/




















































































































































Автор: Яковлев С.В. Краснобородько И.Г. Рогов В.М.
Теги: сточные воды обработка, удаление, использование водоснабжение очистка воды водопотребление
ISBN: 5-274-00174-2
Год: 1987
Текст
Технология
электрохимической
очистки воды
с. В. ЯКОВЛЕВ,
и. Г. КРАСНОБОРОДЬКО
в. М. РОГОВ
Технология
электрохимической
очистки веды
Ленинград
Стройиздат
Ленинградское
1987
отделение
ального раз-
и на период
гкн ресурсе-
н снижения
а ния. Суще-
ий является
•ткн природ-
тревращение
й, протекаю-
учили разви-
чнетки воды,
мпактны, вы-
атации срав-
рообработка
и позволяет
да примесей
1ым является
, не увеличи-
:лючается об-
к количество.
1реимущества
•тодами обра-
электроэнер-
jb, значитель-
ектролнзеров,
в том числе
щии электро-
,ие на основе
рохимических
нологии водо-
'ЧНЫХ вод.
опубликован
:с монографий
которых рас-
(ДОПОДГОТОВКИ.
3
УДК L2S.S31
Яковлев С. В.» Краснобородько И Г., Рогов В. М Гехнолон1Я >л< к-
трохнмнческой очистки воды.—Л.. Стройиздат. Леншнр. отд-пне, I9N7.
.312 с., ил ISBN 5-274-00174-2
В книге изложены теоретические основы. механизм н закономерности книг
тики электродных реакций и процессов превращения примесей, протекающих при
электрохимической очистке водных систем. Представлены классификация, сущ
кисть и перспективные области применения методов электрообработкн загрявнен
пых жидкостей; конструктивные особенности и методика инженерного расчета
аппаратов; примеры практического использования электротокиол <н ин а ра <лнч
пых системах водоподготовки н обезвреживания жидких промышленных отходов
Уделено особое внимание основным проблемам современной технологии и пер
спектнвам развития и совершенствования элсктрообработки природных и сточных
вод.
Предназначена для научных и инженерно технических работников научно-
исследовательских и проектных организаций, а также может быть использована
в качестве учебного пособия для повышения квалификации слушателей хнмнко-
техиологнческнх и инженерно-строительных вузов прн изучении вопросов, свя-
занных с охраной окружающей среды.
Рецензенты—д-р техн, наук В. В. Найденко и д-р хим. паук, лау-
реат премии нм. Л. В. Писаржевского В. Д. Гребенюк
Научное издание
Сергей Васильевич Яковлев
Иван Георгиевич Краснобородько
Владимир Михайлович Рогов
ТЕХНОЛОГИЯ
электрохимической
очистки воды
Зав. редакцией Н. Н. Дмпрова
Редактор В Л. Ануфриева
Оформление художника Н. Г. Всесввтского
Художественный редактор О. В. Сперанская
Технический редактор В. В Живнова
корректоры Т. Б. Верникова и Ю М. Зислин
ИБ 7* 4156
Сдано в набор 29.05.87. Подписано в печать 27.11.87. М 32432. Формат 60xW/>,. Бума! а
тип. 7* I. I арии гура .Литературная.. Печать высокая. Уел. печ. л. 19.5. Уч. над л. 2155
Уел. кр.-отт. 19.5. Изд. 7* 2455Л. Тираж 7200 »кз. Заказ 7* 2148. Цена 3 р.
Стройиздат. Ленинградское отделение I9I01I. Ленинград, пл. Островского. 6
Ленинградская типография 7* 4 ордена Трудового Красного Знамени Ленинградского
объединения «Техническая книга, нм. Евгении Соколовой Союзполнграфпрома при Государ-
ственном комитете СССР по делам издательств, полиграфии и книжной торговли 1'4126.
Ленинград. Социалистическая ул.. 14.
ISBN 5-274-00174-2
„ 3206000000—084 „„ „
Я--------------- 160 88
047(01) 87
© Стройиздат, Ленинградское отделение. ;987
ПРЕДИСЛОВИЕ
В «Основных направлениях экономического и социального раз-
вития народного хозяйства СССР на 1986—1990 годы и на период
до 2000 года> указывается па необходимость разработки ресурсо-
сберегающих технологий, повышения эффективности и снижения
энерго- и металлоемкости технологического оборудования. Суще-
ственным резервом в осуществлении этих мероприятий является
использование электрического тока в технологии очистки природ-
ных н сточных вод, при котором происходит прямое превращение
электрической энергии в энергию химических реакций, протекаю-
щих в растворе с большой скоростью.
В настоящее время методы этектрообработки получили разви-
тие как эффективные и прогрессивные в техно погни очистки воды.
Установки по реализации этих методов достаточно компактны, вы-
сокопроизводительны, процессы управления и эксплуатации срав-
нительно просто автоматизируются. Кроме того, электрообработка
при правильном сочетании ее с другими способами позволяет
успешно очищать природные и сточные воды от ряда примесей
различного состава и дисперсности. Весьма позитивным является
также и то, что при электрообработке, как правило, не увеличи-
вается солевой состав очищенной воды и нередко исключается об-
разование осадков или значительно уменьшается их количество.
Все это обеспечивает в ряде случаев существенные преимущества
электрохимических методов перед традиционными методами обра-
ботки воды.
Постоянно прогрессирующее снижение стоимости электроэнер-
гии, увеличение количества и мощности ее источников, значитель-
ные успехи в области конструктивных разработок электролизеров,
появление новых электротехнических материалов, в том числе
малоизнашивающихся стойких к анодной поляризации электро-
дов, позволяет считать, что установки, действующие на основе
принципов воздействия электрофизических и электрохимических
факторов, найдут особо широкое применение в технологии водо-
подготовки и локальной очистки промышленных сточных вод.
В технической литературе за последние 10 лет опубликован
ряд представляющих значительный научный интерес монографий
по электрообработке дисперсных систем, каждая из которых рас-
сматривает отдельные технологические процессы водоподготовки.
Г 3
бл шрующиеся н основном ил теоретических основах флотации пли
коллоидной химии Однако любой меюд электрообработки снизан
с комплексом сложных физических явлений и в первую очередь
с первичным актом протекания электрохимической реакции па
ранит ра «дела фаз «электрод — растворэ, который зачастую яв-
ляется отетствеппым за все процессы, происходящие на элек-
тродах и в объеме обрабатываемой жидкости. Этому чрезвычайно
важному вопросу в специальной литературе по электрохимиче-
ской очистке поды не уделяется, к сожалению, должного внима-
ния, в то время как все явления и превращения веществ при на-
ложении электрического поля на дисперсные системы связаны
общей теорией электродных процессов, электрокинетических явле-
ний и взаимодействия дисперсной фазы с электрогенерированными
продуктами электролиза.
Кроме того, до настоящего времени еще не разработана стро-
гая классификация методов электрообработки, не принята общая
терминология процессов, реализующих в аппаратах по очистке
водных систем весь сложный комплекс различных физико-хими-
ческих механизмов, что порождает определенные трудности при
трактовке основных теоретических положений и интерпретации
обсуждаемых результатов экспериментальных исследований.
В книге, предлагаемой широкому кругу специалистов, рабо-
тающих в области охраны окружающей среды, впервые изложен
обобщенный систематизированный материал, освещающий основ-
ные проблемы современной технологии, достижения теории и
практики, перспективы развития и совершенствования электрооб-
работки природных и сточных вод. Авторами поставлена цель —
дать логическую классификацию методов электрообработки, учи-
тывающую своеобразие эффектов, определяющих превалирующее
направление в различных технологических приемах использова-
ния электрического тока для очистки загрязненных жидкостей.
Книга написана С. В. Яковлевым и И. Г. Краснобородько,
а глава 2 и п. 3.4, 4.1, 4.2.1, 4.2.2, 4.2.4, 4.3.1, 4 4.1, 5.2.1, 5.2.3, 6.3,
6.7 — совместно с В. М. Роговым.
В книге использованы исследования и разработки, выполнен-
ные В. Д. Дмитриевым, Н. И. Рукобратским, О. В. Смирновым,
Е. С. Светашовой, Л. Н. Губановым, В. Л. Филипчуком, Р. С. Са-
финым, В. Н. Анопольским, М. Т. Никифоровым. В. В. Кузнецо-
вым, Д. Н. Пластуновым и др.
Авторы приносят благодарность проф. В. В. Найдепко (заве-
дующему кафедрой водоснабжения и канализации ГИСИ) и проф.
В. Д. Гребенюку (заведующему отделом электрохимических ме-
тодов деминерализации воды ИКХиХВ АН УССР) за ценные за-
мечания, сделанные при рецензировании рукописи.
Авторы будут благодарны за все замечания и пожелания, ка-
сающиеся улучшения содержания книги.
ОСНОВНЫЕ ОБОЗНАЧЕНИЯ
Аг—константа Гамакера
А», А к—электрохимический, химический эквивалент вещества
А — работа
а — радиус частицы, константа уравнения Тафеля
а', ас' — активность и средняя активность электролита
Bi — ионная эквивалентная электропроводность
Вт. Вт”, Вт" — выход по току вещества, соответствеиио анодный и ка-
тодный выход по току
Ь — константа уравнения Тафеля (2,303 RT/zF)
С, Се, Си—текущая, исходная и конечная концентрации вещества
в растворе
Сж', Сщ’ — концентрация кислоты и щелочи
Ст — удельная теплоемкость воды
D — коэффициент диффузии
Dn, D4 — диаметр пузырька, частицы
Дт — расход тока (удельное количество электричества по объ-
ему обрабатываемой жидкости)
d — расстояние между частицами
du — дипольный момент
Ег, Екр — напряженность н критическая напряженность электриче-
ского поля
£р — напряжение разложения
е — заряд электрона
F — постоянная Фарадея
F —энергия Гельмгольца
Fn — площадь поперечного сечения аппарата
f —частота внешнего электрического поля
/», fn — активная поверхность электрода, мембраны
fn, fn,— частота столкновений соответственно отиородных и разно-
родных частиц
Г, Гр — газосодержаннс электролита, объемное газосодержание
G — средний градиент скорости
AG —хбыль свободной энергии Гиббса
ц — ускорение свободного падения
Н — энтальпия
Н, — рабочая высота аппарата
/in. Л» — высота слоя пены, электрода
/ — сила тока
I, 1г — плотность тока и объемная плотность тока в электролите
/г. In — анодная, катодная плотность тока
->
J — плотность потока вещества
Кр, Кл. Кп, Кг, Кх — константы равновесия, диссоциации, нестойкости, гидро-
лиза, электропроводности
Кв — полное произведение воды
к, Л — постоянная Больцмана, константа скорости реакции
L — длина аппарата
/•. /м — расстояние между электродами, мембранами
М — атомная масса; масса вещества
Me — металл
т—масса вещества (воды), обрабатываемая в единицу вре-
мени
V* — число Авогадро
Ni, — число молекул, суммарное число ионов
W+, N~ — число положительных и отрицательных ионов
Мп. Мч — число пузырьков, частиц
л,, лЛ, лЛ — число переноса, соответственно, катионов и анионов
Р,, Аь Рп — толщина электрода, диафрагмы, диэлектрических прокла-
док
Q — расход воды
q — заряд частицы, нона
<7» - количество электричества
qr — скорость подачи газа в жидкость
R — универсальная газовая постоянная
R, — электрическое сопротивление раствора
г, гв Гч — радиус иона, средний радиус пузырька, частицы
S — энтропия
S., $м — общая площадь электродов, мембран
Т — абсолютная температура
t, t» — время; продолжительность электролиза
U — энергия
U, — напряжение электролиза
Уи — объем иа 1 моль растворенного вещества (разбавление)
Уг, Уж, V„ — объем газа, жидкости, пузырька
Ур — рабочий объем аппарата
v —скорость электрохимической реакции
— скорость фильтрования воды
V/, V/1 — абсолютная, относительная подвижность ионов
_уп, v„ — скорость движения катионов, анионов
«Ж, «п—средняя скорость движения жидкости, подъема пузырь-
ков
1Уо, 1Ум, IFr, 1У,п — полная энергия; энергия молекулярного, электростатиче-
ского и липоль-лнпольиого взаимодействия частиц
ПГ.л — расход электроэнергии
г — заряд иоиа. валентность
Э — эффект (степень) извлечения примесей
а, ап — степень диссоциации, превращения
«г — удельный расход газа иа единицу извлекаемых примесей
Р —степень гидролиза
T>i, Уп — коэффициент активности анионов н катионов
ус — средний коэффициент активности
у,, у. — обз-емиый вес воды и удельный вес электролита
уа — поляризуемость частицы
бо, б(. Од — общая толщина ЦЭС, толщина плотной н диффузной ча-
стей ДЭС
ея —диэлектрическая проницаемость среды
С —эдектрокинетическнй потенциал
т] — вязкость среды
»Ъ, ’h- Пя- Пф—перенапряжение общее, электрохимической стадии, диф-
фузии, разряда и фазовое
О — коэффициент диализа
к —постоянная Дюбая — Гюкксля
Хп — электропроводность электролита
? . Ао — эквивалентная электропроводность; то же, при бесконеч-
ном разбавлении
ц, — термодинамический потенциал
8
и» ионная сила раствора
цо — динамический коэффициент вязкости жидкости
v кинематический коэффициент вязкости
|»«. Рч, р/. Рг — плотности жидкости, твердых частиц, газа, газовой
эмульсин
р, — удельное сопротивление электролита
о — поверхностное натяжение
о, — поверхностная плотность заряда; заряд поверхности элек-
трода
Сг—коэффициент, учитывающий газосодержа ине электролита
ф. фо. ф* — абсолютный, стандартный и равновесный потенциал
Фи*, фо* — равновесный потенциал катода, анода
Афи. Аф, — катодное, анодное перенапряжение (полярнзаиня)
Vo. Ve. V, — полный скачок потенциала, скачок потенциала в плотной,
диффузной части ДЭС
I
ГЛАВА I
ЭЛЕКТРОХИМИЧЕСКАЯ ТЕХНОЛОГИЯ
В СИСТЕМАХ ВОДОПОДГОТОВКИ
И ОЧИСТКИ сточных вод
1.1. ЭЛЕКТРОХИМИЧЕСКИЕ ПРОЦЕССЫ И ИХ ОРГАНИЗАЦИЯ
Электрохимия относится к области знаний о взаимных превра-
щениях химического и электрического видов энергии и об исполь-
зовании этих превращений. Электрохимические процессы широко
применяются в настоящее время в различных областях современ-
ной техники, составляя основу прикладной электрохимии: элек-
трометаллургию, гальванотехнику, электросинтез органических и
неорганических соединений, производство химических источников
тока, химотронику, электрохимические методы контроля и ана-
лиза и т. д. Далеко не последняя по своему практическому значе-
нию область прикладной электрохимии относится к разработке
методов электрообработки загрязненных водных систем, в кото-
рой уже сейчас достигнуты значительные успехи, позволяющие
предвидеть в будущем более широкую реализацию электрохими-
ческой технологии для решения стоящих перед человечеством эко-
логических проблем.
Электрохимическая очистка загрязненных природных и сточ-
ных вод основана на использовании электрической энергии при
проведении процессов электролиза водных растворов электроли-
тов.
Электролиз осуществляется в системах, состоящих из следую-
щих элементов:
раствора элек1ролита— проводника второго рода, в котором
вещества диссоциированы на ионы (загрязненные воды являются
растворами электролитов, так как в них всегда присутствуют
ионы в той или иной концентрации);
электродов — проводников первого рода, погруженных в рас-
твор электролита;
внешнего источника тока;
токоподводов—металлических проводников первого рода, со-
единяющих электроды с источником тока.
Рассмотрим кратко механизм прохождения тока через рас-
8
Рнс. 1.1. Принципиальная
схема электролитической
ванны
1 — «под: 2 — источник ЭДС;
3 — катод
Рис. 1.2. Принципиальная
Схема гальванического эле-
мента
I — положительный электрод;
2 — потребитель тока; S — от-
рицательный электрод
творы электролитов. По современным воззрениям, электрический
ток в металлических проводниках — это поток электронов, пере-
двигающихся от отрицательного полюса источника тока к поло-
жительному. Если соединить полюсы источника тока металличе-
ским проводником, то источник тока, подобно насосу, засасывает
электроны через положительный полюс и выталкивает их в про-
водник через отрицательный полюс.
Положим, что в раствор электролита опущены два электрода
(рис. 1.1), соединенные с полюсами источника тока н, следова-
тельно, заряженные один положительно (анод), другой отрица-
тельно (катод).
Вода сама по себе является плохим проводником, и электроны
не могут перемещаться в ней так, как в металлическом провод-
нике, однако находящиеся в растворе ионы, образующиеся при
диссоциации электролита, передвигаются по двум противополож-
ным направлениям: положительные ионы (катионы) двигаются
к катоду, отрицательные (анионы)—к аноду. Достигая катода,
катионы получают от него недостающие им электроны и стано-
вятся нейтральными атомами iliii группой атомов (молекулами).
Одновременно с этим анионы отдают аноду спои «лишние» элек-
троны, тоже переходя в нейтральные атомы или группы атомов.
Непрерывный переход электронов с катодов на катионы и с анио-
нов на аноды поддерживает движение электронов в проводах, со-
единяющих полюсы источника тока с электродами. При этом
число электронов, получаемых анодом, равно числу электронов,
передаваемых за то же время катодом, т. е. во внешней цепи ток
идет так же, как он шел бы, если бы электроны непосредственно
проходили через раствор. В сущности же па границе раздела элек-
трод— раствор происходит переход от электронной проводимости
к ионной, причем прохождение электрического тока через провод-
ники второго рода сопровождается выделением на электродах
продуктов электрохимических реакций, т. е. продуктов взаимо-
действия ионов и электронов. Реакция между ионом и электроном
иа границе раздела электрод — раствор определяет превращение
электрической энергии в химическую.
Ч
Электрохимические процессы, как правило, осуществляются
или за счет подведенной извне электрической энергии, или при
ее получении.
Химические превращения в растворе электролита за счет внеш-
ней электрической энергии происходят в электролизерах или элек-
тролитических ваннах (см. рис. 1.1). В таких системах часть рас-
твора электролита, находящаяся у анода, называется анолитом,
находящаяся у катода — католитом.
Генерация электрической энергии за счет протекающих в рас-
творе электролита химических превращений происходит в галь-
ванических элементах или химических источниках тока (рис. 1.2).
Здесь электрод, направляющий электроны во внешнюю цепь, на-
зывается отрицательным полюсом элемента, принимающий элек-
троны из внешней цепи — положительным.
В общем случае при электролизе протекают окислительно-вос-
становительные процессы: на аноде — потеря электронов (окисле-
ние), на катоде — приобретение электронов (восстановление).
Однако механизм электрохимических реакций существенно отли-
чается от обычных химических превращений веществ.
Отличительной особенностью электрохимической реакции по
сравнению с химической является пространственное разделение
электрохимической реакции на два сопряженных процесса (две
сопряженные электродные реакции).
Пространственное разделение электрохимических реакций при-
дает им ряд специфических качеств, которых нет у обычных
химических реакций. Так. условием протекания обычных гомоген-
ных химических реакций в растворе является взаимодействие
реагирующих компонентов (молекул, атомов, ионов) при их столк-
новении друг с другом в любой точке раствора. В момент столкно-
вения становится возможным переход электронов с одного веще-
ства на другое Совершится ли этот переход или нет, зависит от
запаса энергии реагирующих веществ и ее соотношения с энер-
гией активации, которая является функцией природы химической
реакции. Следовательно, необходимость контакта реагирующих
частиц в растворе — первая характерная особенность гомогенного
химического процесса. Вторая особенность заключается в том, что
путь электрона при этом оказывается очень малым, длина его не
превышает радиуса атома или молекулы.
При химических реакциях в растворах место встречи и на-
правление эпектроппых переходов ориентированы в реакционном
объеме любым образом или при любых взаимных положениях
реагирующих частиц, что является третьей характерной особенно-
стью химического процесса, отличающегося хаотичностью, беспо-
рядочностью столкновения между частицами и иенаправлепно-
стыо электронных переходов.
При электрохимическом процессе условия протекания химиче-
ской реакции необходимо изменить так, чтобы электронные пере-
ходы были бы не беспорядочны, а совершались в одном опреде-
ленном направлении г. с необходимо создать пространственную
направленность электронных переходов. Кроме того, при яъм про
цессе, как уже было сказано, необходимо расчленение электрохи-
мической реакции на дна электродных акта, а также существен-
ное увеличение пути электронных переходов до макроскопических
размеров, так как использование энергии электрического тока воз-
можно лишь в том случае, если путь электронов велик по сравне-
нию с размерами атомов. Однако путь электронного перехода не
может быть большим, если реагирующие частицы контактируют
друг с другом. Поэтому необходимо еще и пространственное раз-
деление участников реакции, нс допускающее непосредственного
их взаимодействия.
Таким образом, при проведении электролиза в отличие от про-
текания химических реакций необходимо разделение реагентов и
образование гетерогенной системы, в которой переход электронов
от одной группы атомов или молекул к другой осуществляется
через металлические проводники электроды.
Прохождение электрического тока через реакционное про-
странство осуществляется как непосредственными участниками
электрохимической реакции (если они присутствуют в ионизиро-
ванном состоянии), так и специально добавляемыми соедине-
ниями (электролитами), обладающими высокой ионной проводи-
мостью.
В основе всякого электролиза лежат процессы разложения ве-
ществ или получения новых продуктов на границе электрод — рас-
твор при помощи электрического тока. При электролизе в зависи-
мости от фазово-дисперсного состояния компонентов, содержа-
щихся в растворе, могут также наблюдаться явления, связанные
с электрокинетическими свойствами коллоидов. При этом могут
протекать электрохимические реакции восстановления и окисле-
ния, сопровождающиеся образованием твердых или газообразных
продуктов, процессы восстановления н окисления без выделения
самостоятельной фазы и процессы, сопровождающиеся растворе-
нием материала электродов.
В объеме раствора, подвергающегося электролизу, за счет
электродных процессов происходит изменение активной реакции
(pH) среды и окислительно-восстановительного потенциала (Eh)
системы, а также фазово-дисперсные превращения примесей воды.
Практическое значение приобретает область химических реак-
ций, протекающих в электрических разрядах, при которых изме-
няются гидродинамические свойства дисперсной фазы и обеспечи-
вается глубокая минерализация органических соединений.
Все явления и превращения веществ при различных приемах
использования электролиза связаны общей теорией процессов,
происходящих на электродах и в объеме электролита. Между
электролизом же и явлениями, происходящими в электрических
разрядах, имеется настолько большая и принципиальная разница,
что кроме внешнего представления о превращениях, связанных
с расходом электрической энергии, общего в области теории, про-
мышленного осуществления и конструктивного оформления аппа-
11
ратуры у них нет. 1ем не менее нее известные способы обработки
жидких дисперсных сред с неволь юнанием элсюрпческого тока
следует относить к электрохимическим методам, так как наряду
с многочисленными процессами изменения элсктрокинстических
свойств веществ в растворах электролитов обязательно протекают
и химические процессы.
1J. ХИМИЧЕСКОЕ ВОЗДЕЙСТВИЕ ЭЛЕКТРИЧЕСКОГО ТОКА.
ЗАКОНЫ ФАРАДЕЯ
Впервые взаимосвязь химических и электрических явлений
была замечена в XVIII в. В 1756 г. М. В. Ломоносов в своей ра,-
боте «Теория электричества, составленная ио математическому
методу» писал: «Так как главным образом химия выведывает
внутреннее строение тел, то без нее труден, даже невозможен до-
ступ во внутренности их, и без химии этот доступ закрыт для [вы-
яснения] истинной причины электричества».
Существенный вклад в раскрытие представлений о взаимо-
связи электрических и химических явлений внес своими блестя-
щими исследованиями М. Фарадей, который в 1832—1834 гг. па
основании многочисленных опытов формулирует законы электро-
химического разложения. Им впервые установлено, что между ко-
личеством электричества, прошедшего через границу электрод —
электролит, и количеством вещества, претерпевшего те или иные
химические изменения на этой границе, существует определенная
пропорциональная и количественная зависимость.
Первый закон Фарадея определяет прямую пропорциональ-
ность химического действия электрического тока, т. е. количе-
ства вещества, прореагировавшего па электроде, к абсолютному
количеству прошедшего через систему электричества:
Am = A3t3. (1.1)
Коэффициент Аа представляет собой количество граммов ве-
щества, прореагировавшего при прохождении единицы количества
электричества, н называется электрохимическим эквивалентом,
т. е. Д,=Ат при </,=//,= 1Д • ч. При выделении на электроде га-
зообразных веществ в выражение для А3 вместо массы может вхо-
дить объем.
Второй закон Фарадея отражает связь между количеством про-
реагировавшего на границе электрод — электролит вещества и его
химической природой и формулируется следующим образом: при
прохождении определенного количества электричества через элек-
тролит количества различных веществ, прореагировавших (выде-
лившихся) иа электродах \т, пропорциональны их химическим
эквивалентам Дх:
Ат,'ЛХ1 \mt/AXi= \т3!Аха = . . . = const. (1.2)
Химический эквивалент — это отношение атомной массы ка-
кого-либо элемента М к его валентности г, т. с. Ах M/z.
12
Таблица 1.1. Атомные массы н электрохимические эквиваленты
Элемент Заряд Атомная месса Коли- чество вещест- ва, выде- ляемого 3600 Кл, г Элемент Заряд Атомная масса Коли- чество вещест- ва. выде- ляемого 3600 Кл, г
Алюминий 3 26,98 0,3356 Никель 2 58,71 1,0954
Водород 1 1,008 0,0376 Платина 2 195,09 3,6437
Железо 2 55,85 1,0424 Платина 4 195,09 1,8219
Железо 3 55,85 0,6949 Ртуть 1 200,59 7,4882
Калий 1 39,10 1,4595 Ртуть 2 200,59 3,7441
Кальций 2 40,08 0,7480 Свинец 2 207,19 3,8673
Кислород 2 16,00 0,2986 Сера 2 32,06 0,5983
Кобальт 2 58,93 1,1000 Серебро I 107,87 4,0269
Марганец 2 54,94 1,0252 Титан 2 47,90 0,8920
Медь 1 63,55 2,3729 Хлор 1 35,45 1,3236
Медь 2 63,55 1,1864 Хром 3 51,996 0,6471
Мышьяк 3 74,92 0,9321 Хром 6 51,996 0,3236
Натрий 1 22,99 0,8585 Цинк 2 65,37 1,2202
Многочисленными экспериментальными данными было уста-
новлено, что при прохождении одного и того же количества элек-
тричества, равного 96 500 Кл (или 26,8 А-ч), на электродах выде-
ляются одни и те же количества вещества, численно равные их
химическим эквивалентам. Это количество электричества — по-
стоянная Фарадея F.
Объединив первый и второй законы Фарадея, получим, что
электрохимические эквиваленты пропорциональны их химическим
эквивалентам. Согласно объединенному закону Фарадея количе-
ство электричества, равное 1 F, всегда изменяет электрохимически
1 г-экв вещества независимо от его природы, величины тока,
формы и материала электрода, конструкции электролизера.
Математическая запись объединенного закона Фарадея имеет
следующий вид:
Ат = 1 /96500 И, М/г. (1.3)
Сопоставляя уравнения (1.1) и (1.3), получим связь между
электрохимическим и химическим эквивалентами:
А, = 1/96500 М/г = Ax/F. (1.4)
На практике электрохимические эквиваленты чаще всего отно-
сят не к постоянной Фарадея F, а к I А-ч. В таком случае элек-
трохимический эквивалент выразится как М/г-26,8. Некоторые
значения этих величин для элементов, наиболее часто встречаю-
щихся при электрообработке водных систем, приведены в табл. 1.1.
Законы Фарадея принадлежат к числу наиболее точных зако-
нов природы и не имеют исключений в применении к процессам,
протекающим иа границе электрод — раствор, т. е. к границе
между проводниками первого и второю рода (к границам со сме-
13
hi ан ной ионно-электронной проводимостью, например, при про-
хождении тока через газы, они неприменимы). Однако при прове-
дении па практике процессов электролиза встречаются кажу-
щиеся отклонения от этих законов.
Причинами таких отклонений могут являться: совместный раз-
ряд на электроде различных ионов, наличие в системе вщимодей-
ствня продуктов электродных реакций между собой или с элек-
тролитом (вторичные реакции), катодное восстановление анодных
продуктов в бездиафрагмениом электролизере, утечки тока, ко-
роткие замыкания и другие затруднения.
Так, например, если проводить электролиз раствора сульфата
цинка и серной кислоты с целью катодного выделения цинка, то
при пропускании 1 А • ч осаждается на электроде не 1 22 г цинка,
как следовало бы по закону Фарадея, а меньшее количество.
Точно так же, если подвергать электролизу растворы хлоридов
(к примеру, NaCl), то в результате прохождения 1 А-ч на аноде
образуется не 1,32 г газообразного хлора, а несколько меньшее
его количество.
В первом случае в электролите присутствуют ионы Н+, Zn2+,
ОН~ и SO«, поэтому одновременно с разрядом Zn2+ на катоде
идет разряд Н+-ионов: Zn2+ + 2e-*-Zn; 2Н1- + 2е->Н2. Если опреде-
лить количество не только осевшего цинка, по и выделившегося
газообразного водорода, то их суммарное количество точно будет
соответствовать закону Фарадея.
Во втором электролите присутствуют ионы Na+, Н+, С1_ и
ОН-, и на аноде одновременно с выделением газообразного хлора
(2С1-—2е-+С12) будут разряжаться и гидроксильные ионы или
молекулы воды с образованием кислорода (2ОН"—2е-*Н2О +
+ ‘/2О2; Н2О—2е->-2 Н++V2O2), поэтому в данном случае часть
тока расходуется и на образование кислорода. Кроме того, выде-
ленный на аноде хлор может частично снова переходить в рас-
твор вследствие вторичных химических реакций: С12 + Н2Очэ=
чэ=НС1О + НС1 илн С12 + 0Н"5=±НСЮ+С1". При осуществлении
процесса в бездиафрагмениом электролизере образующийся на
аноде хлор может частично реагировать с раствором едкого натра,
проникающим из катодного пространства, с получением гипохло-
рита: Cl2-f-2NaOH„^NaCIO + NaCl + H2O. Гипохлорит натрия по
мере накопления начинает подвергаться электролизу, при этом об-
разуется хлорат и кислород: 6СЮ"+6ОН"—6е->2СЮ-+4С1~ +
+ 1 */2 О2 + 3 Н 2О.
Если целевым продуктом при электролизе раовора поварен-
ной соли считать анодное выделение газообра того хлора, то его
образующееся количество, вследствие протекания указанных по-
бочных электрохимических реакций и вторичных химических ре-
акций в объеме электролша, не будет, естественно, соответство-
вать затрачиваемому согласно законам Фарадея количеству про-
пущенного электричества. Но если учесть часть тока, которая
14
расходуется на все побочные электрохимические реакции, то по-
лученные результаты не будут противоречить этим законам.
Чтобы учесть влияние побочных параллельных и вторичных
реакций, введено понятие выхода по току, которое характеризует
полезное использование тока и дает ту часть количества .протек-
шего электричества, которая приходится на долю целевой элек-
тродной реакции:
Вт = Аипр/Ляц., (1.5)
где ДгПор и Лт, — количество граммов основного целевого продукта, соответ-
ственно выделившегося к которое должно было выделиться теоретически по
закону Фарадея при пропускании одного и того же количества электричества.
Выход по току можно представить также в виде отношения:
Вт = <7т/«7пр. 0-6)
где qx и <7пр — соответственно теоретическое количество электричества, кото-
рое должно было быть затрачено на проведение данного процесса по закону
Фарадея, н действительные затраты электричества на единицу целевого про-
дукта.
Выход по току всегда относится к определенной реакции па
электроде. Следует различать катодный (Втк) и анодный (Вта)
вы хоты по току. Суммирование выходов по току всех одновре-
менно протекающих иа электроде реакций должно всегда дать
100 %.
1.3. ВОЗМОЖНЫЕ ОБЛАСТИ РЕАЛИЗАЦИИ
ЭЛЕКТРОХИМИЧЕСКИХ ПРОЦЕССОВ
В ТЕХНОЛОГИИ ОЧИСТКИ ВОДЫ
Основными процессами па электродах при электролизе водных
растворов являются: на катоде— выделение водорода, разряд ме-
таллических ионов с электрохимическим выделением (осажде-
нием) металлов или восстановление веществ без выделения само-
стоятельной фа<ы, иа аноде — выделение кислорода, галогенов,
окисление веществ без выделения самостоятельной фазы или
электролитическое растворение металла электрода.
Рассмотрим некоторые примеры использования электрохими-
ческих процессов в системах водоподготовки и очистки сточных
вод.
Выделяющиеся иа катоде пузырьки водорода способны транс-
портировать нерастворимые примеси из объема жидкости на ее
поверхность: поднимаясь в потоке жидкости, они сталкиваются
с нерастворимыми частицами, прилипают к ним и флотируют нх
на поверхность. Использование электролиза для получения газо-
вых пузырьков находит применение при электрофлотационной очи-
стке сточных вол от взвешенных, коллоидных и эмульгированных
загрязнений, а также при пенной сепарации поверхностно-актив-
ных веществ.
К катодным процессам разряда металлических ионов, сопро-
вождаемым выделением (осаждением) твердой фазы, относятся
15
так называемые процессы электрокрнсталли lamin металлов, име-
ющие место при очистке сточных вод от попои цинка, никеля,
меди, кадмия и других (например, для сточных вол гальваниче-
ских производств). Электрокрнсталлизация металлов существенно
отличается от обычного возникновения и роста кристаллов в жид-
кости, что объясняется прежде всего влиянием сил внешнего
электрического поля. Специальным подбором материала и формы
электродов, условий и режима электролиза можно существенно
влиять на структуру катодных осадков.
Примером катодного процесса восстановления без выделения
самостоятельной фазы может служить процесс ионной переза-
рядки металлов и их оксидов. При этом разряд ионов па катоде
сопровождается актом присоединения электронов с изменением
только лишь валентности элементов: Мп3+ + е->-Мп2+, МпОч--)-
+ e-*-MnOt2-; Fe’+—е-+Ге2ь и т. д.
Катодные процессы могут быть также использованы для вос-
становления органических соединений по вышеуказанному меха-
низму. В ряде случаев катодное восстановление органического ве-
щества состоит из двух последовательных стадий [5]: разряда
ионов водорода с образованием атомарного водорода и химиче-
ского восстановления органического соединения атомарным водо-
родом. Часто в электрохимической реакции па катоде участвуют
непосредственно молекулы органического вещества, превращаясь
в органические анионы R + e-»-R- При этом второй стадией про-
цесса будет нейтрали<ация аниона с образованием продукта гид-
рирования R~ + H+->-RH. Возможно также одновременное участие
в разряде попа водорода и молекулы органического вещества R +
+H++C-+RH Использование процесса катодного восстановления
органических примесей в технологии водоочистки целесообразно
в том случае, когда прямое анодное окисление этих примесей тре-
бует больших затрат электроэнергии, а образующиеся катодные
продукты восстановления нетоксичны (или малотоксичны) или
легко подвергаются дальнейшей окислительной деструкции [33, 45].
Примером такого процесса служит электрохимическая очистка
сточных вод от органических нитросоединений (нитробензола,
нитротолуола и т. п.) [33], которую проводят в электролизере
с инертной диафрагмой. При этом сточную воду предварительно
пропускают через катодную камеру, где происходит электрохими-
ческое восстановление широсоедипеиий до ампиосоедииеппй, а за-
тем обрабатывают в анодной камере для окисления полученных
аминов до нетоксичных продуктов.
В некоторых случаях растворенные в воде органические соеди-
нения могут восстанавливаться на катоде с образованием нерас-
творимых в воде продуктов, выпадающих в осадок. Этот процесс
также находит применение в технологии очистки промстоков, на-
пример при электролизе сточных вод производства антрахнно-
сульфокислоты, являющейся продуктом для синтеза многих кра-
сителей [33]. Аптрахпносульфокнслота восстанавливается на жнд-
16
ком ртутном катоде до нерастворимого в воде антрахинона, кото-
рый выпадает в осадок и затем утилизируется в основном произ-
водстве.
Все многообразие анодных реакций можно разделить на два
процесса, происходящих на нерастворимых и растворимых анодах.
Типичными процессами на анодах из материалов, не подвер-
гающихся электрохимическому растворению, в зависимости от
солевого состава раствора и условий электролиза являются про-
цессы выделения кислорода и хлора. Возможно также образова-
ние перекиси водорода, озона и других соединений. Получаю-
щиеся при этом анодные продукты являются сильными окислите-
лями, обладающими особенно большим запасом химической
энергии в момент их образования, поэтому они широко исполь-
зуются для обеззараживания и очистки природных и сточных вод
от органических загрязнений.
Электролиз растворов поваренной соли или естественных хло-
ридных растворов — подземных минерализованных вод и морской
воды — используется для получения гипохлорита натрия как вы-
сокоэффективного дезинфектанта.
Основное значение в процессах электрохимической очистки
сточных вод с использованием нерастворимых электродов имеют
процессы анодного окисления органических примесей, многие из
которых могут подвергаться глубокому деструктивному распаду
вплоть до образования углекислого газа, воды, азота, аммиака и
других газообразных продуктов. В некоторых случаях анодное
окисление органических и неорганических соединений приводит
к образованию нетоксичных и малотокснчных продуктов.
При использовании анодов из железа, алюминия, меди, цинка
и других металлов происходит их электролитическое растворение
с переходом в раствор ионов этих металлов, которые затем при
гидролизе образуют нерастворимые гидроксилы. Свежеобразован-
ные гидроксиды обладают повышенной адсорбционной активно-
стью к коллоидным и взвешенным частицам. Этот процесс, назы-
ваемый в общем случае электрокоагуляцией, широко используется
при очистке воды от нерастворимых примесей, эмульгированных,
а иногда и растворенных соединений, способных сорбироваться на
хлопьях электрогенерированных коагулянтов (гидроксидов ме-
таллов).
При электрокоагуляции загрязненных жидкостей могут проте-
кать и другие электрохимические, физико-химические и химиче-
ские процессы, происходящие в следующей последовательности:
электрофоретическое концентрирование, т. е. направленное дви-
жение дисперсии как свободно заряженных частиц и концентри-
рование их у поверхности электродов; электролитическое раство-
рение анодов и образование гидроксидов металла; поляризацион-
ная коагуляция дисперсных частиц; упаковка первичных агрега-
тов и флокуляционная коагуляция; флотация образовавшихся
агрегатов пузырьками электролитических газов [20]. Все эти про-
цессы могут обеспечить высокую степень очистки жидкостей, со-
17
держащих загрязнения в различных фазово-дисперсных состоя-
ниях.
В коллоидных системах часть ионов адсорбирована коллоид-
ными частицами, которые при этом получают тот или иной заряд;
при наложении постоянного электрического поля заряженные кол-
лоидные частицы перемещаются электрофоретически к одному из
электродов и осаждаются иа нем. Электрофоретические явления,
наблюдаемые при наложении на дисперсные водные системы од-
нородных и неоднородных электрических полей, могут быть
использованы для очистки высококонцентрированных стоков,
а также при обезвоживании осадков [20].
При электролизе растворов движению заряженных частиц
в одном направлении соответствует движение жидкости в обрат-
ном направлении; это становится особенно заметным, если задер-
жать твердые частицы фильтром или диафрагмой. Такие про-
цессы, основанные на электроосмотических явлениях, могут наптн
применение при электрофнльтроваиии воды, загрязненной нерас-
творимыми примесями, а также при уплотнении обводненных
осадков [30].
Специфической разновидностью электрохимической обработки
воды является электродиализ — процесс электролиза с использо-
ванием полупроницаемых мембран, образующих между электро-
дами отдельные камеры; при этом можно осуществлять сепара-
цию ионов солей, образование и концентрирование минеральных
и органических кислот, а также едких щелочей. Электродиализ
применяется для опреснения соленых н солоноватых вод, а также
для удаления растворенных солей из шахтных и других сточных
вод. Электродиализаторы с использованием электрохимически ак-
тивных ионообменных мембран рекомендуются [18, 105] для реге-
нерации ценных продуктов пз высококонцентрированных про-
мышленных сточных вод (отработанных технологических рас-
творов).
Большое значение в технологии водоочистки приобретают про-
цессы электрокорректнрования pH и Eli, позволяющие изменять
валентное состояние примесей воды и приводить к взаимодейст-
вию последних и изменению их фазово-дисперсного состояния. Это
обеспечивает во многих случаях высокоэффективную очистку жид-
костей от растворенных и коллоидных загрязнений [92].
В постеднее время проводятся всесторонние исследования по
выявлению возможности и целесообразности использования элек-
трического разряда для очистки и обеззараживания природных и
сточных вод. Получены положительные результаты по использо-
ванию подводных электрических разрядов в качестве источника
ударных волн для изменения гидродинамических свойств дисперс-
ной фазы с улучшением ее седиментационных характеристик [14,
20]. При этом обеспечивается также глубокая минерализация ор-
ганических соединений и хороший бактерицидный эффект, объяс-
няющиеся окисляющим действием образующихся при разряде
свободных радикалов и их продуктов, а также воздействием пдаз-
18
мепной составляющей разряда и сопутствующими ему кавитаци-
онными явлениями.
Важным аспектом прикладной электрохимии является исполь-
зование электрохимических методов анализа природных и сточ-
ных вод (кулонометрия, полярография, амперометрическое и
потенциометрическое титрование, изотахофорез и др.). Электро-
химические методы анализа используют закономерности электро-
химических явлений. Такне преимущества этих методов, как бы-
строта, высокая точность, надежность, экономичность, возмож-
ность полной автоматизации процессов отбора проб и проведения
анализов в небольшом объеме жидкости, делают незаменимыми
электрохимические методы анализа при осуществлении контроля
за технологическими процессами водоочистки и наблюдении за
состоянием поверхностных источников. Методами электрохимиче-
ского анализа определяют отдельные элементы, ионы или веще-
ства, некоторые общие характеристики воды, например такие, как
удельная электропроводность, pH, Eh, химическое поглощение
кислорода (ХПК), биохимическое потребление кислорода (ВПК),
жесткость воды и др.
Таким образом, электрохимические процессы находят в на-
стоящее время н могут найти в дальнейшем достаточно широкое
применение в технологии водоподготовки и очистки сточных вод.
Электрохимические методы в соответствии с общепринятой
классификацией относятся к физико-химическим методам очистки
водных систем. Они отличаются многостадийностью и относитель-
ной сложностью происходящих в аппаратах водоочистки физико-
химических явлений. Механизм и скорости протекания отдельных
стадий зависят от многих факторов, выявление влияния и правиль-
ный учет которых необходимы для оптимального конструирова-
ния аппаратов электрообработки и рационального ведения техно-
логических процессов очистки воды.
ГЛАВА 2
ЭЛЕКТРОЛИТИЧЕСКИЕ СВОЙСТВА ВОДЫ
И ВОДНЫХ РАСТВОРОВ
2.1. ЭЛЕКТРОЛИТИЧЕСКАЯ ДИССОЦИАЦИЯ ВОДНЫХ СИСТЕМ
Водные системы, подвергающиеся обработке электрическим
током, включают непосредственно воду как растворитель, при-
месные вещества, которые могут находиться в воде в различной
степени дисперсности, а также продукты электролиза, концентра-
19
Рис. 2.1. Поведение примесей воды под воздействием электрического поля
цня, состав и дисперсность которых зависят от продолжительно-
сти и способа обработки.
Чистая вода, являясь слабым электролитом, относится к пло-
хим проводникам тока (степень диссоциации ее на ноны ничтожно
мала), поэтому перепое зарядов в воде при электролизе осущест-
вляется в основном за счет присутствующих в ней примесей или
накапливающихся продуктов электролиза.
Примеси в воде могут находиться в грубодисперсном, коллоид-
ном, молекулярном н ионном состояниях. Вещества во всех четы-
рех группах степени дисперсности взаимодействуют с электриче-
ским полем, однако в переносе зарядов преимущественно участ-
вуют только частицы, находящиеся в растворе в коллоидном и
ионном состояниях (рис. 2.1). Соотношение зарядов, переносимых
этими формами, определяется их концентрациями и зарядом от-
дельных частиц. В связи с существенным преобладанием числа
частиц в ионном состоянии основная доля зарядов переносится
этими частицами.
Вещества (частицы), обладающие ионной проводимостью, на-
зывают электролитами. Сильные электролиты полностью диссо-
циируют на ноны, слабые — лишь частично.
Согласно теории Аррениуса одной из основных характеристик
раствора является степень диссоциации растворенного вещества
a=NtINt, (2.1)
где Ni — число молекул, распавшихся на ноны, Ni — общее число молекул
растворенного пещества.
При «= 0 диссоциация отсутствует, при «= 1 все молекулы
диссоциируют на ноны. Степень диссоциации зависит от природы
растворенного вещества и рас1ворнтеля, копцеп1рацни раствора и
20
температуры. Величина а для слабых электролитов составляет
0,01—0,02, для сильных—0,8—0,99.
Диссоциация, так же как и любая другая химическая реакция,
характеризуется константой равновесия, называемой константой
диссоциации, которая зависит от температуры раствора и типа
растворителя. Так, при диссоциации серной кислоты при Т=25 °C
Кя = [Н+]2 [SO2-]/[H2SOj =1,2- IO"12. (2.2)
Зависимость между степенью и константой диссоциации опре-
деляется законом разбавления Оствальда:
Кд = агС/(1 - а) = а«/(। - а) (I /Км). (2.3)
Так как Ka=const, то при разбавлении раствора изменяется
степень диссоциации в соответствии с уравнением (2.3). При вы-
соких концентрациях сильные электролиты начинают проявлять
свойства слабых, что в соответствии с теорией Дюбая — Гюккеля
объясняется электростатическим взаимодействием ионов. Для
учета этого фактора в уравнения, характеризующие равновесие
реакции в растворах, вместо концентраций компонентов вводят
заменяющую их величину, называемую активностью, которая свя-
зана с концентрацией соотношением
а' = уС. (2.4)
Для многокомпонентных растворов используют среднюю ак-
тивность электролита <ic' и средний коэффициент активности ус,
определяемые по формулам:
< = («Г (2.5)
Yc = (YKW+Y.A/’),/jV-- (2-6)
Для од| о-двухзарядного электролита
а'= С (ук уа)‘2 - Сус. (2.7)
Коэффициенты активности можно определить по упрощенной
формуле Девиса [7]:
1«У*= — 2*0.511 Vpp/О + 15 Vi^T) + 0,2ррг’. (2-8)
Ионная сила раствора определяется по уравнению
1‘Р- 0.526.-г?. (2.9)
Средний коэффициент активности может быть найден на осно-
вании табличных данных, приведенных в справочной литературе
для ряда растворов [57]. Значения у, для некоторых химических
реагентов, применяемых в технологин обработки воды, приведены
и табл. 2.1.
21
Таблица 2.1. Средние коэффициенты активности некоторых растворов
реагентов при 25 С
Концентра- ция, моль/дм3 AI,(SO.), FeCI, H,SO, НС1 NaOH NaCI MrS< >.
0,001 - 0,80 0,830 0,9656 0,965
0,005 —— 0,65 0,639 0,9285 — 0,927 —
0,01 — 0,59 0,544 0,9048 -— 0,902 0,40
0,05 — 0,47 0,340 0,8404 0,818 0,819 0,22
0.1 0,035 0,41 0,265 0,796 0,766 0,778 0,18
0,5 0,014 0,35 0,154 0,757 0,693 0,682 0,068
1.0 0,018 0,42 0,130 0,809 0,679 0,658 0,049
2.2. ИОННОЕ РАВНОВЕСИЕ В РАСТВОРАХ ЭЛЕКТРОЛИТОВ.
ГИДРОЛИЗ
Как уже отмечалось, при растворении вещества в воде проте-
кает электрохимическая диссоциация. Однако процесс диссоциа-
ции является обратимым, так как катионы и анноны, встречаясь
в растворе, вновь соединяются в молекулы, т. е. происходит про-
цесс моляризации. Как н во всяком обратимом процессе, здесь
устанавливается равновесие, характеризующееся в общем случае
константой равновесия
Кр= [А] [В] [АВ]. (2.10)
Вода, хотя и в незначительной степени, но диссоцннр)ет на
ионы, в результате чего устанавливается равновесие:
2Н1О^Н»О+ + ОН-. (2.11)
Данное равновесие описывается уравнением ионного произве-
дения воды:
K. = [HSO+]|OH-]. (2.12)
Ионное произведение воды при 22 °C равно 10-14. В нейтраль-
ных средах [НзО+]=[ОН“]= 10’. При [Н3О+]>[ОН_] раствор
имеет кислую реакцию, при [НзО+]<[ОН_]— щелочную. Для ха-
рактеристики раствора достаточно знать концентрацию [НзО+],
которую удобно выражать при помощи водородного показателя
pH = — lg[H,O+]. (2.13)
При диссоциации вещества на ионы равновесие характеризу-
ется константой диссоциации, о которой упоминалось выше.
В случае диссоциации комплексных частиц константу диссо-
циации называют константой нестойкости комплекса (Кн), кото-
рую используют как характеристику устойчивости любой ком-
плексной частицы в растворе независимо от того, какие лиганды
она отщепляет. Так, для АЩБСМз, являющегося нестойким гид-
роксокомплексом, в растворе существуют ионы |AI (SO<) (ОН2)4]+,
[Al (SO4)s(OH2)2] и др. И процессы диссоциации характеризу-
22
Таблица 2.2. Произведение растворимости некоторых труднорастворимых
в воде веществ
Вещество Т. ’С ПР Вещество Т. °C ПР
А1 (ОН), 20 1,52-10-» СаСО, 15 9,9-10-»
Fe (ОН), 18 4,8-10-» СаСО, 25 4,8-10_»
Fe (ОН), 18 3,8-10-* CaSO4 25 6,26-10-»
Mg (ОН), 25 5,5-10-« CaS04-2H,O 25 1,3-10-»
FeS 25 3,7-10-“ AgCl 26 1,7-10-»
FeSO, 20 2,5-10-» Са (ОН), 18 5,47-10—•
ются следующими значениями Кн (молекулы растворителя
в уравнениях опущены):
[Al (SO4)2]“ [Al (SO4)]+ + SO*", K„ = 1.26 IO-2;
[Al (SO4)]+x* Al34- + SO2", K' = 6,3 -10~4.
Аналогично для железа
[FcCl3]- [FeCl2]+ + Cl', K„ = 10;
[FeCl?]+rt [FeCl|2+ + Cl-, К'= 2.24-10~*;
(2 14)
(2-15)
(2.16)
(2-17)
(2.18)
(FeCI]2+rt Fe^ + Cl-,
K„ == 3,54 JO-2.
Константы диссоциации, в том числе и константы нестойкости,
можно заменить десятичным логарифмом, взятым с обратным зна-
ком, т. е. рКд = —1g Кд. Для различных растворов эти величины
приведены в справочной литературе [57].
При растворении труднорастворимых веществ, когда устанав-
ливается равновесие между твердой фазой и насыщенным раство-
ром, используют произведение растворимости (табл. 2.2):
ПР=1А+][В-]. (2.19)
Химическое взаимодействие примесей с водой обусловливает
протекание процесса гидролиза, приводящего к связыванию водо-
родных или гидроксильных ионов с образованием труднораство-
римых продуктов. Это приводит к нарушению равновесия между
водородными и гидроксильными ионами, т. е. к изменению pH.
Гидролизу подвержены соли слабых кислот и слабых оснований,
слабых кислот и сильных оснований, сильных кислот и слабых
оснований. Соли, образованные сильной кислотой и сильным осно-
ванием, гидролизу не подвергаются. Гидролиз протекает медленно,
что объясняет изменение во времени pH растворов гидролитиче-
ски расщепляемых солей.
Величина гидролиза .характеризуется степенью и константой
гидролиза. Степень гидролиза р определяется отношением числа
молекул соли, подвергшихся гидролизу, к общему числу молекул
23
Таблица 2.3.Зависимости, связывающие константу гидролиза Кг с ионными
произведением воды Кв и константой диссоциации слабой кислоты (основания)
KJ(K-)
Слабая кислота и сильное основание Сильная кислота и слабое основание Слабая кислота и слабое основание
кг= кв/к* кг = к„/к? в А кг= кв/(к;к«)
Кг = рс/ (I - р) Кг = Р=С/ (I — Р) к, = pMi -Р)«
соли в растворе. Так как процесс гидролиза обратим, то равнове-
сие любого процесса, например
Ме,+ + Н,О Me (OH)(Z~’>♦ + Н+, (2.20)
определяется константой гидролиза Кг:
Кг= (ОН>и—i)+ CH+/CMe(z4-i). (2-21)
Константа гидролиза связана с ионным произведением воды,
константой диссоциации кислоты (основания) и степенью гидро-
лиза зависимостями, приведенными в табл. 2.3.
Гидролизу подвергаются катионы тех металлов, которые обра-
зуют слабые основания. К ним относятся подавляющее большин-
ство металлов периодической системы элементов, а также катион
NII«+. Практически не подвергаются гидролизу только ионы ще-
лочных металлов 1 i+, К+, Na+, Rb+, Cs+ и щелочно-земельных ме-
таллов Cd2+, Cr**, Ва2+. Все эти ионы образуют с гидроксид-
ионами ОН~ сильные основания.
Степень гидролиза р зависит от температуры, концентрации
раствора и pH среды. Повышение температуры, усиливая диссо-
циацию воды увеличивает концентрацию водородных и гидро-
ксильных ионов. При гидролизе солей, образованных слабым
основанием и сильной кислотой или сильным основанием и слабой
кислотой, имеет значение и понижение концентрации раствора.
Поэтому для усиления гидролиза следует пользоваться разбав-
ленными растворами с более высокой температурой. Если какой-
нибудь продукт гидролиза выделяется из раствора, выпадая
в осадок, или улетучивается, то гидролиз протекает практически
с полным разложением исходной соли.
Соль, образованная сильной кислотой и слабым оспонаннем,
гидролизуется в большей степени при более высоких pH, а соль,
образованная сильным основанием и слабой кислотой,— при бо-
лее низких pH (табл. 2.4).
Значение pH, при котором начинает выпадать в осадок гидро-
ксид металла, называется pH гидратообразовання (pH,). Вели-
чина рНг тесно связана с произведением растворимости катиона
солн.
21
Таблица 2.4. Определение величины pH растворов при гидролизе
Химическое вещество
Сильная кислота
Сильное основание
Слабая кислота
Слабое основание
Слабая кислота + соль
слабой кислоты
Слабое основание + соль
слабого основания
Соль слабой кислоты и
сильного основания
Соль сильной кислоты и
слабого основания
Соль слабой кислоты и
слабого основания
Соль сильной кислоты и
сильного основания
Аналитические зависимости, характеризующие
изменение pH при концентрации вещества. г-*кв/л
|н+| = с;: pH = -igc;
|°Н-| = <Ц; pOH=-lgc;
pH = - l/2(lg к; + lg cj 1 /2(рК* - 1g <)
pH = 14 + l/2(lg К? + lg Q 14 + l/2(lg сщ-
-PK-)
pH = lg (C'c/CK) - lg KJ = PK« + lg (С’с/С'щ)
pH = 7 — 1/2 lg K* + l/21gC;=l4-|pK£ +
+ lg(CXu)|
pH =7-1/2 lg Кд— 1/2 IgC; = 7 + l/2pK“+
+ i/2 ige;
pH = 7 + 1/2 lg Кд — 1/2 lg Cj = 7 — l/2pK^ -
—1/2 IgC'
pH = 7 - 1/2 lg K£ + 1/2 lg K* = 7 + l/2pK* -
- l/2pK£
pH не изменяется
Концентрация водородных ионов, соответствующая началу об-
разования гидроксидов в зависимости от концентрации металла и
произведения растворимости гидроксида, выражается уравне-
нием:
pH = 14 + 1/(» 1g ПРМе (ОН)х+) - 1/(т lg Me»*). (2-22)
Увеличение концентрации металла (соли) приводит к умень-
шению pH, т. е. сдвигает рНг в область более кислых значений.
В качестве примера рассмотрим процесс гидролиза, который
может протекать у поверхности растворяющегося стального анода
в растворах различных нейтральных солей. Выделяющиеся ионы
Fe2+ частично окисляются кислородом воздуха до Fe3+. Исполь-
зуя константу гидролиза ионов Fe2+ и Fe3+ (IO-9,5 и 5-10~3 соот-
ветственно), можно рассчитать pH среды в прнанодном простран-
стве [77].
При гидролизе Fe2+ по реакции Fe2++H2O5=tFeOH' + H+ по-
лучим: Кг = СРеОН_ • Сн+/Сре,+= 10—®-5, Среон-= • Величина pH
даже при CFe«+ = 1 моль/л не превышает 4,75. Если же
увеличивать концентрацию ионов Fe2+ до достижения pH гидра-
25
тообразойания с выделением осадка l'e(Oll)2, то pit среды ста-
билизируется около значения ~9, так как
I1PFe(Oll),“ С,ц 1 10 *»; CFe>. cnll
сОН-=т/* ,0~“ М'л; рОН = 5 и pH = 14 —5 *».
Однако даже незначительное количество ионов Fe3+ в растворе
приводит к сильному подкислению среды за счет гидролиза этих
ионов:
(Fe»+ Ц- Н3О+ FeOH’+ + Н+);
К, = Среон«+ + Сн+/С’Ре,+ = 5-10-». Среоп*»
откуда
Сн+ = д/^i СРе,+ = ^/5-10 ’ CFt,+ .
При CFe»+ =1 моль/л pH = 2,0.
Аналогичные расчеты могут быть выполнены н для других
случаев гидролиза.
2.3. ПОДВИЖНОСТЬ И ЧИСЛА ПЕРЕНОСА ИОНОВ
Ионы участвуют в тепловом движении, обеспечивающем хао-
тическое перемещение частиц в произвольном направлении. При
этом в межэлектродном объеме раствора устанавливается средняя
концентрация электролита.
Направленное движение ионов в растворе возникает при про-
хождении электрического тока, который вызывает также ускоре
нпе движения ионов. Этому ускорению противодействуют силы
вязкости, а также релаксационные и электрофоретические явле-
ния, возникающие при движении иона [100]. Поэтому ионы двига-
ются в растворе с постоянной скоростью, обусловленной балансом
этих сил. Хотя в переносе тока участвуют все ионы, присутствую-
щие в растворе, только строго определенные из них разряжаются
на электродах. Во всех случаях количество электронов, участвую-
щих в электродных реакциях, компенсируется количеством элек-
тронов, принесенных ионами к границе раздела фаз до выполне-
ния условия электропейтралыюсти.
Следует отменив, что концентрация ионов, участвующих в пе-
реносе, у катода и анода будет неодинакова из-за разности скоро-
стей различных ионов в растворе. Убыль концентрации в прн-
катодном \СК и нрпанодном АСа слоях связана со скоростью дви-
жения попов следующим соотношением:
ДСК/АС, = (Св - Ск)/(С# — С,) = vjva. (2.23)
Скорость движения иона в см-с-1 при градиенте потенциала,
равном единице, т. е. при напряженности поля В-см-1, принято
называть абсолютной подвижностью:
vi = zje/(6nrq).
(2.24)
26
Таблица 2.5. Относительные подвижности (10* См-м’экв “*) некоторых
ионон при бесконечном разбавлении
Пид нона Относительные подвижности при Т. °C
0 5 15 18 25 35 45 55 100
н+ 225,0 250,1 300,6 315,0 349,8 397,0 441,4 483,1 630
Na+ 26,5 30,3 39,8 42,8 50,1 61.5 73,7 86,9 145
К+ 40,7 46,7 59,7 63,9 73,5 88,2 103,5 119,3 195
nh+ 40,2 — — 63,9 73,5 88,7 — — 180
Mg’+ 28,9 — — 44,9 53,0 — — — 165
Са’+ 31,2 — 47,0 50,7 59,5 73,3 88,2 — 180
Fe‘+ 28,0 — — 44,5 53,5 — — — —
Fe’+ — — — — 68,0 — — — —
ЛР+ 29,0 — — — 63.0 — — — —
он- 105,0 —. — 171,0 198.3 — — — 450
ci- 41.0 47,5 61,4 66,0 76,3 92.2 108.9 126.4 212
NO- 40,0 — — 62,3 71.5 85,5 — — 195
so’- 41.0 — — 68.4 80.0 — — — 260
(2.25)
Так как «Va=F (где Л'А— число Авогадро), то
V, = г< F /(6л/т;Л а)
В электрохимии чаше всего используют понятие относительной
подвижности иоиов
= F о( = 2tF2/(6nrt]/VA). (2 26)
Относительные подвижности иоиов зависят от концентрации
раствора, температуры и природы растворителя. В расчетах при-
нимается радиус гидратированного иона, который тем больше,
чем меньше его истинный радиус. Подвижности некоторых ионов,
наиболее часто присутствующих в природных н сточных водах,
приведены в табл. 2.5.
Следует отметить аномальную подвижность ионов Н+ и ОН-,
объясняемую трансляционным механизмом обмена протонами.
В переносе электричества через растворы электролитов участ-
вуют все ионы, однако вследствие различия их скоростей, кон-
центраций и зарядов доля их участия в переносе различна. По-
этому в электрохимии введено понятие чисел переноса: }
= ЦПо-
Число переноса показывает долю участия данного сорта ионов
qt в переносе электричества через раствор. Ион, двигающийся
с большей скоростью, переносит большее количество электриче-
ства. Очевидно, сумма чисел переноса всех ионов, присутствую-
щих в растворе, равна единице, т. е. Для бинарного элек-
I ролнта п“ 4- л"= 1.
27
Рис. 2.2. Распределение концентрации ионов в межэлектродиом пространстве
при перемешивании жидкости (а) и в неподвижной жидкости (б)
Количество электричества, переносимое через единицу пло-
щади раствора в единицу времени, есть плотность тока:
= «к + '- Е С>к + (2.28)
I I
Используя величину степени диссоциации «С=Ск=Са. по-
лучим
i=ac(£o°+£t,°), (2.29)
откуда 1И = пI < и la = n?i.
Величина п, лежит в пределах от 0 до 1 и зависит от состава
и концентрации раствора.
В результате транспорта ионов в объеме межэлектродного про-
странства возникает градиент концентрации в тонком приэлек-
тродном слое толщиной 0,1—0,5 мм, который в значительной сте-
пени зависит от гидродинамической обстановки в межэлектрод-
ном зазоре. Теоретически при полном отсутствии перемешивания
концентрация транспортируемого иона изменяется линейно в про-
межутке между электродами (рис. 2.2). При разряде иона па
электроде концентрация его в тонком прпэлектродпом слое может
резко падать, несмотря на повышенную концентрацию вблизи
электрода. Однако при возникновении градиента концентрации
неизбежно проявляется эффект диффузии, стремящийся выров-
нять концентрацию в растворе. Окончательное распределение
концентрации иоиов определяется суммой тепловых, электриче-
ских и диффузионных эффектов в жидкости.
2.4. ЭЛЕКТРОПРОВОДНОСТЬ ЭЛЕКТРОЛИТОВ
Определение электропроводности электролитов важно с точки
зрения количественной оценки степени и константы диссоциации,
межионных н других сил в расп оре. С другой стороны, эта ве.тн-
28
чина определяет омическое сопротивление электролита, что позво-
ляет вычислить падение напряжения в электролите и напряжение
электролиза.
Сопротивление раствора определяется аналогично металличе-
ским проводникам:
/?, = р,/ (2.30)
где L — длина прохождения тока в растворе; F. — сечение слоя электролита,
м«
Величина, обратная сопротивлению (1//?.<), называется элек-
трической проводимостью н выражается в сименсах (См).
С точки зрения характеристики электролита более важен по-
казатель электропроводности (См • м-'):
хй = 1 /р, = l,/(R ,F,). (2.31)
Используя закон Ома,
хЛ = = НЕ, (2 32)
(i принимается в А/м2, Еэ— в В/м).
Теоретически чистая вода имеет электропроводность 0,038- 10—®,
талая (дождевая) вода—(14-5)-10-5, природная вода средней
минерализации (2004-500 мг/л) — (54-10) •10-Б, сточная вода —
10~*-s-10-1 Ом_|«см_|.
Величина xr различных растворов электролитов зависит от их
ионного состава и концентрации. Зависимость xr=/(C) характе-
ризуется максимумом, что объясняется увеличением межиониого
взаимодействия в электролите при повышении его концентрации.
Для большинства электролитов такой максимум наблюдается при
концентрациях 100— 200 г/л, что ие характерно для природных и
сточных вод. Для слабоконцентрированиых солей (0,5—5 г/л) мо-
жет быть принят линейный характер зависимости электропровод-
ности от концентрации.
При оценке проводимости раствора используют также экви-
валентную электропроводность, характеризующую электропровод-
ность раствора, в объеме которого содержится 1 г-экв электро-
лита.
Эквивалентная X, и удельная x/t электропроводности связаны
соотношением (м2/Ом • г-экв):
X, = хй/С, (2.33)
где С — концентрация раствора, г-экв/м*.
Эквивалентную электропроводность рассчитывают из экспери-
ментально определяемых значений х« с учетом концентрации С
раствора электролита. Если концентрация раствора выражена
в молях, то эквивалентная электропроводность характеризуется
как молярная и выражается в м2/Ом-моль.
29
Зависимости хп и Л, от температуры описываются эмпириче-
скими уравнениями. Так, зависимость х» от температуры Т опре-
деляется уравнением Кольрауша (Ом ‘-см ')
«Л = »<т-25-с(1+атА7’ + ₽тд7”), (2.34)
где ат и рт — эмпирические коэффициенты; ДТ-Г—25 *С.
Зависимость Х:, от Т определяется аналогичным уравнением
(см2/Ом • г-экв):
Л, = ХГ-о (1 + а^Т + ₽ТП. (2 35)
Так как рт<^ат, то зависимости носят практически линейный
характер в широком интервале температур. Для сильных кислот
ат~0,01б4, для щелочей ат5»0,0190, для солей ат~ 0 0220.
В процессе электролиза образуется газовая фаза, заполняющая
часть объема межэлектродного пространства, поэтому электро-
проводность раствора изменяется в зависимости от газссодержа-
ния и в присутствии газовой фазы может быть вычислена по фор-
муле
4 = <’г*л. (2-36)
где Хд — электропроводность раствора электролита без газовой фазы; <тг —
коэффициент, учитывающие газосодержанне электролита:
аг=(1 —Г)32, (2.37)
или по формуле В. П Машовца:
I/O 0.785Г — Г). (2.38)
Для среднего гззосодержанпя в межэлектродном пространстве
(Г = 0.014-0,04) влияние этой фа <ы на электропроводность незна-
чительно и может не учитываться в практических расчетах. Од-
нако при малых расстояниях между электродами (/л<5 мм) Г
может резко возрастать и это влияние окажется более заметным.
Для сильных электролитов зависимость от С определяется
правилом Ko.il раута:
?, = Ло-Кх VC. (2 39)
Константа Кх определяется по формуле Кх = Ах + В?>-0, где
Ах и Вх—величины, зависящие от температуры, вязкости рас-
твора и диэлектрической проницаемости среды. Для одпо-двухза-
рядного электролита при 25 еС Кх= 1,914-10-* + 7.258-10~3 >.о.
Для слабых электролитов, если принять, что «= (Лэ/Ло). и ис-
пользовать константу диссоциации Кд, то Zo и Лэ могут быть пай
дены из формулы:
Кд = Х,Кх/[Хо(Х0-Хэ)]. (2 40)
Относительную подвижность ионов v,° называют ионной экви-
валентной электропроводностью и выражают в Ом-1 • м2 • г-экв.
30
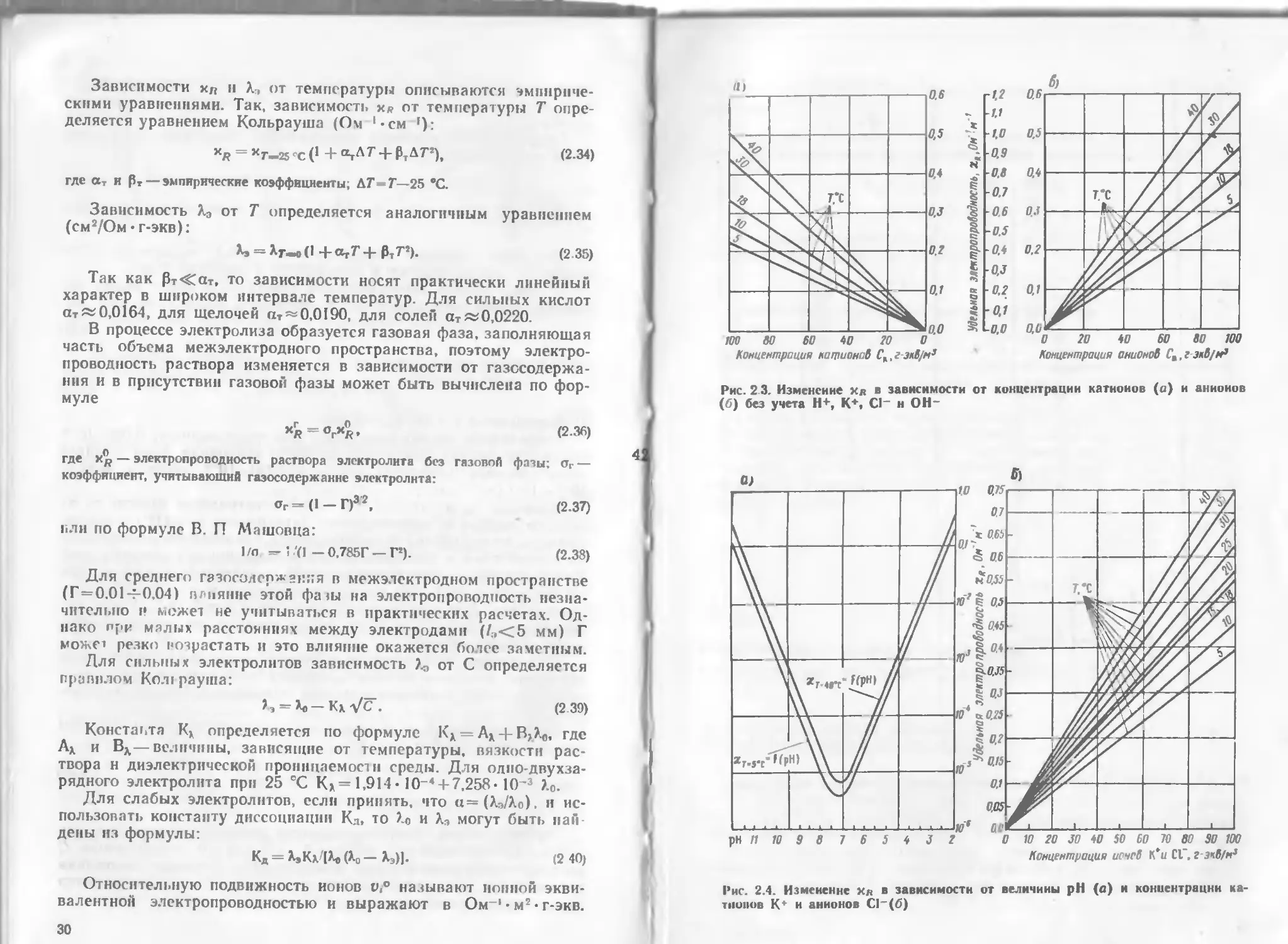
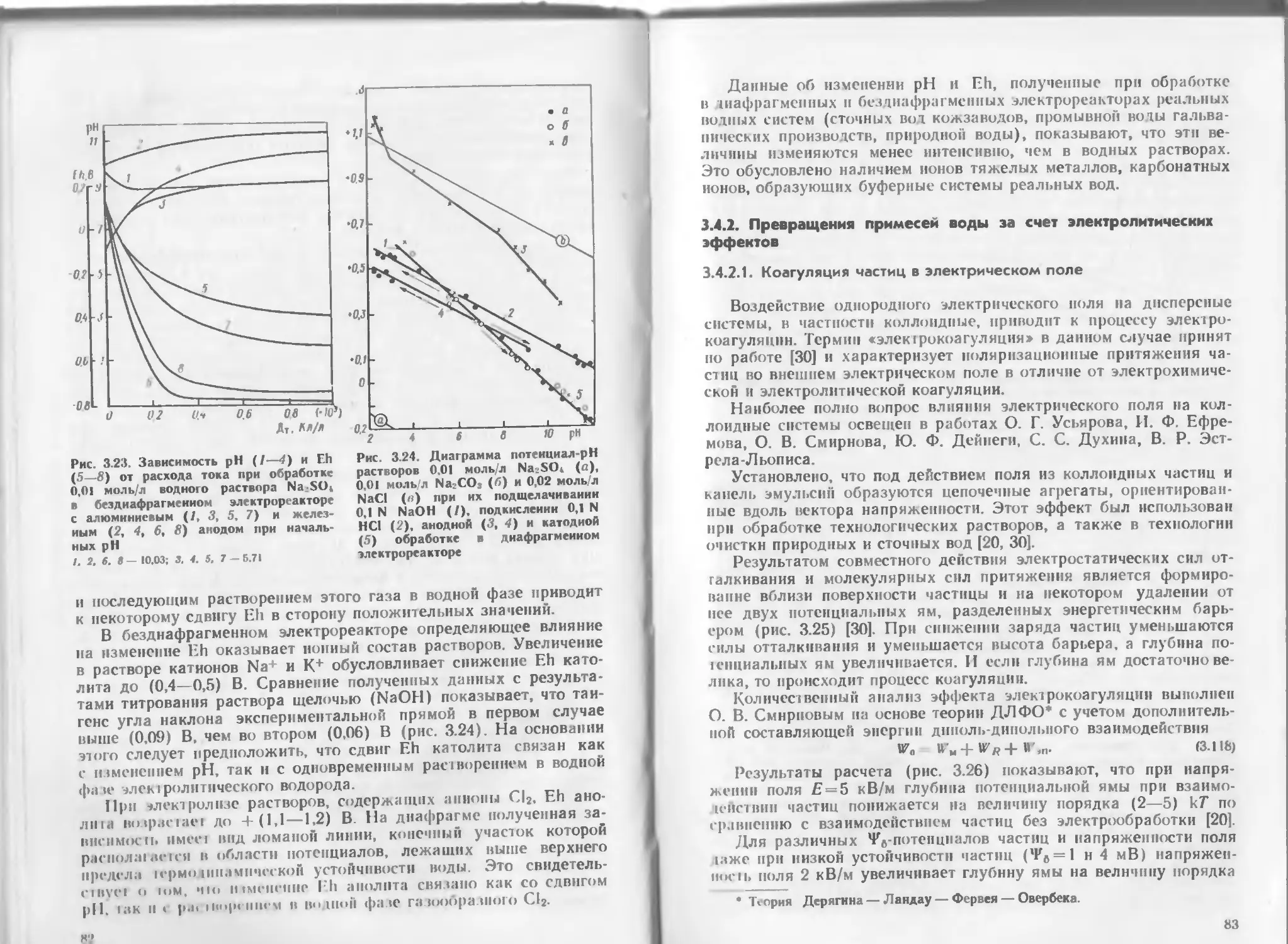
Рис. 2 3. Изменение хв в зависимости от концентрации катионов (а) и анионов
(б) без учета Н+, К*, С1_ н ОН-
Рис. 2.4. Изменение Хя в зависимости от величины pH (о) и концентрации ка-
тионов К* и анионов С1(б)
Расстояние между электродами
4.5
АО
3.5
5.0
25
2,0
1,0- Ключ
э 0,05-1
Ответ
0.5-1
Номогримна
8
J.O-,
2,5-
2.0-
1,5-
1.0-
0.3 -
08-
0,7 -
0.6-
0.5-
0,4 -
0.2-
0,15-
0.1-
80 -|
70-
60-
5и-
40-
30-
25-
20-
15
400
350
300
250
500
200-
150 -
100-
75 -
50-
25-
10-1
0.1-
0.05-
Рис. 2.5. Номограмма расчета омического падения напряжения в электролите
Значения эквивалентной электропроводности могут быть найдены
из формул:
,-а(В«+в;);
-еу г в?“.
(2.41)
(2.42)
32
где В* ; В" — ионные эквивалентные электропроводности при бесконечном
разведении.
Используя относительную подвижность ионов и,°, числа пере-
носа при бесконечном разбавлении можно представить в виде:
«к. = BF/( + й?“) = (2*3)
Ч = В?1(В? + в;-) = Bj»/\). (2.44)
Для любой концентрации раствора сильного и слабого элек-
тролитов при бесконечном разба ленив
п* п*«В*!\. (2.45)
Для слабого электролита любой концентрации
П? = п‘ = аВ?/\). (2.46)
Определение электропроводности электролита производится
с помощью мостов сопротивления, питаемых переменным током
с частотой 500- 1000 Гц.
При невозможности экспериментального определения электро-
проводность может быть приближенно рассчитана на основании
вышеприведенных зависимостей. Такой расчет представлен в виде
номограмм (рис. 2.3—2.5), позволяющих вычислить на основа-
нии данных солевого состава и концентрации с учетом pH и тем-
пературы раствора
ГЛАВА 3
ТЕОРЕТИЧЕСКИЕ ОСНОВЫ ПРОЦЕССОВ,
ПРОТЕКАЮЩИХ ПРИ ЭЛЕКТРОЛИЗЕ ВОДНЫХ СИСТЕМ
3.1. НЕКОТОРЫЕ АСПЕКТЫ ХИМИЧЕСКОЙ
И ЭЛЕКТРОХИМИЧЕСКОЙ ТЕРМОДИНАМИКИ
Объектами изучения термодинамики являются процессы, в ко-
торых происходит изменение химического состава, структуры или
фазово-дисперсного состояния компонентов системы за счет вза-
имных переходов различных форм энергии.
Важнейшими взаимосвязанными понятиями термодинамике
являются энергия (U), теплота (Q) и работа (Л). При этом раз-
личают внутреннюю и внешнюю энергию, прикладываемую к си-
стеме.
2 Заказ М 2148
33
При химических реакциях происходят изменения внутренней
энергии, которые обусловлены переходами элекпронов от одних
веществ к другим или изменением состояния электронов в атомах
реагирующих веществ. Такне изменения внутренней энергии про-
являются в виде выделения или поглощения тепла.
Абсолютное значение внутренней энергии в какой-либо си-
стеме измерить невозможно, однако опытным путем удается оп-
ределить изменение внутренней энергии системы при переходе ее
из одного состояния в другое. Внутренняя энергия является
функцией состояния системы, т. е. зависит от ее начального и ко-
нечного состояния н не зависит от пути процесса.
В отличие от внутренней энергии теплота и работа не явля-
ются функциями состояния, а характеризуют только процесс.
Соотношение между теплотой и работой при изменении общего
запаса энергии системы устанавливается первым началом тер-
модинамики: Al/=Q—А, т. е. увеличение внутренней энергии си-
стемы равно теплоте, сообщенной системе, за вычетом работы,
произведенной системой.
Технологические процессы и химические превращения в ок-
ружающей среде чаще всего совершаются при постоянном давле-
нии и температуре (изобарно-нзотермнческие процессы). В этом
случае тепловой эффект равен изменению эшальпни снстемыД//:
Qp. Т = Икон — Нплч, (3.1)
где Н — энтальпия—новая функция состояния, которая является комбина-
цией параметров в виде суммы: t/4-pV.
В химических процессах одновременно изменяются энергети-
ческий запас системы и степень беспорядка. Если реакция проте-
кает при 7"=const и p = const, то критерием направленности про-
цесса служит энергия Гиббса (О') — изобарный потенциал, ха-
рактер изменения которого позволяет судить о принципиальной
возможности или невозможности осуществления процесса и под-
чиняется уравнению
AG = ДЯ — TAS, (3.2)
где S — энтропия, т. е. функция состояния системы, оценивающая степень ее
беспорядка.
Использование изобарного потенциала дает количественную
оценку движущей силе процесса. Чем больше но абсолютной ве-
личине значение ДО, тем больше движущая сила реакции.
Другими критериями направленности процессов могут служить
изохорный (энергия Гельмгольца), термодинамический, химиче-
ский и электрохимический потенциалы.
Энергия Гельмгольца (F)—функция состояния системы, оп-
ределяемая разностью между внутренней энергией и произведе-
нием термодинамической температуры на энтропию, т. е. F=
= U—TS при условии F=f(T, V).
Термодинамические потенциалы U, Н, F, G — это функции,
убыль которых в равновесном процессе, протекающем при сохра-
34
пепин постоянства значений соответствующей пары параметров
(S н V; S и р; Т н V; Т и р), равна полной работе, произведен-
ной системой, за вычетом работы, совершенной против внешнего
давления. Связь между термодинамическими потенциалами и их
юмнературным коэффициентом дается уравнениями Гиббса—
Гельмгольца:
AG = ЛЯ + Т (dbGldT)p, (3.3)
AF = Д(/ + Т (d\F/dT)v (3.4)
Наряду с характеристическими функциями в термодинамике
используются частные производные внутренней энергии, энталь-
пии, энергий Гельмгольца н Гиббса по числу молен t-го компо-
нента, участвующих в химическом процессе, так называемые
химические потенциалы, которые характеризуют запас энергии
отдельных компонентов системы (раствора). Через величину хи-
мического потенциала (р,) может быть выражена максимальная
полезная работа химической реакции:
<ЮР, Т Ер dNi, или dFTt у = (3.5)
Химический потенциал в идеальных системах (идеальных рае-
шорах пли идеальных газах) зависит от концензрации:
Hi- pO-f-KT ln(C,Y,), (З.ь)
lie р" — химический потенциал i-ro компонента при С<=1, называемый стан-
ллртным химическим потенциалом и являющийся функцией Т (при p=consl).
Выражение (3.6) является уравнением для химического по-
1епцпала незаряженных частиц. Для характеристики энергии за-
ряженных частиц вместо химического потенциала используют так
называемый электрохимический потенциал, который включает
также запас электрической энергии и записывается в виде:
)‘эх = pt + RT In (CfTr) + zt F ф, (3.7)
l ie zt— запас электрической энергии (I моль вещества); <р — электрический
потенциал данной фазы по отношению к бесконечно удаленной точке, лежа-
щей п вакууме.
Для характеристики термодинамического состояния электро-
химических систем применяют связь между напряжением си-
стемы и максимальной полезной работой процесса [93].
Известно, что максимальная работа \Д, которую может со-
вершить система, равна убыли энергии Гиббса. В электрохими-
ческой системе эта максимальная работа равна произведению
плнряженпя системы Е на количество прошедшего электрпче-
ci на гГ:
\Л zF’f -AG, (3 8)
। ic / .химическое напряжение на равновесной электрохимической системе
и in ее 11ектродвнжущая сила (ЭДС).
Понятие «электродвижущая сила» следует отличать от «элек-
ipipiecKoro напряжения системы (ячейки)», определяемого экс-
iirpiiMeiiia.ii.iio. При прохож Тепин электрического тока через
35
систем} напряжение должно быть равно величине U3. ЭДС си-
стемы Е относится только к состоянию равновесия и нс является
функцией силы тока. Если система находится в электрохимиче-
ском равновесии, то Е=*—U3= — SCIzE.
Дифференцированием выражения (3.8) по температуре можно
определить изменение энтропии из температурного коэффициента
химического напряжения системы:
гР(ЭЕ/Э7)0= — (dSGIdT)p. (3.9)
Подставив в уравнение (3.9) выражение убыли энергии Гнббса
и произведя несложные преобразования, получим
Е = — (А/7/z F) 4- Т (дЕ1дТ)р. (3.10)
Так как изотермическая и обратимая электрическая работы
являются полными дифференциалами параметров р и Т, то при
p=const полученное уравнение можно переписать в виде:
Е - (Л//р/г F) + Т tdEldT)p. (3.11)
Совершенно аналогичное выражение можно получить, если
процесс идет изотермически н обратимо при постоянном объеме:
Еу= —(MivlzE)+T(dEldT)v. (3.12)
В этих уравнениях \Н относится к количеству вещества, всту-
пившему в реакцию при прохождении zV количества электриче-
ства, т. е. к 1 молю.
Электрическая работа, совершаемая системой, может быть
больше, меньше или равна изменению энтальпии в -зависимости
от знака температурного коэффициента химического напряже-
ния. Если (dE/dT)p=0, то Е=—\H/zF и электрическая работа
точно равна изменению энтальпии Это уравнение называется
уравнением Томсона. Его можно применять для ориентировочных
расчетов химического напряжения, т. е. ЭДС системы.
3.2. ПРОЦЕССЫ НА ГРАНИЦЕ ЭЛЕКТРОД — РАСТВОР
3.2.1. Электродные потенциалы
Протекание электрохимического процесса сопряжено с обме-
ном заряженными частицами между двумя фазами, у которых
происходит перераспределение электрических зарядов и. следова-
тельно, возникает скачок электрического потенциала на границе
раздела фаз. Появление скачков потенциалов является следст-
вием стремления всякой системы к термодинамическому равно
веспю.
Рассмотрим явление возникновения скачка потенциала на
примере цинкового электрода в растворе сульфата цинка.
При опускании металлической пластинки в раствор своих
ионов металл и раствор начинают взаимодействовать, в резуль-
тате чего происходит переход ионов металла в раствор в обратно.
36
Чтобы вывести иои цинка из кристаллической решетки, надо
затратить работу, равную энергии связи ионов в кристалличе-
ской решетке, т. е. работу выхода иона из металла UM. Для того
чтобы вывести ион цинка из раствора, необходимо затратить ра-
боту, равную энергии гидратации, т. е. энергии связи этого иоиа
с молекулами воды 1/р. Если энергия связи ионов в кристалличе-
ской решетке металла меньше, чем энергия гидратации этих
ионов т. е. как в цинке, то при погружении металла
н раствор ионы металла будут высвобождаться из кристалличе-
ской решетки и переходить в раствор.
Такой переход ионов металла в раствор будет иметь место
до тех пор, пока энергии ионов металла на электроде и в рас-
творе не выравняются, т. е. не сравняются значения соответст-
вующих электрохимических потенциалов. При этом в системе
устанавливается динамическое равновесие, при котором переход
попов цинка в раствор продолжается, но одновременно и с той
же скоростью происходит обратный их переход из раствора
в твердую фазу.
При равенстве скоростей прямого и обратного процессов со-
стояние электрода и раствора не изменяется и электрод нахо-
дится в равновесии с раствором.
У поверхности отрицательно заряженного электрода возни-
кает обкладка из положительно заряженных ионов и, следова-
тельно, скачок электрического потенциала (рис. 3.1) [53].
Скачки потенциалов на границе электрод — раствор могут воз-
никать и в тех случаях, когда ионы материала электрода не
\частвуют в переносе зарядов. Поверхность некоторых металлов
(платина, золото и др.), имеющих прочную кристаллическую pe-
ine жу, может не посылать свои ионы в раствор, а адсорбировать
молекулы, атомы и ионы веществ, способных окисляться или вос-
ешнавливаться, т. е. отдавать или приобретать электроны. Так,
нлирпмер, если погрузить платину в раствор, содержащий ионы
подорола. и продувать через раствор водород, то _на поверхности
иллиты возможно протекание реакции: 2Н++2е->-Н2. Это так
пл 1ываемый водородный электрод.
Возможны случаи, когда обе формы (окисленная или восста-
новленная) находятся в растворе. Например, если опустить пла-
iiniy в раствор солн трехвалентного железа FeCI3, не содержа-
щий восстановителей, то источником электронов для протекания
pc.iKiinii восстановления (Fe3+ -f-е->Fe’+) будет поверхность пла-
iiiiiH. которая получает избыточный положительный заряд, а ра-
< шор отрицательный.
{.ряженная поверхность электрода вместе с прилегающими
к нему противоположно заряженными ионамн образует двойной
• icKipiniecKHH слон (ДЭС).
В простейшем случае ДЭС состоит из ионов одного знака,
р mi юженпых в один ряд, избыточная концентрация которых
пики hiельно общей их концентрации в растворе сохраняется
37
a)
tlemann Pacmlop
Рис. 3.1. Распределение по-
ложительных ионов (а) и
скачка потенциала (б)
у отрицательно заряжен-
ного электрода
лишь на весьма малом расстоянии от поверхности электрода,
равном их радиусу. За этим плотным рядом ионов эквивалент-
ные концентрации катионов и анионов равны (Ск=Са). Измене-
ние их в непосредственной близости от поверхности электрода
в достаточно концентрированных растворах происходит резким
скачком. Но в сильно разбавленных растворах электролитов, ка
кнмн являются большинство категорий природных и сточных вод,
изменение концентрации анионов и катионов происходит не рез-
ким скачком, а более плавно, постепенно приближаясь к равен
ству эквивалентных концентраций при достаточном удалении
от плотного слоя закрепленных па электроде иоиов (рис. 3.2).
Простое строение двойного слоя с резким скачком концентра-
ций ионов к эквивалентному их значению за пределами плотной
части нарушается тепловым движением молекул растворителя и
ионов, а также взаимным отталкиванием разноименных ионов.
В результате ДЭС приобретает размытое (диффузное) строение,
общая толщина которого бо при этом слагается из толщины
плотной части и толщины бл размытой части: бо = ^4-бд. Ве-
личина б, зависит от природы и особенно от концентрации элек-
тролита В чистой воде бд—10~® см, в то время как в концентри-
рованных растворах бд очень мала и 6о~бф (6ф равна радиусу
сольватированных ионов плотной части порядка нескольких анг-
стремов).
С ростом температуры происходит увеличение энергии тепло-
вого движения и возрастает толщина диффузионной части ДЭС.
При одинаковой температуре и концентрации днффузность слоя
с увеличением заряда ионов (т. е. их валентности) уменьшается.
Повышение напряженности внешнего электрического поля сжи-
мает диффузную часть за счет увеличения общего скачка потен-
циала электрод — раствор.
Распределение скачков потенциала у отрицательно заряжен-
ного электрода было показано на рис. 3.1; оно имеет моею в тех
случаях, когда заряд плотной части двойного слоя не превышает
заряда электрода и обусловлен электростатическим притяжением
иоиов к электроду. Однако известно, что ряд попов способен при-
тягиваться к поверхности электрода вследствие ио только элек-
тростатического взаимодейс1вия разноименных зарядов, ио и на-
38
Рис. 3.2. Изменение концентрации катио-
нов и анионов в ДЭС у отрицательно за-
ряженного электрода
С и С а — концентрации катионов и анионов,
mm — поверхность электрода; п’п' — граница
плотной части двойного слоя; пп — граница
диффузной части двойного слоя; х— расстояние
от электрода; 6О> и вд — толщина соот-
ветственно общая, плотной и диффузной частей
ДЭС
Рис. 3.4. Изменение потенциала в ДЭС при
специфической адсорбции катионов (о) и
анионов (б)
— изменение потенциала в диффузной части
двойного слоя; фа— абсолютный скачок потенциала
иа границе электрода с раствором; / — изменение
потенциала при отсутствии специфической адсорб-
ции
Рис. 3.3. Строение ДЭС при
< нецифической адсорбции ка-
тионов («) и анионов (б)
Рис. 3.5. Схема установки по изме-
рению потенциалов
1 — стеклянная трубка с ртутью; 3 — си-
фон; 3 — металлический электрод, у ко-
торого измеряется потенциал; 4 — плати
пированная платиновая пластина; 5 —
стеклянный сосуд; 3 — жидкостный за-
твор; 7 — ацетатный буферный раствор
личин особых, специфических сил адсорбционного характера.
В таких случаях суммарный заряд ионов в плотной части ДЭС
больше, чем заряд электрода. Часть ионов, плотно прилегающих
к iKiiiepxHocTii электрода, в свою очередь притягивает из рас-
ihop.i поны того же знака, что и заряд электрода, и в диффузной
ч.к in двойного слоя появляется избыток ионов с зарядом того
ы Ш.1КП, что и заряд электрода. Строение двойного слоя, от-
39
вечающее случаю специфической адсорбции катионов и анионов,
показано на рис. 3.3, а изменение при этом потенциала с рас-
стоянием х от поверхности электрода — на рис. 3.4.
Как видно из рис. 3.4, при специфической адсорбции скачки
потенциала в плотной и диффузной частях ДЭС имеют противо-
положные таки. Вверх от начала координат отложены положи-
тельные, а вниз — отрицательные значения потенциала. За нуль
принят потенциал раствора в области, где Са = Ск. Из рисунка
следует также, что адсорбция не влияет на суммарный потенциал
<ра между электродом и раствором, имеющий термодинамическое
равновесие.
Условию термодинамического равновесия при погружении ме-
таллического электрода в раствор его потеициалообразующих
ионов соответствует скачок потенциала между электродом и рас-
твором, устанавливающийся при равенстве скоростей прямой и
обратной реакций и являющийся мерой максимальной работы
процесса, самопроизвольно совершающегося на границе фаз, и
называется абсолютным равновесным потенциалом электрода
в данном растворе.
Абсолютный скачок потенциала на границе «электрод — рас-
твор» определяется уравнением
Ф = ф' - Ф* = [(rf - rf')/i F]-(RT/2 F) In (a0K/aRed), (3.13)
или
Ф = Ф» + (RT/Z F) In (aOx/aRed), (3.14)
где ф' и ф" — электрический потенциал соответственно металла и раствора;
И? н И, — стандартные химические потенциалы ионов металла соответ-
ственно на электроде и в растворе, т. е. ионов металла в восстановленной и
окисленной форме; а01 и a Red —соответственно активность окисленной н вос-
становленной форм ионов металла; ф» — стандартный потенциал реакции, про-
текающей на электроде.
Уравнение (3.14), называемое уравнением Нернста, устанав-
ливает зависимость равновесного потенциала электрода относи-
тельно раствора, в который он погружен, от температуры и ак-
тивности окисленных и восстановленных форм вещества, участ-
вующих в электрохимической реакции.
Так как величины стандартных химических потенциалов неиз-
вестны, то неизвестна и величина <р0, следовательно, абсолютный
скачок потенциала <р рассчитать нельзя. Нельзя его и измерить
экспериментально.
Для сопоставления равновесных потенциалов различных элек-
тродов условились измерять их относительно так называемого
нормального или стандартного водородного электрода, представ-
ляющего собой платину, покрытую слоем губчатой платины и
опущенную в раствор кислоты с активностью ионов водорода
ан+ = 1 при продувании через раствор пузырьков водорода под
парциальным давлением рн =0,1 МПа. Установка но измерению
40
потенциала электрода по водородной шкале представлена на
рис. 3.5.
В соответствии с уравнением Нернста равновесный потенциал
водородного электрода
Фн+н,= Фо 4* (/?77zF)Jn (эн+^Рн,)- (3.15)
Согласно этому уравнению потенциал водородного электрода
определяется не только активностью водородных ионов, но и
парциальным давлением газообразного водорода рн , которое
здесь заменяет величину активности восстановленной формы ве-
щества. Для стандартного потенциала водородного электрода
Ф|!*/Н, = Ф-
Таким образом, потенциалом любого неизвестного электрода
но водородной шкале является разность потенциалов между ис-
следуемым и водородным электродом, находящимся в стандарт-
ных условиях.
Равновесный потенциал металлического электрода, на кото-
ром протекает реакция Me1 +ze^tMe, согласно уравнению (3.14)
Фме*+/ме = Фо + (/?7'(zF)ln (аМе**^*Ме)- (3.16)
Так как активность чистого твердого вещества аме при задан-
ной температуре постоянна, то ее условно принимают равной
единице, и выражение (3.16 ) примет вид:
ФМег+ Me = Фо + (Я Т г F) 1п аМег» (3.17)
Из уравнения (3.16) следует, что потенциал металлического
электрода, на котором установилось равновесие, зависит от ак-
тивности только катионов металла. Ионы, непосредственно опре-
деляющие величину электродного потенциала, называются потеи-
цналоопределяющими. Как видно из уравнения (3.17), потенциал
1лектрода является стандартным, когда активность потенциало-
определяющих ионов в растворе равна единице.
Значения стандартных потенциалов ряда электродных реак-
ций приведены в табл. 3.1, пользуясь которыми можноспомощью
сравнения Нернста рассчитать равновесный потенциал электрода
и растворе определенного состава.
Применение водородного электрода для измерения электрод-
ного потенциала не всегда целесообразно, так как для него тре-
буется водород высокой чистоты. Поэтому на практике, как
правило, пользуются различными электродами сравнения с из-
иестиым (по водородной шкале) значением потенциала, вычисляя
кием неизвестное значение потенциала электродной реакции. Для
шмереинй в водной среде применяют электроды второго рода,
состоящие из металла, покрытого малорастворимым соединением
«того металла (соль, оксид или гидроксид), и погруженные в рас-
тор, содержащий анионы малорастворимого соединения. К та-
ким электродам относят каломельный, хлорсеребряный, ртутно-
сульфатпый и некоторые другие. Так, например, хлорсеребрянын
41
Таблица 3.1. Стандартные электродные потенциалы в водных растворах
при 25 °C
Электродная реакция Стандартный потенциал, В Электродная реакция Стандартный потенциал, В
А1-*-А1’+ + Зс —1,66 Си -► Си*+ + 2е +0,345
Zn -► Zn*+ + 2е —0,763 _ 4ОН — 4г -► 2Н О+О, {-0,401
Fc -► Fe,+ + 2е - 0,440 Си -» Си+ + е +0,521
Cd — Cd’+ + 2e —0,403 2Hg — Hg|+ + 2ё +0,798
Co -► Co»+ + 2c - 0,277 Ag -* Ag+ + 2k> +0,799
Fe -► Fe*+ + 3c -0,036 2C1 — 2e — Cl, + 1,360
H, -> 2H + 2e 0,000 Au -*• Au,+ + 3e + 1,50
Примечание. — водородный электрод; кислородный электрод.
и каломельный электроды сравнения и насыщенном растворе KCI
приобретают устойчивый электродный потенциал ( + 0,200 В для
хлорсеребряного и +0,248 В для каломельного). При пересчете
значений потенциалов, полученных с использованием данных
электродов сравнения, проводят алгебраическое суммирование
определяемых величин соответственно с величинами потенциалов
электродов сравнения (В):
для хлорсеребряиого электрода
<Рн*/н, = Фхс = 0.200; (3.18)
для каломельного электрода
Ф°н*/н, < + °’2™’ <3|9>
Если стандартные потенциалы каких-либо реакций непосред-
ственно определить не удается, то производят косвенное опреде-
ление в соответствии с правилом Лютера:
гФсум=22 ~ Фсум 22 zi' (з.2О)
где z н — число электронов, участвующих соответственно в суммарной н
промежуточных реакциях; <р°ум н <р° — стандартные потенциалы соответ-
ственно суммарной и промежуточных реакций.
3.2.2. Окислительно-восстановительное равновесие
и устойчивость водных растворов
При протекании окислительно-восстановительных реакций
возникает потенциал между восстановленной и окисленной фор-
мой вещества. Например, при погружении металла в раствор его
соли, как уже отмечалось, возникает по1еиниал между ионами
42
металла, перешедшим в раствор, и самим металлом, т. е. пропс
ходит электрохимический процесс:
Окисление , _
Me , Ме*+ + и.
Восстановление
Направление окислительно-восстановительных реакций, воз-
можность их протекания и одновременного существования в рас-
творах различных соединений позволяют определять значения
стандартных потенциалов (табл. 3.1), составленных в порядке
возрастания их положительных величин, т. е. отвечающих элек-
। рохимнческому ряду металлов (так называемому ряду напря-
жений).
Окисленная форма вещества с большим потенциалом явля-
ется окислителем для восстановленных форм с более низким по-
(еицналом, и, наоборот, восстановленная форма вещества явля-
ется восстановителем для окисленных форм с более высоким по-
тенциалом. Таким образом, пользуясь стандартным потенциалом,
можно определить, в каком направлении пойдет реакция, и рас-
считать ее окислительно-восстановительный потенциал, величина
которого равна разности потенциалов окислителя и восстано-
вителя.
Окислительно-восстановительный потенциал химических ре-
акций в растворах можно определить на практике также по
уравнению Нернста (3.14), пользуясь отношением концентраций
ионов водорода и молекулярного водорода. Окислительно-восста-
новительный потенциал (Eh) для этой пары примет следующий
вид:
Eh - (RT/zF) 1пЦН+р/|Н21). (3.21)
При подстановке всех констант и замене натуральных лога-
рифмов десятичными (1п = lg/0,4343) получим следующее выра-
жение:
Eh = 0,0591g (|Н+ИН21). (3.22)
Если отрицательный логарифм концентрации водородных
ионов обозначим через pH, а отрицательный логарифм концен-
трации молекулярного водорода, выраженный в атмосферах, че-
рез гНг, то после соответствующих преобразований уравнение
(3.22) примет еще более простой вид:
Eh = 0,059 (г Н, — 2рН), (3.23)
о I куда
Г Н, = (Eh/0,059) 4- 2рН. (3 24)
При этом Eh — характеристика свободной энергии окнслн-
(елыю-восстаиовителыюй системы, a rHj—показатель окислн-
ельно-восстановительных свойств среды: чем меньше гН2, тем
лете идут процессы восстановления; увеличение гН2 и, соответ-
сгиеино, уменьшение давления молекулярного водорода характе-
ризует протекание окислительных процессов.
43
Стандартные потенциалы используются также при решении
многих проблем, связанных с химическим равновесием в раство-
рах. По величине стандартных потенциалов относительно водо-
родного н кислородного электродов судят о термодинамической
устойчивости соединений в воде.
Водные системы, в которых размещены электроды с потенциа-
лом более отрицательным, чем потенциал водородного электрода,
термодинамически неустойчивы. Реакции в таких системах дол-
жны протекать самопроизвольно в сторону получения более окис-
ленных форм с одновременным разложением воды и выделением
нз нее газообразных продуктов электролиза.
Системы, в которых применены электроды с потенциалами
более положительными, чем у равновесного кислородного элек-
трода ( + 0,401 В), также термодинамически неустойчивы и дол-
жны разлагать воду с выделением газообразного кислорода.
Принципиальная возможность получения равновесного потен-
циала какой-либо электрохимической реакции определяется со-
отношением равновесного потенциала электрохимической реак-
ции с участием молекул воды.
Для характеристики термодинамической устойчивости элек-
трохимических систем в водных средах пользуются диаграм-
мами (р—pH, называемыми диаграммами Пурбе. При построении
такой диаграммы иа оси ординат откладывают равновесные по-
тенциалы реакций взаимодействия с ионами, образующимися при
диссоциации воды, а по оси абсцисс — pH (рис. 3.6). Линия ab
на рисунке выражает зависимость потенциала водородного, а ли-
ния cd — кислородного электрода от pH среды.
Зависимость потенциала водородного электрода от активно-
сти ионов водорода в растворе в соответствии с уравнением
Нернста (3.14) дается выражением:
Фн*/н, — Фо. (Н+ н,) + zF)ln а^+, (3.25)
которое с учетом того, что pH = —lg а,|+ можно переписать в во-
дородной шкале потенциалов в виде:
Фн+/н,= —(RTzF) 2,ЗрН,
или при 25 °C
Фн*н.= -0.059РИ. (3.26)
Для потенциала кислородного электрода при 25 °C уравнение
имеет вид:
Фо>/ОН- = 0 401 — 0,0591g аон_, (3.27)
или с учетом ионного произведения воды Кв = [а’н+][аон-]
Фо,/он- = 1 ’23 — 0,059рН. (3.28)
Представленные иа диаграмме Пурбе (см. рис. 3.6) зависи-
мости (3.26) и (3.28), изображенные прямыми линиями с накло-
нами, отвечающими коэффициентам при величине pH, делят диа-
44
Рис. 3.6. Диаграмма термодинамиче-
ской устойчивости воды
Рис. 37 Упрощенная диаграмма по-
тенцнал-рН для системы Гс— Н.О
грамму на области термодинамического состояния. Область
потенциалов, заключенная между этими линиями, является об-
ластью электрохимической устойчивости воды. Область, лежа-
щая ниже линии ab равновесных потенциалов водородного элек-
1рода, соответствует зоне разложения воды с выделением водо-
рода. Область же, расположенная выше линии cd равновесных
потенциалов кислородного электрода, соответствует зоне разло-
жения воды с выделением кислорода. Уравнения электрохимиче-
ских реакций разложения воды в зависимости от активности
среды приведены соответственно на диаграммах ab и cd.
Большое значение приобретают диаграммы применительно
к системам металл — вода, возможность образования в которых
комплексных ионов и гидроксидов, изменяющих свой состав в за-
висимости от pH, значительно усложняет взаимодействие с рас-
творителем.
Рассмотрим это на примере [5] упрощенной диаграммы Пурбе
для систем Fe—Н2О (рис. 3.7), чаще всего используемой при
электрокоагуляции загрязненных жидкостей. На этой диаграмме
учитываются лишь гидратированные формы оксидов и наиболее
важные в коррозионном аспекте реакции в рассматриваемой си-
стеме. На иен представлены далеко не все варианты образования
возможных продуктов взаимодействия железа с водой.
Сетка линий делит диаграмму иа зоны преобладания опреде-
ленных компонентов и фаз. Так, ниже ломаной линии 1—2—3
располагается область, охватывающая те значения потенциалов
п pH, при которых железо существует в металлическом состоя-
нии и является устойчивым. Эта область называется зоной тер-
модинамической устойчивости железа. Выше линии 1 преобла-
даю! гидратированные двухвалентные ионы железа, выше липни
7, Я гидроксиды двухвалентного железа, выше линии 3 попы
45
HFeOj. Область, расположенная выше ломаной линии 10—4—7—
2—8—6, является вероятной зоной пассивности железа, в которой
преобладают твердые его соединения, защищающие железо oi
дальнейшего разрушения. На диаграмме выделяются также две
области потенциалов и pH, внутри которых железо должно под-
вергаться растворению. Одна из них заключена между левой
осью ординат и ломаной линией 1—7—4—10. Металл в пределах
этой области должен растворяться в водных растворах с обра-
зованием ионов Fe2+ или (выше ливни 9) ионов Fe3+. Другая об-
ласть неустойчивого состояния железа с образованием в водных
растворах комплексных анионов HFeOj" заключена между ло-
маной линией 3—8—6 и правой осью ординат.
Таким образом, диаграммы Пурбе позволяют оценить термо-
динамически вероятное поведение металлов в водных растворах,
установить пределы потенциалов и pH, внутри которых данный
металл должен быть устойчив, выяснить химическую природу
продуктов коррозии и обосновать, за счет какой реакции (выде-
ления водорода или восстановления кислорода) идет растворе-
ние металла.
При использовании диаграмм Пурбе необходимо иметь ввиду,
что они указывают лишь на термодинамическую возможность
или невозможность того или иного процесса. В реальных усло-
виях не исключены отклонения, и протекающие процессы могут
быть из-за кинетических ограничений иными, чем те, которые сле-
дуют из общих термодинамических соотношений. Однако даже
упрощенная диаграмма Пурбе позволяет составить представле-
ние о том, какие сведения о поведении металлов способен дать
термодинамический подход к рассмотрению проблем электрокоа-
гуляционной очистки жидкостей при выборе опшмальиых режи-
мов процесса.
3.3. МЕХАНИЗМ И КИНЕТИКА ЭЛЕКТРОДНЫХ ПРОЦЕССОВ
3.3.1. Понятие о лимитирующей стадии и скорости процесса
Любая электрохимическая реакция является сложным процес-
сом и может состоять из отдельных элементарных последователь-
ных (или параллельных) электрохимических и химических ста-
дий, включающих подход участника реакции из объема раствора
к внешней границе ДЭС и далее через размытую его часть к по-
верхности электрода. Эта стадия протекает в основном за счет
диффузии под действием градиента концентрации. Далее сле-
дует собственно электродная реакция, заключающаяся в осво-
бождении или присоединении электронов (окисление или восста-
новление), которая может протекать в одну пли несколько ста-
дий. За собственно электрохимическим превращением происходит
удаление продуктов реакции. Стадия, которая определяет ско-
рость электрохимического превращения, является лимитирующей
или замедленной.
46
Рис. 3.8 Катодная поляризационная
кривая, выражающая электрохимиче-
скую и диффузионную кинетику
Говоря о замедленности какой-либо стадии, подразумеваем,
что в данных условиях общая суммарная скорость электрохими-
ческого превращения не может превышать скорости медленной
стадии. Так, при замедленной электродной реакции и быстрой
диффузии скорость последней оказывается ограниченной вслед-
ствие медленного убывания реагирующего вещества, что не со-
здает достаточного градиента концентрации, необходимого для
развития диффузии.
В зависимости от того, какая стадия является медленной, раз-
личают электрохимическую и диффузионную кинетику электрод-
ных реакций. Чисто электрохимическая или чисто диффузионная
кинетика осуществляется только в предельных случаях большого
неравенства скоростей этих процессов. На рис. 3.8 схематически
приведена кривая, иллюстрирующая области протекания элек-
тродного процесса. При малых отклонениях потенциала от его
равновесного значения процесс протекает в области электрохи-
мической кинетики. По мере удаления от равновесного значения
потенциала скорость реакции возрастает и диффузия уже начи-
нает тормозить ее, не успевая поставлять достаточного числа ча-
стиц в единицу времени к поверхности электрода. При достнже
пни определенного момента, когда скорость диффузии вырастает
до своего максимального значения, наступают условия так на-
зываемого предельного тока, когда увеличение сдвига потенциала
уже не может вызвать увеличения скорости реакции. Очевидно,
что в этих условиях dCx/d<p=O.
Таким образом, выявление и установление природы и зако-
номерностей замедленной стадии являются важнейшими зада-
чами кинетики электродных реакций.
Скорость электрохимической реакции, подобно скорости хими-
ческой реакции, показывает число химических превращений на
। ранице электрод — электролит, приводящих к образованию про-
дуктов реакции на единице поверхности раздела фаз за единицу
времени:
V— ± (т,—ffi|)/(/i—6)-!/Зэ = ± Affi/Л/ 1/3,. (3.29)
Разность (гиг—т>) выражает среднюю скорость реакции о
в интервале времени (/2—Л)-
Выразив количество прореагировавшего вещества Д/п через
количество прошедшего электричества q.„ в соответствии с зако
47
ном Фарадея нетрудно показать, что скорость реакции может
быть выражена величиной плотности катодного пли анодного
токов:
(3.30)
Скорость электрохимической реакции (или плотность тока)
зависит от концентрации и температуры раствора электролита,
условий перемешивания, материала электрода, адсорбции на
электроде веществ из раствора и т. д,
В любой равновесной электродной системе скорости самопро-
извольных катодного и анодного процессов равны между собой
и соответствуют плотности тока обмена (io). Чем выше io, тем
труднее сместить положение равновесия электрохимической ре-
акции путем внешней поляризации электрода. Для большинства
металлов <о = 10-8-?-105 А/см2, а для инертных металлов (Pt, Au,
Ag) io«0.
3.3.2. Поляризационные явления
в электрохимических процессах
Разность между потенциалом электрода под током и его рав-
новесным потенциалом (Д<р=<р«—ф°) называется электродной по-
ляризацией. Принято различать катодную поляризацию, т. с. сме-
щение потенциала в отрицательную сторону при наложении
катодного тока, и анодную поляризацию, т. е. смещение потен-
циала в положительную сторону при наложении анодного тока
Согласно определению катодная поляризация всегда отрица
тельна: Афк = ф.—ф°<0, а анодная всегда положительна: Аф.,=
==<Р,—Ф°>0.
Потенциал электрода (ф,) и электродная поляризация (Aq)
являются прежде всего функциями силы тока (плотности тока).
Анализируя формы зависимости потенциала от плотности тока,
поляризационных кривых для различного состава раствора, тем-
пературы и других физико-химических параметров, в некоторых
случаях можно получить достаточно полное представление о при-
роде электродного процесса.
Непосредственной причиной поляризации является замедлен-
ность тон или иной стадии. Если известна причина замедленной
стадии, то вместо термина «поляризация» принято употреблять
«перенапряжение» (т]П), т. е. поляризация электрода, обусловлен-
ная замедленностью протекания определенной стадии суммарного
электродного процесса.
В зависимости от природы замедленной стадии различают не-
сколько видов или причин перенапряжения: диффузионное т)д
(стадия перенапряжения реагирующих частиц); реакционное i)P
(стадия химического превращения); электрохимическое т)э (ста-
дия переноса электронов); фазовое г)ф (стадия построения пли
разрушения кристаллической решетки).
В общем случае поляризация является суммой всех видов пе
рснапряжения:
VT Пд + ’Ь4-П?+’М>- (3.31)
В частном случае, если один из видов перенапряжения мак-
симален, то он и определяет поляризацию электрода. Однако еле
дует иметь в виду, что уравнение (3.31), представляющее элект-
родную поляризацию как простую сумму различных видов пере-
напряжения, является формальным, так как все эти виды нельзя
рассматривать как совершенно не связанные величины.
Умение выявить вид перенапряжения и оценить его значение
на практике позволяет целенаправленно провести процесс и сни-
зить затраты на получение какого-либо вещества.
3.3.3. Напряжение разложения водных растворов электролитов
Разложение веществ под действием электрического тока про-
исходит лишь в определенных условиях, и электролиз может на-
чаться после того, как приложенная извне разность потенциалов
превышает величину разности обратимых потенциалов электро-
дов. Максимальная разность потенциалов, которую необходимо
приложить, чтобы начался электролиз, называется напряжением
разложения электролитов. Во многих случаях, для того чтобы
начался электролиз, необходимо приложить извне разность по-
тенциалов большую, чем разность равновесных потенциалов элек-
тродов, на величину перенапряжения:
= ф. - Фк + ДФа + ДФк- (3 32)
Очевидно, что нрн отсутствии перенапряжения на электродах
напряжение разложения равно разности равновесных потеициа
лов электродов.
Рассмотрим более подробно разложение растворов, электро-
лиз которых сопровождается разложением воды: катодный про-
цесс (Н2О—2с-»-Н2\ анодный процесс (Н2О—4е->О2-|-4Н+).
Для равновесного потенциала анода можно записать:
Ф. = ф" + (RT/4F) In ([Н2О]2 [О2]/[ОН-]4). (3.33)
Так как р = 1, a [OH~]=Kb/[H+]. то
<«тлр). ш nW"*! . + + ,„ (|| +|.
Та ' к4 2F к2 F
(3.34)
Равновесный потенциал катода
<рк (PT/2F) ln(IH+]’'IHil)= (RTFF) 1п[Н+1. (3 35)
Напряжение разложения такого раствора после подстановки
(3 31) и (3.35) в (3.33) может быть вычислено по формуле
^р = Фа-Фк Ф0 (РГГ) 1п([Н2О]/К2). (3 36)
40
Если учесть, что при 25 °C К»= 1,27-10 й, <р°=+0,401 В, и
принять активность воды равной единице, получим Ер= 1,229 В.
Из уравнения (3.36) видно, что напряжение разложения рас-
творов, электролиз которых сопровождается разложением воды,
не зависит от состава раствора, т. е. напряжение разложения
воды будет практически одинаковым в растворах различной кис-
лотности. Напряжение разложения может быть определено бо-
лее или менее точно лишь при уделении на электродах чистых
твердых веществ. В случаях, если при электролизе образуются
газы, то напряжение разложения зависит от формы и размеров
электродов, чистоты поверхности, условий удаления газов и дру-
гих факторов, трудно поддающихся учету. Из всего вышеизло-
женного следует, что величина разложения не может служить
мерой однозначности для любого электролита при различных ус-
ловиях.
3.3.4. Кинетические закономерности стадии
разряда — ионизации
Перенос заряда через границу раздела между электродом и
раствором п связанные с этим нарушения электродного равно-
весия являются чисто электрохимическими явлениями и пред
ставляют наибольший интерес в кинетике электродных реакций.
Общий вид уравнений стадии непосредственно электрохими-
ческого превращения для реакции, в которой окисленная форма
ох принимает п электронов и превращается в восстановленную
форму (ox + ne-»-Red), выглядит следующим образом [26]:
скорость катодной реакции (восстановление)
(аг F ф° \ / аг F Лфк Ч ( ₽г F ф*
---------I ехР I----------I СХР I —---——
RT J \ RT J \ RT
(3.37)
скорость анодной реакции (окисление)
где а, Р — коэффициенты распределения, характеризующие долю энергии
ДЭС. действующего на катодную (а) и анодную (Р) реакции, называемые
коэффициентами переноса; фд<а)—скачок потенциала в диффузионной части
ДЭС; kt, к> — константа скорости соответственно анодной и катодной реакций
Уравнения (3.37) и (3.38) могут описывать скорость любой
электродной реакции, а не только рассмотренного процесса окис-
ления — восстановления.
Рассмотрев простейший случай, когда отсутствует диффузион-
ная часть двойного слоя (т. е. <рд=0), и учтя, что (Лц>=-= <р(—<г°),
уравнения (3.37) и (3.38) будут иметь следующий вид:
|к = Jt| Со exp (—azFtfIRTy, (3.39)
ia = кС„ exp (Рг F ф R Т). (3.40)
50
Рис. 3.9. Поляризационные кривые
/ частные; 2 суммарная
Эти уравнения представлены графически на рнс. 3.9: при
ф->-—оо ia->-0, tK-+ + oo (катодная поляризация); при ф->оо ia->
-*—оо, «1(->0 (анодная поляризация); при <р = <р° ia = iK=io, где
io — ток обмена; при ф>ф° суммарная скорость окисления ia =
= <о—<к; при<р<ф° суммарная скорость восстановления iK = io—ia.
Уравнения (3.39) и (3.40) являются функциональными урав-
нениями электрохимической кинетики и носят название уравне-
ний замедленного разряда.
Если поляризация достаточно высока по сравнению с вели-
чиной равновесного электродного потенциала, то величины <рк
и фа в уравнениях могут быть заменены на значения электрод-
ной поляризации VpK и Дфа и тогда эти уравнения примут вид:
»к = *i Сохехр (— ou F Лфи 'ЯТ’); (3.41)
= kt CRed exp фг F b<f3'RT). (3.42)
Логарифмируя данные выражения и учитывая, что в частном
случае (при выявленной замедленной стадии) поляризация явля-
ется перенапряжением, получим
1)к = Лфк = RTI(cuF) [ln*, + lnCoxl — КЛТ.ссгF) 1g(3.43)
Афа= (РГфтР) |ln*1-lnCRedl+(/?7’/pjF)lg/a. (3.44)
Объединив постоянные величины в константы перенапряжения
а и Ь, получим уравнения:
Пк = — АФк = а + frig <«; (3 45)
Па = — Дфа = а + ь 1g «а. (3.46)
Эти уравнения впервые выведены Тафелем при изучении пе-
ренапряжения водорода, нх теоретическое обоснование получено
значительно позже на основании исследований А. Н. Фрумкина,
М. Фольмера и др.
Константа а изменяется в широких пределах в зависимости от
природы преобразуемого на электроде вещества, материала и со-
стояния поверхности электрода и других факторов. Если г=
= 1 А/см2, то т) = о, т. е. величина а определяет перенапряжение
при i= 1 А/см2.
51
Опытные данные часто показывают, чю при а=р=0,5 вели-
чина />=0,059/(0,5 z)=0,H8/z. Это означает, что при увеличении
плотности тока в 10 раз перенапряжение возрастает иа 0,118 В.
Величина Ь практически ие зависит от материала электрода и
является характеристикой самого электрохимического процесса.
Коэффициент а определяется главным образом физической при-
родой электрода.
Следует отметить, что уравнения Тафеля справедливы только
в области величин поляризующих токов выше 10*® А/см2. Зави-
симость <р от I при меньших значениях токов носи г линейный,
а ие логарифмический характер.
3.3.5. Массолеренос и основные уравнения
диффузионной кинетики
При исследовании электрохимических реакций и выявлении
механизма процессов, как уже было показано (см. п. 3.3.1), су-
щественную роль играют процессы переноса вещества к поверх-
ности электрода, т. е. массолеренос.
Перенос вещества из глубины раствора к поверхности элек-
трода осуществляется несколькими путями, наиболее важным из
которых является молекулярная диффузия. Другим способом до-
ставки реагирующих частиц является перенос иод действием
электрического поля, называемый миграцией, которая наблюда-
ется только для заряженных частиц. Наконец, третьим пу<ем до-
ставки вещества к поверхности электрода является конвекция,
т. е. перенос вещества вместе с потоком движущейся жидкости.
В соответствии с рассмотренными путями переноса вещества
суммарный поток вещества к поверхности электрода (Jz) скла-
дывается из потоков диффузии (/д), миграции (/м) и конвекции
(/к). Рассмотрим влияние различных потоков на протекание
электродных процессов.
Молекулярная диффузия и предельный ток. В равновесных
условиях концентрация вещества, во всех точках раствора за пре-
делами ДЭС одинакова. При пропускании тока вблизи электрода
происходит нарушение равновесия (за счет расходования веще-
ства в электродной реакции) и возникает разница в концентра-
ции частиц у поверхности электрода и в объеме раствора, что и
приводит к диффузии вещества из глубины раствора к наружной
границе ДЭС, а продуктов реакции — в глубь раствора.
Удельная скорость диффузии вещества в слое, прилегающем
к электроду, описывается согласно закону Фика:
dmldt = — D (dC/dx). (3.47)
При линейном изменении концентрации с изменением расстоя-
ния (х) от поверхности электрода величину dC/d\ можно заме-
нить иа ДС/Дх, причем ДС<0. Тогда
dm/dt w D (ДС/Дх). (3.48)
52
Рис. 3.10. Распределение эквивалент-
ной концентрации катионов (Си),
анионов (С.) и электролита в це-
лом (С) вблизи поверхности элек-
трода тт
б» — толщина двойного слов, включая
и диффузионную его часть. дд — толщина
диффузионного слоя, в котором Ск — Са;
С» — концентрация в глубине раствора
На рис. 3.10 представлено распределение концентрации ка-
тионов и анионов вблизи электрода с отрицательным потенциа-
лом. Концентрация в точке Ь равна общей концентрации в глу-
бине раствора, но больше, чем в точке а. При линейном падении
концентрации в пределах толщины диффузионного слоя градиент
концентрации будет равен (Со—Са)/бл. При стационарной диф-
фузии (dC/d/ = 0 и dC/dt=const), когда величина бд стабилизиро-
валась, уравнение (3.48) запишется:
dmldt = D ДС/6д = D (Со — С,)/6Д. (3.49)
Для скорости электродного процесса, контролируемого диф-
фузией, диффузионную плотность тока можно записать:
«д = г F dmldt. (3.50)
Тогда
1Л = г FD (Со — С,)/6Д. (3.51)
Если скорость электродной реакции очень велика, то Ct~0
(каждый ион, переносимый диффузией, немедленно вступает
в реакцию), и величина (л примет максимально возможное зна-
чение, называемое предельным током диффузии (1„д):
гдр = zFD СО/6Д. (3.52)
Из уравнений (3.51) и (3.52) имеем:
(3.53)
Для определения поляризации (перенапряжения) воспользу-
емся уравнением Нернста:
ф = Фо + (К Т!г F) • In Сп. (3.54)
При прохождении тока потенциал электрода будет опреде-
ляться концентрацией реагирующего вещества у поверхности
(Q:
Ф/ - фо + (R T,z F) • In Са. (3.55)
53
Таким образом,
Лф Чд ф.-ф (RT/>F) 1п(С,/Сп). (3.56)
Подставив (3.53) в (3.56), получим выражение для перена-
пряжения стационарной диффузии:
Чд — (R7'/zF)ln(l —|д/|дР), (3.57)
или
‘ж = ‘д₽ D — ех₽ (V F RT)] (3-5«)
Уравнение (3.58) выражает скорость электродного процесса
при замедленной диффузии, причем оно справедливо при избытке
фонового электролита, т. е. когда миграцией практически можно
пренебречь. Это уравнение удобно представить в виде графика
(рис. 3.11).
При малых перенапряжениях экспоненту можно
разложить в ряд и, ограничившись двумя первыми числами, по-
лучить
•д’ *iP-(VFR7')’ <3-59»
откуда следует, что в области малых перенапряжении имеется
линейная зависимость между тд и т]д.
При больших отрицательных т)д экспонентой можно прене
бречь по сравнению с единицей и iB=inni'.
Миграция частиц под действием электрического поля. Как
уже указывалось, ток, обусловливающий перенос заряженных
частиц, называется миграционным током (iM). При элсктроокнс-
ленпи миграция способствует подходу анионов и замедляет под-
ход катионов. В случае элекгровоссгаиовлеппя наблюдается об-
ратная картина.
В первом приближении можно считать, что молекулярная
диффузия и электрический перенос ионов, обусловливающие об-
щий ток диффузии, являются независимыми [100]. Тогда общий
ток будет равен: i = in+iM. Долю тока, переносимого нонами,
можно выразить через числа переноса (iM = i«;), тогда
« = »д+»пь или <д = «(1—л,). (3 60)
Для катодной поляризации и восстановления катионов можно
записать:
*д = «к (1 — Пк) = Ма- (3.61)
Для анодной поляризации и окисления анионов:
«д = «а (1 — Ла) = 1ЛП к. (3 62)
Однако в некоторых случаях электрический перенос ионов
не только не способствует поступлению реагирующего вещества
к электроду, а, наоборот, препятствует ему (например, окисле-
ние на аноде I’e2* до 1'е3+), и в общем виде уравнение для об-
54
Рис. 3.11. Зависимость тока от перенапряжения при
разряде ионов металла на одноименном металле
Рис. 3.12. Распределение скоростей течения жидко-
сти при обтекании тонкой плоской пластины
Х’О Х*1 X
щсго тока будет иметь следующий вид: i — Знак « + » в слу-
чае, если потоки молекулярной диффузии и миграции совпадают,
а знак <—» — когда противоположны.
Конвективный перенос вещества. Предельное значение тока,
создаваемого диффузией и миграцией, относительно невелико
вследствие малых скоростей потоков Чтобы повысить эту вели-
чину, необходимо проводить перемешивание раствора, т. е. уве-
личивать перенос вещества совместно с потоком движущейся
жидкости, называемый конвективным переносом, однако переме-
шивание может возникнуть и в естественных условиях. Так, из-
менение концентрации приводит к изменению плотности рас-
твора и возникновению потоков жидкости. Изменение плотности
может вызвать также выделение газа на электродах и разогрев
электролита.
В соответствии с теорией конвективной диффузии [83] слой
раствора, прилегающий к поверхности электрода, где происходит
основное изменение концентрации, расположен внутри другого
слоя, называемого граничным слоем Прандтля, где скорость двн
жен и я жидкости постепенно увеличивается по мере удаления от
электрода.
55
Толщина слоя Праидтля fin может быть оценена на примере
обтекания тонкой плоской пластины жидкостью (рис. 3.12). Не-
обходимо подчеркнуть, что бп является функцией расстояния /
от начала пластины.
В слое Прандтля следует выделить толщину слоя Нернста
бц, в котором концентрация диффундируемого вещества понижа-
ется от значения Со в глубине раствора до значения С6 , которое
характерно для раствора непосредственно около границы фаз,
где устанавливается стационарный градиент концентрации, ха-
рактерный для диффузии в неперемешиваемом ограниченном
объеме. Толщина слоя Прандтля в ламинарном режиме [83].
лп ~ « f/VRe. (3.63)
где Re—число Рейнольдса: при обтекании плоской пластины Re =/p^/v.
Значение би для этих же условий можно определить из соот-
ношения
бн/вп « (D/v)l/3 « S^1'3, (3.64)
где Sr—число Шмидта, равное отношению чисел Пекле и Рейнольдса (число
Пекле Ре = /c^/D); в водном растворе би*>би/10.
Поток вещества в условиях конвективной диффузии /КОив=
= Сож. Это показывает, что за единицу времени через единицу
поверхности проходит такое количество вещества, которое содер-
жится в столбе жидкости с площадью основания, равной еди-
нице, и высотой, равной расстоянию, преодолеваемому частицей
за единицу времени (численно равной скорости движения жидко-
сти о»). Приняв, что div о = 0 и dpldt = —div 7, получим
dCfdi = — grad С ё* . (3.65)
В стационарном состоянии
grad С = (Со — Се)'вн- (3.66)
Важнейшим количественным приложением теории конвектив-
ной диффузии является вращающийся дисковый электрод, глав-
ным свойством которого является равнодоступность поверхности
[83]. Таким образом, толщина диффузионного слоя на любых
участках диска является одинаковой, а плотность тока — посто-
янной в каждой точке поверхности. Для величины бп на вращаю-
щемся дисковом электроде получена следующая зависимость:
бп 1.61D1V 2, (3.67)
или с учетом (3.52)
|"р = 0.62г FD2/3v-,,6w,/2C0. (3.68)
где w — угловая скорость вращения дискового электрода.
Вращающийся дисковый электрод находит широчайшее при-
менение при установлении замедленной стадии процесса. В са-
56
мом деле, если процесс лимитируется диффузией, то зависимость
тока от л/w должна быть прямолинейной и проходить через на
чало координат.
Если скорость процесса контролируется кинетикой, то ток
не зависит от подвода вещества и от скорости вращения диска.
С помощью измерений на вращающемся дисковом электроде
можно но предельному току вычислить коэффициенты диффузии
отдельных ионов или молекул. При известном коэффициенте диф-
фузии (вычисленном, например, из подвижностей) можно по ве-
личине »дРопределить число электронов (z), участвующих в элек-
тродном процессе. Это имеет практическую ценность при уста-
новлении механизма реакций, протекающих с участием органиче-
ских веществ.
3.3.6. Кинетика основных электродных процессов,
протекающих при очистке природных и сточных вод*
Процессы электрохимической очистки природных и сточных
вод практически всегда протекают с выделением газов иа элек-
тродах и образованием новой фазы (растворение аиода либо вы-
деление металлов на катоде). Поэтому знание механизма и ки
иетических закономерностей отдельных электродных процессов
позволяет технологически рационально организовать тот или
иной процесс, снизить затраты на очистку и т. д.
3.3.6.1. Катодные процессы
Выделение водорода. Катодное выделение водорода всегда
имеет место при электролизе водных растворов электролитов.
Реакция катодного восстановления водорода протекает на неко
торых металлах со значительным перенапряжением, существенно
превышающим перенапряжение многих других электродных ре-
акций. Величина водородного перенапряжения зависит от мно-
гих факторов, и в первую очередь от состава раствора, мате-
риала катода и состояния его поверхности, плотности тока и тем-
пературы. Рассмотрим возможные пути выделения водорода из
кислых и щелочных растворов.
В кислых растворах восстановление водородного нона можно
изобразить схемой I: а) Н3О++е->-Надс + Н2О; б) Надс + Надс->Н2.
На первой стадии восстановления (а) происходят разряд ка-
тиона с потерей гидратной оболочки и адсорбция на поверхности
электрода. Вторая стадия (б) заключается в образовании моле-
кул Н2 путем рекомбинации ионов Надс.
В щелочных растворах, где концентрация Н3О+ ничтожно
мала, разряд протекает непосредственно из молекул воды по
схеме II: а) Н2О+е-*-Надс + ОН_; б) Hanc + Hw->H2. Именно
* Материал подготовлен совместно с панд. техн, паук В. В. Кузнецовым
57
Рис. 3.13. Перенапряжение восстановле-
нии водорода на ртутном катоде при
•к — 10 1 А/см", в зависимости от pH
раствора
стадии (а) в обоих случаях являются медленными и определяют
общую скорость выделения водорода.
Для водородного перенапряжения в зависимости от того,
в какой среде протекает процесс, получены следующие урав-
нения:
в кислой среде
п= — 0,1181gЛ 4-0.059 pH 4-te + 0.118lg«K; (3.69)
в щелочной среде
п= — 0,1181g* — 0,059рН— Ifrj + O.ll81g/K. (3.70)
Анализ этих уравнений показывает, что в щелочных раство-
рах перенапряжение выделения водорода уменьшается с ростом
pH, а в кислых растворах оно, наоборот, возрастает с ростом
pH. Эти выводы подтверждаются экспериментальными данными
(рис. 3.13).
Перенапряжение водорода снижается с ростом температуры
приблизительно на 2—4 мВ на 1 С. Зависимость перенапряже-
ния водорода от плотности тока (при значительном сдвиге по-
тенциала от его равновесного значения) описывается известным
уравнением Тафеля (3.45).
Условно принято различать металлы с высоким перенапряже-
нием выделения водорода (Hg, Pb, Cd, Zn, Sn), средним (Си, Ag,
Fe, Ni, Co, Pd) и низким (PI, Ru) [93].
Константа а, вычисленная но опытным данным, совпадает
с теоретической величиной. Предлогарифмический коэффициент
b для большинства металлов колеблется от 0,1 до 0,12 В [100].
Очень сильное влияние на перенапряжение выделения водо-
рода оказывает присутствие в растворе поверхностно-активных
веществ (ПАВ), влияющих на 'Гд-потенциал. Адсорбция поверх-
ностно-акгпвных анионов смещает ^-потенциал в более отрица-
тельную с i орону; при этом концентрация Н+ в плотной части
ДЭС и скорость процесса возрастают, а величина цц2 снижается.
Однако следует заметить, что при больших смещениях потен-
циала, достигающих потенциала десорбции, влияние анионов пре-
кращается. Поверхностио-акгивные катионы приводят к обрат-
58
пому эффекту. Неионогенные ПАВ в зависимости от их природы
могут повышать или понижать перенапряжение.
В процессах очистки природных и сточных вод перенапряже-
ние выделения водорода имеет большое практическое значение,
особенно при электрофлотацнн, где процесс протекает в основ-
ном за счет транспорта загрязнении (в виде пузырьков водорода
на поверхности жидкости). В этом случае высокое перенапря-
жение вредно. Однако высокое перенапряжение иногда и полезно,
например при электрохимической очистке сточных вод от ионов
тяжелых металлов. Здесь высокое водородное перенапряжение
необходимо для уменьшения затрат электроэнергии иа побочный
процесс, т. е. для повышения селективности электрохимической
реакции.
Электровосстановление металлов. Электрохимическая очистка
сточных вод от иоиов тяжелых металлов в настоящее время
осуществляется в основном электрокоагуляцнонным методом
с применением растворимых анодов [102], однако в последнее
время получили распространение методы катодного осаждения
металлов с последующей их регенерацией.
Электролитическое выделение металлов из гидратированных
ионов с образованием катодного осадка описывается следующим
уравнением:
Мс»+ х Н,О4-гё->|Ме)+хН,О. (3.71)
Катодное выделение металлов является наиболее сложным
разделом электрохимической кинетики, что связано с образова-
нием новой фазы (осадка) иа электроде, с непрерывным обнов-
лением поверхности катода и ее энергетической неоднородностью.
При изучении кинетики этой реакции практически всегда нужно
учитывать протекание сопряженной реакции выделения водорода,
причем большее значение имеет не величина перенапряжения
водорода, а механизм выделения его на металле [5]. Кроме того,
ряд электроотрицательных металлов вообще не может быть вы-
делен из водных растворов. Со 100%-ным выходом по току
можно выделить металлы, обладающие достаточно электрополо-
жительным равновесным потенциалом.
При электроосаждеиии величина перенапряжения в первую
очередь зависит от природы разряжаемого металла. Среди них
можно выделить металлы с очень низким металлическим перена-
пряжением (не более тысячных долей волыа) —Hg, Ag, Pb, Cd,
Sn. К другой группе относятся металлы, имеющие перенапряже-
ние порядка сотых вольта — Bi, Си, Zn. Наибольшим перенапря-
жением обладают металлы третьей группы — Со, Fe, Ni, у кото-
рых оно достигает порядка нескольких десятых вольта.
Кроме всего прочего, существенную роль играет природа
аннона соли. Обычно перенапряжение уменьшается при переходе
от одного аниона к другому в следующем ряду: POj~>NOr>
>С1ОГ >С1 >Bi >J . На кинетику процесса влияет также нали-
чие ПАВ.
59
На катодное восстановление оказывает влияние подщелачива-
ние раствора вблизи электрода из-за параллельного выделения во
дорода, чго способствует гидролизу солей металлов с образова
нием основных солен и гидроксидов, которые могут влиять на про-
цесс электроосаждеиня и включаться в катодный осадок.
3.3.6.2. Анодные процессы
В данном разделе рассмотрены кинетические аспекты протека-
ния анодных процессов выделения кислорода и хлора, имеющие
важнейшее значение при разработке методов электрохимической
деструкции, закономерности анодного растворения металлов как
основы электрокоагуляцпонного способа очистки сточных вод.
Электролитическое выделение кислорода. Как уже ранее было
показано (см. п. 3.3.3). выделение кислорода на аноде является
неотъемлемой частью процесса разложения воды и имеет не мень-
шее практическое значение, чем выделение водорода.
По современным теоретическим воззрениям, процесс анодного
выделения кислорода в зависимости от pH среды протекает по
следующим путям [5]: 4ОНу*О2 + 2Н2О + 4е= (в щелочной);
2112О^О2 + 4Н+ + 4е= (в кислой, где концентрация ионов ОН"
мала, разряду подвергается молекула воды). Эти уравнения ото-
бражают лишь в общей суммарной форме процесс выделения кис-
лорода. На самом же деле ои протекает более сложно, так как
вне зависимости от pH в реакции принимают участие 4е, что при-
водит к появлению нескольких электрохимических стадий, каждая
из которых может быть замедленной и контролировать скорость
всего процесса.
Предложены различные схемы выделения кислорода через ста-
дии образования поверхностных оксидов, радикалов ОН’, атомар-
ного кислорода и др Однако ни одна из них не может полностью
объяснить закономерностей протекания электрохимического выде-
ления кислорода в зависимости от различных факторов.
Сложности протекания рассматриваемой реакции обусловлены
многими факторами, и в первую очередь окислением электрода,
т. е. образованием поверхностных оксидов, которые изменяют
cipoeime ПЭС, вследствие чего приходится учитывать падение по-
тенциала н в слое оксида
Кроме того, известно, что выделение газообразного кислорода
in растворов кислот происходит при потенциале анода более по-
ложительном. чем равновесный потенциал кислородного элек-
трода (+1,23 В при аН4. = 1 и 25 °C), на величину кислородного пе-
ренапряжения при данной плотности тока. Однако это справед-
ливо лишь для ряда металлов (Pt, Pd, Ан) н оксидных электро-
дов (ОРГА, ОКТА. ТДМА). стойких в растворах кислот в усло-
виях анодной поляризации. Для большинства других металлов
вместо выделения кислорода происходит их анодное растворение
или окисление. В щелочных растворах могут быть применены Fe,
60
Ni, Cd и другие, так как равновесный потенциал кислорода у них
менее положителен ( + 0,401 В при аон_= 1 и 25 °C). Эмпирически
установлено, что в области средних плотностей тока (=»10 3А/см2)
перенапряжение кислорода при выделении из щелочных раство-
ров растет в следующем ряду металлов: Fe, Ni, Cd, Pb, Au, Pt.
Таким образом, подводя итог обсуждаемых данных, можно кон-
статировать многообразие форм и путей протекания реакции элек-
трохимического выделения кислорода, сложные зависимости от
многих факторов, что очень затрудняет выявление кинетических
закономерностей процесса.
Выделение хлора. Изучение кинетических закономерностей вы-
деления хлора имеет принципиально важное значение при разра-
ботке методов электрохимической деструкции органических ве
ществ [51, 73] н синтеза «активного хлора» как эффективного
дезинфицирующего агента в очистке природных и сточных вод
[63]
При электролизе чистых водных растворов поваренной соли иа
аноде возможны выделение хлора 2Cl~ + 2e^C\j_ и сопряженный
процесс выделения кислорода 2H2O^tO2 + 4n‘ +4е.
Стандартный потенциал первой реакции +1,359 В, а второй
+ 1,23 В. Из этих величин, казалось бы, основным процессом при
электролизе должно быть выделение кислорода, по вследствие вы-
сокого кислородного перенапряжения иа многих металлах проис-
ходит выделение хлора.
Относительные скорости выделения кислорода и хлора сущест-
венным образом зависят от материала анода, концентрации С1_-
нонов. pH и температуры электролита. Для практических целей
важно, чтобы вы телепне хлора происходило с минимальным пере-
напряжением. а кислорода — с наибольшим. Лучше других под-
ходят аноды 1Й благородных металлов — платина, платинирован-
ная платина, платинированный титановый анод (ПТА) н так
называемые малонзпашпваемые аноды, представляющие собой
титановую основу с нанесенным иа иее поверхностным слоем
окнслов металлов. Наиболее перспективны для этих целей ОРТА
и ОКТА, причем последний более экономичен.
Особенностью электрохимической очистки сточных вод с ис-
пользованием нерастворимых анодов является низкая концентра-
ция С)~-ноиов в растворе и, как следствие, конкурирующие про-
цессы выделения хлора и кислорода.
При электролизе пнзкокопцептрнрованных растворов поварен-
ной соли выделяющийся на аноде хлор растворяется в электро-
лите с образованием соляной и хлорноватистой кислот, причем
последняя реагирует с прнкатодной щелочью с образованием ги-
похлорита согласно следующим уравнениям-
Cl,+ H2Ort НПО + HCI; (372)
нею -+ на + 2ОН т‘2Н2О+С1- + С1О-. (3.73)
fit
Рис. 3.14. Общая (/) и частная (2) поляри-
зационные кривые образования гипохлорита
на ПТА в 2 %-ном растворе NaCI при 25 °C
Однако образование гпнохлорша может протекать и по элек-
трохимическому механизму:
СЮ Ц- ОН~ — 2>«*С1О~ + Н,О. (3.74)
На рис. 3.14 приведены общие и частные поляризационные кри-
вые образования гипохлорита на ПТА в2%-ном растворе при
25 °C [122]. Из сопоставления этих кривых следует, чю с увели-
чением потенциала доля тока, идущая па образование гипохло-
рита, возрастает и становится максимальной при достижения по-
тенциала 2,1 В. В области потенциалов о г 1,55 до 1,75 В медлен-
ной является электрохимическая реакция образования гипохло-
рита, когда большая часть его образуется за счет реакции (3.73).
В области же потенциалов 1,75—1,90 В скорость электрохимиче-
ской реакции образования СЮ возрастает быстрее, чем скорость
реакции образования хлора, а доля тока, идущая на образование
гипохлорита, возрастает. Это продолжается до потенциала 2,1 В.
после чего она остается постоянной. Предположительно, что при
потенциалах выше 2,1 В образование СЮ на платине в разбав-
ленных растворах хлорида происходит по электрохимическому ме
ханпзму [реакция (3.74)].
Аналогичные зависимости (рис. 3.15) получены при изучении
поляризационных кривых на раз
NaCI с добавками органических
веществ [51, 73]. В области плот-
ностей тока, применяемых при
очистке сточных вод, поляриза-
ционные кривые па ОРТА имеют
угол наклона, равный 40—45 мВ,
что свидетельствует о преиму-
щественном выделении хлора иа
аноде. В присутствии органиче-
ских веществ потенциал анода
несколько повышается и наблю-
Рис 3.15. Анодные поляризационные
кривые
а — раствор 5 г/л NaCI; б — то же. и 0.1 г/л
красителя кислотною фиолетового С; о — то
же, н 0.1 г/л прогресса; л —то же. и 0.1 г/л
хлорофоса. / — ОРТА 2—ОКТА; 3 — ТЛМА:
4 — активный слой 9!» % МпО +5 г ('О О,;
5 графитовый анод
х анодах в растворе 5 г/л
дается увеличение угла наклона поляризационной кривой, что
объясняется затруднением процесса разряда С1 -попов вслед-
ствие адсорбции органических веществ.
По своим электрохимическим характеристикам ОКТЛ близок
к ОРТА. Для ТДМА и анодов, покрытых смесью окислов МпО« +
Ч-СОзОч, поляризационная кривая сдвинута в сторону более поло-
жительных потенциалов по сравнению с соответствующими для
ОРТА и ОКТА и углы наклона колеблются от 80 до 115 мВ, что
свидетельствует о преимущественном выделении кислорода.
Методом стационарных поляризационных измерений на вра-
щающемся дисковом электроде ОРТА [31] в растворах xMNaCl +
+ (5,13М—x)NaC!O4 установлено, что в отсутствии перемешива-
ния скорость анодного процесса лимитируется в основном диффу-
зией молекул хлора от электрода. При интенсивном перемешива-
нии раствора процесс протекает в области смешанной кинетики.
Порядок анодной реакции по С1_-иоиу равен 2, по С12 — нулю,
а тафелевский наклон, отвечающий истинной кинетике анодного
процесса, равен 35 мВ, т. е. близок к 2,3/?T/zF На основании этих
данных сделай вывод, что в области анодных и небольших катод-
ных потенциалов процесс протекает в основном по механизму
Фольмера — Тафеля с лимитирующей стадией рекомбинации.
Таким образом, варьируя различными параметрами электро-
лиза, меняя состав раствора и подбирая анодный материал, можно
даже в сильно разбавленных растворах NaCI значительно снизить
перенапряжение выделения хлора и уменьшить долю тока, иду-
щую иа побочное выделение кислорода
Анодное растворение и пассивность металлов. Процессы анод-
ного растворения металлов лежат в основе широко распространен-
ного метода электролитической коагуляции. В качестве анодов
в основном применяют железо н алюминий, поэтому представляет
интерес рассмотреть поведение этих металлов при анодной поля-
ризации.
Анодное растворение по своей природе и последовательности
протекания отдельных стадий противоположно катодному осажде-
нию металлов, поэтому часто наблюдается сходство в поведении
многих металлов при их растворении и катодном осаждении. Од-
нако анодный процесс более сложен и сопровождается такими яв-
лениями, как анодная пассивация, т. е. резкое снижение тока при
достижении определенного потенциала.
Исследование анодного растворения металлов затрудняется
тем, что происходит непрерывное изменение истинной поверхности
электрода вследствие энергетически более выгодного растворения
кристаллографических граней, выступов и др.
В общем случае анодное повеление металлов зависит от их
природы, состава электролита, состояния поверхности и величины
поляризации.
Для большинства металлов в определенном интервале потен-
циалов скорость растворения в растворах электролитов монотонно
растет по мере смещения потенциала в анодную сторону (рис.
63
Рис. 3.16 Различные зависи-
мости логарифма плотности
тока от потенциала при анод-
ном растворении металлов
г, нА. сн!
Рис. 3.17. Анодные поляризационные кривые рас-
творения алюминия в растворах NaCI различной
концентрации
I — 0.001 моль/л: 3 — 0.01 моль/л; 3 — 0.1 моль/л; 4 —
0.5 моль/л: 5 — 1.0 моль/л
Рис. 3.18. Анодные поляри-
зационные кривые раство
рения жечеза в 0,5 и lltSOt
I — зона активного растворе-
ния: 11 — переходная обл сть;
111 — область пассивации; IV —
область анодного выделения
кислорода
3.16). Площадка на кривой отвечает предельному току диффузии
растворяющегося металла в случае растворения металлов из
амальгам.
Совершенно иная картина наблюдается при растворении же-
леза или алюминия (рис. 3.17, 3.18) [104]. Здесь при достижении
потенциала около —1,4 В для алюминия и нулевого потенциала
для железа наблюдается незначительное падение плотности тока
(скорости реакции) при дальнейшей поляризации
Рассмотрим более подробно анодную поляризационную кри-
вую растворения железа На пей отчетливо видны четыре харак
териых участка: I — область активного растворения: II — пере
ходная область из активного состояния в пассивное; 111 — область
пассивации; IV — область анодного выделения кислорода. Для ха-
рактеристики потеицпостатической кривой особенно важны точки
64
перехода из одного состояния в другое. Потенциал, при котором
начинается переход из активного состояния в пассивное, называ-
ется потенциалом пассивации. При шленциале <раКт железо уже
пассивно. Потенциал ф,кт называют обычно потенциалом актива-
ции или фладе-потеицналом, отрицательнее которого металл пере-
ходит в область активного растворения. И последней характерной
точкой на кривой является потенциал анодного выделения кисло-
рода. Одиако при некоторых определенных условиях на ме-
талле происходит не выделение кислорода, а наоборот, его даль-
нейшее растворение, т. е он переходит в область траиспас-
сивностн.
Пассивность металлов объясняется образованием на поверх-
ности анода фазовой, окисной или гндроксидной пленок, отделяю-
щих металл от окружающей среды и препятствующих его раство-
рению (пленочная теория пассивности).
Более эффскгивсн адсорбционно-электрохимический механизм
пассивирования, связанный с образованием на поверхности ме-
талла мопослоя или даже долей моиослоя адсорбированного кис-
лорода, который изменяет энергетическое состояние поверхности
атомов, блокирует активные центры растворения, изменяет строе-
ние ДЭС. Однако обе теории пассивности не исключают друг
друга.
Активирующее действие на металл оказывает анионный состав
электролита По своей активирующей способности анионы распо-
лагаются в следующий ряд Cl->Br->J >F“>С1Оч >ОН—>
>SO<_. Таким образом, вводя в раствор поваренную соль (что и
делают иа практике), можно значительно увеличить время работы
апода до пассивации. Другим не меиее эффективным путем акти-
вирования анодов является переполюсовка, т. е. очистка электрода
выделяющимся водородом.
3.3.6.3. Редокси-процессы
(электрохимическое восстановление и окисление)
Для электрохимической очистки природных и сточных вод от
органических загрязнений важное значение имеют процессы элек-
трохимического окисления, так как именно они приводят к разру-
шению основной углерод углеродной цепи органических веществ
и обеспечивают глубокую cieneHi. минерализации, вплоть до воды
и СО2. Катодное восстановление, хотя и имеет определенное зна-
чение, но больше распространено в органической препаративной
химии.
Реакции электровосстановления в водных средах связаны с ме-
ханизмом выделения водорода на электроде и поэтому говорят
о различных по своей природе восстанавливающих агентах: элек-
тропах, атомах водорода и нонах водорода [5]
Иа ход процесса восстановления влияет характер заместителя
(электроотрицательные и электроположительные группы) и его
положение в восстанавливаемой молекуле. В качестве примера
3 Такт М 2148
65
рассмотрим электрохимическое восстановление нитробензола
в амииобепзол, протекающее по схеме
XII ЮН
(3.75)
Образующиеся аминосоединения значительно легче окисля-
ются, чем исходные нитросоедппеппя На практике сточные воды,
содержащие интросоедипепия, подвергаются обработке сначала
в катодной камере электровосстаповлеиием, а затем в анодной,
где происходит окисление аминосоедпиеинй.
Процессы электроокнслення протекают значительно труднее,
нежели катодные процессы. При электроокислении органических
соединений необходимо учитывать энергетическую неоднородность
металла, участие частиц ОНадс. В некоторых случаях окисление
может протекать по электронному механизму, т. е. органическое
вещество, адсорбируясь на аноде, отдает электроны с одновремен-
ной или предшествующей дегидратацией: R— Н — e-*-R' +Н .
Дальнейшее превращение радикала /?' определяется его реакци-
онной способностью. При электроокислении формальдегида на
платиновом аноде наряду с СО2 были обнаружены Н2СО и
НСООН или НСОО , что означает протекание реакции через про-
межуточное образование Н2СО и НСООН или НСОО [26].
Интересны данные по окислению пропилепглнколя на ОРТА
[36], в продуктах деструкции которого идентифицированы СО2 и
CHjCOOH. Предполагается, что реакции окисления протекают без
предварительной хемосорбции пропилепглнколя па ОРТА, причем
медленной стадией является отрыв электрона от молекулы пропн
ленгликоля. Кроме того, не исключено протекание процесса по
другим маршрутам.
Таким образом, процессы электровосстановлепия и окисления
зависят от очень многих факторов, таких как химическое строение
органических веществ, возможность адсорбции иа электродах, па-
раметры электролиза, присутствие посторонних ионов и прочее,
что бывает практически очень трудно учесть при интерпретации
экспериментальных данных.
3.4. ПРОЦЕССЫ, ПРОТЕКАЮЩИЕ В ОБЪЕМЕ ЭЛЕКТРОЛИТА
В объеме электролита под влиянием электрического поля про-
текают сложные физико-химические процессы (рис. 3.19), в ре-
зультате которых происходит ряд превращений, таких как коагу-
ляция и флокуляция коллоидных частиц, изменение валентности
иоиов при окислительно-восстановительных реакциях с участием
примесей воды, формирование твердой, газообразной н жидкой
фаз, деструкция сложных органических соединений и т. д. Изме-
Рис. 3.19. Основные процессы, протекающие в объеме электролита, и методы
их осуществления
пенни в объеме электролита оказывают также существенное влия-
ние па электродные реакции, особенно в таких разбавленных си-
i юмах, какими являются природные и сточные воды.
Ниже рассмотрены более подробно теоретические аспекты про-
цессов, наблюдаемых в объеме электролита при наложении внеш-
него электрического поля.
г
67
3.4.1. Электролитические эффекты, возникающие в жидкости
при электрических воздействиях
3.4.1.1. Воздействие однородных и неоднородных
электрических полей
Воздействие электрического поля иа дисперсные системы обус-
ловливает целый комплекс неравновесных электроноверхностных
явлений, приводящих к качественному изменению компонентов си-
стемы. К «акнм эффектам, имеющим важное значение для очи-
стки загрязненных жидкостей, относятся:
электроорнентационпын эффект — ориентация частиц и цепо-
чечных агрегатов вдоль вектора внешнего ноля;
поляризационная коагуляция — безреагентный процесс агреги-
рования частиц в электрическом ноле, определяющий неравновес-
ные поляризационные силы взаимодействия между частицами дис-
персной фазы.
Поляризация и ориентация дисперсных частиц во внешнем
электрическом поле определяются их" электрическими свойствами
и свойствами дисперсной среды, в первую очередь диэлектриче-
ской проницаемостью. Под действием поля может происходить по-
ляризация самой частицы или ее ДЭС. В результате этого возни-
кает индуцированный дипольный момент (ПДМ) коллоидных ча-
стиц, который обусловливает днполь-лнполыюе их притяжение.
ПДМ в не слишком сильных полях является линейной функцией
поля: dm=ynEp, где уп — поляризуемость частицы.
Ориентация частиц под действием поля может быть связана
как с поляризацией частицы, так н с наличием у нее постоянного
дипольного момента.
Поляризационную коагуляцию авторы [29, 119] определили как
взаимодействие двух частиц в неоднородных полях их ПДМ.
В случае ноиостабилпзпрованпых дисперсных систем (природные
и сточные воды) устойчивость к коагуляции определяется балан-
сом двух основных факторов ноноэлектростатнческого отталкива-
ния двойных слоев и молекулярного притяжения. Во внешнем
электрическом поле к этим двум факторам добавляется третий —
поляризационное взаимодействие частиц. На основе развитой
С. С. Духппым и его сотрудниками диффузионно-электростатиче-
ской теории поляризации ДЭС [29, 30] полечен критерий необра-
тимой коагуляции, связывающий величину критической напряжен-
ности поля Екр, приводящая к образованию нераснадающпхся аг-
реотов после снятия поля, с величиной поверхностного заряда
частиц:
Е ьр = 4ло,/ео. (3.76)
где а, — поверхностная плотность заряда.
Силовое воздействие электрического поля обеспечивает также
миграцию (|рапспорт) заряженных части в жидкости. В первую
очередь это транспорт ионов (электродиализ), коллоидных частиц
68
(электрофорез, электроосмос), а также дпполофорсз—квадратич-
ный но величине внешнего ноля дрейф частицы, обусловленный
действием недопородного электрического ноля па ПДМ заряжен-
ной частицы. Количественные закономерности учета этого явления
для ионов приведены в главе 2,
В технологии обработки воды наиболее характерен транспорт
коллоидных частиц, для которых возникающие электрические силы
соизмеримы с силон тяжести. Электрические силы возникают за
счет существования заряда на поверхности частиц и взаимодей-
ствия его с электрическим полем.
По аналогии с границей раздела электрод — раствор (см.
и. 3.2.1) в ДЭС коллоидной частицы различают плотную часть, ко-
торая включает в себя адсорбированные ноны, жестко скреплен-
ные с поверхностью частицы, и диффузионную, где характер рас-
пределения ионов определяется действием электрических сил и
теплового движения.
Заряд частицы в соответствии с классическими представле-
ниями косвенно характеризуется величиной ^-потенциала. Исполь-
зуя эту величину, можно рассчитать по формуле Смолуховского
скорость движения коллоидной частицы в электрическом ноле:
^эф=(Г1Х/(4лт1)]Е. (3.77)
Формула справедлива при ха^>300, т. е для частиц большого
размера с тонким ДЭС
Уравнение, полученное Гюккелем, позволяет определять оЭф
при xa<g.l, т. е. для частиц малого размера с толстым ДЭС:
= ко? (6лт))]С. (3 78)
В интервале 1«;ха«:300 определение о,ф целесобразно прово-
дить по уравнению Генри:
е»ф= [ТоС'(6лЦ)]Е I (иа). (3 79)
Коэффициент f (ха) изменяется от I до 1,5 и может быть ориен-
тировочно определен по следующим данным:
ха 0,1 0,3 1 3 5 10 20 5» 100
1.0 1,01 1,03 1,10 1,16 1,25 1,34 1.12 1,16
Дпполофорсз, характеризуемый выделением во внешнем элск-
грнчсском поле иа поверхности раздела фаз свободных и связан-
ных поляризационных зарядов [27, 119], определяется двумя фак-
юрамп: воздействием неоднородного электрического поля на ПДМ
час I ины, величина которого определяется балансом свободных и
связанных поляризационных зарядов частицы и ДЭС, и элсктро-
осмо1пчсской компонентой, определяемой концентрационной поля
рнзацпей ДЭС частицы в неоднородном поле. При этом отмечено.
69
что вторая компонента, расчет которой наиболее сложен, в боль-
шинстве реальных систем существенно меньше нервом и, соответ-
ствию, скорость дннолофореза достаточно точно может быть рас-
считана по формулам [119]:
~ К (Re у’) а2/у5; (3.80)
Rc ft = Re у*/о3 = [ - 1/2 + 3 (ch f в/2 - 1) (1 + x0)/(a2 + ₽2) б']. (3.81)
где
a, = |4ch (Ve 2) 4- хо] (1 + х„); р, = х, (4ch ($<j/2) + хо +
+ x0|2ch(^6 2) + xa]|; 6'= а, 4-ХоР(; К е» М»/(12лц);
х0 = Уя*//(2Р) ; М = U3rtrt (rt — г,).
Yo* — поляризуемость частицы; ife — безразмерный потенциал: ^6~VIRT;
у*— расстояние от центра частицы до внутреннего электрода; rt н гг—ра-
диусы внутреннего и наружного электродов.
Диполофорез пропорционален дипольному моменту коллоидной
частицы (dm= Rey*na3E). Направление скорости дннолофореза оп-
ределяется знаком дипольного момента. В общем случае знак ско-
рости дннолофореза зависит от частоты внешнего поля, при изме-
нении которой можно в принципе изменить направление движения
частицы.
Изменение температуры электролита определяется балансом
тепла в электрореакторе (без учета рассеивания тепла в окужаю-
щую среду)
Лт (U, — Ер) - тст\Т (3.82)
Используя соотношения Дг = //(?; Vo=SJ,'. /=S»i, после не-
сложных преобразований получим
АТ Дт(Д/,-ЕР)'(?лП i(U,-Ep) (ТвСтМ. (3.83)
или с учетом электропроводности раствора хЛ
ЛГ = [i7(Ybct)1 [(iМ - (Ер /,)|. (3.84)
Из уравнения (3.84) следует, что при повышении плотности
юка происходит более интенсивный нагрев обрабатываемой жид-
кости, так как U3=f(i).
Для расходов тока в пределах 500—1000 Кл/л изменение тем-
пературы несущественно, однако при больших расходах тока, ха-
рактерных для концентрированных систем (осадков н шламов
сточных вод и т. п.), повышение температуры может быть значи-
тельным (до +20 °C). При повышении температуры па 10—20 °C
(расходы тока 3000—5000 Кл/л) возможно ускорение химических
реакций при превращении ионных примесей, денатурация белков
с последующей коагуляцией, повышение растворимости солей.
70
3.4.1.2. Воздействие продуктов электродных реакций
Весьма существенным фактором, определяющим состояние
электролита в межэлектродном пространстве, являются продукты
электродных реакций. При миграции продуктов в глубь раствора
происходит их химическое взаимодействие с примесями или водой.
Это в первую очередь окислительно-восстановительные и прото-
литические реакции, приводящие к изменению pH и Eh обрабаты-
ваемого раствора.
Генерация вещества за счет электродных реакций приводит
к увеличению его локальных концентраций, смещению химиче-
ского равновесия и, как следствие этого, к фазовым превраще-
ниям в жидкости.
При электролизе образуются растворимые продукты, а также
газовая и твердая фазы. Образование жидкой фазы при электро-
лизе происходит в редких случаях в сугубо специфических ус-
ловиях.
Растворимые продукты электролиза изменяют в основном про-
толитические и окислительно-восстановительные свойства ра-
створа, которые, в свою очередь, изменяют энергетическое состоя-
ние примесей. Так, если изменение валентного состояния про-
дуктов реакций и примесей не обеспечивает прямое превращение
последних, то существенно изменяет условия взаимодействия, тем
самым ускоряя превращения примесей в десятки и сотни раз.
Учитывая незначительную растворимость гидроксидов метал-
лов (твердая фаза) и водорода или кислорода (газовая фаза),
можно считать, что ионная сила раствора и поверхностное натя-
жение в процессе электролиза изменяются незначительно. Для ма-
лых межэлектродных зазоров (/,<3 мм) и высоких плотностей
юка (»яз1000 А/м2) возможно изменение плотности жидкости, од-
нако на практике величина газосодержапия ие превышает 1 %,
что несущественно сказывается на этом параметре. При электро-
лизе возможны различные направления изменения величины pH
в объеме раствора в зависимости от ионного состава электролита
и вида применяемых электродов.
При этом предположим, что в растворе отсутствуют вещества,
склонные к превращениям при изменении pH (Са2+, Mg2+ ит.п.).
В системе Pt+|AKBp, H2O|~Pt (где АКВР—молекула вещества, со-
держащая Лр+— катионы с потенциалами выделения более отри-
шпельиыми, чем потенциал водородного электрода, и Вк~—анионы
с потенциалами выделения более положительными, чем потен-
циал кислородного электрода) pH среды изменяется в соответ-
С1ВНИ с уравнением электродных балансов:
—— ЛкВр-----— Н,О->—Л (ОН)р+— WKB + —Hi + —О,. (3.85)
кр 2 р к 2 4
В прпкатодной зоне образуется щелочь, в приаиодиой — экви-
пален i ное количество кислоты. При смешивании продуктов реак-
71
пий происходит их взаимная нейтрализация, что не изменяет pH
среды, при разделении диафрагмой — повышение pH католита и
снижение pH анолита.
В системе Pt4 |СК£>Р, НгО|~Р1 (где CKDP— молекула вещества,
содержащая Ср — катионы с потенциалами выделения менее от-
рицательными, чем потенциал водородного электрода, hDk~—ани-
оны с потенциалами выделения менее положительными, чем по-
тенциал кислородного электрода) pH среды не изменяется:
---— cKDp — С® + — D". (3.86)
кр р к
На аноде и катоде разряжаются эквивалентные количества ка-
тионов Ср+ и анионов DK~, что не изменяет pH среды в приэлек-
тродных слоях и соответственно в объеме электролита.
В системе Pt+|AKDP, H2O| Pt (где ЛКОР— молекула вещества,
содержащая Др+—катионы с потенциалами выделения более от-
рицательными, чем потенциал водородного электрода, и DK~—
анионы с потенциалами выделения менее положительными, чем
потенциал кислородного электрода):
---— AKDP — Н,О — — ‘А (ОН)р + — D« + — Н». (3.87)
кр р к 2
На аноде разряжаются ионы D* па катоде выделяется водо-
род. В прикатодной зоне образуется щелочь, что приводит к повы-
шению pH католита при разделении продуктов электродных ре-
акций диафрагмой, а также всего объема электролита при их сме-
шении.
В системе Pt+1СКВР, H2O|_Pt (где СКВР — молекула вещества,
содержащая Ср+— катионы с потенциалами выделения меиее от-
рицательными, чем потенциал водородного электрода, и Вк~
анноны с потенциалами выделения более положительными, чем по-
тенциал кислородного электрода):
---?-СкВр_— Н,О —— НКВ + — Св + — О,. (3.88)
кр 2 к р 4
На катоде разряжаются иоиы Ср+, иа аноде выделяется кис-
лород. В прианодной зоне образуется кислота, что приводит к сни-
жению pH всего объема электролита при смешении продуктов
электродных реакций или аиолнта прн их разделении диафрагмой.
Обязательным условием изменения pH при электролизе с при-
менением нерастворимых анодов является наличие и иоде катио-
нов с потенциалами выделения меиее отрицательными и анионов
с потенциала мп выделения менее положительными, чем, соответ-
ственно, потенциалы водородного и кислородного электродов.
В нриэлек!родных зонах при выделении кислорода и водорода об-
разуется кислоi.i в ше.ючь Прн разделении продуктов приэлек-
тродных зон ди.тфр.н млмн можно получать распюры, обладающие
кислотными и щелочными свойствами в соответствии с вышепри-
веденными уравнениями.
При применении растворимых анодов Р+ (Fe, Al, Си, Mg и др.)
в системе Р+|Лц£р, НгО|~Р (где AKLP—молекула вещества, со-
держащая Лр+— катионы с потенциалами выделения более отри-
цательными, чем потенциал водородного электрода, и LK~—ани-
оны с потенциалами выделения более положительными, чем по-
тенциал ионизации растворимого анода):
— Me + Н,О-----— Мек Ьт----— ЛкГр-* — А (ОН)р -f-J- Н»‘ <3 89>
m тк рк р 2
На аноде образуются ионы Ря+, иа катоде выделяется во-
дород. В прикатодной зоне образуется щелочь, что приводит к по-
вышению pH католита при разделении продуктов электродных ре-
акций или всего объема прн их смешении.
В двухкамерном электрореакторе с неактивной диафрагмой ко-
личественная оценка изменения pH раствора, содержащего раз-
личные ионы, начальные концентрации которых в анолите Ci",
а в католите С<*, может быть произведена на основании уравне-
ния, описывающего изменение концентрации i-го иона в анолите
или католите в процессе электролиза:
Л
где ACJm(km), АС*э(кэ), ACJ" — изменение концентрации 1-го иона в анолите
(или католите) соответственно за счет миграции, электродных реакций, а также
химических превращений в объеме электролита
Изменение концентрации ионов в результате миграции через
диафрагму за время t
Ac>(K-) = //(FVe(K))fn<<«. (3 91)
о
Используя величину анодного или катодного расхода тока
/Ц<“> = 7//У1(К), получим
«г
АС*ы (км) = F~‘ ( п^Д?<*>. (3.92)
о
Для выражения изменения концентрации ионов в аполиге или
католите за счет электродных реакций используем закон Фарадея:
АС»э ("э) = В”((к)W/(FV.-W) = В?™Д?<K>/F. (3.93)
Типичными реакциями в объеме электролита, в результате ко-
юрых изменяются кажущиеся концентрации иоиов в анолите или
каюлите, являются реакции образования или диссоциации ком-
плексных ионов (например, НСОГ), малодпссоцннрованных соеди-
нений II.О, Mg(OH)2, СаСОз и др.), а также окнслителыю-восега-
73
иовнтельиые реакции (например, Fen— e^Fe3*). Необходимо
учитывать, что если в реакции участвуют два вида ионов, то их
концентрации изменяются в эквивалентных количествах. Так как
один ион может участвовать в нескольких реакциях, то в формуле
(3.91) учитывается суммарное изменение концентрации.
С учетом (3.91) и (3.92) формула (3.90) приводится к виду
CJ <*> = С? <“*> ±
( В« «Одд. <К> ± В» <К»Д» (Ю/F ± у дСхп
b 1 1 /Ь1!
(3.94)
При электролизе раствора, содержащего а различных катионов
и b анионов, в двухкамерном диафрагменном электролизере числа
переноса для i-го катиона и /-го аннона определяются по фор-
мулам:
(3.95)
(3.96)
где е, и V*—подвижности /-го катиона и /-го аннона. Ом-1-см1; CjaT> t—
концентрация /-го катиона в анолнте; Сан_— концентрация /-го аниона
в католите.
Уравнения (3.94) — (3.96) совместно с уравнениями констант
химического равновесия, записанными для ионов, участвующих
в химических реакциях в объеме электролита (уравнения произве-
дения растворимостей, констант нестойкости комплексных ионов,
ионного произведения воды, констант диссоциаций), описывают
изменение pH среды с разделением продуктов электродных реак-
ций. Модель учитывает начальную концентрацию веществ в обра-
батываемом растворе, объемы анодного и катодного пространства,
миграцию веществ через неактивную диафрагму при изменяю-
щихся числах переноса, химические превращения веществ на элек-
тродах и в объеме электролита.
В случае описания процесса электролиза раствора, содержа-
щего большое число ионов, участвующих в электродных реакциях,
а закже в химических реакциях в объеме электролита, возникают
рудпосш при решении системы этих уравнений. В простейшем
случае изменение кислотности и щелочности раствора, содержа-
щего ионы Na', SO’ , IP и ОН-, в двухкамерном диафрагмен-
ном элек । рол и icpe с iiepaci воримымн электродами и неактивной
днпфр.п мин опи< ын.к'и'и c.ie;i\ кицимп уравнениями
74
анолит
— ^Ne+ nNe* d (3.97)
1
CH + ^||-"”c~ nH* ^Л" F (39H)
г о
1 '^T
C* , C*« 2 4 — f n .,_ d.4“; (3 99)
i/2so* i/2so*— f ) i/iso; T
0
. At
^oh ^oh-H—("on-— •)<* V + h.o! (3 100)
католит
AT
^n»+ = cnb*H—jz~.f я№+ (З.Ю1)
1
Ch* —ch*+-^-| (”н+ ~ О^Д? + А^н O'. (3.102)
At
C" 2_ = C* 2----------— f « 2 ; (3 103)
I/2SO* I/2SOJ F J l/2SO*~’ ' ’
. At
coh- = coh------z-) nOH- t1^ ~~ Acii о» (3.104)
г о
где ЛСн.о и ДСн,о—увеличение концентрации ионов Н+ и ОН
в анолите и католите за счет диссоциации молекул води.
Учитывая постоянство ионного произведения воды Кв для ано-
ппа и католита, а также то, что число переноса иона п, опреде-
ляет долю тока, переносимую данным видом иона, получим си-
нему уравнений:
С^С&Н_=К„: суо.схн. = кв; (3 105)
где
У Ст,, с* v
‘ 1 Na+ Ne+
4-С" v 4-Ск , 0 , 4-CK V 1
Н* Н* I/2SO*- I/2SOJ-1 ОН- ОН-’
73
Из уравнений (3.98), (3.100) н (3.102). (3.104) нетрудно найти
изменение кислотности анолита и щелочности католита:
Дт
ЛК» = (Сн+ — сн+) — (сон- — сон~) = “V I (1~”н* ~ яон-) =
г о
-тТ <3IW’
о 4 * '
Л,,1К - (COH-“COH-)~(C?I+~CH+)- ('~ЯН+ —яОН-)<,-Г1т —
1 Д’1
= — ( (п Л+ » 2-^Д?- С3107)
F J \ Ne* I/2SOZ )
и
Для приближенного описания изменения кислотности анолита
и щелочности католита можно использовать уравнения, аналогич-
ные формуле (3.93):
ДКа = В«срД;/Р; Л1ЦК - (3.108)
где В^р — средняя (эффективная) величина выхода по току кислотности
(щелочности) за время /, соответствующее некоторому анодному расходу тока
Д£ и катодному Д£|
ВтЧ> д» f (%♦ + ”i/2soj~)
Мт о 4 z
= ДК J (%.♦ "l/ZSoJ-)^’
Изменение величины pH раствора:
в анодной зоне [99]
рп.= -1к('Ка + 10-р,,и);
в катодной зоне
ю-«
рН„ = — 1g--------—----------
10 (pH,, — 14) + ЛК»
(3.109)
(3.110)
(3.111)
Расчет с помощью полученных уравнении показывает, что по-
вышение концентрации Na2SO< (солесодержапня) в растворе при-
водит к росту предельных значений кислотности (щелочности), на-
капливаемых в анолите (католите). При этом в системе с аннон-
обмепной диафрагмой максимальное значение кислотности (ще-
лочности), достигаемое в анолите (католите) при электролизе,
только на 3- 4 % нише, чем в системе с неактивной диафрагмой.
В то же время в chcicmc с катнонообменнон диафрагмой максн-
76
Рис. 3 20 Зависимость кислотно-
сти анолита (щелочности като-
лита) от расхода тока ори об-
работке раствора Na SO. различ-
ной концентрации
I — 0.01 и; 2 — 0,02 и: 3-0.05 и; 4 —
0.1 н
мальное значение щелочно-
сти (кислотности) уже на
30 40 % меньше, чем в си-
стеме с ашюнообменной
диафрагмой. Такое явление
можно объяснить разли-
чием подвижностей конкурирующих ионов Н , Na+ н ОН ,
1/2S0J-, участвующих в переносе тока через диафрагму.
Как показывают расчеты, для повышения pH воды от 6 и выше
можно использовать кат ионообменную диафрагму, а для сниже-
ния pH от 8 и ниже — апиопообменпую. Электрохимическое изме-
нение pH воды в переходной области от 3—4 до 10—11 можно осу-
ществлять с использованием неактивной диафрагмы.
Во всех рассмотренных выше случаях наблюдалось постоянное
снижение мгновенного и среднего выхода по току кислотности
(щелочности) при увеличении расхода тока. Для случая обра-
ботки в диафрагменном электролизере раствора Na2SO« с различ-
ными начальными значениями pH раствора в камерах данная за-
кономерность (рис. 3.20) иногда нарушается. Так, максимальный
Рнс. 3.21. Зависимость мгновенного
(/ 4) и среднего (Г—4') выхода
ни току кислотности (щелочности)
от расхода тока при обработке рас-
нора N.i SO» различной концентра-
ции
I I <101 и; 2 2' —0.02 и; 3.3' —0 05 и.
г 4' 0.1 и
Рис. 3.22. Расчетная зависимость изме-
нения pH при обработке воды в без-
диафрагмениом электролизере с желез-
ным анодом при начальных pH
/ — 9.0; 2 — 6.0; 2 — 4.0: 4 — 3.6; 5 — 3.0; в —
2.5
77
выход по току наблюдается, когда в обеих камерах электролизера
производили истирал изящно растворов. При достижении точки эк-
вивалентности выход по току кислотности (щелочности) равен 1,0.
Минимальный выход по току характерен для случая обработки
анолита с начальным значением pH менее 7,0 и католита —
более 7,0.
Отклонение экспериментальных данных от теоретических кри-
вых составляет 0,5—14 % для расхода тока 0—5.0- 103 кл/л и воз-
растает с увеличением расхода тока (рис. 3.21). Несовпадение ре-
зультатов, особенно при высоких значениях расхода тока, можно
объяснить присутствием процесса электроосмоса, приводящего
к уменьшению объема анолита и увеличению объема католита на
2—11 %, а также наличием в обрабатываемой жидкости раство-
ренной углекислоты, которая попадает в анолит в результате окис-
ления графитового анода, а в католит—в результате контакта
с атмосферным воздухом — п оказывает существенное влияние на
изменение кислотности (щелочности).
При применении растворимых электродов изменение pH в без-
днафрагменном электрорсакторе описывается зависимостями [116]:
ГМеп+1 К2
|Меп+]с — -------------Ь----; (3.112)
KZ + К. ([Н+]н - Л [Ме"+]в + [н+]ду
[Ц1+1и=—lMe»+]. + |H+b,)[—^5—+1H+U--------- [Меп+|с| = К.;
(л И 1Н+]. п J
(3.113)
(11+1р - (Н+]и [Мсп+1в + [Н + 1д. (3.114)
п
Решая эту систему уравнений, можно определить концентра-
цию [Н ]р ниже pH гндратообразовапия. Выше этого pH соот-
ношение между [Men+]fOH-]; ” [Me (ОН)„] характеризуется про-
изведением растворимости. Для учета концентрации недпссоцпи
ровапиых молекул гидроксида металла в воде указанное соотно-
шение можно записать, используя константу нестойкости гидро-
ксокомплексон металла К»- Совместное решение новых уравнении
позволяет определить [Н *]р выше рНг.
Как показывают расчеты (рис. 3.22) по приведенным уравне-
ниям, при электролизе воды с растворимыми анодами величина
pH попытается до 9,5—9,8. Однако конечное значение pH зависит
не только от концентрации выделившихся ионов металла, но и от
начального pH воды. Так, прн электролизе кислых растворов ко-
нечная величина pH не превышает 8,5—8,8.
Разряд па электродах ионов Н\ ОН' и молекул воды, приво-
дящий к изменению pH среды, можно рассматривать также под
углом зрения окислительно-восстановительного взаимодействия,
поскольку при электролизе донором электронов служит катод, а их
78
акцептором — анод. Естественно, что в таком случае эти взаимо-
действия будут оказывать влияние на Eh воды.
При обработке водных растворов в диафрагменном и бездиаф-
рагменном электролизере с нерастворимыми анодами изменение
pH среды будет вызывать соответствующее обратное изменение
Eh среды по уравнению Eh =—0,0591 pH.
В то же время при использовании растворимых анодов, в част-
ности железных, вследствие образования ионов Fe2+ изменение Eh
также будет происходить за счет системы Fe3+/Fe2+. Рассмотрим
эту систему, приняв постоянными величины pH образования гид-
роксидов железа.
При рН<1,5 иоиы Fe2+ и Fe3* не образуют гидроксидов. По-
этому до данного значения pH величина Eh воды не зависит от из-
менения активной реакции среды:
Eh = 0.771 + 0,0591 lg ([Fe’+] (Fe’+J). (3.115)
Образование Fe(OH)3 начинается прн рН^1,5, a Fe(OH)2 —
при рН>6,3. Используя уравнение (3.115) и произведение раство-
римости Fe(OH)3, величину Eh воды в этом интервале pH можно
определить:
Eh - 0,771 4- 0,0591 lg , — (.041 — o,177pH — 0,0591 lg [Fe»+]-
К3 [Fez+]
(3.116)
С учетом (3.116) и произведения растворимости Fe(OH)2 ве-
личину Eh воды запишем:
1-Fc (ОН)-> (Н +]»
Eh 0,771 + 0,05911g----—”----— =0,28 — 0,0591 pH. (3.117)
/-Fe(OH)*ni + l1
Следовательно, при pH^6,3 величина Eh среды снижается
только в зависимости от повышения pH воды. Конечное значение
Eh среды достигает в этом случае величины —(0,6-^0,8) В. Для
реальных водных систем, имеющих открытый контакт с воздухом,
снижение Eh аналогично. В данном случае конечное значение Eh
достигает потенциалов —(0,4—0,8) В, превышая потенциалы ниж-
него предела области термодинамической устойчивости воды.
Подобным образом можно оценить влияние на величину Eh
среды и других ионов переменной валентности, образующихся при
ионизации растворимых анодов.
Обобщенные теоретические данные о возможных направлениях
и величинах электрохимического изменения Eh прн различных ва-
риантах электролиза воды приведены в табл. 3.2.
Экспериментальная проверка теоретических положений пока-
зала, что изменение pH растворов в безднафрагменном электроре-
акторе практически не зависит от материала катода. Прн повы-
шении плотности тока процесс изменения pH несколько интен-
сифицируется. Использование растворимых анодов приводит
к повышению pH растворов, причем наибольшее изменение pH па-
«• itoiaeicH в электрореакторе с железными анодами. В эюм случае
79
ко
максимальное повыше-
ние величины актив-
ной реакции (до 9,5—
10,5) происходит в рас-
творах с начальными
значениями pH не
менее 5,2—5,8. При
начальных рН=3,24-
4-5,2 конечная вели-
чина активной реак-
ции не превышает
6,8—7,3 (см. рис. 3.22).
При использовании
алюминиевых анодов
pH растворов повыша-
ется только до 9,0—
9,1 вследствие амфо-
терности образую-
щегося при электро-
лизе гидроксида алю-
миния. По этой же
причине в исходных
щелочных растворах
pH среды снижается
на 1,0—1,1 единицы.
При обработке рас-
творов в бездиафраг-
мсппом электрореак-
торе с нераствори-
мыми анодами направ-
ление изменения pH
определяется катион-
ным и анионным со-
ставом растворов. Так,
катионы К+, Na+ и
анионы СггОГ, SO«-
не влияют на измене-
ние pH. В то же время
наличие в растворе ка-
тиона Сн2+ приводит
к снижению pH от
5.3- 5,9 до 2.9 3,6.
При электролизе рас-
твора, содержащего
анионы Cl~, SO«~, pH
увеличивается па 1.0—
1.8 единицы В свою
очередь, наличие в рас-
творе аннона СО/’ замедляет изменение величины pH. При
электролизе растворов с применением графитовых анодов pH сни-
жается от 8,7—10,3 до 6,0—6.5. Это явление можно объяснить ра-
створением диоксида углерода, который образуется в результате
окисления графита кислородом.
При обработке растворов в диафрагменном электрореакторе
изменение pH (щелочности и кислотности) католита и анолита
зависит о г материала диафрагмы, используемой для разделения
продуктов электродных реакций.
В электрореакторе с катионообменной диафрагмой изменение
этих показателей меньше, чем в э.тектрореакторе с анионообмен-
пой или неактивной (целлофановой) диафрагмами. Так, для ка-
тпонообменной диафрагмы повышение щелочности католита на
10—15 %, а кислотности анолита иа 20—30 % меньше, чем для
целлофановой диафрагмы. Кроме того, если в электрореакторе
с анионообменной диафрагмой повышение кислотности па 8—15%
меньше, чем в электрореакторе с целлофановой диафрагмой, то
повышение щелочности в обоих случаях одинаково. При этом для
целлофановой диафрагмы изменение щелочности католита и кис-
лотности анолита в среднем только на 8—15% больше, чем
в электрореакторе с неактивной тканевой (хлориновой) диаф-
рагмой.
Существенное влияние на изменение pH оказывают начальные
значения pH и солесодержание раствора. Так, при применении не-
активной диафрагмы наиболее эффективное изменение pH като-
лита и анолита наблюдается при начальных pH растворов в пре-
делах (2,8—3,2)-> (10,8—11,2). В свою очередь, от солесодержа-
ния зависит выход по току кислотности н щелочности, а также их
максимальное значение в католите и анолите. Например, в элек-
трореакторе с целлофановой диафрагмой величина изменения кис-
лотности (щелочности) на начальном участке изменения pH при
росте солесодержания от 0,5 до 2 г/л приводит к увеличению вы-
хода по току кислоты и щелочи от 0,70—0,85 до 0,87—0,95. Мак-
симальное значение кислотности (щелочности), достигаемое в ка-
толите (анолите) при указанном выходе по току, возрастает от
4,0—4,7 до 8,0—10,5 мг-экв/л.
При использовании хлориновой диафрагмы плотность тока
в пределах 30—300 А/м2 не влияет па изменение щелочности,
а в пределах 150—300 А/м2—на изменение кислотности. В то же
время снижение плотности тока до 30 50 А/м2 приводит к неко-
торому замедлению процесса изменения кислотности.
Обработка растворов в безднафрагменпом электрореакторе
с растворимыми железными анодами приводит к сдвигу величины
I'h среды до (0,55—0,80) В, а с алюминиевыми анодами — до
(0,40—0,55) В (рис. 3.23). Применение нерастворимых анодов
обусловливает снижение Eh среды на (0.1—0,2) В независимо от
ионного состава раствора. Исключение составляют растворы, с<>-
ц-ржащие ионы С1~, разряд которых на аноде с образованием С1а
81
Рис. 3.23. Зависимость pH (/—4) и F.h
(5—8) от расхода тока при обработке
0,01 моль/л водного раствора Na SOt
в бездиафрагмеииом электрореакторе
с алюминиевым (/, 3, 5, 7) и желез-
ным (2, 4, 6, 8) анодом при началь-
ных pH
/. 2. S. 8 — 10.03; 3. 1. S. 1 — Б.71
Рис. 3.24. Диаграмма потеициал-рН
растворов 0.01 моль/л Na2SOt (а),
0,01 моль/л Na2CO3 (б) и 0,02 моль/л
NaCI (в) при их подщелачивании
0,1 N NaOH (/), подкислении 0,1 N
НС! (2), анодной (3, 4) и катодной
(5) обработке в диафрагменном
электрореа кторе
и последующим растворением этого газа в водной фазе приводит
к некоторому сдвигу Eh в сторону положительных значений.
В безднафрагменном электрореакторе определяющее влияние
на изменение Eh оказывает ионный состав растворов. Увеличение
в растворе катионов Na+ и К+ обусловливает снижение Eh като-
лита до (0,4—0,5) В. Сравнение полученных данных с результа-
тами титрования раствора щелочью (NaOH) показывает, что тан-
генс угла наклона экспериментальной прямой в первом случае
выше (0,09) В, чем во втором (0,06) В (рнс. 3.24). На основании
этого следует предположить, что сдвиг Eh католита связан как
с тмснсппем pH, так и с одновременным растворением в водной
фазе элеыролнтического водорода
При электролизе растворов, содержащих анионы С1г, Eh ано-
лша iioipaciaei до +(1.1 —1,2) В. На диафрагме полученная за-
внснмосп. iimcci вид ломаной линии, конечный участок которой
раскола)аеiси в области потенциалов, лежащих выше верхнего
предела |грмо пшамической устойчивости воды. Это свидетель-
cmvei о юм. чю iiiMciiciine Eh анолита связано как со сдвигом
pH. 1ак и < |>.н ।н«>р« пнем в иодной фазе газообразного С1;.
к?
Данные об изменении pH и Eh, полученные при обработке
в тиафраг мепных и бездиафрагмснпых электрореакторах реальных
нотных систем (сточных вод кожза видов, промывной воды гальва-
нических производств, природной воды), показывают, что эти ве-
личины изменяются менее интенсивно, чем в водных растворах.
Это обусловлено наличием ионов тяжелых металлов, карбонатных
ионов, образующих буферные системы реальных вод.
3.4.2. Превращения примесей воды за счет электролитических
эффектов
3.4.2.1. Коагуляция частиц в электрическом поле
Воздействие однородного электрического поля на дисперсные
системы, в частности коллоидные, приводит к процессу электро-
коагуляции Термин «элек!рокоагуляцня» в данном случае принят
но работе [30] и характеризует поляризационные притяжения ча-
стиц во внешнем электрическом поле в отличие от электрохимиче-
ской и электролитической коагуляции.
Наиболее полно вопрос влияния электрического поля на кол-
лоидные системы освещен в работах О. Г. Усьярова, И. Ф Ефре-
мова, О. В. Смирнова, Ю. Ф. Дейнеги, С. С. Духина, В. Р. Эст-
рела-Льописа.
Установлено, что под действием поля из коллоидных частиц и
капель эмульсий образуются цепочечные агрегаты, ориентирован-
ные вдоль вектора напряженности. Этот эффект был использован
при обработке технологических растворов, а также в технологии
очистки природных и сточных вод [20, 30].
Результатом совместного действия электростатических сил от-
талкивания и молекулярных сил притяжения является формиро-
вание вблизи поверхности частицы и на некотором удалении от
нее двух потенциальных ям, разделенных энергетическим барь-
ером (рис. 3.25) [30]. При снижении заряда частиц уменьшаются
силы отталкивания и уменьшается высота барьера, а глубина по-
тенциальных ям увеличивается. И если глубина ям достаточно ве-
лика, то происходит процесс коагуляции.
Количественный анализ эффекта электрокоагуляции выполнен
О. В. Смирновым на основе теории ДЛФО* с учетом дополнитель-
ной составляющей энергии диноль-дипольпого взаимодействия
W'o + »'.п. (3.118)
Результаты расчета (рнс. 3.26) показывают, что при напря-
жении поля Е=5 кВ/м глубина потенциальной ямы при взаимо-
icHciBHH частиц понижается на величину порядка (2—5) кГ по
сравнению с взаимодействием частиц без электрообработки [20].
Для различных Чгв-потепцналов частиц и напряженности поля
т.чже при низкой устойчивости частиц (1Ев = 1 н 4 мВ) напряжен-
ное и, поля 2 кВ/м увеличивает глубину ямы на величину порядка
• Теория Дерягина — Ландау — Фервея— Овербека.
«3
w/kT
Рис 3 25. Потенциальные кривые
энергии взаимодействия частиц
/ — прн отсутствии поля, с неглубокой
потенциальной ямой; 2 — при наложе-
нии поли, с углубленной потенциальной
ямой (получена сложением кривых I
и 3); 3 — диполь-дипольного притяже-
ния
Рис 3 26 Глубины потенци-
альной ямы при напряжен-
ности внешнего электриче-
ского поля
/ — О кВ/м; 2 — 6 кВ/м; 3 —
10 кВ/м
Рис. 3.27. Изменение оптической
плотности раствора латекса после
обработки в бездиафрагменной
ячейке с алюминиевыми (/, 2) и
||>афнтовымн (.7, 4) электродами
при и 1ПГ1ННТИ тока
1.1 А я’. 4-100 А/м'
Ionian. м,и
Рис. 3.28. Изменение опти-
ческой плотности золя ла-
текса, обработанного в
анодной и катодной зоне
электрической ячейки с цел-
лофановой диафрагмой и
графитовыми электродами
при времени воздействия
тока
I — 0.6 мин; 2 — 1.6 мин; 3—
2.6 мин. 1 - Б мин; 5 — 7.5 мин;
/. 2. 3.1, 5 — анодная эона: /'.
У. У. 1', S' —катодная зона
кТ, что явно недостаточно для эффективной коагуляции частиц,
особенно с высоким значением Ч'в-потенциала. Значительный
эффект действия ноля проявляется прн напряженностях 10 и
30 кВ/м, однако такие напряженности поля довольно редко ис-
пользуются в технологии очистки воды.
Анализ данных по изучению влияния электрического поля на
дисперсные системы во многих случаях показывает, что не всегда
учитывается вклад электрохимической и электролитической со-
ставляющих воздействия поля. Так, например, изучение влияния
напряженности поля с применением алюминиевых электродов при
воздействии продуктов электродных реакций на обрабатываемый
объем сточных вод производства полистирола [20] не позволяет
оцепить вклад каждой составляющей в процессе очистки.
Сравнительная оценка вклада составляющих электрохимиче-
ского процесса до настоящего времени в литературе отсутство-
вала, поэтому была произведена оценка влияния напряженности
поля в одно-, двух- и трехкамерпом электрореакторе с графито-
выми электродами в статических условиях. В качестве дисперсной
системы использовался золь монодисперсного латекса с размером
частиц 0,78±0,2 мкм, приготовленного по методике Kotera [133].
Плотность тока иа электродах поддерживалась в пределах 50-4-
-4-100 А/м2, расход тока—100-4-5000 Кл/л. Материал диафрагмы—
целлофан. В качестве фонового индифферентного электролита, по-
вышающего солесодержание раствора, использовали Na2SO< с кон-
центрацией (1.5-4-З.О) • 10 3 моль/л.
Комплексное воздействие электрического тока на золь латекса
как для нерастворимых (графитовых), так и для растворимых
(алюминиевых) электродов характеризуется снижением оптиче-
ской плотности золя, что свидетельствует об изменениях в дис-
персной системе (рис. 3.27). Эффективность воздействия раствори-
мых электродов значительно выше, чем нерастворимых, при оди-
наковых расходах тока, особенно в конечной стадии обработки.
Исследуемые величины плотности тока оказывают несущественное
влияние иа изменение оптической плотности.
Обработка золя в двухкамерном электролизере с разделением
продуктов катодной и анодной реакций показала, что вклад элек-
трохимической составляющей в коагуляцию может быть сущест-
венным, особенно при больших расходах тока, т. е. при значитель-
ном изменении pH н Eh золя. Однако снижение оптической плот-
ности в катодной камере происходит более иитепсивио, чем в анод-
ной, что противоречит теоретическим закономерностям (рис. 3.28).
Учитывая высокий отрицательный заряд частиц латекса (35—
10 мВ), можно было предположить его снижение в анодной зоне
за счет уменьшения pH и соответствующее улучшение условий
коагуляции, что не подтверждается экспериментальными данными.
Наблюдаемое явление позволяет предположить существенное
влияние электрического поля на поведение системы. Однако при
воздействии поля могут возникнуть два эффекта: транспорт ча-
сти и концентрирование нх в определенном объеме ячейки без
85
дисперсного превращения; агрегация частиц в жидкости за счет
дпиоль-дппольпого взаимодействия.
Для оценки этих эффектов и возможности протекания необра-
тимых дисперсных превращений в золе латекса под действием
ноля изучено влияние электрообработки в однокамерном электро-
лизере с графитовыми электродами с последующей экспозицией
золя для наблюдения за изменением его оптической плотности. За
период экспозиции оптическая плотность золя не изменяется, что
говорит об отсутствии необратимых дисперсных изменений в си-
стеме, хотя оптическая плотность снижается с увеличением вре-
мени обработки.
Такая закономерность указывает на существенную роль транс-
порта частиц за счет электрофореза. Увеличение плотности тока
(соответственно, и напряженности ноля) при Дт=соп5( приводит
к интенсификации процесса переноса частиц, что подтверждает
вышеуказанные предпосылки.
При электролизе с алюминиевыми электродами характерно не-
обратимое дисперсное изменение золя латекса во времени после
снятия напряжения. Отсюда нетрудно сделать вывод о преиму-
щественном влиянии ионов растворяющегося металла на дисперс-
ные превращения золя латекса. Увеличение плотности тока, как и
в предыдущем случае, ускоряет процесс коагуляции частиц, что
указывает иа синергический эффект продуктов электродных реак-
ций и поля.
Данный опыт не позволяет оценить «чистое» влияние поля
из-за образующейся газовой фазы, изменяющей гидродинамиче-
скую обстановку в электролитических камерах и способствующей
ускорению коагуляции за счет перемешивания.
Электролиз золя в трехкамерной электрохимической ячейке
с нерастворимыми электродами, препятствующей миграции про-
дуктов электродных реакций в среднюю (нейтральную) зону
ячейки, показал, что под действием поля не происходит сущест-
венных (в пределах ошибки опыта) дисперсных изменений частиц
золя (рис. 3.29). Визуально отмечено интенсивное осаждение ча-
стиц па диафрагме, обращенной к анодной зоне, что подтверж-
дает полученные данные по электрофоретической подвижности.
После смены полярности электродов частицы золя мигрировали
в раствор и оптическая плотность его соответствовала исходной.
Незначительное уменьшение оптической плотности в анодной зоне
можно объяснить осаждением частиц золя на электроде, однако
этот процесс затруднен из-за срыва частиц с поверхности выде-
ляющимся кислородом при принятой достаточно высокой плотно-
сти тока на элеш родах.
I стесч пенно, в данном опыте не учтена миграция ионов S0«
и Na' из нейтральной зоны и соответствующее снижение солесо-
держания. Однако изменение концентрации ионов SO^ и Na+
в пределах концентраций 10 54-10 2 моль/л изменяет величину
^-потенциала частиц латекса па 10—15% [133], что не отразится
существенно на стабильности системы.
8Г>
Рис. 3.29. Изменение оптической плотности
золя латекса при обработке его в трехкамер-
ной электролитической ячейке с целлофано-
выми диафрагмами и графитовыми электро-
дами при времени воздействия тока
/. /" — 3 мни; 2, 2". 2" — 10 мни; /, 2 — анод-
ная зона; /', 2* — катодная «она; /*, 2* — нейтраль-
ная зона
Рис. 3.30. Изменение опти-
ческой плотности золя ла-
текса, обработанного в ка-
тодной (/, 2) и анодной
(3, 4) зоне электролитиче-
ской ячейки с целлофано-
вой диафрагмой и графи-
товыми электродами при
плотности тока
I. 3 — 60 AJ*'; 2. 4-100 А/м’
Кривые оптической плотности, полученные при электрообра-
ботке золя в двухкамерной диафрагменной ячейке с графитовыми
электродами, не обнаружили существенного влияния продуктов
анодной и катодной реакций на дисперсные изменения золя
(рис. 3.30). Если в катодной зоне плотность тока не оказывает
значительного влияния на снижение оптической плотности, то
в анодной зоне наблюдается выраженное уменьшение электрофо-
реза при увеличении плотности тока, что подтверждает предполо-
жение о влиянии газовой фазы па перемешивание электролита,
транспорт и осаждение частиц па электроде.
3.4.2.2. Агрегация частиц за счет
продуктов электролиза
Как уже указывалось, растворимые продукты электролиза из-
меняют в основном окнелшелыю-восстановительные и протолити-
ческие свойства жидкости. Веледе ише сдвига pH н Eli происхо-
дит, в свою очередь, изменение фазово-диснерспого состояния
веществ.
1. Изменение формы существования непосредственно примесей
воды согласно термодинамическим диаграммам Пурбе [130]. Так,
молекулярную серу согласно диаграмме S — НО можно нолучшь
при значениях рН = —24-2 в узкой области значений Eh ()4-0,1 В.
87
При этом моляризацня идет без участия воды и других веществ,
находящихся в воде.
2. Изменение формы существования примесей за счет взаимо-
действия с продуктами диссоциации воды ври изменении pH. Так,
образование гидроксидов тяжелых металлов происходит при на-
личии избытка ионов ОН за счет повышения pH воды. Примером
взаимодействия серы с ионами Нь при снижении pH может слу-
жить образование молекулярного сероводорода в кислой среде.
Изменение Eh раствора при взаимодействиях может изменить,
во-первых, валентное состояние непосредственно примесей для
создания условий их взаимодействия с водой или другими вещест-
вами, и, во-вторых, валентное состояние воды и других химиче-
ских соединений, которые могут взаимодействовать с извлекаемой
примесью. Примером такого взаимодействия может служить обра-
зование малорастворимого FeS согласно диаграмме Пурбе. При
этом, если Fe2+ рассматривать как примесь, которая должна быть
удалена, а серу как вспомогательное вещество, присутствующее
в воде, то удаление железа в виде FeS возможно при снижении
Eh до значений менее 0,2 В с целью перевода серы в форму S2-
для взаимодействия с Fe2+. Конечно, при оценке такого взаимо-
действия учтена кинетика процесса и трудность перевода SO«~
в S2-, однако такой подход является достаточно достоверным для
выбора направления регулирования pH и Eh с целью превраще-
ния примесей воды.
3. Изменение строения границы раздела фаз при агрегации
примесей воды. При этом может происходить: поверхностная дис-
социация примесного вещества за счет изменения pH и Eh; ион-
ный обмен на поверхности раздела за счет изменения концентра-
ции ионов Н+ и ОН в растворе; изменение валентного состояния
попов на поверхности раздела; образование нерастворимых соеди-
нений с нонами, входящими во внутреннюю н внешнюю обкладки
ДЭС по схеме, описанной в вышеприведенных пунктах.
В свою очередь, влияние pH и Eh на границу раздела фаз
может быть отнесено и к электродным процессам, где может из-
меняться не только протекание электрохимических реакций, но и
протекание фазово-дисперсных превращений. Так, при повышении
pH электролита интенсифицируется процесс растворения алюми
пиевого катода, который протекает, как объясняют некоторые ис-
следователи, по химическому механизму [78]. Копнен грация избы-
точной щелочности на границе раздела «катод — электролит»
приводит к ионизации алюминия без участия электрической со-
ставляющей процесса. Аналогично может протекать химическая
коррозия анода при снижении pH в прнанодиой области при по-
тенциалах ниже потенциала ионизации алюминия.
В лшер.тре указывается на существенное влияние pH при
крнс1аллп I.HIIIII гидроксидов металлов с концентрацией попов ОН "
в растворе выше протвелспни растворимости [31]. Так, предпола-
гается, что Boip.n i.iiun- величины pH среды (от 4 до 7) способ-
стнует увеличению степени полимеризации гндроксокомплексов
алюминия по схеме
AI (ОН)+^ А16 (ОН)^ А110 (ОН)«+ А124 (ОН)^+- AIm (OI 1)'8+. (3.119)
Кроме того, с изменением pH изменяется сорбция одно- и двух-
валентных анионов (Cl , SO< и др.) иа гндроксикомплексах, но-
сящая ионообменный характер. Включение в структуру анионов
различной природы, естественно, будет отражаться па характере
полимеризации. Аналогичные примеры могут быть приведены и
для других гидролизующихся металлов.
Конкретным примером превращения примесей (кристаллиза-
ция) при изменении pH является, в частности, подщелачивание
природной воды в катодной камере диафрагменного электрореак-
тора, что приводит к образованию в католите нерастворимых кар-
бонатных и гндрокспдных соединений Са2+ и Mg2+. В результате
этого концентрация попов кальция в католите снижается от 4,8—
4,2 до 0,5—0,4 мг-экв/л, ионов магния — от 1,6—2,0 до 1,2—
0,7 мг-экв/л, а общая жесткость — от 6,8—5,8 до 1,6—1,1 мг-экв/л.
Кроме того, щелочность католита уменьшается на 0,8 —
1,0 мг-экв/л.
В анолите также наблюдается снижение концентрации ионов
Mg2+ до 2,0—1,6 мг-экв/л, Са2+— до 1,0—0,8 мг-экв/л и общей
жесткости — до 1,4—1,8 мг-экв/л. Это связано с миграцией ионов
кальция и магния из анодной камеры в катодную через диаф-
рагму. Соответственно при подкислении анолита щелочность его
уменьшается, а кислотность возрастает от 1,2—1,6 до 4,8—
5,8 мг-экв/л.
Подщелачивание в катодной камере промывной воды гальва-
нических производств приводит к образованию гидроксидов тяже-
лых металлов, что снижает их концентрацию (табл. 3.3).
Аналогичным примером влияния pH иа образование твердой
фазы может служить получение гидроксида магния из морской
воды. Основное влияние на процесс оказывает расход тока,
а также величина солесодержания. Подщелачивание католита
происходит практически с одинаковой интенсивностью, в то время
как подкисление анолита — скачкообразно.
Интенсивность гндратообразования зависит в основном от рас-
хода тока и начальной концентрации магния в воде. При началь-
ной концентрации магния 500 мг/л расход тока, необходимый для
связывания 40—65 мг/л магния, превышает 800 Кл/л. Увеличение
расхода тока до 1000 Кл/л позволяет связывать 80—125 мг/л маг-
ния. Одновременно со снижением концентрации магния в воде при
электролизе снижается жесткость воды и содержание кальция.
С помощью полного факторного эксперимента получены урав-
нения регрессии, описывающие изменение концентрации магния
в католите при электролизе в диафрагменном электрореакторе,
хлоридов, магния и кальция в анолите, pH католита (табл. 3.4),
а также общей жесткости католита и анолита. Математическое
89
Таблица 3.3. Зависимость pH и концентраций ионов тяжелых металлов от
расхода тока при обработке промыниой воды гальванического цеха в катодной камере
диафрагменного электрореактора
Наименование показателя Расход ток л. Кл/л
0 360 540 720 1080
Активная реакция pH 7,38 8,92 9,26 9 39 10,21
Медь, мг/л 860 7,30 3,75 2,66 0,68
Цинк, мг/л 24,10 0,36 0,21 0,18 0,58
Никель, мг. л 12,00 9,60 2,68 1,14 0,87
описание процесса произведено в окрестности точки факторного
пространства с координатами: Дт=1400 Кл/л (*i), См<=1,25 г/л
(х2) и i=300 Л/м2 (х3).
Формирование твердой фазы в объеме электролита зависит от
направления протекания катодных и анодных процессов (табл. 3.5)
и проходит последовательно в несколько стадий.
При формировании гидроксидов металлов последние обра-
зуют аква комплексы типа [А1(Н2О)3- (ОП)з], [A1(H2O)5-OH]2+mt. п
за счет ступенчатого отщепления протонов от молекул воды. Прн
этом происходит полимеризация гидроксокомплексов с образо-
ванием многозарядного полимеризованного нона типа AbfOH)*,
А14(ОН)<+, А17(ОН)«+ А1|3(ОН)з+ Fe3(OH)J+ и др. [7]. Для
катионов А13+ и Fe3* характерна сильная тенденция к образова-
нию соединений не только с ионами гидроксила, ио и с другими
анионами и группами: фосфатными, сульфатными, карбоксиль-
ными и др. Так, предполагается наряду с формированием
A1(OH)SO4, AI2(OH)SO4 образование соединений типа
[AI(H2O)5OH]SO4, Ale(OH)12, (SO4)3, [Л1Р2О7], [FeHPO4]+ Ана-
логичные соединения образуют и другие металлы.
Форма соединений в большой степени зависит от pH раствора
Диаграммы процентного содержания разных форм железа и алю-
миния в зависимости от рн приведены в работе [7].
Таблица 3.4. Уравнения регрессии, описывающие образование гидроксида
магния при электролизе морской воды в диафрагмеииом электрореакторе
Описываемое изменение Уравнения регрессии
Концентрация магния в католите Концентрация магния в анолите Общая жесткость католита Общая жесткость анолита pH католита Концентрация кальция в анолите Концентрация хлоридов в анолите Ух = 1010,44 — I64.09JC, 4 703.61х, yt = 1163,51 —20,13X14 687,5х, у, = 86,94 - 13,81X1 + 15Я.56х, + + l,81xixt yt = 99,81 — 1,7х! + 56,59х, j,B = 9.59 4- о,44Х| — 0,36х, 4- 0.14х, у, = 93,01 4- 2.81х, у, = 9373,96 4- 22104,66л, 4- 421.86х,х,
90
Т а б л и ц а 3.5. Пути формирования твердой фазы в электрохимических реакторах
Электродный процесс Формирующаяся твердая фаза Условна формированаа
Анодное (катодное) рас- творение металла Г идрокс иды металлов Наличие соответствующей концентрации ионов ОН-
Генерация ОН”-ноиов в растворе То же Наличие попов тяжелых ме- таллов в обрабатываемой ЖИДКОСТИ
То же Нерастворимые соедине- ния солей жесткости Наличие ионов кальция и магния
Изменение pH (Eh) рас- твора при соответству- ющих электродных реакциях Кристаллизация веще- ства, присутствующего в растворе Наличие веществ, способных переходить в черастворн- мую форму (сера и дру- гие)
Рост полимеризованного иона приводит к образованию фазы,
которая для гидроксидов металлов представляет на первой стадии
аморфные структуры, проходящие в последующем стадию кри-
сталлизации [8]. До настоящего времени нет единого мнения о на-
чале кристаллизации, однако пре тиолагается, что процесс кри-
сталлизации начинается сразу же после образования гидроксидов,
дальнейшая агрегация которых подчиняется основным закономер-
ностям теории устойчивости и коагуляции дисперсных систем. Су-
щественным отличием образования коагуляционных структур гид-
роксидов следует считать формирование на первой стадии сплош-
ной пространственной сетки, затем ее разрыв иод влиянием гидро-
динамических воздействий и процесс старения на второй стадии.
Для описания процесса образования твердой фазы, в част-
ности гидроксидов металлов, используют правило постоянства
произведения активности пли произведения растворимости. Од
пако некоторые исследователи предлагают отказаться от этой
характеристики вследствие ее непостоянства при старении ги-
дроксидов.
Несмотря на то что в справочной литературе приводятся ве-
личины произведения растворимости для свежих и «старых» ги-
дроксидов, исследованиями установлено, что полный гидролиз —
процесс медленный, и устойчивое состояние равновесия, напри-
мер при гидролизе солей железа, достигается через пять-шесть
месяцев.
Старение характеризуется уменьшением кажущегося объема
осадка, улучшением его седиментационных, реологических и
фильтрующих свойств, ухудшением сорбционной и каталитиче-
ской способности и растворимости [7]. Кроме того, структурный
анализ показывает, что в первоначальном аморфном осадке со
временем обнаруживается кристаллическая фаза, общее содер-
жание воды (связанной и адсорбированной), кзк правило, умепь-
91
шается. захваченные при возникновении осадка примеси (в ча-
стности, аииопы) снова переходят в раствор.
На скорость старения гидроксидов влияют pH, температура
и особенно примеси, присутствующие в растворе и захватывае-
мые осадком. С ростом pH н температуры старение свежеосаж-
денного гидроксида железа протекает интенсивнее. Роль приме-
сей двояка: они либо тормозят старение (соли многовалентных ме-
таллов, кремневая кислота и др.), либо существенно его ускоряют
(растворенный кислород и т. п.).
Учитывая вышеприведенные закономерности формирования и
растворения твердой фазы в объеме раствора, можно оценить
условия ее поведения при электрообработке.
1. Формирование и растворение твердой фазы в электриче-
ском поле, что может привести к изменению ее структуры при
формировании и ускорению растворения за счет поляризации
частиц.
2. Формирование различных структур твердой фазы в меж-
электродпом пространстве за счет перераспределения концент-
рации иоиов при их миграции в электрическом поле, а также за
счет электродных процессов, в частности изменения pH и Eh.
3. Последовательный ввод соединений, формирующих твер-
дую фазу, при потоке жидкости в межэлектродпом пространстве.
4. Перемешивание жидкости в межэлектродпом пространстве
электрохимическими газами, способными создавать различную ин-
тенсивность массоперепоса как во времени, так и в объеме
раствора
При формировании твердой фазы в объеме электролита воз-
можно взаимодействие ее с примесями воды коллоидной степени
дисперсности, приводящее к агрегации частиц.
Как правило, образование твердой фазы в электрореакторах
происходит за счет реакций гидролиза продуктов анодного и ка-
тодного растворения металла электродов. Такими продуктами
являются гидроксиды алюминия, железа, магния и других ме-
таллов. Размер частиц гидроксидов зависит от условий их фор-
мирования и в большинстве случаев лежит в пределах 1,6—
2.5 мкм [7], хотя имеются частицы и значительно больших разме-
ров— 10—30 мкм. По мере кристаллизации размер частиц гидро-
ксидов уменьшается [8].
Взаимодействие формирующейся твердой фазы с примесями
воды может происходить по двум механизмам: гетсрокоагуля-
цнн частиц гидроксидов металлов и примесей воды (при этом
твердая фаза гидроксидов и примесей может иметь как одноимен-
ные, так и разноименные заряды); гомокоагуляцнн частиц при-
месей за счет изменения ионной силы раствора, pH и т. п. при
формировании твердой фазы.
Количественная оценка взаимодействия частиц по данным
механизмам производилась иа основании теории ДЛФО при ус-
ловии, что напряженность поля до 2 кВ/м в элекзрореакторе не
оказывает существенного влияния на коагуляционные процессы.
82
Рис. 3.32. Диаграмма устойчивости ча-
стиц латекса при гомокоагуляции
/ —Цр —100; 2—цр-10; 3— цр-5; 4 —рр
Рис. 3.31. Энер1ия взаимодействия
при гетерокоагуляции частиц ла-
текса и гидроксида магния при зна-
чениях pH
I. 4 — 9.6: 2. 5—11.0; 3, 5-12.0; Аг —
_|0~u Дж; Ci-0.8 мкм, 01-1,4 мкм
(1-3); а,-0.2 мкм (1—£)
Расчеты производились для золя латекса с диаметром частиц
1,6 мкм и ^-потенциалом, изменяющимся в пределах —(20?
-4-100) мВ, а также для частиц гидроксидов магния с потенциа-
лом (104-40) мВ и даметром частиц 0,4, 1,6, 2,8 мкм при различ-
ных фиксированных значениях pH = 3,04-12,0. При этом пред-
полагалось, что исходный золь устойчив в широком диапазоне pH.
Выполненные расчеты суммарной энергии (рис. 3.31) показы-
вают, что для всех случаев взаимодействия разноименно заря-
женных частиц ионно-электростатнческая составляющая способ-
ствует притяжению частиц и значительно превышает энергию мо-
лекулярных сил Ван-дер-Ваальса. Суммарная энергия притяже-
ния выше для меньших значений величин pH и возрастает с уве-
личением радиуса взаимодействующих частиц. Следует отметить,
что с уменьшением потенциала положительно заряженных частиц
электростатическая составляющая, а следовательно, и суммарная
энергия значительно снижаются, н при пулевом потенциале ча-
стиц эта составляющая равна нулю. Взаимодействие происходит
только за счет сил молекулярного притяжения. Отсюда следует
предположить, что скорость флокуляции должна возрастать с ро-
стом потенциалов частиц противоположного знака и при более
низких значениях pH (для вышеуказанных пределов).
При взаимодействии разнородных частиц одинакового знака,
отличающихся величиной потенциалов и размеров, характерно их
отталкивание в широком диапазоне pH при величине ^-потенциа-
лов выше 20 мВ и константы Гамакера 1 • 10-20 Дж.
93
Обобщенная диаграмма устойчивости частиц для второго ме-
ханизма взаимодействия, построенная па основании расчетных
данных энергии взаимодействия в зависимости от изменяющейся
ионной силы раствора за счет введения нона металла в раствор
и величины pH (рис. 3.32), показывает, что гомокоагуляция ча-
стиц (без учета образования гидроксидов) протекает при очень
низких (<3) и высоких (>11) значениях pH, а также при до-
статочно высоких концентрациях вводимого иона металла
(10 3-:-20 1 моль/л) Такне условия являются нехарактерными
для работы применяемых электрореакторов, так как расход тока
для вытелепия такого количества ионов металла в раствор ие
превышает 100 Кл/л.
В остальной области pH и концентраций металла устойчи-
вость золя высока и возрастает по мерс приближения pH к ней-
тральным значениям и снижения концентрации металла в рас-
творе. Обобщая проведенные расчеты, можно отметить положе-
ния, приведенные ниже.
1. Гомокоагуляцня частиц не наблюдается в широкой области
pH и концентрации электрогеперпруемых ионов металла при от-
сутствии формирования твердой фазы в растворе.
2. При формировании твердой фазы в растворе одного знака
с частицами примесей происходит гетерокоагуляцня при сниже-
нии зарядов частиц. Скорость коагуляции возрастает при увели-
чении ионной силы раствора за счет изменения pH пли ввода
электролита, а также константы Гамакера. Коагуляция, как пра-
вило, наблюдается во вторичном энергетическом минимуме, что
указывает на неустойчивость образующихся агрегатов. Условия
взаимодействия должны оцениваться в каждом конкретном
случае.
3. При формировании в растворе твердой фазы разного знака
с частицами примесей наблюдается гетерокоагуляция частиц
в широком диапазоне pH и концентрации попов металла, опреде-
ляющих в основном условия формирования твердой фазы.
Исходя из вышесказанного, целесообразно во всех случаях
в растворе формировать при данных условиях (pH, ноппая сила
и т. п.) частицы твердой фазы, имеющие разноименный знак
с частицами примесей волы при этих же условиях.
Такое условие выполнимо путем соответствующего подбора
материала растворяющегося электрода, посылающего ионы в рас-
твор. При этом следует учитывать взаимное расположение точек
пулевого заряда примесей и частиц формирующейся твердой
фазы (рнс. 3.33).
Как правило, окислы металлов положительны в нейтральной
области pH и должны хорошо взаимодействовать с частицами при-
месей. имеющих отрицательный заряд. Энергия взаимодействия
зависит от величины зарядов разноименно заряженных частиц со-
гласно вышеприведенным расчетам.
Формирование газовой фазы в электрохимических реакторах
происходит за счет продуктов катодной и анодной реакций Под
94
Рис. 3.33. Оптимальная область pH (заштрихована) для ге-
терокоагуляции частиц примесей (II) и твердой фазы гидро-
лизующегося металла (М) в зависимости от их точек нуле-
вого заряда
рНр и рН“ — точки нулевого зарада соответственно примеси н
гидроксида металла
влиянием диффузионных и гидродинамических факторов пу-
зырьки газа рассеиваются в объеме межэлектродного прост-
ранства, образуя газовую эмульсию. Химический состав газо-
вой эмульсии зависит в основном от ионного состава водного
95
раствора. При электролизе природных и сточных вод в электро-
реакторах могу г образовываться пузырьки кислорода, водорода и
хлора. Растворимость кислорода и водорода при нормальном
давлении их 0,1 МПа незначительна по сравнению с хлором, по-
этому газоиасыщенне объема жидкости в межэлектродпом про-
странстве происходит за счет пузырьков кислорода и водорода.
Общепринято характеризовать газовую эмульсию величиной газо-
содержания:
Г £Иг/(грг+у,,)- (3.120)
В электрохимических реакторах эту величину удобнее выра-
жать в следующем виде:
Г-<?,/еп. (3-121)
Скорость подачи газа зависит от электрических параметров
электролиза и геометрических размеров электрореактора. Ско-
рость всплывания пузырьков возрастает при восходящем дви-
жении жидкости и коалесценции пузырьков или уменьшается при
нисходящем движении жидкости и закреплении на пузырьках
частиц с рч>рп. Так как трудно учесть влияние коалесценции
пузырьков на газосодержапие, то обычно предполагается, что
она отсутствует в объеме электролита.
При движении газовой эмульсин двухфа шый поток наряду
с истинным газосодержапием характеризуется объемным расход-
ным газосодержапнем:
ГР = <7г/(?г + Ьж)- (3.122)
С учетом соотношения (3.121)
Гр = —-------------- (3.123)
сп (Г vn/t>M + О
От объемного расходного газосодержання зависит профиль
скоростей жидкости в газожидкостном потоке.
Для системы «азот—вода> Э. В. Сенькиной получено
= 1 + 2,25 Гр, (3.124)
где ф, — коэффициент, учитывающий наличие профиля скорости жидкости
в газожидкостном потоке.
Так как при движении жидкости в межэлектродном прост-
ранстве устанавливается параболический профиль скорости, за-
висящий существенно от расстояния между электродами, то учет
коэффициента ф0 применительно к электрохимическим реакторам
следует считать обязательным.
В электрохимических реакторах образуются полиднсперсиые
газовые эмульсии, однако в теоретических расчетах предполага-
ется, что пузырьки монодисперсны и не изменяют своего размера
при взаимодействии. Средний диаметр пузырьков принимается
в пределах 0,054-0,08 мм. Для таких пузырьков характерен лами-
96
)Ро
(3.127)
парный режим движения (Reel), так как максимальный их раз
мер, при котором Re=l,
D„= У 18^Re/(p^g) = 3J 7 180,012-1/(12 981)«O,O12 cm. (3.125)
Формула для расчета D„ получена из зависимостей:
Rc = t'nDnPo/Po " bn = DnPo£/(18H))- (3.126)
Определение u„ произведено с использованием формулы Сто-
кса без учета поправки Адамара — Рычннского — Бонда, так как
при Re<20 пузырьки всплывают, как твердые шарики, с плотно-
стью, равной плотности газа [95]. В стесненных условиях скорость
всплывания уменьшается и зависит от газосодержапия
— 9/2Г|,3 + 9/2Г6/3— ЗГ2
18ро (3 + 2Г6'3)
Расчеты по формуле (3.127) показывают, что наиболее интен-
сивно скорость подъема пузырьков изменяется при Г<10%. При
Г=3,5 % скорость снижается в 2 раза по сравнению со скоростью
свободного всплывания пузырьков.
Газосодержапие оказывает существенное влияние на плот-
ность тока при значениях Г=0,1 и выше. Так как такое газосо-
держание труднодостигаемо для известных конструкций электро-
реакторов, то влиянием Г на i для Г=0,01—0,1 и 1 = 10—
200 А/м2 можно пренебречь.
Среднее газосодержапие в меж- и надэлектродном простран-
ствах электрореактора с вертикальными плоскопараллельными
электродами определяется с помощью зависимостей, связываю-
щих электрические параметры электролиза, удельное газовыделе-
ние н конструктивные размеры электрореактора (табл. 3.6) [2].
Эти формулы могут быть использованы для определения как сред-
него газосодержапия, так и размеров электродной системы по за-
данному газосодержанию. Для расчета скорости движения жид-
кости использовано выражение
йж = *МДт<»)- (3.128)
Коэффициент фж неявно связан с Г н может быть определен
по зависимости [2]
Фж = Фр/(1-Г). (3.129)
Так как от Г зависит ип (3.127), то истинное газосодержапие
может быть определено только методом последовательных при-
ближений.
Для вертикального расположения электродов в электрореак
торах характерно изменение газосодержапия в межэлектродпом
пространстве по высоте электродов, это не учтено формулами
4 Зака1 № 2148
97
Таблица 3.6. Формулы для расчета среднего газосодержания в электрореакторе
вертикальными плоскопараллельными электродами
Электроревктор
Условвя электролиза
Расчетные формулы
Межэлектрод-
ное простран-
ство
Надэлектрод-
иый объем
Неподвижная жидкость
Восходящее движение жид-
кости
Нисходящее движение жид-
кости:
при оп/гж < 4>ж
прн оп/ож > *ж
Неподвижная жидкость
Восходящее движение жид-
кости
Мо' .
v„l,
2 (рпЛ>ж + Тж)
Д??о _ .
2 (1рж — »п/»ж) ’
_ Д1*° ;
2 (vt,/v* — Ч’ж)
*<й<?0‘(А0 -М.
Дт?0
»п/»Ж + *ж
Примечание. hQt Л, — высота соответственно влектрореактора и электродов, м;
Q —удельное гавовцделенае. ма/Кл; <—плотность тока. А/м1; оп — скорость всплывания
коллектива пузырьков, м/с; I* —расстояние между электродами, м; Дт—расход тока,
Кл/м1; ож. «ж — средняя скорость движения воды соответственно в межэлектродном и над-
электродном пространствах, м/с; фж — коэффициент, учитывающий отношение истинной
скорости движения жидкости к средней, ф^— коэффициент, учитывающий отношение объ-
емов воды з меж- н над электродном пространствах.
табл. 3.7. В условиях электролиза неподвижной жидкости можно
использовать формулу [81]
Г =------------. (3.130)
273 [рг pH? (Л, — ft)) vnls
где рг —давление под слоем жидкости; Л- расстояние от нижнего края элек
тродов до расчетного слоя, р. — плотность газовой эмульсии р.—ро (1 Г)
При расчете на нормальные условия формула (3.130) упро
тается:
Г Ч1/(с„/9). (3131)
Лилли< полученных теоретических звисимостей показывает,
•по па га.юсодержанне существенно влияют электрические пара-
метры процесса и форма электрореактора. При электролизе ие-
подвпжиоп жидкости Г растет по мере увеличения плотности
юка и высоты >.'и Kipopc.iKiopa В движущемся потоке для коп-
ок
Т а б л и ц а 3.7. Удельное газовыделенне в электрохимических реакторах
Содержание Nat), г/л Удельное газовыделенне (10~* мУКл) при плотности тока А/м1 Среднее удельное газовыделенне, 10-* м’/Кл
25 50 100 150 200
0 0.139 0,138 0.142 0,158 0,160 0,148
1 0,132 0 135 0 144 0,144 0,147 0,140
5 0,128 0,127 0,132 0,138 0 143 0,134
10 0.127 0.312 0,134 0,138 0,137 0,134
центрирования пузырьков необходимо увеличивать расход тока.
Прн восходящем направлении движения жидкости повышение Г
происходит в развитых по вертикали электрореакторах, а при
нисходящем — в плоских.
Концентрирование пузырьков может быть достигнуто также
введенном в надэлекгроднук» часть электрореактора дополнитель-
ного сопротивления, уменьшающего сечение в месте формирова-
ния так называемого «тазового» слоя [2]. Для определения
в этом электрореакторе Г получена следующая зависимость:
Г = КСГН/,УО, (3-132)
где Кс — коэффициент, учитывающий соотношение скоростей подъема пузырь-
коп при отсутствии и наличии перфорированной диафрагмы в электрореак-
торе, Г„ газосотсржаннс в иадэлсктродном объеме при отсутствии перфо-
рированной диафрагмы; fn — площадь соответственно сечения надэлсктрод-
iioii части элсктрорсактора н отверстия в диафрагме.
Экспериментальные данные подтверждают теоретические вы-
во in о пропорциональности скорости выделения электролитиче-
ских газов плотности тока Однако следует учесть, что наряду
с увеличением плотности тока возрастает количество электриче-
ства. проходящего через раствор. Эта величина определяет об-
щий объем выделившегося газа, который при постоянном расходе
тока мало .зависит от его плотности. Рассчитанные по экспери-
ментальным данным значения удельного газоны деления нахо-
дятся в пределах (0,127—0,160)* 10 6 м3/Кл (табл. 3.7).
Теоретическое газовыделенне прн отсутствии хлоридов состав-
1ясг 0,187-10 6 м*/Кл, что в среднем па 20 % выше экспернмеп-
1.1 п.ных значений. Данная поправка может учитываться в прак-
П1ЧССК-ЦХ расчетах в процессе определения объемов выделяю-
щихся газов при электролизе.
Газосодержапие, как и объем газа, в значительной степени за-
висит от плотности тока (рис. 3.31). Снижение газосодержания
при увеличении высоты наполнения связано с интенсивной коа-
лесценцией пузырьков и увеличением скорости нх выделения из
кн (кости Этот эффект наблюдается особенно заметно при вы-
coie наполнения более 50 см.
При размещении в иадэлектродпом объеме диафрагмы повы-
1Н.К1СЯ газосодержапие обрабатываемой жидкости, что подтверж
** 99
Рис 3.34. Зависимость гдзосодсржа-
ния Г от плотности тока i при вы-
соте наполнения электрореактора
/ — 43 си; 2 — 76 см; 3 — 110 си
Рис 3.35. Зависимость газосодер-
жаиия Г от плотности тока i для
электрорсактора с перфорирован-
ной диафрагмой ({„/{„— 10) при
высоте наполнения ПО см (1—3)
и 76 см (/) и высоте размещения
диафрагмы 65 см (/), 45 см (2, 4)
и 25 см (•?)
дает теоретические предпосылки (рнс. 3.35). Так, при плотности
тока 200 А/м2 можно достичь газосотержапия Г = 0,03, однако без
применения диафрагмы эта величина может быть получена при
плотности тока 700 А/м2.
При образовании газовой фазы в объеме реактора характерны
три типа взаимодействия, обусловливающие агрегацию: гомо-
взаимодействие «газ — газ», приводящее к укрупнению пузырь-
ков газа (гомокоагуляция пузырьков газа); гомовзаимодействие
«примесь—примесь», приводящее к укрупнению частиц примесей
(гомокоагуляцня частиц примесей); гетеровзапмодействие «при-
месь—газ», приводящее к образованию комплексов (гетерокоагу-
ляция частиц примесей и пузырьков газа).
Если рассматривать образующийся флотокомплекс при гете
ровзанмодействин как независимую частицу, обладающую но-
выми свойствами по отношению к системе (плотность, диаметр н
т. п.), то можно предполагать, то все три типа взаимодействия
приводят к превращениям примесей.
Если при движении газовой фазы в межэлектродном прост-
ранстве неоднородность гидродинамического поля жидкости воз-
никает в основном вблизи поверхности пузырьков, а вклад неод-
нородности поля, вызванный параболическим распределением
скорости в межэлектродном пространстве, невелик, то можно счи-
тать, что взаимодействия в объеме реактора вызваны движущи-
мися пузырьками газа. Таким образом, в объеме жидкости за
счет неоднородности нолей жидкости вблизи пузырьков возни-
кает гомокоагуляция пузырьков газа, частиц примесей (ортокн-
петическая гомокоагуляцня) и гетерокоагуляция частиц примеси
и пузырьков газа (ортокипетическая гетерокоагуляцня).
100
В общем случае частота столкновений на одну частицу при
гидродинамическом взаимодействии для однородных частиц оп-
ределяется уравнением [7]
/ч = (32 3)Д\Л\С, (3 133)
а для разнородных частиц — уравнением
/ч, - (1 6)ЛГЧЛГП (D, - Dn)3 С (3.134)
Нетрудно видеть, что частота столкновений определяется ди-
аметром (диаметрами для полиднсперсных частиц) и числом ча-
стиц и градиентом скорости. Как правило, начальная концент-
рация и диаметр частиц примесей являются заданными для
начальных условий, и поэтому регулируемыми параметрами явля-
ются число пузырьков N„, определяемое величиной газонасыще-
ния Г в электрореакторе, диаметр пузырьков Dn, который зависит
от электрических параметров процесса и конструктивного выпол-
нения электродов, градиент скорости G, который зависит, в свою
очередь, от газоиасыщения Г н диаметра всплывающих пузырь-
ков. Таким образом, увеличение газоиасыщения и диаметра пу-
зырьков привозит к интенсификации процесса гомо- и гетерокоа-
гуляции. Одпако, с другой стороны, для гетерогенных систем типа
<пузырек частица», в которых диаметры взаимодействующих
частиц отличаются друг от друга, показано, что максимальное
взаимодействие характерно при Du — D4, т. е. оптимальные усло-
вия закрепления частиц на пузырьке выполняются, если их диа-
метры равны, что подтверждено теоретическими и эксперимен-
тальными исследованиями.
Теоретический анализ, выполненный в работе [56], позволяет
учитывать вышеуказанные особенности коагуляции в присутст-
вии газовой фазы, приведенные ниже.
1. Уровень неоднородностей гидродинамического поля опре-
теляется значением локального градиента скорости жидкости G,
который, в свою очередь, зависит от объемной доли газовой фазы
Г. а также от формы кривой распределения пузырьков по разме-
рам. При этом G имеет максимум при Г=0,08 в случае стоксов-
ского режима всплывания пузырьков (Re<l), при объемной кон-
центрации последних из интервала (0,05—0,25), а предпочти-
10.1ЫЮ из интервала (0.08 0,16), и при Г = 0,124-0,16 в случае,
если пузырьки более крупные и всплывают при I<Re<40. С уве-
личением уровня полиднсперсносди пузырьков, т. е. с расшире-
нием максимума их распределения по радиусам, величина G до-
вольно быстро растет за счет появления относительно большого
числа крупных пузырьков.
2. Ортокнпетическая гомокоагуляцня является конкурирую-
щей по сравнению с гетерокоагуляциеп, если объемная концент-
рация частиц C4^0,l£j/ZK (где Ея— эффективность захвата ча-
сти пузырьком, a ZK — эффективность гомокоагуляции частиц
п i я дро динамическом поле).
101
Рис. 3.36. Определение градиента ско-
рости Gn в межэлектродном простран-
стве электрореактора в зависимости от
газосодержапия Г и радиуса пузырь-
ков га
Рнс. 3.37. Определение градиента ско-
рости Gn в над электрод ном простран-
стве электрореактора в зависимости от
его электрических параметров
I. I -fn-100 см’: 2. Г~ fn-25o см*; 3.
3' — Fn-500 см’; < Г-Г„-1000 см»; /. 2.
3. / — при Лн -0.И615 см’/Кл; Г. 2*. S'. t —
прн •4qj — 0.0301 см’/Кл
3. Для интенсификации гомокоагулянии и, следовательно, для
процесса в целом целесообразно барботаж суспензии проводить
пузырьками, имеющими довольно широкий спектр размеров, так
как ортокииетическая гетерокоагуляция мелких частиц происхо-
дит лучше прн использовании сравнительно мелких пузырьков,
а ортокииетическая гомокоагуляция — при использовании срав-
нительно крупных. В связи с этим весь процесс обработки в элек-
трореакторе целесообразно проводить в две стадии: вначале срав-
нительно короткое время барботировать объем крупными пузырь-
ками, а затем к образовавшимся агрегатам добавлять мелкие пу-
зырьки.
Учитывая, что скорость коагуляции частиц определяется ло-
кальным градиентом скорости О, который, в свою очередь, харак-
1еризуе1ся величиной газосодержапия жидкости Г, а последний
параметр может быть легко изменен при изменении электрн
ческих ii.ni конструктивных параметров электрореактора, оче-
видно, чю повышение эффективности превращений в электроре-
акторе всиможпо за счет повышения газосодержапия (рис. 3.36).
При но iniiKiioBciiHii коивекшвиых потоков н межэлектродпом
npoci panel не при неравномерном распределении газовой фазы
градиеп ! <корост и может быть вычислен по зависимости G«
102
= 632 V<7r (гле </,—расход газа, отнесенный к единице площади
меж электро шого пространства, см/с).
Построенные графические зависимости (рис. 3.37) позволяют
легко найти параметры электрореактора в зависимости от опти-
мального градиента скорости G, требуемого для осуществления
превращений.
Таким образом, корректно учитывая составляющие электро-
химического процесса и их влияние на превращение примесей, мо-
жно добиться его существенной интенсификации при значитель-
ном снижении расхода электроэнергии на электролиз.
3.4.3. Электрический разряд в жидкости * *
При повышении напряженности электрического поля выше
пробойной происходит электрический разряд. Несмотря па то, что
в последнее время значительно возрос интерес к обработке дис-
персий электрическим разрядом, теории, описывающие разряд
в жидкостях, в основном посят качественный характер и рассмат-
ривают лишь отдельные стороны процесса, не объясняя его
в целом.
Процессы, происходящие в зоне разряда, зависят от парамет-
ров разрядного контура, формы подаваемого импульса и других
его характеристик, а также от физико-химических показателен
дисперсии.
Разряд в жидкости представляет собон сложный процесс, со-
провождающийся ионизацией молекул вещества, химическими ре-
акциями, образованием радикальных продуктов, способствующих
окислению дисперсной фазы, нарушением устойчивости системы.
Электрический разряд во времени протекает в три стадии:
1) предыскровая стадия, в течение которой происходит прора-
стание стримеров и смыкание промежутка проводящим мости-
ком; 2) стадия электрического пробоя — яркая вспышка, при ко-
торой образуется главный капал; 3) стадия дугового разряда (но-
слеразрядпые явления).
Выход первичных электронов, вызывающих ионизацию, опре-
деляется двумя факторами: ударной ионизацией и автоэлектрон-
ной эмиссией.
С увеличением температуры проводящего мостика может про-
исходить тепловая ионизация: при соударении атомов и молекул,
находящихся в возбужденном тепловом движении, образуются
ионы и наблюдается излучение. Излучение, в свою очередь,
может вызвать фотоионизацию, при которой кванты света погло-
щаются молекулами с образованием иоиов и электронов. На
нрсдыскровой стадии протекает небольшой ток, зависящий от нро-
иошмости гидродисперсии. Интенсивность ионизационных про-
• Млюрпал нодютовлеп совместно с капд. техн, паук II. 11. Pvuofipai-
• КИМ
цессов, возникающих па этон стадии разряда, сильно возрастает
н вызывает вспышку. Капал разряда образуется плазмой — га-
зообразной системой, состоящей из элеюронов, ионов, возбужден-
ных и нейтральных атомов. Расширению разогретой плазмы пре-
пятствует инерция окружающей среды, что приводит к кавитаци
оиным процессам, выравнивающим концентрации дисперсной
фазы ио объему обрабатываемой жидкости
На стадии электрического разряда возможно плавление элект-
рода, образование высокодисперсных капель металла электрода
из его паров, которые, окисляясь, переходят в оксиды, гидророк-
сиды и т. д.
При значительной мощности, вводимой в разрядный контур,
возможно сжимающее действие магнитных полей на острие элект-
рода, что влияет иа характер генерирования частиц металла
с электрода.
Уравнение баланса энергий при электрическом разряде опре-
деляется выражением
Uс = UK 4- -|- UT 4- иы, (3-135)
где Uс — энергия, накопленная в конденсаторе; UK — внутренняя энергия ка-
нала; Uuett — энергия радиационных потерь, обусловленных высокой темпера-
турой плазменного канала; Ut — потери на нагрев; UM — энергия ударной
волны.
Возможность реализации электрического разряда для целей
водоочистки изучена в работах О. М. Спиваковой, О В. Смирнова,
Л. Н. Губанова, Н. И. Рукобратского и др.
Исследования выявили решающее влияние низковольтной
проводимости на процессы, протекающие в воде при наложении
электрического поля, достаточного для получения высоковольт-
ного разряда. В коллоидных системах подвижность ионов и ча-
стиц имеет один и тот же порядок, а проводимость системы свя-
зана и с концентрацией дисперсной фазы, которая может изме-
няться, например, из-за образования в растворе продуктов
ионизации материала электродов.
Поступление в дисперсную среду продуктов разрушения элект-
родов происходит в результате испарения металла электрода под
действием резистивного нагревания, электрохимического травле-
ния, отрыва металла электрода в сильном электрическом поле
попдеромоторными силами Количество металла, уносимого
с электро ia в течение одного импульса разряда конденсатора,
можно опре (слить как [25]:
М„ А-аиэ/п/Г ЗСт^пл~Г()^"л 4- ч„ 1, (3.136)
/I К. J
где А'а— коэффициент, учитывающий влияние диаметра токопроводящего
стержня на эрозию; Ua—эквивалентный энергетический потенциал; /„— иите-
i
ipa.i от модуля разрядного тока: /п — i (t)ldl; с-,' — угольная теплоем
о
кость материала стержневого электрода; Тил. То — температура электрода со-
104
ответственно, плавления н начальная. <?пл. ?п — удельная теплота соответ-
ственно плавления и испарения материала электрода; К»—коэффициент вн-
броса материала за одни разряд.
Продукты разрушения электродов, поступая в дисперсионную
среду, в зависимости от коллоидно-химических свойств системы,
могут вызывать гетерокоагуляцню, а в некоторых случаях, на-
оборот, гетеростабилизацию. Рассматривая влияние электриче-
ского разряда на дисперсные системы, авторы в работе [20] при-
водят «общий критерий эффективности электрообработки элект-
рическим разрядом»:
[М»дЕпр)1</Р. (3 137)
где /р — время разряда, vn — подвижность дисперсных частиц; Е„р— пробив-
ная напряженность поля.
Для изменения свойств гидроднсперсии рекомендуется обра-
батывать систему импульсами с длительностью, превышающей
время релаксации ионной сферы, что соответствует правой части
выражения (3.137).
Рассматривая электрический разряд как обеззараживающий
агент, можно выделить следующие факторы, вызывающие унич-
тожение бактериальных тел: радиационное и тепловое излуче-
ния; радикальные продукты; сильные электрические поля, вызы-
вающие нарушение ионного транспорта через оболочку микроб-
ного тела; ударные волны.
Необратимое агрегирование микроорганизмов в электрическом
поле позволяет отделить их па фильтре очистки от грубодисперс-
ных взвесей. Присутствие радикальных продуктов после воздейст-
вия электрическим разрядом может привести к консервации воды.
Анализ механизма рассмотренных процессов, протекающих
при воздействии электрических полей и разрядов в дисперсных
системах, показал следующее:
при выборе оптимального режима электрообработки гидро-
дисперсий необходим учет совокупности поляризационных эффек-
тов (поляризационная коагуляция, электростабнлнзация, электро-
ориеитнроваппый эффект и др.);
основными параметрами электрического поля, определяющими
эффект очистки и обеззараживания водных систем, являются на-
пряженность и частота;
воздействие электрическим ра<рядом малой мощности явля-
ется универсальным с точки зрения улучшения широкого диапа-
зона физико-химических, бактериологических и органолептиче-
ских показателей качества воды.
ГЛАВА 4
МЕТОДЫ И АППАРАТЫ
ЭЛЕКТРОХИМИЧЕСКОЙ ОЧИСТКИ ВОДЫ
4.1. КЛАССИФИКАЦИЯ МЕТОДОВ
Основываясь на фундаментальных законах физической хи-
мии, электрохимии и химической технологии, а также учитывая
основные положения существующих классификаций [40, 44, 541,
электрохимические методы обработки воды можно разделить иа
три основные группы (рис. 4.1).
Первая группа методов обеспечивает изменение физико-хи-
мических и фазово-дисперсных характеристик загрязнений
с целью их обезвреживания или более быстрого извлечения из
воды. Превращение примесей может протекать через ряд после-
довательных стадий, начиная с электронного уровня взаимодей-
ствия растворимых соединений и заканчивая изменением каких-
либо электроповерхпостных или объемных характеристик грубо-
дпсперспых веществ. Взаимодействие примесей прн наложении
па дисперсную систему электрического поля является достаточно
сложным процессом. Здесь наряду с воздействием поля сущест-
венное влияние оказывают продукты электродных реакций, а так-
же окислительно-восстановительные реакции па электродах. Если
процессы разделения в основном протекают в объеме электро-
лита, то процессы превращения могут протекать как в объеме,
так и па границе олектрод—раствор». При этом следует обра-
тить внимание на различные кинетические закономерности этих
процессов, связанные с разнообразием физико-химических и ги-
дродинамических характеристик прнэлектродпого слоя и меж-
электродного объема. Теоретические аспекты этого вопроса изу-
чены еще недостаточно полно, что сдерживает разработку новых,
более совершенных конструкций элсктрореакторов.
Основы превращения примесей воды под воздействием электри-
ческого тока заложены в работах советских ученых: С. В. Яков-
лева, Л. Л. Кульского, С. С. Духнпа, И. С. Лаврова, О. В. Смир-
нова, В. В. Найденко, Л. Н. Губанова, В. Д. Дмитриева, В. Е. Ген-
кипа. И. Г. Красиобородько, Е. С. Светашовой, В. М. Рогова.
В. Р. Эе грела Льониса, В. Л. Слинчепко, П. П. Строкача,
Е. И. Сайгака. О. М. Спиваковой, 3. Я. Ярославского, В. В. Ко-
валева, В. Ф. Малько, М. М. Назаряпа, В. А. Чаптурия, Р. Ш. Ша-
феева, а также зарубежных исследователей: Пола (Pohl). Флей-
шмаиа (Fleishman), Ньюмена (Newman), Лазорко (Lazorko)
и др.
Из электрохимических методов превращения веществ наиболее
широко разработай п внедряется в практику метод электрокоагу-
ляции (ВНИИ ВОДГЕО. ИКХиХВ АН УССР. ЛИСИ. УИИВХ).
106
Рис. 4.1. Классификация электрохимических методов обработки воды
Получил развитие метод электрохимической деструкции .тля обес-
цвечивания красителей и окисления ipynix органических приме-
сей (ЛИСП, Госпипх.торнроект, 1IIII ЮПИ К), а также ме
тод анодного окисления и катодного восстановления (ВНИИ
ВОДГЕО). Интенсивное развитие в последние годы получил ме-
тод диафрагменного электролиза с целью корректирования pH
и Eh воды и последующего превращения примесей, теоретическое
обоснование и технологическое оформление которого выполнено
в УИИВ\ под руководством В. М. Рогова.
Однако теория электрохимических методов превращения тре-
бует обобщения экспериментальных результатов с целью выбора
дальнейшего направления исследований и совершенствования
конструктивного оформления электроаппаратов.
Вторая группа методов предназначена для концентрирования
примесей в локальном объеме электролита бед существенного из-
менения фазово-дисперсиых пли физико-химических свойств из-
влекаемых веществ. Разделение примесей и воды происходит в ос-
новном за счет флотации электрогеперируемыми пузырьками га-
зов или силового действия электрического поля, обеспечивающего
транспорт заряженных частиц в жидкости. Возможен также тран-
спорт незаряженных частиц в жидкости при закреплении их па
специально вводимом носителе, обладающем определенным за-
рядом. Такое направление более широко используется в магнит-
ном разделении, однако электрическое разделение с вспомога-
тельным носителем имеет большие перспективы.
Электрическое разделение в технологии очистки воды широко
исследовано в работах В. А. Клячко, Н. П. Гпусниа, В. Д. Гре-
бенюка, И. Н. Медведева, Г. Г. Первова, И. II. Гвоздика,
Б. М. Матова, А. А. Мамакова, К. М. Салдадзе, А. II. Мацпева,
К). М. Ласкова, Е. В. Алексеева, Е. И. Апельципой, В. Н. Поно-
маревой, а также Гамакера (Иашаксг), Куна (Kuhn), Пнккета
(Pickett), Алтана (Allan) и др.
Из электрохимических методов разделения наибольшее рас-
пространение получил метод электродиализа с целью обессоли-
вания природных и промышленных сточных вод. Разработаны и
серийно освоены аппараты для электродиализного опреснения
воды. Другие методы пока не вышли из стадии лабораторных
или опытно-промышленных испытаний. Осваиваются промышлен-
ностью аппараты электрофильтроваиня воды с целью удержания
микроорганизмов для нужд медицинской, пищевой и других от-
раслей промышленности. Имеется положительный опыт эксплу-
атации электрофлотациоппых установок для очистки ряда катего-
рий промышленных сточных вод.
Третью группу составляют комбинированные методы, кото-
рые предполагают совмещение одного или нескольких методов
превращения и разделения загрязнений в одном аппарате.
Наиболее широко разработан (УИИВХ, ЛИСИ, Харьковским
политехническим институтом — ХПИ) и используется па прак-
тике метод электрофлотокоагуляцин (элемрокоагулянии флота-
108
пни). Основы метода были заложены в работах Б .4. .Матова н
\ Л. Мамакова, теоретическое обоснование выполнено V II. Мац
новым и В. М Роговым, развитие метод получил в работах
Г. В. Иванова. Ю. А. Феофанова. В Д. Ющенко, М. М. Иазаряна,
В Т. Ефимова п других исследователей.
Достаточно перспективным методом глубокой минерализации
трудпоокислнемых органических загрязнении является разраба-
тываемая в ЛИСИ под руководством И Г. Красиобородько ком
оиппроваипая электрокаталитическая очистка сточных вод. сов-
мещающая процессы электролиза с использованием нераствори-
мых анодов и гетерогенного катализа [46. 48, 49]
Комплекс электрических воздействии, предложенный
О. В. Смирновым и др [20], предполагает использование в одном
аппарате методов электрокоагуляции, электрофлотацин, электро-
ратряда малой мощности, ультразвуковых воздействий, электро-
обеззараживания воды и др. Комплексное применение такой со-
вокупности методов целесообразно при глубокой очистке воды для
автономных объектов, технология которой исследована Н. И. Ру-
кобратским, Т. Г Тюняткнпоп [94, 108] и др. Выбор соответству-
ющей совокупности методов при комплексном электрическом воз-
действии зависит от исходного и требуемого качества очищенной
воды.
Электроосаж теине как комбинированный метод включает ме-
тоды разделения электродиализ п электроосмос, а также ме-
тоды превращения электровосстаповлепие, электроокислеиие,
электрокоагуляция. Теоретические и прикладные основы метода
разработаны О. М. Меркушевым [65], Ю. Ф. Дейнегой [27] и др.
Однако эти исследования выполнены в условиях электрофорети-
ческих «чистых» покрытии. Для технологических задач водоочи-
стки этот метод, исследованный В. Е. Генкиным [18], начинает
внедряться применительно к электроосаждению металлов пз сточ-
ных вод с последующей нх регенерацией.
Электроиопообмеипый метод предполагает электрическое ре-
гулирование свойств ионообменных и сорбционных материалов,
что позволяет во многих случаях осуществить их полную регене-
рацию. В этом направлении ишестпы работы В. Д. Гребенюка,
В. В. Славинского и др.
Находит применение обеззараживание поды методом прямого
электролиза или с получением электролитического хлора (НИИ
КВОН, ПКБ АКХ ИМ. К. Д Памфилова, ИКХнХВ ЛИ УССР),
разрабатываемое Г. Я. Медришсм, \. Л Те (пневой, Д. .1. Басиным,
Г. И. Сайгаком и др.
Па основании предложенной классификации методов электро-
ооработки следует осуществлять и выбор типа электрореактора,
который определяется, в первую очередь, видом генерируемого
электролитического эффекта в жидкости (рис. 4.2).
В зависимости от электролитического эффекта подбирается
материал и принимаются соответствующая конфигурация и
к<нк |рукция электродов, камеры электролиза, а также другие
109
Рнс. 4.2. Основные виды электролитического эффекта, возникающего в электрохимических реакторах
!♦] Растворимые
эл™трМ -AHePamicP^
Электрореакторы
для
обработки Воды
♦| Малоизнашибаеные
Рис 4.3. Технологические и конструктивные разновидности алектрореакторов
технологические п конструктивные особенности аппаратов
(рис. 4.3).
Гидродинамическая обстановка в электрореакторе зависит от
его конструкции и фазово-дисперсного состояния продуктов элект-
родных реакций и извлекаемых загрязнений.
Соединение электродов определяется электрическими парамет-
рами процесса электролиза и существующим выпрямительным
оборудованием, что пошоляет максимально использовать его
КПД.
Выбор материала электродов зависит в первую очередь от
вида получаемых продуктов электродных реакций и их взаимо-
действия между собой и примесями воды. При нейтрализующем
влиянии продуктов электродных реакций или их ингибирующем
действии на процесс очистки продукты отделяются от объема об-
рабатываемой воды диафрагмами. Тип диафрагмы определяется
ионным составом обрабатываемой воды, наличием органических
и механических примесей, устойчивостью и быстрым изнашива-
нием.
Конструкция электрореактора должна выбираться в каждом
конкретном случае с обязательным учетом исходного состава и
концентрации загрязнений, а также с учетом взаимодействия их
между собой и продуктами электролиза.
Ниже рассматриваются методы электрообработки воды и кон-
струкции аппаратов для их реализации, которые находят в па
стоящее время иаибольи ее применение или являются, по нашему
мнению, достаточно перспективными.
4.2. МЕТОДЫ ПРЕВРАЩЕНИЯ ВЕЩЕСТВ
4.2.1. Электрокоагуляция
Электрокоагуляция как метод превращения примесей колло-
идной степени дисперсности до грубодисперспого состояния осно-
вывается па множестве физико-химических процессов, протека-
ющих в жидкости под воздействием электрического тока
(рис. 4.4).
Как и любой другой коагутяцпоииый процесс, электрокоагу-
ляция может протекап> за счет энергетических изменений иа гра-
нице раздела <частпца—жидкость» и изменения кинетических
факторов. Для частиц с низкой устойчивостью (£<20 мВ) кинети-
ческие факторы играют превалирующую роль, для частиц с вы-
сокой устойчивостью (t>20 мВ) требуется нарушение равнове-
сия иа границе раздела с целью снижения заряда частиц, измене-
ния голшипы ДЭС или адсорбциопно-сольватных слоев на
поверхности.
Учитывая ранее развиваемые представления о механизме
электрокоагуляции [20, 27, 28, 69, 78], последняя характеризуется
протеканием процессов, приведенных ниже.
I. Электростатическая (поляризационная) коагуляция — ди-
поль дипольное в шимо действие коллоидных частиц за счет даль-
112
Рис. 4.4. Основные пути осуществления процесса электрокоагуляции
нодействующих сил притяжения, возникающих при наложении
электрического поля напряженностью Е3.
2. Электрохимическая коагуляция — взаимодействие частиц
при изменении их заряда или толщины ДЭС за счет изменения
физико-химических свойств раствора в межэлектродном объеме
или приэлектродпых слоях
3 Электролитическая коагуляция -взаимодействие частиц
при введении потенциалообразующих ионов металлов за счет
электрохимического растворения электродов.
4. Гидродинамическая коагуляция—слипание частиц за счет
увеличения числа их столкновений при перемешивании жидкости
в электрореакторе. Перемешивание жидкости может осуществ-
ляться как продуктами электрохимических реакций (электролиз-
ные газы, конвективные потоки и т д). так и за счет конструктив-
ных приемов выполнения электродных систем.
5. Концентрационная коагуляция — увеличение числа столкно-
вений частиц, приводящих к их слипанию, за счет повышения
локальных концентраций частиц в межэлектродном обтеме при
их транспорте, осаждении па электродах и т. п.
Влияние вышеприведенных составляющих электрохимического
процесса па коагуляцию частиц определяется их размерами, элек-
трическими свойствами, материалом электродов, технологиче-
скими параметрами электролиза, ионным составом воды и т. л.
В каждом конкретном случае при выборе метола очистки следует
производить учет этих факторов, чтобы избежать ошибочных
представлений о механизме электрокоагуляции.
4.2.1.1. Поляризационная коагуляция
Многочисленными исследованиями установлено, что в поляр-
ных жидких средах между частицами, находящимися в электрн
веском поле, возникает притяжение, которое проявляется па рас-
стоянии, в 2—3 раза превышающем их размер. В результате об-
разуются линейные цепочки-агрегаты, ориентированные вдопь
поля. Это связано с деформацией н поляризацией ДЭС.
талыю освещено в работах [27, 30].
Были получены выражения для результирующей сплы
действия частиц при елабозаряженной поверхности:
при г.,>х 1
Р лг„ Ar/d'’—+?ха) Р
Ч (еп^)] №/(кГх2)]ГгХ
1 л (/>/„+ <f)<
прн Г„С X’1
в Л,/43 - (v^n/r,) х/( I + ёка) -I
I"8 Ч-) 1 faW)].pftC2n/(kTx2)]|r’F;
I________L_________________~.
Я Йс^ч + d)*
что де-
взаимо-
(4 1)
(4 2)
где rt — эффектпвныП радикс протппопоноп; уР(—коэффициент, учитываю-
шнй аномальную подвижность попов.
При выводе этих у равнении собственная поляризация частиц
вследствие ее малой величины не учитывалась [30].
Энергия поляризационного взаимодействия существенно зави-
сит от напряженности поля и размера частиц Поэтому увеличе-
ние напряженности поля определяет резкое уменьшение барьера
отталкивания частиц и ускорение коагуляционных процессов. При
выключении поля происходит пептизация таких систем, что не
всегда приемлемо с точки зрения очистки воды.
Напряженность поля зависит от конструкции электродов.
Если для однородных полей эта величина определяется доста-
точно просто, то для неоднородных следует учитывать их кон-
фигурацию и взаимное расположение [20].
В неоднородных полях за счет явлений дннолофореза коагу-
ляция наблюдается в местах концентрирования коллоидных ча-
стиц, что обычно происходит па кромках и боковых поверхностях
электродов.
Как было показано в и. 3.4.2, значительный эффект действия
поля проявляется при £э> 104-30 кВ/м, что редко используется
в технологии очистки воды из-за сложности аппаратурного
оформления и высокой стоимости процесса.
Электрокоптактпая коагуляция, протекающая в толще фильт-
рующей загрузки при наложении электрического поля, основана
па концепции частичной поляризации гранул загрузки под дейст-
вием поля напряженностью Е, и частиц примесей, что в конеч-
ном итоге приво шт к диполь дипольному взаимодействию в со-
ответствии с ранее приведенным механизмом. Предполагается,
что под действием поля с критической напряженностью £кр на-
блюдается разрыв сплошности ДЭС с образованием «активных»
участков [30], за счет которых происходит слипание частиц при-
месей и фильтрующей загрузки.
4.2.1.2. Электрохимическая коагуляция
Причиной электрохимической коагуляции следует считать
в первую очередь изменение физико-химических свойств раствора,
в частности рП и I 11. при электролизе. Даже в бездиафрагмеп-
ных электрореакторах нарактерно подкисление и подщелачивание
раствора в при электродных слоях хотя и малой тол-
щины (Л = 0.1 0,5 мм), по (остаточной тля пребывания коллоид-
ной частицы в этом слое в течение времени, необходимого для
изменения ее поверхностных свойств.
Изменение pH обеспечивает, как правило, замещение попон
в слое Штерна или диссоциацию поверхностных групп, что при-
водит к изменению заряда частицы и дальнейшему изменению
условии коагуляции в соответствии с теорией ДЛФО Аналогич-
ное изменение заряда поверхности происходит при изменении Eli
раствора, так как здесь протекают реакции окисления — восста
новления на । ранние раздела, приводящие к электронному из-
бытку на поверхности или дефициту электронов. Прн этом до-
полнительная величина поверхностного заряда, обусловленная
присутствием свободных его носителей, зависит от величины из-
бытка или дефицита электронов па поверхности частицы и в объ-
еме раствора, дебаевского радиуса экранирования, полупровод-
никовых свойств частицы [115].
Таким образом, снижение устойчивости коллоидной системы
прн изменении pH и Eh происходит по нейтрализационному ме-
ханизму [111]. С другой стороны, перенос ионов в электрическом
ноле приводит к их перераспределению в объеме электролита и,
соответственно, влияет па ДЭС. Задача о перераспределении
концентраций в электрохимической ячейке, в частности у по-
верхности катода, обсуждалась в литературе неоднократно [27,
74]. Так, при рассмотрении двух компонент переноса — электро-
миграционной /, и диффузионной ?2 — было получено решение
[27], позволяющее оцепить распределение концентрации ионов во
времени на расстоянии I по направлению к электроду от границы
диффузного слоя Исходя из условия, что полный поток ионов
Jo - 71 + 7а-= |//(F 5э)|лОц + D (dC/dx). (4 3)
а время установления стационарного линейного градиента диф-
фузии f0=/2/:t2D, решение уравнения (4.3) прн времени G/fo^l
может быть представлено в виде:
2/Vn ['"Me+/(FS,)]V£L> (4 4)
При t3> 1,510
- Co = pMe*/(DFSs)j /1(1- 8/л« Jexp | - n«D (4/*)]), (4.5)
а прн G/(0>I
Cx-( - c0 = ['"Me*/(»FS»)V- (4 6)
Последнее выражение соответствует стационарному течению
реакции иа электроде, при котором весь образовавшийся на нем
продукт диффундирует в раствор.
Уравнения (4.4) — (46) удобны тем, что позволяют вычислить
концентрацию попа па расстоянии / от электродов прн любом
времени электролиза С- При попадании коллоидной частицы
в приэлектродный слой с повышенной концентрацией противо-
ионов возможно сжатие ДЭС, что обеспечивает коагуляцию ча-
стиц по концентрационному механизму [111].
4.2.1.3. Электролитическая коагуляция
Коагуляция частиц в этом случае обусловлена введением по-
ложительных мпого.зарятпых ионов металла, гидролизующихся
в воде с образованием гидроксидов и других промежуточных со-
116
единении Механизм лого процесса точно не установлен. В на-
стоящее время существуют две основные концепции теоретиче-
ского его обоснования.
внедрение многозарядпых попов (АР+, Fe3b, Fe2+ и др.)
в слой Штерна с последующим изменением заряда поверхности
частицы за счет специфической сорбции, что приводит к сниже-
нию заряда частицы и, соответственно, коагуляции;
образование малорастворимых соединений попов металла
с компонентами раствора, которые взаимодействуют с коллоид
нымн частицами, имеющими противоположный заряд поверх
пости.
Необходимо отметить, что обе концепции теоретически и эк-
спериментально недостаточно обоснованы. Однако многочислен-
ными экспериментами доказано, что скорость коагуляции значи-
тельно выше при формировании малорастворимых соединений.
По-внднмому, здесь следует предположить о преимущественном
влиянии адгезионного механизма процесса противоположно заря-
женных поверхностей частиц и гидроксидов
Исходя из таких предпосылок процесс электролитической коа-
гуляции можно разделить на следующие стадии: генерация ио-
нов металла на поверхности электрода; миграция ионов металла
с поверхности в объем раствора; образование малорастворимых
соединении металла с компонентами раствора; адгезия коллоид-
ных частиц примесей н образовавшихся малорастворнмых сое-
динений.
Генерация ионов металла па поверхности электродов может
происходить как прн анодной, так и при катодной поляризации.
Так, например, ноны алюминия генерируют на поверхности като-
да при высоких значениях pH прикатодного слоя [78]. Генерация
ионов других металлов может быть осуществлена при созда-
нии определенных физико-химических характеристик прикатод
пого слоя.
Механизм ионизации металла достаточно сложен и не уста-
новлен окончательно. Однако согласно современным воззрениям
анодное растворение металлов идет через образование комплекса
металла с ионами раствора в несколько последовательных ста-
щи, например [32].
специфическая адсорбция анионов па поверхности металла
Me 4-mA- (MeAm)(^; (4 7)
электрохимическая стадия, определяющая скорость про-
цесса — переход комплекса в раствор
(McAm)(";)-*(MeAm)^-") + пё; (48)
распад комплекса на простые ионы или продукты гидролиза
(MeAm)(n-m) л Ме£одн) + m А^оди) . (4 9)
Данный механизм обобщает ряд схем анодного растворения
металлов, в частности железа, предложенных Хойслером, Бокси-
сом, Крнстпансепом, Ротиняном и др. [32].
117
Стадийность протекания процесса растворения обусловливает
правомочность применения icopiiii фа юво-дпсиерсиых превраще
пий к оценке lamioro процесса. Поэтому, учитывая < собеиности
превращения веществ, анодное растворение железа можно пред-
ставить по модифицированной схеме, предложенной Л. II. Ант-
роповым и Ю V Савгира [32] при рИ'>2:
I Ье | ОН .. 1еО11(1Д() ; е.
2 1еОНа»дт | т>11 -• Fc (ОН).(.1Дсг. (J
3. Fc (Of I)2 (адс) -»• Fe (Of 1) (ноли)-
4 Гс(ОН)ш(всдн)^1еОН+ I-OH ;
5. IeOII + --Fe:+ 4- OH .
В данной схеме стадией, определяющей скорость реакции, по-
впдпмому, является стадия 3, связанная с отрывом молекулы
I е(ОН)2 от кристаллической решетки и транспортом ее в объем
электролита. Большая вероятность такого механизма определя-
ется меньшими энергетическими затратами отрыва молекулы
Fc(OH)2 по сравнению с FcOI 1ыдс>. который по существу яв-
ляется положительным однозарядным ионом, взаимодействующим
электростатически с кристаллической решеткой. При образова-
нии Ье(ОП)2(адс) взаимодействие нона |-'е2+ с решеткой осуще-
ствляется посредством физических связен (Ван дер Ваальсово
взаимодействие). Отрыв такой структуры oi поверхности металла
облегчен и может быть вызван гидродинамическими и другими
факторами. Аналогично может быть рассмотрен процесс специ-
фической адсорбции С1~ с последующим образованном FeCI2 и
FeCh. Депассивирующее действие хлоридов в этом случае может
быть объяснено полнотой протекания стадий 4 и 5.
При создании в приэлектродном слое условии, препятствую-
щих диссоциации комплекса на простые ноны, а также при от-
сутствии механических воздействий, благоприятствующих отрыву
молекулы от поверхности электрода (перемешивание жидкости,
движение газовых пузырьков у электрода, вибрация электродов
и т. и ), молекула закрепляется па поверхности за счет физиче-
ских связей, образуя мопомолекулярнын, а »атем и полимолеку-
лирный слой. Электропроводность границы раздела такого слоя и
поверхности электрода резко на (ает за счет отсутствия химиче-
ских связен между кристаллической решеткой электрода и моле-
кулами формирующего слоя, что приводит к резкому падению
тока, проходящего через электродную ячейку.
Таким образом, дтя эффективного протекания процесса анод-
ного растворения металлов должны соблюдаться следующие ус-
ловия:
I) возможное и. специфической сорбции анионов, образующих
комплексы с ионами кристаллической решетки металла;
2) способность перераспределения электронов между сорбиру-
ющимися анионами и ионами кристаллической решетки металла
(мнимый разряд иона);
1 IX
3) достаточная концентрация специфически сорбирующихся
анионов для образования комплексов (молекул) на поверхности
электрода;
I) возможность отрыва комплекса (молекулы) от поверхности
электрода;
5) высокая скорость ионизации в прнэлектротиом слое, т. е.
распад комплекса (молекулы) па простые ионы.
Как видно, условие (4) выполняется при выполнении усло-
вия (5) в связи с нарушением концентрационного равновесия мо-
лекул иа поверхности электрода и в обд.еме приэлектрод-
пого слоя.
При низкой скорости диссоциации концентрация молекул
в нриэлектродном слое возрастает, что приводит к диффузии их
с поверхности электрода. В этом случае решающим условием яв-
ляется условие (4), связанное с отводом образовавшихся моле-
кул из приэлектродного слоя в объем раствора под воздействием
механических или гидродинамических факторов. Для этих целей
разработан ряд конструктивных мер, используемых па практике:
ультразвуковая очистка электродов, вибрация, встряхивание, вы-
сокая скорость движения воды в межэлектродпом пространстве
при напорном режиме или за счет рециркуляции, одпопоточпая
схема движения воды, механическая очистка электродов скреб-
ками, продувка межэлектродиого пространства газом, вращаю-
щийся растворимый электрод и др.
Однако во всех случаях устойчивость процесса анодного рас
творения определяется наличием таких специфических сорбиру-
ющихся попов, которые на последней стадии обеспечивают высо-
кую скорость диссоциации металла. Это подтверждается теоре-
тически. например известными диаграммами Пурбе для железа
(см. рис. 3.7) и алюминия (104], в которых указаны области пас
енвпости металла, совпадающие с областями образования мало-
растворимых соединений продуктов растворения с компонентами
раствора, а также практически, например для анодного раство-
рения железа прн рН<3, в присутствии повышенных концентра-
ций хлоридов при pH = 34-10 и в других случаях.
Соотношение между химическими и электрохимическими про-
цессами растворения металлов существенно зависит от pH обра-
батываемой воды. В нейтральной области pH химическое раство-
рение алюминиевого анода составляет 20 %, катода — 50%,элек
три химическое растворение анода — 90 — 100%. Суммарный выход
по току минимален в диапазоне рП-54-8 и резко возрастает
в кислой и особенно щелочной средах, достигая 200 %
|7«].
Наиболее енлыюе влияние па «кажущийся» выход по току
оказывает pH при химическом растворении катода в щелочной
среде. Для железа подобные зависимости отсутствуют, однако
следует предположить аналогичный механизм растворения. Так.
при изучении растворения стальных электродов получены зпаче
пня критической плотности тока в зависимости от pH среды
i i<>
Рис. 4.6. Влияние плотности тока
на скорость растворения алюми-
ния при поляризации
/. 2 — соответственно полной и ка-
тодной
Рис. 4.5. Зависимости выхода алюминия по
току Вт от температуры воды при плотно-
сти тока
/ 2 мА/сч’: 2 —Б мА/см*; Э — 10 мА/см*
в пределах 6-? 12, выше которых наблюдается пассивация желез-
ного анода [9]
для стали
iKp =- 31.07 — 2,45 pH;
для чугуна
1кр 32.5 —2,33 pH.
(4.11)
(4 12)
При понижении pH ниже 6 скорость растворения возрастает,
при этом чугунный электрод растворяется аиодно легче, чем
стальной.
При повышении температуры от 0 до 80 °C выход по току
алюминия увеличивается, особенно в диапазоне от 2 до 30 °C
(рис. 4.5). При этом активация поверхности электрода наблю-
дается в диапазоне плотности тока от 1 до 4 мА/см2; при более
высоких плотностях тока с повышением температуры выход по
току алюминия снижается.
Ионный состав воды оказывает сложное влияние на процесс
растворения электродов. Так, растворение алюминиевых анодов
интенсифицируется при повышении концентрации С1_-попов, вы-
ход но току превышает теоретические значения на 21—29%.
В присутствии СОз“-ионов наблюдается торможение анодного
процесса, выход алюминия по току не превышает 63 %. SO? -
ионы замедляют активирующее действие С1 "-ионов, и тем в боль-
шей степени, чем больше соотношение между SO; - и С1“-ио-
пами. Особенно низкий выход по току алюминия наблюдается
в гидрокарбопатпых средах и составляет для концентрации 2.5;
5 и 10 мг экв/л соответственно 24, 16,3 и 12,2 % [78]. Однако вве-
дение лаже незначительных количеств С1~-нонов в воду, содер-
жащую SO?- ионы, активирует процесс растворения алюминия.
Гитрокарбонашые ионы не оказывают такого угнетающего лей-
ствия па Cl попы, как сульфатные. И лишь превышение кон
пентрации Н< О„’-ионов по оiшипению к CI нонам более, чем
в 5 раз, незначительно уменьшает их активирующее действие.
Введение в воду катионов Na+, К и Mg2t не оказывает сколько-
нибудь заметного влияния на анодный выход алюминия по току.
При катодном растворении алюминия анионный состав ока-
зывает незначительное влияние па выход по току. В бикарбопат-
иых и сульфатных растворах катодное растворение не ухудша-
ется, а даже интенсифицируется. Однако ионы Са2+ и Mg2h угне-
тают процесс катодного растворения из-за образования на
поверхности катода карбонатных отложений. Присутствие ионов
Nav активирует процесс, и при их содержании 25 % от общего
катионного состава выход по току составляет не менее 100 %.
В присутствии ионов С1 и НСОз происходит депассивация
железных электродов. Сернистый натрий оказывает пассивирую-
щее действие на растворение стального электрода [9].
Оптимальная анодная плотность тока для алюминия лежит
в пределах 24-15 мА/см2, катодная — в пределах 14-3,5 мА/см2
(рис. 4.6). Для железа увеличение скорости растворения наблю-
дается в пределах плотности тока 14-10 мА/см2 [9J. Повышение
плотности тока выше 12 мА/см2 ведет к пассивации железного
анода.
Скорость движения воды относительно электродов в пределах
1,24-400 м/ч не оказывает влияния на выход по току, однако вы-
сокая скорость движения воды препятствует образованию отло-
жений па поверхности.
При выполнении всех условий протекания процесса растворе-
ния металлов необходимо учитывать вторичное образование ма-
лорастворимых продуктов в объеме электролита, которые могут
транспортироваться к поверхности электрода за счет диффузион-
ных, гидродинамических и электрических факторов. Данный эф-
фект может оказать существенное влияние на процесс электро-
лиза при низких плотностях тока на электродах, малом солесо-
держанин, отсутствии перемешивания электролита и т. п.
Анодное растворение алюминия в кислой среде дает гидрати-
рованный ион [А1-хН2О]3+, в слабокислой и нейтральной среде —
труднорастворимый гидроксид [А1 (ОН)3(Н2О)з]°, а в щелочной —
гидратированный ион А1 (ОН)~-хН2О. Гидроксид алюминия об-
разуется при pH>4, а алюминат — при pH = 6,8. Минимальная
растворимость гидроксида наблюдается при рН = 6,8.
К наиболее вероятным формам соединений железа, присутст-
вующим в растворе, следует отнести [Гез(ОН)4]2" Распределение
различных форм железа в растворе определяется, как и для алю-
миния, величиной pH [7]. Минимальная растворимость окенгидра-
тов Fe2+ происходит при рП = 9,7, Fe3*' — при pH = 4,1.
Свойства образующихся нерастворимых соединений металла,
растворяющегося при электролизе, зависят в первую очередь от
ионного состава раствора, величины pH и времени старения.
121
Исследования, проведенные рснтгеноструктурным и радиоспектре
скоппческими методами, показали, что электрохимически нолу-
ченнын окснгпдра) алюминия находится в более далеком от
термодинамического равновесия состоянии, чем химически полу-
ченный оксигидрат, и имеет в связи с этим более высокую внут-
реннюю и поверхностную энергию [106].
В процессе старения оксигидрат алюминия последовательно
проходит через ряд модификаций, в частности бемит — байерит
гидраргиллит. Сорбционная способность алюминия падает в этой
же последовательности. Это объясняется тем, что последние две
модификации включают большую часть ОН -группы во внутрен-
ней сфере гидроксокомплексов. Для образующихся соединений
характерны процессы оляции и оксоляции в соответствии с гидро-
ксокомплексной теорией. Железо, как и алюминии, в процессе
старения проходит также ряд модификаций, приводящих к обра
зоваиию устойчивых форм оксидов железа.
Сорбционная способность электрохимически генерируемых ги-
дроксидов алюминия и железа несколько выше, чем химически
полученных. Отмечена повышенная сорбционная способность гм
фоксидов по отношению к красителям [97], сульфатам и хлорн
дам [43], что сказывается иа коагулирующих свойствах образую-
щейся твердой фазы.
Разновидностью электролитической коагуляции является пред-
ложенная В. Д Дмитриевым электрореагентпая коагуляция, при
которой взаимодействие примесей воды осуществляется под дей-
ствием электрического поля за счет введения пониженных по
сравнению с расчетными дозами химических реагентов. При этом
возможно использование растворимых электродов или нераство-
римых анодов (графит, ОРТА, ОКТА и других), а также раство-
римых катодов (алюминий и другие).
С введением коагулянта повышается концентрация электро-
лита и соответственно его электропроводность, что снижает энер-
гетические затраты на процесс электролиза. Кроме того, химиче-
ски образующиеся гидроксиды металлов под воздействием элек-
трического поля приобретают более активные свойства, чем при
обычной реагентной коагуляции. Следует также отметить благо-
приятное совмесыюе действие химически и электрохимически ге-
нерируемых гидроксидов металлов за счет процесса конденсации
последних па «затравке>, образующейся при гидролизе коагу-
лянта. Сродство структур улучшает молекулярное и адсорбцион-
ное взаимодействие между ними.
Механизм коагуляции частиц в присутствии электрохимически
полученной твердой фазы описывается основными закономерно-
стями общеизвестной теории ДЛФО. Взаимодействие отрица-
тельно заряженных частиц примесей и в большинстве случаев
положительно заряженных гидроксидов обеспечивает эффектив-
ную их коагуляцию в относительно широком диапазоне pH.
122
4.2.1.4. Гидродинамическая коагуляция
Процесс определяется числом столкновений частиц в единицу
времени при влиянии гидродинамических факторов в соответствии
с уравнением Смолуховского [7]. При постоянной концентрации
частиц число столкновений определяется градиентом скорости Ci,
зависящим от скорости движения жидкости в межэлектродном
пространстве, газосодержания и конструктивного исполнения элек-
трореактора. Продолжительность перемешивания определяет в ос-
новном размер и плотность образующихся агрегатов. Как правило,
для коллоидных систем, коагулируемых оксигидратами металлов,
применима ортокпнетическая градиентная коагуляция, обеспечи-
вающая максимальное сохранение структуры и прочности скоагу-
лпрованных агрегатов.
При сохранении постоянным критерия Кэмпа градиентная
коагуляция позволяет получать низкие значения градиента ско-
рости (7=10 20 с *. При турбулентной коагуляции G превышает
шачення 100 с *, что зачастую приводит к разрушению агрегатов,
включающих рыхлые структуры гидрокси юн металлов. Так. на-
пример, при коагуляции золя, состоящего из смеси иопов тяже-
лых металлов при очистке сточных вод гальванического производ-
ства в присутствии СПАВ, градиент скорости G, приводящий
к уменьшению диаметра частиц при перемешивании в 2 раза, со-
ставляет 20 30 с что является оптимумом для коагуляции
большинства коллоидных систем друшх категорий сточных вод.
4.2.1.5. Концентрационная коагуляция
Данный вид коагуляции протекает при создании локальных
градиентов концентрации частиц в приэлекгродных слоях пли на
шафрагме в результате транспорта заряженных коллоидных ча-
стиц в электрическом поле. Скорость переноса частиц и их кон-
центрация могут быть определены на основании зависимостей,
описывающих электрофоретические и электроосмотические явле-
ния в жидкости [5]. Используя уравнение Смолуховского для пол-
ного числа взаимодействий в единице обт>ема жидкости в еди-
ницу времени
я=(32 3)^GD®, (4.13)
нетрудно показать, что при низкой концентрации частиц Л\ в еди-
нице объема и их постоянном начальном размере скорость ко-
агуляции определяется градиентом скорости G (рис. 4.7). При
увеличении числа частиц сказывается их преимущественное влия-
ние на скорость коагуляции.
Следует указать особо иа обратимость концентрационной ко-
агуляции. При смене полярности э юктродов, снятии напряже-
ния пли интенсивном перемешивании жидкости золь ппсстаиав
ливаст своп первоначальные свопе та.
123
4.2.1.6. Аппаратурное оформление процессов электрокоагуляции
Из применяемых конструкций аппаратов для электрокоагуля-
ции с растворимыми электродами наиболее широко внедрены без-
диафрагменпые электрокоагуляторы пакетного типа (рис. 4.8).
Пакет растворимых электродов собирается с помощью специаль-
ных шпилек и распорных втулок. Это обеспечивает технологич-
ность сборки электродной системы и жесткость конструкции. Од-
нако эксплуатация таких аппаратов показала их низкую надеж-
ность в работе из-за забивания продуктами электрохимических
реакций участков прохождения соединительных шпилек, а также
всего межэлектродиого пространства при расстоянии между элек-
тродами менее 6—8 мм. Более удачным техническим решением
следует считать электрокоагуляторы пазного типа (рис. 4.9). Элек-
тродный пакет в таком аппарате укрепляется на рамс, в которой
фрезеруются специальные пазы для фиксации электродов.
Расположение электродов может быть продольным и попереч-
ным по отношению к лотку для слива обработанной воды. При
поперечном расположении токоподводы устраиваются по бокам
электрокоагулятора вне зоны выделения водорода, что повышает
безопасность обслуживания. Расстояние между электродами сле-
дует принимать не менее 8—10 мм для предотвращения забива-
ния межэлектродного пространства. В нижней части электрокоа-
гулятора следует предусмотреть конический бункер для сбора
окалины, шлама и гидроксидов металлов, выпавших в процессе
работы аппарата.
При повышенных плотностях тока на электродах (>30—
50 Л/м2) в верхней части скапливается флотационный шлам, ко-
торый может удаляться с поверхности вручную, под вакуумом,
сдуваться воздухом пли увлекаться потоком жидкости С целью
интенсификации процесса в УИНВХ предложен электрокоагуля-
тор с растворимыми электродами и «газовым слоем». позволяю-
щим повысить газосодержание обрабатываемой жидкости в 2—
3 раза (рис. 4.10). В реакторе принято нисходящее движение жид-
кости в межэлектродпом пространстве, что позволило классифи-
цировать газовые пузырьки и транспортировать в смежно распо-
ложенную камеру «газового слоя» пузырьки диаметром 30 —
80 мкм
В камере «газового слоя» образуется эмульсия с газосодер-
жаннем Г 0.01 . 0,03, что позволяет более эффективно образовы-
вать флотокомнлсксы. За счет применения такого реактора эффект
флотации возрос с 30—40 до 60— 70% без введения дополни-
тельной газовой фазы. При этом влажность снимаемого шлама
но флотационной зоне понизилась с 93—97 до 90—91 %. Основ-
ными недостатками предлагаемых конструкций являются необ-
ходимость использования листового металла и пассивация элек-
тродов.
С целью предо!вращения пассивации рядом исследователей
предложены механические скребки и щетки для снятия отложе-
124
/
Рис. 4.7. Области преимущественного
влияния градиента скорости G (1)
и числа частиц N (2) на скорость
коагуляции
Рис. 4.8. Электродная система пакет-
ного типа
/ — токораспределительные пластины; г —
стягивающие шпильки
Рис. 4.9. Электродная система палюго тина
7 — электроды; 2 — налы; 3 — рама
Рис 4.10. Электрореактор с «пазовым»
слоем
1 — подача воды: 2 — электродная система; 3 —
газовый карман; 4 — камера флотораздслсння;
5 — подача щелочи: 6 — отвод осадка
Рис. 4.11. Электрореактор колон-
ного типа
/ — корпус аппарата: 2 — растворимые
электроды; 3 — перегородка; 4 — кол-
лектор; 5 — пластины
Рис. 4.12. Стружечный электрокоа-
гулятор
/ — катод: 2 — нерастворимый анод; 3 —
металлическая стружка
Рис. 4.13. Стружечный электрокоагу-
лятор с непроточной анодной (ка-
тодной) камерой
/ — диафрагма; 2 —катод (анод): 3—
проючная камера; 4 — непроточная ка-
мера; 5 — iicpaci порнмыЛ л под (клтод),
ъ металлическая стружка
ши, наложение однородного переменного тока различной частоты
или магнитною поля и засыпка магнитных материалов в меж-
j.icki родное iipoiтраново, применение импульсного электриче-
ского тока для питания электродных систем, использование вра-
щающихся электродов. Однако эти меры эффективны в отдельных
конкретных случаях для относительно постоянного состава при-
родных и сточных под. При колеблющемся составе сточных вод
возникают неблагоприятные взаимодействия химических ингреди-
ентов в межэлектродпом пространстве, приводящие к пассивации
или образованию отложении. Такие моменты могут повторяться
периодически или спонтанно, что может привести к выходу из
строя электродной системы, нормально функционирующей в те-
чение длительного времени.
Более надежным приемом можно считать поддержание в меж-
электродном объеме условий, предотвращающих образование мало-
или нерастворимых соединений. Для выбора таких условий
целесообразно использовать диаграммы Пурбе [104], описывающие
область коррозии и пассивации металла в водных средах в за-
висимости от pH и Eh. Как подтвердили экспериментальные ис-
следования, для железа и алюминия необходимо поддерживать
pH менее 4 или более 10 для активации поверхности электрода,
а также редокси-потенциал ниже —(0,2-5-0,4) В. Подтверждением
этих предпосылок являются электрокоагуляторы, предложенные
Вильнюсским ПКБ механизации и автоматизации, использующие
в качестве рабочего раствора кислые или щелочные электролиты
гальванических или других производств.
Электродами может быть листовой металл или металлическая
стружка, загружаемая в специальные кассеты с графитовым то-
коподводом. Наработанный таким образом коагулянт собирается
в емкости, откуда дозируется в обрабатываемую воду. Электро-
коагуляторы обеспечивают стабильный выход металла при низких
затратах электроэнергии. К недостаткам таких аппаратов следует
отнести пониженную коагулирующую и сорбционную способность
коагулянта в связи с длительным его храпением, а также повы-
шенное солесодержание обрабатываемой воды при использован пн
концентрированных электролитов, нс всегда имеющихся иа том
пли ином предприятии
Электрокоагуляторы колонного типа (рис. 4.11), разработанные
в .ХПИ [69], имеют те же преимущества, что и аппараты Виль-
нюсского ПКБ. Электролит подается в нижнюю часть аппарата,
|де размещены электроды. В падэлсктродное пространство по-
дается обрабатываемая вода, которая смешивается с образую-
щимся в электродной камере коагулянтом и насыщается элек-
тролитическим газом. Для предотвращения попадания обрабаты-
ваемой воды в электродную камеру предложено над электродной
системой устраивать запорный обратный клапан. По мнению ав-
торов, такие аппараты позволяю! предотвратить пассивацию элск-
।родов. Модификацией аппарата колонного типа может являться
устройство ионообменной диафрагмы между анодами и катодами
12;
Горьковским НСИ для устранения пассивации электронов
предложено осуществлять процесс электрокоагуляции в напорном
гидродинамическом режиме работы аппаратов, что препятствует
достижению концентрационного равновесия молекул на поверх-
ности электрода и в объеме приэлектродного слоя и обеспечивает
высокую скорость их диффузии с поверхности электрода в глубь
обрабатываемого раствора
Перспективным направлением в разработке элекгрокоагуля-
торов следует считать примепеппе отходов производства, в част-
ности металлической стружки для генерации коагулянта Наибо-
лее совершенные конструкции стружечных аппаратов разработаны
Дальневосточным филиалом ВНИИ ВОДГЕО (рис. 4.12) [а. с.
700468 (СССР), 112]. Над слоем стружки в этих аппаратах распо-
лагается диэлектрическая перфорированная пластина, сверху ко-
торой размещен катод, а в пространство между ними подается
обрабатываемая вода. Главным недостатком таких электрореак-
торов является зашламление межстружечного пространства.
С целью предотвращения зашламления предложено встряхивание
стружки, периодические промывки водой или кислотой.
Элсктрокоагулятор, предложенный в УННВХ, позволяет из-
бежать зашламлеиня межстружечного пространства путем раство-
рения стружки в непроточной анодной камере диафрагменного
электролизера (рис 4.13). Выход железа ио сравнению с извест-
ными стружечными реакторами возрастает на 30—50%. Раство-
рение металла происходит в анодной зоне при рН = 4ч-5. Ионы
металла мигрируют в катодную зону под действием поля Кон-
центрация металла в катодной зоне зависит от расхода обраба-
тываемой воды.
Другой разновидностью диафрагменных электрокоагуляторов,
разработанных в УИИВХ, являются аппараты, совмещающие про-
цессы изменения pH и Eli обрабатываемой воды и растворения
металлической стружки. Анодное (катодное) растворение металла
происходит в кислой (щеточной) среде, получаемой за счет пред-
варительного подкисления (подщелачивания) обрабатываемой
воды в электролизере с неактивной диафрагмой. Это позволяет
не только отказаться от применения концентрированных электро-
литов, но и получать щелочь для нейтрализации кислотности рас-
твора, обрабатываемого в анодной зоне. Исследованы различные
конструкции диафрагменных элсктрокоагуляторов с засыпкой
анодной (катодной) зоны металлической стружкой (рис. 4.14)
Анодная камера электрореактора б разделена дополнительно на
две зоны перфорированной пластиной. В одной из них происхо-
дит подкисление жидкости, в другой производится растворение
стружки.
Наибольшее абсолютное количество растворенного железа об-
разуется в реакторе б и составляет 125 мг/л прн начальном рН = 2
и концентрации хлори юв 5-10-2 моль/л (табл. 4.1). Это обуслов-
лено тем, что растворение происходит как по химическому, так и
по электрохимическому механизму. В реакторе в отмечено более
128
Рис. 4.14. Варианты стружечных электрокоагу-
ляторов с предварительным изменением pH воды
а — размещение стружки в анодной зоне электрореактора; б — последовательное подкисле-
ние н растворение стружки в анодной зоне; в — растворение стружкн электрохимически
подкисленной (под щели ценной) водой; а — размещение стружкн в анодной зоне электро-
реактора с диафрагмой в виде перфорированной диэлектрической пластины
/ — стружка; 2 — диафрагма; 3 — решетка
низкое превращение железа, в среднем иа 20—30 % при рН = 2
и на 50—60 % при рН = 3.
Абсолютная величина превращения не превышает 115 мг/л
при концентрации хлоридов 5«10~2 моль/л и рН = 2. Превращение
железа в реакторе а иа 30 % выше, чем в реакторе г, что указы-
вает на положительную роль диафрагмы.
Изменение pH раствора существенно влияет иа выход железа
и зависит от типа реактора. Для реактора б снижение pH от 3
до 2 вызывает увеличение этой величины на 30—50%, для реак-
Таблица 4.1. Абсолютные значения превращения железа в зависимости от
величины pH и концентрации хлоридов в стружечных реакторах Сре
Тип реактора Показатели Концентрации хлоридов, моль/л
110-» 6 ю-» 1-10-» 3 I0-* 5 10-«
а CFe, мг/л 45 30 47.5 33,5 51
рн 5,1 6,1 4,9 4,9 5.4
б Сре, мг/л 120/81 95/81 105/53 150/76 125/88
в Сре, мг/л 71/21 55/18 77/23 90/32 115/47
е Сре, мг/л 27 30 24 30 27,2
рн 6.8 8,1 6,95 7,65 8,2
Примечание В числителе приведены значения Сре для рИ — 2,0. в знамена
тел<—для pH «3,0
6 Заклз N 2118
129
тора в эта величина возрастает в 2—3 раза снмбатно с увеличе-
нием концентрации хлоридов.
В реакторах а и г не удается достичь снижения pH до выше-
указанных величин В реакторе а pH снижается до 4,9 6,1 при
начальном значении 6,5, а в реакторе г эта величина повышается
до значений 6,8—8,2. В свою очередь, выход железа в реакторе а
в 1,5—2 раза выше, чем в реакторе г.
Стабильность работы реакторов а, б и в обеспечивается при
концентрации хлоридов более 1 -10 1 моль/л, при этом выход же-
леза увеличивается па 20 % при росте концентрации хлоридов
до 5-10-2 моль/л. Стабильность работы реактора г не обеспечи-
вается даже при высоких концентрациях хлоридов из-за образо-
вания гидроксидов железа и забивания межэлектродиого про-
странства. Промывка реактора позволяет поддерживать нормаль-
ный режим его работы.
Обсуждаемые результаты дают основание предположить, что
стабильная генерация ионов металлов возможна при снижении
величины pH раствора, контактирующего с растворяющимся ме-
таллом, до значений, обеспечивающих предотвращение образова-
ния нерастворимых соединений на границе «электрод — раствор».
Повышение концентрации иона-денассиватора интенсифицирует
процесс растворения за счет увеличения числа переноса тока дан-
ным ионом и вытеснения им с поверхности электрода ионов, спо-
собных образовывать нерастворимые соединения с генерируемым
ионом металла. Оценка такого взаимодействия возможна с по-
мощью термодинамических диаграмм Пурбе.
Другие мероприятия обеспечивают только замедление про-
цесса пассивации п требуют их регулярного повторения через оп-
ределенные интервалы времени (периодическая чистка электро-
дов, встряхивание стружки, обработка в кислоте и т. п.).
Формирование твердой фаты следует производить вне границы
«электрод — раствор», в частности в межэлектродиом объеме рас-
твора в отдельном реакторе для предотвращения взаимодействия
твердой фазы с поверхностью электрода.
Процесс электрокоагуляции может быть значительно интенси-
фицирован ври повышении температуры обрабатываемой воты до
60—80 °C и анодной плотности тока 2—2,25 А/дм2. Прн этом
твердая фаза гидроксидов меняет свою структуру и нриобре
тает ферромагнитные свойства [41]. Удельная магнитная восприим-
чивость осадка равна (24-3) • 10 2 см3/г, что характерно для фер-
рпмагпетитов. Гидравлическая крупность частиц осадка увеличи-
вается в 2 -3 раза по сравнению с электрокоагуляциоппым осад-
ком и составляет 0,6—0,8 мм/с. Данный метод, предложенный
ВПТИЭМП (г Кишинев), позволяет извлекать ионы тяжелых ме-
таллов на 90—95 %, нефтепродукты па 80—90%, ПАВ — на 40—
60 % и уменьшать жесткость — на 10—20%.
В последнее время получил распространение гальваннкоагуля-
цнонный метод очистки воды и генерации коагулянта, разрабо-
танный iiHCTiiiyioM Казмеханобр» (г. Алма-Ата). Генерация коа-
130
Рис 1.15. Гл.1ьван1>коагулнционныЛ
барабан
/ — подача воды; 3 — корпус; 3 — опорные
кольца; 4 — перемешивающие устройства.
5 — перфорированный щит: 6 — сборный
бак; 7 — отвод очищенной воды: ® — ре-
дуктор с электродвигателем
гулянта производится во вращающихся частично погруженных
в обрабатываемую воду барабанах, в которые помещается смесь
железной (атомитиевой) и медной стружки, образующая гальвано-
пару (рис. 4.15) Соотношение объемов железной и медной
стружки — 2,5:1, оптимальнее pH обработки — 2-ь4, скорость
вращения барабана — 304-60 об/ч.
Вращение барабана обеспечивает контакт стружки с воздухом
и очищает ее поверхность от продуктов реакций. Для реализации
гальванокоагуляцнонного метода разработаны аппараты различ-
ных модификаций, которые применяются для очистки воды от
попов тяжелых металлов. Барабан длиной 3,5 м и диаметром
1,2 м обеспечивает очистку сточных вод с расходом 2—4 м3/ч.
При производительности 10 м3/ч длина барабана достигает 10 м.
Расход электроэнергии иа очистку 1 м3 воды составляет 1 —
2 кВт-ч Аппараты работают устойчиво на различных видах сто-
ков, не требуют затрат электроэнергии на электролиз. К недо-
статкам аппаратов следует отнести большие габаритные размеры,
шачительный шум и вибрацию, невозможность регулирования па-
раметров процесса.
Оценивая как в теоретическом, так и в прикладном аспекте
юстигнутые успехи в разработке метода электрокоагуляции, сле-
дует, однако, отметить, что до настоящего времени еще недоста-
точно полно изучено влияние ионного состава обрабатываемой
жидкости на растворение стальных электродов, не разработаны
оптимальные конструкции аппаратов, обеспечивающие надежную
и устойчивую нх работу и учитывающие все составляющие дан-
ного многостадийного электрохимического процесса. Работы
в этом направлении по совершенствованию высокоэффективной
юхцологии элсктрокоагуляционной очистки воды должны быть
продолжены.
4.2.2. Электрокорректирование pH и Eh
Изменение pH и F.h в объеме раствора происходит за счет
i.ieKiродных процессов. В прикатодпом слое происходит подще-
ычивапие электролита при разряде па катоде ионов или моле-
кул воды, в прианодпом слое раствора — подкисление среды за
сче! разряда попов ОН или молекул воды на поверхности анода.
I ели катодный или анодный процесс исключает разряд моле-
к.1 поды, то происходит накопление только ионов Н* или ОН
131
Рнс. -I.J6. Распределение ионов в бездиафрагменном (<i) и диафрагменном (б)
электрореакторах
/ — приаиодный слой; 2 — нейтральный слой; 3 — лрикатодный слой (подщелачивание);
Г —анолит (кислая среда); 6 — католит (щелочная среда)
Так, например, при анодном растворении железа, характеризую-
щемся образованием ионов Fe2+ и переходом их в раствор, про-
исходит только подщелачивание. Однако подщелачивание рас-
твора ограничено из-за протекания реакций гидролиза с образо-
ванием малорастворимого соединения: Fe2++2OH_-*-Fe(OH)j.
Аналогично при разряде иа катоде вместо молекул воды других
ионов, например меди, золота и т. д., будет происходить подкис-
ление раствора.
Толщина приэлектродных слоев с резко выраженными кис-
лотно-основными свойствами составляет 100—500 мкм. Концентра-
ция ионов Н+ и ОН- соответственно составляет 1—10 и 1013—
10й г-ион/л [93].
Из прикатодного или приаподного слоя ионы ОН и Н+ мигри-
руют в объем раствора за счет диффузии или конвекции, а также
гранспорта этих ионов в электрическом поле, участвующих в пе-
реносе тока. Так как продукты анодной и катодной реакций об-
разуются в эквивалентном количестве, то при миграции этих ионов
в объеме раствора происходит их химическое взаимодействие
с образованием вновь молекул НгО в соответствии с ионным рав-
новесием воды. В результате этого процесса активная реакция
среды становится нейтральной или не отличается от первона-
чальной (рнс. 4.16). Поэтому при необходимости изменения кис-
лотно-основных свойств раствора стремятся подавить транспорт
ионов Н+ и ОН в объеме обрабатываемой жидкости. Это осу-
ществляется путем разделения продуктов анодной и катодной ре-
акций инертными или ионообменными диафрагмами, снижением
чисел переноса ионов Н+ и ОН- за счет повышения концентрации
индифферентных ионов-переиосчиков тока (К+, Na+, SO4' и др.)
Однако во всех случаях следует учитывать, что числа переноса
ионов Н*- и ОН непрерывно растут по мере накопления кислот -
132
Рис. 4.17 Изменение выхода по току кис-
лотности (щелочности) от величины pH
раствора Na:Sot в электрореакторе с не-
активной диафрагмой при концентрации
соли
/ — в.о г/л: ? — 1,0 г/л: з — o.os г/л
пости и щелочности раствора, что ограничивает рост величины
pH из-за переноса ионов в противоположные камеры и их раз
ряда на электродах или из-за химического взаимодействия с об-
разованием молекул воды. Выход по току кислотности (щелоч-
ное ги) в этом случае резко падает и в значительной степени за-
висит от концентрации индифферентных ионов в растворе
(рнс. 4.17).
Диффузия и конвекция между камерами в диафрагменных
электрореакторах практически подавлена при отсутствии перепада
\ровней воды в камерах. В бездиафрагмеииых аппаратах толщина
приэлектродных слоев и степень изменения pH в приэлектродном
пространстве определяется градиентом концентрации и интенсив-
ностью перемешивания жидкости. Даже при отсутствии механи-
ческого перемешивания последнее осуществляется за счет выде-
ления электролитических газов на электродах и увлечения с ними
приэлектродных слоев жидкости.
Существенным фактором, тормозящим накопление кислотно-
сгн (щелочности) в растворе за счет электрохимических реакций,
следует считать химические превращения веществ в растворе при
их гидролизе. Как уже отмечалось, при гидролизе солей металлов
тормозится накопление щелочности, наличие бикарбопатных ионов
обеспечивает буферные свойства раствора за счет сдвига угле-
кислого равновесия и т. п В каждом конкретном случае эти фак-
юры должны быть учтены в зависимости от химического состава
н< х<> нюго раствора.
Величина Eh изменяется аналогично изменению величины pH
< 1ой разницей, что иа поверхности электродов и в объеме рас-
твора происходят электронные изменения, а в растворе накапли
и.цогся продукты окислительно-восстановительных реакций. Чис-
к-нно величина Eli в общей форме может быть определена по из-
юм гному уравнению Нернста (см. 3.11)
Условием нейтральности воды при 25 °C является поддержа-
ние р11*=7,0 и Eh=+0,404 В. При изменении pH величина Eh
чн< юн воды также изменяется в соответствии с уравнением
Eh 0,817 — 0,059рН. (414)
Как видно, в диапазоне изменения величины pH- 04-14 сдвиг
lli । оставляет достаточно значительную величину — 0,886 В
133
Существенное влияние на ветчину Eh раствора оказывают
ноны металлов переменной валентности, выделяющиеся в раствор
при ионизации металлических анодов (катодов). В первую оче-
редь к ним следует отнести ионы Ге2 , преимущественно выде-
ляющиеся прн растворении железных анодов. Ионы Fe2+. обладаю-
щие восстановительными свойствами, находятся в растворе в рав-
новесии с попами Fe‘ и молекулярным кислородом, обладающим
окислительными свойствами. При этом величина Eh достигает
0,61 В при pl 1 = 6,3 и 1,07 В — при pH =14, превышая таким об-
разом потенциалы нижнего предела устойчивости воды. Ионы
хрома Сгв+, выделяющиеся при использовании анодов из хромо-
никелевых сталей, придают раствору окислительные свойства.
Наибольшим сдвиг в область положительных значений Eh>
>(1,04-1,2) В, превышающих нотспнна.’ы верхнего предела устой-
чивости воды, обеспечивают ноны хлора, разряжающиеся иа аноде.
За счет хорошей растворимости в воде газообразного хлора
в слабокислых и нейтральных растворах образуется ион гипохло-
рита, а при высоких pH — хлорноватистая кшлота. Эти соедине-
ния должны окислять воду до тех пор, пока Eh нс сдвинется в об-
ласть термодинамической устойчивости. Однако из-за малой ско-
рости процесса окисления «хлорная» вода сохраняет высокие зна-
чения Eh длительное время. Расчетные данные показывают, что
при растворении 1 моль/л С12 можно повысить Eh до 4-1,395 В,
при растворении 10 8 моль/л СИ — до 4-1,155 В. Таким образом,
несмотря на малый выход но току, газообразный хлор сдви-
гает Eh водной фазы в область высоких положительных зна-
чений.
Менее существенно влияют на величину Fh выделяющиеся при
электролизе кислород и водород. Как установлено расчетами,
величину Eh воды, нейтральной с точки зрения окислительно-вос-
становительного взаимодействия, можно повысить за счет рас-
творения кислорода только па 0,526 В (до верхнего предела
устойчивости 4-0,814 В), а за счет растворения водорода — по-
низить на 0,9 В (до нижнего предела устойчивости 0.414 В)
Разумеется, если жидкость содержит вещества, легко окисляю-
щиеся кислородом пли восстанавливающиеся водородом, то при
сдвиге потенциала от равновесного достижение предельных зна-
чений I h возможно только после их полного окисления или вос-
становления.
Величина Eh природных н сточных вод, подвергающихся элек-
тролизу. зависит от многих факторов: температуры раствора и ее
изменения, перемешивания электролита, продолжительности кон-
такта с воздухом, координаты точки отбора пробы для оценки
этого 'пачепня. Так, например, в надэлсктродном пространстве
бездпафра! меппого электрореактора с нерастворимыми электро-
дами величина Fh существенно изменяется по высоте. Это объяс-
няется преимущественным растворением водоро ia в жидкости и
его высокой реакционной активностью, которая па дает по мере
растворения кпелоро ьа и хлора
134
Для электрокорректирования pH и Eh применяют как бездпа-
фрагменные, так и диафрагменные электролизеры.
Бездиафрагменные электролизеры используются в тех случаях,
когда процессы, протекающие на поверхности рабочего электрода,
необратимы или изменение свойств раствора в приэлектрод-
ных зонах не влияет отрицательно на выход требуемых про-
дуктов.
Диафрагменные электролизеры обеспечивают разделение про-
дуктов электродных реакций, смешение которых исключает эф-
фект изменения pH и Eh. Как правило, разделение продуктов
реакций осуществляется с помощью ионообменных или инертных
диафрагм. Разделение можно производить также без диафрагм,
отбирая католит и анолит непосредственно из приэлектродных
юн.
Наиболее широко в практике водоочистки используется без-
тиафрагменный электролиз с нерастворимыми электродами в при-
сутствии хлор-попов [46, 48, 50], при котором редоксн-потенцпал
обрабатываемой воды повышается до значений 0,9—1,1 В за счет
образования молекулярного хлора, хорошо растворяющегося
в воде. Этот метод нашел также применение для окисления про-
стых и комплексных цианидов [102]. Электролиз цианистых сточ-
ных вод производится в ваннах без диафрагмы с использованием
графитовых, свинцовых, магнетитовых и других нерастворимых
анодов до образования цианатов, а затем карбонатов, газообраз-
ною азота и аммиака. На катоде может непосредственно восста-
навливаться металл, входящий в состав комплексных цианидов
Аналогично производится окисление фенолов, сульфидов, меркап-
1апов, меркаптидов, красителей [33].
При растворении железных анодов в бездиафрагмениом элек-
тролизере происходит снижение Eh среды до значений — (0,4—
0,8) В за счет перехода в раствор ионов двухвалентного же-
icia, обладающего восстановительными свойствами. Это позво-
гяет восстанавливать шестивалентный хром в сточных водах галь-
ванического производства [102], меховых фабрик [55] и др.
Как правило, величина pH в бездиафрагменных электролизе-
рах с нерастворимыми электродами изменяется незначительно,
и греднем на 0,1—0,5 единиц. Более интенсивно это изменение
наблюдается в электролизерах с растворимыми алюминиевыми и
। слезными анодами, когда величина pH может повышаться до
10,2 прн расходах тока 1000—2000 Кл/л. Однако в пределах
широко используемых расходов тока 100 500 Кл/л величина pH
и (меняется на 0,3—1,0, что часто бывает недостаточно для осу-
ществления превращений примесей, как, например, при осажде-
нии ионов тяжелых металлов.
> 1ектрохимнческое окисление сульфидов, меркаптанов, кра-
> ni* ien может производиться также в двухкамерном электроли-
|« ре, катодное пространство коюрою заполняется щелочью, а диа-
||||>н|м.1 выполняется из хлориновой, фгоролоновой ткани, катпопо-
• u-iiiKHo материала, щелочностей кого асбеста.
135
Электрохимическая технология целенаправленного изменения
pH и Eh реальных водных систем как самостоятельный метод
начала развиваться в СССР с 1969 г. в области обогащения по-
лезных ископаемых. Благодаря работам И. Н. Плаксина,
Р. Ш Шафеева, В. А. Чантурия и других этот метод применяется
для интенсификации процесса флотации руд и очистки отработан-
ных промышленных растворов горнообогатительных предприятий
[58, 115]. В процессе катодной и анодной обработки флотореа-
генты (жидкое стекло, сернистый натрий, ксантогенат и др.) улуч-
шают свои свойства и оказывают активирующее или депрессирую-
щее воздействие на флотацию минеральных частиц, обеспечивая
более высокую степень их извлечения.
Электровосстаиовлеиие в диафрагменных электролизерах по-
лучило распространение для интенсификации селективного разде-
ления минералов при одновременном снижении расхода флото-
реагеитов [115]. Обработка оборотной воды, используемой для
приготовления флотационных реагентов, улучшает ее химические
свойства и, в конечном счете, повышает технологические показа-
тели обогащения руд. Так, электровосстаиовлеиие оборотной воды
позволяет сократить расход собирателей иа 20 % и повысить ка-
чество концентрата на 2,2—5 % при затратах электроэнергии иа
обработку 2,5—4,0 кВт-ч/м3.
В Институте прикладной физики АН СССР разработан метод
и оригинальные аппараты для электрохимической обработки
пульпы н флотационных реагентов с одновременным избиратель-
ным воздействием на иих электролитических газов и сопутствую-
щим изменением pH [58]. Этот метод получил также широкое рас-
пространение для очистки промышленных стоков. Так, во
ВНИИВпроект разработай комплексный метод очистки сточных
вод вискозного производства от цинка, железа, сероуглерода, се-
роводорода с помощью избирательной электрофлотации водоро-
дом с сопутствующим изменением pH.
Интересны работы, выполненные в этом же направлении ис-
следователями Фейном и Мендиа [126]. Очистка хозбытовых сточ-
ных вод по этому методу осуществляется в смеси с морской во-
дой в бездиафрагмеиной установке с разделением продуктов элек-
।родных реакций (рис. 4.18), представляющей собой емкость, на
дне которой уложены графитовые аноды. Катоды расположены
на высоте 10 см выше анодов. Смесь морской и сточной вод в со-
отношении 1 :20—1 :40 протекает над катодом, электрохимически
подщелачивается до pH = 10,4, что приводит к образованию в жид-
кости гидроксидов магния и кальция, которые реагируют с амми-
аком и фосфатами и сорбируют взвешенные вещества. Одновре-
менно происходит флотация взвеси пузырьками электролитиче-
ского водорода, выделяющегося иа катодах. Чистая морская вода,
выполняющая роль диафрагмы, направляется между катодом и
анодом. Снижение ВПК в таких установках составляет 70—80 %,
фосфора — 90 %, аммонийного азота — 80 %, мутности — 60 % при
затратах электроэнергии 0,2—0,4 кВт-ч/м3.
136
Рнс. 4.18. Установка для электролитической очистки бытовых сточных вод со-
вместно с морской водой
/ — скребок; 2 — электролизная камера; 3 — лоток для сбора шлама; < — бак морской
воды; 5 — катод; 6 — анод
Оригинальное устройство для подкисления воды предложено
фирмой «Magna Corporation» [131]. Принцип работы этого устрой-
ства состоит в том, что вода, разделяясь на два потока, проходит
через кольцевые электроды со скоростью, превышающей подвиж-
ность водородных и гидроксильных ионов. Католит сбрасывается,
а аиолит, составляющий около 99 % потока воды, используется
в циркуляционных системах охлаждения.
В Японии, ФРГ и других странах в последние годы получили
распространение бытовые диафрагменные электролизеры для об-
работки питьевых вод с целью улучшения их органолептических
и химических свойств. Один из таких электролизеров (рис. 4.19)
ныполнен в форме стакана, в котором закреплен катод 7 из ме-
таллической фольги. Во внутренней емкости 6 из пористого ма-
териала, играющего роль диафрагмы, установлен анод 4. Внеш-
ний корпус 5 имеет съемную крышку 1 с встроенным электриче-
ским регулятором и выпрямителем. Исходная вода заливается
в электролизер через вороику 3. Подобного тина аппараты при-
меняются в нашей стране для получения «активированной» воды,
н< пользуемой в медицине, при бурении скважин, в сельском
хо»яйстве и т. д. Физико-химические и
биологические свойства «активированной»
воды не изучены еще в достаточной сте
пени, что мешает ее целенаправленному
использованию.
В УИИВХ с 1971 г. ведутся научные
и 1ыскап11я по разработке методов и аппа
р.пов электрохимического регулирования
pH и Eh природных и сточных вод с целью
<>|д.1!1ия безреагептных схем очистки [91,
I’m 4 10 Схема диафрагменного электролизера для
«ip.BwiKH питьевых вод
137
Рнс. 4.20. Диафрагменный электрокорректор pH безнапорного типа
/ — боковые стенки. 2 — электродные блоки; Л — катодные камеры; вводные камеры.
5 — лоток для сборе анолита; 6 распределительная система исходно!) воды; 7 — лоток
для сбора католита; 8 — катод; 9—анод; 10 — диафрагме
110]. На основании этих исследований разработаны различные
модификации промышленных аппаратов (электрокорректоров
pH), внедренных па ряде предприятии страны. Базовый электро-
корректор pH состоит из катодных и анодных камер, разделен-
ных электродными блоками-модулями, подводящих и отводящих
лотков и арматуры (рис 4 20).
Конструктивной особенностью блока э>ектродов является то,
что он состоит из перфорированных катодов и инертных анодов
(ОРТА. ОКТА, графит, нержавеющая сталь и др). При этом
отверстия в электродах выполнены соосно, а сами электроды и
диафрагма плотно прижаты друг к другу с изоляцией внутренней
поверхности электродов специальным материалом (лаки, краски
и др). В этом случае работа электродного блока обеспечивается
наружными поверхностями электродов, тем самым предотвраща-
ется забивание меж электродного пространства и диафрагмы не-
растворимыми продуктами электродных реакций. Это стабилизи-
рует работу электрокорректоров в условиях электролиза воды со
сложным ионно молекулярным составом. Электрокорректоры могут
быть выполнены фильтропресспого или емкостного типов При
обработке воды с высоким солссодержаннем (I—5 г/л) возможно
обычное размещение электродов или устройство блоков-модулей
без диафрагмы с уменьшением размеров отверстий в электродах
до I 1 мм Вместо диафрагмы можно использовать сплошную
диэлектрическую пластину, не доходящую до дна корпуса
аппарата.
С целью использования окислительных пли восстановительных
свойств элекгрол!ИlinerKiix газов, выделяющихся в элекгрокор-
ректорах. они мпп i быть уловлены и направлены в анодную или
13Я
катодную зону [а. с. 893884 (СССР)]. Так, например, восстанов-
ление Ст1’ в анодной камере диафрагменного электрокоррскюра
с растворимыми железными элем родами более интенсивно про-
текает при подаче из катодной камеры водорода, обладающего
восстановительными свойствами. Эффект обезвреживания хрома
возрастает па 20—30%.
Комбинация электродов в анодной или катодной камере поз-
воляет целенаправленно изменять физико-химические свойства
раствора. Предварительное подкисление воды перед анодным рас-
творением стального электрода обеспечивает стабилизацию его
работы, снижает расход железа на восстановление Сгв+. Эффект
очистки ог взвешенных веществ н нефтепродуктов также повыша-
ется на 20—30 % при размещении дополнительного алюминиевого
анода в катодной камере [а. с. 565889 (СССР), а. с. 827404
(СССР)].
Технологические параметры работы электрокорректоров зави-
сят от солевого состава воды и конструкции электродной системы.
Прп обработке природных вод, а также слабоминерализованных
сточных вод основные параметры работы аппарата находятся
в пределах, указанных ниже. Области изменения параметров pH
и 1:11 прп различных способах обработки воды представлены
в |92]
Средние параметры работы ззектр-корректора pH
при обработке слабоиинера зизованных вод
Плотность тока, Мм® ... 30—50
Напряжение электролиза, В 12 24
Расход тока, Клб1 .... 250 480
Расход электроэнергии, кВт-ч/м’ 1,2—2,0
pH католита . . II,2 11,8
pH апатита................................ 6,5—8,2
Соотношение расходов анолита и католита (0,84-?0,88) : (0,124-0.16)
Электрохимическое корректирование pH и Eh имеет сущест-
венные преимущества перед химическим. Кроме исключения ввода
нфнцнтных реагентов, предотвращающих загрязнение воды кати-
онными н анионными остатками кислот и щелочей, преимущест-
вом является также и то, что в результате электролиза изменя-
юня многие физико-химические .характеристики воды, обеспечи-
вающие ее «активность». Такая вода обладает лучшими обезжи-
ривающими п моющими свойс1вамп, биологически активна и мо-
40-1 использоваться но самому широкому назначению. Однако
юн1с|рукцн11 электрокорректоров pH п Eh еще достаточно сложны,
i»ic\ Ищуки малоиз||ашнва1О1цмеся аноды, работающие прп низ-
ких 1|1аченпях pH. Электроды из нержавеющей стали недолго-
iiviiiu н растворяются с вы юлением Сг6* в раствор при псноль-
'»||.|П11н хромоникелевых сталей. II тем не менее даже существую-
... конструкции аппаратов могут найти достаточно широкое
,‘>п n'lK iiiie и практике водоочистки.
139
4.2.3. Электрохимическая деструкция
4.2.3.1. Общие положения
В последние годы все большее распространение в различных
областях народного хозяйства получает электролиз водных рас-
творов с целью электрохимического окисления и восстановления
как органических, так и неорганических соединений.
В настоящее время накоплен огромный экспериментальный ма-
териал по осуществлению электрооргапических реакций. Однако
реализация их на практике сдерживается недостаточной изучен-
ностью механизма и кинетики протекания редокси-процессов.
Кроме того, получающиеся при электролизе окислители и восста-
новители обладают особенно большим запасом химической энер-
гии в момент их образования, что значительно усложняет ход
электросинтеза органических соединений, так как бывает трудно
направить течение процесса в сторону получения основного то-
варного продукта или остановить реакции редокс-превращений на
желаемой стадии При этом могут получаться продукты более глу-
бокого окисления или даже деструктивного распада в результате
разрушения достаточно прочных связен основной углерод-уг-
леродиой цепи, вплоть ю образования углекислого газа и
воды.
При очистке сточных вод это обстоятельство, наоборот, явля-
ется весьма позитивным, так как в основные задачи обезврежива-
ния промстоков входит необходимость деструкции загрязняющих
компонентов до более простых и безвредных продуктов.
Учитывая эти факторы, можно полагать, что процессы прямого
электрохимического окисления или восстановления могут найти
широкое применение в области очистки сточных вод с целью глу-
бокой минерализации содержащихся в них трудноокисляемых ор-
ганических загрязнений.
Реакции электрохимического окисления и восстановления вклю-
чают в себя широкий круг процессов — от простейшей ионной
перезарядки до сложных превращений, лежащих в основе орга-
нического синтеза.
Механизм катодного восстановления органических соединений
существенным образом зависит от потенциала катода, относи-
тельно влияния которого на ход электродного процесса было пред-
ложено несколько теорий. Наиболее приемлемым является по-
ложение, утверждающее, что с повышением катодного потенци-
ала увеличивается потенциальная энергия и восстановительная
способность атомов водорода, выделяющихся па поверхности
электрода
Другая точка зрения исходит из того, что металлы с низким
перенапряжением образуют прочную металл-водородную связь,
а с высоким перенапряжением — относительно слабую Это пред-
ставление, как отмечает Аллен [67], можно распространить и на
протоны. Так, восстановлен не кетона до спирта па ртутном ка-
140
тоде при относительно низком потенциале может быть представ-
лено следующим образом:
'О,е
й I
Me —-> R,C:
Н +
(4.15)
R*CHOH
Заряженная изомерная форма притягивается к катоду, где
атом кислорода, имеющий неподеленную пару электронов, соеди-
няется с водородом В то же время благодаря прочности связи
металл — протон заряженная молекула при относительно низком
катодном потенциале удерживается на электроде в течение вре-
мени, достаточного для перехода двух электронов, в результате
чего образуется спирт.
Процесс прн более высоком потенциале катода идет следую-
щим образом:
ОН он
I I
R2C----CR,.
(4-16)
При этом заряженная форма благодаря слабости связи ме-
алл — протон удерживается па катоде лишь в течение времени,
их*! атомного для перехода только одного электрона к углероду
. недостающим электроном, в результате чего образуется свобод-
ный радикал, который, соединяясь с другим таким же радикалом,
образует пинакон
11срвая теория, основанная па учете потенциальной энергии
ра 1ряжеппого водорода, не объясняет реакционную активность
органических соединений. Взаимодействие заряженной формы ор-
глннческого соединения с электродом позволяет объяснить про-
цесс также и в условиях образования свободных радикалов.
Образование радикалов происходит и при анодных реакциях
> 1екзроокисления. Поэтому при изучении механизма электродных
процессов, протекающих прн очистке жидкостей от органических
.и ря шенцй, необходимо учитывать возможность образования сно-
бо шых радикалов в качестве промежуточных продуктов реакции.
I । । iioii цели можно применить метод вращающегося дисковою
141
электрода с кольцом, что позволяет фиксировать радикалы, имею-
щие время жизни всего 10 - 10 2 с.
Анодное окисление во многих отношениях очень сходно с дей-
ствием сильною окисляющего агента. Однако механизм окисли-
тельного процесса по сравнению с катодным процессом восста-
новления является более сложным и пока еще окончательно не
установленным.
Наибольшее значение в процессах водоочистки приобретает
анодное окисление органических загрязнении с использованием
нерастворимых электродов. Здесь представляется интересным рас-
смотреть механизм, предложенный Глесстоном и Хнкклингом (67],
согласно которому предполагается, что в водном растворе гид-
роксильные ионы разряжаются при низком потенциале, образуя
гидроксильные радикалы, которые, соединяясь, дают перекись во-
юрода. Перекись водорода может затем реагировать с органиче-
ским соединением, вызывая окисление, или разлагаться, давая
кислород и воду:
Окисление
2011 — 2е —2ОН-—Н2О2 (4.17)
\ н,о+1/20,
Реакция может происходить либо с участием гидроксильных
радикалов, являющихся промежуточными продуктами, либо в ка-
честве сильного окисляющего агента кислорода.
К сожалению, не существует общего объяснения для всех из-
вестных электрохимических реакций, поэтому в каждом отдель-
ном случае необходимо специальное изучение механизма окисле-
ния органического соединения.
Сведения по анодному окислению различных органических со-
единении, содержащиеся в многочисленных литературных источ-
никах, позволяют определить возможность и перспективность при-
менения электрохимического способа для глубокой деструкции
трудноокисляемых органических загрязнений. Так, при электро-
лизе иа медном аноде формальдегида, часто встречающегося в не-
которых категориях промышленных сточных вод, анодная реакция
окисления формальдегида протекает с образованием водорода:
НСНО +О-* СО, 4- Н2 -Ь 2е. (4.18)
Возможны также анодные реакции постадинного превращения
более сложных ароматических соединений до относительно про-
стых остаточных продуктов, например окисление фенантрена (67]:
Окисление фенантрена происходит последовательно до фенан-
трахииона дифеновой кислоты и, наконец, до бензойной кислоты.
142
Выход при этом конечного продукта значительно повышается при
добавлении переносчиков кислорода, например Cu(SO4)2.
Большинство исследователей утверждают, что природа продук-
тов, получаемых при окислении органических соединений, в зна-
чительной степени зависит от плотности тока. Это утверждение
вполне резонно, так как оно сводится к тому, что направление
реакции зависит от потенциала электрода.
Следует также отметить, что механизмы электрохимического
окисления (нли восстановления) органических и неорганических
веществ зависят от материала электродов, природы подвергаемых
электролизу исходных продуктов, температуры и состава элек-
тролита (активной реакции среды, присутствия посторонних ве-
ществ ионного нли молекулярного типа, наличия ингибиторов, по-
верхностно-активных веществ и т. п.).
Прн этом, кроме стадий, протекающих па границе раздела
электрод — электролит, существенное значение имеют химические
превращения, совершающиеся в объеме обрабатываемого рас-
твора. Учитывая это обстоятельство, процесс электрохимической
деструкции органических загрязнений может быть значительно
интенсифицирован при наличии в обрабатываемом стоке С1_-нонов.
Интенсификация происходит вследствие образования при элек-
|ролнзе так называемого «активного» хлора (а. х.) — водного
раствора, содержащего хлор и продукты его гидролиза (С12,
IIOCI, С12О, СЮ , СЮз~), являющиеся сильными окислителями
в соотношениях, определяющихся условиями процесса.
При электролизе растворов, содержащих О -ионы, па аноде,
как было показано в н. 3.3.6, возможно протекание реакций вы-
(е.чепия и хлора, и кислорода. Для того чтобы па аноде преиму-
щественно выделялся хлор, нужно подобрать материал анола, па
ко юром хлор выделяется с минимальным, а кислород с макси-
мальным перенапряжением.
Существенное влияние па выход по току хлора (Вт0) оказы-
iiaci концентрация хлоридов. Зависимость Вт° от концентрации
\'а<1 показана па рис. 4 21. Повышение анодной плотности тока
при низких концентрациях NaCI резко увеличивает Вт°, поэтому
с увеличением плотности тока следует ожидать увеличения ско-
рое in окисления органических загрязнений. Однако необходимо
амегить, что, поскольку между величиной тока и напряжением
в растворе существует известная зависимость, выражаемая зако-
ном Ома (Ut—IR}, увеличение / неизбежно приведет к увеличе-
нию U„ и. следовательно, возрастут затраты электроэнергии на
обработку воды Таким образом, выбор оптимальных величин
плотностей тока и напряжения между электродами при электро-
химической очистке воды весьма существен
Как известно, химические реакции значительно ускоряются при
iioitijiiieiiHii температуры и перемешивании растворов. Повышение
1гмпсратуры уменьшает, кроме того, поляризацию; так, повыше
.. |емиературы на 5 °C сдвигает поляризационную кривую вы
нпия хлора в сторону более электроотрицательных нотенцп-
мз
Рис. 4.21. Зависимость выхода по току
хлора от концентрации NaCI при плотно-
сти тока
/ — 1000 А/м*; 2 — 250 А/м’
алов, т. е. облегчает процесс обра-
зования а. х. С другой стороны,
с ростом температуры уменьшается
растворимость хлора и увеличива-
ется его непроизводительный вы-
нос в атмосферу. Что же касается
перемешивания, то влияние этого
фактора в большей степени на-
блюдается в тех случаях, когда
концентрации веществ, вступающих в реакцию, малы и лимитиру-
ющей стадиен процесса является скорость диффузии этих веществ
в зону реакции.
Перемешивание способствует также отводу в раствор образую-
щихся на электродах веществ, в частности деструктпрующих
агентов — хлора, кислорода, озона и т. п., что ускоряет их взаимо-
действие с загрязнениями сточных вод. Поэтому в рассматривае-
мом процессе следует ожидать существенного влияния этих двух
факторов на ход электрохимической очистки воды.
Прн разработке технологии электрохимической деструкции ор-
ганических загрязнений необходимо учитывать закономерности
процессов, протекающих при электролизе. Рассмотрим основные
факторы, влияющие на ход процесса электрохимической деструк-
ции загрязнений.
4.2.3.2. Выбор типа электродов
Правильный выбор материала электродов является важным
условием успешного проведения процесса очистки воды.
В качестве катодов можно использовать практически любые
материалы, обладающие достаточной электропроводностью: же-
лезо, нержавеющую сталь.
Выбору типа анодных материалов для осуществления процесса
электрохимической деструкции органических загрязнений необхо-
димо уделять особое внимание. Основная трудность прн этом
возникает вследствие того, что большинство металлов в условиях
анодной поляризации растворяется нли пассивируется.
Для изготовления анодов обычно применяют графит. Однако
графитовые аноды из-за значительной пористости впитывают
большие количества раствора соли. Поэтому действие электриче-
ского тока проявляется не только на поверхности анода, но и
в самом электроде, что создает благоприятные условия для его
разрушения. Износ графитовых анодов, например, при электрохи-
мической очистке фенолсодержащнх сточных вод составляет ~ 15 г
па 1 м3 обрабатываемого стока [45] Все это значительно услож-
няет процесс очистки вследствие необходимости последующей
114
фильтрации стока для удаления тонкодиспергнроваиных частиц
графита, остающихся в воде, частой достаточно трудоемкой за-
мены электродов, корректировки электрических параметров из-за
изменения размеров и формы анодов.
Для частичного устранения этих недостатков производят пред-
варительную пропитку графитовых анодов растительными мас-
лами, фенолформальдегидными смолами, полимерами винила и
другими. Такая обработка позволяет увеличить срок службы гра-
фитовых анодов, однако этим не решается задача создания не-
разрушающсгося анода, вследствие чего в последние годы возрос
интерес к замене графита малонзнашивающпмися анодами. Ос-
новой для этого послужили успехи в производстве таких металлов,
как титан, тантал и цирконий, которые могут служить в качестве
токопроводящей основы для активного покрытия, например, из
окислов металлов платиновой группы, не подвергающихся корро-
зионному разрушению при анодной поляризации [121]. Это позво-
ляет использовать в качестве анода тонкий слой гальванически
осажденного иа титане активного покрытия, который создает анод-
поработающую поверхность электрода, определяя величину потен-
циала электрода и его электрохимические показатели.
В последнее время при электролитическом производстве хлора
п его кислородных соединений все большее распространение нахо-
дят малоизнашивающиеся аноды па титановой основе; в качестве
основы также рекомендуются цирконий, тантал и ниобий [113,
121], по эти материалы являются труднодоступными для практи-
ческих целей. Для активного покрытия используются или реко-
мендуются магнетит, сплавы на основе серебра, платипоиридие-
вые [ИЗ], оксиды железа, свинца, марганца, кобальта и палладия
[39, 113, 121]. В технологии водоочистки наибольшее распрост-
ранение нашли металлоокисные аноды ОРТА — титановая основа
с активным поверхностным покрытием смесью изоморфных окне-
лов рутения и титана. Толщина слоя покрытия не более 10 мк,
истинная поверхность его в 750—800 раз превосходит видимую.
Возможность применения анодов ОРТА для очистки сточных
вод методом электрохимической деструкции подтверждена в ряде
работ, выполненных в ЛИСП [24, 45, 51, 97]. Аноды содержали
в составе активного покрытия соответственно 30 и 70 мол.% оки-
сей рутения и титана. Покрытие поверхности титана окислами
указанного состава осуществлялось по методике, разработанной
Госниихлорпроектом. Поляризационные кривые, снятые на ОРТА
в растворах с низкой концентрацией Cl-ионов (до 5 г/л
по NaCI) в интервале характерных при очистке сточных вод плот-
ностей тока от 1-10“® до 5-10-2 А/см2, были представлены ранее
на рис. 3.15.
Потенциал анодов ОРТА при плотности тока 0,2 А/см2 и
7=60 °C в области концентрации NaCI 300—30 г/л, т. е. в усло-
виях, соответствующих пронзвочетву хлора и хлорпродуктов, под-
держивается стационарным 1,42 1,45 В относительно нормаль-
ного водородного электрода (н. в. э.). Снижение концентрации
145
if, в
/ , ’ П —------------------------------—-*-------'‘J* или*-
о I
/J I । I I I I I I. I 1 1.1.1 .1 L J 1 l.J.l I 1.1 J
10 20 J1 чО t. /71-
Рис. 4.22. И «менение стационарного потенциала <| анода ОРТЛ от продолжи-
тельности электролиза /э при содержании 0,1 r/л раствора красителя и 5,0 г л
NaCI
I — кислотный синий 2К. 2 — прямой черный 3
NaCI при указанной плотности тока приводит к ускоренному из-
носу активного покрытия, быстром) росту потенциала, вплоть до
разрушения титановой основы. При потенциалах выше 1,5 В на-
блюдается изменение хода анодной поляризационной кривой, ко-
торое обусловлено также ускоренным износом активного покры-
тия, вероятно, вследствие перехода R11O2 в газообразный R11O4.
Указанные условия ведения процесса существенно отличаются
от оптимальных параметров очистки сточных вод, поэтому иссле-
дования работы [97], проведенные в целью выявления стойкости
анодов ОРТА при электролизе растворов красителей с концентра-
цией NaCI до 10 г/л и при анодной плотности тока 2 Л/дм2, имеют
большое практическое значение.
Потенциал ОРТА прп его поляризации указанной плотностью
тока в течение длительного срока (рис. 4.22) остается практиче-
ски постоянным и равным 1,40—1,45 В относительно и. в. э., что
свидетельствует об этектрохимической стойкости этих анодов
в условиях очистки сточных вод. Опыт эксплуатации иромышлеи
пых сооружений иа Саратовской экспериментальной фабрике
спортивного трикотажа в течение более чем 6 лет подтверждает
возможность длите тьмой работы ОРТЛ в системах водоочистки.
Аноды за весь этот срок эксплуатации при ведении технологиче-
ского процесса электрохимической очистки сточных вод с плотно-
стью тока 1,5—2,0 А/дм2 и концентрацией NaCI до 3 г/л работали
надежно п не изменили структуры поверхностного слоя. Однако
следует указать на наличие в составе поверхностного слоя анодов
ОРТЛ окисла благородного металла—рутения, что делает эти
электроды труднодоступными для практического испотьзовання.
В настоящее время ведутся всесторонние поиски новых анод-
ных материалов, не содержащих в составе активного покрытия
окисла рутения или содержащих меньшее его количество, чем
в ОРТЛ, путем замены части окисла рутения па окислы не-
благородных металлов например Мп, Fe, Со
Значительный интерес с точки зрения экономичности и доступ-
ности материалов представляют разрабатываемые в хлорной
промышленности окненокобальтовые (ОКТА) и двуокиспомар
гапцовые (ТДМА) анты на титановой основе, а также аноды
с активным покрышем из смеси окнелов марганца и кобальта
116
95 % МпО2+5 % Co Oi, так как они вообще ие содержа] блаю-
родиых металлов.
Поляризационные кривые, снятые на этих электродах [97|
в растворах, моделирующих сточные воды красильно-отделочных
производств и предприятии бытовой химии, а также в чистых
растворах хлоридов (см. рис. 3.15), для ОКТА оказались не
сколько сдвинутыми в сторону более положительных потенция
лов по сравнению с соответствующими ОРТА, однако углы на-
клона зависимостей <| —lg i в исследуемой области плотностей
тока па анодах ОРТА и ОКТА практически совпали в чистых
растворах NaCI 40—45 мВ. Влияние органических добавок па
электрохимическое поведение ОКТА значительно слабее наблю-
даемого для ОРТА, что можно объяснить меньшей адсорбцион-
ной способностью ОКТА при соответствующих ему более поло-
жительных потенциалах. Отклонение от тафелевскон завиенмо-
С1И, наблюдаемое при повышении плотности тока, по-видимому,
связано с влиянием омических составляющих, включенных в из-
меряемую величину потенциала. Таким образом, по своим элек-
трохимическим характеристикам электроды ОКТА весьма близки
к ОРТА н могут быть применены в условиях очистки сточных
вод.
На рис. 3.15 представлены также поляризационные кривые,
полученные па анодах ТДМА, поверхностный слой которых со-
держит двуокись марганца в качестве основного компонента.
Поляризационные кривые, соответствующие этим анодам, значи-
тельно сдвинуты в сторону положительных потенциалов, и их
углы наклона близки к наблюдаемым па графитовом аноде, что
говорит о преимущественном выделении кислорода. Этого и сле-
довало ожидать, учитывая то, что перенапряжение выделения
хлора на двуокиси марганца значительно выше, чем иа двуокиси
рутения, п близко к перенапряжению на графите. Несмотря на
эго, при использовании ТДМА наблюдалось повышение эффекта
очистки сточных вод, содержащих органические загрязнения при
одновременном уменьшении концентрации а. х., что объясняется
каталитической активностью двуокиси марганца по отношению
к процессам окисления.
Всесторонние исследования по изучению электродов ТДМА
и ОКТА с целью установления возможности их использования
для очистки промышленных сточных вод были выполнены
в ЛИСИ [66, 73] совместно с Днепропетровским химико-техноло-
гическим институтом (ДХТИ). В качестве подложки использо-
ван титан марки ВТ-1-0 толщиной 3,5 мм, перфорированный
с обеих сторон в шахматном порядке лупками диаметром 7—
9 мм и глубиной 1,0—1,5 мм. Пластины подвергали пескоструй
пой обработке и затем методом термодинамического разложе
пня нитратных солей кобальта и марганца послойно наносили ак
явный слой электродов. Способ изготовления электродов, раз-
работанный ДХТИ, довольно прост, производство их может быи>
opiавизовано в небольших мастерских на любом предприятии.
117
Рис. 4.23. Экономические показатели электролитического получении активного
хлора (а. х.) с использованием анодов ОКТ4 и ОРТА
1—5 для ОКТА; /' 5' дли ОРТА, 1. Г — общий выход а. х. (С х); 2, 2'— выход
а. х. по току (В*'х ); 3. 3“ степень превращения хлоридов в а. х. 4. 4' —рас-
ход электроэнергии 5. 5' — затраты ив получение 1 кг в. х. (Зв х)
Анализ поляризационных кривых этих анодов в высококон-
центрированных сульфатных и сульфат-хлоридных растворах
различных красителей показал, что при определенных потен-
циалах существуют горизонтальные площадки на кривых зави-
симости ф—lg I, свидетельствующие о прохождении окислитель-
ных процессов. Однако процессы окисления непосредственно на
аноде, а также образующимся кислородом идут очень медленно
[66]. В присутствии же хлоридов такие площадки отсутствуют,
но наблюдается интенсивное обесцвечивание красителей. По-ви-
днмому, разряд С1 -ионов на аподе подавляет другие анодные
процессы, а окисление красителей происходит за счет образую-
щегося а. х. Наибольшая скорость окисления наблюдается прн
применении анодов ОКТА по сравнению с ТДМА и даже с ОРТА.
При выборе типа электродов, помимо дефицитности мате-
риалов покрытия токопроводящей основы, следует оценивать и
экономические факторы процессов очистки сточных вод: расход
электроэнергии (W'a.,). степень превращения хлоридов в актив-
ный хлор (XrI С jCa_), дозу поваренной соли (Диаа) и сум-
марные затраты на электроэнергию и приобретен не NaCI для по-
лучения I кг активированного хлора (За. х.)- Сравнительные дан-
ные ио экономической оценке этих факторов для анодов ОРТА
и ОКТА представлены на рнс. 4.23. При расчете экономических
показателей принята стоимость электроэнергии по одноставоч-
ному тарифу в среднем по территории РСФСР 1,5 коп. за
1 кВт-ч. Цена 1 т поваренной соли принята по работе [86] с уче-
том погрузочно-разгрузочных работ и наценки, равной 10 руб.
14Я
Более высокий выход а. х. при одинаковых параметрах обра
ботки наблюдается на ОКТА. Характер изменения всех кривых
одинаков. Максимальная степень превращения хлоридов в а. х.
обнаруживается при концентрации CI ионов в растворе ~ 1 г/л.
Это дает основание считать, что повышение концентрации пова-
ренной соли в исходной сточной воде выше указанной величины
не всегда целесообразно.
Из рис. 4.23 также видно, что затраты электроэнергии на по
лучение а. х. при применении анодов ОКТА выше по сравнению
с ОРТА, что связано с большим падением напряжения на самом
электроде ОКТА. Однако общие затраты (За. х.) на ОКТА ниже,
что объясняется свойствами этих электродов, отмеченных нами
ранее, т. е. более высоким выходом а. х. в растворах с низкой
концентрацией хлоридов.
Таким образом, все обсужденные выше данные исследований
свидетельствуют о перспективности и целесообразности исполь-
зования для электрохимической очистки сточных вод ряда новых
малонзпашивающихся анодов, не содержащих в своем составе
окислов благородных металлов. И тем не менее вопросам изу-
чения их стойкости в каждом конкретном технологическом про-
цессе водоочистки должно быть уделено особое внимание.
4.2.3.3. Влияние условий электролиза на окисление
органических загрязнений
Электролиз воды сопровождается окислительными процес-
сами на аноде н восстановител! ными на катоде. Как уже отме-
чалось, в зависимости от типа электрода и условий его работы
па аноде могут выделяться: кислород — в результате разложе-
ния гидроксил-ионов, хлор — при разряде хлорид-иопов — или мо-
жет происходить адсорбция и окисление органических соединений
и других анионов. На катоде в основном происходит вос-
становление водорода и выделение его в виде газа, а также вос-
становление катионных групп в молекуле органических соедине-
ний. Образующийся атомарный кислород частично вступает
в окислительную реакцию с органическими веществами, ассоции-
рует в молекулы и растворяется в воде, а избыток его выделя-
ется в газообразном состоянии. Хлор в сплыюкнслой среде в виде
газа выделяется в атмосферу, а в слабокислой, нейтральной и ще-
лочной средах быстро гидратируется н в зависимости от pH обра
зует хлорноватистую кислоту или гипохлорид-ион (рис. 4.24) 1107],
Хлор и его кислородные соединения вступают в окислительно-вое
становительную реакцию с органическими веществами в объеме
раствора. Прн электролизе хлорнтных растворов высшие окнелы
хлора — хлорит, хлорат и перхлорат могут образовываться только
при продолжительной обработке (121], поэтому в нашем случае
термин «активный» хлор (а. х.) включает в себя молекулярнын
и атомарный хлор, хлорноватистую кислоту и гниохлорит-ион.
149
Рис. 4.24. Соотношение содержания
различных форм хлора в воде в за-
висимости от pH
Таким образом, при элект-
ролизе волы минерализация
органических примесей про-
исходит в основном тремя
способами: в результате реак-
ции непосредственно на элект-
родах, окислением аноднооб-
разующимся кислородом и
окислением активным хлором.
Для оценки эффективности
этих способов очистки рас-
смотрим подробнее все три направления.
Окисление органических загрязнений непосредственно па
аноде эффективно происходи г на платиновых либо платиниро-
ванных электродах [62, I2IJ. Данный процесс энергоемок, а пла-
тина является очень дорогим материалом, что резко ограничи-
вает возможность практического использования способа анод-
ного окисления органических примесей воды.
Основным электродным материалом для электрохимического
получения кислорода являются никель и никелированная сталь,
^ля осуществления процесса требуется очень чистый щелочной
электролит [121]. Так, при электролизе сильиощелочных сточных
вод производства пасты «Фантазия» [73] никелевые электроды
даже при малых плотностях тока интенсивно растворяются, чем
обусловливают невозможность осуществления данного метода
очистки. Другим электродом, обладающим относительно низким
потенциалом выделения кислорода, является ГДМЛ. Однако эф-
фективная очистка сточных вод с ТДМЛ происходит только
в присутствии С1 -попов. Известно также [а. с. 684021 (СССР)],
что при заполнении межэдектродпого пространства гранулиро-
ванным пиролюзитом минерализация органических соединений
происходит и без добавления поваренной соли. Прн этом наблю-
дается интенсивное разрушение пиролюзита, сопровождающееся
повышенным расходом электроэнергии. Вследствие указанных
причин окисление органических веществ анодновыделяемым кис-
лородом практически трудно осуществимо.
Па многих типах электродов потенциал перенапряжения вы-
деления хлора ниже, чем кислорода, поэтому в присутствии хло-
ридов основным анодным процессом является выделение хлора.
В рабою [66] установлено, что в отсутствии хлоридов в суль-
фатных растворах разрушение красителей почти не происходит
на таких электродах, как ОРТА, ОКТА, ТДМА и других, а в хло-
ридиы.х растворах наблюдается быстрое обесцвечивание органи-
ческих красителей. Поэтому для очистки сточных вод электрохи-
мическим способом наиболее эффективным является процесс
электролиза в растворах с относительно высоким (>1 г/л) со-
150
держанием хлоридов и с низким потенциалом перенапряжения
выделения хлора на электродах.
Одновременно следует отметить, что литературные источники
дают противоречивые данные по условиям ведения процесса. На-
пример. в работе [45] установлено, что на общин эффект обесцве-
чивания сточных вод красильно-отделочных производств актив-
ная реакция среды прн pH = 24-10 влияет незначительно. В ра-
боте [24] специальными исследованиями показано,что наибольшее
окисление органики наблюдается в присутствии хлорнова-
тистой кислоты. Автор приходит к выводу, что прн электролизе
интенсивное окисление органических загрязнений происходит
в более широком диапазоне pH, чем диапазон pH, соответствую-
щий состоянию хлора в виде хлорноватистой кислоты. Ряд ав-
торов [II, 16] отмечают, что наибольшая скорость окисления ор-
ганических загрязнений прн электролизе происходит в кислой
среде.
Расхождение во мнениях относительно влияния pH па про-
цесс очистки, вероятно, объясняется природой окислителей, полу-
чаемых при электролизе, и различными кинетическими парамет-
рами ведения окислительных процессов в электролитических ги-
похлоритных растворах.
Для более конкретной оценки влияния различных факторов
иа ход реакции окисления остановимся подробнее на минерали-
зации органических соединений в гнпохлорнтных растворах и рас-
смо1рнм теоретические аспекты процессов, происходящих при
электролизе.
4.2.3.4. Окислительно-восстановительные процессы
в растворах гипохлоритов
Наиболее полно свойства растворов хлора и его кислородных
соединений изучены Т. А. Тумановой и И. Е. Флисом [107]. В их
работах задача исследований сводилась в основном к отбелке
целлюлозы, текстильных п других материалов. Авторами уста-
новлено, что участниками окислительных процессов могут быть
все компоненты растворов кислородных соединений хлора. Доля
же участия этих компонентов в окислительных процессах зави-
сит от pH и природы восстановителя. Максимум скорости окне
лепня в нейтральных растворах гипохлоритов обусловлен только
свойствами окислительной системы. Показано, что реакции, про-
текающие в растворах а. х., относятся к типу реакций, катализи-
руемых в слабокпслой н нейтральной средах ионами Н+ и ОН~,
которые в окислительно-восстановительных реакциях могут быть
vnacTiniKaMH процесса и катализаторами его. В среде, близкой
к нейтральной, иекатализнруемых реакций нет. Кроме того, реак-
ции, протекающие в растворах гипохлоритов, следует отнести
к типу реакций общего кислотно-основного катализа, так как
HCIO н СЮ являются сопряженными кислотой и основанием:
НСЮ+ Н,О« НЛО+ + CIO-; CIO- -|- НгО« HCIO + ОН . (4 20)
151
Необходимо учитывать, что в щелочной среде катализ ионами
Н+ н ОН отсутствует, а подвижность ионов СЮ- в этой среде
значительно меньше, чем в кислой и нейтральной [107].
И. Е. Флис и Т. А. Туманова пришли к выводу, что в раство-
рах кислородных соединений хлора окислительные процессы обус-
ловлены действием атомарного кислорода, получающегося в ре-
зультате разложения а. х. В чистых растворах гипохлоритов
реакция сопровождается образованием хлоратов. Образование
кислорода в результате разложения HCIO термодинамически ме-
нее вероятно, чем такой же процесс разложения CIO-. При совмест-
ном присутствии в растворе хлорноватистой кислоты и гипохло-
рит-иона разложение их с образованием атомарного кислорода
термодинамически более вероятно, чем процесс образования кис-
лорода в результате разложения одного из компонентов.
В чистых растворах гипохлорит наиболее устойчив в щелоч-
ной среде при рН^Ю, а в нейтральной, кислой и слабощелочной
средах разлагается с образованием кислорода, хлорида и хло-
рата. Устойчивость гипохлоритных систем также зависит от кон-
центрации а. х. Наиболее устойчивыми являются щелочные рас-
творы с Са. х.<0,5 моль/л [107], а электрохимически полученных
растворов — по более 2,5—3,0 г/л [63].
В нейтральной и слабокислой средах разложение а. х. проис-
ходит по механизму [107]:
СЮ-4- НСЮ — О24-2С1- + Н+; НСЮ2С1О-— СЮ^ 4-2С1~ 4- Н+.
(421)
В кислой среде разложение идет по следующим реакциям:
2HCIO —2С1-4-О24-2Н+; ЗНС1О — СЮ-4-2С1-4- ЗН+. (4.22)
А. Ю. Прокопчиком [87] разложение гипохлорита в щелочной
среде также рассматривается постаднйно из двух последователь-
ных реакций:
хлорид-хлоратного типа
CIO- -I- СЮ-ClOf 4- CI-; CIQ- CIO- ** CIO^ Cl-; (4 23)
х.юрпд-кнелородного типа
CIO-4-CIO- — 2CI-4-O,. (4.24)
Самоокисление гнпохлоритпой системы и образование хло-
рата— сравнительно медленный процесс. Условия, при которых
разложение гипохлорита с образованием кислорода значительно
ускоряется, приводят к быстрому уменьшению концентрации тех
составляющих, окисление которых ведет к образованию хлората.
Скорость кислородного разложения а. х. значительно увеличи-
вается с повышением температуры [87], при наличии в растворах
легкоокпеляемых органических веществ и катализаторов [107]
В присутствии легкоокпеляемых веществ разложение а. х. сопро-
вождается шиеиенвиым ее окислением, образование хлората пол-
ностью подавляется. При наличии катализатора разложения а. х.
152
образование хлората не обнаруживается, значительно увеличи-
вается содержание кислорода в системе, избыток которого выде-
ляется нз раствора в виде газа. Если же в растворе а. х. нахо-
дятся трудноокисляемые органические соединения, то скорость
кислородного разложения а. х. увеличивается незначительно и
наблюдается образование хлоратов. Образующийся кислород при
этом расходуется на окисление [107].
При очистке воды активным хлором без катализаторов следует
рассматривать соотношение трех процессов: саморазложения,
окисления органических загрязнений и образования хлорирован-
ных продуктов.
Процесс саморазложения а. х. с образованием хлоратов проис-
ходит только в чистых растворах а. х. и в присутствии трудно-
окисляемых органических веществ. Самое интенсивное окисление
происходит в слабокислой, нейтральной и слабощелочной средах,
а в сильнокислой среде процесс сопровождается образованием
хлорпроизводных продуктов [107, 109].
В присутствии катализаторов могут восстанавливаться даже
хлораты и, вероятно, участвовать в окислительном процессе [114].
Поэтому можно предположить, что атомарный кислород получа-
ется каталитическим разложением а. х. во всем диапазоне pH,
в котором хлор находится в растворе в виде кислородных сое-
динений.
В щелочной среде скорость переноса кислорода в 50 раз
меньше, чем в нейтральной и слабокислой средах, что отражается
па процессе окисления [107]. Этим объясняются выводы ряда ав-
торов о том, что в г ппохлорптных растворах наибольший эффект
разрушения органических примесей наблюдается в слабокислой
среде. Так, например, В. М. Филиппов [109] связывает окисли-
тельные процессы с участием хлорноватистой кислоты и описы-
вает окисление красителей следующим выражением: органические
прнмеси (красители и полупродукты)+NaClO (НС1О)-»-про-
дукты окисления (карбоновые кислоты)+N2+Na2SO4-i-CO2 +
+ NaCl (HCI).
Приведенная формула объединяет и другие уравнения, из-
вестные в литературе и отражающие окисление органических ве-
ществ под действием а. х. при его разложении на атомарный
кислород и хлорнд-иопы. Атомарный кислород участвует в окис-
лительном процессе, а хлорнд-иои остается в растворе.
Ниже рассмотрен процесс электрохимической деструкции
с точки зрения положений, изложенных в данном разделе.
4.2.3.5. Окислительно-восстановительные процессы
при электролизе хлоридных растворов
В результате электродных процессов в прнанодном слое
всегда наблюдается кислая среда, а в прикатодном — щелочная,
поэтому в межэлектродном пространстве от анода к катоду pH
изменяется от кислого к щелочному, независимо от реакции нс-
153
ходиого раствора, т. е. в межэлектродном пространстве всегда
есть слон с нейтральным pH. Существование нейтральных слоев
к постоянная ионизация воды в электрохимических гипохлорит-
ных растворах создают благоприятные условия для окислительно-
восстановительных процессов, катализируемых ионами Н+ и ОН~.
Если учесть, что наибольшая скорость кислород-хлоридиого вос-
становления а. х. приходится на нейтральную среду, то отсюда
следует, что электролиз загрязненной воды имеет определенные
преимущества перед обычным хлорированием.
Существенным фактором, обеспечивающим высокие скорости
окисления в электролитических аппаратах, является многократное
участие хлорид-иона в цикле: сначала разряд иа аноде и гидро-
лиз с образованием а. х., затем восстановление а. х. с образова-
нием атомарного кислорода в хлорид-иона. Атомарный кислород
как наиболее сильный окислитель вовлекается в окисление при-
месей. Хлорид-пон вновь разряжается на аноде. Такой процесс
может продолжаться до тех пор, пока скорости образования а. х.
и его восстановления будут равны, а в растворе не будет нака-
пливаться а. х. С уменьшением концентрации веществ, катали-
зирующих восстановление а. х„ начинается постепенный рост
концентрации а. х., а скорость ею образования будет снижаться
вследствие уменьшения концентрации хлорпл-нонов в растворе.
Сущность явления поясним на примере электролиза модель-
ного хлоридного раствора красителя прямого черного 2С с ис-
пользованием анодов ОРТА и катодов из стали Ст. 3 (рис. 4.25)
[71] В течение некоторого промежутка времени от начала элек-
тролиза а. х. не обнаруживается (0—/). При этом время возрас-
тает прямо пропорционально концентрации органического загряз-
нения, т. е. прн двукратном увеличении концентрации красителя
оно возрастает в 2 раза. Затем концентрация а. х. растет почти
с такой же скоростью, что и в чистом растворе хлоридов, кривые
идут практически параллельно друг другу и имеют прямолиней-
ное очертание. В точках // кривые роста концентрации а. х. в рас-
творах красителей начинают отклоняться от прямой, для кривой
2 этот момент наступает при меиьшей Ся В дальнейшем вновь
наблюдается восстановление части а. х„ но меиее интенсивно, не-
жели в отрезок времени 0—1. В то же время в период времени
О—/ происходит резкое снижение ХПК (рис. 4.25,6). Затем ско-
рость окисления плавно уменьшается на определенном уровне.
Дальнейшая электрообработка, хотя и влечет рост концентрации
а. х. в растворе, но не сопровождается углублением степени окис-
ления органических компонентов.
Эти явления относительно ле1 ко проанализировать в свете
изложенных выше положений. В начальный период времени лег-
коокпеляемые органические примеси полностью восстанавливают
образующийся а. х., после чего начинается накопление окисли-
теля. Оставшиеся в воде трудноокисляемые органические соеди-
нения не могут восстанавливать а. х. вследствие недостаточного
окислительного шиенппа ы системы Когда концентрация а. х.
151
Рис. 4.25. Кинетика изменения концентрации остаточного активного хлора (и)
и эффекта очистки (6) растворов красителей ПЧ-2С с концентрацией поварен-
> ой соли 5 г/л при концентрации красителей
/ — 0.0; 2—100 мг/л; 3. У, 3* —200 мг/л; 4'. 4’— 400 мг/л; У, 4‘ эффект очистки по
АПК; У. 4"- эффект обесцвечивания
достигает необходимой величины для создания достаточною по-
тенциала, начинается окисление химически более стойких соеди-
ненна. Вероятно, чем глубже необходимая степень очистки, тем
требуется большая избыточная концентрация окислителя и, соот-
ветственно, остаточною а. х. Очевидно, за этим должна следо-
вать необходимость дехлорирования очищенной воды.
Таким образом, для эффективной очистки загрязненных вод
электрохимическим окислением требуется создать следующие ус-
ловия: достаточную концентрацию хлоридов, определенную ско-
рость протока в межэлектродном пространстве, необходимую про-
должительность обработки.
Большинство сточных вод имеют низкую концентрацию хло-
ридов и сбрасываются, как правило, в пресные поверхностные
водоемы. Поэтому чрезмерное увеличение дозы вводимой пова-
ренной соли вызовет изменение солевого состава поверхностного
источника н рост затрат на приобретение солп. С другой сто-
роны, уменьшение дозы хлоридов в обрабатываемой воде по-
вышает расход электроэнергии па обработку. Следовательно, при
реализации процесса электрохимической деструкции встает во-
прос о получении достаточного количества а. х. при минимальном
расходе поваренной соли и с наименьшими энергозатратами При
очистке воды от трудноокпеляемых органических загрязнении, как
было показано выше, необходимо создавать определенный избы-
ток а. х., что вызывает 1О11ол1штелы1ыс затраты как па получение
а. х„ так п па сю дехлорирование. Эти затраты можно уменьшить
при использовании катализаторов восстановления а. х. Различные
варианты использования катализаторов тля интенсификации про-
цесса электролиза рассмотрены в разделе электрокаталитической
очистки сточных вод (см. п. 4 4 2).
Процесс электрохимической деструкции органических загряз-
нений находит широкое применение в технологии очистки раз
тпчпых категории промышленных сточных вод. В книге [88] пред
Павловы многочисленные примеры практического использования
155
этого метода для очистки сточных вод в химической промышлен-
ности. Авторы отмечают, что электрохимическая очистка во мно-
гих случаях экономически более выгодна, чем другие методы обез-
вреживания промстоков. Так, затраты на электроокнелеиие фено-
лов в 2 раза меньше стоимости озонирования и в 5 раз меньше
адсорбционного метода
Институтом ВНИИ ВОДГЕО [18] разработана основанная на
анодном окислении и катодном восстановлении технология очистки
сточных вод от фенолов, роданидов, нитросоединсний, формаль-
дегида. метанола, азокрасителей, енмазпна, цианурхлорида, про-
изводных антрахинона, этиленгликоля, 2,4-Д-кислоты, перекисных
органических соединений, серосодержащих и прочих органических
загрязнений, присутствующих в сточных водах предприятий хими-
ческой, нефтехимической и других отраслей промышленности. От-
мечается, что при очистке сточных вод производства активных
азокрасителей достигается глубокое обесцвечивание стоков, зна-
чительное снижение содержания в них органических веществ, ре-
генерация едкой щелочи и получение газообразного хлора.
В настоящее время имеется положительный опыт эксплуата-
ции промышленных сооружений по электрохимической деструкции
органических загрязнений, содержащихся в сточных водах кра-
сильно-отделочных производств и предприятий бытовой химии
[48. 52].
Успешной реализации рассматриваемой технологии способст-
вует тщательный анализ кинетических закономерностей меха-
низма процесса очистки сточных вод с целью выявления рабо-
чих режимов эксплуатации сооружений, а также правильный
выбор электродных материалов и оптимальной конструкции элек-
тролизеров.
4.2.3.6. Конструкции аппаратов
для электрохимической деструкции органических загрязнений
Процесс электрохимической деструкции может осуществляться
в электролизерах с диафрагмой и без диафрагмы. Наличие диаф-
рагмы между электродами приводит к значительному повыше-
нию напряжения на аппарате и, следовательно, к увеличению
расхода электроэнергии. Следует иметь также в виду, что электро-
лизеры с диафрагмами представляют более сложную конструк-
цию; применяемые в промышленности материалы для разделе-
ния анодных и катодных процессов не обладают 100%-ной селек-
тивностью, что не позволяет полностью вести процесс в требуемых
режимах электрохимических реакций; диафрагмы в процессе
работы и «меняют свою структуру, а также забиваются продук-
тами электролиза или механическими примесями электролитов,
что вызывает необходимость их замены или регенерации. Приме-
нение диафрагменных электролизеров обусловливается в некото-
рых случаях специфическими свойствами содержащихся загряз-
нений. Так, очистку стоков от нитросоединений целесообразно
156
Рис. 4.26. Аппарат для электрохимической деструкции загрязнений
проводить в две стадии: на первой осуществляется восстановле-
ние нитропродуктов в катодной камере до амнносоединений, а на
второй — окисление полученных аминов в анодной камере до не-
токсичных продуктов.
Конструкции бездиафрагмепиых электроокислнтелей чрезвы-
чайно разнообразны. Ниже представлены наиболее совершенные,
на наш взгляд, аппараты для электрохимического окисления ор-
ганических загрязнении.
Электроокнслнтель [а. с. 812736 (СССР)] (рис. 4.26) разрабо
таи ГПИ-1 МЛП СССР и Госнпихпорнроектом на основании тех
пологическнх исследований, проведенных в ЛИСИ [45]. Аппарат
состоит из 16 электролизных ячеек, ограниченных вертикально
расположенными электродами, которые разделены изолирующими
прокладками 2, имеющими П-образную форму. Прокладки разме-
щены в нижней части аппарата, образуя закрытые с трех сторон
ячейки. К крайним электродам 6 н 10, выполняющим роль като-
дов, внизу приварены штуцеры для подачи исходной 8 и отвода
очищенной жидкости 7. К электроду 10 по соединяется также
переливная труба 9 с уровнемером Вверху к электродам прива
рены плавки с отверстиями для крепления крышки 4.
Аноды 11 п последующие катоды 12 выполнены из листа с со-
ответствующими отверстиями / для перепуска сточной жидкости.
Все электроды имеют группу отверстий в боковой части для пот
соединения шины, питающей электролизер током. Электроды и
прокладки собираются в единый пакет при помощи шпилек.
157
Для создания жесткости аппарата шпильки проходят через
специальную раму, представляющую собой сварную конструк-
цию П-обра той формы, выполненную из швеллера и приварен-
ной к нему пластины. Внизу аппарат имеет лапы 5 для крепле-
ния его к фундаменту.
Крышка снабжена фланцем для крепления к электролизеру
при помощи болтов. Наверху крышки предусмотрены два шту-
цера 3: один — для входа воздуха с целью разбавления электро-
литических газов, другой—для отвода разбавленных газов.
Материал катодов — конструкционная сталь Ст. 3, материал
анодов — титан ВТ1-0, покрытый двуокисью рутения ОРТА В ка-
честве анодов могут быть использованы ОКТА или ТДМА.
Работает аппарат следующим образом. Сточная вода через
штуцер 8 поступает в первую ячейку и движется справа налево,
затем через отверстия 1 в электроде попадает во вторую ячейку,
далее в третью и т. д. Таким образом получается зигзагообразный
ход обрабатываемой жидкости в горизонтальной плоскости. Вы-
ходит очищенная сточная вода из электролизера через штуцер 7.
Электролизер имеет следующие технические характеристики:
нагрузку по току — 3200 А, напряжение- 5,85 В. плотность то
ка—200 А/м2. межэлектродное расстояние — 7 мм. габаритные
размеры (длинахширинаХвысота) — 1400x625x1425 мм, рас-
ход сточной жидкости—10 м3/ч, скорость движения в межэлек-
тродном пространстве— 1600 м/ч.
С целью более полного использования окислительной способ-
ности электрогенерированных продуктов электролиза предложен
[а. с. 1033446 (СССР)] колонный аппарат (рис 4.27). имеющий
в верхней части электролизера доплонителыпю камеру с насад-
ками из колец Рашига. Электроды аппарата расположены гори-
зонтально и выполнены из просечного листа, нс растворимого при
анодной поляризации материала. Увеличенная зона контакта за
счет цилиндрической камеры 4 с насадкой 3 способствует более
полному растворению образующегося а. х., что уменьшает проскок
газообразного хлора и, следовательно, его потери. Горизонтально
расположенный набор анодов и катодов из просечного листа обес-
печивает равномерное распределение выделившихся про ivktob
электролиза и улучшает их массообмен с обрабатываемой жидко-
стью. Просечки в электродах выполнены под углом к их по
верхности При сборке аппарата электроды располагаются таким
образом, что просечки каждого последующего электрода направ-
ляются в противоположную сторону. При прохождении сточной
жидкости через такой пакет электродов происходит интенсивное
перемешивание за счет резкого изменения направления движения
потока.
Аппарат был испытан иа натурных сточных водах производ-
ства аминной соли 2,4 Д кислоты следующего состава, г/л
NaCI - 90.0, 2.1-Д кислоты — 2,0; хлорфеполы — 0,7; органический
углерод— 1.0. Электролиз осуществлялся прп токовой нагрузке
5.0 А, анодной плотности тока 750 А^м2. напряжении — 3,2 В.
15Я
Рис. 4.27. Колонный электроокислитель для
высококонцентрированных сточных вод
1.2 — патрубки соответственно для подачв воздуха
и отвода разбавленных газов, 3 — насадка: 1 —
к жтактная камера; 5 — изолирующие прокладки.
6 — катоды, 7 — аноды; 8 — соединительные шпильки
pH — 4—6, Т — 90—95 °C, количестве
катализатора (гидроксида кобаль-
та)—1,25 г/л в пересчете на ион ко-
бальта, расходе сточных вод —
0,6 л/ч. После очистки сточная вода
содержала 0,06 г/л органических при-
месей. Затраты электроэнергии при
этом составили 30 кВт-ч/мл.
Недостатками рассматриваемого
аппарата являются существенные за-
траты электроэнергии и безвозврат-
ные потери большого количества ка-
тализатора. Этот аппарат целесооб-
разно применять при очистке неболь-
ших количеств высококонцентрпро
ванных по органическим загрязне
пням промышленных сточных вод.
С целью исключения безвозвратных
потерь катализатора может быть ре-
комендован комбинированный спо-
соб обработки сточных вод с использованием гетерогенных гра
нулированпых катализаторов (см. и. 4.4.2).
Отмеченные в данном разделе значительные преимущества ме-
тода электрохимической деструкции загрязнений по сравнению
с ipynniH методами очистки приведут, по-видимому, в ближайшие
годы к его широкому применению для обезвреживания сточных
вод, содержащих трудпоокисляемые органические соединения. Ис
пользование процессов электроокнсленпя и восстановления с об-
разованием растворенных в воде или газообразных нетоксичных
или малотоксичных продуктов упрощает технологическую схему
очистки сточных вод, снижает эксплуатационные затраты и, как
уже указывалось, позволяет интенсифицировать процессы вслед-
ствие как большой химической активности электрогенерированных
окислителей и восстановителей в момент их образования, так и
возможности непосредственного редокс-превращепия загрязнений
па электродах. Важным достоинством метода электрохимической
деструкции является также то, что этот метод в большинстве слу-
чаев почти полностью снимает проблему осадков и реагентов.
4.2.4. Электрокристаллизация
Процесс электрокристаллнзацнн характеризуется возникнове-
нием твердой фазы па поверхности электродов или в объеме рас-
твора прп протекании электродных реакций. Обычно этот процесс
159
происходит при электролизе гомогенных систем па поверхности
электрода за счет разряда ионов Наиболее часто в практике во-
доочистки используется катодное восстановление ионов металлов,
в результате которого образуется кристаллическая твердая фаза.
Кроме того, в прикатодной зоне за счет подщелачивания рас-
твора формируются кристаллы СаСОз, CaSO<, Mg(OH)2, которые,
взаимодействуя с подложкой (электродом), образуют кристалли-
ческие осадки.
Рассмотрим образование металлического кристалла иа инерт-
ном катоде. При отсутствии поляризации раствор находится
в равновесии с электродом и повышение концентрации восстанав-
ливаемого иона приведет к образованию кристалла соли в рас-
творе в соответствии с законами химического равновесия. Для
появления зародыша кристалла на электроде необходимо, чтобы
произошло восстановление иона, т. е. электродный процесс.
Работа, которую надо совершить для образования кристалла
на поверхности, определяется по формуле
32a3V2 , пе.
Л -=---------, (4 25)
R*T* In (С. С,)2
где Vk — объем кристалла; С» и С, — концентрация соли соответственно в ис-
ходном н насыщенном растворе.
Из формулы (4.25) видно, что работа образования зародыша
уменьшается с ростом пересыщения Со/С,. При уменьшении ра-
боты образования зародыша повышается вероятность его возник-
новения:
<о Вехр[ — AI(RT)], (4.26)
где В — постоянная величина.
Как уже отмечалось, простое повышение концентрации ионов
в растворе приведет к образованию кристаллов соли в объеме.
Поэтому необходимо создать пересыщение по восстановленному
иону, что осуществляется прн сдвиге потенциала электрода в от-
рицательную сторону от равновесного. Если работа отклонения от
равновесия (работа пересыщения), рассчитанная иа 1 моль, равна
RTlgCo/Ca, то работа отклонения потенциала от равновесного
(работа катодной поляризации), рассчитанная па 1 г-иои, будет
равна A<pzF. Подставив эту величину в уравнения (4.25) и (4.26),
получим
А = 32o3V2 /[?F2 (Д<р)2] = B/(Aq>)2. (427)
Вероятность возникновения зародыша
со В ехр [ — В /(А<р)т]. (4.28)
Выражения (4 27) и (4.28) показывают, что рост поляриза-
ции (\<р) уменьшает работу образования зародыша и повышает
вероятность его возникновения.
Место на инертном катоде, в котором зарождается центр кри-
сталлизации, не может быть предугадано заранее. Активные ме-
160
Рис. 4.29. Схематические поляриза-
ционные кривые для совместного
восстановления металла (/) и во-
дорода (2)
Рис. 4.28. Схема роста двухмерного
зародыша
1—3 — направления роста зародышей кри-
сталлизации
ста, где образуются зародыши, характеризуются нарушениями
сплошности поверхности, искажениями кристаллической решетки
и другими дефектами.
Рост кристаллов происходит в соответствии со схемой
(рис. 4.28) сравнительно толстыми слоями, видимыми в микро-
скоп. Обычно новый слой возникает у вершины (угла) кристалла
и оттуда распространяется но поверхности примерно с постоянной
скоростью. Движение раствора может вызывать искривление
фронта роста слоев, что указывает па влияние изменения кон-
центрации раствора у фронта роста. Существенную роль на рост
кристаллов оказывает адсорбция посторонних примесей на по-
верхности электрода. Так, при перерывах в электроосаждении
возможна пассивация электрода посторонними примесями с по-
следующим нарушением образования слоя осадка при включении
тока. С учетом сложного состава природных и сточных вод пред-
ставляет интерес совместное восстановление катионов на катоде.
Прн совместном разряде нескольких видов ионов скорости вос-
становления каждого из них отвечает определенная плотность
тока [100], вследствие чего выход по току требуемой реакции оп-
ределяется как Вт=1</ь, где ь,—эффективная плотность тока на
электроде.
Так как в водных растворах при наличии катионов металла
непременно имеются и катионы водорода, то неизбежно должно
осуществляться нх совместное восстановление па катоде. Это мо-
жет происходить в том случае, если равновесный потенциал ме-
талла в данном растворе отрнщпельпес равновесного потенциала
водорода, а также если он положительнее, но катод достаточно
сильно поляризован (рис. 4.29). Выделение водорода может при-
вести к подщелачиванию раствора, образованию гидроксидов и
основных солен металлов, которые покрывают поверхность ка-
тода и препятствуют осаждению осадка.
6
Зака > М 2118
1Г>1
Рис. 4.30. Схематические по-
ляризационные кривые при
выделении двух металлов
/—металл I; 2 — металл II;
3 — суммарная поляризацион-
ная кривая
Рис 4.31. Извлечение металтов из сточных
вод по методу «Chenielec»
/ — электролизер; 2 — шевдоожиженный стой
стеклянных шариков; 3 — катод; 4 — сборный
резервуар; 5 — насос
При совместном разряде двух или нескольких металлов сум-
марные поляризационные кривые зависят от поляризационных
кривых каждого металла (рис. 4.30). В некоторых случаях, когда
на электроде образуется сплав двух металлов, суммарная поля-
ризационная кривая может не быть суммой поляризационных
кривых отдельных металлов.
Процесс электрокристаллнзацин широко применяется прн из-
влечении металлов из сточных вод гальванических цехов [102|.
Концентрированные медь-, никель-, ципкосодержащие электро-
литы направляют в электролизную бе «диафрагменную ванну, где
в качестве анода используют уголь или нержавеющую сталь,
а в качестве катода — металл, ноны которого извлекают из рас-
твора. Когда при электролизе толщина осаждаемого металла до-
стигает 0,1—0,3 мм, процесс прекращают н металл снимают
с электродов механическим или химическим способом. Модифици-
рованный метод «Химэлектро>, разработанный фирмой BEWT
(Англия), позволяет извлекать металлы из промывных вод в элек-
трохимическом аппарате с псевдоожиженным слоем из стеклян-
ных шариков диаметром 0,5—2,0 мм (рис. 4.31).
Промывные воды направляются в сборный резервуар и далее
насосом перекачиваются в электролизер, в котором снижение за-
™узкн ирон шодится восходящим потоком жидкости. Металлы
Cd, Ni, Zn, Ап и другие осаждаются иа катоде, который перио-
дически удаляется с целью его регенерации, осуществляемой
анодным растворением металла с поверхности катода в ваннах
гальванопокрытий. Расход электроэнергии па I т извлеченного
металла состанляет 6000—10 000, на перекачку жидкости — 1500—
10000 кВг. Размеры реактора — 0,5x0,6x0,75 м, площадь элек
тродов — 3,3 м2.
Утилизация благородных .металлов (Au, Pt, Pd и др.) из про-
мывных вод осуществляется методом электрокристаллнзацин на
If >2
волокнистых угольных электродах с развитой нонерхнос1ыо [12]
В качестве анода служит платинированный титан, в качестве
катода — графитовый сетчатый электрод, на котором осаждаются
благородные металлы. Аноды и катоды разделены ионообменной
диафрагмой. Анодное пространство заполняется раствором сер-
ной кислоты. После осаждения металлов катоды могут быть ре-
генерированы путем анодного растворения металла с поверхно-
сти, количество регенераций — 5—10. При сжигании катодного ма-
териала можно выделить чистый металл, свободный от примесей.
В оборотных системах водоснабжения промпредприятнй ис-
пользуется метод электрокристаллнзацин солей жесткости па
поверхности графитовых катодов при плотности тока 30—50 А/ма
с целью предотвращения накнпеобразования и отложений в тех-
нологическом оборудовании. Для интенсификации процесса и кри-
сталлизации солен в межэлектродном объеме размещается на-
садка из стекловолокна.
Болес интенсивно электрокрнсталлизацня солей жесткости про-
текает в катодном пространстве диафрагменного электрореактора
[а. с. 814881 (СССР)]. Умягчение природной воды осуществляется
в этом случае с использованием электрохимического корректиро-
вания pH, которое по сущности химических процессов, происхо-
дящих при удалении солей жесткости, во многом аналогично из-
весткованию. В то же время применение электрокорректировання
pH в сравнении с известкованием позволяет: исключить увеличе-
ние общего солесодержания умягчаемой воды, избежать повыше-
ния остаточной жесткости воды за счет ввода избыточного коли-
чества извести, производить одновременно стабилизационную об-
работку воды, отказаться от использования химических реагентов
п реагентного хозяйства.
Технологическая схема умягчения воды представлена на
рис. 1.32. Очищенная на фильтре вода, имеющая щелочную реак-
цию, смешивается с кислым анолитом, что обеспечивает стабили-
зационную обработку всего объема природной воды. Вода от
промывки фильтров после отделения взвеси на специальных со-
оружениях используется повторно для промынкн или на собствен-
ные нужды очистной станции. Схема внедрена в Кишиневском
НПО «Мнкропровод» с целью подготовки воды для промывных
операций гальванического производства. Основные параметры
умягчения воды с применением электрохимического корректиро-
вания pH н ее качественные показатели приведены ниже:
расход тока на обработку вою и катодных камерах,
Кл/л.................................... . . . . .
обидой расход тока, Кл/л . .
соотношение расходов католита н анолита .
плотность тока на электродах, А/м*................
напряжение электролиза, В ........................
расход электроэнергии. кВт-ч/м3
жесткость умягченной воды. мг-жв/л................
жесткость исходной воды. мгжв/л ..................
2WX1 321X1
IIXX) I31M)
(7 8) (3 2)
25 35
18—24
5.5—6,8
1.4 1.6
4.0 7,0
Рис. 4.32. Технологическая схема умягчения природной воды
/—элсктрокиррсктор pH; 2 — катодная камера; 3— анодная камера; 4 — кристаллизатор-
отстойннк; 5 — узел обработки промывной воды фильтра; 6 — фильтр; 7 — бак промыв-
ной воды
Аналогичная схема предложена институтом горного дела
СО АН СССР (76], в нее дополнительно включен осветли-
тель, обеспечивающий предварительное извлечение солей кальция
и магния перед поступлением на фильтр. Умягчение воды проис-
ходит в диафрагменном электролизере (рис. 4.33), представляю-
щем собой корпус 12, разделенный с помощью перегородок 13 и
8 на три секции: коллектор для ввода воды 3, рабочее простран-
ство 4 и пеносборппк 5. В рабочем пространстве вертикально
размещены титановые катоды 14 и анодные камеры 15; послед-
ние изготовлены из кислотостойкого диэлектрика в виде сборной
рамки, в которую помещается графитовый анод. Разделение
анода и катода осуществляется с помощью диафрагмы 16, изго-
товленной из ткани ТЛФ-4. Исходная вода подается через патру-
бок / в коллектор 3, из которого с помощью отражателя 10 че-
pei щель И поступает в рабочее пространство 4. Для создания
восходящего потока в рабочем пространстве часть волы в элек-
тролизер поступает через нижний патрубок 9. Для обеспечения
возможности протекания электрохимического процесса исходная
вода (30 - 40% общего расхода) подается через патрубок 2
в анодную камеру 15. Умягченная вода через окно 7 поступает
в сборный коллектор 6 и затем отводится из аппарата.
Производительность установки 5 м3/ч, скорость движения воды
составляет: в анодных камерах — 0,31—0,42 м/мнн, в зазоре ме-
жду диафрагмой и катодом—0,12—0 8 м/мин. В процессе опыт-
но-промышленных испытаний установлено, что из воды с жест-
костью 14,5 16.7 мг-экв/л анолит получается с жесткостью 1,1 —
1,5 мг-экн/л и pH 2,5ч-3. а католит — с жесткостью 0.6 -
161
Рнс. 4.33. Схема диафрагменного электролизера для умягчения волы
I мг-экв/л и pH = 10,54-11,0. Жесткость смеси анолита и католита
составляла 0,8—1,2 мг-экв/л, затраты электроэнергии —
3,8 кВт-ч/м9! Анализ осадка показал, что в нем содержится
СаСО3, М«{(ОН)2 и частично Fe2O3-H2O. Применение этой уста-
новки позволило увеличить рабочий цикл Na-катиопптовых
фильтров.
Рассматриваемая технология позволяет проводить кристал-
лизацию солей жесткости без введения реагентов, что упрощает
аппаратурное оформление процесса, а также уменьшает объемы
образующихся твердых отходов. Ранее упоминаемое осаждение
металлов на электродах обеспечивает получение чистого про-
екта и использование его в качестве сырья при производстве
различных технических изделии. Однако скорость процессовэлек-
ipoxiiMiniecKoii кристаллизации относительно низка, что сущест-
венным образом сказывается на расходе электроэнергии при
электролизе.
Специфические особенности данного метода требуют тщатель-
ного его изучения и всесторонних производственных испытаний.
Кроме того, необходима разработка специальных конструкций бо-
iee совершенных устройств для выделения кристаллизующихся
солей из очищенной воды.
4.3. МЕТОДЫ РАЗДЕЛЕНИЯ ВЕЩЕСТВ
4.3.1. Электрофлотация
При электрофлотации электролитически полученные газовые
пузырьки, всплывая в объеме жидкости, взаимодействуют с ча-
стицами загрязнений, в результате чего происходит их взаимное
165
слипание, обусловленное уменьшением поверхностной энергии
флотируемой частицы н пузырька газа на границе раздела фаз
«жидкость—газ». Плотность образующихся агрегатов меньше
плотности воды, что обусловливает их транспорт на поверхность
жидкости и накопление там флотошлама, который периодически
удаляется из сооружения.
Элсктрофлотация широко исполыуется в практике очистки за-
грязненных жидкостей, наибольшее применение она находит
в технологии обработки сточных вод» Еще в 1951 г. процесс элек-
трофлотацни использовали для очистки сточных вод в г. Горь-
ком. Исследованиями, проведенными Н. В. Политковской, было
установлено, что по эффективности этот метол равноценен обра-
ботке городских сточных вод в аэротенках па неполную очистку,
экономичнее и проще в эксплуатации, чем биохимические способы
аэрации илн бнофнльтрации.
В настоящее время на основании глубоких теоретических раз-
работок процесса флотации, выполненных П. Л. Ребнндером,
Л. Н. Фрумкиным и В И. Классеном, а также иа основе извест-
ных экспериментальных работ по электрофлотации [1, 58, 60, 61
и др.] способ получил развитие и рекомендуется к широкому
внедрению для очистки сточных вод в различных отраслях про-
мышленности.
Физико-химические процессы, имеющие место в электрофлота-
ционпых аппаратах очистки воды, включают в себя электроли-
тическую генерацию газовых пузырьков, адгезию газовых пузырь-
ков и частиц загрязнений, транспортирование образовавшихся
агрегатов «пузырек газа — частица загрязнений» на поверхность
обрабатываемой жидкости. ]
Важной и часто определяющей стадией электрофлота иконного
процесса является адгезия газовых пузырьков и частиц загряз-
нений, которая происходит па молекулярном уровне. Сближение
пузыпька и частицы осуществляется под действием внешних гид-
родинамических сил, а когда расстояние между ними уменьшается
до 10 6 мм, начинают действовать молекулярные силы. При этом
акт прилипания частицы к пузырьку сопровождается резким
уменьшением поверхностной энергии пограничных слоев и воз-
никновением сил, стремящихся уменьшить поверхность смачи-
вания.
Процесс флотации протекает тем успешнее, чем больше общая
поверхность газовых пузырьков и чем больше площадь контакта
их с флотируемыми частицами. В системах с одинаковой сте-
пенью । азонаполнепня жидкости суммарная поверхность более
мелких пузырьков будет больше, а расстояние между частицами
н пузырьками меньше, что повышает вероятность их столкно-
вения. J
Основную роль в процессе электрофлотации выполняют пу-
зырьки Boiopoia. выделяющиеся на катоде. Механизм и кинетика
катодного выделения водорода подробно рассматривались
в п. 3.3.6 1 Было показано, что размер и интенсивность образо-
1ы>
вания пузырьков водорода зависят от состава и температуры
электролита, поверхностного натяжения на границе раздела фаз
«электрод— раствор», материала электродов, их формы и шеро-
ховатости поверхности, плотности тока. Изменяя перечисленные
параметры, можно регулировать размер и интенсивность выделе-
ния пузырьков газов при электролизе, т. е. корректировать в зави-
симости от характера загрязнений технологический процесс
очистки воды.
Размер пузырьков газа, выделяющихся на электродах, зависит
от соотношения сил, воздействующих на пузырьки в момент их
образования и роста: поверхностного натяжения и гидростатиче-
ской. Первая тем более прочно удерживает пузырьки на электроде,
чем больше периметр, по которому пузырек крепится к поверх-
ности, вторая пропорциональна в основном объему пузырька.
Фундаментальные исследования, проведенные Б. Н. Кабано-
вым и А. Н. Фрумкиным, показали, что размеры и форму пузырь-
ков, выделяющихся на электродах, можно однозначно определять
краевым углом смачивания, характеризующим величину поверх-
ностного натяжения на трехфазпой границе «электрод — рас-
твор— газ» и определяющим условие равновесия поверхностных
сил взаимодействующих фаз. Уравнение, описывающее условие
равновесия пузырька на поверхности электрода, имеет следую-
щий вид:
4° sin G = V„gp0 4- nd„/[4 (2o/rn — hngp0)], (4.29)
где dK, hn и ra—соответственно диаметр основания, высота и радиус кри-
визны пузырька.
Отрыв пузырька от поверхности электрода происходит тогда,
когда сила гидростатического поднятия превышает удерживаю-
щую силу поверхностного натяжения. Следовательно, равновесие
пузырька определяется действием только капиллярных сил и сил
тяжести.
Величина поверхностного натяжения зависит от потенциала
электрода и корректируется максимумом па электрокапиллярпой
кривой вблизи точки нулевого заряда электрода. Размер пузырь-
ков в момент их отрыва от электрода зависит от величины крае-
вого угла (рис. 4.34). Кроме того, на кинетику роста и отрыва
пузырьков водорода оказывает влияние электрическое поле. За
счет избытка ионов ОН в прпкатодном слое пузырьки водорода
приобретают отрицательный заряд, что обусловливает их оттал-
кивание от поверхности электрода. В местах значительных высту-
пов па поверхности электрода наблюдается неравномерность
»1сктрнческого поля и большая его напряженность, что обеспе-
чивает быстрый рост и отрыв мелких пузырьков. Чем выше на-
пряженность поля и величина заряда, тем больше пондеромотор-
ные силы, отрывающие пузырек от электрода, и тем мельче nv-
ojpi.Kii. Этим объясняется также влияние плотности тока па
1к '1ичнну пузырьков.
1Г.7
Рис. 4.34. Зависимость величины пу-
зырьков газа от величины краевого
угла смачивания в момент отрыва
на электродах
/ — на Ag; 2 — на Pt; 3 — иа Hg
Рнс. 4.35. Эмпирическое рас-
пределение размеров выде-
ляющихся пузырьков водо-
рода по величине их диа-
метров в за исимости ог
кривизны поверхности про-
волочного катода
Влияние поверхности электрода и его кривизны па количество
и размер образующихся пузырьков электролитического водорода
иа катодах из проволоки изучено Б. М. Матовым [60]. Установ-
лено, что величина отрывного диаметра пузырька оказывает су-
щественное влияние на эффективность электрофлотационпого
способа очистки жидкостей. Выявлено также, что при повышении
степени дисперсности пузырьков, т. е. с уменьшением d0, растет
эффективность электрофлотации взвешенных частиц органиче-
ского происхождения. Степень дисперсности пузырьков зависит,
в свою очередь, от параметров проволочного катода: материала
и кривизны поверхности, величины, обратной его радиусу. Экспе
римептальпые данные но измерению tfo и эмпирическое их рас-
пределение представлены на рис. 4.35. Из графика видно, что
с увеличением криви шы поверхности электрода уменьшается не
только среднее значение диаметров пузырьков, но и дисперсия эм
лирического распределения, т. е. сокращается диапазон разброса
пузырьков по величине его диаметра.
Газонаполнепие обрабатываемой жидкости как известно,
влияет па эффективность процесса флотации. Распределение объ-
ема образующегося газа на катодах различного диаметра суще-
ственным образом зависит от кри-
визны поверхности электродов (рис.
4.36). Чем меньше диаметр проволоч-
ного катода, тем ниже перенапряже-
ние выделения водорода, мельче об-
разующиеся пузырьки и выше газо-
наполнение системы.
Рис. 4.36. Распределение относительного объ-
ема газа Vr по величине диаметра электро-
геиерируемых пузырьков при диаметре ка-
тодов
/ — 0.2 мм; 2- 0.4 мм; 3 1.5 мм, 4 — 2.0 мм; 5 —
10 мм
Таблиц 4.2. Влияние размеров ячейки
сеткн (днаметра стержней) иа эффективность
удаления ПАВ при исходной’концентрации
100 мг/л, iK = 2 А/дм1, /э = 20 мин, pH =
= 7,0
Диаметр стержня, мм Обработанный раствор Эффект очистки, %
Спав. мг/л ₽Н
1 40,8 8,15 59,2
2 43,4 780 56,6
3 44 6 7,60 554
4 46,0 7,55 54,0
катодной
ячеек
размеров
(431)
При электрофлотации
целесообразно применять
электроды с развитой по-
верхностью в виде метал-
лических сеток, пористых
металлокерамических ма-
териалов, насадок пли
с перфорацией токопрово-
дящей основы. Так, приме-
нение сеток из нержавею-
щей стали оказалось более
эффективным по сравнению
с графитовыми вертикально
расположенными пластин-
чатыми катодами прнэлек-
трофлотацин сточных вод,
содержащих ПАВ [52]. Уменьшение
сетки, соответствующих диаметру стержней, увеличивает степень
очистки сточных вод (табл. 4.2).
Общее количество газовых пузырьков, выделяющихся иа ка-
тоде, можно определить по абсолютному количеству электриче-
ства, прошедшего через систему. Тогда в соответствии с первым
законом Фарадея (1.1) масса образующихся пузырьков водорода
Ат = А/;, А,//э. (4.30)
Выразим теперь Аш через плотность р„ и средний объем од-
ного пузырька Ун или через его средний радиус г„:
Аш=рпУЛп = 4/3(л?прп^).
По (ставив в полученное уравнение значение Am из (4.30) и
решив его относительно N„, получим
Лп = 3/4[Аэ//,/(л?прп)].
Можно определить число пузырьков со средним радиусом г,
постоянно находящихся в единице объема жидкости, используя
соотношение n=Nn/HS или с учетом (4 32):
«п = 3/4[Аэ/Г,/(л^РпЯф5ф)],
где п — концентрация пузырьков водорода в электрофлотацнонной установке;
//* — высота слоя обрабатываемой жидкости над электродным блоком; S* —
н'ютадь поперечного сечения установки в горизонтальной плоскости
Так как скорость электрохимической реакции принято отно-
сить к единице рабочей площади электрода, которая равна в дан-
ном случае проекции катода на горизонтальную площадь камеры
>лектрофлотации, и определять ее как плотность тока, то можно
тлпнеать:
(4 32)
(433)
iK = //5ф, откуда f = /к5ф.
(4 34)
164
Тогда, подставив в уравнение (4.33) /, выраженную через
плотность катодного тока, получим
"п = 3/4р,ГЛ/(лг^пНф)]. (4.35)
Все величины. входящие в эту формулу, кроме /к, являются
посточ”пыми, так как рассматриваемый процесс осуществляется
в одном и том же аппарате и при заданной продолжительности
обработки. Следовательно, (4.35) можно переписать в таком виде
лп=Аф/к. (4 36)
где Аф — постоянная, значение которой включает в себя все неизменяющиеся
параметры процесса, т. е.
Аф=3'4[А,Г,/(№пРп«ф)]- И .37)
Формула (4.36) показывает, что концентрация пузырьков во-
дорода в электрофлотаторе зависит только от катодной плотно-
сти тока. Таким образом, изменяя плотность тока, можно регу-
лировать величину лп, а следовательно, степень насыщения
жидкости пузырьками водорода, т. е. корректировать процесс
электрофлотациоппой очистки загрязненных жидкостей.
Для скорости подъема газовых пузырьков, получаемых вэлек-
трофлотацнонных аппаратах, Б. М Матов рекомендует исполь-
зовать следующие формулы, хорошо согласующиеся с экспери-
ментальными данными:
при диаметре пузырьков do>0,l мм
»п = g'nPjWy. (4 38)
при de <0,1 мм
«п=₽'-пРп/(9п) + ЗСгп, (4 39)
где С -постоянная, определяемая экспериментальным путем н зависящая от
температуры н состава сточных вод: для масложнросодержащнх сточных вод
С=2/3 г/(с-см»),
Газонасыщение жидкостей над электродным блоком опреде-
ляется по выражению
Г = Л»1'к(э/(Рп7/ф) А э*к/(Рпс'п). (4.40)
Степень насыщения жидкости пузырьками водорода прямо про-
порциональна катодной плотности тока и обратно пропорцио-
нальна плотности пузырьков водорода и скорости их подъема.
Стоящие в знаменателе величины, так же как и электрохимиче-
ский эквивалент водорода А3, являются постоянными для обра-
батываемой той пли иной системы и характеризуют ее физико-
механические свойства. Однако при плотности тока, превышаю-
щей оптимальное значение, эффект флотации может снижаться.
Это объясняется нарушением оптимального гидродинамического
режима всплывания флотокомплексов при избытке газовых пу-
зырьков, разрушением пенного слоя и «вторичным загрязнением»
очищенной воды.
170
Оптимальная плотность тока зависит от физико-химических
свойств системы и обычно при очистке сточных вод от нераство-
римых примесей ие превышает 3 А/дм2.
Б. М. Матов, используя методы подобия, вывел аналитическую
зависимость концентрации пузырьков в единице объема жидкости
от ряда параметров [60]:
Сн2 = /Ро£п^ (4.41)
\ Рг «₽Z J
где Ьф — произведение электрохимического эквивалента электролитического
<аза на выход по току.
Постоянной плотности тока соответствует оптимальная высота
слоя обрабатываемой жидкости Но. Если в электрофлотационной
установке жидкость обрабатывается в слое, высота которого
меньше Но, то соответственно возрастает удельный расход элек-
троэнергии. Увеличение высоты слоя сверх Но не влияет на удель-
ный расход электроэнергии, а приводит к тому, что в установке
возникает дополнительный объем, расположенный между пеной
и уровнем Но. Этот объем ие может рационально использоваться,
так как прилипание газовых пузырьков к частицам происходит
главным образом в слое жидкости высотой Но. Таким образом,
общая рабочая высота электрофлотационной установки должна
определяться как сумма двух величин: оптимальной высоты слоя
обрабатываемой жидкости Но и высоты слоя пены Л.
Изменение концентрации нерастворимых частиц по высоте ра-
бочей камеры электрофлотатора описывается дифференциальным
уравнением [60]
дСч С||П<> п д1Сч д*Сч .
(442>
где Сч — концентрация свободных нерастворимых частиц, к которым еще не
прилипли пузырьки газа; /— отношение среднего объема одной частицы
к среднему объему пузырьков; ло п — концентрация пузырьков, соответствую-
щая оптимальной плотности тока; р и /?«- коэффициенты пропорциональ-
ности.
Уравнение показывает распределение нерастворимых частиц
по высоте //„ в зависимости от времени, которое при введении не-
которых допущений с помощью метода разделения переменных
описывает процесс электрофлотацпи в следующем виде:
Сч - С.ехр ( — R, —-— cos —-— ft (4.43)
I 4Я2 J 2//0
Скорость электрофлотации в значительной степени зависит от
температуры обрабатываемой жидкости. Повышение температуры
способствует уменьшению перенапряжения водорода примерно на
2 3 мВ па каждый градус, с повышением температуры от 20 до
/О КО °C оно снижается для большинства металлов на 30—10 %.
При этом уменьшается вязкость жидкости и поверхностное натя-
жение на границе фаз, что интенсифицирует процесс разделения
171
Уравнение регрессии для эффекта обезжиривания сточных вод
мясокомбинатов в зависимости от температуры обрабатываемой
жидкости в пределах 10—60 °C [60] имеет вид:
Э = 3,2104 (Г/Го)1-2'6 г“|,6762Г/го, (4.44)
где То — оптимальная температура для электрофлотацнп жиров.
При То=30°С для эмульсин из свиного и топленого жиров
зависимость (4.44) имеет следующий вид;
Э = — 0,037’ + 2,457 4- 48. (4.45)
Аналогичные данные получены также в ряде других работ
[37, 120], выполненных под руководством Г. В. Иванова в ЛИСИ.
Электрофлотацпя находит применение для очистки сточных
вод нефтепромыслов, нефтебаз, нефтеперерабатывающих заводов
[105], кожевенных заводов [55], меховых фабрик [55, 61], целлю-
лозно-бумажных [88] и электрохимических [18] производств, тек-
стильных [1, 16, 24], пищевых [37, 58, 60, 79] предприятий и дру-
гих, а также при разделении и уплотнении активного ила после
аэротенков на биологических очистных сооружениях [60, 123]. Эф-
фекты очистки могут составлять: по нефтепродуктам — до 90%,
по взвешенным веществам — до 70%, по жирам — 80%; детер-
генты могут быть удалены на 60—70 %.
Перспективным направлением является ионная электрофлота-
цня прн очистке сточных вод и извлечение как металлов из раз-
бавленных растворов, так и различных ценных веществ из мор-
ской воды. При соответствующих условиях возможно разделять
ионы различных элементов, имеющие одинаковые по величине и
знаку заряды.
Продолжительность электрофлотациопнон обработки сточных
вод может варьироваться в зависимости от вида загрязнений
в достаточно широких пределах (от нескольких минут до 30—
40 мни), при этом и^вл<1 кВт-ч/м3; /7о = 0,5-т-1,5 м, /8=0,5ч-
4-2,0 см, до 3 А/дм2.
Конструкции аппаратов для электрофлотацпонпой очистки
достаточно просты. Электроды могут выполняться в виде пластин,
располагаемых па дне аппарата горизонтально или вертикально,
занимая практически всю площадь днища (рис. 4.37) с целью
предотвращения циркуляционных потоков, препятствующих фло-
тированию загрязнений.
Недостажами такой конструкции являются налипание па элек-
тродах различных эмульсий, жиров, тяжелых минеральных ча-
стиц и свя <анный с этим непроизводительный расход электроэнер-
гии, а также необходимость периодического отключения уста-
новки для очистки электродов или даже замены электродного
блока.
Для предо!вращения образования отложений предлагаются
различные модификации схем размещения электродов в аппарате.
Одна из них нредс! анлена на рис. 4.38. Аноды 3 из графита или
172
Рис 4.37 Электрофлотатор с горизон-
тальными электродами
I — карман для сбора пени. 3— псногои
мое устройство; J — полупогружная перего-
родка; 1. 5 —отводящий и подающий тру-
бопроводы; 6 — катод; 7 — анод
другого электролитически стойкого материала выполнены в виде
отдельных трехгранных призм, расположенных в шахматном по-
рядке, а катоды 5—в виде отдельных проволочных сеток, изогну-
тых под углом н расположенных над призмами анодов парал-
лельно нх граням. Для подвода электрического тока в призмы
запрессованы токопроводящие втулки 4. В верхней части аппа-
рата расположен наклонный желоб 1 для сбора и отвода ценного
конденсата, в котором помещен трубопровод острого пара 2 для
гашения пены, а в нижней части — конусное днище для сбора
осадка.
Сотрудниками Кишиневского политехнического института раз-
работан оптимальный вариант секционированной рабочей емко-
сти электрофлотатора (рнс. 4.39).
Из рабочей секции этого аппарата жидкость переходит в до-
полнительную безэлектродную секцию, которая предназначена для
того, чтобы жидкость полностью освободить от пузырьков газа.
В этой секции удаляются взвешенные частицы с прилипшими пу-
зырьками, которые не всплыли в рабочей зоне.
Такне электрофлотаторы изготовляет опытно-ремонтный меха-
нический завод Мпнмясмолпрома МССР Аппараты предназначены
для обработки сточных вод мясокомбинатов с целью их обезжи-
ривания.
I’m I 1.4 Злектрофлотатор с комбинированным расположением электродов
173
Рис 4 39 Элсктрофлотатор с сек-
ционированием рабочей емкости
I. 2. 4. S, 6 — соответственно приемная
рабочая, безэлектродная. промежуточ
пая н отводящая секции; 3 — слой пе-
ны; 7 - выпуск осадка и опорожнен не
Рис. 4 40 Электросату-
ратор
/ вантуз; 2 пидвояя
щий штуцер; 3 - манометр.
4 — анод; 5 катод; 6 —
отводящий штуцер. 7 — по
репускные окна; 0 тур
бу inзатор
Союзводокапалпроектом разработаны электрофлотаторы про-
изводительностью 50—100 мл/ч [105], которые позволяю) в отра-
ботанных моющих растворах нефтебаз снижать копнен)рацию
нефтепродуктов с 45—50 г/л до 1 мг/л и механических примесей —
с 8—9 г/л до 350 мг/л.
Метод электрофлотацин имеет ряд существенных преимуществ
по сравнению с другими способами флотации сточных вод: про-
стота изготовления аппаратов и несложность их обслуживания;
возможность регулирования степени очистки жидкое)и в зависи-
мости от фазово-дисперсного состояния загрязнений путем изме-
нения только одного параметра (плотности тока) в технологиче-
ском процессе; выокая степень дисперсности газовых пузырьков,
обеспечивающая эффективность прилипания к ним нерастворимых
примесей; отсутствие вращающихся частей в рабочей зоне аппа-
ратов, гарантирующее надежность их работы и исключающее пе-
ремешивание обрабатываемой жидкости и измельчение содержа
щпхея в пен нзвешенпых частиц; дополнительная мннералп тация
органических загря шепни с одновременным обеззараживанием
сточных вод за счет образующихся на аноде проломов электро-
лиза— атомарного кислорода и активного хлора.
Однако приме псине этого меюда связано с необходимостью
предварительной очистки сточных вод от грубодпсперспых загряз-
171
пенни; в некоторых случаях требуется также и очистка поверхно-
сти электродов н межэлектродиого пространства от механических
примесей и замасливающих веществ. Кроме того, электрофлота-
цпя не всегда обеспечивает требуемую степень очистки сточных
вод, что вызывает необходимость интенсификации процесса путем
дополнительного применения коагулянтов или насыщения обраба-
шваемой жидкости газами в напорных электролитических сату-
раторах./Последнее является более прогрессивным решением, для
реализации которого разработаны различные конструкции аппа-
ратов. Одна из них, предложенная ГИСИ [70], показана на
рис. 4.40.
В электросатураторе насыщение жидкости газами осуществля-
ется путем растворения в ней электролитически генерируемых па
аноде и катоде газов. В качестве анодов применяются ОРТА, ка-
юдами служит нержавеющая сталь. Оптимальная анодная плот-
ность тока 5 А/дм2, напряжение 9—10 В.
Установки по напорной флотации с использованием электро-
литического способа насыщения жидкости газами должны вклю-
чать последовательно расположенные и соединенные между собой
трубопроводами усреднитель-накопитель, насосы, электролитиче-
ский напорный сатуратор и камеру флотации. Из приемного ре-
«ервуара усреднителя насосом под давлением 0,3—0,5 МПа ис-
ходная вода подается в электросатуратор,где в течение 15—20 с
происходит насыщение жидкости электролитическими газами. Вы-
сокая степень массообмеиа жидкости и газа достигается за счет
интенсивной турбулизации и циркуляции газожидкостного по-
тока через отверстия, выполненные в нижней части электродов,
и рассеивания потока конусным турбулизатором.
Из электросатуратора насыщенная газами жидкость дроссе-
лируется во флотационную камеру, где за счет снижения давле-
ния до атмосферного происходит выделение растворенных в воде
газов п флотирование загрязнений в поверхностный слой жидко-
сти, откуда пенный продукт направляется на дальнейшую обра-
ботку.
Применение в технологии флотационной очистки напорного
«лектросатуратора позволяет повысить степень извлечения неф-
1спродуктов и взвешенных веществ, обеспечивая устойчивую и
надежную работу всей установки в целом.
4.3.2. Электродиализ *
Разделение и концентрирование ионных примесей в воде осу-
И1гс1вляется методом электродиализа. Сущность метода заключа-
г1ся в использовании направленного движения попов в растворе
в гоответсгвпн со знаками их зарядов под действием разности
пок-пциалов, приложенной к электродам.
М.<г<|Н1ал подготовлен совместно с проф. В. Д. Дмитриевым.
17'.
в)
Рис. 4.41. Схемы процессов электро-
диализа раствора NazSOi
а — без мембран; б. в — с использова-
нием соответственно одной н двух мем-
бран
Рис. 4.42. Схема изменения концен-
трации растворов в ячейках трехка-
мерного аппарата для электрохими-
ческого обессолиаания воды
Принцип метода можно наглядно пояснить па примере элект-
ролиза раствора Na2SO* в электрохимических ячейках с приме-
нением и без применения мембран (рис. 4.41).
При пропускании электрического тока через электролит, не
разделенный мембранами, сконцентрированные у электродов ноны
Na+ и SO*- диффундируют в глубь раствора, что приводит к вы-
равниванию их концентрации по всему объему. Введение одной
мембраны позволяет разделить ионы Na+ и SO*- по камерам
с получением кислоты и щелочи.
Разделение аппарата на три отделения с помощью двух се-
лективных мембран, из которых два крайних являются электрод-
ными камерами, концентрирующими соответствующие попы, поз-
воляет в среднем отделении получать очищенную воду. Таким об-
разом, прп обессоливай и и воды процесс электродиализа можно
осуществляв, в аппарате с числом отделений не менее трех.
Процесс электродиализа объединяет явления электролиза,
включающего разряд ионов и их транспорт к поверхности элект-
родов, диализа и осмоса.
Электролиз. В растворе NaCI электрические заряды от катода
к аноду переносятся ионами ОП и С1_, а от анода к катоду —
попами Н в Na'. При прохождении II7 электричества на катоде
176
выделяется 1 г-экв Н2, на аноде — 1 г-экв С12. Ионы Na4 и Cl-
уходят из среднего отделения в электродные камеры, а ноны Н*
и ОН~ поступают соответственно из анолита и католита в среднее
отделение, что приводит к опреснению воды в этом отделении.
Наличие мембран влияет на числа переноса ионов по сравне-
нию с их значениями в свободном объеме раствора. Зная числа
переноса соответствующих ионов, можно определить изменение
концентрации растворов в трех пространствах электролизера.
На рис. 4.42 представлена схема электролиза раствора NaCI
в трехкамерпом аппарате с неактивными мембранами; стрелками
показано число грамм-эквивалентов перенесенных ионов при про-
хождения через рассматриваемую систему 1F электричества.
Диализ. Через мембраны может происходить диффузия как
электролита из среднего отделения в электродные камеры, так и
кислоты н щелочи в обратном направлении. Количество вещества,
днффундируемое в единицу времени, зависит, как известно, от
коэффициента диффузии и разности концентраций по граням мем-
браны.
Кроме того, по мере увеличения щелочности и кислотности
в электродных камерах в процессе переноса начинают в бблыней
степени принимать участие Н+ и ОН”-иоиы, образующие в сред-
нем отделении воду. Это приводит к замедлению переноса ионов
соли. Если в среднее отделение не было обратного поступления
попов, то 1 Кл прошедшего электричества позволит удалить из
воды 1 г-экв электролита (рис. 4.42). В практических условиях
часть тока расходуется на обратный перенос ионов, а также по-
вторный вывод этих ионов из среднего отделения, что снижает
выход по току.
При высоких концентрациях солей в трехкамерном электро-
диализаторе с инертными мембранами максимальный выход по
току ие превышает 20 %. Повышение эффективности обессолива-
ния воды может быть достигнуто за счет высоких иопоселектив-
пости и проводимости мембран, малой скорости свободной
диффузии, низкой осмотической проницаемости мембран и т. п. Та-
кими качествами обладают ионообменные мембраны с фиксиро-
ванными зарядами: мембраны, имеющие отрицательный заряд и
проницаемые для катионов (катиопитовые), и мембраны, имею-
щие положительный заряд и проницаемые для анионов (аниони-
говые).
Ионитовые мембраны изготавливаются из органических ионо-
обменных смол без основы и на основе капроновых, нейлоновых и
ipvnix тканей. Ионообменные смолы (катиопитовые и аинопнто-
пые) представляют собой практически нерастворимые в воде соли,
кислоты или основания, активные группы которых способны к об-
менным реакциям в растворах.
В зависимости от способа получения и строения ионообменные
мембраны делят на гомогенные, гетерогенные и пропиточные. Го-
miпенные ионитовые мембраны, являющиеся наиболее совершен-
ными по своей обменной способности, достаточно широко приме-
177
Рис. 4.43. Принцип электрохимического метола опреснения волы
а. 6 — трехкамерная ванна соответственно с неактивными и селективными мембранами;
в — многокамерная ванна с селективными мембранами; / — анод; 2 — катод; 3. / — соот-
ветственно анноннтовая и катионптовая мембраны; В — вода; Р - рассол
няются при электродиализе. По способу получения они делятся
на четыре группы: поликопденсациоиные, нолнмеризацнонные, эк-
ипированные и нленкообразующие.
Гетерогенные ионитовые мембраны нзготавливаюк’я прессован-
ными и пропиточными; последние являются наиболее простыми
в технологическом отношении, обладают достаточно высокой се-
лективностью и электропроводностью.
Особым типом мембран, выпускаемых нашей промышлен-
ностью, являются биполярные диафрагмы, на катионитовой сто-
роне которых используются мембраны МК-40, а на анпопитовой —
МЛ-40 или МА-41 Л. Отдельные характеристики ионитовых мем-
бран, выпускаемых отечественной промышленностью, приведены
в работе [33].
Стоимость выпускаемых мембран как в СССР, так и за рубе-
жом пока еще очень высокая. Например, I м2 гетерогенных мем-
бран фирмы «Рум п Хааз» (США) стоит 10 руб., а гомогенных
фирмы «Асахи Касей» (Япония) —до 30 руб.
На рис. 1.43 показан принцип работы аппаратов обессоливания,
с различными ишамп мембран. При применении ионообменных
мембран Kai попы под действием постоянного тока, двигаясь к ка-
тоду, проник.и срез катноинтовые мембраны, но задержива-
ются а11По11И1овымн, а анноппты. двигаясь в направлении анода,
проходят через аппинитовые мембраны, но задерживаются катио-
питовыми. Благодаря селективной ионной проводимости, а также
высокому coiipoi явлению диффузии электрохимически активных
17К
мембран применение их позволяет повысить коэффициент выхода
по току до 85—95 %.
Электроосмос. В капиллярах мембран, находящихся в электрн
песком поле, могут происходить электроосмотические явления,т.е.
движение жидкости относительно неподвижной перегородки, несу-
щей заряд. Направление переноса жидкости определяйся знаком
заряда стенки капилляров мембраны; если анодная мембрана
заряжена положительно, то будет происходить перенос жидкости
из среднего отделения в анодную камеру, а прн отрицательном
ее заряде — перенос жидкости в обратном направлении. Электро-
осмос изменяет не только объем расшора в среднем отделении
электродиализатора, по приводит и к изменению его концентрации,
гак как электроосмотически переносится не только чистая вода, но
и раствор электролитов.
Процессы, протекающие при электродиализе, подчиняются за-
кону Фарадея, который, как известно, устанавливает, что для пе-
реноса (выделения, отложения) 1 г-экв вещества требуется за-
тратить 96 500 Кл электричества (Кл)
q9 = 96500 (Со — Ск). (4.46)
Если выразить количество электричества через силу тока и
время, то (4.46) примет вид (А-ч).
</, - //, 26.8 (Со - Ск). (4.47)
В реальном процессе расход электроэнергии значшельно пре-
вышает расчетные значения. Это связано со многими явлениями:
неполной селективностью мембран, диффузней католита и ано-
лита из электродных камер обратно в камеру опреснения, кото-
рая усиливается прн возрастании перепада концентрации солей
в электродных и средних камерах; бесполезным переносом ионов
II1 ОН ; преодолением омических сопрошвлений растворов и
мембран; протеканием электродных процессов; утечкой тока; пе-
ремещением воды в результате электроосмоса; переносом воды
движущимися попами н другими причинами.
Скорость удаления ионов из среднего отделения прн электро-
диализе выражается уравнением [101]
— de <//= V-1 (//96500 + 0дДС5м). (4.4Я)
Из уравнения (4.48) может быть определено время
/, = ------р--1g Г 7 4 <J6500C>gjAl_l. (4.49)
0.43430д«и L / 4- 96500С,0д5м J
При выводе уравнения (4.49) предполагается, что количество
тлектричества, проходящее прн электролизе, полностью расходу-
йся только на удаление ионов элем рол и га. Па самом деле прн
•лектролизе в электродных камерах появляется кислота и щелочь,
чю не может быть устранено даже прн интенсивном промывании
(Лектродных камер дистиллированной водой. Поэтому в переносе
> к-ктричества через анодную и катодную мембраны будут принп-
M.iii. участие пе только анионы и катионы электролита, удаляе-
179
мые из среднего отделения, но также Н+ и ОН~, поступающие
из электродных камер. По этой причине не все затрачиваемое ко-
личество электричества эффективно используется для удаляемых
из среднего отделения иоиов. Выход по току при электродиализе
определяется по формуле
Вт = 96500 (Со — Q) Рр/(/Т,). (4.50)
Экспериментально определение Вт производится на основании
изучения изменения концентрации попов в жидкости в средней
камере в процессе элск1родналпза в зависимости от количества
прошедшего электричества.
Прогрессивным решением использования процесса электродиа-
лиза является опреснение воды с одновременным получением вы-
сококонцентрированного рассола в одном аппарате, позволяющее
перейти к 'комплексной переработке минерализованных вод, так
как высокая концентрация солей в рассоле облегчает его утили-
зацию [21]. Однако при этом увеличивается вероятность образо-
вания трудпорастворпмых соединений в примембранпых слоях и
в теле мембран. В. Д. Гребенюком с сотрудниками рекомендуется
при концентрировании рассола производить проверку работы элек-
тродиализатора па возможность отложения СаБОч с помощью не-
равенства [75]:
*'Tca2+[Ca2+]flM Yso2_[SOj-]eb < HPCis^, (4 51)
где k' — степень концентрирования рассола; а — коэффициент, учитывающий
распределение тока по высоте аппарата; Ь— коэффициент, учитывающий уве-
личение концентрации рассола в прныембранном слое с отдающей мембраны
Учитывая, прн цр=0,2...2,0 у2«0,26/(цр+0,1), получим
уравнение для определения допустимой концентрация кальция
при концентрировании:
[Са2+] ПРС15О« (рр 4- 0, l)/(0,026*'2a2b2[SO2-y). (4-52)
При а=2, Ь = 2, ПРС15О =6,1 • 10~5 и Л' = 20 уравнение (4.52)
приобретает вид:
[Са2+] sS 3.5 1(Г7 (Ир + 0,1)/[SO;-J. (4.53)
Уравнение (4.53) может быть применено для проверки обра-
зования отложений других малорастворимых соединений с под-
становкой соответствующих концентраций иоиов.
В Институте коллоидной химии и химии воды (ИКХнХВ)
ЛП УССР создан электродиализный опреснитель-концентратор
ЭД-60 [21], в котором достигнута полная герметизация рассоль-
ного тракта по отношению к опреснительному, обеспечивающая
минимализацню утечек тока по рассолу и предотвращение обра-
зования осадков труднорастворимых соединений в камерах кон-
центрирования. Испытание этого аппарата в схеме комплексной
переработки шахтных вод [75] подтвердило принципиальную воз-
180
можность осуществления процесса опреснения с одновременным
получением высококонцентрированного рассола в одном электро-
диализаторе при невысоких затратах электроэнергии (1,45 кВт-ч
па 1 кг удаленной соли) и достаточно высоком выходе по току
(82%). При напряжении на электродах 70 В, плотности тока
0,8 А/дм2 и скорости потока раствора 0,3 см/с был получен рассол
с общим солесодержанпем 168 г/л, в том числе NaCI — 90 г/л,
Na2SO<— 77 г/л, Са2+— 115 мг/л.
В настоящее время, как правило, конструируются многока-
мерные электродиализаторы с большом числом пар катнопо- и
аниопообмеппых мембран (рис. 4.43, в). Такне установки пред-
ставляют собой ванны, состоящие обычно из 100—200 гидравли-
ческих камер, которые могут быть соединены последовательно
пли параллельно с горизонтальной или вертикальной циркуля-
цией воды. Селективность мембран создает условия, при которых
нз нечетных камер пн катионы, пи анионы не могут пройти в со
седине камеры, вследствие того что знак пх заряда совпадает со
знаком соответственно катионоактивных и апноноактивных мем-
бран. Поэтому в четных камерах происходит процесс опреснения,
а в нечетных, наоборот, концентрация солей, в результате чего
в этих камерах образуется рассол. Обессоленная вода и концент-
рированный раствор отводятся по соответствующим линиям. Ще-
лочной раствор из катодной камеры и кислый из анодной могут
отводиться самостоятельно по отдельным линиям для дальней-
шего использования пли же, если такой необходимости пет, под
соединяться к липин рассола.
Электродиализные опреснительные установки достаточно боль-
ших производительностей эксплуатируются в ряде стран с суточ
ным расходом до 12 000 м3/сут.
В пашей стране первая электродиализная установка, разрабо-
апная ВНИИ ВОДГЕО н установленная на пароходе «Тула»
в 1958 г., опресняла воду Черного моря и имела производитель-
ность 12 м3/сут. Удельный расход энергии на этой установке, рабо-
авшей на первой партии мембран МК 40 и МА-40, составил 18—
20 кВт-ч/м3 прн исходном солесодержапии воды 17,7 г-экв/л, об-
щей жесткости 66,5 мг-экв/л, конечном солесодержапии 2—4 г/л
и жесткости 0,5—0,8 мг-экв/л.
В последующем были разработаны установки для сельского
хозяйства (стационарные и передвижные) типа ЭОСХ,
ЭОУ-НИИМП, ЭОУ «Бархан» и др. Производительность этих
хстановок находится в широких пределах — от 0,03 до 320 м3/ч.
В настоящее время промышленные электродиализные установки
пша ЭДУМ изготавливаются и поставляются па предприятия по
индивидуальным заказам Алма-Атинским электромеханическим
заводом (АЭМЗ).
Опреснительная установка состоит нз блока электродиализа-
юров, насосов, баков опресненной воды (дплюата), баков рас
гола (концентрата), кислотного бака, блока нереполюсовкп н
пульта управления.
IHI
Электродиализаторы опреснительных установок конструиру-
ются в основном но типу фильтра-пресса. Они представляют собой
чередующиеся дилюатные и рассольные камеры, отделенные друг
от друга рамками нз диэлектрика, в которые вмонтированы мем-
браны. В разделительных рамках проштампованы отверстия, ко-
торые прп сборке образуют каналы для подвода и отвода опрес-
няемой воды п вывода рассола. Прп помощи торцевых плит,
в которых укреплены анод и катод аппарата, производят сжатие
рамок и мембран для обеспечения необходимого уплотнения,
предотвращающего утечку воды.
Разделительные рамки вынускаклся лабиршпового н прокла-
дочного типов. У аппарата с лабиринтовыми рамками коэффи-
циент использования площади ионообменной мембраны равен
0,5—0,6, а у прокладочных — 0,6—0,7.
В установках большой производительности используют аппа-
раты с рамками прокладочного типа, позволяющие добиться сни-
жения давления па входе в аппарат и уменьшить расход электро-
энергии на перекачивание воды н рассола, в установках малой
производительное!и — с рамками лабиринтового типа, монтаж ко-
торых более прост.
Турбулизация потока в камерах с целью интенсификации мас-
сопереноса достигается устройством сепараторов-турбулизаторов
для камер прокладочного типа или увеличением скорости потока
п постановкой направляющих перегородок в виде поперечного мо-
стика в камерах лабиринтового типа. Аппараты с рамками про-
кладочного типа имеют горизонтальную ось электрического поля,
а лабиринтового типа — вертикальную.
В электродиализных аппаратах применяются химически стой-
кие электроды из платинированного титана (титан марки ВТ-1-1,
толщина покрытия платиной 1,5 мкм), реже — нз нержавеющей
стали или графита.
Электродиализные установки ЭОУ-НИИМП с аппаратом «Род-
ник-3» производительностью 25—50 м3/сут серийно выпускаются
ПО «Тамбовмаш». Аппараты ЭДУ-1000 производительностью до
25 м’/ч изготавливаются Гос. союзным заводом «Двигатель», а бо-
лее мощные аппараты ЭХО М 5000x200 производительностью до
1500 м3/сут — АЭМЗ. В последние готы ГПИ Промстройпроект
разработал крупногабаритный электродиализный аппарат
ЭДА 1500x1000 производительностью 70 80 м’/ч и станцию оп-
реснения воды—7500 м’/cyi [80]. Союзводоканалпроект подгото-
вил проектную документацию па строительство опреснительной
станции производительностью 11 000 м3/сут. Технологические ха-
рактеристики отечественных электродиализаторов приведены
в справочном пособии [ 13]
Процесс опреснения па электродиализных установках осуще-
ствляется по ipeM основным схемам: прямоточной, циркуляцион-
ной порционного действия и циркуляционной непрерывного дей-
ствия.
Прп прямоточной схеме соленую воду пропускают по опресни-
IH2
каленая Вода
Рис. 144. Схема прямоточной электродиализной опреснительной установки
а — одноступенчатой; б — многоступенчатой / — бак соленой воды. 3 — бак рассола; 3 —
•зсктроднализная ванна; 4 — насос
тельному контуру один раз и при этом солесодержание снижа-
ется до заданной концентрации. Эти схемы бывают одноступен-
чатыми и многоступенчатыми (рис. 4 44). Для уменьшения со-
противления мембранного пакета рассол частично повторно воз-
вращается в цикл установки. Рециркуляции может также подпер-
еться н днлюат, но в этом случае при большом количестве
рециркуляций возрастают энергетические потери из-за снижения
средней концентрации опресняемого потока.
В циркуляционных установках порционного действия соленую
поду пропускают многократно через камеры электродиализного
аппарата до получения заданной степени опреснения исходной
во 1ы. В этих установках скорость потока обрабатываемой воды
может быть выше, чем в прямоточных. С целью исключения уве-
личения перепада концентраций в камерах воду рассольных ка-
мер разбавляют опресняемой водой (рнс. 4.45) Циркуляция рас-
сола и дплюата происходит через вспомогательные резервуары.
При получении заданной степени снижения солесодержанпя оп-
ресняемая вода отводится к потребителю н производится новое
«аполненис резервуаров. Рассол также подвергается продувке и
Соленая вода
I щ 4.45. Схема циркуляционной
порционной электродиализной опрес-
ни i гл ьной установки
! I нс частично опресненной воды; 3 —
рассола; 3 — насос; 4 — элсктродиа-
• < панна
Рис. 4 46 Схема циркуляционной элек-
тродиализной опреснительной установки
I — баки опресненной и частично опреснен-
ной воды: 3 — бак рассола; 3—насос; 4 —
электродиализная ванна
1X3
Рис. 4.47. Схема распределения потоков жидкости в электродиализаторе
а — параллельная; б — последовательная; в — комбинированная; / — насыщаемый поток;
2 — опресняемый поток
разбавлению опресняемой водой, с тем чтобы его концентрация
не более чем в 3—4 раза превышала концентрацию соли в воде,
проходящей через рабочие камеры.
Недостатком этой схемы является трудность автоматизации
процесса. Эти трудности вызываются постоянным изменением
электросопротивления из-за нестационарное™ режима опреснения
и необходимостью в связи с этим регулирования напряжения, что
ipe6yer дополнительного оборудования.
В циркуляционных системах непрерывного действия про-
цесс опреснения сводится к циркуляции через камеры электро-
диализного аппарата нли только рассола, или рассола и днлюата
с непрерывной продувкой рассола (рис. 4.46). Контур рассола вы-
полняется замкнутым и с частичным его сбросом.
В циркуляционных схемах имеет место повышенный расход
электроэнергии на повторную перекачку рассола и днлюата.
Оптимизация работы электродиализных аппаратов связана
с выбором технологических схем опреснения воды, которые опре-
деляются распределением потоков жидкости в камерах аппара-
тов. Различают три гидравлических вида нотокораспределения
воды: параллельное, последовательное и комбинированное
(рнс. 4.47).
Прн параллельном потокораспределепни опресняемая вода и
концентрирующая соли вода движутся параллельно по дилюат-
ным и рассольным камерам электродиализного аппарата. Такой
вид движения воды обычно применяется в установках, работаю-
щих по порционной циркуляционной схеме опреснения.
При последовательном движении всей опресняемой воды и
концентрирующей соли воды потоки их последовательно прохо-
дят все днлюатные п рассольные камеры. Основным недостатком
такой схемы являегся создание большого перепада концентраций
184
no обе стороны мембран, так как с одной стороны мембраны
находится опресненная вода, а с другой — рассол с концентра-
цией в 2—2,5 раза большей, чем у исходной воды. В таких усло-
виях селективность мембран снижается, и это особенно заметно
у мембран, находящихся на концевых участках движения пото-
ков воды. По этой причине понижается выход по току и умень-
шается срок службы мембран.
Для улучшения условий работы мембран принимают скорости
потоков равными. Из-за этого коэффициент извлечения пресной
воды в аппаратах составляет лишь 40—45%, тогда как по парал-
лельной схеме — 60 % и более. Аппараты с последовательным
движением потока к тому же имеют большее гидравлическое со-
противление. Несмотря иа отмеченные недостатки, последователь-
ные системы имеют и значительные достоинства: непрерывность
процесса, стабильность режима, простота схемы автоматизации,
малогабаритность. Аппараты такого типа разрабатываются
в СССР под руководством проф. И. Н Медведева в НПО «Пласт-
массы», который уже создал несколько опреснителей подобного
типа.
Комбинированные системы распределения потоков жидкости
в многокамерных аппаратах (рис. 4.47, а) предполагают парал-
лельное движение воды по всем камерам рассола и последова-
тельное движение опресняемого потока по днлюатиым камерам.
По этой схеме достигается более высокий эффект опреснения при
одинаковых линейных скоростях электролита в камерах опресне-
ния и рассола. Разность конечных концентраций опресненного и
концентрированного ноюков на выходе из аппарата при комби-
нированной системе в 2 раза меньше, чем при последовательной.
При прочих равных условиях расход электроэнергии в аппаратах
с данной системой в 1,5 раза ниже, чем с последовательной, а вы-
ход по току достигает 90—95 %. Такого типа системы отличаются
большим количеством сбрасываемого рассола и применяются
главным образом па судовых установках.
Для нормальной эксплуатации электродиализных установок
необходима предварительная подготовка воды, заключающаяся
в удалении грубодисперсиых и коллоидных примесей, солей каль-
ция и магния, поливалентных и «отравляющих» ионов, имеющих
малую подвижность. Так, рекомендуется не допускать в исходной
воде солесодержання железа >0,05 мг/л, марганца >0,05 мг/л,
взвешенных веществ >1,5 мг/л, окисляемости >5 мг/л, цветности
выше 20 град, общей жесткости >40 мг-экв/л.
Борьбу с отложениями в электродиализных аппаратах ведут
нугем подкисления промывной воды, подаваемой в катодную ка-
меру, или исходной воды, промывкой мембран кислотой, спиже
пнем степени концентрирования рассола или частичным умягче-
нием исходной воды.
В настоящее время в СССР накоплен значительный опыт экс-
плуатации электродиализных установок как для опреснения прн
родных вод, так и для обессоливания промышленных стоков
1.45
ванн, фосфорной, серной
Рис. 4.48. Зависимости стоимости опреснения
воды различными методами
1 — ионный обмен; 3 — электродиализ. 2 коп./кВт • я;
3 — электродиализ. I коп./кВт • я; 4 — дистилляция
S - 10* м’/сут.; S — дистилляция I • 10* м"/сут.
Во ВНИИ ВОДГЕО [18] разрабо-
таны электродна ти.зные методы реге-
нерации серной кислоты и металли-
ческого железа, азотной и плавико-
вой кислот из отработанных раство-
ров, образующихся при травлении
сталей, хромовой кислоты из отрабо-
танных электролитов гальванических
и хромовой кислот из отработанных по-
лировочных растворов, едкой щелочи и кристаллического гидро-
ксида алюминия из отработанных щелочных растворов травления
алюминиевых изделий и др. Метод позволяет повторно использо-
вать в производстве ценные продукты и деминерализованную
воду, а токсичные катионы металлов сконцентрировать в виде
осадка гидроксидов, объем которого после обезвоживания зна-
чительно меньше, чем при использовании реагентных методов
очистки сточных вод.
Стоимость процесса опреснения, как отмечается в работе [72],
зависит от солесодержання исходной воды, производительности
установки и местных условий (стоимости топлива, электроэнергии,
реагентов). На рис. 4.48 приведены ориентировочные данные
о стоимости опреснения воды различными методами [59]. Расчеты
выполнялись для установок производительностью 2000 м3/сут прн
стоимости тепла, равной 2 руб. за 1 Мкал, электроэнергии—1 н
2 коп. за 1 кВт-ч, серной кислоты — 25 руб. за 1 т, соды —
20 руб. за 1 т. Анализ приведенных данных позволяет определить
оптимальную область использования того или иного метода.
Технико-экономическая оценка результатов эксплуатации опыт-
но-промышленных установок по очистке сточных вод показывает,
что метод электродиализа значительно дешевле химических спо-
собов обработки. Экономия прн этом достигается в основном за
счет утилизации ценных компонентов.
4.3.3. Электрофорез *
Электрофоретический метод, основанный па транспорте заря-
женных частиц в электрическом поле, может быть применен во
многих случаях для концентрирования примесей, по особенно це-
лесообразен для агрегат явных устойчивых коллоидных загрязне-
ний, имеющих высокое значение электрокипетического потенциала,
когда обработка воды другими способами (например, коагуля-
цией) не дает хороших результатов. Высокая эффективность ме-
* Материал iio.iiornii.ieii еопместио с кант. техн, паук II. II. Рукобратскпм.
186
тода подтверждена при очистке воды от золей, суспензий, эмуль-
сий, бактериальных загрязнений.
Эффективность электрофореза прежде всего зависит от вели-
чины напряженности внешнего электрического поля Е-, и элект-
рокннетического потенциала £. Поэтому более предпочтительно
электрофоретическое разделение с предварительной подзарядкой
полиэлектролитом частиц дисперсной фазы е целью повышения
величины ^-потенциала для обеспечения большей скорости их дви-
жения [116] Преимуществом этого приема является возможность
использования электрических полей сравнительно низкой напря-
женности н управления процессом разделения фаз путем регули-
рования электроповерхностных свойств дисперсных частиц. Так,
например, для увеличения поверхностного заряда частиц неорга-
нических материалов (каолин, тальк, доломит и др.) в дисперсии
вводились небольшие добавки (0,2—0,4 мае. %) полиэлектро-
лнта — зарядчика — натриевой соли карбоксиметилпеллюлозы
(NavKMU). применяемой как загуститель воднодисперсионных
красок.
С помощью HCI или NaOH в дисперсиях с постоянной кон-
центрацией NavKMU создавались различные значения pH. Уста-
новлено, что во всех исследованных системах t-потеициал частиц
зависит от величины pH и достигает максимума прн рН = 7,54-8,5.
Таким образом, изменяя величину pH, можно управлять электро-
кпнетпческнмн свойствами водных дисперсных систем, содержа-
щих полнэлектролпт типа (NavKMIl).
Электрофорез с плоскопараллельными никелевыми или алю-
миниевыми электродами (рабочая поверхность 25-28 мм2), рас-
положенными па расстоянии 17 мм друг от друга, при напряжен-
ности поля 17,7—35,3 В/см приводит к образованию па аноде
электрофоретического осадка, толщина которого увеличивается
в направлении сверху вниз. Осадок частично отщепляется от элек-
|рода за счет электролитического газовыделення и в форме круп-
ных хлопьев постепенно собирается на дне ячейки. Прн высоких
шачениях силы тока образование электрофоретического осадка
может и не наблюдаться, а в приэлектродном пространстве про-
исходит коагуляция электрохимического и коллоидно-химического
характера. Однако и в этом случае очевиден определяющий вклад
электрофореза, обеспечивающего транспорт частиц в локальную
«шу повышенной концентрации электролита, где происходит про-
цесс концентрационной коагуляции нерастворимых примесей.
При оценке светопропускания обработанной суспензии уста-
новлено, что в ряде случаев наблюдается определенная взанмо-
• вязь между эффектом осветления п исходными значениями pH
обрабатываемых систем (рпс. 4.49). Степень очистки возрастает
с увеличением pH, что можно объяснить соответствующим изме-
нением t-потенциала частиц прн переходе от кислой среды к то-
ниной. Практически полное разделение фаз в гндроднсперснях
«полипа достигается прн времени электровоздействня 60 с и на-
пряженности поля 35 В/см, если рН„гт>7.
187
Рис. 4.49. Зависимость светопропускання /
(%) водных суспензий каолина, обрабо-
танных в электрическом поле напряжен-
ностью 35 В см, от значения pH при про-
дол жительности воздействия
/ — 15 с, 2 — 30 с. 3 — 45 с; 4 — 60 с
На рис. 4.50 и 4.51 (кривая 2) приведены зависимости свето-
пропускаиня электрообработанных дисперсий каолина от напря-
жения на электродах при фиксированном времени электрообра-
боткн. Наклон кривых 3—4 (рис. 4.50) резко увеличивается, если
время экспозиции превышает 30 с, а при /=120 с и /73=50 В до-
стигается практически полная сепарация (рнс. 4.51, кривая 2).
Оптимальными условиями разделения фаз в изученных систе-
мах являются U,= 50 = 60 В при / = 604-120 с. Таким образом,
продолжительность электрообработки в предлагаемом способе,
основанном на искусственном продуцировании поверхностного за-
ряда на частицах дисперсных загрязнений, значительно меньше,
чем в других способах [116].
Практическое использование электрофореза для очистки воды
связано с созданием многокамерного аппарата (рис. 4.52) [6]. Оп
Рис 1.50. Зависимость
светопропускання J
(%) I и.'фодиспсрсин
каолина (рНигх 7,4) от
напряжения на электро-
дах U3 при продолжи-
тельности обработки
I—4 — апалогптии рис. 1.10
Рис. 4 51 Зависимость све-
тонропускання J (%) тилро-
диснерсий каолина (рНи,,
7,6) от продолжительности
электрообработки при на-
пряжении на электродах
30 В (/) и продолжитель-
ности электрообработки
120 с (2)
188
Рис. 4.52. Схема многокамерного
электрофоретического аппарата
I — подача исходной воды; 2 — отвод
промывной воды и газообразных пузырь-
ков злсктролнза; 3 — электродная ка-
мера; 4 — работ я камера; 5 — отвод очи-
щенной воды, 6 — заполнение и про-
мывка электродных камер; 7 — отвод
концентрированной фракции; Л — анод;
К — катод; М — мембрана; Ф — фильтр
состоит нз рамок для распределения воды, мембран и фильтров,
зажатых между двумя пластинами, в которые вмонтированы
электроды — платиновый анод н графитовый катод. Вся система
стягивается болтами. В торцевых пластинах имеются отверстия
со штуцерами для подвода и отвода воды, выполненные таким об-
разом, чтобы обеспечить наиболее полное удаление воздуха и га-
зообразных продуктов электролиза прн заполнении аппарата и во
время его работы. Камеры, образуемые рамками, отделены друг
от друга мембранами, способными пропускать ионы солей и за-
держивать коллоидные частицы. Каждая камера разделена фильт-
ром на два отделения — исходной и очищенной воды. В качестве
фильтров могут применяться разнообразные пористые неметалли-
ческие материалы, включая фильтровальную бумагу и нитроцел-
люлозные фильтры.
При создании электрического ноля отрицательно заряженные
взвешенные и коллоидные частицы движутся к аиоду через
фильтр в направлении, противоположном движению воды. При
условии, что скорость электрофоретического переноса частиц оэ
больше скорости фильтрования воды Оф (т. е. u$<va=U.,E), ча-
стицы загрязнений проходят через фильтр, в результате чего
в правой части камеры 4 остается очищенная вода, которая отво-
дится по трубопроводам 2.
Перед мембранами (М) в отделениях исходной воды каждой
камеры происходит концентрирование коллоидных частиц. Под
действием потока исходной воды (а также под действием силы
тяжести, если узягр>т»оды) загрязнения удаляются по трубопро-
водам 3.
В многокамерном аппарате экономически выгодно применять
возможно более тонкие камеры, так как с уменьшением толщины
камеры (при одинаковом количестве их в аппарате) снижается
напряженно па электродах, а это. в свою очередь, влияет иа мощ-
ность потребляемой энергии.
Нормальная эксплуатация аппаратов обеспечивается при по-
стоянстве расхода и ламинарностн потока воды вдоль мембран
и фильтров. Эти условия могут быть нарушены прн прогибании
мембран в результате наличия избыточного давления со стороны
189
камер исходной воды Для исключения этого явления в камерах
чистой воды перед мембранами устанавливаются подложки из
лавсановой сетки
В качестве материала для мембран могут применяться цел-
лофан, пергамент, ионитовые мембраны и др. Наиболее предпоч-
тителен целлофан, обладающий малым электрическим сопротив-
лением и достаточной прочностью прн длительной эксплуатации
аппаратов.
Рамки камер выполняются из листового винипласта толщи-
ной 1,6—2,0 мм. Между рамками, мембранами и фильтрами мои
тируются прокладки пз паронита толщиной 0,6 0 9 мм и вини-
пласта 1,0—2,0 мм. Чередование тонких листов пароинта и не-
электропроводного винипласта исключает утечки тока за счет
гигроскопичности паронита.
Подача и отвод воды нз камеры должны осуществляться как
можно ближе к мембране и дальше от фильтра. Крайние элект-
родные камеры для обеспечения как малого электрического сопро-
тивления, так и надежного удаления газов из них должны иметь
толщину порядка 4—5 мм. Отсутствие нли недостаточная про-
дувка воды из аппарата прн его эксплуатации приводят к посте-
пенному накоплению загрязнений в отделении исходной воды и,
соответственно, снижают эффект очистки вследствие диффузии
коллоидных частиц в отделение очищенной воды через фильтр.
Продувка должна быть не менее 15% от общего расхода воды
через аппарат [6]. Образование щелочного (католита) и кислого
(анолнта) растворов в электродных камерах 5 электрофоретиче-
ского аппарата и диффузия ионов Н+ и ОН- через мембраны в ра-
бочие камеры приводят к изменению величины pH очищенной
воды. Непрерывная промывка электродных камер позволяет регу-
лировать pH воды в целях ее стабилизации. Промывка катодной
камеры необходима также с целью предотвращения образования
отложений на катоде СаСО. и Мс(ОН)2. Прн образовании таких
отложений их удаление возможно пропуском через катодную ка-
меру анолита.
Электрофорез является достаточно перспективным методом
для очистки цветных вод с малым солесодержанпем [6]. Пара-
метры процесса очистки природных вод практически не зависят
от изменения концентрации гумусовых загрязнений в исходной
воде. Так. затраты электроэнергии прн очистке воды с цветностью
до 150 град, составляют 0,6 кВт-ч/м3, а с цветностью 30—
10 град - 0,5 кВт'ч/м3.
Повышение солссодержания исходной воды приводит к уве-
личению расхода электроэнергии на ее очистку вследствие увели-
чения элек1ропроводности воды и возрастания плотное!и тока
для поддержания заданной оптимальной напряженности электри-
ческого поля. Потребность в электроэнергии может быть опре-
делена по формуле (кВт-ч/м3) [6J:
Г,я (4.54)
190
где р —количество удаляемых коллоидов в частях от исходного количества;
d толщина камеры, см; /' — коэффициент пи.тярн jauiiii камеры (обычно
1+2); о»ф — электрофоретическая подвижность коллоидов, см-/(В с); N —
число параллельно работающих камер; 5$ — площадь фильтра в каждой ка-
мере, см*.
Электрофорез для очистки воды может применяться с целью
удаления различных компонентов: гуминовых веществ, минераль-
ных глинистых частиц, ПАВ, детергентов, окиелов железа, водо-
рослей, бактерий, эмульсий масел и других коллоидных загряз-
нений. Он может быть использован также для доочистки бытовых
сточных вод, прошедших предварительно биологическую очистку.
Для обесцвечивания природных вод может быть рекомендо-
вана технологическая схема, включающая мпкрофильтры, элект-
рофоретическую и бактерицидную установки.
4.3.4. Электрофильтрование
В основе данного процесса водоочистки лежат эффекты воз-
действия на движущиеся с потоком жидкости частицы примесей
однородных и неоднородных электрических полей, обеспечиваю-
щих их отделение от воды и формирование в виде осадка у по-
верхности различных коллекторов. При этом в одном аппарате
реализуются совместно во времени и пространстве электрическое
воздействие иа дисперсную фазу и фильтрование дисперсионной
среды. Роль электрического воздейовия сводится в данном слу-
чае в основном к транспортировке заряженных коллоидных частиц
к поверхности коллекторов, вблизи которых происходит концент-
рирование частиц и создаются благоприятные условия для их коа-
гуляции.
Это прогрессивное технологическое решение получило разви-
тие в фундаментальных исследованиях, проводимых в ИКХиХВ
АН УССР под руководством профессоров С. С. Духина.В.Д Гре-
бенюка, П. И Гвоздика [17, 19, 22, 30].
Различают два направления в электрофильтровапии: с приме-
нением проводящих и непроводящих коллекторов. Основное пре-
имущество применения проводящих коллекторов — высокая на-
пряженность поля непосредственно у поверхности коллектора, что
обеспечивает электрофоретический транспорт частиц к поверхно-
сти коллектора и необходимое условие формирования осадка. За
время прохождения через фильтр частицы должны не только
успеть достичь поверхности коллектора, но и сформировать
осадок.
Осадок может быть твердым нли жидким (текучим), что оп-
ределяется двумя противодействующими факторами: межчастич-
пым взаимодействием, обеспечивающим формирование твердого
осадка, и тангенциальным течением воды вдоль поверхности кол-
лектора, которое может привести осадок в текучее состояние.
Основная трудность технологии электрофильтрования состоит
в том, что для обеспечения поляризационной коагуляции необхо-
J01
димо электрическое поле высокой напряженности, при которой
удельный расход электроэнергии становится неприемлемо высо-
ким. Эту трудность удается преодолеть, используя пространственно
неоднородные поля. В области максимума поля можно достичь
необходимой для коагуляции величины напряженности поля, в то
время как но объему среднее значение поля невелико; при этом
область максимума поля создается вблизи коллектора, т. е. там,
где формируется осадок.
При фильтровании в неоднородных полях обычно в качестве
коллектора используют проводники первого рода. Такой прием
реализован в конструкциях мембранных электрофильтров [30] и
при применении гранулированного ионита [19].
При электрофильтроваиии через непроводящие коллекторы ме-
ханизм фиксации частиц в теле фильтра обусловлен концентриро-
ванием частиц под действием диполофореза в области максимума
или минимума поля. Если индуцированный дипольный момент ча-
стицы ориентирован по нолю, диполофорез вызывает концентри-
рование частиц в области максимума поля, при ориентации про-
тив поля — в области минимума. При использовании постоянного
электрического поля эффекты фокусировки обусловлены совмест-
ным действием электро- и диполофореза.
Неоднородные электрические поля в жидкости можно созда-
вать с помощью как электродов различной конфигурации, так и
разнообразных диэлектрических загрузок или перегородок. Част-
ным случаем таких нолей являются неоднородные поля, возни-
кающие в окрестности щелей или ячеек сеток [14].
В работе [98] получены выражения критической напряженно-
сти воля:
для случая ячейки тороидальной конфигурации
£кр = я/73 (£//<’ )(1 - 2а//); (4.55)
для случая дырочной конфигурации
£кр = )(»“ 2af/^i)3. (4-56)
где /—расстояние между центрами противолежащих волокон; гч — радиус
коллоидной частицы; а—радиус волокна; h— расстояние между центрами
отверстий; щ — радиус отверстия.
Прн £=30 МВ и гч = 1 мкм из формулы (4.55) получим, что
для удерживания данного размера коллоидной частицы при до-
пустимых внешних полях порядка 80 В/см и //гч= 100 необходимо,
чтобы величина, характеризующая просвет между волокнами (1—
2а//), была по более 0,4. В этом случае /=0,1 мм, а радиус во-
локна а 0,03 мм. Тогда ширина просвета между волокнами
в сетке должна быть порядка 0,04 мм, что в 20 раз больше диа-
метра удерживаемой частицы. Ясно, что прн увеличении радиуса
частицы величину внешнего ноля можно уменьшить, а просвет
между волокнами в сетке — увеличить.
Выражение (4.66) показывает, что величина критического поля
существенно зависит ог расстояния между краями соседних отвср-
192
ci ин при тех же значениях ^-потенциала, радиуса частицы гч it
О1НОШСННЯ /|/гч= ЮО Эта оценка позволяет заключить, чю сетки
рассмотренного вида наиболее выгодны при проектировании элек-
|рофильтров. Минимальная напряженность поля, необходимая
для удерживания частиц Еэ=11,5 В/см при 1—2а//=0,4.
Следует отметить, что достигнутый в последние годы прогресс
и юории новых электрокнпегических явлений открывает широкие
возможности в реализации высокоэффективной технологии элект-
рофильтроваиия различных дисперсных систем. Однако внедре-
нию этой технологии мешает в настоящее время еще много нере-
шенных проблем прикладного характера, и в первую очередь
несовершенство аппаратурного оформления данного процесса и
необходимость разработки эффективных конструкторско-техноло-
гических приемов удаления образующегося осадка.
4.4. КОМБИНИРОВАННЫЕ МЕТОДЫ
4.4.1. Электрофлотокоагуляция
Метод электрофлотокоагуляции (электрокоагуляции — флота-
ции) позволяет одновременно осуществлять два процесса [58, 91];
изменение дисперсного состояния примесей за счет их коагуляции
под действием электрического поля продуктов электродных реак-
ций и закрепление пузырьков электролитического 1аза па поверх-
ности коагулирующих частиц, что обеспечивает их последующую
флотацию. Непременным условием протекания процесса электро-
флотокоагуляции является закрепление пузырьков газа иа поверх-
ности частиц в состоянии «in statu nascendi», т. е. в момент их
оиразования. В этом случае эффективность флотационного про-
цесса возрастает в 1,5—2,0 раза.
Особенно эффективен метод при анодном (катодном) раство-
рении металла электродов. Это объясняется тем, что коагулирую-
щая активность электрогенерированпых реагентов в отличие от
коагулянтов, полученных при гидролизе солей металлов, зиачи-
1ельно выше [43, 58]. Коагуляция частиц с одновременным закреп-
лением пузырьков газа может происходить в межэлектродиом
пространстве, где дополнительно используется воздействие элек-
грического поля [91], или в надэлектродном объеме, куда траис-
норшруются продукты электродных реакций [69]. В любом слу-
чае первый прием всегда эффективнее из-за воздействия поля и
более высокой «активности» взаимодействующих частиц, однако
его применение ограничивается возможным нарушением работы
элск!родных спсгем прн сложном поппо-молекулярпом составе
природных и СРОЧНЫХ вод.
Основными стадиями электрофлотокоагуляционного процесса
являются: 1) генерация электролитического коагулянта; 2) гене-
рация электролитического газа; 3) коагуляция примесей; 4) за-
крепление электролитического газа па поверхности скоагу.шро-
l.ncai № 211Я 193
ианных частиц (образование флотокомплексов); 5) всплывание
флотокомнлсксов
Стадии 3 и 4 протекают одновременно, предшествующие им
стадии 1 и 2 могут протекать параллельно или последовательно
в зависимости от конструктивных особенностей электродной си-
стемы. Как правило, стадия 5 протекает после завершения стадий
1—4 н осуществляется в отдельной камере. Эффективность фло-
тации зависит от соотношения объема образующегося электроли-
тического газа к массе коагулируемых частиц и электрогенериро-
ванного коагулянта. Объем образующегося газа Уго при электро-
лизе определяется в соответствии с уравнением (6.18), а требуе-
мый объем газа — из выражения
у; = аг(МЦ-М5), (4 57)
где <хг—удельный расход газа на единицу извлекаемых примесей, определяе-
мый экспериментально нлн на основании обобщенных данных, полученных
А. И Мацневым [61] н другими исследователями, Aftx н Мгт — соответственно
масса извлекаемых примесей и масса твердой фазы, образующейся прн гене-
рации электролитического коагулянта, которые находятся нз уравнений:
МТ=С0^о0рЭ; (4 58)
М>КмХеА«еДт; (4 59)
здесь /овр — продолжительность стадии образования флотошлама; КМе —
коэффициент пересчета на твердую массу гидроксидов по химическим реак-
циям гпдратообразовання: КА| = 2.89; KFe,» =- 1,62; КРе1>=1,91.
Если объем газа недостаточен для обеспечения необходимого
эффекта очистки, то производится дополнительное газонасыще-
иие за счет устройства дополнительной электродной системы
с нерастворимыми электродами, обеспечивающими требуемое га-
зоиасыщение. Увеличение газоиасыщения может быть произве-
дено конструктивным путем с использованием камер «газового
слоя>, предложенных в УИИВХ [4]. В последнем случае ие увели-
чиваются энергетические затраты на процесс. Электрофлотокоагу-
ляторы, как правило, выполняются двухкамерными. Во второй ка-
мере производится выделение образовавшихся флотокомплексов
из воды. Электрореакторы располагаются смежно или внутри фло-
тора(делителя, который бывает горизонтального или вертикаль-
ного типа (рис. 4.53). Во флоторазделителе с вертикальным на-
правлением движения воды смежное размещение элсктрореакюра
допускается, если площадь поверхности раздели геля нс превы-
шает 5 К) м2. В противном случае возникают сложности по рав
номерному распределению очищаемой воды по объему флотораз-
де л ш ел я.
Эффект очистки па флотокоагуляцнопных установках зависит
от исходной копцешрацни загрязнений, характера их взаимодей-
ствия с образующимся коагулянтом, их конструкции и других
факторов. При исходной концентрации твердой фазы 300 500 мг/л
остаточная концентрация составляет 20 -50 мг/л (габл. 4.3) и по-
194
Гис 4 53. Схемы двухкамерных электрофлотокпагуляципнных установок гори-
|<>11галы<О1О (и) и вертикального (б) типов
|1лпредслитсльныЛ логок (трубопровод); 3 — камера коагуляции; 3 — электродная ен
ч. < — флоторяздслитсль; 5 — скребковый транспортер. 6 — лоток для сбора шлама;
ток сбора очищенной воды; в — трубопровод отвода осадка; / — исходная вода;
II очищенная вол л; Ш — осадок
iii4in.iv।ся при съеме шлама с поверхности, выпуске осадка, рез-
ким колебании концентрации загрязнений на входе в установку.
Поэтому, как правило, перед установками для элекгрофлотацнн
пре (усматривается предварительная очистка от грубых примесей
и чсргдиепне концентрации солевого состава, а после установок —
<л фильтрования для доочистки от взвешенных веществ
(|>п 1.54). Так, применение фильтра с плавающей пенонолнети-
l>n'ibHoii загрузкой для доочистки стоков конезавода после элект-
риф ююкоагуляцнн позволило получить дополнительный зффект
•••И1С1КИ ог взвешенных веществ на 75%, хрома н железа-
пл • >() %.
11.1 рис. 4.55 приведена разработанная УИИВХ и Черномор-
hiiM HIIKIi компактная установка, включающая флотораздели-
I ь, смежно расположенный электрореактор и размещенный ниже
кгч<ры флотации фильтр с плавающей загрузкой [4] Установка
195
предназначена для очистки
малых количеств (до
20 м3/ч) нефтесодержащих
вод с концентрацией неф-
тепродуктов 300—500 мг/л.
Электродная система на-
ходится ниже уровня воды
в установке, благодаря
чему в межэлектродном
пространстве за счет со-
противления, создаваемого
распределительной систе-
мой, формируется газовый
слой. Флотируемые нефте-
продукты собираются на
ограниченной поверхности
флоторазделнтеля в верти-
кальном трубопроводе,
служащем для вывода из
установки электролизных
газов Гидравлическая на-
грузка па поверхность ка-
меры флотации, как н ско-
рость фильтрования, со-
ставляет 1.5—5 м3/ч-м2.
Т а б л н ц а 4.3. Эффективность очистки
сточных вол кожзавода метолом
электрокоагуляции флотации
Показатели загрязнений, мг/л Исходная поде Очищенная вода Эффект очистки.
Взвешен- ные веще- ства 300 -580 350 20—47 42 88,0 %
хпк 702—1390 800 39—370 200 75.0 %
Сульфиды 37—300 120 1,0-53 15 87,5
Хром _7—300 120 0,1—39 1.0 99,2
ПМ5 3—148 35 1,4—87 15 57,1
Железо — 0,1—5,2 0,5 —
Примечание. В числителе прияедеиы
пределы колебаний, в знаменателе—средние зна-
чения.
Минимальные значения расчетных параметров принимаются
прн средней крупности частиц нефтепродуктов 8—10 мкм. макси-
мальные— 30—40 мкм. Плотность тока принимается 50—200 А/м2,
расход тока 2,8 • 10’->3,6 •! О5 Кл/м3, объемный расход газа —
0.040—0 054 м3/м3. Остаточная концентрация нефтепродуктов со-
ставляет 1—5 мг/л и зависит от вида извлекаемых нефтепродук-
тов. Для очистки воды, содержащей нефтепродукты и взвешенные
вещества, разработана установка, приведенная на рис. 4.56. От-
личительной ее чертой является наличие осадочной части для
взвешенных частиц с встроенными блоками тонкослойных пла-
стин. Подобное решение позволяет в одной установке извлекать
нефтепродукты флотацией, механические примеси — отстаиванием
с последующей доочисткой воды фильтрованном. Недостатком
установки является малый объем надфильтрового пространства,
в связи с чем требуется дополнительная емкость промывной воды.
В блоках электрореакторов установок, приведенных на рис.
4.55 и 4 56. реализовано восходящее направленно движения
жидкости Эффективная флотация прн использовании таких реак-
торов возможна только при размещении электродной системы
ниже уровня воды не менее чем па 0,4—0,5 м.
При электрофлотокоагуляцни с растворимыми электродами
практически отсутствует возможность создания падежного кон-
такта их с проводниками тока над уровнем воды. Поэтому прн
использовании растворимых электродов контакты следует выпол-
196
I
Рис. 4.54. Технологическая
схема очистки воды с при-
менением электрофлотокоа-
суляционных установок
/ — узел очистки от грубых примесей; 2 — узел предварительного осветления (отстойник
или флотатор); 3 — усреднитель; 4— элсктрофлотокоагулятор; 5— фильтр; 6 — узел обра-
ботки шлама и осадка; / — очищаемая вода; 11 — воздух; /// — очищенная вода; IV —
осадок я шлам; V — промывная вода фильтров; VI — обезвоженные твердые отходы
Pik 4 55. Установка для очистки
по гы от нефтепродуктов
I кгрпреактор с «газовым слоем»;
1М|М1ЛНан система с перлетвори
••электродами. 3 — флоторазде.тн
• и 4 нгфк борное устройстве»; .•» —
и <11|>.1влню|цис перегородки; 6 —
ыр плавающей загрузкой; 7 —
|Я <1п тема очищенной воды; 8—
для сбора очищенной воды; 9 —
!• • I - тема промывной воды
Рис 1.56. Схема электрофлотокоагу-
ляционнон установки для очистки
воды от нефтепродуктов и механи-
ческих примесей
/ - «зектрорелктор с «газовым слоем»;
2 -электродная система; 3 —камера «га*
ювеио < доя»; 4 — флот о разделит ель; 5 —
нефтесборное устройство; 6 — струена-
иравляющие перегородки; 7 — сборная
система очищенной воды; 8— фильтр;
9 камера флокуляции; 10 — тоимослой
ныЙ отстойник. // — сборная система
к.-.’Дкт и п;юмы1>> й поды
5
Рис. 4.58. Схема алсктрокоа-
гулятора-флотатора верти-
кального тина с встроенным
пенополистирольным филь-
тром
/ — сборный лоток очищенной
воды; 2 — элсктрокоагулятор; 3 —
юна флотации; 4 — электродный
блок; 5 зона отстаивания: 6 —
периферийный пенополистирольный
фильтр; 7 — отвод осадка: 8» 9 —
соответственно подача исходной
и отвод очищенной воды
Рис. 4.57. Схема электрофлотикиагуляци-
оиной установки для очистки воды от
ионов тяжелых метлтлов
/ — э.чскт pope актор с «газовым слоем»; 2—
«лгктродная система: 3— камера «га юного
слоя»; 4 — флотороактор; 5 — флотор аздел ит ел Ь;
6 струенапрааляющие перегородки; 7 — тонко
слойный отстойник; 8 -- сборная система очи-
щенной воды; 9 — фильтр; 10 — гндромеханнчс
скнй фтокулятор; // — камера флокуляции.
12 — система отвода осадка и промывной воды
пять сухими, т. е. не затапливая электродную систему. В таких
элсктрореакторах движение воды целесообразно принимать нис-
ходящим. На рис. 4.57 показана разработанная УИИВХ и ЛОМО
установка, в которой электрореактор с нисходящим движением
воды размещен смежно с флоторазделителем. Установка предна-
значена для очистки воды or попов тяжелых металлов, нефтепро-
дуктов, ПАВ, органических примесей и т. и. Особенностью
установки является наличие дополнительного флотореактора, рас-
положенного в центре флоторазделителя и соединенного с элект-
рорсакгором. Флотском пл ексы в данной конструкции установки
образу инея как в электрореакторе, так и в флотореакторе.
При необходимое! и в флотореактор может подаваться часть
очищенной воды после насыщения ее воздухом под избыточным
давлением. Вторым дополнительным элементом установки явля-
ется гидромеханический флокулятор, установленный между фло-
торазделигелем и отстойной частью и предназначенный для укруп-
нения части । и дроке ндов в режиме градиентной коагуляции. При
напряжении электролиза 10 16 В. расходе электроэнергии 0,5—
198
1() кВт»ч/м4, расходе тока
100- 300 Кл/л обеспечнва-
екя высокий эффект
очистки от различных при-
месей в промывных водах
I альвапическнх цехов
(табл. 4.4).
В ЛИСП разработана
усьпювка для локальной
очистки сточных вод, со-
щржащпх трудноокпеляе-
мые органические вещества
(рис. 4.58), представляю-
щая собой вертикальный
О1СЮЙНПК, внутри которого
yciaiioB.ien электрокоагу-
лягор. Цилиндрическая
Таблица 4 4. Эффективность очистки
промывных вод гальванических цехов
иа комбинированной
электрофлотокоагуляцнонной установке
Показатели загрязнений, мг/л Исходная вода Очищен- ная вода Эффект очнеткн.
MjTIIOCTb 250 3—10 96,0
Хром (VI) 20-30 0,05-0,1 99,5
Хром (111) 30-50 0.1—0,3 99,0
Никель 15-20 0,3—0,5 96 0
Мель 10 15 0,3-0,8 92,0
Цинк 20—30 0,5—0,8 96,0
Кадмий 10-20 0,3- 0,5 98,5
Нефтенродх кты 100—300 0.8—1,0 96,6
ПАВ 1—5 0,5—1,0 50,0
вертикальная перегородка делит аппарат на центральную и пери-
ферийную зоны: центральная зона выполняет роль флотатора и
(истойника; в периферийной зоне для повышения эффективности
очистки сточных вод от взвешенных веществ устроен пенополисти-
рольный фильтр.
Производительность установки с диаметром корпуса 3 м, высо-
кий рабочей камеры 2 м и конусной части 0,9 м составляет 2,5 м3/ч.
Параметры обработки: концентрация СГ попов в сточной воде
I г/л, плотность тока в электрокоагуляторе 50—75 А/дм2, напря-
жение на электродах 6 -8 В, продолжительность электрообра-
бо!кн — до 10 мин, флотации и отстаивания —1,5 ч, фильтро-
цпкла 8—10 ч.
Установка была испытана при очистке высококонцентрирован-
ных сточных вод красильно-отделочного цеха с исходными пока-
циелямп загрязнений: кратность разбавления ИК= 1:10004-
1:3000, ХПК—4004-1500 мг/л, взвешенные вещества —
250 мг/л, ПАВ = 2004-400 мг/л. Эффекты очистки составляют: по
обесцвечиванию — до 98%, по ХПК — до 60%, по взвешенным
веществам— до 95%, по ПАВ — до 80%. Затраты электроэнергии
па очистку 1,2 кВт-ч/м3.
целью уменьшения расхода потребляемого металла на элект-
рокоагуляцию и сокращения затрат электроэнергии, а также по-
нышення эффективности очистки разрабо!ан ряд комбинирован-
ных аппаратов.
Электрофлотокоагуляционная установка (ЭФКУ, рис. 4.59),
предложенная ЛИСИ [37, 120], включает три камеры, разделен-
ные перегородками.
В первой камере происходит электрофлотациопное извлечение
и । очищаемой жидкости основного количества грубодиснергиро
ii.niiii.ix жиров и взвешенных веществ. В электродном блоке этой
камеры аноды выполнены нз графита Г-4, а катоды нз липовой
in ржавеющей стали Х18П9Т.
100
Рис. 4.59. Электрофлотокоагуляционная
установка ЭФКУ
/ — корпус; 2 — подающая труба; 3 — пекогон;
4 — перегородки между камерами; 5 — ведущий
вал пекогон а*. б — полу погруженная перегородка;
7 — треугольный водослив; 8 — трубопровод опо-
рожнения; 9. 10» 12 — электродные блоки соот-
ветственно третьей, второй н первой камер;
// трубопровод выпуска осадка
Рис. 4.60. Электрофлотокоагулятор колон-
ною типа
/ — камера флотации; 2 — лоток для сбора
пены; 3 — сбор очищенной воды: 4 — камера ко
а>уляцнн; 5 — корпус; б — растворимые элек-
троды: 7 — обратный клапан; 8 — нерастворимые
электроды
Вторая камера, основная, представляет собой электрокоагуля-
тор, в котором происходит анодное растворение металлических
электродов н образование хлопьев коагулянта. Электродный блок
этой камеры состоит из 36 дюралюминиевых пластин марки Д-16А.
Расположение электродов, как и в первой камере, вертикальное.
Третья камера, вторичный электрофлотатор, предназначена для
удаления не всплывших иа поверхность второй камеры хлопьев
с адсорбированными загрязнениями. Электродный блок в пей рас-
положен горизонтально и состоит нз анодной графитовой пла-
стины и катодной сетки (2,5x2,5 мм).
Для сбора грубодпсперспых оседающих примесей, не подвер-
гающихся флотации, а также предотвращения засорений ими меж-
200
электродного пространства между первой и второй камерами уст-
роена иловая камера, выполненная в виде треугольной призмы.
Образующаяся на поверхности пена удаляется пеногопом 4
и лоток н далее по трубопроводу 3 отводится от установки. Для
гашения пены и побуждения движения флотокопденсата по лотку
в последнем уложен напорный дырчатый трубопровод.
ЭФКУ рекомендуется [37] для очистки сточных вод мясоком-
бинатов, рыбоперерабатывающих предприятий, молокозаводов, су-
ton рыбного флота и т. д.
При плотности тока в камерах электрофлотации 10 мА/см2 и
камерах электрокоагуляции 2,5 мА/см2 эффект удаления жира
н взвешенных веществ из сточных вод мясокомбинатов состав-
ляет 93—99 %, а снижение величин ХПК и БПКполп— 75—80 %.
Остаточные концентрации загрязнений не превышают соответст-
венно 30, 60 и 500 мг/л. Затраты электроэнергии — 0,6 кВт-ч/м3.
Расход металла 8 г/м3 при применении дюралюминиевых элект-
родов и 20 г/м3—при железных электродах.
Как известно, существенными недостатками электрофлотоко-
.нуляции, препятствующими ее успешному внедрению в практику
водоочистки. являются процессы пассивации поверхности раство-
римых электродов и адсорбции иа них посторонних примесей, ко-
и>рые резко снижают скорость анодного растворения металлов.
В Харьковском политехническом институте М. М. Назаряном,
В. Т. Ефимовым и другими сотрудниками предложены различные
модификации электрофлотокоагуляционных аппаратов колонного
iiiiia [69], исключающие отмеченные недостатки.
Процесс получения электрогенерировапного коагулянта осуще-
< шляется в этих аппаратах в отдельном электродном блоке с чи-
< ним раствором электролита, расположенном в нижней части
нпарата соосно с рабочими камерами флотации и отстаивания.
>к» позволяет значительно повысить работоспособность электро-
топ и исключить адсорбцию на них посторонних примесей. В ка-
ше i не электролита, подаваемого в блок электрокоагулятора, слу-
кн । предварительно очищенная от примесей вода с добавкой в пе-
риод пуска аппарата в работу NaCI или НС1 в количестве 0,1—
<1 1 г/л.
Болес совершенным является колонпый электрокоагулятор
юполпительной зоной флотации, расположенной. в отстойной
к(мере и ограниченной с одной стороны системой нерастворимых
перфорированных электродов, а с другой — кольцевой перегород-
• пй (рис 4 60).
Наличие в электрокоагуляторс конической перегородки и об-
piinoio клапана полностью исключает проникновение обрабаты
•• мой жидкости в блок с растворимыми анодами Винтообразные
ы приводят во вращение обратный клапан, что способствует
in ремешиваиию сточной воды с электролитом, содержащим про
nVKtij растворения электродов. Коническая перегородка, выпол-
ш иная по профилю сужающегося потока, исключает образование
। о1Пных зон и обеспечивает постоянный отвод электролнтпче-
201
ских газов в камеры флотации, гарантируя взрывобезопасную ра-
боту аппарата.
Для предотвращения уплотнения образующейся пены и свое-
временного ее удаления из отстойной камеры рекомендуется [а. с.
914506 (СССР)] устанавливать вращающийся в горизонтальной
плоскости трубопровод с раструбом, воздушным эжектором и
вставкой в виде винта Архимеда для засасывания и перемещения
пены по трубопроводу к циклону, где происходит разделение газа
и жидкой фракции отходов. Воздух, подаваемый в эжекторную
систему, удаляет образующуюся пену и разбавляет электролити-
ческие газы до взрывобезопасной концентрации. Полная безопас-
ность работы аппарата обеспечивается автоматической блокиров-
кой подачи воздуха в эжектор с подводимым па электроды
электрическим током. При прекращении подачи сжатого воздуха
токовая нагрузка на электродные блоки отключается.
Электрофлотокоагуляционные установки не нашли в целом
должного распространения в практике водоочистки. Одной из при-
чин этого является применение конструкций, не соответствующих
требованиям эксплуатации, что приводит к частому выходу из
строя электродной системы и отдельных узлов установки Опре-
деленные трудности возникают также при конструировании уста-
новок большой производительности, что обусловлено их гидроди-
намическим несовершенством. В ряде случаев установки исполь-
зуют без учета состава и концентрации примесей в воде, а в тех-
нологическую схему не включают аппараты для предварительной
ее очистки.
При применении электрореакторов с растворимыми электро-
дами большое влияние на процесс оказывает колебание концен-
трации примесей, а также адсорбция па них посторонних веществ,
что нарушает работу электродных систем. Требуют дальнейшего
совершенствования системы сбора и удаления образующихся осад-
ков и флотационного шлама. Решение этих вопросов позволит
перейти к широкому внедрению этих перспективных устройств.
4.4.2. Электрокаталитическая очистка*
Прн рассмотрении процесса электрохимической деструкции
(см. п. 4.2.3) было указано, что степень окисления органических
загрязнений может быть повышена за счет дополнительного ввода
в обрабатываемую воду поваренной соли, прн электролизе кото-
рой образуется активный хлор, обладающий достаточно высоким
окислительным потенциалом. Отмечалось также, что для деструк-
ции трудноокнсляемой органики необходимо создание избытка ак-
тивного хлора, что, однако, не всегда обеспечивает требуемый эф-
фект очистки, по вызывает необходимость дехлорирования и по-
вышает затраты на очистку сточных вод. Снизить затраты и повы-
* Материал подготовлен совместно с канд. техн наук М Т. Никифо-
роним.
202
ешь эффективность очистки можно при использовании ка1алпз<1
юров восшаповлепия активного хлора — активированных углей,
пдрокендов и оксидов металлов переменной валентное in. Наи-
большей эффективностью обладают соединении металлов, изме-
няющие свою валентность в широком интервале, например мар-
.ища.
Высокая окислительная способносп> расiворов активного хлора
и присутствии марганца также отмечается в работе [68]. При срав-
iiciiiiii действия смеси гидроксидов металлов Мп—Fe, Мп—Сг и
1е—Сг оказалось, что наибольшей активностью обладает смесь
। идроксидов Мп—Сг. Марганец по сравнению с другими метал-
лами обладает способностью менять свою валентность на 4 и 5
единиц (от Мп2+ до Мп71), оставаясь устойчивым в водном рас-
пюре. Энергетические затраты при последующем окислении иона
марганца мало зависят от степени его окисления [99], т. е. нон
марганца может одновременно взаимодействовать с несколькими
молекулами активного хлора. Этим можно объяснить высокую ак-
тивность марганца. По аналогии с марганцем можно предполо-
жит, что ионы хрома должны быть более активными, чем ионы
Со21, Cu2t\ Fe2+, Ni2' и др. По-видимому, этим объясняется высо-
кая активность смеси гидроксидов марганца и хрома [109]. Веро-
ятио также, что попы металлов переменной валентности, встреча-
ющиеся в некоторых категориях сточных вод, катализируют про-
цесс электрохимического окисления органических загрязнений.
Процесс электрохимической деструкции с каталитическим окис-
лением органических примесей осуществляют двумя методами: вве-
teiiiicM в электродное пространство вместе с потоком жидкости
подвижного пли гранулированного неподвижного катализатора и
ипсдепием катализатора в поток после электролиза в специаль-
ном реакторе.
Преимуществом первой схемы является возможность много-
кратного использования хлорид-иона в том же реакторе, поскольку
л юмарный кислород немедленно вступает в реакцию окисления
и । за своей термодинамической неустойчивости, а хлорнд-ион
опять разряжается на аноде и образует активный хлор. Концент-
рация хлорид-нонов в растворе остается почти неизменной, хотя
они непрерывно участвуют в окислительно-восстановительных про-
цессах, благодаря чему увеличивается общий выход активного
илора. Используя подвижные катализаторы, в поток жидкости
обычно вводят металлы переменной валентности в воде легкораст-
норяющпхея солей нли гидроксидов. Таким же образом нснользу-
юи и порошкообразные активированные угли.
Тля очистки различных категорий сточных вод расход катали-
aiopoB но активному веществу достигает 40 кг/м3 [а. с. 1011548
|‘ < < Р), 16, 68, 109, 118], поэтому если катализаторы не возвра-
ти и. повторно в цикл очистки, то себестоимость обработки жидко-
• in будет высокая. Снизить себестоимость можно созданием до-
йн 111111 ельных узлов улавливания и регенерации каталпз-i юров.
•I I, и свою очередь, усложняет технологическую схему.
203
Размещение гранулированных катализаторов, активированных
углей или материалов, содержащих оксиды металлов переменной
валентности, в межэлектродном пространстве несколько упрощает
технологическую схему очистки. Следует также отметить, что
в межэлектродном пространстве гранулы катализатора работают
как биполярные микроэлектроды, вследствие чего катализаторы
разрушаются и уносятся вместе с потоком очищенной жидкости.
Возможно забивание нор между гранулами и уменьшение про-
пускной способное! и электролизера.
Основным недостатком этого метода является разрушение ак-
тивного слоя анодных пластин, выполненных с применением не-
благородных металлов. В последнее время, как уже было сказано,
для электролиза рекомендуются аноды на титановой основе с по-
крытиями из оксидов кобальта и марганца, магнеппа и других
металлов. Обычно эти покрытия слабо связаны с титановой осно-
вой, из-за чего создается значительное сопротивление переходного
слоя между основой и покрытием.
При попадании в покрытие атомарный кислород, образующийся
при восстановлении активного хлора, может достигать титановой
основы, окислять ее и увеличивать сопротивление переходного
слоя. Анод начинает нагреваться, и вследствие разницы коэффи-
циентов температурного расширения >итанивой основы и оксид-
ного покрытия происходит расслоение и разрушение актвпого
слоя. Постепенно анод выходит нз строя [121], поэтому введение
катализатора в межэлектродпое пространство ио вышеуказанным
причинам нежелательно.
По второму варианту катализ осуществляется в отдельном ре-
акторе. В этом случае электролизер может служи!ь для минера-
лизации легкоокпеляемых органических загрязнений и получения
активного хлора в необходимом количестве. Катализ в отдельном
реакторе можно проводить как с растворенным, так и с гранули-
рованным катализатором. Растворенные и мелкодиспергнрован-
ные катализаторы также требуют улавливания и возврата. Про-
цесс технологически проще выполняется с гранулированными
катализаторами. Известны два основных варианта контакта гра-
нулированной загрузки с потоком жидкости — в виде насыпных
устройств и в виде взвешенного слоя.
Данные химической технологии свидетельствуют, что наибо-
лее эффективным является контакт во взвешенном слое катализа-
тора. Следует учесть, что окислительно-восстановительные реак-
ции могут протекать по радикально-цепному механизму. Длина
цепи радикалыю-цеппых реакций в растворах кислородных соеди-
нений хлора небольшая, поэтому необходимо стремиться к умень-
шению слоя обрабатываемой жидкости. Этому в определенной
мере способствует неподвижный слой гранулированного катализа-
тора, обеспечивающий высокую турбулизацию потока жидкости.
В реакторе с гранулированной загрузкой окислению подверга-
ется оставшаяся трудпоокнеляемая органика и обеспечивается де-
хлорирование очищенной воды, поэтому оптимальное сочетание
204
электролиза и последующего катализа может обеспечть высокую
эффективность очистки при минимальных затратах элсктроэпер-
i ин. Такая технологическая схема является наиболее перспек-
1НВ1ЮЙ.
И. Е. Флис [107] установил, что в гипохлоритных растворах ка-
галитически образующийся атомарный кислород интенсивно взаи-
модействует с трудноокисляемыми органическими соединениями.
Выделение кислорода в атмосферу уменьшается, а скорость вос-
становления гипохлорита при этом почти такая же, как и в отсут-
eiBiic органики. В качестве катализаторов в исследованиях ис-
пользованы свежеосаждепная окись кобальта н хлорид кобальта.
Существуют различные трактовки механизма каталитического
разложения активного хлора. С использованием гидроксидов ме-
1аллов переменной валентности процесс катилити icckcto разло-
жения можно рассматривать по следующему механизму [87]:
CIO- -j- 2Со (ОН), 4- Н»О -► С1~ 4- 2Со (ОН),; (4.60)
4Со (ОН), -► 4Со (ОН), 4-0,4- 2Н.0 (4 61)
Ряд авюров указывают [64, 107], что в присутствии каталнза-
оров возможно образование свободных радикалов С1О‘, СГ, не-
ус юпчивость которых может вызывать цепные реакции. В обра-
юиании радикалов участвуют как хлорноватистая кислота, так
и 11111охлорит-иои. Так, например, образование СЮ*- радикалов
п расI ворах гипохлоритов под действием Со + сшмулируется не-
прерывной реакцией [107].
CIO- 4- Соа+ -► СЮ* 4- Со,+; (4 62)
НСЮ“ 4- Со3+СЮ* 4-Со*+ 4- Н+. (4 63)
Активные радикалы СЮ* могут способствовать реакциям об-
ра «онаиня атомарного кислорода и ОН"-радикала:
процесс, определяющий скорость разложения гипохлорита:
2СЮ- 4- СЮ* 4- ОН- — ЗСГ 4- ОН * 4- 3/20,; (4 64)
быстрый процесс выделения атомарного кислорода:
ОН* 4-ОН*-^ Н3О4-О; (4.65)
менее вероятный процесс продолжения развития цепи в силь-
пощелочпой среде:
ОН* 4-СЮ--►СЮ’4-ОН-. (4.66)
Реакции (4.64) и (4.65) могут идти параллельно, каждая из
iinopijx в конечном итоге сопровождается подкислением рас-
iiiupa, что в действительности имеет место.
В присутствии органических соединений подкисление раство-
рен при каталитическом восстановлении активного хлора иезиа-
нк 1ЫЮ, а кислые растворы в иекоюрых случаях, наоборот, при-
•<ip< 1.1Ю1 более высокое значение pH [73]. Это объясняется тем,
Ч1> и ЧНС1ЫХ растворах активного хлора в радикально ценные
205
Рнс. 4.61. Комбинированный аппарат для
электрокаталитической очистки сточных
вод
реакции iiiitchchbiio вовлекаются
ионы гидроксила, что приводит к
подкислению. При наличии в рас-
Iворах органических веществ ради-
калы взаимодействуют с ними н в
меньшей степени за |рагиваюгся
гидроксил-попы, цепь быстрее пре-
рывается и в реакцию вовлекае!ся
большое количество молекул актив-
ного хлора. Вследствие этого рас-
творы подкисляются в меньшей
степени, интенсивно восстанавлива-
ется активный хлор и окисляются
за счет образующегося атомарного кислорода органические соеди-
нения.
Электрокаталитическая очистка сточных вод является новой и
еще мало исследованной, особенно в области использования не-
дорогих и доступных катализаторов, в том числе отходов про-
мышленности, содержащих оксиды металлов переменной валент-
ности. Однако растущее в последнее время количество публикаций
по данному вопросу, позволяющих оценить состояние исследова-
ний в этой области, дает основание для определенного оптимизма.
Так, например, в ЛИСП исследован метод электрокатали-
тической очистки сючпых вод от красителей, поверхностно-актив-
ных и текстильно-вспомогательных веществ, прн котором в каче-
стве катализа юра рекомендуется использовать гранулированный
активированный пиролюзит — отход термического производства
диоксида марганца.
Для реализации этого метода очистки разработан комбиниро-
ванный аппарат (рис. 4.61), который имеет два блока электродов
6 с нерастворимыми анодами и камеру доочистки 4 с гранулиро-
ванной каталитической загрузкой 8. Вода подается в аппарат
с двух сторон через штуцеры 7, последовательно проходит по
электролшпческпм ячейкам н переливается через верхнюю кромку
катодных пластин в камеру 4. Если в процессе электролиза воз-
можно образование над электродами слоя пены, как, например,
при электрофлотацпи поверхностно-активных веществ, то эта йена
наклонными козырьками крышки 1 вытесняется также в камеру
4. Местная приточно-вытяжная вентиляция 3, предназначенная
для обеспечения взрывобезопасностн, используется для интенси-
фикации пеногашения за счет создания направленной струн воз-
духа над слоем иены из щелевого впускного капала 2.
Струя поступающего воздуха изменяет направление движения
пенного слоя, прижимает его к стенке камеры, по которой сте-
кает очищенная вода из электродного блока. Потоки воздуха и
воды, двигаясь параллельно друг другу, создаю! местное разре-
200
жеиие, что ускоряет дегазацию пены, которая быстро гасшся па
поверхности гранулированной загрузки. Поступающие в камеру
доочистки потоки воды вместе с пеной распределяются сетчатым
устройством 5 по поверхности загрузки 8.
При прохождении воды и пеиокопдеисата через загрузку про-
исходит восстановление активного хлора, прн котором образую
щийся атомарный кислород окисляет находящиеся в жидкости
остаточные органические за) рязнепия. Очищенная водт собирается
в пространстве между ложным днищем 9 и дном камеры 4 и от
водится через патрубок 10.
Опытно-промышленные испытания этого аппарата производн-
1ел1>иостью 10 м3/ч при очистке сточных вод красильного цеха
показали высокую его эффективность [50]. Технико-экономические
показатели аппарата следующие: площадь анодов—16 м2. габа-
ритные размеры—1,4X1.4X2,3 м, нагрузка по току — 2400 Л,
напряжение питания — 8 В, потребляемая мощность—19,2 кВт,
себестоимость очистки I м3 общего стока красильного цеха —
8.5 коп.
4.4.3. Обработка комплексом
электрических воздействий *
Комплекс электрических воздействий (КЭВ) — сочетание воз-
действий электрических полей напряженностью порядка пробой-
ной— 1-т-ЮО кВ/см и напряженностью электрохимической и поля-
1>п «ационпой коагуляции —14-100 В/см [14. 20]. При обработке
с пробойной напряженноегью в жидкости происходит электриче-
ский разряд малой мощности (ЭРММ), который получают от ис-
точника импульсного напряжения до 3 кВ Основным преимуще-
< том обработки ЭРММ по сравнению с высоковольтным импульс-
ным электрическим разрядом (ВИЭР) является то. что ЭРММ
получают прн более низком напряжении, а это позволяет нсполь-
|ои.1ть источники питания с уменьшенными энергомассогабарит-
пыми характеристиками, имеющими более высокий КПД.
С помощью обработки ЭРММ можно одновременно обеззара-
। икать воду до кондиций питьевой, окислять органические загряз-
III иия и коагулировать дисперсную фазу, при этом в аппаратур-
ном оформлении он значительно проще, чем ВИЭР.
Сочетание с обработкой в электрическом поле напряженностью
I 10П В/см позволяет осуществлять процессы коагуляции иан-
|» ice полно, в результате чего достигается высокое качество
ни ки по фнзнко-химическим, органолептическим и бактерноло-
прнткпм показателям.
Метод КЭВ может быть использован при создании водоочист-
I <1 малогабаритного оборудования, предназначенного для
мт mil и обеззараживания природных и сточных вод, особенно
। । нижеиия токсичности и канцерогенности питьевой воды
pii.ii по! готовлен совместно с капл техн паук II И Рскобр.ц
207
Прн создании водоочистных установок с ЭРММ одним из важ-
ных условий является выбор критических параметров частоты
следования импульсов напряжения и напряженности поля в ка-
нале обработки аппарата.
Два основных фактора определяют эффективность электро-
обработки дисперсий электрическим разрядом: во-первых, при
разряде интенсифицируются процессы окисления органических ве-
ществ и происходит обеззараживание; во вторых, в электрическом
поле импульса высокой напряженности резко возрастает интен-
сивность коагуляционных процессов, определяемых поляризацион-
ным притяжением между частицами дисперсной фазы.
Таким образом, частота следования импульсов должна под-
бираться из условия максимальной эффективности этих про-
цессов.
Для любой дисперсной системы экспериментально всегда воз-
можно определить критическую напряженность Екр, при которой
происходит пробой. Величина этой напряженности определяет
скорость, с которой поп радиусом г0 преодолевает расстояние до
соседнего попа, соизмеримое с длиной свободного пробега. Так,
Гюккель решил задачу об электрофорезе попа, окруженного слоем
противоионов. Формула Гюккеля для скорости нона имеет вид:
Гэф егЕэ'(6лт)г„). (4 67)
Соответственно нетрудно получить оценку сверху частоты
внешнего поля
Лплх =- >/(н = ггЕ,/(6лт]гв1н) (4.68)
Прн длительности импульсов, равной ион успевает пройти
расстояние до соседнего нона и производит соударение с нейт-
ральными молекулами, которое вызывает ионизацию последних,
если напряженность поля достаточно высока, чтобы обеспечить
соответствующую кинетическую энергию иона, т. е. при ЕЯ=Е11Р.
Развитие лавинообразного процесса завершается возникновением
электрического разряда, причем время возникновения разряда за-
висит от начальной концентрации ионов и лимитируется длитель-
ностью свободного пробега иона до соседнего.
Таблица 4 5. Зависимость /тлх от концентрации электролитов при Екр =
= 15 кВ'см
С. моль/л’ т. см 'т.х- иГц
N»* г — 0.98А Са"+ г — |,06А АГ* г, 0 67А
0.0001 2.56-10“» 5,1 9.4 22,4
п.001 1.1Я-ю- • 109 21 48
0.01 5.5-10-» 23.5 4.3,5 10.3
0.1 2.56 -10—’ 51 94 224
20Я
Таблица 4.6. Зависимость fminj и fmax3 от размера частиц и концентрации
ионов
а. см (mln,- Го С. моль/л х. Ом см*1 (гп»х,
10-* 44 0,0001 1,08-10-* 22.5-10*
ю-« 1 4(Х> 0.001 1.06-ю-« 22,5-10*
5-10* 17 600 0.01 1.02-10-3 22,5-10’
10~* 4.4-КР 0.1 0,92-10-* 22.5-10"
Как показывают оценочные расчеты (табл. 4.5), возможно по-
лучение электрических разрядов малой мощности импульсами
с высокими частотами следования.
Оптимальная частота для обеспечения максимума поляриза-
ционного взаимодействия определяется условием: 2D/a2<f<x2Z>
Сделав подстановку D=fc77(6ni]r0) и*2 8ле2“£4С//(еокТ), получаем
неравенство:
2к Т (r>ni)TnflZ) < f &~NА 2^С,/(бт)г0е0) 1 000. (4 69)
Подставляя численные значения констант для водного раствора
однозарядного электролита (го=О,98Л), получим:
4.4 10 ‘-'о 22.4 1()*С, (4 70)
а2
Результаты расчета граничных частот /т1П2 и /тах2 представ-
кч1Ы в табл. 4.6 при различных значениях а и с, из которой видно,
что в широкой области изменения параметров имеет место
/- ,x2я5/шах.
Частота, при которой наблюдаемы эффект электростабнлиза-
IUIII, определяется условием fr-r<g.2D/d2. Сравнение frr и /тш2 по-
казывает, ЧТО f,nln2»f<T-
Таким образом, в целях создания оптимальных условий проте-
кания процессов окисления, обеззараживания, агрегирования и
концентрирования дисперсной фазы реальной гидроднсперсии ча-
юта следования импульсов определяется по выражению, согласно
которому fmax^lO8, a fmin выбирается в зависимости от размеров
шсперсных частиц, причем с уменьшением размера частота fmin
возрастает. Кроме того, проведение электрического разряда на по-
вышенных частотах позволит существенно уменьшить массогаба-
1>п1пые характеристики аппаратуры.
При обработке воды импульсом напряжения оптимальной ча-
1Ю1Ы следования можно коагулировать частицы примесей, если
пеш ня полярн <ацио1111ого притяжения U> превышает энергстпче
। кий барьер отталкивания (рис. 4.62) при —Ur.:
UF i^d^lR*. (4.71)
|<(м|- дипольный момент- | dM | — a6FrJ— I '2 4- Зб^ЛЦ- l)|; R— расстоя-
... между центрами частиц: /?«=2лЧ-Л; Л —расстояние между попермин гимн
। <1411111. соетнпяютей их центры.
‘.’09
Рис. 4 62. Зависимость энергии взаимодействия заряженных
частиц I/, (if—2,78) от отношения Л/о 1я—3 10 5 см;
2—а— 10*’см
U3,*T
240
0 002 0,04 0.06 0.08 0,1 h/a
Расчет минимальной напря-
женности поля целесообразно
производить для статистически
вероятного размера частиц, кото-
рый определяют на основании
исследования их дифференциаль-
ного распределения по размерам.
В табл. 4.7 представлены ре-
зультаты расчетов минимальной
напряженности поля для частиц
размером а=10 5 см и я=3х
X 10 s см прн С = фв =70 мВ; xw=4,8-10-4Ом-см-1; х=2,7-10*см;
А =. 1,5- 10~18 эрг.
Как видно из таблицы, для эффективной коагуляции частиц
размером ~ I05 см по поляризационному механизму напряженность
поля должна быть £тщ=10 кВ/см.
Метод комплексного электрического воздействия использован
при разработке ряда малогабаритных водоочистных установок.
Установка (рис. 4.63) предназначена для очистки и обеззаражи-
вания природных и сточных вод объектов небольшой производи-
тельности. Технические характеристики этой установки следующие:
производительное и»—10 л/ч, мощность (максимальная) — 1 кВт,
масса — 40 кг, габаритные размеры — 800X495X500 мм.
Установка состоит из блока электроочистителя, где реализу-
ется воздействие электрического разряда малой мощности и неод-
нородного электрического поля, фильтра с загрузкой из пенополи-
уретана. блока питания и насоса.
В зависимости от физико-химического
состава исходной воды и требований
к качеству очищенной волы в уста-
новке предусмотрена возможность
работы камер разряда и коагуляции
как отдельно, так и в последователь-
ном их соединении.
Установка позволяет получить
питьевую воду высокого качества из
природной воды, трудно поддаю-
щейся очистке традиционными мето-
дами (например, болотной), улуч-
шает физико-химические и органо-
лептические показатели душевой
сточной воды, гарантирует надежное
Рис. 4 63 Установка язя очистки воды с ис-
полыовапнем КЭВ
210
обеззараживание до кончицнй Таблица 4.7. Величина ГmIn
питьевой воды. Энергозатраты в зависимости от размера частиц
в зависимости от качества пс- ______________________________
ходиой воды составляют от 0,1 до 5 кВт • ч/м’. а, см ДО. кГ emln- »*"
4.4.4. Электроосаждение 10 • 71. кихю
Элек1роосажденне является 310-' 221 2 Ъ50
широко распространенным про-
цессом в технологии очистки воды, под коюрым понимается вза-
имодействие заряженных коллоидных частиц, мигрирующих
в электрическом поле, с подложкой, в качестве которой могут
служить в первую очередь катод н анод. Следует различать два
вида осаждения на электродах: электрофоретическое — взаимо-
действие коллоидных частиц с поверхностью электрода с образо-
ванием осадка при воздействии однородного поля; днполофоре-
тнчсское — образование осадка при воздействии неоднородного
злсктрического поля.
Как правило, электроосаждение характеризуется совокупностью
ряда процессов, в частности транспортом частиц к поверхности
подложки (электрода), агрегацией частиц в приэлектродном
слое, контактным взаимодействием частиц с поверхностью элект-
рода, электроосмотическим уплотнением осадка на поверхности
злектрода и др.
Одним из основных условий осуществления процесса осажде-
ния является обеспечение существенного преобладания сил куло-
новской (поляризационной) природы над гравитационными си-
лами. Размер частиц при этом не должен превышать 3—5 мкм.
В противном случае осаждение следует проводить при интенсив-
ном перемешивании
< Kopocib элекгроосаждепня, толщина и плотность осадка за-
писи! ог устойчивое in и концентрации коллоидных частиц. Устой
чпвые к коагуляции частицы быстрее осаждаются, образуя более
пло1ные агрегаты.
При осаждении нз разбавленных коллоидных споем харак-
|<-рпы два процесса: электрофорез частиц п последующее взапмо-
и иовие с электродом. При осаждении из концентрированных сп-
оем, где частицы образуют объемно-пространственную структуру
и жидкости, на первый план выступают явления электроосмоса.
Роль электроосмоса возрастает с увеличением концентрации дис-
пгрсной фазы и соотвеювенно жесткости пространственной струк-
|уры.
Взаимодействие частиц с подложкой определяется электрохи-
мическим и электростатическим механизмами. Оба подхода пра-
1ШЛЫ1Ы, по в каждом случае нх необходимо применять в конкрет-
ной спсгеме. Например, при осаждении на электродах с большим
юном обмена в приэлектродном пространстве протекают сущест-
••• IIIU.IC концентрационные изменения, приводящие к изменению
211
Ьис. 4.64. Схема распределения концен-
трации частиц при элсктрпосажденнп
в межэлектродном объеме ванны
pH и Eh, коагуляции частиц и т. п.
В данном случае электрохими-
ческий механизм является преоб-
ладающим. Однако при малых то-
ках обмена поляризация элек-
трода достаточно высока и элек-
троосаждепне следует оценивать
с позиций взаимодействия про-
тивоположно заряженных частиц и электрода в квазиэлектроста-
тпческом поле. Первый подход широко развит Ю. Ф. Дейнегой,
второй — II. Ф. Ефремовым и О. М. Меркушевым.
Количество осажденного вещества при электроосажденин за-
висит от многих факторов: m=f(E3, l,, С,„ 1э). Этн зависимости
имеют линейный характер и для разбавленных систем (при Сн~
~0,14-0,3%) могут быть описаны уравнением электрофореза. На-
пряженность поля поддерживается, как правило, в пределах 5—
30 кВ/м, что значительно выше напряженности ноля прн электро
химической обработке природных и сточных вод. Область линей-
ной зависимости во времени ие превышает 10—30 с. При /э>154-
: 30 с количество оседающих частиц достигает постоянного значе-
ния, что объясняется полной седиментацией частиц на электроде.
Во всех случаях существует крш пческое время /о, ио истече
нии которого наступает осаждение частиц иа электроде [20]. Для
агрегативпо неустойчивых систем с низким значением £-потепци
ала скорость электроосаждеиия резко падает. Зависимость т от
C,i несколько отличается от линейной, возрастает более круто и
превышает рассчитанную по уравнению электрофореза в 1,5—
2 раза.
Распределение концентрацип частиц в меж электродном про-
странстве прн электроосаждении характеризуется образованием
ряда зов (рис. 4.64) [65]. В прикатодпом слое образуется слой
осадка с постоянной концентрацией — зона АБ. Следующий
слой — БВ представляет собой дисперсную систему с повышен-
ной концентрацией частиц по сравнению с объемом жидкости —
слой ВГ. У противоположного электрода образуется слой чистой
воды /'Д (кривая /). Скачок концентрации частиц иа границе
слоев АБ и БВ объясняется коагуляционным характером взаимо-
действия частиц в осадке. Установлено, что на этой границе (кри-
вая 2) по истечении определенного промежутка времени возможно
осуществление просветленного слоя воды Б'В'. Жидкость в слое
движется вверх и при более высоких концентрациях частиц со-
единяется со слоем ГД. Это явление объясняется электроосмоти-
ческим перемещением среды.
Для ориентировочного расчета электроосаждения смеси частиц
различной природы и заряда используется формула, описывающая
электрофоре» в жидкости:
212
tn — [5Э/Эго£л/(4Л1|)| Cn£. (4 2)
Как уже указывалось, эта формула дает заниженные резуль-
таты в 1,5—2,0 раза. О. М. Меркушев [65] предложил учитывать
электроосмотическую составляющую уплотнения осадка после его
образования. В этом случае
"I Р»СН/, (г'эф + Нос), 14.73)
• ле и е„с — скорости соответственно электрофоретического и элсктроосмо-
Н1чсского уплотнения и перемещения частиц:
Сое — 4 9 Ко£е/(л»1НС (4 74)
Тогда, объединяя зависимости (4.72) — (4.74), получим
т 0.7 [ee£,,(ni))] (>,<»£„£. (4 75)
Приняв ео=ЙО, т] = 0,01 для разбавленных дисперсных систем
и вводя коэффициент аОс, учитывающий особенности осаждения
примесей из природных и сточных вод, в окончательном виде
получим:
Ш =— ClocT- (4-70)
В неоднородном постоянном поле наряду с электрофорезом
имеет место динолофорез, связанный с действием неоднородного
поля на дипольный момент поляризованной частицы. Игнориро-
вание дннолофореза оправданно при невысоких напряженностях
поля или при низкой поляризуемости частиц.
Сравнение электрофоретической и диполофоретической сил
в режиме постоянных напряжений и тока для различной геометрии
внешнего поля может быть выполнено по зависимостям, приве-
денным в работе [20].
Диполофоретические силы резко возрастают с увеличением раз-
мера частицы, и для крупных агрегатов они становятся соизмери-
мыми с электрофоретической силой даже на значительном удале-
нии от центрального электрода. Так как направление диполо-
фореза зависит от знака наведенного дипольного момента, то
дпполофорез для одних систем будет усиливать электрофорез, для
других — уменьшать скорость осаждения и, соответственно, выход
осадка по сравнению со случаем чисто электрофоретического
осаждения.
Процесс электроосаждення в технологии очистки воды играет
июякую роль. С одной стороны, это достаточно эффективный про-
цесс концентрирования загрязнений с одновременной их коагуля-
цией, что позволяет, например, обезвоживать осадки без дополпи-
1ельпого ввода реагентов [а. с. 648539 (СССР)], концентрировать
нефтепродукты, латексы и целлюлозу [20] в сточных водах и т. п.
< другой стороны, образование осадков на поверхности раствори-
мых электродов приводит к изменению электрических параметров
лек1 родной системы, падению силы тока и выходу по току про-
емов анодной (катодной) реакций.
Устройство, приведенное на рис. 4.65, позволяет удалять ча-
1ННЫ латекса из сточных вод [20]. Отрицательно заряженные
ищи перемещаются к положиюльно заряженному электроду 2
213
Рис. 4 .65. Устройство для удале-
ния твердых частии из водных рас-
творов
/ — раствор латекса; 2 — металличе-
ский сеточный а иод-транспортер; 3 —
катод
Рис 4.66. Электроочмститсль с
трехфазпым электропитанием
/ — корпус; 2 осадительный элек
трот; 3 — центральный электрод; 4
сборный коллектор
и осаждаются па нем. Осевший иа сеточном электроде латекс сду-
вается воздухом в специальный бункер.
Для выделения твердых частиц из жидкости с помощью трех-
фазного тока предложено устройство, в котором осадительные
электроды выполнены пустотелыми (рнс. 4.66) [20]. Жидкость
заполняет корпус 1 устройства и движется вдоль электродов 2 и
Л. Находясь под действием неоднородного поля, частицы коагулн
руют и осаждаются на электродах 2.
При обезвоживашш гидроокисных осадков электроосажденпе
можно осуществлять с использованием различных материалов
электродов (табл. 4.8).
Наиболее успешно электрообезвожнваине гидроксидов метал-
лов протекает с применением латунных и алюминиевых электро-
дов. Угольные электроды обеспечивают значительно большую про-
должительность обезвож (ваиия, чем алюминиевые (рис 4.67).
Для проведения процесса электрообезвоживания предлагается
конструкция ячейки, в которой катод выполнен в виде корыта,
а анод—в виде вращающегося барабана [20]. Осадок в процессе
обезвоживания налипает на поверхность барабана, с которого счи-
щается с помощью ножа (рис. 4.68). Для улучшения процесса в об-
рабатываемую суспензию (осадок первичных отстойников и актнв-
Таблица 48 Результаты обезвоживания осадков
Материал адектродои Влажность осадка, при напряжен нм электролиза, В
— 20 40 50 60 70
Нержавеющая сталь 44 — — — 93 —
Уголь 'II 9з 92 92 <эо 91
Алюминий 14 •12 90 86 86 89
Лату иь <И 92 87 82 82 83
214
Рис. 4.67. Кривые скорости фильтрования при
электрообработке осадков в зависимости от
материала электродов и напряжения элек-
тролиза
а — угольные электроды; б — алюминиевые элек-
троды; 1 — 0 В; 2 — 20 В: 3 — 40 В; 4 — 50 В; S —
Ы) В; б — 70 В
Рис. 4.68. Аппарат для электрообезвоживания
осадка
пый ил) добавляют полиакриламид в количестве 0,5—3,0 мг/л
|а. с. 648539 (СССР)]. Время обработки — 3—6 мин. Влажность
осадка снижается с 80 до 71 %. При этом наблюдается полная
стерилизация осадков.
Несмотря на достаточную простоту и эффективность метода
электроосажденпя, ему присущи такие недостатки, как трудность
осаждения частиц с низким зарядом, коррозия арматуры и обору-
дования, сложность удаления образующегося осадка на поверх-
ности электродов или фильтрующих диаграмм. Низкая скорость
транспорта частиц в жидкости, особенно при их высокой концент-
рации, приводит к неоправданному увеличению объема очистных
сооружений.
4.4.5. Электрохимическое обеззараживание
При реализации электрохимических способов водоочистки на-
ряду с достигаемыми санитарно-гигиеническими показателями
обеспечивается в той нли иной степени обеззараживание жидкости,
так как электролиз водных растворов сопровождается образова-
нием в объеме электролита сильных дезинфектантов. Это отно-
сится также и к воздействию иа гидросистемы электрических раз-
рядов, вызывающих уничтожение бактериальных тел за счет теп-
лового и радиационного излучения, образования радикальных
продуктов, влияния сильных электрических полей, ударных воли и
Других факторов, что может привести к консервации воды. Надо
jkciiiic электрического поля на обрабатываемую жидкость может
215
вызывать необратимое агрегирование микроорганизмов, что позво-
ляет отделить их на фильтре очистки от грубодисперсиых при-
месей.
Электрохимическое получение хлора из хлоридных растворов
с последующим его растворением в объеме электролита приводит
к образованию в соответствии с уравнениями (3.72) — (3.74) бак-
терицидных агентов HCIO и СЮ“. которые успешно использу-
ются для обеззараживания питьевых и сточных вод. В последние
годы наибольшее применение для этих целей находят электролиз-
ные установки получения на месте потребления гипохлорита на-
трия (NaClO) из естественных хлоридных растворов—подземных
минерализованных вод и морской воды, а также нз искусственных
растворов хлористого натрия.
Электролитически полученный гипохлорит натрия более акти-
вен, чем такой же реагент, полученный химическим путем. Это
объясняется тем, что в природных электролитах имеются соедине-
ния йода, брома и другие, которые при электролизе образуют силь-
ные окислители.
Выход по току электролитического хлора возрастает при по-
вышении плотности тока и концентрации NaCI в электролите,
снижении его температуры. Весьма существенное значение имеет
характер движения электролита. При электролизе без перемеши-
вания прианодиого слоя в растворе гипохлорит натрия накаплива-
ется более высокой концентрации, чем с перемешиванием из-за
снижения диффузии ионов CIO к аноду и их последующего раз-
ряда.
Электроды работают в условиях контакта с химически актив-
ными веществами, что требует стойкости их к износу и разруше-
нию. Прн этом используют в качестве катодного материала сталь,
графит, титан, в качестве анодного—графит, магнетит, платину
и комбинированные материалы: платину — ирридий, ОРТЛ, пла-
тину— титан и др. Наибольший практический интерес для про-
цесса получения гипохлорита натрия имеют эзектроды с актив-
ным покрытием на основе двуокиси рутения (ОРТЛ). Срок службы
таких электродов составляет до 4 лет и более При концепт рации
хлоридов в растворе 30—35 г/л рекомендуемая плотность тока для
таких электродов должна быть не выше 1000 Л/м2 при папряже
нии около 5 В. Прн снижении содержания хлоридов в рассоле до
7—15 г/л плотность тока должна быть снижена до 700 А/м2 прн
напряжении 6—7 В [63].
На практике применяют два способа электролитического обез-
зараживания- получением гипохлорита натрия электролитическим
путем с последующим введением его в обрабатываемую воду и
прямым электролизом. Наибольшее распространение получил пер-
вый способ, позволяющий уменьшить габариты оборудования для
электролиза и повысить надежность его работы.
Получение гипохлорита натрия электролизом производят п пе-
риодическом или непрерывном режимах Прн периодическом ре-
жиме работы исходный раствор соли заливают в непроточный
216
Электролизер, проводят электролиз до достижения требуемой кон-
центрации гипохлорита натрия, а затем раствор сливают в бак-
накопитель, откуда его дозируют в обрабатываемую жидкость
Для осуществления этого процесса НИИ КВОВ совместно с ПКБ
ЛКХ им. К. Д. Памфилова разработаны, а машиностроительным
заводом «Коммунальник» серийно выпускаются электролизные
установки непроточного типа марки ЭН производи тельностью 1—
100 кг активного хлора в сутки [63, 105|. Основные параметры ра
боты этих установок приведены ниже.
ЭН-1 ЭН-5 ЭН-25 ЭН-100
Производительность актив- ного хлора, кг/сут . . . 1 5 25 100
Ьдельиыи расход соли на 1 кг активного хлора, кг 12 15 12 15 8 9 8 9
Продолжительность цикла электролиза, ч .... 0,075- 0,9 Я 9 10 12 5 6
Рекомендуемое число цик- лов в сутки 3 -5 2 2 3 4
Концентрация активного хлора в растворе, г/л у 6-8 10 12 10 12
Рабочее напряжение па ван- не, В ........ 40 42 40 -42 55 -65 220 -230
Рабочий ток, А .... 55 65 55 65 130 140 4(H) 4.50
V icTbiiui'i расход электро- wiepi ин па 1 кг активного хлора, кВт-ч 7 -9 7 9 8—10 10 12
Стоимость, тис. руб. . . 2,6 2,9 3,4 8,9
Проточный режим предусматривает непрерывную обработку
раствора солн в одном или нескольких последовательно соединен-
ных электролизерах с одновременным дозированием его в обраба-
тываемую воду. Однако установки с таким режимом работы не
получили широкого распространения из-за значительных потерь
активного хлора и трудностей в эксплуатации.
В ИКХнХВ АН УССР предложены электролитические гппохло
рнтиые установки непрерывного (КГ-12, КГ-13) и периодического
(КГ-14 п КГ-15) действия [54]. Технологическая схема установки
КГ-13 представлена на рис. 4.69. Приготовленный в баках 4
10 %-ный раствор поваренной соли насосом 3 перекачивается в ра-
бочий бачок 2, откуда подается по трубе 5 в сифонный бачок /,
обеспечивающий поступление рассола определенными порциями и
осуществляющий разрыв его струи, чем предотвращается утечка
юка через электролит. Рассол нз сифонного бачка поступает в рас-
пределительный бачок 8, из которого стекает в приемные воронки
7 электролизеров 6. Последние представляют собой бездиафраг-
меиные ванны, в которых катодом служит корпус, а в качестве
анодов использованы электроды хлорных ванн. Дальнейшее раз-
витие метода направлено па снижение удельного расхода солн
при получении I кг активного хлора, предотвращение отложеннп
солей жесткости на электродах, регулирование температурною
режима электролиза с целью его оптимизации.
217
рассола на выходе 60—70 г/л, что
Рис. 4 69 Технологическая схема
стационарной гипохлоритной уста-
новки непрерывного действия КГ-13
Источником водоснабже-
ния некоторых автономных
объектов может быть пить-
евая вода, получаемая в ре-
зультате дистилляционного
опреснения солевых вод н по-
следующей нх минерализации.
Современные опреснительные
установки дистилляционного
типа имеют солесодержание
по сравнению с солесодержа-
иием морской (океанской) воды превосходит данный показатель
в 1,5—2,0 раза.
В связи с изложенным представляет особый интерес возмож-
ность использования для получения гипохлорита в качестве соле-
содержащего раствора повышенной концентрации — рассола от оп-
реснительных установок, который используется одновременно для
минерализации и обеззараживания дистиллята.
По данной технологической схеме возможно создание компакт-
ной малоэнергоемкой системы бытового водоснабжения.
ГЛАВА 5
ЭЛЕКТРОХИМИЧЕСКАЯ ТЕХНОЛОГИЯ
ВОДОПОДГОТОВКИ И ОЧИСТКИ СТОЧНЫХ вод
5.1. ПОДГОТОВКА ПРИРОДНЫХ ВОД
ДЛЯ ЦЕЛЕЙ ВОДОСНАБЖЕНИЯ
5.1.1. Электрокоагуляция при обработке природных вод*
Элек рокоагуляния может использоваться для осветления, обес-
цвечивания, обескремнивания, обезжелезивания, обескислорожива-
ния, частичного умягчения н других видов обработки воды [28,
54]. Наиболее широкое применение электрокоагуляция нашла при
осветлении и обесцвечивании воды — снижении цветности, обуслов-
ленной, как правило, весьма разнообразным составом органиче-
* Материал по потоп теп совместно с проф. В Д Дмитриевым.
218
ских примесей, формирующихся при участии почвенного и торфя-
ного гумуса, планктона, высшей водной растительности, продуктов
жизнедеятельности н распада флоры и фауны водных бассейнов,
а также органических загрязнений, вносимых в воду хозяйствен-
ными и производственными стоками.
В большинстве случаев окраска природных вод, особенно Се-
веро-Запада и Севера европейской части СССР, вызвана наличием
в воде гумусовых веществ, которые находятся в виде соединений
различной дисперсности — от взвесей и коллоидов до истинных
растворов. В основной массе гумусовые вещества представляют
гуминовые кислоты и фульвокнелоты. Эти группы гумусовых ве-
ществ в наибольшей степени затрудняют технологические процессы
водоподготовки прн обесцвечивании высокоцветных природных
вод.
В рекомендуемой СНиП 2.04.02—84 формуле по определению
взвеси массовое выражение цветности установлено в виде зависи-
мости М = 0,25 мг/л по цветности. В других литературных источни-
ках величина массового количества взвеси, обусловливающая
1 град, цветности, приводится равной 0,20—0,88 мг/л. Такое рас-
хождение объясняется различием молекулярного веса гуминовых,
апокреиовых и креповых кислот н, следовательно, различной коа-
гулируемостью этих фракций.
Анализ фракционного состава органических примесей, создаю-
щих цветность, показывает, что в среднем 1 град, цветности гуми-
новых кисло) эквивалентен 0,066 мг/л. 1 град, для апокреиовых
кислот — 0,096 мг/л и 1 град, для креповых кислот — 0 821 мг/л;
соответственно 1 мг/л вещества создает цветность, по усредненным
данным, 24,1, 12,6 и 1,9 град.
Как известно, в составе природных вод более 50 % веществ,
обусловливающих цветность, приходится на креповые кислоты.
В то же время при любом виде коагуляции нз воды в первую оче-
редь удаляются гуминовые и апокреновые кислоты. Следовательно,
прп преобладании в воде креповых кислот основная масса орга-
нических веществ прп реагентной коагуляции не удаляется. В связи
с этим принятое измерение органических веществ условными гра-
дусами не отвечает реальному содержанию их в воде и, па наш
взгляд, на современном этапе развития техники очистки высоко-
Рнс. 5.1. Зависимость эффекта обесцвечива-
ния воды от плотности токл и количества
(оз. Разлив)
/ - в осенний период Пнсх-50 гряд.; 2 в шм
ний период Писх > 100 град: 3 в весенний пав»
док Цисх 150 град: /—в летний период 11и< -
ч0 град.
219
Рис. 5.2. Зависимость изменения
окнсляемости Ок и содержания же-
леза от плотности тока и количества
электричества (оз. Разлив)
/ — в зимний период (ОкИсх-20 мгО:/л);
2 — в весенний неводок Юкисх —
— 15 мг О>/л); 3 —в зимний период
(Гсисх “1.8 мг/л); 4— в весенний па-
водок (FeMCX “2.2 мг’л)
Рис. 5.3. Снижение цветности воды
в зависимости от дозы peaieiiTa и
режима электрокоагулированив
/, Г —при Инсх “50 грвд : 2, 2" — при
Иисх “150 град.. I. 2 —при непрерыв-
ном «лсктрокоагулнровании. /'. 2 при
прерывистом элсктрокоагулироианин
цветных вод необходимо переходить к новым характеристикам,
отражающим в полной мере фракционный состав органических
веществ, обусловливающих цветность воды.
Вода большинства поверхностных источников обладает, по-
мимо высокой цветности, низкими мутностью и солесодсржапием,
а также повышенной агрессивностью. При таких показателях ка-
чества воды коагулирование ее химическими реагентами по всегда
дает результаты, отвечающие требованиям стандарта на пшьевую
воду.
Эффективным приемом совершенствования технологического
процесса водоочистки является электрокоагуляция, которая может
осуществляться при неренолюсовке и прерывистой подаче электри-
ческого тока.
Электрокоагуляция при фиксированном времени переполюсовки
(30 мин) гарантирует высокую эффективность этого метода в раз-
ные месяцы н сезоны года. При этом степень обецвечивания воды
не всегда пропорциональна исходной цветности н удельному коли-
220
честву электричества (рис. 5.1).
Это касается и других показате- 1
лей исходного качества воды: 1
перманганатной окисляемости,
содержания железа (рис. 5.2),
а также pH, щелочности, жест-
кости и т. д.
Достаточно высокий н прак-
тически одинаковый эффект
обесцвечивания воды с различ-
ной исходной цветностью в осен-
ний, зимний и весенний периоды
года, равной соответственно 50,
100 и 150 град, достигается при
количестве электричества 36 Кл/л,
стандар+а на питьевую воду. Однако в летний период при ЦцСх =
= 854-95 град, требования стандарта при прохождении через
воду того же количества электричества не достигаются. Очевидно,
такое различие в обработке воды может быть объяснено только
различным составом органических примесей, создающих цвет-
ность. Отмечая отсутствие прямой связи между цветностью и
Таблица 51. Влияние режимоя
прерывистой электрокоагуляции н
обесцвечивание волы (Иисх:= 100 грал.
। = 10 Azm*)
Режим электролиз!» ч Доза АР+. г/м’ Цветность фильтратя, гряд.
1 0,5 3,34 4,5
1 : 1 3,34 lfi,5
! • 2 3,34 23,0
0,5 0,5’ 1.67 5,0
0,5: 1 1.67 21,5
что обеспечивает
требования
удельным количеством электричества, следует указать также на
ряд обстоятельств, характеризующих особенности таких органи-
ческих соединений.
В летний период резко ухудшаются органолептические пока-
затели воды и заметно повышается содержание кроновых кислот,
а содержание гуминовых кислот понижается. Хотя общая цвет-
ность в этот период, как правило, ниже, чем в паводковый и даже
зимний периоды, условия очистки воды значительно ухудшаются
по сравнению с остальными сезонами года. В этот период добиться
снижения цветности до стандартного показателя удается только
при <7э=9О Кл/л или при предварительном воздействии хлора
позами до 2 мг/л и qt=36 Кл/л.
Прерывистое электрокоагулированне — технологический про-
цесс обесцвечивания, при котором время от времени прекраща-
ется поступление электрогенерирова иного реагента (металла)
в воду. Процесс в этом случае происходит в условиях контакта
воды с ранее образовавшимися при электрокоагулированни хлопь-
ями гидроксидов.
Установлено [54], что адсорбционные свойства гидроксида
в обычных условиях коагулирования используются ие полностью,
а лишь па 50—60%, поэтому прекращение подачи коагулянта не
сразу вызывает повышение остаточной цветности воды.
Прн прерывистой электрокоагуляции оптимальным может быть
принято соотношение времени подачи и перерыва в часах электри-
ческого тока, р?вное 0,5- 0,5 (табл. 5.1).
Обесцвечивание воды оз. Разлив с разной исходной цветностью
при различных способах электрокоагулироваиия (рис. 5.3) по-
казывает, что прерывистая обработка не ухудшает эффекта
Таблица 5.2. Характеристика промывных вол
Интенсивность промывки, л/с-м Цветность, грид Взвешенные вещества, мг/л Железо, мг/л ОстаточиыП алюминиП. мг/л
12 90.0 5,0 1,09 0,60
92.0 4.3 0,99 0,12
13 90.0 2.0 0,87 0.30
90.0 1,83 0,85 0,04
89,0 1.80 0,82 0,19
15 88,5 1,56 0,80 0,01
16 89.5 1,62 0,83 0,18
88,5 1,53 0,80 0,01
Примечание. В числителе даны показатели промывной воды при непрерывной
электрокоагуляции, в знаменателе — при прерывистой.
очистки, а, наоборот, увеличивает глубину обесцвечивания воды.
Кроме того, такой прием обеспечивает экономию металла и сокра-
щает на 50 % расход электроэнергии. Наряду с этим в последую-
щих фильтрационных сооружениях наблюдается более медленный
рост потерь напора н увеличивается продолжительность фильтро-
цикла Это, в свою очередь, допускает повышение скорости фильт-
рования и сокращает расход воды на собственные нужды.
Общие потери напора в фильтре высотой загрузки 1,0 м и раз-
мером фракций песка 0,5—2,0 мм при движении воды снизу вверх
н с длительностью фильтроцикла 10 ч составляют 0,9 м прн непре-
рывной электрокоагуляции и 0,52 м — при прерывистой, причем
наиболее интенсивный прирост потерь напора имеет место в ниж-
нем слое фильтра.
Продолжительность фильтроцикла прн прерывистом электро-
коагулнрованпи увеличивается в 1,5—2,0 раза, что позволяет со-
кратить число промывок фильтра. Отмывка загрузки при непре
рывпом и прерывистом электрокоагулпровапни завершается в ос-
новном в одно и то же время. С точки зрения оценки промывки
филырз но качеству промывных вод (табл. 5.2) можно считать
что 6-минут пая промывка песчаной загрузки с интенсивностью
13 л/с-м2 нрн электрокоагулпровапни является вполне падежной.
При удалении цветности с использованием в качестве коагу-
лирующих ионов Fe2+ и F*1 возникает ряд особенностей.
Прп реагентной коагуляции природной воды солями Fe2+ основ-
ной задачей является перевод гидрата закиси железа в гидра!
окиси. Окисление закисной формы железа в окисную с приемле-
мым для практики временем (до 15 мин) происходит только при
значениях рП —7,5-=-9. тогда как при рП = 6,5 : 7 на этот процесс
требуется от 20 до 90 мин. Применение солей трехвалентного же-
леза [FeCI3. Fe(SO<h. хлорированного купороса] для коагулпрова
ния воды без ее подщелачивания возможно в том случае, если pH
после добавления коагулянта не становится ниже 5. Прн этом для
222
начала и завершения процесса коагуляции требуется избыток щ<
лочиосп! ие менее 1,5 мг-экв/л.
Если в воде находятся органические соединения и их комп-
лексы, в которых содержится железо, то эти соединения не всегда
могут быть удалены с применением коагуляции и тем более при
обработке солями железа. В этом случае нередко в процессе вве-
дения в воду коагулянта образуется большое число мелкодисперс-
ных зародышей, которые совершенно не оседают в отстойниках
и плохо задерживаются. Во всех случаях ввод в природную воду
катионов Ее2^ и Fe3 приводит к тому, что в результате взаимо-
действия их с органическими соединениями образуются сильно
окрашенные комплексы, вследствие чего происходит повышение
цветности обработанной воды по сравнению с исходной. Это явле-
ние, наблюдаемое прп обработке высокоцветных мягких вод со-
лями железа, называют «возвратом цветностиэ, вызываемым пере-
ходом окрашенных соединений двухвалентного железа в еще более
окрашенные соединения трехвалентного железа, хотя необходимо
заметить, что и соли двухвалентного железа способны образовы-
вать с гуминовыми веществами устойчивые малорастворимые
формы [7]. Отсюда следует, что если соли железа хорошо коагу-
лируют при повышенных pH, то органические соединения, обра-
зующие цветность, приобретают наиболее устойчивые формы. В то
же время они лучше коагулируют при пониженных pH = 5-^-6,5.
В этом нрот нворсчпн и заключается одна из причин, объясняющая
совершенство коагулирования органических примесей солями
железа.
Сказанное об особенностях процессов при применении солей
железа в качестве коагулянтов в полной мере относится и к элект-
рокоагуляции с использованием железных электродов.
Прн электролитическом растворении железа происходит обра-
зование хорошо коагулируемых хлопьев темно-зеленого цвета —
1'е(ОН)2, которые за счет растворенного в воде кислорода могут
окисляться до Fe(OHh. Для окисления 1 мг Fe2+ требуется тео-
ретически 0,143 мг растворенного кислорода или 0,42—0,5 мг
хлора.
В воде с Т— 15 °C содержится ~ 10 мг/л кислорода. Такого
количества кислорода достаточно для окисления в трехвалентпое
железо 70 мг/л двухвалентного, которое может быть получено
электролитически прн </я=240-Е 260 Кл/л Следовательно, наряду
с формированием Fe(OH)3 в воде после полного использования
кислорода образуется Fe(OH)2, служащий резервом дополнитель-
ного снижения цветное! п за счет восстановительных его свойств.
Гак, полное или частичное удаление нз воды кислорода перед
злек1рокоагуляцней, к примеру, вакуумной дегазацией, приведет
к эффективному обесцвечиванию с меньшими энергозатратами
(рис 5.4) Здесь следует заметить, что в первый момент происхо
ни увеличение цветности, а не снижение се. Это может быть объ-
яснено наличием в воде остаточного кислорода, который окис-
ляет двухвалентное железо н создает условия для образования
223
Рис. 5.4. Влияние предварительного удаления
кислорода иа снижение цветности воды при ко-
личестве электричества
/ — 40 Кл/л; » — 60 Кл/л; Л-80 Кл/л; 4—100 Кл/л
множества зародышей и комплексных
соединений с органическими вещест-
вами.
Определенный интерес прн обесцве-
чивании воды представляет одновремен-
ное воздействие электрического тока и
химических реагентов [28]. Введение реа-
гентов-коагулянтов снижает несколько
pH, увеличивает электропроводность,
уменьшает напряжение между электро-
дами и создает условия для первона-
чального образования гидроксидов. Воз-
действие электрического поля улучшает
взаимодействие между образующимися
нонами гидроксида и коллоидными при-
родными частицами воды. В этом случае создаются наиболее бла-
гоприятные условия по значению pH для коагулирования гидро-
фильных органических веществ: если раствор AlfSOOs понижает
pH, то анодное растворение алюминия в кислой и нейтральной
средах повышает его (рис. 5.5).
Практическая реализация комплексного воздействия процесса
электролиза с нерастворимыми электродами (графит —графит,
ОРТЛ -графит, ОРТА — нержавеющая сталь) с примененном
в качестве химического реагента сернокислого алюминия показы-
вает, что обесцвечивание воды с исходной цветностью 90—
108 град, до стандартных требований (рис. 5.6) достигается при
</,= 154-20 Кл/л; /, = 64-12 А/м2 в дозе коагулянта Дк=0,5ДР (где
Др—расчетная доза по СНиП 2.04.02—84), в то время как ре-
Рис. 5.5 Игмснепис pH рас-
творов NaCI при электро-
коагуляции с исполыонапи-
ем алюминиевых анодов
Рис. 5.6. Зависимость снижения
цветности воды от количества элек-
тричества и дозы Ah(S(h)3
/ —Дк 0.37Др-10 мг/л; ?-Лк О.4.ТЛр
15 мг/л; 3 Дк 0.5Д- 20 мг л
Л р
224
Таблица 53. Анодный выход алюминия по току при Дк = (0,0 : 0,254-0.5)Др
Количество электричества, Кл/л Выход металла, к, при плотности тока. А/м’
2.5 5.0 10 50
1800 874-824-70 1134-1024-98 1174-1154-110 1274-1244-107
3600 824-784-67 1054-954-93 1194-114 : 108 1284-1194-105
5400 784-754-64 1024-934-90 1224-1104-102 1304-113—104
альиый расход AhfSOib до достижения Ц<20 град, составлял
50—55 мг/л вмесю расчетного 40—45 мг/л.
Использование растворимых алюминиевых анодов с предвари-
тельным введением раствора коагулянта обеспечивает снижение
цветности до 4—8 град, при ^»=20 Кл/л, t.=5 А/м2 и Дк=20 мг/л.
При этом образование крупных хлопьев и их осаждение заверша-
ются при /<20 мин, тогда как прн обычном химическом коагули-
ровании той же дозой Als(SO*)3 процесс седиментации длится бо-
лее 40—50 мин.
Анодный выход алюминия (табл 5.3) ингибируется влиянием
ионов SO*2-, поэтому для данного процесса более эффективным
является применение нерастворимых анодов и алюминиевых ка-
тодов (графит — алюминий, ОРТА — алюминий). Оптимальными
параме1рами для обесцвечивания воды с ЦИСх<100 град, следует
считать: при Дк = 0,25ДР— необходимое количество электричества
20 Кл/л, при Дк= 0,37 ДР— 15 Кл/л и при Дк = 0 5Др— 10 Кл/л;
при плотности тока t=!0 А/дм2 эффект обесцвечивания снижа-
ется. Влияние изменения температуры на обесцвечивание воды и
выход алюминия сказывается несущественно. Катодный выход
алюминия показан на рис. 5.7.
Схемы очистных сооружений при обработке природных вод
с применением электрокоагуляции (рис. 5.8) в значительной мере
зависят от стадии проектирования — для вновь строящихся или
реконструируемых сооружений.
Прн реконструкции электрокоагуляционное устройство может
монтироваться в смесителях, а в некоторых случаях и в других
сооружениях, например в отстойниках, осветлителях со взвешен-
ным слоем. На рис. 5,8, в показан вариант размещения блока
электродов в осветлителе коридорного типа. В осветлителях с под-
донным шламоуплотнителем пакеты электродов могут устанавли-
ваться в центральной камере.
Для вновь проектируемых сооружений схемы могут быть упро-
щены: установка пакетов электродов осуществляется в специаль-
ных электролитических ваннах (рис. 5.8,а), после которых вода
поступает на отстаивание или фильтрующие устройства.
Так как при электролизе имеет место выделение газов, то это
явление должно в полной мере использоваться для флотирования
примесей. Этот в данном случае благоприятный эффект может
8 Заказ № 2148
225
Рис. 5.7. Зависимость катодного выхода алю-
миния от pH среды при плотности тока
/ — 10 А/м’; 2 — 20 А/м’; 3 — 30 А/м*; < — 40 А/м*:
5 — 50 А/м’
улучшить процесс отделения диспер-
сий от воды и значительно снизить
грязевую нагрузку на фильтрующие
устройства. Флотирующий эффект
также должен использоваться и в ре-
конструируемых сооружениях.
Применение электролитической и
электрореагентной коагуляции вно-
сит существенное изменение в техно-
логию обработки воды. Выделение
пены на поверхности воды, удаление
и обработка ее становится неотъем
лемой технологической стадией в общем процессе водоподготовки.
Приготовление электролитического раствора и его дозирование
может осуществляться путем пропуска части или всей обрабаты-
ваемой воды через электролизер, а также приготовлением электро-
литического раствора требуемой концентрации в отдельной не-
проточной ванне н последующим введением его в обрабатывае-
мую воду (рис. 5.8, б). В этом случае подготовка его осуществля-
ется в воде с высоким солесодержаннем, что резко снижает на-
пряжение между электродами и уменьшает, соответственно, рас-
ход электроэнергии.
Рис. 5.8. Принципиальные схемы обработки воды электрокоагуляцией
а — схема расположения сооружения; б — схема ввода реагента в обрабатываемую воду;
в — расположение электродов и схема движения воды в осветлителе со взвешенным
слоем при его реконструкции; / — подача воды; 2 — электролизер; 3 — пакет электродов;
4 — дырчатые перегородки; 5 — полупогружеииая перегородка; 6, 7 — варианты подачи
воды нз электролизера на фильтр; в —фильтр; 9 — лотки для сбора пены
22Ь
При злсктрореагептной коагуляции химический реагент гото-
вится и дозируется по общепринятым схемам: после смешения
воды направляется в электролитические аппараты, причем коли
чесгво подаваемой воды на аппарат может регулироваться и ко-
лебаться от 50 до 100 %.
К преимуществам электрокоагуляции при очистке природных
вод следует отнести исключение реагентного хозяйства и необхо
димость доставки коагулянтов, что особенно важно для отда ien-
ных малодоступных районов страны, сопутствующее снижению
содержания ионов кальция и магния на 15—20%, сульфатных
ионов — на 8—12 %, хлоридных — на 3—15%. Активность элек-
|рокоагулироваиного коагулянта значительно выше, чем получен-
ного реагентным путем. Повышение pH при электролизе природ
ных вод во многих случаях достаточно для завершения процесса
гидролиза. Высокий бактерицидный эффект электрического тока
снижает расход реагентов на обеззараживание воды.
В то же время электрокоагуляция предъявляет новые требо-
вания к технологии обработки воды и изменяет сложившиеся
в настоящее время представления. К ним относятся: необходи-
мость наличия электрооборудования (выпрямители тока, электро-
щиты, электрокабель, электроарматура и т. п.), подготовка более
квалифицированного персонала, расход фондируемых материалов
(листового алюминия, железа), периодическая замена электрод-
ного блока.
Электрокоагулнрованне природных вод должно найти широкое
применение в отдаленных районах СССР и особенно в районах
Крайнего Севера, куда затруднена транспортировка материалов
При этом методе массовый расход веществ, применяемых для коа-
|уляцпи. снижается в 25—50 раз по сравнению с реагентной коа-
1} ляцией.
5.1 2. Подготовка воды с применением сильных
электрических воздействии '
В настоящее время все большее внимание уделяется вопро-
сам освоения богатств Мирового океана и природных ресурсов
Крайнего Севера, Сибири и Дальнего Востока. При решении этих
л дач возникает потребность снабжения небольших контингентов
нолей доброкачественной питьевой водой. Водоснабжение боль-
шинства автономных объектов (морские и речные суда, вахтен-
ные поселки и т. д.) осуществляется <возпмой» водой, качество
которой при длительном хранении значительно ухудшается из за
выделяемых в воду веществ разрушения антикоррозионных по-
кры1ин н продуктов коррозии материалов цистерн н трубопро-
вода. а также жизнедеятельности микроорганизмов и продуктов
и моаболизма.
'I л сериал подготовлен совместно с капд. техн, наук Н. И Р^кобратскнм
227
Рнс. 5.9. Схема лабораторной установки по исследованию КЭВ
/ — формирователь импульсов; 2 — высоковольтный трансформатор; 3 — усилитель мощно-
сти; 4 — блок питания. 5 — задающий генератор; ( — мультивибратор; 7 —емкость очи-
щенной воды; в —фильтр с полиуретановой загрузкой; S—разрядная камера; 10 — ем-
кость исходной воды, // — осциллограф Н 117; 12 — осциллограф С-15
Специфические условия автономных объектов не позволяют
использовать для водоподготовки традиционные реагентные ме-
тоды. Одним из прогрессивных методов очистки воды, обеспечи
вающих комплекс операций (осветление, обесцвечивание, обез
железивапие, обеззараживание), является комбинированная обра-
ботка сильными электрическими воздействиями (КЭВ), под
которой понимается обработка импульсным электрическим полем
и разрядом малой мощности (см. п. 4.4.3).
Разработанная в ЛИСИ экспериментальная установка по реа-
лизации этой технологии (рис. 5.9) состоит из камеры электри-
ческих воздействий, фильтра с полиуретановой загрузкой и комп-
лекса аппаратуры для получения электрических воздействий и
контроля их параметров. Источник питания обеспечивает подачу
на электроды камеры пучков импульсов напряжением 0,7—1,2 кВ
и частотой 10 кГц со скважностью 2. Электроды изготовляются
из стали (Х-18 Н 9Т) и вольфрама (BA 1А). Производительность
установки варьируется от 1 до 9 л/ч, и, соответственно, скорость
потока в канале камеры электрических воздействий изменяется
от 1,7 до 15,3 см/с. Конструкция фильтра позволяет производить
фильтрацию воды от периферии к центру.
Результаты работы этой установки с амплитудой разрядного
тока от 60 до 120 мА, представленные в табл. 5.4, подтверждают,
что обработка воды по предложенной технологической схеме по-
зволяет осветлять и обесцвечивать ее до требований
ГОСТ 2874—82 «Вода питьевая». Незначительная эрозия стали
используемых электродов при рекомендуемых режимах обра-
ботки воды с исходным содержанием железа 0,05 мг/л (табл. 5.5)
свндетельс1вуст о преимущественно силовом воздействии элек
трнческого поля па процессы коагуляции частиц, так как влияние
продуктов электрохимического растворения электродов при этом
несущественное.
228
Таблица 5.4. Некоторые показатели
качества исходной и обработанной воды
в аппарате с использованием КЭВ
Скорость воды в канале электро- обработки, см/с Покзззтели качеств* воды
Мутность, мг/л Цветность, град- Железо, мг/л
1,7 2,3/0,2 30 1 1,3 0.05
5,1 3,7/0 1 60 1 3,4 0.07
10,2 3.4/0.2 52'2 2.9/0,08
15,3 4,7/1.2 30 10 2,8/0,30
Примечание- В числителе приведены
показатели исходной воды, в знаменателе — очи-
щенной.
Т а б л и ц а 5.5. Эрозия стального
электрода в камере электрических
воздействий
Скорость • потоке РОДЫ в канале элек- трообработки, см/с Концентрация железа в обработан- ной воде, мг/л
'иод Катод
1,7 0,11 0,10
5,1 0,06 0,05
10,2 <0,05 <0,05
15,3 <0,05 <0,05
Для консервации запасов «возимой» воды обычно предусмат-
ривается ее серебрение или хлорирование, но введение электро
литического серебра или хлора не исключает необходимость очи
стки воды от продуктов разрушения антикоррозионного покрытия
и коррозии металла резервуара, поэтому перед употреблением
закую воду также необходимо осветлять или обесцвечивать
При обработке в блоке КЭВ питьевой воды, предварительно
насыщенной ионами серебра в концентрации до 0,72 мг/л, сниже-
ние содержания серебра почти не наблюдается. В то же время
пропуск хлорированной водопроводной воды через установку свн-
(етельствует о значительном снижении концентрации активного
хлора (табл. 5.6).
Таким образом, обработка воды но двухступенчатой техноло-
тческой схеме (электрические воздействия и фильтрование)
обеспечивает осветление, обесцвечивание, обезжелезивание и
(схлорнроваиис воды до кондиций питьевой прп незначительном
расходе материала электродов При этом не наблюдается сорб-
ция электролитического серебра на фильтрующей загрузке.
Прн обработке силь-
ными электрическими воз-
юйствнямн (напряжен-
ность электрического поля
равняется или несколько
меньше пробойной вели-
чины) открывается воз-
можность получения де-
•ннфектантов из водной
среды, которые как ot-
ic.паю, так н в сочетании
воздействием сильного
(Лектрнческого поля прн
поляг к обеззараживанию
п<> ты
Tfa б л н ц а 5 6. Концентрация активного
хлора до и после обработки КЭВ
Скорость поток а воды •*’* в канале обработки, см'с Концентрация активного хлора в воде, мг/л
В ИСХОД- НОМ в обработанной
после камеры электро- воздей- ствий после камеры электровоз- действий и фильтр
1.7 5,3 0,68 0,30
5,1 5,7 1,00 0,50
10.2 5,1 1,30 0.70
22П
Рис. 5.10. Технологический об-
разец малогабаритною блока
очистки питьевой воды
Вклад электрофизических процессов сводится к транспорти-
рованию, концентрированию и удержанию микробных тел в при
электродных областях, где концентрация дезинфектантов паи
большая. Сильное электрическое поле воздействует на клетку,
вызывая поляризацию ее структуры, изменение мембранных и
концентрационных потенциалов, пробой мембраны, внутренний
электрофорез. Совокупность физических и химических бактери-
цидных факторов создает благоприятные условия для инактива-
ции микробных тел.
При обработке воды электрическим разрядом малой мощно-
сти (ЭРММ) перечень обеззараживающих факторов расширяется
за счет сопутствующих разряду процессов радиационного н теп-
лового излучений, получения окислителей и радикальных продук-
тов и ударной волны. Воздействие электрического разряда
с целью обеззараживания питьевой воды было реализовано при
создании технологического образца малогабаритного блока про
изводительностью до 6 л/ч (рис. 5.10), который состоит из гидрав-
лического аккумулятора, предназначенного для приема порции
жидкости и последующей подачи в камеру обработки ЭРММ.
фильтрующих элементов иа основе пенополиуретана, регулирую-
щей и переключающей арматуры.
Разрядная камера выполнена в виде последовательно чере-
дующихся друг с другом каналов обработки п ячеек смешения,
вдоль которых проходит стержневой электрод (нержавеющая
сталь), служащий протнвоэлектродом. Игольчатый электрод нз
вольфрама расположен в каналах обработки перпендикулярно
направлению движения жидкости. Напряжение на электроды ка-
мер подается из узла электропитания, состоящего нз задающего
генератора, управляемого мультивибратором, и повышающего
трансформатора.
Узел электропитания мощностью 100 Вт обеспечивает потуче
пне импульсного напряжения с амплитудной до 800 В и частотой
10 кГц, причем импульсы подаются пучками, что достигается
включением в схему мультивибратора для управления работой
задающего генератора. Поролоновые фильтры состоят из распо-
ложенных внутри корпуса поджатых круглых пластин из пенопо
лиуретана, через которые осуществляется процесс фильтрации.
230
рабочей
электрода
переключе-
Таблица 5.7 Обеззараживание
длительно хранящейся волы в блоке
ЭРММ произвол и тел ьностью 3 л/ч
Дополнительно ин фи цироваи- иая культуре Число тест-микробов в 1_мл исследуемой воды
ИСХОДНОЙ обработан- ной
Кишечная 8,8-10» 20,0±9,6
палочка 8,9-10* 9,4-10* 43,0±14,6 40,0± 12,1
Стафилококк 8,5-10» 8,9-10» 9,2-10» 32,5±7,3 30,0±9,7 50,0±9,7
результаты экспе-
длнтельно храня-
естественной обсе-
и дополнительно
При срабатывании
част н стержневого
предусматривается
ние на следующий (игольчатый)
электрод.
Некоторые
риментов иа
щенся воде с
менепностью
инфицированной культурами кн
щечной палочки и стафилококка,
представленные в табл. 5.7, сви-
детельствуют о высокой эффек-
тивности инактивации вегетатив-
ных форм микроорганизмов. При
бактериальной обсемененности
длительно хранящейся воды спорами бацилл ангракоида 3,3 • 1054-
4-6,2-106 эффективность обеззараживания составляла 98,07—
99,98 %. Электрический разряд малой мощности с последующим
фильтрованием обеспечивает не только высокую эффективность
обеззараживания длительно хранящейся воды, но и улучшение
ее органолептических и химических показателей качества до тре-
бований ГОСТ 2874—82. Так, при обработке модельной длительно
хранящейся воды в блоке ЭРММ производительностью 6 л/ч мут-
ность снижается с 3,54-15,0 до 0,24-1,3 мг/л, цветность — с 404-70
до 54-6 град., содержание железа — с 1,04-3,5 до 0,14-0,15 мг/л.
Приведенные санитарно-гигиенические показатели убедительно
характеризуют токсикологическую безвредность воды, подвергну-
той ЭРММ. Однако учитывая длительное употребление воды кол-
лективами автономных объектов, необходимо убедиться и в ее
онкологической безопасности, т. е. отсутствии опухолеродных
свойств. Исследованиями методом Эймса мутагенности н опреде-
ляемой непосредственно через нее канцерогенности воды с саль-
монеллой тнфинурпум установлено, что количество мутантов
в пробах, подвергнутых электрообработке, не превышает частоту
мутаций в контроле (30—40 колоний для штамма ТА-98 и 120-
150 -для штамма ТА-100). Таким образом, вода, обработанная
сильными электрическими воздействиями, не обладает мутаген-
ной. а следовательно, и канцерогенной активностью.
I.J. ОЧИСТКА СТОЧНЫХ вод
S.2.I. Технологические воды
гальванических производств
Вода в цехах гальванопокрытий расходуется на приготовле-
ние з.1ектролитов, обезжиривающих и травильных растворов, на
промывку деталей, охлаждение ванн н выпрямителей тока Ос-
новной водопотребляющеп операцией является промывка изде-
231
лин, от эффективности которой зависит качество и надежность
гальванопокрытий. Образующиеся при этом промывные сточные
воды, а ыкже отработанные концентрированные технологические
paciворы поступают в канализационную сеть промпредприятий.
По содержанию загрязнений сточные воды гальванических
производств делятся на: щелочные — от обезжиривания изделий;
кислотные — от предварительного травления изделий, кислотного
меднения, никелирования, кислотного цинкования; циансодержа-
щие— от процессов цианистого меднения, цианистого цинкования
и других операций; хромсодержащие — от процессов хромирова-
ния, пассивации, травления в хромпике и других операций; фтор-
содержащие— от процессов травления и оловянирования.
Промывные сточные воды от проточных ванн поступают в ка-
нализацию постоянно, режим сброса отработанных концентриро-
ванных растворов из основных ванн периодический.
Отработанные концентрированные электролиты, содержащие
кислоту и щелочь, направляются, как правило, в отдельные ем-
кости-накопители и используются как реагенты для нейтрализа-
ции сточных вод. Отработанные хром- и циансодержащие растворы,
а также сточные воды, содержащие другие ценные веще-
ства, должны подвергаться регенерации и возвращаться в техно-
логический процесс. При невозможности повторного их использо-
вания они обезвреживаются раздельно илн совместно с промыв-
ными сточными водами.
Выбор метода очистки зависит от состава и режима поступле-
ния сточных вод, концентрации загрязнений, необходимости и
возможности повторного использования очищенной воды [90, 102].
Особое внимание при этом уделяется электрохимическим методам
очистки, которые можно разделить на четыре основные категории:
электрокоагуляция с использованием растворимых железных
анодов для очистки промывных сточных вод, содержащих соеди-
нения шестивалентного хрома и другие ионы тяжелых металлов;
электрохимическое восстановление Сг*+ ионов с применением
нерастворимых анодов;
электрохимическое окисление цианидов;
электродиализ для очистки промывных вод и регенерации от-
работанных электролитов.
Прн электрокоагуляционном методе происходит химическое
восстановление Сг’+ ионами Fe2+, образующимися в процессе
электролитического растворения анодов, а также гидрозакисью
железа Fe(OH)2, образующейся в обрабатываемой воде при взаи-
модействии Fe2+ и ОН~-ионов при pH^5,5, согласно следующим
химическим реакциям:
Сг^-+ 6Fe2+-F MU+ —ZCr'+ ^GFc^+7Н2О; (5.1)
Сг2о£ ! (,Г-(О11) . 4-7Н ,О 2Сг (ОН)Я 4-GFe (ОН)3 | 2ОНЛ (5 2)
232
Дополнительно происходят катодные электрохимические про-
цессы восстановления ионов хрома:
Сг2°?~ + ,*н+ + 6«-*2Сг8+ + 7Н2О; (5 3)
СгО^- + 4Н2О + Зё-*- Сг (ОН)3 4- 5ОН~. (5 4)
Для выбора оптимальных технологических режимов электро
коагуляции необходимо по стехиометрическим соотношениям ис-
ходного содержания Сгв+ и требуемого расхода железа рассчи-
тывать в соответствии с законом Фарадея теоретическое коли-
чество электричества (gj). При одновременном присутствии Сг®1
и ионов тяжелых металлов q3 определяется исходными концент-
рациями этих компонентов и расходом сточных вод. При этом за
необходимое </, принимается наибольшее q3, рассчитанное для уда
лепия каждого из компонентов в отдельности и увеличенное на
20 % [90].
Количество электричества, затрачиваемое на восстановление
1 мг Сгв+, существенно зависит от исходного pH обрабатываемой
воды. Ниже представлены рекомендуемые значения этих величин:
рн ....................
2,0 3,0 4,0 5,0
6,0 7,0
<7Уд. Кл/(мг-л) ..............
7—9 13—15 17—19 19-21
21—23 23-25
При снижении pH исходной воды от 7,0 до 3,0—3,5 спи
жается в 1,3—1,5 раза. Это позволяет использовать кислотный ре
«•рн для уменьшения расхода электроэнергии иа восстановление
хрома.
Рекомендуемые анодные плотности тока составляют для кон-
центрированных растворов 0,64-1,5 А/дм2, для слабокопцептри
рованпых (<100 мг/л)—0,154-0,8 А/дм2.
Прн электролизе обрабатываемый раствор подщелачивается,
ык как концентрация водородных ионов в результате вышенриве
генных реакций, а также электрохимического выделения газооб-
p.i того водорода уменьшается, что благоприятствует коагуляции
гидроксидов хрома и железа с образованием хлопьев, на кото-
рых происходит адсорбция других примесей, содержащихся
и сточных водах. Прирост величины pH может составлять 1—4
гдиницы, что зависит от исходных концентраций Сг*^ и исходной
лк । нвной реакции среды.
С целью создания оптимальных условий для лучшей седнмен
i.iiiiiii образующихся гидроксидов металлов осуществляют кор-
рек iкровку активной реакции сточных вод после электрокоагу-
И1Н11ониой обработки до рН = 8ч-10. Выпадающий прн этом
л юк характеризуется рыхлой хлопьевидной структурой и высо
• »»ft влажностью. Объем осадка зависит от исходной концентра
нпн с’г®*- и ионов других металлов и составляет после 2 часового
пн питания 6—15% от объема очищаемого стока при исходной
।парной концентрации ионов тяжелых металлов 50 150 мг/л
233
Таблица 5.8. Результаты очистки
гальваностоков в колонном электролизере
Продолжи- тельность обработки. МИИ Содержание тяжелых металлов, мг/л, в неходком (числитель) и очищенном (знаменатель) стоке при плотности тока. А/см*
0,015 0,030
3 300 200 300 200
30 44 21,5 36
5 300 200 300 200
1,2 2,2 0,55 1.7
10 300 200 300 200
Следы 0,2 Следы Следы
{80]. После 24-часового уп-
лотнения объем осадка
уменьшается на 20—30%,
влажность его прн этом де-
стигает 98,2—98,5%.
Метод электрохимиче-
ской коагуляции целесооб-
разно применять прн со-
держании Сг®+ до 150 мг/л,
так как увеличение его
концентрации вызывает
возрастание удельного рас-
хода металла и электро-
энергии, а также пассива-
цию железных анодов,
вследствие чего ухудшается
их электролитическое рас-
творение и снижается выход по току. Для предотвращения пас-
сивации предлагается использование анодов из меди или ее спла-
вов, добавление в сточные воды некоторого избытка NaCI. Депас-
сивация электродов может производиться также механическим и
химическим способами или с применением переменного электри-
ческого тока.
Электроды, как правило, изготовляются из низкоуглеродистой
стали, по с целью повышения эффективности водоочистки и улуч-
шения отделения образующейся твердой фазы возможно приме-
нение железоалюмипневых электродов [42]. При электрокоагуля
ции фторсодержащнх сточных вод применяют алюминиевые элек-
троды [42]. В последние годы предложено несколько конструкций
электролизеров с засыпными анодами из отходов металлообра-
ботки [а. с. 700468 (СССР), 20, 112].
Принципиальная схема электрокоагуляциоппой очистки вклю
чает электролизер с растворимыми анодами, снабженный вытяж-
ной вентиляцией, емкости пли аппараты для корректирования ве-
личины pH, а также сооружения осветления воды.
Электрообработка жидкости обычно осуществляется в плас-
тинчатых электролизерах вертикального исполнения с подводом
очищаемой жидкости снизу вверх.
Расстояние между электродами составляет 6—12 мм, толщина
электродов — 3—8 мм, соотношение анодной и катодной площа-
дей— 1 : 1. скорость воды— 1—2 м/мии.
Удаление образующегося осадка гидроксидов осуществляется
в отстойниках, флотаторах-отстойниках, флотаторах-осветлите-
лях. Для уменьшения объема осадка в обрабатываемую воду до-
зируют раствор полиакриламида в количестве 30 мг/л.
Доочистка жидкости производится иа песчаных и пенополи-
стирольных фильтрах, в электрофлотаторах, обессоливанием на
ионообменных фильтрах.
Авторами монографии |69| предложено вести очистку стоков,
Рис. 5.11. Технологическая схема очистки сточных вод гальвапоцехов с электро-
химическим корректированием pH н
содержащих ноны тяжелых металлов, в колонных электролизерах
цилиндрической формы (рис. 4.11). Эффективность очистки в за-
висимости от концентрации вредных примесей и режимов работы
аппарата этого типа приведена в табл. 5.8.
В УИИВХ [92] разработана технологическая схема очистки
промывной воды гальванических производств от ионов тяжелых
металлов с электрохимической нейтрализацией очищенной воды.
Особенностью схемы (рис. 5.11) является смешение всех катего-
рий промывных вод (за исключением циансодержащих), что по
зволяет в большинстве случаев предотвратить пассивацию желез-
ных анодов, вызываемую присутствием шестивалентного хрома.
Допустимые концентрации хрома (Ст*), поступающего на
очистку в зависимости от общего солесодержания воды, приве-
дены ниже.
< >бщее солесодержание, мг/л 80—150 150 200 200 300 300—400 400 500
[опускаемая концентрация Ст*Г, мг/л 5 10 20 30 40
Общее солесодержание, мг/л 500- 700 700 -900 900—1200 1200—1500
(опускаемая концентракня <т*+, мг/л 60 70 90
Промывную воду нз гальванического цеха (см. рис. 5.11) по-
д.но| в усреднитель /, где за 3—5 ч обеспечивается стабильность
• е лемропроводностп и гомогенизации состава. Затем с помощью
насосов 2 воду направляю! в электрореактор 3, в котором про
п< ходит выделение с поверхности анодов в раствор ионов Ге71.
(аиавливающих Ств+ до Ст’4, а также насыщение жидкости
п\ оирькамн катодного образующегося водорода.
235
934
Для интенсификации процесса были применены электрореак-
торы с «газовым слоем» (см. рис. 4.57), позволившие повысить
эффект флотации по гидроксидам до 70—90%. При этом одно-
временно нз воды извлекаются СПАВ, красители и нефтепро
дукты. В камеру «газового слоя» из бака-накопителя католита 6
дозируют раствор щелочи, получаемой в электрокорректоре pH 7,
для повышения активной реакции воды с целью перевода метал-
лов в гидроксидные соединения.
Разделение твердой и жидкой фаз во флотаторе-осветлителе 4
проводят осаждением хлопьев гидроксидов и их флотацией обра-
зующимся электролитическим водородом. Остаточную взвесь, ire
извлеченную во флотаторе-осветлителе, задерживают на филь-
тре 5. Очищенную от нонов тяжелых металлов воду с рН=9,84-
4-10,2 направляют в анодные камеры 9 электролизера для ней-
трализации. Часть воды из расчета соотношения расходов като-
лита и анолита (1,24-1,6) : (8,84-8,4) подают в катодные ка-
меры 8.
В анодных камерах вода подкисляется до pH = 7,54-8,0, после
чего анолит (84—88% от общего расхода воды) через промежу-
точную емкость 10 насосом 2' возвращают иа повторное исполь-
зование. В катодных камерах вода дополнительно подщелачива-
ется до рН= 11,54-11,6, после чего щелочной католит (12—16 %
от общего расхода воды) направляют в бак 6, откуда дозируют
во флотатор-осветлнтель 4 для осаждения гидроксидов металлов.
Шлам имеет низкое удельное сопротивление и легко обезво-
живается.
Осадок и шлам, образующиеся во флотаторе осветлителе, пе-
риодически сбрасывают в илоуплотнитель 12 и далее обезвожи-
вают на вакуум-фильтре 11. Воду от промывки вакуум-фильтра
возвращают в усреднитель для повторной очистки совместно с ис-
ходной водой.
Параметры работы основных сооружений технологической
схемы (рнс. 5.11) следующие: в электрореакторе r* = 30-i-40 А/м2,
1/э=9-т-12 В, <7э= 144-18 Кл на 1 мг Сг*+, и7эл = 0,34-1,0 кВт-ч/м3;
во флотаторе осветлителе /Осв=304-40 мин, U3= 1,84-2,0 м/ч, ин-
тенсивность промывки 20—25 л-с-'-м-2; в электрокорректоре
pH 1 = 304-50 А/м2, U3= 124-24 В, Дт = 2504-480 Кл/л, №,„=0,5-5-
г-1,0 кВт-ч/м3, рНк.т= 11,24-11,8, рН.„ = 6,54-8,2.
Данная технологическая схема внедрена на ряде предприятий,
в частности в ЛОМО (г. Ленинград) и на опытном заводе «Пром-
связь» (г. Талдом).
Очищенная вода используется в замкнутом цикле, что позво-
ляет сэкономить 70—90 % воды, потребляемой гальваническим
нроп«водсгвом. Показатели качества очистки воды на этих пред-
прия1иях приведены в табл. 5.9.
Метод электрохимического восстановления хрома с примене-
нием нерастворимых анодов используется для обезвреживания сто-
ков с большими концентрациями Сг6* — более 2 г/л. В присут-
ствии ионов Fe3' катодный процесс восстановления Сг’+ до Сгя+.
236
Таблица 5.9. Показатели качества очищенной воды при применении
технологической схемы с электрохимическим корректированием pH
Показатели Концентрация примеси в воде Х|н|н к 1 Очистки, ч
исходная очищенная
Мутность, мг/л 250 3—10 96,0
Цветность, град- 50—100 20 U
Хром (Сг*+), мпл 20-30 0,05 0,1 ‘><1,5
Хром (Cr’+k мг/л Никель, мг/л 30-50 0,1—0,3 99.(1
10—20 0.3—0,5 96,0
Медь, мг/л 10—15 0,3—0,8 92.0
Цнпк, мг/л 20—30 0,5—0,8 96,0
Нефтепродукты, мг/л 10—100 0,5—1,0 90,0
11 \В, мг/л 1—5 0,5-1,0 50,0
Активная реакция 3,5-9,0 6,5—8,5 —
интенсифицируется и одновременно ингибируется анодный про
цесс окисления Сг г до Сг6С Электролиз ведется в безднафрш
менном напорном аппарате периодического действия со евнпцо
ними анодами или РЬ—Sb-сплава.
После электрохимической обработки к сточным водам дооав
ляются щелочные реагенты для осаждения Сг3+ в виде Сг (ОН) ;
выход Сг3+ по току составляет 70—90 %.
Электрохимическое выделение металлов из промывных вод
гальванического производства имеет определенные трудности иг
за их относительно небольших концентраций (обычно <1 г/л)
Катодный выход металлов по току, как правило, небольшой.
Существенно интенсифицировать процессы очистки удалось
с применением электродов с развитой поверхностью 1103]. Кроме
того, в результате интенсивного перемешивания или быстрого про
пускания жидкости через объемный электрод создают турбулеш
ное движение электролита, что значительно уменьшает толщину
диффузионного слоя и увеличивает коэффициент массонере
носа [82].
По конструкции электролизеры для выделения металлов н i
разбавленных растворов можно условно разделить на пять групп
с проточными пористыми или объемно-насыпными катодами,
с псевдоожиженными электродами; с плоскими пластинчатыми
пли сетчатыми электродами и загрузкой из инертных м;нер|ы
.юн; с вращающимся катодом; типа «шведского пирога» (Swiss
loll).
Объемно насыпные проточные катоды, как правило, состоя)
in токопроводящей сетки или пластины, контактирующих с ны
рикамн или гранулами из токопроводящего материала — элеюро
графита, меди, свинца, серебра или с металлизированными стек
лянными или пластмассовыми шариками. Исследованию и и<
пользованию электролизеров такого типа посвящено большое
г лнчество работ [82, 128 и др.].
237
Значительное количество работ относится к применению леев
доожнжеииых электродов. Псевдоожиженные катоды состоят из
металлических или металлизированных шариков или гранул, ко-
торые загружаются на металлическую токоподводящую сетку
[82] и прн интенсивном прокачивании жидкости снизу вверх на-
чинают двигаться и распределяться во всем объеме электроли-
зера. Этим достигается большая рабочая поверхность катода и
создается интенсивное перемешивание и турбулентное движение
жидкости.
Электролизеры типа плоских пластин с инертной загрузкой из-
готовляются английской фирмой «Electricity Council Research
Centre T. H. Chemeles cell». В электролизер помещается несколько
пар сетчатых электродов, между которыми загружены стеклян-
ные шарики. Движение потока электролита регулируется таким
образом, чтобы шарики распределялись равномерно во всем его
объеме. Металл выделяется иа сетчатых катодах [82, 125J.
Использование электролизеров с катодами в виде вращаю-
щихся цилиндров апробировано в работающем в iioieiniBociэти-
ческом режиме аппарате, катодом которого являлся цилиндр нз
нержавеющей стали с диаметром и высотой около 6 см, вращаю
щнйся со скоростью 1200 мин 1 [82].
Электролизер тина Swiss-roll представляет собой свернутый
на металлическом стержне в длинный рулон «бутерброд» из двух
электродов и сепаратора из неэлектронроводного материала
между ними. Этот рулой помещается в цилиндр таким образом,
чтобы внешний электрод н металлический цилиндр контактиро-
вали друг с другом. Электрический ток подводится к электродам
через металлический цилиндр и центральный металлический стер-
жень. Движение электролита осуществляется в направлении, па-
раллельном центральному стержню [82]. Электролизеры такого
типа имеют следующие достоинства: относительно большую удеть-
иую поверхность, возможность использования дешевых материалов
для их изготовления, компактность и простоту конструкции, вы
сокую производительность благодаря использованию больших
скоростей массопереноса, однородность распределения потен-
циала, достижение высоких выходов по току.
Метот электрохимического окисления цианидов применяется
для обезвреживания любых циансодержащнх сточных вод и от-
работанных растворов с концентрациями цианидов более 200 мг/л
[90|.
Прн электролизе циансодержащнх стоков на аноде происхо-
дит электрохимическое окисление CN~-hohob, комплексных ани-
онов [Cu(CNb|a- [Zn(CN)4]2-, [Cd(CNh]2- и т. п. с образова-
нием цианат ионов; по мере накопления в сточных водах цианат-
ионов происходит их частичное электрохимическое окисление иа
аноде с образованием нетоксичных газообразных продуктов —
СОо. No:
CN + 2OII- — 2г -* CNO- + Н2О; (5.5)
238
(Cu (CN),]*- 4- 6ОН- — 6г — Си+ + 3CNO- + ЗН,О; (5 6)
2CNO- 4- 4ОН -> 2СОД 4- Nrf 4- 2Н2О 4- бё. (5.7)
На катоде происходит разряд Н-ионов с образованием озо
образного водорода, а также разряд ионов Cu2+, Zn2+, Cd2+, об-
разующихся при диссоциации комплексных ионов.
Для активации процесса необходима добавка 5—10 %-ного
раствора хлористого натрия [90, 132]. При этом происходят сле-
дующие реакции:
2С1-—Cl,4-CN-4-2OH--bCNO-4-2С1-4-2Н,О (5.8)
Применяются аноды из графитированного угля или из магне-
1ита или двуокиси свинца. Процесс можно интенсифицировать за
счет использования проточных пластинчатых (ОРТА, ОКТА)
электродов. Перспективным является использование объемно-по-
ристых электродов из углеграфнтовых материалов [10, 13].
Электролиз циансодержащнх стоков осуществляется в откры-
iwx бездиафрагменных электролизерах непрерывного или перио-
дического действия.
Анодная плотность тока принимается 0,5—2,0 А/дм2. Степень
очистки стоков от цианидов достигает 100 %. В виде катодных
осадков утилизируется до 80 % общего количества металла, со
держащегося в сточных водах. Остальное количество металла
удаляется в виде гидроксидов.
Метод электродиализа целесообразно примени ib для очистки
с юков после отдельных видов покрытия. Это дает возможность
повторно не только использовать очищенную воду, но и утилизи-
ровать сконцентрированные вещества.
Принципиальная схема очистки сточных вод методом элек-
фодналнза приведена на рис. 5.12. После ванны покрытия про
мывные воды прн наличии механических примесей направляются
па фильтр, заполненный активированным углем, а прн отсутствии
примесей — сразу в электродиализатор. Периодически электро-
диализный комплекс промывается серной кислотой. Эта операция
позволяет содержать аппарат в хорошем рабочем состоянии и из-
бегать избыточного сопротивления от образования пленки солей
на мембранах.
Очистка сточных вод в электродиализаторах ведется при еле
дующих условиях: pH = 44-9, концентрация ионов металлов —
0.1 : 5,0 г/л, плотность тока — 0,84-1,8 А/дм2, скорость потока —
0.5 : 0,7 л/мин, температура— 18:30 °C.
К настоящему времени ВНИИ ВОДГЕО разработан способ
рс|снерации отработанных растворов хромовой кислоты, который
<н ушествляется в двухкамерном электродиализаторе с катионн-
loiioii мембраной, причем отработанный раствор хромовой кис
ины помещают в анодную камеру (в катодную камеру поме
щ.1Ю1 3—5%-иый раствор серной кислоты) В процессе электро
л > попы Ec1h, Си2*-, Сг1’ н других металлов, загрязняющие рас
239
Рис 5 12. Принципиальная схема очистки сточных вод методом электродиализа
! — усреднитель; 2 — насос; 3 — фильтр с активированным углей; 4 — электродиализатор;
5 — измеритель расхода; б — отвод очищенной воды; 7 — отвод рассола
творы, переходят из анодной камеры в катодную н концентриру-
ются в ней. В анодной камере происходит также частичное
окисление Сг3+ до СГдО2-. Поверхность катодов покрывается
осадками металлов (железо, медь и др.). Электролиз проводят
при 1 = 5004-1000 А/м2 с применением свинцовых анодов и сталь
пых катодов. Расход электроэнергии в зависимости от степени
загрязнения растворов составляет 800—1200 кВт-ч/м3. Растворы
хромовой кислоты после удаления из них основной массы приме-
сей могут быть возвращены в производство для повторного ис-
пользования, а катодные осадки металлов — использованы для
технических целей [102].
В институте ВНППИЧерметэиергоочистка разработан способ
регенерации кислых растворов после травления сталей, содержа-
щих в себе соли Fe2+, путем электродиализа в трехкамерном
аппарате с двумя аиионитовыми мембранами. С целью увеличе-
ния скорости регенерации растворов дополнительно вводят окис-
литель в количестве, обеспечивающем окисление 60—70 % Fe2+.
Способ позволяет вернуть регенерированную кислоту в травиль-
ный цикл и извлечь металлы в виде кристаллических осадков,
не вызывающих зарастания мембран [а. с. 762912 (СССР)].
Лля регенерации кислот из фторсодержащих травильных рас-
творов предложено перед электродиализатором нейтрализовать
стоки аммиаком до рН = 94-10, а электродиализ производить прн
силе тока 2—10 А и напряжении 10—40 В. На катоде осаждается
никель, растворенный в стоке. Степень регенерации кислот со-
ставляет 63—65%, никеля — 85—87 % [а. с. 1105515 (СССР)].
Испытания по использованию электродиализного метода очи-
стки промывных вод процесса никелирования в сочетании с кас-
кадным способом промывки показали [82]. что степень очистки
промывных вод составляет 90%. Условия испытаний: площадь
мембраны — 10 м2, рабочий ток — 8 А. рабочее напряжение —
60 В, выход по току — 90%, расход электричества — примерно
5,5 кВт«ч/кг никеля, производительность установки по никелю —
170—200 г/ч. Прн этом получается концентрат, содержащий
500 мг/л Ni Он используется как добавка к электролитам, а очи
щепная вода расходуется на различные технологические нужды.
240
Рис. 5.13. Технологическая схема очистки стоков гальванического цеха с при-
менением электрохимических методов
/ — сборник цнаисодержащих стоков; 3 — сборник хромсодержащих стоков; 3 — сборник
кислотно-щелочных стоков; 4 — электролизер с нерастворимыми электродами: 5 —элек-
трокоагулятор; 6 — нейтрализатор; 7 отстойник; 8 — очищенная вода; 9 — вакуум фильтр;
/О — обезвоженный осадок
Возможность очистки электродиализом сточных вод, образую-
щихся в процессе промывки деталей после операций цианистого
кадмирования, меднения и цинкования, исследовалась в аппарате
с использованием мембран МКК-1 и МАК-1 с мембранным рас-
стоянием 0,3 мм. Применялись катод из нержавеющей стали и
анод ТДМА. При сравнительно небольшом расходе электроэнер-
гии концентрация цианидов снижалась с 15—40 до 2—4 мг/л.
Очистка сточных вод гальванических цехов с применением элек-
|рохнмических методов ведется, как уже было указано, но от
дельным видам стоков. Как правило, промывные воды разделя-
ю1ся иа три потока: кислотно-щелочные, циансодержащие и хром-
содержащие (рис. 5.13).
Кислотно-щелочные стоки, минуя электролизер, направляются
и нейтрализатор, где происходит осаждение катионов металлов и
юведеиие pH стока до значения 7,5—8,5 с помощью 5-10 %
щелочи или известкового молока. Цнансодержащне и хромсодер-
ж.нцне стоки проходят электрохимическую очистку в различных
(лектролизерах, после которых они поступают в тот же нейтра-
ли нт юр, куда были направлены технологические кислотно-щелоч-
ные сточные воды.
При обезвреживании концентрированных стоков они собнра-
юн-я в отдельные емкости-накопители, откуда дозируются на
ичнегку вместе с промывными водами или подвергаются самосто
тельной очистке.
Использование в практике очистки сточных вод гальваииче
< кн\ производств электрохимических методов позволило приме
нин> более рациональные объемно-планировочные решения, кото
рые обеспечивают значительную экономию в стоимости строп
и п.ства и снижении площади для очистных сооружении,
гпрощают технологическую схему н эксплуатацию сооружений,
толяют частично или полностью отказаться от реагентного хо-
I'oiriiia.
241
$.2.2. Сточные воды предприятий бытовой химии
На предприятиях битовой химии в весьма широком ассорти
менте осуществляется производство товаров народного потреб-
ления: синтетических моющих средств (СМС), отбеливающих,
подкрахмаливающих, чистящих н дезинфицирующих препаратов,
чернил, туши, акварельных и гуашевых красок, инсектицидов и
пестицидов, пятновыводителей, средств но уходу за автомобилями
и других, в результате чего образуются разные по составу весьма
загрязненные сточные воды.
В настоящее время наибозее рациональным и достаточно
эффективным методом очистки рассматриваемых сточных вод яв-
ляется электрохимическая обработка [48, 52, 73, 97].
Ннже представлены принципиальные схемы и технологические
режимы электрохимической очистки некоторых наиболее харак-
терных категорий сточных вод предприятий бытовой химии.
Очистка сточных вод производства черной туши. Основным
загрязнением сточных вод является тонкодиспергнрованная сажа
в концентрации до 175 мг/л, удаление которой возможно комп-
лексным воздействием различных электрохимических процессов:
электрокоагуляции, электрофлотации, электрофореза и т. п. [97]
Так, электрокоагуляционная обработка стока в течение 15 мин
прн ia = 0,5 А/дм2 в присутствии 600 мг/л NaCI приводит к пол-
ному удалению сажн. В процессе обработки дисперсные частицы
всплывают; отстаивается обработанный сток очень плохо (про-
должительность отстаивания доходит до 5 ч). поэтому для интен-
сификации процесса очистки целесообразна двухстаднйная обра-
ботка: электрокоагуляция — электрофлотацня, совместное при-
менение которых позволяет уменьшить расход электродов на
20—38%.
Оптимальные параметры двухступенчатой очистки: /а в ка-
мере электрокоагуляции — 0,5 А/дм2, в камере электрофлота-
ции»—1 А/дм2; /э— соответственно 7—10 н 10—15 мин. Расход
металла — до 0,8 г/л, общий расход электроэнергии — 0,8—
1,0 кВт-ч/м3. Эффекты очистки составляют: но обесцвечиванию —
99%. по ХПК —76%.
При проектировании рекомендуется применять аппараты го-
ризонтального типа, в которых камера электрофлотации может
быть совмещена с камерой электрокоагуляции. Аппараты обору-
дуются приспособлениями для удаления всплывшей сажи Пен-
ный продукт можно обрабатывать иа вакуум фильтрах, пресс-
фнльтрах и далее вывозить в отвал или повторно использовать.
Очистка сточных вод производства гуашей. Данные промстоки
являются устойчивыми коллоидными системами, содержащими
тонко растертые пастообразные смеси минеральных и органиче-
ских пигментов, а также пластифицирующие (декстрин, глице-
рин), диспергирующие (препарат НФ) и антисептические (фенол)
добавки. Концентрация пигментов в стоке достигает 100—200 мг/л.
Тш обезвреживания таких сточных вод выявлена целесообраз
242
iiocib элекгрокоагуляционной очистки в аппаратах с плоскими
j.ickiродами из стали Ст. 3 с последующим О'стаиваннем пли
фло!ацией [52]. Выбор оптимальных технологических параме!рои
>лек1рокоагуляции был осуществлен с использованием регрессн
онного математического анализа, в результате которого получены
информация о влиянии различных факторов и уравнение про
цесса, позволяющее рассчитывать эффект очистки сточных вод
Э = 86,73 + 4,46Та + 8,71/э (5 9)
(та — в А/дмI 2; /э— в мин).
Оптимальными являются следующие параметры обработки:
стадии элек!рокоагуляции — /, = 0,54-0,75 А/дм2; /,=74-12 мни,
расход материала электродов — 80-Т-120 г/м3; рН = 6,54-8,0; ста
дин осветления — продолжительность отстаивания 454-60 мни,
элекгрофлотации—154-20 мин. Эффекты очистки составляют
при этом: по обесцвечиванию — 99,6-г-99,9 %, по ХПК— 70%, ио
феиотам — 90 %.
Очистка сточных вод производства чернил и цветной туши.
В этих производствах образуются интенсивно окрашенные сточ
пые воды, содержащие до 150 -170 мг/л красителей, очистка ко
юрых может быть достигнута путем элек1рокоагуляцин или элек-
трохимической деструкции.
Изменение анодной плотности тока с 5 до 20 мА/дм2 позво-
ляет довести эффект электрокоагуляционного обесцвечивания
до 99,6 % ври продолжительности обработки стока 30 мин. Од-
нако содержание органических загрязнений ио ХПК снижается
нсего на 32%. Значительно повысить эффект очистки от органп
ческнх соединений даже при увеличении анодной плотности тока
то 50 мА/дм2 при электрокоагуляции не удается.
Учитывая образование большого количества осадка (до 20 %
•и объема стока), значительный расход металла, низкий эффект
очистки по ХПК, рекомендовать электрокоагуляцию для очистки
тайной категории сточных вод нецелесообразно.
Электрохимическое обесцвечивание стоков с использованием
малонзнашивающихся стойких анодов обеспечивает глубокое
окисление органических красителей и позволяет устранить пере
•тс ц-нпые недостатки.
В работах [24, 45J* путем тонкослойной хроматографии. УФ
. пекгроскопин, полярографии, количественного элементарного
'ЦП .отческого анализа и анализа выделяющихся газов установ-
ки общий баланс веществ при оптимальных параметрах элек-
ipoхимического обесцвечивания сточных вод, который показал,
чю основная часть органических соединений (60—70%) пол-
..... минерализуется до СО,. N2, Nil-., SO42- и Н2О, а часть
гр.н-нтелей разрушается с образованием незначительного колн-
I |сс.1слона1П1Я по выявлению продуктов электрохимической деструкции
. ,,т. lei'i выполнены в ЛИСИ совместно с ЛГУ нм. А. А. Жданова
243
чества (5—6%) от исходной концентрации ароматических соеди-
нений.
В работе [47] показана принципиальная возможность даль-
нейшей биологической доочистки сточных вод, прошедших ста-
дию электрохимической обработки, и определены пределы токси-
кологического влияния этих стоков на комплекс микроорганизмов,
ведущих процесс биохимической деструкции.
С. В. Яковлевым и др. [124] дан математический анализ про-
цесса электролиза модельных растворов красителей прямого чи-
сто голубого и кислотного ярко-синего антрахинонового, которые
являются наиболее характерными представителями красителей,
часто применяющихся в технологии рассматриваемых произ-
водств. Показано, что эффект очистки сточных вод (ап) зависит
от режима электролиза (Дт) и исходной концентрации загрязне-
ний по ХПК (Со):
an 1 — 15,2ехр (ЬСс)/[С0 Дт + 15,2ехр (ЬС0)J, (5.10)
где Ь — коэффициент константы скорости окисления красителей, равный тан-
генсу угла наклона прямой в координатах 1п( !/£)—Со, обработкой экспери-
ментальных данных установлено, что при электролизе растворов красителей,
содержащих 2 г/л NaCl, b=8,035- 10-’.
Зависимость (5.10) можно использовать для расчета электро-
лизеров (см. и. 6.5); ее область применения соответствует значе-
нию ХПК= 1004-600 мг/л, что характерно для сточных вод не
только рассматриваемых производств, но и других предприятий,
к примеру текстильной и трикотажной промышленности.
Высокая степень электрохимического обесцвечивания сточных
вод подтверждена при длительной (более 9 лет) эксплуатации
промышленной установки цеха чернил Лужского химзавода |48],
в результате которой выявлена технологическая эффективность
очистки промстоков по основным показателям (табл. 5.10). Элек-
трообработка производилась при ia=l,0+l,5 А/см2, /э=54-7 мин,
концентрации NaCl — 24-3 г/л, расход электроэнергии при этом
составлял 1—2 кВт-ч/м3.
Эффективность обесцвечивания стоков достигает в среднем
99,6 % при стабильном снижении органических загрязнений
(ХПК) на 70—80%. Осадок в очищенном стоке не образуется,
pH стабилизируется, концентрация взвешенных веществ умень-
шается. Очищенная вода отвечает всем требованиям, необходимым
для направления промстоков на централизованные станции аэра-
ции. а также может быть повторно использована в производстве.
Очистка сточных вод производства СМС. Содержание ПАВ
в сточных водах производства жидких и пастообразных СМС ко-
леблется в пределах 0,3—1,5 г/л, а в стоках производства порош
кообразных СМС превышает 1,5 г/л. достигая в некоторых слу
чаях 4 г/л [52, 971.
Эффективность удаления ПАВ прн этектрофлотацпи зависит
от пехотной концентрации, повышение которой свыше 100 мг/л
244
Таблица 5.10. Электрохимическая очистка сточных под производства чернил
на опытно-промышленной установке
Показатели загрязнений Сточная жидкость
исходная очищенная
Интенсивность окраски 1 : 512 :-1 : 4096 1 : 8-М : 24
I . 2048 1 : 10
Активная реакция 7,5 8,0
ХПК, мг/л 1635 2970 320-510
2000 330
Взвешенные вещества, мг/л 50—180 0—40
95 29
Сухой остаток, г/л 0,2—5,2 0,41 0,9—1,6 1.5
Фенолы, мг/л 2—7 0,1—0,9
Хлориды, мг/л 500 800 700
Активный хлор, мг/л 14—31
20
Примечание. В числителе приведены пределы колебаний, в знаменателе — сред-
ние значения.
снижает степень их извлечения, что связано с мицеллообразова-
пнем [89]. Увеличивается при этом и объем флотоконденсата: так,
при исходной концентрации ПАВ, равной 300 мг/л, эта величина
может достигать 9—10 % объема обрабатываемого стока. Увели
чсние слоя флотируемой жидкости с 0,2 до 1,2 м незначительно
влияет на эффект удаления ПАВ, однако при этом в 6 раз сокра
шлются удельные энергозатраты. Поэтому высоту слоя Но сле-
дует выбирать в пределах 1—1,2 м.
При оптимальной плотности тока 2—3 А/дм2 и продолжитель-
ное in обработки 20—30 мин можно удалить из сточных вод 50—
53 % ПАВ. Образующийся при электрофлотации пенный кондсн-
г.н в зависимости от конкретных условии можно подвергать двух
< (уненной электрохимической обработке 116, 97], обрабатывать
природными сорбентами, сжигать [52].
Электрокоагуляционная обработка исследована на модельных
p.ieiворах аииоиоактнвного препарата «Прогресс М 20» с исход
иым содержанием 1, 2 и 4 г/л
Применение стальных анодов и обработка воды в течение
1(1 мин позволяет снизить содержание ПАВ в среднем на 55%.
При увеличении ia с 1,5 до 3 А/дм2 эффект очистки повышается
ш 58% при =20 мин, однако в этом случае возрастают расходы
• '|скгроэнергив, превышающие 10 кВт*ч/м\
245
При применении кальцийсодержащих электролитов наблю-
шется некоторое повышение эффективности удалении ПАВ [а. с.
789406 (СССР)], которое повышается также и при катодном
растворении алюминия [а. с. 1011549 (СССР), 73]. Установлено
[73], что при проведении процесса с железным анодом и алюми-
ниевым катодом достигается наиболее высокая эффективность
удаления ПАВ. Оптимальными параметрами процесса являются:
та = 754-100 А/м2, /,= 1,54-3,0 мин. Эффект очистки от АПАВ до-
стигает 96,6 %, при расходе электроэнергии — 0,54-0,7 кВт-ч/м3.
Возможность электрохимической деструкции ПАВ с исполь-
зованием малонзнашивающихся анодов ОРТА изучена иа раство-
рах иеноногеиных веществ: ОП-Ю (100—300 мг/л), сопла OF
(1—4 г/л) и аиноноактивного препарата сПрогресс М 20» (100—
300 мг/л).
Обработка растворов ОП 10 в течение 5—15 мин позволяет
снизи1ь содержание ПАВ на 62—85 8% (в среднем 80%). Даль
неГнисе увеличение продолжительности обработки приводит к не-
значительному росту эффекта очистки
Оптимальными параметрами процесса электрохимической
деструкции сточных вод, содержащих до 0,1 г/л НПАВ и 0,1 —
0,2 г/л АПАВ, при плотности тока 2 А/дм2 являются: CNaci = 3r/.'i,
/,= 15-т-20 мин. При содержании ПАВ свыше 1 г/л продолжитель-
ность обработки следует увеличить до 60 мии.
В определенных случаях более рациональной является ком-
бинированная элек1рокаталнтическая деструкция поверхностно-
активных веществ. Применение этой технологии [49] подтвердило
высокую эффективность очистки растворов анионоактивного пре
парата Marlon (алкилбензолсульфонат натрия) фирмы Hiilst
(ФРГ), позволило выявить кинетические закономерности и мате-
матически описать суммарный процесс окисления ПАВ во взаи-
мосвязи технологических параметров как электролиза, так и по-
следующей стадии каталитической деструкции.
Суммарная степень превращения (окисления) ПАВ описыва-
ется зависимостью:
ап I — IC.fJfe, Дт 4-Л.) Vkt/Q+11-1. (5.11)
где А>| и ki — константы скорости соответственно окисления в электролизере,
л/Л-ч, и каталитическом реакторе, мии-1; — объем катализатора, л; Q —
расход сточных вод, л/мии.
Выведенные в работе [49] зависимости можно использовать
при инженерном расчете комбинированного аппарата (рис. 4.61)
элсктроката.111П1ческой очистки сточных вод от ПАВ.
В ЛИСИ разработаны технологические схемы очистки сточ-
ных вод предирия1нн бытовой химии от ПАВ в зависимости от
исходной концеп(рации загрязнений (рис. 5.14).
При производстве жидких и пастообразных СМС образуются
сточные воды, в которых, как уже было указано, концентрация
ПАВ может достигать I г/л и выше. Очистку таких промстоков
рекомендуется производить по схеме 1. Удаление ПАВ из сточ
246
Схема 2
Рис. 5.14. Технологические схемы очистки сточных вод от ПАВ
/ — усреднитель. 2 — электрокоагулятор; 3 — отстойник; 4 — аппарат элсктрокаталитнче-
ск<Л очистки. 5 — реагентное хозяйство; 6 — узел обезвоживания осадка; 7 — электрофло-
1ятор, 8 — накопитель пенного конденсата; 9 — электролизер с нерастворимыми электро*
да ми
ных вод по этой схеме составляет до 98%, на стадии электро
коагуляции — 85 %. При этом ХПК снижается на 80 95%, взве-
шенные вещества—на 80—90%, а фосфаты — на 70%.
Очищенные по данной схеме сточные воды могут быть накрав
тепы в городскую канализацию или непосредственно, или после
разбавления сточными водами других производств.
При производстве чистящих средств, жидких «лбелнвателей,
< редств для подкрахмаливания и аппретирующих образуются
11 очные воды, содержащие до 300 мг/л ПАВ Такие сточные воды
после усреднения по количеству и составу направляются в элек-
|рофлотатор, затем — в комбинированный аппарат электроката
прической очистки (схема 2). Перед электрофлотатором, аиало-
। пчпо первой схеме, вводится раствор хлористого натрия. Обра
бо1<1пный сток может быть выпущен в городскую канализацию.
Общий эффект очистки по ХПК составляет 80—85%, от ПАВ —
'•>% (на стадии флотации — 55%). Образующаяся в электро-
ф io гаторе пена после ее гашения обрабатывается в электроко
нгуляторе и после отстаивания направляется в самостоятельный
<1сктролизер с нерастворимыми электродами, и далее — в усред
•опель для совместной обработки с исходными сточными водами
< о тержаиие ПАВ в пенном конденсате (после его обработки) не
п|>< iii.iniaci их концентрации в исходном стоке.
247
В поверхностных стоках предприятий бытовой химии могу г
содержаться ПЛВ от 200 мг/л (при наличии производства жидких
и пастообразных СМС) до 2 г/л (при производстве порошкооб
разных СМС). Кроме того, в них присутствуют нефтепродукты,
масла и взвешенные вещества, способные засорять канализацион-
ные сети. Поэтому поверхностные стоки в зависимости от содер-
жания в них ПАВ следует очищать по соответствующим схемам,
представленным на рис. 5.14. Очистные сооружения поверхност
ных стоков могут быть или автономными, или объединены с соо-
ружениями для очистки стоков производства СМС.
Эффекты очистки этих стоков следует принимать: от ПАВ —
в соответствии с принятыми технологическими схемами, по нефте-
продуктам и маслам — 90—95%, по взвешенным веществам — до
90%.
5.2.3. Нефтесодержащие воды *
Нефтепродукты являются наиболее распространенными за
грязняющимн веществами. Большое количество этих загрязнений
содержится в стоках автотранспортных и авторемонтных предпри-
ятий, предприятий железнодорожного транспорта и сельхозтех-
ники, нефтебаз, перекачивающих станций и наливных пунктов.
При очистке сточных вод возникают определенные трудности,
связанные с выделением из них эмульгированной части иефте- и
маслопродуктов, которые образуют с водой нестабилизирован-
ные, слабостабилизнрованиые и сильностабилизированные эмуль-
сии.
Электрофлотацня является одним нз наиболее эффективных
способов очистки воды от нефте и маслопродуктов [58 60], оиа
осуществляется в аппаратах с нерас 1воримымн или растворимыми
электродами. В первом случае перед электрофлотацией часто про-
изводится реагентная подготовка очищаемой воды.'.
При использовании нерастворимых электродов эффективность
флотации зависит от крупности извлекаемых капель. Например,
если степень извлечения частиц диаметром 18 мкм составляет
62,5%, то диаметром 10 мкм — 23,3 % [84] Частицы нефтепро-
дуктов диаметром 5 мкм электрофлотацией практически ие из-
влекаются, а диаметром более 22 мкм удаттются достаточно
эффективно. В зависимости от состава эмульскн рекомендуется
га = 34-6 А/дм2 и /э = 3-5-бО мин. Концентрация нефтепродуктов
снижается при этом с 68—372 до 3—10 мг/л [84], что по эффек-
тивности иракшчески равноценно реагентной напорной флотации
Безреагентная электрофлотацня рекомендуется также для
очистки силыюстабилизированных эмульсий, в частности обезжи-
ривающих растворов [а. с. 1073180 (СССР)]. Эти растворы, содер-
жащие 100—120 г/л щелочей, после электрофлотации использу-
* Материал подготовлен совместно с канд. техн наук В. Н. Лпопольским
248
ются повторно (концентрация масел при эффекте очистки НО -
90 % снижается до 1.00-150 мг/л, а содержание механических
примесей — на 75—85 %).
Более высокие показатели очистки электрофлотацией дости-
гаются предварительной обработкой стабилизированных эмуль-
сий реагентами. При использовании коагулянтов (известь, квасцы,
соли железа и т. п.) концентрация масел в очищенной воде на-
ходится в пределах 5—20 мг/л. Для очистки щелочных растворов
от масел в качестве коагулирующего реагента предлагается ис-
пользовать соли магния, что упрощает технологию очистки.
Значительно шире и глубже исследована технология очистки
воды от нефте- и маслопродуктов электрофлотацией с использо-
ванием растворимых электродов. Параметры процесса сущест-
венно зависят от состава и свойств очищаемой воды. Так, очистка
маслосодержащнх стоков цеха горячей прокатки происходит ка-
чественно при («-0,74-1,0 А/дм2 и /з=10 мин, а очистка отрабо-
танных эмульсий цехов холодной прокатки—при ia=0,5-?2,7 А/дм2
и (»<20 мин.
Кроме электрических параметров, иа степень извлечения
нефте- и маслопродуктов большое влияние оказывают аппаратур-
ное оформление и гидравлические параметры процесса электро-
флотации. Так, например, предложенный фирмой «Форд Моторе»
электрофлотатор с противоточным движением воды и газовых пу-
зырьков, а также с вращающимся подающим и сборным устрой-
ством позволяет более равномерно распределять воду в объеме
аппарата и повысить эффективность очистки [84].
В настоящее время актуальной проблемой является разра-
ботка эффективных приемов безреагентпой интенсификации э.тек-
। рофлотациониой очистки нефте и маслосодержащнх вод. Полу-
чены положительные результаты при очистке этих вод с наложе-
нием электромагнитного излучения в области видимого света,
которое способствует укрупнению частиц. Разработана магнито-
•лектрическая флотационная установка [85], в которой процесс
интенсифицируется в результате перемешивания очищаемой воды
пузырьками газа при изменении полярности электродов через
каждые 15 с.
Электрокоагуляция-фло-ация (ЭК-Ф) как способ очистки
нефте- и маслосодержащнх вод изучена значительно меньше, чем
। юкгрофлотация. Первые исследования были проведены
и УИИВХ применительно к очистке нефтесодержащих вод судов
IM
Высокий эффект может быть достигнут при применении алю-
миниевых электродов. Концентрация дизтоплива в этом случае
снижается с 100—500 до 40 мг/л, а содержание мазута — с 200 до
10 мг/л. Наибольший эффект получен при использовании комби-
нированной системы алюминиевых и графитовых электродов. Так,
при очистке моющего раствора концентрацией 200 мг/л дизтоп-
HIH.1 и 0,15% детергента МЛ-72 в очищенной воде содержалось
11 мг/л нефтепродуктов, в то время как при раздельном исноль-
249
зованнн алюминиевых и графитовых электродов — соответственно
27 и 84 мг/л. Плотность тока принимается 150—200 А/м2, при
этом в зависимости от солесодержания расход электроэнергии
составляет 0,2—1,2 кВт-ч/м3. Для сравнения следует отметить,
чго прн очистке моющих растворов коагуляцией с напорной фло-
тацией в очищенной воде в зависимости от концентрации детер-
гента содержалось 30—70 мг/л нефтепродуктов (начальная кон-
центрация — около 240 мг/л, доза сернокислого алюминия —
400 мг/л).
Применение нерастворимых электродов для очистки нестаби-
лизированных эмульсий предпочтительнее, несмотря на рост при
этом энергозатрат. Увеличение срока службы электродов, умень-
шение объема пены и упрощение технологии ее обработки компен-
сирует дополнительный расход электроэнергии. Вместе с тем ана-
логично другим способам флотации эффективность очистки эмуль-
сий ЭК-Ф с нерастворимыми электродами существенно зависит
от дисперсности извлекаемых капель. По расчетам, при скорости
разделения 1 мм/с и объемном расходе газа 0,01 м3/м3 из очищае-
мой воды может быть извлечено 40—55 мг/л нефтепродуктов
с диаметром частиц 10 мкм и до 5 г/л частиц крупностью 50 мкм
при условии полного использования газовой фазы. С этой целью
целесообразно применять электрореакторы с «газовым слоем»
(см. рис. 4.10, 4.55 и 4.56).
Испытания установки с такими электрореакторами [4] вы-
явили некоторые особенности очистки воды от нефтепродуктов.
Так, эффективность очистки во флоторазделителе зависит пре-
имущественно от дисперсности и концентрации частиц нефтепро-
дуктов. Эффект извлечения растет при увеличении их крупности
и количества без изменения параметров очистки, остаточная кон-
центрация нефтепродуктов при этом составляет 1—5 мг/л.
О влиянии «газового слоя» на эффективность очистки можно
судить из того, что при элекТрофлотации эффект извлечения ча-
стиц диаметром до 14 мкм составляет менее 50 % [4]. а в уста-
новке с «газовым слоем» он значительно выше — достигает 70—
80%.
Газосодержапие обрабатываемой жидкости увеличивается бо-
лее, чем в 2 раза, при неизменных электрических параметрах про-
цесса (расход и плотность тока иа электродах) и достигает 0,5—
1,0%, тогда как в обычных электрофлотационных аппаратах сте-
пень насыщения жидкости пузырьками при изменении плотности
тока в пределах 100—300 А/м2 не превышает 0,04—0,13%.
Для нефтесодержащих вод морских судов характерно нали-
чие значительного (0,5—2,0 г/л) количества ионов магния. Обра-
ботку таких вод перед флотационным раздепением предложено
проводить в катодной зоне диафрагменного электрореактора, где
в результате подщелачивания образуется гидроокись магния, яв-
ляющаяся эффективным сорбентом нефтепродуктов [а. с. 739004
(СССР)]. Пенный продукт обрабатывается в анодной зоне элек-
трореактора, в которой в результате подкисления происходит рас-
250
Рис. 5.15. Схема обезвреживания отработанных СОЖ при наличии солей
магния:
/ диафрагменный электролизер; 2 — флотатор: 3 — фильтр; 4 —сборник шлама и про-
мывной воды. 5 — сборник нефтепродуктов
ворение гидроокиси и отделение нефтепродуктов (рис. 5.15). При
расходе тока 8- 105 Кл/м3 и начальной концентрации 50 мг/л
в гидроокись переходит 40—65 мг/л магния. Увеличение расхода
юка до 106 Кл/м3 вызывает рост начальной концентрации до 80—
125 мг/л. Разработанный способ является перспективным при очи-
тке нефтесодержащпх вод от мойки емкостей морских судов и
анкеров.
Для обезвреживания сильностабилнзированных эмульсий типа
отработанных смазочио охлаждающих жидкостей (СОЖ), мою
тих II друц|х растворов применима схема с предварительным под-
кислением жидкости соляной кислотой до рН = 0,54-5,5 и после-
1ующей электрокоагуляцией с алюминиевыми электродами и от-
стаиванием, предложенная Харьковским отделом ВНИИ ВОДГГО
|(>9| Модификация схемы с дополнительным применением гидро-
цнклонов и электрофлотаторов выполнена институтом <Казме
х.нюбр».
Обработка отработанных СОЖ с исходными ХПК = 5-5-21 г/л
но рекомендуемой ВНИИ ВОД ГЕО схеме обеспечивает ее енн
жевие на 80—95%. Более высокий эффект очистки достигается
применением колонных электрофлотокоагуляторов. Очищенная
ГОЖ при этом удовлетворяет требованиям технических условий
к воде для приготовления рабочих растворов. Однако после не-
скольких циклов циркуляции очищенные отработанные СОЖ не
г оответствуют требованию по коррозии в связи с накоплением
и воде различных форм хлора. Предложенная технологическая
схема без использования кислоты для очистки СОЖ (рис 5.16)
позволяет многократно применять эмульсию в технологическом
цикле.
Из других способов электрохимической очистки нефтесодер-
жащих вод следует отметить применение неоднородного электри
веского поля, создаваемого системой стальных электродов, рас-
положенных перпендикулярно друг к другу. Электрические воздев
СНИ1Я также интенсифицируют очистку нефтесодержащих вод
в гндроцнклонах (рис. 5.17). Модификацией конструкции явля-
251
Рис. 6.16. Технологическая схема регенерации обработанной эмульсии «Укри-
нол-1» с использованием колонных электрокоагуляторов
/ — электрокоагулятор; 2 — циклон; 3, 4—сборники соответственно отходов и отработан
ной эмульсии; 5 — ротаметры; 6, 7 — сборники соответственно электролита и очищенной
воды
Рис. 5.17. Схема гидроцикюиа
с использованием неоднородного
электрического пиля
/ — патрубок отвода нефтепродуктов
(анод); 2 — диэлектрические вставки;
3 — корпус. 4 ™ подающий трубопро-
вод; 5 — осадочная часть; 6 — катод
Рис. 5.18. Схема судовой установки для
очистки нефтесодержащих вод в неод-
нородном электрическом поле
Г — сборник отстоя; 1 — цистерна для от-
стоя; 3 — фотоплотномер; 4 — преобразова-
тель частоты; 5 — генератор; g— камера-раз-
делитель; 7 — подача исходной эмульсин;
я — насос
ется применение системы электродов типа «Коаксиальные ци-
линдры» |20|.
Типовая схема судовой очистной установки льяльных вод
с применением вышеуказанных аппаратов приведена на рнс. 5.18.
После отстаивания иода поступает в камеру-разделитель, где
252
смонтированы перпендикулярно друг к другу плоские электроды.
Учьтразвуковые излучатели для интенсификации процесса распо-
ложены в камере-разделителе с таким расчетом, чтобы излучения
были направлены вдоль потока.
•едует отметить, что до настоящего времени отсутствует еди-
ное мнение об оптимальной конструкции электроаппаратов для
очистки нефтесодержащих вод из-за недостаточного опыта экс-
плуатации промышленных установок. Однако первые полученные
результаты подтверждают целесообразность внедрения в прак-
тику очистки нефте- и маслосодержащих вод электроустановок.
ГЛАВА 6
МЕТОДИКА РАСЧЕТА АППАРАТОВ
ДЛЯ ЭЛЕКТРООБРАБОТКИ П. ИРОДНЫХ И СТОЧНЫХ ВОД
6.1. ОСНОВНЫЕ ТЕХНОЛОГИЧЕСКИЕ ПАРАМЕТРЫ
ПРОЦЕССОВ ЭЛЕКТРОХИМИЧЕСКОЙ ВОДООЧИСТКИ
Несмотря па существенные различия в механизме и кинети-
ческих закономерностях применяемых на практике методов элек-
трохимической очистки загрязненных жидкостей, а также на
конструктивные различия аппаратов для электрообработки
воды, некоторые технологические показатели и отдельные тех-
нические указания по их расчету являются общими. Основные
из них рассмотрены ниже.
1. Важнейшим показателем процесса электрохимической очи-
стки загрязненных жидкостей является расход электроэнергии,
юобходимый для достижения требуемой степени очистки еди-
ницы объема. Удельный расход электроэнергии в общем случае
определяется по формуле
(7эл = <7,С/о«.ц/(1 ОООПвХГ) = /(/oem'(100Ch]BQ). (6.!)
me qa — количество прошедшего электричества, А - ч; / — сила тока. А;
— напряжение на электролизере. В; т)« — КПД выпрямительного устрой-
ств; 1Г и Q — соответственно объем, м3, и расход обрабатываемой жидкости,
м’/ч
2. Обозначив вторую важнейшую характеристику электрохи-
мического процесса как расход тока (удельное количество элект-
ричества, приходящееся на единицу объема обрабатываемой
жичкости, т. е. Дт = 9э/Уж = //’>/Уж, или Дт=//(?), получим сле-
дующее выражение для расхода электроэнергии:
(Гэл Дт Г/общ/(1000|]в). (6.2)
253
По физическому смыслу величина Дт определяет дозу про-
дуктов электродных реакций (т) на единицу объема жидкости,
что эквивалентно дозе реагента при химической обработке. Эта
величина легко устанавливается экспериментально, ие требует
применения специальных методик для определения концентра-
ции электрохимически генерируемого вещества в жидкости (Сп),
а также выхода по току (Вт). Взаимосвязь этих величии элект-
рохимического процесса находится в соотношении
С„ = т ВТДТ. (6.3)
3. Плотность тока i определяет скорость электродных реак-
ции и является характеристикой процессов иа границе раздела
«электрод—раствор». Величина i не влияет на превращения
в объеме электролита. В последнем случае превалирующее вли-
яние оказывает расход тока.
Влияние плотности и расхода тока па процесс превращения
примесей в объеме обрабатываемой жидкости следует определять
при Дт=const с целью получения достоверных результатов Ве-
личина i, как правило, определяет выход по току той или иной
электродной реакции, поэтому для сохранения Св=const в об-
рабатываемом объеме необходимо изменять величину Дт путем
варьирования плотности тока и времени обработки.
4. Общее напряжение па электролизере (/oCiu можно разделить
на следующие составляющие: равновесные электродные потен-
циалы <ра и фк: перенапряжения иа электродах т]а и т|к и падения
напряжения на преодоление электрического сопротивления в ра-
створе Up, диафрагме С/д, электродах U, и UK, токоподводящнх
шинах и,п п контактах [/КОнт:
и общ = фл + Фк + Ча + Чк + Up + l/д + ^а+^к + ^ш + ^коит- (6 4)
В бездиафрагменных электролизерах Ua исключается.
4.1. Равновесные потенциалы электродов (фа и <рк) определя-
ются протекающими иа них реакциями и могут быть рассчитаны
по термодинамическим данным для каждой электродной ре-
акции.
Например, при электролизе кислых растворов иа катоде раз-
ряжаются ионы водорода: 2Н+ + 2е—‘Н2, а на аноде — молекулы
воды: П2О—2е-*2Н++ */2О2.
Термодинамические функции воды и ее составляющих изве-
стны с большой точностью. Так, стандартные величины свобод-
ной энергии ЛС|5 с для Н2, Н+ и О2 равны нулю, для Н2О =
«=—56.69 ккал-моль-1, для ОН =—37,585 ккал-моль-1.
Расчет изменения изобарного потенциала в процессе проте-
кающих реакций производится алгебраическим суммированием
значений \G° для продуктов реакции (со знаком плюс) и исход-
ных веществ (со знаком минус):
для первой реакции
^Gol, - Дб° + 0:
254
Рис. 6.1. Увеличение сопротивления электролита в зави-
симости от газонаполнеиия
Г
для второй реакции
AG® 2AG®, + + 1/2AG®, - AG®, о = - 56.69 ккал-моль
Из рассчитанных значений изменения изобарного потенциала
н 1Йдем стандартные потенциалы этих процессов:
То.(II) - Л°?/(2 23.07) = 0.0; <р0. (OJ -0°/(2 23.07) 1.23 В. (6.5)
1де 23,07- коэффициент перехода от тепловых к электрическим единицам.
Стандартные потенциалы электродных реакций соответст-
вуют нормальным условиям (/-*=0,1 МПа. Т=15°С, активность
электролита а -1) и могут быть найдены по таблицам [104].
На практике условия электролиза отличаются от стандарт-
ных, поэтому при определении равновесных потенциалов делают
поправку иа температуру и учитывают активность электролита.
Для рассматриваемых процессов равновесные потенциалы могут
быть рассчитаны по уравнениям Нернста:
Тн, =- <Ро. (Н,) + 2-3 (RT/F) 'й- 2.3 (RT/F) pH = - 0.058рН; (6 6)
Ч?) - Ч'о (Н ) + 2.3 (RTfF) 1g =1,23— 2.3 (RT!F) pH = 1,23 — 0.058PH
(6-7)
4.2. Падение напряжения в растворе UP для наиболее распро-
страненного случая взаимно параллельных плоских электродов
рассчитывается но формуле (В)
Up = (i7xR)/3Or (6 8)
При этом аг вычисляется по (2.37) или (2 38) Газосодержание
Г при определении ог может быть найдено по (4.40) или по фор-
муле
Г= <7гЯэ/(2/,?п), (6 9)
где <?г — количество газа, выделяющееся с единицы поверхности электродов
Графическая связь между величиной газонаполнеиия и уве-
личением сопротивления электролита изображена на рис. 6.1.
При расчете электрофлота горов, электродная система кото-
рых состоит из сетчатого катода и параллельно расположенного
плоского анода (см. рис. 6.2), падение напряжения в обрабаты-
ваемом растворе можно приближенно рассчитывать по формуле
В П Машовца-
Up 14.6«к/(хяф lg (6 d) + i./xw (Z, - 6/2)] К. (6. Ю)
255
Рис. 6.2. Схема электродной системы из сетчатого катода
и плоского анода
4.3. Электрическое сопротивление диафрагмы ил равно сопро-
тивлению электролита в ее порах и может быть рассчитано по
формуле В. В. Стендера:
t/д = Рэбд₽*/(Уп5д) - Рэ/*/(уп5д6д), (6.11)
где (3 — коэффициент извилистости пор (//бя), показывающий, во сколько раз
действительная длина поры больше толщины диафрагмы; уп— объемная пори-
стость в долях единицы.
4.4. Потери напряжения на преодоление сопротивления элек-
тродов (Un и Ult) подводящих шин (t/ш) рассчитывают по за-
кону Ома как для проводников первого рода (В):
t/np= (прРпр/пр/^пр, (6 12)
где /Пр — величина тока, протекающего по проводнику. А; рПр — удельное со-
противление материала проводника, Ом-мм1-м“' (величина рлр приведена
в табл. 6 1); /Пр — длина проводника, м; Звр — поперечное сечение провод-
ника, мм2.
4.5. Падение напряжения в контактах определяется по фор-
муле
(6.13)
(/ КОНТ — /контРкопт«
О’коит и рконт — величины соответственно в А/мм2 и Ом-мм2).
Величина рКОит зависит от удельного давления в контакте, ха-
рактера обработки поверхности материала и температуры кон-
такта. При удельном давлении 50 кг/см2 и температуре 25 °C
Ркоит принимает следующие значения: для контакта «медь—
медь» — 0,05, «алюминий — алюминий» — 0,5, «графит — медь» —
1,25, «титан—медь» — 0,1.
4.6. На практике общее напряжение <7оСщ может быть опре-
делено па основании экспериментально полученных зависимостей
между напряжением и плотностью тока на электродах (вольт-
амперных характеристик), построенных для конкретного вида
сточных вод и выбранного материала электродов при определен-
ном межэлектродном рас-
стоянии. Определение мож-
но произвести графически
Таблица 6.1. Удельное сопротивление
и допускаемая плотность в токопроводах
Материал Удельное электрическое сопротивление. Оимн’-и-' Допускаемая плотность тока. А-мм-*
Алюминий 0,026- 0,029 2,0
Медь 0,017-0,018 3,0
Сталь 0,103—0,140 1.5
Цннк 0,053—0,062 —
Латунь 0,031 0,079 —-
или аналитически по урав-
нению регрессии С/общ = с/ +
+d, где с и d — эмпириче-
ские коэффициенты, опре-
деляемые при математиче-
ской обработке данных
вольтамперных характери-
стик. В табл. 6.2 представ-
лены эти зависимости, ус-
тановленные при электро-
256
Рис. 6.3. Схемы соединения электродов
а. 6, в — соответственно монополярная, би-
полярная и комбинированная схемы
коагуляционной обработке неко-
торых категорий промышленных
сточных вод [51].
Соединение электродов в ка-
мерах основных блоков электро-
обработки воды возможно по
моиополярпой, биполярной и
комбинированной схемам (рис.
6.3.). Наиболее целесообразно
применение комбинированной схемы (комбинированное соедине-
ние нескольких блоков по моно- или биполярной схеме) с целью
снижения общего потребляемого тока электроустановок н мак-
симального использования КПД выпрямительных агрегатов.
При низком напряжении электролиза (2—-5 В) возможно по-
следовательное соединение отдельных блоков электродной си-
стемы (рис. 6.3, а) с целью получения суммарного напряжения,
равного напряжению на клеммах применяемого выпрямителя.
В этом случае падение напряжения на электродной системе сла-
гается из падения напряжений на каждом блоке при неизменной
силе тока (В)
V» 1/14-17,+ . . . +Un = £ Un. (6.14)
0-1
пе Ut, Ut, .... Un—падение напряжения в блоках электродной системы
Прн одинаковых размерах блоков (В)
U3=NUn. (6.15)
гте .V — число послетоватсльно соединенных блоков электретной системы
(рнс. 6.3, в).
Формулы (6.14) и (6.15) справедливы также для биполярной
схемы соединения электродов, при этом (7|, и2........Un составляют
Т а б л н ц а 6.2. Зависимости между плотностью тока и напряжением иа
электродах при электрокоагуляции сточных вод (межэлектродиое расстояние 2 см)
Категория сточных вод Материал Аналитическая зависимость
Общин сток меховых фабрик Ст.0 изп = 0,514т + 0,51
Сырьевой сток меховых фабрик Ст.0 </,л 0,544i + 0.41
Общий сток кожзаводов СтО = 0.29i + 0,79
Общий сток кожзавода без золь- ника СтЗ U эя — 0,33< + 0,55
Общий сток молокозаводов Алюминий U,n = 16,6т + 5,0
Общий сток заводов искусственной технической кожи Алюминии U3n = 70,01 + 0,1
Общий сток мясокомбинатов Дюр - алюминий U3„ 0,42т + 0,83
'/гЭ Заказ Ж 2(48
257
падение напряжения на каждой ячейке электролиза, a N равно
числу ячеек («—1).
Питание электродной системы выпрямленным током произ-
водится от источников постоянного или выпрямленного тока,
обеспечивающих электрические параметры процесса очистки. Об-
щее напряжение иа электролитическую ячейку при электрохими-
ческой очистке не должно превышать 36 В с целью обеспечения
правил техники безопасности. Исключение составляют способы
обработки воды электрическими разрядами, при реализации ко-
торых напряжение в рабочей камере может достигать 40 кВ и
более [20]. При этом необходимо учитывать особые требования
по технике безопасности электроустановок.
5. Величина тока, который необходимо пропускать через
электролизер, может быть принята по выбранному типу выпря-
мительного устройства, определена по заданной анодной плотно-
сти тока с учетом общей площади анодов или ориентировочно
найдена по формуле
/ Дт(? (С0-Ск)<?л;. (6.16)
где Со и Ск — концентрация загрязняющего компонента в исходных я очищен-
ных водах, г/м3: Д/ — теоретическое количество электричества, необходимое
для очистки жидкости от I г загрязнения, Кл.
6. Вычисленные значения и 7 позволяют подобрать не-
обходимый тип выпрямительных устройств, в качестве которых
рекомендуется использовать агрегаты серии ВАК н ВАКР [15].
Источник постоянного электрического тока должен быть ci аб-
жеи регулировочным устройством, обеспечивающим изменение
величины тока и напряжения в электрической цепи, а также
устройством для периодического реверсирования тока.
7 Требуемая установленная мощность составляет (кВт)
РсбЩ 7<бщ1/(б1Ц- (6. 17)
8. При проектировании установок для электрохимической вс-
доочпетки важным вопросом является обеспечение требуемой
степени вентиляции производственного помещения, так как водо-
род, выделяющийся при электролизе на катоде, может образовы
вать с воздухом взрывоопасную смесь. Нижний предел взрывае-
мости соответствует 4.0 об. % водорода в воздухе. В соответствии
с требованиями СНиП П-М.2—72 предельно допустимая взрыво-
опасная концентрация (ПДВК) водорода в производственном
помещении нрнннмается 10% от нижнего предела взрываемости,
т. е. 0,4 об. %. Вследствие этого ориентировочный расчет требуе-
мой степени вентиляции осуществляется в последовательности,
приведенной ниже [90].
А Объем водорода, выделяющейся га катодах при нормаль-
ном давлении, может быть определен по формуле (м3/ч)
vri, ~ в? Аэ 1 <273 + т>273- (6 ,3)
ие В* — катодный выход по току водорода: В* =0,9 0.95; А* —объем-
ный электрохимический экнивалснт водорода- Д* =0,00042 м’/(Л-ч).
258
Б. Количество водорода, растворяющегося в воде, может быть
найдено по формуле (м3/ч)
21,4(273+ Т) Q 273 103, (6.19)
не 21.4— объем водорода, растворяющегося в 1 м’ воды при Т=0 °C и дав-
лении 0,1 МПа.
Б. Объем водорода, выделяющегося непосредственно в атмо-
сферу, составляет (м3/ч)
VaHT - Vh ~ VH (6 20)
Г. Электролизер должен быть снабжен отдельным вентиля-
ционным устройством, если в результате выделения водорода его
содержание в воздухе производственного помещения может до-
стигать ПДВК, т. е. 0,4 об. %.
Д. При расчете возможной концентрации водорода в воз-
14 \с производственного помещения учитываются общий объем
помещения, кратность смены в нем воздуха в течение I ч, до-
стигаемая в результате естественной или уже действующей в по-
мещении механической приточной вентиляции. Концентрация во-
дорода рассчитывается по формуле
Си VJ,/(A'lipVlip) 100 %, (6 21)
г ю Л'пр — кратность смены воздуха в производственном помещении в течение
I ч, I io> — объем пронзвоктвенпого помещения, MJ.
Расчет системы вентиляции следует проводить в соответст-
вии с главой СНиП II 33—75.
I Прн оборудовании электролизера автономным вытяжным
вентиляционным устройством его производительность может быть
определена нз условия разбавления выделяющегося водорода
воздухом до концентрации его в отсасываемом воздухе (<<0,4
об. %). В соответствии с этим производительность вентилятора
составит (м3/ч)
<?„ (300 + 350) у* . (6.22)
Ж. При выполнении требований, указанных в пи. Г- Е, здание,
в котором размещена электролизная установка, по СНиП П-Л1.2—
72 может быть отнесено в категории Д (невзрывоопасное).
9. Прн проектировании электрохимических систем водоочп-
с!кн важным вопросом является расчет теплового баланса, для
составления которого нужны данные баланса напряжения, ма-
териального баланса, а также значения температур исходной
воды, получаемых продуктов электролиза, наружных поверхно-
ciefl аппаратов и окружающей их среды.
Приход тепла составляется из трех статен.
I. Количество тепла, вносимого в электролизер обрабатывае-
мой водой и засасываемым воздухом, может быть определено по
данным расхода воды, количеству поступающего воздуха, их те
плоемкостн и температуры. Теплоемкость воды ст прн всех
1 л*
259
температурах «1 Ккал/л. Газовая постоянная R для воздуха со-
ставляет 288 Дж/(кг«град)
2. Тепло, выделяемое в электролизере электрическим током
(джоулево тепло), является основной статьей в общем приходе
тепла. Оно может быть рассчитано по формуле (кДж/ч)
<?Т = 3.617/((У,— 1,4Н>. (6.23)
где 3,617 — пересчетный коэффициент от электрических к тепловым единицам,
1,48 — величина напряжения, расходуемая иа разложение воды, В.
3. Тепло, выделяющееся при прохождении химических реак-
ций и побочных процессов, включает: а) теплоту сгорания ано-
дов (в случае применения графитовых электродов эта теплота
составляет 394 Дж на 1 г-моль для образовавшейся газообраз-
ной углекислоты СО2 и 419 Дж — для растворенной); б) теплоту
взаимодействия с водой выделяющегося в электролизере водо-
рода (определяется по количеству водорода в электролитиче-
ском газе: иа 1 моль Н2 выделяется около 100 кДж); в) теплоту,
выделяющуюся при взаимодействии продуктов электролиза (на-
пример, в случае электролиза поваренной соли теплоту взаимо-
действия хлора и щелочи с образованием хлорноватистой и хлор-
новатой соли около анода и теплоту восстановления хлорновати-
стой соли па катоде и т, п.). Приведенные составляющие могут
ие учитываться, так как эти величины обычно невелики.
Расход тепла состоит нз двух основных статей (рассчитыва-
ется по общепринятым методикам)
1. Теп то, выносимое уходящими продуктами (хлором, водо-
родом, водяными парами, кислородом, углекислотой и т д.),
а также очищенной водой, образующимися осадками и флото-
шламом, днлюатом и рассолом (в процессе электродиализа)
и т. п.
2. Тепло, расходующееся на излучение и конвекцию. Расчет
теплового баланса обязателен при проведении процесса электро-
диализа. так как при этом следует особо обратить внимание иа
то, чтобы плотность тока ие увеличивалась до такой величины,
когда может начаться перегрев и прожог мембран. Изготовляе-
мые в настоящее время мембраны допускают максимальную тем-
пературу 60 °C.
Предельная плотность тока по отношению к джоулевому
теплу зависит от электрического сопротивления и теплопроводно-
сти мембран, от коэффициента теплопередачи между мембра-
ной н протекающим раствором и от температуры раствора. На-
пример, при электродиализе воды с большим содержанием рас-
творимых солен количество подводимой электрической энергии
на единицу объема жидкости может быть настолько большим,
что вызовет значительное повышение температуры растворов
Повышение температуры воды может быть определено по
формуле
&T = Ql'(Ac1qCBy3), (6 24)
где ДГт — удельная теплоемкость; фсв— секундный расход воды; уа — плот-
ность электролита
260
Для отвода тепла от электродиализных аппаратов предусмат-
ривают охлаждение одного нз потоков (дилюата или рассола) и
регулирование температуры всей системы Для этого устанавлп
иают теплообменник в гидравлической системе ЭДУ вне элект-
родиализного аппарата.
Таким образом, в соответствии с указаниями данного раздела
технологический расчет электроаппаратов для водоочистки сво-
дится к определению основных геометрических размеров рабо-
чих камер и всей установки в целом, а также к определению
всех эксплуатационных характеристик процесса общего напря-
жения и силы тока на отдельных электродных блоках с целью
подбора типа выпрямительных устройств; расхода потребляемой
электроэнергии; продолжительности эксплуатации растворимых
электродных материалов до полной их замены; объема образу-
ющихся осадков и флотошлама, необходимой степени вентиля-
ции пропзвотственного помещения; количества выделяемого при
электролизе тепла для оценки теплового баланса процесса очи-
стки воды.
Ниже приведены частные методики расчетов некоторых типов
электроаппаратов, позволяющие определить массогабарнтные их
характеристики и основные эксплуатационные параметры Рас-
чет же общего напряжения на электродных блоках, определение
мощности и расхода потребляемой электроэнергии, количества
выделяемого водорода для установления необходимой степени
вентиляции, но i6<>р соответствующих выпрямительных устройств
и составление теплового баланса производятся по указаниям на-
стоящего раздела.
6.2. РАСЧЕТ ЭЛЕКТРОФЛОТАТОРОВ
Основными исходными данными для расчета следует прини-
мать: производительность установки Q (м3/ч), начальную и ко-
нечную концентрации загрязняющего компонента Со н Ск (г/м3),
1сктронроводность хи, вязкость I), плотность ро обрабатываемой
жидкости.
На основании предварительных экспериментальных исследо-
ваний выявляют следующие оптимальные параметры: плотность
катодного тока iK (А/м2), высоту рабочего слоя жидкости Но (м),
средний радиус пузырьков водорода г„ (м), продол ж итедыюсть
обработки / (ч), плотность газа р/.
В табл. 6.3 представлены оптимальные параметры процесса
электрофлота цпн некоторых категорий сточных вод.
Методика расчета электрофлотаторов должна учитывать кон-
струкции электродных блоков и их расположение в рабочих ка-
мерах аппаратов.
Ниже представлены методики расчетов электрофлотацнонных
аппаратов, имеющих горизонтально расположенные электроды
из плоского анода и катодной сеткп и вертикально расположен-
ные плоскопараллельные пластины.
«I
Зака» № 3148
261
Таблица 6.3. Оптимальные параметры и эффективность
электрофлотацнонной очистки сточных вод
Источники образования сточных вод Показатели
'»• МИН 4.' А дм- материал электродов (А — анод. К — катод)
Молокозаводы 10 0,5 Л — ОРТА, К —не- ржавеющая сетка
Мясокомбинаты 15 1.5 Л — графит, К — не- ржавеющая сетка
Рыбоперерабатывающие заводы (от тузтука) 10 0,5 То же
Красильно-отделочные фабрики Заводы бытовой химии: 30 1.5-2,0 Л — ОРТА, К — не- ржавеющая сетка
производства синтетических мою- щих средств 20-30 2-3 То же
производства черной туши (после предварительной электрокоагуля- ции) 10—15 1,0 >
Продолжение
Источники образования сточных вод Эффекты очистки. •.
ПО В1НГ1ШИ- иым веществам ПО жирам по ПАВ по обес- цвечива- нию
Молокозаводы 52 60 — —
Мясокомбинаты 74 «0 — —
Рыбоперерабатывающие заводы (от тузлука) 71 75 —
Красильно-отделочные фабрики Заводы бытовой химии: 59 — 65 40
производства синтетических мою- щих средств — — 55 —
производства черной tjiuh (после предварптслыюй электрокоагуля- ции) 98 До 100
6.2.1. Расчет электрофлотаторов
с горизонтальными плоским анодом
и сетчатым катодом
Методика расчета разработана па основании анализа и ре-
шения дифференциального уравнения (4.42) процесса электро-
флотацнн Б М Матовым для очистки масло- и жнросодержа-
щнх сточных вод.
1 Вычисляется средняя скорость подъема пузырьков в жид-
кости v„ по формулам (4.38) или (4.39).
2. Определяется концентрация пузырьков водорода в жидко-
сти С„ из уравнения (441).
3. Находят значение параметра процесса
*о= яо"п/(т'СнРо). (6 25)
где у'—отношение среднего объема нерастворимых частиц к среднему объему
пузырьков водорода.
4. Принимая во внимание, что высота //() и длина аппарата
1-ф при обработке дайной жидкой неоднородной системы оста-
ются постоянными при заданной производительности Q, опреде-
ляют ширину аппарата
Вф = (4Qb2 In C0[cosn//0/(2L^)] Ск) /[Яо/?^2 (/.ф - /г)]. (6.26)
где Ьг и /г — расстояния соответственно от боковой и продольной стенки аппа-
рата ю края электродного блока, равные 0,03 + 0,05 м.
5. По найденным габаритам аппарата вычисляют рабочую
площадь, которая в электрофлотационпом аппарате должна быть
равной площади поперечного сечения рабочей камеры
= Вф(-ф- (627)
6. Рассчитывается величина постоянного электрического тока,
необходимая для питания аппарата,
I iKF* (6.28)
6.2.2. Расчет электрофлотатора с вертикальными
плоскопараллельными электродами
1. Рабочий объем аппарата
V* = <?/,. (6.29)
2. Площадь камеры флотации
(6.30)
г ic Н, принимается равной 0,8—1.1 м.
3. Общая высота электрофлотатора
77ф 7/д Ц-/ц/i2, (6.31)
। и* Л| и /о — высоты соответственно слоя пены (hi —0.05 + 0,15 м) и борта аппа-
рата над уровнем пены (с учетом размещения устройств для удаления пены
h3 • 0.2+0,4 м).
4. Длина камеры флотации
бф=/7фВф, (6 32)
lie — ширина аппарата: прн производительности до 10 м’/ч вф — 0,8 +
1.0 м, до 20 м’/ч бф=1,5+2,0 м, до 50 м’/ч вф = 3,0ч-3,5 и.
Для более удобной компоновки I ф = Зн-4 Вф-
5. Общая длина аппарата
l-о £ф+£„ (6 33)
no li длина сборного кармана очищенной жидкости: L, =0 15+0,3 .».
263
G. Число электродов (пластинчатых вертикально расположен-
ных у дна):
анодов
па = (вф - 2a + Ь. + 2/э) (2/э + Ь, + />*); (6.34)
катодов
лк = (®Ф — 2в — 21J (2/э -}- Ь| + Ь2), (6.35)
где a — расстояние от стенки аппарата до края электрода: а=0,034-0,05 м;
hi — толщина катодов- ft|=0,3-s-0,5 мм; Ь2— толщина анодов: для графита
/>2 = 0.024-0,04 м, для ОРТА и других металлов fcj=0,l-i-0,5 см; /• — межэлек-
тродное пространство: /а=0,05т-0,02 м.
7. Активная площадь одного вертикального электрода без
учета площади торцов (м2)
/, = 2ЛЭЛ„ (6.36)
где Л3—высота электрода: Л»=0,14-0.15 м; Ь.— ширина электрода (принима-
ется равной /.♦, т. е 0,1).
8. Активная площадь горизонтально расположенных электро-
дов (м2)
/1 = /к (Дф-0,1)(й-0.1). (6.37)
9. Сила тока иа электрофлотатор (А)
/ = «ropV- 1 (6 38)
10. .Масса электродной системы (т)
М 4 W.. (6 39)
где и уДе — плотность соответственно катодного и анодного материала,
т/м’ (д..я графита -у=1,5).
II. Продолжительность работы электродной системы может
быть определена для случая применения графитовых анодов
(сут):
т МА/ 24 7гр/) Пр l.5#yia/(24<7rr/), (6.40)
где чр — коэффициент использования графитовых анодов: Чр~0,84-0,9; qrt>—
пзпо». графита: $гр=83 мг/(А-ч).
6.3. РАСЧЕТ ЭЛЕКТРОКОАГУЛЯТОРОВ
Данная методика может быть использована при расчете
электрореакторов, применяемых для агрегации дисперсных ча-
стиц и молярнзацин примесей за счет взаимодействия их с про-
дуктами растворимых электродов и формирования твердой
фазы в объеме обрабатываемой воды. Методика применима при
расчете элекгрокоагуляторов с растворимыми электродами,
а также электрокрнсталлизаторов, базирующихся на использова-
нии твердой фазы для ускорения процесса кристаллизации. При
этом предполагается, что действие электрического поля является
сопутствующим и учитывается величиной расхода тока.
261
Рис. 6.4. Расчетная схема электрокоагулятора
Исходными параметрами для расчета устаговок следует при-
нимать: производительность установки Q (м3/с). расход тока
Чт (Кл/м3), плотность тока на электродах (, (Л/м2). Расчетная
схема электрореактора представлена на рис. 6.4.
Последовательность выполнения расчетов приведена ниже.
I. Объем камеры электролиза V'» определяется нз условия
размещения электродной системы, обеспечивающей необходимый
расход тока с целью получения требуемой дозы электрогенернру-
емого вещества.
Так как при обработке воды в бездиафрагменнох! электроре-
акторе используются одновременно продукты анодной и катод-
ной реакций, то суммарная доза вещества, образующегося в объ-
еме обрабатываемой жидкости (г/м3)
д:-д:+д; (b^+bx)^. <6 41,
। ,. ga(K) — вь1хо1 по току данного вещества А на аноде (катоде); Дт
расход тока, который определяется экспериментально или вычисляется по тре-
буемой дозе вещества (для некоторых категорий природных и сточных вод
*тн величины приведены в табл 6 4)
Прн этом необходимо обеспечить условие: > Дв, где
Чт—требуемая юза вещества для осуществления превраще-
ний примесей, определяемая экспериментально для различных
категорий вод или может быть принята ориентировочно по дозе
вещества с применением химических реагентов [СНиП 2.01.02—
К1].
2. Сила тока, необходимая для выделения в раствор Д/ ве-
щее та (Л)
/Р Дт<2 (6 42)
3. Активная поверхность электродов (м2)
5, Дт<? (3.6«,). (6 43)
265
Г а б л и ц а 6.4. Параметры работы эаектрокоагуляторов при очистке некоторых категории сточных под
Расстояние между электродами /э. мм О СО ОО Щ Р to О О Т О tC сч сч сч сч — сч — сч — | сч —
Материал электродов « .= .= 2 • X X X (, X » о о о о s о 5 s S. © t-н йь SE (-5ESrab^f-x£ О О и и 2 <_>2£o.<J2s « < <5 <’|В
Расход металла электродов Д*. г/м" 8 8 8 о о — о о 8 гч. еч ci Is- — — г* сч — — II II 1 1 1 1 1 1 8 8 8 8 n § — ® сч — — J
Напря- женность электро- лиза, В in lO хг СТ II II со со сч сч ir сч ю ш сч сч сч — — оо— — — — 1 1 II 1 11 оо О сч сч о оО оо
Расход электро- энергии ®эл* кВт ч/м О О СЧ 00 Ю tO О tO tO tO xf* xy 7 7 7 7’ 7 7 77 7 77 ubco -<r CO CO о О OU3 ЧГ СЧСЧ — o' о" о о — — — - О с'с
© W я -< Л © X Сх£ 88 88 о 8 о 88 о 8S 77 7 7 7 7 ® 77 77 3 8 88 8 SS8S8S??
Расход тока Дт Кл л s 8 —8 о 8 * §8 ? SS СЧ Ж — СО О чу >Л О4< tn «Л — | | || | 8 3 S3 Й 83 §8 2 £2 — ю со— сч со — со — — —
Рекомендуе- мая величина pH обраба- тываемой воды tn 2 2 2 2 г. о со 00 00 — оооо 1 I II 1 ' 1 М । 1^ со 00 00 оо со оо со ь- с^- 07 г--с -
Категория обрабатываемых вод Конезаводы: общий сток сощии сток оез золь- ника Меховые фабрики: общий сток доочистка после реа- гентной обработки Фаорики искусственной технической кожи Мясокомбинаты Молокозаводы Нефтесодержащие стоки судов Гальванические цехи Доочистка биологически очищенных сточных вод
Плотность тока на электродах ь принимается с учетом мак-
симального выхода по току данного вещества в раствор на осно-
вании экспериментально полученных вольтамперных зависимо-
стей. При отсутствии таких данных можно воспользоваться упро-
щенной эмпирической зависимостью эффективности очистки от
плотности тока при постоянных значениях его расхода.
4. Расчет конструктивных размеров электрореактора произ-
водится в следующей последовательности:
а) Площадь поперечного сечения аппарата (м2)
<?/^ж = В.А. (6-44)
где Ви — ширина реактора; LH — длина реактора; — скорость движения
в межэлектродпом пространстве.
б) Приняв отношение Вк : LK= 1 : 1, найдем размеры реактора
в плане (м)
= = (6.45)
в) Число пластин, размещаемых в камере на ширине LK,
Лэ = (f-к — 1з)'1Рз /э). (6.46)
lie I, и Рл—соответственно межэлектродное расстояние и толщина электро-
дов (см. рис. 6.4): lt — ориентировочно можно принимать по табл. 6.4. Р,=
54-8 мм.
Принимаем число п3 равным целому числу и уточняем длину
камеры (м):
^-к («э—1)/эЧ-Р»яэ- (6.47)
г) Размер одной пластины реактора (м2)
/э = Мэ S,(n,— 1), (648)
п, соответственно, высота пластины (м)
Величины Лэ н Ь3 могут
равными кратным ширине и
из которого будут изготов-
лены электроды.
д) Уточняем скорое1ь
ишженпя воды в межэлек-
родном пространстве для
многопоточной схемы (м/с)
4 <?,[(«,-1)^,1 (6.50)
или с помощью номограм-
мы (рис. 6.5).
Для одиопоточной схе-
мы н.,=2 вместо л.,— I сле-
пет использовать выраже-
ние (л,— 1)/А\., где А’г —
число секций с многопоточ-
ным движением воды.
Лэ = /э ь3. (6.49)
быть скорректированы с размерами,
длине стандартных листов металла.
Таблица 6.5. Рекомендуемые скорости
движения воды в межэлектродном
пространстве электрореакторов, м ч
Схема движения воды Мини- мальная Средн я я Макси- мальная
Восходящее движение, Нисходящее движе- ние: 20 30 40 100
при использова- нии газовой фазы 20 50—80 100
бет использова- ния газовой фазы 10 10- 20 100
Гор нзоптальное дви- жение 20 20 100
267
Рис. 6.5. Номограмма для расчета скорости движения воды в меж электродном
пространстве этектрореактора с плоскопараллельными электродами
Рассчитанное значение сравнивается с рекомендуемыми
значениями, приведенными в табл. 6.5 Прн невыполнении соот-
ношения BK:LK н значений рекомендуемой скорости и» изменяют
величины b3, h3 и повторяют расчет.
ж) Рабочий объем камеры электролиза (м3)
V3 = b3h3l3(n3—I)-|-бэРэ^э- (6.51)
з) Строительный объем аппарата (м)
= V, + 4 V„ BnLnHn + (l/3)BnZ.nHOc H B„LnH„. (6 52)
где В — строительная ширина реактора В ;“Дк-Ь26, (6.,—величина зазора
между электродной системой и стенками аппарата ба=54-10 мм), La — строи-
тельная длина реактора: Ln = Д.и + 26 , 11а— строительная высота реактора;
-(1,14-1,2) Л»; Яос—высота осадочной части, м (принимается конструктивно
прн угле наклона конуса 45—60е); Ня — высота нейтральной зоны, м: //н—
(0.24-0,3) Л».
5. Масса электродной системы электрореактора (т)
М, = ТМе/вРэ"'». (6.53)
где уме — плотность материала электродов, т/м3: для железа ум»—7,86, для
алюминия ум «—2,58
6. Масса металла электродной системы с растворимыми
электродами, которая может быть использована при электро-
лизе (т)
Мр -= ПрМэ, (6.54)
где т]р — коэффициент использования материала электродов: ц-. —0.84-0.9.
268
Расход металла злскпродоб Гододои расход неполна м"^трод
Расход пока Д,, Л ч/м3 Расход пока Л.. 4 " /н3
Рис. 6.6. Номо|рамма определения расхода металла электродов при электро-
химической очистке воды
Рис 6.7. Номограмма для определения основных конструктивных параметров
>1ектрореакторов в зависимости от расхода обрабатываемой воды и основных
центрических параметров их работы
7. Продолжительность работы электродной системы с раство-
римыми электродами (сут.)
тэ Мр 10 24<?сут. (6 55)
Для приведенной методики расчета составлены номограммы
(см. рис. 6.6, 6.7), позволяющие графически выполнить расчет
электрокоа гул яторов.
«.4. РАСЧЕТ ЭЛЕКТРОФЛОТОКОАГУЛЯТОРОВ
Исходными данными для расчета являются: расход сточных
вод Q (м3/ч), активная реакция среды pH, концентрация хлорн
дов в сточных водах [С1 ] (г/л), начальные и конечные концент-
рации загрязнений Со и Сь (мг/л или г/м3), плотность р0 (г/см3),
электропроводность (Ом 1 -см_|), вязкость ц (мПа-с) жид-
кости, плотность pi дисперсной фазы (г/см3), температура воды
Т (°C).
На основании предварительных экспериментальных исследо-
ваний определяют следующие оптимальные параметры процесса:
плотности тока на электродах (А/см2), удельный расход рас-
творимых металлических электродов Хтк (г/м3), выход по току
металла Вт.
6.4.1. Расчет колонных электрофлотокоагуляторов
Основные положения и алгоритм расчета представлены в ра-
боте [69]. Схема аппарата показана па рис. 4.60
1. Продолжительность процесса полной коагуляции определя-
ется по формуле (ч)
/к -= 3 4[i^ \ (R ГС0)|. (6.56)
где R — сфера притяжения коагулирующих частиц загрязнений, равная сумме
их радпчеов (ri-j-r-).
Продолжительность полной коагуляции /к можно также опре-
делить па основании экспериментальных кинетических кривых
изменения концентрации загрязнений при действии электрогене-
рироваииых гидроксидов металлов.
2. Рабочий объем камеры электрокоагуляции (м3)
v; QtK. (6 57)
3 Основные размеры электрокоагуляцноиной камеры (м)
высота
//к (,,58>
где —линейная скорость движения воды в элсктрокоагу-ляциоипой ка-
мере— совмещенной камере х.топьеобразовання и флотации 0.01 м/с;
диаметр
(6 59)
270
4. Диаметр штуцера для ввода исходной воды (м)
= л/4<?(лгж)> (6.<J<>>
где г* — скорость движения воды в подводящем трубопроводе при движе-
нии воды за счет гидростатического напора =0,84-1,2 м/с, при движе-
нии воды самотеком =0.34-0,6 м/с.
5. Остаточная концентрация нерастворимых частиц в камере
электрокоагуляции
Ск = Со (1 + Ф С0/к). (6 61)
где Ф — константа скорости коагуляции.
Ф = 4 3[R Т/(Пгч,Ул)|. (6.62)
6. Рабочий объем зоны электрофлотации (м3)
Г* = (?(ф. (6.63)
где (ф— продолжительность пребывания воды во флотационной камере (ф =
7. Основные размеры электрофлотационной камеры (м):
высота
(6G4)
гте /ф — скорость движения воды во флотационной камере; (ф- 0,0054-0,2 м/с;
диаметр
Рф 2д/^*/(лЯф). (6.65)
8. Площадь кольцевых нерастворимых электродов флотаци-
онной камеры (м2)
5эф=4/л(°ф-Ок)- №66)
Электроды располагаются иа глубине 0,5—1 м под уровнем
жидкости в зависимости от концентрации примесей в зоне фло-
1 а цни.
9. Величина тока иа нерастворимых электродах, установлен-
ных в камере флотации (А),
/эф “ 5»ф1ф, (6 67 )
। ie (ф — плотность тока на электродах в камере электрофлотацин рекоменду-
йся [69] (ф=0,014-0,02 А/см2.
10 Величина тока па электродах блока получения электро-
1сиерирован11ого коагулянта (А)
/к ?УЯ(? (С> os)
II. Удельное количество электричества (А-ч/м3)
4уд“Лтк/(/’’вт). (,;'-''1
I |с \шк — удельный расход электрогенернроваипого коагулянта, г/м, оирс-
. чып экспериментальным путем па основе исследования ibhciimoitii
271
степени очистки сточных вод от дозы коагулянта; Л* — электрохимический экви-
валент металла анода г/(Л-ч).
12. Суммарная площадь анодов блока электрогенсрацнн ко-
агулянта (м2)
S,- /K/i, XmKQ/(Aa3fiTi,), (6.70)
где ia — плотность анодного тока: для колонных ЭФК «• “0.014-0,05 А/смг.
13. Толщина анодов с учетом их износа на 80 % » заданного
срока непрерывной эксплуатации (мм)
Йа ЛпрТзад 0,8, (6./I)
где Лпр — толщина слоя аноднорастворяемого металла с единицы поверхности
электрода в единицу времени; тэая — заданная продолжительность эксплуата-
ции анодов прн 8-часовом рабочем дне: т3«д=44-6 месяцев.
Толщина слоя Лпр рассчитывается по уравнению (мм)
Лпр Л»₽т'.т- <6-72>
где Лэ— линейный электрохимический эквивалент металла, мм/(Л-мпн)
Аэ А* • 10 (рме.$1,); рме — плотность металла, г/см’.
Значения линейного электрохимического эквивалента состав-
ляют: для алюминия — 0,0207, для железа — 0,0220 мм/(А-мин)
14. Количество растворимых электродов прн принятом квад-
ратном сечении камеры электрогенератора коагулянта с разме-
ром сторон, равным £>к,
и, = DK (Ла +1, + 2Pn + 1), (6.73)
где /3— межэтектродиое расстояние: /а—0,0154-0.02 м; Ра — толщина диэлек-
трической прокладки подбирается пз технологических соображений РО*»54-
4-10 мм.
15. Площадь одного электрода (м2)
S. п3. (6 74)
16. Высота электродов (м)
h3 = /, b,. (6.75)
где Ьл— ширина и.ыстип электро юв (1>к- 2РП), м.
Верхняя граница растворимых электродов располагается иа
2ч-3 DK ниже места ввода очищаемой воды в камеру смешения
с электрогеиернрованпым коагулянтом.
6.4.2. Расчет электрофлотокоагуляционной установки
горизонтального типа
Методика разработана для расчета ЭФКУ по очистке сточ-
ных вод мясокомбинатов, молокозаводов, рыбоперерабатываю-
щих предприятий [37] и может быть использована и для других
категорий промстоков при исходном содержании загрязнений, нс
превышающих следующих показателей: жира — I г/л, взвешен-
272
пых веществ — 2 г/л, БПКп—1,4 г/л. Схема установки показа-
на на рис. 4.59.
Технические параметры ЭФКУ: производительность — до
• >0 м3/ч; тип — трехкамериая; вид — прямоугольная; носледова
гельиость расположения рабочих камер: электроф нотация —
электрокоагуляция — электрофлотация; процесс обработки — не-
прерывный; высота слоя обрабатываемой воды — 0,84-0,9 м;
электрический ток — постоянный; плотность тока: в камере элек-
|рокоагуляции — 54-7,5 А/м2, в камерах электрофлотацпи —
1004-150 А/м2 (в пересчете па горизонтальную площадь камеры);
ширина аппарата при его производительности до 10 м3/ч -0,84-
4-1,0 м, до 20 м3/ч— 1,54-2,0 м и до 50 м3/ч — 3,04-3.5 м.
Расчет производится в последовательности, приведенной ниже.
1. Определяется продолжительность обработки сточных вод
в каждой камере и в целом по аппарату с учетом извлекаемых
загрязнений по формулам:
для жира и взвешенных веществ
Ск/С0 - [I + 1,76а, (Сл/С0)'-4 /,] 1 [1 + 0,3a2 (С, /С,.)0-33/,]"3-4 X
X [ 1 + 0,25а3 (Ск С6)-°-3 /3] 1; (6.76)
по снижению величины ХПК
Ск/Со = (I 4- 0,36а, (С,, С0)2 /,]-1. [ 1 + 0,34а2 (С,JCrf 6,]-з,4 х
> [1+0,ЗЗа3(Ск/С/)°-8/3]-1, (6.77)
где Со. Ск — содержание загрязнений в исходной сточной воде из аппарата,
мг/л; Cfv Ct2 — содержание загрязнений в жидкости после первой и вто-
рой камер, мг/л; G, /г, 6 — продолжительность обработки сточных вод соот-
ветственно в первой, второй и третьей камерах элктрофлотокоагулятора: а,,
«ь «з—коэффициенты, зависящие от плотности тока на электродах. pH. кон-
центрации хлоридов в сточных водах и высоты слоя обрабатываемой воды.
Величины этих коэффициентов для каждой камеры сле-
ющие:
ai = «i/«is 12.13- 1,13 (рН„ рН,)|(0,18|С1 ] |С!+0.S2) X
(1,455 - 0,455 (//„ //„.,)]. (6.78)
। тс it — плотность тока в первой камере этектрофлотацни в пересчете на го-
рнюнтальную площадь камеры, мА/см2; Ги-15 мА/см2; pH, = 7.0; [Cl-]oi=
0,8 г/л; //□,•=0,8 м.
Для дюралюминиевых электродов
a, ij/ij.*. (|, 79)
Для железных электродов
а» «2 «з,1. (6.80)
lie i2i,“2,5 мА/см2; «зл=3,5 мА/см2;
= «з «и| 1.0 — 0,6(рНо pH,)] (0.87-1-0.13 [С1 ] ICI-]U.,). (6.81)
Продолжительность процесса в отдельных камерах, по данным
|.37]. целесообразно назначать в соотношении /|: /2 : h= 1,0 : 0,6 :
1,1, а учитывая, что /, = «з=15 мА/см2, формулы (6.76) и (6.77)
|ожи<> упростить:
273
для жира и взвешенных веществ
СКСО (I + 0,044а/в)_5-4; (6.82)
по снижению величины ХПК
Ск Со (I + 0.019а/в)“5-4, (6.83)
где to — общая продолжительность пребывания сточных вод в рабочем объеме
ЭФКУ; а—прн высоте обрабатываемой жидкости 0.8—1.1 м для сточных вод
мясокомбинатов, величина pH которых находится в пределах 6,8—7,5 н кон-
центрация хлоридов составляет 0,8—1,5 r/л, зависит от плотности тока н ма-
териала электродов во второй камере электрокоагуляции: согласно формулам
(6.79) н (6.80) для дюралюминиевых электродов a=«2/«ji, для железных
электродов a «2/1'3, j.
При расчете зависимости между ХПК и БПК сточных вод мя-
сокох бинатов рекомендуется принимать: для исходной сточной
жидкости БПКп = 0,75 ХПК; для жидкости, очищенной электро-
флотокоагуляцией, БПКп=0,70 ХПК- Для других категорий сточ-
ных вод эти зависимости следует устанавливать эксперимен-
тально.
Ю. А. Феофановым рекомендуется [79] при расчете ЭФКУ для
локальной обработки сточных вод рыбоперерабатывающих пред-
приятий использовать полученные экспериментальные зависимо-
сти требуемой степени очистки от пр полжнтельностн процесса
электрофлотацнн и электрокоагуляции. Так, прн очистке тузлу-
ков (сточных вод от процессов посола рыбы) методом электро-
флотации следует пользоваться формулами:
Эв,в = 73 (1 - е-°-345'ф); (6.84)
Эж = 78 (1 — е—0,345/ф), (6.85)
где Эвэ». Эж — эффект очистки тузлуков по взвешенным веществам и жирам.
%; /ф— продолжительность процесса электрофлотацнн. мни.
При обработке тузлуков в электрокоагуляторах с последую-
щим отстаиванием или элсктрофлотацней эффекты очистки со-
ставляют:
Э„зв 95(1 -е-о-б'к): (6.86)
Эж = 94.3 (1 — ё~ °-55/к), (6.87)
где t„ — продолжительность электрокоагуляции, мни.
Оптимальная плотность тока принимается при этом для ка-
мер электрофлотацнн п электрокоагуляции 5 мА/см2, выход ме-
талла по току в камерах электрокоагуляции — 0.83.
Если продолжительность обработки стоков прн удалении
жира, взвешенных веществ и снижении величин ХПК и БПК
(в случае, если ХПК и БПК также лимитируются санитарными
органами) получается различная, то за расчетную принимается
наибольшая продолжительность обработки стоков.
274
2. Общий рабочий объем ЭФКУ (м3)
Vp = Qt п, (6.88)
в том числе для каждой камеры
V| = Qtt п; V2 = Qt2 п; 4 э Qt3 п. (6.89)
где п— количество ЭФКУ (принимается не менее двух)
3. Объем пловон камеры (м3)
V„ - (0,15 4- 0,2) (У, + 1'2). (6.90)
4. Рабочая высота аппарата (м)
H0 = hl + hi, (6.91)
где hi — высота слоя осветляемой жидкости, считая от нижней кромки элек-
тродного блока до слоя пены: Л1 0,8—1.1 м; Л2— высота слоя пены: Л2=
-0,054-0,1 м.
5. Полная высота аппарата (м)
Ни = H0 + h3 + ht. (6.92)
где Л3 — расстояние от кромки борта аппарата до слоя пены: Л2=0,24-0,3 м:
Л* — расстояние от нижней кромки электродов до дна аппарата: Л4 = 0,1 4-0.15 м.
6. Площадь зеркала воды в каждой камере (м2) F^Vi'IP, F2=V2H; F3=\’3tl. (6.93)
7. Длина каждой камеры (м) li- Г, В-, L2 - Г2 В; L3 F3 В. (6.94)
8. Общая длина ЭФКУ (м) Ft> 7-1 + L2 -|- L3 -|- L|, (6.95)
где l-i — длина камеры для сбора осветленной жидкости: 7.4=0,154-0,3 м.
Для более удобной компоновки ЭФКУ отношение ширины ап-
парата к его длине рекомендуется принимать 1 : (34-4), т. е.
Lo= (3-5-4) В, м.
9. Дотки для сбора осветленной жидкости и пены, а также
подводящие н отводящие коммуникации рассчитываются исходя
нз расхода сточной воды в ЭФ КУ-
10. Сила тока в первой камере (А)
7| = *lropf 1- (6 9( )
11. Число электродов в первой камере (шт.):
п. (В —2a-t-p“ -|-2Г,)/(2/, (6.97)
пк = (В-2а-Р*) (2/, • Р* • Р’), (6.98)
। те а — расстояние от стенки камеры до крайнего электрода: а=0.003 - 0.005 м;
толщина катодных электродов: Р, =0.0034-0.005 м; Р* —тол-
щина анодных электродов: Р* =0.024-0.04 м; /□—меж электрод ное простран-
ство: /э=0.0154-0,02 м.
Эти формулы справедливы в том случае, когда два крайних
электрода будут являться катодами, что уменьшает действие
275
конвективных токов на корпус аппарата, т. е. уменьшает его кор-
розию.
12. Размеры электродов в первой камере рекомендуется при-
нимать следующими: длину l\=L\—0,1, высоту й| =0,14-0,15 м.
Активная площадь анодного электрода, если пренебречь пло-
щадью торцов анода, составит (м2)
- 2Л,/,. (6.99)
Если площадь двух крайних электродов fKp=A|/i. м2, то истин-
ная плотность тока в первой камере (А/м2)
«I Л Иа (Па-1)1 (6 100)
13. Сила тока во второй камере (А)
/, = aTQ. (6 101)
Расход тока можно определить по формуле Дт = It IV или при-
мять по экспериментальным данным:
(,, А м! 5 15 25 35 50 75
Дт, А-ч м3 6,75 20,25 33,75 47,25 67,5 100,25
14. Количество электродов во второй камере составит
и., = (В-2а + /,) (Рэ< -J- /э), (6.102)
где Р,2 — толщина электродов: Р,2 0,0054-0,006 м.
15. Активная площадь одного электрода в камере электроко-
агуляции (м2)
/1 = 2/2й2. (6.103)
или
1г /2 («2Л2). (6 104)
где !г — длина электрода. (0,1—0.15), Л2—высота электрода,-м.
Общая активная площадь всех электродов во второй камере
SL., (1 2)/2п2 (б 105)
16. Расход металлических электродов (г/м3)
9ме «т"еА''г/2/(?, «•> 106)
Цля Fe3+—А''1' 0,6949, для Fe'+—.4?е 1.0424, для А13+-А,'е =
= 0.3356 г/(Л-ч).
Выход металла по току В}'е может быть определен по эмпи-
рическим формулам (мА/см2):
для дюралюминиевых электродов
BTU 0,015pH-|-0,C44i2 0,0325[С1“] 0,644; (6.107)
для железных электродов
В*’ O.OIfipH 4- 0,14i2 } 0.(7 [Cl ] I 0,674. (6.108)
276
Приведенные формулы справедливы при plI>3,0; [CI ]>
>0,3 г/л и 1'2 <7,5 мА/см2 (монополяриое соединение электродов).
Эти условия отвечают реальной характеристике сточных вод мя-
сокомбинатов и оптимальной плотности тока в коагуляционной
камере ЭФКУ.
17. Сила тока в третьей камере (А)
7з = <эР®. (G 109)
18. Активная площадь горизонтальных электродов в третьей
камере (м2)
/а3 = /кя = (^з—0,!)(в—0.1). (6 110)
19. Масса электродной системы (т):
для первой камеры
•"i = TiMa.pJ. тЛ«кр“. (6Н1)
гте уз—плотность катодного материала, т/м; yi— плотность графита: у( —
1,5 т/м;
для второй камеры
М* = УзАл2РЭ1, (6 112)
где уз—плотность металла для железа Уз=7,86, для дюралюминия у3—
=2,58 т/м;
для третьей камеры
где Р^—толщина анода: P9j =0,044-0.06 м; Р9кс — толщина катодной
сетки ( иля упрощения считаем, что сетка представляет собой сплошную ме-
таллическую пластину), м
20. Продолжительность работы электродной системы опреде-
тяется аналогично (6.40) и составляет (сут.):
в первой камере
xl = MI/a«a,/>;i/(24<7Tp/l'); (6.114)
во второй камере
Ъ= Чр". (24Д/2); (6.Ц5)
в третьей камере
тз ’1рТ!/аР"э>/(24<?гр/3). (6.116)
21. Количество образующейся при очистке воды пены зависит
от плотности тока и времени обработки. Оно может быть под-
считано по эмпирической формуле в процентах от расхода сточ-
ных вод:
П = (0,028«, +0,41«2 -p0,014i3)$33. (6.117)
Формула выведена для соотношения : /2: /3= 1,0:0,6 : 1,4
22. Количество флотоконцентрата можно определять с по-
мощью коэффициента гашения пены ак, равного отношению обь-
277
ема флотоконцептрата к объему пены. Коэффициент ап прн пол-
ном разрушении пены равен 0.3. В среднем в оптимальном ре-
жиме работы ЭФКУ количество пены равно 3,88 %, а флотокои-
пентрата — 1,16 % от расхода сточных вод.
Для принудительного разрушения пены целесообразно при-
нимать метод, согласно которому мешалка с двумя рядами ло-
паток на вертикальном валу вращается с частотой 30—40 мин-1.
При этом полное разр\ шенне пены наступает через 15—30 мин
160].
23. При оптимальном режиме работы ЭФКУ флотокопцептрат
характеризуется следующими показателями содержание жиров —
41.9 г/л, содержание взвешенных веществ — 80.1 г/л, влаж-
ность— 904-95%. зольность—154-18%, плотность—1,012 т/м3.
6.5. РАСЧЕТ АППАРАТОВ
ЭЛЕКТРОХИМИЧЕСКОЙ ДЕСТРУКЦИИ
Электроаппараты для деструкции загрязнений рекомендуется
применять прн очистке сточных вод, содержащих растворимые
химически стойкие органические соединения. Пример конструк-
тивного оформления аппаратов показан па рнс. 4 26.
В табл 6.6 представлены оптимальные параметры электро-
химической деструкции некоторых категорий сточных вод в элек
тролизере с нерастворимыми металлоокпсиымн титановыми ано-
дами (ОРТА, ОКТА ТДМА) и катодами Ст3 или нержавеющей
стали.
Расчет аппаратов может быть произведен различными спосо-
бами.
Первый способ, предложенный ЛИСИ и Госпннхлорпроектом.
базируется на предварительном выборе выпрямительного устрой
ства, в соответствии с выходными электротехническими показате-
лями которого производится весь расчет. Этот способ не учиты-
вает кинетические закономерности окисления органических загряз-
нений в электролизере и поэтому применяется для ориентировоч-
ных расчетов.
Ниже представчеп пример расчета электролизера для очистки
сточных вод краснльно отделочных фабрик по первому способу.
1. С целью обеспечения максимальной производительности
электролизера принимаем одни из мощных выпрямителей, вы-
пускаемых отечественной промышленностью, ВАКГ-12/6—3200
2. Опреде ieime производительности электролизера
а) Производительность по гипохлориту натрия
<?NaCIO ~3200 1,4 0,7 3100 г ч 3,1 кг ч, (6.118)
пе / — iiarpyiKa на электролизер по току: / = 3200 А; А,— электрохимиче-
ский эквивалент гипохлорита натрия: Д9 —1,386 г/(А-ч); Вт — выход по
току, устанавливаемый экспериментально: для рассматриваемой категории
сточных вот Вт -0.7.
278
Т а б л н ц а 6.6. Оптимальные параметры процесса электрохимической деструкции
Сточная вода Пока «ателн
/э, мин А дм- cNaCI> ’ л
Красильно-отделочных фабрик: общий сток 1-2 0,5—1,5 2
силыюзагрязненный поток (отрабо- 1,5-2,5 1,5-2,0 2
тайные красильные растворы) Флогошлам после электрокоагуляции- 30 2,0 20
флотации (16) Заводов бытовой химии: производства чернил и цветовой туши 0,5—1,5 1,5 2
производства хлорофоса 5—10 5,0 20
Заводов тонкого органического синтеза: производства иафтасолола 60 5,0 Г*
производства компоненты «Пурпур- 120 5,0 40
ная--6» Производства органических красителей: активного алого 120 2,0 35
прямого черного 2 С 120 2,0 150
Продолжение
Сточная иода Эффекты очистки, %
по обес- цвечива- нию xnriK по ПАВ по фе- нолу по хлорофосу
Красильно-отделочных фабрик общин сток 97 85 80
силыюзагрязненный поток (отрабо- 98,5 87 80 100
тайные красильные растворы) Флотошлам после электрокоагуляции- 99 85 85 -
флотации (16) Заводов бытовой химии: производства чернил и цветной tyiiiii Ди 100 90
производства хлорофоса 95 — — До 100
Заводов тонкого органического синтеза: производства иафтасолола 90 100 -
производства компоненты «Пурпур- 85 — -— —
ная-6» Производства органических красителей: активного алого 92
прямого черного 2 С 99 80 — — —
б) Производительность по активному хлору
Qa х. 71NaCIO MNaCIO 71-3,174,5 2,95 кг,ч, (6 119)
пс Мхасю — мол. масса гипохлорита натрия; 1 г-мол. гипохлорита натрия
соответствует 35-2 — 70 г. активного хлора.
в) Производительность электролизера по водороду можно оп-
ределить по (6 18) или по формуле (м3/ч)
22 43.4^7в’’ (1 ' 0.003667 )/(1000Мц), (6.120)
где Л” —электрохимический эквивалент водорода: А*1 —0,0376 г/(А-ч);
Втн— выход по току: В— 0,9-j-0.98; Мн — мол. масса водорота; 7» —
температура электролита, °C.
По величине определяется необходимая степень вентиля-
ции производственного помещения (см. п. 6.1.8).
3 Определение удельного расхода электроэнергии
а) Напряжение на электролизере UO6m определяется по фор-
муле (6.4). В результате проведенных исследований сумма (фа +
+<Рк + ца + т]к) па анодах ОРТА и катодах Ст.З прн Cn3ci в элект-
ролите до 5 г/л и i‘a = 2 Л/дм2 оценивается не более 4 В.
Падение напряжения в электролите в соответствии с форму-
лой (6.8) составит
t/р = ЧРэ/эОг = 0,02 1.1 10М.7-1,2= 1,85 В (6.121)
где <« — анодная плотность тока: по табл. 6 6 i»—0,02 А/см1; ра— удельное
сопротивление электролита, Ом-с.м; 1г—межэлектродное расстояние: по кон-
структивным соображениям /.—0,7 см; ог — коэффициент газоиаполнення:
пг= 1,2.
Так как удельная электропроводность общего стока кра-
сильно-отделочных фабрик после добавления 5 г/л NaCI, по
данным исследований ЛИСИ, составляет 0.8-10 24-10 3 Ом-1Х
Хсм-1, то
рэ = х*1 = (0,8-10-2)-1 = 1.1 102 Ом см. (6 122)
Напряжение на электродах составляет
Uобщ = 4 + 1,85 = 5,85 В. (6.123)
Величину падения напряжения в материалах анода и катода,
а также падение напряжения в контактах, подводящих ток
к электродам, в данном расчете не учитываем, считая сопротив-
ление метал тческнх проводников в электролизере ничтожно
малым.
б) Удельный расход электроэнергии:
на 1 м3 очищенного стока определяется по формуле (6.1);
на 1 кг активного хлора
U7*/ = 3,2-5,85 2.95 = 6,5 кВт ч кг. (6.124)
4 Определение общей рабочей поверхности анодов
S. = ///.= 3200 200 = 16 м». (6.125)
5. Количество анодных пластин
и» = S. (6 126)
где h, н /—соответственно высота и длина рабочей поверхности анода, м
(назначаются нз соотношения А.//< —1/2); т — число рабочих сторон анода
(принимается равным двум).
Выбирая по конструктивным соображениям йл = 0,8 м и /а =
= 1,2 м, определяем общее количество анодных пластин:
па 16'(0,8 1,2 -2) = 8 (6 127)
280
6. Число ячеек электролизера
N = 2ла 10. (fi 128)
7. Продолжительность пребывания сточных вод в электроли-
зере (ч)
ts = laN/v, (6.129)
где v — скорость движения сточных вод в межэлектродном пространстве: и—
-104-20 м/ч.
8. Тепловой расчет
а) Тепло, вытеляющееся в электролизере, по (6.23)
QT = 3,617/(£/об<ц—Ер) = 3,617-3200 (5,85 — 2,1) = 43 424 кДж/ч. (6.130)
где Ер — напряжение разложения хлорида натрия: прн /» —2А/дм* Ер—2,1 В.
б) Нагрев электролита в электролизере определяется по
(6.24). Ввиду небольшой концентрации NaCI в сточных водах
(до 5 г/л) значения теплоемкости сг и плотности электролита у„,
входящие в формулу (6.24), принимаются как для воды (ст=
= 10 ккал/(кг-°С), уэ=1,1*103 кг/м3).
9. По полученным параметрам электролизера и характеристи-
кам очищаемой жидкости производится расчет ожидаемого эф-
фекта очистки по ХПК и по ПАВ, а также определяется концент-
рация остаточного активного хлора [73]
'пчв 2.3 + 0,945Эхпк; С*пк = Со/(1 + 1.912Cg-3«—О-25/’-'*); (6.131)
С, X. =* 1 -7 Ю-V-02(рН)0-38^-07. (6 132)
Если при электролизе не достигается требуемая степень очи-
стки сточных вод, то необходимо предусмотреть реактор ката-
литической доочистки с гранулированной (размеры гранул 2—
5 мм) загрузкой из активированного пиролюзита илн других
специально подобранных катализаторов. Для этого может быть
использован комбинированный аппарат (см. рнс. 4 61) с совме-
щенной камерой доочистки или отдельно стоящий реактор.
Прн концентрации остаточного активного хлора 100—150 мг/л
высоту каталитической загрузки можно принимать 0,5—0,7 м,
при концентрации 150—300 мг/л—до 1,2 м. Максимальная вы-
сота загрузки принимается при наличии в обрабатываемом стоке
поверхностно-активных веществ. Если при расчете концентрации
остаточного активного хлора получена более 300 мг/л, то необ-
ходимо пересчитать продолжительность электрообработки, под-
ставив в выражение (6.132) концентрацию остаточного активного
хлора, равную 300 мг/л.
Площадь поверхности каталитического реактора определя-
ется но гидравлической нагрузке, равной 9—10 м3/ч. Расстояние
от верха электродов до поверхности загрузки в комбинированном
ппнарате должно быть не менее 0,4 м.
В юрой способ, разработанный ЛИСИ, используется для уже
щученных категорий сточных вод, когда известны механизм и
кнне1нческне закономерности процесса электрохимической дест-
Ю
Закя! М 2148
281
рукции органических загрязнений, т. е. когда выявлены зависи-
мости эффекта очистки от режима электролиза (Дт) и исход-
ного значения загрязнений (Со)
Рассмотрим пример расчета станции электрохимической очи-
стки сточных вод цеха крашения полушерстяных тканей кислот-
ными и прямыми красителями.
Исходные данные. Суточный расход стоков цеха крашения —
800 м3, требуемая степень очистки (Э) — 55 % по ХПК, среднее
значение ХПК очищаемой жидкости (Со) — 240 мг/л.
Для расчета используются кинетические зависимости про-
цесса очистки сточных вод, рассмотренные в п. 5.2.2.
1. По формуле (5.10) определим потребное количество элект-
ричества, необходимое для обеспечения требуемой степени очи-
стки сточных вод,
Дт ап-15,2схр(М?о) |С„(1 — а,,)) - 0.55 15.2ехр (8,035-10-’ 240)/[240(1 —
— 0,55)J - 0,532 А ч/л. (6 133)
2. Вычисляем требуемую силу тока при заданном часовом
расходе сточных вод 33,3 м3 (800/24):
I 0,532-33,3 1000- 17 715,6 А (6.134)
3. Для обеспечения требуемой силы тока выбираем шесть вы-
прямителей ВАКГ-12/6—3200 с /ВЫПр = 3200 А, работающих авто-
номно на один электролизер.
4. Определяем расход очищаемой сточной воды на один элект-
ролизер
Q' = /выпр (Дт 1000) = 3200/(0.532 1000) « 6 м»,ч. (6.135)
При этом общая производительность станции составит 6-6=
= 36 м3/ч>33,3 м3/ч, что обеспечивает очистку сточных вод цеха
крашения. Для нормальной эксплуатации очистных сооружений
следует зарезервировать еще одни аппарат и один выпрямитель.
5. Назначаем рабочую анодную плотность тока »а = 200 А/м2,
тогда общая поверхность анодов в одном аппарате составит
5а /выпр/«. 3200/200 16 м« (6.136)
6. Принимаем количество (л.,) анодных пластин в аппарате,
равное семи, тогда с учетом необходимости нх мопополярпого
соединения, т. е. при двусторонней работе, площадь аиода с каж-
дой стороны составит
/в = S./(nB 2) = 16/(7 2) = 1,15 м*. (6 137)
что не превышает стандартной площади титановых листов.
Далее аналогично первому способу определяется продолжи-
тельность пребывания сточных вод в электролизере (6.129), про-
изводится тепловой расчет (6.130) и рассчитывается необходи-
мая степень вентиляции в соответствии с п. 6.1.8 (в целях под-
бора соответствующего вентиляционного устройства). При этом
имеющиеся в крышке электролизера щелевые каналы для впуска
свежего воздуха рассчитываются на скорость движения воздуха
3—6 м/с.
282
Данная методика расчета может быть использована также и
для других классов органических красителей, имеющих анало-
гичное химическое строение и обладающих сродством к окисле-
нию активным хлором (например, для триазиповых, дисперсных
и других типов красителей).
Третий способ заключается в определении основных разме-
ров аппарата по оптимальной продолжительности (выявляется
экспериментально) процесса очистки сточных вод, а также в под-
боре выпрямительных устройств по вычисленным в соответствии
с указаниями п. 6 1 значениями (Л,бЩ, Р. Этот способ ана-
логичен способу расчета электрокоагуляторов и электрофлотато-
ров (см. п. 6.2 и 6.3).
6 6. РАСЧЕТ ЭЛЕКТРОДИАЛИЗАТОРОВ*
При проведении технологического расчета электродиализных
установок исходными данными являются количество опресняе-
мой воды, ее солесодержанне и солесодержанне рассола, при этом
предполагается, что будут заранее выбраны конструкции серийно
выпускаемых аппаратов (т. е. известны площади мембран, их
марки, тип корпусных рамок, аппаратов и т. п.).
В распоряжении проектировщика должны быть также данные
о выходе по току, установленные на основании результатов пред-
варительных исследований па малогабаритном лабораторном
электродиализаторе с натурной водой
1. Выход по току для нейтральных растворов может быть оп-
ределен по формуле (4 50) или с достаточной для практических
расчетов точностью—из уравнения удельного расхода электри-
ческой энергии на электродиализ:
Вт 2».8(С„-Скиад) (лГ,л) 10«, (6.138)
| ле 26.8 — количество ампер часов, необходимых для переноса 1 г-экв соли;
Со и С„ - солесодержанне соответственно исходной и обессоленной воды;
(Ал— полное напряжение на электродах аппарата; «—число парных камер
в аппарате. 1Г»Л удельный расход электроэнергии
При обработке растворов с низким или высоким pH выход
по току определяется по упрощенной формуле
Вт •= 1 — (п“~ + й;+) — FD/(,, (6 139)
~К-- I
1 *е Л, И П/—числа переноса попов в соответствующих мембранах
2. Определяемыми величинами для прямоточных аппаратов
являются требуемое число ступеней опреснения, количество па
рлллельных аппаратов в каждой ступени и число рабочих ячеек,
|ребуемое напряжение и сила постоянного тока.
При расчете циркуляционных установок определяется число
параллельных аппаратов и число рабочих ячеек в них, папря-<
жеине на электродах, сила постоянного тока в разные циклы оп
• Материал подготовлен совместно с проф В. Д. Дмитриевым
10» 283
реснепия и величины циркуляционных расходов днлюата и рас-
сола.
2.1. Число ячеек определяется по формуле
ляч = 26.Ж?АС(/М Вт«,). (6 140)
где АС— снижение концентрации солей, мг-экв/л: для циркуляционных уста-
новок АС—Со—С„, для каждой ступени прямоточных установок ДС—С.х—
—С,ы«; /и — рабочая площадь (нетто) мембраны, м2.
Для любой ступени прямоточной установки С>Ых=пС„, где
а — коэффициент снижения концентрации дилюата:
!g(1/a) = 4.5/.B/(K'/>n). (6 141)
где L — длина пути, проходимого шлюатом в камере ЭД аппарата, м; К'—
коэффициент, характеризующий деполяризационные свойства сепараторов: для
где L — длина пути, проходимого дилюатом в камере ЭД аппарата м; К'—
— 2-10*, для капроновых плетеных сепараторов К'“3-10*; Р„ — толщина
корпусной рамки, равная расстоянию между мембранами в камере электродиа-
лизного аппарата, м.
Выход по току Вт в зависимости от исходного солесодержа-
пия Со может быть ориентировочно принят:
С01 г-экв/л 0,01 0,02 0,05 0,100 0,200 0.5
Вт 0,89 0,88 0.86 0,840 0,825 0.8
2.2. Расчетная плотность тока «Р должна быть меньше пре-
дельного значения плотности тока lm> (ip<inp) и равняться опти-
мальному значению i”nT, которая определяется технико-экономи-
ческим расчетом.
Для прямоточных и циркуляционных установок производи-
тельностью до 500 м®/ сут с аппаратами прокладочного типа при
рабочей площади мембран до 0,5 м2 значения /£пт принимаются
по данным табл. 6.7,
Предельное значение плотности тока <ПР может быть опреде-
лено по формуле [101]
inp = FD C„Ml(nf- - пг) RemPe,/3. (6 142)
где Ь, т и /экв — соответственно константы и эквивалентное расстояние между
мембранами, значения которых приведены в табл. 6.8; и щ — числа пере-
носа С1~-ноиов в мембране и растворе при исходной концентрация Со.
При расчете критерия Re в формуле (6.142) подставляется ве-
личина Пэки — эквивалентная скорость в камере с турбулизатором,
равная произведению скорости в свободной камере на коэф-
фициент ее стеснения ес- На мембранах МА-40 и МК-40 выяв-
лены две характерные области движения воды в камерах с се-
параторами. Первая из них прн Re<ReK₽ представляет собой об-
ласти ламинарного, параллельно-струйного режима движения,
прн котором для всех сепараторов т = 0,5. Вторая область при
Re>ReKP представляет собой область режима переходного к тур-
булентному. При этом режиме т возрастает от 0,5 до 1,0.
284
Г а б л и ц а 6 7 Оптимальные плотности тока
Солесодсрж вине исходно!) воды, Св, г/л Стоимость электроэнергии, коп./кВт-ч Стоимость мембран, руб/м Расчетная оптимальная плотность ток». т”пт. А/м’
для циркуля- ционной ЭДУ для 1 ступени пр ямоточноП многоступенча- той ЭДУ
15 1 6 12 90 130 250 360
2 6 12 70 90 230 360
7,5 1 6 12 80 Ю0 2<Х) 280
3 6 12 60 80 170 230
2,5 1 6 12 60 80 110 150
3 6 12 50 60 80 110
Примечание. Значения
(1ШХ). изготовленными методом
оров и* капрона /°пт следует
<рПТ даны для камер с
просечки — вытяжки; при
уменьшать иа 10 •».
сепараторами из винипласта
применении плетеных сспара-
Если установлено, что величина <п><пр, то общее число камер
следует разделить на несколько параллельно работающих аппа-
рлтов, в каждом из которых будет п'яч камер, т. е. п'яч =n„4/N,
г те N — число аппаратов.
Расчетные плотности тока по ступеням прямоточной уста-
новки определяются по формуле
h'it il'ii «Л — = 1/а,
(6 143)
• <i, it. it, it и т. д.— расчетные оптимальные плотности тока на аппарате
нерпой, второй, третьей и других ступеней; а — коэффициент снижения кон-
центрации дилюата [см. (6141)1
2.3. Число ячеек ляч в каждом из параллельно работающих
ишаратов не должно превышать 200—250 штук. Тогда количе-
ство параллельно работающих аппаратов N в циркуляцпоиио-
285
Таблица 6.8. Рабочие параметры электродиализных ванн, зависящие от
конструкции камеры и типа сепараторов турбулизаторов, но Л. Д. Ушакову (ВНИИ
ВОДГЕО1
Конструкция камеры т ь Расстоя- ние между мембра- нами, мм ^ЭКВ* см 1 Кекр Крити- ческая ско- рость. икр* см/с Потеря давления при икр на I м длины камеры
Для камер лабиринтного типа без сепаратора Для камер прокладочно- го типа: 0,54 30 2 1 0,385 0,196 2320 2320 69 135 26 196
с сепаратором из пер- форированного вини- пласта (гофры попе- рек потока) 0.43 3 2 0.203 92 5.2 2,63
то же (гофры вдоль потока) 0.5 0,45 2 0,203 220 12,4 24.6
с сепаратором, изго- товленным методом просечки — вытяжки 0,67 2.5 1 0,105 26,5 2,88 78
порционной установке и в каждой ступени прямоточной уста-
новки определится как
N = n„4/n'„4. (6.144)
2.4. Напряжение Ujn па электродах электродиализных аппа-
ратов
^эл = <4 + «яч’Гм + «р^я/яч • (6-145)
где (7, — падение напряжения на электро ной системе, равное 3—5 В; гяч —
омическое сопротивление ячейки. Ом; <гм — мембранный потенциал ячейки
с учетом концентрационной поляризации (В).
Чм 4>n + iflg(Cp/Cn). (6 146)
здесь Ср и Ся — соответственно расчетная концентрация рассола и днлюата
в аппарате, мг-экв/л: Ср—(3-5-4) Со, а Ся определяется из выражений
для любой ступени прямоточной многоступенчатой установки
Сд= (Свх-Свых) |2.31g(CBX'CBUX)I; (6.147)
для циркуляционной установки
Сд (Со - Ск) [2,31g (Со С„)|; (6 148,
Ч * it if — коэффициенты, значения которых принимаются в зависимости от
температуры обрабатываемого раствора:
Т,‘С . 10 15 18 20 25 30
ф® 0,087 0,089 0,090 0,091 0,093 0,095
♦ 0,081 0,083 0,084 0,085 0,085 0,088
286
Омическое сопротивление ячейки гяч определяется по формуле
(Ом)
Гяч = 1 /м Хд + Prfin + 2|>ы). (6.149)
где рм — среднее удельное поверхностное сопротивление мембран; бл — коэф-
фициент увеличения омического сопротивления камеры сепаратором или лаби-
ринтом; Хя н Хр — удельные электропроводности днлюата и рассола.
Значения 6Л в аппаратах «прокладочной» типа при рц=0,1 см
принимаются в зависимости от выбранного материала: при ис-
пользовании сепараторов из винипласта, изготовленных методом
просечки — вытяжки, рм=1,54; при использовании плетеных сепа-
раторов из капрона рм=1,48. Значения рм для гетерогенных мем-
бран МК-40 и МА-40 равны 20-?-30 Ом-см2.
Величина удельной электропроводности исходной воды (при
7'=18 °C) определяется при проведении химического анализа.
При отсутствии таких данных удельная электропроводность мо-
жет быть определена по эмпирической формуле
Х„ С₽ 8300. (6.150)
|дс Х|» — удельная электропроводность прн Г—18 “С, Ом-’-см-1; С—кон-
центрация солей днлюата или рассола, мг-экв/л; р — коэффициент, зависящий
от отношения содержания сульфата к общему количеству анионов ХДц; значе-
ние р может быть определено по следующим данным:
SO2-/ZAii 0,2 0,2—0,4 0.4 -0,6 0,6—0,8 0,8—1,0
₽ 0,94—0,92 0,92 -0,89 0,89—0,87 0,87—0,84 0,84 -0,81
Удельная электропроводность при температуре, отличной от
температуры Т= 18 °C, определяется по формуле
Хг Х„|1 +0,02(7+ 18)]. (6.151)
При этом необходимо учитывать, что дплюат и рассол при
однократном прохождении через электродиализатор нагреваются
па 0,5-s- 1,0 °C за счет джоулева тепла.
2.5. После определения напряжения и сопротивления в аппа-
рат фактическая плотность тока в начале и в конце цикла уточ-
няется по закону Ома:
*э = [^э — (фэ + пячФм)1/(/мпяч'’яч)- (6 152)
<дс ф—электродный потенциал, В.
Прн уточнении плотности тока может оказаться, что опа да-
лека от оптимальной, так как подбор количества аппаратов N
оказался недостаточным. Если поверочный расчет показал, что
плотность тока, вычисленная по формуле (6.142) для начала и
конца цикла и в среднем за цикл, окажется значительно больше
ошпмалыюй, то принятое количество аппаратов надо увеличить и
повторно произвести расчет.
287
3. Производительность насосов, подающих в камеры опресняе-
мую воду н рассол, должна обеспечивать денолялпзующую ско-
рость движения воды в аппарате не менее (м ’/ч) [101];
QH = 3.6lO-4u;PnZMftnn>. (6.153)
где — деполяризующая скорость движения воды, в зависимости от типа
турбулизирующсн сетки принимаемая 2,94-9,3 м/с; ZM— расстояние между
мембранами, см; Ьш — ширина прохода для воды в камере, см.
Величина Ь„ равна расстоянию между внутренними краями
рамки в направлении, перпендикулярном направлению движения
воды, а в лабиринтовой камере — расстоянию между направля-
щими стенками лабиринта.
Критическая скорость потока определяется по формуле (см/с)
‘5«рп=*(‘Г),/т/(с,/т/*в)- <6154>
где С — средняя концентрация солей в опресняемой воде в камере (для цир-
куляционных установок в конце цикла), г-экв/л.
Для гладких камер лабиринтового типа /экв = 4 f/x, где /их —
площадь сечения и смоченный периметр камеры. Для камер с се-
параторами 1эКя определить сложно, поэтому рекомендуется ис-
пользовать данные, приведенные в табл. 6.8.
4. Проверка работы электродиализатора па возможность об-
разования отложений малорастворимых соединений производится
по формулам (5.51)—(4.53) с подстановкой концентраций соот-
ветствующих ионов.
5. Предельно достижимая концентрация рассола рассчитыва-
ется по уравнению [21]
CnK = UBT + LK,CMF)/(FA). (6.155)
где К» —Рк/6к + ₽«/6а; ₽ — коэффициент диффузионного переноса соли через
катноио- н аннонообмениые мембраны, имеющие толщину би и ба; Сл — кон-
центрация днлюата; A~l Wp+fx К.; <оР— количество рассола, вытекающего из
как еры концентрирования за время прохождения 1 Кл электричества.
6. Расход электроэнергии на опреснение воды складывается
из расхода электроэнергии на собственно электродиализ и
на прокачивание через ЭДА опресняемой воды, рассола и воды
для промывки катодного и анодного пространства № (кВт-ч)
[72]
^^=1^^,4/1000, (6.156)
где /ы. потто — площадь одной мембраны, м’; п—число пакетов в аппарате.
Для двух одинаковых насосов с подачей 20 % воды на про-
мывку катодной и анодной камер расход электроэнергии может
быть вычислен но формуле (кВт-ч)
U7"'p = 21,2<?’H/(102n,ri11I), (6.1Б7)
где QH— расход воды, подаваемой одним насосом, л/с; Н — напор, развивае-
мый насосом, м; t). и 1]я1 — КПД соответственно электродвигателя- »],—
«0.954-0,97 и насоса 1)я1»=0,7+0,8.
288
Расход элек1роэнергнп па опреснение I м3 воды (кВт-ч/м3)
= («^т + «^мР)/0. <6 158)
6.7. РАСЧЕТ АППАРАТОВ ЭЛЕКТРОХИМИЧЕСКОГО
КОРРЕКТИРОВАНИЯ pH И Eh
Данная методика может быть применена при расчете электро-
реакторов, используемых для изменения величин pH и Eh раство
ров, а также электроокислителей (восстановителей), в которых
превращение примесей происходит за счет накопления раствори-
мых продуктов электродных реакций в объеме электролита.
Исходными параметрами для расчета аппаратов являются рас-
ход воды (Q, м3/ч) и величина изменения pH (кислотность и
щелочность) или Eh обрабатываемой воды. Расчетная схема ап-
парата приведена на рис. 6.8.
1. Соотношение расходов в общей камере Q,, и вспомогатечь-
ной Qn назначается по следующим соображениям:
а) при сбросе воды нз вспомогательных камер в канализацию
принимается минимальный расход во вспомогательных камерах
Q., но не менее 0,10—0,15 Qp;
б) при использовании воды из вспомогательных камер в про-
цессах очистки или на технологические нужды QB принимается
по расчету необходимого количества воды для использования.
В первом случае расчетный расход в рабочей камере (м3/ч)
Ср = «к<2о- (6.159)
где ак — отношение расхода поды в рабочей камере к общему расхочу почы
п случае протекания по вспомогательной камере минимального расхода почы
а« 0.85-?-0,90.
Расход воды во вспомогательной камере (м3/ч)
(?„= (1-ак)(?.. (6 160)
Во втором случае коэффициент а1( устанавливается исходя из
требуемого расхода воды во вспомогательной камере:
ак=(<?о —<?.)<?« (6 161)
2. Основной расчетной величиной для требуемого изменения
|>П и Eh в рабочей камере является расход тока, определяемый
экспериментально в каждом конкретном случае в связи со слож
постыл ионного состава природных и сточных вод Орнешпро
полно эта величина может быть определена по формуле (Кл/л)
ДР = (Кк-Ки) ЮО/dy = (Щк - Щн) 100/d“. (6.162)
где Кк(Щк) и Кн(Шн) — требуемая соответственно конечная и начальная кн
гчотность (щелочность) воды; d^ и d"' удельное изменение соответственно
кислотности и щелочности на 100 Кт/л расхода тока, мг-экв/л (эти величины
принимаются согласно табл 69)
280
Рис. 6.8. Расчетная схема электрореактора диафрагменного типа для коррек-
тирования pH и Eh
Расход тока на обработку воды во вспомогательной камере
(Кл/л)
д;=др<?р/ов. (6.163)
Эта величина может быть использована для оценки измене-
ния величины кислотности (щелочности) и Eh прн предполагае-
мом дальнейшем использовании воды из вспомогательных камер
в технологической схеме очистки воды (табл. 6.10).
Для работы электрокорректора с максимальным выходом по
току желательно, чтобы величины изменения pH, кислотности,
Таблица 6.9 Рекомендуемые величины изменения кислотности (щелочности)
и pH воды
Параметры < бработки Солесодержание, мг/л
50 300 500 2000 700(1
Изменение кислот- ности (щелочно- сти) на 1<Х> Кл/л расхода тока, мг-экв/л 0 18 0.29 П.37 0.55 0.60 0.73 0.88 0.95 0.95 1,0
Пре тельное изме- нение кислотно- сти (щелочно- сти). мг-экв/л 1.8 2.0' 3.6 4.0 5.8 6.5- 13 15 30 35
Диапазон измене- ния pH 4 7 9.7 3.9 10.5 3.6 10 8- 3.0 11.2 2.5 11.7
290
Таблица 6.10. Рекомендуемые величины изменения Eh воды
Параметр обработки Направление изменения Eh
повышение снижение повышение со снижением pH снижение С повышением pH
Предельное изменение Eh, В Изменение Eh на 100 Кл/л расхода тока В + (0,8+1.1) 0,09 +0.13 — (0,1+0,2) - (0,5+0.8) 0,08+0,09 0.08+1.10 + (0,3+0,6) + (1.14-1.3) 0,064 0,09 0,10+0,14 — (0.2+0,4) — (0.6+0.8) 0,10+0,15 0,12+0,16
Примечание. В числителе приведены данные изменения Eh в отсутствии хлори-
дов прн повышении Eh. в знаменателе — при разряде хлоридов. Данные, приведенные
в знаменателе со снижением Eh. получены при размещении растворимого железного
электрода в анодной камере.
щелочности и Eh не превышали предельных значений, приведен-
ных в табл. 6.9 н 6.10.
3. Активная поверхность электродов (катодная или анодная,
м')
•$, = Д?<?р/(3,6|э). (6.164)
Пчотность тока на электродах определяется в зависимости от
ионного состава воды н напряжения электролиза или может быть
принята ориентировочно по следующим экспериментальным
данным:
(Солесодержание, мг/л .... 50 300 500 2000 7000
Плотность тока, А/дм1 .... 10—15 20 30 30 50 80—100 100—200
4. Рабочая поверхность электродов
SP = S3(1—6П). (6 165)
те Лп—степень перфорации электродов: 6П—0,2+0,4.
5. Требуемое количество катодов (п.,к) и анодов (лвп):
при использовании перфорированных анодов
<- (6 '66)
при использовании сплошных анодов
2nJ = — Sp/(b/(), (6.167)
• де Ья— рабочая ширина электродов, м: Ь.=0,7-+0,9 м; h, — рабочая высота
исктродов, равная высоте слоя воды в электрореакторе: й.=0,б4-0,8 м.
6. Габаритная высота электродов (м)
йг ' й3 + Ай. (6 168)
291
7. Габаритная ширина электродов (м)
br b, -f. \Ь, (б 1G9)
где Л/i н ЛЬ — соответственно высота н ширина нерабочей части электродов;
Ah-0.08+0,1 м, ЛЬ = 0,01+0,03 м.
С целью упрощения изготовления электродов и экономии ме-
талла величины Лг и Ьг следует назначать равными (кратными)
размерам стандартных листов материала, идущих на изготовле-
ние электродов при условии соблюдения соотношения br'.hr=l:
: (0,7-+0,8). При использовании стандартных графитовых плит
и качестве анодов габаритную высоту электродов целесообразно
принимать равной высоте графитовых плит.
8. Габаритная площадь электрода (м2)
)г=Мг- (6.170)
9. Количество используемых диафрагм
пд = <. (6171)
Габаритные размеры диафрагм принимаются равными габа-
ритным размерам катодов.
10. Количество электродных камер в электрокорректоре
1¥э = л£. (6.172)
11. Количество рабочих секций
NP= NKIN„ (6.173)
где N, — количество электродных камер в одной секции: Л/,=8-+12.
12. Количество резервных секций N3 может быть принято
в зависимости от количества рабочих секций Np прн надеж-
ности системы не менее 0,82—0,85:
Np, шт. . . Менее 7 7- 12 13—14 Более 16
Л'3, шт. . . . . 1 2 3 4
13. Общее количество секций
N°c=Np + N3. (6.174)
14. Расход воды на одну рабочую секцию (м /ч)
Qc = Qn'Np. (6.175)
15. Гидравлическая нагрузка на электродную камеру электро-
корректора (м3/ч)
qc=2QaN3. (6.176)
16. Удельный расход воды, отбираемый из рабочей электрод-
ной камеры (м3/ч),
<6,77>
292
Таблица 611 Рекомендуемые предельные значения гидравлической нагрузки
и удельных расходов воды в электрокорректоре 1см. рис. 6.8|
Режим работы диафрагмы Схема движения воды Гидравлическая иа|рузка ес. ы* Расход воды, м7ч
*с
Погружной А 0,8—1,2 0,4—0,6 0,4—0.6
Фильтрующий Б 0,6-0,9 0,4—0,6 0,2—0,3
17. Удельный расход воды, отбираемый из вспомогательной
камеры (мл/ч),
?с (6.178)
Если расчетные величины гидравлической нагрузки и удель-
ных расходов воды превышают их предельные значения (табл.
6.11), то увеличивается количество рабочих секций элекгрокоррек-
чора с соответствующим перерасчетом площади и количества
электродов.
Во вспомогательных камерах могут устраиваться электроды
в виде одной сплошной (рис. 6.8, и) нли двух перфорированных
пластин (рис. 6.8, б).
18. Длина одной камеры (м):
при использовании перфорированных электродов во вспомо-
гательной камере
Ьг =₽? + ₽: + (6179)
прн использовании сплошных электродов
£rP = ^ + ^ + 's; (6.180)
длина вспомогательной камеры
Е?=Р“+РЯ Р£ + 2/:, (6.181)
где Рр, Pl, Рл — толщина электрода (катода или анода) соответственно
в рабочей и вспомогательной камерах и толщина диафрагмы: Рр, Р“ — 14-
.1 мм; Рх=0,54-1,0 мм; 1в — расстояние между соседними перфорированными
катодами (анодами) в электродной камере: />=204-40 мм; /“—расстояние
между сплошными анодом (катодом) и диафрагмой: /” — 154-30 мм.
19. Длина одной секции электрокорректора (м):
при использовании перфорированных электродов во вспомога-
е 1ыюй камере
Z-c LrNp-, (6162)
при использовании сплошных анодов
(6 183)
293
20. Габаритная длина электрокорректора (м)
L" Л-р£с + Х;Д|. (6.184)
21. Габаритная ширина электрокорректора (м)
В, Ьг + ЕД«. (6.185)
22. Габаритная высота электрокорректора (м)
II, й. 4- Е Ai. О'* 1«6)
п
где Е Д* — суммарная толщина стенок, перегородок между секциями, изоля-
ционного покрытия, зазоров и пр. (ориентировочно равна 3—5 % от соответ-
ствующих габаритных размеров электрокорректора).
23. Величина тока, необходимая для обработки воды (А),
/р isS3. (•> 1«7)
24 Напряжение электролиза определяется на основании экс-
периментально полученных вотьтамперных характеристик, по-
строенных для обрабатываемых природных и сточных вод в за-
висимости от конструкции электродного блока, которую предпо
латается использовать в электрокорректоре.
В зависимости от величины /р и иэ выбирают тип выпрямитель-
ных устройств, количество которых принимается не менее двух
(рабочих).
В случае поступления па электрокорректор воды, электропро-
водность которой значительно изменяется, необходимо произве-
сти проверку выпрямительного устройства па величину макси-
мального тока.
ГЛАВА 7
ОСНОВНЫЕ ПРОБЛЕМЫ СОВРЕМЕННОЙ ТЕХНОЛОГИИ,
ПЕРСПЕКТИВЫ РАЗВИТИЯ И СОВЕРШЕНСТВОВАНИЯ
ЭЛЕКТРООБРАБОТКИ ПРИРОДНЫХ И СТОЧНЫХ ВОД
Разработка и внедрение электрохимических методов является
прогрессивным направлением в технологии водоподготовки и
очистки сточных вод, которые находят широкое применение как
альтернативные, когда традиционные способы механической, био-
химической и физико-химической обработки воды оказываются
недостаточно эффективными пли не могут быть использованы
291
из-за дефицита производственных площадей, сложности доставки
и использования реагентов, либо по другим причинам. Эти методы
позволяют корректировать физико-химические свойства обраба
тываемой воды, концентрировать и извлекать из нее ценные хи
мические продукты и металлы, обеспечивают глубокую минера-
лизацию органических загрязнений, обладают высоким бактери
цндиым эффектом, значительно упрощают технологические схемы
очистки. Во многих случаях электрохимические методы являются
экологически чистыми, исключающими «вторичное» загрязнение
воды анионными и катионными остатками, характерными для
рсагешных методов [38].
Систематические исследования в области теории электрохими-
ческих процессов и электроповерхностных явлений оказались ис-
ключительно плодотворными. Состояние этих исследований дает
основание для определенного оптимизма и обеспечивает новый уро-
вень развития таких методов применительно к технологии обра-
ботки воды.
К настоящему времени достигнуты значительные успехи
в разработке теоретических основ различных способов электрохи-
мической водоочистки и достижения теории все чаще находят
практическое приложение. Вместе с тем развитие науки ставит
перед исследователями все новые и новые задачи. Некоторые из
них можно сформулировать, анализируя материал, изложенный
в данной книге
Так, следует отметить, что современные теоретические обосно-
вания эффективности различных приемов электрообработкн за-
। рязпеппых водных систем носят зачастую феноменологический
и нолуэмппрический характер без соответствующего изучения и
анализа механизмов и кинетических закономерностей многооб
разных процессов, протекающих как иа границе «электрод—рас-
тор», так и в объеме обрабатываемой жидкости под влиянием
наложенного электрического поля и продуктов электродных реак-
ций, взаимодействующих с примесями воды. Все это затрудняет
досюверпую интерпретацию полученных результатов.
Вместе с тем уже накоплен значительный экспериментальный
материал, объяснение которого требует строгого учета всех воз-
можных кинетических стадии электрохимического процесса: элек-
।родных реакций, образования новой твердой или газообразной
ф.иы, химических многостадийных и параллельных процессов
превращения примесей воды, явлений пассивности электродных
ма1сриалов и адсорбции на них органических соединений. Такой
подход необходим для выявления замедленной, лимитирующей
стадии исследуемого электрохимического процесса и, соответ
с । ценно, для более детального описания и анализа дополни тель-
ных возможностей создания новых и изыскания путей иптепсп
фикации известных технологических приемов электрообработки
iai рязпеппых жидкостей. Поэтому количественное исследование
MiiieiiPiecKiix закономерностей различных электрохимических про
иессов водоочистки представляет собой одну нз основных про
20Г.
блем современной технологии элекгрообработки природных и сточ-
ных вод.
Для дальнейшей оценки представлений о возможностях тех-
нологии электрохимической водоочистки на первый план выдви-
гается задача развития теории фазово-дисперсного и физико-хи-
мического превращения примесей воды под действием электри
ческого тока и продуктов электролиза. Приведенные в книге
примеры практического применения целенаправленного электро-
корректирования pH и Eh обрабатываемых растворов позволяют
наметить пути дальнейшего использования электрохимической тех-
нологии для очистки воды от растворенных н коллоидных загряз-
нений. Регулирование этих показателей обеспечивает изменение
прочности комплексных соединений, окислительно-восстанови-
тельного потенциала, направления и скорости химических реак-
ций, осуществление требуемых химических взаимодействий раз-
личных соединений. Это способствует постановке новых задач при
теоретическом исследовании данной технологии, и в первую оче-
редь по разработке диафрагменного электролиза с применением
инертных мембран.
Известно, что для создания любого технологического процесса
и, следовательно, его аппаратурного оформления необходимо
иметь математическое описание механизма явлений, проис-
ходящих в проектируемом объекте, установить баланс взаимодей-
ствия внутренних и внешних факторов системы и правильно вы-
брать для обоснования рациональности эксплуатации оборудова-
ния и эффективности ведения процесса критерий оптимальности.
Поэтому, изучая экспериментальные данные по выявлению ме-
ханизма и определению скорости процесса электрообработкн, сле-
дует оценивать совместное влияние факторов на эффективность
очистки загрязненных жидкостей во взаимосвязи технологических
режимов электролиза с наблюдаемыми кинетическими закономер-
ностями. Постановка и корректное решение задач математиче-
ского описания механизма явлений, происходящих в электрореак-
торах, позволяет оптимизировать конструктивные параметры
аппаратов и оборудования и разработать более совершенные мето-
дики инженерного расчета и управления технологическими
системами электрохимической водоочистки с применением совре-
менной вычислительной техники.
Важным технологическим аспектом на пути широкого исполь-
зования электрохимических методов для целей водоочистки явля-
ется проблема поиска новых дешевых и доступных электродных
материалов, удовлетворяющих одновременно требованиям высо-
кой активности, селективности, химической устойчивости н эко-
номии. В этой связи представляют большой практический интерес
исследования института электрохимии АН СССР, Днепропетров-
ского химико-технологического института, Госниихлорпроекта и
других организаций по разработке малоизнашиваюшнхся пла
стннчатых. насыпных материалов из различных зернистых мате
риалов (суспензионных), а также пористых, волокнистых и псев-
296
доожнжепных электродов, позволяющих существенным образом
интенсифицировать электродные процессы Так, ианример, прн
менеиие псевдоожиженных электродов — взвесей частиц элек-
тродного материала в растворе, передающих при контакте с то-
коотводящим электродом свой заряд, обеспечивает протека-
ние электродных процессов на границе каждой из частиц
с раствором, что снижает диффузионные ограничения и позво-
ляет сосредоточить в малом объеме большую поверхность для
протекания реакции.
Заслуживает внимания применение пористых матричных си-
стем в качестве элементов электрохимических аппаратов, созда-
ющих во многих случаях предпосылки для реализации новых тех-
нологических решений, существенно более рациональных по срав-
нению с традиционными погружными системами [117]. Наряду
с пористыми электродами в технологии электроводоочистки могут
найти широкое применение диафрагмы, которые препятствуют
смешению электродных продуктов и позволяют значительно
уменьшить межэлектродное расстояние. Комбинированные много-
слойные матричные системы, в которых пористые электроды и
диафрагмы находятся в тесном контакте, можно использовать,
например, для отделения электролитических газов от обрабаты-
ваемого раствора, находящегося в межэлектродном пространстве,
что обеспечивает целенаправленное превращение примесей воды
путем изменения их физико-химических свойств за счет электро-
корректнрования величин pH и Eh.
В пористых матрицах, где электрохимические и химические
процессы происходят внутри пор, благодаря развитой внутренней
поверхности, которая в ряде случаев достигает сотен м2-см~3
объема, можно получить высокие габаритные скорости процесса.
Селективность пористых мембран может быть использована для
рационального конструктивного решения вопроса подачи электро-
геиерированных реагентов или отвода н разделения продуктов
электродных реакций.
С целью экономии листового металла в процессах электроли-
тической коагуляции перспективным следует считать разработку
стружечных диафрагменных электрореакторов, а также гальва-
пореакторов, в которых используются отходы металлорежущих
1ехнологических операций.
Прн выборе электродных материалов для осуществления при-
нятого электрохимического процесса очистки загрязненных жид-
костей необходимо обращать особое внимание на важность изу-
чения поведения электродов в разбавленных водных растворах,
какими являются большинство природных и сточных вод. При ис-
следовании этого вопроса возникает задача разработки более со-
першепных методик снятия поляризационных кривых в динами-
ческих условиях протекания процесса с изменяющимися физико-
химическими свойствами растворов, которые оказывают влияние
на изменение констант диффузии и скоростей электродных ре
акций.
207
Одним из прогрессивных направлений технологии электрохи-
мической очистки сточных вод является глубокая деструкция
сложных органических загрязнений до простейших нетоксичных
или малотоксичных соединений. Имеющийся опыт эксплуатации
промышленных сооружений по электрохимической деструкции ор
ганических красителей, ПАВ, фенолов и других загрязнений под-
тверждает высокую эффективность данной технологии. Однако
широкое внедрение этого способа очистки сдерживается в насто-
ящее время отсутствием серийного выпуска малоизнашивающихся
стойких анодов.
При применении процесса электрохимической деструкции для
очистки различных категорий промышленных сточных вод необ-
ходимо детальное изучение механизма и кинетических законо-
мерностей окисления присутствующих органических загрязнений
с идентификацией остаточных продуктов электролиза и их ток
синологической оценкой. Существенным резервом достижения
более глубокой степени минерализации сложных органических
примесей является совмещение процессов электрохимической де-
струкции и гетерогенного катализа. Комбинированный способ
электрокаталитической деструкции позволяет ие только полнее
использовать окислительные свойства электрогенерированных
агентов, но и создавать благоприятные предпосылки для разра-
ботки безотходных технологий в замкнутых циклах водоснабже-
ния предприятий.
Перспективным направлением в разработке высокоэффек-
тивной технологии обработки воды является исследование воз
действия электрического поля на биологические объекты, в том
числе и па микроорганизмы, осуществляющие процессы биохими-
ческой очистки сточных вод в бпоокислителях и обезвреживания
образующихся осадков в метантенках, перегиивателях и т. п. Из
вестно, что умеренное воздействие электрического поля стимули-
рует рост и жизнедеятельность бактерий, увеличивая их окисли-
тельную способность по отношению к органическим примесям
воды. Это направление выдвигает ряд специфических задач в ис-
следовании данного феноменологического фактора, решение ко-
торых может оказать значительное влияние на интенсификацию
процессов биоокисления органических примесей, содержащихся
как в сточных волах, так и в образующихся осадках.
Обсуждая в целом представленный впервые в данной книге
обобщенный и далеко не полный материал, освещающий совре-
менное состояние вопроса электрохимической очистки воды, сле-
дует отметить, что достигнутый в последние годы прогресс в тео
рип электродных реакций, процессов превращения примесей, про
текающих в объеме электрореакторов за счет целенаправленного
изменения физико-химических свойств обрабатываемой жидкости,
новых электрокппезнческпх явлений па поверхности дисперсных
частиц и других научных разработок, открывает широкие возмож-
ности в реализации да иной высокоэффективной технологии при
менительио к очистке природных и сточных вод. Однако виедре
208
нию этой прогрессивной технологии мешает в насюящее время
еще ряд нерешенных проблем прикладного характера |38, 103|
В технологическом плане к ним относится необходимоеib сии
жепня расхода: материала электродов за счет новых технических
решений и конструктивного совершенствования аппаратов; реа
гентов при электрообработке за счет выявления оптимальных
условий их эффективного использования для процесса очистки
воды, полного и целенаправленного использования газовой фазы
путем улучшения гидродинамических условий работы элекгро-
флотаторов, утилизации продуктов электролиза (металлов при
электродиализной очистке или электроосаждепнп, а также водо-
рода и кислорода) в качестве вторичного сырья для промышлен-
ности. регенерации образующихся гидроксидов с целью возврата
их в качестве коагулянтов и т. и.
Конструкторские проработки должны быть направлены па
поиск оптимальных конструктивных решений и аппаратурного
оформления электрохимических методов очистки воды и обра-
ботки осадков с целью снижения их энергоемкости, па разработку
унифицированных модификаций электрореакторов, малогабарит-
ных выпрямительных устройств с дополнительной градацией ио
силовой нагрузке и мощности, а также систем автоматического
управления и контроля технологического процесса, дистанцион-
ного и экспрессного анализа качества обрабатываемой жидкости.
Применение различных способов электрообработки должно
быть в каждом конкретном случае техники экономически обосно-
вано. Здесь следует указать, что сложившееся мнение о целесо-
ооразпостп использования этих способов только лишь для объек-
1он с небольшим расходом обрабатываемой воды со ссылкой на
1начптелы1ую энергоемкость процессов электрообработки явля-
ется неправомерным.
1 ели корректно учитывать затраты электроэнергии на работу
механизмов и аппаратов по обеспечению и организации всех тех-
нологических операций физпко химических или биологических
способов очистки (производство, приготовление и дозирование
реагентов, сбор, удаление и обезвреживание образующихся осад-
ков и нзбыючной биомассы, аэрация стоков в бпоокислителях,
ак1нвация и регенерация сорбентов при адсорбционной очистке
и I. и.), то эти затраты во многих случаях могут превышать за
рты электроэнергии па прямое ее использование в элеюрореак-
юра.х (см. приложение). Поэтому при сравнении различных мето-
юи водоочистки следует строже учитывать энергозатраты па все
основные и вспомогательные технологические операции, что по
«полит дать глубокую и всестороннюю технико-экономическую
оценку сопоставляемых методов н обосновать выбор наиболее ра-
циональной технологии.
299
ПРИЛОЖЕНИГ
Технико-экономическая оценка методов
электрохимической очистки воды
Целесообразность внедрения методов электрообработки в тех-
нологию очистки природных и сточных вод базируется, как пра-
вило, па высокой экономической эффективности затрат по срав
пенпю с традиционными способами их обработки. При этом ос-
новным условием проведения расчетов сравнительной экономиче-
ской эффективности является правильный выбор базовой техно-
логии, обеспечивающей одинаковую с аналогом степень очистки
воды.
Главным критерием эффективности различных вариантов тех-
нических решений служит минимум приведенных затрат, т. е.
сумма годовых эксплуатационных расходов С, включающих в себя
стоимость израсходованных при очистке воды реагентов, материа-
лов, топлива и электроэнергии, амортизационные отчисления,
транспортные расходы, заработную плату обслуживающего пер-
сонала, и капитальных вложений К с учетом нормативного коэф-
фициента их эффективности Е||(С + ЕНК).
Ниже приведены сравнительные затраты (тыс. руб.) на строи-
тельство н эксплуатацию станций водоподготовки производитель-
ностью 960 м3/суткн с применением коагуляции реагентами
[А12(5Оч)з, СаС12О] и электрокоагуляциониого способа с раство-
римыми стальными (Ст. 3) анодами и алюминиевыми като-
дами [14]:
Показатели затрат Реагентная коагуляция Электроко* агуляцня
Реагенты н транспорт 19,00 —,,
Электроэнергия 90,00 G0.00
Содержание персонала 63,20 10,10
Здания и сооружения 374.44 92,62
Сети 4,84 2,31
Оборудование 154,55 4,18
Текущий ремонт зданий и сооружений, 14,20 2,70
сетей, оборудования
Амортизационные отчисления 39.29 6,12
Сумма эксплуатационных затрат 213,50 82,60
Кап италовложен и я 433,70 99,10
Таким образом, годовая экономическая эффективность от внед-
рения электрокоагуляции при Ен=0,15 составляет:
\Э (С. + ЕнК,) - (Cj + EhKjI - (213,5 + 0.15 433.7)- («2,6 + 0,15 99,1) -=
181,1 тис. руб.
300
Электрохимическая технология отличается, как уже отмеча-
лось, положительными массогабаритпыми характеристиками и ком
п а кт пост ью оборудования, что имеет существенное значение при
реконструкции действующих и строительстве в стесненных усло-
виях новых очистных сооружений.
Рассмотрим на примере реконструкции очистных сооружений
Вырицкого филиала Ленинградского производственного текстиль-
но-галантерейного объединения «Север» обоснование целесообраз-
ности внедрения технологии электрокаталитической очистки сточ-
ных вод красильно-отделочного производства.
До настоящего времени па этом предприятии эксплуатирова-
лись сооружения реагентно-деструктивной прсдочнстки красильных
стоков [48, 105], которые в течение длительного периода (с 1974 г.)
работали эффективно, обеспечивая проектное качество очистки
сточных вод от красителей и ПАВ. Однако сложившиеся специ-
фические условия, а также наличие сульфатов в очищенных сто-
ках выше значений ИДК не позволяли направлять их на обще-
поселковые сооружения биологической очистки. Кроме того, зна-
чшельпый расход реагентов, образование осадка, сложность в экс-
плуатации морально и физически устаревшего водоочистного обо-
рудования требовали принятия качественно новых технологиче-
ских решений прн очистке сточных вод.
Для этих целей предложена разработанная кафедрой канали-
зации ЛИСИ технология электрокаталитической очистки, которая
без существенных дополнительных капитальных затрат обеспечила
повышенные требования к качеству обезвреживания промстоков
перед направлением их на общепоселковые биологические соору-
жения.
Внедрение данной технологии позволяет значительно снизить
taipaTbi на реагенты, электроэнергию, освободить производствен-
ные площади, сократить численность обслуживающего персонала,
упростить схему обработки сточных вод. Особо следует подчерк-
ну и», что при такой обработке не образуется осадок. Это также
является немаловажным преимуществом электрокатализ оческой
1ехпологии.
Основные затраты па очистку сточных вод (Q = 26000 м3/год)
при реконструкции сооружений Вырицкой фабрики составили:
1. Расход электроэнергии иа обработку сточных вод —
19,5 тыс. кВт-ч/год, на перекачку—10,2 тыс. кВт-ч/год, всего
29,7 тыс. кВт-ч/год. Стоимость электроэнергии — 29,7-30 =
891 руб., где 30 — стоимость 1000 кВт-ч, руб., по одноставоч-
ному тарифу.
2. Стоимость поваренной соли 52- 10 = 520 руб., где 52 — годо-
вой расход поваренной соли, т.; 10 — цена поваренной соли с уче
юм доставки и погрузочно разгрузочных работ, руб./т [63].
3. Стоимость гранулированного активированного пиролюзита
при расходе 1 т/год составляет 57 руб. (отпускная цена отходов
производства диоксида марганца).
4. Содержание персонала:
301
Должность Колн честно человек Месячный оклад, руб Г вдовой фонд заработной платы, тыс. руб
Начальник станции 1 165 1.98
Химик лаборант 1 100 1.20
Слесар ь- ремонт и и к 1 150 1.80
Слесарь КИПиА 1 180 2. Ki
Аппаратчик 2 ПО 2.64
Всего по статье:
9.78 тыс руб. в год
5. Здания и сооружения. Для размещения технологического
оборудования электрокаталитической очистки сточных вод ис-
пользуется ’/а часть существующего здаты и оборудования, смет-
ная стоимость которых составляет 237,22/5 = 47,44 pj6., где
237,22 — остаточная стоимость здания и оборудования.
6. Оборудование:
Номера статей прейскурантов, укрупненных показателей Количество н наименова- ние оборудования Сметная стоимость, тыс. руб.
строн- тельио- моитаж- ных работ обору- дования общая ст. и- мость
Прейскурант 24-84, доп 6, поз 08-075 Выпрямительные агре- гаты ВЛКР-12 6 2 шт — 14,06 14.06
— Электролизеры (песта» дартное оборудование). 2 шт. 0.4 7.0 7,40
УСН. ч П, 42 38 Резервуар для мокрого хранения поваренной соли, Рр 5 м’* 0.5 0.4 0.90
Прейскурант 23 03.42. поз. 002 Реактор нз нержавею- щей стали 0,15 1,48 1 63
СУПС Госкомитета, вып II, т 98-6 Контрольно-измери- тельна я аппаратура 2,05 2,25 4,30
Всего по статье: 28.29 тыс руб.
7. Сводный расчет затрат но статьям:
302
Наименование затрат Сумма затрат по очистным сооружениям, тыс. руб.
д» реконст- рукции пссле реке н- струкцни
1. Здания и сооружения 237,22 47.44
2 Оборудование — 28,29
3 Текущий ремонт здания и соору женин (2,2 % от статьи 1) — 1.04
4 Текущий ремонт оборудования (3,8 % от статьи 2) — 1,08
5. Амортизационные отчисления на здания и соору- жения (6 % от статьи 1) — 2,85
6 То же. иа оборудование (12 % от статьи 2) — 3,39
7 Содержание персонала — 9,78
8. Реагенты 12 23 0,597
9 Электроэнергия 12.00 0.891
10 Транспорт 24,41 —
11. Общефабричныс затраты 28,94 —
12. Эксплуатационные затраты (статьи 3 ... 11) 111,46* 9.848
13. Удельные эксплуатационные затраты, руб./м3 4.29 0,76
14 Капитальные вложения (статьи 1 и 2) 237,22’* 75.73
15 Удельные капитальные вложения, руб. /м3 9.1 3,15
• Фактические данные эксплуатации очистных сооружений за 1986 г.
•• Остаточная стоимость всех сооружений совместно с оборудованием.
Ожидаемый экономический эффект от внедрения технологии
электрокаталитической очистки сточных вод при реконструкции
сооружений Вырицкой фабрики составит:
ЛЭ ((4,29 + 0,15-9,1) —(0,76 + 0,15-3,15) • 26000= 114,998 тыс. руб.
в год.
Электрокаталитическая технология является весьма эффектив-
ной и при доочистке биохимически очищенных сточных вод авто-
номных коммунальных объектов [46, 48]. Реализация этой техно-
логии па очистных сооружениях учебно-спортивной базы «Кавго-
лово» в Ленинградской области позволила обеспечить требуемое
достаточно высокое качество очищенных стоков на сброс их в ма-
ломощный и непроточный водоем, расположенный в курортной
зоне. Гак, концентрация органических загрязнений в очищенной
поде составляет по БПКб в среднем 2,0 мг/л, взвешенных ве-
meciB—1,5 мг/л, фосфатов — 0.12 мг/л, аммонийного азота —
0,14 мг/л.
Ниже представлены основные технико-экономические показа-
юлп глубокой доочистки биохимически очищенных сточных вод,
определенные для станции производительностью 200 м3/суткп по
шум наиболее близким по технологической эффективности вари-
антам. По первому варианту сточная вода подвергается электро
лту с использованием нерастворимых анодов в iipiicvTciBiin
300 мг/л С1 -ионов и последующему фильтрованию через слой
303
гранулированного пиролюзита; второй вариант, разработанный
в ГЙСИ, предусматривает механохимическую обработку содой
с добавлением полиакриламида, фильтрацию через песчаную за-
грузку, каталитическое окисление озоном, флотацию и заключи-
тельную стадию фильтрования также через песчаную загрузку.
Показатели Затраты, тыс. руб.
электро- каталмтн ческая Очистка метод гиси
Реагенты 0,57 3,34
Электроэнергия 6,02 7,78
Содержание персонала 4.68 6,63
Здания и сооружения 29,77 41,95
Оборудование 20,79 59,44
Капиталовложения 60,63 120,19
Эксплуатационные затраты 19.73 31,40
Удельные капиталовложения, руб./м3 0,83 1,65
Себестоимость очистки воды, коп./м3 27,0 43,0
Таким образом, из приведенных выше данных по технико-эко-
номической оценке различных процессов очистки воды следует,
что применение электрохимических методов является достаточно
эффективным. Это позволяет надеяться на более широкое их внед-
рение в ближайшее время в технологию водоподготовки и очистки
сточных вод.
СПИСОК ЛИТЕРАТУРЫ
1. Алексеев Е. В. Исследование и интенсификация процесса очистки сточ-
ных аод предприятий шелковой промышленности флотационными и электро-
химическими методамн//Ав ореф дне. ...каид. техн. наук. М 1979.
2. Анопольский В. Н Интенсификация флотационной очистки нефтесодер-
жащнх вод с использованием электрореактора с газовым слоем//Автореф.
Дне.... каид. техн. наук. Л. 1983.
3. Анопольский В Н., Рогов В. М. Интенсификация очистки судовых иеф-
тссодержащих вод методом электрокоагуляции — флотацни//Пути предотвра-
щения загрязнения моря и атмосферы плавсредствами: Материалы по об-
мену опытом. Л.: Судостроение, 1980. Вып. 315. С. 93—95.
4. Анопольский В. п., Рогов В. М., Давидюк Ю. В. Очистка иефтесодер-
жащнх вод в малогабаритной установке, включающей электрореактор с газо-
вым слосм//Хнмия и технология воды. 1983 Т. 5. № 6. G 528—532.
5. Антропов J1. И. Теоретическая электрохимия. М. Высшая школа, 1984.
519 с.
6. Апельцина Е. И. Исследование закономерностей очистки природных вод
электрофоретическим методом//Автореф. дне....каид. техн. наук. М. 1972.
7. Бабенков Е. Д. Очистка воды коагулянтами. М.: Наука, 1977. 356 с.
8 Бажал И. Г., Куриленко О. Д. Переконденсация в дисперсных систе-
мах. Киев: Наукова думка, 1975. 216 с.
9. Баранов А. Н., Леонов С. Б., Сапов В. М. К вопросу об анодном рас-
творении железа пр очистке сточных вод обогатительных фабрик методом
элсктрокоагуляции//Цветиая металлургия. 1977. № 4. С. 3—7.
10. Бек Р. Ю. Электрохимическая очистка промышленных сточных вод от
ксантагеитов и циаиидов//Цветиая металлургия. 1982 № 2. С. 97—99.
11. Белозерова Е. Н. Технология очистки сточных вод валяльно-войлоч-
ною производства//Автореф. дне., каид. техн. наук. Л. 1984.
12. Варенцов В. К. Электролиз с объемно-пористыми проточными элек-
тродами в гидрометаллургии благородных металлов//Изв. СО АН СССР. Серия:
Химические науки. 1984. Вып. 6. № 17. С. 106—120.
13. Варенцов В. К., Белякова 3. Т. Окисление цианид- и роданнт-ноиоа
сточных вод на проточных пластинчатых и волокнистых аиодах//Цаетиая ме-
таллургия. 1983. № 3. С. 103—105.
14. Веселов Ю. С., Лавров И. С.. Рукобратский Н. И. Водоочистное обо-
рудование. Л.: Машиностроение, 1985. 232 с.
15. Выпрямительные агрегаты серии ВАК и ВАКР. М.: Информ электро,
1974.
16. Ганичев С Д Разработка методов обезвреживания флотошлама, об-
разующегося при флотационной очистке сточных вод предприятий текстильной
и трикотажной промышлеииости//Авторсф. дне.... каид. техн. наук. М. 1983.
17. Гвоздяк П. И., Чеховская Т. П. Электроудерживаиие мнкроорганнз-
моа//Микробиология. 1976. Т. 45. № 5. С. 901—905.
18. Генкин В. Е. Электромеханические способы очистки промышленных
сточных вод//Совершенствование систем водоснабжения, очистки сточных вод
и сооружений промышленной гидротехники. Труды ВНИИ ВОДГГО М 1984.
С. 39—42.
305
19 Гнусин И П Гребенюк В Д Электрохимия гранулированных нонн
тов Киев Техника, 1976. 160 с.
20 Грановский М Г, Лавров И С, Смирнов О В. Электрообрабогка
жидкостей. Л 1976. 216 с.
21. Гребенюк В Д. Электродиализ. Киев: Техника, 1976. 159 с.
22. Гребенюк В. Д., Мазо А Л. Обессоливание воды ионитами. М: Хи-
мия, 1980. 256 с.
23 Грушко Я М. Вредные органические соединения в промышленных
сточных водах Справочник 2-е изд., перераб. и доп. Л.: Химия, 1982. 216 с.
24. Губанов Л Н Локальная очистка сточных нод красильно-отделочных
фабрик электрохимическими методами //Авторсф. дне.... канд. техн наук. Л.
1975.
25 Гулый Г А., Малюшевский П П Высоковольтный электрический раз
рид в силовых импульсных системах Кнеа Паукова думка, 1977. 175 с.
26. Дамаскин Б. Б. Петрий О А. Введение в электрохимическую кинетику
М.: Высшая школа, 198-3. 400 с.
27. Дейнега Ю. Ф. Ульберг 3. Р, Эстрела-Льопис В. Р. Электрофорети-
ческое осаждение металлополнмеров. Киеа. Наукова думка, 1976. 254 с.
28 Дмитриев В Д. Методы подготовки воды в условиях Севера. Л
Стройиздат, 1981 120 с.
29. Духин С С, Семенихин Н. М. Теория поляризации двойного слоя и
ее влияние па электрокинетическпе и электрооптические явления и диэлектри-
ческую проницаемость дисперсных систем//Коллоиди. жури. 1970. Т. 32.
С. 360-368.
30 Духин С С., Эстрела Льопис В Р, Жолковский Э. К. Электропоаерх-
иостиые явления и электрофильтроаание Киев: Наукова думка, 1985. 288 с
31. Евдокимов С В., Печорский М М, Городецкий В. В. Кинетика и ме-
ханизм разряда-ионизации хлора иа электродах ОРТА//Тез. докл. 5-го Все-
союзного совещ. <Малонзнашиааемые аноды и применение их в электрохими-
ческих процессах». М: ЦП ВХО им Д И Менделеева, 1984. С 8—9.
32. Жук Н П Курс теории коррозии и защиты металлов. М.’ Металлур-
гия 1976. 472 с.
33. Жуков А. И., Монгайт И. Л., Родзиллер И. Д Методы очистки произ-
водственных сточных вод. М.: Стройиздат, 1977. 136 с.
34. Жукова Л. А. Теория статического и динамического осаждения и соо-
саждення ионов. М.: Эиергоатомиздат, 1981 80 с.
35 Защита аодосмоа от загрязнений сточными водами предприятий чер
ной металл)ргни/Левии Г М, Пантслят Г С., Вайнштейн И Л и др М
Металлургия, 1978. 216 с.
36. Зяблицева М П„ Сафонова Т. Я-, Петрий О. А. Электроокислспне
проппленгликоля иа окисных рутеннево-тнтаиоаых анодах а хлорндиых рас-
солах//Электрохпмия. 1984. Т. XIV. № 1 С 131.
37 Иванов Г В Расчет флотационных установок для жиросодержаших
сточных вод Учебное пособие. Л.: ЛИСИ, 1984. 84 с.
38. Казаринов В Е. Электрохимические аспекты экологических проблем//
Тез. докл. Всесоюзной конф. <Электрохнмня и охрана окружающей среды».
Иркутск, 1984. С. 4.
39 Калиновский Е. А, Жук А П, Бондарь Р У Стойкие аноды для элек-
трохимического хлорирования морской воды//ЖПХ 1980. Т 1И. № 10.
С. 2233 -2237.
40. Кафаров В В Принципы создания безотходных химических производств
М.: Химия. 1982. 288 с.
41. Ковалев В В, Бонд М И. Электромагнитная очистка нагретых сточ-
ных аод//Электрониая обработка материалов. 1982 № I. с. 61—64
42. Ковалев В В, Судварг М. И, Пынзарь В В Повышение эффектив-
ности электрохимической обработки сточных аод с использованием комбини-
рованных желсзоалюминневых элсктродов//Электронная обработка материа-
лов. 1981. № 5. С. 55—59.
43. Коваленко Ю А., Коварский Н. Я-. Кондрикова Н М Сорбционные
саойстаа смешанного окенгндрата Fe (II)—Fe (III) в момент его образова-
ния //Химия и технология воды 1980. Т 2. № 1. С. 8 12.
306
44 Классификации физико-химических мето ion обработки noiuMpo. 1.111
ский 3. Я. Николадзе Г. II, Пальгунов II. II., Долгошкои |> И/Вопнл ре-
сурсы. 1974 Ns 2. С. 120 126.
45. Краснобородько И Г. Разработка методов обесцвечивании снятых шн
красильно-отделочных производств предприятий текстильном и трико ы ж ион
промышлеииостн//Лвтореф дне. канд техн, наук Л. 1974
46. Краснобородько И Г Доочистка сточных вод с целые.....плис вин из
лежиостн защиты поверхностных водоемоа от загрязнения//! loBbiiiicinic и i к-ж
ности систем водообеспечения городов и промышленных предприятий Ленин
града и области. Л.: ЛДНТП, 1985. С. 69—73.
47. Краснобородько И. Г. Токсикологическая опенка электрохнмпчсскон
очистки сточных вод//Текстнльпая промышленность 1986 Nt 4 С. 58 60
48. Краснобородько И Г Деструктивные методы очистки сточных нот//
Водоснабжение и санитарная техника 1986 № 10 С. 4—7.
49. Краснобородько И Г. Кузнецов В В Кинетические закономерности
удаления анноноактнвных ПАВ метолом комбинированной электрокаталитиче-
ской обработкн//Извлечспис из сточных вот н использование ценных веществ
в системах водоотведения Межвуз темат. сб тр Л ЛИСИ, 1986. С 64 70.
50. Краснобородько И Г, Никофоров Л Т. Опытно-промышленная уста-
новка для электрохимической очистки сточных вод комбината тонких н тех-
нических сукои//Текстильная промышленность 1984 № 4 С 56—58.
51. Краснобородько И. Г.. Светашова Е С. Электрохимическая очистка
сточных вод. Учебное пособие. Л.: ЛИСИ. 1978. 89 с.
52. Краснобородько И Г. Спивакова О М. Сафин Р С Схемы капалнзо-
вапия очистка сточных вод предприятий бытовой химии: Обзор и ниф Сер.:
Бытовая химия и проблемы ее развития М ННИТЭХПМ. 1977 45 с
53. Кубасов В Л.. Зарецкий С А Основы электрохимии М Химия, 1985
168 с.
54. Кульский Л А Теоретические основы и технология кондиционирова-
ния воды Киев Наукова думка, 1980 564 с.
55. Ласков Ю М, Федоровская Т. Г. Жмаков Г. Н Очистка сточных вот
предприятий кожевенной и меховой промышленности М • Легкая и пищевая
промышленность, 1984 168 с.
56. Лещов Е С. Рулев Н Н. Рогов В М Ортокннетпческая коагуляция
мелких частиц при их Флотации пузырьками, вспаивающими ппн умеренны»
числах Рейнольдса (1 <Ре<40)//Хнмпя и технология воды. 1983. Т 5 № 2.
С 142 147
57 Лурье Ю К) Справочник по аналитической химии М Химия, 1979
180 с
58 Ма.макол А А. Современное состояние н перспективы применения
э 1ектролитической фтотанни веществ Ч. I. Кишинев: Штнннна. 1975 134с
59. Мартынова О И. Водоподготовка. Процессы и аппараты М \том
и пат, 1977
60 Матов Б М Электрофлотаиноиная очистка сточных вод. Кишинев:
Кзртя Молдоаеняскэ. 1982. 170 с.
61 Мацнев А И. Очистка сточных вод флотацией. Кпеп: Будивелышк.
1976 1.32 с
62. Машников И В Исследование сопряженных реакций каталитического
распада перекиси водорода и окисления форматьтегита па графитах с метал-
лическим покрытнемЛЛвтореф дне . канд. техн наук. М 1976
63 Медриш Г. Я, ТеАшева А. 4. Багин Л Л Обеззаражпнапис i№H«»i-
иых н сточных вод с использованием электролиза. М: Стройиздат, 1982 80 с
61. Меньшикова Б Н. Захарова Т Л . Кириллова М II Фишки xiimh ic
скне основы процесса отделочного производства 1 Легкая и пищевая про
мыштенность, 1982 280 с
65 Меркушев О М. Электроосаждеиие гстеросуспеизнй//Лисперсныс си-
стемы и их поведение в электрических и магинтиых полях: Межвуз сб тр
•V? I Л. ЛИСИ, 1976. С 56—82.
66 Металлоокнсные аноды для обесцвечивания сточных вот. содержащих
красителн/Сорокендя В С, Бондарь Р У, Краснобородько И Г и др//Тез
докл 5-го Всесоюзного совет. «Металлоизнашиваютиеся аноды н применение
307
их в электрохимических процессах». М.: IUIBXO им. Д 11 Менделеева. 1984
С 63—64
67 Мильтон Дж Аллен Электродные процессы в органической химии Л,
Госхимнздат, 1961. 180 с.
68. Мулина О. А., Мацкевич Е. С., Кульский Л. А Интенсификация про-
цессов электроокисления органических примесей природных и сточных вод//
Интенсификация очистки приротных и сточных вод Тез. докл Всесоюзи
научно-техн, конф Ровно УИИВХ 1983. С. 112
69 Назарян М М. Ефи иов В Т Электрокоагуляторы для очистки про-
мышленных стоков. Харьков: Вита школа. 1983. 144 с.
70. Найденко В В, Алексеев В И.. Губанов Л. Н. Электросатурання прн
флотационной очистке сточных вод//Хнмня и технология ноты 1986 Т. 8 № 3
С 84—85
71. НаМенко В В. Губанов Л Н. Чернышева В И Технология очистки
промышленных сточных вод: электрокоагуляция, электрофлотацня, электролиз.
Горький: ГИСИ, 1980 С. 31—32.
72. Найденко В В, Губанов Л И. Чернышева В И Технология очистки
промышленных сточных вод Электродиализ Горький: ГИСИ. 1980 36 с.
73 Никифоров М Т Локальная физико-химическая очистка сточных вод
от красителей и ПАВ//Автореф. дне. .. канд. техн наук. Л. 1984.
74 Ньюмен Дж. Электрохимические системы. М Мир. 1977 462 с.
75. Опреснение умягченной воды электродиализом с одновременным полу-
чением высококонцентрированного рассола/Гребснюк В Д. Писарук В. И
Стрижак И П и др//Химия и технология воды 1980. Т 2. № 1
С. 36-38.
76. Опытно-промышленные испытания установки для умягчения волы
электрохимическим способом/Бочкарев Г Р, Попов Л В. Ростовцев В И
и др //Водоснабжение и санитарная техника 1982. .V» 4 С. 7—8
77 Основы теории и практики электрохимической обработки металлов и
сплавов/Щербак М В, Толстая Т А., Анисимов А П., Постаиогов В. X. М.:
Машиностроение, 1981. 263 с.
78 Очистка воды электрокоагуляннсй/Кульскнй Л А.. Строкач П. П,
Слипчепко В А Сайгак F И Киев Будпведышк. 1978 П2 с.
79 Очистка сточных вод предприятий мясной и молочной промышлешю-
сти/Шнфрнн С М„ Иванов Г В.. Мищуков Б. Г.. Феофанов Ю А М.: Легкая
и пищевая промышленность, 1981. 272 с.
80 Нервов Г Г Опреснение соленых н солоноватых вод и их использо-
вание в народном хозяйстве//Совери1енствование систем водоснабжения, очи-
стки сточных вол и сооружений промышленной гидротехники. Труды ВНИИ
ВОЛГГО М, 1984 С. 19 23
81 Перепелкин Е С., Матвеев В С Газовые эмульсии. Л.: Химия, 1979.
200 с
82 Пичага А К. Ляуксминас В А Электрохимические методы регенера-
ции металлов из водных растворов//Материалы семинара <3ашита окружаю-
щей среды и техника безопасности в гальваническом производстае». М.. 1982.
С. 54—59.
83. Плесков Ю В, Филиновский В Ю Вращающийся дисковый электрод
М • Наука, 1972 344 с
84 Пономарев В. Г Иоахимис Э Г. МонгаПт И Л. Очистка сточных вод
нефтеперерабатывающих заводов М Химия, 1985 256 с
85 Предотвращение загрязнения природной среды отходами нефтепродук-
тов и масел/Внснапуу Л.. Каазик М.. Кирш А.. Балащенко Г. Таллин: Эст.
ИИИНТИ, 1981 20 с.
86 Прейскурант 003 Розничные пены на поваренную соль. Постановление
Госкомитета цен от 14 12 76. № 626 М.: Прейскураитиздат. 1977 8 с
87 Прокопчик А Ю. Разложение некоторых окислителей в щелочной
среле//Автореф. дне. . . д-ра хим наук. Вильнюс, 1963.
88 Проскуряков В А . Шмидт Л И Очистка сточных вод в химической
промышленности Л Химия. 1977. 463 с.
89 Пушкарев В В, Трофимов Л И Физико-химические особенности очи
стки сточных вод от ПАВ М : Химия, 1975 143 с.
398
90. Рекомеидаинн по проектированию подоснабження и капали laiinii цехов
гальванопокрытий М ГПИ Сантехпроект, 1981 151 с.
91. Рогов В. М Применение электроагуляцин— флотации для очистки
сточных вод. содержащих высокоднсперсиые загрязпения//Лвтореф дне
каид. техн. наук. Новочеркасск, 1973
92. Рогов В. М,, Филипчук В Л. Применение электрохимического изме-
нения величин pH и Eh в технологии очистки воды//Химия и технология воды
1983. Т 5. № 1 С 45—49.
93. Ротинян А. Л, Тихонов К Л.. Шошина И А Теоретическая электро-
химия. Л : Химия, 1981 421 с.
94. Рукобратский Н. И. Исследование регенерации воды в замкнутых
объемах метолом электрических воздействнй//Лвтореф лис., каид техн,
паук. Л . 1979
95 Рулев И Н Коллективная скорость всплывания пузырьков//Коллоили.
жури 1977 Т. 39. № I С 80-86
96. Рулев Н. Н„ Рогов В. М Двухмерная модель конвективных потоков,
возникающих при мпкрофлотацни//Химия и технология воды. 1983 Т. 5. № 3
С 195—199
97. Сафин Р. С Разработка электрохимических методов очистки сточных
вод основных производств предприятий бытовой хнмии//Антореф. дис.. канд.
техн наук. Л , 1977.
98 Семенихин Н М, Жолковский Э К, Рукобратский И И Оценка
эффекта фильтрования для сеточных электрофильтров</ЖПХ 1980. Т 53
V- 10 С 2237—2243
90 Сененов Н Н Химическая физика Физические основы химических
превращений. ,М.: Знание. 1978 64 с.
100 Скорчеллетти В В Теоретическая электрохимия. Л Химия. 1974 568 с.
101 Слесаренко В И Современные методы опреснения морских и соле-
ных вод. М Энергия, 1973 248 с.
102 Смирнов Д И Генкин В Е Очистка сточных вод в процессах об-
работки металлов. М.: Металлургия, 1980 196 с.
103 Соловьев С Г. Фиошин Af Я, Иванов Е И Основные тенденции
ра 1ИПТНЯ эзектрохпмпческой очистки сточных вод/ГГез докл Всесоюзной конф
еЭлектрохимия н охрана окружающей среды». Иркутск. 1984 С 9
101 Справочник по электрохпмпн/Под рел. А М Сухотина Л Химия.
J0R1 188 с
105 Справочник проектировщика Канализация населенных мест и про-
мышленных ппетпрнятнй/Поз общ. род. В И Самохина 2 е изт.. перераб и
юн М Стройнздат. 1981. 639 с.
106 Структурные особенности окенгнзрата алюминия, синтезированного
> 1ектрохнм11чс<'К11м способом/Бочкарев Г. И.. Зильберман Б. Д.. Лебедев В. Ф.,
Кочни 11 М/^Физико-технические проблемы разработки полезных ископаемых
1970 № 2 С 85—89.
107 Тиманова Т Л Флис И F Физико-химические основы отбелки це.з-
лю.тозы/Под рст К П Мищенко М- Лесная промышленность. 1972 262 с.
108 Тюняткина Т Г.. Рукобратский Н. И Очистка волы в сильных элек-
трических нолях/ТХимия п технология воды. 1980 Т 2. № 3. С 242—245
109 Филиппов В М Исследование и создание промышленного способа
очистки сточных вод производства активных красителей активным хлором://
Зптореф. дне. ... канд. техн, паск Новочебоксарск. 1970
ПО Филипчик В. Л. Электрохимическое изменение величин pH и Fit прн
очистке волы, содержащей ионы тяжелых металлов//Автореф. дне. .. канд.
техн паук Киев. 1980
III Фролов Ю Г Курс коллоидной химии Поверхностные явления н
лисперснмс системы М Химия, 1082 400 с
112. Харитонов И В Разработка электролизеров с засыпанными анолами
из отходов металлообработки для очистки сточных вод гальванических про-
изводств//Автореф дне . канд техн, наук М. 1983
113 Химическая промышленность: Обзорная информ Гер Хлорная про
мы111лен11ость- Производство гипохлорита за рубежом М НИИТЭХИМ. 1980
309
114 Царьков Л. В. Очистка минерализованных сточных вод от некоторых
органических примесей rerepoi енпо-ката литическим окислением с целью и
полыоваиия в производстве хлора и каустической соды//Лвтореф. лис.
каид. техн. наук. М., 1980.
115 Чантурия В А, Шафеев Р Ш Химия поверхностных явлений прн
флотации М Недра. 1977 191 с
116. Электрофоретическое разделение фаз в I идродисперсиях неоргаииче
скнх матерналов/Ковылов А Е., Гфремов И Ф. Рукобратский И И и Др.//
ЖПХ 1981 № I. С. 81 87
117. Электрохимические процессы в системах с пористыми матрицами'
Ксенжек О С., Шембсдь Г М Калиновский Г.. А. и др. Киев: Вита школа.
1983. 219 с.
118. Эндюськин П. П. Селезенкин С В. Дюмасв К. И Электрохимиче-
ская очистка сточных вод производств органических красителен//ЖПХ 1983
Т VI. № 5 С 1167—1169
119 Эстрела-Льппис В. Р Теория лнполофореза и электрокоагуляции
в дисперсных снстемах//Лптореф. дне., каид. техн, наук Киев. 1977.
120 Ющенко В Д Исследования по предварительной очистке сточных
вод мясокомбинатов методом электроф дотации— электрокоагуляции// Хвтореф.
дне.. . каид. техи наук Л . 1975
121. Якименко Л. М Электрохимические процессы в химический промыш-
ленности. М.: Химия. 1981. 280 с.
122 Якименко Л М. Серышев Г А Электрохимические процессы в хи-
мической промышленности: Электрохимический синтез неорганических соедв
нений. М Химия. 1984. 160 с
123 Яковлев Л И.. Шостенко А Ю Глущенко Л Ф Исследование воз-
можности нптенсифнкац|||! процессов биологической очистки сточных вод пп".
нзводств химических реактивов путем взаимодействия электрического подя
активный ил//О«истка сточных вод и газовых выбросов в витаминной промыт
лснпости М .. 1962. С. 56—62
124 Яковлев С В, Краснобородько И Г, Кузнецов В В. Кинетически"
<акономерностп окисления органических красителей при электрохимшн^к"**
очистке сточных вод. Изв. вузов «Строительство и архитектура». 1986 К» 9
С 96 100
125 Balke Е М. Arpade S D Method Гог the recovery of mercury and
other liea» v medallions from a lnguid stream Pal 4208258. USA
126 Foun E Tlekfrolv lick Kloakkrensing «Stads-oj hav neingenioren». 1968
hd 94 № 6 p 101 104
127 Jloi S. X'akatHiira J., Kuwahara T Elektrodialilic recovery process of
melal finishing waste waler Water life Proc. Ini Conor Dcsalin and Water
Re-Use К C. Chanaiasappa Met. Nice, 1979 Vol. 3 Teancck N 1 1980 P 383—
389
128 Marcantonia P. I Apparatus for electrochemical remo al of heavy me-
tall such as chromium from dilute waste water. Streams using flow through
porous electrodes Pat. 4292160. USA
129. Methodes electrolitimies d'epnration dos eanx residnaires. Elimination
des sidfachtifs de svnlhesse/Bersit C. laskiemez II. Tran Manh-Sung G Sand-
eel Г Trih CFBEDFAU 1979. 32 V- 424 P 79 88
130 Pourbaix M Atlas d'equilibres cletrochcnii<|iic. Paris. Plenum Press.
1963
131 Rohshach Sihon II Ekctrolilic treatmenl of cooling Waler system Pat
138.3097. England.
132. Veeraranhaean R Dambal R P Electrochemical process of treatment.
<>f piatig effluent I Electrochem Soc India. 1982 31 2. P 27 32
13.3. Wishart A I Gregory I. Filtration of 1 afex particles through Bed of
fine alumina fibres. «Deposition and filtration of particles from gases and li
quids». Simposium Loughborough Univers.. England, 6-8 September, 1978
P 143 153
310
о I .'I Ml .ПЕН HE
I Ipv iik.к>11 не...................................................... 3
< >< ионные обозначения 5
I i а в a I Электрохимическая технология в системах водоподготовки
и очистки сточных вод
I 1 .Электрохимические процессы и их организация........................ 8
I ’.* Химическое аоздействне электрического тока. Законы Фарадея ... 12
I I Во1можныс области реализации электрохимических процессов в техполо-
iiiii очистки воды . : ..................................... 15
* I । а и л 2. Электролитические свойства воды и водных растворов 19
I Электролитическая диссоциация водных систем ............ 19
’ Ионное равновесие в растворах электролитов. Гидролиз 22
' 1 Подвижность н числа переноса ионов .................... 26
’• I Электропроводность электролитов ........................ ... 28
I а и а 3. Теоретические основы процессов, протекающих прн электролизе
водных систем
И Некоторые аспекты химической и электрохимической термодинамики 33
12. Процессы на границе электрод раствор . 36
3.2.1 Электродные потенциалы ......................36
3 2.2. Окислительно-восстановительное равновесие и устойчивость вод-
ных растаоров 42
1.3 Механизм и кинетика электродных процессов 46
3.3.1. Понятие о лимитирующей стадии и скорости процесса 46
3.3.2. Поляризационные явления в электрохимических процессах 48
3.3.3. Напряжение разложения водных растворов электролитов 49
3.3.4. Кинетические закономерности стадии разряда ионизации 50
3.3.5. Массоперенос и основные уравнения диффузионной кинетики . 52
3.3.6. Кинетика основных электродных процессов, протекающих при очи-
стке природных и сточных вод 57
3.3.6.1. Катодные процессы 57
3.3.6.2. Анодные процессы........................................ 60
3.3.6.3. Редоксн-процессы (электрохимическое восстановление н окис-
ление) 65
3 1. Процессы, протекающие в объеме электролита . . 66
3.4.1 Электролитические эффекты, нолникакнцне н жидкости при элек-
трических воздействиях ... . ... 68
3 4 1.1 Воздействие однородных и неоднородных электрических полей <*8
3.4.1.2. Воздействие продуктов электродных реакций................71
3.4.2. Превращения примесей поды за счет электролитических эффектов 83
3.4.2.1 Коагуляция частиц а электрическом поле ..... 83
3.4.2.2. Агрегация частиц за счет продуктов электролиза 87
3.4.3. Электрический разряд в жидкости ..................... . . , 103
Глава 4 Методы и аппараты электрохимической очистки воды
1.1. Классификация методов ; . . . 108
1.2. Методы превращения веществ 112
4.2.1 Электрокоагуляция 112
4.2.1.1. Поляризационная коагуляция 114
4 2.1.2. Электрохимическая коагуляция 115
4 2.1.3. Электролитическая коагуляция ... 116
4.2.1.4. Гидродинамическая коагуляция........................... 123
311
4.2.1.5. Концентрационная коагуляция.................... ... 123
•1.2.1.6. Аппаратурное оформление процессов электрокоагуляции . 124
4.2.2. Элсктрокоррсктиронанне pH и Eli . 131
4.2.3. Электрохимическая деструкция ............................ . 140
4.2.З.1. Общие положения . . . НО
4.2.3 2. Выбор типа электродов ..................... 144
4.2.3.3. Влияние условии электролиза иа окисление органических за-
грязнений .......................................................149
4.2.3.4. Окислительно-восстановительные процессы в растворах гипо-
хлоритов .................. ..............................151
4.2.3.5. Окислительно-восстановительные процессы при электролизе
хлорндных растворов..............................................153
4.2.3 6. Конструкции аппаратов для электрохимической деструкции ор-
ганических загрязнений . 156
4.2.4. Электрокрнсталлизация..................................... .159
4.3. Методы разделения веществ ... . 165
4.3.1. Электрофлотацня . . . . . 165
4.3.2. Электродиализ .......................................... - 175
4.3.3. Электрофорез .......... . . . . 186
4.3.4. Электрофпльтрованне ... ... .191
1.4. Комбинированные методы........................................ . 193
4.4.1. Элсктрофлотокоагуляция . . ............... . . 193
4.4.2. Электрокаталитическая очистка.............................. 202
4.4.3. Обработка комплексом электрических воздействий..............207
4.4.4. Электроосаждение .... . . 211
4.4.5. Электрохнмннеское обеззараживание ..... 215
Глааа 5. Электрохимическая технология водоподготовки и очистки
сточных вод
5.1. Подготовка природных вод для целей водоснабжения . 218
5.1.1. Электрокоагуляция при обработке природных вод.............. 216
5.1.2. Подготовка аоды с применением сильных электрических воздействий 227
5.2. Очистка сточных аод ............................... .... 231
5 2.1 Технологические воды гальванических производств . 231
5.2.2. Сточные воды предприятий бытовой химки..................... 242
5.2.3. Нефтесодержащие воды....................................... 246
Глава 6. Методика расчета аппаратов для электрообработки
природных и сточных вод
6.1. Основные технологические параметры процессов электрохимической водо-
очистки ........................................................... • 253
6.2. Расчет электрофлотатороа........................................ 261
6.2.1. Расчет электрофлотаторов с горизонтальным и плоским анодом н
сетчатым катодом ...........................................
6.2.2. Расчет электрофлотатора с вертикальными плоскопараллельными
электродами........................................ : . . . 263
6.3. Расчет этсктрокоагуляторов...................................... 264
6.4. Расчет электрофлотокоагуляторов ..... . . . 2/0
6.4.1. Расчет колонных электрофлотокоагуляторов....................270
6.4.2. Расчет электрофлотокоагуляциониой установки горизонтального типа 272
6.5. Расчет аппаратов электрохимической деструкции . . 276
6.6. Расчет лектродиализаторов....................................... 263
6 7. Расчет аппаратов электрохимического корректирования pH и Eh 269
Глава 7. Основные проблемы современной технологии, перспективы
развития и совершенствования электрообработки природных
и сточных вод
Приложение......................................................... 300
Список литературы.....................................................305
312