции нулевого
порядка имеет вид 10.19:
[А] = [А]0-*/ (10.19)
где [А] — концентрация А в момент времени /, [А]0 — начальная
концентрация А.
График зависимости [А] от / представляет собой прямую, и константа
скорости к определяется по тангенсу угла наклона прямой.
Пример 10.3 Бромирование ацетона
Бром реагирует с ацетоном в присутствии соляной кислоты по
следующему уравнению:
|| + Вг2 > || + НВг
Н3С СН3 Н3С СН2Вг
За ходом реакции можно наблюдать с помощью вид./УФ-спектро-
скопии; водный раствор брома поглощает свет с длиной волны
395 нм.
Кинетическое уравнение имеет вид
Скорость реакции = -d[Br2]/df = fr[Br2]m[CH3C(0)CH3]n[H+]P
а) Реакция проводится в водном растворе и исходные концентрации
ацетона, соляной кислоты и брома равны 1,6, 0,4 и 0,0041 моль/л
соответственно. Экспериментальные результаты приведены ниже в
таблице. Определите порядок реакции по Вг2.
Время, с
[Br2] 10~3, моль/л
0
4,1
10
3,85
20
3,6
30
3,3
40
3,1
50
2,85
60
2,6
б) Если кислота и ацетон по-прежнему присутствуют в избытке, но
их концентрации изменяются, скорость реакции меняется
следующим образом:
[Ацетон], моль/л
[Н+], моль/л
[Вг2]0, моль/л
Относительная скорость реакции8
1,6
0,4
0,0041
1
1,6
0,2
0,0041
0,5
0,8
0,4
0,0041
0,5
0,8
0,2
0,0041
0,25
8 По данным работы BirkJ.P., Walters D.L., Journal of Chemical Education, 1992, vol.
69, p. 585.
Используйте эти данные вместе с данными (а) для вывода общего
кинетического уравнения.
508 10. Кинетика химических реакций
Решение
а) Условия проведения реакции таковы, что и кислота, и ацетон присутствуют в
большом избытке, поэтому скорость реакции можно записать в виде
-d[Br2]/oV=^[Br2r
Поскольку нам ничего не известно о возможной зависимости скорости от [Вг2],
попробуем построить график зависимости [Вг2] от времени:
-§ 0,0045
§
£ 0,004
0,0035
0,003
0,0025
0 20 40 60
Время, с
Так как график представляет собой прямую, можно сделать вывод, что
скорость не зависит от концентрации брома в растворе и реакция имеет нулевой
порядок по Вг2.
Таким образом, зависимость кинетического уравнения от концентрации
брома имеет следующий вид:
-d[Br2]/d/=^[Br2]° = ^
б) Общее кинетическое уравнение имеет вид
-d[Br2]/d/ = *[Вг2]т[СН3С(0)СН3],|[Н+]'
но так как т = 0, можно записать:
-d[Br2]/d/= к [СН3С(0)СН3]я[Н+Р
Из данных, приведенных в таблице, можно видеть, что скорость реакции
уменьшается
• вдвое, если [Н+] уменьшается вдвое;
• вдвое, если [СН3С(0)СН3] уменьшается вдвое;
• в четыре раза, если и [Н+], и [СН3С(0)СН3] уменьшаются вдвое.
Можно сделать вывод, что скорость реакции прямо пропорциональна и [Н+],
и [СН3С(0)СН3]. Следовательно, реакция имеет первый порядок по каждому из
этих реагентов.
Кинетическое уравнение имеет следующий вид:
-d[Br2]/df= *[CH3C(0)CH3][H+]
Возникает вопрос: как определить общую константу скорости к?.
Из задания (а) известно, что и [СН3С(0)СН3], и [Н+] можно считать
постоянными в ходе реакции. [Вопрос: Определите процентное изменение
концентраций ацетона и Н+ к моменту, когда Вг2 прореагирует полностью. Можно ли
считать верным положение о неизменности концентраций ацетона и Н"*"?]
10.5 Интегральная форма кинетических уравнений 509
Таким образом, для данного эксперимента
-d[Br2]/d/= A:[CH3C(0)CH3][H+] = А: 1,6-0,4
Тангенс угла наклона прямой в задании (а) равен К\
^ = 2,5- 10"5моль/л-с
Объединяя два уравнения для скорости реакции, получаем:
-d[Br2]/d/= *' = *• 1,6 0,4
Следовательно, можно найти общую костанту скорости к:
к = А7(1,6 • 0,4) = 2,5 • 10"5/(1,6 • 0,4) = 3,9 • К)"5л/мольс
[Вопрос: Почему единицы измерения к отличаются от единиц измерения к??]
Реакции первого порядка по А
Интегральная форма кинетического уравнения для реакции первого порядка
имеет вид
ln[A] = ln[A]0 - kt (10.20)
График зависимости 1п[А] от / представляет собой прямую, свободный член
которой равен 1п[А]0. Константа скорости к определяется по тангенсу угла
наклона прямой.
Пример 10.4 Этерификация трифторуксусной кислоты
Бензиловый спирт реагирует с трифторуксусной кислотой согласно
следующему уравнению:
С6Н5СН2ОН + CF3C02H > CF3C02CH2C6H5 + H20
Бензиловый Трифторуксусная Бензилтрифторацетат Вода
спирт кислота
Это пример общей реакции
Спирт + Карбоновая кислота > Сложный эфир + Вода
За скоростью расходования спирта можно наблюдать с помощью
спектроскопии ЯМР 1Н по изменению интегральной интенсивности
сигнала от протонов группы СН2. Нижеприведенные данные
получены при 310 К. Кислота в реакционной смеси присутствовала в
большом избытке. Докажите, что реакция имеет первый порядок по
спирту, и определите константу реакции псевдопервого порядка.
Время, мин
[Спирт], моль/ла
0
1,00
6,5
0,67
10
0,57
15
0,41
20
0,30
25
0,23
30 40
0,14 0,07
50
0,04
а По данным работы Minter D.E., Villareal M.C., Journal of Chemical Education, 1985,
vol. 62, p. 911.
510
10. Кинетика химических реакций
Решение
Кинетическое уравнение реакции можно записать в виде:
Скорость реакции = -d[C6H5CH2OH]/oV = ^[C6H5CH2OH]n[CF3C02H]w
Но так как кислота присутствует в большом избытке, то
-d[C6H5CH2OH]/d/= */[С6Н5СН2ОН]я
Необходимо подвердить, что п = 1 и определить константу реакции
псевдопервого порядка к?. Проще всего это сделать, построив график зависимости
1п[Спирт] от времени.
Построим таблицу с необходимыми данными:
Время, мин
1п[Спирт]
0
0
6,5
-0,40
10
-0,562
15
-0,892
20
-1,204
25
-1,470
30
-1,966
40
-2,659
50
-3,219
График зависимости 1п[Спирт] от времени представляет собой прямую:
40 50 60
Время, мин
Это соответствует интегральному уравнению для реакции первого порядка:
1п[Спирт] = 1п[Спирт]0 - kft
и подверждает, что реакция имеет первый порядок по бензиновому спирту.
Константа скорости реакции псевдопервого порядка определяется из
наклона прямой:
к = 0,066 мин"1, или 1,1 • 10"3 с"1.
Реакции второго порядка по А
Интегральная форма кинетического уравнения для реакции второго порядка
по А имеет вид
1/[А] = 1/[А]0 + */ (10.21)
График зависимости 1/[А] от / представляет собой прямую, свободный член
которой равен 1/[А]0. Константа скорости к определяется по тангенсу угла
наклона прямой.
10.5 Интегральная форма кинетических уравнений 511
Пример 10.5 Реакция между иодом и гексеном-1
Гексен-1 реагирует с l2 в уксусной кислоте с образованием 1,2-дии-
одгексана. Зависимость скорости реакции от [l2] исследована в
условиях большого избытка гексена-1. Нижеприведенные данные
получены при 298 К. Определите порядок реакции по l2 и запишите
кинетическое уравнение реакции. Можно ли на основании
полученных результатов исключить участие уксусной кислоты в
лимитирующей стадии процесса?
Время, с 0 1000 2000 3000 4000 5000 6000 7000 8000
[12],моль/ла 0,02 0,0156 0,0128 0,0109 0,0094 0,0083 0,0075 0,0068 0,0062
аПо данным работы Field К. IV., Wilder D, UtzA., Kolb K.E., Journal of Chemical
Education, 1987, vol. 64, p. 269.
Решение
Кинетическое уравнение реакции можно записать в виде:
-d[I2]/d/ = А:[12]т[Гексен-1 ]"
Но так как гексен-1 присутствует в большом избытке, имеем:
-d[l2]/d/ = ^[l2r
Построим график зависимости [12] от времени
§ 0,02
0,015
0,01
0,005
0 2500 5000 7500
Время, с
Поскольку этот график нелинеен, очевидно, что т * 0, но возможно, что т = 1
или 2. Однако возможность того, что реакция имеет первый порядок,
отбрасывается на основании данных времени полупревращения. Время, необходимое
для того, чтобы исходная концентрация уменьшилась вдвое, составляет -3500 с.
Если бы реакция имела первый порядок по 12, то по прошествии следующих
3500 с концентрация 12 снова уменьшилась бы вдвое. Можно видеть, что это не
так. [Вопрос: Дайте приближенную оценку второго времени полупревращения.]
Далее проверим, подчиняется ли реакция уравнению второго порядка. Из
интегральной формы кинетического уравнения имеем
1/[12] = 1/[121о + ет
поэтому необходимо построить график зависимости 1/[12] от времени.
512
10. Кинетика химических реакций
Время, с
1/[12], л/моль
0
50
1000
64
2000
78
3000
92
4000
106
5000
120
6000
134
7000
148
8000
162
5000 7500
Время, с
График линеен и это подтверждает, что реакция имеет второй порядок по 12.
Следовательно, кинетическое уравнение реакции псевдовторого порядка
записывается в виде
-d[I2]/d/=*-[I2]2
и значение к можно определить по тангенсу угла наклона прямой.
Тангенс угла наклона = к! = 0,014 л/мольс
Участие уксусной кислоты не может быть исключено, хотя она и является
растворителем, так как она присутствует в большом избытке.
ЗАВИСИМОСТЬ СКОРОСТИ РЕАКЦИИ ОТ ТЕМПЕРАТУРЫ:
УРАВНЕНИЕ АРРЕНИУСА
Распределение Больцмана и энергия активации
^споеяеление Больи- ^ид больцмановского распределения молекул по энергиям за-
*шъчг>-г**, noon i iq висит от температуры. На рис. 10.6 показан типичный вид рас-
мэнэ. см. рэзд. ■. iо
пределения для одного и того же образца при двух различных
температурах. Число молекул в образце при нагревании остается
постоянным, поэтому площадь под обеими кривыми должна быть одинакова.
Однако при повышении температуры максимум кривой смещается в сторону
более высоких энергий. Это означает, что с повышением температуры
увеличивается число молекул с более высокой энергией.
Реакция может протекать только при столкновении молекул, обладающих
► определенной минимальной энергией Еа « Аакт#. Для данной
Внутренняя энергия и реакции энергия активации (рис. Ю.7) имеет постоянное зна-
энтальпия: чение; Еа практически не зависит от температуры. На рис. Ю.6
см. разд. 3.5 схематически показана величина Е. Видно, что число молекул,
10.6. Зависимость скорости реакции от температуры 513
Более низкая температура
Более высокая температура
Энергия
Энергия активации
Рис. 10.6. Больцмановское распределение молекул по энергиям при двух различных
температурах. Если число молекул остается неизменным, площади под кривыми
равны. Величина энергии активации реакции не зависит от температуры.
Заштрихованная область под кривой 2 имеет много большую площадь, чем заштрихованная область
под кривой /.
о.
X
Рис. 10.7. Профиль одностадийной
реакции, на котором указана величина Ел.
Реагенты
Продукты
Координата реакции
обладающих этой или более высокой энергией, резко возрастает с ростом
температуры. Это означает, что доля молекул, которые могут принимать
участие в реакции, увеличивается, а следовательно, скорость реакции
возрастает. Как правило, увеличение температуры на 10 К приводит к возрастанию
скорости реакции приблизительно в два раза (ниже это утверждение
обсуждается с критической точки зрения).
Уравнение Аррениуса
Количественная связь между скоростью реакции и температурой задается
соотношением Аррениуса (уравнение 10.22). Следует отметить, что в уравнение
Аррениуса входит энергия активации. В разд. 3.5 обсуждалось различие меж-
Акт"
(10.22)
ду значениями энергии и энтальпии, здесь же принимается, что Е
In к = In Л -EJRT
где Еа измеряется в кДж/моль, к — константа скорости, А — частотный
фактор, R — универсальная газовая постоянная (8,314 • 10~3 кДж/моль-К); Т —
температура (К).
514
10. Кинетика химических реакций
Частотный фактор (предэкспоненциальный множитель) А можно считать
постоянным для данной реакции. Он измеряется в тех же единицах, что и
константа скорости, и связан с частотой столкновений молекул реагентов
между собой и их взаимной ориентацией в пространстве*.
Типичные значения энергий активации для некоторых процессов приведены
в табл. 10.1. Эти данные показывают, что катализатор влияет на величину Еа
химической реакции.
Согласно уравнению Аррениуса
In к = In A - EJRT
где к - константа скорости, А - частотный фактор, R - универсальная
газовая постоянная, Г- температура, Еа измеряется в кДж/моль.
Таблица 10.1. Типичные значения энергии активации. (Обратите внимание, что
наличие катализатора влияет на величину Е)
Реакция
2Н202 -> 2Н20 + 02
2Н202 -> 2Н20 + 02
2NOC1 -> 2NO + С12
N0 + 03 -> N02 + 02
2Н1->Н2 + 12
2Н1->Н2 + 12
2Н1->Н2 + 12
2NH3->N2 + 3H2
2NH3->N2 + 3H2
С2Н5Вг + ОН" -> С2Н5ОН + Вг"
Примечания
Без катализатора
Ферментативный катализ
Без катализатора
Катализатор — золото
Катализатор - платина
Без катализатора
Катализатор — вольфрам
Реакция в водном растворе
Еа, кДж/моль
79
23
100
10,5
185
121
59
335
162
90
Пример 10.6 Определение энергии активации реакции
разложения органического пероксида
Разложение органического пероксида ROOR является реакцией
первого порядка по пероксиду. Константа скорости реакции
зависит от температуры следующим образом:
Температ
*,С"1
ура, К
410
0,0193
417
0,0398
426
0,0830
436
0,2170
Определите энергию активации этой реакции
(Я = 8,314 10-3кДж/мольК)
Величина частотного фактора А связана с энтропией образования активированного комплека AiS*
соотношением: А = ехр[Д*Уу/?7]. Согласно ряду других моделей, частотный фактор зависит от частоты
нулевого колебательного уровня переходного состояния (поэтому он и получил свое название). -
Прим. перев.
10.6. Зависимость скорости реакции от температуры
Решение
Согласно уравнению Аррениуса, зависимость к от Г имеет вид
\nk=\nA-Ea/RT
поэтому необходимо построить график зависимости In А: от \/Т.
515
\/т, к-1
In к
0,00244
-3,948
0,00240
-3,224
0,00235
-2,489
0,00229
-1,528
In А:
0,0023 0,00235 0,0024 0,00245
1/Г, К"1
£а можно определить из тангенса угла наклона прямой:
-EJR = -15960
£а = 15960 • 8,314 • 10"3 = 132,7 кДж/моль
Помимо возможности расчета энергии активации уравнение Аррениуса
позволяет предсказать изменение скорости реакции в зависимости от
температуры. Если предположить, что для выбранной реакции величины А и Еа
постоянны, можно записать уравнение 10.23, которое преобразуется в
уравнение 10.24, где кп — константа скорости реакции при температуре Тп.
In к2 - In кх = [In A -EJRT2\ - [In A -EJRT{[ = (EJR)\\/TX -\/T2] (10.23)
In (*2А,) = (Ea/R)[\/T{ -\/T2] (10.24)
Последнее выражение дает отношение констант k2/kv и соотношение
скоростей реакций можно определить как показано в примере 10.7.
Внимание/ Реакции не обязятельно подчиняются уравнению Аррениуса в
широком интервале температур, поэтому уравнение 10.24 следует применять с
осторожностью.
516
10. Кинетика химических реакций
Пример 10.7. Зависимость скорости реакции от температуры
Энергия активации реакции гидроксид-иона с бромэтаном с
образованием этанола равна 90 кДж/моль. Во сколько раз увеличится
скорость реакции при увеличении температуры от 295 до 305 К?
(Я = 8,314 10~3 кДж/моль К)
Решение
Чем больше значение &, тем выше скорость реакции. Согласно уравнению Ар-
рениуса
\n(k2/kx) = {EJR)[\/Tx-\/T2\
In (k2/kx) = [90/(8,314-10"3)][l/295 -1/305] = 1,20
k2/kx = 3,32
Таким образом, константа скорости возрастает в 3,32 раза при увеличении
температуры от 295 до 305 К.
Информация о кинетическом уравнении отсутствует, но если две реакции
проводятся при одинаковых исходных концентрациях реагентов, отношение
скоростей должно равняться отношению констант скорости:
(Скорость 1)/(Скорость 2) = ^,[С2Н5Вг]т[ОН-]7А:2[С2Н5Вг],"[ОН-]л
В начале данного раздела утверждалось, что повышение температуры на 10 К
приводит к возрастанию скорости реакции приблизительно вдвое. Однако из
уравнения 10.24 можно видеть, что изменение скорости реакции зависит от
величины Еа. На рис. 10.8 показано, как увеличение температуры от 300 до
310 К влияет на скорость реакции при значениях Еа, изменяющихся от 10 до
150 кДж/моль. Увеличение скорости реакции примерно в два раза
наблюдается только для значений Еа ~ 50 кДж/моль; такое значение типично для
многих реакций, протекающих в водных растворах.
6
5
4
3
2
1
0 50 100 150
£а, кДж/моль
Рис. 10.8. При увеличении температуры от 300 до 310 К отношение констант
скоростей k2/k{ меняется в зависимости от величины энергии активации. Примерное
удвоение скорости реакции наблюдается только при значениях Еа ~ 50 кДж/моль (см.
упражнение 10.13 в разд. 10.13).
10.7. Катализ и автокатализ
517
КАТАЛИЗ И АВТОКАТАЛИЗ
Катализатором называется вещество, которое оказывает влияние на скорость
реакции, но само при этом не расходуется. Катализатор может ускорять
реакцию или замедлять ее. Последнее явление известно как отрицательный
катализ. Оно играет важную роль в стабилизации многих
промышленно-важных материалов. Примеры некоторых катализируемых реакций приведены в
табл. 10.1.
Катализатор - вещество, которое ускоряет или замедляет скорость
реакции, но само при этом не расходуется.
Интересное явление наблюдается, когда один из продуктов реакции
способен выступать в роли ее катализатора. Такая реакция называется
автокаталитической. В исходной смеси реакции, протекающей по уравнению 10.25,
присутствуют только реагенты А и В; продукт D катализирует данную
реакцию, и с появлением D (уравнение 10.26) скорость реакции начинает
возрастать.
Исходная реакция: А + В -> С + D (10.25)
D
Автокатализ: А + В > С + D (10.26)
На рис. 10.9 показано изменение концентрации Мп04" в ходе реакции с ди-
метиламином. Вначале реакция развивается относительно медленно, но по
мере накопления продуктов в растворе скорость расходования перманганат-
ионов увеличивается. Данная реакция катализируется ионами Мп2+, которые
образуются при восстановлении перманганат-ионов.
В качестве биохимического примера рассмотрим превращение трипсино-
гена в трипсин, катализируемое трипсином. На рис. 10.10 показан процесс
образования трипсина в ходе реакции как функция времени. S-образная
кинетическая кривая характерна для автокаталитических реакций - по мере
увеличения концентрации катализатора в системе, скорость процесса
возрастает.
Рис. 10.9. Зависимость концентрации
перманганат-ионов от времени в ходе
автокаталитической реакции между Me2NH
и Мп04~, протекающей в водном
растворе. Катализатор реакции — ионы Мп2+.
[Mata-Perez F., Perez-Benito J.F., Journal of
Chemical Education, 1987, vol. 64, p. 925.]
- 4
10 20
Время, мин
10. Кинетика химических реакций
I
s
Рис. 10.10. График зависимости
концентрации трипсина от времени в ходе
реакции гидролиза трипсиногена до
трипсина. Реакция катализируется продуктом.
, S-образный вид кинетической кривой ха-
Время рактерен для автокаталитических
реакций.
ОБРАТИМЫЕ РЕАКЦИИ
До сих пор обсуждались только реакции, протекающие в одном
направлении, но во многих случаях прямая реакция сопровождается обратной
реакцией. В данном разделе кратко рассмотрено кинетическое уравнение для
процесса, состоящего из двух противоположных реакций первого порядка.
Рассмотрим реакцию 10.27, в которой константы скорости прямой и
обратной реакций равны k] и k_] соответственно.
*,
А( 1 В (10.27)
*-.
Измеряемая скорость расходования А не подчиняется простому уравнению
реакции первого порядка, поскольку в ходе процесса образуется В, а из В
снова образуется А по реакции первого порядка. Скорость прямой реакции
определяется уравнением
Скорость реакции = -d[A]/d/= £,[A] - А:_,[В] (10.28)
Предположим, что в исходной смеси присутствует только соединение А,
тогда начальная скорость реакции равна ^j[A]0 (по уравнению 10.8, если
обозначить исходную концентрацию А как [А]0).
Концентрация В в момент времени / равна разности концентраций А в
моменты времени 0 и / (уравнение 10.29). Обратите внимание, что это
соотношение зависит от стехиометрии реакции; в данном случае из одного моля
А образуется один моль В, и наоборот.
[В] = [А]0-[А] (10.29)
Таким образом, можно переписать кинетическое уравнение в виде уравнений
10.30 и 10.31.
Скорость реакции = -d[A]/d/= ^[А] - *_,{[А]0 - [А]} (10.30)
Скорость реакции = -d[A]/d/= (А:, + А;_,)[А] - £_,[А]0 (10.31)
Поскольку [А]0 — постоянная величина, второй член в правой части 10.31
также не меняется во времени. Таким образом, реакция имеет первый поря-
10.8. Обратимые реакции
519
док по А, и график зависимости скорости реакции от [А] представляет собой
прямую. Общий вид уравнения 10.31
у= ах + b
и, следовательно, тангенс угла наклона линейной зависимости скорости от
[А] равен (/:, +£_,). Значение к_х можно определить исходя из величины
свободного члена, поскольку начальная концентрация [А]0 известна. Таким
образом, можно определить обе константы скорости.
Пример 10.8. Обратимая реакция
Ниже приведены экспериментальные данные по реакции:
1?В
*-1
Исходная концентрация А равна 0,05 моль/л. Определите
константы скорости кл\лк_л.
[А], моль/л
Скорость - 104, моль/л-мин
0,045
8,97
0,04
7,94
0,03
5,88
0,02
3,82
0,01
1,76
Данная обратимая реакция подчиняется следующему кинетическому
уравнению:
-d[A]/d/=(A:l+A:_l)[A]-A:_l[A]0
(см. уравнение 10.31), общий вид которого: у = ах + Ь.
График зависимости скорости реакции от [А] представляет собой прямую,
тангенс угла наклона которой а = (£, +/:_,), а свободный член b = -Л:_,[А]0.
10
Р 4
I
о
о L
О 0,01 0,02 0,03 0,04 0,05
(А), моль/л
Тангенс угла наклона а = (Л, + *_,) = 2,06 • 10~2 мин-1.
С помощью экстраполяции определяем свободный член:
b = -£_i[A]0 = 0,3 • 10~4 моль/лмин.
Из значения свободного члена:
*_, = 0,3 • 10-4/[А]0 = 0,3 • 10-4/0,05 = 6 • 10"4 мин"1
520
10. Кинетика химических реакций
Подставляя значение к_х в выражение для тангенса, можно найти кх:
кх = (2,06 • 10"2) - к_х = (2,06 • 1(H) - (6 • 10"4) = 0,02 мин"1
(Обратите внимание, что константа скорости обратной реакции много меньше
константы скорости прямой реакции. Что изменится для случая к_{ > &,?)
НЭ9 МОЛЕКУЛЯРНОСТЬ
Ранее изучались свойства больших ансамблей частиц; в данном разделе
химические реакции рассмотрены на молекулярном уровне, что позволит
обсуждать микроскопический механизм реакции. Следует подчеркнуть важное
обстоятельство — скорость реакции можно измерить экспериментально, и
механизм реакции предлагается на основании этих измерений. Механизм
реакции не может быть «доказан», — в лучшем случае, он находится в согласии с
имеющимися кинетическими данными.
Элементарные стадии реакции
Обсуждение кинетических уравнений базировалось на на двух реакциях,
задаваемых уравнениями 10.10 и 10.11. Во многих случаях полный
реакционный путь от реагентов до продуктов включает несколько стадий, каждая из
которых называется элементарной стадией реакции. Например, цепные
реакции, описанные в разд. 8.11, включают элементарные стадии зарождения,
развития и обрыва цепи. Последовательность элементарных стадий
составляет механизм реакции. Механизм может включать образование одного или
нескольких промежуточных продуктов, как показано на рис. 10.11 для
реакции А -> D.
Молекулярность. Моно- и бимолекулярные реакции
Молекулярностью элементарной стадии реакции называется число молекул или
атомов реагентов, принимающих участие в этой стадии. Большинство
элементарных стадий протекают с участием одной или двух частиц, и их
называют моно- и бимолекулярными стадиями соответственно.
А > в > с > D
Реагент Промежуточ- Промежу- Конечный
ный продукт точный продукт
с*
S
с
JQ
X
(Г)
Координата реакции
Рис. 10.11. Профиль реакции А —> D. Механизм включает три элементарные стадии,
протекающие с образованием двух промежуточных продуктов В и С.
продукт
10.10. Микроскопический механизм реакции
521
Молекулярность элементарной стадии реакции - число молекул или
атомов реагентов, принимающих участие в этой стадии реакции.
15и 17
Уравнения 10.32 и 10.33 - примеры мономолекулярных реакций. Первая из
них представляет собой реакцию разложения (за счет гомолитического
разрыва связи), а вторая — реакцию изомеризации.
Вг2->2Вг* • (10.32)
С1 Н Н Н
с=с > с=с о°-33)
/ \ / \
Н С1 С1 С1
В случае бимолекулярной стадии, происходит столкновение двух частиц с
образованием продуктов. Реагирующие частицы могут быть одинаковы, а могут
отличаться друг от друга:
2N02->N204 (10.34)
СН3СН2Вг + ОН" -> СН3СН2ОН + Вг" (10.35)
►" В разд. 10.10 обсуждается взаимосвязь между элементарными
Механизмы реакций: стадиями и суммарным механизмом реакции, а в разд. 10.11
см. также гл. 14, рассмотрена реакция между Н2 и Вг2, включающая по меньшей
мере пять элементарных стадий.
Общее кинетическое уравнение
Для некоторой реакции одна из элементарных стадий является медленной
(или лимитирующей). Молекулярность лимитирующей стадии (ЛС)
определяет наблюдаемую кинетику всей реакции в целом. Именно поэтому
множители, присутствующие в кинетическом уравнении, могут не соответствовать
стехиометрии многостадийной реакции.
Во многих случаях кинетические уравнения имеют сложный вид из-за
того, что частицы, участвующие в лимитирующей стадии, представляют собой
промежуточные продукты, а не исходные реагенты. Как правило, скорость
реакции выражают через концентрации исходных веществ или продуктов, а
не через концентрации промежуточных продуктов. Пример сложной
реакции такого типа обсуждается в разд. 10.11.
МИКРОСКОПИЧЕСКИЙ МЕХАНИЗМ РЕАКЦИИ
И ПРИБЛИЖЕНИЕ СТАЦИОНАРНОГО СОСТОЯНИЯ
Итак, элементарные стадии играют большую роль в формировании
механизма реакции. В данном разделе обсуждается вывод кинетического уравнения
многостадийной реакции, выраженного через измеряемые концентрации
исходных реагентов или продуктов реакции.
522
10. Кинетика химических реакций
Кинетическое уравнение для моно- и бимолекулярных стадий
Ранее в данной главе обсуждалось использование экспериментальных
данных для определения общего кинетического уравнения (см. разд. 10.4). Также
было показано, что суммарная реакция складывается из элементарных
стадий, которые представляют собой моно- или бимолекулярные реакции.
Целью кинетических исследований является установление механизма реакции.
Для решения этой проблемы вначале записывают предполагаемый
механизм, т. е. последовательность элементарных стадий, а затем выводят
кинетическое уравнение, соотвествующее этому механизму. Если кинетическое
уравнение находится в согласии с экспериментальными данными,
предложенный механизм, возможно, верен (хотя доказать этого нельзя).
Необходимо обратить особое внимание на следующее ключевое
обстоятельство. Хотя кинетическое уравнение многостадийной реакции нельзя
вывести исходя из ее стехиометрии, кинетическое уравнение элементарной
стадии должно соответствовать стехиометрии этой стадии.
Кинетическое уравнение элементарной стадии реакции
соответствует стехиометрии этой стадии, но общее кинетическое уравнение
многостадийной реакции нельзя вывести исходя из стехиометрии этой
многостадийной реакции.
В случае мономолекулярной стадии, например при распаде С12
(уравнение 10.36), скорость реакции прямо пропорциональна концентрации
реагента, так как в этой стадии участвует одна молекула реагента. Распад С12
происходит при возбуждении молекулы (термическом или фотолитиче-
ском), сопровождаемом разрывом связи CI—C1. Скорость реакции распада
С12 с образованием двух радикалов задается уравнением 10.37
С12->2СГ (10.36)
Скорость реакции = к[С\2] (10.37)
В случае бимолекулярной стадии реакция происходит при столкновении двух
молекул, и, следовательно, скорость реакции зависит от концентрации обоих
веществ. Пример бимолекулярного процесса - димеризация N02. Скорость
димеризации описывается уравнением 10.38. Обратите внимание на то, что
квадратичная зависимость означает участие двух молекул в этом процессе.
Скорость реакции = &[N02]2 (10.38)
Если в бимолекулярной реакции участвуют две различные молекулы, это
отражается при записи кинетического уравнения. Например, при
столкновении радикала кислорода с молекулой озона образуется молекулярный
кислород (уравнение 10.39), и скорость этого процесса зависит и от [0#], и от [03]
(уравнение 10.40).
0#+03->202 (10.39)
Скорость реакции = А:[0*][03]
(10.40)
10.10. Микроскопический механизм реакции
523
Для мономолекулярной стадии А -> В: Скорость реакции = /с[А].
Для бимолекулярной стадии 2А —> В: Скорость реакции = /с[А]2.
Для мономолекулярной стадии А + В -* С: Скорость реакции = /с[А][В].
Общее кинетическое уравнение для двухстадийной реакции
Выше подразумевалось, что реакция включает лишь одну элементарную
стадию. Что произойдет, если реакция содержит более одной стадии?
Рассмотрим реакцию А —> С, протекающую с образованием промежуточного
соединения В; каждая из стадий характеризуется собственной константой
скорости, как показано в уравнениях 10.41 и 10.42.
А ■—>В (10.41)
В —>С (10.42)
Рассмотрим следствия из возможных соотношений между к{ и к2.
Случай 1: кл « к2
В этом случае скорость образования В из А много меньше, чем скорость
превращения В в С. Это означает, что как только соединение В образуется, оно
сразу же переходит в С. Общая скорость реакции определяется лимитирую-
^- щей стадией (ЛС), т. е. реакцией А —> В
Величина к:
см. разд. 10.2
Скорость реакции = к] [А] (10.43)
Случай 2: кл » к2
В этом случае лимитирующей стадией является образование соединения С из
промежуточного продукта В, поэтому скорость реакции определяется
уравнением 10.44.
Скорость реакции = £,[В] (10.44)
Теперь возникает проблема. «Теоретическая» скорость реакции выражается
через концентрацию В, тогда как в эксперименте измеряются концентрации
исходного реагента А или конечного продукта С. Как правило, прямое
наблюдение за изменением концентрации промежуточного продукта во времени не
представляется возможным. Во многих случаях промежуточные продукты
присутствуют в исчезающе малых концентрациях и не могут быть
зафиксированы. Это обстоятельство используется в подходе, известном как приближение
стационарного состояния. В этом случае предполагается, что в ходе реакции
концентрация В большую часть времени поддерживается на постоянном,
стационарном уровне (уравнение 10.45). Очевидно, это приближение не
справедливо в самом начале реакции, как показано на рис. 10.12.
d[B]/d/=-d[B]/d/ = 0 (10.45)
524
10. Кинетика химических реакций
Время
Рис. 10.12. Реакция А -» С
протекает с образованием промежуточного
продукта В. Приближение
стационарного состояния предполагает, что
концентрация В поддерживается на
постоянном, стационарном уровне. Это
предположение не выполняется в
начале реакции.
Приближение стационарного состояния утверждает, что
концентрация промежуточного продукта В в основном постоянна в ходе
реакции.
d[B]/df=-d[B]/df=0
Как можно использовать это приближение? Промежуточное вещество В
образуется из А в реакции с константой скорости к{ и расходуется с
образованием С в реакции с константой к2. Таким образом, скорость образования В
можно записать в виде уравнения 10.46, преобразование которого позволяет
выразить концентрацию промежуточного продукта В (уравнения 10.47 и 10.48).
d[B]/d/= *,[А] - А:2[В] = 0 (10.46)
*,[А] = А:2[В] (10.47)
[В] = *,[А]Л2 (10.48)
Из уравнения 10.44 следует, что скорость реакции зависит от концентрации
В. Таким образом, объединяя уравнения 10.44 и 10.48, можно получить новое
выражение для скорости реакции:
Скорость реакции = А:2[В] = к2(к{[К\/к2) = к{[А] (10.49)
Важное свойство этого уравнения состоит в том, что оно содержит
экспериментально измеряемую концентрацию реагента А.
Общее кинетическое уравнение для реакции
с конкурирующими стадиями
Рассмотрим двухстадийное образование С из А, механизм которого усложнен
обратной реакцией образования А из промежуточного продукта В (уравнения
10.50—10.52). Элементарные стадии 10.51 и 10.52 называют конкурирующими
стадиями.
10.11. Радикальные цепные реакции
525
*1
*2
->В (10.50)
->С (10.51)
В !->А (10.52)
Скорость реакции можно определить экспериментально, наблюдая за
образованием С (уравнение 10.53) или за расходованием А; здесь
рассматривается только первый случай.
Скорость реакции = d[C]/d/= к2[В] (10.53)
Приближение стационарного состояния позволяет приравнять к нулю
скорость изменения [В] (уравнение 10.54) и выразить концентрацию В через
экспериментально измеряемую концентрацию А (уравнения 10.55 и 10.56).
d[B]/d/ = А:,[А] - *2[В] - А:_,[В] = 0 (10.54)
*,[А] = *2[В] + *_,[В] = (к2 + *_,)[В] (10.55)
т^к^Щ + к^) (10.56)
Объединяя уравнения 10.53 и 10.56, можно получить общее кинетическое
уравнение 10.57, которое показывает, что скорость реакции зависит только от
[А]. Однако следует отметить, что константа скорости к* включает
составляющие из всех трех элементарных стадий.
Скорость реакции = d[CJ/d/= *2*,[А]/(*2 + *_,) = К [А] (10.57)
где*' = *2*|/(*2 + *-1)-
РАДИКАЛЬНЫЕ ЦЕПНЫЕ РЕАКЦИИ
Кинетика радикальных цепных реакций довольно сложна, но данный вопрос
представляет существенный практический интерес. Примерами радикаль-
►" ных цепных реакций могут служить химические реакции в пла-
Радикальные цепные мени, в атмосфере, а также взрывы. В данном разделе показана
реакции: сложная природа реакций, протекающих с участием радика-
см. разд. 8.11 лов
Неразветвленные цепные реакции
Цепная реакция включает стадии зарождения и развития цепи, на которых
происходит образование радикалов, а также стадию обрыва цепи, на которой
радикалы удаляются из системы. Если во время реакций развития цепи
Не происходит увеличения общего количества радикалов, то цепная реакция
называется неразветвленной (см. уравнение 10.58).
СН3* + С12 -> СН3С1 + С1*
(10.58)
526
10. Кинетика химических реакций
В неразветвленной цепной реакции количество радикалов не
увеличивается.
Разветвленные цепные реакции
В некоторых радикальных реакциях на стадии развития цепи может
происходить увеличение числа радикалов. Уравнение 10.59 иллюстрирует реакцию
между радикалом Н* и молекулой кислорода, в которой образуются радикал
гидроксила и атом кислорода (бирадикал)*.
Н# + 0,->ОН* + #0#
(10.59)
Процессы такого рода приводят к разветвлению цепи, так как увеличение
числа радикалов в системе означает увеличение числа возможных
радикальных стадий. Скорость реакции резко возрастает, в результате чего может
произойти взрыв. Примерами разветвленных цепных реакций могут служить
газофазное окисление углеводородов (горение) и реакция между Н2 и 02 с
образованием воды, инициируемая искрой.
В разветвленной цепной реакции количество радикалов
увеличивается входе реакции.
Различие между неразветвленными и разветвленными цепными реакциями
проиллюстрировано на рис. 10.13.
Рис. 10.13. Различие между неразветвленной и разветвленной цепными реакциями:
а — неразветвленная цепная реакция, в которой на каждой стадии развития цепи
расходуется и образуется по одному радикалу; б — разветвленная цепная реакция,
в которой на каждой стадии (после образования первичного радикала) образуется
два радикала.
Строго говоря, данная реакция не приводит к разветвлению цепи, так как молекула кислорода 02
также является бирадикалом. — Прим. перев.
10.11 Радикальные цепные реакции
527
Реакция между водородом и бромом
Газообразные Н2 и Вг2 реагируют при 500 К с образованием бромоводорода.
Эта реакция протекает по неразветвленному цепному механизму. Стадии
зарождения, развития и обрыва цепи иллюстрируются уравнениями
10.60-10.62 и 10.64. Реакция 10.63 называется стадией ингибирования, так
как при этом расходуется часть конечного продукта НВг, хотя и образуется
новый радикал. Обратите внимание, что реакция 10.63 обратна реакции
развития цепи 10.61, а стадия обрыва цепи 10.64 обратна стадии зарождения
цепи 10.60.
к.
(10.60)
(10.61)
(10.62)
(10.63)
(10.64)
Этот механизм находится в согласии с экспериментально наблюдаемой
кинетической зависимостью, подчиняющейся уравнению 10.65, вывод
которого исходя из указанных элементарных стадий с помощью приближения
стационарного состояния приводится в дополнении 10.4.
(к \- -
[-ПВг2]2[Н2]
к_х ) L 2J L 2J
Скорость реакции = = (10.65)
d/ A:3[Br2] + А:_2[НВг]
В данной реакции возможны и другие элементарные стадии, например,
стадия зарождения цепи за счет гомолитического разрыва связи Н—Н в Н2 или
стадия ингибирования по реакции НВг с радикалом Вг*. Однако
соответствие между уравнением 10.65 и экспериментальными данными указывает на
то, что эти стадии не играют существенной роли.
вг2 L_
Rr* 4- Н
Н* + Вг2-
НВг+Н#-
* i
2Вг#
-> 2Вг#
к2
v uDr _i_ Н*
*3
— > НВг+Вг#
i-> Вг* + Н2
—-> Вг2
Дополнение 10.4. Использование приближения стационарного состояния
Элементарные стадии 10.60—10.64 составляют неразветвленный цепной механизм
реакции между Н2 и Вг2 с образованием НВг. Кинетическое уравнение, согласующееся с
экспериментальными данными, можно вывести как это показано ниже.
Н Вг образуется в реакциях 10.61 и 10.62 и расходуется в реакции 10.63.
Следовательно, скорость образования НВг записывается следующим образом:
d[HBr]/d/= *2[Вг-][Н2] + *3[Н-][Вг2] - *_2[Н-][Вг-] (I)
Согласно приближению стационарного состояния:
d[Br']/d/= d[H"|/d/= 0 (См_ продолжение)
528 10. Кинетика химических реакций
(Продолжение)
d[Br*]/d/ = 2*,[Вг2] - *2[Вг#][Н2] + *3[Н#][Вг2] + ^_2[Н*][НВг] - 2А:_,[Вг#]2 = О (II)
и
d[H*]/d/= *2[Вг*][Н2] - *3[Н-][Вг2] - *_2[Н*][НВг] = О (III)
Сложение уравнений (II) и (III) приводит к сокращению трех членов, и можно записать:
2*,[Br2]-2*_i[Br#]2 = 0
Следовательно:
[Вг-]2 = 2Л1[Вг21/2А:_1
[Br4 = <V*-i>l/2lBr2ll/2 (iv)
Из уравнения (II) можно выразить [Н*]:
[НЧ = *2[Вг*][Н2]/(Л:з[Вг2] +*_2[НВг])
Подставляя в это выражение [Вг#] из уравнения (IV) получаем:
[Н-] = к2(кх/к_х)^ [Вг2]1/2[н2]/(А:з[Вг2] +*_2[НВг]) (V)
Теперь подставим выражения для [Вг*] и [Н#] из уравнений (IV) и (V) в уравнение (I).
Множитель [Н#] в двух последних членах уравнения (I) можно вынести за скобки, тогда:
d[HBr]/d/= А:2[Вг#][Н2] + *3[Н#]{*3[Вг2] ~ *-2[НВг]> =
[Н2][Вг2]
2
= к2 М- Р[Вг2]2[Н2] + L__i {А:3[Вг2] - *_2[НВг]}
\ -1 / ^ гаг 1 -I- ^ ri-mri
= *.■-''&
^т;
1 ' *3[Br2l + к_2 [HBr]
WAHl(l+^Br2] + ^[HBr]|
- u ( *« Рп»г Ан 1 ^[Br2l + *-2 [НВг1 + *з [BrJ - *-2 [НВг]
- к2 ^—] [Вг2] [Н2] ______
- * ( *• РгВг Лн 1 2^[Вг2]
-*2trr)[Br2|[HJ ,з[Вг2] + ,2[НВг]
1 Urn. i2r
A:3[Br2] + А:_2 [НВг]
Итоговое кинетическое уравнение можно переписать в более простой форме:
d[HBr]/d/= fc[Br2]3/2[H2]/([Br2] +^[HBr])
где к = 2к2(кх/к_{)^2 и к! = £_2Д3.
10.12. Заключение
529
ЩЦа ЗАКЛЮЧЕНИЕ
В данной главе обсуждается ряд вопросов кинетики химических реакций и вводятся основные
уравнения. Важнейшая часть данной главы - проработка решенных примеров, поскольку приобретение
навыков обработки экспериментальных данных, получаемых при изучении скоростей химических
реакций, играет ключевую роль в понимании данной темы. Следует отметить, что во многих случаях
существует несколько возможных путей решения той или иной проблемы. Например, чтобы доказать, что
реакция А -> В имеет первый порядок, можно использовать следующие подходы:
• использовать метод времени полупревращения;
• использовать кинетическое уравнение в интегральной форме (проверить линейность графика
зависимости In [А] от времени);
• построить касательные к графику зависимости [А] от времени и проверить линейность графика
зависимости скорости реакции (изменение тангенсов углов наклона касательных) от [А].
Знаете ли вы, что означают следующие термины?
• Скорость реакции •
• Лимитирующая стадия •
• Кинетическое уравнение •
• Константа скорости •
• Порядок реакции •
• Время полупревращения •
• Период полураспада •
• Псевдопервый порядок •
• Интегральная форма кине- •
тического уравнения •
Теперь вы должны уметь:
Распределение Больцмана
Энергия активации
Уравнение Аррениуса
Катализатор
Автокатализ
Элементарная стадия
Молекулярность
Механизм реакции
Мономолекулярная стадия
Бимолекулярная стадия
Приближение
стационарного состояния
Цепная реакция
Неразветвленная цепная
реакция
Разветвленная цепная
реакция
объяснить, что такое скрость реакции и
кинетическое уравнение реакции;
различать порядок реакции по некоторому
реагенту и общий порядок реакции;
записать дифференциальные формы
кинетического уравнения реакции А -> Продукты для
случаев реакций нулевого, первого и второго
порядков по А;
объяснить, почему единицы измерения константы
скорости реакции не всегда одинаковы;
записать (или вывести) интегральные формы
кинетического уравнения реакции А -> Продукты
для случаев реакций нулевого, первого и
второго порядков по А;
обрабатывать экспериментальные данные для
определения нулевого, первого или второго
порядка реакции по определенному реагенту;
определять константы скорости реакций из
экспериментальных данных;
описать способы проведения эксперимента в
условиях псевдо-п-го порядка и понимать, для
чего необходимы такие условия;
различать константу скорости реакции псевдо-
п-го порядка и общую константу скорости
реакции; уметь определять их из
экспериментальных данных;
вывести дифференциальную форму
кинетического уравнения для простой обратимой
реакции;
объяснить, почему скорость реакции зависит от
температуры;
записать уравнение Аррениуса;
использовать зависимость константы скорости
реакции от температуры для определения
энергии активации реакций;
кратко объяснить, почему катализатор
изменяет скорость реакции;
объяснить, что означает понятие «автокатализ»;
обсудить значение терминов «элементарная
стадия», «мономолекулярная стадия» и
«бимолекулярная стадия»;
записать кинетические уравнения для моно- и
бимолекулярной стадий;
связывать последовательность (двух или трех)
элементарных стадий в единый механизм
реакции и использовать детали механизма для
вывода общего кинетического уравнения;
различать неразветвленные и разветвленные
цепные реакции;
обсудить применимость приближения
стационарного состояния и объяснить, для чего оно
необходимо;
связывать скорость простой цепной реакции
с ее механизмом (и, возможно, уметь
выводить общее кинетическое уравнение для этих
случаев).
530
10. Кинетика химических реакций
ВЯЕ УПРАЖНЕНИЯ
10.1. а) Данные на рис. 10.2,а относятся к
реакции А —> Продукты. Определите
скорость этой реакции, б) Рис. 10.2,5
соответствует другой реакции
А -> Продукты, в которой скорость
зависит от времени. Определите
скорость реакции в моменты времени г,
и /2, указанные на графике.
10.2. Для реакции А —> Продукты,
имеющей нулевой порядок по А,
схематически изобразите а) зависимость
скорости реакции от времени;
б) зависимость [А] от времени.
Повторите упражнение для реакций
первого и второго порядка по А.
10.3. Докажите, что для реакции
А -> Продукты, имеющей первый
порядок по А, константа скорости имеет
размерность с-1. Докажите, что для
реакции второго порядка по А константа
скорости имеет размерность л/мольс
10.4. Что означают следующие термины:
а) скорость реакции; б)
дифференциальная и интегральная форма
кинетического уравнения; в) порядок
реакции (как общий, так и порядок
по данному реагенту); г) константа
скорости реакции, константа
скорости псевдо-л-го порядка и общая
константа скорости; д) энергия
активации; е) катализ и автокатализ;
ж) мономолекулярная реакция;
з) бимолекулярная реакция.
10.5. Железо(Ш) окисляет иодид-ионы по
следующему уравнению:
2Fe3+ +21" -> 2Fe2+ + I2
Запишите кинетическое уравнение
этой реакции, если удвоение
концентрации иодид-ионов приводит к
увеличению скорости реакции в
четыре раза, а удвоение концентрации
железа(Ш) приводит к удвоению
скорости реакции.
10.6. 2-Хлор-2-метилпропан реагируете
водой по уравнению
Ме3СС1 + Н20 -> Ме3С(ОН) + НС1
Изменения концентрации Ме3СС1 в
ходе эксперимента, проводимого
при 285 К, приведены в таблице.
[Allen A., Haughey A.J., Hernandez У.,
Ireton S., Journal of Chemical
Education, 1991, vol. 68, p.609.]
Время, с [Me3CClJ-104, моль/л
20 2,29
25 1,38
30 0,90
34,5 0,61
39,5 0,42
44,5 0,26
а) Определите порядок этой реакции
по Ме3СС1.
б) Вычислите константу скорости в
предположении, что кинетическое
уравнение имеет вид
Скорость = А:[Ме3СС1]".
в) Означает ли уравнение,
приведенное в п. (б), что в лимитирующей
стадии участвует только Ме3СС1?
10.7. Кинетика реакции между алкенами и
12 зависит от природы алкена и
растворителя. Приведенные ниже
данные относятся к реакции пентена-1 с
12 в двух различных растворителях
[Field K.W., Wilder D , Viz A., Kolb K.E.,
Journal of Chemical Education, 1987,
vol. 64, p. 269]; в обоих случаях алкен
присутствует в большом избытке.
Определите порядок по иоду и
константы скорости псевдо-л-го порядка
для обеих реакций.
Реакция I: растворитель — 1,2-ди-
хлорэтан.
Время, с [IJ, моль/л
~~¥ ~о7оУоо
1000 0,0152
2000 0,0115
3000 0,0087
4000 0,0066
5000 0,0050
6000 0,0038
7000 0,0029
8000 0,0022
10.13. Упражнения
531
Реакция II: растворитель — уксусная
кислота.
Время, с
О
1000
2000
3000
4000
5000
6000
7000
8000
[12], моль/л
""одшГ
0,0163
0,0137
0,0119
0,0105
0,0093
0,0084
0,0077
0,0071
10.8. Хлорид фенилдиазония гидролизует-
ся по уравнению:
+ на + N
Скорость реакции можно
исследовать по скорости образования азота.
Объем выделившегося азота
позволяет определить количество
исходного реагента, израсходованного в
процессе реакции. С
использованием приведенных данных определите
порядок реакции по хлориду
фенилдиазония. Реакция проводится в
водном растворе. Общий объем
азота, выделившегося за время реакции,
равен 40 мл.
Время, мин
0
5
10
20
30
40
50
60
Объем N2>
0
8,8
15,7
25,3
31,1
34,5
36,7
38,0
10.9. Ниже приведена реакция димериза-
ции циклического диенона I:
о
Соединение I поглощает свет при
460 нм (емакс = 225 л/мольсм).
Скорость реакции исследовали методом
колориметрии (длина пути луча в
кювете = 1 см). С использованием
приведенных данных по оптической
плотности определите: а) порядок
реакции по I; б) константу скорости.
Время, мин
0
5
10
20
30
35
45
55
75
120
Оптическая плотность0
1,20
1,10
1,00
0,81
0,72
0,67
0,59
0,54
0,45
0,33
а Пл данным работы Weiss H.M., Touchette К.,
Journal of Chemical Education, 1990, vol. 67, p. 707.
10.10.Фенолфталеин широко используется
как кислотно-основной индикатор.
В области рН < 8 он бесцветен, а его
формулу условно можно представить
как Н2Р При более высоких
значениях рН он приобретает красную
окраску и присутствует в форме [Р]2~.
В сильнощелочных растворах
интенсивность окраски уменьшается из-за
протекания реакции:
Р2- + ОН" -> РОН3"
Исследование кинетики этой
реакции методом колориметрии (длина
пути луча в кювете = 1 см)
позволило получить следующие данные
[Nicholson L., Journal of Chemical
Education, 1989, vol. 66, p. 725]:
Эксперимент 1: [OH~] = 0,2 моль/л
Время, мин Оптическая плотность
0 0,560
1 0,454
2 0,361
3 0,292
4 0,228
532
10. Кинетика химических реакций
Эксперимент 2: [ОН~] = 0,1 моль/л
Время, мин Оптическая плотность
0,560
0,449
0,361
0,301
0,247
Эксперимент 3: [ОН-] = 0,05 моль/л
Время, мин
Оптическая плотность
0
3
6
9
12
0,560
0,468
0,415
0,368
0,313
а) Почему было проведено более
одного эксперимента и почему
именно три, а не два?
б) Каков порядок реакции по Р2~?
в) Каков порядок реакции по гидро-
ксид-иону?
г) Какие еще данные необходимы
для определения общей константы
скорости реакции?
10.11. Какие из следующих параметров
будут меняться для реакции
А —> В + С при повышении
температуры от 290 до 320 К:
а) скорость реакции; б) константа
скорости; в) £а; г) [А]0 - [А]?
10.12.Пероксид водорода разлагается по
уравнению 2Н202 -> 2Н20 + 02.
Константа скорости реакции зависит
от температуры следующим образом:
Температура, К к, с'1
295 4,93 • 10"4
298 6,56 • Ю-4
305 1,40 Ю-3
310 2,36 Ю-3
320 6,12 Ю-3
Определите энергию активации этой
реакции.
10.13. На рис. 10.8 показано, как меняется
отношение к2/к1 при изменении
энергии активации реакции в случае
увеличения температуры от 300 до
310 К. Определите отношения к2/кх
для значений Ег 10, 30, 50, 70, 90,
110, 130 и 150 кДж/моль для
изменения температуры а) от 320 до 330 К и
б) от 420 до 430 К. Постройте
графики зависимостей к2/кх от £а. В чем
существенное отличие графиков,
построенных вами, от рис. 10.8?
Прокомментируйте утверждение:
«при повышении температуры на 10
К скорость реакции увеличивается
приблизительно в два раза» с
критической точки зрения.
10.14.а) Кратко объясните, что вы
понимаете под приближением
стационарного состояния и укажите условия
его применимости.
б) Для решения этого подпункта
необходимо проработать дополнение
10.4. Механизм разложения озона
можно представить следующим
образом:
О,
-» 02 + О*
v-l
*о
з
>20,
02 +0* -
03 + 0#
С использованием приближения
стационарного состояния покажите,
что общее кинетическое уравнение
этой реакции задается выражением:
Скорость реакции = -d[03]/d/ =
2*1*2[03]2/(*_1[021 + А:2[Оз])
СОДЕРЖАНИЕ
Предисловие редактора перевода 5
Предисловие авторов к русскому изданию 6
Предисловие 7
Список обозначений 9
ОСНОВНЫЕ ПОНЯТИЯ
1.1 Что такое химия и почему она важна 11
1.2 Что такое ИЮПАК? 12
1.3 Единицы СИ 13
1.4 Протоны, электроны и нейтроны 16
1.5 Химические элементы 17
1.6 Состояние вещества 19
1.7 Атомы и изотопы 21
1.8 Моль и постоянная Авогадро 25
1.9 Газовые законы и идеальные газы 25
1.10 Периодическая система химических элементов 30
1.11 Радикалы и ионы 32
1.12 Молекулы и химические соединения: образование связи 34
1.13 Молекулы и химические соединения: относительная молекулярная
масса и число молей 35
1.14 Концентрации растворов 36
1.15 Стехиометрия реакции 37
1.16 Окисление, восстановление и степень окисления 38
1.17 Термохимия 41
1.18 Больцмановское распределение энергий молекул 45
1.19 Термодинамическая и кинетическая стабильность 46
1.20 Энтальпии плавления и испарения 47
1.21 Межмолекулярные взаимодействия 47
1.22 Константы равновесия и принцип Ле Шателье 48
1.23 Эмпирические, молекулярные и структурные формулы 52
1.24 Номенклатура химических соединений 53
1.25 Заключение 59
1.26 Упражнения 59
534
Содержание
АТОМЫ И АТОМНАЯ СТРУКТУРА
2.1 Значение электронов 62
2.2 Классическое описание строения атома 62
2.3 Классическая модель строения атома - модель Бора 63
2.4 Квант 65
2.5 Корпускулярно-волновой дуализм 65
2.6 Принцип неопределенности 66
2.7 Уравнение Шредингера 67
2.8 Плотность вероятности 70
2.9 Функция радиального распределения 4nr2R(r)2 71
2.10 Квантовые числа 72
2.11 Атомные орбитали 75
2.12 Типы орбиталей и главное квантовое число 79
2.13 Подробнее о функции радиального распределения 80
2.14 Решение уравнения Шредингера для атома водорода 82
2.15 Проникновение орбитали к ядру и экранирование 84
2.16 Атомный спектр водорода и правила отбора 87
2.17 Многоэлектронные атомы 91
2.18 Принцип последовательного заполнения наинизших орбиталей 92
2.19 Электронная конфигурация 94
2.20 Правило октета 97
2.21 Одноатомные газы 98
2.22 Периодичность 103
2.23 Заключение 105
2.24 Упражнения 106
ГОМОЯДЕРНАЯ КОВАЛЕНТНАЯ СВЯЗЬ
3.1 Введение 107
3.2 Измерение межъядерных расстояний 109
3.3 Ковалентный радиус атома 113
3.4 Энергия связи. Образование двухатомной молекулы Н2 115
3.5 Энергия связи 118
3.6 Стандартная энтальпия атомизации элемента 121
3.7 Определение энтальпии связей по стандартным теплотам
образования 124
3.8 Природа ковалентной связи в Н2 126
3.9 Структуры Льюиса для молекулы водорода 126
3.10 Задача описания электронов в молекулах 128
3.11 Теория валентных связей 129
3.12 Метод молекулярных орбиталей 131
3.13 Какие свойства молекулы Н2 могут быть предсказаны теорией
валентных связей и методом молекулярных орбиталей? 137
Содержание
535
3.14 Гомоядерные двухатомные молекулы s-элементов -
элементов второго периода 138
3.15 Перекрывание р-орбиталей 140
3.16 Порядок связи 142
3.17 Взаимосвязь между кратностью связи,ее длиной и энергией
разрыва 143
3.18 Гомоядерные двухатомные молекулы р-элементов второго периода:
F2n02 143
3.19 Смешивание орбиталей и о-л-взаимодействие 147
3.20 Гомоядерные двухатомные молекулы р-элементов второго периода:
В2, С2 и N2 150
3.21 Периодические закономерности в свойствах двухатомных
гомоядерных молекул элементов второго периода 151
3.22 Двухатомные частицы 02, [02]+, [02]" и [02]2_ 156
3.23 Двухатомные гомоядерные молекулы. Изменение свойств
по группам 157
3.24 Заключение 162
3.25 Упражнения 163
ГЕТЕРОЯДЕРНЫЕ ДВУХАТОМНЫЕ МОЛЕКУЛЫ
4.1 Введение 165
4.2 Структуры Льюиса для HF, LiF и LiH 166
4.3 Применение теории валентных связей для объяснения образования
молекул HF, LiF и LiH 166
4.4 Применение метода молекулярных орбиталей для описания
связывания в гетероядерных двухтомных молекулах 169
4.5 Применение метода молекулярных орбиталей для описания
образования связи в LiH, LiF и HF 171
4.6 Энтальпии диссоциации гетероядерной связи 176
4.7 Электроотрицательность по Полингу 179
4.8 Зависимость электроотрицательности от степени окисления и
порядка связи 181
4.9 Обзор связывания в HF: выводы 182
4.10 Другие шкалы измерения электроорицательности 182
4.11 Полярные двухатомные молекулы 184
4.12 Изоэлектронные частицы 186
4.13 Описание образования связи в СО при помощи структур Льюиса и
теории валентных связей 188
4.14 Описание образования связи в моноксиде углерода методом
молекулярных орбиталей 190
4.15 Ионы CN" и NO+, изоэлектронные СО 194
4.16 Частицы NO+, NO, NO" 196
4.17 Заключение 197
4.18 Упражнения 198
536
Содержание
МНОГОАТОМНЫЕ МОЛЕКУЛЫ:
ФОРМА И ХИМИЧЕСКАЯ СВЯЗЬ
5.1 Введение 199
5.2 Геометрия трехатомных молекул 202
5.3 Молекулы с числом атомов более трех линейной или
угловой геометрии 206
5.4 Геометрия молекул, содержащих р-элементы второго периода 207
5.5 р-Элементы третьего, четвертого и пятого периодов 210
5.6 Модель отталкивания электронных пар валентной оболочки
(теория Гиллеспи) 214
5.7 Недостатки теории Гиллеспи 222
5.8 Модель Кеперта 224
5.9 Применение модели Кеперта 225
5.10 Исключение из модели Кеперта: плоскоквадратная геометрия 227
5.11 Геометрическая изомерия 228
5.12 Две близкие по энергии структуры: тригональная бипирамида и
квадратная пирамида 233
5.13 Форма и дипольные моменты молекул 236
5.14 Углерод имеет лишь три типа координационного окружения 240
5.15 Форма молекул и правило октета 242
5.16 Расширение октета 246
5.17 Теория валентных связей: резонансные структуры 247
5.18 Теория валентных связей и гибридизация 250
5.19 Гибридизация и форма молекул, содержащих р-элементы 255
5.20 Гибридизация: роль негибридных орбиталей 257
5.21 Теория молекулярных орбиталей и многоатомные молекулы 261
5.22 Сравнение результатов теории ВС и метода МО при описании
связывания в молекуле метана 264
5.23 Заключение 265
5.24 Упражнения 266
ИОНЫ
6.1 Введение 267
6.2 Карты электронной плотности 269
6.3 Энергия ионизации 272
6.4 Тенденции в изменении энергий ионизации 273
6.5 Сродство к электрону 279
6.6 Электростатические взаимодействия между ионами 282
6.7 Ионные решетки 285
6.8 Структурный тип хлорида натрия (каменной соли) 287
6.9 Определение стехиометрии соединения по строению элементарной
ячейки на примере NaCI 288
Содержание
537
6.10 Структурный тип хлорида цезия 291
6.11 Структуный тип фторида кальция (флюорита) 291
6.12 Структурный тип рутила 293
6.13 Структурные типы сульфида цинка(И) 294
6.14 Размеры ионов 296
6.15 Энергия решетки: чисто ионная модель 302
6.16 Энергия решетки: экспериментальные данные 307
6.17 Сравнение значений энергии решетки, определенных по уравнению
Борна-Ланде и по циклу Борна-Габера 308
6.18 Поляризация ионов 310
6.19 Заключение 311
6.20 Упражнения 311
СТРУКТУРА ТВЕРДЫХ ПРОСТЫХ ВЕЩЕСТВ
7.1 Введение 313
7.2 Плотнейшие шаровые упаковки 313
7.3 Простая и объемноцентрированная кубические шаровые упаковки 320
7.4 Сходство и различие между плотнейшими и неплотнейшими
упаковками 323
7.5 Кристаллические и аморфные твердые тела 323
7.6 Твердые простые вещества, образованные элементами
группы 18 324
7.7 Твердые простые вещества, состоящие из двухатомных молекул 325
7.8 Молекулярные твердые простые вещества, образованные
элементами групп 15 и 16 327
7.9 Молекулярные аллотропные модификации углерода: С60 331
7.10 Твердые вещества, образующие бесконечные ковалентные
кристаллические решетки 335
7.11 Структуры металлов при 298 К 343
7.12 Металлический радиус 345
7.13 Металлическая связь 346
7.14 Заключение 351
7.15 Упражнения 351
АЛКАНЫ, АЛКЕНЫ И АЛКИНЫ
8.1 Введение 353
8.2 Номенклатура органических соединений 354
8.3 Первичные, вторичные, третичные и четвертичные атомы
углерода 362
8.4 Структурная изомерия 362
8.5 Конформации 365
538
Содержание
8.6 Хиральные молекулы 369
8.7 Основные особенности механизмов реакций 373
8.8 Физические свойства алканов 377
8.9 Промышленная переработка углеводородов 380
8.10 Реакции алканов 382
8.11 Хлорирование метана. Радикальная цепная реакция 385
8.12 Хлорирование пропана и 2-метилпропана 387
8.13 Реакции алкенов. 1. Реакции окисления и присоединения 389
8.14 Механизм электрофильного присоединения 395
8.15 Реакции алкенов. 2. Радикальное замещение 401
8.16 Реакции алкенов. 3. Радикальная полимеризация 402
8.17 Реакции алкенов. 4. Миграция двойной связи и изомеризация
алкенов 405
8.18 Реакции алкенов. 5. Гидроборирование 410
8.19 Реакции алкинов. 1. Присоединение 411
8.20 Реакции алкинов. 2. Алкины как кислоты 413
8.21 Реакции алкинов. 3. Димеризация 414
8.22 Заключение 415
8.23 Упражнения 416
СПЕКТРОСКОПИЯ
9.1 Что такое спектроскопия? 419
9.2 Спектр электромагнитного излучения и спектроскопические методы 422
9.3 Шкала времени 423
9.4 Закон Ламберта-Бера 424
9.5 Колориметрия 426
9.6 Колебательная спектроскопия. 1. Двухатомные молекулы 428
9.7 Колебательная спектроскопия. 2. Многоатомные молекулы 434
9.8 Использование ИК-спектроскопии в аналитических целях 437
9.9 Электронная спектроскопия. 1. Электронные переходы в молекулах,
вакуумный УФ и выбор растворителя 446
9.10 Электронная спектроскопия. 2. я-Сопряжение 450
9.11 Электронная спектроскопия. 3. Видимая область спектра 457
9.12 Спектроскопия ядерного магнитного резонанса. 1. Теоретические
основы 460
9.13 Спектроскопия ядерного магнитного резонанса. 2. Молекулы с
единственным типом химического окружения 466
9.14 Спектроскопия ядерного магнитного резонанса. 3. Молекулы с
различными типами химического окружения 467
9.15 Спектроскопия ядерного магнитного резонанса
4. Спектры ЯМР1Н 471
9.16 Спектроскопия ядерного магнитного резонанса. 5. Спин-спиновое
взаимодействие ядерных спинов ядер с /= 1/2 474
Содержание
539
9.17 Заключение 485
9.18 Упражнения 486
И КИНЕТИКА ХИМИЧЕСКИХ РЕАКЦИЙ
10.1 Введение 489
10.2 Кинетические уравнения. Зависимость скорости от концентрации 493
10.3 Как определить порядок реакции? 495
10.4 Кинетические уравнения для реакций с участием нескольких
реагентов 501
10.5 Интегральная форма кинетических уравнений 503
10.6 Зависимость скорости реакции от температуры:
уравнение Аррениуса 512
10.7 Катализ и автокатализ 517
10.8 Обратимые реакции 518
10.9 Молекулярность 520
10.10 Микроскопический механизм реакции и приближение
стационарного состояния 521
10.11 Радикальные цепные реакции 525
10.12 Заключение 529
10.13 Упражнения 530
СОВРЕМЕННЫЙ
КУРС
ОБЩЕЙ ХИМИИ
СОДЕРЖАНИЕ ТОМА 2
ВОДОРОД И s-ЭЛЕМЕНТЫ
АТОМЫ И АТОМНАЯ СТРУКТУРА
р-ЭЛЕМЕНТЫ И ВЫСОКИЕ СТЕПЕНИ ОКИСЛЕНИЯ
d-ЭЛЕМЕНТОВ
ПОЛЯРНЫЕ ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
ЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
КООРДИНАЦИОННЫЕ СОЕДИНЕНИЯ d-ЭЛЕМЕНТОВ
КАРБОНИЛЬНЫЕ СОЕДИНЕНИЯ
ПРИЛОЖЕНИЯ
ОТВЕТЫ К УПРАЖНЕНИЯМ
Учебное издание
Катрин Е. Хаускрофт, Эдвин К. Констебл
СОВРЕМЕННЫЙ КУРС ОБЩЕЙ ХИМИИ
Том 1
Руководитель проекта канд. хим. наук Т. И. Почкаева.
Ведущий редактор И. С. Беленькая.
Художник Инфантэ П. Технический редактор Е. В. Денюкова.
Оригинал-макет подготовлен О. Д. Эшлиман, С. Н. Лаврентьевой
и В. В. Левтоновым.
Лицензия ЛР № 010174 от 20.05.97 г.
Подписано в печать 30.10.2001. Формат 70 х 100/16. Бумага офсетная. Печать офсетная.
Гарнитура NewtonC. Объем 17 бум. л. Усл. печ. л. 44,20. Уч.-изд. л. 39,33.
Изд. № 3/9669. Тираж 5000 экз. Зак. №448
Издательство «Мир»
Министерства РФ по делам печати,
телерадиовещания и средств массовых коммуникаций.
107996, ГСП-6, Москва, 1-й Рижский пер., 2.
Диапозитивы изготовлены в издательстве «Мир»
Отпечатано с готовых диапозитивов в ОАО «Внешторгиздат»
127576, г. Москва, Илимская ул., 7.
Периодическая система элементов Д.И. Менделеева
1
1
н
1.008
3
Li
6.94
11
Na
22.99
19
К
39.10
37
Rb
85.47
55
Cs
132.91
87
Fr
223
2
4
Be
9.01
12
Mg
24.31
20
Ca
40.08
38
Sr
87.62
56
Ba
137.34
88
Ra
226.03
1
■
3
21
Sc
44.96
39
Y
88.91
La-Lu
Ac-Lr
■
4
22
Ti
47.90
40
Zr
91.22
72
Hf
178.49
104
Ш
5
23
V
50.94
41
Nb
92.91
73
Ta
180.95
105
1
H
1.008
6 7
24
Cr
52.01
42
Mo
95.94
74
w
183.85
106
25
Mn
54.94
43
Tc
98.91
75
Re
186.2
107
-<
-<
-<
8
26
Fe
55.85
44
Ru
101.07
76
Os
190.2
108
Атомный номер
Z
Символ элемента
атомная масса Аг
9 10 11
27
Со
58.93
45
Rh
102.91
77
lr
192.2
109
28
Ni
58.69
46
Pd
106.4
78
Pt
195.08
29
Cu
63.54
47
Aq
107.87
79
Au
196.97
12
30
Zn
65.37
48
Cd
112.40
80
Hq
200.59
13
5
В
10.81
13
At
26.98
31
Ga
69.72
49
In
114.82
81
TI
204.37
14
6
С
12.01
14
Si
28.09
32
Ge
72.59
50
Sn
118.71
82
Pb
207.19
15
7
N
14.01
15
P
30.97
33
As
74.92
51
Sb
121.75
83
Bi
208.98
16
8
О
16.00
16
S
32.06
34
Se
78.96
52
Те
127.60
84
Po
210
17
9
F
19.00
17
CI
35.45
35
Br
79.91
53
I
126.90
85
At
210
18
2
He
4.00
10
Ne
20.18
18
Ar
39.95
36
Kr
83.80
54
Xe
131.30
86
Rn
222
.
Лантаниды
57
La
138.91
58
Ce
140.12
59
Pr
140.91
60
Nd
144.24
61
Pm
146.92
62
Sm
150.35
63
Eu
151.96
64
Gd
157.25
65
Tb
158.92
66
Dy
162.50
67
Ho
164.93
68
Er
167.26
69
Tm
168.93
70
Yb
173.04
71
Lu
174.97
Aki
89
Ac
227.03
90
Th
232.04
91
Pa
231.04
92
и
238.03
93
Np
237.05
94
Pu
239.05
95
Am
241.06
96
Cm
244.07
97
Bk
249.08
98
Cf
252.08
99
Es
252.09
100
Fm
257.10
101
Md
258.10
102
No
259
103
Lr
262
0)r^h-L00>C0CDh-C0C4Jini^inOC\j4-O^tC0i-C0Or^
in^tOCO'-C7)OmO)tDNC«30){DO)0)0)00)OOT-0)
ОЮт-ч-СМГ^СМО)СО
O^OOJi-COCMOCM
ОЮ^О^С0СМГ^Ч^^^ОГ^С0С0Г^С^С0СМС01^ОС00)1ЛСМСМОЮ'»-СМГ^
CM -г- y- CM i- -г- CM T- 4- г- CNJ г- CM CM C\J T- i- CM T-
OI^^C\J(>J^COI^i-OOi-^COtN^COC\JOO)CDC\JOinh-0)h-TtincOOOO)00
CO CO Tf CD CO CO i- Tt CM CO LO 00 Г^ If) CO *fr CsJ O) CD О) О *- CO i- CM If) If) CO Tt O) CO
ШП Z3 E П <l> 0>0»-.0__ТОФ.ОО._.сЕ £ w_ — к- И в> с l
XCECEcyDQ-(y)cy}<C0cyD(/)l-l-l-l-l-l-»-l-O3LLQ.lJLLLCJCJOONNl
.0
JjliisHllnHbii
s о -
о
Q- -г * w j.
Q) i а о то
5-9-1 an5
о о
BB)(D(D(l)Sh>5S§©JsO>5u5S?teu»flii
о о о о о о о о н к ь к к ь к к > >в в в в * х =г=ги
О b,s
* 3s
QUO
S >S Q.
J" (0 0)
CO О) О О h- i-
CT) О 00 CO О О)
^th-r-^-TtO^CMO^^tCOinO)!-
0)0)С00)1Г)1-0)0)0)СМт-ОС0СЛ
т- СОЮ ч-СМ^ОО 1-
NCM^OO 0)0)0 0 СМ О)
СОаЭСО^^ШСМСО^^^СОС01Л^СМ^-ОГ^СОСМО)СООСОЮО)ООСОт-СОСМСОСМ
Ю CM CO CO ^t СО СО 1^СМЮС0Ю0)Г^СМ^СМС0Ю0)Ю^0)О0>С0г"^^с/5С>1СМС0О
т- СМ 1- СМ т-
CMt-t-i-i-CMCMt-7-CMCMCMi-i-
1^^С0^С0Г^С0С01-СМ1Л0)т-СМС0т-ООС0С0^СМОС0С0С0^-^0)^-1-С0С01П1Л
СМ ■»- СО If) О) If) О h--r-CMCMOTfC0T-C0i-0)CMTtOinr^-^'^.0)C0L0C00)C0C0r^Tf
Oc7)^XO-3Z3±lJ25o22<ZZZZZZZc7)OQ.5_Q_Q.Q.Q-CLCECE0C0C
1- СО СО СО Ю
О О О) О О)
^- 1- СО 1- ч- Tf СО О If) If) СМ О) О О) СО О СО If) I4- СМ О ^t 1- О О СО СО О
СОООСОО)0)0)ОСОСМГ^^01ЯО)ЮО)СОО)000)СМОО)^-1-000
^Г^Ю1-0)ОГч^О)О)ОО)О00'1-С0Г^0)С0^-СМ^-СМ"»-1ПСО^-СОСМС0С0СМ0)СМОС0
т- CM CM ^t СО у- СО ^ 1- 1^ If) О СО If) СО h» h- СО СО LO If) О) 1- СМ О) h- СО т- СО If) ^f 1-
СМ СМ СМ у- СМ СМ у- у- у- i- i- ч- ,- i- у- т- i- i- CM
Г^СЛС0ЮС0ЮС0^1^1ПЮС0С0^^^^СМСМСМГ^СОС0СО0)О)С0Г-ОО)00О)С0ОС0
СО "г- О) ч- СО If) О) СО СМ СО h- СО СО Г^ СО СО СО СО CM h- Tt Ю h- Is- CO Tt T- O) CM
O—Ew+^COO).* »_ ._ ^"ОТОч-ФФО>чЗфЗ^
z<<<<<DQmmmm>mi$ooiiOiQmu.<£_
.Q "D м- TO _
ii
s s g
QS1
о 5.
x d
«Сс55о.отоФФоаяхоото*ЯяФФО^т50105-£ртогатото^
Физические постоянные
Физическая постоянная
Символ
Вел
i и размерность в СИ
Число Авогадро
Константа Больцмана
Постоянная Фарадея
Универсальная газовая постоянная
Постоянная Планка
Молярный объем идеального газа при 10^ Па (1 бар)
и 273 К
Скорость света в вакууме
Масса покоя электрона
Заряд электрона
Масса покоя протона
Масса покоя нейтрона
Диэлектрическая проницаемость вакуума
Гравитационная постоянная
Нормальное ускорение свободного падения
Отношение длины окружности к диаметру круга
"А
к
F
R
h
V
те
е
тр
тп
е0
G
9
п
6,022 136 7 1023 моль"1
1,380 658-Ю-23 Дж-К"1
9,648 530 9 104 Клмоль"1
8,314 510 ДжК"1 моль"1
6,626 075 5 Ю"34 Дж с
0,022 771 08 м3моль-1 = 22,711 08 дм3 моль-1
2,997 924 58-Ю8 м с"1
9,109 389 7 Ю"31 кг
1,602 177 33 10"19IOi
1,672 623 1 Ю"27 кг
1,674 928 6 10"27 кг
8,854 187 186-Ю"12 Ф-м"1
6,672 59-10"11 м3кг-1с"2
9,806 65 мс"2
3,141 592 653 59
Перевод единиц
Давление в стандартном состоянии
Внесистемная единица (вне-СИ) давления
Энергия
Ю5Па= 102кПа= 1 бар
1 атм = 101 325 Па
1 эВ = 96,4853 кДжмоль
-1
Периодическая система элементов Д.И. Менделеева
1
1
н
1.008
3
Li
6.94
11
Na
22.99
19
К
39.10
37
Rb
85.47
55
Cs
132.91
87
Fr
223
4
Be
9.01
12
Mg
24.31
20
Ca
40.08
38
Sr
87.62
56
Ba
137.34
88
Ra
226.03
3
21
Sc
44.96
39
Y
88.91
La-Lu
Ac-Lr
4
22
Ti
47.90
40
Zr
91.22
72
Hf
178.49
104
5
23
V
50.94
41
Nb
92.91
73
Ta
180.95
105
l 1
H
1.008
6 7
24
Cr
52.01
42
Mo
95.94
74
w
183.85
106
25
Mn
54.94
43
Tc
98.91
75
Re
186.2
107
*<
-<
-<
8
26
Fe
55.85
44
Ru
101.07
76
Os
190.2
108
Атомный номер
Z
Символ элемента
Относительная
атомная масса Аг
9 10 11
27
Со
58.93
45
Rh
102.91
77
lr
192.2
109
28
Ni
58.69
46
Pd
106.4
78
Pt
195.08
29
Cu
63.54
47
Ag
107.87
79
Au
196.97
12
30
Zn
65.37
48
Cd
112.40
80
Hg
200.59
13
5
В
10.81
13
Al
26.98
31
Ga
69.72
49
In
114.82
81
TI
204.37
14
6
С
12.01
14
Si
28.09
32
Ge
72.59
50
Sn
118.71
82
Pb
207.19
15
7
N
14.01
15
P
30.97
33
As
74.92
51
Sb
121.75
83
Bi
208.98
16
8
О
16.00
16
S
32.06
34
Se
78.96
52
Те
127.60
84
Po
210
17
9
F
19.00
17
CI
35.45
35
Br
79.91
53
I
126.90
85
At
210
18
2
He
4.00
10
Ne
20.18
18
Ar
39.95
36
Kr
83.80
54
Xe
131.30
86
Rn
222
Актиниды
57
La
1 138.91
89
1 Ac
1 227.03
58
Ce
140.12
90
Th
232.04
59
Pr
140.91
91
Pa
231.04
60
Nd
144.24
92
и
238.03
61
Pm
146.92
93
Np
237.05
62
Sm
150.35
94
Pu
239.05
63
Eu
151.96
95
Am
241.06
64
Gd
157.25
96
Cm
244.07
65
Tb
158.92
97
Bk
249.08
66
Dy
162.50
98
Cf
252.08
67
Ho
164.93
99
Es
252.09
68
Er
167.26
100
Fm
257.10
69
Tm
168.93
101
Md
258.10
70
Yb
173.04
102
No
259
71
Lu
174.97
103
Lr
262
Лучшие книги мира-
в издательстве «мир»
Имеются в продаже:
о
"Я
ел
X)
^
8g
i «л
(N ел
^Е
*. <N 2
о °° ..
*чО
03 00
СО (N
о 1о
У 5 у
I:
Серия «Теоретические основы химии»
Новинка!
Бейдер Р. АТОМЫ В МОЛЕКУЛАХ: Квантовая теория:
Пер. с англ. — 532 с, ил.
Излагаемая в книге теория позволяет определить место
атома в общей схеме квантовой механики. Законы квантовой
механики в применении к связанному атому дают
возможность сформулировать необходимые и достаточные условия
химической связи между атомами. Изложение строится на
применении топологической теории, что не совсем обычно для
традиционной теоретической химии; в основу положены современные
методы вычислительной математики.
Благодаря такому подходу химические результаты физических
измерений можно представить в графическом виде или в виде
ограниченного набора конкретных численных характеристик. Это открывает
хорошие перспективы для широкого применения теории Бейдера в
различных областях химии и в смежных дисциплинах.
Для студентов старших курсов университетов, технических вузов,
аспирантов и научных работников.
Новинка!
Степанов И.Ф. КВАНТОВАЯ МЕХАНИКА И КВАНТОВАЯ
ХИМИЯ. - 519 с, ил.
т
Y^^g Учебное издание соответствует программе учебных курсов
т^^^Ш университетов. Подробно излагаются основные положения
^■М^Н квантовой теории и ее химические приложения.
Представлена основная система приближений, вводимых при
рассмотрении молекулярных задач, а также принципы, отвечающие этим
приближениям при применении квантовохимических методов.
Обсуждаются прикладные и более специальные задачи квантовой химии.
Каждая глава снабжена набором вопросов и задач для самостоятельной
работы.
Для студентов университетов и технических вузов.
Лучшие книги мира-
В ИЗДАТЕЛЬСТВЕ «МИР»
Имеются в продаже:
Ласло П. ЛОГИКА ОРГАНИЧЕСКОГО СИНТЕЗА. В 2-х
томах: Пер. с франц. — 429 с, ил.
Книга профессора знаменитой Политехнической школы
концентрирует внимание на самом главном в
синтетической органической химии и в то же время открывает
множество «окон» в «большую» химию, ориентируя на
самостоятельную работу. В т. 1 излагаются основы органического синтеза,
теоретические сведения, рассматриваются в общем виде реакции
различных типов, дается концепция ретросинтетического анализа. В т. 2
приведены примеры синтезов, обсуждаемых в общем виде в т. 1, а также
сведения из области ферментного катализа, химии биологически
активных и технологически важных соединений.
Для студентов всех уровней, изучающих органическую химию,
аспирантов, преподавателей вузов и средних учебных специальных
учреждений, химиков-органиков.
Новинка!
»mvniiii:ifea«H Дорохова Е.Н., Прохорова Г.В. ЗАДАЧИ
АНАЛИТИЧЕСКОЙ ХИМИИ. - 267 с.
дачи и вопросы»)
И ВОПРОСЫ ПО
ил. — (Серия «За-
М Основная цель данной книги — детальное обсуждение
решений типовых задач, относящихся к важнейшим
теоретическим разделам аналитической химии. В каждой главе в
кратком теоретическом введении дается подробный вывод важнейших
формул, обсуждаются границы их применимости и приводятся
расчетные примеры.
Включенные в книгу задачи и вопросы могут быть использованы
преподавателями в качестве модельных при составлении контрольных работ
и экзаменационных билетов, а также студентами - для самоподготовки.
Для студентов университетов и технических вузов.
Эмсли Дж. ЭЛЕМЕНТЫ: Пер. с англ. — 256 с, ил.
ЭЛеМеНТЫ Справочник, который не имеет аналогов в мировой научной
литературе. Каждый разворот книги — один химический
элемент. Приводятся разнообразные и весьма ценные данные о
всех элементах: химические и физические свойства, интерес-
1 ные сведения об истории открытия, биологической роли и др.
Для школьников, студентов, аспирантов, преподавателей, научных
работников.
о
"Я
С/3
Х>
3
а
Ъ
8g
сч ел
в 00 О
о °° ..
cd oo
CQ (N
vo 3
U Yoo
I—« С '
24 3 ^
о я©
т. *w
8.Ё i
3
Книги издательства «Мир»
можно приобрести по издательским ценам
в рекламно-коммерческом центре издательства по адресу:
Москва, 1-й Рижский пер., д. 2
Тел.: (095) 286-83-88, 286-25-50, 286-82-33
Проезд: метро «Рижская», далее авт. 714 до остановки
«1-й Рижский переулок»
проспект Мира
или в магазине «Транспортная книга»:
ул. Садовая-Спасская, 21/1, стр. 1
(м. «Красные ворота»), тел.: (095) 262-25-13, 262-13-19
Иногородним покупателям (в пределах России)
книги высылаются наложенным платежом
ЗАКАЗЫ
с указанием автора, названия книги, количества экземпляров,
ФИО и адреса получателя следует направлять в коммерческую службу:
обычной почтой: 107996, ГСП-6, Москва, 1-й Рижский пер., д. 2
факсом: (095) 286-84-55
по электронной почте: victor@mir.msk.su
по сети Internet: http://www.mir-pubs.dol.ru
КНИГИ ИЗДАТЕЛЬСТВА «МИР»
также можно приобрести в следующих крупнейших магазинах:
Москва
• ГУП «Объединенный центр «Московский Дом книги»-ул. Новый Арбат, д. 8.
Тел.: (095) 290-45-07
•ТД «Библио-глобус» - ул. Мясницкая, д. 6. Тел.: (095) 928-43-51
• «Дом технической книги» — Ленинский пр., д. 40. Тел.: (095) 137-60-38
• «Медицинская книга» - Комсомольский пр., д. 25. Тел.: (095) 245-39-33
• ДК «Молодая гвардия» — ул. Б.Полянка, д. 28. Тел.: (095) 238-50-01
Санкт- Петербург
• ГУП КТ «Санкт-Петербургский Дом книги»—Невский пр., д. 28. Тел.: (812) 219-49-15
• ГУП КТ «Техническая книга» - ул. Пушкинская, д. 6. Тел.: (812) 164-65-65
Новосибирск
•«Топ-книга»- ул. Арбузова, д. 1/11. Тел.: (3832) 36-10-28
Екатеринбург
• «Дом книги»-ул. Валека, д. 12. Тел.: (3432) 59-42-00
• «Книжный магазин №14»— ул.Челюскинцев, д. 23. Тел.: (3432) 53-24-89
I ЗАРУБЕЖНЫЙ I
УЧЕБНИК
СОВРЕМЕННЫЙ
КУРС
ОБЩЕЙ ХИМИИ
Учебник «Современный курс общей химии» написан
на основе 18-летнего опыта преподавания химии в
лучших школах, колледжах и университетах, в том
числе в Кембридже.
В настоящее время оба автора работают в
Швейцарии в Базельском университете. Катрин Хаускрофт
читает лекции в Институте неорганической химии, а
профессор Эдвин Констебл является директором этого
института.
Доктор Хаускрофт проводит теоретические и
экспериментальные исследования в области химии твердого
тела и молекулярных структур кластеров металлов.
Она является автором более 100 научных работ.
Профессор Констебл специализируется в области
неорганической и органической химии, в частности
супрамолекулярной химии. Им опубликовано свыше
200 работ. Оба автора - известные ученые; они
являются членами редколлегии международного журнала
Coordination Chemistry Reviews, а профессор Эдвин
Констебл - также редактором журнала Chemical
Communications.