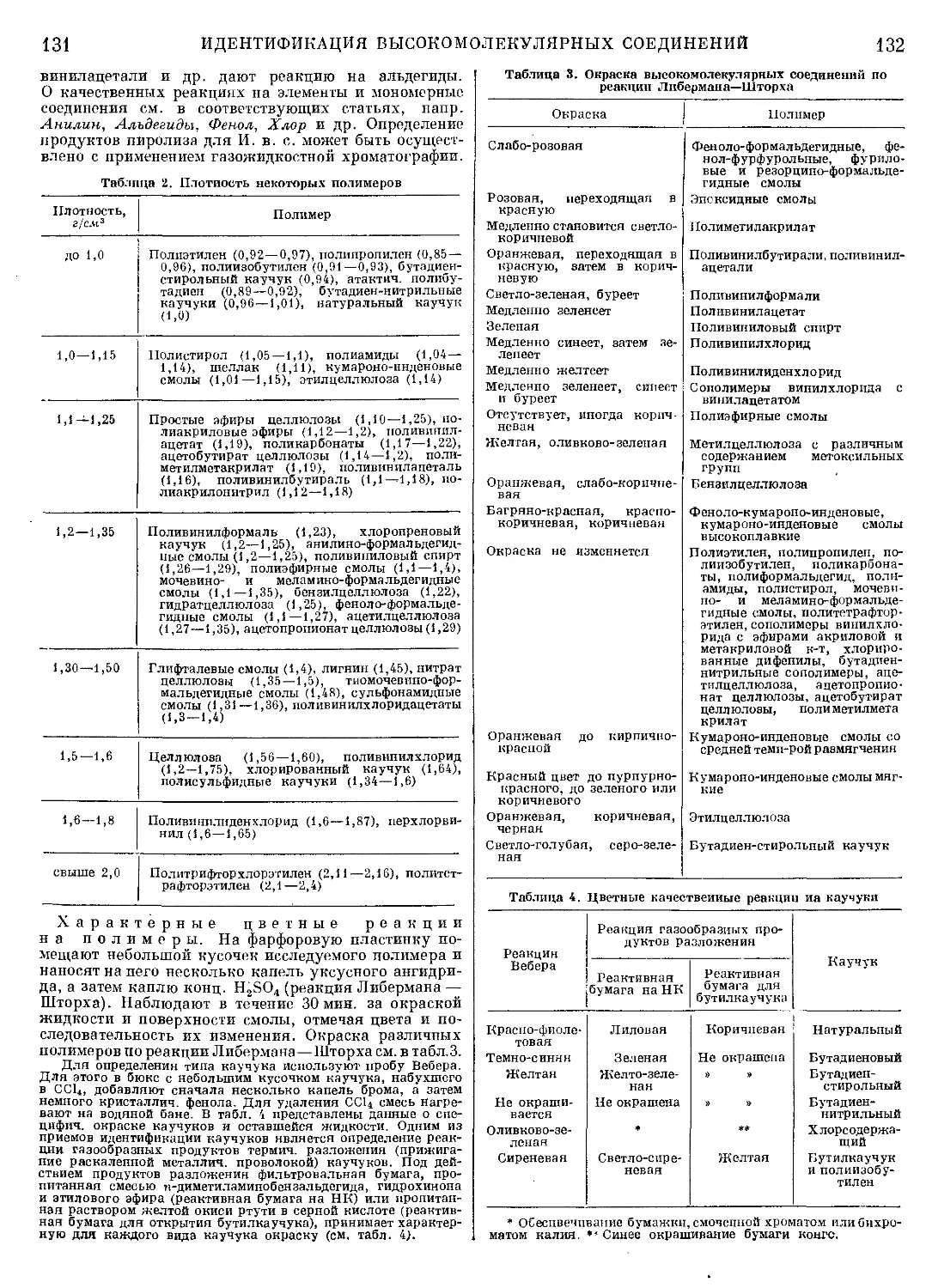
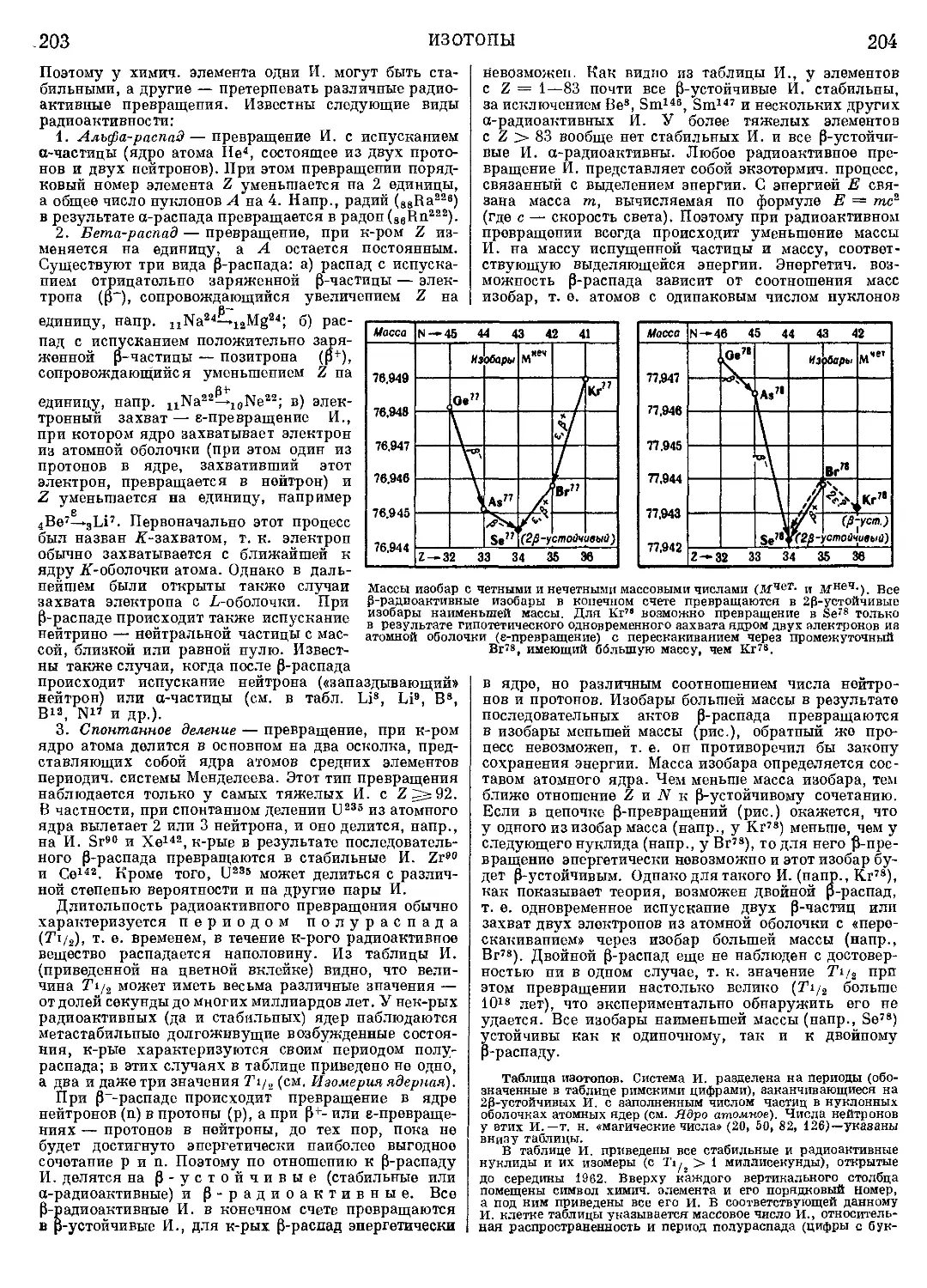
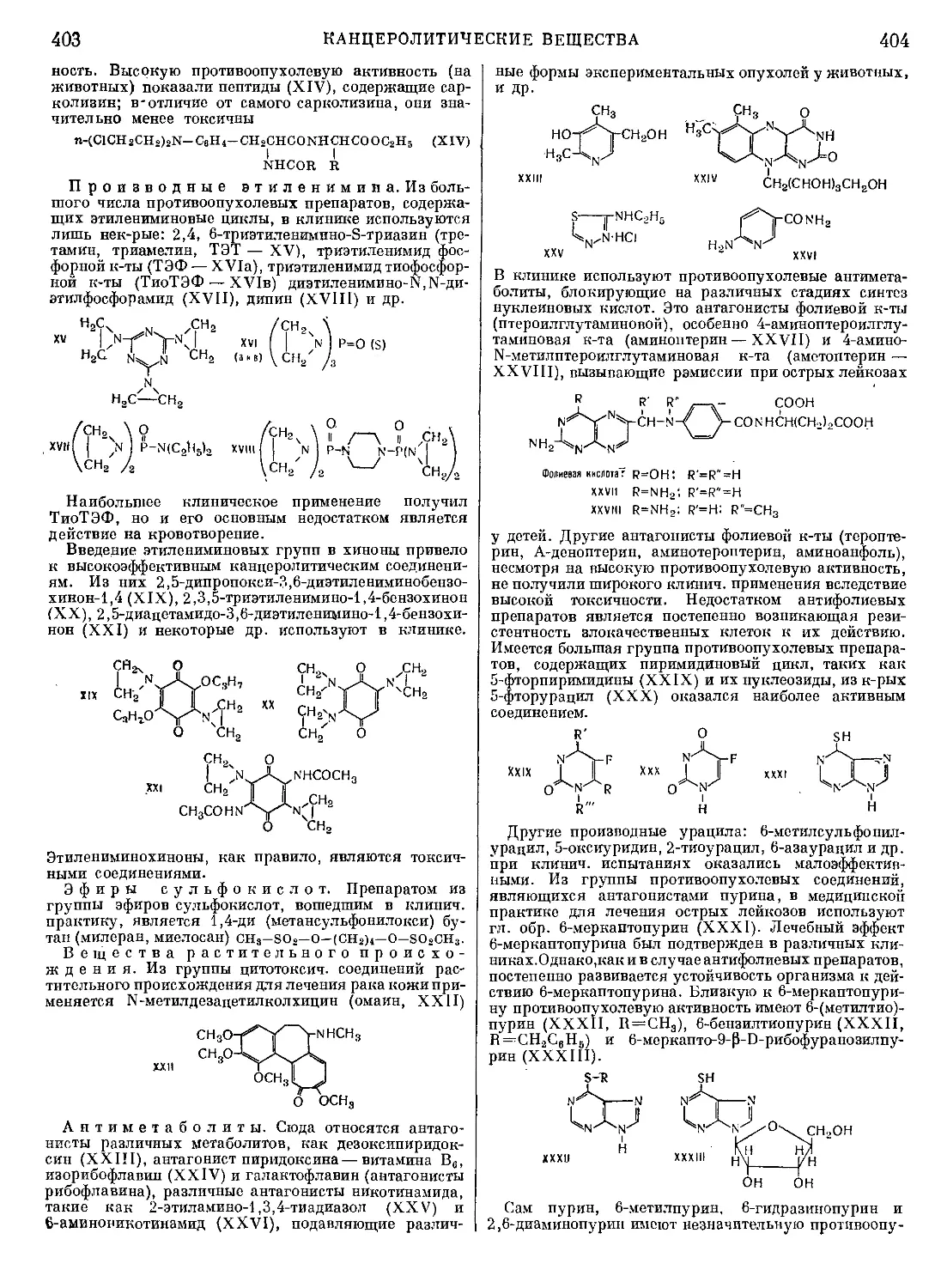
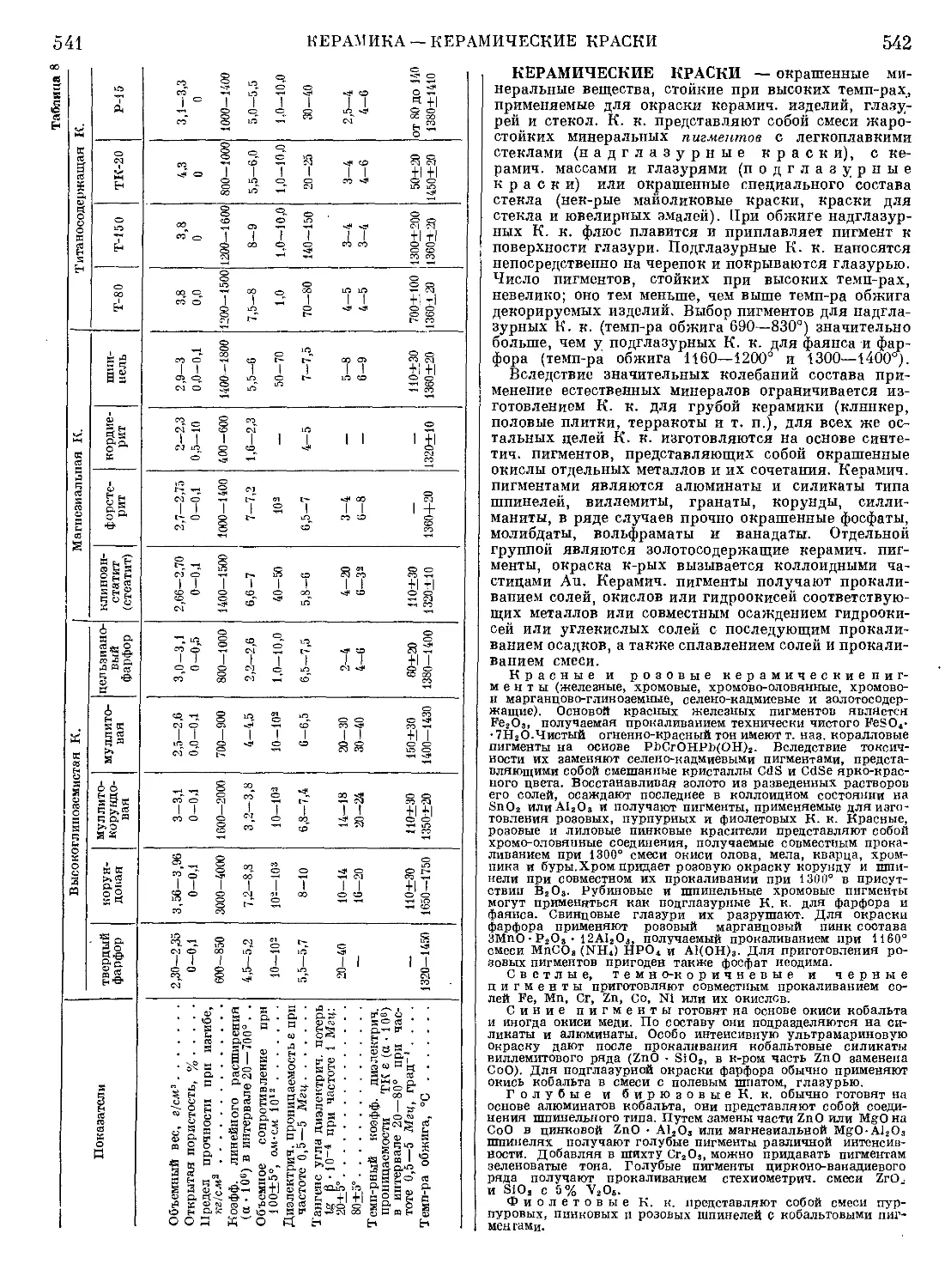
/
















































































































































































































































































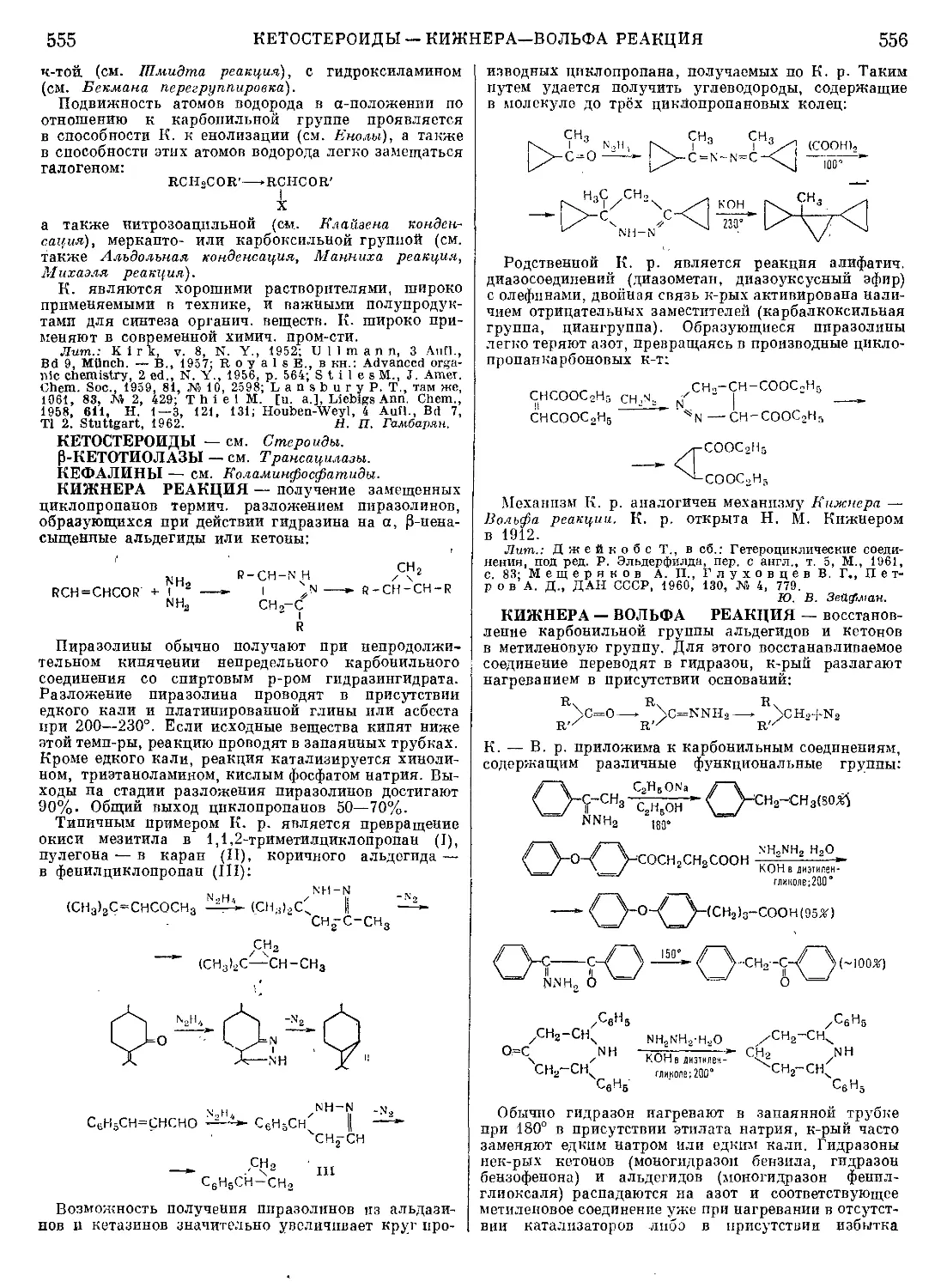
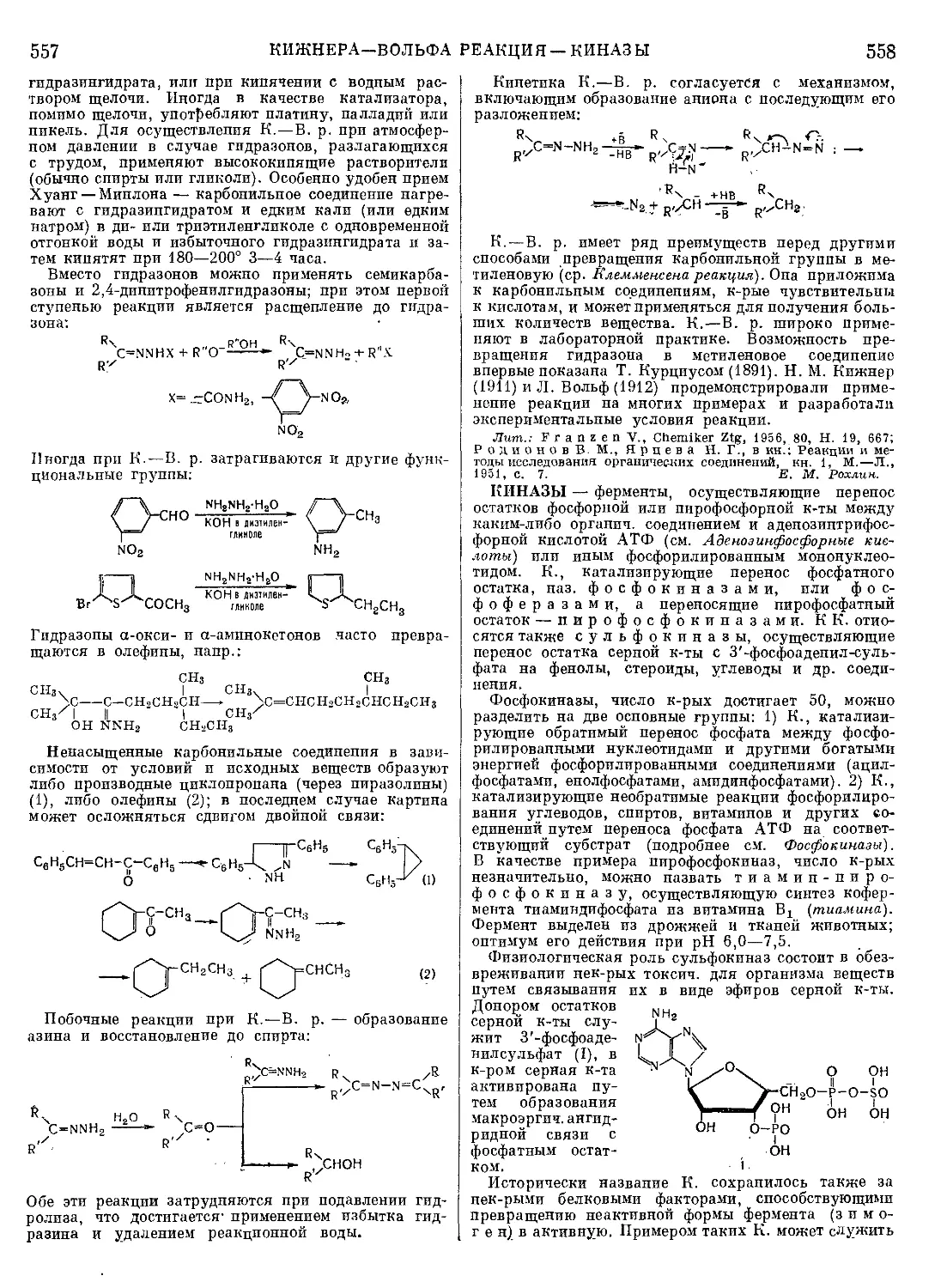








































































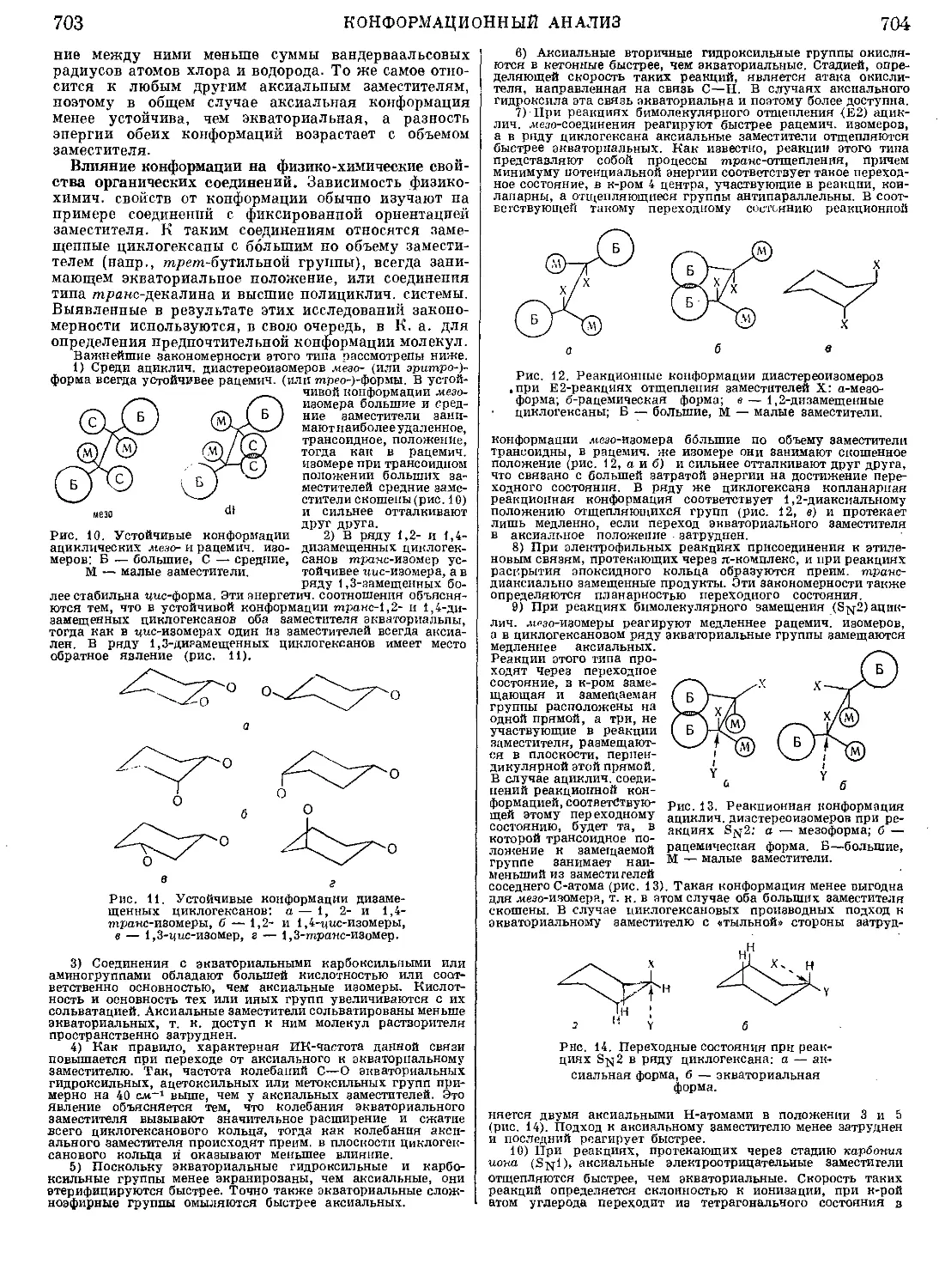










































































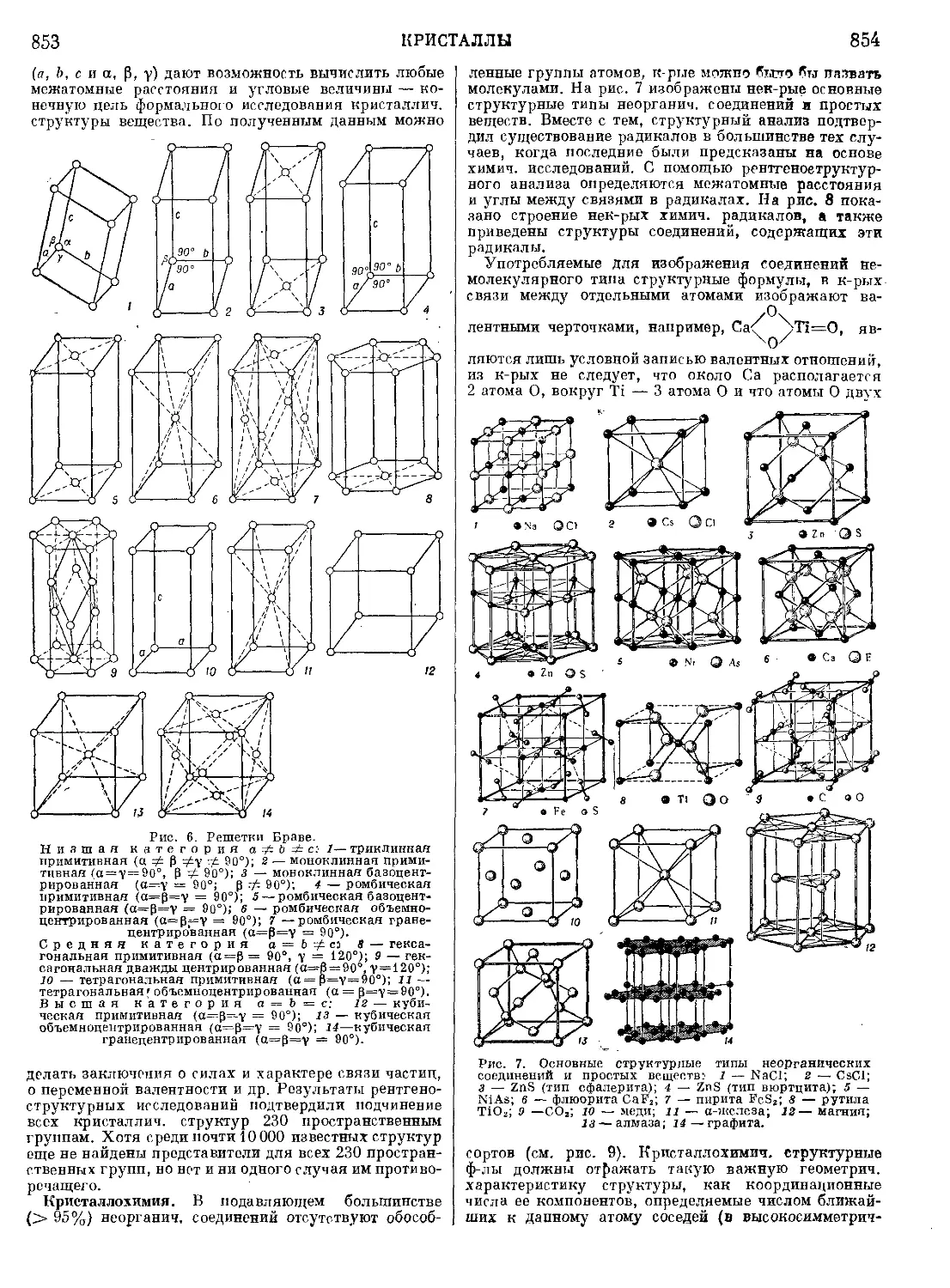























































































































Текст
КРАТКАЯ
ХИМИЧЕСКАЯ
ЭНЦИКЛОПЕДИЯ
РЕДАКЦИОННАЯ КОЛЛЕГИЯ
И. Л. КНУНЯНЦ (главный редактор), Г. Я. БАХАРОВСКИЙ (зам. главно-
го редактора), А. И. БУСЕВ, Я. М. ВАРШАВСКИЙ, Н. И. ГЕЛЬПЕРПН,
В. И. ДОЛИН, В. А. КИРЕЕВ, Г. А. МЕЕРСОН, А. Н. МУРИН, С. А.
ПОГОДИН, П. А. РЕБИНДЕР, Г. Л. СЛОНИМСКИЙ, М. И. ТЕМКИН,
Д. А. ЭПШТЕЙН
2
Малоновый эфир
ГОСУДАРСТВЕННОЕ НАУЧНОЕ ИЗДАТЕЛЬСТВО
«СОВЕТСКАЯ ЭНЦИКЛОПЕДИЯ»
ГОСУДАРСТВЕННОЕ НАУЧНОЕ ИЗДАТЕЛЬСТВО
«СОВЕТСКАЯ ЭНЦИКЛОПЕДИЯ»
ЭНЦИКЛОПЕДИИ
СЛОВАРИ
СПРАВОЧНИКИ
НАУЧНЫЙ СОВЕТ ИЗДАТЕЛЬСТВА
Л П. АЛЕКСАНДРОВ, Д. А. АРЗУМАНЯН, А. В. АРЦИХОВСКИЙ,
Н. В. БАРАНОВ, А. А. БЛАГОНРАВОВ, Н. Н. БОГОЛЮБОВ,
Б. А. ВВЕДЕНСКИЙ (председатель Научного Совета), Б. М. ВУЛ,
Г. II. ГОЛЯКОВ, И. Л. КНУНЯНЦ, Ф. В. КОНСТАНТИНОВ,
Б. В. КУКАРКИН, Ф. Н. ПЕТРОВ, В. М, ПОЛЕВОЙ, А. И. РЕВИН
(заместитель председателя Научного Совета), Н. М. СИСАКЯН,
А. А. СУРКОВ, Л. С. ШАУМЯН (заместитель председателя Научного
Совета)
МОСКВА . 1963
54 (03)
К 78
РЕДАКТОРЫ-КОНСУЛЬТАНТЫ
А. К. БАБКО, В. Н. БЕЛОВ, П. П. БУДНИКОВ, В. Н. БУКИН, А. И. ГОЛЬ-
БИНДЕР, Л. В. ГОРДОН, Б. А. ДОГАДКИН, М. А. ЕЛЬЯШЕВИЧ, Д. Д. ЗЫ-
КОВ, А. В. КИСЕЛЕВ, А. И. КОРОЛЕВ, А. Д. КУЗОВКОВ, О. Ю. МАГИДСОН,
Н. Н. МЕЛЬНИКОВ, И. И. НОВИКОВ, А. Б. ПАКШВЕР, Н. Г. ПУЧКОВ,
А. С. ХОХЛОВ, А. И. ШЕРЕШЕВСКИИ, Н. М. ЭМАНУЭЛЬ, В. А. ЯКОВЛЕВ
РЕДАКЦИЯ КРАТКОЙ ХИМИЧЕСКОЙ ЭНЦИКЛОПЕДИИ
Заведующий редакцией — Г. Я. БАХАРОВСКИИ; научные редакторы —
Д. Н. ВАСКЕВИЧ, Р. В. ГАРКОВЕНКО, 3. И. ГОДИН, Н. И. МОСТОВЕНКО;
редактор —М. Е. ТРУХАНОВА; литературные редакторы —Л. Д. КИРИЛЛОВА,
Ю. А. ГОРЬКОВ, В. Л. КИСЛОВ; младшие редакторы — В. А. ЛЕБЕДЕВА,
В. А. СОЛОМЕННИКОВА
Редактор-библиограф — Е. И. ЖАРОВА
Художественный редактор — И. Н. САХАРОВА
Технический редактор — И. Д. КУЛИДЖАНОВА
Корректор — А. В. МАСЛОВА
Указатель составил — А. Б. ДМИТРИЕВ
ОТ РЕДАКЦИИ
Международный союз чистой и прикладной химии принял (август 1961) новую единую шкалу
атомных весов вместо существовавших ранее двух шкал: физической и химической. Как известно,
основой химической шкалы являлась кислородная единица, равная 1/16 массы атома природного кис-
лорода, состоящего из смеси трех изотопов: О16, О17, О18. В основу физической шкалы была положена
масса изотопа кислорода О16 = 16. В основе новой шкалы лежит значение 12 для массы изотопа угле-
рода С12. Между новой шкалой атомных весов (Ас) и прежней химической шкалой (А ) существует
следующее соотношение: А{ = Ах • 1,000043.
Ниже приводятся таблицы атомных весов по углеродной шкале.
МЕЖДУНАРОДНЫЕ АТОМНЫЕ ВЕСА НА 1962 г.
(по порядковым номерам)
Поряд- ковый номер Название элемента Символ Новое значение Старое значение Поряд- ковый номер Название элемента Символ Новое значение Старое значение
1 Водород н 1,00797 н-0,00001 1,0080 50 51 Олово Сурьма Sn Sb 118,69 121,75 118,70 121.76
2 Гелий Не 4,0026 4,003 52 Теллур Те 127,60 127,61
3 Литий Li 6,939 6,940 53 Иод J 126,9044 126,91
4 Бериллий Be 9,9122 9,013 54 Ксенон Хе 131,30 131.30
5 Бор в 10,811 +0,003 10,82 55 56 Цезий Барий Сз Ва 132,905 137,34 132,91 137,36
6 Углерод с 12,01115 +0,00005 12,011 57 58 Лантан Церий La Се 138,91 140,12 138,92 140,13
7 Азот N 14,0067 14,008 59 Празеодим Рг 140,907 140,92
8 Кислород О 15,9994 +0,0001 16 60 61 Неодим Прометий Nd Pm 144,24 144,27 [147]
9 Фтор F 18,9984 19,00 62 Самарий Sm 150,35 150,35
10 Неон Ne 20,183 20,183 63 Европий Eu 151,96 152,0
11 Натрий Na 22,9898 22,991 64 Гадолиний Gd 157,25 157,26
12 Магний Mg 24,312 24,32 65 Тербий Tb 158,924 158,93
13 Алюминий Al 26,9815 26,98 66 Диспрозий Dy 162,50 162,51
14 Кремний Si 28,086 +0,001 28,09 67 68 Гольмий Эрбий Ho Er 164,930 167,26 164,94 167,26
15 Фосфор p 30,9738 30,975 69 Тулий Tu 168,934 168,94
16 Сера s 32,064 +0,003 32,066 +0,003 70 71 Иттербий Лютеций Yb Lu 173,04 174,97 173,04 174.99
17 Хлор Cl 35,453 35,457 72 Гафний Hl 178,49 178.50
18 Аргон Ar 39,948 39,944 73 Тантал Ta 180,948 180,95
19 Калий К 39,102 39,100 40,08 74 Вольфрам W 183,85 183,86
20 Кальций Ca 40,08 75 Рений Re 186,2 186,22
21 Скандий Sc 44,956 44,96 76 Осмий Os 190,2 190,2
22 Титан Ti 47,90 47,90 77 Иридий Ir 192,2 192,2
23 Ванадий V 50,942 50,95 78 Платина Pt 195,09 195,09
24 Хром Cr 51,996 52,01 79 Золото Au 196,967 197,0
25 Марганец Mn 54,9381 54,94 80 Ртуть Hg 200,59 200,61
26 Железо Fe 55,847 55,85 81 Таллий Ti 204,37 204,39
27 Кобальт Co 58,9332 58,94 82 Свинец Pb 207,19 207,21
28 Никель N1 58,71 58.71 83 Висмут Bi 208,980 209,00
29 Медь Cu 63,54 63,54 84 Полоний Po [210)
30 Цинк Zn 65,37 65,38 85 Астат At [210]
31 Галлий Ga 69,72 69,72 86 Радон Rn [222]
32 Германий Ge 72,59 72,60 87 Франций Fr [223]
33 34 Мышьяк Селен As Se 74,9216 78,96 74,91 78,96 88 89 Радий Актиний Ra Ac [226] [227] 232.05
35 Бром Br 79,909 79,916 90 Торий Th 232,038
36 Криптон Kr 83,80 83,80 91 Протактиний Pa [23Ц
37 Рубидий Rb 85,47 85,48 92 Уран u 238,03 238,07
38 Стронций Sr 87,62 87,63 93 Нептуний Np 237]
39 Иттрий Y 88,905 88,92 94 Плутоний Pu 242]
40 Цирконий Zr 91.22 91,22 95 Америций Am 243]
41 Ниобий Nb 92,906 92,91 96 Кюрий Cm 247]
42 Молибден Mo 95,94 95.95 97 Берклий Bk 247]
43 Технеций Tc [97] 98 Калифорний Ct 249]
44 Рутений Ru 161,07 101,1 99 Эйнштейний Es 254]
45 Родий Rh 102,905 102,91 100 Фермий Fm 253]
46 Палладий Pd 106,4 106,4 101 Менделевий Md 256]
47 Серебро Ag 107,870 107,880 102 Нобелий No 1255]
48 49 Кадмий Индий Cd In 112,40 114,82 112,41 114,82 103 Лауренсий Lw
МЕЖДУНАРОДНЫЕ АТОМНЫЕ ВЕСА НА 1962 г.
(по алфавиту)
Название Сим- Поряд- Атомный Название Сим- Поряд- Атомный Название Сим- Поряд- Атомный
элемента вол номер вес элемента вол номер вес элемента вол номер вес
Азот N 7 14,0067 Кислород О 8 15,9994 Рений Re 186,2
Актиний Ас 89 [227] Кобальт Со 27 58,9332 Родий Rh 45 102,905
Алюминий А1 13 26,9815 Кремний Si 14 28,086 Ртуть Hg 80 200,59
Америций Ат 95 [243] Криптон Кг 36 83,80 Рубидий Rb 37 85,47
Аргон Аг 18 39,948 Ксенон Хе 54 131,30 Рутений Ru 44 101,07
Астат At 85 [210] Кюрий Ст 96 [247] Самарий Sm 62 150,35'
Барий Ва 56 137,34 Лантан La 57 138,91 Свинец Pb 82 207,19
Бериллий Be 4 9,0122 Лауренсий Lw 103 Селен Se 34 78,96
Беркелий Bk 97 [247] Литий Li 3 6,939 Сера s 16 32,064
Бор В 5 10,811 Лютеций Lu 71 174,97 Серебро Ag 47 107,870
Бром Вг 35 79,909 Магний Mg 12 24,312 Скандий Sc 21 44,956
Ванадий V 23 50,942 Марганец Mn 25 54,9381 Стронций Sr 38 87,62
Висмут Bi 83 208,980 Медь Cu 29 63,54 Сурьма Sb 51 121.75
Водород н 1 1,00797 Менделевий Md 101 [256] Таллий T1 81 204,37
Вольфрам W 74 183,85 Молибден Mo 42 95,94 Тантал Ta 73 180,948
Гадолиний Gd 64 157,25 Мышьяк As 33 74,9216 Теллур Те 52 127,60
Галлий Ga 31 69,72 Натрий Na и 22.9898 Тербий Tb 65 158,924
Гафнии щ 72 178,49 Неодим Nd 60 144,24 Технецкий TC 43 [97]
Гелий Не 2 4,0026 Неон Ne 10 20,183 Титан Ti 22 47,90
Германий Ge 32 72,59 Нептуний Np 93 [237] Торий Th 90 232,038
Гольмий Но 67 164,930 Никель Ni 28 58,71 Тулий Tu 69 168,934
Диспрозий Dy 66 162,50 Ниобий Nb 41 92,906 Углерод c 6 12,01115
Европий Ей 63 151,96 Нобелий No 102 [255] Уран и 92 238,03
Железо Fe 26 55,847 Олово Sn 50 118,69 Фермий Fm 100 [253]
Золото Au 79 196,967 Осмий Os 76 190,2 Фосфор p 15 30,9738
Индий In 49 114,82 Палладий Pd 46 106,4 Франций Fr 87 [223]
Иод J 53 126,9044 Платина Pt 78 195,09 Фтор F 9 18,9984
Иридий Ir 77 192,2 Плутоний Pu 94 [242] Хлор Cl 17 35,453
Иттербий Yb 70 173,04 Полоний Po 84 [210] Хром Cr 24 51,996
Иттрий Y 39 88,905 Празеодим Pr 59 140,907 Цезий Cs 55 132,905
К адмий Cd 48 112,40 Прометий Pm 61 [147] Церий Ce 58 140,12
Калий К 19 39,102 Протактиний Pa 91 [231] Цинк Zn 30 65,37
Калифорний Cf 98 [249] Радий Ra 88 [226] Цирконий Zr 40 91,22
Кальций Ca 20 40,08 Радон Rn 86 [222] Эйнштейний Es 99 [254]
Эрбий Er 68 167,26
ДОПОЛНИТЕЛЬНЫЙ СПИСОК НЕКОТОРЫХ ПЕРИОДИЧЕСКИХ ИЗДАНИЙ
Вестн. техн, и экон, информации—Вестник технической и
экономической информации (СССР)
Изв. Высш. уч. зав. Хим. и химия, технол .— Известия
Высших учебных заведений СССР. Химия и химическая
технология
Гласи, хем. друш. — Гласник хемическог друштва (Юго-
славия)
Химия и индустрия (Болгария)
Acta biochim. Polon. — Acta Biochimica Polonica (Поль-
ша)
Akta Chim. Akad. Scie. Hung. — Akta Chimica Akade-
miaae Scientiarum Hungaricae (Венгрия)
Arch. Kem. — Archiv za Kemiju (Югославия)
Bull. Acad. Polon. Sci. Ser. Chim. — Bulletin de TAcademie
Polonaise de Sciences. Sdrie de Sciences Chimiques (Поль-
ша)
Bull. Chem. Soc. Japan — Bulletin of the Chemical Society
of Japan (Япония)
Chem. Anal. — Chemia Analityczna (Польша)
Chem. prdmysl — Chemicky prdmysl (Чехословакия)
Chem. stosow. — Chemia stosowana (Польша)
Chem. — Chernik (Польша)
Finska kemist. medd. — Finska kemistamfundets medde-
1 an den (Финляндия)
Koll. Chechosl. chem. commun. — Koilection of Chechos-
lovak Chemical Communication (Чехословакия)
Magyar Kem. fol. — Magyar Kdmiai foJydirat (Венгрия)
Magyar kdm. lap. — Magyar kcmiai lapja (Венгрия)
Post, biochem. — Postepy biochemii (Польша)
Przem. Chem. — Przemysl Chemizny (Польша)
Rev. chim. — Revista de Chimie (Румыния)
Roczn. chem. — Roczniki Chemii (Польша)
Sci. abs. Chem. a Chem. Techn. — Science abstracts of
China. Ser. Chemistry and Chemical Technology (Китай)
Suomen Kemist. — Suomen Kemistilehti (Финляндия)
Stud, cercet. biochim. — Studii §i cercStari de biochimie
(Румыния)
Stud, cercet. chim. — Studii §i cercetari de chimie (Румыния)
Wadom. Chem. — Wiadomsci Chemiczne ( Польша)
ЖАРОПРОЧНОСТЬ — свойство конструкционного
материала сохранять высокую сопротивляемость пла-
стин. деформированию при значительном повышении
темп-ры. В связи с бурным развитием новой высоко-
температурной техники (газовые турбины, реактивные
двигатели, ракетные установки и т. п.) Ж. становится
одним из важнейших свойств, определяющих эксплуа-
тационные качества материала. Для оценки Ж. мате-
риала пользуются различными условными характе-
ристиками. Наиболее употребительными из них
являются: 1) предел ползучести в кГ /мм2 — напряже-
ние, вызывающее суммарную деформацию в 1% за
определенное время, напр. за 1000 часов. Обозначе-
ние: а™0 , где ос — предел ползучести; дробный
Ч юоо
индекс 1/1000 показывает деформацию в процентах
и время в часах, верхний индекс 700 указывает
температуру испытания; 2) длительная прочность
в кГ/мм2 — напряжение, вызывающее разрушение
при заданной темп-ре за определенное время. Обозна-
чение: где <т9 — длительная прочность, ниж-
ний индекс — время до разрушения в часах, верх-
ний индекс — темп-pa испытания.
Чистые металлы, как правило, резко снижают свои
прочностные характеристики с повышением темп-ры
в результате возрастания тепловой подвижности ато-
мов и связанного с этим более легкого перемещения
дислокаций в решетке [см. Дислокации (в кри-
сталлах)]. Так, например, критич. скалывающее
напряжение монокристаллов кадмия снижается в 5—
6 раз при повышении темп-ры с —196° до 250°,
а предел текучести монокристаллов цинка в том же
интервале темп-p снижается в 4—5 раз. В поликри-
сталлич. структурах с ростом темп-ры также облег-
чается деформация в отдельных зернах, но, кроме
того, возникает относительное перемещение зерен,
быстро приводящее к разрушению металла. Вместе
с тем в поликристаллич. металлах, находящихся под
напряжением, при определенной темп-ре (равной
примерно 0,4 абс. темп-ры плавления) развивается
самопроизвольный процесс рекристаллизации — пе-
рестройка кристаллитной структуры, способствующий
резкому возрастанию скорости пластич. течения.
Сплавы оказывают более высокое сопротивление
пластич. деформации при повышенных темп-pax, т. к.
наличие в решетке чужеродных атомов создает поле
упругих напряжений, препятствующее перемещению
дислокаций. Кроме того, нек-рые присадки к основ-
ному металлу укрепляют границы зерен и затрудняют
процесс рекристаллизации, что также повышает жа-
ропрочность,-
Простейшая классификация жаропрочных материа-
лов м. б. дана на основе тех рабочих темп-p, для к-рых
предназначены эти материалы. Для темп-p до 350° —
обычные конструкционные углеродистые стали и
алюминиевые сплавы; для 350—500° — слабо ле-
гированные стали ферритного и перлитного классов
и титановые сплавы; для 500—650® — стали аусте-
нитного класса и для 650—800° — сплавы на никеле-
вой и кобальтовой основе. Обычные конструкционные
углеродистые стали, обработанные на высокую проч-
ность (закалка и низкий отпуск), не обнаруживают
ползучести и успешно работают при темп-pax до
350°. Стали ферритного и перлитного классов, леги-
рованные хромом, молибденом или кремнием, приме-
няют в котлостроении, в установках для крекинга
нефти, для клапанов двигателей внутреннего сгора-
ния и т. д.
Наиболее распространены следующие марки этих
сталей (табл. 1).
Таблица 1
Марка стали Состав стали в %
с Ст S1 Мо
15ХМ 0,1—0,2 0,8—1,1 0,4 0,3—0,6
Х6М 0,15 5,0-6,5 0,5 0,4—0,6
СХ8 0,35—0,45 9,0—10,5 1,9-2,6 —
СХ8М 0,35-0,5 8,0—9,5 2,2—3,2 0,7—0,9
СХ12 0,4—0,5 10,0—12,0 3,4-4,7 —
Первые две марки представляют собой типичные
малолегированные жаропрочные стали для котло-
турбостроения. Три последние марки, имеющие общее
наименование сильхромы, применяют в произ-ве дви-
гателей внутреннего сгорания. Аустенитные жаро-
прочные стали применяют для изготовления лопаток
газовых турбин, деталей реактивных двигателей и т. п.
Наиболее типичные марки этих сталей приведены
в табл. 2.
Таблица 2
Марка Состав стали в %
стали с S1 Мп Сг N1 W Мо 11 Nb
Я1 <0,14 < 1,2 <0,7 16—20 8-11
Я1Т <0,14 <1,2 <0,7 16-20 8—11 — 0,8
Я1МО <0,07 <0,9 <0,7 18 10 — 2,5 —
Я1Н6 < 1,14 < 1,0 < 1,5 18 10 — 1,5
ЭИ69 0,4-0,5 <0,8 <0,7 13-15 13-15 2—2,7 ОД—0,4 —-
ЭИ257 <0,2 <0,2 <0,8 <0,7 14 14 2,5 0,3 — —
ЭИ123 <0,8 <0,7 14 14 2,5 1,5 —
Тимке- <0,1 <0,8 <0,7 16 25 — 6 — —
наллой Тинидур <0,1 <0,8 <0,7 15 30 — — 2 —
Все эти аустенитные стали содержат аустенит и
карбиды. В результате термообработки карбидная
фаза составляет от 4 до 7% всего объема металла.
Ж. этих сталей существенно зависит от темп-ры за-
калки. Наибольшая Ж. достигается при закалке от
1150—1200°.
Никелевые и кобальтовые сплавы применяются для
изготовления лопаток турбин реактивных двигателей
и других деталей, требующих нагрева до 800—900®
15
ЖАРОПРОЧНОСТЬ — ЖЕЛАТИНА
16
при наличии напряженного состояния. Составы не-
которых применяемых для этой цели сплавов приве-
дены в табл. 3.
Таблица 3
Группа Марка сплава Состав сплава в %
С Сг NI Со TI А1 | Мо | Fe
Никелевая ХН80 (нихром) ХН75 (инконель) ХН80Т (нимоник) 0,1 0,1 0,1 20 15 20 80 75 76 — 3 1 — 10
Кобальтовая Виталлиум 0,25 27 3 - —
Экспериментально были найдены присадки, повы-
шающие Ж. различных металлов (за исключением
стали и никеля, о к-рых сказано выше). Для меди
такими присадками являются никель с кремнием,
хромом или кобальтом; для алюминия — медь, никель
или магний; для олова и его сплавов — мышьяк кад-
мий и для цинка и его сплавов — магний или медь.
Нек-рые общие положения, к-рым должны удовлет-
ворять все жаропрочные сплавы независимо от их
состава и структуры, сводятся к следующим. Для
машин и аппаратов, работающихпри высоких темп-рах,
наиболее подходящими являются сплавы с относи-
тельно крупным зерном и совершенно свободные от
остаточных напряжений; сплав должен быть по воз-
можности однородным. Поэтому, как правило, литые
детали обнаруживают большее сопротивление дефор-
мирующим силам при повышенных темп-рах, чем
детали, предварительно прошедшие холодную обра-
ботку. Особенно это справедливо при рабочих
темп-рах, близких к плавлению сплава. В связи с этим
детали, предназначенные для работы при высоких
темп-рах и подвергшиеся механич. обработке, должны
пройти предварительный отжиг при темп-ре, превы-
шающей эксплуатационную.
В нек-рых случаях жаропрочные сплавы подвер-
гают дополнительной обработке — нанесению на их
поверхность жаростойкого, т. е. стойкого
по отношению к газовой коррозии при высоких
темп-рах металла или сплава. Материалом длн таких
покрытий служат кремний (силицирование), алюми-
ний (алитирование), хром (хромирование) или титан
(титанирование). В нек-рых случаях используются
комбинированные покрытия из нескольких элемен-
тов. Достигаемая при этом защита жаропрочного мате-
риала значительно уменьшает потери от газовой
коррозии и предохраняет его от образования корро-
зионных трещин, быстро приводящих к разрушению
уже при весьма малых напряжениях (см. Коррозия
металлов).
Важной группой жаропрочных материалов являются
керметы. Сравнительно высокой Ж. обладают неорга-
нич. полимерные материалы на основе кремния.
Значительное распространение в различных отрас-
лях пром-сти, связанных с использованием высоких
темп-p, получил неметаллич. жаропрочный материал,
т. н. жаростойкий бетон. Он используется
в качестве футеровки обжиговых печей, в облицовке
и изоляции паровых котлов, кожухов регенераторов
мартеновских печей, в футеровке реакционных печей
химич. пром-сти и т. п. Жаростойкий бетон состоит
в основном из заполнителя и связки, а в нек-рых слу-
чаях и тонкомолотой минеральной добавки (кварцевый
песок, шамот, доменный шлак и др.), уменьшающей
падение прочности бетона при нагревании.
Связующим в жаростойком бетоне служат цементы:
портландский, шлако-портландский, пуццолановый,
глиноземистый, высокоглиноземистый, доломитовый
водоустойчивый, периклазовый, бариево-моноалюми-
натный, а также жидкое стекло с добавкой кремне-
фтористого натрия. Размер зерен заполнителей в раз-
ных бетонах колеблется от 2—3 до 20—35 мм и дости-
гает иногда 40—60 мм. Наиболее распространены
следующие комбинации заполнителей и цемента:
шамотный или шамотно-каолиновый заполнитель (для
рабочих темп-p не более 1100—1200°) на портландце-
менте; хромитовый порошок с портландским (до 1300°)
или глиноземистым (1400°, иногда 1500°) цементом,
хромомагнезитовый заполнитель с глиноземистым
цементом (1600°), высокоглииоземистый шамот с высо-
коглиноземистым цементом (до 1400—1700°).
Лит.: Гуляев А, П., Металловедение, 2 изд., М.,
1951; Фридман Я. Б., Механические свойства металлов,
2 изд., М., 1952; Мортон К. Смит, Основы физики ме-
таллов, пер. с англ., М., 1959; Кузнецов В. Д., Физика
твердого тела, т. 2, [Томск], 1941; Сб. научных докладов по
теории жаропрочности, М., 1961; Бернштейн М. Л.,
Стали и сплавы для работы при высоких температурах, М.,
1956; Будников П. П. [идр.], Технология керамики
и огнеупоров, М., 1954; Некрасов К. Д., Жароупорный
бетон, М., 1957; Технология и свойства жароупорных бетонов,
М., 1959 (Тр. и.-и. ин-та бетона и железобетона, вып. 7):
Корнилов И. И., Физико-химические основы жаропроч-
ности сплавов, М., 1961.
ЖАСМОН СпН1вО, мол. в. 164,24 — бесцветная
жидкость с запахом жасмина; т. кип. 134—135°/12 мм;
df 0,9437; ng 1,4979. Ж. дает
реакции, характерные для
кетонов. Ж. выделен из ма- ['-''4*Ч-сн2СН=СНСН2СНа
ела цветов жасмина. Синтезы |_____Lq
Ж. ввиду чрезвычайной гро-
моздкости, малых выходов и труднодоступное™ исход-
ного сырья не имеют промышленного значения.
Ж. можно применять в парфюмерных композициях
вместо натурального жасминного масла, но ввиду ма-
лой доступности в пром-сти используют его аналог,
т. н. дигидрожасмон, обладающий схожим запахом.
А. А. Зеленецкая.
ЖЕЛАТИНА — продукт переработки коллагена,
распространенного в природе белкового вещества,
образующего главную составную часть соединитель-
ной ткани позвоночных, особенно в коже, оссеине
костей и в сухожилиях. По аминокислотному и эле-
ментарному составу Ж. близка к коллагену. Глав-
нейшие к-ты: глицин (ок. 27%), пролин (ок. 16%),
оксипролин (ок. 14%), глутаминовая к-та (ок. 12%),
аргинин (ок. 9%), лизин (ок. 5%). Элементарный
состав Ж.: 48,7—51,5% С; 6,5—7,2% Н; 17,5—18,8%
N; 24,2—26,8% О; 0,3—0,7% S. В Ж. ок. 15% Н2О
и ок. 1% золы. Лучшие сорта Ж. слабо окрашены
в желтый цвет; d 1,3—1,4; nD 1,5; средний мол. в.
ок. 60000; благодаря наличию в Ж. кислых (карбок-
сильных) и основных (амино) групп она имеет амфо-
терный характер. Ж., полученная но «щелочному»
способу, имеет изоэлектрич. точку при pH 4,8—5,1,
а полученная по «кислотному» способу — при pH
ок. 9. Ж. набухает в ноде и при нагревании раство-
ряется; при охлаждении р-р Ж. образует студень
(гель), к-рый при нагревании опять переходит в р-р.
Темп-pa застудневания и прочность студня зависят
от концентрации р-ра и качества Ж. Основными кри-
териями качества Ж. являются вязкость р-ра, проч-
ность студня, темп-pa его плавления и застуднева-
ния, измеренные при определенных условиях. В конц.
р-рах нек-рых веществ (наир., роданистого калия,
бензолсульфоната натрия и др.) Ж. растворяется на
холоду. Эти же вещества препятствуют образованию
студня. Под действием дубителей Ж. теряет способ-
ность набухать в воде и растворяться.
Основным сырьем для произ-ва Ж. служат кости
крупного рогатого скота, отходы кожененного про-
из-ва (обрезки шкур, мездра) и сухожилия. В Японии
для этой цели с недавнего времени применяют также
17
ЖЕЛАТИНИРОВАНИЕ — ЖЕЛЕЗА ОКИСЛИ И ГИДРООКИСИ
18
содержащие коллаген отходы китобойного промысла.
Ж. может быть получена из кожи, чешуи и плавательных
пузырей рыб. Такая Ж., однако, дает слабый студень
и используется только как клей. Из костей сначала
получают оссеин; для этого, после их обезжщэива-
ния горячей водой или органич. растворителями (бензи-
ном, дихлорэтаном), удаляют всю минеральную часть
(углекислый и фосфорнокислый кальций) 4—6%-ным
р-ром соляной к-ты. Отработанную к-ту используют
для получения двузамещенного фосфата кальция
(преципитата). Для последующей обработки полу-
ченного т. обр. оссеина применяют при нормальной
темп-ре известковое молоко. Для оссеина этот про-
цесс (золка) длится один-два месяца, для кожевенного
сырья — до шести месяцев. После золки сырье нейтра-
лизуют соляной или фосфорной к-той до pH ок. 6 и
промывают. Подготовленное сырье варят при 55—60°
до тех пор, пока концентрация Ж. в р-ре (бульоне)
не достигнет 5—7%. Затем бульон сливают и сырье
заливают новой порцией воды. Так поступают нес-
колько раз, пока из сырья не будет извлечена вся Ж.
Для каждой последующей порции воды темп-ру варки
повышают на 5—10°. Лучшие сорта Ж. (фотографи-
ческую, пищевую) получают из первых двух-трех
бульонов. Бульоны фильтруют, упаривают в вакуум-
аппаратах, а затем студенят и студни сушат. В послед-
нее время начинает распространяться «кислотный»
способ произ-ва Ж. При этом исключена золка. Сырье
обрабатывают короткое время раствором к-ты для
снижения pH прибл. до 4. В этих условиях варка
идет быстро и при умеренных темп-рах.
В зависимости от степени чистоты и качества разли-
чают фотография., пищевую и техник. Ж. Первую при-
меняют в произ-ве фото- и кинопленок, фотопласти-
нок и фотобумаги. Пищевую Ж. используют в кули-
нарии, в кондитерском деле, в виноделии и пивоваре-
нии; техническую — в бумагоделательной, полигра-
фия. и в др. отраслях пром-сти. Ж. применяется также
в медицине (напр., как кровеостанавливающее сред-
ство) и в качестве питательной среды для культиви-
рования бактерий.
Лит.: Коваль В. Д., Производство желатины, М.,
1951; Белки, под ред. Г. Нейрата и К. Бэйли, пер. с англ.,
т. 1—3, М., 1956—59; Sauer Е., Chemie und Fabrikation
der tlerischen Leime und der Gelatine, B., 1958.
B.A. Бекунов.
ЖЕЛАТИНИРОВАНИЕ — см. Застудневание (рас-
творов высокомолекулярных соединений).
ЖЕЛЕЗА КАРБИДЫ — см. Железа сплавы.
ЖЕЛЕЗА НИТРАТЫ — азотнокислые соли 2- и
3-валентного железа. Нитрат 2-валентно-
го железа (ферронитрат) из водных р-ров при
темп-рах от —28° до —12° кристаллизуется в виде
Fe(NO3)2-9H2O, выше —12°—в виде Fe(NO3)2-6H2O —
ромбич. светло-зеленых кристаллов, плавящихся
с разложением при 60,5°. Растворимость в воде
81,8 г на 100 г Н2О при 20°. Образуется при действии
разб. HNO3 (с плоти, ниже 1,034) на железо. Нит-
рат 3-в алентного железа (ферринит-
рат) известен в форме кристаллогидратов с 9 и 6 моле-
кулами воды; Fe(NO3)3 • 9Н2О — бледно-фиолетовые
моноклинные призмы, плоти. 1,684 (21°), т. пл.
47,2°, при нагревании выше 50° разлагается;
Fe(NO3)3 • 6Н2О — прозрачные кристаллы кубич. фор-
мы, т. пл. 35—40°. Обе соли гигроскопичны и на воз-
духе расплываются, хорошо растворимы в воде
[87,3 г Fe(NO3)3 • 9Н2О в 100 г воды при 25°]. Р-ры
сильно гидролизованы, из конц. р-ров осаждаются
основные соли. В технике Ж. н. получают действием
HNO3 с плоти, ок. 1,115 на железные стружки или
обрезки кровельного железа. Из насыщенного р-ра
выпадает смесь 6- и 9-водного кристаллогидратов.
Применяется как протрава при крашении хлопчатобу-
мажных тканей и как утяжелитель шелка.
ЖЕЛЕЗА ОКИСЛЫ И ГИДРООКИСИ — соедине-
ния 2- и 3-валентного железа с кислородом; закись
FeO, закись-окись Fe3O4 и окись Fe2O3 и соответствую-
щие гидроокиси Fe(OH)2 и Fe(OH)3. Об образовании
кислородных соединений железа при его окислении
в различных условиях см. Железо. Свободная желез-
ная к-та H2FeO4 и отвечающий ей ангидрид •— трех-
окись железа (FeO3) не получены. Выделены соли же-
лезной кислоты — ферраты, общей формы Me2FeO4
(где Me — одновалентный металл), — исключительно
сильные окислители.
Железа закись FeO — черные кристал-
лы, кубич. решетка с периодом а = 4,299 А, плоти.
5,7; т. пл. 1368°. Теплота образования ДЯ°2в8 =
= —63,7 ккал/моль. В воде практически нерастворима,
хорошо растворима в кислотах. Легко окисляется,
пирофорна; после прокаливания становится химиче-
ски менее активной. Закись железа получают восста-
новлением Fe,O3 водородом или нагреванием окса-
лата железа FeC2O4 • 2Н2О в атмосфере азота; продукт
реакции содержит примеси Fe и С. При действии
щелочей на соли Fe (П) выпадает гидроокись
2-валентного железа Fe(OH)2 в отсутствии О2 в виде
объемистого бледно-зеленого осадка. При соосаждении
с изоморфной Mg(OH)2 может быть получена кристал-
лин. форма Fe(OH)2 — бледно-зеленые кристаллы,
гексагональная решетка, а = 3,24 А, с = 4,47 А;
плоти. 3,4. Теплота образования ДЯ°2в8 — — 135,8
ккал/молъ. Легко окисляется до Fe(OH)3. При 150—
200° разлагается: 4Fe(OH)2 = Fe 4- Fe3O44- 4Н2О.
В воде Fe(OH)2 трудно растворима. Легко раствори-
ма в кислотах и р-ре NH4C1.
Железа закис ь-о к и с ь Fe3O4— черные
кристаллы, кубич. решетка, а => 3,380 А, плоти.
5,2, ферромагнитна. При 1538° разлагается. Теплота
образования ДЛ°298 = —267,0 ккал/молъ. Раство-
ряется в кислотах, образуя смесь солей Fe2+ и Fe3+.
При нагревании на воздухе окисляется до Fe2O3.
Закись-окись железа получают действием водяного
пара на раскаленный металл, частичным окислением
закисных соединений, восстановлением окиси железа
закисью или металлом. Природная закись-окись желе-
за (магнетит) служит сырьем для получения железа.
Железа окись Fe2O3 существует в трех
модификациях: парамагнитная a-Fe2O3 и ферро-
магнитные y-Fe2O3 и 6-Fe2O3. Наиболее устой-
чива a-Fe2O3 — кристаллы от темно-красного до
черно-фиолетового цвета, гексагональная решетка
с параметрами а = 5,025 А, с = 13,735 А и а= 55°14'
и плоти. 5,25. Теплота образования ДЯ°2в8 =
= —196,5 ккал/молъ. Плавится при 1562° (с разл.).
y-Fe2O3 — коричневые кристаллы кубич. системы,
а = 8,339 А, плоти. 4,69. Как а-, так и y-Fe2O3
образуются на поверхности железа при его окис-
лении. Модификация 6-Fe2O3 может быть получена
при окислении р-ров солей железа в щелочном р-ре.
Окись железа растворима в кислотах. Может быть
получена прокаливанием Fe(OH)3 или железного
купороса, окислением FeS2 и др. способами. Природ-
ная a-Fe2O3 — гематит, служит сырьем для получе-
ния железа.
Железа (Ш) гидроокись Fe(OH)3—
красно-коричневые кристаллы, кубич. решетка, а =
= 5,71, плоти. 3,4—3,9. Теплота образования кристал-
лич. модификации Д//°298 = —197,0 ккал/молъ. Рас-
творимость при 18° 0,000048 г в 100 г воды; хорошо
растворима в кислотах. Fe(OH)3, осажденная щелочью
из р-ра Fe (Ш), представляет собой гидрогель; гид-
розоль получают добавлением гидрогеля к раство-
ру FeCl3. В пром-сти Fe(OH)3 получают взаимодей-
ствием FeCI2 с мелом и окислением осадка воздухом
или непосредственным окислением FeCO3 воздухом.
Fe(OH)3 обладает слабо выраженной кислотной функ-
19
ЖЕЛЕЗА СПЛАВЫ
20
звуковых лент.
19
21
23
25
26
29
30
33
цией. Соответствующие соли — ферриты, ана-
логичны по составу алюминатам и производятся от
одноосновной железистой к-ты HFeO2, не выделенной
в свободном состоянии. Известны ферриты щелочных,
щелочноземельных и нек-рых др. металлов. Интересно,
что отдельные, сходные по соста-
ву ферриты обладают различ-
ными структурами кристаллов
и различными магнитными свой-
ствами.
Поскольку окислы железа яв-
ляются основным сырьем по-
лучения железа, представляет
большой интерес их термич. дис-
социация. Темп-pa диссоциации
FeO (pg, = 0,21 атм) равна2500°.
Давление О2 над FeO при 750°
равно 1,602 • 10-17 мм рт. ст., а
при 950°— 1,49 • 10-11 мм рт. ст.
Fe2O3 диссоциирует при не очень
высоких темп-рах (1350—1500°).
Давление О2 над Fe2O3 при 427°
равно 2,241 • 10-26 мм рт. ст.
Давление О2 над Fe3O4 при 725°
равно 4,25 • 10~17 мм рт. ст.
Окислы и гидроокиси железа
применяют в качестве пигмен-
тов (см. Пигменты железоокис-
ные). Ферромагнитные окислы
железа, а также ферриты приме-
няют в электротехнике, в част-
ности в производстве магнитных звуковых лент.
Лит.: П озин М. Е., Технология минеральных солей,
2 изд., Л., 1961; см. лит. при ст. Железо.
ЖЕЛЕЗА СПЛАВЫ. Содержание:
I. Система железо—углерод ...
II. Углеродистые стали.....
III. Чугуны ...............
IV, Легированные стали.....
V. Конструкционные стали ....
VI. Инструментальные стали . . .
VII. Стали с особыми свойствами
VIII. Ферросплавы ...........
Железа сплавы — металлич. сплавы на основе
железа. До начала 19 в. к Ж. с. относили преимущ.
сплавы Fe — С (с примесями Si, Мп, S, Р), получившие
название сталей и чугунов. Все возрастающие требо-
вания техники к металлич. материалам, прежде всего
в отношении их механич. свойств, жаропрочности,
коррозионной стойкости в различных агрессивных
средах, привели к созданию новых Ж. с., содержащих
Ci, Ni, Si, Mo, W и др. В настоящее время к Ж. с.
относят: углеродистые стали, чугуны, легированные
стали, содержащие, кроме С, другие элементы, и
стали с особыми физико-химич. и механич. свойствами.
Кроме того, в черной металлургии для введения в сталь
легирующих элементов применяются особые Ж. с.,
получившие название ферросплавов. Значение Ж. с.
для современной техники следует из того, что 95%
всей металлич. продукции составляет сталь и чугун
и только 5% — сплавы цветных металлов.
I. Система железо—углерод. Наиболее изучена
важнейшая для практики часть системы Fe—С от
0 до 6,67% С* (рис. 1). В этой области за компоненты
системы можно принять Fe и Fe3C — карбид железа,
или цементит. Железо может находиться в двух кри-
сталлич. модификациях: a-Fe (феррит) с объемно-
центрированной кубич. решеткой и y-Fe (аустенит)
с гранецентрированной кубич. решеткой. a-Fe устой-
чиво в двух интервалах темп-p: ниже 910° и от 1400°
до 1539° — темп-ры плавления Fe. Диаметр наиболь-
ших пор решетки a-Fe составляет 0,62 А, что, по-види-
Здесь и далее указаны весовые проценты.
мому, недостаточно для размещения в этих порах ато-
мов С. Наблюдающаяся весьма малая растворимость С
в a-Fe (не более 0,04%) в основном связана с наличием
дефектов решетки, особенно по границам зерен. у-Fe
устойчиво в интервале 910—1400°. В y-Fe диаметр
Рис. 1. Диаграмма равновесия системы железо—углерод.
наибольших пор составляет 1,02 А, что дает возмож-
ность атомам С разместиться в этих порах, но при этом
параметр решетки увеличивается и тем в большей мере,
чем выше концентрация С. Максимальная раствори-
мость С в y-Fe ок, 2,0% (1130°).
Карбид железа FegC наиболее тверд (Нъ >
> 800 кГ/мм2) и наиболее хрупок из всех фаз и струк-
турных составляющих системы Fe—Fe3C. По твердости
Fe3C занимает среднее положение между корундом и
алмазом. Кристаллич. структура Fe3C очень сложна
и может быть представлена рядом октаэдров, оси
к-рых расположены друг к другу под нек-рыми углами.
В вершинах октаэдров расположены атомы Fe, а внут-
ри каждого октаэдра находится атом С. Плотность
Fe3C равна 7,82. Ок. 1100° цементит распадается
с выделением графита. При обычной темп-ре Fe3C
обладает слабыми ферромагнитными свойствами, исче-
зающими при 217°. Цементит способен образовывать
твердые р-ры замещения: атомы углерода могут заме-
щаться азотом, кислородом, а атомы железа — мар-
ганцем, хромом, вольфрамом и др. элементами. В рас-
плавленном состоянии Fe и С смешиваются (до 6,67%С)
во всех отношениях. Прибавление С к Fe понижает
темп-ру ликвидуса до 1130°, что соответствует эвтек-
тич. точке С при 4,3% С. Далее линия ликвидуса
повышается до точки D, отвечающей темп-ре плавле-
ния FegC (табл. 1).
Таблица 1
Точки Темп-ра, °C Вес. % С Точки Темп-ра, °C Вес. % С
А 1539 0,0 D 1600 6,67
В 1485 0,5 G 910 0,0
н 1485 0,1 Р 723 0,04
I 1485 0,16 8 723 0,8
N 1400 0,0 К 723 6,67
Е ИЗО 2,0 Q 600 0,01
С ИЗО 4,3 L 600 6,67
F ИЗО 6,67 м 768 0,0
Линия ABCD является ликвидусом (L) системы,
линия AHIECF — солидусом (S). При содержании
21
ЖЕЛЕЗА СПЛАВЫ
22
Вес. % С
Структура
1. От 0 до 0,01
2. От 0,01 до 0,03
Феррит: a-Fe (С) — твердый р-р углерода в железе
Феррит и третичный цементит;
a-Fe (С) Fe3CJn
феррит цементит
третичный
Феррит, третич. цементит и перлит;
a-Fe (С) + Fe3Cm + [а-Fe (С) + Fe3C]
феррит цементит феррит цементит
третичный перлита перлита
перлит
Вторичный цементит и перлит;
FesC„+ [a-Fe (С) + Fe3C]
цементит феррит цементит
вторичный перлита перлита
перлит
Перлит, вторичный цементит и ледебурит?
[a-Fe (С) + Fe3C] + Fe3CM + {[а-Fe (С) + Fe3C] + Fe3C}
перлит цементит перлит — продукт цементит
вторичный распада аустенита эвтектики
продукт'распада ледебурит
аустенита
Первичный цементит и ледебурит;
Fe3Cj -J- {[a-Fe (С) -J- Fe3Cj -J- F5C}
цементит перлит — продукт цементит
первичный распада аустенита эвтектики
ледебурит
3. От 0,03 до 0,8
4. От 0,8 до 2,0
5. От 2,0 до 4,3
6. От 4,3 ДО 6,67
1—4—углеродистые стали; 5—доэвтектические чугуны; 6
тические чугуны.
свыше 2,0% С по схеме L —>y-Fe -f- Fe3C (горизон-
таль ECF, 1130°) кристаллизуется эвтектич. смесь
аустенита с цементитом — ледебурит. При со-
держании свыше 0,64% С и 723° (горизонталь PSK)
протекает эвтектоидный распад у-Fe —>a-Fe 4- РвзС,
в результате чего образуется смесь феррита с цементи-
том — перлит. Точка S отвечает эвтектоидному
составу с содержанием углерода 0,8%. Слева от
точки S наряду с перлитом имеется избыточный фер-
рит, справа от точки S (ок. 2% С), кроме перлита,
образуется избыточный цементит. В зависимости от
условий образования цементита различают цементит
первичный, выделяющийся при кристаллизации, вто-
ричный, образующийся из пересыщенного аустенита, и
третичный, выделяющийся из пересыщенного феррита.
В соответствии с диаграммой, стали и чугуны, равно-
весно охлажденные до комнатной темп-ры, состоят из
след, структурных составляющих (табл. 2).
При высокой скорости охлаждения (закалке) стали
распад аустенита на феррит и перлит не успевает
осуществиться и образуется пересыщенный твердый
р-р углерода в a-Fe, т. паз. мартенсит. Вслед-
ствие внедрения в решетку a-Fe избыточных атомов
углерода она искажается и переходит из кубической
в тетрагональную, причем степень тетрагональности
увеличивается с увеличением содержания углерода.
Мартенситное превращение начинается при опреде-
ленной темп-ре и по мере понижения темп-ры количе-
ство мартенсита в стали увеличивается; кристаллы
мартенсита растут с очень большой скоростью, изме-
рить к-рую пока не удалось. Темп-ра, при к-рой прак-
тически заканчивается мартенситное превращение,
зависит от содержания углерода — чем выше концент-
рация углерода, тем при более низкой темп-ре завер-
шается этот процесс. Мартенситное превращение
сопровождается значительными объемными изменени-
ями в стали, что часто приводит к образованию тре-
щин и к короблению.
II. Углеродистые стали. Сплавы Fe—С, содержащие
до 2,0% С, паз. углеродистыми сталями. Обычная
углеродистая сталь содержит, кроме того, еще
0,3—0,7% Мн, 0,2—0,4% Si, 0,01—0,05% Р и
Таблица 2
0,01—0,04% S. Марганец и кремний вво-
дятся в сталь при ее производстве, а фос-
фор и сера неизбежно попадают из шихто-
вых материалов. Помимо этих элементов,
в сталь попадают в качестве примесей ки-
слород, азот и водород. Могут присутство-
вать также и другие элементы, к-рые нахо-
дятся в местных рудах или шихтовых
материалах. Так, напр., керченские руды
содержат мышьяк и ванадий, халилов-
ские — хром и никель, тагильские — медь
и т. д. Т. обр., обычные углеродистые стали
могут содержать малые количества 8—10
элементов. Важнейшим элементом углеро-
дистых сталей является углерод, опреде-
ляющий их структуру и свойства.
Для обозначения марок углеродистых
сталей принята буквенно-цифровая система.
Электротехнич. малоуглеродистая сталь —
железо типа армко (см. Железо) — обозна-
чается буквой А или Э. При обозначении
мало- и среднеуглеродистых сталей числа
показывают среднее содержание углерода
в сотых долях процента (напр., Ст 08, Ст 20,
Ст 45 и т. п.), а при обозначении высоко-
углеродистых сталей — в десятых долях
процента. Кроме того, при обозначении
высокоуглеродистых марок сталей перед
цифрой ставится буква У (напр., У8, У9
— заэвтек- и т. д.). Если сталь содержит минималь-
ные примеси серы и фосфора, а также
соблюдены все условия металлургия, производства,
в конце марки стали ставится буква А (напр., 10А, 20А,
45А, У8А, У9А и т. п.). Такие стали являются высоко-
качественными. В табл. 3 приводятся составы нек-рых
сталей, а в табл. 4 — их механич. свойства.
Таблица 3
Марка стали Содержание элементов а,%
с S1 Мп S Р
А г? 0,025 =S0,30 я? 0,035 0,025 0,015
08 0,05-0,12 == о,15 ==0Д5 0,040 0,040
10 0,05—0,15 0,17-0,37 0,35—0,65 0,045 0,045
20А 0,15-0,25 0,17-0,37 0,35—0,65 0,035 0,040
35 0Д0—0,40 0,17-0,37 ОД —0,8 0,045 0,045
45 0(40—0,50 0,17-0,37 ОД —од 0,045 0,045
45А 0,40-0,50 0,17—0,37 ОД -0,8 0,035 0,035
а Остальное’- Fe Таблица 4
Марка стали Вид и состояние полуфабрикатов Механич. свойства (не менее)
кГ/мм* к Г/мм* б, % ф> % нв, кГ/мм*
А Прутки горячеката- ные (малоуглеро- дистые) 27 12 26 60 77-103
08 Листы отожженные 28—38 18 26 60 76-113
10 Прутки горячекатаные 32 18 30 55 95-143
20А Листы отожженные 40-55 22 24 53 111—152
35 Поковки и прутки нормализованные или отожженные 52 28 15 45 143-187
45 Поковки и прутки нормализованные или отожженные 60 32 13 40 170-229
45А Прутки для рас- чалок закаленные и отпущенные 70-85 — и 55 187-241
Обозначения: <тб — прочность (временное сопротивление раз-
рыву); <т0 8 — предел текучести; б — удлинение перед раз-
рывом; ф — сужение в шейке перед разрывом; Hg— твердость
по Бринеллю.
23
ЖЕЛЕЗА СПЛАВЫ
24
Во многих случаях углеродистая сталь используется
в машиностроении без специальной термин, обработки
в т. наз. «сыром» виде — после горячей прокатки.
Механич. свойства такой
Рис. 2. Влияние углерода на ме-
ханические свойства стали.
Нв — твердость по Бринеллю,
ав — предел прочности, ф — от-
носительное сужение, 6 — отно-
сительное удлинение, ая — удар-
ная вязкость.
в термически обработанном
ном состоянии ее свойства
, стали в сильной степени
зависят от содержания
углерода, как это видно
из рис. 2. Литая сталь
обладает более низкими
механич. свойствами. Го-
рячекатаная сталь ши-
роко используется для
производства разных ма-
шин, станков, строите-
льных металлоконструк-
ций, предметов домаш-
него обихода и т. д.
Для повышения прочно-
сти отдельных наиболее
ответственных деталей ма-
шин и механизмов угле-
родистые стали, иду-
щие на их изготовление,
подвергают термической
обработке — закалке и
последующему отпуску.
В табл. 5 приведены ме-
ханич. свойства стали 45
состоянии (в горячеката-
даны в табл. 4).
Таблица 5
Термич. обработка Механич. свойства
темп-ра закалки, °C охлаждаю- щая среда темп-ра отпуска, °C кГ/мм* ff0.2 , кГ/мм* б, % ф. %
850 вода 450 100 85 10 40
850 » 500 85 75 12 45
850 550 80 65 16 50
850 » 600 75 60 25 55
Углеродистые стали применяются также для изго-
товления режущего инструмента. Для этих целей
используются стали с высоким содержанием углерода
от 0,7 до 1,4% (марки стали от У7А до У13А). После
закалки стали У7А, У8А и У9А приобретают струк-
туру мартенсита, асталиУЮА, У11А, У12АиУ13А —
структуру мартенсит 4- цементит. Наличие мартен-
сита вызывает повышенную хрупкость из-за значи-
тельных искажений решетки, поэтому после закалки
необходим отпуск, темп-pa к-рого определяется вели-
чиной твердости, к-рой должен обладать инструмент.
III. Чугуны — сплавы Fe, содержащие больше
2,0% С (обычно от 2 до 4%). Углерод в чугунах может
находиться в связанном состоянии в виде цементита
или в структурно-свободном состоянии в виде графита.
Основным отличием чугунов от сталей является нали-
чие в них ледебурита и свободного графита. Сущест-
вуют две точки зрения на процесс образования графита
в чугуне: 1) графит выделяется непосредственно из
жидкого чугуна во время кристаллизации и при рас-
паде аустенита; 2) образованию графита предшествует
процесс выделения из жидкого чугуна и аустенита
цементита, к-рый в дальнейшем распадается на
железо и графит. В кристаллохимии, отношении вто-
рая точка зрения более обоснована. S, Ni, Al, CunTi
ускоряют процесс графитизации; Сг, Мп, W, Mo, S
и О2, наоборот, затрудняют его.
Различают белый, серый и ковкий чугун. Белый
чугун при обычной темп-ре содержит перлит и
цементит. Весь углерод в белом чугуне находится
в связанном состоянии. Серый чугун содержит
графит, включения к-рого имеют форму тонких пла-
стинок. Ковкий чугун так же, как и серый,
содержит графит, но графитовые включения имеют
сферич. форму, что повышает пластичность и проч-
ность чугуна. Во всех марках чугуна содержится
Si, Мп, S и Р, т. е. те же примеси, что и в углеродистых
сталях, но в больших количествах. Кроме того,
в чугун часто вводят Сг, Ni, Си, W, Мо и др. элементы.
Такие чугуны наз. легированными.
Особенно сильно влияет на процесс графитизации
кремний. Чем больше кремния в чугуне, тем полнее
распад цементита. Содержание кремния в чугунах
колеблется от 0,3—0,5% до 3—5%. Фосфор не влияет
на процесс графитизации, но улучшает жидкотеку-
честь чугуна, что объясняется образованием легко-
плавкой фосфидной эвтектики с т. пл. 950°. Чугуны,
содержащие фосфор, легче плавятся, медленно осты-
вают и хорошо заполняют самые мелкие изгибы формы.
Поэтому повышенное содержание фосфора имеют
чугуны, применяющиеся для художественного литья.
"Хромистый чугун (26—30% Сг) обладает повы-
шенной прочностью (гв 60 кГ/мм‘) и стойкостью в агрессив-
ных средах. Применяется в химич. аппаратостроении.
Медистый чугун (2—2,5% Си) обладает повышен-
ной прочностью (’„ 50—55 кГ/мм2), используется для из-
готовления коленчатых валов, маховиков и станин.
Титановый чугун (ок. 0,7% Ti) обладает анти-
фрикционными свойствами в паре с другими металлами. При-
меняется как антифрикционный сплав.
Алюминиевый чугун (20—24% А1) обладает
высокой стойкостью к газовой коррозии при повышенных
темп-рах (до 1000°). Используется для изготовления печной
арматуры.
Высококремнистый чугун (5—6% Si) об-
ладает высоким сопротивлением ползучести до 900°. Приме-
няется в нагревательных системах.
Высокомарганцовый чугун (7—12% Мп)
обладает малой магнитной проницаемостью и высоким электро-
сопротивлением. Применяется в электротехнике.
Хромоникелевый чугун (0,2—1,5% Сг, 0,2—
4% Ni) обладает высокой прочностью, износостойкостью и
стойкостью к газовой коррозии. Применяется в двигателях
внутреннего сгорания, локомотивах и т. д.
В машиностроении детали со сплошной структурой
белого чугуна не используются; обычно применяются
детали, у к-рых поверхность имеет структуру белого
чугуна, а сердцевина — серого, что достигается спе-
циальной термин, обработкой. Высокая твердость
отбеленной поверхности (350—400 Нв) придает изде-
лиям большую стойкость против износа. Такие чугуны
с отбеленной поверхностью используются для изготов-
ления колес товарных вагонов, прокатных валков,
волочильных досок, плужных лемехов и т. д. Серый
и ковкий чугуны получили широкое распространение
в автостроении, вагоностроении, при произ-ве стан-
ков, с.-х. машин и т. д.
Таблица 6
Сорт чугуна ° в » кГ!мм* Нв, кГ/мм*
Серый чугун с 3,0—3,5% С: Ферритно-графитный с тонким графитом То же, с грубым графитом ФерритнОперлитно-графитный 12—18 10-15 100—120 100-120
12-20 100—200
Перлитно-графитный с тонким графитом 28—38 180-220
То же, с грубым графитом 20-32 180—220
Ковкий чугун с 2,0—2,7% С Термически обработанный сорбитный 30-45 110—140
чугун до 60 и выше 250—300
Чугун с шаровидным сферич. графитом 50-80 150-300
Большая часть белого чугуна, получаемого домен-
ной плавкой, предназначается для передела в сталь,
а нек-рая часть — для получения ковкого чугуна.
Серый чугун благодаря высоким литейным свойствам
(низкая темп-pa кристаллизации, жидкотекучесть, ма-
лая усадка и др.) является основным Ж. с. для литья
и широко применяется в машиностроении. Механич.
свойства серых чугунов зависят от их структурных
составляющих, количества и формы выделений гра-
25
ЖЕЛЕЗА СПЛАВЫ
26
фита. В табл. 6 приведены нек-рые свойства серых
и ковких чугунов в зависимости от содержания С и
различных структурных составляющих.
IV. Легированные стали. Стали, содержащие раз-
личные элементы, заметно изменяющие их свойства,
наз. легированными. К легирующим элементам отно-
сятся: Сг, Ni, Мп, Si, W, Mo, V, Со, Ti, Nb, Al, N,
Zr, В, Та и Си. Легированные стали обладают высо-
кими механич. и физико-химич. свойствами.
Твердые р-ры замещения с железом образуют Сг,
Ni, Мп, W, Mo, V, Со, Ti, Nb, Al и Zr. Если коли-
чество введенных элементов превышает их предел
растворимости, то образуются интерметаллич. соеди-
нения и,т. обр., в структуре, наряду с твердым раство-
ром предельной концентрации, появляются новые
фазы — соединения легирующего элемента с железом.
Неограниченной растворимостью в y-Fe при доста-
точно высокой темп-ре обладают лишь никель, ко-
бальт и металлы группы платины, а в a-Fe — только
хром и ванадий. При медленном охлаждении непре-
рывные твердые р-ры этих двойных систем, в опреде-
ленном интервале концентраций, образуют соедине-
ния: FeNi3, FeCo, FeCr и FeV. Марганец, вольфрам,
молибден, титан, ниобий, алюминий и цирконий обра-
зуют с железом твердые р-ры замещения с ограничен-
ной растворимостью. Твердые р-ры внедрения обра-
зуют углерод, бор и азот.
По отношению к углероду легирующие элементы
в железных сплавах можно разделить на две основные
группы: 1) элементы, не образующие устойчивых
карбидов (Ni, Si, Со, Al и Си); 2) карбидообразующие
элементы (Сг, Мп, W, Mo, V, Ti, Nb, Та и Zr) (см.
Карбиды). Если количество углерода в стали недоста-
точно для образования карбидов со всеми присутст-
вующими карбидообразующими элементами, то кар-
биды образуют лишь те элементы, к-рые обладают
наибольшим химич. сродством к углероду. Другие
элементы остаются в этом случае в твердом р-ре или
в виде металлич. соединений. По степени возрастаю-
щего сродства легирующих элементов к углероду
они располагаются в ряд: Fe —>• Мп —► Сг —»• W —<•
— Мо — Та — V — Zr — Ti — Nb.
Установлено, что в сталях могут образовываться
следующие карбиды (как простые, так и сложные —
двойные): Fe3C, MnsC, Сг7Сд, Сг23С6, WC, W2C, Fe2W2C,
Fe6W6C, МоС, Мо2С, Fe2Mo2C, Fe6Mo6C, ТаС, Та2С,
VC, ZrC, NbC, TiC. В нек-рых из этих карбидов угле-
род располагается в узлах сложной кристаллич.
решетки, образуя химич. соединения с металлом,
как, напр., цементит. К числу таких карбидов отно-
сятся: Мп3С, Сг7Сд, Cr23C6, Fe2W2C, Fe2Mo2C. В дру-
гих карбидах углерод располагается в междуузлиях
простой металлич. решетки, как в обычных твердых
р-рах внедрения, и поэтому такие карбиды представ-
ляют собой фазы внедрения. К их числу относятся:
WC, W2C, МоС, Мо2С, ТаС, Та2С, VC, ZrC, NbC, TiC.
Особенностью фаз внедрения является то, что они
кристаллизуются со значительным недостатком угле-
рода против стехиометрия, состава. Напр., в VC содер-
жится не 50, а всего лишь 30—35 ат. % углерода.
Наряду с рассмотренными карбидами в сталях обра-
зуется легированный цементит. Карбидообразующие
элементы прежде всего растворяются в цементите
и образуют легированный цементит типа (Fe,Cr)3C,
(Fe, Мп)3С и т. д. При большом содержании легирую-
щих элементов они образуют свои карбиды, к-рые,
в свою очередь, растворяют железо.
Классификация легированных сталей.
Легированные стали обычно классифицируют по химич. со-
ставу, микроструктуре и применению.
По химическому составу различают следующие стали:
низколегированные с общим содержанием легирующих эле-
ментов не выше 2,5%; среднелегированные — от 2,5 до 10%
легирующих элементов, и высоколегированные — выше 10%
легирующих элементов. По микроструктуре в отожженном
состоянии различают среднеуглеродистые и высокоуглероди-
стые легированные стали и делят их на доэвтектоидные, за-
эвтектоидные и ледебуритные. Кроме того, при небольшом
содержании углерода и высоком содержании легирующих
элементов, сужающих область у-твердых растворов так, что
она может почти или совершенно исчезнуть (хром, вольфрам,
молибден, ванадий и др.), структура стали может представлять
собой только один a-твердый р-р. Такие стали наз. полуфер-
ритными или ферритными. Следовательно, в этом случае стали
по микроструктуре делятся на пять классов: доэвтектоидные,
заэвтектоидные, ледебуритные, ферритные и полуферритные.
При высоком содержании легирующих элементов, расширяю-
щих область у-твердых растворов (никель, марганец и др.),
структура у-твердого р-ра может почти или полностью сохра-
ниться в стали, т. е. f^a-переход будет частично или полно-
стью предотвращен. Такие стали наз. полуаустенитными или
аустенитными. Следовательно, и в этом случае стали делятся
на пять классов: доэвтектоидные, заэвтектоидные, ледебурит-
ные, аустенитные и полуаустенитные. Стали, содержащие
большое количество карбидообразующих элементов, прй до-
статочно высоком содержании углерода могут быть выделены
в особый класс — карбидный. Структура сталей карбидного
класса состоит из основного структурного фона (перлита, мар-
тенсита, аустенита) и избыточных карбидов.
По применению легированные стали делятся на три
группы: А. Конструкционные стали,
применяемые для изготовления деталей машин. В зави-
симости от условий работы конструкционные стали,
в свою очередь, делят на цементируемые и улучшае-
мые. Б. Инструментальные стали, при-
меняемые для изготовления различного инструмента.
Эта группа сталей объединяет: 1) углеродистую
инструментальную, 2) легированную инструменталь-
ную, 3) штамповую и 4) быстрорежущую. В. Ста-
ли с особыми свойствами. Такими
сталями являются: 1) нержавеющие и кислотоупор-
ные, 2) жаропрочные и жаростойкие, 3) износоустой-
чивые, 4) с особым тепловым расширением, 5) с осо-
быми магнитными свойствами, 6) высокого электро-
сопротивления и т. д.
Для обозначения марок легированных сталей по
ГОСТ принята буквенно-цифровая система. Буквами
обозначают легирующие элементы: никель — Н;
хром — X; молибден — М; вольфрам — В; ванадий —
Ф; кобальт — К; кремний — С; марганец — Г; медь —
Д; фосфор — П; титан — Т; алюминий — Ю. Циф-
рами показывают содержание углерода и легирую-
щих элементов. Первые две цифры в начале обоз-
начения марки указывают среднее содержание угле-
рода в сотых долях процента. Цифры, стоящие после
буквы, — примерное содержание легирующего эле-
мента в процентах. Напр., сталь марки 12Г2А перед-
нем содержит: 0,12% С, 2,0% Мп. Буква А показы-
вает, что сталь чиста по сере и по фосфору, а также
соблюдены все условия металлургии, произ-ва высоко-
качественной стали (см. выше). Нек-рые легированные
стали выделены в особые группы и обозначаются
следующими буквами: хромистые нержавеющие ста-
ли — Ж; хромоникелевые нержавеющие стали — Я;
быстрорежущие стали — Р; шарикоподшипниковые
стали —Ш; магнитные стали— Е. Легированные стали,
выплавляемые на заводе «Электросталь», помимо
стандартного обозначения, имеют, свои названия.
Они маркируются буквами ЭИ (электросталевское
исследование) и номером стали, напр. ЭИ-100,
ЭИ-445 и т. д. Ниже приводится краткая характери-
стика легированных сталей.
V. Конструкционные стали. От стали, идущей на
изготовление деталей машин, требуются гл. обр. высо-
кие механич. свойства. Они должны обладать доста-
точной пластичностью и вязкостью, чтобы успешно
противостоять динамич. и ударным нагрузкам и вместе
с тем высокой прочностью и выносливостью, особенно
в условиях усталостного нагружения. Простая угле-
родистая сталь не всегда может удовлетворить этим
требованиям и в таких случаях используются легиро-
ванные стали. Положительное влияние легирующих
элементов сказывается прежде всего на т. наз. про-
27
ЖЕЛЕЗА СПЛАВЫ
28
каливаемости стали, т. е. на выравнивании механич.
свойств деталей после закалки и отпуска по всему
объему даже при значительных размерах деталей.
Вместе с тем легирующие элементы, растворяясь в це-
ментите, препятствуют его коагуляции при нагреве
и тем способствуют повышению прочности, т. к. гру-
бодисперсный цементит снижает прочность. Кроме
того, распределяясь по границам зерен твердого
р-ра, легирующие элементы затрудняют собиратель-
ную рекристаллизацию металла и тем самым препят-
ствуют росту зерна, что также приводит к возраста-
нию прочности.
Никелевые стали. Никель образует с же-
лезом непрерывный ряд твердых р-ров и расширяет
температурный интервал существования аустенита.
Уже малые добавки никеля (до 4—5%) значительно
увеличивают прокаливаемость стали и повышают ее
коррозионную устойчивость в агрессивных средах.
Высоконикелевые сплавы обладают особыми физич.
свойствами. Сплав Fe с 36% Ni и 0,15—0,25% С
(инвар) имеет минимальный коэфф, линейного
расширения и практически не расширяется в интер-
вале темп-p от — 100° до + 1С0°. Инвар широко
применяется в приборостроении для изготовления
эталонов, деталей часовых механизмов, барографов,
альтиметров и прочих приборов, к-рые с изменением
темп-ры должны сохранять свои размеры. Сплав Fe
с 46% Ni и ок. 0,15% С имеет такой же коэфф, линей-
ного расширения, как у платины и стекла; он наз.
платинитом и применяется вместо платины
для электродов лампочек накаливания.
Хромистые стали. Хром, в отличие от
никеля, сужает область у-твердого р-ра и расширяет
область a-твердого р-ра. Предельное содержание
Сг, при к-ром существует еще у-твердый р-р, равно
13%. При малом содержании хрома наряду с а-твер-
дым р-ром хрома в железе присутствует также леги-
рованный цементит (Fe, Сг)3С. При увеличении кон-
центрации хрома образуется карбид (Сг, Fe),C3, при
содержании хрома более 10% — карбид (Сг, Fe)23C6
и при содержании хрома более 28% — металлич.
соединение FeCr (о-фаза). Добавка хрома повышает
твердость и прочность стали, не снижая пластичности.
Однако увеличение содержания хрома выше 1,0—
1,5% снижает ударную вязкость. Увеличение содер-
жания хрома до 4—5% наиболее резко повышает
твердость закаленной стали, тогда как свойства ото-
жженной стали изменяются незначительно. Следова-
тельно, наиболее резкое воздействие на твердость
и прочность стали оказывает хром, на-
ходящийся в мартенсите, а не в феррите
или карбидах. Хром повышает коррозион-
ную стойкость в атмосферных условиях
и сопротивляемость стали газовой кор-
розии при высоких температурах. При
больших концентрациях хрома на поверх-
ности стали образуется тонкая окисная
пленка (Сг2О3), препятствующая развитию
процесса коррозии в атмосферных усло-
виях, а также в кислотах, особенно в азотной.
Хромистые стали получили широкое распростра-
нение для изготовления мерительных, режущих и
пневматич. инструментов, штампов и валиков для
холодной прокатки, ролико- и шарикоподшипников,
постоянных магнитов, хирургия, инструмента и т. п.
Кроме того, хромистые стали широко распространены
как теплоустойчивые и коррозионностойкие матери-
алы в нефтяной и химич. пром-сти. Наиболее распро-
странены след, марки хромистых сталей: 15ХА с со-
держанием ок. 0,15% С и 0,7—1,0% Сг; 38ХА с со-
держанием ок. 0,38% С и 0,8—1,1% Сг и ШХ15
(шарикоподшипниковая) с содержанием ~0,9—1,1% С
и 1,3—1,65% Сг.
Хромоникелевые стали. Одновремен-
ное введение Сг и Ni весьма благоприятно влияет на
строение и свойства стали. Сталь приобретает высо-
кую твердость и прочность, достаточную вязкость и
пластичность, хорошую прокаливаемость, однородное
строение. Совместное присутствие Сг и Ni улучшает
также физич. и химич. свойства стали. Поэтому хромо-
никелевые стали более распространены, чем другие
легированные стали. Содержание хрома и никеля
в конструкционных сталях обычно изменяется в пре-
делах: от 0,6 до 1,5% Сг, от 1,0 до 4% Ni при содержа-
нии от 0,10 до 0,40% С. В табл. 7 приведены составы
нек-рых хромоникелевых сталей.
%, Таблица 7
Марка стали Содержание элементов а, %
с S1 МП Сг Ni
13Н2А 0,10-0,16 0,17-0,37 0,30-0,60 0,2 —0,5 1,7 -2,2
12ХНЗА 0,10-0,16 0,17—0,37 0,30—0.60 0,60—0,90 2,25—2,75
12Х2Н4А 0,10—0,15 0,17—0,37 0,30—0,60 1,25-1,75 3,25—3,75
20ХНЗА 0,17—0,25 0,17—0,37 0,30—0,60 0,60-0,90 2,75-3,25
37XH3A 0,33—0,41 0,17-0,37 0,25—0,55 1,20—1,60 3,00—3,50
а Остальное — Fe.
Дополнительное введение W и Мо в хромоникелевые
стали значительно улучшает их механич. и другие
свойства. С вольфрамом железо образует ограниченные
твердые р-ры и металлич. соединения Fe,W6 и Fe2W.
Вольфрам сужает область у-твердых р-ров; при 6%
у-область замыкается. С углеродом вольфрам образует
стойкие карбиды WC, W2C, Fe2W2C и FeeWeC. Воль-
фрам сильно обедняет твердый р-р углеродом, препят-
ствует росту зерна аустенита при нагревании, умень-
шает чувствительность стали к перегреву. Перлит
вольфрамовой стали имеет очень тонкое, а мартенсит —
мелкоигольчатое строение. После термич. обработки
вольфрамовые стали приобретают повышенную твер-
дость, прочность и высокую ударную вязкость. Воль-
фрам добавляется к конструкционным хромоникеле-
вым сталям и к жаропрочным сталям, а также является
основным легирующим элементом быстрорежущих
сталей.
С молибденом железо образует также ограниченные
твердые р-ры и металлич. соединения Fe,Mo6 и FeMo.
Молибден сужает область у-твердых р-ров; при 5%
Мо у-область замыкается. С углеродом молибден
образует карбиды: МоС, Мо2С, Fe2Mo2C и Fe6Mo6C.
Составы нек-рых сложнолегированных сталей, содер-
жащих W и Мо, приводятся в табл. 8.
Таблица 8
Марка стали Содержание элементов а,%
с S1 Мп Сг N1 W МО
18ХНВА 0,14—0,21 0,17—0,37 0,25-0,55 1,35-1,65 4,0—4,5 0,8-1,2
25 X НВ А 0 £1—0,28 0,17—0,37 0,25—0,55 1,35—1,65 4,0—4,5 0,8-1,2 —
40ХНМА 0,36—0,44 0,17-0,37 0,50—0,80 0,60—0,90 1,25-1,75 — 0,15—0,25
а Остальное — Fe.
X р омом олибденоалюминиевая
сталь. Алюминий с железом образует ограниченные
твердые р-ры и металлич. соединения: Fe3AI, FeAl,
FeAl2, Fe2AlB, FeAl3. Растворимость Al в a-Fe при
обычной темп-ре 32%. Область у-твердых р-ров в при-
сутствии А1 сужается; при 1% А1 у-область замы-
кается. Алюминий является лучшим раскислителем
стали. Он добавляется в ванну незадолго до выпуска
стали из печи, а также вводится в ковш или жолоб.
В небольших количествах, до 0,1%, А1 добавляется
во все конструкционные и инструментальные стали,
от к-рых требуется зерно малой величины. Особенно
сильно А1 повышает стойкость стали против газовой
коррозии при высоких темп-рах. Сплавы для постоян-
29
ЖЕЛЕЗА СПЛАВЫ
30
ных магнитов, имеющие высокие магнитные свойства,
содержат 12—15% А1. Хромомолибденоалюминиевые
стали широко применяются для изготовления деталей
машин, от к-рых требуется большая стойкость про-
тив износа и истирания при обычных и повышенных
темп-рах.
Xромомарг анцо в о кремни ст а я сталь
(хромансиль). Одна из наиболее распространен-
ных марок этой стали — ЭОХГСА содержит в среднем
0,3% С и по 1% Сг, Мп и Si. Марганец образует с
железом твердые р-ры и расширяет область у-твер-
дых р-ров. Растворимость марганца в a-железе огра-
ничена. С углеродом марганец образует карбид
Мп3С, изоморфный цементиту Fe3C. Кремний образует
с железом твердые р-ры и металлич. соединения —
Fe3Si2, FeSi, FeSi2. Растворимость кремния в а-железе
при обычной темп-ре составляет 14%. Особенно за-
метно кремний повышает пределы упругости и теку-
чести, что позволяет широко применять кремнистые
стали для изготовления пружин. Сталь хромансиль
представляет большой интерес для машинострое-
ния, т. к. она обладает хорошей свариваемостью,
высокими механич. свойствами, хорошей прокаливае-
мостью, удовлетворительной обрабатываемостью ре-
жущими инструментами и высокой пластичностью
в отожженном состоянии. Из хромансиля изготовляют
полуфабрикаты в виде листов, труб, профилей, поко-
вок, прутков и проволоки. В табл. 9 приведены со-
ставы хромомарганцовокремнистых сталей.
Таблица 9
Марка стали Содержание элементов а, %
с Si Мп Сг
25ХГСА . . . • . 0,22—0,29 0,90—1.20 0,80—1,10 0.80-1,10
ЗОХГСА 0,28-0,35 0,90—1,20 0,80—1,10 0,80-1,10
а Остальное — Fe.
VI. Инструментальные стали. Инструменты по сво-
ему назначению можно разделить на три основные
группы: режущие, мерительные и ударно-штампо-
вые. Основным требованием к инструментальной
стали является продолжительность сохранения режу-
щей кромки — лезвия инструмента, изнашивающейся
при резании, причем на износ работает очень тонкая
полоска металла. Чтобы эта полоска металла была
устойчива против истирания, она должна обладать
высокой твердостью и жаропрочностью. Ударно-
штамповый инструмент испытывает ударные нагрузки,
распределяемые по сравнительно большой поверхно-
сти. Здесь важную роль играет ударная вязкость
материала. Наилучшей штамповой сталью, очевидно,
будет такая сталь, в к-рой оптимальным образом
сочетаются твердость и вязкость.
Легированная инструментальная сталь в большин-
стве случаев содержит 1—3% легирующих элементов.
В нек-рых сталях содержание легирующих элемен-
тов доходит до 12—14%. Содержание 0,8—1,3% угле-
рода обеспечивает высокую твердость стали для режу-
щего инструмента. Основным легирующим элементом
инструментальных легированных сталей является
хром. Введение 0,3—0,5% Сг уже заметно увеличивает
прокаливаемость стали и износостойкость. Иногда
в качестве улучшающих присадок в инструментальные
стали вводят вольфрам и ванадий. Вольфрам повы-
шает сопротивление стали износу и уменьшает чув-
ствительность к перегреву. Но вольфрам дорог, поэ-
тому в практике инструментальные стали, легирован-
ные W, стремятся заменить хромистыми. Высокого
сопротивления износу можно достигнуть введением в
сталь, содержащей ~ 1,5% С, до 12% Сг; при этом об-
разуется большое количество карбидов типа (CrFe)7C3,
к-рые сильно повышают износостойкость стали. Со-
ставы нек-рых легированных инструментальных ста-
лей приведены в табл. 10.
Таблица 10
Марка стали Содержание элементов а,%
с Мп Si СГ W V
X 1,0-1,15 ==0,4 <0,4 1,3-1,6
хг 1,3-1,5 0,45-0,7 <0,4 1,3-1,6 —
9ХС 0,85-0,95 < 0,4 1,2-1,6 0,95—1,25
хвг 0,9—1,05 0,8-1,1 < 0,4 0,9-1,2 1,2-1,5 __
Х12Ф1 1,45-1,70 ==0,4 <0,4 11,0—12,5 — 0,7-0,9
а Остальное — Fe.
Быстрорежущая сталь должна сохранять свои
режущие свойства при больших скоростях резания, т. е.
до темп-р 600—700°. Основными легирующими элементами
быстрорежущей стали являются хром и вольфрам. Другие
элементы, вводимые в быстрорежущую сталь, как-то: ванадий,
молибден и кобальт играют роль добавок, улучшающих свой-
ства. Наиболее распространенной является высоковольфрамо-
вая быстрорежущая сталь марки Р18; ее состав приво-
дится в табл. 11. _ , ..
Таблица И
Марка стали Содержание элементов а,%
С Мп Si Сг W V
Р18(РФ1) 0,68—0,80 <0,4 <0,4 3,8-4,8 17,5-18,5 1,0-1,4
а Остальное — Fe.
По структуре эта сталь относится к ледебуритному классу.
В литом виде она содержит твердый р-р, ледебуритную эвтек-
тику и карбиды. Легирующие элементы в быстрорежущей
стали распределяются между отдельными фазами след, обра-
зом: хром распределен примерно поровну между ферритом
и карбидной фазой, вольфрам и ванадий присутствуют в стали
в виде карбидов. Сумма карбидов типа (Ст, Fe)? Са, Fe2W2C
и VC достигает 30—35%. Наличие большого количества кар-
бидообразующих элементов, к-рые связывают весь С, приводит
к повышению износостойкости стали при повышенных темп-рах.
Термин, обработка быстрорежущей стали состоит в закалке
с темп-рой 1270—1290° и последующем отпуске при 540—560°.
Наряду с рассмотренной маркой быстрорежущей стали
в практике используются и низколегированные быстрорежу-
щие стали с пониженным содержанием дорогостоящего воль-
фрама. Эти стали, по сравнению со сталью марки Р18, обла-
дают несколько пониженными режущими свойствами. Однако
режущие свойства низколегированных быстрорежущих сталей
оказываются в ряде случаев вполне достаточными, и они
успешно применяются в различных отраслях пром-сти. В табл.
12 приведены наиболее распространенные марки низколегиро-
ванных быстрорежущих сталей.
Таблица 12
Марка стали Содержание элементов а, %
с Мп S1 | Ст W V
Р9 . . . . 0,85—0,95 <0,45 ^0,45 4,0—4,6 8,5—10,0 2,0-2,6
ЭИ347 • . 6,7—0,8 <0,45 <0,45 4,0-4,6 8,5-10,0 1,4-1,7
а Остальное — Fe.
VII. Стали с особыми свойствами. Нержавею-
щие стали. Углеродистые и низколегирован-
ные стали не обладают сколько-нибудь значительной
коррозионной стойкостью, т *
т. к. образующаяся на них Таблица 13
окисная пленка не изоли-
рует металл от химиче-
ского воздействия среды.
Только введение в сталь
ок. 12% Сг делает ее ус-
тойчивой против коррозии
в атмосфере, кислотах, ще-
лочах, р-рах солей. Все
стали, содержащие 12% Сг
Содержание
элементов а, %
I с | Сг
Марка
стали
Ж1
Ж2
Х17
Х27
<0,14
0,15—0,23
0,12
0,15
12,5—14,5
12,5-143
16-18
26—29
а Остальное — Fe.
и более, являются нержа-
веющими. В табл. 13 приведены составы нек-рых
наиболее употребительных марок хромистых нержа-
веющих сталей.
31
ЖЕЛЕЗА СПЛАВЫ
32
('тали Ж1 и Ж2 наиболее дешевы и применяются
без предварительной термин, обработки. Стали Х17
и Х27 относятся к сталям ферритного класса, не имею-
щим a-превращений при изменении темп-ры.
Эти стали используются в основном как тара для
сильных к-т. Введение достаточного количества Ni
в 18%-ную хромистую сталь переводит ее в аустенит-
ное состояние при всех темп-pax. Тем самым обеспе-
чиваются повышение прочности и предела текучести,
а также более высокая коррозионная устойчивость.
Стали с 18% Сг и 8% Ni (18-8) получили как нержа-
веющие наиболее широкое распространение в машино-
строении, в изделиях широкого потребления, а так-
же в архитектуре и скульптуре. При обычной темп-ре
эти стали имеют аустенитную структуру. При содер-
жании С более 0,04% он образует карбидную фазу
(Сг, Fe)23Ce, расположенную но границам аустенит-
ных зерен. При нагревании до 1100° карбиды целиком
растворяются в аустените и при быстром охлаждении
до 800°; при 25% Сг жаростойкость повышается до
900—950°. Элементы, наиболее успешно повышающие
жаростойкость стали (Сг, Si, Al), не являются одно-
временно оптимальными с точки зрения увеличения
жаропрочности. Для рабочих темп-p не выше 300—
350° наиболее пригодны низколегированные конструк-
ционные стали, содержащие Сг, Ni и Мо и обработан-
ные на высокую прочность (закалка и низкий отпуск);
в этом интервале темп-p они практически не подвер-
жены ползучести и достаточно стойки против газовой
коррозии. В интервале рабочих темп-р 350—500°
следует применять стали перлитного или ферритного
класса, содержащие больше Сг и Si.
Для интервала рабочих температур 500—650° ис-
пользуются высоколегированные стали аустенитного
класса. В таблице 14 приведены составы наиболее
широко используемых сталей этого класса. Волее
подробно о жаропрочных свойствах Ж. с. см. Жаро-
прочность.
Таблица 14
Содержание элементов а, %
ixicipna стали С Мп Si Сг N1 W Мо V Т1 другие элементы
ЭИ69 0,4—0,5 =5 0,7 0,7-0,8 17—15 13-15 . 2—2,7 0,2-0,4
ЭИ388 .... 0,38—0,47 6,0—8,0 0,9-1,4 14-10 6,0-8,0 0.5-0,8 1,4-1,8 —
ЭИ395 .... < 0,12 1,0—2,0 0,5-1,0 15—17,5 24-27 — 5,5—7,0 — 0,1-0,2% N
ЭИ415 .... 0,16—0,24 0,25-0,6 ==0,4 2,4—3,3 з? 0,5 0,3—0,5 0,35-0,55 0,6-0,8 — —
ЭИ417 .... =~0,18 =5 1,5 5? 1,0 22—25 17-20 — —
ЭИ481 .... 0,34—0,40 7,5—9,5 0,3-0,8 11.5—13,5 7,0-9,0 — 1,1—1,4 1,25-1,55 <0,12 0,4—0,8% Nb
ВЛ745У . . . < 0,3 0,6—1,2 <0,8 18—24 45-50 7—9 — 0,01 % В
22-11-2,5 . . 0,15—0,25 0,6—1,0 0,8-1,5 21-25 9-12 2,5-3,5 0,15—0,2 0,1-0,2 0,05—0,2 —
а Остальное — Fe.
не выделяются вновь, образуя пересыщенный твердый
р-р углерода в аустените. Этот пересыщенный твер-
дый р-р устойчив до 500—550°. Но при более высоких
темп-pax вновь начинается выделение карбидной фазы
по границам зерен, что приводит к появлению весьма
опасной интеркристаллитной коррозии. Процесс кор-
розии очень быстро распространяется по границам
зерен в глубь металла и приводит к полному его раз-
рушению. Интеркристаллитная коррозия возникает
в стали 18-8 лишь после нагрева до 500—700° при
наличии избытка углерода над пределом раствори-
мости (0,04%). Склонность к интеркристаллитной
коррозии в этих сталях устраняется введением в сплав
сильных карбидообразователей, связывающих угле-
род. Такими свойствами обладают титан и ниобий.
Эти элементы вводятся в таком количестве, чтобы свя-
зать весь углерод в сплаве. Нержавеющие стали легко
упрочняются при холодной обработке. Так, при про-
катке с обжатием в 40% предел прочности стали
18-8 повышается с 60 кГ/мм2 до 120 кГ/мм2, а предел
текучести — с 25 кГ/мм2 до 100 кГ/мм2.
Жаропрочные и жаростойкие
стали — сложнолегированные стали, обладающие
значительным сопротивлением пластич. деформации
и разрушению при высоких темп-pax (жаропроч-
ность) и стойкие против газовой коррозии в этих
условиях (жаростойкость). В большинстве
случаев к этим сталям, работающим при высоких
темп-pax, предъявляются одновременно требования
жаропрочности и жаростойкости. Для создания
жаростойкости необходимо легировать сталь такими
элементами, к-рые при высоких темп-pax окисляются
энергичнее, чем железо, и образуют при этом плотные
защитные пленки окислов. Такими элементами яв-
ляются хром, кремний, алюминий и никель. Количе-
ство хрома в жаростойкой стали в значительной мере
определяет максимальную темп-ру, при к-рой она
может работать без интенсивного окисления. Так,
напр., сталь, содержащая 1% Si и 15% Сг, жаростойка
Элементы, повышающие темп-ру рекристаллизации,
прежде всего Мо и W, увеличивают жаропрочность.
1 ат. % Мо повышает темп-ру рекристаллизации на 115°,
а 1 ат. % W — на 240°. Темп-ра рекристаллизации, а сле-
довательно, и жаропрочность в присутствии Мо и W
повышается вследствие блокировки ими границ зерен
основного металла. Углерод, связывая молибден и
вольфрам в карбиды, уменьшает содержание этих
элементов в твердом р-ре и тем самым понижает жаро-
прочность. Поэтому прибавление Ti, Nb, Та, к-рые,
связывая углерод в карбиды, увеличивают содержа-
ние Мо и W, повышающих темп-ру рекристаллиза-
ции, увеличивает жаропрочность. Обычно жаропроч-
ные стали аустенитного класса содержат ок. 0,1% С,
т. е. минимально возможное по условиям их произ-
водства количество. Аустенитные жаропрочные ста-
ли применяют в производстве лопаток газовых тур-
бин, деталей реактивных двигателей, клапанов мото-
ров и т. д.
Износоустойчивые стали. Устойчи-
вость против износа достигается обычно приданием
высокой поверхностной твердости путем цемен-
тации (науглероживания) или азотирова-
ния (насыщения поверхностного слоя стали азо-
том). Исключительно износоустойчива при сравни-
тельно невысокой поверхностной твердости (//в =
= 200—250) сталь Г13А с 1,0—1,3% С и 11—
14% Мп, обладающая большой способностью к упроч-
нению вследствие измельчения зерна под действием
пластич. деформации. Из стали Г13А делаются пере-
водные стрелки на железнодорожных путях, детали
камнедробилок, черпаки экскаваторов и т. д.
Магнитные сплавы. Материал для изго-
товления постоянных магнитов должен обладать
высокими значениями коэрцитивной силы Нс и оста-
точной индукции Вг (магнитной индукции, остающейся
в материале после его намагничивания и снятия маг-
нитного поля) и постоянством этих величин во вре-
33
ЖЕЛЕЗА СПЛАВЫ
34
мени. Для небольших по размеру магнитов исполь-
зуется сталь У10-У12, к-рая после закалки дает Нс =
= 60—65 эрстед (Ое) и Вг = 8000—8500 гаусс (G).
Отпуск снижает Нс, однако отпуск при низкой темп-ре
(150—200°) все же необходим, т. к. магнитные свой-
ства закаленных и неотпугценных сталей изменяются
вследствие старения, Для изготовления более круп-
ных магнитов к углеродистым сталям добавляется
1,5—3,5% Сг, что увеличивает прокаливаемость стали.
Магнитные характеристики при этом существенно не
меняются.
Для изготовления мощных постоянных магнитов
используются сплавы железа с никелем и алюминием.
В сплаве Fe с 22—34% Ni и И—14% А1 можно полу-
чить коэрцитивную силу до 400—500 Ое при остаточ-
ной индукции 6000— 7000 G. В Ж. с., подвергаемые
переменному намагничиванию, вводится Si. Т. наз.
трансформаторная сталь содержит ок. 4% Si и мини-
мальное количество других элементов. Si раство-
ряется в Fe, повышая его электросопротивление, чем
уменьшает потери на токи Фуко и, кроме того, раскис-
ляет Fe, чем способствует уменьшению коэрцитивной
силы. В тех случаях, когда от магнитомягкого мате-
риала требуется также и повышенная вязкость, упо-
требляют так назыв >емую динамную сталь, являющую-
ся безуглеродистым Ж. с. с 1% Si. По магнитным
свойствам динамная сталь уступает трансформатор-
ной, но она обладает большей пластичностью и вяз-
костью.
VIII. Ферросплавы. Сплавы Fe с другими элемен-
тами, используемые как промежуточные шихтовые
материалы в произ-ве легированных сталей, наз. фер-
росплавами. Их получают восстановлением руд,
концентратов руд и чистых окислов металлов углеро-
дом или металлами. Восстановление углеродом произ-
водят в доменных или электрич. печах, а металлами —
в специальных шахтах. Последний способ называет-
ся внепечным, или металлотермическим; восстановите-
лем обычно служат алюминий (см. Алюминотермия),
реже кремний и др. Наиболее дешевым способом
получения ферросплавов является производство их
в открытых (реже закрытых) дуговых электрич. печах
с непрерывными самоспекающимися углеродистыми
электродами.
Ферробор содержит от 5 до 20% В. Его полу-
чают из борсодержащих материалов (борсодержащих
минералов, борного ангидрида) в смеси с окислами
железа путем восстановления их углеродом, крем-
нием или алюминием. Восстановление проводят
в дуговых печах или внепечным способом. При восста-
новлении углеродом и кремнием ферробор содержит
повышенное количество восстановителей. При восста-
новлении борного ангидрида и окиси железа алюми-
нием получаемый ферробор имеет пониженное со-
держание алюминия. Состав ферробора, получаемого
из боратных руд, приведен в табл. 15.
Таблица 15
Способ получения Содержание элементов**, %
в А1 Si С S
Дуговая печь 12—13 3—5 3—4 0.10 0,02
13—14 4-6 3-4 0.02 0.01
а Остальное — Fe.
Ферробор обычно применяют как раскислитель при
плавке легированных сталей, для модифицирования
сталей с целью повышения механич. и технологич.
свойств. В последнее время разработаны марки ста-
лей, специально легированных бором (до 1—5%),
для атомной энергетики.
Феррованадий содержит от 35 до 80% V.
Его получают восстановлением окислов ванадия
углеродом, кремнием или алюминием. Основным
сырьем для получения феррованадия служит пяти-
окись ванадия, получаемая из концентратов ванадие-
вых руд или из железных руд с повышенным содержа-
нием ванадия. Ванадийсодержащие руды или кон-
центраты вначале плавят в доменной печи для полу-
чения чугуна с повышенным содержанием ванадия
(до 0,4—0,5% V). Затем этот чугун перерабатывают
в сталеплавильных печах (мартен, конвертор) с окис-
лением ванадия и обогащением получаемого при
этом шлака окислами ванадия. Такие шлаки подвер-
гают окислительному обжигу в присутствии солей
щелочных металлов; при этом образуются хорошо
растворимые соединения ванадия — ванадаты нат-
рия и калия. После выщелачивания и разделения рас-
творимых соединений ванадия осаждается продукт,
в к-ром содержится 80—95% V2O6. Пятиокись ванадия
в виде предварительно просушенных и сплавленных
слитков используется для получения феррованадия
в дуговых печах закрытого типа или внепечным метал-
лотермии. способом. Основные реакции восстановле-
ния окислов ванадия углеродом или алюминием:
V»O3 + 5С = 2V + 5СО; 3V2O3 + 10А1 = 6V + 5А1»О3.
Так как, кроме окислов ванадия, в шихте присутствуют
окислы железа, то при этом вместо чистого ванадия
получается его сплав с Fe, состава: 35—80% V, не
менее 3,5% Si, не более 2% А1, 1,0% С, 0,25% Р,
0,15% S, остальное — Fe.
Ферровольфрам содержит 65—80% W.
Его получают из вольфрамовых концентратов, содер-
жащих 50% и выше WO3. Наиболее распространено
получение ферровольфрама одновременным восстанов-
лением окислов вольфрама и железа углеродом в элект-
рич. дуговых печах. При внепечном способе в качестве
восстановителей иногда применяют алюминий и сили-
коалюминий. Основные реакции при этих процессах:
WO3 + ЗС = W + ЗСО; WO8 + 2А1 = W + A1SOS
2WO3 + 3Si = 2W + 3S1OS
Состав получаемого этими способами ферроволь-
фрама приведен в табл. 16.
Таблица 16
Содержание элементов а, %
Марка W Si Мп Си s i р С
не менее не более
В 1 70 0,4 0,2 0,25 0,08 0,05 0,5
В 2 70 0,5 0,4 0,30 0,10 0,8 0,8
В 3 65 1,0 0,7 0,30 0,10 0,12 0,8
ВнепечноЙ спо« до
соб6 80 0,45 0,50 — — — —
а Остальное — Fe. 6 Содержит также 0,50% А1.
Ферромарганец содержит не менее 70% Мп.
Основными методами его получения являются: домен-
ный процесс, электротермич. и металлотермич. спо-
собы. При выплавке ферромарганца в доменных пе-
чах в качестве шихтовых материалов применяются
марганцовая руда, кокс и известняк (как флюс). Вос-
становление окислов Мп при высоких темп-рах сво-
дится к след, реакциям:
2MnO + 2С = 2Mn + 2СО; 6МпО + 8С = 2Мп3С -|- 6СО
2MnO + Si = 2Mn + S1O2; 6MnO + 4А1 = Мп + 2А12О3
При восстановлении углеродом получается ферромар-
ганец с высоким содержанием С и повышенным Р.
Состав ферромарганца, получаемого этим способом:
не менее 70% Мп, 6—7,0% С, до 2% Si, не менее
0,35%, Р, 0,03% Si, остальное Fe.
35
ЖЕЛЕЗА СПЛАВЫ
36
В дуговых электрич. печах при восстановлении
марганцовой руды коксом образуется электротермия,
ферромарганец, богатый углеродом. Чтобы понизить
его содержание, в шихту вводят силикомарганец, т. е.
сплав Мп с Si и Fe, получаемый восстановлением соот-
ветствующих окислов в электрич. печах. Электро-
термия. ферромарганец содержит не менее 70% Мп;
примесей не более: 2% Si, 0,38% Р, 0,03% S; содержа-
ние С: менее 0,5% в низкоуглеродистом, 0,5—1,5%
в среднеуглеродистом, до 7% в углеродистом.
Ферромолибден содержит не менее 55%
Мо. Его получают внепечным методом восстановления
молибденитового концентрата кремнием — так назы-
ваемым силикотермическим методом. Иногда в каче-
стве восстановителя применяют А1, сплавы Al с Si
(силикоалюминий) и др. Основные реакции восста-
новления:
МоОз 4- ЗС = Мо + 300; 2МоО3 -J- 3SI = 2Мо 4- 3SiO2
МоОз 4- 2А1 = Мо 4- AI2O3
Состав полученного этим методом ферромолибдена
приведен в табл. 17.
Таблица 17
Марка Содержание элементов3, %
Мо Si С I S | Р
не менее не более
Мо 1 Мо 2 Мо 3 55 55 55 1,0 1,5 2,0 0,10 0,15 0,20 0,10 0,15 0,20 0,10 0,15 0,20
а Остальное — Fe.
Феррониобий содержит 30—75% Nb. Его
получают внепечным способом, восстановлением нио-
биевого концентрата или пятиокиси ниобия кремнием
или алюминием. Т. к. в ниобиевых концентратах часто
содержится довольно много тантала, то получаемый
из этих видов сырья феррониобий содержит также и
тантал. Основные реакции восстановления пятиокиси
ниобия кремнием или алюминием:
2Nb2O5 4- 5Si = 4Nb 4- 5S1O2
6Nb2O 5 4- 20A1 = 12Nb 4- ЮА12О3
Получаемый этим способом феррониобий содержит
не менее 50% Nb 4- Та, не более 10—11,5% Si,
7,0% Ti, 7,0% Al, 0,15—0,17% Р, 0,03—0,5% S,
0,12% С, остальное — Fe.
Ферросилиций содержит: от 9 до 15%
Si (малокремнистый), 43—50% Si (среднекремнистый)
и 72—95% Si (высококремнистый). В качестве восста-
новителей при получении ферросилиция применяют
древесный уголь, нефтяной или металлургия, кокс,
иногда каменный уголь. Основные реакции восстанов-
ления и образования ферросилиция:
SiO2 4-2С = Si 4-2СО; FeO 4-С = Fe 4-СО
Fe 4- SI = FeSi
Составы промышленных марок ферросилиция при-
ведены в табл. 18.
Таблица 18
Способ получения Марка Содержание элементов3, %
Si Мп Сг Р S1
не более
Доменный Э лектротермиче- ский ....... ФС1 ФС2 СиЭО Си75 Си45 13 и более 9-13 87-95 72-78 43—50 3,0 3,0 0,5 0,7 0,8 0,2 0,5 0,5 0,15 0,15 0,04 0,05 0,05 0,04 0,04 0,04 0,04 0,04
а Остальное — Fe.
Ферротитан содержит 15—25% Ti, а в от-
дельных марках до 40—50%. Его получают из концент-
ратов титановых руд гл. обр. алюминотермия, восста-
новлением по реакции:
ЗТ1О2 4- 4А1 = ЗТ1 4- 2А12О3
Восстановление можно вести внепечным способом,
а также в электрич. печах закрытого типа. Наряду
с Ti при этом восстановляются и другие окислы, избы-
ток алюминия переходит в ферротитан. Состав полу-
чаемых этим способом промышленных ферротитанов
приведен в табл. 19.
Таблица 19
Содержание элементов3. %
Ti Si | Al | Си | С | S | Р
не более
Марка
Ти1 18 3,5 5,0 3,0 0,2 0,05 0,05
Ти2 18 5,0 5,0 3,0 0,2 0,08 0,08
ТиЗ 18 6,0 8,00 4,0 0,2 0,10 0,10
а Остальное — Fe.
Если восстановителем служит алюминий с повышен-
ным содержанием меди (сплавы типа дуралюмин),
то ферротитан содержит медь. В последнее время
открываются перспективы получения ферротитана
с высоким содержанием титана за счет применения
отходов произ-ва титана и титановых сплавов.
Феррохром содержит 60—85% Сг. Основные
примеси: С, Si, S, Р. Технология получения ферро-
хрома зависит от его назначения. Углеродистый фер-
рохром производят в шахтной электропечи или в до-
менной печи; феррохром с весьма низким содержанием
С (не выше 0,02—0,03%) получают внепечным спосо-
бом — алюминотермия, восстановлением концентра-
тов хромистого железняка FeCrO2 или смеси окисей
Сг и Fe. Основные реакции восстановления хромистых
руд:
14Сг2О3 4- 54С = 4Сг7С3 4- 42СО
2Сг20з 4“ 6С = 4Сг 4' 6СО
2FeO 4- 2С = 2Fe 4- 2СО
Карбиды хрома, хром и железо, сплавляясь, обра-
зуют углеродистый феррохром с содержанием до
6—8% С. Для получения малоуглеродистого ферро-
хрома из высокоуглеродистого проводят окислитель-
ную плавку в электрич. печи с продувкой воздуха
или кислорода; при этом избыточный С окисляется
хромистой рудой. Алюминотермия, способ получения
безуглеродистого феррохрома сводится к реакции:
2Сг2О3 4" 4А1 == 4Сг 4- 2А120з; 3FeO 4- 2А1 = 3Fe 4" А120з
Этот метод может быть применен для получения
как феррохрома из обогащенных хромистых руд, так
и металлич. Сг технич. чистоты из окиси Сг. Составы
различных марок феррохрома приведены в табл. 20.
Таблица 20
Марки феррохрома Содержание элементов3, %
С Сг не менее Si р 1 S
не б олее
Бевуглероднстый . . до 0,06 65 1-1,5 0,06 0,04
Малоуглеродистый . 0,16—0,50 60 1,5—2,0 0,06 0.04
Среднеуглеродистый 0,51—4,0 60 2,5-3,0 0,10 004
Высокоуглеродистый 4,1-8,0 65 3,0—5,0 0,07 0,04
а Остальное — Fe-
Кроме этих основных ферросплавов, широко исполь-
зуемых в пром-сти, можно указать еще два сплава,
37
ЖЕЛЕЗА СУЛЬФАТЫ — ЖЕЛЕЗА СУЛЬФИДЫ
38
имеющих ограниченное применение. Это ферроцирко-
ний и феррофосфор. Ферроцирконий —
сплав железа с цирконием, получаемый в электродуго-
вых печах. Применяется для раскисления сталей,
нейтрализации вредного влияния азота в стали и вве-
дения циркония в сталь. Феррофосфор —
сплав железа с фосфором, получаемый восстановле-
нием апатитов или фосфоритов в доменных или элект-
рич. печах. Применяется для легирования фосфором
нек-рых марок сталей, а также для получения в ваг-
ранках литейных чугунов с повышенной жидкотеку-
честью.
Лит.: Б о ч в а р А. А., Металловедение, 5 изд., М.,
1956; Гуляев А. П., Металловедение, 3 изд., М., 1956;
Богачев И. Н., Металлография чугуна, М.—Свердловск,
1952; Корнилов И. И., Железные сплавы, т. 1—3, М.—
Л., 1945—56 (Ин-т общ. и неорган. химии); Химушин
Ф. Ф., Нержавеющие, кислотоупорные и жароупорные стали,
М., 1945; Елютин В. П., Павлов Ю. А. и Левин
Б. Е., Производство ферросплавов, М., 1951; Машинострое-
ние. Энциклопедический справочник, т. 3, М., 1948; Металло-
ведение и термическая обработка. Библиографический спра-
вочник 1860—1947, сост. М. И. Мышкина и М. А. Раевская,
М., 1952. И. И. Корнилов, В. И. Лихтман.
ЖЕЛЕЗА СУЛЬФАТЫ — сернокислые соли 2-
и 3-валентного железа.
Сульфат 2-валентного железа
(ферросульфат) FeSO4 в пределах от —1,82° до 56,8°
кристаллизуется из водных растворов в форме
FeSO4 • 7Н2О, наз. в технике железным ку-
поросом (зеленым купоросом). FeSO4-7H2O—голу-
бовато-зеленые моноклинные кристаллы с параметра-
ми решетки: а = 14,02 А, Ъ — 6,50 А, с = 11,01 А,
р = 105°34', плоти. 1,899 (15°); на воздухе выветри-
вается и окисляется, окрашиваясь в желтый цвет
вследствие образования на поверхности Fe(OH)SO4.
От 56,8° до 64,0° стабилен FeSO4 4Н2О — зеленого
цвета, выше 64,0° — белый FeSO4 • Н2О. Последний
полностью обезвоживается при 300°, разлагаясь при
этом с выделением SO2, SO3 и образованием основного
сульфата окиси железа. Безводный FeSO4 — белый
порошок, может быть получен обезвоживанием кри-
сталлогидратов в токе Н2; чистый препарат выделить
трудно. Давление диссоциации FeSO4 по реакции
2FeSO4 ZZ Fe2O3 -1- SO2 4- SO3 равно (в мм рт. ст.)'.
254 (614°), 600 (650°). В отсутствии кислорода FeSO4
разлагается с достаточно большой интенсивностью
и полнотой при 700°; в токе воздуха его разложение
значительно ускоряется в присутствии восстановите-
лей — угля и пирита (смесь из 82,3% FeSO4 • Н2О,
15,2% FeS2 и 2,5% угля разлагается в токе воздуха
при 600° за 40—60 мин. на 98%). Теплота образования
FeSO4 Д77»88 = —220,5 ккал!моль.
Растворимость в воде (в %) в расчете на безводную
соль: 14,91 (—1,8°); 21,01 (20°); 35,06 (56,7°); 35,57
(64°); 27,15 (90,1°). В присутствии свободной H2SO4
растворимость уменьшается. В водном р-ре FeSO4
гидролизуется с выделением осадка основной соли,
а в присутствии кислорода окисляется до Fe2(SO4)3.
В водном р-ре FeSO4 восстановляет Си (И) до солей
Си (I), соли золота и серебра — до металлов; нитраты
и нитриты — до NO, с к-рым FeSO4 образует окра-
шенный комплекс FeSO4 • NO (качественная реакция
на ионы NO? и NO’j). Восстановление перманганата
или бихромата калия р-ром FeSO4 в присутствии
H2SO4 используется для количественного определения
железа. Сульфат 2-валентного железа образует
с сульфатами щелочных металлов и аммония двойные
соли, более стойкие против окисления, напр. соль
Мора (NH4)2SO4 • FeSO4 • 6Н2О —светло-зеленые
кристаллы, растворимые в воде.
В пром-сти железный купорос производится как
побочный продукт на металлообрабатывающих заво-
дах из травильных р-ров, к-рые получаются при обра-
ботке стальных изделий с целью удаления с них ока-
лины перед дальнейшей обработкой поверхности.
Кристаллич. железный купорос м. б. выделен из тра-
вильных р-ров при охлаждении их до —5°, —10°
или выпариванием с последующей кристаллизацией
при охлаждении до 20—25°. Железный купорос
используют для борьбы с вредителями садов и слиз-
нями, для уничтожения мхов, лишайников и грибных
спор, а также для питания растений и борьбы с засо-
лением почв; его применяют также в текстильной
пром-сти, для приготовления чернил и минеральных
красок, для консервации древесины, как реактив
в химич. лабораториях и т. д. Отбросные р-ры суль-
фатов железа перерабатывают на изоляционный мате-
риал — феррон или ферригипс, представляющий
собой смесь гидратов окислов железа и гипса с напол-
нителем. Соль Мора используется в аналитич. химии
как восстановитель.
Сульфат 3-валентного . железа
(феррисульфат) Fe2(SO4)3 — ромбические кристаллы,
плотн. 3,1 (18°), очень гигроскопичны, на воздухе
расплываются. Из водных р-ров кристаллизуется
с 12, 10, 9, 7, 6 и 3 молекулами Н2О. Десятиводный
кристаллогидрат и безводная соль встречаются в
природе в виде минералов квенстетита и коквимби-
та. Давление диссоциации Fe2(SO4)3 по реакции
Fe2(SO4)3 7^ Fe2O3 4- 3SO3 (в мм рт. ст.)'. 70 (614°),
149 (650°). Fe2(SO4)3 • 9Н2О — желтые кристаллы,
гексагональна^ решетка, а = 10,85 А, с = 17,03 А;
плотн. 2,1. При 20° в 100 г воды растворяется 440 г
Fe2(SO4)3 • 9Н2О. В водных р-рах сульфат окиси
железа сильно гидролизуется, особенно при повышен-
ных темп-pax. С сульфатами щелочных металлов и
аммония образует двойные соли — железные
квасцы Me[Fe(SO4)2] • 12Н2О, из которых наибо-
лее распространены железо-аммониевые
квасцы. NH4[Fe(SO4)2] • 12Н2О — фиолетовые кри-
сталлы октаэдрической формы с плотн. 1,71, т. пл.
40°; растворимость (в г на 100 г воды): 124 (25°),
400 (100°); при 150° теряют воду, при 480—490° пре-
вращаются в Fe2(SO4)3.
Сульфат окиси железа обычно получают растворе-
нием окиси железа в 75—80%-ной H2SO4. Растворы
Fe2(SO4)3 могут быть получены окислением серно-
го колчедана азотной кислотой: 2FeS2 4- 10HN03 =
= Fe2(SO4)3 4- H2SO4 4- 4Н2О 4- 10NO. Сульфат окиси
железа используют гл. обр. в качестве коагулянта
при очистке воды, для травления алюминия, меди и
других металлов и пр. Р-р Fe2(SO4)3 способен рас-
творять Cu2S и CuS, что находит применение при гид-
рометаллургия. извлечении меди. Железо-аммониевые
квасцы применяют как аналитич. реактив.
Лит.: П о з и н М. Е., Технология минеральных солей,
2 изд., Л., 1961.
ЖЕЛЕЗА СУЛЬФИДЫ — соединения железа с
серой.
Сернистое железо (моносульфид железа)
FeS — черные кристаллы с металлич. блеском, гекса-
гональная решетка, плотн. ок. 4,8, т. пл. 1193°. Теп-
лота образования ДТ/»88 = —22,72 ккал/молъ. FeS
образуется при нагревании железа с серой; выпадает
также при прибавлении (NH4)2S к р-рам солей Fe (П)
в виде черного осадка, практически нерастворимого
в воде, но легко растворимого в разб. к-тах. На
воздухе влажный осадок FeS легко окисляется до
FeSO4. Продажное сернистое железо (всегда содержа-
щее небольшой избыток Fe) служит для получения
сероводорода.
Двусернистое железо (дисульфид же-
леза) FeS2 — медно-желтые кристаллы с металлич.
блеском. В природе минерал FeS2 встречается в двух
разновидностях: марказит — ромбич. кристаллы,
плотн. 4,86, и пирит (железный колчедан) — ку-
бич. кристаллы, плотн. 5,03. Выше 450° марказит
переходит в пирит. Теплоты образования марказита
39
ЖЕЛЕЗА ХЛОРИДЫ-ЖЕЛЕЗО
40
и пирита ДЯ288 соответственно равны —36,88 и
— 42,52 ккал/молъ. FeS2 растворим в HNO3. На воз-
духе сгорает до Fe2O3 и SO2. Может быть получен про-
каливанием FeCl3 в токе H2S. Природный FeS2 слу-
жит сырьем для получения серной к-ты, серы и желез-
ного купороса.
Серное железо (трисульфид железа)
Fe2S3 — черное нерастворимое в воде вещество, плоти.
4,3. Осаждается сульфидами из р-ров Fe(III). Неустой-
чивое соединение, во влажном состоянии на воздухе
быстро разрушается с образованием Fe(OH)3 и сво-
бодной серы.
ЖЕЛЕЗА ХЛОРИДЫ — соединения железа с хло-
ром, FeCl2 и FeCl3.
Железо хлористое FeCl2 — зеленовато-
серые гексагональные кристаллы, а = 3,57 А, с =
= 17,51 А; плоти. 2,98; т. пл. 672°, т. кип. 1026°. Теплота
образования Д7/»88 =— 81,5 ккал/молъ. На воздухе
желтеет вследствие окисления. Растворимость в воде
40,5% (20°). Водные р-ры слабо гидролизованы. Из
р-ра железа в соляной к-те выделяется FeCl2 -6Н2О —
голубовато-зеленые моноклинные кристаллы с плоти.
1,93. При температурах ниже 12,3° FeCl2-6H2O пе-
реходит в FeCl2 • 4Н2О, при 76,5° он превращается
в FeCl2-2H2O, который при 120° переходит в
FeCl2-H2O. С хлоридами щелочных металлов FeCl2
образует 3 типа комплексов: Me[FeC]3], Me2[FeCl4] и
Me4[FeCle], Получают FeCl2 растворением железа или
его окислов в соляной к-те (в частности, при трав-
лении стальных изделий соляной к-той). Окислением
FeCl2 получают FeCl3 — одну из наиболее важных
солей железа.
Железо хлорное FeCl3 — сильно гигроско-
пичные коричневато-черные кристаллы,„ гексагональ-
ная решетка, а = 5,92 А, с = 17,26 А; плоти. 2,8;
т. пл. 309°, т. кип. 319°. Теплота образования А//2!|8 =
= — 96,8 ккал/молъ. Выше 500° под уменьшенным дав-
лением разлагается на FeCl2 и С12. Растворимость FeCl3
в воде 47,9% (20°). В разбавленных р-рах гидроли-
зуется до Fe(OH)2+, а в очень разбавленных — до
Fe(OH)|. Хлорное железо образует ряд сильно гиг-
роскопичных кристаллогидратов, расплывающихся на
воздухе; из них плавятся конгруэнтно: FeCl3-6H2O
при 37°, FeCl3 • 3,5Н2О при 32,5°, FeCl3 • 2,5Н2О
при 56°, FeC]3 • 2Н2О при 73,5°. Из р-ров могут быть
выделены основные соли — оксихлориды. FeCl3 с
солями щелочных металлов образует комплексы типа
Me[FeCl4] и Me3[FeCle],
Хлорное железо образуется при нагревании железа
с хлором. В больших количествах оно получается как
побочный продукт в произ-ве TiCl4, А1С13 и др. горя-
чим хлорированием руд. FeCl3 можно получить также
хлорированием или окислением раствора (или суспен-
зии) FeCl2 с последующим выпариванием р-ра FeCl3
до концентрации, при к-рой он, остывая, затвердевает
в виде FeCl3 • 6Н2О. Применяют FeCl3 как коагулянт
при очистке воды, как протраву для крашения тканей,
как катализатор в органич. синтезе, а также для при-
готовления других солей железа и железных пигмен-
тов и проч.
Лит..- П о з и н М. Е., Технология минеральных солей,
2 изд., Л., 1961.
ЖЕЛЕЗИСТАЯ КИСЛОТА HFeO2 — не получен-
ная в свободном состоянии очень слабая однооснов-
ная кислота 3-валентного железа; образует соли —
ферриты (см. Железа окислы и гидроокиси).
ЖЕЛЕЗИСТОСИНЕРОДИСТАЯ КИСЛОТА
H4[Fe(CN)e] — комплексная кислота 2-валентного же-
леза; бесцветные кристаллы, хорошо растворимые
в воде и спирте. В сухом состоянии Ж. к. устойчива,
но в присутствии влаги частично окисляется на воз-
духе и постепенно синеет. Из солей Ж. к. наиболее
важен железистосинеродистый калий K4[Fe(CN)e].
ЖЕЛЕЗИСТОСИНЕРОДИСТЫЙ КАЛИЙ
K4[Fe(CN)e] — то же, что желтая кровяная соль,
или калия гексацианоферроапг.
ЖЕЛЕЗНАЯ КИСЛОТА H2FeO4 — не получен-
ная в свободном состоянии кислота 6-валентного же-
леза. Ее соли — ферраты, являются сильными окис-
лителями (см. Железо}.
ЖЕЛЕЗНАЯ ЛАЗУРЬ (берлинская ла-
зурь, милори, парижская синяя) —
минеральный пигмент, представляющий собой желез-
ную соль железистосинеродистой к-ты с постоянной
примесью K4Fe(CN)e, K3Fe(CN)e и воды. Состав тех-
нич. Ж. л. обычно выражают следующей ф-лой:
KxFey[Fe(CN)e]z • п Н2О. Химич, состав: 4—10% ка-
лия, 56—68% Fe(CN)e, ок. 20% железа(вне радикала),
3—17% Н2О; плоти. 1,79—1,92. Цвет лазури в зависи-
мости от состава и условий получения меняется от
темно-синего с бронзовым блеском до светло-синего.
Для приготовления Ж. л. «белый осадок», полученный
при взаимодействии р-ров железного купороса и жел-
той кровяной соли, окисляют бертолетовой солью или
хромпиком. Возможно также окисление кислородом
воздуха. С целью повышения щелочестойкости лазури
можно заменить от 9 до 11% железного купороса сер-
нокислым никелем. Далее полученный осадок лазури
промывают, фильтруют, сушат при 65—70° и измель-
чают.
Ж. л. нерастворима в воде, слабых минеральных
к-тах, спирте, эфире, но легко разрушается даже
слабыми щелочами, при этом цвет из синего становится
бурым. При нагревании до 170—180° цвет Ж. л.
не меняется, но свыше 200—280° она разрушается.
Кристаллизуется Ж. л. в кубической системе. Размер
частиц не превышает 0,1—0,2 р. Вследствие высо-
кой дисперсности Ж. л. очень интенсивна, укрыви-
стость составляет 10—20 г/м2, маслоемкость 40—50%.
Ж. л. относительно светостойка, но не коррозионно-
стойка.
В смеси со свинцовыми или цинковыми кронами
дает пигмент зеленого цвета. Применяется в лакокра-
сочной пром-сти при приготовлении масляных, эмале-
вых, нитролаковых и др. красок синих и зеленых то-
нов, кроме того, применяется при приготовлении
полиграфия, красок.
Лит..-Б е л е н ь к и й Е. Ф. и Р и с к и н И. В., Химия
и технология пигментов, 3 изд., Л., 1960. Н. С. Рааудова.
ЖЕЛЕЗНЫЕ КВАСЦЫ — см. Железа сульфаты.
ЖЕЛЕЗНЫЙ КУПОРОС — см. Железа сульфаты.
ЖЕЛЕЗО (Ferrum) Fe — химич. элемент VIII гр.
периодич. системы Менделеева; п. н. 26, ат. в. 55,85.
Состоит из четырех стабильных изотопов: Fe54 (5,84%),
Fe66 (91,68%), Fe57 (2,17%) и Fe58 (0,31%). Из искус-
ственно радиоактивных изотопов важнейшие, приме-
няемые как меченые атомы, Fe55 (Ti/2 = 2,94 года) и
Fe59 (Ti/z = 45,1 дня). Конфигурация внешних элект-
ронов атома 3rfe4s2. Энергия ионизации Fe°—>Fe+—►
—>-Fe21—>Fe3r составляет (в эв): 7,87; 16,11; 43,43.
Металлическое Ж. известно с древнейших времен; начало
его применения в истории материальной культуры относится
к 8—6 вв. до н. э.; с этим временем связано возникновение
и развитие металлургии Ж. — т. наз. «железный век».
Железо в природе является одним из
самых распространенных элементов, его содержание
в земной коре составляет 5,10 вес. % и из металлов оно
уступает в этом отношении только алюминию. В земной
коре Ж. в свободном состоянии изредка встречается
в виде т. наз. самородного железа (фер-
рит). Различают самородное Ж. метеорное — космич.
происхождения (см. Метеориты) и теллурическое —
земного происхождения. Образование теллурич. Ж.
связано с процессами застывания основных и ультра-
основных магм — при наличии в магме углерода Ж.
восстановляется из окислов и сульфидов. В отличие от
41
ЖЕЛЕЗО
42
метеорного Ж., обычно содержащего значительные
примеси Ni и Со, теллурич. Ж. является почти чистым
Ж. с незначительными примесями: до 0,6% Ni, иногда
до 2%; до 0,3% Со, 0,4% Си, 0,1% Pt. Практич. зна-
чения самородное Ж. не имеет.
Ж. входит в состав многих минералов, из к-рых
слагаются месторождения железных руд. Важнейшие
рудные минералы Ж.: магнетит (магнитный
железняк) РвзО4 (содержит до 72,4% Ж.), гема-
тит (железный блеск, красный железняк) Fe2O3
(до 70% Ж.), гетит HFeO2 и гидрогетит
(лимонит) HFeO2 • aq (ок. 62% Ж.), сидерит
FeCO3 (48,2% Ж.). Наряду с полезными примеся-
ми — Мп, Сг, Ni, Ti, V, Со — железные руды содер-
жат и вредные примеси — серу, фосфор и др. Ж.
входит в состав многих природных силикатов, значи-
тельные скопления к-рых могут иметь промышлен-
ное значение. Месторождения железных руд обра-
зуются в различных геология, условиях; с этим
связано разнообразие состава руд и условий их за-
легания.
Железные руды разделяются на след,
промышленные типы: 1) Бурые железня-
ки — руды водной окиси Ж. (главный минерал —
гидрогетит), залегающие в осадочных породах и со-
держащие 55—30% Ж. и меньше. К месторождениям
этого типа относятся Керченское, Лисаковское и др.
2) Красные железняки, или гематитовые
руды (главный минерал — гематит, иногда с магнети-
том), приуроченные гл. обр. к метаморфизованным
месторождениям. Содержание Ж. в богатых гематито-
вых рудах составляет 51—66% при небольшом коли-
честве вредных примесей (серы и фосфора). Крупней-
шие месторождения этих руд в СССР — Криворож-
ский бассейн, Белгородское, Ангаро-Питский бассейн
и др., в США — район Верхнего оз. и т. д. 3) М а г-
нитные железняки, или магнетитовые, кон-
тактово-метасоматич. руды, главным рудным минера-
лом к-рых является магнетит, иногда окисленный (мар-
тит). Содержание Ж. в богатых магнетитовых рудах
колеблется от 50 до 65%. К этому типу принадлежат
месторождения гор Магнитной, Высокой и Благодати,
Горно-Шорское, Соколовское, Сарбайское, Ангаро-
Илимского бассейна в СССР, Айронспринг в США и
др. Особую разновидность магнетитовых руд представ-
ляют титано-магнетитовые руды, являющиеся комп-
лексными железо-титано-ванадиевыми рудами. 4) С и-
деритовые, или карбонатные, осадочные руды
(шпатовые железняки) с содержанием в среднем 30—
35% Ж. 5) Силикатные осадочные железные
руды, содержащие 25—40% Ж. В нек-рых районах
(напр., в Актюбинской области) встречаются комплекс-
ные железные руды, содержащие наряду с Ж. ни-
кель, кобальт и хром.
Физические и химические свойства. Ж. — блестя-
щий, серебристо-белый пластичный металл. Легко
подвергается ковке, прокатке и другим видам обра-
ботки в горячем и холодном состояниях. Физич. свой-
ства Ж. зависят от его чистоты. Приведенные ниже
данные относятся к чистейшему Ж. с общим содер-
жанием примесей менее 0,01%. Ж. имеет две кристал-
лин. модификации, а и у. Ниже 910° устойчиво а-Ж.,
обладающее объемно-центрированной кубич. решет-
кой, а = 2,86645 А (20°) и плоти. 7,874 (20°). Между
910° и 1400° устойчиво у-Ж. с гранецентрированной
кубич. решеткой с параметром а = 3,64 а . Выше
1400° у-Ж. переходит в 6-Ж. со структурой а-модифи-
кации (а = 2,94 А), устойчивое до темп-ры плавления.
Характерным свойством а-Ж. является ферромагне-
тизм. При темп-рах выше 769° (точка Кюри) ферромаг-
нетизм исчезает при сохранении структуры а-Ж.
Парамагнитное Ж., устойчивое в интервале 769—
910°, иногда наз. (3-Ж.
Атомный радиус Ж. 1,26 А; ионные радиусы Fe2+
0,80 A; Fe3+ 0,67 А. Т. пл. 1539°, т. кип. ок. 3200°.
Теплота плавления 3,64 ккал/г-атом, теплота испаре-
ния 81,3 ккал)г-атом (т. кип.); термич. коэфф, линей-
ного расширения 11,7 • 10 е (20°); теплопроводность
0,177 кал)см град сек (25°). Теплоемкость Ж. зави-
сит от его структуры и сложным образом изменяется
с темп-рой. При 20° атомная теплоемкость Ср =»
6 кал/г-атом град. Начиная с 250° теплоемкость
резко возрастает, достигая в точке Кюри (769°) мак-
симума — ок. 18 кал/г-атом град, затем круто сни-
жается, образуя перелом при ат^у-превращении.
Средняя уд. теплоемкость Ж. в интервале 0—1000“
0,153 кал/г град. Уд. электросопротивление 9,7-
• 10-6 ом • см (20°), термич. коэфф, электросопро-
тивления 6,51 • IO’ 3 (0—100°). При исследовании Ж.
до 0,75° К сверхпроводимость не была обнаружена.
Модуль нормальной упругости Ж. (модуль Юнга)
19—21 • 103 кГ/мм?', температурный коэфф, модуля
Юнга 4 10-6; модуль сдвига 8,4 • 103 кГ/мм2', кратко-
временная прочность (на разрыв) 17—21 кГ/мм2', отно-
сительное удлинение 45—55%; твердость по Бринеллю
35—45 кГ/мм2', предел текучести при растяжении
(о'о,2) 10 кГ/мм2. Ударная вязкость 30 кГм/см2.
По химич. свойствам Ж. как переходный элемент
близок к соседним элементам той же группы перио-
дич. системы — никелю я кобальту. В соединениях Ж.
чаще всего 2- и 3-валентно, но известны также валент-
ности 1, 4 и 6. Для высших валентных состояний Ж.
характерны кислотные свойства. Ж., особенно 3-ва-
лентное, склонно к комплексообразованию. В химия,
отношении Ж. — металл средней активности. В сухом
воздухе при нагревании до 150—200° на поверхности
компактного Ж. образуется тонкая защитная окисная
пленка, предохраняющая его от дальнейшего окисле-
ния. Эта пленка образуется в результате быстрой ад-
сорбции кислорода, и толщина ее не превосходит
30—40 А. Во влажном воздухе Ж. легко окисляется
и покрывается ржавчиной, состоящей гл. обр. из
гидратированной окиси Ж. (см. Коррозия металлов).
Из кислородных соединений Ж. наиболее характер-
ны окислы двух- и трехвалентного Ж. Известны
следующие окислы Ж.: закись FeO, закись-окись
Fe3O4 и окись Fe2O3, существующая в двух модифика-
циях: а и у. Закись Ж. FeO образуется при окислении
Ж. выше 570°. В этих условиях фаза FeO является
устойчивой и расположена на поверхности металла.
Над ней располагаются последовательно слой Fe3O4
и a-Fe2O3. Переход от Fe3O4 к a-Fe2O3 осуществляется
через промежуточный этап образования окиси y-Fe2O3,
обладающей той же кубич. структурой, что и Fe3O4.
Однако y-Fe2O3 претерпевает полиморфное превраще-
ние и переходит в устойчивую модификацию a-Fe2O3
ромбоэдрич. структуры. При понижении темп-ры
(ниже 570°) фаза FeO распадается на Fe Д- Fe3O4
и на поверхности металла остается Fe^a и над ней
пленка a-Fe2O3. Этот распад происходит достаточно
быстро и практически не зависит от скорости охлажде-
ния Ж. Цвета побежалости, образующиеся на поверх-
ности Ж. при наличии градиента темп-ры вдоль по-
верхности, обязаны только пленке a-Fe2O3, т. к. пленка
Fe3O4 непрозрачна для видимого света. Образующаяся
на Ж. при низких температурах (до 200°) в сухом
воздухе тончайшая окисная пленка представляет
собой, по-видимому. y-Fe2O3, но, возможно, и Fe3O4.
Отсутствие ясности в этом вопросе связано с большими
трудностями идентификации этих окислов, т. к. они
обладают одной кристаллич. структурой, а пленка
весьма тонка (до 40 А). При темп-рах выше 200°
можно с уверенностью утверждать, что образующаяся
на Ж. окисная пленка представляет собой Fe3O4.
Тонкий слой Fe3O4 на поверхности Ж. обладает высо-
кими защитными свойствами против окисления и по-
43
ЖЕЛЕЗО
44
этому в ряде случаев создание этого поверхностного
слоя (воронение) применяется для защиты Ж. от кор-
розии. О свойствах н применении FeO, Fe3O4, Fe2O3
и соответствующих гидроокисей см. Железа окислы
и гидроокиси.
При взаимодействии Ж. с галогенами или с галоге-
новодородами образуются галогениды Ж. Известны
все галогениды 2- и 3-валентного Ж., за исключением
иодида Fe (III). Из них наибольшее практич. значение
имеют железа хлориды. Непосредственное соединение
Ж. с серой приводит к образованию моносульфида
FeS и дисульфида FeS2. Из водных р-ров солей Fe
(III) возможно получение неустойчивого Fe2S3 (см.
Железа сульфиды).
Ж., как и его ближайшие аналоги — кобальт и
никель, способно поглощать водород; поглоще-
ние водорода наблюдается при травлении кис-
лотами и в процессе катодного выделения Ж. при
электролизе. В последнем случае водород проникает
в Ж. в виде протонов, чем и объясняется его глубокая
проникающая способность. Адсорбируясь на дефектах
структуры — дислокациях, границах блоков мозаики
и границах зерен, водород действует как сильное по-
верхностно-активное вещество и резко снижает
прочность и пластичность Ж. (т. наз. водородная
хрупкость). С повышением темп-ры этот эффект исче-
зает необратимо, по-видимому, в связи с образованием
молекулярного водорода. Водород, поглощенный Ж.
в твердом состоянии, находится в Ж. в виде твердых
растворов внедрения. При комнатной темп-ре раство-
римость водорода в a-Fe можно считать < 0,005%.
При нагревании степень поглощения Ж. водорода
возрастает. Полиморфное az^y-превращение Ж.
или плавление Ж. вызывает скачкообразное изме-
нение растворимости в нем водорода. Ниже при-
водится растворимость водорода в Ж. в зависимости
от темп-ры:
Темп-ра, СС . . . 409 620 775 852 904 930 1033 1350 1550 жидк.
Объем водорода на один объем Ж. 0,091 0,103 0,176 0,231 0,335 0,377 0,460 0,821 2,10
Большая часть поглощенного водорода может быть
удалена отжигом Ж. при 850—900°.
Вопрос образования гидридов Ж. и их точные
стехиометрии, составы (FeH, FeH2, FeH3 и FeHe)
нельзя считать окончательно решенным. Имеющиеся
в литературе данные позволяют привести нек-рые
сведения о свойствах этих гидридов: FeH —гранулы
серого цвета, устойчив до 150°; FeH2 — темно-серое
вещество, устойчив под слоем эфира до 50°, при 53—
55,6° переходит в FeH; FeH3 — гранулы черного цве-
та, выше 58—60° переходит в FeH; вычисленные по
измерению упругости диссоциации теплоты образо-
вания гидридов FeH, FeH2 и FeH3 составляют: —10,77;
—7,18 и —9,365 ккал/моль соответственно.
Ж. взаимодействует с азотом; при малых концентра-
циях азот поглощается Ж. и дает твердые растворы
внедрения; при больших концентрациях азота обра-
зуются нитриды Ж., отвечающие составам
Fe4N и Fe2N. В соответствии с диаграммой состояния
системы Fe—N, растворимость азота в a-Fe при эвтек-
тоидной темп-ре (590°) равна ~0,4 ат.%, а в y-Fe
при второй эвтектоидной темп-ре (650°) она равна
10,3 ат.%; при повышении темп-ры растворимость
азота в a-Fe резко повышается (от ~0,02 вес.% при
300° до 0,1 вес.% при 590°), а растворимость в y-Fe,
наоборот, понижается. Максимальная темп-ра устой-
чивого состояния соединения Fe4N составляет 680°,
а для Fe2N не установлена.
При взаимодействии Ж. с углеродом образуются
твердые р-ры внедрения в a-Fe и y-Fe, получившие
названия соответственно феррит и аусте-
нит. При больших концентрациях углерода обра-
зуются карбиды Ж. Согласно диаграмме состоя-
ния системы Fe—С, предельная растворимость угле-
рода в a-Fe при комнатной темп-ре весьма низкая
(s:';0,008 вес.%), по мере повышения темп-ры эта рас-
творимость возрастает н при эвтектоидной темп-ре
(723°) составляет ~0,02 вес.%; его растворимость
в y-Fe значительно выше, чем в a-Fe и при эвтектич.
темп-ре (1147°) составляет 2,06 вес.%. Карбид железа
Fe3C (цементит) неустойчив. Он образуется в про-
цессе кристаллизации Ж. при наличии избытка угле-
рода и сохраняется в структуре сплава при быстром
охлаждении; Fe3C имеет ромбоэдрич. структуру. При
медленном охлаждении или при последующем нагреве
и выдержке в условиях высоких темп-p Fe3C разла-
гается на графит и твердый р-р углерода или в a-Fe
или в y-Fe (в зависимости от темп-ры нагрева). Имеют-
ся нек-рые указания на существование карбида железа
состава Fe2C с очень узкой областью гомогенности.
С фосфором Ж. также активно взаимодействует; при
малых концентрациях он дает с а- и y-Fe ограничен-
ныц твердые р-ры, при больших концентрациях —
соединения. Наиболее устойчивы фосфиды: Fe3P,
т. пл. 1200°, Fe2P, т. пл. 1350° и др. Сплавы Ж. с фос-
фором могут быть изготовлены непосредственным
сплавлением компонентов, термин, восстановлением
водородом нек-рых фосфатов (FePO4, Fe2P2O7 и др.),
а также электролизом соответствующих соединений
Ж. и фосфора. С многими металлами Ж. образует
твердые р-ры и соединения (см. Железа сплавы).
При нагревании железного порошка, полученного
восстановлением из FeO (водородом), в струе СО при
150—200° и давлении ок. 100 атм образуется к ар -
б о н и л Ж. состава Fe (СО)5. Он представляет собой
желтоватую жидкость (т. пл. —20°, т. кип.
103°), растворимую в бензоле и эфире и не-
растворимую в воде. Пентакарбонил Ж. при-
меняется для получения порошка химически
чистого Ж. Из пентакарбонила Ж. могут быть
получены твердые малоустойчивые Fe2(CO)9 и
Гез(СО)12 (см. также Карбонилы металлов).
Взаимодействие Ж. с водяным паром при вы-
соких темп-рах ведет к образованию окислов Ж. и
водорода по уравнениям:
4НаО + ЗЕе ~ ГезО4 4- 4На (ниже 570°)
НаО 4 Fe FeO 4- На (выше 570°)
На использовании второй реакции основан один из
технич. методов получения водорода, ныне устаревший.
Нормальный электродный потенциал Ж. в водной
среде для реакции Fez^Fe2+ 4- 2е, £0 = —0,44 в и
для реакции Fej2Fe3+ -4 Зе, Ео = —0,036 в. В разб.
к-тах Ж. легко растворяется с образованием ионов
Fe2+. Конц, азотной и серной к-тами пассивируется
благодаря образованию тончайшей окисной пленки.
В щелочах, за исключением их конц. горячих р-ров,
Ж. не растворяется.
Своеобразно действие на Ж. азотной к-ты разных
концентраций. Холодная разб. азотная к-та (плотн.
ниже 1,034) растворяет Ж. с образованием нитрата
2-валентного Ж. без выделения водорода, сама восста-
навливаясь при этом до аммиака. Кислота плотно-
стью до 1,115 также дает нитрат 2-валентного Ж., но
при этом продуктами восстановления азотной к-ты
являются азот и закись азота. Кислота плотностью
выше 1,115 образует нитрат трехвалентного Ж. с вы-
делением газообразных окислов азота. Наконец, конц.
к-та плотн. 1,45 и выше растворяет Ж. и пассивирует
его, после чего оно перестает растворяться и в разб.
азотной к-те, вытеснять медь из раствора ее сульфата
45
ЖЕЛЕЗО
46
и т. д. О наиболее важных солях 2- и 3-валентного Ж.
см. Железа сульфаты, Железа хлориды, Железа нит-
раты.
Соли 2-валентного Ж. в водных р-рах и в виде
кристаллогидратов имеют бледно-зеленый цвет. Р-ры
вследствие гидролиза показывают кислую реакцию;
на воздухе постепенно окисляются до Fe (III). Устой-
чивость р-ров солей Fe (II) повышается в присутствии
избытка кислот, тормозящих гидролиз, а вследствие
этого и окисление. Более устойчивы двойные соли Fe
(II) (напр., Me2SO4 • FeSO4 • 6Н2О, где Me — одноза-
рядный ион металла или аммония) и комплексные
соли. Из комплексных солей наиболее известны
гексацианоферраты (II), или гексацианоферроаты,
получающиеся всегда при взаимодействии ионов
Fe2+ и CN- без промежуточного образования циани-
да Fe (II). Гексацианоферраты (II) не ядовиты, так
как группа CN-, содержащаяся в них, сильно свя-
зана в комплекс. Гексацианоферраты (II) щелочных
и щелочноземельных металлов, за исключением ба-
риевой соли, легко растворяются в воде. Из них прак-
тически наиболее важен калия гексацианоферроапг
K4[Fe(CN)e] —т. наз. желтая кровяная соль.
Соли 3-валентного Ж. обычно получаются
путем окисления солей Fe (II) или при растворении
свежеосажденной гидроокиси Fe (III) в кислотах.
Вследствие гидролиза соли Fe (III) в водных р-рах
обнаруживают сильно кислую реакцию. При этом
Fe (III) образует различные полимерные многоядер-
ные оксоионы, напр. [Fe4O3]e+. При взаимодействии
растворимых солей Fe (III) с оксалатом аммония
образуется коричневый осадок оксалата Fe (III),
при избытке реактива — раствор комплексной соли
(NH4)3[Fe(C2O4)3]. Этот раствор под действием света
претерпевает внутреннее окисление-восстановление;
Fe (III) превращается в Fe (II), а ион С2О42+ окисляется
в углекислый газ; реакция может служить для измере-
ния количества энергии света и используется при све-
токопировке чертежей.
Фосфат FePO4 образуется в виде желтовато-белого
осадка при действии Na2HPO4 на растворимые соли
Fe (III). Образование тонкой пленки фосфатов на
поверхности Ж. используется для защиты его от
атмосферной коррозии. Под действием Н3РО4 образует-
ся растворимое бесцветное комплексное соединение,
содержащее анионы [Fe(PO4)3]6-. Взаимодействие
свежеосажденной гидроокиси Fe (III) с уксусной
кислотой приводит к образованию темно-красной
растворимой комплексной соли, содержащей катион
[ Fe3(OH)2(CH3COO)e]+; эта же соль получается при дей-
ствии CH3COONa на растворимые соли Fe (III). Гидро-
лиз комплексной соли дает красно-коричневый осадок
соединения Fe3O3(OH)2CH3COO.
При взаимодействии солей Fe (III) с роданид-ионами
образуется растворимое соединение Fe(SCN)3 кроваво-
красного цвета, извлекаемое эфиром. При
избытке иона SCN- возникают комплекс-
ные соединения вплоть до [ Fe(SCN)e]3-,
все растворимые в воде и окрашенные
в различные оттенки красного цвета.
Носителем кроваво-красной окраски в вод-
ных р-рах без большого избытка родани-
да является в основном катион FeSC N2+.
Простой цианид 3-валентного Ж. неизве-
стен. Напротив, гексацианоферраты (III),
или гексацианоферриаты, хорошо изуче-
ны. Наибольшее практич. значение имеет калия
гексацианоферриат K3Fe(CN)e (т. наз. красная кро-
вяная соль). Гексацианоферриаты менее устойчивы
в водной среде, нежели гексацианоферроаты, по-
этому ядовиты. Раствор K3[Fe(CN)e] действует окис-
ляющим образом на ион Fe (II), согласно реакции
Fe(CN)r4-Fe2<-+[Fe(CN)e]~4-Fe3+.
При окислении хлором в щелочной среде соединений
Fe (III) получаются соединения Fe (VI) — фер-
раты (VI) или просто ферраты. По химич. поведению
они близки хроматам и обладают более сильными окис-
лительными свойствами, чем перманганаты; наиболее
растворимый из них K2FeO4. В кислом р-ре ферраты
(VI) выделяют кислород и дают соли Fe (III). При
постепенном разложении ферратов (VI) в щелочной
среде получены нек-рые ферраты (IV).
Аналитическое определение. Ка-
чественно Fe (III) можно определить по образованию
синего осадка с KJFe(CN)e] («берлинская лазурь»)
или по образованию кроваво-красного окрашивания
с р-ром роданида калия (аммония); Fe (II) определяют
по образованию темно-синего осадка с K3[Fe(CN)e]
(«турнбуллева синь») или по образованию красного
окрашивания с раствором а,а'-дипиридила. Каче-
ственное и количественное определение Ж. можно
проводить спектральным методом. При определении
Ж. весовыми методами в качестве осадителей приме-
няют аммиак, формиат аммония, ацетат аммония,
купферон, неокупферон и оксихинолин. Весовой фор-
мой служит Fe2O3. Титриметрич. методы определения
Ж. основаны на титровании Fe (II) различными окис-
лителями [КМпО4, К2Сг2О7,соли Се (IV) и др.]; вос-
становление Fe (III) до Fe (II) можно провести с ис-
пользованием в качестве восстановителей SnCl2,
SO2, H2S, Zn. Можно также применять методы титро-
вания непосредственно Fe (III) различными восстано-
вителями, как, напр., SnCl2, солями Ti (III) или солями
Сг (II), иодометрически и т. д. Имеются также методы
комплексометрия, и фотометрия, титрования Ж.
Фотометрия, методы определения Fe (III) основаны
на реакциях с роданидом, салициловой и сульфосали-
циловой к-той и др. реактивами; Fe (II) — с о-фенант-
ролином, а,а'-дипиридилом, диметилглиоксимом и др.
Имеются полярография., амперометрия., кулономет-
рия. и др. методы определения Ж. Для отделения Ж.
от других элементов используют различные варианты
его гидролитич. осаждения; экстракционные методы
(напр., экстракция HFeCl4 эфиром из солянокислой
среды).
Получение. Ж. в чистом виде получают различными
методами: электролизом водных р-ров его солей, тер-
мин. разложением в вакууме пентакарбонила Ж.
и др. Технически чистое Ж. — «Армко железо», «ВИТ>
и др. марки, а также технич. Ж. — нек-рые марки
низкоуглеродистых сталей (содержащие 0,10—0,12%
С), производят в мартеновских печах (см. Пирометал-
лургия). В последнее время большое значение приоб-
рело прямое восстановление Ж. из окислов в твердом
состоянии.
В таблице приводится содержание примесей в некото-
рых марках Ж., получаемых приведенными выше ме-
тодами:
Марка железа Содержание примесей в вес. %
С S1 Мп N1 S р О N сумма при- месей в %
Технич. Ж. . . . 0,12 0,20 03 0,5 0,04 0,04 не опреде- лены 1,4
Армко Ж 0,02 0/33 0335 0,14 0,02 0,015 то же 0,27 0,0207
Чистейшее Ж. . 0,001 0,002 0,002 0,007 0,004 0,001 0,003 10,0003
Карбонильное Ж. 0,00016 Химическими методами не определяются 030016
Все эти методы получения чистого Ж., за исключе-
нием мартеновского, будучи пока дорогими, приме-
няются гл. обр. для исследовательских целей и
произ-ва специальных изделий. Одной из разновид-
ностей технически чистого Ж. является сварочное-
Ж., получаемое различными способами в виде тесто-
образных частиц рафинированного Ж. с включениями
47
ЖЕЛЕЗО
48
незначительного количества равномерно распределен-
ного шлака.
Основным промышленным способом получения Ж.
служит произ-во его в виде различных сплавов с уг-
леродом — чугунов и углеродистых сталей. Чугуны
получают доменным процессом, а стали — мартенов-
ским, конверторным и электроплавильным процес-
сами. В доменном процессе в качестве основных ших-
товых материалов участвуют: железная руда, кокс и
известняк в пропорциях, необходимых для восстанов-
ления окислов Ж. в руде углеродом и разделения
расплавленных чугуна и шлака. В домну подается
воздух или, для ускорения процесса, кислород (кисло-
родное дутье). Углерод кокса окисляется кислородом:
С + О-. = СО2; С + СО2 = 2СО
Образующаяся при этом закись углерода и углерод
кокса восстанавливают окислы Ж.:
3Fe2O з 4- СО = 2Fe3O4 -гСОз; Fe3O ; 4- СО = 3FeO 4~ СОз
FeO 4- СО = Fe 4- СО2; FeO + С = Fe 4- СО
Поскольку указанные реакции протекают при из-
бытке углерода, восстановленное Ж. сплавляется
с углеродом и образуется чугун со значительно более
низкой темп-рой плавления, чем чистое Ж. Чугун
эвтектич. состава (с 4,3 вес. % С) плавится при 1135°,
а Ж. при 1539°. Расплавленные низкоплавкие чугун
и шлак собираются в горне доменной печи и перио-
дически выпускаются через специальные отверстия.
Т. к. в шихтовых материалах (руде и коксе) содержатся
и другие примеси, то получаемый чугун не является
двойным сплавом Ж. с углеродом; в него переходят
кремний, марганец, сера, фосфор и другие примеси,
одни из к-рых полезны (Si, Мп), а другие вредны (S, Р).
При плавке комплексных руд, содержащих никель,
кобальт, хром (см. выше), в доменных печах или элект-
ропечах получаются легированные этими элемента-
ми чугуны — так называемые природнолегированные
чугуны.
Чугуны по назначению разделяются на передельный
и литейный. Передельный чугун, состав-
ляющий ок. 80% всего производимого чугуна, служит
полупродуктом, идущим на последующую переработку
в углеродистые и др. стали. Литейный чугун
применяется для произ-ва чугунных отливок. Хро-
мисто-никелевые легированные чугуны используются
или для дальнейшего извлечения из них никеля, или
для изготовления малолегированных никелевых и
хромо-никелевых сталей.
Способы передела чугуна — мартеновский, конвер-
торный и электроплавильный, сводятся к удалению
избыточного углерода и вредных примесей (серы, фос-
фора) путем их окисления (выжигания) и к доводке
содержания легирующих элементов до заданного
путем добавления их при плавке. Предельно допусти-
мое содержание вредных примесей и необходимое
содержание легирующих элементов установлены для
каждой марки стали. Выжигание примесей идет за
счет кислорода воздуха или добавляемых в печь окис-
лов железа (железного лома, окалины, железной руды).
В зависимости от конкретных способов получения
углеродистые стали классифицируются следующим
образом: 1) основная мартеновская
сталь — получается в мартеновских печах с ос-
новным подом печи; 2) кислая мартенов-
ская сталь — в печах с кислым подом с набивкой
кремнеземистого песка; 3) конверторная
сталь — выжиганием примесей в конверторах,
продуванием воздуха или кислорода через расплав-
ленный чугун; 4) электросталь — плавкой
в дуговых и индукционных печах.
Сложность металлургич. процесса получения Ж.
и сталей, включающего доменный процесс и передел
чугуна, является причиной постоянного развития и
усовершенствования т. наз. метода прямого получения
Ж. из железных руд, существующего многие столе-
тия. Еще Д. И. Менделеев (в 1899) предсказывал, что
придет со временем опять пора искать способы прямого
получения железа и стали из руд, минуя чугун. Начи-
ная примерно с 1930 гг., в нашей стране, так же как
и в других странах, выполнены крупные исследования
в этом направлении и достигнуты к настоящему вре-
мени существенные результаты. Проблема получения
Ж. этим способом сводится к решению двух задач:
1) восстановления Ж. из окислов без значительного
науглероживания его и загрязнения примесями
и 2) отделения получаемого Ж. от пустой породы.
В качестве восстановителя при этом применяются
твердый углерод или газы — водород, природный газ
и др. По температурному режиму этот способ получе-
ния Ж. может быть разделен на три группы: 1) вос-
становление при сравнительно невысоких темп-рах
(ок. 1100—1150°) с получением губчатого Ж.; 2) вос-
становление во вращающихся печах с темп-рой про-
цесса 1300—1350° и с получением сваренных частиц
Ж. в виде крица и тестообразного шлака; 3) восстанов-
ление с получением конечного продукта в жидкой
фазе. Получаемое этими способами губчатое Ж.
потребляется в широких масштабах в качестве ших-
тового материала для плавки сталей в мартеновских
и электропечах, для получения порошкообразного
Ж. и других целей.
Применение. Ж. — главный и основной металл для
современной техники. Применение чистого Ж. огра-
ничено, т. к. по своим механич. свойствам оно не удо-
влетворяет ряду требований к конструкционным ма-
териалам. Чистое Ж., сварочное Ж. и Ж., получаемое
методами порошковой металлургии, отличается высо-
кими пластич. свойствами, хорошо сваривается и
имеет высокие магнитные свойства. Поэтому оно при-
меняется в тех областях техники, где требуются высо-
кие пластич. свойства металла и особые физические
свойства.
Наибольшее применение Ж. находит в виде углеро-
дистых и легированных другими элементами сталей и
сплавов и специальных марок чугунов. Углеродисты-
ми сталями наз. все сплавы Ж. с углеродом, содержа-
щие до 2% С; сплавы с более высоким содержанием
углерода относятся к чугунам. В зависимости от на-
значения и областей применения различают: ^кон-
струкционные стали — применяемые для
строительства зданий, мостов, судов, вагонов, разных
сооружений и машин; 2) инструменталь-
ные стали — для изготовления различных ин-
струментов. Эта группа сталей делится на углероди-
стые и легированные инструментальные стали, бы-
строрежущие стали и др.; 3) стали с особы-
ми физическими свойствами — а) не-
ржавеющие, коррозионностойкие, б) жаростойкие
и жаропрочные, в) электротехнические (магнитные,
стали с высокой электропроводностью и с высоким
электросопротивлением), г) стали с особыми физич.
свойствами, напр. малым тепловым расширением
и др. Все эти группы сталей по назначению находят
широкое применение в самых разнообразных отрас-
лях народного хозяйства и новых областях техники
(см. Железа сплавы).
Лит.: Mellor, v. 12, 13, 14, L., 1942; т о ж е, v. 12 13,
14, L., 1953; Pascal, v. 18, Р., 1959; G m е I i n, 8 A.U11.
Syst.-Nummer 59, Eisen, B.. 1929 —59; llllmann, 3 Aull.,
Bd 6, Munch. —B., 1955, S. 261—428; Славинский M. П.,
Физико-химические свойства элементов, M., 1952; Кэмп
Д. М. и Ф р е н с и с К. Б., Производство и обработка стали,
пер. с англ., под редакцией И. П. Бардина, 5 изд., М., 1946;
Hansen М., A n d е г к о К., Constitution of binary alloys,
2 ed., N. Y., 1958; Тимошенко H. H., Прямое восстанов-
ление железных руд, М., 1959; Гудремон Э., Специаль-
ные стали, пер. с нем., т. 1, М., 1959; Б о з о р т Р., Ферро-
магнетизм, пер. с англ., М., 1956; Г и л л е б р а н д В. Ф. [и
49
ЖЕЛЕЗООКИСНЫЕ ПИГМЕНТЫ — ЖЕЛЧНЫЕ ПИГМЕНТЫ
50
др.], Практическое руководство по неорганическому анализу,
пер. с англ., М., 1957, с 402; Анализ черных металлов, под
ред. С. В. Липина, М.—Свердловск, 1951; Берг И. А., Тех-
ника безопасности в черной металлургии, М., 1954.
ЖЕЛЕЗООКИСНЫЕ ПИГМЕНТЫ — см. Пиг-
менты железоокисные.
ЖЕЛЕЗОСИНЕРОДИСТАЯ КИСЛОТА Н3 [Fe(CN)e]-
комплексная кислота 3-валентного железа; кристаллы
бурого цвета. Свободная Ж. к. может быть получена
взаимодействием конц. р-ров K3[Fe(CN)e] и НС1.
Из солей Ж. к. наиболее важен железосинеродистый
калий K3[Fe(CN)e].
ЖЕЛЕЗОСИНЕРОДИСТЫЙ КАЛИЙ K3[Fe(CN)eJ—
то же, что красная кровяная соль, или Калия гек-
сацианоферриат.
ЖЕЛТЫЙ ФЕРМЕНТ ВАРБУРГА — см. Флави-
новые кислоты
ЖЕЛЧНЫЕ КИСЛОТЫ — группа стероидных к-т,
входящих в состав желчи. Почти все Ж. к. являют-
ся производными не встречаю-
щейся в природе холановой
кислоты. Сведения о строении,
физических свойствах и источ-
никах выделения важнейших
природных Ж. к. приведены
в таблице.
Название кислоты Положение и конфи- гурация ОН-групп Т. пл., 'С [a]D Источник выделения
Литохолевая . . . За 184—186 + 32° Желчь челове- ка, быка, кро- лика, желч- ные камни
а-Гиодезоксихолевая За,6а 196-197 + 8° Желчь свияьи
^-Гиодезоксихолевая X енодезоксихоле- вая (антроподезо- ксихолевая, гал- лодезоксихоле- ЗР ,6а 189-190 + 5° Желчь свияьи
вая) Урсодезоксихоле- За,7а 140 4-11° Желчь челове- ка, быка, гу- ся, курицы, медведя,мор- ской свинки
вая За, 70 203 4-57° Желчь медве- дя
Дезоксихолевая . . За,12а 176-177 4-53° Желчь челове- ка, быка, оленя, соба- ки, овцы, ко- зы, антило- пы, кролика
Холевая За,7а,12а 196—198 4-37° Желчь челове- ка, быка ов- цы, козы, ан- тилопы
З-Фокохолевая . . . а - Лагодезоксихоле- 3, 7, 23* 222—223 4- 27° Желчь тюленя, моржа
вая f - Лагодезоксихоле- За,12* 156—157 4-80° Желчь кроли- ка
вая Не уста- новлены 213 4-37° То же
Нутриахолевая . . Буфодезоксихоле- То же 198 — Желчь бобра
вая 3, 7, 8, 12-Тетраокси- » » Аморфна — Желчь жабы
холевая 3, 7. 8. 12* 223—225 — Желчь кроли- ка
‘Конфигурация не установлена.
Ж. к. содержатся в желчи человека и животных
в виде пептидоподобных производных аминокислот.
Так, важнейшая Ж. к. — холевая кисло-
та — находится в желчи в виде Na-солей амидов гли-
кокола (гликохолевая к-та) и таурина (таурохолевая
к-та). Свободные Ж. к. получают щелочным гидроли-
зом твердого вещества желчи. Продукт гидролиза
желчи быка состоит в основном из холевой, дезокси-
холевой и незначительных количеств хенодезоксихо-
левой и литохолевой к-т.
Образование Ж. к. происходит в печени и, по-ви-
димому, связано с обменом холестерина, т. к. при
введении животным дейтерохолестерина удается вы-
делить холевую к-ту, содержащую дейтерий. Большое
физиология, значение имеют комплексные соединения
Ж. к. с разнообразными органическими веществами, в
частности растворимые в воде комплексы дезокси-
холевой к-ты (так называемые холеиновые кислоты).
Комплексы Ж. к. с жирами и жирными к-тами игра-
ют большую роль в жировом обмене (см. Обмен ве-
ществ).
Из природных Ж. к. синтетич. методами получен
ряд производных Ж. к. (различные окси-, диокси- и
триоксихолановые к-ты, 3 fj, 7 а-диокси-12-кетохолано-
вая к-та и др.). При нагревании Ж. к. в вакууме про-
исходит их дегидратация с образованием ненасыщен-
ных Ж. к. (а- и [3- литохоленовая к-та, Д5-холеновая
к-та, апохоленовая к-та, 3,12-диокси-Д14-холеновая
к-та, 3,12-диокси-Д7,14-холадиеновая к-та и др.).
При расщеплении Ж. к. и ненасыщенных Ж. к. по-
лучены так называемые билиановые к-ты — производ-
ные холановой к-ты, в к-рых одно из колец рас-
крыто.
Ж. к. применяют для синтеза стероидных гормонов,
содержащих атом кислорода при €„; в частности,
разработаны промышленные методы превращения
холевой и дезоксихолевой к-т в кортизон.
Лит.: Ф и з е р Л., Ф и з е р М., Химия природных со-
единений фенантренового ряда, пер. с англ., М.—Л., 1953;
Назаров И. Н., Бергельсон Л. Д., Химия стеро-
идных гормонов, М., 1955; Fieser L. F., Fleser М.,
Steroids, N. Y.—L., [1959]; Lettrd H., Inhoffen
H. H., T sch each e R., Uber Sterine, Gallens8uren und
verwandte Naturstofte, Bd 1—2, Stuttgart, 1954—59; H e f t-
maun E., Mosettig E., Biochemistry of steroids, N. Y.,
[I960]; H a s 1 e w о о d G. A. D„ Physiol. Revs, 1955, 35,
№ 1, 178; Bergstrom S., Rec. Chem. Progr. 1955, 16,
№ 2, 63. Л. Д. Бергельсон.
ЖЕЛЧНЫЕ ПИГМЕНТЫ — гетероциклич. пир-
рольные соединения; образуются в печени в результате
распада гемоглобина и миоглобина. В структуру Ж. п.
(А) входят 4 пиррольных кольца, соединенных 3 ато-
мами углерода.
I
о н 6 в
А
Полностью насыщенные соединения по а, б и в паз.
биланами, соединения, имеющие 1 двойную связь
(в одном из положений а, б или в),— биленами (би-
лиенами), 2 двойные связи — биладиенами (билидие-
нами), 3 двойные связи — билатриенами (билитрис-
нами). В Ж. п. по месту расположения углеродных
атомов 1, 3, 6 и 7 расположены группы СН3, по месту
4 и 5 — остатки пропионовой к-ты — СН2СН2СООН,
по месту 2 и 8 — винильная или этильная группа.
В положении 1' и 8' находятся гидроксильные группы.
Многообразие различных Ж. п. может быть система-
тизировано по числу двойных связей в положениях
а, б и в и в пиррольный кольцах I — IV, а также по
нахождению в положении 2 и 8 винильных или этиль-
ных групп. В желчи человека и животных в основном
находятся 2 Ж. п.: желто-коричневый — билирубин,
и зеленый — биливердин (Б) — C33H34OeN4,
мол. в. 582,67, т. пл. 300°, слабо растворим в СН3ОН,
СНС13 и др., не дает реакции с диазореактивом Эрлиха
51
ЖЕСТКОСТЬ ВОДЫ
52
(см. ниже). В желчи птиц и амфибий находится почти
исключительно биливердин, к-рый обнаружен также
в виде включений в протоплазме нек-рых простейших.
СООН СООН
Б
Ж. п. с металлами образуют соли, причем соли тяже-
лых и щелочноземельных металлов нерастворимы
в воде (соли Са являются составной частью желчных
камней). В организме Ж. п. восстанавливаются до-
уробилина, уробилиногена и др. Взаимосвязь меж-
ду отдельными Ж. п. можно представить следующей
схемой:
>2Н . .ЮН , *2Н
биливердин-»- билирубин -•- уробилин уробилиноген
-2Н
Ж. п. дают цветные реакции: 1) с конц. HNO3, со-
держащей HNO2 (реакция Гмелина); в по-
граничном слое между Ж. п. и окисляющей смесью
последовательно появляются сине-зеленая, фиолето-
вая и желтая окраски; 2) с диазореактивом Эрлиха
(сульфаниловая кислота 4- NaNO2 -ф- НС1) — реакция
Ван-ден-Берга, и др.
В организме взрослого человека образуется до
280 мг Ж. п. в сутки; при нек-рых заболеваниях их
количество значительно увеличивается.
Лит.: Степаненко Б. Н., в кн.: Усп. соврем, био-
химии, т. 1, М., 1947, с. 269; Асатиани В. С., Биохими-
ческая фотометрия, М., 1957; Балаковский С. Д.
и Б а л а х о в с к и й И. С., Методы химического анализа
крови, 3 изд., М., 1953; Lemberg R. and Legge
J. W., Hematin compounds and bile pigments, N. Y.—L.,
194*9; Kleiner I. S., Human biochemistry, St. Louis,
[1946], p. 197; Gray С. H., Kulczycka A., Nichol-
son D. C., J. Chem. Soc., 1961, June, p. 2276; Klat-
skin G., Bile pigment metabolism, Ann. Rev. Med., 1961,
12, p. 211. И. 3. Сергиенко.
ЖЕСТКОСТЬ ВОДЫ — свойство природной воды,
определяемое присутствием в ней растворенных солей
кальция и магния. Различают жесткость кальциевую,
связанную с присутствием солей кальция, и магние-
вую, зависящую от содержания в воде солей магния.
Суммарное содержание этих солей в воде наз. общей
Ж. в., подразделяемой на карбонатную жест-
кость — концентрацию в воде двууглекислых (и угле-
кислых 1П)и рН>8,3) солей кальция и магния, и
некарбонатную жесткость — концентрацию в
воде кальциевых и магниевых солей сильных кислот.
При длительном кипячении воды из нее выделяется
углекислый газ и выпадает остаток, состоящий преим.
из карбоната кальция; Ж. в. при этом уменьшается.
Поэтому иногда применяют термин «временная
жесткость», понимая под этим присутствие дву-
углекислых солей, Удаляемых из воды при ее ки-
пячении в течение 1 часа. Оставшаяся после кипяче-
ния Ж. в. называется постоянной жест-
костью.
В СССР Ж. в. выражают в миллиграмм-эквивален-
тах на 1 л (1 мг-акв Са2+ = 20,04 мг', 1 мг-акв Mg2+ =
= 12,16 мг). Ж. в., сильно умягченной для питания
ею паровых котлов высокого давления, выражают
в микрограмм-эквивалентах на 1 л (1 мкг-экв =
= 0,001 мг-акв). В других странах Ж. в. измеряют
в градусах жесткости, например в Германии 1° жест-
кости выражает содержание СаО в сотых долях грам-
ма в 1 л воды; в Англии Ж. в. измеряют в граду-
сах жесткости, выражающих содержание СаСО3 в
гранах (1 гран = 0,0648 г) в 1 галлоне (4,546 л)
воды; во Франции 1° жесткости равен 1 г СаСО3 в
100 000 г воды. В табл. 1 приведены сравнения единиц измерения жесткости воды.
Таблица 1
Мг-экв/л* Немецкие градусы Английские градусы Французские градусы
1.00 2,80 3,50 5,00
0,36 1,00 1,25 1,79
0,29 0,80 1,00 1,43
0,20 0,56 0,70 1,00
* Принятая в СССР единица измерения.
Ж. в. чаще всего определяют титрованием р-ром
двунатриевой соли этилендиаминтетрауксусной к-ты
(комплексон III, трилон Б, версен, ЭДТА) в щелочной
среде. Проводя это титрование со специальным инди-
катором (хромоген черный специальный ЕТ-ОС, эрио-
хром черный Т), находят общую Ж. в.; пользуясь дру-
гим специальным индикатором (мурексид), определяют
кальциевую Ж. в.; магниевую Ж. в. рассчитывают
по разности между этими двумя определениями. Если
содержание кальция и магния в воде было определено
другими способами, общую Ж/ в. можно вычислить
по формуле:
_ [Са2+] , [Mgs+1
-иг-экв 20,04 + 12,16
в к-рой Ж мг-акв — общая жесткость воды в мг-акв/л.
Ж. в. природной колеблется в широких пределах;
она различна в разных водах, а в одном и том же водо-
источнике величина ее изменяется по временам
года. Вода с жесткостью менее 4 мг-акв)л характери-
зуется как мягкая, от 4 до 8 мг-акв/л — сред-
нейжесткости, от 8 до 12 мг-акв/л — жест-
кая, выше 12 мг-акв) л— очень жесткая.
Ж. в. главнейших рек СССР в летний период приведе-
на в табл. 2.
Таблица 2
Наимено- вание реки Пункт Ж. в. в мг-экв/л
общая карбо- натная некарбо- натная
Аму-Дарья г. Турткуль 4,7 2,3 2,4
Волга г. Вольск 5,9 3,5 2,4
Вятка г. Киров 2,4 2,4 0,0
Днепр с. Разумовка 3,7 3,2 0,5
Дон ст. Аксайская .... 5,6 4,3 1,3
Енисей г. Красноярск .... 1,3 1,2 0,1
Иртыш г. Омск 1,6 1,3 0,3
Калаус с. Петровское (Став- ропольский край) . 46,3 6,2 40,1
Кама г. Чистополь 5,8 3,1 2,7
Кубань хутор Тиховский . . 2,1 1,8 0,3
Лена с. Кюсюр (Якутская АССР) 1,2 1,1 0,1
Москва с. Татарово 4,2 4,1 0,1
Нева с. Ивановское .... 0,5 0,5 0,0
Обь г. Новосибирск . . . 1,6 1,4 0,2
Ока г. Муром 3,9 0,6 3,3
Печора с. Усть-Дильма . . . 0,4 0,4 0,0
Сев. Двина деревня Звоз (Архан- гельская об л.) . . . 2,9 2,0 0,9
В поверхностных водоисточниках Ж. в. достигает
наибольшего значения в конце зимы, а наименьшего
в период паводка. Так, в р. Волге у г. Горького мак-
симальная Ж. в. бывает в марте (4,3 мг-акв)л), мини-
мальная — в мае (0,5 мг-акв/л). В поверхностных водо-
источниках обычно преобладает карбонатная Ж. в.
(70—80% общей Ж. в.). Магниевая Ж. в. редко пре-
вышает 30% общей Ж. в., однако в нек-рых районах
(Донбасс, Кривой Рог) магниевая Ж. в. достигает
60% общей Ж. в. Жесткость подземных вод, особенно
артезианских колодцев, более постоянна и меньше
изменяется в течение года.
53
ЖИВИЦА —ЖИДКИЕ КРИСТАЛЛЫ
54
Жесткость морской воды: Черного моря — кальцие-
вая 12 мг-акв/л, магниевая 53,5 .иг-акв/л, общая
65,5 мг-экв!л\ Каспийского моря — кальциевая 36,4
мг-акв/л, магниевая 30 мг-акв/л, общая 66,4 мг-акв/л',
Аральского моря — кальциевая 28 мг-акв!л, магниевая
40 мг-акв/л, общая 68 мг-акв/л', океанов — кальцие-
вая 22,5 мг-экв/л, магниевая 108 мг-акв/л, общая
130,5 мг-акв/л.
При питании жесткой водой котлов на их поверх-
ности нагрева отлагается накипь, вследствие чего
уменьшается теплопроводность металла, что приво-
дит к перерасходу топлива и перегреву металлич.
котлов. Пользование жесткой водой в текстильной
пром-сти, в прачечных и в быту для стирки белья обык-
новенным мылом ведет к перерасходу мыла и ухудше-
нию качества тканей; мыло сначала расходуется на
связывание солей, обусловливающих Ж. в., и начинает
давать пену только после того, как эти соли будут
переведены в нерастворимый осадок. Величина потери
мыла при стирке в воде, имеющей 2 мг-акь/л жестко-
сти, равна 1,2 г мыла на 1 л воды, или 24% от общего
расхода мыла; при Ж. в. в 6 мг-акв/л потеря составляет
3,2 г мыла, или 64% от общего расхода мыла. Качество
тканей, стираемых в жесткой воде, а также качество
тканей, при отделке к-рых применяется жесткая
вода, ухудшается вследствие осаждения иа поверх-
ности нитей тканей кальциевых и магниевых солей
жирных к-т мыла. Следует отметить, что синтетич.
моющие вещества (ОП-7, ОП-Ю, некаль, сульфанол
и т. п.) не образуют с ионами кальция и магния нерас-
творимых солей и потому на их моющей способности
Ж. в. не отражается.
Большая магнезиальная Ж. в. придает воде горько-
ватый вкус и оказывает послабляющее действие на
кишечник человека. Поэтому, как правило, в питьевой
воде не должно содержаться более 100 мг[л магния.
Общая Ж. в., подаваемой хозяйственно-питьевыми
водопроводами, при наличии устройства для осветле-
ния и умягчения воды не должна превышать
14,4 мг-акв/л (ГОСТ 2874—54). О способах борьбы
с Ж. в. см. Водоподготовка.
Лит. см. при ст. Водоподготовка. Ю. Ю. Лурье.
ЖИВИЦА (терпентин) — смола, выделяющаяся при
подсочке или повреждении стволов хвойных деревьев.
Свежая, неизмененная Ж. сосны обыкновенной (Pinus
Silvestris) — прозрачная, светлая, вязкая жидкость,
содержащая 35—38% скипидара (терпентинного мас-
ла) и 62—65% кислот смоляных. На воздухе Ж. по-
степенно изменяет окраску и кристаллизуется вслед-
ствие окисления и испарения скипидара. Промышлен-
ная Ж. содержит 16—20% скипидара, к-рый состоит
гл. обр. из смеси терпенов состава С10Н1в (в основном
а- и fl-пиненов, Д3-карена и др.). Кристаллич. часть
Ж. составляют преим. смоляные кислоты с общей ф-лой
С20Н30О2 (левопимаровая, декстропимаровая, абиети-
новая, неоабиетиновая и др.) продукты их окисле-
ния и — в значительно меньших количествах — ре-
зены (высокомолекулярные инертные углеводороды).
Ж. — основное сырье для произ-ва скипидара и кани-
фоли. На основе терпенов и смоляных к-т произво-
дится синтез камфары, терпинеола, эфиров канифоли
и др. химич. продуктов.
Лит.: Васечкин В. С., Технология экстрактивных
веществ дерева, М.—Л., 1953; Медников Ф. А., Подсочка
леса, М,—Л., 1955; Бардышев И. И. [и др.], ДАН
БССР, 1960, 4, М 10, 421. А. Г. Соколов. ,
ЖИДКИЕ КРИСТАЛЛЫ — термодинамически ус-
тойчивое фазовое состояние веществ, промежуточное
по своим свойствам между жидким состоянием и кри-
сталлическим. Известно ок. 3000 органич. веществ,
способных к образованию Ж. к. Все они имеют моле-
кулы удлиненной формы, причем наличие у молекул
боковых ответвлений сокращает область существова-
ния Ж. к. Открыты Ж. к. Рейницеромв 1889. Первыми
aW'f'J Iifmtwm
6
а
Характер расположения моле-
кул в Ж. к.: а — нематическая
форма, б — смектическая форма.
веществами, полученными в жидкокристаллич. со-
стоянии, были холестерилбензоат, пара-азоксифенетол,
пара-азоксианизол, этиловый эфир пара-азоксибен-
зойной к-ты.
На диаграмме состояния Ж. к. всегда имеют четкую
замкнутую область устойчивого существования. Так,
кристаллич. холестерилбензоат переходит в жидко-
кристаллич. состояние при 145°, превращаясь в мут-
ную на вид жидкость, а при 179° переходит в обычный
прозрачный расплав. Мутность жидкокристаллич.
фазы обусловлена тем, что она состоит из большого
числа оптически анизотропных областей, беспорядочно
ориентированных по отношению друг к другу. Как
и обычные жидкости, жидкокристаллич. тела обладают
текучестью и образуют капли, способные сливаться
друг с другом. Однако для жидких кристаллов харак-
терна анизотропия оптич., лектрич., магнитных и
эластич. свойств, присущая кристаллич. состоянию
вещества. Анизотропные свойства Ж. к. являются
следствием определенной взаимной ориентации длин-
ных молекул, а не внут-
ренним свойством самих
молекул. Известны две ос-
новные структурные раз-
новидности жидких кри-
сталлов (см. рисунок):
а) Нематическая
форма (напр., у пара-
азоксианизола), в кото-
рой молекулы вытянуты
параллельно друг другу,
упакованы в боковом на-
правлении с сохранением
лишь ближнего порядка; продольные сдвиги молекул
не упорядочены, б) Смектическая форма
(напр., олеат аммония), в к-рой молекулы образуют
слои, располагаясь перпендикулярно к плоскости
этих слоев; боковая упаковка молекул в слое,
как правило, характеризуется ближним порядком.
Нек-рые вещества способны образовывать обе разно-
видности, обнаруживая фазовый переход из одной
в другую.
В отличие от жидкости, где форма капель (на к-рые
не действуют внешние силы) всегда сферична,
у нек-рых веществ в жидкокристаллич. состоянии
капли имеют продолговатую форму. При выпадении
из расплава нематич.*кристаллы имеют сферич. форму,
в то время как смектич. жидкие кристаллы приобре-
тают удлиненную форму. Как следствие слоистости
смектич. жидких кристаллов, у них иногда можно
наблюдать ступенчатую форму капель. Вещества
в жидкокристаллич. состоянии удавалось ориентиро-
вать магнитными полями т. обр., что все молекулы
устанавливались вдоль направления магнитных сило-
вых линий. Нек-рые коллоидные системы, напр.
водные растворы мыл, дают образования типа Ж. к.,
называемые лиотропными Ж. к. По мере увеличения
количества растворителя система становится сначала
смектич., затем нематич. и, наконец, переходит в изо-
тропную жидкость. В смектич. мыльных р-рах моле-
кулы мыла образуют двойные слои, обращенные по-
лярными группами к воде, к-рая выполняет роль
прослойки между этими двойными слоями. Наличие
такой структуры объясняет моющее действие мыльных
р-ров.
Ж. к. — не единственное мезоморфное (т. е. про-
межуточное по своей структуре) фазовое состояние
вещества. Его следует отличать от ротационно-
кристаллического (или газокристалличе-
ского) состояния, структура к-рого характеризуется
трехмерной кристаллич. решеткой центров молекул
и беспорядочными поворотами молекул около этих
I центров из-за наличия свободы вращения.
55
ЖИДКИЕ СИСТЕМЫ
56
Исследование Ж. к. имеет важное значение для
теории строения вещества и представляет большой
интерес для техники, медицины, биологии.
Лит.: Disc. Faraday Soc., 1958,25, 1—79; Чистяков
И. г., Кристаллография, 1960, 5, вып. 6, 962—76.
Ю. В. Мнюх.
ЖИДКИЕ СИСТЕМЫ — физико-химия системы,
находящиеся в жидком состоянии в определенном
интервале темп-p при любых соотношениях компо-
нентов. Наиболее подробно изучены двойные системы
(двухкомпонентные, или бинарные).
Взаимная растворимость жид-
костей. Компоненты Ж. с. при данной темп-ре
или в данном интервале темп-p могут: смешиваться
друг с другом в любых отношениях, как, напр.,
вода и спирт; обладать ограниченной взаимной рас-
творимостью, напр. вода и этиловый эфир (при 20°
вода растворяет 6,48 вес.% эфира, а эфир — 1,2%
воды); быть нерастворимыми друг в друге, точнее,
практически нерастворимыми, как, например, вода и
парафиновое масло (при 16° оно растворяет всего
0,003% воды).
На рис. 1, а изображена диаграмма растворимости,
компоненты к-роп А п В обладают ограниченной вза-
имной растворимостью, причем их растворимость
с повышением темп-ры возрастает. По оси абсцисс
Рис. 1. Диаграмма взаимной растворимости двух жидкостей:
а — с верхней критич. точкой; б — с нижней критич.
точкой; в — с верхней и нижней критич. точками.
отложена концентрация х компонента#., по оси орди-
нат — темп-pa I. Если при темп-ре Д постепенно при
перемешивании прибавлять жидкость В к жидкости А
(или наоборот), то по достижении состава, отвечаю-
щего точке S (или точке U), в системе наблюдается
расслоение, т. е. образование двух жидких слоев,
один из к-рых является насыщенным р-ром
В в А, а другой — насыщенным р-ром А в В.
При повышении темп-ры концентрация
обоих насыщенных р-ров увеличивается,
в связи с чем изображающие их состав и
состояние точки (М и N при г2, О и Р при г3)
сближаются и, наконец, сливаются при
некоторой температуре гк, которая наз. кри-
тич. темп-рой растворения, а отвечающая
ей точка К — критич. точкой растворения
(или просто критич. точкой). Точки, подоб-
ные S и U, отвечающие двум растворам,
находящимся в равновесии при одной
и той же температуре, наз. сопряжен-
н ы м и, а соединяющая их прямая SU — к о н -
нодой (также связующей прямой, или яодой). Кри-
вая SMOKPNU, являющаяся геометрия, местом со-
пряженных точек, наз. бинодальной кри-
вой, или кривой расслоения. Если разделить пополам
конноды SU, MN и ОР, то точки деления будут ле-
жать на прямой, пересекающей бинодальную кривую
в точке К (при условии, что состав выражен в весо-
вых процентах); в этом заключается правило прямо-
линейного диаметра В. Ф. Алексеева. При критич.
темп-ре оба находящихся в равновесии р-ра стано-
вятся тождественными. Выше критич. темп-ры ком-
поненты системы смешиваются друг с другом во всех
отношениях. Часть плоскости диаграммы, лежащая
вне бинодальной кривой, наз. областью, или полем
гомогенности, а часть, охватываемая этой кривой, —
областью, или полем гетерогенности (расслоения).
Если точка, соответствующая данной смеси, попадает
в область гетерогенности (напр., точка К, изобража-
ющая состав смеси, содержащей АВ процентов В при
темп-ре t2 ), то смесь распадается на два раствора,
к-рым отвечают точки М и N. Если же точка, соот-
ветствующая данной смеси, попадает в область гомо-
генности, то смесь представляет собой один р-р. Ж. с.,
подобные описанным выше, называются системами
с верхней критич. точкой, к-рая является максималь-
ной точкой бинодальной кривой. Напр., система
вода — фенол имеет верхнюю критич. точку при
65,85° и 34 вес. % фенола.
Диаграмма растворимости для случая, когда взаимная
растворимость двух жидкостей с повышением темп-ры умень-
шается, изображена на рис. 1, б. Это т. и. системы с нижней
критич. точкой К, к-рая достигается при понижении темп-ры.
Область расслоения (гетерогенности) в таких Ж. с. лежит
выше критич. точки, являющейся минимальной точкой бино-
дальной кривой. Напр., система триэтиламин—вода имеет
нижнюю критич. точку при 19,1° и 31,6 вес. % триэтиламина.
На рис. 1, в изображена диаграмма растворимости Ж. с. с
двумя критич. точками: нижней ТС, и верхней К2. В этой
системе расслоение возможно лишь в нек-ром интервале
темп-p (от 1, до (2), бинодальная кривая замкнута. Напр.,
система вода — никотин имеет верхнюю критич. точку при
208’ и нижнюю — при 60,8 .
Давление пара. Идеальные Ж. с., т. е.
образующиеся из компонентов без изменения объема
и без выделения или поглощения теплоты, подчи-
няются обобщенному закону Рауля, согласно к-рому
при постоянной темп-ре давление пара любого ком-
понента пропорционально выраженной в мольных
долях его концентрации в жидкости, причем коэфф,
пропорциональности является давление пара чистого
компонента. Поэтому общее давление пара идеаль-
ной Ж. с. — величина аддитивная; для двойной
Ж. с. оно изображается в зависимости от мольной
концентрации жидкости х прямой линией РАРВ
(рис. 2, а). Давление пара в зависимости от его со-
става изображается гиперболой РАСРВ, обращенной
вогнутостью вверх и лежащей под указанной прямой
(рис. 2, а). Линия, изображающая давление пара,
в зависимости от состава жидкости, наз. линией
жидкости, а изображающая то же давление в за-
Рис. 2. Изотермические диаграммы давления пара двойных Ж. с.: а — для
идеальной системы; б — без экстремумов; в — с максимумом; г — е ми-
нимумом.
висимости от состава пара — линией пара.
Для неидеальных двойных Ж. с. при неограниченной
взаимной растворимости компонентов следует раз-
личать 3 случая: экстремум (т. е. максимум или мини-
мум) на линиях жидкости и пара отсутствует (рис. 2,6);
обе эти линии имеют максимум (рис. 2, е); обе эти
линии имеют минимум (2, г).
Давление пара двойных Ж. с. подчиняется законам
Д. П. Коновалова: 1) Пар относительно богаче тем
компонентом, прибавление к-рого повышает общее
давление пара системы. 2) Экстремуму на кривой
давления пара двойной Ж. с. отвечают жидкость и
пар одинакового состава. Ж. с., имеющие экстремум
давления пара, наз. азеотропными смесями.
57
ЖИДКИЕ СИСТЕМ-Ы — ЖИДКИЕ УДОБРЕНИЯ
58
На рис. 2, б, в, г изображены изотермич. (при по-
стоянной темп-ре) диаграммы давления пара двойных
Ж. с. Линия жидкости обозначена буквой Z, линия
пара — буквой g. Под линией пара лежит поле
пара, точки к-рого изображают состав и темп-ру
пара в отсутствии жидкости. Над линией жидкости
лежит поле жидкости, точки к-рого изобра-
жают состав и темп-ру жидкости без пара. Смесям
жидкости и пара, находящимся в равновесии, соот-
ветствует поле, лежащее между линиями жидкости
и пара; напр., точка h2 на рис. 2, б изображает смесь
пара, отвечающего точке с и жидкости, отвечающей
точке d.
Отношение количества пара и жидкости в смеси опреде-
ляется отношением отрезков Ми ch2 по правилу рычага. Пар
состава h начинает конденсироваться, когда его давление
достигает величины, изображаемой точкой ht. Появлению
первой капли жидкости отвечает точка /, конденсация закан-
чивается, когда давление достигает величины, изображаемой
точкой h3 (последнему пузырьку пара отвечает точка 1). Диаг-
раммы давления пара Ж. с., в к-рых происходит расслоение,
характеризуются тем, что давление пара двух жидких слоев,
находящихся в равновесии, одинаково.
Температуры кипения. Жидкость заки-
пает тогда, когда давление ее пара становится равным
внешнему давлению; т. обр. большему давлению пара
жидкости отвечает более низкая темп-ра кипения.
На диаграммах состав — темп-ра кипения тоже име-
ются кривая жидкости и кривая пара, иногда наз.
кривой кипения и кривой конден-
сации, т. к. они дают темп-ры начала н конда кипе-
ния. Кривая кипения изображает зависимость темп-ры
начала кипения Ж. с. от состава жидкости, а кривая
конденсации — от состава пара. Первая кривая лежит
под второй, что обратно их расположению на изотер-
мич. диаграммах состав — давление пара. Если на
диаграммах давления пара кривая повышается, то на
диаграммах темп-p кипения
соответствующая кривая по-
нижается (и обратно). Кри-
вым с максимумом на диаг-
раммах давления пара отве-
чают кривые с минимумом
на диаграммах температур
кипения и обратно, причем
составы, отвечающие этим
максимумам и минимумам,
не одинаковы. Ж. с., отве-
чающие экстремумам темп-р
кипения, наз. постоян-
Рис. 3. Диаграмма состав—
темп-ра кипения системы
азот—кислород при давле-
нии 760 мм рт. ст.
но кипящими сме-
сями, т. к. они перегоняются без разложения и ки-
пят при постоянной темп-ре. Из законов Коновалова
следует, что пар относительно богаче тем компонен-
том, добавление к-рого понижает темп-ру кипения.
Диаграммы темп-p кипения очень
важны для теории и расчета процес-
сов перегонки и ректификации Ж. с.
На рис. 3 изображена не имеющая экс-
тремумов диаграмма темп-p кипения систе-
мы кислород — азот, дающая теоретич.
основу для получения кислорода и азота
из жидкого воздуха.
Вязкость. Для изучения двой-
ных Ж. с. особенно важна вязкость.
Она сильно зависит от темп-ры; по-
» >—- в этому вязкость измеряют для всей
Рис. 4 типы диа- системы при постоянной темп-ре и
грамм состав — результаты изображают на изотер-
вязкость. мич. диаграммах вязкости (изотер-
мах вязкости), дающих зависимость
коэфф, вязкости Г| от состава х (обычно в мольных
процентах). Английский химик А. Дунстан в 1906
установил три класса изотерм вязкости двойных
Ж. с.: 1) Близкие к прямым кривые, обращенные
выпуклостью к оси состава, для систем без химич.
соединений и без диссоциации компонентов (рис. 4, /).
2) Кривые со значительной выпуклостью, обращен-
ной к оси состава (или даже с минимумом), для си-
стем с диссоциацией (в частности, ассоциированного)
компонента (рис. 4, II). 3) Кривые, обращенные
вогнутостью к оси со-
става (или даже с мак-
симумом), для систем
с образованием не-
сколько диссоцииро-
ванного химич. соеди-
нения (рис. 4, III).
Н. С. Курнаков и
С. Ф. Жемчужный в
1912 открыли принци-
пиально новый класс
изотерм вязкости для
системы с образова-
нием недиссоцииро-
ванного соединения.
В этом случае изотер-
ма вязкости (рис.4, IV)
состоит из двух вет-
вей, пересекающихся Рис. 5. Изотермы вязкости системы
в сингулярной точке вода—бромаль.
А, положение кото-
рой соответствует составу химич. соединения. При
повышении температуры вязкость соединения умень-
шается, как правило, сильнее, чем вязкость компо-
нентов, поэтому сингуляр-
ная точка становится не-
сколько менее резко выра-
женной, но сохраняется
до темп-ры начинающейся
диссоциации соединения;
точка S также не смещает-
ся в сторону, т. к. состав
соединения не зависит от
темп-ры.
На рисунке 5 изображена
диаграмма" вязкости системы
вода — бромаль с образовани-
ем диссоциированного соеди-
нения. При понижении тем-
пературы максимум становит-
ся более резким и приближает-
ся к ординате, соответствую-
щей соединению 1:1. На рис.6
изображены диаграммы вязко-
сти и плотности системы алли-
ловое горчичное масло — ме-
тиланилин с образованием
недиссоциированного соедине-
ния аллил-метил-фенил-тио-
мочевины. Изотермы вязкости
состоят из двух ветвей, пере-
секающихся в отвечающих со-
ставу соединения 1:1 сингу-
лярных точках, к-рые сохра-
Рис. 6. Изотермы вязкости (h>
и плотности (d) системы ме-
тиланилин — аллиловое гор-
чичное масло.
няются и при повышении тем-
пературы. Изотермы плотности d тоже состоят иа двух ветвей,
пересекающихся в сингулярных точках.
Лит.: Менделеев Д. И., Избр. соч., т. 3, Л., 1934;
его же, Соч., т. 4, Л., 1937; его же, Растворы, М.,
1959; Коновалов Д., Об упругости пара растворов,
3 изд., Л., 1928; Курнаков Н. С. и Жемчужный
С. Ф., Внутреннее трение двойных систем, в кн.: Курнаков
Н. С., Избранные труды, т. 1, М., 1960; Курнаков Н. С.,
Введение в физико-химический анализ, там же; Аносов
В. Я. и Погодин С. А., Основные начала физико-хи-
мического анализа, М.—Л., 1947; Вревский М. С.,
Работа по теории растворов, М.—Л., 1953; Timmermans
J., Les solutions concentrees. Theorie et applications aux me-
langes binalres de composes organlque, P., 1936.
В. Я. Аносов.
ЖИДКИЕ УДОБРЕНИЯ — водные р-ры удобри-
тельных веществ, аммиака или жидкий аммиак. Наи-
большее значение имеют, азотные Ж. у. и сложные
Ж. у. Последние имеют в своем составе два или три
питательных для растений элемента (азот, фосфор,
калий). Ж. у. характеризуются концентрацией (%)
59
ЖИДКОСТИ
60
и соотношением содержащихся в них питательных
элементов (N : РгО8 : КгО). В состав Ж. у. вводят
иногда микроэлементы, гербициды, инсектициды и
ростовые вещества. Главнейшие жидкие азотные удоб-
рения: аммиачная вода, жидкий аммиак и аммиакаты.
Аммиачная вода — 25%-ный р-р аммиака,
содержит ок. 20%N. Жидкий аммиак — са-
мое концентрированное азотное удобрение, содержит
ок. 82% N; не удобен для применения вследствие вы-
сокого давления паров и необходимости хранения его
под давлением 30—35 атм. Аммиакаты — ком-
плексные соединения, образующиеся при насыщении
аммиаком водных растворов солей аммиачной селитры,
кальциевой селитры, мочевины или их смесей; содер-
жат, 30—45% N. Аммиакаты, применяемые в качест-
ве Ж. у.,—светлые жидкости,плотн. 1—1,25. Давление
паров над аммиакатами значительно ниже, чем над
жидким аммиаком, и их хранят и перевозят в цистер-
нах или баллонах, рассчитанных на небольшое давле-
ние. Жидкие сложные удобрения могут иметь разно-
образный состав. Ж. у. вносят в почву специальными
машинами для подкормки растений. Произ-во Ж. у.
экономичнее произ-ва твердых удобрений; недостат-
ком их является необходимость сооружения резервуа-
ров большой емкости. Некоторые Ж. у. (аммиак, амми-
акаты) применяют также для усреднения свободной
кислотности (аммонизации) суперфосфата и смешан-
ных удобрений.
Лит.: Клевке В. А., Поляков Н. Н., А р с е н ь-
е в а Л. 3., Технология азотных удобрений, М., 1956; Клев-
ке В. А., Кильман Я. И., Ж. прикл. хим., 1959, 32,
№ 8, 1649; Турчин Ф. В., Удобрение и урожай, 1956,
№ 2, 20; Клевке В. А., Кантор А. С., Хим. пром-сть,
1959, № 6. В. А. Плевке.
ЖИДКОСТИ — тела, находящиеся в агрегатном
состоянии (жидком состоянии), промежуточном между
твердым и газовым состояниями. По своей высокой
плотности и малой сжимаемости, по наличию сильного
межмолекулярного взаимодействия Ж., будучи кон-
денсированными системами, близки к твердым телам
и существенно отличаются от газов. Наряду с этим,
изотропность, текучесть (т. е. способность легко из-
менять внешнюю форму под действием малых нагрузок)
приближают их к газам. Вследствие текучести под
действием внешних сил легко изменяется собственная
форма Ж. (форма шара, соответствующая наименьшей
поверхности при данном объеме). Форму шара Ж.
принимают под действием молекулярных сил, про-
являющихся в поверхностном натяжении. Текучесть
Ж., вызывая релаксацию касательных напряжений
в них, позволяет обнаруживать упругость формы Ж.
только при очень малой продолжительности действия
сдвигающей силы. Вследствие высокой плотности,
вязкость Ж., в отличие от газов, резко падает с повы-
шением темп-ры (у газов она при этом возрастает).
Область существования Ж. ограничена со стороны
низких темп-p переходом в твердое или стеклообраз-
ное состояние. Для каждого вещества характерна
критич. темп-ра, выше к-рой Ж. не может существо-
вать в равновесии с собственным паром (см. Крити-
ческое состояние).
Как правило, вещества имеют только одну жидкую
модификацию. Исключением являются нек-рые веще-
ства, у к-рых существует как нормальная жидкая
фаза, так и анизотропные фазы — т. е. жидкие кри-
сталлы, а также гелий, к-рый может находиться в двух
жидких фазах.
Агрегатное состояние вещества (твердое, жидкое
или газообразное) определяется физич. условиями,
в к-рых оно находится в основном значениями
темп-ры Т и давления р. Характерным параметром
является отношение е средней потенциальной энергии
молекулы к ее средней кинетич. энергии. Это соотно-
шение зависит от темп-ры и давления. Для твердых тел
(при темп-pax, достаточно далеких от темп-ры плавле-
нии) е>1. Иными словами, в твердых телах силы сцеп-
ления между молекулами удерживают их вблизи рав-
новесных положений, несмотря на тепловое движение,
стремящееся установить хаотич. распределение частиц.
В газах имеет место обратный предельный случай —
силы притяжения недостаточны, чтобы удерживать
молекулы друг возле друга, вследствие чего положе-
ния и скорости их распределены хаотически. Соответ-
ственно этому в газах е мало по сравнению с единицей.
В Ж. оба эти фактора примерно уравновешивают друг
друга е~1. Однако при темп-pax ниже критической-
притяжение преобладает. Именно этим обусловлены
два характерных признака жидкого состояния — мо-
лекулярное давление и связанное с ним поверхност-
ное натяжение, на границе с насыщенным паром (см.
также Поверхностные явления).
Изучение рассеяния рентгеновских лучей, нейтро-
нов и электронов показывает, что Ж. обладают свое-
образной молекулярной структурой: ближайшие со-
седи каждой молекулы Ж. в среднем располагаются
в к.-л. определенном порядке, так что число ближай-
ших соседей и их взаимное расположение в среднем
для всех молекул одинаковы, т. е. в Ж. существует
т. н. ближний порядок. Этот порядок про-
является в том, что число соседей молекулы и их вза-
имное расположение в среднем для всех молекул оди-
наково. Иногда это среднее ’ взаимное расположение
молекул такое же, как и в кристаллич. состоянии.
Структура и физич. свойства Ж. зависят от химич.
индивидуальности образующих ее частиц и от харак-
тера и интенсивности сил, действующих между ними.
Интерпретация рентгенография, данных практиче-
ски возможна только для т. н. простых Ж., состоящих
из сферически симметричных, неполярных частиц
с ненаправленными и ненасыщающимися силами
взаимодействия.. К ним относятся сжиженные инерт-
ные газы с чисто ван-дер-ваальсовским взаимодей-
ствием и жидкие металлы, в к-рых осуществляемая
свободными электронами металлич. связь также нена-
правлена и ненасыщаема, как и ван-дер-ваальсово
взаимодействие, а металлич. ионы сферически сим-
метричны и обладают электронной структурой, ана-
логичной электронной структуре атомов инертных
газов. Приближенно можно считать простыми некото-
рые двух- и многоатомные неполярные Ж. (напр.,
СвНв, СС14).
Типичная непростая Ж. — вода. Направленные и
насыщающиеся водородные связи между молекулами,
реализующие большую часть сил сцепления, приводят
к тому, что молекулы воды в среднем сохраняют при-
мерно тетраэдрич. взаимную координацию, близкую
к координации, существующей во льду. Однако одно-
временно происходит сильное растяжение и изгибание
водородных связей; кроме того, имеется заметная доля
разорванных водородных связей и много «дислоциро-
ванных» молекул, заполняющих собой пустоты тетра-
эдрич. структуры. Поэтому дальний порядок в си-
стеме отсутствует и трансляционной повторяемости
истинной, мгновенной структуры нет (тогда как в
среднем макроскопические свойства жидкости оди-
наковы во всех точках жидкой среды). Еще сложнее
обстоит дело с другими непростыми Ж., состоящими
из многоатомных молекул со сложными взаимодей-
ствиями (ассоциирование молекул в комплексы и др.).
Тепловое движение молекул Ж., согласно Я. И.
Френкелю, состоит из нерегулярных колебаний со
средней частотой 1/т0, близкой к частотам колебаний
атомов в кристаллич. телах, и амплитудой, определяе-
мой размерами «свободного объема», предоставленного
данной частице ее соседями. Центр колебаний опре-
деляется полем соседних частиц и смещается вместе
с их смещением за время т на расстояние порядка
61
жидкости — ЖИРЫ
62
молекулярных размеров, поэтому, в отличие от кри-
сталлов, положения равновесия в Ж. временны и
неустойчивы, В связи с большой плотностью и силь-
ным взаимодействием частиц в Ж., их перемещения
совершаются преимущ. в виде более или менее редких
активированных скачков с преодолением потенциаль-
ного барьера, разделяющего две возможные области
колебаний одной частицы. В случае несферич. молекул
к колебаниям и скачкам должны быть добавлены
вращения и вращательные качания частиц. Время т,
соответствующее времени «оседлой жизни» молекулы
во временном положении равновесия между двумя
активированными скачками, связано с величиной
т0 соотношением: т ~ ехр(7?//сТ), где Е — соответ-
ствующая энергия активации. Средний период колеба-
ний молекул т0 имеет порядок 10~12 сек. Время т
может оказаться самым различным, но всегда т>>т0;
юно зависит от природы Ж. и от Е[кТ, уменьшаясь
с ростом темп-ры. Для маловязких Ж, т~10 л1 сек;
оно растет с ростом вязкости, достигая порядка часов
или даже суток у стекол.
Непрерывно и в большом числе совершающиеся
переходы с места на место определяют сильно выра-
женную сам,одиффуаию частиц Ж., а также основное
свойство ИС — текучесть. Под действием постоянной
внешней силы появляется преимущественная направ-
ленность скачков частиц Ж. вдоль направления дей-
ствия силы, т. е. возникает поток частиц в этом же
направлении. Если величина приложенной силы
достаточно мала, то частота скачков 1 /т не изменяется.
Существенно статистич. механизм этого процесса при-
водит к тому, что возникающий поток Ж. пропорцио-
нален приложенной силе, что обусловливает тем самым
конечную величину коэфф, вязкости; из-за актива-
ционного механизма перескоков темп-рная зависи-
мость вязкости описывается обычной экспоненциаль-
ной формулой с энергией активации, определяющейся
характером межмолекулярного взаимодействия (см.
Вязкость'). Если же приложенная внешняя сила дей-
ствует очень кратковременно и по интенсивности
очень велика, то это может привести к нарушению
прочности Ж. в виде «трещин», «поломок» и т. д., как
и кристалла.
Феноменологически механич. свойства Ж. описы-
ваются системой дифференциальных ур-ний в частных
производных, из к-рых особо важным является ур-ние
Навье-Стокса. Исследование этих ур-ний при соот-
ветствующих начальных и граничных условиях яв-
ляется предметом гидромеханики. Феноменология,
описание термодинамич. свойств дается ур-нием состоя-
ния р = /(7, г>), связывающим давление р с темп-рой
Т и уд. объемом v. Наряду с уравнениями состояния,
определенными строгими методами, существует боль-
шое число полуэмпирич, уравнений состояния. Наи-
более простым в то же время теоретически обоснован-
ным является ур-ние Ван-дер-Ваальса, Оно каче-
ственно описывает не только равновесные свойства
газов и жидкостей, но, будучи дополнено термоди-
намическими соотношениями, и фазовые переходы
жидкость — пар (см. Испарение}. Зная ур-ние состоя-
ния, можно вычислить термодинамич. характери-
стики: Ж.: теплоемкость, сжимаемость и т. д. Вдали
от критич. точки коэфф, сжимаемости и теплового
расширения не очень чувствительны к давлению.
Однако сжимаемость медленно уменьшается с увели-
чением давления.
Электропроводность чистых Ж. обычно мала, за
исключением жидких металлов и электролитов. Ве-
личина диэлектрин, проницаемости зависит от струк-
туры Ж. и полярности составляющих ее молекул.
Специфик, особенности жидкого состояния — интен-
сивное взаимодействие частиц при их большой неу-
порядоченности — очень затрудняют теоретич. ана-
лиз проблем. Теории газового и кристаллин, состояний
вещества в качестве предельных случаев рассматри-
вают идеальный газ — конечное разрежение частиц
при их полной неупорядоченности, и идеальный кри-
сталл — совершенную упорядоченность частиц при их
большой плотности. Для Ж. не существует никакой
простой и легко обозримой модели, к-рая могла бы
быть принята за «нулевое приближение», в связи
с чем при построении теории Ж. возникают серьезные
трудности.
Современная строгая теория описывает структуру
Ж., равно как и все ее физич. свойства, набором функ-
ций распределения положений групп частиц, т. е.
функций, определяющих вероятности того или иного
взаимного расположения молекул относительно друг
друга. Теория не нуждается ни в каких дополнитель-
ных гипотезах о структуре Ж., ее задача — изучение
этих функций распределения. Эта теория получила
развитие гл. обр, в работах Дж, Кирквуда, Н. Н. Бо-
голюбова, М, Борна и X. Грина.
Лит.: Френкель Я. И., Кинетическая теория жид-
костей, М.—Л., 1945; Фишер И. 3., Статистическая теория
жидкостей, М., 1961; Гиршфельдер Д ж., Кертисс
Ч,, Берд Р,, Молекулярная теория газов и жидкостей, пер.
с англ.,.М., 1961; Данилов В. И., Строение и кристалли-
зация жидкости. [Сборник], Киев, 1956; Корифеи ьд
М„ Упругость и прочность жидкостей, М.—Л., 1951.
ЖИРНЫЕ КИСЛОТЫ — см. Карбоновые кислоты
и Высшие жирные кислоты.
ЖИРОВОЙ ОБМЕН — см. Обмен веществ.
ЖИРЫ — полные сложные эфиры глицерина и
жирных кислот (триглицериды) общей формулы:
а СН3—О—СО—R
I
₽ СН — О—СО—R'
I
а’ СН3—О—СО—R"
где R, R' и R" — радикалы жирных к-т.
Наиболее часто в образовании жиров участ-
вуют: стеариновая СН3(СН2)1вСООН, пальмитино-
вая СН3(СН2)14СООН, а также олеиновая кислоты
СН3(СН2)7СН = СН(СН2)7СООН. Кроме того, в состав
природных Ж, входят также и др. жирные к-ты, наибо-
лее важные из к-рых приведены в ст. Высшие жирные
кислоты. Кислоты, образующие природные Ж., как
правило, имеют неразветвленное строение и содержат
четное число атомов углерода. Природные Ж. являются
смешанными триглицеридами; в их состав входят ра-
дикалы различных жирных к-т, напр. а-олео-0-паль-
митостеарин. Если в положениях а и а' находятся
остатки различных к-т, то такие смешанные тригли-
цериды оптически активны. Кроме триглицеридов,
в природных Ж. всегда присутствуют различные при-
меси: свободные жирные к-ты, моно- и диглицериды,
фосфатиды, стерины, витамины и др.
Известно более 1300 различных Ж, В основе клас-
сификации Ж. лежат признаки, связанные с их проис-
хождением, составом и свойствами. По происхождению
природные Ж. подразделяются на животные и расти-
тельные; растительные Ж, чаще наз. маслами; те и дру-
гие различаются составом и физико-химич. свойствами.
Большинство растительных масел (кроме пальмо-
вых) имеет жидкую консистенцию; характерной осо-
бенностью многих из них является способность к вы-
сыханию с образованием на поверхности, на к-рую на-
несено масло, твердых и эластич. пленок. Способность
масел к высыханию зависит от степени непредельно-
сти кислотных радикалов, входящих в состав масел,
К высыхающим маслам относятся: тунговое, льня-
ное, конопляное, маковое, ореховое; к полувысыхаю-
щим — подсолнечное, соевое, хлопковое; к невысы-
хающим — оливковое, арахисовое, миндальное и др.
Пальмовые масла — кокосовое, пальмовое, пальмо-
ядровое, имеют твердую консистенцию и относятся
к невысыхающим маслам.
63
ЖИРЫ
64
Большинство животных Ж. имеет твердую или ма-
зеобразную консистенцию и неспособны к высыханию.
Характерным признаком растительных Ж. является
присутствие в них в качестве примеси фитостеринов
(ситостерин, эргостерин и др.), в то время как в жи-
вотных Ж. фитостерины отсутствуют, а содержатся
зоостерины (преим. холестерин).
Источники получения Ж. см. в ст. Жиры животные
и Жиры растительные.
Физические свойства Ж. При комнатной темп-ре
Ж. представляют собой твердые, мазеобразные или
жидкие вещества. Консистенция Ж. определяется их
составом: Ж. с высоким содержанием предельных к-т
имеют более высокую темп-ру застывания и при обыч-
ной темп-ре находятся в твердом состоянии; Ж., в
к-рых преобладают непредельные к-ты, являются жид-
костями. Ж. не имеют строго определенной темп-ры
плавления вследствие того, что они состоят из смеси
различных триглицеридов. Наиболее низкую точку
плавления имеет ореховое масло (—27°), наиболее
высокую — баранье сало (4-55°). Для нек-рых три-
глицеридов характерно наличие двух темп-p плавле-
ния, что, по-видимому, связано с диморфизмом. Так,
трипальмитин плавится при 45°, при дальнейшем
нагревании затвердевает и вновь плавится при 65°;
тристеарин также имеет две точки плавления (55° и
72°). Температура застывания Ж. обычно на несколько
градусов ниже темп-ры их плавления. Т. к. темп-ра
застывания Ж. нерезко выражена, то на практике часто
определяют темп-ру застывания не самого Ж., а его
жирных к-т. Эта величина называется «т и т р о м» Ж.
Ж. кипят только в очень высоком вакууме при
остаточном давлении меньше К) 3 мм', тристеарин
253°/0,001 мм, трилаурин 188°/0,001 мм. При нагре-
вании до 250—350° при атмосферном давлении Ж. раз-
лагаются с образованием летучих веществ (акролеина).
Темп-ра, при к-рой начинается выделение дыма, наз.
температурой дымоотделения; она
зависит от наличия в Ж. свободных жирных к-т.
Плотность всех Ж., за исключением трибутирина,
обычно колеблется в пределах 0,91—0,97. При повы-
шении темп-ры на 1° плотность Ж. в среднем умень-
шается на 0,0007. Вязкость Ж. при нагревании пони-
жается. Ж. сильно преломляют световые лучи; при
40° коэфф, преломления равен 1,456—1,472; с повыше-
нием темп-ры на 1° коэфф, преломления уменьшается
в среднем на 0,0004. Ж. принадлежат к числу веществ
с низким поверхностным натяжением; они легко про-
никают в капилляры; для большинства Ж. поверхно-
стное натяжение на границе с воздухом имеет величину
порядка 30—37 дин!см.
Чистые Ж. — плохие проводники электричества.
Электропроводность Ж. увеличивается при прогорка-
нии, а также при возрастании в них содержания сво-
бодных жирных к-т. Диэлектрич. проницаемость Ж.
составляет 3,0—3,2. Теплопроводность Ж. при 20° ок.
0,0004 кал)сек см. Теплоемкость Ж. колеблется от
0,3 до 0,5 ккал/кг град и увеличивается с повышением
темп-ры. Теплота плавления Ж. имеет величину по-
рядка 29—35 ккал/кг. Темп-ра вспышки большинства
Ж. лежит в пределах 170—350°, самовоспламенения —
выше 350°.
В воде Ж. практически нерастворимы, но образуют
с ней эмульсии. Количество Ж., эмульгирующегося
в 100 г воды (без добавления специальных эмульгато-
ров), составляет для свиного и конского Ж. ок. 50 мг,
для говяжьего — ок. 10 мг. К хорошо эмульгируемым
Ж. относятся касторовое масло и олеомаргарин.
Способность Ж. к образованию стойких эмульсий осо-
бенно возрастает в присутствии различных детерген-
тов (мыла, белки, желчные кислоты и др.). Примером
такой эмульсии может служить молоко. Способность
Ж. растворять воду незначительна; она несколько
увеличивается при повышении темп-ры. В свином Ж.
при 40—100° растворяется от 0,15 до 0,45% воды.
Характерной особенностью Ж. является их способ-
ность поглощать и прочно удерживать различные
пахучие вещества, на чем основан один из способов
извлечения душистых веществ из цветов. Ж. хорошо
растворимы в органич. растворителях: эфире, бензоле,
бензине, CS2, хлороформе, четыреххлористом углероде
и др. хлорзамещенных углеводородах. Одним из луч-
ших растворителей для Ж. является трихлорэтилен,
в к-ром Ж. растворяются в соотношении 1 : 3,5. Рас-
творимость Ж. в спирте, в том числе и горячем, незна-
чительна и повышается с увеличением содержания в
них свободных жирных к-т. Жидкие Ж. обычно раство-
ряются в органич. растворителях легче, чем твердые.
Химические свойства Ж. В химич. отношении чистые
триглицериды, в особенности триглицериды предель-
ных к-т, довольно инертные вещества, способные лишь
к ограниченному числу превращений, характерных
для всех сложных эфиров. Триглицериды, содержа-
щие непредельные к-ты, способны также вступать
в реакции, характерные для соединений с двойной
связью. При воздействии на триглицериды водяного
пара происходит их омыление с образованием свобод-
ных жирных к-т и глицерина:
CIbOCOR сн,он
I I
CHOCOR -I- ЗН2О 72 СНОН + 3RCOOH
I I
CHqOCOR сн2он
Омыление триглицеридов значительно ускоряется
в присутствии щелочей; при этом образуются соли
жирных к-т, на чем основан промышленный способ
получения мыла (подробнее см. Расщепление жиров).
Ж. могут быть также расщеплены кипячением их
в спирте (алкоголиз Ж.), что приводит к образованию
глицерина и эфиров жирных к-т:
CH2OCOR СН3ОН
CHOCOR + ЗС3Н5ОН 22 СНОН +3RCOOC2H5
I I
CH3OCOR СН2ОН
Алкоголиз Ж. протекает намного легче, чем гидролиз,
поскольку в спирте Ж. более растворимы, чем в воде,
и реакция в этом случае идет в гомогенной среде.
Алкоголиз, так же как и гидролиз, ускоряется ката-
лизаторами (газообразный НС1). Триглицериды спо-
собны также к реакциям ацидолиза (замена одного кис-
лотного остатка в молекуле триглицерида на другой
при нагревании в присутствии соответствующей сво-
бодной жирной к-ты) и переэтерификации (взаимный
обмен кислотными остатками между двумя сложными
эфирами). Переэтерификация может происходить ме-
жду различными триглицеридами, между триглице-
ридом и к.-л. другим сложным эфиром, а также внутри
молекулы одного и того же триглицерида, напр.:
CH3OCOR' CH3OCOR"
! I
CHOCOR" 22 CHOCOR’
iH2OCOR’” CH2OCOR"'
Переэтерификацией широко пользуются в лабора-
торной практике для выделения входящих в состав Ж.
жирных к-т в виде перегоняемых метиловых эфиров.
Гидрирование Ж., в состав к-рых входят непредель-
ные жирные к-ты, происходит по двойным связям; по
этим же связям могут присоединяться галогены и
серная к-та. В последнем случае образуются эфиры
серной к-ты:
-СН2-СН=СН—СН3----f- H3SO4 —►
-> —СН3—CH2-CH(OSO3OH)—сн2-
Наконец, ненасыщенные кислотные остатки могут под-
вергаться окислению.
65
жиры
66
• Синтез триглицеридов был впервые осуществлен
в 1854 М. Бертло. Простые триглицериды могут быть
получены по общим реакциям синтеза сложных эфи-
ров (непосредственное взаимодействие глицерина с
жирными к-тами, синтезы, основанные на применении
хлорангидридов жирных к-т и на реакции галогенгид-
ринов глицерина с солями жирных к-т). Для полу-
чения смешанных триглицеридов с заранее намечен-
ным положением кислотных остатков необходимо при
введении кислотного остатка в определенное положе-
ние глицерина защищать две остальные оксигруппы,
напр.:
а СНоОН СН3 а СН2-0 .СН3
I 1 * )с€
₽ СНОН 4- со -► ₽ СН -о/ \СНв
I I I 3
а' СН2ОН СН3 а' СН2ОН
Несмотря на относительную химич, устойчивость
чистых триглицеридов, природные Ж. при хранении,
особенно на открытом воздухе, быстро портятся —
прогоркают. К числу факторов, вызывающих прогор-
кание, относятся: частичное расщепление Ж. под
действием содержащегося в них активного фермента —
липазы, приводящее к появлению свободных, в том
числе низкомолекулярных летучих к-т; окисление
жирных к-т, в особенности непредельных, под дей-
ствием кислорода воздуха, а также образование пере-
кисных соединений, оксикислот, альдегидов и кетонов
в результате жизнедеятельности нек-рых микроорга-
низмов. Прогоркшие Ж. обладают специфич, запахом
и, как правило, неприятным вкусом. Ж. предохраняют
от прогоркания путем добавления антиоксидантов,
роль к-рых играет также содержащийся в природных
Ж. витамин Е.
Важнейшими качественными реакциями на Ж. являются
проба на акролеин, образующийся при нагревании Ж. до 320 —
340°, а также проба на способность вещества омылиться с об-
разованием жирных к-т. Элементарный состав Ж. колеблется
в узких пределах: содержание водорода от 10,9 до 12%, угле-
рода от 74 до 78%. Элементарный количественный анализ Ж.
практически не производится вследствие незначительной раз-
ницы в элементарном составе различных триглицеридов; кроме
того, природные Ж. являются смесями различных триглице-
ридов. Поэтому Ж. характеризуются с помощью особых кон-
стант, или чисел, выражающих, обычно в условных единицах,
количественное содержание в исследуемом Ж. тех или иных
составных частей или химически однородных структурных
единиц. Так, содержание свободных жирных к-т в Ж. харак-
теризуется кислотным числом, выражаемым в мг КОН, необ-
ходимого для нейтрализации свободных кислот, содержащих-
ся в 1 а данного Ж. Общее количество как свободных, так и
связанных жирных к-т в Ж. характеризуется омыления числом,
представляющим собой количество мг КОН, необходимого для
нейтрализации свободных жирных к-т и омыления триглице-
ридов, входящих в состав 1 г исследуемого Ж. Содержание
триглицеридов в Ж. характеризуется эфирным числом;
оно выражается количеством мг КОН, необходимого для омы-
ления триглицеридов, входящих в состав Ж., и представляет
собой разность между числом омыления и кислотным числом.
Содержание в Ж. нелетучих и нерастворимых в воде кислот
(в сумме с неомыляемыми веществами) характеризуется чис-
лом Ген ер а, выражающим в процентах количество
указанных веществ, образующихся при омылении 100 г иссле-
дуемого Ж. Содержание в Ж. летучих, растворимых в воде
кислот характеризуется числом Рейхерт а—М е й с-
с л я, обозначающим количество мл 0,1 н. щелочи, необхо-
димой для нейтрализации этих кислот, выделенных из 5 г
омыленного Ж. Аналогичным образом число Поленске
характеризует количество содержащихся в Ж. летучих, но
нерастворимых в воде кислот. Содержание оксикислот в Ж.
характеризуется ацетильным числом, к-рое выра-
жается количеством мг КОН, необходимого для нейтрализа-
ции уксусной к-ты, образующейся при омылении 1 г предвари-
тельно ацетилированного Ж. Определение ацетильного числа
основано на том, что при обработке Ж. уксусным ангидридом
гидроксильные группы присутствующих в Ж. оксикислот
ацетилируются.
Общая степень непредельности Ж. характеризуется иод-
ным числом, к-рое выражается количеством граммов галогена
(в пересчете на иод), присоединяющегося к 100 г исследуемого
Ж. Для характеристики содержания в Ж. ненасыщенных к-т
используется также родановое число, выражаемое
количеством г иода, эквивалентным количеству родана, при-
соединяющегося к 100 г Ж. (родан присоединяется не к каж-
дой двойной связи в молекуле жирной к-ты: из двух связей
линолевой к-ты родан присоединяется только к одной, из трех
двойных связей линоленовой к-ты — к двум). Сравнение иод-
2 К. X. Э. т. 2
ного и роданового числа дает возможность количественно рас-
считать содержание каждой из ненасыщенных к-т (олеиновой,
линолевой и линоленовой). При сложном составе Ж. для таких
расчетов необходимы дополнительные показатели: процентное
содержание высокомолекулярных насыщенных к-т, определя-
емое по методу Бертрама, игексабромное число,
характеризующее содержание триеновых к-т (типа линолено-
вой) и выражаемое количеством гексабромстеариновой к-ты,
получаемой из 100 г Ж. Более точное определение состава
жирных к-т в исследуемом Ж. может быть произведено мето-
дами количественной хроматографии на бумаге или газовой
хроматографии. Для количественного определения ди-, три-
и полиеновых к-т применяют спектрофотометрии, методы.
Разделение Ж. на индивидуальные триглицериды может
быть произведено методом молекулярной дистилляции, кри-
сталлизацией из растворителей и окислением ненасыщенных
глицеридов; из них лишь первый дает возможность выделить
нек-рые индивидуальные триглицериды, остальные пригодны
лишь для разделения Ж. на более или менее узкие фракции род-
ственных глицеридов.
Биологическая роль жиров. Ж. — одна из основных
групп органич. веществ, входящих наряду с белками
и углеводами в состав всех растительных и животных
клеток. В организме животных различают запасной
н протоплазматич. Ж. Запасной Ж. откладывается
в специальных жировых депо — в подкожной клет-
чатке и в сальниках; в таком виде организм запасает
энергию, к-рая расходуется при голодании и вновь
накапливается при достаточном или повышенном пи-
тании. Протоплазматич. Ж. структурно связаны с бел-
ками и углеводами и входят в состав протоплазмы
клеток. Эти Ж. непосредственно связаны с осущест-
влением клеткой своих основных биологич. и химич.
Функций. Эпергетич. ценность Ж. исключительно
велика; при окислении 1 г Ж. до СО2 и Н2О выделяется
9,3 ккал, т. е. в 2 раза больше, чем при окислении
белков или углеводов. Важную роль играют Ж.
в процессах теплорегуляции животных организмов.
Находясь в подкожной клетчатке, Ж., благодаря
плохой теплопроводности, предохраняют организм
от переохлаждения; особенное значение это имеет для
морских животных (киты, тюлени). Одновременно
Ж. являются своеобразной и выгодной формой, в к-роп
организм запасает воду. Будучи одними из наиболее
богатых водородом органич. соединений, Ж. образуют
при окислении ок. 140—150 мл воды на 100 г Ж. Это
играет исключительное значение для животных сухих
и пустынных областей; в частности, Ж. в горбах вер-
блюдов является таким источником воды для орга-
низма животного.
Ж. — необходимая составная часть пищи. Норма
суточного потребления Ж. взрослым человеком 60—
70 г. Пищевая ценность Ж. определяется не только их
высокой калорийностью, но также и наличием в при-
родных Ж. таких биологически ценных веществ, как
фосфолипиды, стерины, витамины A, D, Е и др. К
числу витаминов относятся также входящие в состав
Ж. ненасыщенные к-ты: линолевая, линоленовая и
арахидоновая.
Ж., поступающие в организм с пищей, подвергаются
в тонком кишечнике расщеплению (перевариванию)
на глицерин и жирные к-ты под действием фермента
липа.зы, выделяемого поджелудочной железой. Рас-
щеплению предшествует эмульгирование Ж., к-рому
способствуют соли желчных кислот (холевой литохб-
левой, дезоксихолевой); в этом процессе принимают
участие и присутствующие в кишечнике жирные к-ты.
В процессе расщепления Ж. образуется смесь глице-
рина, жирных к-т, а также моно- и диглицеридов,
к-рая всасывается стенками кишечника. При этом
глицерин как водорастворимое соединение всасывается
непосредственно, а нерастворимые жирные к-ты вса-
сываются в виде растворимых в воде комплексов с
желчными к-тами, т.н. холеиновых кислот.
В эпителии кишечника всосавшиеся холеиновые к-ты
распадаются и из освободившихся жирных к-т и
всосавшегося глицерина вновь синтезируются три-
глицериды, специфические для данного организма.
67
ЖИРЫ —ЖИРЫ ЖИВОТНЫЕ
68
Образующиеся Ж. поступают в кровь и в основной
своей массе откладываются в виде запасного Ж.
в жировых депо, откуда Ж., по мере надобности, до-
ставляются кровью к местам использования.
Распад Ж. в тканях начинается с их гидролиза
под действием тканевых липаз; образующийся при
гидролизе глицерин фосфорилируется, окисляется до
фосфодиоксиацетона, к-рый под действием фермента
изомеразы изомеризуется в 3-фосфоглицериновый аль-
дегид:
сн2он сн2он
I АТФ I — 2Н
снон снон ---------------.
I I
СН2ОН СН2ОРО(ОН)2
фосфоглицерин
СН2ОН СНО
I I
—►СО снон
I I
СН2ОРО(ОН)2 СН2ОРО(ОН)2
фосфодиоксиацетон 3-фосфоглицери-
новыи альдегид
3-фосфоглицериновый альдегид, являющийся одним
из промежуточных продуктов процесса гликолиза,
может далее окислиться до Н2О и СО2 или же вклю-
читься в синтез углеводов. Жирные к-ты в процессе
обмена активируются, образуя ацилпроизводное ко-
фермента А;
R — СН2СН2СООН + HS — КоА + АТФ -►
—►В, — СН2СН2 — СО — S — КоА + АМФ + пирофосфат
Активированные жирные к-ты претерпевают про-
цесс т. наз. р-окисления, включающий: дегидрирова-
ние в положении Д2~3, приводящее к р-ненасыщенной
к-те; гидратирование по двойной связи, приводящее
к р-оксикислоте; дегидрирование, приводящее к fl-ке-
токислоте; расщепление последней с участием еще
одной молекулы кофермента А, что приводит к обра-
зованию одной молекулы активированной уксусной
к-ты (ацетил-КоА) и одной молекулы активированной
жирной к-ты, углеродная цепочка к-рой укорочена
на два атома С по сравнению с исходной жирной к-той:
S—КоА S-KoA S—КоА
Со (Jo СО
1 сн2 — 2Н | + Н2 О | -2Н
► СН — СН2
сн2 <Лн СНОН
1 R R R
S—КоА 1 СО I HSKoA — СН2 ► 1 СО 1 R S—КоА + S—КоА 1 1 СО со 1 1 СН3 R
Активированная уксусная к-та в виде ацетил-КоА
может быть далее окислена до СО2 и Н2О в цикле
трикарбоновых кислот, а активиро-
ванная жирная к-та может повторять
описанный выше ряд превращений
до тех пор, пока она полностью не
распадется до уксусной к-ты. В орга-
низме происходят не только процессы
окисления и распада Ж., но также и
процессы их синтеза. Ж. синтезируются
в организме из продуктов обмена угле-
водов и белков. Необходимый для син-
теза Ж. глицерин образуется в про-
цессе гликолиза. В синтезе жирных
кислот центральное место принадлежит
ацетил-КоА и малонил-КоА.
Поскольку ацетил-КоА образуется не только в ре-
зультате окисления жирных к-т, но также и из угле-
водов в процессе гликолиза и из нек-рых аминокислот,
то создается связь между обменом белков и углеводов
и обменом Ж.
Применение Ж. — см. Жиры животные и Жиры
растительные.
Лит.: Зиновьев А. А., Химия жиров, М., 1952;
Тютюнников Б. Н., Ю хновс кий Г. Л., Марк-
ман А. Л., Технология переработки жиров, М., 1950;
Либерман С. Г., Петровский В. Й Справочник
по производству животных жиров, 2 изд., М., 1956; Р и х е А.,
Основы технологии органических веществ, пер. с нем., М.,
1959, с. 392 — 409; Fruton J. S., Simmonds S., Ge-
neral biochemistry, 2 ed., N. Y., 1958; Deuel H. J., The li-
pids. Their chemistry and biochemistry, v. 1—3, N. Y.—L.,
1951—57; Progress in the chemistry of fats and other lipids,
ed. R. T. Holman, W. O. Lundberg, T. Malkin, v. 1—5, L.,
1952—58; Черкасова Л. С.,МережинскийМ. Ф.,
Обмен жиров и липидов, Минск, 1961. В. Б. Спиричев.
ЖИРЫ ЖИВОТНЫЕ — природные продукты, по-
лучаемые из жировых тканей различных животных;
состоят гл. обр. из триглицеридов высших однооснов-
ных жирных к-т. Ж. ж. делятся на твердые и жидкие.
Твердые Ж. ж. содержатся в тканях наземных
животных; для них характерно высокое содержание
глицеридов насыщенных к-т, гл. обр. пальмитиновой
и стеариновой, и незначительное содержание глицери-
дов непредельных к-т, гл. обр. олеиновой. Иодное
число большинства Ж. ж. этой группы колеблется
в пределах 35—85.
Жидкие Ж. ж. содержатся в тканях морских
млекопитающих и рыб, а также в костях и копытах
наземных животных. Характерная особенность жи-
ров морского зверя и рыб — наличие в них глицери-
дов высоконепредельных жирных к-т (с 4, 5 и 6 двой-
ными связями). Для них характерно высокое иодное
число (150—200) и ограниченная способность к высы-
ханию. Особое место среди Ж. ж. занимает масло
коровье. В табл. 1 и 2 приведен жирнокислотный состав
важнейших Ж. ж., в табл. 3 и 4 — их основные физич.
и химич. свойства. Содержание влаги в Ж. ж. колеб-
лется в пределах 0,2—1,0%.
Получение Ж. ж. Сырьем для таких пищевых Ж. ж.,
как говяжий, бараний, свиной и конский, служит
сало-сырец, получаемое при разделке туш животных.
Наиболее богаты жирами подкожная жировая ткань
и ткань, окружающей внутренние органы: почки,
кишечник, желудок. Содержание жира в этих тканях
достигает 82—83%. Основным методом получения
жиров является вытапливание. Перед вытапливанием
жировые ткани отделяют от мяса, сортируют, промы-
вают водой и измельчают. В процессе вытапливания
стенки клеток жировой ткани разрываются, жир
расплавляется и отделяется от клеточных структур,
влаги и белковых веществ. Различают два основных
метода жиротопления: мокрый и сухой. При мокром
методе жировую ткань обрабатывают водой и водяным
паром в автоклаве под давлением до 3—4 ат. При
сухом методе жиры выплавляют на открытом огне
или в обогреваемых котлах; испаряющаяся при этом
влага удаляется в атмосферу или отсасывается (в ва-
Таблица 1
Наимено- вание жира Триглицериды насыщенных кислот, % Триглицериды ненасыщенных кислот, %
миристи- новой пальмити- новой стеарино- вой Д9’10 -тет- радецено- вой Д9'10 -гек- садецено- вой олеи- новой лино- левой
Говяжий Бараний Свиной* Конский 2-2,5 2-4 0,8-1,1 27-29 25—27 25—30,4 29 24—29 25-31 12,2-17,9 7 0,4-0,6 0,2 0,1—0,2 2,4-2,7 1,3 1,5-2,7 43—44 36-43 41,2—48,1 55 2-5,0 3-4 5,7-7,8 7
• Кроме того, содержится 2% триглицеридов ариходоновой и линоленовой к-т.
69
ЖИРЫ ЖИВОТНЫЕ
70
Таблица 2
Наимено- вание жира Триглицериды насы- щенных кислот, % Триглицериды ненасыщенных кислот, %
С14 С10 018 С20-22 014 Сю С18 С20 С22
Тресковый (печень) . 5,8 8,4 0,6 0,2 20 (2,3) 29,1 (2,8) 24,4 (6) 9,6 (6)
Сельдиа . . 7,0 11,7 0,8 0,1 1,2(2) 11,8 (2,4) 19,6 (3,5) 25,9 (5,2) 21,6 (4,3)
Тюлений® . 4,7 12,1 2,9 0,1 3,3(2) 19,2 (2,2) 31,8 (2,7) 12,9 (5,7) 13,4 (10,6)
Китовый . . 9,2 15,6 1,9 0,6 2,5 (2) 13,9 (2,1) 37,2 (2,4) 12,0 (7,1) 7,1 (9,4)
а Кроме того, содержит 0,1% триглицеридов насыщенных к-т (Ct») и 0,1%
глицеридов ненасыщенных к-т (С24). “ Кроме того, содержит 0,5% триглицери-
дов ненасыщенных к-т (С»4).
Примечание: Цифры в скобках характеризуют степень ненасыщенности
к-т, обозначая среднее количество атомов Н21 необходимое для полного гидриро-
вания к-т этой группы.
кууме). При щелочном способе выплавки жиров, имею-
щем довольно узкую область применения, в частности
при переработке мездры, жировое сырье обрабаты-
вают в 1,75%-ном р-ре щелочи, при этом происходит
растворение белковых веществ и выделение свободных
жиров. По окончании вытапливания расплавленный
жир сливают в отстойники для отделения от воды и
белков, а шквару перетапливают для дополнитель-
ного выделения жира и прессуют. Прессованная
шквара используется на корм скоту.
Таблица 3
Наименование жира | Плотность Т.пл., °C Т. заст., °C 40 nD
Говяжий 0,937-0,957 42-52 34-38 1,4566-1,4583
Бараний 0,937—0,961 44—55 34-35 1,4566—1.4583
Свиной 0,915—0,923 28—48 22—32 1,4577-1,4609
Конский 0,916—0,922 29-43 22-37 ——
Сельди 0,918-0,931 — — 1.4701-1,4747
Китовый 0,910—0,928 — 1,463-1,471
Тресковый 0,919—0,929 1,477-1,483
(при 20°)
Тюлений 0,910—0,929 — — —
Жиры, получаемые вытапливанием, обычно содер-
жат различные примеси: частицы шквары, влагу,
и др. Кроме того, при использовании несвежего сы-
рья в них могут присутствовать свободные жирные
к-ты, а также вещества, придающие неприятный запах
и темную окраску. Для улучшения качества таких
жиров их подвергают дальнейшей очистке путем до-
полнительного отстаивания или рафинирования. Пи-
щевые жиры, получаемые из свежего сырья, обычно
не нуждаются в рафинировании.
Таблица 4
Наимено- вание жира Числа
омыле- ния Генера Рейхер- та—Мейс- сля иодное родано- вое ацетиль- ное . Полен- ске
Говяжий 193-200 95—96 0,2-0,6 32-47 29—40 2,7-3,6
Бараний 191—206 94—95,5 0,6-0,7 35—46 30—39 0,3—0,5
Свиной 193—200 95—96 0,3—0,9 46-66 44-52 2,6 0,4—0,6
Конский 195-204 — 71—86 51,8 __
Сельди 179—194 95—96 123-146 —
Тресковый 175-196 — — 150-175 —
Китовый 175-220 93,5-96,6 94—145 — __
Тюлений 170-195 — — 165—190 — — —
Жиры морского зверя и рыб. Ме-
дицинский рыбий жир получают из пе-
чени и подкожного сала китов, а также из печени
трески, пикши, колючей акулы и ската. Вытопка
жира производится мокрым способом с помощью ост-
2*
рого пара под давлением или в ва-
куум-аппаратах с внешним обогревом.
Вытопленный жир промывают соле-
вым р-ром и горячей водой, центри-
фугируют, охлаждают и фильтруют
для отделения от твердой фракции.
Рыбий жир сохраняют в закупоренном
состоянии в заполненных доверху со-
судах, чтобы предотвратить его от окис-
ления кислородом воздуха.
Технические жиры (во р-
в а н ь) получают из подкожного сала,
внутренностей, костей и мяса морских
животных (китообразных, моржей, тю-
леней) путем вытапливания с помощью
пара под давлением. Для получения
технич. жира из рыбьего сырья (мало-
мерная рыба, рыба, механически по-
врежденная, отходы рыбоконсервного и
филейного производства и т. п.) применяют сухое и мо-
крое жиротопление, а также прессование и экстракцию.
Жиры из внутренностей рыб выделяют также фермен-
тативным способом, при к-ром для расщепления соеди-
нительной ткани и освобождения жиров используются
ферменты, содержащиеся в пищеварительном аппарате
рыб. На механизированных предприятиях рыбное
сырье перерабатывают в зависимости от содержания
в нем жира на прессовых (при наличии в сырье не
менее 6% Ж.) или на экстракционных установках.
Экстракцию тощего рыбного сырья или муки, полу-
ченной с прессовых установок, производят бензином
или трихлорэтиленом. Добыча китового жира обычно
производится на китобойных базах, представляющих
собой плавучие заводы; сырье перерабатывается по-
точным методом. После разделки туши кита гладкое
покровное сало перерабатывают при 100—110° и раз-
ряжении до 600 мм рт. ст. с целью получения меди-
цинского жира. Полосовое сало и кости кита после
предварительной распиловки перерабатывают в жи-
ротопочных котлах острым паром под давлением 4 ат.
Полученная разваренная масса поступает в жиро-
отделители, где жир отделяется от воды и белковых
веществ. После отстаивания в отстойниках жир очи-
щается на жировых сепараторах и сливается в танки
для хранения. Мясо и шквара перерабатываются
на корм скоту.
Для произ-ва костных жиров, содержание
к-рых составляет 6—8% от веса костей, используют
позвонки, бедренные, берцовые и головные кости.
Извлечение жира из кости производят тепловым или
холодным способами. Выварка костей тепловым спо-
собом производится в открытых котлах (при 80—85°)
или в автоклавах при давлении 4—5 ат. Для очистки
жира от механич. примесей и влаги его про-
пускают через фильтрпресс и сепарируют.
Более экономичным и совершенным является
холодный (импульсный) способ извлечения
жира из костей. При этом способе жир извле-
кают из измельченной кости холодной водой,
приводимой в вихреобразное движение в спе-
циальных аппаратах. Холодный метод позво-
ляет извлечь до 80% жира, содержащегося
в костях, в то время как при вытапливании
в автоклаве выход составляет60%, а в откры-
тых котлах 40—50%. Кроме того, жир, полу-
чаемый холодным способом, отличается более
высоким качеством. Полное извлечение жира
из костей достигается экстрагированием
растворителями. Однако экстракционные
органич.
методы используются только для получения технич.
жира.
Ж. ж. и продукты их переработки применяются
в качестве продуктов питания, а также в различных
71
ЖИРЫ РАСТИТЕЛЬНЫЕ
72
отраслях народного хозяйства. К пищевым Ж. ж,
относятся масло коровье, говяжий, бараний, свиной
и др. Жиры морских млекопитающих и рыб из-за
присущего им неприятного запаха используются в ка-
честве пищевых продуктов только в переработанном
гидрогенизированном виде (в качестве составной части
маргарина). Богатый витаминами рыбий жир приме-
няется в качестве средства против рахита, куриной
слепоты и др. заболеваний у детей, для лечения ожо-
гов, язв и инфицированных ран (в виде составной
части различных мазей), а также для получения вы-
сококонцентрированных очищенных препаратов ви-
таминов А и D, В технике значительное количество
Ж. ж. применяется для варки мыла. Одним из важных
способов переработки Ж. ж. является гидрогенизация
(см, Гидрогенизация жиров), гл. обр, жиров морских
животных и рыб.
Лит. см. при ст. Жиры, В. Б. Спиричев.
ЖИРЫ РАСТИТЕЛЬНЫЕ (масла растительные
жирные) — природные продукты, добываемые из се-
мян и мякоти плодов различных растений и состоящие
гл. обр, (на 95—98%) из полных сложных эфиров гли-
церина (глицеридов), насыщенных и ненасыщенных
высших одноосновных жирных к-т. Кроме глицери-
дов, в Ж. р. присутствуют: свободные жирные к-ты
(обычно 1—2%), фосфатиды (гл. обр. лецитины, ке-
фалины и инозитфосфатиды) 0,05—3%, растительные
мерины (фитостерины) — в свободном виде или в фор-
ме стеридов жирных к-т 0,3—0,5% (в маслах злако-
вых растений до 1,5% и больше); незначительные
количества пигментов (каротины, ксантофиллы, обус-
ловливающие желтую окраску большинства масел;
хлорофилл, окрашивающий, напр., конопляное масло
в зеленоватый цвет и др,); витамин Е до 0,5%. Отдель-
ные Ж. р. содержат также дубящие вещества, алка-
лоиды, гликозиды, слизи, эфирные масла и пр,, что
соответственно отражается на их вкусовых и ароматич.
качествах, а иногда обусловливает их токсич. или ле-
чебные свойства.
Содержание Ж. р. в семенах и плодах растений
колеблется в широких пределах (от 2—3 до 70% и
более). Масла, содержащиеся в растительных клет-
ках, являются структурным элементом протоплазмы
и запасным питательным веществом, расходуемым по
мере надобности, в особенности в период прорастания.
Жирнокислотный состав растительных масел специ-
фичен для каждого данного вида растений. Масла,
близкие по систематич, положению видов, имеют близ-
кий жирнокислотный состав, Хотя естественная клас-
сификация масел в деталях не разработана, однако
для отдельных родов и семейств можно отметить ха-
рактерные показатели состава масел. Так, для масел
семейства пальмовых характерно большое содержа-
ние низко- и среднемолекулярных к-т (от капроновой
до лауриновой); для лавровых — присутствие лаури-
новой к-ты; для мускатных — миристиновой, для
крестоцветных — эруковой и бегеновой, для зонтич-
ных — петрозелиновой, для розоцветных — парина-
ровой, для бобовых — арахиновой и лигноцериновой,
для молочайных — элеостеариновой и т. д. Характери-
стика свойств и состав нек-рых Ж. р. даны в табл. 1 и 2.
Технич. классификация НС р. основана на учете
их жирнокпслотного состава и таких физико-химич.
показателей, как плотность и отношение к окисляю-
щему действию кислорода воздуха (способность обра-
зовывать высыхающие пленки). В соответствии с этой
классификацией Ж. р. подразделяют на две группы.
1. Твердые: а) не содержащие летучих к-т (масло
какао); б) содержащие летучие к-ты (кокосовое масло).
2. Жидкие: а) содержащие оксикислоты, не высы-
хающие и не образующие пленок (касторовое масло);
б) содержащие гл, обр. кислоты с одной двойной
связью (олеиновую, эруковую) типа оливкового масла
и высыхающие с трудом; в) содержащие гл. обр.
кислоты с двумя раздельными двойными связями
(липолевую) типа макового масла и высыхающие
с умеренной скоростью; г) содержащие гл, обр. ки-
слоты с тремя раздельными двойными связями (лино-
леновую) типа льняного масла, высыхающие быстро;
д) содержащие значительное количество кислот с со-
пряженными двойными связями (элеостеариновую)
типа тунгового масла, высыхающие чрезвычайно
быстро.
Приемы извлечения Ж. р. для каждого вида мас-
личного сырья имеют своп особенности, однако в су-
щественном они сводятся к двум типам операций —
прессованию и экстракции, При любом способе пере-
работки семена очищают от всякого рода примесей
сепараторами и пневматич. очистителями, после чего
в зависимости от вида сырья оно подвергается следую-
щим видам обработки: 1) Сырье, шелуха к-рого легко
отделяется от ядра семени (семена подсолнечника,
хлопчатника, арахиса и др.), обрабатывают на спе-
циальных машинах, где отделяют оболочку от ядра.
После этого ядерную фракцию семян, содержащую
нек-рое количество шелухи, размалывают на вальце-
вых станках. 2) Семена, у к-рых оболочка прочно
связана с ядром (лен, рапс и др,), размалывают цели-
ком. Измельченный материал (мятка) первого и вто-
рого вида увлажняют, подвергают тепловой обра-
ботке (при 100—115°) в жаровнях. Прожаренный
материал (мезга) прессуют в шнек-прессах, причем
высокомасличное сырье прессуют дважды; сначала на
форпрессах, а затем — после вторичного прожари-
вания — па прессах окончательного отжима.
При экстракционном извлечении масла семена
проходят те же подготовительные операции, что и
при прессовом извлечении, вплоть до измельчения.
Затем мятку обрабатывают нагретым до 50—55° рас-
творителем в экстракторах до возможно полного из-
влечения масла. В качестве растворителей обычно
применяют легкий бензин (т. кип. 65—85°), технич.
гексан или (реже) этанол. Из полученного раствора
масла в растворителе (крепостью 10—25%) отгоняют
бензин, к-рый возвращается в процесс, а масло охла-
ждают и фильтруют. Жмых (шрот) после экстракции
также подвергают тепловой обработке глухим и ост-
рым паром для удаления растворителя.
Высокомасличное сырье чаще всего перерабаты-
вают комбинированным методом: сначала на прессах
отжимают большую часть масла, 65—80%, а затем
измельченный форпрессовый жмых экстрагируют.
Качество получаемого масла зависит в основном от
качества перерабатываемого сырья и температурных
условий ведения процесса. Форпрессовое масло, по-
лучаемое при более низких темп-рах, является луч-
шим по качеству. Экстракционное масло применяется
в пищу только после специальной очистки, Сырые
масла, полученные тем или иным методом, содержат
нек-рые количества примесей, подлежащих удалению.
Для этого Ж. р, подвергают рафинированию.
Ж, р. находят многообразные формы применения;
основное применение—использование для пищевых
целей. Масла подсолнечное, хлопковое, оливковое,
арахисовое, соевое и др. потребляются непосредст-
венно в пищу в натуральном (после рафинации) и
в гидрированном виде (в маргарине и в кухонных
жирах); вводятся в состав соусов, майонезов и пр,;
применяются в произ-ве овощных и рыбных консер-
вов, шоколадов (масло какао), кремов, халвы и мно-
гочисленных иных кондитерских изделий. Высыхаю-
щие масла являются основным сырьем в произ-ве
пленкообразователей (олиф, лаков). До настоящего
времени Ж. р. в натуральном и гидрированном виде
(см. Гидрогенизация жиров) занимают важнейшее
место в составе жирового набора, из к-рого готовятся
73
74
ЖИРЫ РАСТИТЕЛЬНЫЕ
Таблица 1. Основные свойства жиров (масел) растительных
Масло Цвет Маслич- ность семян (плодов). % Плот- ность при 15°, г/см3 Темпе- ратура заст., °C 40 nD Число омыле- ния Число Генера Число Рейхерта- Мейссля Число Полен- ске Иодное число Родано- вое число
Абрикосовое a Светло-желтый 40-51 (в ядре) 0,918 -20 1,4646 188-198 95-96 0,1 0,3 96—109 —
Арахисовое Бесцветный до крас- новато-бурого 29-39 0,91 -0,96 -3 1,4634 188—197 94-96 0,4-1,6 0,3 83-105 70-72
Буковое Светло-желтый 23—29 0,921 -17 1,4730 (15°) 191—196 95 <0,1 — 104—111 —
Горчичное Светло-желтый до светло-бурого 33—38 0,918 -15 1,4659 170-183,8 94-96,5 0,3-0,9 — 92 — 123 80- 86
Какао® Желтоватый 48—57 0.9G0 21,5-27 1,4569 192—196 95-96 0,1-0,4 0,5-1,0 34-38 32—35
Касторовое® От бесцветного до темно-желтого 40-55 0,962 —10 до —18 1,4745 176-187 96,1 0,2-0,3 - 81-90 81,6
Кокосовое1, Белый до желтоватого 57-75 0.925 19—23 1,4497 246—268 86-92 4-7 8,5—11 8-10 8,2-9,6
Конопляное Желто зеленый 28-35 0,929 -27 1.4517 (25°) 190—194 92—93 2,0 — 140-143 —
Кориандровое Зеленовато-бурый 19-21 0,926 —4 1.4704 (30°) 190 — — 94-100 —
Лаллеманции Светло-желтый 27-33 0,934 -34 до —35 181—185 93,3 1,6 — 162 —
Льняное Желтый до бурого 28-44 0,933 -18 ДО —27 1,4840 (15°) 191—195 95 — — 174-183 —
Маисовое (ку- курузное) Золотисто-желтый 15—40 (в зародыше; 0,924 —10 до —15 1,4745 (23°) 188—193 92-96 0,3-2,5 — 117—123 77-77,5
Маковое^ Золотисто-желтый 45—60 0,925 -15 до —20 1.4751 (25“) 189-198 95-95,5 — — 131—143 78,7
Миндальное Желтый 45—54 0,917 -10 ДО —21 1,4672 189—195 96-97 0,2—0,5 0,2—1,0 92—102 82-85
Оливковое Золотисто-желтый 40-70 0,917 до —6 1,4635 185—196 95-96 о,1-оз — 80-85 76,5
Ореховое Зеленовато-желтый 40-65 (в ядре) 0,925 2/ 1,4710 188-197 95,4 0—3,2 1,6 143—162 —
Пальмовое6 Темно-желтый до тем- но-красного 51—67 0,923 31-41 1,4545 196-210 94-98 0,4—1.9 0,4-0,6 51—57 —
Пальмоядро- воеж Белый до желтова- того 46-53 0,930 19—24 1,4516 240-257 89-93 4-7 8,5—11 12—16 11,5-15
Перилловое Светло-желтый до темно-желтого 41-45 0,931 — 1,4745 187—197 95—96 — — 180-196 —
Персиковое Золотисто-желтый 32-45 (в ядре) 0,920 -20 до -23 1,4645 189—195 94 0-0,1 0,3-0,8 92—110 —
Подсолнечное Светло-желтый 28-48 0,924 -16 до —18 1,4680 186—194 95 0,3-1,0 0,3 127—136 80 -83
Рапсовое Бурый, после рафи- нирования светло- желтый 33-45 0,914 -4 до —10 1,4650 172—175 94—96 0,1-0,8 — 94—106 77,8
Сафлоровое3 Желтый 30-32 0,925 —13 до —20 1,4685 187—194 94—96 0,2-1,6 0,6 138-150 —
Сезамовое (кунжутное) С ветло-желтый 35—55 0,922 -3 до —6 1.4708 (25°) 187—195 95-96 0,1-0,5 0,15 103-112 75-77
Сливовое Золотисто-желтый до буроватого 40-50 (в ядре) 0,918 ДО —17 1,4635 188-198 96-97 0,8 0,7 91-104 —
Сурепное Бурый 33-40 0,918 -8 — 173-181 — — — 105 — 122 —
Соевое11 Светло-желтый до темно-желтого 15-26 0,928 -8 до-18 1,4678 188—195 94—96 — 124-133 83,7
Тунговоек С ветло-желтый до оранжевого 36 0,940 -17 ДО—21 1,520 (25°) 188-197 95-97 0,4-0,7 — 154—176 78-87
Хлопковоел Красно-бурый до буро- черного; после ра- финирования жел- тый 16—25 0,920 —1 до —6 1,4634 194—196 95—96 0,2—1,0 — 103—111 62-67
аТ. пл. минус 16—20°, гексабромное число 32—57; 6 т. пл. 33—35°; в [“Id= +7,6-+9,7°; вязкость по Энглеру (Ei#)
139—140°; г т. пл. 24—27°; Д т. пл. 2°; е т. пл. 27—30°; жт. пл. 25—30°; 3 т. пл. минус 5°; и т. пл. минус 7—8°; к вязкость но
Энглеру (Е20) 32—39°; л при 10° и ниже выпадает белый осадок (хлопковый стеарин).
* Таблицы, 1 и 2 составлены по след, материалам: Иванов С. Л., Учение о растительных маслах, М., 1924; Hand-
buch Ubbelohdes der Chemie und Technologie der Ole und Fette, Lpz., 1932; G г if n Ad., Haiden W., Analyse der Fette
und Wasche, Bd 2, B., 1929; Jamieson G., Vegetable fats and oils, 2 ed., N. Y., 1943; Ekey E. W., Vegetable fats and
oils, N. Y., 1954.
75
ЖИРЫ РАСТИТЕЛЬНЫЕ - ЖИРЫ РАСЩЕПЛЕННЫЕ
76
Таблица 2. Жирные кислоты, входящие в состав Ж. р. (в %)
Масла Мири- стиновая С14 Пальми- тиновая Сю Стеари- новая Cig Арахи- новая Сю Бегеновая и лигноце- риновая С22—С24 Олеино- вая С18 Эруко- вая С22 Лин о- левая 018 Линоле- новая 018
Абрикосовое 5,3 14,3
Арахисовое 6-11 4,5-6,2 2,3-4,9 1,9-3,1 40-66 —— 18-33 —
Буковое — 4,9 3,5 — — 76,7 — 9,2 0,4
Горчичное 0,5 — — — 4,9 25—28 50 14,5-20 3
Какао — 23-25 31-34,5 — — 39—43 — 2 —-
Касторовоеа — — 3 — — 3-9 — 2-3 —
Кокосовое^ 16,5—20 4,3-7,5 0,8-5 — — 2-10,3 1
Конопляное — 4 э — — 14 — 65 16
Кориандровое13 — 8 - — — 32,0 1 —
Лаллеманции — 6 — — 93 э
Льняное — 8- -10 —— — 5—20 — 25—50 21-45
Маисовое (кукурузное) . . . — 7,7 3,5—3,6 0,4 0,2 44,8-45,4 — 41-48 —
Маковое — 4,6 2,6 — — 28,3 — 58,5 —
Миндальное 1,5-5,4 — — 80-83,7 — 14.8-16 —
Оливковое следы 7-10 2-4 0,1-0,2 — 70—87 — 4-12 —.
Ореховое — 7,0 — 13-14 — 72,5-77,2 3,2
Пальмовое — 32—40 8-10 — — ок. 50 — — —
Пальмоядровое 12-16 7-9 1-7 — — 4-16 — 1 —
Перилловое — 6 -8 — — 14-20 12-18 65—70
Персиковое — 15,6 — — — — — —
Подсолнечное — 9 - — — 33,3 — 39,8 —
Рапсовое 1.5 — 1.6 1,5 . — ок. 20 56—65 14 2-3
Сафлоровоег — 10-11.8 следы — — — — — —
Сез’амовое (кунжутное) . . . — 7,7 4,6 0,4 0,04 48,1 — 36,8 —
СливовоеД — —
Соевое — 2,4-6,8 4,4-7,3 0,4—1 — 32-35,6 — 51,5-57 2-3
Сурепное — 2 — — 2 20,5 47 25,5 2
Тунговое® — 3,7 1,2 — — 10-15 — — —
Хлопковое 0,3-0,5 20—22 2 0,1-0,6 — 30 ,э—За ,2 — 41,7-44 —
а Помимо указанных жирных к-т, в состав касторового масла входит 80—85% рицинолевой к-ты; ®в состав
кокосового и пальмоядрового масел входят также (соответственно): 10,7—22,2% и 8—13% капроновой, каприловой
и каприновой к-т (С&—Сю), 45—51% и 50—55% лауриновой к-ты (Сю); в в состав кориандрового масла входит также
53% петрозелиновой к-ты; г содержание ненасыщенных к-т 88,2—90%; Д содержание ненасыщенных к-т ок. 70%;
е в состав тунгового масла входит также ок. 80% элеостеариновой к-ты.
мыла хозяйственные, туалетные и специального назна-
чения, а также косметич. изделия; лишь в самое по-
следнее время они начинают вытесняться синтетич.
жирными к-тами и жирозаменителями. Ж. р. наряду
с ворванями применяются для жирования кож. Ка-
сторовое масло, отличающееся высокой вязкостью,
применяется в качестве смазочного; благодаря прису-
щему ему специфич. действию, оно используется и
в качестве лекарственного препарата. Кроме касто-
рового масла, в фармации применяется чаульмугровое
масло, к-рому приписывается противолепрозное дей-
ствие, миндальное и нек-рые «косточковые» масла
(напр., персиковое) для инъекций совместно с жирорас-
творимыми лекарственными препаратами. Методом рас-
щепления Ж. р. (см. Расщепление жиров) разделяются
на глицерин и жирные к-ты, далее используемые в мы-
ловарении или фракционируемые на стеарин и олеин.
Лит.: Зиновьев А. А., Химия жиров, М., 1952;
Голдовский А. М., Теоретические основы производства
растительных масел, М., 1958; Шарапов Н. И., Маслич-
ные растения и маслообразовательный процесс, М.—Л.,
1959; Е с к е у Е. W., Vegetable fats and oils, N. Y., 1954;
Hll ditch T. P., The chemical constitution of natural
fats, 3 ed., L., 1956; К a u f m a n n H. P., Analyse der Fette
und Fettprodukte, B. [u. a.], 1958; см. также при ст. Жиры.
А. Л. Маркман.
ЖИРЫ РАСЩЕПЛЕННЫЕ — см. Расщепление
жиров.
ЗАЖИГАТЕЛЬНЫЕ СОСТАВЫ — пиротехнич. со-
ставы, а также горючие вещества или их смеси, при-
меняемые для снаряжения зажигательных бомб, сна-
рядов, мин и т. п. К 3. с. относят также огнеметные
смеси. 3. с. делятся на 2 группы: с окислителями —
термиты и термитно-зажигательные составы, а также
кислородсодержащие соли (КС1О4 и др.); без окисли-
телей: сгорающие за счет кислорода воздуха — жид-
кие нефтепродукты (бензин, керосин и др.), отверж-
денные и загущенные жидкие нефтепродукты и др.
горючие (напр., напалм), магниевый сплав «электрон»,
белый фосфор, его соединения и сплавы, прочие 3. с.,
в том числе и самовоспламеняющиеся при соприкос-
новении с водой или воздухом (напр., металлич. нат-
рий и др.).
Все 3. с. должны безотказно воспламеняться от
воспламенительных составов, иметь большую теплоту
и темп-ру горения, гореть со скоростью, наиболее
выгодной в условиях их применения, обладать воз-
можно большей площадью соприкосновения с поджи-
гаемым объектом (текучесть 3. с. или образование при
горении легкоплавких шлаков). Скорость горения
3. с. зависит от состава, а также и от конструкции
боеприпасов; так, сгорание 3. с. в зажигательных пу-
лях происходит почти мгновенно, скорость горения
запрессованных термитных составов составляет неск.
мм/сек, а жидкие нефтепродукты горят еще медлен-
нее. В таблице приведены основные показатели неко-
торых 3. с.
Зажигательные
составы
Железо-алюминиевый термит
Сплав «электрон».........
Фосфор белый.............
Напалм ..................
Теплота горения, ккал/г Темп-ра горения, °C Плотность, 2/СЛ1*
0,8 2500 -—3,2 (спрессов.)
6,0 ^2000 1,8
5,8 -4300 1,8
10 ~ 900 0,8-0,9
Эффективность действия 3. с. определяется количе-
ством теплоты, переданной при горении поджигае-
мому материалу. Следует учитывать, что 3. с. яв-
ляются только инициаторами горения, поэтому в слу-
чаях, когда нет притока достаточного количества воз-
духа к поджигаемому материалу, использование 3. с.
не приводит к цели.
Лит.: Шидловский А. А., Основы пиротехники,
2 изд., М., 1954; Горлов А. П., Зажигательные средства,
2 изд., М.—Л., 1943; Демидов П. Г., Основы горения
веществ, М., 1951. А. А. Шидловский.
ЗАЙЦЕВА ПРАВИЛО — порядок отщепления эле-
ментов галогеноводородных к-т от моногалогенопроиз-
водных; согласно 3. п., водород отщепляется от наи-
менее гидрогенизованного атома углерода, напр.:
(СН3)2СН-СНГ—СНз (СН3)2С—сн-сн3
[а не (СН3)2СН-СН = СН2].
Исследование кинетики отщепления галогеноводо-
родов действием спиртового р-ра КОН на галоген-
алкилы показало, что этот процесс протекает как
бимолекулярно (Е2), так и мономолекулярно (Ej).
В обоих случаях соблюдается 3. п. Так из соединений
типа R—СН2—СНХ—СН3 в случае механизма Ej
могли бы возникнуть два олефина:
r-ch2-ch -Сн3—R-Ch2-ch-ch3+x
X
r-ch=ch-zc^h-*-rch2-ch-ch3—
н
1
—
R-C^CH»CHS
U
У формы I имеется сопряжение минимум с тремя
С—Н-связями, тогда как у формы II — только с двумя.
Поэтому I устойчивее и его образование термодинами-
чески более выгодно, чем образование II. При меха-
низме Е3 мыслимо образование двух энергетически
равноценных комплексов:
5 но н Н-он5“
К-СН-СН-СНз и к-сн2-сн-Сн2
Р’ Xs-
III IV
Поэтому образование R—СН=СН—СН3 может быть
объяснено только тем, что этот олефин обладает мень-
шей энергией и поэтому устойчивее. При образовании
ментенов, как это установлено экспериментально,
3. п. строго не соблюдается. Правило было сформу-
лировано А. М. Зайцевым в 1875 (см. также Марков-
никова правило).
Лит.: Реутов О. А., Теоретические проблвмы органи-
ческой химии, М., 1956, с. ИЗ. Р. Р. Галле.
ЗАКРЕПЛЯЮЩИЕ ВЕЩЕСТВА (в текстильной
промышленности) — вещества, применяемые для при-
дания окраскам волокнистых материалов устойчиво-
сти к действию света, воды, пота, к стирке и др. За-
крепление окрасок особенно необходимо для прямых
и кислотных красителей. Условно 3. в. можно подраз-
делить на 3 группы:
1. Катионактивные вещества, дейст-
вие к-рых основано на образовании малорастворимой
или нерастворимой в воде соли, напр. с прямыми кра-
сителями:
[Kp-SO8]-Na++[R-NH8]+Cl--»NaCl+[Kp-SO8]-[R-NH8]l-
краситель высокомолекуляр- нерастворимая соль
ная соль амина амина и красителя
Катионактивные вещества повышают устойчивость
окраски к воде, а в некоторых случаях к кислотам
и щелочам, но не улучшают или улучшают очень
мало прочность окраски к стирке. К катионактив-
79
ЗАКРЕПЛЯЮЩИЕ ВЕЩЕСТВА - ЗАНДМЕЙЕРА РЕАКЦИЯ
80
ным 3. в. относятся стеариламидометилпиридиний
(C17H35CONHCH2i>IC5H5)Cl', додецилфосфониевые и
сульфониевые соединения, например (соответственно)
С12Н26Р(СН3)3 и CI2H25S(CH3)2 и др. Если длина цепи
увеличивается, а конечная группа остается той же
самой, то несколько увеличивается и прочность
окраски к мокрым обработкам; одновременно наблю-
дается нек-рое смягчение ткани.
2. Поверхностно - неактивные чет-
вертичные аммониевые основания
эффективнее предыдущих по повышению устойчивости
окрасок к стирке; они меньше влияют на оттенок
окраски и меньше понижают светоустойчивость кра-
сителя. Соединения этой группы разнообразны по
строению, К ним относятся различные продукты
конденсации хлорпарафпнов с полиэтиленполиами-
нами, напр. солидоген BSE,
[R-NH-(C2H4NH)n-C2H4-NH2-CH3]+SO2CH3-.
Упрочнение окрасок, прежде всего к стирке, дости-
гается применением продуктов сочетания азотсодер-
жащих оснований или их солен с ароматич, или гете-
роциклич. соединениями, напр. тригликолевый эфир
триметилолмеламина — препарат 105 (I), солидоген
BS (II) и др.
NHCH2O(CHa)2OH
с
N^'-N
1 II
HO(CH2).,OCH2HN-C4 C-NHCH2O(CH2)2OH
Известны и др, препараты этой группы,
3. Синтетические смолы, раство-
римые в воде (преконденсат ы), сооб-
щающие особенно высокую устойчивость к стирке,
действию воды и пота. Почти все 3, в., применяемые
в наст, время, относятся именно к этой группе. Сниже-
ние устойчивости к действию света устраняется путем
одновременной обработки солями меди, В качестве
3. в. применяют растворимые в воде продукты конден-
сации формальдегида с дициандиамидом, гуанидином,
бигуанидином и др. В водных р-рах эти вещества
диссоциируют на ионы, катионом является смоляной
остаток. Закрепляющее действие веществ этой группы
основано на образовании нерастворимых солей между
катионами смолы и анионом красителя. К 3. в. этой
группы относятся препараты ДЦУ (III) и ДЦМ (IV),
последний является медным комплексом закрепителя
ДЦУ:
/ N HCONH.A + /
I I |ОЧ
H.,N“C Д>СН3
I О
' / г
III
NHCONH /NHCONHI2+ 1“
C=NH2-"Cu—H2N=C I ''C~CH3
1 1 / \o' „
fj-------------N< ' 12 n
-H2C Л CH2-
Препарат ДЦУ — продукт конденсации формальде-
гида с дициандиамидом, при 20° — вязкая прозрачная
жидкость серовато-зеленого цвета, хорошо раство-
ряется в горячей воде, 2%-ной теплой уксусной и
муравьиной к-тах. Этот закрепитель не повышает
прочности окрасок к действию света," а в нек-рых
случаях даже снижает ее. Закрепитель ДЦМ устра-
няет эти недостатки. Ацетаты меди и хрома не дают
осадка с раствором ДЦУ и могут применяться совме-
стно с ним. Образованию осадка способствуют элект-
ролиты и смачиватели. Для повышения прочности
окрасок основными красителями к действию света
применяют фосфорно-молпбденовую к-ту.
Лит.: Фротшер Г., Химия и физическая химия тек-
стильных вспомогательных материалов, пер. с нем., т. 1, М.,
1958, с. 143; Хвала А., Химические вспомогательные
вещества в текстильной промышленности, пер. с нем., М.—Л.,
1948, с. 377; Б о г о с л о в с к и й Б. М., Л а п т е в Н. Г.,
Химия красителей, М., 1957, с. 406; В еп катар аман К,
Химия синтетических красителей, пер. с англ., т. 1, Л., 1956,
с. 675; Садов Ф. И., Корчагин М. В., МатецкиЙ
А. II., Химическая технология волокнистых материалов,
2 изд., М., 1956, с. 505. П. В. Мориганов, Б. Н, Мельников.
ЗАМЕНИМЫЕ АМИНОКИСЛОТЫ — аминоки-
слоты, содержание, к-рых в пище не обязательно для
развития, роста и поддержания нормального физио-
логия. состояния животных и пек-рых микроорганиз-
мов. При отсутствии 3, а. в пище происходит их син-
тез в организме животного из других аминокислот пли
из небелковых компонентов. Резкой границы между
3. а. и незаменимыми аминокислотами не существует,
поскольку потребность организма в определенных
аминокислотах зависит от вида животного, наличия
в пище витаминов, аминокислот-антагонистов и др.
факторов. Для человека и большинства животных
3. а, являются: аланин, аргинин, аспарагиновая к-та,
гистидин, глицин, глутаминовая к-та, океппролин,
пролин, серин, тирозин и цистин. Незаменимы для
всех видов животных: валин, изолейцип, лейцин, ли-
зин, метионин, треонин, триптофан, фенилаланин.
Подробнее см. Незаменимые аминокислоты.
Лит.: Майстер А., Биохимия аминокислот, пер.
с англ., М., 1961. Л. И. Линееич.
ЗАНДМЕЙЕРА РЕАКЦИЯ — замена диазогруппы
в ароматич. диазосоединении галогеном (или псев-
догалогеном, напр, CN-группой) в присутствии соли
одновалентной меди
Си2Х2
ArN2X------► АгХ -J- N2
Для замены диазогруппы галогеном (хлор, бром)
обычно прибавляют соответствующую соль закисной
меди к р-ру соли диазонпя в галогенводородпой к-те.
Замена на нитрильную группу происходит в щелочной
среде при обработке диазосоединенпя водным р-ром
цианистых солей одновалентной меди и натрия или
калия, Кинетика замены диазогруппы галогеном со-
гласуется с механизмом:
ArN2Cl X ArN+ + CI'; CuCl + Cl* ~ CuCl-
медл. быстр.
ArN£ -I- CuCl---► [ArN+ • CuCl-] --► ArCl + N2 CuCl
Скорость замены диазогруппы хлором в фенилдиазо-
нии и его пара-замещенных уменьшается в ряду:
NO2 > Cl > Н > СН3 > ОСН3, что подтверждает та-
кой механизм. Побочно образуются азосоединения и
диарплы:
Ar—N=N—Ar + Ns 4- 2CuC12
2 [ArN+ CuCl-] С
- *Аг—Аг + 2N2 + 2CuCl2
Т. к. 3. р. осуществляют в водной среде, часто проис-
ходит побочное образование фенолов:
ArN2 ArOH-|-N2
П случае замены нитрильной группой в реакции уча-
ствует комплексное соединение цианистой меди с циа-
нидом щелочного металла:
Ai-N2C1 4- Na [Cu(CN)2] —► ArCN 4- CuCN 4- N2 4- NaCl
3. p, широко применяется для получения арилбро-
мпдов, арплхлоридов и нитрилов арилкарбоновых
81
ЗАПОЛНИТЕЛИ ПОРИСТЫЕ — ЗАСТУДНЕВАНИЕ
82
к-т из соответствующих диазосоединений, т. е. в ко-
нечном счете из ароматич. и гетероциклпч. аминов.
Арплнодиды образуются при простом нагревании ди-
азосоединенип с р*-ром иодида калия. Арплфториды
получают из дпазосоединений, гл. обр, по Шиманна
реакции, Среди важнейших технич. продуктов, полу-
чаемых по 3, р., следует назвать 1,8-хлорнафталин-
сульфокислоту, хлорпроизводиые толуола, замещен-
ные о-цианфенилтиогликолевые кислоты, 1,8-цианна-
фталинсульфокислоту, к-рые являются полупродук-
тами в синтезе органич. красителей.
Реакция открыта в 1884 Т. Зандмейером.
Лит.: Ворожцов Н. Н., Основы синтеза промежуточ-
ных продуктов и красителей, 4 изд., М., 1955; Surrey A. R.,
Name reactions In organic chemistry, N. Y., 1954; Hodgson
H. H., Chem. Revs, 1947, 40. № 2, 251. E. M. Рохлин.
ЗАПОЛНИТЕЛИ ПОРИСТЫЕ (заполнители лег-
кие) — природные или искусственные минеральные
сыпучие материалы, применяемые для получения лег-
ких бетонов, железобетона и строительных р-ров
с пониженным объемным весом, 3. п, разделяются на
гравий (щебень) с величиной зерна 5—40 мм и объем-
ным насыпным весом до 1000 кг/,м3 и песок с величи-
ной зерна до 5 мм и объемным насыпным весом до
1200 кг/м3. 3, п. по показателям объемного веса
делятся на марки от «100» до «1000» (объемный насып-
ной вес от 100 до 1000 кг/лг3) для гравия и песка; для
песка допускается также марка «1200». К природным
3. п. относятся горные породы: пемза, туфы, ракушеч-
ники, вулканические шлаки и пеплы. К искусствен-
ным 3. п. относятся: отходы промышленности (метал-
лургия. и топливные шлаки) и специально изготовляе-
мые — керамзит, шлаковая пемза, гранулированный
шлак, аглопорит, вспученный перлит (обсидиан).
Искусственные 3. п. получаются вспучиванием
(поризацией) огненно-жидких шлаков (шлаковая пем-
за) и минеральных расплавов или обжигом легко-
плавких глин во вращающихся печах (керамзит) и на
агломерационных решетках (аглопорит), а также
вулканических водосодержащих стекол (вспученный
перлит); аглопорит может быть получен также спе-
канием топливных шлаков, зол ТЭЦ и др. Керам-
зит получают обжигом при 1100—1200° гранул,
сформованных из легкоплавкой глины или щебня из
предварительно дробленного глинистого сланца, со-
держащих 6—10% окислов железа и органич. примеси.
Марка керамзита допускается в пределах от «250» до
«600», прочность при сжатии от 6 до 30 кг/см3. Шла-
ковую пемзу изготовляют поризацией расплав-
ленных шлаков (водой и др.) в бассейнах и в машинах
специальной конструкции с последующим дроблением
и рассевом. Щебень из шлаковой пемзы обычно имеет
марку от «400» до «800», прочность при сжатии от 4
до 20 кг/с.«2. Наиболее легкий 3, п, — вспученный
перлит, получают обжигом во взвешенном состоя-
нии в коротких вращающихся и др. печах (в т.ч. в ки-
пящем слое) из вулканич. стекол с содержанием
SiO2 — 65—75% и воды — до 8%. Ряд перлитов тре-
бует обжига в две стадии, из к-рых первая состоит
в нагреве при 250—450° в течение 5—30 мин-, вторая —
в обжиге при 1000 — 1200° продолжительностью до
1 мин. Объемный насыпной вес перлитового щебня —
в пределах 250—400 кг/м3, прочность при сжатии от 10
до 25 кг/слг2; насыпной вес перлитового песка 50—
250 кг/м3. Кроме объемного веса и прочности 3. п,,
значение имеют характер пор (открытые, закрытые,
крупные, мелкие), водопоглощенпе, теплопроводность,
морозостойкость. 3. п, применяются для изготовления
теплоизоляционных, конструктивно-теплоизоляцион-
ных и конструктивных бетонов, для штукатурных
тепло- и звукоизоляционных р-ров, засыпок и пр.
Лит.: Легкие бетоны на пористых заполнителях. Сб. статей,
под ред. И. А. Попова, М., 1957; Шлаковые заполнители и бе-
тоны на их основе. [Сб. трудов], Харьков, 1958; «Строительные
материалы», 1961, К 8; На льянос И. Н., Мерзляк
А. Н., Вермикулит и перлит—пористые заполнители для тепло-
изоляционных изделий и бетонов, М., 1961; ОнацкийС. П.,
Производство керамзита, М., 1962. А. И. Полинковская.
ЗАРИН (изопропиловый эфир фторангидрида ме-
тилфосфиновой кислоты) мол. в. 140,1 — бесцветная
подвижная жидкость; т. пл. ок. минус
СзН7°\р^° 54°; т. кип. 151,5°; 1,094; 1,383;
сн / \р давление насыщенного пара 1,43 мм
3 рт. ст, при 20°, летучесть (максималь-
ная концентрация) 11,3 мг/л при 20°; смешивается
с водой во всех отношениях и растворяется в органич.
растворителях. 3. медленно гидролизуется водой,
реакция носит автокаталитич. характер; скорость
гидролиза увеличивается в присутствии минеральных
к-т и особенно щелочей. В результате гидролиза обра-
зуются изопропиловый эфир метилфосфиновой к-ты
или ее соли. 3. энергично взаимодействует с водными
р-рами аммиака и аминов с образованием соответ-
ствующих солей изопропилового эфира метилфосфино-
вой к-ты. Процесс получения 3. складывается из
следующих стадий:
РС13 + 3ROH + 3R'N -* P(OR)3 + 3R’N • НС1
ГСН3 OR] OR
. P(OR)3 + CH3C1 — )p(or -► CH3P^OR + RC1
L Cl 'ORJ XO
OR
CH3P^OR + PC13 -> CH3POCI2 + 2POCI3 + RCI
xo
OCH(CH3)a
CH3POCb + (CH3)»CHOH -> CH3P<C1
xo
OCH(CH3)2 OCH(CH3)2
CH3PXCI + KF -> CH3P%F 4- KC1
о 'o
3. относится к судорожно-паралитич. OB с резко
выраженным миотич. действием (сужение зрачка).
Смертельная концентрация 3, ок, 0,2 мг/л при минут-
ной экспозиции; концентрация ок. 0,002 мг/л вызы-
вает сильнейший миоз; попадание в капельно-жидком
состоянии на кожу вызывает общее отравление. За-
щитой от 3. служат противогаз и защитная одежда.
Дегазация 3. может быть осуществлена водными рас-
творами NH3 и аминов.
'Лит.: Сартори М., Усп. химии, 1954, 23, № 1; Сон-
дерс Б., Химия и токсикология органических соединений
фосфора и фтора, пер. с англ., М., 1961. Р. Н. Стерлин.
ЗАСТУДНЕВАНИЕ (растворов высокомолекуляр-
ных соединений) — процесс непрерывного увеличе-
ния вязкости растворов высокомолекулярных соедине-
ний, сопровождающийся непрерывным нарастанием
эластич. свойств. 3. приводит к затвердеванию всей
системы в однородную нетекучую эластичную массу —
студень — в результате образования структурной
сетки полимера, пронизывающей весь объем системы
и удерживающей растворитель. 3. зависит от природы
высокополнмера, растворителя и темп-ры. Основной
причиной 3. является усиление взаимодействия между
макромолекулами полимера, находящимися в раство-
ре, или между их агрегатами вследствие частичного
понижения растворимости полимера (или каких-либо
его функциональных групп) в растворителе.
3. может быть вызвано либо изменением темп-ры,
либо изменением состава растворителя при данной
темп-ре (введением осадителя). В результате этого
между отдельными активными группами, входящими
в состав макромолекулярных цепей, парциальная
растворимость к-рых понижена, возникают локальные
межмолекулярные связи. Образуется структурная
сетка, пронизывающая весь раствор и удерживающая
растворитель. Влияние растворимости на 3. сказы-
вается в том, что 3. может происходить как при пони-
жении, так и при повышении темп-ры, в зависимости
от того, понижается или повышается растворимость
83
ЗАУРАНОВЫЕ ЭЛЕМЕНТЫ - ЗАЩИТА ИНДИВИДУАЛЬНАЯ
84
полимера с изменением темп-ры. Важным условием
3. является достижение определенной критич. кон-
центрации раствора, характерной для каждой данной
системы полимер — растворитель, определяемой соот-
ношением между числом внутри- и межмолекулярных
связей. Величина критич. концентрации зависит от
гибкости полимерной цепи в растворе.
Процесс 3. является самопроизвольным, идущим
с уменьшением свободной энергии системы, причем
как теплосодержание, так и энтропия системы в про-
цессе 3. уменьшаются. 3. обратимо, но переход рас-
твора в студень не является фазовым переходом. Са-
мопроизвольное 3. раствора не является конечной
стадией изменения системы во времени.’3. — кинетич.
процесс, спонтанно развивающийся до наступления
равновесного состояния, сопровождающегося разде-
лением ранее однофазной системы на две фазы —
равновесный студень постоянного состава и раствор
высокомолекулярного вещества, находящийся в тер-
модинамич. равновесии со студнем (синерезис
студня). Если концентрация раствора соответ-
ствует равновесной концентрации студня, то 3. не со-
провождается синерезисом. На характер процесса 3.
существенное влияние оказывают полидисперсность
полимера и наличие даже незначительных примесей.
Последние приводят к тому, что неравновесное со-
стояние может сохраняться в течение длительного
отрезка времени.
3. может быть вызвано также введением в раствор
веществ, способных к химич. взаимодействию с ма-
кромолекулами, в результате чего пространственная
сетка возникает за счет образования химич. связей
между макромолекулами. В этом случае процесс 3.
уже не является обратимым.
Лит..- Л и п а т о в Ю. С., Прошлякова Н. Ф.,
Усп. химии, 1961, 30, вып. 4, 517—31. Ю. С. Липатов.
ЗАУРАНОВЫЕ ЭЛЕМЕНТЫ — см. Трансурано-
вые алементы.
ЗАЩИТА ИНДИВИДУАЛЬНАЯ (промышленная)—
защита организма (органов дыхания, глаз, кожных по-
кровов) от вредного воздействия паров, газов, жидких
веществ, пыли и др. при работе на химич. предприятиях.
Защита органов дыхания. На органы дыхания мо-
гут оказывать вредное действие пыль, пары, туман и
газы разных веществ, Для
защиты от пыли применяют
респираторы со специаль-
ными фильтрами. В качестве
последних служат волокни-
стые материалы в виде фет-
ра, гофрированной бумаги,
рыхлого картона или тка-
ней из натуральных и синте-
тических волокон. Респира-
торы (за исключением рес-
пираторных повязок) обыч-
Рис. 1. Респиратор ПРБ-5. но снабжены выдыхатель-
ным клапаном. На рис. 1
показан респиратор марки ПРБ-5. Характеристика
основных типов респираторов, выпускаемых в
СССР, приведена в табл. 1.
Наиболее распространены респираторы ПРБ-5 и
РПП-57, к-рые защищают от пыли синтетич. красите-
лей, ультрамарина, суперфосфата и др. В пром-сти
широко пользуются повязкой типа ШБ-1 (т. н. лепе-
сток), обладающей высокой защитной способностью
и малым весом.
Для защиты от вредных газов, тумана и паров при-
меняют промышленные противогазы,
состоящие из шлема-маски (лицевая часть) и коробки
с поглотителем. В табл. 2 приведена характеристика и
указана отличительная окраска коробок промышлен-
ных противогазов (см. также Противогазы).
Таблица 1
Марка респиратора Вес, г Площадь фильтра, Сопротивле- ние вдоху, мм вод. ст. С (по масляно- му туману), °/ /о
РПП-57 275 600 3,5 до 5
Ф-46-К* 310 500 4,0 4-8
ПРБ-5 300 410 6,0 5-6
ПРШ-2-59 800 800 3-4 10-12
ШБ-1 (респиратор, повязка) .... 11 280 3,0 1,6-6,0
* Респиратор Ф-46-К снабжен сменными патронами с по-
глотителями для защиты от небольших концентраций различ-
ных паров и газов: патрон А — от паров органич. веществ;
Б — от кислых газов (сернистый газ, сероводород и др.);
Г — от паров ртути; КД — от смеси аммиака с сероводородом.
•• Коэффициент, показывающий число частиц масляного
тумана (в %), прошедших через фильтр респиратора.
Таблица 2
Марка короб- ки Цвет коробки Поглощаемый компонент Сопротивле- ние вдоху, мм вод. ст. (не более)
А Коричне- вый Пары органич. жидкостей (бензин, керосин, ацетон, толуол, ксилол, нитро- соединения, бензол и его гомологи и др.) 18
В Желтый Кислые пары, Cl, SO2, окнс- лы азота, H2S, НС1, СОС12 и др. 18
г Черно-жел- тый Пары ртути 18
Е Черный AsH3, PHS 18
кд Серый NH3, H2S (в смеси и в от- дельности) 18
со Белый Окись углерода 21
м Красный Все газы и пары, перечис- ленные выше (но с мень- шим временем защитного действия) 21
БКФ Защитный Кислые газы и AsH3; дым, пыль и туман 25
КВ — Окислы азота и аммиак 26
П-2 Красный с белой поло- сой Карбонилы никеля и же- леза, аэрозоли никеля, железа, окись углерода 25
Примечание. Если противогаз снабжен также про-
тиводымным фильтром, на его коробку наносится белая про-
дольная полоса.
При работах, связанных с осмотром или промывани-
ем емкостей, напр. нефтяных, кислотных и др. цистерн,
отсеков нефтеналивных судов и т. п., где количество
кислорода в воздухе ниже 16%, применяются гл. обр.
изолирующие приборы типа шланговых противогазов,
представляющие собой шлем-ма-
ску, соединенную со шлангом
для подачи свежего воздуха. Эти
противогазы бывают самовсасы-
вающие (ПШ-1) с максимально
допустимой длиной шланга 9 м
или с принудительной подачей
(ПШ-2, ДПА-5). В последних све-
жий воздух для дыхания подает-
ся при помощи переносной воз-
духодувки центробежного типа
с ручным или электр омеханич.
приводом.
Защита глаз. Для этой цели в
условиях химич. цехов обычно
Рис. 2. Наголовный
щиток с прозрачным
экраном.
применяют очки закрытого типа
(ГОСТ 9802—61). Недостаток этих очков — нек-рое
ограничение поля зрения и запотевание стекол. Для за-
щиты всего лица от брызг (напр., при разливе кислот)
85
ЗАЩИТА ИНДИВИДУАЛЬНАЯ - ЗАЩИТА МЕТАЛЛОВ
86
применяют наголовные щитки (рис. 2), состоящие из
фибрового наголовника и укрепленного на нем откид-
ного экрана из прозрачной негорючей пластмассы.
От агрессивных паров, газов или пыли надежно защи-
щают герметичные очки типа ПО-1 или ПО-2 с резино-
вой оправой (ГОСТ 9496—61) и специальными вкла-
дышами из незапотевающей пленки.
Защитная одежда (спецодежда). В зависимости от
вида продукции, изготовляемой химич. предприя-
тиями (кислоты, щелочи, галогены, органич. раство-
рители, синтетич. красители, синтетич. каучук и др.),
и условий труда применяют различные типы защит-
ной одежды (как по покрою, так и по типу тканей).
Так, для защиты от разбавлен-
Q ных кислот рекомендуются гру-
бошерстное сукно или хлопча-
тобумажные ткани (молескин,
J i 1 диагональ) с кислотозащитной
I 'i ijM пропиткой из синтетич. смол;
от конц. кислот — материал
Wj ШХВ-30, состоящий из смеси
«Ьд грубой шерсти и синтетич.
rj 1' ) кислотостойких волокон (хлор-
уА УУ винилового, лавсанового и др.)
уд у с пропиткой; от щелочей —
VVz брезент, плотные хлопчатобу-
.г/Д мажные ткани, клеенка, т. н.
1 пластикат и др.; от нефтепро-
— ViA? дуктов, масел, растворителей,
„ „ лаков и красок — хлопчато-
РИ° 3комбинеВаонЫЛЬНЫЙ бумажные ткани, пропитанные
нитрильным каучуком или его
смесью с хлорвиниловыми смолами и др. Стойкими
к действию сильных к-т, щелочей и органич. раство-
рителей являются также ткани с полиэтиленовым
пленочным покрытием. На рис. 3 приведен комбинезон
из специального молескина для защиты от проник-
новения пыли (см. также Защитная одежда).
Защита открытых участков кожи (лицо, шея, руки)
достигается также применением профилактиче-
ских паст (мазей). Эти пасты делятся на гидро-
фильные (защита от жиров и масел, нефтепродуктов,
лаков, смол, различных углеводородов и др. органич.
веществ) и гидрофобные (для защиты от воды, водно-
масляных эмульсий, водных р-ров к-т, щелочей, со-
лей и др.).
Гидрофильные пасты содержат веще-
ства, легко смачивающиеся водой н растворимые
в ней. Пасты этой группы изготовляют на крахмаль-
ной и мыльной основе. В состав пленкообразующих
паст, напр. противопековой пасты, могут входить же-
латина или крахмал (основной компонент), глицерин
(мягчитель), белая глина, окись цинка, а также кра-
ситель «конго красный» (в качестве светофильтра для
задержания ультрафиолетовых лучей) и др.
Гидрофобные мази делятся на мази с при-
менением жиров и масел и на пленкообразующие
мази. В первом случае в качестве основы применяют
окись цинка, воск, стеарин и растительное или мине-
ральное масло и др. (напр., цинкстеаратные мази).
Пленкообразующие мази содержат в качестве основы
нитроцеллюлозу, воск, шеллак, кремнийорганич. со-
единения (силиконовые пасты) и др.
Для защиты кожи от органич. растворителей, аро-
матич. углеводородов, нефтепродуктов, лаков, красок,
смол, хлорированных углеводородов и др. можно
пользоваться мазями или пастами: ХИОТ-6, ПМ-1,
ПЭР-1, Я ЛОТ, Селисского; для защиты от воды, вод-
ных р-ров кислот, солей, составов для хромирования,
никелирования и др. применяют мази или пасты:
цинкстеаратные № 1 и № 2, ИЭР-2, Чумакова, сили-
коновые; для защиты от кам.-уг. смол, пека, битума
и др. пользуются пастами: противопековыми ХИОТ
или Шапиро, ЦНИЛ-ГИС 1, 3, 6, пастой Гавриловой.
Почти все защитные мази и пасты легко смываются
горячей водой. Для удаления загрязняющих веществ,
трудно смываемых водой с мылом, применяют специ-
альные моющие средства и очистители кожи. К таким
поверхностно-активным веществам относятся: суль-
фонат ы—сульфоэфиры высокомолекулярных спир-
тов, сульфоно л—алкилбензосульфонат, препа-
раты «ОП» —полигликолевые эфиры алкилфенолов
и др. средства.
Защитные рукавицы и перчатки
применяют для защиты от кислот, щелочей и др. ед-
ких жидкостей; обычно для этих целей используют
перчатки из найритового каучука или полихлорви-
нила, а также рукавицы типа «КР», сшитые из мягких
хлопчатобумажных тканей и покрытые (путем мака-
ния) стойким к крепким кислотам н щелочам пленко-
образующим составом из синтетич. каучуков и смол.
Спецобувь. Для защиты от к-т и щелочей
применяются кислото- и щелочестойкие резиновые
сапоги, а также резиновые чуни. Кроме того, в раз-
личных специализированных произ-вах применяют
обувь целевого назначения: направо взрывоопасных
цехах, для работ с органич. растворителями, сжирами,
нефтепродуктами и т. д. См. также Противохимическая
защита, Дегазация, Защитная одежда, Противогазы.
Лит.: Найман И. М., Полонский 3. Б.,
Хабаров П. Г., Средства индивидуальной защиты на
производстве, 2 изд., М., 1958; Найман И. М., Защита
глаз на производстве, М., 1961; Торопов С. А., Защита
органов дыхания на производстве, [М.], 1954; О р е х о в. А. П.,
Полонский 3. Б., Хабаров П. Г., Спеподежда
и спецобувь, [М.1, 1955. И. М. Найман.
ЗАЩИТА МЕТАЛЛОВ электрохимическая — за-
щита металлов от коррозии при помощи искусственной
катодной поляризации поверхности металла. Поляри-
зацию поверхности производят наложением электрич.
тока, к-рый сдвигает потенциал поверхности до значе-
ний потенциала анода разомкнутой цепи. Катодная
защита применима в случае, если защищаемый металл
окружен проводящей средой (электролитом, почвой,
бетоном и т. д.), по к-рой может распространяться
электрич. ток, и неприменима против атмосферной и
газовой коррозии, а также коррозии неэлектролитами.
Различают две основные системы защиты: с наложен-
ным током и протекторную (защита автономными ано-
дами, гальванич. анодами, жертвенными анодами).
При катодной защите с наложен-
ным током последний подводят от внешнего
источника; постоянный ток подается от положитель-
ного полюса по проводу в специальное анодное зазем-
ление или анодное устройство, через к-рые он посту-
пает в среду, воздействующую на металл. Переходя
из среды на защищаемый металл, ток поляризует его;
Рис. 1. Защита трубопровода: 1—трубопровод;
2 — заземление; 3 — источник тока; 4 — точка дре-
нажа; 5 — повреждения изоляции.
затем ток из защищаемой конструкции через точку
дренажа и соединительный провод поступает к отри-
цательному полюсу источника тока. Общая схема за-
щиты различных объектов приведена на рис. 1, 2, 3.
При протекторной системе катод-
ной защиты необходимый для поляризации за-
щищаемой поверхности ток возникает за счет образо-
вания гальванич. пары, в к-рой роль протектора
87
ЗАЩИТА МЕТАЛЛОВ
88
(анода) играет металл более электроотрицательный,
чем защищаемый, а катодом служит защищаемый ме-
талл. Схема протекторной защиты различных объектов
приведена на рис. 4, 5 и 6.
4 — источник тока; 5 — лебедка.
При помощи катодной защиты теоретически можно
защищать все металлы, подверженные электрохимия,
коррозии. Практически, однако, наибольшее распро-
странение этот метод нашел для предупреждения кор-
Рис. 3. Защи-
та резервуара:
1 — резервуар;
2 — анод; з —
источник тока.
розии стали и свинца. Имеются сведе-
ния о защите этим способом алюминия
и его сплавов, бронзовых изделий и др.
При правильном выполнении защиты
потери металла обычно сокращаются на
90—98%. Технич. трудности при катод-
ной защите сводятся главным образом
к необходимости достаточно равномер-
ного распределения тока по защищае-
мой поверхности и наличия токопрово-
дящей среды, в окружении к-рой нахо-
дится защищаемый металл. При помощи
катодной защиты предохраняют от кор-
розии подземные и подводные трубопро-
воды, силовые кабели и кабели связи,
внутренние поверхности и днища резервуаров, химиче-
скую и теплообменЕхую аппаратуру, морские суда, буи,
сваи, эстакады и др. Крите-
рием полной защиты чаще
всего принимают величину за-
вис. 4. Защита трубопровода: 1 — трубопровод;
2 — протектор; 3 — соединительный провод; 4 — спе-
циальная засыпка.
щитного потенциала труба — почва или металл — элек-
тролит, равную —0,85 в (при измерении по медно-
сульфатному неполяризующемуся электроду). При
наличии в действующей
Рис. 5. Защита судна (аппа-
рата и т. п.): 1 — защищае-
мый лист металла; 2 — про-
тектор; 3 — изолирующая
подкладка; 4 — соединитель-
ный болт с гайкой; 5 — изо-
лирующая заливка.
пластинке.
среде (почве, воде и т. д.)
сульфатвосстанавливающих
бактерий, т. е. при опасно-
сти биокоррозии, величину
защитного потенциала ре-
комендуется принимать рав-
ной —0,95 в. Защиту кон-
тролируют обычно при по-
мощи контрольных пласти-
нок, устанавливаемых на
защищаемой конструкции
парами; одна из пластинок
включается в защитную схе-
му, а вторая остается вне
схемы. Через нек-рое, заранее установленное время
обе пластинки взвешивают и таким образом опреде-
ляют степень замедления коррозии на защищенной
Электрохимия, способом можно защищать как от-
крытые металлич. поверхности, так и изолированные
или окрашенные. Защита открытых поверхностей
обычно требует больших расходов защитного тока,
поэтому в большинстве случаев электро-
химия. защиту применяют совместно
с изоляцией защищаемых поверхностей
различными покрытиями (преим. высоко-
диэлектрическими). Максимальный за-
щитный потенциал для стальных изоли-
рованных поверхностей обычно ко-
леблется в пределах минус 1,2 — 1,5 в,
реже —2,0 в.
Посредством катодной защиты можно
также предотвратить коррозию, вызы-
ваемую блуждающими токами, которые
возникают вследствие плохого состоя-
ния промышленных и коммунальных
цепей постоянного тока (трамвая, элек-
трической ж. д., электролизных систем).
Для подавления этих токов методами
катодной защиты необходимо применять
Рис. 6. Защи-
та резервуара:
1 — резервуар;
2 — протектор;
3 — соедини-
тельный про-
вод; 4 — корро-
зионная среда.
повышенные
плотности тока и повышенные потенциалы.
Основными частями установки катодной защиты являются:
источник постоянного тока; анодное заземление или анодное
устройство; соединительные провода или кабели; щиток или
ящик с регулирующими, контрольными и измерительными
приборами; специальные узлы на защищаемом сооружении
(герметич. вводы проводов, электроизолирующие соединения,
контрольные образцы, контрольные выводы — проводники
для измерений и т. п.); измерительная установка для кон-
троля степени защиты. Выбор источника тока зависит от при-
нятой системы защиты, необходимой мощности и местных усло-
вий. В большинстве случаев мощность установок бывает в
пределах 1,5—2,0 кет, при предельном напряжении в 40 в
и величине защитного тока до 200 а. В нек-рых случаях мощ-
ность отдельных установок измеряется всего сотнями ватт;
в редких случаях мощность установок может достигать 7,5 —
8,0 кет. Для катодной защиты с наложенным током при мощ-
ностях тока до 1,5 кет чаще всего применяют селеновые вы-
прямители (применение выпрямителей оправдывается, если
источник их питания удален не больше чем на 2 км от установки).
При больших мощностях применяют генераторы, приводимые
от' электродвигателей или двигателей внутреннего сгорания
различного рода. При малых мощностях в качестве источника
тока используются аккумуляторы и гальваноэлементы.
Для подачи тока в окружающую среду с целью катодной
поляризации защищаемой конструкции при катодной защите
в почве устраивают заземления, а в жидких средах — анодные
устройства. Наиболее часто для этой цели применяют различ-
ное бросовое железо (старые трубы, рельсы, швеллеры и т. п.).
Более долговечные заземления выполняются из оцинкованного
железа, графита, прессованного магнетита и др. При малых
габаритах анодного устройства применяют серебряные или
титановые аноды, покрытые платино-палладиевым сплавом
и позволяющие иметь высокие плотности тока на поверхности
анода.
Протекторную защиту применяют обычно при небольших
мощностях установок; защита производится с помощью отдель-
ных протекторов, устанавливаемых т. обр., чтобы создать рав-
номерную плотность тока на защищаемой площади. В качестве
материала для протекторов применяют металлы более электро-
отрицательные, чем защищаемый. Так, для стали используют
цинк, магний, алюминий и
их сплавы. Нередко, напр.
при защите в морской воде,
анодам придают специаль-
ную форму для того, чтобы
вначале достичь наибольшей
плотности тока и быстрого
создания защитной пленки
на защищаемой поверхности.
К электрохимия, мето-
дам защиты относится
и т. п. э л е к т р и ч е-
с к и й дренаж, к-рый
применяется для защиты
подземных линий от
блуждающих токов. Этот
способ заключается в со-
единении защищаемого
сооружения (трубопро-
вода, кабеля и т. д.) и линии, являющейся источником
блуждающих токов (рельсы), проводником (рис. 7;
стрелками показан путь блуждающих токов), по к-рому
Рис. 7. Схема электрического
дренажа: 1 — рельсы электриче-
ской железной дороги; 2 — элек-
тродренажное устройство; з — за-
щищаемый трубопровод; 4 — по-
ляризующее реле; 5 — отсасы-
вающий фидер; 6 — регулирую-
щее сопротивление.
£9
ЗАЩИТА ОТ ИЗЛУЧЕНИЙ РАДИОАКТИВНЫХ ВЕЩЕСТВ
90
блуждающий ток, собирающийся на защищаемой под-
ъемной линии, поступает обратно в основную цепь тока.
При такой схеме на защищаемой линии устраняются
анодные зоны (участки выхода тока в окружающую
среду), а вся она оказывается катодно-поляризованной
и, следовательно, защищенной от электрохимия, корро-
зии. Регулируя сопротивление, установленное на со-
единении, можно добиться оптимальных защитных ус-
ловий на сооружении. К электрохимия, защите следует
отнести также анодную, применяемую для защиты
аппаратуры из нержавеющей стали. Схема защиты
в этом случае аналогична изображенной на рис. 3,
но с обратной полярностью. Положительный полюс
источника тока, присоединенный к корпусу защищае-
мого сосуда, обеспечивает анодную поляризацию
в местах контакта с электролитом с образованием за-
щитной окисной пленки, пассивирующей металл. Об-
ратно к источнику ток возвращается через катод,
специально помещаемый в электролит.
Лит.: Томашов Н. Д., Теория коррозии и защиты
металлов, М., 1959; Приту л а В. А., Катодная защита
трубопроводов и резервуаров, М.—Л., 1950; его же, За-
шита заводских подземных трубопроводов от коррозии, М.,
1961; Спирин А. А., Цекун Н. А., Салам-заде
М. М., Электрическая защита подземных металлических со-
оружений от коррозии, Баку, 1954; Трифель М. С,,
Алиев К. А., Защита и эксплуатация газовых сетей,
Баку, 1956; Притула В. А., Электрическая защита от
коррозии подземных металлических сооружений, М.—Л.,
1958; Цикерман Л. Я., Борьба с коррозией подземных
металлических трубопроводов, М.—Л., 1951; Справочник по
транспорту газов, М., 1954; Труды Всесоюзного совещания
по борьбе с морской коррозией металлов, Баку, 1958; Теория
и практика противокоррозийной защиты подземных сооруже-
ний, М., 1958; Рубенчик Л. И., Микроорганизмы как
фактор коррозии бетонЪв и металлов, Киев, 1950; Ершов
И. М., Защита подземных сооружений от коррозии, вызываемой
блуждающими токами, М., 1948; П р и т у л а В. А., Катод-
ная защита заводской аппаратуры, М., 1954; Францевич
И. Н., Хрущева Е. В., Францевич-Заблу-
довская Т. Ф., Катодная защита магистральных газо-
проводов, Киев, 1949; Правила защиты подземных металли-
ческих сооружений от коррозии, СН 28—58, М., 1959; [Д о-
р о ш е н к о П. Г.], Руководство по электрическим измере-
ниям и защите трубопроводов от коррозии, вызываемой блуж-
дающими токами, М., 1956; Ж у к Н. П., Коррозия и защита
металлов. Расчеты, М., 1957; Глазков В. И., Доро-
шенко П. Г., Котик В. Г., Защита магистральных
трубопроводов от подземной коррозии, М., 1960; Цикер-
ман Л. Я., Никольский К. К,.Разумов Л. Д.,
Расчет катодной защиты трубопроводов, М 1958; Стри-
же в с к и й И. В., Т о м л я н о в и ч Д. К., Блуждающие
токи и электрические методы защиты от коррозии, М., 1957;
Притула В. А., Борьба с коррозией танкеров, М., 1957;
Juchniewicz R., Katodowd, protektorowa i anodowa
ochrona metali w technice, Warszawa, 1960. В. А. Притула.
ЗАЩИТА ОТ ИЗЛУЧЕНИЙ РАДИОАКТИВНЫХ
ВЕЩЕСТВ и других излучений высо-
ких энергий (а, Р, у, нейтронов и т. д.) — сни-
жение уровня активности, воздействующей на чело-
века, до неопасной для здоровья дозы. Биология,
действие этих излучений особенно опасно тем, что,
во-первых, оно не воспринимается непосредственно
органами чувств человека, а, во-вторых, обладает
свойством аккумулятивпостп, т. е. постепенного на-
копления необратимых вредных изменений в орга-
низме, могущих привести в конечном итоге к его ги-
бели (при воздействии на организм в общем итоге
порядка сотен рентген). Исходя из этого, разработаны
предельно допустимые нормы доз облучения (см. Доза
ионизирующего излучения), не приносящих ощутимого
вреда здоровью человека при длительной его работе
с излучениями. Для различных видов излучений эти
дозы соответствуют их относительной биологич. эф-
фективности (ОБЭ), т. е. числу, показывающему, во
сколько раз предельно допустимая доза для данного
излучения должна быть уменьшена по сравнению
с у-излучением. Предельно допустимые дозы и ОБЭ,
а также соответствующие им мощности доз (доза в еди-
ницу времени) или интенсивности потоков частиц
(число частиц, поглощаемых 1 см2 поверхности в сек)
приведены в табл. 1. Суммарная, предельно допусти-
мая доза за все время работы человека (в возрасте Л
лет) с излучениями по действующим нормам не должна
превышать величины 5 (JV — 18) биологич. эквивален-
тов рентгена (бэр = ~R). Защита зависит от вида
излучений и связана с их физич. свойствами.
Таблица 1. Предельно допустимые дозы облучения
Виды излучения Допус- тимая до- за за не- делю, фэр Относи- тельная биол. эф- фектив- ность, обэ Мощность дозы или поток при 36-часовой рабочей неделе
а-Чзстицы и про- тоны 0,01 10
р-Частицы . . . 0,1 1 20 р-частиц
7-Лучи Тепловые ней- троны 0,1 0,033 1 3 слса • сек 2,8 миллирентгена(мрУ
час 750 нейтронов см3 • сек
Быстрые нейтро- ны (—10 Мэе) 0,01 10 20 нейтронов
С.И1 • сек
Таблица 2. Длина пробега (R) а- н (3-частиц в воздухе
и алюминии (в с.ч)
Ех, Мэв (воздух) «« (алюминий) Eg, Мэв R3 (воздух) R3 (алюминий)
4 2,5 0.0015 0,01 0,13 0,00006
5 3,5 0,0020 0,05 2,91 0,00144
6 4,6 0,0027 0,1 10,1 0,00500
8 7,4 0,0040 0,5 119 0,0593
10 10,6 0,0056 1 306 0,159
5 1900 0,924
10 3900 1,92
Поскольку а-частицы заданной энергии (£„) ха-
рактеризуются определенной длиной пробега (ВД
в различных средах (см. табл. 2), то их вредное воз-
действие можно полностью устранить, используя
слой защитных материалов, превышающий длину
пробега.
Радиоактивные вещества испускают а-частицы в ди-
апазоне энергий от 4 до 11 Мэв. Таким образом,
согласно соответствующим данным по длинам пробега,
защититься от а-частиц не представляет никакого
труда: слой воздуха в 15 с.и уже предохраняет от
их вредного действия. Однако представляет опасность
работа с a-активными летучими веществами, а также
с открытыми нелетучими препаратами (см. Сцилларда—
Чалмерса эффект), поскольку высокая биологич.
активность а-частиц приводит к тяжелым последст-
виям при их попадании в организм. Поэтому работать
с открытыми препаратами большой активности раз-
решается только в закрытых герметпч. боксах в ре-
зиновых перчатках, к-рые предохраняют руки от
действия а-излученпя и мягкого р-пзлучения.
P-Частицы, как и а-частицы, имеют определенную
длину пробега в поглощающих материалах (см.
табл. 2). Используя толщины защитных материалов,
превышающие пробеги Р-частиц, можно устранить их
вредное воздействие. Радиоактивные вещества испу-
скают Р-частицы с энергией до 3 Мэв и выше. Вредное
действие p-излучения весьма значительно, и поэтому
следует особенно тщательно защищаться от него при
работе с открытыми препаратами в боксах. Как следует
из длин пробегов, стеклянная стенка бокса толщиной
в 2—3 .и.и может оказаться недостаточной для погло-
щения Р-частиц, в связи с чем прошедшие через стенки
Р-частицы могут вызвать опасные ожоги глаз и дру-
гих органов работающего. При поглощении р-частиц
91
ЗАЩИТА ОТ ИЗЛУЧЕНИЙ РАДИОАКТИВНЫХ ВЕЩЕСТВ
92
защитными материалами возникает тормозное рентге-
новское излучение (внешнее тормозное излучение).
Кроме того, все Р-излучатели испускают гамма-лучи
малой интенсивности (внутреннее тормозное излуче-
ние). Оба эти излучения становятся заметными при
очень сильных p-источниках. Приближенно тормоз-
ное излучение /р p-источников можно оценивать по
формуле: 1р = 1,23 • 10~4 (Z 3) _Е| Мэе[распад-, здесь
Z — средний атомный номер материала защиты, Ер —
максимальная энергия р-частиц в Мэе, коэфф. 3 учи-
тывает внутреннее тормозное излучение р-источника.
При прохождении через вещество у-лучи, в отличие
от а- и р-частиц, не характеризуются определенной
длиной пробега.
Поглощение 7-лучей определяется логарифмическим за-
коном: In — = цг, где Ро — исходная мощность дозы у-излу-
чения, Р — мощность излучения после прохождения через
слой г материала, ц — коэфф, ослабления, зависящий от вида
материала и энергии излучения (см. табл. 3); он соответствует
обратной величине среднего пробега I. (ц = т. е. среднего
значения величины г при изменении Р от Р„ до 0. Исходя из
закона поглощения, в защитных экранах возможно не полное
поглощение у-излучения, а лишь ослабление его до безопасной
мощности дозы Р. Для оценки необходимой толщины защиты
в заданном месте определяют кратность (А) превышения
мощности дозы 7-излучения (Р„) против допустимой мощности
дозы (Г): А = Ро/Р, а затем по закону поглощения вычисляют
толщину слоя защиты г. При этом удобно пользоваться поня-
тием слоя половинного ослабления D = 0,693/ц, тогда г =
= 3,33 Dig А.
Эта толщина защиты будет недостаточной вследствие воз-
никновения вторичных у-лучей в защитном материале. Вторич-
ные у-лучи можно Оценить с помощью т. иаз. фактора накоп-
ления В, к-рый показывает, во сколько Раз доза за защитой
будет больше нормы при данном значении цг (произведение
коэфф, накопления на первоначально найденную толщину
защиты). В таблице 4 приведены значения В для у-лучей с
энергией Еу = 1 Мэв в воде, алюминии (бетоне), железе (чу-
гуне) и свинце.
Таблица 3. Коэффициент ослабления р. (с.и~1) при поглощении
7-излучения
Еу, Мэв Вода , Бетон . ( 2,2—) \ CM3 J Алюминий ( 2,7-М 1 см31 Чугун (7,2-М 1 см31 Свинец f Л у см3 1
0,5 0,097 0,186 0,228 0,6 1,74
1 0,0706 0,135 0,166 0,43 0,79
3,0 0,0396 0,079 0,094 0,26 0,46
Таблица 4. Фактор накопления при поглощении 7-нзлучения
Материал защиты Р-от (при E-,=i Мэв)
2 4 10 15
Фактор накопления В
Вода 3,5 7,2 15 24 45
Алюминий 3,3 6,6 13 21 38
Железо 2,9 5,4 10 16 28
Свинец 1,7 2,3 3 3,7 4,8
у-Лучи радиоактивных веществ являются весьма
проникающими, и для их достаточного ослабления при-
ходится применять довольно толстые слои защитных
материалов. При работе с радиоактивными р-рами
в химич. шкафах для защиты от у-лучей чаще всего
применяются свинцовые кирпичи размером 5 X 10 X
X 10 см3 или 5 X 10 X 20 ел»3, а также свинцовые
стекла. После сооружения защиты желательно про-
верить интенсивность у- и P-излучения на рабочем
месте соответствующими дозиметрами (см. Дозимет-
рия). В особенности это касается P-излучения, т. к.
р-частицы сильно рассеиваются, особенно тяжелыми
материалами, и могут создавать повышенный фон
Р-частиц, несмотря на то, что прямой пучок р-частиц
и у-лучей достаточно ослаблен свинцовыми кирпи-
чами.
Помимо а-, р- и у-лучей, радиоактивные препараты
могут испускать нейтроны (напр., источники, упако-
ванные в стеклянные контейнеры, содержащие при-
месь бериллия, бора и др. легких материалов, а также
специальные нейтронные источники). Нейтроны по-
глощаются веществом по тому же закону, что и у-излу-
чение, причем коэфф, р. = п<у, где п — число ядер
в единице объема поглотителя, а о — сечение захвата
ядра. Поэтому расчет защиты от нейтронов аналоги-
чен соответствующему расчету в случае у-излучения.
Защита от нейтронов, испускаемых радиоактивными
препаратами, осуществляется обычно водой или пара-
фином, замедляющими быстрые нейтроны. Отметим,
что слой воды в 7 см или парафина в 6 см умень-
шает плотность быстрых нейтронов примерно в
2,7 раза. От медленных (тепловых) нейтронов защи-
щаются листовым кадмием или боросодержащимп
экранами. Контейнеры для хранения и транспорти-
ровки нейтронных источников изготовляются из смеси
парафина с бурой или борной к-той, сильно поглощаю-
щими медленные нейтроны. В качестве защитного
материала от излучений крупных источников (реак-
торы, ускорители, кобальтовые источники) служит
обычно бетон (обычный и специальный).
Лит.: Гусев Н. Г., Справочник по радиоактивным
излучениям и защите, М., 1956; Гусев Н. Г., Машко-
в и ч В. П., О б в и н ц е в Г. В., Гамма-излучение радио-
активных изотопов и продуктов деления, М., 1958; Горш-
ков!’. В., Гамма.-излучение Радиоактивных тел и элементы
расчета защиты от излучения, М.—Л., 1959; Защита ядерных
реакторов, пер. с англ., М., 1958; Радиационная дозиметрия,
под ред. Дж. Хайна и Г. Браунелла, пер. с англ., М., 1958;
Комаровский А. Н., Строительные материалы для
защиты от излучений ядерных реакторов и ускорителей, М.,
1958; Санитарные правила работы с радиоактивными вещест-
вами и источниками ионизирующих излучений, М., i960;
Правила перевозки радиоактивных веществ, М., 1961; А р-
д а ш н и к о в С. Н. [ид р.], Зашита от радиоактивных излу-
чений, М., 1961; Гусев Н, Г. [ид р.1, Защита от излучений
протяженных источников, М., 1961; Егер Р., Дозиметрия
и защита от излучений, пер. с нем., М., 1961. Г. В. Горшков.
ЗАЩИТНАЯ ОБРАБОТКА ДРЕВЕСИНЫ — про-
изводится с целью увеличения срока службы лесома-
териалов, древесных плит й различных изделий из
древесины. Наибольшее значение имеет защита дре-
весины от воспламенения и гниения. 3. о. д. произ-
водится защитными веществами — антипиренами, ан-
тисептиками и др. — путем пропитки древесины или
ее покрытия этими веществами. Защитные вещества
могут быть введены в древесину следующим образом:
1) окраской древесины огнестойкими веществами или
обмазкой ее антисептич. пастами, из к-рых антисеп-
тики диффундируют в сырую древесину; 2) вымачи-
ванием древесины в пропиточных р-рах; 3) введением
масел или р-ров в древесину под вакуумом и давле-
нием. Наибольшее значение имеет пропитка древесины
под избыточным давлением в пропиточных цилиндрах
(автоклавах) методами полного, ограниченного или
частичного поглощения. При пропитке методом пол-
ного поглощения из древесины под вакуумом удаляют
воздух, засасывают в пропиточный цилиндр пропи-
точную жидкость и вводят ее в древесину под давле-
нием 7—25 ат. При пропитке методом ограниченного
или частичного поглощения в цилиндре сначала со-
здают воздушное давление для сжатия воздуха в дре-
весине, затем закачивают жидкость и пропитывают
древесину под повышенным давлением, после чего
создают вакуум в цилиндре для удаления из древе-
сины излишка пропиточной жидкости. В цилиндрах
легко пропитываются заболонные и спелодревесные
лиственные породы (береза, бук, липа, клен и др.),
труднее — ядровые породы (сосна, лиственница и др.)
и наиболее трудно спелодревесные хвойные породы
(ель, пихта). Пропитку древесины антисептиками и
93
ЗАЩИТНАЯ ОДЕЖДА —ЗАЩИТНОЕ ДЕЙСТВИЕ
94
антипиренами обычно производят на деревопропиточ-
ных заводах.
Для защиты древесины от огня их об-
рабатывают антипиренами, к-рые легко плавятся при
нагревании и покрывают поверхность дерева плотной
пленкой, преграждающей доступ к нему кислорода.
Нек-рые антипирены разлагаются с выделением ббль-
шого количества негорючих газов, что также препят-
ствует доступу к древесине кислорода.
Пропитанная антипиренами древесина не воспла-
меняется, не тлеет и не поддерживает горения, посте-
пенно разрушаясь под влиянием высокотемператур-
ного нагрева извне. Наиболее распространенными
антипиренами являются фосфорнокислый и сернокис-
лый аммоний (и их смесь), бура и др., к-рые вводят
в количестве 5—8% сухого огнезащитного состава по
отношению к весу пропитываемого дерева. Времен-
ную защиту от огня дают огнезащитные краски на
жидком стекле. Обработку лесоматериалов антипире-
нами обычно производят в готовом изделии.
Защита древесины от гниения до-
стигается гл. обр. путем применения антисептиков,
среди к-рых различают масляные (масла каменно-
угольное, сланцевое и др.) и водорастворимые (NaF,
уралит, ZnCl2 и др.). В качестве антисептиков приме-
няют также синтетич. продукты (хлорфенолы и др.),
а в нек-рых случаях и различные мышьяковые пре-
параты. Для 3. о. д. часто применяют смеси антисеп-
тиков, каждый из к-рых берется в количестве, мини-
мально необходимом для прекращения жизнедеятель-
ности грибов, вызывающих гниение древесины. Для
профилактич. защиты древесины от морских древо-
точцев ее пропитывают ядовитыми для них вещест-
вами (кам.-уг. масла и др.). Норма поглощения анти-
септика зависит от его свойств, способа консервиро-
вания, породы дерева и др. и обычно колеблется в пре-
делах (кг/м3 древесины):
а) для масляных антисептиков: дуба и листвен-
ницы 45—60, сосны и ели 65—110, бука и березы
145—170;
б) для водорастворимых антисептиков 4—7 (в пе-
ресчете на чистую 100%-ную соль).
Средний срок службы (в годах) неконсервирован-
ных и пропитанных антисептиками железнодорожных
шпал и опор связи из дерева различных пород:
Порода Неконсер- вирован- ная древе- сина Древесина, пропитан- ная антисептиками
водораство- римыми масляны- ми
Сосна 5-6 12-14 18—23
Ель 4-5 10-12 14-16
Бук 2—3 14-18 25—30
Лит.: Лекторский Д. Н., Защитная обработка
древесины, М., 1951; Г о р ш и н С. Н., Тр. Ин-та леса АН
СССР, 1950, т. 6; Петри В. Н., Дулькин А. Л.,
Разрушители древесины, Свердловск, 1950; Голдин М. М.,
Антисептическая защита деревянных конструкций, 2 изд.,
М„ 1951; ГОСТ 5430—50. Г. И. Новицкий.
ЗАЩИТНАЯ ОДЕЖДА противохимическая — сред-
ства защиты кожных покровов от кожно-нарывных и
нек-рых общеядовитых отравляющих веществ (ОВ),
радиоактивных веществ и бактериальных средств, а
также ограниченной защиты от проникающей радиа-
ции и светового излучения. По принципу защитного
действия 3. о. может быть изолирующего или фильт-
рующего типа.
3. о. изолирующего типа изготовляется
из резины, прорезиненной или проолифенной ткани,
специальной пропитанной бумаги или др. синтетич.
высокомолекулярных материалов, через к-рые ско-
рость проникания ОВ крайне незначительна. По кон- ]
струкции изолирующая 3. о. может быть герметичной
(костюм, комбинезоны) и негерметичной (плащи, на-
кидки, фартуки и т. п.).
Герметичная 3. о. применяется для длитель-
ной защиты кожи от капель и паров ОВ, от радиоак-
тивных веществ (радиоактивной пыли) и бактериаль-
ных средств. Она используется при действии на зара-
женной местности, при выполнении дегазационных,
дезактивационных и дезинфекционных работ. Ком-
плект герметичной 3. о. состоит из комбинезона или
костюма из прорезиненной или проолифенной ткани,
резиновых сапог, резиновых перчаток. Для большей
герметичности прилегания капюшона к голове исполь-
зуется хлопчатобумажный подшлемник. Комплект
весит 5—6 кг.
Защитные комбинезоны изготовляются 3 ростов:
1-й для лиц ростом меньше 165 см; 2-й •— от 165 до
172 см и 3-й — более 172 см. Резиновые сапоги изго-
товляются от 38 до 44 размера (вкл.); резиновые пер-
чатки — одного размера. Защитный костюм состоит из
куртки, брюк, резиновых сапог, резиновых перчаток
и хлопчатобумажного подшлемника; вес ок. 5—6 кг.
Наряду с описанными защитными комбинезонами и
костюмами имеются легкие защитные костюмы, изго-
товленные из прорезиненной ткани. Эти костюмы вы-
полнены заодно с защитными чулками и перчатками;
они надеваются поверх обычной одежды; вес комплекта
3,5—5 кг. Одним из существенных недостатков такой
3. о. является необходимость строго регламенти-
ровать время пребывания в ней, особенно в летних ус-
ловиях. Допустимые сроки непрерывного пребы-
вания герметичной 3. о.: при 30° и выше не бо-
лее 15—20 мин при 25—29° 30 мин при 15—19° до
1,5—2 час.
Негерметичная 3. о. применяется для пре-
дохранения обычной одежды (обмундирования), сна-
ряжения и открытых частей тела от заражения кап-
лями отравляющих веществ, радиоактивными вещест-
вами и бактериальными средствами. Комплект такой
одежды состоит из бумажной накидки, пропитанной
специальным составом, защитных чулок из прорези-
ненной или проолифенной ткани и защитных перча-
ток из прорезиненной ткани. Защитная бумажная
накидка имеет вид плаща без рукавов; вес накидки
ок. 300 г; она изготовляется одного размера и после
использования уничтожается.
З.о. фильтрующего типа обеспечивает
защиту от паров ОВ и представляет собой обычное
обмундирование, пропитанное (импрегнированное)
специальными веществами. Защитное действие импрег-
нированного обмундирования основано на поглоще-
нии паров ОВ компонентами пропитки. Наибольшее
распространение получили импрегнаты абсорбцион-
ного и хемосорбционного типа. В качестве пропитки
для импрегнатов абсорбционного типа применяют
сульфированное касторовое масло, а для импрегнатов
хемосорбционного типа—хлорамины, напр. дихлор-
амин Т. По санитарно-гигиенич. свойствам импрегниро-
ванное обмундирование почти не отличается от обыч-
ного и его можно носить постоянно.
Лит.: Вознесенский А. Д., Индивидуальные
средства противохимической защиты, М., 1960; Кокосов
Б. В., Противоатомная, противохимическая, противобакте-
риологическая защита солдата в бою, М., 1957.
С. Н. Васильев.
ЗАЩИТНОЕ ДЕЙСТВИЕ (в радиационной хи-
мии) — понижение нек-рыми веществами (защитными
добавками) радиационно-химич. разложения других
веществ. 3. д. обусловлено взаимодействием защитных
добавок со свободными радикалами, ионами или воз-
бужденными молекулами, образующимися при дей-
ствии ионизирующего излучения на вещества и спо-
собными реагировать с молекулами защитных веществ.
Напр., при радиолизе водного р-ра метиленового го-
95
ЗАЩИТНЫЕ КОЛЛОИДЫ-ЗАЩИТНЫЕ ПОКРЫТИЯ
96
лубого обесцвечивание последнего можно предотвра-
тить добавлением глюкозы, взаимодействующей с гид-
роксильными радикалами, образующимися при ра-
диология. разложении воды. В отсутствии глюкозы
гидроксильные радикалы окисляют краситель.
В нек-рых случаях 3. д. связано с процессом само-
ингибирования (самозащита). При этом ско-
рость разложения вещества уменьшается в присут-
ствии продуктов его превращения. Так, при действии
у-лучей на разб. водный р-р хлорфенолового красного
этот краситель обесцвечивается в результате взаимо-
действия с продуктами радиолиза воды. Однако воз-
никающие при этом промежуточные продукты реаги-
руют с радикалами, образующимися из воды, и за-
медляют, т. обр., процесс обесцвечивания красителя.
Часто 3. д. вызвано передачей заряда или энергии
возбуждения от защищаемого вещества к защитной
добавке. Если защитная добавка обладает низкой
радиолитич. устойчивостью, то ее молекулы, дезак-
тивируя молекулы защищаемого вещества, распа-
даются сами («жертвенная» защита). При высокой
радиолитич. устойчивости защитной добавки ее мо-
лекулы могут в значительной степени рассеивать по-
лучаемую энергию не разлагаясь (защита типа
«губки»). Так, при радиолизе смеси циклогексан —
циклогексен осуществляется жертвенная защита пер-
вого компонента вторым. В смеси циклогексан — бен-
зол энергия возбуждения, передаваемая от циклогек-
сана к бензолу, в основном превращается в теплоту,
не приводя к химич. распаду бензола. 3. д. обычно
используется для подавления нежелательных радиа-
ционно-химич. процессов.
Нек-рые вещества оказывают 3. д. на живые орга-
низмы при облучении. Детально механизм действия
таких веществ в большинстве случаев невыяснен.
Лит.: Радиационная химия. Сб. статей, пер. с англ., М.,
1953; Burton М., L i р s k 1 S., Mechanisms of protec-
tion in radiolysis of organic systems, J. Phys. Chem., 1957,
61, № 11, 1461 (обзорная статья, содержит библиографию).
В. А. Лронгауз.
ЗАЩИТНЫЕ КОЛЛОИДЫ — вещества, к-рые при
добавлении в малых количествах к лиофобпым золям
и дисперсиям резко повышают их агрегативную
устойчивость и особенно их устойчивость против коа-
гулирующего действия электролитов. Действие 3. к.
(защитное действие) обеспечивает длитель-
ное существование золей и других дисперсий в боль-
ших концентрациях, чем в незащищенном состоянии.
3. к. являются обычно высокомолекулярные вещества,
растворимые в дисперсионной среде, напр. белковые
вещества, дубители (танниды), гликозиды, полисаха-
риды. Раньше эти вещества считали лиофильными
коллоидами, отсюда и название-— 3. к. К 3. к. в пря-
мом смысле относятся поверхностно-активные ве-
щества типа мыл, образующие в дисперсионной среде
лиофильные коллоидные растворы. Для таких 3. к.
характерно проявление моющего действия. Защитное
действие обычно вызывается адсорбцией 3. к. на
поверхности частиц дисперсной фазы золей с возник-
новением структурированных адсорбционно-сольват-
ных оболочек, имеющих лиофильную поверхность и
предохраняющих частицы от коагуляции, или же,
кроме того, связано с образованием пространствен-
ной структуры студня (геля) в объеме дисперсионной
среды. 3. к. являются обычно сильными стабилизато-
рами дисперсных систем — суспензий, эмульсий и
пен — и обусловливают практически полную агрега-
тивную устойчивость их по отношению к коагуляции и
коалесценции. Нек-рые золи после защиты становятся
обратимыми, напр. медицинский препарат протаргол,
растворимый в воде, представляет собой защищенное
металлич. серебро в коллоидном состоянии. Защитное
действие зависит от химич. природы 3. к. й поверх-
ности р; зд?ла частица — среда; количественно оно
условно характеризуется т. наз. золотым ч и с-
л о м, т. е. числом миллиграммов 3. к., достаточным,
чтобы воспрепятствовать перемене красного цвета
в фиолетовый у 10 мл гидрозоля золота (0,006% кон-
центрации, полученного по методу Зигмонди) от
коагулирующего действия 1 мл 10%-ного р-ра хло-
ристого натрия. Защитное действие играет важную
роль в физиология, процессах и в технологии многих
ПроИЗ-В. П. А. Ребиндер.
ЗАЩИТНЫЕ ПОКРЫТИЯ — слои, искусственно
создаваемые на поверхности металлич. изделий и
сооружений с целью предохранения их от коррозии
(см. Коррозия металлов). Если наряду с защитой от
коррозии покрытие служит также для декоративных
целей, его называют защитно-декоративным. В зави-
симости от материала 3. п. подразделяют на несколько
групп.
Металлические покрытия. Материалами для метал-
лич. 3. п. могут быть как чистые металлы (цинк,
кадмий, алюминий, никель, медь, хром, серебро и
др.), так и их сплавы (латунь, бронза и др.). Для
получения металлич. 3. п. широко применяются
гальванич. способы, нанесение 3. п. горячим и диф-
фузионным способами, а также металлизацией.
Гальванические 3. п. — тонкие прочно
сцепленные с освовным материалом металлич. покры-
тия, получаемые электролитич. осаждением металлов
на защищаемой поверхности (см. Гальванотехника).
Защитное действие гальванич. покрытий при их
плотной мелкокристаллич. структуре определяется
в основном их толщиной, которая, в свою очередь,
зависит от условий работы покрываемых изделий.
Различают легкие, средние и жесткие условия.
К легким относят атмосферу закрытых сухих (до 80%
относительной влажности) помещений; к средним —
наружную атмосферу с колебаниями темп-ры от
—50 до —50°; к жестким — те же условия, что и
к средним, с одновременным воздействием различных
загрязнений, пыли и др.
Минимальная толщина цинковых покрытий для
защиты стальных изделий в зависимости от условий
работы изделий устанавливается (соответственно)
5, 15 и 30 мк. Для изделий из меди и медных сплавов
толщина никелевого покрытия устанавливается соот-
ветственно 5, 10 и 15 мк. Для защиты изделий от
потускнения дополнительно наносится слой хрома
толщиной до 1 мк. При защитно-декоративном покры-
тии стальных изделий суммарная толщина меди +
никель должна быть (соответственно) 10, 30 и
50 .мк; изделия из цинкового сплава рекомендуется
покрывать слоями такой же толщины.
Толщина оловянных покрытий при защите от
атмосферной коррозии обычно равна 10—15 мк;
применение промежуточного слоя (медь или латунь)
позволяет снизить эту толщину примерно вдвое.
Толщина оловянного покрытия на белой жести
равна 1—3 мк. Толщина свинцовых покрытий, при-
меняемых для защиты от серной к-ты, сернистых
газов и различных сернокислых соединений, обычно
измеряется сотыми, а в некоторых случаях десятыми
долями мм. Хромовые покрытия, применяемые на-
ряду с защитой от механич. износа также для защиты
стали от коррозии, имеют толщину 50, 100 и 200 мк.
Лит.: Лайнер В. И., Кудрявцев Н. Т., Основы
гальваностегии, ч. 1—2, 3 изд., М., 1953—57.
В. И. Лайнер.
Защитные покрытия, получаемые
горячим способом. 1) Цинкование —
проводится путем погружения предварительно обез-
жиренных, травленых и промытых железных изделий
(листов, труб) сначала в слой флюса (NH4C1, ZnCl?
и глицерин), а затем в расплавленный пинк, в к-рый
Обычно добавляют 0,15—0,2% Al, 1—3% Sn и др.
97
ЗАЩИТНЫЕ ПОКРЫТИЯ
98
составляющие. Вес цинкового покрытия 300—700 г/м2,
толщина образующегося слоя 20—30 мк. 2) Луже-
ние — по технологии сходно с цинкованием; вес
покрытия 27—45 г/.ц2, толщина 3—5 мк. Применяется
для покрытия консервных банок, молочной посуды,
пищеварных котлов, медных кабелей и др. 3) П о-
крытие свинцом — по технологии сходно
с цинкованием. В ванну, кроме свинца, обычно до-
бавляют 10—25% олова (реже сурьму или кадмий).
Применяется для обработки стальной проволоки,
в качестве оболочек на кабелях, для покрытия сосудов
в химич. пром-сти. Для тары и емкостей, предназна-
ченных для пищевых продуктов, освинцевание не
применяется, т. к. соли свинца ядовиты. 4) Али-
тирование — проводится при 700—800° в сплаве
алюминия с железом (8—9% железа) в продолжении
1—30 мин; поверх сплава — слой флюса (ZnCI2,
NaCl, криолит). Технология алитирования анало-
гична цинкованию. Толщина образующегося на
железе диффузионного слоя (сплав железа и алюми-
ния) 20—150 ми. Алитирование применяется для
обработки труб, листов, инструмента для литья
цветных металлов, головок клапанов и др.; повы-
шает окалиностойкость и коррозионную устойчи-
вость стали и чугуна. Горячие способы получения
3. п. применяются также для нанесения эмалевых
слоев ва поверхность железных и чугунных изделий
(химич. аппаратура, посуда, трубы) путем расплав-
ления смесей нек-рых окислов и неорганич. солей
(подробнее см. Эмали).
Защитные покрытия, получаемые
диффузионным способом. ^Азотирова-
ние— проводится в среде аммиака при 600—650°
в продолжении 40—60 мин (реже при 800—820°
в течение 5—10 мин). На поверхности стали обра-
зуется тонкий (0,01—0,02 мм) нитридный слой (Fe2N,
Fe4N), обладающий повышенной хрупкостью на
углах деталей. Азотирование применяется для обра-
ботки болтов, тяг, вентилей, деталей приборов и др.
Азотированный слой устойчив во влажной воздушной
атмосфере, воде, бензине, неочищенном масле, пере-
гретом паре и др.; неустойчив — в кислотах и мор-
ской воде. 2) Цинкование — проводится при
380—390° в печах с вращающимися ретортами или
в ящиках, куда вводится порошок цинковой пыли.
В ретортных печах слой толщиной 15—20 мк дости-
гается за 1 час (в ящиках за 2 часа). Коррозионная
устойчивость обработанных т. о. деталей не уступает
коррозионной устойчивости гальванически обрабо-
танных деталей, причем стоимость диффузионной
обработки ниже гальванической. При пассивировании
деталей, подвергнутых диффузионному цинкованию,
их коррозионная устойчивость повышается. При
диффузионном цинковании стали при 750—900° в ат-
мосфере паров цинка обработанные детали устойчивы
при 500—550° в газовой среде, содержащей H2S.
3) Алитирование — проводится при 950—
1050°: а) в порошкообразной смеси, состоящей из
99% ферроалюминиевого сплава и 1% NH4C1 или
50% А1, 49% А12О3 и 1% NH4C1; б) в порошке фер-
роалюминиевого сплава в среде Н2 НС1; в) метал-
лизацией стали алюминием с последующим диффу-
зионным отжигом. Слой глубиной 0,5—0,8 .ч.м дости-
гается за 5—10 час. Алитирование повышает окали-
ностойкость обработанных деталей; применяется для
труб и деталей теплообменников, коллекторов дизе-
лей, клапанов автомобилей и др. 4) Хромиро-
вание — проводится при 950—1050°: а) в порошко-
образной смеси, состоящей из 60% феррохрома,
39% А12О3 и 1% NH4C1; б) в порошке хрома или фер-
рохрома в среде Н2 НС1 или в вакууме. Слой глу-
биной 0,1 мм на стали образуется в течение 5—10 ча-
сов. Хромированные детали обладают окалиностой-
костью (до 800°), повышенной устойчивостью в азот-
ной и уксусной к-тах, р-ре NaCl, а также в Н2О2.
Диффузионное хромирование применяется для труб
теплообменников, электродов автомобильных свечей,
различных промышленных деталей и др. 5) Сили-
цирование — проводится при 950—1050°: а) в по-
рошкообразной смеси, состоящей из 60% ферросили-
ция или карбида кремния, 39% А12О3 и 1% NH4C1;
б) в среде хлора в ретортной печи, в к-рую загру-
жают обрабатываемые стальные детали, куски фер-
росилиция или карбида кремния. Обработанные де-
тали кислотоупорны (особенно в азотной, серной и
соляной к-тах), коррозионноустойчивы в простой и
морской воде и обладают повышенной износоустойчи-
востью. Силицирование повышает также окалино-
стойкость некоторых металлов и сплавов (особенно
молибдена); применяется для обработки стальных де-
талей химич. машиностроения и деталей, работающих
в морской воде.
Существенное повышение коррозионной устойчи-
вости и кислотоупорности стали может быть достиг-
нуто также бериллизацией, титанированием, воль-
фрамированием, борированием, молибденированием
и др., но эти процессы по разным причинам не полу-
чили пока промышленного применения.
Металлизация — способ получения метал-
лич. защитных покрытий на различных сооружениях
(мосты, детали судов, большие баки и др.); при этом
способе расплавленный металл распыляется (пуль-
веризуется) с помощью особого прибора — металли-
затора. Частички металла, вылетающие с большой
скоростью из сопла аппарата, осаждаются на поверх-
ности изделия и создают 3. п. Металлизацию широко
применяют для получения цинковых, свинцовых и
алюминиевых покрытий.
Лит.: Бахвалов Г. Т., Ту рк опекая А. В.,
Коррозия и защита металлов, М., 1947; Металловедение и
термическая обработка стали. Справочник, т. 2, М„ 1962;
Ямпольский А. М., Защитные покрытия металлов,
Л., 1958. А. Н. Минкевич.
Неметаллические покрытия. Эти покрытия можно
подразделить на следующие основные группы: лако-
красочные, пластмассовые и гуммированные.
Лакокрасочные 3. п. — широко приме-
няются для защиты металлов от коррозии и придания
изделиям красивого внешнего вида. Наиболее часто
эти 3. п. наносятся на изделия и конструкции (станки,
ж.-д. вагоны, мосты, автомашины, холодильники,
железные кровли и др.), подвергающиеся коррозии
на воздухе как внутри, так и вне помещений; кроме
того, лакокрасочными 3. п. окрашивают химич.
аппаратуру, а также подводную и надводную частй
речных и морских судов. Покрытия создают нанесе-
нием на поверхности изделий нескольких (двух
и более) слоев лакокрасочных материалов. Поверх-
ность предварительно тщательно подготовляют: уда-
ляют окалину и ржавчину, жировые вещества и влагу.
Для улучшения сцепления с покрытием применяют
обработку металлич. песком из закаленного чугуйа
или взвесью кварцевого песка в воде (гидропеско-
струйная обработка); иногда создают пористый под-
слой путем химич. или электрохимия, обработки
(фосфатирование, анодирование). Нижний слой, не-
посредственно соприкасающийся с поверхностью ме-
талла, называется грунтовочным, верхние
слои — покровными. 1'рунтовочный слой дол-
жен надежно сцепляться с металлом и обладать хоро-
шими антикоррозийными свойствами. Для этого
в грунтовочный слой вводят в качестве пленкообра-
зующих веществ алкидные, алкилфенольные, эпок-
сидные смолы, растительные масла, а также пигменты
(железный и свинцовый сурик — для грунтов по чер-
ным металлам, и цинковый крон — для грунтов по
цветным металлам); с помощью фосфатирующих
99
ЗАЩИТНЫЕ ПОКРЫТИЯ
100
грунтов производят одновременное фосфатирование
и грунтование поверхности. Хорошими антикорро-
зийными свойствами обладают протекторные грунты,
содержащие до 95% цинковой пыли и ок. 5% плен-
кообразующего вещества. Такие грунты защищают
металл электрохимически, подобно металлич. цинко-
вым покрытиям. Между грунтовочными и покровными
слоями при необходимости наносят промежуточные
слои (для выравнивания поверхности — шпаклевоч-
ные слои, для хорошего сцепления между слоями —
переходные слои и др.).
Наиболее распространенными лакокрасочными ма-
териалами для покровных слоев являются алкидные,
алкидно-мочевинные и алкидно-меламиновые эмали,
а также перхлорвиниловые эмали. При окраске гру-
зовых автомобилей, деталей приборов и др. применяют
быстросохнущие нитроцеллюлозные эмали. В ка-
честве термостойких применяют лакокрасочные по-
крытия на основе кремнийорганич. смол, к-рые выдер-
живают нагрев 250—300°, а при их пигментировании
алюминиевой пудрой — до 400—450°. Алюминиевая
пудра, помимо повышения термостойкости покрытий,
замедляет их старение, т. к. хорошо отражает свето-
вые лучи (в частности, разрушающе действующие
УФ-лучи). Хорошей стойкостью по отношению ко
многим химич. реагентам обладают покрытия на
основе феноло-формальдегидных, эпоксидных и пер-
хлорвиниловых смол, сополимера винил- и вини-
лиденхлоридов.
Лакокрасочные 3. п. наносят на окрашиваемые
поверхности кистью, окунанием, обливанием и рас-
пылением сжатым воздухом (пульверизацией). По-
следний метод — высокопроизводительный и позво-
ляет наносить материалы, обладающие различной
скоростью высыхания. Сушка покрытий производится
без нагрева (холодная) или с нагревом (горячая).
Горячую сушку проводят в конвекционных сушиль-
ных камерах, а также ускоренными способами с по-
мощью ИК-лучей или вихревых токов, возникающих
при электромагнитной индукции.
Лакокрасочные 3. п. можно наносить в электрич.
поле высокого напряжения (ок. 100 тыс. в); частицы
краски получают при этом электрич. заряд и притяги-
ваются к противоположно заряженной поверхности
изделий. Лакокрасочные 3. п. можно наносить также
путем безвоздушного распыления; циркулирующая
в замкнутом пространстве нагретая краска распы-
ляется под действием гидравлич. давления, создавае-
мого насосом и парами низкокипящей части раство-
рителей. Последние методы позволяют уменьшить
потери краски и сократить расход растворителей,
а также повысить производительность.
Лит.: Дринберг А. Я , Гуревич Е. С., Ти-
хомиров А. В., Технология неметаллических покрытий,
Л., 1957; Неметаллические материалы и их применение в авиа-
строении, под ред. И. П. Лосева и Е. Б. Тростянекой, М„
1958, гл. 10; Л юб имо в Б. В., Специальные лакокрасоч-
ные покрытия в машиностроении, М.—Л., 1959.
М. М. Гольдберг.
Пластмассовые 3. п. — применяются для
защиты металлич. поверхностей (химич. аппаратуры
и др.) от коррозии. Перед нанесением покрытия
металлич. поверхности должны быть предварительно
подготовлены (зачистка выступающих частей, песко-
струйная обработка с последующим удалением пыли).
Для получения 3. п. из пластич. масс могут быть
использованы следующие материалы на основе поли-
винилхлорида: 1) винипластовая пленка (толщиной
0,5—1 мм); 2) пластикат (толщиной 1—3 мм); 3) пла-
стифицированное поливинилхлоридное покрытие (тол-
щиной 2—3 мм), получаемое горячим напылением.
На подготовленные поверхности склеиваемых материа-
лов наносят по 2 слоя клея на основе смеси перхлор-
вициловой и эпоксидной смол (с промежуточной
подсушкой на воздухе до 90 мин), после чего покрыв-
ной материал накладывают на защищаемую поверх-
ность и прижимают под давлением 1-—2 ksJcm2'. Отвер-
ждение заканчивается через 48 часов. Пластифици-
рованные полихлорвиниловые покрытия можно полу-
чать методом пламенного напыления пасты (поли-
хлорвиниловая смола плюс пластификатор 1 : 1)
на поверхности, предварительно покрытые 10—
13%-ным перхлорвиниловым клеем; производится
при помощи специально предназначенного для этих
целей аппарата. Для футеровки полиизобутиленом
пригодны клеи № 8 и 88.
Защита небольших металлич. поверхностей может
быть осуществлена путем их футеровки фаолитом,
для чего подготовленную поверхность покрывают
бакелитовым лаком и накладывают нагретую до 60°
фаолитовую пластину или равномерный слой фаоли-
товой замазки. Фаолит плотно прижимает к металлу
и изделие подвергают термообработке в течение
30 час при 60—130°. После отверждения фаолита
производят зачистку заусениц, подмазку щелей и ла-
кировку бакелитовым лаком и снова подвергают тер-
мообработке в течение 12—13 час при 18—130° для
отверждения лакового покрытия. Футеровать фаоли-
том большие металлич. поверхности не следует из-за
большой разницы коэфф, расширения фаолита и
металла, а также вследствие усадки фаолита в про-
цессе его отверждения.
В качестве антикоррозийного 3. п. применяется
асбовиниловая масса, состоящая из 65,5% этиноле-
вого лака, 28—34% антофиллитового асбеста и 6—8%
хризотилового асбеста. Очищенную поверхность грун-
туют разбавленным (100 вес. ч. массы и 100 вес. ч.
лака) или жидким (25 вес. ч. измельченного асбеста
и 100 вес. ч. лака) асбовинилом, после чего наносят
1—4 слоя (в зависимости от агрессивности среды)
асбовинила в следующем порядке: слой лака, содер-
жащего 25% минерального наполнителя, слой асбо-
винила. Продолжительность сушки слоя лака 3 часа,
слоя асбовинила 24—72 часа (в зависимости от тол-
щины слоя). После нанесения каждого слоя покрытие
уплотняют и укатывают валиками. Полимеризация
массы производится в течение 50 час при 40—130°;
на крупногабаритном оборудовании покрытие отвер-
ждается при 15—20° в продолжение 30 суток. Асбо-
виниловое покрытие устойчиво к большинству агрес-
сивных сред (за исключением окислителей и нек-рых
органич. растворителей) при темп-pax до 100°.
Для защиты от щелочных и слабокислых сред могут
быть применены композиции состава: 100 вес. ч. эпок-
сидной смолы, 20—25 вес. ч. пластификатора, 12 вес. ч.
отвердителя и 40 вес. ч. наполнителя или пигмента.
В качестве отвердителя можно использовать полиэти-
ленполиамин, гексаметилендиамин или фенолоформ-
альдегидную [смолу. Более дешевое покрытие такого
типа может быть получено на основе эпоксидной
смолы и дегтя.
Металлич. поверхности могут быть защищены также
покрытиями на основе порошкообразных материалов,
к-рые наносят пламенным, вихревым, струйным и др.
методами. Сущность способа пламенного напыления
заключается в том, что частички термопластич. мате-
риала, проходя через воздушно-ацетиленовое пламя,
размягчаются и, попадая на нагретую поверхность,
сплавляются на ней в сплошное покрытие. При вихре-
вом методе изделие, нагретое до темп-ры, превышаю-
щей темп-ру плавления термопластического мате-
риала, погружается во взвесь порошка в воздухе или
инертном газе (в псевдоожиженный порошок). Ча-
стички термопластичного материала, соприкасаясь с
нагретой поверхностью изделия, оплавляются и обра-
зуют покрытие. В табл. 1 указаны порошкообразные
материалы, пригодные для получения таких покрытий.
101
ЗАЩИТНЫЕ ПОКРЫТИЯ
102
Таблица 1
Наименование материала Температура предваритель- ного нагрева, °C
при пламен- ном способе при вихревом способе
Полиэтилен низкого давления . 200 240—340
Поликапролактам 200—210 240-370
Полиамид П-68 200—210 240—370
Полиамид АК-7 — 280—370
Полипропилен 160 220—250
Поливинилбутираль 220 260—350
Пластмасса ПФН-12 200 —
Фторопласты (3, ЗМ и др.). . 270 —
Покрытия на основе полиэтилена, полипропилена,
фторопластов могут быть применены для защиты от
воздействия кислот и щелочей; полиамиды, поливи-
нилбутираль устойчивы к воздействию растворителей;
полиамидные покрытия, кроме того, являются изно-
соустойчивыми. Широкое распространение полу-
чают также 3. п. из ткани, пропитанной битуминоз-
ными составами (для защиты подземных трубопро-
водов), и др.
Лит.: Егоров И. А., Фаолит и его применение в хи-
мической промышленности, М., 1956 (Коррозия в химических
производствах и способы защиты, вып. 6); II о л я к о в К. А.,
Неметаллические химически стойкие материалы, 2 изд.,
М.—Л., 1952; Бакланов Н. А., В а ш и н Г. 3., Хи-
мическое оборудование из винипласта, М., 1956; Шр ад ерВ.,
Обработка и сварка пластических масс, пер. с нем., 4 изд.,
М., 1960; Полякова К. К. иАвгустов Ю. А., Горя-
чее напыление пластических масс, в кн.: Конструкционные
неметаллические материалы и коррозия металлов. Сб. ст. № 1 7,
М., 1954; Нанесение покрытий способом газопламенного на-
пыления. Справочные материалы по газопламенной обработке
металлов, вып. 15, М., 1958; Самосатский Н. Н.,
Карпов А. А., Газопламенное напыление пластических
масс, Л., 1960; Августов Ю. А., Нанесение пластмас-
совых покрытий на металлические изделия методом погруже-
ния в псевдоожиженный порошок, Химическое машинострое-
ние, 1960, № 2; Я к о в л е в А. Д., Алексеева Е. А.,
Му лин Ю. А., Получение покрытий из порошкообразных
смол во взвешенном слое, Л., 1961; Клинов И. Я., Опыт
применения асбовинила, в кн.: Защита химического оборудо-
вания от коррозии, М., 1960. Ю. А. Августов.
Гуммированные 3. п. (покрытия из резины
и эбонита) — применяются для защиты металлич.
аппаратуры от кислот, щелочей и растворов солей.
Эбониты обладают большей стойкостью, чем резины,
однако невысокая темп-ра размягчения эбонитов
(60—70°) и хрупкость ограничивают их применение.
Химич, стойкость резиновых и эбонитовых покрытий
зависит от концентрации среды, ее темп-ры, состава
резины или эбонита, прочности их крепления к ме-
таллу и др.
Нек-рые сведения по химич. стойкости резин и
эбонитов приводятся в табл. 2.
Таблица 2
Наименование среды Концентрация среды, при ко- торой сохра- няется стой- кость покры- тия, % Темп-ра среды, °C
Серная к-та до 50% до 65
Соляная к-та любая то же
Фосфорная к-та до 85% » »
Едкие натр, калий любая » »
Известковое молоко » » »
Белильная известь » » »
Хлористый цинк ДО 50% »
Покрытия из мягких резин применяются также для
защиты аппаратуры от слабой азотной к-ты (до 5%)
при20°, уксусной к-ты (до 15%), водных р-ров аммиака
и т. п. В воде, особенно при повышенной темп-ре,
резины набухают; к органич. соединениям резины
менее стойки, чем к неорганическим. Резиновые
покрытия разрушаются царской водкой, гипохлори-
тами, сероуглеродом, бензолом и др.
Процесс гуммирования состоит из подготовки по-
верхности металла (очистка от ржавчины, окалины,
масел и др. загрязнений), покрытия ее клеем, нало-
жения резиновой или эбонитовой смеси, ее прикатки
и вулканизации. Аппаратуру гуммируют следующими
способами: а) покрытием невулканизов энными эбо-
нитовыми или резиновыми смесями (с помощью
клеев), с последующей вулканизацией покрытия;
б) покрытием аппаратуры (с помощью клеев) вулка-
низованной резиной.
Конструкции покрытий, производимых по первому
способу, могут быть: однослойными (один слой эбо-
нита или полуэбонита); двуслойными (слой резины
с подслоем эбонита или эбонита с подслоем резины);
трехслойными (эбонит — резина — эбонит или рези-
на — эбонит — резина). Эбонит или полуэбонит не-
посредственно крепятся к металлу. Для крепления
мягкой резины применяют термопреновый клей. При
гуммировании вулканизованной резиной для креп-
ления пользуются термопреновым клеем или клеем
88-Н. Иногда применяют комбинированные покрытия
из вулканизованной резины с последующим нало-
жением на нее керамич. плиток.
Гуммированию могут быть подвергнуты аппараты
(ванны, центрифуги, цистерны), их детали (вентили,
трубы, колена), изготовленные из стали, чугуна и
сплавов легких металлов. Аппараты из меди или
бронзы перед гуммированием лудят. Гуммируемые
аппараты должны быть жесткими, чтобы избежать
деформаций и нарушения целостности покрытия.
Крупные аппараты делаются разъемными; неразъем-
ные — должны иметь люки. Швы должны быть свар-
ными в стык, углы закруглены. Трубы небольшо-
го диаметра гуммируются отрезками длиной не более
1 .и; колена должны быть изогнуты под углом не ме-
нее 90°.
Готовые покрытия аппаратов Осматривают, высту-
кивают и испытывают на электропробой посредством
искрового индуктора током напряжением от 10 до
40 тыс. в. Сроки службы резиновых и эбонитовых
покрытий для нек-рых сред приведены в табл. 3.
Таблица 3
Наименование среды Кон- центра- ция, % Темпе- рату- ра, °C Вид покры- тия Пример- ный срок службы, лет Вид аппаратуры
Соляная к-та До 36 25 Резина 2—3 Трубопроводы
» » Рассол +влаж- Любая 25 » 3—5 Ж.-д. цистер- ны
ный хлор — 80—90 Эбонит 1-2 Для производ- ства хлора
Серная к-та До 50 До 50 » До 7 Закрытая ем- кость
Уксусная к-та 10 20 До 6 То же
Едкий натр Кон- цент- риров. До 50 * 5—6 » »
Так как при гуммировании аппаратуры приме-
няются клеи на бензине, помещения для этих работ
должны быть огнестойкими, снабжены мощной при-
точно-вытяжной вентиляцией и необходимыми про-
тивопожарными средствами.
Лит.: Бирюков И. В., Технология обкладки хими-
ческой аппаратуры резиновыми смесями, Л.—М., 1952; Б а-
л а л а е в Г. А., в кн.: Неметаллические химически стойкие
покрытия аппаратуры и строительных конструкций [Сборник!,
М., 1951; Клинов И. Я., Коррозия химической аппа-
ратуры и коррозионнрстойкие материалы, 3 изд., М., 1960;
II о л я к о в К. А. [и др.], Коррозия и химически стойкие
материалы, М,—Л., 1953; Жеребков С. К., Крепление
резины к металлам, М., 1956. С. К. Жеребков.
103
ЗЕАКСАНТИН - ЗЕИН
104
ЗЕАКСАНТИН — см. Каротиноиды.
ЗЕЕМАНА ЯВЛЕНИЕ — расщепление спектраль-
ных линий атомов, молекул и кристаллов в магнитном
поле на составляющие. Явление обусловлено расщеп-
лением на тесно расположенные подуровни уровней
энергии, переходы между к-рыми дают эти линии;
термин 3. я. применяют и к расщеплению самих
уровней энергии. Наряду с переходами между под-
уровнями двух различных уровней, дающими
составляющие спектральной линии, возможны пере-
ходы и между подуровнями одного и того же
уровня — магнитный резонанс, который
наблюдается методами радиоспектроскопии.
3. я. открыто П. Зееманом в 1896 при исследовании
спектра свечения паров натрия. Для наблюдения
3. я. источник света, испускающий линейчатый спектр,
располагается между полюсами мощного электро-
магнита; при этом каждая спектральная линия рас-
щепляется на несколько составляющих. Расщепление
весьма незначительно и для магнитных полей в 20—
30 тыс. эрстед достигает лишь нескольких десятых
долей ангстрема. Поэтому при исследовании 3. я.
пользуются спектральными приборами высокой раз-
решающей силы — чаще интерференционными спект-
роскопами. Составляющие, на к-рые расщепляется
спектральная линия, поляризованы. Если наблюдение
производится поперек магнитного поля, то состав-
ляющие поляризованы линейно; если наблюдение
производится вдоль магнитного поля, то составляю-
щие поляризованы по кругу в плоскости, перпенди-
кулярной к направлению магнитного поля.
В простейшем случае (рис. 1) одна составляющая
спектральной линии расположена в том же месте,
где располагалась первоначаль-
ная линия, и две другие — симмет-
рично по обе стороны от нее. Все
три составляющие обнаруживают-
ся лишь при наблюдении поперек
магнитного поля, причем для
средней составляющей колебания
электрич. вектора параллельны
линиям магнитной напряженно-
сти (л-компонента), а для край-
них^— перпендикулярны к линиям
магнитной напряженности (п-ком-
понента). При наблюдении вдоль
поля видны (благодаря попереч-
ности световых колебаний) лишь
две крайние составляющие; со-
ставляющая, смещенная в сторону больших частот
(в фиолетовую сторону спектра), поляризована по
кругу против часовой стрелки, а смещенная в сто-
рону меньших частот (в красную сторону спектра) —
по часовой стрелке. Указанное расщепление на три
составляющие называется простым, или нормаль-
ным, 3. я. Впоследствии выяснилось, что простое рас-
щепление спектральных линий на три составляющие
наблюдается только в некоторых частных случаях,
а также в очень сильных магнитных полях. Обычно
спектральные линии расщепляются не на три, а на
большее число составляющих (иногда до 20 и больше).
Составляющие расположены симметрично относи-
тельно первоначальной линии и поляризованы так
же, как и в случае простого расщепления, на три
составляющие (п- и о-компоненты). Такое расщепле-
ние называют сложным (или аномальным) 3. я.
Расстояние Av между составляющими в сложном
3. я. равно рациональной дроби от расстояния,
вычисленного для простого расщепления; Av = Я Av0,
где q и i — целые числа.
Современная теория 3. я. для атомов исходит из квантовой
теории строения атома. Энергия атома, находящегося в маг-
нитном поле И и имеющего магнитный момент ц, Е =
— Ео — Нин где Но— энергия атома в отсутствии поля,
|Лд — проекция ц на направление Н. Полный магнитный мо-
мент атома и связан с его механич. моментом ЛГ:
р. = — g (е/2шес) ЛГ
(О
где g(e/2m,c) — магнетомеханпч. (или гиромагнитное) отно-
шение, g — т. и. множитель Ланде (фактор магнитного рас-
щепления, или g-фактор), с и те — заряд и масса электрона
соответственно, с — скорость света. По общим законам кван-
товой механики проекция Мдполного момента ЛГ на направле-
ние поля может принимать только дискретные значения Мд=
= тЛ/2ст; m = J, J — 1.... —J, где J — квантовое число,
определяющее полный момент количества движения атома.
При данном J возможны 2J + 1 различных значений магнит-
ного квантового числа «п. Б результате энергия атома в маг-
нитном поле равна
E=E0 + gmp.BH (2)
где ив — <Л/2тес — магнетон Бора. Ур-ние (2) пока-
зывает, что уровень энергии атома с заданным значением J
расщепляется в магнитном поле на 2J +1 подуровней. Рас-
стояние между подуровнями равно gMB Н, что при g — 1 дает
(в единицах E/hc) 4,69*10~5 Н см~*ъ H/2Q ООО см-i. Величина
g зависит от квантовых чисел, характеризующих уровень
энергии атома. Расщепление спектральных линий определяет-
ся расщеплением комбинирующих уровней энергии атома
в магнитном поле и правилами отбора Am = 0,+1. Излу-
чаемые линии при Ат = 0 поляризованы параллельно*маг-
нитному полю (л-компоненты); при Ат = ±1 — по кругу,
в плоскости, перпендикулярной магнитному полю (о-компо-
ненты).
Типичный пример расщепления уровней энергии
и соответствующего расщепления спектральной линии
приведен на рис. 2»
Исследование карти-
ны зеемановского рас-
щепления позволяет
определить характери-
стики комбинирующих
уровней, что имеет боль-
шое значение для ин-
терпретации атомных
спектров.
Наблюдение 3. я. в
молекулярных спектрах
имеет меньшее значе-
ние, так как расшиф-
□ г ст Мда л а
Ь) Н^о
File. 2.
ровка электронных пе-
ре ходов молекул произ- °) 0
водится главным обра-
зом по вращательной
структуре. Кроме того, наблюдение 3. я. в полосатых
спектрах представляет большие экспериментальные
трудности из-за сложности расщепления и близости
линий вращательной структуры. 3. я. в молекуляр-
ных спектрах также исследуется методами радио-
спектроскопии. 3. я. можно наблюдать и в спектрах
кристаллов в том случае, когда эти спектры имеют
выраженную дискретную структуру.
Изложенное выше относится как к испусканию,
так и к поглощению света, т. е. применимо и к т. наз.
обратному 3. я. (для линий поглощения).
Лит: Ландсберг Г. С., Оптика, 4 изд., М., 1957
(Общий.курс физики, т. 3); Е л ь я ш е в и ч М. А., Атомная
и молекулярная спектроскопия, М., 1962; Горди Б.,
Смит Б. и Трамбарул о Р , Радиоспектроскопия,
пер. с англ., М., 1955; Ельяшевич М. А., Спектры ред-
ких земель, М., 1953; Архангельская В. А., Фео-
фи л о в II. II., Оптика и спектроскопия, 1958, т. 4, вып. 5,
с. 602; Handbuch der Physik, Bd 28, Б. [u. a.], 1957, S. 253—60,
296—325.
ЗЕИН — белок группы проламинов, содержится
в зернах кукурузы (3—7%). 3. нерастворим в воде
и водных солевых р-рах, хорошо растворим в спирте.
При электрофорезе и ультрацентрифугировании 3.
разделяется на несколько фракций. Изоэлектриче-
ская точка 3. находится при pH 6,2, коэфф, седимен-
тации 1,9 S (единиц Сведберга), мол. в. ок. 50 000.
Ориентировочный химич. состав гидролизатов 3.
(в %): общий азот 16,2; аммиак 3,0; аланин 11,5;
серин 7,8; треонин 3,0; валин 3,0; лейцин 24,0; изо-
105
ЗЕЛЕНОЕ МАСЛО —ЗЕМЛИ ОТБЕЛИВАЮЩИЕ
106
лейцин 7,4; метионин 2,3; цистин 1,0; пролин 10,5;
февилаланин 6,5; тирозин 5,3; гистидин 1,7; арги-
нин 1,8; аспарагиновая к-та 5,7; глутаминовая к-та
27,0. В 3. не обнаружены остатки таких аминокислот,
как глицин, триптофан и лизин. Получают 3. экстра-
гированием кукурузной муки 90%-ным спиртом,
затем спирт отгоняют в вакууме, остаток растворяют
в этаноле и полученный раствор выливают в большое
по объему количество ацетона. При этом 3. выпадает
в осадок.
Лит.: Белки, под ред. Г. Нейрата и К. Вейли, пер. с англ.,
т. 3, ч. 1, М., 1958, с. 198, 210. В. О. Шпикитер.
ЗЕЛЕНОЕ МАСЛО — смесь высокомолекулярных
полициклич. углеводородов, выделяемых ректифи-
кацией из продуктов пиролиза нефти и ее дистиллятов.
Темп-ра начала перегонки 3. м. не ниже 150°; до
350° должно отгоняться 95%. Более тяжелая фрак-
ция продуктов пиролиза, из к-рой до 350° отгоняется
65%, называется бурым маслом. Плотность
зеленого и бурого масел 0,9—1,0; химич. состав
этих масел может меняться в широких пределах.
Содержание нафталина в 3. м. допускается не более 5%
(для бурого масла не более 2%). 3. м. и бурое масло
применяются гл. обр. как сырье для произ-ва лампо-
вой сажи, как поглотительное масло, а также для
приготовления инсектицидов. При деструктивной
гидрогенизации 3. м. получаются высокооктановые
бензины с высоким содержанием низших ароматич.
углеводородов. Ранее 3. м. называли также антраце-
новое масло (см. Каменноугольная смола).
Лит. СМ. при ст. Вавелин. Л. С. Поваров.
ЗЕЛИНСКОГО - СТАДНИКОВА РЕАКЦИЯ -
метод сивтеза алифатич., алициклич. и ароматич.
а-аминокислот из соответствующих альдегидов и
кетонов:
R R\ /МНютг л R. ,СООН
>С=О + KCN + NHjCl -* )С< >С<
R’z RZ 'CN R’z 'NH.
где R и R' = H пли углеводородный радикал. Реак-
ция представляет собой усовершенствование циангид-
ринного синтеза аминокислот по Штреккеру (1850):
R\c—о NH:i' HCN R\C/CN И?.О. R\C/COOH
R.Z или NHSCN R’Z \NHS R'/ \nh2
В отличие от метода Штреккера, с использованием
легколетучей и токсичной HCN, в 3. — С. р. при-
менена нелетучая смесь KCN и NH4C1, благодаря
чему упростились условия реакции и повысились
выходы конечных продуктов. 3. — С. р. — универ-
сальный метод, применимый к синтезу любых а-ами-
нокислот. Предложенный авторами механизм реак-
ции предполагает следующие последовательные ста-
дии:
KCN + н2о — HCN -р кон
NH1C1 4- КОН ->• NH3 4- КС1 4- Н.,0
NH3
RR’CO 4- HCN -► RR’C(OH)CN--> RR’C(NH»)CN
H2O
RR’C(NH2)CN----> RR'C(NHa)COOH
Связывание карбонильным соединением HCN и NH,
сдвигает цепь равновесий вправо, в результате чего
реакция идет до конца. Необходимость присутствия
воды для 3. — С. р., доказанная ее авторами, подтвер-
ждает эту схему. Реакция проводится в водных
средах при темп-рах 0-1-20° и заканчивается за неск.
часов. С выходами 60—90% получаются а-амино-
нитрилы, далее гидролизуемые в а-аминокислоты.
3. — С. р. была распространена на синтез N-заме-
щенных а-аминокислот с использованием хлоргидра-
тов первичных аминов вместо NH4C1.
Лит.: Зелинский Н. Д., Собрание трудов, т. 1, М.,
1954. А. Ф. Бочков.
ЗЕМЛИ ОТБЕЛИВАЮЩИЕ — вещества минераль-
ного происхождения, обладающие способностью обес-
цвечивать разнообразные минеральные, растительные
и животные продукты. К 3. о. относятся: отбеливаю-
щие коллоидные гливы, состоящие гл. обр. из мине-
ралов монтмориллонитовой группы, образовавшиеся
в результате выветривания изверженных пород, и
диатомовые земли — кизельгур или инфузорная
земля, состоящие из остатков микроводорослей —
диатомей. Близки к диатомовым землям по составу
трепелы и опоки, являющиеся, вероятно, продуктами
превращения диатомитов. Состав отбеливающих глин
колеблется в пределах: 40—72% SiO2, 5—33% А12О3,
1,25—14,9% Fe3O4, 0,0—6,5% CaO, 0,0—8,3% MgO,
4,3—15% H3O. Состав диатомовых земель: 80—90%
SiO2, 1,5—9% Al203, остальное — окислы Fe, Mg, Cg.
3. о. типа глин подразделяются на фуллеровы
земли (флоридины) и бентониты. Первые обладают
достаточной активностью в естественном виде и перед
применением подвергаются лишь термич. активации
(прокаливание при 150—400° для удаления содер-
жащейся в них адсорбированной влаги; оптимальная
темп-ра прокаливания определяется для каждой
глины отдельно). Промышленные месторождения 3. о.
этого типа находятся во многих районах СССР,
напр. гумбрин в Грузии, нальчикин в Кабардинской
АССР.
Бентониты в естественном виде обладают малой
активностью и перед употреблением нуждаются
в кислотной активации, производимой обработкой
глин разб. серной или соляной к-той при нагревании
и перемешивании. В результате активации их отбе-
ливающая способность в 2—4 раза превышает эффек-
тивность лучших сортов естественных глин. Пред-
полагается, что подобная активация приводит к гид-
ратации алюмосиликатов и к частичному удалению
из них ряда катионов (К, Na, Mg, Са). Это, наряду
с нек-рым изменением пористой структуры и химич.
состава, способствует образованию на поверхности
глин активных центров. Наиболее хорошо поддаются
кислотной активации малонабухающие бентониты,
содержащие способные к обмену ионы кальция и
магния; сильно набухающие в воде бентониты, со-
держащие ионообменный натрий, активируются хуже.
Месторождения бентонитов широко распространены,
напр. асканит в Грузии, гиляби и ханларит в АзССР,
кил в Крыму.
3. о. типа опок активации почти не поддаются.
Из земель этого типа наибольшее применение имеет
зикеевская земля (ст. Зикеево ок. Брянска) и землп
ряда месторождений между Волгой и Уралом, напр.
Волково-Паршинского, Филатовского, Краснополян-
ского, Привольского и др.
Отбеливающее действие связано в значительной
мере с адсорбционными свойствами высокоразвитой
внутренней поверхности. Уд. поверхность глин ко-
леблется в пределах 10—200 м-/г и более. Однако
избирательность и необратимость адсорбции, нейтра-
лизующие и каталитич. свойства 3. о. типа глин,
показывают, что здесь имеет место не только физич.
адсорбция. Активность 3. о. связывается и с химич.
свойствами поверхности, в частности с наличием на по-
верхности глин соединений типа алюмокремневыхк-т,
а также с кислыми свойствами глин. Действительно,
многие глины, обладающие хорошей отбеливающей
способностью, показывают кислую реакцию. Однако
ряд активных глин в естественном виде имеет щелоч-
ную реакцию, напр. флоридин (pH 8,6), гумбрин
(рН7,3), нальчикин (pH 8,1). Активность глин связы-
вается также с наличием у них кристаллич. струк-
туры монтмориллонитового типа. Существенным для
активных глин является наличие в их составе прочно
связанной конституционной воды, полное удаление
107
ЗЕМЛЯНОЙ ВОСК - ЗИМОЗАН
108
к-рой при темп-ре выше 750®, происходящее необра-
тимо й с трудом, приводит к разрушению кристаллич.
структуры, спеканию и почти полной потере ими
активности.
3. о. применяются гл. обр. для очистки смазочных
и специальных масел, а также для их регенерации.
3. о. селективно адсорбируют асфальтово-смолистые
вещества, иногда в количестве 9—12% от веса сор-
бента, а также другие кислородные, сернистые и
ненасыщенные соединения; они обладают также
способностью нейтрализовать серную к-ту, адсорби-
ровать сульфокислоты и эфиры, образующиеся при
сернокислотной очистке масел и растворенные в масле.
Кроме нефтеперерабатывающей пром-сти, 3. о. при-
меняются при рафинировании растительных масел,
для очистки саломаса, осветления вина и пива, в пар-
фюмерной и фармацевтич. пром-сти, а также в качестве
наполнителей и стабилизаторов в произ-ве резины,
бумаги, тканей («сукновальные глины») и т. д. Харак-
терной для отбеливающих глин является способность
к ионному обмену и каталитич. активность в отно-
шении полимеризации олефинов, а при более высокой
темп-ре — крекинга углеводородов, благодаря чему
глины применяются как катализаторы крекинга,
а также как носители катализаторов в других про-
цессах. Диатомиты используются также как носители,
напр. для приготовления кобальт-ториевого катали-
затора для синтеза по Фишеру—Тропшу. Отработан-
ные 3. о. регенерируют экстракцией растворителями
или путем обжига, однако первоначальная активность
обычно не восстанавливается; их можно также исполь-
зовать как сырье в цементном и кирпичном произ-
водстве.
Лит.: Зульфугаров 3. Г., Исследование физико-
химических свойств и отбеливающей способности глин место-
рождений Азербайджанской ССР и гумбрина, Ваку, 1957;
Быков В. Т., Сорбционные свойства и структура отбели-
вающих земель, Владивосток, 1953; Геологический словарь,
т. 1—2, М., 1955. В. В. Щекин.
ЗЕМЛЯНОЙ ВОСК — см. Озокерит.
ЗИМАЗА — сложная ферментная система, к-рая
входит в состав бесклеточного дрожжевого сока;
катализирует спиртовое брожение глюкозы.
Впервые 3. получена Э. и Г. Бухнерами в 1897
растиранием дрожжей с чистым песком и отжиманием
полученной массы под давлением; выделением 3.
была впервые продемонстрирована возможность осу-
ществления ферментативного процесса вне живой
клетки. 3. (дрожжевой сок), полученная по способу
Бухнеров, представляет собой опалесцирующую жид-
кость слабо-желтого цвета со слабокислой реакцией;
в ней содержится до 10% растворенных веществ
белковой и небелковой природы. С помощью диализа
3. может быть разделена на 2 компонента, каждый
из к-рых в отдельности не обладает ферментативной
активностью. Недиализирующийся компонент являет-
ся смесью высокомолекулярных белковых веществ,
не устойчивых к нагреванию, и называется апо-
зимазой. Диализирующийся термостабильный
компонент, названный козимазой, представляет
собой смесь различных низкомолекулярных в-в не-
белковой природы — коферментов. 3. включает более
20 ферментов, к-рые катализируют различные частные
реакции процесса брожения, в том числе: гексоки-
назу, изомеразу фосфоглюкозы, 6-фосфофруктокиназу
(см. Киназы), альдолазу, енолазу, карбоксилазу, алко-
гольдегидрогеназу (см. Дегидрогеназы) и др., а также
ряд коферментов, среди них аденозиптрифосфорную
к-ту (АТФ) и дифосфопиридиннуклеотид (ДПН). Все
ферменты, входящие в состав 3., выделены в кри-
сталлич. или высокоочищенном состоянии и изучены.
Лит.: Нейландс Д. и Штумпф П., Очерки по
химии ферментов, пер. с англ., М., 1958; The enzymes, ed.
J. В. Summer, К. Myrback, V. 2, pt. 1, N. Y., 1951, p. 687.
В. Б. Спиричев,
ЗИМОГЕНЫ (проферменты) — неактивные пред-
шественники ферментов, превращающиеся в активные
ферменты в результате структурных изменений. 3.
чаще всего встречаются среди протеиназ (пепсин,
ренин, трипсин, химотрипсин и карбоксипепти-
даза А), гидролитически расщепляющих белковые ве-
щества в пищеварительном тракте животных. Эти
ферменты вырабатываются клетками слизистой же-
лудка, кишечника или поджелудочной железой и
выделяются в желудочно-кишечный тракт в виде 3.
(пепсиногена, прореннина, трипсиногена, химотрип-
синогена и прокарбоксипептидазы). Такой способ
образования и выделения протеиназ является при-
способлением, защищающим клетки и ткани орга-
низма от самопереваривания их ферментами, выра-
батываемыми в этих клетках.
Превращение 3. в активные ферменты происходит
каталитически под действием ферментов либо ионов
водорода. Так, напр., трипсиноген превращается
в трипсин под действием специального фермента
кишечного сока энтерокиназы или самого трипсина.
Пепсиноген активируется ионами водорода или самим
пепсином. Поскольку превращение этих 3. катали-
зируется теми же ферментами, к-рые в результате
этого превращения образуются, то активация яв-
ляется аутокаталитич. процессом, скорость к-рого
возрастает во времени.
Механизм активации 3. состоит в расщеплении
одной или нескольких пептидных связей в молекуле 3.
с отщеплением или без отщепления определенного
участка или участков полипептидной цепи, в резуль-
тате чего происходит внутримолекулярная пере-
стройка, приводящая к демаскированию или форми-
рованию активного центра фермента. Так, превра-
щение трипсиногена в трипсин сводится к гидролизу
одной пептидной связи и отщеплению с N-концевого
участка полипептидной цепи шестичленного пептида
На]\-вал.-асп.-асп.-асп.-асп.-лиз.-ОН:
Н2К-вал.-(асп.)4-лиз.-изол.-Л. —►
—> Н2К-вал.-(асп.)4-лиз.-ОН -J- HoN-изол.-R
где R — остальная часть молекулы трипсина. Вслед-
ствие отщепления этого гексапептида структура бел-
ковой молекулы изменяется таким образом, что
сближаются ранее пространственно разобщенные
функциональные группы, входящие в состав актив-
ного центра фермента, в частности остатки серина
и гистидина.
Кроме протеиназ, в форме 3. продуцируются также
некоторые ферменты, принимающие участие в про-
цессе свертывания крови, в частности протромбин,
превращающийся в тромбин под действием тромбо-
киназы, и плазминоген, превращающийся в плазмин
под действием плазминокиназы.
Лит.: Диксон М., Уэбб Э., Ферменты, пер. с англ.,
М., 1961, с. 510; Н е й р а т Г., Структура белков и действие
ферментов, в кн.; Современные проблемы биофизики, перевод,
т. 1, М., 1961, стр. 238; Збарский Б. И., Иванов
И. И., Мардашев С. Р., Биологическая химия, 3 изд.,
М., 1960, с. 311. В. Б. Спиричев.
ЗИМОЗАН — смесь нерастворимых в воде полиса-
харидов клеточных оболочек дрожжей. Содержание
полисахаридов в препаратах 3. составляет в среднем
70—80%; кроме того, в них содержатся также азотис-
тые вещества, в составе к-рых обнаружены амино-
кислоты (лизин, аргинин и др.), и гексозамины.
В препаратах 3. обнаружены также фосфор, ионы
Mg, Са и др. металлов. Углеводы 3. представлены
полимерами глюкозы (глюканами), маннозы (манна-
нами) и гетерополисахаридами — глгокоманнанами
с различным соотношением глюкозы и маннозы.
Соотношение полисахаридов в 3. и их состав зависят
от рас дрожжей и метода получения препаратов.
Препараты 3. — гигроскопич. белый порошок, не-
растворимый в воде, спирте, соляной к-те и щелочах;
109
ЗИНИНА РЕАКЦИЯ-ЗОЛОТА ГАЛОГЕНИДЫ
110.
в серной к-те 3. частично растворяется. При комнат-
ной темп-ре 3. сильно набухает в воде и водных р-рах
кислот и щелочей. После нагревания со щелочью
из препарата 3. можно извлечь водой растворимую
фракцию, содержащую глюкоманнаны; остаток со-
держит нерастворимый глюкан. При нагревании
с кислотами в жестких условиях полисахариды 3.
гидролизуются до глюкозы и маннозы. 3. образует
с пропердином (белковый компонент крови, играю-
щий большую роль в естественном иммунитете орга-
низма) нерастворимый комплекс (PZ), к-рый инакти-
вирует в определенных условиях одну из фракций
плазмы крови, обладающую бактерицидной актив-
ностью (третий компонент комплемента С\). Комплекс
PZ разлагается в среде с высокой ионной силой на 3.
и пропердин.
Получают 3. обработкой дрожжей протеолитич.
ферментом трипсином (по Пиллемеру) с последующей
очисткой нерастворимого остатка 3. извлечением
его водой и спиртом или фенол-водной экстракцией
дрожжей. 3. используют для выделения пропердина
из сыворотки или плазмы по методу Пиллемера и его
модификациям, в основе к-рых лежит реакция обра-
зования пропердин-зимозанового комплекса и для
определения титра пропердина в крови.
Лит.: Багдасаров А. А., Ч е р т к о в И. Л., Рау-
ше н б а х М. О., Пропердиновая система организма, М., 1961;
Рутберг Р., в кн.: Углеводы и углеводный обмен в животном
и растительном организмах, М., 1959, с. 155; Р i Пеш er L.,
[а. о.], J. Exp. Med., 1956, 103, М 1. Л. И. Линееич.
ЗИНИНА РЕАКЦИЯ — метод получения арома-
тич. аминов восстановлением нитросоединений;
ArNO2 + 3H2S -> ArNH2 + 3S + 2H2O
В качестве восстановителей первоначально исполь-
зовали сероводород и сернистый аммоний. Этим
способом впервые были получены а-нафтиламин,
анилин, к-рый до этого получали из индиго, а также
л1-аминобензойная к-та и .и-фенилендиамин. На этих
примерах Зининым была доказана общность откры-
той им реакции. 3. р. открыла простой путь для
синтеза ароматич. аминов из легкодоступных нитро-
соединений и сделала ароматич. амины доступным
промышленным сырьем. Значительно позже (1854)
в качестве восстановителя было предложено пользо-
ваться чугунными стружками в кислой среде (см.
Бешана восстановление), к-рые применяют при полу-
чении ароматич. аминов и до сих пор. Сернистый
аммоний и сернистый натрий применяют в лабора-
тории и в пром-сти в качестве восстановителей для
получения аминов ряда антрахинона и для частич-
ного восстановления ди- и полинитросоединений.
Реакция открыта Н. Н. Зининым в 1842.
Лит.: Порай-Кошиц А. Е., Усп. хим., 1943, 12,
вып. 2, с. 94; Ф и г у р о в с к и й Н. А., Соловьев Ю. М.,
Н. Н. Зинин, М., 1957. Н. Д. Кулешова.
ЗОЛИ — коллоидные растворы, т. е. двухфазные
микрогетерогенные дисперсные системы с предельно
высокой дисперсностью. В противоположность гелям,
частицы дисперсной фазы в 3. не связаны в простран-
ственную структуру, а свободно участвуют в интенсив-
ном броуновском движении. Частицы дисперсной фазы
в 3. наз. мицеллами. Они состоят из инактивного по
отношению к среде ядра и защитной адсорбционно-
сольватной оболочки, к-рая включает и двойной
электрич. слой ионов на поверхности. Размеры
дисперсных частиц 3. (по порядку величины) —
от 10~5 до 10“’ см.
3. различаются по характеру дисперсионной сре-
ды — гидрозоли — 3. в воде, органозо-
ли — 3. в органич. жидкости, алкозоли—
в спирте, бензозоли — в бензоле и т. д. Аэро-
золями (правильнее — аэродисперсиями) обычно наз.
не только коллоидно-дисперсные, но и грубодисперс-
ные системы в газовой среде (воздухе). Нек-рые ми-
нералы, цветные стекла, содержащие распределен-
ные в твердой среде мельчайшие частицы других
твердых фаз, наз. иногда твердыми 3. Если подобные
3. или коллоидные дисперсии в расплавах солей,
в металлах, стеклах образуются при высоких темп-рах,
они наз. пирозолями. Такие дисперсные системы,
как эмульсии, пены, суспензии, которые хотя и имеют
многие характерные свойства коллоидных систем,
но отличаются сравнительно большой величиной
частиц дисперсной фазы (10~5—10 3 см и больше),
к 3. обычно не относят.
Все 3. делятся на две большие группы — лио-
фобные и лиофильные 3. Лиофобные 3.,
в частности гидрофобные (напр., гидрозоли металлов
платины, золота, серебра, сульфидов), являются
термодинамически неравновесными, агрегативно не-
устойчивыми дисперсными системами, способными
к агрегации диспергированных частиц — коагуляции.
Такие 3. поэтому не могут быть получены в концент-
рированном виде и коагулируют при введении малых
добавок электролитов, при повышении темп-ры и т. д.
В отличие от них, в лиофильных 3. (3. мыл, краси-
телей), дисперсная фаза к-рых обладает на границе
с дисперсионной средой весьма малой удельной по-
верхностной энергией, частицы сильно сольватиро-
ваны средой. Такие 3. агрегативно устойчивы и тер-
модинамически равновесны. К лиофильным 3. при-
мыкают самопроизвольно образующиеся, а потому
предельно высокодисперсные, эмульсии, включая и
критич. эмульсии и туманы, возникающие вблизи
критич. темп-ры смешения двух жидких фаз или
жидкости и пара. Раньше лиофильными 3. считали
также растворы высокомолекулярных соединений.
В отличие от частиц в грубодисперсных системах —
суспензиях, эмульсиях, пенах, ядру мицелл 3. часто
нельзя приписать определенное агрегатное состоя-
ние. Это относится особенно к лиофильным 3., тогда
как в лиофобных 3. частицы, напр. в гидрозолях
металлов, — нормальные кристаллики предельно ма-
лых размеров (10-6—10-7 см) с почти неизменными
параметрами решетки. О различиях частиц лиофиль-
ных и лиофобных 3. СМ. Мицеллы. П. А. Ребиндер.
ЗОЛОТА ГАЛОГЕНИДЫ — соединения золота
с галогенами, соответствующие его валентностям
4-1 и -|-3 (моно- и тригалогениды). Оба ряда соеди-
нений непрочны.
Моногалогениды золота окрашены в раз-
личные оттенки желтого цвета, в воде практически
нерастворимы; растворимы в конц. р-рах галогено-
водородных к-т и их солей с образованием комплекс-
ного аниона [АиГ4]_: ЗАиГ -|- МеГ=Ме[АиГ4] 2Аи.
Моногалогениды разлагаются либо путем диспропор-
ционирования: ЗАиГ=АиГ3 -4- 2Аи, либо — отщеп-
ления галогена: 2АиГ=2Аи -f- Г2. Монофторид AuF
известен лишь в парах. Плотности AuCl, AuBr, AuJ
соответственно равны 7,8; 7,9; 8,25; теплоты образо-
вания А77298 (ккал/молъ) имеют значения: —8,4;
—4,4; -4—0,2 соответственно. Получаются моногало-
гениды при разложении соответствующих тригалоге-
нидов: AuCl — при 185—200°; AuBr — при 140°;
AuJ — при обычной темп-ре.
Тригалогениды золота растворимы в воде;
при взаимодействии с галогеноводородными к-тами
и их солями образуют комплексные кислоты Н[АиГ4]
и соли — аураты Ме[АиГ4]; разлагаются по схеме
АиГ3=АиГ 4~ Г2. Золото фторное AuF3 —
оранжевый порошок; легко гидролизуется в воде и
40%-ном р-ре HF. Темп-ра разложения на элементы
500°. Получают нагреванием при 180° AuBrF6, к-рый
образуется при растворении Au в BrF3. Золото
хлорное АиС13 — красные игольчатые гигроско-
пич. кристаллы, плотн. 4,67; т. пл. 288° (под давле-
нием хлора); в ларах димерно (Ап2С16). Теплота обра-
Ill
ЗОЛОТА СПЛАВЫ-ЗОЛОТО
112
зования —28,3 ккал/моль. Растворимость
АиС]3 : 68 г в 100 г Н2О; при растворении в воде
образуется акнокислота Н,[АиОС13]; растворимо в
эфире. Образует комплексные соли — хлораураты
(см. Золотохлористоводородная кислота). Получают
хлорированием золота при 180°. Используют при
извлечении золота из руд (см. Золото). Золото
бромное AuBr3— темно-коричневый порошок, рас-
творим в воде и эфире. Теплота образования ДН298 ==
=—13,0 ккал!молъ. Получают действием брома на
свежеосажденное золото при комнатной темп-ре.
При обработке золота бромной водой, содержащей
КВт, образуется К[АпВг4] — бромаурат калия. 3 о-
л о то иодное AuJ3 — темно-зеленый, почти чер-
ный порошок; в воде нерастворимо, разлагается при
обычной темп-ре с образованием AuJ. Получают
действием раствора KJ на раствор АиС13. Иодаурат
K[AuJ4] более устойчив, чемАи!3, но также легко
разлагается.
Галогениды золота легко образуют соединения
с галогенидами других элементов, аммиаком, органич.
соединениями и т. д.
Лит." Щук ар ев С. А., Оранская М. А., Ц и н-
пиус В. М., Ж. неорг. . химии, 1956, 1. вып. 5, 881;
Щ у к а р е в С. А., [и др.1, там же, 1958, 3, вып. 7, 1478.
ЗОЛОТА СПЛАВЫ — сплавы, главным компонен-
том к-рых является золото. С серебром золото обра-
зует непрерывные твердые растворы I типа (см.
Двойные системы), темп-pa начала кристаллизации
возрастает от Ag к Ан. Указания на существование
ниже 800° фазы, представляющей твердый р-р со-
единения AgAu в своих компонентах (60—71 вес. %
Au), подтверждается частичной упорядоченностью
структуры кристаллич. решетки; при 20—40 вес. % Ag
твердый раствор имеет зеленовато-желтый цвет, при
50% — бледно-желтый. Сплавы Ag с Au мягки и
ковки. С палладием золото образует непрерывные
твердые р-ры I типа. С медью золото дает непрерыв-
ные твердые р-ры III типа с минимумом при 884°
и 18 вес. % Си. После длительного отжига при 500—
670° твердые р-ры Au—Си претерпевают превращения
с образованием фаз, имеющих сингулярные точки
на ординатах, отвечающих составам CuAu и Cu3Au.
Этим впервые была доказана возможность превра-
щения твердых р-ров в химич. соединения. Сплавы
Au—Си имеют красновато-желтый цвет. Отжиг (ниже
425—450°) придает сплавам, содержащим металлид
CuAu, твердость, хрупкость, а закалка — мягкость,
пластичность. Сплавы Au—Си более тверды и упруги,
менее ковки, чем золото. С платиной золото образует
ограниченные твердые р-ры IV типа. Сплавы, содер-
жащие прибл. до 20 вес. % Pt, представляют твердые
р-ры Pt в Au (<Xj), а сплавы, содержащие прибл. выше
97 вес. % Pt, — твердые р-ры Au в Pt (с^). При со-
держании более 20 вес. % и менее 97 вес. % Pt твер-
дый р-р (а) при охлаждении распадается с образова-
нием смеси двух твердых р-ров; из твердого р-ра при
темп-ре ниже 1000° выделяется фаза (возможно
PtAu3), образующая твердые р-ры со своими компо-
нентами.
Система Au—Sb характеризуется "О ди им соеди-
нением AuSb2, плавящимся с разложением при 460°;
при 25 вес. % Sb оно образует с Au эвтектику с т. пл.
360°. Твердый р-р Sb в Au при 500—600° содержит
0,7 вес. % Sb; ниже 200° растворимость в твердом
состоянии отсутствует. Сплавы Au с 0,1% Sb совер-
шенно непригодны для обработки, и последняя
возможна лишь начиная с содержания меньше
0,1%. То же наблюдается при небольших примесях
Bi и РЬ. Со свинцом золото образует соединения
Au2Pb и AuPb2, плавящиеся с разложением соответ-
ственно при 418° и 254°; AuPb2 и РЬ образуют эвтек-
тику прибл. при 85 вес. % РЬ, с т. пл. 215°. С оловом
золото дает соединения AuSn4 и AuSn2, плавящиеся
с разложением при 252° и 309°, а также AuSn с т. пл.
418°. Максимальная растворимость Sn в Au при
498 ± 10° составляет 4,2 вес. %. Растворимость Au
в Sn при 200° ок. 0,3 вес. %. Кроме того, известны
следующие а у р и д ы, т. е. соединения Au с другими
металлами: MgAu, Mg2Au, Mg6Au2, Mg3Au; In2Au,
InAu; ZnAu3, ZnAu, Zn3Au; MnAu3, MnAu2, MnAu,
Mn3Au; Te2Au; Na2Au, NaAu2; UAu3, U2Au3; Th3Au6,
ThAu3; Ti3Au, TiAu, TiAu2, TiAue; ZrAu3; Pr2Au,
PrAu, PrAu3; La2Au, LaAu, LaAuj, LaAu3. О сплавах
Au—Hg см. Амальгамы.
Сплавление золота с металлами, к-рые образуют
с ним твердые р-ры, имеет целью повышение прочности
и твердости и экономию золота. 3. с. применяют для
электрич. контактов, в зубопротезном деле, ювелир-
ном производстве и др. Содержание в них золота
выражают пробой.
Лит.: К У Р и а к о в Н. С., Собрание избранных работ,
т. 2, М. — Л., 1939; Плаксин И. Н., Металлургия благо-
родных металлов, М., 1958; Hansen М., Anderko К.,
Constitution of binary alloys, 2 ed., N. Y., 1958.
И. H. Плаксин.
ЗОЛОТО (Aurum) Au — химич. элемент I гр. перио-
дической системы Менделеева; п. н. 79, ат. в. 196,967;
относится к благородным металлам. Известен один
устойчивый изотоп Au197. Из искусственно радиоак-
тивных изотопов наиболее важны Au195 (Ух/2 =
= 185 дней) и Au198 (Ti/, — 2,686 дня). Конфигура-
ция внешних электронов атома 3.: 5d10 6s1. Энергия
ионизации Аи° —» Аи+ 9,22 эв.
3., встречающееся в природе в свободном состоя-
нии, иногда в виде крупных самородков, отличаю-
щихся цветом, блеском и ковкостью, несомненно,
было известно человеку уже в древнейшие времена;
возможно, что оно явилось первым металлом, к-рый
человечество стало применять для своих потребностей.
Имеются данные о добывании 3. и изготовлении из
него различных изделий (украшений, утвари, при-
надлежностей религиозного культа) в Египте (3500—
3000 лет до н. э.), Индии, Индокитае (2000—1500 лет
до н. э.).
3. относится к числу рассеянных металлов. Содер-
жание 3. в горных породах 5 • 10~7%, в водах рек
и океанов 0,01—0,05 лг/m, оно обнаружено в тканях
и крови живых организмов. Промышленные скопле-
ния, в к-рых 3. находится в виде руд кореннйх и
россыпных месторождений, встречаются редко. Ос-
новной формой нахождения 3. в природе является
самородное золото, содержащее серебро
(5—30%), медь (от небольшой примеси до 20%),
реже — металлы платиновой группы, селен и др.
Химич, соединения 3. в природе редки, встречаются
теллуриды (AuTe.J. К золотосодержащим минералам
относятся: самородное 3., электрум (AuAg), меди-
стое 3., теллуриды 3. (калаверит, кренерит, сильва-
нит и др.). В рудах 3. присутствует в виде вкраплений
размерами б. ч. 1000—0,1 мк, иногда находят само-
родки до нескольких десятков кг. Форма зерен 3.
в рудах разнообразна: листочки, уплощенные кон-
креции, сфероиды, октаэдры, палочки, иногда кри-
сталлич. агрегаты и сростки, а также коллоидные
частицы и их скопления. Поверхность частиц 3.
иногда покрыта пленками водных окислов железа
и окислов др. металлов, сульфида ’серебра и т. д.,
к-рые могут затруднить или снизить его извлечение.
Промышленными являются руды, содержащие 4 — 8 г
3. и выше на 1 т руды, а при массовой отработке
месторождения и дешевых методах переработки руд-
ной массы — 2 г\т. Весьма часто 3. добывается как
спутник меди и свинца из медных и полиметалпич.
руд. Золоторудные месторождения СССР находятся
в Восточной и Западной Сибири, гл. обр. в Забай-
калье, Якутии, Приморье, а также в Красноярском
из
ЗОЛОТО
114
крае, Казахстане, на Урале. Наиболее крупные
зарубежные месторождения — в Ю. Африке (более
50% мировой добычи 3.), Канаде, США, Австралии.
Мировая добыча 3. из золотых руд и в качестве
спутника цветных металлов составляет ок. 90%,
остальное — из россыпей.
Физические и химические свойства. 3. — металл
желтого цвета; имеет гранецентрированную кубич.
кристаллич. решетку, а = 4,0704 А. Атомный радиус
1,44 А; ионный радиус Ац+ 1,37 А. Плотн. 19,32;
т. пл. 1 063°; т. кип. 2947° (по Эйкену). Теплота
плавления 3,06 ккал/г-атом', теплота испарения
при т. кип. 81,8 ккал/г-атом; уд. теплоемкость
0,0316 мл/г град (0—100°); термин, коэфф, линей-
ного расширения 14,2 • ПГ6 (0—100°); уд. теплопро-
водность 0,744 кал/см сек град (0°); уд. электросо-
противление 2,25 • 10 6 ом см (20°); термин, коэфф,
электросопротивления 0,00396 (0—100°); электросо-
противление 3. резко падает при глубоком охлажде-
нии. Металлич. 3. диамагнитно с атомной восприим-
чивостью при комнатной темп-ре —30 • 10 s. 3. весьма
тягучий и ковкий металл; может быть проковано или
прокатано в листки толщиной 8 10'5 мм, просвечи-
вающие синевато-зеленым цветом; модуль упругости
7 900 кГ/мм2; предел прочности при растяжении
14 кГ/мм2; относительное удлинение 30—50%; суже-
ние площади поперечного сечения 90%. Твердость 3.
(в отожженном состоянии): по Бринеллю 18 кГ/мм2,
по Моосу 2,5.
Химически 3. мало деятельно. Вследствие наиболь-
шего сродства к электрону оно является самым бла-
городным металлом. На воздухе 3. не изменяется
и несет лишь тончайший адсорбционный слой кисло-
рода, устанавливаемый оптич. исследованиями. С гало-
генами в отсутствии влаги 3. без нагревания не
взаимодействует. С кислородом, водородом, азотом
ц углеродом не реагирует даже при очень высоких
темп-рах. При нагревании порошка 3. до 140—150°
в атмосфере хлора образуется хлорное 3. АпС13,
а затем при 180—190° — хлористое 3. АпС1 (см.
Золота галогениды). С многими металлами 3. образует
сплавы (см. Золота сплавы). На легком образовании
амальгамы 3. основан один из методов извлечения 3.
из горных пород. Электродный потенциал 3. в водных
р-рах: для реакции Au°=Au+4~e> £'о=1>68 в, для
реакции Au°=Au3+-j- Зе, Ео = 1,50 в. 3. не раство-
ряется ни в щелочах, ни в таких кислотах, как серная,
соляная, азотная, плавиковая, а также в органич.
к-тах, но растворимо в смесях к-т: соляной с азотной,
серной с марганцовой, серной с азотной, а также
в горячей селеновой к-те. Хорошо растворяет 3.
царская водка: Au -j- HNO3 ЗНС1= AuCl3 -j- NO -|-
2Н2О; из раствора 3. в царской водке после осто-
рожного выпаривания выделяются желтые кристаллы
комплексной эолотохлористоводородной кислоты
НАпС!4 • ЗН2О. В водных р-рах цианидов (Na, К, Са)
при доступе кислорода (воздуха) или других окисли-
телей 3. растворяется с образованием комплексного
иона: 2Au -j- 4CN~ Н2О -j- J/2O2 (раствор) =
= 2[Au(CN)2] ‘ 2ОН~, что лежит в основе важней-
шего промышленного способа извлечения 3. из руд
(см. Гидрометаллургия).
В химич. соединениях 3. одно- и трехвалентно.
Соединения 3. непрочны, легко восстанавливаются
до металла. О кислы AujO и Ап2О3 могут быть
получены косвенным путем, при нагревании соот-
ветствующих гидроокисей АпОН и Ац(ОН)3. По-
следние, в свою очередь, образуются действием на
хлориды 3. карбонатами щелочных металлов или
гидроокисью магнпя. Закись 3. Ап2О — серо-фиоле-
товый порошок, выше 200° распадается на элементы.
Окись 3. Ап2О3 — черно-бурый порошок, легко вос-
станавливается в струе Н, и СО при слабом нагре-
вании. При действии щелочей на Ап(ОН) и Ап(ОН)3
образуются соли, в к-рых 3. входит в состав аниона,
соответственно ауриты и а ура ты. Ауриты в
свободном состоянии не выделены. Аураты [напри-
мер, желтый КАпО2-ЗН3О; зеленый Ва(АпО2)2• 5Н2О)
получены в твердом виде. Образование ауратов в
растворах может протекать различно, в зависимости
от щелочности среды: Au(OHl3 -j- ОН~=Аи(ОН)~,;
Ап(ОН)3 4- 2ОН-=Ап(ОН)?-.
Кислородные соединения 3. неустойчивы, легко
образуют взрывчатые смеси. Соединение окиси 3.
с аммиаком Au2O3(NH3)4 — т. наз. гремучее
золото, может быть получено действием NH3 или
(NH4)2CO3 на раствор АпС13. Гремучее золото взры-
вается при 145°, иногда — неожиданно; без взрыва
растворяется в цианистых щелочах; разлагается йод
действием сероводорода и хлористого олова.
Соли Au неустойчивы и довольно быстро разлат
гаются: 3Au+=Au3 -j- 2Ап, поэтому на практике при-
меняются редко. Они, однако, легко образуют комп-
лексные соединения (аммиакаты, напр. AuCl • NH3,
AnCl • 12NH3 и др., цианиды K[Au(CN)2] и проч.),
которые, как правило, значительно устойчивее прос-
тых солей и отличаются гораздо большей раство-
римостью в воде. Соли Ап3г можно получить рас-
творением Ап(ОН)3 в кислотах. Для них наиболее
характерна резко выраженная способность к обра-
зованию комплексных анионов, напр. при взаимодей-
ствии АпС13 с водой образуется коричнево-красный
р-р аквокислоты Н2[ОАпС13]. Благодаря образованию
аниона [AuCIJ успешно идет процесс т. наз. хлори-
нации — извлечения 3. из руд действием хлора
в присутствии нек-рого количества воды.
Сульфиды при непосредственном сплавлении 3.
с серой не образуются. Темно-коричневый порошок
Au2S получают пропусканием H2S через подкисленный
р-р цианида или хлорида 3. Черный Au2S3 — обра-
боткой сухого Li[AuCl4] сероводородом при 10°.
При нагревании сульфиды 3. разлагаются; Au2S3
начинает разлагаться ок. 100°, окончательный распад
на элементы происходит ок. 240°. При темп-ре выше
50° 3. довольно хорошо растворяется в р-рах гидро-
сульфидов щелочных металлов с образованием
Me[AuS], а при понижении темп-ры снова выделяется,
что имеет значение для геохимии 3.
При разложении комплексных цианидов однова-
лентного 3. (см. выше), напр. Na[Au(CN)2] к-тами, об-
разуется цианид AuCN — лимонно-желтый поро-
шок. На свету в сухом воздухе AuCN не изменяется,
а во влажном — зеленеет; нерастворим в воде; не
разлагается большинством кислот даже при нагре-
вании до кипения. AuCN образует имеющие большое
значение в гидрометаллургии 3. комплексные циани-
стые соли 3. — K[Au(CN)2], Ca[Au(CN)2]2 и др.,
довольно хорошо растворимые в воде. Цианид трех-
валентного 3. Au(CN)3 • ЗН2О и его комплексные про-
изводные, напр. K[Au(CN4] • 1,5 Н2О, мало устойчи-
вы, легко разлагаются с образованием металлич. З.и
практич. значения не имеют. Отвечающую им свобод-
ную золотосинеродистую к-ту можно полу-
чить в виде бесцветных кристаллов H[Au(CN)4] • ЗН2О
действием HCN на раствор АпС13. Золотосинероди-
стая к-та хорошо растворима в воде, спирте, эфире.
При ее прокаливании образуется металлическое 3.
Известен роданид одновалентного 3. AuSCN, не-
растворимый в воде и органических растворителях.
При 140° AuSCN разлагается, при действии воды
переходит в более устойчивый роданид трехвалент-
ного 3. Au(SCN)3. Последний образует комплексную
к-ту H[Au(SCN)4] • 2Н2О и соответствующие ей соли.
Нитраты и сульфаты 3. не выделены. Допускают
возможность образования нитрата одновалентного 3.
Известна комплексная кислота H[Au(NO3)4], обра-
115
ЗОЛОТО
не
зующая нитроаураты щелочных металлов, аммония
и таллия, напр. K[Au(NO3)4]. При анодном окислении
3. в р-ре H2SO4 допускают образование Au2SO4,
быстро превращающегося в Аи(ОН)3. В противопо-
ложность сульфату, селенат 3-валентного 3. Au2(SeO4)3
известен и может быть выделен из р-ра 3. в горячей
безводной селеновой к-те. При растворении 3. в сла-
бокислых р-рах тиомочевины небольшой концентра-
ции в присутствии окислителей образуется тиокар-
бамид 3. AuCl • 2CS(NH2)2. При восстановлении разб.
р-ров солей 3., а также при электрич. распылении ме-
талла в воде образуются коллоидные р-ры 3., окраска
к-рых зависит от степени дисперсности частиц.
Аналитическое определение 3. про-
водится как методами пробирного анализа,
так и новыми методами аналитич. химии. При опре-
делении 3. в рудах, сплавах и др. объектах часто
пользуются основным методом пробирного анализа —
анализом «сухим путем», состоящим в плавлении
навески руды с флюсами и коллектором (свинец).
При этом получают сплав свинца с 3. и серебром,
к-рое обычно сопутствует 3. Полученный сплав под-
вергают окислительному плавлению в пористой огне-
упорной чашечке (капели), всасывающей окись свинца.
Остающийся сплав 3. с серебром (требуется преобла-
дание серебра по отношению к 3. в сплаве в 2—3
раза, а иногда и более) разделяют обработкой разб.
азотной к-той, растворяющей серебро. 3. после
промывки, сушки и прокаливания взвешивают. Иногда
вместо свинца 3. сплавляют с медью или кадмием.
При анализе растворов, содержащих 3., для аналитич.
химии важны в основном соединения Аи(Ш). Каче-
ственно 3. может быть открыто добавлением NaOH,
к-рый осаждает красно-бурую гидроокись, раствори-
мую в избытке щелочи. Чувствительной реакцией
на 3. является образование пятна с солянокислым
парарозанилином. 3. легко может быть восстановлено
до металла различными агентами (солянокислым или
сернокислым гидразином, гидрохиноном, щавелевой
к-той, FeSO4, SnCl2, аскорбиновой к-той и др.), что
также может использоваться как для качественного
обнаружения 3., так и для его количественного опре-
деления весовым методом в виде металла. Имеются
титриметрич. методы определения 3., основанные на
восстановлении Au(III) и последующем титровании
избытка восстановителя. Может быть проведено
потенциометрия, титрование р-ра Аи(Ш) р-ром SnCI2
или Си2С12. Фотометрически 3. определяют по оран-
жевой окраске бромаурат-иона, а также по интенсив-
ной окраске соединений 3. с различными органич.
реагентами (а-толуидином, бензидином, я-диметил-
аминобензилиденроданином и др.). Имеются также
полярографические, спектральные и др. методы опре-
деления 3. Для отделения 3. от группы платиновых
металлов могут быть использованы методы, основан-
ные на восстановлении 3. до металла. Небольшие
количества 3. от платиновых металлов отделяют
экстракцией АиС13 эфиром или этилацетатом из
солянокислых р-ров, применяют также экстракцию
изопропиловым эфиром из 2М раствора НВт. Следы
3. можно выделить на коллекторе (напр., SnCl2 или
теллуре) с последующим фотометрия, определением
с я-диметиламинобензилиденроданином. Этот реагент
используется также для Оценки содержания 3. в био-
логия. материалах.
Получение. Извлечение 3. из руд и песков россып-
ных месторождений производится гравитационным
и флотационным обогащением (см. Обогащение полез-
ных ископаемых, Флотация), амальгамацией и циа-
нированием, а также при выплавке и последующем
рафинировании свинца и меди (см. Пробирный ана-
лиз). Руды коренных месторождений дробят и измель-
чают. Крупные частицы 3. предварительно извлекают
гравитационным обогащением (отсадочными маши-
нами, шлюзами, гидравлич. ловушками) или амаль-
гамацией, т. е. смачиванием ртутью (см. Амальгамы).
Амальгамацию чаще применяют для извлечения из
гравитационного концентрата. В случае мелких
частиц 3. ртуть пропитывает, а в случае крупных —
поверхностно амальгамирует их. Амальгаму после
съема из аппаратов (шлюзы, бочки, амальгаматоры)
очищают, промывают и отжимают под давлением
(фильтруют) в прессе. Твердый остаток подвергают
отгонке в реторте (лучше при небольшом вакууме —
для гарантии безопасности) сначала при невысокой
темп-ре, а под конец — при 800°. Остаток 3. в реторте
плавят и аффинируют на специальных заводах.
Наиболее широко распространенным способом извле-
чения является цианирование, введенное
в промышленную практику в начале 90-х гг. 19 в.
Измельченную руду или концентрат обрабатывают
водным р-ром цианистого натрия с перемешиванием
в течение 6—48 часов или просачиванием раствора
в течение 5—7 суток с последующей промывкой.
Значительная часть 3. может быть извлечена в цикле
измельчения в мельнице-классификаторе, если послед-
нее производится в цианистом р-ре. Интенсификация
выщелачивания цианистым р-ром достигается повы-
шением количества кислорода, растворенного в водной
фазе, и другими приемами. Для успешного цианиро-
вания вводят защитные щелочи — Са(ОН)2 или NaOH
(от 0,02 до 0,05%), к-рые предохраняют цианид от
гидролиза и разложения кислотой и солями тяжелых
металлов, возникающими при окислении сульфидов.
После фильтрации цианистых золотосодержащих
р-ров из них осаждают 3. цинком. Для улучшения
этого процесса (для более полного осаждения металла
и снижения расхода цинковой пыли или цинковой
стружки) проводится предварительное удаление кис-
лорода из раствора. Осаждение 3. цинком основано
на реакции: 2Au(CN)j -f- Zn=Zn(CN)|_ -[- 2Аи; одно-
временно происходит растворение нек-рого коли-
чества цинка, с чем связано удаление остающегося
в растворе кислорода и выделение небольшого коли-
чества водорода. Цинк предварительно обрабатывается
раствором уксуснокислого или азотнокислого свинца;
образующийся на поверхности цинка рыхлый слон
свинца улучшает катодную деполяризацию. Получен-
ный осадок 3. обрабатывают р-ром серной к-ты и
плавят, полученный металл направляют.в аффинаж.
К другим возможным методам осаждения из циани-
стых р-ров относится применение полуактивирован-
ного древесного угля или ионообменных смол. Пока-
зана возможность извлечения 3. из щелочных циа-
нистых р-ров жидкостной экстракцией с использо-
ванием для этого органич. растворителей.
В зависимости от состава руд применяется ряд
специальных схем, в к-рых сочетаются флотация,
обжиг и гидрометаллургич. процессы для извлече-
ния различных компонентов (3., медь, уран и др.).
Основными видами добычи песков россыпного 3.
являются дражный, гидравлический и экскаватор-
ный. При добыче другими способами извлечение
производится на стационарных передвижных или
плавучих промывочных установках. Извлечение 3.
из песков россыпных месторождений сводится к пред-
варительному разрыхлению и последующей промывке
водой на шлюзе, представляющем наклонный желоб,
покрытый перфорированными железными листами
(дражные шлюзы), деревянными трафаретами или
пеньковыми коврами, на к-рых и задерживаются
частицы 3. Более совершенным способом является
отсадка, представляющая обогащение по плотности
в пульсирующей струе воды.
Применение 3. в настоящее время преим. имеет
валютные цели. В условиях социалистич. экономики
117
ЗОЛОТООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ — ЗОНГОРИН
118
3. является денежным металлом и выполняет функ-
цию меры стоимости. Наряду с другими товарами
оно служит одним из средств обеспечения советской
валюты. Покупная цена 1 г чистого золота установ-
лена в новом масштабе цен 1 рубль. Другим примене-
нием 3. является изготовление ювелирных изделий.
В сплаве с платиной 3. используется в произ-ве
химически стойкой аппаратуры, в сплавах с плати-
ной и серебром — в электротехнике, в виде химич.
препаратов — в фотографии (тонирование). Препа-
раты 3. повышают защитные свойства организма
и иногда применяются при лечении инфекционного
полиартрита, волчанки, туберкулеза легких и гор-
тани. Однако в последнее время, в связи с введением
в практику других фармацевтич. препаратов, приме-
нение 3. для медицинских целей сократилось. Золотом
в сплавах с другими металлами широко пользуются
для целей протезирования, особенно в зубоврачеб-
ной практике. Коллоидные р-ры радиоактивного 3.
нашли применение в радиотерапии.
Лит.: Плаксин И. Н., Металлургия благородных
металлов, М., 1958; его же, Опробование и пробирный ана-
лиз, М., 1947; Звягинцев О. Е., Аффинаж золота, се-
ребра и металлов платиновой группы, 3 изд., М., 1945; Нев-
ский Б. В., Обогащение россыпей, М., 1947; Базилев-
ский В. М., Вторичные драгоценные металлы (серебро, зо-
лото, платина), М., 1947;;Ф ер см ан А. Е., Геохимия, т. 4,
Л., 1939; Звягинцев О. Е., Геохимия золота, М. — Л.,
1941; Gmelin, 8 Aufl., Syst.-Nummer 62, Gold, Lfg. 1—3,
В., 1950—54; Mellor, v. 3, L. [a. o.], 1946; то же, v. 3,
1952; Pascal, t. 3, P., 1957; Ullman, 3 Aufl., Bd 8,
Munch. — B., 1957, S. 253—307; Kirk, v. 7, N. Y., 1951,
P. 274—88; Плаксин И. H., Ласкорин Б. H., Шив-
рин Г. H., Жидкостная экстракция цианистых комплексов
золота и серебра из цианистых растворов, Цветные металлы,
1961, № 9, 20—23; их же, ДАН СССР, 1961, 139, № 5,
1170 — 72. И. Н. Плаксин.
ЗОЛОТООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ —
известны все три возможных типа: RAuX2, R2AuX и
R3Au. Моногалогениды алкилзолота могут быть полу-
чены из диалкилзолотогалогенидов:
(C2H9)2AuBr -J- Вг2 —► С2НзАиВг2 4- CgHgBr
Безводное хлорное золото металлирует ароматич.
соединения, напр.
С0Н0 -(- AuClg —► CgHgAuClg
При этом заместители 1-го рода в ароматич. ядре
ускоряют реакцию, но понижают устойчивость обра-
зующих 3. с.; противоположное влияние оказывают
заместители 2-го рода. Галогениды диалкилзолота
получаются при действии магнийорганич. соединений
на бромное или хлорное золото:
АпВг3 4- 2 CeH5CH2MgBr —> (CeH5CH2)2AuBr -f- 2 MgBr2
Нек-рые триалкильные соединения золота синтези-
рованы с помощью литийорганич. соединений при
низкой темп-ре (—60°):
АиВГд -J- ЗСН3Ы —► (СНз)зАи ЗЫВг
3. с. разлагаются на свету и при нагревании с вы-
делением металлич. золота. Термически наименее
стойки полнозамещенные 3. с.; так, триметилзолото
разлагается уже при —40°:
2(СНз)3Аи —► 3C2Hfl -J- 2Ан
еще менее стабильно триэтилзолото. Соединения
типа RAuCL2 разрушаются под действием даже сла-
бых восстановителей с образованием золотого зер-
кала. Наиболее устойчивы соединения типа R2AuX;
они представляют собой, как правило, твердые, плохо
растворимые соединения, пассивные в отношении
кислот, слабых щелочей л восстановителей. 3. с.
дают комплексы с аминами; эти комплексы обыч-
но имеют определенные температуры плавления или
разложения, например NH2CH2CH2NH2 • 2R2AuX;
NH2CH2CH2NH2. • 2(CH3)3Au. 3. с. могут быть исполь-
зованы для идентификации аминов, диаминов и ами-
нотиоспиртов.
Лит.: Kharasch М. S., Isbell Н. S., J. Amer.
Chem. Soc., 1931, 53, № 7, 2701; их же, там же, 1931, 53,
8, 3053; Gilman Н., Woods L. А., там же, 1948,
70, М 2, 550; Ewens R. W. G., Gibson С. S., J. Chem.
Soc., 1949, February, 431. H. А. Несмеянов.
ЗОЛОТОХЛОРИСТОВОДОРОДНАЯ КИСЛОТА
H[AuC14] — устойчивое и наиболее распространенное
соединение 3-валентного золота. Кристаллизуется в ви-
де длинных светло-желтых игл состава Н[АиС14]-4Н2О,
расплывающихся во влажном воздухе; в сухом воз-
духе устойчив гидрат Н[АиС13] • ЗН2О. Теплота обра-
зования безводной 3. к. Д//298 = —77,8 ккал/маль.
Ион [ АиС14]~ значительно прочнее аналогичных ионов,
образованных другими трехзарядными катионами,
константа нестойкости 5 10’22. При нагревании до
120° 3. к. превращается в АиС13. 3. к. легко раство-
ряется в спирте, эфире. Получают 3. к. растворе-
нием Ап в царской водке, содержащей большой из-
быток соляной кислоты. Из солянокислых раство-
ров хорошо экстрагируется эфиром. 3. к. образует
соли — хлораураты, легко растворимые в воде.
Как сама 3. к., так и ее соли легко восстанавливаются
до металлич. золота. Na[AuCl4] • 2Н2О (т. наз. з о-
лотая соль) кристаллизуется в виде желтых
ромбич. столбиков или табличек, растворима
в эфире; применяется для окрашивания фотоснимков.
К[АиС14] • 2Н2О~ светло-желтые ромбич. таблички,
легко теряет кристаллизационную воду. Наряду с 3. к.
известны аналогичные кислоты Н[АиЙт4] и H[AuJ4],
ЗОМАН (пинаколиновый эфир фторангидрида метил-
фосфиновой кислоты) (СН3)3С — СН (СН3)
СН3/ \F’
мол. в. 182 — бесцветная жидкость; т. пл. ок.
—80°; т. кип. 42° (0,2 мм); d™ 1,013; п% 1,408; лету-
честь (максимальная концентрация) при 20° ок.
3 л»г/л. 3. ограниченно растворим в воде, хорошо
растворяется в органич. растворителях, очень мед-
ленно гидролизуется водой (реакция носит автоката-
литич. характер). Скорость гидролиза значительно
увеличивается в присутствии минеральных к-т и
особенно щелочей; в результате гидролиза образуется
соответственно пинаколиновый эфир метилфосфино-
вой к-ты или ее соль. 3. энергично реагирует с вод-
ными р-рами аммиака и аминов; может быть синте-
зирован аналогично аарину. 3. относится к судо-
рожно-паралитическим ОВ с резко выраженным
миотич. действием (сужение зрачка); смертельная
концентрация ок. 0,02 .иг/л при экспозиции 1 мин;
концентрация ок. 0,0002 .иг/л вызывает сильнейший
миоз, при действии на кожу в капельно-Жидком и
парообразном состоянии вызывает общее отравление.
Защитой от 3. служит противогаз и защитная одежда.
Для дегазации 3. могут быть использованы упомяну-
тые реакции (гидролиз, реакции с водными р-рами
NH3 и аминов и др.).
Лит.: Сондерс Б., Химия и токсикология органиче-
ских соединений фосфора и фтора, пер. с англ., М., 1961.
Р. Н. Стерлин.
ЗОНГОРИН—C21H29O3N, мол. в. 343,47 — алкало-
ид, находящийся в корнях аконита джунгарского
Aconitum soonqoricum Slapf,
9 произрастающего в Средней
/X.____сн Азии, в верховьях Таласа,
ОН . [ I Lruj" мелкоигольчатые кристаллы,
т.пл. 201—203°, [a]D=—193,89°
(абс. спирт). 3. легко раство-
ряется в хлороформе, мети-
ловом и этиловом спиртах,
труднее — в эфире; трудно растворим в петролейном
эфире и почти нерастворим в воде. Дает кристалли-
ческие соли: хлоргидрат, т. пл. 287—258°; нитрат,
т. пл. 230—23Г (с разлож.); иодметилат, т. пл.
209—210°; бромгидрат, т, пл. 260—261° (с разлож.);
119
ЗОННАЯ ПЛАВКА
120
перхлорат, т. пл. 236 -237° (с разл.). 3. дает семи-
карбазон. 3. — ненасыщенное основание; при ката-
литич. гидрировании присоединяет 2 атома водоро-
да. Иодметилат 3. при нагревании с КОН в р-ре
метилового спирта образует дезоснование, легко да-
ющее подметилат. Выделен 3. в 1929 С. Ю. Юндсовым.
См. также Аконитовые алкалоиды.
• ЗОННАЯ ПЛАВКА (зонная перекри-
сталлизация) — один из методов разделения
в глубокой очистки веществ. Иногда этот метод наз.
зонной очисткой. Кроме того, 3. п. можно использо-
вать для таких целей, как выращивание монокристал-
лов, создание тех или иных распределений концент-
рации веществ, как, напр., выравнивание состава,
создание градиентов концентрации и т. п.
3. п. осуществляется расплавлением небольших
участков твердой загрузки, называемых «зонами»,
и последовательным продвижением расплавленных
зон одна за другой в выбранном направлении вдоль
загрузки (рис. 1). При этом на поверхностях раздела
твердой и жидкой фаз совершаются следующие про-
цессы: на фронте кристаллизации происходит кри-
сталлизация материала, на фронте плавления —
,подпитка зоны исходным мате-
S ? 2 риалом. Если вдоль загрузки
\
Рис. 1. Схема зонной плавки: 1 — ло-
4 —* дочка; 2 — загрузка, подпитываю-
щая расплавленную зону; 3 — рас-
плавленная зона; 4 — кольцевой нагреватель; 5 — закристал-
лизовавшаяся часть загрузки позади зоны. Стрелкой указано
направление движения зоны.
продвигается несколько расплавленных зон, то под-
питка происходит материалом, закристаллизовавшим-
ся при прохождении предыдущей зоны. Очистка
происходит за счет различного распределения веществ
между соприкасающимися твердой и жидкой фазами.
Важной характеристикой процесса разделения ве-
ществ при кристаллизации является коэфф, распре-
деления К, представляющий собой отношение кон-
центраций примесей в твердой и жидкой фазах
(А=СТВ /Сдаидк ). Те примеси, к-рые имеют К < 1,
преимущественно концентрируются в расплаве и при
3. п.' уносятся расплавленными зонами в ту часть
загрузки, где зоны заканчивают свое движение. Та-
ким образом ведет себя большинство примесей. При-
меси, имеющие К > 1, при кристаллизации преим.
концентрируются в твердой фазе, и в результате мно-
гократной 3. п. (расплавленные зоны продвигаются
вдоль слитка много раз в одном направлении) этими
примесями оказывается загрязнена та часть загрузки,
откуда расплавленные зоны начинают свое движение.
На эффективности очистки существенно сказывается
скорость движения зон. При повышенных скоростях
примеси скапливаются в непосредственной близости
от фронта кристаллизации, не успевают диффузион-
ным путем равномерно распределиться по всей рас-
плавленной зоне (при К < 1), и тем самым эффектив-
ность процесса снижается. Практически скорости
движения зон не превышают значений 0,1 —
2,0 мм/мин. Значительной интенсификации процесса
3. п. удается достичь путем принудительного переме-
шивания расплава в зоне. Для этой цели лучше
всего использовать электромагнитное перемешива-
ние. Кроме того, для достижения глубокой очистки
требуется, как правило, большое число проходов
в одном направлении (от 10—15 до нескольких сот).
При этом создается т. наз. предельное рас-
пределение примесей, соответствующее наиболь-
шей достижимой очистке.
Техника.осуществления 3. п. в большинстве слу-
чаев очень проста. В связи с необходимостью преду-
преждать возможное загрязнение очищаемого мате-
риала его изолируют от окружающей атмосферы,
помещая в кварцевые трубы или стальной кожух.
Процесс обычно ведется в вакууме или в защитной
атмосфере. Создание вакуума может вызвать разло-
жение очищаемого вещества за счет испарения, что
нежелательно в случае 3. п. разлагающихся соеди-
нений и благоприятно в случае очистки веществ
от легколетучих примесей. Конденсация примесей
на очищенных участках устраняется током защит-
ного газа, направленного в сторону движения рас-
плавленных зон. Лодочки могут иметь поперечное
сечение различной формы и изготовляются из мате-
риала, к-рый не взаимодействует с очищаемым ве-
ществом и не содержит примесей, могущих загряз-
нить очищаемый материал. Напр., при 3. п. германия,
сурьмы, алюминия и др. используются лодочки из
графита высокой чистоты, а при 3. п. кремния, тел-
лура, антимонидов и т. п. — лодочки из плавле-
ного кварца и т. д. Для уменьшения смачивания и
сцепления внутренние стенки лодочки могут подвер-
гаться специальной обработке или покрытиям. На-
грев для создания расплавленных зон производят
различными способами: нагревателями сопротивле-
ния, токами высокой частоты, пламенем сжигаемого
газообразного или жидкого топлива, электрич. раз-
рядом (дуговая 3. п. и 3. п. электронной
бомбардировкой), излучением (фокусиров-
кой излучения ламп накаливания или солнечной
энергии). Если на концах слитка очищаемого мате-
риала разместить холодильники, с помощью к-рых
станет возможным регулировать их темп-ру, то
можно осуществить 3. п. за счет джоулева тепла,
выделяемого электрич. током, проходящим по за-
грузке в осевом направлении. Для создания более
узких расплавленных зон используют специальные
холодильники, размещаемые с различных сторон
нагревателя. Расплавленные зоны вдоль загрузки
перемещают либо движением слитка относительно на-
гревателей, либо движением нагревателей относитель-
но слитка. Вследствие различия уд. объемов веществ
в твердом и жидком состоянии может происхо-
дить массоперенос вдоль загрузки, и для его предот-
вращения лодочкам придается определенный на-
клон.
Процесс 3. п. является малопроизводительным.
Совершенствование применяемой для 3. п. аппаратуры
идет по пути сокращения продолжительности про-
цесса. Для более полного использования нагрева-
телей твердой загрузке при-
дают форму разомкнутого коль-
ца или спирали (рис. 2) и пере-
мещают вдоль загрузки одно-
временно несколько распла-
вленных зон. Наиболее удачен
вариант, при к-ром несколько
нагревателей создают в слитке
расплавленные зоны и медлен-
но перемещают их все в одном
направлении, а затем путем
быстрого возвращения нагре-
вателей в исходное положе-
ние расплавленная зона, «вы-
пущенная» одним из нагре-
вателей, «подхватывается» дру-
гим (3. п. с «подхвате м»)
и продолжает продвигаться в
том же направлении.
3. п. высокореакционных веществ ведется бести-
гельным методом: слиток располагается вертикально
и закрепляется сверху и снизу; расплавленный уча-
сток такого слитка удерживается между твердыми
участками силами поверхностного натяжения и может
перемещаться вдоль слитка как вверх, так и вниз
з
г
Рис. 2. Схема зонной
плавки слитка, спираль-
ной формы (вид сверху):
i — твердая загрузка;
г — нагреватель (распо-
ложен сверху); з — рас-
плавленные зоны. Стрел-
ками указаны направле-
ния движения зон.
121
ЗОННАЯ ПЛАВКА —ЗООЦИДЫ
122
(метод «пл а в а ю щ е й» зон ы). Вращение
твердых частей слитка относительно друг друга вокруг
вертикальной оси обеспечивает перемешивание рас-
плава в зоне. Если слиток снабжают продольными
тонкостенными ребрами, неплавящимися вследствие
большой теплоотдачи через них, то это создает до-
полнительную поддержку расплавленной зоне (м е-
т о д 3. п. «в к л е т к е»). Кроме того, поддержка
зоны иногда осуществляется путем использования
взаимодействия индуцированных и индуктирующих
токов или пропускаемого через слиток и зону тока
с внешним магнитным полем (3. п. с магнитным
подвешиванием).
Важным направлением совершенствования про-
цесса 3. п. является создание непрерывных вариантов,
отличительная особенность к-рых — отсутствие необ-
ходимости периодически загружать и разгружать
контейнер. В настоящее время предложено два спо-
соба осуществления подобных процессов.
При зонно-пустотном методе (рис. 3) материал
в ходе самой 3. п. перемещается в вертикально расположенных
колоннах за счет создания «пустот» (полостей) в месте вывода
и движения этих полостей к месту по-
дачи питающего материала. При дви-
жении нагревателя снизу вверх над по-
лостью происходит плавление материа-
ла, а под расплавленной зоной — кри-
сталлизация. За один проход нагрева-
теля происходит перемещение материа-
ла сверху вниз колонны на размер
полости. В месте вывода загрязненного
продукта происходит слив обогащенной
примесями расплавленной зоны. При
дальнейшем продвижении нагревателя
вверх идет процесс проплавления проб-
ки из твердой загрузки, полость барбо-
тирует через расплав в питателе, и соот-
ветствующее количество загрузки по-
ступает в колонну. Количество мате-
риала в питателе требуется пополнять.
По колонне одновременно продвигается
несколько создаваемых нагревателями
расплавленных зон.
очищенного продукта
5 — выпуск чистого
примесями продукта.
Рис. 3. Схема аппарата для непрерыв-
ной «зонно-пустотной» плавки: J — на-
греватель; 2 — расплавленная зона;
3 — полость; 4 — питание аппарата;
продукта; 6 — выпуск обогащенного
Стрелками указано направление движе-
ния зон (снизу вверх).
При зонно-транспортном методе (рис. 4)
используется способность расплавленной зоны перемещать
материал, если содержащий его
непрерывной «зонно-транс-
портной» плавки: 1 — нагрева-
тель; 2 — расплавленная зона;
3 — питание аппарата; 4 — вы-
пуск чистого продукта; 5 — вы-
пуск обогащенного примесями
продукта. Стрелками указано
направление движения зон.
контейнер находится в на-
клонном положении. Движу-
щая сила создается за счет
разности уровней твердой за-
грузки, поступающей в зону,
и кристаллизующейся зоны.
Контейнер представляет со-
бой продолговатый лоток
с отверстиями на каждом
конце. Через отверстие для
отбора очищенного материа-
ла происходит слив зоны,
когда нагреватель начинает
продвижение расплавленной
зоны вдоль лотка. Загряз-
ненный материал сливается
через отверстие в противо-
положном конце лотка, ко-
гда нагреватель заканчивает
продвижение зоны. Пита-
ние аппарата осуществляет-
ся в наивысшей точке лотка, чтобы движение материала про-
исходило в противоположных направлениях от этого места.
В ряде случаев в предварительно очищенный мате-
риал требуется вводить специальные примеси (леги-
рующие добавки). Обычно в этих случаях необходима
исключительная однородность распределения легиро-
ванного материала. Эта однородность относительно
просто достигается применением зонного вы-
равнивания. Выравнивание концентрации при-
меси достигается в результате достаточного числа
проходов зоны попеременно в прямом и обратном
направлениях вдоль загрузки. На практике леги-
рующую примесь обычно вводят в жидкую зону в на-
чальный момент, после чего зона «разносит» примесь
по всей загрузке. 3. п. смеси порошков были впервые
получены гомогенные сплавы германия с. кремнием
без .применения длительных (в течение нескольких
месяцев) отжигов при повышенной темп-ре. Зонное
выравнивание, проводимое с особым регулированием
длины расплавленных зон и скорости их движения,
позволяет получать особые типы распределения
примесей в материале. Таким образом, напр., можно
получить материал с линейно увеличивающейся
концентрацией нужной примеси по длине монокри-
сталла. Для изготовления полупроводниковых диодов
или триодов иногда требуется создание более резких
изменений концентрации примесей или соотношений
концентраций примесей (создание перехо-
д о в), различающихся по своему электрофизич.
воздействию на основной материал. Эта задача ре-
шается с помощью 3. п. путем резкого изменения
состава расплавленной зоны, изменения ее длины
или изменения скорости ее перемещения.
Впервые 3. п. была применена в связи с необхо-
димостью глубокой очистки таких полупроводниковых
элементов, как германий и кремний, к-рые к тому же
требовалось получить в монокристаллич. виде. Затем
этот метод был использован для очистки других эле-
ментов и соединений, нашедших применение в полу-
проводниковой технике. В настоящее время для
удовлетворения потребностей электро- и радиотех-
ники, атомной энергетики и других областей науки
и произ-ва применение 3. п. значительно расшири-
лось. Почти для всех полупроводников и металлов,
а также многих неорганич. и органич. соединений
3. п. позволила успешно вести очистку от примесей.
3. п. пока представляет собой лабораторный метод,
к-рый все чаще используется как крупно-лаборатор-
ный, однако ему, очевидно, предстоит стать и про-
мышленным методом. При этом должны быть решены
многие задачи интенсификации и автоматич. регули-
рования этого процесса.
Лит.: Петров Д. А., в сб.: Вопросы теории и исследо-
вания полупроводников и процессов полупроводниковой ме-
таллургии, М., 1955; Методы получения чистых металлов. Сб.
переводов, М., 1957, с. 171—389; Шашков Ю. М., Метал-
лургия полупроводников, М., 1960; Пфанн В. Дж., Зон-
ная плавка, пер. с англ., М., 1960; Парр Н. Д., Зонная
очистка и ее техника, пер. с англ., М., 1962; В и г д о р о-
в и ч В. Н., Вольпян А. Е., Изв. высших учебных заве-
дений. Цветная металлургия, 1960, 3, № 3. 126—35; Ро-
зин К. М., Вигдорович В. Н., Крестовни-
ков А. Н., Изв. АН СССР. Отд. техн. наук. Металлургия
и топливо, 1961, № 6, 56—73. В. Н. Вигдорович.
ЗООЦИДЫ — химические средства борьбы с гры-
зунами; различные неорганич. и органич. соединения.
Из неорганич. соединений-для борьбы с грызунами
(мыши, крысы, суслики и т. п.) используют фосфид
цинка Zn3P2, сульфат таллия T12SO4, углекислый ба-
рий и некоторые соединения мышьяка. Из органи-
ческих соединений получили практич. применение
такие вещества, как фторацетат натрия и бария,
фторацетамид и фторацетанилид, а-нафтилтиомочеви-
на, тетраметилендисульфотетрамин, 2-хлор-4-диметил-
амино-6-метилпиримидин (кас.трикс), 4-хлорфе-
нилдиазотиомочевина (промурит), 3, 4-дихлор-
фенилдиазотиомочевина (хлорпромурит), про-
изводные 4-оксикумарина и индандиона. Для борьбы
с грызунами начали использовать также нек-рые ин-
сектициды— ДДТ, токсафен, эндрин и др. Токсичность
важнейших 3. для грызунов приведена в таблице.
Одним из старейших 3., применявшихся еще в сред-
ние века, является морской лук, действующее начало
к-рого гликозид скиллиразид (1). Большое
внимание уделяется изучению и использованию раз-
личных производных оксикумарина и индандиона,
! известных под названием антикоагулянтов. Из этой
123
ЗООЦИДЫ-ЗРИТЕЛЬНЫЙ ПУРПУР
124
Соединение Токсичность для грызу- нов лд50, мг/кг Соединение Токсичность для грызу- нов ЛД50, мг/кг
Фторацетат нат- рия Сульфат таллия Фосфид цинка . . Барий углекис- лый Промурит .... Кастрикс .... а-Н афтилтиомоче- вина ...... 0,1-10 15-25 40-75 400-1500 1-5 1-5 7-250 Скиллиразид . . Варфарин .... Кумахлор .... Фумарин .... Пив аль Гетраметиленди- сульфотетра- мин 0,5-0,7 60 1200 400 60—120 0,5-1
группы практич. применение нашли: 3,3''-мети-
лен-бис-4-оксикумарин (д и к у м а р о л); 3-(1-фе-
нил-2-ацетилэтил)-4-оксикумарин (в а р ф а р и н);
3-(2-п-хлорфенил-2-ацетилэтил)-4-оксикумарин (ку-
махлор); 3-(1-фурил-2-ацетилэтил)-4-оксикумарин
(ф у м а р и н); 2-пивалилиндандион-1,3 (п и в а л ь);
2-бензоилиндандион-1,3 и др. Активными антикоагу-
лянтами 3. являются также нек-рые 3, 4-дигидропи-
ранокумарины (II).
3. чаще всего применяют для борьбы с грызунами
в виде приманок (0,1—2% к пищевым продуктам).
Для получения приманок на зерне добавляют не-
большие количества веществ, способствующих при-
липанию 3. В нек-рых случаях для борьбы с грызу-
нами 3. используют в смеси с маслами; такие препа-
раты загрязняют шкурку грызуна, и после слизыва-
ния препарата животное погибает.
Лит.: Mtinchberg Р., Chem. Ztg., 1954, 78, 5;
Zblrovsky М., MyS k a J., Insektlcidy fungicidy, ro-
denticidy, Praha, 1957. H. H. Мельников.
ЗРИТЕЛЬНЫЙ ПУРПУР — см. Родопсин.
и
ИВАНОВА РЕАКЦИЯ — метод получения а-заме-
щенных арилуксусных к-т из их галогенмагниевых
производных (реагентов Иванова) по схеме магнийор-
ганич. синтеза. Реагенты Иванова образуются при дей-
ствии двойного избытка алифатич. магнийорганич. со-
единения на арилуксусную к-ту либо эквивалентно-
го количества магнийорганич. соединения на Na-соль
соответствующей к-ты. Наиболее высокие выходы
были получены с применением u30-C3H7MgCl:
R'
Ar-C-COONa(MgX)RMg?-
I
H
R' о
Ar—С—i-ONa(MgX)
MgX
генты Иванова приводит к смеси бензилкетонов и
дифенилалкилоксиглутаровых к-т:
C6H5-C-COOMgCl
MgCl
R-C-Cl
и
О
zcooMgci
с6н5-сн -----------
C-R
II
о
свн5-сн-соон
R-C-OH
С6Н5-СН-СООЦ
-со2
C6H5-CH2-C-R —-
II и
о
вп5'
M8Ct
C8H5CHCOOMgCl
Ar—фенил, о- и п-хлорфенил, 2-и 3-тиенил; R'—арил, алкил,
аралкил, феноксигруппа.
Галогенмагниевое производное фенилацетата нат-
рия (или галогенмагния) реагирует с алифатич.
или ароматич. кетонами и альдегидами с обра-
зованием соответственно ди- или монозамещенных
троповых к-т; при этом последовательно действуют
соответствующим реагентом, а затем кйслотой:
Окиси олефинов вступают в И. р. по обычной схеме
магнийорганич. синтеза:
1. СН2-СНа; 2. Н+
С8Н5—CH—COONa------------------
(СН3Т2С=б
C6Hs-CHCOONa
MgCl
СН2О
euxoa 80% О’ теоретач
свнв-сн-соон
(СН3)2С-ОВ
с6н5-сн-соон
СН2ОН
MgCl
-->НО-СН2СН2-СН-СООН
I
С8н5
Действие реагентов Иванова на у-бутиролактон
и у-валеролактон приводит к соответствующим про-
изводным тетрагидрофурана:
а, P-Непредельные кетоны взаимодействуют с реа-
гентами Иванова по двум направлениям:
ОН
о r-^R-CH=CH-C~CH-COOH
c6H8-CHCOOMgci , 2.присом1)НД С°Н5
MgCl
l-*-R-CH-CH2-C-R'
I II
I о
с6н5-сн-соон
1.4 - присоединение
CeH5-CHCOOMgCl—
M^Cl
В И. р. удалось ввести также алкилизоцианаты и
алкилизотиоцианаты:
Карбонизацией галогенмагниевых производных арил-
уксусных к-т удалось получить арилмалоновые к-ты,
напр.:
C8H5-CH-COOMgCl---C-°2: 2' Н+.С8Н5СН(СООН)2
(выход 56% от теоретич.)
C6H5-CHCOOMgCI
MgCl
Действие галогенангидридов карбоновых к-т на реа-
свн5-сн-соон
с=о
NH-R
С6Н5-СН-СООН
C=s
NH-R
127
ИДЕАЛЬНЫЕ РАСТВОРЫ
128
Реагенты Иванова подвергаются окислительной ди-
меризации под влиянием брома или кислорода. Осо-
бенно легко эта реакция протекает в случае галоген-
магниевых производных гидратроповых к-т:
сн3
I
2 C0H5-C-COOMgX
MgX
сн3
I
[О] СоН5—с—соон
С(,Н;-С- соон
I
сн8
Образование и реакции алифатич. реагентов Иванова
наблюдались на примере винилуксусной к-ты:
сна=сн-сн2-соон
2 ИЗО-CgHjMgCl
сн,
II
сн
сн-соон
сн3=сн-сн-COOMgCl
ОН
MgCi
о но^сн2-сн=сн-соон
Образование производного кротоновой кислоты свя-
зывают с наличием равновесия
CH2=CH-CH-COOMgCli=CH2-CH=CH-COOMgCl
I I
MgCl MgCl
И. р. была открыта Д. Ивановым в 1931.
Лит.; I v а п о f f D., S р a s о f f A., Bull. Soc. chlm.
France, 1931, 49—50, ser. 4, K« 1, 19; K« 3, 371 — 79; 1932, 51 —
52. № 10, 1321—325; Bllcke F., Rat telson H.,
J.Amer. Chem. Soc., 1952, 74, K‘7. 1730; В 11 c k e F., Z 1 n n e s
H., там же, 1955, 77, JOS 18, 4849, № 20, 5399; Blieke F.,
Wright P. E., J. Organ. Chem., 1960, 25, 693.
Л. С. Герман.
ИДЕАЛЬНЫЕ РАСТВОРЫ — см. Растворы.
ИДЕНТИФИКАЦИЯ — установление тождества
неизвестного соединения с другим известным. Для
этого сопоставляют физико-химич. константы, свой-
ства и реакции обоих веществ. Перед И. вещество
очищают перегонкой или перекристаллизацией, пос-
ле чего проводят предварительное его исследование:
сопоставляют агрегатное состояние, цвет, вязкость,
потемнение в р-рах, проводят испытание растворимо-
сти в воде, органич. растворителях, основаниях и
кислотах, определяют реакцию, горючесть и др.
свойства.
И. неорганич. веществ включает открытие ка-
тионов и анионов на основе характерных реакций;
иногда устанавливают в исследуемом в-ве содержание
или соотношение ионов (комплексных) и определяют
физико-химич. и. физич. характеристики вещества:
константу диссоциации, теплопроводность, теплоем-
кость, электропроводность, характер кристаллов,
спектры поглощения и испускания и др.
Для И. органич. соединений определяют физич.
константы: плотность, показатель преломления, вяз-
кость, спектры поглощения и испускания и др.,
мол. вес и эквивалентный вес по отношению к одной
или нескольким .группам или элементам. На основе
характерных реакций обнаруживают функциональные
группы и принадлежность к разным классам, напр.
альдегиды — по образованию серебряного зеркала;
по отношению к азотистой к-те различают первичные,
вторичные и третичные амины. Важной характе-
ристикой является темп-ра плавления смешанной
пробы исследуемого вещества с веществом известного
строения; если не наблюдается депрессии темп-ры
плавления, то можно говорить о тождестве обоих
соединений. Далее вещество идентифицируют по кон-
стантам его производных, напр. кислоты идентифи-
цируют по темп-ре плавления или кипения их амидов,
эфиров, нитрилов и т. д. Во многих случаях для И.
проводят реакции, позволяющие отличить исследуемое
в-во от других аналогичных, напр. при нагревании
р-ра щавелевой к-ты с резорцином и прибавлении
после охлаждения конц. H2SO4 появляется синее
кольцо в месте соприкосновения жидкостей; при ки-
пячении — темно-зеленое окрашивание, по охлажде-
нии — светлое желто-зеленое. Реакции не мешают
муравьиная, уксусная, молочная, винная, лимонная,
малеиновая, янтарная и бензойная к-ты.
Лит.; Вайбель С., Идентификация органических со-
единений, пер. с англ., М., 1957; Бауер К., Анализ ор-
ганических соединений, пер. с нем., 2 изд., М., 1953;
Мейер Г., Анализ и определение органических соедине-
ний, пер. с нем., Л., 1937. Д. И. Кузнецов.
ИДЕНТИФИКАЦИЯ ВЫСОКОМОЛЕКУЛЯРНЫХ
СОЕДИНЕНИЙ — установление идентичности (тож-
дественности) исследуемого вещества с известным вы-
сокомолекулярным соединением. Определение свойств
для установления природы исследуемого полимер-
ного вещества проводят обычно в следующей по-
следовательности: поведение полимера в пламени,
определение плотности, исследование продуктов де-
струкции на содержание отдельных элементов или
низкомолекулярных веществ, проведение характер-
ных цветных реакций, определение растворимости и
элементарного состава. Для установления природы по-
лимера иногда достаточно определить два или три
показателя, например для фторопластов характерны
внешний вид, плотность и содержание фтора; для
полиэтилена — внешний вид, плотность и элементар-
ный состав; для поливинилхлорида — содержание
хлора и растворимость. Для более полной характе-
ристики проводят количественный элементарный ана-
лиз, а также реакции, позволяющие установить строе-
ние исследуемого вещества; определяют темп-ру
плавления, мол. вес, фракционный состав, строение
с применением рентгеноструктурного анализа и т. д.
Для установления состава сополимеров применяют
инфракрасную спектроскопию и др. методы.
Горение полимеров. Наблюдение по-
ведения полимерных веществ в синем конусе пламени
горелки (окраска пламени, степень горючести поли-
мера, запах продуктов горения) — один из важных
приемов идентификации. Характеристика поведения
нек-рых типов полимеров в пламени дана в табл. 1.
Определение плотности полимеров
проводят с помощью пикнометра (см. также Денси-
метрия}. Данные о плотности полимеров представ-
лены в табл. 2.
Растворимость полимеров проверяют
при комнатной темп-ре или при нагревании (с обрат-
ным холодильником). При определении растворимости
необходимо учитывать, что у полимеров она зависит
не только от мол. веса, но и от многих других факторов.
Для исследования продуктов деструкции
небольшой кусочек исследуемого полимера помещают
в реторту из тугоплавкого стекла, быстро нагревают
ее внутренним синим конусом пламени горелки.
Газообразные продукты деструкции собирают и опре-
деляют присутствие отдельных элементов и мономеров.
Так, реакцию на хлор дают поливинилхлорид и его
сополимеры, поливинилиденхлорид, перхлорвинило-
вая смола и другие хлорсодержащие полимеры. Азот
содержит мочевино- и меламино-формальдегидные смо-
лы, полиамиды, полиуретаны, полиакрилонитрил,
анилино-формальдегидные смолы и др. полимеры.
Реакцию на фтор дают политетрафторэтилен, поли-
трифторхлорэтилен, реакцию на кремний — кремний-
органич. полимеры. Фенол обнаруживается в про-
дуктах деструкции феноло-формальдегидных, феноло-
фурфурольных смол. Полимеры, полученные конден-
сацией альдегидов с фенолами, аминами и другими
соединениями, а также полиэфирные и эпоксидные
смолы, продукты полимеризации альдегидов, поли-
129
ИДЕНТИФИКАЦИЯ ВЫСОКОМОЛЕКУЛЯРНЫХ СОЕДИНЕНИЙ
130
Таблица 1. Поведение высокомолекулярных соединений при внесении в пламя н характеристика
продуктов пиролиза
Пламя Запах Реакция продуктов пиролиза Полимер
Отношение к нагреванию
Плавится, горит хорошо, продолжает гореть при уда- лении из пламени То же » » » » » » Светящееся, внутри окра- шено в синий цвет Коптящее Яркое Парафина То же Парафина, сладковатый Формальдегида Полиэтилен Полипропилен Полиизобутилен Поликарбонаты Полиформальдегид
» » Синеватое, с желтыми краями Жженого рога Щелочная Полиамиды
V • То же Резкий Полиуретаны
» » Горит, продолжает гореть при удалении из пламени С желто-зеленой каймой и с искрами Яркое, коптящее Уксусной к-ты, жженой бумаги Сладковатый цветочный Кислая Ацетилцеллюлоза Полистирол
То же Светящееся Сладковатый, жира Кислая Полиэфирные смолы
» » » » То же Древесного угля Формальдегида » Феноло-фурфурольные и фуриЛовые смолы Резорцино-формальде- гидные
» » Коптящее Специфический Кислая Эпоксидные смолы
» » Желтое Аммиака, анилина, фор- мальдегида Щелочная Анилин ©-формальде- гидные смолы
» » Обычное. Образуется белая зола Формальдегида К р емн ий ор ганичес кие соединения
» » Светящееся, синеватое Сильный, цветочно-пло- довый Кислая Полиакрилаты
в » Светящееся, слегка коптит То же Кислая Полиметакрилаты
» » Голубое, иногда с неболь- шой белой верхушкой Слабый, чуть сладкова- тый, затем формальде- гида » Поливинилформаль
» » Синеватое с желтыми краями Прогорклого масла или сыра Кислая Поливинилбутираль
» » » » Светящееся Светящееся, синеватое Уксусной к-ты Специфический, жира и рыбы » Поливинилацетат Поливиниловый спирт
» » Яркое, белое, быстрое горение Окислов азота Кислая Нитроцеллюлоза
V 1» 9 9 » » С искрами, яркое, желто- белое Яркое Окружено желто-зеленой каймой Светящееся, коптящее Масляной к-ты, жже- ной бумаги Жженой бумаги Сладковатый, жженой бумаги Горьких миндалей 9 Ацетобутират целлюлозы Целлюлоза Этилцеллюлоза Бензйлцеллюлоза
Горит, при удалении из пламени гаснет Ярко-зеленое Резкий, хлористого во- дорода Кислая Поливинилхлорид
То же, много черной золы То же Сладковатый, затем рез- кий, хлористого водорода » Поливинилиденхлорид
Загорается с трудом, со- храняет форму при нагре- Белое Рыбы, формальдегида Щелочная Меламино-формальде- гидная смола
вании
Обугливание, по краям бе- лый налет, сохраняет форму Загорается с трудом Не горит, разлагается Белое Аммиака, формальдегида Фенола и формальдегида Резкий Щелочная » Мочевино-формальде- гидная смола Феноло-формальдегидная смола Политетрафторэтилен
Горит, гаснет при удалении Желтоватое Пригорелого молока Щелочная Казеин
из пламени
Горит, продолжает гореть при удалении из пламени Яркое, коптящее Резкий, скипидара Нейтраль- ная Натуральный каучук
То же » » Резкий Нейтраль- ная или слабокис- лая Бутадиеновый и бута- диенстирольный каучук
3 К. X. Э. т. 2
131
ИДЕНТИФИКАЦИЯ ВЫСОКОМОЛЕКУЛЯРНЫХ СОЕДИНЕНИЙ
132
винилацетали и др. дают реакцию на альдегиды.
О качественных реакциях на элементы и мономерные
соединения см. в соответствующих статьях, напр.
Анилин, Альдегиды, Фенол, Хлор и др. Определение
продуктов пиролиза для И. в. с. может быть осущест-
влено с применением газожидкостной хроматографии.
Таблица 2. Плотность некоторых полимеров
Плотность, г/с.и3 Полимер
до 1,0 Полиэтилен (0,92—0,97), полипропилен (0,85— 0,96), полиизобутилен (0,91—0,93), бутадиен- стирольный каучук (0,94), атактич. полибу- тадиен (0,89—0,92), бутадиен-нитрильные каучуки (0,96—1,01), натуральный каучук (1,0)
1,0 — 1,15 Полистирол (1,05—1,1), полиамиды (1,04— 1,14), шеллак (1,11), кумароно-ннденовые смолы (1,01—1,15), этилцеллюлоза (1,14)
1,1—1,25 Простые эфиры целлюлозы (1,10—1,25), по- лиакриловые эфиры (1,12—1,2), поливинил- ацетат (1,19), поликарбонаты (1,17—1,22), ацетобутират целлюлозы (1,14—1,2), поли- метилметакрилат (1,19), поливинилацеталь (1,16), поливинилбутираль (1,1—1,18), по- лиакрилонитрил (1,12—1,18)
1,2—1,35 Поливинилформаль (1,23), хлоропреновый каучук (1,2—1,25), анилино-формальдегид- ные смолы (1,2—1,25), поливиниловый спирт (1,26—1,29), полиэфирные смолы (1,1—1,4), мочевино- и меламино-формальдегидные смолы (1,1 — 1,35), бензилцеллюлоза (1,22), гидратцеллюлоза (1,25), феноло-формальде- гидные смолы (1,1—1,27), ацетилцеллюлоза (1,27—1,35), ацетопропионат целлюлозы (1,29)
1,30—1,50 Глифталевые смолы (1,4), лигнин (1,45), нитрат целлюлозы (1,35—1,5), тиомочевино-фор- мальдегидные смолы (1,48), сульфонамидные смолы (1,31—1,36), поливинилхлоридацетаты (1,3-1,4)
1,5 —1,6 Целлюлоза (1,56—1,60), поливинилхлорид (1,2—1,75), хлорированный каучук (1,64), полисульфидные каучуки (1,34—1,6)
1,6 —1,8 Поливиштлиденхлорид (1,6—1,87), перхлорви- нил (1,6—1,65)
свыше 2,0 Политрифторхлорэтилен (2,11—2,16), политет- рафторэтилен (2,1—2,4)
Характерные цветные реакции
на полимеры. На фарфоровую пластинку по-
мещают небольшой кусочек исследуемого полимера и
наносят на пего несколько капель уксусного ангидри-
да, а затем каплю конц. H2SO4 (реакция Либермана —
Шторха). Наблюдают в течение 30 мин. за окраской
жидкости и поверхности смолы, отмечая цвета и по-
следовательность их изменения. Окраска различных
полимеров по реакции Либермана—Шторха см. в табл.З.
Для определения типа каучука используют пробу Вебера.
Для этого в бюкс с небольшим кусочком каучука, набухшего
в СС14, добавляют сначала несколько капель брома, а затем
немного кристаллич. фенола. Для удаления СС14 смесь нагре-
вают на водяной бане. В табл. 4 представлены данные о спе-
цифич. окраске каучуков и оставшейся жидкости. Одним из
приемов идентификации каучуков является определение реак-
ции газообразных продуктов термин, разложения (прижига-
ние раскаленной металлич. проволокой) каучуков. Под дей-
ствием продуктов разложения фильтровальная бумага, про-
питанная смесью n-диметиламинобензальдегида, гидрохинона
и этилового эфира (реактивная бумага на НК) или пропитан-
ная раствором желтой окиси ртути в серной кислоте (реактив-
ная бумага для открытия бутилкаучука), принимает характер-
ную для каждого вида каучука окраску (см. табл. 4).
Таблица 3. Окраска высокомолекулярных соединений по
реакции Либермана—Шторха
Окраска Полимер
Слабо-розовая Розовая, переходящая в красную Медленно становится светло- коричневой Оранжевая, переходящая в красную, затем в корич- невую Светло-зеленая, буреет Медленно зеленеет Зеленая Медленно синеет, затем зе- ленеет Медленно желтеет Медленно зеленеет, синеет и буреет Отсутствует, иногда корич- невая Желтая, оливково-зеленая Оранжевая, слабо-коричне- вая Багряно-красная, красно- коричневая, коричневая Окраска не изменяется Оранжевая до кирпично- красной Красный цвет до пурпурно- красного, до зеленого или коричневого Оранжевая, коричневая, черная Светло-голубая, серо-зеле- ная Феноло-формальдегидные, фе- нол-фурфурольные, фурило- вые и резорцино-формальде- гидные смолы Эпоксидные смолы Полиметилакрилат Поливинилбутирали,поливинил- ацетали Поливинилформали Поливинилацетат Поливиниловый спирт Поливинилхлорид Поливинилиденхлорид Сополимеры винилхлорида с винилацетатом Полиэфирные смолы Метилцеллюлоза с различным содержанием метоксильных групп Бензилцеллюлоза Феноло-кумароно-инденовые, кумароно-инденовые смолы высокоплавкие Полиэтилен, полипропилен, по- лиизобутилен, поликарбона- ты, полиформальдегид, поли- амиды, полистирол, мочеви- но- и меламино-формальде- гидные смолы, политетрафтор- этилен, сополимеры винилхло- рида с эфирами акриловой и метакриловой к-т, хлориро- ванные дифенилы, бутадиен- нитрильные сополимеры, аце- тилцеллюлоза, ацетопропио- нат целлюлозы, ацетобутират целлюлозы, поли метилмета крилат Кумароно-инденовые смолы со средней темп-рой размягчения Кумароно-инденовые смолы мяг- кие Этилцеллюлоза Бутадиен-стирольный каучук
Таблица 4. Цветные качественные реакции иа каучуки
Реакция Вебера Реакция газообразных про- дуктов разложения Каучук
Реактивная бумага на НК Реактивная бумага для бутилкаучука
Красно-фиоле- товая Лиловая Коричневая Натуральный
Темно-синяя Зеленая Не окрашена Бутадиеновый
Желтая Желто-зеле- ная » » Бутадиен- стирольный
Не окраши- вается Не окрашена » » Бутадиен- нитрильный
Оливково-зе- леная ♦ ** Хлорсодержа- щий
Сиреневая Светло-сире- невая Желтая Бутилкаучук и полиизобу- тилен
* Обесцвечивание бумажки, смоченной хроматом или бихро-
матом калия. »» Синее окрашивание бумаги конге.
133
ИДОЗА - ИЗАТИН
134
О химич. методах И. в. с. см. также Элементарный
анализ, Функциональный анализ. Об определении мол.
веса см. Молекулярный вес высокомолекулярных соеди-
нений.
Об идентификации различных видов полимеров см.
в соответствующих статьях (напр., Белки, Полистирол,
Полиамиды п т. д.).
Лит.: Лосев И. П., Федотова О. Я., Практикум
по химии высокополимерных соединений, М., 1959; К о р-
ш а к В. В., Р а ф и к о в С. Р., Синтез и исследование вы-
сокомолекулярных соединений, М., 1949; Методы исследова-
ния полимеров, пер. с англ., М., 1961; В а й б е л ь С., Иден-
тификация органических соединений, пер. с англ., М., 1957;
К и в и л и с С.С., Техника измерения плотности жидкостей
и твердых тел, М., 1959; Лазарев А. И., Сорокин М. Ф.,
Синтетические смолы для лаков, М., 1953; Доброволь-
ская Н. Н„ Проворов В. Н„ Хераскова Е. П.,
в кн.: Труды НИИ резиновых и латексных изделий. Сб.
Ki 1, М., 1958, 140; К р е ш к о в А. II. [и др.], Анализ крем-
нийоргаиических соединений. М., 1954.
Ubaldinl I., Ind. plast. mod., 1956, 8, № 8, 45; II u s-
mann J., Lab.-Praxis, 1958, 10, № 8, 116; 1957, 9, № 7, 80;
Gel In R e n ё, Offic. matures plast., 1957, 4, Л5 41, 57;
Hummel D., Kunststoff — Rundschau, 1958, 5, H. 3, 85;
его же, Kunststoffe, 1956, 46, H. 10, 442; Roff W. J.,
Fibres, plastics; and rubbers. A handbook of the common poly-
mers, L., 1958; Feigl F., Anger V., Mod. Plast., 1960, 37,
№ 9, 147, 191; P a 1 i t S. R., Kunststoffe, i960, 50, H. 9, 513,
P a r r 1 s s W. H,. H о 1 1 a n d P. D., Brit. Plast., 1960, 33,
№ 8, 372; Jones R., Moyles A. F., Nature, 1961, 189,
Xi 4760, 222.
ИДОЗА C6H12O6, мол. в. 180,16 — одна из восьми
изомерных альдогексоз.
сно
I
носн
I
неон
I
носн
I
неон
I
сн2он
Открытая альдегидная
форма D-идозы
сн2он
Циклическая пиранозная
форма D-идозы
В кристаллич. виде И. не выделена; D-И. и L-И.
получены в виде сиропа; для D-H.[a]D = -f-53°
(равновесная система в воде). И. хорошо растворима
в воде, плохо — в большинстве органич. растворите-
лей. И. восстанавливает р-ры Фелинга и р-р AgNO3,
образует производные за счет альдегидной группы
(фенилозазон и др.). Озазон И. идентичен озазону
гулозы и сорбозы. При окислении альдегидной группы
D- и L-И. получаются одноосновные D- и L-и д о н о-
в ы е к-ты. При окислении и альдегидной, и первичной
спиртовой групп D- и L-И. образуются двухосновные
D- и L-и досахарные, или идаровые к-ты.
При восстановлении D-И. образуется шестиатомный
спирт D-и д и т (D-ид ито л), при восстановлении
L-И. — его антипод L- и д и т (L-и дитол, со р-
б и е р и т). При действии на И. щелочей она частично
изомеризуется в кетозу (сорбозу). При действии разб.
кислот из D- и L-И. образуются ангидриды соот-
ветственно D- и L-и д о з а н ы. Дрожжами И. не
сбраживается.
D- и L-И. получены синтетически; D-идоза — при
гидролизе полисахарида варианозы (наравне с D-
глюкозой, D-галактозой и L-альтрозой). L-И. встре-
чается в природе в виде производных — сорбиерита
и L-идуроновой к-ты, к-рая входит в состав нек-рых
физиологически активных полисахаридов, напр. хон-
дроитин-сульфата или дерман-сульфата; И. полу-
чается при гидролизе продукта восстановления этого
полисахарида боргидридом.
Лит.; The carbohydrates chemistry, biochemistry, phy-
siology. Ed. W. P i g m a n, N. Y„ 1957; P 1 g m a n w. W.,
Goepp R. M., Chemistry of carbohydrates, N. Y„ 1948;
Pigman W„ Nuslzawa K., Tsuikl S., Annual
Rev Biochem., 1959, 28, 25. Л. И. Линееич.
ИЕРВИН C27H3IO3N, мол. вес 427,27 — один из
главных алкалоидов растений, распространенных
в умеренном климате,
относящихся к раз-
личным видам чеме-
рицы (род Veratrum,
семейства Liliaceae).
Содержание И. в вы-
сушенных частях ра-
стения и различных видах чемерицы колеблется от
0,02% до 0,5%. Основание кристаллизуется из спирта
(призмы); т. пл. 243—245°; [a]D25 = —147° (в спирте);
хлоргидрат, т. пл. 330—334°; иодгидрат, т. пл.
302—305°; пикрат, т. пл. 247—284°. При стоянии
растворенного И. в метиловом спирте, насыщен-
ном НС1, превращается в изоиервин, т. пл.
114—116°. При внутривенном введении И. мышам
ЛД — 9,3 mcJks.
Лит.: Орехов А. П., Химия алкалоидов, 2 изд., М.,
1955, с. 710; Генри Т. А., Химия растительных алкалои-
дов, пер. с англ., М„ 1956, с. 735; Fried J„ К 11 n g s-
berg A., J. Amer. Chem. Soc., i953, 75, № 20, 4929;
В о i t H. G., Ergebnlsse der Alkaloid — Chemie bis 1960 unter
besonderer Berilcksichtigung der Fortschritte seit 1950, B.,
1960. А. Д. Кузовков.
ИЗАДРИН [алудрин, изопреналин, изупрел, хлор-
гидрат 1-(3', 4'-диоксифенил)-2-изопропиламиноэтано-
ла] СПН18О3МС1,
бесцветна7’ к^ H°-Q-CH0HCH2NHCH(СН3)2 ' НС!
Сталлы; растворим ' ДТ
в воде (1:3)испир- nu
те (1 :50), нерастворим в хлороформе и эфире; т. пл.
167—169°. Водные р-ры И. розовеют при действии света
и воздуха. При прибавлении к раствору И. (10 мг в 5 мл
Н2О) 1 капли р-ра FeCl3 появляется интенсивное зе-
леное окрашивание, переходящее при стоянии в олив-
ково-зеленое. И. получают конденсацией 3,4-ди-
окси-а-хлорацетофенона с изопропиламином и вос-
становлением образующегося при этом 3,4-диокси-
а-изопропиламиноацетофенона;
3,4-(ОН)2С8Н3СОСН2С1 + (CH3)SCHNH3 ->
н2
->-3,4-(OH)2CaH3COCH,NHCH(CH3)2 ->-
->• 3,4-(OH)2CeH3CHOHCH2NHCH(CH3)2
Подобно адреналину, И. обладает сильным брон-
хорасширяющим действием. В отличие от адреналина,
сосудосуживающее действие И. выражено очень
слабо. И. применяют ингаляционно для купирования
приступов бронхиальной астмы, а также при аст-
моидных и эмфизематозных бронхитах.
Лит..- Машковский М. Д., Лекарственные средства,
4 изд., М„ 1960; Epitome of the pharmacopeia of the United
States and National Formulary with comments, 10 ed., Phil. —
Montreal, [19551. А. И. Травин.
ИЗАТИН [лактам изатиновой (о-аминофенилгли-
оксалевой) кислоты] C8H6O2N, мол. в. 147,13 —
желто-красные призматич. кристаллы; т. пл. 203,5°;
растворим в ацетоне, бензоле, метиловом спирте
и горячей воде; мало растворим в холодной воде
и эфире. И. проявляет как слабоосновные, так и
кислотные свойства. Он образует с хлорной к-той
устойчивую соль состава C8H6O2N • НС1О4 • 2Н2О.
С другой стороны, И. взаимодействует с NaOH и КОН
с образованием солей:
В различных реакциях соли И. образуют производные,
соответствующие лактамной (I) или лактимной (II)
3»
135
ИЗАТИН
133
форме. Так, при действии йодистого метила на сереб-
ряную соль И. образуется О-метиловый эфир И. (III),
О
ОСН3
а при метилировании
натриевой или калие-
вой соли И.— 1-метил-
изатин (IV).
Ацилирование И.
приводит к 1-ацильным
производным. Карбо-
ния NH-группы электрофильный реагент вступает
в положение 5, а затем 7 бензольного ядра. Так был
получен 5-хлор-, 5-бром-, 5,7-дибро.м-, 5-нитроизатин,
5-изатинсул.ьфокислота и др. И. окисляет а-амино-
кислоты в альдегиды (Штреккера. реакция).
Впервые И. был получен окислением индиго хро-
мовой или азотной к-той. От оксиндола можно перейти
к II. галогенированием до 3,3-дигалогеноксиндола
и последующим гидролизом:
нильная группа в положении 3 изатинового цикла
вступает в обычные для кетонов реакции. При взаимо-
действии И. с гидроксиламином, фенилгидразипом
и т. п. реагентами образуется 3-изатиноксим, 3-иза-
тинфенилгидразон и другие соответствующие произ-
водные; синильная к-та дает 3-циангидрин И.
Под влиянием катализаторов И. конденсируется
с различными
V
ароматич. соединениями, образуя
3,3-замещенные оксиндолы, на-
пример (V).
С тиофеном в присутствии
серной к-ты И. образует индо-
фенин (см. Индофениновая реак-
Индолы, содержащие в бензольном ядре электроот-
рицательный заместитель, образуют И. при окислении
хромовой к-той, однако незамещенный И. таким путем
получить нельзя. Широко применяется для получения
И. синтез Зандмейера, позволяющий получать И.
с различными заместителями в бензольном ядре:
И. конденсируется с соеди-
нениями, содержащими актив-
ную метиленовую группу.
В мягких условиях в первую
очередь образуются производные диоксиндола (VI),
+ CCI3CH(OH)2+NH2OH
NH
затем превращающиеся в производные алкилиденокс-
индола (VII). От последних возможен переход или
к производным индола, напр. к триптамину (VIII),
или к производным хинолина (IX).
CH„CN
+ I 2
СООС2Н5
Второй метод Зандмейера — получение И. из произ-
водных тиомочевины:
C6H5NH4cs KCN _
С6Н5Г<Н' РЬС°3
При конденсации И. с кетонами в конц. щелочи
происходит размыкание изатинового цикла и обра-
зуется 4-хинолинкарбоновая к-та {Пфитцингера, ре-
акция). Нагревание раствора И. в щелочи приводит
к размыканию цикла; образуются соли изатиновой
к-ты; при подкислении они вновь образуют И.:
CO-COONa
nh2
Размыкание изатинового цикла происходит также при
окислении И., напр. при действии хромовой к-ты
образуется ангидрид
изатовой кислоты (X),
к-рый превращается
затем в антраниловую
к-ту. Хотя 2-карбо-
нильная группа И.
явлиется амидной, И.
свойственны нек-рые реакции а, р-дикетонов, напр. с
о-фенилендиамином И.образует индолхиноксалин (XI).
И. вступает в реакции нитрования, галогенирования,
сульфирования. В результате ориентирующего влия-
c6H5N
'СС\ — ,
C6H5NH (NHa)3S
Для получения N-замещенных И. используется
метод Штолле:
И. может быть получен также из о-нитробензоил-
хлорпда:
GCOCOOH
NHg
И. и а-изатинанилид являются ценными полупро-
дуктами в произ-ве кубовых индигоидных красителей.
11. применяют для открытия и фотометрия, определе-
ния тиофена в бензоле, пиррола, индикана и меркап-
танов в воздухе, для открытия Си и Ag.
Изатин -р-оксим C3H6O2N2, мол. в. 162,15-
зол отисто-желтые иглы, т. пл. 202° (с разл.), плохо
растворяется в воде и эфире, лучше —
[ ||--i=NOH в этиловом спирте, нерастворим в
^ОН бензоле; получают взаимодействием
N И. с гидроксиламином И водно-спир-
товой среде. Применяют для обнаружения U, Ag,
Со, Си, Fe, Pb, Ni и др. элементов; для определения
урава. При взаимодействии спиртового раствора
137
ИЗАФЕНИН — ИЗМЕЛЬЧЕНИЕ
138
изатин-[}-оксима с раствором соли ртути (II) или
уранила выпадает оранжевый осадок.
Лит.: Д ж у л и е н П., М е й е р Э., II р и н т и Э., в кн.?
Гетероциклические соединения, пер. с англ., т. 3, М., 1954, с.
150; X ю к к е л ь В., Теоретические основы органической хи-
мии, пер. с нем., т. 1, М., 1955, с. 237. М. Н. Преображенская.
С=О
I
КН
ИЗАФЕНИН (изацен, диаце- „г„гн
тилдифенилизатин) — белый по- ^нз°СО ОСОСН.
рошок со слабым запахом ук-
сусной кислоты; т. пл. 242°;
нерастворим в воде и спир-
те, растворим в кипящей ук-
сусной кислоте. При нагревании
И. в 0,5 н. спиртовом растворе
КОН появляется красное окра-
шивание. Изафенин получают следующим образом:
Свн5он
н2зо4
(СН3СО)2О -------С(С6Н4ОСОСН3)2
L X J>o
I
н
И. применяют в качестве слабительного средства.
Лит.: Государственная фармакопея СССР, 8 изд., М.,
1952; Машковский М. Д.» Лекарственные средства,
4 изд., М., 1960. А. С. Елина.
ИЗВЕСТНЯКИ — осадочные горные породы, со-
стоящие в основном из минерала кальцита
(СаСО3). Содержат переменное количество различных
примесей. Средний химич. состав: 5,19% SiO2; 0,06%
TiO2; 0,81% А1.3ОЭ; 0,54% Fe2O3; 7,89% MgO; 42,57%
CaO; 0,05% Na2O; 0,33% K2O; 0,77% H2O; 0,04%
P2O5; 41,54% CO2; 0,05% SO3; 0,23% Сорг. По мере
увеличения примеси карбоната магния И. переходят
через ряд промежуточных разновидностей в доло-
миты; с увеличением содержания глинистых частиц—
в мергели, а затем в известковистые глины; с ро-
стом содержания грубых обломочных частиц—в пес-
чаники. При перекристаллизации под воздействием
высокой темп-ры И. превращаются в мрамор.
II. .чаще всего образуются на дне морей в результате
накопления органич. остатков (напр., раковин) либо
путем химич. осаждения СаСО3 из морской воды.
Гораздо реже встречаются И., отложившиеся на дне
пресноводных водоемов. И. залегают обычно в виде
пластов. Они широко распространены среди отло-
жений различных геологич. систем, преим. последо-
кембрийских, слагая приблизительно 20% от общего
количества осадочных пород. И. используются как
строительные материалы, сырье для произ-ва извести
и известковых цементов, в качестве флюсов при
выплавке чугуна из железных руд, для известкования
кислых подзолистых почв, для получения углекислого
газа СО2, широко используемого в произ-ве соды,
в литографском произ-ве, для скульптурных работ
И т.'Д. А. Б. Ронов.
ИЗВЕСТЬ — вяжущее вещество, состоящее в ос-
новном из кальция окиси СаО, получаемое обжигом
известняка или мела. Чистая И. — продукт белого
цвета, мало растворима в воде (0,13% при 20°),
с повышением темп-ры растворимость падает; плотн.
3,4, т. пл. 2585°; при взаимодействии СаО с водой
образуется Са(ОН)2. Водный раствор И,—извест-
ковая вода — обладает щелочными свойствами.
Различают И. воздушную и гидравлическую.
Воздушную И. получают при обжиге из-
вестняков с малым содержанием глины (до 6%);
темп-ра обжига 1000—1200°. По виду обработки И.
делится на негашеную (комовую и молотую) и гаше-
ную (порошок — пушонку и тесто). По содержанию
примеси окиси магния в воздушной И. различают:
маломагнезиальную (до 5% MgO), магнезиальную
(5—20% MgO) и высокомагнезиальную (20—40%
MgO). По скорости гашения И. различают: быстро-
гасящуюся (до 20 мин) и медленногасящуюся (свыше
20 мин). И. бывает 1-, 2- и 3-го сортов; содержание
активных СаО и MgO (минимальная активность)
в негашеной И. должна соответственно быть не менее
85, 70 и 60%., содержание непогасившихся зерен —
не более 10, 20, 25%: минимальная активность 1-,
2- и 3-го сортов гашеной И.—пушонки, соответственно
67, 55, 50%, влажность — не более 5%. Тонкость
помола негашеной и гашеной И.: остаток на сите
№ 063 не более 2%, на сите № 009 не более 10%.
Прочность известково-песчаного раствора с гашеной
И. 5—10 кг/см1, с молотой негашеной И. 5—50 кг/сл«2.
Обжиг известняка и др. продуктов для получения
И. производится в известковообжиговых печах (шахт-
ных или вращающихся); при этом углекислый кальций
разлагается на СаО и СО8. Реакция протекает с по-
глощением тепла (425 ккал на 1 кг СаСО3). При га-
шении И. водой образуется Са(ОН)2, к-рый сравни-
тельно легко соединяется с углекислым газом воздуха
(особенно в присутствии влаги) с образованием СаСО3.
Воздушную И. можно превращать в порошок не только
помолом, но и путем гашения водой. Однако молотая
порошкообразная негашеная II. имеет ряд преиму-
ществ перед гашеной (ускоренное схватывание, твер-
дение и сушка). При помоле негашеной И. к ней до-
бавляют карбонатные породы (карбонатная И.),
а также другие минеральные добавки (шлаки, золы,
трепел, кварцевый песок, вулканические породы
и ДР-)-
Воздушная И. широко применяется в строительстве,
в химич. пром-сти (получение хлорной извести, соды
и др.), металлургии, сахарном и кожевенном
произ-вах, в с. х-ве, водоочистке и т. п.; в строи-
тельстве применяют в смеси с 2—4 частями песка.
И. употребляют и в смеси с различными вяжущими
веществами (портландцемент, строительный гипс) и
добавками. И. широко используют для произ-ва
силикатных автоклавных изделий и различных строи-
тельных деталей.
Гидравлическую И. получают обжигом
известняков с большим содержанием глины (6—20%)
при 900—1100°. Образующиеся при этом силикаты
(2СаО • SiO2), алюминаты (СаО • А12О3 • 5СаО • ЗА12О3)
и ферриты кальция (2СаО • Fe2O3) придают этой И.
способность длительно сохранять прочность в воде
(после предварительного твердения на воздухе);
гидравлич. И. отличается большей, по сравнению
с воздушной, прочностью, но меньшей пластичностью.
Гидравлич. И. используется в строительстве для тех
же надобностей, что и воздушная, а также для изго-
товления нек-рых видов бетона и растворов для кладки
сооружений, подвергающихся действию воды.
Лит.: Б у т т Ю. М., Технология цемента и других вяжу-
щих материалов, 3 изд., М., 1956; Юнг В. Н., Основы тех-
нологии вяжущих веществ, М., 1951; Осин Б. В., Негаше-
ная известь как новое вяжущее вещество, М., 1954.
Ю. М. Бутт.
ИЗМЕЛЬЧЕНИЕ — механическое разрушение твер-
дых тел с образованием мелких частиц измельченно-
го продукта. В отличие от дробления — полу-
чения крупных кусков продукта, И. приводит к обра-
зованию частиц размером порядка миллиметра и
мельче. По размерам (дисперсности) образующихся
частиц различают грубое (0,1—1 мм), среднее (0,01 —
0,1 мм) и тонкое (мельче 0,01 .и.и) И. Наиболее тонкое
И. (диспергирование) твердых тел эффективно осу-
ществляется в активной среде и приводит к образо-
ванию дисперсных систем — суспензии или, в пре-
деле, коллоидного р-ра, тонкого порошка (пудры,
139
ИЗМЕЛЬЧЕНИЕ
140
пыли). Задача И. — увеличение дисперсности твер-
дого материала. Часто при И. стремятся получить
продукт определённого гранулометрия, состава, раз-
делить агрегаты на первичные частицы, придать ча-
стицам продукта форму чешуек, хлопьев, зерен и т. п.
Основной характеристикой И. является степень
И. — отношение среднего размера зерен исходного
материала к среднему размеру частиц измельченного
продукта. Иногда степень И. характеризуется также
отношением уд. поверхностей или средних весов
частиц исходного и измельченного материалов. Сред-
ний размер б частиц, особенно при тонком И. и в слу-
чае полидисперсного материала, может быть оценен
по удельной поверхности продукта (см. Дисперс-
ность). Повышение степени И. сырья и полупродуктов
ведет к улучшению однородности смеси, к ускорению
гетерогенных реакций (произ-во удобрений, двуокиси
титана и др.), снижению применяемых темп-p и дав-
лений (варка стекла, синтез новых материалов),
сокращению расхода дефицитных составляющих, улуч-
шению структуры и повышению прочности материалов
(цементные бетоны, твердые сплавы, тонкая керамика,
огнеупоры и др.), повышению интенсивности и кра-
сящей способности красителей и пигментов, активности
катализаторов и т. д. В ряде случаев при чрезмерно
тонком И. твердых материалов возникают произ-
водственные трудности, связанные с увеличением
газо- и водопоглощения, образованием пористых
структур, препятствующих перемешиванию и уплот-
нению (см. Структурообрааование в дисперсных си-
стемах). Для преодоления этих затруднений в ре-
цептуру смеси иногда вводят добавки — поверхностно-
активные вещества, а также вносят изменения в па-
раметры последующих операций технология, процесса
(темп-ра, давление и др.).
И. материалов производят раздавливанием, уда-
ром, раскалыванием, истиранием и т. п. В современ-
ных измельчителях обычно сочетается два или более
видов таких воздействий. В зависимости от твердости
(табл. 1) и прочности измельчаемого материала, его
структуры, влажности, крупности, требуемой сте-
пени И. применяют различные методы и средства И.
(табл. 2, рис. 1, 2).
Таблица 1. Шкала твердости
Минерал Показатель по Моосу Практическая оценка
Тальк 1 Очень мягкий
Каменная соль . . , 2
Кальцит 3 Мягкие
Флюорит 4
Апатит 5
Полевой шпат .... 6 Твердые
Кварц 7
Топаз 8
Корунд 9 Очень
Алмаз 10 твердые
Для дробления и И. наиболее твердых материалов,
как правило, используют машины, в к-рых основными
являются раздавливающие и ударные воздействия.
Хрупкие материалы хорошо измельчаются также
раскалыванием, мягкие — истиранием, а при боль-
ших скоростях воздействий — ударом; при И. волок-
нистых материалов эффективны разрывающие уси-
лия. Для получения тончайших порошков и высоко-
дисперсных суспензий целесообразно применять из-
мельчители, характеризуемые высокой энергонапря-
женностью процесса И., — вибрационные, планетар-
ные, кавитационные и струйные мельницы.
И. может быть периодическим и непрерывным.
Периодический процесс И. исполь-
зуется при малых масштабах произ-ва, т. к. он срав-
нительно мало экономичен, сопровождается повышен-
ным нагреванием и агрегированием обрабатываемого
материала и характеризуется содержанием в измель-
Рис. 1. Основные виды дробилок и мельниц грубого из-
мельчения. а — щековая дробилка: 1 — неподвижная
щека; 2 — подвижная щека; б — конусная (гирационная)
дробилка: 1 — неподвижный конус; 2 — эксцентрично
вращающийся конус; 3 — вертикальный вал; в — валко-
вая дробилка; г — ударно-центробежная дробилка (мель-
ница); 0 — дезинтегратор.
ченном продукте переизмельченных и недоизмель-
ченных фракций. Непрерывное И. может
проходить в открытом и замкнутом цикле с после-
дующей или предварительной классификацией. При
открытом цикле И. также получается продукт
«широкого» гранулометрия, состава (содержащий зна-
чительные количества мелких и крупных фракций).
Наилучшие показатели по качеству продукта и энерго-
затратам достигаются при И. в замкнутом цикле,
с непрерывным отбором тонкой фракции, причем
схема с предварительной классификацией, как пра-
вило, применяется при содержании в исходном ма-
териале не менее 30—40% требуемой тонкой фракции.
С целью экономии энергии степень И. должна со-
ответствовать оптимальному значению требуемой дис-
персности; при высокой степени И. процесс проводят
ступенчато (последовательно в мельницах предвари-
тельного, среднего и тонкого И.). В зависимости от
требований произ-ва и свойств измельчаемого ма-
териала применяют сухое и мокрое И. Сухое И.
производится обычно в воздушной среде, а при пере-
работке окисляющихся, огне-, взрывоопасных и ток-
сичных материалов — в инертных газах. Мокрое И.
ведется в воде и в других жидкостях; использованием
добавок поверхностно-активных веществ можно по-
лучить особо высокодисперсные продукты.
Для выражения зависимости между затратами энергии и
результатами И. предложен ряд гипотез и эмпирич. соотно-
шений. По первому из них (Риттингер), справедливому для
достаточно тонкого И., уд. работа А, затрачиваемая при И.,
пропорциональна приросту уд. поверхности AS измельчаемого
материала:
А = KgAS кГ • см/кг (1)
где Kg — коэфф., выражающий затрату работы на создание
единицы поверхности.
По другому соотношению (Кирпичев, Кик), к-рое применимо
для грубого И. или дробления,
S
А = е In кГ • см/кг (2)
Ьо
где е = Р^/2Е — коэфф., выражающий работу предельной
упругой деформации Рп на единицу объема разрушаемого тела;
Е — модуль упругости, кГ/см*; S и 80 — уд. поверхности
измельченного и исходного материала.
141
ИЗМЕЛЬЧЕНИЕ
142
Неудовлетворительное выполнение на опыте соотношений
(1) и (2), особенно в области высокой дисперсности, привело
к появлению ряда других эмпирия, выражений, напр. (Бонд);
. . / 10 10 \ „
А= Ао ( — ----_z 1 кГ см/кг
Wp V FJ
(3)
где — Ао работа в квт-ч/т, необходимая для И. матери-
ала от теоретически бесконечно большого размера до 80%
прохода через сито 100 мк; Р и F — диаметры услов-
ных частиц продукта И. и исходного материала в мк, соот-
ветствующие размерам сит, через к-рые проходит 80% ма-
териала.
Соотношения (1), (2), (3) и ряд других математически обоб-
щаются (Рундквист, Чарльз) выражением вида
, . , dx
dA=b —
Хп
где X — линейный размер частицы, b и п — коэфф., зависящие
от свойств материала и метода И.
На основе исследования процесса мокрого И. кварца в виб-
рационной мельнице с учетом действия активной среды выве-
дено следующее уравнение (Ходаков), являющееся интеграль-
ной формой ранее предложенного выражения (Ребиндер):
А °= 4 Pj 1П < + Са + Pzsm) 1“ -§-°1 кГ см/кг (5)
A I U О Q \ U / -О!
где К — кпд измельчителя; е — плотность энергии предель-
ных упругих деформаций; с — коэфф, формы частиц; а — по-
стоянная; I — толщина слоя в с.и; р — средняя по толщине
слоя плотность энергии предельных пластич. деформаций,
кГ смА/ке; Sm — уд. поверхность предельно измельченного
порошка, см2/см“.
Из этого выражения при дроблении и грубом И. следует
соотношение (2), при достаточно тонком И. — соотношение (1),
а при очень тонком И.:
Sm- S.
А = ?lSm In -=---£ кГ . см/кг (6)
Из выражения (6) получено для этого случая след, уравнение
кинетики И.:
S = Sm (1 - е“ к^) (7)
где К2 — константа скорости диспергирования; t — продол-
жительность процесса.
Практически для расчета измельчителей разных типов
используются различные эмпирич. зависимости. Так, для наи-
более распространенного в технике измельчителя — шаровой
мельницы, производительность
Q — KiLDmm, кГ/час (8)
где К). — коэфф., зависящий от вцда измельчаемого материала
и параметров процесса измельчения; L *— длина барабана мель-
ницы; D — его диаметр и т — показатель степени, равный
2,4—2,6.
Мощность W, потребляемая шаровой мельницей, также
может быть рассчитана по приближенным эмпирич. формулам
типа:
VV === K$G V D кет (9)
где К2 — коэфф., зависящий от степени заполнения мельницы
измельчающими телами, числа оборотов и пр., G — вес измель-
чающих тел.
Рис. 2. Основные виды мельниц, а — Барабанная шаровая: 1 — корпус; 2 — бронированные плиты.
Мельница заполнена на 35—40% объема мелющими телами; измельчаемый материал располагается в межшаровом про
странстве. И. производится ударом и истиранием под действием падающих, вращающихся и скользящих мелюших тел.
б — Барабанная бесшаровая: 1 — корпус; 2 — приводная шестерня; з — диафрагма. Принцип ее дей-
ствия аналогичен шаровой, роль мелющих тел играют крупные куски измельчаемого материала, в — Вибрацион-
ная: 1 — электродвигатель; 2 — корпус с мелющими телами; з — дебаланс. Корпус мельницы совершает частые кру-
говые колебания в вертикальной плоскости; мелющие тела вращаются, сталкиваются, скользят по стенкам корпуса.
Кроме того, вся загрузка перемещается вокруг центральной трубы корпуса. Мелющие тела занимают 80—95% объема
корпуса, измельчаемый материал — все межшаровое пространство, з — Планетарная: 1 — планетарный привод;
2 — барабаны. Барабаны с мелющими телами и измельчаемым материалом одновременно вращаются вокруг своих
осей и центрального вала; в результате возникают большие ускорения, что обусловливает весьма интенсивное И.
б — Кольцевая: 1 — размольное кольцо; 2 — каток; з — крестовина; 4 — вертикальный вал. Измельчаемый ма-
териал попадает в зазор между горизонтальным и вертикальным кольцом и катящимися по его внутренней поверхности
крупными цилиндрами или шарами; разгрузка мельницы обычно пневматическая, е — Истирающая: 1 — прием-
ный бункер; 2 — питатель; з — жернова; 4 — приводной вал. И. материала происходит между неподвижным и вра-
щающимся дисками (жерновами). Исходный материал из приемного бункера подается питателем к центру вращающегося
диска, проходит в зазоре между жерновами к их периферии, ссыпается в нижяюю часть корпуса и оттуда — в прием-
ник готового продукта, ж — Пневматическая: 1 — разгонная труба; 2 — отбойная плита; з — классификатор.
Исходный материал подхватывается в разгонной трубе воздухом, нагнетаемым воздуходувкой, доводится до скорости
60—70 м/сек, измельчается при ударах об отбойную плиту и разделяется в воздушном классификаторе на тонкую
фракцию, к-рая отделяется в циклоне и фильтре, и на грубую фракцию, возвращаемую на доизмельчеяие. з — Струй-
ная: 1 — сопло; 2 — размольная камера; з — камера предварительной классификации. И. происходит за счет энергии
потока газа (напр., воздуха), сжатого в компрессоре до 7—14 ат, или перегретого пара. Исходный материал поступает
в мельницу в виде крупки и приобретает большую скорость за счет энергии энергоносителя, и — Кавитацион-
ная: 1—ротор; 2 — статор. Суспензия, подлежащая диспергированию, проходит через кольцевой зазор между
ротором и статором, причем сечение прохода все время то увеличивается, то уменьшается, вызывая изменение вели-
чины действующего давления. По окончании цикла готовый продукт отводится через разгрузочный кран.
143
ИЗМЕЛЬЧЕНИЕ - ИЗО
144
Таблица 2. Типы дробилок и мельииц и их технологические характеристики
Тип машин Преобладающий вид воздействия Крупность исходного материала, мм Тонина по- мола гото- вого про- дукта, мм Произво- дитель- ность, т/час, Расход электро- энергии, квт-ч/т Для каких материа- лов используется
Др обилки
Щековые Конусные (гирационные): Раздавливание 150-1800 25-280 Ю—ЮОО 0,2-0,7 Средней и высокой твердости
а) крупного дробления То же 150-1800 25—280 35—3500 0,15—0,5 То же
б) среднего дробления Валковые: » » 25-250 6—37 10—600 0,4-2 » »
а) с гладкими валками » » 6—75 3-15 3-150 0,7-1 Средней твердости и абразивных
б) с рифлеными валками .... Раскалывание, истирание 75-500 50-200 5—1000 0,15-0,4 Хрупких
Молотковые •: а) с жестко ук репленными мо-
лотками Удар 1,6—250 0,3-3 0,2-600 0,4-7 Абразивных
б) с шарнирно-укрепленными 0,06—50 0,05—400
молотками Удар и истирание 0,8—1000 0,7-150 Неабразивных
Мельницы Барабанные: 0,5-2
а) стержневые Истирание и удар 12-25 3-120 0,4-3 Абразивных
б) шаровые, галечные То же 0,6-25 0.08—1 0.5-75 7-15 То же
в) бесшаровые » .» 900 0,3—5 0,5-120 6-50 » »
Вибрационные:, . 0,5-8 5- 100а
а) сухого помола » » 0,1-10 10-50 От мягких до твер- дых абразивных
б) мокрого помола Истирание 0,2-3 0,1-30а 0,01-2 30-150 То же
Планетарные Истирание и удар 0,2-3 0,1—30а 0,01-0,02 20-100 От мягких до срав- нительно твердых
Кольцевые среднеходовые Раздавливание 0,8-25 44—800а 0,02-20 4-150 То же
Ножевые Резание 25-300 0,2—5 0,1-10 7-18 Мягких, волокни- стых, вязких
Истирающие Истирание 10-25 74-800а 0,2-0,5 11-150 Мягких, волокнистых
Пневматические Удар и истирание 2—25 0,1—0,5 0,1-25 25- 50 Ограниченной твер- дости с низкой темп-рой размягче- ния
Струйные То же 0,1-12 1- 30а 0,1-10 б Ограниченной твер- дости и волокнистых
Кавитационные Кавитация и истирание 0,1-0,2 5- 50а 0,1-3 10-100 Ограниченной твер- дости, для повы- шения однородно- сти суспензий
• Дробилки и мельницы; а мк; ® 1—4 кг пара или 6—9 кг сжатого воздуха на 1 кг продукта.
Для расчета производительности мельницы, продолжи-
тельности И. и дисперсности продукта определяют т. н. к о э ф ф.
измельчаемости (размалываемости), к-рый характе-
ризует сопротивляемость материала измельчению в мельнице
данного типа. Этот коэфф, определяется сравнением резуль-
татов, полученных при И. исследуемого и эталонного материала
в одной и той же лабораторной мельнице, для чего сопоста-
вляют: 1) уд. энергозатраты при И. испытуемого и эталонного
материала до одной и той же дисперсности; 2) величины остат-
ков на определенном сите или приростов поверхности при рав-
ных энергозатратах; 3) величины производительности мель-
ницы при доведении проб испытуемого и эталонного материала
до одинаковой дисперсности. Так как задачи И. — увеличение
поверхности, при определении коэфф, измельчаемости целе-
сообразно использовать первый из этих путей, определяя дис-
персность измерением уд.поверхности измельчаемого материа-
ла. Коэфф, измельчаемости зависит от конструктивных особен-
ностей мельницы, параметров И. и поэтому имеет ограничен-
ное значение в пределах опытных данных, полученных приме-
нительно к поставленной конкретной задаче.
Лит.: Андреев С. Е., Зверевич В. В., Пе-
ров В. А., Дробление, измельчение и грохочение полезных
ископаемых, М., 1961; Касаткин А. Г., Основные про-
цессы и аппараты химической технологии, 7 изд., Му 1960;
Левенсон Л. Б., Цигельный П. М., Дрооильно-
сортировочные машины и установки для переработки камен-
ных материалов, м., 1952; Mlttag С., Die Hartzerkleine-
rung, В. [u. а.], 1953; Моргулис М. Л., Вибрационное
измельчение материалов, М., 1957; Олевский В. А.,
Конструкции, расчеты и эксплуатация дробилок, М., 1958;
Перов В. А. иБранд В. Ю., Измельчение руд, М. —
Л., 1950; Ребиндер П. А.,. Ш р е й н е р Л. А., Ж и-
г а ч К. Ф., Понизители твердости в бурении, М. — Л., 1944;
Ромадин В. П., Пылеприготовление, М. —Л., 1953;
Ру.ндквист А. К^ Общая форма законов дробления,
Научно-техн, информ, оюл. н.-и. и проектного ин-та «Меха-
нобр», 1956, Хе 2; Charles R. J., Mining Engng, 1957, 9,
№ 1; Smekal A., Z. Verelnes dtsch. Ingr., 1937, 11, № 46;
Smith J. C., Chem. Engng, 1952, 59, № 8; T а г г a P т А. Ф.,
Справочник по обогащению полезных ископаемых, пер. с англ.,
т. 1—4, М., 1949—52; Циборовский Я., Процессы хи-
мической технологии, пер. с польск., Л., 1958; Хода-
к о в Г. С., Р е б и н д е р П. А., Коллоидн. ж., 1961, 23, Кг 4;
Фадеев В. И., Современное оборудование для дробления
и измельчения руд, Л., 1959; Bond F. С., Brit. Chem. Engng,
1961,6, Кг 6; Chem. Engng, 1962, 69, № 3. M. Л. Моргулис.
ИЗО — приставка, указывающая на наличие раз-
ветвления в жирной цепи органич. соединений. Напри-
мер, углеводород СН3СН(СН3)СН3 называют изобута-
ном, в отличие от изомерно-
го н-бутана СН3СН2СН2СН3
нормального строения. Не-
редко приставке «изо» при-
дают более широкий смысл:
она служит сокращенным
обозначением слова «изо-
мерный», причем речь может
идти об изомерии любого
типа. Так, изованилин (I)
отличается от ванилина (II)
СНО
1
относительным расположе- щ 1у
нием ОСНг и НО-групп; изо-
хинолин (III) изомерен хинолину (IV) с другим поло-
жением N в цикле; изоацетилен С=СН2 представляет
собой изомер ацетилена НС^СН и отличается от по-
145
ИЗОАМИЛАЦЕТАТ — ИЗОДИБЕНЗАНТРОН
146
следнего наличием двойной связи вместо тройной и
содержанием двухвалентного атома С; изокрото-
новая к-та находится в отношении цис-транс-изож-
рии к кротоновой к-те. Приставка «изо» широко при-
меняется для обозначения нек-рых изомеров сравни-
тельно сложно построенных соединений (алкалоидов,
терпенов И т. Д.). я- ф- Комиссаров.
ИЗОАМИЛАЦЕТАТ — см. Амилацетат и изо-
амилацетат.
ИЗОАМИЛОВЫЙ СПИРТ (пентанол-2) — см. Ами-
ловые спи'рты.
ИЗОБАРНЫЙ ПОТЕНЦИАЛ — см. Термоди-
намические потенциалы.
ИЗОБАРЫ РЕАКЦИИ УРАВНЕНИЕ — соотно-
шевие между константой равновесия, темп-рой и теп-
ловым эффектом химич. реакции при постоянном дав-
лении. И. р. у. выводится из изотермы реакции урав-
нения и Гиббса — Гельмгольца уравнения. Оцо являет-
ся вполне строгим, относится как к гомогенным, так и
к гетерогенным реакциям и представляется в виде
/д In Ка\ _________________ дн
\ ОТ /р RT1
где АН — изменение энтальпии в реакции, Т —
темп-ра в °К, R — газовая постоянная, Ка — кон-
станта равновесия реакции, выраженная через ак-
тивности участвующих веществ (в сильно разбав-
ленных р-рах константа равновесия может быть вы-
ражена через концентрации и обозначена ЙГС). В га-
зовых реакциях при условии применимости к ним
законов идеальных газов вместо Ка пользуются кон-
стантой равновесия Кр, выраженной через парциаль-
ные давления участвующих в реакции веществ.
Для практич. расчетов ур-ние (1) обычно должно
быть интегрировано. Если рассматриваемый темпе-
ратурный интервал невелик или если реакция не
сопровождается существенным изменением теплоем-
кости системы, то АН можно принять не зависящим
от темп-ры. Тогда интегрирование приводит к ур-нию
In АГа =-*«- +С (2)
где С — постоянная интегрирования. Из ур-ния (?.)
видно, что In Ка есть линейная функция от 1/т.
При необходимости учесть изменение АН с темп-рой,
его выражают с помощью закона Кирхгофа через
теплоемкости веществ, участвующих в реакции; по-
лученное выражение АН =f (t) подставляют в ур-ние
(1) и интегрируют его. И. р. у. широко применяется
при расчетах химич. равновесий.
Лит. см. при ст. Изотермы реакции уравнение.
В. А. Киреев.
ИЗОБАРЫ ЯДЕРНЫЕ — см. Изотопы.
ИЗОБУТИЛАЦЕТАТ — СМ. Бутилового спирта
эфиры.
ИЗОБУТИЛЕН (СН3)2С =СП2, мол. в. 56,11 —
бесцветный газ при комнатной темп-ре; т.нл. — 140,35°;
т. кип. —6,900°; давление пара: 989,0 мм рт. ст.
(0°); 3,444 ат (30°), 28,41 ат (125°). В пром-сти И.
получают каталитич. дегидрогенизацией изобутана
па окисных катализаторах (Сг2О3 на А12О3, U2OS
и др.) при 500—600°; его производят также дегидра-
тацией изобутанола, получающегося при восстанов-
лении СО водородом, а также дегидратацией первич-
ного изобутанола. Применяют И. для алкилиро-
вания ароматич. соединений и изобутана. И. легко
полимеризуется под действием кислых агентов. При-
меняют И. как компонент при совместной полимериза-
ции с различными соединениями, содержащими оле-
финовые связи. См. также Бутилены, Полиизобу-
тилен. Г. М. Цигуро.
ИЗОВЕРИН (дихлоргидрат N-изоамилкадаверина)
NH2(CH2)5NHCH2CH2CH(CH3)2 • 2НС1, мол. вес
245,24 — бесцветные кристаллы; т. пл. 293—295°
(с разл.); хорошо растворим в воде и спирте, плохо —
в эфире и ацетоне. Основание И., выделенное 0,1 н.
р-ром щелочи в присутствии ацетона, дает с нитро-
пруссидом натрия сиреневое окрашивание, быстро
переходящее в красно-фиолетовое. Количественно
И. определяют путем выделения его основания, от-
гоняемого с водяным паром в течение часа в прием-
ник с 0,1 н. H2SO4; избыток H2SO4 титруют NaOH
по метиловому красному. И. получают из капролак-
тама (I), при действии на к-рый NaOH образуется
6-аминокапроновая к-та (II); N-бензоильное произ-
водное этой кислоты превращают в амид (III) и далее
по реакции Гофмана — в монобензоилкадаверин (IV).
Последний алкилируют бромистым изоамилом; при
омылении полученного продукта (V) образуетсн И.
HN-(CH2)5-CO
C6HtCONH(CH2)5COOH
II
CeH3CONH(CH.,)5CONH2; C6H5CONH(CH.,)5NTH2
111 IV
C„H6CONH(CH2)5NHCH2CH2CH(CHS)S
V
И. по фармакологии, действию близок к алкалоиду
сферофизину, синтетич. аналогом к-рого он является.
Применяют И. в качестве родоускоряющего средства
и для ускорения сокращения мускулатуры матки
в послеродовом периоде.
Дит.: Дозорцева П. М., Фармакология и токсико-
логия, 1955, 18, № 6, 17; Р е м б е з И. Н., Акушерство и
гинекология, 1957, № 3, 37. Л. Н. Яхонтов.
ИЗОДЕКСТРОПИМАРОВАЯ КИСЛОТА С10Н,0О3,
мол. в. 302,44 — бесцветные кристаллы, хорошо
растворимые в большинстве органич. растворителей,
нерастворимы в воде. И. к. (I) вместе с декстропима-
ровой к-той (II) относится к кислотам смоляным
и входит в состав канифоли.
l«]D
Т. пл. ме-
тилового
эфира, “С
Изодекстропимаровая кислота 162,4 0,0 61,5—62
Декстропимаровая кислота . . 219 82,5 69
Обе кислоты, в отличие от других смоляных к-т,
медленно окисляются на воздухе. Соли щелочных
металлов растворимы в воде и спирте, но соли декстро-
пимаровой к-ты в этих растворителях растворимы
труднее, чем такие же соли других смоляных кислот.
Обе к-ты содержатся (в количествах от 10 до 20%)
в бальзамах различных хвойных, в том числе в жи-
вице сосны и ели обыкновенной. И. и. Бардышвв.
ИЗОДИБЕНЗАНТРОН (изовиолантрон) С^Н^Оз,
мол. в. 456,47 — темно-фиолетовый порошок; раст-
воряется в нитробензо-
ле, давая красно-фио-
летовый р-р, обладаю-
щий коричневой флуо-
ресценцией; очень мало
растворим в обычных
органич. растворителях; раствор в конц. серной
к-те — зеленого цвета. И. окисляется, галогенирует-
ся, нитруется гл. обр. в крайних бензольных ядрах.
В пром-сти И. получают щелочным плавлением бен-
зантрона', сплавлением 3-галоген-.исго-бензантронов
147
ИЗОДИМОРФИЗМ — ЙЗОЛЕЙЦИН
148
с КОН при 120—140°; известны и др. способы полу-
тения И. Применяют И. в качестве красителя (ин-
цантрен фиолетовый). Производные И., в частности
галогенозамещенные и метоксипроизводные, также
обладают свойствами красителей и для этой цели
ПРОИЗВОДЯТСЯ прОМ-СТЬЮ. Л. С. Поваров.
ИЗОДИМОРФИЗМ — см. Изоморфизм.
ИЗОИНДОЛЫ — соединения (I), для которых воз-
можна таутомерная структура псевдоизоиндола (II).
Простейшие производные этого класса мало изучены
и трудно доступны. Описан N-метилизоивдол, к-рый
образуется при действии фениллития на бромистый
N, N-диметилизоиндолиний:
К производным И. относятся фталоцианиновые кра-
сители, фталимидины (III) и фталимиды.
Лит. см. при ст. Индол.
ИЗОИОННАЯ ТОЧКА (изопротонная
точка) — концентрация водородных ионов в рас-
творе белка, при к-рой белок поглощает из раствора
одинаковое количество ионов Н+ и ОН-. Это опреде-
ление приложимо не только к белкам, но и к другим
амфотерным полиэлектролитам, напр. к полилизин-
полиглутаминовой к-те, к сополимерам акриловой
к-ты и 2-винилпиридина и др. Вследствие равного
поглощения обоих видов ионов белок или другой
амфотерный полиэлектролит при внесении в такой
раствор не изменяет его pH, что является одним из
методов экспериментального определения И. т. Дру-
гой метод определения И. т. заключается в том, что
к белковому р-ру добавляют различные количества
кислоты или щелочи и измеряют равновесное pH
водородным или стеклянным электродом. По кривым
титрования белков измеряют ход поглощения ионов
Ит и ОН- в зависимости от pH и определяют значение
pH, при к-ром поглощение обоих ионов равно нулю
(кривая поглощения пересекает ось pH).
Существует также много косвенных методов определения
И. т. — по минимуму растворимости, вязкости, набуханию,
оптич. вращению, поглощению кислых и основных красителей,
по максимуму мутности и др. В чистых водных р-рах, в к-рых
заряд белковых молекул определяется только поглощением
ионов Н+ или ОН-, И. т. совпадает с точкой нулевого заряда
молекул или с изоэлектрической точкой. В этом случае положе-
ние И. т. на шкале pH определяется для 1—1-валентного амфо-
терного электролита ур-нием Михаэлиса:
рни. PKa pKb+7Kw
или _______
[Н+]и. т. =^/"Kw
где Ка — константа диссоциации карбоксильной группы,
Кь — то же — для аминогруппы и К — произведение диссо-
циации воды (pKw = 13,9). В белке, где содержится несколько
карбоксильных и аминогрупп с различными значениями кон-
стант диссоциации, положение И. т. определяется Ка и Кь
наиболее сильных групп.
В растворах электролитов при преимущественной
адсорбции белками анионов солей (Cl~, J-, SO42- и др.)
часть ионов ОН- из белка вытесняется в раствор
и, поэтому И. т. смещается в сторону более щелочных
pH, а при преимущественной адсорбции катионов
солей (Ca2+, Mg2+ и др.) и вытеснении части Н+ ионов
из белка в раствор, соответственно, — в сторону более
кислых значений pH. В растворах солей И. т. сме-
щается в противоположном направлении по сравнению
с изоэлектрич. точкой. В большинстве случаев это
смещение не превышает 0,3—0,6 pH. Смещение И. т.
в щелочную сторону (иногда на 0,5 pH) наблюдается
также при денатурации белков. Химич, обработка
белков, приводящая к удалению аминогрупп (дез-
аминирование, обработка формальдегидом и др.),
вызывает значительное смещение И. т. в кислую
сторону (иногда до 2 pH). И. т. является основной
электрохимия, константой белков и протеидов; по-
ложение И. т. для ряда белков см. в ст. Изоэлектри-
ческая точка. Измерения И. т. часто применяются
для характеристики состояния тканевых простых
белков и протеидов (напр., нуклеопротеидов) при
различных физиология, процессах. А. г. пасынский.
ИЗОКСАЗОЛ C3H3ON, мол. в. 69,06 — бесцветная
жидкость с резким запахом, подобным пиридину;
т. кип. 95—95,5°; йр 1,0843; ограниченно
растворим в воде (1 : 6 при 20°), хо- HCj:—3СН
рошо — в органич. растворителях; обла- HC5.,-3N
дает слабыми основными свойствами; °
с хлористым кадмием дает кристаллич. комплекс
C3H3ON • CdCl2. И. обладает ароматич. характером;
однако химич. свойства И. изучены только на примере
его производных (метилизоксазолов и др.). Так,
нитрование 3-метилизоксазола приводит к 4-нитро-
3-метилизоксазолу. Водородом в момент выделения
или каталитически возбужденным ядро И. разры-
вается по связи N—О:
н с-1 !! СНЗ ~~ сн3~с-сн=с-сн3
° О NH2
И. получают взаимодействием NH2OH с пропар-
гиловым альдегидом или диэтилацеталем. Ядро И.
(изоксазолина) входит в состав нек-рых природных
соединений (см., напр., циклосерин). Структурным
изомером И. является оксазол. В- Н. Фросин.
ИЗОЛЕЙЦИН (а-амино-р-метил-н-валериановая ки-
слота, а-амино-Р-метил-6-этилпропионовая кислота)
сн3сн2 сн-сн соон
I । , мол. в. 131,17 — содержит два
CH3NH2
асимметрия, атома углерода; известна в виде четы-
рех оптически активных изомеров, отличающихся по
физич. свойствам.
Изомеры Т. пл., °C Г“1о (в воде) W D |в 20 % НС1)
L-mpeo-И золсйцин О-трео-Изолейцин L-алло-Изолейцин О-алло-Изолейцин 285—286 (с разл.) 283—284 (с разл.) 278 (с разл.) 274—275 (с разл.) 4-10,7° — 10,7° + 14,0° — 14,2° + 40 ,8° —41 ,6° + 38 ,1° —38 ,0°
И. — твердый продукт, растворим в воде, щелочах,
углекислых солях щелочных металлов и в горячей
уксусной к-те; трудно растворим в горячем спирте,
нерастворим в эфире; рй^ 2,36, рй’2 9,68 (при 25°),
р/ 6,02. И. дает общие реакции на аминокислоты
(нингидриновую и др.). Определение И. производят
микробиология, и хроматография, методами. И. —
структурный элемент многих белков растительного
и животного происхождения; может быть выделен из
природных продуктов. Синтетически И. может быть
получен аминированием а-бром-[5-метилвалериановой
к-ты, а также из а-оксиамино-р-метил-н-валериановой
к-ты и др. способами.
Лит. см. при ст. Аминокислоты и ст. Лейцин.
Л. П. Алексеенко.
149
ИЗОЛИРОВАННЫЕ СИСТЕМЫ - ИЗОМЕРИЗАЦИЯ
150
ИЗОЛИРОВАННЫЕ СИСТЕМЫ — системы, ли-
шенные возможности обмена веществом и энергией
с окружающей средой и находящиеся при постоянном
объеме. Последнее условие вызывается тем, что из-
менение объема всегда связано с затратой работы.
Наиболее характерным свойством таких систем яв-
ляется то, что их внутренняя энергия, в соответствии
с законом сохранения энергии, не может изменяться
ни при каких процессах, происходящих в системе.
В И. с., согласно второму закону термодинамики,
необратимые процессы всегда сопровождаются воз-
растанием энтропии системы dS>0, а пределом само-
произвольного их протекания (т. е. протекания без
затраты работы извне) и, следовательно, условием
равновесия является достижение максимального зна-
чения энтропии системы (т. е. условие dS = 0 и
rf25<0).
ИЗОЛОГИЧЕСКИЕ РЯДЫ — группы углеводоро-
дов и их производных с одинаковым числом углерод-
ных атомов в радикале и тождественными функцио-
нальными группами, но с различной степенью не-
насыщенности, т. е. с возрастающим в каждом И. р.
количеством кратных (двойных, тройных) связей
в радикале. Постоянной количественной разностью
между изологами является 2Н или кратная ей вели-
чина. Простейший И. р. углеводородов: этан С2Н6,
этилен С2Н4 и ацетилен С2Н2. Этот ряд, а также ряды
его функциональных производных, напр. пропионо-
вая к-та СН3СН2СООН, акриловая СН2=СНСООН,
пропиоловая СН=ССООН исчерпываются тремя чле-
нами. По мере усложнения углеводородного радика-
ла число изологов в рядах возрастает, например:
С6Н14—2Н С6Н12—2Н — С6Н10—2Н — С6Н8—2Н-*
->С6Нв ... до С6Н2. Число изологов в ряду, следова-
тельно, находится в прямой зависимости от числа
атомов углерода в радикале.
Для определения принадлежности какого-либо со-
единения к определенному И. р. недостаточно принять
во внимание только соотношение С и Н в радикале,
но необходимо учитывать и его структурный тип.
Так, у циклич. соединений (карбо- и гетероцикли-
ческих) существуют свои И. р., напр.: циклогексан
(I) —► циклогексен (II) —>- циклогексадиен (III) —► бен-
зол (IV), если последний изображать как циклогексэ-
^СН2 Сн2 СН2 СН
Н2С СН2 -2Н Н2С CH _2Hj Н2С СН -2Н НС СН
H2C^JCH2 Н2С^^СН НС^Н НЧ/£Н
СН2 СНа СН СН
II III IV
трпен. В химич. отношении различия между изоло-
гами не только количественного порядка, но и в за-
висимости от типа кратных связей и их положения
в молекуле, иногда резко качественного характера.
Так, в полиенах изолированное, кумулированное
и сопряженное расположение двойных связей, при
одной и той же степени ненасыщенности, обусловли-
вает различную реакционную способность и различ-
ный тип реакций присоединения.
Понятие об И. р. впервые было установлено в
1844—45 Ш. Жераром. См. также Гомологические
ряды. р- р. Галле.
ИЗОМЕРАЗЫ — группа ферментов, катализирую-
щих внутримолекулярный перенос водорода.- К ним
относятся И., катализирующие превращение альдозо-
фосфат кетозофосфат (И. триозофосфата, фосфо-
глюкозы, фосфоманнозы, фосфорибозы и др.), и И.,
катализирующие взаимопревращение свободных аль-
доз и кетоз (И. ксилозы, катализирующая превра-
щение D-ксилоза D-ксилулоза; И. маннозы, И.
эритрозы и др.). К И. также относится глиоксалаза I,
катализирующая реакцию: метилглиоксаль -|- HS-глу-
татитон ZZ S-лактилглутатиоя. В эту же группу
входят И., осуществляющие таутомеризацию: тауто-
мераза фенилпирувата (кето-форма д: енольная фор-
ма) и И. стероидов, катализирующая превращение
Д6-3-кетостероиды zt Д4-3-кетостероиды.
К И. относят иногда также ферменты, катализи-
рующие изменение стерич. конфигурации различных
соединений, в частности различные рацемазы, осу-
ществляющие рацемизацию а-амино- и а-оксикислот,
и галактовальденазу, катализирующую обратимое
превращение галактозы в глюкозу, формально пред-
ставляющее собой вальденовское обращение при
четвертом атоме углерода гексозы:
В состоянии равновесия в этой системе содержится
25% галактозы. Коферментом галактовальденазы
является дифосфопиридиннуклёотид (ДПН+). И. трио-
зофосфата катализирует обратимое взаимопревраще-
ние фосфодиоксиацетона и 3-фосфоглицеринового аль-
дегида:
СН^ОН СНО
I I
СО -Z снон
I I
СН2-О-РО(ОН)2 СН2-О-РО(ОН)2
Равновесие в этой системе устанавливается при от-
ношении фосфодиоксиацетона к 3-фосфоглицерино-
вому альдегиду 22 : 1. Оптимум действия при pH
7—8. Константа Михаэлиса для 3-фосфоглицеринового
альдегида ЙГМ = 3,9 • 10~4М; активность очень вы-
сока: 1 моль фермента при 26° катализирует превра-
щение 945 000 молей субстрата в минуту. Активность
фермента подавляется ионами фосфата. Фермент
широко распространен в животных тканях и играет
важную роль в процессе гликолиза, обеспечивая ути-
лизацию фосфодиоксиацетона.
И. фосфоглюкозы также является ферментом глико*
литич. процесса, катализирует обратимое взаимопре-
вращение: В-глюкозо-6-фосфатХВ-фруктозо-6-фосфат;
равновесие устанавливается при содержании 68%
D-глюкозо-б-фосфата и 32% £>-фруктозо-6-фосфата;
оптимум действия при pH 9, актявность исключи-
тельно велика. Фермент очень широко распространен
в природе.
Исследование механизма указанного ферментатив-
ного превращения в присутствии D2O показало, что
изомеризация, по-видимому, происходит через про-
межуточное образование ендиола
НС(ОН)=С(ОН)(СНОН)3СН2ОН.
Лит.: Нейландс Д ж., Шту мп ф П., Очерки по
химии ферментов, пер. с англ., М., 1958, с. 309; Fermente,
Hormone, Vitamine und die Beziehungen dieser Wirkstoffe
zueinander, hrsg. R. Ammon, W. Dirscherl, Bd 1, 3 Aufl., Stutt-
gart, 1959, S. 410; Meyerhof О., в кн.: The enzymes, ed.
by J. B. Summer and K. Myrback, v. 2, pt. 1, N. Y., 1951; Top-
per Y. J., J. Biol. Chem., 1957, 225, № 1, 419.
В. Б. Спиричев.
ИЗОМЕРИЗАЦИЯ — превращение органич. соеди-
нения в соединение другого строения или с иным рас-
положением атомов или групп в пространстве без
изменения состава и молекулярного веса.
Известны изомерные превращения различного типа,
в результате к-рых изменяется углеродный скелет
молекулы, меняются характер функциональных групп
или их положение в молекуле, происходит сужение
или расширение цикла и др.
151
ИЗОМЕРИЗАЦИЯ - ИЗОМЕРИЯ
152
Примером И. с изменением скелета атомов углерода
может служить превращение нормальных парафи-
нов под действием А1С13 в углеводороды изостроения,
напр. СН3СН2СН2СН3 —► (СН3)2СНСН3. К этому же
типу И. относятся расширение или сужение цикла али-
циклич. соединений; так, углеводороды ряда цикло-
пентана при действии А1С13 на холоду изомеризуются
в соотв. циклогексановые углеводороды, напр. метил-
циклопентан (I) в циклогексан (II), см. ниже; при нагре-
вании И. протекает в противоположном направлении.
Алициклич. углеводороды, содержащие более 6 ато-
мов С, в присутствии А1С13 изомеризуются только с су-
жением цикла, напр. циклогептанвметилциклогексан.
Во многих случаях при И. меняется положение
функциональных групп в молекуле. Так, при действии
А1С13 на первичные бромиды образуются вторич-
ные бромиды, например СН3СН2СН2Вг —» (СН3)2СНВг.
Сюда же относятся распространенные случаи И.
с перемещением или перераспределением кратных
связей. Так, нагревание бутена-1 СН3СН2СН=СН2
(550°) ведет к образованию симметричного буте-
на-2 СН3СН=СНСН3; при нагревании со спиртовым
р-ром щелочи бутин-1 СН3СН2С=СН переходит в
бутин-2 СН3С=ССН3; при нагревании изопентина
(СН3)2СНС=СН со спиртовой щелочью образуется
изомерный углеводород (СН3)2С=С=СН2 алленовой
структуры (А. Е. Фаворский).
Можно назвать много изомерных превращений,
ведущих к изменению характера функциональных
групп. Так, а-окиси можно легко превратить с рас-
крытием цикла в альдегиды (напр., окись этилена —►
ацетальдегид). От эфиров фенолов можно легко пе-
рейти к изомерным оксикетонам, напр. С6Н6ОСОСН3-+
--►ге-НОС6 Н4СОСН3 (см. Фриса перегруппировка). Нестой-
кие С-нитрозосоединения легко изомеризуются в ок-
симы, напр. CH3CH(NO)CH3 —► (CH3)2C=NOH. Оксимы
под действием кислотных агентов изомеризуются в ами-
ды к-т: R2C=NOH —► RCONHR (см. Бекмана перегруп-
пировка). Перегруппировка-
U-CH3 циклич. оксимов, например
*" I J превращение циклогексанон-
оксима (III) в е-капролактам
' " О (IV) сопровождается расши-
0 рением цикла.
Во многих случаях удается
' осуществить взаимные пере-
(|| |v ходы между геометрия, изо-
мерами. Так, малеиновая к-та
цис-строения под каталитич. действием НС1 или НВг
изомеризуется в фумаровую к-ту транс-строения.
С другой сто роны, У Ф-облучение водных растворов фу-
маровой к-ты (V) ведет к ее И. в малеиновую к-ту (VI);
в re-ксилол, дающий при окислении терефталевую
к-ту; с другой стороны, фталевую и изофталевую
к-ту можно изомеризовать в терефталевую — один
из исходных продуктов для производства синтетич.
волокна «терилен». Многие перегруппировки пред-
ставляют собой практически важные процессы И.,
напр. Бензидиновая перегруппировка, Бекмана пере-
группировка, Арбузова реакциям яр. Я.Ф.Комиссаров.
ИЗОМЕРИЯ — явление, заключающееся в суще-
ствовании нескольких соединений с одинаковым коли-
честв. и качеств, составом и одинаковым молекуляр-
ным весом, но различающихся по физич. и химич.
свойствам; такие соединения называют изомерами.
И. чрезвычайно распространена в органич. химии
и служит одной из основных причин разнообразия
и многочисленности органич. соединений.
Известны два основных вида И.: структурная и про-
странственная (стереоизомерия). Структурные изо-
меры отличаются порядком связей атомов в моле-
куле. Стереоизомеры при одинаковой структуре
различаются расположением атомов в пространстве.
Структурная изомерия. Этот вид И. подразделяется
на несколько разновидностей: И. скелета, И. поло-
жения и др. Изомерия скелета зависит от
различия в порядке связи углеродных атомов, обра-
зующих скелет молекулы. В случае насыщенных
углеводородов гомологии, ряда СпН2П+2 такая И.
возможна, начиная уже с 4-го члена ряда; помимо
н-бутана СН3—СН2—СН2—СН3, существует и изо-
бутан (СН3)2СНСН3. Для 5-го члена этого ряда, имею-
щего состав С6Н12, возможны 3 изомера — н-пентая
(I), изопентан (II) и тетраметилметан (III), с макси-
мально разветвленной цепью атомов углерода:
сн3-сн2-сн3-сн3-сн3 сн3- сн-сн2-сн3
сн3
сн3-с-сн,
СН3
III
С удлинением ряда число возможных
изомеров очень быстро возрастает.
Так, в случав углеводородов соста-
ва C13H2S возможно существование
802 изомеров, для углеводородов
С20Н42 возможны более 366 000 изомеров и т. д.
В случае циклопарафинов состава С„Н21 И. скелета
может заключаться в различном числе атомов угле-
рода в кольце; напр., метилциклопентан (IV) с 5 ато-
мами С в цикле изомерен циклогексану (V) с 6 атомами
в кольце. Кроме того, существуют циклич. соединения
с боковыми цепями, И. к-рых обусловлена различием
в порядке связей атомов углерода в боковой цепи,
напр. H-nponHn-(VI) и изопропилциклогексан (VII).
н-с-соон н-с-соон
II II
ноос-с-н н-с-соон
V VI
Q-CH3
IV
Ароматич. кетоксимы типа ArC( = NOH)R, существую-
щие в двух стереоизомерных формах, при действии
брома, к-т и других реагентов превращаются в более
стабильную форму, тогда как УФ-облучение ведет к И.
с образованием менее стабильного геометрия, изомера.
Механизмы изомеризационных процессов (нередко
обратимых, с применением разных катализаторов)
могут быть самыми различными. Направление про-
цесса в отдельном случае определяется большей ста-
бильностью образующегося изомера в условиях опыта,
обычной тенденцией к образованию систем с сопря-
женными кратными связями и т. д.
Мвогие процессы И. имеют большое практич. зна-
чение. Так, процессы переработки вефти (крекинг,
пиролиз, риформинг и др.) сопровождаются И. угле-
водородов. Извсства изомеризация о- и .«-ксилола
Другой вид структурной И. — изомерия по-
ложения — состоит в том, что при одинаковом ске-
лете атомов углерода функциональные группы могут
занимать различное положение. Так, в н-пропиловом
спирте СН3СН2СН2ОН спиртовая группа ОН находится
при крайнем первич-
ном атоме С, тогда
как в изомерном изо-
пропиловом спирте
СН3СНОНСН3 группа
ОН связана со сред-
ним вторичным ато-
мом С. Известны 3 изо-
мерных двухатомных фенола с различным относитель-
ным расположением О-Н-групп: пирокатехин (VIII),
резорцин (IX) и гидрохинон (X).
153
ИЗОМЕРИЯ
Структурные изомеры могут, кроме того, разли-
чаться между собой характером функциональных
групп. Примерами могут служить ацетон СН3СОСН3
и пропионовый альдегид СН3СН2СНО, окись этилена
и ацетальдегид и т. д. Естественно, что такие изомеры
гораздо больше различаются физич. и химич. свойст-
вами, чем изомеры положения с одинаковыми функ-
циональными группами.
Особым случаем И. является широко распростра-
ненная в органич. химии таутомерия (равновесная
динамич. И.), когда два изомера, различающиеся
функциональными группами, легко переходят друг
в друга до достижения равновесия, напр.:
СН3СОСН2СООС2Н5 CH;iC;OHe. CHCOOG.H-,
Известны многочисленные случаи, когда различия
в изомерных соединениях объясняются и характером
функциональных групп и порядком связи углеродных
атомов. Так, соединению состава С2Н6О соответствует
как этиловый спирт СН3СН2ОН, так и диметиловый
эфир СН3—О—СН3.
Иногда различают еще одну разновидность струк-
турной И., т. и. метамерию, при к-рой изомерные
соединения отличаются по положению гетероатома
в цепи* напр. метилпропиловый CHSOCH2CH2CH3 и
диэтиловый СН3СН2ОСН2СН3 эфиры; метилпропиламин
CH3NHCH2CH2CH8 и диэтиламин CH3CH2NHCH2CII3
И т. Д.
Пространственная изомерия. К этому виду И. от-
носятся; геометрическая И. (цис-транс- изомерия)
и оптическая И. Геометрическая изо-
мерия наблюдается в соединениях, содержащих
двойные связи или плоские циклы. В случае соеди-
нений с двойной связью возможность геометрич. И.
возникает при наличии двух неодинаковых замести-
телей у каждого из атомов,соединенных двэйнойевязью.
Такая И. обусловлена жесткой конфигурацией по-
добных молекул, где свободное вращение атомов угле-
рода с двойной связью невозможно. Вследствие этого
два заместителя могут расположиться по одну пли по
разные стороны плоскости, к-рую мысленно можно
провести через двойную связь, напр.:
а—С—b а—С-Ь
И II
а-С—b 5 —С—а
цис-форма транс-форма
боксильными группами,
транс-форма):
н-с-соон
fl
н-с-соон
малеиновая к-та
и фумаровая кислота —
ноос-с-н
II
н-с-соон
фумаровая к-та
Геометрич. изомеры заметно отличаются по своим
физич. свойствам: цис-изомеры плавятся ниже, ови
значительно лучше растворимы в воде (табл. 1).
Таблица 1
Соединение Т. пл, °C Раствори- мость при 25° (г/100 г воды) Константа диссоциа- ции при 25°, Ki Теплота сгорания, ккал/моль
Малеиновая к-та
(цис)........ Фумаровая к-та 130 78,8 . 1,17-10~2 326
(транс) 286 • 0,7 9,3-10-4 320
uuc-Коричная к-та 68 14,43 1,4-10-4 1047
транс-Коричная к-та 133 0,1 3,5-10-5 10*0
Геометрич. изомеры существенно различаются и по
дипольным моментам; транс-соединения обычно имеют
дипольные моменты, близкие к нулю, тогда как ди-
польный момент цис-изомеров явно выражен (напр.,
для цис-дихлорэтилена 1,8) £>). Геометрич. изомеры
различаются нек-рыми химич. свойствами; так, обра-
зование ангидрида из к-ты НООС—СН=СН—СООН
происходит только при пространственной близости
карбоксильных групп, т. е. в случае малеиновой к-ты.
Из фумаровой к-ты в жестких условиях можно полу-
чить малеиновый ангидрид вследствие предваритель-
ной изомеризации исходной к-ты. Из двух геометрич.
изомеров один (в большинстве случаев цис-форма)
является более стойким. Нагревание и воздействие
небольших количеств различных реагентов (J2, гало-
геноводородные к-ты, HNO3, РС15 и др.) ведут к полу-
чению более стойкого изомера. С другой сторовы,
облучение УФ-лучами способствует превращению
стойкого стереоизомера в лабильную форму.
Геометрич. И. наблюдается также в случае соеди-
нений с двойной связью между атомами С и N, если
у атома С имеются два различных заместителя. В ка-
честве примеров можно привести альдоксимы, не-
симметричные кетоксимы, гидразоны и др.:
Подобного рода жесткую конфигурацию можно
вообразить, исходя как из квантово-химич. представ-
Рис, 2
ся соединением тетраэдров
ц препятствует свободному
лении о характере л-свя-
зи (см. рисунок в ст.
Двойная связь), так и
из тетраэдрич. моделей
Вант-Гоффа.
Плоскость, проходя-
щая через параллельные
оси р-электронов, пер-
пендикулярна плоско-
сти, в к-рой расположе-
на вся молекула (рис. 1,
где заштрихована пло-
скость л-связи). В тетра-
эдрич. моделях двой-
ная связь осуществляет-
ребрами (рис. 2), что
вращению атомов угле-
с6н5-с-Н
II
НО —N
анти-бензальдонсим
c0n5-c-h
II
N-OH
син-бензальдоксим
Установлена геометрич. И. и в случае соединений
с двойной связью между атомами азота, напр. азо-
соединений, азоксисоединений:
c6h5-n
II
cuu-азобензол
C6H3-N
II
n-c6h5
анти-а зо бензол
C0H5-N=O
A-CeN5
транс-азоксибепзол
Аналогичные отношения наблюдаются и в случае пло-
рода.
Типичным примером цис-транс-изомерии может
служить двухосновная кислота со структурной фор-
мулой НООС—СН=СН—СООН, существующая
в двух изомерных формах (малеиновая кислота —
цис-форма, с пространственно сближенными кар-
скостных циклич. систем, где цикл препятствует сво-
бодному вращению.
Примером может слу-
жить циклопропаи-
дикарбоновая к-та,
существующая в двух
формах (XI и XII).
Цис-форма (XI), в от-
СООН
Н
Н
XI
СООН
li СООН
XII
личие от транс-изо-
мера, подобно малеиновой к-те, легко дает ангидрид.
Транс-форма (XII) не может быть совмещена со своим
зеркальным изображением и поэтому возможно рас-
155
ИЗОМЕРИЯ
156
щеплеиие XII на оптически деятельные формы.
На этом примере видно, что в циклич. соединениях
часто наблюдаются оба вида пространственной И. —
геометрия, и оптич.
Геометрия. И. наблюдается и в случае производных
циклобутана и циклопентана, нек-рых конденсиро-
ванных циклич. соединений, напр. декалина и др.,
содержащих плоское или почти плоское кольцо.
Оптическая изомерия является след-
ствием пространственной асимметрии молекул и
проявляется в способности вращать плоскость по-
ляризованного луча (см. Активность оптическая,
Антиподы оптические).
Нек-рые неорганич. соединения в кристаллич. состоянии
(кварц, хлорат калия и др.) обладают оптич. активностью.
Однако асимметрия подобных соединений обусловлена их
кристаллич. решеткой, а не конфигурацией молекул. При
переходе в раствор или в неупорядоченное жидкое состояние
их оптич. активность исчезает, тогда как оптич. И., вызывае-
мая молекулярным строением, не связана с агрегатным состоя-
нием и с формой кристаллов.
В молекулах оптически деятельных соединений
обычно содержится один или несколько асимметри-
ческих атомов углерода, соединенных с 4 различными
атомами или группами атомов. При наличии одного
асимметрия, атома углерода возникает возможность
образования двух прост-
ранственных изомеров, от- J’ 5
носящихся друг к другу,
как несимметричный пред- / \\ \
мет к своему зеркальному / / аЛ--~ \
изображению, напр.:
Типичными примерами d_MpBa
соединении, содержащих
один асимметрия, атом С, являются молочная кислота
*
СН3СНОНСООН (звездочкой отмечен асимметрия, атом
*
углерода), глицериновый альдегид СН2ОНСНОНСНО
и др., существующие в двух формах. Физич. свойства
(темп-ры кипения и плавления, плотность и т. д.),
а также химич. свойства обоих изомеров одинаковы,
но один из них вращает плоскость поляризации света
влево (1 или (—)-форма), а другой (d или (-(-)-форма)
на такой же угол вправо. Такие изомеры называются
оптич. антиподами.
Растворы равных количеств антиподов лишены
способности вращать плоскость поляризованного луча.
В них образуются непрочные продукты присоедине-
ния антиподов, т. наз. рацемия, соединения, или
рацематы, к-рые по физич. свойствам резко отли-
чаются от оптич. антиподов. Рацематы обычно обоз-
начаются как d,1-формы.
При синтезах, в результате к-рых могут образо-
ваться оптически активные соединения, всегда полу-
чаются рацематы, т. к. шансы образования каждой
из двух активных форм одинаковы. Разработан ряд
способов расщепления рацематов на оптич. антиподы,
основанных на непосредственном разделении асим-
метрия. кристаллов, на преимущественном усвоении
определенными бактериями одной из двух активных
форм и, яаще всего, на химич. разделении при помощи
оптически активных веществ, напр. путем солеобра-
зования.
Несмотря на близость химич. свойств антиподов,
они нередко различаются по физиология, действию.
Так, d-аспарагин NH2COCH2CH(NH2)COOH сладок,
а 1-аспарагин безвкусен; природный 1-адреналин
3,4-(OH)2C6H3CHOHCH2NHCH3 значительно более
активен, чем d-адреналин и др. Это явление объяс-
няется тем, что многие субстраты, с к-рыми реаги-
руют антиподы в организме, также обладают несим-
метрич. строением.
В случае соединений типа abcC—Cdef с двумя
асимметрия, атомами углерода число возможных
изомеров (без учета рацематов) равно .4 (22). Дей-
ствительно, если обозначить эти атомы С с неодина-
ковыми заместителями через А и В и учесть знак
вращения, то возможны следующие 4 изомера:
4-А -А + А -А
+ в -в —в +в
Аналогичным образом при наличии 3 асимметрия-
атомов возможны 8 (23) изомеров и в общем виде
при п асимметрия, атомах число возможных оптич.
изомеров (не считая рацематов) равно 2я. Если нек-рые
из асимметрия, атомов структурно одинаковы, то
число возможных изомеров меньше 2я. Так, в случае
двух асимметрия, атомов углерода с одинаковыми
заместителями возможны следующие три изомера:
+ А —А 4-А
-|- А — А — А
I II III
Соединения типа I и II оптически активны и в рав-
ной мере вращают плоскость поляризованного луча
в противоположных направлениях. В соединении
III одна половина молекулы обладает (-(-^конфигура-
цией, а вторая (—^конфигурацией; молекула про-
странственно симметрична и лишена оптич. активности
вследствие внутренней компенсации. Такие соедине-
ния (мезоформы), в отличие от рацематов, в принципе
нельзя разделить на оптически деятельные формы.
Типичным примером рассматриваемых соединений
являются винные к-ты НООССНОНСНОНСООН.
Для соединений такой структуры установлено суще-
ствование рацемата, D-, L- и мезоформ.
Из табл. 2 видно, что обе активные формы винной
к-ты обладают одинаковыми физич. свойствами, к-рые
сильно отличаются от свойств как рацемата, так и
.иеао-формы.
Таблица 2
Свойство D-форма L-форма Рацемат Мезо- форма
Т. пл., °C 170 170 206 140
Плотность 1,76 1,76 1,687 1,666
Растворимость, г в 100 г воды при 15° 139 139 20,6 125
Константа ионизации: Ki 0,00117 0,00117 0,0011 0,00077
к2 0,000059 0,000059 0,000058 0,000016
Обозначения оптич. антиподов при помощи букв
1- и d-соответственно для левовращающего (—) и
правовращающего (-|-) изомеров во многих случаях
приводит к противоречиям. Так, при осторожном
окислении (-|-)-глицеринового альдегида образуется
левовращающая глицериновая к-та, к-рая может быть
превращена в амид или эфир с правым вращением.
Знак вращения для одного вещества может меняться
в зависимости от темп-ры, концентрации и природы
растворителя. Таким образом, одинаковый знак вра-
щения не может служить подтверждением сходства
пространственной конфигурации. Поэтому в настоя-
щее время применяют прописные буквы D и L для
обозначения конфигураций соединений, связанных
генетич. переходами и обладающих одинаковым про-
странственным строением.
Вопрос об абсолютной конфигура-
ции оптически активных соединений, т. е. о дей-
ствительном их расположении в пространстве, долгое
время оставался неясным. Напр., из двух форм гли-
церинового альдегида форма I была произвольно
обозначена D-формой, а форма II — L-формой. Впо-
следствии на основании данных вращательной дис-
персии и рентгеноструктурного анализа было уста-
новлено, что первоначальное предположение деа-
157
ИЗОМЕРИЯ — ИЗОМЕРИЯ ЯДЕРНАЯ
158
ствительно верно. Аналогичным образом была уста-
новлена абсолютная конфигурация нек-рых амино-
кислот и других оптически активных соединений:
оно сно
н-с-он но—с—н
I I
СН2ОН СН2ОН
D-форма L-форма
Установлено много случаев оптич. И., обусловлен-
ной несимметричностью молекулы в целом при отсут-
ствии асимметрия, атомов. Так, у производных ди-
фенила с заместителями большого размера в орто-
положениях затрудняется или делается
,——к невозможным свободное вращение бен-
\ У~\ / зольных колец вокруг общей оси между
ними. При этом оба кольца располага-
ются в разных плоскостях, молекула при отсутствии
асимметрия, центров не имеет плоскости симметрии
и возможны две формы, относящиеся друг к другу
как предмет к своему зеркальному изображению.
Это явление получило название апгропоизомерии.
Аналогичным образом нет плоскости симметрии и
А А
в замещенных алленах типа ).С=С=С\ , которые
в хв
можно расщепить на антиподы. Такой случай оптич.
И. был предсказан Вант-Гоффом еще в 1875 и под-
твержден в 1935 на примере 1,3-дифенил- или 1,3-ди-
нафтилаллена. Далее, в спирановых структурах
(XIII—XVI) кольца находятся в перпендикулярных
0сх] ОО Сс0 оо
XIII XIV XV XVI
плоскостях, молекула несимметрична и может быть
разделена на оптически деятельные формы.
Оптич. И. наблюдается также у соединений, содержа-
щих вместо асимметрия, атома углерода другие атомы с кова-
лентными связями, направленными к углам тетраэдра (атомы
Si, Sn, N, Р, As, S, Se, Pt и Др.). Кроме того, оптич. И. воз-
можна и в случае атомов октаэдрич. конфигурации (А1, Сг,
Zn, As, Pt и др.) и плоской конфигурации (Ni, Pt, Pd). Приме-
рами подобных соединений, разложенных на оптически дея-
тельные формы, могут служить
с6н5ч ГН2СеН5
>Si<
«-CjHj/ 'CH2C6H1SO3H
С2Н5Ч + ZCH.,CH=CH21
>N< J-
СН/ \CH2CeH5 J
И. различного типа наблюдается и в случае нек-рых
неорганич. соединений. Так; известны О- и N-гидроксил-
аминосульфокислоты HOSO2ONH2 и HOSO2NHOH.
Широко представлена И. в области комплексных
соединений. Гексагидратам хлористого хрома СгС13,
различающимся цветом, легкостью дегидратации
и способностью отщеплять С1_ при реакции с
AgNO3, приписывают строение:
[Сг(ОН2)е]С13 [Сг(ОН2)5С1]С12-Н2О [Сг(ОН2)4С12] С1-2Н2О
фиолетовый светло-зеленый темно-зеленый
Помимо структурной и пространственной И., из-
вестна также особого рода И., т. наз. изомерия пово-
ротная, объясняющаяся большей или меньшей за-
торможенностью свободного вращения даже вокруг
простых связей, напр. в этане. Переход структурных
и пространственных изомеров друг в друга осуще-
ствляется путем разрыва и возникновения связей
между атомами; энергетически неравноценные пово-
ротные изомеры превращаются друг в друга только
за счет свободного вращения. См. также Конформа-
ционный анализ.
Лит.: Хотинский Е. С., Стереохимия, Харь-
ков, 1950; Виттит Г., Стереохимия, пер. [с нем.], М.—Л.,
1934; Бартон Д., в об.: Перспективы развития органиче-
ской химии, пер. с нем. и англ., М., 1959; S h г 1 n е г R. L.,
Adams Й., Marvel С. S., в кн.: Organic chemistry, v. 1,
N. Y. —L., 1943. Я. Ф. Комиссаров.
ИЗОМЕРИЯ ПОВОРОТНАЯ — явление, заклю-
чающееся в существовании изомеров, образующихся
при несвободном внутреннем вращении групп атомов
вокруг ординарной связи. Подробнее см. Конформа-
ционный анализ.
ИЗОМЕРИЯ ЯДЕРНАЯ — существование атомов
с ядрами в метастабильном (относительно долгоживу-
щем) состоянии. Атомы с одинаковым составом ядра,
но находящиеся в различных энергетич. состояниях и
потому имеющие различные ядерные свойства, наз.
ядерными изомерами. И. я. была открыта в 1921
О. Ханом, показавшим, что при бета-распаде изотопа
тория Th234 (UX,) образуются ядра атомов изотопа
протактиния Ра234 с различными периодами полу-
распада:
их2 (Т1у2 = 1,175 JKUH) и UZ (Т1уо = 6,66 час)
И. я. у радиоактивных изотопов стабильных элемен-
тов была открыта в 1935—36 И. В. Курчатовым с со-
трудниками при исследовании искусственной радио-
активности у брома.
Причина И. я. состоит в следующем. При бомбар-
дировке атомного ядра различными частицами или
в результате ядерных превращений ядро может полу-
чить дополнительное количество энергии и перейтп
из нормального (основного) состояния с наименьшей
энергией на какое-нибудь из дискретных энергетич.
состояний с большей энергией, называемых возбуж-
денными уровнями ядра. Обычно время жизни в воз-
бужденном состоянии очень мало и ядро переходит,
иногда через ряд промежуточных уровней с меньшей
энергией, в основное состояние ядра. Однако у ряда
атомных ядер существуют один или два метастабиль-
ных уровня, на к-рых ядро может находиться отно-
сительно долгое время. В этом случае и будет наблю-
даться И. я. Т. к. различие между возбужденными и
метастабильными состояниями заключается только
во времени жизни ядра, то четкой границы между
ними нельзя провести. Поэтому к изомерам обычно
относят ядра с периодами полураспада, к-рые еще
можно экспериментально измерить (Ti/2 = 10 '12 —
10~14 сек, в нек-рых случаях до 10~17 сек) или выби-
рают какое-нибудь произвольное значение в каче-
стве нижней границы Ti/2. Так, на цветной вклейке
в ст. Изотопы помещены все изомеры стабильных и
радиоактивных ядер с Ti/2 > 1 миллисек. Из таблицы
видно, что Т>/2 у изомеров может иметь самые раз-
личные значения: от очень малых до нескольких лет.
Для того чтобы время жизни ядра в метастабильном
состоянии было достаточно большим, нужно, чтобы
между метастабильным
состоянием и уровнем,
на который переходит
ядро, была бы сравни-
тельно небольшая раз-
ность энергии и боль-
шая разность механич.
моментов (спинов).
Атомное ядро в мета-
стабильном состоянии
имеет избыток энергии
по сравнению с ядром
в основном состоянии,
поэтому и масса изоме-
ра больше массы ядра в
нормальном состоянии
(рис.) на ДМ = &Е1сг
(где ДЕ — разность энергии между этими состояниями,
ас — скорость света). При изомерном переходе
в основное состояние или на один из промежуточных
уровней ядро освобождается от избытка энергии
159
ИЗОМОЛЯРНЫХ СЕРИЙ МЕТОД
160-
посредством испускания гамма-лучей или передачи
энергии одному из электронов атомной оболочки
(внутренняя конверсия). Ряд изомеров превращается
не посредством изомерного перехода, а путем бета-
распада (с испусканием электрона или позитрона или
путем электронного захвата). Нек-рые изомеры могут
превращаться как путем бета-распада, так и посред-
ством изомерного перехода. Напр., 'из рис. видно,
что 95% атомов изомера (с Ti/2 = 4 час) изотопа
In115 (Ti/a = 6 • 1014 лет) превращается посредством
изомерного перехода в основное состояние In116,
а 5% превращается в изотоп Sn116 с испусканием
электрона (Рт). Кроме того, известны случаи (Ро211,
Ti'2 = 25 сек) а-распада изомера, а для трансурано-
вых элементов, вероятно, возможен и распад изомера
посредством спонтанного деления (см. Радиоактив-
ность). Изомерный переход может сопровождаться
разрушением химич. связей, и это обстоятельство
можно использовать для разделения изомеров.
Изомеры обычно обозначаются символом изотопа
с буквой т около массового числа, указывающей на
то, что ядро находится в метастабильном состоянии.
Можно также отмечать изомер знаком 0 и др. спосо-
бами. Напр., И. In116 можно обозначать символом:
InII5m или О In115. Возможность изомерии у различ-
ных изотопов и время жизни изомера зависят от рас-
пределения и характеристик ядерных уровней, муль-
типольности перехода и степени заполнения нуклон-
ных оболочек в ядре. Поэтому исследование И. я.
имеет большое значение для развития теории строе-
ния атомных ядер.
Лит.: Курчатов И. В. и Русинов Л. И., Изоме-
рия атомных ядер, в кн.: Юбилейный сборник, посвященный
тридцатилетию Великой Октябрьской социалистической рево-
люции, ч. 1, М. — Л., 1947; Корсунский М. И., Изо-
мерия атомных ядер, М., 1954; Гамма-лучи, М. — Л., 1961.
И. П. Селинов.
ИЗОМОЛЯРНЫХ СЕРИЙ МЕТОД (непрерывных
изменений метод, Остромысленского — Жоба метод) —
способ определения состава соединения в р-ре без
выделения в кристаллич. виде (или более точно —
способ нахождения стехиометрия, коэфф, при взаимо-
действии двух или более веществ в р-ре). Часто
используется также для доказательства наличия
химич. взаимодействия между компонентами в р-ре.
Эксперимент заключается в том, что р-ры двух, трех
(или более) компонентов реакции смешивают в раз-
личных отношениях, но при условии, что сумма об-
щих концентраций компонентов остается постоянной;
концентрация других веществ, присутствующих в р-ре,
а также pH и др. условия должны быть постоянными.
Измеряют численные значения к.-л. свойств таких
р-ров в зависимости от состава; экстремальное значе-
ние измеряемого свойства наблюдается при соотно-
шениях концентраций компонентов, соответствую-
щих стехиометрия, коэффициентам ур-ния реакции.
Если один или неск. компонентов также имеют свой-
ства, качественно аналогичные свойствам образую-
щегося соединения, используют различные варианты
измерения отклонения от аддитивности.
В большинстве простых случаев изучают реакцию
между двумя компонентами А и В. Готовят их рас-
творы одинаковой молярной концентрации, далее
готовят серию смесей этих р-ров так, чтобы соотно-
шение объемов р-ров компонентов изменялось, но
суммарный объем оставался постоянным. Напр.,
готовят следующие 11 р-ров;
Раствор А, мл 10 9 8 ................. 1 0
Раствор В, мл 0 i 2................ 9 10
Затем измеряют численные значения к.-л. свойства
каждого раствора серии, напр. оптич. плотность
в определенном участке спектра, электропроводность,
экстрагируемость и т. п. На оси абсцисс откладывают
Рис. I. Диаграмма состав —
свойство для компонентов А
и В. Кривые 1, 2 и 3 соответ-
ствуют образованию соедине-
ний АВ, АВ, и АВ,. Кри-
вая з а рассчитана для случая
отсутствия диссоциации или
разложения соединения АВ,.
S
4 т п в
Рис. 2. Схематическая диаг-
рамма равновесий системы
А — В — растворитель (S).
отношение объемов исходных р-ров Л и В, а на оси
ординат — измеренные численные значения свойств.
Полученные диаграммы «состав — свойство» имеют
максимум (или минимум),
точка находится при таком
соотношении объемов рас-
творов компонентов А и В,
к-рое равно молярному от-
ношению реагирующих в
данных условиях компо-
нентов. Кроме того, полу-
чение кривой с экстре-
мальной точкой при про-
стых стехиометрич. отно-
шениях компонентов яв-
ляется обычно доказатель-
ствомтого, что компоненты
реагируют друг с другом
в данных условиях; кон-
центрация, pH, раствори-
тель и т. п.
Метод в фотометрия, ва-
рианте был разработан
Остромысленским, позже
Жобом, к-рые показали,
что положение экстремума по отношению к оси абсцисс
изомолярной серии не зависит от диссоциации (или
разложения) образующегося в растворе соединения.
Заметная диссоциация или другие причины непол-
ного взаимодействия компонентов А и В в р-ре при-
водят лишь к понижению и размытости максимума
(пунктирная линия 3 а, рис. 1, соответствует пол-
ному образованию, а линия 3 — неполному образо-
ванию того же соединения).
Другой способ обоснования метода и всех выводов
следует из принципов физико-химич. анализа по
Курнакову; при этом уста-
навливается также связь
с др. методами определёния
состава соединения в рас
творе. На рис. 2 схемати-
чески .показана диаграмма
для системы А, В, S, где
S’ — растворитель. В обла-
сти SAmpk доминирует
свободный компонент А и
лишь частично образуется
АВ. В области kpmnrq до-
минирует соединение АВ,
а в области qrnB — соеди-
нение АВ2. Изомолярная
серия соответствует ли-
ниям, параллельным стороне А — В. Линии тр и
пг направлены к вершине 5, но далее изгибаются
параллельно стороне AS при концентрациях, численно
равных ступенчатым константам (K,q....) комплексов
АВ, АВ2......
Метод имеет ряд ограничений. Пользуясь рассма-
триваемым методом, можно найти только коэфф,
уравнения реакции между А и В, но нельзя судить
о составе соединения. Так, при изучении взаимодей-
ствия между сульфатом металла и серной к-той полу-
чается кривая типа 1 (рис. 1). Однако максимум может
быть обусловлен образованием как иона HSO4", так
и комплекса H2[Me(SO4)2]. Далее, при комплексооб-
разовании между А и В обычно образуется неск,
соединений; АВ, АВ2, АВ3, ... В зависимости от
абсолютных и относительных концентраций, а также
от pH и др., в растворе доминирует то или другое
соединение (см. рис. 1). Поэтому, напр., серия а—Ь
будет показывать преимущественно образование АВ,
а серия а'—Ь' (при более конц. р-рах) может указы-
вать либо на соединение АВ, либо на АВ2, или
161
ИЗОМОРФИЗМ - ИЗОНИТРИЛЫ
162
же максимум может получиться при нек-ром про-
межуточном соотношении объемов. Положение мак-
симума по отношению к оси абсцисс может также
зависеть от того, измеряется ли свойство, харак-
терное для АВ или для АВ2. Метод неприменим,
когда в системе протекают гидролиз, полимеризация
и др.
Лит.: 1) Ostromisslensky I., Вег., 1911, 44,
268; 2) J о b Р., Ann. chimie, 1928, 9, 113; 3) Б а б к о А. К.,
Физико-химический анализ комплексных соединений в раст-
ворах, Киев, 1955; 4) Т а н а н а е в И. В., Изв. Сектора физ.-
хим. анализа, 1950, 20, 277; 5) К о м а р ь Н. П., Ж. неорг.
химии, 1956, 1, вып. 6, 1243. А. В. Бабко.
ИЗОМОРФИЗМ — свойство атомов, ионов или мо-
лекул замещать друг друга в кристаллах (частица
на частицу). При этом образуются смешанные
кристаллы переменного состава — твердые растворы
замещения. Общие условия И.: 1) Достаточно малое
различие объемов частиц (атомов, ионов, молекул),
замещающих друг друга в кристалле (не более 15%).
2) Замещающие друг друга частицы должны быть спо-
собны к образованию близкого по характеру типа
химич. связи.
Изоморфно замещающие друг друга атомы и ионы
могут иметь одинаковую валентность (изовалент-
н ы й И., напр. КС1 и КВт) или разную (гетеро-
валентный И., напр. SrSO4 и KBF4; BaSO4 и
КМпО4). В последнем случае идет одновременно
замещение не менее двух видов частиц. В окрестности
замещающей частицы одного вида (напр., иона К)
для компенсации заряда или для создания нуж-
ных геометрических соотношений должны быть заме-
щающие частицы второго вида (ионы MnOf). А. Е.
Ферсман, В. И. Вернадский, В. Соболев составили
ряды изоморфно замещающих друг друга химич. эле-
ментов.
И. — понятие, введенное Митчерлихом в 1819, пер-
воначально означало сходство внешней кристаллич.
формы у веществ, родственных по химич. составу,
напр. KHAsO4 и КНРО4; КН2РО4, KH2AsO4 и
NH4H2PO4. Однацо кристаллы, имеющие сходную
внешнюю форму (т.н. изогональные кри-
сталл ы), не всегда родственны по химич. составу и
наоборот. Исследование внутреннего строения кри-
сталлов показало, что часто вещества, близкие по
химич. составу, имеют одинаковую кристаллич.
структуру, сходное пространственное расположение
частиц — атомов или ионов (изоструктур-
ность), и поэтому — сходную внешнюю форму.
Следовательно, И. в его первоначальном понимании
является непосредственным следствием изоструктур-
ности веществ. Напр., И. в ряду: MgCO3, СоСО3,
FeCO3, ZnCO3, МпСО3, СаСО3 является следствием
принадлежности этого ряда карбонатов к структур-
ному типу кальцита. В. М. Гольдшмидт предложил
называть вещества с одинаковой кристаллич. струк-
турой, т. е. вещества, принадлежащие к одному
структурному типу, изоморфными в широком смысле.
И. в таком понимании все более заменяется термином
«изоструктурность». Изоморфными являются изо-
структурные вещества, образующие твердые р-ры
замещения.
И. присущ далеко не всем изоструктурным веще-
ствам, так как последние могут резко различаться по
химич. природе и размерам частиц.
Напр., изоструктурные кристаллы CsCl и CuZn, принадле-
жащие к одному и тому же структурному типу и имеющие
одинаковую внешнюю форму, не являются изоморфными. Они
ire образуют смешанных кристаллов, кристаллы одного из них
Не могут вызывать кристаллизацию другого. Отсутствие И.
здесь объясняется слишком большой разницей, в величине
атомных радиусов и в характере связи.
• Вещества, образующие иепрерыиный ряд смешанных
кристаллов ~ твердых p-ров (совершенный И.), не-
избежно изоструктурны. Один из примеров совер-
шенного И. — кристаллы квасцов KA1(SO4) 12НаО,
в к-рых ионы К+ могут в любых количествах заме-
щаться ионами Rb+, NH% а ионы А13г — ионами Fe3+,
Сг3', имеющими приблизительно одинаковые размеры.
Нек-рые изоструктурные вещества образуют смешан-
ные кристаллы в ограниченных пределах (несовер-
шенный И., напр. Mg2SiO4 и Ca2SiO4; BaSO4 и КМпО4).
Если неизоструктурные вещества образуют между
собой твердые р-ры в ограниченных пределах, то
такие вещества наз. изоди морфным и. Это
вещества с несовершенным И., например, мине-
рал сфалерит (Zn,Fe,Mn)S содержит изоморфные
примеси Fe и Zn, причем FeS и ZnS являются
неизоструктурными, а следовательно, изодиморф-
ными.
Изодиморфные в-ва иногда могут образовывать
твердые р-ры при любом соотношении компонентов,
создавая видимость существования непрерывного ряда
твердых р-ров. Это происходит, когда непрерывное
изменение размещения атомов относительно друг
друга в результате изменения состава вызывает по-
степенный переход одного структурного типа в дру-
гой. Напр., в системе LiCl—MgCl2 с увеличением со-
держания MgCl2 совершается непрерывный переход
от структурного типа NaCl, свойственного LiCl,
к типу CdCl2, свойственному MgCl2. В этом случае,
как и в ряде других, одновременно с замещением идет
процесс внедрения (вычитания) частиц, т. е. имеет
место И. с заполнением пространства.
В органич. кристаллах «кирпичами», элементами
кристаллич. структуры являются группы из боль-
шого числа атомов — молекулы, стремящиеся образо-
вать плотнейшую упаковку с одновременным сохра-
нением в кристалле возможно более высокой симме-
трии. Если в неорганич. соединениях атомы и ионы
можно рассматривать как сферы и они отличаются
друг от друга в подавляющем большинстве случаев
только объемом, то в органич. соединениях молекулы,
как правило, отличаются и формой. При образовании
твердых р-ров оргапич. веществ идет замена молекулы
на молекулу, при этом химич. природа атомов мало
сказывается на межмолекулярном взаимодействии,
а основную роль играют объемные и геометрия, соот-
ношения (форма молекул).
Если неперекрывающиеся объемы двух молекул имеют
в сумме наименьшее значение Д, а перекрывающиеся — т.
то С = 1 — Д/т наз. степенью И. молекул, и для образования
непрерывных твердых р-ров величина С не должна превы-
шать 0,9.
Сокращение межмолекулярных расстояний в ре-
зультате деформации кристаллич. структуры из-за
различной формы молекул не должно превышать
0,4—0,5 А, а искажение валентных углов тоже должно
быть незначительным. При И. имеет место уменьше-
ние свободной энергии смешанного кристалла с бес-
порядочным распределением частиц примеси по срав-
нению со свободной энергией кристаллов чистых
компонентов.
Лит.: Гольдшмидт В. М., Кристаллохимия, пер.
с нем., М., 1937; Менделеев Д. И., Соч., т. 1, Л., 1937;
Вернадский В. И., Очерки геохимии, 4 изд., М., 1934;
Ферсман А. Е., Геохимия, т. 1, Л., 1934; Соболев В.,
Введение в минералогию силикатов, Львов, 1949; В о к ий Г. Б.,
Кристаллохимия, 2 изд., М., i960; С ау к ов А. А., Геохи-
мия М., 1950; Я н у л о в К. П., в кн.: Труды АН Таджик-
ской ССР. Записки Таджикского отделения Всес. Минерало-
гия. о-ва, 1959, 104, вып, 1, 139; Г о р ю н о в а Н. А., Франк-
Каменецкий В. А., в сб.: Кристаллография, вып. 5,
М„ 1956, с. 51; Белов Н. В., Геохимия, 1960, Кз 6, 551;
Уманский Я. С. [и др.], Физические основы металлове-
дения М., 1955; Китайгородский А. И., Ж. струк-
турной химии, i960, 1, А’ 3, 324. Н. Л. Смирнова.
ИЗОМОЧЕВИНА — см. Мочевина.
ИЗОНИТРИЛЫ (карбиламины) R—N=C — орга-
нич. производные синильной кислоты, отвечающие ее
изомерной форме HN.bG.
163
ИЗОНИТРИЛЫ - ИЗООКТАН
164
Физические свойства некоторых изонитрилов
Изонитрил T. пл., 'С Т. кип., ’С
CHjNC -45 59,6 0,754
C3H3NC -60 78,1 0,742
C3H7NC —- 99,5 —
U30-C4H9NC -66 116 —
C6H3NC 166 0,9775
(с разл.) • (15’)
Химич, свойства И. определяются наличием ярко
выраженной поляризованной связи С—N. Эти соеди-
нения реагируют с солями многих металлов с обра-
зованием комплексных соединений, напр.:
CoJ2 4- 4R-N=C -> Со (RNChJa
Являясь электронными аналогами окиси углерода,
И. легко вытесняют ее из карбонилов железа, никеля
и других металлов:
Fe(CO)3 + Rfec __ZlMCO)1CNR + СО
*Fe(CO)s(CNR)2 + 2СО
И., как правило, устойчивы к действию водных
р-ров щелочей; разб. неорганич. к-ты легко гидроли-
зуют их до первичного амина и муравьиной к-ты
с промежуточным образованием алкилформамида:
+ - НзО Г /н
R-N=C R—N=C<
H+ L ХО1
Действие сероводорода на И. приводит к замещен-
ным тиоформамидам:
R—NSC — £ R —NH -с/
-* R-NH2 + НСООН
С высшими карбоновыми к-тами И. образуют N-заме-
щенные формамиды и ангидриды соответствующих к-т.
Уксусная к-та в этих условиях образует замещенный
ацетамид и окись углерода:
+ — z.0
R-N=C + 2R’C00H -» RNHC^ + R'CO-O-COR'
R—N=C + CHSCOOH -* CHsCONHR 4- CO
Обработка И. хлористым ацетилом с последующим
гидролизом приводит к первичному амину и пиро-
виноградной к-те:
CH3-N^C-f-CHs-C0Cl -►
Н2о
..... н+
chs-nh24-chscocooh <-----
Конденсация И. с аммиаком или первичными ами-
нами в присутствии карбонильных соединений яв-
ляется удобным методом синтеза замещенных амидов
а-аминокислот:
R—NHa 4- R'-C<^° + R"NSC -► RNH—CR'H—CONHR"
Окись ртути окисляет И. до изоцианатов
R—N=C = O; действие серы приводит к изотиоциана-
там R—N=C=S. Хлорированием И. были получены
карбиламинхлориды R—N=CC12. Каталитически воз-
бужденным водородом изонитрильная группа —N=C
восстанавливается до метиламиногруцпы;
+ - Hj(Ni)
R-N=C-------► R--NH-CHS
При повышенной темп-ре И. превращаются в изомер-
ные нитрилы:
+ _ 250’
R—N — C---> R-CHN
Основные методы синтеза И.: 1) Действие галоген-
алкилов (обычно иодистых) на серебряные, ртутные
или свинцовые соли синильной к-ты, напр.:
CHS J 4- AgCN -* CH3-N=C 4- AgJ
Щелочные соли синильной к-ты с галоидными алки-
лами образуют И. лишь в качестве побочных про-
дуктов. 2) Обработка смеси хлороформа или бромо-
форма с первичным амином спиртовым раствором
щелочи:
-ЗНСКНВг) + ~
R-NH2 4- CHCI3(CHBr3) ------► R—N=C
Реакция первичных аминов с галоформными соеди-
нениями, напр. хлороформом, является весьма чув-
ствительным способом качественного определения как
тех, так и других (изонитрильная проба по Гофману),
т. к. образующийся И. легко обнаружить по характер-
ному запаху. 3) Реакция третичных фосфинов с орга-
нич. изотиоцианатами:
R-N=C=S------> R-N= C4-R3P=S
4) Взаимодействие хлорокиси фосфора с N-замещен-
ными формамида в присутствии основания (пиридин,
т/>ет-бутилат калия и др.):
лО
R-NH-C/
-и
РОС13
оснозание
R- NSC
И. являются весьма токсичными соединениями и об-
ладают специфическим отвратительным запахом, об-
наруживаемым в ничтожныхконцентрациях. И. были
открыты А. Гофманом в 1866.
Лит.: Hou ben— Weyl, 4 Aufl., Bd 8, Stuttgart,
1952; Malatesta L., Sacco A., G h 1 e I m 1 S., Gazz.
chim. Ital., 1952, 82, 516; К I ages F. [u. a.], Chem. Ber.,
1952, 85, № 2, 109; U g 1 I., M e у r R., Angew. Chemie, 1958,
70, № 22—23, 702; 1960, 72, № 7—8, 267. Л. С. Герман.
ИЗОНИТРОЗОАЦЕТОН (монооксим метилглио-
ксаля) СН3СО—CH=N—ОН, мол. в. 87,08 — бес-
цветные кристаллы; т. пл. 69°; возгоняется; легко
растворим в воде, эфире, ограниченно — в бензоле,
хлороформе, четыреххлористом углероде, плохо —
в петролейном эфире. И. восстанавливается в амино-
ацетон (напр., каталитич. гидрированием над Pd на
угле в спиртовом р-ре, содержащем НС1):
2Н2
CHSCOCH=N-ОН------► CH3COCH2NH2 4-Н2О
При действии хлора И. дает СН3СОС(С1)~NOH, при
кислотном гидролизе — метилглиоксаль и т. д. По-
лучают И. действием нитрозирующих агентов на
ацетоуксусный эфир:
СН3СОСН2СООС2Н3 4- C3HnONO -»
4-HsO
-> СН3СО-СН-СООС2Н5 -----f
NO
-» CH3COCH=NOH 4- C2H3OH 4- co2
И. реактивирует холинэстеразу, ингибированную
фторфосфатами. В- н. Фросин.
ИЗООКТАН (2,2,4-триметилпентан), молекулярный
вес 114,22, СН3С(СН3)2СН2СН(СН3)СН3 — один из изо-
меров октана; прозрачная бесцветная жидкость, на-
поминающая по запаху легкий бензин прямой гон-
ки; т. пл. —107,38°; т. кип. 99,24°; dsf 0,69192; reg
1,39145; давление пара при 23,4° 47,9 мм; теплотвор-
ная способность 11 450 ккал/ке; теплота испарения
73,50 кал/г (25°); теплота сгорания 1305,29 ккал/молъ
I (25°, р = const). Дает бинарные азеотропы (т. кип.,
165
ИЗОПЕНТАН - ИЗОПРЕН
166
вес. % И.): с бензолом (80,1°; 2,3%), с метанолом
(59,4°; 47%), с этанолом (71,8°; ок. 60%) и с др.
спиртами. И. вместе с другими изомерами октана
содержится в небольших количествах в нефтяных бен-
зинах прямой гонки. При нагревании до 500—570°
И. образует гл. обр. метан и изобутилен с небольшим
количеством этана, этилена, пропилена и водорода.
При каталитич. крекинге И. с сульфидом вольфрама
при 400° и 250 ат или с AlClg и НС1 при 20—140°
главным продуктом распада является изобутан;
с алюмосиликатами при 500° — метан, бутан, изобу-
тан, изобутилен и бутилен, а при 550° также и про-
пилен. Химич, превращения И. (окисление, галоге-
нирование, нитрование, сульфирование и т. д.) не
имеют практич. значения. Расширение произ-ва И.
до крупнопромышленных масштабов связано с необ-
ходимостью обеспечения карбюраторных авиацион-
ных двигателей, работающих по циклу Отто, бензи-
ном с высокими антидетонационными свойствами.
В пром-сти И. получают гидрогенизацией диизобу-
тилена над никелевым, медно-хромовым и др. катали-
заторами или алкилированием изобутана изобутиле-
ном в присутствии серной к-ты, катализаторов, со-
держащих BF3, и др. веществ. В результате получают
технически чистый И. с октановым числом 98—100
или технич. И. с октановым числом 92—97. Антиде-
тонационные свойства И. приняты за 100 единиц
шкалы т. наз. октановых чисел, характеризующих
устойчивость топлив к детонации. Содержание И.
в топливе определяют методами разгонки, ИК-спек-
троскопии, масс-спектроскопии, по спектрам комби-
национного рассеивания света, хромотографически
или термохроматографически (наиболее быстрый и
удобный метод) и др. способами.
Лит.: Selected values of chemical thermodynamic proper-
ties, by F. D. Rossini [a. o.l, Washington, 1952; Химия углево-
дородов нефти, под ред. Б. Т. Брукса [и др.], пер. с англ.,
М., 1958; Моторные топлива, масла и жидкости, под ред.
К. К. Папок и Е. Г. Семенидо, т. 1, 3 изд., М., 1957.
Г. 1И. Цигуро.
ИЗОПЕНТАН — два изомерных углеводорода со-
става С5Н12: 2-метилбутан (диметил этилметан)
СН3СН(СН3)СН2СН3 и 2,2-диметилпропан (тетраметил-
пентан, неопентан), (СН3)4С — бесцветные, подвиж-
ные легколетучие жидкости со слабым запахом.
2-М е т и л бу т а н;т. кип. 27,852°/760 мм; т. пл.
— 159,90°, rf2/ 0,61967; 1,35373; теплота испарения
5,842 ккал! моле (т. кип.); теплота горения (газ)
843,24 ккал!молъ (25°, Н2О жидк.); теплота образова-
ния (жидк.) А/7298 = — 42,85 кал]молъ, свободная
энергия образования (жидк.) — 3,59 ккал]моль (25°);
С” (газ) 0,3935 кал1град-г (25°). При хлорировании
2-метилбутана образуется смесь 1- и 2-хлор-2-метил-
бутана, используемая для получения амиловых спир-
тов, амилнафталинов, меркаптанов и аминов. Нитро-
вание этого углеводорода при 400° дает до 70% моно-
нитрометилбутанов. Кроме того, 2-метилбутан при-
меняют для получения изопрена методом каталитич.
гидрогенизации.
2,2-Д иметилпропан (неопентан): т. кип.
9,503°/760 мм; т. пл. —16,55°; 0,5910 (рНасыщ.);
nD 1,342 (Рнасыщ.)] теплота испарения 5,438 ккал/моль
(т. кип.); теплота горения (газ) 840,49 ккал)моль (25°,Н2О
жидк.); теплота образования (жидк.) ДИ208= — 44,98
кал]молъ, свободная энергия образования (газ)—
3,64 ккал}молъ (25°), Ср (газ) 0,4029 кал! град г (25°).
И. содержится в легких погонах нефти, в природ-
ном газе, в легких продуктах деструктивной перера-
ботки нефти (в смеси с др. углеводородами); может
быть выделен четкой ректификацией, а также получен
каталитич. изомеризацией н-пентана в специальных
условиях. Наибольший практич. интерес представ-
ляет 2-метилбутан, преобладающий в этих продуктах;
напр., в газовом бензине, получаемом из природного
газа, содержится 20—40% т. наз. технич. пентана,
представляющего собой смесь н-пентана и 2-метил-
бутана в отношении 1:1 и до 2% 2,2-диметилпро-
пана. И., обладающий высоким давлением пара и
высоким октановым числом, является ценным компо-
нентом моторных бензинов. Для компаундирования
обычно применяют т. наз. технич. И., добавляемый
к бензину в количестве до 15% и являющийся смесью
изомерных пентанов; выкипает в пределах 24—34°,
давление пара 1140 мм рт. ст. (40°), плоти. 0,620 и
октановое число 90 (по моторному методу).
Лит.: А з и н г е р Ф., Химия и технология парафиновых
углеводородов, пер. с нем., М., 1959. В. В. Щекин.
ИЗОПОЛИКИСЛОТЫ — комплексные кислоты,
анионы к-рых содержат ангидриды только одного
рода. Подробнее см. Изополисоединения.
ИЗОПОЛИСОЕДИНЕНИЯ — комплексные соедине-
ния, в состав комплексного аниона к-рых входят
молекулы ангидрида только одного рода (в отличие
от гетерополисоединений). Способность к образованию
И. характерна для элементов побочных подгрупп
5 и 6 групп периодич. системы (Сг, Мо, W, U, V, Та,
Nb). И. можно представить как производные «про-
стой» кислоты (напр., Н2СгО4), в к-рой О2~ замещены
анионами той же кислоты:
- О "
О Сг О
О
к2
- о
О Сг СгО4
О
Рентгенографически показано, что ионы типа Cr2OV,
Сг3О% и т. п. представляют собой совокупность тетра-
эдровСгО34" с общими вершинами (рис.).
Соответственно И. вольфрама и молиб-
дена построены в виде цепочек, со-
стоящих из тетраэдров и октаэдров
(WO3f и W0V), соединенных одной
или двумя общими вершинами.
И. Мо и W известны только в твер-
дом состоянии и поэтому могут быть
получены только сухим путем, напр.
сплавлением нормальных солей с ангид-
ридами: Na2WO4 -|- WO3 —► Na2W2O7.
Эти И. разлагаются водой с образова-
нием акваполисоединений. И. Сг, V и
в р-ре; они могут быть получены при подкислении
р-ров их простых солей. Так, при добавлении кислоты
к р-ру К2СгО4 образуется бихромат:
K2CrO4 -Т H2SO< —> K2SO4 К2Сг2О7
При подщелачивании р-ра К2Сг2О7 можно получить
К2СгО4. Аналогичные соотношения установлены для
реакций образования в р-ре И. ванадия:
2 [V3O9]3- -I- 2Н+ = [V6O17p- + Н2О
[V0O17p~ + 2ОН- = 2 [V3Oe]3- H2O
Для урана характерно образование в р-рах не про-
стых уранатов, а полиуранатов до типа Me2UeO19
включительно (Me=Na, К):
UO2C12 + NaOH = 2NaCl + Na2U2O7 + ЗН2О
Соответствующие И. Та и Nb (напр., K2Nb6O16,
К2Та4О11( К2Та2О6) могут быть получены сплавле-
нием нормальных солей с окислами. Нек-рые из этих
соединений нерастворимы в воде, другие —- разру-
шаются ею. См. также Комплексные соединения.
Лит.: Гринберг А. А., Введение в химию комплекс-
ных соединений, 2 изд., Л. — М., 1951; Химия координаци-
онных соединений, пер. с англ., М., i960; Lindqvist I.,
Acta chem. scand., 1980, 14, M 4. H. Н. Желиговская.
ИЗОПРЕН (2-метилбутадиен-1,3), мол. в. 68,11,
СН2=С(СН3)—СН=СН2, — ненасыщенный углеводо-
род ряда СпН2п_2, диеновое соединение (диоле-
167
ИЗОПРЕН
• 168
фин) с сопряженными двойными связями; бесцветная
жидкость: т. пл. —146°; т. кип. 34,076°; rf2/ 0,681;
1,4219 (жидк.); полностью растворим в спирте и
диэтиловом эфире; ркрит 55,6 ат; гкрит 400°; давление
паров (мм рт. ст.): 13,6 (—50°), 127,0 (—10°),
201,8(0°), 309,1 (10°), 460,3 (20°), 942,4 (40°). Тепло-
емкость жидкого И. (кал/г град): 0,505 (—3°);
0,516 (7°); 0,527 (17°); 0,536 (25°); теплоемкость паров
И. (кал/г • град): 0,329 (27°); 0,415 (127°); 0,497(227°);
0,679 (527°); 0,723 (627°); скрытая теплота плавления
16,8 кал/г; скрытая теплота испарения (ккал/молъ):
6,57 (13,4°); 6,27 (25°); теплота сгорания (ккал/молъ):
766,3 (газ), 760,0 (жидк.); теплота образования (газ)
при 25° — 18,1 ккал/молъ', вязкость жидкого И.
(спуаз): 0,260 (0°); 0,236 (10°); 0,216 (20°); 0,198 (30°);
т. всп. —48°; темп-ра самовоспламенения 400°; взры-
воопасные концентрации в воздухе 1,66—11,50 об. %;
допустимая концентрация в воздухе рабочих помеще-
ний 0,1 Мг/л.
И. образует двойные азеотропные смеси (см. табл.),
а также тройную азеотропную смесь: 0,5% воды, 7,7%
ацетона, 92% изопрена; т. кип. 32,5°.
Второй компонент Содержание изопрена в азеотропной смеси, вес, % Т. кип азео- тропной смеси, °C
Этиловый спирт 97 32,65
Метиловый спирт 95 29,5
Ацетон < . . . 80 30,5
Метилформиат 90,8 22,5
Бромистый этил » . . . 53,0 32,4
Этилформиат 76,0 32,5
Изопентан 8,0 27,7
н-Пентан . < 90,0 33,8
Окись этилена 52,0 33,2
Окись пропилена 40,0 31,6
Изопропилнитрит . 69,0 33,0
Диэтиловый эфир 52,0 33,2
И. вступает в большинство реакций, характерных
для олефинов; легко протекают гидрирование, гало-
генирование, гидрогалогепирование, образование ме-
таллоорганич. соединений (со щелочными металлами)
и др.
Наиболее интересными являются реакции присо-
единения и полимеризации. И., в зависимости от
условий, способен образовывать линейные или раз-
ветвленные цис- и транс-полимеры — изопреновые
каучуки. В случае взаимодействия только с двумя
одновалентными атомами или радикалами присоеди-
нение происходит преим. к двум крайним атомам
в положении 1,4 с образованием двойной связи
между 2 и 3 атомами углерода. При повышенной
темп-ре И. способен образовывать циклические димеры.
Напр., водород в момент выделения присоединяется
почти полностью в положении 1,4. В зависимости
от условий реакции и применяемых катализаторов
присоединение может происходить также и в положе-
нии 1,2. Гидрирование И. молекулярным водоро-
дом в присутствии платипы дает смесь изопентанов
с присоединением водорода как в положении 1,4,
так и в 1,2 и в 3,4. Бром также присоединяется в трех
положениях. С НВт И. дает продукт следующего
строения:
СН3
СН3-С=СН-СН2Вг
И. способен вступать в реакцию полимеризации
также с соединениями винилового ряда. Характерной
реакцией И. является присоединение малеинового
ангидрида; эта реакция служит основой для его
количественного определения.
К промышленным методам получения И. относятся
следующие: 1) Так наз. диоксановый метод,
состоящий йз двух стадий. Вначале из изобутилена и
формальдегида при температуре 25—65° образуется
4,4-диметилдиоксан-1,3:
СН3
СН3\ „ I
>С=СН2 + 2СН,0 ->• СН3-С----О---СН2
CHSZ | |
СН2- СН2- о
Катализаторами этой реакции могут служить водные
р-ры хлористого цинка, фосфорновольфрамовой, фос-
форной, соляной и серной к-т и фтористый бор. За-
тем 4,4-диметилдиоксан-1,3 превращают в И.:
СНз—С----О---СН2 -► СН2==С--СН=СН2+ СН2О + Н2О
сн2-сн2- о
Процесс проводят в парах при повышенной темп-ре
в контактных аппаратах над твердым катализатором.
Выделившийся при расщеплении диоксана формаль-
дегид вновь возвращается в процесс.
2) Наиболее перспективным является дегидри-
рование изоамиленов или изопентана. Наличие
значительных количеств пентанов в попутных газах
нефтедобычи и низкая их стоимость делают этот метод
экономически выгодным. Целесообразно использова-
ние также изоамиленов, содержащихся в крекинг-
бензине. Изопентан можно дегидрировать в одну или
в две стадии; при одностадийном дегидрировании
выход И. ниже. При двухстадийном методе изопентан
вначале дегидрируют до изоамиленов:
СИ, СНз
I I
СН,- СН СН2-СН3 — Н2 + СНз-СН-СН=СН2
СНз СНз
I I
или СН3—С=СН—СН3 или С2Н=С—СН2—СН3
Процесс проводят над твердым катализатором при
550—600°. В качестве катализатора может быть
использована окцсь хрома, нанесенная на окись алю-
миния и промотированная нек-рыми добавками. Про-
цесс осуществляется в движущемся слое гранули-
рованного катализатора, образующего с поступаю-
щими парами изопентана псевдоожиженный слой.
Непрерывный ввод в реакционную зону нагретого
в регенераторе катализатора обеспечивает подачу
реакционного тепла. Выделение изоамиленов и изо-
пентана из реакционной смеси, вследствие близости
их темп-p кипения, является трудной задачей, к-рая
решается с помощью экстрактивной и азеотропной
дистилляции.
Все три изоамилена при последующем дегидрирова-
нии образуют И.:
иэо-С5Н10 -► СН2 = С(СН3)-СН = СН2 + Н2
Реакцию проводят при пониженном парциальном
давлении углеводородов и при повышенной темп-ре.
Понижение парциального давления углеводородов
может быть достигнуто путем разбавления их водя-
ным паром, к-рый одновременно является тепло-
носителем, обеспечивающим поступление тепла в
реакционную зону.
3) Метод А. Е. Фаворского, основанный на взаимо-
действии ацетилена с ацетоном в присутствии мёлко-
раздробленного безводного КОН с образованием
диметилацетиленилкарбинола:
сн3. сн3.
СН^СН + >С=О — >С(ОН)-СНСН
CHSZ СН,<
169
ИЗОПРЕНОВЫЙ КАУЧУК
170
к-рый далее гидрируют в мягких условиях в диметил-
винилкарбинол:
сн3
СНЗЧ I
>С(ОН)-С7:СН + Н2 СН3-С(ОН)-СН=СН2
СНзХ
Последняя стадия — дегидратация — проводится в па-
ровой фазе над твердым катализатором:
СНз СНз
I I
СН3-С(ОЫ)-СН=СН2 сн»=с-сн=сн» + Н2О
Метод не получил промышленного развития вслед-
ствие трудностей регенерации КОН, расход к-рого
на первой стадии весьма значителен.
Для полимеризации требуется И. очень высокой
степени чистоты, в связи с чем применяют сложные
методы его выделения и очистки. И. применяют для
получения синтетич. каучуков как самостоятельно,
так и в смеси с другими мономерами (напр., при полу-
чении бутилкаучука).
Количественное определение И. основано на реак-
циях присоединения брома, водорода или (преиму-
щественно) малеинового ангидрида. См. также Изо-
преновый каучук.
Лит.: Смирнов Н. И., Синтетические каучуки, 2 изд.>
Л — м„ 1954; Фаворский А. Е., Сб. избр. трудов,
М. — Л., 1940; Литвин О. Б., Основы технологии синтеза
каучуков, М., 1959. А. К. Никитин.
ИЗОПРЕНОВЫЙ КАУЧУК (СКИ, СКИ - 3, натсин,
корал, америпол Эс-Эн, полиизопрен) — синтетич. кау-
чук, получаемый полимеризацией изопрена под дейст-
вием катализаторов — металлич. лития, литийорганич.
соединений, перекисных соединений или каталитич.
комплексных систем, к-рые образуются при взаимодей-
ствии триалкилалюминия с галогенидами тяжелых ме-
таллов (напр., триэтилалюминия с четыреххлористым
и треххлористым титаном). Полимеризацию проводят
при 20—-50° в среде растворителя или без него. При-
менение растворителя (н-гептан, н-гексан, н-пентан)
позволяет равномернее распределять катализатор,
лучше отводить реакционное тепло. Исключительное
значение имеет высокая степень очистки изопрена и
растворителя от примесей, разрушающих катализатор
и вызывающих образование нежелательных структур.
Поэтому исходную смесь тщательно осушают с по-
мощью силикагеля или активной окиси алюминия и
очищают от кислородсодержащих соединений. Про-
цесс проводят при интенсивном перемешивании и
непрерывном отводе тепла водой или холодильным
рассолом. Содержание полимера доводят до 15—25%
(вязкость 15%-ного р-ра 100 000 спуаз), после чего
в реакционную массу вводят дезактиватор катализа-
тора (напр., кислоты) и противостаритель. Раствори-
тель может быть удален в червячном прессе, по вы-
ходе из к-рого И. к. является готовым продуктом.
При полимеризации молекулы изопрена
СНз
сн2=с-сн=сн2
12 3 4
могут соединяться друг с другом по любой двойной
связи, в результате чего получаются полимеры с раз-
ными положениями звеньев в цепи макромолекулы:
а)1,4—1,4 (т. наз. «голова к хвосту», см. I); б) 1,1—4,4
(т'.' наз. «голова к голове» — «хвост к хвосту», см. II);
в),1,4—1,2 (см. III); г) 4,4—3,1 (см. IV)
(I) СНз СНз - СНз
I I I
-Н2С-С=СН-СН2-СН2-С=СН-СН2-СН2-С=СН - сн2 -
1234 1234 1 23 4
(II) СНз CH., СНз
I I I
-Н,,С-С-СИ- СН.,-СН.,-СН= С-СН2-СН2-С=СН-СН2-
1234 4 321 123 4
СНз СН, СН3
I I I
(III ) - СН,—С=СН-СНг-СН.>-С-СН»-С=СН-СП^-
‘21 1’2 3 4
1 2 3 4 1 ЗСН
II
4CII,
СН, СНз
I |
(IV ) -СН,- С=СН- СН,-СН,- СН=С-СП, -СН,-СН —
‘31
1 2 3 4 4 3 2 1 4 2С-СНз
II
1СН,
Соотношение структур И. к. в зависимости от при-
меняемых катализаторов приведено в табл. 1.
Таблица 1
Содержание звеньев в Нату- раль- ный кау- чук Комплексные катализаторы Катализа- тор литий
макромолекуле И. к., % ски-з америпол Эс-Эн СКИ ко- рал
цис-1,4 97,8 92-99 . . 91 65 94
транс-1,4 0 0 8 25 0
1,2 0 0-3 0 1 0
3,4 2,2 1-3 1 6 6
Наиболее высокими механич. свойствами обладает
полимер изопрена, состоящий из макромолекул кон-
фигурации 1,4—1,4, при условии, что большинство
звеньев находится в г^ис-форме. Регулярность в по-
строении макромолекулярной цепи обеспечивает вы-
сокую прочность И. к. Однако строгая регулярность
приводит к образованию высококристаллич. твердых
полимеров (см. Изотактические полимеры). Наличие
в полимерной изотактич. стереорегулярной цепи
небольшого количества атактич. сегментов вызывает
нек-рую разупорядоченность структуры И. к. и сни-
жает эффект кристаллизации, что обеспечивает полу-
чение эластичного полимера с высокими физико-ме-
ханич. показателями. В зависимости от применяемых
катализаторов и условий проведения полимеризации
образуются И. к. различной структуры. Более регу-
лярно построенные И. к. — СКИ-3 и америпол Эс-Эн,
полученные с комплексными катализаторами, дают
характерную для натурального каучука рентгено-
грамму, указывающую на наличие кристаллин, фазы,
и имеют темп-ру стеклования, равную тёмп-ре стек-
лования натурального каучука — минус 70°. Менее
регулярный И. к. — СКИ, способен кристаллизо-
ваться только в растянутом состоянии и, так же как
И. к. корал (оба получаются в присутствии катали-
затора — металлич. лития), имеет более высокую
темп-ру стеклования — минус 68°.
Основной особенностью И. к., выгодно отличаю-
щей его от других синтетич. каучуков общего назначе-
ния, является высокая прочность вулканизатов без
применения активных (усиливающих) наполнителей,
хорошо сохраняющаяся при повышении темп-ры до
100°. В отличие от других синтетич. каучуков, И. к.,
подобно натуральному каучуку, обладает высокой
клейкостью. По прочностным показателям И. к.
практически равноценны натуральному каучуку и
лишь незначительно уступают ему в эластичности.
В связи с большим содержанием звеньев ifuc-1,4 и
высокой эластичностью в резинах из И. к. наблю-
дается низкое теплообразование при многократных
деформациях, что. очень важно для работы резино-
вых изделий, особенно автомобильных шин, Основные
свойства вулканизатов И. к. представлены в табл. 2.
171
ИЗОПРОПИЛБЕНЗОЛ - ИЗОПРОПИЛОВЫЙ СПИРТ
172
Таблица 2
Показатели Свойства ненаполненных вулканизатов Свойства сажевых вулканизатов (30 в. ч. сажи на 100 в. ч.. каучука)
натураль- ный каучук комплексные катализаторы катализатор литий натураль- ный каучук комплексные катализаторы катализа- тор литий
ски-з америпол Эс-Эн ски корал ски-з америпол Эс-Эн ски
Предел прочности при разрыве, кз/слф1: при 20° 250-350 200 250—300 266 230—320 268 325—380 314 350 300-380
» 100° 170—200 120-170 80—100 149 200 200 167 110-150
Относительное удлинение, %: при 20° % . . . 800-850 850 900 1100—1200 870 750—850 750 790 950-1050
» 100° 1000 950—1000 855 1000-1100 960 900 850 930 900—1000
Эластичность по отскоку, %: при 20’ 70—72 66-69 68 65-70 65 50 48 46 50
» 100е 78—81 75-78 76 75-79 71 67 55 53 64
Теплообразование, °C: при постоянной деформации 40% 50 49 52 80 89 89 80
при постоянной нагрузке 165 кз 75 75 — 74 - 92 100 — 88
По нек-рым механич. свойствам резины из И. к.
уступают резинам из натурального каучука (более
низкое сопротивление раздиру, несколько меньшая
температуростойкость). И. к. относится к классу
каучуков общего назначения, и из него могут изго-
товляться всевозможные резиновые изделия, к к-рым
не предъявляется требований по специальным свой-
ствам (маслобензостойкость, газонепроницаемость, хи-
мич. стойкость, термостойкость и др.).
Ходимость автомобильных шин, изготовленных из
И. к., приближается к ходимости шин из натураль-
ного каучука. Наряду с другими стереорегулярными
каучуками (ifuc-бутадиеновый, сополимер этилена
с пропиленом и др.) И. к. является лучшим полиме-
ром, обладающим наиболее высоким комплексом ди-
намико-эластич. свойств.
Лит.: Субботин С. А., Самолетова В. В.,
Знаменская А. К., Хим. пром-сть, 1956, № 7, 21;
Рейх В. Н. [и др.], Каучук и резина, 1960, М 3, 1; Синте-
тический каучук, под ред. Г. С. Уитби, пер. с англ., Л., 1957;
Яшунская Ф. И., Маркович Г. А., Каучук и ре-
зина, 1959, МП, 1;Волдырев И. И. [и др.], Хим. наука
и пром-сть, 1957, 2, № 3, 391; Новые каучуки. Со. переводов,
под ред. В. Ф. Евстратова, Ф. И. Яшунской, М., 1958.
А. К. Никитин.
ИЗОПРОПИЛБЕНЗОЛ (кумол) СвН6СН(СН3)2, мол.
в. 120,19 — бесцветная жидкость с ароматичным запа-
хом; т. пл. —96,03°; т. кип. 152,39°; d'-!j 0,8618;
п'р 1,49130; смешивается со спиртом, эфиром, ацето-
ном, бензолом, хлороформом; плохо растворим в воде
(при 20° менее 0,01%); горит и образует взрывоопас-
ные паро-воздушные смеси; т. всп. 38°; ядовит (в боль-
шей степени, нежели бензол и толуол), при ингаляции
вызывает острые и хронич. поражения кроветворных
органов (костного мозга, селезенки). И. — типичное
ароматич. соединение: легко алкилируется, хлорирует-
ся, сульфируется и нитруется. Получают И. жидко-
фазным (в присутствии А1С13) или парофазным (в при-
сутствии фосфорной к-ты, нанесенной на кизельгур)
алкилированием бензола пропиленом (пропиленовой
фракцией газов пиролиза или крекинга углеводоро-
дов). И. содержится в «легком бензоле», образую-
щемся при коксовании угля или ароматизации нефти.
Используют И. в основном как растворитель лаков и
красок, как высокооктановую добавку к авиационным
бензинам и исходное вещество при произ-ве фенола и
ацетона жидкофазным каталитич. окислением И. кис-
лородом воздуха и последующим разложением гидро-
перекиси И. серной к-той.
Лит.: Kirk, v. 1, N. Y., 1952, р. 543; Лазарев Н. В.,
Химические вредные вещества в промышленности, ч. 1, Л. —
М„ 1951, с. 100; Безуглов П. Т., Справочная таблица
огнеопасных веществ, 3 изд., М. —Л., 1950, с. 55.
Г. А. Сокольский.
ИЗОПРОПИЛОВЫЙ СПИРТ (пропанол-2, изопро-
панол) СН3СНОНСН3, мол. в. 60,09 — бесцветная
жидкость с характерным запахом; т. пл. —89,5°;
т. кип. 82,40°; d-% 0,7851; zij'j 1,3776; давление пара
(.«.« рт. ст.): 10 (2,4°), 40 (23,8°), 100 (39,5°),
400 (67,8°), 1020,7 (90°); вязкость (20°) 2,43 спуаа;
поверхностное натяжение (20°)’21,7 дин]см\ гкрит 234,9°;
Ркрит. 53 ат> УД- теплоемкость (кал/г): 0,563 (0°),
0,677 (30°), 0,740 (50°); теплота плавления (при
т. пл.) 21,08 кал/г; скрытая теплота испарения (при
т. кип.) 160 кал/г; теплота сгорания 7 970 кал/с;
темп-ра вспышки в закрытой чашке 11,7°; темп-ра
самовоспламенения 456°; нижний предел взрывае-
мости в воздухе (25°) 2,5%. И. с. смешивается с водой
и органич. растворителями во всех отношениях; обра-
зует с водой азеотропную смесь (12,6%, т. кип.
80,3°). И. с. обладает большим наркотич. действием,
чем этиловый спирт; токсичность его в 2 раза превы-
шает токсичность этилового спирта.
И. с. обладает всеми свойствами спиртов жирного
ряда: образует простые и сложные эфиры; гидро-
ксильную группу можно заменить галогеном; реаги-
рует с реактивом Гриньяра и т. д. При дегидрирова-
нии или неполном окислении образуется ацетон; оба
процесса являются промышленными (см. далее).
С ароматич. соединениями И. с. конденсируется в при-
сутствии H2SO4 с образованием производных, напр.
изопропилбензола, изопропилтолуола и др.
В пром-сти И. с. получают гидратацией пропилена.
Известны два варианта процесса: сернокислотная ги-
дратация пропилена и прямая гидратация. В первом
случае для гидратации применяют 80—90%-ную
H2SO4 (давление 5—10 ат, темп-ра 25—50°). Процесс
основан на образовании изопропилсерной к-ты и
диизопропилсульфата:
СН3СН=СН2-(-H2SO4 (CH3)2CHOSO2OH
2СН3СН=СН2 + H2SO4 [(CH3)2CH]2SOi
и последующем гидролизе их при нагревании с водой,
напр.:
(CH3)2CHOSO2OH (СН3)2СНОН + H2SO4
[(CH3)2CH]»SO4 2(СН3)2СНОН -J-H2SO4
Прямую гидратацию пропилена осуществляют с жид-
ким или твердым катализатором. Жидким катализа-
тором является разб. р-р II2SO4 (15 ат, 200°); катали-
173
ИЗОПРОПИЛОВЫЙ ЭФИР-ИЗОТАКТИЧЕСКИЕ ПОЛИМЕРЫ
174
затор в паровой фазе — смесь окиси вольфрама, акти-
ватора и окиси цинка, нанесенная на силикагель.
В качестве сырья используют пропан-пропиленовую
фракцию газов крекинга, а также пропиленовую
фракцию газов пиролиза нефти. Известны другие
способы получения И. с.: окисление парафиновых
углеводородов воздухом, каталитич. гидрирование
СО, ферментация сахаристых веществ и др.
В пром-сти И. с. применяют гл. обр. для получения
ацетона; для этого пользуются дегидрированием или
неполным окислением. Дегидрирование ведут в паро-
вой или жидкой фазе; в паровой фазе катализатором
является окись цинка,нанесенная на пемзу(380—400°),
в жидкой фазе — никель Ренея, диспергированный
в исходном спирте. Окисление проводят в паровой
фазе (серебряный катализатор, 500—600°) или в жид-
кой (давление, 90—140°). И. с. хорошо растворяет
многие эфирные масла, нек-рые синтетич. смолы, алка-
лоиды и др.; при этом снижается огнеопасность
произ-ва. И. с. используют в качестве антифриза.
В лабораторной практике И. с. применяют для введе-
ния в молекулу изопропильной группы.
И. с. образует пурпурно-красную окраску при
окислении пробы 1%-ным р-ром К2С.г2О7 в присут-
ствии нитропруссида натрия и хлористого аммония.
Количественно И. с. определяют гидроксиламинным
методом по количеству ацетона, образовавшегося
при окислении пробы хромовой смесью.
Лит.: Kirk, v. 11, N. Y., 1953, p. 182.
А. В. Арбатский.
ИЗОПРОПИЛОВЫЙ ЭФИР (диизопропиловый
эфир) (СН3)2СН—О—СН(СН3)2, мол. в. 102,17 — бес-
цветная подвижная жидкость с характерным эфир-
ным запахом; т. пл. —85,89°; т. кип. 68,47°; 0,7244;
Лд 1,3681; давление паров (.я.и рт. ст.): 44,3 (0°);
74,5 (10°); 120,2(20°); 186,8 (30°); 760,0 (68,47°),
уд. теплоемкость 0,528 кал/г град (30°); теплота
испарения 68,2 кал/г', теплота сгорания 9380 кал/г-,
теплота образования 841 кал/г-, темп-ра вспышки
—22,5°; темп-ра самовоспламенения 443°; пределы
взрываемости в воздухе 1,1—4,5 об. % (100°). В воде
растворяется 0,94 вес. % при 20° И. э.; в И. э. рас-
творяется 0,55 вес.% воды. И. э. образует азеотроп
с водой (99,5 вес.% И. э., 62,2°), с изопропиловым
спиртом И. э. дает характерные реакции эфиров;
окисляется, хлорируется, бромируется и т. п.; обра-
зует перекиси, гидролизующиеся до Н2О2, ацетона и
изопропилового спирта. Получают И. э. при гидрата-
ции пропилена до изопропилового спирта; процесс
может быть направлен в сторону образования И. э.;
может быть получен дегидратацией изопропилового
спирта в присутствии серной к-ты. Применяют И. э.
как растворитель масел, жиров, органич. к-т и др.,
а также в качестве компонента моторного топлива
для повышения его октанового числа.
ИЗОСАФРОЛ (3,4-метилендиокс.ипропенилбензол)
С10Н10О2, мол. в. 162,2 — бесцветная маслообразная
жидкость; т. кип. 149°/20 мм; d1", 1,122;
nsp 1,5763. Известен в виде цис- и
р |Г° т/>анс-изомеров. Для транс-И.: т. пл.
L J 6,7—6,8°; т. кип. 253°/760 мм, 111—
Т 112°/6 мм; d-.J' 1,1160; zi'n7 1.5746. И. хо-
СН=СНСН3 ' ’ ° % <
рошо растворим в спирте, эфире, бензо-
ле, нерастворим в воде. Хромовой к-той И. окисляет-
ся в пиперонал. И. в природных маслах не найден;
получают изомеризацией сафрола при нагревании по-
следнего со спиртовым р-ром К ОН. Количественно
может быть определен роданированием. И. — исход-
ный продукт для получении гелиотропина.
А. А. Зеленсцхая.
ИЗОСТИЛЬБЕН — см. Стильбен.
ИЗОТАКТИЧЕСКИЕ ПОЛИМЕРЫ — один из ви-
дов полимеров стереорегулярных, характеризующийся
наличием в каждом мономерном звене макромолекулы,
по крайней мере, одного асимметрия, атома, входя-
щего в основную цепь, причем повторяющиеся одно-
типные асимметрия, атомы на достаточно длинных
участках молекулярных цепей имеют одинаковые
пространственные конфигурации (только d или
только 1). И. п. получают стереоспецифич. полимери-
зацией несимметрич. мономеров винилового ряда,
диенов, окисей олефинов и др. В принципе И. п. мо-
гут быть получены из любого мономера, если при его
полимеризации или поликонденсации в каждом звене
полимерной цепи возникает асимметрия, атом, вхо-
дящий в основную цепь.
Получение И. п. проводят в присутствии стерео-
специфических катализаторов. Для получения изо-
тактич. полиолефинов обычно применяют гетероген-
ные катализаторы Циглера—Натта.
Если изобразить основную цепь И. п. в виде вытя-
нутого плоского зигзага (рис. 1), то все боковые заме-
стители X или Y у асимметрия, атомов расположатся
Рис. 1.
по одну сторону от плоскости зигзага. Макромолекулы
И. п. могут иметь различные конформации.' Конфор-
мационный набор И. п. в аморфном состоянии или
в растворе в первом приближении не отличается от
конформационного набора соответствующего атакти-
ческого полимера. Одйако регулярность макромоле-
кул И. п. во многих случаях позволяет получить
И. п. в кристаллич. состоянии. Цепи И. п. при кри-
сталлизации принимают конформации, соответствую-
щие плотной упаковке в регулярной трехмерной
решетке при минимуме стерич. напряжений, возни-
кающих благодаря внутримолекулярному взаимодей-
ствию валентно не связанных атомов. Для большин-
ства исследованных И. п. этим условиям отвечают
спиральные конформации, получающиеся при пово-
роте звеньев цепи на определенные углы вокруг орди-
нарных связей. На рис. 2 изображена спиральная
Рис, 2.
конформация цепи изотактич. полипропилена, к-рая
получается при повороте каждого мономерного звена
цепи на 120° относительно предыдущего вокруг
связи С—С. В данном случае в периоде идентичности
вдоль оси спирали укладывается три мономерных
звена, к-рые образуют один виток спирали. Аналогич-
ные конформации цепей возникают в кристаллах изо-
тактич. полистирола, поли-1-пентена, поли-1-гексена
и др. Для кристаллов других И. п. могут быть ха-
рактерны более сложные спиральные конформации.
Способность к кристаллизации является основной
причиной, обусловливающей различия в физико-
механич. свойствах изотактич. и атактич. полимеров
в конденсированной фазе.
Так, атактич. полипропилен в зависимости от молекуляр-
ного веса при обычных темп-pax представляет собой вязкую
жидкость или каучукоподобный пекристаллизующийся мате-
риал с темп-рой стеклования порядка —40°. Изотактич. поли-
пропилен — кристаллизующийся волокнообразующий поли-
мер, т. пл. кристаллов 176°. Атактич. полистирол не кристал-
лизуется, темп-ра стеклования порядка 80°. Изотактич. поли-
175
ИЗОТАКТИЧЕСКИЕ ПОЛИМЕРЫ
173
стирол кристаллизуется, т. пл. 230—240°. Изотактич. поли-
стирол в аморфном состоянии по свойствам очень близок к атак-
тич. полистиролу. Данные об основных свойствах нек-рых
И. п. в конденсированной фазе приведены в табл. 1.
Полимер Формула мономера Пери- од иден- тично- сти, А Число мономерн. звеньев в периоде идентич- ности Т. пл., °C Плот- ность, г/см3
Полипропилен3 . . . сн2=сн—сн3 6,50 3 176 0,92
Полибутен-1 сн2=сн—сн2—сн3 6,50 3 128 0,91
Полипентен-1 .... СН2=СН—(СН2)2—сн3 6,60 80 0,87
Полигексен-1 .... Поли-З-метилбутен-1 СН2=СН—(СН2)3—сн3 СН2=СН—СН—сн3 6,84 4 —55 300 0,90
Поли-4-метил пентен-1 сн3 сн2=сн—сн2—сн—сн3 13,8 7 235 0,83
Поли-4-метилгексен-1 сн3 сн2=сн—сн2—сн—с2н5 14,0 7, 188 0,86
Полистирол б , . . , сн3 сн2=сн—С6Н5 6,65 3 230-240 1,08
Температура стеклования: а 35—40% 6 80—85%
Растворы и кристаллы нек-рых И. п. обладают оптич.
активностью. Присутствие в основной цепи полимера асим-
метрия. атомов одной и той же пространственной конфигура-
ции, однако, не является достаточным условием для проявле-
ния оптич. активности. Так, И. п., полученные из стирола,
пропилена, метилметакрилата и др., не вращают плоскость
поляризации света ни в растворе, ни в твердой фазе. Хотя
макромолекулы этих И. п. в каждом звене имеют асимметрия,
центр (атом углерода, связанный с четырьмя различными за-
местителями: X, Y и двумя полимерными радикалами различ-
ной длины и с различными группами на концах), два из бли-
жайших соседей у каждого из асимметрия, атомов оказы-
ваются одинаковыми (рис. 1). Точки структурного различия
(концы цепей) в общем случае оказываются удаленными от
асимметрия, центров. В связи с этим большинство третичных
асимметрия, атомов углерода не проявляют оптической актив-
ности, поскольку последняя резко падает с увеличением рас-
стояния между асимметрия, центром и точками структурного
различия. Исключение составляют лишь асимметрич. атомы
на концах макромолекул И. п., вклад к-рых в оптическую
активность исчезающе мал. Это обусловливает практически
полное отсутствие оптич. активности авеньев. При образова-
нии спиральных конформаций в твердой фазе И. п. можно
было бы ожидать проявления оптич. активности, обусловлен-
ной асимметрией спиралей. Однако для И. п., не содержащих
асимметрич. центров в боковых заместителях, равновероятно
образование как левых, так и правых спиралей. Поэтому
кристаллич. фаза такого И. п. представляет собой рацемат из
спиралей обоих типов. В нек-рых гетероцепных И. п., таких,
как полипропиленоксид, содержащий только 1 или только d
конфигурации асимметрических атомов, каждый асимметри-
ческий атом углерода в цепи окружен четырьмя различ-
Рис. 3.
ными ближайшими соседями (рис. 3). Растворы и кри-
сталлы подобных полимеров проявляют оптич. активность.
Специальную группу оптически активных полимеров состав-
ляют И. п., получаемые из оптически активных виниловых
мономеров и содержащие, кроме асимметрия, атомов углерода
в основных цепях, асимметрич. центры в каждом из боковых
заместителей, напр. поли-(8)3-метилпентен-1, поли-(в)4-метил-
гексен-1, поли-(8)-5-метилгептен-1 и др. Оптич. активность этих
полимеров обусловлена оптич. активностью отдельных звеньев
и асимметрией формы возникающих спиральных конформаций.
Разновидностью И. и. являются диизотакти-
чески е полимеры, к-рые могут быть полу-
чены из мономеров типа СНХ—CHY, напр. из дейте-
ропропилена HDC=CH(CH3). В каждом мономерном
звене макромолекулы такого полимера содержится
два асимметрич. атома углерода, один из к-рых соеди-
нен с боковым заместителем X, а другой — с боко-
вым заместителем Y. Диизотактич. полимеры, ха-
рактеризующиеся одинаковыми кон-
таблица 1 фигурациями всех асимметрич. ато-
мов (рис. 4, а), наз. эритро-ди-
изотактическими, в отличие
от т р е о-д иизотактических,
у к-рых все асимметрич. атомы,
связанные с боковыми заместителя-
ми X, имеют одинаковые простран-
ственные конфигурации, противо-
положные конфигурациям асиммет-
рич. атомов, связанных с замести-
телями Y (рис. 4, б).
Химич, свойства И. п. также
могут отличаться от химич. свойств
соответствующих синдиотактиче-
ских полимеров или атактических
полимеров. Это обусловлено разли-
чиями во взаимном расположении
и влиянии функциональных групп,
принадлежащих соседним звеньям
макромолекулы. Так, боковые эфир-
ные группы полиметилметакрилата,
расположенные в изо-положении,
гидролизуются значительно быстрее,
чем боковые группы, расположенные в синдио-поло-
жении. Различия в реакционности функциональных
групп могут быть использованы для определения доли
изотактич. звеньев в полимере.
/ Y Y Y Y Y /
/ \> I v I v I v t v I / f ; Г
/• I _^-iC । I *c I I '
/ V H> H? Н/ a
/ H H H H H /
' н н н н H /
X *C J X .cj A /
I c* 6
H H H H H /
Рис. 4.
И. п. применяются в тех же областях, что и др. кристаллизующиеся полимеры, напр. изотактич. полипропилен производится пром-стью для изготов- ления волокон, пленок и разнообразных деталей. В табл. 2 приведены данные об основных механи- ческих свойствах волокон из полипропилена в срав- нении со свойствами других волокон и стальной проволоки. Таблица 2
Материал Плот- ность, г/см3 Предель- ная проч- ность на разрыв, кг/.мм* Удельная прочность, г/денье Раз- рывное удли- нение, % Работа разры- ва, г/денье
Изотактич. поли- пропилен3 . . . Нейлон Высокопрочный нейлон ...... Сталь 0,92 1.14 1.14 7,8 77 54 78 210 9,3 5,3 7,7 3,0 31 26 14 8 1.6 0,9 0,7 . .0,2
а Атактический полипропилен не образует волокон.
Лит.: Натта Дж., Химия и технология полимеров, сб.
переводов, 1957, № 1, 20; X и р л Дж., там же, 1957:, А 1,
96; Э й р и к Ф., Марк X., там же, 1957, № 5, 18; На т т а
Дж., там же, 1957, К» 5, 139; Д а н у с с о Ф„ Морали оД.,
гам же, 1957, №5, 3; Натта, Фарина, Перальдо,
177
ИЗОТЕРМЫ РЕАКЦИИ УРАВНЕНИЕ - ИЗОТИОЦИАНАТЫ
178
там же, 1961, Ml, 28; Г е й л о р д Н., Марк Г. Ф., Линей-
ные стереорегулярные полимеры, пер. с англ., М., 1962.
ИЗОТЕРМЫ РЕАКЦИИ УРАВНЕНИЕ —“выра-
жение, определяющее изменение одного из изотермич.
потенциалов системы при протекании химич. реакции
при заданных (постоянных) концентрациях реаги-
рующих веществ и при постоянной темп-ре. В наи-
более общей форме И. р. у. имеет вид:
&Z = RT (In к'а - In Ка) (1)
где AZ — изменение соответствующего изотермич.
потенциала, Кп — константа равновесия реакции,
выраженная через активности участвующих в реак-
ции веществ, К'а — выражение, составленное вполне
аналогично константе равновесия, но связывающее
активности этих веществ не в условиях равновесия,
а при заданных концентрациях, Т — абсолютная
темп-pa, R — газовая постоянная. Активности ве-
ществ, содержащихся в системе в виде индивидуаль-
ных жидкостей или твердых тел, одинаковы в выра-
жении Ка и в выражении К'а, поэтому в ур-нпи (1)
они сокращаются. В такой форме И. р. у. является
вполне строгим термодинамич. соотношением.
Для газовых реакций при применимости к ним за-
конов идеальных газов и для реакций в очень разб.
р-рах активные массы веществ, участвующих в реак-
ции, можно выразить через их концентрации в молях
на 1 л (или в других соответствующих единицах).
В этом случае для реакции
ЬВ + dD gG + IL (2)
протекающей при постоянном
нимает вид;
объеме, И. р. у. при-
I cr • CT
AF = RT in
I r. b r'a
\ LB'bD
cf, • Сг
. tr L
1П —----.
bD
(3)
где AF — изменение изохорпо-изотермич. потенциала,
Cq, C£, Cb, Сц — концентрации веществ G, L, В, D
в заданных условиях, a CG, CL, Св, CD — концентра-
ции тех же веществ в условиях равновесия при той
же темп-ре.
Для реакций в идеальных газах AZ=AF. В этом
случае И. р. у. может быть выражено через парциаль-
ные давления компонентов вместо их концентраций,
в. форме вполне аналогичной уравнению (3).
В частном случае, когда все активности, содержащиеся
в выражении К'а уравнения (1) для каждого из веществ, равны
единице, уравнение (1) принимает вид:
AZ» = -RT In Ка (4)
Значения AZ, относящиеся к этим условиям, наз. стандарт-
ными и отмечаются верхним индексом (°). Соотношение (4)
дает возможность находить константу равновесия, если изве-
стно AZ''. Для определения AZ» существуют различные пути;
важнейшими из них являются: 1) расчет на основе потенциа-
лов образования (свободных энергий образования) всех ве-
ществ, участвующих в реакции, и 2) измерение электродви-
жущей силы соответствующей гальванич. цепи.
И. р. у. позволяет установить, в каком направле-
нии и до какой степени может происходить рассма-
триваемая химич. реакция при заданных начальных
условиях. Оно часто применяется при расчетах химич.
равновесий.
Лит.: Вант Гофф Я. Г., Очерки по химической дина-
мике, пер. с франц., Л., 1936; Капустинский А. Ф.,
Термодинамика химических реакций и ее применение в неор-
ганической технологии, 2 изд., М. — Л,, 1935; Карапет ь-
янц М. X., Химическая термодинамика, 2 изд., М. —Л.,
1953; Б рейс к йй А. И., Физическая химия, т. 1, 6 изд.,
М. — Л., 1948; Киреев В. А., Курс физической химии,
2 изд., М., 1955. В. А. Киреев.
ИЗОТИОЦИАНАТЫ (горчичные масла) — органиче-
ские N-производные изороданистоводородной кис- ;
лоты R—N=C=S. Многие из И. встречаются в ра- |
стениях в свободном виде или в виде гликозидов,
нек-рые обладают физиологическим действием и на-
ходят применение в медицине. Аллилизотиоцианат
СН2 = СН—СН2—N = C = S является действующим на-
чалом горчицы.
И. образуются в результате перегруппировки тио-
цианатов (роданидов) при нагревании:
R-S-CeN -»• R—N=C=S
Превращение катализируется солями металлов. Осо-
бенно легко изомеризуются аллилроданиды:
ch2=ch-ch2-s-c=n s=c=n-ch2-ch=ch3
Варианты этого метода — образование И. при дей-
ствии S и NaCN на аллилгалогениды:
CH2=CH-CH2Cl + S !- NaCN -» CH2=CH-CH2-N=C=S
и при взаимодействии роданистого водорода с олефи-
нами:
СНа, сна.
)С=СН2 + HS-C = N )CH-CH»-N=C=S
CHaz 1 CHaz
Ряд методов получения И. основан на разложении
различных производных дитиокарбаминовой к-ты,
соли к-рой образуются при взаимодействии первич-
ных аминов с CSa в присутствии оснований:
nh4oh
RNH24-CS2 -» RNHC(=S)SH ------► RNHC(----S)SNHI
a) RNHC(=S)SNH4 + Pb(NOa)2 -*
-> R—N=C=S + NH4NOa + HNOa + PbS
6) RNHC(=S)SNH4 + C1COC1(OC2H5) -►
-► RNHC(=S)SCOC1(OC2H5) -4.
-» R—N=C=S + NH4CI + COS + HC1(OC2H5)
a) RNHC(=S)SNH4 + 4N.aOCI + NaOH -►
R—N=C=S + 3NaCl + NH4C1 + Na2SO4 + H2O
И. могут быть получены из производных тиомочевииы:
ArNHC(=S)NHAr + НС1 -> Ar-N=C=S + ArNH2 . НС1
ArNHC(=S)NHs -> Ar-N=C=S -f- NHS
и из тиофосгеиа и первичных аминов:
RNH3 + CSC12 -»• [RNHC(=S)C1] -► R—N=C=S + HC1
а также присоединением серы к изонитрилам:
r—n=C + S R—N=C=S
Большая часть реакций И. связана с присоедине-
нием веществ с подвижным атомом водорода:
R—N=C=S + НХ -> R— NHC(=S)X
X=OR, OAr, SH, SR, CN, NH2, NHR, NRR', NHNH.,
NaSOa, NaHSO2Ar
Присоединение карбоновых и тиокарбоповых к-т
сопровождается элиминированием COS (соответствен-
но CS2) с образованием амидов к-т:
R-N=C=S+ R'COOH(SH) ->
RNH--C-O(S)-
i II
. s
-COR'
-< R-NH-C-R’4-CO(S)S
Ь-
Взаимодействие И. с ангидридами к-т приводит
к имидам:
R— N=C=S + (СНаСО)2О RN(COCH3)2 + COS
И. присоединяют Na-малоновый эфир:
R-N=C=S4- NaCH(COOC2Hs)3 -с R-NH-C=C(COOC..H5)2
I
SNa
179 ИЗОТОНИЧЕСКИЕ РАСТВОРЫ - ИЗОТОПНОГО РАЗБАВЛЕНИЯ МЕТОД 180
Присоединение реактивов Гриньяра, а также взаимо-
действие с ароматич. углеводородами в присутствии
А1С13, приводит к амидам тиокислот:
н2о
R-N=C=S -I- R'MgX -* R-N-C-R'---► R-NH-C-R'
XMg S
А1С13
R.-N=C=S 4-АгН---► R-NH-C-Ar
s
s
И. восстанавливаются водородом в момент выделения
до первичных аминов и тиоформальдегида или
замещенных метиламинов и сероводорода:
,r-nh2 4- CH2S
r-N=c=s 4- н2<
*R-NHCH, 4- H2S
И. гидролизуются только при нагревании; кислоты и
щелочи ускоряют процесс:
R—N=C=S 4- 2Н2О RNH» + H2S + СО2
Галогенирование И. приводит к карбиламипгалоге-
нидам:
ДО
R-N=C=S + 2С12 R-N=CC12 4- SC12
При взаимодействии И. с окисью ртути образуются
изоцианаты:
R—N=C=S + HgO -> R-N=C=O 4- HgS
И. реагируют c JFB при нагревании в пиридине:
R—N=C=S 4- JF5 -» R-N-S-N-R
I I
CFg CF3
И. способны вступать в многочисленные реакции цик-
лообразования. Так, при действии азида натрия обра-
зуются меркаптотетразолы:
R— N=C=S 4- NaN3 —> R-N---C-SH
I II
N N
Фенилизотиоцианат с PC15 образует 2-хлорбензти-
азол, а с серой — 2-меркаптобензтиазол:
Различные И. обладают бактерицидными, фунгицид-
ными и инсектицидными свойствами. Нек-рые И.
могут быть использованы для получения полиизотио-
цианатов, модифицирования смол, применяемых в
произ-ве еинтетич. волокон и пр.
Лит.: Houben-Weyl, Bd 9, 4 Aufl., Stuttgart, 1955,
S. 876. Б. Л. Дяткин.
ИЗОТОНИЧЕСКИЕ РАСТВОРЫ (и з о о с м о ти-
пе с к и е растворы) — растворы с одинаковым
осмотич. давлением. Обычно изотоническими наз.
растворы, осмотич. давление к-рых равно осмотич.
давлению крови или внутриклеточной жидкости жи-
вых организмов. Изотоничность р-ров может быть
установлена путем измерения их осмотич. давления
или же любой другой характеристики, связанной
с осмотич. давлением определенной зависимостью
(напр., измерением понижения давления паров рас-
творителя). Если р-ры не являются изотоническими,
то р-р с меньшим осмотич. давлением наз. гипото-
ническим, а р-р с большим осмотическим давле-
нием — гипертоническим. И. р., разделен-
ные мембраной, проницаемой для молекул раствори-
теля и непроницаемой для молекул растворенных ве-
ществ, находятся в состоянии равновесия и не изме-
няют своей концентрации.
В живых организмах внутриклеточная жидкость
обычно находится в изотония, равновесии с внекле-
точной жидкостью, в частности с кровью. Это равен-
ство осмотич. давления клеточного содержимого и
окружающего клетки раствора является важным усло-
вием нормального функционирования клетки. При
нарушении этого условия происходит перемещение
воды из клетки в раствор или обратно, что приводит
к нарушению структуры и нормальной жизнедеятель-
ности клетки. Так, при помещении эритроцитов в ги-
потония. р-р происходит их набухание, сопровождаю-
щееся разрывом клеточных оболочек (гемолиз). По-
этому р-ры, вводимые в живой организм в лечебных
или экспериментальных целях или используемые при
работе с изолированными тканями и клеточными куль-
турами, должны быть изотоническими по отношению
к этим объектам. Для теплокровных животных изо-
тоничны 0,85—0,9%-ный р-р NaCl и 4,5—5%-ный р-р
глюкозы. 0,85%-ный р-р NaCl, к-рый называют фи-
зиология. р-ром, применяют для возмещения объема
крови и поднятия кровяного давления после боль-
ших кровопотерь, а также для возмещения объема
жидкости в организме при различных паталогич.
состояниях, связанных с сильным обезвоживанием
организма (ожоговая болезнь, упорные рвоты и т. п.).
И. р. более сложного состава, приближающиеся по
своему составу, pH, а также буферным и др. свой-
ствам к сыворотке крови или другим биологич. жид-
костям, наз. физиологическими рас-
творами. Наиболее широко применяемые физио-
логия. р-ры приведены в таблице (в г на 1 л р-ра).
Компоненты раствора Раствор Рингера3 Раствор Рингера — Локка6 Раствор Рингера — Тироде0
NaCl 6,5 9 8
КС1 0,14 0,42 0,2
СаС12 042 0,24 0,2
NaHCOa 0,2 0,2 04
MgCl, —— 04
NaH8PO< — — 0,05
Глюкоза — 1 1
Дистиллированная вода . . до 1 л ДО 1 Л до 1 л
а Для холоднокровных животных. 6 Для теплокровных
животных.
В состав И. р., используемых в качестве кровезаме-
нителей, обычно вводят также различные высокомоле-
кулярные соединения (коллидин, декстран и др.)
для создания коллоидоосмотич. (онкотического) да-
вления, определяемого присутствием в крови высоко-
молекулярных, гл. обр. белковых, веществ.
Лит.: [А гр аненко В. А.], Препараты крови и крове-
заменители, М., 1956; Biochemist’s handbook, N. Y., 1961,
p. 58. В. В. Спиричев.
ИЗОТОПНОГО РАЗБАВЛЕНИЯ МЕТОД — метод
количественного анализа, основанный на использо-
вании меченых атомов; состоит в том, что к анализи-
руемому элементу в смеси добавляют известное коли-
чество его изотопа (как правило, радиоактивного) и
по концентрации этого изотопа судят о содержании
определяемого элемента или образуемых им соедине-
ний без количественного выделения анализируемого
объекта из смеси. Измерив концентрацию (А) изотопа
в любой пробе (по отношению к данному элементу)
и зная его количество (В), введенное в смесь, опреде-
ляют общее количество элемента в исходной смеси,
равное В/A. Если в сложной смеси соединений необ-
ходимо следить лишь за каким-либо одним из них, то
радиоактивный изотоп вводят в соответствующее со-
единение; при этом обязательным условием является
отсутствие изотопного обмена между данным соеди-
нением и всеми остальными. Наоборот, если целью
анализа является контроль за определенным элемен-
том в смеси его соединений, необходимо наличие пол-
181
ИЗОТОПНЫЕ ИНДИКАТОРЫ
182
кого и быстрого изотопного обмена данного элемента
между всеми его соединениями в смеси. Возможность
применения И. р. м. обусловлена тем, что изотопы
почти всех элементов распределяются равномерно по
всей смеси или отдельным ее компонентам, и эта рав-
номерность не нарушается при протекании различ-
ного рода процессов, в к-рых участвует данный эле-
мент. Основное преимущество И. р. м. состоит в том,
что, благодаря энергетич. особенностям излучения и
наличию специальных методик точного измерения
радиоактивности, отдельные изотопы могут быть
определены в весьма малых количествах. Напр.,
с помощью счетчика Гейгера можно обнаружить
10’16 г J131 и надежно определять количества J131
порядка 10“14 г. Поэтому к элементу примешивают
лишь незначительное по весу количество радиоак-
тивного изотопа.
Применение И. р. м. часто сопряжено с ограниче-
ниями или необходимостью учета возможных искаже-
ний. Это касается гл. обр. искажений, связанных
с изотопным обменом. При использовании в И. р. м.
образцов высокой уд. активности (0,1—1 кюри) не-
обходимо считаться с возможностью искажений, вы-
зываемых радиационно-химич. процессами. Однако
при обычно применяемых уровнях активности (до
1 мкюри) этот эффект очень мал и может не учиты-
ваться. Используя в И. р. м. легкие изотопы, следует
учитывать возможные изотопные эффекты, к-рые могут
приводить к изменению изотопного состава данного
элемента в ходе процесса. В особенности это касается
изотопов водорода (Н1, D и Т), имеющих наибольшее
относительное различие в массах. Точность И. р. м.
определяется чистотой введенного изотопа, его удель-
ной активностью, соотношением количеств веществ,
введенных с индикатором и имевшихся в исследуемой
пробе, ошибками измерения радиоактивного излуче-
ния. В обычно применяющихся условиях И. р. м.
дает точность порядка нескольких процентов от из-
меряемой величины.
И. р. м. нашел широкое применение в химии, фи-
зике, биологии и технике. Главным образом И. р. м.
распространен в аналитич. химии в тех случаях,
когда использование обычных методов затруднительно
или невозможно. Это прежде всего касается измере-
ния малых количеств веществ, напр. при определе-
нии растворимости плохо растворимых веществ или
при измерении давления пара над твердыми веще-
ствами при относительно низкой темп-ре. И. р. м. широ-
ко распространен в масс-спектрометрии. Проведено
большое число работ при помощи И. р. м. по иссле-
дованию кинетики и механизма химич. реакций,
а также по изучению строения различных соединений,
особенно в органич. химии и биохимии. Использова-
ние И. р. м. дало возможность найти пути решения
ряда биологич. проблем, как, напр., обмен веществ
в живом организме. Все большее применение находит
И. р. м. в технике. Он используется для определения
объема жидкости и газа в больших хранилищах,
концентрации примесей в сплавах, веса металла в раз-
ливочных ковшах и т. д.
Лит.: Методы работы с применением радиоактивных ин-
дикаторов, М., 1955; Бродский А. И., Химия изотопов,
2 изд., М., 1957. Е. А. Борисов.
ИЗОТОПНЫЕ ИНДИКАТОРЫ — вещества, имею-
щие отличный от природного изотопный состав и
благодаря этому используемые в качестве меченых для
изучения самых разнообразных процессов. Роль изо-
топной метки выполняют стабильные или радио-
активные изотопы химич. элементов, к-рые легко
могут быть обнаружены и определены количественно.
Высокая чувствительность и специфичность И. и.
позволяют проследить за ними в сложных процессах
перемещения, распределения и превращения веществ
в сколь угодно сложных системах или непосредственно
в живых организмах, что дает возможность судить
о скоростях химич. реакций и о их механизме. В осо-
бенности велика роль И. и. для выяснения вопросов,
изучение к-рых другими методами невозможно или
малоэффективно, напр. для изучения коэфф, самодиф-
фузии различных тел и в частности металлов, для уста-
новления различной степени усвоения растениями
фосфора, внесенного с удобрениями и первоначально
содержавшегося в почве, для наблюдения за процес-
сами жизнедеятельности животных и растительных
организмов, а также для решения многих других
проблем, казавшихся ранее совершенно неразреши-
мыми.
Метод И. и. был впервые предложен Г. Хевеши и
Ф. Панетом в 1913 для определения растворимости
труднорастворимых солей свинца с использованием
в качестве индикатора солей свинца, содержащих его
радиоактивные изотопы. Несколько позже этот метод
применил Вл. И. Спицын при изучении соединений
тория. Широкое использование И. и. стало возмож-
ным после разработки методов выделения изотопов,
открытия искусственной радиоактивности, достиже-
ний в области ядерной техники.
Общая характеристика метода изотопных индика-
торов. В основе метода И. и. лежит предположение
об идентичности физич. и химич. свойств изотопов
(отсутствие заметных изотопных эффектов, обусло-
вленных относительно большим различием в массах
изотопов). При использовании метода необходим учет
возможных реакций изотопного обмена, приводящих
к перераспределению меченых атомов и, следова-
тельно, к потере соединением метки, и радиационных
эффектов, вызванных ионизирующим действием излу-
чения радиоактивных изотопов. Изотоп, используе-
мый в качестве метки, непосредственно вводится
в состав изучаемых химич. соединений. При этом
в большинстве случаев его положение в молекуле
должно быть строго определенным. Могут быть ис-
пользованы как стабильные, так и радиоактивные
изотопы.
Преимуществом стабильных изотопов является их
устойчивость и отсутствие ядерных излучений. Однако
только небольшое число элементов имеет подходящие
стабильные изотопы. Малая доступность последних
и сравнительно сложная техника обнаружения, на-
личие изотопных эффектов в случае легких элемен-
тов — составляют недостатки метода И. и. с приме-
нением стабильных изотопов. Преимуществом радио-
активных изотопов является возможность их получе-
ния практически для всех элементов периодич. си-
стемы, высокая чувствительность, специфичность и
точность определения, простота и доступность изме-
рительной аппаратуры. Радиационные же эффекты
могут быть устранены применением низких индика-
торных концентраций. Поэтому большинство исследо-
ваний, использующих метод И. и., выполнено с ра-
диоактивными изотопами.
В настоящее время известно более 270 стабильных и бо-
лее 1000 радиоактивных изотопов. В качестве стабильных
изотопов чаще всего используют Н3, С13, N1*, О'8 и др. Такие
элементы, как водород, углерод, сера, хлор, свинец, имеют
удобные для использования как стабильные — Н3, С13, S34,
С135, С137, РЬ8»4, так и радиоактивные изотопы—Н3, С11, С14,
S35, С138, РЬ31*. Стабильные И. и. получают обогащением при-
родных изотопных смесей путем многократного повторения
операции разделения (перегонка, диффузия, термодиффузия,
изотопный обмен, электролиз, см. Изотопов разделение), а
также при помощи масс-спектрометрич. установок и по ядер-
ным реакциям.
Для элементов, существующих в природе в виде одного
изотопа (бериллий, фтор, натрий, алюминий, фосфор, иод),
в качестве меченых атомов используются только искусствен-
ные радиоактивные изотопы. Из них наиболее часто применя-
ются Н3, С14, Р3*, S3S, Са4Б, Сг31, Fe33, Со*», Sr83, Zr33, Nb35,
Ag11», J131 и др. Выбор радиоактивного изотопа определяется
его ядерными характеристиками — периодом полураспада,
типом и энергией излучения. Для индикации пригодны ра-
183
ИЗОТОПНЫЕ ИНДИКАТОРЫ
184
диоактивные изотопы, период полураспада к-рых не очень мал,
что позволяет работать в течение времени, необходимого для
эксперимента, но и не очень велик, что дает возможность ра-
ботать с весьма малыми количествами индикатора.
Основным методом анализа стабильных изотопов служит
масс-спектрометрия (чувствительность 10"*% изотопа при
точности 0,1—1% для проб весом в доли Л1г). Все большее
применение находят спектральные методы (ИК и высокоча-
стотные спектры) и парамагнитный резонанс (см. Изотопов
стабильных анализ). Дейтерий, О1в и нек-рые др. изотопы опре-
деляют по изменению показателя преломления, теплопровод-
ности, плотности как самого элементарного вещества, так и его
соединений. Денсиметрич. методом устанавливают изотопный
сос'гав воды с точностью до десятитысячных долей процента.
Количественное определение стабильных изотопов тем менее
надежно, чем тяжелее изотоп.
Радиоактивные изотопы определяют по их излучению при
помощи счетчиков Гейгера или сцинтилляционных счетчиков.
Уд. активность радиоактивных индикаторов обычно выра-
жается в единицах радиоактивности на единицу веса вещества.
Измеряемые активности составляют до 10~* мккюри
(1 мккюри С14 соответствует 2 • 10"7 г этого изотопа; 1 мккюри
рза — з . Ю"1’ г соответствующего). При помощи радиоавто-
графии могут быть обнаружены и отдельные атомы изотопа.
Обычная точность радиометрич. измерений составляет 1—3%,
а в отдельных случаях—6,1 %.
Оценка изотопного состава в любой момент времени про-
водится измерением уд. активности, выражающей относитель-
ное содержание данного изотопа в элементе или долю молекул
данного вИда,’ содержащих в своем составе изотоп, употребляе-
мый в качестве метки;
Все измерения радиоактивных препаратов произ-
водят в идентичных условиях, позволяющих оцени-
вать количество изотопов прямым сравнением изме-
ренной активности. При работе с радиоактивными
изотопами необходимо учитывать особенности радио-
химия. методов исследования, к к-рым относятся:
изменение количества радиоактивного изотопа во
времени; возможность потери вещества вследствие
адсорбции и коллоидообразования; наличие радиа-
ционных эффектов, для уменьшения к-рых стремятся
работать с возможно более низкими концентрациями
радиоактивных изотопов; необходимость соблюдения
правил техники безопасности в соответствии с су-
ществующими нормами (см. Защита от излучений
радиоактивных веществ).
Известны различные способы синтеза меченых соеди-
нений (см. Меченых соединений синтез). Наряду с обыч-
ным химич. синтезом используются реакции изотоп-
ного обмена и биологич. синтез. При метке соедине-
ния двумя изотопами в индикаторных количествах
считают, исходя из статистич. представлений, что
молекул, содержащих одновременно два меченых
изотопа, практически не встречается. В большинстве
случаев изотопная метка занимает определенное поло-
жение в молекуле, напр. используемая в качестве
И. и. пропионовая кислота может быть помечена
следующим образом: С14Н3СН3СООН, СН3С14Н2СООН,
СН3СН2С44ООН.
Пути применения изотопных индикаторов. Имеют-
ся три основных направления использования И. и.
Методом И. и. изучают характер распределе-
ния веществ и пути их перемещения.
И. и. вводят в ту или иную систему и через определен-
ные промежутки времени устанавливают наличие
И. и. в различных частях системы. Наиболее нагляд-
ные картины распределения получаются без разру-
шения образца при помощи радиоавтограмм (см.
Радиоавтография). Используя, напр., радиоавтографы
кристаллов, можно убедиться, распределяется ли
примесь равномерно по всему кристаллу или только
по отдельным его ребрам и граням; радиография
растения показывает, что фосфор, введенный с удоб-
рением, накапливается преим. в растущих частях
растения, Возможность следить за перемещением
И. и. в самых различных процессах используется при
проверке аналитич. и технологич. схем, при изучении
путей и скорости обмена веществ в организме, при
оценке эффективности удобрений) при исследовании
свойств лекарств и т. д.
Другое направление использования И. и. — ко-
личественный анализ. Один из самых
простых и распространенных вариантов метода И. и.—
метод изотопного разбавления, позволяет производить
определения ничтожно малых количеств трудноопре-
деляемых веществ, анализировать сложные смеси,
анализ и разделение к-рых другими методами невоз-
можны (см. Изотопного разбавления метод).
Широкими возможностями отличается активационный ме-
тод анализа (см. Радиоактивационный анализ)^ основанный
на осуществлении ядерных реакций, приводящих под дейст-
вием облучения к образованию определенных радиоактивных
изотопов, т. е. И. и. Образец облучают нейтронами, у-лучами
или заряженными частицами. В зависимости от состава исход-
ного образца в нем образуются те или иные радиоактивные
изотопы, к-рые могут быть идентифицированы и количест-
венно определены по излучению без химич. разложения обг
разца. Использование эталонов с известным содержанием оп-
ределяемого элемента существенно упрощает методику ана-
лиза. Количество образующегося изотопа зависит гл. обр.
от величины эффективного сечения данной реакции и опре-
деляется свойствами ядра, а не электронной оболочки. Этр
обусловливает высокую специфичность активационного ана-
лиза, возможность одновременного определения нескольких,
близких по свойствам, элементов и измерения ничтожных сле-
дов примесей, не открываемых даже спектральным анализом.
Если в результате облучения получают смесь изотопов несколь-
ких элементов, анализ к-рой нельзя произвести с помощью
измерения излучения, то прибегают к химич. разделению
смеси с добавлением носителей. Таким образом могут быть
практически определены одновременно более десяти элементов.
Особенно большое значение этот метод имеет при определении
микроэлементов в металлах, сплавах, минералах, тканях,
быстром контроле технологич. процессов, исследовании архео-'
логич. находок и история, ценностей.
Третьим направлением использования И. и. яв-
ляется выяснение механизма различных
процессов и изучение строения химич.
соединений. Введение изотопной метки в определен-
ное положение молекулы устраняет химич. неразли-
чимость атомов, допуская возможность однознач1
ного выяснения механизма тех или иных реакций',
для к-рых обычные химич. методы описывают только
начальное и конечное состояния. Так, напр., методом
И. и. удалось решить вопрос о механизме гидролиза
сложных эфиров; возможны два пути, дающие одина-
ковые продукты:
О : О :
Я : tR Hi
Т. R-C-O-R' -J-Н О—Н “. R-C-Of H-J-R'-O18H
ИЛИ
О : О :
И. R-C-O-R' + Н-018 !-Н — R-C-O1SH + R'0-i-H
Выбор между алкилкислородным (СО—В') и ацил-
кислородным (С—ОН') механизмом однозначно произ-
водится при использовании воды, обогащенной О18,
в пользу II схемы, если тяжелый кислород обнару-
живается только в кислоте.
При окислении пропионовой к-ты перманганатом
образуются щавелевая к-та и углекислый газ. При
помощи метода И. и. удалось установить, откуда
отщепляется атом углерода, образующий углекислый
газ, — из карбоксильной группы или углеводород-
ной части молекулы. Использование И. и. с меченым
углеродом показало, что возможны оба пути окисле-
ния, причем изменение pH среды изменяет соотноше-
ние между обоими путями окисления.
Метод И. и. используется для выяснения неравно-
ценности связей различных атомов одного и того же
химич. элемента в молекуле вещества. При этом
вводят метку в определенное положение в молекуле
и изучают реакции, в к-рых участвуют не все атомы.
Таким образом было подтверждено, что в кристаллич.
решетке пятихлористого фосфора, состоящего . из
ионов РС1, и РС1 в, связи Р—CI различны. Неравно-
ценными оказались атомы железа в Fe4[Fe (GN)e]3,
что соответствует структурным данным. Неравно-
ценность атомов серы в тиосульфате легко продемои-
185
ИЗОТОПНЫЕ ИНДИКАТОРЫ —ИЗОТОПНЫЕ ЭФФЕКТЫ
188
стрировать, проведя синтез кипячением сульфита
с меченой элементарной серой. После разложения
тиосульфата метка S36 полностью остается в элемен-
тарной сере: Na3SS36O3 —► Na2SO3 S36.
В качестве варианта метода И. и. для выяснения
механизма нек-рых реакций и строения химич. соеди-
нений может быть использован изотопный обмен..
Способность к изотопному обмену определяется строе-
нием молекул и природой заместителей, реакцией
среды, наличием сольватации и ассоциации, окисли-
тельно-восстановительных процессов, катализаторов
и т. д. По зависимости константы скорости изотоп-
ного обмена от темп-ры определяют энергию акти-
вации реакции обмена, что позволяет судить о ха-
рактере химич. связи, ее реакционной способности
и о подвижности атомов и групп.
Области использовании изотопных индикаторов. Все ука-
занные направления применения И. и. широко используются
в различных областях химии, биологии, медицины, техники,
сельского х-ва и т. д. Ниже приводятся отдельные примеры
их использования.
В йеор гания ес кой химии И. и. использова-
лись для доказательства существования неустойчивых соеди-
нений (гидридов свинца, гидратов благородных газов); для
определения состава и строения комплексных соединений,
строения окислов и кислот; для изучения механизма образо-
вания тионатов и политионатов, гетерополисоединений и др.,
механизма реакций с перекисью водорода и процессов ее ката-
литич. разложения и т. д. И. и. принадлежит большая роль
в открытии и установлении места в периодич. системе новых
элементов — технеция, прометия, астатина, трансурановых
элементов.
В аналитической химии И. и. нашли много-
численные применения в анализе редких и рассеянных эле-
ментов, минералов, рудных и горных пород, промышленных
отходов и концентратов. Разработаны методы быстрого и точ-
ного контроля близких по свойствам элементов — Nb и Та,
Zr и Hf, Se и Те, редкоземельных и др. элементов.
-И. и. незаменимы при оценке и усовершенствовании мето-
дов количественного разделения; они дают ответ на вопросы
количественно ли разделение, соосаждаются ли примеси, про-
исходят ли потери при нагревании, прокаливании и т. п.
Благодаря И. и. стала возможной объективная оценка точ-
ности, усовершенствование старых и создание новых анали-
тич. методов; при этом существенно повысились чувствитель-
ность и точность определений, стали возможными надежные
методы определения кларковых количеств элементов в поро-
дах и метеоритах, полупроводниках с содержанием примесей
порядка 10’’—10-10 г/г.
В области органической химии метод И. и.
весьма эффективен при выборе возможных схем процессов.
Напр., путем введения С14 в различные положения молекулы
углеводорода и изучения продуктов окисления была опроверг-
нута теория деструктивного окисления и предложен радикаль-
но-цепной механизм процесса. Существование промежуточ-
ных комплексов в реакции обмена между галогеналкилом и
галогенидионом подтверждает механизм вальденовского обра-
щения, поскольку скорость изотопного обмена совпадает со
скоростью рацемизации. Для объяснения кинетич. особенно-
стей реакции Меншуткина R3N + RX R4 N++ Х~ предпо-
лагалось существование промежуточного комплекса, однако
это не подтвердилось опытами с мечеными тритием алкилиоди-
дами.
В физической химии И. и. используются для
определения коэфф, активности, изучения диффузии и само-
диффузии, определения упругости пара, растворимости, рас-
пределения вещества между фазами, выяснения механизма
реакций, усовершенствования методов приготовления катали-
заторов и т. д. При изучении реакций, протекающих с обра-
зованием промежуточных веществ, предложен кинетич. изо-
топный метод, позволяющий устанавливать путем совместного
решения уравнений кинетики процесса и изменения изотоп-
ного состава, генетич. взаимоотношения веществ, участвующих
в сложных химич. реакциях. Возможности этого метода были
продемонстрированы на примере изучения механизма окисле-
ния метана. При помощи С140 было убедительно доказано
существование активных центров на поверхности никелевого
катализатора. Радиоактивные И. и. используются для опре-
деления поверхности кристаллич. веществ путем изучения
реакций гетерогенного обмена ионов между поверхностью и
раствором, в этом случае получают значение «активной» по-
верхности, что более важно, чем оценка чисто геометрич. по-
верхности. Радиоактивный метод определения поверхности
нашей применение при оценке активной поверхности катали-
заторов и цементов.-
С И. и. изучалась кинетика окислительно-восстановитель-
ных реакций, процессы обмела между металлом и ионами,
применимость закона Нернста при ультрамалых концентра-
циях й'т. д. Частичное замещение в молекулах одних изотоп-
ных атомов на другие вызывает смещение и расщепление
спектральных линий, помогающее расшифровать спектр и
выяснить строение сложных молекул.
В биохимии И. и. используются для выяснения строе-
ния молекул белков и нуклеиновых к-т, а также механизма
синтеза нуклеиновых к-т, белков, жиров и углеводов в живом
организме. С помощью изотонов углерода, кислорода и водо-
рода удалось выяснить механизм нек-рых стадий фотосинтеза.
Установлено, что весь выделяемый растениями кислород полу-
чается из воды, а углерод, поглощающийся растением в виде
углекислого газа, через короткое время, пройдя ряд промежу-
точных стадий, оказывается в сложных органич. соединениях,
являющихся основой жизнедеятельности растений.
В геологии своеобразной разновидностью метода
И. и. является использование изменения изотопного состава
естественных радиоактивных элементов в результате радио-
активных превращений иля благодаря нарушению радиоак-
тивных равновесий в процессах, миграции. Первый эффект
используется в радиоактивных методах определения возраста
геологич. формаций (см. Возраст геологический абсолютный)',
изучение количественных соотношений между членами есте-
ственных радиоактивных семейств дает ценные сведения о пу-
тях миграции, позволяет устанавливать генезис месторож-
дений.
И. и. в технике и промышленности позво-
ляют решать ряд важнейших проблем. В металлургии мето-
дом И. и. установлены факторы, влияющие на процессы ста-
леварения в мартеновских и электрич. печах, изучен процесс
расплавления ферросплавов в жидком металле и распределе-
ния легирующих материалов при выплавке стали, определена
скорость шлакообразования, исследовано движение газов и
шихтовых материалов в доменных печах. Радиоактивные
индикаторы используются при определении прочности состав-
ных частей сплавов, при выявлении причин брака, при ана-
лизе металлов, для контроля процессов термич. обработки
металлов и т. д. В металлообрабатывающей пром-сти И. и. при-
меняются для определения износа материала трущихся по-
верхностей. В нефтяной пром-сти — при исследовании буро-
вых скважин и изучении режима грунтовых вод. При перека-
чивании по нефтепроводам различных нефтепродуктов метят
первые порции каждого вида, чтобы определять границу раз-
дела.
Широкие возможности открываются перед применением
И. и. в биологии, медицине, почвоведении,
сельском хозяйстве. В медицине метод И. и. по-
зволяет успешно изыскивать новые меры борьбы с инфекцион-
ными заболеваниями, проводить диагностику, изучать дейст-
вие лекарственных препаратов, выяснять характер обмена
веществ в нормальных и больных организмах. Ветеринария
использует меченые лекарственные соединения для преду-
преждения и диагностики заболеваний с.-х. животных. В энто-
мологии изотопы фосфора, железа, серы и др. служат для
наблюдения за поведением насекомых, их питанием, мигра-
цией, физиологией, переносом ими различных бактерий,
грибков и вирусов. В рыбном хозяйстве И. и. применяются для
подсчета числа мальков, в водоемах. Использование изотопов
фосфора, азота, серы и др. элементов дало возможность выяс-
нить ряд вопросов, связанных с питанием растений.
Использование изотопов фосфора, углерода и т. наз. мик-
роэлементов позволило рационально подойти к составлению
кормового рациона, изучить динамику движения этих эле-
ментов во всех частях организма животного, исследовать фак-
торы, влияющие на рост домашних животных. При мелиора-
тивных исследованиях И. и. помогают определять влажность
почвы, выяснять режим движения воды и минеральных солей
в грунтах, устанавливать количество промывных вод при от-
мывке засоленных почв. При помощи меченых соединений была
показана возможность измерений объема и веса зеленой массы
растений на корню.
И. и. позволили решить много сложных научных и прак-
тич. вопросов и получить высокий экономич. эффект в ряде
произ-в.
Лйт.: Бродский А. И., Химия изотопов, 2 изд.,
М., 1957; Рог и*н с к и й С. 3., Теоретические основы изо-
топных методов изучения химических реакций, М., 1956;
Использование радиоактивности при химических исследова-
ниях, пер. с англ., М., 1954; X е в е ш и Г., Радиоактивные
индикаторы..., пер. с англ., М., 1950; Изотопы и излучения в
химии, М., 1958 (Тр. Всес. научно-технич. конференции); К о-
м а р С., Радиоактивные изотопы в биологии и сельском
хозяйстве, пер. с англ., М., 1957; Применение меченых атомов
в аналитической химии [Докл. на конференции], М., 1955;
Изотопы в биохимии. Сб. статей, пер. с англ., под ред. А. М. Ку-
зина, М., 1953; Труды международных конференций по мир-
ному использованию атомной энергии, Женева, 1955, 1958,
М., 1957—59; Брода Э., Шенфельд Т., Применение
радиоактивности в технике, пер. с нем., М., 1959; Радиоактив-
ные изотопы и ядерные излучения в народном хозяйстве СССР.
Тр. Всес. совещания 12—16 ацр. i960 г., т. 1—4, М., 1961;
Курсанов А. Л., Меченые атомы в разработке научных
основ питания растений, М., 1954; А р о нов С., Изотопные
методы в б ио химии, пер. е англ.., М., 1959; Применение радио-
активных изотопов в аналитической химии, [Труды комиссии
по аналитической химии, т. 9 (12)], М., 1958; Radioisotopes
in scientific research. Proceedings of the International confe-
rence held in Paris in September, 1957, v. 1—4, L. — N. Y. —
P. — Los Angeles, 1958 (UNESCO). К. Б. Заборенко.
ИЗОТОПНЫЕ ЭФФЕКТЫ — неидентичность
свойств изотопов данного элемента, обусловленная
187
ИЗОТОПНЫЕ ЭФФЕКТЫ
188
различием масс изотопных атомов (атомных весов).
И. э. проявляются в различии любых свойств изото-
пов, кроме радиоактивных. Однако поскольку для
изотопов большинства элементов (за исключением наи-
более легких) относительное различие в атомных весах
изотопов невелико, то И. э. для этих элементов выра-
жены весьма слабо. Даже для легких элементов вто-
рого периода системы Менделеева (Li — Ne) относи-
тельные различия в атомных весах изотопов не пре-
вышают 35%; для третьего периода (Na—Аг) они
не превышают 20%, для четвертого (К.—Кг) и пя-
того (Rb—Хе) периодов— 15%; для более тяжелых
элементов они всегда менее 10%. Лишь для элемен-
тов первого периода (Н и Не) относительные разли-
чия в массах изотопных атомов весьма велики — для
водорода максимальное различие достигает 200%,
а для гелия— 100%.
Неодинаковые атомные веса изотопов обусловли-
вают определенные различия таких свойств изотоп-
ных соединений, как плотность, вязкость, показа-
тель преломления, коэфф, диффузии, уд. заряд ионов
и др. При этом отношение плотностей изотопных
соединений достаточно точно совпадает с отношением
их молекулярных весов, а удельные заряды изотоп-
ных ионов обратно пропорциональны их молекуляр-
ным весам. Кроме того, различие масс изотопных
атомов вызывает изменение уровней поступательной,
вращательной и колебательной энергии молекул
при их изотопном замещении, что приводит к разли-
чию колебательно-вращательных спектров изотопных
соединений. В табл. 1 сопоставлены колебательные
частоты ые (см. Молекулярные спектры) ряда двух-
атомных изотопных молекул.
Молекула й>е(см~!) Молекула a>e (cm-1 )
н 4405,3 HCl35 2988,95
3118,8 DCl38 2143,52
Т2 2546,5 HCl37 2987,5
2359,6 HBr78 2649,67
Na DBr78 1885,95
nF 2279,6 HBr87 2646,6
1580,4 L17H 1405,65
О а Li’D 1055,12
О 2® 1490,0 L1«H 1420,32
«Г 564.9 Na!3H 1170,8
cii7 549,4 Na23D 845,3
323,86 C12Q16 2167,4
C13Q16 2119,2
Brf1 319,84 C12O78 2115,2
jF7 214,36 NHQie 1£ 05,54
jF9 212,69 N15Q16 1872,34
Изменение указанных энергетич. уровней при изо-
топном замещении, в свою очередь, вызывает измене-
ние термодинамич. свойств, таких, как теплоемкость,
теплопроводность, теплоты испарения и плавления,
темп-ры кипения и плавления, давление насыщенного
пара и др. Так, напр., отношение давлений пара Н2
и D2 составляет 2,448 при —251,1°; отношение давле-
ний пара Н2О и D2O составляет 1,148 при 20° и 1,052
при 100°; соответствующее отношение для Н2О16
и Н2О18 составляет 1,009 при 23° и 1,003 при 100°;
для N14H3 и Ni6H3 оно равно 1,0053 при —75,4° и
1,0025 при —33,7°.
Что касается химич. свойств изотопных соединений,
то они остаются в основном неизменными, т. к. масса
атома не влияет на его электронную конфигурацию,
определяющую химич. свойства. Однако термодина-
мич. неравноценность изотопных соединений приво-
дит к неравномерному распределению изотопов при
равновесии изотопного обмена (термодинамич. И. э.),
а также к преимущественной адсорбции одной из
изотопных форм на сорбенте. Кроме того, термодина-
мич. неравноценность исходных изотопных соеди-
нений в сочетании с аналогичной неравноценностью
переходных состояний (активных комплексов) при
химич. реакциях изотопных соединений обусловли-
вает различие в скоростях протекания этих реакций
(кинетич. И. э.). Поскольку термодинамич. икинетич.
И. э. зависят от различий колебательно-вращательных
и поступательных энергетич. уровней изотопных
молекул, то, исходя из данных колебательного спек-
тра этих молекул, можно рассчитать указанные И. э.
статистич. методами. При этом существенно, что
точность расчетных значений термодинамич. И. э.
обычно превышает точность эксперимента. Для ки-
нетич. И. э. расчет носит сугубо ориентировочный
характер из-за отсутствия необходимых спектраль-
ных данных для переходных состояний соответствую-
щих химич. реакций. Термодинамич. И. э., выражен-
ные отклонениями от единицы коэфф, равновесного
распределения изотопов, для изотопного обмена водо-
рода в случае трития и протия могут максимально
достигать при 20° 16—18-кратного значения, а в слу-
чае дейтерия и протия — 8—9-кратного значения;
для изотопного обмена других легких элементов —
таких, как литий, бор, углерод, азот, хлор, эти откло-
нения редко превышают 10%, а в случае более тяже-
лых элементов они обычно не превышают 1%.
Кинетич. И. э., выражающиеся отношением кон-
стант скоростей химич. реакций для различных изо-
топных соединений, в случае изотопов водорода также
могут быть Очень велики. Так, напр., отношение
констант скоростей синтеза бромистого водорода щ
бромистого дейтерия равно 5. Для изотопов всех
других элементов отклонения этого отношения от
единицы никогда не превышают 50%. Так, напр.,
отношение констант скоростей разложения малоно-
вой к-ты, обычной и содержащей изотоп С13, на угле-
кислый газ и уксусную к-ту равно 1,02 (при 138°).
Соответствующее отношение для разложения бромма-
лоновой к-ты, обычной и меченной изотопом С14, на
углекислый газ и бромуксусную кислоту состав-
ляет 1,41 (при 115°). Это отношение для разложе-
ния фталамида, обычного и содержащего изотоп
N1S, на фталимин и аммиак равно 1,016 (при 136°)
и т. д.
В основе использования изотопов в качестве мече-
ных атомов лежит их химич. и физико-химич. иден-
тичность (см. Изотопные индикаторы). Фактически
же всегда имеют место различия в свойствах изотопов,
характеризуемые значениями И. э. Т. обр., знание
И. э. позволяет вносить поправки на различие свойств
изотопов при их использовании в качестве меченых
атомов. Очевидно, что учет соответствующих попра-
вок имеет существенное значение при работе лишь
с изотопами легких элементов и особенно водо-
рода.
Различия в свойствах изотопов позволяют разделять
изотопы и определять их содержание в изотопных
смесях. Любой способ разделения изотопов, а также
количественный анализ стабильных изотопов осно-
ваны на наличии И. э. (при этом метод разделения тем
эффективнее, чем больше соответствующий И. э.,
см. Изотопов разделение и Изотопов стабильных
анализ). Так, напр., ректификационный метод разде-
ления основан на различии в давлениях пара изотоп-
ных соединений. В основе метода центрифугирования
лежит различие плотностей. Диффузионный метод
предполагает различие коэфф, диффузии. Методы
разделения с использованием реакций изотопного
обмена основаны на термодинамич. И. э. Имеются
методы разделения изотопов, основанные на кинетич.
И. э., как, напр., широко распространенный электро-
189
ИЗОТОПНЫЙ ОБМЕН
190
литпч. метод получения тяжелой воды (см. Дейтерий.),
и т. д.
Лит.: Рогинский С. 3., Теоретические основы изотоп-
ных методов изучения химических реакций, М., 1956; Брод-
ский А. И., Химин изотопов, 2 изд., М., 1957; Химин изо-
топов, пер. с англ. Сб. 1—2, М., 1948. С. Э. Вайсберг.
ИЗОТОПНЫЙ ОБМЕН — реакция, единственным
результатом к-рой является перераспределение изото-
пов какого-либо элемента между реагирующими
веществами. При И. о. вещества сохраняют неизмен-
ным свой элементный состав и переходят лишь из
одних изотопных форм в 'другие, напр.:
Н..0 + HD = HDO + Н2; 1ЩН+ + N1SH3 = N1SH+ -)- N1«H3;
(Hg=00)+ -р (Hg203)3+ = (Hg203)+ .J- (Hg200)2+
и t. д. Такие реакции могут протекать также между
различными изотопными формами одного и того же
вещества, напр.: Н2О D2O = 2HDO. И. о. является
частным случаем обмена одинаковыми атомами или
группами атомов; принципиальная возможность та-
кого обмена была предсказана еще Менделеевым
в 1886, но обнаружить это удалось только при по-
мощи изотопов, исследуя реакции И. о. Возможности
проведения этих реакций весьма различны: они могут
протекать в гомогенных условиях (между растворен-
ным веществом и растворителем, между реагирующими
веществами в нейтральном растворителе, в смеси
газов и т. д.), а также в гетерогенных условиях
(между твердым или жидким веществом и нераствори-
мым газом, между газами на поверхности твердого
катализатора и т. д.).
Термодинамика изотопного обмена. Равновесие И. о.
можно характеризовать коэфф, распределения изото-
пов и константой равновесия реакции. Коэфф, распре-
деления наз. величина, показывающая, во сколько
раз отношение равновесных концентраций изотопов
в одном из реагирующих компонентов больше соот-
ветствующего отношения в другом. Константа равно-
весия представляет собой отношение равновесных
концентраций конечных и начальных изотопных
форм реагирующих компонентов. При И. о. между
компонентами АХП и ВХт (где элемент X имеет изо-
топы Х° и X *; А и В — части молекул без элемента X,
а п и т — числа атомов элемента X в молекулах
компонентов) протекает ряд обменных реакций:
О •t’ О О
_ 1 + BXm _ jX j _ AXn _ jXj +
О *
+ _ j-_|_ i Xj _ i
где i = 1, 2, 3,..., n; / = 1, 2, 3,...,m с константами
равновесия. s
[ AX„ _ jX j ] [ BXm _ j jXj_j]
Kii □ * c SA
[AXn—i_|_jXj — ,] [BXm—jXyl
где величины в квадратных скобках являются кон-
центрациями соответствующих изотопных форм. Коэфф,
распределения выражается отношением:
*
([ *]/[«’]) BXm
где индексы АХП и ВХт обозначают компоненты,
для к-рых берутся отношения равновесных концен-
*
траций изотопов [х] и [х°]. Коэфф, распределения а,
помимо экспериментального измерения, может быть
вычислен по спектральным данным методами ста-
тистич. термодинамики. При этом, если колебания
в молекулах гармоничны и все атомы X в каждом из
компонентов равноценны, то:
II(z/shz)A, • (z/shz)A>>
“ “'П (z/shz)B, . (z/shz)B„
где: shz = (1—e~2z)/2e~~z (гиперболический синус),
z = hcti>J2kT, h — Планка постоянная, k — Больц-
мана постоянная, с — скорость света, Т — абсолют-
ная темп-ра, со — основная колебательная частота
(см1), индексы А', А", В' и В" обозначают соответ-
* *
ственно изотопные формы АХ^_4Х4, AXJl_i^_1Xi_1(
*
BX^—j-Xj и BX^j^j Xj_4, П—знак произведе-
ния по всем основным колебательным частотам в мо-
лекуле. Формула Относится к И. о., проводимому
в газовой фазе. Для точных вычислений в расчетные
формулы вводят поправки на ангармоничность коле-
баний в молекулах. Отклонение распределения изото-
пов от равномерного при равновесии И. о., харак-
теризуемое отклонением а от единицы, Очень неве-
лико для изотопов всех элементов, кроме водорода.
Для изотопов водорода, наряду с системами веществ
с малым отклонением а от единицы, часто встречаются
системы с резко неравномерным распределением.
В таблице приведены значения а при 25° для ряда
изотопов и систем веществ (при этом за а взято част-
ное от деления Отношения концентрации тяжелого
изотопа к концентрации легкого для первого из ука-
занных компонентов на это отношение для второго).
Константы равновесия И. о. связаны с величиной а
соотношениями:
где <т — число симметрии молекулы, т. е. общее
число эквивалентных положений, к-рые может иметь
молекула в результате полных поворотов вокруг
каждой из ее осей симметрии.
Коэффициент распределения изотопов « для нек-рых
изотопов и систем веществ (при 25”)
Системы
И. о. водорода (D и Н1)
Н2О/Н2 3,352
H.O/HL1 7,241
НгО/НС1 2,359
Н2О/НВг 2,881
H2O/HJ 3,603
И. о. водорода (Т 1 HI)
Н2О/Н2 5,353
h2o/hf 1.246
Н2О/НС1 3,340
Н2О/НВг 4,443
H2O/HJ 6,138
И. о. лития (Li7 и Lie)
LiH/Li 1,025
И. о. бора (В11 и В10)
BF3/BC13 1,090
BF3/BC13 (1КИДК.) . . 1,076
BF3/BBr3 (жидк.) . . 1,101
И. о. углерода (C13 и CIS)
со;/со* 1,012
соГ/° 1,023
CO=-/HCN 1,078
соГ/со 1,099
cor/CN- 1,109
Системы
«25°
И. о. азота (N1» и №<)
nh,/n2 1,023
NHt/NHs 1,035
NHf/NO 1,038
NHf/HCN 1,038
Nh+/CN“ 1,039
И. о. кислорода (О18 и О1е)
COj/CO 1,011
CO2/SO4 1,023
CO2/SO2 1.026
CO2/O2 1.033
CO2/H2O 1,047
И. о. хлора (С137 и С133)
C1OJ/C10-...........
cior/cioi...........
сюг/С12.............
сю;/нс1.............
1,035
1,052
1,077
1,080
И. о. брома (Вг81 и Вг7®)
ВгО3 /Вг2
ВгО3/НВг
1 1,007
I 1,007
И. о. иода (Jiao и J12>)
J о3 /J2
1,003
Кинетика изотопного обмена. Специфич. особен-
ность реакций И. о., отличающая их от обычных (эле-
ментных) химич. реакций, состоит в том, что концент-
рации реагирующих компонентов остаются неизменны-
ми, а изменяется лишь их изотопный состав. Эта
особенность приводит к тому, что эти реакции, незави-
симо от их истинного механизма, практически могут
быть описаны кинетич. ур-нием первого порядка:
191
ИЗОТОПНЫЙ ОБМЕН
192
где к — константа скорости реакции, х0, х и хр —
концентрации изотопа в веществе, соответствеяно
в начальный момент, момент времени т и момент
равновесия, а — независимая от х постоянная, опре-
деляемая лишь неизменными в процессе обмена кон-
центрациями компонентов, суммарной концентрацией
изотопа в системе и коэфф, распределения; при этом
зависимость а от концентраций компонентов опреде-
ляется истинным порядком реакции И. о. и может
служить указанием на ее механизм.
Строго говоря, первый порядок кинетич. уравне-
ния соблюдается не всегда, а только в случае выпол-
нения хотя бы одного из трех условий, а именно:
1) близость а к единице, 2) малая концентрация ком-
понента, в к-ром изучается накопление или убыль
изотопа, и 3) малая концентрация изотопа, переход
к-рого исследуется. Однако поскольку для всех изо-
топов, кроме изотопов водорода, равновесное распре-
деление близко к равномерному (а=«1), то для них
И. о. всегда соответствует уравнению первого поряд-
ка. Кроме того, в подавляющем большинстве случаев
имеют дело с малой концентрацией исследуемого
изотопа (особенно это касается радиоактивных изото-
пов). Заметные отклонения от первого порядка могут
наблюдаться лишь в случае изотопного обмена водо-
рода при несоблюдении ни одного из трех указанных
выше условий; при этом отклонения могут быть
настолько велики (в десятки и более раз), что уравне-
ние первого порядка оказывается неприменимым.
В ряде случаев (при наличии в молекулах реагирую-
щих веществ нескольких неравноценных по скорости
обмена атомов данного элемента, при участии в обмене
более двух веществ, при диффузионных затруднениях
обмена, при обмене в потоках веществ и т. д.) имеет
место также кажущееся несоответствие кинетич.
уравнения реакции И. о. первому порядку. Однако
если возможен учет подобных усложняющих обмен
обстоятельств, то кинетич. уравнение суммарного
процесса всегда включает уравнение для неуслож-
ненного И. о., к-рое соответствует первому порядку.
Вопрос о величине скорости реакций И. о. тесно свя-
зан с проблемой механизма этих реакций.
Механизм изотопного обмена. И. о. протекает
по различным механизмам, причем встречаются все
механизмы, присущие элементным химич. реакциям,
и, сверх того, механизмы, не имеющие прямых ана-
логов в обычной химии. И. о. может быть одно-,
двух- и многостадийным, гомогенным и, гетероген-
ным. В его основе могут лежать переходы электро-
нов, ионов, атомов, групп атомов и целых молекул.
В качестве промежуточных стадий реакций И. о.
могут наблюдаться диссоциация молекул на заряжен-
ные или незаряженные частицы, ассоциация отдель-
ных частиц, внутримолекулярные перегруппировки
атомов. Кроме того, И. о. для каждого данного
элемента имеет свои характерные черты. Каждая
из перечисленных характеристик механизмов реак-
ций И. о. может быть положена в основу их клас-
сификации, однако любая из таких классификацион-
ных схем в отдельности не в состоянии отразить все
многообразие механизмов И. о.
Механизм И. о. посредством электронных перехо-
дов соответствует окислительно-восстановительному
процессу. Этот механизм может осуществляться
в тех случаях, когда атомы данного элемента могут
образовывать иопы разного заряда, напр.:
i е I* _ _ *
МпО4~ -г MnO4 ZT Мпог + МпО4
--------е~---.
1 *1 __*
[Fe(CN)ep- + [Fe(CN)B]3~ ZT [Fe(CN)e]3- 4- [Fe(CN)8p-
! e” 1
I* - 1 *
СЮ" -j”. CIOs CIOs "f" CIOs
и т. д., где звездочкой обозначены меченые атомы.
Многочисленны случаи И. о. посредством перехода
простых атомов и ионов; при этом переход происхо-
дит, как правило, после стадии диссоциации, напр.:
1) цепной стадийный гомогенный процесс обмена
в молекулярном водороде:
а) Н2 —>• 2Н, б) Н 4-D2 -► HD + D, в) D2 ->• 2D,
г) D-|-Н2-»• DH-|-Н, 0) Н-|-D —► HD
2) Фотохимии, обмен хлора между С1г и НС1
а) С12 ->• 2CI, б) Cl + HCI ->• НС1 + С1,
в) С12 + С*->- С1С1*+ CI, г) С*+ CI -> С1С*
3) Ионный И. о. в растворах диссоциирующих кис-
лот, оснований и солей, к-рый происходит путем
обычного смешения катионов или анионов:
Н+/В*- -I- К+/ВГ- ZT Н+/ВГ- + К+/ВГ-
РЬ2+/(СН3СОО-)2 + Pb2+/(NO"j)2 ZT
ZT РЬЗ+/(СИ3СОО-)2 + Pb2+/(NOJ)2
Н+/Н8ОГ + D+/C1- ZT D+/HSO? + H+/CI'
и т. д. Очень часто И. о. осуществляется путем пере-
носа изотопов в составе атомных групп: сложных
ионов, радикалов и молекул, напр.:
ВаСО3 4- COj , ' ВаСО3 4- СО3
(n-CH3C6H4)2SS + n-CH3C6H4SH zr
zr (n-CH3ccH4s-) + <n CH3cdH4s-)+
+ (H-) + (n-CH3C6H4S-) ZT (n-CH3C3H4)2SS 4-
4~ n-CH3C3H4SH
N2O3 4- NO2 ZT NO3 4- NO2 4- NOj ZT NNO5.4- no2
[Cr(H2O)c]3+ 4- H2O zr [Cr(H20)5(H2O)]3+ 4- H2O
И. о. иногда является следствием элементных химич.
реакций, представляющих переход атомов или атом-
ных групп, не содержащих данный изотоп, напр.
при взаимном окислении-восстановлении спирта и
альдегида
СН3С: Ну он 4- 8н3сно zr сн3сно 4- сн3сн2он
или при обратимой конверсии окиси углерода:
1 J* I я $
со 4- н2;0.4- со2 zr со2 4- н2 4- со.о• z: со2 4- н2о4-со
Часто классифицируют механизмы реакций И. о.
по последовательности стадий диссоциации и ассоциа-
ции. Выше было дано несколько примеров дйссоциа-
тивного И. о., т. е. обмена, при к-,ром исходные ве-
щества первоначально диссоциируют на более простые
молекулы, иопы, радикалы или атомы. Для ассоциа-
тивного обмена первой стадией является ассоциация
исходных веществ в переходные комплексы или про-
межуточные вещества, а последующая диссоциация
вновь приводит к образованию исходных веществ,
но в иных изотопных формах, напр.:
* * *
Вг~ 4- Вг2 ZT (ВгВгВг)? zT ВгВг -|- Вг-
ND3 4- Н2О ZT ND3HOH ZT ND2H 4- HDO
и т. д. В качестве промежуточной стадии И. о. встре-
чаются иногда таутомерные внутримолекулярные пере-
группировки, напр., при дейтерообмене ацетона
с водой:
СН3СОСН3 4- HDO ZT СН2=СОНСН3 4- HDO zr
zr CH2=CODCH3 4- Н2О zr CH3COCH2D -J- H2O
Ввиду большого многообразия механизмов И. о.
их классификацию и выяснение закономерностей,
обусловливающих порядок величины скорости реак-
ции И. о., проводят обычно отдельно для И. о. дан-
ного элемента. Наиболее изученными являются реак-
193
ИЗОТОПНЫЙ ОБМЕН
194
ции И. о. для водорода, кислорода и галогенов, а также
для углерода, азота и серы.
Изотопный обмен водорода характеризуется
весьма быстрым (часто неизмеримо быстрым) протеканием в тех
случаях, когда обменивающиеся атомы водорода в каждом из
исходных веществ связаны с элементами, имеющими в этих
соединениях свободные электронные пары; это касается по-
давляющего большинства соединений со связями Hal—Н,
О—Н, S—Н, N—Н, Р—Н и т. д. Легкость такого обмена,
протекающего, как правило, без катализаторов, связана, оче-
видно, с возможностью присоединения изотопа водорода (напр.,
в виде иона) к свободной электронной паре элемента с одно-
временным или последующим отщеплением другого атома
Н
водорода, напр. по схеме: D ; NiH4^—►. Для соединений,
Н
в к-рых элемент, связанный с водородом, не обладает свобод-
ной электронной парой (напр., связи С—Н, Si—Н, В—Н,
Н—Н, связь N—Н в ионе NH7 или связь Р—Н в солях фос-
форноватистой к-ты), непосредственный обмен сильно затруд-
нен, поскольку для его протекания требуется предварительное
отщепление водородного атома, что связано со значительной
энергией активации. Такое объяснение различий в скоростях
водородного обмена впервые было дано А. И. Бродским.
Затрудненный И. о. можно часто ускорить использованием
щелочного или кислотного катализа и применением нрклес-
фильных- и электрофильных реагентов. Этим способом осуще-
ствляют водородный обмен в С—Н-связях многих углеводоро-
дов в'среде таких веществ, как жидкий NH3 (особенно в при-
сутствии KNH2), H2SO4, жидкие HF и НВг (особенно в присут-
ствии BF3 или соответственно А1Вг3) и т. д. Часто обмен в свя-
зях Н—Н и С—Н проводят также в газовой фазе па катали-
заторах (Pt, Ni, иногда Fe).
Изотопный обмен кислорода, не считая
случаев ионного обмена в растворах солей, кислот и основа-
ний с одинаковыми анионами, в значительной степени затруд-
нен. Обычно его можно считать быстрым, если он заканчи-
вается за десятки часов при комнатной темп-ре. Основным и
наиболее типичным механизмом кислородного обмена в вод-
ных р-рах является, по-видимому, гидролитич. механизм,
напр,:
2СгО|“ + НаО Cr2Of" 4- ОН“ 4- ОН - Z2
х Crop + Сг03О2"+ ЩО
или
сг2О" 4~ н2о t * CrOj 4~ СгОзО" +2Н^ t * Сг-)ОцО-~ 4- н2о
В водном р-ре за десятки и более часов не обнаружено обмена
в NO-, NO-, CIO-, CIO-, SO|- (не считая H2SO4), SeO£’ ,РО^
НРО|~, Н2РО~, в группах ОН спиртов и фенолов. Медленный
обмен с водой протекает в случае СО|- и НСО - (не считая Н2СО3),
BrO~, JO~, SiO|- и групп СООН~ органич'. к-т; значительно
более быстрый обмен наблюдается в нейтральных растворах
для МоО|~, WO|~, AsO", HAsO|-, СГО|-, Сг2О* ВО~, В4СБ(-}
S2O| ',S2Og~, SeO|_, а также в растворах Н2СО3, H2SO4, НаВО3
и в группе С—О альдегидов и кетонов. Щелочи ускоряют обмен
в Сг2О|~, С1О3 ,JO.7, в альдегидах, кетонах и резко тормозят
его в СгО|~, NO-,“SO|-и S2O|~. Кислоты ускоряют обмен в
СгО-“, CJO.-, NO.-, альдегидах, кетонах и в слабой степени
в SOp, SeOp, CIO-и в органич. к-тах. Между газообразными
О2, Н2О, СО, СО2, SO2 и SO3 кислородный обмен осуществляют
обычно при 300—900° на окисных катализаторах (MgO, СиО,
А12О3, У20б, Мп2О3, Fe2O3, NiO и т. д.), а иногда на Pt, нек-рых
солях, асбесте и т. д.
Изотопный обмен галогенов наиболее по-
дробно исследовал С. 3. Рогинский с сотр. Для этого обмена
весьма распространен ассоциативный механизм с промежуточ-
ным образованием полигалогенидного комплекса. Хлор, бром
и иод почти мгновенно обмениваются с водными р-рами своих
ионов. В кислом водном растворе обмен быстро идет для С12
с СЮ,- и для Вг2 с BrO-f значительно медленнее — для J2
с JO- и вовсе отсутствует дляС12 с С1О-. ВрастворесСЦочень
быстро протекает обмен соответствующих галогенов с галоге-
новодородами, а также с РС13 и РС18, с РВг3, AsBr3, SnBr4 и
медленнее — с СВг4 (с SiBr4 за 6 часов при 100° обмен не обна-
ружен). В бромном р-ре Вг2 легко обменивается с ВВг3, А1Вг3,
Т1Вг3, PBr3, AsBr3, SbBr3 и SnBr4. В пентановом р-ре имеет
место обмен J2 с SbJ3, Пары Вг2 обмениваются с расплавлен-
ными (а в ряде случаев и с кристаллич.) бромидами металлов.
В газовой фазе быстро идет обмен галогеноводородов с галоге-
нами, а также обмен НС1 с парами РС13 и AsCl3, медленнее —
с РОС12, за несколько часов — с SiCI4 и SC12 (обмен НС1 с СС14
отсутствует). Обмен в растворе между анионами галогенов и
анионами С1О~, С1О~, ВгО-hJO- не был обнаружен, за
исключением случая медленного обмена KJ с KJO3 при 200°.
Галогенорганич. соединения большей частью не обмениваются
с галогенами и их анионами или же такой обмен, идет крайне
медленно. Однако СН3Вг сравнительно легко обменивается
с НВг, a CH8J и СН2 = CHCH2J c-HJ. Быстро протекает
обычно обмен органич. галогенидов с Д1На18, а также между
собой в его присутствии.
4 К. X. Э. т. 2
Й з о т о п н ы'й обмен углерода легко идет в р-ре
между СО2 и NaHCO3, между (СНЯСО)2О и CH3COONa или
CH3CH2CH2COONa и между СН-группой аденина и HCONH2
(в последнем случае при нагревании и в присутствии H2SO4);
нет обмена между группой CN- нитрилов и NaCN и между
СО?-и Na2C2O4. На катализаторе (Au, Ag или кварц) при 800°
происходит обмен между газообразными СО2 и СО.
Изотопный обмен азота в растворе между N.
и NO-, NO?, Co(NO2)J"‘, а также NH2OH • НС1 не протекает.
Медленный обмен обнаружен в растворе СС14 и в газовой фазе
между NO2 и N2O6, более быстрый — между NO2 и NO. Прак-
*
тически мгновенно обменивается. NH3 в растворе с NH+. На
активированном железе при 500° быстро протекает в газовой
фазе реакция N2 + N2 2NN.
Изотопный обмен серы характеризуется малой
подвижностью атома- S’ в связях С,—S и О—S и значительной
его подвижностью в координационной сфере иона тиосульфатв.
Быстрый обмен обнаружен в водном р-ре между (NH4)2S и
растворенной серой, между SOq- и одним атомом S в S2O^_,
более медленный — между HS-'и S2O|~. Отсутствует обмен
между SO|-hHS~, SO|-илп82О|-, а также между H2S и цисти-
ном, тиомочевиной или тпогликолевой к-той. Обмен между
SO2 и концентрированной H2SO4 идет лишь при сильном нагре-
вании. При темп-pax диссоциации SO3 протекает И. о.:
2SO3 4~ SO2 , * 2sOo 4“ Оз 4~ Й0о 7 * -J- so2 4- SO3 4“ SO3
Значение метода изотопного обмена. И. о. весьма
широко применяют в различных исследовательских
и препаративных работах, а также в пром-сти. Им
пользуются для разделения природных смесей ста-
бильных изотопов химич. методами, основанными на
неравномерном равновесном распределении изотопов
между веществами (см. Изотопов разделение). Напр.,
Для концентрирования дейтерия при промышленном
получении тяжелой воды применяют реакции И. о.:
HDS + Н2О X H.s + HDO (а30. = 2,31,
*1г0. = 1,86) и HD 4- Нг + HDO («100. = 2,63);
для концентрирования Li6 используется реакция
(Ы“)+ + L1 < Z — (LP)+ + Li®z
I (реакция проводится на цеолите, Z — цеолитовый
радикал, а = 1,022); для концентрирования С13 —
реакция:
C1SN- + HC12N C13N- 4- HC13N (<х = 1,033)
для концентрирования S34 — реакция:
S’«O2 + HS32O7 S32O2 + HS34O.r
п т. д. При исследованиях с мечеными атомами метод
И. о. применяют наряду с синтетич. методами для полу-
чения соединений, меченных по заданному элемент)' (см.
Меченых соединений синтезы). И. о. применяют иногда
для аналитич. целей, а именно для стандартизации изо-
топного состава веществ по одному из элементов при
изотопном анализе этих веществ по другому элементу.
Весьма важное зпачевие имеет метод И. о. при ис-
следованиях, связанных с изучением реакционной
способности веществ и подвижности различных ато-
мов в их молекулах. О реакционной способности и
подвижности атомов судят по скорости протекания
И. о. При этом И. о., в отличие от других химич.
превращений, не изменяет элементного состава ве-
ществ, а следовательно, и электронную структуру их
молекул, к-рая определяет реакционную способность.
Это обстоятельство делает реакции И. о. в известном
смысле модельными для изучения реакционной спо-
собности и подвижности атомов. В ряде случаев при
исследованиях с мечеными атомами И. о. приводит
к перераспределению меченого атома после его перво-
начального перехода в результате исследуемого
химич. процесса, что искажает результат исследо-
вания. В этих случаях необходим учет И. о.
Лит.: Бродский А. И., Химия изотопов, 2 изд., М.,
1957; Рогинский С. 3., Теоретические основы изотоп-
ных методов изучения химических реакций, М., 1956;
Ю р э й Г., Термодинамика реакций изотопного обмена, в кн.:
Химия изотопов, пер. с англ., сб. 1, М., 1948; Варшав-
ский Я. М, Вайсберг С. Э„ Усп, химии, 1957, 26,
вып. 12, 1434. С. Э. Вансберг.
195
ИЗОТОПОВ РАЗДЕЛЕНИЕ
196
ИЗОТОПОВ РАЗДЕЛЕНИЕ — разделение смеси
изотопных веществ на компоненты, содержащие от-
дельные изотопы. Чаще всего И. р. сводится к выделе-
нию из смеси одного из изотопных веществ или просто
к концентрированию этого вещества в смеси. И. р.
является частным случаем разделения веществ, близ-
ких по свойствам (напр., изомерных соединений,
полимеров разного молекулярного веса и т. д.).
Ввиду близости свойств изотопных веществ их разде-
ление весьма трудоемко. Для разделения используют
различия физич. или химич. свойств веществ, обу-
словленные различием в их изотопном составе (см.
Изотопные эффекты). В соответствии с используемым
изотопным эффектом существуют следующие методы
И. р.: диффузия (различие коэфф, диффузии), термо-
диффузия (различие коэфф, термодиффузии), ректифи-
кация (различие давлений пара), химич. обмен (нерав-
номерное распределение изотопов при изотопич. об-
менном равновесии), кинетич. метод (различие кон-
стант скорости химич. реакций), центрифугирование
(различие плотностей) и электромагнитный метод
(различие удельных зарядов ионов).
Разработка методов И. р. была начата одновременно с откры-
тием изотопов. Еще в 1913 Дж. Дж. Томсоном был применен
электромагнитный метод разделения изотопов неона Ne-(> и
Ne--, явившийся также способом их открытия. Будучи усовер-
шенствован, этот метод был использован в дальнейшем (1920)
Ф. Астоном для открытия и разделения изотопов многих эле-
ментов. В 1919 Ф. Линдеманном и Ф. Астоном был предложен
для И. р. метод центрифугирования. В 1932 Г. Герц использо-
вал для разделения изотопов метод диффузии через пористые
перегородки, а в 1934 — метод диффузии в струю пара. Метод
ректификации изотопных смесей был применен в 1931 В. Кезо-
мом и Г. Ван-Дейком для разделения Ne2» и Ne22, а Г. Юри,
Ф. Брикведом и Л. Мэрфи — для концентрирования дейтерия
в жидком водороде. В 1933 Г. Льюис и Р. Макдональд полу-
чили тяжелую воду электролизом (кинетич. метод). В 1935
Г. Юри и Л. Грейфф был предложен для и. р. метод химич.
обмена. В 1938 К. Клузиусом и Г. Диккелем для целей И. р.
был применен термодиффузионный метод.
В связи с потребностью для нужд ядерной энерге-
тики больших количеств таких изотопов, как D и U235,
многие методы И. р. получили, начиная со времени
второй мировой войны, промышленное использова-
ние: метод диффузии — для выделения U235 с приме-
нением газообразного UF6, методы ректификации,
химич. обмена и электролиза для выделения дейтерия.
Промышленное значение имеет также разделение изо-
топов лития. Разделение других изотопов осуществ-
ляется в лабораторном масштабе.
Общие принципы разделения. Однократная опера-
ция И. р. приводит лишь к небольшому обогащению
разделяемой смеси по требуемому изотопу, что свя-
зано с малыми, как правило, значениями изотопных
эффектов. Поэтому для полного выделения или значи-
тельного концентрирования одного из изотопных
веществ операцию разделения многократно повторяют
в ступенчатом разделительном каскаде. Ступень кас-
када представляет собой один или несколько парал-
лельно соединенных разделительных аппаратов; сту-
пени соединены между собой последовательно. По-
скольку исходное содержание выделяемого изотопного
вещества обычно мало, то поток исходной смеси,
проходящей через каскад, очень велик по сравнению
с количеством получаемого продукта. Каскады широко
применяются в химич. технологии и лаборатор-
ной практике для разделения не только изотопных
смесей, однако в случае изотопов они содержат обыч-
но значительно большее число ступеней, чем в дру-
гих случаях. Поэтому современные методы расчета
каскадов разработаны именно в связи с задачей И. р.
Характерные черты каскадного способа разделения
сохраняются независимо от используемого метода И. р.
Поток исходной смеси подается на первую ступень
каскада (см. рис. 1). В результате Операции разделе-
ния он разбивается на два потока: обедненный —
удаляемый из каскада, и обогащенный — подаваемый
на 2-ю ступень. На 2-й ступени обогащенный- поток
вторично подвергается разделению: обогащенный по-
ток 2-й ступени поступает на 3-ю, а ее обедненный по-
ток возвращается на преды-
дущую ступень (1-ю) и т. д.
С последней ступени каска-
да отбирается готовый про-
дукт с требуемой концентра-
цией заданного изотопа.
Поток смеси, протекающий
по каскаду От предыдущих
ступеней к последующим,
наз. прямым, или обогащае-
мым, а протекающий в об-
ратном направлении — воз-
вратным, или обедняемым.
Концентрация изотопа в воз-
вратном потоке с данной
ступени не всегда соответ-
ствует предыдущей сту-
пени, и обедненный поток
может возвращаться не на
I , ‘ ।
|] I;--;
г
I з I
С3У3 1 _______I ^2Х2
I , . 2 '.===)
f__________ I
I " i
Vi I I
----*- прямой пйток L
Одну, а на две ступени ---------------- возвратный воюя G
ниже. х и у концентрации
Степень разделения на _ . „ „ „ „„
14 Рис. 1. Схема каскада раз-
каждОи (i-тои) ступени ха- деления,
рактеризуется коэфф, разде-
ления а — где XinYi — относительные кон-
центрации заданного изотопа соответственно в обо-
гащаемом и обедняемом потоках этой ступени: =
— xji — xit Yi — yili — yit Xi и (1 — xt) — атомные
доли требуемого и отбрасываемого изотопов в обога-
щаемом потоке, и (1 — у|)—соответствующие атом-
ные доли в обедняемом потоке. Коэфф, разделения
зависит от вида изотопов, а также от метода и усло-
вий разделения, но не зависит практически от кон-
центрации изотопа; поэтому при заданных условиях
разделения он одинаков для всех ступеней.
Указанная связь концентраций изотопов в потоках сту-
пени, = xj(l — Vi)/У{(1 — хр = <х, совместно с ур-нием
материального баланса на каждой ступени, Lj_jx|_j +
лииия равновесна.
Рабочая лниия <
Рис. 2. х—у-Диаграмма кас-
кадного разделения.
+ = L{X{ + (где L ий— количества веществ,
составляющих прямой и соответственно возвратный потоки,
i — номер ступени), служит ос-
новой для расчета каскада, т. е.
определения числа ступеней, не-
обходимых для заданного кон-
центрирования. Расчет обычно
проводят начиная с последней
ступени, для которой по приве-
денным двум ур-ниям и исходя
из заданной концентрации про-
дукта определяют две неизвест-
ные концентрации: в прямом
потоке, подаваемом на послед-
нюю ступень с предпоследней,
и в возвратном, идущем с по-
следней ступени на предпослед-
нюю. Используя значения этих
концентраций, проводят анало-
гичный расчет для предпослед-
ней ступени ит. д., пока рассчи-
танное значение концентрации
в поступающем на ступень пря-
мом потоке не совпадет с кон-
центрацией изотопа в исходном
сырье. Такой расчет можно проводить графически, исходя из
т. н. х — р-диаграммы (рис. 2), на которой построены кривая
равновесия [по выражению для коэфф, разделения (а)] и
рабочая линия (по ур-нию материального баланса); число
ступенек, которые можно построить между этими линиями от
точки для исходной концентрации до точки для конечной, со-
ответствует искомому числу ступеней. В зависимости от типа
каскада и распределения в нем потоков существуют также
специальные методы расчета.
Весьма важной характеристикой каскада является
степень извлечения изотопа, т. е. Отношение количе-
ства изотопа в продукте к его количеству в исходном
сырье: у = Lnxn/Loxo. Если из первой ступени кас-
када обедненный поток отбрасывается (см. рис. 1),
197
ИЗОТОПОВ РАЗДЕЛЕНИЕ
198
то степень извлечения мала, т. к. в отбросе содер-
жится значительная часть требуемого изотопа (всего
лишь на величину однократного разделения меньше,
чем в исходном сырье). Такая схема каскада исполь-
зуется в случае больших ресурсов сырья при необ-
ходимости извлечения только одного изотопа. В про-
тивном случае к указанной схеме концентрирующего
каскада пристраивают аналогичную схему другого
каскада, в к-ром концентрируется второй изотоп;
по отношению к первому изотопу эта схема является
исчерпывающей частью общего каскада.
Другой важной характеристикой каскада является
доля отбора со ступени, т. е. отношение отбираемого
со ступени прямого потока к поступающему на нее:
q — Для уменьшения затрат энергии, объема
аппаратуры и периода первоначального накопления
изотопа в системе каскада обычно сокращают потоки
при переходе от более низких ступеней к более высо-
ким (что схематически указано на рис. 1 путем посте-
пенного уменьшения толщины линий, обозначающих
потоки, и размеров ступеней), т. е. применяют кас-
кады с q < 1. Степень такого сокращения зависит от
величины коэфф, разделения а. Если а мало превы-
шает единицу и число ступеней велико, то q лишь
немного меньше единицы, и сокращение потоков вдоль
каскада является почти непрерывным (каскад при-
ближается при этом к т. н. идеальному каскаду). При
существенно большей величине а и меньшем числе
ступеней потоки сокращают весьма резко от ступени
к ступени. Часто, однако, применяют т. н. прямоуголь-
ные каскады, т. е. каскады без сокращения потоков
по ступеням (q = 1). Такими каскадами являются
колонны типа ректификационных, где ступенями
служат тарелки или участки насадки; их применение
оправдано конструктивной простотой, позволяющей
в одном несложном аппарате объединить большое
число ступеней. При этом часто используют каскад,
состоящий из нескольких последовательно соединен-
ных колонн с сокращением потоков от колонны к ко-
лонне («каскад каскадов»).
Характеристика методов разделения. Выбор метода
И. р. зависит, в первую очередь, от величины лежа-
щего в его основе изотопного эффекта, определяющего
значение коэфф, разделения. Для промышленного
получения изотопов важнейшую роль при этом играют
экономия, показатели: затрата энергии, стоимость
аппаратуры, ресурсы сырья, а для лабораторного
И. р. — простота и малые размеры разделительных
установок, надежность их работы и т. д. В таблице
на примере трех видов изотопов (водорода, углерода
и урана) проведено сравнение методов И. р. для водо-
рода, легких и тяжелых элементов в зависимости
от козфф. разделения.
Метод разделения Изотопы
H-D С12—С13 U235—U233
вещество а вещество а вещество a
Электролиз (кинетич. ме- тод) Н2О 5-12
Химич, обмен Н2-Н2О 2,8 HCN—CN 1,013 _а 1,000
Ректификация Н2 1,81 СО 1,01 _а 1,000
Диффузия (через пористые перегородки) Н2 1.20 СН4 1,030 UFe 1,0042
Диффузия (в струю пара) Н2 1,19 СН4 1,016 uf8 1,0022
Термодиффузия Н2 1,052 СН4 1,0026 UF0 —
Центрифугирование .... н2 1,009 сн4 1,009 UF8 1,026
а В любых системах веществ.
Диффузия газов через пористые перегородки
является одним из важнейших методов разделения
тяжелых, а также многих легких изотопов. Приме-
няется в пром-сти для разделения U238 и U235, а также
в лабораторных условиях для разделения различных
изотопов. В основу метода положено различие скоро-
стей теплового движения молекул изотопных веществ.
Исходя из обратно пропорциональной (зависимости
квадрата средней скорости теплового движения моле-
кул от их массы, можно показать, что максимальный
коэфф, разделения изотопных молекул при их диф-
фузии через пористые перегородки определяется
выражением а — УМг/М1г где Л/, и М2 — мол. веса
легкой и соответственно тяжелой изотопных молекул.
Диффузионный разделительный каскад состоит из
многих ячеек (ступеней). Каждая ячейка — камера,
разделенная на две части пористой перегородкой, по
одну сторону к-рой нагнетается насосом газообразная
изотопная смесь. Размер пор — порядка длины сво-
бодного пробега данных молекул при используемом
давлении. Часть смеси проходит через перегородку
и. при этом обогащается легким компонентом, т. е.
скорость его диффузии больше. Из одной части ка-
меры выводится обогащенный поток, из другой — обед-
ненный. Оба потока поступают на соответствующие
ступени каскада для дальнейшего разделения.
Используется также диффузия разделяемой смеси
в к.-л. постороннем газе (или лучше в паре), к-рый
легко потом отделить от смеси конденсацией. Смесь
подается в струю пара, и часть ее с большим содержа-
нием легкого компонента диффундирует против по-
тока пара.
Термодиффузия, т. е. явление изменения
диффузионного равновесия газа при наличии перепада
темп-p, также широко используется в лабораторной
практике И. р. Теория термодиффузии весьма сложна
и базируется на достаточно полной и строгой кинетич.
теории газовых смесей. Термодиффузионное равнове-
сие приводит к разности концентраций молекул в го-
рячей и холодной частях системы; благодаря зави-
симости этого равновесия от масс молекул наблю-
дается частичное разделение изотопной смеси. Мак-
симальный коэфф, разделения выражается величиной
_ _ 4 I 105(Мг-МОгСГа-Т!) R
"r iiepfi + Msjcn + Ts)
где Мг и М2 — мол. веса легкой и тяжелой молекул,
7?т — поправочный коэфф., зависящий от характера
межмолекулярного взаимодействия, Т>_ и Т2 — низ-
шая и высшая темп-ры системы (°К); более тяжелые
молекулы концентрируются в холодной части. Термо-
диффузионный процесс И. р. проводят обычно в пус-
тотелых колоннах с охлаждаемыми стенками и с рас-
каленной проволокой, протянутой в центре вдоль
колонны. Такая колонна в зависимости от ее высоты
фактически равноценна многим последо-
вательно соединенным ступеням. Прямой
и возвратный потоки в колонне обеспе-
чиваются естественными конвекционными
токами (вдоль раскаленной проволоки
ток направлен вверх, а вдоль стенок —
вниз). Между потоками в каждом попе-
речном сечении колонны протекают тер-
модиффузионные процессы, последова-
тельное наложение которых приводит к
накоплению более тяжелого изотопа вни-
зу колонны, а более легкого — на-
верху.
Ректификация жидких изотоп-
ных смесей основана на различии уп-
ругостей пара изотопных веществ. Ко-
эфф. разделения определяется выраже-
нием а = угР1/Р2, где Pi/P2 — отношение давлений
насыщенного пара (большей к меньшей) изотопных
199
ИЗОТОПОВ РАЗДЕЛЕНИЕ - ИЗОТОПОВ СТАБИЛЬНЫХ АНАЛИЗ
200
веществ, п — число атомов изотопа в молекуле веще-
ства (напр., для дейтероаммиака);
а = Pnh3 _ “I/ pNH3 _ -' f ^NH3
^NHsD Г pNHDj Г pNDj
Давления насыщенного пара имеют заметное различие
лишь в случае изотопных веществ с малым мол. весом.
Поэтому методом ректификации пользуются для раз-
деления сравнительно легких изотопов (водорода,
углерода, азота, кислорода и др.), находящихся
в соединениях с небольшим мол. весом (простые газы,
вода, аммиак, окись углерода и др.). Процесс прово-
дят (при темп-ре кипения исходного вещества) в рек-
тификационных колоннах (тарельчатых или насадоч-
ных), включающих много ступеней; колонны соеди-
няют в каскад. Ректификация воды и водорода исполь-
зована в произ-ве тяжелой воды (см. Дейтерий).
Химический обмен — метод И. р., осно-
ванный на неравномерном распределении изотопа
в системе двух веществ при равновесии изотопного
обмена. Коэфф, разделения определяется соответст-
вующими константами равновесия. Так же, как ректи-
фикация, метод химич. обмена применим к легким
изотопам, однако мол. вес соединений не имеет значе-
ния для величины а. Процесс чаще всего проводят
в колоннах. Поскольку обмен происходит между
различными химич. соединениями, то прямой поток
в колонне представлен одним веществом, а возврат-
ный — другим; поэтому на конце колонны необходим
реактор для обращения потоков, т. е. для химич. пре-
вращения одного вещества в другое. В лабораторной
практике метод широко применяется для разделения
многих легких изотопов. Используется иногда ката-
литич. изотопный обмен и изотопный ионный обмен.
В пром-сти химич. обмен применяется при концентри-
ровании дейтерия; при этом для ликвидации дорого-
стоящей стадии обращения потоков использован т. н.
двухтемпературный вариант изотопного обмена между
водой и сероводородом, в к-ром стадия химич. превра-
щения воды в сероводород заменена изотопным обме-
ном между этими же веществами, но при более высо-
кой темп-ре. В произ-ве тяжелой воды применяют
также химич. обмен между водой и водородом в сочета-
нии с электролизом, к-рый представляет собой в этом
случае стадию обращения потоков (см. Дейтерий).
Кинетический метод И. р. представлен
практически единственным химич. процессом — элект-
ролизом, к-рый применяют в основном для концентри-
рования дейтерия. Коэфф, разделения, равный отно-
шению констант скоростей электролитич. разложения
Н2О и В2О, составляет 5—15 (в зависимости от мате-
риала и поверхности катода); D2O разлагается мед-
леннее. Обогащенную тяжелой водой фракцию отби-
рают из электролита, а электролитич. водород пред-
ставляет собой обедненную фракцию. Электролизеры
комбинируются по ступеням общего разделительного
каскада (см. Дейтерий).
Центрифугирование было использовано
для разделения изотопов углерода (в ииде СС14),
криптона, ксенона и урана (в виде UF6), Разделение
осуществляется за счет различия центробежных сил,
действующих на молекулы разных масс. Применяется
противоточная газовая центрифуга, в к-рой смесь
циркулирует, двигаясь вверх, вдоль оси вращения
в центральной части, и вниз — по периферии. Такая
центрифуга — аппарат колонного типа с многократ-
ным повторением элементарного разделительного эф-
фекта (в каждом поперечном сечении) вдоль направ-
ления прямого и возвратного потоков. Коэфф, раз-
деления определяется выражением:
( (М2 — Ml) (Г| — ГЦ I
а = ехр (-----|
где Mi и Л/2 — мол. веса изотопных молекул, и
гг — расстояния потоков от оси вращения, а> — угло-
вая скорость, R — газовая постоянная, Т — абс.
темп-ра. Отсутствие зависимости а от абс. значения
мол. веса существенно при разделении тяжелых изо-
топов. Недостаток метода — низкая производитель-
ность центрифуг и необходимость обеспечения очень
больших угловых скоростей (порядка 60 000 об/мин).
Электромагнитный метод является
единственным абс. методом И. р. в том смысле, что
он обеспечивает полное разделение всех изотопных
веществ смеси в однократном разделительном процессе.
Разделение основано на зависимости отклонения ио-
нов в магнитном и электрич. полях от величины т/е
(т — масса иона, е — его заряд). Разделение происхо-
дит в масс-спектрометре, где ионизованная изотопная
смесь геометрически разобщается на отдельные ион-
ные пучки с одинаковым уд. зарядом (е/т) ионов
в каждом из них (см. Масс-спектрометрия). Из-за
очень малой производительности и сложности разде-
лительного прибора метод применяется лишь для
получения малых количеств очень чистых изотопных
препаратов.
Лит.: Розен А. М., Теория разделения изотопов в ко-
лоннах, М., 1960; Получение изотопов. Тр. Всесоюзной научно-
технической конф, по применению изотопов, М.,1958;Джонс
К. иФерри В., Разделение изотопов методом термодиффу-
зии, пер. с англ., М., 1947; Юрай Г., Химические методы
разделения изотопов, в кн.: Химия изотопов, сб. № 1, М.,
194S; Cohen К., Theory of isotope separation as applied to
the large-scale production of U-35, N. Y.—Toronto—L., 1951.
ИЗОТОПОВ СТАБИЛЬНЫХ АНАЛИЗ — опреде-
ление содержания данных изотопов в элементе или его
соединениях. И. с. а. необходим в случае применения
стабильных изотопных индикаторов (при изучении
физич., химич., биологич. и технологич. процессов),
для контроля разделения и концентрирования изото-
пов, при решении ряда геохимич. задач, напр. опре-
делении геологич. возраста и генезиса пород изотоп-
ными методами, и т. д. Химич, различия изотопов
слишком малы для возможности их применения
в И. с. а. Пользуются зависимостью разных физич.
свойств от изотопного состава (см. Изотопные аффек-
ты) или, реже, специфич. ядерными реакциями, воз-
никающими при воздействии ионизирующих излу-
чений на данный изотоп.
Методы, применяемые для И. с. а., разнообразны
и выбираются в зависимости от задачи, требуемой
точности и аппаратурных возможностей лаборатории.
Наиболее универсальный п распространенный спо-
соб — применение масс-спектрометра (см. Масс-спек-
трометрия) с электрической (или, менее точно, фото-
графической) регистрацией интенсивностей ионных
пучков изотопов, разделенных в электрич. и магнит-
ных полях после ионизации образца электронным уда-
ром или др. методами. Так, этим методом Дж. Томсон
(1912), Ф. Астон (с 1919) и А. Демпстер (с 1922) открыли
изотопию в ряде элементов и впервые измерили их
изотопный состав. Позже были предложены разные
конструкции масс-спектрометров, в частности упро-
щенные модели специально для И. с. а. В обычных
серийных приборах можно определять изотопный
состав с относительной точностью 1—0,1% в образце,
где содержание данного элемента не превышает долей
мг. В более совершенных приборах с применением
дифференциальных методов возможно определение
вариаций изотопного состава до 0,01% и меньше.
Более грубы различные спектральные методы, ос-
нованные на изотопных смещениях энергетич. уров-
ней атомов и молекул. Спектр смеси изотопов пред-
ставляет наложение спектров отдельных изотопов
с соотношениями интенсивностей, отвечающими изо-
топному составу. Чаще всего применяют полосатые
молекулярные спектры в видимой или УФ-области
или (гл, обр. для определения дейтерия в его органич.
201
изотопы
202
соединениях) колебательные ИК-спектры. Пользуют-
ся также вращательными микроволновыми спектрами
в области 104—105 Мгц и спектрами ядерного пара-
магнитного резонанса (см. Радиоспектроскопия).
Для И. с. а. в случае легких элементов широко
пользуются точными измерениями разных физич.
констант из числа тех, к-рые достаточно чувстви-
тельны к изотопному составу и могут быть найдены
без сложного специального оборудования. Для опре-
деления содержания тяжелых изотопов водорода и
кислорода в воде чаще всего применяют разные ден-
симетрия. методики. Наиболее точные и простые
основаны на сравнении темп-p флотации кварцевого
поплавка в анализируемой воде и стандарте, капель
воды в нерастворяющей ее жидкости или поплавка,
наполненного исследуемой водой и погруженного
в воду природного изотопного состава. Этими спо-
собами нетрудно измерить вариации флотационной
темп-ры в 0,001°, что отвечает изменению плотности
воды приблизительно иа 2 • 10~7 г [см3 или содержа-
ния дейтерия на 0,0003% (при 25°). Применяют также j
измерение аналогичными флотационными способами
плотности образца в виде кристаллика исследуемого
вещества или, главным образом для дейтерия и его
летучих соединений, плотности газа с помощью
газовых весов. Для И. с. а. воды можно также изме-
рять показатель преломления, вязкость, давление
или теплопроводность пара, темп-ру замерзания и др.
Для дейтерия и его газообразных соединений хоро-
шим и чувствительным методом служит измерение
теплопроводности в разных видоизменениях мано-
метра Пирани. Большинство перечисленных методик
может быть применено к малым количествам анализи-
руемой пробы, порядка мг и даже меньше.
Очень малые содержания изотопа можно опреде-
лять с большой чувствительностью, но не очень боль-
шой относительной точностью, методом активацион-
ного анализа (см. Радиоактивационный анализ),
напр. О1' по характерному позитронному излучению
F18, образующегося при облучении пробы дейтронами
по реакции О17 (d, п) F18. Отношение Н : D можно
также находить по поглощению медленных нейтро-
нов, для к-рых сечение захвата протонами во много
раз больше, чем дейтронами.
(.'.центральные и масс-спектрометрич. методы имеют
то преимущество, что они не требуют такой тщатель-
ной очистки образца, как при измерении физич. ;
констант, и часто позволяют находить не только :
суммарное содержание данного изотопа, но и соотно- I
шение концентраций молекул с разным изотопным
замещением в образце, напр. C6D6, C6D5H,...,C6He
в дейтеробеязоле.
Лит.: Бродский А. И., Химия изотопов, 2 изд.,
М., 1957; Шатенштейн А. II.[и др.], Изотопный анализ
воды, 2 изд., М., 1957; Киршепбаум И., Тяжелая вода.
Физические свойства и методы анализа, пер. с англ., М., 1953;
Барнард Дж., Современная масс-спектрометрия, пер.
с англ., М., 1957. А. И. Бродский.
ИЗОТОПЫ — разновидности химич. элемента с
одинаковым числом протонов, но различным числом
нейтронов в атомных ядрах; имеют одинаковое число
электронов в атомной оболочке и занимают одно и
то же место в периодам, системе химич. элементов
Менделеева (откуда и название «И.» от греч. iso; —
одинаковый и топос; — место).
В зависимости от состава ядра атомы можно груп-
пировать различным образом. Атомы с различным
числом протонов и нейтронов, но с одинаковым общим
числом частиц (нуклонов) в ядре (Л = const)
наз. изобарами, с одинаковым числом нейтро-
нов (N =* const) — изотонами и с одинаковым
числом протонов (2 = const) — изотопами. В ка-
честве общего названия для всех атомов, отличаю-
щихся составом ядра, применяется (чаще в иностран-
ной литературе) термин «нуклид». Число нуклонов
в ядре (А = Д' -f- Z) наз. массовым числом, т. к. оно
равно округленному до целого числа значению массы
атома И. (в кислородной или углеродной шкале
атомных весов). И. (нуклиды) обозначаются символом
химич. элемента с массовым числом сверху (справа
или слева, напр. РЬ208). Иногда внизу слева указы-
вается число протонов, а справа число нейтронов.
И. были открыты при определении места в периодич. си-
стеме Менделеева радиоактивных веществ, образующихся
при распаде тория и урана. В 1900 было показано, что ионий
Jo (Th230) и торий (Th!3!) при различных радиоактивных
свойствах имеют одинаковые химич. свойства и оптич. спектры;
т. е. являются одним и тем же химич. элементом. Вскоре была
обнаружена химич. идентичность и пек-рых других радиоак-
тивных веществ. Такие вещества Ф.Содди назвал «И.». В 1913
Дж. Томсон опубликовал данные об И. неона — NeM и Ne!1
(окончательно это открытие было доказано в 1919 Ф. Астоном).
Т. обр. было установлено существование И. и у стабильных
элементов. В дальнейшем, в результате систематич. исследо-
ваний Ф. Астона, А. Демпстера, К. Бейнбриджа, А. Нира и др.,
число И. увеличивалось из года в год, и уже к 1950 были от-
крыты почти все стабильные и природные долгоживущие И.
химич. элементов. Благодаря открытию в 1934 супругами И.
и Ф. Жолио-Кюри искусственной радиоактивности стало воз-
можным получение, посредством различных ядерных реакций,
новых радиоактивных И. и элементов, отсутствующих на
Земле вследствие сравнительно короткого времени их жизни.
К 1962 найдено 272 стабильных И. и больше тысячи радиоак-
тивных II.
В природной смеси И. химич. элемента разные И. со-
держатся в различных количествах. Число атомов
данного И., выраженное в процентах к общему числу
атомов всех И. химич. элемента, наз. относи-
тельной распространенностью И.
Напр., бор состоит из двух стабильных И. В10 и В11
с относительной распространенностью соответственно
19% и 81%. Физико-химич. свойства И. почти тож-
дественны, т. к. они в основном зависят от электрон-
ной оболочки атома, одинаковой у всех И. данного
элемента. Поэтому относительная распространенность
И. (а следовательно, и атомный вес элемента) при раз-
личных физико-химич. процессах, протекающих в
природе, почти не меняется. Все же физико-химич.
свойства И. элементов нельзя считать абсолютно
тождественными, так как на них сказывается, хотя
и в небольшой степени, различие в массах атомов
(см. Изотопные эффекты). Наибольшее различие
наблюдается в свойствах II. легких элементов, где
относительное различие масс И. больше, чем у сред-
них и тяжелых элементов. Вследствие этого в природе
происходит, напр., нек-рое перераспределение тяже-
лых и легких 11. водорода и кислорода. Однако
в общем круговороте воды в природе эти различия
выравниваются. Из тождественности изотопного со-
става метеоритного и земного железа, теллура и др.
элементов можно заключить, что в общем средняя
относительная распространенность И. на Земле близка
к соотношению И., возникшему в процессе образова-
ния химич. элементов солнечной системы.
Используя различие в нек-рых свойствах И.,
можно разными способами произвести полное разде-
ление И. или получить химич. элемент с большим
содержанием данного И. (см. Изотопов разделение).
Изучение физико-химич. свойств элементов, обога-
щенных различными И., составляет содержание фи-
зики и химии И. Как радиоактивные, так и стабиль-
ные И. широко применяются в качестве изотопных
индикаторов, а радиоактивные И. — также и как
источники ядерных излучений в самых различных об-
ластях науки и техники. Нек-рые И. урана и плу-
тония (U235, Ри239 и др.) являются «ядерным горю-
чим» (см. Ядерная энергия).
Ядерные превращения и массы изотопов. У разных
И. одного и того же химич. элемента ядерные свой-
ства (тип превращения, время жизни, ядерные мо-
менты и др.) совершенно различны, т. к. атомные
ядра этих И. отличаются по составу и структуре.
.203
изотопы
204
Поэтому у химич. элемента одни И. могут быть ста-
бильными, а другие — претерпевать различные радио-
активные превращения. Известны следующие виды
радиоактивное ти :
1. Альфа-распад— превращение И. с испусканием
а-частицы (ядро атома Не4, состоящее из двух прото-
нов и двух нейтронов). При этом превращении поряд-
ковый номер элемента Z уменьшается на 2 единицы,
а общее число нуклонов А на 4. Напр., радий (88Ra226)
в результате а-распада превращается в радон (88Rn222).
2. Бета-распад—превращение, при к-ром Z из-
меняется на единицу, а А остается постоянным.
Существуют три вида 0-распада: а) распад с испуска-
нием отрицательно заряженной 0-частицы — элек-
трона (0~), сопровождающийся увеличением Z на
единицу, напр. nNa24^->12Mg24; б) рас-
пад с испусканием положительно заря-
женной 0-частицы — позитрона (0+),
сопровождающийся уменьшением Z на
единицу, напр. 11Na22^»10Ne22; в) элек-
тронный захват — е-превращение И.,
при котором ядро захватывает электрон
из атомной оболочки (при этом один из
протонов в ядре, захвативший этот
электрон, превращается в нейтрон) и
Z уменьшается на единицу, например
4Be7?~3Li7. Первоначально этот процесс
был назван /f-захватом, т. к. электрон
обычно захватывается с ближайшей к
ядру /f-оболочки атома. Однако в даль-
нейшем были открыты также случаи
захвата электрона с L-оболочки. При
0-распаде происходит также испускание
нейтрино — нейтральной частицы с мас-
сой, близкой или равной нулю. Извест-
ны также случаи, когда после 0-распада
происходит испускание нейтрона («запаздывающий»
нейтрон) или а-частицы (см. в табл. Li8, Li9, В8,
В12, N17 и др.).
3. Спонтанное деление — превращение, при к-ром
ядро атома делится в основном на два осколка, пред-
ставляющих собой ядра атомов средних элементов
периодич. системы Менделеева. Этот тип превращения
наблюдается только у самых тяжелых И. с Z^92.
В частности, при спонтанном делении U235 из атомного
ядра вылетает 2 или 3 нейтрона, и оно делится, напр.,
на И. Sr90 и Хе142, к-рые в результате последователь-
ного 0-распада превращаются в стабильные И. Zr90
и Се142. Кроме того, U235 может делиться с различ-
ной степенью вероятности и на другие пары И.
Длительность радиоактивного превращения обычно
характеризуется периодом полураспада
(Ti/a), т. е. временем, в течение к-рого радиоактивное
вещество распадается наполовину. Из таблицы И.
(приведенной на цветной вклейке) видно, что вели-
чина Ti/2 может иметь весьма различные значения —
от долей секунды до многих миллиардов лет. У нек-рых
радиоактивных (да и стабильных) ядер наблюдаются
метастабильные долгоживущие возбужденные состоя-
ния, к-рые характеризуются своим периодом полу-
распада; в этих случаях в таблице приведено не одно,
а два и даже три значения Ti/S (см. Изомерия ядерная).
При 0_-распаде происходит превращение в ядре
нейтронов (п) в протоны (р), а при 0+- или е-превраще-
ниях — протонов в нейтроны, до тех пор, пока не
будет достигнуто энергетически наиболее выгодное
сочетание р и п. Поэтому по отношению к 0-распаду
И. делятся на 0-устойчивые (стабильные или
а-радиоактивные) и 0-радиоактивные. Все
0-радиоактивные И. в конечном счете превращаются
в р-устойчивые И., для к-рых 0-раснад энергетически
невозможен. Как видно из таблицы И., у элементов
с Z = 1—83 почти все 0-устойчивые И. стабильны,
за исключением Be8, Sm146, Sm147 и нескольких других
а-радиоактивных И. У более тяжелых элементов
с Z > 83 вообще нет стабильных И. и все 0-устойчи-
вые И. а-радиоактивны. Любое радиоактивное пре-
вращение И. представляет собой экзотермич. процесс,
связанный с выделением энергии. С энергией Е свя-
зана масса т, вычисляемая по формуле Е = тс2
(где с — скорость света). Поэтому при радиоактивном
превращении всегда происходит уменьшение массы
И. на массу испущенной частицы и массу, соответ-
ствующую выделяющейся энергии. Энергетич. воз-
можность 0-распада зависит от соотношения масс
изобар, т. е. атомов с одинаковым числом нуклонов
Массы изобар с четными и нечетными массовыми числами (Мчег- и Мнеч-). Все
fj-радиоактивные изобары в конечном счете превращаются в 20-устойчивые
изобары наименьшей массы. Для Кг’8 возможно превращение в Se78 только
в результате гипотетического одновременного захвата ядром двух электронов из
атомной оболочки (е-превращение) с перескакиванием через промежуточный
Вт78, имеющий бблыпую массу, чем Кг78.
в ядре, но различным соотношением числа нейтро-
нов и протонов. Изобары большей массы в результате
последовательных актов 0-распада превращаются
в изобары меньшей массы (рис.), обратный же про-
цесс невозможен, т. е. он противоречил бы закону
сохранения энергии. Масса изобара определяется сос-
тавом атомного ядра. Чем меньше масса изобара, тем
ближе отношение Z и N к 0-устойчивому сочетанию.
Если в цепочке 0-превращений (рис.) окажется, что
у одного из изобар масса (напр., у Кг78) меньше, чем у
следующего нуклида (напр., у Вг78), то для него 0-пре-
вращение энергетически невозможно и этот изобар бу-
дет 0-устойчивым. Однако для такого И. (напр., Кг78),
как показывает теория, возможен двойной 0-распад,
т. е. одновременное испускание двух 0-частиц или
захват двух электронов из атомной оболочки с «пере-
скакиванием» через изобар большей массы (напр.,
Вг78). Двойной 0-распад еще не наблюден с достовер-
ностью ни в одном случае, т. к. значение Ti/2 при
этом превращении настолько велико (Г‘/2 больше
1018 лет), что экспериментально обнаружить его не
удается. Все изобары наименьшей массы (напр., Se78)
устойчивы как к одиночному, так и к двойному
0-распаду.
Таблица изотопов. Система И. разделена на периоды (обо-
значенные в таблице римскими цифрами), заканчивающиеся на
2Р-устойчивых И. с заполненным числом частиц в нуклонных
оболочках атомных ядер (см. Ядро атомное). Числа нейтронов
у этих И.—т. н. «магические числа» (20, 50, 82, 126)—указаны
внизу таблицы.
В таблице И. приведены все стабильные и радиоактивные
нуклиды и их изомеры (с Ti/г > 1 миллисекунды), открытые
до середины 1982. Вверху каждого вертикального столбца
помещены символ химич. элемента и его порядковый номер,
а под ним приведены все его и. В соответствующей данному
И. клетке таблицы указывается массовое число И., относитель-
ная распространенность и период полураспада (цифры с бук-
205
ИЗОФТАЛЕВАЯ КИСЛОТА —ИЗОЦИАНАТЫ
206
вами: г, или л, — годы, ч — часы, м — минуты, с — секунды,
мс — миллисекунды); цифра с буквой у стабильного И. обо-
значает период полураспада изомера; у радиоактивных И.,
имеющих изомеры, верхняя цифра обозначает период полу-
распада основного состояния, а нижняя—изомерного. [:-устой-
чивые И. отмечены зеленым цветом: полностью закрашенные
клетки обозначают стабильные И., а заштрихованные—^-ра-
диоактивные. В незаштрихованной части указан Ti/2 спон-
танного деления. В верхней части таблицы, над ^-устойчивы-*
ми и. находятся ₽+- и «-радиоактивные И., обозначенные
одним знаком «-)-»; в нижней части таблицы—^“-радиоактив-
ные И. (обозначены «—»). а-Распад у p-радиоактивных И.
ввиду недостатка места в клетках не отмечен. К р-радио-
активным И. отнесены также И., у к-рых, по положению в
системе, должен быть p-распад, но экспериментально наблю-
дался пока только а-распад:
Gd148, Dy154, Dy480, Ро482-288, Го247, a48, At188, 283, At212-214.
At248-248, Rn243, Fr247, 248, 22», 224, Ra243, 248, Ac224-223,
Th223, Pa223, Pa226, U227, Np234, Of244, Es24e, Fm238, E-Yb2»3.
р-Радиоактивные И. c 1 года помещены в белых клет-
ках с утолщенными линиями.
На одной стороне вклейки приведены И. элементов с нечет-
ными Порядковыми номерами ZHe4', а на другой с 2чет-. Такое
разделение принято в связи с тем, что основной закономер-
ностью систематики И. является резкое различие в числе p-ус-
тойчивых И. у элементов Z4eT- и ZHe4-.
Из таблицы видно, что, начиная с кислорода (Z = 8),
все элементы ZHe4- имеют только один или два 2р-устойчивых
И., причем эти И. обладают нечетным массовым числом Анеч-,
а элементы Z4eT- имеют от 3 до 5 20-устойчивых И. как Ачет-,
так и Анеч-. Эта закономерность связана с тем, что после И.
кислорода О1" построение ядер 20-устойчивых И. идет путем
последовательного присоединения сначала двух нейтронов
(2п) или (4п), а затем уже двух протонов (2р). Напр., „0“
-80,’-,01»-,F1"-10Nei!0-1oN21-1„Ne!!2-11Na!3-1!Mg!-‘-... При
ппрр пп р р п
типе построения 2и2р у элемента Z4eT- будет три 20-устойчивых
И. (напр., О1", О1’, О18), а притипе построения 4п2р—пнть2?-ус-
тойчивых И. (напр., Хе128, Хе429, Хе130, Хе134, Хе132). Кроме
20-устойчивых И., у элементов Z4eT- может быть от одного
до четырех И., неустойчивых к 2.:-распаду, но устойчивых к
0-распаду. Эти И. имеют Ачет- (напр., Хе124, Хе128 и Хе134,
Хе131). В таблице 20-устойчивые И. у элементов Z4eT- распо-
ложены таким образом, что на средней горизонтали (0 = 0)
находятся все 20-устойчивые И., соответствующие присоеди-
нению второго нейтрона (напр., О18, Хе139) как при типе
построения 2п2р, так и при типе 4п2р. В случае элементов
ZHe4. на одной горизонтали находятся все 20-устойчивые И.,
соответствующие присоединению первого протона (напр.,
F49, Na23) в основной последовательности 20-устойчивых И.
элементов Z4eT-. Для характеристик «аналогичных И.», рас-
положенных на средней горизонтали, в таблице введено пле-
ядное (изотопное) число 0, показывающее число избыточных
или недостающих нейтронов в ядре по сравнению с числом
нейтронов у 20-устойчивого И., находящегося в средней
строчке таблицы (0 = 0). Внизу таблицы приведены разности
массовых чисел (ДА) между соседними аналогичными И.,
расположенными в одних и тех же горизонтальных строчках.
Если ДА = 4, то это соответствует построению ядер у 20-ус-
тойчивых И. по типу 2п2р, а если ДА = 6, то по типу 4п2р.
Внизу таблицы указано, сколько в периоде ДА = 4. Оказы-
вается, что в каждом периоде (после первого) это число умень-
шается на единицу (5, 4, 3, 2) и эта закономерность дает
возможность предсказать строение плеяды 20-устойчивых И.
у еще неизвестных химич. элементов с Z = 102—106.
Лит.: Астон Ф. В., Масс-спектры и изотопы, пер.
с англ., М„ 1948; Сиборг Г., Перлман И., X о л-
лендер Дж., Таблица изотопов, пер. с англ., М., 1956;
Селинов И. П., Атомные ядра и ндерные превращения,
т. 1 —Таблицы по физике атомного ядра, М. —Л., 1951;
его же, Периодическая система изотопов, М., [1962];
Джелепов Б. С. и Пекер Л. К., Схема распада радио-
активных ядер, М,—Л., 1958. И. П. Селинов.
ИЗОФТАЛЕВАЯ КИСЛОТА (.и-фталевая кислота)
С6Н4(СООН)2, мол. в. 166,13 — одна из. простейших
двухосновных ароматич. кислот, иглы, трудно раство-
римые в воде, т. пл. 348,5°. См. Фталевые кислоты.
ИЗОХИНОЛИН (Р, у-бензопиридин) C9H7N, мол.
вес 129,06, т. пл. 24,6°, т. кип. 242,5°; 1,0986,
zip21!1 1,6243, запах слегка миндальный,
йРп5|| а] напоминает хинолин. И. более сильное
основание, чем хинолин (константа дис-
социации 2 • 10“9). И. — ароматич. со-
единение, вступает в типичные реакции ароматич.
замещения, но проявляет низкую реакционную спо-
собность по отношению к обычным электрофильным
реагентам. Сульфирование И. при 180° дает изохино-
лин-5-сульфокислоту. При нитровании получают 5-ни-
троизохинолин, в более жестких условиях образуется,
вероятно, 5,7-динитроизохинолин. Галогенированием
и меркурированием получают производные И., за-
мещенные в положение 4. Для И. известны реакции
нуклеофильного замещения. И. реагирует с амидом
Na, образуя 1-аминоизохинолин:
При нагревании И. с КОН происходит гидроксили-
рование и образуется изокарбостирил:
Взаимодействием с металлоорганич. соединениями мо-
гут быть получены 1-алкил- и 1-арилизохинолины,
напр. (I). Четвертичные соли И. получаются при
обработке свободного основания галогеналкилами
или алкилсульфатами. Окислением И. получают
смесь фталевой (II) и цинхомероновой (III) к-т:
Qr-COOH
Lcooh
НООСтИ4]
HOOC-l^N
in
Важнейший метод получения И. и его производных
(Бишлера — Напиралъского реакция) заключается в
действии фосфорного ангидрида или хлорангидридов
фосфорной к-ты на р-фенилэтиламиды кислот. Многие
важные алкалоиды являются производными И. (мор-
фин, папаверин, наркотин, сальсолин, эметин И ДР-).
Лит.: Генслер В., в кн.: Гетероциклические соеди-
нения, под ред. Р. Эльдерфилда, пер. с англ., т. 4, М., 1955,
с. 264. Н. В. Гнучев.
ИЗОХОРНЫЙ ПОТЕНЦИАЛ — см. Термодинами-
ческие потенциалы.
ИЗОХОРЫ РЕАКЦИИ УРАВНЕНИЕ — соотно-
шение между константой равновесия, темп-рой и теп-
ловым эффектом химич. реакции при постоянном
объеме. И. р. у. имеет вид:
/ашха\ ли
дТ )v~ RT?
где АН — изменение внутренней энергии в реакции;
Т — темп-ра в °К, Б — газовая постоянная. По вы-
воду и по условиям и формам применения И. р. у.
вполне подобно изобары реакции уравнению.
Лига. см. при ст. Изотермы реакции уравнение.
В. А. Киреев.
ИЗОЦИАНАТЫ — N-производные изоциановой
к-ты R—N=C=O, где R может быть алифатич., аро-
матич., алкилароматич., гетероциклич. или элементо-
органич. радикалом. Физич. свойства важнейших И.
приведены в таблице (стр. 207). •
Наиболее важным методом синтеза И., широко
применяемым в пром-сти, является действие фосгена
на первичные амины или их производные •— хлор-
гидраты, карбаматы и мочевины:
-на
СОС12 4- rnh2--------------------
—2НС1
СОС12 + rnh2 НС1 --------------
-СО2,-НС1 —
СОС12 + RNHCOO- • RNH+-----------
СОС12 + RNHCONHR-----------------
-НС1
-ь [RNHCOC1] ---. RN=C=O
207
ИЗОЦИАНАТЫ
208
Изоцианат Формула Мол. в. T. пл.. °C Т.кпп., °C d-’o 4 20 nD
Этил- C2H5NCO 71.08 60 0,90
н-Вутил- c4h8nco 99,03 -70 114 0,8998 1,4064
1,4-Вутанди- [(CH2)2NCO]2 140,14 -41 106/13 — 1,4522
1,6-Гександи- 1(CH2)3NCO]2 168,19 -67 127/10 1,0460 1,4530
Додецил- c12h25nco 211,34 —29,9 151/15 0,8714 1,4446
Октадецил- c18h37nco 295,49 21,3 178/3,5 0,8556а 1,4468а
Циклогексил- CeHuNCO 125,07 -80 52/10 0,9852 1.4557
Фенил- c„h5nco 119,12 -31,3 48/10 1,9954 1,5362
1-Нафтил- CwH7NCO 169,17 2,4-3 152/20 1,1704 1.6338
к-Толил- C?H-NCO 133,14 — 67/10 1,0596 1,5317
п-Фенилендн- CeHj(NCO)2 160,13 96 110/12 1,4407 —
2,4-Толуиленди- CH3CeH3(NCO), 174,15 21.8 121/10 1,2178 1,5678
1,5-Нафтиленди- .... C ioHe(NCO)2 210,18 126,9 183/10 1,4253 —
4,4'-Дифенилметанди- . . CH2(CBH4NCO)2 250,24 40 156/0,1 1,185° 1,5906°
4,4'»4"-Трифенилметан- три- CH(C4H4NCO)3 367,35 91 — — 1,6150в
a При 30°. 6 При 50”. в Прн 80°.
Фосгенирование свободных оснований проводят при
избытке фосгена в газовой или в жидкой фазе (в по-
следнем случае — в инертном растворителе, напр.
в толуоле). В остальных случаях применяют жидко-
фазное фосгенирование. Сильно основные амины,
напр. гексаметилеидиамин, употребляют обычно в виде
карбаматов.
И. могут быть получены также при взаимодействии:
ди- и моноалкилсульфатов с солями циановой кислоты:
R2SO4 + 2KOCN -* 2RN=C=O + K2SOi
ROSOaOK + KOCN -► RN=C=O -|- K2SOt
галогеналкилов или ацилов с цианатом серебра или (в ди-
метилформамиде) с цианатами щелочных металлов:
RX + MeOCN -► RN=C=O + МеХ
RCOX + MeOCN ->• RCON=C=O -|- МеХ
солей диазония с цианатами щелочных металлов:
Си
ArN2X + KOCN ->• ArN=C=O + КХ + Ns
окпеи ртути с изонитрилами и изотиоцианатами:
RN=C -I- HgO -► RN=C=O + Hg
RN=C=S + HgO -► RN=C=O + HgS
первичных аминов с №,М'-карбонилдиимидазолом:
N==\
RN-C—О + 2 | NH
И. могут быть получены также при отщеплении:
спиртов от уретанов:
-R'OH
RNHCOOR'--------► RN=C=O
аммиака или аминов от замещенных мочевин:
-RNH2
RNHCONHR ------->- RN=C=O
галогенводородов от N-галогеиамидов карбоновых к-т (пере-
группировкз Гофмана):
R-CO-NHX —RN= С=О
азота от ааидов карбоновых к-т (перегруппировка Курциуса):
-N2
R—СО—N3------> RN=C=O
воды от гидроксамовых к-т (перегруппировка Лоссеня):
-Н«О
R—СО—NHOH-------► RN=C=O
Метокспдифторметилизоцианат образуется при пи-
ролизе циклоаддукта тетрафторэтилена и метилового
эфира азодикарбоновой к-ты:
CFj N—СООСНз CF2-N-COOCH3
II 4- II —I I —*
cf2 n-cooch3 cf2-n-cooch3
-> 2CH3OCF2N = C=O
Высокая реакционная способность И. объясняется
наличием системы кумулированных связей—N=C== О
(аналогичной системе связей в кетенах). Ароматич.
И. более реакционноспособны, чем алифатич. Замести-
тели I рода снижают, а II рода повышают актив-
ность И. Весьма характерны для И.
реакции присоединения веществ с по-
движным атомом водорода. Так, гало-
гене водороды взаимодействуют с И.
с образованием весьма термолабиль-
ных галогенангидридов карбаминовых
кислот:
RN=C=O + НХ RNHCOX
И. очень легко присоединяют спирты
с образованием уретанов, а также ам-
миак и амины — с образованием соот-
ветствующих мочевин:
RN=C=O 4- R'OH — RNHCOOR'
RN=C=O + R'R 'NH —> RNHCONRH"
Аналогично присоединяются бисуль-
фит натрия, амиды карбоновых к-т,
сульфамиды:
RN=C=О + NaIiSO3 —> RNHCOSOsNa
RN=C=O + R CO Nib -+ RNHCONHCOR'
RN=C=O + R'SO.,NH2 -► RNHCONHSO2R'
При гидратации И. образуются кар-
баминовые к-ты, к-рые легко теряют
СО2, давая амины. Последние могут взаимодейство-
вать с И. с образованием симметрия, мочёвин:
-со2
RN=C=O + Н.,0 -» RNHCOOH---->
RN=C=O
-* RNH2--------► RNHCONHR
Присоединение к И. карбоновых к-т приводит обычно
к смешанным ангидридам, выделяющим СО2 с образо-
ванием амидов карбоновых к-т:
-со2
RN—0=0 + R'COOH -> RNHCOOCOR'-----► RNHCOR'
Иногда образуются ангидрид карбоновой к-ты и сим-
метрия. дизамещенная мочевина:
2RN=C=O + 2R’COOH -> RNHCONHR + (R СО)>О
Перечисленные реакции присоединения ускоряются со-
лями металлов, третичными аминами и нек-рыми метал-
лоорганич. соединениями, напр. (CH3)2Sn(OCOCH3)2.
В присутствии натрия или алкоголятов И. присоеди-
няют малоновый эфир:
RN=C-О + СН2(СООС2Н5)2 —► RNHCOCH(COOC2H5)2
В результате взаимодействия И. с магнийорганич.
соединениями и последующего гидролиза образуются
амиды к-т:
Н2о
RN=C=O 4- R’MgX -»• [RN=CR'-OMgX]---► RNHCOR’
Конденсация II. с ароматич. углеводородами в присут-
ствии А1С13 приводит к амидам ароматич. карбоно-
вых к-т:
А1С13
R№=C=O + CeHe----► CeH3CONHR
Взаимодействие фепилизоцпаната с дпазометапом дает
р-лактам:
C,II5N=C=O + ch2n» -> cuh5-n —с=о
I I
сн2-сн.
При действии пиридина или фосфинов ароматич. И.
способны димеризоваться с образованием 4-членпых
циклич. димеров I или II:
Ar-N—С=О Ar—N=C-N-Ar
II I ।
О=--С—N-Аг О-С=О
I II
к-рые легко распадаются до мономеров. В присутствии
ацетатов или формиатов щелочных металлов обра-
зуются тримеры—триарилизоцианураты:
Аг
I
м.
ОГСС=О
Ar-S-.c^N~Ar
209
ИЗОЦИАНИНОВЫЕ КРАСИТЕЛИ - ИЗОЭВГЕНОЛ
210
Наиболее важной областью применения И. является
конденсация ди- и полиизоцианатов с полиокси- и по-
лиаминосоединениями с образованием высокомолеку-
лярных продуктов — полиуретанов и полимочевип,
весьма широко используемых современной техникой.
В текстильной и кожевенной пром-сти И. применяют
для обработки тканей и кож с целью придания им
водоотталкивающих свойств. И. используют также
в ряде специальных случаев, напр. 4,4', 4"-три-
фенилметинтриизоцианат— для приготовления клея,
соединяющего резину с металлом и тканью, и т. д.
Поскольку большинство уретанов и мочевин хорошо
кристаллизуется, И. применяются для идентификации
окси- и аминосоединепий. Низшие алифатич. и аро-
матич. моно- и диизоцианаты обладают сильным лак-
рпматорным действием. Многие из них весьма ток-
сичны, вызывая раздражение дыхательных путей,
бронхоастмазм, а в высоких концентрациях — отек
легких.
циклы имеют два общих атома углерода (конденсиро-
ванные ядра), напр. нафталин (VII).
Ароматич. и алициклич. соединения нередко свя-
заны между собой взаимными переходами. Напр.,
гидрированием бензола над мелкораздробленным Ni
можно получить циклогексан (II), хлорированием
бензола при УФ-облучении — гексахлорциклогексан
(III); при гидрировании нафталина (VII) образуются
различные продукты, в том числе тетрагидронафта-
лин (тетралин—VIII), содержащий ароматич. ядро,
конденсированное с алициклич., и декагидронафталин
(декалин, бициклодекан — IX). С другой стороны,
дегидрированием тетралина или декалина можно
получить нафталин. Ароматизация циклопарафинов
также основана на их дегидрировании. При действии
спиртового р-ра щелочи на гексахлорциклогексан
образуется смесь изомерных трихлорбензолов, при-
надлежащих к ароматич. ряду.
Лит..- Ullmann, 3 Aufl., Bd. 9, В., 1957, S. 1.
В. Л. Дя-тнин.
ИЗОЦИАНИНОВЫЕ КРАСИТЕЛИ — см. Цианино-
вые красители.
ИЗОЦИАНУРОВАЯ КИСЛОТА — см. Циануровая
кислота.
ПЗОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ (карбоцик-
лические соединения) — один из основных классов
органич. соединений, характеризующихся содержа-
нием колеп (циклов) из атомов углерода. И. с. отли-
чаются от соединений жирного ряда наличием цик-
лов, а от гетероциклич. соединений — отсутствием
в кольцах к.-л. других атомов, помимо атомов угле-
рода.
И. с. подразделяются на два ряда: ряд алицикли-
ческих соединений и ряд ароматических соединений.
Ароматич. соединения (см. также Ароматические
системы), типичным представителем к-рых является
бензол, содержат замкнутую сопряженную систему
шести л-электронов и обладают особыми характер-
ными химич. свойствами. К соединениям ароматич.
характера, помимо бензола и его производных, отно-
сятся <гек-рые другие И. с., напр. трополоны с 7-член-
ным кольцом, азулены, содержащие конденсированные
7-членные и 5-членные кольца.
К алициклич. соединениям относятся все остальные
обычно делятся на на-
сыщенные алициклич.
соединения (полимети-
леновые соединения,
циклопарафины, цик-
ланы, циклоалканы)и
их производные, напр.
циклопропан (I), цик-
логексан (II), гекса-
хлорциклогексап (III)
и ненасыщенные али-
циклич. соединения,
опентадиен (V).
И. с. могут содержать различное число атомов С
в цикле, различное число циклов в молекуле, связан-
ных между собой тем или иным образом. Так, цикло-
пропан, циклобутан, циклопентан и циклогексан
служат примерами И. с., содержащих 3, 4, 5 и 6 ато-
мов С в цикле. В зависимости от числа колец в моле-
куле различают одноядерные или моноциклич. соеди-
нения, би-, три- и полициклич. соединения. Два
цикла могут быть разделены цепью атомов углерода,
напр. дифенилэтан С„Н6— СН2СН2—С6Н6, или же
связаны между собой непосредственно ординарной
связью, напр. дифенил С0Н6—С0Н6. Кроме того,
оба цикла могут иметь один общий атом углерода
(спираны, спиросоединения), напр. спиро- [3, 4]-октан
(VI). Очень часто, в особенности в ароматич. ряду,
И. с. и их производные имеют большое практич.
значение. К ним относятся терпены, многочисленные
синтетич. красители и лекарственные вещества аро-
матич. ряда. Многие II. с. применяют для получения
синтетич. смол и пластич. масс. д. ф, комиссаров.
ИЗОЦИТРАТДЕГИДРОГЕНАЗА (изолимонной кис-
лоты дегидрогеназа) — см. Цикл трикарбоновых кис-
лот.
ИЗОЭВГЕНОЛ (1-окси-2-метокси-4-пропенилбен-
зол; 2-метокси-4-пропенилфенол) С10Н12О2, мол. в.
164, 20 — бесцветная жидкость, обладающая нежным
запахом гвоздики. И. существует в виде:
трянс-(П) форм. Цис-11. — жидкость;
134—135°/13 мм, 11575 мм,
Транс-11. — кристаллы; т.
пл. 33—34°, т. кип. 141 —
142713 мм, <2^ 1,0844;
1,5784. Технич. 11. — смесь
этих изомеров; обычно—
жидкость, к-рая застывает
при 15—20°; т. кип. 140—
142М6лэ«, 118°/5 иг.ад; rf’g
1,081 — 1,087; 1,572 —1,578. И.плохо
1,0848;
ОН
цис-(1) и
т. кип.
1,5729.
Н СН3
ОСН3
ОН
II
растворим в
воде, хорошо — в обычных органич. растворителях,
гликолях и эфирных маслах; в 50%-ном этиловом спир-
те— в соотношении 1 : 6. И. встречается в эфирных
маслах: илангилапговом, гвоздичном, коллюрии, масле
мускатного ореха и нек-рых др. Из гвоздичного масла
обычно получают цис-форму; из масла эвгенольного
базилика и масла коллюрии — транс-форму.
' И. легко может быть получен нагреванием (190—
195°) эвгенола или содержащего эвгенол эфирного
масла с 50%-ным р-ром КОН с последующим под-
кислением:
Предложены также методы синтеза И. из гваякола
и пирокатехина.
И. легко полимеризуется с образованием диизо-
эвгенола— кристаллич. продукта, т. пл. 180—181°.
При перегонке кристаллич. полимера в вакууме пли
при выпаривании его водноспиртового р-ра образуется
аморфный полимер в виде серой массы или красно-
бурого порошка. И. применяют в парфюмерии, кос-
211
ИЗОЭЛЕКТРИЧЕСКАЯ ТОЧКА —ИЛИДЫ
212
метике, а также для отдушки мыла и нек-рых пищевых
продуктов.
Количественно совместно с эвгенолом И. можно
определять по содержанию гидроксильной группы
ацетилированием в пиридине; для определения И.
в присутствии эвгенола применяют роданирование.
ИЗОЭЛЕКТРИЧЕСКАЯ ТОЧКА^- концентрация
водородных ионов в р-ре белка, амфотерного полиэлек-
тролита или в нек-рых дисперсных системах (коллоид-
ных растворах), при к-рой общее число положитель-
ных зарядов у макромолекул или у частиц дисперсной
фазы равно общему числу их отрицательных зарядов,
вследствие чего при этом pH отсутствует передвижение
частиц в электрич. поле. Для определения И. т.
измеряют pH, при к-ром электрофоретич. подвижность
растворенных макромолекул или дисперсных частиц
равна нулю. Если заряд коллоидных частиц опреде-
ляется не Н+ и ОН"-ионами, а другими, напр.
ионами Agr и J" в золях AgJ, то соответственно изме-
ряют не pH, a pAg (или pj) для характеристики точки
нулевой электрофоретич. подвижности; в этом случае
И. т. называют точкой нулевого заряда. Отсутствие
электрофоретич. подвижности может также наблю-
даться во многих высокоочищенных коллоидных
системах, частицы к-рых практически не имеют
двойного электрич. слоя на своей поверхности, или
при таких концентрациях электролитов, при к-рых
отсутствует диффузная часть двойного слоя, однако
в таких системах отсутствует перезарядка частиц, и
они не имеют И. т. или точки нулевого заряда. Поло-
жение И. т. белков и точки нулевого заряда нек-рых
коллоидных р-ров указано в табл. 1 и табл. 2.
Таблица 1
Белки
Положе-
ние И. т.
(pH)
Белки
Положе-
ние И. т.
(PH)
Яичный альбумин
Сывороточный альбу-
мин .............
Желатина.........
Химотрипсин . . . .
4,71
4,59
5,05
8,6
.Гемоглобин . . . 6,87
₽-Казеин......... 4,50
Цитохром-с . . . 10,6
fJ-Лактоглобулин 5,1
Рибонуклеаза . . 9,4
Таблица 2
Лиофобные коллоиды
(в воде)
Положение точки
нулевого заряда
методами — по минимуму растворимости, оптиче-
ского вращения и др., но наиболее надежными яв-
ляются прямые электрофоретич. измерения. Подобно
изоионной точке, И. т. является основной электро-
химия. константой белков. А. г. Пасынский.
ИЛИДЫ — биполярные ионы, в к-рых положи-
тельно заряженный ониевый атом (N, Р, As, S и др.)
ковалентно связан с отрицательно заряженным ато-
мом углерода. Наиболее характерными представите-
лями И. являются биполярные ионы с ониевым ато-
мом азота, азот-илиды — вещества, содержащие груп-
пировку N—С \ . Большинство азот-И. — нестой-
кие соединения, быстро разлагающиеся на возду-
хе. Многие из них известны лишь как промежуточ-
ные неустойчивые соединения. Особая неустойчи-
вость и реакционная способность азот-И. обусловлены
тем, что между положительно заряженным атомом азо-
та и связанным с ним отрицательно заряженным ато-
мом углерода невозможно образование двойной связи,
т. к. атом азота не способен к образованию пяти кова-
лентных связей.
И., у к-рых ониевым атомом является атом Р, As,
S, отличаются от азот-И. тем, что ониевые атомы этих
элементов способны расширять внешнюю электронную
оболочку до 10 электронов, а в случае серы—до 12 элек-
тронов. Поэтому, в противоположность азот-И.,
между ониевым атомом и соседним атомом углерода
возможна двойная связь. Способность этих атомов
к образованию двойных связей обусловливает умень-
шение полярности молекулы и ее большую устойчи-
вость. Азот-И. могут стабилизоваться, либо вступая
в реакции присоединения, перегруппировки и рас-
щепления, либо за счет делокализации зарядов. Все
азот-И. легко взаимодействуют с кислотами и гало-
геналкилами, образуя соли аммония:
R8N-CR2 -f- АХ -+ r8n-cr2 + х-
I
А
где А = Н, R; X = Hal, анион кислоты.
Многие азот-И. взаимодействуют с водой:
R8N-CR2 + НОН -> R2N-CR2 + он-
н
Нестойкие азот-И., в к-рых карбанионом является
алкильный остаток, расщепляются на олефины и тре-
тичные амины:
AgJ
AgBr
AgCl
pAg 6
pAg 5,7
pAg 4,0
Для чистого водного р-ра белка, в к-ром заряд ма-
кромолекул определяется только ионами Н+ и ОН~,
И. т. совпадает по смыслу и по методам определения
с изоионной точкой. В присутствии солей, когда заряд
белковых макромолекул определяется также ионами
электролитов, обе изоточки изменяются различным
образом. Напр., при преимущественной адсорбции
анионов и щелочном смещении изоионной точки на
белковой макромолекуле остаются избыточные отри-
цательные заряды анионов, сообщающие ей электро-
форетич. подвижность в сторону анода. Для ее ком-
пенсации следует сообщить макромолекуле эквива-
лентное количество положительных зарядов, что мо-
жет быть достигнуто подкислением раствора, т. к.
из более кислого р-ра белок адсорбирует больше
Н+-ионов. Т. обр., И. т. сместится в более кислую
область, т. е. в другом направлении, чем изоионная
точка. При преимущественной адсорбции белком ка-
тиона соли И. т. смещается в щелочном направлении,
а изоионная точка — в кислом. Смещение И. т. в р-рах
солей можно иногда также наблюдать косвенными
Многие неустойчивые азот-И. стабилизуются путем
перегруппировок в третичный амил:
(CH3)2N-CH-C3H5
сн8
К этим перегруппировкам относятся Стивенса пере-
группировка:
+ СвН5Ы
(CH3)2N-CH2C3H5 -----►
I
СН3
-> (CH8)2N-CH-C3H5
СН8
Соммле перегруппировка, идущая с переносом реак-
ционного центра:
+ CgHgLi Г _ +
CeH6CH2N(CH3)2 -----► C3H5CH-N(CH8)2 -►
сн2свн5 L сн2с6н5_
-> o-CHsCeH4CH-N(CH8)a
С«н5
213
ИЛИДЫ—ИЛОСВАЯ РЕАКТИВЫ
214
а также перегруппировка циклических солей ам-
мония.
Чем более устойчив карбанион, входящий в моле-
кулу И., тем более устойчив сам И. В меньшей мере
устойчивость И. зависит от природы радикалов, свя-
занных с аммониевым азотом. Напр., триметиламмо-
нийметилилид известен только в виде комплекса с со-
+ —
лями лития (СН3)3 N—СН2 • LiBr, триметиламмоний-
» флуоренилид (I) получен в свободном
NfChyg состоянии, однако сохраняется толь-
ко в атмосфере азота. Особой устой-
Г J_____|1 \ чивостыо обладают азот-И., в к-рых
' анионом является замещенный цикло-
1 пептадиенил-анион — циклопентадие-
нилиды, относящиеся к классу ароматич. небензоид-
ных соединений (см. Ароматические системы).
Все азот-циклопентадиенилиды отличаются исключи-
тельно высоким для органич. соединений дипольным
моментом. Азот-циклопентадиепилиды растворяются
в кислотах, переходя при этом в соли аммония, и
вновь высаживаются щелочами, приобретая опять
илидцое строение:
Атомы водорода циклопентадиенил-аниона способны
замещаться на дейтерий в нейтральной, щелочной
и слабокислой средах и на галоген при действии бром-
новатистокислого калия в щелочном р-ре. Азот-цикло-
пентадиенилиды растворяются в полярных и не
растворяются в неполярных растворителях. Они
представляют собой ярко окрашенные кристаллич.
продукты, обладающие высокой темп-рой плавления
пли не плавящиеся при нагревании. Типичным пред-
ставителем циклопентадиенилидов является пириди-
нийциклопентадиенилид (II), его дипольный момент
равен 13,6 D.
Известно ок. 10представителей азот-циклопентадиен-
илидов, в том числе триметиламмонийциклопента-
диенилид (III), а также нек-рые алкил-и бензилзаме-
щенные пиридинийциклопентадиенилида.
Известны азот-И., в к-рых положительно заряжен-
ный атом азота и отрицательно заряженный цикло-
пента диенильный остаток разделены системой сопря-
женных связей, напр. (IV), дипольный момент этого
И. равен 9,7 D;
(CH3)3N
III
Свойства И. с ониевым атомом Р, As, S и др. близки
к свойствам азот-И., но менее ярко выражены. Эти
11. более устойчивы и менее реакционноспособны.
Каки азот-И., они реагируют с кислотами и галоген-
алкилами, претерпевают перегруппировки Стивенса
и Соммеле, однако с большим трудом. Наиболее
сильно илидные свойства проявляются у цикло-
пентадиенилидов. Трифенилфосфонийциклопентадиен-
илид (V) — твердое вещество, т. пл. 229—231°,
дипольный момент равен 7 D, сочетается с солями
диазония.
Многие И. с ониевым атомом Р, As, S способны
к реакциям, характерным не для биполярной струк-
туры (илидной), а для структуры с двойной связью
(иленовой). К реакциям такого рода относится Вит-
тига реакция, к-рая была открыта им на примере
реакции трифенплфосфорплепа с бензофеноном. При
этом образуются несимметричный дифенилэтилен и
окись трифенилфосфина:
(С6Н5)3Р=СН2 + (СвИ5)5С=<0 —-
~*(СвН5)3Р=О + (СаН5)2ОСН2
В реакцию Виттига вступают многие фосфорилены.
Кроме фосфориленов, в реакцию Виттига вступают
илены мышьяка и серы; при этом увеличение поляри-
зации связи между ониевым атомом и атомом угле-
рода илена тР=С, и увеличение поляризации
а*
в с=О группе карбонильного соединения способ-
ствуют этой реакции. Азот-И. в реакцию Виттига не
вступают.
Общим способом получения И. является действие
на ониевые соли различных оснований: едких
щелочей, алкоголятов, амидов щелочных металлов,
фениллития:
^>N—С—Н + : В —>• С< + ВН
Лит.: Хэги В., Усп. химии, 1956, 25, вып. 7, 903;
Lloyd D., Sneezum J. S., Tetrahedron, 1958, 3, Ke 3/4,
334; Ramirez F., Levy S., J. Organ. Chem., 1958,
23, № 12, 2035; Виттиг Г., Усп. химии, 1957, 26, вып. 10,
1142; Johnson A. W., La Count R. В., Tetrahedron,
i960, 9, 130. 3. H. Парнес, Н. К. Баранецкая.
ИЛОСВАЯ РЕАКТИВЫ — реактивы, применяемые
для открытия и определения ацетилена, для открытия
окислов азота и азотистой к-ты. И. р. для откры-
тия ацетилена — аммиачный р-р соли закис-
ной меди, полученной восстановлением соли окисной
меди солянокислым гидроксиламином. Для открытия
ацетилена анализируемый газ пропускают через
И. р.; образуется коричнево-красный осадок ацети-
ленида меди С2Си2, а при очень малой концентрации
ацетилена И. р. окрашивается в красный цвет.
Минимальная открываемая концентрация ацетилена
1 : 2,7 • 105 по объему (0,00037 об.%). Присутствие
H2S, СО2 и большого количества кислорода мешает
анализу. И. р. может быть использован для коли-
чественного определения ацетилена. Для этого оса-
док G2Cu2 растворяют в водном р-ре Fe^SO,)^ содер-
жащем H2SO4, и титруют образовавшийся FeSO, раство-
ром КМпО4 или раствором К2Сг2О, в присутствии
дифениламина. Очень малые количества ацетилена
определяют колориметрически, измеряя интенсивность
окраски И. р., появляющейся после взаимодействия
с ацетиленом.
И. р. для открытия окислов азота
и азотистой кислоты — смесь растворов
сульфаниловой к-ты и а-нафтиламина в разб. уксус-
ной к-те. При взаимодействии И. р. с HNO2 или
окислами азота, образующими HNO2 при растворе-
нии в воде, сульфаниловая к-та диазотируется и полу-
чающееся диазосоединение вступает в реакцию азо-
сочетания с а-нафтиламином, в результате чего
образуется азокраситель и раствор окрашивается
в красный цвет. Анализируемый газ пропускают
через сосуд с водой, растворяющей окислы азота, при-
бавляют к полученному раствору И. р. и наблюдают
появление красной окраски. Минимальная открывае-
мая концентрация азотистой к-ты 1 : 109. И. р. пред-
ставляет собой усовершенствованный Грисса реактив,
в к-ром компоненты реактива растворены не в сер-
ной к-те, а в уксусной, что увеличивает скорость азо-
сочетания и развития окраски и, соответственно,
чувствительность реактива.
Лит.: Верль-Лунге, Химико-технические методы
исследования, т. 1, вып. 2, т. 2, ч. 1, вып. 2, Л., 1937; Ерем и-
н а В. Г., Газовый анализ, Л., 1955; Дементьева М. И.,
Анализ углеводородных газов, Л. — М., 1951.
А. А. Черкасский.
215
ИЛЬКОВИЧА УРАВНЕНИЕ -ИМИДГАЛОГЕНИДЫ
216
ИЛЬКОВИЧА УРАВНЕНИЕ — основное уравне-
ние полярографии, выведенное Д. Илькевичем в 1938
для случая диффузии вещества в р-ре к сферич.
электроду (ртутной капле) и устанавливающее прямую
пропорциональность между диффузионным током id
в микроамперах и концентрацией С (миллимоли/литр)
вещества:
= 605»Z)1/2Cm2/3?/e
где п — число электронов, участвующих в электрод-
ной реакции, D — коэфф, диффузии (см2/сек) нона
(или молекулы), обусловливающего эту реакцию,
т — вес ртути (мг), вытекающей из капилляра за
1 секунду, t — период капания ртути (сек). Число-
вой коэфф, включает число Фарадея и, кроме того,
зависит от размера капли в каждый момент ее «жизни».
Величина «605» соответствует среднему значению
тока.
Лит.: Крюкова Т. А., Синякова С. И., Аре-
фьева Т. В., Полярографический анализ, М., 1959.
О. А. Сангина.
ИМИДАЗОЛ (глноксалип, иминазол, 1,3-диазол),
мол. в. 68,08 — твердый продукт; т. пл. 90°, т. кин.
256°; рК а 7,03; дипольный момент 6,2 D; ассоциирован;
aHC-NH хорошо растворим в воде, спирте, умерен-
Ц5 ill но — в эфире.
Ядро И. обладает л-электронным сек-
с ‘ стетом, обусловливающим ароматичность,
хотя и несравненно меньше, чем у бензола. Неподе-
ленная пара электронов одного атома азота может
связывать протон без нарушения л-электронного сек-
стета, причем катион (I) имеет одинаковую груп-
пировку электронов у обоих атомов азота; аппон (II),
образующийся путем отдачи протона, также имеет
два равноценных атома азота;
Поэтому ни у катиона, ни у аниона II. положительный
или отрицательный заряд нельзя приписать опреде-
ленному атому азота. Возможность постоянного пе-
ремещения водорода от одного атома азота к другому
приводит к таутомерному равновесию и отсутствию
изомерных 4- и 5-производных. И. образует соли
и с кислотами и с рядом металлов. Водород имино-
группы алкилируется обычными алкилирующими
агентами (галогеналкилы, диазометан, диметилсуль-
фат); N-алкилимидазолы при пропускании через раска-
ленные трубки • перегруппировываются в 2-алкпл-
имидазолы. Ядро И. обычно расщепляется галоген-
ангидридами к-т в присутствии щелочей (см. Шот-
тена — Баумана реакция):
C6H5COC1, NaOH H<jj NHCOCeHe
НС—NHCOC6HS
N-замещенные соединения при этом не изменяются,
а нек-рые производные И. (напр., гистидин) ацили-
руются. Сильные окислители (азотная и хромовая
к-ты, щелочной р-р перманганата) с трудом окисляют
кольцо И., но окислители перекисного типа легко рас-
щепляют его. И. не восстанавливается амальгамой
ватрия, цинком и соляной к-той, красным фосфором
и иодистоводородной к-той. Хлорирование, бромиро-
вание, фторирование И. протекают легко, иодирова-
ние идет гладко в присутствии окислителей. И. легко
нитруется смесью конц. H2SO4 и HNO3 в положение
4 (5); ввести нитрогруппу в положение 2 не удается.
При сульфировании дымящей H2SO4 также получают
только импдазол-4-сульфокпслоту, И. легко вступает
в сочетание с солями диазония с образованием диазо-
соединений; сочетание происходит преим. в положе-
ние 2; если же последнее занято, реакция направ-
ляется в положение 4 (5). Нагревание И. с 40%-ным
р-ром формальдегида дает смесь 2-окснметил- п
4-окс иметилимидазолов.
Общий метод синтеза И. и его производных заклю-
чается в конденсации 1,2-дикарбонильных соединений
с аммиаком и альдегидами:
R-C---0 NH3 Н R-C-NH
, I + O=C-R"—- II 'c-R'
R-OO NH5
И. получают при взаимодействии глиоксаля с аммиа-
ком и формальдегидом. Вторым общим методом
синтеза II. и его производных является дейст-
вие роданнстоводородной к-ты на а-аминоальдегпды
и аминокетоны. При нагревании с KSC.N п НС1
образуются сульфгидрильные производные имид-
азолов, к-рые при окислении НХ’О2 переходят в II.:
СН3-С=О N-H СНз-C-NH СНз-C-NH
°., «~ „L'C'SB । >
\ Пъ । N шА
NH2 нс ‘
Производные П. часто встречаются в природе и
имеют биологически важное значение (см. Гистамин,
Гистидин, Карнозин, Пилокарпин, Пурин и т. п.);
бензимидазольная группировка входит в состав мо-
лекулы витамина В1г; сочетание имидазольного и тио-
фенового циклов имеется в молекуле биотина, имид-
азольное кольцо содержится в нек-рых циклич.
уреидах (парабановая к-та, гидантоин). Нек-рые
производные И. вызывают сужение кровяных сосудов,
повышение кровяного давления, оказывают противо-
малярийное действие.
Лит..- Гетероциклические соединения, под ред. Р. Эльдср-
фплда, пер. с англ., т. 5, М., 1961. Н. В. Гнуяев..
ИМИДГАЛОГЕНИДЫ — галогенангидриды ги-
потетич. имипокарбоновых к-т. И., не содержащие
заместителя при атоме азота, легко образуются при
действии безводных галогеноводородов (НС1, НВт
и HJ) на нитрилы:
НХ R-C=NH
R-CZN------► ।
X
При действии брома на алифатич. нптрилы образуются
имидбромиды бромзамещенных карбоновых к-т:
Вг., ,кн
C.,H-,C-£N-C,HtBiC -N' J- HBr -► C.HjBr-C-/
xBr
Имидхлорпды, замешенные npji атоме азота, полу-
чают при действии РС15 или SOCI2 на соответствующие
амиды:
С1
SOC1>
С3Н5—С —NH—CgHj------► С8Н3-С=Х--СиН5
РС15
С„Н3-С -NH -СН3--------> С6Н5- C=-N -СНз
II 1
Пмидфториды фторкарбоновых к-т были получены при
действии первичных аминов на фторолефины:
F
C0H5NH2 I
CF->=CFC1-------- Ct-Hs-N=C—CFC1H
Многочисленные реакции И. связаны в первую
очередь с высокой подвижностью атома галогена
галогениминогруппы. Незамещенные при азоте И.
при осторожной обработке холодной водой переходят
217
ИМИДЫ КИСЛОТ - ИМИНОЭФИРЫ
218
в соответствующие нитрилы:
H>0
R—С—NH —
I 0°
Cl
Это позволило Ганчу предположить для незамещенных
при азоте И. структуру [В—C= NH]Cl~. Энергичный
гидролиз И. приводит к амидам соответствующих к-т:
Н2о
R—C=NH —►R-C-NH2
I II
Cl о
нао
R-C=NR’-----> R-C-NHR'
I II
Cl О
И. легко вступают в реакции с соединениями,
держащими подвижный атом водорода:
R— c=NR'
I
Cl
r-C'cn + на
со-
ROH j
R-C=NR’
I
OR
R"NH
NH.OH j
R- C=NR'
I
NHOH
R—С—NR'
I
NR"
И. конденсируются с многоатомными фенолами (ре-
зорцин, флороглюцин) в присутствии кислотных
катализаторов с образованием кетиминов (Г'ёша син-
тез). И. легко восстанавливаются в альдимины прп
действии SnCl2 в абсолютном эфире (см. Стефена
реакция). При повышенных темп-рах N-алкилимид-
хлориды распадаются иа нитрил C6H5C=N и хло-
ристый алкил СН3С1. Имидхлориды алифатич. к-т
легко претерпевают прототропную перегруппировку
с образованием соответствующих N-виниламинов:
\сН—C=NR' — \с=С—NHR
7 I 7 I
С1 С1
Наличие кратной связи, сопряженной со связью C=N,
естественно предотвращает эту перегруппировку.
При действии третичных аминов нек-рыеимидхлориды
удалось превратить в азотистые аналоги кетенов:
<ch3)2ch-c=n^Q-ch3
Cl
СН3
Лит.: Н о u Ь е n - W е у 1. 4 Aufl., Bd 8, Stuttgart, 1952,
S. 673; Stevens C. L., French J. C., J. Amer. Chem.
Soc., 1954, 76, № 17, 4398. Л. С. Герман.
ИМИДЫ кислот — производные кислот, содер-
жащие два ацильных остатка, связанных с имидной
группой (NH). Имиды, являющиеся производными
монокарбоновых к-т, напр. (CH3CO)„NH, наз. также
диациламидами; различают симметрия, и неепмметрич.
днациламиды в зависимости от того, содержат ли они
одинаковые или различные ацильные радикалы.
Известны линейные и циклич. И. к. Наибольшее зна-
чение имеют циклич. И. к. — производные двухос-
новных к-т, напр. фталимид, сахарин (смешанный
имид карбоновой к-ты и сульфокислоты).
Основной способ получения линейных И. к. со-
стоит в ацилировании амидов к-т, их металлич. про-
изводных, иминоэфиров пли изоцианата калия
RCONHa + R'COCl -» RCONHCOR’
RCOR' + (CH3C0),0 -► RCONHCOCH3
NH
KN=C=O + (CH3CO)aO -» (CH3CO)2NK -+
H,O
(CHSCQ)3.N —-> (CH8CQ)2NH + CHaCOOH
Циклич. И. к. легко образуются при действии NH3
на ангидриды двухосновных к-т или при нагревании
таких различных производных этих к-т, как аммо-
нийные соли, кислые амиды и др.
СН2-СО СНг-СО
| 'О + NH3—- | 'NH
снг-со снг-со
'COONHj
(СН,),\ , -----
22coonh4
,.-ср
1 %
'-СО
,-CONHj ,-ср
I --- I 'NH
•-соон ’--со
И. к., в отличие от амидов, совершенно лишены
основных свойств и являются слабыми к-тами. Атом
водорода в И. к. легко замещается атомами металла
или галогена:
/-СО
( >н
''-СО
МеОН,
MeOhT
Н2-СО
>н
н2-со
/~СР ру /-ср
( NMe кх » ( 'NR
-СО '—СО
NaBrO
сн2-ср
I 'NBr
сн2-сб
И. к. обладают более ярко выраженными ацилирую-
щими свойствами и легче гидролизуются, чем амиды
к-т. Подобно амидам к-т, И. к. легко восстанавли-
ваются и реагируют с РС15. Металлич. соли и N-бром-
производные циклич. И. к. широко применяются
в органич. синтезе: в синтезе гетероциклич. соеди-
нений, аминов, аминокислот и пептидов.
Лит.: Traite de chimie organique sous la dir. de V. Gri-
gnard, G. Dupont et R. Locquin, t. 13, P., 1941; Tasc h n e r E
Кос or M., Meier S., Roczniki chemii, 1952, 26, z. 4,
692; Davidson D., Skovronek H., J. Amer. Chem.
Soc., 1958, 80, Л1 2, 376. H. П. Гамбарян.
ИМИЗИН [тофранил, имипрамин, хлоргидрат N-(3-fln-
метилампнопропил)-иминодибензила] C19H24N2 • HCl,
мол. в. 316,88 — бесцветные кристаллы; т. пл. 174—
175°, хорошо растворим в воде, хуже — в спирте, не-
растворим в эфире; pH 1,25%-ного водного р-ра 6—
6,5. По химич. строению И. (I) близок к аминазину
CH3CH2CH2N(CH3)2-HCl Н
При растворении II. в конц. HN03
тенсивная синяя окраска, переходящая в грязно-
зеленую и затем в бурую. Количественно И. опреде-
ляют титрованием р-ром щелочи в среде вода —
спирт — CHCl3. И. получают конденсацией имцно-
дпбензила (II) с З-димотиламинопропилхлоридом в
присутствии NaNII, или CelIsLi, а также иминоди-
бепзила с тозильным эфиром (эфиром
сульфокислоты) 3-дпметиламинопропанола под дей-
ствием C6H0Li. И. применяют в психиатрия, практике
для леяения депрессивных состояний.
Лит.: Schindler W., Н 3 1 1 1 g е г F., Hclv. chim.
acta, 1954, 37, fasc. И, №59, S. 472; Kuhn R Schweiz,
med. Wochenschr., 1957, 87, № 35/36, S. 1135; H u 1 s g e n R.,
Laschtuvka E„ Bayerlein F., Chem. Ber., 1960,
93, № 2, S. 392; Яшунский В. Г. [и др.1, Мед. пром-сть,
1961, № 12, 10. В. Г. Яшунский.
/ZNH
ИМИНОЭФИРЫ (имидоэфиры) R—С/ ,— слож-
к -
ные эфиры гипотетия. иминокарооновых к-т R—с\он
Основным методом синтеза И. является действие
спиртового р-ра галогеноводорода на соответствую-
щий нитрил с последующей обработкой образовав-
появляется ия-
п-толуол-
219
ИММЕРСИОННЫЙ АНАЛИЗ — ИММУНОПОЛИСАХАРИДЫ
220
шейся галогеноводородной соли И. щелочным
гентом (напр., К2СО3) при охлаждении:
pea-
R-CSN
лш2
R-Ckf
СГ- -> R-C- OR'
NH
п. также могут быть получены действием спирта на
иминогалогенид R—CC1=NH. Нитрилы 6-оксикислот
при обработке галогеноводородом в эфирном р-ре об-
разуют галогенгидраты циклич. И. — иминолактонов:
Ароматич. нитрилы, особенно содержащие замести-
тель в орто-положении, либо не образуют И., либо
образуют с большим трудом.
Являясь сильными основаниями, И. легко обра-
зуют соли. Галогеноводородные соли И. гидроли-
зуются в мягких условиях; в зависимости от строения
исходного И. получают либо сложные эфиры, либо
амиды карбоновых к-т:
r-c-nh2 + R'OH
II
о
R-COOR' + NH4C1
Г Z/NHS 1 Н2О ,
R-С< CI-——7
L V)R' j N
Свободные И. более устойчивы к гидролизу. При
действии аммиака или аминов галогенгидраты И.
соответствующих амидинов:
переходят в соли
R—С<^
nh2
OR' -
R”NH2 ,nh2
ci-----> R—c7 ci-
L XNHR"J
И. реагируют с магнийорганич. соединениями с обра-
зованием кетиминов.
Родановые эфиры при действии спиртового р-ра
НС1 переходят в соли соответствующих И., к-рые,
легко отщепляя галогеналкил, образуют тиоуретаны:
ZOR'
^nh2
R-S-CHN -»
R-S-C
Cl- -> r-s-c-nh2
Замещенные при атоме азота И. получают при обра-
ботке кетонов смесью азотистоводородной к-ты, спирта
и галогеноводорода:
СН8.
>с=о
сн7
HN3 + R'OH + HCl
/NCH3
CH3-C^0R, • HCl
Лит.: Хувен И., Методы органической химии, пер.
с нем., т. 4, вып. 1, кн. 1, М., 1949, с. 537. Л. С. Герман.
ИММЕРСИОННЫЙ АНАЛИЗ — метод опреде-
ления показателя преломления вещества, взятого
в виде мелких зерен. Для выполнения анализа
сравнивают под микроскопом показатели прелом-
ления жидкости и погруженного в нее вещества.
На границе двух сред с различными показателями
преломления вследствие интерференции и полного
внутреннего отражения возникает узкая тонкая
световая полоска (полоска Бекке), расположенная
у края кристалла. При подъеме тубуса полоска дви-
жется в сторону вещества с большим показателем
преломления. Показатель преломления вещества дол-
жен оказаться промежуточным между показателями
преломления двух соседних жидкостей.
Для анализа обычно пользуются специальным на-
бором жидкостей, отличающихся на 0,003 единицы
светопреломления. Самая высокопреломляющая жид-
кость — иодистый метилен CH2J2 — имеет показатель
преломления равный 1,74; растворением в нем
серы и иодидов металлов показатель преломления
можно повысить до 1,85. Для определения более
высокопреломляющих веществ в качестве иммер-
сионной среды применяют специальные сплавы,
напр. серы с селеном. Показатель преломления опре-
деляют также по потемнению края кристалла в косом
освещении и др. способами. И. а. широко применяют
в минералогии и петрографии для определения показа-
теля преломления прозрачных минералов; при этом не
обязательно выделять из горной породы зерна испытуе-
мого вещества; определение можно выполнять с малы-
ми по размерам частицами вещества. Пользуясь И. а.,
можно изучать компоненты равновесных систем, про-
дукты химич. технологии; при этом непосредственно
устанавливают, какие химич. соединения входят
в состав исследуемой смеси. И. а. можно использовать
также в качественном микроскопия, анализе.
Лит.: Лодочников В. Н., Основы микроскопиче-
ских методов исследования кристаллического вещества, 2 изд.,
Л., 1932; В о к и й Г. В., Кристалло-оптический анализ, ч. 1,
М. — Л., 1944; Татарский В. В., Кристаллооптика и
иммерсионный метод Определения вещества, Л., 1949; Ме-
ланхолии Н. М., Измерение показателей преломления
под микроскопом иммерсионным методом, М., 1949;Аяше-
л е с О. М., Буракова Т. Н., Микрохимический анализ
на основе кристаллооптики, Л., 1948. Д. Н. Васкевич.
ИММУНОПОЛИСАХАРИДЫ — полисахариды, об-
ладающие свойствами антигенов, т. е. способные
при введении в организм животного стимулировать
в нем возникновение специфич. белков — антител,
и образующие с этими антителами осадки — п р е-
ципитины (см. также Иммунохимия). Почти все
полисахариды микробного (специфич. полисахариды
пневмококков, декстран и др.), растительного (крах-
мал) и животного (полисахариды группы крови)
происхождения наравне с белками обладают анти-
генными свойствами. Полисахаридный компонент
обусловливает антигенные свойства липополисаха-
ридов, к-рые обычно выражены тем сильнее, чем
больше мол. вес полисахаридов. Химич, группиров-
ками, обеспечивающими антигенные свойства И.
(детерминантами), в большинстве случаев
являются повторяющиеся участки молекул И. (напр.,
остатки глюкозы, образующие а-1,6-связц, остатки
целлобионовой к-ты и т. д.). И. — поливалентны по
отношению к антителам, т. е. каждая молекула И.
может связываться с несколькими молекулами анти-
тела, а антитело, в свою очередь, может связывать
несколько молекул антигена; т. обр. возникают круп-
ные конгломераты антигенов с антителами, к-рые
выпадают в осадок. Если какой-либо полисахарид
содержит структурные группировки, сходные с де-
терминантными группами какого-либо И., то он дает
реакцию преципитации со специфич. антителами
этого И. (к р о с с-р е а к ц и я, или перекрест-
ная реакция). Реакция образования комплек-
сов между И. и соответствующими им антителами
тормозится олигосахаридами, структурно сходными
с детерминантами И. В ряде случаев для иммуно-
логия. специфичности И. большое значение имеют его
концевые группы: при отщеплении этих групп от И.
исчезает его способность связываться с соответствую-
щим ему антителом.
Лучше других групп изучены И. микроорганизмов
и, в частности, патогенных микроорганизмов (возбу-
дителей брюшного и сыпного тифов, дизентерии, вос-
паления легких и т. д.). Многие вирулентные микро-
организмы образуют капсулы, в к-рых содержатся
И. В капсулах различных типов пневмококков со-
держатся различные полисахариды. Полисахарид
каждого типа пневмококка вызывает образование
специфич. антитела, но не дает, как правило, пере-
крестной реакции с антителами других типов пневмо-
кокков, т. е. каждый II. специфичен для одного типа
пневмококка. Строение этих специфич. И. полностью
не установлено, но для нек-рых из них известны повто-
ряющиеся участки цепи.
В составе специфич. полисахаридов микроорганиз-
мов обнаружено много редких сахаров: 3-дезокси-
221
ИММУНОХИМИЯ
222
сахаров, сахаров с разветвленной цепью, специфич,
аминосахаров и др. Для выделения И. пользуются
их способностью давать преципитат антиген — анти-
тело и др. методами. Установление однородности и хи-
мич. строения И. производят общими методами, приме-
няемыми в химии полисахаридов (см. Полисахариды).
См. также: Антигены, Антитела, Иммунохимия.
Лит.: Зильбер Л. А., Основы иммунологии, М.,
1958; Успехи стереохимии, пер. с англ., под ред. М. Г. Гоник-
берга, М., 1961; Stacey М., Barkers. A., Polysaccha-
rides of microorganism, Oxf., 1960; D a v i e s D. A. L., в кн.;
Advances in carbohydrate chemistry. Ed. M. L. Wolfrom, v. 15,
N. Y., 1960, p. 271; Blochimie des glucides, p., 1961.
Л. И. Линееич.
ИММУНОХИМИЯ — наука, изучающая химич.
процессы, с к-рыми связана невосприимчивость орга-
низма к инфекционному заражению (иммуни-
тет)^ также повышенная чувствительность к повтор-
ному введению в организм нек-рых веществ. Невос-
приимчивость может быть обусловлена врожденными
особенностями организма (естественный им-
мунитет), а также создана искусственно (при-
обретенный иммунитет). Химич, основы
естественного иммунитета могут существенно разли-
чаться в случае разных инфекции (напр., наличие
у нек-рых организмов специальных ферментов, раз-
рушающих биосубстраты бактерий; наследственно
закрепленная аномалия гемоглобина, пагубно влияю-
щая на возбудителя малярии, и т. п.). Главным пред-
метом изучения И. является приобретенный имму-
нитет, в основе к-рого лежит способность организма,
в ответ на попадание в него ряда веществ (антигенов)
синтезировать специфич. белки — антитела, к-рые
взаимодействуют именно с этими веществами или ве-
ществами, близкими к ним по структуре. Основные
проблемы И.: изучение химич. природы антигенов,
механизма биосинтеза антител, изучение взаимодейст-
вия между антигенами и антителами, создание химич.
методов определения содержания антител и выделения
их из сыворотки в чистом виде, изучение структуры
антител.
Антигенами обычно являются высокомолекулярные
соединения — белки и полисахариды, однако и многие
низкомолекулярные соединения («г а п т е н ы»), при-
соединенные к белкам, становятся антигенами. Клас-
сик. исследования К. Ландштейнера таких конъюги-
рованных антигенов показали, что антигенные свой-
ства гаптенов в сильной мере определяются природой
и положением полярных радикалов в их структуре.
Например, в исследовании, приведенном в таблице,
для реакции была взята сыворотка, в к-рой присутст-
вуют антитела, образовавшиеся при введении л;цвет-
ным комплекса, содержащего jh-H2NC6H4COOH и
.w-H2NC6H4SO8H. При добавлении к этой сыворотке
различных аминов были получены след, результаты:
Амины, введенные животным для получения антител Интенсивность реакции при добавлении к сыворотке ароматич. аминов след.
nh2 0 NHa Ч^Л-соон ^н2 соон Л о о NHa H3C-|^S| Ч^Д-соон NHa
0 + + + 0 + + + + + + + +
0 0 0 — 0 + + + +
(0 — результаты отсутствуют; -(- результат положительный)
Так, антитела, образованные при Введении живот-
ному белка, содержащего иг-аминобензойную к-ту,
совершенно не реагировали с п-аминобензойной к-той,
но давали незначительную реакцию с метаниловой
к-той.
Высокой специфичностью отличаются антитела про-
тив пептидов: антитела против глицил-лейцина слабо
реагируют с глицил-глицином и совершенно не всту-
пают в реакцию с лейцил-глицином. Оптич. изомеры
могут также различаться по антигенным свойствам.
Аналогично этому, в природных высокомолеку-
лярных антигенах специфичность определяется не
всей молекулой, а лишь относительно небольшими
детерминирующими группами, состоящими из 2—4
аминокислотных остатков, или моно- или дисахари-
дов. Так, с помощью иммунохимия, методов обнару-
живаются антигенные различия между инсулинами
быка и лошади, отличающимися тем, что в положе-
ниях 8 и 10 пептидной цепи первого белка находятся
остатки аланина и валина, а во втором белке на их
месте — остатки треонина и им лейцина. Из оболочек
многих бактерий были выделены антигенные поли-
сахариды и в ряде случаев установлена их структура.
При этом было показано, что различия антигенов
в отдельных группах Salnionell связаны с входящими
в структуру моносахаридами: у группы А с паратозой
(3,6-ди-дезокси-П-глюкозой), а у группы В с абек-
возой (3,6-ди-дезокси-П-галактозой). Нек-рые из ан-
тигенов групп крови (напр., обозначаемые, как А, В
и О) оказались мукополисахаридами, а другие (напр.,
М и И) — белками. Антигены эритроцитов групп А и В
содержат глюкозу, галактозу и глюкозамин. При
ферментативном отщеплении галактозы и А^-ацетил-
глюкозамина эти эритроциты теряют свои антигенные
свойства и приобретают антигенные свойства эритро-
цитов другого типа.
Введенные в организм антигены раньше всего об-
наруживаются в ретикуло-эндотелиальных клетках
печени, селезенки, лимфатич. желез, костного мозга,
легких, кожи и др. органов. Там они локализуются
в микросомах и позднее в митохондриях. Меньшие
(но ощутимые) количества антигенов проникают в ядра
клеток.
Антитела появляются в крови животного не сразу
после введения антигена, а через нек-рый латентный
период, длительностью от 3 дней до 4—6 недель.
Образование антител связано не с превращением уже
сформированных молекул белка, а с биосинтезом новых
молекул белка специфич. строения. Этот процесс
происходит гл. обр. в плазматич. клетках селезенки,
легких, лимфатич. желез, костного мозга. Антитела
образуются в микросомах клеток и затем быстро
транспортируются в окружающую клетки среду.
Интенсивность и продолжительность биосинтеза ан-
тител варьируют в зависимости
от природы антигена, способа его
введения и вида животного. По-
вторное введение многих анти-
генов вызывает значительно бо-
лее интенсивный биосинтез анти-
тел, чем первичное. Продолжи-
тельность образования антител
у людей после введения антигена
может варьировать от недель до
десятков лет.
Вопрос о механизме биосин-
теза антител не может считаться
полностью разрешенным. В проб-
леме образования антител са-
мым существенным является
вопрос о том, каким образом в
ответ на введение в организм
антигена с данной детермини-
223
ИММУНОХИМИЯ
224
рующей группой в нем синтезируется белок, имею-
щий строение, подобное или дополнительное к струк-
туре этой группы.
По этому вопросу имеются следующие теории:
1) Антитела целиком или частично образуются из
антигенов (Г. Бухнер, 1893, Г. Смирнов, 1896, В. Ме-
нуоринг, 1930, Г. Рамон, 1930, II. Иордан, 1940).
Несостоятельность этой теории доказана при изуче-
нии соотношения между количеством введенного ан-
тигена и количеством образующихся антител (на
1 молекулу антигена может образоваться более 106
молекул антитела/, сравнении химич. состава анти-
генов и антител, а также применении изотопного
метода. 2) Все образуемые данным организмом анти-
тела (или способность их образовывать) в том или
ином виде пред существуют в организме. Согласно этим
взглядам в организме существуют структуры (теория
«радикалов» или боковых цепей П. Эрлиха, 1906),
молекулы глобулина (теория Н. Ерне, 1955), слу-
чайно имеющие участки, строение к-рых является
дополнительным к строению детерминв рующей группы
антигена, или в организме присутствуют группы
клеток (клоны) с наследственно закрепленной спо-
собностью синтезировать молекулы всех видов анти-
тел (теория селекции клонов Ф. Барнета, 1957—59).
Введение антигена приводит к отбору и резкому уве-
личению числа подобных «радикалов», молекул гло-
булина или клеток. Теории Эрлиха и Ерне не соот-
ветствуют современным представлениям о структуре
и биосинтезе белка. Теория селекции клонов получила
широкое распространение, хотя и не может объяснить,
почему в организме животных существуют клоны
клеток, способные образовывать антитела к синтетич.
соединениям, к-рые не попадали в организм животных
за всю историю развития их вида. 3) Введение анти-
гена животным изменяет синтез белка. Ряд исследо-
вателей признает, что детерминирующая группа анти-
гена, подобно штампу, оказывает влияние на синтез
каждой молекулы антитела, обеспечивая появление
в них участков с дополнительной структурой (мат-
ричные теории). Первоначально сторонники
матричной теории предполагали, что антиген влияет
яа синтез пептидных цепей (Ф. Брейнл и Ф. Гауро-
витц, 1930). В дальнейшем было признано, что анти-
ген влияет не на строение полипептидных цепей, но
на вторичную и третичную структуру образующихся
глобулярных молекул антитела (Л. Полинг и Ф. Гауро-
витц, 1940). Возможно также, что антиген является
штампом, на к-ром через S—S-связи происходит
закрепление вначале лабильной третичной структуры
(Ф. Каруш, 1956). Матричная теория объясняет
возможность образования антител против любых
антигенов, однако и она сталкивается с серьезными
трудностями, так как имеются многочисленные факты,
свидетельствующие в пользу представлений о непря-
мом влиянии антигена на синтез антител. Одна из
теорий, признающих это, исходит из предположения
о возможности влияния введенного антигена на струк-
туру дезоксирибонуклеиновой к-ты (ДНК), что при-
водит к образованию измененных матриц рибону-
клеиновой к-Ты (РНК), осуществляющих синтез
белков, в том числе белков — антигенов (Р. Швит
и Р. Оуен, 1957).
Наибольшее количество антител, образующихся
в организме, содержится во фракции сывороточных
7-глобулинов; они могут присутствовать также и во
фракциях 0- и а-глобулинов. Для лечения и профи-
лактики ряда заболеваний применяют сыворотки
крови иммунизированных животных. Частично очи-
щенные препараты иммунных глобулинов выделяют
из сыворотки иммунизированных животных методами
фракционирования белков. Чистые препараты анти-
тел могут быть получены путем избирательной ад-
сорбции антител нерастворимыми антигенами и от-
щеплением антител от антигенов. В качестве нераст-
воримых антигенов используются бактерии, комплексы
антигенов с целлюлозой или смолами. Этими мето-
дами в нек-рых случаях удается получить препараты
100%-ной чистоты.
Антитела человека имеют мол. в. 160 000 и размер
частиц 236 X 37А, однако в крови обнаруживается
нек-рое количество антител с мол. в. 300 000 —
500 000. Антитела с мол. в. 900 000 присутствуют
в крови нек-рых животных (лошади, коровы, свиньи).
Специфич. способность антител вступать в реакции
с антигенами обусловлена двумя относительно не-
большими участками молекулы антитела — актив-
ными центрами размером в 3—5 аминокислотных
остатков полипептидной цепи. При действии про-
теолитич. ферментов на антитела удается получить
иммунохимически активные фрагменты с мол. весом
ок. 50 000. Активные фрагменты с мол. в. 50 000 и даже
13 000, выделенные из ферментативного гидролизата
кроличьих антител, содержат повышенное количество
триптофана. Эти, а также нек-рые другие данные
дают основание для предположений о важной роли
остатков триптофана в активном центре антител.
Вопрос о механизме реакции антител с антигенами
еще не получил полного разрешения. Несомненна,
однако, существенная роль в этом процессе ионных,
водородных и ван-дер-ваальсовых связей. Реакция
эта, по крайней мере, двухстадийва. Первая стадия
протекает очень быстро и не сопровождается видимыми
изменениями. Она характеризуется довольно сущест-
венным тепловым эффектом А// от 2 до 40 ккал/молъ.
Вторая стадия взаимодействия антигена с антителом
длится часами. В различных системах ояа может про-
текать по-разному, сопровождаясь преципита-
цией (выпадением образовавшегося комплекса в
осадок), агглютинацией (слипанием анти-
генных частичек), лизисом (растворением клеток),
нейтрализацией токсинов и вирусов и т. п.
Иммунохимии, и иммунологии, методы применяются
не только для диагностики заболеваний (выявление
специфичных антител в крови заболевших), но также
и для определения и идентификации белков в слож-
ных смесях по их антигенным свойствам. Важней-
шим иммунохимии, методом, позволяющим опреде-
лять абсолютное содержание антител и. антигенов
в системах, где в результате реакции между этими
веществами выпадает осадок, является метод М. Гей-
дельбергера. Он основан на осаждении антител анти-
геном и определении количества белка, выпавшего
в осадок. Более универсальным является метод опре-
деления абсолютного количества антител по приросту
белка на иммуносорбентах. Чувствительность этих
методов 1—4 мкг азота антител.
Относительное количество антител может быть
определено нахождением наибольшего разведения
содержащей антитела сыворотки (титр разве-
дения), при к-ром еще выявляется действие анти-
тел. Чувствительность иммунобиология, методов мо-
жет быть чрезвычайно большой (до 0,0002 мкг азота
антител).
Очень тонкую дифференцировку сложных белковых
смесей позволяет осуществить качественный иммуно-
химия. метод, основанный на выявлении преципита-
ции в геле, где происходит взаимодействие антигенов
и антител, диффундирующих навстречу друг другу.
Еще большие возможности дает сочетание этого метода
с электрофорезом в агаровом геле (иммуноэлектро-
форетич. метод II. Грабаря). Последний метод позво-
ляет выявить в сыворотке человека более 25 белковых
фракций, различающихся по антигенным свойствам.
Методы И. находят в последнее время применение
для установления эволюционных связей и различий
225
ИМОНИЯ СОЛИ - ИНВЕРСИЯ САХАРОВ
226
между организмами (иммуногенное сходство и раз-
личие белков).
Лит.: Бойд В., Белки в реакциях иммунитета, в кн.:
Белки, под ред. Г. Нейрата и К. Бзйли,т. 3, ч. 1, пер. с англ.,
М., 1958, с. 477; Гауровитц Ф„ Химия и биология бел-
ков, пер. с англ., М., 1953; Грабар П., Биохимия, 1957,
22, вып. 1—2, 49; Гостев В. С., Химия специфического
иммунитета, М„ 1959; Г у р в и ч А. Е., Изучение белковых
антигенов и антител при помощи хроматографии на бумаге,
в кн.; Хроматография, ее теория и применение, М., 1960,
с. 361; его же, Современные представления о биосинтезе
антител, в кн.: Химические основы процессов жизнедеятель-
ности, М., 1962; Зильбер Л. А., Основы иммунологии,
3 изд., М., 1958; Э ф р о и м с о н В. П., Ж. Всес. химич. об-ва
им. Д. И. Менделеева, 1961, Ki 3, 314; Burnet F. М., The
clonal selection theory of acquired immunity, N. Y., 1959;
Bnssard A. D., Annual Rev. Microbiol., 1959, 13, 279;
Haurowlt. г W., Annual Rev. Blochem., 1960, 29. 609;
Landsteiner K., The specificity of serological reactions,
Camb., 1946; I s 1 i k e r H. C., Advances Protein Chem., 1957,
12, 387; К ah at E. A., Blood group substances, N. Y.,1956;
Kabat and Mayer's experimental Immunochemistry, 2 ed.,
Springfield, 1961. A. £.. Гурвич.
ИМОНИЯ СОЛИ — соли замещенных и незаме-
щенных альдиминов и кетиминов, т. е. аммониевые
соли (см. Аммония соединения), в к-рых атом азота
связан двойной связью с одним органич. остатком.
И. с. бывают вторичными (I), третичными (II) и чет-
вертичными (III).
\c=nh2x- ^c=nhrx- ^>c = nr2x-
I II HI
Соли азотсодержащих гетероциклич. аминов типа
пиридина, хинолина и др. также можно рассматри-
вать как И. с.
Наиболее устойчивы четвертичные И. с., хотя
и они легко гидролизуются подой и особенно быстро
щелочами, образуя карбонильные соединения и вто-
ричные амины. Легкость гидролиза сильно зависит от
заместителей, особенно у атома азота. Ароматич.
и разветвленные алифатич. радикалы затрудняют
гидролиз. И. с. мезомерны карбония ионам, чем
обусловливается способность их к реакциям с ну-
клеофильными реагентами: алкоголятами, меркапти-
дами, цианидами, гидридами металлов, литийалки-
лами, реактивами Гриньяра и др.:
4. + + .NR,
R <--» R-C -XHs 1ГС Ml + R'M -> R C<
XR'
где
R'—R"O, R"S, CN, H, R"
Так же реагируют и циклич. И. с.:
При наличии атома водорода у а-атома углерода
II с. находятся в таутомерном равновесии с солями
виниламмония;
R\ +
>C=NRS
R\ + .Н
л С— Nч
R' CH- I XR"
R"
Положение равновесия определяется в основном строе-
нием И. с.
Самыми устойчивыми являются И. с. с комплекс-
ными анионами; такие И. с. можно получать непо-
средственным взаимодействием альдегидов и кетонов
с комплексными аммониевыми солями:
2CtH5CH=CHCHO -|- [RsNHs]SnCir -►
-» [CtH5CH=»CH-CH=NR2]2SnCl’~
(СН3)2С=О 4-[(CH3)2NH2](CeH5)4B- -ь
— [(OH8)2C=N(CH3)a](C3H5)4B-
Даже вторичные II. с. с комплексными анионами
вполне устойчивы, напр.:
CHjx SnCls +
' C-=NOH---► [(CH3)2C=NHa]2SnCi
CH 3Z HC1
Четвертичные II. с., содержащие некомплексные анио-
ны, получаются, аналогично солям аммония, алкили-
рованием Шиффовых оснований галогеналкилами,
диалкилсульфатами и др.:
R. R. + .R'
>C=NR' + R"X -► )C=N< X-
Rz Rz XR"
Другой метод синтеза И. с. основан на отщеплении
нуклеофильной группы от геминальных производных
альдегидов и кетонов (реакция, обратная взаимо-
действию И. с. с нуклеофильными реагентами):
/NRa +
СН2< + Н+ -♦ CH-*=NRa -J- R OH
XOR'
/NR' + _
R..C< 2 + AgNO3 R3C=NR'NO,-J- AgCN
4CN
NR' 4-2HC1 + , +ci2 .NR’,
R2C( ;-----------► RiC=NR.Cl- 4------R2C< -
xnr2 -r'nhhci -r;nci XNR.)
И. с. являются цепными промежуточными соеди-
нениями для синтеза вторичных аминов из первичных:
Rac=0 R”X r. + , 4-НаО R,
RNHa----->RN=CR'----► >N=CR.,X- -------r» )NH
2 R"/ -R'C=OR',/
Образование И.с., вероятно, является промежуточной
стадией во многих органич. реакциях, таких, как
Лейкарта — Валлаха, Манниха реакция, Стивенса
перегруппировка.
Лит.: Н о u b е n - W е у 1, 4 Aufl., Bd 11/2, Stuttgart,
1 958, S. 616; Wittig G., Tochtermann W„ Ber.,
1961, 94, At 6, 1692; L a m c h e n M., [a. o.J, j. Chem. Soc.,
1954, December 4418. H. П. Гамбарян.
ИНВЕРСИЯ САХАРОВ — гидролиз сахарозы
(свекловичного сахара, тростникового сахара), со-
провождающийся изменением направления вращения
плоскости поляризованного луча света р-ром сахара.
Иногда понятие II. с. распространяют на гидролиз
всех олиго- и полисахаридов. Свекловичный сахар,
являясь дисахаридом (а-О-глюкопиранозил-Р-О-фрук-
тофуранозидом), гидролизуется до D-глюкозы и D-
фруктозы:
Сахароза вращает вправо, [a]D = -(-66,5°; при ее
гидролизе образуются эквимолекулярные количества
D-глюкозы, вращающей вправо, [a]D = +59,7° и
D-фруктозы, которая вращает влево, [a]D =—92,3°.
Смесь эквивалентных количеств этих моносахаридов
поворачивает плоскость поляризации света влево,
и знак вращения р-ра сахара т. обр. меняется. Смесь
D-глюкозы и D-фруктозы называется инверт-
н ы м, или инвертированным, сахаром.
Гидролиз сахарозы под влиянием разб. полных
растворов к-т является реакцией первого порядка.
Механизм ферментативного гидролиза зависит от ха-
рактера фермента.
Гидролизующие сахарозу ферменты — инвер-
тазы — делятся на две группы: р-фруктофурано-
зидазы, примером к-рых являются инвертазы дрож-
жей, плесневых грибов и высших растений (ферменты
типа трансфераз), и a-глюкозидазы, к к-рым
227
ИНВЕРТАЗЫ — ИНГИБИТОРЫ
228
относят инвертазу пчел, содержащуюся в меде.
И. с. под влиянием инвертазы дрожжей идет путем
трансгликозилирования (см. Трансгликозилаза). Гид-
ролиз под влиянием ферментов обратим; процесс,
обратный И., называют рив ерсиеп.
И. с. пользуются в пищевой пром-сти с целью пре-
дотвращения кристаллизации сахара в пищевых
продуктах (напр., засахаривания варенья и т. д.),
получения искусственного меда. В медицине инверт-
ный сахар применяется как заменитель глюкозы.
Инвертный сахар используется для произ-ва много-
атомных спиртов (сорбита, манита) и заменителя
глицерина — глицерогена (смеси глицерина,
пропиленглико ля, этиленгликоля, эритрита и гек-
ситов).
а-И нверсия сахаров — изменение по-
ложения в пространстве атома водорода и гидроксиль-
ной группы у углеродного атома альдоз, занимаю-
щего a-положения по отношению к альдегидной груп-
пе; этот процесс называют чаще эпимеризацией. См.
также Лобри де Бруина и Ван Экенгитейна реакции.
Лит.: Р и х е А., Основы технологии органических ве-
ществ, пер. с нем., М., 1959; Вардинская М. С., в кн.:
Углеводы и углеводный обмен в животном и растительном
организмах, М., 1959, с. 230; Handbuch der Pflanzenphysiolo-
gie, hrsg. W. Ruhland, Bd 6, B., 1958. Л. И. Линевич.
ИНВЕРТАЗЫ (инвертин, сахаразы) — ферменты,
гидролитически расщепляющие (инверсия) сахарозу
на D-глюкозу и D-фруктозу. Поскольку сахароза
является одновременно рфруктофуранозпдом и
а-глюкопиранозидом, то ее можно расщепить двумя
типами И. К первому относятся И., являющиеся
p-фруктофуранозидазами, к-рые атакуют молекулу
сахарозы со стороны остатка фруктозы. Для действия
этих ферментов важно наличие неизмененного остатка
фруктозы и p-фруктозидной связи, природа же
афруктона не имеет существенного значения. В связи
с этим И. этого типа могут, помимо сахарозы, рас-
щеплять и другие p-фруктофуранозиды, в частности
рафинозу, но не могут расщеплять такие а-глюко-
пиранозиды, как, напр., мелицитозу, являющуюся
3-(а-глюкопиранозидо)-р-фруктофуранозидо-а-глюко-
пиранозидом. Ко второму типу И. относятся ферменты,
являющиеся а-глюкозидазами. Они атакуют моле-
кулу сахарозы со стороны остатка глюкозы. Для их
действия важно наличие неизмененного остатка глю-
козы и а-глюкозидной связи, природа же аглюкона
не имеет существенной роли. В связи с этим ферменты
этого типа, помимо сахарозы, расщепляют также
другие а-глюкозиды, в частности мелицитозу, но не
расщепляют рафинозу. Помимо гидролиза сахарозы,
И. катализируют также реакции трансгликозилиро-
вания, т. е. переноса остатков моносахаридов от
сахарозы на различные спирты, моно- и дисахариды.
При этом И. первого типа осуществляют перенос
остатка фруктозы, а И. второго типа — глюкозы.
И. широко распространены в растительных, живот-
ных тканях и у микроорганизмов. И. растенпй и
микроорганизмов представлены гл. обр. р-фрукто-
фуранозидазами; И. животных являются а-глюко-
зидазами. Наиболее изучена И. дрожжей (р-ф р у к-
тофуранозидаза). Оптимум действия этого фер-
мента в очищенном состоянии соответствует pH 4,5—
5,5; величина константы Михаэлиса (Ам) 0,016 М
для сахарозы и 0,24 М для рафинозы. Фермент рас-
щепляет сахарозу со скоростью, в 2 раза превышающей
скорость расщепления рафинозы. Активность этой
И. подавляется реагентами на SH-группу, напр.
фенилртутьацетатом и др. Ионы тяжелых металлов:
Hg+, Hg2+, Cu2+,Cd2+, Zn2+ и Pb2+, обратимо подавляют
активность И. При обработке И. иодом образуется
стабильное иодированное производное, т. н. J-ин-
вертаза, ферментативная активность к-рой равна
55% активности природного фермента. J-ипвертаэа
характеризуется такими же величинами Км и pH опти-
мума действия, что и исходная И., отличаясь от нее
нечувствительностью к действию тяжелых металлов.
Определение активности И. основано на измерении
скорости гидролиза сахарозы путем поляриметрия,
определения изменения оптич. вращения раствора
или же определения количества восстанавливающих
сахаров по Фелингу. Чистые препараты И. находят
применение при анализе сахаров.
Лит.: Му г back К., Invertases, в кнл The enzymes,
ed. Р. D. Boyer, H. Lardy, К. MyrbSck, v. 4,2 ed., N. Y. — L.,
1960. В. Б. Спиричев.
ИНВЕРТНЫЙ САХАР — см. Инверсия сахаров.
ИНГИБИТОРЫ — вещества, тормозящие химич.
процессы. Круг веществ, задерживающих химич.
реакции, весьма обширен, а области их применения
разнообразны. Так, И. применяют для задержки,
а иногда и полной остановки цепных реакций, корро-
зионных и всевозможных окислительных процессов
и т. д. Количество добавляемых в реакционную среду
И. колеблется в весьма широких пределах, от тысяч-
ных долей процента (напр., для остановки цепного
процесса На С1а = 2НС1 требуется всего лишь
0,001% NC13) до нескольких процентов (присадки
к нек-рым смазочным маслам).
Различают: И. коррозии металлов, И. окисления
нефтепродуктов, И. полимеризации, И. цепных реак-
ций, И. ферментов и др. В настоящей статье рассмот-
рены И. коррозии (в том числе И. коррозии металлов
нефтепродуктами) и И. окисления нефтепродуктов.
Об И. других процессов см. Полимеризация, Цепные
реакции, Ферменты, а также Антиокислители и
Антиокислители пищевых продуктов.
Ингибиторы коррозии металлов — вещества, сни-
жающие скорость коррозионного разрушения различ-
ных металлич. материалов при добавлении их в кор-
розионную среду. В практике борьбы с коррозией
металлов И. широко применяют гл. обр. в системах,
работающих с постоянным или малообновляемым
объемом растворов, напр. в нек-рых химич. аппаратах,
системах охлаждения, паровых котлах, конденсаци-
онных устройствах и др. Часто И. применяют для
защиты от коррозии металлич. изделий и оборудования
при их длительной консервации, а также для травле-
ния металлов с целью удаления с поверхности ока-
лины и ржавчины и для защиты от коррозии металлич.
емкостей, предназначающихся для перевозки и хра-
нения кислот.
Известно большое число веществ, к-рые можно рас-
сматривать как И. коррозии. Наиболее четко инги-
биторное действие выражено у таких типов соедине-
ний, как амины, азотсодержащие гетероциклич.
соединения, меркаптаны, мочевина, тиомочевина,
сульфиды и альдегиды. Эффективными И. являются
многие природные органич. соединения и продукты
их химич. переработки (напр., сульфированный клей
и др.).
По условиям применения И. коррозии можно разде-
лить на И. для водных р-ров (кислотные, щелочные
и для нейтральных сред) и И. для защиты от атмос-
ферной коррозии («летучие» И.). И. коррозии
иногда применяют и в случае неводных коррозион-
ных растворов, а также для повышения антикорро-
зионных свойств лакокрасочных покрытий, смазок
и цементов.
По современным воззрениям механизм действия
значительного числа И. имеет электрохимии, природу
и заключается в адсорбции И. на корродирующей
поверхности и последующем преимущественном тор-
можении анодных (анодные И.) или катодных про-
цессов (катодные И.). При соизмеримом анодном
и катодном торможении можно также говорить об
И. смешанного типа. К И. часто причисляют также
229
ИНГИБИТОРЫ
230
вещества, понижающие скорость электрохимии, про-
цессов путем снижения агрессивности коррозионной
среды (напр., путем связывания активного кислорода
при котельной коррозии) или путем образования
на поверхности металла фазовых (видимых) защитных
пленок, состоящих обычно из продуктов взаимодей-
ствия И. с раствором, металлом или продуктами его
коррозии (кроющие И.).
Характер адсорбции И. на поверхности металла,
эффективность его действия, а также принадлежность
И. к катодному или анодному типу зависят не только
от его характера, но в значительной степени опреде-
ляются также потенциалом (зарядом) <р поверхности
металла относительно раствора. Величина <р находится
как разность между стационарным потенциалом ме-
талла в данном растворе (е) и потенциалом нулевой
точки поверхности металла (<р0).
При положительном заряде поверхности металла
относительно раствора (<р0 < е) на нем будут адсор-
бироваться И., являющиеся анионами, при отрица-
тельном заряде поверхности (<р0 > е) — И., представ-
ляющие собой катионы, а при заряде поверхности,
близком к нулю (<р0 = в), — И., являющиеся недис-
социированными молекулами. Т. обр., при переходе
к другому металлу, изменении его структуры или при
изменении стационарного потенциала металла, напр.
вследствие изменения раствора или наложения внеш-
ней поляризации, могут изменяться характер адсорб-
ции и, следовательно, характер и эффективность
действия И. Действие И. может также заметно из-
меняться в зависимости от условий — темп-ры, состава
и концентрации коррозионной среды и т. д. Эффек-
тивность действия И. может быть Оценена по формуле:
z = s0/s, где z — величина ингибиторного эффекта,
s0 — скорость растворения металла в неингибирован-
ной коррозионной среде, s — скорость растворения
того же металла в ингибированной коррозионной
среде.
К анодным ингибиторам относятся
такие окислители, как нитраты, нитриты, широко
применяемые в различных Отраслях пром-сти (авиа-
ционной, химической, нефтеперерабатывающей, ма-
шиностроительной и др.). Из большого числа обсле-
дованных окислителей с анионами типа МеО”- наи-
более эффективными И. оказались пертехнат-ионы
(ТсО4)~ и хромат-ионы (СгО4)2 ; практич. применение
нашли гл. обр. хроматы и фосфаты.
Механизм действия окислительных И. определяется
гл. обр. переходом защищаемого металла в устойчивое,
пассивное состояние. Однако действие этих И. более
сложно, нежели простое окисление поверхности ме-
талла, и связывается также в ряде случаев с адсорб-
цией поверхностью металла непосредственно окисли-
тельного аниона. Подтверждением этого является то,
чтц эффективность действия этих И. не связана про-
стой зависимостью с их окислительно-восстановитель-
ным потенциалом (см. Пассивирование металлов).
К анодным И. относятся также производные бен-
зойной к-ты (бензоаты), циклогексиламин, морфолин
и др. органич. вещества, ряд И., тормозящих анодный
процесс вследствие образования на анодных участках
корродирующей поверхности металла кроющих за-
щитных пленок, напр. для железа — добавки в кор-
розионную среду фосфатов, NaOH и Na2CO3, для
алюминия и его сплавов, а также железа — силика-
тов щелочных металлов (жидкого стекла). Анодные
И., будучи добавлены в недостаточном количестве,
в условиях коррозии с катодным контролем могут
вызывать образование отдельных точек глубоких
местных поражений (питтингов), являясь, т. обр.,
«опасными» И.
Катодные ингибиторы уменьшают ско-
рость коррозии за счет снижения эффективности про-
текания катодного процесса. К ним относятся: 1) И.,
повышающие перенапряжение катодного выделения
водорода; такое действие на железо оказывают, напр.,
многие соли мышьяка (AsCI3 и др.) и висмута —
Bi2(SO4)3 и др.; 2) И., экранирующие активные ка-
тоды; напр., по отношению к железу в воде такими И.
являются СаНСО3, ZnSO4, ВеС12идр.; 3) И.—поглоти-
тели катодных деполяризаторов, гл. обр. кислорода
(сульфит натрия, гидразин и др.), нашедшие широкое
применение при борьбе с коррозией котельной аппа-
ратуры. Многие органич. И. наряду с анодным тор-
можением могут также заметно тормозить катодный
процесс как путем увеличения перенапряжения ка-
тодного процесса, так и путем экранирования, вслед-
ствие образования адсорбционных или фазовых
пленок.
В отличие от анодных, катодные И., будучи добав-
лены в раствор в недостаточном количестве для пол-
ного подавления коррозии, не вызывают появления
местной коррозии (при условии, если коррозия про-
исходит с катодным контролем).
Под летучими И. подразумевают вещества,
к-рые, имея высокое давление пара, достаточно быстро
заполняют окружающую атмосферу и, адсорбируясь
на металлич. поверхностях, защищают их от атмос-
ферной коррозии. Большое значение для эффектив-
ности действия летучих И., помимо высокого давле-
ния пара, имеет также способность адсорбированной
пленки И. уменьшать гигроскопичность (создавать
гидрофобность) металлич. поверхности. По своему
составу летучие И. чаще всего являются соединениями
аминов (напр., моноэтаноламина, амиламина, дибу-
тиламина, тризтиламина, циклогекси ламина, изо-
пропиламина, морфолина, дициклогексиламина, ди-
изопропиламина и др.) с азотистой, угольной, бензой-
ной, коричной или хромовой к-тами либо солями
нек-рых гетероциклич. соединений. Механизм дей-
ствия летучих И. имеет также электрохимич. природу.
Летучие И. иногда применяют для защиты металло-
изделий при хранении или транспортировке, помещая
защищаемые детали вместе с И. в замкнутые емкости
(контейнеры); часто летучие И. используют для про-
питки внутреннего слоя упаковочной бумаги, упот-
ребляемой для антикоррозионной консервации метал-
лич. изделий, а также для защиты внутренних по-
верхностей конденсаторов пара, паровых котлов, дви-
гателей внутреннего сгорания и других приборов
и аппаратов при их консервации.
В табл. 1 приведены нек-рые наиболее часто реко-
мендуемые И. коррозии, указаны области их приме-
нения и наиболее вероятный механизм их действия.
Лит.: Томашов Н. Д., Теория коррозии и защиты
металлов, М., 1959; Е vans U. R., The corrosion and oxi-
dation of metals, L., 1960; Антропов Л. И., в кн.: Проб-
лемы коррозии и защиты металлов (Труды V Всес. совещания
по коррозии и защите металлов), М., 1956, с. 79; Розен-
фельд И. Л., Замедлители коррозии в нейтральных средах,
М., 1953; Путилова И. Н., Балезин С. А., Ба-
ра н н и к В. П., Ингибиторы коррозии металлов, М., 1958;
Акользин П. А., Михайлова Н. М., Теплотех-
ника, 1960, № 7, 59; Гинцберг С. А., Шрейдер А. В.,
Ж. прикл. химии, 1960, 33, вып. 7, 1594; АнощенкоИ. П.,
там же, 1957, 30, вып. 3, 393, вып. 4, 523; 1960, 33, вып. 6,
1319; Л о с е в В. В., ДАН СССР, 1953, 88, № 3, 499; Фрум-
кин А. Н. [и др.], Кинетика электродных процессов, М.,
1952; С аг 11 е dg е &. Н., J. Phys. Chem., 1955, 59, № 9, 979:
1956, 60, № 1, 28, 32, № 8, 1037; Hackerman N., M a k-
rides A. C., Industr. and Engng. Chem., 1954, 46, № 3,
523; Розенфельд И. Л., Персианцева В. П.,
Хим. наука и пром-сть, 1958, 3, К 4, 500; Ингибиторы корро-
зии (Сборник ВСНИТО М2 и № 7), [М.], 1957; К о 1 о t у г-
kin J. М., В u n е N. J., Z. phys. Chem., 1960, 214, Н.
5/6, 264; И о ф а 3. А., Вестник МГУ. Сер. матем. ..., 1956, № 2,
139; 1958, Кз 5, 171. Н. Д. Томашов.
Ингибиторы коррозии металлов нефтепродуктами
(антикоррозионные присадки) — вещества, вводимые
в топлива, масла и смазки с целью уменьшения кор-
розии металлов. К топливам обычно добавляют
т. н. И. ржавления, замедляющие коррозию металлов
231 ИНГИБИТОРЫ 232
Таблица !. Некоторые рекомендуемые ингибиторы, области применения и предполагаемый механизм их действия
Области применения Название ингибиторов Металлы, коррозия которых тормозится данным ингибитором Предполагаемый основной механизм действия ингибитора
Борьба с кислотной корро- зией; травление железа и стали в кислотах; защита емкостей при хранении и транспорте кислот Анилин, этпламин, диэтпламин, че- тырехзамещенные аммониевые основания и их производные, па- рафенилендиамин, (Э-нафтиламин, фенил-р-нафтиламин, пиридин, хинолин, нафтохинолин, диме- тилхинолинь дибензилсульфидтио- карбамнд, дибензилсульфоксид, нафтилхинолин - о - толилтиомоче- впна, тиодигликоль, акридин, акрилфлавин, сульфированные нефтяные остатки, сульфирован- ное касторовое масло и др. Железо, низко- и высо- колегированные стали, чугун Анодное торможение или экра- нирование поверхности метал- ла от коррозионной среды
То же As2O3, NaAsOj, соли галогенов (особенно NaJ) Черные металлы и ни- кель Катодное торможение (повыше- ние перенапряжения выделе- ния водорода)
Борьба с коррозией в ней- тральных солевых р-рах и рассолах; холодильная про- мышленность; противообле- денительные и антигрн- зутные р-ры Бикарбонат кальция Все металлы Образование защитных карбо- натных пленок
Хроматы Fe, Al, Zn, Си, латунь Пассивирование
Двузамещенные фосфаты Fe, Zn, Си Образование фосфатных пленок
Гексаметафосфат натрия Fe, Zn, Си, Pb, латунь То же
Сульфит натрия Fe и др. металлы Связывание кислорода
Бензоат натрия Fe, сталь, чугун —
Защита оборудования в при- родной и технической воде; водяные холодильники; оборудование нефтедобы- вающей и горной пром-сти и др. Бикарбонат кальция Fe й др. металлы Образование пленок нераство- римых карбонатов
Са (ОН). Fe, Zn, Си, латунь Подщелачивание илй нейтрали- зация среды; образование за- щитных пленок
Хроматы Fe, Zn, Си, латунь Пассивирование
Двузамещенный фосфат Fe, Zn, Си, латунь Образование нерастворимых фосфатных пленок
Гексаметафосфат Na и Са Fe, Zn, Си, Al, латунь
Ферроцианид Fe, Zn, Си, латунь -
NaOH Fe, сталь, чугун Подщелачивание среды и обра- зование защитных пленок
NaNO3 Fe, Zn, Си, латунь, А1 Пассивирование
Na.SiO, Fe, Zn, Си, латунь Образование силикатных пленок
Сульфит натрия Fe и др. металлы Поглощение кислорода
Бензоат натрия Fe и др. металлы —
Защита от коррозии систем снабжения питьевой водой Бикарбонат кальция Fe и др. металлы Образование защитных пленок
Са (ОН)2 Fe, Zn, Си, латунь Подщелачивание или нейтрали- зация среды и образование за- щитных пленок
Na.SiO, Fe, Zn, Си, латунь Образование защитных пленок
Защита от коррозии паро- вых конденсаторов и ко- тельных систем Двухосновной фосфат 1 1 — •{ Fe, Zn, Си, латунь - Гексаметафосфат Na и Са | J Образование нерастворимых фосфатных пленок
NaOH Fe, Zn, Си, латунь Подщелачивание среды и обра- зование защитных пленок
NH3 Fe, сталь, чугун Повышение pH котельной воды и конденсата
Морфолин, октадециламин | Fe, сталь, чугун Образование защитных гидро- фобных пленок
Борьба с коррозией под дей- ствием влажных нефтепро- дуктов Гидразин Fe, сталь, чугун Поглощение кислорода
Лецитин Fe, Al По-видимому, анодное тормо- жение
Меркаптобензотиазол | Fe, сталь, чугун Анодное торможение
NaNO3 Fe, Zu, Си, латунь
NaNOa Fe, сталь, чугун
233
ИНГИБИТОРЫ
234
Продолжение
Области применения Название ингибиторов Металлы, коррозия которых тормозится данным ингибитором Предполагаемый основной механизм действия ингибитора
Ингибиторы, применяемые как пигменты в лакокра- сочных покрытиях или за- щитных смазках Хроматы Zn и РЬ Fe, Zn, Си, латунь Анодное торможение (пассиви- рование)
Свинцовый сурик и нек-рые орга- нич. И. Fe, сталь, чугун Экранирование и анодное тор- можение
Зашита от атмосферной кор- розии при хранении дета- лей в контейнерах или упаковке в оберточные материалы (летучие И.) Аммиак, морфолин, карбонат цик- логексиламмония, нитрит дицп- нлогексиламмония, бензойнокис- лый амиламин, нитриг дипзо- пропиламмония, карбонат мстил- цпклогсксиламмонпя п др. Железо, сталь, чугун Переход в газовую фазу, ад- сорбции пленкой влаги на по- верхности металла и после- дующее торможение микро- электродных процессов
в присутствии воды, попадающей в топлива в процессе
пх эксплуатации. Эти присадки, являясь поверхност-
но-активными веществами, концентрируются на по-
верхности раздела топливо — металл и защищают по-
следний от контакта с водой. Для этих целей приме-
няют аминопроизводные и аммонийные соли сульфо-
нафтеновых к-т, эфиры жирных к-т и полиспиртов,
полйалкилированный бензосульфонат аммония, диал-
.килортофосфаты, соли аминов и диалкилортофосфор-
ной к-ты в концентрациях 0,004—0,08%. Роль И.
выполняют и содержащиеся в топливах естественные,
а... также добавляемые синтетич. антиокислители и
дезактиваторы металлов.
Для дизельных и котельных топлив, в к-рых со-
держание серы достигает 1—3%, большое значение
имеют И., устраняющие влияние сернистых соеди-
нений на коррозию металлов. В этих случаях к топ-
ливам добавляют нек-рые амины, тормозящие кор-
розию черных металлов; для защиты цветных металлов
(медь) вводят а-нафтиламин, триэтаноламин, анилин,
хинолин, бензиловый спирт, фталевый ангидрид.
Продукты сгорания сернистых соединений — SO.,
и SO3 — дают с водой сернистую и особенно агрессив-
ную серную к-ты. Присадки могут снижать корро-
зионное действие продуктов сгорания сернистых со-
единений; торможением окисления SO2 в SO3 в газовой
фазе, образованием на рабочих поверхностях двигателя
защитных пленок и нейтрализацией окислов серы.
Для этих целей вводят 0,3% нафтената цинка, 1—2%
трибутилфосфата, нитраты и карбонаты щелочных
металлов, а также диэтиламин (1—3%), кубовые
аминные остатки (0,8%). Так, напр., при введении
в топливо, содержащее 1,5% серы, растворимой соли
натрия износ гильз цилиндров снижается на 60—70% .
Высокий эффект дает введение в камеру сгорания дви-
гателей газообразного аммиака (0,1—0,2%, считая
на топливо).
Для мазутов, используемых в качестве топлив для
котельных установок и газовых турбин, большое
значение имеют присадки, устраняющие ванадиевую
коррозию, усиливаемую присутствием в золе топлива
натрия. При сгорании топлив, содержащих ванадий,
образуется пятиокись ванадия, способствующая кор-
розии металлов (напр.: Fe -|- V2O5 —> FeO -j- V2O4,
затем V2O4 4* 1/2Ог —” V2O5 и т. д.). Для устранения
ванадиевой коррозии в топки котельных установок
и в камеры сгорания двигателей отдельно от топлива
вводят соединения кремния — силиконы, кизельгур,
опоки, диатомовую землю, каолин, бентонит, а также
газообразный аммиак. Присадки в виде нафтенатов
бария,_ магния и кальция, магниевых солей синтетич.
жирных к-т (С17—С20), нейтрализованного магнием
окисленного петролатума (0,05—0,2%) добавляются
непосредственно в топлива.
И., вводимые в масла, предназначены для предохра-
нения подшипников от коррозии продуктами окисле-
ния масел. Механизм действия И. состоит в образо-
вании на поверхности металлов защитных пленок
и изменении направления окисления масла в сторону
образования менее агрессивных соединений. В каче-
стве И. применяют трибутилфосфит, трифенилфосфит,
тиофосфорные соединения, органич. сульфиды, осер-
неннос масло, феноляты диалкилфенолсульфпдов и др.
Сравнительная эффективность нек-рых И. приве-
дена в табл. 2.
Таблица 2. Влияние присадок, вводимых в дизельное масто Д-11, на коррозионный износ метанов (на лабораторной установке)
Наименование присадок Содержа- ние инги- битора, % Содержание металла в мас- ле за 10 час работы, .нг/л
РЬ Си' Fe
Масло без присадок 96 19 63,5
Масло с присадками: ЦИАТИМ-339 .... 3 75,5 12,7 36.2
НАМИ-25 3 69,4 8,5 29,5
Применяемые в практике промышленные антикор-
розионные присадки для масел часто обладают и дру-
гими полезными свойствами (моющими, антиокисли-
тельными и др.). Нек-рые И., добавляемые к маслам,
защищают двигатель и от коррозии продуктами сго-
рания сернистых соединений. Так, при применении
масла с 3—3,5% присадки ЦИАТИМ-339 дизельные
двигатели нормально работают на топливах, содержа-
щих до 1% серы. Для этой же цели применяются
«щелочные» присадки, содержащие комплексные ме-
таллорганич. соединения бария и др. металлов, спо-
собные нейтрализовать серную и сернистую к-ты
(содержание присадок в маслах 8—10% и даже 20%).
К маслам, предназначенным для консервации меха-
низмов, добавляются И. ржавления: нек-рые эфиры
непредельных жирных к-т (напр., присадка МТ-6),
защищающие от коррозии сталь и не оказывающие
отрицательного влияния на цветные металлы; соли
нефтяных сульфокислот, нек-рые жирные к-ты, соли
жирных и нафтеновых к-т, азот- и фосфорсодержащие
соединения, защищающие сталь, но агрессивные
к цветным металлам. К нек-рым смазкам, имеющим
невысокие защитные свойства (литиевые, бентонитовые
и др.), добавляют в качестве И. свободные жирные
к-ты, их мыла, нек-рые амины, эфиры, соли нафте-
новых и сульфоновых к-т.
Ингибиторы окисления нефтепродуктов (антиокис-
лители) — вещества, добавляемые к топливам, мас-
лам и смазкам с целью замедления их окисления при
длительном хранении или в процессе применения
в механизмах. Наиболее эффективные И. окисления
для топлив найдены среди ароматич. аминов, фено-
лов, нафтолов, аминофенолов, амицонафтолов, амино
алкилфенолОв и др. И. добавляют к топливам в ко-
личестве 0,001—0,1%. Обычно повышение концен-
трации в указанных пределах улучшает стабиль-
ность топлив. Характеристика нек-рых И. приведена
в табл. 3.
235
ИНГИБИТОРЫ — ИНДАЗОЛ
236
Таблица 3. Характеристика нек-рых ингибиторов
окисления нефтепродуктов
Ингибиторы окисления Топлива и масла и применяемые концент- рации ингибиторов Растворимость
Полифеиолы
Древ есно-смо ль- ный типа Б (смесь фенолов и их эфиров) Автобензины, 0,065—0,1% на крекинг-компонент В нефтепродук- тах ограничен- но, в воде труд- но
Пиролизат дре- весной смолы (смесь фенолов) Автобензины, 0,065—0,1% на крекинг-компонент То же
А м ин офенол ы
Фенил-п-аминофе- нол (параокси- дифениламин) Авиабензины, 0,004— 0,005%; реактивное топливо, 0,01 %; транс- форматорное масло, 0,01—0,1% В нефтепродук- тах очень огра- ниченно, в воде трудно, в бен- золе хорошо
Ал кил фенолы
2,4-Диметил-6- тпрет-бутилфе- нол Авиабензины, 0,0015— 0,003%; реактивные топлива, до 0,015% В топливе неогра- ниченно, в воде весьма трудно (0,02% при 17°)
2,6-Ди-тпрет-бу- тил-4-метил-фе- нол (технич. название ионол, топанол-О, ке- робит) Авиабензины, 0,0015— 0,003%; трансформа- торные масла, 0,1 — 0,8%; реактивные топ- лива, до 0,015%; масла для газовых турбин, 0,2—0,3% В топливе неогра- ниченно, в воде весьма трудно (0,01 % при 20°)
Амины
N, N'-Ди-втор- бутил-п-фени- лен-диамин Авиабензины, 0,0015— 0,003%; реактивные топлива, до 0,015% В нефтепродук- тах растворим, в воде весьма трудно (0,05% при 20°)
Максимальное повышение стабильности топлив на-
блюдается при введении И. в «свежие» топлива; при
повторном добавлении И. в топлива в процессе их
хранения иногда также наблюдается положительный
результат. Часто И. применяют совместно с дезакти-
ваторами металлов (напр., с салицилиденами в кон-
центрациях 0,002—0,005%), устраняющими влияние
металлов на процессы окисления топлив. Эффектив-
ность И. определяется не только их строением, но за-
висит и от условий применения, типа окисляющихся
углеводородов топлив, присутствия в них доба-
вок и т. д.
В качестве И. для автомобильных бензинов, трак-
торных керосинов, реактивных и дизельных топлив,
содержащих компоненты термин, и каталитич. кре-
кинга, применяют полифенолы (напр., антиокисли-
тели древесно-смольные марки Б, пиролизат древес-
ной смолы, фенолы, выделенные из продуктов пере-
работки бурых углей). Алкилфенолы (2,4-диметил-6-
тренг-бутилфенол; 2,6-ди-трет-бутил-4-метилфенол)
и аминофенолы (фенил-парааминофенол) менее эффек-
тивно тормозят процессы смолообразования, чем
полифенолы. С повышением пределов выкипания топ-
лив снижается эффект от добавления к ним И.; наи-
меньшей «приемистостью» к И. обладают дизельные
топлива каталитич. крекинга. Добавление фенольных
И. (0,065—0,1% на крекинг-компонент) позволяет
увеличить допустимый срок хранения автомобильных
бензинов с нескольких месяцев до 1,5 лет и более.
Для этилированных авиационных бензинов, не
содержащих непредельных углеводородов, эффектив-
ными И. являются аминофенолы — фенил-н-амино-
фенол (параоксидифениламин) и бензил-н-аминофе-
нол, а также «экранированные» алкилфенолы — 2,6-
ди-трет-бутил-4-метилфенол, 2,4-диметил-6-трет-
бутилфенол и и-бутил-н-аминофенол, а также нек-рые
амины, напр. N, N'-ди-втор-бутил-н-фенилен-диамин.
Добавление фенил-н-аминофенола (0,004—0,005 вес. %)
позволяет увеличить срок хранения этилированных
авиационных бензинов с нескольких месяцев до
3—4 лет. Фенил-н-аминофенол и ионол вводят и
в этиловую жидкость для ее стабилизации при хра-
нении.
И. можно добавлять также в реактивные топлива
прямой перегонки с целью сохранения их термин,
стабильности (устойчивости топлив против образо-
вания в них осадков при 150—250°) при длительном
хранении. Для трансформаторных и турбинных ма-
сел, являющихся маслами глубокой очистки, могут
применяться И. тех же типов, что и для топлив,
т. е. присадки аминного и фенольного характера.
Наиболее широкое распространение в качестве И.
для трансформаторных и турбинных масел получили
фенилпарааминофенол и ионол (табл. 3), посредством
к-рых стабильность и срок службы трансформаторных
и турбинных масел можно повысить в 2—3 раза.
В качестве И. для моторных масел (автомобильных,
авиационных и дизельных), к-рые содержат большое
количество полициклич. ароматич. и нафтеново-
ароматич. углеводородов и смолистых соединений и
работают при высоких темп-pax (иногда выше 200°),
подвергаясь сильному каталитич. влиянию металлов,
применяют добавки алкилфенольного характера, со-
держащие в молекуле также сульфидную серу и тя-
желый металл, и добавки, содержащие фосфор,
серу и металл. Такие присадки не только тормозят
окисление масел, но и предотвращают каталитич.
воздействие металлич. поверхностей, создавая на
них изолирующие пленки.
В качестве И. для консистентных смазок (см. Пла-
стичные смазки) применяют фенилпарааминофенол,
альдол-а-нафтиламин, фенил-а-нафтиламин, триэта-
ноламин, фенилендиамины, 4-7прет-бутилфенол, 2,6-
ди-трет-бутил-4-метилфенол, а также дитиокарбамат
цинка, мыла нек-рых металлов, фенотиазины, дила-
урил-селенид и др.
Лит.: ЧерножуковН. И., Крейн С. Э., Окис-
ляемость минеральных масел, М., 1955; С а б л ин а 3. А.,
Гуреев А. А., Присадки к моторным топливам, М., 1959;
Лосиков Б. В., Пучков Н. Г., Энглин Б. А.,
Основы применения нефтепродуктов, 2 изд., М., 1959; Мотор-
ные топлива, масла и жидкости, т. 1—2, М., 1957; Petrol.
Refiner, 1959, 38, М 9; Бонер К. Д ж., Производство
и применение консистентных смазок, пер. с англ., М., 1958.
ИНГИБИТОРЫ ФЕРМЕНТОВ — см. Ферменты.
ИНДАЗОЛ (бензпиразол) C,H6N2, мол. в. 118,14 —
твердый продукт; т. пл. 146,5° (кристаллизуется из
горячей воды); т. кип. 260—270°; сублимируется. И.
может существовать в трех таутомерных формах. На
основании спектроскопия, данных для И. и его произ-
I и m
водных, не имеющих заместителей ни у одного из ато-
мов азота, характерна структура III. И.—более слабое
основание (рл 1,3±0,2), чем пиразол. Ароматич. свой-
ства И. по сравнению с пиразолом менее выражены.
Пиразольное ядро И. в достаточной степени устойчиво
к нитрованию; И. нитруется в положение 5. Мягкие
окисляющие агенты (Фелинга реактив) не действуют
на И.; более жесткое окисление приводит к деструк-
тивному распаду. И. восстанавливается каталити-
чески с трудом, но легче, чем бензол. Галогенирование
И. легко проходит и подробно изучено. Ацилирова-
ние И. в зависимости от условий реакции дает либо
стабильные 1-ацилиндазолы, либо лабильные 2-аци-
линдазолы. Алкилированием получают 1- и 2-произ-
водные И. Взаимодействие 1- и 2-алкилиндазолов с
галоидными алкилами приводит к четвертичным солям.
При синтезах И. обычно используют соединения
с уже имеющимся бензольным кольцом, замыкая
237
ИНДАМИНЫ — ИНДАНДИОН
238
пиразольный цикл всеми возможными способами,
напр. нагреванием N-нитрозобензоил-о-толуидина:
С6Н5СООН
5
И. до настоящего времени в природных соединениях
не обнаружен.
Лит. см. при ст. Имидазол. Н. В. Гнучев.
ИНДАМИНЫ — аминопроизводные N-фенилхинон-
диимина (I). Строение И. может быть изображено
двумя общими формулами (А) и (Б), где R—алкил или
И. — глубокоокрашенные (от синевато-красного до
зеленого цветов) кристаллы; хорошо растворимы в
органич. растворителях и минеральных к-тах; в воде
нерастворимы; индамины Б хорошо растворимы в
воде, образуют с минеральными солями (ZnCl2,
HgCI.2, PtCI4) двойные соли. Индамины А окрашены
выше, чем индамины Б; соли индаминов А имеют близ-
кую к индаминам Б окраску. Поглощение света
И. в растворах в значительной степени зависит от
pH среды; увеличение кислотности вызывает гипсо-
хромный сдвиг полосы поглощения. Так, если краси-
тель «зеленый Бинд-
шедлера»(11)в уксусно-
кислом растворе (pH 4)
поглощает в области
ок. 730 ммк (1g е ~
1,1) максимум погло-
щения лежит при 710 ммк (1g е = 4,5). Дальнейший
гипсохромный сдвиг поглощения света наблюдается
при растворении II в конц. HCI (оранжевого цвета)
и конц. H2SO4 (желтого цвета), что связано с присоеди-
нением протона, вначале к центральному азоту, а за-
тем к свободной NR2 группе.
И. — соединения неустойчивые; в щелочной среде
легко гидролизуются с образованием индофенолов,
при кислотном гидролизе получаются и-бензохинон
и производные n-фенилендиамина. Неустойчивость
И. по отношению к кислотам и щелочам ограничивает
их применение в качестве красителей. Аналогично
хинонам И. присоединяют при действии на них вос-
становителей (SnCl2 -j- HCI и др.) два атома водорода
с образованием замещенных 4,4'-диаминодифенил-
амина; последние при окислении (PbO2, Ag2O и др.)
обратно переходят в И. и поэтому могут рассматри-
ваться как лейкосоединения.
И. получают совместным окислением п-фенилен-
диамина и его производных и ароматич. моноамина,
напр. бихроматом натрия, а также взаимодействием
n-нитрозодиалкиланилина с ароматич. аминами. И.
представляют интерес в качестве промежуточных про-
дуктов для синтеза сафранинов. Очевидно, и нек-рые
продукты окисления анилина, образующиеся при
получении красителя, «анилинового черного», имеют
полииндаминовую структуру. Получение окрасок
при крашении меха хинониминовыми красителями
также идет через стадию образования И.
Лит.: Коган И. М., Химия красителей, 3 изд., М.,
1956, с. 321; Венкатараман К., Химия синтети-
ческих красителей, пер. с англ., т. 2, Л., 1957, с. 871 сл.
В. А. Пучков.
ИНДАНДИОН-1,3, мол. в. 146,14 — бесцветные
кристаллы, т. пл. 129 —130°; в щелочах растворяется
с желтой окраской, образуя соли енольной формы.
И.-1,3—наиболее известный кетон группы индана (I).
Получают И. конден-
сацией диэтилфталата _________5СО СО •
с этилацетатом в при- || II 2сН "«« С6н/ >Нг
сутствии металлич. Na СО
и разложением образо-
вавшейся натриевой соли этилового эфира индан-
дионкарбоновой к-ты минеральной к-той:
ZCO°CSHS снвсоос^ СбН; -с-соос2н5 2С
ЧСООС2Н5 ‘ C-ONa
ср
* С6Н4 CHjj
vc6
Две молекулы И. легко конденсируются с образо-
ванием ангидро-бис-И. (б и н д о н а, II).
Последний является хорошим реагентом на первич-
ные амины, нитросоединения и гидразин, его при-
меняют для определе-
ния высокомолекуляр- . хф ZC6H4^
ных аминов, используе- 111 С6н4 С=С СО
мых при флотации ка- ХСО 4ц/
лиевых солей. Окисле- 4 „
ние И. SeO2 дает ин-
датрион. И. конденсируется с альдегидами и хино-
нами; в зависимости от условий образуются или
илидениндандионы, или геминальные дииндандио-
нильные соединения:
А°
СвН^СН2
\ /СО
с=с >6Н4
со
/ф
+С6Нл ZCH2
со
ср
-свн4 сн2
со
со
сн; с6н4
х / со
ДО
сн. zc6H4
, со
Последние при действии водоотнимающих веществ
отщепляют молекулу воды от своих енольных форм
с образованием пиранов. При действии аммиака или
первичных аминов кислород пиранового цикла обме-
нивается на азот и получаются производные дигидро-
пиридинов, часто легко окисляющихся до пиридинов.
Т. обр., исходя из И., альдегида и хинона (кетона),
легко перейти к сложным многоядерным гетероцик-
лич. соединениям:
СО-С5Н4 СО-С6Н4 со-свн4
J. А АвяА г = с
Y %н V % _N!V у W)
z 'с=с'он с = <
со-с6щ со-с6Щ 6о-свн4 ;
И. легко галогенируется, нитруется, сульфируется
и азосочетается по активной метиленовой группе. Из
этих производных наиболее важен 2-нитроиндандион.
И. сульфируется диоксансульфотриоксидом или сер-
ной к-той и уксусным ангидридом, получены И. моно-
и дисульфокислоты. 2-Арилиндандионы получают изо-
меризацией фта лидов:
ДСНАг
с6н4 о
сс
СНдОХа
/ф
СВН4 СНАг
чс6
239
ИНДАНТРЕНОВЫЕ КРАСИТЕЛИ - ИНДЕН
240
Некоторые арилиндандионы являются антикоагулян- I
тами крови и нашли практическое применение, на- |
пример 2-фенилиндандпОн (в Совет-
ском Союзе под названием «фенилин»).
Они легко галогенируются, сульфи-
руются, нитруются ио активной ме-
тиленовой группе; нитросоединения
расщепляются щелочью на фталевую
кислоту и нитрометильное производ-
ное (см. Нитроиндандион). 2-Галогеп-
2-арилпндандиоиы реагируют с ами-
нами. Наибольший интерес представ-
ляют 2-алкиламино-2-фенилиндандио-
ны, проявляющие сильное физиологии.
действие. 2-Метиламино-
2-фенилиндандион предло-
жен как снотворное, 2-этп-
ламино - 2 - фенилииданди-
он — в качестве противо-
судорожного средства.
2-Ацилиндандионы полу-
чаются конденсацией диме-
тилфталата с метилкетона-
ми в присутствии метилата
натрия;
I в И. при окислонип кислородом воздуха. Предель.
| ным продуктом восстановления является антразин (V).
О
VaOCl
II (.зеленоеаю-желтый >
Na2S-rO4 + Zn + Na ОН или HJ + P
О Na
V (бесцветный)
III (темно-синий куб)
a
ONa
IV (иеМо-коричиевый куб)
О
I (синим)
Na2S2O4 + NaOH
ZCOOCH3 ZCQ
С6н' + CH3COR С6Н4 CHCOR
\ Х-. П IN а х /
соосн3 со
Среди них имеются очень сильные антикоагулянты кро-
ви, напр. дифенилацетилпидапдион [R = CH(C6H6)2],
называемый «дипакспн» (за границей), «дифенацин»
(СССР), триметилацетилипдандион [пивалилипдапдиоп
R = С(СН3)3] и др. Дифенацин является наиболее
эффективным антикоагулянтом крови. Последние со-
единения применяют также как средства для борьбы
с вредными грызунами, напр. «ратиндан», содержащий
в качестве действующего начала дифенацин.
Лит.: Циклические 0-дикетоны [Сб. статей], под ред.
Г. Ванага, Рига, 1961. Г. Я. Ваиаг.
ИНДАНТРЕНОВЫЕ КРАСИТЕЛИ — см. Кубо-
вые красители.
ИНДАНТРОН (ипдантрен, №,№-дигидро-1,2,
1',2'-антрахиноназин) C28H14O4N2, мол. в. 442,14 —
синие с металлич. красноватым блеском иглы пли
зеленовато-синие кристаллы (полиморфизм), трудно
растворимые в органич. растворителях: 1 часть И.
растворяется при темп-ре кипения в 500 частях хино-
лина или 5000 частях нитробензола. При нагревании
II. возгоняется. И. не разлагается при нагревании на
воздухе до 470°, выдерживает нагревание с конц.
НС1 до 400° и сплавление с КОН при 300°. И. — сла-
бое основание; с конц. минеральными к-тами образует
легко гидролизующиеся соли.
При действии окислителей, напр. NaOCI, И. (I)
теряет оба атома водорода иминогруппы и переходит
в зеленовато-желтый аптрахиноназин (II). Образо-
вание II происходит также при окислении И. на во-
локне. Этим объясняется непрочность красителя
при обработке окрашенной ткани гипохлоритом. В от-
личие от других полициклич. красителей, содержащих
4 карбонильных группы, для перевода И. в «куб»
достаточно двух атомов водорода (2 эквивалента вос-
становители, напр. Na2S2O4 -j- NaOH). Образующееся
при этом лейкосоединение (III) окрашено в темно-
синий цвет и имеет высокое сродство к хлопку. Кра-
шение И. ведут при 60°. При повышении темп-ры или
восстановлении И. гидросульфитом с добавкой Zn-пы-
лп окраска куба становится желто-коричневой, что
связано с образованием тетрагидропроизводного (IV);
четырехнатриевая соль последнего обладает незначит.
сродством к волокну и лишь частично, превращается
Из реакций электрофильного замещения практи-
ческое значение имеет галогенирование И., так как
при этом улучшается прочность красителя к хлору.
При прямом хлорировании И. в среде C6H5NO2 или
H,SO4 атомы хлора вступают в положения 3,3' и
4,4'; нагревание И. с SO2C12 дает исключительно
3,3'-дихлориндантрон, известный под названием
кубового голубого К. При бромировании И. в среде
H2SO4 при 60° получается 3,3'-диброминдантрон
(ипдантрен голубой ГЦ); при 100° образуется тетра-
бромпроизводное. Нитрование И. сопровождается
реакциями его окисления. Нагревание II. с конц.
HNO3 приводит к образованию тетранитротетраокси-
антрахиноназина; смесь HNO3-|- H2SO4 уже при 0°
окисляет И. в антрахиноназин (II).
В пром-сти И. с выходом 55% получают сплавле-
пием 2-аминоантрахинона со смесью КОН-|- NaOH-|-
4- CHgCOONa при 200—250° в присутствии окисли-
телей (KNO3 и др.). И. образуется с выходом до 70%
при нагревании до 200° 1-аминоантрахииона с феноля-
тами и солями низших алифатич. кислот. И., откры-
тый еще в 1901, до настоящего времени применяется
в крашении хлопка под названием «кубовый синий О»,
«индантреновый синий Р» и др.
Лиш.: Коган И. М., Химия красителей, 3 изд., М„
1 956, с. 562; Венкатараман К., Химия синтетических
красителей, пер. с англ., т. 2, Л., 1957, с. 1068. „ . _
В. А. Пучков.
ИНДЕКС ВЯЗКОСТИ — показатель, характери-
зующий изменение вязкости смазочных масел в за-
висимости от их темп-ры. Подробнее см. Вязкость
нефтепродуктов.
ИНДЕН С9Н8, мол. в. 116,15 — бесцветная жид-
кость; т. пл. —2,59°; т. кип. 182,44°; 0,9957;
Ир 1,5756; т. воспл. 650—655°; парахор
JJ ||| 290,0; криоскопия, константа 7,28^; не-
растворим в воде, растворим в пиридине,
2 СС14, ацетоне, CS2, хорошо — в спирте
и эфире; дает пикрат (т. пл. 98°). И. уже при комнат-
ной темп-ре, особенно легко в присутствии H,SO4,
полимеризуется с образованием полииндена; присо-
единяет бром по месту двойной (2,3) связи; с натрием
образует инденнатрий, к-рый при алкилировании
галогеналкилами дает 1-алкилзамещенные И.:
241
ИНДИВИД ХИМИЧЕСКИЙ—индиго
242
восстановлением в присутствии Ni (200°), Pt (20°)
или натрием в спирте превращается в гидрин-
д е н (индан) — бесцветная жидкость; т. кип.
177°; dl0 0,9645; nfi 1,5381:
При более высокой темп-ре образуется гидриндан.
С альдегидами И. конденсируется подобно цикло-
пентадиену. напр., с бензальдегидом в присутствии
щелочных алкоголятов образуется 1-бензальинден.
И. получают из кам.-уг. смолы. В небольших коли-
чествах И. содержится в нефти, продуктах катали-
тич. крекинга (630—680°, Си), нек-рых эфирных мас-
лах и др. И. применяют для приготовления кумарон-
инденовых (т. наз. паракумароновых) смол.
В. Н. Фросин.
ИНДИВИД ХИМИЧЕСКИЙ — фаза, состоящая
из одного вида вещества (химич. элемента или химич.
соединения). В химич. практике под И. х. подразу-
мевают «химически чистые» вещества — элементарные
простые тела н химич. соединения, состоящие в пре-
деле на 100% из атомов или молекул определенного
вида и обладающие индивидуальным комплексом
постоянных свойств. Поскольку абсолютная чистота
веществ практически не достижима, то, в этом смысле,
понятие И. х. является идеальным. И. х. противопо-
ставляются механич. смеси и однородные фазы — рас-
творы, состоящие из различных И. х.
Лит.: Доклады на совещании по определению понятия
химического соединения, М., 1953; Резолюция совещания по
определению понятия химического соединения, Ж. неорг.
химии, 1956, 1, вып. 7, 1597.
ИНДИГО (индиготин) C16HL0O2N2, мол. в. 262,27 —
кристаллы синего цвета; т. пл. 390—392° (с разл.);
трудно растворяется в боль-
? , шинстве органич. растворите-
-С. /V4 лей (10° мл CeHsN°2 ПРИ 20°
Р Т 3 2С=С^ ,Y fl растворяют 0,0004 г, при т.
кпп- 0,5—1,0 г). В полярных
14 * растворителях (анилин, фе-
0 нол) И. имеет синий цвет, в
кипящем парафине — красный; пары И. пурпурно-
красного цвета. И. легко вступает в реакции электро-
фильного замещения. Ориентация при замещении оп-
ределяется opmo-положением СО- и NH-групп, имею-
щих противоположное ориентирующее влияние. Элек-
тронофильный заместитель вступает в молекулу И.
последовательно в 5,5' и 7,7'-положения, т. е. порто-
и пара-места по отношению к иминогруппе. Замещение
в положения 4 и 6, являющиеся по отношению к СО-
группе орто- и пара-местами, не идет, вследствие дез-
активирующего влияния СО-группы. Поэтому 4- и 6-
замещенные получают синтезом из соответствующих
замещенных бензола.
И. хлорируется и бронируется в среде C6H6NO2 или
H2SO4 с образованием 5-моно-, 5,5'-ди-, 5,5',7-три- и
5',5,7,7'-тетрагалогенопроизводиых, представляющих
собой ценные кубовые красители.
При сульфировании И. получают 5,5'-дисульфоки-
слоту — индигокармин, и далее 5,5',7,7'-тетрасульфо-
производное. При нитровании в молекулу И. вступает
одна, две или три нитрогруппы в зависимости от ко-
личества HNO3. Система двух сопряженных СО-групп
определяет способность И. легко восстанавливаться
(присоединение атомов водорода). Так, при действии
Na2S2O4 или Fe(OH)2, Zn и др. в щелочной среде
И. образует бесцветный р-р лейкоиндиго (I),. Лейко-
индиго обладает значительным сродством к хлопку
(шерсти) и хорошо выбирается (адсорбируется) ок-
рашиваемой тканью; лейкоиндиго под действием
кислорода воздуха на ткани легко переходит в нерас-
творимое И. При осторожном подкислении раствора
лейкопндиго выделяется белый осадок, т. н. белое
И. После смешения с мелассой белое И. применяли
для крашения и печати. При обработке сухого лей-
коипдиго (I) хлорсульфоновой к-той в среде пиридина
образуется сложный эфир—индигозоль (11) (см.
Кубовые красители растворимые'), устойчивый в ще-
лочной и нейтральной средах, но при действии
окислителей в кислой среде превращается в И.
OSO3N’a
OSO3Na
При окислении И. мягкими окислителями (РЬО2,
Ag2O) в инертных растворителях образуется дегид-
роиндиго; в более жестких условиях (КМпО4, озон)
окисление идет с разрывом этиленовой связи и обра-
зованием изатина. При щелочном гидролизе И. полу-
чаются индоксил-2-альдегид и антраниловая кислота,
а при сплавлении с КОН — антраниловая кислота
и анилин.
И. известен в цис- и транс-формах. Считают,
что И., свежеприготовленное окислением куба, яв-
ляется tfuc-формой, к-рая самопроизвольно в течение
нескольких часов переходит в транс-форму. Рентгено-
графии. исследование кристаллов И. показало на-
личие только транс-формы.
Применявшийся в древности краситель античный пурпур
(Тирийский пурпур), добывавшийся . ив моллюсков Мигех
braudaris, представляет собой 6,6'-диброминдпго и может быть
получен синтетически из п-бром-о-нитробензальдегида.
Первый синтез И. осуществлен в 1870 Байером из изатина
через изатинхлорид; при восстановлении последнего (Zn +
+ СНзСООН) образуется И. В дальнейшем было разработано
большое число методов синтеза И., многие из к-рых получили
промышленное значение. На схеме приведены нек-рые епо-
ашранилзеая к-ia. соль
| CiCHgCOONa
NH-CH2COONa
COONa
февилглицин о=карбоновой
щеяочнал плавка
р—
о
Zn,CHaCOOH
индоксил
собы получения И. Развитие промышленного произ-ва син-
тетич. И. оказало огромное влияние на всю химич. пром-сть;
напр., были разработаны методы окисления нафталина до
фталевого ангидрида» получение хлора электролизом и др.
Количественное определение И. основано на его сульфиро-
вании до тетрасульфокислоты и титровании окислителем
(КМпО*) или восстановителем (ТтС12).
И. применялся в качестве красителя, добывался он из ряда
индигоносных растений (Indigofera tinctoria, I. anil, Isatin
tinctoria и др.), содержащих индикан CuHi?Oe-3H8O — гли-
козид индоксила. В настоящее время И. утратило свое исклю-
чительное значение, к-рое оно занимало раньше среди нату-
ральных, а затем синтетич. красителей. И. — дешевый ку-
бовый краситель, применяющийся длн крашения хлопка
и шерсти. Прочность окрасок И. недостаточна к трению и дейст-
вию хлора.
Лит.: Джу л иен П., Мейер Э., Пр инти Э.,
в кн.; Гетероциклические соединения, под ред. Р. Эльдер-
фплда, [Сб. статей], пер. с англ., т. 3, М., 1954, с. 135; К о-
243
ИНДИГОИДНЫЕ КРАСИТЕЛИ —ИНДИЙ
244
ган И. М., Химия красителей, 3 изд., М., 1956; В е н-
катараман К., Химия синтетических красителей, пер.
с англ., т. 2, Л., 1957, с. 1152. В. А. Пучков.
ИНДИГОИДНЫЕ КРАСИТЕЛИ — кубовые кра-
сители, близкие по строению к индиго. К ним отно-
II
сятся замещенные инди-
го, его изомер индиру-
бин (I), их аналоги, где
иминогруппы частично
или полностью замещены
атомами серы, напр. тио-
индиго красный С (II), или другими гетероатомами,
а также соединения, содержащие в молекуле только
один гетероцикл, как это имеет место у тиоиндиго
алого (III).
Рентгеноструктурное исследование кристаллов ин-
диго, тиоиндиго и селениндпго показало наличие
только транс-изомера, что, вероятно, относится также
и к другим И. к. в твердой фазе. Для тиоиндиго, его
замещенных и нек-рых N-замещенных индиго в рас-
творах, по данным исследования спектров, обнаружены
вещества, для к-рых принимается строение ifuc-изоме-
ров. Взаимный переход осуществляется под влиянием
света в зависимости от длины волны.
Для И. к. принята специальная номенклатура, со-
гласно к-рой индиго обозначается 2,2'-бисиндолин-
диго; индирубин (I) — 2,3'-бисиндолиндиго; тиоин-
диго (II) — 2,2'-бистионафтениндиго; а тиоиндиго
алый (III) — 2-тионафтен-1'-аценафтениндиго. Счет
положения заместителей повторяется для каждой
половины молекулы, и, в соответствии с общим прави-
лом, начинается с гетероатома. Торговые названия
Вг
IV
И. к. в СССР для замещенных индиго включают слова
«индиго», для серусодержащих красителей «тиоин-
диго», напр. броминдиго (IV), тиоиндиго красный
С (II), тиоиндиго черный для печати (V).
Типичной для И. к. является группировка
В значительных количествах для печати по тканям
из целлюлозных волокон применяется броминдиго(1У),
получаемое бромированием индиго. В ряду произ-
водных тиоиндиго и смешанных несимметричных И. к.
известны кубовые красители от оранжевого до черного
цвета. Они отличаются хорошей прочностью, не-
сколько уступают по прочности к свету полициклич.
кубовым красителям. М огут красить из слабощелочного
(аммиачного) «куба» и, следовательно, пригодны для
крашения животных волокон и меха. И. к. выпус-
каются в значительных количествах и главное при-
менение находят в печати по тканям из целлюлозных
волокон.
Лит.: Коган И. М., Химия красителей, 3 изд., М.,
1956; Венкатараман К., Химия синтетических кра-
сителей, пер. с англ., т. 2, Л., 195 7. Н. С. Докунихин.
ИНДИГОКАРМИН (динатриевая соль 5,5'-дисуль-
фокислоты индиго) CleH8O8N2S2Na2, мол. вес 466,37 —
синий мелкокристаллич. порошок; плохо растворим в
воде (растворы темно-синего цвета), нерастворим
в спирте. Темно-синий водный раствор И. быстро обес-
цвечивается различными сильными восстановителями.
Применяют И. в
качестве окисли- о
тельно-восстапо- 3 ~Г |Г C=cf J L •
вительного ин- \cQ/\XUSO;i'',a
дикатора (Е =
= 0,296 в при pH = 0), напр. при титровании Fe
(III) оловом (II); как pH-индикатор с переходом ок-
раски от синей к желтой в интервале pH от 11,6
до 14,0; для фотометрия, определения нитратов; как
красивый синий краситель, особенно как субстан-
тивный краситель для шерсти, однако недостаточно
устойчивый к действию света. Применяют И. также
для приготовления чернил.
Лит.: Wei cher F. J., Organic analytical reagents,
v. 4, Toronto — N. Y. ~ L., 1948, p. 505. И. IT. Ефимов.
ИНДИЙ (Indium) In — химич. элемент III гр. пери-
одич. системы Менделеева, п. н. 49, ат. в. 114,82.
Природный И. состоит из двух изотопов с м. ч. ИЗ
(4,33%) и 115 (95,67%); последний изотоп обнаружи-
вает очень слабую ^“-радиоактивность (Ti/a = 61014
лет). Поперечное сечение захвата тепловых нейтронов
190 барн (на атом). Из искусственно радиоактивных
изотопов наиболее важен In114 (Ti/2 = 49 дням).
Конфигурация внешних электронов атома И.: 5s2
5рК Энергия ионизации (в эв):
In0 —> In+ —> Ina+ —> In4+ —> In4+
соответственно равна 5,78; 18,87; 28,04; 58,0.
И. был открыт в 1863 Ф. Рейхом и Т. Рихтером, к-рые при
исследовании цинковой обманки фрейбергского месторожде-
ния обнаружили новые спектральные линии синего цвета
(индиго), принадлежавшие неизвестному до того времени
элементу. Благодаря окраске этих линий вновь открытый
элемент был назван «индием».
И. мало распространен в природе и принадлежит
к числу рассеянных металлов. Содержание его в зем-
ной коре 1 • 10“6 вес%. Собственных минералов не
имеет. Будучи халькофильным элементом, содержится
в виде изоморфной примеси в сульфидных минералах
цинка и свинца, а также минералах, представляющих
собой сульфостаннаты и сульфоантимонаты свинца
и цинка (напр., в килиндрите и франкеите).
Физические и химические свойства. И. — серебри-
сто-белый металл. Кристаллизуется в гранецентриро-
ванной тетрагональной решетке с периодами а =
= 4,583 А, с = 4,936 А. Атомный радиус 1,66 А,
ионные радиусы: 1п+ 1,30 А, 1п3+ 0,92 А. Плоти.
7,31; т. пл. 156,4°; т. кип. 2000° ± 100°. Теплота
плавления 6,8 кал/г, теплота испарения 482 кал/з.
Уд. теплоемкость: твердого (0—150°) 0,057 кал/г-град,
жидкого 0,062 кал/г-град. Термин. коэфф, линейного
расширения 33-10-в (20°). Теплопроводность 0,06
кал)см-сек.град (0—100°). Уд. электрич. сопротивление
9,10-10“в ом-см (23°). Температурный коэфф, электро-
сопротивления (0—100°) 0,00490. Твердость по Бри-
неллю 0,9 кГ)мм2. Предел прочности при растяжении
0,23 кГ/.и.и2. Относительное удлинение 22%. Отно-
сительное сужение поперечного сечения 87%. Предел
прочности при сжатии 0,22 кГ/мм2. Модуль нормаль-
ной упругости 1100 кГ]мм2.
И. — химич. аналог галлия; образует одно-, двух-
и трехвалентные соединения; последние наиболее
устойчивы. При комнатной темп-ре на воздухе не
изменяется. При нагревании выше точки плавления
окисляется до In2O3. С хлором и бромом взаимодей-
ствует при комнатной темп-ре, с иодом — при нагре-
вании, образуя соответственно 1пС13, 1пВг3 и InJ3.
В воде в присутствии воздуха И. медленно корроди-
рует. Растворяется в азотной, серной и соляной к-тах.
Нормальный электродный потенциал И. — 0,336 в.
Из о к и с л о в И. наиболее важны 1п2О3 и 1п2О.
245
ИНДИЙ —ИНДИКАН
246
In2O3 — желтые кристаллы с объемно-центрирован-
ной кубич. решеткой, а = 10,105 А; т. пл. ок. 2 000°;
не растворяется в воде, но растворяется в кислотах;
диссоциирует с образованием 1п2О при нагревании
выше 850°. 1п2О — черные кристаллы, возгоняется
в вакууме (104—10 5 мм рт. ст.) при 650—700°;
хорошо растворяется в соляной к-те. Из р-ров солей
трехвалентного И. щелочами осаждается амфотерная
гидроокись 1п(ОН)3; ее кислотные свойства
выражены в меньшей степени, чем у гидроокиси гал-
лия. В растворах едкого натра гидроокись И. незна-
чительно растворяется с образованием индата натрия,
но эти растворы неустойчивы и разрушаются при
нагревании или длительном стоянии. В р-рах аммиака
гидроокись И. нерастворима (отличие от гидроокиси
галлия). В кислотах 1п(ОН)3 легко растворяется;
произведение растворимости в воде 0,7-10-32 (25°).
Хлорид 1пС13 — бесцветные гигроскопич. кристаллы,
плотн. 3,45, растворимость в воде 33,5 г/л (22°);
давление паров 1пС13 над твердой солью достигает
1 атм при 498°; плавится 1пС13 в запаянной ампуле
при 585°. Средний сульфат In2(SO4)3 — бесцвет-
ная хорошо растворимая в воде соль; плотн. 4,438;
выше 600° разлагается; растворимость в воде 62,1%
(25°). Из сернокислых р-ров, содержащих И. ок.
100 г/л и 10—20% серной кислоты, до 30° кристалли-
зуется гидрат In2(SO4)3 • ЮН20. При 30—60° из этих
р-ров кристаллизуется In2(SO4)3- 6Н2О. Из растворов
с концентрацией серной к-ты более 28% (при 20°)
и более 54% (при 60°) выделяется кислый сульфат
In2(SO4)3 • H2SO4 • 7Н2О, к-рый можно также рас-
сматривать как комплексную соль след, состава:
H[In(SO4)2] • 31/2Н2О. Известен основной сульфат
состава 2 In(OH)SO4 • 5Н2О. Сульфид In2S3 изве-
стен в двух модификациях. Из них a-форма желтого
цвета, гигроскопична; имеет кубич. гранецентриро-
ванную решетку, а = 5,36 А; P-форма красного
цвета, негигроскопична, имеет решетку типа шпи-
нелей, а = 10,72 А; темп-ра перехода а 22 Р ок.
330°. Сульфид плавится при 1050° и заметно субли-
мирует выше 800°; выделяется из слабокислых р-ров
солей И. при пропускании сероводорода, растворяется
в р-рах сернистых щелочей с образованием сульфо-
солей.
Аналитическое определение. Ка-
чественно И. определяют гл. обр. спектральным ме-
тодом; количественно — весовым, колориметрии., поля-
рография. и спектральным методами. Весовыми ме-
тодами И. определяют: а) в виде окиси 1п2О3 после
отделения мешающих элементов, осаждения гидро-
окиси 1п(ОН)з и прокаливания последней при темп-ре
не ниже 1200°; б) в виде металла, осажденного элек-
тролитически на катоде из раствора, содержащего
буферную смесь (с pH 3,5—3,8) виннокислого нат-
рия и соляной к-ты. К олориметрич. определение И.
основано на способности оксихинолята И. давать в
четыреххлористом углероде желто-зеленую окраску;
предварительно отделяют И. от мешающих элементов
путем экстракции бромида И. из р-ра НВг в присут-
ствии в качестве восстановителя Na2S2O3. Полярогра-
фия. определение И. проводят в солянокислом р-ре
(3—6н. НС1); небольшие примеси Си, Cd, Pb, Bi и
значительное количество Zn не мешают.
Получение. Сырьем для получения И. служат от-
ходы и полупродукты свинцово-цинкового и оловян-
ного произ-в, в к-рых содержание И. колеблется от
десятых до тысячных долей процента. Освобождение
И. от основных компонентов сырья: Zn, Pb, Sn, Cd,
Си, Fe и др. — трудоемкий, многостадийный процесс,
состоящий из ряда гл. обр. гидрометаллургия, опе-
раций. Технология обычно складывается из 2 этапов:
1) получения обогащенного И. продукта — концен-
трата и 2) переработки концентрата до металлич. И.
Сырье обрабатывают серной к-той, в результате чего
И. вместе с рядом примесей переходит в раствор и
отделяется от основной массы свинца. Для получения
концентрата И. проводят его осаждение гидролизом
(при pH 3,5—4) и цементацией на цинковой пыли
(т. е. вытеснением И. из раствора металлич. цинком
по реакции: 2In3-b —|— 3Zn° т* 21п° -[- 3Zn2+).
Отделение И. от основной массы цинка, мышьяка,
олова и алюминия достигается обработкой осадков
гидроокисей, содержащих И., щелочами, при этом
гидроокиси перечисленных металлов переходят] в
водорастворимые соединения, а И. остается в осадке;
очистка от меди — выделением ее в осадок цемен-
тацией на железе или на цинке при определенной
кислотности раствора (И. остается в растворе);
очистка от железа — переводом И. в осадок цемен-
тацией его на цинке и гидролитич. осаждением после
восстановления железа до двухвалентной формы, в
результате чего железо остается в растворе [pH вы-
деления Fe(OH)2 выше, чем pH выделения 1п(ОН)3].
Обогащенный И. осадок выщелачивают серной к-той
и проводят доочистку от остатков меди, цинка и кад-
мия, что достигается обработкой раствора аммиаком.
В результате этого перечисленные элементы перехо-
дят в растворимые аммиачные комплексы, а И. —
в нерастворимую гидроокись. Последнюю снова рас-
творяют в серной к-те и для отделения от И. остатков
меди, кадмия, мышьяка проводят осаждение их суль-
фидов из кислого р-ра сероводородом; И. при этом
остается в очищенном р-ре, из к-рого металлич. И.
выделяют либо цементацией на цинке или алюминии,
либо электролизом. Для получения И. высокой чи-
стоты, пригодного для применения в полупроводни-
ковой технике, применяют электрохимия, рафини-
рование, химич. способы очистки, а также зонную
плавку.
Применение. И. используется гл. обр. для антикор-
розионных покрытий и в полупроводниковой технике
при изготовлении германиевых кристаллич. выпрями-
телей. Добавка И. к германию создает в нем дырочную
проводимость. Соединения InSb и InAs также имеют
перспективу применения в качестве полупроводнико-
вых материалов. Нек-рое промышленное значение
имеют легкоплавкие сплавы И. со свинцом, оловом,
кадмием и висмутом, применяемые для припоев и в
системе пожарной сигнализации.
Лит.: Меерсон Г. А., Зеликман А. Н., Ме-
таллургия редких металлов, М., 1955; БлешинскийС. В.,
Абрамова В. Ф., Химия индия, Фрунзе, 1958; Рассеян-
ные металлы (индий, галлий, таллий, рений). Области освоен-
ного и возможного применения, под ред. К. А. Большакова,
М., 1959; Smelin, 8Aufl., Syst.-Nr. 37, Indium, В., 1936;
Mellor, v. 5, L.—N. Y. — Toronto, 1952; U 1 1 m a n, 3
Aufl., Bd 8, MUnch. — B., 1957; Kirk, v. 7, N. Y., 1951.
H. А. Гурович.
ИНДИКАН (З-оксииндол-р-О-глюкозид) C14H17NOe,
мол. в. 295,34 — глюкозид индоксила (3-оксииндола),
содержащийси во мно-
гих растениях рода
Indigofera (1,5—2%),
а также в вайде кра-
сильной (0,5%), вкра-
сильной гречке и в
других индигоносных
растениях. И. (I) — бесцветный продукт, растворим
в воде, плохо растворим в большинстве органических
растворителей; из воды кристаллизуется с ЗН2О,
т. пл. гидрата 57—58°, т. пл. безводного И. 176—
178°; аД(,(,= —77,6° и —65,63°, соответственно безвод-
ный и трихлоргидрат ов воде (для зеленой линии Hg
при длине волны 5461 А).
Кислотами и ферментами Р-глюкозидазами (напр.,
индэмульсином микроорганизмов) гидролизуется до
D-глюкозы и индоксила (II), легко окисляемого кис-
лородом до индикотина (синего индиго — III). Эти
।
247
ИНДИКАТОРНЫЕ ЭЛЕКТРОДЫ — ИНДИКАТОРЫ
248
реакции используют при качественном и количест-
венном определении И.
11 ill
Синтетически И. получен из эфира индоксил-2-кар-
боновой к-ты и тетраацетилглюкозилбромида.
Животный индикан (индоксилсульфат — IV)
CsHeNSO4K, мол. в. 251,31 — эфир индоксила и сер-
ной к-ты, бесцветные кристаллы, хорошо раствори-
мые в воде. При действии кислот животный И. гид-
ролизуется до индоксила и серной к-ты. Животный И.
синтезируется в печени, содержится в незначительных
количествах в крови и выводится из организма с мо-
чой. Животный И. является продуктом обезврежи-
вания в организме ядовитого индола, постоянно
всасывающегося из кишечника, где он образуется
из триптофана под влиянием микроорганизмов.
При нек-рых заболеваниях желудочно-кишечного
тракта (при брюшном тифе, раковых опухолях, и
др. патология, состояниях) содержание И. возрастает
как в крови (индиканемия), так и в моче (индика-
нурия). Методы количественного определения И.
в крови и моче основаны на цветных реакциях, к-рые
дает образующийся из И. индоксил, в частности, на
реакциях превращения индоксила в индиго при дей-
ствии различных окислителей: хлорного железа
(Обермейера реакция), хлорной извести
(проба Яффе) и др.
Лит.: Майстер А., Биохимия аминокислот, пер.
с англ., М., 1961; Каррер П, Курс органической химии,
пер. с нем., Л., 1960; The carbohydrates, N. Y., 1957; Hand-
buch der Pflanzenphyslologle, Bd 6, B. — [u. a.], 1958, 761.
ИНДИКАТОРНЫЕ ЭЛЕКТРОДЫ - см. Электро-
ды сравнения.
ИНДИКАТОРЫ. Содержание:
I. Кислотнотщелочные (pH) индикато[ы........ 248
II. Окислительно-восстановительные индикаторы ... 251
III. Комплексометрические индикаторы........... 252
IV-. Адсорбционные индикаторы.................. 253
V. Хемилюминесцентные индикаторы.............. 254
Индикаторы — органич. и неорганич. вещества,
позволяющие устанавливать достижение конечной
точки при титровании (см. Объемный анализ) по из-
менению цвета, появлению или исчезновению мути,
свечения и т. д. или дающие возможность определять
величину к.-л. физико-химич. показателя среды (pH,
окислительно-восстановительный потенциал). И. обыч-
но вводят в анализируемый р-р в небольших коли-
чествах. Конечная точка титрования, устанавлива-
емая при помощи правильно подобранного И., прак-
тически совпадает с точкой эквивалентности химич.
реакции (рТ — показатель титрования должен быть
равен значению pH в точке эквивалентности). И. мо-
гут быть обратимыми или необратимы-
м и. В первом случае, при изменении соответствую-
щего физико-химич. показателя среды, наблюдаемое
изменение И. около его точки перехода может быть
повторено любое число раз (напр., для подавляющего
большинства pH-индикаторов, применяемых при кис-
лотно-щелочных титрованиях) в одну и другую сто-
рону. Необратимые И. (напр., азосоединения в бро-
матометрии) позволяют только один раз наблюдать
точку перехода, т. к. они подвергаются необратимому
химич. изменению. Иногда при титровании одно из
веществ, принимающих участие в реакции, выполняет
функцию И. Так, при титрованиях перманганатом
нередко обходятся без введения И., т. к. минимальный
избыток перманганата вызывает отчетливое изменение
окраски р-ра в конечной точке титрования. В боль-
шинстве случаев И. вводят в анализируемый р^р
(внутренний И.), а иногда отбирают каплю
р-ра и помещают ее на бумажку, пропитанную И.,
или же смешивают каплю исследуемого р-ра с р-ром
II. на белой фарфоровой пластинке или на фильтро-
вальной бумаге. В этом случае И. называют «вне Ш-
н и м».
I. Кислотно-щелочные (pH) индикаторы. И. этой
группы реагируют на изменение pH-среды, и поэтому
применяются в титриметрич. анализе при реакциях
нейтрализации, а также для колориметрия, определения
pH. Обычно это азосоединения, производные трифе-
нилметана и др. органич. соединения, молекулы к-рых
претерпевают структурные изменения в зависимости
от реакции среды.
Рассматриваемые (по теории Оствальда) И. явля-
ются слабыми кислотами или слабыми основаниями,
окраска к-рых зависит от степени диссоциации (а).
Точка перехода И. зависит от константы диссоциации
К индикатора. При малой концентрации И. справед-
ливо ур-ние:
1g рт” = Р Н — рА, где рК = — 1g К
рА — константа И., численно равная значению pH,
при к-ром в растворе концентрации «кислой» и «ще-
лочной» форм одинаковы. Интервал перехода окраски
И. должен находиться между значениями pH = pA'j- 1.
Обычно изменение окраски И. связано с более
сложными равновесиями, чем это предполагает теория
Оствальда, как напр., переход лактонной формы
фенолфталеина из кислой среды (бесцветный) в хиво-
идную форму в щелочной среде (красный цвет)
и в ион карбоновой к-ты в сильно щелочной среде
(бесцветный). Отсутствие общепринятой теории цвет-
ности ле дает возможности во всех случаях однозначно
установить связь между строением молекул II. и их
цветом. В табл. 1 приведены свойства нек-рых наибо-
лее употребительных в' ацидеметрии и алкалимет-
рии II.
Таблица 1
Индикаторы Характер индикато- ра; О-ос- нованпе, К-кпслота Интер- вал пе- рехода PH Цвет
в кислой среде в щелоч- ной среде
Тропеолин ОО (ди- фениламиноазо-п- бензолсульфонат натрия) 0 1,3-3,0 красный желтый
Метиловый оранже- вый (диметиламинс- азобен зол сульфонат натрия) 0 3,1—4,4 красный желтый
Метиловый красный (диметйламиноазо- бензолкарбонован к-та) 0 4,2-6,3 красный желтый
Феноловый красный (феиолсульфофта- леин) к 6.4-8.0 желтый красный
Фенолфталеин . . . к 8,0—9,8 бесцвет- ный красно-' фиолето- вый
Тимолфталеин .... к 93—10,5 бесцвет- ный синий
На интервал перехода окраски рН-индикаторов
влияют: темп-ра, наличие в р-ре солей, органич.
растворителей, белковых в-в и др. Влияние темп-ры
наиболее значительно для И., являющихся слабыми
основаниями; так, для метилового оранжевого при
комнатной темп-ре окраска изменяется в пределах
pH 3,1—4,4, а при 100° — в пределах pH 2,5—3,7.
249
ИНДИКАТОРЫ
250
При титровании применяют такой. И., чтобы зна-
чение pH анализируемого р-ра в точке эквивалентно-
сти находилось в интервале перехода его окраски.
Это особенно важно длн разбавленных р-ров (коицеи-
трацин меньше 0,01 н.). Для последних необходимо
иметь И. с узким интервалом перехода окраски, сов-
падающим с областью наиболее резкого изменении
pH в конечной точке титровании. Наилучшим И. будет
такой, к-рый дает переход окраски между дополни-
тельными цветами через серый оттенок. Это дости-
гается применением смешанных И. (см. ниже). Кис-
лотно-основные И. служат также длн определения
pH. Длн этого сравнивают цвет анализируемого р-ра,
к к-рому добавлен И., с цветом шкалы, составленной
ив буферных р-ров с известным значением pH, содер-
жащих такой же И. Для большей точности необходимо
исключить т. наз. «солевую ошибку» от присутствия
разных количеств солей в анализируемом и буферных
р-рах.
Значение pH можно установить без применения буферных
р-ров. Метод основан на том, что для одноцветного И. при
равных концентрациях его в р-рах окраска зависит только
от концентрации окрашенной формы (метод Миха-
элиса). В анализируемый р-р добавляют а граммов одно-
цветного И., подходящего по интервалу перехода окраски,
напр. одного из нитрофенолов, е щелочноокрашенной формой.
Окраску сравнивают с эталонными, достаточно щелочными
р-рами, содержащими в таком же объеме воды различные
количества И., находящегося в этом случае полностью в ще-
лочной форме. Если эталонный р-р имеет одинаковую окраску
с испытуемым и содержит в граммов И., то в/а — F — отно-
шение концентрации окрашенной формы И. к общей его кон-
центрации. pH вычисляют по формуле:
pH — рК-Н 1gт-—4
1 — г
Г. А. Левцов.
Смешанные и н д и к а т о р ы — смесь двух
И., обеспечивающая отчетливое изменение окраски
в узком интервале значений pH (до 0,2 единиц pH),
что повышает точность титрования. Пользуясь сме-
шанными И., можно, напр., точно оттитровать уксус-
вую к-ту р-ром аммиака, карбонат натрин в присут-
ствии бикарбоната и др. Длн приготовления такой
смеси подбирают два И. с очень близкими интервалами
перехода, имеющими в этой точке дополнительные
цвета. Вследствие этого в точке перехода наблюдаетсн
резкое изменение окраски, переходящее через серый
цвет. Примеры смешанных И. приведены в табл. 2.
Таблица 2
Компоненты показа- тель тит- рования рТ Переход окраски
Бромкрезоловый зеленый 4- метиловый красный (5:1) 4,3 оранжевый—сине- зеленый
Бромкрезоловый пурпурный 4- бромтимоловый синий (1:1) 6,7 желтый — сине- фиолетовый
Тимоловый синий 4- фенолфта- леин (1 :3) 9,0 желтый — фиоле- товый
Тимолфталеин 4- ализариновый желтый (2:1) 10,2 то же
Применяют также смеси И. с красителем, имеющим
цвет, дополнительный к цвету И. в точке перехода,
напр., смесь метилового желтого и метиленового
голубого (1:1) при рТ = 3,25 меняет окраску от сине-
фиолетовой до зеленой; смесь нейтрального красного
и метиленового голубого (1:1) при рТ = 7,0 изменяет
окраску от фиолетовой до сине-зеленой. Предло-
жено также применять смешанные адсорбционные И.,
напр., для аргентометрия, титрования галогенидов
и роданидов, титрования сульфатов р-ром хлорида
бария, фторидов р-ром нитрата кальцин и т. п. Напр.,
длн определении хлоридов и бромидов рекомендована
смесь флуоресцеина и метиленового голубого (15:20);
переход окраски от светло-зеленой к розовато-фиоле-
товой. А. А. Черкасский.
Флуоресцентные индикаторы — ве-
щества, применяемые для кислотно-основных титро-
ваний мутных или сильно окрашенных р-ров, у к-рых
при освещении УФ-лучами при определенном зна-
чении pH появляется (или исчезает) флуоресценция,
или же изменяется ее цвет или оттенок. И. этого типа
являются обычно многие ароматич. (напр., произ-
водные нафталина) и гетероциклич. соединения (напр.,
производные акридина, флуоресцеина, кумарина и
др.). Изменение величины pH у флуоресцентных И.
приводит к образованию ионов или таутомеров, что
сопровождается изменением спектра флуоресценции.
Полосы поглощения обычно находятся в УФ-части
спектра, а полосы излучения в видимой. В табл. 3
приведены свойства пек-рых флуоресцентных И.
Таблица 3
Индикатор Интервал pH измене нин флуо- ресценции Изменение цвета флуоресценции
4-Этоксиакридон 1,4—3,2 зеленый — синий
Диметилнафтэйродшт .... 3,2—3,6 лиловый — оранжевый
а-Нафтиламин 3,4-4,8 отсутствует синий
Акридин . • 4,8-6,6 зеленый — синий
f-Метилумбеллиферон . . . 5,8—7,5 отсутствует синий
Семикарбазон салицилового альдегида 7,5-8,0 слабо желтый — синий
2-Нафтол-б, 8-дисульфокис- лота 7,4-9,0 отсутствует синий
Эухризин 3R (основание акридинового оранжевого) 8,4-10,4 оранжевый — зеленый
Индикаторы для титрования в не-
водных средах — многие из обычных рН-пн-
дпкаторов, а также нек-рые более специфические.
Таблица 4
Индикатор Определяемые веще- ства, среда Титрую- щий реактив • Переход окраски
Кристаллический фиолетовый (0,5% в ледяной уксус- ной к-те) Основания напр. амины и их пикраты, пуриновые основа- ния, в среде лед. уксусной к-ты нею. синяя — зеленая
Метиловый фио- летовый (0,2% в хлорбензоле) Основания, напр. ал- калоиды в среде лед. уксусной к-ты то же фиолето- вая — синяя
Тимоловый синий (0,3% в метаноле) Кислоты в среде ди- метплформамида или бутиламина. Основа- нии (напр., алкалои- ды) в среде этилен- гликоль 4- изопропи- ловый спирт СН3ОК нею. желтая —- синяя
Азофиолетовый (р-р в бензоле) Кислоты в среде эти- лендиамина CH3OR красная — синяя
п-Оксиазобен- зол 4- азофполето- вый (0,2% в бен- золе) Смесь карбоновой к-ты и фенола в среде ацетонитрила или ацетона СН3ОК бесцвет- ная — жел- тая (аце- тонитрил); красная — синяя (ацетон)
о- Н итр оани лин (0,15 % в бензоле) Фенолы в среде эти- лендиамина СН3ОК желтая — оранжево- красная
* Имеются в виду вещества, реагирующие как основания
в среде ледяной уксусной к-ты и т. п.
Поведение И. в органич. растворителях отличается
от поведения его в воде и пока еще мало изучено;
напр., метиловый красный в воде дает переход окраски
при более высоких значениях pH, чем бромфеноловый
251
ИНДИКАТОРЫ
252
синий; в гликолевом р-ре отношение обратное. В
табл. 4 даны нек-рые наиболее употребительные И.
для титрования в неводных средах и указаны области
их применения. Г. А. Певцов.
Универсальные индикаторы — ин-
дикаторы, многократно изменяющие окраску при
разных значениях pH; представляют собой смеси двух-
цветных и одноцветных кислотно-основных индикато-
ров с разными интервалами перехода, подобранные
так, что при изменении значения pH в широких преде-
лах происходят заметные на глаз изменения окраски.
Один из наиболее распространенных универсальных
И. приготовляют смешением 0,1%-ных спиртовых
р-ров следующих индикаторов: диметиламиноазо-
бензола (15 мл), бромтимолового синего (20 мл),
метилового красного (5 мл), фенолфталеина (20 мл)
и тимолфталеина (20 мл). В зависимости от значения
pH этот универсальный И. имеет окраски при pH:
2 — красно-розовую, 3 — красно-оранжевую, 4 —
оранжевую, 5 — желто-оранжевую, 6 — лимонно-жел-
тую, 7 — желто-зеленую, 8 — зеленую, 9 — сине-
зеленую, 10 — фиолетовую. Описаны также другие
смеси, в частности для определения значений pH в
более узких интервалах. Изменения окрасок универ-
сальных И. происходят менее резко и при больших
интервалах pH, чем у индивидуальных индикаторов.
Поэтому универсальные И. не применяют для титро-
вания, ими пользуются только для приближенного
определения значений pH в широких пределах с точ-
ностью ок. одной единицы pH. Индикаторы часто
наносят на фильтровальную бумагу и применяют
в виде универсальных индикаторных бумаг. Окраску
такой бумаги сравнивают с эталонами цветной шкалы.
См. также Бумаги, реактивные. А- Черкасский.
И. Окислительно-восстановительные (о.-в.) индика-
торы. В эту группу И. входят вещества, к-рые реа-
гируют на изменение окислительно-восстановительно-
го потенциала р-ра и поэтому применяются в титри-
метрич. методах анализа, основанных на реакциях
восстановления-окисления (см. Окисления-восстанов-
ления методы), а также для колориметрии, определе-
ния окислительно-восстановительного потенциала (пре-
имущественно в биологии). Обратимые о.-в. И. могут
существовать в р-ре в двух формах: окисленной (IndOx)
и восстановленной (IndRed), находящихся в равнове-
сии между собой и с анализируемой окислительно-
восстановительной системой:
I °’059 I. [In,1Oxl р, . 0,059 , [Ох]
^0 + — ’g = Ео + “ 'g [Red]
где Ео и — нормальные окислительно-восстанови-
тельные потенциалы И. исистемы, [IndOx] и [IndRea] —
концентрации в р-ре соответствующих форм И.,
[Ох] и [Red] — то же для окислителя и восстанови-
теля, п — число электронов, участвующих в реакции.
Нормальный окислительно-восстановительный по-
тенциал И. измеряют при одинаковой концентрации
обеих форм в р-ре (в вольтах, по отношению к нормаль-
ному водородному электроду). Интервал перехода
окраски равен
Eq ± 0,059/п вольт
Для большинства И. п=2. У производных у, у'-ди-
пиридила, т. наз. виологенов более широкий
переход окраски, т. к. в реакции участвует лишь
один электрон. У большинства о.-в. И. окрашена
только окисленная форма; у виологенов окрашена
восстановленная форма. Если в точке эквивалентности
происходит скачок потенциала между значениями
Е2 и Ег, то подходящий И. имеет нормальный потен-
циал между этими значениями.
При титровании сильными окислителями широко
применяются обратимые И., имеющие сравнительно
высокие величины потенциалов перехода окраски,
напр. дифениламин и его производные. При титро-
вании сильными восстановителями (напр., TiCl3,
V11 и др.) применяют обратимые о.-в. И. с более низ-
кими потенциалами перехода окраски, напр. сафра-
нин, метиленовую синюю. Для колориметрии, опре-
деления окислительно-восстановительного потен-
циала применяют обратимые о.-в. И., расположенные
в ряд по убывающей величине Ео. В этот ряд входят
различные индофенолы (Ео от 4-0,27 до 4-0,12 в
при pH 7), индигосульфонаты (Ео от —0,05 до —0,16 в)
и др. Окисленная форма у них окрашена, восстанов-
ленная—бесцветна. Отличительной особенностью этих
И. является сильная зависимость их Ео от рН-сре-
ды. У виологенов потенциал перехода окраски не
зависит от pH.
Колориметрич. определение окислительно-восста-
новительного потенциала аналогично колориметрич.
определению pH: применяют эталоны с различными
количествами И. в его окрашенной форме, что обычно
соответствует разным отношениям (F) окисленной
формы И. к общему постоянному количеству его. По-
тенциал исследуемого р-ра находят по ур-нию:
e = Eq + 0-^ ig ,-Ц
Ч I п 6 1 _ р
где Ео — нормальный потенциал И. при pH анализи-
руемого р-ра, п — число электронов, к-рое теряет
И. при обратимом окислении. В табл. 5 указаны
свойства о.-в. И., наиболее часто применяемых в тит-
риметрич. анализе.
Таблица 5
Индикатор Нормальный окислительно- восстановит. потенциал при рН=0, е Окраска
восстанов- ленной формы окисленной формы
Сафранин Т 4“ 0»24 бесцветная красная
Индигодисульфонат натрия 4-0,29 » синяя
Метиленовый синий . 4-0,53 >> голубая
Дифениламин .... + 0,76 » фиолетовая
Дифениламиносуль- фонат натрия . . . 4- 0,85 » красно-
Фенилантраниловая кислота 4- 1,08 » фиолетовая то же
о-Фенантролин (фер- роин) 4- 1,14 красная бледно-
Нитрофенантролин (нитроферроин). . + фиолетово- голубая то же
Комплекс «, а'-дипи- ридила с рутением 4- 1,зз красная желтая бесцветная
III. Комплексометрические индикаторы. И. этой
группы применяют для установления конечной точки
при комплексометрии, титровании (см. Комплексо-
метрия). И. образует комплекс с определяемым ио-
ном. Прочность этого комплекса должна быть меньше
прочности соответствующего комплекса металла с
комплексоном, применяемым для титрования. По
мере добавления комплексона ионы определяемого
металла образуют с ним прочное соединение. В ко-
нечной точке титрования, к-рая должна отвечать
точке эквивалентности, соединение И. с металлом
распадается и появляется окраска свободного И.,
характерная для данного значения pH. Так, при
титровании магния комплексоном в присутствии
эриохромчерного Т в буферном р-ре при pH 8—10
красная окраска комплекса И. в конечной точке пере-
ходит в синюю окраску свободного И. Для выпол-
253
ИНДИКАТОРЫ — индоксил
254
нения титрования необходимо соблюдение неравен-
ства:
рС0 4- 2 ==S рА;О рК — 2
где рС0, рК{, рК — соответственно отрицательные ло-
гарифмы начальной концентрации определяемого иона
металла, константы «нестойкости» соединения И.
с металлом и константы нестойкости комплексоната
металла соответственно. Типичными комплексометрич.
И. являются гл. обр. хорошо растворимые в воде
органич. красители, способные образовывать с ионами
металлов интенсивно окрашенные внутрикомплексные
соединения. Наиболее важные из них приведены в
табл. 6.
Таблица 6
Индикатор pH перехода окраски свободного индика- тора Цвет комплекса с металлом
Эриохромчерный Т (1-окси-2-нафтилазо)- 6-нитро-2-нафтол-4- сульфокислоты, Na-соль Пирокатехиновый фиолетовый (3, 3', 4'- триоксифуксон-2"- сульфокислота) Фталеинкомплексон (продукт конденсации крезолфталеина, фор- мальдегида и имино- диуксусной к-ты) Ксиленоловый оран- жевый [3, З'-бис-ди- (карбоксиметил) аминометил-о-крезол- сульфофталеин] ПАН 1 -(2-пиридил-азо)-2- нафтол ПАР 1-(2-пиридил-азо)- резорцин < 6 — винно-красный 7—11 — синий > 11,5 — желто- оранжевый в кислой среде — желтый 9—12 — фио- летовый — красно- фиолетовый < 6,5 — бесцветный 7—10 — розовый > 12 — красный < 5—6 — лимонно- желтый в щелочной среде — красно-фиолетовый желтый < 3 — зеленовато- желтый 3—10 — желтый > 11 — красный При pH 8—10 с Mg винно-крас- ный В слабокисл, среде с В1 и в сла- бощелочной с Си — синий При pH 10—И с В а и Sr темно- фиолетовый; с Са и Mg— красный При pH 2,5-3,5 с Th — красный с Zn при pH < 6 розовый
Изменение окраски при взаимодействии металлохромных
И. с ионами металлов имеет ту же природу, что и у рН-инди-
каторов, часто содержащих также фенольные гидроксилы,
напр. у сульфофталеинов, фталеинов. Отнятие или присоеди-
нение к такой группе протона (или иона металла) вызывает
изменение окраски при наличии сопряжения гидроксила с ос-
тальной частью молекулы. Комплексометрич. И., имеющие
в молекуле такие же группы, как и у комплексона, взаимо-
действуют более сложно, т. к. здесь в образовании хелатного
соединения принимает участие и комплексон и фенольные
гидроксилы И. Весьма полезным для определения кальция
является N, М'-ди(карбоксиметил)аминометилфлуоресцеин,
т. наз. флуорексон. Он представляет «металлофлуоресцентный»
И. В щелочном р-ре следы катионов щелочно-земельных ме-
таллов с этим И. дают сильную зеленую флуоресценцию,
к-рая исчезает при титровании комплексоном.
IV. Адсорбционные индикаторы. Вещества, в при-
сутствии к-рых в конечной точке титрования по
методу осаждения происходит изменение цвета осадка,
наз. адсорбционными И. Нельзя дать общую теорию,
объясняющую единообразно механизм действия по-
добных И. В одних случаях происходит адсорбция
ионов И. или его молекул на поверхности частиц
золя, в других — комплексообразование.
Физич. теория действия адсорбционных И. (Фаянс и др.)
объясняет изменение цвета при взаимодействии анионов
И. с катионами тяжелых металлов (Ag, Hg, Pl и др.) сильной
деформацией электронных оболочек анионов. Адсорбционные
И. обычно являются обратимыми. В конечной точке титрова-
ния избыток титрующего реагента вызывает перезарядку
частиц золя; добавление титруемого иона вызывает появление
заряда первоначального знака, что сопровождается изменением
цвета осадка и т. д.
Адсорбционными И. являются обычно красители
(в том числе многие pH- и о.-в.-И.). Область примене-
ния каждого И. ограничена определенными пределами
pH, а также концентрацией титруемого иона. Сущест-
вует связь между т. наз. «цветными твердофазными»
реакциями и явлениями, происходящими при титро-
вании с адсорбционными И.
Большое применение адсорбционные И. имеют в
аргентометрии и меркурометрии. Напр., при титро-
вании иодид-ионов нитратом серебра в присутствии
эозина (адсорбционный И.) в конечной точке титро-
вания находящиеся в избытке ионы серебра адсор-
бируются на поверхности галогенида серебра и взаи-
модействуют с ионами эозина, образуя окрашенный
в красный цвет эозинат серебра. Ниже приведены
области применения нек-рых адсорбционных И.:
Индикатор Определяемый ион Ион-осадитель
Дибромфлуоресцеин .... Дифенилкарбазон Флуоресцеин Эозин Вг- С1-, J-, CN- С1-, Вг-, J-.CNS- J- в присутствии ci- Ag+ Hgs2+ Ag+ Ag+
V. Хемилюминесцентные индикаторы. Группа ве-
ществ, к-рые высвечивают видимым светом в конечной
точке титрования. Их мощно применять при многих
реакциях нейтрализации и окисления-восстановле-
ния в случае титрования сильноокрашенных раство-
ров. В первом случае их действие основано на
возникновении или исчезновении окислительно-вос-
становительной реакции, сопровождаемой излуче-
нием света при достижении определенного значения
pH р-ра в процессе титрования. Так люминол
(гидразид 3-аминофтал.евой кислоты), добавленный в
титруемый раствор кислоты вместе с гемоглобином и
перекисью водорода, дает свечение при достижении
значения pH 8,0—8,5 при титровании щелочью. Л ю-
ц и г е н и н (нитрат N,N'-диметилдиакридилия) в
аммиачном р-ре в присутствии перекиси водорода
высвечивает при pH 9,0—10,0. Эти же И., а также
силоксен (SieHeO3)n можно применять при окис-
лительно-восстановительных титрованиях различных
восстановителей, напр. перекисью водорода в щелоч-
ной среде. Механизм возникновения свечения мало
изучен (см. Люминесцентный анализ).
Лит.: Кольтгоф И. М., Стенгер В. А., Объем-
ный анализ, пер. с англ., т. 1, М.—Л., 1950; Кольтгоф
И. М., Применение цветных индикаторов к нейтрализацион-
ному анализу и к колориметрическому определению кон-
центрации водородных ионов, пер. [с нем.], 3 изд., Л., 1929;
К а р я к и н Ю. В., Кислотно-основные индикаторы, М.—Л.,
1951; Кольтгоф И. М., ЛайтиненГ. А., Определе-
ние концентрации водородных ионов и электротитрование,
пер. с англ., М., 1947; TomiJek О., Chemical Indicators,
L., 1951; Пршибил Р., Комплексоны в химическом ана-
лизе, пер. с чеш., 2 изд., М., 1960; Б у с е в А. И., Пет-
ренко А. Г., Заводск. лаборатория, 1958, 24, Л, 12, 1449;
Палит Шанти Р., Дас Мехр Натх, Сомаяд-
ж ф л у Г. Р., Неводное титрование, пер. с англ., М., 1958;
Певцов Г. А., Тр. Комис, по аналит. химии. АН СССР,
1947, 1, 53—91; Певцов Г. А., Кошелева Г. Н.,
Заводск. лаборатория, 1952, 18, № 12, 1459; Певцов Г. А.,
Тр. Всес. н.-и. ин-та хим. реактивов, 1958, вып. 22, 95—103;
Кошелева Г. Н., там же, 78—94; ЯцимирскийК. Б.,
Заводск. лаборатория, 1955, 21, .V 10, 1149. Г. А. Певцов.
ИНДИКАТОРЫ УНИВЕРСАЛЬНЫЕ — смеси ин-
дикаторов, многократно меняющие свою окраску
при разных значениях pH. Применяют И. у. для при-
ближенного определения значений pH в широких
пределах. Подробнее см. Индикаторы.
ИНДОКСИЛ (3-кетоиндолин, 3-кетодигидроиндол,
3-оксииндол) C8H7ON, мол. в. 133,14 — светло-жел-
тые нестойкие кристаллы; т. пл. 85°; обладает силь-
ным фекальным запахом. И. растворяется в щелочах
с образованием солей щелочных металлов, из к-рых
действием СО2 можно получить И. При окислении
кислородом воздуха и другими окислителями образует
255
ИНДОЛ
256
индиго, при восстановлении дает индол. Существует
в двух таутомерных формах:
ствие реактивов Гриньяра на И. приводит к N-индол-
магнийгалогенидам (II). Цианэтилирование И. дает
Х-(0-цианэтил)-индол, ацетилирование уксусным анги-
дридом — 1,3-диацетилиндол (Ill). И. чрезвычайно
склонен к реакциям электрофильного замещения, при-
чем заместитель направляется в положение 3 (см. IV).
При ацилировании И. уксусным ангидридом об-
разуются О-(1) или N-(I1) моноацетильные или
О, N-диацетильные (Ill) производные. Метилирование
диметилсульфатом приводит к о-метильному произ-
водному (IV), иодистый метил образует 2,2-диметил-
индокспл:
N-COCH
ОН
N-COCHa
III
з
ОСОСНд
Действие на И. азотистой к-ты приводит к 2-изатинок-
спму. И. конденсируется с альдегидами и кетонами,
образуя индогениды (V). Примером может служить
конденсация И. с изатином, дающая индирубин
(VI) — побочный продукт при окислении И. в индиго.
Конденсация И. с формальдегидом и диалки-
ламинами по Манниху приводит к 3-диалкилами-
ноиндолам (V); формилированпе (СНС13 и щелочь
или РОС13 и метилформанилид) дает 3-индолальдегид
(VI); ацетилирование и азосочетание также дают
продукты замещения в положении 3. Оксалилхло-
рид и И. в среде эфира образуют 3-индолилглиок-
салилхлорид (VI1), действие диазоуйсусного эфира
приводит к 3-индолилуксусной к-те (VI11):
И; находится в моче человека и животных в виде ка-
лиевой соли 3-индоксилсернон к-ты (индикан мочи),
в растениях Indigofera и Isatis tinctoria в виде гли-
козида индикана, откуда он может быть выделен после
гидролиза. Синтетически И. получается из о-нитро-
фенилпропиоловой к-ты:
аС®С-СООС2Н5
no2 ^S0‘
Взаимодействие И. с соединениями, содержащими
двойную связь, активированную электроотрицатель-
ным заместителем (с нитроолефинами, алкилвинилке-
тонами, винилпиридином), приводит к соответствую-
щим 3-замещенным индолам (IX):
Фепилглицин или о-карбоксцфенилглицин при сплав-
лении со щелочью образуют И., превращающийся
затем в индиго.
Лит.: Д ж у л и е н П., Мейер Э., П р и н т и Э.,
в кн.: Гетероциклические соединения, под ред, Р. Эльдар-
фильда, [сб. ст.], пер. с англ., т. 3, М., 1954, с. 139.
М. Н. Преображенская.
ИНДОЛ (2,3-бензопиррол) C8H7N, мол. в. 117,14 —
бесцветные кристаллы; т. пл. 52—53°; т. кип. 253—
254°; теплота сгорания 1021 ккал)молъ-, растворим в
горячей воде, обычных органич. раство-
r 1 рителях и в жидком аммиаке; летуч с
’.>(“) водяным паром; обладает неприятным
запахом, усиливающимся при хранении.
По своим свойствам И. является ароматич. соеди-
нением. Свободная пара электронов у атома азота
сопряжена с л-электронами бензольного и пирроль-
ного ядер, вследствие чего И. не обладает основными
свойствами. Он проявляет слабокислые свойства:
при взаимодействии с натрием в вазелиновом масле
при 200° или с р-ром натрия в жидком аммиаке
образуется N-индолнатрий (1); N-индолкалий обра-
зуется при взаимодействии И. с КОН при 130°. Дей-
+ сн2=снх
К тому же типу реакций относится димеризация И.
(X) в присутствии сухого НС1 в инертном растворителе:
Н
Димер И. (X) в дальнейшем может образовать
тример (XI). В сильноинелой- среде И. неустойчив,
поэтому реакции электро-
фпльного замещения в этой [ |Г
среде — нитрование, суль-
фирование, галогенирование
и др., для И. часто невоз-
можны. С присоединением
протона свободная пара эле-
ктронов азота перестает участвовать в создании проч-
ной ароматической системы. II. теряет ароматич. свой- :
257
ИНДОЛ
258
ства и приближается по свойствам к фенилвиниламмо-
ншо (XII). Вследствие ацидофобности И. для введения
в его молекулу сульфо-, бром- или
GCh=ch-NH нитрогруппы приходится прибе-
3 гать к косвенным методам (сульфи-
х1| рование или галоидирование комп-
лексносвязанным S03 или Вг2).
При действии на И. пиридинсульфотриоксида об-
разуется в зависимости от условий реакции 1- или
2-индолилсульфокислота; диоксандибромид превра-
щает И. в 3-броминдол, хлористый сульфурил дает
3-хлориндол, раствор иода в иодистом калии превра-
щает И. в 3-иодиндол. При действии на И. алкилнит-
ритов или азотистой к-ты образуются производные
индолениновой структуры (XIII). Известны также
а случаи присоединения реагентов по
TN0H двойной связи пиррольного цикла
(в кислых условиях). Вступление
' электрофильных заместителей в бен-
зольное ядро индольного цикла для
самого И. не наблюдается вследствие высокой по-
движности водорода в положениях 2 и 3. Для 2,3-за-
мещепных И. известны случаи образования 5- или
6-иитро- или галогенопроизводных. 4, 5, 6 и 7-Нитро-,
амино-, галоген о- и другие производные И. можно по-
лучить лишь косвенным путем. N-Индолмагнийгало-
генид реагирует обычно с переносом реакционного
центра и образует И., замещенные в положении 3.
И. способен образовывать производные индолени-
повоп (псевдоиндольной) структуры:
RX
СН3
При электролитич. восстановлении И. или при гид-
рировании над хромитом меди или никелем Ренея при
100° и 100 ат образуются индолины. Гидрирование
над никелем Ренея в более жестких условиях приво-
дит к 2-этилциклогексиламину. Гидрирование над
Pt-катализатором дает октагидроиндол. Окисление
И., стабилизованных заместителями в пиррольном
или бензольном ядре, приводит к производным
о-аминофепилалкилкетонов (XIV) или производным
антраниловой к-ты (XV), в нек-рых случаях обра-
зуются замещенные изатины:
COR
NHCOR’
РиК’зйкнл
X
СООН
xv NHCOR"
При окислении самого И. образуется индоксил, но
удается выделить лишь продукты его дальнейших
превращений.
И. содержится в кам.-уг. смоле, откуда его вы-
деляют через индолнатрий или индолкалий, а также
в некоторых эфирных маслах, напр. в масле цве-
тов жасмина (Jasminum grandiflorum). Вместе с
3-метилиндолом — скатолом — он образуется в ки-
шечнике человека и млекопитающих; И. и ска-
тол возникают там в результате расщепления
триптофана гнилостными бактериями. Escherichia
coli и нек-рые другие бактерии используют И. и серин
в синтезе триптофана. Имеются данные о том, что
длительное введение И. в организм вызывает миэло-
идную лейкемию (у мышей).
Разработано большое число методов синтеза И.
и его производных. Наиболее широко применяется
реакция Фишера (циклизация фенилгидразонов, аль-
дегидов или кетонов):
5 К. X. Э. т. 2
Для получения исходных фенилгидразонов часто
используется Джэппа — Клингемана реакция. Для по-
лучения самого И. используют фенилгидразон пиро-
виноградной к-ты:
Другим общим методом синтеза индольных производ-
ных является Вишлера реакция (из а-галоген- или
а-оксикетонов и анилина):
CHXR"
Сам И. этим методом получить нельзя. Широко при-
меняется для синтеза индолов Маделунга реакция'.
СН3
NHCHO
Очень часто пользуются также реакцией Рейссерта —
получение индолов из о-нитрофенилпировиноградной
к-ты через 2-индолилкарбоновые к-ты:
И. и его производные можно получить также вос-
становлением 2, со-динитростиролов:
Разработано много других, часто специфич. методов
синтеза различных производных И. Получают И.
также восстановлением индоксила, а также других
окисленных производных И.
Наиболее характерной качественной реакцией на
И. и многие его производные является красное окра-
шивание с реактивом Эрлиха (спиртово-солянокислый
р-р n-диметиламинобензальдегида), а также реакция
на сосновую лучинку (сосновая лучинка, смоченная
соляной к-той, а потом спиртовым раствором И. окра-
шивается в красный цвет). При хроматографии на
бумаге производных индола используется также их
флуоресценция в УФ-свете. Для количественного
определения можно использовать выделение красного
пикрата И. (в абсолютном эфире). Для нек-рых про-
изводных И. разработаны флуоресцентные методы
количественного определения.
И. служит исходным сырьем в промышленном син-
тезе гетероауксина (3-индолилуксусной к-ты), трипто-
фана, используется в парфюмерной пром-сти для
создания различных композиций. Выяснение огром-
ной биохимич. роли И. и его производных, создание
ценных препаратов, таких, как серотонин, резерпип,
5-окситриптофан и др., привело к еще большему уве-
личению произ-ва И.
Лит.: Джу л иен П., Мейер Э., Пр инти Э.,
в кн.: Гетероциклические соединения, под ред. Ф. Эльдер-
филда, пер. с англ., т. 3, М., 1954, с. 5; Sumpter W.,
Miller F., Heterocyclic compounds with indole and car-
bazole systems, N. Y.— L., 1954; Morton A., The che-
mistry ol heterocyclic compounds, N. Y. — L., 1946.
M. H. Преображенская.
259
ИНДОЛЕНИНОВЫЕ КРАСИТЕЛИ — ИНД'ОФ'ЕНОЛЫ
260
ИНДОЛЕНИНОВЫЕ КРАСИТЕЛИ — группа про-
изводных индоленина, являющегося таутомерной
формой индола. Иногда эти красители наз. индо-
карбоцианинами. И. к., в отличие от других цианино-
вых красителей, являющихся сенсибилизаторами,
применяются для крашения текстильных материалов,
в частности ацетатного шелка и полиакрилнитрило-
вых волокон. Первым технически ценным И. к. был
«астрафлоксин ФФ»:
И. к. являются основными красителями; они окра-
шивают шелк и хлопок (по танниновой протраве)
в красивые чистые тона, более яркие, чем родамины.
Однако светопрочность окрасок на шелке и хлопке
невысокая, так же, как и у родаминов. Колористич.
показатели окрасок на ацетатном шелке и нитроне
значительно выше, чем на «природных» волокнах.
И. к. входят в ассортимент основных красителей для
искусственных и синтетич. волокон, выпускаются под
различными фирменными названиями: астразоны
(ФРГ), севроны (США) И др. В- А- Пучков.
ИНДОЛЕНИНЫ (псевдоиндолы) — производные
таутомерной формы индола. Простейший, не содержа-
„ щий заместителей И. неизвестен. В от-
________? > личие от индолов, И. обладают основ-
TR ными свойствами (образуют соли с кис-
лотами, соли четвертичных оснований
ит. п.). Под действием катализаторов
полимеризуются; восстановление И. приводит к индо-
линам. Алкильные или арильные заместители в пир-
ролениновом ядре И. способны в определенных усло-
виях к миграции из положения 2 в положение 3 и
обратно (перегруппировка Планше). Двойная связь
C=N активна и по этой связи могут присоединяться
амины, уксусный ангидрид, фенилгидразин и др. реа-
генты. Атомы водорода в 2-алкилиндоленинах весьма
подвижны (конденсация с бензальдегидом и т. д.).
И. получают в ряде случаев из производных ин-
дола (реакции с переносом реакционного центра), а
также из фенилгидразонов а, а-дизамещенных альде-
гидов или кетонов:
chr'r
NH-n^C-R
Нек-рые производные индола, напр. (J-индолальде-
гид, существуют в таутомерной индолениновой форме:
Многие из цианиновых красителей являются произ-
водными И.
Лит. см. при ст. Индоксил. М. Н. Преображенская.
ИНДОЛИЛМАСЛЯНАЯ КИСЛОТА — см. Гетеро-
ауксин.
ИНДОЛИН (2,3-дигидроиндол) CSH9N, мол. вес
119,16 — бесцветная, темнеющая при хранении жид-
кость с запахом анилина; т. кип. 228—
1-------з]снг 230°, d420 1,069; 1,5923; слабо рас-
творим в воде, хорошо — в обычных ор-
п ганич. растворителях. Являясь вторич-
ным ароматич. амином, И. образует соли с кислотами,
соли четвертичных аммониевых оснований, N-алкиль-
ные и N-ацильные производные. И. и его производ-
ные легко вступают в реакции электрофильного за-
мещения (галогенирование, нитрование, ацилирова-
ние по Фриделю — Крафтсу, сульфирование и др ),
причем в случае самого И. и его ацильных или алкиль-
ных производных заместители направляются в по-
ложение 5, а в случае солей И. — в положение 6.
Т. обр., ориентация соединений ряда И. в реакциях
электрофильного замещения отличается от ориента-
ции их ближайших аналогов — производных о-толуи-
дина, где основное направляющее влияние оказывает
алкильная группа. И. и его алкильные производные
легко дегидрируются с образованием соответствую-
щих индолов при действии хлоранила, палладиевой
черни, хромита меди и т. д. Получают И. восстановле-
нием индола электролитически или каталитич. гид-
рированием. Индолиновая структура лежит в основе
ряда алкалоидов, напр. физостигмина.
Лит..- Джу л иен П., Мейер Э., Пр инти Э.,
в кн.: Гетероциклические соединения, под ред. Р. Эльдер-
филда, пер. с англ., т. 3, М., 1954, с. 85; К 1 n g F. Е., В а г 1-
trop J. A., Walley R. J., J. Chem. Soc., 1945, 277;
Терентьев А. П., Преображенская М. Н.,
Ж. общ. химии, 1960, 30, вып. 4, 1218.
М. Н. Преображенская.
ИНДОФЕНИНОВАЯ РЕАКЦИЯ — появление ин-
тенсивного синего окрашивания при взаимодействии
изатина (I) с тиофеном (II) в присутствии конц. сер-
ной к-ты, связанное с образованием красителя —
индофенина. Наиболее вероятное строение соответ-
ствует III.
H2SO4
Индофенин C24H14O2N2S2 — синий порошок;
при нагревании не плавится и не возгоняется, а обуг-
ливается; не растворяется в воде и углеводородах,
очень трудно растворяется в спирте, эфире и хлоро-
форме с синей окраской. Индофенин во многих отно-
шениях сходен с индиго; его раствор в серной к-те
синего цвета, при нагревании раствора образуется
дисульфокислота. При действии на иидофенин цинко-
вой пыли в уксусной к-те или в щелочи получается
бесцветный р-р, из к-рого при стоянии на воздухе
снова выделяется в неизмененном виде индофенин.
В. Мейер, пользуясь И. р., открыл в 1882 присут-
ствие тиофена в бензоле, выделенном из кам.-уг.
смолы (в сыром бензоле содержится до 0,05% тио-
фена). Бензол, полученный из бензойной к-ты, не
дает И. р.
Лит.: Габель Ю. О., Гетероциклические соединения,
Л.—М., 1941; Каррер П., Курс органической химии,
пер. с нем., Л., 1960. В. А. Пучков.
ИНДОФЕНОЛЫ — окси- и аминопроизводные N-фе-
нилхинонмоноимина; оксипроизводные (I) иногда на-
зывают «истинными» И., а аминопроизводные (II) —
индоанилинами. Известны также И. производные
1,4-нафтохинонимина. И. — окрашенные кристаллы
(часто с металлич. блеском); растворимы в органич.
растворителях и минеральных к-тах; оксипроиз-
водные растворимы в едких щелочах и соде. Под
воздействием разб. к-т И. расщепляются на хинон и
ароматич. диамин или аминофенол. Фенолиндофенол
(I) в разб. НС1 дает гидрохинон и немного п-аминофе-
нола. И., аналогично бензохинону, присоединяют
261
ИНДУКТОМЕРНЫЙ ЭФФЕКТ-ИНДУКЦИОННЫЙ ЭФФЕКТ
262
бисульфит, тиосульфат, ароматич. амины и др. со-
единения с образованием продуктов замещения в ядре.
При восстановлении, напр. Fe + кислота, Na2S,
Na2S2O4, И. присоединяют 2 атома водорода; при
этом из I образуются п,п'-диоксидифеииламины, а из
II — п,п'-аминооксидифениламины, к-рые при дей-
ствии окислителей (HgO, Ag3O, О2) обратно переходят
в I и II. Обратимый переход окисленной и восстано-
вленной форм И. аналогичен известному для кубовых
красителей переходу:
' + [Н]
краситель ,_РЛ. лейкосоединение
+ [О]
На этом свойстве И. основано их применение в ка-
честве окислительно-восстановительных индикаторов.
Нормальные потенциалы нек-рых И. приведены ниже.
Индофенол
Нормальный
потенциал
(тг0) при 30°, в
Фенолиндофенол .............
о-Бромфенолиндофенол........
о-Хлорфенолиндофеиол . • . . .
2,6-Дибромфенолиндофенол . .
о-Йрезолиндофенол...........
+ 0,649
+ 0,659
+ 0,663
+ 0,668
+ 0,616
Значение окислительно-восстановительного потен-
циала И. в сильной степени зависит от pH среды и
для указанных выше И. может быть определено по
формуле:
IlndowwpjT 1
= Jto + 0,03 1g — цнии2Е, - О.ОбрН, если pH < 8
[1вавосст.1
Некоторые оксипроизводные обратимо изменяют
окраску при переходе из кислой в щелочную среду, что
связано с образованием полярных (анионоидных и
катионоидных) структур типа III, IV и V:
V. сильно-мслая среи
И. в пром-сти получают: 1) взаимодействием п-ни-
трозофенола (соответственно п-нитрозодиалкилани-
лина) с фенолом или окислением (К2Сг2О7, NaOCl)
смеси n-аминофенола (соответственно п-аминодиалкил-
анилина) с фенолом.
Практич. применение в качестве красителя нашел
лишь т. наз. а-нафтоловый синий. И. широко при-
меняют в синтезе оксазиновых: красителей, тиазиновых
красителей и сернистых красителей, в аналитич. хи-
мии как индикаторы и в цветной (трехцветной) фото-
графии.
Лит.: Коган И. М., Химия красителей, 3 изд., М.,
1956; Венкатараман К., Химия синтетических кра-
сителей, пер. с англ., т. 2, Л., 1957; Кольтгоф И. М.,
С т е н г е р В. А., Объемный анализ, пер. с англ., т. 1, М.—Л.,
1950. В. А. Пучков.
ИНДУКТОМЕРНЫЙ ЭФФЕКТ (динамический ин-
дукционный эффект) — поляризация о-связей реа-
гирующих молекул в результате их взаимодействия
во время элементарного акта реакции. В результате
б*
этого взаимодействия изменяются полярности <т-свя-
зей по сравнению с тем, что имеет место при отсутст-
вии взаимодействия. Нет необходимости разграничи-
вать И. э. от индукционного эффекта; это вытекает из
сущности теории переходного состояния, согласно
к-рой активированный комплекс рассматривается по
аналогии с обычными молекулами (см. Переходное
состояние). Скорость реакции определяется разностью
между свободными энергиями активированного ком-
плекса и исходных молекул, к-рая зависит в том числе
и от соответствующей разности взаимодействий, обу-
словленных индукционным эффектом. Под И. э. фак-
тически понимают индукционное влияние в активи-
рованном комплексе; экспериментальному определе-
нию поддается только разность между влияниями в ак-
тивированном и исходном состояниях. Поэтому И. э.
на практике сливается с индукционным аффектом и
вместе с последним учитывается в величинах р*о*,
р *О° ИЛИ р jOj.
Лит.: ИнгольдК. К., Механизм реакций и строение
органических соединений, пер. с англ., М., 1959; Тафт Р. У.,
в кн.: Пространственные эффекты в органической химии, пер.
с англ., М., 1960, гл. 13. В. А. Пальм.
ИНДУКЦИИ ПЕРИОД — интервал времени между
моментом создания условий для реакции и моментом,
когда реакция становится заметной. И. п. наблюдается
в цепных и аутокаталитич. реакциях, а также в про-
цессах, связанных с образованием новой фазы, и мо-
жет составлять от долей секунды (дли реакции
2Н2 + О2 = 2Н2О при 485° и давлении 6—8 мм рт.
ст.) до нескольких месяцев (для распада озонида
калии: 2КО3=2КО3 + О2 при —9° И. п. равен
54 суткам,при —18° равен 205 суткам). Продолжитель-
ность И. п. зависит от темп-ры, концентрации реаги-
рующих веществ, наличия в реакционной смеси по-
сторонних примесей, интенсивности и длительности
освещения (в случае фотохимия, реакций) и др. Тща-
тельно очищенные водород и хлор реагируют без
И. п., наличие в реакционной смеси сотых долей про-
цента О2 или тысячных долей процента NC13 приводит
к появлению И. п. Нек-рые добавки, в зависимости
от темп-ры, могут увеличивать или уменьшать И. п.
(если окислять метан при 340°, добавление к реак-
ционной смеси 0,3% иода увеличивает И. п. с 14 до
135 мин, при повышеиип же темп-ры до 447° добавле-
ние 2% иода сокращает И. п.).
Существенно влияет на наблюдаемый И. п. метод
измерения скорости реакции, напр. увеличение чув-
ствительности метода измерения скорости реакции
2Н2 + О2=2Н2О в 100 раз уменьшает наблюдаемый
И. п. в 5 раз, а увеличение чувствительности в
10 000 раз уменьшает И. п. в 10 раз. Согласно предста-
влениям цепной теории, в течение всего И. п. разви-
вается самоускоряющаяся разветвленная цепная реак-
ция, скорость к-рой растет пропорционально е^
(t — время, <р — постоянная для данной реакции ве-
личина) и может быть отмечена данным прибором лишь
по истечении И. п. Для реакций, скорость к-рых из-
меряется по изменению давления Др реагирующей
смеси, в течение И. п. Др = Aevt, где А — постоян-
. др J
иая величина. Отсюда следует, что т = i In —>
где т — период индукции, Д^т;п — чувствительность
измерительного прибора. И. п. в фотографии см. Про-
явление фотографическое.
Лит.: Семенов Н. Н., О некоторых проблемах хими-
ческой кинетики и реакционной способности, 2 изд., М.,
1,958; Кондратьев В. Н., Кинетика химических газовых
реакций, М., 1958. В. Е. Островский.
ИНДУКЦИОННЫЙ ЭФФЕКТ — взаимное влия-
ние между какими-либо двумя частями моле-
кулы, обусловленное их эффективными электро-
отрицательностями. Величина последних характери-
зует способность данного заместителя X (атом или
263
ИНДУКЦИОННЫЙ ЭФФЕКТ,TIНО
264
группа атомов) поляризовать a-связь, соединяющую
ее с остальной частью молекулы, путем «оттягивания»
или «отталкивания» пары валентных электронов, осу-
ществляющих эту связь. Этим вызываются «смеще-
ние» а-электронной пары в сторону более электроот-
рицательного заместителя, появление эффективных
электрич. зарядов на атомах и дипольные моменты
а-связей.
В структурных формулах направление смещения
а-электронов обозначается стрелкой на соответствую-
щей валентной черте, напр.: 6^Н—-С!6 - Указанная
формула показывает, что атом С1 более электроотри-
цателен, чем атом Н, вследствие чего а-электронная
пара смещена в сторону хлора. Через 6+ и 6_ обозна-
чены соответствующие эффективные заряды на ато-
irfax, меньшие, чем величина элементарного заряда
(электрона или протона).
Влияние эффективных электроотрицательностей мо-
жет проявляться непосредственно, если соответствую-
щие заместители связаны между собой a-связью, либо
через участок молекулы, к-рый находится между ними.
Ниже изображено такое опосредствованное влияние
атома хлора на остальные атомы в молекуле хлор-
уксусной к-ты:
н
I
С1*-С*-СО<— о*-н
Благодаря И. э. атом хлора, непосредственно не
связанный с водородом карбоксильной группы, все же
способствует оттягиванию электронов от атома Н.
Поэтому указанный атом водорода наделен в хлорук-
сусной к-те большим положительным эффективным
зарядом, чем в уксусной к-те. Влияние И. э. быстро
уменьшается с увеличением длины цепи атомов, свя-
зывающей два взаимодействующих заместителя.
Количественная мера эффективных электроотрица-
тельностей различных заместителей может быть уста-
новлена при помощи принципа линейности свободных
энергий (см. также Гаммета уравнение)- Наибольшее
практич. значение для этой цели имеют полярные
константы а* Тафта для алифатич. заместителей и
постоянные а0 для замещенных фенилов. В случае
алифатических заместителей величина а* для группы
СН3 приравнена нулю. Заместители, более электро-
отрицательные, чем СН3, характеризуются положи-
тельными значениями а*, менее электроотрицатель-
ные— отрицательными о*. Следовательно, наиболее
электроотрицательные заместители характеризуются
наибольшими значениями а* и наоборот.
В случае замещенных фенилов а0 приравнена
к нулю для СвН5. В остальном шкала величин а0
должна иметь тот же масштаб, что и а*. В связи с этим
величины а* для замещенных фенилов могут быть
получены из соотношения: а* = а0 + 0,600, где
0,600 — значение а* для С6Н5. Для метазамещенных
фенилов величины о0 практически равны значению
о-мета Гаммета. То же самое относится к пара-заме-
щенным фенилам в случае заместителей с отрицатель-
ном (акцепторным) эффектом полярного сопряжения
(NO2, СООН, SO3H и т. д.). В случае пара-заместите-
лей с положительным (донорным) эффектом поляр-
ного сопряжения (NH2, галогены, ОН и т. д.) а0 > а.
Существует также «ароматическая» шкала индукцион-
ных постоянных Цр к-рые считаются приближенно
равными индукционным составляющим в величинах а
Гаммета и а0 для заместителей X в X—С6Н4. Между
Oj для заместителя X и а* для заместителя X — СН2
существует соотношение: <Jj = 0,450 а*.
Величины а* заместителей, для к-рых эксперимен-
тальные данные отсутствуют, могут быть вычислены,
пользуясь правилами аддитивности и затухания И. э.
Правило аддитивности записывается следующим об-
разом:
°(Х1Х,Х3СНГ) — ’(XtfHj-) + с(Х2СН—) + 3(Х8СНГ)
Правило затухания для всех заместителей с а* > о
выражается формулой:
а(х—)— 2>8°*Х1Сн-)
Для водорода и алифатич. углеводородных радика-
лов:
а*х— 1) — 4>9 (°*х 1СНГ) 0,100)
Приведенные выше выражения эффективных элект-
роотрицательностей содержат, кроме индукционной
составляющей, также и составляющую, обусловлен-
ную эффектом поля (способность к электростатич.
взаимодействию через пространство). Возможно, что
исключение составляют лишь величины а* для али-
фатич. углеводородных радикалов и водорода. '
Величины а*, а° и могут быть использованы для
вычисления составляющих, обусловленных И. э.
в величинах свободных энергий реакции или актива-
ции, теплотах реакций, смещениях спектральных ча-
стот, изменениях потенциала полуволны полярогра-,
фич. восстановления, дипольных моментах и т. д.
В случае соединений типа Х;—Y, где Х; — пере-
менный заместитель, Y — постоянный «реакционный
центр», характеристика к-рого рассматривается; соот-
ветствующие формулы Тафта имеют вид:
APi = p*a?, A/,i = pa° или AZ’i=p1aI
где =Pi — Ра, Р}ПРа — составляющие, обуслов-
ленные И. э. заместителей Х; и Хо соответственно;
Хо — стандартный заместитель [aj(alo, cr{J) = 0].
Применимость этих уравнений не лимитирована огра-
ничениями относительно строения заместителя Х4.
Если интересующая величина; напр. свободная
энергия реакции или активации (выраженная через
логарифмы констант скорости или равновесия), не за-
висит от других факторов, связанных с Х4, кроме
И.э.,то соблюдается уравнение типа Гаммета— Тафта:
lg Мо = ДЛ = Р’Ч* (P’i или Pi’ii)
Величины р*, р и pj характеризуют чувствитель-
ность интересующей нас величины к влиянию И. э.
заместителя Хг‘Положительные значения р *, р и pj
означают, что более электроотрицательные замести-
тели способствуют увеличению величины ДРр и на-
оборот.
Если р*, р или pj известны, то влияние остальных
факторов строения заместителей Х; может быть пай-
fti
дено из разности типа 1g -----р*а^. Таким обра-
зом определяют величины составляющих, обусловлен-
ных неполярным и полярным сопряжениями, гипер-
конъюгациеи и стерич. эффектом. В связи с этим коли-
чественный учет влияния И. э. является предпосылкой
создания более общей количественной теории зависи-
мости перечисленных выше характеристик соедине-
ний типа Х;— Y от строения заместителя Хр
Постоянные а*, а0 и наиболее характерных за-
местителей приведены в таблице. В первой графе
таблицы приведены заместители, во второй — значе-
ния а*, в третьей и четвертой графах — значения
Oj и а0 для ароматич. соединений, замещенных в пара-
положении.
265
ИНДУКЦИЯ ХИМИЧЕСКАЯ —ИНЕРТНЫЕ ГАЗЫ
266
X 3* °I ?o
N(CH3)3+ NO3 CN F Cl Br J CF3 OH OCH3 ... • CH2C1 C+C-R CGH5 H CH=CH-CH-} сыгсвн5. . : CH3 C2H3 U30-C3H7 mpein-Cillg 4* 5,3 4- 3,9 4- 3,6 4- 3,1 4- 2,9 4- 2,8 4-2,36 4-2,6 4- 1,55 4- 1.45 4- 1,05 4- 1.30 4- 0,600 -L- 0,490 4- 0.360 -r 0,215 0,000 - 0,100 — 0,190 — 0,300 4-0.86 4-0,63 --0,58 4-0,52 4-0,47 4- 0,45 4-0,39 --0,41 -- 0,25 -- 0,25 4’0,17 4-0,10 0,00 4- 0,04 - 0.05 - 0,05 — 0,07 + 0,82 + 0,69 + 0,17 4- 0,27 + 0,26 + 0,27 -0,13 - 0,16 0,00 0,00 — 0,15
Лит.: Тафт Р. У. мл., в кн.: Пространственные эффекты
в органической химии, пер. с англ., М., 1960, гл. 13; П а л ь м
В. А., Усп. химии, 1961, 30, вып. 9, 1069; Hine J. S.,
Physical organic chemistry, L. —Toronto, 1956. В. А. Пальм.
ИНДУКЦИЯ ХИМИЧЕСКАЯ — совместное про-
текание двух химич. реакций, из к-рых одна (А + В)
обусловливает или ускоряет непроизвольный вторич-
ный процесс (реакцию А + С). Такие две реакции
являются сопряженными, вещество А наз. акто-
ром, В — индуктором, С — акцепте-
р о м. И. х. характеризуется фактором индукции I,
к-рый указывает, сколько моле-
кул акцептора вступило в реак-
цию в расчете на одну прореагиро-
вавшую молекулу индуктора: I =
= А [С]/Д [В]. Первые подробные
исследования И. х. были проведе-
ны Н. А. Шиловым и Йориссеном
в конце 19 — начале 20 вв. И. х.
наблюдается в тех случаях когда
в обеих реакциях (А + В и А + С)
образуются активные промежуточ-
ные продукты, в качестве к-рых . При 26 amjlt
выступают промежуточные актив-
ные окислительно-восстановительные формы, ком-
плексы, образованные молекулами индуктора и актора
или индуктора и акцептора, свободные радикалы.
Примеры И. х. Соли мышьяковистой к-ты
в водном р-ре практически не окисляются кислородом
воздуха. Напротив, соли сернистой к-ты довольно
быстро окисляются на воздухе. При совместном при-
сутствии окисляются как сульфит-ионы, так и арсенит-
попы:
2SO% + О2 = 2SO% (1)
2AsO% + О2 = 2AsO% (2)
т. е. реакция 1 ипдуцирует реакцию 2. В этом примере
О2 — актор, SO% — индуктор, AsO% —акцептор. Ин-
дукция обусловлена тем, что при окислении сульфита
образуется ион SO%, к-рый окисляет как SO%, так
и AsO% ионы:
SO% + SO% = 2SO%
SO% + AsO% = SO% + A«O%
Мышьяковистая к-та окисляется хромовой, но не
окисляется бромноватой к-той. При совместном про-
текании реакция Н2СгО4 + H3AsO3 индуцирует реак-
цию НВгО3 + H3AsO3. В этом примере H3AsO3 —
актор, Н2СгО4 — индуктор, НВгО3 — акцептор. Реак-
ция 2СО + О2 = 2СО2 практически не идет при темпе-
ратурах и давлениях, при к-рых реакция 2Н2 + О2 =
= 2Н2О протекает быстро. В присутствии водорода
СО окисляется. Здесь О2 — актор, Н2 — индуктор,
СО — акцептор. Индукция обусловлена свободными
радикалами и атомами, образующимися из водорода
и кислорода (Н, ОН, НО2).
Лит..- Шилов Н. А., О сопряженных реакциях окисле-
ния, М., 1905; Jorissen W. Р., Induced oxidation,
Amst. — Га. о.], 1959; Кондратьев В. Н., Кинетика
химических газовых реакций, М., 1958. Е. Т. Денисов.
ИНЕРТНЫЕ ГАЗЫ (благородные газы, редкие
газы) — элементы нулевой группы периодич. системы
Менделеева: гелий, неон, аргон, криптон, ксенон и
радон. Для атомов И. г. характерно наличие устойчи-
вых внешних электронных слоев (у Не — 2 электрона,
у остальных И. г. во внешнем слое — 8), что и обуслов-
ливает их химич. инертность.
И. г. открыты позже большинства прочих элементов.
Гелий был обнаружен в 1868 в солнечном спектре, а позднее
найден в минерале клевеите и в воздухе. В 1894 из воздуха
был выделен Аг, а в 1898 —Ne, Кг и Хе. В 1908 в спектре
газов, находившихся в ампуле с радием, были обнаружены
линии Rn. Позже было показано, что Rn не имеет стабильных
изотопов. В 1903 Д. И. Менделеев включил И. г. в периодич.
систему элементов в виде новой самостоятельной группы.
В природе И. г. образуются в результате различных
ядерных процессов, поэтому их изотопный состав
зависит от происхождения образца. В таблице, при-
ложенной к статье Изотопы, указан изотопный состав
элементов в атмосфере. И. г. присутствует в воздухе
в следующих количествах (объемные проценты):
0,9325% Аг; 0,00161% Ne; 0,00046% Не; 0,000108%
Кг; 0,000008% Хе. Ниже даны первые потенциалы
ионизации и атомные радиусы (по таблице Г. Б. Бо-
кия) И. г. в порядке возрастания атомного веса и их
важнейшие физич. свойства. Другие физич. свойства
см. в статьях об отдельных элементах.
Элемент Ат. вес Ат. радиус, A Потенци* ал иони- зации, в Плотн., г/л(0°С, 760 мм рт.ст,) При 760 леи рт. ст. Критич. состояние
Т. кип., °C т. пл., °C темп-ра, °C давл., кг/см* плотн., К0/Л
He 4,0026 1.22 24,58 0,17846 —268,93 —272,6* -267,90 2,335 0,06930
Ne 20,183 1.60 21,56 0,90035 —245,9 -248,6 —228,6 27,75 0,4835
Ar 39,948 1.92 15,76 1.7839 —185,9 —189,3 -122,43 49,59 0,5308
Kr 83,80 1.9S 14,00 3,745 —153,2 -157,1 - 63,8 55,6 0,909
Xe 131,30 2,18 12.13 5,851 -108,1 -111,8 + 16,59 60,16 1,155
Rn 222 — 10,75 9,96 ок.—63 ок.—71 +104 64,5 — '
Для элементов нулевой группы периодич. системы
характерна исключительная химич. пассивность. Со-
единений с ионной и ковалентной связями. И. г. не
дают. Различными методами получены молекуляр-
ные соединения И. г. Так, Р. Вийяр использовал сжа-
тие Аг до 150 атм над переохлажденной водой. Кри-
сталлизация льда сопровождалась образованием кри-
сталлогидрата Аг • 6Н2О. Аналогичным способом были
получены Хе - 6Н2О и Кг- 6Н2О. Б. А. Никитин
разработал метод изоморфного соосаждения, при
к-ром не требуется создавать большие давления газов,
т. к. истинно изоморфные вещества способны образо-
вывать смешанные кристаллы при любой концентра-
ции компонентов. Аналогами И. г. в кристаллохимич.
отношении являются СН4, H2S, НВг, СО2, SO2 (бли-
зость ван-дер-ваальсовых сил, сходство молекул пр
размерам и форме). Изоморфным соосаждением с
кристаллогидратами H2S, HCl, SO2, СО2 получен
Rn-6Н2О, соосаждением с SO2 выделены Аг- 6Н2О
и Ne- 6Н2О. Никитин с сотрудниками провели также
изоморфное соосаждение соединений R • 2CeHsOH и
R- 2CeHsCH3, где R— SO2, HCl, НВг, H2S, с соответ-
ствующими молекулярными соединениями Rn и Хе.
Выделены также Аг- 3CeH5OH, Rn • ЗС1СвН4ОН,
Хе- ЗСвН5ОН, а также соединения Аг, Кг и Хе с хи-
нолином. Основываясь на различной способности И. г.
образовывать молекулярные соединения, Никитин
разработал метод их разделения. Так, Ne, в отличие
от Аг, не дает кристаллогидрата при соосаждении
с НС1. Перекристаллизацией гидрата SO2 в атмо-
267
ИНЕРТНЫЕ ГАЗЫ — ИНЖЕКЦИЙ
268
сфере И, г. можно количественно отделить Rn от Ne и
Не, Аг от Ne и Не, Аг от Rn, Аг от Не, а также выде-
лить и разделить тяжелые И. г. (Хе, Кг и Rn). По
представлениям Ф. Крамера, образование описанных
соединений происходит следующим образом. В круп-
ноячеистой сетчатой структуре органич. вещества
существуют пустоты, к-рые могут включать при кри-
сталлизации под давлением молекулы И. г. Так, охла-
ждением гидрохинона в атмосфере И. г. при давле-
нии 4—40 атм получены соединения включения гид-
рохинона с Аг, Кг и Хе. Кристаллогидраты И. г.
также рассматриваются как соединения включения.
В идеальной элементарной ячейке гидратов И. г.
содержатся 46 молекул Н2О и 8 молекул газа. Первые
находятся в вершинах пентагондодекаэдров, моле-
кулы газа располагаются внутри. Следовательно,
ф-ла должна быть представлена в виде R 5,75 Н2О.
Для Кг такая ф-ла действительно найдена экспери-
ментально. В структуре других гидратов имеются
незаполненные места, что и приводит к значению
R. 6Н2О. Кроме упомянутых соединений И. г., име-
ются указания на возможность образования следую-
щих: Аг • nBF3 (n = 1, 2, 3, 6, 8, 16), Аг • Hg, Кг • Hg,
а также соединений Не с Р, J, S.
Для точных количественных определений И. г.
необходима высококачественная вакуумная аппара-
тура. Первоначально отделяют химически активные
газы: О2, Н2, СН4, С2Н4, H2S, N2 и др. Отделение Не
и Ne от прочих И. г. достигается адсорбционным мето-
дом. При малых давлениях Не и Ne практически не
адсорбируются активным углем, охлажденным до
темп-ры жидкого воздуха (20,4° К). Прочие И. г.
связываются активным углем и могут быть десорбиро-
ваны нагреванием последнего до 300° С. Для разде-
ления Не и Ne используется либо многократная ад-
сорбция и десорбция на активном угле, либо конден-
сационный метод (при темп-ре жидкого водорода Ne,
в отличие от Не, затвердевает). В нек-рых случаях
разделение Не и Ne не производят, а находят их соот-
ношение по теплопроводности смеси или же с помощью
газовых микровесов. Тяжелую фракцию И. г. разде-
ляют на компоненты многократной адсорбцией и де-
сорбцией при соответствующей темп-ре. Для анализа
И. г. пользуются также масс-спектрометрич. методом,
причем масс-спектрометр служит либо как газоана-
лизатор, либо с его помощью проводится определение
И. г. методом изотопного разбавления. Спектраль-
ный метод может предоставить лишь качественные
данные: интенсивность линий зависит, помимо кон-
центрации, от силы разрядного тока, давления, кон-
центрации компонентов смеси и т. д. При определе-
ниях И, г. используются также соответствующие ра-
диоактивные индикаторы.
Основной источник промышленного получения Не —
подземные гелионосные газы. Применяется метод глу-
бокого охлаждения с конденсацией всех газов, кроме
Не. Дополнительная очистка может проводиться ад-
сорбционным методом. Часть Не добывается из воз-
духа, также конденсационным методом. Прочие И. г.
получаются как побочный продукт на заводах по
произ-ву жидкого кислорода и азота (см. Воздуха
разделение).
И. г. благодаря их специфич. свойствам широко
применяются в технике. В электротехнике И. г. ис-
пользуются для заполнения различных осветительных
устройств: экономичных ламп накаливания, газо-
светных и сигнальных ламп. И. г. применяются также
в различных электронных приборах, в аппаратуре
для регистрации ядерных излучений, в вакуумной
технике, при проведении технология, процессов, тре-
бующих инертной среды, и т. д.
Лит.: Фастовский В. Г., Редкие газы, М.— Л.,
1S40; его же. Криптон и ксенон, М.—Л., 1941; его же,
Разделение газовых смесей, М.—Л., 1947; Кееаом В.,
Гелий, пер. с англ., М., 1949; Крамер Ф., Соединения
включения, пер. с нем., М., 1958; Никитин Б. А., Избран-
ные труды, М.—Л., 1956; U 1 1 m а п п, 3 Aufl., Bd 6, Mun-
chen— В., 1955; G m е 1 i n, 8 Aufl., Syst.-Num. 1, Edelga-
se, Lpz. — B., 1926; Mellor, v. 7, L. — N. Y. — Toronto,
[1952]; P a s с a 1, v. 1, P., 1956; U 1 1 m a n n, 3 Aufl., Bd 6,
MUnch. — B., 1955; К i r k, v. 7, N. Y., 1951; С о о k G. A.,
Argon, helium and the rare gases, v. 1—2, L., 1961.
Э. IC. Герлинг.
ИНЖЕКЦИЯ — процесс непрерывного смешения
двух потоков разных давлений с образованием сме-
шанного потока промежуточного давления. Смешивае-
мые потоки могут быть равнофазными (газы, пары,
капельные жидкости) и разнофазными (газ — жид-
кость или пар — жидкость).
Принцип И. показан на схеме (рис. 1). В сечении
1 — 1 поток газа или жидкости имеет нек-рое давле-
ние Pj и среднюю линейную скорость 7/4. В суженном
сечении 2 скорость пото-
ка, естественно, возра-
стает, вызывая тем са-
мым падение давления
(br2>6r1, Р^Рг), т. е.
рост кинетич. энергии
потока сопровождается
уменьшением его потен-
циальной энергии. Вслед-
ствие падения давления в узком сечении 2 по трубке 3
будет всасываться жидкость, газ или пар из среды с
давлением Р«. При дальнейшем движении смеси (газо-
вой, жидкой или газо-жидкостной) по трубе с расши-
ряющимся живым сечением падение скорости до 7/3
(кинетич. энергии) будет сопровождаться нараста-
нием давления (потенциальной энергии) до величины
Р3, причем Р'<Р3<Р1. Таким образом, в резуль-
тате И. давление всасываемой жидкости или газа
возрастает от Р'., до Р3 за счет падения давления ра-
бочего потока газа или жидкости от Рг до Р3. Поток
всасываемого газа или жидкости наз. инжектируе-
мым, а поток рабочего газа или жидкости — инжекти-
рующим.
И. применяется на практике как для отсасывания
газов, паров и жидкостей из закрытых объектов с це-
лью их эвакуации, так и для сжатия газов и паров
или сообщения избыточного давления жидкости с це-
лью ее нагнетания в различные аппараты и резер-
вуары. В первом случае процесс часто наз. э ж е к-
ц и е й, во втором случае — инжекцией. Применяе-
мые для осуществления этих процессов струйные
приборы наз. соответственно эжекторами и
инжекторами, хотя принципиально они совер-
шенно тождественны. Если в качестве инжектирую-
щего потока применяются пар, газ или вода, то ин-
жекторы наз. соответственно пароструйными,
газоструйными или водоструйным и.
На рис. 2 схематически изображен инжектор для
сжатия газов или паров от давления Р% до давления
Р3 при помощи инжектирующего газового или паро-
вого потока с начальным давлением Pt. Последний
при истечении из сопла 1 расширяется от давления Pt
до давления P's, к-рое несколько меньше Р2, обуслов-
ливая тем самым приток всасываемого газа или пара
по штуцеру 2 в камеру смешения 3. Инжектирующий
поток, проходя с большой скоростью через камеру
269 ИНИЦИАТОРЫ ПОЛИМЕРИЗАЦИИ —ИНИЦИИРУЮЩИЕ ВЗРЫВЧАТЫЕ ВЕЩЕСТВА 270
смешения, увлекает за счет поверхностного трения
определенное количество инжектируемого газа (пара)
и смешивается с ним. При дальнейшем движении сме-
шанного потока через диффузор £ его скорость падает,
а давление за этот счет возрастает до Р3.
Важнейшей характеристикой инжектора является
весовое количество инжектируемого газа (пара или
жидкости), приходящееся на 1 кг инжектирующего
газа (пара или жидкости): gjgi — V кг/кг. Величина
U, наз. коэффициентом инжекции,
зависит от начального состояния (давление, темп-ра,
энтальпия, скорость) инжектирующего потока, от
степени сжатия инжектируемого потока (Р3/Р2), а
также от физич. свойств газов (паров или жидкостей)
в обоих потоках. Как правило, коэфф, инжекции
растет с увеличением давления Рх и с уменьшением
степени сжатия Р3/Рг. Инжекторы для двух жидкост-
ных потоков, а также для разнофазных потоков (газ —
жидкость, пар — жидкость) имеют в принципе устрой-
ство, изображенное на рис. 2; нек-рое различие в их
конфигурации объясняется особенностями агрегат-
ных состояний потоков, обусловливающими разное
соотношение конструктивных размеров.
Выполняя, по существу, функции таких машин, как
насосы для перекачивания жидкостей, вакуум-
насосы для отсасывания газов и паров из замкнутых
объемов, а также компрессоры для сжатия газов и
паров, И. имеют несравненно более простое устройство,
несоизмеримо меньшую металлоемкость и значи-
тельно большую компактность при чрезвычайной
простоте обслуживания. Этим преимуществам И. про-
тивостоит их низкий кпд (расход энергии на инжек-
цию часто в 2—3 раза выше, чем на насосы и компрес-
соры; кпд И. — отношение количества энергии, сооб-
щенной инжектируемому потоку для повышения его
давления и скорости, к количеству энергии, потерян-
ной инжектирующим потоком при его расширении до
состояния смешанного потока). Этот недостаток И.
ограничивает область их применения в пром-сти срав-
нительно немногочисленными случаями, когда затраты
энергии либо не играют определяющей роли, либо
покрываются какими-либо практич. преимуществами.
Так, И. широко применяют для создания или поддер-
жания вакуума в аппаратах при необходимости отса-
сывания паров (или парогазовых смесей) химически
агрессивных веществ, когда использование обычных
вакуум-насосов затруднено из-за коррозии. В каче-
стве инжектирующего потока в этих случаях чаще
всего применяют пар или воду.
И. применяют: для сжатия вторичных паров с це-
лью их использования в выпарных аппаратах, ра-
ботающих по принципу теплового насоса (см. Выпа-
ривание}', в системах синтеза аммиака под высоким
давлением (заменяют циркуляционные компрессоры);
для подъема и перекачивания жидкостей с глубин,
превышающих всасывающую способность жидкост-
ных насосов; при нагревании жидкостей голым паром,
являющимся в этом случае инжектирующим агентом,
обеспечивая хорошее перемешивание нагреваемой
среды и конденсата. Кроме того, И. применяются в си-
стемах пневматич. транспорта, в абсорбционных и
экстракционных аппаратах, создавая хороший кон-
такт контактирующихся фаз (жидкость — газ, жид-
кость — жидкость) и способствуя процессу массооб-
мена.
Лит.: Келлер С. Ю., Инжекторы, Киев, 1954; Ка-
ме н е в П. Н., Гидроэлеваторы и другие струйные аппараты,
М„ 1950; Ложкин А. Н., Трансформаторы тепла, М.—Л.,
1948; Гельперин Н. И., Найдич И. М., Проблема
инжекции, М„ 1939. Н. И. Гельперин.
ИНИЦИАТОРЫ ПОЛИМЕРИЗАЦИИ — см. Поли-
меризации инициаторы.
ИНИЦИИРУЮЩИЕ ВЗРЫВЧАТЫЕ ВЕЩЕСТВА -
соединения, способные легко взрываться от про-
стого начального импульса (действие пламени, тре-
ния, удара). Отличительной особенностью И. в. в.,
наз. также первичными взрывчатыми
веществами (ВВ), является то, что горение их
легко переходит в детонацию в условиях (атмосферное
давление, непрочная оболочка, малые заряды), в к-рых
такой переход для других ВВ, наз. вторичными,
не происходит. Это различие связывают с тем, что
уже при атмосферном давлении химич. превращение
И. в. в. завершается очень быстрое выделением макси-
мального количества тепла и соответственно образова-
нием газов, имеющих высокую темп-ру. Горячие газы
проникают в заряд между частицами И. в. в. на зна-
чительную глубину, поджигают их, что приводит
к быстрому подъему давления и возникновению дето-
национной волны. И. в. в. применяют в военной тех-
нике и взрывном деле в виде малых (доли грамма) за-
рядов, запрессованных до умеренной плотности в тон-
костенные оболочки — т. н. к а и с ю л и-д е т о н а-
т о р ы — вместе с небольшим зарядом вторичного
ВВ. При действии накола или пламени И. в. в. в кап-
сюле детонирует и вызывает детонацию контактирую-
щего с ним заряда вторичного ВВ. Высокую чувстви-
тельность, легкую воспламеняемость и большую ско-
рость химич. превращения И. в. в. используют также
для воспламенения порохов и пиротехнич. составов
с помощью т. и. капсюлей-воспламенителей. В капсю-
лях-воспламенителях применяют смеси И. в. в. с го-
рючими и окислителями, причем величину заряда
(сотые доли грамма) и состав его выбирают так, чтобы
происходила не детонация, а только быстрое сгора-
ние. Важнейшими И. в. в. являются: соли тяжелых
металлов гремучей кислоты HONC, или фульминаты,
напр. гремучая ртуть', соли и др. производные азоти-
стоводородной к-ты, или азиды, напр. азид свинца или
циануртриазид C3N12: соли тяжелых металлов поли-
нитрофенолов, напр. тринитрорезорцинат свинца',
ацетилениды металлов, напр. ацетиленпд серебра
Ag»C2; нек-рые диазосоединения и их соли, напр.
4,6-динитро-2-диазофенол или 2,4-дипитродиазобен-
золперхлорат; нек-рые органические перекиси, на-
пример гексаметилентрппероксиддиамин C6H12N2O0;
производные тетразена H2N—HN—N=NH и тетразола
I----------------,
N= N—NH—CH = N, напр. гуанилнитрозоампнгуа-
нилтетразеп, наз. в технике тетразен. Наибольшее
применение имеют гремучая ртуть, азид свинца, три-
нитрорезорцинат свинца и тетразен.
Сравнительной мерой инициирующей способности
является т. и. предельный заряд — мини-
мальный заряд И. в. в., при взрыве к-рого в опре-
деленных условиях еще возникает детонация одного
из вторичных ВВ. Для тетрила в нек-рых условиях
предельный заряд азида свинца составляет 0,025 г,
а гремучей ртути 0,29 г, для тротила — 0,09 и 0,36 г
соответственно. Чтобы обеспечить возбуждение взрыва
азида свинца, воспламеняемость к-рого сравнительно
мала, в капсюлях-детонаторах поверх заряда этого
И. в. в. помещают тонкий слой тринитрорезорцпната
свинца или смеси последнего с тетразеном, горючими
и окислителями. Сами по себе тетразен и тринитро-
резорцинат свинца обладают слабым инициирующим
действием; их применение, кроме отмеченного выше,
ограничивается приготовлением составов для капсю-
лей-воспламенителей. Многие составы капсюлей-вос-
пламенителей содержат в качестве И. н. в. гремучую
ртуть и ее смеси, напр. с хлоратом калия и сульфи-
дом сурьмы. Ввиду большой опасности возникнове-
ния взрыва, изготовление И. в. в. и обращение с
ними требуют особых мер предосторожности. Пере-
возить И. в. в. можно только в виде готовых кап-
сюлей.
Лит.: Бубнов П. Ф., Инициирующие взрывчатые
вещества и средства инициирования, М., 1940; Горст А. Г.,
*271
ИНОЗИН — ИНОЗИНФОСФОРНЫЕ кислоты
272
Пороха и взрывчатые вещества, 2 изд., М., 1957; Андре-
ев К. К., Беляев А. Ф., Теория взрывчатых веществ, М.,
1960. А. И. Гольбиндер.
ИНОЗИН (9-Р-О-рибофуранозил-гипоксантин, ги-
поксантозин) С10Н12ОБ?<4, мол. в. 268,24— нуклеозид,
содержащий остаток гипоксанти-
на и D-рибозы; бесцветные кри-
сталлы; кристаллизуется с двумя
молекулами воды; т. пл. 218°;
[a]g + 73,6 (2,5% Р-Р в 0,01н.
NaOH); умеренно растворим в
воде; рКЯ1 1,2; рКа., 8,75; рА'Пз
12,33. И. обладает характерным
поглощением в УФ-свете; спектро-
фотометрии. константы И. в УФ-
свете приведены в таблице. При
pH X, MMK E* • 103 ©1 © о «1 01 и 1я ! 1 ©1 © ОО © I 01 01 и 1н 1 ©1 © © © о» С4 и
макс, МИН им. макс. миним. 260 ммк
3 248 223 12,2 3,4 7,1 1,68 0,25 0,025**
7 —— — — 7,4 1.68 0,25 0,025
12 253 224 13,1 2,5 И,7 1,05 0,18 —
• Е — коэффициент молярной экстинкции; • • при pH 2.
кислотном гидролизе И. распадается на гипоксантин
и рибозу. Качественные и количественные определе-
ния И. основаны на цветных реакциях на гипоксан-
тин и рибозу и поглощении И. в УФ-свете; произво-
дятся методами колориметрии, хроматографии и
ионофореза.
И. образуется при химич. или ферментативном от-
щеплении фосфатных остатков от инозинфосфорных
кислот, а также при дезаминировании аденозина (при
действии NaNO2). При конденсации 4,6-диаминопири-
мидина с рибозой в спиртовом р-ре в присутствии
НС1 был получен изомер И. — Э-р-О-рибопиранозил-
гипоксантин, отличающийся от И. тем, что имеет пи-
ранозную структуру сахара.
И. является составной частью инозиновых нуклео-
тидов (см. Инозинфосфорные кислоты), в виде к-рых
он распространен в животных и растительных орга-
низмах.
Лит.: Шабаров а 3. А., Усп. химии, 1959, 28, вып. 4,
369; Vol kin Е., Cohn W. Е., в кн.: Methods of bio-
chemical analysis, v. 1, N. Y., 1954, p. 287; The nucleic acids.
Chemistry and biology, ed. E. Chargaff and J. N. Davidson,
V. 1, N. Y„ 1955. Л. H. Боброва.
ИНОЗИНФОСФОРНЫЕ КИСЛОТЫ (инозиновые
кислоты, гипоксаптозинфосфорные кислоты, 9-p-D-
рибофуранозил-гипдксантпн-5'-фосфорные кислоты) —
нуклеотиды, содер-
жащие остаток ги-
поксантина, D-ри-
бозы и остатки
фосфорной кисло-
ты. Известны И. к.,
содержащие от 1 до
3 остатков фосфор-
ной к-ты в моле-
куле, — инозинмо-
но-, ипозинди- и
инозинтрифосфор-
ные к-ты (ИМФ,
ИДФ,ИТФ).ВИ.к.
биологич. происхо-
ждения фосфат присоединен к 5-му атому углерода
пентозы; при дезаминировании аденозин -2'- и З'-моно-
фосфатов получены соответствующие изомеры инози-
новой к-ты.
И н о з и н-5'-м оно фосфорная кислота
(инозиновая к-та, ИМФ) C10H13N4O8P, мол. в. 348,22 —
сиропообразная жидкость, растворима в воде, нерас-
творима в эфире, незначительно растворима в этаноле;
[a]D = + 36,8° (1,5% р-р в 0,1н.НС1 при 27°),
pK(li 1,54; рКа^ 6,04. В 100 г воды при 16° растворяется
0,25 г бариевой соли ИМФ. При кислотном гидролизе
ИМФ распадается на гипоксантин, рибозу и фосфор-
ную кислоту; в более мягких условиях, напр при
кипячении в водном р-ре, отщепляется пентозофосфат
и гипоксантин. С NH 4, Na+, К+, Ва2+ и Са2+ ИМФ обра-
зует кристаллич. соли. ИМФ м. б. получена из мышеч-
ной ткани, где она образуется в результате фермента-
тивного дезаминирования адениловой к-ты.
Инозин -5'- ди фосфорная кислота
(ИДФ) C10H14N4P2O1i, мол. в. 428,20 — бесцветный
аморфный продукт, растворима в воде, нерастворима
в органич. растворителях. Соли щелочноземельных и
тяжелых металлов ИДФ мало растворимы в воде,
однако более растворимы, чем аналогичные соли ИТФ;
соли щелочных металлов растворимы в воде. При
действии 1н.НС1 на ИДФ в течение 7 мин при 100°
происходит разрыв пирофосфатной связи и практи-
чески полное отщепление фосфатного остатка, нахо-
дящегося в ^-положении. В этих условиях почти не
отщепляется фосфат, находящийся в а-положении.
ИДФ получают ферментативным отщеплением (дей-
ствием миозина) у-фосфата от ИТФ, а также при деза-
минировании (в присутствии HNO2) аденозицдифос-
форной к-ты (АДФ).
Инозин-5'- три фосфорная кислота (ИТФ)
C10H15O14N4P3, мол. в. 508,19— бесцветный аморфный
продукт, растворима в воде, плохо растворима в ор-
ганич. растворителях, соли щелочных металлов рас-
творимы в воде; плохо растворимы в воде и хорошо
растворимы в кислотах соли щелочноземельных и тя-
желых металлов. ИТФ является гипоксантиновым
аналогом аденозинтрифосфорной кислоты, имеет две
молекулы легкогидролизуемого фосфата (р- и у-
фосфаты), отщепляющегося при действии 1н.НС1
при 100° в течение 7 мин. В более жестких условиях
кислотного гидролиза разрывается N-гликозидпая
связь и образуются гипоксантин, рибоза и 3 молекулы
фосфорной к-ты. Гидролиз в слабощелочном р-ре
дает инозиновую к-ту (ИМФ) и фосфат. ИТФ м. б.
получева путем дезаминирования аденозинтрифосфор-
ной к-ты (АТФ) в присутствии HNO2; выделена и оха-
рактеризована в виде бариевой соли, кристаллизую-
щейся с 7 молекулами воды (безводная соль гигро-
скопична). Очистка препаратов ИМФ, ИДФ и ИТФ
производится с помощью ионообменной хромато-
графии.
И. к. имеют максимум поглощения в УФ-свете при
249—250 ммк (в кислой среде); спектры ИМФ, ИДФ и
ИТФ в УФ-свете близки между собой. Коэфф, моляр-
ной экстинкции И. к. при pH 2 и 250 ммк равен 10 900.
И. к. дают цветные качественные реакции на гипо-
ксантин, рибозу и органически связанный фосфат.
Количественные определения И. к. основаны на тех
же цветных реакциях, а также на измерении поглоще-
ния И. к. в УФ-свете. Для качественных и количест-
венных определений пользуются также методами
хроматографии и ионофореза. И. к. содержатся в жи-
вотных и растительных организмах. Являясь макро-
эргич. соединениями, ИТФ и ИДФ принимают уча-
стие в таких ферментативных процессах, как, напр.,
нуклеотидфосфокиназные реакции и декарбоксилиро-
вание щавелевоуксусной к-ты до фосфоенолпировино-
градной к-ты.
Лит.: ВенкстернТ. В., Баев А. А., Биохимия,
1957, 22, вып. 6, 1043; Котельникова А. В., Усп.
совр. биол., 1957, 43, вып. 2, 133; П а с X и н а Т. С., Усп.
биол. химии, т. 3, М., 1958, с. 227; Чепинога О. П., Укр.
биохим. журнал, 1958, 30, Ks 3, 451; Горкин В. 3., Био-
химия, 1958, 23, вып. 1, 106; К 1 е i п г е 1 1 е г A., Biochem.
J., 1942, 36, 729; The nucleic acids, ed. E. Chargatt and J. H.
Davidson, N. Y., 1955; Kiessling W., Biochem. Z., 1934,
273, H. 1-3, 103. Л. H. Боброва.
2 73
.'ИНОЗИТ — ИНОЗИТФОСФАТИДЫ
274
ИНОЗИТ (гексаоксициклогексан) С6Н12Ов, мол. в.
180,162 — шестиатомный циклич. спирт (I), существует
в виде восьми цис-, т/>амс-стереоизомеров, семь из
к-рых представляют собой оптически неактивные
.мезо-формы, а один стереоизомер находится в виде
двух оптич. антиподов и одного рацемата.
Из стереоизомеров И. оптич. недеятельный циклогек-
сан—(1, 2, 3, 5-цис)-гексил, обычно называемый мезо-
инозитом, или .мио-инозитом (II), относится к веще-
ствам с витаминной активностью; он является так-
же ростовым фактором для некоторых культурных
дрожжей.
е-экваторнальные
В молекуле И., имеющей форму «кресла», 6 чере-
дующихся связей атомов углерода кольца с замести-
телями направлены параллельно главной оси симмет-
рии молекулы, т. е. перпендикулярно к основной
плоскости кольца («аксиальные» связи обозначаются
буквой а), а другие 6 чередующихся связей распола-
гаются радиально по отношению к главной оси сим-
метрии («экваториальные» связи обозначаются бук-
вой е). Формула мезо-И. может быть представлена
в виде «кресла» (III) с единственной северо-аксиаль-
ной гидроксильной группой в положении 2, характе-
ризующейся своим особо выступающим положением
в молекуле; см. также ст. Конформационный анализ.
Мезо-И. кристаллизуется в бесцветных моноклинич.
призмах (из воды) ниже 50° с 2 молекулами Н2О;
т. пл. 225—227°; перегоняется в вакууме, обладает
сладким вкусом. Все формы И. хорошо растворимы
в воде. И. свойственны обычные реакции, характер-
ные для полпоксисоедипений с соседними гидрокси-
лами. Мезо-11. получают из фитина — гексафос-
форвого эфира лгеао-И., содержащегося в растениях,
напр. в кукурузе. Он широко распространен в расти-
тельных тканях, находится в мышцах, веществе мозга
(н виде кефалинфосфолипида) и др. органов. В расте-
ниях встречаются: метиловый эфир мезо-И. — се к-
в о й т о л, т. пл. 204°; 5-метиловый эфир L-инозита —
квебрахит, т пл. 191°: 5-метиловып эфир d-ино-
зита — пинит, т. пл. 185—186°.
Лит.: Березовский В. М., Химия витаминов, М.,
1959, с. 117. В. М. Березовский.
ИНОЗИТФОСФАТИДЫ — фосфатиды (фосфоли-
пиды), характеризующиеся наличием в их составе
фосфорного эфира шестиатомного циклич. спирта ино-
зита. В природных И. инозит присутствует в виде
одного из оптически неактивных изомеров — мио-
пнозита (см. Инозит). И. широко распространены
в природе, они встречаются в животных и раститель-
ных тканях, а также в микроорганизмах. Особенно
богаты И. бобы сои и мозговая ткань. И. составляют
ок. 0,4% от чистого веса мозга животных. Природ-
ные И. выделяют путем экстракции и фракционирова-
ния органич. растворителями с последующей очисткой
и разделением на колонках с H2SiO3 или А12О3. И. раз-
личного происхождения отличаются по своему строе-
нию и свойствам. Известные в настоящее время И. мо-
гут быть разделены на три группы. К первой группе
относятся И., в состав к-рых входят инозит, фосфор-
ная к-та, глицерин и жирные к-ты (обычно с числом
атомов углерода С1в или С18) в молярном соотношении
1 : 1 : 1 : 2. В структурном отношении эти И. подобны
кефалинам и лецитинам и могут быть названы диацил-
глицерил фосфорил инозитами:
Другое распространенное название этих И. —моно-
фосфоинозитиды, или фосфатидил-
инозиты. В фосфатидилинозитах фосфорная к-та
связана с гидроксилом в положении 1 лшо-инозита.
Поскольку при этерификации этого гидроксила моле-
кула жио-инозита утрачивает плоскость симметрии,
то фосфат леио-инозита может существовать в двух
стереоизомерных формах. В фосфатидилинозитах при-
сутствует только оптически активный L-лшо-инозит-
1-фосфат. Фосфатидилинозиты выделены иэ печени и
сердца животных, из бобов сои, зародышей пшеницы
и дрожжей- В печени и дрожжах они составляют ок.
90% от всего количества И. Фосфатидилинозиты —
твердые, бесцветные, медленно окисляющиеся на воз-
духе вещества, растворимы в хлороформе, 95%-ном
эфире и в смеси хлороформ — метанол 3:2; нераст-
воримы в ацетоне, этаноле и метаноле. С водой фосфа-
тидилинозиты образуют вязкие гели; pH водных р-ров
5,5—5,8. Природные фосфатидилинозиты оптически
активны, [a]D = 5,5°.
Ко второй группе И. относятся фосфатиды, в состав
к-рых инозит, (фосфорная к-та, глицерин и жирная
кислота входят в молярном соотношении 1 : 2 : 1 : 1.
При кислом гидролизе этих И. образуется л-дифосфат
инозита, что дает основание приписать им следующую
структуру:
О о-
СН2-О-Р---О\__/ОН
RCO-CH „ НО~\ У ОН
। и \ /
СН2-О-Р---СИ 'ОН
О"
Инозитфосфатиды этого типа обычно наз. д и ф о с-
ф о и н о з и т и д а м и; они составляют основную
массу И. головного мозга животных, являются кис-
лыми фосфатидами, и их выделяют в виде кальциевых
или магниевых солей. Дифосфоинозитиды, выделен-
ные из кефалиновой фракции мозга фракционирова-
нием метанолом, представляют собой белый порошок,
неэмульгирующийся водой и нерастворимый в боль-
шинстве органич. растворителей, за исключением
влажного хлороформа. Из тканей мозга выделены
также трифосфоинозитиды, содержащие
на 1 моль инозита 3 моля фосфорной к-ты.
К третьей группе относятся И. более сложного строе-
ния. Многие из них еще не получены в химически
индивидуальном состоянии, и их структура оконча-
тельно не определена. К числу таких И. с установлен-
ным строением следует отнести выделенный из Mico-
bacterium tuberculosis диманнозид диацилглицерофос-
форилинозита, представляющий собой обычный фос-
фатидилинозит, в к-ром кольцо инозита связано гли-
козидными связями с 2 молекулами D-маннозы. Жир-
ными к-тами, входящими в состав этих И., являются
пальмитиновая и туберкулостеариновая (10-метил-
стеариновая). К числу указанных сложных И. отно-
сится также липозитол, выделенный из соевого
масла. До настоящего времени неизвестно, является
ли липозитол химически индивидуальным соедине-
275
ИНСЕКТИЦИДЫ
276
нием или фракцией различных И. и других фосфоли-
пидов. Гидролизом липозитола получены инозит,
фосфорная и олеиновая к-ты, насыщенная жирная
к-та (смесь пальмитиновой и стеариновой), галактоза,
зтаноламин и винная к-та. Липозитол представляет
собой тонкий, жирный, белый порошок, постепенно
окисляющийся на воздухе. Растворим в необеэвожен-
ных петролейном и серном эфирах, в бензоле и хлоро-
форме; нерастворим в метиловом и этиловом спиртах,
ацетоне и диоксане; с водой образует эмульсию.
Примером очень сложного инозитсодержащего фос-
фолипида растительного происхождения может слу-
жить фитогликолипид, выделенный мягким
щелочным гидролизом из фосфатидной фракции семян
нек-рых растений. В состав его входят: фитосфинго-
зин, цереброновая и фосфорная к-ты, инозит, глюку-
роновая к-та, D-глюкозамин, арабиноза и галактоза,
связанные с глюкуроновой к-той, и манноза, связанная
с инозитом. Строение фитогликолипида может быть
представлено следующим образом:
СН3(СН2)13- СНОН-СНОН-СН-СН2-О-РО-И-М
I I I
NH о- Г
I I
СО Г1
I !
СНОН ГА
I
(CH2)2iCHs
М — манноза, И — инозит, Г — глюкуроновая к-та,
Tj — глюкозамин, ГА — галактоза и арабиноза.
Вполне возможно, что фитогликолипид является
лишь частью более сложного нативного липида и об-
разуется из него в процессе гидролиза.
И., подобно другим фосфолипидам, играют весьма
важную роль в биохимич. процессах, протекающих
в живых организмах. Обмен И. в нервной ткани мозга
происходит в 10 раз интенсивнее, чем обмен других
фосфолипидов. Та же картина наблюдается в таких
интенсивно секретирующих белок органах, как под-
желудочная и щитовидная железы. Это дает основа-
ние приписать И. особую роль в осуществлении актив-
ного переноса катионов и белка через клеточные мем-
браны.
Лит.: Трусов В. И., Успехи соврем, биол., 1958,
45, вып. 1, 33; D е u е I Н. J., The lipids, their chemistry and
biochemistry, v. 1, N. Y. — L., 1951, p. 450; H a n a h a n D. J.,
Lipide chemistry, N. Y.—L., [1960], p. 106; Hawthorne
J. N., J. ot Lipid Research, 1960, 1, Ks 4, p. 255.
В. Б. Спиричев.
ИНСЕКТИЦИДЫ (инсектисиды) — химические
средства борьбы с вредными насекомыми. Рациональ-
ная химич. классификация И. отсутствует. Чаще всего
пользуются физиология, классификацией, характе-
ризующей пути проникновения яда в организм насе-
комого. И. разделяют на следующие группы: 1) ки-
шечные, попадающие в организм насекомого через
рот; 2) контактные, попадающие в организм насеко-
мого в результате соприкосновения (контакта) с той
или иной частью его тела; 3) фумиганты, или дыха-
тельные, действующие на насекомых в парообразном
состоянии или в виде аэрозоля и попадающие в орга-
низм через органы дыхания; 4) системные, или внут-
рирастительные, вещества, способные поглощаться
корнями и листьями и перемещаться по растению.
Такие вещества делают растение ядовитым для пара-
зитирующих на нем насекомых. Системные И. иногда
применяют также для борьбы с паразитами живот-
ных, для чего эти И. вводят в организм с пищей (если
они безвредны для животного).
К кишечным И. относится большинство неор-
ганич. соединений, в первую очередь мышьяковистые
(арсенаты: кальция, магния, марганца, свинца, цинка;
арсенит кальция, парижская зелень), соли кремне-
фтористоводородной к-ты, фториды металлов, раство-
римые в воде соли бария и нек-рые др. Из органич.
соединений к таким И. следует отнести тиодифенил-
амин, соли четвертичных аммониевых оснований, про-
тивомольные препараты — эйлан НЦ (I), эйлан ной
(II), митин ФФ (III), эйлан НК (IV) и ирган НТ (V),
применяемые для пропитки шерсти, нек-рые арома-
тич. производные тиомочевцны и др.
сн3/сн3
2-n-ch2conh
Cl
о
Cl
К контактными, относятся такие вещества,
как ДДТ, гексахлоран, хлорциклодиены, (хлориндан,
альдрин, дильдрин и Др-), фосфорорганические инсек-
тициды (0,0-диэтил-0,4-нитрофенилтиофосфат, или па-
ратион, 0,0-диметил-0,4-нитрофенилтиофосфат — ме-
тафос, метилпаратион и др.), пиретрины, севин и др.
Большинство контактных И. обладает и кишечным
действием. Кфумигационным И. относятся
гл. обр. летучие соединения, к-рые при комнатной
темп-ре имеют высокое давление паров. В качестве
фумигантов применяют бромистый метил, дихлорэтан,
хлористый аллил, хлорпикрин, дихлорнитроэтан,
трихлорацетонитрил, акрилонитрил, окись этилена,
сероуглерод и др. Фумигационным действием обла-
дают также многие контактные И. (особенно в жаркую
погоду). Эта группа И. используется преим. для борь-
бы с вредителями зерновых запасов, а также для
обработки почвы с целью борьбы с обитающими в ней
вредителями, в том числе и с нематодами (круглые
черви). При этом часто происходит стерилизация
почвы как от вредителей растений, так и от возбуди-
телей болезней и сорняков. Хорошими стерилизато-
рами почвы являются такие препараты, как N-метил-
дитиокарбамат натрия, 1, 2-дибромэтан, смесь дихлор-
пропена с дихлорпропаном (ДД), 1-хлор-2, 3-дибром-
пропан (немагон) и др.
К препаратам, нарушающим функцию дыхательных
органов, следует также отнести нек-рые пылевидные
препараты, представляющие собой тонко размолотые
силикаты, действие к-рых основано па закупорива-
нии органов дыхания насекомого, в результате чего
насекомое погибает от асфиксии (удушья). Аналогич-
ным действием, в известной мере, обладают минераль-
ные масла, к-рые широко применяют для борьбы с вре-
дителями плодовых деревьев и кустарников, в борьбе
со щитовками, клещамп и др.
277
ИНСТРУМЕНТАЛЬНЫЕ МЕТОДЫ АНАЛИЗА — ИНСУЛИН
278
К системным И., большинство к-рых яв-
ляются фосфорорганич. соединениями (см. Фосфорор-
ганические инсектициды), относятся, напр., О,О-диме-
тил-2-этилмеркаптоэтилтиофосфат (м е т и л м е р к а п-
тофос, метсистокс), О,О-диэтил-2-этилмеркап-
тоэтилтиофосфат (меркаптофо с, систокс,
демето н), О,О-диэтил-2-этилмеркаптоэтилтиофос-
фат (М-74, д и с и с т о н), октаметилтетраамид пиро-
фосфорной к-ты (октаметил, песток с, трэ-
да н), 0,0-диметил-2-этилмеркаптоэтилдитиофосфат
(э к а т и н, М-81, интратион), О,О-диэтилэтил-
меркаптометилдитиофосфат (т и м е т), тетраметилди-
амидофторфосфат (д и м е ф о к с), О.О-диметил-S-
метиламидометилдитиофосфат (рогор, фосфа-
ми д, диметоат). Растворы системных И., в от-
личие от других, быстро и почти полностью всасы-
ваются растением и по сосудистой системе распрост-
раняются по организму растения, делая сок их ядо-
витым для сосущих вредителей. Это отличие обуслов-
ливает ряд преимуществ системных И., напр.: неза-
висимость действия от дождя, ветра и росы; длитель-
ность действия И. на сосущих вредителей (не менее
1—4 недель); относительно малая опасность действия
нек-рых И. (напр., октаметила) на полезных насеко-
мых-опылителей и т. п. Положительным свойством
системных И. является возможность их применения
не только путем опрыскивания, но и внесением в почву
(совместно с семенами или отдельно от них), что позво-
ляет защищать всходы от повреждения насекомыми.
Механизм действия И. на насекомых выяснен не
для всех классов соединений. С большей или меньшей
степенью достоверности он выяснен для органич. сое-
динений фосфора, соединений мышьяка и нек-рых про-
тивомольных препаратов. Одним из важных вопросов
в развитии и усовершенствовании химич. методов
борьбы с вредными насекомыми является преодоле-
ние приобретаемой насекомыми устойчивости («при-
выкания») к определенным группам И.
И. используют в виде порошков (дустов), растворов,
эмульсий, суспензий и т. п.; большинство И. можно
применять наземными и авиационными средствами.
Применение И. позволяет резко сократить потери уро-
жая от вредителей растений (в среднем эффект в 10—
15 раз превышает произведенные затраты).
Лит.: Мельников Н. Н., Ж. Всес. хим. о-ва им.
Д. И. Менделеева, 1960, 5, № 3, 242: Орлов В. И.,
Морозова М. А., Крутицкая М. Н., там же, 1960,
5, № 3, 268; Попов П. В., Справочник по ядохимикатам, М.,
1956; Metcalf R. L., Organic insecticides, 2 ed., N. Y.—L.,
1961; Advances in pest control research, ed. by R. L. Metcalf,
v. 1—4, N.Y.—L., 1957—61; PerkOwW., Die Insektizidc,
Hdlb., 1956; ZbirovskJ M., My Ska J., Insekti-
ci dy, fungicidy, rodenticidy, Praha, 1957; Brown A. W. A.,
Insect control by chemicals, N. Y.—I.., 1951; FUrst H.,
Chemie und Pfianzenschutz, 2 Aufl., B., 1959.
ИНСТРУМЕНТАЛЬНЫЕ МЕТОДЫ АНАЛИЗА8—
количественные апалитич. методы, для выполнения
к-рых требуется электрохимии., оптич., радиохимия,
и иная аппаратура. Иногда И. м. а. не совсем пра-
вильно противопоставляют химич. методам, напр.
гравиметрическим, выполнение к-рых невозможно
без взвешивания па аналитич. весах. К И. м. а. обычно
относят: 1. Электрохимия, методы — потенциомет-
рию, вольтамперометрию и полярографию, амперо-
метрия. и вольтаметрич. титрование, электрогравимет-
рию, электролитич. разделение, кулонометрию, кон-
дуктометрию, высокочастотные методы и др. 2. Ме-
тоды, основанные на эмиссии или абсорбции излуче-
ния: эмиссионный спектральный анализ, абсорбцион-
ный спектральный анализ (фотометрия в УФ, видимой
в ИК-области спектра; см. Фотометрические методы
анализа), в том числе спектрофотометрия, флуоримет-
рия, турбидиметрия, нефелометрия, комбинационное
рассеяние света, рвнтгеноспектральный анализ и др.
3. Масс-спектрометрия, различные методы газового
Схема строения инсулина
В А
фал.
вал. глиц.
acn.-NHa изол.
глут.-NHo вал.
ГИС. глут.
лейц. 7 гЛуТ.-NHa
цис.—S— S—ЦИС.—ЦИС.
глиц. 8.ал. S
сер. 9.сёр. S
гис. 10.вал.-цис.
лейц. сер.
вал. лейц.
глут. тир.
ал. глут.-NHa
лейц. лейц.
тир. глут.
лейц. асц.-NHa
вал. тир.
цис.—S- —S— цис.
глиц. acn.-NH2
Условные обозначе-
ния аминокислот см.
1 т., стр. 180.
анализа, методы, основанные на измерении радиоак2
тивности (напр., Радио активационный анализ). См.
также Амперометрическое титрование.
Лит.: Юинг Г. В., Инструментальные методы хими-
ческого анализа, пер. с англ., М., 1960; Delahay Р.,
Instrumental analysis, N. Y., 1957; его же, Новые приборы
и методы в электрохимии, пер. с англ., М., 1957;
Strobel Н. A., Chemical instrumentation, Reading (Mass.),
1960. А. И. Буеве.
ИНСУЛИН — гормон поджелудочной железы, ре-
гулирующий процессы углеводного обмена и поддер-
жание нормального уровня
сахара (глюкозы) в крови;
вырабатывается в р-клет-
ках своеобразных образова-
ний поджелудочной железы
(т. н. «островков» Лангерган-
са). И.—простой белок; наи-
меньшая, мономерная, струк-
турная субъединица инсули-
на CjjaHj^NgjOjjSg, мол. в.
6000 (5733 на основе дан-
ных об аминокислотном со-
ставе и строении). В водных
р-рах И. существует в виде
более крупных, ассоцииро-
ванных частиц с мол. в.
12 000, 36 000 и 48 000.
Инсулин — первое слож-
ное, биологически активное
вещество, строение к-рого
удалось полностью расшиф-
ровать. Наименьшая струк-
турная субъединица И. с
мол. весом 6000 состоит из
двух полипептидных цепей
А и В (см. схему).
Приведенная схема отно-
сится к И. крупного рога-
того скота. И. животных
других видов несколько от-
личаются по строению, причем различия ка-
саются лишь природы аминокислотных остат-
ков 8, 9 и 10-й цепи А (см. таблицу).
И. получен в кристаллин, состоянии; бес-
цветный продукт, т. пл. 233°. Препараты кри-
сталлин. И. содержат обычно 0,3—0,6% Zn2+.
Высокоочищенные аморфные препараты И.
могут быть получены свободными от Zn2-,
этом они сохраняют свою биологическую актив-
глут.
арг.
глиц.
фал.
фал.
тир.
тре.
прол,
лиз.
ал.
при
ность. Изоэлектрическая точка И. соответствует pH
Вид животного
Аминокислоты
Бык . .
Свинья
Лошадь
Варан .
Кит . .
Номер аминокислотного остатка
8 9 10
. . . -ал.-сер.-вал,-. . .
. . . -тре.-сер.-изол.- . . .
. . . -тре.-глиц.-изол.- . . .
. . . -ал.-глиц.-вал,- . . .
. . . -тре.-сер.-изол,- . . .
5,3—5,4. В области изоэлектрич. точки препараты
И. плохо растворимы в воде (10 мг/л при 5°). И. хо-
рошо растворяется в разб. к-тах и щелочах, а также
в водно-спиртовых р-рах (в частности, в 80%-ном
спирте). И. весьма устойчив в кислых и несколько
менее — в щелочных средах: при pH 2,5 он не утрачи-
вает биология, активности даже при нагревании до
52°. И. устойчив также к действию других денатури-
рующих белки агентов, в частности органич. раство-
рителей. Характерным свойством И. является спо-
собность образовывать фибриллы при нагревании
в сильно кислой среде (100°, pH 2,0).
В химич. отношении И. ведет сЛэбя подобно боль-
шинству веществ белковой природы (дает многие
279
ИНСУЛИН — ИНТЕРФЕРОМЕТРИИ
280
цветные реакции, характерные для белков, в частно-
сти биуретовую, нингидринную, ксантопротеиновую,
Миллона и др.). Восстановители (цистеин, тиогликоле-
вая к-та, восстановленная форма метиленового си-
него и др.) инактивируют И., расщепляя дисульфид-
ные мостики в молекуле гормона в результате восста-
новления их до SH-групп. Окисление И. надмуравьи-
ной к-той также приводит к расщеплению —S—S—
связей и отделению полипептидных цепей А и В друг
от друга, при этом сера остатков цистеина окисляется
до HSO3-rpynn. При ацетилировании в мягких усло-
виях (напр., кетеном) свободных аминогрупп И.
последний не теряет своей активности. В то же время
ацетилирование фенольных гидроксилов тирозиновых
остатков в молекуле И. приводит к полной утрате
гормональной активности, к-рая, однако, вновь восста-
навливается после гидролитич. отщепления ацетиль-
ных группировок. И. легко образует соединения с ио-
нами двухвалентных металлов, особенно с Zn2+, Со2+ и
Cd2+. Большое практич. значение имеет способность И.
образовывать комплексы с нек-рыми белковыми вещест-
вами основного характера, в частности с протаминами.
И. легко гидролизуется протеолитич. ферментами
(трипсином, химотрипсином, пепсином), а также ткане-
выми протеазами и содержащимся в печени фермен-
том инсулиназой.
Физиология, роль И. состоит гл. обр. в регулиро-
вании обмена глюкозы и поддержании ее нормаль-
ного уровня в крови. При недостатке И., в частности
у людей, страдающих диабетом, нарушаются процессы
гликогенообразования и нормального потребления
глюкозы в тканях. В результате в крови резко воз-
растает концентрация глюкозы (гипергликемия) и
значительно усиливается выведение глюкозы с мочой
(глюкозурия). Введение больному'диабетом И. хотя
и не излечивает его, но устраняет все эти симптомы,
уровень сахара в крови возвращается к норме. При
введении чрезмерных количеств И. уровень сахара
в крови падает ниже нормы, что может привести к ги-
погликемия. коме и даже смерти. И. вводят в организм
больного путем инъекции внутримышечно или под-
кожно.
Механизм действия И. окончательно еще не выяс-
нён; имеются указания на то, что И. увеличивает про-
ницаемость клеточных мембран для глюкозы.
И. применяют при сахарном диабете и при лечении
ряда психич. заболеваний (в частности, шизофрении),
с помощью т. н. «инсулиновой комы»; кроме того, И.
иногда назначают при истощении, упадке питания,
поражениях печени и др.
И. для медицинских целей получают из поджелу-
дочной железы свиней или крупного рогатого скота.
Обычно из 1 кг поджелудочной железы крупного ро-
гатого скота получают 2500—3000 международных
единиц (ME) И. Активность препаратов И. определяют
по снижению содержания сахара в крови или по
появлению судорог у мышей. В биохимия, исследова-
ниях содержание И. определяют по его способности
ускорять потребление глюкозы тканью диафрагмы
крысы. За 1 ME активности И. принимают активность
0,04082 мг международного стандартного препарата
И.; это количество И. в течение 3 часов вызывает сни-
жение на 45 мг % содержания сахара в крови здоро-
вого кролика весом 2 кг, не получавшего пищи в те-
чение 24 часов.
Лит.: Машковский М. Д., Лекарственные средства,
4 изд., М., I960; Фер дм ан Д. Л., Биохимия, М., 1959;
Белки. Под ред. Г. Нейрата и К. Бейли, пер. с англ., т. 3,
ч. 1, М., 1958; Лейбсон Л. Г., Сахар крови, М.—Л.,
1962; Hand О., Hormone, Jena, 1959, S. 545; Straub F. В.,
Biocnemie, Budapest, 1960; Fermente. Hormone. Vitamine
und die Beziehungen dieser Wirkstplle zueinander, hrsg. v.
R. Ammon, W. Dirscherl, Bd 2, 3 Autl., Stuttgart, 1960, S. 16;
Betsin Th., Biochemie der Hormone, Lpz., 1959.
В. Б. Спиричев.
ИНТЕГРАЛЬНАЯ ДОЗА — см. Доза ионизирую-
щего излучения.
ИНТЕРМЕТАЛЛИЧЕСКИЕ СОЕДИНЕНИЯ — хи-
мические соединения металлов друг с другом. В
отличие от обычных химич. соединений, И. с. часто не
подчиняются стехиометрии, законам постоянства со-
става и простых кратных отношений. Они относятся
к соединениям, обладающим преимущественно «ме-
таллической'связью». Подробнее см. Металлические
соединения.
ИНТЕРФЕРОМЕТРИЯ — методы различных фи-
зич. измерений, основанные на интерференции света:
сравнение длин и толщин, измерение показателей пре-
ломления, изучение формы и тонкой структуры спект-
ральных линий, измерение угловых диаметров звезд,
контроль качества обработки поверхностей, изучение
диффузии и конвекционных токов и др. В химии И.
применяют гл. обр. для определения концентрации
растворов, состава жидких или газообразных смесей
и примесей к ним. Эти измерения основаны на зави-
симости показателя преломления от состава.
В лабораторной практике пользуются обычно интерферо-
метром Релея конструкции Габера и Леве (рис. 1). Пучок
света ог лзмпочки накаливания или монохроматора через
щель 2 поступает в трубку 1, где расщепляется двойной
щелью .3 на два узких параллельных пучка, проходящих
через одинаковые половинки 4 и 5 двойной кюветы. В оку-
лярной системе 10 оба луча сводятся и дают интерферен-
ционный спектр из системы параллельных полос, наблюда-
емых через цилиндрич. линзу 11.
Если среды, заполняющие 4 и 5 (напр., раствор и чистый
растворитель), имеют разные показатели преломления nt
и п2, то возникает оптич. разность хода обоих лучей и интер-
ференционный спектр сдвигается на т полос, причем Дп =
— rnML, где Дп = п2 — щ, X — длина волны и L — длина
кюветы. Смещение находят сравнением с несмещенным спект-
ром, образованным интерференцией обоих лучей, прошедших
мимо кюветы (Дп = 0). Этот спектр сравнения подводят
вплотную к измеряемому с помощью призмы 9. Для измерения
смещения т служит компенсатор Жамена из двух одинаковых
стеклянных пластинок 6 и 7 на пути обоих лучей. Одна из
них вращается вокруг горизонтальной оси с помощью рычага
и винта 8 со шкалой, так что один луч проходит в пластинке
слой переменной толщины. Вращением винта компенсируют
оптич. разность хода, вызванную разными показателями пре-
ломления в 4 и 5 и возвращают верхний спектр к совпадению
со спектром сравнения. Шкалу винта калибруют в числах
полос или непосредственно в единицах концентрации иссле-
дуемого компонента.
5
Рис. 1. Схема интерферометра Габера—Леве
(фирмы Цейсс): А — вид сбоку, Б — вид
сверху; 1 — коллиматор с входной диафраг-
мой 2 и двойной щелью 3; 4 и 5 — двойная кю-
вета; 6 и 7 — стеклянные пластинки компен-
сатора; 8— винт с барабаном и шкалой отсчета;
9 — призма для проектирования спектра
сравнения; 10 — окуляр с цилиндрической
линзой 11.
При измерениях в белом свете черные полосы сопро-
вождаются цветными каемками (рис. 2, .4), образован-
ными наложением интерференционных полос от раз-
ных длин волн (ширина полосы пропорциональна X).
Средняя нулевая полоса 7, служащая началом от-
счета, отличается отсутствием таких каемок. Для
точных измерений пользуются монохроматич. светом,
при к-ром спектр состоит из гораздо более тонких и
резких черных полос без цветного окаймления
(рис. 2, Б). В этом случае нулевая полоса пе отличима
от остальных, но ее легко найти переключением на
281
ИНТЕРФЕРОМЕТРИЯ — ИНУЛИН
282
белый свет. Из-за разной дисперсии стекла компенса-
тора и измеряемого вещества, через одинаковые про-
межутки (в обычных приборах: порядка 10 полос) воз-
Рис. 2. Интерференционные спектры: А — интерфе-
ренционный спектр в белом свете; верхний — из-
меряемый, нижний — неподвижный спектр сравне-
ния. Цвета каемок обозначены разной штриховкой.
Верхний спектр смещен относительно нижнего на
1—2 полосы (см. полосу 1—1); Б — тот же спектр
в монохроматическом свете.
пикают сдвиги на 1 целую полосу. В местах сдвигов
две соседние полосы имеют одинаковое цветное окай-
мление и неотличимы. Если измеряемые Дге больше
интервала до первого сдвига и если требуемая точ-
ность не допускает ошибки в 1 полосу, то места сдви-
гов должны быть заранее найдены, для чего служат
разные несложные способы, и нужно к отсчету добав-
лять число целых полос, отвечающих числу сдвигов.
И. имеет перед рефрактометрией преимущество уве-
личенной на несколько порядков чувствительности,
что позволяет анализировать газовые смеси и сильно
разб. р-ры. По разным практич. соображениям обычно
ограничивают длины кювет 8 см для жидкостей и
100 см для газов. Т. к. можно измерять смещения до
1/во полосы, то с такими кюветами можно найти раз-
ность Дге до 10-7 единиц для жидкостей и 10~8 единиц
для газов. Это отвечает, напр., изменению концентра-
ции водных р-рон солей на 10-в% несдержанна в возду-
хе влаги на 0,03% или СО2 на 0,008%. Лучшие рефрак-
тометры имеют предел чувствительности 10~Б единиц
для Дге, но их относительная точность несколько выше,
чем у интерферометров. При анализе смесей несколь-
ких компонентов нужно измерять Аге до и после извле-
чения отдельных составных частей или комбиниро-
вать И. с другими физич. измерениями (плотности,
вязкости и др.). Для смеси из к компонентов нужно,
очевидно, к — 1 независимых измерений.
В портативных лабораторных приборах применяют
аутоколлимацию с помощью зеркальца, отражающего
лучи, так что эффективная длина кювета увеличи-
вается вдвое. Имеются специальные интерферометры
для анализа рудничного газа с вмонтированными в них
поглотителями влаги и СО2. Сравнение рудничного
газа с наружным воздухом дает непосредственно по
отсчетам шкалы содержание СН4. И. газов приме-
няется также для определения О2 и СО2 в дымовых
газах, влаги и СО2 в воздухе, паров бензола в коксо-
вом газе, содержания примесей в электролитич. Н2,
испытания чистоты баллонных газов, контроля извле-
чения газов нулевой группы из воздуха, контроля раз-
ных промышленных газов и др. Жидкостная И. при-
меняется для контроля качества водопроводной воды,
определения солености морских вод, проверки титров,
определения примесей к органич. жидкостям, измере-
ния растворимости малорастворимых веществ и' др.
В физико-химич. исследованиях И. применяют
для измерения летучести и давления пара, адсорбции,
скорости диффузии, в химич. кинетике, для измере-
ния рефракции в сильноразбавленных растворах
и др. И. служит также удобным и быстрым методом
определения содержания дейтерия в воде (можно
найти до 0,002% D).
Лит.: Физические методы органической химии, под ред.
А. Вайсбергера, пер. с англ., т. 1, М., 1950, гл. 14; Бро д-
с к и й А. И., в кн.: Труды Всесоюзной конференции по ана-
литической химии, т. 1, М., 1939, 115. А. И. Бродский.
ИНУЛИН — запасный полисахарид растений се-
мейства сложноцветных и колокольчиковых, построен-
ный из остатков D-фруктозы (ок. 96%), связанных
Р-2,1-связями, и D-глюкозы (ок. 6%). Молекула И.
представляет собой неразветвленную цепь, содержа-
щую ок. 35—42 D-фруктофурапозидных остатков; вос-
станавливающий конец этой цепи связан с D-глюкозой
за счет ее полуацетального гидроксила.
Препараты И. различного происхождения неодно-
родны, они содержат ряд полимергомологов со сред-
ним мол. в. 5000—6000. И. нек-рых растений (напр.,
георгин) может содержать 2—3 остатка D-глюкозы во
фруктофуранозиднои цепи. В растениях И. сопровож-
дают полимеры D-фруктозы аналогичного строения,
но с более низким мол. весом — инулиды.
И. — бесцветные двоякопреломляющие кристаллы
(P-И.) или белый аморфный порошок (а-И). И., по-
лученный кристаллизацией из воды, плохо раство-
рим в холодной и хорошо — в горячей воде; И.,
осажденный из водных р-ров спиртом, растворим
в воде лучше. Растворимые в воде формы И. получают,
обрабатывая И. окисью углерода или ацетамидом.
Растворы И. обладают значительной вязкостью;
[a]D = —40°. И. осаждается Са(ОН)2 или Ва(ОН)а
н виде комплексов.
И. — невосстанавливающий полисахарид, иодом не
окрашивается; легко гидролизуется при нагревании
с водой, а также неорганич. и органич. (уксусной,
винной) к-тами. При гидролизе к-тами из И. обра-
зуется D-фруктоза, D-глюкоза и ди-О-фруктозоангид-
риды. При неполном гидролизе И. образуется дисаха-
рид ин ул ибиоза (l-P-D-фруктофуранозил-фрук-
тоза). Гидролиз И. может идти под влиянием фер-
ментов Р-фруктофуранозидаз, напр. инвертазы дрож-
жей и нек-рых плесневых грибов. Специфич. фер-
менты, гидролизующие И., наз. инулазами
(и н у л и н азами). Фермент инулосах а р а за
принимает участие как в гидролизе, так и в биосин-
тезе И.
И. получают из корней инулиноносных растений
(топинамбура, или земляной груши, георгинов, цико-
рия, кок-сагыза и др.) извлечением водой с последую-
щим вымораживанием или осаждением спиртом.
И. легко усваивается человеком и животными; многие
инулиноносные растения используются как кормо-
вые культуры, в бродильной и пищевой пром-сти.
И. применяется при клинич. исследованиях деятель-
283
ИНФРАКРАСНАЯ СПЕКТРОСКОПИЯ
284
ности почек; может быть использован для произ-ва
фруктозы.
Лит.: Роминський!. Р., Фруктоза та 1иул1Н, Ктв,
1 959; Whistler R. L., Smart С. L., Polysaccharide
chemistry, N. Y., 1953; Handbuch der Ptlanzenphysiologie,
Bd 6, B., 1958;Colloque sur la biochimie des glucides, [Actes.P.l,
1961; Clinical physiology. Ed. E. J. Moran Campbell, C. J.
Dickinson, Oxf., 1960. Л. И. Линееич.
ИНФРАКРАСНАЯ СПЕКТРОСКОПИЯ — раздел
спектроскопии, охватывающий длинноволновую об-
ласть спектра, границы которой условны. Она начи-
нается сразу же за красным концом видимого спектра
и далеко вклинивается в микроволновую область, где
ее граница в настоящее время находится ок. X =
= 2,5 мм.
Методы И. с., главным образом абсорбционной,
применяют для изучения и практического использо-
вания вращательно-колебательного движения сво-
бодных или взаимодействующих молекул (сжатые
газы или конденсированная фаза вещества); харак-
тер движения определяется строением и состоянием
молекул.
Спектр поглощения вещества характеризуется рас-
пределением интенсивности в зависимости от длины
волны к или волнового числа v =-^(^вак дается в см),
а при наличии выраженных пиков—их интенсивностью.
Если имеются изолированные линии или полосы, то
для характеристики спектра используется также ин-
тегральная интенсивность всей полосы и ее полуши-
рина (ширина на половине высоты). Понятие интен-
сивности имеет прямой смысл величины потока излу-
чения только для спектров испускания. В поглощении
интенсивностью принято называть оптическую плот-
ность Dу поглощающего слоя, связанную с величиной
падающего потока радиации j0 и прошедшего J (ири
учете потерь на отражение) уравнением:
J = J^exp (— Л v) (1)
В отличие от интенсивности при испускании, опти-
ческая плотность дает ее абсолютную меру, не зави-
сящую от воздействия излучения на вещество. И К-
сцектры поглощения вещества в молекулярной форме
состоят из более или менее широких полос, имеющих
правильное строение и частично налагающихся друг
на друга. Только в случае сравнительно легких сво-
бодных молекул (в газах) они состоят из большого
числа тонких линий (вращательное строение колеба-
тельных полос). В твердых и жидких телах полосы
имеют более простую форму. Их полуширина обычно
составляет от нескольких десятых см-1 до двух-
трех десятков. Лишь в случае сильной водородной
связи она может достигать нескольких десятков и
даже сотен см~г. В этих случаях полоса обычно не
является простой. Истинные значения интенсивности
и полуширины полосы могут сильно искажаться под
влиянием различных инструментальных факторов,
учет которых необходим при документации спектров.
Для характеристики вещества по спектру погло-
щения недостаточно знания величины оптич. плот-
ности, т. к. эта величина является сложной и зависит
не только от коэфф, поглощения Ку (при данном зна-
чении м), но и от толщины поглощающего слоя d и
концентрации вещества С в образце: Dv = KyCd.
На основании закона Бугера — Бэра (см. Погло-
щение света): Jv = JOvexp (KvCd) можно по экспери-
ментально найденным значениям Jv и JOv определить
любую из трех величин, стоящих в показателе сте-
пени, если две другие известны.
Строение молекул, характер их движения, взаимо-
действие между ними, в частности образование более
или менее устойчивых ассоциатов, находят отражение
ц спектральном распределении Kv и могут быть изу-
чены с помощью ИК-спектров. По числу и положе-
нию пиков на спектрограмме можно судить о природе
вещества (качественный анализ), а по интенсивности
полос „ о его количестве (количественный анализ).
Эффективность И. с. определяется следующими осо-
бенностями: 1) Универсальностью; круг органич. со-
единений, для к-рых применимы И. с., практически
неограничен; он включает даже такие трудные объ-
екты, как каменные угли и смолы. С водными р-рами
работа практически невозможна вследствие сильного
поглощения самой воды в очень широкой области.
2) Характеристичностью ИК-спектров, в особенности
в средней части (от 1500 саг1 до нескольких сот ел-1),
служащей индивидуальной характеристикой веще-
ства. Отдельные узкие части спектра в более длинно-
волновой области характерны для различных струк-
турных элементов молекулы (отдельных групп ато-
мов или кратных связей), часто определяющих их
химич еские свойства. 3) Избирательностью. Пользуясь
ИК-сп ектрами, имеющими узкие полосы, можно
легко отличать спектры близких по строению моле-
кул, иногда не различимых по другим физико-хими-
ческим свойствам. 4) Большой чувствительностью;
ничтожные примеси, не улавливаемые другими ме-
тодами, можно обнаружить И. с. Чувствительность
этого метода во много раз выше Л чем в спектрах
комбинационного рассеяния света; она на порядок
меньше, чем в случае электронных спектров, но более
избирательна. 5) Как и в случае комбинационного
рассеяния света сравнительно большой простотой ин-
терпретации. Колебательные спектры во многих слу-
чаях хорошо изучены и даже могут быть рассчитаны
и предсказаны. ИК-спектры характеристичны для
того состояния вещества, в к-ром они изучаются также
и другими методами, напр. физико-химическими.
В этом смысле они выгодно отличаются от УФ-спект-
ров, при образовании к-рых молекулы переходят в воз-
бужденное, часто более активное состояние.
Необходимый для измерений узкий участок ПК-из-
лучения может быть выделен с помощью более или
менее узкополосных фильтров. Обычно же для этой
цели применяют спектрофотометры, основной частью
которых является монохроматор. Для работы в ИК-
области пользуются призмами из различных материа-
лов, в зависимости от используемого участка спектра.
Вести работу выгоднее недалеко от длинноволновой
границы поглощения материала призмы. В таблице
приведены данные, характеризующие наиболее вы-
годный участок спектра длн различных призм.
Материал призмы
Область пропуска-
ния, мк
Стекло .........................
Кварц...........................
Фторид лития (L1F)..............
Флуорит (CaF2)..................
Каменная соль (NaCl)............
Бромид калия (КВг)..............
Иодид цезия (CsJ)...............
от видимой до 2,5
от видимой до 3,5
2,5-6,0
3,0—8,0
6—15
10-25
.25—50
Более совершенным пнструментом является дифрак-
ционная решетка. Она требует предварительной грубой
монохроматизации, но превосходит призму по разре-
шающей силе, свободна от вредного поглощения при-
месей в материале призмы и более удобна в работе.
Лит.: Чулановский В. М., Введение в молекуляр-
ный спектральный анализ, 2 изд., М.—Л., 1951; Леконт Ж.,
Инфракрасное излучение, пер. с франц., М., 1958; Бел-
лами А., Инфракрасные спектры молекул, пер. с англ., М.,
1957; Герцберг Г., Колебательные и вращательные спектры
многоатомных молекул, пер. с англ., М., 1949; Кросс А.,
Введение в практическую инфракрасную спектроскопию,
пер. с англ., М., 1961; К б s s 1 е г I., Methoden der Infrarot-
Spektroskopie in der chemischen Analyse, Lpz., 1961; L a w-
son К, E., Infrared absorption of inorganic substances, N. Y.,
1961; Ullmann, 3 Anti., Bd 2/1, Milneh. — B„ 1961 (An-
wendung physikalischer und physikalisch-ehemiseher Methoden
im Laboratorium). В. M. Чулановский.
285
ИОД
286
ИОД (Jodum) J — химич. элемент VII гр. периодич.
системы Менделеева; относится к галогенам; п. н.
53, ат. в. 126,9044. Природный И. состоит из одного
стабильного изотопа с м. ч. 127. Из искусственно
радиоактивных изотопов И. важнейшие I131 (Т =
=8 дням), J133 (Г1/2 =22 часам) и др. Конфигурация
внешних электронов атома 5s25/>5. Энергия ионизации
(в эв): J°—»J+—»J2+ соответственно равна 10,45 и 19,01;
сродство к электрону J°->J" 3,28 эв. Атомный радиус
1,36 А, ионный радиус I- 2,20 А. Молекула И. двух-
атомна. Теплота диссоциации J2—-2J 35,5 ккал/молъ,
степень диссоциации 2,8% (при 1000° К), 89,5% (при
2000° К). И. открыт в 1811 Б. Куртуа. Название эле-
мента связано с цветом его паров (от греч. loeifirjs —
фиолетовый). Содержание И. в земной коре 3 • 10~5
вес.%. Рассеянный И. выщелачивается водами из маг-
матич. горных пород и концентрируется организмами,
напр. водорослями. Промышленные количества И.
встречаются в водах нефтяных месторождений и селит-
репных отложениях.
Физические и химические свойства. И. — кристаллы
черно-серого цвета с фиолетовым металлич. блеском.
Имеет ромбич. решетку с периодами а = 7,250 А,
b = 9,772 А, с = 4,774 А; плотность кристаллич.
И. 4,940 (20°), жидкого 3,960 (120°). Т. пл. 113,5°,
т. кип. 184,35°. При обычной темп-ре И. испаряется
с образованием фиолетовых паров, обладающих рез-
ким запахом. Теплота плавления И. 14,85 кал/г;
теплота сублимации 56,94 кал/г (113,6°); теплота
испарения жидкого И. 39,28 кал/г; гкрит 553°, ркрит
116 атм. Давление пара И. (мм. рт. ст.): 0,31 (25°),
90,5 (113,8°). Уд. теплоемкость (кал/г • град) твердого
И. 0,05058 + 4,688 • 10-5 г (25—113,6°), жидкого
0,0756 (113,6—184°). Вязкость жидкого И. (спуаа):
2,268 (116°), 1,414 (185°). Диэлектрич. проницаемость
твердого И. 10,3 (23°), жидкого 11,08 (118°). Уд.
электропроводность (oat-1 • cat1): твердого И. 1,7 • 10-’
(25°), жидкого 4,48 • 10~6 (138,2°J. И. растворим в боль-
шинстве органич. растворителей. Растворы И. в угле-
водородах, их хлористых и бромистых производных,
нитросоединениях и сероуглероде окрашены в фиоле-
товый цвет, а в органич. растворителях, содержащих
кислород, азот, серу и иод, — в коричневый. Раство-
римость И. в воде (г/л): 0,1620 (0°), 0,3395 (25°) и
0,9566 (60°).
Главные валентности И.: —1 (иодиды), +5 (йодаты)
и +7 (перйодаты). Известны также соединения с ва-
лентностью -4-1 (гипоиодиты) и 4-3. По реакционной
способности И. уступает хлору и брому. Со многими
элементами И. непосредственно не взаимодействует
(углерод, азот, кислород, сера, селен), с нек-рыми
вступает в реакции только при повышенных темп-рах
(водород, кремний и многие металлы). Из неметаллов
И. легко реагирует с фосфором, мышьяком, фтором,
хлором, бромом. С металлами энергично соединяется
в присутствии влаги (в отсутствии влаги металлы
при обычной темп-ре реагируют лишь с поверхности).
В водных р-рах И. частично гидролизуется: J2 4-
4- H/feHJO +• J- 4. Н+; с водными р-рами щелочей
(и карбонатов) идет реакция: 3J2 4- 6NaOH = 5NaJ 4-
4- NaJO3 4- ЗН2О. И. легко выделяется в свободном
состоянии из растворов его солей даже при действии
слабых окислителей, напр. солей Fe (III) и Си (II).
Сильные окислители, напр. хлор и гипохлорит, окис-
ляют в водных р-рах И. до йодноватой к-ты HJO3.
Элементарный И. легко восстанавливается сероводо-
родом, тиосульфатом, сульфитом и др. восстановите-
лями до J-. С водным р-ром аммиака И. образует
взрывчатый иодистый азот NJ3.
Соединения И. с одно-и двухвалентными металлами
представляют собой типичные соли. Все иодиды,
кроме AgJ, Cu2J2 и Hg2J2, хорошо растворимы в воде.
Многие иодиды растворимы в полярных органич. рас-
творителях (спирты, кетоны, эфиры). Иодиды метал-
лов III, IV, V и VI групп, так же как и иодиды не-
металлов (Si, Р, As, Sb), легкоплавки и растворимы
даже в неполярных растворителях. В соединениях
с И. многие металлы не проявляют своей высшей
валентности. Для многих иодидов характерно образо-
вание комплексных ионов (напр., HgJ|~, PbJ| ).
Пятиокись иода J2O6 получается при обез-
воживании йодноватой к-ты; представляет собой бе-
лые кристаллы, разлагающиеся при 300°. Другие
окислы иода малостойки. Иодноватистая
кислота HJO может существовать только в очень
разб. р-рах. Она диссоциирует как кислота HJO^H+4-
4- JO- и как основание JOH=J> 4- ОН-. Соли ее
известны только в растворах. Производные JOH
относительно устойчивы, в особенности при образо-
вании комплексов с пиридином [напр., JCN, JCNS,
J(C6H6N) NO3 и др ]. Кислота, соответствующая трех-
валентному И.—HJO2, и ее соли неизвестны. Не суще-
ствует в свободном состоянии и гидроокись трехвалент-
ного И. J(OH)3, но известны соли, являющиеся ее
производными (нитрат, перхлорат, фосфат, ацетат,
сульфат). Йодноватая кислота HJO3 по-
лучается в водных р-рах при окислении И. хлором,
конц. азотной к-той и другими сильными окислите-
лями или же по обменной реакции между иодновато-
кислым барием и серной к-той. После отгонки воды
в вакууме она может быть получена в виде стекловид-
ных кристаллов, плавящихся при 110°. Соли йодно-
ватой к-ты — йодаты — весьма устойчивы и раз-
лагаются только выше 400°; сильные окислители.
При взаимодействии с иодидами в присутствии кислоты
йодаты выделяют элементарный И.:
NaJOs 4- 5NaJ 4- 3H2SO4 = 3J2 4- 3Na»SOt 4- 3H2O.
Йодноватокислый калий KJO3 — кри-
сталлы, растворимые в воде (8,13 г на 100 г Н2О при
20°). Получается при взаимодействии И. с гидрооки-
сью калия или при окислении И. перманганатом или
хлоратом калия, используется как окислитель при
химич. анализах. Иодная кислота HJO4
известна в виде гидрата HJO4-2H2O — кристаллы,
т. пл. 122°; выше этой темп-ры разлагаются с выделе-
нием кислорода. Иодная к-та, в отличие от хлорной,
является слабой кислотой. Образует наряду с нормаль-
ными солями MeJO4 (метаперйодаты) также Me3JO3
(ортопериодаты) и Me3H2JOe (парапериодаты). Пара-
периодат калия K3H2JOe получается при окислении
II. в щелочном р-ре хлором или бромом. Применяется
в аналитич. химии как окислитель. И. образует сое-
динения со всеми остальными галогенами: с фтором —
JF6 и JF7, с хлором—JC1 и JC13 и с бромом — JBr. При
растворении И. в растворах иодистых солей обра-
зуются полииодиды MeJs, MeJ6 и т. п.
Аналитическое определение. Для
качественного обнаружения И. раствор подкис-
ляют серной к-той, выделяют элементарный И. нитри-
том и экстрагируют органич. растворителем, напр.
хлороформом. В присутствии И. хлороформенный
слой окрашивается в розово-фиолетовый цвет. Коли-
чественное определение элементарного И. основано на
реакции его с тиосульфатом. При отсутствии других
галогенов иод-ион определяют титрованием AgNO3.
Для количественного определения И. в присутствии
хлоридов или бромидов используют окисление J~ до
J° нитритом или другим окислителем и экстракцию
органич. растворителем; по другому методу иодид
окисляют гипохлоритом или гипобромитом до йодата
и после разрушения избытка окислителя добавляют
иодистый калий и кислоту и титруют выделившийся
элементарный И. тиосульфатом (см. также Галогенов
определение).
287
ИОДАЛЛИЛУРОТРОПИН - ИОДБЕНЗОЛ
288
Получение. И. добывают из вод нефтяных место-
рождений, морских водорослей и маточников селит-
ренного произ-ва. Воду, содержащую 0,001—0,01%
И. в виде иодидов, подкисляют серной к-той до pH
2,5—3,5 и обрабатывают хлором или раствором нит-
рита натрия для выделения элементарного И., к-рый
затем адсорбируют активным углем или выдувают
воздухом. Насыщенный И. уголь промывают р-ром
NaOH, причем образуется р-р NaJ 4. NaJO3. Из этого
р-ра при действии серной к-ты и окислителя (хлор,
бертолетова соль и др.) выделяют элементарный И,
По другому способу воздух, содержащий пары И.,
смешивают с сернистым ангидридом. Из образовав-
шейся в присутствии влаги смеси иодистоводородной
и серной к-т выделяют И. действием хлора. Водоросли
сжигают, золу выщелачивают водой и из полученного
р-ра выделяют И. под действием окислителей. Маточ-
ники селитренного произ-ва, содержащие И. в виде
NaJO3, для выделения элементарного И. обрабаты-
вают сернистым ангидридом.
Техника безопасности. И. ядовит, при обращении
с ним соблюдают меры предосторожности. Пары И.
действуют раздражающе на слизистые оболочки. Пре-
дельно допустимая концентрация И. в воздухе
0,001 мг/л. При частом действии И. на кожу возможны
дерматиты. Попавший на кожу И. смывают р-ром
тиосульфата натрия или соды.
Применение. И. и его соединения используют гл.
обр. в медицине и как реактивы в химич. лаборато-
риях. В пром-сти И. применяют незначительно (напр.,
при получении высокочистых металлов термич. раз-
ложением иодидов, как катализатор и др.). Радиоак-
тивные изотопы И. нашли применение в медицине и
биологии гл. обр. при исследовании функций и лече-
нии заболеваний щитовидной железы. При патологич.
повышении функции щитовидной железы лечение
I131 основано на разрушающем действии его р-излу-
чения на секреторные клетки.
Лит.: К с е н з е н к о В. И. иСтасиневичД. С.,
Технология брома и иода, М., 1960; Денисович Б. П.,
Иод и его производство, М., 1938; Орлов И. Е., Методы
анализа рапы, буровых вод и контроль, производства иода
и брома, М., 1939; П о з и н М. Е., Технология минеральных
солей, 2 изд., Л., 1961, гл. VIII; ОшеИп, 8 Autl., Syst.-
Nr. 8. Jod, В., 1931—33, 1954; M e 1 1 о r, v. 2, L —[а. о.], 1946;
то же, V. 2, 1952; Mellor, suppl., v. 2, pt 1, L. — la. 0.1,
1956; Ullmann, 3 Autl., Bd 9, Miln'chen — B., 1957, S.
124—46; Kirk, v. 7, N. Y., 1951, p. 942—72.
Д. С. Стасин свич.
ИОДАЛЛИЛУРОТРОПИН (иодаллилгексаметилен-
тетрамин) C8H1-N4J, мол. вес 308,18 —белые кри-
сталлы, без запаха; т. пл.
/ 2\ 148°; хороню растворим в
\ n воде и спирте, нерастворим
CH2H2CZI- Сн2=Снсн21 в эфире и хлороформе. Го-
товят И. смешением экви-
молекулярных количеств
^2/ 2 йодистого аллила и гекса-
N метилентетрамина в среде
СНС13. Применяют И. как реагент на кадмий (обра-
зуется белый осадок), при определении показателя
преломления минералов.
Лит.: Welcher F. J., Organic analytical reagents,
V. 3, Toronto — N. Y. — L., 1947, p. 121. И. П. Ефимов.
ИОДАТОМЕТРИЯ — титриметрический метод ко-
личественного анализа, основанный на применении
в качестне рабочего р-ра йодата калия (окислитель)
К1О3. Восстановление до иодида в слабокпслом
р-ре происходит через стадию выделения свобод-
ного иода, к-рый является фактическим окислителем.
Иодат калия применяют для определения восстанови-
телей (двухвалентного олова, аскорбиновой к-ты)
в слабокислом р-ре в присутствии избытка иодида
калия, а также для определения концентрации силь-
ных к-т. Индикатором в этих случаях является крах-
мал. В сильно солянокислых р-рах (не меаее 3 и. НС1)
н2.
иодат калия количественно восстанавливается раз-
личными восстановителями (J3, J~, As3+, N2H4, SOV,
SCN~,T1+) до монохлорида иода. Конец реакции
устанавливают по исчезновению красной окраски
хлороформа, не растворяющего образующийся моно-
хлорид иода при добавлении избытка йодата калия
к р-ру, содержащему иод. И. используется при опре-
делении меди после осаждения ее в виде Cu3(SCN)2,
цинка после осаждения в виде Zn [Hg(SCN)4], а также
нек-рых элементов (Hg, TI, Bi), осаждаемых Рейнеке
солью.
Лит.: Кольтхоф И. М. и Сендэл Е. Б., Ко-
личественный анализ, пер. с англ., 3 изд., М., 1948; Кольт-
г о ф И. М. [н др.], Объемный анализ, пер. с англ., т. 3, М.,
1961. В. Г. Типцова.
ИОДАЦЕТАМИД (амид иодуксусной кислоты)
JCH2CONH2, мол. в. 184,97 — кристаллич. продукт,
т. пл. 95° (из воды или СНС13); хорошо растворим в го-
рячей воде; обладает сильным слезоточивым дейст-
вием. И. получают обработкой хлорацетамида K.J
или NaJ в спирте. Подобно иодуксусной кислоте,
И. является специфич. реагентом на SH-группы, об-
разуя с тиоловыми соединениями S-карбоксиметиль-
ные производные. Наряду с иодуксусной к-той при-
меняется в биохимич. исследованиях в качестве инги-
битора SH-ферментов и для количественного определе-
ния SH-групп в белках.
Лит. см. при ст. Надуксусная кислота. В. Б. Спиричев.
ИОДБЕНЗОЛ — продукт замещения атомов водо-
рода в бензоле атомами иода. Известны все возможные
соединения, образующиеся в результате последова-
тельного замещения, от моноиодбензола CeH6J до
гексаиодбензола C6J6. Практич. интерес представляет
гл. обр. моноиодбензол (далее иодбензол).
Иодбензол C6H6J, мол. в. 204,02 — бесцвет-
ная или слегка желтая жидкость с эфирным запахом;
т. пл.—31,4°; т. кип. 188,6°; 1,832; reg 1,6210;
растворимость в воде 0,3 г/л при 20°: хорошо раство-
ряется в спирте, эфире, хлороформе, бензоле; обладает
слабым наркотич. и местным раздражающим действием.
Для И., как и для всех галогенобензолов, харак-
терны реакции электрофильного замещения водоро-
дов ядра преим. в пара- и орто-положения; при этом
пассивирующее влияние атома иода в реакциях за-
мещения проявляется в меньшей степени, чем в случае
хлора и брома. Так, при нитровании И. азотной к-той
образуется смесь, содержащая 59% ге-нитроиодбен-
зола и 41% о-изомера; относительные скорости нитро-
вания бензола, И., бром- и хлорбензолов составляют
1:0, 18 : 0,030 : 0,033. При сульфировании образуются:
ге-иодбензолсульфокислота, ге-дииодбензол и бензол-
сульфокислота (при использовании серной к-ты),
бис-ге-иодфенилсульфон (при использовании олеума
или хлорсульфоновой к-ты). При галогенировании
образуются ге- и o-иодгалогенобензолы и 3,5-дигало-
гениодбензол.
И. легче, чем другие галогенобензолы, в присутствии
порошкообразной меди и при нагревании арилирует
едкие щелочи, алкоголяты, меркаптиды, соли слабых
к-т (цианиды, феноляты, соли карбоновых к-т), ам-
миак, амины и др., напр.:
C0H5J + KCN-<- CaH3CN + KJ
И., как и другие иодароматич. соединения, харак-
теризуется легкой окисляемостью с образованием
неустойчивых продуктов (см. Галогенониевые соедине-
ния). Так, при действии на И. перекиси водорода или
надсерной к-ты образуется иодозобензол, при дейст-
вии озона или органич. перекисей — иодобензол,
при действии хлора в безводной среде — фенил иодид-
дихлорид:
CeHsJ-
; KasaO8
СНцСОбОН
ci3 ~"
с0н5ю
CoHg 1Оа
С0Нд1С1а
289
ИОДИСТЫЙ ВОДОРОД — ИОДОЗОБЕНЗОЙНЫЕ КИСЛОТЫ
290
И. получают иодированием бензола в присутст-
вии окислителей, необходимых для связывания
йодистого водорода: азотной к-ты (наиболее эффек-
тивно), иодной к-ты и ее ангидрида, дымящей серной
к-ты, треххлористого железа, окиси ртути. Наиболее
чистый И. получают диазотированием анилина и
последующим разложением фенилдиазонийхлорида
в присутствии йодистого калия:
c6h3nh2 c6h5n£ci ->- C8H5J
И. используют как промежуточный препарат в ор-
ганич. синтезе.
Дииодбензол C6H4J2, мол. в. 329,93, суще-
ствует в трех изомерных формах. Все изомеры трудно
растворимы в воде, растворимы в спирте, эфире.
Изомер орто (1,2)|мета (1,3) пара (1,4)
Т. пл., °C 27’ 40е 129,4°
Т. кип., °C 286—287’ 285’ 285’
Теплота плавления, кал/г . . . 10,2 11,55 16,21
Диэлектрин, проницаемость, s. 1,27 1,69 0
о- и .и-Дииодбепзолы получают из соответствующих
диазотированных иоданилипов'разложением их HJ;
n-дииодбензол образуется вместе с И. при кипяче-
нии бензола с иодом и персульфатом натрия в уксус-
ной к-те. Дииодбензолы ограниченно применяют
в органич. синтезе.
Полииодбензолы — бесцветные или слегка
желтые кристаллы с эфирным запахом; хорошо раст-
воряются в большинстве органич. растворителей.
ИОДИСТЫЙ ВОДОРОД HJ — бесцветный1'’удуш-
ливый газ, сильно дымящий на воздухе; т. пл. —50,9°;
т. кип. — 35,4°, %рит 151°, ркрит 82 атм. Давление
пара (в атм) равно: 3,97 (0°), 37,68 (100°). И. в. — тер-
мически нестойкое соединение, при повышенных тем-
пературах диссоциирует на иод и водород, реакция
обратима. Теплота образования газообразного И. в.
Д/7%8 = 6,20 ккал/молъ. Уд. теплоемкость жидкого
И. в. 0,1106 кал/г-град (—37°), газообразного
0,0545 кал/г-град (25°). Молекула И. в. имеет поляр-
ную связь. Растворимость в воде (г HJ на 100 г Н2О
при 760 мм рт. ст.): 234 (10°), 132,5 (127°).
Плотность водных растворов
HJ, 0/100 а раствора 10 20 30 40 50 60 65
1,0751 1,1649 1,2737 1,4029 1,5600 1,7700 1,9010
Раствор НJ в воде — иодистоводородная
кислот а, бесцветная жидкость с резким запахом.
Ее азеотропная смесь содержит 56,9% HJ и кипит
при 127° (при 760 мм рт. ст.). Продажная иодисто-
водородная к-та содержит ок. 47% HJ и имеет плот-
ность 1,5. Иодистоводородная к-та является сильной
к-той. И. в. — сильный восстановитель; окисляется
уже при стоянии на воздухе с выделением элементар-
ного иода. При попадании на кожу вызывает зуд и
воспаление. И. в. получают взаимодействием паров
иода с водородом над катализатором — платиниро-
ванным асбестом при 500°. В лаборатории И. в.
обычно получают взаимодействием Р J3 с водой. Исполь-
зуется в химич. лабораториях как реактив, а также
для приготовления различных иодистых производных.
Лит. см. при ст. Иод, Д, С. Стасиневич.
ИОДИСТЫЙ МЕТИЛ CH3J, мол. в. 141,96 — тяже-
лая, бесцветная, окрашивающаяся иа свету жид-
кость с характерным запахом; т. пл. — 66,45°,
т. кип. 42,6°/760 мм; dwi 2,27899; п), 1,53306; теплота
сгорания 194,7 ккал/молъ (р = const), 193,25 ккал/молъ
(»=const); в 100 г воды растворяется 1,4 г (20°), раство-
рим в этаноле, эфире и СС14; образует постояннокипя-
щую смесь (39°) с СН3ОН (93% И. м.). Получают И. м.
взаимодействием метанола с иодом и красным фосфо-
ром или диметилсульфата с KJ; широко применяется
в органич. синтезе как метилирующий агент; с боль-
шинством третичных азотистых оснований образует
иодметилаты; легко взаимодействует с металлич. маг-
нием, образуя магнийиодметил (реактив Гриньяра, см.
Гриньяра реакция, Активный водород, Алкилирование).
Н. С. Вульфсон.
ИОДИСТЫЙ МЕТИЛЕН СН2К, мол. в. 267,85 —
тяжелая жидкость; т. пл. 6°, т. кип. 181°/760 мм (с
частичным разложением), 66—70° /И—12 мм; dlQ 3,325;
ng 1,7425; в 100 г воды растворяется 1,419 г (20°);
легко растворим в спирте и эфире; теплота сгора-
ния 178,4 ккал/молъ (р = const), 178,1 ккал/молъ
(и = const). Получают И. м. частичным восстановле-
нием йодоформа HJ или мышьяковистокислым нат-
рием, напр.: СН J3 -ф- Na3AsO3 -ф- NaOH — СН2 J2 -ф-
+ NaJ -ф- Na3AsO4; известны и другие способы получе-
ния. С магнием И. м. образует CH2(MgJ)2, с ртутью
JCH2HgJ и CH2(HgJ)2; при фотохимия, окислении —
СН2О, НСООН, СО и .Т2О5; с малоновым эфиром и
избытком этилата натрия образует метиленмалоновый
•эфир (С2Н5ООС)2С=СН2. Применяют И. м. для раз-
деления минералов по плотности и для различных син-
тетич. целей. н. с. Вульфсон.
ИОДИСТЫЙ ЭТИЛ (иодэтан) C2H5J, мол. вес
155,97 — тяжелая, бесцветная, окрашивающаяся на
свету жидкость с характерным запахом; т. пл. —111,1°,
т. кип. 72,30°/760 мм, d? 1,93706; ng 1,5133; в 100 г
воды растворяется 0,403 г (20°); теплота сгорания
356,0 ккал/молъ (p=const); 355,4 ккал/молъ (v — const).
Получают И. э. медленной перегонкой спирта с HJ;
взаимодействием этанола с иодом и красным фосфором
или кипячением этилового эфира п-толуолсульфокис-
лоты с раствором KJ. Применяют И. э. в органич.
синтезе в качестве этилирующего средства; взаимо-
действуя с третичными азотистыми основаниями, дает
иодэтилаты; легко взаимодействует с металлич. маг-
нием, образуя магнийиодэтил (см. Гриньяра реакция,
Алкилирование). н. С. Вульфсон.
ИОДНОЕ ЧИСЛО — количество иода в граммах,
присоединяющегося к 100 г органич. вещества. И. ч.
характеризует содержание в непредельном соедине-
нии двойных связей, по к-рым присоединяется иод.
При определении И. ч. непосредственно измеряют ко-
личество присоединяющегося иода, а иногда устана-
вливают количество присоединяющегося брома и
вычисляют эквивалентное ему количество иода. И. ч.
широко пользуются гл. обр. при анализе жиров, жир-
ных к-т и при определении содержания реагирующих
с иодом примесей в ароматич. углеводородах.
Лит. см. при ст. Бромное число. А. А. Черкасский.
ИОДОГНОСТ — см. Сулъфопиридааин.
ИОДОЗОБЕНЗОЙНЫЕ КИСЛОТЫ OJC6H4COOH,
мол. в. 264,03 — кристаллы; известны 3 изомера И. к.:
орто, мета и пара.
о-И одозобензойная кислота кристал-
лизуется из воды в виде пластинок, т. пл. 223—225°
(с разл.); плохо растворима в холодной воде и эфире,
растворима в щелочах; константа диссоциации К —
= 6-10“7. При действии SO2 или KJ в кислой среде
легко восстанавливается до о-иодбензойной к-ты.
Количественно о-И. к. определяют иодометрич. ме-
тодом, основанным на определении J2, образующегося
в стехиометрия, соотношении при взаимодействии
о-И. к. с К J. При pH 7,0 о-И. к. легко окисляет
SH-группы цистеина и белков, превращая их в дисуль-
фидные группировки:
2RSH + OJC6H4COOH-.RSSR + JCeH4COOH % Н2О
В связи с этим о-И. к. подавляет активность многих
ферментов, содержащих свободные SH-группы (алко- •
291
ИОДОЗОБЕНЗОЛ — ЙОДОФОРМ
292
гольдегидрогеназу, дегидрогеназу 3-фосфоглицерино-
вого альдегида, мышечную фосфорилазу и др.).
о-И. к. получают окислением о-иодбензойной к-ты
КМпО4 в кислой среде, окислением иодбензойной
к-ты дымящей HNO3 или обработкой иодбензойной
к-ты газообразным С12 в уксусной к-те с последующим
щелочным омылением образующегося хлорида. о-И. к.
применяют в биохимич. исследованиях в качестве спе-
цифич.ингибитора SH-ферментов, а также для количе-
ственного определения свободных SH-групп в белках.
м-И одозобензойная кислота, т. пл.
185°, обладает интенсивным запахом, нерастворима
в СНС13 и бензоле, слабо растворима в спирте и эфире.
n-И одозобензойная кислота, т. пл.
265—270° (из разб. спирта); обладает интенсивным
запахом; нерастворима в СНС13, бензоле и спирте.
Химич, свойства и способы получения м-И. к. и n-И. к.
сходны со свойствами и способами получения о-И. к.
Лит. см. при ст. Иодуксусная кислота. В. Б. Спиричев.
ИОДОЗОБЕНЗОЛ С6Н5JO, мол. в. 220,02 — белые
кристаллы; т. пл. 210° (со взрывом); плохо растворим
в холодной воде (<1 г/л),.в эфире, бензоле, ацетоне,
хлороформе; лучше растворяется в горячей воде;
хорошо — в спирте; ядовит. При перегонке И. с водя-
ным паром или при долговременном хранении раз-
лагается с образованием смеси иодбензола и иодобен-
зола:
2CtH5JO -► с6н5.т + CcH5JO2
И. обладает основными свойствами и образует
соли уже при растворении в разб. кислотах, напр.
фенилиодидхромат C6H5JCrO4 — желтые кристаллы,
разлагающиеся при нагревании; фенилиодид-
дихлорид C6H5JC12— желтые кристаллы с т.
пл. 120—121° (с разл.), плохо растворимые в воде,
эфире, сероуглероде и хорошо — в бензоле, хлоро-
форме, ледяной уксусной к-те.
И. при окислении в кислой среде (хлорноватистой
или надсерной к-тами) превращается в иодобензол:
c6h5jo + нею -> CeH5JO2 + на
Иодобензол — белые прозрачные кристаллы;
т. взрывного разл. 227—228°; растворимость в воде:
0,274% (14°) и 1,173% (99°); плохо растворяется
в эфире и хорошо — в бензоле, ацетоне, хлороформе;
ядовит; сильный окислитель.
При восстановлении И., напр. сернистым ангид-
ридом, образуется иодбензол. При конденсации И.
с ароматич. соединениями металлов, с ароматич. угле-
водородами в присутствии конц. серной к-ты или
иодобензолом в присутствии влажной окиси серебра
образуются производные иодония (см. Г алогеноние-
вые соединения):
Н«О
СвН5.ТО + CeH5MgBr -► (C6H5)2JOH
CcH5.rO 4~ CgHg 4“ H2SO4 —► (CgHcJg JOSO2OH 4~ H2o
G5H5.ro + C6H5JO2 + AgOH -> (C6H5)2JOH + AgJOa
И. образуется при окислении иодбензола озоном,
органич. перекисями или щелочными гипохлоритами,
а также при обработке фенилиодиддихлорида водными
р-рами едких щелочей:
CeHsJCh + 2NaOH -t- C6H5JO 4- 2NaCl + H2O
II. и его производные используются в качестве про-
межуточных препаратов в лабораторной практике.
7 - ИОД - 8 - ОКСИХИНОЛИН 5 - СУЛЬФОКИСЛОТА
(феррон, лоретин) C9HeO4NSJ, мол. в. 351,13 —
желтый мелкокристаллич. порошок без
запаха; в 100 г воды растворяется 0,2 г
(15°), 0,6 г (95°); плохо растворим в спир-
те, нерастворим в эфире, бензоле, хло-
роформе и др. органич. растворителях.
Растворяется в серной к-те. Водные р-ры
окрашены в желтый цвет, с солями трехвалентного
щелеза окрашиваются в зеленый цвет. Получают ре-
актив из калиевой соли 8-оксихиполин-5-сульфокис-
лоты действием KJ, хлорной извести и НС1. При-
меняют И. для фотометрия, определения железа и
фтора; для осаждения Са. Sr, Ba, Th.
Препарат в смеси с NaHCO3 (3 : 1) наз. «ятрен»
и используют в медицине как антисептик. Ятрен
обладает избирательным действием по отношению
к возбудителю амебной дизентерии; его применяют
также как дезинфицирующее средство при ранениях,
ожогах и т. д. И. п. Ефимов.
ИОДОЛИПОЛ — см. Рентгеноконтрастные пре-
параты.
ИОДОМЕТРИЯ — титриметрический метод опре-
деления веществ, обладающих окислительными или
восстановительными свойствами, с использованием
процессов, связанных с превращением свободного
иода J2 в ионы J" или обратно: J2 -f- 2ez42J_, а также
определения кислот на основании реакции: JO ., 4~
5J- 6Н^3.12 4- ЗН2О. Нормальный потенциал
системы J2/2.T (£о = 0,58 в при pH = 0 и 25°)
находится примерно в середине ряда окислительно-
восстановительных потенциалов. В отличие от КМпО4
и К2Сг2О7, иод является сравнительно более слабым
окислителем, но ионы обладают более сильными
восстановительными свойствами, чем Мп21- и Сг3+.
Систему J2/2J используют как для определения вос-
становителей (Ев < -}-0,58 в) с помощью титрования
р-рами J2 (титр к-рых предварительно устанавливают
по тиосульфату), так и окислителей: 1) Избыток KJ и
кислоты 4- определяемый окислитель—►выделение эк-
вивалентного определяемому окислителю количества
иода (J2). 2) Титрование J2 тиосульфатом: J2 4"
4- 2Na2S2O3i7:2NaJ 4- Na2S4O6 (методы, основанные
иа титровании р-ром тиосульфата, иногда называют
тиосульфатометрическими). Точку эквивалентности
при иодометрич. определениях устанавливают по
появлению в конце титрования слабо-желтой окраски
свободного иода или по появлению окрашивания
слоя добавленного хлороформа; чаще для этой цели
применяют 0,1%-ные растворы крахмала; определения
следует выполнять на холоду, т. к. при повышенных
темп-pax возможно частичное улетучивание иода, а
также понижение чувствительности крахмала как
индикатора; pH р-ра не должен превышать 9, т. к.
в сильно щелочных р-рах образуется гипоиодид:
J2 4" 2О1Т~ 72.10 ~ 4~ "Ь Н2О, обладающий более вы-
соким окислительно-восстановительным потенциалом,
чем иод, и способный окислять S2O.5“ до SO^-Ионы Н+,
образующиеся в процессе реакции, связывают с по-
мощью Na2HCO3. Обычно необходимо присутствие
избытка KJ для переведения образующегося J2 в рас-
творимое соединение (KJ 4- а также для
ускорения медленно протекающих окислительно-вос-
становительных процессов. Реакционную смесь перед
титрованием иода необходимо выдерживать в темном
месте 3—5 минут. Иодометрич. методы широко при-
меняются для определения кислот, активного хлора
в хлорной извести, меди в купоросе, Fe3+, AsO;%
SnCl2, металлов в порошках, частично содержащих
окислы (напр., цинковая пыль), нек-рых органич.
соединений и т. д.
Лит.: Гиллебранд В. Ф. [и др.], Практическое ру-
ководство по неорганическому анализу, пер. с англ,, М., 1957;
Бабко А. К. и Пятницкий И. В., Количественный
анализ, М., 1956; Алексеев В. Н., Количественный
анализ, М., 1954; Кольтгоф И. М. [и др.], Объемный
анализ, пер. с англ., т. 3, М.—Л., 1960. Н. А. Канаев.
ЙОДОФОРМ CHJ3, мол. в. 393,78 — желтые гекса-
гональные пластинки (из ацетона) с характерным
неприятным запахом; т. пл. 119°; легко возгоняется,
летуч с водяным паром; растворим в хлороформе,
уксусной к-те. В 100 г растворителя содержится:
в воде 0,01 г (25°); в этаноле 1,5 г (15°); в эфире
13,6 г (25°); нерастворим в бензоле. Получают И. вза-
293
ЙОДОФОРМНАЯ реакция —иодфенол
294
имодействием спирта или ацетона с р-ром гипоиодита
натрия (р-р иода в NaOH или Na2CO3). Реакция обра-
зования И. имеет аналитич. значение для количест-
венного определения спирта, ацетона, других метил-
кетонов, ацетальдегида, молочной к-ты и др. (йодо-
формная реакция). В пром-сти И. получают электро-
лизом спиртового р-ра K.J или NaJ. Применяют
И. в качестве антисептика. Н. с. Вульфсон.
йодоформная реакция — метод качествен-
ного и количественного определения органич. соедине-
ний, содержащих ацетильную группу, связанную с во-
дородом, алкильными или арильными радикалами.
Метод заключается в действии иода и щелочи на ис-
следуемое соединение и идентификации образующе-
гося йодоформа:
CHgCOR + 3J2 + 4NaOH-»
CHgJ-|-RCOONa + 3NaJ + ЗН2О (1)
Кроме ацетальдегида и метилкетонов, И. р. дают
также соединения, содержащие группы, к-рые окис-
ляются или гидролизуются в ацетильную (иодацетиль-
ную) при действии реактива. И. р. положительна,
напр., для этилового спирта и метилкарбинолон (2),
этилацетата (3), дибензоилметана (4):
CHgCHR J2 + Na°H CHgCOR — СН Jg -J- RCOONa (2)
OH
ch8cooc2h3 —^2^-rcH8CHsOH^±N12ScHJ8 (3)
(ceHgCO)2CH2 J2 + Na05 (CgH3C0)2CJ2 —
~* и и* in a
- C6H5COCHJ2 + NaOg CHJg + CgllgCOONa (4)
a, p-Непредельные кетоны и Р-кетоспирты, не со-
держащие ацетильной группы, дают И. р. в результате
гидролиза на- альдегид и метилкетон:
CgHyCH = СНСОСаНд —5 СдН;СН - СН2СОС2Нда
ОН
s± СдН7СНО+СН8СОС2Нд
СН3 И. р. не удается осуществить для
/—метилкетонов с пространственно за-
Н3С~/ VCOC.Hj трудненными радикалами, напр. I,
'=\ а также в тех случаях, когда соеди-
СН3 нение, содержащее СН3СО-группу,
* быстро гидролизуется, образуя веще-
ство, неспособное иодироваться, напр.
сн8сосн2соон сн8соон
При проведении И. р. к щелочному воднодиоксанр-
вому р-ру исследуемого вещества добавляют иод
в р-ре иодида калия. Образующийся йодоформ опре-
деляют либо весовым, либо объемным методом. При-
меняя несколько измененный реактив, состоящий из
цианида калия, иода и водного р-ра аммиака, можно
определять метилкетоны в присутствии метилкарби-
нолов.
Механизм И. р. включает медленное превращение
кетона в енолят-ион (5), иодирование, приводящее
к образованию трииодкетона (6) и гидролиз трииод-
кетона с образованием йодоформа (7):
CHgCOR 4-6Н- -<• [CH2COR]--|-Н2О (5)
[CH»COR]--|-3J2 — JgCCOR+ 2HJ + J- (6)
JgCCOR + он-- CHJg + RCOO- (7)
Найдено, что И. р. дают простые виниловые эфиры
(II), виниллактамы (III), n-бензохиноны (IV), гидро-
хинон, резорцин и нек-рые другие соединения.
О
CH2=CHOAlk CH2=CH-N-C=O r JL r-
сснг)„ 1 J
Л III IV
о
И. p. широко используется для доказательства
строения органич. соединений, а также для количест-
венного определения ацетона (напр., в метаноле) или
винилпирролидона.
И. р. была разработана Либеном в 1870.
Лит.: Шрайнер Р., Фьюсон Р., Систематический
качественный анализ органических соединений, пер. с англ.,
М., 1950, с. 143; Booth Н., Saunders В. С., Chem.
and Ind., 1950, № 51, 824; Шостаковский М. Ф.,
Сидельковская Ф. П., Зеленская М. Г., Изв.
АН СССР. ОХН, 1956, № 5, 615; Gillis В. Т., J. Organ.
Chem., 1959, 24. № 7, 1027. Ю. В. Зейфман.
ИОДУКСУСНАЯ КИСЛОТА JCH2COOH, мол. в.
185,96 — бесцветные ромбич. формы кристаллы (из
воды или петролейного эфира), т. пл. 83°, 2,2694,
rf4130 2,1893; константа диссоциации в воде: К =
= 7,0-10“4 (18°) и 7,4-10“4 (26°); поверхностное на-
тяжение 38,63 дин/см (85°), 33,41 дин/см (130°); рас-
творима в воде и спирте, слабо растворима в эфире.
И. к. сильно раздражает слизистые оболочки, на
коже вызывает сильные ожоги. И. к. легко реагирует
с SH-группами тиоглпколевой к-ты, цистеина, глу-
татиона и белков, образуя алкилированные производ-
ные этих веществ:
RSH + jch2cooh->.rsch2cooh + HJ
С цистеином И. к. образует S-карбоксиметилцистеин.
И. к. необратимо тормозит действие многих ферментов,
содержащих свободные SH-группы (алкогольдегидро-
геназу, дегидрогеназу 3-фосфоглицеринового альде-
гида, креатйнкиназу, мышечную фосфорилазу и др.).
Будучи сильным ингибитором SH-ферментов, И. к.
тормозит процесс гликолиза в мышцах и спиртового
брожения в дрожжах.
И. к. получают действием К J или NaJ на хлор- или
бромуксусную к-ту при 50° в водном р-ре или в аце-
тоне. И. к. применяют в биохимич. исследованиях
в качестве специфич. ингибитора ферментов, содер-
жащих SH-группы, а также для количественного опре-
деления остатков цистеина в белках. В аналогичных
целях применяют также производные И. к.: иод-
ацетамид и этиловый эфир, являющиеся еще более
активными реагентами на SH-группы.
Лит.: Диксон М., Уэбб Э., Ферменты, пер. с англ.,
М., 1961, с. 364; Cole R. D., Stein W. Н., MooreS.,
J. Biol. Chem., 1958, 233, № 6, 1359; Methods of biochemical
analysis, v. 1, N. Y. — L., 1954, p. 1. В. Б. Спиричев.
ИОДФЕНОЛ JCeH4OH, мол. вес 220,02 — иголь-
чатые кристаллы с запахом фенола.
Изомеры И. Т. пл., 'С Т. кнп., °C .ил. df Устойчивость
орто 43” 1867160 1,8757 (80°) разл. ва воздухе
мета 40° — — —
пара 94° — 1,8573 (112°) разл. на воздухе
Все три И. перегоняются с водяным паром; плохо
растворимы в воде; хорошо растворяются в спирте,
эфире, ацетоне, бензоле, сероуглероде; по физиология,
действию подобны фенолу. Все три изомера обладают
кислыми свойствами в большей степени, чем фенол.
Легко ацилируются галогенангидридами минераль-
ных, карбоновых и сульфокислот с образованием соот-
ветствующих сложных эфиров: о-иодфенилбензоат,
т. пл. 34°; .и-иодфенилацетат, т. пл. 38°; бис-л-иод-
фенилкарбонат, т. пл. 193°.
Электрофильное замещение атомов водорода ядра
в И. затруднено по сравнению с фенолом и иодбензо-
лом. При этом ориентирующее влияние фенольного
гидроксила (см. Фенол) преобладает над влиянием
атома иода; так, при нитровании о-И. образуются
4- или 6-нитрозамещенные; при иодировании орто-
и пара-И. — 2,4-дииодфенол. Все три И. арилируют
едкие щелочи и соли слабых к-т (цианиды, соли кар-
боновых к-т, феноляты и др.). При сплавлении с ед-
295
ИОДЦИАН — ИОНИЗАЦИИ ПОТЕЙЦЙА’Д
296
кими щелочами при низкой темп-ре образуются со-
ответствующие диоксибензолы, при высокой темп-ре —
резорцин.
Получают И. сплавлением соответствующих иод-
бензолсульфояатов с твердыми едкими щелочами,
а также диазотированием иоданилинов и последующим
разложением образующихся иодфенилдиазониевых
оснований, п- и о-И. получают иодированием фенола
в водном р-ре (процессы описывают уравнениями
первого порядка: о = к [J+] [СвН5О-]).
И. используют в качестве промежуточных препара-
тов, н частности для приготовления диоксибензолов
и дииодфенолов. 2,5-Дииодфенол является исходным
продуктом для синтеза тетраиодфенолфталеина (кон-
трастное вещество для рентгенология, исследований
мочеполовых органов и при тестировании функций
печени).
Полииодфенолы:
2,4-J,CeHaOH, т. пл. 72°
2,4,6-J'3C0H. ОН, т. пл. 158’
3,5,6-J8CeH2OH, т. пл. 114°
Лит.: Ивгольд К. К., Механизм реакций и строение
органических соединений, пер. с англ., М., 1959, с. 234.
Г. А. Сокольский.
ИОДЦИАН — см. Галогеноцианы.
ИОНИЗАЦИИ ПОТЕНЦИАЛ — наименьший по-
тенциал, необходимый для удаления электрона из
атомной системы (атома, иона, молекулы, радикала).
И. п. относят обычно к основным (низшим) энергетич.
состояниям исходной и конечной систем. И. п. свя-
зан с энергией ионизации (Е) (т. е. с энергией, необ-
ходимой для удаления электрона) соотношением:
Е — el, где 1 — 11. п., е — заряд электрона. И. п.
выражают в вольтах, и он численно равен Е, выражен-
ной в электрон-вольтах.
Наиболее полно изучены И. п. атомов и одноатом-
ных ионов. Химич, элементу с порядковым номером Z
соответствует Z различных значений И. п., т. к. его
атом содержит Z электронов. При этом все И. п.,
кроме 1-го, относятся уже не к нейтральному атому,
а к соответствующему иону (2-й Н.п. — к одно-
зарядному, 3-й — к двухзарядному и т. д.). В связи
с увеличением заряда атомного остатка, получающе-
гося при последовательном удалении электронов,
притяжение электронов возрастает. Поэтому, в част-
ности, каждый последующий И. п. больше предыду-
щего. Кроме того, при переходе от одной электронной
оболочки атома к другой обнаруживаются резкие
скачки в возрастании И. п., т. к. электроны более
близких к ядру оболочек прочнее снязаны с ним и
требуют для удаления из атома большей энергии.
Если допустить (в первом приближении) полное
экранирование той части заряда ядра, к-рая равна
по величине заряду электронов в атомном остатке,
можно представить И. п. выражением: Ц = ie2/2r,
где г — среднее расстояние отрываемого 1-того элек-
трона от ядра, меняющееся скачками при переходе
от одной электронной оболочки к другой. Т. обр.,
измерение всех И. п. атома является важнейшим кри-
терием для установления его электронной конфигу-
рации. Первый И. п. (Zj) атома является количест-
венной характеристикой так называемой Электрополо-
/,в
жительности элемента, играющеп
важнейшую роль в определении
его химич. свойств. Первые И. п.
элементов находятся н периодич.
зависимости от их порядкового
номера Z (см. рис.). Значения И. п.
не
20
10
1
Cs
О ГО 20 30 40 50 60 70 80 Z 90
химич. элементов приведены в статьях об этих элемен-
тах. Первые три И. п. элементов приведены в табл. 1.
В случае молекул, многоатомных ионов и радикалов
определение И. п. осложняется тем, что энергетич.
состояние продукта ионизации обычно не соответ-
ствует его низшему колебательному уровню. В соот-
ветствии с принципом Франка — Кондона (согласно
к-рому электронный переход, изменяя потенциальную
энергию атомной системы, не влияет непосредственно
на положение ядер) удаление электрона из много-
атомной системы приводит к образованию иона
с исходными межъядерными расстояниями, обычно
не соответствующими равновесным для основного
колебательного состояния данного иона. Поэтому
Таблица 1. Потенциалы ионизации элементов (в вольтах)
Z Эле- мент 1-Й 2-й 3-й Z Эле- мент 1-Й 2-Й 3-й Z Эле- мент 1-Й 2-й 3-й
1 н 13,595 28 Nt 7,633 18,15 35,16 55 Cs 3,893 25,1
2 Не 24.581 54,403 29 Cu 7,724 20,29 36,83 56 Ba 5,210 10,001
3 Li 5,390 75,619 122,419 30 Zn 9,391 17.96 39,70 57 La 5,61 11,43 19,17
4 Be 9,320 18,206 153,850 31 Ga 6,00 20,51 30,70 58 Ce 5,60
5 В 8,296 25,149 37,920 32 Ge 7,88 15,93 34,21 59 Pr 5,48
6 С 11,256 24,376 47,871 33 As 9,81 18,63 28,34 60 Nd 5,51
7 N 14,53 29,593 47,426 34 Se 9,75 21,5 32 65 Tb 5,98
8 О 13,614 35,108 54,886 35 Br 11,84 21,6 35,9 68 Er 6,08
9 F 17,418 34,98 62,646 36 Kr 13,996 24,56 36,9 72 Hf 7 14,9
Ю Ne 21,559 41,07 63,5 37 Rb 4,176 27,5 40 73 Ta 7,7 16,2
11 Na 5,138 47,29 71,65 38 Sr 5,692 11,027 74 w 7,98 17,7
12 Mg 7,644 15,031 80,12 39 Y 6,38 12,23 20,5 75 Re 7,87 16,6
13 Al 5,984 18,823 28,44 40 Zr 6,84 13,13 22,98 76 Os 8,7 17
14 Si 8,149 16,34 33,46 41 Nb 6,88 14,32 25,04 77 Ir 9
15 p 10,484 19,72 30,156 42 Mo 7,10 16,15 27,13 78 Pt 9,0 18,56
16 s 10,357 23,4 35,0 43 Tc 7,45 15,26 79 Au 9,22 20,05
17 Cl 13,01 23,80 39,90 44 Ru • 7,364 16,76 28,46 80 Hg 10,43 18,751 34,2
18 Ar 15.755 27,62 40,90 45 Rh 7,46 18.07 31,05 81 TI 6,106 20,42 29,8
19 К 4,339 31,81 4& ' 46 Pd 8,33 19,42 32,92 82 Pb 7,415 15,028 31,93
20 Ca 6,111 11,868 51,21 47 Ag 7,574 21,48 34,82 83 Bl 7,287 16,68 25,56
21 Sc 6,54 12,80 24,75 48 Cd 8,991 16,904 37,47 84 Po 8,43
22 Ti 6,82 13,57 27,47 49 In 5,785 18,86 28,03 86 Rn 10,746
23 V 6,74 14,65 29,31 50 Sn 7,342 14,628 30,49 88 Ra 5,277 10,144
24 Cr 6,764 16,49 30,95 51 Sb 8,639 16,5 25,3 89 Ac 6,9 12,1 20
25 Mn 7,432 15,636 33,69 52 Те 9,01 18,6 31 90 Th 6,95
26 Fe 7,87 16.18 30,643 53 J 10,454 19,09 92 и 6,08
27 Co 7,86 17,05 33,49 54 Xe 12,127 21,2 32,1
297
ИОНИЗАЦИИтПОТЕНЦИАЛ - ИОНИТОВЫЕ ДИАФРАГМЫ
298
для многоатомных систем пользуются понятием т. н.
вертикального И. п., соответствующего разности анер-
гий основного колебательного состояния исходной
частицы и возбужденного колебательного состояния
образующегося иона с исходными межъядерными
расстояниями. Этот И. п. больше обычного (или адиа-
батического) на величину разности между возбужден-
ным и основным колебательными уровнями иона.
Напр., адиабатич. И. п. Н2 равен 15,427 е, тогда как
вертикальный И. п. составляет 16,0 в.
В табл. 2 приведены значения И. п. нек-рых моле-
кул и радикалов.
Таблица 2. Потенциалы ионизации некоторых молекул
и радикалов
Молекулы и радикалы Потенциалы ионизации, в Метод определения
н2 15,427 Спектроскопический
сн3 9,86 -г 0,02 Электронный удар
сн4 13,04 » »
С2Н3 9,45 + 0,05 » »
С2Н4 10,516 ±0,01 Фотоионизация
С2Н5 8,80 -г 0,05 Электронный удар
С2Н9 11,65 » »
9,73 + 0,01 Фотоионизация
11,08 »
9,61 + 0,02
10,63 4- 0,03 »
С,-Ню 9,50 + 0,02 »
С 5 Н 10,55 Электронный удар
с,н3 9,90 » »
С«н6 9,245 + 0,01 Фотоионизация
Нафталин 8,12 + 0,02 »
Дифенил 8,3 + 0,1 Электронный удар
Полиметилен 10,15 Экстраполяция
О_, 12,075 + 0,01 Фотоионизация
он 13,18 + 0,1 Электронный удар
Н2О 12,59 + 0,01 Фотоионизация
со 14,01 + 0,01 »
со2 13,79 + 0,01 »
сн3он 10,85 + 0,02 »
НСОСНз 10,1811 + 0,0007 Спектроскопия
С2Н-,ОН 10,50 + 0,05 Фотоионизация
СД1.ОН 8,50 + 0,01 »
N2 15,60 + 0,01 Электронный удар
NH, 10,15 + 0,01 Фотоионизация
NO 9,25 4-0,02 »
n2h4 9,56 + 0,03 »
f2 15,83 ±0,05 Электронный удар
HF 15,77 + 0,02 » »
Cl2 11,48 + 0,01 Фотоионизация
HC1 12,74 + 0,01 »
J2 9,28 4- 0,02 »
IIJ 10,25 Электронный удар
Br2 10,55 + 0,02 Фотоионизация
HBr 10,62 4- 0,01 »
H2S 10,46 + 0,01 »
SF« 19,3 Электронный удар
И. п. может быть непосредственно измерен при
поипзации ударом бомбардирующей частицы, исходя
из зависимости: Ка = el, где Л'о — мини-
мальная энергия частицы, вызывающей ионизацию,
т — ее масса, М — масса ионизируемой атомной
системы. Если бомбардирующей частицей является
электрон, для к-рого т < М, то К = el. Подвер-
гая атомы, молекулы, радикалы бомбардировке быст-
рыми электронами и измеряя количество образую-
щихся иопов (ионный ток г) при различных энергиях
К (эв) бомбардирующих электронов, можно найти Ка
и, следовательно, I путем экстраполяции кривой
i = / (К) к нулю ( 1—>0). В простейших устройствах
электроны, испускаемые раскаленным катодом, уско-
ряются соответствующим потенциалом и направляют-
ся в ионизационную камеру, содержащую исследуе-
мый газ при давлении 10~6—10”8 мм рт. ст. Обра-
зующиеся ионы удаляются из камеры слабым электрич.
полем, затем ускоряются и направляются в анализа-
тор, где разделяются по массам и затем регистри-
руются. Основной экспериментальной процедурой
определения И. п. является регистрация ионного
тока в зависимости от ускоряющего электроны напря-
жения. При этом получаются т. наз. кривые появле-
ния ионов, по порогу к-рых и определяются И. п.
Необходимо отметить, что абсолютное значение энер-
гии электронов определяется, помимо ускоряющего
потенциала, приложенного извне, неконтролируемым
непосредственно контактным потенциалом между като-
дом и ионизационной камерой. Поэтому во всех слу-
чаях приходится сравнивать измеряемый И. п. с из-
вестным значением И. п. калибровочного газа (обыч-
но благородного газа), вводимого в ионизационную
камеру одновременно с исследуемым. Точность опре-
деления И. п. с помощью электронной бомбардиров-
ки достигает 0,01 эв.
При определении И. п. радикалов последние обычно
получают пиролизом галогенозамещенных молекул,
легко теряющих атом галогена. Пиролиз проводят
в непосредственной близости от области ионизации,
так что радикалы попадают в нее практически без
столкновений. Разработана также методика, позво-
ляющая получать радикалы путем диссоциации моле-
кул, под действием электронного удара, а затем иссле-
довать их, ионизируя полученные радикалы вторым
электронным ударом. В тех случаях, когда по каким-
либо причинам не удается получить «газ», состоящий
из свободных радикалов, для определения их И. п.
пользуются косвенными методами.
В случае ионизации светом (фотоионизации) И. п.
находят по выражению hva = el, где h — постоян-
ная Планка, v0 — граничная частота фотоионизации
(или фотоэлектрич. эффекта). Обычно удается весьма
сильно монохроматизировать ионизирующий луч и
тем самым повысить точность определения И. п.
(до 0,001 эв). При термин, ионизации газа в пламени ис-
х2 / 2лт\3/2/т -r\5/o ' el/kT
пользуют зависимость1-^^р=(-р-) (кУ) е
где х —степень ионизации (отношение концентрации
иопов к общей концентрации частиц, находимое по
электропроводности пламени), р— давление газа, т—
масса электрона, к — постоянная Больцмана, Т —
абсолютная темп-ра. Наиболее точным методом опре-
деления И. п. является спектроскопический, к-рый
дает энергетич- уровни исходного и конечного про-
дуктов ионизации и тем самым энергию ионизации,
а следовательно, И. п. При этом получают значение
адиабатич. И. п. Однако спектроскопия применима
в основном для атомов и простейших молекул, тогда
как для сложных молекул и радикалов в большинстве
случаев она не может дать полезных результатов из-за
трудности расшифровки спектров.
См. также Ионы (в газах).
Лит.: Кондратьев В. Н., Структура атомов и мо-
лекул, 2 изд., М„ 1959; Шпольский Э. В., Атомная
физика, т. 1—2 М.—Л., 1951; Мо ore Ch., Atomic energy
levels, v. 3, Wash., 1958; Энергии разрыва химических связей.
Справочник, под ред. В. Н. Кондратьева, М., 1962.
Е. Л. Фратевич, С. Э. Вайсберг.
ИОНИТОВЫЕ ДИАФРАГМЫ — нерастворимые
полиэлектролиты, изготовляемые в виде пластин и
отличающиеся избирательной иопопроницаемостью.
По типу ионогецных групп различают катионитовые
(кислотные группы) и апионитовые (основные группы)
диафрагмы; по методу изготовления — гомогенные
и гетерогенные И. д. Опущенная в р-р электролита,
находящегося в поле постоянного тока, катионитовая
И. д. проницаема для катионов, а анионитовая —
для анионов р-ра. Селективность, т. е. проницаемость
И. д. только для ионов одного знака заряда, дости-
гает 93—98%, возрастая по мере снижения набухае-
мости диафрагмы. Электрич. сопротивление И. д.
определяется степенью ионизации йоногенных групп
и их концентрацией, а также концентрацией электро-
лита в растворе. Гомогенные И. д. получают поли-
299
ИОНИТОВЫЕ СИТА - ИОНИТЫ
300
конденсацией мономеров, содержащих ионогенные
группы, при режимах, предупреждающих образова-
ние трещин, или прививкой полистирола к пленке
полиэтилена с последующим присоединением ионо-
генных групп к звеньям полистирола. Гетерогенные
И. д. обычно изготовляют смешением тонкоизмельчеп-
ного ионита с эластомером (полиэтилен, полипропи-
лен) или с ненаполненной резиновой смесью. Смесь
вальцуют, каландрируют и прессуют в пластины
(часто для повышения прочности смесь напрессовы-
вают на ткань). Содержание ионита в И. д. достигает
обычно 60—70%. С повышением содержания ионита
возрастают электропроводимость и пабухаемость И. д.,
снижаются селективность и механич. прочность.
Электроиопитовые установки используют для удале-
ния солей из растворов, в том числе из морской воды
или из вод засоленных озер (напр., удаления из них
радиоактивных ионов). Электроионитовое обессоли-
вание целесообразно применять в тех случаях, когда
начальная концентрация солей составляет 3—20 г/л
и более и остаточная концентрация допустима ок.
0,5 г/л (эту часть солей проще удалить па ионито-
вых фильтрах). Для повышения производительности
электроионитовую установку делают многокамерной
и воду пропускают одновременно через несколько
десятков камер. Каждая камера ограничена от сосед-
них катионитовой и аиионитовой диафрагмами. Элек-
троионитовый метод применяется и для очистки ор-
ганич. веществ от минеральных примесей и для полу-
чения кислот или щелочей из их солей.
Лит.: Исследования в области промышленного примене-
ния сорбентов. Сб. ст., под ред. К. В. Чмутова, М., 1961;
Л аскор ин Б. Н., Смирнова Н. М., Г а н т м а н
М. Н., Ионообменные мембраны и их применение, М., 1961;
см. также лит. в ст. Иониты. Е. Б. Тростянская.
ИОНИТОВЫЕ СИТА — иониты с кристаллич. или
сетчатой макромолекулярной структурой, в к-рых
поногенные группы доступны только для ионов опре-
деленного размера. Природные и многие синтетич.
минеральные иониты, напр. алюмосиликаты, слабо
набухают в водпых средах и могут иметь регулярную
кристаллич. структуру, создаваемую ионами кремния
или алюминия (см. Молекулярные сита)- Незакреп-
ленными в кристаллич. решетке являются ионы ще-
лочных или щелочноземельных элементов. Эти ионы
мигрируют в узких каналах кристаллич. решетки и
могут обмениваться с ионами, находящимися в рас-
творе, но только в том случае, если диаметр послед-
них в гидратированном состоянии столь мал, что
позволяет им проникать в каналы кристаллич. ре-
шеток. Этим свойством кристаллич. ионитов поль-
зуются для избирательного извлечения малых ионов
из раствора и «отсеивания» более крупных. Струк-
туру кристаллов синтетич. алюмосиликатов можно
несколько изменять, варьируя этим доступность их
подвижных ионов.
Органич. иониты, даже получаемые методом сополи-
меризации, не имеют жестких регулярных структур.
На-внешней поверхности сферич. гранул находится
лишь ничтожная часть ионогенных групп; доступ-
ность остальных достигается набухаемостью сетча-
того сополимера. Изменяя соотношение основного
мономера и мостикообразующего, можно регулировать
размеры макромолекулярной сетки и проницаемость
ионитовых гранул, облегчая избирательное извлече-
ние ионов из р-ров. Полимерные И. с. применяются
для отделения антибиотиков (стрептомицин, тетра-
циклин, пенициллин и др.) или витаминов от мине-
ральных солей, для разделения на отдельные фракции
полимерных ионов и т. д.
Лит. см. при ст. Иониты, а также: Самсонов Г. В.,
Сорбция и хроматография антибиотиков, М.—Л., 1960.
Е. Б. Тростянская.
ИОНИТЫ — твердые, практически нерастворимые
в воде и органич. растворителях материалы (поли-
электролиты), способные к ионному обмену. И. могут
быть минерального и органич. происхождения, при-
родные и синтетич. Подавляющее большинство И.
представляет собой высокомолекулярные соединения
с сетчатой, или пространственной, структурой.
По типу ионогенных групп в составе И. последние
разделяют на нерастворимые кислоты — катио-
ниты и нерастворимые основания — а н и о н и-
т ы. По степени ионизации И. делят на сильно- и
слабокислотные катиониты и сильно- и слабоосновные
аниониты. К сильнокислотным относят катиониты,
содержащие, напр., сульфогруппы, остатки фосфор-
ной или фосфиновой кислот, к слабокислотным — со-
держащие карбоксильные, сульфгидрильные, окси-
фенильные группы. В сильноосновиых анионитах
имеются группы аммониевых или сульфониевых осно-
ваний, в слабоосновных — аминогруппы различной
степени замещения. И. могут одновременно содержать
различные кислотные или различные основные груп-
пы. Такие И. наз. полифупкциоиальными, в отличие
от монофункциональных, содержащих только один
тип ионогенных групп. Катиониты обладают способ-
ностью к обмену катионов своих ионогенных групп
па натионы растворенных солей или водородные
ионы к-т;
R (SO3H) г- R (SO3) . . . mH+
n 0 m n m
R (SOjJ* . . . mH+ + mNaCl R (SO3)~ . . . mNa+ + mHCl
72 772 71 7)2
Аниониты обменивают анионы своих ионогенных
групп на анионы солей или кислот, находящихся
в растворе:
R„(NrJ0H~) ffl;±Rn(NR3) + . тиОН-
I О4 J Но * /1' °'
Rn(NR3)% . . mOH- -J- mNaCl —-
—- Rn(NR3)% . . mCl~-4-mNaOH
Обменная способность И. определяется встряхива-
нием навески И. с титрованными р-рами к-ты (или
щелочи), а также фильтрованием этого раствора через
слой И., с последующим титрованием отфильтрованной
пробы р-ра щелочью (или к-той). Обменная емкость
И. обычно выражается в миллиграмм-эквивалентах
поглощенного иона на 1 г (или единицу объема)
И. Высокоемкие И. могут поглощать 6—10 мг-экв
иона на 1 г И.
Природные минеральные катиониты принадлежат
к группе алюмосиликатов; цеолиты, глины, глауко-
ниты (железоалюмосиликаты калия) и др. К природ-
ным минеральным анионитам относятся, напри-
мер, апатиты [Ca5(PO4)a]F или гидроксиапатиты
[Са5(РО4)3] ОН, в к-рых фтор- или гидроксил-иопы
могут быть заменены хлор-, сульфат- или фосфат-
ионами. Обменная способность этих И. невелика,
к тому же зерна мало набухают в воде, а пористость
их незначительна, поэтому в обмен вступает лишь
та часть ионогенных групп, к-рая находится на внеш-
ней поверхности частиц. Природные минеральные
И. обменивают ионы в почвах, регулируя подачу
ионов солей к растениям.
К синтетич. минеральным И. относятся слабо-
кислотные алюмосиликаты — пермутиты. Обменная
емкость пермутитов невелика, но в щелочных средам
может достигнуть 1,5—2 мг-экв/г. Фосфат циркония,
гидроокиси циркония, тория, ванадия также являютс;
И. Степень ионизации фосфата циркония изменяете;
в зависимости от среды. При рН7 обменная емкостт
равна 1,35 мг-экв/г, при рН12 — достигает 4,5 мг-экв/г
Указанные гидроокиси в кислых средах являютш
анионитами с обменной способностью до 1,2 мг-экв/г
В щелочных средах они становятся слабокислотным!
катионитами с обменной емкостью ок. 0,75—
0,8 мг-экв/г. Синтетич. минеральные И. набухаю'
в воде, что облегчает участие всех ионогенных труп;
301
иониты
302
зерна в реакциях ионного обмена, однако их низкая
обменная емкость и малая степень ионизации ограни-
чивают области их практич. применения рамками лабо-
раторных исследований и теми случаями, когда обмен
необходимо проводить при повышенных температурах.
Природными И. органич. происхождения являются
гуминовые к-ты почв, принимающие участие в регу-
лировании питания растений, и гуминовые к-ты уг-
лей, особенно бурых. Гуминовые к-ты относятся
к слабокислотным полиэлектролитам. Для усиления
кислотных свойств и обменной емкости угли, измель-
ченные в мелкие зерна, сульфируют в избытке олеума.
Сульфоугли являются наиболее дешевыми полиэлек-
тролитами, содержащими сильно- и слабокислотные
группы, но их обменная емкость в нейтральных сре-
дах невелика, мала их химич. стойкость и механич.
прочность зерен.
Наибольшее значение имеют искусственные поли-
мерные И. (ионообменные смолы), отличающиеся
высокой поглотительной способностью, механич. проч-
ностью и химич. устойчивостью. Полимерные И. пред-
ставляют собой аморфные полимеры с сетчатой струк-
турой. Присутствие в макромолекулах ионогенных
групп придает полимеру гидрофильность. Поскольку
цепочки макромолекул полимера в ионитовых смо-
лах «сшиты» друг с другом в пространственную сетку,
растворитель вызывает только набухание смолы, вели-
чина к-рого определяется структурой полимера, типом
и концентрацией ионогенных групп в нем и составом
раствора электролита.
Синтез полимерных И. производят чаще всего: 1) поли-
конденсацией или полимеризацией мономеров, содержащих
ионогенные группы, с образованием сетчатого полимера;
2) присоединением ионогенных групп к отдельным звеньям
ранее синтезированного сетчатого полимера; 3) присоединением
ионогенных групп к звеньям синтетич. линейного полимера
с одновременным превращением его в сетчатый полимер.
Первоначально полимерные И. получали только по первому
способу, напр. из фенолсульфокислоты, образующей в смеси
с фенолом сетчатый полимер при поликонденсации с формаль-
дегидом:
ОН он
я’ |^j| + + я”СН20 ---—
SO3H
он он он
2,4-Диоксибензойная к-та, оксифеноксиуксусная к-та,
монооксифенилфосфат образуют нерастворимые полимеры при
взаимодействии с формальдегидом; смеси меламина с дициан-
диамидом или с гуанидином также вступают в реакцию с фор-
мальдегидом, образуя аниониты; аниониты получают и при
взаимодействии эпихлоргидрина с полиэтиленполиамипом:-
NH (СН2)2 NH (6н2)2 NH' NH (Н2С)2 (СН2)2
NH - 1- С1СН2СНСН2 +- NH N - ch2-ch-ch2-n
(СН2)2 xoz (СН2)2 (Н2С)2 6н (СН2)2
Куски смол измельчают до требуемых размеров (0,25—1,5 мм).
Более удобно использовать И., куски к-рых имеют сферич.
форму, что достигается использованием метода суспензионной
поликонденсации. И., получаемые поликонденсацией, нан
правило, полифункциональны, сетчатость их структуры трудно
регулировать. К тому же их механич. прочность и химич.
устойчивость недостаточны. Поэтому все И. изготовляют
гл. обр. полимеризацией. Сетчатая структура полимера обра-
зуется при сополимеризации мономерного электролита или
мономера, к н-рому впоследствии можно присоединить ионо-
генные группы, с мономером, имеющим две двойные связи
и выполняющим поэтому функцию мостинообразователя.
Соотношением основного компонента и мостинообразователя
регулируют сетчатость сополимера и его набухаемость. Сопо-
лимеры химически устойчивы и механически достаточно
прочны. Суспензионной полимеризацией получают сополимер
в виде сферич. гранул. Примером сополимеризации моно-
меров, содержащих ионогенныё группы, служат способы
получения слабокислотного катионита сополимеризацией
метакриловой к-ты с дивинилбензолом:
СН3
сн2=с
соон
сн3 сн3
-сн2-с-сн2—сн-сн2-с-
СООН соон
-СН2-СН-
Присоединением ионогенных групп к сополимерам получают
И. разнообразных типов. Для этой цели чаще всего исполь-
зуют сополимеры стирола с дивинилбензолом. Гранулы такого
сополимера сульфируют, получая сульфокислоты:
к ним присоединяют группы фосфинистой или фосфиновой к-ты:
+н,о
-СН2-СН- + PCl3 + AICI- —— -СН2~СН- -------*
PCI,? AlCl®
ОН
Хлорметилируя сополимер хлорметиловым эфиром или смесью
парафор.ма и хлористого водорода, получают промежуточный
продукт, к-рый переводят в монофункциональный И. любой
основности:
или в слабокислотный катионит:
В сополимер стирола и дивинила вводят ионогенные группы,
одновременно превращая его в сетчатый' сополимер.
Присоединяя к звеньям полимеров сульфгидрильные группы,
получают И., способные и к реакциям электронного обмена
(окислительно-восстановительным реакциям); присоединяя од-
новременно кислотные и аминогруппы, придают И. амфотерные
свойства; близким расположением в звеньях полимера ионо-
генпых групп и полярных создают условия для образования
комплексов с поглощаемым ионом:
.Ме2+
-CHs-CH-+aciCH2cooH —*-~-chs-ch-----
СН2
NH2
г сн2соон
ch2-nC
СН2СООН
_£Н2сОО*
чСНаСОО‘
•Me®*
303
Ни]|
304
иониты
Свойства ионитов
Типы ионитов и их наименование Емкость в мг-экб!г Набу- хаемость в воде, % Химич, стойкость и механич. прочность Термо-
Ионогенные группы рк NaOH ♦ НС1 *• Nad * Nad *• стой- кость, °C
Катиониты .
Синтетич. органич. иониты: Сульфоугли, цеокерб £>Он -R СООН, -SO3H — 1,9-2,2 1,3-2,7 25—60 Низкая щелочно- и кислотостойкость, прочность слабая ДО 70
Сульфо-феноло-формальде- • гидные: КУ-1, КУ-5 и др. а> ОН р!<1 РК2 = 2,2 = 10,0 4,0-5,0 1,6-2,8 60-100 Низкая стойкость в окислит, и щелоч- ной среде, удовле- творит. прочность до 80
$о3н
Сульфосополистирольные: КУ-2, СДВ-3, СБС и др. б> рК1 = 1,4 3,0—4,9 2,8-4,75 20—250 Высокая химич. стой- кость и хорошая прочность 120-140
Фосфорсодержащие: РФ (I) ОН -СЕоАк чон РА'1 рк2 рк3 II II II SO 6,8 2,3-3,8 30-40 Низкая стойкость в щелочных и окис- лит. средах, высо- кая прочность 70-80
СФ, КФ (11) Q р-он 'он РК, рА« = 3,4 = 7,1 8,8—10,0 4,0—5,0 20-60 Высокая химич. стой- кость и механич. прочность 130—150
Карбоксилеодержащие: КБ-4, КМ и др. в> (III) -RCOOII PK1 = 6,5 8,5-10,5 0,6-1,0 до 300 Высокая химич. стой- кость, удовлетво- рит. прочность 150-180
КФУ, КР (IV) -^^-осн2соон он 6-8 0,8-1,2 40—60 Разрушается в щелоч- ных р-рах (pH вы- ше 2). высокая проч- ность 150-160
КС (V) Q-COOH рК1 = 5,7 5-6 - ДО 70 Высокая химич. стой- кость и механич. прочность, малая набухаемость 180-200
Аниониты
Синтетич. органич. аниониты Слабоосновные: Амины различной степени замещения 1,6-4,0 0,25 —0,40 20-40 Разрушается в окис- лителях, прочность удовлетворительная 60—90
ТМ, Вофатиты М и др. г) Среднеосновные: ММГ, ЭДЭ-10 11 и др. Я) То я е 4,2-9,5 1,5-2,2 40-80 То же 60—90
Сильноосновные монофунк- циональные: -R1N+OH’ рК 4, 1 от до 2,3-3,8 1,6-2,4 20—100 Разрушается в окис- лителях, прочность хорошая 40—70
АСД, АВ-17 и др. е>
Электрообмепивающие: КСГ и др. chssh но-^2Уон 4,4 Восстано- вит. спо- собность по иону железа 380 мг!г 30-40 Высокая механич. прочность, устойчи- вость в многократ- ных циклах окис- ления-восстановле- ния
Биполярные: СДВ + ВП АСД + ВМА Retardion 11А8 Синтетич. неорганич. /—\ /“Л Z Vso3h и n -r4n+ои- и -R'COOH 3,8-4,2 + + 3,0-3,5 2,3-2,8 + + 3,5—4,5 — — В ысокая механич. прочность, удовлет- ворит. химич. стой- кость 50-70
ИОНИТЫ: Цирконий фосфат PK1 = 6,2 4,5 1,35 — Разрушается в щелоч- ных р-рах (pH вы- ше 2). высокая проч- ность 300-350
Вофатит,
гг тч т» м п и о w и я- я_р г’-иячынают что к этим группам также относятся катиониты: а) МСФ, СКФ, СН, _
Р F КС Амбермйт JR-100 и 105, Дауэкс-30, Дуалайт С-3. Ионикс-200, Стайонит ФК Леватит PN, Цеокерб 215, Дайайон S
К-I;’ б)С’смПХлайт Ш-120 'Жони™: n5’;
А», Ж5 <^Z q-3^™t L150 и L-165; 0^-28, АСБ, АСМ, Дауэкс-1, Дауэкс-2,
'Пермутитгб,. Амберлайт АЧОО, Церолит F, F, Дайайон SA-100.________________________________________________
* По Na+ для катионитов. * • По С1 для анйонитов.
305
ИОНИТЫ — ИОННАЯ АТМОСФЕРА
306
Полимеризуя, напр., винилпиридин внутри гранул сульфо-
катионита или метакриловую к-ту в гранулах высокооснов-
ного анионита, создают биполярные И., поглощающие од-
новременно катионы и анионы из р-ров и десорбирующие их
вытеснением водой.
Способность И. вступать н ионный обмен с находя-
щимися н растворе электролитами широко исполь-
зуется в технике. Поглощение ионов из р-ра сопро-
вождается строго эквивалентным вытеснением по-
движных ионон И. Скорость установления ионообмен-
ного равновесия определяется степенью ионизации
И. н данной среде и скоростью диффузии ионов в р-ре
и в зернах поглотителя. С повышением степени набу-
хания И. и с понижением размера зерен скорость
устанонления обменного равновесия повышается. В на-
бухающих и сильно ионизированных И. процесс
замещения всех подвижных ионов длится 2—5 сек.
Поэтому извлекать ионы из р-ра можно дииампч.
методом, т. е. фильтрацией его через колонну, запол-
ненную зернами И. Для извлечения солей из р-ров
применяют последовательно соединенные фильтры,
одни из к-рых заполнены катионитом, а другие
анионитом. По мере передвижения р-ра по колонне
с катионитом происходит поглощение катионов солей
и вытеснение ионов водорода. В анионитовой колонне
анионы солей вытесняют гидроксильные группы анио-
нита. Через нек-рый промежуток времени, опреде-
ляемый обменной способностью И. при данном pH
раствора и концентрацией в нем ионов, поглощение
солей прекращается и фильтры подвергают регене-
рации, промывая катионитовые колонны р-ром кис-
лоты, а анионитовые — р-ром щелочи. Для удале-
ния из р-ров таких слабых к-т, как угольная или
кремневая, применяют высокоосновные аниониты.
Ионитовыми колоннами целесообразно пользоваться
в случае обессоливания разб. р-ров (до 1,5—2 г/л).
Для полного удаления следов электролитов из р-ра
применяют смешанные фильтры, т. е. колонны, за-
полненные смесью апионптовых и катионитовых зерен,
имеющих различную плотность. Перед регенерацией
аниониты и катиониты отделяют друг от друга фло-
тацией. Для удаления из р-ра кислорода воздуха
пользуются электронообменивающими И., периоди-
чески регенерируя их р-ром гидросульфита натрия.
Метод ионообменной сорбции применяют для умяг-
чения или обессоливания воды, удаления солей из са-
харных сиропов, молока, вин, растворов фруктозы,
из р-ров дубильных веществ, продуктов гидролиза
отходов с.-х. сырья, из р-ров различных лекарствен-
ных препаратов (антибиотиков, витаминов, алкало-
идов), для удаления ионов кальция из плазмы крови
перед ее консервацией, для очистки от минеральных
ионон р-ров органич. реактивов, для очистки сточных
нод от фенола, хрома, никеля и др., а также для
концентрирования ценных ионов, находящихся в
микродозах в р-ре, н том числе для извлечения ионов
из сливных нод гальванич. цехов, для улавливания
и концентрирования радиоактивных ионов, ионов
меди из сточных вод медноаммпачного произ-ва искус-
ственного шелка.
Одной из важнейших областей применения И. яв-
ляется ионообменная хроматография, т. е. разделе-
ние сложной смеси электролитов в разб. р-ре. Разде-
ление производят методами избирательного вытесне-
ния или избирательного поглощения. В первом слу-
чае используют монофункциональные И. с высокой
степенью ионизации и наибольшей скоростью уста-
новления ионообменного равновесия. Подбирая вытес-
нитель, образующий с одним из ионов наиболее проч-
ные соединения (часто комплексные), производят
избирательное вытеснение иона. Для избирательного
поглощения применяют И., наиболее прочно соеди-
няющийся с ионами одного из веществ, находящихся
в р-ре (желательно И., способный образовывать клеш-
невидные комплексы с ионами этого вещества). Уста-
новлено, напр., что в кислых р-рах И., содержащие
оксифенильные группы, избирательно, реагируют с
ионами ванадия, висмута, ртути, олова, свинца. Карбо-
ксильные И. образуют более прочные соединения
с поливалентными ионами, чем с одновалентными, за
исключением ионов водорода. Фосфорнокислые И.
избирательно поглощают ионы лития, урана, уранила,
железа, ванадия. Высокоосновные аниониты извле-
кают ионы хлора в присутствии гидроксильных или
сульфатиопон, отделяют ионы иода от ионон брома
и хлора.
Разработаны методы извлечения и разделения разно-
образных ионон. И. могут служить и н качестве сильйо-
полярных поглотителен, извлекая органич. вещества
из р-ров с низкой диэлектрич. проницаемостью. СуЛЬ-
фокатиониты извлекают амины из органич. раствори-
телей. Аниониты извлекают нитро-,нитрил- и галоген-
замещенные соединения. Вытеснение с ионита погло-
щенных им соединений производят растворителями
с высокой диэлектрич. проницаемостью. И. предло-
жены н качестве лечебных средств для изменения pH
среды н организме, для извлечения радиоактивных
ионон из желудочно-кишечного тракта.
И. применяют в качестве высокоактивных катали-
заторов процессов этерификации, гидратации,, эфири-
зации и дегидратации, с помощью к-рых можно при-
водить синтез непрерывным методом. Характеристики
нек-рых из наиболее распространенных И. приведены
н таблице.
Лит.: Ионный обмен и его применение. [Сб. статей],
М., 1959; Хроматография, ее теория и применение. Труды
Всес. совещания по хроматографии, JL, 1960; С а л д а д з е
К. М., Пашков А. Б., т и т о в В. С., Ионообменные
высокомолекулярные соединения, М., 1960; Grlessbach R.,
Austauschadsorptlon in Theorle und Praxis, B., 1957; Kn-
n 1 n R., Myers R. T., Ion exchange resins, N. Y. — L.,
1951; Rabek T. I., Teoretyczne podstawy syntezy poll-
elektrolitdw i wymleniaczy jonowych, Warsz., 1960; Гель-
фер их Ф., Иониты, пер. с нем., М., 1962; Иссле-
дования в области промышленного применения сорбентов.
Сб. ст., под ред. К. В. Чмутова, М., 1961. Е. Б. Тростянская.
ИОННАЯ АТМОСФЕРА (ионное облако) — элек-
трич. поле, возникающее вокруг каждого данного
(«центрального») иона в растворе или расплаве;
это поле создается вследствие среднестатистич. но
времени распределения окружающих ионон под влия-
нием взаимодействия их зарядов и теплового движе-
ния; важнейшее понятие теории сильных электроли-
тов. Распределение подчиняется Максвелла — Больц-
мана закону распределение. При отсутствии внешнего
электрич. поля И. а. имеет сферич. симметрию, при-
чем плотность электрич. заряда и потенциал н раз-
личных точках И. а. уменьшаются с увеличением
расстояния до центрального иона и могут быть вы-
числены теоретически. Суммарный заряд И. а. по
величине ранен и по знаку противоположен заряду
центрального иона.
Согласно теории сильных электролитов, электрич.
взаимодействие центрального иона и его И. а. экви-
валентно взаимодействию этого иона с электриче-
ски заряженной сферой, имеющей заряд И. а. и ра-
диус _
га = 1,98 • 10-10 У~ см,
где е — диэлектрич. проницаемость, Т — абсолютная
темп-ра, I — ионная сила раствора, га наз. радиусом
И. а.; га уменьшается с ростом концентрации электро-
лита и зависит от валентности ионон. При 25° в иод-
ных р-рах, имеющих концентрации 0,1; 0,01 и
0,001 М[л, величины га соответстненно ранны (н А);
у электролитов 1—1 (типа NaCl) 9,64; 30,5;, .96,4;
у электролитов 1—2 и 2—1 (типов СаС12 и K2SO4)
5,58; 19,3; 55,8; у электролитов 2—2 (типа CuSO4)
4,82; 15,3; 48,2 и у электролитов 1—3 и 3 — 1 (типов
307
ИОННАЯ СВЯЗЬ-ИОННЫЙ ОБМЕН
308
FeCla и К3РО4) 3,94; 13,6; 39,4. Существование И. а.
является причиной эффектов Вина, Фалькенхагена
и оказывает значительное влияние на электропро-
водность, кинетику ионных реакций, ассоциацию
ионов, на величины активностей и на другие свойства
р-ров электролитов. Учет влияния И. а. на свойства
электролитов составляет основу количественной тео-
рии сильных электролитов, к-рая я строгой форме раз-
вита лишь для разб. р-рон.
Лит. см. при ст. Мойная сила раствора.
Н. Е. Хомутов.
ИОННАЯ СВЯЗЬ — см. Химическая связь.
ИОННАЯ СИЛА РАСТВОРА — мера интенсив-
ности электрич. поля, создаваемого ионами в растворе.
И. с. р. (Z) равна полусумме произведений моляль-
ностей каждого нова mi (число грамм-ионов н 1000 г
растворителя) на квадрат его валентности Z4, т. е.
I = Так, н растворе, содержащем 0,01 моля
K2SO4, 0,01 моля KNO3 и 1000 г воды, она равна:
7=1 (0,03- Z*K + 0,01 • Z^04 + 0,01 • ZJjo,) = 0,04.
Величина И. с. р. существенно определяет термо-
дпнамич. и кинетич. свойства электролитов, я том
числе коэфф, активности, осмотич. коэфф., константы
скорости ионных реакций и др. свойства. Точная
количественная связь И. с. р. со снойстнами электро-
литов установлена теоретически только для разб.
р-рон. Она характеризуется наличием линейной зави-
симости многих свойств (или их логарифмов) от УI.
Теоретически установлено, что н весьма разб. р-рах,
имеющих одинаковые величины И. с. р., коэфф,
активности к.-л. электролита имеет постоянное зна-
чение, зависящее только от величины И. с. р., но не
зависящее от природы других, находящихся в р-ре
электролитов. Это положение, называемое пра-
вилом ионной силы, есть предельный закон.
В водных р-рах оно точно соблюдается приблизи-
тельно до Z = 0,02—0,05, а н неводных растворителях
с малой диэлектрич. проницаемостью область его
применимости ограничивается очень разб. р-рами.
Правило ионной силы широко используется н расче-
тах коэфф, активности. Известна приближенная
полуэмпирич. связь ряда термодинамич. свойств
р-роя электролитов (напр., коэфф, активности) с
И. с. р. в полном диапазоне концентраций.
В теории электролитов И. с. р. часто выражают
через молярные концентрации В этом случае
соответствующий параметр Г = наз. иональ-
ной концентрацией.
Лит.: Изгарышев Н. А., Горбачев С. В.,
Курс теоретической электрохимии, М.—Л., 1951; Измай-
лов Н. А., Электрохимия растворов, Харьков, 1959; X а р -
и ед Г., Оуэн Б., Физическая химия растворов электро-
литов, пер. с англ., М., 1952; Г лес с тон С., Введение
в электрохимию, пер. с англ., М., 1951. Н. Е. Хомутов.
ИОННЫЙ ВЫХОД — величина, показывающая,
какое количество молекул возникает (или распадается)
на каждую пару ионон, образовавшихся при погло-
щении веществом ионизирующего излучения. И. н.
обозначается отношением М/N, где М — число всех
образовавшихся (или разложившихся) молекул, N —
число всех пар ионон. И. н. используется как сравни-
тельная характеристика радиационно-химических ре-
акций.
ИОННЫЙ ОБМЕН — обмен ионон между двумя
электролитами. И. о. может происходить как в гомо-
генной среде (истинный р-р нескольких электролитов),
так и н гетерогенной, н к-рой один из электролитов
является, напр., твердым (при контакте р-ра электро-
лита с почвой, ионообменными сорбентами, структур-
ными элементами клетки и пр.). Нередко под терми-
ном «И. о.» понимают именно гетерогенный И. о.
между жидкой и твердой фазами; такие процессы
со сложным и своеобразным механизмом широко ис-
пользуют н науке и технике.
И. о. был открыт в 50-х гг. 19 в. (еще до создания теории
электролитич. диссоциации) для почв, компоненты к-рых
интенсивно обмениваются с ионами почвенного р-ра, вследст-
вие чего изменяется состав р-ра при внесении в него пробы
почвы. Дальнейшее развитие учения об И. о. в почвах связано
с именем К. К. Гедройца, к-рый изучил поглотительную спо-
собность почв и ввел понятие почвенного поглощающего
комплекса.
В качестве ионообменных сорбентов применяются
как сорбенты минерального происхождения (алюмо-
силикаты, силикаты, гидрат окиси алюминия, фос-
фат циркония и др.), так и органич. сорбенты (феноло-
формальдегидные, полистирольные и др. катиониты
с различными функциональными группами, амино-
формальдегидные, полиаминоные и др. аниониты— см.
Иониты).
Можно считать установленным, что И. о. происхо-
дит в эквивалентных отношениях и в большинстве
случаен является обратимым.
В литературе распространены н основном три
точки зрения на механизм И. о. Первая из них сво-
дится к т. н. мембранному равновесию, т. е. процессу,
протекающему при наличии мембраны, прони-
цаемой для одних ионон и непроницаемой для других;
аналогия с такой мембраной связана с неподвижностью
одного из ионон (аниона или катиона) твердого сор-
бента и невозможностью его перехода н жидкую
фазу. При мембранном равновесии ионные произве-
дения для свободных ионон по обе стороны мембраны
равны, что приводит к выводу о том, что константа
равновесия И. о. равна единице. Другая точка зрения —
осмотич. теория И. о. — сводится к рассмотрению
термодинамики осмотич. явлений при проникновении
жидкой фазы н сорбент и его набухании. Наиболее же
распространенным является взгляд на И. о. как на
гетерогенную химич. реакцию двойного обмена типа:
2RH 4- СаС12 2RH 4- Сам- 4- 2С1~ RsCa 4- 2НС1
При расчете константы равновесия этой гетерогенной
реакции (константы обмена) следует учитывать коэф-
фициенты активности веществ в растворе и в сорбенте.
Как правило, константы обмена возрастают с увели-
чением заряда иона; на сульфокатионите константа
обмена однозарядных ионов на водород близка к еди-
нице, а трехзарядных —• к 10. У ионон данной вели-
чины заряда и н не слишком конц. р-рах она возрастает
с увеличением истинного (негидратированного) радиу-
са иона; так, на монофункциональных сульфокатио-
нитах константа обмена лития на водород равна при-
мерно 0,6, а цезия —• ок. 2. Иониты с аномально высо-
ким по отношению к данному иону ионообменным
сродством носят название селективных; избиратель-
ность достигается введением в иониты специальных,
обычно слабооснонных функциональных групп, не-
редко образующих с выделяемым ионом комплексное
соединение. Большие по размерам ионы (витамины,
антибиотики и пр.) или даже обычные ионы при по-
глощении ионообменными сорбентами очень плотной
структуры обмениваются в малой степени, иногда —
лишь на внешней поверхности зерен сорбента (т. наз.
экстракристаллически й обмен).
В остальных случаях обычно н И. о. участвуют все
способные к обмену группы сорбента. Поэтому ионит,
н отличие, напр., от активного угля, можно рассматри-
вать как гомогенную фазу безотносительно к его
структуре. Вследствие этого, н частности, обменная
емкость ионообменных сорбентов не зависит от их
зернения.
В отличие от гомогенного И. о., для к-рого харак-
терна нулевая энергия активации и, следовательно,
практически мгновенная скорость, гетерогенный И. о.
является сравнительно медленным; кинетика гетеро-
генного И. о. имеет диффузионный характер за исклю-
309
ИОННЫЙ ОБМЕН-ИОННЫЙ РАДИУС
310
чением И. о. на сорбентах с очень слабо ионизован-
ными функциональными группами. Вследствие этого
скорость И. о. убывает с увеличением размера зерен
сорбента, более резко — в случае внутридиффузион-
ной кинетики (диффузия внутри зерен сорбента),
отчетливо проявляющейся в конц. растворах (более
0,1 М). Коэфф, внутренней диффузии ионов в ионитах
имеет порядок 10 е см2/сек и убывает с увеличением
зарядов ионов и степени сшитости ионита. В случае
внешнедиффузионной кинетики (диффузия в граничном
слое между зернами и жидкой фазой), отчетливо прояв-
ляющейся в разб. р-рах (менее 0,001 М), скорость об-
мена вследствие турбулизации потока возрастает с уве-
личением скорости обтекания зерен сорбента раствором.
Различают 3 способа осуществления процесса И. о.:
статический, динамический и хроматографический.
Первый из них состоит в однократном контакте ионо-
обменного сорбента с р-ром электролита. Он имеет
значение во многих природных процессах (поглоще-
ние ионов почвами и пр.), а также при изучении равно-
весных соотношений; иногда используется в техноло-
гии для извлечения одного из компонентов смеси
с резко повышенной сорбируемостью (напр., при
извлечении урана анионитами из карбонатных или
сульфатных р-ров). Обычно же в технологии и в ана-
литич. практике статич. процессы не применяются
вследствие принципиальной невозможности полного
извлечения (удаления) компонента. В динамич. усло-
виях исходный р-р пропускают через слой (колонку)
ионообменного сорбента в одном направлении. При
этом вследствие удаления током раствора продуктов
ионообменной реакции достигается количественное
извлечение компонента из раствора и полное исполь-
зование обменной емкости участков слоя сорбента,
расположенных первыми по току р-ра. Однако в ди-
намич. процессе отделяемый компонент появляется
за слоем сорбента раньше, чем полностью исполь-
зуется обменная емкость сорбента. Величина потери
обменной емкости слоя, зависящая от кинетич. пара-
метров (скорости течения р-ра, зернения сорбента
и пр.), иногда доходит до 10—20% от общей емкости
слоя. Динамич. И. о. широко применяется при очистке
р-ров и извлечении из них ценных компонентов.
Ионообменную хроматографию (см. Хроматогра-
фия), широко применяемую при разделении смесей
близких по свойствам ионов, напр. смеси редко-
земельных элементов, изотопов, антибиотиков и пр.,
осуществляют введением в колонку небольшого (по
сравнению с обменной емкостью слоя) количества
анализируемой смеси и промыванием колонки спе-
циально подобранным растворителем.
И. о. имеет большое значение в различных отраслях тех-
ники. Исторически первым и до настоящего времени наиболее
значительным по масштабу использования ионообменных
сорбентов является водоподготовка — умягчение и обессоли-
вание воды; при этом достигается высокая степень очистки
воды, напр. на двухколоночной установке (катионит и анионит)
можно получить воду с удельным сопротивлением порядка
10> ом. Первым по току р-ра помещают сравнительно сильно-
основной ионит. При необходимости получения воды со зна-
чительно меньшей электропроводностью за раздельными ко-
лоннами ставят колонку со смешанным слоем катионита
и анионита. При деминерализации засоленных вод перед
колоннами целесообразно установить электродиализатор с
ионообменными мембранами.
Широкое использование нашел И. о. в гидрометаллургии:
извлечение благородных, цветных и редких металлов (серебро,
медь, *никель, хром и др.) из сбросных р-ров на катионитных
или анионитных колоннах, а также хроматография, разделе-
ние близких по свойствам элементов (редкоземельные эле-
менты, гафний и цирконий, ниобий, тантал и др.). Ионообмен-
ные сорбенты используют также для очистки отбросных р-ров
от химически вредных (фенолы и др. ионогенные органич.
соединения) и радиоактивных веществ. Удаление иояов каль-
ция методом И. о. позволяет на 5—10% уменьшить потери
при произ-ве сахара из сахарной свеклы, получать хорошо
сохраняющуюся консервированную кровь и приготовлять
«грудное» молоко из коровьего. И. о. применяют в аналитич.
химии для удаления мешающих определению ионов (напр.,
при определении сульфатов или фосфатов в присутствии ка-
тионов металлов). Весьма широко распространейа в аналитич.
химии ионообменная хроматография, к-рая обеспечивает
количественное разделение смесей близких по свойствам
компонентов при их анализе. Ионообменную хроматографию
все более широко применяют при получении чистых органич.
препаратов (карбоновые к-ты, аминокислоты, алкалоиды,
витамины, антибиотики и пр.).
И. о. имеет большое значение в процессах жизнедеятель-
ности растений и организмов. Явление избирательного И. о.
лежит в основе усвоения минеральных веществ клетками кор-
ней высших растений и питания микроорганизмов и клеток
водорослей; вероятно, именно процессы И. о. определяют
избирательную проницаемость клеток.
Лит.: Ионный обмен и его применение, М., 1959; Сам-
сонов Г. В., Хроматография, Л., 1955; Ионообменная
технология. [Сб. статей], под ред. Ф. Находа и Дж. Шуберта,
пер. с англ., М., 1959; Ионообменные смолы в медицине и био-
логии, пер. с англ., М., 1956; Самуэльсон О., Приме-
нение ионного обмена в аналитической химии, пер. с англ.,
М., 1955; Гельферих Ф., Иониты, пер. с нем., М., 1962.
М. М. Сенявин.
ИОННЫЙ РАДИУС — понятие, принятое в кри-
сталлохимии для обозначения размеров шарообразных
ионов и вычисления межатомных расстояний в ионных
соединениях. Использование понятия «И. р.» основано
на принципе аддитивности, т. е. на предположении,
что размеры ионов не зависят от состава молекул
и кристаллов, в к-рые они входят. Поэтому для созда-
ния системы ионных радиусов достаточно знать раз-
мер хотя бы одного иона. Но поскольку из экспери-
мента всегда известно лишь значение длины связи,
разделение межатомного расстояния на части, соот-
ветствующие отдельным ионам, сопряжено с дополни-
тельными допущениями. Все системы ионных радиу-
сов, появившиеся за последние 35—40 лет, в зависи-
мости от исходных точек можно грубо разбить на две
большие группы — на «теоретические» и «эмпириче-
ские». Такое разделение весьма условно, т. к. строгое
вычисление И. р. бессмысленно (квантово-механически
можно рассчитать лишь расстояние от ядра иона до
точки, соответствующей максимуму электронной вол-
новой функции) и все теоретич. значения базируются
в той или иной мере на эмпирич. величинах И. р.
Начало эмпирич. направлению в расчетах И. р.
было положено Ланде, к-рый, исходя из идеи кон-
такта анионов в кристаллич. структурах, определил
прямо из размеров ячейки радиусы нек-рых анионов.
Однако анионный контакт имеет место только в
случае маленьких, а потому сильно поляризующих
катионов, и полученные таким путем радиусы анио-
нов занижены (радиусы катионов получаются соот-
ветственно завышенными). Поэтому В. Гольдшмидт
предпочел положить в основу своей системы И. р.
кислорода и фтора (1,32А и 1,ЗЗА), вычисленные
Вазашерной из рефрактометрических данных. Работа
Вазашерны не может считаться правильной, и потому
приведенные выше числа должны рассматриваться
как постулаты. Очевидная ошибка состоит в заниже-
нии И. р. кислорода.
Более правильное определение яонной рефракции,
а затем и радиуса кислорода привело Кордеса к зна-
чению г02- = 1,35А. Н. В. Белов и Г. В. Бокий исполь-
зуют для вычисления величину 1 ,ЗбА, Л. Аренс —
1,40А и В. Захариазен — 1,4бА. Хотя теоретически
последние значения более предпочтительны, наилуч-
шее соответствие результатов расчета с экспериментом
имеет место в системе Белова — Бокия. Поэтому
приведенные в табл, значения радиусов анионов
взяты из последней системы, для ионов серы и селена
приводятся среднеарифметич. значения из таблиц
Белова — Бокия и Захариазена.
Теоретич. направление работ по вычислению И. р.
было начато исследованием Л. Полинга, к-рый с по-
мощью квантовой механики (положив, правда, в
основу значение г02- = 1,40А) вывел значения радиу-
сов почти для всех элементов. Далее следует отме-
тить работы Е. Кордеса, К. Стоккара, К. Б. Яцимир-
ского, А. Ф. Капустинского, С. С. Бацанова и снова
311
ИОННЫЙ РАДИУС —иононы
312
Кордеса. В этих работах развивались либо идеи
Полинга (Кордес), либо использовались в различ-
ных вариантах формулы атомной физики. В работе
Бацанова произведено вычисление И. р. электрополо-
жительных элементов из их ковалентных радиусов
с помощью концепции электроотрицательностей. Тео-
ретически рассчитанные И. р., как правило, очень
близки к эмпирически определенным величинам и
поэтому в табл, приводятся значения радиусов
катионов по системе Белова — Бокия с дополнения-
ми: И. р. лантанидов и актинидов по Захариазену,
а металлов VIII группы — по Баранову.
Следует, однако, отметить одно видимое противоре-
чие. Именно, понятие «И. р.» имеет только тот смысл,
что сумма таких радиусов должна равняться межатом-
ному расстоянию. Кроме того, элементарные энерге-
тич. соображения показывают, что идеальных ионов
Величины И. р. сильно зависят от координационных
чисел и приведенные в табл. 1 и 2 значения соответ-
ствуют координационному числу КЧ-6 (структура
типа NaCl).
Лит.: Lands A., Z. Phys., 1920, 1, М 3, 191; Gold-
schmidt V. М., Trans. Faraday Soc., 1929, 25, 253; W а -
s a s t j e r n a J. A., Z. Phys. Chem., 1922, 101, H. 3/4, 193;
Бацанов С. С., Структурная рефрактометрия, M., 1959,
с. 44; Kordes Е.. Z. Phys. Chem., 1939, 44, 249, 327;
Бокий Г. Б., Введение в кристаллохимию, М., 1954;
Ahrens L. Н., Geochim. et cosmochim. acta, 1952, 2,
155; Захариазен В., в кн.: Актиниды, под ред. Г. Си-
борга и Дж. Каца, пер. с англ., М., 1955, с. 623; Pauling L.,
Z. Kristallogr., 1928, 67, Н. 3/4, 377; Kordes Е., Z. Phys.
Chem., 1940, 48, 91; Stockar К., Ilelv. chim. acta, 1950,
33, 1409; Яцимирский К. Б., Ж. общ. химии, 1953,
23, вып. 2, 180; Капустинский А. Ф., Ж. неорг.
химии, 1956, 1, вып. 1, 82; Б а ц а н о в С. С., Ж. физ. хи-
мии, 1956, 30, вып. 12, 2640; Kordes Е., Naturwissenschaf-
ten, 1960, 47, Н. 20, 463; Zachariasen W. Н., Z. Kris-
tallogr., 1931, 80, Н. 3/4, 137. С. С. Бацанов. i
Таблица ионных радиусов
1’ Пе‘ 1 риоды Подгруппы
1а Па Illa IVa | Va Via j Vila | i VIIIa| | Ib j IIb | IIIb | IVb | Vb j VIb VIIb Villa6
1 H 1+ 0,00 1-1,36 He 0 1.22
2 : L1 1+ 0,68 Be 2+0,34 В 3+ 0,21 C 4+ 0,20 4- 2,60 1 N 5+ 0,15 3~ 1,48 0 6+ 0,09 2- 1,36 F 7+ 0,07 1- 1,33 Ne 0 1.60
3 Na 1+ 0,98 Mg 2+ 0,74 Al 3+ 0,57 SI 4+ 0,39 p 5+ 0,35 3- 1,86 s 6+ 0,30 2- 1,86 Cl 7+ 0,26 1- 1,81 Ar 0 1,92
'4 К 1+1,33 Ca 2+ 1,04 Sc 3+ 0,83 Ti 2+ 0,78 3+ 0,69 4+ 0,64 V 2+ 0,72 3+ 0,67 4+ 0,61 5+ 0,59a Сг 2+ 0,83 3+ 0,64 6+ 0,52a Mn 2+ 0,91 3+ 0,70 4+ 0,52 7+ 0.46 Fe 2+ 0,80 3+ 0,73 Co 2+ 0.80 3+ 0,72 Ni 2+ 0,79 3+ 0,72 Cu 1+0,98 2+0,80 Zn 2+ 0,83 Ga 3+ 0,62 Ge 2+ 0,65 4+ 0,44 As 3+0,69 5т 0,47 3- 1,91 Se 4+ 0,69 6+ 0,35 2~ 1,98 Br 7+ 0,39 1- 1,96 Kr 0 1,93
5 Rb 1+ 1,49 Sr 2+ 1,20 Y 3+ 0,97 Zr 4-1- 0,82 Nb 4+ 0,74a 5+ 0,66 Mo 4+ 0,68 6+ 0,65 Tc 7+ 0,56 Ru 2+ 0,85 3+ 0,77 4+ 0,71 Rh 3+ 0,78 4+ 0,71 Pd 2+ 0,88 4+ 0,73 Ag 1+ 1,13 Cd 2+ 0,99 Jn 1+ 1,30 0,92 Sn 2+ 1,02 4+ 0,67 Sb 3+ 0,90 5+ 0,62 3~ 2,08 Те 44 0,89 3+ 0,56 j- 2,22 J 7+ 0,50 1- 2,20 Xe 0 2,18
6 Cs 1+ 1,65 Ba 2+ 1,38 La 3+ 1,04 Ш 4+0,82 Ta 5+ 0,66 W 4+ 0,68 6+ 0,65 Re 4+ 0,72 7+ 0,56 Os 2+ 0,89 3+ 0,81 4+ 0,75 Ir 2+ 0,89 3+0,81 4+ 0,75 Pt 2+ 0.90 4+ 0,76 Au 1+ 1,37 31' 0,85 Hg 2+ 1,12 TI 1+ 1,36 3+ 1,05 Pb 2+ 1,26 4+ 0,76 Bi 3+ 1,20 5+ 0,74 Po At 7+ 0,62a Rn
7 Fr 1+ 1.75 Ra 2+ 1,44 Ac 3+ 1,11
Лантаниды Ce 3^ 1,00 4+ 0,93 Pr 3+ 1.00 4+ 0,92 Nd 3+ 0,99 4+ 0,90 Pm 3+ 0,98 Sm 2+ 1,11 3+ 0,97 Eu 2+ 1,09 3+ 0,96 Gd 3+ 0,94 Tb 3+ 0,92 4+ 0,84 Dy 3+ 0,91 Ho 3+ 0,89 Er 3+ 0,87 Tu 3+ 0,86 Yb 3*- 0,85 Lu 3+ 0,84
Актиниды Th 3+ 1,08 4+ 0,94s Pa 3 г 1.05 4+ 0,96 5+ 0,90 U 3+ 1.03 4+ 0,93 5+ 0,87 6+ 0,83 Np 3+ 1,01 4+ 0,92 5+ 0,88 6+ 0,82 Pu 3+ 1,00 4+ 0,90 5+ 0,87 6+ 0,81 Am 3+ 0,99 4+ 0,89 5+ 0,86 6+ 0,80 Cm Bk Cl Es Fin Md N 102 Lw
а По Аренсу. 6 Для инертных газов даны значения атомных радиусов.
в соединениях, по-видимому, не бывает. Однако
теоретич. значения радиусов, в общем, хорошо согла-
суются с эмпирич. значениями. Решение этого про-
тиворечия заключается в том, что размер атома по
мере отрыва от него электронов очень быстро умень-
шается до размера катиона, соответствующего макси-
мальной положительной валентности элемента (напр.,
радиус серы S+ практически равен радиусу Se+,
однако из-за постоянных значений радиусов анионов
этот эффект не всегда можно обнаружить в системах
ионных радиусов). Поэтому-то вывод значений И. р.
из межатомных расстояний не чисто ионных веществ
все-таки не сильно отличается от теоретич. величин.
ИОНОНЫ — группа производных циклогексена,
кетоны, содержащие кетогруппу в боковой цепи
в составе радикала R’. К И. относят собственно И.
(а- и fl-изомеры); метилиононы — н-а-, н-^-, изо-а-
и пзо-р-метилиононы, ирон, а-, (5- и у-изомеры (для
каждого изомера возможна цис- и транс-изомерия,
а для а- и у-пронов — также циклпч. стереоизомерия).
Для И. характерен следующий углеродный скелет
молекулы:
Н3С_СН3
[fHSr-CH’C-COR
is 3. ft’
313
иононы —ионы
314
Отдельные иононные препараты различаются поло-
жением двойной связи в цикле, а также радикалами.
И. обладает запахом кедра, а в разведении — цветов
фиалки (а-ионон имеет более нежный запах, чем
P-Изомер). Метилионон также имеет запах фиалки.
Из изомерных метилиононов изо-а-метилионон обла-
дает наиболее нежным запахом, в парфюмерии он
наз. «И р а л и я»; ирон также имеет запах цветов
фиалки, хотя в них и не обнаружен. И. обладают
высокими парфюмерными качествами, входят в состав
композиций духов с запахом фиалки и сирени. И. и
метилионон получают синтетически; ирон входит
в состав эфирного масла, выделяемого из корней
ириса.
Все И. вступают в реакции, характерные для ке-
тонов: при восстановлении карбонильной группы обра-
зуются соответствующие спирты; возможно избира-
тельное гидрирование по двойным связям, к-рое
проходит ступенчато с образованием дигидро- или
тетрагидропроизводных; образуют семикарбазоны,
тиосемикарбазоны и т. п.
Синтез И. основан на конденсации цитраля (I)
с ацетоном в присутствии щелочных агентов в псевдо-
ионон (II); последний изомеризуется в И. (III)
влиянием кислых агентов:
ПОД
Кетон Положе* ние двой- ной связи в цикле R' R" Т кип., ° С/мм dl° 20 nD
а-ИОНОН 3,4 Н СН3 129—129,5/11; 80-82/1 0,9291 1,4987
З-Ионон 2,3 Н СН3 134-135/12; 88—90/1 0,9434 1,5192
н-а-Метилионон 3,4 Н CgHa 125-126/9 0,9262 1,49692
н-р-Метйлионон 2,3 Н С2н5 133/9 0,9387 1,51532
изо-а-Метилионон .... («Иралия») 3,4 сн3 СНз 121—122/9; 103—104/2 0,9345 1,50188
изо-р-Метилиопон .... 2,3 сн3 СНз 124-125/9; 105-106/2 0,9355 1,50580
а-ИрОНа 3,4 н СНз
>Ироиа 2,3 II СНз 144/16 0,9367 1,5011
1-Ирона> 6 н СНз
а Константы ирона приведены для смеси всех трех изомеров.6 В т-ироне
в положении 3—метиленовая группа, в положении 6 — метильная группа.
Разработано несколько схем полного синтеза псевдо-
п он он а из ацетона и ацетилена. Относительные коли-
чества а- и f-изомеров в смеси, образующейся, при
циклизации псевдоионона, зависят от примененного
циклизирующего агента, его концентрации, а также
от темп-]эы реакции, а- и Р-изомеры разделяют ректи-
фикацией на высокоэффективных колонках и др.
способами. Качественно а-ионон определяют по крас-
ному окрашиванию р-ра с несколькими каплями
20%-ного спиртового р-ра NaOH, р-ионон не дает
этой реакции. Со смесью соляной и уксусной к-т
Р-ионон дает красное окрашивание. Количественно об-
щее содержание И. определяют с гидроксиламином;
содержание каждого из них в смесях — по показа-
телю преломления, спектрофотометрически (в спирто-
вом р-ре для а-ионона 1тах = 227—228 ммк,Е =
= 14150; для Р-ионона соответственно 295 ммк и
10700) или газо-жидкостной хроматографией.
Метилионон в производственных условиях полу-
чают конденсацией цитраля (I) с метилэтилкетоном
в присутствии щелочных агентов. Образующуюся
смесь н-псевдометилионона (IV) и иао-псевдометил-
ионона (V) циклизуют до смеси соответственно н-а- и
H-f-метилиононов или uao-а- и uao-f-метилионойов:
, ,СН3
:С сн-сно сн3С0Сан5
Н3СХ ,СН3
ai-CH=CHCOCaHs
сн3
IV
Н3С
Н3С
СНЭ
. ,С||з
iC сн-сн=с-сосн3
II
^Асн3
V
Разработан синтетич. способ получения ирона, однако
пока его не получают в больших количествах.
Лит.: Белов В. Н. [и др.], Химия и технология ду-
шистых веществ, М.,.1953, с. 124; Усп. химии, 1957, 26, вып. 1,
96; Белов В. Н., Д а е в Н. А., С к в о р ц о в а Н. И.,
Ж. Всесоюзного хим. о-ва им. Д. И. Менделеева, 1960,
5, вып. 4, 362; Kulesza J., G 6 г a J., Т у с z к о-
w s к 1 A., Chemla 1 technologla zwlqzkow zapachowych, War-
szawa, 1961. H. И. Скворцова.
ИОНООБМЕННОЕ СРОДСТВО — см. Ионный
обмен.
ИОНЫ — заряженные частицы, представляющие
собой атомы или группы химически связанных атомов
с избытком или недостатком электронов. В зависимости
от количества избыточных или недостающих электро-
нов они могут быть одно-, двух- и многозарядными.
И. образуются путем отрыва от атомов
или молекул (или присоединения к ним)
электронов, протонов или других ионов
(ионизация). Являясь химически актив-
ными частицами, И. вступают в различ-
ные реакции с атомами, молекулами и
между собой, причем эти реакции зача-
стую являются начальными или проме-
жуточными стадиями самых разнообраз-
ных химических процессов. И. могут обра-
зовываться и существовать в веществах,
находящихся в любых агрегатных состоя-
ниях. Наличие их н газовой фазе при-
водит к специфическим химич. реакциям
и физич. свойствам. В растворах И. обра-
зуются в результате электролитической
диссоциации и обусловливают свойства
электролитов. Они делятся на два типа:
катионы — положительно заряжен-
ные И. (при электролизе направляющиеся
к катоду) и анионы — отрицательно
заряженные И. (направляющиеся к ано-
ду). В твердых телах И. часто образуют кристаллин,
решетку кристаллов. Кроме того, из них состоят
т. н. твердые электролиты. Ниже описаны И. в газо-
вой фазе. О И. в растворах см. Электролитическая
диссоциация, Электролиты, Кислоты и основания,
Катализ кислотно-основной, Ионный обмен. О И. в
твердых телах см. Ионный обмен, Кристаллы.
ИОНЫ (в газах). Содержание:
I. Элементарные процессы образования ионов в газах 315
II. Реакции ионов с молекулами ........... 317
III. Рекомбинация ионов .................. 319
IV. Роль ионов в различных системах ...... 321
Образование заметных концентраций И. в газах
происходит либо при очень высоких темп-рах, либо
в результате воздействия на газы квантов высокой
315
ИОНЫ
316
энергии или быстрых частиц. Вследствие этого газо-
вые И. не играют заметной роли в основных процессах
химич. технологии. Однако в связи со все возрастаю-
щей ролью радиационных явлений, в первую очередь
в ядерной технике, с повышением интереса к новым
технич. использованиям электроразрядной плазмы
и с бурным развитием работ, касающихся верхних
слоев атмосферы, в последние годы резко возрос
интерес к изучению химии И. в газах.
1. Элементарные процессы образования иоиов в га-
зах. Образование ионов при ударе
электрона о молекулу (см. также Иони-
зации потенциал). Если энергия электрона равна
энергии ионизации молекулы или превышает ее, то
при ударе электрона о молекулу с определенной
вероятностью образуется положительный молеку-
лярный И. С повышением энергии электрона вероят-
ность ионизации растет, вначале линейно с энергией,
достигая максимума в области энергий, соответствую-
щих 3—5 потенциалам ионизации. Наряду с процес-
сами ионизации при ударе электрона о молекулу про-
исходят процессы диссоциативной ионизации, напр..
СН4 + е -> СН+ + Н + 2е
Такой процесс становится возможным, когда энергия
электрона достигает соответствующего порога. Так,
в приведенном выше примере этот порог (называется
потенциалом появления иона СН3+) равен сумме
D (СН3 — Н) -j- J (СН3), где J — энергия ионизации
радикала, D — энергия диссоциации.
В зависимости от структуры и энергетики моле-
кулы соотношение между вероятностями различных
путей диссоциативной ионизации бывает весьма раз-
личным. Когда энергия электрона достигает 70—
100 эв, это соотношение уже перестает заметно ме-
няться с энергией электронов. В табл. 1 представ-
лены относительные вероятности образования различ-
ных И. при бомбардировке электронами с энергией
70 зв. За 100 принята максимальная вероятность обра-
зования И. Можно видеть, что в случае наиболее
распространенных двухатомных молекул ионизация
в основном не сопровождается диссоциацией; слабо
диссоциируют при ионизации ароматич. молекулы;
молекулы насыщенных углеводородов, напротив, пре-
терпевают в основном диссоциативную ионизацию.
Таблица 1. Относительные вероятности образования ионов
Молекула Ионы и вероятности нх образования
n2 N2+ (100), N+ (4,3)
СО СО+ (100), С+ (4,7), О+ (1,7)
с2н. С2Н4+ (100), С2Нз4- (65), С2Н2+ (62), С2Н+ (11,7), С2+ (3,7), СН2+ (6,3), СН+ (3,5)
Н-С»НН) С4Н10+ (12,3), С3Н7+(100), С3Н5+ (27,8), С3Н3+ (12,5), С2Н5+ (44,2), С2Н3+ (37,1)
свнв СвНв+ (100), СвН3+ (14), С4Н4+ (19), С3Н3+ (14)
Вероятность процесса ионизации количественно
обычно выражается величиной сечения иони-
зации, принятой при описании столкновения
любых частиц. Этот термин обозначает эффективную
площадь поперечного сечения молекулы, в к-рую
должен попасть электрон, чтобы процесс ионизации
произошел. Для процессов ионизации электронами
с энергией 100—300 эв сечение ионизации имеет вели-
чины в пределах 10*1®—10~14 см2 и обычно пропор-
ционально геометрия, размерам молекул. Зная сече-
ние ионизации а, можно рассчитать количество ионов
7Vb образующихся в слое газа толщиной d в резуль-
тате прохождения через него Ие электронов:
Nj = ondWe
В этой формуле п — концентрация молекул в газе.
При столкновении электронов со многими молеку-
лами с определенной вероятностью образуются и от-
рицательные И.; при этом также возможна недиссо-
циативная и диссоциативная ионизация. Примерами
соответствующих процессов являются:
SF3 + е —► SF1T
СС14 + е -> СС13 + Cl-
CO + е -* С+ + О- + е
Молекулярные отрицательные И. образуются в
результате захвата электрона. При этом выделяется
энергия, равная величине сродства молекулы к
электрону. В том случае, если энергия, передан-
ная отрицательному И. в результате присоединения
электрона к молекуле, выделится в виде энергии
излучения или (при ионизации в газе с повышен-
ным давлением) будет передана другой молекуле
при столкновении с ней, молекулярный отрицатель-
ный И. будет стабильным. Если же такие стабилизи-
рующие ион процессы не успеют произойти, то он
распадется либо на нейтральный и отрицательно заря-
жённый осколок, либо на исходные частицы — моле-
кулу и электрон.
У нек-рых молекул энергия, выделяющаяся при
присоединении электрона, переходит во внутреннюю
энергию образующегося отрицательного И., к-рый
при этом остается стабильным. Однако такой переход
энергии может проходить только в том случае, если
энергия электронов, сталкивающихся с молекулой,
имеет строго определенное значение. При столкнове-
нии электронов с другой энергией молекулярные
отрицательные И. практически не образуются. Опи-
санные процессы образования отрицательных И. наз.
резонансными. Образование отрицательных И. исполь-
зуется, напр., для установления шкалы абсолют-
ных значений энергий электронов при исследова-
ниях методом электронного удара. Чаще всего при
этом используется ионизация электронами моле-
кул SF6. Образование ионов SFs максимально в том
случае, если энергия электронов равна 0 ± 0,01 эв.
Отрицательные И. могут образовываться также в ре-
зультате диссоциации возбужденных электронным
ударом молекул на положительный и отрицательный
осколки.
Фотоионизация. Когда энергия кванта
света превосходит энергию ионизации атома или
молекулы, взаимодействие кванта света с нейтральной
частицей с определенным сечением приводит к обра-
зованию И. Дальнейшее увеличение энергии кванта
может приводить к диссоциативной ионизации моле-
кулы. Зависимость эффективного сечения фотоиони-
зации от энергии квантов отличается от соответствую-
щей зависимости в случае ионизации электронным
ударом. Квант света с максимальной вероятностью
ионизирует атом (а во многих случаях и молекулу),
когда его энергия равна энергии ионизации или
немного (на 0,1—1 эв) превосходит ее. Эта осо-
бенность ионизации квантами света позволяет
точно определять сами потенциалы ионизации мо-
лекул.
Ионизация при соударениях ней-
тральных частиц. Ионизация в этом случае
может происходить, если относительная энергия дви-
жения частиц превосходит энергию ионизации. Счи-
талось, что вероятность ионизации будет только очень
медленно расти с увеличением кинетич. энергии
частиц, достигая максимума при энергии, на несколь-
ко порядков превосходящей пороговую энергию иони-
зации. Однако эксперименты последнего времени
показывают, что, по-видимому, во многих случаях
сечение ионизации растет с энергией гораздо быстрее,
достигая величины порядка 10*1’—10-18 см2 при
энергии, в 3—100 раз превосходящей пороговое зна-
317
ИОНЫ
318
чение. Примером ионизации такого рода является
процесс:
К + О2 -> К+ + О2 + е
Большую роль в процессах ионизации при столкно-
вении нейтральных частиц играет, по-видимому, об-
разование утяжеленных ионов; напр.,
Аг + Аг —► Аг+ + е; Н2 + Н2 —> Н+ + Н + е
Для осуществления таких процессов требуется меньше
энергии, чем при простой ионизации, т. к. при обра-
зовании более тяжелых, чем исходные, частиц выде-
ляется нек-рая энергия.
Ионизация на поверхности. Про-
цесс образования И., как правило, резко облегчаетсп,
когда нейтральная частица находится на поверхности
твердого тела. При образовании положительного И.
на поверхности металла происходит отрыв электрона
от молекулы и переход его к металлу. В этом послед-
нем акте выделяется энергия, равная по абсолютной
величине работе выхода электрона из металла. Т. обр.,
суммарная затрата энергии на образование И. на
поверхности равна 1 — <р, где I — энергия иониза-
ции, а <р — работа выхода. Отношение числа И.
к числу нейтральных частиц в пучке, летящем с на-
гретой поверхности, будет равно: exp [— (Z—ф)/Я Г],
где R — газовая постоянная, а Т — абс. темп-ра
поверхности. Для веществ с низкими потенциалами
ионизации (тяжелые щелочные металлы) легко до-
стигаются условия, когда удар атома о поверхность
приводит к его ионизации с вероятностью, равной
единице.
Ионизация на поверхности в случае молекул пред-
ставляет более сложное явление, в к-ром большую
роль должны играть процессы диссоциации с адсорб-
цией нейтрального осколка, а также ионно-молеку-
лярные взаимодействия. При адсорбции на поверх-
ности металла электроотрицательных атомов послед-
ние могут присоединять к себе электрон, выходящий
из металла. При этом на вырывание электрона из
металла расходуется энергия ср, а при прилипании
электрона к атому выделяется энергия е, равная
энергии сродства атома к электрону. Т. обр., затрата
энергии на образование отрицательного И. на по-
верхности будет равна <р — е. Отношение числа отри-
цательных И. к числу нейтральных атомов, летящих
с нагретой поверхности, будет в этом случае равно
ехр [—(ф — е)ЛТ]. Исследование эмиссии отрицатель-
ных ионов с поверхности позволяет определить важ-
ную энергетич. характеристику атомов — величину
их сродства к электрону.
Ионизация в сильном электриче-
ском поле. Когда нейтральная частица попадает
в электрич. поле такой величины, что перепад потен-
циала на расстоянии, равном по порядку величины
размерам электронной оболочки, становится равным
потенциалу ионизации молекулы, может произойти
ее ионизация. Такие условия создаются обычно вблизи
очень тонких острий, к к-рым приложено высокое
напряжение.
II. Реакции ионов с молекулами. Столкновения И.
с молекулами могут приводить к ионно-моле-
кулярным реакциям. Основными типами
ионно-молекулярных элементарных реакций в случае
медленных частиц являются перезарядка и
переход тяжелых частиц. Перезарядкой
наз. процесс, происходящий при столкновении И. и
нейтральной частицы (атома или молекулы) и приво-
дящий к тому, что заряд И. переходит к нейтральной
частице. Переход тяжелой частицы наиболее часто
происходит при столкновении водородсодержащих
ионов или молекул. При этом переходящей к иону
или от иона частицей является либо атом Н, либо
протон, либо гидридный ион Н~. Примеры таких
реакций:
NH ь + С2Нв -э- NH3 + С2Н+
Хе+ + С2Н4 — Хе + С2Н+
Хе+ + С2Н4 -> Хе 4- С2Н+ + Н
Хе+ + С2Н4 Хе + C2Ht + Н,
1
I перезарядка
)
Н|+Н2-Н+ + Н
СН+ + сн4 —>• СН+ + снз переход тяжелых
частиц
Аг+ + Н2 -> АгН+ + Н
Н2О+ + H2S ->- HSO+ + HS •
Реакции с переходом тяжелых частиц протекают
обычно с образованием сравнительно долгоживущего
промежуточного возбужденного комплекса — ассо-
циата столкнувшихся И. и молекулы. В зависимости
от свойств этого комплекса и давления газа, комплекс
либо диссоциирует на те или другие осколки до того,
как с ним столкнется еще одна молекула, либо же,
если время жизни относительно велико, «доживает»
до такого столкновения. Одним из результатов столк-
новения может быть стабилизация комплекса вслед-
ствие передачи энергии возбуждения молекуле, с
к-рой он столкнулся. Другим результатом столкнове-
ния может быть следующая ионно-молекулярная реак-
ция. Напр., при ионизации этилена при давлении
~0,03 мм рт. ст. первичные И. С2Н4+ сталкиваются
с молекулами С2Н4, образуя вторичные И. С3Н3+.
Последние И. снова вступают в реакцию: С3Н3+ +
+ С2Н4-»С4Н5+ + СН2. Образовавшаяся частица С4Н5+
может, по-видимому, рекомбинировать с электроном:
С4Н5+ + е —> С4Н5 (см. ниже процессы рекомбина-
ции). В этом случае последовательность ионно-моле-
кулярных элементарных реакций является ионной
цепной реакцией, приводящей к образованию слож-
ной частицы С4Н5. В случае газа, состоящего из
полярных молекул, продуктом многократных после-
довательных столкновений И. с этими молекулами
являются т. наз. «сольваты» (в литературе встречаются
и другие названия таких сольватов — «клостеры»,
«гроздья»). Так, напр., в тлеющем разряде в парах
воды основной вид И. Н3О+-пН2О, где п— 0—4.
Такие частицы при рекомбинации с электроном,
естественно, диссоциируют, напр., след, образом:
Н3О+ • пН2О 4- е —► Н -J- (n + 1) Н2О
Константы скорости ионно-молекулярных реакций
с переходом тяжелых частиц, как правило, весьма
велики и достигают (при комнатной темп-ре) значений
10 9 см^/сек, т. е. приблизительно на порядок больше,
чем у самых быстрых реакций между нейтральными
частицами. Это связано с тем, что большое число
ионно-молекулярных реакций протекает без энергии
активации, а также с тем, что образование промежу-
точного комплекса в таких реакциях происходит
цод действием силы притяжения между И. и поляри-
зованной им молекулой. Такая сила является, как
правило, значительно более дальнодействующей, чем
силы взаимодействия между нейтральными частицами,
Константа скорости ионно-молекулярной элементар-
ной реакции с переходом тяжелой частицы, не имею-
щей энергии активации, практически не зависит от
темп-ры в весьма широком диапазоне значений по-
следней.
Реакции перезарядки имеют столь же большие (или
даже несколько большие) константы скорости, когда
перенос электрона не требует затраты энергии или
ее выделения, т. е. когда тепловой эффект этого пере-
носа равен нулю. В этом случае перезарядка наз.
резонансной. Так, напр., с большим эффективным
ионы
320
310
сечением происходит перезарядка атомных ионов
на одноименных атомах:
Ari- + Аг Аг + Аг+
и т. п. С другой стороны, известно, что большим
сечением обладает процесс:
Ari- + СН4 -> Ar + СН+ +Н
экзотермичпый на 32 ккал/моль. Это значит, что на
самом деле он протекает через две стадии:
Ari- + СН4 -> Ar + CHI- и СН+ _ СН- + Н
причем первая стадия, т. е. собственно перенос
электрона, является термонейтральной. Имеются ос-
нования полагать, что определенную роль в реакциях
перезарядки также может играть образование проме-
жуточного комплекса. При увеличении энергии отно-
сительного движения И. и нейтральной частицы
могут происходить процессы нерезонансной, в том
числе эндотермич. перезарядки. Примером может
служить процесс Хе+ + С2Н4 -+Хе4- CHt + СН2,
к-рый эндотермичен на 6,5 эв, однако при энергии
И., равной 100 эв, идет с большим сечением. При еще
больших энергиях И. становятся возможными про-
цессы ионизации, в к-рых вырывание электрона нз
молекулы происходит за счет кинетич. энергии И. При-
мером может служить ионизация водорода быстрыми
ионами Н+ (энергия их равна 10 кэз):
Н+ + Н2 -> II j + Н+ + е
Реакции перехода тяжелых частиц с увеличением
энергии относительного движения до 5—10 эв, по-
видимому, становятся мало вероятными. Зато начи-
нает расти вероятность процессов ударной диссоциа-
ции. При высокой энергии И. происходит такой,
напр., процесс: HJ + Аг —> Н 4- Н+ 4- Аг.
III. Рекомбинация ионов. Когда отрицательный
И. или электрон сталкивается с положительным И.,
происходит рекомбинация, в результате к-рой обра-
зуются нейтральные частицы. Скорость рекомбина-
ции И. в общем случае выражается формулой, харак-
терной для любых бимолекулярных процессов:
---— = kN^N~
dt
где Л+ и N~ — концентрации положительных и отри-
цательных И. (в 1/сл«3), к — коЪфф. рекомбинации
(в см31сек). (В дальнейшем индекс при коэфф, к будет
.указывать на тип рекомбинации, напр. — коэфф,
рекомбинации И. с электронами, \ — коэфф, реком-
бинации положительного И. с отрицательным И.).
Если положительные И. находятся в газе при низком
давлении, когда нет других частиц, способных захва-
тить электроны, и давление газа мало, то основным
процессом рекомбинации будет рекомбинация И.
с электронами. При этом положительный И. захваты-
вает электрон, а энергия рекомбинации выделяется
в виде кванта (радиационный захват электрона).
Если относительная кинетич. энергия электрона при
его столкновении с И. пренебрежимо мала, то энер-
гия кванта hv = el, где I — потенциал ионизации;
если же электрон имел энергию eU, то энергия кван-
та hv = е (I 4~ U). Коэффициент рекомбинации И. с
электронами ке возрастает с уменьшением энергии
электронов.
При рекомбинации с электронами И., содержащих
несколько атомов, выделяющаяся энергия может при-
вести к диссоциации И. на нейтральные осколки,
напр., 4~ е —> 2Н. В общем случае коэфф, реком-
бинации может быть выражен через т — время потери
возбуждения нейтральной частицей, образующейся
сразу же в процессе рекомбинации: ке«г аа/х, где
а — «размер» И. в см, а — сечение столкновения
электрона с II. При радиационном захвате
тг 4;= 10 8 сек, что приводит к величинам ке <
sg 10'1° см3{сек. Диссоциация возбужденной молеку-
лярной частицы, образовавшейся после «прилипания»
электрона к И., может произойти за время, равное
длительности одного колебания (10—13 сек)-, резкое
уменьшение величины т в этом случае объясняет
сравнительно большие величины Ле (до 10~в см3/сек).
В табл. 2 приведены величины коэфф, рекомбинации
И. с электронами Лр, измеренные в различных газах.
Таблица 2. Коэффициенты рекомбинации ионов
с электронами ke
Газ Давление, мм рт. ст. Средняя энергия электронов, ив /?е, сл<3/сек
Не 1 0.03 1,7 10-8
Ne 15-30 0,03 2 • IO"’
Аг 15-30 0.03 3•10“7*
Н.7 3-12 0.03 2,5 • 10-е
n2 2—5 0,03 1.4 • 10-е
02 2—20 0,03 2,7 • 10-7
Cs 0,01-0,1 0,15 (3-4) • 10-ю
Hg 0,3 0.15 ~ 2. • 10—io
* Сравнительно большой коэфф, рекомбинации ионов
в инертных газах объясняется образованием утяжеленных
ионов Аг? и т. п.
При повышенном давлении в газах начинает играть
роль рекомбинация И. с электронами с участием тре-
тьей частицы. Роль третьей частицы, к-рой может
быть атом, молекула, И. или электрон, заключается
в том, что третья частица, находясь вблизи рекомби-
нирующего И., берет на себя избыточную кинетич.
энергию электрона, снижает таким образом его ско-
рость и делает более вероятной его рекомбинацию;
третья частица может также принимать на себя энер-
гию, выделяющуюся в результате рекомбинации.
В газе, в к-ром имеется много молекул, обладающих
положительным сродством к электрону, свободные
электроны будут существовать весьма малое время.
Это связано с тем, что электроны, сталкиваясь с та-
кими молекулами, образуют отрицательные И. Если
время жизни электрона до его захвата молекулой
мало по сравнению со временем жизни И. до его ре-
комбинации с электроном, то основным процессом
рекомбинации И. будет процесс, в к-ром участвуют
положительные и отрицательные И. Следует отметить,
что в этом случае константа скорости рекомбинации ki
сильно зависит от относительной скорости двух П.
При большой относительной скорости положитель-
ный и отрицательный И. могут пролететь друг мимо
друга, даже в том случае, если они сближаются до
небольших расстояний.
При больших давлениях — атмосферном и выше —
процесс рекомбинации может рассматриваться как
диффузионное движение разноименных И. друг к другу
под действием их электрич. поля. При таком рассмот-
рении коэфф, рекомбинации выражается через подвиж-
ности И. и определяется выражением: ki = 4ле (|х+4~
4- |т~), где цА и |Л_— подвижности положительных
п отрицательных И. Экспериментальные значения^,
приведенные в табл. 3, обычно хорошо согласуются
с вычисленными по последней формуле.
Таблица 3. Коэффициенты рекомбинации ионов, kj,
в собственном газе при 760 мм рт. ст. и О С
Газ
fet • 10-е,
см3/сек
hj • Ю—з,
см3, сек
О3...............
Воздух...........
СО...............
1,5
1.4
0,85
SO2.............
N2O.............
Н2О.............
1.4
1.4
0.9»
» При 100°С.
321
ИОНЫ
322
Одним из процессов гибели И., особенно сущест-
венным при низкой концентрации И., является ре-
комбинация на стенке. При низких давлениях, когда
длина пробега И. больше или соизмерима с разме-
рами сосуда, содержащего газ, время жизни И. в от-
сутствии электрич. и магнитных полей определяется
просто временем пробега его между стенками. При
больших давлениях время жизни определяется време-
нем диффузии И. к стенкам и равно т=»х2/2£>, где
х — расстояние между стенками, D — коэфф, диффу-
зии И. Если в газе имеется одинаковая концентрация
положительных И. и электронов (или отрицательных
И.), то диффузия их к стенкам происходит с одинако-
вой скоростью (т. наз. амбиполярная диффузия).
Коэфф, амбиполярной диффузии определяется сред-
ним значением подвижностей диффундирующих поло-
жительных и отрицательных И. (или электронов)
цср = где р — подвижности. Коэфф, диф-
фузии связан с подвижностью соотношением D =
= —ц, где Т — темп-ра диффундирующих частиц,
R — газовая постоянная.
IV. Роль ионов в различных системах. Ионы
в радиационной химии и фотохи-
мии. В радиационной химии газов И. играют зна-
чительную, а в нек-рых случаях решающую роль
в механизме происходящих химич. превращений.
Это связано с тем, что радиационный выход (см.
Ионный выход), равный 3—4 парам на 100 эв погло-
щенной энергии, близок по величине к выходу пер-
вичных продуктов возбуждения, а также с тем, что
ионно-молекулярные реакции имеют очень большие
константы скорости. Как правило, стадия ионно-
молекулярных реакций успевает осуществиться до
рекомбинации И. Основными ионно-молекулярными
реакциями в радиационной химии являются реакции
перехода тяжелых частиц и реакции перезарядки.
Доказано, напр., что радиационное гидрирование
малых примесей олефинов в смесях инертных газов
с водородом является результатом образования ато-
мов Н по след, схеме:
Аг —► Аг+ + е; Аг+ г Н» —>• ArHt- -р н
АгН+ г е —' Ar ;- Н
При радиационном окислении азота важнейшую
роль играет процесс:
N+ + °2 —► NO+ + NO
а при радиационном крекинге метана — процесс:
CHt + сн4 -► сн+ + сн3
При фотохимия, реакциях ионные процессы в газах
могут начинать играть роль для большинства веществ
только в далекой УФ-области. Поэтому, за исключе-
нием специальных исследований процесса фотоиони-
зации, этот процесс в земных условиях практически
не играет роли в фотохимии газов.
Ионы в электрических разрядах.
Образование заряженных частиц в электрич. разря-
дах, определяя процесс переноса электричества,
играет, по-видимому, весьма малую роль в химич.
превращениях в разрядах (тлеющие разряды и дуги
при давлениях 0,1 — 10э мм рт. ст.). Это связано
с тем, что темп-ра электронов в таких разрядах обычно
не превышает (4—5) • 104°К. Этим темп-рам соответ-
ствуют кинетич. энергии электронов 4—5 эв. Такая
темп-ра достаточна, чтобы большинство электронов
было способно осуществлять процессы возбуждения
или диссоциации через возбуждение, но лишь сравни-
тельно малая доля их способна осуществлять иониза-
цию. Так, напр., в тлеющем разряде в водороде при
0,1—1 мм рт. ст. (трубка Вуда), применяемом для
получения атомарного водорода, при плотности тока
10 1 а/см2 концентрация атомов водорода может быть
6 К. X. Э. т. 2
доведена почти до 100%, а степень ионизации при этом
будет меньше 1%. В электрич. разрядах при низких
давлениях (10-2 мм рт. ст. и ниже), особенно осу-
ществляемых с помощью введения в газ пучка быст-
рых электронов, ионные процессы начинают играть
фактически такую же роль, как и при радиационно-
химич. превращениях.
Ионы в газах при высоких темпе-
ратурах. Известно, что в пламенах образуются
концентрации И., достаточные, чтобы получить зна-
чительную электропроводность. Эти И., концентра-
ция к-рых в отсутствии специальных добавок дости-
гает 1010—1011 ионов/см3, не играют существенной
роли в механизме самого горения. Однако сам факт
их образования важен для ряда приложений. Поэтому
вопросы механизма ионизации в пламенах привлекают
внимание многих исследователей. Главными ионами,
обусловливающими электропроводность пламен, яв-
ляются И., образование к-рых требует наименьшей
энергии, напр. ионы Н3О+. Влияние примесей органич.
веществ на электропроводность водородных пламен
используется в одном из методов регистрации газов
на выходе хроматография, колонок (пламенно-иони-
зационный детектор). В ряде случаев, когда стремятся
получить особо высокую степень ионизации в пла-
менах, напр., при разработке магнитогидродинамич.
преобразователей тепловой энергии в электрическую,
к газовой смеси специально добавляют легко иопизую-
щиеся в-ва (щелочные металлы).
Условия, соответствующие весьма высоким разо-
гревай воздуха, создаются при прохождении через
атмосферу метеорных тел, ракет и спутников. В ре-
зультате возникает сильная ионизация, играющая
большую роль в ряде технич. и научных задач.
Ионы в атмосфере Земли. И. в атмо-
сфере образуются под действием космич. излучения,
излучения радиоактивных элементов, находящихся
в земной коре и в воздухе, УФ-нзлучения Солнца
(в верхних слоях атмосферы). В атмосфере присут-
ствуют И. обоих знаков. Среди них имеются как
легкие И. (Of, NJ, О+ и др.), обладающие большой
подвижностью (порядка единиц см3/в- сек), так и
тяжелые (в основном те же И., но окруженные мо-
лекулами воды); подвижность таких И. мала
(~ 10 3 см3!в* сек). Концентрация И. в атмосфере
изменяется с высотой — возле поверхности Земли
концентрация легких И. составляет в среднем ок.
500 частиц/см3, а тяжелых — до 104 частицIсм3.
Присутствие заряженных частиц в атмосфере обуслов-
ливает ее электропроводность, которая возрастает
с высотой от 10-leojn-1cjn"1 у поверхности Земли
до 10 4 о.ч1 • смг1 в ионосфере. Наиболее ионизованные
слои в ионосфере расположены на высоте от 100 км
до 300 км. Концентрация И. и электронов в этой об-
ласти достигает 10е частиц/см3, что составляет 10 4—
10-е часть от полной концентрации частиц. Относи-
тельная доля электронов, отрицательных и положи-
тельных, и нейтральных атомов в ионосфере изме-
няется в зависимости от интенсивности УФ-излучения
Солнца. Эти изменения связаны с проходящими в ионо-
сфере процессами ионизации, диссоциации, рекомбина-
ции и ионно-молекулярными реакциями. В нижних
слоях ионосферы основными И. являются NO+; на
высоте 100—120 км наряду с И. NO+ в заметном ко-
личестве появляются И. Ot. На высотах 140—150 км
в составе ионосферы появляются И. О+, к-рые на
высотах, ббльших 200 км, становятся преобладающими.
Лит.: Месси Г., Б а р х о п Е., Электронные и ионные
столкновения, пер. с англ., М., 1958; Энгель А., Ионизо-
ванные газы, пер. с англ., М., 1959; Field F. Н., Frank-
lin J. L., Electron impact phenomena and the properties of
gaseous ions, N. Y., 1957; Бучельникова H. C.,
Отрицательные ионы, «УФН», 1958, т. 65, вып. 3, с. 351;
Миртов Б. А., Газовый состав атмосферы Земли и методы
его анализа, М., 1961. В. Л. Тальроае, Е. Л. Франкевич.
323
ИОНЫ-РАДИКАЛЫ — ИОЦИЧА РЕАКЦИЯ
324
ИОНЫ-РАДИКАЛЫ — частицы, несущие одно-
временно заряд (ионы) и имеющие неспаренный
электрон (свободный радикал). И.-р. могут образо-
ваться из валентно-насыщенных частиц в результате
захвата или отдачи одного электрона:
В4-е-->В- В: — е_-> В+
К числу неорганич. И.-р. относятся устойчивые ионы
металлов переменной валентности (Си2+, Сг3+, Мп2+,
Fe3+, V2+) и такие анионы-радикалы, как SO7,
к-рый при рекомбинации дает валентно-насыщенный
анион дитионовой к-ты;
so^ + -Cu2+ -> . so3 + Cu+
2 -so? s2ob
В органич. И.-р. носителями неспаренного электро-
на и ионного заряда могут быть как гетероатом, так
и атом углерода; обычно они стабилизуются за счет
сопряжения и рассредоточения радикального и ион-
ного зарядов. К числу органич. И.-р. относятся ка-
тионы и анионы мерихиноидных солей и нек-рые дру-
гие устойчивые соединения, а также неустойчивые
частицы, промежуточное образование к-рых предпола-
гается в ряде реакций. Свободные радикалы, в к-рых
имеется семиполярная связь, напр. двуокись азота,
дифенилокись азота, можно рассматривать как би-
полярные И.-р.:
+/0~ CeHs. +
•N< )N-0~
хо ceHZ
Такие частицы, как правило, проявляют двойственную
реакционную способность;
/0
+ -0 «R-NC
R-.. .Nf < 40
\0-’R-0-N=0
Пример устойчивых И.-р. — ионы триариламминия.
Соли триариламминия образуются при окислении
триариламинов и тетраарилгидразинов:
Вг2
(CeH6)3N: -> [(CeH3)3N-]+[Br ... Вг2]-
(n-CHsCeH4)3N: ----------
(O2N)3CeH2OH
-> [(n-CH3CeH4)3N-]+ [(O3N)3C0H2O]-
.. .. C104
(n-CH3CeH4)2N—N(CeH4CH3-w)2 --►
->[(п-СНзСвН4)2Н-Н(СвН4СНз-п)2]+С1ОГ
Триариламин здесь проявляет свойства благородного
металла, к-рый образует соли с к-тами в присутствии
окислителей. Соли триариламминия имеют глубокую
окраску и являются парамагнитными соединениями.
Триариламин превращается в И.-р. за счет отдачи
одного электрона; триарилбор, наоборот, присоеди-
няет электрон, образуя анион триарилбора. Так,
трифенилбор взаимодействует с натрием, превра-
щаясь в соответствующую натриевую соль, в к-рой
атом бора является носителем как неспаренного
электрона, так и отрицательного заряда:
(СвН5)3В + -Na -* [(CeH3)3B.]-Na+
Радикальный характер аниона триарилбора подтвер-
ждается его реакциями, напр.;
2 [(СвН5)3В.]-Na+ + СН3ОН [(CeHs)3BH]-Na++
+ [(CeH5)3BOCH3]-Na+
Промежуточное образование И.-р. предполагается,
напр., при ацилионовой конденсации сложных эфиров:
2R-C-0R’+ 2 -Na -> 2 FR-C-OrI Na+ ->
& L о- J
R'O OR'
I I
R-C-C-R
I I
-o o-
2Na+ ------►
—2R’0Na
R-C-C-R
Й II 2Na
4-2HOH
2Na+------->
- 2NaOH
R-C-CH-R
& ОН
И.-р., подобно незаряженным свободным радика-
лам, способны инициировать цепные реакции, напр.
полимеризацию'.
R"
R.
R.
R'-
R" R
+ -so4
>с----с-
iso? 4'R"'
R R"
-* ~ >C-----C---C---C- —* и T. д.
Rf/I II I
0S03 R"' R' R'"
См. также Ионы и Радикалы свободные.
Лит.: Хюк к ель В., Теоретические основы органи-
ческой химии, пер. с нем т. 1, М., 1955, с. 156; Семенов
Н. Н., О некоторых проблемах химической кинетики и реак-
ционной способности, 2 изд., М., 1958, с. 256. Е. М. Рохлин.
ЙОХИМБИН (квебрахин) C21H2eO3N2, мол. в.
354,45 — алкалоид коры дерева Corynanthe yohimbe
Shum, сем. маревых (Rubia-
ceae), произрастающего в цент-
ральной части Африки, и коры
южноамериканского растения
Quebracho Ыапсо, сем. кутро-
вых; бесцветные кристаллы с
т. пл. 235—237° (из разб. спир-
та)! [а]р = +105° (пиридин);
легко растворим в спирте и
R
6
э
10
и
12
Е
N
I
Н
Н-'
14
N
D
н
1э
18
И
он
СН3ООС i,
хлороформе, трудно — в эфире. Кристаллич. соли:
хлоргидрат, т. пл. 300—302° (с разл.); нитрат,
т. пл. 269—270°; тартрат (6Н2О), т. пл. 213°
и, вторично, 278°; иодметилат (1Н2О), т. пл. 250°.
По строению И. близок к резерпину. Известны алка-
лоиды, стереозомерныеИ.: аллоиохимбин, р-иохимбин,
псевдоиохимбин, раувольсцин, 3-эпи- а-иохимбин, рау-
химбин (коринантин), изораухимбин и др. Строение
И. подтверждено полным синтезом этого алкалоида.
И. снижает кровяное давление и возбуждает дыха-
ние; в больших дозах вызывает паралич дыхатель-
ного центра, в умеренных — прилив крови к коже и
слизистым оболочкам и возбуждает половую деятель-
ность. Обладает симпатолитич. и слабым местноане-
стетич. действием. Применяют в виде хлоргидрата
в ветеринарии для повышения производительной
способности и лечения импотенции быков и жеребцов,
и иногда в медицине как гипотенсивное.
Лит.: Яхонтов Л. Н., Усп. химии, 1957, 26, вып. 2,
239; Tamelen Е. van [а. о.], J. Amer. Chem. Soc., 1953,
80, № 18, 5006. А. Е. Васильев.
ИОЦИЧА РЕАКЦИЯ — взаимодействие ацетиле-
новых углеводородов с магнийорганич. соединениями
с образованием алкенилмагнийгалогенидов:
RC=CH + R'MgX -> RCSCMgX + R'H
Этинилмагиийбромид находится в равновесии с ди-
магнийдибромацетиленом и ацетиленом:
2HC=CMgBr 77 BrMgC^CMgBr 4- HCHCH
В зависимости от условий реакции можно сдвигать
равновесие в желаемом направлении и получать в
основном как продукты моно-, так и дизамещения.
Магнийгалогеннроизводные ацетиленов (реактивы
Иоцича) менее активны, чем реактивы Гриньяра,
но все же легко вступают во многие характерные
для магнийорганич. производных реакции, что откры-
вает широкие пути синтеза разнообразных органич.
соединений. Реакция с галогеналкилами или диалкил-
сульфатами приводит к ацетиленовым углеводородам;
RC=CMgX -ь R'X -+ RC=CR'
С дицианом NCGN или хлорцианом C1GN образуются
нитрилы ацетиленового ряда:
Rt&CMgX -+ RCHCCN
G эфирами муравьиной к-ты НСООС2Н5 или орто-
325
ИПРАЗИД — ИРИДИЙ
326
муравьиным эфиром HC(OR)3 — ацетиленовые альде-
гиды
RCEZCMgX RC=CCHO
Конденсация с хлорангидридами или ангидридами
кислот приводит к ацетиленовым кетонам;
RC=CMgX ->• RCHCCOR'
а с СО2 к ацетиленовым кислотам. Ацетиленовые
эфиры лучше всего получать взаимодействием реак-
тивов Иоцича с ацеталями и а-галогенэфирами:
RR'C(OC2H6)a
RCHCMgX------------- RC=C-CRR'OC2H5
xCRR'OC2H6
Наиболее изучена реакция реактивов Иоцича с аль-
дегидами и кетонами:
RRCO + RCaCMgX -> RC=CC(OH)RR'
причем с незамещенным ацетиленом, в зависимости от
условий реакции, образуются преим. либо этинильные
спирты RR'C (ОН)С = СН (при введении ацетилена
под давлением), либо у-гликоли общей формулы
RR'C (ОН) С = СС (ОН) RR'. Реакция альдегидов и
кетонов с магнийбромэтоксиацетиленом приводит к
синтезу р-оксиальдегидов, р-оксикислот и аф-непре-
дельных кислот.
он
R\ C2H5OC=CMgBr R, I
)CO —----------► >C-C^COC2H5->
R’z R'z
OH OH _
Pd(H2) R. I % l /U
------->- У-СН=СНОС2Н3 -> >C-CH2CC
R'/ R'z xh
OH
H2O Ry I Ry
------->• )C-CH2COOC2H6 -> >C=CHCOOC2H5
HCI-----R'Z R'z
Реакция с магнийбромэтоксиацетиленом широко ис-
пользуется также в синтезе сложных природных со-
единений: витамина А, каротиноидов, гормонов коры
надпочечников и др. Реакция открыта Ж. И. Иоцичем
в 1902.
Лит.: Бергельсон Л. Д., в кн.: Реакции и методы
исследования органических соединений, кн. 4, М., 1956.
Н. Гамбарян.
ИПРАЗИД (марсилид, ипрониазид, 1-изоникотино-
ил-2-изопропилгидразин) C9H13ONS, молекулярный вес
179,22 — бесцветные кри-
О„ , сталлы: т. пл. 110—114°;
CONHNHCmcH3)s хорошо растворим в воде,
спиртах, хлороформе, пло-
хо — в бензоле, эфире, СС14; т. пл. дихлоргидрата
И. 227—228°. С нитропруссидом натрия при подщела-
чивании И. дает красно-оранжевое окрашивание,
с сульфатом меди — изумрудно-зеленое окрашивание
с одновременным выделением пузырьков газа; при
нагревании раствор приобретает желтую окраску
с зеленоватым оттенком. Количественное определение
И. основано на потенциометрия, титровании нитри-
том натрия точной навески И., растворенной в смеси
ледяной уксусной и конц. соляной к-т (7 : 5). И. полу-
чают при взаимодействии гидразида изоникотиновой
к-ты с ацетоном в изопропаноле с последующим гидри-
рованием реакционной массы в присутствии платино-
вого катализатора:
N
LONHNHj— N( Y-CONHN-ClCHah
ONH№£H(CH3)8
И. является эффективным ингибитором моноамино-
оксидазы; применяется при лечении депрессивных
состояний, а также для улучшения терапевтич. эф-
фекта алкалоидов раувольфии. В отличие от обычных
стимуляторов нервной системы (кофеин, фенамин,
пиридрол), действие И. развивается постепенно и
сохраняется длительное время.
Лит.: Машковский М. Д., Ж. невропатол. и пси-
хиатрии, 1959, 59, вып. 4, 385; Fox Н. Н., Gibas J. Т.,
J. Organ. Chem., 1953, 18, № 8, 998; McMillan Fr. H.
[a. о.J, J. Amer. Pharmac. Assoc., 1953, 42, № 8, 457.
Л. H. Яхонтов.
ИПРИТ — см. В, Дихлордиатилсулъфид.
ИРИДИЙ (Iridium) Ir — химич. элемент VIII гр.
периодич. системы Менделеева, принадлежит к плати-
новым металлам-, п. н. 77, ат. в. 192,2. Природный
И. — смесь двух стабильных изотопов: 1г191 (38,5%)
и 1г1эз (61,5%). Поперечное сечение поглощения тепло-
вых нейтронов атомом И. 440 ± 20 барн. Из искус-
ственно радиоактивных изотопов важнейшие 1г192
(7\/2 = 74,37 дней) и 1г194 {Ух/я — 19 час.); первый
из них применяется как источник у-излучения в гамма-
дефектоскопии. Конфигурация внешних электронов
атома И. 5d’6s2. Энергия ионизации (в эв):1г° —>• 1г+
ок. 9,2; 1г+ —► 1г2+ 16 (вероятно).
И. открыт в 1804 Теннантом на основании еще ранее
замеченного изменения цвета его соединений при вос-
становлении Ir(IV) — Ir(III). Назван «И.» от греч.
ipig — радуга, вследствие разнообразия окраски его
солей. Химия И. и методы разделения платиновых ме-
таллов детально изучены К. К. Клаусом (1841—44),
чистый И. выделил М. Козицкий (1840—41). Содер-
жание И. в земной коре 1 • 10~’ вес. %. В самородном
состоянии находится редко, чаще — в виде минера-
лов группы осмистого иридия, встречающихся как
самостоятельно, так и в самородной платине. В при-
родных условиях И. и осмий изоморфно замещают
друг друга, образуя твердые р-ры; реже в состав
твердых р-ров входят также рутений и родий. К мине-
ралам этой группы относятся: невьянскит, или
осмистый и р и д и й (Ir, Os), сысертскит,
или иридистый осмий (Os, 1г), родиевый
невьянскит (Ir, Os, Rh), рутениевый невьянскит
(Ir, Os, Ru), родиевый сысертскит (Os, Ir, Rh), руте-
ниевый сысертскит {Os, Ir, Ru). Наиболее широко
распространен невьянскит — хрупкий минерал оло-
вянно-белого или светло-серого цвета с металлич.
блеском; плотн. 17,0—21,0; твердость ок. 6—7. Мине-
ралы группы осмистого И. встречаются в корен-
ных и россыпных месторождениях платины и золо-
та — на Урале и в Сибири (СССР), в Калифорнии
(США), на острове Тасмания, в Трансваале (Ю. Афри-
ка) и др.
Физические и химические свойства. И. — сереб-
ристо-белый, очень твердый и хрупкий металл;
кристаллизуется в гранецентрированной кубич. ре-
шетке, а = 3,8312 А. Атомный радиус 1,35 А; ионный
радиус 1г4+ 0,75 А. Плотн. 22,4 (18°), т. пл. 2 410°;
т. кип. 5 300° (вероятно). Уд. теплоемкость 0,0312
кал/г- град (0—100°); термич. коэфф. линей-
ного расширения 6,5- 10~в (0—100°); уд. электро-
сопротивление 5,40 мком • см (25°); температурный
коэфф, электросопротивления 39,25- 10~4. Металлич.
И. слабо парамагнитен с атомной магнитной воспри-
имчивостью 35- 10~« (при комнатной темп-ре). Ме-
ханич. свойства И. (при комнатной темп-ре): модуль
нормальной упругости 52 000 кГ/мм?-, предел проч-
ности при растяжении 23 кГ)ммг-, относительное удли-
нение 2%, твердость по Бринеллю 164 кГ/мм*. При
высокой темп-ре прочность и упругость И. значи-
тельно ниже, пластич. свойства и относительное
удлинение не изменяются.
И., будучи благородным металлом, весьма устой-
чив по отношению к химич. воздействиям. В виде
6’
327
ИРИДИЙ
328
компактного металла И. нерастворим не только в обыч-
ных кислотах, но и в царской водке; в состоянии
весьма тонкого измельчения (напр., после сплавле-
ния с цинком) медленно растворяется в последней.
Обычными методами растворения порошка И. яв-
ляются хлорирование его в смеси с NaCl и щелочно-
окйслительная плавка с NaOH -f- NaNOa или Na2O2
с последующим растворением сплава в НС1. Губча-
тый И. может быть переведен в раствор электролити-
чески, с использованием переменного тока. С кисло-
родом взаимодействует лишь выше 600°, с фтором,
хлором, серой — при темп-ре красного каления.
В соединениях обычно 3- и 4-валентен, редко 1-,
2- и 6-валентен. Известны окислы 3-, 4- и 6-валент-
ного И. Низшие окислы 1г2О и 1гО неустойчивы и
не выделены. 1г2Оа (получаемый сплавлением метал-
лического И. с KHSO4) разлагается при нагревании
до 400° по реакции 21г2Оа = 31гО2 + 1г. Гидро-
окись 1г(ОН)а образуется при действии щелочи па
раствор Naa [1гС1е] в атмосфере инертного газа. На
воздухе окисляется с образованием синего гидрата
окиси 1гО2 • 2Н2О. При нагревании И. на воздухе
или в токе О2 образуется 1гО2; при темп-ре свыше
1100° происходит частичная диссоциация окисла
1гО2 —► 1г -|- О2. Шестиокись 1гОа выявлена лишь
в соединениях со щелочами типа 4Na2O- 1гОа. При
нагревании И. в токе хлора образуются, в зависи-
мости от температурных условий, хлориды И.:
IrCl, 1гС12 и 1гС1а. При давлении хлора 760 мм рт. ст.
их темп-ры диссоциации следующие: 1гС1а < 763°,
1гС12 > 763° и < 773°; IrCl > 773° и <798°. IrCl —
медного цвета, 1гС12 — серого, IrCl — зеленого. Хло-
риды И. нерастворимы в кислотах и разб. щелочах.
При нагревании 1гС1а в атмосфере хлора под давле-
нием можно полечить 1гС14 — неустойчивое соедине-
ние. Известны также соединения И. с другими гало-
генами, напр. при нагревании И. с фтором обра-
зуются желтые кристаллы IrFe. Известны суль-
фиды И.: IrS, Ir2Sa, IrS2, Ir3S8. Нагреванием И.
с серой при 700° получают IrS — черный порошок,
нерастворимый в воде и кислотах. При действии серо-
водорода на раствор Na3IrCJe медленно (даже при
100°) образуется коричневый осадок гидросульфида
Ir (SH)a • 2Н2О. Гидросульфид неустойчив и легко
разлагается, что используется для получения хими-
чески чистого И. При действии H2S на раствор
Na2 [ 1гС1в] образуется соединение состава Ir2S3 •
• 3H2S.
И. образует многочисленные комплексные
соединения. Координационное число этих со-
единений, как правило, равно 6. Особенно большое
значение имеют комплексные хлориды 3- и 4-валент-
ного И. — хлориридиты (иридий-3-гексахло-
риды) и хлориридаты (нридий-4-гексахло-
риды). Хлориридиты могут существовать в двух фор-
мах: Меа [ 1гС1е] или Ме2 [1г (Н2О) С15]; хлориридаты —
в одной форме: Ме2 [1гС1е]. Хлориридатыщелочных
металлов (за исключением лития и натрия) малораство-
римы в воде; наиболее трудно растворим хлориридат
цезия. Растворимость хлориридата аммония в воде
(ок. 0,9%) резко снижается (0,4%) в конц. р-ре хло-
ристого аммония, что используется в аффинаже пла-
тиновых металлов. Хлориридат натрия может быть
получен при темп-ре красного каления действием
хлора (или хлорирующего агента, напр. СС14) на
смесь тонкого порошка И. с NaCl. Этот способ приме-
няют для извлечения хлором платиновых металлов из
концентрата. При выпаривании разб. р-ров хлорири-
даты частично восстанавливаются до хлориридитов.
Последние образуются также при восстановлении
хлориридатов сероводородом, сернистым газом, окса-
латами, сахарами и др. В водном р-ре хлор-
иридиты легко переходят в аквопентахлориридиты
Ме2 [1гН2ОС15]. Во избежание этого следует вводить
в раствор избыток щелочного хлорида. Кроме ука-
занных комплексных соединений, И. образует весьма
устойчивые комплексы с NH3, NO2, SO2£. Гексани-
триты типа Na3 [Ir (NO3)6] используются в аффинаже
и анализе. Известен ряд соединений типа: [1гАе] Х3
(где А — NH3, пиридин, органические амины, X —
галоген), а также соединения переходных рядов
[Ч-Л1 хз-я и Ме3_я [1гХв.пА„], где Me-К,
Na, NH4, например [Ir(NH3)4Cl2]Cl, Me2[IrNH3Cl5],
Получен ряд смешанных комплексных нитрито-,
хлоро- и аминосульфитов типа: Na5 [Ir(SOa)2 Cl4],
К4 [ Ir(SO3)(N О2)3]. При действии концентрированной
серной кислоты на Ка[1гС1в] или (NH4)a [1гС1в] в
присутствии K2SO4 или (NH4)2SO4 образуются соеди-
нения зеленого цвета: Ка [Ir(SO4)a]H2O или аммоний-
ные соли двух кислот — H2[Ir(SO4)2(OH)H2O] и
Н3 [Ir (SO4)2 (ОН)2]. При растворении 1г2Оа • хН20
в серной кислоте в атмосфере инертного газа обра-
зуется сульфат Ir2(SO4)a • хН20. В присутствии суль-
фатов К, Cs, NH4 из раствора этого соединения могут
быть выделены желтые соли состава Me2SO4 •
• Ir (SO4)3 • 24Н2О. Строение и свойства сульфатов
И. мало изучены.
Аналитическое определение. Ка-
чественно И. можно обнаружить по реакциям соеди-
нений Ir(IV). H2S осаждает коричневый сульфид
IrS2, растворимый в сульфиде аммония. При добавле-
нии щелочи к раствору соли Ir(IV) раствор окра-
шивается в голубой цвет, что обусловлено обра-
зованием коллоидного раствора гидрата окиси Ir (IV).
NH4C1 осаждает темно-красный гексахлориридат
аммония (NH4)2IrCle. Металлический цинк восста-
навливает И. до металла (металлич. И. имеет серую
окраску).
При выпаривании раствора соединений И. с конц.
H2SO4 или НаРО4 в присутствии окислителей (HNOa,
НС1О4) образуются комплексные сульфаты или фос-
фаты Ir(IV), окрашивающие раствор в сине - фио-
летовый цвет. И. может быть также обнаружен
по реакции хлористого И. с бензидином, при к-рой
сразу же появляется синее окрашивание [Au (III)
и окислители должны отсутствовать]. В резуль-
тате умеренного восстановления соли окиси И. пе-
реходят в соли закиси, окрашенные в зеленый
цвет.
Количественно И. определяют весовым методом
в виде металла с использованием в качестве осади-
теля 2-меркаптобензотиазола (тиомочевины). Осадок
прокаливается до металла в токе водорода (И. можно
также выделять в осадок в виде гидроокиси, сульфида
или хлориридата аммония). Спектрофотометрически
И. может быть определен измерением интенсивности
сине-фиолетовой окраски, получаемой при действии
на раствор соли Ir(IV) при нагревании смеси хлорной,
фосфорной и азотной к-т. Потенциометрич. методы
определения И. основаны на титровании раствора
хлориридата восстановителями: Си2С12, гидрохиноном,
аскорбиновой кислотой. Отделение неблагородных
металлов от И. можно производить гидролитич.
осаждением из растворов, содержащих платиновые
металлы в виде комплексных нитритов, а также с по-
мощью ионообменных смол типа КУ-2. От Pt Ir
может быть отделен гидролитич. осаждением в при-
сутствии бромата. Pd количественно отделяется от 1г
осаждением диметилглиоксимом. Отделение Rh от 1г
достигается восстановлением родия солями Ti (III),
осаждением родия меркаптобензотиазолом в присут-
ствии восстановителей и с помощью нитритно-суль-
фидного метода.
Получение. И. добывается совместно с платиной
из коренных, сульфидных (медно-никелевых) и рос-
сыпных месторождений, а также из золотых россыпей
329
ИРИДИЙ — ИСКОПАЕМОЕ ТВЕРДОЕ ТОПЛИВО
330
и золотых руд в форме минералов — осмистого ири-
дия, иридистого осмия и др. При металлургия, пере-
работке медно-никелевых руд И. переходит в кон-
центрат магнитной сепарации и в шлам электролиза
никеля. Аффинаж И. основан на осаждении и разде-
лении химич. соединений платиновых металлов в за-
висимости от их растворимости, летучести и др.
свойств (см. Платиновые металлы).
При аффинаже (метод И. И. Черняева) отделение
платины от И. производится действием сахаров. При
растворении сырой платины или шлама в царской
водке платина и И. находятся в четырехвалентном
состоянии. Четырехвалентный И. при восстановле-
нии сахарами (фруктозой, тростниковым сахаром; не-
сколько медленнее — глюкозой) переходит в трех-
валентный, перестает быть изоморфным платине и
может быть отделен от хлороплатината (платина из
хлороплатината до соединений низшей валентности
восстанавливается значительно труднее). Для осаж-
дения из раствора И. в дальнейшем должен быть
окислен (азотной кислотой). В форме комплексного
или простого иона И. может быть извлечен из р-ра
ионообменными смолами.
При действии насыщенным р-ром хлористого аммо-
ния на раствор четырехвалентного И. выпадает темно-
красный мелкокристаллич. осадок хлориридата аммо-
ния: Н2[1гС16] + 2NH4C1 = (NH4)2[IrCl6] + 2НС1. При
прокаливании (NH4)2[ 1гС16] в атмосфере водорода полу-
чается чистый И. Выделение И. из иридистого р-ра
можно также осуществить по нитритно-сульфидному
методу. И. переводят в гексанитрит Na3 [Ir (NO2)6]
обработкой нейтрализованного р-ра азотистокислым
натрием.
Остальные металлы осаждают сероводородом и
сульфиды отфильтровывают. Гексанитрит в фильт-
рате разлагают соляной к-той при доведении раст-
вора до сильнокислой реакции, а затем полностью
переводят И. в 4-валентное состояние азотной
к-той и осаждают в виде хлориридата аммония.
Известен ряд работ по применению ионообменных
смол для извлечения И. из солянокислых и1 др.
р-ров.
Иногда И. при аффинаже и анализе извлекают •
плавкой на свинцовый сплав (см. Пробирный анализ,
Свинец). Для лучшего коллектирования в свинце
должно присутствовать серебро, вводимое в шихту
пробирной плавки в виде смеси хлористого серебра
с содой. Дальнейшее разделение производят азотной
к-той, после действия к-рой в осадке остаются И.,
родий, рутений и осмий. И. остается в осадке и после
дальнейшего (последовательного) сплавления с KHSO4
и затем с КОН + KNO3, тогда как другие плати-
новые металлы переходят в растворимые соеди-
нения.
Применение. Чистый И. применяется редко, напр.
(в виде исключения) для тиглей, дающих возмож-
ность изучать реакции при 2 000—2 300°. Электро-
технич. сплавы платины с И. (2,5—30% 1г) представ-
ляют наиболее надежные материалы для разрывных
контактов и потенциометров автоматически управ-
ляемой аппаратуры. Сплавы И. используются для
электродов (90% Pt, 10% 1г), термопарных прово-'
лок (40% 1г + 60% Rh, 90% 1г + 10% Ru). Весьма
высокая твердость и коррозионная стойкость природ-
ного осмистого И. позволяют применять его для ответ-
ственных измерительных приборов и мореходных
инструментов; для тех же целей применяется сплав
II. с вольфрамом (дешевле и удобнее в произ-ве).
И. часто служит добавкой (до 4%) к платине для по-
вышения твердости при изготовлении изделий. И. раза
в 4,5—5 дороже золота.
Лит.: Гринберг А. А., Введение в химию комплекс-
ных соединений, 2 изд., Л. — М., 1951; Рудницкий А. А.,
Платиноиды, М., 1959; Звягинцев О. Е., Аффинаж
золота, серебра и металлов платиновой группы, 3 изд., М.,
1945; Б е т е х т и н А. Г., Минералогия, М., 1960; Плак-
син И. Н., Металлургия благородных металлов, М., 1958;
его же, Опробование и пробирный анализ, М., 1947;
Gmelltt, 8 Aufl., Syst. — Num. 67 — Iridium, В., 1939;
Pascal, v. 19, P., 1958, p. 405—575; Mellor, v. 15,
L., 1947; то же, v. 15, 1953; Kirk, v. 10, N. Y., 1953, p.
819—859; Rare metals handbook, ed. C. Hampel, 2 ed., L.,
1961, p. 321—23; Г и л л e б p а н д В. Ф. [и др.], Практи-
ческое руководство по неорганическому анализу, пер. с англ.,
М., 1957, с. 367. И. Н. Плаксин.
ИСКОПАЕМОЕ ТВЕРДОЕ ТОПЛИВО — естест-
венные твердые горючие вещества органич. происхож-
дения, образовавшиеся из отмерших растений и планк-
тонов в результате жизнедеятельности микроорга-
низмов. В крупных месторождениях (бассейнах)
могут залегать миллиарды тонн топлива. II. т. т.
всегда содержит минеральные примеси, количество
к-рых колеблется в широких пределах. Все И. т. т.
разделяются на сапропелиты и гумолиты, различаю-
щиеся материалом, из к-рого они образовались.
Сапропелиты образуются при восстанови-
тельном разложении остатков низших организмов —
сапропеля (гниющего ила). К этим И. т. т. отно-
сятся, напр., горючие битуминозные сланцы, бог-
хеды и др. Гумолиты образовались при
окислительном разложении остатков высших расте-
ний; они, в свою очередь, делятся на гумиты,
состоящие в основном из гумусовых веществ (торф,
бурый и каменный уголь, антрацит), и л и п то-
био л и т ы, образовавшиеся из стойких элемен-
тов низших растений (спор, пыльцы, смоляных те-
лец и др.). Наибольшее значение из И. т. т. имеют
гумиты.
Существующие гипотезы происхождения И. т т.
можно разделить на два направления: абиогенные и
биогенные. Согласно абиогенной гипотезе,
И. т. т. рассматриваются как продукт превращения
веществ, входящих в состав растений — углеводов,
лигнина, белков и т. д. Сторонники биогенной
гипотезы считают, что И. т. т. образуются син-
тетически из продуктов, получающихся в результате
разложения растительных остатков за счет жизне-
деятельности микроорганизмов. Наблюдения над при-
родными явлениями с учетом свойств растений за-
ставляют отдать предпочтение второй точке зрения.
Происхождение наиболее изученного вида И. т. т. —
гумитов — представляется в следующем виде. На-
чало распада растительных остатков — процесс чисто
биогенный. В его основе лежат энзиматич.реакции
(деструкции, гидролиза, окисления, восстановления),
в результате к-рых образуются продукты, частично
используемые микроорганизмами для питания и раз-
множения; другая часть вступает в реакции полиме-
ризации, поликонденсации, взаимодействия с дру-
гими веществами. При этом образуются новые вещества,
разного строения, свойств и состава и, в частности,
гуминовые — основа гумитов. Эти синтезы, к-рые
можно рассматривать как комбинации биогенных и
абиогенных процессов, продолжаются до завершения
образования буроугольной залежи, т. е. до покрытия
органич. отложений минеральными осадочными поро-
дами. Превращения бурых углей в каменные под
воздействием темп-ры, давления, среды, в к-рой они
совершаются, следует рассматривать как процессы
абиогенные, подчиняющиеся известным физико-химич.
законам.
И. т. т. по природным запасам составляет более
97% всех горючих ископаемых (см. Топливо). Геоло-
гии. запасы только бурых и каменных углей по СССР
составляют 8 669,51 млрд. т\ тем самым Совет-
ский Союз обеспечен И. т. т. на несколько тысяче-
летий.
Качество различных видов И. т. т. характеризуется
следующими показателями (в %);
331
ИСПАРЕНИЕ
332
Вид
И. т. т.
Влага
гигроско-
пическая,
W
Зола, Ас
с
Элементарный состав
(на органич. вещество)
н
N | S
Торф...........
Бурый уголь . .
Каменный уголь
Антрацит . . . .
Сапропеди . . . .
Сапропелита . .
Сланцы........
Липтобиолиты. .
12
5-12
1-5
2
10
2-3
3-5
1-10
0,5-26,0
6,0—40,0
1,5—30,0
1,0—15,0
30—80,0
50—40,0
40—70
2-38
53-62
63-75
76-95
90-96
49—60
63—82
56-78
66—82
5-6,5
4,5-6,5.
3,5-6,3
1,3—3,0
6—8,0
7—11,0
6—10,0
6—10,0
1-4,0
0,5-1,5
1,2-2,5
• До 1,3
2,0-5,7
0,5—2,6
0,2-1,0
1,0
0,1—0,5
}о,3-12
10,3—
/ 3,0
0,5—7,0
вают адиабатическим И., а темп-ру <м — темпе-
ратурой адиабатич. И., или темп-рой мокрого
термометра (т. к. эту темп-ру показывает мок-
рый термометр психрометра). При ненасыщен-
ной парогазовой смеси т. е. жидкость
может быть охлаждена при И. ниже темп-ры
газовой среды, с к-рой она соприкасается.
Скорость И. определяется коли-
чеством пара, образующегося за единицу
времени на единице поверхности жидко-
сти (г!сек • см2, или моль!сек - см2). Ско-
9—25 рость И. зависит от соотношения числа
____ молекул жидкости, оторвавшихся с ее по-
о
29-40
14-32
1,5-17
1-2
25-40
5-22
12-36
И. т. т. — основа современной энергетики, энерге-
тич. сырье черной и цветной металлургии, а про-
дукты его переработки — исходные материалы мно-
гих отраслей химич. пром-сти.
Литл Стадников Г. Л., Происхождение углей
и нефти, 3 изд., М., 1937; Веселовский В. С., Хими-
ческая природа горючих ископаемых, М., 1955;А р о н о в С. Г.,
Нестеренко Л. Л., Химия твердых горючих, ископае-
мых, Харьков, 1960; К у харен к о Т. А., Химия и ге-
незис ископаемых углей, М., I960; Francis W., Coal,
Its formation and composition, L., [19541; Van Kreve-
1 e n D. W. and Schy er J., Coal Science, Amst. — L. —
N. Y., 1957. H. M. Караваев.
ИСПАРЕНИЕ (парообразование) — переход ве-
щества из конденсированной (твердой или жидкой)
фазы в газообразную. И. твердого тела наз. сублима-
цией, а парообразование в объеме жидкости — кипе-
нием. Обычно под И. понимают парообразование, про-
исходящее на свободной поверхности жидкости при
темп-ре ниже точки кипения при данном давлении.
И. происходит в результате теплового движения
молекул жидкости, скорость к-рых колеблется в ши-
роких пределах, сильно отклоняясь в обе стороны от
ее среднего значения. Часть молекул, обладающих
достаточно большой кинетич. энергией, вырывается
из поверхностного слоя жидкости в газовую среду.
Избыточная энергия теряемых жидкостью молекул
затрачивается на преодоление сил сцепления между
молекулами и работу расширения (увеличения объема)
при превращении жидкости в пар. И. — эндотермич.
процесс. Если к жидкости не подводится соответ-
ствующее количество энергии (тепла) извне, то в ре-
зультате И. она охлаждается (испарительное
охлаждение). Количество теплоты, поглощаю-
щейся при изотермич. процессе И., наз. тепло-
той фазового перехода (И. или паро-
образования), или скрытой теплотой И.
Различают молярную теплоту И. (теплота И. 1 моля
жидкости, кал!молъ) и весовую уд. теплоту И. (кал) г).
Теплота И. зависит от темп-ры и с повышением по-
следней уменьшается, особенно резко вблизи критич.
точки, при достижении к-рой теплота И. обращается
в нуль.
Вследствие необходимости подвода к поверхности жид-
кости теплоты фазового перехода и различия, как правило,
темп-p жидкости и газовой среды, процессу И. всегда сопутст-
вует, кроме массообмепа (переноса пара в газовой фазе),
также теплообмен. Направление и интенсивность потоков
тепла в жидкой и газовой фазах зависят от соотношения между
темп-рами основной массы жидкости (ж, поверхности раздела
фаз 1Гр и гааовой среды tr (см. рис.). Потон тепла Qa, возни-
кающий в результате конвективного теплообмена, может быть
направлен как от жидкости к газовой среде (при tГр.Хг), так
и от газовой среды к жидкости (при iyp.^r)- От основной
массы жидкости к ее поверхности подводится теплота в коли-
честве Q — Qp ±Q<p где Qp — теплота, пошедшая на паро-
образование. При испарительном охлаждении, пока не до-
стигнут предел охлаждения жидкости, всегда Qpi»Qa, т. е.
Qi»0. При trp,= tr конвективный теплообмен отсутствует
(Qa = 0), но если газовая среда не насыщена паром, И. и ох-
лаждение жидкости не прекращаются. Теоретич. предел ох-
лаждения жидкости достигается при ее темп-ре 1Ж = <гр.= tM,
которой отвечает Qa = Qp. Дальнейшее И. происходит при
постоянной темп-ре жидкости tM. В этом случае процесс назы-
верхности вследствие теплового движе-
ния, и числа молекул пара, поглощенных жидкостью
при ударах о ее поверхность. Молекулы образовав-
шегося пара проникают в газовую среду в резуль-
тате диффузии и конвекции. С возрастанием темп-ры
жидкости, то есть с ростом интенсивности теплового
движения ее молекул, скорость И. увеличивается.
Общее количество испарившейся жидкости возрастает
пропорционально площади ее свободной поверхности
(зеркала И.).
Распределение температур и направление потоков
тепла при различных режимах испарительного охла-
ждения жидкости.
Скорость И. наиболее высока при И. в вакууме
и неограниченном объеме, когда влияние существова-
ния молекул в газовой фазе весьма мало и молекулы
образовавшегося пара могут беспрепятственно уда-
ляться от поверхности жидкости. В ограниченном
стенками замкнутом пространстве, не содержащем
других паров или газов, И. жидкости без подвода
тепла извне и удаления образующегося пара приво-
дит к понижению темп-ры и повышению давления
до тех пор, пока между жидкостью и паром не уста-
навливается термодинамич. равновесие. Последнее
носит динамич. характер и определяется тем, что
количество молекул пара, поглощаемых жидкостью
(конденсирующихся) при ударах о ее поверхность,
становится равным количеству молекул, вырываю-
щихся из жидкости.
При отсутствии термодинамич. равновесия между
жидкостью и ее паром, но при наличии над поверх-
ностью жидкости относительно плотного газа (или
пара другого вещества), процесс И. существенно
замедляется и в том случае, если пар, проникающий
в газовую среду, постоянно удаляется с движущейся
парогазовой смесью. Вследствие отражения от моле-
кул инертного газа число молекул пара, вновь попа-
дающих на поверхность жидкости и частью погло-
щаемых ею, возрастает, а остальные молекулы обра-
зовавшегося пара проникают в газовую среду благо-
даря диффузии и конвекции. При обычных давлениях
333
ИСПАРЕНИЕ
334
парогазовой смеси и концентрациях в ней пара ско-
рость удаления этих молекул весьма мала по сравне-
нию со скоростью испускания молекул- жидкостью,
в результате чего у ее поверхности образуется тонкий
слой парогазовой смеси, практически насыщенный
паром.
Нарушение термодинамич. равновесия между жид-
костью и ее паром, приводящее к И., связано, согласно
кинетич. теории, с образованием на границе раздела
скачка давления и темп-ры. Кроме того, при неравно-
мерном распределении в газовой фазе темп-ры и
парциальных давлений компонентов возникают, по-
мимо обычных явлений теплопроводности и диффузии,
вторичные явления: термодиффузия и диффузионная
теплопроводность. Однако при практич. расчетах
И. этими дополнительными эффектами можно пре-
небрегать вследствие их незначительности и прини-
мать (если кривизна поверхности не слишком мала),
что парциальное давление пара у поверхности раз-
дела равно давлению насыщения при темп-ре поверх-
ности жидкости. При очень малых радиусах кривизны
поверхности И. (напр., И. очень малых капелек жид-
кости) нужно еще учитывать влияние поверхностного
натяжения жидкости, благодаря к-рому равновесное
давление пара над поверхностью раздела выше дав-
ления насыщенного пара той же жидкости над
плоской поверхностью.
Подобно тому как интенсивность теплообмена за-
висит от разности темп-p, интенсивность массообмена
при И. зависит от разности химических потенциалов
пара у поверхности раздела и в основной массе паро-
газовой смеси. Но при возможности пренебрегать
вторичными молекулярными явлениями разность хи-
мич. потенциалов может быть заменена разностью
парциальных давлений пара или разностью влаго-
содержаний газа. В соответствии с этим количество
испарившейся жидкости определяется для элемента
ее поверхности dF формулой:
^И = ₽р(^п. гр.-рп)^ =
= (®rp. ~ *) dF Укг1'шс\ (1)
где Рр — коэфф, массоотдачи, кг[м* • час • am-, Pnvp
и Рп — парциальные давления пара у поверхности
раздела фаз и в основной массе парогазовой смеси,
ат; Рх — коэфф, массоотдачи, отнесенный к разности
влагосодержаний газа, кг/м? • час; жГр и х — влаго-
содержания (т. е. содержания пара на 1 кг абсолютно
сухого газа) у поверхности раздела и в основной
массе парогазовой смеси, кг/кг.
Общее количество тепла, отдаваемого жидкостью
при И., составляет:
— “ж (гж ~~ ггр? dF =
= rdGn + аг (ггр — zr) dF [ккал/час] (2)
где аж — коэфф, теплоотдачи от основной массы жид-
кости к ее поверхности, ккал)мг • час • град; г — yj\.
теплота И., ккал!кг; аг — коэфф, теплоотдачи со
стороны газовой фазы, ккал/м2 • час • град.
Когда испаряющаяся жидкость интенсивно пере-
мешивается (напр., при стекании ее по каким-либо
поверхностям в аппарате), темп-ра массы жидкости
приблизительно равна темц-ре поверхности раздела
фаз (ггр «=«ж) и при рассмотрении процесса И. можно
принимать во внимание только тепло- и массообмен
в газовой фазе.
Коэфф. аг и рр зависят от формы и геометрич. размеров
поверхности И., характера движения парогазовой смеси (сво-
бодное или вынужденное, ламинарное или турбулентное),
давления, темп-ры, физич. свойств жидкости и газа и кон-
центраций компонентов в парогазовой смеси. Зависимость
их от указанных факторов определяется обычно опытным пу-
тем. Если коэфф, тепло- и массоотдачи и разности парциаль-
ных давлений и темп-p изменяются по поверхности И., при
расчетах берут их средние значения или расчет производится
последовательно для отдельных участков поверхности И.,
в пределах к-рых эти величины можно принимать постоянными.
Во многих практич. случаях И. происходит при малой кон-
центрации пара в парогазовой смеси и относительно малых
разностях парциальных давлений пара и темп-p. Если, кроме
того, тепловой и диффузионный критерии Прандтпя (Pr = v/a
и Ргд = v/D, где v и a — коэфф, кинематич. вязкости и темпе-
ратуропроводности смеси, D — коэфф, диффузии) для газовой
среды равны между собой или мало различаются по величине,
то можно в указанных случаях с достаточным приближением
принимать, что существует аналогия между процессами тепло-
и массообмена в газовой фазе. При этом коэфф. аг и рр оказы-
ваются связанными между собой простым соотношением
ar/₽p= XRnT/D или, если отнести коэффициент массоотдачи
к разности влагосодержаний:
аг ср
(где Rn — газовая постоянная для активного компонента
смеси — пара, Т — абс. темп-ра газовой среды и с'р — уд.
теплоемкость парогазовой смеси, отнесенная к 1 кг содержа-
щегося в ней инертного газа). Это обстоятельство позволяет
выразить количество теплоты, отдаваемой жидкостью при И.,
как
Q = Ря • Д(ср. * F (3)
где ДгСр. — средняя разность энтальпий парогазовой смеси
у поверхности раздела и в основной ее массе, ккал/кг. Расчет
процесса И. при помощи ур-ния (3) мало отличается от обыч-
ного расчета теплообмена.
Когда условия существования приближенной аналогии
между тепло- и массообменом не удовлетворяются, приведен-
ными соотношениями для коэфф, тепло- и массоотдачи, а также
ур-нием (3) пользоваться нельзя. В этих случаях, характери-
зующихся в основном значительной плотностью поперечного,
т. е. нормального к поверхности жидкости, потока пара, не-
обходимо учитывать влияние последнего на условия И. По-
перечный поток вещества (пара) влияет на И. непосредственно
в результате переноса им энергии (тепла) и косвенно — в ре-
зультате вызываемого им изменения гидродинамич. условий.
При И. бинарных жидких смесей, а тем более жидко-
стей, содержащих более двух компонентов, все ука-
занные закономерности И. сильно усложняются вслед-
ствие того, что давление насыщенных паров зависит
в этих случаях не только от темп-ры, но и от концен-
траций компонентов в смеси, а теплота И. смеси за-
висит, кроме темп-ры и теплот И. компонентов, также
от состава смеси и теплот растворения. Составы жид-
кой и паровой фаз смеси, находящихся между собой
в равновесии, различны и зависят от темп-ры. При И.
бинарной жидкой смеси образующийся пар относи-
тельно богаче более летучим компонентом, исключая
только смеси, у к-рых кривые состояния имеют экстре-
мум (максимум или минимум) и к-рые в точке экстре-
мума испаряются как чистая жидкость (см. Азеотроп-
ные смеси). В общем случае И. смесей сопровождается
изменением во времени парциальных давлений паров,
концентраций компонентов в жидкой и паровой фа-
зах, теплоты и скорости И.
Так как общее количество испарившейся жидкости
возрастает с увеличением поверхности контакта жид-
кой и газовой фаз, конструкции испарителей преду-
сматривают обычно возможно большее увеличение
поверхности И. Достигается это созданием большого
зеркала жидкости, раздроблением ее на мелкие капли
или сливом жидкости в форме тонкой пленки по к.-л.
поверхностям. Увеличение интенсивности тепло- и
массообмена при И достигается повышением скорости
газовой среды, омывающей поверхность жидкости, п
выбором более благоприятной формы этой поверхно-
сти. Повышением скорости газовой среды можно
пользоваться лишь в определенных ограниченных
пределах, т. к. с увеличением этой скорости растет
гидравлич. сопротивление аппарата.
Испарительное охлаждение жидкости часто наблю-
дается в естественных условиях, а также широко ис-
полвзуется в промышленной практике, в частности
для охлаждения циркуляционной воды в замкнутых
(оборотных) системах водоснабжения. Для испари-
тельного охлаждения воды применяются пруды-охла-
335 ИСТОЧНИКИ ЯДЕРНЫХ ИЗЛУЧЕНИЙ - ИТАКОНОВАЯ КИСЛОТА
336
дители, брызгальные бассейны и градирни, в к-рых
вода вступает в непосредственный контакт с атмосфер-
ным воздухом. И. используется в холодильных уста-
новках. И. чистых и многокомпонентных жидкостей
наблюдается при многих технология, процессах: горе-
нии жидких топлив, сушке влажных материалов,
выпаривании р-ров, кристаллизации, подогреве и
увлажнении воздуха в кондиционерах, охлаждении
газов в скрубберах и т. д.
Лит.: Берман Л. Д., Испарительное охлаждение
циркуляционной воды, 2 изд., М.—Л., 1957; Г и р ш ф е л ь-
дер Д., Кертисс Ч. и Берд Р., Молекулярная
теория газов и жидкостей, пер. с англ., М., 1961; К а р а-
петьянц М. X., Химическая термодинамика, 2 изд.,
М., 1953; Лыков А. В., Теория сушки, М.—Л., 1950;
Фукс Н. А., Испарение и рост капель в газообразной
среде, М., 1958; Эккерт Э. Р., Д р е й к Р. М., Теория
тепло- и массообмена, пер. с англ., М.—Л., 1961.
Л. Д. Берман.
ИСТОЧНИКИ ЯДЕРНЫХ ИЗЛУЧЕНИЙ — уст-
ройства, к-рые могут быть использованы для облучения
различных объектов у-лучамн, ускоренными электро-
нами, Р-частицами, нейтронами, протонами и более
тяжелыми частицами. И. я. и. применяются с целью
воздействия на физико-химич. или биология, свойства
объектов, а также на протекающие в этих объектах
процессы. Потоки частиц и у-кваптов могут быть
получены от изотопных источников, от ускорителей
частиц и ядерпых реакторов. Природа излучения, его
энергия и интенсивность являются основными харак-
теристиками И. я. и., т. к. ими определяются важней-
шие условия процесса облучения; доза, мощность
дозы, глубина и равномерность облучения. Сущест-
венными являются также форма и габариты облуча-
теля.
Из изотопных источников излу-
чений наиболее широкое применение в качестве
источника у-излучения получил Со60, приготовляемый
в ядерном реакторе по реакции Со69 (п, у) Со60. Ис-
пользуется также Cs137, выделяемый из продуктов
деления (осколков) тяжелых ядер. Важной проблемой
является использование у- и P-излучений смеси оскол-
ков в виде «концентратов», получающихся после хи-
мич. переработки тепловыделяющих элементов ядер-
ного реактора (ТВЭЛ). Эффективность этих И. я. и.
определяется временем, прошедшим от момента оста-
новки реактора до их использования в радиационном
аппарате, и возможностью получения «концентратов»
с необходимой уд. активностью. При использовании
изотопных источников у-излучепия (в частности,
Со60) с большой уд. активностью удается создать мощ-
ности поглощенных доз до 103—104 pad/сек. В качестве
источников ^-излучения применяют: Sr90, выделяемый
из продуктов деления урана и превращающийся в до-
чернин радиоактивный изотон Y90, также являющийся
Р-излучателс-м; Р32, приготовляемый в ядерном реак-
торе по реакции Р31 (н, у)Р32, и др. В качестве источни-
ков a-излучения используются Rn, Ро210. Изотопные
источники применяются в установках, предназначен-
ных для облучения разнообразных объектов (универ-
сальные установки), или в специализированных аппа-
ратах, предназначенных для проведения определенных
радиационно-химич. процессов. Преимущества изо-
топных источников излучений состоят в их простоте
и безотказности в эксплуатации; время действия
практически определяется периодом полураспада
изотопа.
Из ускорителей частиц для облучения
используются ускорительные трубки и генераторы
Ван-де-Граафа (дающие электроны, протоны, дейт-
роны), бетатроны и мощные рентгеновские аппараты
(тормозное — рентгеновское излучение), циклотроны
(а-частицы, протоны, дейтроны), линейные ускори-
тели и динамитроиы (электроны, тормозное излучение)
и др Последние создают мощность дозы в пучке элект-
ронов ~103 р!сек и в пучке тормозного излучения до
Ю3—104 р/сек.
Ядерные реакторы как И. я. и. могут
быть использованы в нескольких направлениях.
Проведение радиационно-химич. процесса при непо-
средственном контакте реагирующего вещества с деля-
щимся материалом позволяет использовать кинетич.
энергию осколков деления урана. Эта энергия состав-
ляет основную часть мощности реактора, поэтому такой
вариант использования реактора эффективен для энер-
гоемких радиационно-химич. реакций. Однако реа-
лизация этого способа связана с необходимостью
решения довольно сложной технич. проблемы очистки
продуктов реакции от радиоактивных загрязнений.
Применение смешанного потока нейтронов и у-излу-
чепия, а также использование энергии частиц, обра-
зующихся в результате ядерных реакций на легких
ядрах, в меньшей степени связано с радиоактивными
загрязнениями, но позволяет использовать только
небольшую часть энергии деления. Использование
у-излучения отработанных ТВЭЛ во время их вы-
держки, а также радиационных контуров (РК),
в к-рых какое-либо вещество (в частном случае деля-
щийся изотоп) активируется в реакторе и отдает
энергию у-излучения в установке для облучения, не
приводит к загрязнению реагирующих продуктов ра-
диоактивностью.
Эффективность использования у-излучения ТВЭЛ
определяется длительностью работы ТВЭЛ в ядерном
реакторе и в радиационно-химич. установке, мощ-
ностью реактора, приходящейся на ТВЭЛ, временем,
затрачиваемым на транспортировку ТВЭЛ из реактора
в радиационную установку, а также габаритами и
конструкцией ТВЭЛ (самопоглощение в облучателе).
Применение ТВЭЛ и РК, хотя и не столь эффективно
в смысле использования атомной энергии, как исполь-
зование кинетич. энергии осколков, но достаточно
для осуществления высокоэффективных радиационно-
химич. процессов, протекающих по цепному механиз-
му с большими выходами. Использование реактора
как источника излучений является перспективным
также и в связи с этими возможностями.
В настоящее время строятся РК, в к-рых рабочим
веществом служит сплав Jn—Ga (23% Jn) или
Jn—Ga—Sn (25% Jn, 62%, Ga), имеющие темп-ру
плавления соответственно 16° и 5°. Ок. 95%, радиа-
ционной мощности у-излучения таких сплавов обус-
ловлено изотопом Jn113, образующимся по реакции
JnI15(n, у) Jn113, при прохождении сплава вблизи
активной зоны ядерного реактора.
Лит.: Труды Всесоюзной научно-технической конферен-
ции по применению радиоактивных и стабильных изотопов
и излучений в народном хозяйстве и науке. Получение изо-
топов. Мощные гамма-установки. Радиометрия и дозиметрия,
М., 1958; Б регер А. X., Источники ядерных излучений
и их применение в радиационно-химических процессах, М.,
1960; Рябухин Ю. С. иБрегер А. X., Атомная энер-
гия, 1958, 5, № 5, 533; 1959, 7, К 2, 129; Б и б е р г ал ь
А. В., Синицын В. И., Лещинский Н. И., Изо-
топные гамма-установки, М., 1960; Large radiation sources in
Industry, v. 1—2, Vienna, 1960. ’ A. X. Epeiep.
ИТАКОНОВАЯ КИСЛОТА, мол. в. 130,10 — т. пл.
167—168°; константа диссоциации (25°): Л'1=],5-101;
К, = 2,2 10~3. И. к. (I) легко при стоянии полиме-
ризуется; при нагревании выше плавления обра-
зует цитраконовый ангидрид (II). Перманганат калия
окисляет И. к. до оксипираконовой кислоты (III).
НООС-С-СН2СООН СНз-с — с=о
|| \о ноос
сн-с=о Г’
сн2
II
/Сн2—с=о
hoz \сн2-о
I
И. к. присоединяет по двойной связи
галогены и галогеноводороды (против
правила Марковиикова). В отличие от ее
ангидрида И. к. не вступает в реакцию
Ш
н2с-с^°
I >°
Н2с=с-с^о
IV
337
ИТТЕРБИЙ — ИТТРИЙ
338
диенового синтеза. При обработке И. к. хлористым
тионилом образуется ангидрид (IV). И. к. получают при
ферментации р-ров сахара с помощью Aspergillus
itaconicus; может быть получена нагреванием лимон-
ной кислоты. Нек-рое применение находят сополи-
меры эфиров И. к. с метакрилатом, стиролом и др.;
применяется также в произ-ве синтетич. моющих
средств.
Ангидрид И. к., т. пл. 68°; диметиловый эфир, т. кип. 208°;
диэтиловый эфир, т. кип. 155°/95 Л1Л1. Н. А. Несмеянов.
ИТТЕРБИЙ (Ytterbium) Yb — химич. элемент с
п. н. 70, относится к лантанидам.
ИТТРИЙ (Yttrium) Y — химич. элемент III гр.
периодич. системы Менделеева; п. н. 39, ат. в. 88,905.
Природный И. состоит из одного стабильного изо-
топа Y89. Сечение захвата тепловых нейтронов
1,21 ± 0,06 барн (на атом). Из искусственно радио-
активных изотопов важнейшие Y91 (Т1/« = 57,5 дней)
и уэо (Ti/з = 64,24 часа), образующиеся при делении
урана и тория как дочерние продукты Sr91 и Sr90, и др.
Конфигурация внешних электронов атома И. 4d 5s2.
Энергии ионизации (в эв); Y°—>-Y+—>-Y2+-*Y3+ соот-
ветственно равны 6,38; 12,40; 20,49. В 1794 Ю. Гадо-
лин обнаружил в минерале из Иттербю (Швеция)
неизвестную «землю», к-рую в 1797 А. Экеберг назвал
иттриевой землей. Впоследствии из этого минерала
были выделены окислы редкоземельных элементов —
У, Eu, Gd, Tb, Dy, Но, Er, Tu, Yb и Lu. Металлич.
(нечистый) И. впервые получил Ф. Вёлер в 1828.
Среднее содержание И. в земной коре 2,8 • 10"3
вес. %. И. входит в состав многих редкоземельных
минералов и в природе всегда встречается совместно
с редкоземельными элементами иттриевой группы
(особенно Dy, Gd, Er, Yb), а также в нек-рых минера-
лах совместно с элементами цериевой группы. Глав-
йейшими минералами, содержащими И. и редкоземель-
ные элементы иттриевой группы, являются: ксенотим
YP04, фергюсонит (Y, Er, Се, U) • (Nb, Та, Ti)O4, эв-
ксенит (Y, U)(Nb, Ti)2O6, гадолинит Y2FeBe2Si.2O10,
браннерит (U, Са, Fe, Y)3Ti6Oi6, иттропаризит
Ca(Y, Ce).2[CO3]3F2, иттрофлюорит (Са, Y, Ce)F2_3, та-
леиит Y,[Si2O7], иттриалит (Y, Th)2[Si2O,].
Основными промышленными типами месторождений
И. являются: россыпи, содержащие ксенотим, эвксе-
нит и фергюсонит, гранитные пегматиты с ксеноти-
мом и титано-тантало-ниобатами И., а также гидротер-
мальные месторождения, связанные с субщелочными
гранитоидами, содержащие ксенотим и иттропаризит
в ассоциации с ферриторитом. Кроме того, значи-
тельное количество И. может быть получено попутно
при переработке нек-рых руд урана (золото-браннери-
товые конгломераты, уравсодержагцие фосфориты,
угли), тория (ксенотим-ферриторитовые месторожде-
ния), ниобия и тантала (фергюсонитовые, эвксенито-
вые, самарскитовые руды). Главнейшими месторо-
ждениями И. являются эвксенитовые россыпи шт. Ай-
дахо в США и золото-браннеритовые конгломераты
района Блайнд-Ривер—Алгона в Канаде.
Физические и химические свойства. Свободный И. —
металл с гексагональной плотноупакованной структу-
рой; параметры решетки: а = 3,6474 А, с = 5,7306 А.
Атомный радиус 1,81 А, ионный радиус Y3+ 0,97 А.
Плотн. 4,472, т. пл. 1 525 ± 25°, т. кип. 3 025°. И.
имеет и высокотемпературную модификацию (темп-ра
перехода 1470—1490°) с объемноцентрированной ку-
бич. решеткой, а = 4,11 А. Теплота плавления
4,1 ккал/г-атом, теплота испарения 80 ккал/г-атом.
Уд. теплоемкость (в кал/г-граду. 0,074 (50°); 0,081
(400°) и 0,124 (1000°); средний коэфф, линейного рас-
ширения 9,3 10“в (25—1000°), теплопроводность (в
кал/см сек - граду 0,023 (0°); 0,026 (200°) и 0,047
(1000°); электросопротивление 69 ± 3 мком • см (25°).
Чистый И. — мягкий металл; по своей прочности и
упругим свойствам он занимает промежуточное поло-
жение между А1 и Ti. Твердость по Бринеллю 45—
50 кГ/мм2', модуль упругости 6328—7000 кГ/мм2',
модуль сдвига 2672 кГ/мм2, коэфф. Пуассона 0,265,
коэфф, сжимаемости 2,09 • 10fl см’ЧкГ.
Компактный металл медленно окисляется в кипящей
воде, легко растворяется в обычных к-тах, медленно —
в уксусной и более или менее инертен к плавиковой
к-те. При окислении И. на воздухе в интервале темп-р
370—425° образуется черпая плотно пристающая
окисная пленка; интенсивное окисление начинается
выше 760°. И. легко взаимодействует с галогенами,
с водородом образует в интервале 315—1540° устой-
чивые металлич. гидриды различного состава. При
760° И. соединяется с азотом с образованием серовато-
черного нитрида YN. При определенных темп-pax И.
взаимодействует также с углеродом, серой, фосфором
и т. д. О к и с ь И. Y2O3 — бесцветные кристаллы,
кубич. решетка, а = 10,61 А, плотн. 5,85, т. пл. 2415°,
т. кип. 4300°. Гигроскопична и поглощает из воздуха
СО2. Имеет слабоосповные свойства; практически
нерастворима в воде (8 • 10“6 моль!л), растворяется
в минеральных к-тах. Окись И. диамагнитна. Наблю-
дающаяся небольшая парамагнитность окиси И. свя-
зана с присутствием следов парамагнитных лантанид-
ных ионов. Гидроокись Y(OH)3 осаждается из р-ров
нитратов при pH 6,78, имеет основной характер. Про-
изведение растворимости 1 • 10“24 (18°) и 8,1 • 10“23
(25°). При стоянии на воздухе Y(OH'3 постепенно пре-
вращается в карбонат.
Хлорид, нитрат, ацетат, сульфат И. растворимы
в воде; фторид, карбонат, фосфат, оксалат, феррициа-
нид мало растворимы, Поны Y3гв растворах бесцветны.
Большинство солей И. кристаллизуется со значитель-
ным количеством молекул воды; таковы карбонат
Y2(CO3)3 • ЗН2О, феррицианид Y[Fe(CN)6] • 4Н2О, хло-
рид и нитрат — YCJ3 • 6Н2О и Y(NO3)3 • 6Н2О, суль-
фат Y2(SO4)3 8Н2О, оксалат Y2(C.2O4)3 • 9Н2О. Многие
соли И. с солями одновалентных металлов обра-
зуют двойные и комплексные соли, напр. Me[Y(SO4)2]
и Meg[Y(SO4)3], Me[Y(CO3)„] и Me3[Y(C2O4)3] • Н2О.
Известны соединения И. с лимонной к-той— Y(H2Cit'3,
молочной к-той — Y(C3H6O3)3, а также этиленди-
аминтетраацетаты, нитрилтриацетаты (Y2X3) и т. п.,
в форме к-рых можно более или менее успешно отделить
И. от лантанидов ионным обменом на сульфокатиони-
тах. Фтористые калий, рубидий и цезий образуют
с фтористым И. комплексные соединения типа крио-
лита K3(YF6). В разведенных р-рах соли И. гидро-
лизуются с образованием основных солей. При про-
каливании кислородсодержащие соли И. переходят в
основные соли, оксисоли, а затем в окислы.
Важное отличие И. от лантанидов состоит в том, что
по свойствам своих соединений И. не следует строго
за каким-либо одним из лантанидов, а занимает среди
них то одно, то другое место. Напр., при кристаллиза-
ции броматов И. располагается в ряду Но—Y—Ег;
при хроматографии нитратов на y-ALOs — в ряду
Y—La—Pr—Nd; в случае кристаллизации карбонатов
(при 0°) — в ряду Yb—Lu—Y, и т. д. Такая особен-
ность II. позволяет, перемещая его относительно
элементов цериевой и иттриевой подгрупп, добиваться
более эффективного удаления примесей лантанидов из
препаратов И.
Аналитическое определение й. в
чистых солях не представляет больших затруднений
и может быть проведено как химич., так и физич.
(напр., спектральными) методами анализа Характер-
ными качественными реакциями на И. является обра-
зование осадков Y(OH)3, YF3 и У2(СгО4)3, нераствори-
мых в избытке осадителя. Количественно И. в чистых
препаратах может быть определен осаждением в виде
гидроокиси или оксалата с последующим прокали-
339
ИТТРИЙ - ихтиол
340
ванием до Y2O3 (весовая форма). Осаждение И. в виде
оксалата используется для весового, объемного, по-
тенциометрического и др. методов, к-рые проводятся
так же, как и при определении суммы редкоземель-
ных элементов.
Для определения И. в смеси с др. элементами (также
и др. редкоземельными элементами) используют в ос-
новном физич. методы анализа: эмиссионный спект-
ральный анализ, рентгеноспектральный анализ, а
также радиохимии, методы.
Получение. Исходные чистые соединения И. полу-
чают разделением соединений редкоземельных элемен-
тов. Если перерабатывается смесь иттриевых земель,
вначале выделяют 90—95%-ный концентрат И. Для
этого может быть использован метод феррицианидного
осаждения, осаждение по основности p-ром NH4OH
(разб. или конц.) из нитратного р-ра, осаждение двой-
ных сульфатов, хроматов, оксалатов из щелочных
р-ров в присутствии комплексонов (трилона Б или А), а
также экстракционные методы разделения, напр. с три-
бутилфосфатом. Концентрат И. доочищают затем до
необходимой степени чистоты. Получение препара-
тов И. очень высокой чистоты представляет труд-
ную задачу и требует затраты огромного времени.
Описан способ получения окиси И. высокой чистоты,
включающий операции: кристаллизацию карбонатов
при 0°, экстракцию нитратов н-пентаноном, хромато-
графию нитратов на у-А1.2О3 (двухкратную). В ре-
зультате такой длительной обработки из 660 г 90%-ного
концентрата выделен 1,2 г окиси; достигнутая чистота
составляет 99,9985%. Окись И. чистотой 99,9996%
была получена фракционным осаждением феррициа-
нида (30 осаждений) и двойного сульфата И. и калия
(2 осаждения). Задача еще более усложняется, если
И. необходимо извлекать из смеси его с элементами
цериевой подгруппы.
Элементарный И. (как и большинство металлов
иттриевой подгруппы — Ег, Но, Dy, Tu) получают
в виде компактного металла необходимой степени
чистоты двумя способами: восстановлением фторида И.
кальцием в присутствии магния («губчатый метод»);
восстановлением хлорида И. литием.
Применение. И. относится к легким металлам и
имеет малое сечение захвата тепловых нейтронов. Это
привлекает к металлу внимание как к возможному кон-
струкционному материалу в атомной технике, а также
в авиации. Окись И. (чистота к-рой, по литературным
данным, должна составлять 99,99999) употребляется для
изготовления иттриевых ферритов, применяемых в
радиоэлектронике, в слуховых приборах, ячейках
памяти счетно-решающнх устройств и т. п. Изотоп
Y90 употребляется при лечении опухолей. В сочетании
с другими редкоземельными элементами металлич. И.
применяется для легирования нек-рых сплавов.
Лит.: Vickery R. С., The chemistry of yttrium and
scandium, Oxf.-[a. o.J, i960; его же, Analytical chemistry
of the rare earths, v. 3, N. Y. — P., 1961; G i I 1 e о M. A.,
G e I I e r S., Phys. Rev., 1958, 110, ser. 2,№ 1, 73; Fischer W.,
Nieman К. E., Z. anorgan. und allgem. Chem., 1956, 283,
H. 1—6, 96; Проблемы современной металлургии. Сб. перево-
дов, К« 1, 2, 4, М., 1961; Борисенко Л. Ф., Скандий,
М., 1961; См. также лит. при ст. Лантанийы.
3. Ф. Андреева.
ИХТИОЛ — смесь аммониевой соли сульфокислот
сланцевых масел с водой, сульфатом аммония и непро-
сульфированными маслами; красно-бурая, прозрачная
(в тонком слое), вязкая жидкость со своеобразным
запахом. И. растворяется в воде, при растворении
в спирте выпадает осадок сульфата аммония, С глице-
рином смешивается во всех отношениях. И. обычно
содержит до 45% воды, 14% серы (на сухое в-во);
вязкость условная (по Энглеру) при 80° не более 9.
И. получают из смол, образующихся при газификации
и полукоксовании сланцев. Получаемая при дистил-
ляции смолы фракция масел с пределами выкипания
220—400° обрабатывается 23%-ным р-ром NaOH (для
освобождения от фенолов), а затем разб. H2SO4 (от
пиридиновых оснований). Полученное масло после
тщательной промывки сульфируют олеумом, отделяют
образовавшиеся сульфокислоты от непросульфиро-
ванных масел и продуктов полимеризации, промывают
сульфокислоты водой, обрабатывают 25%-нои аммиач-
ной водой и упаривают. И. применяют как наружное
средство при накожных заболеваниях и ожогах
(компонент мази Вишневского), а также в качестве
антисептика. Б. С. Ичиксон.
КАДАВЕРИН (пентаметилендиамин, 1,5-диамиио-
пентан) H2N(CH2)5NH2, мол. в. 102,18 — бесцветная
сиропообразная дымящая на воздухе жидкость;
т. пл. —21°; т. кип. 178—180,5°/760 с?}| 0,8846,
0,873; п^’е 1,45889; 1,46776; К = 7,3- 10~4(25о);
Т]25 0,02350 пуаз. К. хорошо растворим в воде, спир-
те, плохо — в эфире; образует гидрат C5H14N2 • 2Н2О,
соли, например C5H14N2 • 2НС1; последняя при на-
гревании разлагается на NH4C1 и хлоргидрат пипе-
ридина. Химич, свойства К. определяются наличием
2 аминогрупп; реакции с HNO2, ацилирующими аген-
тами и т. д. аналогичны реакциям моноаминов, напр.
H2N(CH2)5NH2 + 2HNO2 -* НО(СН2)5ОН + 2N2 + 2Н2О
К. может быть получен всеми способами, известными
для моноаминов, напр. Габриеля реакцией из 1,5-ди-
хлорпентана, восстановлением динитрила глутаровой
к-ты и т. д., а также щелочным гидролизом лизина:
H2N(CH2)4CH(NH2)-.COOH----» H2N(CH2)5NH2
—со2
К. встречается в природе; он найден в спорынье,мухо-
море; содержится в сыре, пивных дрожжах; находится
в моче и экскрементах людей, больных холерой ицисти-
нурией. Наряду с путресцином и др. органич. осно-
ваниями, т. наз. птомаинами, К. образуется при
гниении белковых веществ. в- н- Фросин.
КАДИОНЫ — группа органич. реактивов, приме-
няемых в основном для обнаружения и определения
кадмия. Все К. содержат по две азогруппы. Практич.
применение нашли л-нитродиазоаминоазобензол (I) и
динатриевая соль л-нитродиазоаминоазобензолдисуль-
фокислоты (II).
O2NCeH4-N=N-NHCeH4-N=NCeH4
I
SOsNa
02N-^~~^-N=N-NH-C6H/-N=N~^y-s63^A
II
К а д и о н (I) CuHuOjN,, мол. в. 346,35 — оранжевые
игольчатые кристаллы, темнеющие при нагревании, т. пл. 197°
(с разл.), нерастворим в воде, мало растворим в спирте, лучше
растворяется в бензоле и ацетоне, легко растворим в спирто-
вых растворах щелочей с образованием слабой пурпурной
окраски, в присутствии минеральных к-т быстро разлагается.
Получают К. (I) диазотированием п-нитроанилина с после-
дующим азосочетанием с n-аминоазобензолом; применяют
для обнаружения кадмия (при взаимодействии спиртового
р-ра К. с р-ром сопи кадмия и последующем подщелачи-
вании выпадает красный осадок), для открытия магния и
для фотометрического определения кадмия.
К а д и о н (II), или кадион ИРЕ A, C18H12O,N,S2Na2,
моп. в. 550,45 — оранжево-желтый мелкокристаппич. поро-
шок, растворимый в воде; 0,1%-ный водный р-р оранжевого
цвета. При действии сопяной к-ты раствор реактива обесцве-
чивается, при действии щелочи образуется фиолетовая ок-
раска. В щелочной среде (рН^9,0) образует с кадмием окра-
шенное соединение; в зависимости от количества кадмия
окраска раствора меняется от бурого до оранжево-розового
цвета. Получают К. ИРЕА диазотированием п-нитроанилин-о-
сульфокислоАт с последующим взаимодействием с 4-амино-
азобензоп-4'-супьфокиспотой. Применяют для открытия и фо-
тометрии. определения кадмия.
Лит.: Welch er F. J., Organic analytical reagents,
V. 4, N. Y., 1948; Вещества высокой чистоты и реактивы. Сб.
статей, вып. 23, М., 1959 (Тр. Всес. н.-и. ин-та химич. реак-
тивов). И. П. Ефимов.
КАДМИЕВЫЕ ПИГМЕНТЫ — см. Пигменты кад-
миевые.
КАДМИЙ (Cadmium) Cd — химич. элемент II гр.
периодич. системы Менделеева; п. и. 48, ат. в. 112,40.
Природный К. состоит из смеси восьми стабильных
изотопов с массовыми числами: 106 (1,215%), 108
(0,875%), 110 (12,39%), 111 (12,75%), 112 (24,07%),
113 (12,26%), 114 (28,86%), 116 (7,58%). Получены
искусственные радиоактивные изотопы К.; из них
наиболее важны: Cd109 (Г1/2 = 470 дней), изомеры
Cd113 (Г1/2 = 5,1 года) и Cd115 (Ti/2=43 дня), а также
Cdlis (Г1/2 == 53 часа). Поперечное сечение поглощения
тепловых нейтронов атомом К. 2400 барн. В основном
нейтроны поглощает изотоп Cdlis, сечение захвата для
к-рого составляет 25000 барн. Конфигурация внешних
электронов атома К.: 4d105s2. Энергия ионизации
(в эв): Cd°—>-Cd+—>Cd2+->Cds+ соответственно 8,99;
16,905; 35,0.
К. открыт в 1817 Ф. Штромейером в карбонате цинка;
название Й. произведено отгреч. слова хабц1а, или хавце1а,
к-рым обозначалась цинковая руда (rajjMeft). Независимо от
Штромейера и почти одновременно с ним Й. был открыт в
цинке и его соединениях др. учеными.
Среднее содержание К. в земной коре 1,3 • 10~*
вес. %. К. образует минералы: гринокит CdS
(77,7% Cd) и очень редкие, мало изученные, отавит
CdCOs и монтепонит CdO (87,5% Cd). Примесь К.
(тысячные доли %) обычно имеется в гидротермальных
рудах, где он присутствует в сфалерите, галените и др.,
гл. обр. сульфидных минералах. Повышенное содер-
жание К. (до 1—1,5%) характерно для маложелези-
стых сфалеритов. Практич. интерес как источники К.
представляют первичные и окисленные руды многих
колчеданно-полиметаллич. и полиметаллич. месторо-
ждений.
Физические и химические свойства. К. — серебри-
сто-белый металл, мягкий, ковкий и тягучий. Палочки
К. при сгибании издают треск (подобно олову), что
позволяет ориентировочно судить о чистоте металла
(в случае загрязненного примесями К. это явление
не наблюдается). К. кристаллизуется в гексагональной
решетке, а = 2,97311 А, с = 5,60694 А (при 25°);
отношение с/а = 1,8859 сильно отклоняется от иде-
ального для гексагональной шаровой упаковки
(с/а = 1,663). Аллотропия, модификаций К. не имеет.
Ат. радиус 1,56 А, ионный радиус Cd2+ 0,99 А. Плот-
ность 8,65 (20°); т. пл. 321°; т. кип. 767°. Теплота
плавления 13,2 кал/г; теплота испарения 286,4 кал/г'
уд. теплоемкость (в кал/г • град): 0,0632 (321—700°);
0,055 (25°); термин, коэфф, линейного расширения
29,8 • 1О-0 (25°); теплопроводность (кал/см • сек град):
0,105 (358°); 0,119 (435°). Уд. электросопротивление
343
КАДМИЙ — КАДМИЙОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
344
7,4 мком см (20°); термич. коэфф, электросопро-
тивления 4,3 • 10-3 (0—100°). Модуль упругости
5300 кПммг; предел прочности на растяжение
6,4 кГ!ммг; предел упругости 0,3 кГ/мм?; предел
текучести 1 кГ!мм2; относительное удлинение 20%;
относительное сужение 50%; твердость по Бринеллю
16,0 кГ1ммг, по Моосу 2,0.
В химич. соединениях К. всегда 2-валентен. В чи-
стом влажном воздухе на поверхности К. медленно
образуется тонкая пленка CdO, защищающая металл
от дальнейшего окисления. При сильном нагревании
на воздухе К. сгорает в CdO бурого цвета. Галогены
легко окисляют К. в галогениды (см. Кадмия галоге-
ниды). К. медленно растворяется в HCI, H2SO4 и др.
неорганич. к-тах, легче всего — в HNO3. В отличие от
Zn, в щелочах К. не растворяется. Соли К. общей
формулы CdX2 характеризуются наличием катиона
Cd2+; они бесцветны (если анион бесцветен), за исклю-
чением CdS, к-рый в зависимости от условий осаждения
имеет цвет от бледно-желтого до красновато-желтого
(см. Кадмия сульфид). Р-ры солей К. сильно гидро-
лизованы, имеют кислую реакцию. Едкие щелочи
осаждают из р-ров солей К. белую гидроокись Cd(OH).,,
нерастворимую в избытке осадителя, по легко раство-
римую в NH4OH с образованием комплексного катиона
[Cd(NH3)4]2+. Труднорастворимый цианид Cd(CN)2
с избытком KCN образует комплексные цианиды типов
Me2[Cd(CN)4] и Me4[Cd(CN)el, легко растворимые в во-
де. Н и т р а т К. Cd(NO3)2 легко растворим в воде
(127 г в 100 г Н2О при 18°); из р-ров кристаллизуются
гидраты: Cd(NO3)2 • 9Н2О (ниже 0°); Cd(NO?)2 • 4Н2О
(от 0° до 59,5°); выше 59,5° устойчив только безводный
Gd(NOa),. Сульфат К. CdSO4 также растворим
в воде (76,2 г в 100 г Н2О при 18°). Он получается при
растворении кадмия, CdO или CdCO3 в разб. H2SO4;
из р-ра выпадает гидрат 3CdSO4 . 8Н2О, устойчивый
от —18° до 74,5°; выше этой темп-ры устойчив гид-
рат CdSO4 • Н2О; известен Метастабильный гидрат
CdSO4 • 7Н2О. "Карбонат К. CdCO3 осаждается
р-ром Na2CO3 из р-ров солей К.; при 357° разлагается
нацело на CdO и СО2.
Аналитическое определение. При
систематич. ходе качественного анализа CdS остается
в осадке сульфидов IV группы вместе с HgS, PbS,
CuS, Bi2S3. После отделения HgS конц. HNO3, р-р нит-
ратов Cd, РЬ, Си, Bi выпаривают с H2SO4 до обильных
паров SO3, остаток по охлаждении разбавляют водой.
Отфильтровав от PbSO4, прибавляют к фильтрату из-
быток NH4OH; в осадок выпадает В1(ОН)3,вр-ре оста-
ются Си н Cd в виде комплексных катионов. Прибавив
KCN до обесцвечивания р-ра, в пего пропускают H2S;
в присутствии К. образуется желтый осадок CdS,
т. к. H2S не осаждает комплексных цианидов меди.
В присутствии Hg, РЬ, Си, Bi можно открыть К., на-
неся на фильтровальную бумагу, смоченную р-ром
дифенилкарбазида, каплю испытуемого р-ра (кислот-
ность к-рого предварительно понижена добавкой
твердого NaC2H3O2); появление фиолетово-синего
окрашивания указывает на присутствие К. Весовым
путем К. определяют в виде CdSO4 (что требует отде-
ления К. от всех других металлов) или электролизом
из цианидного р-ра. При серийных определениях
лучше всего пользоваться быстрым и достаточно точ-
ным полярографии, методом. Спектрографически К.
может быть определен с относительной погрешно-
стью ±5%.
Получение. Сырьем для получения К. служат по-
бочные продукты переработки цинковых, свинцово-
цинковых и медно-цинковых руд: медно-кадмиевые
кеки, пыль обжигательных и дестилляционных печей,
бегхаузов и котрелей, кеки литопонных заводов. Содер-
жание К. в этих продуктах составляет от 0,2—0,5%
до 6—7% и выше. Для извлечения К. названные про-
дукты (иногда после окислительного обжига) обраба-
тываются разб. H2S04; в р-р переходят Cd и Си, к-рые
затем осаждаются цинковой пылью. Полученную
кадмиевую губку (50—58% Cd, 10—20% Zn, осталь-
ное Fe + Си) подвергают окислению посредством
выдержки в теплом и влажном помещении в течение
2—3 недель, после чего из нее извлекают К. при
помощи разб. H2S04 (70—80 г/л). Иногда, не окисляя
губки, ее обрабатывают непосредственно 75%-ной
H2SO4, в к-рую пропускают острый пар. Из р-ров
CdS04 осаждают К. электролизом при 40° на кад-
миевых катодах; аноды свинцовые. Снятые с катодов
листы электролитич. К. после сушки переплавляют
в железных котлах под слоем расплавленного NaOH
при темп-ре 330—350°. К. отливают в форме палочек
или чушек. Электролитич. К. содержит до 0,02%
примесей.
Все растворимые в воде и разб. к-тах соединения К.
ядовиты. Чаще всего отравления происходят при вды-
хании «дыма» его окиси; при этом наблюдается кашель,
головная боль, затруднение дыхания, воспаление
легких и отек их со смертельным исходом. Меры пре-
досторожности — хорошая вентиляция, ношение ре-
спираторов или защитных масок. При приеме внутрь
соли К. вызывают рвоту, благодаря чему яд удаляется
из желудка.
Применение. Благодаря большому сечению захвата
тепловых нейтронов из К. изготовляют регулирующие
и компенсационные стержни, а также аварийные
стоп-стержни ядерных реакторов. К. применяют в ще-
лочных аккумуляторах (см. Химические источники
тока), для приготовления кадмия сплавов И- для
кадмирования (см. Гальванотехника). Сернистый К.—
прочная желтая краска (см. Кадмия сульфид). Серно-
кислый К. и амальгама К. применяются в Вестона
нормальном элементе.
Лит.: Gmelin, 8 Aufl., Syst. — Num. 33 — Cadmium,
Lpz. — B., 1959; Mellor, v. 4, L. — N. Y. — Toronto,
1946; то же, v. 4,1952;U 1 1 m a n n, 3 Aufl., Bd 4, Milnch. —
B., 1953, S. 804—27; Bare metals handbook, ed. C. Hampel.
2 ed., L., 1961, p. 82—92; Маковецкая M. А., Металлур-
гия кадмия, в кн.: Справочник металлурга по цветным метал-
лам, т. 2, под ред Н. Н. Мурача, М., 1947, . С. А. Погодин.
КАДМИЙОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ —
нестойкие соединения общей формулы R2Cd, как
правило, бесцветные сильно преломляющие свет жид-
кости. В индивидуальном состоянии известно лишь
несколько К. с., обычно их не выделяют из растворов.
К. с. получают действием магнийорганич. соединений
на соли галогенидов кадмия:
CdBr2 + 2C2H5MgBr —>- Cd(C2H3)2 -J- 2MgBr2
Кадмий способен вытеснять ртуть из ее диарильных
соединений при достаточном нагревании последних
с порошком металла:
Hg(CeH3)2 + Cd -Г Hg -ф (CsH3)2Cd
По химич. свойствам К. с. напоминают цинкорганич.
соединения; они быстро окисляются на воздухе, раз-
лагаются водой, почти не взаимодействуют со слож-
ными эфирами, но реагируют с хлорангидридами к-т,
альдегидами и значительно более вяло с кетонами,
напр.:
/°
CeH3C^ + (C6H3)2Cd -> CeH3CH(OH)CeH3
ЧН
/z° z°
RCf + (C6H3)aCd -> RC/
XC1 хс6н5
В связи с такой реакционной способностью К. с. мо-
гут быть использованы в органич. синтезе в тех
случаях, когда магнийорганич. соединения оказы-
ваются слишком активными, напр. при синтезе кето-
нов, а-хлоркетонов и кетокислот из хлорангидридов
345
КАДМИЯ ГАЛОГЕНИДЫ—КАЗЕИН
346
или ангидридов соответствующих кислот и в др.
синтезах:
2С1СНаСОС1 4- (CtHgJssCd -> 2С1СН.СОС!Н<| + CdCl2
/О
,С% их
2СвН4< >О + R»Cd -> [ROC-CeHj-COO ]2Cd--->-
с<о
-> 2R0C—СеЩ-С00Н + CdX2
BrMgfCHaJjMgBr + CdBr2 -» -j—(CHsJiCd—। + 2MgBr2
-(CH2)4Cd-, + 2ROOC(CH2)nCOCl ->
ROOC(CH2)nCO(CH2)4CO(CH2)„COOR+ CdCl2
См. также Цинкорганические соединения и Магнийор-
ганические соединения. Н- А- Несмянов.
КАДМИЯ ГАЛОГЕНИДЫ — соединения Cd с га-
логенами общей формулы CdX2 (где X = F, Cl, Вг, J).
Получают действием галогенов на Cd, а также раство-
рением Cd, CdO или CdCO3 в галогеноводородных
к-тах. Все К. г. бесцветны, хорошо растворимы в воде
(кроме CdFA и легко образуют аутокомплексные
соли, напр. Cd[CdJ3]2, а также двойные комплексные
соли, преим. типов Me[CdX3], Me2[CdX4] и Me4[CdX4];
Me — одновалентный металл.
Фтористый кадмий CdF2 — мелкокри-
сталлич. иглы, плотн. 6,6, т. пл. 528°, т. кип. ок.
1100°, растворимость в воде 4,3% (25°); гидратов не
образует. Хлористый кадмий CdCl2 — ли-
сточки с перламутровым блеском, плотн. 4,05, т. пл.
568°, т. кип. 975°. Гигроскопичен, легко растворим
в воде: 57,35% (20°), 59,52% (100°). Из водных р-ров
кристаллизуются гидраты: CdCl2 • 4Н2О (ниже —5°),
CdCl2 • 2,5Н2О (между —5° и +34°), CdCJ2 • Н2О
(выше 34°). Применяется в гальванотехнике. Бро-
мистый кадмий CdBr2 — мелкие таблитчатые
кристаллы, плотн. 5,28, т. пл. 567°, т. кип. 810°.
Растворимость в воде: 48,9% (18°), 61,6% (100°);
ниже 36° кристаллизуется гидрат CdBr2 • 4Н2О,
переходящий при 36° в CdBr2. Н2О. Применяется
в фотографии. Иодистый кадмий CdJ2 —
кристаллы, в виде гексагональных табличек, плотн.
5 67, т. пл. 390°, т. кип. 900°, растворимость в воде:
45,8% (20°), 56,08% (100°), гидратов не образует.
Применяется В фотографии. С. А. Погодин.
КАДМИЯ ОКИСЬ CdO — кристаллы от светло-
коричневого до темно-бурого цвета (в зависимости
от способа приготовления), с решеткой типа NaCl;
т. пл. 8,26; при 700° возгоняется не плавясь; давление
пара достигает 760 мм рт. ст. при 1500°. В воде и
р-рах щелочей нерастворима, растворяется в к-тах,
в р-рах солей аммония и KCN. При прокаливании
с Fe2Os при 800° образует соединение CdFe2O4 типа
шпинели. Восстановление К. о. посредством С или СО
начинается ок. 300—400°. При восстановлении сперва
получается недокись Cd2O. Получают К. о. дистилля-
цией Cd в графитовой реторте и сжиганием его пара
в струе воздуха, а также прокаливанием Cd(NO3)2
или CdCO3. Применяют К. о. в гальванотехнике, как
катализатор при гидрировании жиров или как до-
бавку в светящиеся составы.
Гидроокись Cd(OH)2 получают осаждением
кипящего р-ра Cd(NO3)2 едкой щелочью при переме-
шивании; осадок многократно декантируют горячей
водой, отфильтровывают через асбест и промывают
горячей водой. Cd(OH)2 присоединяет СО2 из воздуха,
легко растворима в к-тах и аммиаке; при нагревании
теряет воду. Применяют в гальванотехнике и в про-
из-ве Cd—Ni-аккумуляторов. С. А. Погодин.
КАДМИЯ СПЛАВЫ — металлич. системы, в к-рых
Cd является либо основным компонентом, либо опреде-
ляет те или иные практически важные свойства сплава.
Cd служит основой нек-рых подшипниковых сплавов
для тяжелонагруженных авиационных, автомобильных
и судовых двигателей. Примером является сплав Cd
с ок. 1 %Ni или с 0,6% Си и 0,7% Ag. Сплавы Си с Cd
(ок. 1% Cd, иногда с небольшими добавками Be,
Sn, Zn) под названием кадмиевой бронзы служат для
изготовления телеграфных, телефонных и троллейных
проводов, т. к. по сравнению с чистой медью обладают
большей прочностью, твердостью и износостойкостью
при несколько пониженной электропроводности. Спла-
вы Cd с нек-рыми металлами, имеющими низкую
темп-ру плавления, применяют как легкоплавкие при-
пои; таковы тройные эвтектики: 8,15% Cd, 51,65% Bi,
40,2% Pb (т. пл. 21,5°); 31,2% Cd, 40,9% Bi, 27,9% Sn
(т. пл. 103°); 18% Cd, 32% Pb, 50% Sn (т. пл. 145°);
24,8% Cd, 73, 1% Sn, 2,1% Zn (т. пл. 164°). Еще ниже
плавятся четверные К. с., а именно: сплав Вуда (50%
Bi, 25% Pb, 12,5% Sn и 12,5% Cd, т. пл. ок. 70°)
и очень близкий к нему по составу сплав Липовица
(50% Bi, 27% Pb, 13% Sn, 10% Cd, т. пл. ок. 70°);
наиболее легкоплавки сплавы: 46,8% Bi, 26,0% Pb,
9,4% Cd и 6,3% Hg (т. пл. ок. 62°); 40,95% Bi, 22,10%
Pb, 10,65% Sn, 8,20% Cd, 18,10% In (т. пл. 47°).
Легкоплавкие К. с. применяют для спайки стекла
с металлом в автоматич. огнетушителях (спринклерах),
для предохранителей в паросиловых установках, для
тонких и сложных отливок в гипсовых формах, ана-
томия. слепков и т. п. С многими металлами Cd
образует химич. соединения (кадмиды), напр. LiCd
(т. пл. 549°), NaCd2 (т. пл. 384°), кристаллизующиеся
непосредственно из жидкости; соединения MgCd,
MgCd3 и Mg3Cd образуются при превращении твердых
р-ров Cd—Mg,
Лит. см. при ст. Висмута сплавы и Кадмий.
С. А. Погодин.
КАДМИЯ СУЛЬФИД (сернистый кадмий) CdS —
кубич. или гексагональные кристаллы от светло-жел-
того до оранжево-красного цвета, плотн. 4,8. При
нагревании на воздухе окисляется до CdS04 и CdO;
в атмосфере N2 ок. 1000° возгоняется. В воде практи-
чески нерастворим (6 • 10~15 мол/л)-, устойчив в р-рах
щелочей и разб. к-тах, растворяется в конц. НС1,
HN03, H2S04. Сероводород осаждает К. с. из кислых
р-ров, содержащих ион Cd2+. Применяют в масляных
красках для живописи (кадмиевая желтая) и для
получения цветных стекол. В технике К. с. приготов-
ляют прокаливанием смесей CdO, CdCO3 или CdC2O4
С серой. С. А. Погодин.
КАЗЕИН — фосфопротеид, главный белковый ком-
понент молока. Мол. вес К., по имеющимся данным,
колеблется в пределах от 30 000 до 400 000. К. прак-
тически нерастворим в воде и органич. растворителях,
растворяется в водных р-рах солей и разб. щелочей,
иэ'К-рых выпадает в осадок при подкислении. Изоэлек-
трическая точка К. при pH 4,6—4,7. Аминокислотный
состав К. из коровьего молока (в г на 100 г белка):
3,2 Серин............. 6.3
2,0 Треонин........... 4,9
7,2 Цистин............ 0,3
9,2 Метионин........... 2,8
6,1 Аргинии.......... 4,1
10,6 Гистидин......... 3,1
5,0 Лизин............ 8,2
6,3 Аспарагиновая к-та . . 7,1
1,7 Глютаминовая к-та . . 22,4
Аланин . . .
Глицин . . .
Валин ....
Лейцин . . .
Изолейцин .
Пролин . . .
Фенилаланин
Тирозин . . .
Триптофан .
К. содержит ок. 1% фосфора, входящего в состав
фосфорной к-ты, этерифицирующей оксигруппы окси-
аминокислот К. Выделяют К. из молока, в к-ром он
содержится в количестве 2,5—4,5%. Способы выделе-
ния К. основаны на осаждении этого белка в кислой
среде. К. не является однородным белком и может
быть разделен на ряд фракций, названных a-, 0- и
у-казеинами. Эти фракции отличаются друг от друга
по аминокислотному составу и электрофоретич. под-
вижности. В состав К. из коровьего молока вхоДят
75% а-К., 22% 0-К., 3% у-К. '
КАКАО МАСЛО-КАЛИЙ
348
347
Образование сгустка К. при створаживании молока
иногда приписывают превращению т. н. казеиногена
в К. или К. в т. наз. параказеин. Механизм этих
превращений точно не установлен.
К. имеет большое значение как пищевой продукт,
являясь гл. составной частью творога и сыра. К. —
полноценный белок, т. к. содержит все необходимые
организму аминокислоты. К. используется для из-
готовления пластмасс, красок, клеев, искусственного
волокна.
Лит.: Лисовская Н. П., Ливанова Н. Б.,
Фосфопротеины, М., 1960, с. 17—28; Ин их о в Г. С., Био-
химия молока, М., 1956, с. 25—44; Мак-Микин X. Т.,
Белки молока, в кн.: Белки, под ред. Г. Нейрата и К. Бэйли,
пер. с англ., т. 3, ч. 1, М., 1958, с. 692—707. В. О. Шпикитер.
КАКАО МАСЛО—см. Жиры растительные-
КАКОДИЛА СОЕДИНЕНИЯ — группа мышьяк-
органических соединений, в состав к-рых входит
остаток диметиларсина (CH3)2As —, т. наз. к а к fl-
fl и л. К. с. — бесцветные жидкости или твердые в-ва;
плохо растворимы в воде, хорошо — в спирте и эфире;
обладают исключительно неприятным запахом; очень
ядовиты; нек-рые из них (тетраметилдиарсин, диметил-
арсин и др.) самовоспламеняются на воздухе.
Соединения Мол. в. Т. КИП.
Дикакодип (тетраметипдиарсин)а (CH3)2As—As (СН,)а 209,96 170°
Окись какодила (диметиларсин- оксид) (CH3)aAs—О—As (СН3)2 . 225,96 149-51° 1,486 (4°)
Хлористый какодил® (диметилхпор- арсин) (СНа)а AsCl 140,44 106,5—107° 1,504 (12°) 1,213
Какодиповый водород (диметилар- син) (CHa)aAsH 105,99 35,6°/747 лол
Цианистый какодил (диметилциая- арсия)в (СНа)а As—CN 131,00 140’ (20°)
а Т. пл. — 6°. 6 Не растворяется в эфире. в Т. пл. 33°.
Гетраметилдиарсин (дикакодил) и диметиларсиноксид
окись какодила) входят в состав т. наз. «жидкости
Кадэ», образующейся при совместном прокаливании
5елого мышьяка (мышьяковистого ангидрида) и ацета-
та калия:
«Ж идкость Кадэ»
[ Диканодил (55,9%) 1
Окись какодила (40%) 4-
Полимеры (2,6%) J
+ K2COS + СО2 + СН4 (следы) As (следы)
Механизм этой реакции (Кадэ, 1760) окончательно
ie выяснен. Долгое время «жидкость Кадэ» являлась
единственным источником К. с.; так, из «жидкости
4адэ» (или окиси К.) действием конц. соляной к-ты
юлучают диметилхлорарсин (хлористый какодил);
три восстановлении последнего водородом в момент
отделения образуется диметиларсин (какодиловый
юдород):
(СН3)2А?С1 -► (CHs)2AsH
взаимодействие «жидкости Кадэ» с безводной си-
щльной к-той приводит к диметилцианарсину (циа-
истому какодилу):
[(CH3)2As]2O + 2HCN ->• 2(CH3)2As—CN + Н2О
i настоящее время К. с. могут быть синтезированы
ругими способами (см. в ст. Мышьякорганические со-
чинения). При окислении К. с. образуется какодиловая
цимети л арсиновая) к-та (CH3)2As(O)OH, напр.;
лО
2(CHa)2AsCl + О2 + 2Н2О 2(CH3)2As% + 2НС1
ХОН
Какодиловая кислота (мол. в. 137,99) —
есцветные лишенные запаха кристаллы; т. пл. 200°; хорошо
астворима в воде и спирте; не разлагается даже при действии
царской водки и др. сильных окислителей; соли, напр.
(CH3)2A8(O)ONa • ЗН2О (мол. в. 214,02), применяют в меди-
цине. Исследования Кадэ, Бунзена в области К. с. имели
большое значение для развития теории химич. строения.
В. Н. Фросин.
КАЛИ ЕДКОЕ — см. Калия гидроокись.
КАЛИЙ (Kalium) К — химич. элемент I гр. пе-
риодич. системы Менделеева, принадлежит к подгруп-
пе щелочных металлов, п. н. 19, ат. в. 39,102. При-
родный К. состоит из 3 изотопов: К39 (93,08%), К.40
(0,0119%) и К41 (6,76%). Слабо радиоактивный изотоп
К40 (Т»/2 = 1,32 • 10е лет) распадается двумя путями:
88% атомов образуют в результате ^“-распада Са40,
12% путем A-захвата превращаются в Аг40. Попереч-
ное сечение поглощения тепловых нейтронов атомом
К. (природная смесь изотопов) 1,97 барн. Из искус-
ственных радиоактивных изотопов наиболее важен
К43 (Т»/а = 12,52 года), применяющийся как радио-
активный индикатор в химии, медицине и биологии.
Конфигурация внешних электронов атома К. is.
Энергия ионизации (в эв): К °—.К+—.К2+—►К3+-*К4+
соответственно равна 4,34; 31,8; 46,0; 60,9. Металлич.
К. был впервые выделен Дэви в 1807 электролизом
твердого, слегка влажного К.ОН. Позднее Гей-Люссак
и Тенар получили К. в больших количествах, прокали-
вая едкое кали с углем в стальной трубке.
Содержание К. в земной коре составляет 2,6 вес. %.
В свободном состоянии К. вследствие своей большой
химич. активности в природе не встречается. Он входит
в состав многих породообразующих минералов, из
к-рых важнейшими являются полевые пшаты и слюды.
В результате действия воды и углекислого газа на
горные породы, содержащие К., он переходит в раство-
римые соединения, к-рые частью уносятся в моря,
частью удерживаются почвой. К числу важнейших
минералов К. относятся: сильвин КС1 (порода, со-
стоящая из сильвина и галита NaCl, носит название
сильвинит), карналлит КС1 • MgCl2 • 6Н2О, каинит
КС1 • MgSO4 • ЗН2О, лангбейнит K2SO4 • 2MgSO4 и
др., образующие большие скопления т. наз. калий-
ных солей. Залежи калийных солей были впервые
обнаружены и изучены в Германии (Стассфуртское
месторождение). Самое крупное в мире месторождение
калийных солей находится в СССР, близ Соликамска.
Соли К. содержатся также в морской воде, рапе мно-
гих соляных озер и в подземных рассолах. В огром-
ных количествах К. встречается также в силикатах —
нефелине, лейците и др.
Физические и химические свойства. К. — серебри-
сто-белый металл, в виде тонких пластин кажется
фиолетовым, кристаллизуется в объемноцентриро-
ванной кубич. решетке с параметром а = 5,33 А.
Аллотропии, превращений у К. не обнаружено. Атом-
ный радиус 2,36 А; ионный радиус К+ l,33jA. Плотн.
0,862 (20°); плотность жидкого К. определяется урав-
нением: dt = 0,826—2,22 • 10~4 (—62,5°). Т. пл.
63,55°; т. кип. 760°. Теплота плавления 14,63 кал/г',
теплота испарения 490 кал/г (при т. кип.). Уд. тепло-
емкость = 0,180 кал/г-град (0—100°); Ср твер-
дого К. можно рассчитать по ур-нию; Ср = 0,1728 4"
4- 14,2 • 10~6г; жидкого — по ур-нию: Ср = 0,213—
—7,57 • 10-6г 4- 7,52 • 10"3г3 (в интервале 63—800°).
Термин, коэфф, линейного расширения 8,33 - 10~®
(0—-50°). Уд. теплопроводность в кал/см- сек - град
твердого К. 0,232 (20,9°), жидкого 0,1073 (200°).
К. обладает высокой электропроводностью. Уд. элек-
тросопротивление (в ом- см): твердого К. 7,118- 10-в
(20°); жидкого 15,49 (100°); 21,80 (200°); 28,20 (300°).
Термин, коэфф, электросопротивления твердого К.
5,8 • 10“6 (20°). Вязкость жидкого К. в спуазах:
0,515 (69,5°); 0,258 (250°); 0,191 (400°); 0,136 (700°).
Поверхностное натяжение К. 80—86 дин/см в интер-
вале от т. пл. до 250°. Давление паров К. в мм рт. ст.з
349
КАЛИЙ
350
1 (342°), 10 (443°), 200 (581°), 400 (696°), 760 (760°).
Пары К. при низких темп-рах зеленого цвета, а при
высоких — фиолетового. Наряду с одноатомными мо-
лекулами в парах содержится и нек-рое количество
двухатомных. Диэлектрин, проницаемость твердого К.
1,19 (0°), а паров 1,00092 (500° и 10 мм рт. ст.). Ме-
таллич. К. парамагнитен. Уд. магнитная восприим-
чивость худ = —0,62 • 10~8 (при комнатной темп-ре).
К. — мягкий металл, его можно прессовать и прока-
тывать в холодном состоянии; легко режется ножом
и сохраняет свою пластичность даже при низких
темп-рах. Твердость К. по Бринеллю 0,037 кГ/мм2.
Химически К. очень активен. На воздухе быстро
окисляется (при нагревании загорается); поэтому
обычно его хранят под слоем бензина, керосина или
минерального масла. Нормальный электродный по-
тенциал К. равен — 2,92 в. В химич. соединениях К.
одновалентен. При взаимодействии К. с сухим возду-
хом образуется окись К2О, бесцветные кристал-
лы, плотн. 2,32. Теплота образования ДН°а98 =
= —86,4 ккал{моль. При 300—400° разлагается без
плавления на металлич. К. и перекись К2О2. С водой
К2О образует калия гидроокись КОН. С водородом
при низких темп-рах К. практически не взаимо-
действует и может быть расплавлен в атмосфере На.
Начиная с 200°, заметно поглощает водород, при-
чем образуется гидрид КД — белые кристаллы
с кубич. решеткой, а = 5,712 А, плотн. 1,52. Теплота
образования ДН°а98 = —13,6 ккал[молъ. При нагрева-
нии разлагается на элементы без плавления; давление
диссоциации (в мм рт. ст.) составляет: 7,31 (300°),
760 (411°). Гидрид калия весьма реакционноспособен;
на воздухе воспламеняется, очень чувствителен к
влаге (КН + Н2О—»КОН + Н2); сильный восстано-
витель (восстанавливает окислы металлов и др.).
К. энергично соединяется с галогенами с образова-
нием соответствующих солей (см. Калия фторид,
Калия хлорид, Калия бромид, Калия иодид). Так,
в атмосфере фтора К. воспламеняется и сгорает. Рас-
плавленный К. сгорает в хлоре (твердый К. слабо
взаимодействует с сухим хлором и практически не
взаимодействует с жидким). С жидким бромом К. со-
единяется со взрывом. Реакция между К. и иодом также
может сопровождаться взрывом, если их прессовать
совместно. Уже при слабом нагревании начинается
реакция между К. и серой, причем образуется калия
сульфид KaS. При нагревании К. соединяется также
с селеном и теллуром. Азот не взаимодействует с К.
даже под давлением и при нагревании до высоких
темп-p и в этих условиях может быть использован
как защитная атмосфера. При действии на К. азота
в электрич. разряде могут образоваться азид К.
KN3 и, в значительно меньших количествах, нит-
рид К. K3N. Азид К. — блестящие бесцветные кри-
сталлы, плотн. 2,056, т. пл. 352°. При повышенных
темп-рах К. взаимодействует с углеродом (графитом),
причем выше 200° графит поглощает до 40% К. (от
веса графита). При 300° образуется соединение КС8,
а при 360°—КС18. Известны также карбиды К2С2 и
КНС2, получаемые косвенным путем. С металлами К.
дает многочисленные соединения.
В системе калий — натрий образуется
соединение Na2K (46,0 вес. % К.), плавящееся инкон-
груэнтно при 7°. Эвтектич. точка лежит при —12,5°
и 77,2 вес. % К. При комнатной темп-ре сплавы с со-
держанием 40—90 вес. % К. представляют собой се-
ребристо-белые жидкости. Ввиду близости свойств К.
и натрия такие физич. свойства их жидких сплавов,
как плотность, теплоемкость, вязкость, давление па-
ров, плавно изменяются с изменением состава и могут
быть с достаточной для практич. целей точностью рас-
считаны путем интерполяции. Электросопротивление
и теплопроводность сплавов К—Na выше соответ-
ствующих величин для К. и натрия. Уд. электросопро-
тивление сплава с 78% К. равно (в мком • см)\
45,63 (100°) и 104,51 (800°). Теплопроводность сплава
с 77,7% К. равна (в вт/см- град) 0,238 (150°) и 0,255
(700°). Сплавы К. с натрием отличаются высокой
химич. активностью, самовоспламеняются на воздухе.
Получаются сплавлением металлов в инертной атмо-
сфере, а также при действии металлич. натрия на КОН
или КС1.
С неорганич. соединениями К. взаимодействует
как сильный восстановитель. Реакция с водой:
К + НаО-*КОН + 1/2На энергично идет уже при
—100°, а при комнатной темп-ре сопровождается взры-
вом и воспламенением К. Во влажном воздухе К. пре-
вращается сначала в КОН, а затем в К2СО3. Бурно,
иногда со взрывом, реагирует с растворами кислот
с образованием соответствующих солей (см. Калия
карбонат, Калия сульфат и т. д.). Реакция с жидкой
безводной HaFa сопровождается горением, с газообраз-
ными НС1, НВг и HJ — протекает сравнительно
медленно. Будучи растворен в жидком аммиаке (ра-
створ имеет темно-синий цвет), К. медленно взаи-
модействует с ним: К + NH3—>KNH2 -f- 1/2На. Раст-
воры К. в аммиаке используют как сильный восстано-
витель. К. энергично взаимодействует с окислами
азота, а при высоких темп-рах — с СО и СО2. При
высоких темп-рах К. восстанавливает ВаО3 до бора,
А12О3 до А1 и SiO2 до свободного кремния (разрушение
стекла начинается выше 350—400°). Сульфаты, суль-
фиты, нитраты, нитриты, карбонаты и фосфаты при
нагревании с К. восстанавливаются с образованием
КаО и окислов соответствующих металлов (или сво-
бодных металлов). Окислы Hg, Ag, Ni, РЬ, Со, Sn,
Си, Сг, Bi, Ti и др. восстанавливаются до свободных
металлов.
По отношению к органич. соединениям К. весьма
реакционноспособен. Так, К. еще более энергично,
чем натрий, взаимодействует со спиртами, образуя
соответствующие алкоголяты. Уже на холоду реаги-
рует с ацетиленом (при длительном действии обра-
зуется КНС2), а при 500—600° с этиленом: С2Н4 +
+ 2К-»КаСа + 2На. Вызывает реакции полимериза-
ции олефинов и диолефинов. Насыщенные алифатич.
и ароматич. углеводороды при низких темп-рах инерт-
ны по отношению к К., с галогеналкилами и арилами
взаимодействует с образованием калийалкилов и соот-
ветственно калийарилов.
Аналитическое определение. Ка-
чественно К. определяют по фиолетовому окрашива-
нию пламени; по образованию белого кристаллич.
осадка кислой виннокалиевой соли; по характерным
линиям спектра: двойной красной линии 7699 А и
7665 А и двойной фиолетовой линии 4047 А и 4044 А.
Качественное определение К. можно провести также
с помощью методов, используемых для его количе-
ственного определения. Количественно К. опреде-
ляют осаждением в виде перхлората, хлороплатината,
кобальтинитрита, тетрафенилбората, а также в виде
соединения с дипикриламин’ом и т. д. Чаще всего ко-
личественное определение К. проводят спектрогра-
фически или пламенно-фотометрич. методом.
Получение. Для промышленного получения К.
обычно применяется обменная реакция между ме-
таллич. Na и КОН или же KG1 по реакциям: КОН +
+ Na = К + NaOH, и соответственно КС1 + Na=
= NaCl + К. Взаимодействие между расплавленной
КОН и жидким натрием осуществляется противотоком
в тарельчатой реакционной колонке из никеля при
380—440°. При получении К. из хлорида через рас-
плавленную соль при 760—800° пропускают пары
натрия. Выделяющиеся пары К. конденсируют. Те же
способы могут быть использованы для получения
сплавов К. с натрием. Перспективными являются
351
КАЛИЙНЫЕ УДОБРЕНИЯ — КАЛИЙОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
352
вакуум-термич. способы получения К., основанные
на реакциях:
6КС1 + 2А1 + 4СаО = 6К + ЗСаС1» + СаО А12О3
или
4КС1 + SI + 4СаО = 4К + 2СаС13 + 2СаО • ЙО2
идущих при темп-ре выше 900°. Получение К. электро-
лизом расплавленных КС1 или КОН не нашло широ-
кого распространения из-за низкого выхода по току и
трудности создания безопасного аппаратурного офор-
мления процесса. Представляет интерес способ, осно-
ванный на получении сплава РЬ—К электролизом
К2СО3 или КС1 с расплавленным свинцовым катодом
и отгонке К. из сплава.
Техника безопасности. При работе с К. необходимо
иметь в виду его высокую реакционную способность
и прежде всего свойство загораться при попадании
влагп. Все работы с К. должны проводиться в резино-
вых перчатках и в защитных очках или лучше в
масках, полностью закрывающих лицо. С большими
количествами К. рекомендуется работать в специаль-
ных камерах, заполненных инертным газом (азот,
аргон). Для тушения горящего К. применяют пова-
ренную соль или соду.
Применение. Основное применение металлич. К. —
получение перекиси калия К2О4, применяющейся для
регенерации кислорода. К. служит катализатором
при получении нек-рых видов синтетич. каучука.
Сплав К. с натрием используется как жидкометаллич.
теплоноситель в атомных реакторах и как восстано-
витель в произ-ве титана. В лабораториях сплав К.
с натрием применяют для осушки газов и освобожде-
ния их от кислорода. О применении солей К. см.
Калийные удобрения и статьи об отдельных солях.
Лит.: Алабышев А. Ф. [и др.], Натрий и калий, Л.,
1959, с. 259—379; Баймаков Ю. В., Электролиз в ме-
таллургии, т. 2, М., 1944, с. 116—33; Г у с ь к о в В. М.[идр.],
Ж. прикл. химии, 1959, 32, вып. 6, 1373; Gmelln, 8 Aull.,
Syst. — Num. 22, Kalium, В., 1936—38; то же, Kalium, An-
hangband, 1942, Nachdruck 1953; M e 1 1 о r, v. 2, L. — N. Y. —
Toronto, 1946; то ж e, v. 2,1952; U 1 1 m a n n, 3 Aufl., Bd 9,
Munch.— B., 1957, S. 229 — 40; Kirk v. 1, N. Y., 1947, p.
430—55; то ж e, v. 11, N. Y., 1953, p. 12—28; Кочеш-
ков К. А. и Талалаева T. В., Синтетические методы
в области металлоорганических соединений лития, натрия,
калия, рубидия и цезия, М., 1949; Жидкометаллические теп-
лоносители (натрий и натриево-калисвый сплав), пер. с англ.,
М., 1958; Андреев П. А., Канаев А. А., Федо-
рович Е. Д., Жидкометаллические теплоносители ядерных
реакторов, Л., 1959.
КАЛИЙНЫЕ УДОБРЕНИЯ — вещества, исполь-
зуемые для питания с.-х. растений, основным пита-
тельным элементом к-рых является калий. Содержа-
ние калия в удобрениях исчисляют в процентах К2О.
В качестве К. у. применяют сырые природные калий-
ные соли (гл. обр. сильвинит, в меньшей степени каи-
нит), продукты их переработки (хлористый калий,
40—30%-ные калийные соли, сульфат калия, сульфат
калия-магния) и золу растений. Наиболее распро-
страненными К. у. являются хлористый калий и
40—30%-ные калийные соли. Содержащиеся в К. у.
калийные соли растворимы в воде.
Сильвинит шКС1 + nNaCl с примесью солей
магния, кальция и др.' содержит 14—18% К2О и
ЗГ—40% Na2O; гигроскопичен.
Каинит КС1 • MgSO4 • ЗН2О с примесью NaCl,
CaSO4, MgSO4 и др.; содержит 10—12% К2О, 7—
10% MgO, ок. 35% Na2O и ок 40% С1.
Хлористый калий КС1 — технич. продукт,
идущий на удобрение, содержит 52—60%К20. Полу-
чают КС1 в основном из сильвинита методами галур-
гии или флотации, или комбинированными способами.
Метод галургии основан на различной раствори-
мости КС1 и NaCl в зависимости от темп-ры. С повышением
темп-ры растворимость КС1 значительно возрастает, а раство-
римость NaCl изменяется очень мало. При совместном при-
сутствии обеих солей растворимость NaCl с повышением
темп-ры падает, а растворимость КС1 возрастает. Сильвинит
обрабатывают при повышенной темп-ре насыпанным на ко-
лоду р-ром обеих солей. При этом р-р дополнительно насы-
щается КС1, а часть NaCl переходит в осадок и отделяется
фильтрованием. Из охлажденного р-ра выкристаллизовы-
вается только КС1, кристаллы к-рого отделяют от маточного
р-ра на центрифугах и высушивают. Маточный р-р применяют
для растворения КС1 из новой порции сильвинита.
Метод флотации основан на различной смачи-
ваемости водой сильвина (КС1) и каменной соли (NaCl). Фло-
тация калийных солей проводится в насыщенных р-рах сырых
калийных солей. Получаемый флотацией КС1 состоит из более
крупных кристаллов, чем получаемый методом галургии,
и меньше слеживается. Хлористый калий можно получать
и комбинированным методом флотации и гидроциклона. Метод
гидроциклона основан на том, что в среде тяжелых суспензий
под действием возникающих центробежных сип сильвин
и каменная соль разделяются по плотности.
30—40%-н ые калийные соли (содержат
соответственно ок. 30% и ок. 40% К2О) получают
смешением хлористого калия с сырыми калийнымй
солями.
Сульфат калия K2SO4 — технич. продукт,
идущий на удобрение, содержит 45—52% К2О; не
гигроскопичен и не слеживается.
В СССР сульфат калин получают переработкой каинитоланг-
бейнитовой руды, содержащей сульфаты калия и магния.
Растворимые минералы извлекаются горячими оборотными
р-рами; конц. р-р осветляют и из него получают K2SO4 методом
обменного разложения КС1 и MgSO4. Сульфат калия вследст-
вие его сравнительно невысокой растворимости выпадает
в осадок, его отфильтровывают и высушивают, а сильно раст-
воримый MgCl2 остается в р-ре.
K2SO4 является хорошим К. у. для всех культур,
по т. к. он дороже KG1, его применяют преим. для куль-
тур, на к-рые отрицательно действует хлор (табак,
виноград, чай, эфиромасличные, цитрусовые и др.).
Сульфат калия-магния (шёнит) пред-
ставляет собой двойную соль сернокислых калия и
магния K2SO4 • MgS04, технич. продукт, содержит
24—27% К2О и 8—11% MgO. В СССР получают при
переработке каинитолангбейнитовой руды.
Зола — минеральный остаток от сжигания расти-
тельных веществ, содержащий различные количества
поташа К2СО3. В золе подсолнечника содержится 30—•
40% К2О, ок. 25% — в золе гречихи, ок. 20% — ржа-
ной соломы, 13—15% — березы и вяза, 3—4% — ели,
в смешанной древесной золе — 7—10% К2О. В золе
содержится также 1—12% Р2ОВ и 6—60% СаО, хлор
отсутствует. Зола особенно эффективна на кислых
почвах.
Потребность в К. у. особенно проявляется на дегра-
дированных и выщелоченных черноземах и на дер-
ново-подзолистых, легких песчаных и супесчаных,
торфянисто-болотных и луговых почвах. Для боль-
шинства культур К. у. вносят из расчета 45—60 кг/га
К2О. Для культур с повышенной потребностью в калии
(свекла, картофель, табак и др.) и на почвах, бедных
калием, дозы К. у. увеличивают до 90—120 кг/га
К2О. Особенно нуждаются в К; у. картофель, конопля,
лен, свекла, табак и многие овощные культуры.
Лит.: Андреичев А. Н., Нудельман А. Б.,
Добыча и переработка калийных солей, М., 1960; Вольф-
кович С. И., Из истории советской калийной промышлен-
ности, в кн.: Материалы по истории отечественной химии,
М.—Л., 1950; П о з и н М. Е., Технология минеральных
солей, Л., 1961; Справочник по минеральным удобрениям.
Теория и практика применения, М., 1960; Прянишни-
ков Д. Н., Избранные сочинения, т. 1, Агрохимия, М.,
1952; Чириков Ф. В., Агрохимия калия и фосфора, М.,
1956; Мандель Р. А., Ж. прикл. химии, 1962, 35, № 1,1.
А. И. Шерешевский.
КАЛИЙОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ. Чрез-
вычайно реакционноспособные соединения, более,
чем соответствующие соединения натрия; известны
только в растворе. К. с. загораются на воздухе, раз-
лагаются спиртами, водой и реагируют с большинством
растворителей;
RK-|-H2O RH + KOH; RK + СН3ОН-> RH + СН3ОК
С2Н5К + (С2Н5)2О -» С3Н8 + с3н4 + С2Н-,ОК
При получении К. с. в качестве растворителя исполь-
зуют петролейный эфир и работают в атмосфере тща-
тельно очищенного азота (С6Н6К может быть получен в
353
КАЛИПНОН — КАЛИЯ ГЕКСАЦИАНОФЕРРОАТ
354
бензоле, СвНвСН2К — в толуоле и т. д.). О выходе К. с.
судят по выходу соответствующей кислоты после
обработки реакционной смеси твердой СО2. Наиболее
устойчивые К. с. образуются при действии металлич.
калия или амида калия на углеводороды с подвижным
атомом водорода; таковы, напр., ацетиленид калия и
трифенилметилкалий:
2СНЕСН + 2К -* 2СН-СК + Hs
(CcH5)sCH+ KNHS -► (CeH5)sCK + NHS
К. с. могут быть получены принципиально теми же
методами, что и натрийорганич., однако относительное
значение этих методов изменяется. Так, существенную
роль играет расщепление простых эфиров под дейст-
вием калия:
СвН5СН2ОСН3 + 2К -> с6н5сн2к + СН3ОК
тогда как реакция калия с галогеналкилами протекает
с более низкими выходами, чем та же реакция с на-
трием:
н-С5НцС1 + 2К -♦ м-С5НцК + КС1
Калий легко вытесняет ртуть из ртутьорганич. со-
единений:
(w-C4H8)2Hg + 2К 2н-С4Н0К + Hg
К. с. сравнительно быстро металлируют ароматич.
углеводороды с образованием ароматич. соединений
калия:
M-СаНцК + С6Н0 M-C5H1S + СеН5К
толуол металлируется в боковую цепь:
СвН5СН3 + СвН5К -♦ CeH5CHsK + CeHe
К. с. присоединяются к непредельным углеводородам:
СеН5СН=СНг+СвН5(СН3)2СК-
- С3Н5СН(К)СН2С(СН3)2СаН5
Реакции К. с., кроме вышеупомянутых, мало изучены.
Они редко применяются в органич. синтезе, что объяс-
няется неудобством работы с ними, а также тем, что
в большинстве случаев они могут быть заменены на-
трий- или литийоргаиич. соединениями. См. Натрий-
органические соединения, Шорыгина реакция.
Лит..: Кочешков К. А. и Талалаева Т. В.,
Синтетические методы в области металлоорганических сое-
динений лития, натрия, калия, рубидия и цезия, М.—Л.,
1949 (Синтет. методы в области металлоорганич. соединений,
вып. 1, ч. 1-—5). Н. А. Несмеянов.
КАЛИПНОН (баротал, калипнеттен, 5-этил-5-кро-
тилбдрбитуровая кислота) C10H14N2O3, мол. в. 210,24—
СН3-СН=СН-СН2 CO-NH
х /
л
С2Н6 CO-NH
белые кристаллы горь-
кого вкуса, т. пл. 110—
С=о И5°; трудно растворим
' в воде, растворяется в
спирте, эфире, ацетоне
и разб. щелочах. Водный р-р К. имеет кислую ре-
акцию по метиловому красному; К. обесцвечивает
бромную воду. К. представляет собой смесь равных
количеств цис- и транс-формы Щис-форма, т. пл.
106—108°; транс-форма, т. пл. 118—120°). При дей-
ствии NaOC4H9 может быть получена Na-соль К. При
действии на К. СаС12 и Co(NO3)3 в щелочном р-ре
появляется сине-фиолетовое окрашивание. Количест-
венно К. определяют титрованием щелочью в спирте.
К. может быть получен конденсацией бромистого
кротила с Na-солью 5-этилбарбитуровой к-ты в присут-
ствии NaOOCCH3:
Na CO-NH
CH2-CH=CH-CH2Br + c 'co—
C2H6 CO-NH"'
К. можно также получить из этилкротилмалонового
эфира и мочевины.
К. — снотворное средство, иногда применяется так-
же как противосудорожное средство при эпилепсии.
По быстроте снотворного эффекта и продолжитель-
ности действия К. занимает промежуточное место
между этаминалом и барбамилом.
Лит.: Elbert G., Dtsch. Gesundheitswesen, 1953, 8,
Н. 40, 1216; Wolf Н. J., там же, 1954, 9, Н. 24, 767;
HoIImann Н. [и. а.], там же, 1951, в, Н. 50, 1438.
В. Г. Яшунский.
КАЛИФОРНИЙ (Californium) Cf — химич. эле-
мент с п. и. 98 из семейства актинидов.
КАЛИЯ БОРГИДРИД КВН4 — бесцветные кри-
сталлы кубич. системы, а = 6,7272 А; плотн. 1,175,
теплота образования ДН°298 = —57,5 ккал/моль. Раз-
лагается без плавления ок. 500° с выделением водоро-
да. В сухом воздухе К. б. стоек до 350°, при более вы-
сокой темп-ре окисляется до КВО2 и Н2О, от огня
воспламеняется и спокойно сгорает; негигроскопичен;
растворим в воде (19,3 г на 100 г Н2О при 20°), жидком
аммиаке (20,0 г на 100 г NH3 при —30°), метиловом и
этиловом спиртах. Водные р-ры разлагаются выше
50° с выделением водорода. Щелочные растворы стойки
до 100° и выше. К. б. энергично разлагается кисло-
тами с выделением Н2 и B2He. С галогенидами бора
реагирует с образованием диборана. К. б. — сильный
восстановитель; селективно восстанавливает карбо-
нильные и нек-рые другие группы, не затрагивая двой-
ных связей. Получение К. б. основано на взаимодей-
ствии масляной суспензии NaH с В(ОСН3)3 и разложе-
нии полученной смеси NaBH4 и NaOCH3 конц. р-ром
КОН. Применяется в химико-фармацевтич. пром-сти,
а также в лабораторных органич. синтезах.
Лит.: Самсонов Г. В. [и др.], Бор, его соединения
и сплавы, Киев, I960, с. 109; Гейлорд Н., Восстановле-
ние комплексными гидридами металлов, пер. с англ., М., 1959,
с. 24; Kir k, Suppl., v. 1, N. Y., 1957, p. 507 (см. также лит.
при ст. Калий). Д. С. Стасиневич.
КАЛИЯ БРОМИД (калий бромистый) КВг — бес-
цветные кристаллы с кубич. решеткой, а = 6,596 А;
плотн. 2,75; т. пл. 730°; т. кип. 1380°. Теплота образо-
вания ДН°293 = —93,73 ккал[молъ. Растворимость
в воде (г на 100 г Н2О): 54,0 (0°); 65,6 (20°); 105 (100°).
Криогидратная точка —12,6° (45,7 г КВг на 100 г
Н2О). Насыщенный р-р, содержащий ок. 110 г КВг
на 100 г Н2О, кипит при 112°. Плотность водных ра-
створов:
%КВг........ 10 20 30 40
Й20 ........ 1,0740 1,1601 1,2593 1,3746
Растворим в жидком аммиаке, метиловом спирте.
Образует непрерывный ряд твердых растворов с КС1.
Получают К. б. действием Вг2 на аммиачный р-р КОН
или обменной реакцией между FeBr2 и К2СО3; при-
меняют в медицине и фотографии.
Лит.: Ксензенко В. И. иСтасиневичД. С.,
Технология брома и иода, М., 1960. См. также лит. при ст.
Калий. Д. С. Стасиневич.
КАЛИЯ ГЕКСАЦИАНОФЕРРИАТ (железосинеро-
дистый калий, феррицианид калия, красная кровяная
соль) K3[Fe(CN)e], мол. в. 329,26 — темно-красные
ромбич. кристаллы; растворимость в воде26,8% (10°);
водный р-р зеленовато-желтой окраски; на свету по-
степенно разлагается с образованием KJFe(CN)e].
В щелочной среде К. г. — сильный окислитель (осо-
бенно при нагревании). При нагревании с разб. H2SO4
разлагается с выделением синильной кислоты, при
действии конц. H2SO4 выделяется окись углерода.
Получают К. г. окислением K4[Fe(CN)e] хлором в соля-
нокислой среде; применяют для обнаружения Fea+,
с к-рым К. г. образует темно-синий осадок турнбуле-
вой сини Fe3[Fe(CN)e]2; эта реакция используется при
светокопировании. К. г. образует осадки с Ag+, Cs+,
Соа+, Nia+ и др. Применяют К. г. также в фотографии.
И. П. Ефимов.
КАЛИЯ ГЕКСАЦИАНОФЕРРОАТ (железисто-
синеродистый калий, ферроцианид калия, синькали,
желтая кровяная соль) K4[Fe(CN)e] ЗН2О, мол.
вес 422,41 — светло-желтые кристаллы, устойчивые
355
КАЛИЯ ГИДРООКИСЬ —КАЛИЯ ПЕРМАНГАНАТ
356
на воздухе. При нагревании до 100° К. г. теряет
кристаллизационную воду и белеет, при сильном про-
каливании разлагается с выделением азота; в воде
растворяется 22,4% (20°); нерастворим в спирте, эфи-
ре. При продолжительном хранении (особенно на
солнечном свету) водный раствор К. г. постепенно
разлагается; в присутствии кислот и кислых солей
(особенно при нагревании) медленно разлагается с вы-
делением синильной к-ты. Получают К. г. из очисти-
тельной массы газовых заводов, содержащих циани-
стые соединения, а также взаимодействием цианплава
(черного цианида) с FeSO4 с последующей обработкой
раствора КС1 и NaaCO3. Ранее К. г. получали сплав-
лением нек-рых отходов боен, напр. крови, с КаСО3
и железными опилками (отсюда произошло и назва-
ние «желтая кровяная соль»). Применяют К. г.
для обнаружения ионов Fe3+, с к-рыми К. г. дает синий
осадок берлинской лазури Fe4[Fe(CN)e]3 (см. Железная
лазурь), а также Си, Cd, Со; для определения ряда
катионов титриметрическими методами с использова-
нием визуальных и физико-химич. методов индикации
конечной точки (амперометрии, потенциометрии и др.).
В пром-сти К. г. применяют для произ-ва красок,
K3[Fe(CN)e] и др. веществ, для цементации сталей
И Т. Д. И. П. Ефимов.
КАЛИЯ ГИДРООКИСЬ (едкое кали) КОН — бес-
цветные кристаллы; при обычной темп-ре устойчива
ромбич. решетка (а-КОН), а = 4,03 А, Ь = 11,4 А,
с = 3,95 А; выше 248° устойчива кубич. решетка
(Р-КОН), а — 5,79 А; плотн. 2,12 (25°); т. пл. 380°,
т. кип. 1320°. Теплота образования ДН°а98 =
— —101,78 ккал/моль. Технич. продукт представляет
собой белую твердую непрозрачную массу с лучистым
изломом. Растворимость в воде (г на 100 г НаО):
97 (0°); 112 (20°); 178 (100°). Плотность водных рас-
творов:
% КОН.......... 10 20 30 40 50
dis............ 1,0918 1,1884 1,2905 1,3991 1,5143
Из 45—55%-ных растворов при (—25°) — (+30°)
кристаллизуется КОН • 2НаО. При более высокой кон-
центрации выше 30° кристаллизуется КОН • Н2О.
Известны также КОН - 1,5 НаО и КОН - 4НаО. Про-
дажный продукт содержит 90—92°/0 КОН и представ-
ляет собой смесь безводного КОН и КОН • НаО.
К. г. гигроскопична. При ее соприкосновении с водой
выделяется большое количество тепла. Водные р-ры
имеют сильнощелочную реакцию. К. г. поглощает
СОа из воздуха, превращаясь в калия карбонат.
Действует на кожу и слизистые оболочки прижигаю-
щим образом. Особенно опасно попадание даже малей-
ших количеств К. г. в глаза. Все работы с нею должны
производиться в защитных очках и резиновых пер-
чатках. К. г. разрушает кожу, бумагу и др. материалы
органич. происхождения. К. г. получают электроли-
зом р-ров КС1, обычно с применением ртутных като-
дов, что дает продукт высокой чистоты, не содержащий
примеси хлоридов. Применяют в произ-ве жидких
мыл, как исходный продукт для получения различных
солей калия и т. д.
Лит.: Виллитер Ж., Промышленный электролиз
водных растворов, пер. с нем., М., 1959. См. также пит. при
СТ. Калий. Д. С. Стасиневич.
КАЛИЯ ИО ДИД (калий иодистый) KJ — бесцвет-
ные кристаллы с кубич. решеткой, а = 7,066 А;
плотн. 3,115; т. пл. 686°; т. кип. 1324°. Теплота обра-
зования ДН°а98 = —78,31 ккал!молъ. Растворимость
в воде (г на 100 г НаО): 127,8 (0°); 144,5 (20°); 208
(100°). Криогидратная точка —23° (109 е KJ на 100 г
НаО). Насыщенный раствор кипит при 118,4° и со-
держит 221 г KJ на 100 г НаО. Плотность водных
растворов:
%KJ............. 10 20 28 40 50 60
dao ............ 1,0761 1,1660 1,2487 1,3959 1,5458 1,7310
Растворим в метиловом и этиловом спиртах. На
свету окисляется кислородом воздуха с выделением
иода, поэтому К. и. хранят в склянках темного стекла.
Получается при взаимодействии FeJa с КаСО3. При-
меняется в медицине и как реактив при химич. ана-
лизах.
Лит.: Ксензенко В. И., Стасиневич Д. С..
Технология брома и иода, М., 1960; Денисович В. П.,
Иод и его производство, М., 1938. См. также пит. при ст. Калий.
Д. С. Стасиневич.
КАЛИЯ КАРБОНАТ (калий углекислый, поташ)
КаС03 — бесцветные кристаллы; плотн. 2,418; т. пл.
891°; теплота образования ДН°а98 = —273,93 ккал/моль.
Очень гигроскопичен. Растворимость в воде (г на
100 г НаО): 105,5 (0°); 110,5 (20°); 155,7 (100°). Плот-
ность водных р-ров:
% К2СО3 .......... 10 20 28 40 50
d2»............... 1,0904 1,1898 1,2756 1,4141 1,5404
Образует гидрат КаСО3 • 1,5НаО — блестящие стекло-
видные кристаллы, полностью теряющие воду при
130—160°. К.к.производят карбонизациейр-ров КОН,
полученных электролитич. путем, или же суспензии
MgCO3 в растворе КС1, а также как побочный продукт
при переработке нефелина на глинозем. Раньше К. к.
добывали из растительной золы. Применяют в сте-
кольном и мыловаренном произ-вах, а также как
исходный продукт для получения различных калиевых
соединений и как калийное удобрение.
Лит.: П о з и н М. Е., Технология минеральных солей,
2 изд., Л., 1961. См. также пит. при ст. Калий.
Д. С. Стасиневич.
КАЛИЯ НИТРАТ (калий азотнокислый, калийная
селитра) KN03 — бесцветные ромбич. кристаллы,
а = 6,45 А, b = 9,17 А, с = 5,42 А; плотн. 2,11.
Выше 127,8° устойчива ромбоэдрич. модификация;
т. пл. 336°, теплота образования ДН°а98 = —117,76
ккал/моль. Растворимость К. н. в воде (г на 100 г
НаО): 31,6 (20°); 245 (100°); насыщенный раствор
кипит при 118°; криогидратная точка —2,85° (12,2 г
KNO3 на 100 г НаО). Плотность водных р-ров:
% KNO„......... 10 20
d2O.............. 1,0627 1,1326
При нагревании выше темп-ры плавления К. н. разла-
гается с выделением кислорода; смеси К. н. с органич.
веществами легко воспламеняются и интенсивно сго-
рают. В природе К. н. образуется при разложении орга-
нич. веществ в результате жизнедеятельности нитри-
фицирующих бактерий. Получается обменной реакцией
между NaNO3 и КС1 или при действии HNO3 или
нитрозных газов на КаСО3 или КС1. Применяется
как удобрение, а также в пиротехнике, при консерви-
ровании мясных продуктов, в стекольном произ-ве,
для приготовления черного пороха.
Лит.: П о з и н М. Е., Технология минеральных солей,
2 изд.. Л., 1961; М и н и о в и ч М. А., Соли азотной кислоты,
М.—Л., 1946. См. также лит. Прист. Калий, д. С. Стасиневич.
КАЛИЯ ПЕРМАНГАНАТ (калий марганцовокис-
лый) КМпО4 — темно-фиолетовые ромбич. кристаллы,
параметры „решетки а = 7,4114 А, b — 9,0992 А,
с = 5,7076 А; плотн. 2,703; теплота образования
ДН°а93 = —194,4 ккал/моль-, разлагается без плавле-
ния выше 240° с выделением кислорода. Растворимость
К. п. в воде (г на 100 г НаО): 2,83 (0°); 6,4 (20°); 22,2
(60°); растворы имеют красно-фиолетовый цвет; раст-
ворим в метиловом спирте, уксусной к-те и ацетоне.
К. п. сильный окислитель (в кислой среде Мп7+ вос-
станавливается до Мп2+, в щелочной — до Мп4+),
взрывается при обработке конц. HaSO4, а также при со-
прикосновении с органич. веществами. Получают К. п.
сплавлением пиролюзита с КОН при доступе воздуха
и дальнейшим электролитич. окислением образовав-
шегося КаМпО4. Применяют в промышленном орга-
357
КАЛИЯ ПЕРСУЛЬФАТ —КАЛИЯ СУЛЬФИД
358
нич. синтезе как окислитель, в медицине и ветерина-
рии и как химич. реактив.
Лит.: По зин М. Е., Технология минеральных солей,
2 иад., Л., 1961; U 1 1 m а п п, 3 Aufl., Bd 12, Munch. — В.,
1960, S. 229. Д. С. Стаеиневич.
КАЛИЯ ПЕРСУЛЬФАТ (калий надсернокислый)
KaSaOa, мол. в. 270,33 — бесцветные кристаллы (мел-
кие призмы, при медленной кристаллизации — большие
пластинчатые кристаллы) или кристаллич. порошок;
растворимость в воде 4,49% (20°). При нагревании
уже ниже 100° К. п. начинает выделять Оа. При дей-
ствии солей нек-рых металлов на растворы KaSaOa
последний разлагается на HaSO4, KaSO4 и Оа, причем
металлы, способные окисляться до мярокисей (как
Мп, Ni, Со, РЬ), в присутствии щелочей образуют чер-
ные осадки перекисей. К. п. может быть получен
по любой из реакций:
(NHHsSsOs + 2KCI---------------->• KsSsOa + 2NH4C1
(в присутствии КОН)
(NHiJgSsOe -f- 2КОН —► KaSaOa -j- 2NHB -f- 2НаО
Применяют К. п. как окислитель при фотометрия,
определении Мп; для разложения соединений железа,
содержащих хром; как окислитель в органич. синтезе,
органич. и неорганич. анализе и т. д. и, п. Ефимов.
КАЛИЯ ПЕРХЛОРАТ (калий хлорнокислый)
КСЮ, — бесцветные кристаллы ромбич. системы,
а = 7,24 А, b = 8,85 А, с = 5,66 А; плотн. 2,524;
выше 299,5° превращается в кубич. модификацию,
а = 7,50 А; т. пл. 610°; теплота образования ДЯ°а98 =
= —103,6 ккал/молъ-, трудно растворим в воде (1,8 г
на 100 г НаО при 20°); нерастворим в органич. раство-
рителях. Устойчив на воздухе. Выше темп-ры плавле-
ния разлагается на КС1 и Оа. К. п. является более
сильным окислителем, чем хлорат калия, и в то же
время более стоек при хранении (не взрывается от
трения и удара). Получается обменной реакцией между
NaC104 и КС1. Применяется для приготовления без-
опасных взрывчатых веществ.
Лит.; Позин М. Е., Технология минеральных солей,-
2 изд., М., 1961; Perchlorates, ed. J. С. Schumacher,
N. Y. — L., 1960. См. также лит. при ст. Калий.
Д. С. Стаеиневич.
КАЛИЯ РОДАНИД (калий роданистый) KCNS
бесцветные расплывающиеся гигроскопич. кристаллы,
плотн. 1,886; т. пл. 173,2°; т. кип.'(с разложением)
500°. В 100 г воды при 0° растворяется 177,2 г К. р.,
растворение сопровождается сильным понижением
темп-ры раствора. Легко растворим в спирте. К. р.
обычно получают сплавлением цианида калия с серой:
KCN+ S= KCNS. Применяют, как и NH4CNS, в ана-
литич. химии для обнаружения железа, кобальта и др.
элементов и в др. областях (см. Аммония роданид).
КАЛИЯ СИЛИКАТЫ — калиевые соли кремне-
вых кислот. Известны следующие К. с.: КаО • SiOa
(или KaSiO3) — метасиликат калия, КаО • 2SiOa
(KaSiaOB) — бисиликат калия, КаО • 4SiOa (K2Si4O9) —
тетрасиликат налия. Эти силикаты образуют гид-
ратные формы: KaSiO8 • 0,5 НаО, K2SiO3- Н2О,
KaSi2OB • НаО и KaSi4O9 • НаО. Метасиликат
калия образует кристаллы ромбич. системы,
двуосные, оптически положительные, показатели пре-
ломления ng 1,528, пр 1,520; т. пл. 976°. Виси л и -
кат калия имеет пластинчатую форму кристал-
лов ромбич. системы; кристаллы двуосные, оптически
отрицательные, показатели преломления ng 1,513,
Пр 1,503; т. пл. 1045°. Для бисиликата калия обнару-
жены два превращения в кристаллич. состоянии —
при 560° и 250°. Тетрасиликат калия
образует кристаллы в виде двойных пластинок, дву-
осные, оптически положительные, ng 1,482, пр 1,477.
При 592° тетрасиликат претерпевает энантиотропное
превращение, сходное с а—0-превращением кварца;
т. пл. 770°. Плотность тетрасиликата калия 2,335
(20°). Интересно, что стекло того же состава имеет
значительно более высокую плотность — 2,384. Все
описанные безводные К. с. получаются при кристал-
лизации стекол соответствующего состава. Силикаты
калия по своим свойствам сходны с силикатами нат-
рия (см. Натрия силикаты). Все К. с., за исключением
тетрасиликата, растворяются в воде, образуя коллоид-
ные р-ры практически любой концентрации. Концент-
рированные р-ры силикатов калия (см. Стекло раст-
воримое) имеют меньшее практич. значение, чем ра-
створы силикатов натрия.
Лит..- Ай л ер Р. К., Коллоидная химия кремнезема
и силикатов, пер. с англ., М., 1959; Евстропьев К. С.,
Торопов Н. А., Химия кремния и физическая химия
силикатов, 2 изд., М., 1956; Григорьев П. Н., Мат-
веев М. А., Растворимое стекло, М., 1956; Vail J. G.,
Soluble silicates, v. 1—2, N. Y., 1952. В. П. Борзаковский.
КАЛИЯ СУЛЬФАТ (калий сернокислый) K2SO4 —
бесцветные ромбич. кристаллы, а = 7,42 А, b =
= 10,01 А, с = 5,73 А; плотн. 2,66; выше 585° устой-
чива гексагональная модификация; т. пл. 1074°;
т. кип. 2> 2000°; диэлектрическая проницаемость 5,87
(18°); теплоемкость 0,18 кал/г- град-, теплота образо-
вания ДЯ°а98 = —342,66 ккал/молъ. Растворимость
К. с. в воде (г на 100 г Н2О): 7,35 (0°); 11,1 (20°);
14,8 (40°); 24,2 (100°). Криогидратная точка—1,51°
(7,1 г KaSO4 на 100 г НаО). Насыщенный р-р Кипит при
101,4° и содержит 24,3 г KaSO4 на 100 г НаО. Плот-
ность водных р-ров при 20°: 1,0393 (5%), 1,0817 (10%).
Нерастворим в органич. растворителях. Образует мно-
гочисленные двойные соли, в том числе квасцы — двой-
ные соли К. с. с сульфатами трехвалентных металлов.
Двойные соли K3Na(SO4)a (глазерит), KaSO4 • MgSO4-
• 6Н2О (шенит), KaSO4- MgSO4- 4НаО (леонит),
KaSO4- 2MgSO4 (лангбейнит), K2SO4- MgSO4-2CaSO4-
• 2НаО (полигалит) встречаются в месторождениях
калийных солей. К. с. получают обменной реакцией
между К.С1 и MgSO4 или HaSO4, а также при прокали-
вании лангбейнита с углем: KaSO4 • 2MgSO4 + С=
= K2SO4 + 2MgO + СОа -f- 2SOa. Применяют как
удобрение, а также как исходный продукт для полу-
чения квасцов и др. солей.
Калия бисульфат KHSO4 — бесцветные
ромбич. кристаллы, плоти. 2,3, т. пл. ок. 200°. Очень
гигроскопичен. Растворы имеют сильно кислую реак-
цию. При 300—400° отщепляет воду, превращаясь
в смесь KaSO4 + K2SaO7. Получают действием HaSO4
на КС1. Применяют как флюс в металлургии и в ана-
литич. химии и как сульфирующий агент в анилокра-
сочной пром-сти.
Лит.; Позин М. Е., Технология минеральных солей,
2 изд., Л., 1961. См. также лит. при ст. Калий.
Д. С. Стаеиневич.
КАЛИЯ СУЛЬФИД (калий сернистый) KaS — бес-
цветные кристаллы кубич. системы, а = 7,391 А;
плотн. 1,80; т. пл. 471°; теплота образования ДН°а98 =
= —100 ккал/молъ. При нагревании желтеет, расплав-
ленный — красного цвета. К. с. гигроскопичен, хо-
рошо растворим в воде, образует кристаллогидраты
с 5 и 12 молекулами Н2О; в водных р-рах сильно гид-
ролизован. Легко окисляется кислородом воздуха до
тиосульфата калия KaS2O3. При поджигании сгорает
с образованием SO2. При кипячении водного р-ра К. с.
с избытком серы получаются полисульфиды
калия KaSa, K2S3) K2S4, K2SB. При насыщении водного
р-ра КОН сероводородом образуется калия ги-
дросульфид (кислый сернистый калий) KHS —
бесцветные кристаллы, гексагональная решетка с па-
раметрами а = 4,95 А и с = 9,94 А; т. пл. 455°. Теп-
лота образования Д77°ава = —63,2 ккал/молъ. Хорошо
растворим в воде; при кипячении водного р-ра выде-
ляется H2S.
К. с. получают сплавлением поташа с серой без
доступа воздуха. Применяют в фотографии. Продукт
359
КАЛИЯ СУЛЬФИТ - КАЛИЯ ХЛОРИД
360
от красно-коричневого до желто-зеленого цвета, по-
лученный при спекании КаСО3 с избытком серы при
доступе воздуха, содержащий примесь полисульфидов
и тиосульфата, называется серной печенью.
Лит. см. при ст. Калий. Д. С. Стасиневич.
КАЛИЯ СУЛЬФИТ (калий сернистокислый)
KaSO3— бесцветные кристаллы гексагональной систе-
мы; теплота образования &H°2as = —266,9 ккал/моль.
Растворимость в воде (в %):’ 51,5 (0°); 52,9 (90°).
Растворы сульфита калия из-за гидролиза име-
ют щелочную реакцию; весьма гигроскопичен, рас-
плывается на воздухе; образует кристаллогидрат
KaSO3 • 2НаО. Сухой К. с. стоек на воздухе, в растворе
быстро окисляется. При прокаливании разлагается
на KaSO4 и KaS. Получают К. с. пропусканием SOa
в раствор КОН или КаСО3. Применяют в текстильной
пром-сти как восстановитель.
Калия бисульфит KHSO3 — бесцветные
кристаллы; легко растворим в воде; при нагревании
превращается в KaSaO6. К. с. получают при взаимодей-
ствии кипящего р-ра КаСО3 или КОН с SOa. Приме-
няют для отбелки различных материалов (солома,
дубильные экстракты), а также в текстильной пром-сти
при крашении и печатании.
Калия пиросульфит (калия метабисуль-
фит) KaSaO5 — бесцветные моноклинные кристаллы,
плотность 2,34; теплота образования А77°а9а =
= —362,6 ккал]моль, разлагается при нагревании до
190°; не гигроскопичен. Растворимость в воде 30,9%
(20°); водные р-ры имеют кислую реакцию и выделяют
SOa. Пиросульфит получают насыщением кипящего
р-ра KaSO3 избытком SOa с последующей кристаллиза-
цией при охлаждении. Применяют в текстильной
пром-сти и фотографии.
Лит. см. при ст. Калий. Д. С. Стасиневич.
КАЛИЯ ФОСФАТЫ — калиевые соли мета-, пиро-
или ортофосфорной к-ты, соответственно мета-, пиро-
или ортофосфаты калия. Практич. значение имеет
гл. обр. метафосфат калия КРО3 как высококонцен-
трированное безбалластное фосфорно-калийное удоб-
рение (чистый метафосфат калия содержит 60,11%
РаО6 и 39,89% КаО). Метафосфат калия получают раз-
ложением хлористого калия при высокой темп-ре
ортофосфорной к-той. В качестве побочного продукта
получается соляная к-та. В зависимости от темп-ры
процесса, степени дегидратации и введения небольших
добавок солей можно получать метафосфат в водо-
или цитратнорастворимой форме, а при необходимости
и в нерастворимой форме, т. е. с различной подвижно-
стью в почве. Пирофосфат калия К4РаО7 и триполи-
фосфат калия К5Р3О1о применяют в качестве компо-
нентов жидких моющих средств.
Лит.: Вольфкович С. И. и Б а н ь щ и к о в а Т. А.,
Модифицирование метафосфата калия посредством добавок
солей, Ж. прикл. химии, 1959, 32, вып. 5, 941—47; Брон-
ников А. X. иИмханицкаяС. М., Исследование
свойств метафосфатов калия, там же, 1960, 33, № 8, 1733—39;
Бронников А. X., Веселов В. К., О некоторых
свойствах метафосфата калия, там те, 1962, 35, № 4, 739.
А. И. Шерешевский.
КАЛИЯ ФТОРИД (калий фтористый) KF — бес-
цветные прозрачные кристаллы, расплывающиеся на
воздухе: кристаллизуется в гранецентрированной
кубич. решетке, а — 5,344 А; плотн. 2,505; т. пл.
857°; т. кип. ок. 1500°. Теплота образования ДЯ°а9а =
= —134,46 ккал/моль. Растворимость 92,3 г на 100 г
НаО (18°). Из водных р-ров от 17,7° до 45° кристалли-
зуется гидрат K.F- 2НаО — гигроскопичные бесцвет-
ные кристаллы, плавящиеся при 41°; от —21,8°
до 17,7° кристаллизуется KF- 4НаО с т. пл. 19°.
Получают К. ф. растворением КОН или КаСО3 в плави-
ковой к-те. Применяют как составную часть электроли-
та при получении металлич. калия, при изготовлении
кислотоупорных замазок и флюсов для пайки и сварки,
для введения фтора в органич. соединения.
Калия бифторид KHFa — бесцветные
кристаллы; плотн. 2,35; т. пл. 239°. Известен во многих
модификациях. При нагревании до 400—500° отщеп-
ляет HF; гигроскопичен; растворяется в воде с сильно
кислой реакцией; водный р-р разъедает стекло. Раст-
воримость в воде (г на 100 г НаО): 39,2 (20°); 114,0
(80°). Бифторид калия получают растворением КОН
или КаСО3 в плавиковой к-те. Применяют для получе-
ния элементарного фтора, как компонент флюсов для
пайки серебра, для получения матовой поверхности
стекла, консервирования древесины и др.
Известны также соединения KF- 2HF, 2KF- 5HF,
KF - 3HF и KF - 4HF, легкоплавкие кристаллич.
продукты, используемые как электролиты при полу-
чении элементарного фтора.
Лит.: Рысс И. Г., Химия фтора и его неорганических
соединений, М., 1956; Фтор и его соединения, [сб. ст.], пер.
с англ., т. 1, М., 1953. См. также лит. при ст. Калий.
Д. С. Стасиневич.
КАЛИЯ ХЛОРАТ (хлорноватокислый калий, бер-
толетова соль) КС1О3 — бесцветные кристаллы, моно-
клинная решетка, а = 7,047 А, b = 5,585 А, с =
= 4,647 А, а = 108°46; плотн. 2,32; т. пл. 356°;
теплота образования Д77°а98 = —93,50 ккал/моль.
Растворимость К. х. в воде (г на 100 г НаО): 7,3 (20°);
56,2 (100°); насыщенный р-р кипит при 104,2° и содер-
жит 61,5 г КС1О3 на .100 г НаО. При нагревании К. х.
разлагается с выделением кислорода. Разложение
катализируется двуокисью марганца. Расплавленный
К. х. в присутствии органич. веществ взрывается са-
мопроизвольно, а смеси сухого К. х. с органич. веще-
ствами — от трения и удара. К. х. ядовит. Его полу-
чают электролизом р-ра КС1 или при обработке хлори-
стым калием раствора хлората кальция, полученного
действием хлора на известковое молоко при 60—70°.
Применяют в спичечном произ-ве и пиротехнике.
Лит.: Ш р а й б м а н С. С., Производство бертолетовой
соли и других хлоратов, М., 1938; П о з ин М. Е., Техно-
логия минеральных солей, 2 изд., Л., 196.1.
Д. С. Стасиневич.
КАЛИЯ ХЛОРИД (калий хлористый) К.С1 — бес-
цветные кристаллы с кубич. гранецентрированной ре-
шеткой, а = 6,290 А; плотн. 1,989; т. пл. 790°; т. кип.
ок. 1500°; диэлектрич. проницаемость 4,7 (20°); теп-
лоемкость 0,16 кал/г- град, твердость по Моосу
2—2,5. Теплота образования ДЯ°а9а = —104,3
ккал/моль. Растворимость К. х. в воде (в г на 100 г
НаО): 28,1 (0°); 34,3 (20°); 40,3 (40°); 56,2 (100°). Крио-
гидратная точка —10,7° (24,6 г КС1 на 100 г НаО).
Насыщенный водный р-р кипит при 108,59° и содер-
жит 58,4 г КС1 на 100 г НаО. Плотность водных ра-
створов:
% КС!........... 10 20 24
dso....... 1,0633 1,1328 1,1623
Немного растворим в жидком аммиаке и метиловом
спирте, нерастворим в большинстве органич. раство-
рителей.
К. х. встречается в природе в виде минерала силь-
вина. Сырьем для получения К. х. служит природный
сильвинит, представляющий собой смесь сильвина
с галитом NaCl, содержащую обычно 20—40% КС1,
а также минерал карналлит КС1 • MgCla • 6НаО.
Получение К. х. из сильвинита основано на том, что
растворимость КС1 в воде значительно увеличивается
при повышении темп-ры, в то время как растворимость
NaCl с темп-рой изменяется мало, а при одновременном
насыщении К.С1 уменьшается с повышением темп-ры.
Раствор, насыщенный обеими солями, содержит при
20° 10,4% КС1 и 20,7% NaCl, а при 100° 21,7% КС1
и 16,8% NaCl. Сидьвинитовую породу обрабатывают
горячим щелоком, при этом КС1 переходит в раствор,
a NaCl остается нерастворенным. При охлаждении
щелока К. х. кристаллизуется, а маточник возвра-
щается на растворение новой порции сильвинита.
361
КАЛИЯ ХРОМАТ —КАЛОРИМЕТРИЯ
362
Можно разделить сильвинит на КС1 и NaCl при по-
мощи флотации. Получение К. х. из карналлита осно-
вано на расщеплении этой двойной соли водой или
щелоком определенного состава, в результате чего
MgCla переходит в раствор, а К. х. остается в осадке.
К. х. применяется как удобрение (см. Калийные удоб-
рения), а также как исходное сырье для получения
КОН и калиевых солей.
Лит.: АндреичевА. Н. иНудельман А. Б.,
Добыча и переработка калийных солей, М., 1960; П о-
з и я М. Е., Технология минеральных солей, 2 изд., Л., 1961.
См. также лит. при ст. Калий. Д. С. Стасиневич.
КАЛИЯ ХРОМАТ (калий хромовокислый)
К2СгО4 — желтые ромбич. кристаллы, а = 7,61 А,
Ъ = 10,40 А, с = 5,92 А; плотн. 2,74; т. пл. 984°; теп-
лота образования А77о2в8 = —330,49 ккал/молъ. К. х.
негигроскопичен; растворимость в воде (г на 100 г
НаО): 63,0 (20°); 79,2 7100°); криогидратная точка
—11,35°(55 г К2СгО4на 100 г НаО). При действии кис-
лот превращается в КаСгаО7. Получают действием ще-
лочи на КаСгаО7. Применяют как химич. реактив.
Калия бихромат (калий двухромовокис-
лый, калиевый хромпик) КаСгаО, — оранжево-крас-
ные триклинные кристаллы, а = 7,50 А, 6 = 14,37 А,
с =7,38 А, а = 112°34', 0 = 90°5Г, у = 95°22';
плотн. 2,684. Превращается при 236,8° в моноклин-
ную форму, плавящуюся при 398°. Выше 610° разла-
гается на КаСгО4, СгаО3 и кислород. Теплота образо-
вания А77°ав8 = —485,90 ккал/молъ. Негигроскопи-
чен; растворимость в воде (г на 100 г НаО): 4,68 (0°);
12,3 (20°); 103 (100°). Сильный окислитель; ядовит.
Получают обменной реакцией между КС1 или K2SO4
и NaaC^Oj. Технич. применение КаСгаО7 не велико,
т. к. в большинстве случаев выгоднее применять более
дешевый NaaCraO7. Применяют в спичечной пром-сти,
пиротехнике и фотографии, а также для изготовле-
ния цинковой желтой (см. Пигменты) и как химич.
реактив.
Лит.: П о з и н М. Е., Технология минеральных солей,
2 изд., Л., 1961; Д ымчиши н Д. А., Производство хро-
мовых солей, М.—Л., 1934; Ullmann, 3 Aufl., Bd 5,
Mtinch. — В., 1954, S. 587. См. также лит. при ст. Калий.
КАЛИЯ ХРОМОРОДАНИД (роданохромиат ка-
лия) K3[Cr(SCN)e] • 4НаО, мол. в. 589,88 — блестящие
темно-красные кристаллы; устойчив на воздухе, при
нагревании выше 110° теряет кристаллизационную
воду; хорошо растворим в воде и в спирте. При взаи-
модействии с солями висмута в подкисленном р-ре
выпадает нерастворимый осадок кирпично-красного
цвета Bi[Ci(SCN)e]. Получают К. х. при взаимодей-
ствии солей 3-валентного хрома с избытком роданида
калия. Применяют К. х. для обнаружения Bi капель-
ной реакцией (открываемый минимум 0,4 мкг Bi),
для весового и объемного определения висмута.
КАЛИЯ ЦИАНИД (калий цианистый) KCN —
бесцветные кристаллы; кристаллич. решетка гране-
центрированная кубйч., а = 6,523 А; плоти. 1,56;
т. пл. 634,5°; теплота образования Д77°ав8 =
= —26,90 ккал/молъ. Выше 600° без доступа воздуха
возгоняется (без разложения), но в присутствии воз-
духа и СО2 окисляется при более низкой темп-ре до
цианата калия КС NO. Расплывается во влажном воз-
духе. Растворимость в воде (г на 100 г НаО): 71,6
(25°); 122,8 (103,3°); водный р-р вследствие гидролиза
имеет щелочную реакцию и выделяет HCN. Константа
гидролиза 2,54- 10-5(25°). Влажный К. ц. полностью
разлагается углекислым газом. В водном р-ре при ки-
пячении постепенно разлагается с образованием NH3
и НСООК. Растворим в метиловом и этиловом спиртах.
Получается взаимодействием КОН с HCN. Применяет-
ся в гальванотехйике, а также для азотирования стали.
Очень ядовит.
Лит.: Ullmann, 3 Aufl., Bd 5, Munch. —В., 1954,
S. 648. См. также лит. при ст. Калий. Д. С. Стасиневич.
КАЛИЯ ЭТИЛКСАНТОГЕНАТ (этилксантогено-
/ZS
вокислый калий) СаН6ОС7 , мол. в. 160,3 — желто-
ватые кристаллы, разлагающиеся во влажном воздухе;
в 100 г воды растворяется 88 г (0°) (водный р-р имеет
сильно щелочную реакцию); растворим в спирте,
плохо растворяется в эфире; при действии минераль-
ных к-т разлагается. Получают К. э. действием серо-
углерода на алкоголят калия. Применяют К. э. при
флотационном обогащении металлич. руд; для обна-
ружения и колич. определения металлов группы серо-
водорода и сульфида аммония: Си, Ag, Ni, Hg, Мо
и др;; для осаждения белков; при определении алка-
лоидов; для синтетич. целей. Хранить К. э. необхо-
димо в закрытых банках оранжевого стекла.
И. П. Ефимов.
КАЛОМЕЛЬНЫЙ ЭЛЕКТРОД — один из наиболее
распространенных электродов сравнения, представ-
ляющий собой металлич. ртуть в контакте с ионами
Hg|+ в растворе КС1. Источником ионов Hg|+ является
HgaCla — каломель, растворимость к-рой, а следо-
вательно, и концентрация ионов ртути в растворе
зависят от концентрации раствора КС1. Чем выше кон-
центрация последнего, тем сильнее сдвинуто равнове-
сие влево:
Hg2ci2 Hg;+ + 2С1-
и тем отрицательнее, следовательно, потенциал К. э.,
зависящий от концентрации ионов ртути в растворе.
К. э. применяют с различной концентрацией хлорида
калия—0,1н., 1н. и насыщенные. Ниже приведены
значения потенциалов К. э. относительно нормального
водородного электрода при 25° С:
Концентрация КС1, М 0,1 1,0 насыщенный
Потенциал К. з.,в +0,3365 +0,2828 +0,2438
Чаще всего пользуются насыщенным К. э. в связи
с простотой его приготовления. Растворимость КС1
330
320
ЗЮ
300
290
280
270
260
250
240
230
220'—------.—.----------------------
$ Ю 15 20 2$ 30 35 40 45 50 С
0.1 к.к.3
зависит от темп-ры; поэто- £.м,
му потенциал насыщенного $4о|—
К. э. сильнее зависит от
последней, чем потенциал
других К. э. (см. рис.).
При приготовлении К. з. на-
носят на слой ртути, налитой
на дно соответствующего сосу-
да, слой пасты, состоящей из
каломели, растертой с метал-
лич. ртутью (во избежание окис-
ления Hg|+); затем в сосуд на-
ливают р-.рКС1 нужной концент-
рации; этим же р-ром поль-
зуются для приготовления ука-
занной выше пасты. Форма со-
судов для К. з. весьма разнооб-
разна и зависит от условий, в к-рых будет работать этот элект-
род. Напр., для полярография, и амперометрия, работ при-
меняют сосуд, обеспечивающий большую поверхность ртути
(во избежание поляризации); для работ, не связанных с про-
хождением тока, напр. для обычных, потенциометрия, изме-
рений, поверхность ртути не имеет значения и К. э. можно
придать любую форму, удобную для работы. О. А. Сангина.
Насьиц.
КАЛОРИМЕТРИЯ — совокупность методов изме-
рения количества теплоты, выделяющейся или погло-
щаемой в различных физич. или химич. процессах.
Методы К. применяют при определении теплоемкости,
тепловых эффектов химич. реакций, теплот фазовых
переходов, растворения, смачивания, адсорбции, ра-
диоактивного распада и др. Калориметрии, методы
играют важнейшую роль в определении термодина-
мич. параметров химич. реакций и фазовых перехо-
дов и термодинамич. свойств различных веществ
(энтропии, изотермич. потенциалов и др.). Методы
К. широко применяются в пром-сти для определения
теплотворной способности топлива и т. д. Данные К.
имеют большое практич. значение для составления
тепловых балансов, напр. при проектировании про-
363
КАЛОРИМЕТРИЯ
364
мышленных предприятий, и широко используются
в химич. технологии, металлургии, теплотехнике
и др. отраслях народного хозяйства.
Определение количества выделившейся (или по-
глотившейся) в том или ином процессе теплоты про-
водят обычно в специальном приборе — калори-
метре. Совокупность частей калориметра, между
к-рыми распределяется все тепло, подлежащее изме-
рению, наз. калориметрической систе-
мой. Калориметрия, измерения, как правило, со-
стоят в наблюдении за изменением температуры ка-
лориметрия. системы во время опыта*.
Устройство калориметра в основном
определяется характером изуяаемого процесса, его
продолжительностью, велияиной теплового эффекта,
сопровождающего процесс, темп-рой, при к-рои про-
изводятся измерения, и требуемой тояностью изме-
рений. Калориметрия, измерения производятся в са-
мых различных условиях. Так, область темп-p про-
стирается от 0,1 до ~4000°К; длительность изуяаемых
процессов колеблется от долей секунды до нескольких
суток, а колияество измеряемого тепла — от ~10-5 ка-
лории до неск. тысяя калорий. Тояность измерений
определяется задаяей исследования и во многих слу-
яаях является весьма высокой — порядка 0,1—0,01%.
Большое разнообразие задач, ставящихся при ка-
лориметрия. измерениях, и условий проведения этих
измерений обусловливает наличие большого числа
различных типов калориметров. Устройство калори-
метров настолько разнообразно, что всеобъемлющая
классификация их чрезвычайно затруднительна. От-
дельные, наиболее распространенные типы калори-
метров описаны ниже. Большинство из них относит-
ся к калориметрам с переменной
т емп-рой. Количество теплоты Q, полученное
таким калориметром во время опыта, вычисляется
по ф-ле Q = Н М, где Я — тепловое зна-
чение калориметра, т. е. количество теп-
лоты, необходимое для нагревания калориметра на 1°,
а Дг — изменение его темп-ры в опыте. Это изменение
обычно составляет величину 1—3° и в прецизионных
работах должно измеряться с высокой точностью.
Для измерения темп-ры калориметра обычно исполь-
зуются ртутные термометры, термометры сопротив-
ления или термопары, а при высоких темп-рах —
оптич. пирометры. Часто употребляются специальные
калориметрии, термометры, обладающие высокой чув-
ствительностью. Значение Н определяется Или спе-
циальными опытами, в к-рых в калориметр вводится
известное количество теплоты и измеряется Дг,
или же расчетом, по теплоемкости всех тел, входящих
в калориметр. Второй способ является менее точным
и в последнее время применяется редко. Для опре-
деления Н нагревом (или охлаждением) калориметра
известное количество теплоты Q вводится или с по-
мощью нагревателя, питаемого электрич. током, или
с помощью процесса, тепловой эффект к-рого хорошо
известен (напр., теплота сгорания бензойной к-ты,
теплота растворения хлористого калия и т. д.). Опре-
деление Н вводом известного количества теплоты
может быть произведено с высокой точностью (до
0,01%, а иногда и выше). Очень существенно, что этот
способ позволяет измерять темп-ру калориметра
в условных единицах. Наиболее благоприятным
является случай, когда при определении неизвестного
количества теплоты Qx и при определении теплового
значения калориметра Н в опытах совпадают началь-
ные и конечные темп-ры; в этом случае требуется лишь
воспроизводимость показаний термометра и отпадает
* Нередко термин «калориметрическая система» заменяют
термином «калориметр», к-рый, т. обр., употребляется в двух
значениях. В данной статье в целях сокращения также принята
такая вамева, когда это не может вызвать недоумений.
необходимость его тщательной градуировки. Полное
совпадение наблюдается редко, но на практике почти
во всех случаях, когда Дг при определении Н и Qx
достаточно близки, а опыты начинались при одной
и той же темп-ре, большая часть термометрия, попра-
вок не вводится.
Калориметрический опыт обычно со-
стоит из трех периодов. В начальном периоде (до
ввода измеряемого тепла в калориметр) происходит
равномерное изменение темп-ры калориметра вследст-
вие теплообмена с окружающей средой (оболочкой)
и побочных процессов в самом калориметре (мешалка
и т. д.). Это изменение, отнесенное к единице времени,
обычно наз. температурным ходом кало-
риметра. Главный период начинается С момента ввода
тепла в калориметр и характеризуется быстрым
и неравномерным изменением темп-ры калориметра.
Конечный период наступает, когда ввод измеряемого
тепла и его распределение в калориметре заканчи-
ваются, и температурный ход калориметра снова
становится постоянным.
При определении изменения темп-ры во время
калориметрия, опыта влияние окружающей среды
и посторонних процессов, происходящих в калори-
метре (таких, как трение мешалки или пропускание
тока через термометр сопротивления), должно строго
учитываться. Для этого к наблюдаемому в опыте из-
менению темп-ры вводится поправка на Теп-
л о о б м е н. Для точного определения поправки
на теплообмен калориметры обычно изолируют от
внешней среды оболочками, темп-pa к-рых контроли-
руется заданным образом и к-рые, как правило,
бывают или изотермическими, или адиабатными.
В прецизионных работах постоянство темп-ры изо-
термич. оболочки поддерживается с точностью ±0,001°;
с такой же точностью нередко осуществляются адиа-
батные условия.
В калориметрах с изотермич. оболочкой во время
опыта во всех трех периодах производится измерение
темп-ры калориметра через равные промежутки вре-
мени (т. е. снимается кривая темп-ра — время).
Для вычисления поправки на теплообмен по этим
данным предложен ряд формул. Наиболее часто при-
меняется формула Реньо — Пфаундлера — Усова, ос-
нованная на законе охлаждения Ньютона. Эта фор-
мула учитывает теплообмен с окружающей средой
и полностью исключает влияние постоянного во вре-
мени подвода тепла, обусловленного, напр., трением
мешалки или током, проходящим через термометр
сопротивления. Поправка на теплообмен в случае
калориметра с изотермич. оболочкой нередко бывает
значительной (1—4% от всей величины Дг), но вы-
числяется весьма точно. Калориметры с изотермич.
оболочкой обычно применяются при определении
теплот сравнительно быстрых процессов (продолжи-
тельность главного периода 10—20 мин).
В калориметрах с адиабатной оболочкой во всех
трех периодах калориметрии, опыта производится
измерение разности темп-p оболочки и калориметра.
Темп-ра калориметра обычно измеряется только
в начальном и конечном периодах опыта. Темпера-
турный ход калориметра в этих периодах должен быть
равен нулю или близок к нулю (незначительный тем-
пературный ход зачастую может иметь место и при
равенстве темп-p калориметра и оболочки вследствие
неполного устранения влияния внешней среды адиа-
батной оболочкой). Поправка на теплообмен в кало-
риметрах с адиабатной оболочкой значительно меньше
по величине, чем в калориметрах с изотермич. обо-
лочкой, и обусловлена как небольшими разностями
между темп-рой оболочки и темп-рой калориметра,
так и незначительным температурным ходом калори-
метра. Калориметры с адиабатными оболочками часто
365.
КАЛОРИМЕТРИЯ
366
применяют при определении теплот медленных про-
цессов.
Наиболее часто для работы при комнатных темп-рах
употребляются калориметры с жидкостью (обычно
водой), наз. иногда обыкновенными калориметрами.
Жидкостные калориметры различных
конструкций с успехом применяют при определениях
теплот сгорания, теплот растворения, нейтрализации
и других химич. реакций, при определении теплоем-
костей твердых и жидких тел, теплот испарения и т. д.
Калориметрия, сосуд, имеющий вместимость от 50
до 4000 мл, изготовляется чаще всего из металла
с хорошей теплопроводностью (медь, серебро, плати-
на). Нередко также в качестве калориметрия, сосуда
употребляются стеклянные сосуды Дьюара.
На рис. 1 изображен жидкостный калориметр, предназна-
ченный для определения теплот сгорания. В калориметрия.
сосуд 1 помещается взвешен-
ное количество воды, термо-
метр 2 и нагреватель з, пред-
назначенный для подогрева
калориметрия, жидкости до
темп-ры опыта (в других слу-
чаях нагреватель применяется
также и для определения теп-
лового значения калориметра).
Мешалка калориметра 5 долж-
на хорошо перемешивать кало-
риметрия. жидкость при не-
большой и постоянной во вре-
мени теплоте трения. Кроме
того, в сосуд 1 полностью по-
гружена калориметрия, бом-
Рис. 1. Калориметр для опре-
деления теплот сгорания: 1 —
калориметрический сосуд; 2 и
7 — термометры калориметра
и оболочки; з и S — нагреватели калориметра и оболочки;
4 — калориметрия, бомба; 5 и 6 — мешалки; 9 — водяная изо-
термич. оболочка, окружающая калориметрия, систему.
ба 4, служащая для сожжения веществ в избытке кислорода
(обычно при давлении 30 атм). Калориметрическ ая
бомба представляет собой толстостенный стальной стакан
объемом 250—300 мл и должна выдерживать давление 100—
150 атм (в нек-рых случаях применяют бомбы, имеющие зна-
чительно меньший объем). Известно много конструкций ка-
лориметрия. бомб, одна из к-рых изображена на рис. 2. Бомба
, изготовлена из нержавеющей стали и
а состоит из двух частей, соединяемых
лЖЗ накидной гайкой 2. При повышении
давления в бомбе ее верхняя часть
прижимает кольцевую резиновую про-
: кладку 7 к нижнему срезу накидной
: гайки 2, что обеспечивает надежную
герметизацию бомбы. Чем больше дав-
ление внутри бомбы, тем надежнее ее
Рис. 2. а) Самоуплотняющаяся калориметрия, бомба:
1—конический клапан для промывания бомбы кислоро-
дом; 2 — накидная гайка; з и в — клеммы; 4 — конический
клапан; 5 — сальник; 7 — уплотняющаяся прокладка (ре-
зина); 3 — стержни для подвода тока; 9 — брикетка
сжигаемого вещества, б) Деталь самоуплотняющейся
бомбы.
герметизация. Для зажигания вещества на его брикетку на-
кладывается железная проволочка, сгорающая при включении
в цепь источника тока. В других случаях в брикетку вещества
впрессовывается хлопчатобумажная нить, загорающаяся от
раскаленной током платиновой проволочки. Клеммы з и 6
служат для присоединения источника тока. Одна из них (з) не
изолирована от корпуса бомбы. Расположение изолированного
ввода снизу бомбы под чашечкой предохраняет изоляцию и
входной клапан от действия высокой темп-ры.
Измерения теплот сгорания органич. веществ, в ка-
лориметрия. бомбе могут проводиться С очень высокой
точностью (около 0,01%). Значительные затруднения
возникают при определении теплот сгорания соеди-
нений, содержащих галогены или серу, т. к. образую-
щиеся продукты обычно содержат эти элементы в раз-
личной степени окисления. В этих случаях, длн того
чтобы довести систему до строго определенного ко-
нечного состояния, в бомбу вводят дополнительные
вещества, обычно восстановители или окислители,
а для ускорения процесса восстановления или окис-
ления зачастую производится энергичное перемеши-
вание содержимого бомбы во время калориметрия,
опыта путем приведения бомбы в движение с помощью
специального механизма (качающиеся или вращаю-
щиеся бомбы). Для пересчета измеренных в бомбе
теплот сгорания к стандартному состоянию (изо-
термич. теплота сгорания при 1 атм и 25°С) разра-
ботана система поправок.
В жидкостных калориметрах иного назначения
в калориметрия, сосуд часто помещаются другие
приборы для проведения изучаемой реакции. Такими
приборами могут быть, напр., камера с горелкой для
определения теплот сгорания при давлении, близком
к атмосферному, в струе кислорода или фтора, реак-
ционные камеры для проведения в калориметре реак-
ций галогенирования, гидрогенизации и т. д. Устрой-
ство таких камер определяется характером изучаемой
реакции. В ряде случаев, напр. при определениях
теплот растворения, нейтрализации, смешения и др.,
изучаемая реакция проводится непосредственно в ка-
лориметрия. сосуде. В этих случаях в калориметрия,
сосуд обычно помещается ампула из стекла или к.-л.
иного материала, содержащая твердое или жидкое
вещество и разрушаемая в процессе проведения опыта
(в начале главного периода).
В нек-рых жидкостных калориметрах вместо воды
используются другие жидкости — бензол, этиловый
спирт, жидкий аммиак, расплавленное олово и т. д.
Применение других жидкостей, как правило, вызы-
вается или необходимостью использовать их в ка-
честве компонента изучаемой реакции, или необхо-
димостью повысить температурный интервал приме-
нения жидкостного калориметра. При обычных же
условиях и в области темп-p, близких к комнатным,
калориметр с водой является, безусловно, самым
распространенным из жидкостных калориметров. Не-
достатком такого калориметра, помимо ограничен-
ности температурного интервала его применения,
является так1ке испарение воды во время опыта, к-рбе
может служить серьезным и трудно устранимым источ-
ником погрешностей, особенно при наиболее точных
измерениях. В некоторых случаях, для того чтобы
предотвратить испарение жидкости, прибегают к гер-
метизации калориметра. Однако герметич. калори-
метры сложнее по устройству и пока еще не получили
широкого распространения.
В массивном калориметре вместо
калориметрия, жидкости употребляются блоки из
металла с хорошей теплопроводностью
(обычно из меди, алюминия, серебра). Масса блока
определяется задачей исследования и в различных ка-
лориметрах колеблется от 150 г до 32 кг. Внешняя по-
верхность блока тщательно полируется для умень-
шения тепловой радиации. Для размещения термо-
' метра, нагревателя и тех или иных реакционных
сосудов (или ампул) блок обычно имеет соответствую-
щие ячейки. Иногда термометр н нагреватель разме-
щаются на внешней поверхности блока. Темп-ра
блока может измеряться ртутным термометром, но
чаще в массивных калориметрах применяются термо-
метры сопротивления, к-рые могут сочетать небольшие
габариты с высокой чувствительностью. Нередко для
367
КАЛОРИМЕТРИЯ
368
измерения темп-ры в массивных калориметрах при-
меняют термисторы или батареи термопар. Массивные
калориметры очень часто применяют для определения
средних теплоемкостей в широком интервале темп-р
от 0° и почти до 3000° методом смешения.
Одив из таких массивных калориметров изображен на
рис. 3. Существенной частью массивного калориметра является
печь для нагрева исследуемых образцов до заданной темп-ры.
Рис. 3. Схема массивного калориметра для
определения средних теплоемкостей: 1— зри-
тельная труба; 2 — ртутный термометр тер-
мостата; 3 — термометр сопротивления; 4 —
нагреватель термостата; 5 — побочный спай
термопары; 6 — медный блок с ячейкой для
приема ампулы; 7 — оболочка; S — мешалка;
9 — печь; 10 — ампула с образцом; 11 — про-
тивовес.
Образец помещается в ампулу (10) из тугоплавкого мате-
риала, к-рая после нагрева в печи сбрасывается в медный блок
(в) и плотно входит при этом в его конусообразную ячейку.
Темп-ра образца перед сбрасыванием измеряется термопарой
или оптич. пирометром. Для измерения темп-ры массивного
блока, изготовленного из электролитич. меди, служит плати-
новый термометр сопротивления, навитый на его боковую по-
верхность. Ртутный термометр 2, термометр сопротивления з
и нагреватель 4 служат для автоматич. регулировки темп-ры
термостата и для контроля за ее постоянством.
Кроме средних теплоемкостей, в массивных кало-
риметрах определяются теплоты сгорания, испарения,
адсорбции и т. д. Основными преимуществами мас-
сивных калориметров являются возможность их
использования в широком диапазоне темп-p и от-
сутствие погрешностей, связанных с испарением
калориметрия, жидкости. Значительным недостатком
массивного калориметра является более медленное
выравнивание темп-ры калориметрической системы
по сравнению с жидкостным калориметром. Возник-
новение в связи с этим погрешностей существенно
уменьшается, если опыты по определению теплового
значения калориметра проводятся в тех же условиях,
что и опыты по определению неизвестного теплового
аффекта.
Во многих случаях калориметрич. сосудом является
тонкостенный контейнер (ампула), в к-рый
помещается исследуемое вещество. Такой калори-
метр-контейнер обычно имеет сравнительно
небольшие размеры (как правило, в пределах 10—
150 мл) и изготовляется из металла с высокой тепло-
проводностью и небольшой теплоемкостью (медь,
серебро). При исследовании веществ с высокой реак-
ционной способностью материалом контейнера часто
является золото, платина или нержавеющая сталь.
Термометр и нагреватель можно размещать на внеш-
ней поверхности контейнера, но чаще их вставляют
в специальные ячейки. Темп-ра обычно измеряется
термометром сопротивления, к-рый должен быть
тщательно проградуирован. Калориметры-контейнеры
чаще всего применяют при измерении истинных те-
плоемкостей в широком интервале темп-p (от 0,1°К
до ~1400°К) и теплот фазовых переходов, а также
в ряде случаев при измерении теплот растворения,
испарения, смачивания и т. д. При работе с калори-
метром-контейнером вследствие его сравнительно ма-
лых размеров и очень небольшой теплоемкости (осо-
бенно при низких темп-рах) особое значение приобре-
тает его изоляция от внешней среды. При низких
темп-рах для улучшения теплоизоляции, кроме си-
стемы изотермич. или адиабатных оболочек, окру-
жающих калориметрич. сосуд, применяют высокий
вакуум. В этом случае давление в пространстве,
окружающем контейвер и оболочки, во время кало-
риметрич. измерений составляет обычно 10“6—10 6 льн
рт. ст.
Опыты обычно проводят методом периодич. ввода
тепла, т. е. работа ведется классич. методом, при
к-ром опыт делится на три периода (см. выше). Однако
нередко при определении теплоемкостей при высоких
темп-рах применяется методика определений с непре-
рывным вводом тепла. При этом методе калориметрич.
система находится в адиабатных условиях и изме-
ряются две величины: количество тепла, полученное
системой за известный промежуток времени, и соот-
ветствующий подъем темп-ры. Метод непрерывного
ввода тепла дает возможность быстро измеряй,
теплоемкости в широком интервале темп-p. Сущест-
венным недостатком метода является отсутствие
теплового равновесия в калориметрич. системе во
время измерений, что может привести к значительным
градиентам температурного поля и
к некоторому искажению темп-ры,
к к-рой должны быть отнесены из-
меренные значения теплоемкости
образца. На рисунке 4 показано уст-
ройство калориметра - контейнера
для низких температур. Калориметр
предназначен для определения ис-
тинных теплоемкостей методом пе-
риодич. ввода тепла. В некоторых
случаях калориметр-контейнер ок-
ружают несколькими адиабатными
оболочками с целью свести до ми-
нимума влияние внешней среды.
При измерениях тепловых эффек-
тов, а также и теплоемкостей с ус-
пехом применяют двойной ка-
лориметр, имеющий две совер-
шенно одинаковые калориметрич.
системы. В качестве таких систем
Рис. 4. Адиабатный калориметр для оп-
ределения истинной теплоемкости при
темп-рах 12—300°К. Схематический раз-
рез прибора; 1 — контейнер для веще-
ства; 2 — адиабатная оболочка; 3 — крышка оболочки; 4 —
цилиндр для подогрева подводящих проводов до темп-ры обо-
лочки; 5 —вакуумный стакан.
могут быть использованы сосуды, наполненные жид-
костью (водой), как в жидкостных калориметрах,
металлич. блоки, как и в массивном калориметре,
или тонкостенные контейнеры, наполненные иссле-
дуемым веществом. Обе калориметрич. системы двойно-
го калориметра находятся при одной и той же тем-
пературе и имеют одинаковый теплообмен с окру-
жающей средой. При таком устройстве отпадает не-
обходимость учета поправки на теплообмен; вместо
этой поправки вводится небольшая по величине по-
369
КАЛОРИМЕТРИЯ - КАЛЬЦИЙ
370
дуемой реакции (Qx),
правка на неидентичность блоков, к-рая определяется
специальными опытами. При определении тепловых
эффектов экзотермич. реакций в один из блоков вво-
дят неизвестное количество теплоты — теплоты иссле-
а в другой — известное коли-
чество теплоты (Q), которое
подбирается т. обр. (напр.,
путем изменения мощности
тока в электронагревателе),
чтобы темп-ры обоих блоков
были равны в продолжение
всего опыта; при этом усло-
вии Qx = Q. Примером тако-
го двойного калориметра яв-
ляется калориметр, пред-
назначенный для измерения
теплот полимеризации и изу-
чения кинетики выделения
тепла в реакциях полимери-
зации (рис. 5).
Рис. 5. Двойной массивный кало-
риметр для изучения кинетики
реакций полимеризации по мощ-
ности выделения тепла; 1 и 2 —
массивные серебряные полуцилиндры; з и 4 — стальные диски
для крепления; 5 — изоляционные втулки; в — термометры
сопротивления; 1 — нагреватели; 8 — конусообразные ячейки
для помещения ампул; а — термопары; 10 — трубки для ввода
ампул.
При проведении опытов с этим калориметром (рис. 5)
оболочку с двумя массивными серебряными блоками 1 и 2
помещают в масляный термостат и выдерживают при темп-ре
реакции (200—240°). Затем в один из блоков вносят ампулу,
содержащую исследуемое вещество или смесь веществ, а в
другой — такую же ампулу, содержащую нейтральное веще-
ство равной теплоемкости (напр., рассчитанное количество
хлористого калия). Тепло, выделяющееся в первом блоке, ком-
пенсируется нагревом второго блока электрич. током. Для конт-
роля равенства темп-p служат термопары S. Кривая мощ-
ность тока — время отражает кинетику выделения тепла в
реакции, а теплота реакции может быть вычислена интегри-
рованием. Двойные калориметры часто применяют и при изме-
рении теплоемкостей. При этих оп-
ределениях один из блоков напол-
няют веществом с известной тепло-
емкостью, а в другой помещают ис-
Ю следуемое вещество. Во время опы-
та плавной регулировкой устанав-
ливают такое соотношение мощно-
стей тока в нагревателях обоих бло-
ков, при к-ром имеет место равен-
ство темп-p блоков во время нагрева.
В двойном калориметре была, напр.,
определена теплоемкость многих
водных р-ров по отношению к теп-
лоемкости воды с очень высокой
точностью (порядка 0,01 %).
Калориметры с по-
стоянной температу-
рой отличаются от всех опи-
санных типов калориметров
тем, что калориметрич. система
во время опыта не претерпевает
изменения темп-ры и все сооб-
Рис. 6. Ледяной калориметр: 1 — ле-
дяная ванна; 2 — уровень льда в
ванне; з — стеклянные сосуды с ме-
таллич. крышками, герметически
примазанными вакуумной замазкой;
4 — трубка для ввода образца; S —
змеевик, по к-рому подается в трубку медленный ток углекис-
лого газа, предупреждающий проникновение водяных па-
ров; 6 — объем, занятый водой в измерительном сосуде; 7—объ-
ем, занимаемый льдом, к-рый намораживается на трубку 4 пог-
ружением в нее пробирки с сухим льдом; 8 — экраны, затруд-
няющие конвекцию образующейся вокруг трубки 4 воды; S —
нагреватель, служащий для градуировйи калориметра; 10 —
стакан с ртутью.
щаемое ей тепло идет на изменение агрегатного состоя-
ния вещества. В К. нередко применяют калориметры
с плавящимся твердым телом (лед, дифениловый
эфир, нафталин) (рис. 6). Количество введенного
в калориметрич. систему тепла определяется в таких
калориметрах по количеству твердого вещества,
перешедшего в жидкое состояние при темп-ре сосу-
ществования фаз. Количество расплавившегося ве-
щества обычно устанавливается по изменению веса
ртути в стакане (10), к-рое наблюдается вследствие
изменения объема системы твердое тело — жидкость,
или же, в случае малых тепловых эффектов, по изме-
нению уровня ртути. Калориметры с плавящимся
твердым телом обладают высокой чувствительностью
и часто применяются при измерении теплот продол-
жительных процессов (до нескольких суток). Не-
удобством такого калориметра является необходимость
проводить все измерения при темп-ре плавления
вещества.
Иногда калориметрич. опыт проводится при по-
стоянной темп-ре и в водяном или массивном калори-
метре. Отрицательный тепловой эффект при этом
компенсируется электрич. нагревом (компенсацион-
ный метод);, неизвестное количество тепла рассчиты-
вается по мощности тока и времени его пропускания.
Небольшая поправка вводится на изменение темп-ры
во время опыта, к-рое имеет место из-за неточности
компенсации; однако это изменение обычно можно
сделать очень незначительным. Такой метод нередко
применяется, напр., при определении теплот испаре-
ния; испарение жидкости при этом интенсифицируется
продуванием через нее воздуха или инертного газа.
В ряде случаев в К. используются методы, в к-рых
измерение количества тепла основано на измерении
мощности теплового потока между калориметрич.
системой и окружающей ее оболочкой. Калориметры
Кальве, в к-рых используется этот принцип, приме-
нялись в последнее время для измерения теплот раз-
бавления, этерификации, омыления сложных эфиров,
исследования реакций с участием высокомолекуляр-
ных соединений и для изучения ряда биология, про-
цессов. Эти калориметры являются очень сложными
приборами, но имеют высокую чувствительность
(~0,1 мквт) при практически неограниченной про-
должительности процесса. Значительная часть (до
95%) экзотермич. теплового эффекта компенсируется
в таких калориметрах эффектом Пельтье. Некомпен-
сированная часть тепла определяется измерением
с помощью особым образом расположенных термопар
теплового потока от внешней поверхности калори-
метра к оболочке. Высокая чувствительность калори-
метра требует уничтожения влияния температурных
колебаний термостата. Это достигается симметричным
расположением двух спаренных калориметрич. систем
в блоке из материала с очень хорошей теплопровод-
ностью и соединением их навстречу друг другу.
Лит.: Попов М. М., Термометрия и калориметрия,
2 изд., М., 1954; Experimental thermochemistry, ed. F. D.
Rossini, N. Y.—L., 1956; Experimental thermochemistry, ed.
H. A. Skinner, v. 2, N. Y.—L., 1962; Ro t h W. A.,B ecke г F,
Kalorimetrische Methoden zur Bestimmung chemischer Reak-
tionswarmen, Braunschweig. 1956; SwietoslawskiW., Mic-
rocalorimetry, N. Y., 1946; Э й т e л ь В., Термохимия сили-
катов, пер. с англ., М., 1957; Кубашевский О.,
Эванс Э., Термохимия в металлургии, пер. с англ., М.,
1954; Cal vet Е., Prat Н., Microcalorimetrie, Р., 1956;
Skinner Н. A., Modern aspects of thermochemistry, L., 1958;
Физические методы органической химии, под ред. А. Вайсбер-
гера. пер. с англ., т. 2,М., 1952, с. 69—189; Стрелков П. Г.
[и д р.], Ж. физ. химии, 1954, 28, вып. 3, 459; Шмидт Н. Е.,
Соколов В. А., Ж. неорг. химии, 1960, 5, вып. 8, 1641;
Armstrong G. Т., Jessup R. S., Combustion calo-
rimetry with fluorine: constant pressure flame calorimetry,
J. Res. Nat. Bur. Standards, 1960, 64A, 1, 49—59.
В. П. Колесов.
КАЛЬЦИЙ (Calcium) Ca— химия, элемент II гр.
периодич. системы Менделеева; принадлежит к ще-
лочноземельным металлам; п. и. 20, ат. в. 40,08.
Природный К. состоит из смеси 6 стабильных изо-
топов с м. ч.: 40, 42, 43, 44, 46, 48, из к-рых наиболее
371
КАЛЬЦИЙ
372
распространен Са40 (96,97%). Поперечное сечение
поглощения тепловых нейтронов атомом К. 0,43 барн.
Из искусственно радиоактивных изотопов наиболее
важен Са46 (Ti/3 = 163,5 дня). Внешняя электронная
оболочка атома К. имеет строение 4s2. Энергии иони-
зации (в эв)'. Са° -► Са+ -► Са2+ —► Сая+ —► Са4+ —< Са5+
соответственно составляют: 6,11; 11,87; 51,21; 66,7;
84,39.
Природные соединения К. — известняк, мрамор, гипс,
а также известь, получаемая обжигом известняка, уже в древ-
ности применялись как строительные материалы. Металлич.
К. впервые выделен в 1808 Дэви после отгонки ртути из амаль-
гамы К., полученной электролизом. Название «К.» дано от
латинского calx — известь.
По распространенности вприроде К. занимает
5-е место. Его содержание в земной коре 3,6 вес.%.
Основные минералы К. входят в состав осадочных
и метаморфич. горных пород. Наиболее распрост-
ранены известняки и мел — продукты жизнедеятель-
ности морских и беспозвоночных животных с извест-
ковым скелетом. Известняки состоят в основном
из минерала кальцита СаСО,, содержащего
в качестве примесей Mg, Fe, Мп (до 8%), реже — Zn
(до 2%), Sr. Кальцит — бесцветные или окрашенные
примесями кристаллы тригональной сингонии; плотн.
2,6—2,8; твердость 3; хрупок. Прозрачный кальцит
(исландский шпат), минерал гидротермаль-
ного происхождения, встречается в крупных моно-
кристаллах среди эффузивных изверженных пород.
Реже встречается арагон ит, минерал того же со-
става; кристаллы ромбич. сингонии; плотн. 2,9—3,0;
твердость 3,5—4,0; хрупок (см. Кальция карбонат).
Мел — плотный, полуземлистый известняк белого,
реже желтоватого цвета, содержащий до 99% СаСО3;
плотн. 2,65—2,70; твердость меньше 1. Мрамор —
метаморфич. горная порода, состоящая в основном
из зерен кальцита или доломита, окрашенная в раз-
личные цвета; плотн. 2,72—2,85; твердость 3,0—3,5.
Широко распространенным минералом осадочных
пород является ангидрит CaS04, образующийся
при кристаллизации солевых р-ров озерных и мор-
ских бассейнов. Часто встречается в соляных место-
рождениях. Ангидрит — кристаллы ромбич. сингонии;
плотн. 2,8—3,0; твердость 3,0—3,5. При гидратации
ангидритовых залежей под действием поверхностных
водных р-ров и при испарении соленых озер и мелко-
водных заливов образуется гипс CaSO4 • 2Н2О; син-
гония моноклинная; плотн. 2,3; твердость 1,5; весьма
хрупок. Флюорит (плавиковый шпат)
CaF2 образуется в основном при гидротермальных
процессах и часто встречается как спутник рудных
металлич. минералов в жилах. Иногда содержит
в виде изоморфной примеси С1, а также примеси би-
туминозных веществ, Fe2O3, редких земель и, из-
редка, урана; сингония кубическая; плотн. 3,0—3,2;
твердость 4; хрупок; часто флюоресцентен. Весьма
распространенным минералом, содержащим К., яв-
ляется апатит Са5(РО4)3 (F, С1).
Кальций входит в состав многих осадочных и ме-
таморфических пород — доломитов, песчаников,
сланцев и др., водных алюмосиликатов (цеолитов)
и рудных минералов — перовскита СаТЮ3, по-
веллита СаМоО4, шеелита CaWO4, гидроборацита
MgCaBeOn • 6Н2О, колеманита Ca2BeOn • 5Н2О, пиро-
хлора (Na, Са, ...)2(Nb, Ti, ...)2Oe[F, ОН] и др. Мор-
ская вода содержит 0,042% К.
Основные месторождения известняка в СССР на-
ходятся иа Украине, в Карелии, Прибалтийских
республиках, Подмосковье, Поволжье, в бассейне
Дона и на западном склоне Урала. Наиболее важные
меловые отложения в СССР имеются в районе Бел-
города, Славянска, Воронежа, Днепровско-Донецкой
впадины, Поволжья и Белоруссии, наиболее важные
месторождения мрамора красивых расцветок — на I
Урале, в Карелии, Грузии и Армении, Узбекистане
и Забайкалье. Крупнейшие месторождения гипса
встречаются в Архангельской, Московской, Горь-
ковской, Пермской, Псковской, Донецкой и мп. др.
областях, наиболее крупное месторождение флюо-
рита — в Забайкалье.
Физические и химические свойства. Свободный К. —
серебристо-белый металл. Известны 2 его аллотропия,
модификации: низкотемпературная a-форма с гра-
нецентрированной кубич. решеткой, а = 5,56 А,
плотн. 1,54 (20°) и высокотемпературная 0-форма
с гексагональной решеткой; темп-ра перехода а —> 0
464° ± 4°. Ат. радиус 1,97 А, ионный радиус Са2т
1,04 А. Т. пл. 851°, т. кип. 1482°. Теплота плавления
2,23 ккал/г-атом', теплота испарения 36,7 ккал/г-атом
(т. кип.); уд. теплоемкость 0,149 кал/г-град (0—100°);
термин, коэфф, линейного расширения 22-10-6 (0—
300°); теплопроводность 0,3 кал/см-град-сек (20°);
уд. сопротивление (в мком см)'. 3,43 (0°); 4, 6 (20°);
термин, коэфф, электросопротивления 4,57 • 10 3
(20°). Модуль упругости 2600 кГ/мм2', предел проч-
ности при растяжении 6 кГ/ммг\ предел упругости
0,4 кГ/мм?', предел текучести 3,8 кГ/мм2\ относитель-
ное удлинение 50%; твердость по Бринеллю 20—
30 кГ/мм2. К. достаточно высокой чистоты пластичен,
хорошо прессуется и может быть прокатай в листы.
Хорошо поддается обработке резанием.
В соединениях К. двухвалентен (известны, одпако,
соединения CaF, CaCl и т. п., формально отвечающие
одновалентному К.). Химически К. очень активен
и принадлежит к наиболее электроотрицательным ме-
таллам; его нормальный электродный потенциал равен
—2,84 в. Будучи энергичным восстановителем, К.
вытесняет почти все металлы из их окислов, сульфидов
и галогенидов. При обычной темп-ре К. легко взаимо-
действует с кислородом и влагой воздуха (поэтому его
хранят в минеральном масле или в герметически за-
крытых сосудах). При нагревании на воздухе или
в кислороде воспламеняется, образуя основной окисел
СаО, дающий с водой сильное основание Са(0Н)2
(см. Кальция окись). Известны также перекиси К.:
бесцветная СаО2 и желтая СаО4 — сильные окисли-
тели. К. разлагает СО2, а при 550—650° — даже СО.
С холодной водой взаимодействует сначала быстро,
затем реакция замедляется из-за образования пленки
Са(ОН)2. Энергично взаимодействует с горячей водой
и кислотами (кроме конц. HNO3) с выделением Н2.
На холоду К. взаимодействует с хлором и бромом
только в присутствии влаги. При темп-pax выше 400°
идет энергичное образование СаС12 и CaBr2. С фтором
К. реагирует на холоду. Расплавленные галогениды
образуют с металлич. К. субсоединения состава
CaCl, CaJ, CaF, устойчивые при темп-pax выше точек
плавления соответствующих галогенидов 2-валент-
ного К. (см. Кальция галогениды). При нагревании К.
с серой образуется кальция сульфид CaS, а при дейст-
вии S на CaS — полисульфиды CaS2, CaS4, CaS5 и др.
При взаимодействии с сухим Н2 при 300—400° К.
образует кальция гидрид СаН2 — ионное соединение,
в к-ром водород является анионом. Реакция обратима,
но уже при 150—170° идет в сторону образования
СаН2 со значительной скоростью. СаН2 — энергичный
восстановитель. Выделяющийся при его диссоциации
водород химически более активен, чем молекуляр-
ный Н2; поэтому СаН2 восстанавливает тугоплавкие
металлы из их окислов. Предполагается существова-
ние гидрида СаН или образование твердых р-ров
между Са и СаН2. Один объем К. способен поглотить
850 объемов водорода. При 500° К. взаимодействует
с N3 с образованием нитрида Ca3N2. Эвтектика Са
и Ca3N2, содержащая 0,57—0,76% N, плавится при
780°. Ca3N2 разлагается водой: Ca3N2 -|- 6Н2О =
= 3 Са(ОН)2-|- 2NHS. При взаимодействии К. с жид-
373
КАЛЬЦИЙ
374
ким или газообразным аммиаком на холоду обра-
зуется комплексный аммиакат Ca(NH3)3,
представляющий собой кристаллы с золотистым блес-
ком, отличающиеся очень высокой электропровод-
ностью. На воздухе Ca(NH3)3 самовоспламеняется.
При хранении в вакууме превращается в амид
Ca(NH2)2. При нагревании в среде аммиака К. обра-
зует Ca3N2 и СаН2. При нагревании Ca(NH2)2 в ва-
кууме образуется пернитрид Ca3N4. Нагреванием К.
с фосфором без доступа воздуха получают фосфид
Са3Р2, плавящийся выше 1600°. Са3Р2 стоек по отно-
шению к крепким HNO3 и H2S04, но разлагается водой.
При нагревании К. с графитом образуется кальция
карбид CaCj. В технике CaCj, используемый для про-
изводства ацетилена и кальция цианамида CaCN2, полу-
чают по реакции: СаО -|- ЗС = СаС2 -|- СО. С крем-
нием К. образует силициды Ca2Si, CaSi, CaSi2 (в тех-
нике CaSi2 получают нагреванием СаО, Si и С). При
взаимодействии CaSi и CaSi2 с N2 образуется CaSiN2.
Известен борид К. состава СаВ3. В сплавах К. дает
интерметаллич. соединения с Al, Ag, Au, Си, Li,
Mg, Ni, Pb, Sn, Sb, TI, Zn. О важнейших солях К. см.
Кальция карбонат, Кальция нитрат, Кальция суль-
фат, Кальция фосфаты.
Аналитическое определение. Не-
растворимые соединения К. обрабатывают НС1 или
сплавляют с Na2CO3 с последующим растворением
плава в кислоте. Все катионы, кроме щелочных ме-
таллов, Mg, Ва и Sr, отделяют от К. осаждением при
помощи H2S, (NH4)2S или NH40H. От магния К.
можно отделить двукратным осаждением СаС2О4 -Н2О
или осаждением СаМоО4 из кипящего слабокислого
или слабощелочного р-ра. Из прокаленной смеси
MgO и СаО последнюю можно количественно отделить
экстракцией 30%-ным р-ром сахара. От Ва и Sr К.
отделяют экстракцией Ca(NO3)2 из смеси сухих
нитратов спирто-эфирной смесью или концентриро-
ванной HNO3. Другой метод состоит в том, что из
смеси хлоридов Са, Ва и Sr осаждают ВаСгО4, затем
осаждают Са и Sr в виде карбонатов, к-рые превра-
щают в нитраты и разделяют спирто-эфирной смесью.
Для определения К. CaC^Oi • Н2О обрабатывают
H2SO4 и титруют освобожденную Н2СгО4 р-ром КМпО4.
При весовых методах СаСгО4 • Н2О превращают
в СаСО3, CaS04 или CaF2. Имеется колориметрия,
способ определения К. Качественно К. обнаруживают
осаждением углекислым газом в виде СаСО3 или по
оранжево-красному окрашиванию пламени горелки.
Получение. Промышленное значение для произ-ва
метаЛлич. К. имеет только чистый известняк. К.
получают электролизом расплавленного СаС)2 или
смесей СаС12 и CaF2 или СаС12 и К.С1, либо металло-
термия. восстановлением СаО в вакууме. Для при-
готовления СаС1.2, предназначенного для электролиза,
известняк обрабатывают р-ром НС1, полученный р-р
выпаривают и подвергают СаС12 • 6Н2О обезвожива-
нию, к-рое заканчивают плавлением при 850—900°
в присутствии NH4C1 (для предотвращения гидро-
лиза). Электролиз ведут при 800° и местном перегреве
до 850° у катода для выделения жидкого К. Раство-
римость К. в электролите достигает 17 вес. %. По-
этому применяют способ осаждения К. на т. наз.
катоде касания, при этом выделившийся жидкий
металл застывает на конце опущенного в электролит
вертикального охлаждаемого водой стального катода;
постепенно поднимая катод, наращивают на его конце
кальциевую штангу длиной до 500 мм. Чистота ка-
тодного К. составляет всего 97—98%. Для получения
чистого металла его необходимо возгонять в вакууме.
Значительно более экономичен способ электролиза
с жидким медно-кальциевым катодом, поскольку
растворение К: в электролите при его сплавлении
с медью резко уменьшается. Сплавы с медью, содер-
жащие до 65% К., имеют плотн. до 2,1, благодаря
чему они удерживаются на дне ванны. Электролитом
служит расплав, содержащий 80—85% СаС12 и 20—
15% КС1. Электролиз ведут при 650—715° в чугунной
ванне, футерованной снаружи огнеупорным кирпи-
чом и заключенной в стальной кожух. Анодом слу-
жит пакет из графитовых блоков. Ванны включаются
в серию последовательно. Сплав, содержащий 62—
65% К., из ванны вычерпывают или удаляют вакуум-
ковшом. К. отгоняют из сплава в стальной реторте,
установленной в электропечи, при 950—1000° и ва-
кууме 0,1—10~3 .«л рт. ст. Пары К. конденсируются
в верхней части реторты на стенках входящей в нее
закрытой трубы, охлаждаемой водой. Отгонку ведут
до содержания в сплаве 30—35% К.; остаток от воз-
гонки возвращают на электролиз для использования
в качестве жидкого катода. Возогнанный К. содержит
менее 0,5% примесей.
Алюминотермия, восстановление К. в вакууме осно-
вано на реакции: бСаО -|- 2А1 — ЗСа -|- ЗСаО • А12О3.
Вакуум необходим для снижения температуры
восстановления и предотвращения взаимодействия
паров К. с азотом и кислородом. Сырьем служит
возможно более чистый известняк, в частности с ми-
нимальными примесями соединений магния и щелоч-
ных металлов. Известняк обжигают «намертво»
при 1200°, известь размалывают, смешивают с алю-
миниевым порошком и брикетируют при давлении
150 кГ/см?. Брикеты загружают в горизонтальные
реторты из хромоникелевой стали, обогреваемые га-
зом или элементами сопротивления. Восстановление
протекает при 1200° и вакууме 0,01—0,02 мм рт. ст.
Пары К. конденсируются на поверхности железной
гильзы, вставленной в Выступающую из печи часть
реторты. Друзы кристаллов К. раздавливают на
гидравлич. прессе или непосредственно прокатывают
на листовой металл. Из хорошего известняка полу-
чают К. чистотой 98—99%. В последние годы делают
попытки получить К. термин, диссоциацией СаС2
при высоких темп-рах в вакууме.
Применение. В виде чистого металла К. используют
для восстановления из соединений металлич. U, Th,
Ст, V, Zr, Cs, Rb и нек-рых редкоземельных металлов,
для очистки РЬ от Bi, для раскисления сталей, ни-
келя, бронз и др. сплавов, десульфуризации нефте-
продуктов, обезвоживания органич. жидкостей, для
очистки аргона от примеси азота и в качестве погло-
тителя газов в высоковакуумных приборах, напр.
в электронных трубках. Большое применение в тех-
нике получили антифрикционные сплавы К. с РЬ
(содержащие Na) и сплавы с РЬ, используемые для
изготовления аккумуляторных пластин и оболочки
электрич. кабелей. Присадка К. к алюминиевым спла-
вам улучшает их обрабатываемость и электропровод-
ность. Связыванием К. в СаН2 с последующим разло-
жением гидрида водой пользуются как экономичным
средством получения и транспортировки Н2 в поле-
вых условиях.
Широкое применение находят и минералы К. Так,
основное количество добываемого известняка исполь-
зуется в произ-ве извести, цемента, силикатного
кирпича или непосредственно как строительный ма-
териал (ракушечник), а также в металлургии как флюс,
в химич. пром-сти — для получения карбида кальция,
соды, едкого натра, хлорной извести, удобрений,
в произ-ве сахара и стекла. Мел применяют гл. обр.
как белый пигмент в малярном деле и лакокрасочной
технике и в качестве наполнителя в произ-ве резины,
писчей бумаги, клеенки и обоев; мрамор — как ар-
хитектурно-строительный и облицовочный материал,
для изготовления электротехнич. распределительных
щитов и панелей и др. Исландскйй шпат благодаря
его высокому двупреломлению применяют для из-
375
КАЛЬЦИЙ ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ - КАЛЬЦИЯ ГАЛОГЕНИДЫ
376
готовления оптич. поляризационных приборов. Гипс
и ангидрит — в произ-ве вяжущих материалов для
строительства, строительных деталей, формовочного
гипса, в качестве добавок к портландцементам, на-
полнителей, в нроиз-ве бумаги, красок и эмалей.
Молотый гипс используют непосредственно в сель-
ском х-ве. Флюорит в основном применяют как ком-
понент флюсов в металлургии, как минерализатор
в произ-ве цементов, для получения плавиковой к-ты
и ее солей, в частности криолита и фтористого алю-
миния, а также в керамич. пром-сти при изготовлении
эмалей и глазурей. Прозрачные кристаллы флюорита
используют в оптике для изготовления линз в объек-
тивах микроскопов. Главная область применения
апатитов и фосфоритов — произ-во минеральных удоб-
рений. О разнообразном применении солей К. см.
соответствующие статьи.
Лит.: Доронин Н. А., Металлургия кальция, М.,
1959; Mantell С. L., Hardy С. Н., Calcium, metal-
lurgy and technology, N. Y., 1946; G m e 1 i n, 8 Aufl., System-
Nummer28, Calcium, B., 1956; Mellor, v. 3, L.—N. Y. —
Toronto, 1946; т о ж e, v. 3, 1952; U 1 1 m a n n, 3 Aufl., Bd 4,
MUnchen—B., 1953; Pascal, t. 4, P., 1958; Rare metals
handbook, ed., C. A. Hampel, 2 ed., L., 1961; Гиллебранд
В. Ф. [и др.], Практическое руководство по неорганическому
анализу, пер. с англ., М., 1957; Беляев А. И., Жем-
чужина Е. А., Фирсанова Л. А., Физическая
химия расплавленных солей, М., 1957; Бетехтин А. Г.,
Курс минералогии, 3 изд., М., 1961;Торопов Н. А. и
Б у л а к Л. Н., Курс минералогии и петрографии, М., 1953.
А. Я. Фишер.
КАЛЬЦИЙОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ —
органические соединения, содержащие связь угле-
род — кальций. За исключением полученного не-
давно диметилкальция, индивидуальные К. с. до
сих пор не получены, хотя образование их в растворе
известно уже давно. Кальций медленно реагирует
с иодистыми алкилами в эфире, однако удовлетвори-
тельные выходы несимметричных соединений типа
RCaJ достигаются только в случае R = С3Н5. Реак-
ция Са RX —► RCaX, где X = Cl, Вг, J, гладко
протекает в эфире, если кальций перед реакцией ак-
тивируют нагреванием со ртутью и реакцию
проводят ниже 0°. Примесь магния катализирует
процесс, по-видимому, вследствие частичного обра-
зования реактива Гриньяра. Йодистый метил в среде
пиридина с металлич. кальцием образует нераство-
римый комплекс с пиридином (СН3)2Са • C5H5N;
комплекс теряет пиридип в вакууме, образуя твердый
диметилкальций. Кальций не вытесняет ртуть из
ртутьдиалкилов, а при нагревании с цинкдиал-
килами дает комплексы типа CaZnR4. По химич.
свойствам К. с. в большей степени напоминают ли-
тийорганич. соединения, чем реактивы Грипьяра;
в частности, К. с. заметно расщепляют простые эфиры.
Алкилкальцийгалогениды с СО2 дают соответствую-
щие кетоны, третичпые спирты и лишь следы кислот,
однако фенилкальцийиодид гладко карбоксилируется
до бензойной к-ты. При взаимодействии несимметрич-
ных К. с. с хлористым бензоилом образуются соот-
ветствующие карбинолы. К. с. бурно реагируют
с водой и легко окисляются на воздухе; в инертной
среде (СН3)2Са устойчив вплоть до 400°.
Лит.: Ro chow Е. G., Hurd D. Т., Lewis
R. N., The chemistry of organometallic compounds, N. Y.—L.,
1957; Payne D. । A., Sanderson R. T., J. Amer.
Chem. Soc., 1958, 80, 5324; В г у c e-S mlth D., Skinner
A. C., Chemistry and Industry, 1960, M 35, 1106.
О. Ю. Охлобыстин.
КАЛЬЦИНИРОВАННАЯ СОДА — техническое на-
звание безводного натрия карбоната.
КАЛЬЦИЯ АРСЕНАТ И АРСЕНИТ — кальциевые
соли соответственно мышьяковой и мышьяковистой
к-т Ca3(AsO4)2 и Ca(AsO2)2, применяемые в качестве
средств борьбы с вредителями с.-х. растений. Технич.
арсенат кальция — белый или светло-серый
исслеживающийся и хорошо распиливающийся тонкий
порошок. Состоит из смеси Ca3(AsO4)2 (с 57% As3O6)
и Ca3(AsO4)2 СаО (с 50,7% As2O5); в воде мало раст-
ворим (растворимость отдельных образцов от 0,25
до 0,75 г в 100 г воды); хорошо растворим в соляной
и азотной к-тах. В пром-сти арсенат кальция полу-
чают: 1) действуя известковым молоком на р-р ар-
сената натрия, полученный окислением р-ра арсенита
натрия кислородом воздуха в присутствии катализа-
тора (обычно CuSO4); 2) действуя известковым мо-
локом на мышьяковую к-ту, полученную по реакции;
As2O3 + 2Н2О + 2HNO3 -► 2H3AsO4 + NO + NO3
Способ 2 дает продукт, обладающий необходимой для
приготовления концентрированных суспензий дис-
персностью, пригодных для опрыскивания с само-
летов. Арсенат кальция гидролизуется до H3AsO4,
на чем и основано его использование как инсекти-
цида. Технич. арсенит кальция — белый
или сероватый не слеживающийся пылевидный по-
рошок; содержит не менее 62% As2O3; в воде трудно
растворим. Арсенит кальция получают взаимодейст-
вием Аз2О3 с известковым молоком. В отличне от
широко используемого арсената кальция арсенит
применяют ограниченно, т. к. он сильно обжигает
растения. Оба препарата ядовиты для человека
и с.-х. животных.
Лит.: П о з и н М. Е., Технология минеральных солей,
2 изд., Л., 1961; Ефимов А. Л., Справочник по приме-
нению ядов для борьбы е вредителями и болезнями сельско-
хозяйственных растений, 4 изд., М., 1951. с. А. Погодин.
КАЛЬЦИЯ ГАЛОГЕНИДЫ — соединения кальция
с галогенами, отвечающие 2-валентному Са (нормаль-
ные) и одновалентному Са (т. наз. субгалогениды).
Свойства CaF2 резко отличны от свойств остальных
галогенидов 2-валентного Са. Так, фторид нераство-
рим не только в воде, но и в разб. к-тах, тогда как
другие К. г. гигроскопичны, хорошо растворяются
в воде и из р-ров выделяются в виде кристаллогидра-
тов. Эти свойства усиливаются в ряду СаС12 —► СаВг2 —►
-* CaJ2. Аналогичен и порядок понижения темп-р
плавления. Расплавленные К. г. образуют с металлич.
Са субгалогениды, устойчивые выше темп-р
плавления соответствующих нормальных галогенидов.
Кальция фторид CaF2 — бесцветные ку-
бич. кристаллы, а = 5,462 А; плотность 3,180; т. пл.
1360°; т. кип. 2500°; теплота образования Д77°08 =
=.—290,3 киал/л«оль.Теплоемкость15,86 ккал!молъ-град
(25°). Показатель преломления 1,43385, твердость
по Моосу 4. Практически нерастворим в воде. При
действии серной к-ты разлагается с выделением HF.
В природе CaF2 встречается в виде минерала флюо-
рита (плавиковый шпат), входит в состав апатита.
Может быть получен действием NaF или NH4F на
р-р СаС12. Применяется как исходное сырье для по-
лучения HF и других фтористых соединений и в ме-
таллургии как флюс.
Кальция хлорид СаС1, — бесцветные ром-
бич. кристаллы, а = 6,24 А, b ='б,43 А, с = 4,20 А;
плотность 2,512; т. пл. 772°; т. кип. 1600°; теплота
образования А77208 = —190,0 ккал/моль. Сильно ги-
гроскопичен и энергично поглощает водяные пары,
сначала образуя твердые гидраты, а затем расплы-
ваясь в жидкость. Давление паров воды над сплав-
ленным СаС12 0,36 мм рт. ст. (при 20°). Раствори-
мость в воде (г на 100 г Н2О): 49,6 (0°); 74(20°); 154 (99°).
Плотность водных р-ров:
%СаС12 ... 10 20 30 40
дао........ 1,0835 1,1775 1,2816 1,3957
Темп-ры кипения р-ров СаС12: 40,8 %-ный при 120°;
50%-ныйпри 130°; 75%-ный при 175°. Темп-ры за-
мерзания водных р-ров: 20%-ный при —18,57°;
30%-ный при —48°. СаС12 образует ряд кристалло-
гидратов. При охлаждении конц. р-ров выпадает
СаС12 • 6Н2О, к-рый при 30,1° плавится в кристалли-
377
КАЛЬЦИЯ ГИДРИД-КАЛЬЦИЯ КАРБИД
378
зационной воде и переходит в СаС12 • 4Н2О, затем
в СаС12 • 2Н2О (при 45,1°) и в СаС12 • Н2О (при 175,5°).
Полностью хлорид кальция обезвоживается выше 250°.
При обезвоживании К. х. частично гидролизуется
с образованием СаО и НС1. При смешении СаС12 • 6Н2О
(58,8%) со снегом или мелко истолченным льдом
(41,2%) темп-ра понижается до —55° (криогидратная
точка). Безводный К. х. растворяется в воде со зна-
чительным выделением тепла; СаС12 • 6Н2О — с по-
глощением тепла. К. х. растворим в низших спиртах
и жидком аммиаке, образуя с ними сольваты, а также
в ацетоне.
Получают К. х. как побочный продукт в произ-ве соды
по аммиачному способу, а также в произ-ве бертолетовой
соли КС1О3. Применяют как сырье для получения
металлич. кальция, как высушивающее средство
для газов и жидкостей. Растворы К. х. используют как
жидкости с низкой темп-рой замерзания в холодиль-
ных системах, а также для поливки дорог — с целью
предотвращения их пыления, как добавку к бетону
и т. д. В водяных банях р-ры К. х. служат для под-
держания определенной темп-ры, от 105° (20% без-
водного К. х.) до 178° (75%). К. х. применяется также
в медицине.
Кальция бромид СаВг2 — бесцветные
ромбич. кристаллы, а = 6,55 А, Ъ = 6,88 А, с =
= 4,34 А, плотность 3,353; т. пл. 760°, т. кип. 810°.
Теплота образования Д77°298 = —161,3 ккал/молъ.
Легко растворим в воде (594 г на 100 г Н2О при 0°)
и спирте. Из воды кристаллизуется в виде гидрата
СаВг2 • 6Н2О, плавящегося при 38,2°. Известны также
гидраты с 5, 4, 3, 2,Р/2 и 1 молекулами ПЛ). Давление
паров воды пад безводным СаВг2 0,14 мм рт. ст. (25°).
Растворим в жидком аммиаке и этиловом спирте
и образует с ними ряд сольватов. Получают СаВг2
взаимодействием брома с известковым молоком в при-
сутствии аммиака или же растворением гидроокиси
или карбоната кальция в бромистоводородной к-те.
Применяют в медицине и фотографии.
Кальция иодид CaJ2 — бесцветные гекса-
гональные кристаллы, а = 4,48 А, с = 6,96 А; плот-
ность 3,956; т. пл. 575°, т. кип. 718°; теплота обра-
зования ДЯ°298 = —127,8 ккал/молъ, легко растворим
в воде (757 г на 100 г Н2О при 0°). Очень гигроско-
пичен. Из воды кристаллизуется в виде CaJ2 • 6Н2О.
Известны также кристаллогидраты с 7, 4 и 3 моле-
кулами Н2О. Получается аналогично СаВг2.
Лит.: Позин М. Е., Технология минеральных солей,
2 изд., Л., 1961; Р ы с с И. Г., Химия фтора и его неоргани-
ческих соединений, М., 1956 (по СаЕ2). Д. С. Стаеиневич.
КАЛЬЦИЯ ГИДРИД СаН2 — бесцветные крис-
таллы, ромбич. решетка, а = 5,936 А, Ь = 6,838 А,
с — 3,600 А; плотн. 1,902; т. пл.>1000°. Теплота
образования Д7/°29й = —45,1 ккал/молъ. Давление
диссоциации (в мм рт. ст.): 0,1 (600°); 760 (990°).
К. г. разлагается водой и спиртами с выделением
водорода, однако реакция протекает спокойно и вы-
деляющегося тепла недостаточно для воспламенения
образующегося водорода. В обычных неорганич.
и органич. растворителях К. г. нерастворим. Раство-
ряется в расплавленном КОН и эвтектич. смеси КС! -|-
LiCl. С сухим кислородом К. г. не реагирует до
400°, но при поджигании спокойно сгорает. С азотом
К. г. взаимодействует при 500°, образуя нитрид каль-
ция. К. г. — сильный восстановитель; он восстанавли-
вает окислы до металлов, сульфаты до сульфидов
и т. д. Получают К. г. взаимодействием металлич.
кальция (в виде стружек) с водородом при 500—700°
и давлении, близком к атмосферному. Применяют при
получении чистых металлов (титан, цирконий, нио-
бий, тантал) из их окислов, для удаления следов
влаги из органич. жидкостей (трансформаторное
масло, эфиры), для высушивания газов, а также
в аналитич. химии для определения воды в органич.
жидкостях и в кристаллогидратах.
Лит..- X В р д Д., Введение в химию гидридов, пер.
с англ., М., 1955. Д. С. Стаеиневич.
КАЛЬЦИЯ КАРБИД СаС., — соединение кальция
с углеродом; один из наиболее важных карбидов,
применяемых в технике. Впервые получен в 1862
Ф. Вёлером нагреванием сплава цинка и кальция с
углем. Промышленный электротермия, способ произ-ва
К. к. за рубежом был осуществлен в 1895—96,
в России во время первой мировой войны. Химически
чистый К. к. бесцветен. Цвет технич. К. к. изменяется
в зависимости от состава — от светло-бурых до чер-
ных тонов. К. к. имеет несколько кристаллич. моди-
фикаций. В интервале 25—447° К. к. образует тетра-
гональную решетку, а выше 447° — кубическую.
Плотность твердого чистого К. к. 2,2. Плотность рас-
плавленного технич. К. к. (при 2000°) с содержанием
80% СаС2 1,84. Средняя уд. теплоемкость чистого
К. к. в кал/г град: 0,22 (20—325°); 0,24 (20—500°);
0,275 (20—725°); 0,28 (0—2000°). Уд. электропровод-
ность расплавленного 70%-ного К. к. при 1850°
составляет ок. 0,4—4,0 о.и1- см 1 и является электро-
литической. Т. пл. чистого К? к. ок. 2300°; теплота
плавления 120 кал/г.
Диаграмма плавкости системы СаС2 — СаО (рис. 1)
показывает существование двух эвтектик: первой —
при содержании 68%
СаС2 и темп-ре 1750°,
и второй — при 35,6%
СаС2 и 1800°. 'Между
этими двумя эвтекти-
ками имеется макси-
мум при 52,4% СаС2
и 1980°, почти в точ-
ности отвечающий со-
ставу СаС2 - СаО. На-
личие в К. к. при-
месей (SiO2, А12О3,
Содержание CaCg,|>
Рис. 1. Диаграмма плавкости
системы СаС2 — СаО.
MgO) существенно понижает его темп-ру плавления.
Теплота образования К. к. Д77°298 = —15,0 ккал/молъ.
К. к. взаимодействует с водой с выделением аце-
тилена: СаС2 4- 2Н2О = С2Н2 % Са(ОН)2. Для от-
вода большого количества выделяющейся теплоты
(30,4 ккал/молъ) процесс проводят в избытке воды.
Азот при нагревании взаимодействует с К. к., обра-
зуя цианамид кальция: СаС2 Na — Ca(CN)2.
К. к. получают по реакции: СаО -|- ЗС = СаС2 -f-
-|- СО. Эта реакция протекает с поглощением большого
количества тепла (108 ккал/молъ) и практически на-
чинается при 1700—1800°. При повышении темп-ры
до 2200° начинается испарение К. к. и разложение
его на металлич. кальций и углерод. Темп-ру процесса
поддерживают в пределах 1900—1950°. Процесс ведут
в электрич. печах. Сырье перед загрузкой в печь
дробят, отбирая после отсева куски извести размером
60—80 лш и углеродистого материала размером 25—
30 мм. Количественное соотношение реагентов опре-
деляется заданным «литражом» (литражом иаз. коли-
чество ацетилена в л, приведенных к 20° и 760 мм
рт. ст., получаемое при полном разложении 1 кг
продукта водой). С увеличением количества углерода
в шихте литраж карбида повышается, хотя выход
самого К. к. при этом уменьшается. При высоком
содержании в шихте извести выход К. к. увеличивается,
а его литраж снижается. Обычно применяют шихту,
содержащую 40—50% углерода.
Для снижения содержания вредных примесей
и балласта в К. к. используют более чистые исходные
материалы. Известняк, из к-рого получают известь,
должен содержать не менее 97% СаСО3. Суммарное
содержание в известняке MgO -|- А12О3 -|- Fe2O3 не
должно превышать 2%, а содержание Р и S — соот-
379
КАЛЬЦИЯ КАРБОНАТ —КАЛЬЦИЯ ОКИСЬ
380
ветственно 0,008 и 0,1%. В качестве восстановителя
применяют антрацит и кокс, выделяющие при нагре-
вании минимальное количество газообразных и ле-
тучих веществ. Кокс должен содержать не более
0,04% Р; 1,5% S; 5—10% золы; 6—8% летучих
веществ; 3—5% влаги. К. к. обычно получают в трех-
фазных электропечах.
Большинство действующих карбидных печей от-
крыто сверху, выделяющаяся СО по выходе из печи
сгорает до СО2. Использование образующейся СО
как горючего газа или сырья для органического
синтеза снижает стоимость К. к. и улучшает усло-
вия труда. В связи с этим разработаны конструк-
ции закрытых печей с отбором окиси углерода.
Наиболее эффективными являются закрытые элек-
тродуговые печи с вращающейся ванной; такое
устройство способствует рыхлению шихты и уско-
ряет ее расплавление, защищает подину печи от про-
жигания, обеспечивает равномерность износа футе-
ровки стен и пода печи, противодействует образованию
пустот и подвисаиий в шихте.
На рис. 2 показана схема закрытой вращающейся печи.
Цилиндрич. ванна печи покоится на опорных колоннах, уста-
новленных на железобетонном поворотном столе. Стол вращает-
ся вокруг центральной оси на катках, движущихся по круго-
вому рельсу. Ванна, вра-
щаясь, совершает либо
полный оборот вокруг
своей центральной оси,
либр осциллирует (ка-
чается) на 120°. Загру-
вочное устройство вра-
щающейся закрытой печи
полностью механизиро-
вано. После взвешивания
на автоматич. весах и пе-
ремешивания шихта по-
ступает в симметрично
расположенные над печью
бункеры, а оттуда по за-
грузочным желобам и ру-
кавам, окружающим каж-
дый электрод, загружает-
ся в ванну печи. В печи
поддерживается повы-
шенное давление; . уро-
вень шихты в ней изме-
няют в соответствии с
мощностью питающей се-
ти, что расширяет диапа-
зон рабочих мощностей.
На крупных заводах расплавленный К. к. выпускают из
печи непосредственно во вращающийся грануляционный ба-
рабан с наружным водяным охлаждением. Из барабанов,
заполненных азотом, через 18—22 мин выгружают куски
охлажденного К. к. Такая система снижает потери и исключает
необходимость дальнейшего дробления.
Разработан способ получения К. к. в доменных печах с кис-
лородным дутьем без применения электроэнергии. Кокс и обож-
женная известь подаются в шахту, нижняя часть к-рой футеро-
вана блоками из графитированного углерода. Дутье кислородом
производится через водоохлаждаемые фурмы, расположенные
на небольшой высоте над подом. Загружаемая шихта прогре-
вается реакционными газами, к-рые на выходе из печи имеют
темп-ру всего ок. 200°.
Литраж химически чистого К. к. составляет
377,73 л/кг (20°С, 760 мм рт. ст.). Литраж технич.
К. к. (65—80% СаС2 и 10—25% СаО, остальное фер-
росилиций и др. примеси), в зависимости от размера
кусков, колеблется в пределах 245—280 л/кг.
К. к. широко применяют в технике, гл. обр. для
произ-ва ацетилена, кальция цианамида и восста-
новления щелочных металлов.
Лит.: Вольфкович С. И., Роговиц 3. А.,
Руденко Ю. П. иШманенков И. В., Общая
химическая технология, т. 2, М., 1950; Кузнецов Л. А.,
Проивводство карбида кальция, М.—Л., 1950; Мороз
Л. И., Новиков Б. М., Вести, технической и экономи-
ческой информации, 1959, К» 3, 80;Стрижевский
И. И., Гузов С. Г. и Ковальский В. А., Ацети-
леновые станции, 2 изд., М., 1959; Туров Ю. Я., Ю Д о-
в и ч Е. Е., Ж. Всес. хим. о-ва им. Д. И. Менделеева,
1962, 7, № 1, 81. И. И. Стрижевский.
КАЛЬЦИЯ КАРБОНАТ (кальций углекислый)
СаСО3 — встречается в двух кристаллич. формах:
кальцит и арагонит. Кальцит образует бесцветные
Рис. 2. Схема закрытой вращающейся
печи для производства карбида каль-
ция.
гексагональные кристаллы, а = 4,983 А, с = 17,02 А,
плотность 2,711; арагонит — бесцветные кристаллы
ромбич. системы, а = 5,72 А, Ъ = 7,94 А, с = 4,94 А;
плотность 2,93. Кальцит широко распространен в при-
роде (главный породообразующий минерал карбо-
натных пород — мела, известняка, мрамора), ара-
гонит встречается реже. Теплоты образования каль-
цита и арагонита (—А77°298! в ккал/молъ) соответствен-
но равны; 288,45 и 288,49.
При натщевании СаСО3 разлагается без плавления
на СаО и СО2, Давление диссоциации (в мм рт. ст.):
6 (650°); 25 (700°); 201 (800°); 760 (885°). Может быть
расплавлен только при очень высоком давлении СО2
при темп-ре выше 1300°. Растворимость К. к. в воде
незначительна (при 18°); для кальцита 14 мг/л, для
арагонита 15 мг/л', в присутствии СО2 растворимость
резко повышается вследствие образования растворимого
в воде бикарбоната кальция Са(НСО3)2. При пар-
циальном давлении над раствором СО2, равном 1 атм,
при 25° в 1 л р-ра содержится 1,08 г CaCOs. Легко
растворяется в кислотах с выделением СО2. В органич.
растворителях нерастворим. Природный К. к. при-
меняется как строительный материал (известняк,
мрамор) и как исходный продукт для получения
извести-, молотый природный К. к. (мел) — в строи-
тельном деле, а также как наполнитель для резиновых
смесей, бумаги, линолеума. В ряде отраслей техники
применяется осажденный К. к., полученный при
взаимодействии известкового молока с СО2 или при
взаимодействии СаС1., и Na2CO3 в р-ре. Он мягче
и тоньше, чем молотый К. к. Применяется как на-
полнитель для бумаги, резины, в производстве ги-
гиенич. и косметич. средств и т. п. Кальция
бикарбонат (кальций двууглекислый) Са(НСО3)2
известен только в водных р-рах. При кипячении рас-
творов разлагается на СаСО3 и СО2.
Лит. см. при ст. Кальций. Д. С. Стасиневич.
КАЛЬЦИЯ НИТРАТ (кальций азотнокислый, каль-
циевая селитра) — при обычных темп-рах выделяется
в виде Ca(NO3)2 • 4Н2О — бесцветных кристаллов,
т. пл. 42,7°. Известны и другие кристаллогидраты
К. н. Выше 51,6° кристаллизуется безводная соль —
кристаллы кубич. системы, а = 7,62 А, плотность2,36.
Безводная соль плавится при 561°, однако уже пря
500° начинается ее разложение с потерей кислорода
и образованием Ca(NO2)2, к-рый далее распадается
на СаО и N02. Теплота образования безводной соли
Д^°298 = —224,0 ккал/моль. Растворимость в воде
(е на 100 г Н2О); 127 (20°), 355 (51,6°). Насыщенный
р-р кипит при 151° и содержит 78,4% Ca(NOs)2.
Криогидратная точка —28° [78,6 г Ca(NOs)2 на
100 е Н20]. Как безводная соль, так и кристалло-
гидраты К. н. гигроскопичны, поэтому К. н. хранят
без доступа влаги. Получают К. н. растворением
известняка в азотной к-те или поглощением нитроз»
пых газов известковым молоком. Полученный р-р
упаривают до концентрации Ca(NOs)2 73—82% и ох-
лаждают, применяя в качестве затравки для кристал-
лизации немного азотнокислого аммония. Приме-
няют К. н. как азотное удобрение.
Лит.: П о з и н М. Е., Технология минеральных солей,
2 ивд., Л., 1961. Д. С. Стасиневич.
КАЛЬЦИЯ ОКИСЬ СаО — бесцветные кристаллы,
кубич. решетка, а = 4,812 А; плотн. 3,4; т. пл.
2585°; начинает размягчаться при 1800°. Теплота
образования ДЯа293 = —151,9 ккал/молъ. Уд. теп-
лоемкость 0,188 кал/г • град (20°). Технич. продукт
(негашеная известь) представляет собой белое по-
ристое вещество. К. о. жадно реагирует с водой с вы-
делением большого количества тепла (15,6 ккал/молъ)
и образованием Са (ОН)2. Кристаллич. К. о., полу-
ченная прокаливанием кальция карбоната при очень
высоких темп-рах, реагирует с водой очень медленно.
381
КАЛЬЦИЯ СУЛЬФАТ— КАЛЬЦИЯ ФОСФАТЫ
382
Кальция гидроокись Са(О Н)2 — бес-
цветные кристаллы, гексагональная решетка, а =»
= 3,64 А, с = 4,85 А; плотн. 2,24. При нагревании
отщепляет воду, превращаясь в СаО. Давление дис-
социации (мм рт.ст.): 100 (350°), 760 (547°). Теплота
образования Д77°298 = —235,80 ккал/моль. Уд. теп-
лоемкость 0,261 кал/г град. Растворимость в воде
(г на 100 г Н2О): 0,165 (20°); 0,077 (100°); в присутствии
солей растворимость сильно повышается. Технич.
продукт (гашеная известь, или пушон-
ка) — белый пушистый порошок. Суспензия К. г.
в воде наз. известковым молоком, а вод-
ный р-р — известковой водой. Гидроокись
кальция — сильное основание, поглощает СО2 из
воздуха, причем выпадает СаСО3. О получении и при-
менении СаО и Са(ОН)2 см. Известь.
Д. С. Стасиневич.
КАЛЬЦИЯ СУЛЬФАТ (кальций сернокислый)
CaSO4 — бесцветные кристаллы, существующие в виде
нескольких модификаций. Природный минерал а н-
г и д р и т (т. наз. нерастворимый ангидрит) кристал-
лизуется в ромбич. системе с периодами решетки:
а = 6,95 А, Ъ = 6,96 А, с = 6,21 А; плотность 2,899—
2,985; твердость по Моосу 3—4. Т. наз. растворимый
ангидрит, получаемый при обезвоживании гипса,
известен в двух формах, а и р, кристаллизующихся
в гексагональной системе и отличающихся величиной
кристалликов; a-форма, получаемая при медленном
обезвоживании, — крупнокристаллическая, плотн.
2,587; p-форма, получаемая при быстром обезвожи-
вании, — мелкокристаллическая (размеры кристал-
ликов менее К)'4 мм), плотность 2,484. Теплоты
образования (—Д77°298, в ккал/моль): нерастворимый
ангидрит 342,42; растворимый ангидрит 340,27 (а)
и 339,21 (Р). Превращение растворимого в нераство-
римый ангидрит происходит выше 400°. При 1180°
устойчива моноклинная модификация К. а., плавящая-
ся при 1420°, однако уже при более низких темп-рах
(ок. 960°) начинается разложение CaSO4 на СаО,
SO2 и О2. В присутствии SiO2 и, в меньшей степени,
Fe2O3 и А12О3 темп-ра начала разложения еще ниже.
При прокаливании с углем К. с. восстанавливается
до CaS.
Нерастворимый ангидрит негигроскопичен и не-
растворим в воде. Растворимый ангидрит жадно при-
тягивает влагу (вследствие чего применяется для
высушивания) и растворяется в воде. Растворимость
в воде (г на 100 г Н2О): 0,176 (0°); 0,2036 (20°); 0,200
(60°); 0,067 (100°). Присутствие К. с. обусловливает
постоянную жесткость воды. Растворимость К. с.
в воде повышается в присутствии солей (NaCl, MgCl2),
а еще сильнее — в присутствии соляной и азотной
кислот, К. с. образует кристаллогидраты с !/2 и 2
молекулами Н2О. Ниже 60° стабильной фазой в со-
прикосновении с водными р-рами является CaSO4 •
•2Н2О, выше 60° — безводный CaSO4. Р-р, насыщен-
ный по отношению к CaSO4 . Х/2Н2О и содержащий
0,885 г CaSO4 на 100 г Н2О, метастабилен.
Дигидрат CaSO4 • 2Н2О (широко распространенный
минерал гипс) рристаллизуется в моноклинной
системе, а — 6,28 А, Ъ = 15,15 А, с = 5,67 А, 0 =«
<= 114°12'; плотность 2,314—2,328; твердость по
Моосу 1,5. Теплота образования ДЯ°298 = —483,06
ккал/моль. Полугидрат CaSO4-1/2H2O (полуобожжён-
ный гипс) кристаллизуется в гексагональной систе-
ме, а = 6,28 А, с = 6,24 А. Известен в двух модифи-
кациях, различающихся по величине Яристалли-
ков и плотности. Крупнокристаллическая а-форма,
получаемая при медленном обезвоживании гипса,
имеет плотность 2,72—2,73; мелкокристаллич. fj-фор-
ма, получаемая при быстром обезвоживании, имеет
плотность 2,67—2,68. Теплота образования —Д77°298
(в ккал/моль): 376,47 (а) и 375,97 (Р). Будучи замешан
в кашицу с водой, полуобожженный гипс быстро
твердеет, превращаясь в CaSO4. О применении К. с.
см. Вяжущие материалы гипсовые.
Лит.: Термодинамические свойства гипса и продуктов его
дегидратации, перевод, М., 1949; Ullmann, 3 Aufl., Bd 8,
В., 1957, S. 97—132. См. также лит. при ст. Вяжущие материалы
гипсовые. Д. С. Стасиневич.
КАЛЬЦИЯ СУЛЬФИД (кальций сернистый) CaS —
бесцветные кристаллы, кубич. решетка, а = 5,697 А,
плотность 2,58, т. пл. > 2000°, теплота образования
ДЯ°293 = —115,3 ккал/моль. Почти нерастворим в
воде, но медленно разлагается ею с образованием
Са(ОН)2 и Ca(SH)2. При кипячении водной суспен-
зии CaS с серой образуются оранжевые полисульфиды
CaS4 и CaS6, растворимые в воде. Влажный CaS
постепенно окисляется на воздухе, превращаясь
в CaS2O3. В присутствии нек-рых микропримесей
(V, Bi, Мп) прокаленный К. с. способен фосфоресци-
ровать. К. с. получают при прокаливании сульфата
кальция с углем или сульфида натрия с известняком.
Он образуется также при очистке газов от сероводо-
рода при помощи извести. Применяют для приго-
товления светящихся препаратов, а также в кожевен-
ной пром-сти для удаления волос со шкур. Каль-
ция гидросульфид Ca(SH)2 при обычных
условиях образует гидрат Ca(SH)2 - 6Н2О — бесцвет-
ные или желтоватые кристаллы, легко растворимые
в воде и спирте. Получают при пропускании серово-
дорода в известковое молоко. При нагревании р-ра
постепенно разлагается с отщеплением H2S. При
упаривании р-ра досуха превращается в CaS. При-
меняют в производстве искусственного волокна,
Д. С. Стасиневич.
КАЛЬЦИЯ СУЛЬФИТ CaSO3 — из водных р-ров
кристаллизуется в виде CaSO3 • 1/2Н2О — бесцвет-
ных кристаллов. В воде очень мало растворим
(0,043 г/л при 13—100°). В присутствии SO2 раствори-
мость повышается из-за образования кислых солей.
При нагревании до 150° теряет кристаллизационную
воду. При более высокой темп-ре разлагается на
CaSO4 и CaS. На воздухе легко окисляется, превра-
щаясь в CaSO4. Образуется при очистке газов от
двуокиси серы при помощи извести.
Кальция бисульфит Ca(HSO8)2 известен
только в р-рах. Образуется при пропускании SO2
в известковое молоко или суспензию СаСО3; р-ры
содержат 2,7—3,5% SO2, в т. ч. 52—65% в свободном
состоянии. Применяют в бумажной пром-сти при
получении целлюлозы из древесины.
Лит.: П о з ин М. Е., Технология минеральных солей,
2 изд., Л., 1961.
КАЛЬЦИЯ ФОСФАТЫ — кальциевые соли фос-
форных кислот. Наибольшее практич. значение имеют
соли ортофосфорной к-ты — ортофосфаты кальция —
трикальцийфосфат, дикальцийфосфат и монокальций-
фосфат. Трикальцийфосфат Са3(РО4)2 —
бесцветные гексагональные кристаллы, плотность
3,14, т. пл. 1670°. В воде при 20° почти нерастворим
(0,0025%). Легко взаимодействует с кислотами, даже
слабыми, с образованием кислых солей, значительно
лучше растворимых. Широко распространенные фос-
фатные минералы представляют собой двойные соли
Са3(РО4)2 с CaF2 (апатит) или Са(ОН)2 (гидроксил-
апатит). Трикальцийфосфат входит также в состав
костей. Трикальцийфосфат (как природный — в виде
костяного угля и костяной золы, так и полученный
из суперфосфата) применяют для подкормки скота
и птиц. Используется также для очистки сахарного
сиропа, в произ-ве керамики и стекла, для приготовле-
ния зубных паст и порошков, абразивов и др.
Дикальцийфосфат СаНРО4 — бесцветные
триклинные кристаллы, плотность 2,89, кристалли-
зуется из водных р-ров выше 36°. Ниже 36° кристал-
лизуется дигидрат СаНРО4 • 2Н2О — моноклинные
S83
КАЛЬЦИЯ ЦИАНАМИД— КАМЕДИ
384
кристаллы с плотностью 2,31; растворимость в воде
(е дигидрата на 100 г Н2О): 0,025 (0°), 0,133 (60°).
Хорошо растворим в водных р-рах лимоннокислого
аммония. Дикальцийфосфат получают взаимодейст-
вием фосфорной к-ты с известью. Применяется под
названием преципитата как фосфорное удобрение,
а также для подкормки скота.
Монокальцийфосфат Са(Н2РО4)2 — бес-
цветные гигроскопичные кристаллы. Йолучают взаи-
модействием фосфорной к-ты с известью или действием
фосфорной к-ты на апатит или фосфориты (т. н. двой-
ной суперфосфат). При действии серной к-ты на апатит
или фосфориты получается смесь монокальцийфосфата
и гипса (суперфосфат). В зависимости от условий осаж-
дения может быть получена как безводная соль, так
и (обычно) моногидрат Са(Н2РО4)2 • Н2О — кристаллы
с триклинной решеткой, плотн. 2,22. Применяют
как удобрение, а также в хлебопечении, благодаря
начинающейся в холодном тесте и завершающейся
во время выпечки реакции:
ЗСа(Н2РО4)2 • Н2О + 8NaHCO3=Ca3(PO4)2 +
+ 4Na2HPO4 + 8СО2 + 11Н2О
выделение СО» вызывает подъем теста.
Прокаливанием СаНРО4 2Н2О при 900° получают
пирофосфат кальция Са2Р2О7, применяе-
мый в качестве мягкого абразива и в составе зубных
порошков.
Лит.: П о а и н М. Е., Технология минеральных солей,
2 изд., Л., 1961; Ваггаман В., Фосфорная кислота, фос-
фаты и фосфорные удобрения, пер. с англ., М., 1957. См. также
лит. при Ст. Фосфор, Фосфорные удобрения.
Д. С. Стасиневич.
КАЛЬЦИЯ ЦИАНАМИД CaCN2 — бесцветные кри-
сталлы, .гексагональная решетка, а = 3,67 А, с =
= 14,85 А, а = 39°55'; плотн. 2,29; т. пл. 1340°,
однако при 1150° начинается возгонка. Стандартная'
теплота образования ДН°298 = —84 ккал/молъ. Раст-
воримость К. ц. в воде 2,5 г/100 мл Н2О (25°). В спирте
нерастворим Йри действии минеральных к-т на вод-
ный р-р К. ц. образуется свободный цианамид H2CN2,
превращающийся в дальнейшем в мочевину: H2CN2
-|- Н„О = CO(NII2)2. Технич. продукт вследствие на-
личия примесей (9—13% углерода) имеет серовато-
черный пвет и содержит 55—65% CaCN2, что соот-
ветствует примерно 19—23% N. Плотность К. ц.,
содержащего 62,1% CaCN2, равна 2,3.
К. ц. получают азотированием кальция карбида:
СаС., 4- N2 = CaCN2 С. В этом процессе в качестве
промежуточного продукта образуется цианистый каль-
ций: CaC2 N2 — Ca(CN)2, к-рый затем разлагается:
Ca(CN)2 = CaCN2 -ф- С. Реакция азотирования кар-
бида кальция обратима и экзотермична (выделяется
72 ккал/моль). Она протекает с оптимальной ско-
ростью и полнотой при 1000—1150°. Реакционную
массу нагревают до этой темп-ры, к-рая поддержи-
вается за счет выделяющегося тепла. Добавки к шихте
катализаторов: CaCN2 (до 20% от веса карбида),
CaF2 (до 2%) и СаС12 (до 5%) позволяют снизить
темп-ру азотирования. Используемый в процессе
технич. карбид кальция должен содержать 80—85%
СаСг, азот — не менее 99,8% N2. Для достижения
хорошего контакта между твердой фазой и азотом
дробленый карбид кальция подвергают размолу
в трубчатой карбидной мельнице в атмосфере азота.
Для азотирования карбида применяют преим. реторт-
ные печи периодич. действия с электрообогревом,
реже — печи непрерывного действия.
Ретортная печь (см. рис.) состоит из цилиндрич. стального
кожуха 1, футерованного шамотным кирпичом 2. Пространство
з между кожухом и футеровкой заполнено инфузорной землей.
В верхней части печь имеет чугунное кольцо 4 с песочным за-
твором, к-рое укреплено в футеровке печи. В песочном затворе
устанавливается крышка 5, закрывающая внутреннюю часть
печи. На нек-ром расстоянии от дна печи расположены штуцеры
для присоединения труб в, по к-рым поступает азот. В крышке
печи находится труба для выпуска газов и масляный манометр.
В центре крышки и днища имеются круглые отверстия 7 для
установки нагревательного угольного электрода. Загрузка
шихты производится в «ретортах» из плотной бумаги или .кар-
тона; в более крупных печах шихту засыпают непосредственно
в печь, выложенную изнутри
слоем бумаги. После установки
в печи электрода в нее подают
азот под избыточным давлением
в 60—70 лии масляного столба.
В процессе азотирования шихта
спекается и образуется плот-
ный блок, к-рый вынимают из
печи при помощи крана и пере-
носят в отделение охлаждения.
После охлаждения производят
дробление и размол. В цехе
азотирования устанавливается
неск. десятков, а иногда и сотен
таких печей. Менее распростра-
нены канальные и трубчатые
вращающиеся печи непрерывно-
го действия. Размолотый циан-
амид, идущий на удобрение, под-
вергают далее гашению водой и
умасливанию минеральным мае-
Схема ретортной цианамид-
ной печи.
лом для разложения не вступив-
шего в реакцию карбида и уменьшения пылеобразования. На
нек-рых заводах Й. ц. обрабатывают р-ром азотнокислого
кальция, после чего гранулируют во вращающемся барабане.
Содержание азота в К. ц. должно быть не менее
18,5—20%; содержание карбида не более 0,2—2%.
Освоено промышленное произ-во К. ц. в псевдо-
ожиженном слое. В установку вводится пылевидная
смесь СаСг, CaCN2 и CaF2, приведенная в псевдоожи-
женное состояние с помощью азота. Процесс проводят
в горизонтальной вращающейся печи при 1150°.
По сравнению со способом азотирования в обычной
вращающейся печи этот способ более экономичен.
Кроме электротермия, способа получения К. ц. из
карбида кальция, нек-рое применение имеет способ
получения «белого пианамида» по реакции: СаО -4-
+ 2HCN = CaCN2 -|- СО -ф- П2, при 700—900°. В
СССР был впервые разработан способ получения бе-
лого цианамида по реакции: СаСО3 -|- 2NH3 =
= CaCN2 -|- ЗН2О. Получаемый таким способом К. ц.
содержит до 28% N.
Произ-во К. ц. из карбида было начато в 1905
и резко возросло в годы первой мировой войны, так
как К. ц. использовался для получения аммиака:
CaCN2 -(- ЗН2О = СаСО3 Ц- 2NH3, и азотной к-ты.
В связи с развитием производства синтетического
аммиака К. ц. потерял свое значение как источник
связанного азота. К. ц. применяют гл. обр. в качестве
азотного удобрения, для предуборочного удаления
листьев хлопчатника (дефолиация), для получения
цианистых соединений (цианистого плава), дициан-
диамида, гуанидина, нитрогуанидина, меламина и др.
Лит.: Общая химическая технология, т. 2,М., 1959; Куз-
нецов Л. А., Производство карбида кальция, М.—Л.,
1950; Марковский Л. Я., Оршанский Д. Л. и
Прянишников В. П., Химическая электротермия,
Л.—М., 1952; КазарянП. Е., Ж. Всес. хим. о-ва им. Д. И.
.Менделеева, 1962, 7, М 1, 87. И. И. Стрижевский.
КАМЕДИ (гумми) — вещества или смеси веществ
углеводного характера, способные в воде набухать и
образовывать вязкие р-ры или дисперсии. К. содержатся
в эксудатах растений, к-рые образуются при механи-
ческих повреждениях или заболеваниях растений;
в эндосперме семян нек-рых растений и в водорос-
лях. К. эндосперма семян и водорослей иногда наз.
слизями. К К. относят также полисахариды, полу-
чаемые модифицированием природных полисахаридов,
напр. клетчатки, крахмала и др.
К. делят на 3 группы: 1) нейтральные; 2) кислые К.,
кислотность к-рых обусловлена наличием в них
остатков уроновых к-т; 3) кислые К., кислотность
к-рых обусловлена наличием эфирносвязанных ос-
татков серной к-ты.
Нейтральные К. представлены в основном галак-
томаннанами — полисахаридами, построенны-
ми из остатков D-маннозы и D-галактозы, и г л ю-
385
КАМЕННОУГОЛЬНАЯ СМОЛА
386
ко маннанами — полисахаридами, построенны-
ми из остатков D-маннозы и D-глюкозы. Предста-
вителями К. этого типа являются к а р о б а и,
г у а р а н, галактоманнаны верблюжьей травы, лю-
церны, клевера, кофейных бобов, сои. Галактоманнаны
различных растений отличаются соотношением га-
лактозных и маннозных остатков, строением и мол.
весом, однако макромолекулы большинства галакто-
маннанов представляют собой длинные цепи, пост-
роенные из маннопиранозных остатков, при этом
галактопиранозные остатки образуют короткие бо-
ковые ветви. Напр., гуаран содержит маннозу и галак-
тозу в соотношениях 2:1, макромолекула его пред-
ставлена линейной цепью, построенной из остатков
маннозы, к каждому второму остатку к-рой присо-
единено боковое звено остатка галактозы:
. н н сн2
н но I
Повторяющийся участок макромолекулы гуарама
В каробане соотношение галактозы и маннозы 1:3—
1:4, в его макромолекуле полиманнозидная цепь со-
держит меньше боковых ответвлений в виде остатков
галактозы. Глюкоманнаны встречаются в растениях
семейства лилейных, орхидных и др. Строение их
изучено мало.
Кислые К., содержащие уроновую к-ту, представ-
лены полисахаридами эксудатов растений (аравий-
ская К., или гуммиарабик) и водорослей. (См., напр.,
Альгиновые кислоты).
Кислые К., содержащие остатки серной к-ты, пред-
ставлены полисахаридами водорослей: агаром (см.
Агар), карагеенаном, фукоидином и др.
Карагеенан содержит остатки D-галактозы и L-ra-
лактозы, в состав фукоидина входят L-фукоза, D-га-
лактоза, D-ксилоза. Во всех этих К. серная к-та эте-
рифицирует спиртовые гидроксильные группы сахаров.
Синтетич. К. получают введением остатков серной
к-ты и др. групп в амилозу и др. полисахариды.
К. — бесцветные в-ва. В воде нек-рые К. сильно
набухают, другие образуют вязкие растворы, легко
превращающиеся в студни. Линейные К. легко образуют
пленки, но не обладают клеящими свойствами. Раз-
ветвленные К. не образуют пленок^но обладают высо-
кими клеящими свойствами. Вязкость р-ров нейтраль-
ных К. изменяется незначительно в широких пределах
pH, вязкость кислых К. зависит от pH. О химич.
свойствах К. см. ст. Полисахариды.
К. применяют в пищевой, бумажной, текстильной,
фармацевтической, в горной и др. отраслях пром-сти
в качестве клеев, стабилизаторов, для получения
вязких р-ров, искусственного волокна, пленок, на-
полнителей, взрывчатых веществ.
Лит.: Степаненко В. Н., Усп. химии, 1961, 30,
вып. 5, 626; Whistler R. L., в кн.: Colloque sur la blo-
chlmle des glucldes, P., 1961, p. 151; Industrial gums. Poly-
saccharides and their derivatives. Ed. R. L. Whistler, N. Y.,
1959; Hirst E. L., Jones J. K. N., в кн.: Handbuch
der ptlanzenphysiologie, Bd VI, B. — Gottingen—Heidelberg,
1958, S. 500; Wh 1 s t 1 e r R. L„ Smart G h. L., Poly-
saccharide chemistry, N. Y., 1953. Л. II. Линевич.
КАМЕННОУГОЛЬНАЯ СМОЛА (коксовая смола,
каменноугольный деготь) — вязкая черная жидкость
с характерным фенольным запахом; получается на
коксохимия, и газовых заводах при коксовании ка-
7 к. X. Э. т. 2
менных углей (выход К. с. ок. 3% от каменного угля).
Свойства К. с., полученных из каменных углей, в боль-
шей степени определяются условиями пиролиза, чем
природой этих продуктов. К. с. — сложная смесь
циклич. и гетероциклич. соединений и их производных
(из К. с. выделено более 400 различных индивидуаль-
ных соединений); практически наиболее важные из
них приведены в табл. 1.
Таблица 1
Наименование Т. кип., °C Темп-ра кристал- лизации, °C Содержа- ние, %
Нафталин 218 80,3 5,0—12,9
Тионафтен . . . : ...... 219,9 32,0 0,1—1,0
1-Метилнафталин 244,6 -30,0 0Л—1,4
2-Метилнафталин 241,0 34,5 1.2-1.8
Аценафтен 279 96 1.0-2,0
Флуорен 297,9 115 0,8—1,6
Фенантрен 336,8 99,2 4.0—6,0
Антрацен 351 216.6 0,8-1.8
Карбазол 355 245 0,8—2,0
Пирен 393,5 150 1.0—2,0
Хризен 440,7 255 0,6—1,5
Фенолы — — 1,5-3,5
Пиридин и его гомологи . . — — 0,2—0.S
Хинолин и его гомологи. . — — 0,3—03
К. с. представляет собой смесь истинных и переох-
лажденных растворов с коллоидными системами.
Состав и физич. свойства К. с., как указано выше,
в определенной мере зависят от природы исходных
каменных углей и технологич. условий коксования.
Менее метаморфизированные угли дают большие вы-
ходы смолы с большим содержанием фенолов. Кок-
сование при . повышенных против нормы темп-рах
и в условиях перегрева стен и подсводового прост-
ранства коксовых печей дает более пиролизованную
К. с., что характеризуется высоким содержанием
в ней нафталина и нерастворимых в толуоле составных
частей, более высокой плотностью и пониженным
содержанием фенолов. В зависимости от степени пи-
ролиза плотность К. с. колеблется в пределах 1,12—
1,25; наиболее характерны К. с. плотностью 1,175—
1,185, содержащие ок. 10% нафталина и до 2%
фенолов.
Фракционный состав К. с. характеризуется кривой
ее разгонки по истинным темп-рам кипения. Кривые
однократного испарения К. с. характеризуют соот-
ношение количества паров (отгона) и жидкости, на-
ходящихся в равновесии при различных темп-рах
испарения смолы под различным давлением. Эти кри-
вые описываются ур-нием: t = А—В (174,5—g),
где t — темп-ра однократного испарения; А — кон-
станта (по данным различных авторов, колеблется
в пределах 683—745); В — коэфф., зависящий от
давления (при 760 мм равен 2,4, при 100 мм — 3,1);
g — процент отгона.
Вязкость К. с. и ее фракций может быть рассчитана
по ур-нию:
lg lg (v +0,8) = ^-^ IgT
где v — вязкость в сантистоксах, Т — темп-ра в гра-
дусах Кельвина, К и — константы, значения к-рых
приведены в табл. 2.
Таблица 2
Продукт К K1
Легкое масло 9,32 4,0
Фенольная фракция 10,35 4,3
Нафталиновая фракция . . . 10,37 4,3
Поглотительная фракция . . . 10,25 4,2
Антраценовая фракции .... 11,07 4,4
Антраценовое масло 12,65 5,0
Каменноугольная смола . . . 13,80 5,33
387
КАМЕННОУГОЛЬНАЯ СМОЛА - КАМПСА РЕАКЦИЯ
388
Истинная уд. теплоемкость К. с. может быть рас-
считана по ур-нию:
с = | [0,302 + 0,000656 (1,8 г + 32)]
где t — темп-ра в °C; d — плотность при 20°. Изме-
нение плотности веса смол с темп-рой может быть
рассчитано по ур-иию:
d. == d20 [ 1 — 0,0007 (t — 20)]
Теплота сгорания К. с. ок. 9120 ккал/кг; т. всп-
ок. 100°.
Переработка К. с. заключается в разделении ее
перегонкой на фракции и пек и дальнейшей перера-
ботке полученных фракций. Незначительная часть
К. с. применяется в непереработанной виде в дорож-
ном строительстве, строительной пром-сти и для
энергетич. целей, однако, поскольку при этом те-
ряются нек-рые ценные ее составные части, часто для
перечисленных целей применяется не сырая кам.-уг.
смола, а препарированная, получаемая при перера-
ботке К. с.
Перед переработкой К. с. предварительно осво-
бождают от воды отстаиванием, затем ее подвергают
разгонке на трубчатых установках непрерывного
действия с ректификационными колоннами. На ри-
сунке дана схема такой установки. В табл. 3 приве-
дены отбираемые фракции, их выход и пределы выки-
пания.
Таблица 3
Фракция Выход, % Пределы вы- кипания, °C
Легкое масло 0,2-0,8 до 170
Фенольная 1,2-3,0 170-210
Нафталиновая 9,0—11,0 210-230
Поглотительная 7,0—10,0 230-270
Антраценовая I 15,0-18,0 270—350
Антраценовая II Пек каменноугольный (тем- 5-8,0 320-369
пература размягчения 65—75°) 50-60 —
Легкое масло сходно по составу с сырым бензолом
п перерабатывается вместе с ним. Фенольная фракция
Р?1
rffjll—
9 1
о
е:
iB-
е
is-
ai
сз
Схема одноколонной атмосферной
ректификации каменноугольной смо-
лы: 1 — сборник сырой смолы; 2 —
сборник обезвоженной смолы; 3 —
испаритель первой ступени; 4—труб-
чатая печь; 5 — испаритель второй
ступени; 6 — теплообменник; 1 —
фракционная колонна; 8 — холо-
дильники; 9 — сепаратор; 10 — от-
парная колонна.
тификацией которого получают
содержит до 40% фе-
нолов, 8% оснований
и 30% нафталина.
Нейтральная обезнаф-
талипенная часть фе-
нольной фракции со-
держит инден, кума-
рон, высшие гомологи
бензола; применяется
для получения кума-
роновых смол. Нафта-
линовая фракция со-
держит до 83% нафта-
лина, до 10% сопро-
вождающих его ве-
ществ (тионафтен, ин-
дол, метилнафталины),
до 4% крезолов и
ксиленолов, до 3%
оснований. Фракцию
перерабатывают кри-
сталлизацией при ох-
лаждении и после-
дующем горячем прес-
совании. Таким путем
выделяют прессован-
ный нафталин, очист-
кой и повторной рек-
чистый кристаллич.
нафталин. Выход прессованного нафталина 5—8%
Is
Лен
(от смолы). Поглотительная фракция после отмывки
фенолов и хинолиновых оснований служит для улав-
ливания сырого бензола из коксового газа, а также
для получения других технич. кам.-уг. масел. Из
поглотительного масла выделяют также содержа-
щиеся в нем аценафтен, флуорен, индол, метил-
и диметилнафталины. Антраценовая I фракция ох-
лаждается до 30—40°; при этом из нее выделяется
кристаллич. осадок — сырой антрацен, состоящий
гл. обр. из антрацена, карбазола, фенантрена и со-
провождающих их веществ. Получаемое после отде-
ления сырого антрацена антраценовое масло является
исходным продуктом для получения шпалопропиточ-
ного и других высококипящих кам.-уг. масел. Антра-
ценовая II фракция применяется для энергетич. це-
лей, для приготовления препарированных смол,
а также сажи.
В табл. 4 приведены нек-рыс физико-химич. дан-
ные фракций К. с.
Таблица 4
Наименование фракций Й20 Средняя темп-ра кипения, °C Тепло- емкость при 100°, кал/г Скрытая теплота испа- рения, кал/г
Легкое масло 0,900 142 0,45 108,0
Фенольная 0,980 186 0,44 80,0
Нафталиновая 1,040 218 0,42 74,0
Поглотительная 1,060 268 0,40 70,0
Антраценовая I 1,090 337 0,39 54,0
Антраценовая II 1,110 378 0,38 50,2
Пек 1,267 — 0,34 —
Кроме описанной, наиболее распространенной схемы
переработки К. с., имеются другие эффективные
схемы.
Лит.: Белов К. А., Переработка химических продуктов
коксования, Харьков — М., 1949; Брон Я. А., С а т а н о fl-
ски й С. Я., Трубчатые агрегаты для перегонки каменноуголь-
ной смолы, Харьков, 1961; Липлавк И. Л., Физико-хи-
мические свойства химических продуктов коксования камен-
ных углей, Свердловск—М., 1954; Литвиненко М. С.,
Носалевич И. М., Химические продукты коксования
для производства полимерных материалов, Харьков, 1962.
И. М. Носалевич.
КАМЕННОУГОЛЬНЫЕ МАСЛА — см. Масла
каменноугольные технические.
КАМПСА РЕАКЦИЯ — образование оксихиноли-
нов внутримолекулярной конденсацией о-ациламипо-
ацетофенонов под действием оснований:
L Т СНз
%X^NH-CO-CH3
он-
70Я 20Х
Ациламиноацетофенон обрабатывают спиртовым р-ром
этилата натрия или водным р-ром едкого натра. При
подкислении полученного р-ра выделяют оксихино-
лины. С помощью К. р. могут быть получены и много-
ядерные гетероциклич. соединения, содержащие окси-
хинолиновую группировку атомов:
^усосн2сн2 Na0H
X/^NHCOCH/ н2°
389
КАМФАН —КАМФЕН
390
К. р. открыта Бишлером и Хоуэллом (1893), но
подробно разработана Р. Кампсом в 1899.
Лит.: Эльдерфилд Р., в кн.: Гетероциклические
соединения, пер. с англ., т. 4, М., 1955, с. 45; Surrey A. R.,
Name reactions in organic chemistry, N. Y., 1954, p. 25.
E. M. Рохлин.
КАМФАН (1,7,7-триметилбицикло-[1, 2,2]-гептан),
мол. вес 138,25 — белые кристаллы (гексагональ-
ные пластинки или призмы); чрезвычайно
Ф летуч (даже в твердом состоянии); по запаху
напоминает борнеол; т. пл. 158—159°; т. кип.
160—161°/760 мм; оптически недеятелен; хо-
рошо растворим в этиловом эфире, этилацетате, ху-
же — в метиловом и этиловом спиртах и петролейном
эфире. К. чрезвычайно стоек к нагреванию и окисли-
телям, на него не действует конц. H2SO4 при 180°.
При действии на К. разб. HNO3 (145—150°) обра-
зуются стереоизомерные питрокамфаны: а-нитрокам-
фан (I), т. пл. 128—130°, и а'-нитрокамфан (II),
т. пл. 146—147°. При хлориро-
е.-.Н вании К. в р-ре СС14 вначале
\\'О2 Г--|ХН образуется борнилхлорид, а в
XE-'J дальнейшем — 2,3-дихлоркам-
I II фан. Обмен водорода на дейте-
рий в К. протекает по той же
схеме. К. пока не найден в природе; его получают
восстановлением хлористого борнила металлич. Na
в спирте или каталитич. восстановлением борнилена,
или же сухой перегонкой гидразона камфары в при-
сутствии безводного КОН.
Лит.: Simonsen J. L., The terpenes, v. 2, 12 ed.],
Camb., 1949. И. И. Вардыгиев.
СН3
КАМФАРА (1,7,7-триметилбицикло-[ 1,2,2 ]-гепта-
нон-2) С10Н16О, мол. в. 152,23 — полупрозрачные
кристаллы белого цвета с характер-
ным запахом; весьма летуча на воз-
духе, легко возгоняется и перего-
няется с водяным паром; флюоресци-
рует в УФ-свете слабо-коричневым
цветом, легко растворяется в орга-
нич. растворителях: 100% вС2Н5ОН,
173% в (С2Н5)2О, 300% в СНС13,
растворяется в CS2, С6Нв, СН3ОН,
СН3СООН, СН3СОСН3, в конц. НС1, HNO3, 0,1%
в Н2О. При растирании с фенолом, ментолом и ти-
молом К. образует густые, прозрачные жидкости
(эвтектич. смеси). К. бывает в виде 1-, d- и d,1-форм.
Т. кип. 209.Г7759 мм, т. пл. d- и 1-форм 178,5—179°
и d,1-формы 178—178,5°; а?18 0,985, 1,000; [a]D
К. изменяется в зависимости от природы растворителя
и концентрации раствора: в абс. спирте [а]р = 4-41,4°
(С = 1%), 4-44,8° (С = 20%), 4-48,4° (С = 50%).
Наибольшее значение [a]D для 1-К. —44,22° (абс.
этанол) или —42,76° (95%-ный этанол). Теплота сго-
рания 1414 ккал/молъ (о = const); теплоемкость
0,5 кал/г, теплота испарения 92,5 кал/г.
При окислении К. азотной к-той получаются кис-
лоты: камфарная С10Н16О4, т. пл. 187° (активная)
и 204—205° (неактивная), камфаноновая С8Н14О3
и камфароновая С8Н14О8. При нагревании К. с
КОН образуется смесь изокамфолевой и камфоле-
вой кислот с преобладанием последней. При дейст-
вии РС16 и PJ5 при нагревании К. превращается
в л-цимол. Если реакцию проводить осторожно,
в контролируемых условиях, получается 2,2-дихлор-
камфан (т. пл. 155—156°), к-рый претерпевает пере-
группировку Наметкина (см. Камфеновые перегруп-
пировки) и превращается в 2,4-дихлоркамфан. При
действии брома на К. образуется а-бромкамфара
(т. пл. 76°), при действии хлора — а-хлоркамфара
(т. пл. 94°). При гидрировании К. с Pt-чернью в
СН3СООН получается смесь: 90% изоборнеола
и 10% борнеола.
К. реагирует с металлич. натрием с образованием
весьма реакционноспособной натрийкамфары, строе-
ние к-рой точно не установлено, но ее широко исполь-
зуют для синтеза. К. не реагирует с бисульфитом
натрия, но взаимодействует с другими реагентами на
карбонильную группу. Оксим d-K.—иглы или призмы,
т. пл. 119,5°, т. кип. 249—254° (с разл.); семикарба-
зон d-K. — иглы, т. пл. 247,8°; фенилгидразон
d-K. — т. кип. 210°/17 мм, растворим в С2Н5ОН.
Камфара широко распространена в природе, входит
в состав многих эфирпых масел: базиликового, по-
лыни, камфарного шалфея, скипидаров, полученных
из древесины различных сосен (т. наз. «осмольных»),
хвойных эфирных масел и т. д. К. содержится в масле,
получаемом из древесины, листьев и корней камфар-
ного лавра, произрастающего на о. Тайвань, Вьетнаме
и Японии. Из этого масла получают т. н. натураль-
ную d-K.
В пром-сти К. (d, 1-форма) синтезируют из скипи-
дара или его основного компонента a-пинена (I).
Присоединяя к последнему НС1, получают хлористый
борнил (II), к-рый при нагревании со щелочами или
солями жирных к-т превращается в камфен (III). При
действии на камфен смесью ледяной уксусной и сер-
ной к-т получают ацетат изоборнеола (IV) и небольшое
количество ацетата борнеола, из к-рых после омыле-
ния и дегидрирования образуется К. В пром-сти кам-
фен получают каталитич. изомеризацией а-пинена
(чаще с ТЮ2). Оптически деятельную 1-К. в СССР
получают из хвойного эфирного масла пихты сибир-
ской, к-рая содержит до 40% борнеола и борнил-
ацетата, 20% камфена и 10% a-пинена. Предложены
другие способы синтеза К. из а- и (J-пиненов.
К. широко применяют в пром-сти, она служит
пластификатором при получении целлулоида и пленок
па его основе; ее добавляют (ок. 1%) к бездымному
пороху для придания ему устойчивости при хранении
(флегматизатор); используют для борьбы с молью
и комарами в помещениях. В медицине К. применяют
как средство, усиливающее сердечную деятельность;
К. возбуждает центральную нервную систему, осо-
бенно дыхательный и сосудодвигательные центры.
Применяют К. чаще в масляных р-рах, к-рые вводят
под кожу при упадке сердечной деятельности и угне-
тении дыхания, а также в качестве жаропонижающего,
антисептического и болеутоляющего средства.
Лит.: Рудаков Г. А., Химия и технология камфары,
М.—Л., 1961; Simonsen 3. L., The terpens, v. 2, 2 ed.,
Camb., 1949. И. И. Еардъсшев.
КАМФАРНОЕ МАСЛО — см. Эфирные масла.
КАМФЕН (3,3-диметил-2-метилен-бицикло- [1,2,2]-
гептан), мол. в. 136,23 — белые кристаллы; летуч
на воздухе, обладает характерным камфарным запа-
хом; т. пл. 51—52° (48°); т. кип. 160—161°/760 мм,
52°/17 мм; d'l* 0,8422; п% 1,5514 (1,4562); MRD 44,02
7*
391
КАМФЕН — КАМФЕНОВЫЕ ПЕРЕГРУППИРОВКИ
392
. хорошо растворим
R = CH3
[a]D от 0° до ±103,9° (в эфире), ±99,6° (в спирте),
в диэтиловом эфире, бензоле,
хуже — в этиловом спирте, не-
растворим в воде. При гидри-
ровании К. на Pt-катализаторе
при обычной темп-ре образует-
ся изокамфан С10Н18 (т. кип.
165—166°/760 мм, т. пл. 63—
64,5°). При гидратации К. в оп-
ределенных условиях получают
камфенгидрат, чаще же этим путем получают смесь изо-
борнеола и борнеола. При действии на К. органич. к-т
(муравьиной, уксусной и др.) при участии катализато-
ров (H2SO4, ZnCl2) получают в основном сложные эфиры
изоборпеола, омыление к-рых приводит к изоборнео-
лу; реакция служит для количественного определения
К в смесях его с другими терпеновыми углеводорода-
ми, к-рые в этих условиях не этерифицируются.
При действии на К. НС1 получают равновесную
смесь изомерных хлоридов: камфенгидрохлорид (1),
борнилгидрохлорид (II) и изоборнилгидрохлорид (III)
с преобладанием пос-
леднего. При действии
на К. НВт и особенно
HJ образуются в ос-
новном галогенопро-
изводные изоборнила.
При окислении К.
КМпО4 получают камфенгликольС10Н18О2, камфенила-
новый альдегид С10Н18О, камфениловую к-ту С10Н1вО3,
камфенилон С9Н14О и камфеновую к-ту С10Н18О4.
Изучение продуктов окисления позволило Вагнеру
дать правильную формулу строения К. Окисление
К. хромовой к-той приводит в основном к камфаре
и небольшому количеству камфарной к-ты. Окисление
озоном и кислородом воздуха дает камфенилон. При
действии на К. гидроперекиси бепзоила получается
окись К. (т. кип. 90—92°/20 мм, т. пл. 86—88°).
При хлорировании и бромировании водород метиле-
новой группы К. замещается галогеном, а двойная
связь сохраняется. При действии HNO3 получают
нитропроизводные К. Присоединение НОС1 к К.
дает продукт с т. пл. 93°. Триоксиметилен замещает
водород ъ метиленовой группе К. на СН2ОН (полу-
чается со-оксиметилкамфен).
К. содержится в небольших количествах в скипи-
дарах различных хвойных, особенно в хвойных эфир-
ных маслах, напр. масле Abies sibirica Ledeb, откуда
его можно выделить методом ректификации и вымора-
живания. Содержится также в кипарисовом, лаван-
довом, лимонном, фенхельном, валериановом и др.
маслах. К. чаще получают синтетически изомериза-
цией а- и р-пиненов при воздействии на них катали-
заторов — активных глин, TiO2 и др. Выход К. при
этом достигает более 70%. Дегидратация изоборнеола,
дегидрохлорирование борнил- и изоборнилхлорида
приводит в основном к К. Все перечисленные выше
методы получения К. приводят к образованию раз-
личного состава смеси изомерных углеводородов, на-
ходящихся в равновесном состоянии: трициклена,
борнилена и К. с преобладанием последнего. Состав
смеси зависит от условий реакции. Наиболее чистый
К. получается при действии HNO2 на борниламин.
Имеются и другие способы получения К.
К. широко применяют в пром-сти: промежуточный
продукт при синтезе камфары. Хлорированием К.
получают весьма эффективные инсектициды. В СССР
эти продукты известны под названием «хлорфен»,
«полихлоркамфен»; в США — «токсафен».
Лит.: Simonsen J. L., The terpenes, v. 2, 2 ed.,
Cambridge, 1949; Пигул евский Г. В., Химия терпе-
нов, Л., 1949; Г о р я е в М. И., Характеристика химических
соединений, входящих в состав эфирных масел, Алма-Ата,
1953. И. И. Вардышев.
КАМФЕНОВЫЕ ПЕРЕГРУППИРОВКИ — изо-
меризация соединений терпенового ряда. Известны
т. наз. К. п. первого и второго рода.
К. п. I рода открыта Е. Е. Вагнером. Позднее над выясне-
нием механизма этой перегруппировки много работал Меер-
вейн. Поэтому К. п. I рода наз. часто перегруппировкой Ваг-
нера, Вагнера — Меервейна и, наконец, ретропинаколиновой
перегруппировкой.
Перегруппировка Вагнера — Меервейна в терпеновом
ряду аналогична перегруппировке при дегидратации
вторичного пинаколинового спирта в тетраметилэти-
лен (ретропинакалиновая перегруппировка). Гидра-
тация тетраметилэтилена, приводящая к образованию
пинаколинового спирта, наз. пинаколиновой перегруп-
пировкой. (Подробнее см. Пинаколиновая и ретропи-
наколиндвая перегруппировки).
Ретропинаколиновая перегруппировка
Н3С СН3 г Н3С СН3
\ / \ / I ПзЦ /ЬНз
Н3С-С-С-Н Н3С-С-С-СН3 >С=С<
H3CZ Чн L но/ Ч J НзС XcHs
Пинаколиновая перегруппировка
Аналогичный переход вторичных спиртов борнеола
(I) и изоборнеола (II) в камфен (III) (ретропинаколи-
новая перегруппировка) и обратный переход — от
камфена к изоборнеолу (пинаколиновая перегруппи-
ровка), по Вагнеру, приведен ниже:
Примерами К. п. I рода может служить переход от
пинена к борнилгидрохлориду, а также гидратация
пипена в борнеол и фенхиловый спирт и др. При
К. п. I рода должны получаться оптически деятельные
соединения, т. к. один асимметрич. атом не затраги-
вается реакциями. На самом же деле при указанных
реакциях получаются чаще всего оптически недея-
тельные или .малодеятельные в-ва. Для объяснения
этого явления С. С. Наметкин предложил схему, к-рая
называется К. п. II рода, или перегруппировкой На-'
меткина.
К. п. II рода обусловливает рецемизацию в кам-
фенном ряду. Напр., при дегидратации d-изобор-
неола (IV) в результате К. п. I рода образуется 1-кам-
фенгидрат (V), а затем 1-камфен (VI). В то же время,
в результате одновременных перегруппировок вна-
чале I рода, а затем II рода образуется d-камфен-
гидрат (VII), а затем d-камфен (VIII). В зависимости
от преобладания той или другой перегруппировки
получаются препараты камфейа, имеющие различную
оптич. деятельность, или оптически недеятельный
камфен.
393
КАМФЕНОВЫЕ ПЕРЕГРУППИРОВКИ — КАНАМИЦИН
394
Одновременно с перегруппировками I и
рацемизация камфена протекает частично
оптически недеятельный трициклен (IX):
Сн3 СН3
Н,С С._ ИС
I I 1'ОН (-Н,О)
|Н3С—С—СН3| -------—1н3с-с-сн3
Н2С | СН2 Н2с | СН2
I I IX
н н
В последнее время механизм К. п. I и II рода изоб-
ражают в виде превращений карбопия иона, напр.
перегруппировка I рода борнеола (I) в камфен (IID
сн3
^С
Н2С^ ^с
(ОНО 1
сн
II
и
рода
через
сн3
0Н
н2с Г
I ’ I н
|н3с-с-сн3|
1 сн.
н2С.
I I I ’ н 1рода
|н3с—с—сн3 —*
Н2С\ XII,
Y .
н
СН3
С^
сн
Н2С
I I ЮН-)
|н3с-с-сн3| "
Н2С I сн2
I X
н
Н3СХ .он
с'С
сн
I (-НОН)
fl 2С
|н3с-с-сн3|
Н2С I С
I XI
н
| XII
н
сн,
II
с\
Н2С----1 -—СИ
|н3С-С-СН3|
Н2с I сн2
I III
н
теоретических основ органической химии, Л., 1959; X ю к-
к е л ь В., Теоретические основы органической химии, пер.
с нем., т. 1, М., 1955; Перспективы развития органической
химии, под ред. А. Тодда, пер. с англ, и нем., М., 1959.
И. И. Бардышев.
КАНАМИЦИН (канамицин А) С]8НзвК40ц — анти-
биотик, основание, мол. в. 468 (444 и 427 по Расту,
490 по Зигнеру), вычисленный мол. в. — 484,31.
Молекула К. состоит из остатков 2 изомерных
аминогексоз (З-амипо-З-дезокси-В-глгокозы и 6-амино-
6-дезокси-В-глюкозы), глюкозидно связанных с ос-
татком молекулы мезо-1,3-диамипо-4,5,6-трпоксп-
цпклогексапа (2-дезокспстрептамина):
К образует соли с различными кислотами. Сернокис-
лая соль хорошо кристаллизуется из водно-метаноль-
иого р-ра
СН2
и
С
в
З2с
|н3с-с-сн3|
Г сн3
виде моногидрата (С^НзоГ^Оц • H2SO4-
• Н2О), имеющего форму неправильных
призм; т. разл. выше 250°; [а]^ = -|-121
(1%, вода); хорошо растворима в воде,
нерастворима в метиловом спирте и
других органич. растворителях. Удале-
ние воды из кристаллич. соли дости-
гается лишь при 170° в вакууме.
К. не имеет характерных полос по-
глощения в УФ-части спектра, ИК-
спектр типичен для полиоксиполи-
н
амипосоединений. К. дает положительные реакции
с нингидрином, реактивами Молйша, Эльсона—Мор-
гана и отрицательные с реактивами Сакагучи, на
редуцирующие сахара и па образование мальтола.
Все атомы азота в молекуле К. входят в состав амино-
групп и определяются по Ван-Слайку; [а]^ = —|—146°
(С = 1%; 0,1 н. H2SO4). К. стабилен в водном р-ре,
особенно при нейтральном и щелочном значении pH.
Выдерживание водных р-ров антибиотика в автоклаве
при pH 7,0 (1 час, 120°) снижает биологич. активность
па 10%. При нагревании К. в р-ре 6 н. соляной
к-ты в течение 45 мин наблюдается полная потеря
активности; нагревание в 40%-ной H2SO4 (100 мин,
100°) приводит к образованию вещества фурфуроло-
подобного типа, имеющего максимум поглощения
в УФ-части спектра при 275—280 ммк.
К. образуется при выращивании Streptomyces
canamyceticus (27—29°; pH 7,0) на среде, содержащей
различные питательные вещества (крахмал, соевая
мука и др.). К. извлекают из культуральной жидкости
методом ионообменной сорбции при использовании
карбоксильных, сульфо- и др. катионитов, а также
экстракционным методом с применением переносчи-
ков (жирных к-т, хлорфенолов и др.)- Десорби-
руют К. водными р-рами кислот или аммиака; выде-
ляют в виде хорошо кристаллизующейся сернокислой
соли.
Хроматография, изучением культуральной жид-
кости и.препаратов К. в системе и-бутанол — вода —
n-толуолсульфокислота было установлено, что наряду
с К. образуется еще два антибиотика, обозначенных
канамицинами В и С. Канамицин В в отличие от ка-
происходит через карбония иопы борнила (X) и кам-
,фенила (XI) и камфенгидрат (XII).
' Лит.: Реутов О. А., Теоретические проблемы орга-
нической химии, М., 1956; Темникова Т. И., Курс
памнцина А не образует при кислотном гидролизе
соединения, подобного фурфуролу. Содержание К.
определяют биологич. методом. Описан быстрый ме-
тод определения К. по Ван-Слайку.
395
КАНИФОЛЬ
396
К. подавляет рост многих патогенных аэробных
грамположительных, грамотрицательных и мико-
бактерий. Однако нек-рые пневмококки, часть штам-
мов стрептококков и почти все анаэробные микроор-
ганизмы, дрожжи и грибы устойчивы к К. По дейст-
вию на туберкулезную палочку К. почти идентичен
стрептомицину. К. обладает относительно слабой
токсичностью ЛД60 2 90— 340 мг/кг при внутривенном
введении мышам. Клииич. испытания показали его
высокую эффективность при лечении заболеваний
(сепсисов, перитонитов и др.), вызванных стафило-
кокками, особенно устойчивых к другим антибиоти-
кам. Он оказался весьма действенным в большинстве
тяжелых случаев сибирской язвы, а также при ле-
чении многих случаев острых и хронич. инфекций
мочевыводящих путей. Хорошие результаты получены
при лечении гонореи у мужчин. К. выделен в 1957.
Лит.: Химия антибиотиков, т. 1,- 3 изд., Мл 1961, с. 730.
С. М. Мамиофе.
КАНИФОЛЬ — твердая, хрупкая, стеклообразная
прозрачная смола светло-желтого (реже темно-крас-
ного) цвета, составная часть смолистых веществ хвой-
ных деревьев, остающаяся после отгонки из смолистых
веществ скипидара. К. хорошо растворяется в эфире,
абс. спирте, ацетоне, скипидаре и бензоле, хуже —
в бензине, керосине и фурфуроле; не растворяется
в воде. Т. размягч. К. 52—70°; т. кип. 250°/5 мм',
dl°0 1,007—1,085; теплоемкость 0,54 ввал/вг; тепло-
проводность 0,11 ккал/м час град', скрытая теплота
плавления 15,8 ккал/кг; коэфф, расширения расплав-
ленной К. 0,055; теплотворность 9074—9171 ккал/кг;
кислотное число 150—175.
К. состоит из кислот смоляных (80—95%), имеющих
общую формулу С18Н28СООН, и неомыляемых веществ
(5—12%), к-рые содержат метилхавикол, стильбе-
новые производные, терпеновые димеры, альдегиды
и смесь гидрофенантреновых углеводородов. С мине-
ральными и органич. основаниями К. дает соли —
Канифольное мыло (напр., C19H29COONa),
а с различными гидроксилсодержащими соедине-
ниями — эфиры канифоли и др. ценные
продукты. Конденсацией К. с резольной феноло-фор-
мальдегидной смолой при 290° получают а л ь б е р-
т о л и; при их этерификации различными спиртами
образуется ряд ценных смол.
В зависимости от вида сырья и способа получения
различают следующие виды К.: живичную, экстрак-
ционную и талловую. Живичную К. получают
отгонкой скипидара из очищенной живицы. Этот вид
К. содержит 5—7% неомыляемых веществ и практи-
чески не содержит жирных к-т. Экстракцион-
ную К. получают экстракцией органич. раствори-
телями (чаще всего бензином) измельченной смолистой
древесины хвойных деревьев (обычно сосны обыкно-
венной). Этот вид К. отличается от живичной более
темным цветом, имеет пониженное кислотное число
(150—155), т. размягч. 52—58°, содержание жирных
к-т до 12%. При осветлении экстракционной К. ее
свойства и ценности аналогичны живичной. Талло-
вую К. получают фракционной перегонкой талло-
вого масла.
Для придания К. стойкости к действию света и ат-
мосферным воздействиям ее подвергают различным
химич. превращениям: гидрируют с применением
катализаторов и давления; нагревают до 200—300°
с платиновым катализатором (при этом происходит
перераспределение водорода в молекулах смоляных
к-т); обрабатывают конц. H2SO4 р-ры К. в бензине;
окисляют кислородом воздуха при нагревании и т. д.
В результате получают т. наз. модифициро-
ванную К.: гидрированную, состоящую
в основном из дигидросмоляных к-т (С18Н31СООН);
диспропорционированную, состоящую
из смеси дигидро-, тетрагидро- и дегидросмоляных к-т
(С19Н31СООН, С19Н33СООН и С19Н27СООН); пол и-
ме р и зов а н н у ю, состоящую в основном из
димера смоляных к-т (С18Н28СООН)3; окислен-
ную — из оксисмоляных к-т и их полимеров; суль-
фированную — из сульфосмоля ных к-т
(C19H28SO3H), конденсированную (с ма-
леиновой или фумаровой к-тами) — из аддукта лево-
пимаровой к-ты и малеинового ангидрида, и др.
К. — один из важнейших продуктов лесохимия,
пром-сти (см. Лесохимия), применяется, напр., для
проклейки бумаги,приприготовлениимыла, лаков, сур-
гуча, линолеума, замазок, мазей, пластырей, липких
веществ, изоляции электрич. кабелей, смазочных ма-
сел, пластич. масс, фунгицидов и т. п.
В пром-сти в основном применяется К., получен-
ная из смолистых веществ сосны; в последние годы К.
также получают из смолистых веществ других хвой-
ных пород, напр., ели обыкновенной, кедра и сибир-
ской лиственницы, крымской сосны и др.
Канифольное мыло — средние (или кис-
лые) натриевые (реже калиевые) соли К. (обычной
или модифицированной, предпочтительно гидрирован-
ной или диспропорционированной); аморфный гидро-
фильный продукт, легко образующий прозрачные кол-
лоидные р-ры с органич. растворителями и углево-
дородами; хорошо растворяется в воде, растворяет
жиры, обладает большой моющей способностью,
хорошо мылится и пенится, размягчает мыла, изго-
товленные на основе твердых жиров, и способствует
их ценообразованию. Соли К. других групп элементов
(кроме первой) гидрофобны, не растворяются в воде,
но хорошо растворяются в неполярных органич.
растворителях (особенно в углеводородах). Кислые
соли Са, Zn, Al имеют т. размягч. ок. 120°, более
тверды, чем К., и применяются в качестве сиккативов
и для повышения твердости лаковых пленок; соли
Мп и Со используют лишь в качестве сиккативов.
Канифольное мыло широко применяют в различных
отраслях пром-сти: в мыловарении, где К. (до 10%)
добавляют к жирам; для приготовления клеев, для
проклейки бумаги; в качестве стабилизатора эмуль-
сий в произ-ве синтетич. каучука, напр. при эмуль-
сионной сополимеризации бутадиена и стирола.
Канифоли эфиры — сложные эфиры К.
и гидроксилсодержащих веществ. Эфиры К. и одно-
атомных спиртов — вязкие, высококипящие жид-
кости (т. кип. 210—300° при 4 мм). Эфиры К. и этилен-
и диэтиленгликолей, глицерина, пентаэритрита и кси-
лита — твердые смолы с т. размягч. (соответственно):
45°, 70°, 85° и 71°. Глицериновый эфир К., к-рый
часто называют эфиром гар-
пиуса (I), состоит в основном из
эфира абиетиновой кислоты. Эфиры
К. хорошо растворяются в неполяр-
ных растворителях (бензине, бензоле,
скипидаре и др.), трудно омыляются,
стойки па воздухе и хорошо совме-
щаются с основными пигментами. Качество эфиров
гидрированной, диспропорционированной и полиме-
ризованной К. лучше соответствующих эфиров обыч-
ной К.
Эфиры К. получают прямой этерификацией К. при
250—300° (в присутствии катализаторов и без них),
причем этерификация К. с многоатомными спиртами
проходит легче, чем с одноатомными. При обработке
эфиров К. малеиновым ангидридом или фумаровой
к-той при 180° (или при обработке К. этими реаген-
тами, а затем спиртами) получают канифольно-ма-
леиновые или канифольно-фумаровые смолы.
Лит.: Комшилов Н. Ф., Состав канифоли и строение
смоляных кислот сосны и ели, М.—Л., 1955; Васечкин
В. С., Технология экстрактивных веществ дерева, М.—Л.,
1953; Медников Ф. А., • Подсочка леса, М,—Л., 1955;
СН2ОСО—Ci9H->9
I
СНОСО-С19Н.,9
I
СН2ОСО-С19Н29
I
397
КАННИЦЦАРО РЕАКЦИЯ— КАНЦЕРОГЕННЫЕ ВЕЩЕСТВА
398
Ш а м п е т ь е Г., Рабате Г., Химия лаков, красок и
пигментов, пер. с франц., т. 1, М., 1960; Ш л я н и н В. Н.,
Лесохимическая промышленность в 1959—1965 гг., М., 1960;
Сборник стандартов лесохимической промышленности, [М.],
1950. И. И. Вардышев.
КАННИЦЦАРО РЕАКЦИЯ — окислительно-вос-
становительное диспропорционирование двух молекул
альдегида в соответствующие спирт и кислоту:
-ОН
2RCOH-----> RCOOH + RCH2OH
Превращение происходит при обработке альдегида
конц. водным или водноспиртовым р-ром щелочи.
К. р. характерна для ароматич. и гетероциклич.
альдегидов, а также для алифатич. альдегидов, не
содержащих водорода у атома углерода, связанного
с альдегидной группой:
СН3 СН3 СНз
I 1 I
НО—С—СОН -> IIO-C-CIROII -I- НО-С-СООН
(*:нз СНз СНз
Электроноакцепторные заместители в бензальдегиде
ускоряют К. р., а электронодопорные — замедляют ее:
n-(CH3)2N < П-СН3О < n-R с Н С n-Cl < jh-C1 < jh-NO2
Такой порядок влияния заместителей согласуется
с первой стадией механизма, предлагаемого для К. р.:
/0-
rcoh 4- -он —>• R-CH
^он
(-ОН он
% о I // I
но-с~н + с=о—- НО-С + н-с-о —-
Il II
R R R R
—- RCOO' + RCH2OH
Атом водорода альдегидной группы непосредственно
переходит от одной молекулы альдегида к другой,
не обмениваясь с протоном среды, что доказывается
получением не содержащего дейтерия бензилового
спирта из бензальдегида и NaOD в тяжелой воде и,
наоборот, превращением дейтеробензальдегида в
дважды дейтерированный бензиловый спирт:
CeHsCOD + он -> c8h5cd2oh + с8н5соо-
К. р., протекающая с участием двух различных аль-
дегидов, наз. перекрестной реакцией Канниццаро.
В качестве восстанавливающего агента обычно при-
меняется большой избыток формальдегида:
Альдегиды, содержащие в a-положении атомы водо-
рода, сначала оксиметилируют, а далее восстанавли-
вают:
сн2он
сн2о / -он
RCH2CHO-----> RC-CHO 4- СН2О---->
\и2 ОН
СН20Н
I
-> К-С-СН2ОН 4- нсоо-
СН20Н
Таким путем, в частности, получают имеющий про-
мышленное значение пентаэритрит.
Внутримолекулярная К. р. характерна для гли-
оксаля и а-кетоальдегидов, превращающихся в при-
сутствии щелочей в а-оксикислоты:
R R ОН it ОН ЧО*
Внутримолекулярная К. р. аналогична бензиловой
перегруппировке ароматич. дикетонов в диарилокси-
карбоновые к-ты. Реакция была открыта С. Канниц-
царо в 1853.
Лит.: Гейсман Т. А., в кн.: Органические реакции,
сб. 2, пер. с. англ., М., 1950, с. 106; Hauser С. R., Н а ш-
rick Р. J., Stewart А. Т., J. Organ. Chem., 1956,
21, № 2, 260. Н. ГГ. Гамбарян.
КАНОНИЧЕСКОЕ РАСПРЕДЕЛЕНИЕ — см.
Гиббса распределение.
КАНТАРИДИН (ангидрид,
3,6-эпокси-1,2-диметил-
циклогександикарбоновой-1,2 кислоты)
мол. в. 196,20—бесцветные
прозрачные кристаллы, т.
возг. 85°, т. пл. 212—213° СНз
(в запаянном капилляре); r/W"c2o
1 вес. ч. К. растворяется в I ? Л-сб
30 000 вес. ч. холодной и сн3
15 000 вес. ч. горячей во-
CioH1204,
О
ды, 1100 вес. ч. спирта,
760 вес. ч. эфира, 150 вес. ч. этилацетата, 55 вес. ч.
СНС13, 40 вес. ч. ацетона. К. растворяется при нагре-
вании в щелочах с образованием солей кантариди-
новой к-ты. Колориметрич. метод определения К.
основан на возникновении коричневого окрашивания
при нагревании пробы исследуемого р-ра с 1 каплей
10%-ного водного р-ра СН2О и 4—5 мл H2SO4 (чувст-
вительность 0,01 мг). К. является действующим на-
чалом яда шпапских мушек (Cantharis vesicatori а)
и нек-рых других видов насекомых. Синтез К. очень
труден и многостадиен (1953), поэтому К. по-прежнему
получают экстракцией высушенных и растертых
в порошок насекомых; иа 100 г мушек выделяют 0,3—
1,5 г К.
Применение К. в медицине и фармакологии основано
на его раздражающем кожу действии. К. — сильный
яд, ЛД для кошек и собак — ок. 1 мг/кг', при приеме
внутрь 30 мг наступает быстрая смерть, более
малые дозы вызывают болезни почек, изменение
в крови и т. д. Взаимодействие К. с этилендиамином
приводит к соединению C12HJ8O3N2, к-рое образует
о золотом и др. тяжелыми металлами соли, обладаю-
щие сильным бактерицидным действием. К. оказы-
вает ингибирующее влияние на ферментативное бро-
жение. В. Н. Фросин.
КАНЦЕРОГЕННЫЕ ВЕЩЕСТВА — органич. ве-
щества (нек-рые многоядерные ароматич. углеводо-
роды и их производные), обладающие способностью
вызывать рак при нанесении на кожу или при инъек-
ции под кожу животных.
Известны профессиональные раковые заболевания людей,
к-рые по роду своих занятий находятся в постоянном соприкос-
новении с различными продуктами. Примерами таких заболе-
ваний служат рак кожи трубочистов, шахтеров, кочегаров,
ткачей, лиц, имеющих дело со смазочными маслами, рабочих
кам.-уг. пром-сти; рак мочевого пузыря рабочих анилинокра-
, сочной пром-сти и т. п. Наблюдения над раковыми заболева-
ниями в результате Контакта с кам.-уг. смолой и продуктами
ее переработки возбудили интерес к исследованиям различных
веществ, с помощью к-рых можно вызвать раковые заболевания
искусственно.
В 1915 ЯМагива и Ишикава удалось вызвать опухоли кожи
длительным смазыванием уха кролика кам.-уг. смолой. С этого
момента началось интенсивное изучение активности смол раз-
личного происхождения и на протяжении многих лет «дег-
тярный рак» оказывался в центре внимания экспериментаторов-
онкологов. Следующим этапом изучения экспериментального
рака явились работы Хигера (1930), показавшего, что раз-
личные канцерогенные образцы смол обнаруживают характер-
ный спектр флуоресценции углеводородов ряда 1,2-бензан-
трацена (I). В связи с этим были синтезированы и подвергнуты
биологич. испытанию различные конденсированные ароматич.
углеводороды, из к-рых 1,2,5,6-дибензантрацен (II) и нек-рые
гомологи 1,2-беязантрацена оказались способными вызывать
рак при нанесении на кожу животных. Вскоре затем удалось,
используя флуоресцентный метод анализа, выделить из кам.-уг.
дегтя индивидуальный углеводород — 3 ^-бензпирен (III),
оказавшийся основным канцерогенным ингредиентом дегтя.
Открытие канцерогенного начала дегтя позволило не
только объяснить причину возникновения «дегтярного рака»,
но и допустить участие ароматич. углеводородов в возникно-
вении других форм злокачественных образований у человека.
399
КАНЦЕРОГЕННЫЕ ВЕЩЕСТВА
400
Особенное значение для этиологии рака приобрело открытие
высокой канцерогенной активности 20-метилхолантрена (IV),
к-рый удалось получить из дезоксихолевой кислоты — одной
из желчных кислот.
Открытие канцерогенной активности нек-рых про-
изводных 1,2-бензантрацена повлекло за собой ин-
тенсивное развитие исследований по синтезу различ-
ных типов многоядерных ароматич. углеводородов
и их биологич. испытанию, а также испытанию кан-
церогенной активности представителей других классов
оргапич. соединений. В результате исследований было
установлено, что канцерогенной активностью обла-
дают не только различные многоядерные ароматич.
углеводороды и их производные, но и мпогоядерные
гетероциклич. соединения, ароматич. амины, азо-
красители, половые гормоны и их синтетич. аналоги,
а также (весьма слабо) нек-рые простейшие оргапич.
соединении, как, напр., диэтиленгликоль, л-бензо-
хинон. Злокачественные новообразования вызываются
также многими неорганич. соединениями, в особен-
ности радиоактивными — радием, бромистым радием,
мезаторием, окисью тория, солями радиоактивного
изотопа Се144 и др.
Значительный экспериментальный материал позво-
ляет сделать нек-рые выводы о связи между строением
оргапич. соединений и их канцерогенной активностью.
Простейшие ароматич. углеводороды — бензол и на-
фталин — безвредны, однако аминопроизводное на-
фталина — Р-нафтиламин — вызывает опухоли моче-
вого пузыря у собак, если это вещество ежедневно,
в течение продолжительного времени, прибавлять
к пище в количестве 1—100 мг. Действием Р-нафтил-
амина вызывается рак мочевого пузыря рабочих
анилинокрасочной пром-сти.
Установлено, что активность высших полициклич.
углеводородов связана с определенным числом бен-
зольных колец в .системе, ниже и выше к-рого кан-
церогенная активность резко падает или совсем ис-
чезает. Но даже и для соединений с оптимальным по
активности числом колец подчас самые незначитель-
ные изменения структуры сопровождаются резкими
изменениями в активности, приводя к возникновению
или потере ее. Трпциклич. углеводороды — антрапен
и фенантрен — оказались неактивными, однако 9,10-
диметилантрацен обладает очень слабой активностью.
Из тетрациклин, углеводородов — нафтацен (V),
пирен (VI) и трифенилен (VII) — совершенно псак-
VI» I*
тпвны, 1,2-бепзантрацен (I) и хризен (VIII) очень
слабо активны и только 3,4-бепзфенантреп (IX)
обладает умеренной активностью, вызывая, у 30%
подопытных мышей по истечении года рак кожи при
смачивании 0,3%-пым бензольным р-ром дважды
в неделю. Введение алкильных заместителей в моле-
кулу 1,2-бензантрацена в определенные положения
приводит к появлению активности, в нек-рых случаях
весьма значительной. Одним из самых активных кан-
церогенных соединений являются 9,10-ДИметил-1,2-
бензантрацен (X), который по своей активности
и доступности может быть рекомендован для экспе-
риментальных исследований по раку. Смазывание
кожи мышей 0,1%-ным бензольным р-ром этого ве-
щества приводит к образованию рака у всех живот-
ных, проживших более 3,5 месяцев. Углеводород
вызывает саркому на месте введения под кожу уже
при применении 0,1 мг, тогда как оптимальной дозой
для большинства канцерогенных углеводородов яв-
ляется 5 — 10 мг.
Весьма высокой активностью обладают холантрея
(XI) и 20-метилхолантрен (IV), широко применяемые
в экспериментальной онкологии. При смазывании
0,3%-ным бензольным р-ром 20-метилхолаптрена у
17 мышей из 50 образовался рак кожи, причем первая
опухоль возникла на 49-й день. Из 15 возможных угле-
водородов, содержащих систему из пяти конденсиро-
ванных бензольных ядер, были испытаны 12. Из них
только два обладают канцерогенной активностью —
1,2,5,6-дибензантрацен (II) и 3,4-бензпирен. Первый
углеводород слабо активен, тогда как 3,4-бензплрен
относится к числу наиболее сильных канцероген-
ных соединений. При нанесении па кожу мышам
0,3%-ного бензольного р-ра углеводорода у 24 из 30
возник рак кожи через 5 месяцев.
Вещество Доза, мг Процент заболева- ний сар- комой Средний латентный период, недели Продол- житель- ность ’ опыта
1,2-бензантрацен . . 1 30 23 месяцев
20-метилхолантрен . 1, 2, 5, 6-дибензан- трацен 1 63 16 26 недель
0,02 56 — 22 месяца
3,4-бензпирен .... 9,10-диметил-1,2-бен- 0,15 52 24,4 39 недель
зантрацен • . . . . 2 20 В течение 18 34 недели
* Мыши породы СС 57.
В таблице приведены данные по испытанию канцерогенной
активности углеводородов путем инъекции под кожу соответ-
ствующего соединения на мышах одной породы — С 57 black (для
первых четырех соединений). Для 9,10-диметил-1,2-бензантра-
цена приведены данные по испытанию на мышах СС 57, устой-
чивых к воздействию канцерогенных соединений, но в сопоста-
вимой дозе. Не для всех опубликованных случаев имеются
данные о среднем латентном периоде. Среди исследований по
канцерогенной активности указанных углеводородов нет
таких, к-рые позволили бы составить четкую таблицу актив-
ности, хотя эти углеводороды изучались многими авторами
(405 статей по 20-метилхолантрену, 75 — по 1,2,5,6-дибенз-
антрацену, 125 — по 3,4-бензпирену, 157 — по 9,10-диме-
ти л-1,2-бензантрацену).
Из исследованных 6-, 7- и 8-циклич. конденсирован- •
яых углеводородов ни один не оказался активным.
Канцерогенной активностью обладают пек-рые сте-
роидные соединения. Невысокую активность прояв-
ляет дсзокспхолевая к-та. Бензоат эстрона, дппрэ-
401
КАНЦЕРОЛИТИЧЕСКИЕ ВЕЩЕСТВА
402
ппонат и бензоат эстрадиола, прогестерон и нек-рые
другие соединения при общей дозе в несколько мил-
лиграммов вызывают у мышей рак сосков.
Способностью вызывать злокачественные опухоли
обладают многие многоядерные гетероциклич. сое-
динения. К их числу относятся 1, 2, 5, 6-дибензакри-
дин (XII), 5,9-диметил-3,4-беизакридин (XIII), 1,2,5,
6-дибевзкарбазол (XIV), 4,9-диметил-5,6-бензтиофан-
трен (XV). Нек-рые из них очень активны.
Канцерогенной активностью обладают многие бис-
Р-хлорэтилампны при испытании методом инъекции.
Среди этой группы соединений особенно активен
гидрохлорид метил-ди-(2-хлорэтил)-амина (э м б и-
х и и), вызывающий опухоли у всех подопытных
мышей при дозе в 0,1 мг в течение 16 недель. Актив-
ны также различные производные этилеиимина и
Р, Р'-дихлорэтилсульфид (иприт).
Специфич. способность вызывать опухоли печени
проявляют нек-рые азокрасители, напр. о-амино-
азотолуол, прибавляемый к пище в количестве 0,1%,
приводит к образованию опухолей печени у 25 крыс
из 160 в течение 196 дней. Активны также п-диметил-
аминоазобевзол, N-метил-п-аминоазобензол, 4-метил-
4'-диметиламиноазобензол.
Лит.: Михайлов Б. М., Изв. АН СССР. Отд. хим.
наук, 1946, № 6, 619; Ш а б а д Л. М., Очерки эксперимен-
тальной онкологии, М., 1947; Badger G. М., Brit. J.
Cancer, 1956, 10, № 2, 330; Survey of compounds which have
been tested for carcinogenic activity, Wash., 1957.
Б. M. Михайлов.
КАНЦЕРОЛИТИЧЕСКИЕ ВЕЩЕСТВА — со-
единения, способные тормозить рост и размножение
злокачественных клеток и в конечном счете вести
к их ликвидации в организме. Канцеролитич. актив-
ность обнаружена у большой группы веществ природ-
ного происхождения, у антибиотиков, гормонов и
различных соединений, обладающих алкилирующими
свойствами.
За последние 10—15 лет на животных была испы-
тана противоопухолевая активность десятков тысяч
веществ. Многие из них, оказавшиеся активными
в эксперименте на животных, при проверке в клинике
на человеке дали отрицательные результаты.
Наиболее обстоятельно исследованы азотные ана-
логи иприта (I), явившиеся родоначальниками не-
скольких' групп родственных противоопухолевых со-
единений, обладающих алкилирующими свойствами;
к таким соединениям относятся производные этиле-
нпмина (II), эпоксидов (III), эфиров сульфокис-
лот (IV) и др.
R-N(CH2CH2C1)2 (I)
/СН2\ \
, | >N РО (II)
\СН2Х /з
R' R"
I I
R-CH-C--С--СН—R"' (III)
R—SO2—О-(СН.,)П-О-SO2-R
(IV)
Установлено, что высокая биологич. активность
алкилирующих соединений связана прежде всего с их
способностью алкилировать различные нуклеофильные
центры белка и нуклеиновых к-т (ОН, SH, NH,,
СООН и др. группы).
Азотные аналоги иприта. Одним
из первых препаратов, применявшихся в клинике,
является хлоргидрат метил-бис-([5-хлорэтил)амипа
(эмбихина—V), синтезированный еще в 1935.
Основным недостатком эмбихина и других простей-
ших хлорэтиламинов (новоэмбихин — VI, трихлортри-
этиламин — VII и др.) является высокая токсичность:
CH3N(CH2CH2C1)2-HC1 (V)
CH3CHC1CH2N(CH2CH2C1)2.HC1 (VI)
N(CH2CH2C1)3-HC1 (VII)
Действие этих веществ слабо дифференцировано по
отношению к раковым и нормальным клеткам, след-
ствием чего при их применении, как правило, является
поражение клеток костного мозга. Действующие дозы
препаратов близки к токсическим; препараты эффек-
тивны гл. обр. при лечении злокачественных заболе-
ваний кровэтворной системы и малоактивны при ле-
чении истинных опухолей. Японскими учеными для
лечения злокачественных образований предложена
N-окись эмбихина (нитромин)
CH3N(CH2CH2C1)2 • НС1
1 (VIII)
о
Терапевтич. индекс нитромина (отношение максималь-
но переносимой дозы к минимально эффективной)
в 40 раз выше, чем у эмбихина, а токсичность в 10 раз
меньше при большей терапевтич. активности. Высокую
противоопухолевую активность при лечении нек-рых
злокачественных заболеваний человека показал «эндок-
сан» (IX):
.nh-ch2.
(C1CH2CH2)2N-P< >СН2 (IX)
|| \о--СН2/
о
Препарат хорошо переносится больными, но оказы-
вает побочное действие на костный мозг.
В 1953 в Англии была синтезирована серия жирно-
ароматич. кислот, содержащих хлорэтиламиногруп-
пы. Из них п-ди-(2-хлорэтил)аминофенилмасляная
к-та (хлорамбуцил, СВ 1348, лейкеран) (X, п = 3)
оказалась наиболее активной. Высокую противоопу-
холевую активность на животных показали также ами-
нокислоты (XI), ацилированные хлорамбуцилом.
n-(ClCH2CH2)2N-CeH4-(CH2)nCOOH (X)
n-(ClCH2CH2N)2N—CeH7-(CH2)3CONHCH(R)COOR' (XI)
Близкую к хлорамбуцилу противоопухолевую ак-
тивность в эксперименте на животных имеет синтези-
рованный в 1960 диэтиловый эфир ;г-ди(2-хлорэтил)-
аминобензилфосфиновой к-ты (XII)
(ClCH2CH2)2N-CeH4-CH2PO(OC2H5)2 (XII)
Мутагенная ди-(2-хлорэтил)аминогруппа была введена-
в ряд природных соединений, таких как сахара, ами-
нокислоты, гетероциклические соединения. Из синте-
зированных соединений допан [4-метил-5-ди-(2-хлор-
этил)-аминоурацил] и «дегранол» [1,6-ди-(2-хлорэтил-
амино)-1,6-ди-дезокси-(1-маннит, дихлоргидрат] ис-
пользуются в клинике.
В 1954 одновременно в СССР и Англии был синтези-
рован п-ди[2-хлорэтиламино-фенилаланин], DL-фор-
ма — сарколивин] L-форма — мелфалан (XIII)], оказав-
шийся активным по отношению к нек-рым истинным
опухолям человека.
n-(C10H2CH2)»N-CeH4-CH2CH-C00H (XIII)
I
nh2
Орто-изомер сарколизина имеет в эксперименте на
животных более высокую противоопухолевую актив-
403
КАНЦЕРОЛИТИЧЕСКИЕ ВЕЩЕСТВА
404
ность. Высокую противоопухолевую активность (на
животных) показали пептиды (XIV), содержащие сар-
колизин; в-отличие от самого сарколизина, они зна-
чительно менее токсичны
n-(ClCH2CH2)2N-CeH4-CH2CHCONHCHCOOC2H5 (XIV)
I I
NHCOR R
Производные этиленимипа. Из боль-
шого числа противоопухолевых препаратов, содержа-
щих этилениминовые циклы, в клинике используются
лишь нек-рые: 2,4, б-триэтиленимино-Э-триазин (тре-
тамин, триамелин, ТЭТ — XV), триэтиленимид фос-
форной к-ты (ТЭФ — XVIa), триэтиленимид тиофосфор-
ной к-ты (ТиоТЭФ — XVIb) диэтиленимино-N,N-ди-
этилфосфорамид (XVII), дипин (XVIII) и др.
xvi
(а и в)
/СН2 \ О
. xvill | \ I P-N(C2l<5)2
\сн2 /г
СН2у \
| 4N Р=О (S)
сн2
Наибольшее клиническое применение получил
ТиоТЭФ, но и его основным недостатком является
действие на кровотворение.
Введение этилениминовых групп в хиноны привело
к высокоэффективным канцеролитическим соединени-
ям. Из них 2,5-дипропокси-3,6-диэтилениминобензо-
хинон-1,4 (XIX), 2,3,5-триэтиленимино-1,4-бензохинон
(XX), 2,5-диацетамидо-3,6-диэтиленимино-1,4-бензохи-
нон (XXI) и некоторые др. используют в клинике.
Си г»
Этилениминохиноны, как правило, являются токсич-
ными соединениями.
Эфиры сульфокислот. Препаратом из
группы эфиров сульфокислот, вошедшим в клинич.
практику, является 1,4-ди (метансульфонилокси) бу-
тан (милеран, миелосан) ch3-so2—о—(Сн2)4—о—so2ch3.
Вещества растительного происхо-
ждения. Из группы цитотоксич. соединений рас-
тительного происхождения для лечения рака кожи при-
меняется N-метилдезацетилколхицин (омаин, XXII)
Анти метаболиты. Сюда относятся антаго-
нисты различных метаболитов, как дезоксипиридок-
син (XXIII), антагонист пиридоксина—витамина Вй,
изорибофлавин (XXIV) и галактофлавин (антагонисты
рибофлавина), различные антагонисты никотинамида,
такие как 2-этиламино-1,3,4-тиадиазол (XXV) и
6-аминоникотинамид (XXVI), подавляющие различ-
ные формы экспериментальных опухолей у животных,
и др.
XXIII
В клинике используют противоопухолевые антимета-
болиты, блокирующие на различных стадиях синтез
нуклеиновых кислот. Это антагонисты фолиевой к-ты
(птероилглутаминовой), особенно 4-аминоптероилглу-
таминовая к-та (аминоптерин—XXVII) и 4-амино-
N-метилптероилглутаминовая к-та (амотоптерин —•
XXVIII), вызывающие рэмиссии при острых лейкозах
R r- R" ___ соон
VcONHCH(CH2)2COOH
nhAnAn^J -
Фолиевая кислота г р=ОН! R'=R"=H
XXVII R=NH2t R-R"=H
XXVIII R=NH2; R'=H: R"=CH3
у детей. Другие антагонисты фолиевой к-ты (теропте-
рин, А-деноптерин, аминотероптерин, аминоанфоль),
несмотря на высокую противоопухолевую активность,
не получили широкого клинич. применения вследствие
высокой токсичности. Недостатком антифолиевых
препаратов является постепенно возникающая рези-
стентность злокачественных клеток к их действию.
Имеется большая группа противоопухолевых препара-
тов, содержащих пиримидиновый цикл, таких как
5-фторпиримидины (XXIX) и их нуклеозиды, из к-рых
5-фторурацил (XXX) оказался наиболее активным
соединением.
XXIX
XXXI
SH
Н
Другие производные урацила: 6-метилсульфонил-
урацил, 5-оксиуридин, 2-тиоурацил, 6-азаурацил и др.
при клинич. испытаниях оказались малоэффектив-
ными. Из группы противоопухолевых соединений,
являющихся антагонистами пурина, в медицинской
практике для лечения острых лейкозов используют
гл. обр. 6-меркаптопурин (XXXI). Лечебный эффект
6-меркаптопурина был подтвержден в различных кли-
никах.Однако,как и в случае антифолиевых препаратов,
постепенно развивается устойчивость организма к дей-
ствию 6-меркаптопурина. Близкую к 6-меркаптопури-
ну противоопухолевую активность имеют б-(метилтио)-
пурин (XXXII, R = CH3), 6-бензилтиопурин (XXXII,
R=CH2CeH5) и 6-меркапто-9-(1-О-рибофуранозилпу-
рин (XXXIII).
Сам пурин, 6-метилпурин, 6-гидразинопурин и
2,6-диаминопурин имеют незначительную противоопу-
405
КАНЦЕРОЛИТИЧЕСКИЕ ВЕЩЕСТВА— КАПЕЛЬНЫЙ АНАЛИЗ
406
холевую активность. Производные аденина: 6-амино-
2-меркапто пурин, 2-(метилтио)-аденин и изогуанин —
активны лишь в максимально переносимых дозах.
Противоопухолевая активность 6-хлорпурина, 6-тио-
гуанина и 6-тиогуанозина не превосходит активности
6-меркаптопурина.
Антибиотики. Известные в настоящее время
противоопухолевые антибиотики, как правило, дей-
ствуют на экспериментальные опухоли животных и
только в отдельных'случаях—на опухоли человека. От-
личительной особенностью почти всех противоопухоле-
вых антибиотиков является их высокая токсичность.
Известна группа противоопухолевых антибиотиков,
являющихся продуктами жизнедеятельности различ-
ных актиномицетов. Это — актиномицины А, В, С, D,
F, I, К и X. Большинство из них не являются
индивидуальными химическими соединениями, а
представляют собой смеси близких по структуре ве-
ществ. Актиномицины С и D используются в клинике.
Японскими учеными выделено и испытано неск.
десятков противоопухолевых антибиотиков. Описаны
митомицин С, плюрамицин и другие, среди к-рых мито-
мицин С дал хорошие результаты при испытании в
клинике на лейкозах. Другие антибиотики не были
широко исследованы в клинике вследствие своей вы-
сокой токсичности. Малоэффективным в отношении
опухолей человека оказался
сн2=с—сн—соон также саркомицин (2-мети-
I I лен-3 - кетоциклопентанкарбо-
xxxiv ?°/Н2 новая к-та — XXXIV).
сн3 В Советском Союзе выде-
лены противоопухолевые ан-
тибиотики аурантин, актиноксаптин, фумагиллин и
неоцид — антибиотик бактериального происхождения.
Гормоны. Исторически первым направлени-
ем в химиотерапии злокачественных новообразо-
ваний является гор-
XXXV —Сн—мональное направле-
HO-L J С2н5 C,h5LJ-oh ние- Успешно исполь-
" зуется в клинике груп-
па женских половых
гормонов, таких как
эстрадиол, синэстрол
(XXXV), диатилстиль-
бастрол (XXXVI), ди-
фосфат диэтилстильбэстрола («хонваш — XXXVII)
и др. эстрогены. Сам хонван не обладает противо-
с2н5
xxxvn (НО)2р_о_Ц^!1 с2н5 Ч^Д-о-рюн^
о о
опухолевой активностью, но, попадая в опухолевую
клетку, гидролизуется, образуя активный диэтилстиль-
бэстрол.
Из мужских половых гормонов используются тесто-
стерон (XXXVIII), мепгилтестостерон и нек-рые их
производные.
"• ОН Наряду с половыми гор-
JJsCi монами применяется группа
н [ j кортикостероидов. Это кор-
xxxviii 1 тивон и его производные:
| Т | дигпдрокортизон, предни-
зон, преднизолон, фторгид-
рокортизон И др.
Однако рэмиссии, вызываемые кортикостероидами,
непродолжительны. Вследствие развивающейся устой-
чивости организма к действию этих препаратов, по-
степенно они теряют свою противоопухолевую актив-
ность. Наряду с кортизоном в случае острых лейкозов
используется также адренокортикотропный гормон.
Гормонотерапия не является индивидуальным мето-
дом лечения злокачественных заболеваний, а ис-
пользуется обычно параллельно с другими методами.
Лит.: Ф а р бе р С. (и др.1, в кн.: Успехи в изучении рака,
пер. с англ., т. 4, М., 1958, с. 11—84; Сток Ч., там же, т. 2,
М., 1956, с. 439—510; Чернов В. А., Мед. пром-сть СССР,
1959, № 4, 17; его же, там же, 1959, №5; Ларионов
Л. Ф., Хим. наука и пром-сть, 1956; 1, № 4, 433; его же,
Вести. АМН СССР, 1958, М2, 8; Вермель Е. М., Совре-
менные проблемы онкологии, 1956, № 4 (73), 3; Кнунянц
И. Л., Голубева Н. Е., Кильдишева О. В.,
Успехи соврем, биол., 1960, 50, вып. 2 (5), 167; П е р е в о д-
чикова Н. И., Лекарственные методы лечения злока-
чественных опухолей, М., 1961; S t о с k J. А., в кн.: Clba Foun-
dation Symposium on amino acids and peptides with antlme-
tabollc activity, L., 1958, p. 89—103; Annals of the New York
Academy of Sciences, 1958, 68, art. 3; Unio internat. contra
cancrum, Acta 1959, 15, № 1, bis № 1. H. E. Голубева.
КАОЛИН — тонкодисперсная пластичная порода,
продукт выветривания полевых шпатов, слюд, грани-
тов и др. горных пород; состоит в основном из мине-
рала каолинита — гидросиликата алюминия
А12О3 2SiO2 . 2Н2О — и различных примесей (кварца,
полевого шпата, слюды и др.). К. — мелкие гидро-
фильные частицы пластинчатого строения, .залегают
в виде пластов, линз и пр.; делится на первичные и
вторичные. Первые образовались на месте выветри-
вания и содержат до 45—60% кварца, вторые — пере-
отложенные первичные К.; во вторичных К. кварц
и остатки др. минералов практически отсутствуют.
Содержание частиц разного размера в одном из луч-
ших советских К. (Просяновском):
Размер частиц, леи Содержа- ние, %
0,25 -0,05 0,28
0,05-0,01 12,28
0,01 —0,005 33,2
0,005—0.001 19,56
Менее 0,001 34,68
Химический состав этого
К. (вес. %): 65—69,7 S1O2,
0-0,4 Т1О2, 21,7—26,4
А1ц0)з, 0,8—1 Fe2Oa, 0,4—
0,7 СаО, 0,1—0,3 MgO,
0,27—0,83 К2О, 0,03—0,21
Na2O; потери при прока-
ливании 4,9—7,9.
Чистый каолинит — частицы белого цвета, твердость
по Моосу 1,2, плотн. 2,5—2,6, обычно образует
хорошо выраженные пластинчатые гексагональные
кристаллы с четкими гранями, углы между к-рыми
равны 120°, толшина частиц пластинок 0,1—3 мк. Ки-
слоты, за исключением плавиковой, при обычной
темп-ре незначительно действуют на каолинит; при
Кипячении с конц. H2SO4 он разрушается с образова-
нием A12(SO4)3 и кремниевой к-ты. При сплавлении
со щелочью или содой каолинит разрушается пол-
ностью; в интервале 430—550° из каолинита удаляется
большая часть химически связанной воды, остатки
ее удаляются при 900—1000°. Для удаления из К.
различных примесей его, как правило, подвергают
обогащению, обычно мокрому (отмучивание), заклю-
чающемуся в пропускании водно-каолиновой суспен-
зии (с добавкой пептизатора) через систему отстой-
ников, где примеси осаждаются, а суспензию К. под-
вергают коагуляции известковым молоком или хлори-
дом кальция и обезвоживают на фильтрпрессах.
Огнеупорность каолина ок. 1750°. К. применяют
гл. обр. в керамич. пром-сти (огнеупорная, фарфоро-
вая, фаянсовая), а также в бумажной, резиновой и др.
Лит.: Самойлов В. Ф., Мельников И. И.,
Каолин, вып. 2, М., 1951; Торопов Н. А., Пула к
Л. Н., Курс минералогии и петрографии с основами геологии,
М., 1953; К у к о л е в Г. В., Химия кремния и физическая
химия силикатов, М., 1951. Н. М. Павлушкин.
КАПЕЛЬНЫЙ АНАЛИЗ — качественный или по-
луколичественный химич. анализ, в к-ром исследуе-
мый р-р и реагенты берут в количестве нескольких
капель; реакцию выполняют на фильтровальной бумаге
или капельной пластинке, реже — в микропробирке.
По количеству применяемых для анализа материалов
и реагентов К. а. занимает промежуточное положение
между классич. пробирочным анализом и микроанали-
зом с применением микроскопа. Вследствие скорости
и удобства работы, высокой чувствительности и изба-
407
КАПЕЛЬНЫЙ АНАЛИЗ
408
рательностп К. а. широко применяется для идентифи-
кации различных неорганич. и органич. веществ и
для контроля их чистоты, для быстрого полевого
анализа руд и минералов, для различных видов тех-
пич. и биохимич. анализа, при исследовательских
работах и для многих других целей. Отдельные реак-
ции К. а. эпизодически использовались давно, по
широкое распространение К. а. получил только
с 20-х гг. 20 в., после того как Н. А. Тананаев
и Ф. Файгль опубликовали результаты своих фунда-
ментальных исследований по К. а.
Техника работы в К. а. заключается в следующем. На ку-
сочек фильтровальной бумаги или на фарфоровую пластинку
с небольшими углублениями помещают каплю исследуемого
р-ра. Если применяют зернышко твердого препарата, то его
вначале растворяют в углублении капельной пластинки или
в микропробирке в подходящем реагенте. Далее прибавляют
по капле растворы требующихся реагентов и наблюдают ре-
зультат. На фильтровальной бумаге образуются окрашенные
пятна, на капельной пластинке — окрашенные р-ры или осад-
ки. В К. а. широко применяются бумаги реактивные; т. к.
в такую бумагу реагент уже введен, то техника выполнения
реакции часто сводится только к помещению на реактивную бу-
магу капли анализируемого р-ра и наблюдению появляющегося
эффекта. В К. а., в частности в электрография, методе, при-
меняют также желатинированную бумагу. Иногда в К. а.
прибегают к нагреванию. Незначительное нагревание можно
выполнить и на фильтровальной бумаге, держа ее над пламенем
микрогорелки.
Реже реакции К. а. выполняются в микропробирках или
в мпкротиглях. В этом случае можно экстрагировать, нагре-
вать (уже до значительных темп-р), прокаливать, сплавлять
и выполнять другие операции обычного пробирочного анализа.
Такие реакции отличаются от соответствующих реакций про-
бирочного анализа только масштабом, что, однако, приводит
к экономии реагентов и времени.
Приборы и посуда, используемые в К. а., отличаются край-
ней простотой. Помимо небольших кусочков фильтровальной
бумаги, широко применяют стеклянные капилляры для пере-
несения капель анализируемого р-ра и реактивов, тонкие
стеклянные палочки для перемешивания, фарфоровые пла-
стинки с углублениями для выполнения реакций, реже —
микропробирки. Нагревание или выпаривание производят
на микрогорелке.
Применяемые в К. а. растворы реагентов хранят в малень-
ких склянках, пробки к-рых снабжены капиллярами. В К. а.
широко используются газообразные реагенты: аммиак, бром,
пары кислот. Для обработки такими реагентами полученное
па фильтровальной бумаге пятно держат несколько секунд
над склянкой с концентрированным аммиаком, соляной к-той
или бромной водой.
К. а. применяется гл. обр. для качественного откры-
тия ионов или отдельных веществ. Путем сравнения
интенсивности окраски пятен па фильтровальной бу-
маге со стандартом можно выполнить и полуколиче-
ствепные определения («капельная ко по р и-
м е т р и я»). Этот прием широко используется для
быстрой маркировки сплавов и даже для быстрого их
анализа (см. Весстружковый метод анализа). Выпол-
нение колич. определений облегчают, ограничивая
площадь растекания помещаемых на бумагу капель.
Для этого на бумаге расплавленным парафином печа-
тают кольца с определенной внутренней поверх-
ностью, обычно 1 см2 (метод Ягоды). Внутрь этого
кольца вносят капли растворов и реагенты.
Использование в К. а. фильтровальной бумаги
для выполнения реакций не только обеспечивает удоб-
ство и простоту работы, по и устраняет трудности,
связанные с созданием точных значений pH. Доста-
точно нанести на фильтровальную бумагу рядом капли
кислоты и подкисляемого р-ра, чтобы в зоне их впиты-
вания образовались места с любым промежуточным
значением pH. В месте с подходящим значением pH
произойдет нужная реакция. Аналогично достигаются
необходимые соотношения между реагентом и анали-
зируемым р-ром. Если анализируемый раствор содер-
жит осадок, его не требуется отфильтровывать. На
фильтровальную бумагу наносят каплю раствора с
осадком и ждут, пока капля впитается. После этого на
зону, лишенную осадка, наносят реагент. При необ-
ходимости более тщательного фильтрования каплю
раствора с осадком наносят на сложенные вместе
2—3 слоя бумаги и реакцию выполняют на втором
или третьем слое. При выполнении реакций на филь-
тровальной бумаге в какой-то мере происходит разделе-
ние растворенных веществ, хотя и не столь полное,
как в хроматографии на бумаге. Малый расход реа-
гентов позволяет применять в К. а. дорогие или мало-
доступные реагенты.
В неорганич. К. а. существуют варианты система-
тич. хода анализа, аналогичные ходу обычного проби-
рочного анализа. Для К. а. характерно широкое при-
менение реакций, позволяющих обнаруживать опреде-
ляемый элемент непосредственно в присутствии мно-
гих других элементов, без их отделения. Для этой
цели применяют высокочувствительные реагенты,
преим. органические, с высокой избирательностью
действия. Отдельные реакции являются даже специ-
фическими. Высокая избирательность многих приме-
няемых в К. а. органич. реагентов объясняется нали-
чием в их молекулах т. наз. характерных атомных
группировок, к-рые раньше иногда назывались спе-
цифическими. Реагенты, имеющие такие группировки,
при подходящих значениях pH, с одними элементами
реагируют, а с другими нет. При других значениях
pH число реагирующих элементов может быть иным
(подробнее см. Органические реагенты).
Перечень нек-рых часто применяемых в неорганическом
К. а. реагентов с указанием элементов, для к-рых разработаны
ценные реакции их обнаружения при использовании этих
реагентов, приведен в таблице.
Реагенты Элементы Реагенты Элементы
Ализарин, ализа- рин S Антразо Арсеназо .... Бензидин .... Бензидин+молиб- дат аммония. . Бензоипоксим . . Д иметилглио к сим Дипикриламин . n-Д имети ламипо- бензилиденро- данин а, а'-Дипиридил . Дитизон Дпфенилкарбазид Иодид калия . . Метиловый фио- летовый .... Al, In, Zr IV Sn, Те, Pb U, Th, лан- тан иды и др, Au, Си, Сг, Мп, Т1 Ge, Р Си Ni, Fe, Pd К, Rb, Cs Ag, Au, Си, Hg, Pd Fe11 Ag, Си, Hg, Pb, Zn и др. Cr, Cd, Hg Bi, TI, Pd Sb, TI, Hg, Zn, W а-Нитрозо-р-наф- тол ₽-Нитрозо-а-наф- тол Родамин В . . . . Роданид калия . Родизонат натрия Рубеановодород- ная кислота . . Тетраметилдиами- нодифенилметан Тиомочевина . . Титановый жел- тый Торон о-Фенантролин . Ферроцианид ка- лия Хинализарин . . Хромотроповая кислота .... Этилксантогенат калия Со Zr Sb, Tl,Zn, W Co, Fe, Mo, W Ba, Sr, Pb Co, Cu,Ni Pb, Mn Bi Mg Th Fe Fe, U Be Ti, U Mo
Используемые в К. а. реакции характеризуются
весьма высокой чувствительностью, обусловленной
применяемыми реагентами и особенностями капель-
ного выполнения реакций. На фильтровальной бумаге
продукты реакций часто оказываются локализован-
ными на небольшой поверхности и поэтому даже мини-
мальные их количества хорошо заметны. Для уменьше-
ния поверхности окрашенных пятен реакцию иногда
выполняют па острых кончиках бумажных полосок.
Чувствительность капельных реакций характери-
зуют открываемым минимумом или предельным
разбавлением (D). Открываемый минимум
обычно составляет 0,1—0,01 мкг в неорганич. К. а.
и 1—0,1 мкг в органическом. Предельное разбавление,
выражаемое в частях раствора, в к-ром присутствует
1 часть открываемого соединения, может варьировать
в очень широких пределах, доходя до 1 : 10е—
1 : 10е. В неорганич. К. а. открываемый минимум и
предельное разбавление принято выражать в пересчете
на элемент.
В органич. К. а. разработаны методы качествен-
ного обнаружения С, Н, О, N, S, галогенов и др. эле-
ментов и методы обнаружения многих органич- со-
409
КАПИЛЛЯРНАЯ КОНДЕНСАЦИЯ
410
единений по заключающимся в их молекулах функ-
циональным группам: ОН, СНО, СООН, NO, NO2,
NH2 и др.
Для соединений почти всех классов существуют
удобные, чувствительные и избирательные капельные
реакции. Известны также высокоизбирательные реак-
ции идентификации различных органич. соединений.
Длн улучшения избирательности здесь иногда при-
бегают к комбинации нескольких реакций или к их
комбинации с простыми методами отделения.
К. а. широко используется для решения чрезвы-
чайно разнообразных задач.
Лит.: Тананаев Н. А., Капельный метод, 6 изд.,
М.—Л., 1954; Ф а й г л ь Ф., Капельный анализ. Теорети-
ческие основания и практическое выполнение, пер. с нем.,
М„ 1937; его же, Капельный анализ органических
веществ, пер. с англ., М., 1962; Реакции и реактивы для ка-
чественного анализа неорганических соединений, пер. с франц.,
М.—Л., 1950; КульбергЛ. М., А л ь т е р з о н Г. С.,
Вельтман Р. П., Капельный анализ, М.—Л., 1951;
Тананаев Н. А., Бесстружковый метод, Свердловск —
М., 1958; Feigl F. [а. о.], Spot tests in'inorganic analysis,
Amst., 1958; J a g о d a H., Mikrochemie, 1938, 24, 117; его
же, Industr. and Engng Chem., 1937, 9, M 2, 79.
В. И. Кузнецов.
КАПИЛЛЯРНАЯ КОНДЕНСАЦИЯ — конденса-
ция пара в жидкость в порах при давлениях р,
меньших нормального давления насыщенного пара
ps', происходит наряду с адсорбцией и завершает сорб-
цию в пористых телах. В случае тонкопористых адсор-
бентов (молекулярные сита, тонкопористые силика-
гели, алюмогели, активные угли, цеолиты и т. п.) поры
заполняются сильно адсорбирующимся веществом уже
при низких относительных давлениях пара р!рз-
У крупнопористых адсорбентов (силикагели, алюмо-
силикагели и др.) при адсорбции на стенках пор об-
разуется адсорбционная пленка (моно- и полимолеку-
лярная), поверхность к-рой в сужениях пор вогнута.
Давление насыщенного пара над вогнутыми менисками
жидкости меньше ps, поэтому над вогнутыми поверх-
ностями адсорбционной пленки в порах конденсация
пара в жидкость начинается при р/ра <. 1.
Условие механич. равновесия поверхности раздела:
фаза I (пар) — фаза 2 (жидкость)
— P'dv'— P"dv" -|- ads' = 0 (1)
где Р' и Р" — гидростатич. давление в фазах 1 и 2,
v' и о" — объемы этих фаз, о — межфазное (поверх-
ностное) натяжение и s' — поверхность раздела фаз
Т-. к. в замкнутой системе
dv” =— dv', a ds' = s’ (l/pt + l/p2) dn
где pi и p2 — главные радиусы кривизны и dn —
смещение по нормали к поверхности s', то из (1) сле-
дует основное ур-ние теории капиллярности
Р'-/’" = <’(1/р1 + 1/р8) (2)
причем центры кривизны лежат внутри фазы 1 (пара).
Гидростатич. давление Р связано с химическим потен-
циалом р. ур-нием:
др'дР = v (3)
где v — мольный объем в соответствующей фазе. При
равновесии р.' = р" и dfi' = dy." — RTdlnp, где
R — газовая постоянная, Т — абс. темп-ра, ар —
давление пара. Отсюда с учетом (2)
_ va (1/Р1 + 1/Ра)
р^р3е RT (4)
где v — мольный объем жидкости, ps — давление
пара над вогнутой поверхностью. Это ур-ние Томсона
(Кельвина) дает зависимость р над поверхностью во-
гнутого мениска от его кривизны, показывая, что
с ростом кривизны мениска в тонких капиллярах р
уменьшается, для выпуклой же поверхности—нао-
борот. Для шаровидного мениска рх = рг — r!2 cos 0,
а при полном смачивании pt — р2 — г/2, где 0 —
угол смачивания, г — радиус круглого капилляра,
поэтому формула Томсона для такого мениска дает:
__ 2va
гшЯТ
Рш = Pse (5>
где гш — радиус мениска. Для цилиндрич. мениска
кривизна вдоль оси цилиндра равна нулю, поэтому
Рц = Лв
вс
ГцНТ
(6)
Т. обр., давление пара над цилиндрич. мениском
радиуса гц понижается меньше (в показателе степени
нет двойки), чем над шаровым мениском. С этим свя-
зано явление капиллярно-конденса-
ционного гистерезиса.
При К. к. в порах воронкообразной формы (рис. 1, а)
в результате адсорбции на стенках пор образуются
пленки с вогнутой поверхностью. В наиболее узкой
части поры образуется ша-
ровидный мениск радиуса
гш. Когда давление пара над
этой поверхностью р дости-
_________________ 2га .
гает значения pse rnjRT,
пар станет относительно
этой поверхности насыщен-
ным, и начинается К. к.
Эта К. к. приводит к увели-
чению гш, а, следовательно,
для ее продолжения надо
увеличивать р (рис. 1, б).
При десорбции процесс пой-
дет тем же путем в обратном
направлении. Если пора
цилиндр ической
ф о р м ы с одного конца
закрыта (рис. 1, в), то
Рис. 1. Простейшие формы пор
и сорбционных пленок (слева)
и соответствующие изотермы
сорбции и десорбции (справа);
а и б — для воронкообразной поры, «иг — для круглой ци-
линдрической поры с закрытым концом, д и е — для круглой
цилиндрической поры с открытыми концами.
у этого конца образуется шаровидный мениск. Здесь
при К. к. лшне изменяется, р— тоже; изотерма К. к.
будет вертикальной (рис. 1, г). Десорбция будет про-
исходить тем же путем. Если же цилиндрич. пора
открыта с обоих концов (рис. 1, д), шаровидный
мениск при адсорбции не образуется, и К. к. начи-
нается на вогнутом цилиндрич. мениске пленки, по-
крывающей стенки капилляра, при давлении пара
_ га
ри = psti Уирт. К. к. в этом случае приводитк увели-
чению толщины пленки, т. е. к уменьшению г , по-
этому при давлении р весь капилляр сразу заполнится
жидкостью. Изотерма в адсорбционном направлении
будет вертикальной при р = рц(см. рис. 1, е). После
заполнения капилляра на его концах образуются
шаровидные мениски, соответствующие рш = рц, т. е.
с той же кривизной, что и цилиндрич. мениск, следо-
вательно, с радиусом гш= 2гц. При дальнейшем повы-
шении р от рц до ps кривизна этих менисков будет
уменьшаться до нуля. При десорбции вначале про-
цесс пойдет обратимо: в устья капилляра будут вдав-
ливаться шаровидные мениски растущей кривизны.
411
КАПИЛЛЯРНАЯ КОНДЕНСАЦИЯ — КАПИЛЛЯРНЫЕ ЯВЛЕНИЯ
412
Однако при р = рц эти мениски прорваться еще не
могут, поэтому капилляр еще останется заполненным,
десорбционная ветвь разойдется с адсорбционной, и
радиус шаровидного мениска будет продолжать умень-
2га
шаться. Только при р = рш = pse гшкт радиус ша-
ровидного мениска сделается равным радиусу адсорб-
ционной пленки в цилиндре, мениски прорвутся и
вся капиллярно-сконденсированная жидкость испа-
рится. Т. обр., десорбционная ветвь вертикально опу-
• стится до обратимой изотермы полимолекулярной
адсорбции при рш < ра, т. е. на сорбционно-десорб-
ционной изотерме получается петля гистере-
зиса (рис. 1, е).
В адсорбентах глобулярной структуры поры имеют
сложную форму с сужениями у мест контактов глобул,
расширениями в средней чцсти и
горлами — «окнами», ведущими
из одной «элементарной поры»
в другую. На рисунке 2 показа-
на схема такой поры при чи-
сле касаний одинаковых глобул,
равном 6. В клиновидных ча-
стях поры К. к. начинается об-
ратимо, как в случае воронко-
образных пор (рис. 1, а и 1, б).
Однако продвигающиеся к гор-
лам такой поры мениски при
их слиянии друг с другом об-
разуют в этих горлах мениски
кольцеобразной формы. Эти гор-
ла вместе с более широкими про-
странствами между глобулами при
_ ‘а
р, близком к >рц= pse rnItT, где гц—радиус кольце-
образного (приблизительно цилиндрич.) мениска
в горле, заполняются скачком, подобно цилиндрич.
поре с открытыми концами (рис. 1, д и 1, е). При десорб-
ции в горлах пор между глобулами образуются мениски
шаровидной формы, к-рые прорываются лишь при
2га
Рис. 2. Схема поры,
Образованной пра-
вильной упаковкой
шаровидных частиц
равных размеров с
числом касаний 6.
давлении рш — pse rmRT, т. е. при рш < рц. Это при-
водит к возникновению петли гистерезиса. Реальные
адсорбенты не обладают столь однородной структурой,
поэтому их поры заполняются (или опоражниваются)
Рис. 3, а — изотерма адсорбции и
десорбции пара однороднокрупно-
пористым силикагелем; б — соответ-
ствующая зависимость объема пор
от эффективного радиуса их горл;
в — кривая распределения пор по
эффективным радиусам их горл.
не одновременно, что
выражается в накло-
не ветвей петли гис-
терезиса.
На риС. 3, а пока-
зана изотерма адсорб-
ции пара бензола в
порах довольно одно-
роднокрупнопористо-
го силикагеля.
При сорбции обра-
зуются шаровидные
мениски (рис. 1, айв)
и цилиндрич. (рис. 1,
д), а при десорбции —
только шаровидные
(рис. 1, а, б и д). По-
этому десорбционную
ветвь изотермы используют для определения эффек-
тивных размеров пор, т. е. размеров, эквивалент-
ных круглым цилиндрич. порам. Каждая точка
изотермы дает значения адсорбции а и plps. Умно-
жая а на V, находят заполненный жидкостью
объем пор w, а подставляя в формулу Томсона (4)
соответствующие значения р!р&, находят эффективный
радиус шаровидного мениска в поре гш. Радиус поры
г находят, прибавляя к гш толщину адсорбционного
слоя т, к-рую определяют при том же р/р3 из изотермы
абсолютных (отнесенных к единице поверхности)
величин адсорбции, полученной для непористого ад-
сорбента той же природы. Строят графич. зависимость
w от г, называемую структурной кривой адсорбента.
Находя производную dwldr, получают зависимость
dwldr от г, т. е. кривую распределения
объема пор адсорбента по значениям эффективных
радиусов. На рис. 3, б и 3, в показаны структурная
кривая и кривая распределения для силикагеля, полу-
ченные из десорбционной ветви изотермы. Кривая рас-
пределения показывает, что этот силикагель является
довольно однороднопористым. Наиболее вероятные
размеры пор, т. е. размеры горл между глобулами
(см. рис. 2), составляют в этом случае около 100 А.
В действительности К. к. налагается на адсорбцию.
Полимолекулярная адсорбция приводит к сужению
горл пор, так что даже в случае очень рыхлых упаковок
твердых частиц достаточно малых размеров наблю-
дается капиллярно-конденсационный гистерезис. К. к.
обыуно начинается, когда радиус мениска ок. 10—
15 А, в зависимости от размеров молекул адсорбата.
Модифицирование химическое поверхности пористых
адсорбентов влияет гл. обр. лишь на адсорбционную
составляющую суммарного процесса сорбции, если
модифицированная поверхность полностью смачи-
вается, или также и на К. к., если в результате моди-
фицирования смачивание поверхности ухудшается.
По адсорбционно-десорбционной изотерме опреде-
ляют также величину поверхности пленки, на к-рой
происходит К. к. Теплота К. к. больше теплоты обыч-
ной конденсации.
Лит.: БрунауерС., Адсорбция газов и паров, пер. с
англ., М., 1948; Методы исследования структуры высокодис-
персных и пористых тел. Тр. совещания 1951 г. и Тр. совещания
1956 г., М., 1953, М., 1958; ИсирикянА. А., Киселев
А. В., Ж. физ. химии, 1957, 31, Ali 12, 2635; 1960, 34, А8 10,
2146; Аристов Б. Г., Карнаухов А. П., Кисе-
лев А. В., там же, 1962, 36, N 10, Н; Аристов Б. Г.
[и др.], там же, 1962, 36, .Vo 12; Р а д у ш к е в и ч Л. В.,
Изв. АН СССР. Отд. хим. наук, 1958, № 3, 285; АВ 4, 403;
1961, АВ 5,756; АВ 6,984. А. В. Киселев.
КАПИЛЛЯРНЫЕ ЯВЛЕНИЯ — явления, вызы-
ваемые влиянием поверхностного натяжения на обра-
зование, движение и равновесные формы поверхностей
раздела фаз жидкости и пара (двух не смешивающих-
ся жидкостей). В более широком смысле К. я. на-
зывают все поверхностные явления на границах раз-
дела соприкасающихся тел (в поверхностных слоях).
К. я. обусловлены мо-
лекулярными силами
взаимодействия. Они
связаны с искрив-
лением поверхности
жидкости вблизи твер-
дой стенки (см. рису-
нок) вследствие смачи-
вания (вогнутый ме-
ниск) или несмачива-
ния (выпуклый ме-
ниск). На таких ис-
кривленных жидких
поверхностях со сред-
ним радиусом кривиз-
ны г поверхностное Явление капиллярности: а) жид-
натяжение т. е уд- кость смачивает стенки капилляра;
свободная ’ поверхйо- б> жидкостьк не^смач.шает стенок
стная энергия о, вы-
зывает возникновение добавочного давления (по
сравнению с тем, которое испытывает жидкость
с плоской поверхностью) — капиллярного
давления ра = 2 о/r, направленного в сторону
радиуса кривизны. Для выпуклой поверхности ра —
413
КАПИЛЛЯРНЫЙ ЯВЛЕНИЯ — е-КАПРОЛАКТАМ
414
положительно, для вогнутой — отрицательно. Ка-
пиллярное давление — причина ряда важнейших
К. я. Таковы прежде всего явления поднятия и всасы-
вания жидкостей в узкие трубки (капилляры) или
поры со смачиваемыми стенками и опускание жидко-
стей в таких трубках или выталкивание жидкостей из
узких пор в случае несмачивания. Капиллярное
давление в узкой трубке в общем случае промежуточ-
ного смачивания при краевом угле в, определяю-
г0 2тсоь0 г,
щем кривизну мениска г = Ра = ———' Здесь
г0 — радиус трубки (капилляра). В предельном слу-
чае полного смачивания cos S —» -4-1 и р~ = — =
1 * То
=тах, а при полном несмачивании cos S —> — 1>
Р° ~~ го’
К. я. и, в частности, капиллярные равновесия,
напр. равновесная высота поднятия h жидкости
в смачиваемом полностью капилляре, позволяют опре-
делять поверхностное натяжение жидкости па гра-
нице с воздухом (паром). При полном смачивании
высота капиллярного поднятия обратно пропорцио-
нальна радиусу капилляра и не зависит от материала
его стенок. Из условий гидростатич. равновесия при
капиллярном поднятии следует, что произведение
из высоты поднятия на радиус кривизны мениска rh,
а при полном смачивании — на их радиус капилляра,
является физико-химич. константой двух соприка-
сающихся фаз (жидкости и пара или двух несмеши-
вающихся жидкостей). Эта константа имеет размер-
ность площади и зависит только от поверхностного
межфазного натяжения Oj2 и разности плотностей
обеих фаз р1( р2. Она наз. капиллярной по-
стоянной (а2):
аг _ 2°12— —rh = const t
g (pi — Pa)
Для воды на границе с паром при 20° сг = 72,8 эрг!см2,
а2 = 14,9 .и.и2.
К. я. определяют равновесные формы жидких по-
верхностей раздела, напр. в простейшем случае,
когда на жидкость не действуют внешние силы, она
принимает форму шара. К. я. определяют также формы
движущихся жидких поверхностей и условия движе-
ния вязких жидкостей вблизи поверхностей раздела
(капиллярная гидродинамика). Основное значение
в этой группе К. я. имеют образование, распростране-
ние и затухание на поверхности жидкости или на гра-
нице двух жидкостей капиллярных волн, т. е. коротких
поверхностных волн (ряби).
С К. я. связаны образование новой фазы в первона-
чально относительно неустойчивой (метастабильной)
среде, конденсация паров, кипение жидкостей, кри-
сталлизация и др. При этом К. я. определяют возник-
новение высоких пересыщений пара без образования
капелек жидкости, перегрева жидкости выше точки
кипения, переохлаждения расплавов или пересыщения
растворов без выделения кристалликов. С этим свя-
заны повышенное давление насыщенного пара или
повышенная растворимость искривленных поверхно-
стей — малых капелек или кристалликов, и, наоборот,
пониженное давление пара в маленьких пузырьках
внутри жидкости. Эти явления вызваны капиллярным
давлением и играют важную роль в технике, а также
при образовании атмосферных осадков. К. я. обуслов-
ливают и капиллярную конденсацию, т. е. конденса-
цию паров в узких порах пористых тел (сорбентов),
смачиваемых данной жидкостью, при давлениях пара
ниже насыщения. Явления смачивания и флотации
(прилипания малых тяжелых частиц к пузырькам
газов в жидкой среде вследствие неполного сма-
чивания) — К. я., имеющие важное практич. зна-
чение.
Теория К. я. в наст, время развилась в общую тео-
рию поверхностных явлений, в к-рой было установлено
решающее влияние физико-химич. факторов (химич.
состава, структуры, адсорбции, темп-ры) на К. я.,
и приобрела важное значение в молекулярной физике
и коллоидной химии.
Лит.: Гроиека И. С., Собрание сочинений, М., 1952,
с. 27; Лев и ч В. Г., Физико-химическая гидродинамика,
М., 1952; Гиббс Дж. В., Термодинамические работы, пер.
с англ., М.—Л., 1950; Адам Н. К., Физика и химия по-
верхностей, пер. с англ., М.—Л., 1947. П. А. Ребиндер.
КАПРИЛОВАЯ КИСЛОТА (октановая кислота)
СН3(СН.,)6СООН, мол. в. 144,21 — т. пл. 16,63°;
т. кип. 239,37760 мм, 124°/10лм»; с?1° 0,9089; 1,4280.
К. к. обладает общими свойствами предельных кар-
боновых кислот. Она содержится в виде триглицерида
(трикаприлина) в нек-рых невысыхающих маслах (ко-
косовое, пальмоядерное, бабассу) в количестве 3—8%.
Свинцовая соль К. к. (т. пл. 83—84°) нерастворима в
воде, медная (т. пл. 264—266°) и цинковая (т. пл. 135—
136°) соли плохо растворимы. Производные К. К.: ме-
тиловый эфир, т. кип. 193—194°; этиловый эфир, т. кип.
207—208'7753 мм; триглицерид, т. пл. 8°; п2£ 1,44817;
хлорангидрид, т. КИП. 195—196°. н. А. Несмеянов.
КАПРИЛОВЫЙ АЛЬДЕГИД (октаналь), мол. в.
128,08, СН3(СН2)вСНО — бесцветная жидкость
с характерным запахом; т. кип. 167—1707760 мм,
65711 мм.; 0,821; Пр 1,4217. К. а. плохо растворим
в воде, хорошо — в спирте и эфире; перегоняется
с водяным паром Встречается К. а. в природе — вхо-
дит в состав эфирных масел (лимонного, розового и др.).
К. а. обладает всеми свойствами, характерными для
насыщенных алифатич. альдегидов. В пром-сти К. а.
получают окислением октилового спирта. К. а. бла-
годаря своему приятному запаху в разб. состоянии
применяют в Парфюмерии. Н. П. Гамбарян.
КАПРИНОВАЯ КИСЛОТА (декановая кислота)
СН3(СН2)8СООН, мол. в. 172,26 —т. пл. 31,3°;
т. кип. 2707760 мм, 170°/25 мм; di0 0,8858; 1,42855;
в 100 г воды растворяется 0,015 г (20°); кислотное
число 325,7. К. к. обладает общими свойствами пре-
дельных к-т алифатич. ряда (см. Карбоновые кислоты).
Содержится К. к. в небольшом количестве в виде три-
глицерида в коровьем и кокосовом маслах; медная
соль ее нерастворима в воде, магниевая и Калиевая
соли хорошо растворимы. Метиловый эфир К. к.,
т. кип. 224°; этиловый эфир; т. кип. 243—245°; три-
глицерид (С33Н62О6), т. пл. 31°; амид, т. пл. 108°; хлор-
ангидрид, т. КИП. 114°/15 мм. К. А. Несмеянов.
е-КАПРОЛАКТАМ (лактам е-аминокапроновой ки-
слоты, 2-оксигексаметиленимин), мол. в. 113,16 — бе-
лые кристаллы; т. пл. 68—69°, т. кип.
СН2-СН2—С=О 262,57760 мм рт. ст., 139712 мм
I рт. ст.; п-£ 1,4768; хорошо растворим в
I | воде (525 г в 100 г воды), спирте, эфире,
сн2—сн2—сн2 бензоле, хлороформе и др. органич.
растворителях.
К. — циклический амид е-аминокапроновой к-ты
и может быть получен при нагревании последней выше
темп-ры плавления (210—220°). Водные растворы к-т
и щелочей вызывают обратный процесс — гидролиз К.
до е-аминокапроновой к-ты. При нагревании (250—
260°) в присутствии небольших количеств воды, спир-
та, аминов, органич. к-т и нек-рых др. соединений К.
полимеризуется с образованием полиамидной смолы,
из к-рой получают волокно «капрон». К. является
продуктом многотоннажного производства. Промыш-
ленные способы получения К. основаны на использо-
вании в качестве исходного сырья гл. обр. бензола.
Все эти способы включают стадию получения полу-
продукта — циклогексаноноксима, к-рый превра-
щают в К. обработкой серной к-той или олеумом (пере-
группировка Бекмана). В наиболее распространенном
415
е-КАПРОЛАКТАМ
416
способе исходят из фенола, к-рый перерабатывают
в К. по схеме:
Гидрирование фенола на никелевом катализаторе
(135—150°) и дегидрирование циклогексанола в цик-
логексанон на цинк-железном катализаторе (400—
450°) протекают с высокими выходами (95—97%).
Циклогексаноноксим в заводской практике получают
обработкой циклогексанона водным р-ром гидроксил-
аминсульфата при 85—95° и pH 3; для нейтрализации
выделяющейся H2SO4 в реактор непрерывно подается
газообразный аммиак. Циклогексаноноксим (сырец)
в расплавленном состоянии или в р-ре инертного ор-
ганического растворителя обрабатывают олеумом,
содержащим 18—25% свободного SO3. Продукт пе-
регруппировки нейтрализуют аммиаком и техниче-
ский К. очищают дистилляцией или ректификацией
(см. схему).
Схема непрерывной изомеризации циклогексаноноксима
в капролактам: 1 —аппарат для плавления циклогекса-
ноноксима; 2 — аппарат для проведения перегруппи-
ровки; з — нейтрализатор; 4 — аппарат для отделе-
ния лактама от раствора соли; 5 — аппарат для от-
гонки воды от лактама; 6 — колонка для дистилляции;
1 — приемник для чистого лактама.
Для получения К. высокого качества последний эк-
страгируют из продукта перегруппировки либо бен-
золом, либо вначале трихлорэтиленом, азатем из р-ра
в трихлорэтилене — водой. Дистилляцию или ректи-
фикацию экстракта рекомендуют сочетать с пере-
кристаллизацией, с обработкой сорбентами, ионооб-
менными смолами, окислителями, восстановителями
и т. п.
Технологически важная группа способов получения
К. основана на гидрировании бензола до циклогексана
и переработке последнего различными способами
в циклогексаноноксим и далее в К. К ним относится
NHoOH
----»- капролактам
способ, основанный па окислении циклогексана воз-
духом до смеси циклогексанола и циклогексанона.
Жидкофазное окисление циклогексана осуществляют
при 120—130° и давлении 15—20 ат в присут-
ствии катализаторов — нафтената Со или стеарата
Мп. Степень превращения циклогексана 10—12%,
суммарный выход циклогексанола и циклогексанона
(1 : 1) 65—75%. Одновременно получается адипи-
новая к-та — 0,5 т на 1 т К. Из реакционной смеси
циклогексанон выделяют ректификацией и превраща-
ют в К. Циклогексанол с целью увеличения выхода
циклогексанона дегидрируют на Zn—Fe катализа-
торе. По другому способу циклогексан нитруют азот-
ной к-той, образующийся нитроциклогексан восста-
навливают в циклогексаноноксим и последний изоме-
ризуют в К.:
—»- капропа«и«
Нитрование циклогексана осуществляют при повышен-
ных темп-ре и давлении. Процесс протекает в адиаба-
тич. условиях и заканчивается за короткое время.
При этом образуются нитроциклогексан и адипиновая
к-та с суммарным выходом около 80%. Количество
адипиновой к-ты составляет ок. 0,4 т на 1 т К.
Нитроциклогексан восстанавливают в циклогексанон-
оксим водородом в присутствии жидкого аммиака и
металлич. меди. Процесс осуществляется непрерывно
при 80—130° и давлении 170—200 ат с высоким вы-
ходом (85—90%).
Наиболее эффективный способ получения К. осно-
ван на фотохимия, нитрозировании циклогексана,
приводящим непосредственно к получению хлоргид-
рата циклогексаноноксима:
NOH" НС1 __________ ИМППП1«Т«И
пап рил в я I о»
Нитрозирование проводят при облучении р-ра NOC1
в циклогексане видимым светом (450—550 ммк)
и непрерывном насыщении р-ра газообразным НС1.
Полученный хлоргидрат циклогексаноноксима пе-
рерабатывают в собственно оксим и затем в К., либо
непосредственно направляют на перегруппировку.
К группе способов, включающих стадию получе-
ния циклогексаноноксима, следует отнести и полу-
чение К. из анилина. Способ заключается в восста-
новлении анилина до циклогексиламина, окислении
последнего до циклогексанона и циклогексанола и
дальнейшей переработке, как описано выше. Однако
этот способ технологически менее эффективен. Эффек-
тивность его может быть существенно повышена пря-
мым окислением циклогексиламина в циклогекса-
ноноксим.
Отдельно от описанной группы способов стоит син-
тез К. из толуола:
Выходы по отдельным стадиям весьма высоки и вслед-
ствие доступности толуола метод представляет прак-
тич. интерес.
К. может быть получен также из неароматических
продуктов (фурфурола, ацетилена, окиси этилена.
417
КАПРОН —КАРАН
418
пропилена, бутадиена и др.):
НС — СН О
Il II <
. нс с-с,
V
нс=сн
н
НС^ССН2ОН
н
н2с-сн2 н2с=с'
\0' . , 'сн3
х \ / с.сн2сн=снснаС|
Сн2=СН—СН=СН2
В плодах красного (испанского) перца
Capsicum Annum К. содержится в ко-
личестве около 0,03%; кристаллизует-
ся из петролейного эфира в виде бесцвет-
ных табличек, т. кип. 210—22070,01 мл,
ch2=chcn
НС—СН
II и
НС сн
v
НОН2СС^ССН2ОН
НО(СН2)4ОН
н2с-сн2
н2с сн2
сГ
СКСН2)4С1
NC(CH214CN
h2n(ch2;5cn
ncch2ch=chch2cn
но
СН2 NHCO(CH2)4 СН=СНСН(СН3)2
/СНа-СНз-С^О
н2сч |
СН2-СН2~NH
осн3
т. пл. 64—65°; почти нерастворим в хо-
лодной воде, легко растворим в эфи-
ре, спирте, бензоле, а также, благо-
даря фенольной группе, — в едких ще-
лочах. При гидролизе в жестких ус-
ловиях К. расщепляется па вапилил-
амин и метилноненовую (дециленовую)
кислоту.
Строение К., установленное в 1919—
1920, было подтверждено в 1930 синте-
Все эти схемы синтеза включают стадию получения
общего полупродукта — адиподинитрила. Превраще-
ние последнего в е-аминокапронитрил и затем в К.
освоено пока только в лабораторном масштабе. Однако
в перспективе использование неароматич. сырья мо-
жет представлять известный интерес особенно по схеме,
включающей стадию гидрирующей димеризации акри-
лонитрила.
Для аналитического определения К. используют
полярографии, метод или метод, основанный на титро-
вании формальдегидного производного е-аминокапро-
новой к-ты.
Лит.: Современные методы синтеза мономеров для гетеро-
цепных волокнообразующих полимеров. Сб. статей, под ред.
И. Л. Кнунянца, М., 1961; Хопфф Г., Мюллер А.,
Венгер Ф., Полиамиды, пер. с нем., М., 1958; Волокна
из синтетических полимеров, под ред. Р. Хилла, пер. с англ.,
М., 1957; Кларе Г., Химия и технология полиамидных
волокон, пер. с нем., М., 1956.
Е. В. Генкина, В. Н. Фросин.
КАПРОН — см. Полиамидные волокна.
КАПРОНОВАЯ КИСЛОТА (н-гексановая кислота)
СН3(СН2)4СООН, мол. в. 116,16— т. пл. —3,9°; т. кип.
205,35°; d1& 0,9314; Ид 1,4170; в 100 г воды раство-
ряется 0,886 г (20°); кислотное число 483,0. К. к. об-
ладает общими свойствами предельных алифатич. к-т
(см, Карбоновые' кислоты). К. к. находится в виде эфи-
ров в эфирных маслах и в виде глицерида во многих
жирах и маслах; образуется при маслянокислом бро-
жении сахара; может быть получена окислением лау-
риновой к-ты. Хлорангидрид К. к., т. кип. 151—153°.
Эфиры К. к. обладают фруктовым запахом и могут
применяться в производстве фруктовых эссенций.
В табл, приведены свойства некоторых эфиров К. к.,
которые могут быть использованы для этих целей,
а также для идентификации К. к.
Эфир Т. кип., °C dso
Метиловый 151.5 0,8893 а 1,4067
Этиловый 167 0,8732 6 —
н-Пропиловый 187,5 0,8587 1.4140
w-Бутиловый 207,7 0,8569 1,4188
н-Амиловый 226,2 0.8553 1,4228
н-Гексиловый 245,4 0.8541 1,4264
н-Гептиловый 260 0.8533 1,4234
н-Октиловый 275,2 0,8526 1,4326
Трикапроин (триглицерид) — 0,9867 6 1,4427
а При 15°. 6 При 20°.
Н. А. Несмеянов.
КАПСАИЦИН (ванилиламид 8-метилнонен-6-овой
к-ты) C18H27NO3 — действующее начало различных
видов стручкового перца Capsicum (сем. Solanacrae).
зом из ваиилиламина и хлорангидрида дециленовой
к-ты, полученной по схеме:
(CH3)2CHCH2ZnJ 4- С1СО(СН2)4СООС2Н5 ->
восстановление
-> (СН8)2СНСН2СО(СН2)4СООН --------------►
НВг
(СН3)2СНСН2СНОН(СН2)4СООН----►
хинолин
-> (СН3)2СНСН2СНВг(СН2)4СООН------>•
-> (СН3)2СНСН=СН(СН2)4СООН
Действующим началом различных видов черного
перца является пиперин, принадлежащий, как и К.,
к классу амидов к-т.
К. содержится в спиртовой настойке стручкового
перца, применяющейся в медицине как раздражающее
и отвлекающее средство.
Лит.: Орехов А. П., Химия алкалоидов, 2 изд., М.,
1955, с. 687; Nelson Е. К., J. Amer. Chem. Soc., 1919,
41, № 7, 1115, Ni 9, 1472; 1920, 42, N 3, 597; Spath E.,
DarllngS. F., Ber., 1930, 63, № 3, 737. Я. Ф. Комиссаров.
К АПСЮ Л Ь - ДЕТОН ATOP — см. Средства иници-
ирования.
КАПТАКС (2-меркаптобензтиазол) C7H5NS2, мол. в.
167,25 — желтый порошок; т. пл. 179°; хорошо рас-
творим в спирте, эфире, ледяной уксусной к-те, нерас-
творим в воде; имеет горький вкус.
В растворах щелочей и карбонатов
щелочных металлов К. растворяется Г Y с—SH
с образованием соответствующих со- 5 х
лей; конц. щелочи при повышенной
температуре расщепляют его с образованием солей
о-аминотиофенола. При окислении К. образуется аль-
такс. К. получают в пром-сти нагреванием анилина
с серой и сероуглеродом под давлением или из р-нитро-
хлорбензола и гидросульфита натрия с последующей
обработкой сероуглеродом (без давления):
К., а также его цинковая соль и другие производные,
напр. 2-бензтиазолил-Х,Х-диэтилсульфенамид, широко
используются в качестве ускорителей вулканизацпи
каучука (см. Вулканизации ускорители}. К. применяют
также для обнаружения и весового определения
Bi, Cd, Со, Си, Au, Pb, Hg, Ni, Т1 и Zn и как исход-
ный продукт для синтеза цианиновых красителей.
Е. М. Рохлин.
КАРАН (4, 7, 7-триметилбицикло-[0, 1,4]-гептан)
С10Н18, мол. в. 138,25 — сраввительно мало изучен-
419
КАРБАЗОЛ — КАРБАНИОН
420
ныи углеводород. По положению заместителей при С4
различают г{нс(1)- и траис(П)-формы К. Препарат,
содержащий в основном цнс-К., имеет константы длн
а- и 1-форм соответственно:
и 168,5^/760 мм; d-° 0,8412
'4
т. кип. 169,5°/759 мм
и 0,8398; ng 1,4567 и
1,4552; MRd 44,71 и
44,69; [a]D=+57,64°,
—47,06°. Препарат,со-
держащий в основном
транс-К., имеет: т.
кип. 166°/749 мм; dl°
0,8317, ng 1,4511, MRD 44,76; [a]D = —15,48°. Трех-
членное кольцо К. может раскрываться с образованием
м- или п-мептана и их смесей. Так, при гидрировании
К. при 160° с катализатором—палладированпым
углем, образуется в основном n-ментан. При действии
на К. галогеноводородных к-т, напр. НВг, полу-
чаются галогеноментаны, напр. 8-бром-л«-ментап, т.
кип. 123—124°/29 мм. Галогены разрывают 3-членпое
кольцо К. с образованием соответствующих дигало-
генопроизводных, напр. 2,8-дибром-.м-ментана.
К. можно получать по Кижнеру — сухой пере-
гонкой со щелочью гидразонов терпеновых кетонов
(пулегона и карона); гидрированием непредельных
терпеновых углеводородов — каренов-, из А3-карена
получается в основном г^ис-К., а из А4-карена —
транс-К.
Лит.: Simonsen J. L., The terpenes, v. 2, [2 ed.],
Camb., 1949; Рудаков Г. А., Марчевский A. T.,
в кн.: Сборник статей по общей химии, т. 2, М.—Л., 1 953,
с. 1433. И. И. Бардышев.
КАРБАЗОЛ (дибензопиррол) CI2H9N, мол. вес
167,21 — кристаллы; т. пл. 245—247°; т. кип. 354—
355°; пикрат, т. пл. 185—186°; произ-
водное с тринитробензолом, т. пл.
[г Дз |ы1 J 166°, нерастворим в воде, растворим
в органич. растворителях: 0,92 г в
100 мл спирта (1,4°), 3,1 г в 100 мл
эфира (30°), 5,3 г в 100 мл бензола (50°), 11,1 г
в 100 мл ацетона (30°). К. дает цветные реакции,
характерные для пиррольного кольца. К. — сла-
бое основание, соли образует плохо; при окис-
лении образует дикарбазилы: 9,9' (с КМпО4) или
3,3' (с Na2Cr2O7-|-H2SO4). Восстанавливается К. с
большим трудом. С Sn-|-HCl К. дает 1,2,3,4,10,11-гек-
сагидрокарбазол. Каталитич. гидрирование К. в же-
стких условиях над различными катализаторами при-
водит к смеси веществ с различной степенью насыщен-
ности, вплоть до додекагидрокарбазола. Замещение
в К. у N-атома производят либо непосредственно,
либо через калиевое производное (см., напр., N-cuhua-
карбазол). Обработка N-металлированного К. СО2
приводит к карбазол-9-карбоновой к-те. Вступая
в Фриделя — Крафтса реакцию, К. образует 3,6-за-
мещенные производные. Известны различные способы
размыкания колец К. Нитрозирование К. приводит
к 9-нитрозопроизводному, к-рое может перегруппиро-
вываться в 3-нитрозокарбазол. При нитровании К.
дает смесь нитропрбизводных, вплоть до тетранитро-
карбазопа, но в среде нитробензола образуется 3-нит-
рокарбазол. При галогенировании К. дает 3- и 3,6-про-
изводные, а при сульфировании — 3-карбазолсуль-
фокислоту или 1,3,6,7-карбазолтетрасульфокислоту.
Основным источником К. служит антраценовое
масло из кам.-уг. смолы, но очистка полученного
таким путем К. очень сложна. От антрацена К. отде-
ляют в виде Na-соли, нелетучей и нерастворимой
в нек-рых органич. растворителях. Синтетически К.
можно получить из о-аминодифенила, из фенантрена
(через 2,2'-динитродифе11ил-6,6'-дикарбоповую к-ту),
по Гребе—Ульману из 2-аминодифенпламина и т. д.
К. находит применение в произ-ве гидроидных краси-
телей, а его нитро- и хлорпроизводные используются
как инсектициды.
Лит.: Фрейденберг В., в кн.: Гетероциклические
соединения, под ред. Р. Эльдерфилда, пер. с англ., т. 3, М.,
1954» с. 231. А. Е. Васильев.
КАРБАМИДНЫЕ ПЛАСТИКИ — см. Амино-
пласты.
КАРБАМИНОВАЯ КИСЛОТА H2NCOOH, мол. в.
61,04 — кислый амид угольной кислоты, в свободном
виде неизвестна, известны производные К. к.: эфиры
(уретаны), амид (мочевина), гидразид (семикарбазид),
соли (карбаматы), галогенангидриды и т. п.
Аммониевая соль К. к. H2NCOONHi — наиболее
доступный карбамат; гигроскопич. кристаллы, хорошо раст-
воряются в воде, плохо — в жидком аммиаке; разлагается па
СО2 и NH3 при 59°; получают прямым взаимодействием СО2
и NH3; промежуточный продукт (без выделения) в произ-ве
мочевины, в к-рую превращается при нагревании под давле-
нием:
HoNCOONHi -► H2NCONH3 + H2O
Содержится в продажном карбонате аммония; применяется
для получения других карбаматов.
Калиевая соль К. к. H2NCOOK — кристаллы,
легко растворяются в воде; при нагревании образует цианат
калия:
H3NCOOK -► КОСМЧ-Н2О
Кальциевая соль К. к. (H2NCOO)2Ca — хорошо
растворяется в воде, раствор при стоянии мутнеет (образуется
карбонат кальция), при нагревании превращается в кальция
цианамид:
(H2NCOO)3Ca -> CaNCN + 2НаО + СО2
Хлорангидрид К. к. (карбамилхлорид) H2NCOC1 —
бесцветная, резкопахнущая жидкость, т. кип. 61—62°; разла-
гается на циануровую кислоту (НОСК)зИ хлористый водород
уже при стоянии, легче — при нагревании; получают действием
фосгена на хлористый аммоний или на аммиак при повышенной
темп-ре:
NHdC14-COCl2 -► H2NCOC14-2HC1
NH3 + COCh -► HoNCOCl + НС1
Применяется для введения группы CONH2 в органич. соеди-
нения, напр. с помощью Фриделя—Крафтса реакции. Со спир-
тами образует уретаны, с аммиаком, аминами и амидами —
мочевину или ее производные.
Азид К. к. H2NCON3 — т. пл. 94°, легко взрывает.
Лит.: U 11m апп, 3 aufl., Bd 5, Munch.—В., 1954, S. 70.
Е. М. Рохлин.
КАРБАНИОН — ион, содержащий трехковалент-
ный отрицательно заряженный углерод ^С: Нали-
чие отрицательного заряда и свободной пары электро-
нов приводит к тому, что К. малоустойчив, т. е. обла-
дает большой реакционной способностью, к-ран про-
является в сильной нуклеофильности. К. легко при-
соединяет протон и другие электрофильные реагенты:
^С: + А+ -> ^С-А
Атом углерода имеет сравнительно малое сродство
к электрону, что следует из его положения в периодич.
системе. Все элементы, находящиеся правее углерода,
обладают большим сродством к электрону, чем угле-
род. Подавляющее число элементов-органогенов (Э)
расположено правее углерода; поэтому связь С—3
обычно поляризована т. обр., что плотность электрон-
ного облака больше у Э, т. е. /С—Э_. Обратное на-
правление поляризации связи С—Э имеет место
г- г+
в случае связи С—Н и особенно С — металл. Те со-
единения, в к-рых связь С—3 сильно поляризована,
так, что у С имеется избыточная плотность электрон-
ного облака, во многом напоминают в своем поведении
К.; они обладают высокой нуклеофильностью, их
растворы имеют дипольные моменты. Натрий- и ка-
лийорганич. соединения большей частью, подобно со-
лям, представляют собой кристаллич. вещества с вы-
сокой темп-рой плавления (иногда разлагаются не
плавясь), нерастворимы в неполярных растворителях.
Нек-рые щелочнооргапич. соединения проводят элект-
рич. ток и являются истинными солями (натрий- и
421
КАРБАНИОН
422
калий три- и дифенилметаны), большинство же щелоч-
ноор ганич. соединений обладает лишь сильнополяри-
зованной ковалентной связью.
Стабильный кинетически независимый К. образует-
ся в тех случаях, когда отрицательный заряд дело-
кализован, т. е. распределен между несколькими
атомами. К таким К. относятся трифенилметиланион,
циклопентадиениланион, дифенилметиланион, флуо-
рениланион и нек-рые другие. Особый интерес пред-
ставляет циклопентадиениланион, стабильность к-рого
обусловлена его ароматичностью (см. Ароматические
системы).
В химии К. особое место занимают т. н. илиды —
биполярные ионы, в к-рых целый или частичный поло-
жительный заряд находитсн на ониевом атоме (N, Р,
О, S), а отрицательный — на углероде. Наиболее
устойчивые илиды — циклопентадиенилиды, т. е.
биполярные ионы, в к-рых анионом нвляется устой-
чивый циклопентадиениланион. Очень характерен
по свойствам циклопептадиенилид пиридиния (I) —
стабильное кристаллич. вещество, неплавящееся до
360°; растворы имеют необычайно большой для орга-
нич. соединения диполь-
/ \ ный момент (13,5 D).
\ / Известны также флуореп-
/ V /7Т1 ] Ч илиды. Флуоренилид
\ J1 I I А \ / пиридиния (II)—менее
/ \ стойкое соединение, чем
4 ) циклопентадиенилид пи-
' '—' и ридипия (I), однако он
существует в токе азота. Илиды, содержащие али-
фатич. анион, чрезвычайно нестойки; об их образо-
вании судят по продуктам реакции. Известен лишь
триметиламмонийметилид, устойчивый в виде комп-
лекса с бромидом лития: (CH3)3N(CH2)LiBr.
Наличие электроноакцепторного заместителя у ато-
ма углерода должно повышать устойчивость К. Од-
нако ацетоуксусный и малоновый эфиры, ацетилаце-
тон и т. п. соединения, содержащие у одного атома
углерода два сильных электроноакцепторных замести-
теля, отрывая протон, образуют не К., как это можно
было ожидать, а анион енола, содержащий отрица-
тельный заряд в основном па атоме кислорода:
Н
СН3—С=С—С—сн3
<i- а
При наличии связи С—Н склонность соединения
к образованию К. выражается в легкости, с к-рой оно
отщепляет протон, т. е. в «кислотности» этого веще-
ства. Протонная подвижность водорода проявляется
наиболее отчетливо в склонности вещества вступать
в реакцию изотопного обмена водорода под действием
основания и в его способности вступать в реакцию
металлирования. В зависимости от степени протонной
подвижности водорода замещение его на дейтерий
проходит под действием оснований различной силы:
чем больше протоннан подвижность водорода, тем
менее сильное основание может быть применено для
реакции. Первым актом реакции водородного обмена
явлнетсн отрыв протона:
^.С-н + в -* + нв+
Второй акт—взаимодействие К. с донором дейтерия,
напр. дейтероспиртом:
-С: + DOR -* — CD -f- OR
Эти акты могут протекать один за другим через обра-
зование истинного К., а могут накладываться: друг на
друга во времени. В процессе установления этих двух
кислотно-основных равновесий происходит распределе-
ние дейтерия между обоими компонентами. Так, цик-
лопентадиен вступает в реакцию водородного обмена
с C^HgOD в присутствии органич. основания, ацети-
лен — под действием водной щелочи, жирноарома-
тич., ароматич., этиленовые углеводороды вступают
в реакцию водородного обмена при действии различ-
ных концентраций раствора амида калия в жидком
аммиаке.
Замещение водорода металлом для разных углево-
дородов происходит под действием: водных щелочей,
сплавления со щелочами, с металлом, различных
щелочноорганич. соединений — Шорыгина реакция.
Реакция Шорыгина заключается во взаимодействии
углеводорода с натрийпроизводным другого углево-
дорода с образованием нового углеводорода и нового
натрийпроизводного:
RH + R'Na -> RNa + R’H
Эта реакпин аналогична вытеснению сильной к-той
более слабой из ее соли; она позволяет сравнивать
кислотность различных углеводородов, для к-рых уста-
новлен ряд уменьшающейся кислотности: флуоре-
ден > циклопентадиен > инден > перинафтен >
флуорен > трифенилметан > дифенилметан >> то-
луол > бепзол > метан.
Конант и Уэланд вычислили условные константы
диссоциации по отношению к метиловому спирту
(А = 1). Для фенилацетилена, индена, фенилфлуо-
репа К = 21, флуорена — 25, трифенилметана — 33,
дифенилметана, дифепилметилэтилена — 36, изо-
пропилбензола — 37.
Несмотря на то, что существование К. доказано
только в немногих случаях, представление о проме-
жуточном образовании К. привлекается довольно
часто для объяснении механизма органич. реакций-
Так, реакции Стивенса (1) и Соммеле (2), заключаю-
щиеся в перегруппировке солей аммония в амины,
можно рассматривать как изомеризации промежуточно
образующегося биполярного иона:
C6H5CH2-N(CH3)2 Сб"5-* CeH5CH-MCH3)2 —-
СНз СН3
Предполагается, что полимеризация олефинов, про-
исходящая под действием щелочных металлов, метал-
лоорганич. соединений и других сильных оснований,
протекает с участием К. — анионная поли-
меризация:
зарождение цепи: Na+R~ 4-->/С—-J-Na+
R
Лит.: Темникова Т. И., Курс теоретических основ
органической химии, Л., 1959; Шатенштейн А. И.,
423
КАРБЕНЫ - КАРБИДЫ
424
Изотопный обмен и замещение водорода в органических соеди-
нениях в свете теории кислот и оснований, М., 1960; А 1 е-
xander Е. R..Principles of ionic organic reactions, N. Y.,
1950. 3. H. Парнес.
КАРБЕНЫ — неустойчивые соединения двухва-
лентного углерода, простейшим представителем к-рых
является метилен СН2: Согласно последним данным,
основное состояние метилена триплетно; об электрон-
ном строении других К. достоверных сведений не
имеется. Имеются данные о том, что дихлоркарбен
является жидкостью, диспропорционирующейся при
темп-ре кипения (—20°) на гекса хлор бензол и гекса-
хлорэтан, а в присутствии воздуха — окисляющейся
в фосген.
Интерес к К. вызывается их промежуточным обра-
зованием в самых различных органич. реакциях.
Свободный метилен образуется в результате термин,
или фотолитич. расщепления диазометана или ке-
тена, либо а-элиминированием галогеноводорода или
2 атомов галогена:
CH,N2 -* СН,: + N2; СН2=СО -► СН2: + СО
Г Zn/Cu
СН3Х —► СН2: 4-НХ; СН2Х2------► СН2: + Х2
Доказательством образования метилена служит полу-
чение продуктов вторичных реакций, в к-рые всту-
пает чрезвычайно реакционноспособный метилен, напр.
приведенных ниже этилена, замещенных циклопропана
и др.:
пСН2: 4- Те —> (ТеСН2)п
СН2: + СН2: -♦ СН,=СН2
СН,: + /С=С<^ -----С/
^CH2Z
СН,: 4-RH -► RCH3
Аналогичными путями могут быть получены и за-
мещенные, зачастую очень сложные карбены:
R, R4 R\ ,R
)CN2 -> ;C: -> >C=C<
R'- R'/ R’/ Xr-
ZR
RCOCR'N, -> RCOCR' -> O=C=C<
XR'
ZR' .. ,R
RCOC(-H -> RCOCR' -> O=C=C<
\x XR’
R, R\
' CIICIL.X -> >CHCH: -► RCH=CHR’
R'Z ‘ R'/
Наиболее достоверно доказано промежуточное образо-
вание дигалогенокарбенов в следующих реакциях:
НСХ3 4- в- -> СХ2: + НВ + х-
CCl3COONa -> СС12: + СО2 + NaCl
RONa
СС13СООС3Н5------> СС1,: + NaCl 4. ROCOOC,H5
2RONa
CC13COCC13 --2CC1,: 4- ROCOOR + 2NaCl
(CH3)3SnCF3 -► (CH3)3SnF 4- CF2:
Дигалогеиокарбены проявляют ясно выраженные
электрофильные свойства и легко реагируют с раз-
личными нуклеофильными реагентами, такими как
гидроксильные, алкоксильные, фенолятные и тиофено-
лятные ионы. Особый интерес представляют реакции
присоединения дигалогенокарбенов к кратным свя-
зям. Эти новые реакции имеют широкое распростра-
нение и делают доступными разнообразные трехчлен-
ные циклич. соединения, дальнейшие превращения
к-рых представляют существенный интерес:
— С:
сх2
Mg
с
AgNO3
.С=С^С
С=С- С:
сх
X он
СвН5С^СС0Н5 + СХ: -> СвН5С=ССвН6 -► СвН5С=ССвН5
\1Х2
с
О
CeH3CH=NCsH5 4- СХ,: -> CeH5CH-NCeH3 ->
сх2
-> CeH5CHXCONHC(,H5
R2C=C(OR)2 4- :СХ2 —> R,C—C(OR), -> R,C=CXCOOR
^CXs
Моногалогенокарбены способны присоединяться и
к ароматич. двойной связи с образованием солей
тропилия:
СН2Х2 -^:СИХ
снх
X'
Привлечение гипотезы о промежуточном образовании
карбенов помогает выяснению механизмов ряда реак-
ций (Фаворского, Вольфа, Реймера — Тиманна и др.).
Лит.: Кнунянц И. Л., Гамбарян Н. П.,
Рохлин Е. М., Усп. хим., 195 8, 27,вып. 12, 1361; S с h m е 1 fr-
eer М., SchrSter Н., Angew. Chem., 1960, 72, 10,
349. Н. И. Гамбарян.
КАРБИДЫ — соединения металлов, а также не-
которых неметаллов с углеродом. К. — твердые при
нормальных условиях вещества, нерастворимые без
разложения ни в одном из известных растворителей.
По характеру межатомных связей К. можно разбить
на 4 группы.
1. Солеобразные соединения. К.
этого типа содержат анионы углерода и катионы ме-
таллов. Их образуют гл. обр. элементы I, II, III и
отчасти IV, V и VI групп периодич. системы. Эта
группа К. может быть разделена на три подгруппы,
а) К., не содержащие связи между углеродными ато-
мами в одном анионе. Эти К. можно представить как
продукты замещения атомов водорода в СН4 металла-
ми; при взаимодействии с нагретой водой или разб.
к-тами они разлагаются с выделением метана. Таковы
Ве2С с кубич. антифлюоритной кристаллич. решет-
кой и А14С3, обладающий ромбоэдрич. слоистой решет-
кой с цепями из углеродных атомов, б) К. с анионами
[С.=С.]2, т. наз. ацетилениды. Эти К. могут быть
представлены как продукты замещения водорода
в С2Н2 металлами; водой и разб. к-тами разлагаются
либо с выделением С2Н2 (К. элементов I и II групп
периодич. системы, а также А1,С6 и Ce-Xo), или с вы-
делением смеси С2Н2 с другими углеводородами и
иногда с Н2 (К. редкоземельных элементов). К. этой
группы кристаллизуются в тетрагональной решетке
типа СаСг- Примерами их служит Ха2С2 и др. К. ще-
лочных металлов: Cu2C2, Ag2C2, Ап2С2, ВеС2, MgC2,
СаС2, SrC,, ВаС2, ZnC2, CdC2, HgC2, A]2Ce, Се2С6,
YC2, LaC2, СеС2, PrC2, NdC2, SmC2. К ацетиленидам
относят также и ThCj, VC2, UCj, разлагаемые
водой и разб. к-тами с выделением смеси С,Н2 с дру-
гими углеводородами; однако строение, например,
ThG2 и иСг можно представить и как Th4+(C=C)4-
и U4+(C=C4-. Нек-рые из ацетиленидов не стойки и
в сухом виде взрывают при ударе или нагревании
425
КАРБИДЫ
426
(Ci:2C2, Ag2C2, Au2C2, HgC2). в) Mg2C3 — единственный
пример К. с анионом [С=С=С]4водой разлагается
с образованием метплацетилена СН3—С=С—Н.
Свойства солеобразных К. представлены в табл. 1.
Таблица 1. Свойства некоторых солеобразных карбидов
Кар- бид Т. пл., °C Плотн., г/см3 Тип кристаллич. решетки Теплота образова- ния ДН° , 298’ ккал/моль
Ве.,С 2150 (диссоц.) 2,44 кубич. антифлюорит- ного типа —
А14С3 > 2800 2,99 ромбоэдрич. —30,9
СаС2 2300 2,22 тетрагональная гра- иецентрированная -15,0
MgC2 — 2,07 то же —21,0
ВаС2 > 1780 3,81 » » — 5,0
СеС2 — 5,23 » » —
ЬаС2 — 5,02 » » —
тьс2 2655 9,6 моноклинная -44,8
ис2 2350 11,28 тетрагональная объемноцентрирован- ная —33,8
2. К. с ковалентной связью. Типичными
представителями этой группы являются «гигантские
молекулы» SiC и В4С(В12С5). Оба эти К. отличаются
высокой твердостью (см. табл. 2); они тугоплавки,
химически весьма инертны, жаростойки. Кристаллич.
структура SiC, близкая к алмазу, имеет несколько
видоизменений плотной кубич. или гексагональной
упаковки (см. Кремния карбид). Кристаллы К. бора
имеют ромбоэдрич. элементарную ячейку, в к-рой
атомы бора образуют группу В12, а атомы углерода,
располагаясь в наибольших пустотах каркаса В12,
образуют линейную группу С3. Т. обр., правильной
формулой К. бора следует считать В12С3. Состав К.
бора может изменяться от В12С3(В4С) до В13С.,(Вв,5С)
за счет того, что один из трех атомов углерода в эле-
ментарной ячейке может быть заменен атомом бора.
В тройной системе В—С—Si имеются сплавы, твер-
дость к-рых еще выше, чем у В4С (микротвердость до
7000 кГ/мм2 и выше).
Таблица 2. Свойства некоторых ковалентных карбидов
Карбид Т. пл., °C еИ'Э/З ‘•Н1О1ГЦ Тип кристаллич. решетки Теплота образо- вания дп° . 298 * ккал/моль Микро- твердость, кГ/мм3
S1C 2200 (разл.) 3,2 имеется кубич. и неск. типов гексаг. модифи- каций — 3500
В4С(В12Са) 2250 (пе- ритектич. разл.) 2,52 ромбоэдрич. -14,0 5000
Близким к группе ковалентных К. предположи-
тельно считают и Сг3С.2, имеющий непрерывные зигза-
гообразные цепочки углеродных атомов в ромбич.
кристаллич. структуре. Однако по электропроводности
и другим свойствам К. хрома близки к металлоподоб-
ным К. (см. ниже). Кроме СГдС,, хром образует еще
Сг23Св(Сг4С) с гранецентрированной кубич. структу-
рой, Сг7С3 с гексагональной структурой, и предполо-
жительно СгС, стабильный возможно выше 2000°.
К. хрома отличаются высокой твердостью (см. табл. 3),
стойкостью против кислот и против окисления на воз-
духе до 1000—1100°. Существуют и аналогичные К.
марганца, изоморфные с К. хрома: Мп23Св и Мп,С3.
3. К. — фазы внедрения. Кристаллич.
структуры К. этого типа представляют собой решетки
из атомов металлов переходных групп, в поры к-рых
внедрены атомы углерода, при отношении атомных
радиусов углерода и металла 0,59 гс : гме 0,41
(по Хеггу). Из этой группы К. особо важное значение
имеют весьма твердые и жаропрочные. К. тугоплав-
ких металлов: TiC, ZrC, VC, NbC, ТаС, Mo2C, W2C,
WC (по нек-рым воззрениям, WC не относится к фазам
внедрения, но близок к ним). При внедрении атомов
Таблица 3. Свойства некоторых металлоподобных карбидов
Исходные металлы Карбиды
ме- талл т. пл., °C тип кристаллич. решетки а карбид т. пл., °C плот- ность, г/см3 тип кристаллич. решетки теплота образования ДН° » 298 ккал/моль микро- твердость, кГ/мм2
TI 1660 а’Т1 1 гекс. Т1Сб 3140 4,92 кубич. гранецентр. —43,9 2850- 3000
Zr 1830 а-Zr 1 ПЛОТНО- ZrC 3500 6,70 то же -44,1 2830
HI 2222 а-Ht j унак. Н1С 3890 12,67 » » — 2800
V 1900 кубич. объемноцентр. VC 2830 5,46 » » —28 2090
Nb 2500 то же NbC 3500 7,82 » » —33,7 2050
Та 2850 » » ТаС 3880 14,47 » » —38,5 1550-1800
Мо 26‘>0 » » Мо2С 2690 8,9 4,2 1480
W 3410 » » W2C 2700 17,3 | гекс, плотноупак. 7,1
WC 2600 1 гекс. 9,1 1730
(перитектич. разл.)
Мн 1244 а-Мп । кубич. объем- Mn3CB 1520 6,89 ромбич. -23 —
Fe 1539 a-Fe j ноцентр. Fe3C 1650 7,67 » 5,4 1080
Сг 1875 Cr3C2 1900 6,70 » —21,0 1300
(перитектич. разл.)
Cr7C3 1680 6,92 гекс. —42,5 —
(перитектич. разл.)
СЛазС/д 1520 6,99 кубич. гранецентр. -16,6 —
(Cr4C) (перитектич. разл.)
а Сокращения: гекс. — гексагональная, плотноупак. — плотноупаковаиная, объемноцентр. — объемноцентрироваиная,
грапецеитр. — гранецентрированная.
с TiC, ZrC, HfC, "VC, NbC, ТаС — формулы предельного насыщения углеродом. Эти К. способны образовывать «струк-
туры вычитания», т. е. решетки с дефицитом часта углеродных атомов, что создает нек-рые области гомогенности, прилегаю-
щие к предельному составу МеС.
в Мп образует еще Мп7С3 и Мп23Св — изоморфные соответствующим К. хрома.
427
КАРБИДЫ — КАРБОАНГИДРАЗА
428
углерода между атомами металла последние несколько
раздвигаются, в результате чего происходит незначи-
тельная перестройка кристаллич. решетки металла
(см. табл. 3) и развиваются громадные внутренние
давления, что приводит к переходу части внешних
валентных электронов от углеродных атомов на нена-
сыщенный электронный rf-уровень атомов переходных
металлов (по Я. С. Уманскому). Т. обр., межатомные
связи в К. — фазах внедрения, имеют металлич.
характер, причем в этих связях принимают участие и
электроны атомов углерода. Указанные К. обладают
рядом металлич. свойств (высокая электропровод-
ность, положительный термич. коэфф, электросопро-
тивления, активное взаимодействие с металламии др.).
К. данной группы обладают также весьма высокой
твердостью и жаропрочностью, что объясняется, с од-
ной стороны, заклиниванием плоскостей скольжения
в решетках металлов внедренными углеродными ато-
мами, а с другой — высокой прочностью связей между
самими металлич. атомами, проявляющейся в высоких
темп-рах плавления исходных металлов. Высокое срод-
ство углерода к большинству тугоплавких металлов
приводит к тому, что темп-ры плавления их К. выше
точек плавления самих металлов (кроме WC) — см.
табл. 3. Образовавие дополнительных связей металл —
углерод служит причиной и нек-рого дополнительного
упрочнения решетки. Однако заклинивание плоско-
стей скольжения, приводящее к устранению пластич-
ности, создает высокую хрупкость К. Кислотами К.
тугоплавких металлов не разлагаются; окисляются
и растворяются только в горячей смеси HF-j- HNO3
и в горячих р-рах или расплавах щелочей на воздухе
или с добавкой окислителя. К. тугоплавких металлов —
IV и V гр. периодич. системы, особенно TiC, обладают
высокой жаростойкостью — активно окисляться они
начинают только выше 1100—1200°. Объясняется это
образованием плотной защитной пленки низших
окислов, образующих твердые растворы с карбидной
основой.
4. К. типа Fe3C. Структуру, близкую к фазам
внедрения, но иного, более сложного типа имеют К.
группы Fe3C (Mn3C, Fe3C, Со3С, Ni3C). В Fe3C (ромбич.
решетка) атомы Fe образуют трехгранные призмы,
в центре к-рых расположены изолированные атомы С
(см. Железа сплавы). Подобная же структура и у Мп3С.
Решетки Со3С и Ni3C (весьма неустойчивые метаста-
бильные К.) несколько иного типа. Имеются данные,
что атомы металлов в этих К. расположены ио гекса-
гональной решетке, т. е. они близки к структуре ме-
тастабильного e-Fe3C и к аналогичной е-фазе в системе
Fe—N. К. группы Fe3C также обладают рядом метал-
лил. свойств (положительный температурный коэфф,
электросопротивления и др.), однако характер меж-
атомных связей металл—углерод не вполне выяснен.
Эти К., в отличие от К. тугоплавких металлов, раст-
воряются в разб. к-тах с выделением смеси углеводо-
родов с водородом и не обладают такими высокими
темп-рами плавления и твердостью, как тугоплавкие К.
Методы получения. Наиболее распространенным
методом получения К. служит прокаливание при высо-
ких темп-рах смеси соответствующего металла или
его окисла с углем в атмосфере инертного (Аг, Не)
или восстановительного (СО, Н2) газа. При получении
К. из таких трудно восстанавливаемых окислов, как
СаО, SiO2, В2О3, TiO2, ZrO2, ВеО и др., применяют
темп-ры ок. 2000° и выше; WC получают прокалива-
нием смеси W -j- С при 1400—1500°. Ряд К., в том
числе и такие неустойчивые, как Си2С», Ag2C2, Na2C2,
получают обработкой углеводородом (для указанных
трех К. — ацетиленом) р-ров солей (Си, Ag) или
металла (Na — при 220°).
Применение. К. имеют большое значение в ряде
отраслей техники. Одна из наиболее важных областей
применения К. — произ-во чугунов и сталей. Чугун п
углеродистая сталь обязаны своими твердостью и из-
носоустойчивостью и другими свойствами наличию
включений цементита Fe3C. Легированные стали,
в частности инструментальные, содержат ряд других
простых и сложных К., как, например, (Fe, Сг)3С;
(Fe, Сг)7Сй; (Ст, Mo).,3Ce; (Fe, Ст, Mo).3SCe; Fe^k^C;
Fe.2W2C и др. Дисперсные выделения К. упрочняют
сталь, повышают ее жаропрочность и износоустойчи-
вость (подробнее см. Железа сплавы). В последние деся-
тилетия начали производить особо твердые сплавы на
основе твердых тугоплавких К. (WC, такие сложные
К., как TiC—WC, TiC—ТаС—WC и т. п.) с неболь-
шими добавками (3—15%) цементирующего вязкого
металла (Со). Такие твердые карбидные сплавы изго-
товляют методами порошковой металлургии (спека-
нием брикетов из смеси порошков). Эти сплавы более
производительны, жаропрочны и износостойки, чем
инструментальные легированные стали. К. вольфрама,
хрома, марганца входят также в состав литых твердых
сплавов на кобальтовой и железной основах.
Твердые литые К. вольфрама без добавок (эвтек-
тик. сплав W2C—WC) используют для оснащения
рабочих поверхностей долот нефтяного бурения и др.
деталей и инструментов. К. бора и кремния широко
применяют в качестве абразивных и шлифующих
материалов. Кремния карбид служит также в качестве
основы высокотемпературных нагревательных эле-
ментов (стержней) электрич. печей (силитовые печи).
Из солеобразных К. весьма широко применяют каль-
ция карбид СаС2 для получения ацетилена, кальций-
цианамида и для других целей. К. используются также
в качестве восстановителей, раскислителей, катализа-
торов для нек-рых химич. процессов, как материалы
электрич. контактов, разрядников, в качестве антикор-
розионных, жаропрочных и жаростойких материа-
лов и т. п.
Лит.: Бокий Г. Б., Введение в кристаллохимию,
М., 1954; Ормонт Б. Ф., Структуры неорганических
веществ, М.—Л., 1950; S 1 d g w 1 с k N. V., The chemical ele-
ments and their compounds, v. 1, Oxford, 1950, p. 519 (Carbi-
des); Mellor, v. 5, N. Y. — [a. o.], 1946; то же, v. 5,
1952; Самсонов Г. В. иУманский Я. С., Твердые
соединения тугоплавких металлов, М., 1957;Киэффер
Р., Шварцкопф П., Твердые сплавы, пер. с нем., М.,
1958; Меерсон Г. А. иУманский Я. С., Известия
сектора физ.-хим. анализа Ин-та общей и неорганич. химии
АН СССР, 1953, 22, с. 104 — 10. Г. А. Меерсон.
КАРБИЛАМИНЫ — см. Изонитрилы.
КАРБИН — гербицид для борьбы с овсюгом в посе-
вах пшеницы. Действующим началом К. являются
4-хлорбутиниловый эфир л -х л о р-
фенилкарбаминовой кислоты (4-
хлорбутинил-М-З - хлорфен и л кар-
бамат, барбан, хлорина т) СцЩОгМСЛг,
мол. в. 258,11 — бесцветные кристаллы, т. пл. 78°,
нестойкий продукт (легко разлагается при нагрева-
нии; особенно быстро разлагается в присутствии со-
лей железа). В состав К. входит 11,8—12% действую-
щего начала, ок. 10% эмульгатора, дихлорбензол,
растворитель и стабилизатор (эпихлоргидрин глице-
рина).
4-хлорбутиниловый эфир л-хлорфенилкарбаминовой
к-ты получают след, образом:
ClCeHsNCO + НОСН,С=ССН2ОН ->
-> ClCeHsNHCOOCH2C=CCH2OH
CICflHjNHCOOСН2С-ССН2ОН -J- SOC12 —►
-> ClC0HdNHCOOCH2CSCCH3Cl + SO» + HCI
Для борьбы с овсюгом К. применяют в фазе развития
у растений 2—3 листьев; нормы расхода 300—800 г!га
(по действующему началу), однако при расходе свыше
500 г!гп возможно повреждение пшеницы.
Н. Н. Мельников.
КАРБОАНГИДРАЗА (угольная ангидраза) — фер-
мент, катализирующий обратимую реакцию образова-
429
КАРБОГИДРАЗЫ — КАРБОДИИМИДЫ
430
ния и распада угольной к-ты: Н2О -f- СО2 НСО3 -|-
-|- Н+; представляет собой Zn-металлоэнзим с мол. в.
34 000; изоэлектрич. точка соответствует pH 5,3.
Ион Zn2+ в К. связан с апоферментом (термо-
лабильной белковой частью активного комплекса фер-
мента — протеида) очень прочно и может быть отделен
диализом только в присутствии комплексообразова-
телей. Диализованный фермент, не содержащий Zn2+,
практически не обладает активностью, но может быть
активирован добавлением ионов Zn2+, а также (в мень-
шей степени) ионами Со2+, Fe2+ и Ni2+.
1 моль фермента при pH 7,3 и 0° катализирует обра-
зование 9,6 • 10’ молей СО2 в минуту. Активность К.
подавляется сульфидами, азидами, цианидами, а
также ионами Cu2+, Ag2+, Au2+, Hg2+, Особенно силь-
ное и избирательное угнетающее действие на К. ока-
зывают сульфамиды: тиофен-2-сульфамид тормозит
действие К. на 50% в концентрации 10-5 М. На этой
способности избирательно тормозить активность К.
основано лечебное применение ряда сульфамидов и,
в частности, 2-ацетиламино-1,3,4-тиодиазол-5-сульф-
амида (диакарба), используемого в качестве диуретин,
средства и при лечении глаукомы.
К. содержится только в животных тканях: у позво-
ночных — в эритроцитах, в слизистой желудка, под-
желудочной железе, слюнной железе, почках, тканях
внутреннего уха, хрусталике, яйцепроводах птиц и др.;
у беспозвоночных — в кишечной трубке, в тканях ме-
дуз и актиний. Физиологии, роль К. состоит в осво-
бождении организма животного от углекислого газа,
образующегося в процессе тканевого дыхания; кроме
того, К. принимает участие в образовании соляной
к-ты в желудке, бикарбонатов поджелудочного сока
и слюны. В яйцепроводах птиц К. играет большую
роль в образовании скорлупы яиц. К. способствует
десорбции воды в мочевых канальцах; угнетение ак-
тивности К. почки вызывает повышенное образование
мочи.
Лит.: Крепе Е. М., Усп. совр. биологии, 1944, 17,
вып. 2, с. 125; Methods in enzymology, ed.S. P., Colowick, N. O.
Kaplan, v. 2, N. Y., 1955, p. 836; Gr о о r H. van, Carbonic
anhydrase, Enzymologia, 1948, 13, 73; Keller H. [a. o.],
Biochem. Biophys. Res. Commons, 1960, 3, № 1, 24.
В. Б. Спиричев.
КАРБОГИДРАЗЫ — большая группа ферментов,
катализирующих обратимую реакцию гидролитич.
расщепления гликозидных связей в гликозидах, а
также в олигосахаридах и полисахаридах:
О хН ,--Оч ZH
!( + НОН - ‘ + R —он
'-С OR ''-о он
.1 I
Многие К. способны также катализировать трапс-
гликозилирование, т. е. перенос гликозильного остат-
ка с одного сахара на другой. В зависимости от суб-
стратной специфичности К. обычно подразделяют на
гликозидазы и полиазы. Первые гидролизуют гликозид-
ные связи в гликозидах и олигосахаридах, но не дей-
ствуют на полисахариды. Примером их могут служить
мальтаза, 8-фруктофуранозидаза, эмульсин. Полиазы
расщепляют гликозидные связи как в поли-, так и
в олигосахаридах. К их числу относятся а- и Р-ами-
лазы, целлюлаза и др. Известна и другая классифика-
ция, согласно к-рой все ферменты, гидролизующие
гликозидную связь, в том числе и нуклеозидазы, объ-
единяют под общим названием гликозидаз. Специфич-
ность действия К. определяется гл. обр. конфигура-
цией расщепляемой гликозидной связи (а- или В-),
а также природой гликозидного остатка.
К. широко распространены в природе; они входят
в состав пищеварительных соков, присутствуют в клет-
ках животных, растений и микроорганизмов. К. пище-
варительных соков животных, расщепляя полиса-
хариды пищи до моносахаридов, способствуют их
всасыванию и усвоению организмом. В растениях К.,
гидролизуя полисахариды, превращают запасные
нерастворимые углеводы в удобную для транспортиро-
вания растворимую форму.
Лит.: Диксон М., Уэбб Э., Ферменты, пер. о
англ., М., 1961; Fermente, Hormone, Vitamine und die Bezle-
hungen dieser Wirkstoffe zuelnander, hrsg. v. R. Ammon,
W. Dirscherl, Bd 1, 3 Aufl., Stuttg., 1959. В. В. Спиричев.
КАРБОДИИМИДЫ — органические производные
гипотетич. симметрич. формы цианамида RN=C=NR‘.
К. получают отщеплением сероводорода от 1\т,М'-диза-
мещенных тиомочевин действием HgO, РЬО, As2O3
или, если R и R' — алкилы, NaOCl:
—H2S
RNH-CS-NHR'-----. RN=C=NR'
Система кумулированных связей — N=C = N—• по-
добна системе связей в кетенах = С=С=О и про-
являет аналогичные свойства. К. легко присоединяют
вещества с подвижным атомом водорода с образова-
нием: в случае воды — М,]\'-дизамещенных мочевин
(1), спиртов и фенолов — тризамещенных изомоче-
вин (2), аминов — тризамещенных гуанидинов (3),
сероводорода — М,]\'-дизамещенных тиомочевин (4),
карбоновых кислот —ацилмочевин (5):
RN—C=NR + Н2О -о RNH—СО—NHR (1)
RN=C=NR + R’OH -► RN=C—NHR (2)
OR’
RN=C=NR + R’NHj -► RN=C—NHR (3)
NHR'
RN=C=NR 4- H2S -<• RNH-CS-NHR (4)
RN=C=NR 4-R'COOH -> R'CO-NR-CO—NHR (5)
В ряде случаев взаимодействие карбоновых кислот
с К. приводит к соответствующим ангидридам:
2R'COOH4- RN=C=NR -о (R'CO)2O 4- RNH—СО—NHR
Аналогичным образом К. присоединяют HCN и мало-
новый эфир:
HCN
RNH—C=NR
I
CN
RNH-C=NR
I
CH(COOC2H3)2
rn=c=nr-
СН2(СООС2Н5)2
К. применяют в качестве дегидратирующих агентов
для создания сложноэфирных, амидных, лактамных и
пептидных связей:
RCOOH -(- R’OH 4- CeHiiN=C=NCeHii -►
-о RCOOR' 4- CeHnNHCONHCeHn
RCOOH 4- R'NH2 -(- CeHnN=C=NCeHii -►
-► RCONHR' -(- C6HnNHCONHCeHn
I I
—C С-COOH 4- CeHiiN=C=NCeHn -►
i I
nh2
I I
-► —C--C- -(-CsHuNHCONHCsHh
I I
NH—C=O
RCOOH 4- R'CHNH2COOH 4- CeHnN=C=NCeHii -►
-<• RCONHCHR'COOH 4-CeHiiNHCONHCeHn
В частности, с помощью К. впервые был замкнут
лактамный цикл в пенициллоиновых к-тах с обра-
зованием пенициллинов. К. применены для реакций
фосфорилирования:
О
-Н2О II NaOH
RO-PO(OH)2 4- R’OH-----► RO—РО—OR'-----►
I
OH
-► R’O-PO(OH)3
431
КАРБОИДЫ —КАРБОКСИЛАТНЫЕ КАУЧУКИ
432
и для получения эфиров пирофосфорной кислоты:
О О
zO —Н20 II II
2(RO)2P<(-----> (RO)2P-O-P(OR)2
ХОН
С использованием К. удается осуществить селективную
деструкцию пептидов по схеме:
CHSCONHCH2CONHCH2COOH + RN=C=NR ->
NaOH
CH3CONHCH>CONHCHaCONRCONHR--------►
CHsCONHCH2COOH + NH2CH2CONRCONHR
Лит.: Ho ube n-We 11, 4 aufl., Bd 8, Stuttg., 1952,
S. 178; Khorana H. G., J. Chem. Soc., 1952, 2081;
Sheehan J. C., Hess G. P., J. Amer. Chem. Soc., 1955,
77, Ns 4, 1067; G 1 1 h a m P. T., T e n e r G. M., Chem.
Ind., 1959, M 17, 542. В. Л. Дяткин.
КАРБОИДЫ — нерастворимые в горячем бензоле
высокоассоциированные продукты сложной малоизу-
ченной структуры, содержащиеся в продуктах термич.
переработки твердых топлив (нефтяных гудронах,
асфальтах, смолах), а также образующиеся при воз-
действии тепла и окислителей на угли, нефти и смолы
в процессе их переработки. По внешнему виду К.
похожи на асфальтены.
Основными факторами, определяющими образова-
ние К., являются продолжительность и интенсивность
нагрева, химич. состав исходного продукта и действие
химич. агентов, ускоряющих или тормозящих про-
цессы карбоидообразования. Образование К. играет
большую роль при крекинге нефтяного сырья. Глав-
ным источником карбоидообразования при этом яв-
ляются ароматич. углеводороды. Этот процесс может
служить типичным примером т. наз. консеку-
тивных реакций (т. е. реакций, при к-рых
конечный продукт образуется не сразу, а через боль-
шее или меньшее число промежуточных соединений).
Наиболее просто К. образуются на основе ароматич.
углеводородов, не имеющих боковых цепей (бензол,
нафталин, фенантрен и т. д.). Молекулы ароматич.
углеводородов конденсируются, образуя продукты
с постепенно увеличивающимся мол. весом. Указан-
ный процесс схематически может быть представлен
следующим образом:
ароматич. 1 продукты I я-лЯЛЬТрны-* К
углеводороды / конденсации j асфальтены к.
Конечными высокоассоциированными продуктами
дальнейшего термич. разложения углеводородов по
приведенной схеме являются коксы.
Лит.: Наметкин С, С., Химия нефти, [3 изд.],
М., 1955; Тиличеев М. Д., Химия крекинга, М.—Л.,
1941; Веселовский В. С., Химическая природа го-
рючих ископаемых, М., 1955. А. М. Кунин.
КАРБОКСИГЕМОГЛОБИН — см. Гемоглобины.
КАРБОКСИЛАЗА — фермент, катализирующий
практически необратимую реакцию декарбоксилиро-
вания пировиноградной к-ты с образованием ацеталь-
дегида:
CHsCOCOOH -> chscho:+ со2
К. декарбоксилирует также высшие гомологи пирови-
ноградной к-ты — вплоть до а-кетоглутаровой. В ка-
честве побочного продукта реакции наряду с альдеги-
дом обычно образуются ацилоины, в частности при
декарбоксилировании пировиноградной к-ты — аце-
тоин, образование к-рого может быть представлено
следующим образом:
2СН3СОСООН -> CHSCH(OH)COCHS + 2CO2
В состав молекулы активной К., помимо белкового
апофермента, входят тиаминдифосфат (кокарбокси-
лаза), играющий роль кофермента, и ион Mg2+. Оба
кофактора необходимы для проявления К. фермента-
тивной активности. О механизме действия К. см.
Тиамин ди фосфат.
Активность К. тормозится п-хлормеркурбензоатом,
катионами тяжелых металлов, ацетальдегидом, взятым
в высоких концентрациях, а также нек-рыми анало-
гами тиаминдифосфата, в частности окситиаминдифос-
фатом. К. играет существенную роль при спиртовом
брожении, она присутствует в дрожжах и нек-рых
других микроорганизмах, а также в высших расте-
ниях; в животных тканях К. отсутствует. Наиболее
высокоочищенные препараты К. получены из дрожжей
и из пшеничных зародышей. Мол. вес К. из дрожжей
ок. 75 000, из пшеничных зародышей ок. 1 000 000;
изоэлектрич. точка дрожжевой К. соответствует
pH 5,1; 1 моль фермента при 30° катализирует превра-
щение 103 молей пировиноградной к-ты в минуту. Оп-
тимум pH для ферментативной активности соответ-
ствует 6,0.
Лит.: Болдуин Э., Основы динамической биохимии,
пер. с англ., М., 1949, с. 180JMethods in enzymology, ed. ty
S. P. Colowlck, N. O. Kaplan, v. 1, N. Y., 1955, p. 460; Fermente,
Hormone, Vltamine und die Beziehungen dleser Wlrkstoffe
zuelnander, hrsg. v. P. Ammon und W.Dirscherl, Bd 1, 3 Aufl.,
Stuttg., 1959. В. Б. Спиричев.
КАРБОКСИЛАТНЫЕ КАУЧУКИ — полимеры
бутадиена, изопрена, сополимеры бутадиена со стиро-
лом, бутадиена с нитрилом акриловой к-ты и др.,
содержащие в молекулярной цепи небольшое число
остатков метакриловой к-ты. Наибольшее практич.
значение имеют К. к., являющиеся сополимерами
бутадиена со стиролом или с нитрилом акриловой
к-ты. К. к. получают сополимеризацией различных
мономеров с метакриловой к-той в водной эмульсии
при 5° в кислой среде; в щелочных средах метакрило-
вая к-та переходит в водную фазу и не участвует
в реакции. Для инициирования применяют обратимые
окислительно-восстановительные системы, сущест-
венно ускоряющие процесс полимеризации. Содержа-
ние в цепи небольшого числа остатков метакриловой
к-ты (одна карбоксильная группа на 200—300 угле-
родных атомов основной цепи, что соответствует
1—2 вес. % метакриловой к-ты) не оказывает заметного
влияния на темп-ру стеклования полимеров. Нек-рое
повышение темп-ры стеклования наблюдается при со-
держании кислоты более 3%.
Вулканизуют К. к. в основном окислами двухвалент-
ных металлов (MgO, СаО и др.), к-рые, реагируя с кар-
боксильными группами сополимеров, образуют про-
странственную вулканизационную сетку; это дает
возможность получать ненаполненные резины (без
активных усилителей), по прочности не уступающие
резинам из натурального каучука и значительно пре-
восходящие в этом отношении вулканизаты из бута-
диен-стирольного каучука общего назначения. Эти
резины характеризуются повышенной способностью
к ориентации молекулярных цепей; при растяжении
эффект ориентации возрастает с повышением модуля
резины (напряжение, вызывающее заданное относи-
тельное удлинение), что достигается как увеличением
числа карбоксильных групп в цепи, так и увеличением
количества вводимых окислов металлов. Монотонное
возрастание ориентации и прочности с повышением
модуля резины в К. к. свидетельствует о том, что эти
эффекты вызваны не структурой полимера, а специфич-
ностью вулканизационной сетки. Ненаполненные
резины из К. к., вулканизованные с помощью серы и
ускорителей вулканизации в отсутствие окислов
металлов, имеют весьма низкую механич. прочность
(ок. 20 кг/см2} и не проявляют ориентационного эф-
фекта в рентгенограмме. Резины из бутадиен-стироль-
ного К. к. отличаются хорошим сопротивлением тепло-
вому старению (в связи с отсутствием серы и полисуль-
фидных связей), высокими износостойкостью и стой-
костью к разрастанию порезов и повышенной эластич-
ностью. Лучшими показателями обладают бутадиен-
стирольные К. к. с содержанием 1—2% метакриловой
433
КАРБОКСИЛАТНЫЕ КАУЧУКИ — КАРБОКСИЛИРОВАНИЕ
434
к-ты (см. табл. 1, где данные в колонках 1 относятся
к бутадиен-стирольному каучуку холодной полимери-
зации, а 2 — к бутадиен-стирольному К. к. того же
типа с 1,5% метакриловой к-ты).
Таблица 1
Показатели Ненапслнепные резины Сажевые резины (30 вес. ч. газовой канальной сажи)
2 1 2
Модуль при 300% растяже-
нии, кг/см3 ........ 13 30-45 35 70—90
Предел прочности при раз-
рыве, кг/см3 ........ 66 300-400 236 350-450
Относительное удлинение, % 810 850 750 800
Остаточное удлинение, % 16 24 27 61
Эластичность по отскоку, % 57 67—70 59 32
Введение сажи и др. активных наполнителей не
приводит к усилению резин из К. к., как это наблю-
дается для большинства синтетич. каучуков. Проколы
и надрезы, нанесенные на образцы резины из К. к.,
практически не разрастаются после многократной
деформации (360 тыс. циклов), в то время как резины
из обычного бутадиен-стирольного каучука при этом
разрушаются после 130—140 тыс. циклов.
Бутадиен-нитрильный К. к. обычно вырабатывается
с содержанием 3% метакриловой к-ты. Резины на
основе этого каучука имеют по сравнению с резинами
из обычных бутадиен-нитрильных каучуков более
высокие показатели прочности и твердости, сопротив-
ления раздиру, эластичности, морозостойкости, сопро-
тивления истиранию и высокую стойкость к удару при
низких темп-рах (см. табл. 2, где данные в колонках
1 относятся к бутадиен-нитрильному каучуку, содер-
жащему 26% нитрила акриловой к-ты, а в колонках
2 — к этому же каучуку, содержащему дополнительно
3% метакриловой к-ты).
Таблица 2
Показатели Ненаполнепные резины Сажевые резины (30 вес. ч. ка- нальной сажи)
1 2 1 2
Модуль при 300% растяже- нии, кг/смг 14 60-70 86 110—140
Предел прочности при раз- рыве, кг/см2 ........ 80 400—500 343 380-450
Относительное удлинение, % 870 700—800 615 650—750
Остаточное удлинение, % 16 10-20 61 10—20
Эластичность по отскоку, % — 45-50 32 40-45
Сопротивление раздиру, кг/см ............ - 37* 36* 48*
*30% нитрила акриловой к-ты.
Резины, полученные на основе К. к. с применением
в качестве вулканизующих агентов только окислов
двухвалентных металлов, проявляют заметную теку-
честь при темп-рах порядка 100° и выше и особенно
при многократных деформациях. Текучесть резины
может быть предотвращена созданием дополнительных
связей путем введения в резиновую смесь, кроме
окислов, также небольших количеств тиурама, серы
и др. веществ, образующих дополнительные связи
с макромолекулами полимера. Серьезным недостатком
К. к. является преждевременная подвулканизация
сырых резиновых смесей при использовании в качестве
вулканизующих агентов окислов двухвалентных ме-
таллов. Это связано с реакциями солеобразования,
к-рые г начинают протекать частично уже в процессе
смешения и при дальнейшей переработке. Определен-
ными приемами это явление может быть значительно
ослаблено. Большое применение во многих отраслях
техники находят латексы синтетические карбоксил-
содержащих полимеров (бутадиен-стирольные и бута-
диен- нитрильные).
Лит.: Долгоплоск Б. А. [и др.], Каучук и резина,
1957, N» 3, И; № 6, 1; 1958, № 8, 1; Каучук и резина, 1959,
М 5, 57. А. К. Никитин.
КАРБОКСИЛИРОВАНИЕ — непосредственное вве-
дение карбоксильной группы в органич. соединения
действием СО2. Особенно легко карбоксилируются
металлоорганич. соединения. Так, если в раствор сме-
шанного магнийорганич. соединения пропускать СО2
или ввести твердую СО2, то после гидролиза обра-
зуется соответствующая карбоновая к-та:
RMgX +со2 RCOOMgX ->• RCOOH
Аналогичным образом можно карбоксилировать
литий- и другие металлоорганич. соединения. Так,
для получения 2-пиридилуксусной к-ты переводят
а-пиколин в пиколиллитий, прибавляют избыток
твердой СО2 и из образовавшейся соли выделяют сво-
бодную кислоту:
СН2СОО1<
-СН2СООН
На К. основан один из главных способов получения
кислот ацетиленового ряда:
СО,
RC=CNa----► RC_CCOONa -» RC=CCOOH
Большое практич. значение имеет способ получения
ароматич. оксикислот карбоксилированием фенолятов
натрия по Кольбе; так, в частности, получают салици-
ловую кислоту:
C6H5ONa + CO2 -► o-HOC6H4COONa
Аналогичным образом К. л1-амипофенола можно полу-
чить n-аминосалициловую к-ту. Один из методов син-
теза ароматич. к-т состоит в действии СО2 на арома-
тич. соединения в присутствии хлористого алюминия:
A1CI,
С8НдС1-|-СО2---► п-С1С6Н4СООН
К. имеет препаративное значение и, кроме того,
применяется для выяснения направления образования
металлоорганич. соединений, напр. реактивов Гринь-
яра, при наличии нескольких атомов галогенов (часто
различных) в молекуле.
К. играет большую роль в нек-рых ферментативных
биологич. процессах. Ферменты, катализирующие
К., получили название карбоксилаз. При фотосинтезе
первичным акцептором СО2 служит рибулезо-1,5-ди-
фосфат; образующееся соединение распадается на две
молекулы 3-фосфоглицериновой к-ты:
CH2OPOSH2 1 CH2OPOSH2
со 1 нос-соон CH.OPOSH2
н<1он + со2 СО -► 2 СНОН
неон неон соон
H2COPOSH2 H2COPOSH2
Подобную же роль играет рибулезодифосфат и прп
связывании СО2 в процессе хемосинтеза (серные бак-
терии).
Способность гетеротрофных организмов использо-
вать СО2 для синтеза органич. веществ была впервые
открыта А. Ф. Лебедевым в 1921. Благодаря более
поздним исследованиям с помощью изотопных инди-
каторов обнаружено многостороннее значение реак-
ций К. у гетеротрофных организмов (микробов и
животных) в разнообразных биосинтезах: жирных к-т,
углеводов, аминокислот, пуриновых и пиримидиво'-
435
КАРБОКСИЛИРОВАНИЕ — КАРБОКСИМЕТИЛЦЕЛЛЮЛОЗА
436
вых оснований и др. Весьма существенным видом
реакций этого рода является обратимое восстанови-
тельное К. пировиноградной к-ты с участием восстанов-
ленной формы трифосфопиридиннуклеотида ТПН[Н]
(1), а также образование С4-дикар боновых к-т К. фосфо-
енолпировиноградной к-ты в щавелевоуксусную,
к-рое в животных тканях осуществляется по ур-нию
(2), где ипозиндифосфат (ИДФ) служит акцептором
фосфата:
СООН
СН. 1 сн2 н+ -» 1 снон СООН
со -р со2 + тпн[н] + 1
СООН СООН
сн2 COPOs Н2 + СО2 + ИДФ - 1 сн2 2 I + ИТФ
СООН со СООН (инозинтрифосфат)
(2)
Приведенные выше реакции К. обеспечивают беспе-
ребойное течение цикла трикарбоновых кислот, по-
полняя убыль его членов, постоянно отвлекаемых
в русло азотистого обмена (а-кетоглутаровая к-та,
сукцинил-КоА, щавелевоуксусная к-та). В зеленых
растениях (листья шпината) образование щавелевоук-
сусной к-ты К. из фосфоенолпирувата протекает иначе:
СООН
СН2 |
1 СН2
COPOSH2 4- СО2 —► | 4- неорганич. фосфат
I СО
СООН |
СООН
Особенностью этого варианта К. является его необ-
ратимость; в результате улавливается СО2, выделяю-
щаяся при дыхании растений в темноте.
Во многих реакциях К. в качестве простетич. группы
соответствующих карбоксилаз функционирует биотин,
принимающий прямое участие в переводе СО2 в «актив-
ную» форму. К подобным реакциям К. относятся:
а) К. ацетил-КоА до малонил-КоА:
СН3СО—SKoA + СО2 + АТФ
HOOC-CH2-CO-SKoA + АДФ + Р
Этой реакцией открывается цепь превращений, при-
водящая за счет энергии распада аденозинтрифосфата
(АТФ), к синтезу высших жирных к-т. СО2 играет ката-
литич. роль, не входя в состав конечного продукта
синтеза — жирной к-ты, т. к. на следующем после К.
этапе.— конденсации малонил-КоА с ацил-Ко.А —
происходит одновременно отщепление новообразо-
ванной карбоксильной группы в виде СО2 (см. также
Декарбоксилирование), что делает всю цепь реакций
синтеза жирных к-т необратимой:
KoA-S-CO-CH3-COOH + КоА—S—COR -о
-о KoA-S-CO-CH2-COR + СО2 + KoASH
б) К.р-метилкротонил-КоА в р-метилглутаконил-КоА
(см. Декарбоксилирование}.
в) К. пропионил-КоА до метилмалонил-КоА —
промежуточного этапа на пути превращения пропио-
новой к-ты в янтарную:
CHsCH2-CO-S-KoA + СО2 + АТФ 7^
— CHsCH(COOH)-CO-S-KoA + АДФ + Р
Далее происходит изомеризация метилмалонил-КоА
в сукцинил-КоА, катализируемая ферментом изоме-
разой, простетич. группой к-рой оказалось производ-
ное витамина В12 — кобаламиновый коэнзим:
CHsCH(COOH)-CO-S-KoA KoA-S-CO-CH2-COOH
При этой изомеризации перемещается карбоксильная
группа, связанная с КоА. Указанные реакции вводят
остаток пропионовой к-ты в цикл трикарбоновых к-т.
Установлено (Люнен, 1959), что СОг должна быть
предварительно активирована путем образования
промежуточного соединения с биотином. СО2 присое-
диняется к одному из атомов азота, находящихся
рядом е карбонильной группой биотина. В результате
продукт присоединения приобретает свойства ангид-
рида к-ты. С—N-связь должна быть поляризована
в направлении азота так, что СО2-группа может функ-
ционировать как ацилирую-
щий агент по отношению
к карбаниону ацил-КоА.
Этот механизм, по-виднмо-
му, действителен также и
для К. ацетил-КоА и про-
пионил-КоА.
СО„
ОС—N XNH
“° н<р-----|н
Н2Сх^СН(СН2)4СООН
В 1960 обнаружен новый тип К. — транс-к а р-
боксилирование (Вуд). Из пропионовокислых
бактерий выделена ферментная система, катализи-
рующая перенос карбоксильной группы с метилмало-
нил-КоА на пировиноградную к-ту с образованием
соответственно пропионил-КоА и щавелевоуксуспоп
к-ты:
CH3CH(CHS)-СО—S—КоА + СН3СОСООН —
д: СН3СН2СООН + ноос-сн2-сосоон
Лит: Н о u b е n - W е у 1, 4 Aufl., Bd 8, Stuttg., 1952,
S. 359; Лебедев А. Ф., Иав. Донского ун-та, 1021, кн. 1,
25; Lynen F., J. Cellular and Compar. Physiol., 1959,
54, Suppl. 1; Lynen F. [u. a.], Angew. Chemie, 1959,
71, № 15/16, 481; Wood H. G., Stjernholm R.,
Proc. Nat. Acad. Sci. USA, 1961, 47, Д8 3, 289.
Я. Ф. Комиссаров, В. С. Шапот.
КАРБОКСИЛЬНАЯ ГРУППА — см. Карбоновые
кислоты.
КАРБОКСИМЕТИЛЦЕЛЛЮЛОЗА — простой эфир
целлюлозы и гликолевой кислоты; общая формула
[С6Н7О2(ОН)з_ж(ОСН2СООН)ж]. К. получают при обра-
ботке щелочной целлюлозы (хлопковой или древесной)
монохлоруксусной к-той или ее натриевой солью.
В технич. типах К. содержится от 0,5 до 1,2 карб-
оксиметильных групп на элементарное звено макро-
молекулы целлюлозы. Определение степени замеще-
ния проводят путем осаждения медной соли К. с по-
следующим определением количества связанной меди,
а также титрованием (потенциометрич. или в присут-
ствии индикатора). В реакции карбоксиметилирова-
ния первичные гидроксильные группы элементарного
звена макромолекулы целлюлозы примерно вдвое
более реакционноспособны, чем гидроксильные груп-
пы, находящиеся у вторичных атомов углерода. К.
в форме свободной к-ты нерастворима в воде и органич.
растворителях; растворяется в водных слабощелочных
р-рах (рНок. 11), образуя вязкие р-ры натриевой солиК.
Главные области применения К. и Na-соли К. —
произ-во моющих веществ, нефтяная, фармацевтич.,
текстильная, пищевая и др. отрасли пром-сти. Воз-
можность использования Na-соли в качестве моющего
средства связана с адсорбцией полимерного аниона К.
на целлюлозном волокне, что увеличивает электро-
статич. отталкивание от волокна грязевых частиц,
в большинстве случаев также несущих отрицательный
заряд. Стабилизация глинистых суспензий при введе-
нии Na-соли К. (основное направление ее использова-
ния в нефтяной пром-сти) осуществляется за счет
образования структурированной защитной пленки.
В текстильной пром-сти К. используют для шлихто-
вания нитей основы, а также как загустку для печат-
ных красок. Карбоксиметилирование хлопчатобумаж-
ных тканей применяется для модифицирования их
свойств (напр., с целью придания ионообменных
свойств).
Лит.: Роговин 3. А., Шорыгина Н. Н., Хи-
мия целлюлозы и ее спутников, М.—Л., 1953, с. 497—98;
Петропавловский Г. А., Ж. прикл. химии, 1959,
32, вып. 2, 241; Reid J. D., Daul G. С., Text. Res.
J., 1947, 17, № 10, 554; Hoffpaulr C. L., Guthrie
J. D., там же, 1950, 20, 617. Л. С. Гальбрайх.
437
КАРБОКСИПЕПТИДАЗА А —КАРБОНАТЫ
438
КАРБОКСИПЕПТИДАЗА А — протеолитический
фермент, относящийся к группе С-копцевых экзопеп-
тидаз (см. Пептидазы)', катализирует гидролитич.
отщепление С-копцевых аминокислотных остатков
в белках и пептидах:
R R R
I I I
H2NCHCO -(HNCHCO)ft -HNCHCOOH + Н2О ->
R R R
I I I
-> H2NCHCO-(HNCHCO)n_1-HNCHCOOH +
R
I
+ h2nchcooh
Необходимым условием действия К. А является нали-
чие у отщепляемого аминокислотного остатка сво-
бодной а-карбоксилыюй группы. Активность К. А
особенно велика по отношению к субстратам с арома-
тич. С-концевыми аминокислотными остатками (фал.,
тир., трнп.) или алифатич. остатками с разветвленной
боковой цепью (лейц., изол.). К. А не расщепляет'белки
или пептиды, в которых С-концевыми аминокислотными
остатками являются остатки двухосновных амино-
кислот (арг., лиз.) или пролина. К. А гидролизует
также нек-рые синтетич. эфиры оксикислот.
К. А входит в состав желудочно-кишечного сока
высших животных и играет важную физиологии, роль
в процессе пищеварения, обеспечивая расщепление
продуктов первичного переваривания белков до амино-
кислот. К. А вырабатывается поджелудочной железой
в виде неактивного зимогена — прокарбоксипептида-
зы — и активируется в кишечнике трипсином.
К. А, выделенная из поджелудочной железы быка
в кристаллич. состоянии, представляет собой белок
с мол. в. 34 000 и [а]д = —18°. Молекула ее состоит
из одной полипептидной цепи, включающей 310 ами-
нокислотных остатков, с остатками аспарагина в ка-
честве С- и N-концевых аминокислот. Изоэлектрич.
точка фермента лежит при pH 6,0, оптимум действия—
при pH 7,5. К. А представляет собой типичный метал-
лоэпзим, содержащий на 1 моль белка 1 грамм-атом
Zn2+. Ион цинка можно отделить от белка длительным
диализом против буферных р-ров, содержащих такие
металлосвязывающие комплексообразующие соедине-
ния, как 1,10-фенантролин; при этом К. А утрачивает
свою ферментативную активность. При добавлении
к неактивному белковому апоферменту Zn2+ актив-
ность полностью восстанавливается. Ферментативная
активность может быть восстановлена также другими
металлами первой переходной группы, которые по
способности активировать апофермент располагаются
в след, порядке: Со2+ > Ni2+ > Zn2+ > Mn2+ > Fe2+.
Активность Со-содержащей К. А примерно в2 раза вы-
ше активности Zn-содержащей.
Замена в К. А иона Zn2~на ионы Сй2+или Hg2+npn-
водит к изменению субстратной специфичности фермен-
та, выражающемуся в полной утрате пептидазной ак-
тивности и усилении эстеразной активности. Актив-
ный центр К. А образован ионом Zn2+, комплексно свя-
занным с SH-группой единственного остатка цистеина,
а также атомом азота имидазольного кольца гисти-
дина или же а-аминогруппы какой-то другой амино-
кислоты, входящей в состав молекулы К. А. В связи
с тем, что в активный центр К. А входит ион ме-
талла, ее активность эффективно подавляется таки-
ми металлосвязывающими комплексообразователями,
как 1,10-фенантролин, 8-оксихинолин-5-сульфокисло-
та, а,а'-дипиридпл, комплексон III (версен), диэтилди-
тиокарбамат, меркаптоэтапол, оксалат, цитрат и др.
Торможение К. А нек-рыми из этих соединений может
быть предупреждено добавлением Си2+ или Fe2+.
Конкурентными ингибиторами К. А являются 0-фенил-
пропионовая к-та и нек-рые а-амииокпелоты.
Из поджелудочной железы выделен также фермент,
названный карбоксипептидазой В, гидро-
лизующий белки и пептиды с двухосновными С-конце-
выми аминокислотными остатками лизином или арги-
нином.
Свойство К. А катализировать последовательное
отщепление отдельных аминокислотных остатков от
С-конца молекул полипептидов и белков широко ис-
пользуется в научных исследованиях для расшиф-
ровки химич. строения этих веществ.
Лит.: Валле Б. Л., Труды V Международного био-
химического конгресса, симпозиум IV, тетрадь 3, М., 1961,
с. 33; Ne ur at НН., в кн.: Theenzymes, 2 ed., v. 4, N. Y.—
L., 1960, p. 11—36. В. Б. Спиричев.
КАРБОЛИНЕУМ — препарат, па основе сланце-
вой или торфяной (реже каменноугольной) смолы,
применяемый в виде водной эмульсии (приготовляемой
в мягкой теплой воде) для борьбы с вредителями садо-
вых культур.
Исходным продуктом для приготовления слан-
ц е вчо г о К. является фракция (205—350°) сланцевой
смолы; в эту фракцию, нагретую до 55—60°, медленно
добавляют при помешивании талловое масло, после
растворения к-рого постепенно вводят 40%-ный
водный р-р едкого натра. Полученную реакционную
смесь интенсивно перемешивают в течение 3 часов
при 55—60°, затем к ней добавляют небольшое коли-
чество дистиллированной воды. На 1 т К. расходуется
860 кг сланцевой фракции, 80 кг таллового масла и
13 кг едкого натра (в пересчете на 100%-ный). Сланце-
вый К. применяют гл. обр. для уничтожения яиц ябло-
невой тли, зимней пяденицы, яблоневой медяницы
и др. вредителей плодовых деревьев.
Т орфянойК. готовят на основе фракции (220—
320°) торфяного дегтя и эмульгатора (зеленое мыло,
канифольное мыло, технич. мыло, а также на смесях
указанных продуктов с нейтрализованным газойле-
вым контактом). Препарат имеет следующий состав:
9—10% фенолов, 2—3% органич. оснований, 46—
47% нейтральных масел, 38—42% эмульгатора и
до 2% едкого натра. Действующим началом в торфя-
ном К., кроме фенолов, являются органич. осно-
вания и ароматич. углеводороды, содержащиеся в
исходных торфяных маслах. Торфяной К. — эффек-
тивное средство борьбы с многочисленными вреди-
телями садовых, огородных и полеводческих культур
(луковой мухой, паутинным клещиком, сливовой и
яблоневой тлей, смородинным клещиком, тлей па
посевах овса, сладкого люпина и др.).
Лит.: Труды научно-технического совещания по приме-
нению сланцевых продуктов для борьбы с вредителями, бо-
лезнями и сорняками сельскохозяйственных растений, Таллин,
1956; Хорсфолл Д. Г., Фунгисиды и их действие, пер.
С англ., М., 1948; Труды Московского Торфяного ин-та, 1955,
вып. 3. А. М. Кунин.
КАРВОЛОВАЯ КИСЛОТА — см. Фенол.
КАРВОЛОВОЕ МАСЛО — см. Фенольные масла.
КАРБОНАТЫ — соли угольной кислоты Н2СО3.
Наибольшее значение имеют нормальные К. (с анио-
ном COV) и кислые (или бикарбонаты, с аниопом
HCOj). Из нормальных К. в воде растворимы только
соли щелочных металлов, аммония и таллия. В резуль-
тате значительного гидролиза растворы их показы-
вают щелочную реакцию. Наиболее трудно раство-
римы нормальные К. кальция, стронция, бария и
свинца (2-валентного). Все бикарбонаты, напротив,
хорошо растворимы в воде. При реакциях обменного
разложения осадки нормальных К. образуются лишь
тогда, когда растворимость их значительно меньше,
чем соответствующих гидроокисей. Это имеет место
для кальция и его аналогов, скандия и его аналогов,
лантанидов, а также Ag+, РЬ2+, Мп2+иСй2+. Остальные
катионы при взаимодействии с растворимыми К. дают
либо основные углекислые соли (большинство), либо
даже свободные гидроокиси (напр., Als+, Fe3+, Crs+).
439
КАРБОНИЛЫ МЕТАЛЛОВ
440
Для ряда катионов (многих 3-валентпых, Hg2+, Cu2+ и
др.) нормальные К. вообще не могут быть получены,
т. к. они термически неустойчивы уже при обычных
условиях.
Образование кристаллогидратов для К. мало харак-
терно, они известны лишь у производных немногих
элементен (Na, Sc, Y, La и лантанидов). При нагрева-
нии безводных К. они, как правило, разлагаются еще
до достижения точки плавления. Исключение пред-
ставляют К. щелочных металлов и таллия. Бикарбо-
наты при нагревании переходят в нормальные К.:
2NaHCO3= Na2CO3 -ф- Н2О -ф- СО2. Сильными кисло-
тами нормальные и средние К. разлагаются с выделе-
нием СО2.
В природе нормальные К. широко распространены,
составляя одну из групп минералов. Из яих наиболее
обычными являются безводные К. из ряда кальцита
(кристаллы тригональной системы) и арагонита (ром-
бич. системы). Главнейшие из них: ряд кальцита —
кальцит СаСО4, магнезит MgCO3, сидерит FfCO3,
родохрозит МпСО3, смитсонит ZnCO3; ряд арагонита —
арагонит СаСО3, витерит ВаСО3, стронцианит SrCO3,
церуссит РЬСО3 и др. Многие природные К. содержат
сложный катион из днух или более металлов, изо-
морфно замещающих друг друга, папр. анкерит
Ca(Mg, Fe)[CO3]2 и др. Распространены также двойные
соли определенного состава с ограниченной смеси-
мостью, напр. доломит СаСО3 • MgCO3. Из водных К.
следует отметить малахит СиСО3 • Си[ОН]2 и азурит
2СнСО3 • Си[ОН]2. Скопления К. кальция н магния
образуют мощные толщи осадочных и метаморфич.
горных пород (известняки, мраморы, доломиты). В зо-
нах окисления рудных месторождений в результате
воздействия вод, содержащих СО2, образуются много-
численные К. цинка, свинца, меди и других тяжелых
металлов. Многие природные К. являются весьма цен-
ными металлическими рудами', таковы К. цинка,
свинца, меди, железа, марганца и др. К. кальция,
магния, бария и др. представляют важное нерудное
сырье, употребляющееся в строительном деле, в виде
огнеупорен (магнезит), в химич. пром-сти, оптике
й т. д. Из синтетич. К. в технике широко применяется
сода (углекислая Na2CO3 и двууглекислая NaHCO3).
Бикарбонаты выполняют важную физиология, роль,
являясь буферными веществами (см. Буферные рас-
творы), регулирующими постоянство реакции крови.
Лит.: Некрасов Б. В., Курс общей химии, 13 изд.,
М„ 1961.
КАРБОНИЛЫ МЕТАЛЛОВ — соединения метал-
лов с окисью углерода общей формулы (Me)m(CO)n.
Впервые был открыт карбонил никеля Ni(CO)-i
(Монд, 1890). С тех пор получены карбонилы многих
металлов и нек-рых неметаллов. Карбонилы переход-
ных металлов отличаются значительной устойчивостью
и отсутствием дипольного момента. Карбонилы щелоч-
ных металлов [МеСО]2 загораются на воздухе, нераст-
воримы в органич. растворителях и обладают ярко
выраженным полярным характером. Для меди, се-
ребра и золота известны лишь карбоиилгалогениды,
Ме(СО)Х, устойчивые только в атмосфере окиси угле-
рода.
Наиболее важны и изучены карбйнилы пере-
ходных металлов — «истинные карбонилы»:
VI гр. — Сг(СО)в, W(CO)o, Мо(СО)«
VII гр. — Мп2(СО)1о, Re(CO)!0
VIII гр. - Fe(CO)s, Fe2(CO)a, Fe3(CO)42, Со2(СО)8
Co4(CO)I2, Ni(CO)4, Ru(CO)3, Ru2(CO)8, Ru3(CO)12, Rh2(CO)8
Rh4(CO)n, Os(CO)3, Os2(CO)8, Ir2(CO)8
K. m. могут быть «одноядерными» и «мпогоядсрни-
ми» в зависимости от числа атомов металла в моле-
куле; известны также смешанные карбонилы, напр.
{Fe(CO)4]Hg; [Co(CO)4]2Zn. Связь между атомами угле-
рода и кислорода в одноядерных карбонилах близка
по характеру к связи в окиси углерода; расстояние
С—О в окиси углерода лишь немного меньше. В одно-
ядерных карбонилах металл непосредственно связан
с атомами углерода СО-групя, причем атомы металла,
кислорода и углерода расположены на одной прямой.
Карбонилы переходных металлов имеют различную
пространственную структуру, так, Ni(CO)4 построен
тетраэдрически, Сг(СО)в октаэдрически, a Fe(CO 5
имеет структуру тригональной бипирамиды. Атомы
углерода и металла связаны между собой за счет
пары электронов, предоставляемой СО-груипой; при
этом электронная оболочка металла заполняется до
стабильной оболочки соответствующего инертного
газа. Увеличение отрицательного заряда на металле
компенсируется участием rf-электронов металла в об-
разовании Me—С-связи так что последняя приобретает
частично двоесвязный («полуторный») характер (рас-
стояние Me—С примерно на 0,16 А меньше суммы ко-
валентных радиусов). Такое участие (/-электронов
в образовании связи возможно только для переходных
металлов, поскольку они обладают недостроенным
(/-уровнем.
Металлы с нечетным атомным номером образуют
только многоядерные карбонилы; последние известны
и для иек-рых металлов с четным атомным номером,
напр. для железа. Строение Fe2(CO)9 и Со2(Со)8 отра-
жают следующие формулы:
\ О
II
ОС с со
ОС —Fe----Fe —СО
ос \с^ хсо
схо
II
О
ОС с со
ОС-Со Со—СО
ОС с со
II
о
Т. о., в мвогоядерных карбонилах имеется ковалент-
ная связь металл — металл и присутствуют СО-группы
двух типов; СО-группы, связанные с двумя атомамп
металла, близки по своей природе к карбонильным
группам в кетонах.
Известно много соединений переходных металлов,
близких по строению к карбонилам; одни из них яв-
ляются переходной ступенью от К. м. к чисто неорга-
нич. соединениям (таковы, напр., нитрозилкарбонилы
металлов и нитрозильные соединения); другие, на-
против, служат мостом, связывающим К. м. с «сэнд-
вичсвыми» соединениями типа ферроцена.
При действии на К. м. окиси азота, аминов, фосфи-
нов и др. часть СО-групп замещается:
Fe(CO)3 + 2NO -> Fe(CO)2(NO)2 + ЗСО
Соединениями такого рода являются:
[(C0H5)3P]2N1(CO)2; Cr(CO)i(Py)2; Fe(Py)3Fe(CO)4
где (Ру — пиридин). Как правило, две нитрозильные
группы замещают три карбонпльпыс и предоставляют
но три электрона для связи с металлом. Амины и
фосфины дают для образования связи свои свободные
электронные пары и поэтому эквивалентны одной
карбонильной группе.
К соединениям, переходным между К. м. и дицикло-
пенгадиенилытыми соединениями металлов, можно
отнести такие, как циклопеитадиенилмаргаиецтри-
карбонил С6Н6Мп(СО)3, а также (C5H5W)2(CO)6,
(С5Н6Мо)2(СО)„ (C5H5Fe)2(CO)4 п др. Получено значи-
тельное число смешанных карбоиильно-олефиновых
соединений переходных металлов, где в качестве оле-
финов обычно выступают диены.
Большинство К. м. — кристаллич. продукты, толь-
ко Ni(CO)4, Fe(CO)3, Ru(CO)5 и Os(CO)5 — жидкости.
К. м. диамагнитны, не имеют или обладают лишь не-
большим дипольным моментом, хорошо растворимы
441
КАРБОНИЛЫ МЕТАЛЛОВ—КАРБОНИЯ ИОН
442
в органич. растворителях, весьма летучи, чрезвычайно
токсичны, однако не обладают кумулятивным дей-
ствием. При действии галогенов на нек-рые К. м.
в мягких условиях образуются карбонилгалогеииды,
причем два атома галогена заменяют одну СО-группу:
Fe(CO)5 -J- Xs ->• Fe(CO)4X2
Карбонилы хрома, молибдена и вольфрама разру-
шаются при обработке галогенами с образованием
галогенидов металлов. При нагревании выше опреде-
ленной темп-ры К. м. разлагаются с выделением окиси
углерода и металла в мелкодисперсном состоянии.
Нагревание иек-рых К. м. под давлением водорода
приводит к карбопилгидридам:
[Со(СО)4]2 -|- Н2 —► 2Со(СО)4Н
Последние получаются также при действии растворов
щелочей на нек-рые карбонилы; в случае кобальта,
напр. при действии разб. р-ра щелочи:
[Со(СО)4]2 Т Н2О —► НСо(СО)4 4- Со(ОН)2
Для получения карбонилгидрида железа требуется
более концентрированный р-р щелочи; при этом сна-
чала образуется соль, из к-рой подкислением
можно получить сам карбонилгидрид:
НС1
Fe(CO)s -|-3NaOH -► NaHFe(CO)4 —♦ H2Fe(CO)4
Карбонилгидрид никеля удалось получить восстанов-
лением карбонила никеля натрием в жидком аммиаке:
2N1(CO)4 + 3Na -J-2NH, -► [HN1(CO)S]2 4-
+ 2NaNH2 + NaCO + CO
Карбонилгидриды растворяются в растворах щелочей
с образованием солей карбонилгидридов; в жидком
аммиаке они дают аммонийные соли карбонилгидридов
К. м. могут быть получены следующими общими
способами:
1) Действием СО на галогениды или сульфиды ме-
таллов при повышенных темп-рах и высоком давле-
нии. Особенно гладко идет реакция в присутствии
меди, роль к-рой сводится к связыванию галогена;
CoJ2 -j- СО —► Co(CO)4J2
2Со(СО)4J2 4- 4Cu —► 4CuJ 4- £Со(СО)4]2
2) Действием СО на металлы при повышенных давле-
нии и темп-ре. При нормальном давлении эта реак-
ция идет только с никелем и железом.
3) Взаимодействием СО с безводным хлоридом ме-
талла в присутствии реактива Гриньяра (роль послед-
него сводится, по-видимому, к роли восстановителя).
Этим способом можно получать гексакарбонилы хрома,
молибдена и вольфрама. Магнийорганич. соединения
в этой реакции могут быть с успехом заменены мелко-
дисперсным цинком.
Наибольшее технич. значение имеют карбонилы
никеля, кобальта и железа. Карбонилы применяют
для получения чистых металлов, образующихся при
их термин, разложении. Напр., извлечение никеля
из руд в виде тетракарбонила позволяет очистить его
от примесей других металлов, а также серы и пр.
Термин, разложение карбонилов кобальта, никеля и
хрома используется для нанесения металлич. покры-
тий, особенно на поверхности сложной формы. Кар-
бонилы железа, кобальта и никеля (особенно послед-
него) применяют в качестве катализаторов важней-
ших химич. процессов. Их используют при синтезе
карбоновых К;Т и их производных из олефинов;
акриловых к-т и их производных из ацетилена:
Ni(CO)4
CHSCH 4- со + Н2О-------► сн2=снсоон
Карбонил кобальта является катализатором гидро-
формилирования:
[Со(СО)4]2 4- Н2 ->• 2Со(СО)4Н
4Со(СО)4Н 4- 4С2Н4 4- 2Н2 -о 4CHsCH2CHO -|- [Co(CO)s]4
[Co(CO)s]4 4“ 4СО —► 2 [Со(СО)4]2
К. м. являются катализаторами и в ряде других
синтезов; они — хорошие антидетонаторы моторного
топлива, однако при их сгорании образуются трудно-
удалимые окис.чы. Нек-рые карбонилы используются
для получения совершенно чистой окиси углерода.
Лит.: Белозерский Н. А., Карбонилы металлов,
М., 1958; Химия координационных соединений, под ред. Дж.
Бейлар, Д. Буш, пер. с англ., М., 1960; U 1 Imann, 3 Aufl.,
Bd 12, Munchen—В., 1960, S. 312. H. А. Несмеянов.
КАРБОНИЛЬНАЯ ГРУППА — см. Альдегиды и
Кетоны.
КАРБОНИЯ ИОН (карбониевый ион, ион карбо-
ния) — ион, содержащий положительно заряженный
трехковалентный углерод. Вальден дока- +
зал (1902) существование трифенилмегил- R'-C-R"
катиона. Меервейн (1927) и Уитмор (1932)
ввели представление о промежуточных не-
стабильных К. и. для объяснения механизма ряда ре-
акций органич. соединений. Благодаря наличию поло-
жительного заряда и незаполненной p-орбиты К. и.
обладает большой реакционной способностью, а следо-
вательно, малой устойчивостью. Устойчивость К. и.
зависит от того, находится ли положительный заряд на
одном атоме углерода или же он распределен (дело-
кализован) в результате сопряжения и гииерконъюга-
ции между несколькими атомами углерода. Так, устой-
чивость алифатич. К. и. зависит от того, находится лп
положительный заряд у первичного, вторичного или
третичного атома углерода. При этом вследствие
гиперконъюгации устойчивость К. и. увеличивается
в ряду СН3—СН2 < СН3— СН—СН3 < СН3—С(СН3)2.
Устойчивость К. и. растет с накоплением фенильных
групп у карбоииевого центра:
Особой устойчивостью обладают небензоидные аро-
матич. К. и., т. е. карбоциклич. ионы, в к-рых поло-
жительный заряд равно распределен между всеми
атомами углерода в цикле, напр. ион тропилия По-
ложительный заряд может быть распределен также
между атомом углерода и другими атомами соедине-
ния (напр., кислорода, азота), обладающими свобод-
ной парой электронов:
RvP.
с-он
R
сн2-6)+
I СН
сн2-6>
Мерой стабильности К. и. может служить энергия
образования его из соответствующего радикала. Эта
энергия равна:
Радикал CHS С2Н5 Н-С3Н7 mpem-C4H9 СзН5СН2
Энергия, ккал 230 201 183 159 178
В качестве меры стабильности может быть также ис-
пользована константа равновесия реакции между К. и.
и растворителем. Чем более устойчив К. и., тем труд-
нее он будет вступать во взаимодействие с раствори-
телем. Если растворителем служит вода, то наблю-
дается кислотно-основное равновесие:
R+ 4- 2Н2О 7^ R0H 4- HSO+
Чем меньше значение константы равновесия
АА, = [R0Hrp?f~—, тем выше стабильность К. и.;
° [R+]
рКа (т. е. — log Аа) для иона тропилия равен 4,75; для
(С„Н5)3С+ 6,63; для (СвН5)2СН 13,3.
Образование иона карбонил.
1) При гетеролитич. разрыве связи С—А, при к-ром
атом А (или группа атомов) отрывается с парой элект-
ронов, К. и. может образоваться в виде аниона. Об-
443
КАРБОНИЯ ИОН
444
разуется К. и. гл. обр. в растворах, т. к. в этом случае
значительная энергия, необходимая для гетеролитич.
разрыва связи, в той или иной мере компенсируется
энергией сольватации образующихся ионов. Поэтому
увеличение полярности растворителя и его сольва-
тирующих свойств способствует образованию К. и.
Отрыв аниона может осуществляться действием солей
координационно ненасыщенных металлов (А1С13, FeCl3,
ZnCl2 и т. д.) на галогеналкилы:
R' R'
R'Oc-Cl 4- Aids -► R"^C 4- А1С1Т
R"'Z R",Z
Отрыв гидридиона (реакция окисления) может
происходить при действии окислителей (конц. H2SO4,
координационно-ненасыщенных солей металлов), а
также действием других К. и.:
/ R,\ + /
R"3C—Н + С<- -> R"Ac 4- Н-С^-
R"'Z \ R,„Z \
2) К. и. может образоваться при распаде ониевых со-
единений'. е
R \ + R’\ +
R"AN_ch2C6H5 -► r"An + СН2С6Н5
R",Z R,„Z
3) К. и. может образоваться присоединением про-
тона (или другого электрофильного заместителя)
к кратным связям:
Н
>=< 'с-с''
+ н+----
с=о с'он
Z
Свойства иона карбония. 1) К. и.
обладает секстетом электронов у карбониевого центра,
с я/>2-гибридизацией, вследствие чего имеет плоскост-
ное тригональное строение. Атака К. и. анионом рав-
новероятна с обеих сторон плоскости. Поэтому, если
из оптически активной молекулы образовался К. и.
с положительным зарядом у асимметрия, центра, то
молекула, образующаяся при присоединении аниона
к К. и., будет рацемической. Т. обр., рацемизация
указывает на образование К. и. в ходе реакции (метод
стереохимической индикации).
2) Наличие положительного заряда и недостроен-
ность электронной оболочки у К. и. определяют его
высокую электрофильность, которая выражается в
том, что К. и. легко присоединяет анноны, молекулы,
содержащие атомы с неподеленной парой электронов,
молекулы с кратными связями, взаимодействует с раз-
личными соединениями, образуя продукты алкилиро-
вания в местах с наибольшей электронной плотностью.
Напр.:
4с+ОН —> ^с-он
с=с
3) При стабилизации К. и. может отщепиться про-
тон с образованием олефина (процесс, обратный полу-
—- zc=cC + '
чению К. и.). Первичный и вторичный К. и. могут
изомеризоваться путем внутримолекулярного переме-
щения гидрид-иона или группы атомов с парой элект-
ронов к карбониевому центру в третичный К. и.
(CHS)2C$H2 -<• (CHS)2£-CHS
н
Стабильные К. и. дают кристаллич. соли, устойчивые
в обычных условиях. Эти соли нерастворимы в непо-
лярных растворителях и растворимы в большинстве
полярных. Растворы солей дают обычные реакции на
анион; органич. катионы могут быть обнаружены
путем перевода их в нерастворимые соли. Для обнару-
жения стабильных К. и. применяют обычные методы,
существующие для неорганич. солей. Наиболее упот-
ребительными методами являются электропровод-
ность, криоскопия, спектроскопия и др. При малых
концентрациях, к-рые обычно характерны для неста-
бильных промежуточно образующихся К. и., прямые
доказательства существования К. и. не могут быть
применены. В этих случаях прибегают к косвенным
методам (стереохимия, индикация, кинетич. метод,
метод меченых атомов).
Кинетическим методом определяют число частиц, участву-
ющих в самой медленной стадии процесса. Если при гетероли-
тич. разрыве связи реакция является мономолекулярной Sjyl,
то это свидетельствует о промежуточном образовании неста-
бильного К. и. Скорость образования стабильных К. и. может
быть соизмеримой и даже превышать скорость их взаимодей-
ствия. Поэтому реакции с участием стабильных К. и. могут
проходить по механизмам Sjy 1 и Sjy2. Метод меченых атомов
может дать различные сведения о поведении К. и.; в частности,
К. и. способны вступать в реакцию изотопного обмена водо-
рода с водородными атомами растворителя, обладающими
протонной подвижностью.
Установлено, что многие реакции идут с промежу-
точным образованием К. и. Это — реакции типа пина-
колиновых и ретропинаколиновых перегруппировок,
Демьянова перегруппировки, Вагнера и Меервейна
(см. Камфеновые перегруппировки)', изомеризация алка-
нов, ионная полимеризация олефинов, Фриделя —
Крафтса реакция и мп. др.
Широкое изучение К. и. привело к представлениям
о различных видах К. и. Если анион находится близко
от К. и. и имеет общую с ним сольватную оболочку,
он экранирует К. и. и заместитель может вступать
лишь с противоположной стороны, т. е. наблюдается
обращение, а не рацемизация. В этом случае не суще-
ствует кинетически независимого К. и., а промежуточ-
ное образование, наз. «ионной парой». В реак-
циях с промежуточным образованием нестабильных
К. и. скорость процесса определяется стадией обра-
зования К. и. Для таких реакций характерны при-
мерно одинаковые скорости для веществ сходного
строения. Однако оказалось, что скорость гидролиза
трифенилэтилхлорида в 60 000 раз больше скорости
гидролиза неопентилхлорида, хотя обе реакции про-
ходят через промежуточное образование К. и.:
(C6H5)SCCH2C1 Кгидр = 2,3 • 10~3
(CHS)SCCH2C1 КгАдр = 4 • 10-S
Для объяснения таких явлений было высказано
предположение, что в этих случаях образуются т. наз.
«с и н а р т е т и ч е с к и е», или «некласси-
чески е», К. и. В «неклассич.» К. и. положительный
заряд распределен между тремя атомами углерода.
Энергетич. выгодность образования такого К. и. (а следо-
вательно, и большая скорость реакции) обусловлена тем, что
гетеролиз связи С—А и распределение положительного заряда
между тремя центрами происходит одновременно;
R
R /А
'С-С-А —-
В случае, если R = СвН& (ион «фенония»), в рассредоточении
положительного заряда участвует также и ароматич. кольцо.
При атаке неклассич. К. и. анионом могут образоваться
два продукта реакции:
445
КАРБОНОВЫЕ КИСЛОТЫ
446
Если у обоих атомов С, входящих в неклассический К. и.
(Ci и Со), одинаковые заместители, то оба продукта реакции
должны получаться в равных количествах.
Лит.: Темникова Т. И., Курс теоретических основ
органической химии, Л., 1959; Alexander Е. R., Prin-
ciples of ionic organic reactions, N. Y.—L., 1950; Бетел
Д., Голд В., Усп. химии, 1960, 29, вып. 1, 106; Streit-
wieser A., Chem. Revs, 1956, 56, №4, 571.
3. Н. Парное, И. С. Ахрем.
КАРБОНОВЫЕ КИСЛОТЫ — класс органич. со-
единений, содержащих карбоксильную группу (кар-
боксил) ~с\он' В зависимости от радикала,
связанного с карбоксилом, различают алифатич. (пре-
дельные и непредельные), алициклич., ароматич. и
гетероциклич. к-ты. По количеству карбоксильных
групп в соединении к-ты могут быть: одно-, двух- и
многоосновными; при введении в молекулу кислот
других функциональных групп (напр., —ОН, =СО,
—NH2 и др.) образуются окси-, кето-, аминокислоты
и другие классы соединений.
Как правило, для обозначения кислот пользуются
тривиальными названиями (уксусная, салициловая,
мезаконовая и т. д.). По Женевской номенклатуре
к названию углеводорода добавляют окончание «овая»
и слово «кислота»; непосредственно за оконча-
нием следует цифра, указывающая на место поло-
жения функциональной группы: 2-метилбутановая-
4-кислота' СН3СН(СН3)СН2СООН, 8-хлорбктановая
к-та С1СН2(СН2)6СООН. Рациональная номенкла-
тура рассматривает К. к. как производные простей-
ших к-т того или иного ряда. Так, масляная
к-та СН3(СН2)2СООН может быть названа этилуксус-
ной, С6Н5СН2СООН — фенилуксусной, кротоновая
СН3СН=СН—СООН—0-метилакрйловой и т. д. Часто,
особенно в случае алициклич. и гетероциклич. к-т,
к названию радикала добавляют слова «карбоновая
.------------------ГСООН
кислота», напр. II — Р-пирролкарбоновая
N
к-та, НООС—С=С—СООН — ацетилендикарбоновая
к-та и т. д.
К. к. — слабые к-ты, константы диссоциации боль-
шинства из них лежат между 1,4 • 10-5 и 1 • 10~4.
Однако сила К. к. существенно зависит от электро-
фильности радикала, связанного с карбоксилом. Так,
бензойная к-та несколько сильнее кислот предельного
ряда, а пропиоловая к-та НС=С—СООН значительно
сильнее пропионовой СН3СН2СООН. Введение электро-
ноакцепторных (электрофильных) заместителей (—NO2,
О
, Cl и др.) в положение, соседнее с карбокси-
лом, резко повышает константу диссоциации. Так,
циануксусная в 200 раз, а трихлоруксусная в 700 раз
сильнее уксусной к-ты. Такого же рода влияние эти
заместители' в бензольном ядре оказывают на кон-
станты диссоциации бензойных к-т.
Все К. к. способны вытеснять угольную к-ту (СО2)
из растворов ее солей. В солях К. к. (по существу
в анионе) оба атома кислорода карбоксильной
группы равноценны, что обычно отображают Го
в следующей записи: R-C'
К. к. способны обменивать гидроксил кар- Со’
боксильной группы на нек-рые другие груп-
пы. При этерификации К. к, спиртами в присутствии
минеральных к-т образуются сложные эфиры; слож-
ные эфиры могут быть получены также при взаимо-
действии К. к. с алифатич. диазосоедипепиями:
RCOOH -|-CH2N2 -о RCOOCHs -|- N2
С галогенидами фосфора, напр. РС15, хлористым тио-
нилом и сульфурилом, К. к. дают хлорангидриды
RCOC1, а при нагревании с водоотнимающими сред-
ствами — ангидриды (ВСО2)О. Ангидриды получа-
ются также по реакции:
СН3СОС1 + CH3COONa'—* (СН3СО)2О -|- NaCl
Сухой перегонкой аммониевых солей получают амиды
К. к., к-рые образуются также при взаимодействии
хлор ангидридов с аммиаком. Используя вместо аммиа-
ка амины, можно синтезировать замещенные амиды к-т:
CH3COONH4 -» CH3CONH2 -I- Н.,0
СН3СОС1 + NH(C2H5)2 -» CH3CON(C.>H5)2>-|- NaCl
Нитрилы К. к. могут быть получены нагреванием ами-
дов с водоотнимающими средствами:
Р2О5
CH3CONH2-----► CH3C=N
При прокаливании в присутствии щелочей К. к. де-
карбоксилируются:
C6H5CH2COONa -» С6Н5СН3 -|- Na2CO3
Легче декарбоксилируются (как в кислой, так и
в щелочной среде) дикарбоновые к-ты с близко рас-
положенными карбоксильными группами (щавелевая,
малоновая) и кислоты, содержащие в а- или Р-поло-
жении другие электроноакцепторные группировки
(напр., —NO2, —СС13), а также нек-рые ароматич.
и гетероциклич. кислоты. К. к. можно восстановить
(напр., LiAlH4) до соответствующих спиртов и даже
до углеводородов; однако с этой целью чаще восстанав-
ливают эфиры кислот, а не сами кислоты.
Описанные выше свойства связаны с реакционной
способностью самой карбоксильной группы и поэтому
являются общими для всех К. к. С другой стороны,
карбоксильная группа оказывает влияние на осталь-
ную часть молекулы; характер такого влияния тесно
связан с природой радикала в кислоте и специфичен
для каждого класса соединений. Так, в алифатич.
к-тах карбоксильная группа сообщает большую
подвижность атомам водорода в a-положении. Это
проявляется при галогенировании и многих других
реакциях. В непредельных к-тах двойная связь в
a-положении сопряжена с карбоксилом. Вследствие
этого • присоединение галогеноводородов происходит
следующим образом:
£о 3+ д-
СН2=СН —С + н—Вг —»- ВгСН2СН2СООН
4 он
В ароматич. ряду карбоксильная группа является
слабым .мета-ориентаптом; она заметно пассивирует
электрофильное замещение. Ароматич. к-ты сульфи-
руются и нитруются в более жестких условиях, чем
бензол, и не вступают или вступают с трудом в Фри-
деля — Крафтса реакцию. Введение карбоксильной
группы в нек-рые гетероциклы (фуран, пиррол)
сообщает таким соединениям большую стойкость
к окислению и действию минеральных к-т; это же от-
носится к трех- и четырехчленным циклам.
К. к. (особенно алифатические) распространены
в природе в свободном виде или в виде производных.
Они входят в состав жиров, эфирных масел, находятся
в мускульной жидкости, выделениях животных,
крови, плодах растений и т. д.
Для многих кислот главным способом получения
служит выделение из природных продуктов (высшие
жирные к-ты, лимонная к-та и пр.). Многие из них
получают сбраживанием сахаристых веществ с по-
мощью бактерий (маслянокислое, молочнокислое,
уксуснокислое брожение и др.).
Синтетически К. к. могут быть получены: 1) Омылением
нитрилов к-т; метод очень важен вследствие легкой до-
ступности нитрилов. 2) Гидролизом других функциональных
производных кислот. 3) Окислением спиртов и альдегидов.
4) Окислением углеводородов. Непредельные соединения
окисляются по месту двойной связи с образованием в общем
случае двух кислот:
CHa(CH2)nCH=CH(CH2)mCHs -►
-» СН3(СН2)ПСООН + СН8(СН2)тСООН
447
КАРБОРУНД — КАРВАКРОЛ
448
Нек-рые дикарбоновые к-ты получают каталитич. окислением
циклич, углеводородов или спиртов:
ОН
нс-соон
нс-соон ; LJ — Н°ОС-(СН2)4-СООН
Ароматич. и нек-рые гетероцик.чич. к-ты могут быть получены
окислением боковых цепей, обычно метильных групп в соот-
ветствующих углеводородах:
С6Н5СН3 — С8Н5СООН
5) Карбоксилированием магний-, литий- и натрийоргаиич.
соединений:
[+Н]
C8H5MgBr + СО2 -> C8H3COOMgBr-----► С8Н5СООН
6) Введением новых заместителей в радикал, связанный с кар-
боксилом:
С1СН2СООН + KCN -> NC-CH2COOH
Высгиие жирные кислоты получают окислением пара-
фина.
К. к. и их производные находят самое разнообразное
применение. н. А. Несмеянов.
КАРБОРУНД — см. Кремния карбид.
КАРБОСТИРИЛ (2-оксихинолин, лактам о-амино-
коричной кислоты) C8H7NO, мол. в. 145,15 — бес-
цветные кристаллы; т. пл. 199—200°; растворимы
в спирте, эфире, горячей воде, в р-рах кислот и щело-
чей; нерастворимы в водном р-ре аммиака. К. дает
коричневатую окраску с хлорным железом. Сходство
УФ-спектров К. и его N-метильного производного
указывает на то, что лактим-лактамное таутомерное
равновесие смещено в сторону лактамной формы:
При действии иодистых алкилов в присутствии ще-
лочи К. превращается в смесь N-алкил- и О-алкилпро-
изводных; из безводной серебряной соли К. полу-
чаются лишь О-алкилпроизводные. С РС15 К. дает
2-хлорхинолин. При нитровании образуется в основ-
ном 6-нитро-К. К. легко гидрируется в дигидрокар-
бостирил или в тетрагидрохинолин и окисляется
в изатин и оксалилантраниловую к-ту:
Важнейшие синтезы К. основаны либо на введении
гидроксильной группы в готовое хинолиновое ядро,
либо на замыкании цикла. Введение гидроксила про-
изводится гидроксилированием хинолина по Чичиба-
бину и при окислении хинолина р-ром хлорной из-
вести. К. можно также получить восстановлением его
N-окиси и удалением алкильной группы при нагрева-
нии 2-алкоксихинолинов с тиофенолом. Синтез К. за-
мыканием кольца происходит при циклизации про-
изводных о-амипокоричпон к-ты и при конденсации
о-ацетаминобеизальдсгида под действием щелочей:
См. также Кнорра реакция и Кампса реакция.
Лит.: Гетероциклические соединения, под ред. Р. Эльдер-
филда, пер. с англ., т. 4, М., 1955. Н. П. Гамбарян.
КАРБОФОС — см. Фосфорорганические инсекти-
циды.
КАРБОХОЛИН (карбампноплхолин, дорил, карб-
аминоилхолипхлорид) C6H15O2N2C1, мол. в. 182,66 —
белый, без запаха и вкуса г + -,
кристаллич. гигроскопич. h2ncoch2ch2N(GH3)3 Cl-
порошок; т. пл. 203—204° L о J
(с разл.); хорошо раство-
рим в воде, труднее в спирте, нерастворим в хлороформе
и эфире. Водные р-ры К., в отличие от ацетилхолина,
устойчивы и имеют нейтральную реакцию.
При действии на водные р-ры К. AgNOs осаждается
AgCl; при нагревании с щелочью появляется запах
аммиака. Количественное определение К. основано
на титровании хлорид-иона по Фольгарду или опре-
делении содержания азота по Кьельдалю. К. может
быть получен, исходя из этиленхлоргидрина:
NHS
С1СН2СН2ОН + СОС12 -> С1СН3СН2ОСОС1 —►
N(CH3)3
-о C1CH2CH2OCONH2------у к
Применяют К. при атонии кишечника и мочевого
пузыря, при ранних формах гипертония, болезни, при
глаукоме и др. заболеваниях.
Лит.: Машковский М. Д., Лекарственные сред-
ства, 4 изд., М., 1960. В. Г. Ягиунский.
КАРБОЦЕПНЫЕ ВЫСОКОМОЛЕКУЛЯРНЫЕ СО-
ЕДИНЕНИЯ — см. Высоко молекулярные соединения.
КАРБОЦИАНИНЫ — см. Цианиновые красители.
КАРВАКРОЛ (2-метил-5-изопропилфенол) С10Н14О,
мол. в. 150,22 — бесцветное густое масло, постепенно
темнеющее при хранении, с характерным
фенольным запахом, напоминающим за-
пах тимола; т. пл. —0,5°; т. кип. 236—
236,57742 мм, 1137Ю мм-, d? 0,976;
/Гд 1,52295; дисперсия Н^,— На 0,0157.
К. растворим в спирте, в эфире. В раст-
ворах щелочей К. образует растворимый
в воде фенолят, к-рый гидролизуется
водой, поэтому К. можно регенерировать из щелочных
растворов отгонкой с водяным паром. При окислении
FeCl3 в водноспиртовом растворе образуется дикар-
вакрол С2оН2802, т. пл. 165—166°. При окислении
хромовой смесью или КМпО4 образует тимохиноп,
т. пл. 45,5°.
При восстановлении водородом в присутствии N1 переходит
в смесь а- и p-карвоментолов Ci8H28O.
К. образует: фенилуретан, т. пл. 138°; а-нафтилуретап,
т. пл. 287—288°; нитрозокарвакрол, т. пл. 153°; бензалыгит-
розокарвакрол, т. пл. 85—87°; n-нитробензоат, т. пл. 50°;
3,5-динитробензоат, т. пл. 76—77°; метиловый эфир, т. кип.
216—217°, d° 0,9543, пд 1,50189; этиловый эфир, т. кип,
235°; уксусный эфир, т. кип. 245 — 248°, n28 1,49128, d25 0,9896.
К. входит в состав многих эфирных масел, напр.,
тминного (Carum carvi L.), перуанского перца (Schi-
mus molle L,), испанского ориганового (Origanum
hirtum L.) и др.-Из испанского ориганового масла
(содержащего до 80% К.) его выделяют обработкой
5%-ным водным р-ром NaOH, фенолят разлагают
H2SO4, продукт разложения дистиллируют. Синтети-
чески К. лучше всего получать нагреванием карвона
с муравьиной, фосфорной или серной к-той. К. —
прекрасное антисептич. средство; применяют в меди-
цине для полосканий.
^,СН
Н3С Сн3
449
КАРВОН — КАРИОФИЛЛЕН
450
СН3
[6*4f=°
сн3
о
Н8С СН3
11
н2с^сн3
О
Лит..- Г о р я е в М. И., Характеристика химических
соединений, входящих в состав эфирных масел, Алма-Ата,
1953; Дюпон Г., Терпентинные масла (скипидары), их
получение, состав и применение в промышленности, пер.
с франц., Л., 1931. И. И. Бардышев.
КАРВОН (1 - метил - 4 - изопропенил - Д6-циклогек-
сен-2-он) С10Н14О, мол. в. 150,21 — маслообразная
жидкость с характерным тминным запахом. К. (I)
существует в виде d-, 1- и d,1-форм. d-K; т. кип.
230°/755'лим; d}8-’ 0,9611; 1,4993; [а]}$ = 4-62,32°;
1-К: т. кип. 230—231°/763 мм; 0,9652; «д
1,4988; [a]J5 = —62,46°; d.l-K: т. кип. 230—231°;
0,9645; пд 1,5003; AMR =-|-0,46.
К. присоединяет галогены,
галогенонодороды, H2S. При
действии на гидробромид К.
спиртовым р-ром КОН он пре-
вращается в циклогептадиено-
вый кетон — эйкарвон (II), т.
кип. 85—87°/12 мм.
Продукт присоединения H2S к К. имеет для d-K. т. пл.
210—211°, [<х]р = +48,71° (в СНС1з); ДЛЯ 1-К. — т. пл. 210—
211°, 1а)р =—48,11° (в СНС13); для d, 1-К. — т. пл. 189—190°.
Семикарбазон d-K. имеет две формы: т. пл. 162—163° и 141 —
142°; семикарбазон d,l-K.; т. пл. 154—156°; фенилгидразон
К., т. пл. 109—110°; оксим d-К., т. пл. 71°, [u]d == +43,9°
(в спирте); 2,4-динитрофенилгидразон, т. пл. 191°; d-карвоксим,
т. пл. 71°, lajp = +43,9° (в спирте); d, 1-форма, т. пл. 92—93°.
К. — составная часть многих эфирных масел (ук-
ропного, тминного, мятного и др.), откуда его и полу-
чают. К. находится в продуктах окисления лимонена и
карвеола; может быть получен синтетически из лимо-
нена. К. нашел применение в фармакопее и в ликерном
произ-ве как заменитель тминного масла.
Лит.: Simonsen J. L., The terpenes, у. 1, 2 ed.,
Camb., 1949; Бурно К., Терпены, пер. с нем., [Л.], 1933.
И. И. Бардышев.
КАРДАМОНОВОЕ МАСЛО — см. Эфирные масла.
КАРДИОТРАСТ (артериодов, диодон, диодраст)
С,,Н,Й J,N90r, мол. в.510,10 — диэтаноламиновая соль
16 2 2 3,5-дииод - 4-пири-
дон-1-уксусной к-ты;
хорошо растворима в
N - CHgCOONHg ' (CHgCHgOH)g воде, растворы про-
z зрачны, бесцветны,
pH 6.8—7,4. При
действии восстанав-
ливающих средств отщепляется иод; при подкислении
водных р-ров выпадает кристаллич. осадок 3,5-дииод-
4-пиридон-1-уксусной к-ты, т. пл. 233—236° (с разл.)
При обработке р-ра К. щелочью и сульфатом меди по-
является темно-синее окрашивание. Количественно К.
определяют нагреванием с Zn в присутствии щелочи
с последующей нейтрализацией СН3СООН и титро-
ванием выделившегося иода. ” ~
4-пиридона след, образом:
О О
ClCHgCOOH
mJ ОН' ’
J
J
. К. получают из солей
J NH(CH2CH2OH)2
—------------•- К.
CHgCOOH
К. в виде 35, 50 и 75%-ных р-ров применяют как конт-
растное средство для рентгенология исследований по-
чек, мочевого пузыря, мочеточников, почечных лоха-
нок, а также кровеносных сосудов и сердца.
Лит.: Baker W., Briggs A. S., J. Soc. Chem. Ind.
Trans. Commune., 1943, 62, M 11, 189; Hackman R. H.,
Lemberg R., там же, 1946,65, № 7, 204. В. Г. Яшунский.
КАРЕН С10 Н1е, мол. в. 136,23 — из- есн3
гестны четыре изомера, отличающиеся 1
положением двойной связи: 2,3-Д2-К.;
3,4-Д’-К.; 4,5-Д«-К.; 4,8-₽-К.
Все К. весьма реакционноспособны,
быстро окисляются кислородом воздуха
8 К. X. Э. т. 2
аСН3
юСН3
Свойства Д8*Карена Д3-Каренб Д4-Каренв р-Каренг
Т. кип., °С/Л4Л1 160—161/758 169,5—170,5/760 167,5—168,0/760 72,5/20
d2» 03618 0,8645 0,8616 0,8729
SO пр 1,4698 1,4723 1,4747 1,4927»
lain, град. -108,6 +18,0 4-89,37 4-1235
MR eD 44,07 44,15 44,49 45,47
а 4,7,7-Триметилбицикло-(0,1,4)-гептен-2. 6 4,7,7-Триметил-
бицикло-(0,1,4)-гептен-3. в 4,7,7-Триметилбицикло-(0,1,4)-геп-
тен-4. г 7,7-Диметил-4-метиленбицикло-(0,1,4)-гептан. д При
22°. е Молярная рефракция.
и другими окислителями, легко изомеризуются при
воздействии кислых агентов с раскрытием трехчлен-
ного кольца, сравнительно стойки к нагреванию.
При действии TiO2, СН3СООН, H2SO4 в определен-
ных условиях Д3-К. частично изомеризуется в Д4-К.
При гидрировании в мягких условиях все они пре-
вращаются в смесь цис- и транс-каравов. К. хранят
только в запаянных ампулах, в темноте и в присут-
ствии антиокислители, напр. пирокатехина.
Д2-К. не образует характерных кристаллич. производных.
Д3-К. образует нитрозат, т. пл. 147,5° (с разл.); каренгликоль,
т. пл. 70—71°; нитрозохлорид, т. пл. 101—102° (с разл.),
к-рый, в свою очередь, образует нитрометиламин, т. пл. 180°;
моногидрохлорид, т. кип. 77—83°/7 мм; исчерпывающее гидро-
хлорирование приводит к смеси дигидрохлоридов дипентена
и сильвестрена. Для Д4-К. пока не получены к.-л. кристаллич.
производные. При окислении растворами КМпО. он превра-
щается в жидкую 1,1-диметил-2-7-кетооутилциклопропан-3-кар-
боновуюк-ту CioHieOs, семикарбазон к-рой имеет т. пл. 182—
183°. В-К. при окислении образует кетон — (7,7-диметилби-
цикло-[0,1,4]-гептанон-4), т. кип. 54°/2 мм; d3jj 0,9080; пр
1,4845; семикарбазон кетона плавится при 191°
Получают Д3- и Д4-К. фракционированной пере-
гонкой эфирных масел. Д3-К. содержится в скипида-
рах хвойных, напр. Pinus Silvestris, Picea excelsa
и др.; Д4-К. — в масле травы Andropogon Iwarancusa
(Индия); Д2-К. и 0-К. в природе не найдены, их полу-
чают синтетически. Наибольшее практич значение
имеет Д3-К. (содержится в отечественном скипидаре
ок. 15%). К. — флотореагент, получают окислением
фракций скипидара, содержащих К., кислородом воз-
духа при нагревании; возможно применение гидропере-
киси карана в качестве инициатора полимеризации.
Лит.: Никитин В. М., Химия терпенов и смоляных
кислот, М.—Л., 1952; П и г у ле в с к и й Г. В., Химия тер-
пенов, Л., 1949; Kuczyiiski Н., Chabudziiiski
Z., Roczniki chemii, 1958, 32, z. 1, s. 49. И. И. Бардышев.
КАРИОФИЛЛЕН С1бН24, мол. в. 204,35 — бесцветная
вязкая жидкость со слабым запахом; т. кип. 259—261°;
на воздухе сравнительно быстро окисляется и при
этом осмоляется. В воде не растворяется. Хорошо рас-
творяется в неполярных ор-
ганич. растворителях. К. — би-
циклич. сесквитерпеновый уг-
леводород, встречается в а-,
fi- н у-формах. Строение а- и
у-форм недостаточно установ-
лено. Наиболее вероятное стро-
ение 0-К.:
Физические свойства изомерных кариофилленов
сн2
VCH;
Н3С\ х-
с—СН
Н3С"] |
Н2С—сн сн
Un g
HgC СН3
Свойства a-Кариофил- лен (гуму- лен) р-Карио- филлен у-Кариофил- лен (изока- риофиллен)
Т. КИП., °С/Л1Л€ . . . Плотность lain, град nD 118-19/10 132-4/16 d23 03933 4-1,7—4,2 1,5001(20°) 118-19/9,7 129-30/14 df 0,9052 -9,54-9,4 1,5009(17°) 125/14,5 dj8 0,8994 —26,24-21,9 1,4966 (19°)
451
КАРИУСА МЕТОД —КАРНО ЦИКЛ
452
Наиболее изучен 0-К. При озонировании он дает
кетон С10Н1вО, т. кип. 73—75°/9 мм, дикетон С10Н20О3,
т. кип. 137—142°/9.и.и; кетоальдегид С14Н23О3, т. кип.
181—184°/13лии и кето-и дикетокислоты. При окисле-
нии КМпО4 получается гликоль С14Н22О», т. пл. 120°.
Гидрирование с катализатором Ренея приводит к ди-
гидрокариофиллену; гидратации по Вальбауму —
к получению p-кариофилленового спирта с т. пл. 96°.
Смесь а-, 0- и у-К. встречается во многих эфирных
маслах: гвоздичном, из тысячелистника, лавандовом,
хмелевом, коричном и др. К. лучше всего получать
из гвоздичного масла. Обработкой масла р-ром ще-
лочи удаляют из него эвгенол, остаток перегоняют
с водяным паром. Масло насыщают хлороводородом,
кристаллизуют дигидрохлорид К., а затем разла-
гают его метилатом натрия. Фракционной перегонкой
в вакууме, а затем хроматографически разделяют изо-
мерные формы К.
Лит.: Пигулевский Г. В., Химия терпенов, Л.,
1949; Горяев М. И., Характеристика химических соеди-
нений, входящих в состав эфирных масел, Алма-Ата, 1953;
Simonsen J. L. and Barton D. H. R., The
terpenes, 3, 2 ed., Camb., 1952. И. И. Бардышев.
КАРИУСА МЕТОД — способ количественного оп-
ределении содержания нек-рых элементов, преим.
галогенов и серы, в органич. соединениях. Метод ос-
нован на окислительном разложении органич. веще-
ства при нагревании его в течение неск. часов до 250—
350° с конц. HNO3 в запаянной трубке. При этом гало-
гены количественно образуют галогеноводородные
к-ты, а сера окисляется до серной к-ты. Ионы гало-
гена или сульфат-ион могут быть определены различ-
ными способами, применяющимися в неорганич. ана-
лизе: весовым, титриметрич., потенциометрич., нефе-
лометрич. и др. Галогены часто определяют весовым
способом в виде солей серебра, а серу — в виде суль-
фата барин. При определении галогеновв трубку вводят
до ее запаивании необходимое количество кристал-
лич. нитрата серебра. Осаждение галогенного серебра
происходит в процессе разложения органич. вещества.
После вскрытии трубки ее содержимое разбавляют
водой и определяют образовавшееся галогенное се-
ребро. Метод более надежен дли определения хлора
и брома, чем иода; неприменим дли анализа полигало-
генных соединений. Разложение по Кариусу исполь-
зуют также для определении в органич. веществах
мышьяка, селена, теллура, фосфора. В настоящее
время К. м. в значительной мере вытеснен другими
более совершенными способами. См. Галогенов опре-
деление. Впервые метод был опубликован Л. Кариу-
сом в 1860.
Лит.: Мейер Г., Анализ и определение строения орга-
нических веществ, Харьков—Киев, 1935; Гаттерман Л.,
Виланд Г., Практические работы по органической химии,
пер. с нем., 5 изд., М.—Л., 1948; Pregl-Roth, Quanti-
tative organische Mikroanalyse, 7 Aufl., W., 1958.
А. А. Черкасский.
КАРМИНОВАЯ
492,38 — красные
КИСЛОТА С33Н20О13, мол. вес
иглы, темнеют при 130° и разла-
гаются ок. 250°; растворяется
в воде. К. к. — производное
р тетраокисантрахинона (в к-ром
он строение R = С6НиО5 точно
не установлено). К. к. является
красящим в-вом кошенили —
красители, ранее применяв-
шегося для крашения шерсти и шелка. Коше-
ниль — высушенные и растертые женские особи
насекомых-червецов вида Coccus cacti, живущих на
мексиканских кактусах. К. к. получают экстрагиро-
ванием кошенили водой (кошениль содержит 10%
К. к.). При кипячении кошенили с водой в присутст-
вии кйасцов выпадает осадок, состоящий в основном из
алюминиевой соли К. к., т. наз. кармина, к-рый
применяют в косметике, для приготовлении аква-
рельных красок, дли подкрашивании кондитерских
изделий и нек-рых препаратов при микроскопии,
исследованиях. Известна также дубовая кошениль,
получаемая из насекомых вида Coccus ilicis, живущих
на дубах, произрастающих в средиземноморской
части Европы. Красящее в-во дубовой кошенили —
кермесован кислота — отличается от К. к.
ТОЛЬКО остатком R = СОСН3. В. Н. Фросин.
КАРНАУБСКИЙ ВОСК — см. Воски.
КАРНИТИН (новаин, витамин Вт), мол. в. 161,21,
(CH3)3NCH,CHOHCH3COO~ — бетаин-у-амино-р-окси-
масляной к-ты; кристаллич. продукт; известен в
двух формах: D, L-К., т. пл. 195—197° (с разл.), очень
гигроскопичен, легко растворяется в воде и этаноле.
L-K. — кристаллич. в-во, [а]^ = —23,5°. Водные
растворы К. обладают сильно выраженными щелоч-
ными свойствами; с НС1, HgCl3, HAuCJ4, H2PtCle и
др. образуют соли: C7H16O3NHC1, C7H15O3N • HgCl2,
C7H15O3N-PtClj и т. д. При взаимодействии D, L-K.
с НС1 образуется D, L-карнитингидрохлорид, кристал-
лизующийся из этанола в виде звездообразных скопле-
ний, т. пл. 196° (с разл.); хорошо растворим в водей го-
рячем этаноле,плохо растворимв ацетоне,не растворяет-
ся в эфире. L-K.c НС1 образует L-карнитингидрохло-
рид, кристаллы,!, пл. 137—139°; [а]у =-(-20,4°. Полу-
чены этиловые эфиры карнитинхлорида C9H20O3NCl
и карнитинрейнеката C9H20O3N • C4HeNeS4Cr • Н2О;
ацетилкарнитинхлорида C9H17O4N • HCJ; ацетилкар-
нитинхлораурата C9H17O4N • HAuC14.
К. обнаружен в бактериях, растениях, животных.
Наибольшие количества его находятся в мышечной
ткани позвоночных и беспозвоночных животных. Спо-
собность ряда живых организмов к биосинтезу К.,
его высокая биологич. активность по отношению к от-
дельным видам насекомых наряду с необходимостью
введении его в пищу для насекомых, к-рые не спо-
собны к биосинтезу К., позволили отнести его к вита-
минам. Вероятно, К. участвует в различных процессах,
протекающих в организме, причем для нек-рых из
них достаточно микроколичеств К., дли других необ-
ходимы большие количества его.
К. извлекают из водной или спиртовой вытяжки
мясного экстракта фенолом, затем при прибавлении
эфира и воды К. переводят в водный р-р, очищают
активным углем и хроматографируют на колонках с
А13О3. Описан ряд синтезов К., исходи из эпихлор-
гидрина, NaCN, (CH3)3N и т. д.
К. открыт В. С. Гулевичем и Р. Кринбергом в 1905;
витаминные свойства обнаружены в 1947.
Лит.: Гулевич В. С., ЖРФХО, 1926, 58, 610; Fraen-
kel G., FriedmanS., Carnitine. Vitamins and hormones,
v. 15, N. Y., 1957, p. 74; T p у ф а н о в А. В., Биохимия и
физиология витаминов и антивитаминов, М., 1960, с. 571;
Del tour G., Broekhuysen J., Dierickx L.,
Clin. chim. acta, 1960, 5, К» 2, p. 181; В г о e k h u у s e n J.,
DeltourO., Ann. biol. clin., 1961, 19, № 7—9, 549.
Э. Д. Михлин.
КАРНО ЦИКЛ — круговой тепловой процесс, в ре-
зультате к-рого нек-рое количество тепла термодина-
мически обратимым способом переносится от горячего
тела к холодному. Процесс должен совершаться
т. обр., чтобы тела, между к-рыми происходит непо-
средственный обмен энергией, находились при одина-
ковой темп-ре, т. е. и горячее и холодное тела счи-
таются настолько большими тепловыми резервуарами,
что темп-ра первого при отнятии и темп-ра второго
при прибавлении рассматриваемого количества тепла
ощутимо не изменяются. Для этого необходимо вспо-
могательное «рабочее тело», к-рое сначала при более
высокой темп-ре Тг приводится в соприкосновение
с горячим телом и изотермически получает от него
указанное количество тепла. Затем оно адиабатиче-
ски охлаждается до темп-ры Т\, отдавая при этой
темп-ре тепло холодному телу с темп-рой 1\, и, нако-
453
КАРНОЗИН - КАРОТИНОИДЫ
454
нец, адиабатически возвращается в начальное состоя-
ние. При расширениях, связанных с этими процессами,
рабочее тело совершает внешнюю работу. Можно
показать, что наибольший возможный кпд тепловой
машины, использующей для своей работы данный
перепад темп-ры между двумя телами, равен отноше-
нию ———-. Эта величина имеет тем большее значе-
1 2
ние, чем выше Т„ и ниже Тх. К. ц, был впервые рас-
смотрен С. Карно в 1824 в связи с вопросом о кпд
тепловых машин.
КАРНОЗИН (0-а лани л-L-r и с т и д и н), мол.
в. 226,2, C9H14N4O3 — дипептид, построенный из
аминокислот L-гиствди-
НС=С-СН2-СН СООН на и 0-аланина.
' Открыт в 1900 В. С. Гу-
СН I - ' левичеми С. Амираджиби
ОС—(Cfig)?-NH2 в мясном экстракте. Со-
держится в скелетных
мышцах позвоночных животных в свободном виде
(0,1—0,15% от веса ткани) и в виде метил-производно-
го — ансерина. В небольших количествах К. обнаружен
в сердечной мышце, гладких мышцах, печени, крови
и слизистой желудка. К. — бесцветные кристаллы,
т. пл. 260° (с разл.), хорошо растворим в воде, нераст-
ворим в этаноле и эфире. К. является амфотерным
электролитом с изоэлектрич. точкой при pH 7,6—7,7;
[а]о == 4-20,5° (2%-ный р-р в воде). При взаимодейст-
вии с нингидрином дает цветную реакцию. Для иден-
тификации К.может быть использована его медная соль,
плохо растворимая в воде, темп-ра пл. 220°. При дейст-
вии кислот и щелочей, а также ферментативным пу-
тем К. гидролизуется с образованием гистидина и
0-аланива. Ферменты, гидролизующие К., называются
к а р н о з и н а а а м и; они обнаружены в селе-
зенке, печени и почках нек-рых животных. Ферменты
желудочно-кишечного тракта человека К. не гидроли-
зуют. При действии на К. хлорокиси фосфора в щелоч-
ной среде может бытьполучен дифосфат К. — макро-
эргическое соединение. Получают К. из конц. водных
экстрактов мышц в виде ртутной соли, к-рую затем
очищают и разлагают сероводородом. Синтетически
К. может быть получен несколькими способами, ис-
ходя из гистидина и р-аланина, напр. конденсацией
метилового эфира L-гистидина и азида карбобен-
зокси-р-аланина с последующим отщеплением ме-
тильной и карбобензоксигруппы от образующегося
продукта. Биологич. роль К. точно не установ-
лена. Возможно, что К.-фосфат служит донором фос-
фата при нек-рых реакциях биологич. фосфорилиро-
вания.
Лит.: Северин С. Е., Распространение, превращения
в организме и биологическое значение карнозина и ансерина,
в кн.: Успехи биологической химии, под ред. В. Н. Ореховича,
т. 2, М., 1954, с. 355—77; Майстер А., Биохимия амино-
кислот, пер. с англ.,М., 1961; Biochemical preparations, v. 4,
N. Y.—L., 1955, p. 38; P e г г у S. V„ Annual Rev. Biochem.,
1961, 30, 473.
КАРОТИНОИДЫ — пигменты от желтого до крас-
ного цвета, встречающиеся в тканях растений, многих
грибках, бактериях и водорослях; относятся к группе
сильно ненасыщенных углеводородов терпенового
ряда. Характерным признаком К. является большое
число сопряженных двойных связей, что объединяет
их в группу полиенов и обусловливает абсорбционные
спектры в видимой и УФ-частях спектра. К. раство-
римы в жирах и липоидах, поэтому их часто называют
липохромами. К. лучше всего разделяют
хроматография, адсорбцией. В организме животных
К. не синтезируются, а поступают с растительной
пищей. Нек-рые К. (а-, 0- и у-каротины, криптоксан-
тин и др.) являются провитаминами А, т. к. могут
преобразовываться в организме животного в вита-
мин А. С концентрированной H2SO4, СС13СООН,
AsCl3, 8ЬС13 К. дают интенсивное окрашивание, к-рое
8*
обусловлено, по-видимому, образованием неустойчи-
вых карбониевых солей.
Известно более 70 К., большинство к-рых содержит
40 атомов углерода. Молекулы К. построены по еди-
ному структурному принципу и, по-видимому, имеют
между собой определенную генетич. связь. Главными
представителями К. являются а-, 0- и у-к а р о т и н ы
С40Н6в. а-Каротин (I) — темно-красные иглы, т. пл.
187—188°, оптически активен, [а]£°а = -(-385° (бензол).
Максимум абсорбции 478 и 447,5 ммк (бензин). 0-Ка-
ротин — кристаллы темно-рубинового цвета с метал-
лич. блеском, т. пл. 181—182°, максимумы поглоще-
ния в УФ-свете при 273, 453 и 481 ммк, коэфф, экстинк-
ции (A}%J соответственно 383, 2592 и 2268 (гексан).
Строение 0-каротина (II) отличается от а-каротина (I)
лишь положением двойной связи в алициклич. груп-
пировке Б (II). у-Каротин — кристаллы, т. пл. 178°,
отличается от а-каротина строением группировки
Б (Ш). а-, ₽- и у-Каротины легко растворяются в хло-
роформе, сероуглероде и бензоле, меньше в серном и
петролейном эфирах, жирах и маслах; в спирте прак-
тически нерастворимы. Каротины легко присоединяют
кислород воздуха, особенно на свету и при нагрева-
нии; в результате окисления образуются бесцветные
продукты. Качественно и количественно каротины
определяют по максимумам абсорбции или колоримет-
рически.
Н3С_СН3 СН3
'(СН=СН-С=СН)2-СН
СН3
СН3
н3с^сн3 СН3 ||
(СН = СН-С=СН)2-СН
Наиболее широко распространенный 0-каротин
известен в виде изомера, обладающего транс-конфигу-
рацией по всем двойным связям (наиболее стабильная
форма); эта форма может подвергаться транс-цис-
изомеризации в растворах под действием света, при
нагревании, а также при добавлении иода и кислоты,
давая сложную равновесную смесь изомеров. 0-Каро-
тин с хлороформенным р-ром хлористой сурьмы дает
характерное синее окрашивание с максимумом погло-
щения 590 ммк. 0-Каротин получают экстрагирова-
нием сушеной моркови (содержание до 1 г на 1 кг),
люцерны, гречихи и др. растительных материалов
органич. растворителями и избирательной адсорбцией
на активированном меле, окиси магния или фосфате
кальция. Важным промышленным источником полу-
чения 0-каротина является пальмовое масло. Воз-
можно получение каротина микробиология, путем.
Синтетически 0-каротин может быть получен, напр.,
из 0-ионона (см. Иононы), виниловых эфиров и аце-
тилена.
Потребность организма в витамине А в значитель-
ной степени удовлетворяется за счет природных ка-
ротинов. Каротины применяют при витаминизации
455
КАРОТИНЫ — КАТАЛИЗ
456
пищи и кормов животных, лечении поражений кож-
ных покровов И в качестве пищевого красителя.
Производными каротинов являются различные кислород-
содержащие растительные пигменты; к ним относятся; Крип-
токсантин (моноокси-В-каротин) СдеЩеО, т. пл. 169°.
Зеаксантин C40H3eOs (IV) — красящее вещество куку-
рузных зерен, кристаллич. продукт, т. пл. 215,5°, почти не-
растворим в петролейном эфире, растворим в спирте. Зеаксан-
тин — производное 0-каротина, отличается от последнего
присутствием гидроксильных групп в алициклич. группи-
ровках А и Б. Дипальмитиновый эфир зеаксантина — ф и з а-
л и н — воскоподобный пигмент, распространенный в плодах
и ягодах. Ксантофил CjoHgeOz — кристаллы, т. пл. 193°,
1а1Са = + (этилацетат), находится в цветах и листьях
в свободном и этерифицированном состоянии. При действии
окислителей образует эпоксид (V), к-рый является широко
распространенным в природе продуктом присоединения кисло-
рода по двойной связи 0-иононного кольца.
Помимо каротинов и их производных, к К. относятся;
Ликопин CjoHjq (VI) — пигмент томатов, т. пл. 174—175°,
хорошо растворим в сероуглероде, хлороформе, горячем бен-
золе, почти нерастворим в спиртах; отличается от а- и 0-каро-
тинов строением группировок А и Б. Получен синтетич. путем.
Биксин (VII) — монометиловый эфир дикарбоновой к-ты —
норбиксина;
СНзСОО—(СН=СНС=СН)2—СН=СН— (СН=СНС=СН)2- СООН
сн3 Jhs
встречается в вегетативных органах растений в виде лабиль-
ного (т. пл. 196°) и стабильного (т. пл. 202°) изомеров. Пре-
параты биксина применяют для подкрашивания продуктов пи-
щевой пром-сти (масло, маргарин). Кроцетин СгоН^СЦ—
дикарбоновая к-та, пигмент шафрана.
Лит.: Г у д в и н Т., Сравнительная биохимия каротинои-
дов, пер. с англ., М., 1954; Савинов Б. Г., Каротин
(провитамин А) и получение его препаратов, ’Киев, 1948;
К а р р е р П., Курс органической химии, пер. с нем., Л., 1960;
Каггег Р., Jucker Е., Carotinoide, Basel, 1948 (Lehrbilcher
und Monographlen aus dem Gebiete dec exakten Wissenschaften.
Chemische Reihe, Bd 3); I s 1 e г O. [u. a.], Helv. chim. acta,
1956, 39, 249; Goodwin T. W., Advances Enzymol. and
Related Subjects Biochem., 1959, 21. Л. А. Вакулова,
Г. И. Самохвалов.
КАРОТИНЫ — см. Каротиноиды.
КАСТОРОВОЕ МАСЛО — см. Жиры растительные.
КАТАЛАЗА — фермент, катализирующий разло-
жение перекиси водорода на воду и молекулярный
кислород: 2Н2О2 —► 2Н2О -|- О2. Со значительно мень-
шей скоростью К. расщепляет монозамещенные про-
изводные Н2О2; метил- и этилпероксид. В определен-
ных условиях (низкая концентрация Н2О2) К. ча-
стично проявляет пероксидазную активность (см.
Пероксидазы), окисляя за счет Н2О2 метиловый и
этиловый спирты, формальдегид и нитриты.
Кристаллич. К., выделенная из печени животных,
представляет собой гемопротеид с мол. в. 225 000—
248 000 и изоэлектрич. точкой, соответствующей
pH 5,4—5,7. Спектр К., подобно спектрам других,
родственных ей гемопротеидов, характеризуется ин-
тенсивной полосой поглощения при 400—409 ммк
(линия Соре), а также максимумами при 620—629,
536—544, 500—506 и 280—286 ммк. Простетич. груп-
пой К. является про-
тогематин — гидро-
окись феррипрото-
порфирина IX.
В молекулу К. жи-
вотного происхожде-
ния входят 4 протоге-
матина, в К. из листьев
шпината содержится,
по-видимому, лишь
2 простетич. группы.
Простетич. группа в К. очень прочно связана с белком
и отделить ее, напр. путем диализа, не удается.
К. — один из наиболее активных ферментов: одна
молекула К. разлагает в минуту при 0° до 5 • 10е мо-
лекул Н2О2. Активность К. может быть измерена
полнрографически, манометрически — по объему вы-
деляющегося О2 или перманганатометрически — тит-
рованием неразложивщейся Н2О2. Максимальной
НООССН2СН, СН2СН»СООН
активностью К. обладает при 0—10° (при более высо-
ких темп-рах начинает сказываться инактивирующее
действие на фермент Н2О2). Оптимум pH для актив-
ности К. лежит в области 6,0—8,0. Активность К.
подавляется такими ингибиторами, как сульфиды,
цианиды, азиды и гидроксиламин. Кроме того, актив-
ность К. специфически угнетается анионами ряда
кислот (нитрат, сульфат, хлорид, ацетат, фосфат),
к-рые вытесняют связанный с железом гидроксил
в простетич. группе К. Вероятный механизм катали-
тич. действия К. может быть представлен следующим
образом:
K.-FeOH-(H2O2s==iK.-FeOOH4-H2O.
K.-FeOOH+H2O2i=tK.-FeOH+H2O+Os
Биологическая роль К. состоит в защите организма
от ядовитого действия Н2О2, образующейся при функ-
ционировании ряда флавопротеиновых оксидаз. К. ши-
роко распространена в животных тканях, наиболее
богаты ею печень и эритроциты, в к-рых содержание
фермента составляет 0,1—0,2% сухого веса; в расте-
ниях К. содержится в меньших количествах. У ряда
анаэробных микроорганизмов К. совершенно отсут-
ствует.
Лит.: Михлин Д. М., Биохимия клеточного дыхания,
М., I960; Нейландс Дж., Ш т у м п ф П., Очерки по хи-
мии ферментов, пер. с англ., М., 1958; The Enzymes, ed.
J. В. Summer, К. Myrback, v. 2, pt 1, N. Y., 1951, p. 397.
В. Б. Спиричев.
КАТАЛИЗ. Co держание:
Введение ....................................456
Классификация каталитических процессов.......458
Избирательность действия катализаторов ..... 45Э
Промотирование и отравление катализаторов....460
Открытие явлений катализа....................460
Важнейшие каталитические процессы в промышлен-
ности ....................................461
Катализ в живой природе......................463
Введение. Катализ — изменение скорости химич.
реакции в присутствии веществ (к а т а л и з а т о-
р о в), многократно вступающих в промежуточное
химич. взаимодействие с участниками реакции, но
восстанавливающих после каждого цикла промежуточ-
ных взаимодействий свой химич. состав. Если про-
межуточное взаимодействие с катализатором делает
возможным новый реакционный путь, по к-рому та
же реакция протекает с большей скоростью, чем без
катализатора, то явление наз. положительным
катализом и его нередко отождествляют с об-
щим понятием К. Возможен и обратный случай, наз.
отрицательным катализом, когда при-
сутствие катализатора исключает один из возможных
без него путей реакции и оставляет лишь более мед-
ленные, в результате чего реакция замедляется или
даже практически полностью подавляется. (См. Анти-
катализаторы, Антиокислители, Ингибиторы). Яв-
ления К. распространены в природе и широко при-
меняются в технике. Каталитич. воздействию может
быть подвержено большинство химич. реакций. Круг
веществ, выполняющих роль катализаторов, весьма
велик.
Характерной особенностью К. является сохранение
катализатором своего состава в процессе реакции.
Количество реагирующего вещества, к-рое может испы-
тать превращение в присутствии определенного коли-
чества катализатора, не ограничивается какими-либо
стехиометрия, соотношениями и может быть очень
большим. Так, в большинстве технич. каталитич. про-
цессов небольшое количество катализатора способст-
вует превращению весьма значительных количеств
реагирующих веществ. Напр., одна весовая часть
катализатора в произ-ве серной к-ты вызывает прев-
ращения 104 вес. ч. реагирующего вещества, при
окислении нафталина во фталевый ангидрид 103,
в произ-ве азотной к-ты окислением аммиака 10е.
Явления К. не связаны с изменением свободной
энергии катализатора. Этим каталитич. реакции от-
457
КАТАЛИЗ
458
личаются от индуцируемых или сопряженных реак-
ций, при к-рых одна реакция вызывается или уско-
ряется другой реакцией, т. е. необратимым хими-
ческим превращением определенного вещества — ин-
дуктора первой реакции. Из этого не следует, что при
течении каталитич. реакции катализатор не претер-
певает никаких изменений. Во многих случаях наблю-
даются изменения структуры катализатора, а иногда и
его состава в результате взаимодействия с примесями
или даже основными компонентами реакционной смеси.
Характерным для катализа является то, что все эти
изменения представляют собой побочные процессы, ни
в коей мере не обусловливающие каталитич. действия.
Возможность ускорения химич. реакций и напра-
вления их в сторону образования желаемых продук-
тов без затраты энергии и катализатора и определяет
исключительно большое практич. значение К. Так как
каталитич. действие не связано с изменением свободной
энергии катализатора, то следовательно и смещение
положения равновесия химич. реакции под воздейст-
вием катализаторов невозможно. Следовательно,вблизи
состояния равновесия катализаторы должны в равной
степени ускорять как прямую, так и обратную реак-
ции. При удалении от состояния равновесия это усло-
вие может и не выполняться.
Воздействие катализатора на химич. реакцию
осуществляется путем промежуточного химич. вза-
имодействия его с реагирующими веществами. Вза-
имодействие с катализатором открывает новый ре-
акционный путь, обычно более сложный по числу
стадий, на к-ром катализатор входит в состав актив-
ного комплекса по крайней мере одной из стадий.
В соответствии с этим катализатор можно определить
как вещество, изменяющее скорость химич. реакции,
участвуя в образовании активного комплекса одной
из стадий данной реакции, и восстанавливающее свой
химич. состав при его превращении.
Большая скорость реакции по новому реакцион-
ному пути при положительном К. может быть обуслов-
лена двумя причинами: 1) меньшей высотой энергетич.
барьера нового, более сложного, реакционного пути,
возникающего при участии катализатора;2) возможно-
стью осуществления реакции при участии катализато-
ра по цепному механизму. Основным фактором, опре-
деляющим скорость химич. превращения, является
анергия активации (£) — разность энергий активного
комплекса и исходных реагирующих молекул. Если
предположить, что протекание химич. реакции не на-
рушает равновесного распределения энергии между
молекулами, то вероятность образования активного
комплекса, а следовательно, и скорость реакции в пер-
вом приближении пропорциональна ехр (—EjRT),
где R — газовая постоянная, Т — абс. темп-ра.
Отсюда следует, что скорость реакции тем больше, чем
меньше Е, и вследствие экс-
___________ поненциальной зависимости
возрастает значительно даже
Л при небольшом снижении Е.
£' Tr’Vk'Xl1 к" \ На рисунке представлено
1/ft\ \ изменение энергии реакцион-
Н/ ‘ т \ \|/ ной системы вдоль пу-
А \ у ти реакции без ката-
iii\® лизатора (кривая I)
______________________________________ \ и при участии ката-
______________________с лизатора (кривая II).
Точки 1 и 2 отвечают
Энергетический регистр реакцией- пппп,.ктям ппомга.-v-
него пути. А — энергия исходных продуктам промежу
веществ; В — промежуточного сое- точного взаимодеист-
динения; С — конечных продуктов, вия. Если энергия ак-
тивации всех стадий
реакционного пути с участием катализатора (Ег) ниже
энергии активации реакции без катализатора (£'),
то участие катализатора приведет к значительному
увеличению скорости; условие положительного ката-
лиза Е" <_ Е'. При этом механизме каталитич. дей-
ствия концентрация активных комплексов лимити-
рующей стадии не превышает равновесного значения
по отношению к концентрации реагирующих веществ.
Активность катализатора зависит от характера про-
межуточного взаимодействия, определяющего свой-
ства активных комплексов всех стадий, а следова-
тельно, и энергетич. рельеф реакционного пути. Кри-
вая II с двумя максимумами отвечает образованию
одного промежуточного* продукта (число стадий и
промежуточных продуктов может быть и значительно
большим, но катализатор может не участвовать во
всех стадиях). Энергия активации образования про-
межуточного соединения может быть больше, чем
энергия активации образования конечных продуктов
в присутствии катализатора. При этом взаимодействие
реагирующих веществ с катализатором не приводит
к образованию стабильной формы промежуточного
соединения; но и в этом случае катализатор входит
в состав активного комплекса и взаимодействие реа-
гирующих веществ с катализатором определяет реак-
ционный путь (кривая III).
Во многих случаях К. введение катализатора вы-
зывает цепную реакцию. При этом ускорение дости-
гается благодаря появлению богатых* энергией частиц
в процессе самой реакции, причем их концентрация
может превосходить равновесную. Напр., каталитич.
действие воды на окисление окиси углерода связано
с развитием реакционных цепей, с* участием гидро-
ксилов и атомов водорода. Отрицательный К. часто
связан с прекращением цепной реакции вследствие
обрыва цепей при взаимодействии отрицательного
катализатора с активными частицами. Примером
может служить замедляющее влияние кислорода на
соединение водорода с хлором. Отличие отрицатель-
ного К. от химич. ингибирования заключается в том,
что катализатор многократно вступает во взаимо-
действие с активными частицами, восстанавливая
свой состав после каждого акта обрыва цепи, тогда
как ингибитор после акта взаимодействия с актив-
ными частицами выбывает из реакции. В обоих рас-
смотренных случаях каталитич. ускорения химич.
реакций новый реакционный путь становится возмож-
ным в результате промежуточного химич. взаимо-
действия реагирующих веществ с катализатором.
Классификация каталитических процессов. Природа
химич. взаимодействия реагирующих в-в с ..катализа-
тором весьма разнообразна. Обычно различают по
характеру промежуточного взаимодействия две группы
каталитич. реакций — кислотно-основного и окисли-
тельно-восстановительного К. В реакциях первой
группы имеет место промежуточное кислотно-основ-
ное взаимодействие реагирующих в-в с катализато-
ром, т. е. переход протона от катализатора к одному
из реагирующих веществ или, наоборот, от реагиру-
ющего вещества к катализатору. При последующих
стадиях каталитич. реакции протон перемещается
в обратном направлении и катализатор восстанавли-
вает свой состав. При катализе апротонными кисло-
тами взаимодействие осуществляется через свободную
пару электронов реагирующего в-ва. Примерами
кислотно-основного катализа могут служить: гидро-
лиз сложных эфиров, ускоряемый кислотами, гидрата-
ция олефинов в присутствии фосфорно-кислотных
катализаторов, изомеризация и крекинг углеводоро-
дов на алюмосиликатных катализаторах и мн. др.
реакции. Для многих реакций кислотно-основного К.
удалось установить связь между кислотностью ката-
лизатора и его каталитич. активностью (подробнее см.
Катализ кислотно-основной).
При реакциях окислительно-восстановительного К.
промежуточное взаимодействие. связано с электрон-.
459
КАТАЛИЗ
460
ными переходами между катализатором и реагирую-
щими в-вами. К этой группе каталитич. реакций
относятся окисление двуокиси серы в трехокись в
произ-ве серной к-ты, окисление аммиака до окиси
азота при получении азотной к-ты, многочисленные
реакции парциального окисления органич. соедине-
ний, напр. этилена в окись этилена, нафталина во
фталевый ангидрид, гидрогенизации, дегидрогениза-
ция, циклизация и ароматизация углеводородов, раз-
ложение перекиси водорода, восстановление и окис-
ление ионов металлов в растворах и мн. др. реакции.
Каталитич. активностью в отношении окислительно-
восстановительных реакций преим. обладают металлы
4, 5 и 6-го периодов системы Д. И. Менделеева с недо-
строенной d-оболочкой электронов и их соединения.
Рассмотренные группы далеко не охватывают все раз-
нообразие каталитич. реакций. Во многих случаях
промежуточное взаимодействие не связано с измене-
нием заряда. Возможно также сочетание электронных
и протонных переходов в отдельных стадиях катали-
тич. реакций.
Конкретные механизмы каталитич. реакций очень
многообразны и пока лишь в немногих случаях более
или менее твердо установлены. Различают гомоген-
ный и гетерогенный К. При гомогенном К. ка-
тализатор и реагирующие в-ва образуют одну одно-
родную систему. Примерами гомогенного К. может
служить каталитич. окисление СО до СО2 в присутст-
вии паров воды или ускорение гидролиза раствори-
мых углеводов в водном р-ре в присутствии кислоты.
В первом случае катализатор и катализуемые в-ва
образуют однородную газовую фазу, во втором —
однородный, или же истинный, р-р; границы раз-
дела между катализатором и реагирующими в-вами
отсутствуют. При гетерогенном К. катали-
затор и реагирующие в-ва находятся в разных фазах
и отделены границей раздела. Чаще всего при гетеро-
генном К. катализатором является твердое тело, а
реагирующие в-ва находятся в газовой или жидкой
фазе; примером служит окисление аммиака в присут-
ствии металлич. платины или разложение растворен-
ной перекиси водорода под влиянием нерастворимого
в воде порошка двуокиси марганца. Отдельно следует
упомянуть микрогетерогенный К., при
к-ром катализатор находится в коллоидном или в вы-
сокомолекулярном состоянии. Примерами этого рода
К. могут служить биокаталитич. процессы, в к-рых
катализатррами являются ферменты, ускоряющие
многие химич. реакции, существенные для протекания
жизненных процессов (подробнее см. Ферменты). Гете-
рогенные каталитич. процессы часто называют кон-
тактными, а твердые катализаторы — контакт-
ными веществами или просто контактами.
Избирательность действия катализаторов. Сложные
каталитич. процессы могут протекать по нескольким
термодинамически возможным направлениям с обра-
зованием иногда большого числа различных продук-
тов. Преобладающее течение реакции по тому или
иному направлению зависит от добавленного катали-
затора, причем не всегда ускоряется процесс, термо-
динамически самый выгодный из нескольких возмож-
ных. Так, в присутствии окиси алюминия или окиси
тория этиловый спирт разлагается преим. на этилен и
воду: С2Н5ОН = С2Н4-(- Н2О. В присутствии се-
ребра, меди и др. металлов практически имеет место
только дегидрогенизация спирта: С2Н5ОН =
= СН3СНО Н2, с образованием уксусного альде-
гида. В присутствии многих других катализаторов обе
реакции идут одновременно. Существуют катализа-
торы, на к-рых этиловый спирт превращается в зна-
чительной степени в дивинил, эфир и т. д. Наряду
с активностью важной характеристикой катализатора
является поэтому избирательность действия, оцени-
ваемая относительным выходом определенного про-
дукта. Особенно резко выражена избирательность
действия у сложных и очень активных органич. ката-
лизаторов — ферментов, ускоряющих определенные
превращения только одного вещества или узкой груп-
пы веществ одинакового строения.
Промотирование и отравление катализаторов. Ка-
талитич. активность смеси катализаторов обычно не
аддитивна и иногда значительно превосходит актив-
ность компонентов, взятых в отдельности. Увеличение
активности часто наблюдается и при добавлении к ка-
тализатору веществ, к-рые сами по себе не обладают
заметной каталитич. активностью. Такие вещества
наз. промоторами, или активаторами. Так, каталитич.
активность V2O6 по отношению к реакции окисления
SO2 повышается в сотни раз при добавлении неболь-
ших количеств сульфатов щелочных металлов. Сме-
шанные и промотированные катализаторы широко
применяются в технике.
Практич. использованию каталитич. процессов
часто препятствует снижение активности катализатора
при воздействии на него веществ, называемых ката-
литич. ядами (см. Каталитические яды). Это явление
принято называть отравлением катализа-
торов. Различают обратимое отравление, при
к-ром после прекращения подачи яда активность вос-
станавливается, и необратимое, при к-ром требуется
специальная обработка — регенерация катализатора,
для полного или частичного восстановления первона-
чальной активности. При гомогенном К. отравление
может вызываться химич. взаимодействием яда с ка-
тализатором с образованием каталитически неактив-
ного или малоактивного соединения. Нек-рые случаи
отрицательного К. удалось объяснить отравлением
первоначально присутствующего положительного ка-
тализатора. В гетерогенном К. возможно отравление
вследствие блокировки поверхности катализатора
молекулами яда, к-рый в ряде случаев может образо-
вывать химически стойкое и неактивное соединение
с поверхностью катализатора. В органич. катализе
почти всегда имеет место постепенное снижение ак-
тивности твердых катализаторов в результате покры-
тия поверхности труднолетучими продуктами полиме-
ризации и углистой массой. На значение этого явле-
ния особое внимание обратил Н. Д. Зелинский. Так,
напр., при крекинге углеводородов на алюмосиликат-
ных катализаторах их активность быстро снижается
вследствие отложения на поверхности кокса или
смолы. Кроме блокировки, причиной отравления
твердых катализаторов может быть изменение реак-
ционной способности поверхности катализатора под
влиянием яда.
Для удлинения срока службы катализатора в про-
мышленных условиях в большинстве случаев произво-
дят тщательную очистку реагирующих веществ от
примесей, являющихся каталитич. ядами (напр.,
в произ-ве серной к-ты сернистый газ очищают от сое-
динений мышьяка, в произ-ве аммиака освобождают
газы от сернистых соединений и окиси углерода,
и т. д.). Известны случаи, когда одно и то же вещество
может служить промотором или ядом в зависимости
от его количества и способа введения в катализатор.
Эти явления называются модифицированием
катализаторов.
Открытие явлений катализа. Первые научные сведения
о К. относятся к началу 19 в. В 1806 Н. Клеман и Ш. Дезорм
открыли каталитич. действие окислов азота на окисление
SOs в камерном процессе получения H2SOi. В 1811 К. С. Кирх-
гоф открыл способность разб. к-т вызывать превращение
крахмала в сахар (глюкозу); в 1814 им же было установлено,
что эту реакцию может катализировать диастаза из ячменного
солода, Чем было положено начало изучению биологич. ката-
лизаторов — ферментов. В 1818 последовали открытия Л. Те-
нара ускоряющего действия большого числа твердых тел на
разложение р-ров перекиси водорода, и Г. Дэви — способности
паров спирта и эфира окисляться кислородом на платине.
461
КАТАЛИЗ
462
И. Дёберейнер в 1822 установил, что водород и кислород
соединяются на платине при обычной темп-ре, а Э. Митчерлих
в 1833 нашел, что спирт в присутствии H2SO4 превращается
в эфир. Затем последовало открытие и ряда других примеров
резкого положительного действия веществ на скорость или
возникновение химич. реакций. Это привело к установлению
существования особой группы явлений, названных Митчер-
лихом контактными (1833) и И. Берцелиусом каталитическими
(1835). К тому же периоду относятся первые гипотезы о при-
роде каталитич. действия.
Успехи органич. К. неразрывно связаны с развитием теории
строения органич. соединений Бутлерова. В 1869—74 А.М. Бут-
леров заложил основы гидратационного К., превратив оле-
фины в спирты путем присоединения воды в присутствии сер-
ной к-ты, а также открыл каталитич. полимеризацию непре-
дельных углеводородов в присутствии IbSOj, Н3РО4, BF3 и
других катализаторов. Обе эти реакции нашли широкое про-
мышленное применение. В 1871 М. М. Зайцев впервые приме-
нил каталитич. гидрирование водородом (в присутствии пал-
ладия) при восстановлении питросоединений в амины. В 1881
М. Г. Кучеров открыл реакцию гидратации ацетиленовых
углеводородов при каталитич. действии солей окиси ртути,
широко применяемую в пром-сти для получения ацетальдегида
из ацетилена. В. Н. Ипатьев впервые показал большую эффек-
тивность сочетания катализа с высокими давлениями, положив
тем самым начало большому и важному для техники разделу
К. под давлением (синтеза аммиака, метанола, гидрирование
органич. соединений).
Важнейшие каталитические процессы в промышлен-
ности. Первым промышленным произ-вом, основан-
ным на использовании гетерогенного К., явился про-
цесс Дикона — получение хлора окислением хлори-
стого водорода кислородом воздуха при 400° в при-
сутствии солей Си, нанесенных на шамот. После освое-
ния пром-стью электролиза NaCl процесс Дикона был
оставлен. В последние годы, в связи с появлением
значительных ресурсов избыточного хлористого водо-
рода, вновь пробудился интерес к его каталитич.
окислению, и были разработаны новые катализаторы,
активные при более низких темп-рах. В конце 19 в.
был создан контактный процесс произ-ва серной ки-
слоты, основанный на окислении SO2 кислородом в
присутствии Pt, нанесенной на различные носители.
В 20-х гг. 20 в. для произ-ва серной к-ты были разра-
ботаны ванадиевые катализаторы, представляющие
собой V2O5, нанесенную в смеси с K2SO4 на различ-
ные носители; с применением этого катализатора
ежегодно вырабатывают десятки миллионов тонн
HaSO4-. С помощью катализаторов (металлич. железо,
промотированное окислами калия и алюминия) уда-
лось преодолеть химич. инертность элементарного
азота и осуществить при высоких давлениях синтез
аммиака. Одновременно было осуществлено получе-
ние азотной кислоты путем окисления аммиака на
платиновых сетках или нек-рых окисных катализато-
рах. Синтез аммиака является основным источником
получения соединений азота для произ-ва удобре-
ний, взрывчатых веществ и других химич. продук-
тов. Получение водорода для синтеза аммиака и
других химич. произ-в также осуществляется с по-
мощью каталитич. процессов, а именно взаимодей-
ствия окиси углерода с водяным паром в присут-
ствии окисножелезных катализаторов и взаимодей-
ствия СН4 с водяным паром и О2 в присутствии раз-
личных никелевых катализаторов.
Особенно большую роль сыграли каталитич. ме-
тоды в развитии процессов органич. синтеза. Широкое
промышленное использование получило отверждение
жиров путем каталитич. гидрогенизации соединений,
содержащих двойные связи. Для этого процесса было
предложено большое число различных катализаторов,
гл. обр. на основе металлич. никеля. Крупным успе-
хом явились разработка и реализация произ-ва кау-
чука синтетического, основанного на превращении
этилового спирта в дивинил. Каталитич. методами
производятся и все остальные мономеры, исполь-
зуемые в произ-ве синтетич. каучука, — стирол, изо-
бутилен, изопрен и т. п.
В 30-х гг. 20 в в Геамании было построено большое
число крупных заводов для произ-ва моторного топ-
лива путем каталитич. гидрирования угля и смолы,
а также синтеза углеводородов из водяного газа, полу-
чаемого при газификации углей. Для осуществления
этих процессов были разработаны весьма сложные
катализаторы на основе сульфидов вольфрама, молиб-
дена, никеля и др. металлов — для процессов гидри-
рования, и металлич. кобальта, железа или никеля
с различными добавками — для процессов синтеза
из окиси углерода и водорода. Сейчас эти процессы
временно утратили свое значение, но во время второй
мировой войны они служили основным источником
снабжения Германии горючим.
В конце 30-х гг. каталитич. методы за короткий
срок заняли господствующее положение в технологии
нефтепереработки. С помощью каталитич. крекинга
углеводородов нефти на алюмосиликатных катализа-
торах ежегодно производятся сотни миллионов тонн
высококачественного моторного топлива. Для улуч-
шения качества бензинов в пром-сти переработки
нефти широко используются каталитич. реакции
циклизации и ароматизации углеводородов на пла-
тине, окиси молибдена или окиси хрома, нанесенных
на окись алюминия. Очень распространена и ката-
литич. десульфуризация нефтепродуктов — разложе-
ние органич. соединений, содержащих серу, с выделе-
нием сероводорода, осуществляемая на катализаторе,
содержащем кобальт и окись молибдена, нанесенные
на окись алюминия.
Гидратация этилена на фосфорнокислотных ката-
лизаторах является основным и наиболее экономич-
ным методом получения этилового спирта. Ценным
продуктом является окись этилена, образующаяся
при окислении этилена на серебряных катализаторах.
Каталитич. методы позволяют использовать пропилен
для получения изопропилового спирта, ацетона, ак-
ролеина, нитрила акриловой к-ты, продуктов алкили-
рования. Путем дегидрирования на окиснохромовых
катализаторах бутана, бутиленов, изопентана произ-
водятся в больших масштабах основные мономеры для
производства синтетич. каучука — дивинил и изо-
прен. Упомянутые уже выше реакции каталитич. аро-
матизации используются для производства из нефти
бензола, толуола и других ароматических углеводо-
родов.
Каталитич. методы используются не только для
получения мономеров для произ-ва синтетич. каучу-
ков, синтетич. волокон и других высокополимер'ных
материалов, но и для осуществления самого процесса
полимеризации. В последнее время применение спе-
циальных катализаторов (см. Катализаторы Циг-
лера — Натта) позволило решить проблему стерео-
специфич. полимеризации. Этим положено начало
развития новой области применения К., когда подбор
специфически действующего катализатора дает воз-
можность не только ускорять реакцию и направлять
ее в сторону получения продукта требуемого состава,
но и регулировать детальное строение этого продукта-.
Катализатор как бы выполняет роль программируют
щего устройства, матрицы, определяющей сложное
строение получаемого вещества. Новым направле-
нием использования К. является каталитич. очистка
технологич. газов путем превращения вредных за-
грязнений в безвредные или легко удаляемые (см.
Газов очистка). Таким путем производят очистку
технологич. газов от серусодержащих органич. сое-
динений, О2, СО и СО2, С2Н2 и др. В случае очистки
водорода каталитич. методы позволяют снизить со-
держание нек-рых примесей до одной десятимиллиард-
ной. К. может быть использован для обезврежива-
ния отходящих газов промышленности и транспор-
та (в том числе и автомобильного), а также для
очистки сточных вод, загрязненных органическими
веществами.
463
КАТАЛИЗ ГЕТЕРОГЕННЫЙ
464
Катализ в живой природе. К. играет ведущую роль
в химич. превращениях не только в пром-сти, но и
в живой природе. Вся сложная система управления
жизненными процессами в организмах основана на К.
Сложные комплексы химич. превращений, обуслов-
ливающие брожение, дыхание, пищеварение, синтез
белков и других соединений, преобразование химич.
энергии в механическую и т. п., осуществляются с по-
мощью ферментов (наз. также энзимами) — катали-
заторов белковой природы, образующихся в живых
телах. По нек-рым свойствам ферменты существенно
превосходят промышленные катализаторы. В послед-
нее время широко ведутся исследования синтетич.
органич. катализаторов — органич. полупроводников,
комплексных соединений, хелатных полимеров и др.,
характеризующихся более простым составом и строе-
нием по сравнению с ферментами, но моделирующих
в известной степени их действие. Подробнее см. Фер-
ментативные процессы.
Лит.: Зелинский Н. Д., Избранные труды, т. 2,
М.—Л., 1941; Зелинский Н. Д. и Баландин А. А.,
Катализ в органической химии, в кн.: Советская химия за
25 лет, М.—Л., 1944; Д о л г о в Б. Н., Катализ в органической
химии, 2 изд., Л., 1959; Проблемы кинетики и катализа. Сб.
переводных ст., т. 1—10, М,—Л., 1931—1960; Боресков
Г. К., Катализ в производстве серной кислоты, М.—Л., 1954;
Вопросы химической кинетики, катализа и реакционной спо-
собности. Доклады к Всес. совещанию по химич. кинетике и
реакционной способности, М., 1955; Emmett Р. Н., Cata-
lysis, V. 1—7, N. Y., 1954—60. Г. К. Боресков.
КАТАЛИЗ ГЕТЕРОГЕННЫЙ. Содержание:
Введение ......................................463
Мультиплетная теория катализа..................464
Кислотно-основной гетерогенный катализ . . ;...465
Окислительно-восстановительный гетерогенный катализ.
Катализ на полупроводниках.................... . 466
Катализ на металлах............................468
Катализ и кристаллическая структура............469
Промоторы и яды в гетерогенном катализе........470
Промышленное применение гетерогенного катализа . . 471
Кинетика гетерогенного катализа................472
Введение. Катализ гетерогенный — изменение ско-
рости реакции под воздействием катализатора, об-
разующего самостоятельную фазу (отделенную от
реагирующей системы границей раздела). Наиболее
распространены случаи ускорения твердыми катали-
заторами реакций в газовой или жидкой фазах. При
К. г., как и во всех прочих каталитич. процессах, из-
менение скорости реакции связано с промежуточным
химич. взаимодействием реагирующих веществ с ка-
тализатором, вызывающим изменение реакционного
пути. Для твердых катализаторов промежуточное
химич. взаимодействие ограничивается поверхностью
катализатора и не приводит к образованию промежу-
точных соединений в форме отдельных фаз. Это по-
верхностное взаимодействие обычно отождествляют
с хемосорбцией, предполагая, что адсорбируемые мо-
лекулы связываются с поверхностными атомами ката-
лизатора силами химич. связи.
Ускорение реакции при К. г. в большинстве слу-
чаев связано со снижением энергетич. барьеров всех
стадий нового реакционного пути, открываемого про-
межуточным взаимодействием с катализатором. Вы-
сказывалось мнение, что при К. г. ускорение реакций
вызывается возникновением цепного процесса на по-
верхности катализатора (Н. Н. Семенов, В. В. Воевод-
ский). Экспериментально пока не удалось доказать
сколько-нибудь широкое распространение этого меха-
низма, за исключением реакций, приводящих к обра-
зованию цепей при синтезе углеводородов из СО и
Н4, при полимеризации олефинов и гидрополимериза-
ции СО с олефинами (Я. Т. Эйдус). В отдельных слу-
чаях действие твердых катализаторов сопровождается
возникновением цепной реакции в реакционном про-
странстве (объеме). Активные частицы, вызывающие
продолжение реакции в объеме, образуются в ре-
зультате превращения реагирующих веществ па по-
верхности катализатора. Эти процессы иногда назы-
вают гетерогенно-гомогенным ката-
лизом (М. В. Поляков, А. А. Ковальский). Давно
известны и явления отрицательного катализа твердыми
поверхностями, прекращающими цепную реакцию
в результате обрыва на них объемных цепей.
Проявление химич. сил при поверхностном взаимо-
действии характеризуется рядом специфич. особен-
ностей, связанных с тем, что одним из участников
взаимодействия является твердое тело со сложной
электронной структурой. Активность катализатора,
определяемая изменением скорости реакции под его
воздействием, зависит от энергии промежуточного по-
верхностного взаимодействия, природы возникающих
и разрываемых связей и возможности образования
нескольких связей реагирующих веществ на опреде-
ленных расстояниях. Последний фактор имеет суще-
ственное значение при химич. превращениях слож-
ных молекул. Возникновение нескольких связей
между реагирующими веществами и катализатором на
определенных расстояниях друг от друга возможно
лишь при строгом соответствии между пространствен-
ным расположением атомов и связей в реагирующих
молекулах и геометрия, параметрами кристаллич.
решетки катализатора.
Мультиплетная теория катализа. Высказанное выше поло-
жение явилось основой структурной части' мультиплетной
теории катализа А. А. Баландина. Эта теория развивает пред-
ставление Н. Д. Зелинского о значении для катализа деформа-
ции реагирующих молекул под действием силового поля по-
верхности катализатора. Предполагается, что при каталитич.
реакции образуется мультиплетный комплекс, включающий
как атомы реагирующих веществ, между к-рыми разрываются
существующие связи и образуются новые химич. связи в про-
цессе реакции, так и определенное число атомов поверхности
катализатора. Так, для реакции перераспределения связей
между атомами А, В, С и Ь предложена мультиплетная схема
(рис. 1). Здесь К — атомы катализатора. Рамкой обведена т. н.
Рис. 1. Дублетная схема реакции:
АВ + CD AD + ВС
индексная группа, т. е. группа реагирующих атомов, сопри-
касающихся с катализатором. Согласно мультиплетной теории,
легкость протекания реакции должна зависеть от простран-
ственного соотношения между адсорбированной молекулой и
строением поверхности, т. е. должна определяться геометрии,
соответствием, обусловливающим минимальное напряжение
связей, к-рые остаются в молекуле или возникают вновь при
взаимодействии ее с элементами поверхности. Для реакции
дегидрирования циклогексана предполагаемся промежуточное
образование секстетного комплекса, в к-ром участвуют 6 ато-
мов поверхности катализатора (рис. 2),
Рис. 2. Секстетиая модель дегидрогенизации
циклогексана.
Из этой схемы вытенает, в хорошем согласии с опытом,
что активными катализаторами дегидрирования циклогексана
могут быть металлы, на поверхности к-рых находятся окта-
эдрич. грани с расстоянием мец^цу ближайшими атомами в пре-
делах от 2,48 A (N1) до 2,77 A (Pt). Геометрическое соответ-
ствие является необходимым, но недостаточным условием для
К. г. Так, напр., медь, образующая гранецентрированиые
кубич. кристаллы с расстоянием между ближайшими атомами
равным 2,56 А (т. е. лежащим между соответствующими рас-
стояниями для наиболее активных катализаторов — никеля
и платины), совершенно неактивна в отношении реакций гидри-
рования бензола и дегидрирования пиклогексаяа. Мульти-
плетная теория объясняет эю необходимостью наряду с гео-
465
КАТАЛИЗ ГЕТЕРОГЕННЫЙ
466
метрич. соответствием соблюдения энергетич. соответствия.
Последнее требует, чтобы энергия связи атомов реагирующих
веществ с катализатором отвечала бы определенному опти-
мальному значению, при к-ром эндотермичность стадий обра-
зования мультиплетного комплекса и его разложения была бы
минимальной. Для рассмотренной выше реакции перераспре-
деления связей между атомами А, В и С, D энергия образо-
вания мультиплетного комплекса:
E1 = — (Qab + Осо) + (Qak + Qbk + Qck + Оок)
а его распада:
= Qad + Qbc - (Qak + Qbk + Qck + Qdk)
Здесь Qjj соответствующие энергии связи.
С увеличением энергии связей реагирующих веществ с ка-
тализатором(2С/Х) величина Ei возрастает, а Е2 уменьшается.
Предполагается, при определенных допущениях, что энергия
активации каталитич. реакции составляет нек-рую долю
(0,75) от абс. значения теплового эффекта наиболее эндотермич.
стадии. Минимальное значение энергии активации и соответ-
ственно наибольшая величина скорости реакции должны
достигаться при вполне определенном значении энергии связей
реагирующих веществ с катализатором (SQjjj). Это отражает
более общее положение, высказывавшееся независимо от муль-
типлетной теории, заключающееся в том, что энергия проме-
жуточного взаимодействия реагирующих веществ с катализа-
тором не должна быть ни слишком малой, т. к. при этом будет
мала скорость первой стадии взаимодействия, ни слишком
большой, т. к. это приведет к высокой эндотермичности, а
следовательно, и к высокой энергии активации последующих
стадий, связанных с, отрывом атомов реагирующих веществ
от катализатора.
Мультиплетная теория катализа не позволяет Оценивать
теоретич. значения Q/)f,a следовательно, и предвидеть влияние
свойств катализатора на эти величины. Определение Qjk из
экспериментальных данных связано с очень большими погреш-
ностями, и распространение полученных значений на другие
соединения с теми же атомами в индексной группе — нена-
дежно. Тем не менее, в отдельных случаях на основе принципа
энергетич. соответствия удалось объяснить последовательность
протекания каталитич. превращений различных соединений.
Кислотно-основной гетерогенный катализ. В К. г.,
так же как и в катализе гомогенном, целесообразно
рассмотреть отдельно реакции с промежуточным взаи-
модействием кислотно-основного характера, связан-
ного с переходами протона, и окислительно-восстано-
вительные реакции, при к-рых взаимодействие реаги-
рующих веществ с катализатором обусловлено элект-
ронными переходами. К первой группе часто относят
реакции, катализируемые кислотами, нанесенными на
твердые носители, напр. гидратацию или полимериза-
цию олефинов на фосфорнокислотных катализаторах.
Эти процессы по существу псевдогетерогенны, т. к.
каталитич. реакция протекает в жидкой гомогенной
пленке кислоты, покрывающей инертный носитель.
Хорошо известны, однако, действительно твердые
катализаторы, обладающие активностью в отношении
реакций кислотно-основного катализа. К их числу
принадлежат алюмосиликатные катализаторы кре-
кинга углеводородов, кремне-циркониевые катализа-
торы превращения спиртов, вольфрамовые катализа-
торы гидратации олефинов, окись алюминия и т. п.
Активность этих катализаторов также обусловлена
их кислотными свойствами, к-рые удалось непосред-
ственно измерить. Функция кислотности нек-рых
твердых катализаторов, напр. алюмосиликатных или
кремнециркониевых, того же порядка, что и конц.
фосфорной к-ты. К числу твердых кислотных катали-
заторов надо отнести и высокополимерные синтетич.
смолы — катиониты в протонной форме, получившие
в последнее время довольно широкое применение.
Известны случаи значительного каталитич. ускорения
превращений различных органич. веществ твердыми
основаниями. Количественная связь между катали-
тич. активностью и кислотностью твердых катализа-
торов довольно сложна и различна для разных реак-
ций- Это связано с различной основностью реагирую-
щих веществ, а также и с тем обстоятельством, что
промежуточное взаимодействие при многих реакциях
^'Является чисто кислотно-основным и его энергия
определяется не только кислотными, но и другими
свойствами катализатора.
Окислительно-восстановительный гетерогенный ка-
тализ. Катализ на полупроводниках. В случае окис-
лительно-восстановительных реакций промежуточное
химич. взаимодействие обусловливается определен-
ным взаимодействием электронов катализатора и реа-
гирующих веществ. Характер этого взаимодействия
в значительной степени зависит от электронной струк-
туры твердого катализатора. Приближенное кванто-
вомеханич. рассмотрение показывает, что уровни
энергии, отвечающие отдельным атомам или ионам,
при их сближении до расстояний, характерных для
твердого тела, расщепляются и образуют энергетич.
зоны. Эти зоны могут перекрываться или разделяться
запрещенными промежутками. Металлы характери-
зуются отсутствием запрещенной зоны. У изоляторов
заполненная электронами зона отделена от следую-
щей более высокой свободной зоны широким интерва-
лом запрещенных уровней энергии. У полупроводни-
ков этот интервал невелик или внутри его находятся
локальные электронные уровни, на к-рые электроны
могут переходить из нижней заполненной зоны (ак-
цепторные уровни дырочных полупроводников), либо
уровни, с к-рых возможен переход в верхнюю свобод-
ную зону (донорные уровни электронных полупро-
водников, подробнее см. Полупроводники). Полупро-
водниковыми свойствами обладают многие катализа-
торы — окислы, сульфиды, металлоорганич. поли-
меры.
Трактовка на основе зонной теории промежуточногов
взаимодействия при К. г. для полупроводниковых
катализаторов была впервые дана Ф. Ф. Волькен-
штейном. Свободные электроны или дырки (отсутствие
электрона в заполненной зоне) рассматриваются как
свободные валентности твердого катализатора, уча-
ствующие в поверхностном взаимодействии с реаги-
рующими веществами. Различаются «слабая» связь,
осуществляемая без участия свободного электрона
или дырки катализатора, при к-рой хемосорбирован-
ная частица остается электрически нейтральной, и
«прочная» связь, в к-рой свободный электрон или
дырка принимают непосредственное участие. При этом
адсорбированная частица может связывать свободный
электрон, что приводит к отрицательному заряжению
поверхности (акцепторная связь), или связывать
дырку, т. е. отдавать электрон, в результате чего по-
верхность заряжается положительно (донорная связь).
Различные формы адсорбции могут переходить друг
в друга в результате соответствующих электронных
переходов. Концентрации частиц, адсорбированных
в различной форме, зависят от положения уровня
Ферми (уровня химич. потенциала электрона) на
поверхности, определяющего работу выхода элект-
рона. Так, напр., при повышении уровня Ферми (сни-
жении работы выхода электрона) облегчается хемо-
сорбция с помощью донорной связи и затрудняется
осуществление акцепторной связи. Хемосорбирован-
ные частицы в различных формах обладают различной
реакционной способностью. Скорость каталитич. ре-
акции определяется, т. обр., концентрацией хемосор-
бированных частиц в определенной форме и может
меняться в зависимости от положения уровня Ферми.
Правильнее рассматривать влияние положения
уровня Ферми не на равновесные концентрации хемо-
сорбированных частиц в различных формах, а на кон-
центрацию активных комплексов лимитирующей ста-
дии реакционного пути, связанной с образованием
или превращением заряженных хемосорбированных
частиц (С. 3. Рогинский). Если лимитирующая ста-
дия связана с переходом электрона между катализа-
тором и реагирующим веществом, то изменение энер-
гии активного комплекса этой стадии, а следовательно,
и энергии активации реакции, равно нек-рой доле
от изменения работы выхода (Г. К. Боресков), Многие
467
КАТАЛИЗ ГЕТЕРОГЕННЫЙ
468
авторы не рассматривают возможности «слабой» хемо-
сорбции и учитывают только образование на поверх-
ности заряженных хемосорбированных частиц (Ха-
уффе).
Рассматриваемые представления позволяют связать
каталитич. активность полупроводников с другими
свойствами, определяемыми их электронной структу-
рой — работой выхода электронов, электропроводно-
стью, энергией активации электропроводности и др.
Во многих случаях наличие такой связи было экспе-
риментально доказано. Как известно, многие свой-
ства примесных полупроводников, в частности элект-
ропроводность и работа выхода, могут меняться в ши-
роких пределах путем введения добавок. Так, введе-
ние в состав NiO небольшой добавки Li2O значи-
тельно увеличивает электропроводность и повышает
работу выхода; оказалось также, что значительно
возрастает и каталитич. активность в отношении реак-
ций разложения гидразина и закиси азота. Добавка
1п3О3 оказывает противоположное действие как на
электропроводность и работу выхода, так и на ката-
Литич. активность в отношении указанных реакций
(Н. П. Кейер). Добавки сульфатов щелочных металлов
к V2O5 резко повышают как электропроводность, так
и каталитич. активность в отношении реакций окисле-
ния SO2, Н2 и гомомолекулярного изотопного обмена
кислорода. В большинстве случаев соотношения ока-
зались, однако, более сложными и часто плохо согла-
сующимися с выводами теории. Так, добавки 1п2О3
снижают, a Li2O повышают электропроводность в ре-
акции окисления СО на упомянутых выше катализа-
торах из NiO. Введение в Ge добавок Ga и Sb, изме-
нивших концентрацию свободных электронов и дырок
на 8 порядков, не привело к изменению каталитич.
активности в отношении реакции изотопного обмена
в молекулярном водороде. Отсюда следует, что адсорб-
ция водорода на Ge протекает без участия свободных
электронов или дырок из объема полупроводника.
Каталитич. активность полупроводников зависит,
т. обр., не только от полупроводниковых свойств —
электропроводности, положения уровня Ферми и др.
Примером может служить хемосорбция со связыванием
электрона (акцепторная связь). Теплота этого про-
цесса: Л , W
q = А — 9 -р W
где А — сродство сорбируемой молекулы к электрону,
не зависящее от свойств катализатора; <р — работа
выхода электрона катализатора; W — энергия, вы-
деляющаяся при приближении сорбируемой частицы
из бесконечности к месту сорбции на поверхности
катализатора. Эта энергия, включающая как куло-
новские, так и обменные составляющие, весьма слож-
ным образом зависит от электронной структуры ката-
лизатора и сорбируемой частицы и не может быть
определена в рамках рассматриваемых теорий. При
вариации свойств катализатора изменение теплоты и
энергии активации сорбциибудутопределяться.т.обр.,
не только положением уровня Ферми, определяющим
работу выхода, но и изменением величины W. Поэтому,
исходя только из полупроводниковых свойств, нельзя,
вообще говоря, предсказать каталитич. активность.
Только в тех случаях, когда изменения W настолько
малы, что ими можно пренебречь по сравнению с из-
мёнёниями <р, напр. при введении очень малых доба-
вок, сильно влияющих на положение уровня Ферми,
изложенная теория позволяет с определенной степенью
надежности предвидеть изменение каталитических
свойств.
Наряду с зонной теорией в трактовке явлений К. г.
на полупроводниках используется и другой прибли-
женный метод, при к-ром каталитич. активность со-
поставляют с электронной структурой отдельных
йоиов или атомов, слагающих твердый катализатор,
пренебрегая их взаимодействием. Экспериментально
установлено, что высокая каталитич. активность в от-
ношении окислительно-восстановительных реакций
преим. свойственна металлам и ионам с незаполнен-
ными d-оболочками. В последнее время Д. Дауденом
была сделана попытка привлечь к расчету взаимодей-
ствия реагирующих веществ с катализатором, содер-
жащим ионы переходных металлов, теорию кристал-
лич. поля, уже давно успешно используемую в химии
комплексных соединений. Сущность эффекта кристал-
лич. поля заключается в том, что вырожденный d-уро-
вень катиона переходного металла в электрич. поле,
создаваемом окружающими отрицательными ионами и
поляризованными молекулами, расщепляется на не-
сколько уровней. Это расщепление зависит как от
силы поля, так и от конфигурации окружающих лиган-
дов. В случае октаэдрич. окружения образуются
3 орбиты с энергией ниже основного уровня и 2 орбиты
с энергией, превышающей основной уровень. В твер-
дых катализаторах, являющихся соединениями пере-
ходных металлов, отрицательными лигандами могут
быть ионы кислорода, серы и т. п. Ионы металла,
расположенные на поверхности катализатора, окру-
жены меньшим числом лигандов, чем внутренние
ионы. Хемосорбция реагирующих веществ увеличивает
число лигандов, окружающих поверхностный кати-
он, в результате чего увеличивается расщепление
d-уровня. В результате расщепления часть электронов
располагается на более низких уровнях, что приводит
к стабилизации системы. Если в результате хемосорб-
ции возникает октаэдрич.
конфигурация, то наи-
большая стабилизация
достигается при 3 и 8
электронах в d-оболочке
катиона, если же число
электронов равно 0, 5 или
10, то энергия стабили-
зации равна нулю (рис. 3).
В соответствии с этой
кривой меняется и теп-
лота сорбции, а следова-
тельно, и скорости сорб-
ции, десорбции и химич.
превращения хемосорби-
рованных частиц, опре-
деляющие скорость ка-
талитич. реакции.
Экспериментальные данные еще недостаточны для
проверки значения эффекта кристаллич. поля в К. г.
Величина эффекта, оцениваемая по оптич. данным,
достаточно велика, и учет его необходим. Надо за-
метить, однако, что стабилизация, вызываемая кри-
сталлич. полем, представляет собой лишь поправку
к энергии взаимодействия, зависящей более сложным
образом от электронной структуры катализатора и
реагирующих веществ. Это ограничивает возможность
предвидения каталитич. действия на основе изложен-
ных представлений.
Катализ на металлах. Еще большие трудности пред-
ставляет теоретич. трактовка каталитич. активности
металлов. Применение зонной теории в этом случае
менее обосновано вследствие высокой концентрации
свободных электронов в металлах. Совокупность экс-
периментальных данных показывает, что наиболее
широко как по числу катализируемых реакций, так и
по величине активности, каталитич. свойства прояв-
ляются у металлов длинных периодов системы Мен-
делеева, гл. обр. в пределах VI, VII, VIII и 16 групп.
Отсюда следует, что основным фактором, определяю-
щим активность металлов, является конфигурация
d-оболочки, постепенно заполняемая в атомах метал-
лов длинных периодов. У металлов, расположенных
энергия стабилизации
12345 6789 10
1,исло </-= электронов •
Рис. 3. Энергия стабилизации
кристаллич. полем в зависимо-
сти от числа электронов в d-обо-
лочке при условии сохранения
принципа максимальной муль-
типлетности.
469
КАТАЛИЗ ГЕТЕРОГЕННЫЙ
470
3d + 4s
Сг Fe Со Ni Со
Рис. 4. Удельная катали-
тич. активность металлов
IV периода в отношении
реакций:
1. Н2 + D2-± 2HD;
2. 2Н2 + О2 ss 2Н2О:
3. N, 4- ЗН-, =г 2NH
в начале периода, в кристаллич. состоянии в металлич.
связи участвуют все s- и d-электроны. С увеличением
числа электронов, начиная примерно с группы VI,
связывающие d-орбиты оказываются заполненными,
и электроны входят преим. в атомные d-орбиты. Это
проявляется в переходе через экстремальное значение
в пределах длинных периодов таких свойств металлов,
как сжимаемость, теплота сублимации, темп-ра плав-
ления и др. Для каталитически активных металлов
характерно наличие вакантных атомных d-орбит.
В рамках зонной теории это отвечает наличию незапол-
ненной d-зоны. Первые металлы длинных периодов
весьма энергично взаимодействуют с многими газами,
но вследствие малой энергии связи между атомами
в металле область устойчивости металлич. состояния
ограничена и поверхностное взаимодействие легко
приводит к фазовому превращению. Каталитич. ак-
тивность наиболее резко выражена у металлов с сум-
марным числом s- и d-электронов, превышающим
число электронов, участвующих в металлич. связи.
Сочетание прочности связи, обеспечивающей устой-
чивость фазы металла, с наличием несвязанных элект-
ронов на атомных d-орбитах,
открывает широкие возмож-
ности поверхностного взаимо-
действия, существенного для
протекания каталитич. реак-
ций. Максимуму каталитич.
активности отвечает опреде-
ленное число электронов на
d-орбитах, различное для раз-
ных реакций (рис. 4). Пока
не удалось дать сколько-ни-
будь строгий расчет взаимо-
действия с участием d-элек-
. тронов металла. Предпола-
гается, что при хемосорбции
Н2, N2, СО, С2Н4 и нек-рых
других газов образуется ко-
валентная связь с участием
d-электронов металла. Как
следует из рис. 4, для диссо-
циативной хемосорбции азо-
та и водорода оптимальным
является различное число
d-электронов, что, возможно,
связано с различной валентностью водорода и азота.
В ряде случаев связь осуществляется путем перехода
неподеленной пары электронов хемосорбируемой мо-
лекулы на d-уровни металла.
удобным методом исследования зависимости ката-
литич. активности металлов от их электронной струк-
туры является изучение каталитич. свойств сплавов.
В сплавах, содержащих переходные металлы, может
происходить перераспределение электронов, приво-
дящее к изменению числа d-электронов на атомных
орбитах. Ряд исследователей обнаружил линейную
зависимость каталитич. активности сплавов от числа
дырок в d-зоне. Во многих работах найдена, однако,
более сложная зависимость. Исследования сплавов
замещения типа Юм-Розери показало, что по мере
роста электронной концентрации и заполнения зоны
Бриллюена наблюдается снижение каталитич. актив-
ности в отношении реакций, лимитирующая стадия
к-рых связана с переходом электрона от реагирующего
вещества к катализатору. В нек-рых случаях наблю-
дается резкое повышение каталитич. активности спла-
вов на границе двухфазной области.
Катализ и кристаллическая структура. Основным
фактором, определяющим электронную структуру и
активность твердых катализаторов, является химич.
состав. Одно время широким распространением поль-
зовались теории о существовании особых структур,
необходимых для проявления каталитич. активности.
Нек-рые авторы отождествляют их с ребрами и углами
кристаллов и с различными физич. нарушениями
кристаллич. решетки. С. 3. Рогинский указывает на
решающую роль избыточной свободной энергии ката-
лизатора в образовании каталитически активных
структур. Н. И. Кобозев полагает, что каталитич. ак-
тивность присуща только аморфным образованиям —
«ансамблям», состоящим из небольшого числа атомов,
расположенных на поверхности кристаллов катализа-
тора или носителя. Кристаллич. фаза, по мнению
Кобозева, лишена каталитич. активности. В проти-
воположность этим взглядам для многих катализато-
ров — Fe, Ni, Pt, Fe3O4, y-A]2O3, MgO, ZnO и др.,
установлено, что при неизменном составе уд. катали-
тич. активность, отнесенная к единице поверхности,
приблизительно постоянна при изменении в широких
пределах крупности кристаллов, условий приготовле-
ния и термич. обработки.
Различные кристаллич. модификации одного и того
же вещества могут обладать сильно отличающейся уд.
каталитич. активностью вследствие изменения харак-
тера связей внутри кристалла и электронной струк-
туры. Так, переход у-А12О3 в а-А12О3 в результате
прогрева при высоких темп-рах на несколько поряд-
ков снижает удельную каталитич. активность этого
катализатора и превращает его из активного дегид-
рирующего в малоактивный дегидрирующий. Не-
большие различия уд. каталитич. активности могут
быть связаны и с разным развитием отдельных граней
кристаллов, отличающихся по хемосорбционным и
каталитич. свойствам.
Промоторы и яды в гетерогенном катализе. Приме-
няемые в пром-сти катализаторы обычно содержат
наряду с основным каталитически активным компонен-
том различные добавки, играющие роль промото-
ров. Механизм промотирования твердых катализа-
торов весьма разнообразен. Добавки могут вступать
с основным компонентом в химич. взаимодействие,
образуя продукты, обладающие повышенной ката-
литич. активностью; образовывать твердые растворы,
меняющие электронную структуру каталитически
активного компонента в сторону, благоприятную для
повышения каталитич. активности; изменять условия
взаимодействия с реагирующими веществами в местах
контакта фаз основного компонента катализатора и
промотора; увеличивать дисперсность и величину до-
ступной поверхности каталитически активного ком-
понента; препятствовать рекристаллизации основного
компонента; ускорять отдельные стадии сложного
каталитич. процесса, способствуя направлению реак-
ции в сторону образования требуемого продукта,
и т. п.
С целью увеличения доступной поверхности и пре-
дохранения катализатора от рекристаллизации часто
применяют носители (трегеры) — инертные мате-
риалы с развитой поверхностью, на к-рую наносится
каталитически активное вещество. Во многих случаях
носитель вступает в определенное взаимодействие
с катализатором, более или менее глубоко воздейст-
вуя на его каталитич. свойства.
Каталитические яды, содержащиеся в виде приме-
сей в реакционной смеси или образующиеся как по-
бочный продукт каталитич. реакции, часто снижают
активность твердых катализаторов в результате бло-
кировки поверхности каталитически активного ком-
понента. Во многих случаях яды взаимодействуют
с каталитически активным компонентом, изменяя его
каталитич. свойства. Несмотря на противоположный
результат, механизм воздействия ядов на катализатор
в этом случае сходен с действием промоторов. Одно и
то же вещество может поэтому, в зависимости от коли-
чества, являться и промотором, и ядом. Так, добавле-
471
КАТАЛИЗ ГЕТЕРОГЕННЫЙ
472
ние к серебряному катализатору окисления этилена
малых количеств хлора повышает скорость образова-
ния окиси этилена, большие же количества хлора резко
снижают каталитич. активность серебра в отношении
этой реакции. Вещества, оказывающие на катализа-
торы такое двойственное действие, иногда наз. моди-
фикаторами. О промоторах см. также статью
Катализ.
Кинетика гетерогенного катализа. Скорость реак-
ций К. г. определяется скоростью наиболее медлен-
ного промежуточного этапа. В промежуточных этапах
при К. г. участвуют хемосорбированные частицы,
концентрации к-рых, вообще говоря, зависят от объем-
ных концентраций всех компонентов системы, взаимо-
действующих с поверхностью катализатора. Хиншель-
вудом на основе работ Лэнгмюра была построена схема
возможных форм кинетич. уравнений, основанная
на предположении, что поверхностные концентрации
реагирующих веществ являются равновесными по от-
ношению к их объемным концентрациям и что энергии
хемосорбции на всех активных участках поверхности
одинаковы и не зависят от заполнения. Полученные
уравнения могут быть представлены в следующей об-
щей форме:
где к — константа скорости реакции, не зависящая
от состава реакционной смеси, Ci — объемные концент-
рации всех компонентов; bi — адсорбционные коэфф.;
п; — число молекул компонента J, участвующих в об-
разовании активного комплекса лимитирующей ста-
дии реакции из объемной фазы; mi — то же в хемо-
сорбированном состоянии.
На этой основе были выведены кинетич. уравне-
ния для ряда реакций К. г. (дегидратация, гидроге-
низация, дегидрогенизация и др.) в удовлетворитель-
ном соответствии с экспериментальными данными.
Однако в большинстве случаев определяемые экспе-
риментально кинетич. зависимости плохо уклады-
ваются в эту форму. На основании разнообразных кос-
венных данных была установлена зависимость энер-
гии поверхностных связей от заполнения поверхности
хемосорбированными частицами (М. И. Темкин,
С. 3. Рогинский). Исходя из линейной зависимости
изменения энергии хемосорбции с заполнением по-
верхности, были выведены степенные кинетич. ур-ния
с дробными показателями, хорошо описывающие
экспериментальные данные по кинетике синтеза ам-
миака, конверсии окиси углерода (см. Водород), окис-
ления двуокиси серы (см. Серная кислота) и ряда
других практически важных реакций. Следующим
этапом развития кинетики реакций К. г. явился
учет воздействия реакционной смеси на состав, а сле-
довательно, и свойства катализатора. Таким путем
удалось объяснить особенности кинетики нек-рых
окислительных реакций (Г. К. Боресков).
Изложенные закономерности описывают кинетику
собственно химич. превращения, не осложненного
влиянием Щ)оцессов переноса реагирующих веществ
и тепла (область химич. кинетики). Промышленные
катализаторы обычно представляют собой пористые
зерна с развитой внутренней поверхностью, в десятки
тысяч раз превышающей наружную поверхность. При
достаточно большой скорости химич. превращения
наблюдаемая скорость каталитич. процесса зависит
от скорости диффузионного переноса реагирующих
веществ внутрь зерен и продуктов реакции в обратном
направлении. Большое значение при этом имеет пори-
стая структура катализатора (объем и размеры пор),
влияющая как на активность, так и на избиратель-
гость катализатора. Для каждого каталитич. про-
цесса, в зависимости от условий его проведения^
кинетич. зависимостей и удельной каталитич. актив-
ности катализатора, может быть установлена опти-
мальная пористая структура, обеспечивающая наи-
большую скорость реакции. Влияние переноса внутри
зерен изменяет форму кинетич. ур-ния. При значив
тельном влиянии процессов переноса, концентрации
реагирующих веществ в центре зерен катализатора
близки к равновесным (область внутренней диффузии),
и наблюдаемый порядок реакции по компоненту,
диффузия к-рого лимитирует процесс, становится
средним между действительным и первым, а по всем
остальным компонентам снижается в два раза. На-
блюдаемая энергия активации становится в два раза
ниже действительной.
Для очень быстрых реакций наблюдаемая скорость
определяется скоростью переноса реагирующего ве-
щества к наружной поверхности зерен (область внеш-
ней диффузии). Концентрации реагирующих веществ
у наружной поверхности зерен в этом случае близки
к равновесным, порядок реакции близок к первому и
наблюдаемая энергия активации очень мала. Вслед-
ствие относительно медленного переноса тепла реак-
ции, темп-ра поверхности катализатора в этом слу-
чае отличается от темп-ры реакционной смеси и для
экзотермич. реакций близка к темп-ре адиабатич. ра-
зогрева при полном превращении. Во внешне-диффузи-
онной области протекают промышленные реакции
окисления аммиака, окисления спиртов, поверхност-
ного горения. В этой области активность катализа-
тора не влияет на наблюдаемую скорость реакции,
а лишь определяет границы условий осуществления
реакции в области внешней диффузии.
Каталитич. активность твердых катализаторов равна
изменению скорости реакции при введении катализа-
тора, отнесенному к единице его поверхности. Для
определения каталитич. активности используются
различные методы измерения скорости реакций (см.
Кинетика химическая).
Промышленное применение гетерогенного катализа.
Промышленные реакции К. г. в газовых системах
обычно осуществляют в неподвижном слое зернистого
катализатора, через к-рый проходит реакционная
смесь. За последние 20 лет получили распространение
контактные аппараты с псевдоожиженным слоем
катализатора, поддерживаемом во взвешенном со-
стоянии подымающимся потоком реакционной смеси.
Преимущество катализа в псевдоожиженном слое
заключается в полноте выравнивания темп-ры в слое,
высоком коэфф, теплопередачи между слоем и поверх-
ностями теплообмена, легкости непрерывной смены
катализатора, возможности применения мелкозерни-
стых катализаторов. Основным недостатком является
истираемость катализатора и необходимость специ-
альных приспособлений для отделения его от газового
потока. Катализ в псевдоожиженном слое целесооб-
разно использовать в случаях частой смены катали-
затора для регенерации, необходимости отвода боль-
ших количеств тепла реакции и, в нек-рых случаях,
для увеличения степени использования внутренней
поверхности зерен путем значительного уменьшения
их размеров.
Основной задачей конструирования контактных
аппаратов является реализация оптимального темпе-
ратурного режима, обеспечивающего максимальную
скорость реакции и выход требуемого продукта. Оп-
тимальный температурный режим определяется киие-
тич. закономерностями каталитич. реакции; для обра-
тимых экзотермич. реакций он отвечает определен-
ному снижению темп-ры с ростом степени превраще-
ния; для параллельных реакций, из к-рых дающая
полезный продукт характеризуется меньшей энергией
активации, темп-ра должна повышаться с ростом
473
КАТАЛИЗ ГОМОГЕННЫЙ
474
степени превращения и т. п. Современные расчетные
методы, основанные на математич. моделировании,
позволяют устанавливать точные оптимальные значе-
ния основных параметров каталитич. процесса и
в нек-рых случаях непосредственно переходить от
данных лабораторного исследования к проектирова-
нию промышленных аппаратов, минуя промежуточ-
ные стадии опытных и полузаводских установок.
(Г. К. Боресков, М. Г. Слинько). Реализация оптималь-
ного температурного режима достигается промежуточ-
ным теплообменом между слоями катализатора, внут-
ренним теплообменом в слое или промежуточным вве-
дением реакционной смеси или разбавителя требуе-
мой темп-ры.
Реакции К. г. с жидкими реакционными смесями
проводят либо со взвешенным катализатором, отде-
ляемым в дальнейшем от реакционной смеси фильтра-
цией или центрифугированием, либо пропусканием
реакционной смеси через неподвижный слой крупно-
зернистого катализатора.
Лит.: Зелинский Н. Д. и Баландин А. А.,
Катализ в органической химии, в кн.: Советская химия за
25 лет, М.—Л., 1944; Гетерогенный катализ в химической
промышленности. Материалы Всес. совещ. 1953 г., М., 1955;
Рогинский С. 3., Адсорбция и катализ на неоднородных
поверхностях, М.—Л., 1948; Волькенштейн Ф. Ф.,
Электронная теория катализа на полупроводниках, М., 1960;
Боресков Г. К., Катализ в производстве серной кис-
лоты, М.—Л., 1954; Проблемы кинетики и катализа. Сб.,
т. 1—10, 1931—60; Катализ. Сб. переводов, М., 1953, 1960
(из: Advances In Catalysis, v. 1—13, N.—Y.—L., 1948—62);
Emmett P. H., Catalysis, v. 1—7, N. Y„ 1954—60;
Жермен Ж., Гетерогенный катализ, пер. с франц., М.,
1961. Г. К. Боресков.
КАТАЛИЗ ГОМОГЕННЫЙ — изменение скорости
или возбуждение химич. реакции с помощью катали-
заторов, к-рые находятся в молекулярно- или ато-
марно-дисперсном состоянии и образуют однородную
(как правило, газовую или жидкую) систему с реа-
гирующим в-вом. Скорость гомогенной каталитич.
реакции чаще всего пропорциональна концентрации
катализатора. Для простейшего случая превращения
А —• в + с + . . .
в присутствии катализатора (Кат.) можно написать
схему
1) А + Кат. = А Кат. 2) А Кат. = В + С + . . . + Кат.
Если скорость процесса определяется первой стадией,
т. е. взаимодействием реагента с катализатором, ки-
нетич. ур-ние будет иметь вид:
-^3 = А1[А] [Кат.]
где — константа скорости первой реакции. Если
же определяющая стадия — вторая, т. е. распад
промежуточного соединения реагента с катализато-
ром, ур-ние скорости реакции будет:
-^^ = fc2K[A] [Кат.]
где к2 — константа скорости второй реакции, К —
константа равновесия первой реакции, равная
[А Кат.]/[А] [Кат.}.
В обоих случаях будет наблюдаться пропорцио-
нальность концентрации реагирующего в-ва и ката-
лизатора. Напр., разложение ацетальдегида
СН3СНО=СН4-)-СО без катализатора начинается
в>цпе 500°, в присутствии же J2 реакция идет уже при
400°, причем скорость ее w = k [СН3СНО] [ J2],
Специальными опытами было показано, что концен-
трация J2 в ходе реакции не изменяется, т. е. J2 по-
стоянно регенерируется и, следовательно, является
катализатором. При разложении СН3СНО в присут-
ствии малых добавок кислорода (О,О5°/о) было найдено
их сильное ускоряющее действие, причем начальная
скорость реакции была пропорциональна [О2}1/2.
В этом случае кислород является не катализатором,
а инициатором реакционной цепи и расходуется в ходе
реакции:
СН3СН0+02=Н02-4-СН3СО •
Н02-+СН3СН0=Н2О2+СН3СО-
СНзСО-=СНз-+СО,- СН3.-|-СН3СНО=СН3+СН3СО.
При катализе сложными органич. молекулами могут
наблюдаться отклонения от первого порядка реакции
по концентрации катализатора (дробный порядок
или даже порядок выше 1-го). Элементарный меха-
низм действия катализатора различен в радикаль-
ных, ионных и молекулярных реакциях.
Радикальные реакции характеризуются
тем, что катализатором для них служит источник
активных промежуточных частиц, богатых энергий,
обеспечивающих развитие цепной реакции. Напр.,
ускоряющее действие паров воды на реакцию окисле-
ния СО заключается в образовании активных частиц:
атомов Н- и радикалов ОН-, поддерживающих цепную
реакцию:
Н-+02=0Н-+0,- ОН-+СО=СО2+Н.; СО+О=СО2
Скорость реакции: w = к [НаО] [СО]. По радикаль-
ному механизму, по-видимому, протекает и приведен-
ная выше реакция разложения СН3СНО в присут-
ствии J2, хотя промежуточных активных частил в этом
случае установить не удалось.
Радикальные реакции являются преобладающим
случаем К. г. в газовой фазе. В р-рах по радикаль-
ному механизму протекает часто катализ ионами
переменной валентности. Напр., по последним дан-
ным Барба, Баксендаля и др., каталитич действие
ионов железа в реакции разложения водных р ров
перекиси водорода можно объяснить совокупностью
след, реакций:
Fea++H2O2=Fe»++OH-+OH-; Fe’+4-ОН- --Fe !+- ОН~
ОН-+Н2О2=НО2-+Н2О; Fe ++НО2-=Ре”--|-НО
FeS++HO2-=O2+H++FeS+; Fe3++HLO2=Fe +-1-НО, Н+
Суммарным результатом является разложение пере-
киси водорода: 2Н2О2—.2Н2О-)~ О2. Добавление к р-ру
Н2О2 ингибиторов, захватывающих свободные ра-
дикалы, тормозит реакцию. Этим доказывается ра-
дикальный механизм реакции. Скорость реакции про-
порциональна концентрации солей железа. Согласно
этому механизму, реакция является истинно катали-
тической, т. к. ионы Fe многократно вступают в про-
межуточное взаимодействие с участниками реакции.
Более старый механизм Габера и Вейсса отводит
ионам Fe лишь роль инициирования цепей
Fe2++H2O2=OH-+OH-+Fe»t; Fe3++H2O2=Fe2++H2O-+Н+
и их обрыва' по реакции ионов Fe со свободными
радикалами; сама же цепь развивается, по реакциям:
он-+н2о2=но2-+н2о; но2-+н2о2=н2о+он-+о2
т. е. образование конечного продукта О2 происходит
без участия ионов Fe.
Приведенный пример указывает на трудность от-
несения вещества, ускоряющего радикальный про-
цесс, к категории катализаторов (в строгом понятии
этого слова) или инициаторов. Не случайно поэтому
в литературе инициаторы гомогенных радикальных
реакций часто наз. катализаторами. Напр., в прак-
тически важных процессах жидкофазного гомоген-
ного окисления парафинов и других органич. соеди-
нений, добавки стеаратов и др. солей Со, Мп, Fe
или Си, ускоряющих реакцию, относят к катализа-
торам. Экспериментально же было показано, что эти
вещества расходуются лишь в начальном периоде
реакции, т. е. являются инициаторами. Так, при
окислении бензальдегида в присутствии ацетата
475
КАТАЛИЗ ГОМОГЕННЫЙ
476
кобальта последний зарождает радикалы, напр., по
схеме:
С eH5CHO+Co3+=Coa++CeH5CO -+н+
после чего идет реакция без его участия:
CuH5Co-+Oa=CeH5COOO-
СвН5СООО-+С6Н5СНО=СсН5СО-+СвН5СОООН
В ионных реакциях промежуточными активны-
ми веществами являются ионы. Особенно распростра-
нен катализ кислотно-основной в р-рах; катализатора-
ми являются ионы Н3О+ и ОН-. Ионы переменной ва-
лентности не всегда вызывают радикальную реак-
цию. Одна и та же реакция при одном pH может
идти как радикальная, при другом — как ионная.
При катализе анионами, содержащими металлы пе-
ременной валентности, иногда удается выделить
кинетически устойчивые промежуточные соединения.
Напр., в реакции разложения Н2О2 под действием
иона молибдата МоО^-:
МоОЗ—|-2Н2О2^=±М0О|-+2Н2О; МоО|-—- Моо®-+Оа
было выделено промежуточное вещество, отвечающее
аниону МоО|~.
• Координационно-комплексный к а-
т а л и з является важным классом К. г., близким
к кислотно-основному катализу. Напр., реакция де-
карбоксилирования щавелево-уксусной к-ты может
ускоряться ионом Н+ или ионом металла, связываю-
щим кислоту в комплекс:
О=С---0—СН2—СОд - »О==С—C^CHg-4-COg
/ / \
О О” о о
'хСи=+'/ \Си/
Ионы Н+ уменьшают отрицательный заряд и уве-
личивают легкость акцептирования остающейся мо-
лекулой пары электронов из отщепляющейся карбо-
ксильной группы. Однако еще лучше эту функцию
выполнит многозарядный ион металла, связывающий
анион дикетокислоты в прочный комплекс, как ука-
зано на схеме. Для реакции декарбоксилирования
найден порядок каталитич. активности ионов:
Als+>Fes+>Cua+>Fea+>Zna+>Mg2+>Mn2+>Caa+
примерно соответствующий их способности к ком-
плексообразованию. В общем, ионы металлов будут
катализировать реакции, ускоряемые кислотами, если
при этом образуются промежуточные хелатные со-
единения. В этом случае ион металла более эффекти-
вен, чем Н+. Если же возможна одна точка присоеди-
нения, тогда каталитически более активен Н+. Ка-
талитич. свойства иона металла обусловлены его
поляризующим действием, т. е. отношением квадрата
его заряда к радиусу е2/г. За счет поляризации
снижается активационный барьер реакции. Иногда
бывает, однако, что хорошие комплексообразователи
с большим е2/г каталитически мало активны. Напр.,
А13+ и Ве2+ не влияют на скорость бромирования аце-
тоуксусного эфира, активными же катализаторами
для этого процесса являются ионы — более сильные
комплексообразователи: Со2+, Cu2+, Ni2+ и др. Для
объяснения отклонений от прямой связи каталитич.
активности иона с отношением е2/г в последнее время
(Ф. Басоло, Р. Пирсон) выдвинуто представление
о том влиянии, к-рое оказывают изменения коорди-
нации лигандов (аддендов) вокруг комплексного иона
переходного элемента на его реакционную способ-
ность. Согласно этим взглядам, для каталитич. реак-
ции может быть существенна конфигурация орбит
d-электронов иона-катализатора. В поле лигандов,
к-рыми могут быть в частном случае и молекулы ве-
ществ, претерпевающих каталитич. превращения,
происходит расщепление d-уровней этого иопа на
подуровни, причем электроны стремятся занять наи-
более низкие d-подуровни. Всякое изменение коорди-
нации вокруг этих ионов вызывает изменение запол-
нения d-подуровней. Напротив, изменение координации
вокруг непереходных элементов, а также ионов со
структурой d° (Sc3+, Ti4+) и d6 (Fe3+, Mn2+) не вызы-
вает изменений электронной конфигурации. Поэтому
каталитич. свойства ионов переходных металлов
должны отличаться от ионов непереходных металлов
и ионов со структурой d°, d6. В энергию активации
первых будет входить со знаком «-|-» или «—» энергия
перехода d-электронов на более низкие подуровни.
В соответствии с этим ионы переходных металлов
оказываются каталитически менее активны или более
активны, чем иопы непереходных металлов. Кон-
кретное указание о направлении (-]- или —) этой
зависимости определяется строением активированного
комплекса в той или иной реакции. Кроме того, важ-
ное значение имеет просто геометрич. размер иона.
Возможно, что в вышеприведенном примере (броми-
рование эфира) ион Ве2+ просто слишком мал для
того, чтобы разместить на себе 2 лиганды в активи-
рованном комплексе.
Широко распространенпый в пром-сти К. г. хло-
ристым алюминием также относится к координацион-
но-комплексному катализу. А1С13, присоединяясь
с помощью иона А13+ к углеводороду, напр., в реак-
ции алкилирования, сильно поляризует связь С—С
и облегчает т. о. реагирование. В гомогенных ком-
плексных катализаторах полимеризации, напр., при
катализе полимеризации пропилена с помощью
Ti(C5H5)2Cl2 -{- А1(С2Н5)2С1, такую роль выполняет
ион титана.
Каталитич. действие солей ртути в реакции гидра-
тации ацетилена (см. Кучерова реакция) объясняет-
ся образованием нестойких промежуточных л-ком-
плексов:
Hg2+
Н\ /ОН
Н3о+Н-С=с-Н—►Hg2+ + /С=С\ —СН3СНО
11 и
Комплексы Ag+c С2Н2 более прочны и каталитически
мало активны. Ион металла может образовывать со-
единение не только с л-связью, но и с о-связью реа-
гирующей молекулы. По-видимому, каталитич. дей-
ствие иона серебра в реакции гомогенного каталитич.
гидрирования обусловлено образованием молеку-
лярного соединения Ag+ с Н2. В дальнейшем идет
реакция:
Ag+
/\ -}-Ag+=2AgH+
Н -Н
Комплекс AgH+ непосредственно восстанавливает
(гидрирует) субстрат.
В живых организмах активаторами биокатализа-
торов — ферментов также служат ионы-комплексо-
образователи: Mn2+, Fe2+, Fe3+, Со2+, Ni2+, Cu2+,
Mg2+, Mo (точное валентное состояние последнего
неизвестно). Комплексы этих ионов с органич. моле-
кулами катализируют многие окислительно-восста-
новительные реакции и реакции гидролиза. Напр.,
Mg2+ входит в состав хлорофилла, Fe2+ — в состав
гемоглобина, Fe3+ — в состав каталазы и т. д. Ката-
литич. активность таких комплексных соединений
часто во много раз превышает каталитич’. активность
изолированных ионов. В ряде случаев была обнару-
жена зависимость каталитич. активности этих ионов
от природы лигандов (М. Кэлвин, В. Лангенбек,
Л. А. Николаев). Особенно сильно активируют азот-
содержащие лиганды. Несмотря на большое различие
констант скорости при катализе комплексами одного
и того же иопа с различными лигандами, в реакциях
этого типа часто наблюдается постоянство энергии
активации. Увеличение же к происходит за счет
множителя к0 в уравнении Аррениуса.
477
КАТАЛИЗ ИОНООБМЕННЫЙ — КАТАЛИЗ КИСЛОТНО-ОСНОВНОЙ
478
Молекулярные гомогенно-ката-
литические реакции встречаются, по-ви-
димому, реже, чем радикальные и ионные. Образова-
ние молекулярных соединений при газовом К. г.
не доказано. При катализе комплексными соедине-
ниями, разобранном выше, во многих случаях ка-
талитич. активность не изменяется при переходе от
одного растворителя к другому, т. е. не наблюдается
зависимости энергии активации от диэлектрич. про-
ницаемости среды; здесь, очевидно, также образуются
молекулярные соединения без разделения зарядов.
В последнее время Е. А. Шилов, Дж. Суэйн, Я. К. Сыр-
кин показали, что активными промежуточными веще-
ствами в случае молекулярных реакций могут быть
кольчатые донорно-акцепторные комплексы исходных
веществ с катализатором. Напр., 2-оксипиридин,
имеющий донорную и акцепторную группы, оказался
катализатором мутаротации тетраметилглюкозы, при-
чем более активным, чем эквивалентная смесь фенола
и пиридина. Активированный комплекс принимает
форму кольца
Мктнвяро”
'ванный *
комплекс
/С-СХ С-С /
~ сх с' Сх *
н-с-о н н-с о н
/ II I
он он
н о »•
1 оксимртин / н О
(катализатор) й —С N —
С О С С катапизатор
Чс = с/ \-z'
Особенно часто встречаются, по-нидимому, 6-членные
комплексы, в к-рых напряжение валентных углов
минимально; напр. механизм образования сложных
эфиров под действием катализатора — недиссоции-
рованной к-ты НА можно представить след, образом:
Rg
СК /О-Н.
>С< >А
R/ ЧО-Н'
I
н
Rg
о, ,о-нч
>А-
R/ \О-НХ
I
н
Чередование рвущихся и образующихся связей в одном
комплексе обеспечивает снижение энергии активации.
Липг.г Кондратьев Б. Н., Кинетика химических
газовых реакций, М., 1958; Семенов И. Н., О некоторых
проблемах химической кинетики и реакционной способности,
2 изд., М., 1958; Вопросы химической кинетики, катализа и
реакционной способности, М., 1955; Катализ. Исследование
гомогенных процессов, пер. с англ., М., 1957; Проблемы кине-
тики и катализа, т. 10, М., 1960; Martell А. Е., Cal-
vin М., Chemistry of metal-chelate compounds, N. Y., [1953];
В a s о 1 о F., Pearson R. G-., Mechanisms of inorganic
reactions, N. Y., 1958; Frost A. A., Pearson R. G.,
Kinetics and mechanism. A study of homogeneous chemical
reactions, 2 ed., N. Y. — L., [1961]. О. В. Крылов.
КАТАЛИЗ ИОНООБМЕННЫЙ — частный случай
кислотно-основного катализа. Наиболее широкое рас-
пространение получил катализ водородными формами
катионитов, к-рый по природе катализирующих ионов
можно считать разновидностью кислотного катализа
в гомогенной среде. Катализ анионитами по механизму
аналогичен основному катализу в гомогенной среде.
Несмотря на сходство механизма реакций, скорость
их протекания в условиях кислотно-основного ката-
лиза и катализа ионитами (ионообменными смолами)
может быть весьма различной. Отношение константы
скорости данной реакции на ионите к константе ско-
рости в условиях кислотно-основного катализа при
эквивалентной концентрации катализирующих ионов
наз. эффективностью катализа на ионитах. Ее зна-
чение может быть равно, больше или меньше 1 и
зависит от свойств реагирующих веществ (в т. ч.
размеров их молекул), природы растворителя (влияю-
щего на коэфф, распределения реагирующих молекул
между ионитом и жидкой фазой), и свойств ионита
(степени поперечной связанности, обменной емкости,
природы активных групп, состояния влажности). При
диффузионных осложнениях эффективность катализа
ионитами зависит также от размера зерен ионита.
Катализ ионитами широко применяют в синтетич. органич.
химии. На катионитах (как правило, сильнокислых сульфо-
катионитах) осуществлены многие реакции, имеющие прак-
тич. значение, напр.: этерификация — синтез этилацетата,
этилцеллозольва и др.; алкилирование — синтез третичного
бутил- и октилфенолов, метилбензилфенола и др.; дегидрата-
ция — напр. получение изобутилена иэ триметилкарбинола,
а-метилстирола из диметилфенилкарбинола; гидратация —
напр. синтез этиленгликоля и др.; гидролиз — инверсия са-
хара и др. углеводов; полимеризация — синтез паральдегида,
низкомолекулярных олефинов и диенов; конденсация — напр.
получение окиси мезитила. Известны и другие реакции,
осуществляемые на катионитах, напр. разложение гидропере-
киси кумола до ацетона и фенола. В нек-рых случаяжна основ-
ной процесс накладываются побочные реакции. Так, гидрата-
ция третичных амиленов может проходить одновременно с их
полимеризацией и изомеризацией.
Реакции, катализируемые в гомогенных условиях катио-
нами металлов, ускоряются катионитами в соответствующих
ионных формах. Так, на Hg-форме катионита протекает гидра-
тация ацетилена и фенилацетилена, а также конденсация аце-
тилена и фенола в 4,4-диоксифенилэтан. Значительно менее
исследован катализ анионитами, что в основном обусловлено
их весьма ограниченной химич. и термич. устойчивостью.
Главной областью применения анионитов являются реакции
конденсации: альдольной, кротоновой, бензоиновой, реакции
Кновенагеля и Пехмана. Известно применение анионитов как
катализаторов циклизации, напр. синтез 2,3-диметилиндола;
реакции присоединения, напр. синтез циангидрина; гидрата-
ции, напр. получение гликолей.
В нек-рых синтезах применяют одновременно катионит и
анионит, действующие независимо друг от друга, напр. при
синтезе кротонового альдегида.
Исходным продуктом является паральдегид, к-рый в кон-
такте с катионитом деполимеризуется до ацетальдегида, кон-
денсирующегося затем под действием одновременно присут-
ствующего анионита в альдоль и кротоновый альдегид.
Медленно протекающие реакции катализа иони-
тами обычно проводят в статич. условиях — переме-
шиванием ионита с превращающимися в-вами или
кипячением смеси с обратным холодильником. Быстро
протекающие реакции выполняются в потоке реаги-
рующих веществ; неподвижный катализатор нахо-
дится в экстракторе аппарата Сокслета или в ионито-
вой колонке. Преимущества катализа ионитами; про-
стота аппаратуры, легкость отделения ионообменной
смолы от катализата, возможность многократного
использования ионита, высокая чистота продуктов ре-
акции, селективность действия ионита, возможность
получения веществ, неустойчивых в условиях гомо-
генного кислотно-основного катализа.
Липг.: С а с м а н С., в сб.: Ионный обмен, пер. с англ.,
М., 1951, с. 270; Helfferrich F., lonenaustauscher, Bd. 1, В.,
1959, S. 448; G 1 е n a t R., Chimie et Industrie, 1956, 75,№ 2,292;
Naumann G., Chem. Technik, 1959, И 1, 18; По-
лянский H. Г., МаркевичС. M., Вести, техн, и
экономической информ., 1961, № 2, 28 (НИИТЭХИМ); По-
лянский Н. Г., Усп. химии, 1962, 31, вып. 9.
Н. Г. Полянский.
КАТАЛИЗ КИСЛОТНО-ОСНОВНОЙ — ускорение
гетеролитич. реакций кислотами и основаниями. Такая
способность кислот и оснований — один из призна-
ков, характеризующих этот класс химич. соединений.
Классич. теория К. к.-о. (В. Оствальд, С. Аррениус)
приписывала каталитич. действие исключительно ионам
водорода и гидроксила. Позднее было установлено,
что реакции, ускоряемые ионами Н+ и ОН-, катали-
зируются также недиссоциированными молекулами
кислот и оснований и различными другими ионами,
напр. ионом NHJ (общий кислотно-основной’катализ).
Известны, однако, реакции специфич. катализа только
ионами водорода (инверсия сахара, гидролиз ацета-
лей и др.) или только ионами гидроксила (напр.,
разложение нитрозотриацетонамина).
Теория общего К. к.-о. является составной частью
теории кислот и оснований Дж. Н. Бренстеда. Эта
теория определяет кислоту как донор, а основание —
как акцептор протона; кислотами и основаниями мо-
гут быть электронейтральные молекулы, катионы и
479
КАТАЛИЗ КИСЛОТНО-ОСНОВНОЙ
480
анионы с различным нарядом. Кислотно-основное
взаимодействие в средах с высокой диэлектрич. про-
ницаемостью состоит в передаче протона от кислоты
НА к основанию В с образованием новой пары —
кислоты ВН+ и основания А-:
HA+Bs=±BH++A- (1)
Напр.
HCl + NH3t=(NH4)+ + ci-
кислота основа- кислота основа-
ние ние
К. к.-о. обусловлен протолитич. реакцией (перенос
протона) между субстратом, выступающим в роли
слабого основания или слабой к-ты, и катализатором,
к-рым могут быть соединения, удовлетворяющие опре-
делению кислот или оснований по Бренстеду. Возмож-
ность К. к.-о. в многочисленных реакциях органич.
синтеза определяется способностью органич. соеди-
нений проявлять кислотные или основные свойства.
Предполагается, что первой ступенью протолитич.
реакции является образование молекулярного соеди-
нения кислоты с основанием за счет водородной связи
(И. Кольтгоф, К. Ингольд, Н. А. Измайлов, Н. Д. Со-
колов). В зависимости от свойств растворителя, кис-
лотности и основности реагентов, реакция может
остановиться на этой стадии, либо привести к образо-
ванию ионных пар, либо завершиться их диссоциа-
цией на ионы. Протолитич. реакция (присоединение
или отщепление протона), как стадия каталитич. про-
цесса, приводит к перераспределению электронов в мо-
лекуле субстрата и тем самым к образованию проме-
жуточных соединений с повышенной реакционной
способностью (карбониевые ионы, карбанионы, по-
лярные комплексы). Эта специфич. стадия реакции
обусловливает снижение энергии активации и уско-
рение процесса в целом. Дальнейшее превращение
активных промежуточных соединений ведет к серии
реакций с образованием конечных продуктов.
С излагаемой точки зрения реакции К. к.-о. обя-
зательно включают стадию переноса протона из одного
места молекулы в другое и потому требуют присут-
ствия как доноров, так и акцепторов протонов, что
отвечает след, общим схемам:
Катализ кислотой
НХ+НА=НХН++А-
в+нхн+=±вн++хн
BH++A-^=t В+НА
Катализ основанием
B+HXz=BH++X-
Х-|-НА=ХН+А-
ВН++А-^=В+НА
В этих схемах НХ — субстрат, НА — кислота, В и
А-— основания, НХН+и Х~—ионизованные формы
субстрата, ХН — продукт реакции. Примером может
служить кетоенольная таутомерия, определяющая,
в частности, протекание известной реакции иодиро-
вания ацетона. В результате протолитич. реакций
кетон переходит в енольную форму:
Н3С-С=О Н3С-£- 0Н4-А-
| +НА^=± |
СНз СН3
В+Н3С-С-ОН=гВН++Н2С=С- ОН
I I
сн3 сн3
Енольная форма легко реагирует с иодом.
Согласно М. Поляни, присоединение протона и его
отщепление в другом месте молекулы субстрата про-
исходит в едином акте, т. е. активированный комплекс
состоит из трех реагентов. Большинство исследова-
телей принимают, однако, стадийную схему, что
в большей мере согласуется с результатами кинетич.
исследований.
Другой пример катализа кислотами — реакция
гидратации олефинов в соответствующие спирты:
RCH=CH24-HA^RCII2-CH24-A~
RCHa-^Ha+HaO^iRCHa-CHa-OHa
RCH2CH2-OH2+A- z=t R- СН2-СН2ОН+НА
Предположение о том, что реакция идет через проме-
жуточное образование карбониевого (ВСН2СН2) и
оксониевого (ВСН2СН2ОН2) ионов находится в со-
гласии с результатами кинетич. исследований.
Каталпз основаниями можно проиллюстрировать
реакцией обратимой альдольной конденсации аль-
дегидов и кетонов:
I I
>СН—С=О4-Вг±>С= С—О-4-ВН+
I I III
>С=С—0-4- >СН— С=О^>СН— С— С— 0=0
i-1
III I'll
> сн-с-с—с =о+вн+ >СН-С—С—.0=0
Многие реакции карбонильных соединений, катали-
зируемые основанием, проходят через стадию иони-
\ । в . |
зации типа: >СН—С = О=* >с=с—0~
/ вн+х
Наблюдаемая константа скорости реакций общего
К. к.-о. слагается из произведений констант катали-
тич. действия всех кислот и оснований, присутствую-
щих в реакционной смеси, в том числе растворителя
(£s) и его протонизованной формы (^SH+) на их кон-
центрацию:
k = + feSH+ [SH+] + feB [В] + feHA [НА] + h A- [A-]
Это уравнение написано без учета солевых эффектов.
Бренстед предложил уравнения, связывающие кон-
станты каталитич. действия кислот (&НА) и основа-
ний (£в) с константами их диссоциации КНА и Къ-.
feHA „ [9 кНА\а feB „ , 2Г ,1-а
-у- = gha —-— ) и -у = °в <₽кв)
Коэфф. GHA, Gb и а зависят от темп-ры, природы
растворителя и природы реакции, причем 0 < а < 1.
Коэфф, р и q — статистич. поправки, введенные Брен-
стедом для учета основности кислоты (р) и числа
мест у основания, к к-рым может присоединиться
протон (7). Эти уравнения подтверждены многочис-
ленными экспериментальными исследованиями. Урав-
нение Бренстеда выражает связь между кинетич. и
термодинамич. характеристиками реакций и имеет
общее значение, выходящее за пределы области
К. к.-о. Теоретич. интерпретация этого уравнения
была дана Поляни.
Константа диссоциации не пригодна для сравнения
силы кислот в разных растворителях, в том числе
в растворах кислот разной концентрации. Для харак-
теристики протоно-донорных свойств различных кис-
лотных сред с высокой диэлектрич. проницаемостью
Л. Р. Гамметт и А. Дейроп предложили приближен-
ную термодинамич. функцию Но, названную ими
функцией кислотности:
„ , , /в „ , свн+
Но = — ig “н+ -= рК - 1g —р—-
/вн+ св
Здесь рК = — IgX, где К — константа равновесия
реакции ВН+ В -f- Н+, ен+ — активность протона,
Св и Свн+ — концентрации основания и его протопи-
зованной формы, /в и /вн+ — их коэфф, активности,
/в
Произведение а„+-----= h0 наз. кислотностью,
/вн+
На = — 1g Ло. В разб. водном р-ре /в//вн+ = 1 и
На = pH, т. е. функция Но — естественное продол-
жение шкалы pH в область концентрированных р-ров
кислот. Измерение Но сводится к измерению степени
ионизации основания с известным рК. С помощью
серии оснований — индикаторов, ионизирующихся
в средах с различной кислотностью, можно измерить
481
КАТАЛИЗ кислотно-основноЛ
482
Но в любом диапазоне концентраций кислоты при
условии, чтобы области ионизации индикаторов пере-
крывались и отношение /в//вн+ было постоянно для
всех индикаторов. Измерения Но были выполнены для
многих кислотных сред (HaSO4—НаО, Н3РО4—Н2О,
НС1—Н2О и др.) в широком диапазоне концентраций,
вплоть до безводных.
Если в ходе реакции кислотного катализа осуще-
ствляется равновесие (1), то концентрация протонизо-
ванной формы субстрата будет:
свн+=к^Г0Св,
где Сд=Св <-'вн+ — суммарная концентрация суб-
страта. Если стадией, определяющей скорость реак-
ции, является дальнейшее превращение однократно
протонизованной формы субстрата, то скорость реак-
ции ш = к1Свн+ = Св, где к4 — константа
скорости лимитирующей стадии. При условии К /г0,
т. е. Свн+ •< Св, наблюдаемая константа скорости
к — kihg/K. Это отвечает уравнению IgA -|- Но = const,
предложенному Гамметтом, к-рое было подтверждено
на примерах многих реакций. Оно является выраже-
нием связи между скоростью реакции и протоно-до-
норными свойствами кислоты-катализатора.
Реакции К. к.-о. протекают не только в растворах,
по также на поверхности твердых катализаторов,
обладающих свойствами кислот и оснований (алюмо-
силикаты, ионообменные смолы и др-)- Перенос
протона от кислоты к основанию является частным
случаем акцепторно-донорного взаимодействия общего
типа, т. е. образования координационной связи между
акцепторами и донорами электронных пар и гетеро-
литич. расщепления таких связей. С этих позиций
Г. Н. Льюис характеризует кислоту как акцептор,
а основание — как донор электронной пары, включая
в класс кпслот и оснований широкий круг веществ,
электронное строение к-рых определяет их акцептор-
но-донорные свойства. Во взаимодействии с акцепто-
ром может участвовать не только неподеленная пара
электронов донора, но также вся система л-электро-
нов ароматич. ядра.
Многие соединении, удовлетворяющие определению
кислот по Льюису, обладают каталитич. действием
в отношении ряда реакций, катализируемых протон-
ными кислотами (алкилирование ароматич. углеводо-
родов, изомеризация, гидрогалогенирование и поли-
меризация олефинов и др.). Катализаторами этих
реакций часто служат BF3, А1С13, HgCla, SnCl4 и др.,
электронное строение к-рых придает им свойства
акцепторов электронных пар. Примером катализа
акцепторами электронных пар может служить реак-
ция алкилирования ароматич. соединений олефинами,
катализируемая хлористым алюминием. Ее механизм
обычно представляют схемой:
li1 9.' н С|
R-C::CH2 + Al:Cl R—СН-С:AJ:Cl
Cl H Cl
основание кислота
основание . кислота
Cl
: АГ.С1
Cl
н
CH-CH2:A1C13 а—у сдвиг
R
протона
CH-CH,
I 3+AlCl'3
Хотя действительный механизм процесса более сло-
жен, приведенная схема в общих чертах отражает
механизм каталитич. действия апротонных акцепторов
электронных пар, характеризуя их как специфич.
комплексообразователей. Определенное сходство ме-
ханизмов каталитич. действия апротонных акцепто-
ров электронных пар и протонных кислот видно из
сопоставления приведенной выше схемы и схемы меха-
низма той же реакции при катализе протонными
кислотами:
С этой точки зрения явления К. к.-о. отнюдь не
ограничиваются реакциями с участием доноров и
акцепторов протова. Однако взаимодействие между
акцепторами и донорами электронных пар -— процесс
более сложной природы, чем переход протона от кис-
лоты к основанию. Это связано с резко выраженным
влиянием особенностей электронного строения, состава
и структуры льюисовских кислот и оснований на
характер акцепторно-донорного взаимодействия. По
этой причине льюисовские кислоты и основания ха-
рактеризуются исключительной специфичностью ка-
талитич. действии.
К. к.-о. приобрел за последние годы исключительно
важное практич. значение в химич. процессах, осу-
ществляемых в промышленном масштабе. К числу
таких важнейших процессов относятся гидратация и
изомеризация олефинов, этерификация спиртов, ни-
трование углеводородов, гидролиз крахмала и дру-
гих полисахаридов, алкилирование ароматич. соеди-
нений, каталитич. крекинг нефти, синтез высокомо-
лекулярных соединений методами ионной полимери-
зации и др. Процесс парофазной гидратации этилена
в этиловый спирт, являющийся основным источником
синтетич. этилового спирта, осуществляется с исполь-
зованием в качестве катализатора фосфорной к-ты,
нанесенной на пористые силикатные носители. Ана-
логичные катализаторы применяются при парофаз-
ном алкилировании бензола олефинами. Катализа-
торами алкилирования ароматич. соединений в жид-
кой фазе служат хлористый алюминий или фтористый
бор. Широкое применение в качестве катализаторов
процесса полимеризации нек-рых непредельных угле-
водородов получили фтористый бор, хлорное олово
и др. Напр., полимеризация изобутилена при ката-
литич. действии BF3 протекает с очень большой ско-
ростью при весьма низких темп-рах (ок. —100°).
Для каталитич. крекинга нефти используют алюмо-
силикатные катализаторы, поверхность к-рых обла-
дает кислотными свойствами. Большая практич. зна-
чимость К. к.-о. определила интенсивное развитие
исследований в последние годы в области практич.
использования кислот и оснований как катализаторов
различных процессов и в направлении выявления
закономерностей и механизма каталитич. действия
этого класса соединений.
Лит.: ШатенштейнА. И., Теории кислот и оснований,
М.—Л., 1949; БеллР., Кислотно-основной катализ и строе-
ние молекул, в сб.: Катализ. Исследование гомогенных про-
цессов, пер. с англ., под ред. А. А. Баландина и А. М. Рубин-
штейн, М„ 1957, с. 4—66; Ки лпатрик М., Кислотный
и основной катализ, там же, 67—95; Людер В. и Ц у ф-
фанти С., Электронная теория кислот и оснований,
пер. с англ., М., 1950; Long F. А., Р a n 1 М. A., Chem.
Revs, 1957, 57, 935; Чирков Н. М., в сб.: Проблемы кине-
тики и катализа, 1960, 10, 255; Топчиева К. В., там
же, 1960, 10, 247; Шатенштейн А. И., Изотопный обмен
и замещение водорода в органических соединениях в свете тео-
I рии кислот и оснований, М., I960. А. И. Гельбштейн.
483
КАТАЛИЗАТОРЫ
484
КАТАЛИЗАТОРЫ — вещества, изменяющие ско-
рость химич. реакции. Основное требование, предъ-
являемое к К.,— высокая каталитич. активность в
расчете на единицу его веса (1 г) или поверхности
(1 л«2). Единой теории подбора катализатора
не существует. Многие К., широко применяемые
в пром-сти (напр., железный К. синтеза аммиака),
были подобраны чисто эмпирически, путем много-
численных проб. Однако за последнее время пред-
ставления о механизме катализа продвинулись на-
столько, что оказалось возможным сформулировать
нек-рые принципы подбора К., пригодные для огра-
ниченного круга реакций. Принимая за основу ту
нли иную теорию действия К., стремятся найти
к.-л. независимую, хорошо изученную характери-
стику К. (параметр кристаллич. решетки, заряд и
радиус иопа, электропроводность, кислотность и т. д.),
к-рую на основе этой теории удается связать простым
соотношением с каталитич. активностью. При этом
для начальной формулировки принципов подбора К.
обычно накапливают сведения о каталитич. актив-
ности различных веществ, на основании этих данных
устанавливают связь каталитич. активности с неза-
висимыми параметрами этих веществ, после чего
переходят •к поискам К. среди неизученных веществ.
Наиболее плодотворны представления, учитывающие
химич. взаимодействие между К. и реагирующим ве-
ществом. Поэтому определяющим фактором в подборе
К. часто является периодич. система элементов Мен-
делеева.
Для кислотно-основных каталитич. реакций типич-
ными К. при гомогенном катализе служат раствори-
мые кислоты и основания, при гетерогенном ката-
лизе — твердые тела, обладающие кислотными (син-
тетич. и природные алюмосиликаты, А12О3, TiO2,
ThO2, реже силикагель) и основными (СаО, ВаО,
MgO" CaNH2) свойствами. Чем больше кислотность
или основность вещества (см. Катализ кислотно-
основной), тем больше при прочих равных условиях
для реакций этого класса и его каталитич. активность.
В реакциях, катализируемых к-тами Бренстеда (см.
Кислоты и основания), напр. в крекинге углеводоро-
дов на алюмосиликатах, в ряде случаев удалось уста-
новить линейное увеличение каталитич. активности
с ростом числа подвижных протонов на поверхности
К. В реакциях, катализируемых кислотами Льюиса,
напр. полимеризации, активными К. могут быть
понные соединения металлов с большим отношением
квадрата заряда к радиусу е2/г или ионные соединения
переходных металлов. Ионы таких соединений обла-
дают высокой комплексообразующей способностью.
Напр., реакции полимеризации олефинов катализи-
руются галогенидами титана или ванадия, связан-
ными с алкилами алюминия (т. н. К. Циглера и Натта):
ТгС13 + А1(С2Н5)3, TiCl3 + Al (изо-С3Н,)3 и т. д.,
а также окислами хрома и молибдена; реакции поли-
меризации окисей олефинов ускоряются катализато-
рами ВеО, MgO, А12О3, соединениями железа. Общим
для этих К. является наличие ионов Al3+, Ti3+, Ве2+,
реэ+ и т. д., обладающих высоким отношением е2/г
и высокой комплексообразующей способностью.
Для окислительно-восстановительных каталитич.
реакций наиболее распространенными К. являются
переходные металлы: Pt, Pd, Ni, Fe, Co и их соедине-
ния: окислы (V2O5, МпО2, МоО3, Сг2О3), сульфиды
(MoS2, WS2, CoS), шпинели, а также полупроводники,
не имеющие в своем составе переходных элементов
(ZnO, CdO, ZnS). Для полупроводников часто наблю-
дается увеличение каталитич. активности с ростом
их электропроводности, к-рая в области примесной
проводимости возрастает с количеством введенной
примеси — промотора, а в области собственной про-
водимости — с уменьшением ширины запрещенной
зоны полупроводника. Для нек-рых реакций на полу-
проводниках можно наблюдать симбатность (согласо-
ванность) изменения каталитич. активности с изме-
нением работы выхода или с изменением интенсивности
окраски. Напр., в реакциях окисления углеводородов
и разложения нестойких кислородсодержащих соеди-
нений (N2O, Н2О2, КС1О3) каталитически активны
окрашенные окислы NiO, Со3О4, CdO и сульфиды
CdS, NiS и др., а бесцветные окислы А12О3, SiO2,
СаО мало активны. Это объясняется тем, что оба
свойства — каталитич. активность в окислительно-
восстановительных реакциях и окраска — обуслов-
лены наличием в твердом теле электронов, энергия
возбуждения к-рых сравнительно невысока (<: 3 эв).
К. для нек-рых реакций являются переходные ме-
таллы или их соединения, обладающие определенной
конфигурацией d-оболочки (напр., для синтеза NH3
наиболее активны металлы с электронной структу-
рой . de, для гидрирования и дегидрирования — da
и т. д.). Удобным критерием для подбора К. является
параметр кристаллич. решетки твердого тела и ее
структура не только потому, что для нек-рых реакций
сложных органич. соединений необходимо геометрич.
соответствие между адсорбирующимися молекулами
и К. (А. А. Баландин), но и потому, что геометрич.
структура решетки определяется электронным строе-
нием слагающих ее атомов или ионов.
Кроме активности и избиратель-
ности (см. Катализ)—свойств, определяемых химич.
составом, применяемые в пром-сти К. должны обла-
дать высокой поверхностью, достаточной механич.
прочностью, малой распыляемостью, малой спекае-
мостью, сопротивляемостью контактным ядам, в ряде
случаев — легкостью регенерации и т. д. Поэтому
за стадией подбора обычно следует разработка метода
приготовления катализатора в фор-
ме, удобной для практич. использования. Для обра-
зования максимальной поверхности К. готовят в ви-
де порошков, высокодисперсных тел или тел с высо-
копористой структурой. Окисные К. чаще всего полу-
чают осаждением гидроокисей из р-ров их солей с по-
следующим их прокаливанием, а также непосредствен-
ным термин, разложением карбонатов, оксалатов,
ацетатов и др. солей. Металлические К. чаще всего
готовят восстановлением их окислов водородом или
другими газами. «Скелетные» никелевые К., употреб-
ляемые для реакций гидрогенизации, получают сплав-
лением Ni с А1 с последующим выщелачиванием А1
и образованием высокопористой структуры.
Согласно теории пересыщения, по С. 3. Рогинскому,
чтобы получить активный К., необходимо вести про-
цесс его приготовления в условиях, максимально
удаленных от равновесных. Напр., для получения
активной NiO разлагают NiCO3 при непрерывном
удалении СО2, так что давление СО2 значительно
ниже равновесного. При темп-рах проведения ка-
тализа дисперсная структура К. обычно термодина-
мически неустойчива. В процессе работы часто про-
исходит рекристаллизация К., сопровождающаяся
их спеканием и уменьшением поверхности.
Для лучшего распределения и предотвращения
спекания К., а в случае дорогих К.’ (Pt, Pd) и для
экономии, их наносят на носители, или т р е-
г е р ы, — пористые или высокодисперсные вещества:
силикагель, активный уголь, асбест, пемзу и т. д.
Следует указать, что Pt и Pd, являющиеся наиболее
активными из известных гетерогенных К., помимо
высокой стоимости, обладают другими отрицатель-
ными качествами — легкой отравляемостью катали-
тич. ядами и трудностью регенерации. Поэтому в ряде
лабораторий ведутся поиски менее активных, но более
дешевых и устойчивых к ядам К. Напр., стремятся
заменить платиновые К. окисления аммиака окисно-
485
КАТАЛИЗАТОРЫ ПОЛИМЕРИЗАЦИИ — КАТАЛИТИЧЕСКИЕ ЯДЫ
488
CL R
ci-
свободные или адсорбированные на гетерогенной по-
верхности радикалы являются активными центрами
К. Ц.—Н. Согласно другой точке зрения, механизм
полимеризации на К. Ц.—Н. — координационно-ион-
ный. Активными центрами являются комплексы,
в состав к-рых входят атомы двух, как правило, раз-
ных металлов (напр., Ti и А1). Активация и стереоспе-
цифич. Присоединение молекул мономера осущест-
вляется в результате их взаимодействия с обоими
атомами металла в переходном комплексе:
С1
; >А1 +СН2=СН
сн2
СН-СН|
СНз
41
СК .R--—А1(
>Т1+С _ + ’>СН2 —►
СГ ^СН2—CHZ I
| сн-сн3
СНз |
C1\tiAai/C1
в технике
массы, которая
кобальтовыми. Иногда сырье до поступления на основ-
ной К. пропускают через т. н. форконтакт, на к-ром
в результате адсорбции или каталитич. реакции уда-
ляются вредные примеси, напр. содержащие серу или
кислород в синтезе аммиака. Целям предотвращения
спекания служат в нек-рых случаях структурооб-
разующие промоторы, напр. добавка А12О3 к желез-
ному К. синтеза аммиака. Для повышения механич.
прочности, предотвращения уноса реагирующими
в-вами и уменьшения гидравлич. сопротивления по-
току реагентов К. применяют в виде таб-
леток или гранул. Если, однако, при
этом каталитич. реакция осложнена яв-
лениями диффузии, применение больших
кусков К. может оказаться невыгодным
из-за уменьшения общего выхода проду-
кта и изменения избирательности дейст-
вия К. (сы. Катализ гетерогенный). Иногда
применяют К. в виде пылевидной
поддерживается во взвешенном слое движением па-
ров или газов реагента — т. н. флюид-процесс, или
катализ в псевдоожиженном слое (см. Псевдоожи-
жение).
Лит. см. при ст.: Катализ, Катализ гомогенный, Катализ
гетерогенный, Катализ кислотно-основной. О. В. Крылов.
КАТАЛИЗАТОРЫ ПОЛИМЕРИЗАЦИИ — см. По-
лимеризации катализаторы.
КАТАЛИЗАТОРЫ ЦИГЛЕРА — НАТТА — группа
гетерогенных или гомогенных катализаторов стерео-
специфич. полимеризации мономеров винилового и
дивинилового рядов. К. Ц.—Н. представляют собой
продукты взаимодействия галогенидов, оксигалоге-
нидов, ацетилацетонатов, алкоголятов, окисей и др.
соединений переходных металлов IV—VIII групп
периодич. системы (Ti, Zr, V, Сг, Мо, W, U, Со и др ),
наз. катализаторами, с металлалкилами, гид-
ридами, амидами и др. соединениями металлов I—
III группы периодич. системы, наз. сокатали-
заторами.
Для получения К. Ц.—Н. компоненты смешиваются
при комнатной или повышенной темп-ре в углеводород-
ных средах. Наиболее активные К. Ц.—Н. образуются,
если компоненты смешивают в присутствии мономера.
Однако для получения К. Ц.—Н., обладающих высо-
кой стереоспецифичностью при полимеризации а-оле-
финов, компоненты смешивают в отсутствие моно-
мера и каталитич. комплексу дают «созреть», нагре-
вая его нек-рое время перед введением в реакцию.
Соотношение компонентов и порядок их смешивания
оказывают существенное влияние на активность,
стереорегулирующее действие К. Ц.—Н., мол. веси
структуру образующихся полимеров. Напр., при
полимеризации этилена на К. Ц.—Н., получаемом из
TiCL и алкилалюмйния, уменьшение соотношения
Ti/Al в катализаторе приводит к заметному возра-
станию скорости полимеризации и мол. веса поли-
мера. Если при полимеризации изопрена молярное
отношение Ti/Al в К. Ц.—Н. равно 0,5—1, образуется
регулярйый 1,4-ТрЗнс-полиизопрен, а при отношении
Ti/Al, равном 2—3, получается регулярный 1,4-цис-
пол иизопрен.
Механизм полимеризации на К. Ц.—Н. не полностью
выяснен. При взаимодействии компонентов катализа-
тора, напр. галогенидов титана с алкилами алюминия,
имеет место сложный комплекс реакций, включаю-
щий восстановление и комплексообразование. Так,
восстановление TiCl4 идет через промежуточную ста-
дию образования алкилгалогенидов титана, к-рые
распадаются с образованием радикалов и треххло-
ристого титана, выпадающего в осадок: TiCl4 -|-
4- A1R3 ->- RTiCl3 + A1R2C1; RTiCl3 ->- R• -|- TiCl3.
Возможно п более глубокое восстановление соедине-
ний Ti. Предполагают, что образующиеся при этом
СН2
I
СН-СНз
I
сна
I
СН—СНз
Молекулы
каталитический комплекс ориентируют-
ся, что
ность присоединения. Если каталитические __
хемосорбированы на кристаллической поверхности
нерастворимых в реакционной среде соединений пере-
ходных металлов, то в этом случае в стереорегу-
лировании несомненную роль играет также ориен-
тирующее влияние гетерогенной поверхности, хотя
непосредственной связи между параметрами кристал-
лич. решетки катализатора и структурными па-
раметрами цепей образующегося полимера обычно
не наблюдается.
К. Ц.—Н. применяют для получения линейных и
стереорегулярных полимеров. При полимеризации
этилена катализаторами служат комбинации: TiCl4 -|-
-]-A1R3; TiCl4-|-A1R2C1 и др. При полимеризации
а-олефинов обычно образуются изотактические поли-
меры. Наиболее распространенным катализатором
для этой группы мономеров является комбинация
TiCl3 -]- A1R3. При использовании стереомодифика-
торов, напр. Р(С6Н5)3, удается также получить син-
диотактические полимеры а-олефинов. При полиме-
ризации диенов на К. Ц.—Н. можно получать регуляр-
ные полимеры различного строения. Напр., при по-
лимеризации бутадиена на катализаторе A1R3 -|- Т1С14
образуется 1,4-цис-полибутадиен,- на катализаторе
LiAlH4 -|- TiJ4—1,4-транс-полибутадиен и 1,2-по-
либутадиен на катализаторе LiAlH4-|- TiCl4. К. Ц.—Н.
можно использовать и для сополимеризации, напр.,
этилена с пропиленом. Для полимеризации полярных
мономеров на К. Ц.—Н. необходимо, помимо катализа-
тора и сокаталиЗатора, вводить в реакционную си-
стему комплексообразователь, напр. эфир или амин,
или использовать соединения переходных метал-
лов с менее выраженной склонностью к комплексо-
образованию, напр. МпС12. Таким путем удается
получать стереорегулярные полимеры винилхлори-
да, метилметакрилата и других полярных моно-
меров.
Из полимеров, получаемых наК.Ц.—Н., промыш-
ленное значение приобрели полиэтилен, изотактич.
полипропилен, а также 1,4-цис-полибутадиен и 1,4-
цис-полиизопрен, не уступающие по свойствам на-
туральному каучуку.
К. Ц.—Н. названы по имени нем. химика К. Циглера, от-
крывшего возможность получения полиэтилена в присутствии
комбинаций из перечисленных выше веществ, и итал. химика
Дж. Натта, к-рый вскоре после открытия Циглера, исполь-
зуя соединения переходных металлов в низших валентных
состояниях в сочетании с алкилалюминием. Впервые осуще-
ствил стереоспецифическую полимеризацию замещенных
а-олефинов.
Лит.: Гейлорд Н., М а р к Г., Линейные и стереоре-
гулярные полимеры, пер. с англ., М., 1962; Натта Дж.,веб.з
Химия и технология полимеров, 1960, № 7—8. В. П. Зубов.
КАТАЛИТИЧЕСКИЕ ЯДЫ (контактные яды) —
вещества, воздействие к-рых на катализаторы (обычно
мономера при вхождении в
юскии комплекс ориентиру:
обеспечивает стереоспецифич-
комплексы
487
КАТАЛИТИЧЕСКИЕ ЯДЫ — КАТЕПСИНЫ
488
на твердые катализаторы, или контакты) приводит
к «отравлению» катализатора, т. е. к снижению его
каталитич. активности или к полному прекращению
каталитич. действия. Причина отравления лежит в ад-
сорбции К. я. на активных центрах катализатора.
При этом встречаются разные виды зависимости ско-
рости реакции от количества введенного К. я. Если
молекула К. я. адсорбируется на активном центре,
не воздействуя на соседние активные центры, она,
т. обр., просто препятствует доступу реагирующих
молекул к этому активному центру. Следовательно,
при таком отравлении блокировкой чаще всего наблю-
дается линейная зависимость каталитич. активности
от количества введенного контактного яда. Примером
служит действие азотистых оснований, напр. хино-
лина, на алюмосиликатные катализаторы крекинга:
каждая молекула хинолина адсорбируется на одном
активном центре алюмосиликата, вероятно, на его
гидроксильных группах, имеющих подвижный про-
тон, вытесняет реагирующее вещество — углеводо-
род и не влияет на соседние активные центры. В дру-
гих случаях действие К. я. может характеризоваться
не линейной, а, напр., гиперболич. зависимостью,
т. е. первые порции К. я. сильно отравляют поверх-
ность катализатора, а при введении следующих пор-
ций К. я. каталитич. активность уменьшается мед-
ленно. Напр., в реакции разложения Н2О2 на Pt
адсорбция только одной молекулы HgCJ2 на 3000 по-
верхностных атомов Pt сразу же снижает каталитич.
активность Pt в 7 раз. Такие эффекты наблюдаются на
металлич. и полупроводниковых катализаторах (см.
Катализ гетерогенный'). Одно из возможных их объяс-
нений заключается в изменении работы выхода <р элек-
трона из катализатора. Согласно электронной
теории катализа, это изменение Д<р может повы-
сить энергию активации каталитической реакции и,
т. о., понизить ее скорость. Встречаются случаи,
когда К. я. в очень малых количествах, наоборот,
активируют катализатор. Напр., анион AsO|_ в ма-
лых количествах активирует платину по отношению
к гидрированию коричной к-ты, а в больших количе-
ствах отравляет ее. Этот вид действия малых ко-
личеств примеси на активность катализатора наз.
модифицированием. В случае сложных
каталитич. реакций К. я. могут действовать изби-
рательно, т. е. подавлять одну из реакций и пе
влиять на другую реакцию на том же катализаторе.
Напр., при каталитич. окислении С2Н4 на Ag добавки
соединений галогенов (HCI, CH3J и др.) отравляют
реакцию глубокого окисления С2Н4 до СО2 и Н2О и
не влияют (или мало влияют) па выход продукта
мягкого окисления — окиси этилена С2Н4О. На этом
основан метод повышения избирательности се-
ребряного катализатора в процессе получения окиси
этилена.
Отравление может быть обратимым и не-
обратимым. В реакции синтеза аммиака на
железном катализаторе кислород и его соединения
(Н2О, СО и др.) отравляют Fe обратимо; в этом слу-
чае воздействие свежей, тщательно очищенной смеси
N2 -|- Н2 вытесняет К. я. с активных центров и сни-
мает отравление. Соединения серы (H2S, CS2) отрав-
ляют Fe необратимо, т. е. действием свежей смеси не
удается восстановить активность катализатора. Для
предотвращения отравления реагирующую смесь,
поступающую на катализатор, подвергают тщатель-
ной очистке. Иногда применяют т. н. ф о р к о н-
такты, — в этом случае К. я. претерпевают на
катализаторе химич. изменения, способствующие пре-
вращению их в неактивную форму. В реакциях орга-
нич. соединений (крекинга, дегидрирования, изоме-
ризации) отравление катализаторов часто происходит
в результате образования полимерной пленки (т. н.
«кокса»), покрывающей поверхность. Для удаления ее
цикл катализа сменяется циклом регенерации, при
к-рой катализатор продувают воздухом, водяным па-
ром, реже водородом, для перевода «кокса» в СО2
или в другие летучие соединения. Напр., при крекинге
углеводородов «кокс» с поверхности алюмосиликата
выжигают продувкой воздуха. Для регенерации
катализаторов, необратимо отравленных К. я., напр.
Pt, отравленной соединениями S или As, приходится
извлекать катализаторы из реактора и промывать их
растворителями, удаляющими К. я.
В нек-рых случаях одни и те же вещества могут
быть К. я. по отношению к одним реакциям и катали-
заторам и не отравлять другие. Напр., кислород от-
равляет железные катализаторы синтеза аммиака, но
активирует платиновые катализаторы окисления во-
дорода. К числу наиболее распространенных К. я.
для металлич. катализаторов относятся соединения,
являющиеся донорами пары электронов; обычно,
это вещества, содержащие в своем составе кислород
(Н2О, СО, СО2), серу (H2S, CS2, CjHjSH и др.), Se,
Те, N, Р, As, Sb, а также непредельные соединения
(С2Н4, С2Н2)иионы металлов: Cu2+, Sn2+, Hg2+, Fe2+,
Со2+, Ni2+. На полупроводники могут действовать те
же К. я., однако полупроводники менее склонны к от-
равлению. Поэтому на практике активные металлич.
катализаторы заменяют часто менее активными, но
более стойкими к К. я. полупроводниками. Напр.,
в реакциях гидрирования и дегидрирования приме-
няют сульфиды: NiS, WS2, CoS, к-рые не отравляются
сернистыми примесями. Кислотные катализаторы
обычно отравляются примесями оснований, а основ-
ные катализаторы — примесями кислот.
Лит. СМ. при ст. Катализ, Катализ гетерогенный.
О. В. Крылов.
КАТЕПСИНЫ — высокомолекулярные соединения
белковой природы, тканевые внутриклеточные про-
теолитич. ферменты, катализирующие гидролиз пеп-
тидной связи в пептидах и белках. К. широко рас-
пространены в животных и растительных тканях и
в микроорганизмах. К. классифицируются по харак-
теру и специфичности их ферментативного действия.
К. I, II и V (А, В и С) являются эндопептида-
зами, т. к. они способны гидролизовать внутренние
пептидные связи в молекулах белков и пептидов.
К. III и IV по характеру ферментативного действия
относятся к экзопептидазам, т. к. они дей-
ствуют только на соединения, содержащие одну или
несколько свободных концевых полярных групп,
таких, как а-карбоксил или а-аминогруппа, и гидро-
лизуют пептидные связи, образованные концевыми
аминокислотами.
К. I по специфичности действия сходен с пепсином.
Его специфич. синтетич. субстратом является карбо-
бензокси-Ь-глутамил-Ь-тирозип. Амидирование а-кар-
боксильной группы в синтетич. субстратах тормозит
К. I так же, как и наличие свободной а-аминогруппы
вблизи от гидролизуемой связи. Оптимум действия
К. I при pH 5,6. В отличие от других К., К. I не акти-
визируется соединениями, содержащими сульф-
гидрильные группы и не угнетается тиоловыми ядами.
Специфичность К. II близка к специфичности трип-
сина; К. II наиболее быстро гидролизует пептидные
связи, включающие а-карбоксильные группы арги-
нина и лизина. К. V, подобно химотрипсину, наиболее
быстро расщепляет пептидные связи, включающие
карбоксил ароматич. аминокислот. К. III обладает
специфичностью панкреатич. карбоксипептидазы.
К. IV является аминопептидазой и вызывает последо-
вательное отщепление аминокислотных остатков с
аминного конца полипептидной цепи. Все К., кроме
К. I, активируются соединениями, содержащими
сульфгидрильные группы (цистеин, глютатион, H2S
489
КАТЕХИНЫ — КАТИОНОТРОПИЯ
490
и др.)- Нанек-рые К. активирующее действие оказывают
такие восстановители, как аскорбиновая к-та и HCN.
Тиоловые яды (иодацетат, иодацетамид, п-хлорморку-
рийбензойная к-та и др.) ингибируют действие К.
Помимо реакций гидролиза, К. способны катализи-
ровать реакции транспептидации и переамидирова-
ния, ведущие к удлинению пептидной цепи и имею-
щие большое значение в процессах биосинтеза белков.
Лит.: Methods in enzymology, ed. S. P. Colowick, N. O. Kap-
lan, v. 2, N. Y., 1955, p. 64; The enzymes, ed. J. B. Summer,
K. Myrback, v. 1, N. Y., 1951, p. 875. См. также лит. к ст. Фер-
менты. О. В. Казакова.
КАТЕХИНЫ— природные вещества группы флавана.
.11 IV
При: R = R' = Н — катехин и эпикатехин (I) (3,5,7,
3’, 4'-пентаоксифлаваны); R — ОН; R’ = Н — галлокатехин
и эпигаллокатехин (3, 5, 7, 3', 4’, 5'-гексаоксифлаваны);
R = Н; В'=галлоил (II) — катехингаллат и эпикатехингал-
лат; R = ОН; R'-галлоил (II) — галлокатехингаллат и эпи-
галлокатехингаллат.
Молекула К. содержит 2 асимметрия, атома угле-
рода; для каждого К. известны 4 оптически активных
изомера и две рацемич. смеси. В последнее время
установлена абсолютная конфигурация (+ )-катехипа
(III) и (+ )-эпикатехина (IV). К. — бесцветные кри-
сталлы с вяжущим вкусом (катехин и эпикатехин
почти безвкусны), хорошо растворимы в воде, мета-
ноле, этаноле, уксусноэтиловом эфире, хуже — во
влажном серном эфире, нерастворимы в петролейном
эфире, С6Н6, СНС13, СН2С12 и СС14; легко окисляются
на солнечном свету и при нагревании. При действии
кислот К. превращаются в нерастворимые продукты
полимеризации, т. и. дубильные красные (флобафены),
со щелочами образуют меланиноподобные темноокра-
шенные продукты. При осторожном сплавлении с твер-
дым КОН в атмосфере N2 К. расщепляются с образо-
ванием флороглюцина, галловой и протокатеховой
кислот. При нагревании (+)-катехина с водой при
125° он окисляется; при этом происходит его эпиме-
ризация с образованием смеси (±)-катехина, (^-эпи-
катехина и (4-)-эпикатехина. Аналогично ведет себя
(—)-эпикатехин. При действии на водные или водно-
спиртовые р-ры К. основным и средним уксуснокис-
лым свинцом они осаждаются, при действии желатины
К. осаждаются неполностью; частично К. связываются
белками. В отличие от большинства прочих природ-
ных фенольных соединений, гликозидные формы К.
в растениях не найдены.
К. окрашиваются: О,1о/о-ным этанольным р-ром
FeCl3 — катехин и эпикатехин в зеленый цвет,
остальные — в синий; I.O’/o-hhm р-ром ванилипа
в конц. НС1 — в красный или малиновый цвет;
р-ром KCN, подкисленным разб. H2SO4,— в оранже-
вый цвет; диазотированными n-нитроанилином или
сульфаниловой к-той — в оранжево-красный цвет.
При смешении ацетонового р-ра К. с равным объемом
1%-ной суспензии надсернокислого калия в конц.
H2SO4 образуется соответствующий антоцианидин
с красно-фиолетовой окраской. Количественное опре-
деление К. основано на их предварительном разде-
лении при помощи хроматографии на бумаге [наиболее
употребительные растворители: смесь н-бутанол —
СН3СООН — Н2О (40 : 12 : 28)и2%-ная СН3СООН],
извлечении с хроматограмм этанолом и последующем
спектрофотометрировании одним из принятых для
фенольных соединений методов (чаще всего исполь-
зуют реакцию с ванилином). Посредством распредели-
тельной хроматографии на колонках крупнопори-
стого силикагеля К. могут быть разделены и количе-
ственно извлечены влажным серным эфиром или
смесью уксусноэтиловый эфир — СС14 (2 : 1).
К. широко распространены в растениях (за исклю-
чением галлоильных производных, к-рые встречаются
значительно реже). Особенно большие количества К.
содержатся в чайном растении (до 20—25%), в вино-
градной лозе (гл. обр. в косточках и кожице) и бобах
какао. К. получают из растительного сырья, гл.
обр. из чайного растения экстракцией уксусно-этило-
вым эфиром, осаждением хлороформом и последую-
щим хроматографированием на силикагеле. Синтез К.
в свободном виде до сих пор не осуществлен вследст-
вие их нестойкости, однако восстановлением соответ-
ствующего флавонола (пентаметилкверцетина) или
антоцианидина (пентаметилцианидина) можно полу-
чить пентаметиловый эфир (±)-эпикатехина.
К. —: биологически высокоактивные вещества; они
регулируют проницаемость мельчайших кровеносных
сосудов (капилляров) и увеличивают упругость их
стенок, а также способствуют более эффективному
использованию организмом аскорбиновой к-ты. По-
этому К. относят к веществам, обладающим Р-вита-
минной активностью, и используют для лечения разно-
образных заболеваний, связанных с нарушениями
функций капилляров.Окислительные превращения К.
играют важную роль в технологии ряда пищевкусовых
произ-в [ферментация чая, виноделие, обжаривание
какао], поскольку продукты окисления обладают
специфич. вкусом и окраской.
Лит.: Биохимические методы анализа растений, пер.
[с нем.), М., 1960; 3 апр ометов М. Н., Успехи соврем,
биол., 1958, 45, вып. 2, 203; его же, в сб.: Хроматография,
ее теория и применение, М., 1960; его же, Биохимия,
1952, 17, вып. 1, 97; Гетероциклические соединения, пер.
сангл.,т. 2, М., 1954; Freundenberg К.,в кн.: Fortschritte
der Chemie organischer Naturslolfe, Bd 16, W., 1958.
M. H. Запрометов.
КАТИОНИТЫ — см. Иониты.
КАТИОНОТРОПИЯ (катионотропные превраще-
ния) — молекулярные перегруппировки, сопровож-
дающиеся миграцией положительно заряженного ато-
ма или группы атомов:
R R
С^С=^ [с-с-с] + R’=J=ir cic-c
Катионотропные превращения облегчаются в том
случае, если промежуточно образующиеся катион и
анион достаточно устойчивы; способность соединения
к катионотропным превращениям увеличивается с ро-
стом ионизирующей способности растворителя. Этот
механизм часто имеет место в случае прототро-
пии — наиболее важного и изученного вида катио-
нотропии. В малополярных растворителях механизм
прототропных превращений может быть иной, вну-
тримолекулярный: перемещение радикала (в виде
положительно заряженной частицы) происходит без
предварительной диссоциации:
R^* R. р
С-С^с с^с^с
По этому механизму, напр., протекает перегруппи-
ровка аллиловых эфиров фенолов (см. Клайаена пере-
группировка).
Лит.: Ин гольд К. К., Механизм реакций и строение
органических соединений, пер. с англ., М., 1959; Реу-
тов О. А., Теоретические проблемы органической химии,
М., 1956. О. А. Реутов.
491
КАТИОНЫ — КАУЧУК НАТУРАЛЬНЫЙ
492
КАТИОНЫ — см. Ионы.
КАУСТИЧЕСКАЯ СОДА — техническое название
натрия гидроокиси (едкого натра).
КАУЧУК НАТУРАЛЬНЫЙ (НК) — высокоэла-
стичный материал растительного происхождения, при-
меняемый гл. обр. для изготовления резины и резино-
вых изделий. НК содержится в млечном соке каучу-
коносных растений или в виде отдельных включений
в клетках их коры и листьев. Товарный НК добы-
вается почти исключительно из млечного сока бра-
зильской гевеи.
Каучук стал известен в Европе после того, как Ш. де Кон-
дамин в 1738 представил в Парижскую академию наук образцы
НК, изделия из него и описания способов добычи НК из бра-
зильской гевеи в странах Южной Америки. Промышленное при-
менение НК стало возможно после нахождения доступных
растворителей (Макинтош, 1823) и в особенности после откры-
тия процессов пластикации и вулканизации (Гэнкок, 1843,
Гудьир, 1839). Состав и генетич. связь НК с изопреном уста-
новлены Вильямсом (1860) и Бушарда (1879). Строение НК
исследовано в работах Гарриеса, Штаудингера и др. Штау-
дингером разработаны представления о НК как линейном
высокомолекулярном полимере изопрена. Обширные иссле-
дования вулканизации каучука принадлежат Веберу, Остро-
мысленскому, Бызову, Фармеру, Догадкину и др. Исследо-
ванию физич. свойств и разработке теории эластичности посвя-
щены работы Гута, Уолла, Кобеко, Александрова, Трелоара
и др.
Млечный сок, наз. латексом (см. Латекс натураль-
ный), добывается подсочкой деревьев, достигших пя-
тилетнего возраста. Каучук в латексе находится в виде
взвешенных в воде шарообразных или грушевидных
частиц — глобул. Для получения НК млечный сок
на месте добычи подвергают желатинированию (или
коагуляции), добавляя муравьиную или уксусную
к-ту. Образующийся рыхлый сгусток (гель) промывают
водой и прокатывают на вальцах в листы, к-рые сушат
и обычно коптят в камерах, наполняемых дымом. Коп-
чение придает НК устойчивость против окисления и
предотвращает развитие в них бактерий. Готовые
листы НК более или менее прозрачны и имеют янтар-
ный цвет; такой НК наз. смокед ш и т. Менее
распространен т. наз. светлый креп. При его
получении к млечному соку перед коагулированием
для отбелки прибавляют бисульфит натрия; промы-
тые и отвальцованные листы сушат на открытом воз-
духе или в камерах. Листы этого сорта НК не про-
зрачны и имеют кремовый оттенок. Состав обоих видов
товарного НК почти одинаков, они содержат (в %):
93—94 собственно НК; 2,4—2,9 белков; 0,3 сахаров;
2,5—3 смол; 0,3 золы. Помимо этих основных видов,
применяются другие, менее ценные сорта НК, напр.
пара-каучук, добываемый кустарным способом из
дикорастущей гевеи. Выпускаются также каучуки
специального назначения, напр. освобожденный от
белков путем ферментативной обработки.
В химич. отношении чистый НК представляет собой
высокомолекулярный непредельный углеводород со-
става (С5Н8)П и рассматривается как полимер изо-
прена, хотя действительный генезис его в растении
еще не выяснен. Основной структурной группировкой
макромолекулы каучука является изопентеновая (изо-
СН3
I
преновая) группа —СН2—С=СН—СН2— (в цис-
изомерной форме). Более тысячи таких групп, соеди-
ненных последовательно между собой в положении
4—1, образуют длинную нитевидную гибкую макро-
молекулу каучука. НК представляет собой смесь
макромолекул разной длины. Средний мол. в. НК от
150 тыс. до 500 тыс., что соответствует длине
10 000—40 000 „А, при поперечном сечении макромо-
лекулы ок. 3 А. Эти особенности макромолекул НК
определяют его физич. и механич. свойства, в част-
ности самое ценное технич. свойство — высокую эла-
стичность (см. Гибкость цепных молекул, Эластичность
полимеров, Эластомеры). НК растворяется в жир-
ных и ароматич. углеводородах и их производных,
напр. в бензине, бензоле, хлороформе, сероуглероде
и т. д., образуя вязкие растворы, к-рые применяют в
качестве клея. Перед растворением НК набухает,
увеличиваясь в объеме до 1000%. В воде, спирте,
ацетоне, жирных к-тах и др. ассоциированных
жидкостях НК практически не набухает и не рас-
творяется.
Будучи ненасыщенным соединением, он взаимодей-
ствует с галогенами, причем наряду с их присоедине-
нием по месту двойных связей происходит и замеще-
ние водорода атомами галогенов. Продукт взаимо-
действия НК с хлором имеет состав (С5НвС14)п. Этот
хлоркаучук применяют в произ-ве огнестойких лаков,
а также клеев для приклеивания резины к металлу.
При каталитич. действии водорода (катализатор —
платиновая чернь) образуется насыщенный гидро-
каучук состава (C5Hi0)n, употребляемый как замени-
тель гуттаперчи и в качестве присадки к смазочным
маслам. Газообразный хлористый водород с НК обра-
зует каучука гидрохлорид состава (С5Н9С1)П, исполь-
зуемый в качестве пластмассы и материала для изго-
товления прочной упаковки для пищевых продуктов.
Галогенопроизводные могут в дальнейшем превра-
щаться в еще более сложные производные НК. Так,
дибромид (CjH8Br2)n конденсацией с фенолом при
80—160° в присутствии хлористого алюминия обра-
зует оксифснилкаучук, реагирующий с солями диазо-
ния, причем сочетание происходит в орто-положении
к гидроксильной группе:
СН3
-сн2-с — CH-CH2J
c6h5-n=n-C^J1 C^-n=n-c6h5
он он
Подобно фенолу, с бромидом НК реагирует анилин.
Образующийся диаминодифенилкаучук может далее
диазотироваться, а диазониевые производные — соче-
таться с фенолами и аминами. Получаемые вещества
растворимы в полярных растворителях и окрашены
в различные цвета. Взаимодействие НК с галогенами
почти всегда сопровождается циклизацией — образо-
ванием внутримолекулярных кольцевых структур.
Особенно интенсивно циклизация идет под действием
водородных к-т (серная, хлорсульфоновая). Проме-
жуточной стадией реакции является образование
карбониевого иона:
Сн3 СНз
~сн2-с=сн-сн2-сн2-с=сн-сн2~ + н+ —»-
^СН2—СН2 ZCH2-CH2
СН2 ^С-СН3 СН2 ‘'с-СНз
—»-~сн2-с+ сн-сн2~—- ~сн2-с—сн-сн2~——
СН3 СНз
ZCH2-CH2
сн2 /-сн3
—~сн2-с—с-сн2~ + н*
сн3
В пределе до 80% НК может переходить в циклич.
форму. Циклокаучук является превосходным свя-
зующим.
НК взаимодействует со многими соединениями,
способными реагировать с двойными связями (малеи-
новый ангидрид, тиокислоты, тиоспирты, меркаптаны,
сероводород и т. д.). Во многих случаях эти реакции
имеют радикальный характер и инициируются пере-
кисями и др. источниками свободных радикалов. При
493
КАУЧУК НАТУРАЛЬНЫЙ
494
этом имеет место не только присоединение реагирую-
щего вещества к двойным связям НК, но и отрыв
водорода от а-метиленовой группы макромолекуляр-
ной цепи каучука. Такой процесс наблюдается, напр.,
при взаимодействии НК с малеиновым ангидридом
в присутствии перекиси бензоила:
СНд
свн5соо + ~сна-с-сн-сн2------
СНз
-С6Н5СООН ,~СН-С-СН-СНг.
СН=СН
I I
со со
'чо/
СНз СН3
---•“ -СН-С=СН-СН2- + ~CHa-C = CH-CH.i-•“
СН Си
I I
со со
чо/
СНз СНз
—*-~сн-с=сн-снй~ + ~сн-с=сн3-сн2~
сн-сн
I I
со со
хо/
Тиокарбоновые к-ты (тиобензойная, бис-тиоадипи-
новая) присоединяются к НК при вальцевании при
комнатной темп-ре. Эти реакции используются для
модифицирования НК, напр. для понижения темп-ры
кристаллизации. Инициирование нек-рых реакций НК
может происходить в результате механич. воздействия
при низких (комнатная и ниже) темп-рах, вызываю-
щего образование свободных полимерных радикалов
вследствие разрыва макромолекулярных цепочек.
Образующиеся свободные макрорадикалы активно
взаимодействуют друг с другом и с низкомолекуляр-
ными соединениями. Так, присоединение к НК малеи-
нового ангидрида происходит и без участия перекиси,
если НК вальцевать с малеиновым ангидридом при
темп-ре не выше 4-10°.
Под воздействием ряда веществ, напр. TiCl4, про-
исходит изомеризация НК — частичное превращение
из цис- в транс-форму.
Практически все реакции НК сопровождаются
структурными изменениями: разрывом макромоле-
кулярных цепей и соединением («сшиванием») их
в сложные сетчатые системы, что приводит к существен-
ному изменению физич. и механич. свойств каучука —
растворимости, прочности, эластичности и т. д. Струк-
турные изменения имеют место и при взаимодействии
НК с кислородом воздуха и др. окисляющими аген-
тами. Уже при комнатной темп-ре кислород присоеди-
няется к НК, вызывая окислительную деструкцию. Это
взаимодействие лежит в основе т. н. старения
каучука и резины, вызывающего изменение
свойств резиновых изделий при их хранении и эксплуа-
тации (уменьшение прочности и эластичности, появле-
ние липкости, хрупкости и т. п.). Соли металлов
с переменной валентностью (железо, марганец), а
также нек-рые органич. соединения (альдегиды, мер-
каптаны) ускоряют окисление; аминосоединения,
спирты и фенолы — задерживают. Последние при-
меняются в качестве противостарителей резины. При
взаимодействии с озоном НК превращается в озонид
(С5Н8О8)П — неустойчивое соединение, распадающееся
с образованием левулиновой кислоты и левулинового
альдегида. Взаимодействие НК с озоном, содержа-
щимся в воздухе, составляет одну из причин появле-
ния трещин на поверхности резиновых изделий при
их хранении и эксплуатации. При нагревании выше
200° НК разлагается с образованием различных низ-
комолекулярных углеводородов, среди к-рых всегда
находится изопрен. Облучение светом с длиной волны
короче 4000 А вызывает деструкцию макромолекул
НК и выделение газообразного водорода. Этот про-
цесс также имеет место при старении резиновых изде-
лий на свету.
Огромное практич. значение имеет взаимодействие
НК с серой, хлористой серой, органич. перекисями
и другими веществами, вызывающими вулканизацию.
Вулканизация приводит к образованию сетчатых
структур, в к-рых длинные макромолекулы каучука
соединены («сшиты») между собой атомами серы или
другого вулканизующего агента. Технически наиболее
ценным свойством НК и особенно его вулканизатов
является высокая эластичность. Мягкие вулканизаты
(резины) из НК способны при комнатной темп-ре
обратимо растягиваться более чем на 1000% и имеют
при этом сопротивление разрыву до 350 кг/см? (исход-
ного сечения). В отличие от кристаллич. тел, дефор-
мация НК в пределах 100—200% растяжения не
сопровождается изменением объема, а следовательно,
и изменением внутренней энергии. В основном эла-
стичность НК сопровождается уменьшением энтропии
при растяжении и увеличением ее при обратном со-
кращении. Поскольку высокая эластичность НК свя-
зана с тепловым движением его гибких макромоле-
кул, она может проявляться в той области темп-р,
в к-рой это движение достаточно интенсивно. При
темп-ре ок. —70° НК утрачивает эластичность даже
при очень медленных воздействиях и становится
хрупким; выше 80—100° НК пластичен, т. к. при этой
темп-ре возникает возможность перемещения отдель-
ных нитевидных макромолекул относительно друг
друга. Величина деформации НК зависит не только
от величины механич. напряжения, но и от длитель-
ности его действия (см. Механические свойства поли-
меров). При коротком действии силы участки макро-
молекул НК не успевают перегруппировываться, и
высокая эластичность не проявляется; каучук ведет
себя при этом как твердое тело. Чем выше темп-ра,
тем короче период релаксации, необходимый для уста-
новления равновесия между силой и деформацией.
При комнатной темп-ре высокая эластичность НК
проявляется, если продолжительность действия силы
(в одном направлении) не менее одной стотысячной
доли секунды.
Растяжение НК, как и всех эластомеров, сопрово-
ждается выделением тепла, обратный процесс — его
поглощением. Тепловой эффект возрастает с увели-
чением деформации, и при 700% удлинения состав-
ляет около 6 кал[г. Основная часть выделяющегося
тепла соответствует тепловому эффекту перехода НК
при растяжении в кристаллич. состояние. Необрати-
мая часть теплового эффекта при растяжении, обус-
ловленная механич. гистерезисом, является причиной
разогрева резиновых изделий при их эксплуатации
в условиях многократных деформаций и сильно
влияет на их прочность и износ. Так, темп-ра массив-
ных резиновых шин при больших скоростях автомо-
биля может достигнуть 100—200°.
Обычно НК находится в аморфном состоянии.
Однако при длительном хранении возможна кристал-
лизация (см. Кристаллическое состояние полимеров).
С наибольшей скоростью кристаллизация происходит
приблизительно при —25°, но и в этом случае кри-
сталлизуется не больше 40% всей массы НК; темп-ра
плавления кристаллов ок. 11°, но заметно зависит от
условий кристаллизации. Растяжение НК также вы-
зывает кристаллизацию. Глубина кристаллизации
возрастает с увеличением деформации растяжения,
достигая в пределе 50% при удлинении в 700%.
495
КАУЧУК СИНТЕТИЧЕСКИЙ
496
Это явление обратимо. Возникновение кристаллич.
фазы при растяжении каучука существенно увели-
чивает прочность НК и его вулканизатов.
В технич. отношении большой интерес представ-
ляют электрич. свойства НК. Диэлектрич. проницае-
мость его (и ненаполненных вулканизатов) составляет
ок. 2,5. В качестве электроизоляционного материала
применяют мягкие вулканизаты, а также эбонит.
Широко используется также газо- и водонепроницае-
мость НК. Чистый каучук практически для воды
непроницаем, коэфф, диффузии , паров воды через
пленку НК при 20° составляет 8- 10~s г/час. Коэфф,
диффузии воздуха 1,21 • 10 е г/час. Основные физич.
константы НК приведены в табл. 1.
Константы и единицы измерения Чистый каучук Технич. каучук Мягкий вулкани- зат с 2% серы Эбонит
Плотность, г/см3 Теплопроводность, 'кал-сек~х,см~1град~1 . . . Теплоемкость Ср кал-г-'-град-1 Сжимаемость, бар 1 • Диэлектрич. проницаемость (при частоте 1000 гц) .................... tg угла диэлектрич. потерь (при частоте 1000 гц) Электропроводность, ом-'-смг1 0,906 0,449 53,7 • 10-е ' 2,37 1,6 • Ю-з 23 • 10-13 0,911 32 • 10-5 2,45 1.8 • Ю-з 420 • 10-18 0,923 34 • 10 5 0,510 51,0 • 10-е 2,68 1,8 • Ю-з 13 • 10-1S 1,173 39 • 10—5 0,341 24,3 -10-е 2,82 5,1 • Ю-з 15 • 10-18
Высокая эластичность НК, водо- и газонепроницае-
мость, высокие электроизоляционные свойства, устой-
чивость против многих агрессивных сред обусловли-
вает его чрезвычайно широкое применение во всех
областях техники и быта. Главная масса НК перера-
батывается в резину (вулканизат). В сыром виде
используется не более 1% добываемого НК (резино-
вый клей, креповая подошва). Добывается и перера-
батывается в год более 2 000 000 т НК. Свыше 60%
НК используется для изготовления автомобильных
шин. Данные о росте мирового произ-ва НК в сравне-
нии с синтетич. каучуком (СК) см. в табл. 2.
Таблица 2
Годы НК, m СК, т | Годы НК, т СК, т
1939 1 105 000 15 000 1954 1 765 000 740 000
1941 1 240 000 72 500 1956 1 912 000 1 230 000
1945 262 500 865 000 1958 1 957 000 1 242 000
1948 1951 1 422 500 1 500 000 480 000 812 000 1960 2 000 100 1 892 000
Основная масса НК добывается в Индонезии, Ма-
лайской Федерации, на Цейлоне, во Вьетнаме и др.,
где имеются обширные плантации бразильской гевеи.
Лит.: Догадкин Б. А., Химия и физика каучука,
М.—Л., 1947; Бызов Б. В., Природный каучук, Л., 1932.
Б. А. Догадкин.
КАУЧУК СИНТЕТИЧЕСКИЙ (СК) — высокопо-
лимерные материалы — эластомеры, предназначенные
для изготовления резины. К. с. обычно получают
полимеризацией и сополимеризацией различных не-
предельных соединений; для получения нек-рых К. с.
применяется также поликонденсация соответствующих
бифункциональных производных углеводородов.
Решающую роль в разработке методов синтеза СК сыграли
исследования русских химиков: С. В. Лебедева, А. Е. Фавор-
ского, И. Л. Кондакова и др. На основе работ Лебедева
в 1931—32 в СССР было впервые начато широкое промышлен-
ное произ-во СК из этилового спирта (натрий-бутадиеновый
СК). Важные исследования по синтезу каучуков были выпол-
нены Г. Бушарда, Г. Ньюландом, У. Карозерсом и др. зару-
бежными химиками.
Исходные мономеры, на основе полимеризации или
поликонденсации к-рых образуются СК, часто назы-
вают каучукогенами. В Качестве каучукоге-
нов используются бутадиен, стирол, изопрен, хлоро-
прен, изобутилен, нитрил акриловой к-ты, а-метил-
стирол и др. Полимеризацию каучукогенов произво-
дят в массе мономера (или смеси мономеров) либо
в эмульсии. В последнем случае СК получается сна-
чала в виде латекса синтетического и выделяется из
него Коагуляцией. Подобно каучуку натуральному,
СК имеют длинные макромолекулярные цепи, иногда
разветвленные, со средним мол. весом, равным сот-
ням тысяч и даже миллионам. Макромолекулярные
цепи построены из повторяющихся группировок, соот-
ветствующих тому мономеру, из к-рого получен СК.
Полимерные цепи СК в большинстве случаев имеют
двойные связи, благодаря к-рым при вулканизации
образуется пространственная сетка,
та лица 1 обеспечивающая характерные для
резин физико-механич. свойства.
Нек-рые виды СК (напр., поли-
изобутилен, силиконовый каучук
и др.)—полностью предельные со-
единения, и потому вулканизацию
их проводят отличными от обыч-
ной серной вулканизации приема-
ми (применение оргаяич. перекисей,
аминов и др ).
Первоначально вырабатывался
преимущественно только один вид
СК — натрийбутадиеновый каучук;
в настоящее время выпускается
около десяти видов и большое чис-
ло разновидностей СК. Первоначально этиловый
спирт был почти единственным источником для
произ-ва СК. В дальнейшем главным и наиболее
экономически выгодным видом сырья сталв попутные
нефтяные газы и газы нефтепереработки. На этом
сырье развивается современное произ-во СК в США,
СССР и др. странах. Легкие углеводороды попутных
нефтяных газов и газов крекинга (н-бутан, изобутан,
пентаны и др.) перерабатываются каталитич. дегидри-
рованием в бутадиен и изопрен. Бутадиен произво-
дится на основе нефтяных газов как через синтетич.
этиловый спирт, получаемый из этилена, так и непо-
средственно дегидрированием н-бутана. В сырьевом
балансе произ-ва СК существенное место занимает
также этиловый спирт, получаемый гидролизом дре-
весины. В Германии было организовано произ-во
бутадиена из каменного угля (карбид кальция —
ацетилен — ацетальдегид — ацетальдоль — бутилен-
гликоль — 1,3-бутадиен или по другому способу:
ацетилен — формальдегид — бутиндиол — бутиленгли-
коль—1,4-бутадиен). Нефтяные газы и природный газ
служат основным сырьем для получения не только
бутадиена, но и других каучукогенов (изопрен, хлоро-
прен, винилиденфторид и др ).
Произ-во и потребление СК из года в год увеличи-
вается. В 1959 произ-во СК (без стран социалистич.
лагеря) достигло 1,6 млн. т; увеличение выработки
по сравнению с 1955 составило 49%. За тот же период
произ-во натурального каучука увеличилось лишь на
5% и составило в 1960 2 млн. т.
В странах Западной Европы доля СК составляет
ок. 30% от общего объема потребления каучука,
в США — ок. 65%.
В начале СК рассматривался как заменитель нату-
рального каучука; после второй мировой войны он
приобрел самостоятельное место в произ-ве резино-
вых изделий, причем отдельные его виды по ряду
технич. свойств значительно превосходят натураль-
ный каучук (напр., по стойкости к действию агрес-
сивных сред и растворителей, термостойкости, сопро-
тивлению истиранию, газонепроницаемости, свето-
и озоностойкости и др). Синтезированные в по-
следнее время каучуки регулярной структуры по
497
КАУЧУК СИНТЕТИЧЕСКИЙ
498
комплексу эластич. свойств близки к натуральному
каучуку.
Произ-во СК характеризуется высокими экономии,
показателями. Разработанные в последнее время про-
цессы и аппаратура для получения мономеров и
эластомеров, а также мощная сырьевая база позво-
ляют создавать крупное механизированное и автома-
тизированное произ-во, что сокращает уд. капиталь-
ные затраты при строительстве заводов и существенно
снижает себестоимость. К числу преимуществ СК по
сравнению с натуральным каучуком относятся также
высокая производительность труда в процессе его
получения, лучшие условия труда и независимость от
климатич. и географии, условий.
В отличие от натурального каучука, имеющего в
своем составе природные защитные вещества, при
переработке СК в резину требуется вводить специаль-
ные вещества — противостарители (антиоксиданты),
предохраняющие от окислительных процессов, к-рые
приводят к деструкции или структурированию поли-
мера. Большинство известных СК (за исключением
цис-изопренового, хлоропренового, бутилкаучука и
нек-рых других) без введения усилителей образуют
вулканизаты резин с невысокой механич. прочностью
(до 50 кг/см2). Для получения резин из СК с хорошими
физико-механич. показателями необходимо при изго-
товлении резиновой смеси вводить активные напол-
нители, усиливающие механич. прочность вулкани-
затов. При этом неизбежно уменьшается эластич-
ность. В качестве активных наполнителей применяют
высокодисперсные в-ва, обладающие высокоразвитой
поверхностью: углеродные сажи, активную кремне-
кислоту (белая ража, аэросил), активную окись алю-
миния, каолин, мел и др. Для получения резин с вы-
сокой прочностью без применения активных напол-
нителей необходимы СК с регулярно построенными
Молекулярными цепями, с правильным чередованием
звеньев и с преимущественным преобладанием цис-
формы, как это имеет место в натуральном каучуке.
СК делятся на два больших класса: а) универсаль-
ные каучуки общего назначения для изготовления
массовых резиновых изделий — автомобильных шин,
транспортерных лент, рукавов, резиновой обуви и
ряда резиновых технич. изделий; б) каучуки со спе-
циальными технич. свойствами, применяемые при
изготовлении резиновых изделий, предназначенных
для работы в особых условиях — в среде растворите-
лей или агрессивных жидкостей, при высоких
темп-рах, под воздействием озона и ультрафиолетовых
лучей и др.
Для получения каучуков со специальными свой-
ствами применяются (обычно при сополимеризации
с бутадиеном) мономеры преим. винилового ряда, со-
держащие полярные группировки (нитрил акриловой
к-ты, винилиденхлорид, метилвинилпиридин и др.).
Нек-рые каучуки этого класса получают из одного или
нескольких мономеров, содержащих также полярные
атомы (хлоропрен, диметилдихлорсилан, винилиден-
фторид, гексафторпропилен и др.).
Для каучуков общего, массового назначения, в ка-
честве каучукогена применяют бутадиен, самостоя-
тельно или в смеси со стиролом или а-метилстиролом.
В связи с успешным синтезом изопренового каучука,
по комплексу эластич. свойств приближающегося к на-
туральному, в последние годы возрос интерес к
изопрену.
Промышленное значение приобрели следующие уни-
версальные каучуки общего назначения: натрий-бута-
диеновый каучук, получаемый полимеризацией бу-
тадиена в массе в присутствии щелочных металлов, и
бутадиен-стиролънъгй каучук, получаемый совместной
полимеризацией бутадиена и стирола или а-метилсти-
рола в водной эмульсии. К этому же классу каучуков
относятся синтезированные в последнее время стерео-
регулярные каучуки — изопреновый, цис-полибута-
диеновый и сополимер этилена с пропиленом.
Как известно, радикальная полимеризация вызы-
вает образование полимеров с той или иной степенью
разветвленности, с двойными связями в боковых це-
пях, что ведет к ухудшению эластич. свойств резин.
В отличие от этого, стереоспецифич. полимеризация
с применением комплексных катализаторов, создает
возможность регулировать и создавать заданное
пространственное расположение звеньев в макромоле-
кулах полимера с преимущественным образованием
линейных структур. При этом удается получать эла-
стомеры (из бутадиена, а также изопрена) с высоким
содержанием в макромолекулах звеньев, имеющих
цис-конфигурацию (до 99%), соединенных между собой
в положении 1,4, благодаря чему воспроизводится
комплекс эластич. свойств натурального каучука.
Отличительной особенностью стереорегулярных СК
является высокая механич. прочность изготовленных
на их основе резин без применения активных усили-
вающих наполнителей.
К СК со специальными технич. свойствами относятся
морозостойкие, маслобензостойкие, термостойкие, ка-
учуки, стойкие к действию света, озона и агрессивных
сред, каучуки с пониженной газопроницаемостью,
с повышенным сопротивлением истиранию и др.
Морозостойкие СК получают на основе бутадиена.
Полимеризация его в массе с помощью особых ката-
лизаторов, приготовленных с применением щелочных
металлов, приводит к получению каучука с темп-рой
стеклования —70°. Сополимеризацией бутадиена с не-
большими количествами стирола (10%) в водной
эмульсии получают каучук с темп-рой стеклования
минус 75—80°.
Устойчивы к действию масел и бензинов хлоропре-
новый каучук, бутадиен-нитрильный каучук, поли-
сульфидные Каучуки (см. Тиокол), бутадиен-метил-
винилпиридиновый каучук. Стойкость их к действию
углеводородов определяется наличием в цепях макро-
молекул полярных атомов и групп Cl, CN, S и др.
К термостойким СК относятся: силиконовые (см.
Кремнийорганические каучуки), фторкаучуки. Термо- и
бензостойкостью обладают фторкаучуки, а также фтор-
силиконовые эластомеры, состоящие из характерных
R R
I I
для силиконовых каучуков цепочек —Si—О—Si—О—,
R R
в боковых органич. радикалах к-рых атомы водорода
полностью или частично замещены фтором. Эти кау-
чуки, сохраняя все свойства кремнийорганич. каучу-
ков в отношении термо- и морозостойкости, обладают
также хорошей стойкостью к воздействию топлив и
масел. Фторкаучуки исключительно устойчивы к дей-
ствию различных агрессивных сред.
Пониженной газопроницаемостью и химич. стой-
костью обладает бутилкаучук. Он отличается повы-
шенной сопротивляемостью к действию УФ-лучей,
озона, химич. реагентов и стойкостью к тепловому
старению в воздушной и кислородной среде. Еще более
высокой химич. стойкостью отличается продукт поли-
меризации изобутилена — полиизобутилен. К числу
химически стойких эластомеров, способных к вулка-
низации и образованию резины с хорошими физико-
механич. показателями, относится сульфохлорирован-
ный полиэтилен. Полиуретановые эластомеры отли-
чаются исключительным сопротивлением истиранию,
намного превышающим сопротивление истиранию
натурального каучука.
Все большее значение и разнообразное применение
приобретают синтетические латексы, в к-рых поли-
499
КАУЧУКА ГИДРОХЛОРИД — КАЧЕСТВЕННЫЙ АНАЛИЗ
500
меры находятся в высокодисперсном состоянии. По-
лучают латексы эмульсионной полимеризацией. При-
менение эластомеров в виде латексов позволяет решать
сложные технич. задачи, в ряде случаев существенно
повышает физико-механич. свойства получаемых резин
и упрощает технологию изготовления резиновых изде-
лий. Ряд изделий, таких, напр., как тонкостенные,
производится только из латексов.
В результате проводимых в больших масштабах ис-
следований с каждым годом расширяется ассортимент
СК и в пром-сть внедряются все новые виды эластоме-
ров, обладающих новыми ценными свойствами.
Об идентификации СК см. ст. Идентификация
высокомолекулярных соединений.
Лит.: Лебедев С. В., Жизнь и труды, Л., 1938; Д о-
г а д к и н Б. А., Химия и физика каучука, М.—Л., 1947;
Смирнов Н. И., Синтетические каучуки, 2 изд., Л.,
1954; Л и т в и н О. Б., Основы технологии синтеза каучуков,
М„ 1959; Бородина И. В., Никитина. К., Техниче-
ские свойства советских синтетических каучуков, Л.—М.,
1952; Новые каучуки. Сб. переводов,-М., 1958; Синтетический
каучук, под ред. Г. Уитби, пер. с англ., М., 1957; Ф е д о-
р е и к о Н. П., С а в и н с к ий Э. С., Очерки по экономике
химической промышленности СССР, М., 1960, с. 220—21; U 1 1-
m а п п, 3 Aufl., Bd, 9, Mlinch.—В., 1957, S. 321—424.
КАУЧУКА ГИДРОХЛОРИД — продукт взаимо-
действия НС1 с натуральным или синтетич. каучуком.
Гидрохлорид натурального каучука может быть по-
лучен пропусканием влажного НС1 через раствор на-
турального каучука в хлороформе. Вязкость раствора
после пропускания НС1 резко падает. Из раствора
К. г. осаждается спиртом в виде белых нитей, превра-
щающихся затем в порошок, по составу близкий
к (С5Н8С1)П. К. г. растворим в бензоле, хлороформе,
нерастворим в спирте и эфире; уже при 40° разла-
гается с выделением НС1 (полного удаления НС1 из
К. г. нагреванием произвести не удается даже при
130° в вакууме). При восстановлении К. г. водородом
образуются циклич. продукты с мол. в. от 2 до
10 тыс., растворимые в обычных растворителях кау-
чука, по свойствам напоминающие циклокаучук, по-
лучающийся при термин, воздействии на каучук
в инертной среде. Циклокаучук получается также
при нагревании гидрохлорида натурального каучука
при 120—130°, причем образуются 6-членные кольца,
расположенные в цепи макромолекулы К. г. Образо-
вание 6-членных циклов может произойти в резуль-
тате отщепления атома водорода из а-метиленовой
группы и хлора от третичного атома углерода.
хСН2---СН8 хСН2-СН2
СН3-С-С1 ZCH8 —— CHg-C^-Cl ZCH8
~СН8 Ci-C-CH2~ '-4CH—c-ch8~
CH3 CH3
Синтетич. полимеры изопрена так же, как и на-
туральный каучук, легко реагируют с НС1, а поли-
меры бутадиена, сополимеры бутадиена со стиролом
и нитрилом акриловой к-ты присоединяют НС1 в более
жестких условиях. Реакцию проводят в смеси диоксана
и толуола, насыщенной хлористым водородом при
—10° и нагретой под давлением до 70—100°. Присо-
единенный хлор менее реакционноспособен, чем в ги-
дрохлориде натурального каучука. Количество при-
соединенного хлора, кроме типа каучука, опреде-
ляется также растворителем, в к-ром происходило
гидрохлорирование. Теоретич. содержание хлора в
полностью гидрохлорированном натуральном кау-
чуке составляет 33,97%, что отвечает формуле
(С5Н8С1)Ж, а в эмульсионном полиизопрене 28,14%,
или 83% от теоретически возможного, что обуслов-
лено наличием в полимере боковых винильных групп
в положении 1—2. Полиизопрен, полимеризованный
в присутствии металлич. натрия, присоединяет 63%
НС1 от теоретич. количества, а полиизопрен, полу-
ченный с алфиновым катализатором (продуктом со-
четания алкоголятов вторичных спиртов и натрий-
алкилов с NaCl), — 77%. Эти результаты представ-
ляют интерес при изучении строения каучуков.
Гидрохлорид натурального каучука после вызрева-
ния превращается в твердый кристаллич. продукт.
При нагревании выше 90° он становится эластичным.
Если при этой темп-ре пленку гидрохлорида растя-
нуть и охладить, то сохранится ориентированная
структура растянутого материала, обеспечивающая
его высокую прочность. В таком виде К. г. применяют
для приготовления пленок (плайофильм), напр. для
упаковки пищевых продуктов. Малая влагопроницае-
мость и химич. стойкость позволяют применять К. г.
для различных лаковых покрытий.
Лит.: Догадкин Б. А., Химия и физика каучука,
М.—Л., 1947; Синтетический каучук, под ред. Г. С. Уитби,
пер. с англ., М., 1957. В. А. Шершнев.
КАУЧУКИ МАСЛО- И САЖЕНАПОЛНЕННЫЕ —
смеси синтетических эмульсионных каучуков (в основ-
ном дивинилстирольного типа) с маслами, играющими
роль пластификаторов, или с сажами. Масляные
каучуки, как и саженаполненные, приготовляют вве-
дением в латексы синтетические эмульсии масла или
водных дисперсий саж с последующей коагуляцией
латексно-масляной или латексно-сажевой смеси. Вве-
дение масел в каучук улучшает обрабатываемость
смесей и удешевляет их, снижая содержание каучука
в резиновых изделиях. Масла способствуют умень-
шению теплообразования в резинах, работающих
при многократных деформациях, что снижает само-
разогревание изделий в эксплуатации. В связи с этим
масляные каучуки находят широкое применение
в автошинах и резино-технич. изделиях. В основном
применяют различные нефтяные масла с преоблада-
нием парафино-нафтеновых или ароматич. углеводо-
родов в зависимости от назначения резин. Ароматич.
масла меньше снижают прочностные свойства резино-
вых изделий, чем парафино-нафтеновые, но последние
придают большую эластичность резинам в широком
интервале темп-p. Наиболее распространенными ти-
пами являются смеси с 25 вес. ч. и 37,5 вес. ч. масел
иа 100 вес. ч. каучука. Применение этих каучуков
в сочетании с каучуками без масла позволяет полу-
чать резины с необходимым комплексом свойств.
Введение саж в каучук на стадии выделения его
из латекса имеет ряд преимуществ по сравнению с вве-
дением сажи в твердый каучук: повышается однород-
ность распределения сажи в смеси, улучшается со-
противление разрыву, износостойкость, усталостная
выносливость резин при многократных деформациях
и др., повышается производительность смесительного
оборудования на 40—50%, снижается общий расход
электроэнергии на изготовление резиновых смесей на
20—30%. Выпускаются также т. наз. саже-масляные
каучуки, представляющие собой саженаполненные
каучуки с различными дозировками масел.
Лит.: Синтетический каучук, под ред. Г. С. Уитби, пер.
с англ., М., 1957; DinsmoreR. Р., Trans. Instn. Rubber
Ind., 1952, 28, Ki 4, 171; Enslin O., Kautschuk und Gummi,
1953, 6, К» 6, 160; Swart G. H„ P f a 11 E. S., W e 1 n-
stock К. V., India Rubber World, 1951, 124, № 3, 309;
Weinstock К. V., Storey E. B., S wee ley J. S.,
Industr. and Engng Chem., 1953, 45, № 5, 1035; К a-
л а у с A. E. [и др.], Хим. пром-сть, 1956, А» 5, с. 267; и х ж е,
там же, 1956, № 8, с. 449; A d a m s J. W., Messer W. E.,
Howland L. H., Industr. and Engng Chem., 1951, 43, Ks 3,
754; Samuels M. E., Rubber Age, 1960, 86, Ns 4, 649 —
653. В. И. Гусева.
КАЧЕСТВЕННЫЙ АНАЛИЗ — один из важней-
ших разделов аналитической химии; совокупность
химич., физико-химич. и физич. методов, применяе-
мых для обнаружения и идентификации элементов,
радикалов и соединений, входящих в анализируемое
вещество или в смесь веществ. В классич. К. а. ис-
501
КАЧЕСТВЕННЫЙ АНАЛИЗ
502
пользуются гл. обр. легко выполнимые, характерные
для данного элемента химич. реакции, сопровождае-
мые к.-л. легко наблюдаемыми эффектами (появление
или исчезновение окрашивания, выделение или рас-
творение осадка, выделение газа и т. п.). Используе-
мые для обнаружения реакции должны быть по воз-
можности специфическими (селективными) и обладать
достаточно высокой чувствительностью.
Классич. анализ неорганич. веществ производят
т. наз. «сухим» или «мокрым» путем. Анализ сухим
путем наиболее часто применяют для предварительных
испытаний и при исследовании минералов. Он вклю-
чает прежде всего испытание на окрашивание пламени
газовой горелки, в к-рое на платиновой игле впосят
исследуемое вещество, смоченное 7,5 н. р-ром соля-
ной к-ты. Таким путем могут быть обнаружены
Hg(I), Т1(1), РЬ, Си, As, Sb, Se, Те, Ga, Ва, Sr, Са,
Li, Na, К, Rb, Cs, ион CN~. Для открытия ряда эле-
ментов применяют также фотометрию пламени, спек-
тральные, рентгеноспектральные, полярография, и др.
методы. При подборе подходящего источника возбуж-
дения и соответствующего прибора спектральный ме-
тод позволяет обнаруживать большинство элементов.
Другим классич. методом предварительного испы-
тания сухим путем является получение перлов буры
или фосфорной соли (NaNH4PO4 • 4Н8О) со следами
анализируемого вещества при нагревании их после-
довательно в окислительном и в восстановительном
пламени. По окраске перлов можно судить о наличии
Ti, V, Сг, Мп, Fe, Со, Ni, Си, Мо, W, Nb, Се, U.
Иногда нагревают небольшую пробу анализиру-
емого материала в закрытой или открытой стеклянной
трубке и наблюдают образующиеся на холодных ча-
стях трубки возгоны или распознают выделяющиеся
газы по запаху или другим путем. При нагревании
исследуемого вещества на платиновой или фарфоровой
пластинке обращают внимание на изменение цвета;
прокаливают исследуемое вещество с углем в при-
сутствии карбоната натрия и иногда нитрата кобальта
и цианида калия и наблюдают цвет образующихся
продуктов реакции и возгонов, металлич. корольков.
Перечисленные методы позволяют обнаруживать Sb,
As, Мо, РЬ, Bi, Zn, Cd, Hg, Fe, Ni, Co, Mn, Ag, Sn,
Cu, Au, Pt, S, Se, а также бораты, фосфаты, силикаты,
сульфиды As и Sb, хлориды ртути (I и II), тиосоли
As и Sb, пирит, соли аммония, алюминия, цинка,
магния, окислы As, Sb и Sn (IV).
К. а. мокрым путем (в растворах) осуществляют
макро-, полумикро-, микро- и ультрамикрометодами,
к-рые отличаются друг от друга гл. обр. количеством
взятого для исследования материала и объемом
раствора и соответственно этому — техникой работы
(см. таблицу).
Метод Количество ве- щества, взято- го для полного анализа, мг Объем рас- твора, мл
Классический макрометод. . . Полумикрометод Микрометод Ультрамикрометод >100 от 100 до 10 от 10 до 0,1 <0,1 >5 от 5 до 0,5 от О',5 до 0,05 <0,05
Характерным для полумикрометода является при-
менение капельниц для дозирования и переноса раство-
ров и жидкостей, центрифугирование вместо филь-
трования (или фильтрование через особые фильтрую-
щие трубки с ватными тампонами), промывание осад-
ков в центрифужных стаканчиках. В полумикроана-
лизе широко применяют микрокристаллоскопич. ре-
акции (см. Микрокристаллоскопия), капельные ре-
акции на фильтровальной бумаге или на капельных
пластинках из стекла или фарфора (см. Капельный
анализ), а также реакции с образованием газообраз-
ных продуктов в микрогазовой камере.
Наибольшее практич. значение в химич. К. а.
имеют специальные реакции, позволяющие обнару-
живать данный элемент даже при небольших его кон-
центрациях в присутствии большого числа других
элементов. Помимо селективности, важнейшей ха-
рактеристикой реакции является ее чувствительность,
выражаемая наименьшим количеством обнаружива-
емого элемента [открываемый минимум в микрограм-
мах (мкг, цг, у)] или же наименьшей концентрацией
раствора, при к-рой данный элемент еще может быть
однозначно обнаружен без предварительного обога-
щения (предельная концентрация). Ме-
ждународным соглашением установлены след, ве-
личины стандартных объемов раствора (Е) в различ-
ных методах: реакция в микропробирке — 1 мл\
капельная реакция — 0,03 мл\ реакция на предмет-
ном стекле — 0,01 мл.
Неорганич. К. а. в водных р-рах основан на ионных
реакциях и позволяет обнаруживать элементы в форме
катионов или анионов. Соответственно он разделяется
на анализ катионов и анализ анионов. В классич.
схеме неорганич. К. а., созданной еще в первой поло-
вине 19 в., катионы с помощью групповых реагентов
разделяют на группы, к-рые, в свою очередь, подраз-
деляются на подгруппы: внутри подгрупп возможно
непосредственное обнаружение соответственных ка-
тионов. Анионы не имеют общеустановленного разде-
ления на группы, хотя и для них предложены схемы
группового разделения.
Разделение ионов на группы и подгруппы основано
на использовании их главнейших химич. свойств,
в том числе кислотно-основных, комплексообразую-
щих и отчасти окислительно-восстановительных.
Аналитич. классификация выражает закономерность
главных химич. свойств ионов. Поэтому она до сих
пор остается важнейшим элементом в специальном
образовании химиков и основой учебного курса К. а.
Вместе с тем она сохраняет и практич. значение для
предварительного испытания анализируемых смесей
при установлении присутствия групп ионов и для
правильного выбора последующих методов анализа.
Имеется несколько разновидностей классич. классификации
катионов, основанной на свойствах сульфидов, хлоридов,
гидроокисей и карбонатов; наиболее всеобъемлющей является
следующая.
1-я группа — катионы, осаждаемые сероводородом
в кислой среде при pH менее 3. Она разделяется на 3 под-
группы: а) катионы, образующие малорастворимые хлориды
и потому осаждаемые при подкислении соляной к-той, — Ag,
Hg (I), TI (I) и Pb (частично); б) катионы, не образующие тио-
солей, сульфиды к-рых поэтому не растворяются в полисуль-
фиде аммония, — Hg (II), Pb, Си, Ru, Rh, Pd, Os, Bi, Cd;
в) катионы, образующие растворимые тиосоли при действии
полисульфида аммония, — As, Sb, Sn, Au, Ir, Pt, Se, Те, Ge,
Mo, W и V. 2-я группа — катионы, осаждаемые сероводо-
родом в щелочной среде, в частности сульфидом аммония
в аммиачной среде. Она также разделяется на 3 подгруппы:
а) редкоземельные элементы, Sc, Y, Th, образующие нерас-
творимые оксалаты в умеренно кислой среде; б) элементы,
сульфиды и гидроокиси к-рых ие растворяются в 3 и. НС1
при нагревании, — Со, Ni, Re, Ti, Nb, Та; в) элементы, оса-
ждаемые после отделения подгруппы «б» аммиаком в присут-
ствии хлорида аммония, — Al3+, Fe3+, Сг3+, Be, U, Zr, HI,
Ga, In, Tl3+, а также Zn2+ и Mn (II), переходящие при этой
операции в раствор. 3-я группа — катионы, осаждаемые
карбонатом в нейтральной или в щелочной среде, в частности
карбонатом аммрния в аммиачной среде (сульфиды и гидро-
окиси их растворимы в воде), — Ra2+, Ва2+, Sr2+, Са2+.
4-я группа — катионы, не осаждаемые названными реа-
гентами в указанных выше условиях, — L1+, Na+, К+, Rb+,
Cs+, Mg+, ион NH+.
Разделение анионов производится после удаления катио-
нов тяжелых металлов кипячением анализируемого р-ра с из-
бытком соды на след, группы: 1-я группа — анионы, оса-
ждаемые катионом серебра в азотнокислой среде, — С1_, Вг_,
J-, JO~, CN-, SCN-, [Fe(CN)o]3-, [Fe(CN)0]«~. 2-я г р у п п а—
анионы, осаждаемые катионом кальция в уксуснокислой
среде, —SO|- F-, РО3~, Р2О«-, РО~, С2О2~, МоО|-, WO2-,
VO-, ВО'*,[Fe(CN)j]4-. 3-я группа — анионы, осаждаемые
503
КВАЗИКОМПЛЕКСНЫЕ СОЕДИНЕНИЯ
504
катионом бария в солянокислой среде, — S1F2-. 4-я
группа — анионы-восстановители, окисляющиеся разб.
p-ром перманганата в сернокислой среде,— SO*-, S-~,
S.O2-, NO-, CN-, SCN-, Вг, J-, AsO^-, C2OJ-, [Fe(CN)cl4’.
5-я группа — анионы-окислители, восстанавливающиеся
раствором иодида калия в солянокислой среде, — Сг^О^-»
AsO]~, JO:, NO-, СЮ;, ВгОг, S2O* [Fe (CN)6]3“, MnO;.
Для обнаружения катионов и анионов широго
применяют различные органич. реактивы.
Теория неорганич. К. а. представляет собой учение
об управлении равновесиями реакций и рассматри-
вает гомогенное, гетерогенное и окислительно-восста-
новительное равновесие, а также кинетику соответ-
ствующих процессов применительно к задачам К. а.
Органич. К., а. резко отличается от неорганич.
анализа. Подавляющее большинство органич. соеди-
нений имеет ковалентный характер и потому каждое
из них должно идентифицироваться индивидуально.
Для этого сначала проводят реакции, определяющие
принадлежность соединения к к.-л. классу органич.
соединений, а затем — реакции, характерные для
данного соединения. В органич. К. а. смесь веществ
первоначально разделяют, основываясь на их разной
летучести, растворимости или сорбции. К легколе-
тучим относят вещества с т. кип. ниже 160°, к трудно-
летучим — ст. кип. выше 160°. Затем вещества
разделяют по классам согласно их растворимости,
преим. в воде и эфире. Наконец, применяют групповые
реакции, с помощью которых устанавливают присут-
ствие классов химич. соединений (спирты, фенолы,
кислоты, амины и проч.). Некоторые химич. реакции
г.озволяют перевести малоразличимую смесь веществ
в вещества с достаточно различными физич. свой-
ствами, что дает возможность отделять их далее по-
средством дистилляции или растворением. Напр.,
можно превратить смесь поликарбоновых к-т и ами-
нокислот в летучие сложные эфиры, сравнительно
легко разделяемые. При идентификации выделенного
чистого вещества большое значение имеет элементар-
ный К. а., проводимый обычными методами для от-
крытия углерода, водорода, азота, серы, галогенов,
фосфора, мышьяка и металлов, а также испытание
основных физич. свойств (темп-р плавления и ки-
пения, растворимости и определение молекулярного
веса). См. также Элементарный анализ, Функциональ-
ный анализ.
Лит..: Блок Н. И., Качественный химический анализ,
М.—Л., 1952; К ля ч ко Ю. А., Ш а п и р о С. А., Курс хими-
ческого качественного анализа, М., I960; Алимарин И. П.,
Архангельская В. Н., Качественный полумикроана-
лиз, 2 изд., М.—JL, 1952; Тананаев Н. А., Капельный
метод, 6 изд., М.—Л., 1954; F е 1 g 1 F., Spot tests In inorganic
analysis, N. Y., 1958; Ф а й г л ь Ф., Капельный анализ ор-
ганических веществ, пер. с англ., М., 1962; Алимарин
И. П., Петрикова М. Н., Неорганический ультрамикро-
аналмз, М., 1960; Коренман И. М., Микрокристал-
лоскопия, М.—Л., 1947; Реакции и реактивы для качествен-
ного анализа неорганических соединений, пер. с франц., под
ред. А. С. Комаровского, М.—Л., 1950; Reactlfs pour 1’analyse
qualitative minerale (4 rapport), P., 1950; Chariot G.,
Thdorie et methode nouvelles d’analyse qualitative, 3 dd., P.,
1949; Nieuwenburg C. J. van, Ligten J. W. L. van,
Qualitative chemisciie Analyse, W., 1959; Staudinger H.,
Anleitung zur organischen qualitativen Analyse, 6 Aufl., B.,
1955 (cobm. c W. Kern); В а й б e л ь С., Идентификация
органических соединений, пер. с англ., М., 1957; Шрай-
нер Р., Фьюсон Р., Систематический качественный
анализ органических соединений, пер. с англ., М., 1950;
Бауер К., Анализ органических соединений пер. с нем.,
2 изд.,- М., 1953. Ю. А. Клячко.
КВАЗИКОМПЛЕКСНЫЕ СОЕДИНЕНИЯ — ор-
ганические соединения металлов, содержащие гало-
гены, окси-, алкокси-, ацетокси- и другие группы
в P-положении по отношению к металлу, способные
легко элиминировать непредельное сочинение при
нагревании или действии различных реагентов. К. с.
легко образуются при взаимодействии органич. со-
единений, содержащих кратные связи, с солями многих
непереходных металлов; это наиболее важный способ
получения К. с., напр.:
SbCl5 + CHSCH --—— (ClCH=CH)3SbCl2
Н2о .
CH8=CH8+HgCl8----- HOCH8CH2HgCl
+ н‘<ососнА—U^h,OCOcB!
Нек-рыеК. с. были получены обменными реакциями:
2(ClCH=CH)2Hg+TlCl3--. (ClCH=CH)2TlCl+2CHCl=CHHgCl
Для перехода от одного К. с. к другому могут быть
использованы реакции диспропорционирования, сим-
метризации и другие:
(ClCH=CH)2SbCl+SbCl3 —► 2CICH=CHSbCl3
(HOCH2CH2)2Hg+HgCl2---► 2HOCH2CH2HgCl
ClCH=CHHgCl —JU. (ClCH=CH)2Hg
Типы К. с. весьма разнообразны, они могут содержать
винильную связь с металлом (аддукты солей металлов
с ацетиленом) и не содержать этой связи (аддукты
с олефинами); могут содержать число радикалов, рав-
ное валентности металла и меньше, напр.:
(XCH=CH)3Sb, (XCH=CH)2SbX, XCH=CHSbX2
и т. п. К. с. известны для ряда непереходных металлов
(Hg, Sn, Pb, As, Sb, Bi, TI и др.) и нек-рых неметал-
лов, напр. для иода. Естественно, что каждый тип
этих соединений имеет свою специфику; особенно
заметно меняются их свойства при переходе от одного
металла к другому.
При элиминировании К. с. ведут себя так, как если
бы они представляли собой координационные ком-
плексы типа:
снеесн сн2=сн2
СНзСН-HgCl HgCI2 SbClg
сн=сн сн2=сн2
При нагревании К. с. более или менее гладко разла-
гаются:
(ClCH=CH)2Hg --. CHEECH4-ClCH=CHHgCl
ClCH=CHHgCl —. СН ~CH^HgCl2
Еще легче происходит разложение с выделением не-
предельного соединения под действием реагентов,
способных связывать ион металла, напр. в виде ус-
тойчивого комплекса. Для ртутных К. с. агентами
такого рода являются иодид калия, тиосульфат нат-
рия, цианид калия, фосфины и т. д.:
CICH=CHHgCI+3KJ —. KHgJ34-2KC14-CHEECH
К. с. сурьмы и мышьяка особенно легко разлагаются
при действии щелочей и сернистых щелочей
2 (ClCH2CH2)3SbCl24-12NaOH —. 3C2HJ-10NaCl+
+Na2H2Sb2O7-|-5H2O
Обычно к полному элиминированию непредельного
соединения приводит действие реактива Гриньяра:
ClCH=CHHgCl-|-C2H5HgX —. C2H2+MgXCl+C2H-oHgCl
С другой стороны, К. с. обнаруживают свойства
истинных металлоорганич. соединений, напр., в сле-
дующих реакциях:
ClC2H2HgCl+HCl —. ClCH=CHa-|-HgCl2
ClC2H2HgCl+J.3 —. ClCH=CHJ+HgCU
2ClC,H2HgC14-SnCl2-. (ClCH=CH)2SnCl2+HgCl2
HOCH2CH2HgCl+CeH5COCl —. C0H5COOCH2CHsHgCl 4- HCI
2ClCH=CHHgCl —(ClCH=CH)2Hg4-HgCl2
Такую двойственную реакционную способность
можно было бы объяснить таутомерией типа:
сн=сн С1
Hg z=± (С1СН—CH)2Hg
СНЕЕСН Cl
А Б
505
КВАНТОВАЯ МЕХАНИКА
506
Было, однако, показано, что для аддуктов солей ме-
таллов с ацетиленом (и соответствующих полноза-
мещенных металлоорганич. соединений) характерна
обычная геометрич. стереоизомерия (что доказывает
наличие двойной связи в соединении), напр.:
н, .н нч yHgCl
>с=с< )с=с<
Clz XHgCl С1Х хн
цис транс
Тем самым отвергается комплексное строение
(типа А) и доказывается невозможность таутомерного
равновесия между структурами А и Б (если бы такое
равновесие имело место, то в растворах неизбежно
происходило бы взаимное превращение цис- и транс-
изомеров через таутомерную форму А). Подобные сте-
реохимии. отношения характерны и для аналогичных
К. с. других металлов. Было установлено, что конфи-
гурация органич. остатка не изменяется при вытес-
нении одного металла другим из квазикомплексного
соединения, а также при симметризации и рассиммет-
ризации; т. о., входящий металл стереоспецифично
становится на место уходящего (правило Несмеянова—
Борисова). Это можно иллюстрировать следующей
схемой:
Что касается квазикомплексных свойств, то они,
очевидно, связаны с ярко выраженным сопряжением
a-связей С1—С и С—Me (а-а-сопряжение). В момент
реакции (под действием агента или при нагревании)
происходит сдвиг электронной плотности:
Сц) ZH
С=сч ’
Hz HgC>
Cl* Н
zc=cz —*.CHSCH
н' HgCl + HgCla
Лит.: Несмеянов A. H., Фрейдлина Р. X.,
Борисов А. Е., в кн.: Юбилейный сборник, посвященный
тридцатилетию Великой Октябрьской социалистической рево-
люции, ч. 1, М., 1947, с. 658; Несмеянов А. Н., Бори-
сова. Е., ДАН СССР, 1948, 60, № 1, 67; Несмеянов А. Н.,
Борисов А. Е., Шепелева Р. И., Изв. АН СССР.
Отд. хим. наук, 1949, 6, 582; Несмеянов А. Н., Б о-
р и с о в А. Е., Абрамова А. Н., там же, 1949, 570; Н е-
смеянов А. Н., БорисовА. Е., Гуськова А. Н.,
там же, 1945, К* 6, 639; Несмеянов А. Н., Кочет-
ков Н. К., там же, 1949, № 3, 305; Несмеянов А. Н.,
Фрейдлина Р. X., Ж. общ. химии, 1937, 7, вып. 22,
2748. И. А. Несмеянов.
КВАНТОВАЯ МЕХАНИКА. Содержание:
Корпускулярно-волновая природа микрочастиц. Соотно-
шения де-Бройля.............................506
Соотношения неопределенностей................508
Состояние микрочастицы. Волновая функция.....509
Уравнение Шредингера. Операторы..............513
Стационарные связанные состояния.............515
Квантовые системы............................519
Квантовые переходы...........................522
Применения квантовой механики................522
Квантовая механика — физическая теория, иссле-
дующая общие закономерности движения и взаимо-
действия частиц очень малой массы — микрочастиц
(элементарных частиц, атомных ядер, атомов и мо-
лекул); теоретич. основа современной физики и хи-
мии. К. м, возникла гл. обр. в связи с необходимостью
преодолеть противоречивость и недостаточность бо-
ровской теории атома. Она была создана в 1924—26 гг.
трудами Л. де-Бройля, Э. Шредингера, В. Гейзен-
берга и П. Дирака. Важнейшую роль в ее история.
подготовке и последующей разработке сыграли иссле-
дования М. Планка, А. Эйнштейна, Н. Бора, М. Борна
и др.
Корпускулярно-волновая природа микрочастиц. Со-
отношения де-Бройля. Опыт показывает,что движе-
ние микрочастиц происходит по законам, отличным
от законов классической механики: микрочастице
присущи некоторые свойства корпускул (частиц) и
некоторые свойства волн. С одной стороны, электрон
(или протон, заряженный мезон) движется и дей-
ствует подобно корпус-
куле: в камере Вильсона
он оставляет след, похо-
жий на траекторию ча-
стицы (рис. 1), в столкно-
вениях участвует как
целое (его энергия и им-
пульс связаны таким же
соотношением, как и у
обычной частицы в клас-
сич. механике). Поэтому
до 20-х гг. электроны
рассматривались кай кор-
Рис. 1. Следы электронов в ка-
мере Вильсона, помещенной в
магнитное поле: прямой след —
быстрых элентронов, искривлен-
ный — медленных.
пускулы. Вместе с тем
при их движении обнару-
живаются такие типично
волновые явления, как
интерференция и дифрак-
ция. Если пропустить монохроматич. пучок быстрых
электронов (т. е. пучок электронов одинаковой энер-
гии) через тонкую (0,01—0,1 мк) кристаллич. пла-
стинку (рис. 2, а), то на экране позади пластинки
появятся интерференционные кольца или полосы;
Рис. 2. Картина дифракции: а — дифракция электро-
нов; б — дифракция рентгеновских лучей.
возникающая картина дифракции похожа на кар-
тину, получающуюся при дифракции рентгеновых
лучей (рис. 2, б). Измерив расстояние между полосами
на картине дифракции, можно определить длину вол-
ны X, соответствующей данному пучку электронов. Т.
обр., электроны (так же как и протоны, нейтроны и др.
микрочастицы) обладают свойствами и частиц и волн.
Двойственную природу обнаруживает также элек-
тромагнитное излучение. Дифракция и интерферен-
ция света (и радиоволн) служат убедительным дока-
зательством его волновой природы. Вместе с тем
взаимодействие излучения с частицами вещества до-
казывает, что излучению присущи также свойства
частиц, что его можно рассматривать как «газ» фото-
нов. Именно монохроматич. излучение испускается
и поглощается атомами или молекулами дискретно,
отдельными «порциями», его энергия и импульс из-
меняются скачкообразно. При столкновении со сво-
бодным электроном фотон ведет себя подобно частице,
он передает электрону часть своей энергии и импульса.
Связь между энергией Е и импульсом р отдельной
микрочастицы (массы т), с одной стороны, и ее вол-
507
КВАНТОВАЯ МЕХАНИКА
508
новыми характеристиками — частотой v и длиной
волны X — с другой, находит свое выражение в со-
отношениях де-Бройля:
Е — h'i. р — тс = й/Х (1)
v — скорость частицы, h — квант действия (постоян-
ная Планка), равный 6,62 • 10-27 эрг • сек (действие —
механич. величина, имеющая размерность произведе-
ния энергии на время или импульса на координату).
Это — универсальные соотношения,
справедливые для микрочастиц любой природы. Из
соотношений (1) следует, что длина де-бройлевской
волны тем меньше, чем больше масса частицы (при
данной скорости) или скорость частицы данной массы
(т. е. чем больше ее энергия). Так, напр., длина де-
бройлевской волны электрона с энергией 150 эв
равна ~ 1 А. Поскольку период кристаллич. решетки
имеет тот же порядок величины, она может служить
дифракционной решеткой для электронов, т. е. с по-
мощью кристаллич. решетки можно измерить длину
де-бройлевской волны такого электрона. Длина же
де-бройлевской волны массивной частицы, напр. пы-
линки с массой 10-6 г, движущейся со скоростью
ок. 1 м/сек, X ~ 10'22с.и (10 14А). Волновые особен-
ности движения такого объекта ни в чем не прояв-
ляются.
С точки зрения классич. физики проявление волно-
вых и корпускулярных свойств в движении одного
и того же объекта исключается. В самом деле, обычная
частица (точнее, ее центр инерции) движется по опре-
деленной траектории, обладая в каждой точке опре-
деленной скоростью (импульсом); к волне, т. е. к про-
цессу распространения периодич.возмущения в сплош-
ной среде — понятие траекторного движения непри-
менимо. С другой стороны, волне присуща периодич-
ность в пространстве и времени, «согласованность»
фаз в разных частях пространства; вследствие этого
при наложении действий различных участков волно-
вого фронта имеет место интерференция. Частица же
действует всегда как целое.
Очевидно, двойственная природа микрочастиц, на-
личие у них и волновых и корпускулярных свойств
означает, что микрочастицу нельзя рассматривать ни
как обычную (классическую) частицу, ни как обыч-
ную волну.
Двойственность микрочастиц можно уяснить, рас-
смотрев подробнее ее проявления на опыте. Особенно
наглядны опыты, в к-рых через дифракционную ре-
шетку (кристаллич. пленку) пропускался электрон-
ный пучок столь слабой интенсивности, что электроны
проходили по одному. Попадая на фотопластин-
— ку, помещенную за решеткой,
, . электроны оставляют точечные
следы (рисунок 3); эти сле-
‘ • • , ды распределены на фотопла-
. стинке неравномерно, и после
" ' • , • ’ , прохождения немногих элек-
• , _ . . / ’ тронов дифракционные полосы
> • еще не обнаруживаются. Лишь
• постепенно, после прохожде-
• • . , ния многих электронов, выри-
..........*........ совывается картина дифракции
Рис. 3. Картина дифрак- и выявляется их волновая ири-
дии после прохождения рода. В те места, где интен-
через дифракционную ре- сивность волны больше, по-
шетку немногих влек- падает большая часть электро-
нов (вероятность попадания
в такие места каждого электрона больше); в места,
где интенсивность волны равна нулю, электроны во-
обще не попадают. Обычная же волна дает картину
дифракции не постепенно, а сразу. Т. обр., каждый
электрон действует на фотопластинку локально (в од-
ной точке), подобно частице, но движется так, что
попадает с наибольшей вероятностью в места макси-
мумов интенсивности волны. Поэтому волновые свой-
ства электрона непосредственно обнаруживаются в ре-
зультате многих одинаковых опытов над совокуп-
ностью электронов, находящихся в совершенно оди-
наковых условиях; волновые свойства микрочастицы
проявляются статистически.
Соотношения неопределенностей. В силу двойствен-
ной природы движение микрочастицы отличается от
движения массивной частицы. Микрочастица одно-
временно не обладает определенными значениями коор-
динаты и импульса, т. е. не движется по траектории
с определенной скоростью в каждой ее точке.
Между неопределенностью координаты Ад и не-
определенностью составляющей импульса по этой
координате Apq имеет место соотношение неопределен-
ностей:
Ад • Apq ~ Й, где Й = й/2л (2)
Это означает, что чем определеннее значение коор-
динаты g микрочастицы в данных физич. условиях
(чем меньше Ад), тем более неопределенным является
импульс (тем больше Apq), и наоборот. Отсюда сле-
дует, что невозможно одним и тем же эксперимен-
тальным устройством одновременно измерить с любой
точностью значения р и д; чем точнее данным классом
приборов (напр., микроскопом) можно измерить
д, тем менее точно измеряется р, и наоборот. Такой
результат обусловлен взаимодействием микрочастицы
с классич. прибором. Вследствие атомизма действия,
прибор, измеряющий местоположение микрочастицы,
«локализующий» ее (напр., микроскоп), в процессе
измерения «размывает» ее импульс (делает его более
или менее неопределенным); прибор, измеряющий им-
пульс частицы, в процессе измерения «размывает» ее
координату.
Однако не следует смешивать понятия «неопреде-
ленность величины» и «неточность измерения». Любое
взаимодействие электрона со значительно более мас-
сивной частицей (напр., атомным ядром), ограничи-
вающее область локализации электрона, создает не-
определенность его импульса (см. ниже), хотя ядро
и не является макроприбором. Существенно лишь,
чтобы «прибор» был достаточно массивным по срав-
нению с микрочастицей.
Если масса частицы т велика по сравнению с h, то
соотношения неопределенностей становятся непри-
менимыми и движение частицы описывается классич.
законами.
Имеет место также соотношение неопределенно-
стей для энергии Е частицы в данном ее состоянии и
моментом времени t, в к-рый измеряется это значение
энергии:
AE-At^H (3)
Чем определеннее энергия микрочастицы (чем
меньше АЕ), тем менее определенно время измерения
ее (тем больше At), и наоборот. Это значит, что для
определения энергии с точностью АЕ необходим
интервал времени At, по меньшей мере равный h/AE-,
с другой стороны, если система находится в нек-ром
состоянии в течение ограниченного времени At, то
неопределенность ее энергии АЕ в этом состоянии по
меньшей мере равна Й/At.
Величина неопределенности к.-л. физич. величины
(импульса, координаты, энергии) может быть охарак-
теризована с помощью к.-л. меры статистич. разброса
ее возможных значений, получающегося при после-
дующем точном ее измерении. Напр., если неопреде-
ленность координаты электрона равна Ад, то при
последующем точном измерении координаты ее зна-
чения будут лежать в нек-ром интервале qi и д,-|- Ад.
Такой разброс значений будет получен на опыте,
509
КВАНТОВАЯ МЕХАНИКА
510
если точно измерить координаты множества электро-
нов, находящихся в таком же состоянии, как и дан-
ный электрон (с одной и той же неопределенностью
координаты, равной А?). При этом вероятность полу-
чить то или иное точное значение координаты, вообще
говоря, будет различна.
Соотношения неопределенностей позволяют давать
приближенные оценки, напр., области локализации
микрочастицы или ее импульса, или времени жизни
ее состояния. Напр., на основании соотношения
неопределенностей для энергии и времени, зная время
жизни возбужденного состояния атома (или молекулы),
можно оценить ширину испускаемой при переходе
спектральной линии, т. е. неопределенность энергии
испускаемых фотонов.
Состояние микрочастицы. Волновая функция. Из
соотношения неопределенностей следует, что движение
микрочастицы не может быть описано таким же об-
разом, как движение массивной (классической) ча-
стицы. В самом деле, состояние движения классич.
частицы (данной массы т) в каждый момент задается
значениями ее координаты н скорости v (или импульса
р — mv). Соответственно, макрочастица в данном
состоянии обладает определенными значениями любых
динамич. величин — кинетич. энергии тг?2/2 = р2/2ш
(являющейся функцией импульса), потенциальной
энергии U (х, у, z) (являющейся функцией координат),
вектора момента количества движения М (и всех
его составляющих), зависящего и от скорости и от
координаты, и т. д.
В противоположность этому для микрочастиц могут
иметь определенные значения не все эти величины, а
лишь часть их; значения другой части величин
остаются неопределенными. Напр., свободный элек-
трон может обладать определенным значением им-
пульса, но его координата в этом случае будет совер-
шенно неопределенной; неопределенность координаты
свободного электрона в данном случае проявляется
в том, что при точном ее измерении она с равной ве-
роятностью принимает любое значение. Электрон,
связанный в атоме, может иметь лишь определенные
значения 3 величин — энергии, момента количества
движения и одной из проекций момента (см. ниже).
«Набор» значений этих величин — значений энергии,
момента и проекции момента — однозначно опреде-
ляет состояние электрона в атоме; они зависят от
целых и полуцелых квантовых чисел, ха-
рактеризующих данное состояние. Остальные же
динамич. характеристики атомарного электрона —
координата, импульс, кинетическая и потенциальная
энергия в отдельности, две другие проекции момен-
та количества движения — остаются неопределенны-
ми; в случае связанного состояния вероятности воз-
можных значений этих величин, получающихся при
их точном измерении, распределяются по строго
определенным законам, характерным для данного со-
стояния.
Таким образом, каждое состояние атомарного элек-
трона характеризуется не только определенной сово-
купностью (набором) квантовых чисел, но и определен-
ным распределением вероятностей
возможных значений координаты при ее измерении,
либо определенным распределением вероятностей воз-
можных значений импульса и т. д. Это — различные
способы характеристики одного и того же состояния;
зная одну из этих характеристик состояния, можно
рассчитать остальные. Следовательно, состояние элек-
трона описывается не числами, задающими его коор-
динаты и скорость в данный момент времени, как это
делается в классич. механике, а нек-рой функцией,
характеризующей закон распределения вероятностей
возможных значений к.-л. физич. величины, чаще все-
го координаты, Эта функция 4‘(z,y,z,Z) от координат
х, у, z и времени t носит название волновой
функции (или пси-функции).
Волновая функция названа так потому, что в ней
находит выражение волновая природа движения
электрона. Например, волновая функция свободного
электрона, обладающего определенным значением
импульса и энергии, отображает бегущую плоскую
волну с заданными значениями длины волны X и
частоты v:
ф (г, t) = Ae2!Xi(r/K-v‘>
выражая ). и v через импульс р и энергию Е с помощью
соотношений де-Бройля, можно волновую функцию
свободного электрона (с определенным импульсом)
записать так:
ф(г, t)=Aei/h-(pr-Et) (4)
Бегущая плоская волна безгранична, она не име-
ет ни начала, ни конца (рисунок 4). Интенсивность
такой ф-волны, изме-
ряемая квадратом ее
амплитуды, постоянна
во всем пространстве;
она не зависит от ко-
ординаты. Но интен-
сивность ф-волны в
каждой точке (как это
было показано выше,
при анализе корпуску-
лярно-волновой при-
Положение волны
в момент времени t
Положение волны в момент
времени t,=t
Рис. 4. ^’-функция свободного элек-
трона, обладающего определенным
значением импульса.
роды микрочастиц) характеризует вероятность обнару-
жить электрон вблизи этой точки. Значит, свободный
электрон в данном состоянии (с определенным значе-
нием импульса) можно с равной вероятностью обна-
ружить в любой точке пространства вдоль оси х.
В действительности, представление об электроне
с совершенно неопределенным местоположением
крайне абстрактно; на опыте всегда имеют дело с элек-
тронами, локализованными в каждый момент в нек-рой
ограниченной области. Поэтому состояние свободного
электрона более точно отображается другой ф-функ-
цией, носящей название волнового пакета
(рис. 5, а). Волновой пакет движется со скоростью v,
Рис. 5. Волновой пакет: а — зависимость амплитуды ф-
фуикции от х; б — кривая интенсивности (ф2) волнового
пакета; в — вероятности локализации электрона в различ-
ных точках пространства, занимаемого волновым пакетом
(вероятность пропорциональна плотности точек); г — рас-
пределение вероятностей значений импульса электрона,
состояние к-рого описывается волновым пакетом.
равной скорости электрона. Амплитуда волнового
пакета отлична от нуля в нек-рой конечной области
между точками х0 -|- Дх/2 и х0 — Дх/2; интенсивность
этой ф-волны показана на рис. 5, б. Это значит, что
вероятность обнаружить электрон отлична от нуля
только для значений координаты, расположенной
между хй — Дх/2 и т0 4- Дгг/2, причем максимум ве-
511
КВАНТОВАЯ МЕХАНИКА
512
роятпости падает на значение хп (рис. 5, в).«Волновой
пакет» так назван потому, что он может быть получен
наложением друг на друга совокупности («пакета»)
плоских волн с несколько различающейся длиной
волны, значения к-рой лежат между — ДЛ/2, и
Ло ДХ/2. В К. м. это означает, что значение импульса
электрона, локализованного в области волнового
пакета, неопределенно; при измерении значения им-
пульса оказываются между. р0 — \р/2 и р0 -|- Др/2
(рис. 5, г), причем величина разброса значений им-
пульса Др тем больше, чем меньше Дх (чем уже вол-
новой пакет), и наоборот, — чем больше Дх (чем шире
волновой пакет), тем меньше Др.
Волновая функция электрона в атоме водорода,
находящегося в s-состоянии с квантовым числом
п = 1 (см. Атом), в координатной форме (не содер-
жащей зависимости от времени) записывается так:
ф(х, у, z) = Ae~r/a (5)
где г — расстояние электрона от центра атома, а —
постоянная, равная наиболее вероятному расстоянию
электрона от ядра — т. наз. воровскому pa-
fl и у с у. Это — уравнение стоячей волны с ампли-
тудой, зависящей от координат. Атомарный электрон
в данном состоянии может быть обнаружен (локали-
зован) с наибольшей вероятностью вблизи воровской
орбиты. Однако оп может быть обнаружен (хотя и
с меньшей вероятностью) и вне этой орбиты.
Таким образом, волновая функция ф(х, у, z, (),
в отличие от обычной волны, дает статистич. картину
поведения электрона. Интенсивность ф-волны в каж-
дой точке, выражаемая квадратом амплитуды ф-функ-
ции в малой области пространства dx = dxdydz
(точнее, квадратом модуля, или абс. значения |ф|Мт),
характеризует вероятность локализа-
ции электрона вблизи этой точки в области dx.
Величину |ф|2, являющуюся отношением вероятности
к величине области локализации, называют п л о'т-
ностью вероятности в данной точке. Если
взять квадрат амплитуды ф-функции электрона в ато-
ме, умноженный на — объем бесконечно тонкого
слоя на расстоянии г от ядра, — то мы получим для
данного состояния распределение вероятностей ло-
кализации электрона в атоме на разных расстояниях
от центра (рис. 6, а); сами же значения ф2 в разных
алектрона в атоме водорода (в низшем 8-состоянии) в за-
висимости от г (а-боровский радиус); б — зависимость
плотности вероятности (фг) от г; в — электронное облако
электрона в этом состоянии.
точках атома дают плотность вероятности локализа-
ции электрона вблизи этих точек (рис. 6, б и 6, в).
Амплитуду ф-функции можно умножить на любое
число, так как существенно лишь отношение ампли-
туд. Обычно амплитуду ф-функции выбирают так,
чтобы она была нормирована на 1, т. е. ^фМт = 1,
иначе говоря так, чтобы сумма всех вероятностей
локализации электрона в к.-л. точке пространства
была равна 1 (такая нормировка возможна, если ин-
теграл не расходится).
Таким образом, волновая функция характеризует
состояние микрообъекта заданием вероятностей всех
возможных, значений его координат (вообще любой
динамич. величины). Она дает полную информацию
о возможных значениях любых динамич. величин в
данном состоянии. В частности, зная волновую функ-
цию ф{ данного состояния электрона,можно опреде-
лить среднее значение любой динамической величи-
ны £ в данном состоянии; именно, среднее значение
£ = ^ф? £ф^т, где ф* — комплексно сопряженная ф4.
Необходимо подчеркнуть еще раз, что микрочастица
в данном состоянии не обладает определенными зна-
чениями всех динамич. величин, подобно макрочастице.
Электрон, связанный в атоме, вовсе не имеет в каждый
момент определенное значение координаты или им-
пульса. Если бы атомарный электрон в каждый мо-
мент имел бы определенные координату и импульс,
то он двигался бы по определенной траектории и кван-
тование энергии и момента (см. ниже) было бы необъяс-
нимо, ибо непонятно, почему одни траектории атомар-
ного электрона «разрешены», а другие «запрещены»;
это и было необъяснимой загадкой в теории атома
Бора. Согласно К. м., электрон в атоме приобретает
те или иные возможные значения координаты только
при измерении, при зондировании атома жесткими
рентгеновскими лучами или быстрыми электронами,
иначе говоря, при его взаимодействии (столкновении)
с жестким фотоном или быстрым электроном; при
этом столкновении он локализуется в небольшой
области атома вблизи нек-рой точки. В результате
такого столкновения электрон выбрасывается из
атома; следовательно, измерение координаты вообще
уничтожает связанное состояние атомарного элек-
трона. Но, повторив много раз этот опыт с атомами,
находящимися в совершенно одинаковых условиях,
мы получаем распределение возможных значений
мест локализации электрона в атоме, т. е. вероятности
различных значений его координаты; такая совокуп-
ность опытов дает статистику мест локализации элек-
трона в атоме, — как говорят, вид электрон-
ного облака, к-рое и описывается ф-функцией
данного состояния, — точнее, квадратом ее модуля;
наглядно говоря, электронное облако характеризует
распределение «электронной плотности» в атоме.
Опыты по зондированию электронного облака атома
(или распределения его эффективного заряда, равного
еф2, где е — заряд электрона) действительно пред-
принимались и подтвердили правильность теоретнч.
расчетов, произведенных на основе К. м.
Из сказанного следует; что процесс измерения в
К. м. обладает специфич. особеннестями. Если измеряе-
мая величина имеет в данном состоянии микрочасти-
цы определенное значение, то ее можно измерить,
не нарушая зто состояние. Если же измеряемая вели-
чина не имеет в данном состоянии определенного
значения, то в процессе ее измерения состояние микро-
частицы с определенной вероятностью преобразуется
в одно из таких состояний, в к-ром данная величина
имеет определенное значение. Напр., процесс изме-
рения импульса электрона, связанного в атоме, пре-
образует данное связанное состояние в одно из сво-
бодных (с определенным импульсом).
Следует отметить нек-рые особенности •ф-функции. Если
одно и то же состояние микрочастицы может быть описано
двумя (или несколькими) волновыми функциями ф, и ф2, то
оно может быть описано и их суммой (или разностью), т. е.
ф = ф, ± ф2. Это свойство в К. м. называется принципом
суперпозиции. Он играет важную роль в теории квантовых
систем. Зная амплитуду и фазу волновой функции в коорди-
натном представлении ф (х, у, z, (), можно путем преобразо-
вания ф-функции получить ее выражение в других предста-
влениях, напр. как функцию от импульса.
513
КВАНТОВАЯ МЕХАНИКА
514
Состояние квантовой системы не всегда может быть оха-
рактеризовано волновой функцией. Например, если внешние
условия, в к-рых-.существует система, сами меняются беспо-
рядочно, то состояние характеризуется более сложным обра-
зом (с помощью т. наз. матрицы плотности). Однако для выяс-
нения основных черт микрообъектов достаточно рассмотреть
т. наз. чистое состояние микрочастицы, описываемое волновой
функцией.
То обстоятельство, что состояние микрочастицы
характеризуется волновой функцией, относящейся
к конечной области пространства, влечет за собой
еще одну существенную особенность квантовых со-
стояний. Именно в К. м. стационарные связанные
состояния (напр., состояния электрона, связанного
с определенным атомом или молекулой) кванто-
ваны, их энергии образуют определенную дискрет-
ную (прерывную) последовательность, характерную
для данных внешних условий, в к-рых находится
электрон. В данном (неизменяющемся) внешнем поле,
в к-ром движется электрон, при определенных взаимо-
действиях его с другими частицами, связанный элек-
трон может находиться не в любых состояниях, а
только в нек-рых, характеризующихся определенными
значениями квантовых чисел. При изменении внеш-
них условий (напр., внешнего поля) изменяется вся
совокупность «дозволенных» состояний. Иначе об-
стоит дело со свободным электроном, не принадлежа-
щим определенной системе (определенному атому или
молекуле). Его состояния образуют непрерывную
совокупность, они могут различаться друг от друга
сколь угодно мало.
Уравнение Шредингера. Операторы. Основная за-
дача К. м. заключается в том, чтобы определить все
возможные состояния микрочастицы, находящейся
в данных физич. условиях, подвергающейся действию
заданного внешнего поля. Необходимо найти вид
ф-функций, характеризующих эти состояния, и опреде-
лить возможные значения динамич. величин в каждом
состоянии. Особый интерес представляет определение
стационарных состояний и нахождение соответствую-
щих им значений полной энергии для электрона,
связанного в атоме, молекуле или твердом теле
(кристалле).
Чтобы решить эту задачу, необходимо прежде
всего установить основное дифференциальное волновое
ур-ние К. м., играющее такую же роль в К. м., ка-
кую играет 2-й закон Ньютона (и выражающее его
дифференциальное ур-ние механики) в классич. тео-
рии. Это ур-ние К. м. не может быть выведено фор-
мально; оно было найдено Э. Шредингером на основе
аналогии с нек-рыми соотношениями классич. меха-
ники и называется по его имени.
Основное ур-ние К. м. — ур-ние Шредингера для
ф-функции — имеет такой общий вид;
_4 Эф ft2 / Э2ф . 0£ф‘ . 02ф\ । tj .,
Л Ot 2т \Эх2 ЙУ2 Sz2 ) + и * ( '
Оно связывает изменения ф-функций во времени (пер-
вую производную ф по времени) и в пространстве
(вторые производные ф по координатам) с внешними
взаимодействиями микрочастицы, обычно с потен-
циальной энергией и частицы во внешнем поле. Од-
нако U может содержать, кроме потенциальной энер-
гии, также члены, характеризующие и др. взаимодей-
ствия микрочастицы.
Для стационарных состояний (с постоянными зна-
чениями энергии Е) ур-ние Шредингера приобретает
более простой вид;
_ _L 021 L -U = Еж (7)
2m 0x2 п ду2 т QZ2 ) П- Y т \ /
Решая ур-ние Шредингера, находят вид ф-функций,
характеризующих возможные состояния микроча-
стицы в данных условиях, и соответствующие им зна-
чения энергии.
9 К. X. Э. т. 2
Решения ур-ния Шредингера, т. е. возможные
ф-функции, должны удовлетворять определенным
(т. наз. естественным) условиям, а именно: ф-функ-
ция должна быть конечной, однозначной и непрерыв-
ной во всем пространстве. Только ф-функции, удо-
влетворяющие этим требованиям, могут описывать
реальные состояния микрочастицы. Это следует из
самого смысла ф-функции, о к-ром говорилось выше.
Для того чтобы рассчитать распределение какой-либо
физич. величины, соответствующее данной ф-функции, кад
последней производят определенную последовательность дей-
ствий (Операций), характерную именно для данной величины;
такая последовательность операций наз. оператором данной
величины и обозначается соответствующей буквой. В К. м.
применяется математик, аппарат линейных операторов, ис-
пользуемый в теории колебаний. Этот математик, аппарат
находится в соответствии с физич. содержанием К. м., в част-
ности со спецификой квантово-механич. измерения (см. выше).
Если в состоянии, характеризуемом данной ф-функцией,
рассматриваемая физич. величина имеет определенное значе-
ние, то, подействовав на ф-функцию соответствующим этой
величине оператором L (т. е. произведя над ней определенную
последовательность действий), мы получим ту же ф-функцию,
но умноженную на нек-рое число это число и дает зна-
чение искомой величины в данном состоянии. Это можно за-
писать так: Ьф = ?.;ф.
Такие ф-функции наз. собственными функциями опера-
тора L, а полученные числа \ — собственными значениями
оператора L. Это соответствует тому факту, что измерение
величины, имеющей определенное значение в данном состоя-
нии микрочастицы, возможно без изменения этого состояния.
Как уже было сказано, состояние, в к-ром импульс имеет
определенное значение Pi(cocлагающимир1Ж> piy, pj2). описы-
вается ф-функцией бегущей плоской волны;
, K^PlXx + PlVv+Pi^~Elt
фр(х,У,г,г) = Аеп 1 (4)
Обозначим оператор вектора импульса буквой Р, а его сла-
гающие через Рж, Ру, Р2 Подействовав этим оператором на
ф-функцию (4), мы должны получить ту же функцию, умно-
женную на число, равное значению импульса pt в этом со-
стоянии; напр., для слагающей импульса по оси х ; Рж фр(х,
у, z, () = р1Ж фр (х, у, z, ()• Очевидно, последовательность опе-
рации над указанной выше ф-функцией (4), к-рая даст нужный
результат, состоит в том, чтобы взять частную производную
Л
по координате х и умножить ее на т. е.
„ ... д
Рх=1'1^ <8’
Это и есть явный вид оператора слагающей импульса. Таким же
образом получается и вид операторов для других составляю-
щих импульса, т. е.
(8’> Р^Л(£ (8">
Из этой же формулы следует, что оператор энергии Н имеет
вид:
О)
Наоборот, зная вид оператора импульса Р и подействовав им
на неизвестную еще ф-функцию, можно иайти вид ф-функции
состояния с определенным импульсом. Для этого нужно ре-
шить соответствующее дифференциальное ур-ние. Решением
этого дифференциального ур-ния и является (4).
Если в состоянии, описываемом ф-функцией, импульс не
имеет определенного значения, то с помощью оператора им-
пульса можно найти среднее значение импульса в этом состоя-
нии, а именно:
4-оо
р = J ф* (эс, у» 2) Рф (х, у, z) dxdydz (10)
— оо
Оператором Q координаты является простое умножение
ф-функции на эту координату.
Последовательное применение двух операторов наз. их
произведением. Операторам присуща важная особенность,
к-рая не имеет места для обычных чисел. Вообще говоря, они
не подчиняются принципу коммутативности: последовательное
применение двух операторов М и N, т. е. произведение опера-
торов MN не равно NM; для операторов существенно, в каком
порядке они применяются. Если MN NM, то эти операторы
паз. некоммутирующими; если MN = NM,* то они-коммути-
руют друг с другом. В частности, оператор импульса Р и
оператор соответствующей координаты Q не коммутируют:
PQ — QP«A
(11)
515
КВАНТОВАЯ МЕХАНИКА
516
Это соотношение наз. перестановочным. В некомму-
тивности операторов координаты и импульса находят отра-
жение соотношения неопределенностей. Именно, если опера-
торы коммутируют, то соответствующие им физич. величины
могут иметь одновременно определенное значение; если опе-
раторы не коммутируют, то соответствующие величины не
могут одновременно иметь определенное значение.
Наибольший интерес представляют операторы момента и
энергии. Оказывается, что для определения их связи с опера-
торами импульса и координаты необходимо поступать по ана-
логии с классич. механикой. Как известно, момент количества
движения связан с импульсом и координатой так: М = [г/»];
в соответствии с этим
или
M = [QP]
(12)
Можно показать, что определенные значения в связанном
.состоянии имеют только операторы момента и одной из его
составляющих; другие же две составляющие оператора момента
в данном состоянии не имеют
определенного значения.
Таким же образом находят вид
оператора энергии. В классич.
механике полная энергия Е ча-
стицы массы т связана с ее им-
пульсом и координатами след,
образом:
£=^+г7(х>у>2)
Первый член представляет собой
кинетическую энергию, второй —
потенциальную энергию частицы
во внешнем поле. В К. м. опера-
тор энергии, называемый г а-
мильтонианом, связан с
операторами импульса и коорди-
наты таким же образом:
H = P-«/2m+U(Q) = -^(-0-s- +
Я2 Л2 \
+ ajr + aS-) + и (ж> у> 4 (13)
Оператор энергии может включать, кроме потенциальной
энергии, и члены, характеризующие другие типы взаимодей-
ствия.
Подействовав данным оператором'энергии, или гамильтониа-
ном, на ф-функцию, описывающую состояние электрона в атоме
или молекуле, мы получим собственное значение энергии в этом
состоянии: Нф = Е{ф или в развернутом виде:
Л2 /с)2ф , С)2ф , 02ф\
~ 2т \dxi~ + dtf + ) + U (Ж’ У> ~ Ei *
то есть уравнение Шредингера для стационарных состояний
квантовых систем.
Стационарные связанные состояния. Связанные со-
стояния электрона, принадлежащего определенному
атому или молекуле, представляют особый интерес
для физики и химии. Такие состояния имеют место,
когда энергия электрона отрицательна (т. е. когда его
средняя кинетич. энергия меньше средней потен-
циальной; при этом потенциальная энергия электрона
на бесконечности полагается равной нулю). Как уже
было отмечено, совокупность связанных состоянии,
в к-рых может находиться электрон в заданном поле,
образует прерывный ряд; они квантованы. Энергия
этих состояний может принимать лишь определенные
значения (дискретные уровни энергии); ди-
скретными являются также и возможные значения
момента количества движения и одной из его проек-
ций. При переходе системы из одного состояния
в другое квантовые числа изменяются скачком.
Нахождение решений ур-ния Шредингера для таких
состояний позволяет определить возможные значения
энергии, форму электронного облака, а на этой ос-
нове — физич. свойства микросистем.
Простейшим (и очень важным) случаем связанного
состояния микрочастицы является состояние, в к-ром
частица массы т совершает малые колебания около
состояния равновесия, происходящие под действием
квазиупругой силы, — так наз. линейный гармо-
нический осциллятор. Примером осцил-
лятора могут служить малые колебания атомов в двух-
атомной молекуле. Интересно сопоставить поведение
гармонии, осциллятора в классич. механике ив К. м.,
т. к. это дает возможность вскрыть ряд специфич.
свойств квантовых процессов.
На рис. 7, а показана зависимость потенциальной энергии
U (х) осциллятора от смещения частицы из положения равно-
весия х : U (х) — (со — круговая частота колебаний).
Классич. осциллятор может совершать колебания с любой
энергией Е; его энергия может равняться и нулю, т. е. он мо-
жет и покоиться в состоянии равновесия (вточке х = 0).
Очевидно, колеблющаяся частица может находиться только
в таких точках, для к-рых потенциальная энергия U меньше
ее полной энергии Е; кривая OU определяет пределы возмож-
ных значений х, т. к. при выходе частицы за эти пределы (об-
ласть II на рис.) энергия осциллятора оказалась бы меньше
потенциальной энергии, что невозможно (в этом случае ки-
нетич. энергия стала бы отрицательной, а скорость частицы —
мнимой). Область II, в к-рую не может проникнуть частица,
ограничена кривой OU\ эта область наз. потенциаль-
Рис. 7. Гармонический осциллятор: а — сопоставление состояний осциллятора в квантовой
и классич. механике (согласование ф-функции внутри потенциальной ямы и вне ее возмож-
но только для функции, изображенной сплошной линией, но невозможно для ф-функции,
изображенной пунктиром); б — ход ф2 при малых (п = 0,1,2) и больших (п = 10) кванто-
вых числах. Штриховые линии изображают распределение вероятностей (р ) обнаружения
классической частицы в разных точках х в состояниях с такой же энергией.
ным барьером. Область I наз. потен и и а л ь н о й
ямой. Таким образом, движение колеблющейся макроча-
стицы может совершаться вдоль любой прямой PQ (т. е. с лю-
бой энергией). Если определить вероятность местонахождения
колеблющейся макрочастицы в разных точках (по длительности
ее пребывания вблизи каждой точки), то распределение ве-
роятностей имеет вид, изображенный на рис. 7, б пунктирными
линиями. Из этих кривых видно, что наиболее велика вероят-
ность обнаружить колеблющуюся макрочастицу вблизи точек
поворота, где скорость ее равна нулю.
Совершенно иная картина получается в К. м. Решения
ур-ния Шредингера для этой задачи представляют собой
дискретную последовательность ф-функций; микрочастица
может находиться не в любых стационарных состояниях, а
лишь в таких, энергия которых квантована и принимает зна-
чения;
Еп=йю(п + 1/2) (14)
где п может быть любым целым положительным числом, вклю-
чая нуль (т. с. п равно 0, 1, 2, ...). На рис. 7, б показаны
ф-функции (точнее, ф2) для нескольких «дозволенных» состоя-
ний (для п — 0,1, 2, 10). Из рисунка видно, что распределение
вероятностей локализации микрочастицы резко отличается
от распределения вероятности местонахождения классич. ча-
стицы; напр., в состоянии с п = О наибольшую вероятность
имеет локализация колеблющейся микрочастицы вблизи по-
ложения равновесия (х — 0). Только для больших чисел п
распределение вероятностей приближается к классич. распре-
делению (напр., для п = 10). Следует обратить внимание на
то, что даже в низшем энергетич. состоянии, при п = 0,
энергия квантового осциллятора равна не нулю, a Vs Лео;
это — т. наз. нулевая энергия. Ес значение находится
в согласии с соотношением неопределенности (2).
Энергия квантового осциллятора, следовательно, при лю-
бом переходе от одного стационарного состояния в другое
изменяется на одну и ту же величину Лео. Как доказывается
в К. м., возможны только такие переходы осциллятора, для
к-рых п изменяется на ± 1. Таковы особенности простейшей
квантовой системы — осциллятора.
В классич. физике также известны процессы с прерывно
изменяющимися частотами — это стоячие волны в ограничен-
ных средах (струне, мембране и т. п.) (рис. 8). Там прерыв-
ность частот обусловлена наличием жестких границ, в к-рых
могут совершаться лишь нек-рые стационарные колебательные
процессы; можно сказать, что в таких системах дозволенные
517
КВАНТОВАЯ МЕХАНИКА
518
стационарные колебания «согласованы» с границами области.
В квантовых системах (в осцилляторе, атоме, молекуле и т. д.)
дискретность ряда возможных состояний обусловлена тем, что
электрон в своем движении в каждый момент испытывает воз-
действие поля не в одной точке (по-
Рис. 8. Стоячие волны
на струне.
скольку он не движется по траекто-
рии), а во всей области системы (атома
или молекулы); можно сказать, что
состояние связанного электрона в каж-
дый момент как бы согласовано с хо-
дом потенциала поля в атоме (моле-
куле) в целом. Математически ди-
скретность решений ур-ний Шрединге-
ра обусловлена тем, что ф-функцин,
к-рая должна быть однозначной, ко-
нечной и непрерывной во всем про-
странстве, вместе с тем подчиняется
определенным «граничным» условиям:
за пределами потенциальной ямы ф
быстро спадает до нуля. Согласование
хода ф-фуниции внутри потенциаль-
ной ямы и за ее пределами возможно
лишь для дискретной последователь-
ности волновых функций (рис. 7, а). Т. обр., квантование со-
стояний связанного электрона представляет собой естествен-
ное следствие его двойственной природы. Напомним в связи
с этим, что в теории атома Бора квантование состояний было
чужеродным элементом.
Необходимо указать на две особенности стацио-
нарных квантовых состояний, неизвестные классич.
физике. Во-первых, наличие в квантовых системах
нулевой энергии — наименьшего значения энергии,
к-рой может обладать такая система (атом, молекула,
твердое тело); значение нулевой энергии характерно
для данной системы. Отнять эту энергию от системы
можно только разрушив ее. Наличие нулевой энергии
у атома объясняет факт особой устойчивости атома
как динамич. системы. Нулевая энергия атомов умень-
шается при ослаблении связи электрона с ядром и
возрастает при ее усилении. Наличие нулевой энергии
не сказывается на спектрах излучения, так как ча-
стоты спектральных линий определяются разностью
уровней энергии; однако реальность нулевой энер-
гии может быть доказана как непосредственными
экспериментами (напр., рассеянием рентгеновых лу-
чей в кристаллах при низких темп-рах), так и рядом
следствий, оправдывающихся на опыте.
Во-вторых, амплитуда ф-функции связанного со-
стояния отлична от нуля не только в пределах потен-
циального барьера, связывающего электрон, но и за
его пределами (см., напр., рис. 7). В классич. физике
локализация частицы за пределами барьера невоз-
можна, т. к. это противоречит закону сохранения
энергии. В К. м. это имеет место и не приводит к про-
тиворечию с законом сохранения энергии. Локали-
зация электрона за пределами потенциального барьера
обусловлена его волновой природой — электронная
волна «просачивается» на небольшое расстояние за
пределы барьера, постепенно затухая (подобно тому,
как световая волна «просачивается» через отражаю-
щую поверхность на расстояние порядка длины волны,
отражении). «Просачи-
вание» ф-волны за по-
тенциальный барьер
играет важную роль
в физике и химии.
Это явление высту-
пает с особенной на-
глядностью, если на
связанный электрон
(в атоме, молекуле,
кристалле) действует
даже при полном внутреннем
Рис. 9. ф-Функция электрона «про- также внешнее поле,
сачивающегося» через потенциаль- стремящееся «ВЫТЯ-
ный барьер. нуть» электрон из си-
стемы. В этом случае
потенциальный барьер имеет определенную высоту ОН
(рис. 9); с той стороны, где действует внешнее поле,
толщина барьера становится конечной. Однако даже
в том случае, когда энергия электрона остается
меньше высоты барьера, т. е. недостаточна, чтобы
преодолеть противодействие поля, препятствующего
его вылету, электрон не остается всегда связанным
с системой. Возникает конечная вероятность проса-
чивания его за барьер и вылета из системы; ф-волна
выходит за пределы барьера (рис. 9). Это явление,
наз. туннельным эффектом, свидетель-
ствует о волновой природе электрона. Туннельный
эффект лежит в основе многих явлений, напр. а-ра-
диоактивности; он сказывается при конденсации
атомов в твердое тело (кристалл); при сближении ато-
мов на их валентные электроны действует электрич.
поле соседних ядер и (если структура решетки этому
благоприятствует) эти электроны, перескакивая от
атома к атому, начинают мигрировать по всему объему
тела. Происходит делокализация внешних электро-
нов атомов.
Рассмотрим теперь подробнее различные типы ста-
ционарных состояний электрона в атоме, причем
ограничимся простейшим случаем водородоподобных
атомов. В отличие от осциллятора, стационарные
состояния атома характеризуются набором кванто-
вых чисел п, I, mi, определяющих значения энергии,
момента количества движения и проекции момента
на к.-л. направление; четвертое квантовое число ms
определяет значение проекции спина электрона на
это же направление (см. Атом). На рис. 10,а пока-
0 2 4 6 8а
0 5 10 15а г
0 6 12 18а
'Рис. 10. Вероятности локализации электрона в атоме во-
дорода в разных состояниях: а — ход ф2 для s-состояний
атома водорода (пунктир) и распределение вероятностей
локализации электрона в этих состояниях (сплошная
штриховка); б — ход ф2 для 2 р-, Зр- и 3 d-состояний
(пунктир) и распределения вероятностей локализации
электрона в этих состояниях (сплошная штриховка).
заны пунктиром электронные облака (ф2), а сплош-
ной штриховкой — распределения вероятностей лока-
лизации электрона на разных расстояниях г в s-со-
стояниях (при п = 1, 2, 3), в которых момент элек-
трона равен нулю. Электронные облака в s-состоя-
ниях сферически симметричны и отличны от нуля
в центре (ядре); они имеют п—1 узловые поверхности,
где ф2 равно нулю. Для них характерна «статичность»,
отсутствие в атоме орбитального тока и, соответст-
венно, магнитного момента. В других состояниях
орбитальный момент отличен от нуля. Как уже от-
мечалось (см. также Атом), значения момента кван-
тованы, они определяются орбитальным квантовым
числом I, могущим принимать значения 0, 1, 2...
п —1 (s-, р-, d-состояния и т. д.). Абс. величина мо-
мента Л/ = Z(Z-|-1). Квантуется также и проекция
момента на к.-л. направление, определяемое кван-
товым числом т, могущим принимать значения —I,
—Z-J-1, ...—1,0, -|-1, ..., I. Квадраты амплитуд
волновых функций и распределения вероятностей ло-
кализации для нек-рых р- и d-состояний показаны
на рис. 10,6. В состояниях с моментом, отличным от
нуля, электронное облако уже не обладает сферич.
симметрией; плотность облака различна в разных
направлениях и зависит от абс. значения т; ф2 равно
нулю в центре. Если проекция момента на нек-рое
направление z также отлична от нуля, то вокруг
этого направления возникает орбитальный электрон-
9»
519
КВАНТОВАЯ МЕХАНИКА
520
ный ток и атом приобретает магнитный момент р,
« еЛш
пропорциональный механическому: и = -----, где с —
^•те с
скорость света, те — масса электрона. Величина р
для т'= 1 является наименьшим магнитным моментом
и наз. магнетоном Бора; она равна 9 • 1О-21
единиц CGSM. Выделенное направление z определяется,
напр., внешним магнитным полем, действующим па
атом.
Важной особенностью состояний водородоподобных
атомов является их вырождение: уровни энер-
гии определяются только квантовым числом п и од-
ному и тому же уровню энергии соответствуют пг
различных ф-функций (при учете спина 2п2); 2п2 наз.
степенью, или кратностью, вырождения. При задан-
ном значении п энергия водородоподобного атома
не зависит от формы его электронного облака; напр.,
энергии низших p-состояний получаются равными
энергии 2$-состояния. Однако независимость энергии
от значений I (вырождение по Z) имеет место только
для водородоподобных атомов и обусловлена сферич.
симметрией кулоновского поля ядра. При отсутствии
подобной симметрии поля (напр., при движении
валентного электрона в поле остова более сложного
атома, когда происходит деформация остова и нару-
шение симметрии) вырождение по I исчезает, как
говорят, снимается; в этом случае состояниям с раз-
личными моментами соответствуют различные уровни
энергии. В действительности снятие вырождения
имеет место и для водородоподобных атомов; у них
наблюдается т. наз. тонкая структура уров-
ней: уровни энергии с заданными п (при п 2)
«расщеплены» — s-, р-, (/-состояниям соответствуют
несколько различные энергии. Это т. наз. естествен-
ное расщепление уровней обусловлено тем, что элек-
трон обладает собственным (неорбитальным) меха-
нич. моментом — спином — и соответственно сам по
себе является элементарным магнитиком; взаимодей-
ствие спина и орбитального движения и приводит
к указанному весьма малому различию энергии раз-
ных состояний.
Дальнейшее расщепление уровней энергии проис-
ходит во внешних полях. Уровни энергии расще-
пляются, если поместить атом во внешнее однородное
магнитное поле (см. Зеемана явление)-, при этом рас-
щепление является полным; различным состояниям
в магнитном поле соответствуют различные энергии
(вырождение снимается полностью).
Частичное расщепление уровней энергии атома
происходит и под воздействием постоянного внешнего
электрич. поля (т. наз. эффект Штарка). Это — спе-
цифически квантовый эффект; классич. физика не
могла объяснить это явление. Как показывает К. м.,
электрич. поле действует по-разному на электронные
облака в состояниях с различным абс. значением
квантового числа т;. Особенно сильно влияние
электрич. полей соседних атомов. Учет этого явления
существен при трактовке т. наз. валентных состоя-
ний атома (т. е. состояний атома, являющегося частью
молекулы).
Квантовые системы. До сих пор речь шла о движе-
нии одного электрона во внешнем поле. Между тем
все атомы, более сложные, чем атом водорода (начи-
ная с атома гелия), являются многоэлектронными
системами, — равно как и все молекулы. К. м. рас-
сматривает систему, состоящую из п частиц, как одну
частицу с 3 п степенями свободы (а с учетом спина —
с степенями свободы). Квантовая система подчи-
няется тем же законам сохранения (энергии, вмпульса,
момента), что и классич. система. Однако квантовая
система отличается от классической тем, что ее в общем
случае нельзя рассматривать как совокупность ча-
стиц, каждая из к-рых обладает индивидуальным
состоянием (своей ф-функцией). Строго говоря, ф-функ-
цию можно приписать только системе в целом. Эта
особенность квантовых систем резко выявляется при
рассмотрении совокупности одинаковых частиц, напр.
электронов. Поэтому многоэлектронные атомы, мо-
лекулы, твердые тела обладают новыми специфич.
квантовыми особенностями, не присущими отдельным
электронам.
Основой К. м. многих частиц является принцип
тождественности одинаковых микрочастиц.
Если в классич. статистич. физике перестановка мест
(координат) двух одинаковых частиц (или их энер-
гетич. состояний) считается реальным изменением,
то в К. м. два таких состояния считаются тождествен-
ными, т. е. одним и тем же состоянием. Напр., если
в молекуле водорода пара электронов меняется ме-
стами, или если в атоме гелия оба электрона обмени-
ваются своими состояниями, то К. м. не рассматривает
это как действительное изменение состояния; ф-функ-
ция системы остается той же (или меняет свой знак,
что не приводит к изменению вида ф2 — электронного
облака). Следовательно, в К. м. важно лишь число
электронов в каждом состоянии (т. наз. числа
заполнения), а не то, какие именно электроны
в них находятся.
Приближенно волновая функция системы частиц
может быть представлена в виде произведения вол-
новых функций отдельных частиц, находящихся в
своих индивидуальных (парциальных) состояниях.
Напр., для 2 частиц общая волновая функция си-
стемы ф (rb г2), где J’j и г2 обозначают совокупность
координат соответственно 1-й и 2-й частиц, м. б.
приближенно представлена как произведение волно-
вых функций ф4 (rt) и фй (г2) одночастичных состоя-
ний.
Ф(гг »’2) = Ф/ (»’1)ФЙ(»’2) (15)
Такой способ представления волновой функции
системы объясняется тем, что ф(Г1,г2) определяет
вероятность локализации обеих частиц, а ф;(гх) и
ф^(г2) — вероятности локализации отдельных ча-
стиц, вероятность же сложного события равна произ-
ведению вероятностей составляющих его простых со-
бытий.
Однако принцип тождественности требует, чтобы
общая ф-функция обоих электронов была выражена
через ф-функции отдельных электронов в виде суммы
(или разности) произведений, отличающихся переста-
новкой электронов, а именно:
ф (»\, Г2) = ф. (т\) фй (г2) ±: ф. (г2) ф& (rt) (16)
В самом деле, нетрудно убедиться, что перестановка
местами обоих электронов при таком представлении
приводит либо к неизменности выражения ф-функции
системы (в случае суммы произведений), либо к изме-
нению знака ф (в случае разности произведений).
В первом случае ф-функция системы наз. с и м-
м е т р и ч н о й, во втором — антисиммет-
ричной.
Существенным свойством квантовых систем является
то, что их состояния могут описываться либо симмет-
ричными, либо антиметричными ф-функциями. Суще-
ствуют два класса микрочастиц — частицы Бозе
и частицы Ферми. Число Бозе-частиц в ка-
ждом парциальном квантовом состоянии (характе-
ризуемом определенным набором квантовых чисел
для каждой частицы) может быть любым. К этому
классу микрочастиц относятся частицы с целочислен-
ным спином 0, 1, 2... (напр., ядра гелия — а-частицы,
фотоны и др.). Ферми-частицы подчиняются иному
закону распределения: в каждом квантовом состоянии
521
КВАНТОВАЯ МЕХАНИКА
522
для микрочастицы может находиться только одна
частица. Последнее положение наз. принципом
Паули. К Фермп частицам относятся микроча-
стицы с полуцелым спином, в том числе с половинным
(электроны, протоны, нейтроны, нейтрино). Следо-
вательно, в квантовой системе Ферми-частиц не может
быть 2 (или более) электронов, характеризуемых одной
и той же четверкой квантовых чисел; существует
корреляция между состояниями различных электро-
нов, принадлежащих к одной системе. Это определяет
особенности их статистики (см. Квантовая статистика').
Как доказывается в К. м., системы Ферми-частиц,
подчиняющиеся принципу Паули, могут находиться
только в состояниях, описываемых антисимметрич-
ными волновыми функциями; системы Бозе-частиц —
только в состояниях, описываемых симметричными
волновыми функциями. При этом ф-функции системы
и отдельных частиц следует рассматривать как зави-
сящие не только от координат, но и от спинов частиц.
Принцип тождественности одинаковых микрочастиц
и связанный с ним принцип Паули приводят к ряду
важнейших результатов в физике и химии. Принцип
Паули дает возможность понять сущность периодич.
закона Д. И. Менделеева (см. Атом). Здесь следует
остановиться подробнее на одном применении прин-
ципа тождественности — на теории ковалентной хи-
мия. связи, необъяснимой с классич. точки зрения.
Простейший пример ковалентной связи — соединение
двух атомов Н в молекулу. Как уже было отмечено,
волновая функция обоих электронов молекулы пред-
ставляет собой сумму или разность произведений
.одночастичных ф-функций от координат электронов
м от их спинов; она должна быть в целом антисиммет-
ричной (см. выше). При этом каждую ф-функцию
можно представить как произведение двух частей:
1-я часть зависит только от координат, 2-я только от
спинов. Если спины обоих электронов антипарал-
лельны, то спиновая часть волновой функции антисим-
метрична; следовательно, координатная часть ф-функ-
ции должна быть симметричной, она имеет вид:
ф (п, rs) = фв (щ) ф* (г2) 4- фа (га) ф* (rt)
Здесь фа и фь — волновые функции электрона, дви-
жущегося соответственно относительно ядра а или в,
т. е. атомарные волновые функции. Электронные об-
лака обоих электронов в данном случае накладываются
друг на друга (интерферируют) так, что они образуют
единое электронное облако молекулы, изображенное
Рис. И. К характеристике условий образования молеку-
лы Н2: а — ход тр (вверху) и общее электронное облако
молекулы Н2 (внизу) в случае антипараллельности спи-
нов обоих электронов; б — ход у3 (вверху) и электронное
облако обоих электронов (внизу) в случае параллельных
спинов.
на рис. И, а; плотность электронного облака оказы-
вается наибольшей между ядрами.
Внутренняя энергия молекулы Н2 складывается
из положительной энергии кулоновского отталкива-
ния обоих ядер и отталкивания электронов друг от
друга и отрицательной энергии притяжения электро-
нов к обоим ядрам. Вследствие наложения волновых
функций (электронных облаков) обоих электронов
в выражении энергии появляется дополнительный
член, обусловленный интерференцией (наложением)
волновых функций обоих электронов; этот член —
обменная энергия — в рассматриваемом
случае антипараллельных спинов отрицателен, он
характеризует т. наз. обменное взаимодействие элек-
тронов в молекуле (последние, локализуясь во всей
молекуле, как бы непрерывно обмениваются местами).
Обменное взаимодействие не является каким-то спе-
цифпч. полем, а характеризует действие обычного
электрич. (кулоновского) поля; оно, как отмечено
выше, появляется вследствие наложения друг на
друга волновых функций обоих электронов.
Если спины обоих электронов параллельны, то
спиновая часть ф-функции симметрична, а их коор-
динатная волновая функция антисимметрична; сум-
марное электронное облако имеет вид, изображенный
на рис. И, б. В данном случае электронная плотность
между ядрами уменьшается, что приводит ко все
большему возрастанию энергии отталкивания обоих
атомов при их сближении. Член обменной энергии
в данном случае положителен. Поэтому молекула Н.,
при параллельности спинов обоих электронов образо-
ваться не может.
Обменное взаимодействие существенно не только
для понимания природы химич. связи. Оно дает воз-
можность также объяснить уровни энергии много-
электронных атомов, природу ферромагнетизма и
много других явлений.
Квантовые переходы. При изменении внешнего
поля (внешних макроскопии, условий) со временем
состояние квантовой системы изменяется. Такое из*
менение следует отличать от того, к-рое испытывает
микросистема при измерении (т. е. при внешнем воз-
действии, совершающемся при сохранении внешнего
поля). При изменении внешнего поля вообще меняется
вся совокупность возможных состояний («набор»
ф-функций). Если известно начальное состояние
квантовой системы в какой-нибудь момент времени,
то в принципе с помощью ур-ния Шредингера можно
найти состояние системы в любой последующий мо-
мент. Однако осуществить решение этой задачи очень
трудно. В К. м. обычно находят решение для случая
слабых внешних переменных полей, в частности та-
ких переменных полей, к-рые действуют лишь в те-
чение определенного промежутка времени. При этом
применяются методы теории возмущений. Примером
может служить воздействие на квантовую систему
(атом, молекулу) электромагнитного излучения. Если
атом (молекула) подвергается воздействию слабого
кратковременного поля излучения, то «набор» воз-
можных стационарных состояний не изменяется, а
имеют место лишь переходы из одного состояния
в другое. К. м. дает возможность определить, какие
именно переходы возможны, и рассчитать их вероят-
ности.
Применения квантовой механики. Выше были из-
ложены основные положения т. наз. нерелятивйстскон
К. м., применимой к движению электронов сравни-
тельно небольшой энергии. При исследовании со-
стояний электронов большой энергии применимо
релятивистское обобщение К. м., данное П. Дираком
и объясняющее наличие у электрона спина, к-рый
из ур-ний нерелятивистской К. м. непосредственно
не получается; явления же, связанные с превраще-
ниями элементарных частиц, исследуются квантовой
теорией поля. Однако и нерелятивистская К. м. слу-
жит основой теории многих атомных и молекулярных
процессов. Этим определяется ее огромное значение
523
КВАНТОВАЯ СТАТИСТИКА
524
для раскрытия строения материи и объяснения ее
свойств.
К. м. позволила объяснить строение и свойства
атомов, атомные спектры, рассеяние света атомами.
На ее основе была создана теория молекулы и раскрыта
природа химической связи, разработана теория мо-
лекулярных спектров. К. м. дала возможность создать
теорию твердого тела, объясняющую его электрич.,
магнитные и оптич. свойства; с помощью К. м. уда-
лось понять природу металлич. состояния, полупро-
водников, ферромагнетизма и множества других явле-
ний. К. м. позволяет выяснить закономерности ядер-
ных реакций, радиационные свойства ядра, а-радио-
активность и т. д.
Познавательные возможности К. м. объясняются
отнюдь не тем, что ее ур-ния легко разрешимы для
всех перечисленных задач. Напротив, точное коли-
чественное решение ур-ния Шредингера даже для
атома возможно лишь для простейшей задачи —
для стационарных состояний атома с одним элек-
троном. В более сложных случаях применяются
различные приближенные методы: приближение
Томаса—Ферми — для атомов с большим числом
электронов, приближение Фока — Хартри (метод са-
мосогласованного поля) — для точного расчета уров-
ней энергии. В каждой области применения К. м.
разработаны свои приближенные методы (см. Кванто-
вая химия, Квантовая статистика). Эвристическое
значение К. м. очень велико и при полуколичествен-
ном рассмотрении различных явлений оно обусло-
влено более глубоким пониманием природы движения
и взаимодействия микроскопия, частиц материи,
раскрытием закономерностей микроявлений, необъяс-
нимых классич. механикой.
Однако К. м. не отгорожена абсолютно от классич.
механики. В тех случаях, когда можно пренебречь
атомизмом действия и положить h—>0, закономер-
ности К. м. переходят в закономерности классич.
механики.
Лит.: Гайтлер В., Элементарная квантовая механика,
пер. с англ., М., 1948; Веселов М. Д., Элементарная кван-
товая теория атомов и молекул, 2 изд., М.—Л., 1962; Бло-
хинцев Д. И., Основы квантовой механики, 3 изд., М.,
1961;Соколов А. А., ЛоскутовЮ. М., Тернов
И. М., Квантовая механика, М., 1962; Шифф Л., Квантовая
механика, пер. с англ., М., 1957; Бом Д., Квантовая теория,
пер. с англ., М., 1961; Ландау Л. Д. иЛифшицЕ. М.,
Квантовая механика, ч. 1, М.—Л., 1948 (Теоретич. физ., т. 5).
КВАНТОВАЯ СТАТИСТИКА — статистическая фи-
зика систем частиц, подчиняющихся законам кванто-
вой механики. Существенные особенности К. с. по
сравнению с классич. статистикой вытекают из осо-
бенностей квантово-механич. описания частиц (см.
Квантовая механика).
Создание К. с. было вызвано противоречиями, с
которыми столкнулась классич. статистика, оказав-
шаяся не в состоянии объяснить нек-рые физич. и
химич. свойства систем (представляющих собой со-
вокупность молекул, атомов, элементарных частиц),
поведение к-рых определяется законами квантовой
механики. Так, согласно классич. статистике, тепло-
емкость твердых тел является постоянной величиной
(см. Дюлонга и Пти закон), между тем опыт показы-
вает уменьшение теплоемкости до нуля при прибли-
жении темп-ры к абс. нулю. Резкое противоречие
с опытом получалось при применении методов классич.
статистики к излучению т. наз. абсолютно черного
тела. Полная энергия такого излучения получалась
бесконечно большой. К. с. разрешила эти, а также
другие противоречия классич. статистики (см. ниже).
Основное значение для К. с. имеют два положения
квантовой механики: дискретность энергетич. состоя-
ний системы и принцип неразличимости одинаковых
частиц. Из принципиальной неразличимости одина-
ковых частиц следует, что вероятность к.-л. конфи-
гурации системы (пропорциональная квадрату модуля
волновой функции ф (1, 2, ..., К) |2) не должна ме-
няться при перестановках частиц, т. е. квадрат модуля
волновой функции должен быть симметричным по
отношению к своим аргументам (1, 2, ..., 7V). Отсюда
для самой волновой функции ф(1, 2, ..., К) имеются
две возможности: она либо симметрична по отношению
к перестановкам координат частищ либо антисиммет-
рична (т. е. меняет знак при перестановках любой
пары частиц). Из уравнения Шредингера следует,
что это свойство должно сохраняться во времени.
О частицах, описываемых антисимметричными функ-
циями, говорят, как о подчиняющихся статисти-
ке Ферми— Дирака (или, короче, статистике
Ферми), а о частицах, описываемых симметричными
функциями, — как о подчиняющихся статисти-
ке Бозе — Эйнштейна (или, короче, стати-
стике Бозе). Статистика Бозе была предложена
Ш. Бозе (1924) для световых квантов, а затем обоб-
щена А. Эйнштейном (1924). Статистика Ферми была
предложена Э. Ферми (1925) для электронов, а ее
связь с квантовой механикой была выяснена П. Ди-
раком (1926). Паули теоретически обосновал получен-
ное на опыте правило, согласно к-рому частицы, об-
ладающие полуцелым спином, подчиняются стати-
стике Ферми, а частицы с целым спином — статистике
Бозе. Большинство элементарных частиц — электро-
ны, позитроны, протоны, нейтроны, ц-мезоны — под-
чиняются статистике Ферми. Фотоны, л-мезоны, а
также сложные частицы, состоящие из четного числа
элементарных частиц, подчиняются статистике Бозе.
Вероятность распределения системы по состояниям
в К. с. определяется так же, как в классич. статистике,
Гиббса распределением- Жп= exp [(/’—E^kT], где п—
номер состояния, Еп—значение энергии в этом состоя-
нии, к — Больцмана постоянная, Т — абс. темп-ра. Ве-
личина F является свободной энергией системы и
выражается через т. наз. сумму состояний
<2=^ехр(—Еп/кТ)е.лец. образом:77 = —кТInQ. Сумми-
п
рование в сумме состояний проводится по всем возмож-
ным состояниям системы. Следует указать на двойст-
венный характер усреднения при нахождении средних
значений величин в К. с. Оно включает в себя как
усреднение, связанное с вероятностным характером
квантового описания состояний отдельной частицы,
так и статистич. усреднение по системе частиц. Функ-
ция распределения в К. с. характеризует лишь вероят-
ность найти систему в нек-ром квантовом состоянии,
без всякого указания на значения координат и им-
пульсов частиц. В силу принципа неопределенности
Гайзенберга речь может идти лишь о распределении
вероятностей отдельно для координат и отдельно для
импульсов.
Характерные особенности К. с. отчетливо видны
при ее применении к простейшему объекту — идеаль-
ному газу (системе любых невзаимодействующих
частиц). Макроскопич. состояние идеального газа
может быть охарактеризовано числом частиц, находя-
щихся на различных энергетич. уровнях. Наиболее
вероятное распределение частиц по энергетич. уровням
(равновесное распределение) зависит от рода стати-
стики, к-рой описываются частицы. В случае спра-
ведливости статистики Бозе (Бозе-газ) в каждом кван-
товом состоянии может находиться любое число ча-
стиц. Статистич. вес макроскопич. состояния системы,
когда энергетич. уровню соответствует состояний
и в них находится я- частиц, равен числу способов,
к-рыми можно распределить ni частиц по ячейкам.
Это число равно:
ДГ4 = (gi -|- nz — 1) !/(g-f — 1)!n; I
525
КВАНТОВАЯ СТАТИСТИКА —КВАНТОВАЯ ХИМИЯ
526
Условие максимума энтропии системы, выраженной
через логарифм ДГ4, при дополнительных условиях
постоянства энергии и числа частиц приводит к след,
равновесному распределению, наз. распределением
Бозе:
пг = ^/(еа + ₽е>-1) (1)
где а =-----, и — химич. потенциал, Р = 1 )кТ.
В случае газа из частиц, подчиняющихся стати-
стике Ферми (Ферми-газ), должен удовлетворяться
Паули принцип, к-рый требует, чтобы в каждом со-
стоянии находилось не более одной частицы. Число
способов распределения ni частиц по g, состояниям
(не более чем по одной в каждом) есть число сочетаний
из g£ элементов по пг, т. е. ДГ4 = gt !/rei!(gi —- ге4)!
Так же, как и в статистике Бозе, отсюда выводится
распределение Ферми:
п; = £г/(е* + + 1) (2)
Различие между
статистиками нагляд-
но видно на примере
числа возможных рас-
пределений двух ча-
стиц по трем кванто-
вым состояниям (см.
схему). В классиче-
ской статистике части-
цы различимы, это от-
ражено в обозначении
их символами а и Ъ.
В К. с. ввиду нераз-
личимости частиц они
обозначены одним
символом а. В тех слу-
чаях, когда число состояний намного превышает число
частиц, то есть 1, квантовые распределения
(1—2) переходят в классич. закон распределения
Больцмана:
— (а 4- Зе.)
п. = в ' ~ к г
При заданной темп-ре условием применимости ста-
тистики Больцмана является достаточная разрежен-
ность газа. С понижением темп-ры становятся суще-
ственны квантовые эффекты и наступает явление
т. наз. вырождения. Критерий вырождения
кТ < —
m
(4)
где h — Планка постоянная, Лт — общее число частиц,
V — занимаемый объем и т — масса частицы. Как
следует из условия (4), вырождению способствуют
след, факторы: малая масса частиц, большая плот-
ность газа, низкая темп-ра. Для обычных газов
темп-ра вырождения (темп-ра, при к-рой проявляются
квантовые эффекты) очень низка, фактически газы при
этой темп-ре не существуют, т. к. переходят в твердую
фазу. Для электронного газа в металлах вследствие
малости массы электрона темп-ра вырождения, наобо-
рот, высока, порядка нескольких тысяч градусов.
Поэтому к электронному газу, как правило, применима
статистика Ферми.
Поскольку в статистике Ферми в одном состоянии
может находиться не более одной частицы, Ферми-газ
даже при Т = 0 обладает конечной энергией. Эта
энергия для JV частиц Ферми-газа отвечает заполнению
Л7/2 низших уровней системы (на каждом уровне могут
находиться два электрона с противоположными ори-
ентациями спинов). С повышением темп-ры часть ча-
стиц переходит на более высокие уровни. Для элек-
тронов в металле вплоть до очень высоких темп-р
число электронов, перешедших на более высокие
уровни, мало по сравнению с общим числом электро-
нов. Поэтому вклад теплоемкости электронного газа
в полную теплоемкость тела оказывается малым по
сравнению с вкладом от теплового движения атомов.
Применение К. с., т. обр., позволило естественным
путем объяснить отсутствие заметной теплоемкости
электронного газа, находившееся в противоречии
с выводами классич. статистики. В случае Бозе-газа
при понижении темп-ры до абс. нуля все частицы
должны перейти на уровень с нулевой энергией
(явление «конденсации Бозе—Эйнштейна»). Это об-
стоятельство является одним из отправных при объяс-
нении таких интересных явлений, как сверхтекучесть
и сверхпроводимость (в последнем случае электроны
образуют пары, подчиняющиеся статистике Бозе).
К. с. послужила основой для создания квантовой
теории твердых тел и электронной теории металлов.
Из положений К. с. непосредственно следует формула
Планка для распределения энергии излучения абсо-
лютно черного тела, полностью подтвердившаяся на
опыте. Из прочих областей приложения К. с. следует
назвать статистич. модель атома Томаса—Ферми, ста-
тистич. теорию ядра и др. Законы К. с. лежат в основе
статистич. термодинамики, позволяющей вычислять
термодинамич. характеристики химич. реакций.
Лит.: Ландау Л. Д., Лифшиц Е. М., Статистиче-
ская физика (классическая и квантовая), М., 1951 (Теоретич.
фиа., т. 4); Л е в и ч В. Г., Введение в статистическую физику,
2 изд., М.—Л., 1954; ГлесстонС., Теоретическая химия,
пер. с англ., М., 1950; Френкель Я. И., Статистическая
физика, М.—Л., 1948; Ферми Э., Молекулы и кристаллы,
пер. с нем., М., 1947; X и н ч и н А. Я., Математические осно-
вания квантовой статистики, М.—Л., 1951. И. Г. Каплан.
КВАНТОВАЯ ХИМИЯ — учение о строении и
физико-химич. свойствах молекул (а также других
химич. объектов исследования: ионов, радикалов,
комплексов), основанное на представлениях современ-
ной квантовой теории и, в частности, на применении
квантовой механики. К. х. охватывает учение о при-
роде химич. связи, о валентности, об электронной
структуре молекул, о спектроскопических (оптиче-
ских и радиочастотных), электрических и магнитных
свойствах молекул, о силах взаимодействия и реак-
ционной способности молекул. К. х. является погранич-
ной теоретич. дисциплиной на стыке физики и химии
и тесно смыкается с соответствующими разделами
физики (с теоретической атомной и молекулярной
физикой). Пределы применимости К. х. определяются
пределами применимости теоретического, расчетного
подхода к проблемам химии, а основой ее математич.
аппарата служит математич. аппарат квантовой ме-
ханики.
В принципе вся совокупность сведений о свойствах
молекул могла бы быть получена путем достаточно
точного решения уравнения Шредингера для соответ-
ствующих систем ядер и электронов. Однако в случае
сколько-нибудь сложных молекул это наталкивается
на практически непреодолимые математич. трудности,
связанные с необходимостью решения волнового
уравнения в многомерном пространстве при неразде-
ляющихся переменных (единственной молекулярной
задачей, где переменные в ур-нии Шредингера разде-
ляются, и ур-ние, таким образом, сводится к совокуп-
ности ур-ний, каждое из к-рых содержит лишь одну
независимую переменную, является одноэлектронный
молекулярный ион Н»). Это приводит к необходимости
использования в К. х. при рассмотрении электронной
структуры молекул приближенных расчетных мето-
дов, а в ряде случаев — полуколичественных или
качественно-описательных методов. При разработке
таких методов опираются не только на математич.
соображения, но и на фактич. материал химии.
В большинстве случаев для химии существенно ос-
новное состояние молекулы, т. е. стационарное состоя-
ние, обладающее наинизшей энергией. В К. х. ши-
527
КВАНТОВАЯ ХИМИЯ
528
роко используется замена уравнения Шредингера
соответствующим вариационным принципом, согласно
к-рому точное значение полной энергии Е основного
состояния молекулы равно минимуму математич.
ожидания энергии Ё (называемого также средней
энергией), вычисленного с любой нормированной
волновой функцией Ф: Е = min Ё, Ё = Ф*Н ФЛ,
где Н—оператор энергии системы (Ф* — функция,
комплексно сопряженная с Ф). ФункцияФ, при к-рой Ё
принимает наименьшее значение, является точной
волновой функцией основного состояния. В качестве
приближенного значения энергии можно использовать
Ё : Е^Е, если форма «пробной» функции Ф выбрана
так, чтобы при соответствующих минимуму Ё значе-
ниях параметров, входящих в Ф, эта функция доста-
точно близко воспроизводила истинную волновую
функцию системы. Всегда выполняется неравенство
Возможно распространение вариационного
принципа и на возбужденные состояния. Точность
расчета энергии, а также других характеристик
системы по вариационному методу повышается при
увеличении числа варьируемых параметров в функций
Ф, а при их достаточно большом числе и надлежащем
выборе Ф она может (в принципе) стать сколь угодно
высокой. Так, напр., с помощью вариационного
метода при различных способах выбора варьируемых
параметров произведено ок. 60 различных расчетов
основного состояния молекулы Н2. При усложнении
варьируемой волновой функции за счет роста числа
параметров наблюдается непрерывное сближение тео-
рии с опытом. Описанный ниже первый расчет Г. Гайт-
лера и Ф. Лондона дал для энергии диссоциации Н2
(без учета нулевой колебательной энергии ядер) зна-
чение D — 3,20 эв, вариационный расчет с 13 пара-
метрами дал результат D = 4,72 эв, а недавний рас-
чет с 50 параметрами, выполненный с помощью элек-
тронно-счетной машины, привел к значению D -=
= 4,746 эв, согласующемуся со спектроскопия, зна-
чением D — (4,7466 ± 0,0007) эв.
Вариационный принцип, отвечающий уравнению
Шредингера, применяется в двух наиболее распростра-
ненных методах приближенного расчета электронной
структуры молекул: методе валентных свя-
зей (электронных пар) и методе молеку-
лярных орбит (сокращенно М О). Последний
обычно используется в форме метода линей-
ных комбинаций атомных орбит
(Л КАО).
Как в методе валентных связей, так и в методе МО-
ЛКАО приближенные волновые функции молекул
конструируются по определенным правилам (своим
для каждого метода) из одноэлектронных атомных
волновых функций, т. н. атомных орбит, и могут со-
держать варьируемые параметры. Уточнение этих
методов стирает различие между ними, но в первона-
чальной простой форме они не совпадают.
Метод валентных связей является развитием под-
хода, К-рый был применен в 1927 Гайтнером и Лондо-
ном к первому квантово-механич. расчету молекулы
Н2, имевшему решающее значение для возникновения
и дальнейшего развития К. х. Этот расчет исходил из
электронных волновых функций системы, состоящей из
двух удаленных друг от друга атомов Н (атомов а
и Ъ), каждый из к-рых находится в основном состоя-
нии с энергией Ео. Существуют две простые волновые
функции, пригодные для описания такой системы
и удовлетворяющие требованию симметрии квадрата
волновой функции, вытекающему из физич. смысла
задачи: ф+ = ,\+[фа( 1)фь(2) + фа(2)фь(1)] и ф. =
=Аг_[%(1)Ф£,(2) — ^2)^(1)], где фа и ф4 — соответ-
ственно волновые функции электрона в основном
состоянии атома а или атома Ъ, а' цифры 1 или 2 обо-
значают совокупность пространственных координат
первого или второго электрона (величины х1, у1,
zr или х2, у„, z2). Коэфф. JV+n Л’._ обеспечивают норми-
ровку функций ф_. и ф„: = 1/ ]/2(1 + А2) ; =
= 1/]/2(1 —А2), где через А обозначен т. н. инте-
грал перекрывания волновых функций атомов а и Ъ:
S = ' гДе = dzi- Принцип
Паули утверждает, что полная волновая функция,
включающая наряду с пространственными и спиновые
координаты электронов, должна быть антисимметрична
по отношению к перестановкам электронов. Из этого
следует, что ф+ описывает т. н. синглетное электрон-
ное состояние, в к-ром спины электронов антипарал-
лельны, а функция ф — т. н. триплетное состояние,
в к-ром спины электронов параллельны. Функции
фи и ф суть точные решения ур-ния Шредингера
для двух электронов в поле двух протонов а и Ъ
только при бесконечно большом расстоянии R между
ядрами. Тем не менее эти функции позволяют вычис-
лить приближенные значения энергии низшего син-
глетного (Е_) и низшего триплетного (Е_) состояний
системы и при конечных значениях R. Такой расчет
дает; А+ = 2Еа + (С + А)/(1 + А2) и Е_ = 2Еа +
+ (С — А)/(1 — А2). Через С и А обозначены следую-
щие интегралы:
С = ( Фа О Г-д- + ~ -г—] Фь (2) dndr3
J J I ~12 г ц, г„ал
а -- «2 И Фа (1) Фь (1) [ 4 + -- - -Л ~ — ] Фа (2) Фь (2) d4d-.2
J J L к М2 Пв %аJ
Обозначения: е — заряд электрона; г12, г1д и л2Д —
соответственно расстояния: между первым и вторым
электронами; первым электроном и ядром Ъ; вторым
электроном и ядром а.
Интеграл С (т. н. кулоновский интеграл) представ-
ляет собой сумму следующих членов: энергии от-
талкивания протонов; энергии отталкивания элек-
трона, движущегося у ядра а, от другого электрона,
движущегося у ядра Ь; энергии притяжения электрона,
движущегося у ядра а, к ядру Ь; энергии притяжения
электрона, движущегося у ядра Ь, к ядру а. Инте-
грал А (т. н. обменный интеграл) не имеет такого
наглядного толкования. Его возникновение в вы-
ражении для энергии обязано тождественности элек-
тронов. При расстояниях R, близких к равновесному
междуядерному расстоянию Ra в Н2, имеет место:
С + А < 0; С — А > 0; < 2Е„-, Е_ > 2Е0. По-
следние два неравенства означают, что система из
двух атомов Н, электронные спины к-рых антипа-
раллельны, является энергетически более устойчивой,
чем система из двух изолированных атомов Н, и что
для параллельной ориентации электронных спинов
имеет место обратное. При сближении двух атомов Н
с антипараллельными спинами возникают силы при-
тяжения, а при их сближении с параллельными спи-
нами — силы отталкивания. Качественное различие
в характере сил, действующих между атомами Н в
синглетном и в триплетном состояниях, можно понять
из рассмотрения распределения пространственной
плотности вероятности обнаружения электрона (со-
кращенно—электронной плотности) в этих состояниях.
Волновая функция синглетного состояния ф+ соответ-
ствует повышению электронной плотности в области
между ядрами по сравнению с суммой электронных
плотностей двух независимых атомов Н. Такое повы-
шение электронной плотности приводит к возникно-
вению сил, стягивающих ядра (вплоть до равновес-
ного расстояния Ro, при к-ром силы притяжения урав-
новешиваются силами отталкивания ядер). В трип-
летном состоянии, наоборот, электронная плотность
529
КВАНТОВАЯ. ХИМИЯ
530
в области между ядрами понижена и кулоновское
отталкивание ядер преобладает над силами притя-
жения.
Расчет .молекулы Н2 послужил убедительным до-
казательством применимости квантовой механики
к проблеме химич. связи и показал, что силы, приводя-
щие к образованию химич. связи, имеют в своей основе
обычные силы электростатич. взаимодействия ядер
и электронов. Однако результат действия этих сил
существенным образом зависит от характера распреде-
ления плотности валентных электронов в области
между ядрами, к-рый, в свою очередь, в силу свойств
симметрии электронной волновой функции по отно-
шению к перестановкам пар электронов (принцип
Паули) определяется взаимной ориентацией спинов
валентных электронов.
В своем общем виде, разработанном Слейтером,
Полингом и др., метод валентных связей опирается
на результат расчета молекулы Н2, согласно к-рому
химич. связь образуется парой электронов с проти-
воположными спинами. В основном состоянии мо-
лекулы должно быть образовано максимально воз-
можное число химич. связей, поэтому при четном
числе электронов общий спин должен быть равен 0
(синглетное состояние). Приближенная волновая функ-
ция молекулы строится в виде линейной комбинации
волновых функций H'fc, соответствующих различным
возможным способам связывания атомных орбит
N
электронными парами: Ф = У] СкФк-
Полный набор линейно независимых функций
(не включающий ионных состояний) может быть
получен на основе теоремы Ю. Б. Румера: обозначе-
ния орбит а, & и т. д. надо расположить на замкнутой
выпуклой кривой (это расположение может не отве-
чать расположению ядер в молекуле) и провести
всеми возможными способами непересекающиеся пря-
мые между соединенными друг с другом орбитами.
Напр., при четырех электронах на четырех орбитах
а, Ь, с, d имеются две возможности образования элек-
а—Ь а b
тронных пар и | I . Каждой такой диаграмме,
d—с d с
называемой валентной структурой, отвечает функция
’Г^,, волновая функция к-рой записывается как линей-
ная комбинация произведений атомных орбит с уче-
том спаривания электронов и требований принципа
Паули. Весовые коэфф. ск (k = 1, 2, ..., А) при функ-
циях отдельных валентных схем определяются из
условия минимума математического ожидания энер-
гии. При составлении валентных структур (и соот-
ветствующих им функций ’Г|;) имеется возможность
учета возбужденных электронных состояний атомов,
переходов электронов между атомами с образованием
ионов и т. д.; точность приближенной волновой
функции Ф повышается при увеличении числа членов
в сумме. В практич. расчетах, однако, в целях упро-
щения ограничиваются по возможности небольшим
числом валентных структур, выбирая наиболее выгод-
ные энергетически. Так, напр., волновая функция ф+,
использованная в первом расчете молекулы Н2,
соответствует учету только одной валентной струк-
туры: На — Нь. Наряду с функцией ф+ можно рас-
сматривать функции, соответствующие ионным ва-
лентным схемам: На ...HS и На ... Нь, к-рые с точ-
ностью до нормировочных множителей будут иметь
вид: фа(1)фа(2) и -фь(1)'фь(2), и написать уточненную
приближенную волновую функцию основного син-
глетного состояния в виде Ф = С1ф++ с2[фа(1)фа(2) +
+ ф6(1)фь(2)]. Определение отношения коэфф,
из вариационного принципа для энергии приводит к
улучшенному по сравнению с первоначальным расче-
том Гайтлера и Лондона значению энергии диссоциа-
ции молекулы водорода: Т) = 4,02 эв.
Электронная структура большинства типов соеди-
нений качественно удовлетворительно описывается
с помощью лишь одной, основной валентной струк-
туры, а добавление в волновую функцию слагаемых,
отвечающих другим структурам, носит характер по-
правок (как и в случае молекулы Н2). Электронное
строение таких соединений с разумной степенью точ-
ности соответствует схеме электронных пар, распо-
ложенных либо в отдельных связях (пары связываю-
щих электронов), либо у отдельных атомов (т. н.
неподеленные электронные пары); их называют соеди-
нениями с локализованными связями; основная ва-
лентная структура отвечает при этом классич. струк-
турной формуле. В тех случаях, когда при данном
расположении атомов, образующих молекулу, воз-
можны две или более равноценные, или приближенно
равноценные энергетически, валентные структуры,
строение соединения не может быть представлено до-
статочно правильно одной определенной картиной
распределения валентных штрихов. При составлении
даже приближенной электронной волновой функции
такой молекулы с «делокализованными связями»
недостаточен учет только одного слагаемого в выра-
жении волновой функции, соответствующего одной
из валентных структур. Типичным примером является
молекула бензола, самая простая качественно пра-
вильная волновая функция к-рой в методе валентных
связей является суммой двух слагаемых, соответствую-
щих двум структурам Кекуле. Лучшая степень при-
ближения может быть достигнута введением в варьи-
руемую волновую функцию слагаемых, соответствую-
щих т. н. структурам Дьюара (в к-рых одна из связей
атомов углерода пересекает бензольное кольцо по
диагонали), ионным валентным схемам, валентным
схемам с возбужденными состояниями атомов и т. д.
Одно из достоинств метода валентных связей, с точки
зрения теоретич. химии, заключается в том, что этот
метод дает наиболее близкий квантово-механич. эк-
вивалент картины химич. связей по отношению к
классич. теории химич. строения. Вместе с тем он
указывает естественный путь расширения этой тео-
рии, позволяющий описать соединения с т. наз.
«делокализованными» связями, не укладывающиеся
в классяч. структурную схему с помощью представ-
ления о суперпозиции валентных структур (Л. По-
линг) .
Метод молекулярных орбит, развитый Р. С. Малли-
кеном, Э. Гюккелем и др. авторами, несколько дальше
отходит от классических химич. представлений, чем
метод валентных связей. Состояние к.-л. электрона
номер i в молекуле описывается волновой функцией
зависящей только от координат этого одного электро-
на — т. н. молекулярной орбитой. Волновая функция
молекулы в целом принимается, в простейшей форме
метода, равной произведению молекулярных орбит.
Принцип Паули учитывается тем, что на каждой
орбите помещается не более двух электронов, причем
два электрона на одной орбите имеют противополож-
ные спины. В основном состоянии молекулярные
орбиты заполняются последовательно электронами,
начиная с энергетически наиболее выгодной. Энергия
в простейшем варианте метода молекулярных орбит
вычисляется как сумма энергий отдельных электронов.
Приближение ЛКАО выражается равенством:
= (i> + cib*b (i> + cic*c <*> + •••
где фа, фь и т. д. — атомные орбиты, cia, cib и т. д. —
коэфф., к-рые находят с помощью вариационного
531
КВАНТОВАЯ ХИМИЯ
532
принципа или, в частных случаях, из соображений
симметрии.
Простейшей исходной задачей метода МО, играю-
щей в нем ту же роль, какую в методе валентных свя-
зей играет задача о молекуле Н2, является задача
о молекулярном ионе Н+. В приближении Л КАО из
орбит фа и соответствующих основному состоя-
нию атома водорода, могут быть получены 2 мо-
лекулярные орбиты, удовлетворяющие требованиям
симметрии: =^,(4,,+фь) и Гц = Лп(фо — фь),
где А^иАГц— нормирующие множители. Соответ-
ствующая энергия электрона дается равенствами
Я1 = Я0+(а+ р)/(1 + ^) и
где а — кулоновский интеграл: а = е
С / 1 11
Р — т. н. резонансный интеграл: р= е2 1фа%с?т
(здесь использованы те же обозначения, что и выше,
при рассмотрении молекулы Н2 по Гайт леру и Лон-
дону). Т. к. при значениях R, близких к равновес-
ному, в Hj а < 0 и Р < 0, а кроме того | р | > | а |,
то Ет < Ео, а £2 > Ео. Это означает, что лишь при
электроне на орбите I образуется стабильный ион
Н^, электрон на орбите II ведет не к притяжению,
а к отталкиванию ядер. Орбиты, обладающие свой-
ствами орбиты ITj, называются связывающими, а ор-
биты, обладающие свойствами орбиты Ф’л,—разрых-
ляющими.
Молекула Н2 в основном состоянии содержит два
электрона с противоположными спинами на связываю-
щей орбите, так что волновая функция молекулы
дается равенством
Г.МО = *1 (О + О)! 1Фа (2) + (2)]
Раскрывая скобки и сравнивая 'Гмо и ф+ метода Гайт-
лера и Лондона, убеждаемся, что '1'мо содержит наряду
с членами, являющимися слагаемыми ф+, также члены,
отвечающие ионным состояниям, причем все члены
имеют равный вес. В действительности, вес ионных
состояний меньше веса слагаемых ф+: в то время
как метод Гайтлера и Лондона полностью пре-
небрегает ионными членами, метод МО-ЛКАО, на-
оборот, переоценивает их значение.
В двухатомных молекулах с большим числом элек-
тронов часть электронов помещается на связывающих,
часть на разрыхляющих орбитах. Кратность связи
определяется избытком числа связывающих электро-
нов над числом разрыхляющих из расчета по два
электрона на одну связь. Напр., в молекуле N2
10 электронов на связывающих и 4_электрона на раз-
рыхляющих орбитах, кратность связи ~-=3,
в согласии с химич. ф-лой N=N. Можно также счи-
тать, что электроны замкнутых внутренних оболочек
(напр., К-оболочек атомов N в молекуле N2) находятся
на атомных орбитах и лишь валентные электроны —
на молекулярных орбитах. В случае многоатомных
молекул с локализованными связями каждая связь
может быть описана как образованная парой электро-
нов на молекулярной орбите, охватывающей два
атома. При делокализованных связях молекулярные
орбиты охватывают несколько атомов. Напр., в мо-
лекуле бензола 6 электронов двигаются по молеку-
лярным орбитам, каждая из к-рых охватывает все
6 атомов С, остальные электроны образуют локали-
зованные связи. Расчет энергии систем с большим
числом подвижных (делокализованных) электронов
выполняется по методу МО-ЛКАО проще, чем по
методу валентных связей.
В случае сложных молекул обычно не удается про-
извести точные расчеты всех интегралов, входящих
в выражение энергии молекулы. По этой причине
имеют большое прикладное значение полуэмпирич.
варианты метода валентных связей и метода МО,
в к-рых для определения значений части молекуляр-
ных интегралов используются опытные данные. Из
такого рода полуэмпирич. методов заслуживают вни-
мания метод «объединенного атома» и метод «атомов
в молекулах». Эти методы основаны на рассмотрении
непрерывной зависимости электронной энергии мо-
лекулы от расстояния между ядрами. Если все между-
ядерные расстояния в молекуле мысленно устремить
к нулю, то электронная оболочка молекулы переходит
в электронную оболочку т. н. объединенного атома,
заряд ядра к-рого равен сумме зарядов ядер атомов,
составляющих молекулу. В методе объединенного
атома электронная волновая функция молекулы раз-
лагается в ряд по взаимно ортогональным волновым
функциям различных состояний объединенного атома,
ядро к-рого мысленно помещается в центр тяжести
положительных зарядов ядер в молекуле. В расчете
энергии молекулы при определении значений ряда
интегралов используются спектроскопия, данные об
энергии термов объединенного атома. В методе «атомов
в молекулах» электронная волновая функция моле-
кулы разлагается в ряд по волновым функциям, опи-
сывающим различные состояния продуктов диссоци-
ации молекулы (атомов или ионов), а в расчете энер-
гии молекулы используются опытные значения энер-
гии этих продуктов. Привлечение экспериментальных
данных атомной спектроскопии позволяет в методе
«объединенного атома» и в методе «атомов в молекулах»
в значительной мере уменьшить ошибки, связанные
с неточностями в учете взаимной зависимости в дви-
жении различных электронов (т. н. эффектов элек-
тронной корреляции). Однако расчеты по этим мето-
дам могут привести к другим, трудно контролируемым
погрешностям, что является серьезным ограничением
их применимости.
В теории комплексных соединений переходных
элементов нашла широкое применение т. н. теория
поля лигандов, тесно связанная с квантово-механич.
теорией атомных спектров ионов-комплексообразова-
телей и с общей теорией симметрии (теорией групп).
В теории поля лигандов образование комплексного
соединения рассматривается как результат электро-
статич. взаимодействия между центральным ионом
переходного элемента и лигандами. Под действием
электростатического поля лигандов (моделируемого
обычно в виде поля точечных зарядов или точечных
диполей), обладающего кубической (или более низ-
кой) симметрией, происходит расщепление rf-уровней
центрального иона, к-рое вызывает стабилизацию
комплекса. Теория поля лигандов оказалась пригод-
ной для объяснения ряда закономерностей электрон-
ной структуры комплексных соединений, а также их
оптических и магнитных свойств. Для более точного
описания электронной структуры комплексных соеди-
нений чисто электростатич. теория поля лигандов
дополняется с учетом возможности образования в
известной мере ковалентных связей между централь-
ным ионом и лигандами; такая уточненная теория
использует представления о гибридизации волновых
функций центрального иона и представляет собой
синтез теории поля лигандов либо с методом валент-
ных схем, либо с методом молекулярных орбит.
Заслуживают внимания также модельные методы
К. х., в к-рых для описания электронной структуры
сложных молекул используются простые модели,
отражающие важнейшие особенности электронной
структуры реальных объектов. Типичным примером
такого рода является модель свободных электронов
для л-электронов в сопряженных и ароматич. угле-
водородах. В простейшем варианте этой модели
533
КВАНТОВЫЕ ЧИСЛА—КВЕРЦЕТИН
534
принимается, что полектроны свободно движутся
вдоль цепочки сопряженных связей, подобно свобод-
ным электронам в одномерном потенциальном ящике
длины L с гладким дном и с бесконечно высокими
стенками. Одномерные волновые функции и уровни
энергии электронов легко вычисляются:
, , , 1 /" 2 п-х „ n-x-h-
Tsin~T’ En=~^L
(n = 1, 2, ... —номер уровня энергии). Такая про-
стая и наглядная модель оказалась весьма полезной;
по широте области своих приложений (рассмотрение
электронных и колебательных спектров сопряженных
молекул, их электрич. и магнитных свойств, сил меж-
молекулярного взаимодействия и реакционной спо-
собности) и по качеству получаемых с ее помощью
результатов она не уступает методу ЛКАО.
Современное развитие К. х. характеризуется раз-
нообразием направлений. Широкое применение элек-
тронно-вычислительной техники позволяет произво-
дить все более точные расчеты электронной структуры
небольших (прежде всего, двухатомных) молекул,
приближая уровень точности теоретич. расчетов к
экспериментальному уровню. В случае сложных мо-
лекул и комплексных соединений ширится применение
полуэмпирич. расчетных методов, а также качест-
венно-описательных методов рассмотрения электрон-
ной структуры; наряду с различными областями фи-
зич. химии эти методы находят все более широкое
применение при исследовании электронных аспектов
биохимии. Развитие радиоспектроскопия, методов
исследования строения молекул сопровождается тео-
ретич. расчетами ряда тонких характеристик электрон-
ной структуры (расчетами магнитного экранирования
ядер, квадрупольной связи ядер с электронной обо-
лочкой, спин-спинового взаимодействия ядер через
электронную оболочку, распределения плотности не-
спаренного электронного спина в радикалах, взаимо-
действия электронного спина с ядерными спинами
и т. д.).
Основные представления и методы К. х. применя-
ются все более широкими кругами ученых-химиков
в разнообразных областях химич. и физико-химич.
исследований.
Лит.: Весело’в М. Г., Элементарная квантовая тео-
рия атомов и молекул, 2 изд., М., 1962; Гайт л ер В.,
Элементарная квантовая механика, пер. с англ., М., 1948;
Helt lerW., Elementary wave mechanics -with applications
to quantum chemistry, 2 ed., Oxf., 1956; Coulson Ch. A.,
Valence, Oxf., 1952; Козман У., Введение в квантовую
химию, пер. с англ., М., 1960; ЭйрингГ., У олтер Дж.,
Кимбалл Д ж., Квантовая химия, пер. с англ., М., 1948;
Preuss Н., Die Methoden der Molekhlphysik und ihre An-
wendungsbereiche, B., 1959; Coulson Ch. A., Revs. Mod.
Phys., 1960, 32, 170; Сыркин Я. К., Д я т к и н а М. Е.,
«Вести. АН СССР», 1959, № 6, 13; М с Le a n A. D., W е 1 s s А.,
Yoshimine М., Revs. Mod. Phys., 1960,32, №2, 211;
Kuhn H., Fortschr. der Chemie der organischen Naturstoffe,
1959, 17, 404. T. К. Ребане.
КВАНТОВЫЕ ЧИСЛА — см. Атом.
КВАРЦ — минерал, одна из кристаллич. модифи-
каций кремнезема SiO2. См. Кремния окислы.
КВАРЦЕВОЕ СТЕКЛО — см. Стекло кварцевое.
КВАСЦЫ — кристаллогидраты двойных сернокис-
лых солей общей ф-лы MeJSO4 • Me!ii(SO4)3 • 24Н2О
или Me!MeTn(SO4)2 • 12Н2О, где Me1 — одновалент-
Hbm(Na, К, Rb, Cs,NH4, Т1идр.), аМе 111 — трехвалент-
ный металл (Al, Ga, In, V, Сг, Мп, Fe, Со, Rh, 1г
и др.). Трехвалентные редкоземельные элементы К. не
образуют. К. относят к комплексным соединениям с
очень небольшой степенью устойчивости комплексных
ионов. Результаты рентгеновского анализа говорят
в пользу структуры [MeI(H2O)6](SO4)2[MeIII(H2O)e].
Все К. хорошо кристаллизуются в кубич. системе
и изоморфны, обладают вяжущим и кпслым вку-
сом (отсюда название «К.», данное в 15 в.). При
обычных условиях К. вполне устойчивы. На ус-
тойчивость К. сильнее влияет находящийся в их
составе одновалентный катион, чем трехвалентный.
В ряду Na, К, Rb, Cs устойчивость К. с одним и тем
же 3-валентным катионом повышается; нек-рые нат-
риевые К. неизвестны. При нагревании К. теряют
кристаллизационную воду и образуются т. наз.
жженые квасцы. В воде К. хорошо растворимы, причем
растворимость в ряду Na, К, Rb, Cs уменьшается;
влияние трехвалентных катионов и в этом случае
проявляется значительно слабее. В разб. водных
р-рах К. практически нацело распадаются на простые
ионы:
KA1(SO4)2^=K++AI’++2SO2-
К. могут быть получены смешением горячих водных
р-ров сернокислых солей одно- и трехвалентных ме-
таллов. Очистку К., выпадающих при остывании рас-
твора, производят перекристаллизацией. Наиболее
широко известные К.: алюмо-калиевые (алюминиевые)
K2SO4- A12(SO4)3- 24Н2О— бесцветные кристаллы;
хромово-калиевые (хромовые) K2SO4- Cr2(SO4)3 •
• 24Н2О — сине-фиолетовые кристаллы; железо-ам-
мониевые (железные) (NH4)2SO4- Fe2(SO4)3 • 24Н2О —
кристаллы светло-фиолетового цвета. Употребляют
преим. алюмо-калиевые К.: они применяются как
дубящее средство в кожевенной и фотопромышлен-
ности, в качестве протравы при крашении тканей;
в бумажной пром-сти для проклеивания бумаги, в
медицине как средство, оказывающее вяжущее, под-
сушивающее и дезинфицирующее действие на сли-
зистые оболочки и кожу (см. также Алюминия суль-
фат, Железа сульфаты, Хрома сульфаты').
Лит.: Позин М. Е., Технология минеральных солей,
2 изд., Л., 1961.
КВЁРЦЕТИН (3, 5, 7, 3', 4'-пентаоксифлавон) мол.
вес 302,24, С15Н10О7 — один из наиболее важных
и широко распространенных красителей группы фла-
вона. К. может быть выделен из коры Quercus tin-
ctoria, экстракт этой коры — кверцитрон — содержит
гликозид кверцитрин, к-рый при кипячении с кис-
лотами распадается на рамнозу и собственно крася-
щее вещество — К. В виде гликозида К. содержится
также во многих желтых цветах и листьях различных
деревьев. Частично в свободном виде, частично в форме
различных гликозидов К. найден в хмеле, чае, ко-
журе лука, цветах желтофиоли, желтой мать-мачехи,
красной розы и др.; 7-метиловый эфир К. — рамне-
тин — содержится в виде гликозида в ягодах нек-рых
видов крушины (Rhamnus); эти ягоды употреблялись
для крашения под названием «авиньонских зерен»,
или «грушки» — «желтой ягоды». К. в чистом состоя-
нии выделяют перекристаллизацией из спирта с двумя
молекулами воды в виде лимонно-желтых игл. Без-
водный К. плавится njiH 313—314° (с разл.). Почти
нерастворим в холодной воде, слабо — в горячей воде,
эфире и хлороформе, легко растворяется в ледяной
уксусной к-те и кипящем спирте. Растворим в разб.
щелочах, щелочные растворы К. при длительном
стоянии на воздухе постепенно окисляются (цвет
раствора переходит из желтого в темно-бурый). В
конц. H2SO4 К. растворяется с образованием ярко-
желтого р-ра, интенсивно флуоресцирующего зеле-
ным цветом. При восстановлении Mg с НС1 К. (I)
дает цианидин (II):
К. образует комплексные соединения со многими эле-
ментами. Соединения К. с элементами третьей группы
535
КЕРАМЗИТ — КЕРАМИКА
536
(Al, Ga, In, TI) интенсивно флуоресцируют в
УФ-свете. Эти свойства К. широко использованы в ана-
литич. химии. К. применяют для обнаружения Fe111
и UVI; для флуорометрия, определения А1; для фото-
метрия. определения Zr, Th, Ge, Sn, Mo и др.
Лит.: The chemistry of flavonoid compounds, ed. T. B. Geis-
sman, L., 1961. И. П. Ефимов.
КЕРАМЗИТ — см. Заполнители пористые.
КЕРАМИКА — изделия и материалы, изготовля-
емые соответствующей обработкой глиняного сырья,
а также разнообразных минеральных композиций,
вклюяающих природные минералы, окислы металлов
и др. высокоогнеупорные соединения с последующим
обжигом отформованного и высушенного полуфаб-
риката. В зависимости от плотности, обусловленной
химия, и гранулометрия, составом исходных сырьевых
материалов и степенью обжига, керамия. изделия и
материалы подразделяются на пористые (водопогло-
щение более 5%) и спекшиеся (водопоглощенпе менее
5%). По структуре материала изделий разлияают зер-
нистую (грубую) К., имеющую яеткое зернистое строе-
ние, иногда с неоднородной структурой (стеновые
материалы, огнеупоры), и тонкую К. с однородной,
тонкозернистой структурой и равномерно окрашенным
материалом (фарфор, технияеская К., фаянс).
В зависимости от области применения К. подраз-
деляют на строительную, огнеупорные материалы,
химияески стойкую (см. Химически стойкие матери-
алы), бытовую (хозяйственный фарфор и фаянс,
художественно-декоративные изделия и др.) и техни-
яескую (электро-, радио- и др. виды К.).
Процесс изготовления керамия. изделий состоит
из обработки сырья и приготовления керамия. массы,
формования, сушки и обжига изделий. Керамия.
изделия изготовляют методами пластия. формования,
полусухого прессования и отливки в формах. Наи-
большее распространение, в яастности при изготов-
лении строительной К., полуяил метод пластия. фор-
мования на специальных прессах. Подготовка пла-
стинной формовочной массы заключается в дроблении
и перемешивании глины с отощающими материалами,
увлажнении и проминке массы до получения однород-
ного пластичного теста. Полученную пластичную
массу формуют и сушат. Изделия из тонкой К. фор-
муют из пластичных, жидких и порошкообразных
масс; при этом в качестве одного из компонентов
применяют глинистые материалы. Отливка изделий
из жидкой массы производится в гипсовых формах;
этот способ получил наибольшее распространение
при производстве полых изделий крупных размеров
или сложной формы. Изготовление изделий из по-
рошкообразных масс производят прессованием па
прессах различной конструкции. В массы из непла-
стичного сырья добавляют органич. термопластич-
ные связующие вещества (парафин, воск и т. п.) и
формуют изделия методом горячего литья в металлич.
формах или прессованием. Полученные керамия. изде-
лия подвергают сушке и обжигу в специальных су-
шилках и печах. Нек-рые керамия. изделия покры-
вают глазурью, декорируют (украшают рисунками)
и т. п. Продолжительность обжига керамия. массы
колеблется от нескольких часов (мелкие изделия)
до нескольких суток (массивные огнеупорные изделия).
При этом в массе протекают сложные физико-
химич. процессы (дегидратация, диссоциация, поли-
морфные превращения, реакции окисления и восста-
новления и др.) с образованием в ряде случаев стек-
ловидного расплава, связывающего зерна более огне-
упорных составных частей в прочный монолитный
материал; обжиг ведется при темп-ре от 900° (строи-
тельный кирпич) до 2000° (специальные высокоогне-
упорные изделия). Этот процесс называется спека-
нием; он может проходить при низких или высоких
темп-рах (низкоспекающиеся или высокоспекающиеся
материалы). Степень спекания повышается с повыше-
нием темп-ры (до определенного предела), при этом
снижается пористость и уменьшаются размеры изде-
лия (усадка), увеличиваются его прочность, химич.
стойкость и диэлектрич. свойства. Кроме температу-
ры, на степень спекания влияет содержание в массе
флюсирующих добавок или минерализаторов. Процесс
спекания при обжиге тесно связан с химич. реакциями
и полиморфными превращениями, протекающими
в кристаллич. веществах. Напр., в изделиях, изготов-
ляемых на основе алюмосиликатного сырья (глина,
каолин), существенное значение при обжиге имеет
образование муллита:
4-ОЛЛ_I 9ЛЛ’
3 [AbO3-2SiO2-H2O]—------~3AJ2O:r2SiO2-h4SiOj4-6H2O
каолинит муллит
Характер и интенсивность указанной реакции за-
висят от природы исходного сырья, наличия приме-
сей, темп-ры обжига и газовой среды. Образовав-
шийся муллит придает керамия. изделиям повышен-
ную химия., механич. и термин, стойкость. Во многих
случаях процессы спекания К. протекают с участием
жидкой фазы, образующейся из основных кристаллич.
фаз и добавок окислов, способствующих образованию
эвтектик в процессе обжига. Наличие жидкой фазы,
взаимодействующей с кристаллич. фазами спекаемой
К., способствует более интенсивному заполнению пор
и ускорению процесса спекания. В качестве добавок
для образования жидкой фазы используются плавни
(полевошпатовые породы, нефелиновый сиенит, нек-рые
окислы и соли щелочноземельных металлов).
Керамика строительная. По назначению изделия строитель-
ной К. делятся на стеновые, кровельные, облицовочные (для
наружной и внутренней облицовки), санитарно-технические
и специального назначения.
Стеновые материалы характеризуются грубо-
зернистым строением, относительно высокой пористостью
(водопоглощение не менее 6—8%), достаточной прочностью
(предел прочности при сжатии не менее 75 — 150 кг/см9, при
изгибе—не менее 14—28 кг/см-) и морозостойкостью. К стеновым
материалам относятся: полнотелый, пустотелый и пористо-
пустотелый кирпич и пустотелые камни различных форм и
размеров. Объемный вес изделий: полнотелого кирпича 1,7 —
1,9 m/Ai3, пустотелых изделий 1,3—1,5 т/м* и менее. В качестве
исходного сырья для изготовления стеновых изделий приме-
няют в основном не засоренные известковыми включениями
легкоплавкие глины (содержащие 55—80% S1O2, 7—22%
А12О3) с введением до 20—30% отощающих и выгорающих
добавок (песок, дегидратированная глина, измельченные то-
пливные шлаки, уголь, опилки и др.)- Выгорающие добавки
придают изделиям повышенную пористость, улучшают тепло-
защитные свойства и морозостойкость. Изделия получают
формованием пластичной массы влажностью 18-*-23% на лен-
точных прессах или штампованием порошкообразной массы
с влажностью 9—12% на механич. прессах при уд. давлениях
100 —150 кг/см* с последующими сушкой и обжигом при 900—
1050°. Кирпич и пустотелые камни применяют для кладки стен,
изготовления крупных кирпичных блоков, стеновых панелей,
для сборки междуэтажных перекрытий, устройства огражде-
ний и т. д.
Кровельные материалы (черепица глиняная)
характеризуются мелкозернистым однородным строением,
относительно невысокой пористостью, повышенной морозо-,
огне- и атмосферостойкостью, выдерживают общую нагрузку
на излом не менее 70 кг.
Сырьем для изготовления черепипы являются не засорен-
ные известняком, гипсом п др. вредными примесями легко-
плавкие пластичные глины. В качестве отощающих добавок
(до 20—25%) используются измельченная обожженная глина
(шамот) и кварцевый песок. Формуют черепицу из пластич-
ной массы на ленточных (реже на механических) прессах с
последующими сушкой и обжигом при 900—1080°. Черепица
применяется для покрытия кровель одно- и многоэтажных
зданий.
Облицовочные материалы, применяемые для
наружной облицовки (фасадные керамич. материалы), харак-
теризуются мелкозернистым однородным строением, относи-
тельно невысокой пористостью (водопоглощение не более
10—14%), повышенной морозостойкостью и достаточной проч-
ностью (предел прочности при сжатии не менее 75—150 кг/см2).
Изделия выпускаются обычно светлых тонов (в отдельных
случаях с лицевой поверхностью, покрытой глазурью разных
цветов). К фасадным керамич. материалам относятся: кирпич
и камни лицевые (в том числе профильные, чаше всего пусто-
телые), плиты и плитки (в том числе ковровая керамика —
537
КЕРАМИКА
538
мелкие плитки, наклеенные на бумагу, для облицовки стено-
вых панелей), архитектурно-художественные детали. Сырьем
для фасадных керамич. материалов обычно служат низкоспе-
кающиесн тугоплавкие и огнеупорные глины, часто с введе-
нием отощающей добавки — шамота (до 50%), в нек-рых
случаях вводят минеральный краситель (2—12%). Изделия
формуют из пластичной массы на ленточных прессах или штам-
пуют порошкообразную массу на механич. прессах при уд.
давлении до 200—250 кг/с.ч’. Сушка полуфабриката произво-
дится в туннельных сушилках, обжиг—в туннельных печах при
980—1160°. Фасадные керамич. материалы применяют для
облицовки фасадов, стен вестибюлей, лестничных клеток ка-
менных зданий и сооружений.
Облицовочные материалы длн внутренней облицовки (гла-
зурованные фаянсовые плитки для внутренней облицовки
стен, керамич. плитки длн полов) характеризуются в зависи-
мости от вида изделий след, данными: а) Фаянсовые плитки —
тонкозернистым однородным строением материала, относи-
тельно высокой пористостью (водопоглощение 9—12%); ли-
цевая сторона покрыта глазурью (белой или цветной). Фаян-
совые плитки изготовляют из огнеупорных глин (26—28%),
каолинов (30—31 %), кварцевого песка (19—26%), бон обож-
женных изделий (15—25%); иногда вводят плавни (напр., по-
левой шпат) и др. добавки, б) Плитки для полов — спекшимся
материалом (водопоглощение не более 4%); тонкозернистым
строением, высокой прочностью, износоустойчивостью (потеря
в весе при истирании не более 0,1 г/сл1а) и химич. стойкостью
(кислотостойкость 97—98%). Для изготовления плиток для
полов применяют низкоспекающиеся тугоплавкие и огнеупор-
ные глины (до 82—100%), плавни (0—14%); иногда вводят
также каолин, кварцевый песок и минеральные красители
(2—13%). Оба вида плиток обычно прессуют из тщательно под-
готовленной различными способами (мокрый помол с даль-
нейшим обезвоживанием и др.) порошкообразной массы при
уд. давлении 200—250 кг/см2 с последующей сушкой в конвейер-
ных сушилах и обжигом в туннельных печах при 1100—1260°.
Если глазурь наносится на обожженный материал, произво-
дится повторный обжиг. Плитки применяют для облицовки
стен и настилки полов различных помещений.
Санитарно-технические изделия (глазу-
рованные умывальники, унитазы, смывные бачки и др.) ха-
рактеризуются тонкозернистым однородным строением. Ос-
новные свойства и характеристики санитарно-технич. изделий
приведены в табл. 1.
Таблица 1
Показатели Санитар- ный фаянс Санитар- ный по- луфар- фор Санитар- ный фарфор Шамот- ный фаянс
Водопоглощение, % . . Объемный вес материа- не более 10-12 не более 3-5 0,2-0,5 12-16
ла, г/см3........ Предел прочности: 1,92-1,96 2-2,2 2,25-2,3 1,8-1,9
при сжатии, кг/см2 . . при ударном изгибе, 900-1000 1200—1500 4000 150 -300
кг • см/см2 ...... 1,5-1.8 1,8-2 2-2,3 —
Модуль упругости, кг/см2 Коэфф, линейного рас- ширения (а • 10й) в ин- 2200—2400 3000-4000 5000—6000 —
тервале 20—700° . . . 4,8-6 4-4,8 4-6,5 z-5
Таблица 2
Показатели Каналиэапионные трубы Кислото- упорный кирпич Кислото- упорные плитки
Водопоглощение, %
(не более) Предел прочности, кг/см2: при сжатии (не ме- 9-11 8-12 6-9
нее) при изгибе (не ме- Выдерживают нагрузку 2—3 т/пог. м 150-250 300
нее) К ислотостойкость, Выдерживают внутреннее гид- равлич. давление не менее 2 ат (избыточных) 150
% (не менее) . . . .Термостойкость, ко- 90 92-94 95-98
личество теплосмен 2 2, для тер- мокислото- упорной 8
В качестве сырья для фаянсовых и полуфарфоровых изде-
лий применяют огнеупорные глины (20—30%), каолины (25—
32%), кварцевый песок (25—30%), обожженный бой изделий
(6—16%), плавни (0—20%). В полуфарфоровые массы для луч-
шего спекания материала вводят большее количество плавней
(в основном за счет уменьшения содержания боя изделий,
песка и каолина). Формуют изделия на конвейерах методом
литья водных суспензий (шликер) в гипсовые формы с после-
дующей сушкой полуфабриката, нанесением глазури и об-
жигом при 1240—1280®.
Изделия специального назначения —
глазурованные канализационные трубы, кислотоупорный кир-
пич, кислотоупорные плитки, клинкерный кирпич (мостовой)
и дренажные трубы. Основные свойства нек-рых изделий этого
типа приведены в табл. 2.
Исходным сырьем длн этих изделий служат не содержащие
известняковых, железистых и др. включений тугоплавкие и
огнеупорные низкоспекающиеся глины (содержат 20—40%
AlaOa и не более 65—70% SiOa) с добавкой 33—40% шамота;
для повышения термостойкости при изготовлении термокис-
лотоупорных плиток добавляют 8—12% талька. Изделия
формуют из пластичной массы на трубных прессах, прессуют
порошкообразные массы гидростатически (трубы) или штам-
пуют (кирпич, плитка). Обжиг высушенных изделий с нане-
сенной глазурью производится при 1100—1160° (трубы) и
1180—1280° (кирпич, плитка).
Лит.: Августиник А. И., Керамика, М., 1957;
Справочник по производству строительной керамики, т. 2, М.,
1961; ЗальмангГ., Физико-химические основы керамики,
пер. с нем., М., 1959; Будников П. П, [и др.],Техноло-
гия керамики и огнеупоров, М., 1962. Г. П. Каллига.
Керамика бытовая (хозяйственная). Этот вид К. подразде-
ляется на фаянсовые и фарфоровые изделия, а) Ф*а я н с о-
вые изделия характеризуются мелкозернистым белым
или равномерно окрашенным пористым, непрозрачным мате-
риалом: в неглазуровапном виде водо- и газопроницаемы.
В зависимости от состава фаянс делится на след, группы:
1) глинистый фаянс, содержащий 75—85% тугоплав-
кой и легкоплавкой глины и 15—25% кварцевого песка или
кварца, получил наибольшее распространение при производ-
стве гончарных изделий, архитектурно-строительной керамики
п др.; 2) известковый фаянс, содержащий 5—15 % мела
или доломита, 35—55% глинистого вещества, остальное —
кварцевый песок; применяется преимущественно для изгото-
вления бытовой посуды и небольших художественных изделий;
3) твердый, или полевошпатовый, фаянс, со-
держащий 40—60% глинистого вещества, 25—40% кварца
или кварцевого песка и небольшое количество полевого шпата;
этот тип фаянса имеет наибольшее распространение и при-
меняется в различных областях народного хозяйства. Основ-
ным сырьем для твердого фаянса служат пластичные низко-
спекающиеся огнеупорные глины, обогащенный каолин, квар-
цевый песок; в качестве плавней применяют полевой шпат или
пегматит, к-рый иногда заменяют нефелиновым сиенитом. Сфор-
мованные фаянсовые изделия подвергают двукратному об-
Таблпца 3
Исходные материалы, о/о Хозяйст- венный фаянс Хозяйст- венный фарфор Глазурь для фаянса9 Глазурь длн фар- фора®
Глина пластичная огне-
упорная ........ 50—60 9—15 8-10
Каолин обогащенный . 0-10 27-34 4-8
Кварцевый песок (кварц) 30—35 29-32 21—22 27-30
Полевой шпат ...... 2-4 19-21 27-28 27—36
Бой изделий ...... 4-6 6-10 — 12-27
а Кроме приведенных компонентов, в глазури для фаянса
содержится: 5—7% доломита или мела, 6—7% _ свинцового
сурика, 9—10% соды и 20—21% неошарита. ° В глазури
длн фарфора содержится также 10—15% доломита или мела.
Таблица 4
Показатели Твердый фаянс Полу- фарфор Хозяйст- венный фарфор
Объемный вес, г,слг’ .... 1,8-1.9 2,1-2,3 3-5 2,2—2,35 0,0-0,1
Водопоглощение, % . . . . 9-12
Предел прочности: при сжатии, кг/см2 . . . 1000-1300 1300—2500 4000—4500
при изломе, кг/см2 .... 150—250 350—450 500—600
при ударном изгибе, кг-см/см2 ........ 1,5-1,8 1,8-1,9 1,8-2,0
Коэфф, линейного расшире- ния (а • 109) в интервале 20—700° 6,0—7,0 4,5-5,0 4,5-5.2 0,8-750/0
Белизна (эталон BaSO*). . — —
539
КЕРАМИКА
540
жигу? утильному при 1250—1300’ для закрепления формы
изделий; глазурному, или политому, при 1000—1200’ для
расплавления нанесенной глазури и закрепления ее на изде-
лии. Фаянсовые изделия покрывают легкоплавкой глазурью,
б) Фарфоровые изделия, применяемые преимуще-
ственно в глазурованном виде, характеризуются плотным,
не пропускающим воду и газы материалом, просвечивающим
в тонком слое. Темп-ра обжига политого фарфора 1280—1320°.
Основным сырьем для произ-ва фарфора служат те же мате-
риалы, что и для произ-ва фаянса, но более высокого качества
и в других соотношениях (см. табл. 3). Все фаянсовые и фар-
форовые хозяйственные изделия декорируются надглазурными
и подглазурными керамич. красками. Примерные составы
фаянса и фарфора, а также глазурей к ним приведены в табл. 3,
а основные свойства фарфоро-фаянсовых изделий — в табл. 4.
Техническая керамика характеризуется окрашенным в бе-
лый или желтый цвет плотным, звонким, не пропускающим
жидкости и газы материалом изделий. Различают след, группы
технич. К.: для токов промышленной частоты (низковольтная
и высоковольтная) — тип твердого фарфора; для токов высо-
кой частоты — высокоглиноземистая и магнезиальная; для
конденсаторов — титаносодержащая К.; для зажигательных
свечей — корундовая и др.
Керамика для токов промышленной
частоты (твердый фарфор) изготовляется из обо-
гащенных каолинов (26—28%), высококачественных пластич-
ных маложелезистых огнеупорных глин (14—19%), кварца
или кварцевого песка (22—40%), полевого шпата или пегма-
тита (25—36%). При необходимости повысить прочностные
характеристики фарфора в его состав взамен кварца вводят
технич. глинозем. Твердый фарфор применяется в глазурован-
ном виде для изготовления высоковольтных, телефонно-теле-
графных и установочных изоляторов.
Керамика для токов высокой частоты
подразделяется на высокоглиноземистую и магнезиальную:
а) Высокоглиноземистая К. изготовляется
из специально 'обработанного электроплавленого корунда
(алунда) со средней величиной зерен 1—2 мк или пред-
варительно обожженной при 1380—1450° технич. окиси
алюминия. Высокоглиноземистан К. делится на след, группы:
корундовую (более 96% А12О3), муллито-корун-
довую (75—96% AI2Os) и муллитовую (меньше 75%
A12Os). Высокоглиноземистую К. применяют: корундовую
для вакуумплотных металлокерамич. узлов высокочастотных
ламп и малогабаритных высокочастотных изоляторов; муллито-
корундовую для крупногабаритных высокочастотных изоля-
торов, установочных высокочастотных изоляторов и др.;
муллитовую для всех видов высокочастотных и высоковольт-
ных крупногабаритных изоляторов, где, кроме диэлектрич.
характеристик, требуются повышенная механич. прочность
и термич. стойкость; муллито-корундовую и корундовую К.
применяют, кроме того, для изготовления автомобильных и
авиационных зажигательных свечей, для изготовления вы-
сокоогнеупорных тиглей для химич. и металлургич. целей,
термопарных трубок и др.
К высокоглиноземистой К. относят также цельзиано-
вую К., в состав к-рой входят предварительно синтезирован-
ный цельзиан (ВаО-А12О3 • SiO2), углекислый барий, коа-
лин и глина. Цельзиановую К. применяют для изготовления
катушек индуктивности высокой стабильности, изоляторов и
высокочастотных конденсаторов большой реактивной мощности
или предназначенных для работы при высоких темп-рах. При-
мерный шихтовый состав высокоглиноземистой К. приведен
в табл. 5.
Таблица 5
Исходные материалы, % Тип керамики
корун- доваяа мулли- то-корун- дован а мулли- товая цель- зиано- вая б
Глина пластичная огнеупор- ная 0-2 20—25 18-20
Каолин обогащенный .... — 23-25 26—28 44-50
Окись алюминия или элек- троплавленый корунд (алунд) 97—99 35—50 40-41 24—30
Доломит — 3-5 2-3 3-4
— 5—8 5—7 (йгСОз) 15-20
а В состав корундовой и муллито-корундовой К., кроме
того, входят 0,5—1,5% минерализаторов (MnO«, MgO, СГ2О3,
TiO;, BaO, SrO и др.) и органич. пластификаторы (1,5—3%
декстрина или 3—5 % парафина — при прессовании изделий;
12—16% парафина с добавлением 0,5—1,5% олеиновой к-ты —
при горячем литье под давлением). ® В состав цельзиановой
К. входит также 4—5% ашарита.
б) Магнезиальная К. делится на клиноэястатитовую
(стеатитовую) MgO • SiO2, форстеритовую 2MgO • SiO2, кор-
диеритовую 2MgO • 2А12О3 • 5S1O2 и шпинелевую MgO А12О3.
Основным исходным сырьем для изготовления первых трех
материалов служит природный тальк (3MgO- 4SIO2 • Н2О)
с различными добавками часовярской глины, бентонита,
СаСОз, ВаСО3, ZrOa, А12О3 и кварца. Шпинелевую К. получают
на основе магнезита (MgCO3) и технич. глинозема путем пред-
варительного их спекания. Магнезиальную К. применяют:
стеатитовую — в радиотехнич. аппаратуре (для установочной
изоляции, высоковольтных, высокочастотных натяжных и
опорных изоляторов); форстеритовую — для вакуумных при-
боров; шпинелевую — для конденсаторов низкого напряже-
ния и др.; кордиеритовую — для изоляции нагревательных
приборов и искрогасительных камер. Примерный шихтовый
состав магнезиальной К. приведен в табл. 6.
Таблица 6
Исходные материалы, % Тип керамики
клино- энста- тито- вая * форсте- рито- вая кор- диери- товая шпи- н еле- вая
Тальк необожженный 15-20 38—40
Тальк обожженный 50—55 58—60 ——
Глина пластичная огнеупорная 15-20 5-7 35-37 15-17
Диборат бария —— — — 6,5—7,0
Бентонит 0-5 — — —
Полевой шпат 0-3 —— 0-3
Мел или барий углекислый . . 5—7 5—6 0—3
Техническая окись алюминия 0-5 — 18-20
Углекислый магний (магнезит) — 25—30 ——
Шпинелевый спек (предвари-
тельно полученная шпинель) — — — 72-75
Плавиковый шпат — — — 1,0-1,5
* Стеатитовая.
Титаносодержащая керамика изготовляется в
основном из окиси титана с добавлением окислов нек-рых
металлов (MgO, СаО, ZnO, ВеО, ZrOa). Кроме того, в их со-
став вводят небольшие количества огнеупорной пластичной
глины и бентонита и органич. пластификаторов (декстрина и
тунгового масла). Количество органич. пластификаторов уве-
личивается с уменьшением содержания глинистых материа-
лов в массе.
Свойства титаносодержащей К. определяются соотноше-
нием TiO2 и добавки, а также свойствами образующейся при
взаимодействии Т1О2 и добавки кристаллич. фазы. В частности,
К., содержащая преимущественно Т1О2 (Т-80), характеризуется
малыми диэлектрич. потерями и высокой диэлектрич. прони-
цаемостью; К., содержащая преимущественно ортосиликат
магния 2MgO-TiO2, характеризуется диэлектрич. проницае-
мостью е, порядка 14—18, и небольшим положительным темпе-
ратурным коэфф, диэлектрич. проницаемости (ТКе), порядка
50—100-Ю6; К., содержащая преимущественно минерал
перовскит (СаО-Т1О2) или твердые кристаллы типа пе-
ровскита (ВаО-Т1О2 и др.), отличается очень высокой диэлек-
трич. проницаемостью и обычно является сегнетоэлектри-
ком, а титанаты циркония (ZrO2-TIO2) — малым отрицатель-
ным значением ТКе.
Титаносодержащую К. применяют гл. обр. для изготовле-
ния конденсаторов различных типов. Твердые р-ры типа
ВаОТ1О2 применяют в качестве сегнетоэлектриков. Составы
наиболее распространенных титаносодержащих К. приведены
в табл. 7. Основные свойства технич. К. приведены в табл. 8
(см. стр. 541).
Таблица 7
Исходные материалы, /0 Марки масс
Т-80 Т-150 ТК-20 Р-15
TiOj обожженная 87,3 44,4 32,3 46,5
ZrOa обожженная 3,9 1,1 46,1 5,0
ВаСОз 0,34 — 6,9 —-
Мрамор 0,66 54,5 — 1.0
Магнезия уста — — — 47,5
Плавиковый шпат 1,9 — 0,5 —
Глина часовярскан 5,9 — 9,3 —
Бентонит — — 4,9 —
Лит.: Б у д н и к о в П. П. [и др.], Технология керамики
и огнеупоров, М., 1962; Будников П. П., Г е в о р-
к я н X. О., Фарфор, М., 1955; Зальманг Г., Физико-
химические основы керамики, пер. с нем., М., 1959; Н и-
кулин Н. В., Производство фарфоровых изоляторов,
М.—Л., 1958; Богородицкий Н. П., Фридберг
И. Д., Высокочастотные неорганические диэлектрики, М., 1949;
их ж е, Электрофизические основы высокочастотной керамики,
М.—Л., 1958; Богородицкий Н. П., ПасынковВ. В.,
Материалы в радиоэлектронике, М.—Л., 1961; Палацкий
А., Техническая керамика, пер. с нем., М.—Л., 1959; П о-
лубояринов Д. Н., Балкевнч В. Л., Попил ь-
с к и й Р. Я., Высокоглиноземистые керамические и огне-
упорные материалы, М., 1960. Г. Н, Дудеров,
541
КЕРАМИКА — КЕРАМИЧЕСКИЕ КРАСКИ
542
Высокоглиноземистая К. I Магнезиальная К. I Титаносодержащая
КЕРАМИЧЕСКИЕ КРАСКИ — окрашенные ми-
неральные вещества, стойкие при высоких темп-рах,
применяемые для окраски керамич. изделий, глазу-
рей и стекол. К. к. представляют собой смеси жаро-
стойких минеральных пигментов с легкоплавкими
стеклами (надглазурные краски), с ке-
рамич. массами и глазурями (подглазурные
краски) или окрашенные специального состава
стекла (нек-рые майоликовые краски, краски для
стекла и ювелирных эмалей). При обжиге надглазур-
ных К. к. флюс плавится и приплавляет пигмент к
поверхности глазури. Подглазурные К. к. наносятся
непосредственно на черепок и покрываются глазурью.
Число пигментов, стойких при высоких темп-рах,
невелико; оно тем меньше, чем выше темп-ра обжига
декорируемых изделий. Выбор пигментов для надгла-
зурных К. к. (темп-ра обжига 690—830°) значительно
больше, чем у подглазурных К. к. для фаянса и фар-
фора (темп-ра обжига 1160—1200° и 1300—1400°).
Вследствие значительных колебаний состава при-
менение естественных минералов ограничивается из-
готовлением К. к. для грубой керамики (клинкер,
половые плитки, терракоты и т. п.), для всех же ос-
тальных целей К. к. изготовляются на основе синте-
тич. пигментов, представляющих собой окрашенные
окислы отдельных металлов и их сочетания. Керамич.
пигментами являются алюминаты и силикаты типа
шпинелей, виллемиты, гранаты, корунды, силли-
маниты, в ряде случаев прочно окрашенные фосфаты,
молибдаты, вольфраматы и ванадаты. Отдельной
группой являются золотосодержащие керамич. пиг-
менты, окраска к-рых вызывается коллоидными ча-
стицами Au. Керамич. пигменты получают прокали-
ванием солей, окислов или гидроокисей соответствую-
щих металлов или совместным осаждением гидрооки-
сей или углекислых солей с последующим прокали-
ванием осадков, а также сплавлением солей и прокали-
ванием смеси.
Красные и розовые керамическиепиг-
менты (железные, хромовые, хромово-оловянные, хромово-
и марганцово-глиноземные, селено-кадмиевые и золотосодер-
жащие). Основой красных железных пигментов является
Ре2О3, получаемая прокаливанием технически чистого FeS04*
•7Н2О.Чистый огненно-красный тон имеют т. наз. коралловые
пигменты на основе РЬСгОНРЬ(ОН)2. Вследствие токсич-
ности их заменяют селено-кадмиевыми пигментами, предста-
вляющими собой смешанные кристаллы CdS и CdSe ярко-крас-
ного цвета. Восстанавливая золото из разведенных растворов
его солей, осаждают последнее в коллоидном состоянии на
SnO2 илиА12О8 и получают пигменты, применяемые для изго-
товления розовых, пурпурных и фиолетовых К. к. Красные,
розовые и лиловые пинковые красители представляют собой
хромо-оловянные соединения, получаемые совместным прока-
ливанием при 1300° смеси окиси олова, мела, кварца, хром-
пика и буры. Хром придает розовую окраску корунду и шпи-
нели при совместном их прокаливании при 1300° в присут-
ствии В2О3. Рубиновые и шпинельные хромовые пигменты
могут применяться как подглазурные К. к. для фарфора и
фаянса. Свинцовые глазури их разрушают. Для окраски
фарфора применяют розовый марганцовый пинк состава
ЗМпО • Р2О3 ♦ 12А12О3, получаемый прокаливанием при 1160°
смеси МпСО8 (NH4) НР04 и А1(ОН)3. Для приготовления ро-
зовых пигментов пригоден также фосфат неодима.
Светлые, теми о-к оричневые и черные
пигменты приготовляют совместным прокаливанием со-
лей Fe, Мп, Сг, Zn, Со, Ni или их окислов.
Синие пигменты готовят на основе окиси кобальта
и иногда окиси меди. По составу они подразделяются на си-
ликаты и алюминаты. Особо интенсивную ультрамариновую
окраску дают после прокаливания кобальтовые силикаты
виллемитового ряда (ZnO • SiO2, в к-ром часть ZnO заменена
СоО). Для подглазурной окраски фарфора обычно применяют
окись кобальта в смеси с полевым шпатом, глазурью.
Голубые и бирюзовые К. к. обычно готовят на
основе алюминатов кобальта, они представляют собой соеди-
нения шпинельного типа. Путем замены части ZnO или MgO на
СоО в цинковой ZnO • ALO3 или магнезиальной MgO-Al2O3
шпинелях получают голубые пигменты различной интенсив-
ности. добавляя в шихту Сг2О3, можно придавать пигментам
зеленоватые тона. Голубые пигменты цирконо-ванадиевого
ряда получают прокаливанием стехиометрия, смеси ZrOj
и SiO2 с 5% V2O3.
Фиолетовые К. к. представляют собой смеси пур-
пуровых, пинковых и розовых шпинелей с кобальтовыми пиг-
ментами.
543
КЕРАТИНЫ — КЕРМЕТЫ
544
Основным пигментом для зеленых К. к. является окись
хрома. Яркие зеленые подглазурные К. к. для фаянса дают
пигменты гранатового типа (состав ЗСаО • CraO8-3SiO2)- Для
получения светло-зеленых надглазурных красок обычно сме-
шивают зеленые хромовые пигменты с желтыми сурьмяными.
Основной пигмент для желтых К. к. — неаполитанская
желтая, получаемая сплавлением свинцового сурика, буры,
окисей сурьмы, железа, кварца, калийной селитры. Стойки
к действию высоких темп-р желтые ванадиевые пинки, при-
меняемые в качестве подглазурных К. к. по фарфору.
Для окраски фарфоро-фаянсовых изделий широко приме-
няют аолото, платину и серебро. Эти металлы наносятся на
изделия в виде смеси металлич. порошков с небольшим коли-
чеством легкоплавких стекол. После обжига на керамике эти
препараты покрываются окненой пленкой; для придания бле-
стящего вида пленка удаляется полировкой агатом или плот-
ной стеклянной щеткой. Чаще применяют другой способ. Он
состоит в том:, что на изделия наносят р-ры органич. соедине-
ний драгоценных металлов, т. наз. резинатов, в смеси органич.
растворителей. Для закрепления блестящей пленки, образую-
щейся на изделии после обжига, в эти препараты добавляют
плавень или флюс (соединения висмута, кадмия, хрома,
урана, родия в виде растворов их резинатов). Аналогично
получают люстровые К. к. — растворы резинатов различных
металлов, дающие после обжига на поверхности глазурован-
ных изделий окраску с металлич. блеском.
Лит.: Туманове. Г., ДАН СССР, 1947, 57, № 1, 69;
Будников П. П. [и др.], Технология керамики и огнеупоров,
М., 1962, с. 494—512; Беркман [и др.], Декорирование
фарфора и фаянса, М., 1949; Ullmann, 3 Aufl.. Bd 9,
Munch.— В., 1957, S. 425—46. С. Г. Туманов.
КЕРАТИНЫ — белки группы склеропротеинов. К.
составляют основную массу волос, шерсти, перьев,
ногтей, рогового слоя эпителия и т. п. К. нераство-
римы в воде, разбавленных к-тах и щелочах, этило-
вом спирте, эфире, ацетоне. По данным рентгенострук-
турного анализа, полипептидные цепи К. существуют
в двух формах: вытянутой (fJ-форма) и складчатой
(a-форма). В К. имеется много дисульфидных связей,
обусловливающих нерастворимость этих белков. К.
растворяются при нагревании с водой при 150—200°.
Сульфиды щелочных металлов, тиогликолевая к-та,
цианиды восстанавливают дисульфидные связи К.
При этом получаются более растворимые вещества,
называемые к е р а т е и н а м и. Химич, состав про-
дуктов гидролиза К, шерсти (в процентах, ориенти-
ровочно): аланин 4,1; глицин 6,5; валин 4,6; лейцин
11,3; пролин 9,5; фенилаланин 3,6; тирозин 4,6;
триптофан 1,8; серин 10; треонин 6,4; цистин/2 11,9;
метионин 0,7; аргинин 10,4; гистидин 1,1; лизйн 2,7;
аспарагиновая к-та 7,2; глутаминовая к-та 14,1;
амидный азот 1,2. К. очищают обработкой измель-
ченных роговых тканей органич. растворителями,
водой, затем пепсином и трипсином.
Лит.: ГауровитцФ., Химия и биология белков, пер.
с англ., М., 1953, с. 204; Белки, под ред. Г. Нейрата и К. Бейли,
пер. с англ., т. 3, ч. 2, М., 1959, с. 352. В. О. Шпикитер.
КЕРАТОСУЛЬФАТ — кислый полисахарид ро-
говой оболочки глаза, построенный из остатков N-аце-
тилглюкозамина, галактозы и серной кислоты. От
других полисахаридных компонентов роговицы К.
отличается тем, что не содержит уроновой к-ты; мета-
хроматически окрашивается толуидиновым синим.
Биологич. роль К. точно не установлена.
Лит. см. при Статье Мукополисахариды. Л. И. Липевич.
КЕРМЕТЫ (керамикометаллические материалы) —
гетерогенные композиции из металлов и неметаллов,
сочетающие тугоплавкость, твердость и жаростой-
кость керамики с проводимостью, пластичностью,
термостойкостью и др. свойствами металлов. В ка-
честве неметаллпч. компонентов используются раз-
личные тугоплавкие окислы, металлоподобные со-
единения переходных металлов (карбиды, бориды,
нитриды), нек-рые силициды и др. неметаллич. ве-
щества, отличающиеся химич. стойкостью, высокой
твердостью и высокой темп-рой плавления. В качестве
металлич. составляющей К. используют гл. обр.
металлы и сплавы группы железа (Fe, Ni, Со) и пе-
реходные металлы VI группы (Сг, Wo, W), иногда
легкие металлы (А1 и др.).
К. получают методами порошковой металлургии.
Для получения компактных композиций, сохраняю-
щих и лучшим образом сочетающих денные свойства
исходных компонентов, стремятся обеспечить в К.
прочные межфазовые связи', в образовании к-рых
значительную роль играют адгезия, обволакивание.
При этом существенное значение имеет характер
взаимодействия фаз на поверхности их раздела:
величина и состояние контактной поверхности, по-
верхностное натяжение, а также возможность обра-
зования в процессе изготовления К. тонких, равно-
мерно распределенных прослоек промежуточного со-
става (ограниченные твердые р-ры, соединения типа
шпинелей и др.). В нек-рых случаях при изготовлении
К. металлич. компонент вводят в расплавленном со-
стоянии (пропитка заготовки) или прибегают к ча-
стичному расплавлению металла в процессе спекания
брикета из смеси порошков металлич. и неметаллич.
компонентов (спекание с участием жидкой фазы).
В этих случаях большое значение приобретает сма-
чивание.
Для получения К. прочных и устойчивых в широком
диапазоне темп-р, как правило, необходимы: а) высо-
кая дисперсность и равномерное распределение в
структуре К. металлич. и неметаллич. фаз; б) обра-
зование в структуре К. непрерывного пластичного
металлич. скелета, или основы (сквозная металлич.
связь), или (и) соответственно жесткого неметаллич.
каркаса; в) ограничение вплоть до высоких темп-р
взаимной растворимости компонентов и их химич.
взаимодействия; г) сравнительно близкая величина
коэфф, термич. расширения обоих компонентов в ши-
роком интервале темп-р; д) возможность регулиро-
вания термич. расширения, межфазовой поверхност-
ной энергии и смачиваемости путем варьирования
состава, режима термообработки и добавлением спе-
циальных присадок; е) отсутствие в компонентах К.
вплоть до высоких темп-р полиморфных превращений,
сопровождающихся значительным изменением объема
и свойств компонентов.
По составу различают: окепдные К., содер-
жащие окислы Al, Be, Mg, Zr, Th, U и др.; кар-
бидные К., включающие в свой состав карбиды
переходных металлов (Wo, Ti, Та, Nb, Сг и др.),
а также сложные карбиды (твердые растворы карби-
дов) этих элементов; боридные К., содержащие
преим. бориды тугоплавких металлов (циркония,
титана и др.).
По свойствам и применению различают К.: 1) ис-
пользуемые при высоких температурах (детали газо-
вых турбин, ракетной и реактивной техники, элек-
тропечная арматура, тигли для расплавов и др.). К
этой группе К. относятся композиции: А12О3—Сг;
А12О3—thO3—Сг—Мо; А13О3—W—Сг, а также боль-
шая группа К. на основе карбида титана с Ni, Со,
Сг, Мо, W, А1 и их сплавами; 2) отличающиеся высо-
кой твердостью, износостойкостью, красностойкостью
и используемые в качестве режущих инструментов
я деталей, работающих на износ. К этой группе К.
относятся композиции: WC—Со; WC—TiC—Со и др.
3) К. с особыми физич. свойствами, используемые
в специальных областях техники: в атомных реакто-
рах (тепловыделяющие элементы и др. детали из ком-
позиций UO2—Al; MgO — Ni; А13О3—Сг); в электро-
технике п электронной технике (напр., графит —
медь для электрощеток, ThO3—Мо или TbO2—W
для усиления эмиссионной способности и др.); в тор-
мозных устройствах (нек-рые фрикционные мате-
риалы, содержащие по несколько металлич. и не-
металлич. компонентов: Си, Fe, Pb, SiO, и др.);
в качестве коррозионностойкпх материалов (напр.,
Сг2О3—W—Ni) для изделий, работающих в агрес-
сивных средах.
Наибольшее значение имеют металлокерамич. кар-
бидные твердые сплавы, являющиеся самыми эффек-
545
КЕРОСИН
546
тивными из всех инструментальных материалов,
а также высокотемпературные К. Подбирая состав
и режим обработки, можно добиться весьма высоких
характеристик жаропрочности и жаростойкости К.
Так, сплав К183А состава TiC (60%)—Ni (32%) —
Ст (2,5%)—Мо(3%)—Ai(2,5%) (здесь и далее везде
весовые %) обладает длительной (100 час) прочностью
при 1000° (ок. 20 кг/мм2). Композиция А12О3 (70%) —
Сг (30%) устойчива против окисления до 1200' и
показывает при 1300° прочность на изгиб более
18 кг!мм2. Вполне удовлетворительная прочность этой
композиции может быть улучшена в два раза за счет
увеличения доли металлич. фазы до 66% веса. Стой-
кость против окисления композиций на основе TiC
(с металлич. связующим в виде Ni, Ni—Сг и др.)
можно существенно повысить добавками карбидов
Та, Nb, а также небольших количеств Сг. Основным
недостатком всех К. является их низкая динамич.
прочность. Некоторое повышение ее может быть до-
стигнуто увеличением в их составе доли металла;
иногда прибегают к поверхностному обогащению
металлами, получая т. н. К. переменного состава или
покрывают К. металлами.
Основные способы получения К.: 1. Составление
смеси порошкообразных компонентов К. конечного
состава с последующей ее обработкой: а) прессова-
нием заготовок требуемой формы и последующего
спекания, большей частью с образованием жидкой
фазы. Этот способ применим для К. с небольшим
количеством металлич. компонента; б) горячим прес-
сованием тех же заготовок с последующей термин,
гомогенизацией полученного К. (или без нее); в) вы-
давливанием прессованием или прокаткой смеси
порошков конечного состава с последующим спека-
нием. Во всех этих случаях для К. с относительно
большим количеством металлич. фазы на завершаю-
щей стадии возможно применение горячей или хо-
лодной обработки давлением с соответствующим
улучшением структуры и свойств К. 2. Формование
пористого каркаса — заготовки из порошка туго-
плавкого неметаллич. компонента путем холодного
прессования, умеренного спекания до заданной
плотности с последующей пропиткой этого каркаса
расплавленным металлом без изменения формы заго-
товки. В нек-рых случаях после пропитки проводят
гомогенизирующий отжиг. Этим методом можно также
получать К. с переменным составом в направлении от
поверхности к центру изделия, в частности, с обога-
щением металлом поверхностных слоев. 3. Составле-
ние водной или неводной суспензии (шликера) из
порошкообразных компонентов К. конечного состава
и заливка этой суспензии в пористые, обычно гипсо-
вые, формы. После поглощения влаги стенками формы
в ней остается сформованная заготовка, к-рую затем
сушат, спекают или обжигают для упрочнения.
Лит.: Киффер Р., Шварцкопф П., Твердые
сплавы, [пер. с нем.], М., 1957; С а мс о и о в Г. В., Порт-
ной К. И., Сплавы на основе тугоплавких соединений, М.,
1961; Керметы. Сб., под ред. Дж. Р. Тинклпо, У. Б. Крен-
далла, пер. с англ., М., 1962; Cermets, ed. J. R. Tinkelpaugh,
W. B. Crandal, N. Y. — L., 1960; Третьяков В. Й.,
Металлокерамические твердые сплавы, М., 1962; П и н ес В. Я.,
Ж. Всес. хим. об-ва им. Д. И. Менделеева, 1960, 5, вып. 2;
Августинин А. И., там же. А. К. Натансон.
КЕРОСИН — смесь углеводородов, выкипающая
в интервале темп-р 180—320°, получаемая при прямой
перегонке нефти или крекинге нефтепродуктов. К. —
прозрачная, бесцветная или желтоватая жидкость с
голубым отливом; плотн. 0,775—0,850, вязкость при
20° 1,25—3,5 сст. Темп-ра вспышки К. зависит от
его фракционного состава: чем выше темп-ра начала
кипения К. и меньше в нем легких фракций, тем выше
плотность и темп-ра вспышки. К. имеет низкое давле-
ние паров. В табл. 1 приведены нек-рые константы
К., полученного из нефтей различных месторождений.
Таблица 1
Керосин Начало ки- пения, °C Конец кипе- ния, °C Температура застывания, °C
Сураханский .... 140 300 -56
Биби-Эйбатский . . 159 307 —60
Ишимбаевский . . . 187 330 -31
Нефтедагский .... 200 319 -60
Сызранский 193 313 —26
Грозненский 165 300 -16
Содержание- различных классов углеводородов в
К. зависит от химич. состава нефти, из к-рой получен
К., и технологии его получения. Напр., К. прямой
перегонки из туймазинской нефти содержит: 47%
парафиновых, 17% нафтеновых, 30% ароматических
и 6% непредельных; из сызранской, соответственно:
48%, 44%, 22% и 1%.
К. применяют в качестве топлива для реактивных
двигателей (авиационный), карбюраторных трактор-
ных двигателей (тракторный) и для бытовых иужд
(осветительный). В районах с низкими темп-рами
К. иногда применяют для автомобильных и трактор-
ных дизельных двигателей.
К. авиационный. Содержание ароматич. углеводо-
родов в этих К. не должно превышать 20—25%, непредель-
ных (по иодному числу) 2,4%. Подробнее об авиационных К.
см. Реактивное топливо.
Н. тракторный — фракции прямой перегонки нефти
и их смеси с фракциями термин, крекинга, выкипающие от
110° до 300°. Для этих К. имеют значение углеводороды, обла-
дающие высокой антидетонационной стойкостью. Качество
тракторных К. характеризуется гл. обр. их фракционным
составом и октановым числом. По этим показателям указанные
К. делятся на тракторные, тракторные высокооктановые
и тракторные экспортные. В табл. 2 приведены основные
требования к качеству тракторных К.
Таблица 2
Показатели Трактор- ный К. Трактор- ный высо- кооктано- вый К. Трактор- ный для экспорта К.
Октановое число (не менее) . . Фракционный состав: Перегоняется 10% при темп-ре, 40 45 55
°C (не ниже) 110 но
То же (не выше) ....... Перегоняется 50% при темп-ре, 180 — —
°C (не ниже) Перегоняется 90% при темп-ре, 190 190 —
°C (не ниже) . 240 240 —
То же (не выше) Перегоняется 98%'при темп-ре, 275 — —
сС (не ниже) ........ Перегоняется до 185°, % (не 300 290 —
менее) — — 10
То же до 200° (не менее) . . . — — 50
Конец кипения, °C (не выше) Темп-ра вспышки, в закрытом — — 275
тигле, °с (не более) Фактич. смолы, л<а/100 мл гоп- - — 35
дива (не более) Кислотность, мг КОН/100 мл 40 40 —
(не более) 1,0 4.5 4
Серы, % (не более) 1,0 0.2
Кроме того, в тракторных К. нормируется содержание
(отсутствие) активных сернистых соединений, воды, механич.
примесей и растворимых в воде кислот и щелочей. Для уда-
ления смолистых веществ и нафтеновых к-т тракторные К. очи-
щают щелочью.
К. осветительный получают прямой перегонкой
из малосернистых нефтей, богатых метановыми углеводоро-
дами (грозненской парафиновой, нек-рых бакинских, майкоп-
ских). Осветительный К. должен гореть в лампах сильным
некоптящим пламенем и не образовывать значительного на-
гара. К. осветительный вырабатывается трех сортов (табл. 3):
осветительный, осветительный тяжелый (пиронафт) — Для
освещения помещений с повышенной огнеопасностью, и осве-
тительный для экспорта. В табл. 3 приведены требования
к осветительным К.
Осветительные. К. очищают серной к-той, щедочщр и про-
мывают водой,
547
КЕТЕНЫ
548
Таблица 3
Показатели Освети- тельный К. Освети- тельный тяжелый К. Освети- тельный К. для экспорта
Цвет в марках (не более) . . . Высота некоптящего пламени, 3,0 3,0 2,3
мм (не менее) Фракционный состав: перегоняется до 200°, % (не 20 — 20
менее) — — 18
то же до 270°, % (не менее) . » » до 315°, % » » 70 — —
98 —
Конец кипения, °C (не выше) . Сера, % (не более) Кислотность, мг КОН/100 мл — — 290
0,1 — 0,05
(не более) Темп-ра вспышки в закрытом 1,4 — 1,4
тигле, °C (не ниже) .... Темп-ра помутнения, °C (не 40 90 40
выше) 12 — 15
Лит.: Л о с и к ов Б. В., П уч к о в Н. Г., Энглин Б. А.,
Основы применения нефтепродуктов, 2 изд., М., 1959; Б р у-
сянцев Н. В., Автотракторные топлива и смазочные ма-
териалы, М., 1958; Рагозин Н. А., Реактивные топлива,
М., 1959; Нефтепродукты и продукты переработки твердых
топлив, М., 1961; Технические нормы на нефтепродукты,
16 Изд., М., 1957. А. Д. Фатьянов.
КЕТЕНЫ — органич. соединения, содержащие сис-
тему кумулированных двойных связей С=С и С=О
общей формулы RR'C=C=O. Простейший предста-
витель— кетен (карбометилен) СН2=С=О, т.
пл. —134,6°, т. кип. —41°. Монозамещенные К. на-
зывают также альдокетенами, дизамещенные — ке-
токетенами. Одним из методов получения К. является
пиролиз карбонильных соединений — карбоновых
к-т, их производных, кетонов и т. д., в частности
пиролиз ацетона (700—800°) или уксусной к-ты в
присутствии Н3РО4 (700°) служит промышленным
методом получения кетена:
сн3-с-сн3—►сн2=С=о+сн4
II
о
СН3СООН—►СН2=С=О-4~Н2О
К. могут быть получены по общему методу дегалоге-
нированием галогенангидридов а-галогенкарбоновых
к-т:
RR 'CXCOX-f-Zn->RR'C=C=O4-ZnX2
и дегидрогалогенированием галогенангидридов
содержащих атом водорода в а-положении:
RR'CHCOX-f-NR’--►RR'C=C=04-NR"-HX
В ряде случаев удобным методом получения К. яв-
ляется разложение диазокетонов:
RCN,COR'—-2-[RCCOR']—>RR'=C=O
К. свойственна высокая и разнообразная реакционная
способность.
К. легко присоединяют различные нуклеофильные
реагенты—спирты, амины, меркаптаны и т. п.
с образованием соответствующих производных кар-
боновых к-т; присоединение воды приводит к карбо-
новым к-там. По-видимому, присоединение происхо-
дит по карбонильной группе с последующей изомери-
зацией: ,
R4
С-С=О + НХ
/
R
К-Т,
С=СХтО-Н
/ \ г
R
Rs
£НСОХ
R
Х=ОН, ООН, OR, SH, SR, NH2, NHR, NRR',
RCOO, PR2, AsR2
Аналогичным образом кетен присоединяется к
неорганич. к-там (HNO3, Н3РО4, HaSO4, галогеново-
дородным и др.), напр. СН2=С=О+ Н3РО4 —<-
—► (НО)2Р(О)ОСОСН3. Присоединение магний-, ртуть-
и кадмийорганич. соединений приводит после гид-
ролиза к соответствующим кетонам:
RR'C=C=O+RHMeX—»RR'C=CR"—О—МеХ—►
О
-->RR'CH-С-R”
Трифенилхлорметан присоединяется с образованием
хлорангидрида 0, 0', р"-трифенилпропионовой к-ты:
(СоН5)3СС1Ч-СНз=С=0—>(C0HS)3CCH2COC1
В присутствии BF3 кетен реагирует с соединениями,
содержащими активную эфирную связь. Так, можно
от тетрагидрофурана перейти к е-капролактону:
Другая большая группа превращений К. — реакции
циклодимеризации с участием как С=С, так и С=О
связей. Кето- и альдокетены при нагревании или до-
статочно длительном хранении димеризуются. Часто
К. димеризуются уже в процессе получения. Кето-
кетены претерпевают циклодимеризацию по типу
«голова к хвосту», причем обе молекулы реагируют
С=С-связью с образованием производных циклобу-
тандиона-1,3;
О
11
R Rx хСч ZR
2 zC = C = O ->- ZC4 zCx
R,Z R C XR'
»
О
Димеризация самого кетена с образованием дикетена
происходит по типу «голова к хвосту», причем одна
молекула реагирует связью С=С, а другая — связью
С=О:
сн2=с=о сн2=с—о
—I I
+сн2=с=о сн2—с=о
Альдокетены обычно образуют димеры типа дикетена;
в отдельных случаях отмечено образование произ-
водных циклобутандиона. Реакции циклодимеризации
происходят также при взаимодействии К. с другими
соединениями, содержащими двойные связи >С=С(^,
/С=О, ^>C=N—, —N=O, —N=N. К. реагируют
с альдегидами и кетонами в присутствии эфирата
BF3, ZnCl2, Mg(C104)2 ит. п.с образованием 0-лактонов:
х х ^0-0=0
j>c=c=o+j>c=o—k
Присоединение К. к основаниям Шиффа служит ме-
тодом получения 0-лактамов:
\ х "^С—С=О
\C=C=O+>C=N----< | |
z z J>C-N-
K. легко присоединяются к сопряженной связи С=С
с образованием соединений ряда циклобутанона:
О
В результате присоединения К. к связям N=N и
N=O образуются циклич. системы, напр.:
С0Н3-N—N—CeHs
(С3Н5)2С-С=О
C3H3-N—о
О=С---С(С6Н5)а
549
КЕТИМИНЫ — КЕТОАЛЬДЕГИДЫ
550
Реакция кетена с диазометаном приводит к циклопро-
панону и продуктам его превращений:
сн2.
>С(ОН)2
сн2—c=o+ch2n3
Н2О /
\CH2N2
\ СН2-СН»
\| I
СН2-С=О
Фотохимически кетен разлагается на этилен и СО:
hy
2СН2=С=О—►СН2=СН2+2СО
К. находят самое широкое применение в синтети-
ческой органической химии. Кетен применяют в
промышленности для получения уксусного ангидрида
и дикетена, для приготовления ацетилцеллюлозы
и т. д. Важнейшими производными К. являются их
ацетали, которые образуются при дегидрогалогени-
ровании а-бромацеталей:
^C-ch(OR)2-^S®£> \c=C(OR)2
Вг
отщеплении спиртов от ортоэфиров (пиролитически
или при действии сильных оснований, например
RNa):
/С—C(OR)3 \c=C(OR)2
H
или при действии цинка на а-бромортоэфиры:
^>С-C(OR)s --^-^С=С (OR)2
Вг
Лит.: X енф орд В. Е., 3 ау ер Д. С., в кн.: Органиче-
ские реакции. Сб. 3, пер. с англ., М., 1951, с. НО; Quad-
beck Angew. Chemie, 1956, 68, К: 11, 361; McElvain
S. М., Chem. Revs., 1949, 45, Ms 3, 453; Houben-Weyl,
4 Aufl., Bd 7, T! 2, Stuttgart. 1962. Б. Л. Дяткин.
КЕТИМИНЫ — производные кетонов, в к-рых
кислород карбонильной группы замещен иминогруп-
пой = NH. Название К. производится от названия
соответствующего кетона, напр. С6НВ—С—С6Н6 бен-
II
NH
зофенонимин, или дифенилкетимин. К. простых али-
фатич. кетонов в свободном виде неизвестны, хотя
получены нек-рые соответствующие им комплексные
соли, напр. [(CH3)2C=NH]2.H2SnCl6. Жирноарома-
тич. К. неустойчивы: они легко гидролизуются,
а при хранении, даже при комнатной темп-ре, элими-
нируют аммиак с образованием N-замещенных кети-
минов:
2CH3CH2-C=NH сн3сн2-с-с6н5
CeHs N
I
сн3сн=с-с6н5
Чисто ароматич. К. вполне устойчивы. Особенно
устойчивы К. с объемистыми радикалами при группе
>C=NH. Так, мезитилфенилкетимин при длитель-
ном кипячении с конц. соляной к-той или со щело-
чами гидролизуется лишь частично.
Общими свойствами К. являются гидролиз в кетоны
и восстановление в амины, а также замена атома
водорода при азоте на галоген и алкильные либо
ацильные группы:
R\
>С=О
R'z
)chnh2
R'z
R"COCI
R\
>C=NCOR'
R'z
H2O
н+
HOC1 I
C—NMgX
R\
>C=NC1
R’z
R\
ч —NR
(R O)2SC>4
При наличии а-атома водорода имеет место тауто-
мерное равновесие между К. и енаминами, причем
положение равновесия определяется в основном
строением вещества. В случае К. Р-дикарбонильных
соединений равновесие сильно сдвинуто в сторону
сопряженных енаминов:
о о
II II
CHs-C-CH2-C-R 5=t СНз—С=СН—С—R
II I
NH NH2
Основные методы получения К.: присоединение
металлоорганич. соединений к нитрилам (1); взаимо-
действие реактивов Гриньяра с хлорцианом (2),
конденсация ароматич. кетонов или оснований Шиффа
с аммиаком (3), восстановление оксимов и N-хлор-
иминов (4), см. также Гёша синтез.
R\ н»о R\
R-CSN-f-MeR'—- )C=NMe-i2L. >C=NH (1)
R'z R’z
Cl
R—MgX-f-ClCHN—>R-C=NMgX -R—lgX,^ ^C=NMgX-►
I^R\G=NH (2)
Rz
Arx NIR Ar\ NHi Ar\
Ar>c=oTho7Ar>c=NH—-Ar>c“NC«H^ <3>
R'>C=NOH —b- \=NH «i—i- R'>C=NC1 (4)
Rz Rz —Cl2 Rz
К. применяют в основном как промежуточные соеди-
нения для синтеза кетонов и аминов. К. ароматич.
оксикетонов являются дезактиваторами металлов,
катализирующих деструктивное окисление органич.
веществ. См. также Шиффовы основания.
Н. П. Гамбарян.
КЕТОАЛЬДЕГИДЫ — соединения, в молекуле
к-рых находятся одновременно альдегидная и кетон-
ная группы. В зависимости от взаимного расположе-
ния последних различают a-, f-, у-, 6- и т. д. кето-
альдегиды. Напр.:
-С-С-СНО —с—С—С—сно -С-С-С-С-СНО
I II I II I I II I I
0 0 о
а ₽ Т
Наиболее важным методом получения а-К. яв-
ляется окисление альдегидов или метилкетонов окисью
селена (1); а-К. получают также нитрозированием
производных Р-кетокислот (2), пиролизом ацетатов
бромкетоспиртов (3), синтезом Гриньяра из производ-
ных диэтоксиуксусной кислоты (4) и из бромметил-
кетонов по Крёнке реакции (5):
RCH2CHO (или RCOCH3) — -^RCOCHO (1)
RCH2COCn2COOR'-^—- RCH2COCCOOR’—►
NOH
—►RCHaCOCHO (2)
.OCOCIIs
RCOCH2OCOCHS—►RCOCH< —-
xBr
—»RCOCHO+CH3COBr (3)
(C2H5O)2CHCOX+RMgJ—-(C2H5O)2CHCOR—.HCOCOR (4)
RCOCH2Br^^[RCOCH2NC5H5]+Br-^i^.RCOCH=
=N<CeH5—2-5-RCOCHO (5)
*0
f3-K. получают в основном Клайзена конденсацией
из кетонов и производных муравьиной к-ты:
,сно
RCOCH2R'+HCOOC2H5—►RCOCH^
551
КЕТОАЛЬДЕГИДЫ - КЕТОНЫ
552
у-К. образуются при раскрытии фуранового цикла
в а-замещенных фуранах:
RCO(CH2)2CH(OC2H5)2—* R СО(СН2)3СНО
6-К. получают конденсацией Михаэля из насыщен-
ных альдегидов и а,0-непредельных кетонов или,
наоборот, из насыщенных кетонов и а,0-непредель-
ных альдегидов:
R'—CHCHoCOR”
RCH2CHO+R’CH=CHCOR"--- I
R-CHCHO
R”-CHCH2CHO
RCH2COR'+R"CH==CHCHO—» |
R-CHCOR'
Общими методами синтеза любых К., имеющими,
однако, меньшее значение, являются окисление кето-
спиртон и озонолиз непредельных кетонов либо
соответствующих диолефинов. К. вступают в реак-
ции, характерные для альдегидов и кетонов, причем
в зависимости от реагентов и условий реакции в ней
участвует либо только альдегидная группировка,
либо и альдегидная, и кетонная. К таким реакциям
относятся окисление, восстановление, взаимодей-
ствие с металлоорганич. соединениями, получение
моно- и диоксимов, гидразонов, 2,4-динитрофенилгид-
разонов, семикарбазонов и т. д.
Для а-К. наиболее характерна внутримолекуляр-
ная Канниццаро реакция, приводящая к получению
а-оксикислот (6). Ароматич. а-К. вступают также
в бензоиновую конденсацию (7). При взаимодействии
а-К. с 1,2-диаминами образуются производные пира-
зина (8), с амидинами — производные имидазола (9):
(6)
(7)
(8)
(9)
он' хсоон
RCOCHO ----- RCH
4 ОН
CN"
АгСОСНО ----АгСОСНСОСОАг
I
ОН
0-К. растворяются в щелочах, а также в карбонатах
щелочных металлов, дают устойчивые соли, тит-
руются бромом, окрашиваются в красный цвет в при-
сутствии хлорного железа вследствие того, что они
существуют в основном в енольной форме. Подобно
0-днкетопам, P-К. дают с гидрокспламином произ-
водные изоксазола, а с гидразином — производные
пиразола:
R
rcoch2cho + NH2OH -----_ J I + I
N _ о N — О
R
RCOCHpCHO + R'NH\H2 ---_ 11 + f 'YR
N — S-R' N — N-R'
у-К. при взаимодействии с фенилгидразином образуют
производные фенплдигпдроппридазпна:
♦
RCOCH2CHsCHO + C6H5NHNII2 —- н f^N-СбНа
H^N
R
б-К. под действием этилата натрия легко изомери-
зуются в ё-лактоны:
р-0— ' ^СН2~СН2
RCO(CH2)3CHO ---- R-СН \н„
чо-со'
К. используются в синтезе разнообразных органи-
ческих веществ.
Лит.: Traitd de chimie organlque, sous la dlr. de V. Gri-
gnard ie. a.], t. 7,P., 1950, p.i 461; Chemistry of carbon compounds,
ed. E. H. Rodd, v. 1, pt A., L.—N. Y., 1951, p. 718, v. 3, pt
B., L.— N. Y., 1956, p. 895. H. ft. Гамбарян.
a-KETO ГЛУТАРОВАЯ КИСЛОТА (1-кетоглутаро-
вая кислота, a-оксоглутаровая кислота), мол. в. 146,7,
НООС(СН3)3СОСООН,— кристаллич. продукт белого
цвета, т. пл. 115—116°; хорошо растворима в воде, в
растворах претерпевает енолизацию. КМпО4 и Н3О3
количественно окисляют К. к. (с декарбоксилирова-
нием) до янтарной кислоты. К. к. образует бисуль-.
фитное производное, семикарбазон (т. пл. 220°),,
фенилгидразон (т. пл. 152—153°), 2,4-динитрофенил-
гидразон (т. пл. 220°), оксим, к-рые используют для
идентификации К. к. Определение К. к. производится
колориметрич. и ферментативными методами; коло-
риметрич. определению обычно предшествует ее отде-'
ление от пировиноградной и др. кетокислот методами
экстракции или хроматографии. Для ферментатив-
ного определения К. к. непосредственно в природных
смесях используют дегидрогеназы молочной и янтар->
ной к-т, декарбоксилазу глутаминовой к-ты и транс*
аминазу (аминоферазу) аспарагиновой и глутами-
ловой к-т. г*
К. к. получают конденсацией этилового эфира-
щавелевой к-ты с диэтиловым эфиром янтарной к-ты,-
а также омылением щелочами дибромглутаровой к-ты:
(а, а' или а, 0). Биологич. путем К. к. можно полу-
чать из грибов Aspergillus и Penicillium и нек-рых
штаммов бактерий; напр. промышленный штамм-:
бактерии Pseudomonas fluorescens образует при сбра+.
живанпи 100 г глюкозы 16—17 г К. к. '
К. к. — одна из важнейших природных дикарбо-
новых кетокислот, важный промежуточный продукт
в процессах дыхания и обмена аминокислот, один из
компонентов цикла ди- и трикарбоновых кислот,
содержится в небольших количествах в тканях жи-
вотных и растений, в крови (ок. 20 мг/л), моче (до
450 мг/л), а также в микроорганизмах.
Одним из важнейших превращений К. к. в орга-
низме является ее окислительное декарбоксилирова-
ние в цикле трикарбоновых кислот под действием
сложного ферментного комплекса, называемого дегид-
рогеназой (оксидазой) a-К. к. (a-к е т о г л у т а-
ратдегидрогеназой). К. к. принимает ак-
тивное участие в процессах переаминирований, ката-’
лизируемых трансаминазами (аминоферазами).
Лит.: Майстер А., Биохимия аминокислот, пер. с англ ,
М., 1961, с. 96; Биохимические методы анализа растений,
Гпер. с нем.], М., 1960, с. 390, 441; Синтезы органических
препаратов, сб. 4, М., 1953, с. 284; Methods of biochemical
analysis, v. 5, N. Y., 1957, p. 107; Methods in enzymology,
ed. S. P. Colowlcli, N. O. Kaplan, v. 3, N. Y., 1957, p. 404.'
M. II. Сафонова.
КЕТОЗЫ — см. Моносахариды.
КЕТОКИСЛОТЫ — см. Альдегида кислоты и кето-
кислоты.
КЕТОНЫ — соединения, содержащие карбониль-
ную (кето-) группу, связанную с двумя углеводород-
ными радикалами: II—СО—R'. Различают симмет-
ричные К. с одинаковыми радикалами и несимметрич-
ные или смешанные. К., у к-рых карбонильвая группа
входит в кольцо, наз. циклическими. Низшие алпфа-
тич. К. — подвижные бесцветные жидкости с прпят7
ным запахом, смешивающиеся с водой. С повышением
мол. веса растворимость в воде падает. Все К. раство-
ряются в органич. растворителях.
553
КЕТОНЫ
554
Один из важных промышленных методов получения
К. заключается в дегидрировании или окислении
соответствующих вторичных спиртов:
СН3СИОНСН3--.СНзСОСНз+Нз
CH3CIIOHCH3+O—.СН8СОСН3+Н3О
Дегидрирование вызывается нагреванием спиртов
в присутствии меди и серебра, окисление производят
либо О2 воздуха в присутствии металлич. катализа-
торов (серебро, медь), либо такими окислителями,
как бихроматы или перманганаты щелочных металлов
и МпО3 (см. также Оппенауэра реакция). К. устой-
чивы к дальнейшему окислению, благодаря чему
метод имеет большее применение, чем при синтезе
альдегидов.
Важное значение имеет также получение К. пиро-
лизом металлич. солей карбоновых кислот:
(НСОО)гСа—-RCOR+CaCOs
Лучшие результаты дает пропускание кислоты
через нагретую окись металла. Метод широко при-
меняется для синтеза симметричных К. Удобным
препаративным методом синтеза К. является конден-
сация карбоновых к-т и их производных с металлоор-
ганич. соединениями, напр.:
RCOC14-RL1:—-RCOR'+LICI
К хорошим результатам приводит синтез К. кетонным
расщеплением замещенных ацетоуксусных эфиров'.
RCH2COCH2COOC2HsH-R’BrH-C3H3ONa—-
—.RCH2COCHR'COOC2H3-----RCH2COCH2R'
Этот метод дает возможность синтезировать в лабора-
тории самые разнообразные К.
Новый промышленный метод синтеза низших али-
фатич. К. заключается в катализируемом кислотами
разложении гидроперекисей алкилбензолов:
C0H3CHRR'---C6H3CRR'(OOH)—.C6H6OH+RCOR'
Меньшее значение имеют синтезы К. гидратацией
ацетиленовых углеводородов (см. Кучерова реакция),
гидролизом хлористых алкилиденов, окислением оле-
финов, взаимодействием альдегидов с диазометаном,
перегруппировкой гликолей (см. Пинаколиновая и
ретропинаколиновая перегруппировки), окислением
активных метиленовых групп, гидролизом трифенил-
фосфпнацилалкиленов и пр. См. также Кондакова
реакция, Кротоновая конденсация (синтез непредель-
ных кетонов), Дикмана реакция (синтез циклич. К.),
Фриделя — Крафтса реакция, Фриса перегруппировка
и Геша синтез (синтез ароматич. К.).
Химич, свойства К. во многом аналогичны свойст-
вам альдегидов, но К. менее реакционноспособны.
Реакционную способность К. обычно обусловливает
карбонильная группа, к-рая участвует в реакции или
активирует соединение. Реакции карбонильной группы
обычно условно разделяют на два вида — реакции
присоединения и реакции замещения атома кисло-
рода. В процессах присоединения наиболее важной
стадией (даже в случае реакций, катализируемых
кислотами) является присоединение нуклеофильной
частицы к электрофильному атому углерода карбо-
нильной группы:
Ко'*- R
ХС = О + В--—*- чс-о
R /в
Типичным примером может служить присоединение
HCN с образованием так называемых циангидринов
(оксинитрилов):
он
I
RCOR'+HCN—-R —С—CN
I
R'
Аналогично, с присоединением во всех случаях атома
водорода к атому кислорода К. реагируют с бисуль-
фитом натрия, хлороформом, триалкилсиланами и др.
При реакциях К. с металлоорганич. соединениями
RMgX, ПС=С—Na и др. (см. Реформатского реак-
ция, Иоцича реакция) углеводородный радикал при-
соединяется к углероду карбонильной группы,
а остальная часть молекулы к атому кислорода.
После гидролиза образуются третичные спирты, напр.:
CH3COCH3-t-RMgX—-(CH3)2CROMgX-—
—>(CH3)3CROH+Mg(OH)X
К процессам присоединения к карбонильной группе
К. относятся также реакции с кетенами, олефинами
и ацетиленовыми углеводородами, алнфатич. диазо-
соединениями, эфирами а-галогенокислот (см. Дар-
зана реакция). К этому же типу реакций принадлежит
присоединение двух молей К. друг к другу (см. Аль-
дольная конденсация), а также восстановление К. во
вторичные спирты:
RCOR-pHa—»R2CHOH
Вторичные спирты образуются при каталитич.
гидрировании, а также при восстановлении изопро-
пилатом алюминия (см. Меервейна — Понндорфа—
Берлея реакция), LiAlH4, NaBH4 и др. При вос-
становлении К. металлами в кислой или щелочной
среде обычно образуются продукты гидродимериза-
ции — пинаконы. При реакции а,[3-непредельных
К. с перечисленными агентами атака может направ-
ляться либо на карбонильную группу, либо на акти-
вированную двойную связь (1,4-присоединение):
.ох
,RCH=CH-C<r/
/ 'Y
RCH-CHCOR'-JX Y
Y Y X
4*RCII-CIL--C<' ' —.RCH-CHCOR’
I \ТЭ' I I
Большинство реакций замещений карбонильного кис-
лорода, обычно сопровождаемых отщеплением воды,
также протекает через стадию присоединения. Таковы
многочисленные реакции с соединениями, в молеку-
лах к-рых содержатся подвижные атомы водорода
при азоте:
R.
)CO-bH2NR"-
R'z
Г R. -ОН
I R'Z XNHR'
С—NR"
R
1ЬО*£../
Реакции с аминами, гидразинами -и др. имеют
большое значение для идентификации и выделения К.
в чистом виде, т. к. образующиеся оксимы, гидразоны,
семикарбазоны и пр. являются кристаллич. продук-
тами, при гидролизе к-рых легко регенерируется
свободный кетон. К рассматриваемым реакциям
относятся также взаимодействия К. с РС15 с замеще-
нием атома кислорода двумя атомами С1, восстанов-
ление карбонильной группы до метиленовой (см.
Клемменсена реакция и Кижнера — Вольфа реакция,
замещение атома кислорода группой CHNH3 по Лей-
карта реакции). Активирующее влияние карбониль-
ной группы К. на соседние атомы проявляется в том,
что связь атомов углерода с карбонильной группой
ослабляется и а-атомы водорода становятся подвиж-
ными. Расщепление связи С—С в К. наблюдается
при пиролизе, окислении, восстановлении в жестких
условиях, а также при реакции с азотистоводородноц
555
КЕТОСТЕРОИДЫ — КИЖНЕРА—ВОЛЬФА РЕАКЦИЯ
556
к-той. (см. Шмидта реакция), с гидроксиламином
(см. Бекмана перегруппировка).
Подвижность атомов водорода в a-положении по
отношению к карбонильной группе проявляется
в способности К. к енолизации (см. Енолы), а также
в способности этих атомов водорода легко замещаться
галогеном:
RCH3COR’—►RCHCOR'
i
а также нитрозоацильной (см. Клайзена конден-
сация), меркапто- или карбоксильной группой (см.
также Альдольная конденсация, Манниха реакция,
Михаэля реакция).
К. являются хорошими растворителями, широко
применяемыми в технике, и важными полупродук-
тами для синтеза органич. веществ. К. широко при-
меняют в современной химич. пром-сти.
Лит.: Kirk, v. 8, N. Y., 1952; Ullmann, 3 Aufl.,
Bd 9, Milnch. — B., 1957; Royals E., в кн.; Advanced orga-
nic chemistry, 2 ed., N. Y., 1956, p. 564; Stiles M., J. Amer.
Chem. Soc., 1959, 81, № 10, 2598; L a n sb u г у P. T., там же,
1061, 83, № 2, 429; T h 1 e 1 M. [u. a.], Liebigs Ann. Chem.,
1958, 611, H. 1—3, 121, 131; Houben-Weyl, 4 Aufl., Bd 7,
TI 2. Stuttgart, 1962. H. П. Гамбарян.
КЕТОСТЕРОИДЫ — см. Стероиды.
Р-КЕТОТИОЛАЗЫ — см. Трансацилазы.
КЕФАЛИНЫ — см. Коламинфосфатиды.
КИЖНЕРА РЕАКЦИЯ — получение замещенных
циклопропанов термин, разложением пиразолинов,
образующихся при действии гидразина на а, р-нена-
сыщенные альдегиды или кетоны:
R-CH-NH /СНг
RCH=CHCOR + I 2----* । ---* R-CH-CH-R
NH3 CH j—С
R
Пиразолины обычно получают при непродолжи-
тельном кипячении непредельного карбонильного
соединения со спиртовым р-ром гидразингидрата.
Разложение пиразолина проводят в присутствии
едкого кали и платинированной глины или асбеста
при 200—230°. Если исходные вещества кипят ниже
этой темп-ры, реакцию проводят в запаянных трубках.
Кроме едкого кали, реакция катализируется хиноли-
ном, триэтаноламином, кислым фосфатом натрия. Вы-
ходы на стадии разложения пиразолинов достигают
90%. Общий выход циклопропанов 50—70%.
Типичным примером К. р. является превращение
окиси мезитила в 1,1,2-триметилциклопропан (I),
пулегона — в каран (II), коричного альдегида —
в фенилциклопропан (III):
N Н NH"N -N
(СН3)2С=СНСОСН3 '-А-М- (СНа).рСС. II —*
сн2-с-сн3
/Сх2
(СН3)2С—СН-СН3
с
М.Н4 NH-N -n2
С6Н3СН=СНСНО С6Н3СН || —-
чсн2-сн
—► ,'С\2 ш
с6н5сн-сн3
Возможность получения пиразолинов из альдази-
нов ц кетазинов значительно увеличивает круг про-
изводных циклопропана, получаемых по К. р. Таким
путем удается получить углеводороды, содержащие
в молекуле до трёх циклопропановых колец:
Н3С СН2
'"nh-n'
кон
230°
Родственной К. р. является реакция алифатич.
диазосоединений (диазометан, диазоуксусный эфир)
с олефинами, двойная связь к-рых активирована нали-
чием отрицательных заместителей (карбалкоксильная
группа, циангруппа). Образующиеся пиразолины
легко теряют азот, превращаясь в производные цикло-
пропанкарбоновых к-т:
СНСООС2Н3 CH.N,
СНСООС2Н3
СНо-СН-СООС2Н5
% ” I
—сн-соос2н3
>|-соос2н3
4'-Lcooc2h3
Механизм К. р. аналогичен механизму Кижнера —
Вольфа реакции. К. р. открыта Н. М. Кижнером
в 1912.
Лит.: Джейкобс Т., в сб.: Гетероциклические соеди-
нения, под ред. Р. Эльдерфилда, пер. с англ., т. 5, М., 1961,
с. 83; М е щ е р я к о в А. П., Глуховцев В. Г., П е т-
р о в А. Д„ ДАН СССР, 1960, 130, № 4, 779.
Ю. В. Зейфман.
КИЖНЕРА — ВОЛЬФА РЕАКЦИЯ — восстанов-
ление карбонильной группы альдегидов и кетонов
в метиленовую группу. Для этого восстанавливаемое
соединение переводят в гидразон, к-рый разлагают
нагреванием в присутствии оснований:
R\c=O—► R\c=NNH2—► R\cH.>+N2
R,z/ Rr' R"
K. — В. p. приложима к карбонильным соединениям,
содержащим различные функциональные группы:
/ \ C2H6ONa 6 \
^J-C-CH3- С^5ОН » ^JhCH2-CH3(80^
NNH2 180е
<OCH2CH2COOH
nh2nh2 h2o
KOH в диэтилен
гликоле;200°
(СНг)3-СООН(95Ж)
zCeH3
хсн2-снч
О-c zNH
4CH2-Cl/
CeH5
(-100Ж)
/С3Н3
/СН2~СНХ
СН2 NH
Х''СН2~С1/
с6н5
NH2NH2-H2O
KOH в лизтилен-
гликолв;200°
Обычно гидразон нагревают в запаянной трубке
при 180° в присутствии этилата натрия, к-рый часто
заменяют едким натром или едким кали. Гидразоны
нек-рых кетонов (моногидразоп бензила, гидразон
бензофенона) и альдегидов (моногидразон фенил-
глиоксаля) распадаются на азот и соответствующее
метиленовое соединение уже при нагревании в отсутст-
вии катализаторов либо в присутствии избытка
557
КИЖНЕРА—ВОЛЬФА РЕАКЦИЯ —КИНАЗЫ
558
гидразингидрата, или при кипячении с водным рас-
твором щелочи. Иногда в качестве катализатора,
помимо щелочи, употребляют платину, палладий или
никель. Для осуществления К.—В. р. при атмосфер-
ном давлении в случае гидразонов, разлагающихся
с трудом, применяют высококипящие растворители
(обычно спирты или гликоли). Особенно удобен прием
Хуанг — Минлона — карбонильное соединение нагре-
вают с гидразингидратом и едким кали (или едким
натром) в ди- или триэтиленгликоле с одновременной
отгонкой воды и избыточного гидразингидрата и за-
тем кипятят при 180—200° 3—4 часа.
Вместо гидразонов можно применять семикарба-
зоны и 2,4-динитрофенилгидразоны; при этом первой
ступенью реакции является расщепление до гидра-
зона:
R\ R"OH R\
C=NNHX + R"O~------— C=NNHq + R"X
R-/ R'z
Иногда при К.—В. р. затрагиваются и другие функ-
циональные группы:
NH2NH2-H2O
КОН в ДИ31ИЛВН-
глиноле
В
NH2NHa-HsO
КОН в лиэтилен-
СОСН3 гликоле
сн2сн
Гидразоны а-окси- и а-ампнокетонов часто превра-
щаются в олефины, напр.:
СН3 СН3
СН3, I СНз. |
>С--С—СН»СН»СН—► )С=СНСН2СН2СНСН2СНз
СН3/| II | СНзХ
ОН NNHa СНоСНз
Ненасыщенные карбонильные соединения в зави-
симости от условий и исходных веществ образуют
либо производные циклопропана (через пиразолины)
(1), либо олефины (2); в последнем случае картина
может осложняться сдвигом двойной связи:
Побочные реакции при К.—В. р. — образование
азина и восстановление до спирта:
R
)c=nnh2 R zR
г--------r->n-n=c4r,
Н2О R\
c=nnh2 — с=о—
R< R ’
Rn.
, .СНОН
Rz
Обе эти реакции затрудняются при подавлении гид-
ролиза, что достигается- применением избытка гид-
разина и удалением реакционной воды.
Кинетика К.—В. р. согласуется с механизмом,
включающим образование аниона с последующим его
разложением:
Rs, +б R. Rk*-n О.
R-C=N-NH2^r,zCtN—- R.zci?-N=N : -
H-N'
mN,+ R>H^ >8;
К. — В. р. имеет ряд преимуществ перед другими
способами превращения карбонильной группы в ме-
тиленовую (ср. Клемменсена реакция). Она приложима
к карбонильным соединениям, к-рые чувствительны
к кислотам, и может применяться для получения боль-
ших количеств вещества. К.—В. р. широко приме-
няют в лабораторной практике. Возможность пре-
вращения гидразона в метиленовое соединение
впервые показана Т. Курциусом (1891). Н. М. Кижнер
(1911) и Л. Вольф (1912) продемонстрировали приме-
нение реакции на многих примерах и разработали
экспериментальные условия реакции.
Лит.: Franzen V., Chemiker Ztg, 1956, 80, Н. 19, 667;
Родионов В. М., Ярцева Н. Г., в кн.-. Реакции и ме-
тоды исследования органических соединений, кн. 1, М.—Л.,
1951, с. 7. Е. М. Рохлин.
КИНАЗЫ — ферменты, осуществляющие перенос
остатков фосфорной или пирофосфорной к-ты между
каким-либо органич. соединением и аденозинтрифос-
форной кислотой АТФ (см. Аденозинфосфорные кис-
лоты) или иным фосфорилированным мононуклео-
тидом. К., катализирующие перенос фосфатного
остатка, наз. фосфокиназами, или ф о с-
фоферазами, а переносящие пирофосфатный
остаток — пирофосфокиназами. К К. отно-
сятся также сульфокиназы, осуществляющие
перенос остатка серной к-ты с З'-фосфоаденил-суль-
фата на фенолы, стероиды, углеводы и др. соеди-
нения.
Фосфокиназы, число к-рых достигает 50, можно
разделить на две основные группы: 1) К., катализи-
рующие обратимый перенос фосфата между фосфо-
рилированными нуклеотидами и другими богатыми
энергией фосфорилированными соединениями (ацил-
фосфатами, енолфосфатами, амидинфосфатами). 2) К.,
катализирующие необратимые реакции фосфорилиро-
вания углеводов, спиртов, витаминов и других со-
единений путем переноса фосфата АТФ на соответ-
ствующий субстрат (подробнее см. Фосфокиназы).
В качестве примера пирофосфокиназ, число к-рых
незначительно, можно назвать тиамин-пиро-
фосфокиназу, осуществляющую синтез кофер-
мента тиаминдифосфата из витамина В! (тиамина).
Фермент выделен из дрожжей и тканей животных;
оптимум его действия при pH 6,0—7,5.
Физиологическая роль сульфокиназ состоит в обез-
вреживании нек-рых токсич. для организма веществ
путем связывания
Донором остатков
серной к-ты слу-
жит 3'-фосфоаде-
нил сульфат (I), в
к-ром серная к-та
активирована пу-
тем образования
макроэргич. ангид-
ридной связи с
фосфатным остат-
ком.
их в виде эфиров серной к-ты.
Исторически название К. сохранилось также за
нек-рыми белковыми факторами, способствующими
превращению неактивной формы фермента (зимо-
г е н) в активную. Примером таких К. может служить
559
КИНГА РЕАКЦИЯ-КИНЕТИКА ХИМИЧЕСКАЯ
560
тромбокиназа, катализирующая в присутствии ионов
Са2+ превращение протромбина в тромбин.
Лит.: Диксон М., Уэбб Э., Ферменты, пер. с англ.,
М., 1961; Нейландс Дж., Штумпф П., Очерки по хи-
мии ферментов, пер. с англ., М., 1958; Б олдуинЭ., Основы
динамической биохимии, пер. с англ., М., 1949; см. также при
ст. Ферменты и Фосфокиназы. В. Б. Спиричев.
КИНГА РЕАКЦИЯ — получение четвертичных пи-
ридиниевых солей иодированием метил- и метиленке-
тонов в присутствии пиридина:
NCjHj
I +
ArCOCHs+Js-HICsHaN—-ArCOCH J-+C5H5NH
I I J-
R R
Метод применяется для получения четвертичных
солей из различных кетонов, содержащих ароматпч.
или гетероцпклпч. остатки:
С6Н5СОСН3 + J2 + c5h5n —>- c6h5coch2ncsh5 г
Q-СОСНз
Вместо пиридина могут употребляться другие ге-
тероциклич. основания.
К. р. пригодна не только для метплкетонов, но
н для нек-рых метилзамещенпых гетероцпклпч. со-
единений:
Обычно исходный кетон нагревают на водяной бане
с эквимолярным количеством иода в избытке пири-
дина. Выпавший в осадок продукт отделяют от иод-
гидрата пиридина перекристаллизацией. Иногда
вместо иода можно применять бром.
Механизм К. р. заключается в иодировании метил-
кетона с последующей заменой атома иода на азотсо-
держащий остаток. Самой медленной стадией реакции
является отщепление протона от метилкетона (1):
CeH3COCH8+C6H5Ni=iCeH&COCHa-|-C6H5NH (D
C0H.COCH--^4i*C0H5C°CH.J-^—-C6H5COCHaNC5H5
К. p. с последующим щелочным гидролизом исполь-
зуется для получения кислот из метплкетонов в слу-
чаях, когда не могут применяться другие методы:
Четвертичные пиридиниевые соли, полученные в ре-
зультате К. р., широко используются в качестве
алкилирующих агентов, например в Крёнке реакции.
К. р. открыта Л. К. Кингом в 1944.
Лит.: Krauch Н., Kunz W., Namenreaktionen der
organlschen Chemie, Heidelberg, 1961; King L. C., McWhir-
ter LVl, t XvUWldUU Л» A-*.» j. ЛИНЯ. uutul. OUU., 1 J 40,
70, 239. E. M. Рохлин.
КИНЕКС — CM. Сулъфопиридазии.
КИНЕТИКА ХИМИЧЕСКАЯ. Содержание:
Основные понятия ...........................559
Методы измерения скоростей реакций .........561
Кинетика гомогенных реакций ................562
Кинетика гетерогенных реакций ..............565
Основные понятия. Химическая кинетика — уче-
ние о скоростях химич. реакций; раздел физич.
химии. Реакция может протекать гомогенно, т. е.
в объеме фазы, и гетерогенно, т. е. на границе раз-
дела фаз. Скоростью реакции наз. число
актов реакции, происходящих за единицу времени
в единице объема фазы, — в случае гомогенной
реакции, или на единичной Поверхности раздела—
в случае гетерогенной реакции. Скорости
реакций выражают также различными величинами,
пропорциональными отвечающим приведенному опре-
делению. Напр., скорость гомогенной реакции часто
измеряется изменением числа молей одного из исход-
ных веществ или продуктов реакции и выражается
в единицах моль • см ~3-сек~1. В случае гетерогенной
реакции, протекающей на поверхности твердого тела,
имеющего форму пористых зерен, для практич. целей
рассчитывают скорость реакции на единицу объема
слоя зерен или на единицу веса, а не на единицу
поверхности твердого тела. Особую группу гетеро-
генных реакций представляют электрохимия., или
электродные реакции, происходящие на
границе электронного проводника (металл или полу-
проводник), наз. электродом, и ионного проводника.
В этих реакциях участвуют электроны, подходящие
к границе или уходящие от нее сквозь электронный
проводник. Скорость таких реакций обычно характе-
ризуют плотностью тока, Т. е. отношением си-
лы тока к площади поверхности электрода.
Под кинетикой реакции понимают зави-
симость скорости данной реакции от концентраций
веществ, темп-ры и других параметров, напр. потен-
циала электрода — в электрохимия, реакциях, интен-
сивности света — в фотохимия, реакциях, мощности
дозы излучения — в радиационно-химич. реакциях.
Реакции, выражающиеся простым стехиометрия,
уравнением, фактически часто являются слож-
ными реакциями, т. е. осуществляются
в результате одновременного протекания нескольких
простых реакций, называемых в этом случае ста-
диями (или элементарными реакциями). Продукты
стадий, быстро потребляемые в других стадиях и
поэтому присутствующие в реагирующей системе
лишь в незначительных концентрациях, наз. про-
межуточными веществами. Напр., в го-
могенной газовой реакции: 2NO + Оа = NaO4, скла-
дывающейся из стадий: 2 NO = (NO)a; (NO)a + Оа =*
— Na04, димер окиси азота (NO)a является проме-
жуточным веществом.
Стадиями в более широком смысле могут быть не
только химич. реакции, но также процессы диффузии,
передачи энергии при столкновении молекул и др.
Задача выяснения механизма реакции (в смысле К. х.)
обычно заключается в установлении природы проме-
жуточных продуктов и стадий реакции.
В случае простой обратимой реакции наблюдаемая
скорость реакции
ш == Ш — (1)
где со и со — скорости реакции в прямом и обратном
направлениях. При достижении химич. равновесия
со = 0, т. е. со = со. В сложных обратимых реакциях
обратимы все стадии и при равновесии для каждой
химич. стадии s выполняется равенство со, — со,.
Иногда в реагирующей системе по отношению ко всем
стадиям, кроме одной, наз. лимитирующей
(или стадией, определяющей скорость), практически
наблюдается равновесие. В таких случаях для кине-
тики реакции имеет значение лишь природа лимити-
рующей стадии, но не других стадий.
Широко распространены явления катализа.
Катализатором наз. вещество, резко увеличиваю-
щее скорость реакции пли вызывающее реакцию,
к-рая термодинамически возможна, но в отсутствии
катализатора не происходит. При этом катализатор,
561
КИНЕТИКА ХИМИЧЕСКАЯ
562
вступая в промежуточное химич. взаимодействие
с участниками реакции, освобождается вновь в после-
дующих стадиях и поэтому не расходуется в ходе
реакции или расходуется в количествах, малых по
сравнению с количеством продуктов реакции. В хи-
мич. технологии наибольшее применение находит
гетерогенный катализ — каталитич. дей-
ствие поверхностей твердых тел. Большое биологич.
значение имеют катализаторы живой клетки — фер-
менты (энзимы), являющиеся веществами белковой
природы.
Кинетический метод выяснения меха-
низма реакций сводится к сопоставлению наблюдае-
мой на опыте кинетики реакции с зависимостями,
выводимыми теоретически на основе определенных
предположений о ее механизме. Другие пути изуче-
ния механизма реакций, такие как метод меченых
атомов (см. Изотопные индикаторы), исследования
оптич. активности, применение спектроскопии, масс-
спектрометрии, электронного парамагнитного резо-
нанса для идентификации и измерения концентраций
промежуточных продуктов, существенно дополняют
кинетич. метод. Для выяснения механизма гетероген-
ного катализа большое значение имеют исследования
адсорбционных явлений, а также физич. методы изу-
чения поверхностей — изменения контактных раз-
ностей потенциалов и др.
Методы измерения скоростей реакций. Приемы,
применяемые для изучения кинетики реакций, весьма
разнообразны. Здесь рассмотрены основные методы
измерения скорости тепловых реакций, т. е.
реакций в системах, обменивающихся с окружающей
средой энергией лишь в форме передачи тепла. При
использовании статического метода реак-
цию проводят в замкнутом сосуде при постоянной
темп-ре. Состав системы изменяется со временем,
и. о скорости реакции судят по изменению состава,
Определяемого анализом, или по изменению давления,
если последнее зависит от состава. Применяют также
измерения к.-л. другого свойства реагирующей смеси,
зависящего от состава.
Скорость реакции в статич. системе непрерывно
изменяется со временем вследствие изменения со-
става реагирующей смеси. Для сопоставления с опыт-
ными данными уравнения, выражающие скорость
реакции как функцию концентраций реагирующих
веществ, должны быть проинтегрированы по времени.
Иногда вычисляют скорость реакции путем числен-
ного или графич. дифференцирования, однако часто
это неосуществимо, т. е. операция дифференцирования
предъявляет высокие требования к точности опытных
данных.
Обычно при анализе результатов исследования
кинетики сложной реакции в статич. системе дела-
ют предположение о квазистационарном
протекании реакции. Это означает, что концентрации
промежуточных веществ и скорости стадий в каждый
момент времени принимают практически равными тем
стационарным значениям, к-рые установились бы по
истечении достаточно большого времени в системе
с постоянными значениями концентраций исходных
веществ и продуктов реакции, равными данным
мгновенным концентрациям. В случае реакций с раз-
ветвляющимися цепями (см. Цепные реакции) тече-
ние реакции может быть существенно нестационар-
ным. При этом концентрации промежуточных веществ
растут со временем, и реакция проходит с возрастаю-
щей скоростью, к-рая определяется не только кон-
центрациями исходных веществ и продуктов реакции,
но и временем, протекшим от начала реакции.
Проточный метод исследования кинетики
реакций прост в осуществлении и позволяет исследо-
вать реакции, скорость к-рых слишком велика для
измерения ее статич. методом. В проточной системе
реагирующая смесь протекает с известной постоянной
скоростью сквозь зону реакции (напр., область по-
вышенной темп-ры или объем, заполненный зернами
катализатора). Степень превращения исходных ве-
ществ в продукты определяется по составу отходя-
щей смеси. Реакция проходит стационарно, состав
реагирующей смеси в каждой точке зоны реакции
не зависит от времени, но изменяется от точки к точке.
Для сравнения опытных данных с уравнением кине-
тики реакции последнее должно быть проинтегри-
ровано по объему реакционной зоны. Чтобы облег-
чить эту задачу, обычно предполагают, что осущест-
вляется режим идеального вытесне-
н и я, т. е. принимают, что скорость потока одина-
кова во всех точках любого сечения, перпендикуляр-
ного направлению потока, и что продольное переме-
шивание отсутствует. В случае газовой реакции,
происходящей с изменением числа молекул, при инте-
грировании должно быть учтено изменение объема
реагирующей смеси. Если изменение объема отсут-
ствует или им можно пренебречь, интегрирование
может быть проведено так же, как в статич. системе,
для этого вводится т. н. время контакта
или время пребывания в реакционной зоне, опреде-
ляемое ур-нием:
t = W/U (2)
где W — объем зоны реакции, U — объемный расход
реагирующей смеси (при темп-ре и давлении зоны
реакции), т. е. объем смеси, проходящий через реак-
тор за единицу времени.
Необходимость интегрирования кинетич. ур-ний
устраняется при использовании безградиент-
ных реакторов, позволяющих получить не-
посредственно скорость реакции как функцию кон-
центраций участников реакции при данной темп-ре.
Для исследования гомогенной реакции может быть
применен проточный перемешиваемый
реактор. В реактор, снабженный мощной мешал-
кой, с постоянной скоростью вводятся исходные
вещества, и из него выводится реагирующая смесь
так, чтобы количество реагирующей смеси в реакторе
было постоянно. При установившемся стационарном
состоянии анализ отбираемой смеси дает не завися-
щий от времени состав реагирующей смеси. Скорость
реакции определяется ур-нием:
ш = СПЛУ
(3)
где С — концентрация продукта реакции, и — рас
ход смеси на выходе из реактора, W — объем реактора
Для гетерогенных про-
цессов безградиентнып ре-
актор реализуется в про-
точно-циркуляци-
онной системе (рис.).
Скорость циркуляции долж-
на быть настолько велика,
чтобы состав смеси на вхо-
де и выходе из реактора
практически совпадал. В та-
ком реакторе отсутствуют
проточно - циркуляционная
система с электромагнитным
поршневым иасосом: 1 — ре-
актор в печи; 2 — циркуля-
ционный насос; 3 — клапа-
ны; 4 — ввод исходйой газо-
вой смеси; S — выход реаги-
рующей смеси за реактором;
5'—выход реагирующей сме-
си до реактора.
не только градиенты концен-
траций, но и градиент тем-
пературы, что недостижимо
в простом проточном реак-
торе при реакциях с боль-
шим тепловым эффектом.
Кинетика гомогенных ре-
акций. В идеальных газо-
вых смесях и в идеальных
жидких растворах скорости простых (одностадийных)
реакций подчиняются действующих масс закону.
563
КИНЕТИКА ХИМИЧЕСКАЯ
564
Скорость реакции?
1Ъ -J- тМ -р . . . = <1 Q + rR + • • • W
(I молекул вещества L реагируют с т молекулами
вещества М и т. д.) дается ур-нием:
ш =7 с* с™ . .. (5)
L М
где — концентрация вещества L, т. е. число моле-
кул L в единице объема (или пропорциональная
величина) и т. д., к — константа скорости
реакции.
Подобно этому, скорость обратной реакции:
Ф = ГС9С’1... (6)
Ц К
При равновесии со = со, следовательно:
С™ . . . к
L М
Это уравнение выражает закон действующих масс
для химич. равновесия в идеальных системах; К —
константа равновесия.
Закон действующих масс для скоростей реакций
можно пояснить следующим образом. Чтобы про-
изошел акт реакции, необходимо столкновение моле-
кул исходных веществ, т. е. молекулы должны
сблизиться друг с другом на расстояние порядка
атомных размеров. Вероятность найти в нек-ром
малом объеме в данный момент I молекул вещества
L, т молекул вещества М и т. д. пропорциональна
с£с^..., следовательно,число столкновений в единице
объема за единицу времени пропорционально этой
величине; отсюда вытекает уравнение (5). Известны
случаи, когда каждое столкновение приводит к реак-
ции. В других случаях лишь малая доля столкно-
вений приводит к реакции, потому что реагировать
способны лишь молекулы с энергией много большей,
чем средняя энергия при данной темп-ре. Это не
влияет на применимость уравнения (5), т. к. число
столкновений, приводящих к реакции, пропорцио-
нально общему числу столкновений.
Число молекул, участвующих в элементарном акте
реакции, выражают термины мономолеку-
лярная, бимолекулярная, тримо-
лекулярная реакции. Оно не превышает 3,
т. к. столкновение большего числа молекул мало
вероятно. Приведенная интерпретация уравнения (5),
очевидно, не применима к мономолекулярным реак-
циям. В этих реакциях соблюдение закона действую-
щих масс определяется тем, что сложная молекула
претерпевает химич. превращение (распад, изомери-
зация) не в момент получения необходимой для пре-
вращения энергии, а со значительным запаздыва-
нием. Благодаря этому в ходе реакции устанавли-
вается равновесие между «энергетизир'о-
ванными» (т. е. имеющими необходимую для
реакции энергию) молекулами и исходными молеку-
лами. В результате скорость реакции, пропорцио-
нальная концентрации энергетизированных молекул,
пропорциональна также и концентрации исходных
молекул. В случае газовых мономолекулярных реак-
ций при низких давлениях скорость передачи энергии
при столкновениях молекул недостаточна, поэтому
концентрация энергетизированных молекул меньше
равновесной и закон действующих масс не выпол-
няется.
Кинетика сложных реакций может быть описана
путем применения уравнения (5) к каждой стадии.
Особый, весьма распространенный тип сложных
реакций представляют цепные реакции. Скорости
сложных реакций иногда описывают уравнениями
вида (5). При этом порядок реакции, т. е.
сумма I + т + ... , а также отдельные показатели
I, тит. д., определяемые Опытным путем, могут
не отвечать стехиометрич. уравнению. Напр., реак-
ция 2N3O5 = 2N2C>4 + О2 протекает по уравнению
первого порядка, ее скорость пропорциональна
cn205 (а не cnso=,)-
Зависимость скорости реакции от темп-ры Т опре-
деляется уравнением Аррениуса (1889):
dlnh Е
dT kT* 1 1
где Е — энергия активации, рассчитан-
ная на 1 акт реакции, к — постоянная Больцмана.
Энергия активации равна средней избыточной энер-
гии молекул, вступающих в элементарный акт реак-
ции. Приближенно ее можно отождествить с величи-
ной энергетического барьера для данной реакции,
т. е. минимальной энергией (по отношению к энергии
молекул при О°К), при к-рой акт реакции становится
возможным. ~Е в первом приближении не зависит от Т,
поэтому интегрирование дает:
__, . ьт
_ fe=Ae (9)
где А — величина, в том же приближении, не завися-
щая от Т, называемая предэкспоненциаль-
н ы м, или частотным фактором.
Независимо от механизма реакции справедливо
термодинамич. ур-ние В ант-Гоффа:
“ = -^- (10)
dT КТ* U 1
где &Е — изменение внутренней энергии при 1 акте
реакции. Уравнение (8) и аналогичное уравнение
для к, содержащее энергию активации обратной
реакции Е, дают поэтому, если выполняется уравне-
ние (7),
Е — Е=АЕ (И)
Численные значения 2 и &Е обычно рассчитывают
на 1 моль, т. е. на число актов реакции, равное числу
Авогадро N (N = 6,02 • 1033). Тогда в уравнениях
(8) — (10) к заменяется на газовую постоянную
R = kN. Наблюдаемые на опыте значения Е для
различных реакций лежат в пределах от 0 до
100 ккал/моль.
Простое истолкование уравнения (9) в случае
газовых бимолекулярных реакций достигается с по-
мощью следующей модели. Будем считать реагирую-
щие молекулы шарами, отталкивающимися друг от
друга до тех пор, пока не произойдет соприкоснове-
ние их поверхностей, приводящее к реакции. Пусть
потенциальная энергия в момент соприкосновения
равна Е. Число столкновений с преодолением оттал-
кивания будет, согласно закону распределения Макс-
велла — Больцмана, в е~E/kT раз меньше числа столк-
новений таких же, но не взаимодействующих молекул.
Последнее известно из кинетич. теории газов; при
концентрациях, равных единице (1 молекула в еди-
ничном объеме), оно дается формулой:
Z=A.('2zftTzui±^yA(ri + r2)8 (12)
где г, и г3 — радиусы молекул, тт и т2 — их массы,
о=1, если молекулы различны, и а = 2, если мо-
лекулы одинаковы. Необходимость для осуществле-
ния реакции определенной взаимной ориентации
молекул учитывается с помощью т. н. с т е р и ч е-
ского фактора р(р<^1), в результате
fe*=pZe-®/feT (13)
и А = pZ. Изложенный подход к вычислению со
наз. теорией столкновений. Расчет по
565
КИНЕТИКА ХИМИЧЕСКАЯ
566
>гравненпю (12) дает Z порядка 1010—10 9 c.w3 • моле-
кула1 • сек'1, или 1014—Ю15^.!!3 • моль 1 сек1. Опыт-
ные данные, относящиеся к реакциям между про-
стыми молекулами, приводят к близким значениям А,
напр. для реакции
H24-J2=2HJ
А — 1 • 1014 см? • моль"1 • сек~г (Е = 40 ккал!моль).
В случае сложных молекул получаются много мень-
шие значения А, напр. в реакции присоединения бу-
тадиена к акролеину с образованием тетрагидробенз-
альдегида (реакция Дильса — Альдера):
сн., сн2 /СНа\
и-/ нс сн2
н-с +ьАно = нсх /С<СН0
~^'СН, сн. н
бутадиен акролеин тетрагидробензаль-
дегид
А = 1,5 • 109 слг3-моль 1-сек 1 (Е = 19,7 ккал!молъ),
так что р порядка 10 5. Такие малые стерич. факторы
показывают недостаточность теории столкновений.
Более точное вычисление предэкспоненциального
фактора А для бимолекулярных реакций осуществ-
ляется с помощью метода активированного
комплекса, или переходного состояния. Акти-
вированным комплексом (переходным состоянием)
наз. возникающая в ходе элементарного акта реакции
конфигурация атомов, к-рая отвечает максимальному
значению потенциальной энергии реагирующей си-
стемы, другими словами — вершине потенциального
барьера, преодолеваемого реагирующей системой.
Сущность метода активированного комплекса заклю-
чается в том, что скорость реакции вычисляется
из концентрации активированных комплексов и
скорости движения составляющих активированный
комплекс атомов, находимых методами статистич.
механики.
В случае мономолекулярных реакций простейшая
трактовка использует представление о внутримоле-
кулярном колебании с нек-рой частотой v, при-
водящем к реакции, если энергия, сосредоточенная
в этом колебании, превышает критич. значение Е.
Энергия гармония, колебания складывается из
2 квадратичных членов, поэтому вероятность того,
что она превышает Б, равна (при условии,
что осуществляется распределение молекул по со-
стояниям, отвечающее статистич. равновесию). За
единицу времени происходит v колебаний, следова-
тельно, вероятность превращения молекулы за еди-
ницу времени равна ve~E'lhT. Отсюда следует уравне-
ние (9) с А = V. Опытные данные для большинства
мономолекулярных реакций приводят к значениям А
около 1013—1014 сек1, что примерно соответствует
порядку величины частот колебаний молекул, хотя
и несколько превышает его. И в этом случае согласие
теорви с опытом может быть улучшено при исполь-
зовании метода активированного комплекса.
Уравнение Аррениуса показывает, что протекание
реакции связано с преодолением энергетич. барьера.
Физич. природа этого барьера была разъяснена
Лондоном (1928) на основе квантово-механич. теории
химич. связей. Силы отталкивания, возникающие
при сближении молекул, имеют то же происхождение,
что и силы, связывающие атомы в молекулы. В при-
ближенной квантово-механич. трактовке, известной
как метод валентных связей (метод электронных пар),
они отвечают обменному взаимодействию электронов.
Кинетика гетерогенных реакций. Специфическая
особенность гетерогенных реакций состоит в том,
что процессы переноса исходных веществ
из объема фазы к границе фаз и продуктов реакции
в обратном направлении играют роль стадий реакции.
Степень влияния процессов переноса вещества на
скорость реакции не является неизменной характе-
ристикой реакции, она может существенно изменяться
при переходе из одной области значений параметров,
таких, как темп-ра, давление и др., в другую. Твердые
катализаторы газовых реакций обычно применяются
в форме пористых зерен с сильно развитой внутрен-
ней поверхностью (напр., 10 м?)г и более). Если
диффузия в порах медленна, реакция происходит
с заметной скоростью лишь в прилегающем к наруж-
ной поверхности слое зерна, толщина к-рого мала
по сравнению с размером зерна. В таком случае
говорят, что реакция протекает во в н у т р е н н е й
диффузионной области. Известны также
случаи, когда скорость реакции определяется пере-
носом вещества к наружной поверхности катализатора
или твердого тела, реагирующего с газом, это — слу-
чаи внешнейдиффузионной области.
Если процессы переноса достаточно быстры по срав-
нению со скоростями химич. стадий и поэтому не
влияют на скорость реакции, то говорят, что реакция
протекает вкинетической области. В даль-
нейшем будем предполагать, что гетерогенная реак-
ция протекает в кинетич. области, и рассмотрим
основы кинетики каталитич. реакций на поверхно-
стях твердых тел, а также тесно связанной с ней
кинетики хемосорбции газов на твердых телах
(т. е. адсорбции, вызванной силами химич. сродства).
Простая формулировка закона скорости реакции,
аналогичная закону действующих масс для гомоген-
ных реакций в идеальных системах, получается,
если, следуя Лэнгмюру (1918), принять, что поверх-
ность твердого тела содержит определенное число
одинаковых мест, каждое из к-рых может удерживать
при хемосорбции одну молекулу или атом, причем
взаимное влияние адсорбированных частиц отсут-
ствует (т. е. энергия адсорбции частицы на данном
месте не зависит от того, имеются ли адсорбирован-
ные частицы на других близких местах). Эту модель
можно назвать моделью идеального адсор-
бированного слоя (по аналогии с терминами
«идеальный газ», «идеальный раствор»). В реакцию
могут вступать частицы, адсорбированные на соседних
местах поверхности. Возможна также реакция адсор-
бированной частицы с налетающей на нее частицей
из газовой фазы. В общем случае реакция запишется
i(I)+ ;(!) + ... 4-!L-|-mM + . . , = Р (14)
Здесь I, J и т. д. — вещества, вступающие в реакцию
из адсорбированного состояния, что обозначено скоб-
ками, L, М и т. д. — вещества, вступающие в реак-
цию непосредственно из объема, Р — совокупность
продуктов реакции. В случае идеального адсорбиро-
ванного слоя, а также идеальной объемной фазы,
скорость реакции на поверхности:
ш . . . С^С™ . . . (‘ + i + • • •) (15)
где 6j — степень покрытия поверхности веществом I
(т. е. отношение числа мест, занятых частицами I,
к общему числу мест) и т. д. 60— доля свободной
поверхности, s — число мест поверхности, занимае-
мых одним активированным комплексом. Как и
в случае гомогенных реакций, фактически в элемен-
тарном акте участвует лишь небольшое число молекул.
Уравнение (15) включает процессы адсорбции и
десорбции как частные случаи. Напр., если моле-
кулы Н3 хемосорбируются на поверхности твердого
тела с диссоциацией на атомы, причем каждый атом
занимает место на поверхности, то s = 2 и со=
567
КИНЕТИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
568
Зависимость к в уравнении (15) от темп-ры описы-
вается уравнением (9). Как и в случае гомогенных
реакций, возможно теоретич. вычисление А с по-
мощью метода активированного комплекса. Величины
доли свободной поверхности и степеней покрытия
поверхности исходными веществами, продуктами реак-
ции й другими веществами, присутствующими в си-
стеме, определяются условиями стационарности по-
верхностных концентраций в ходе реакции. Напр.,
для исходного вещества I:
"“(I) = ш (I) + 46)
где Ф(1) и cD^j) — скорости адсорбции и десорбции ве-
щества I, со — скорость реакции по уравнению (1).
В частных случаях возможны упрощения. Часто
в уравнениях вида уравнения (16) пренебрегают
членом, отвечающим гео. Это эквивалентно принятию
поверхностной реакции в качестве лимитирующей
стадии.
Картина идеального адсорбированного слоя в ряде
случаев не передает наблюдаемых зависимостей. Это
объясняется неоднородностью поверх-
ности или взаимным влиянием хе-
мосорбированных атомов или молекул.
Каждый из этих эффектов приводит к уменьшению
энергии связи адсорбированных частиц с поверх-
ностью при увеличении степени покрытия поверх-
ности. Удовлетворительное и в то же время достаточно
простое описание явлений достигается, если допустить
линейную зависимость энергии адсорбции, а также
энергии активации, от степени покрытия поверхности.
Примером получаемых таким путем уравнений может
служить уравнение кинетики синтеза аммиака.
Выше освещены некоторые теоретич. аспекты хи-
мия. кинетики. Необходимо указать, что кинетика
реакций, используемых в химич. технологии, имеет
большое практическое значение, т. к. позволяет осу-
ществить рациональное проектирование аппаратуры,
автоматизацию процессов и т. д. Возможности такого
использования кинетики реакций весьма расшири-
лись в последние годы благодаря прогрессу в области
вычислительной техники, обусловленному развитием
электронных счетных машин.
Лит.: Кондратьев В. Н., Кинетика химических
газовых реакций, М., 1958; Глестон С., Лейдлер К.
и Э п р и н г Г., Теория абсолютных скоростей реакций, пер.
с англ., М., 1948; Семеновы. Н., О некоторых проблемах
химической кинетики и реакционной способности, 2 изд.,
М., 1958; Франк-Каменецкий Л. А., Диффузия и
теплопередача в химической кинетике, М.—Л., 1947; Frost
A. A., Pearson R. G., Kinetics and mechanism, 2 ed.,
N. Y.—L., 1961; В en son S. W., The foundations of chemical
kinetics, N. Y. [a. o.], 1960; Walas S. M., Reaction kinetics
for chemical engineers, N. Y., 1959; Жермен Ж., Гетеро-
генный катализ, пер. с франц., М., 1961, гл. 6.
М. И. Темкин.
КИНЕТИЧЕСКИЕ МЕТОДЫ АНАЛИЗА — ме-
тоды химич. анализа, основанные на зависимости
между скоростью химич. реакции и концентрацией
реагирующих веществ. Определяемое вещество в
К. м. а. может расходоваться в процессе реакции или
же быть ее катализатором. В последнем случае кине-
тич. методы характеризуются очень высокой чувстви-
тельностью.
Реакция, по скорости к-рой производят определе-
ние вещества, наз. индикаторной. Если такая
реакция может быть схематически представлена
ур-нием:
А + В = X + Y (1)
то скорость ее выражается в простейшем случае:
= kab (2)
где к — константа скорости, а а, Ь, х изменяющиеся
в процессе реакции концентрации соответственно
веществ А, В и X. Определяемое вещество (напр., В)
берется в большом избытке по сравнению с А, и кон-
центрация его за время проведения опыта практи-
чески не меняется. Если реакция (1) является гомо-
генно-каталитической, то ее скорость зависит также
от концентрации катализатора:
= (3)
где z — каталитический коэфф, (константа скорости
каталитич. реакции), Пс — функция концентраций
А и В (в простейшем случае их произведение), Ск —
концентрация катализатора.
Скорость реакции находят либо непосредст-
венно на опыте при помощи специальных приборов
или приемов, либо из графика зависимости х от г
(тангенс угла наклона прямой или касательной).
В этом случае концентрацию вещества В или катали-
затора вычисляют по ур-ниям (2) или (3). Концентра-
ции этих веществ могут быть найдены также при
помощи градуировочных (калибровочных) графиков,
построенных в координатах: искомая концентрация —
( . Если за время Дг изменение концентрации г(Аж)
небольшое по сравнению с начальными концентра-
циями А и В, то концентрация катализатора может
быть найдена из ур-ния:
При этом для наблюдения выбирают либо один и тот
же отрезок времени (Дг = const), либо проводят
реакцию до одного и того же изменения концентрации
х (Дг = const). В первом случае имеет место прямая
пропорциональность между Ск и Ах, во втором —
прямая пропорциональность между Ск и .
Если за избранный отрезок времени происходят
существенные изменения концентрации А и В, то
ур-ния (2) и (3) необходимо интегрировать. Инте-
гральная форма ур-ний (2) и (3) используется и тогда,
когда скорость реакции определяется по изменению
концентрации исходных веществ. При использовании
интегральной формы ур-ний (2) и (3) возможны те же
варианты, что и при применении дифференциальной
формы.
Скорость реакции (1) можно измерять при помощи
различных методов: титриметрич., газоволюметрич.,
фотометрич., фототурбидиметрич., полярографич., по-
тенциометрич. и др. Для определения различных
неорганич. веществ используется несколько десятков
каталитич. реакций следующих типов: 1) Окислитель-
но-восстановительные реакции [окисление перекисью
водорода, галогенатами, солями церия (IV) и др.]-.
2) Реакции изотопного обмена между одноименно
заряженными ионами (Се4+ — Се3+, AuClj —СН и др.).
3) Реакции, приводящие к возникновению каталитич.
полярографич. токов. 4) Реакции замещения во внут-
ренней сфере комплексных соединений [напр., заме-
щение CN" в Fe(CN)’ ] водой. 5) Гетерогенно-ката-
литич. реакции.
Для определения нек-рых веществ и для маскировки
мешающих ионов может быть использован ингиби-
торный эффект, связанный с образованием практи-
чески недиссоципрованного соединения между инги-
битором и катализатором (напр., ионы серебра пре-
кращают каталитич. действие иодида).
К. м. а. предложены для определения более чем
30 элементов; характеристика нек-рых из них при-
ведена в табл.
569
КИНУРЕНИН — КИПЕНИЕ
570
Элементы Чувствительность Элементы Чувствительность
минималь- ная кон- центрация, мкг мл откры- ваемый мини- мум, мкг минималь- ная кон- центрация, мкг'мл откры- ваемый мини- мум, мкг
Ванадий ю-< Ртуть 10-2
Вольфрам 10-8 — Рутений — Ю-з
Железо — 10-4 Свинец — 10-2
Золото — 10-S Селен — 10-5
Иод — Ю-з Сера — 10-4
Кобальт — 10-е Серебро — 10-5
Магний — 100 Тантал 10-2 —
Марганец — 10—S Теллур — 10-2
Медь —— Титан —— 0,5
Молибден 10 4 — Торий 0.1 —
Ниобий — 0,5 Уран Ю-з —
Осмий — 11}-з Фтор —
Палладий 10-2 — Хром 10-8 —
Платина 10-2 — Цинк —— —
Рений — Ю-з Цирконий 1 —
Вследствие высокой чувствительности кинетич.
методы могут применяться для определения микро-
элементов, ничтожных концентрации различных при-
месей в металлах и их сплавах, в воде и в веществах
особой чистоты.
Лит.: F е 1 g 1 F., Chemistry of specific, selective and sen-
sitive reactions, N. Y., 1949; Kolthott I. M., Living-
stonR. S„ Ind. Engng. Chem., Analytical ed., 1935, 7, К» 4,
209; Яцимирский К. Б., Заводск. лаборатория, 1955,
21, Ki 12, 1410; его же, Хим. наука и пром-сть, 1959, 4,
М 2, 188; его же, Chem. Listy, 1960, 54, 795.
К. Б. Яцимирский.
КИНУРЕНИН [В-(о-аминобензол)-а-аминопроппо-
новая кислота] C10H12OsN2, мол. в. 208,22 — раство-
рим в воде, этаноле и водном
<^|Г-СОСН„СНСООН • ацетоне; оптически активен. К.
NH2 легко экстрагируется бутано-
лом из слабокислого водного
р-ра; устойчив по отношению к кислотам и быстро
разлагается при нагревании в слабощелочной среде
с отщеплением NHS, СО2 и образованием о-амино-
ацетофенона. На этой реакции основан один из ме-
тодов количественного определения К. Синтез К.
основан на окислении триптофана озоном.
L-Изомер К. впервые выделен в виде труднораство-
римого сульфата из мочи кроликов, получавших
инъекции триптофана. К. — промежуточный продукт
обмена триптофана при его биология, превращении
в никотиновую кислоту. При ферментативном окисле-
нии К. превращается в 3-оксикинуренин. К. и 3-окси-
кинуренин расщепляются ферментом кинуренин-
азой, содержащим пиридоксаль-5-фосфат, с образо-
ванием аланина п антраниловой (соответственно
3-оксиантраниловой) к-ты. При переаминировании К.
образуется соответствующая а-кетокислота, к-рая
самопроизвольно циклизуется в кинуреновую кислоту.
К. является предшественником пигментов глаз насе-
комых — оммохромов.
Лит.: Майстер А., Биохимия аминокислот, пер. с англ.,
'М., 1961; Перспективы развития органической химии, под
. ред. А. Тодда, пер. с англ, и нем., М., 1959; Браувштейн
А. Е., Биохимия аминокислотного обмена, М., 1949.
Е. В. Горяченкова.
КИНУРЕНОВАЯ КИСЛОТА (у-окси-а-хинолин-
карбоновая кислота) C10H7OsN, мол. вес 189,17 —
игольчатые кристаллы, т. пл. 282—
" ОВ 283°. К. к. растворима в горячем
этиловом спирте, мало растворима
L I ]_гппн в г°Рячей в0Де (0,9%); выделена
впервые из мочи собаки при бога-
: той белком пище. К. к. — один из
конечных продуктов обмена триптофана, образует-
ся из продуктов его превращения — L-кин уренина —
в процессе переаминировании (см. Кинуренин).
Лит. см. при ст. Кинуренин. Е. В. Горяченкова.
КИПЕНИЕ — переход жидкости в пар (фазовый
переход 1-го рода) не только путем испарения со
свободной поверхности, но и во всем объеме вслед-
ствие непрерывного образования и роста в жидкой
фазе пузырьков насыщенного пара, внутрь к-рых
происходит испарение жидкости. Пузырьки возни-
кают из зародышей на поверхностях нагрева (стенках
сосуда), быстро растут вследствие испарения в них
жидкости, отрываются от стенок и, продолжая увели-
чиваться в размерах, всплывают. Достигнув свободной
поверхности, пузырьки разрушаются (лопаются), а за-
ключенный в них пар переходит в пространство иад
жидкостью. Пузырьки могут возникать и в объеме
жидкости при наличии в нем внутренних источников
тепла (напр., в ядерных реакторах).
К. возможно во всем темп-рном интервале равно-
весия жидкости с паром (между тройной точкой и
критич. точкой). При заданном постоянном внешнем
давлении (напр., атмосферном) К. происходит при
вполне определенной темп-ре Ts, наз. темпера-
турой кипения. При Ts давление насыщенного
пара над кипящей жидкостью равно внешнему. Для
поддержания процесса К. к жидкости должно подво-
диться количество тепла, равное уд. теплоте фазо-
вого перехода (теплоте парообразова-
н и я) L, умноженной на массу испаряющейся жид-
кости.
В стационарно кипящей жидкости устанавливается
характерное распределение темп-ры. Жидкость у по-
верхности нагрева находится в несколько перегретом
состоянии, что является необходимым условием обра-
зования пузырьков, т. к. находящийся в них пар,
кроме внешнего и гидростатич. давления, испытывает
капиллярное давление ра = 2о/г, определяемое кри-
визной поверхности пузырька 1/г и поверхностным
натяжением жидкости в. Для того чтобы пузырек мог
существовать, темп-ра пара в нем, а также темп-ра
окружающей жидкости, с к-рой пар находится в тер-
модинамич. равновесии, должна быть равна темп-ре
насыщения Т„ при давлении внутри пузырька, иначе
пар в пузырьке тотчас же сконденсируется. Т. обр.,
в пограничном со стенкой слое кипящей жидкости
с темп-рой Тн и перегретом на величину ДУ, радиус
образующихся пузырьков не может быть меньше
г . = ____21 р1
mln (dp/dT)HAT • P1-Pg
где (dp/dT)a — производная, взятая по кривой насы-
щения (ортобарической кривой) и определяемая
Клапейрона — Клаузиуса уравнением-, р2 — плот-
ность жидкости, р2 — плотность пара. Пузырьки
меньших размеров образоваться в данных услови-
ях не могут, т. к. давление пара в них не урав-
новесит капиллярного давления, возрастающего
с уменьшением радиуса пузырька. Так как пузырьки
пара в момент образования должны иметь конечный
радиус, то их возникновение возможно только на
центрах парообразования (гл. обр. шероховатостях
поверхностей нагрева), радиус кривизны которых
г rmin- Жидкости, тщательно очищенные от вклю-
чений (пылинок, растворенных газов и т. д.), способ-
ных стать зародышами пузырьков пара, в сосудах
с гладкими стенками закипают при значительном
перегреве. Величина перегрева определяется разме-
рами тепловых флуктуаций плотности, способных
в данных условиях стать зародышами пузырьков
пара.
Перегретая жидкость вскипает бурно (толчком).
К. сопровождается выбросами жидкости. На практике
для обеспечения равномерности К. в кипящую жид-
кость вводят химически инертные пористые тела
(обрезки стеклянных капилляров, кусочки пемзы,
фарфора и т. д.). Процесс К., кроме перегрева, часто
571
КИПЕНИЕ — КИСЛОРОД
572
сопровождается вспениванием. Поднимающиеся при
К. на поверхность жидкости пузырьки пара лопаются,
достигая поверхности. Но этот процесс может быть
сильно замедлен из-за повышения устойчивости воз-
никающих жидких пленок под влиянием различного
рода загрязнений жидкости растворенными вещест-
вами (солями и органич. соединениями), а также
образующимися взвешенными коллоидными части-
цами. Замедление самопроизвольного разрыва пузырь-
ков при выходе на поверхность приводит к их накоп-
лению на свободной поверхности жидкости, т. е.
к ценообразованию. Это вызывает иногда, при недо-
статочной чистоте котловой воды, опасные явления
пенного переброса в котлах высокого давления и
в ряде случаев лимитирует интенсивность их работы.
Это явление часто затрудняет процессы дистилляции
и выпаривания растворов. Борьба с этим явлением
осуществляется с помощью полной очистки котловой
воды от примесей (напр., перегонкой) или посредством
введения специальных малых добавок пеногасящих
веществ, а также с помощью механич. разрушения
пены. При больших разностях темп-р (темп-рный
напор) по обе стороны стенки (выше нек-рого критич.
значения) пузырьки пара, образующиеся у нагретой
стенки, сливаются в сплошную паровую пленку,
обволакивающую поверхность нагрева и препятствую-
щую прямому соприкосновению ее с жидкостью.
При таком «пленочном режиме» К. теплопередача
резко снижается, что приводит к уменьшению про-
изводительности теплообменных аппаратов. Поэтому
наиболее выгодные режимы К. соответствуют не-
сколько меньшим температурным напорам, при к-рых
пузырьки пара, образующиеся на поверхности на-
грева, успевают отделиться от стенки, не препят-
ствуя омыванию ее жидкостью.
Ts — важная физико-химич. характеристика ве-
щества. Она возрастает с увеличением внешнего
давления и достигает наивысшего значения в кри-
тической точке (Тк, рк), которая определяет наивыс-
шую возможную температуру К. при критическом дав-
лении рк. Д. И. Менделеев назвал Тк температурой
абс. К. Понижение Ts с уменьшением внешнего дав-
ления лежит в основе определения барометрического
давления (с помощью гипсотермометров). Для неас-
социированных жидкостей Ts при 760 мм рт. ст.
приближенно составляет определенную долю Тк, а
именно: TS/TK = 0,64 (правило Гульдберга — Гюи).
Таким образом, нормальная точка К. является
температурой сравнения, т. е. свойства жидко-
стей следует сопоставлять не при одинаковых абсо-
лютных температурах К., а при соответственных
темп-рах, напр. при темп-рах, составляющих одина-
ковую долю от Тк. Установлено, напр., что молярная
теплота парообразования Ls пропорциональна Ts,
т. е. Ls — ktTs, где молярная энтропия кипения
= 20—21 кал!моль град (правило Трутона—Пикте).
А налогичная связь существует между Ts и а — мо-
лярной капиллярной постоянной (Ма2/?1 = 2а М =
= 2avm) неассоциированных жидкостей: Ma2ITs =
= 1,16, а также между поверхностным натяже-
нием о при темп-ре К.: tsvm/Ts = 0,053, где М — мол.
вес жидкости, vm — молярный объем.
Растворение в жидкости нелетучего вещества по-
нижает давление ее насыщенного пара и повышает Ts
Это позволяет определять мол. вес растворенных
веществ по вызываемому ими повышению Ts раство-
рителя (эбулиоскопия). Разностьтемп-р, наз. темп-рной
депрессией, растет с увеличением концентрации
раствора и с ростом внешнего давления. Темп-ры К.
неазеотропных, однородных жидких смесей, распо-
лагаются в интервале темп-р К. компонентов при
том же внешнем давлении. Азеотропные смеси могут
иметь темп-ры К. ниже и выше темп-р К. компо-
нентов, т. е. минимум и максимум темп-ры. Гетеро-
генные жидкие смеси всегда имеют более низкую
темп-ру К., чем их компоненты при том же давлении
(см. Гетероазеотропные смеси). П. А. Ребиндер.
КИПЯЩИЙ слои — см. Псевдоожижение.
КИСЛОРОД (Oxygenium) О—химич. элемент VI гр.
периодич. системы Менделеева; п. н. 8. Прежняя,
кислородная (так называемая химическая) шкала
атомных весов была основана на кислородной единице:
атомный вес природного К. был принят за 16,0000.
В новой, углеродной, шкале атомный вес природного
К. (на 1962) равен 15,9994. В свободном состоянии
при обычных условиях К. — двухатомный газ (О2)
без цвета и запаха.
Предположение о существовании К. как составной части
воздуха и о его роли в процессах горения содержится в китай-
ских манускриптах 8 в., а также в работах Леонардо да Винчи
(1452—1519). Открытие К. приписывается К. Шееле, к-рый
в 1769—70 выделил его при нагревании азотнокислого магния,
селитры и других веществ; в 1774 Дж. Пристли, независимо
от Шееле, получил К. нагреванием окиси ртути. Однако, как
выяснилось позднее, К. был открыт в начале 17 в. К. Дреббелем
и использован в изобретенной им подводной лодке (это откры-
тие держалось в секрете и не оказало влияния на дальнейшие
исследования). В 1775 А. Лавуазье установил состав воздуха
и показал, что К. содержится во многих телах и является
составной частью кислот. Отсюда название oxygenium (от
греч. сь-о- — кислый и fewaco — рождаю), т. е. «образующий
кислоты».
К. является наиболее распространенным элементом
на Земле и составляет 47,2 вес. % земной коры.
В свободном состоянии он содержится в атмосферном
воздухе в количестве 23,15% по весу, или 20,93%
по объему (всего 1,5 • 1015 т). Главная же масса К.
находится в связанном состоянии: в воде (в морской
воде 85,82 вес. %), песке (53 вес. %), глинах
(56 вес. %), горных породах, рудах и т. д. К. входит
в состав всех веществ, из к-рых построены живые
организмы, напр. в человеческом организме содер-
жится ок. 65% К. Убыль К. в атмосфере в результате
процессов окисления, горения, гниения и дыхания
возмещается выделением К. растениями при фото-
синтезе. Кроме того, по-видимому, имеет место фото-
химия. разложение водяного пара в верхних слоях
атмосферы, что, вероятно, играло важную роль
в образовании атмосферного К. до возникновения
жизни на Земле.
Изотопы, атом, молекула. Природный К. состоит
из смеси 3 стабильных изотопов: О18 (99,759%), О17
(0,037%) и O1S (0,204%). Кроме того, искусственно
получены три радиоактивных изотопа К.: О14 (Т^ =
= 76,5 сек), О15 (Т1/2 = 2,1 мин) и О19 (Т1/2 =
= 29,5 сек). Ядра изотопов К. состоят из 8 протонов
и соответственно для О14, О15, О18, О17, О18 и О19 из
6, 7, 8, 9, 10 и 11 нейтронов. Электронная оболочка
атома К. состоит из 2 внутренних и 6 внешних элект-
ронов и выражается формулой: Is2 2s2 2р2х 2ру 2р,
(см. Атом). Два непарных электрона обусловливают
2-валентность К. Возбуждение более высокого ва-
лентного состояния (напр., 4-валентного) для К.,
в отличие от более тяжелых элементов VI группы
(S, Se, Те), сопряжено с переходом электрона на
орбиту с более высоким главным квантовым числом
(и=3) (см. Валентность), что требует большой
затраты энергии, к-рая не компенсируется при обра-
зовании дополнительных связей; этим объясняется
отсутствие более высокой валентности К. Энергия
ионизации атома К. (О -* О+) равна 313 ккал/г-атом,
а сродство к электрону (О -* О-) 33 ккал[г-атом.
Тетраэдрич. атомный радиус (при коорд. числе 4)
0,66 А; октаэдрич. атомный радиус (при к. ч. 6)
КИСЛОРОД
574
573
0,74 А. Ионный радиус О2"1,36 А, Молекула К. при
обычных темп-ре и давлении двухатомна; при высоких
давлениях спектроскопия, измерения обнаруживают
небольшие количества молекул О4, в тихом разряде
образуются молекулы озона О3. Межатомное расстоя-
ние в молекуле О216 равно 1,2076 А.
Молекула К. обладает необычным строением, де-
тали к-рого полностью еще не выяснены. Прежнее
представление о структуре О = О с двойной связью
опровергается спектроскопия, и магнитными измере-
ниями, показывающими наличие 2 непарных электро-
нов. Молекула К. представляет, следовательно, свое-
образный бирадикал, строение к-рого можно изобра-
зить электронной формулой :О — О: с 1 ординарной
связью и 2 ненасыщенными валентностями. Однако
экспериментальные данные показывают, что энергия
диссоциации молекулы О2 на атомы составляет
118 ккал!моль вместо потребных для разрыва орди-
нарной связи О—0 50 ккал/молъ (напр., в перекиси
водорода). Отсюда вытекает, что в данном случае
непарные электроны упрочняют связь между-атомами
К. Этот особый вид связи трактуется как трехэлектрон-
ная связь. Прототипом ее может служить известный
в спектроскопии молекулярный ион гелия (Не - Не)+,
энергия связи в к-ром составляет ок. 55 ккал/молъ.
На основании этих представлений молекула О2 может
быть приближенно описана формулой :О =- О: с 1 ор-
динарной и 2 трехэлектронными связями. Необычно
то, что ненасыщенная структура представляет основ-
ное состояние молекулы К., а насыщенная струк-
тура 0=0 энергетически менее выгодна и встре-
чается в одном из возбужденных состояний; разность
энергии между основным и возбужденным состоя-
ниями составляет 22 ккал [моль. Энергия ионизации
молекулы К. (О2 —»• OJ) составляет 286 ккал/моль,
а сродство к электрону (О2 —»• О?) 22 ккал/молъ. При
обычной температуре диссоциация молекулярного К.
на атомы ничтожно мала; она становится замет-
ной лишь ок. 1500°, когда для 1 атм парциальное
давление атомарного К. составляет десятые доли
мм рт. ст.; при 5000° наблюдается почти полная
диссоциация.
Физические и химические свойства. К. — бесцвет-
ный газ, сгущающийся при —182,98° и 760 мм рт. ст.
в бледно-синюю жидкость, к-рая при —218,7° затвер-
девает, образуя синие кристаллы гексагональной
системы с периодами: а = 5,75 А; с = 7,59 А. При
0° и 760 мм рт. ст. 1 л К. весит 1,42897 г; плотность
жидкого К. 1,1321 (—182,98°), плотность твердого
1,46 (-252,7°); гкрит —118,84°; ?крит 49,71 атм.
Молярная теплоемкость при 0° (в кал/моль - град.):
Cv = 4,98; Ср = 6,99; Cp/Cw = 1,403. Теплота плав-
ления 3,30 кал/г; теплота испарения 51,0 кал/г.
Теплопроводность 0,000057 кал/сек - см- град (0°).
Диэлектрич. проницаемость газообразного К. 1,000547
(0°), жидкого 1,491. Вязкость 189 мпуаз (0°). В отли-
чие от водорода, азота, хлора и др. элементарных
газов, К. парамагнитен; жидкий К. притягивается
магнитом. Молярная магнитная восприимчивость К.
3340- 10~в абс. единиц (20°); изменяется обратно
пропорционально абс. темп-ре (закон Кюри). Магнит-
ная восприимчивость жидкого и твердого К. дает за-
метные отклонения от этого закона, что указывает на
частичную ассоциацию молекул О2 в диамагнитные
молекулы О4.
Диссоциация молекулярного К. на атомы при обыч-
ной темп-ре ничтожно мала, она становится заметной
лишь ок. 1500°, при 5000° молекулы К. почти пол-
ностью диссоциированы на атомы.
К. мало растворим в воде: при 20° и атмосферном
давлении в 1 см3 воды растворяется 0,031 см3, а при
0°—0,049 см3. Хорошими твердыми поглотителями К.
являются платиновая чернь и активный древесный
уголь. Благородные металлы в расплавленном состоя-
нии поглощают значительные количества К., напр.,
при 960° 1 объем серебра поглощает ок. 22 объемов
К., к-рый при охлаждении почти полностью выде-
ляется.
К. образует соединения со всеми химич. элемен-
тами, кроме инертных газов. С большинством элемен-
тов К. взаимодействует непосредственно, исключение
составляют галогены, золото и платина. С водородом
при обычных темп-рах К. реагирует крайне медленно,
а выше 550° эта реакция идет со взрывом: Н2 + 1/2О2^»
—► Н2О; теплота реакции при 1 атм и 25° ДН°298 =
= —68,3174 ккал/молъ (реакция экзотермична). Со-
единения К. с галогенами (F2O, С12О, С1О2, С1О3, С12Ов,
С12О7, С12О8, Br3O8, J2O5, J2O8 и др.) получены косвен-
ным путем — взаимодействием различных галоген-
ных и кислородных соединений, лишь F2O2 обра-
зуется непосредственно из газообразных О2 и F2
действием тихого разряда при темп-ре ниже —95°.
Заметное взаимодействие К. с серой наблюдается при
повышенных температурах (ок. 360°): S + О2 —>• SO2;
ДЯ°298 = — 70,96 ккал/молъ; аналогично проходит
реакция с Se и Те. При сгорании серы на воздухе в
нек-ром количестве (менее 4%) образуется также SO3:
SO2 + О2 = 2SO3; в достаточной степени эта реак-
ция протекает в присутствии катализатора (Pt) и
применяется в произ-ве серной к-ты. С азотом К. не-
посредственно реагирует лишь при высоких темп-рах
(выше 1200°); !/2 N2 + J/2 О2 NO; реакция эндотер-
мична, ДЯ398 = + 21,600 ккал/молъ; для заметного
сдвига равновесия вправо (с образованием 1—5% NO)
требуется еще большая темп-ра (2000—3000°). Окись
азота легко соединяется с К.: О2 + 2NO = 2NO2.
Кроме NO и NO2, различными способами могут быть
получены окислы N2O, N2O3, N2O5. Непосредственное
взаимодействие К. с фосфором при избытке К. при-
водит к образованию Р2О5, а при недостатке К. —
к образованию Р2О3. Легко соединяется К. с As, Sb
и Bi, образуя As2O3, Sb2O3 и Bi2O3. С аморфным
углеродом К. энергично взаимодействует, образуя
СО2: С + О2 —> СО2; ДН°298 — —94,0518 ккал/молъ;
с алмазом и графитом такая реакция протекает
при 700—800°. При недостатке кислорода происхо-
дит реакция: С + 1/2 О2 —► СО; ДН298 =— 26,4157
ккал/молъ. Энергично протекает реакция К. с крем-
нием: Si —О2 —> SiO2; Д77°298=—217,6 ккал/молъ (новое
значение теплоты образования кварца). К. активно
окисляет почти все металлы (см. Коррозия металлов).
Окиси нек-рых металлов, присоединяя К. без изме-
нения валентности металла, образуют перекисные
соединения, содержащие два или более связанных
между собой атома К. Окиси Na2O и ВаО построены
из ионов металла и иона О2~, перекиси Na2O2 и
ВаО2 включают перекисный ион , супероксиды
NaO2 и КО2 — парамагнитный ион О2~, а озониды
NaO3, КО3, РЬО3 и CsO3 — парамагнитный ион О3”.
Т. обр., известны 5 видов отрицательных ионов К.:
О”, О2, О22 ’, О2_, О3~ (подробнее см. Перекиси и
перекисные соединения).
Скорость окисления зависит от природы окисляе-
мого вещества и от темп-ры, а также от условий
смешения. Нек-рые вещества (окись азота, гемогло-
бин крови, пирогаллол в щелочном р-ре, соединения
одновалентной меди и др.) уже при комнатной темп-ре
с большой скоростью соединяются с К. воздуха;
самопроизвольное окисление при обычной темп-ре
наз. аутооксидацией. К. энергично окисляет органич.
соединения. Выраженной способностью присоединять
К. обладают многие ненасыщенные органич. соеди-
нения с двойной или тройной связями, альдегиды,
фенолы, а также высыхающие масла, скипидар и др.
Скорость реакций окисления, как и других химич.
575
КИСЛОРОДА ОПРЕДЕЛЕНИЕ
576
реакций, сильно увеличивается при повышении темпе-
ратуры. Многие реакции окисления сильно ускоряют-
ся в присутствии катализаторов; напр., в присутствии
дисперсрой платины смесь водорода с К. воспламе-
няется уже при комнатной темп-ре. Особо важную
роль в качестве катализатора окислительных процес-
сов играет вода. Так, в абсолютно сухом К. металлич.
калий сохраняет свою блестящую поверхность; в при-
сутствии же следов влаги — воспламеняется; пламя
окиси углерода, горящей в невысушенном К., при
внесении в атмосферу сухого К. тотчас же гаснет.
Характерной особенностью подавляющего большин-
ства реакций окисления является значительное вы-
деление тепла. Окисление К. питательных веществ
в клетках служит источником энергии живых орга-
низмов .
Получение. В лаборатории К. получают электро-
лизом водного р-ра NaOH с применением никелевых
электродов. Особо чистый К. получается при нагре-
вании перманганата калия: 2КМпО4 = К2МпО4 +
+ МиО2 + О2. В лабораторной практике обычно
используется гл. обр. промышленный К. в баллонах.
В пром-сти К. получают глубоким охлаждением,
сжижением и ректификацией воздуха в специальных
установках (см. Воздуха разделение). Электролизом
воды К. получается только как побочный продукт
при произв-ве водорода. Для проверки чистоты полу-
чаемого К. разработаны специальные методы (см.
Кислорода определение).
Применение. Технич. К. (99—99,5%) используется
в процессах газопламенной обработки металлов, в свар-
ке, кислородной резке, поверхностной закалке, ме-
таллизации и других, а также в медицине, авиации,
на подводных судах и пр. Технология. К. (90—98% О2)
применяют в металлургии, в химич. пром-сти для
получения технология, газов, искусственного жидкого
топлива, смазочных масел, азотной и серной к-т,
соды, метанола, перекисей металлов и других химич.
продуктов. Применение парокислородного дутья в га-
зогенераторах под давлением 28 атм позволяет
получить высококалорийный бытовой газ (4000—
4500 ккал/м^) из бурых углей и торфа. Жидкий К.
находит применение при взрывных работах (см.
Оксиликвиты), в реактивных двигателях и в лабора-
торной практике в качестве хладоагента. См. также
Окисление — восстановление, Перекиси и перекисные
соединен ия.
Лит.: Бродянс кий Б. М., М е е р з о н Ф. II.,
Производство кислорода, М., I960; Глизманенко Д. Л.,
Кислород и его получение, М,— Л., 1951; йшеИп, 8 Aufl.,
Syst.‘Mummer 3, Sauerstoff, Lfg 1, Б., 1943; то же, Syst.-
Nummer 3, Lfg 2, 1953; Syst.-Nummer 3, Lfg 3, 1958; Syst.-
Nummer 3, Lfg 4, 1960; Mellor, v. 1, L.—N. Y. — Toronto,
1946; то же, 1952; К I г k, v. 9, N. Y„ 1952; Pascal, v.
13, P., 1960; Yost D. M.,R u s s e 1 H., Systematic Inorga-
nic chemistry of the fifth-and-sixth group nonmetallic elements,
Oxf., 1946; Налбандян А. Б. и Воеводский В. В.,
Механизм окисления и горения водорода, М.—Л., 1949; Кон-
дратьев Б. И., Свободный гидроксил, М., 1939.
II. Л. Казарновский.
КИСЛОРОДА ОПРЕДЕЛЕНИЕ (качественное и
количественное) — основано на выделении кислорода
из газовых смесей, воды, металлов, а для органич.
соединений — на разложении вещества и определении
кислорода в продуктах пиролиза. В газовом
анализе кислород открывают по окрашиванию
смеси р-ров пирокатехина СеН4(ОН)2 и FeSO4 в интен-
сивно красный цвет или по окрашиванию бесцвет-
ного аммиачного р-ра Си2С12 в синий цвет. Для коли-
чественного определения содержания кислорода в газе
пользуются разными методами в зависимости от его
содержания. Одним из первых методов газового
анализа, применяющимся до настоящего времени,
является поглощение кислорода р-ром пирогаллола
в щелочи, р-ром Na2S2O4 и др. Содержание кислорода
определяют по уменьшению объема газовой смеси.
Этот способ применяют для анализа топочных, про-
мышленных и др. газов. Для непрерывного опреде-
ления кислорода в промышленных газах применяют
автоматич. газоанализаторы, основанные на измере-
нии теплопроводности, магнитной проницаемости и др.
физич. свойств. Напр., пользуясь термомагнитным
газоанализатором, можно определять кислород в газе
в очень широких пределах, от 0 до 100%. Незначи-
тельные концентрации кислорода в газовых смесях
определяют, пользуясь автоматич. газоанализатором,
основанным на измерении теплового эффекта при
сожжении горючего газа, взятого в избытке, с опре-
деляемым количеством кислорода. Известен также
автоматич. полярографич. газоанализатор для опре-
деления содержания кислорода в N2, СО2, С2Н2, С2Н4
и др. в пределах 0—25%, наименьший предел изме-
рения 0—0,1% (см. также Газовый анализ).
Растворенный в воде кислород опреде-
ляют различными методами, напр. добавлением
Мп(ОН)2, к-рая окисляется до Н2МпО3, определяе-
мой иодометрически (метод Винклера):
2Мп(ОН)о + Оо—‘2Н»МпО3 (1)
HsMnO3+2KJ4-4HCl--•2КС1+МпС1з+ЗНгО-Ь,Т3 (2)
Колориметрии, способы определения кислорода в воде
основаны на образовании красной окраски при окисле-
нии восстановленной формы индигокармина или же
на образовании окрашенного в пурпурно-красный
цвет продукта реакции 3,3'-диметилнафтидина и иода,
выделившегося по приведенной выше реакции 2.
В металлах кислород содержится в виде газо-
вых включений, в адсорбированном виде, поверхност-
ных соединениях или в виде окислов. Для его опре-
деления применяют вакуум-плавление .(вакуум-экс-
тракцию) или плавление в среде инертного газа.
Пробу анализируемого металла плавят в предвари-
тельно дегазированной вакуум-печи в присутствии
избытка углерода обычно в графитовом тигле при
темп-ре ок. 1400°. Выделяющиеся газы (СО) откачи-
вают и анализируют обычными методами газового
анализа. Применение платиновой ванны (платинового
тигля) значительно расширило возможности метода;
определение можно проводить при более высокой
темп-ре, вследствие чего увеличивается скорость
восстановления окислов. Метод вакуум-плавления
в платиновой ванне применяется для анализа Be, I:,
Та, Si, Y, В, Си, Сг, Fe, Ti, Zr, Th, Al, Mo, W.
Плавление в инертном газе (азот, аргон и др.) не
требует применения вакуума и в принципе не отли-
чается от вакуум-плавления. Темп-ра плавления
зависит от анализируемого вещества; в большинстве
случаев она не превышает 2000—2400°; анализ про-
водят в графитовых тиглях, нагреваемых индук-
ционно. Метод применяют для определения примеси
кислорода к чистым металлам и элементам, а также
для определения содержания его в окислах и солях,
в том1 числе и в тугоплавких, напр. SiO2, TiO2, U3O8,
окислы Мп и Ag, IJO2, Y и YF3, Zr и его сплавы,
Be, В + В2О3 и др.
Наряду с методами плавления применяют также
прямые методы, основанные на восстановлении нахо-
дящихся в металле окислов или на связывании раст-
воренного кислорода. В алюминиевом методе восста-
новителем является алюминий, содержание кислорода
определяют по количеству образовавшейся окиси
алюминия. В водородном способе — восстановитель
водород, а продукт раскисления — водяные пары.
Наряду с этим применяют след, методы определения
кислорода: 1) отделение металла от окисла амальга-
мированием. Оставшуюся после удаления амаль-
гамы окись металла растворяют и по количеству
металла в р-ре определяют содержание кислорода
(щелочные металлы); 2) дистилляция, заключающаяся
577
КИСЛОРОДНЫЙ ЭЛЕКТРОД— КИСЛОТНОЕ число
578
в отгонке металла от его окисла в вакууме и анализе
остающейся окиси металла химич. способами (щелоч-
ноземельные металлы); 3) бромуглеродный метод,
основанный на реакции: МеО + С + Вг — СО +
+ МеВг2; образовавшийся бромид вымораживают,
а СО определяют обычными методами (Ti, V, Сг, Fe);
г) серный метод, заключающийся во взаимодействии
окислов металлов с парами серы. При высокой темп-ре
свободный металл и металл из его окисла переходят
в сульфид, а кислород связывается с серой в серни-
стый ангидрид, к-рый и определяют (Cu, Zn, Ti,
Zr, Ge). В полупроводниковых материалах и сплавах
кислород определяют также методами изотопного
разбавления и радиоактивационным.
В органических соединениях един-
ственным общепринятым способом определения кисло-
рода является пиролиз органич. вещества и выделение
всего кислорода в виде СО. Органич. в-во разлагают
(800—900°) в кварцевой трубке в токе инертного газа
(азот, аргон, гелий и др.). Летучие кислородсодер-
жащие продукты разложения, проходя через контакт-
ный слой сажи (1120—1150s), количественно разла-
гаются с образованием окиси углерода, к-рая окис-
ляется до СО2 действием пятиокиси иода (120°).
Содержание кислорода вычисляют по количеству
выделившегося иода (титрование тиосульфатом) или
по двуокиси углерода (чаще всего весовым путем,
по привесу аппарата с аскаритом). Метод применим
для веществ, содержащих С, Н, О, N, С1, Вг, J, S.
Имеются многочисленные модификации и усовершен-
ствования, заключающиеся в следующем: 1) пятиокись
иода заменяют медью (200—300°); 2) при анализе
веществ, содержащих серу, в трубку для разложения
вводят медь; 3) для определения СО3 применяют
различные способы, напр. потенциометрии, титрова-
ние Ва(ОН)2 или кондуктометрии, окончание анализа
измерением изменения проводимости поглотительного
р-ра [NaOH, Ва(ОН)2]; 4) количество выделившегося
СО определяют непосредственно, без окисления в СО3,
по изменению теплопроводности газа-носителя; 5) одно-
временно определяют кислород и галогены. Для
этого сначала поглощают галогены, пропусканием
тока газа через р-р щелочи, а затем окисляют СО в СО2
окисью меди. Галогены определяют обычными спо-
собами. Последнее время метод применяют и для
анализа веществ, содержащих другие элементы, глав-
ным образом металлы, для чего предложено использо-
вать водород в качестве газа-носителя или смешивать
навеску с хлоридами серебра или одновалентной
меди.
Разработаны также методы определения кислорода, осно-
ванные на: 1) каталитическом гидрировании газообразным
водородом с превращением всего кислорода органического
вещества в воду; 2) изменении поглощения или рассеивания
испытуемым веществом радиоактивных измерений. Для опре-
деления изотопов кислорода после установления изотопного
равновесия -между испытуемым веществом и другим соедине-
нием, содержащим О16, последнее анализируют на содержание
изотопа кислорода масс-спектрометрически.
Лит.: Еремина Б. Г., Газовый анализ, Л., 1955;
Соколов В. А., Методы анализа газов, М., 1958; Анализ
газов в металлах, М., 1960; Методы определения примесей
в чистых металлах, М., 1960; Freseni us R., J a n a e г G-.,
Handbuch der anatytischen Chemie, Bd 6, B.—Hdlb., 1948;
Fort R., Chiin. anal., 1957, 39, 319, 366; К л я ч к о Ю. А.,
Атласов А. Г., Шапиро М. М., Анализ газов и
включений в стали, М., 1953; Beck Е. J., С 1 а г k F. Е.,
Anatyt. Chem., 1961, 33, № 12, 1767; Б об ране к ий- Б.,
Количественный анализ органических соединений, пер. с
польск., М., 1961; Коршун М. О., Бондарев с кая
Е. А., ДАН СССР, 1956, 110, № 2, 220; Ж. аналит. химии,
1959, 14, вып. 1, 123. И. П. Ефимов, Н. Э. Гельман.
КИСЛОРОДНЫЙ ЭЛЕКТРОД — платиновая пла-
стинка, погруженная в раствор, насыщенный кисло-
родом. На поверхности платины происходит окисли-
тельно-восстановительная реакция:
Оа+2Н2О-4е^—4ОН-
Если считать К. э. термодинамически обратимым, то
можно рассчитать его потенциал по ур-нию:
RT Ро, • 1Н2°]3
fo. — + n7?- in [ОН]»
где <р0 — стандартное значение потенциала, F — число
Фарадея, п — число электронов, pQy — парциальное
давление кислорода.
Выражая [ОН] через ионное произведение воды
Kw и производя соответствующие преобразования,
получаем:
р1/4
То. = ?„ + 0,059 1g + 0,059 1g [Н+]
W
При постоянном давлении кислорода первые. два
члена этого уравнения могут быть. объединены в по-
стоянную величину:
То. = const 0,059 1g [Н+]
Это уравнение показывает, что потенциал К. э.
находится в линейной зависимости от концентрации
Н+, причем тангенс угла наклона прямой в коорди-
натах <ро2—pH составляет 0,059 также, как в случае
водородного электрода.
При давлении кислорода, равном 1 ат, и концен-
трации ОН , равной единице, <р0 имеет величину
—[-0,401 в. Отсюда можно рассчитать значение кон-
станты:
const = + 0,401 + 0,059 1g -fjpfy- 1,23 в
Эту величину можно принять за потенциал кислород-
ного электрода при концентрации Н , равной еди-
нице. Экспериментально не удается воспроизводить
потенциал К. э. с постоянной точностью (в противо-
положность водородному электроду), по-видимому,
вследствие того, что платина »не остается инертной
в присутствии кислорода, а образует окислы, изменяю-
щие состояние ее поверхности и тем самым влияющие
на обратимость электродного процесса.
Лит.: Глесстон С., Введение в электрохимию, пер.
с англ., М., 1951; Скорчеллетти В. В., Теоретическая
электрохимия, Л., 1959. О. А. Сангина.
«КИСЛОТ И ОСНОВАНИИ ТЕОРИИ — см. Кис-
лоты и основания.
КИСЛОТНОЕ ЧИСЛО — величина, характеризую-
щая эквивалентный вес кислоты или смеси кислот,
а также содержание свободных кислот в нек-рых
технич. и природных продуктах. К. ч. равно числу
миллиграммов КОН, расходуемого на нейтрализацию
1 г испытуемого вещества. Для его определения точно
взвешенное количество вещества растворяют в под-
ходящем растворителе. Растворитель предварительно
нейтрализуют р-ром КОН в. присутствии фенолфта-
леина. Приготовленный т. обр. раствор титруют 0,1 н.
водным или спиртовым раствором КОН до появления
розовой окраски. Кислотное число вычисляют по
формуле: К. ч. = 5,61 а/е, где а — количество милли-
литров 0,1 ц. раствора КОН, израсходованного при
титровании, в — навеска испытуемого вещества в
граммах.
Наибольшее значение К. ч. имеет в случае анализа жиров,
качество и технич. характеристика к-рых в значительной мере
определяются содержанием свободных к-т. К. ч. пищевых
жиров от 0,5 до 2,5, а -большинства технич. жиров и расти-
тельных масел — от 2 до 25. При длительном хранении и дей-
ствии воздуха, влаги, света и тепла К. ч. жиров увеличивается.
При анализе смесей жирных к-т в качестве раствори-
телей используют смесь абсолютно сухого спирта
и эфира, при анализе жиров — эфир, бензол и их
смеси со спиртом, при анализе восков — смерь спирта
и ксилола. При титровании водным раств’ором КОН
существенную роль играет присутствие спирта, к-рып
способствует растворению щелочи и образующихся
солей жирных к-т и препятствует гидролизу послед-
10 к. X. Э. т. 2
579 КИСЛОТНО-ОСНОВНОЕ РАВНОВЕСИЕ— КИСЛОТОУПОРНЫЕ ЗАМАЗКИ 580
них. Количество спирта в титруемой смеси должно
превышать объем израсходованного р-ра щелочи не
менее чем в 5 раз.
Лит.: Бауер К., Анализ органических соединений,
2 изд., пер. с нем., М., 1953; Зиновьев А. А., Химия
жиров, М., 1952; Беззубов А. П., Химин жиров, М.,
1956. А. А. Черкасский.
КИСЛОТНО-ОСНОВНОЕ РАВНОВЕСИЕ (в орга-
низме) — соотношение между концентрациями [Н+]
и [ОН~] в тканевых жидкостях, крови и т. п. К.-о. р.
чаще всего выражают через водородный показатель
pH. Состояние К.-о. р. (pH раствора) влияет на направ-
ление и скорость химич. реакций вообще (см. Кислоты
и основания) и, в частности, в живых организмах,
где поддержание постоянства К.-о. р. определяет
нормальное течение всех процессов жизнедеятельности.
Значительная часть тканей организма здорового
человека имеет слабощелочную реакцию; pH боль-
шинства тканевых жидкостей поддерживается на
уровне 7,1—7,4; лишь нек-рые жидкости имеют более
щелочную (напр., секрет толстого кишечника pH
ок. 8) или кислую реакцию (напр., желудочный сок
pH 0,9—1,5). Исключительную роль в обеспечении
стабильности внутренней среды организмов играет
К-о. р. крови. Несмотря на обилие и разнообразие
источников кислот и оснований, поступающих в кровь,
величина pH в ней сохраняется на постоянном уровне
вследствие наличия в ней буферных систем, а также
благодаря различным физиология, механизмам, спо-
собствующим удалению кислот и оснований из орга-
низма. В крови имеется несколько буферных систем:
бикарбонатная (гидрокарбонатная), фосфатная, белки
эритроцитов и плазмы. Состояние К.-о. р. в крови
прежде всего определяется состоянием бикарбонатной
буферной системы: Н2СО3—NaHCO3.
Согласно ур-нию Гендерсона — Гассельбальха, зна-
чение pH крови может быть вычислено по ур-нию:
„ „ |NaHCO3l
pH=pH1+lg[—Q--J
где pH! — постоянная величина.
Поступающие в кровь сильные кислоты (НА)
и сильные основания (МеОН) в первую очередь
взаимодействуют с веществами этой системы:
HA+NaHCO3=NaA+H2CO3
МеОН+Н2СО3=МеНСО3+Н2О
Бикарбонатная система тесно связана с другими
буферными системами и особенно с белками, на долю
к-рых приходится ок. 75% всей буферной емкости
крови. Эта связь с белковыми буферными системами
осуществляется в результате реакции:
Н-СО34-Ме-белок5=±МеНСО8+Н-белок
Т. обр., при поступлении в кровь сильных к-т и осно-
ваний белки участвуют в поддержании постоянства
pH не только путем непосредственного взаимодей-
ствия с ними, но и через бикарбонатную буферную си-
стему. Из белков наибольшее значение здесь имеют
гемоглобин и продукт его окисления — оксигемо-
глобин. Большую роль в регулировании pH крови
играет деятельность дыхательного аппарата, почек
и печени.
При нек-рых заболеваниях почек, сердечно-сосу-
дистой системы, органов дыхания и др. в связи с на-
рушением обменных процессов наблюдаются сдвиги
К.-о. р,: сдвиги в кислую сторону наз. ацидозом,
сдвиги в щелочную — алкалозом. При ацидозе
pH крови иногда уменьшается до 7 и ниже, а при
алкалозе — увеличивается до 7,6 и выше. Как пра-
вило, в начальных стадиях ацидоз и алкалоз проте-
кают без изменения pH и сопровождаются лишь изме-
нением буферной емкости крови.
Лит.: Капланский С. Я., Кислотно-щелочное
равновесий в организме и его регуляция, М.—Л., 1940; Б ла-
де р г р ё и В., Физическая химия в медицине и биологии,
пер. с нем., М., 1951, с. 227. А. II. Бресткин.
КИСЛОТОСТОЙКИЕ МАТЕРИАЛЫ — см. Хими-
чески стойкие материалы.
КИСЛОТОУПОРНЫЕ ЗАМАЗКИ (кислотоупор-
ные цементы, кислотоупорные мастики) — компози-
ции вяжущих веществ, способные переходить из
тестообразного или жидкого состояния в твердое.
К. з. используются в основном как вяжущие при
футеровке химич. аппаратуры стеклянными, графи-
товыми, диабазовыми, керамич. и др. плитками
и с целью создания герметичности в местах соединений
их между собой и для уплотнения швов.
Наибольшее распространение нашли силикатные
К. з. и замазки на органич. основе (т. н. арзамиты),
меньшее — глетоглицериновые, серные, мастики на
основе битумов (т. н. битуминоли) и др.
Силикатные кислотоупорные за-
мазки изготовляются из следующих компонентов:
1) кислотоупорный тонкоизмельченный наполнитель
(андезит, бештаунит, плавленый базальт, плавленый
диабаз и т. п. богатые кремнеземом силикатные мате-
риалы); 2) кремнефтористый натрии, играющий роль
ускорителя твердения; 3) жидкое стекло. В табл. 1
приведены основные физико-механич. свойства наи-
более распространенных силикатных К. з. (диаба-
зовой и андезитовой).
Таблица 1
Замазка
Показатель
диабазовая । андезитовая
Объемный вес, т/м3..............
Темп-ра начала размягчения, °C .
Теплопроводность,
ккал/м • час • град...........
Теплоемкость, ккал/кг • град ....
Коэфф, линейного расширения .
Предел прочности, кг/см2:
при растяжении .............
при сжатии..................
Сцепление, кг/см2:
с бетоном ..................
с диабазом..................
с деревом ..................
с железом...................
с керамикой ................
Кислотостойкость (по методу
ВИОК), %......................
1,88 • 10-5
’ 25-35
180-250
6,6
24,9
27,1
Силикатные К. з. пригодны при темп-ре, не превы-
шающей 800°; они стойки к концентрированным ми-
неральным к-там, исключая плавиковую и фосфорную,
не стойки к щелочам и воде и недостаточно стойки
к разб. минеральным к-там. Силикатные К. з. гото-
вят тщательным смешиванием кислотоупорных на-
полнителей и кремнефтористого натрия с последую-
щим замешиванием их на жидком стекле. Составы
силикатных замазок приводятся в табл. 2.
Таблица 2
Наполнитель Вес наполни теля, г Вес кремне- фтористого натрия, г Жидкое стекло
модуль плот- ность, г/см3 % к весу напол- нителя
базальтовы
ц е м
е
е н т ы
2,50
2,75
3,00
950
950
960 I
50
50
40
36,2
35,5
31,6
1,49
1,46
1,36
Диабазовые и
Измельченный плавлен-
ный базальт или
диабаз ..........
То же............
То же............
Андезитовые и бештаунитов
Измельченные андезиты
или бештауниты . . . 950 50 2,3—2,6
То же............. 960 40 2,6—3,0
ые цементы
1,5-1,45 34-36
1,45-1,38 36-38
581
КИСЛОТЫ ЖИРНЫЕ СИНТЕТИЧЕСКИЕ — КИСЛОТЫ И ОСНОВАНИЯ
582
Кислотоупорные замазки и а органич.
основе (арзамиты) получают смешиванием по-
рошкообразного наполнителя (графит, кремнезем,
сернокислый барий и др.) с катализатором (па-
ратолуолсульфохлорид) и растворителем (феноло-фор-
мальдегидиая смола или смесь феноло-формаль-
дегидной смолы с бензиловым спиртом). В химич.
пром-сти (в кислых и нейтральных средах) в качестве
К. з. чаще всего применяют замазки арзамит-1 и
арзамит-4; первая непригодна при действии плави-
ковой к-ты, вторая стойка к слабым и средним кон-
центрациям HF; кроме того, она теплопроводна.
Арзамиты 1 и 4 разрушаются в кислотах-окислите-
лях. Замазки арзамит затвердевают: при 70° в тече-
ние иеск. минут, при комнатной темп-ре в течение
6 час., при 10° — 3 суток. Нек-рые физико-механич.
свойства замазок арзамит-1 и арзамит-4 приведены
в табл. 3.
Таблица 3
Показатели Замазки
арзамит-1 арзамит-4
Предел прочности, кг/см2:
разрыв 51—54 48-50
сжатие 840 600—700
Усадка, % 0,42 0,037
Сцепляемость, кг/см2'. с железом, защищенным резорцино- феноло-формальдегидной смолой
с графитом 40 40-50
со стеклом 7-7,5 —
с фарфором 11—12 —
с винипластом 24 38
с плиткой из каменного литья . . 17 12
с вулканизованной резиной 1976 . 6—10 —
Проницаемость при гядравлич. давле-
нии 3 ат ............... непрони- цаема непрони- цаема
Коэфф, теплопроводности,
кал/м2 • час град — 18—20
Лит.: Клинов И. Я., Коррозия химической аппа-
ратуры и коррозионностойкие материалы, 3 изд., М., 1980;
Дерешкевич Ю. В., Кислотоупорные сооружения
з химической промышленности, М., 1960; Антикоррозийные
покрытия строительных конструкций и аппаратуры. Справоч-
ное пособие, М., 1959; Смирнов В. К. и Кацнель-
сон С. X., Химически стойкие замазки арзамит и лаки
холодного отверждения, М., 1957. fl. Я. Клинов.
КИСЛОТЫ ЖИРНЫЕ СИНТЕТИЧЕСКИЕ — см.
Высшие жирные кислоты.
КИСЛОТЫ И ОСНОВАНИЯ — классы химич.
соединений. Различные вещества могут выполнять
функции К. и о. лишь в определенного типа реакциях,
называемых кислотно-основными. Т. обр., определе-
ние понятий К. и о. зависит от определения понятия
кислотно-основного процесса, в к-ром реагирующие
между собой К. и о. являются таковыми лишь друг
по отношению к другу, аналогично тому, как это имеет
место в случае окислительно-восстановительных реак-
ций. Единого подхода к классификации веществ,
с точки зрения отнесения их к К. и о., пока нет. Обще-
приняты два таких подхода: по Дж. Н. Бренстеду
и по Г. Н. Льюису. По Бренстеду, кислотой
является донор протона, т. е. реагент, отдающий
протон, а основанием — акцептор протона, т. е.
реагент, присоединяющий протон. Эти реагенты
могут быть как нейтральными молекулами, так и ио-
нами. Сама реакция передачи протона наз. прото-
литической: АН + В~ А' + ВН, где АН —
кислота, а В’ — основание. Поскольку протолитич.
реакция обратима, причем в обратном процессе также
передается протон, то продукты реакции тоже
являются друг по отношению к другу кислотой и ос-
нованием: ВН — кислота (сопряженная с основа-
нием В~), А-—основание (сопряженное с кисло-
той АН). Напр., в реакции: HCI -j- ОН~^д С1~ -ф- НаО,
НС1 и Н2О — кислоты, С1~ и ОН- — соответственно
сопряженные с ними основания; в реакции: H2SO4 +
+ Н2О HSOj + Н3О+, H2SO4 и Н3О+ — кислоты,
HSO4 и Н2О — основания; в реакции: HSO7 + Н2О И
И SO;~ + Н3О+, HSOj и Н3О+ — кислоты, SO;’ и
Н2О — основания; в реакции Nil; + NH 2 2NH3,
NHJ — кислота, NHj — основание, a NH3 выступает
в роли как кислоты (одна молекула), так и основания
(другая молекула) и т. д.
По Льюису, кислота — акцептор электронной
пары, т. е. реагент, присоединяющий пару электро-
нов, а основание — донор электронной пары, т. е.
реагент, отдающий пару электронов. При реакции
между К. и о. пополняется недостаток электронов
у одного из атомов молекулы кислоты, в результате
чего возникает более устойчивая группировка элект-
ронов (в частном случае — октет) и образуется до-
норно-акцепторная ковалентная связь, например:
н F н F
Н : N: +В : F = Н : N : В : F, Где BF3 — КИСЛОТа, а
Н F ii F
NH3 — основание. Т. о., Бренстед и Льюис подходят
к определению К. и о. с разных точек зрения. Если, со-
гласно Бренстеду, кислотные свойства связываются с
наличием протона, то, по Льюису, эти свойства обусло-
вливаются исключительно строением реагирующих мо-
лекул, определяющим их электронно-акцепторные
свойства, и вовсе не связываются с присутствием в них
к.<i. определенного элемента, и в частности водорода,
могущего отщепиться в виде протона. Однако оба
подхода (по Бренстеду и по Льюису) имеют между
собой внутреннюю связь, к-рая Состоит в том, что
сам протон, как и другие льюисовские кислоты, харак-
теризуется большим сродством к электронной паре.
(В современной литературе принято указывать, о ка-
кой кислоте — бренстедовской или льюисовской —
идет речь; отсутствие же таких указаний приводит
к путанице). Кроме этих двух общепринятых подхо-
дов к классификации веществ по кислотно-основным
свойствам, существуют также и нек-рые другие
(теория Э. К. Франклина, взгляды М. И. Усановича,
А. И. Шатенштейна и др ).
К феноменологическим (т. е. внешним) признакам
К. и о. обычно относят: 1) способность К. и о. к взаим-
ной нейтрализации, происходящей быстро и без
заметной энергии активации; 2) способность вытес-
нять более слабые К. и о. из их соединений; 3) взаимо-
действие кислот с металлами с выделением водорода;
4) возможность титрования К. и о. с применением
индикаторов; 5) способность К. и о. катализировать
многие химич. реакции и др. Однако ни один из
феноменология, признаков не является ни необхо-
димым, ни достаточным и поэтому эти признаки не
могут быть положены в основу классификации К. и о.
Напр., бренстедовские К. и о. не удовлетворяют
условию взаимной нейтрализации, т. к. при их взаимо-
действии образуются новые К. и о.; апротонные льюи-
совские к-ты не могут выделять водород при воздейст-
вии металлов и т. д.
Развитие взглядов на кислоты и основания. Понятия
«К. и о.» появились на заре развития химии. В 1778 Лавуазье
выдвинул теорию, объяснявшую особенности кислот наличи-
ем в них кислорода (кислородная теория кислот). Несостоя-
тельность этой теории проявилась в том, что очень многие
кислородсодержащие вещества (окислы металлов, щелочи,
соли и др.) не обладают кислотными свойствами, а с другой
стороны, ряд типичных кислот (плавиковая, соляная, синиль-
ная), как было выяснено Дэви и Гей-Люссаком (1810, 1814),
не содержит кислорода. Берцелиус (1812—19) устранил
первое из этих противоречий, усмотрев причину кислотных и
основных свойств в знаке электрич. заряда окислов: электро-
отрицательные окислы неметаллов образуют кислоты, а элек-
троположительные окислы металлов — основания. В 1814
Дэви предложил считать носителем кислотных свотйтв водород,
входящий в состав всех известных тогда кислот (водородная
теория кислот), а Либих (1833) уточнил эту теорию, отнеся
кислотные свойства за счет не любого водорода, а лишь способ-
ного замещаться металлом.
10*
583
КИСЛОТЫ И ОСНОВАНИЯ
584
После появления теории электролитич. диссоциации Ар-
рениуса (1884, 1887) возникла ионная теория К. ио.: кислотами
были названы соединения, при диссоциации к-рых в водном
р-ре образуются ионы водорода (Н+), а основаниями — соеди-
нения, диссоциирующие с отщеплением иона гидроксила (ОН-)
(см. Электролитическая диссоциацгся). Однако в результате
рцбот по теории р-ров было показано, что диссоциация веществ
происходит вследствие химич. взаимодействия с растворителем,
благодаря чему необходимо учитывать его собственные кис-
лотно-основные свойства; что в водном растворе ион Н- (про-
тон) не может существовать в свободном состоянии — он соль-
ватирован и существует в виде иона гидроксония (ЩСН);
что в неводных растворах также проявляются свойства
Н. и о.
Для неводных р-ров была создана теория т. н. сольвосистем
соединений (Э. Н. Франклин, 1914), по к-рой для каждого
растворителя была введена своя система К. ио., удовлетворяю-
щая условию взаимной нейтрализации По схеме: кислота 4-
— основание = соль 4- растворитель (аммоносистема, суль-
фосиртема, фосгеносистема и т. д.). При этом роль кислоты
приписывалась тому веществу, к-рое отщепляет в р-ре тот же
катион, что и сам диссоциирующий ’ растворитель, а роль
основания — веществу, отщепляющему соответствующий ани-
он; напр., для р-ра в жидком аммиаке (2NH3 NHj' 4- NH-),
в реакции NH4C1 4- KNH^KCl 4- 2NH3, NH,C1 — кислота,
KNHj — основание, a KC1 — соль; для р-ра в жидком SO2
(^SO^SO2^ 4- SO-’' ) в реакции: SOC12 + Cs_SO3^2CsCl 4-
2SO2, SOC12 — кислота, Cs2SO3 — основание, CsCl — соль
и т. д. Теория сольвосистем соединений помогла системати-
зировать многие реакции в неводных р-рах, но от нее отказа-
лись из-за ее крайнего формализма.
В 1923 почти одновременно Дж. Бренстедом, Г. Льюисом
и Т. Лоури были сформулированы основы современной теории
протолитич. кислотно-основного равновесия, обычно связы-
ваемой с именем Бренстеда, разработавшим ее количественно.
Приблизительно тогда же Льюисом был предложен и другой
подход к понятиям К. и о. — с точки зрения электронной
структуры молекулы (см. выше).
Протолитическое кислотно-основное равновесие.
По теории Бренстеда кислотно-основные свойства
веществ определяются термодинамикой протолитич.
реакций, причем термодинамич. активность протона
(ан+) в реакции АН z: А- + Н+ выражает кислотность
реагента АН, тогда как основность реагента А~ вы-
ражается обратной величиной (1/ан+).Для сравнения
кислот по их силе в одном и том же растворителе поль-
зуются константой кислотности:
кАн = (“н1-)[Хщ
где величины, взятые в квадратные скобки, — кон-
центрации соответствующих реагентов (основания А~
и кислоты АН); константе основности соответствует
обратная величина:
„ 1 [АН]
А “н+ [А1
Указанная простейшая реакция отщепления про-
тона АН А--)- Н+практически неосуществима в р-ре,
т. к. для протекания протолитич. реакции необхо-
димо присутствие другого вещества (В ’), способного
принять протон: АН — В.д А~ + ВН. Константа
равновесия последней реакции (константа-
протолиза):
_ [А- ] [ВН] _ КАН
АН,В- [АН][В-] квн
соответствует отношению констант кислотности ве-
ществ АН и ВН и выражает силу кислоты АН по
отношению к силе кислоты ВН в том же растворителе.
Обычно принимают какую-либо из кислот (А0Н) за
стандартную, считая ее константу кислотности еди-
ницей, и оценивают силу других кислот по относи-
тельной константе кислотности
„АоН _ КАН
АН КА0Н
Для прОТОлйтически активного растворителя (S)
удобно выбрать стандартной кислотой протон, соль-
ватированной самим растворителем (т. н. ион лио-
ния SH+), и определять относительную константу
кислотности по константе протолиза реакции АН +
+ S А - + SH+:
кан=кан, s =
[А~] [SII+]
[AH][S]
Для водных р-ров стандартной к-той является ион
гидроксонпя Н3О+ и относительная константа кислот-
ности, напр. соляной к-ты, равна:
„Н3О1 = [С| ] №0+]
НС1 [НС1][Н2О]
При разработке количественной теории, основанной
на применении законов термодинамики к протолитич.
равновесию. Бренстед принял ряд упрощающих
предположений. Так, в теории учитывается только
электростатич. взаимодействие между ионами шаровой
формы с равномерно распределенным зарядом. Из
количественной теории, в частности, следует вывод
о линейной зависимости логарифма константы кислот-
ности (а также основности и протолиза) от обратной
величины диэлектрич.- проницаемости среды (1/е):
in кАН = ш ка + (2ZAH - 1)
где Ка — т. я. термодинамич. константа кислотности,
выраженная, в отличие от через активности
(а не концентрации) и поэтому не зависящая от раст-
ворителя (/fo = lim Л'АН), г — радиус частицы ки-
е—.со
слоты АН, ZAH — число элементарных зарядов (е)
в пей (если АН — нейтральна, то ZAH = 0,) k — по-
стоянная Больцмана, Т - - абс. темп-ра. Из приве-
денной формулы следует также, что для одинаково
заряженных кислот (ZA н = ZAiH = Zy = ...)
в двух различных растворителях (S, и S.,) константы
кислотности связаны условием
аа\й
К®2
.А, Н
К
А3И
S;
А), II
. = const
т. е. существует линейная зависимость констант ки-
слотности (а также основности и протолиза) кислот
в одном растворителе от констант кислотности (соот-
ветственно основности, протолиза) тех же кислот
в другом растворителе; при этом для неодинаково
заряженных кислот получаются различные прямые,
характеризующие эту зависимость.
Выводы теории Бренстеда относятся к разбавленным
р-рам, т. к. опа частично оперирует с концентрациями
К. по., а не их активностями. Многочисленные опыт-
ные данные согласуются с теорией, однако имеется
и ряд отступлений, обусловленных ее приближенным
характером, к-рый связан с учетом только электро-
статич. эффекта междуиопного взаимодействия и не-
учетом специфики химич. взаимодействия между
растворителем и К. и о. Дальнейшее развитие теории
Бренстеда связано с работами Н. А. Измайлова,
в к-рых указанная специфика химич. взаимодействия
учитывается тем, что рассматривается процесс иони-
зации не самих К. и о. в растворителе, а продуктов
их предшествующей ассоциации с молекулами рас-
творителя.
Следует иметь в виду, что теория БренсТеда яв-
ляется термодинамической и рассматривает лишь
равновесие протолитич. реакций, а не их кинетику.
При рассмотрении же кислотно-основного катализа
в первую очередь представляет интерес кинетика
перехода протона от кислоты к веществу, участвую-
щему в катализируемой реакции (или от вещества
к основанию). Однако эмпирически выявлено, что
увеличению константы протолиза, как правило, со-
585
КИСЛОТЫ И ОСНОВАНИЯ
586
путствует рост (правда, в меньшей степени) константы
скорости передачи протона, а следовательно, и ско-
рости катализируемого процесса. Это обстоятельство
выражает полуэмпирическое уравнение Бренстеда:
к= gKa, где к — константа скорости катализи-
руемой реакции, К — константа протолиза (кислот-
ного или основного катализатора с растворителем),
g и а — постоянные для данной реакции величи-
ны (а<1).
Электронная теория кислот и оснований. Теория
протолитич. равновесия очень четко определяет при-
надлежность веществ к классам К. и о. и формули-
рует ряд количественных зависимостей кислотно-
основных свойств. Однако она оказывается недоста-
точной, т. к., связывая эти свойства с наличием про-
тона, оставляет в стороне кислотно-основные свойства
(часто очень ярко выраженные) многих апротонных
веществ — таких, как галогениды бора и алюминия,
четыреххлористое олово и др. Электронная теория
К. и о., основы к-рой были даны Льюисом одновре-
менно с протолитич. теорией, восполняет этот не-
достаток. Отличительным признаком К. и о. по элект-
ронной теории является их взаимная нейтрализация,
осуществляемая путем образования ковалентной связи
между атомом основания, обладающим свободной
парой электронов, и атомом кислоты, в электронную
оболочку к-рого эта пара включается; в продукте
нейтрализации пара электронов обобществлена между
соответствующими атомами. Этим кислотно-основ-
ной процесс отличается от окислительно-восстанови-
тельного, при котором один или несколько электро-
нов полностью передаются от восстановителя к окис-
лителю.
При нейтрализации к.-л. основанием (напр., ионом
С1~) галогенида алюминия или бора (у атомов к-рых
нехватает до октета одной пары электронов) образуется
устойчивая электронная группировка из четырех
пар — октет; ’
:ci: Г ;С ' -
;СГ.А1 + [:С1: ]“ = :С1: А1:С1:
:ci: L -ci: -
При этом фторид бора является настолько сильной
кислотой, что присоединяет даже инертный газ —
аргон:
:F: :F:
:F:B -J-:Ar; = :F:B :Аг:
:F: :F:
В комплексных соединениях олова устойчивая внеш-
няя электронная оболочка центрального атома на-
считывает шесть пар электронов, поэтому нейтрализа-
ция SnCl4 к.-л. основанием (напр., пиридином) вы-
ражается схемой:
С1 С1
Cl:Sh:CH-2C5H5N:=Cl : " : NC5H5
ci Sn
Cl : : NC0H3
ci
Многие катионы металлов, как Li-1', Ве?+, Ag+и др.,
причисляются к кислотам в том случае, когда они
образуют с основаниями — анионами ковалентные
связи, тогда как Cs+ не может быть кислотой, т. к.
он не способен присоединить пару электронов и свя-
зан с анионом чисто кулоновскими силами.
Электронная теория К. и о. не причисляет к ним
бренстедовские кислоты, так как последние не ней-
трализуются основаниями, а образуют лишь новые
бренстедовские К. и о. Для выражения же такой
нейтрализации протонных К. и о., как, например,
НС1 + NHa = NHtCl, в рамках электронной теории
пришлось бы допустить двухвалентность водорода:
Н н
Ii:ci: +;N:H = :ci:H:N:H
Н ” Н
что противоречит теории строения вещества. Сам
протон как частица, легко присоединяющая пару
электронов, относится к сильным кислотам по Льюису.
Бренстедовские же кислоты формально рассматри-
ваются электронной теорией как продукты нейтрали-
зации протона основаниями (напр., НС1 — продукт
нейтрализации Н+ основанием Ci; Н3О+— продукт
нейтрализации Н+ основанием Н2О, и т. д.). При
этом протолитическая реакция АН + В = ВН + А '
соответствует процессу вытеснения основания (А-) из
соединения (АН) более сильным основанием (В~).
В ионизирующем растворителе льюисовские кислоты
повышают концентрацию катионов растворителя,
т. к. присоединяют его анионы в качестве оснований:
SO3+2H2O= Н3О++ HSO7. Аналогично основания
повышают концентрацию анионов растворителя:
С5Н5М + Н2О = ОН- 4~ C3H5NH+. Льюисовские ки-
слоты можно титровать основаниями в присутствии
индикаторов. Напр., SnCl4, ВС13 и др., растворенные
в бензоле, толуоле или хлорбензоле, хорошо тит-
руются триметиламином или пиридином в присутствии
индикаторов: тимолблау, цианина и др. Наконец,
льюисовские К. и о. широко используются в кислотно-
основном катализе.
Характерной особенностью льюисовских К. и о.
является специфичность их взаимодействия. С этим
связана невозможность их расположения в ряд по
силе, т. к. такое расположение зависит от веществ,
принятых за стандарт при сравнении. Напр., при
переходе от реакций с протоном к реакциям с ионом
Ag+ последовательность в силе оснований: CN“>NH3>
>SO?t-, заменяется на другую: C.\>SCH >NH3.
В силу той же специфичности взаимодействия льюи-
совских К. и о. и сложности учета квантово-химич.
эффектов электронная теория не является столь же
четкой и количественной, как теория протолитич.
равновесия. Однако она не только дополняет и расши-
ряет круг веществ с кислотно-основными свойствами,
но и более глубоко вскрывает причины этих свойств,
заложенные в строении веществ.
Практическое значение кислот и оснований. К. и о.
очень широко используются в химии. Это связано
не только с тем, что они охватывают большой круг
веществ, но и с тем, что состояние и поведение раз-
личных веществ в какой-либо среде в большой степени
зависит от свойств среды и, в частности, от кислотно-
основных ее свойств. Увеличение кислотности или
основности среды часто вызывает или усиливает
ионизацию веществ, что в свою очередь приводит
к увеличению их реакционной способности, к изме-
нению механизма взаимодействия и т. д. Все эти
факторы относятся, в первую очередь, к вопросам
кислотно-основного катализа. При этом каталитич.
действие бренстедовских и льюисовских кислот сходно
по своему характеру.
Весьма большую роль кислотно-основные реакции
играют во многих биохимич. процессах, специфич-
ность к-рых нередко определяется очень узким интер-
валом кислотности или основности среды. Важнейшие
соединения, участвующие в биохимич. процессах,
протекающих в живых организмах, — нуклеиновые
кислоты и белки, являются полимерными бренсте-
довскими К. и о.
В аналитич. химии кислотно-основная природа
веществ используется для их количественного опре-
деления путем титрования с применением индикаторов
или электрохимия, способами (потенццфморцрия, кон-
дуктометрия). При испоЛьзовании индццадоров, т. е.
веществ, изменяющих свою окраску в зависимости от
587
КИСЛОТЫ СМОЛЯНЫЕ — КЛАЙЗЕНА КОНДЕНСАЦИЯ
588
кислотности среды, необходимо учитывать их собст-
венные кислотно-основные свойства. Для характе-
ристики кислотности различных сред, включая кон-
центрированные р-ры кислот, пользуются т. н. функ-
цией кислотности Гаммета:
К®[ВН*-]
Ио = - !g -[5Г-
где Кв —------------термодинамич. константа кислот-
а явн+
ности индикатора, ан+, ав и авн+ — активности соот-
ветственно протона, индикатора — в форме основа-
ния (В) и индикатора в форме кислоты (ВН+) в реак-
ции ВН+ В + Н+, — отношение соответст-
вующих концентраций, т. е. степень ионизации инди-
катора. Для разбавленных р-ров, в к-рых активности
равны соответствующим концентрациям, функция
кислотности совпадает с величиной водородного пока-
зателя pH = —1g [Н+], широко используемого для
характеристики кислотности среды в разб. р-рах
(о функции На см. также Катализ кислотно-основной).
В химич. технологии и различных производствах
широко используются кислотно-основные процессы:
нейтрализация, получение солей, гидролиз, раство-
рение солей, кислотно-основной катализ, травление
металлов и т. д.
Лит.: Шатенштейн А. И., Теории кислот и осно-
ваний, М,—Л., 1949; Л ю д е р В., Цуффанти С., Элек-
тронная теория кислот и оснований, пер. с англ., М., 1950;
Bell R. Р., Acids and bases, N. Y., 1956; e г о ж e, The pro-
ton in chemistry, N. Y., 1959; Измайлов H. А., Электро-
химия растворов, Харьков, 1959; Одрит Л. и Клейн-
б е р г Я., Неводные растворители, пер. с англ., М., 1955;
Усанович М. И., Что такое кислоты и основания, Ал-
ма-Ата, 1953; Шатенштейн А. И., Изотопный обмен
и замещение водорода в органических соединениях в свете
теории кислот и оснований, М., 1960.
КИСЛОТЫ
часть (ок. 703:
СООН
СМОЛЯНЫЕ — главная составная
) веществ, экстрагируемых органич.
растворителями из древесины
хвойных пород деревьев; со-
ставляют основную часть жи-
вицы, канифоли, таллового мас-
ла. К смоляным кислотам от-
носят соединения с общей фор-
мулой С10Н20СООН, а так-
и дегидропроизводные: С19Н31СООН,
Б.
Щ3С
/СНз
СН
чсн3
же их гидро-
CiBH38COOH и С19Н17СООН. В живице из отечествен-
ной сосны обыкновенной (Pious Silvestris L.) были
найдены кислоты, строение и нек-рые свойства к-рых
представлены в табл.
Кислоты Т. пл., °C [Ис Чтакс. (спирт) JH.MK аВ
Левопимаровая (I) 150 —280,4 272,5 19,6
Неоабиетиновая (II) .... 167-169 +159 250,5 80
Абиетиновая (III) 174-175 -116 241 85,5
Тетрагидроабиетиновая (IVP 182 +6 — —
Дигидрабиетиновая (V)a’° . 177 +127,6 — —
Дегидроабиетиновая (VI) . . 173—173,5 +62 268И279 2,2 2,4
Палюстровая (VII) 167-169 +72 266 31
d-Пимаровая (VIII)a .... 219 +77 — —
7-изо-й-Пимаровая (1Х)а . . 162—164 0 — —
а УФ-спектр не характерен.
б Двойная связь может быть между атомами углерода
9—14, 13—14 и 7—8; свойства приведены последовательно
для обоих изомеров.
' 1 J
в Уд. коэффициент погашения а = - -- 1g -А- , где с —
концентрация вещества в г/л, I — длина слоя раствора в см,
lg Ja/J — оптическая плотность р-ра.
Все известные К. с. отличаются положенном двой-
ных связей в кольцах или заместителями (см. схему).
СН(СН3)2
СН(СН3)2
С(СН3)2
Кислоты IV, V, VI, VIII и IX являются сравни-
тельно стойкими по отношению к воздействию воздуха
и нагреванию, а все другие К. с. быстро окисляются
на воздухе, легко изомеризуются при действии кислот
и при нагревании и особенно при одновременном воз-
действии кислот и нагревания
(см. схему). К. с. — реакцион-
носпособные соединения. Они об-
разуют многочисленные произ-
водные. Реагируя как кислоты,
они образуют соли, сложные
эфиры, ангидриды, хлорангидриды, амиды, альдегиды
и др. В то же время они реагируют как непредельные
соединения, образуя дегидро-, дигидро- и тетрагид-
рокислоты, моно-, диокси- и тетраоксипроизводные,
галогенопроизводные, димеры и полимеры и др. Кис-
лоты I, II,III, VII реагируют с диеновыми соединения-
ми (напр., с малеиновым ангидридом) с образованием
аддуктов. При нагревании с серой или селеном кислоты
I—VII образуют ретен (X), а кислоты VIII—IX —
пимантрен (XI):
СН3
X XI
Сравнительно хорошо изучен состав смоляных к-т
отечественных хвойных: Pinus Silvestris L., Pinus
cembra L., Pinus taurica Hort, Pinus pithiusa Stev.,
Pinus insignis, Picea excelsa Link., Picea ajauensis
Fisch., Abies sibirica Ldb.
В пром-сти получили применение абиетиновая
и левопимаровая к-ты и их производные. Обычно
в пром-сти применяют сплав всех К. с. — канифоль
и ее производные.
Лит.: Комшилов Н. Ф., Состав канифоли и строение
смоляных кислот сосны и ели, М.—Л., 1955; Ф и з е р Л.
и Ф и з е р М., Химия природных соединений фенантренового
ряда, пер. с англ., М,—Л., 1953; Simonsen J., Barton
D. Н. R., The terpenes, v. 3, Camb., 1952; ШампетьеГ.,
Рабата Г., Химин лаков, красок и пигментов, пер. с
франц., т. 1, М„ 1960, с. 522; См. также статьи: Арбузова Б. А.,
Крестинского В. Н. с corp., Пигулевского Г. В. с сотр., Бар-
дышева И. И. с сотр. и до. в Ж. общ. химии, Ж. прикл.
химии и ДАН СССР за 1935—61. II. II. Бардышев.
КЛАЙЗЕНА КОНДЕНСАЦИЯ — образование 0-кето
(или 0-альдо)-эфиров, -нитрилов или -кетонов взаимо-
действием сложных эфиров с соответствующими про-
изводными кислот или с кетонами в присутствии
щелочных конденсирующих агентов:
RCOOR'+H-c(fx----->RCOC—X
х I
X=COOR", CN', COR
В качестве конденсирующих средств наиболее часто
применяют металлич. натрий, алкоголяты щелочных
металлов в суспензии или в спиртовых р-рах, амид
или гидрид натрия, мезитилмагнипбромид и три-
фенилметилнатрик.
589
КЛАЙЗЕНА ПЕРЕГРУППИРОВКА
590
Классич. пример К. к. — синтез ацетоуксусного
эфира (1). С эфиром щавелевой к ты получаются про-
изводные кетодикарбоновых (2) или дикетодикарбо-
новых (3) к-т, а с эфиром муравьиной к ты — произ-
водные альдокислот (4). Из сложных эфиров и кетонов
образуются 0-дикетоиы (5) или Р-кетоальдегиды (6).
Эфиры кротоновой и сорбиновой к-т, подобно этил-
ацетату, винилогами к-рого опп являются, вступают
в К. к. (7).
СНаСООС»НН СН8СООС2Н5 —> СН3СОСН2СООС2Н6 (1)
CeHs
C2H5OCOCOOC2H54-CoH5CH2CN—»C2H5OCOCOCHCN (2)
орто-аллилфенолов и С-аллильных производных кето-
енолов:
сн2
о чсн о
/Ч HR z4 ?
СН3 _ CH3SCH-CHCH=CH2
С2Н5ООС С2н5оо6
СООС2Н5 НС<СООС<>Н6
I +
СООС2Н5 НС^СООС2Н5
СО-С^-СООС2Й5
СО-С^СООС2Нз
он
с R
сн3 с-снсн=сн2
С2Н5ОоС
НСООС2Нз+СвНзСН2СООС2Н5
СсН5СН
уСООСаНз
ХСИО
СвНзСООСзНз+СНзСОСНг —С0Н3СОСН2СОСН3 (5)
сн2
о сн
I II
сн сн.
Vh2
? ?н
—«►НС нс
12 чсн2-сн2сн=сн2 сн-сн2сн=сн2
HCOOCsH5+CHa(CH=CH)nCOOC2Hs—►
—►НСОСН2(СН=СН)ПСООС2Н5 (7)
Внутримолекулярная К. к. в случае бифункцио-
нальных производных называется Дикмана реак-
цией.
Обычные побочные реакции при К. к. — взаимо-
действие исходных или полученных соединений с кон-
денсирующим средством, напр. аммонолиз сложных
эфиров при применении амида натрия и т. п. При
К. к. щелочной конденсирующий агент взаимодейст-
вует с компонентом, содержащим подвижный водород,
образуя анион I; этот анион присоединяется к слож-
ному эфиру с образованием аниона II, к-рый, от-
щепляя ион алкоголята, превращается в конечный
продукт:
0-
H-C-COrJ^-C-COrJi^k^OO^rcOC-C-OR"—.
I -ыв I I I
R'
I II
Перегруппировку осуществляют нагреванием О-ал-
лильных соединений до 200°, часто в таких инертных
растворителях, как парафиновое масло, тетралин,
дифениловый эфир, диалкиланилины и др., предпоч-
тительно в атмосфере водорода, углекислого газа
или азота. Аллильная группа эфиров фенолов мигри-
рует. почти исключительно в свободное о-положеиие
с «инверсией», заключающейся в присоединении
у-атома углерода аллильной группы к ароматич. ядру
и перемещении двойной связи из (3, у-положения
в а, р-положение:
Даже в случае2,6-ди замещенных аллилфениловых
эфиров промежуточно образуется нестойкий продукт
перегруппировки в о-положение с инверсией, к-рый
далее изомеризуется в стабильный п-продукт:
’ /7°
——.RCOCC;
—R О I \1Г
Реальность образования аниона I подтверждается тем,
что оптически активные сложные эфиры рацемизуются
в присутствии этилата натрия, а также тем, что ско-
рость К. к. соответствует скорости дейтерообмена
сложного эфира в C2H50D в присутствии этилата
натрия. К. к. широко применяется в синтезе раз-
нообразных органич. соединений. Открыта К. к.
в 1863 Гейтером и подробно изучена в 1887 Клай-
зеном.
Лит.: Хаузер Ч. Р., Хадсон Б. Е. мл., в кн.:
Органические реакции, сб. 1, пер. с англ., М., 1948, с. 345;
Ингольд К. К., Механизм реакций и строение органи-
ческих соединений, пер. с англ., М., 1959, с. 632.
Н. П. Гамбарян.
КЛАЙЗЕНА ПЕРЕГРУППИРОВКА — миграция
аллильной или замещенной аллильной группы в алли-
ловых эфирах фенолов или енолов с образованием
Такой механизм реакции подтверждается выделением
аддукта циклогексадиенона с малеиновым ангидридом
при проведении перегруппировки в присутствии по-
следнего.
К. п. легко могут быть подвергнуты и разнообраз-
ные соединения, обладающие группировкой атомов
\с^с— О—С—с=с/.
К. п. находит широкое при-
591 КЛАЙЗЕНА-ШМИДТА РЕАКЦИЯ - КЛАПЕЙРОНА-КЛАУЗИУСА УРАВНЕНИЕ 592
менение в органич. синтезе, в частности в синтезе
природных соединений, как, напр., элемицина, эвге-
нола, кровеацина, апиола укропа и мн. др. Перегруп-
пировка открыта Л. Клайзеном в 1912.
Лит.: Т а р б э л л Д. С., в кн.: Органические реакции, сб.
2, пер. с англ., М., 1950, с. 7; Conroy Н., Firestone
R. A., J. Amer. Chem. Soc., 1956, 78, К» 10, 2290; Schmid
К., Fahrni Р., Schmid Н., Helv. chim. acta, 1 956,
39 708. H. П. Гамбарян.
КЛАЙЗЕНА—ШМИДТА РЕАКЦИЯ—конденсация
ароматич. альдегидов с алифатич. или жирноароматич.
альдегидами и кетонами с образованием а, р-пена-
сыщенных альдегидов или кетонов, напр.:
CbHsCHO+CHsCOCbHs—С0Н6СН=СНСОСН3
К.—Ш. р. является частным примером кротоновой
конденсации и проводится в аналогичных условиях
в присутствии тех же щелочных катализаторов
(гидроокиси металлов, алкоголяты, гидрид натрия
и др.). Известна внутримолекулярная К.—Ш. р., при-
водящая к синтезу кумаронов.
Вг
С2Н5ОН
кон
К.—Ш. р. была использована для введения в декалон-1
ангулярной метильной группы:
Вариантом К.—Ш. р. является получение |5-нитро-
стиролов конденсацией нитроалканов с ароматич.
альдегидами в присутствии щелочей:
Сн3О
С3Н7о
CH3NO2—*
СН,О-Х\-СН
и
qi.oAJ chno2
Наиболее вероятный механизм реакции отображается
схемой:
CH3CORJ-Ii —.[СНОСОК] +вн
О-
ArCHO-HCHs-COR]-—>ArCHCH,COR
—в
он
—.AiCHCFNCOR—-АгСН=СНСОК
К.—Ш. р. очень близка к Клайзена конденсации и
часто носит то же название.
Лит.: Surrey A. R., Name reactions in organic che-
mistry, N. Y.. 1954, p. 35. H. П. Гамбарян..
КЛАПЕЙРОНА УРАВНЕНИЕ (Менделеева — Кла-
пейрона уравнение)—уравнение состояния идеального
газа, устанавливающее связь между объемом V дан-
ной массы (п молей) газа, его давлением/), и темп-рой Т:
pV = 11RT
Здесь R — универсальная газовая постоянная, т. е.
постоянный коэфф., относящийся к одному молю
газа и не зависящий ни от вида газа, ни от условий
его существования (конечно, в пределах примени-
мости закЙ#0в идеальных газов), а только от единиц
измерений.' К. у. непосредственно следует из кинетич.
теории гёййв. Ур-ние в его современном виде было
выведено в 1874 Д. И. Менделеевым путем сочетания
законов Бойля—Мариотта, Гей-Люссака и Авогадро.
В более частном виде (без использования закона
Авогадро) оно было предложено в 1834 Клапейроном
и содержало индивидуальную постоянную, завися-
щую от вида и количества газа.
Как и другие законы идеальных газов, К. у. при-
менимо тем лучше, чем ниже давление и выше темп-ра.
! Для различных газов при одинаковых внешних усло-
виях применимость К. у. неодинакова: обычно
она лучше для веществ обладающих более низкой
темп-рой кипения. Ур-ние широко используется при
различных расчетах свойств газов.
КЛАПЕЙРОНА—КЛАУЗИУСА УРАВНЕНИЕ —
термодинамич. соотношение, характеризующее про-
цессы перехода вещества из одной фазы в другую (ис-
парение, плавление, сублимация, полиморфное пре-
вращение и др.). Установлено в 1834 П. Клапейроном
и в 1850 выведено термодинамически Р. Клаузиусом.
К. — К. у. связывает тепловой эффект L процесса
(теплота испарения, теплота плавления и др.) с изме-
нением объема ДР и соответствующей производной
L=?-^AV (1)
Для процессов испарения и сублимации производная
выражает изменение давления насыщенного пара
р с темп-рой Т; для процессов плавления и полиморф-
’ ного превращения больший интерес представляет
обратная ей величина выражающая изменение
темп-ры перехода Т с давлением р.
В указанной форме К.— К. у. вполне . строго.
Его можно относить к любому количеству вещества,
при условии, что теплота перехода и соответствующее
изменение объема отнесены к одному и тому же
количеству данного вещества. К. — К. у. позволяет
определить характер зависимости равновесного дав-
ления от темп-ры. Можно доказать, что теплота L
положительна (т. е. тепло поглощается), если фазо-
вому превращению способствует повышение темп-ры
(напр., в процессах испарения жидкости). Из К,—К. у.
! dr;
следует, что в таких случаях знак определяется
знаком ДГ: если объем данного количества вещества
при фазовом превращении возрастает, то равновесные
давление и темп-ра меняются в одинаковом направле-
нип I > и 1; если же ооъем данного количества ве-
щества при этом превращении уменьшается, то равно-
весные давление и темп-ра изменяются в противопо-
ложных направлениях Так, напр., из
К. — К. у. следует, что темп-ра кипения повышается при
повышении давления (объем пара больше объема
жидкости), а темп-ра плавления льда (обладающего
меньшей плотностью, чем вода) при повышении
давления понижается.
Наиболее часто К. — К. у. применяется для расчета
процессов испарения и сублимации. В этом случае
для невысоких давлений пара (р<1 атм) К. — К. у.
может быть упрощено следующим путем. Пренебре-
гая объемом жидкости (или твердого тела) по сравне-
нию с объемом того же количества вещества в паро-
образном состоянии и полагая, что к насыщенным
парам применимо ур-ние pV = nRT (где п — число
молей пара при давлении р и темп-ре Т), можно
получить из (1) приближенное уравнение;
L = RT-d-^ (2)
где R — универсальная газовая постоянная. Это
ур-ние не требует знания плотностей сосуществующих
фаз. Для практич. расчетов уравнение (2) б. ч. инте-
593
КЛАРКИ ЭЛЕМЕНТОВ — КЛЕИ
594
грируют. При этом для области невысоких давлений
пара и длн не слишком большого интервала темп-р,
пользуясь допущением о независимости теплоты ис-
парении от темп-ры, получают ур-ние:
1пр = — + С (3)
где С — постоянная интегрирования. Ур-ние (3)
дает возможность, зная давления насыщенного пара
при двух темп-рах, рассчитывать теплоту испарения
и давлении насыщенного пара при других темп-рах
или, зная теплоту испарении и давление насыщенного
пара при к.-н. одной темп-ре, определить р при
других темп-рах. Ур-ния (1), (2) и (3) могут приме-
ниться не только к чистым веществам, но и к рас-
творам, причем как к раствору в целом, так п к от-
дельным компонентам раствора, свнзыван в последнем
случае парциальные давлении насыщенного пара
данного компонента с его парциальной теплотой
испарении.
Лиш..'Киоеев В. А., Курс физической химии, М,—Л.,
1956; К а р а п ет ь я нц М. X., Химическая термодинамика,
2 изд., М.—Л., 19 49; Леонтовнч М. А., Введение в тер-
модинамику, 2 изд., М.—Л., 1952; Левич В., Введение
в статистическую физику, М.—Л., 1950.
КЛАРКИ ЭЛЕМЕНТОВ — числа, выражающие
среднее содержание данного химич. элемента в земной
коре. Чаще эти числа даются в весовых процентах,
иногда в частнх на миллион пли, что то же самое,
в граммах на тонну; последнее удобнее длн более
редких элементов. Кроме К. э. земной коры, поннтпе
«К. э.» применяют длн литосферы и гидросферы, а
иногда и длн осадочных пород; однако нельзн гово-
рить о К. э. длн какой-либо отдельной породы или
определенной территории и т. д,; в этом случае гово-
рит о средней распространенности элемента. Первый
подсчет распространенности деснти главнейших эле-
ментов в земной коре дан в 1889 Ф. У. Кларком,
а для большинства элементов—в 1898 И. Фогтом; даль-
нейшие уточнении и дополнении делались в 1925—30
В. И. Вернадским, в 1923—32 А. Е. Ферсманом,
в 1931—37 В. М. Гольдшмидтом, а в последние годы
К. Ранкамой и Т. Г. Сахамой, А. П. Виноградовым
и др. учеными.
Ниже приведены значении К.э., опубликованные Дж.
Грином в 1953 (левый столбец). Длн удобства цифры
даютсн в граммах па тонну. Эти же значения длн
большинства элементов даны в 1958 Б. Мейсоном;
в скобках приведены данные Мейсона, отличающиеся
от данных Грина. В правом столбце приведены средние
распространенности элементов в горных породах,
опубликованные в 1956 Виноградовым и принятые
Грином в его статье в 1959.
Рассмотрение таблицы кларков позволяет сделать
следующие выводы. 1) Содержание химич. элементов
в земной коре очень неодинаково (так, кислород
в 1,5- 1015 раз более распространен, чем полоний),
причем преобладают элементы малых порядковых
номеров; с увеличением порядкового номера распрост-
раненность убывает. 2) Элементы с четными поряд-
ковыми номерами составлнют 86%, а с нечетными —
14% массы земной коры. 3) Кажущансн «частота»
или «редкость» элемента не соответствует действи-
тельным (так, наир., свинец привычно считать ме-
таллом широко распространенным, т. к. он давно во-
шел в технику п быт; на самом же деле свинец рас-
пространен в 6—10 раз менее,‘чем ванадий—металл,
к-рый обычно считают редким. К. э. служат эталоном
длн суждения о накоплении или рассеянии химич.
элементов. Сравнение среднего содержании к.-л.
элемента в той пли иной горной породе или па дан-
ной территории с его кларком (средним содержанием
длн земной коры в целом) позволнет судить о повы-
шенном или пониженном содержании элемента в каж-
дом случае. Т. обр., К. э. служат критерием длн уста-
новления повышенной металлоносности районов и тер-
риторий (металлогенических провинций). Знание К. э.
необходимо для проведения геохимич. методами по-
исков месторождений полезных ископаемых, в част-
ности месторождений, но выходящих на поверхность.
1 н 1300 (1400) 47 Ag 0.1 0,2
9 Не 0,003 — 48 Cd 0,15 (0.2) 0,13
3 Li 65 (30) 50 49 in 0,11 (0,1) 0,1
4 Be 3,5 (2) 4.2 50 Sn 40 (3) 32
5 В 3 13 51 Sb 1 (0,2) 0,3
6 С 320 230 52 Те 0,0018 (0,002) 0,001
7 N 46,3 (46) ? 53 J 0.3 0,4
8 О 466 000 473 000 54 Xe 2-10—’ __
9 F 700 660 55 Cs 7 10
10 Ne 7-10-’ ? 56 Ba 250 (400) . 640
И Na 28 300 25 000 57 La 19,6 (18) 40
12 Mg 20 900 17 000 58 Ce 24,7 (46) 40
13 Al 81 300 81 000 59 Pr 5.5 (6) 7
14 Si 277 200 291 000 60 Nd 18,1 (24) 30
15 P 1 180 . 900 61 Pm — —
16 S 750 (520) 900 62 Sm 6,5 (7) 4
17 Cl 314(200) 230 63 Eu 1,1 (1) Z-1
18 Ar 0,04 0,04 64 Gd 6,4 (6) 7
19 к 27 000 (25 960) 25 000 65 Th 0,9 z-1
20 Ca 36 300 33000 66 Dv 4,5 (5) 4
21 Sc 5 13 67 Ho 1,2(1)
Ti 4 400 4400 68 Er 2,5 2
23 V 150 (110) 90 69 Tu 0,2 1
Cr 200 117 70 Yb 2,7 (3) 2
Mn 1 000 1000 71 Lu 0,8 1
26 Fe 50 000 46 500 7’. Hf 4,5 (5) 3
27 Co 23 IS 73 Ta 2,1 (2) 2.7
28 Ni 80 100 74 W 1.5.(1) 2?
29 CU 70 (45) 70 75 Re 0,001 0,001
зо Zn 51 (65) 80 76 Os 0,005 (0,001) 0,0001
31 G.a 15 26 77 Ir 0,001 0,001
зз Ge 1.5(2) 78 Pt 0,005 0,005
.33 As 5 (2) 2 79 Au 0,005 0,001
34 Se 0.09 0.01 80 He 0,007 (0.5) 0,06
35 Br 1.62 (3) 1.8 81 TI 1,3(1) 1.7
36 Kr 1,6.10-*» ? 82 Pb 15 16
37 Rb 350 (120) 280 83 Bi 0,2 0,1
.38 Sr 300 (450) 350 84 Po 310-10 2-10-1»
39 Y 17-4 (40) 20 85 At — —
40 Zr 220 (160) 170 86 Rn — 6-10-12
41 Nb 24 20 87 Fr —
42 Mo 2,5 (1) 1.7 88 Ra 1.3*10-6 9-10-’
43 TC ? 89 Ac 3*10-10 5,510-1»
44 Rh 0,005 (0,001) 0,0001? 90 Th 11.5 (10) 13
45 Rh 0.001 0.001? 91 Pa 8*10-7 9-10-7
46 Pd 0,010 0,02 92 U 4 (2) 2,6
Лит.: Ферсман А. Е., Избранные труды, т. 3, М.—
Л., 1955, с. 160—95; Виноградов А. П., Геохимия,
1956, К» 1, 6—52; Green J., Bull. Geol. Soc. America, 1953,
64, № 9, 1001 — 12; его же, там же, 1959, 70, № 9, 1127—
1184; М ason В г., Principles of geochemistry, 2 ed., N.Y., 1958,
p. 1—310. В. В. Щербина.
КЛАТРАТНЫЕ СОЕДИНЕНИЯ — см. Соединения
включения.
КЛЕИ — растворы, дисперсии или расплавы преим.
высокомолекулярных органич. или неорганич. ве-
ществ природных пли синтетич., применяемые длн
соединении (склеивания)различныхматериалов. Склеи-
вание (см. Клеящее действие) основано на явлении
адгезии — прочного прилипании (сцеплении) кленщей
пленки к склеиваемым материалам. Прочность склеи-
вании определнетсн также когезией — прочностью
самой клеящей пленки. Прочное склеивание может
быть достигнуто при использовании кленщего мате-
риала с высокими адгезионными и когезионными
свойствами. Для склеивании необходимо хорошее
смачивание соединяемых поверхностей и их плотное
прилегание друг к другу.
Склеивание происходит в результате затвердевании
К. путем испарении растворители,, застудневании,
а также за счет химич. превращений, полимеризации
пли поликонденсации компонентов К. В результате
этих химич. превращений компоненты К. яасто ста-
новнтсн неплавкими и нерастворимыми. В*процессе за-
твердевании клеевой пленки обычно вода^ает усадка,
отрицательно влпнющая на прочности соединении.
Длн уменьшении усадки в К. вводят -доцолнптели.
Иногда с целью повышения эластичности клеевого
595
КЛЕИ
596
соединения в К. вводят пластификаторы. Остатки
растворителя в клеевом слое способствуют образова-
нию пор и снижают прочность клеевой пленки. Проч-
ность соединения, как правило, повышается с умень-
шением толщины клеевой пленки. Для большинства
К. оптимальная толщина клеевой пленки — 0,05—.
0,25 мм. Пористые материалы (древесина, пенопласты
и др.) и материалы с шероховатой поверхностью бла-
годаря увеличенной поверхности склеивания, а также
за счет «заклинивания» клеящего вещества в склеи-
ваемом материале обладают повышенной прочностью
соединения.
Для получения прочного клеевого соединения по-
верхности, подлежащие склеиванию, обрабатывают
шкуркой или струей песка для придания более
развернутой поверхности, обезжиривают (ацетоном,
спиртом, бензином и др.). Ввиду плохой адгезии из-
вестных К. к таким неполярным (инертным) материа-
лам, как полиолефины, политетрафторэтилен, поли-
этилентерефталат, перед склеиванием их поверхности
подвергают обработке специальными составами (хро-
мовая смесь, раствор металлич. натрия в жидком
аммиаке и др.), в результате чего они приобретают
способность склеиваться обычными К. Максимальная
прочность склеивания обеспечивается правильным
подбором К. для данного вида материала и точным
соблюдением технологии приготовления К. и склеи-
вания. В случае применения К., содержащих рас-
творители, после нанесения их на склеиваемые по-
верхности дают «открытую выдержку» на воздухе,
необходимую для удаления растворителя из клеевой
пленки.
К. применяются для соединения разнообразных
материалов: пластмасс, органич. и силикатного стекла,
натуральной и искусственной кожи, резины, фарфора,
керамики, бумаги, древесины, природных и синтетич.
волокон, стали, серебра, меди, алюминиевых, тита-
новых сплавов и др. металлов, неметаллич. мате-
риалов и их различных сочетаний.
Для испытания К. и клеевых соединений
обычно определяют концентрацию К., его вязкость,
жизнеспособность и прочность склеивания (клеящую
способность). Концентрацию К. находят путем
высушивания навески К. до постоянного веса при
определенной темп-ре (чаще всего 100—120°). Вяз-
кость испытывается с помощью вискозиметров
при 20°. Жизнеспособность К. характе-
ризуется временем, в течение к-рого К. сохраняет
вязкость, удобную для нанесения на склеиваемые
поверхности. Клеящая способность оце-
нивается с помощью испытаний механич. свойств
клеевых соединений на образцах стандартных раз-
меров и формы. Для клеевых соединении металлов
определяют предел прочности при сдвиге и при рав-
номерном и неравномерном отрыве. Прочность клее-
вых соединений металлов при сдвиге в зависимости
от примененного К. и склеиваемого металла колеб-
лется от 100 до 300 кг/см2-, при равномерном отрыве
прочность достигает 800—900 кг/сл«2; при неравно-
мерном отрыве 10—50 кг/см2.
При склеивании органич. стекла, древесины и др.
материалов проводят испытание на скалывание; при
склеивании тканей, теплоизоляции, пленочных мате-
риалов — испытание на отслаивание. Прочность кле-
евых соединений многих неметаллич. материалов
обычно равна прочности склеиваемых материалов или
превышает ее.
По происхождению различают К. природные и син-
тетические.
Природные К. делят на: животные, растительные
и минеральные. Природные К. чаще всего не стойки
к действию влаги, подвержены гниению и относительно
быстро теряют во времени прочностные свойства.
Животные К. — продукты переработки различных
материалов животного происхождения (мездры, костей,
кожи, молока, крови и др.). К. животные разделяются
на глютиновые, казеиновые, альбуминовые и комби-
нированные.
Г лютиновые К. делят на: мездровые, полу-
чаемые из подкожного слоя шкуры животных, кост-
ные — из костей и сухожилий, рыбьи — из плава-
тельных пузырей, чешуи, костей и др. отходов. Глю-
тиновые К. образуют прочные клеевые соединения,
обладающие, однако, малой водоупорностью и загни-
вающие под действием микроорганизмов; применяются
при склеивании древесины.
Сухой глютиновый К. выпускается в виде плиток, таблеток,
чешуек и порошка, а К. с влажностью 50—60% (галерта) —
в виде студня. Клеящая способность, определяеман испытанием
прочности клеевого соединения древесины ясеня или дуба на
скалывание, для лучших сортов мездрового К. составляет не
менее 100 кг/см2. Жизнеспособность р-ров глютиновых К.
ограничивается их недостаточной биология, стойкостью.
Основным веществом казеиновых К. яв-
ляется казеин, получаемый из обезжиренного молока
осаждением творога при действии сычужного фер-
мента или кислот (молочной, серной или соляной).
Водостойкие К. получают обработкой казеина оки-
сями нек-рых тяжелых и щелочноземельных металлов
(чаще всего гидрата окиси кальция, с к-рым казеин
образует нерастворимый в воде казеинат кальция).
Для увеличения жизнеспособности в состав казеи-
новых К. вводят щелочи или натриевые соли. Клея-
щие свойства, водоупорность, вязкость и др. свойства К.
зависят от количества щелочи, извести и воды.
По способу приготовления казеиновые К. разделяются на
жвдкосмешиваемые и порошкообразные. Жидкосмешиваемые
К. готовят на месте потребления. Примерный состав казеино-
вого известково-щелочного К. (в вес. ч.): 100 казеина, 20—30
Са(ОН)г, 8—10 NaOH, 300—400 воды. Необходимо строго
соблюдать порядок смешивания компонентов: набухший в
воде казеин размешивается вначале со щелочью, а затем с га-
шеной известью до получения однородной вязкой массы. К.
водостоек, клеящая способность прц испытании древесины на
скалывание ве менее 130 кг/см2, жизнеспособность 5—7 час.
Порошкообразные К. состоят из сухих компонентов и гото-
вятся на заводах. Примерный состав таких К. (в вес. ч.):
100 казеина, 27 гашеной извести (пушонка), 12 фтористого
натрия, 0,5 медного купороса и 2 керосина. Перед употребле-
нием порошкообразный К. разводят водой. Рабочая жизне-
способность — не менее 4 часов (считая с момента начала раз-
мешивания с водой). Предел прочности клеевого соединения
древесины при скалывании 100 кг/см2 в сухом состоянии и
70 кг/см2 после 24-часового вымачивания в воде. Прочность
казеиновых клеевых соединений выше, чем у глютиновых, яо
стойкость их к атмосферным воздействиям невысока.
Альбуминовые К. в качестве основного
компонента содержат альбумины крови животных.
Примерный состав альбуминового К. (в вес. ч.):
100 альбумина, 900 воды, 7,5 извести гашеной или
негашеной.
Измельченный альбумин замачивают в трехкратном коли-
честве воды и тщательно перемешивают при комнатной темп-ре
в течение 2—3 час., затем добавляют известковое молоко
и оставшуюся воду, нагретую до 26—30°, и все вместе пере-
мешивают до образования однородной желеобразной массы.
Жизнеспособность клеевого р-ра 6—9 час. К. отверждается
при нагревании до 100—'120°. Применяют также К., содержа-
щие аммиак и параформальдегид и отверждающиеся при нор-
мальной темп-ре. Альбуминовые К. образуют более водостой-
кие соединения, чем казеиновые, ио клеевой шов в воде на-
бухает и поражается Микроорганизмами.
Комбинированные К. состоят из альбу-
мина и казеина и обладают более высокой склеиваю-
щей способностью, чем альбуминовые и казеиновые К.
в отдельности. Применяются для горячего склеивания
древесных материалов в прессах.
Растительные К. готовятся на основе различных
камедей, канадского бальзама, крахмала, декстрина,
натурального каучука, гуттаперчи и др. веществ.
Применяются преим. в виде растворов; затвердевают
при испарении растворителя. Основное назначение —•
склеивание бумаги, кожи, текстильных изделий и др.
К минеральным К. относятся асфальтовые, битум-
ные и т. п. композиции.
597
КЛЕИ — КЛЕИ РЕЗИНОВЫЕ
598
Синтетические клеи (смоляные’ К,). Основой
этих К. служат разнообразные синтетич. полимеры
и мономеры. Синтетич. К. представляют собой р-ры
полимеров в органич. растворителях или в мономерах,
а также эмульсии и дисперсии полимеров, или не
содержащие растворителей смолы, отверждающиеся
в присутствии специальных добавок и т. д. Многие
синтетические К. представляют собой композиции
из нескольких полимерных веществ. Это позволяет
сочетать свойства различных полимеров и получать К.
с оптимальными свойствами. К. выпускаются в готовом
к употреблению виде или приготовляются на месте
потребления смешиванием составных частей. Синте-
тич. К. применяют в жидкотекучем состоянии, а также
в виде порошка и пленок. Синтетич. К. на основе
органич. полимеров пригодны для использования до
300—350°; кремнийоргапич. К. имеют более высокую
рабочую темп-ру. Большинство синтетич. К. хорошо
выдерживает воздействие переменных темп-p, они
обычно стойки к действию влаги, масел, жидкого
топлива и различных грибков. При использовании
ограниченно водостойких К. защищают торцы клее-
вых швов специальными лакокрасочными покры-
тиями. Синтетич. К. делят на 3 основные группы:
термореактивные, термопластичные и К. на основе
эластомеров.
1) Термореактивные К. содержат, смолы,
необратимо отверждающиеся при нагревании или
в присутствии специальных отвердителей. К. этого
типа относятся большей частью к числу теплостойких,
они образуют прочные, но жесткие и хрупкие клеевые
швы, К ним относятся К, на основе след, смол: феноло-
формальдегидпых, карбамидных, эпоксидных, поли-
уретановых, полиэфиракрилатных, кремнийоргани-
ческпх и др.
Ф ен о л о-фор м ал ь д ег идные К., не подвергну-
тые модифицированию (ВИАМВ-3, КВ-3, В-31-Ф-9, ЦНИИПС-1,
ЦНИИМОД и др.) применяют гл. обр. с добавками кислых
отвердителей (сульфонафтеновые к-ты — так наз. керосиновый
контакт, и др.) для склеивания древесины, фанеры, древесных
пластиков, пенопластов, органич. стекла и др. материалов
(без нагревания или при 50—6о°). Эти К. готовят гл. обр.
на основе водорастворимых смол резольного типа. Примерный
состав К. (в вес. частях): 100 смолы, 1400—4500 керосинового
контакта, 6—12 ацетона или спирта. Феноло-формальдегидные
К. получают смешением компонентов на месте потребления.
Жизнеспособность 2,5—4 час. Прочность клеевых соеди-
нений древесины (ясеня, дуба) и древесных пластиков при
скалывании — не менее 130 кг/см2. К. водостойки. Феноло-
формальдегидные смолы, модифицированные поливинилацета-
лями (БФ-2, ВФ-4, ВС-ЮТ, ВС-350, Ридакс-Е), каучуками
(ВК-3, ВК-4, ВК-32-200, ВК-32-250, и др.), полиамидами и
другими полимерами, позволяют получать клеевые соедине-
ния, обладающие высокой механической прочностью. Эти К.
пригодны для склеивания металлов и неметаллов в силовых
конструкциях.
Ф ен о л о - по л ив ин и л а ц ета л ьн ы е К. при-
меняют как в виде растворов феноло-формальдегидной смолы
и поливинилацеталя (чаще всего поливинилбутираля) в орга-
нич. растворителе (ВФ, ВС-ЮТ, ВС-350, Ридакс-64 и др.),
так и в виде композиции, состоящих иа р-ра смолы и порошка
поливинилформаля (Ридакс-Е). В последнем случае на по-
верхность склеиваемого материала наносят раствор смолы,
а затем посыпают порошкообразным поливинилформалем.
Применяют также в виде пленок. Склеивание производят при
150—200° под давлением. Прочность клеевых соединений ме-
таллов при сдвиге до 400 кг/см2, теплостойкость от 60° (ВФ-2),
до 200—350° (ВС-ЮТ, ВС-350). Срок хранения раство-
ров — 6 месяцев.
Феноло-каучуковые К. обычно состоят из двух
р-ров: феноло-формальдегидной смолы и резиновой смеси.
Готовят иа месте потребления смешением этих р-ров. Жизне-
способность — до 24 часов. Прочность клеевых соединений
металлов 200—300 кг/см2 при сдвиге. Клеевые соединения
термостойки до 200—300° и отличаются высокой водостой-
костью. Склеивание пленочными К. (ВК-3, ВК-4, ВК-32-200)
производят под давлением при 170—200°. Применение пленоч-
ных К. имеет большие технологич. преимущества при склеи-
вании мало искривленных изделий.
Карбамидные и меламиновые К. (К-17 и Др.)
готовят на основе мочевино- и меламино-формальдегидных
смол. В отличие от феиоло-формальдегидных карбамидные К.
не являются водостойкими и используются преим. в произ-ве
фанеры и при склеивании древесины (без нагревания, а также
и при повышенной темп-ре). К. на основе меламино-формаль-
дегидных смол обладают повышенной водостойкостью. При-
мерный состав мочевино-формальдегидного К. (в вес. частях):
100 смолы, 0,5—2,0 отвердителя (хлористый аммоний); до
10 наполнителя (древесная мука); жизнеспособность обычно
6—10 часов. Режимы склеивания: 3—6 часов при 20° или 3—12
минут при 90—125°. Прочность клеевых соединений древесных
материалов при скалывании (фанера) составляет 15—40 кг/см2
(в сухом состоянии) и 0—30 кг/см2 (после выдержки в воде
при 20° в течение 48 часов).
Эпоксидные К. приготовляют на основе полиэфи-
ров, содержащих в своем составе эпокси-группы (см. Смолы
эпокси&ные). Эпоксидные смолы являются основой К с высо-
кой адгезионной способностью к большинству материалов.
Если в качестве катализаторов применяют вещества основного
характера, напр. амины, эпоксидные К. отверждаются без
нагревания (Л-4, К-153 и др.). Кислые катализаторы (гл. обр.
ангидриды органич. двухосновных к-т) используют при полу-
чении впоксидных К., отверждающихся при нагревании
(ВК-32-ЭМ и др.). Известны твердые эпоксидные К. (в виде
порошка или сформованных прутков) марок Эпоксид-П и ПР.
Аральдит-1 и др. Эпоксидные К. холодного отверждения
обычно не содержат растворителей и состоят (в вес. ч.):
100 жидкой эпоксидной смолы и 6—10 полиамина (гексаме-
тилендиамин, полиэтиленполиамины, триэтилентетрамин и
др.). Жизнеспособность— 0,5—3 часа. Время отверждения—
1—3 суток при 20°. Прочность клеевых соединений метал-
лов при сдвиге 100—150 кг/см2. К. не теплостойки, обладают
невысокой стойкостью к действию воды. К., отверждающиеся
при нагревании (в течение 0,5—1 часа при 180—200°), состоят
из жидкой смолы (100 вес. ч.), в к-рой перед употреблением
растворяют отвердитель (напр., малеиновый ангидрид). Жиз-
неспособность — до 1 суток. Клеевые соединения металлов
имеют прочность 200—300 кг/см2 при сдвиге и до 900 кг/см2
при равномерном отрыве. Теплостойкость — до 100—150°.
Водостойкость удовлетворительная. Для склеивания порош-
ковым К. или К. в виде прутка склеиваемую поверхность
(нагретую до 100—110°) натирают прутком или посыпают по-
рошком с последующим соединением поверхностей и выдер-
живанием при 180—200° под давлением 1—3 кг/см2.
Полиуретановые К. (ПУ-2, ВК-5) используются для
склеивания металлов и большинства неметаллич. материалов
как на холоду, так и при нагревании. Они обычно состоят из
р-ров полиэфира и полинзопианата (напр., толуилендиизоциана-
та). смешиваемых непосредственно перед употреблением. Жизне-
способность 2—4 часа. Прочность клеевых соединений метал-
ла—до 150 кг/см2 при склеивании в течение 4 часов (Ю0—110°).
Теплостойкость—до 60°. Из изоцианатных К. наиболее
широко известен лейкон и т — раствор n, п', п/'-триизоцианата
трифенилметана в органич. растворителе. Применяют для креп-
ления резины к металлу. Модифицированный феноло-формаль-
дегидной смолой полиамидный К. МПФ-1 отличается высокой
эластичностью и хорошими показателями механич. прочности
клеевых соединений и применяется для склеивания металлов
в конструкциях силового назначения; недостаточно водо-
стоек; теплостойкость — до 60°.
К термореактивным К. относятся также? полиэфир-
акрилатные К., применяемые для соединения неме-
таллич. материалов, напр. органич. стекла (ВК-32-70), а также
металлов (ПН-5); кремнийорганические К. (НТ-15,
МАС-1, ИП-Э и др.) служат для склеивания резин ив кремний-
органического каучука, Кремнийорганич, Н. В К-2 и ВК-6
являются высокотеплостойкими (до 1000°) и используются для
соединения металлов и теплостойких неметаллич. материалов.
2) Термопластичные К. (К. на основе
растворов полиамидов, полиакрилатов, поливинилаце-
тата, дополнительно хлорированного поливинилхло-
рида, эфиров целлюлозы и др. полимеров) при нагре-
вании могут размягчаться, при охлаждении — вновь
отвердевать. Эти К. отличаются меньшей прочностью
и меньшей жесткостью, чем термореактивные; при-
меняют для склеивания термопластич. материалов
при нагревании и на холоду. К термопластичным
относятся полиизобутиленовые К., применяемые для
изготовления липких лент.
3) Эластомерные К. — преим. на осно-
ве каучуков, сходные с термопластами, но отли-
чающиеся высокой эластичностью (см. Клеи резиновые).
Лит.: Клеи и технология склеивания. Сб. статей, под
ред. Д. А. Кардашова, М., 1960; Адгезия, клеи, цементы,
припои. Сб., под ред. Н. Дебройиа и Р. Гувипка, пер. с англ.,
М., 1954; Кардашов Д. А. [и др.], Пластические массы,
1959, № 1, 40—49; Эпштейн Г., Склеивание металлов,
пер. с англ., М., 1956; К а р д а щ о в Д. А., Хим. наука и
пром-сть, 1957, 2, № 5; В е р д и н с к и х И. II., Клеи и
склеивание, Киев—М., 1952; Кротова Н. А., О склеива-
нии и прилипании, М., 1956; Благонравова А. А.,
Кардашов Д. А,, Ж. Всео. хим. об-ва им. Д. и. Менде-
леева, 1962, 7, № 2; Klebtechnik. Die Adhfision in Theorie
und Praxis, Stutg,, 1 957; Ullmann, 3 Aufl’., Bd 9, Mun-
clien — B., 1957, S. 578—609. Д-» АрНардащов.
КЛЕИ РЕЗИНОВЫЕ — растворы каучуков (на-
турального или синтетич.) или резиновых смесей
599
КЛЕММЕНСЕНА РЕАКЦИЯ
600
в органич. растворителях (гл. обр. в бензине и бен-
золе). В зависимости от назначения различают:
1) конфекционные клеи для сборки рези-
новых и резинотканевых изделий (обувь, рукава,
гуммирование аппаратуры) с последующей их вулка-
низацией', 2) самовулканизующиеся клеи
для склеивания и ремонта изделий из вулканизован-
ных резин (лодки, аэростаты, костюмы и др.); 3) клеи
для произ-ва прорезиненных тканей; 4) клеи для
произ-ва тонкостенных изделий без шва; 5) клеи для
крепления резины к металлу, дереву, цементу и др.
материалам. К. р. должны обладать: вязкостью,
позволяющей легко их наносить; конфекционной
клейкостью, позволяющей собирать изделие; высокой
прочностью связи деталей в изделии после вулкани-
зации (для конфекционных клеев) и после склеива-
ния (для самовулканизующихся клеев) и достаточной
стабильностью при их перевозке и хранении.
Применяемые для К. р. растворители должны быть
химически инертны по отношению к каучукам и со-
ставным частям резиновых смесей, из к-рых клеи
готовятся. Темп-ра испарения растворителей должна
лежать в определенных пределах, т. к. при быстром
испарении растворителя на слое клея образуется
пленка, затрудняющая дальнейшее испарение раство-
рителя. Кроме того, при быстром испарении раство-
рителя темп-ра клея может понизиться до темп-ры
конденсации влаги воздуха и на пленке клея может
образоваться роса. Это резко снижает клеящие
свойства клеев и прочность склеивания. Растворители
должны иметь минимальную токсичность, не иметь
неприятного запаха и быть наименее огнеопасными.
В Советском Союзе в качестве растворителя для К. р.
применяется «бензин-растворитель» (бензин-галоша)
по ГОСТ 443—56; за границей, кроме бензина,—
бензол. Применение токсичных растворителей (бен-
зола, дихлорэтана и других) в Советском Союзе разре-
шается лишь в исключительных случаях с принятием
ряда мер по технике безопасности. К. р. приготов-
ляют растворением каучука или каучука в смеси
с химикалиями (серой, ускорителем, наполнителем
и др.) в специальных аппаратах — клеемешалках.
Основные характеристики рабочих свойств К. р.: а) кон-
центрация (определяемая по сухому остатку); б) вязкость
(определяемая по времени истечения определенного объема
клея через калиброванное отверстие или временем падения
шарика установленного размера через слой клея определенной
высоты); в) клеящая способность (определяемая прочностью
в кг/см1 связи двух склеенных тканевых полосок при расслое-
нии их на динамометре); г) стабильность клеев при их хране-
нии в определенных условиях. Самовулканизующиеся клеи
содержат ультраускорители, и поэтому вулканизация пленки
клея происходит при обычных темп-рах.и в короткие сроки.
Для предотвращения преждевременной вулканизации эти клеи
готовят двухрастворными: в один раствор каучука входит
ультраускоритель, в другой — сера и активатор. Перед при-
менением клея оба раствора смешивают.
Лит.: Кошелев Ф. Ф. и К л и м о в Н. С., Общая
технология резины, 2 изд., М., 1958; Лепетов В. А.,
Производство резиновых технических изделий, М.—Л., 1946;
Израелит Г. Ш., Механические испытания резины и
каучука, Л.—М., 1949; Догадкин Б. А., Химия и физика
каучука, М., 1947; В о ю ц к и й С. С., Аутогезия и адгезия
высокополимсров, М., 1960. С. Жеребков.
КЛЕММЕНСЕНА РЕАКЦИЯ — замена атома кис-
лорода альдегидной или кетонной группы двумя
атомами водорода при действии амальгамированного
цинка и соляной к-ты:
RCH=O+4[H] HC1.>RCH3
R\c=O+4[H] Zn/HgAjE2LRCH2R'
R'z
К. p. не является суммарным результатом двух по-
следовательных процессов —• восстановления карбо-
нильного .[(^рдинения до соответствующего спирта
п далее —,,-сдцрта до углеводорода (спирты, как пра-
вило, в условиях К. р. не восстанавливаются). К. р.
существенно зависит от перенапряжения водорода
на поверхности Zn/Hg. При разряде протона на по-
верхности металла (Me) адсорбируется водород и об-
разуется поверхностный гидрид МеН, где связь
Me—Н ковалентна. С увеличением потенциала Me
эта связь ионизируется, и в переходном состоянии
при реакции с окислителем водород ведет себя как
гидрид-ио.н, причем перенапрнжение дезактивирует
восстанавливающее действие водорода. Поверхност-
ный гидрид МеН непосредственно не реагирует
с протонами или карбонильными соединениями, т. к.
эти реакции требуют значительной энергии активации.
Восстановлению подвергается сопряженная к-та (I)
карбонильного соединения, к-рая образуется в ре-
зультате присоединения протона по карбонильной
группе. Присоединяя электрон, I переходит в соот-
ветствующий свободный радикал II:
RR'C=O4-H+—.RR'C-ОН — -RR'C-ОН
I II
Затем происходит хемосорбция радикала II поверх-
ностью Me с образованием III, к-рый при действии
протона теряет гидроксил и переходит в карбониевый
ион IV, свободная орбита к-рого заполняется далее
в результате присоединения гидрид-иона (из МеН).
Образующийся V взаимодействует с протоном, давая
углеводород VI:
. (Ме)_ Н+ + МеН
RR'C—ОН-------- (Me)x:RR'COH —>[(Me)x:CRR']—
II III IV
МрН H+
— — (Me;.:CHP.R' _2_^RCH2R’
V VI
Такова схема основной реакции образования углево-
дородов.
Вышеприведенный механизм хорошо объясняет
и побочные процессы — получение пинаконов и оле-
финов. Пинаконы образуются сдваиванием свобод-
ного радикала II:
R4 ,R
2RR'C-ОН—. ус------С'
R'z I 1 XR'
II ОН он
Следующие обстоятельства благоприятствуют этой
реакции: 1) малая величина перенапряжения на по-
верхности Me; 2) высокая концентрация свободных
радикалов на поверхности Me; 3) стабилизация ра-
дикала за счет сопряжения неспарениого электрона.
Олефины в условиях К. р. образуются, если: 1) кон-
центрация поверхностного гидрида МеН на поверх-
ности Me невелика, в результате чего в ионе карбония
(Via) водород или радикал мигрирует из «-положе-
ния; 2) отщепление ОН и (Ме)х от III происходит
синхронно, с образованием карбена VII, где затем
происходит аналогичная миграция водорода или ра-
дикала из а-положения:
/Н
, +/ I , -Me. ,
Mex:RCOHCHaR ---- Me/RC-CHR ---i RCH-CHR
Via
. f I ,
Mex:RCOHCH,R----PC“CHR --------- RCH = CHR
VII
Образование олефинов имеет место также при восста-
новлении нек-рых ароматич. кетонов, где оно при-
водит к образованию энергетически выгодной системы
сопряженных связей.
Образование спиртов в обычных условиях К. р.
не происходит, т. к. для этого требуется большая
энергия активации. Исключениями являются восста-
новление а-кетокислот до а-оксикислот и бензальде-
гида до бензилового спирта. Наиболее широко
601
«КЛЕТКИ» ЭФФЕКТ —КЛЕЯЩЕЕ ДЕЙСТВИЕ
602
К. р. используется для получения углеводородов
из жирноароматич. кетонов. Так, из ацетофенона
получают этилбензол с 80%-ным выходом:
Восстанавливаемые карбонильные соединения могут
иметь другие функциональные группы, связанные
с ароматич. ядром или с алифатич. цепью, напр.
СООН, ОН, СН3О. Обычные осложнения при К. р.
состоят в том, что одновременно с карбонильной
группой гидрируются двойные связи в а, р-положе-
нии к карбонилу, пиррольные и изохинолиновые ядра;
замещается водородом галоген в a-положении к кар-
бонильной группе (реже — в ароматич. ядре) и гид-
роксил в а-оксикислотах и бензоинах. К. р. разра-
ботана в 1913—14 Е. X. Клеммеисеном. Другими
способами превращения альдегидов и кетонов в угле-
водороды являются каталитическое гидрирование
и Кижнера—Вольфа реакция.
Лит.: Мартин Э., в сб.: Органические реакции. Сб. 1,пер.
с англ., М., 1948, с. 194; Staschewski D., Angew.
Chemie. 1959. 71, № 23, 776. Б. Л. Дяткин.
«КЛЕТКИ» ЭФФЕКТ — торможение средой диф-
фузионного рассеяния свободных радикалов, являю-
щихся осколками одной и той же молекулы, приводя-
щее к рекомбинации этих радикалов. В литературе
«К.»э. наз. также эффектом Франка и Рабиновича,
или первичной рекомбинацией, в отличие от реком-
бинации свободных радикалов, образовавшихся при
распаде различных молекул. Молекула, получившая
(в результате соударений или поглощения кванта
света) энергию большую, чем энергия химич. связи,
может распасться на свободные радикалы, к-рые
сразу расходятся на расстояние порядка длины сво-
бодного пробега. В газовой фазе при атмосферном
давлении это расстояние составляет 10 5—10 6 см,
т. е. оно значительно больше размера молекулы
(~4 • 10'8 с.к). В этих условиях вероятность столкно-
вения и рекомбинации радикалов между собой нич-
тожна. В конденсированной же фазе межмолекуляр-
ные расстояния либо одного порядка с размерами
молекул (для низкомолекулярных веществ), либо
даже значительно меньше размеров молекулы (для
полимеров). В этом случае образовавшиеся радикалы
как бы заключены в «клетку» из окружающих моле-
кул среды и нек-рое время после образования нахо-
дятся в непосредственной близости друг к другу.
За это время (1()10—10'11 сек) они претерпевают ряд
последовательных столкновений друг с другом и мо-
гут опять соединиться. По мере увеличения расстоя-
ния'между двумя образовавшимися свободными ра-
дикалами вероятность их встречи, а следовательно,
и первичной рекомбинации уменьшается. Радикалы
вследствие диффузии покидают «клетку». На вероят-
ность первичной рекомбинации влияют свойства
среды, напр. вязкость. В случае, если рекомбинация
происходит при каждом соударении, доля свободных
радикалов, претерпевающих первичную рекомбина-
цию, может достигать ~0,5. В других случаях она
меньше и, в частности, может быть практически равна
нулю. Теоретич. пределы изменения этой величины
подтверждаются экспериментальными данными.
Рассмотренный выше характер первичной реком-
бинации справедлив лишь для химически инертной
(по отношению к радикалам) среды, в к-рой выход
радикалов из «клетки» происходит только за счет
диффузии. Однако возможен и другой механизм вы-
хода радикалов из «клетки» — за счет химич. реак-
ции с молекулами среды
Ri’-J R;X-► Ra+RiX
где R; и R« — первичный и вторичный радикалы,
R2X — молекула среды. Свободная валентность пе-
редается одной из молекул среды, от нее — другой
и т. д. Этот механизм более вероятен в твердой фазе,
где диффузия почти исключена.
Изложенное представление о «К.» э. имеет важное
значение для понимания механизма различных химич.
процессов, происходящих с участием свободных ра-
дикалов. Напр., в процессах свободно-радикальной по-
лимеризации эффективность инициатора полимериза-
ции выражается долей образующихся при его распаде
радикалов, вступивших в реакцию полимеризации. За
время пребывания радикалов в «клетке» вероятность
первичной рекомбинации много больше вероятности
реакции радикала с молекулой мономера, даже если
радикалы находятся в среде чистого мономера (мо-
лярная доля мономера равна 1). Т. обр., эффектив-
ность инициатора не зависит от концентрации моно-
мера в растворе и определяется в основном «К.» э.
«К.» э. проявляется и в процессе деструкции поли-
меров. Образующиеся при нагревании или облучении
в полимере радикалы малых размеров, напр. Н',
СН;, имеют большую вероятность покинуть «клетку»
и вступить в последующие реакции, чем крупные
радикалы. Это обстоятельство ограничивает выход
продуктов деструкции и способствует преимущест-
венному образованию низкомолекулярных веществ.
«К.» э. объясняет также меньшую химич. эффективность
радикалов ОН’ и Н’, образующихся из возбужденных
молекул воды, по сравнению с теми, к-рые возникли
из ионизованных молекул при радиолизе воды. В пер-
вом случае радикалы ОН’ и Н’ заключены в «клетку»,
а во втором — они раздвинуты на несколько молеку-
лярных радиусов в момент их образования.
Лит.: Franck J., Rabinowitsch Е., Trans.
Faraday Soc., 1934, 30, Я» 152, pt. 2, 120; Noyes R. M.,
J. Chem. Phys., 1954, 22, № 8, 1349; J. Amer. Chem. Soc.,
1956, 78, № 3, 1955, 77, № 8, 2042. А. II. Куриленко.
КЛЕТЧАТКА — см. Целлюлоза.
КЛЕШНЕВИДНЫЕ ПОЛИМЕРЫ — см. Коорди-
национные полимеры.
КЛЕШНЕВИДНЫЕ СОЕДИНЕНИЯ — см. Внут-
рикомплексные соединения.
КЛЕЯЩЕЕ ДЕЙСТВИЕ (склеивание) — проявле-
ние способности клеев при затвердевании в контакте
с поверхностями различных твердых тел (материалов)
прочно соединять эти тела. К. д. основано на явлении
адгезии — прочного прилипания прослойки клея к по-
верхностям склеиваемых материалов — в сочетании
с повышением прочности (когезии) самой клеящей
прослойки в процессе ее отвердевания. При разру-
шении прочных клеевых соединений (напр., склеен-
ного силикатного стекла, кварца клеями на основе
синтетич. полимеров) почти всегда наблюдается на-
ряду с адгезионным и когезионный тип разрушения
в объеме клеевой прослойки или склеиваемого мате-
риала (см. Когезия). Поэтому для получения прочных
клеевых соединений необходима не только высокая
клеящая способность высокополимера (адгезия его
к склеиваемым материалам), но и его большая коге-
зионная прочность в отвержденном состоянии. Для
склеивания важное значение имеет смачивание по-
верхности склеиваемого материала (подложки) клеем.
При плохом смачивании субстрата прочность склеи-
вания всегда получается невысокой. Наоборот, обра-
зованию прочных клеевых соединений обычно пред-
шествует хорошее смачивание поверхностей клеем.
Так, напр., синтетич. клеи с активными функциональ-
ными группами в цепных молекулах (—ОН, —СООН,
—NHCO— и др.) практически не смачивают твердых
материалов с низкой поверхностной энергией (поли-
этилен, полипропилен, политетрафтОрэтайен и др.)
и совершенно пе склеивают их. С др^бйй стороны,
твердые тела с высокой поверхностной'ййбргией (ме-
603
КЛЕЯЩЕЕ ДЕЙСТВИЕ-КНЕВЕНАГЕЛЯ РЕАКЦИЯ
604
таллы, кварц, силикатное стекло и др.) хорошо сма-
чиваются перечисленными клеями и при склеивании
образуют весьма прочные соединения. Однако само
по себе смачивание еще не определяет степени при-
липания клея к подложке и прочности получаемых
клеевых соединений.
Интенсивность прилипания определяется силами
притяжения между частицами клея и склеиваемого
материала. При склеивании проявляется действие
ван-дер-ваальсовых сил, водородных связей и сил
химич. сродства (ковалентные и электровалентные
связи, координационные связи и др.). К. д. иногда
связывают с образованием двойного электрич. слоя
при контакте двух различных тел и притяжением
противоположно заряженных частей этого слоя.
Прочность клеевых соединений снижается благо-
даря внутренним напряжениям, возникающим из-за
усадки клея при отверждении или же вследствие раз-
ности коэфф, теплового расширения подложки и от-
вердевшей прослойки клея (т. н. термич. напряже-
ние). Прочное склеивание стали, дуралюмина, титана
и др. металлов и сплавов клеями, в макромолекулах
к-рых содержатся карбоксильные группы (сополи-
меры акриловой и метакриловой к-т, карбоксилсо-
держащие полимеры и др.), связано с образованием
между ними связей ионного типа. Ионные связи могут
образоваться как при взаимодействии с поверхност-
ными окислами, так и с чистыми неокисленными по-
верхностями металлов.
Образование координационных связей наблюдается
при соединении металлов клеями на основе полиами-
дов или диизоцианатов. При контакте полиамид-
ного клея со сталью могут возникать поверхностные
химич. соединения типа (I), в к-ром атом азота из
полиамида делит два своих электрона с атомом железа.
Одновременно между угле-
родом кетогруппы и ато- •
мом кислорода в окисле
железа возникает допол-
нительная ионно-диполь-
ная связь. Таким образом
при склеивании стали по-
лиамидами по существу об-
разуется хелатное соеди-
нение, имеющее координационную и ионно-диполь-
ную связь. Подобное сочетание координационной
и ионно-дипольной связи обеспечивает более прочное
сцепление между клеем и металлом, чем при образо-
вании одной координационной связи. К. д. эпоксидных
смол, отвержденных аминами, по отношению к ме-
таллам переменной валентности также связано с об-
разованием координационной связи. Химические связи
между макромолекулами каучука и медью через
атомы серы образуются также при креплении резины
к латунированным металлич. поверхностям в про-
цессе ее «привулканизации». Ионно-дипольное взаи-
модействие с металлами может иметь место в случае
полимеров, содержащих группы CN в полиакрилонит-
риле, сложноэфирные груп-
I пировки в полиметилмета-
м । крилате или поливинилаце-
/ \ тате и т. д.
Н3С \ >N —Н К. д. высокополимеров
'—' r = Q (изоцианатные клеи, живот-
। ные клеи) по отношению к
О силикатному стеклу может
--------1 поверхность быть настолько высоким
-Si- стекла (прочность на разрыв до
1 300—400 кг/слт2), что разру-
II шение клеевых соединений
часто происходит по клею
или даже по самому стеклу. Это связано со способно-
стью стекла вступать в химич. взаимодействие с мо-
Н О
I U
-CHo-N--------с-сн2-------
°\| °\
— Fe +------Fe + — поверхность
стали
I
лекулами клея. Так, клеи на основе толуилендиизо-
цианатов при взаимодействии с поверхностью стекла
образуют ковалентные связи (II),
Исследования механизма К. д. показали, что в за-
креплении полимерных клеев на твердых поверх-
ностях участвует незначительная часть активных
в отношении адгезии центров. Остальная же часть
активных центров на межфазной поверхности не мо-
жет приблизиться друг к другу на достаточно близкие
расстояния из-за геометрич. затруднений. Высокое
К. д. поэтому может быть обусловлено только воз-
никновением химич. связей между клеем и субстратом.
Сравнительно слабое склеивание (напр., стекла поли-
эфирными смолами, нитроцеллюлозой, поливинил-
хлоридом и др. полимерами) связано с тем, что К. д.
ограничивается в этих случаях только ван-дер-вааль-
совыми силами или водородными связями.
Полиэтилен, полипропилен, политетрафторэтилен,
политрифторхлорэтилен и др. полимеры, обладающие
низкой поверхностной энергией, не склеиваются су-
ществующими клеями. Для склеивания их с метал-
лами и другими материалами поверхности указан-
ных полимеров предварительно подвергают химич.
модифицированию (напр., путем окисления). В ре-
зультате этого в макромолекулах поверхностного слоя
образуются адгезионно-активные функциональные
группы (ОН, СООН, NH2 и т. д.). Химич, модифици-
рование поверхности используется и при склеивании
нек-рых резин (напр., с кожей в произ-ве обуви).
Применение методов химич. обработки поверхности
твердых материалов позволяет решать и обратную
задачу, когда требуется снижение пх адгезионной
способности (устранение прилипания формуемых из-
делий к стеклянным и металлич. формам, борьба
с обледенением твердых поверхностей и т. д.).
Для проявления К. д. существенное значение имеет
также строение пограничного слоя между клеем и под-
ложкой. Резкая граница образуется, напр., при склеи-
вании металлов и др. твердых тел. В этом случае
К. д. связано с физич. и (или) химич. адсорбцией
макромолекул клея на твердой поверхности. Резкая
граница раздела клея и подложки отсутствует, напр.,
при контакте термопластич. полимеров, обладающих
взаимной совместимостью и растворимостью. Благо-
даря микроброуновскому движению при этом проис-
ходит диффузия гибких молекул высокополимера
или их участков из одной фазы в другую, сопровож-
дающаяся исчезновением резкой границы раздела
и образованием переходного слоя с постепенным изме-
нением состава и свойств полимера в этом слое. Взаим-
ное проникновение и перепутывание цепных молекул
способствуют упрочнению связи между двумя различ-
ными полимерами, и адгезия их друг к другу пред-
ставляет уже не поверхностное, а объемное явление.
Прочность склеивания в таких системах аналогична
когезионной прочности полимеров.
Лит.: В о ю ц к и й С. С., Аутогеаия и адгезия высоко-
полимеров, М„ 1960; Эпштейн Г., Склеивание металлов,
пер. с англ., М., 1956; Трепнел Б., Хемосорбция, пер.
с англ., М., 1958; Дерягин Б. В. иКротова Н. А.,
Адгезия. Исследования в области прилипания и клеящего дей-
ствия, М,—Л., 1949; Р у т ц л е р Д. Е., Химия и техноло-
гия полимеров, I960, Кг 10, с. 138 и Кг И, с. 134; Кротова
Н. А., О склеивании и прилипании, М., 1956; Клеи и техно-
логия склеивания, сб. ст. под ред. Д. А. Кардашова, М., 1960;
ШреМнер С. А., Зубов И. И., ДАН СССР, 1959, 124,
№ 5, 1102; их- же, Коллоидн. ж., 1960, 22, вып. 4; В о ю ц-
кий С. С. [и др.], Высокомолекулярные соединения, 1962,
4, Кг 2, 285 и 294. А. Я. Королев.
КНЕВЕНАГЕЛЯ РЕАКЦИЯ — конденсация кар-
бонильных соединений с соединениями, содержащими
активные метиленовые группы в присутствии аммиака
или аминов, с образованием производных этилена;
R\ /X
>с=о-ьснх
R'/
R'\
>NH
R'"/
R
R'
\c-c/X
— U\.
'Y
605
КНЕВЕНАГЕЛЯ РЕАКЦИЯ — КНОРРА РЕАКЦИЯ
бое
Карбонильным компонентом может служить аро-
матич., алифатич. или гетероциклич. альдегид, иногда
кетон; в качестве метиленового компонента, как пра-
вило, применяют малоновую к-ту или ее производные.
При наличии свободной карбоксильной группы в ме-
тиленовом компоненте реакция часто сопровождается
декарбоксилированием:
СвН6СНО+СП2(СООС2Н5)2—.СвН5СН=С(СООСаН5)2-|-Н2О
СвН13СНО+СН2(СООН)—>С0Н13СН=СНСООН-|-СО2-1-Н2О
.соон
С6Н5СНО-|-СН2< —►
XCONH-C0H4-CH3
—.C0H3CH=CHCONH-CoH4-CH3-|-C02-l-H20
CoIIsCHO-|-CHs-CeH4-SOsCH2COOH—-
—-CeH5CH=CH-SO2-Cl)H4CH3+CO2+H2O
Конденсирующими средствами обычно служат ам-
миак и алифатич. первичные или вторичные амины
в количествах не более эквимолекулярного; особенно
часто применяют пиперидин. Иногда заранее готовят
из альдегида и амина соответствующий альдимин или
оксиамин и затем проводят реакцию с метиленовым
компонентом. В качестве растворителя обычно приме-
няют спирт, бензол, пиридин. Темп-ра К. р. обычно
не превышает 100°, выходы, как правило, хорошие.
Конденсация альдегидов с барбитуровой к-той и ее
производными осуществляется при простом нагре-
вании в водном р-ре без конденсирующих средств:
СН3 I-- ..1-СНО ZCO-NHX
I + сн2 со —-
VCO-Nlf
ZCO'-NH4
СН3 I т-С!<=С со
-*СНз I Lo “~М|/
т
Иногда в условиях К. р. образуются оксисоедине-
ния, напр. р-оксикислоты:
СТ3\ пиридин OS's.
>С=О+СН2(СООН)2 \с-сн2--соон
CF/ CFZ I
он
Побочно часто образуются аминосоединения; при
нек-рых условиях (соответствующее строение исход-
ных веществ, применение избытка аммиака или амина,
менее продолжительное нагревание) они оказываются
главными продуктами реакции (см. Манниха реак-
ция, Родионова реакция).
До сих пор нет единого мнения относительно меха-
низма К. р.; вероятно он различен в зависимости от
строения исходных веществ. Наиболее вероятны сле-
дующие схемы реакции;
я>
4-ft"
t. СНгГСООЮ.-^г^СНГСООЮг——-Н-.
-RR’NH»
—'R"-9H-CH(COOH)2^^-
О’
Р"-<рН-СН(СООН).
он
—--------->- r"-Ch=ch~coo'H
—СО2:—Н2О
2.
/Он +СН2ССООН)2
R"“CH — н л
\NRR' н2°
—►Е"-сН-Сн(соон)а --- R"—СН=СН—Соон
| * Nn
NRli'
Первая схема — частный случай обычной интер-
претации механизма реакций карбонильных соеди-
нений с веществами, содержащими активные метиле-
новые группы, в присутствии оснований (см., напр.,
Перкина реакция, Альдольная конденсация). О второй
схеме см. Родионова реакция.
Продукты, получаемые в результате К. р., гл. обр.
производные коричной кислоты, применяются в ка-
честве лекарственных и душистых веществ. К. р.
открыта Э. Кневенагелем в 1896.
Лит.: Surrey A. R., Name reactions In organic che-
mistry, N. Y., 1954, p. 103; В e й г а н д К., Методы экспе-
римента в органической химии, пер. с нем., ч. 2, М., 1952,
е. 459—66; Balas ubramanlan М., В all ah V.,
Science and Culture, 1956, 21, Ki 8, 450—51.
E. M. Рохлин.
КНОРРА РЕАКЦИЯ — методы синтеза азотсо-
держащих гетероциклических соединений:
1. Получение 2-оксихинолинов внутримолекулярной
конденсацией ариламидов fj-кетокарбоновых к-т в при-
сутствии серной к-ты:
Н
Аналогичная циклизация N-алкилариламидов при-
водит к 2-хинолонам:
Ариламид постепенно добавляют к холодной серной
к-те и смесь нагревают до 100° несколько часов.
2. Синтез 5-пиразолонов конденсацией эфиров
^-кетокислот с гидразинами;
СН3-С-Сн2-С* + H2nnhr Г.Чг0'~^2^бн>.:-
о ос2н5
___ н3с-с сн2
Nxn.C = O
R
3. Получение пирролов конденсацией а-амино-
кетонов с кетонами, содержащими а-метиленовую
группу: ,
R'-OO H2C-R’ -2Н2О R't-------------л-R"
R'-CH-NH2 o=c-r” : ” R'A.NJ-R~
H
Побочной реакцией является собственная конденса-
ция а-аминокетона с образованием пиразина:
R-C=O | H2N-CHR'-2Нго» R-(yN^C-R'
g-CH-NHg O=CR" [о]” R'-C^C-R'
В том случае, если R' = R"', a R" = R"”, стадию
получения пиррола можно объединить со стадией
образования а-аминокетона. Кетон R'CH2COR" ча-
стично нитрозируют с образованием а-изонитрозо-
кетона R'C(= NOH)COR" и полученную смесь восста-
навливают цинком в ледяной уксусной к-те. Вариан-
том этого метода является* конденсация,,,диоксимов
а-дикетонов с эфирами ^-кетокислот при действии
607
КОАГУЛЯЦИЯ
608
цинка в ледяной уксусной к-те:
CHS-C=NOH Н2С-СООС2Н5 И____________
CH3-C=NOH О=С~СН3 -H2O:-NH2OH
____ Н3С-Ц--рСООС2Н5
н3с-%Лсн3
Для вторичных а-аминокетонов К. р. приводит
к N-алкилпирролам. Указанные реакции очень широко
применяют для синтезов в ряду пиррола.
4. Получение пирролов из у-дикетонов и аммиака
пли первичных аминов (синтез Пааля — Кнорра):
сн3-с-сн2сн2-с-сн3 + rnh2——*- ГД_СНз
О о ON
R
Реакции разработаны Л. Кнорром в 1886 и сл.
Лит.: Morton A. A., The chemistry of heterocyclic
compounds, N. Y.—L., 1946; Гетероциклические соединения,
под ред. Р. Эльдерфилда, Пер. с англ., т. 1—2, М., 1953—54,
с. 222, 25; Bergstrom F. W, Chem. Revs, 1944, 35, Ki 2,77;
Невеска H., Chemie der Beta-Dlcarbonylverblndungen,
В., 1950, S. 31. Б. Л. Дяткин.
КОАГУЛЯЦИЯ (от лат. coagulum — сгусток) —
слипание частиц в дисперсных и особенно коллоидных
системах с образованием более или менее крупных
агрегатов. К., если она зашла далеко, приводит
к образованию хлопьев, выпадающих в осадок (или
всплывающих), в результате чего число взвешенных
частиц в дисперсной системе становится незначи-
тельным. К. также может привести к возникновению
сплошной, хотя и рыхлой, пространственной струк-
туры — коагуляционной структуры геля (коагеля, см.
также Структурообразование в дисперсных системах).
От К. следует отличать коацервацию — разделение
дисперсной системы с образованием новой фазы
(напр., в виде капель). Слипание частиц эмульсий
нередко переходит в их слияние (см. Коалесценция).
Изменение структуры белков в процессах денатурации
обычно связано с К. их растворов.
К. — проявление термодинамич. неустойчивости дис-
персных систем. При К. эти системы переходят в со-
стояние, более близкое к равновесному, при коалес-
ценции и перекристаллизации частиц уменьшается
общий запас поверхностной энергии.
Различают две стадии К.; 1) потеря агрегативной
устойчивости и слипание частиц (скрытая К.);
2) выпадение (или всплывание) образовавшихся агре-
гатов частиц в осадок (коагулят) — явная К.
О начальной стадии К. в системе судят по резкому
увеличению интенсивности светорассеяния (см. Опти-
ческие свойства коллоидных систем), по изменению
окраски, увеличению вязкости, появлению вязкости
структурной и др. признакам. (Первая стадия К.
не обязательно переходит во вторую). К. может про-
исходить самопроизвольно (автокоагуляция)
под влиянием химич. и физич. процессов, протекаю-
щих в системе во времени (т. н. старение), но, как
правило, К. вызывается внешним воздействием под
влиянием: добавления к дисперсионной среде различ-
ных веществ — коагулянтов (электролитов или не-
электролитов), длительного диализа, электрофореза,
повышения темп-ры, действия света и др. высоко-
частотных электромагнитных колебаний, а также
ультразвуковых колебаний, встряхивания и переме-
шивания. Гидрозоли могут коагулировать при взбал-
тывании с неполярными жидкостями (напр., бензин),
частицы при этом собираются на границе раздела
жидкостей. В аэрозолях (дымах и туманах) частицы
несут на своей поверхности очень малые заряды или
вовсе не зарйжены, из-за чего в аэрозолях всегда идет
самопроизвольная К. В промышленных условиях
К. дымов и др.: аэрозолей производится путем фильтра-
ции их через пористые материалы или с помощью
электрич. полей с высоким градиентом потенциала
(электрофильтр Коттреля и др.).
К. препятствуют электрич.заряд частиц и сольва-
тация их поверхности, а иногда — адсорбция на ней
растворимых и устойчивых в этих условиях крупных,
молекул поверхностно-активных веществ, например’
белков. Для коллоидных систем это последнее явле-
ние получило название коллоидной защиты, к-рая
обусловлена образованием структурно-механич. барье-
ра на поверхности частиц (см. Защитные коллоиды).
Следует, однако, иметь в виду, что добавление высо-
комолекулярных веществ к лиофобным золям может
привести иногда и к уменьшению устойчивости по
отношению к электролитам (сенсибилизация)
и даже к К. Снижение величины электрич. заряда
(или дзета-потенциала) коллоидных частиц и их соль-
ватации (обычно изменения электрических свойств и
сольватации поверхности частиц происходят одно-
временно) вызывает К. (см. Электрические свойства
дисперсных систем).
Скорость К. измеряется числом частиц, сли-
пающихся за единицу времени в единице объема. Она
зависит от темп-ры, а также от природы дисперсной
системы. Величина, обратная скорости К., служит
мерой устойчивости дисперсной системы. Скорость
К. находят, считая частицы, видимые с помощью
ультрамикроскопа, или же, измеряя светорассеяние,
а также и др. методами. Скорость К. определяется
соотношением сил притяжения и отталкивания, дейст-
вующих между частицами на близких расстояниях.
Силы притяжения между сближающимися частицами,
обусловливающие их слипание, являются результатом
межмолекулярных взаимодействий. Силы отталки-
вания могут возникать при перекрытии и сближении
ионных атмосфер двух частиц (влияние электрокине-
тического потенциала) или их сольватных оболочек.
Сопротивление утоныпению и деформации диффуз-
ного слоя и сольватной оболочки объясняют возник-
новением «расклинивающего давления», к-рое оттал-
кивает сближающиеся частицы (Б. В. Дерягин,
П. А. Ребиндер).
В полидисперсных системах К. (о р т о к и н е-
т и ч. К.) происходит быстрее, чем в монодисперсных,
т. к. крупные частицы при оседании увлекают за собой
более мелкие. Форма частиц также влияет на скорость
К., так, напр., удлиненные частицы коагулируют
обычно быстрее, чем шарообразные. В золях с анизо-
диаметрич. частицами (игольчатые, пластинки) коагу-
лят часто приобретает структуру геля, что объяс-
няется локальной ориентацией частиц. Гидрозоли
обычно более устойчивы по отношению к К., чем
органозоли.
Согласно теории быстрой К. (когда каждое столк-
новение частиц приводит к их слипанию и К.), разви-
той М. Смолуховским, у монодисперсных систем
(т. е. систем с одинаковыми по величине частицами)
число всех частиц (одиночных и их агрегатов)
по истечении времени t определяется ур-нием:
где п0 — первоначальное число частиц, Т — время,
за к-рое число частиц убывает в 2 раза (так наз.
время К.), — эмпирич. постоянная, характерная
для каждой системы, зависящая от темп-ры, размера
частиц, их концентрации и вязкости дисперсионной
среды. Ур-ние Смолуховского было экспериментально
подтверждено рядом исследователей. При этом было
найдено, что радиус сферы действия сил притяжения
лишь немногим более, чем диаметр частицы.
Наиболее изучена К., вызываемая электролитами.
Коагулирующее действие электролитов связано со
609
КОАГУЛЯЦИЯ — КОАЦЕРВАЦИЯ
610
сжатием диффузной части двойного электрич. слоя
на поверхности дисперсных частиц (т. е. с уменьше-
нием электрокинетич. потенциала) и одновременным
снижением лиофильности их поверхности (поверх-
ностно-активные неэлектролиты иногда способствуют
К., снижая лиофильность частиц). Наименьшая
концентрация электролита, вызывающая К. за опре-
деленный (обычно короткий) промежуток времени,
наз. порогом К. (у). Для различных электроли-
тов величины порогов К. могут сильно варьировать,
даже если они найдены для одной и той же коагули-
руемой системы. Теоретич. расчетами показано, что
отношения порогов коагуляции у одно-, двух- и трех-
зарядных ионов при прочих равных условиях обратно
пропорциональны шестой степени величины заряда:
1 1 1 „
Yi : Ya : Уз = 1в ; 2в : зв- В среднем можно считать, что
концентрации электролитов, соответствующие порогу
К., с однозарядными ионами в 60 раз выше, чем
с ионами двухзарядными, и приблизительно в 750 раз
выше, чем с ионами трехзарядными (правило Шульца
и Гарди, или правило зиачности). Для ионов одинако-
вой валентности порог К. у определяется их положе-
нием в лиотропном ряду. В ряду одновалентных и,
В меньшей степени, двухвалентных ионов у умень-
шается с увеличением радиуса ионов.
Все эти приближенные закономерности нарушаются,
если добавленные электролиты химически взаимо-
действуют с коллоидными частицами, образуя с
ионами коллоидного раствора нерастворимые соеди-
нения. При К. смесью электролитов их влияние
может складываться (аддитивность), ослабляться (ан-
тагонизм) или усиливаться (синергизм). Трех- и че-
тырехвалентные коагулирующие ионы способны из-
менять знак заряда ядра коллоидных частиц, в связи
с чем при изменении концентрации этих ионов
в коллоидном растворе наблюдаются две зоны К.
и, соответственно, два порога К. (неправиль-
ные ряды К.), обусловленные действием ионов
с разным знаком заряда.
Определение величины порога К. осложняется
явлением привыкания, к-рое состоит в том, что при
добавлении электролита небольшими порциями порог
К. выше, чем при внесении всего электролита сразу.
Наблюдается и обратное явление, когда порог К.
при постепенном введении электролита ниже, чем
при быстром добавлении его (отрицательное привы-
кание) .
К., вызываемая диализом или электродиализом, свя-
зана с удалением ионов, придававших стабильность
коллоидным частицам; К., наблюдаемая при электро-
форезе, также обусловлена в основном изменением
ионного состава дисперсионной среды. При смешении
коллоидных р-ров (особенно, если они содержат
противоположно заряженные частицы) часто наблю-
дается снижение их устойчивости (астабилизация),
приводящее к их взаимной К. Золи гидроокисей
металлов легко коагулируют при повышенной темп-ре.
Течение золей, а также их перемешивание Могут
иногда ускорить их К., но, с другой стороны, слишком
энергичное механич. воздействие на систему может
привести к распаду агрегатов. Радиоактивные излу-
чения вызывают К. золей гидроокисей железа, алю-
миния и др., содержащих положительно заряженные
частицы. Действие у-лучей, рентгеновских лучей
и видимого света на К. связано с теми химич. реак-
циями, к-рые могут происходить в золях под их
влиянием и, в частности, с реакциями окисления —
восстановления. Ультразвук может вызывать К. или,
наоборот, диспергировать капельки в эмульсиях.
К. происходит при облучении ультразвуком концен-
трированных эмульсий (при этом особенно легко идет
К., заканчивающаяся коалесценцией).»и у эмульсий
с крупными капельками жидкости, плотность к-рой
значительно отличается от плотности окружающей
жидкой среды.
В ряде случаев между агрегированными частицами
в коагуляте остаются тончайшие прослойки диспер-
сионной среды. Это определяет малую прочность
коагуляционного сцепления и возможность разделе-
ния агрегатов (свежих осадков, в к-рых еще не про-
изошла перекристаллизация) на первичные частицы,
т. е. обратного перехода коагулята в состояние золя
(см. Пептизация) под влиянием механич. воздействия
(напр., перемешивания) или образования адсорб-
ционных слоев. При долгом хранении таких коагу-
лятов вследствие старения уменьшается их способ-
ность обратно переходить в состояние золя. Если
частицы в коагуляте находятся в тесном контакте
друг с другом, то со временем они срастаются, и К.
необратима (напр., К. золей золота). Чистый коагулят
получается при длительном диализе или электро-
диализе. При необратимой К. ионы коагулирующего
электролита поглощаются коагулятом, вытесняя ионы
из наружной (диффузной) части двойного электри-
ческого слоя коллоидных частиц.
К. очень распространена в природных и приме-
няемых в технике дисперсных системах. Она играет
важную роль в геология, и почвенных процессах,
в биологич. и метеорология, явлениях. Явления К.
представляют интерес в связи с вопросами агрегатив-
ной устойчивости многих дисперсных систем, напр.
почв, коллоидных р-ров, суспензий различных ве-
ществ и в том числе продуктов питания и лекарствен-
ных веществ. К. широко применяют в разнообразных
технологич. процессах (напр., деэмульгирование нефти,
очистка питьевой воды и воздуха с целью освобожде-
ния от взвешенных частиц и бактерий).
Литл Коагуляция коллоидов. Сб. статей, подрец. А. И. Ра-
биновича и П. С. Васильева, М.—Л., 1936; Песков Н. П.,
Физико-химические основы коллоидной науки. 2 изц., М.—Л.,
1934; Наука о коллоидах, под ред. Г. Р. Кройта, пер. с англ.,
т. 1, М., 1955; Пасынский А. Г., Коллоидная химия, М.,
1.95.9; Шелудко А., Коллоидная химия, пер. с болгарок.,
М., 1960; Труды III Всесоюзной конференции по коллоидной
химии, М., 1956; Ребиндер П. А., Коллоидн. ж.,
1958, 20, № 5. II. Н. Путилова.
КОАЛЕСЦЕНЦИЯ — слияние капель жидкости
в газовой среде (туманы) или в другой жидкости
(эмульсии) или пузырьков газа (пара) в жидкости под
влиянием молекулярных сиЛ, проявляющихся в по-
верхностной энергии. К. — самопроизвольный про-
цесс, сопровождающийся при постоянной темп-ре
понижением свободной поверхностной энергии на
величину, пропорциональную убыли поверхности.
Т. обр., К. — предельный случай процесса коагуля-
ции, характерный для дисперсных систем с жидкими,
т. е. легкоподвижными поверхностями раздела (ту-
маны, пены, эмульсии). В случае твердых дисперсных,
систем — концентрированных суспензий (осадков),
мелкозернистых твердых тел (металлов и сплавов) —
процессы собирательной рекристаллизации анало-
гичны К. и состоят в объединении соседних кристал-
ликов в один кристалл в результате тепловой подвиж-
ности атомов (молекул) с исчезновением поверхностей
раздела.
Выпадение дождя после предварительного укрупне-
ния мельчайших капелек изотермич. перегонкой (см.
Капиллярные явления) связано с К. этих капелек
в более крупные, быстро падающие под влиянием силы
тяжести. Практич. значение К. очень велико — преж-
де всего для расслоения эмульсий, напр. при обез-
воживании нефтей. К. при деэмульгировании вызы-
вается разрушением стабилизирующей (защитной)
оболочки эмульгатора на поверхности капель.
П, Ребиндер.
КОАЦЕРВАЦИЯ — расслоение р-ранвшкапельно-
жидкой форме или в виде двух слоев;, при' создании
611
КОБАЛЬТ
612
условий взаимно-ограниченной растворимости компо-
нентов р-ра. К. наблюдается в системах фенол —
вода, анилин — вода и др., при темп-рах ниже кри-
тич. точки, в р-рах комплексных солей кобальта,
кремнекислого натрия и др., нс( особенно подробно
К. изучена на р-рах полимеров желатины, ацетил-
целлюлозы, крахмала и др. Часто под К. понимают
именно явления расслоения в р-рах полимеров.
Напр., К. имеет место в водных р-рах желатины при
добавлении сшп)та или сернокислого натрия; напро-
тив, в р-рах белков — проламинов, хорошо раство-
римых в спирте, К. достигается при добавлении воды.
При pH 1,2—4,8 частицы (макромолекулы) желатины
обладают положительными зарядами; если к такому
р-ру желатины добавить гуммиарабик или крахмало-
фосфорную к-ту, макромолекулы к-рых несут отри-
цательные заряды, то вследствие нейтрализации
зарядов растворимость желатины понизится, и в си-
стеме произойдет К. Аналогично К. имеет место в
р-рах двух белков, макромолекулы к-рых при данной
pH обладают зарядами различного знака (т. е. белков
с сильно различными положениями изоточек), в смеси
р-ров белков, нуклеиновых кислот и др. К. при
взаимодействии двух полимерных в-в иногда назы-
вают комплексной, а при взаимодействии полимера
и низкомолекулярного в-ва (напр., желатины и
спирта) — простой. Во всех случаях К. возникает
в результате взаимно ограниченной растворимости
компонентов раствора. Степень отделения полимеров
при К. очень велика, напр. при К. 1%-ного р-ра
желатины до 93% ее количества входит в состав
капель коацервата (коацерватом обычно называют
обогащенную полимером фазу), тогда как равновесная
жидкость, в к-рой находятся капли К., сильно обед-
нена полимером. К. является термодинамически
равновесным состоянием системы, и при изменении
темп-ры или состава среды степень К. может обратимо
изменяться. Физико-химич. свойства коацерватов
в ряде отношений напоминают соответствующие свой-
ства протоплазмы (к-рая, однако, является термо-
динамически неравновесной системой), что .привле-
кает к ним внимание биологов. К. подробно изучалась
Бунгенберг-де-Йонгом и его школой, а свойства фер-
ментных коацерватов — А. И. Опариным, согласно
взглядам к-рого К. имела большое значение для про-
странственного разделения и организации полимерных
веществ в истории возникновения жизни на Земле.
Лит.: Евреинова Т. Н., Усп. современной биоло-
гии, 1954, 37, вып. 2, 177; Опарин' А. И., Возникновение
жизни на Земле, М., 1957. А. Г. Пасынский.
КОБАЛЬТ (Cobaltum) Со — химич. элемент VIII гр.
периодич. системы Менделеева; п. н. 27, ат. в. 58,9332.
Природный К. состоит из одного стабильного изотопа
Со60. Поперечное сечение поглощения тепловых нейт-
ронов атомом К. 34,8 барн. Из искусственно радио-
активных изотопов важнейшим является Со60 (Ti/% =
= 5,24 года), получаемый по реакции Соб0(я, у) Со60.
Конфигурация внешних электронов атома 3rf74s2.
Энергии ионизации (в эв): Со—► Со+—> Со2+соответ-
ственно равны 7,86; 17,31.
Соединения К. применялись уже в Древнем Египте
и Китае для окраски стекол и глазурей в красивый
и устойчивый синий цвет. Металлич. К. был впервые
получен в 1735 Ю. Брандтом.
Содержание К. в земной коре составляет ок. 0,004
вес. %. В рассеянном состоянии К. встречается во
многих изверженных породах, в морской воде и ми-
неральных источниках. К. содержится в метеоритах.
Он обнаружен также в почве, в растительных и жи-
вотных организмах. Известно до 40 кобальтовых мине-
ралов: арсенидов, сульфидов и сульфоарсенидов, ар-
сенатов и сульфатов, окислов и карбонатов. Из них
наиболее часто встречаются кобальтин и продукты его
окисления — эритрин и асболан. К обальтин
(кобальтовый блеск) CoAsS образует крупные кри-
сталлы кубич. системы с металлич. блеском; содержит
до 35% Со (примесь Fe составляет 5%); твердость
ок. 6, плотн. 6,0—6,4. Эритрин (кобальтовые
цветы) Co3(AsO4)2 • 8Н2О представляет скопления мел-
ких моноклинных кристалликов, ярко окрашенных
в красно-розовый цвет; содержит до 37,5% СоО;
твердость 1,5—2,5, плотн. 2,95. Асболан (асбо-
лит) Со2О3 • СоО - рМпО2 • 7Fe2O3- яН2О дает смо-
листые натечные образования черно-коричневого цве-
та; содержит 4,8—8,6% СоО, с примесью 1—2%
NiO. Чисто кобальтовые руды встречаются сравни-
тельно редко, а их месторождения маломощны. Но
соединения К. встречаются в нек-рых медных, нике-
левых, серебряных, железных, марганцовых и поли-
металлич. рудах, из к-рых К. извлекается в качестве
побочного металла. Наиболее богатые и мощные
месторождения кобальтовых руд известныв республике
Конго (Катанге), Сев. Родезии, Марокко. Имеются
они и в СССР, а также в Бирме, Канаде, Финляндии
и др. странах.
Физические и химические свойства. Компактный
К., полученный восстановлением при высоких тем-
пературах, обладает характерным металлич. блеском,
по цвету напоминает сталь с синеватым отливом, ковок
и пластичен. Но если он получен восстановлением
при низких темп-рах в токе водорода, то представляет
собой .черно-серый порошок, а если восстановителями
служат уголь, крахмал или аммиак, то выделяется
в виде серой губчатой и хрупкой массы. К. существует
в виде двух аллотропных модификаций: при обычных
темп-рах и до 417°±7° устойчив а-Со, обладающий
гексагональной плотноупакованной кристаллич. ре-
шеткой с периодами: а = 2,5017 А, .с = 4,0614 А
и плотн. 8,84. Выше этой темп-ры устойчив |3-Со
с кубич. гранецентрированной решеткой, а = 3,5370 А
и плотн. 8,79. Ат. радиус 1,25 А. Ионные радиусы
Со2+ 0,78 А, Со3+ 0,64 А. Т. пл. 1493°, т. кип. 3100°.
Теплота плавления 62 кал/г-, теплота испарения
1500 кал/г-, уд. теплоемкость 0,1056 кал/г-град (15—
100°); термич. коэфф, линейного расширения 13,36-10 е
(40°); теплопроводность 0,165 кал/см- град-сек (0—
100°). Уд. электросопротивление (в мком-см): 5,68 (0°),
9,30 (100°), 32,20(500°); термич. коэфф, электросо-
противления: 5,31-10 3 (0°), 7,36-10-3 (100°), 13,8-10’3
(500°). К. ферромагнитен и теряет магнитные свойства
при 1121° (точка Кюри). В сильном магнитном поле
скорее намагничивается, чем никель. Механич. свой-
ства К. очень сильно зависят от способа получения
и предварительной обработки. Модуль упругости
20750 кГ/мм2, предел прочности на растяжение
24 кГ/мм2, относительное удлинение 5,0%, твердость
по Бринеллю 124 кГ/мм2 (литого) и 300 кГ/мм2 (осаж-
денного электролитически).
Компактный К. на воздухе не окисляется и лишь
при 300° начинает покрываться тонкой пленкой окиси
(см. Кобальта окислы и гидроокиси). Но порошко-
образный К., полученный восстановлением водородом
при 250°, пирофорен и воспламеняется на воздухе
самопроизвольно. В разб. к-тах — соляной, серной,
азотной — К. медленно растворяется с образованием
солей 2-валентного К., но плавиковая к-та на него
не действует даже при нагревании, а дымящая азотная
быстро пассивирует металл. Царскаи водка и щавеле-
вая к-та растворяют К. при обычной темп-ре, щелочи
реагируют с ним при красном калении. Хлор, бром
и иод взаимодействуют с К. уже при обычной темп-ре,
сера, селен, фосфор, мышьяк и сурьма — при нагре-
вании (см. Кобальта галогениды, Кобальта сульфиды).
К. сплавляется с бором, кремнием, углеродом, обра-
зуя соответственно бориды, силициды, карбиды,
а с металлами дает интерметаллич. соединения и твер-
613
КОБАЛЬТ
614
дые р-ры. Так, с железом К. образует непрерывный
ряд твердых р-ров. В качестве легирующего элемента
в сталях К. расширяет область y-Fe и ускоряет у «
превращение, что благоприятно сказывается на ме-
ханич. свойствах стали (см. Железа сплавы). При
низких температурах компактный К. поглощает 35,
а порошкообразный — до 100 объемов водорода.
При взаимодействии СоС12 и фенилмагнийбромида
с водородом при полном отсутствии воздуха и паров
воды образуется гидрид СоН2 в виде коричневого
порошка. Порошкообразный пирофорный К. при
определенной темп-ре (150—220°) и под давлением
(до 250 атм) соединяется с окисью углерода с обра-
зованием к а р б О'н ила [Со(СО)4]2. Нитриды К.
точно неизвестны, хотя комплексные азиды сущест-
вуют.
В своих соединениях К. может быть 2- и 3-валент-
ным. В подавляющем большинстве простых соеди-
нений К. 2-валентен (см. Кобальта карбонаты,
Кобальта нитрат, Кобальта сульфаты); напротив,
в своих более устойчивых комплексных соединениях
ОН Чаще всего 3-валенТен. Ф- м- Перельман.
В соответствии с принятой номенклатурой, комплекс-
ные производные двухвалентного К. отличают суф-
фиксом «о» (кобальто-), производные трехвалентного
кобальта — суффиксом «и» (кобальти-). Из комп-
лексных соединений двухвалент-
ного К. имеют значение галогениды и роданиды К.
общего состава Ме2[СоХ4], где Me — КС, Na\ NH4+
или катион другого металла или органич. основания,
X — чаще всего SCN~ или С1~. Анионы солей указан-
ного состава подвергаются в водных р-рах ступен-
чатой диссоциации по схеме:
СоХ^—-СоХ'+Х-—>СоХ2+Х-—-СОХ++Х-—-Со2++Х-
Диссоциация уменьшается при введении неводных
растворителей (ацетон и др.). Окрашенные в синий
цвет комплексные анионы Co(SCN)|- и СоС1|~ исполь-
зуются для фотометрия, определения К. Укажем
еще на оксалатокобальтоаты состава Ме4[Со(С2О4)3]
и Ме2[Со(С2О4)2]; вместо С2О|" во внутренней сфере
комплексного соединения могут находиться также
анионы др. органич. кислот — малоновой, янтарной,
винной. Известны гексацианокобальтоаты металлов
состава Me4[Co(CN)e]. Гексаммины двухвалентного
К. — [Co(NH3)ejX2 мало устойчивы и легко превра-
щаются при окислении даже на воздухе в соответст-
вующие соединения трехвалентного К. (см. ниже).
Среди комплексных соединений двухвалентного К.
с органич. лигандами интересны комплексные соеди-
нения с этилендиаминтетрауксусной кислотой (ЭДТА)
и другими комплексонами, которые принадлежат к
классу внутрикомплексных соединений. Атомы К. со-
единены здесь и с атомами кислорода карбоксильных
групп и координационно связаны с атомами азота
иминогрупп. . Образование нескольких пятичленных
колец обусловливает большую устойчивость соеди-
нений этого типа. Комплексообразование с ЭДТА
применяется для аналитич. определения К. объемным
методом. Из других внутрикомплексных соединений
изучены ацетилацетонаты К. Со(СН3СО=СНСОСН3)2,
оксихинолинат Co(CeH9ON)2 • 2Н2О, дифенилтиокар-
базонат (дитизонат) Со SC< , рубеанат
\ \N=NCyrij /2
Co(NHCSCSNH)2; для большинства этих соединений
характерна растворимость в органич. растворителях
(хлороформе, четыреххлористом углероде и др-),
что находит применение при экстракционном отделе-
нии К. от др. элементов и для его фотометрия, опре-
деления.
Известно несколько тысяч комплексных
соединений трехвалентного К. — од-
но- и многоядерных. Особенно многочисленны ам-
миачные комплексные соединения К. — к о б а л ь-
тиаммины (кобальтиаки). Исследование этих
соединений сыграло большую роль в развитии коорди-
национной теории Бернера. Координационное число
К. в кобальтиамминах равно 6. Лигандами могут быть
как нейтральные молекулы (аммиак, вода, органич.
соединения — этилендиамин, пропилендиамин и др.),
так и кислотные радикалы, число и заряд к-рых
определяют суммарный заряд комплексного иона.
Гексаммины К. (т. наз. «лютео» соли, желтого цвета)
содержат катион [Co(NH3)6]3+, в к-ром атом К. окру-
жен шестью атомами азота в виде правильного тетра-
эдра. Они устойчивы как в твердом состоянии, так
и в водных р-рах. Соли кобальтигексаммина полу-
чаются при окислении аммиачных растворов солей
двухвалентного К.
При замещении одной из групп NH3 внутренней
сферы на молекулу воды получаются аквопентаммипы
(«розео» соли, красного цвета) с комплексным катио-
ном [Co(NH3)6H2O]3+. Молекула воды легко замещается
кислотным радикалом, причем получаются соли
ацидопентамминов красного цвета («пурпурео»соли).
Представителем солей этого типа является хлорид
хлоропентаммина [Co(NH3)5C1]C12, к-рый служит ис-
ходным продуктом для синтеза многих др. комплекс-
ных амминов.
Обширным классом комплексных соединений 3-ва-
лептного К. являются соли тетрамминов. Они пред-
ставляют продукты замещения двух молекул аммиака
во внутренней сфере гексамминов либо двумя моле-
кулами воды (диаквотетраммины), либо одной моле-
кулой воды и одним кислотным радикалом (ацидо-
аквотетраммины) или, наконец, двумя кислотными
радикалами (диацидотетраммины). Отличительной осо-
бенностью тетрамминов является их способность к об-
разованию цис- и транс-изомеров. Так, дихлоротетр ам-
мины с комплексным катионом [Co(NH3)4C12]+ обра-
зуют два вида солей — зеленого цвета («празео»)
и фиолетового цвета («виолео»). Изомеры отличаются
по своей устойчивости и реакционной способности.
Описаны также соединения с еще меньшим со-
держанием аммиачных групп во внутренней сфере,
например [Со(ГЧН3)з(Н2О)3]3+, [Co(NH3)2(NO2)4]- и др.
Растворимость кобальтиамминов в воде обычно не-
велика.
Значительной устойчивостью отличаются гекса-
цианокобальтиаты Me3[Co(CN)e], которые, в отличие
от комплексных цианидов двухвалентного никеля,
не разлагаются в щелочных р-рах. Представляют
интерес труднорастворимые гексанитрокобальтиаты
Me3[Co(NO2)e], где Me — К+, Cs+, NH+-, Т1+, Ag+
и др. Образование этих соединений используется
в анализе для отделения К. от никеля и большинства
других элемеитов, а также для его количественного
определения; кроме того, эти соединения приме-
няются для осаждения калия и нек-рых других ка-
тионов.
Из внутрикомплексных соединений трехвалеитного
К. с органич. лигандами наиболее важен а-нитрозо-
Р-нафтолат К. пурпурно-красного цвета, где К.
замещает водород оксигруппы реактива и координа-
ционно связан с азотом нитрозогруппы. Соединения
аналогичного состава получаются также с р-нитрозо-
а-нафтолом, нитрозо-Н-солью и другими производ-
ными этих реактивов. Представителем комплексных
соединений трехвалеитного К. с альдегидами и кето-
нами является ацетилацетонат К. зеленого пвета,
растворимый в хлороформе и др. органич. раствори-
телях.
Аналитическое определение. Длп
качественного обнаружения К. переводят,в малорас-
творимые комплексные соли K3[Co(NQ2)3] желтого
цвета, а-нитрозо-р-нафтолат К. пурпурно-красного
615
КОБАЛЬТ - КОБАЛЬТА ГАЛОГЕНИДЫ
616
цвета, растворимые роданиды К. Me2[Co(SCN)j] синего
цвета (в присутствии ацетона или амилового спирта
или с применением экстракции). Весовые методы
количественного определения К. основаны на выде-
лении последнего в виде CoSO4, Со3О4 или в виде
металла; в последнем случае металлический К. выде-
ляют из р-ра электролизом. Для объемного опреде-
ления аммиачный р-р соли двухвалентного К. тит-
руют раствором K3[Fe(CN)6]; применяют также тит-
рование ЭДТА в присутствии металлохромных инди-
каторов (мурексид и др.). Широко распространены
фотометрия, методы, основанные на измерении' ин-
тенсивности окраски роданидных (пли хлоридных)
комплексных соединений К. или растворимых комп-
лексных соединений с нитрозо-Н-солью. Известны
также полярография, методы определения, основан-
ные на восстановлении трехвалентного К. до двух-
валентного в растворе смеси гидроокиси аммония и
хлористого аммония или смеси пиридина и хлористого
пиридина. Каталитич. действие К. на реакцию между
ализарином и перборатом может быть использовано
для определения ниятожных следов кобальта (доли
микрограмма). И. в. Пятницкий.
Иолучение. Руды, из к-рых извлекается К., весьма
разнообразны по своему химия, и минералогия, со-
ставу, прияем содержание К. в них колеблется в очень
широких пределах — от десятых долей процента до
нескольких процентов. Но пеокисленные руды (суль-
фидные пли мышьяковистые), особенно комплексные,
являются промышленными даже при концентрации
К. 0,15% и менее, т. к. они поддаются обогащению.
Перевод К. в растворимую форму и предварительное
отделение основной массы преобладающих металлов
достигается в результате различных пирометаллур-
гия. процессов: окислительного обжига, шахтной
плавки, конвертирования, спекания с различными
добавками, хлорирования и электролиза. После
перевода К. в раствор его отделяют от захваченных
в процессе переработки и выщелачивания значитель-
ных количеств сопутствующих металлов: серебра,
меди или свинца, или железа, марганца и никеля.
Для очистки К. от этих элементов разработаны соот-
ветствующие гидрометаллургия, процессы. Так, из
сернокислых р-ров серебро и медь удаляют цемента-
цией (или сероводородом, или содой), а свинец —
в виде сульфата. Для отделения железа, марганца и
небольших количеств меди добавляют соду, окислитель
или известковое молоко. Труднее всего отделить К.
от никеля. Обычно используется большая склонность
К. к переходу в 3-валентную форму и к образованию
устойчивых комплексных соединений; с этой целью
р-р обрабатывают в соответствующих условиях хло-
ром, гипохлоритом натрия, перекисью водорода,
перекисью свинца или другими окислителями. Ко-
нечным продуктом гидрометаллургической стадии яв-
ляется Со3О4, из которого получают металлич. К.
различными способами: восстановлением углем, во-
дородом, окисью углерода: алюмотермией и электро-
лизом. Для рафинировки К. применяют электроли-
тич. его осаждение из р-ра сульфата, насыщенного
борной к-той.
За последние годы предложены новые методы
вскрытия кобальтсодержащих руд и отделения ни-
келя от К. Таков, напр., способ, основанный па
выщелачивании неокисленпых руд смесью серной
и азотной к-т в автоклаве. Отделение К. от никеля —
после предварительного окисления солей обоих ме-
таллов — проводят на ионнообменной колонке, с по-
мощью катионита, к-рый прочно адсорбирует 3-ва-
лентпый К., в то время как 2-валентпый никель легко
вымывается аммиачным р-ром. Известны попытки
разделепийЧ'К. и никеля методами жидкостной экст-
ракции б1 ’‘Органич. растворителями. Предложено
также получать металлич. К. взаимодействием его
галогенидов со щелочными или щелочноземельными
металлами, а также из аммиачных комплексов вос-
становлением водородом под давлением 15—20 атм
при 170—200°.
Применение. К. начал широко применяться с пер-
вой четверти 20 в., после открытия твердого спла-
ва—стеллита. В наст, время до 77% всего получае-
мого К. выпускается в виде металла и употребляется
в основном для произ-ва специальных сплавов и
сталей; алнико, пермендур, стеллит, виталиум и мн.
др. (см. Кобальта сплавы). К. и его соединения ис-
пользуются и в других отраслях народного хозяйства.
На основе К. получены превосходные катализаторы
для органич. синтеза, обессеривания нефти, окисле-
ния аммиака, сушки лаков и масляных красок (сик-
кативы). Из соединений К. изготовляют эмали и краски
необычайной стойкости и красоты. За последние годы
радиоактивный изотоп Со60 получил широкое приме-
нение в медицине в борьбе с раком («кобальтовая
пушка»). Наконец, К. применяют в с.х-ве в качестве
микроудобрепия, а также для подкормки животных
(животные, к-рые питаются растениями, выросшими
на лишенной К. почве, страдают анемией).
Лит.: Перельман Ф. М., Зворыкин А. Я.
и Гудима Н. В., Кобальт, М.—Л., 1949; Gmelin,
8 Aufl., Syst.—Num. 58, Kobalt, В., 1930—32; Mellor,
v. 14, L.—N. Y. — Toronto, 1935; то же, v. 14, 1 953; P a s с a 1,
t. 10, P., 1933; то я:е, t. 18, P., 1959; Tillmann, 3 Aufl.,
Bd 9, Miinch. — B., 1957, S. 609—20; Rare metals handbook,
ed. C. A. Hampel, 2 ed., L., 1961; Sidgwick N. V., The
chemical elements and their compounds, v. 2, Oxf., 1951.
Ф. M. Перельман.
КОБАЛЬТА ГАЛОГЕНИДЫ — кобальт образует
многочисленные соединения со всеми галогенами.
Из них наибольший интерес представляют фториды.
Кобальта фториды. При взаимодействии
порошкообразного кобальта с фтором при 500° обра-
зуется смесь CoF2 и CoF3. Чистый CoF2 получают
из СоС12 и плавиковой к-ты при обычной темп-ре,
а также обезвоживанием CoF2 • ЗН2О. Безводный
CoF2 — розовые кристаллы с тетрагональной решет-
кой типа рутила; а = 4,695 А, с = 3,193 А; плотн.
4,43. Теплота образования А/7% = —159 ккал/моль.
CoF„ очень мало растворим в воде — всего 1,4% (25°).
При обычной температуре растворы CoF2 в воде
сравнительно устойчивы; ок. 700° пары воды разла-
гают CoF2 с образованием СоО и H2F2. При взаимо-
действии Со(ОН)2 с разб. H2F2 образуются в зависи-
мости от условий кристаллогидраты с 4,3 и 2 молеку-
лами воды. CoF2 • 4Н2О существует в двух модифика-
циях (а- й (3-); a-CoF2 • 4Н2О — кристаллы красного
цвета, получают из р-ров, содержащих избыток H2F2,
при последующей кристаллизации в вакууме над
H2SO4. При 200° a-форма не обезвоживается полно-
стью и начинает разлагаться; в спирте и др. органич.
растворителях не растворяется. Если осаждение ве-
дут этиловым спиртом, то получают p-CoF2 • 4Н2О —
розовые кристаллы. В отличие от а-формы, f-форма
растворима в H2F2 уже на холоду и при 60° те-
ряет около трех молекул воды. CoF2 • ЗН2О полу-
чают при выпаривании разб. водного р-ра на водяной
байе в виде розовато-красных кристаллов; полностью
обезвоживается в токе азота при 300°. Из более конц.
р-ров и при более высокой темп-ре выпаривания по-
лучают CoF2 • 2Н2О — кристаллы розового цвета.
Известна также кислая соль CoF2 • 5HF 6Н2О.
Наконец, при длительном кипячении CoF, • 2Н2О
с водой образуется основная соль — CoF, • СоО •
• 1/2Н2О. Подобно другим галогенидам кобальта,
CoF2 образует двойные соли с фторидами натрия,
калия и аммония. Фторид 3-валентного кобальта
CoF3 образуется при взаимодействии CoF2 и F2
при 150° в виде бледно-коричневых кристаллов гек-
сагональной решетки с периодами: а — 7,64 А,
617
КОБАЛЬТА ГАЛОГЕНИДЫ — КОБАЛЬТА ОКИСЛЫ И ГИДРООКИСИ
618
с = 3,66 А и плотн. 3,88. Теплота образования
ДЯ288 = —187 ккал/молъ. При анодном окислении
насыщенного р-ра CoF2 в 40%-ной плавиковой к-те
образуется кристаллогидрат CoF3 • 3,5Н2О зеленого
цвета. При нагревании до 250° в токе СО2 CoF3 на-
чинает выделять фтор, при 350° полностью превра-
щается в CoF2. Трифторид кобальта — сильный окис-
литель; он выделяет кислород из воды и хлор из
р-ров соляной к-ты; фторирует мышьяк, фосфор,
углерод, кремний, бром, иод, превращаясь при этом
в CoF2. При фторировании сложных насыщенных угле-
водородов с помощью CoF3 получены химически и
термически устойчивые смазки — жидкие и воскооб-
разные полимеры.
Безводные хлориды, бромиды и ио-
диды 2-валентного кобальта получают из элементов
(прокаливанием порошкообразного Со с хлором, бро-
мом, парами иода) или обезвоживанием их гидратов,
а также другими способами. СоС12 — блестящие
голубые кристаллы, гексагональная решетка, а =
= 3,533А, с = 17,34А; плотн. 3,356; т. пл. 724°,
т. кип. 1049°; весьма гигроскопичен; СоВг2 — крис-
таллы ярко-зеленого цвета, решетка гексагональная,
а = 3,685.4, с — 6,120А; плотн. 4,91; т. пл. 678°;
также сильно гигроскопичен и во влажном воздухе
образует красную жидкость. CoJ2 встречается в двух
изоморфных модификациях. Обычная а-форма — чер-
ные гексагональные кристаллы, а = 3,96А, с = 6,65Д;
плотн. 5,68; гигроскопична и дает с водой розовый
р-р. Будучи нагрета в высоком вакууме, плавится
при 515—520° (без разл.), диссоциирует, начиная
с 540°, а при 570° кипит, частично разлагаясь па Со
и J2. Остальная часть соли возгоняется гл. обр. в виде
а-формы; 1% возгона конденсируется в виде f-формы
ярко-желтого цвета, к-рая дает с водой бесцветный
р-р, устойчивый на холоду. Теплоты образования
СоС12, СоВг, и CoJ2 (—ДН»88, в ккал/молъ) соответст-
венно равны: 77,8; 55,5; 24,4.
Все описанные галогениды кобальта хорошо рас-
творимы в воде и кристаллизуются из водных р-ров
в виде различных кристаллогидратов. Для хлорида
известны СоС12 • 6Н2О, который переходит при 49°
в СоС12 • 4Н2О, при 58° в СоСЦ • 2Н2О, а при 90°
в СоС12 • Н2О. Полное обезвоживание наступает
при 140°. Тетра- и гексагидраты розового цвета;
менее гидратированные соли — сине-фиолетового. Рас-
творимость СоС12 в воде (в г/100 г Н2О): 43,5 (0°),
55,3 (25°), 106,2 (100°); очень хорошо растворим в
спирте, несколько хуже — в ацетоне, ацетонитриле
и др. органич. растворителях. Водный розовый р-р
СоС12 при нагревании, а также при добавлении соля-
пой или серной к-т становится синим. На этом свойстве
основано его применение в качествеТимпатич. чернил.
Известны кристаллогидраты СоВг2 с 6 молекулами
Н2О (устойчивы до 43°), с 4Н2О (до 60°) и с 2Н2О
(выше 60°). СоВг2 • 6Н2О и СоВг2 • 4Н2О — красного
цвета, СоВг2 • 2Н2О —• пурпурно-синего. Большая ги-
гроскопичность СоВг2 наряду с резким изменением
окраски при гидратации (из зеленой в пурпурно-
синюю и красную) делает его пригодным для обна-
ружения следов влаги. Растворимость СоВг2 в воде
превышает растворимость СоС12 и достигает при 60°
226,3 г безводной соли на 100 г Н2О. Иодид образует
при обычной темп-ре темно-красный Со,Т2 • 6Н2О;
из горячих р-ров выделяется зеленый CoJ2 • 4Н2О;
выше 100° появляется CoJ2 • 2Н2О — тоже зеленого
цвета; растворимость безводной соли 210 г/100 г Н2О
(25°). Конц, водный р-р ниже 20° темно-красный,
выше 35° — ярко-зеленый. В органич. растворителях
Со12 образует обычно р-ры синего цвета. СоС12, СоВг2
и CoJ2 дают ряд двойных солей с соответствующими
галогенидами щелочных металлов, ярко-фиолетового
зеленого или синего цвета. В отличие от фтора, хлор,
бром и иод не образуют к.-л. простых галогепидов
3-валентного кобальта.
Лит. см. при ст. Кобальт. Ф. М. Перельман.
КОБАЛЬТА КАРБОНАТЫ — углекислые соли 2-
и 3-валентного кобальта. Безводный СоСО3 встре-
чается в природе в виде минерала розового цвета
(кобальтовый шпат), часто в изоморфной смеси с
MgCO3 и FeCO3. В безводном состоянии СоСО3 полу-
чен длительным нагреванием СоС12 с содой при 140—
150° в запаянной трубке. Безводный СоСО3 — ро-
зовые кристаллы, гексагональная решетка, плотн.
4,13. Теплота образования ДН298 = —172,7 ккал/молъ.
При атмосферном давлении термин, диссоциация на-
чинается ок. 400° (данные не проверены). При 350°
в высоком вакууме СоСО3 полностью разлагается
на СоО и СО2. Растворимость СоСО3 в воде при мини-
мальном давлении СО2 (0,0005 атм) равна 0,011 г/100 г
Н2О (15°). Из р-ров К. к. выделяется в виде нейт-
ральной соли СоСО- • 6Н2О лишь в токе СО2. Если
же вести осаждение в обычной атмосфере, то при до-
бавлении карбоната щелочного металла к р-ру соли
2-валентного Со выделяются аморфные основные
карбонаты переменного состава: 2СоСО3 • ЗСоО-4Н2О,
СоСО3 2СоО • 2Н2О, СоСО3 • ЗСоО • ЗН2О или СоСО3 •
•СоО-5Н2О. Эти гидратированные основные карбонаты,
подобно гидроокиси кобальта, имеют склонность
окисляться на воздухе. С карбонатами натрия, калия
и аммония СоСО3 образует ряд гидратированных
двойных солей типа Ме2Со(СО3)2 • 4Н2О.
При обработке соли 2-валентного Со в присутствии
бикарбоната натрия к.-н. сильным окислителем об-
разуется раствор зеленого цвета. Если окислителем
служит перекись водорода, то в осадке выделяется
зеленый порошок — Со2(СО3)3. По нек-рым данным,
эта соль довольно устойчива в сухом состоянии.
Лит. см. при ст. Кобальт. <£> М. Перельман.
КОБАЛЬТА НИТРАТ Co(NO3)2 — кристаллизует-
ся при низких темп-рах в виде Co(NO3)2 • 9Н2О; при
—21° переходит в Co(NO3)2 • 6Н2О, при 55,5° — в
Co(NO3)2 • ЗН2О, к-рый плавится при 91°, а при более
высокой темп-ре разлагается. Образует с аналогич-
ной никелевой солью смешанные кристаллы. Раство-
римость К. н. в воде (в г/100 г. Н2О): 71,1 (—21°),
98,9 (18°); 338,7 (91°). Водные р-ры всегда розового
цвета. Легко растворим в органич. растворителях —
спиртах, ацетоне; в 100 г метилацетата при 15° его
растворимость достигает 16,3 г. В р-рах известен
и Co(NO3)3, но он крайне неустойчив и быстро пре-
вращается в Co(NO3)2. К. н. широко применяется
в аналитич. химии для количественного определения
др. элементов (см., напр., Натрия кобальтинитрит).
Лит. см. при ст. Кобальт. Ф. М. Перельман.
КОБАЛЬТА ОКИСЛЫ И ГИДРООКИСИ— кисло-
родные соединения кобальта. Из многочисленных окие-
лов Со, описанных в литературе, бесспорно сущест-
вование только трех: СоО, Со3О4 и Со2О3. Как СоО2,
так и низшие окислы Со, по-видимому, не образуются
как определенные химич. соединения. СоО получают
нагреванием металлич. Со на воздухе или прокали-
ванием Со(ОН)2 и СоСО3. Наиболее активная форма
СоО образуется при прокаливании СоСО3 в вакууме
при 350°, в виде очень тонкого порошка оливково-
зеленого цвета. Кристаллич. решетка СоО куби-
ческая, а — 4,25 А; плотн. 5,68. Теплота образования
ДН»98 — —57,2 ккал/молъ. Т. пл. 1935°, при 2800°
начинает разлагаться, теряя кислород; с другой
стороны, при 18° СоО поглощает кислород, адсорби-
руя его до содержания в Со3О4; однако соответствую-
щее изменение кристаллич. решетки наступает лишь
при нагревании; Со3О4 можно получить дакже прп
нагревании Со2О3 • Н2О. Окисел Со3О4 —т|ч®рные кри-
сталлы, изоморфные с магнетитом; рещстуа куби-
ческая, а = 8,09А, плоти. 6,07. Теплота образования
619
КОБАЛЬТА СПЛАВЫ —КОБАЛЬТА СУЛЬФАТЫ
620
ДЯ;98 = —210 ккал/молъ. Со304 при прокаливании
теряет кислород и при 900° превращается в СоО.
Он растворим в расплавленном NaOH, а также в ки-
пящем р-ре соды. Со3.О4 также поверхностно адсорби-
рует кислород (до содержания в Со203). Окисел Со203
не существует в безводном состоянии, но его гидрат
Со203 • Н2О образуется в виде коричневого или чер-
ного порошка при окислении окислов Со перекисями,
бромом, перманганатом калия, а также при осажде-
нии щелочью солей З-валентного Со; его сушат при
150°. Структурная вода удаляется лишь при 300°
с одновременным разложением и частичным выделе-
нием кислорода. Растворимость Со203 • Н2О в воде,
определенная с помощью радиоактивных изотопов,
равна 0,01046 мг/л.
Гидроокись Со(ОН)2 существует в двух модифика-
циях: синей и розовой. Если к р-ру соли 2-валентного
Со добавить на холоду и без доступа воздуха р-р
КОН, то вначале образуется синий осадок Со(ОН)2,
к-рый постепенно (а при нагревании быстро) стано-
вится вначале фиолетовым, а затем розовым. Синяя
модификация более мелкодисперспа и отдает воду
уже при 170°, а розовая не отдает ее полностью .даже
при 300°. Соли никеля, а также галактоза и мальтоза
стабилизируют синюю модификацию, препятствуя ее
превращению в розовую. Произведение растворимости
Со(ОН)2 равно 1,6 • 10 18. Со(ОН)2 — сильное основа-
ние и ее соли многочисленны, стабильны и являются
сильными электролитами. Если барботировать воздух
сквозь суспензию Со(ОН)2 в воде, то она окисляется
и переходит в Со203 • Н2О. Этот процесс идет легко
под действием более сильных окислителей, таких, как
гипохлорит натрия, бром и перекись водорода. В по-
следнем случае предполагалось даже образование
соединения Со02 • пН2О, но выделить его не удалось.
Лит. см. при ст. Кобальт. Ф. М. Перельман.
КОБАЛЬТА СПЛАВЫ — сплавы, в к-рых кобальт
преобладает или составляет значительную часть.
Используются в качестве жаропрочных и жаростой-
ких материалов, при изготовлении постоянных маг-
нитов, в качестве материалов, обладающих высокой
стойкостью в сильно агрессивных средах, а также
для изготовления режущего инструмента.
Типичным представителем жаропрочных и жаро-
стойких К. с. является виталлиум, содержащий
(в вес. %): 65 Со, 28 Сг, 3Ni и 4 Мо. Этот сплав сохра-
няет высокую прочность и практически не подвержен
газовой коррозии до 800—850°. Используется в литом
виде, т. к. с большим трудом обрабатывается. Приме-
няют в газовых турбинах высоких параметров и для
изготовления различных деталей реактивных двига-
телей. Присадка кобальта к алюминиево-пикель-
железным сплавам резко повышает их магнитные
свойства. Типичным представителем таких сплавов
является алнико-24, содержащий (в вес. %): 50 Fe,
24Со, 14 Ni, 9 Al и 3 Си. Постоянные магниты, изго-
товляемые из этого сплава, обладают коэрцитивной
силой до 500 эрстед и остаточной индукцией до 12000
гаусс. Новым классом магнитных сплавов являются
кобальто-платиновые сплавы с содержанием от 40
до 60 ат. % кобальта. Коэрцитивная сила таких
сплавов и их остаточная индукция намного превос-
ходят ныпе используемые магнитные сплавы. Однако
высокая стоимость кобальто-платиновых сплавов су-
щественно ограничивает их применение.
Высокой стойкостью в сильно агрессивных средах
обладает К. с., содержащий (в вес. %): 75 Со, 13 Si,
7 Сг и 3 Мп; в азотной, серной и соляной к-тах он
растворяется меньше, чем платина. Этот сплав ис-
пользуется для изготовления анодов.
Кобальт является основой многих твердых сплавов,
широко используемых для изготовления режущего
инструмента. Литые твердые сплавы на кобальтовой
основе, т. наз. стеллиты, содержат (в вес. %): 45—
60 Со, 5—20 W, 20—35 Сг и от 1 до 2 С. Т. к. высокая
твердость и хрупкость этих сплавов очень затрудняют
их механич. обработку, то их обычно наплавляют
в кислородно-ацетиленовом пламени на рабочую
поверхность штампов или режущую часть инстру-
ментов. В очень важных для металлообрабатывающей
и горной пром-сти металлокерамич. твердых сплавах
содержится (в вес%): в сплавах типа ВК8—92% кар-
бида вольфрама, 8% Со, в Т5К10—85% карбида вольф-
рама, 5% карбида титана и 10% Со и в Т15К6—79%
карбида вольфрама, 15% карбида титана и 6% Со;
кобальт служит связующим материалом, цементи-
рующим частицы карбидов и вместе с тем придающим
этим сплавам нек-рую пластичность. Нек-рые сплавы
кобальта с никелем, вольфрамом, марганцем и др.
элементами имеют термич. коэфф, линейного расши-
рения, практически близкий нулю в широком интер-
вале темп-p. Эти сплавы применяются для изготов-
ления пружин в часовых механизмах, а также для
припоя металла к стеклу.
Свойства различных К. с. еще далеко не полностью
изучены. В настоящее время широко изучаются как
диаграммы состояния двойных и тройных систем на
основе кобальта, так и различные физич. и физико-
химич. свойства таких сплавов.
Лит. см. при ст. Кобальт. Ф. М. Перельман.
КОБАЛЬТА СУЛЬФАТЫ — сернокислые соли 2-
и 3-валептного кобальта. CoS04 7Н2О встречается
в природе в виде минерала биберита, карминно-крас-
ного цвета. Безводный CoS04 образуется при окисле-
нии сульфида кобальта па воздухе или окиси кобальта
в токе SO2. Он представляет собой розовые гигроско-
пичные кристаллы, решетка ромбич., а — 6,71 А,
Ь = 8,45 А, с — 4,65 А; плотн. 1,924. Теплота об-
разования Д77298 = —2 0 7,5 ккал/молъ. При нагре-
вании выше 600° CoS04 начинает диссоциировать,
а при 735° полностью распадается на СоО и SO2
Гидратированный сульфат кобальта получают путем
взаимодействия окиси, гидроокиси или карбоната ко-
бальта с серной кислотой. CoS04 • 7Н2О устойчив
до 41,5°, выше этой темп-ры переходит в CoS04 • 6Н2О,
к-рый при 71° превращается в CoS04 • Н2О. Все
3 гидрата моноклинны и изоморфны с аналогичными
солями никеля или магния. Известны также мета-
стабильные тетра- и дигидраты. Растворимость CoS04
в воде при 25° составляет 39,3 г на 100 г Н2О и до 100°
повышается с темп-рой, а затем падает и при 145°
становится меньше, чем при 15°. В присутствии сер-
ной к-ты растворимость CoSO4 резко понижается
и падает до нуля в 55%-ной H2SO4 при 70°. Так же
ничтожна его растворимость в спирте. В водных
р-рах CoS04 образует с сульфатами никеля и меди
непрерывные ряды твердых растворов, а с суль-
фатами калия и аммония — шестиводные двойные
соли, значительно более растворимые в воде, чем
аналогичные соли никеля. Известна основная соль
CoS04. ЗСо(ОН)2.
При анодном окислении р-ра CoS04 в 8н. H2SO4
или при «окислении озоном или фтором образуются
синие кристаллы сульфата 3-валентного кобальта
Co2(S04)3- 18Н2О, устойчивого в разб. H2SO4 и на
холоду, но разлагающегося в воде (а также при на-
гревании до 30°) с выделением кислорода. Сульфат
3-валептпого кобальта — сильный окислитель: вы-
тесняет хлор из НС1, иод из KJ, окисляет этиловый
спирт и муравьиную к-ту. С сульфатами калия, руби-
дия, цезия, аммония получены кобальтовые квасцы
MeCo(S04)2 • 12Н2О — неустойчивые в воде темно-
синие кристаллы. Водные р-ры CoS04 применяют при
гидрометаллургия, переработке кобальтсодержащего
сырья с целью извлечения кобальта.
Лит. см. при ст Кобальт. Ф. М. Перельман.
621
КОБАЛЬТА СУЛЬФИДЫ — КОДЕГИДРОГЕНАЗЫ
622
КОБАЛЬТА СУЛЬФИДЫ — соединения кобальта
с серой: CoS, Co3S4, Co2S3 и CoS2. Из них CoS встре-
чается в природе в виде довольно редкого минерала
джепурита; Co3S4 составляет основную часть мине-
ралов группы линнеита; CoS2 (кобальт-пирит) встре-
чается гл. обр. в виде смешанного минерала, в к-ром
Со частью замещен на Ni, Си, или Fe. Сплавлением
кобальта с серой в токе СО2 при 450—500° или с гип-
сом и щелочью, или восстановлением сульфата ко-
бальта получают безводный CoS — кристаллы сталь-
ного цвета, иногда с красноватым оттенком, решетка
гексагональная, а = 3,385 А, с = 5,213 А; плотн.
5,45. Теплота образования = —202 ккал/моль
(для кристаллич.). Плавится при 1160° (с разл.).
На воздухе начинает окисляться при 684° с превра-
щением в CoSO4. Сульфид CoS, полученный действием
H2S па водные р-ры солей 2-валентного Со, имеет
черный цвет; свежеосажденный растворим в кислотах,
но при стоянии переходит в нерастворимую форму.
Растворимость CoS в воде 5,4 • 10 е молъ/л (при pH
5,0). Синтетич. Co3S4 получен прокаливанием CoSO4
в токе H2S при 400—500° в виде черно-серого порошка;
кристаллизуется в граиецентрировапной кубич. ре-
шетке с периодом в пределах а = 9,398—9,458 А.
При 680° он разлагается на CoS и CoS2. Дисульфид
CoS2 имеет кубич. решетку, а = 5,524 А. При про-
каливании СоСО3 с избытком серы и поташа обра-
зуется Co2S3 в виде серого кристаллич. вещества, не-
растворимого в воде и очень слабо реагирующего
с сильными кислотами. При действии сероводородом
на водные р-ры аммиачных комплексов кобальта
также получают Co2S3, но он неустойчив и при нагре-
вании выше 450° переходит в смесь моно- и дисуль-
фидов. Co4S3 применяют в качестве катализатора при
гидрогенизации алифатич. полисульфидов, эфиров
тиокислот, тиокарбонилов и др. органич. сульфидов.
Лит. см. при ст. Кобальт. Ф. М. Перельман.
КОВАЛЕНТНАЯ СВЯЗЬ — см. Химическая связь.
КОВАЛЕНТНЫЙ РАДИУС — радиус, приписывае-
мый атомам данного элемента, чтобы передать межъ-
ядерные расстояния в молекулах или кристаллах,
считая ковалентно соединенные атомы соприкасаю-
щимися. Оказывается возможным выбрать значения
К. р. для связей данной кратности (одинарной, двой-
ной, тройной) так, чтобы длины связей, вычисленные
как суммы К. р., совпадали в большинстве случаев
с опытными величинами, полученными рентгеногра-
фии., электронографии, и другими методами, с точ-
ностью 0,02—0,03 А. К. р. применимы для вычисле-
ния межатомных расстояний и при частично ионном
характере связи, но если ионный характер преобла-
дает, то для этой цели используют ионные радиусы.
Более точные результаты можно получить при учете
разности электроотрицательности атомов, образующих
ковалентную свнзь.
Ниже приведена таблица К. р. по Полингу. Ее
применение иллюстрируют след, примеры (величины
в А). Связь С—Н вычислена 1,07, опыт: 1,07 (CH3Ci,
HCN), 1,09 (СН4), 1,06 (СН3ОН). Связь С—О вычис-
лена 1,43, опыт: 1,43 (СН3ОН), 1,44 (СН2—СН2О).
Ковалентные радиусы (А): I — в одинарной, II — в двойной,
III — в тройной связи
Атом I П Ш Атом I II
Н 30 Ci 0,99
в 0,88 0,76 0,68 As 1,21 1.11
с 0,77 0,67 0,60 Se 1,17 1,07 .
N 0,70 0,60 0,55 Вг 1,14 —
О 0,06 0,55 Sb 1,41 1,31
F 0,64 — Те 1,37 1,27
Si 1,17 1.07 1.00 1 J 1,33 —
р 1.10 1,00 0,93
S 1.04 0,94
ОН
ОН
Если вследствие резопанса валентных структур
данная связь имеет кратность, промежуточную между
одинарной и двойной или двойной и тройной, то
и длина связи принимает промежуточное значение.
Напр., в молекуле бензола связь соседних атомов
является промежуточной между одинарной и двойной;
сумма К. р. для связи С—С 1,54, для связи С=С
1,34; наблюденная длина связи 1,39 (в А).
КОГЕЗИЯ (сцепление) — притяжение между мо-
лекулами • (атомами, ионами) в объеме данного тела.
Вещества в конденсированном состоянии — в виде
твердых и жидких тел — вследствие предельно малых
расстояний между образующими их молекулами,
ионами и атомами характеризуются высокой К.,
в газообразном состоянии — весьма малой К. В от-
личие от адгезии, К. характеризует идеальную проч-
ность тела, определяемую силой сцепления образую-
щих его молекул, атомов, ионов. (См. также Межмо-
лекулярное взаимодействие').
КОДЕГИДРОГЕНАЗЫ — коферменты многочислен-
ных анаэробных дегидрогеназ, являющиеся промежу-
точными переносчиками водорода во многих важ-
нейших биохимии, окислительно-восстановительных
процессах. Имеются несколько К.: 1) К о д е г и д р о-
г о п. аза I (Koi, пикотинамидадениидинуклеотид,
дифосфопиридипнуклеотид, козимаза) — динуклеотид
аденина и никотинамида. Окисленную форму коде-
гидрогеназы I принято обозначать Koi, или ДПН,
восстановленную — Ко1-Н2, или ДПН-Н. Два изо-
мера Koi, а- и (3- отличаются конфигурацией глико-
зидной связи в никотин-
амидрибозидной части
молекулы. Каталитиче-
ски активным является
только (3-изомер. Обыч-
но в препаратах Koi,
получаемых из биоло-
гич. объектов, наряду
с (3-изомером содержит-
ся 10—15% неактивно-
го а-изомера. 2) Ко-
дегидрогена за
Il (KoII, никотинамид-
аденин динуклеотид фос-
фат, трифосфопиридип-
нуклеотид) отличается
от Ко! наличием тре-
тьего остатка фосфор-
ной к-ты в положении
2' рибозы адениловой
части молекулы. Окис-
ленную форму иодегид-
рогеназы II обозначают
KoII, или ТПП, восста-
новленную — KoII-Н»,
или ТПН-Н.
Ни одна К. не полу-
чена в кристаллич. со-
стоянии. Получаемые
из биологич. источни-
ков препараты К. высо-
кой степени чистоты ха-
рактеризуются следую-
щими свойствами: a) Koi, Р-изомер (Р ДПН), мол. в.
663,43 — белый аморфный порошок, хорошо раство-
римый в спирте и ацетоне. С тяжелыми металлами
образует нерастворимые в воде соли. Хининовая
соль (2 молекулы Ко! и 3 молекулы хинина) кристал-
лизуется из воды в виде игл, т. пл. 162—170°; (а]д
Р-пзомера = —34,8° (в воде); [а] д а-цзомера =
— +14,3° (в воде), б) KoII, мол. в. 743,42 — бесцвет-
ный аморфный порошок, легко 'растворим в воде и
Д П Н.
Ко' 1
ОН 0-Р(ОН)2
6
'Т П И. Kb II
Р-изомер (РДПН), мол. в.
623
КОДЕГИДРОГЕНАЗЫ — КОДЕИН
624
метаноле, нерастворим в спирте, эфире, этилацетате.
С тяжелыми металлами, подобно Koi, образует нерас-
творимые соли.
Окисленные К. довольно устойчивы в кислых р-рах
И быстро разрушаются в щелочах; восстановленные
К. относительно устойчивы в щелочных р-рах и не-
устойчивы в кислотах. УФ-спектр окисленных К.
характеризуетсн наличием интенсивного пика по--
глощенпн при 260 ммк (е = 16,5-103); у восстанов-
ленных форм К. появляется дополнительный, ха-
рактерный пик при 340 ммк (в = 4,5—6,3-103). Это
свойство К. широко используется для спектрофото-
метрия. определения активности различных дегидро-
геназ и изучения кинетики реакций, сопряженных
с окислением или восстановлением К. Наиболее чув-
ствительным методом количественного определения
Ко! и KoII является флуорометрия, метод, посред-
ством к-рого можно определить содержание каждой
К., как в восстановленной, так и в окисленной форме
при концентрации их до 10 8 Л/.
Ко! является коферментом более 40 различных де-
гидрогеназ, среди к-рых ферменты гликолитич. цикла
(дегидрогеназа 3-фосфоглицеринового альдегида, лак-
тикодегидрогеназа, алкогольдегидрогеназа), фермен-
ты цикла трйкарбоновых к-т (дегидрогеназа яблочной
к-ты, дегидрогеназа а-кетоглутаровой к-ты) и фер-
менты, входящие в систему окисления жирных к-т.
Число ферментов, требующих КоП, менее значительно.
Из них наиболее важными являются дегидрогеназа
изолимонной кислоты (цикл трйкарбоновых кислот),
а также дегидрогеназы глюкозо-6-фосфата и 6-фосфо-
глюпоновой кислоты, относящиеся к системе апото-
мического (гексозомопофосфатного) окисления угле-
водов.
Коферментная специфичность дегидрогеназ, как
правило, очень высока: ферменты, требующие Koi,
не функционируют с KoII и наоборот; исключение
составляет дегидрогеназа L-глутаминовой к-ты, ко-
ферментом к-рой могут служить как Koi, так и КоП.
К. и белковая часть соответствующих ферментов
связаны за счет пиридиновой части молекулы кофер-
мента и активных SH-групп белка. Эта связь очень
непрочна и легко диссоциирует. Легкость, с к-рой К.
отщепляются от соответствующих дегидрогеназ, яв-
ляется характерной особенностью, позволяющей им
осуществлять роль подвижных промежуточных пере-
носчиков водорода, объединяющих и связывающих
между собой различные окислительно-восстановитель-
ные процессы. Эта роль К. может быть схематически
представлена следующим образом:
В реакции (1)Ко1 восстанавливается за счет окисления
субстрата АН2; в реакции (2) восстановленный Ко1-Н2
вновь окисляется до Koi, восстанавливая субстрат
В. Каждая из этих реакций катализируется особым
ферментом. За один оборот цикла Koi переносит от
одного субстрата к другому два атома водорода. Ме-
ханизм переноса сводится к обратимому восстановле-
нию пиридинового кольца за счет присоединения к
нему 2 электронов и 1 протона, отщепляемых от суб-
страта (второй протон остается в растворе), в резугль-
тате чего при восстановлении кодегидрогеназы раствор
подкисляется, а при окислении — подщелачивается:
Ферментативный перенос водорода от субстрата на
К. и от К. на субстрат стереоспецифичен. С помощью
дейтерированных субстратов показано, что при вос-
становлении К. атом водорода присоединяетсн в по-
ложение 4 пиридинового кольца, располагаясь в
зависимости от специфичности дегидрогеназы только
над или только под плоскостью пиридинового кольца.
При окислении К. водород отщепляется с этой же
стороны плоскости кольца. Такой стереоспецифич-
ности, естественно, не наблюдается при химич. вос-
становлении К. Каталитич. активность К. весьма
значительна: 1 молекула Ко! в лактикодегидрогеназе
осуществляет в минуту 2-104 элементарных актов
переноса.
Биохимич. значимость К. исключительно велика.
Они являются первым звеном в цепи промежуточных
переносчиков водорода в процессе биологич. окисле-
нии. Воспринимая водород непосредственно от окис-
ляющихся субстратов (углеводов, аминокислот, жир-
ных и карбоновых к-т), К. передают его следующим
переносчикам с более высоким окислительно-восста-
новительным потенциалом, обычно флавопротеидам
(см. Флавиновые коферменты). Изменение свободной
энергии при переносе водорода с К. на флавины со-
ставляет 10 ккал/моль. Эта энергия аккумулируется
за счет сопряженного процесса окислительного фос-
форилирования в виде макроэргич. фосфатных связей
адепозинтрифосфорной к-ты (АТФ). К. осуществляют
также перенос водорода между различными окисляю-
щимися и восстанавливающимися субстратами. Тако-
ва роль Ко! в гликолитич. цикле и в ряде других ре-
акций, напр. в превращении тестостерона в андро-
стерон. Восстановление Ко! в стенке желудка служит
источником соляной к-ты желудочного сока. Нако-
нец, К. принимают участие в процессе фотосинтеза.
К. присутствуют во всех растительных и животных
тканях, а также в микроорганизмах. Содержание
Ко! обычно в 5—10 раз выше, чем КоП. Основным
источником для получения Ко! являются пекарские
дрожжи (из 10 кг дрожжей получают 250 мг Koi), а
для получения КоП — печень овцы или свиньи.
К. выделяют экстракцией теплой водой, осаждением
солями Ag или Hg и хроматографией на ионообменных
смолах. Обычно этими способами получают окислен-
ные формы К. Восстановленные К. могут быть при-
готовлены из окисленных К. химич. или фермента-
тивным восстановлением.
Лит.: М п х л п и Д. М., Биохимия клеточного дыхания,
М., 1 ОБО; Нейландс Д., III т у м п ф П., Очепки по химии
ферментов, пер. с англ., М., 1958; Диксон М., Уэбб Е.,
Ферменты, пер. с англ., М., 1961; Б е р е з о в с к и й В. М.,
Химия витаминов, М., 1959; Methods in .enzymology, ed. S. P.
Colowick, N. O. Kaplan, v. 3, N. Y., 1957,p. 876; The enzymes,
2 ed, v. 3, N. Y., 1960. ' В. Б. Спиричев.
КОДЕИН C18H2IO3N, мол. в. 299,28 — один из
несозревших плодов
алкалоидов опия, млечного сока
снотворного мака Papaver som-
niferum'L. Содержание алка-
лоидов в опии 20—25%, из'
них К. 0,2—2% от сухого веса
опия. Выделение К. из смеси
алкалоидов производится мно-
гократной экстракцией. К. —
белый кристаллич. порошок
(моногидрат); т. пл. 155° (для без-
водного вещества), [a]D-= —137,7°(в спирте) и—111,5;
(в хлороформе). Умеренно растворим в воде: 1:120
(25°), 1:60 (80°) и 1:17 (ТОО0), в водном р-ре аммиака
1:68 (15,5°), эфире 1:75 (15,5°) и 1:50 (25°), хорошо
растворим в спирте 1:2 (25°) и хлороформе 1:0,5
(25°); лучше, чем морфии растворим в анизоле 1:6,5
(16°) и холодном бензоле 1:13, но почти нераство-
рим в щелочи. Важнейшие соли К.: хлоргидрат,
т. нл. 264°, хорошо растворим в горячей воде; суль-
625
КОЖА
фат (H2SO, • 5Н2О), т. пл. 278° (с разл.); фосфат
(Н3РО4- 1,Н112О), т. пл. 235° (с разл.), хорошо рас-
творим в воде 1:2,5 (25°).
Бущучи ароматическим О-метилморфином, К. впол-
не аналогичен ему по химич. свойствам, за исключе-
нием свойств, обусловленных наличием фенольного
гидроксила в морфине. Полный синтез К. — см.
Морфин. Частичный синтез К. метилированием мор-
фина, лучше всего с помощью [C6H6N(CH3)3OH],
проводится в производственном масштабе.
По физиологии, действию К. аналогичен морфину,
но метилирование фенольного гидроксила снижает
наркотич. и анальгетич. действие К. Применяется для
успокоения кашля (сильно снижает возбудимость
кашлевого центра) и, в сочетании с другими медика-
ментами, — как успокаивающее средство. Фосфат
К. используется аналогично. В медицине также при-
меняются производные К. — гидрокодон (7,8-дигид-
рокодеинон-6, против кашля активнее, чем К.) и
текодин (7,8-дигидро-14-оксикодеинон-6, заменитель
морфина).
Лит. см. при ст. АлкалоиЭы. А. Е. Васильев.
КОЖА — материал, изготовляемый из среднего
слоя шкур животных. К. называют и сами шкуры,
являющиеся сырьем кожевенного производства. Шку-
ра животного состоит из трех слоев: наружного — эпи-
дермиса, среднего — дермы, нижнего — подкожной
клетчатки. Дерма образована тесно переплетаю-
щимися пучками волокон соединительной ткани, со-
стоящей в основном из белкового вещества — колла-
гена. Промежутки между пучками волокон и волок-
нами заполнены межволоконным белковым веществом,
растворимым в воде и щелочных растворах. Дерма
имеет верхний, т. наз. сосочковый, слой, где сосредо-
точены железы и волосяные образования, и нижний —
сетчатый слой, состоящий преимущественно из соеди-
нительной ткани. В дерме различают следующие
структурные элементы: пучки волокон диаметром от
30 до 200 р, волокна — диаметром от 1 до 3 р и фиб-
риллы — от 0,1 до 0,05 р. В процессах кожевенного
производства из шкуры удаляется межволоконное
вещество, эпидермис и подкожная клетчатка. Дерма
подвергается физико-химич. и механич. обработке.
Выделанная К. представляет собой дерму животной
шкуры, сохранившую в основных чертах ее волокни-
стую структуру, но физико-механич. и химич. свой-
ства дермы претерпевают существенные изменения
в зависимости от назначения К.
В зависимости от назначения различают четыре
класса К.: 1) обувные, 2) шорно-седельные, 3) техни-
ческие и 4) одежно-галантерейные. Каждый класс
подразделяется на группы в соответствии с более уз-
ким назначением. Так, напр., обувные К. делятся на
К. для низа обуви и К. для верха обуви. К. классифи-
цируют также по видам сырья, видам дубления, спо-
собу и характеру отделки, конфигурации, толщине,
площади.
Свойства К. зависят, прежде всего, от ее микрострук-
туры и изменения волокон дермы в процессах дубле-
ния и наполнения. В зависимости от вида сырья и
метода выделки микроструктура К. характеризуется
рядом признаков, 'К к-рым относятся: регулярность
сплетения пучков волокон, угол сплетения, компакт-
ность, извитость пучков волокон. Химич, состав К.
характеризуется содержанием гольевого вещества
(прежде всего, коллагена), дубящих веществ (мине-
ральных и органич., связанных и вымываемых), воды,
жиров и минеральных веществ. Разница в химич.
составе К. в зависимости от метода выделки и ее наз-
начения можетбыть значительной. Так, напр., в К. для
верха обуви содержание гольевого вещества при влаж-
ности 18% составляет 50—70%, связанных дубящих
626
веществ — 3,5—7%; в К. для низа обуви, соответ-
ственно, — 28—50% и 20—30%.
В структуре К. пет отдельных изолированных во-
локон, все волокна разветвляются и переплетаются,
образуя сплошную сетку. Выделанная К. имеет поэ-
тому большую пористость (достигающую в ряде слу-
чаев 70—75% от общего объема К.). Большая пори-
стость К. и достаточно высокая гигроскопичность
определяют такие важные гигиенич. свойства К.,
как высокая паро- и воздухопроницаемость, а также
высокие теплозащитные свойства (в сухом состоянии).
Коэффициент теплопроводности подошвенной К.
0,118—0,135 ккал/м2час • град (у микропористой ре-
зины — 0,186). Вследствие высокой пористости, у К.,
в отличие от других материалов, различают истинную
плотность и кажущуюся (ее часто наз. просто плот-
ностью). Первая — это уд. вес плотного вещества К.,
тогда как вторая — вес единицы объема К. с учетом
пор. Истинная плотность зависит, гл. обр., от состава
К., и значение ее колеблется от 1,3 до 1,9 г/см3. Ка-
жущаяся плотность зависит от пористости К., к-рая,
в свою очередь, зависит от вида сырья и метода вы-
делки К. Максимальную кажущуюся плотность имеет
К. для низа обуви (0,99—1,24), минимальную — жи-
ровая замша (0,23—0,42).
Отношение К. к воде зависит от микроструктуры,
пористости, способности К. смачиваться и поглощать
воду. Характеристиками отношения К. к воде яв-
ляются влагоемкость (намокаемость), водопроницае-
мость и водопромокаемость. В результате сильного
намокания К. утрачивает теплозащитные свойства,
при высыхании часто становится жесткой и ломкой.
Предел прочности К. при растяжении колеблется
от 1,0 до 8,0 кг/мм2. Величина относительного удлине-
ния, характеризующего тягучесть К., при нагрузке
1 кг/мм2 составляет от 3 до 70% в зависимости от
характера и назначения К.; обладает явно выражен-
ными релаксационными свойствами. Одной из харак-
теристик качества К. является сопротивление ее раз-
рушающим усилиям в процессе сжатия. Разрушающая
нагрузка при деформации сжатия примерно в 10 раз
превышает предел прочности при растяжении и соста-
вляет для подошвенной К. хром-растительного Дубле-
ния 20 кг/мм2. В процессе эксплуатации К. подвер-
гается многократно повторяющимся деформациям
изгиба, растяжения, сжатия, а также истиранию. К.
выдерживает свыше 1 млн. изгибов. Поведение К^ при
повышенной темп-ре обычно характеризуют темп-рой
сваривания, при к-рой обводненная К. необратимо из-
меняет свои размеры и свойства. К. жирового дуб-
ления имеет темп-ру сваривания 65°, растительного
дубления — от 70 до 85°, хромового — от 80 до 130°,
формальдегидного — 90°. Разрушение сухой К. на-
чинается при темп-ре около 210°.
К. вырабатывают из кожевенного сырья (шкур
животных) путем сложных химич. воздействий и
механич. обработки. Основные операции превращения
сырой шкуры в выделанную К. делятся на три группы:
подготовительные, дубильные и отделочные.
Подготовительные операции имеют целью выделе-
ние дермы из шкуры и изменение ее микроструктуры.
Первой подготовительной операцией является от-
мока — погружение шкуры в воду, чистую или содер-
жащую химич. реагенты (например, Na2S, Na2SO3,
NaSiF6, щелочи и др.) и бактерии, с целью удаления
загрязнений, консервирующих веществ и раство-
римых белков. В процессе отмоки дерма набухает
и разрыхляется. Отмоченные шкуры подготовляют
к удалению волос и эпидермиса (обезволашива-
нию). Для этого чаще всего используют два способа:
1) намазь — нанесение на нижнюю поверхность
шкуры кашицеобразной смеси сернистого натрия,
извести, воды и выдерживание шкуры в таком виде
627
КОЖА —КОЖА ИСКУССТВЕННАЯ
628
несколько часов; 2) золение — погружение шкуры
в водную суспензию извести (известковое молоко)
часто с добавкой сернистого натрия, соды. При такой
обработке ослабляется связь волоса и эпидермиса с
дермой, изменяется структура коллагена, омыляготся
жиры, содержащиеся в шкуре, растворяются меж-
волокоппые вещества. Волос удаляют па волосогои-
ных машинах, после чего па мездрильных машинах
удаляют подкожную клетчатку. Шкуры, подготовлен-
ные первым способом, после снятия волоса обычно
подвергают дополнительному золению в растворе
сернистого натрия для разрушения остатков корней
волоса, волосяных сумок и эпидермиса и изменения
структуры коллагена. Обезволошенную шкуру с из-
мененной микроструктурой наз. гольем. Лицевой
слой голья обычно подвергают чистке с целью удаления
продуктов распада белков и жиров (гнейста) из сосоч-
кового слоя. Для изготовления отдельных видов К.
голье разделяют па двоильно-леиточных машинах
(двоение). Двоение производят иногда и после дуб-
ления.
В золеном голье коллагеновые волокна в на-
бухшем состоянии содержат солеобразно связанный
кальций, капиллярно поглощенный и сорбированный
кальций в виде гидрата окиси. В сосочковом слое
дермы, в волосяных каналах, в жировых и потовых
железах содержатся продукты распада белков шкуры,
пе удаленные при чистке. Эти вещества могут увели-
чить жесткость готовой К. Поэтому такое голье
обеззоливают — нейтрализуют щелочи и удаляют
кальциевые соединения либо при помощи р-ров аммо-
нийных солеи или кислот, либо комбинируя то и дру-
гое. После обеззоливания голье обычно подвергают
обработке ферментами (мягчение). Под действием фер-
ментов происходит частичный распад коллагена, раз-
рыхление структурных элементов лицевого слоя,
растворение продуктов разрушения волосяных сумок,
растворение и удаление из дермы остатков межволо-
конных и других белков, дальнейшее омыление жи-
ров. В результате этой обработки К. приобретает
мягкость. Для мягчения обычно применяют искус-
ственные мягчители, изготовляемые из поджелудоч-
ной железы крупного рогатого скота (ороион, технич.
панкреатин) или из нек-рых плесневых грибков (ори-
зон). Мягченное голье подвергают затем обработке
кпслотно-солевыми смесями (пикелевание) для даль-
нейшего разрыхления структурных элементов колла-
гена и подкисления голья.
После подготовительных операций К. подвергается
дублению с помощью дубящих веществ. Молекуляр-
ные цепи и волокна коллагена связываются молеку-
лами дубителя в пространственную структуру с боль-
шим числом поперечных связей (водородных, кова-
лентных, электровалентных). Выдубленная К. при-
обретает характерные свойства: уменьшение дефор-
мируемости обводненной дермы; сохранение пористо-
сти К. в процессе сушки; уменьшение склеиваемостп;
повышение прочности при растяжении; уменьшение
влагоемкости при набухании в воде; повышение тер-
мостойкости и др.
Отделкой придают К. соответствующий внешний вид
(цвет, гладкость) и определенные физико-мехапич.
свойства. К отделочным операциям относятся раз-
водка, строжка, крашение (см. Крашение кожи), жи-
рование, сушка, увлажнение, тяжка, шлифование,
покрывное крашение, лощение, прессование, про-
катка.
К. как материал, обладающий специфическими фи-
зпко-механич. свойствами, применяется для изго-
товления . разнообразных предметов потребления
(обувь, одежда, галантерейные изделия п др.) и в тех-
нике (приводные ремни, детали машин в текстильной
пром-сти И Т. Д.).
Отходы кожевенного производства используются
для других отраслей пром-сти: мездра — для приго-
товления желатины и клея, шерсть -- для валяльНО-
войлочпого производства, хромовая стружка — для
изготовления кожи искусственной.
Лит.: Химин кожевенного и мехового производства,
(под ред. II. В. Чернова], М., 1957; Технология кожи и меха,
под ред. II. В. Чернова, М., 1959; Справочник кожевника,
т. 1--3, М., 1 952—54; Химин и технология кожи, пер. с англ.,
•г. 1 — 2, М., 1960—62. И. С. Афонская.
КОЖА ИСКУССТВЕННАЯ — материалы, изго-
тавливаемые из полимерных веществ для замены на-
туральной кожи. I?. и. по составу разделяют на ма-
териалы: резиновые, проклеенные волокнистые, пле-
ночные н ткани с покрытием. По назначению К. и.
подразделяются на обувные, одежные, технические и
галантерейные и др.
Резиновые материалы вырабатываются двух типов:
монолитные (с плотностью выше 1) и пористые (с
плотностью нпже 1, наир. 0,2—0,4). Применяются
они преимущественно для низа обуви (подошвднные
резины). К. и. па основе резины изготовляют обычными
методами резинового производства, заключающимися
в смешении каучука с ингредиентами (наполнители,
мягчители, вулканизующие агенты, противостари-
тели, порообразователй и др.) и вулканизации (см.
Резина, Резины пористые). Пористые виды К. и. выра-
батываются также па основе других материалов,
напр. полиуретанов (поролон). Порообразующим ве-
ществом в этом случае является углекислый газ,
выделяющийся при поликонденсации толуилен-изо-
циопата с полиэфирами в присутствии воды. Порис-
тые К. и. в 3—5 раз легче кожи, имеют высокие
теплоизоляционные и амортизационные свойства,
высокое сопротивление старению, сохраняют эластич-
ность в зимних условиях.
К. п. на волокнистой основе готовят из прочесов
хлопка пли из прошитой волокнистой основы путем
проклейки их поливинилхлоридом или латексами в
процессе прессования с последующим наложением
полиамидной млн другой отделки. Эти заменители
по технич. свойствам близки к натуральной коже,
по по гпгиенич. свойствам уступают ей.
Для изготовления К. и. типа картонов волокнистую
массу, полученную путем размола целлюлозы или
кожевенных краснодубиых отходов, отходов хромовой
кожи и др. волокнистых материалов, проклеивают
латексами (см. Латексы синтетические) или кани-
фольно-битумпой дисперсией. Чаще всего используют
синтетич. латексы на основе дивинилхлорвинилиде-
пового ка}’чука (ДВХБ-70). Из этой волокнистой
массы отливают листы на гусс-машинах (однослой-
ный отлив) или на круглосеточных и столовосеточных
машинах (многослойный отлив). Полученные листы
прессуют, сушат. К. и. этого типа обычно исполь-
зуются для изготовления задников и стелек для обуви.
Подошвенную К. и. (пл асткож а) вырабатывают
путем склеивания смоло-каучуковыми клеями ко-
жевенного волокна с последующей вулканизацией
пластин пласткожи. К. и. типа картонов по своей
структуре похожи на натуральную кожу, но имеют
малую пористость.
Для получения К. и. на тканевой основе ткани
покрывают пленкой из каучука или пластиков. Для
этого используют растворы бутадиенового каучука,
синтетич. латексы, пасты из поливинилхлоридной
смолы и др.плеикообразующие полимеры с последую-
щей отделкой. К. и. этого вида являются кирза,
получаемая из ткани с резиновым покрытием, а к-
р и п и т (тоже ткань с резиновым покрытием, но еще
отделанная дисперсиями акриловых смол). Ш ар-
го л и н, т е к с т о в и и и т и искусственная ла-
ковая кожа представляют собой ткани с покрытием,
их используют для верха обуви. Для подкладки обуви
629
КОЗИМАЗА — КОКС
630
применяют искусственный футор, изготовляемый
из начесанной футорной байки,, пропитанной диспер-
сиями синтетич. каучука (латексами) и подвергнутой
вулканизации. Для этой же цели применяют подкла-
дочный пористый текстовинит. К. и. для одежды тоже
вырабатывается на ткани с .покрытием из поливинил-
хлоридной смолы или с резиновым покрытием. Для
галантерейных изделий используются павинол
(с поливинилхлоридным покрытием), г р а л е к с
(резиновое покрытие). Обивочными К. и. являются
автобим и текстовинит, дерматин
(ткань с нитроцеллюлозным покрытием).
Искусственную обувную замшу готовят путем
покрытия ткани поливинилхлоридной пастой, затем
клеем из растворов перхлорвинила и мочевино-фор-
мальдегидной смолы. На полученную липкую поверх-
ность наносится в электрич. поле мелко нарезанное
вискозное волокно. Замшеобразный ворс на материале
образуется вследствие ориентации волоконец в элек-
трич. поле. Другой способ получения искусственной
замши состоит в покрытии слоя поливинилхлоридной
пасты на ткани порошком сульфата натрия. После
обработки на каландре избыток соли вымывается во-
дой. К. и. на тканях имеет высокое сопротивление
надрыву, мягкость. Но у нее малы паропроницаемость,
сопротивление истиранию и стойкость к многократ-
ному изгибу. Для придания К. и. гигиенич. свойств
в покрывную массу, наносимую на ткань, вводят
волокно, порообразующие вещества (бикарбонат нат-
рия и др ), нейтральные соли (с последующим их вы-
мыванием из пленки), различные гидрофильные до-
бавки и т. д.
Пленочные К. и. (обычно это пленки из поливи-
нилхлоридной смолы, содержащие стабилизаторы,
пластификаторы и пигменты) применяют в галан-
терейной промышленности.
К. и., хотя и уступает натуральной коже по ряду
свойств (особенно в гигиенич. отношении), имеет и
существенные преимущества. К. и. дешевле натураль-
ной, более однородна по качеству, чем натуральная,
выпускается в виде многометражных кусков, и по-
этому более удобна в производстве. Применение К. и.
создает больше возможностей для автоматизации
производства.
Лит.: Зайончковский А. Д., Искусственная
кожа, М., 1960; Технология искусственной кожи, под ред.
С. А. Павлова, М., 1958; Авилов А. А., Искусственная
кожа, М., 1961.
КОЗИМАЗА — см. Кодегидрогенаа'ы.
КОКАИН Ci,H2iO4N (I) алкалоид, получаемый из
листьев кустарника кока (Erytroxylon coca) семей-
ства Erytroxylaceae, произрастающего в Южной Аме-
рике и на острове Ява. Бесцветные кристаллы, из
спирта кристаллизуется в виде моноклинных призм;
т. пл. 98°, т. кип. 187—188°/0,1 мм; [а]д = —15,8°.
Плохо растворим в воде (1:700), легко — в органич.
растворителях. С кислотами дает хорошо кристалли-
зующиеся соли, из к-рых гигроскопич. хлоргидрат,
т. пл. 200—202°; [а]р =—71,95° (6Н2О), хорошо
растворим в воде (2:1) и спирте (1:2,6) и нерастворим
в эфире; применяется в медицине. Другие применя-
емые соли К.: нитрат (2Н2О), т. пл. 58—63°, периодид
(J2), т. пл. 161°, формиат, т. пл. 42° и т. д.
По своему строению природный К. — 20-карбо-
метокси-ЗР-бензоилокситропан или, иначе, метиловый
-J---Г-СООН
K-CHy-QH
а
эфир бензоилэкгонина. При гидролизе легко расщеп-
ляется на экгонин (II), СН3ОН и С3Н5СООН. Строение
экгонина и К. подтверждено многочисленными син-
тезами. В Erytroxylon coca К. содержится вместе
с другими близкими по строению алкалоидами, в
состав к-рых вместо остатка С8Н5СООН входят остатки
других кислот. Длн получении К. всю сумму алкало-
идов гидролизуют и образовавшийся экгонин после-
довательно метилируют и бензоилируют в К. Воз-
можно получение К. из тропиновых производных,
к-рые входят в состав алкалоидов, добываемых из
отечественного сырья. Ниже приведен разработанный
в СССР полный синтез К. по схеме, близкой к био-
генетич. Общий выход идентичного природному К.,
исходя из фурана, 4,15%, а выход (±) кокаина, заме-
няющего природный, 5,47%. Наиболее сложным эта-
пом синтеза является восстановление- метилового
эфира тропинонкарбоновой к-ты в метиловый эфир
С, СН3ОН
Р Вг2
Нг _
Ni(CKeneniJ
осщ
осн3
h-(hci)
сн2сно ch3nh2-hci + снсоосн;
| —। c-ок
CH2CHO CH,COOK
____COOCH3
N-CHJ=° —•
COOCH3
-OH —►dl-l
III
экгонина (Ш). В зависимости от условий и способа
восстановления могут образоваться 4 стереоизомер-
ных рацемических экгонина, один из к-рых, получа-
емый восстановлением амальгамой натрия, при бен-
зоилировании дает К., идентичный природному, сте-
реохимия к-рого соответствует формуле I.
К. — один из основных представителей местно-
анестезирующих веществ, т. е. веществ, понижающих
или полностью подавляющих возбудимость чувстви-
тельных нервных окончаний и тормозящих проведе-
ние возбуждения по нервным волокнам. При нанесе-
нии на слизистые оболочки и при введении под кожу
вызывает местную потерю чувствительности. При
всасывании вызывает эйфорию, возбуждение, а затем
торможение нервной системы. При повторном при-
менении К. может развиться болезненное пристрастие
к нему (кокаинизм). Своим действием кокаин обязан
/N—(С)п—X—СОАг (для К. п = 3, Х=О), т. наз.
«анестезиофорной» группировке, входящей в состав его
молекулы. Многочисленные синтетич. местные ане-
стетики, содержащие эту группировку, более простые
и менее токсичные, чем К., ограничили область при-
менения последнего, гл. обр. офтальмологией. Высшая
(разовая и суточная) доза К. — 0,03 г для взрослых.
Лит..- Базилевская Г. И. [и др.1, Ж. общ. химии,
1958, 28, вып. 4, 1097; 1960, 30, вып. 5, 1458.
А. Е. Васильев.
КОКОСОВОЕ МАСЛО — см. Жиры растительные.
КОКС — твердый остаток, образующийся при на-
гревании различных топлив до высоких температур
без доступа воздуха (см. Коксование). В зависимости
от исходного топлива различают следующие виды
промышленного К.: каменноугольный, электродный
пековый и нефтяной.
Кокс каменноугольный — спекшийся твёрдый про-
дукт, образующийся при нагревании нек-рых углей
631
кокс
632
в коксовых печах до 900—1050е; куски К. имеют удли-
ненную, более или менее правильную форму (отно-
шение длины к ширине, напр., у К. из донецких углей
1,2—1,8), цвет от блестяще-серебристого до светло-
серого или матово-серого. Пористость К. обычно ко-
леблется в пределах 49—53%; истинная плотность
1,80—1,95, насыпной вес 400—500 кг)м3. К. произ-
водится гл. обр. для металлургия, целей. По харак-
теру использования К. подразделяется на Домен-
ный и литейный (ваграночный). Основной
потребитель К. — доменные печи, в к-рых он одно-
временно является топливом и восстановителем же-
лезной руды. Расход К. на 1 т чугуна ок. 0,8 т; по
мере совершенствования доменного процесса расход
К. снижается за счет подвода в доменную печь других
топлив, напр. природного газа. К. применяется также
в качестве генераторного топлива, обычно в виде
кусков размером 10—25 мм. Коксовая мелочь исполь-
зуется для агломерации руд.
Химич, свойства К. определяются следующими
показателями: 1) Степень обуглероженности — по
выходу летучих веществ (0,7—1,2% на горючую массу)
п элементарному составу (свыше 96,5% углерода);
теплота сгорания горючей массы К. ок. 8000 ккал/кг,
темп-ра воспламенения 520—600°. 2) Содержание вла-
ги составляет 2—4% (не более 5%) при тушении его
водой и до 0,5% при сухом тушении (инертными га-
зами). 3) Зольность К. обычно не превышает 10—11%;
зольность доменного К. 9—10,5%. Содержание крем-
незема и глинозема в коксовой золе обычно 60—85%.
В процессе доменной плавки золу переводят известью
в шлак. 4) Содержание серы в литейном К. не должно
превышать 1,2%; в доменном К., получаемом из до-
нецких углей, обычно содержится 1,5—1,9% S, а из
кузнецких 0,4—0,5%. Большое содержание серы
в К. загрязняет чугун; для удаления серы со шлаком
увеличивают расход флюсов (при увеличении содер-
жания серы в К. на 1% расход флюсов и кокса —
каждого в отдельности — увеличивается примерно
на 12%). 5) Содержание фосфора в К. имеет важное
значение при выплавке чугуна; напр., при выплавке
бессемеровского чугуна количество фосфора не дол-
жно превышать 0,015%. При повышенном содержа-
нии фосфора чугун становится хрупким.
К физико-механич. свойствам К. относятся:
1) Прочность — наиболее важный показатель
качества К. — характеризует способность К. проти-
востоять разрушению под воздействием многократ-
ных усилий по пути от коксовых печей к доменной
печи и гл. обр. в самой доменной печи. Механич.
прочность К. при разных условиях нагрузки харак-
теризуется: дробимостью, истираемостью, сопротив-
лением раздавливанию, термин, стойкостью. Проч-
ность К. может быть также охарактеризована его
трещиноватостью, обусловливающей образование кок-
совой мелочи. Прочность К. обычно определяется
барабанной пробой.
В’железный барабан помещают 410 пг К. кусками более
25 л1м; барабан вращают в течение 15 мин со скоростью
10 об/мин. Образующаяся при этом мелочь просыпается, а вес
остатка представляет показатель барабанной пробы. Доменный
К. из донецких углей должен давать остаток в барабане не
менее 330 кг, а из кузнецких, карагандинских и кизеловских
углей — 300—325 кг. Механич. прочность К. можно опреде-
лять и др. способами (напр., по ГОСТ 8929—58).
2) Ситовый состав — для доменной плавки
К. должен быть крупнее 25 или 40 мм; содержание
мелочи (0—25 мм) не должно превышать 4% (ГОСТ
2014—53). Ситовый состав К. может быть охарактери-
зован по ф-ле:
ь_______(40 - 80)
Г О 80) + (25 - 40)
где (25Ц-‘4о) — процентное содержание класса круп-
ностью 125—40 мм; (40—80) — процентное содержа-
ние класса крупностью 40—80 мм; (> 80) — про-
центное содержание класса крупностью более 80 мм.
Чем выше к, тем равномернее кусковатость К. и тем
более он проницаем для газов доменной печи.
Основными физико-химич. свойствами К. являются
горючесть и реакционная способность, скорость горе-
ния К. (С -j- О2 = СО2) и скорость восстановления
двуокиси углерода (С + СО2 = 2СО). Доменный К.
должен быстро сгорать у фурм, хорошо восстанавли- ,
вать СО2 в СО в средней части шахты печи и с трудом
вступать во взаимодействие при пониженных темп-рах
в верхней части печи (во избежание бесполезного
расхода углерода). В противоположность этому при
горении литейного К. по возможности не должна
образовываться окись углерода (при полном сгорании
выделяется на 3240 ккал/кг больше тепла, чем при
сгорании до окиси углерода). Различия в.реакционной
способности К., резко проявляющиеся при низких
темп-рах, сглаживаются при темп-рах свыше 1000°.
На величину реакционной способности К. влияют:
пористость, свойства исходного угля, состав золы,
темп-ра и продолжительность коксования. Методы
исследования горючести и реакционной способности
К. основаны на непосредственном воздействии кис-
лорода и двуокиси углерода на К.
Лит. см. при с г. Коксование. А. А. Агроскин.
Кокс электродный пековый — богатый углеродом
твердый остаток, получаемый при разложении кам,-
уг. пека; наряду с К. нефтяным К. пековый является
основным сырьем для производства электродов. К.
пековый — очень прочные куски стального (серого)
цвета, истинная плотность 1,95—2,09, уд. электросо-
противление 200—250 ом мм?/м; зольность, являю-
щаяся основной характеристикой сортности этого
вида К., как правило, не превышает 0,3%. Выход
летучих колеблется в пределах 0,6—0,9%; содержа-
ние серы 0,6—0,7% (для заводов Юга) и 0,3—0,5%
(для заводов Востока). Элементарный состав К. пе-
кового (в %): 96,5—97,6 углерода, 0,4—0,5 водорода,
0,5—0,6 серы, 1,0—1,2 кислорода; примерный состав
золы (в %): 30—31 Si О.-, 22—23 ALO3, 24—26 Fe2O3,
3—11 СаО, 2—3 MgO и 12—13 SO3.
Для получения К. электродного пекового высоко-
плавкий кам.-уг. пек (темп-ра размягчения 140—160°)
подвергают коксованию в камерных динасовых печах.
Высокоплавкий пек отличается от среднетемператур-
ного повышенным коксовым остатком (65—67% вме-
сто 50—55%), большим содержанием веществ, не-
растворимых в толуоле (48—55% вместо 18—20%),
и меньшим выходом летучих (52—49% вместо 67—
62%). По темп-рным интервалам коксование пека
в печах распадается на несколько стадий, из к-рых
наиболее важной является интервал 450—550°; в
этом интервале происходит дистилляция легко кипя-
щих фракций, разложение основной массы пека с об-
разованием газообразных и тяжелых продуктов, за-
твердевание утяжеленного остатка и образование
полукокса. При дальнейшем нагревании пека (выше
550°) из него выделяются летучие в-ва (преим. богатые
водородом) и в массе К. появляются усадочные тре-
щины. Процесс заканчивается, когда темп-ра в центре
коксового пирога достигнет 900—1000°, прекращается
усадка и К. отходит от стен камеры. Газо- и парооб-
разные продукты, выделяющиеся при коксовании
пека, охлаждают, смола конденсируется, а газ очи-
щается и поступает в газопровод для дальнейшего ис-
пользования (для обогрева печей и др.). Полученная
пеко-коксовая смола подвергается такой же обработке
воздухом, как и среднетемпературный пек с целью
перевода ее в высокоплавкий пек, используемый для
получения К. электродного пекового. Готовый К.
из камер охлаждается в тушильном вагоне водой и
поступает на коксосортпровку. Существуют и. другие
633
КОКС — КОКСОВАНИЕ
634
менее распространенные способы произ-ва К. электрод-
НОГО пекового. м. А. Степаненко.
Кокс нефтяной — твердый остаток вторичной пере-
работки тяжелых продуктов термин, процессов пере-
работки нефти или нефтепродуктов. Элементарный
состав К. нефтяного (в %): 90—95 углерода, 4—6 во-
дорода, 0,1—2 серы, 0,1—0,8 золы. В Советском
Союзе К. нефтяной вырабатывается следующих ма-
рок: а) К. нефтяной пиролизный электродный —
КНПЭ; б) К. нефтяной крекинговый электродный —
КНКЭ; в) К. нефтяной — КН; г) К. нефтяной пиролиз-
ный специальный — КНПС; д) К. нефтяной крекинго-
вый специальный — КИКС. Основные показатели
качества К. нефтяного различных марок приведены
в таблице.
Показатели КНПЭ I КНКЭ Марка кокса
КН кнпса кнкс | кипе6
Содержание влаги, % (не более) 3,0 3,0 3,0 2,0 2,0 3,0
Содержание золы, % (не более) 0,3 0,8 0,5 0.3 0,5 0.3
Содержание серы, % (не более) 1,0 1,0 1,0 0,8 0,8 0,4
Выход летучих веществ, % (не более) 7,0 7,0 7,0 5,0 5,0 7,0
Содержание окиси кремния, % (не более) 0,07 — — 0,07 0,07 —
Содержание окиси железа, % 0,08 офз 0,08
(не более) — — —
Истинная плотность после про- каливания при 1300° в тече- ние 5 часов;
не менее 2,01 2,08 — 2,04 2,03 2.04
не более — — — — 2.08
Механич. прочность на исти-
раемость, % (не более).... 13,0 8,0 — 13;0 8,0 13,0
Мелочи (менее 25 лм,), % (не 4,0 4.0
более) 4,0 1.0 4,0 4,0
Размер кусков, мм -- — — 25—150 25-150
а По ТУ 470—53; ° по ТУ МГХП 253 — 59
К. нефтяной получают: 1) В металлич. коксовых
кубах; процесс периодический, металлоемкий, обеспе-
чивает высокое качество К. 2) В необогреваемых ка-
мерах (цикличный процесс); сырье для коксования
предварительно нагревают в трубчатой печи до 470—
500° и закачивают в полые металлич. камеры, в к-рых
испаряются керосино-газойлевые фракции. Тяжелые
остатки сырья превращаются в сплошной массив К.,
к-рый охлаждают паром и водой, затем струей воды
под давлением 150 ат К. измельчают и вымывают из
камеры; цикл повторяется. Получаемый при этом К.
содержит значительное количество летучих веществ
и в таком виде непригоден для электродной пром-сти;
используется в шихте с К., получаемым в коксовых
кубах. 3). В камерах с движущимся твердым теплоноси-
телем (непрерывный контактный процесс). Сырье —
гудрон или крекинг-остаток предварительно нагре-
вается до 380° и поступает в ректификационную ко-
лонну, где происходит отгон светлых фракций; одно-
временно внизу колонны сырье смешивается с цирку-
лирующим возвратным тяжелым продуктом коксо-
вания. После колонны смесь сырья и циркулирующего
продукта коксования поступает в печь для нагрева,
а затем в смеситель реактора, смешивается с горячим
теплоносителем — гранулированным К., и поступает
в камеру коксования, где нагревается до 515—520°.
В смесителе из сырого продукта испаряются легкие
части, неиспаряющаяся часть коксуется на поверх-
ности теплоносителя (гранулированного К.). Тепло-
носитель после продувки паром выводится из реак-
тора и делится на товарный К. и возвратный теплоно-
ситель. Теплоноситель, охладившийся в реакторе,
элеватором (эрлифтом) передается в бункер-нагрева-
тель, где теплоноситель нагревается до 560—570°.
Пары и газы, образовавшиеся в реакторе, выводятся
из него и подвергаются ректификации и конденсации.
Жидкие продукты после стабилизации и очистки
используются для приготовления из них различных
товарных продуктов. Весовое отношение основного
теплоносителя (гранулированного К.) к сырью со-
ставляет 11,5:1; время контакта 9 ,и«и.
Лит.: Обрядчиков С. Н., Технология нефти, ч. 2.
3 изд., М.—Л., 1952; Авдеева О. М., Левинтер
М. X., Исследование процесса коксования в толстом слое,
в кн.: Шестая научно-техническая конференция, 1951, М.—Л.,
1952 (Московский нефт. ин-т им. Губкина); Степанен-
ко М. А., Брон Я. А., Кулаков Н. К., Производ-
ство пекового кокса, Харьков, 1961; Красюков А. Ф.
1и др.], Замедленное коксование тяжелых нефтяных остат-
ков. Тр. Башкирок, н.-п. ин-та по переработке нефти, 1959,
вып. 1; Красюков А. Ф., Цинь к о А. В., Изу-
чение механических свойств нефтяных коксов, там же;
Красюков А. Ф., Кудряшова М. С., Электри-
ческие свойства нефтяного кокса, там же, 1960, вып. 3; Кра-
сюк о в А. Ф., Истинная плотность нефтяного кокса, там
же; Красюков А. Ф., [и др.], Некоторые вопросы ме-
ханизма коксования тяжелых нефтяных остатков, там же;
Кунин Н. Ф., Шулепов С. В., ДАН СССР, 1955,
104. № 3. В. П. Калашников.
КОКСОВАНИЕ — метод переработки топлив, пре-
им. углей, заключающийся в нагревании их без до-
ступа воздуха до 900—1050°; топливо при этом раз-
лагается с образованием летучих веществ и твердого
остатка — кокса.
Процесс К. состоит из нескольких стадий: до 250°
испаряется влага и выделяется СО2 и СО; ок. 300°
начинается выделение паров смолы и образование
пирогенетич. влаги. Выше 350° спекающиеся угли
переходят в тестообразное, пластвч. состояние, вяз-
кость массы постепенно уменьшается. Пластич. масса
цементирует угольные зерна, к-рые сращиваются;
ок. 500° пластич. масса начинает бурно разлагаться
с выделением первичных летучих продуктов (газа и
смолы) и затвердевает, образуя твердый продукт —
полукокс. Спекаемость коксуемого угля определяет-
ся вязкостью пластич. массы, давлением выделяющихся
газов, насыпным весом угля, температурными гра-
ницами пластичности и уд. поверхностью зерен. При
нагревании до 700° главную роль играют поликонден-
сациопные процессы; полукокс подвергается дальней-
шему разложению, причем из него выделяется гл.
обр. водород; одновременно массив полукокса дает
усадку. В полукоксе образуются трещины, разделяю-
щие его на куски разной величины. Выше 700° про-
исходит преим. упрочнение кокса. Первичные лету-
чие продукты, соприкасаясь с раскаленным коксом
и нагретыми стенками и сводом печи, подвергаются
пиролизу и превращаются в сложную смесь паров (с
преобладанием соединений ароматич. ряда) и газов,
содержащих водород и метан. Преобладающая часть
серы исходных углей и все минеральные вещества
остаются в коксе. Входящие в состав угля элементы
распределяются в продуктах К., напр., следующим
образом (в весовых процентах от содержания элемента
в исходном угле):
Наименование С н N о S
Уголь 100,00 100,00 100,00 100,00 100,00
Кокс 85,91 6,86 45,00 4,50 60,60
Газ 9,25 63,83 40,19 33,26 ——
Смола 3,28 8,86 4,41 1,11 21,85
Бензол 1,45 1,90 —
Аммиак —— 1,09 10,40 Л
Пирогенетич. вода . . . — 13,64 61,00
Сероводород — 0,64 — — 17,55
Коксуемость углей, т. е. их способность давать
металлургия, кокс, обычно оценивается в, лаборатор-
ных условиях при помощи т. н. пластометрич. метода
i Л. М. Сапожникова (ГОСТ 1186—48). Мерилом кок-
635
КОКСОВАНИЕ — КОКСОХИМИЧЕСКОЕ ПРОИЗВОДСТВО
636
суемости служит толщина пластич. слоя (в л.«),
определяемая в строго регламентированных условиях.
Сырьем для К. служат смеси каменных углей (шихты),
дающие прочный кусковой кокс. Шихты для К. из
донецких углей имеют примерно следующий состав:
20% газовых углей, 40% жирных, 20% коксовых
и 20% отощепных спекающихся. Расширение сырьевой
базы К. осуществляется путем увеличения насыпного
веса шихты; в шихтах используются газовые и другие
слабо спекающиеся угли. При введении углей с высо-
ким выходом летучих веществ избыток последних
компенсируют введением в шихту отощающих доба-
вок (бурый уголь, полукокс, коксовая мелочь, ан-
трацит, железная руда). Более радикально эта задача
решается разработкой новой технологии К., в част-
ности путем предварительного брикетирования угля
пли быстрого нагрева его в первой стадии К. и формо-
вания в пластич. состоянии. Для снижения зольности
и сернистости кокса угли предварительно обога-
щают (если зольность их превышает 7%) на углеобо-
гатительных фабриках. Для обогащения угля при-
меняются гл. обр. гравитационные процессы, основан-
ные на различии в плотности угля и породы, и фло-
тация, основанная на различии в смачиваемости угля
п породы. Угольные смеси для К. составляются из
угольных концентратов и частично из малозольных
углей. Для большей прочности и однородности кокса
смесь углей перед К. измельчают, причем содержа-
ние класса ниже 3 мм составляет 90—95%.
К. проводится в коксовых печах (см. рис.), состоя-
щих из камеры коксования прямоугольного сечелия
(длина 13—14 м, ширина 0,4 м, высота 4,5—5,0 м),
Схема коксовой печи и движения газов в ней:
1 — камеры коксования; 2 — газовоздушные
клапаны; з — кодовые воздухоподводнщие ка-
налы; 4 — регенераторы; 5 — газопроводы кок-
сового газа; в — каналы отопительных простен-
ков; 7 —сборный канал продуктов горения; в —
газопроводы доменного газа.
в к-рую сверху загружается уголь, и отопительных
простенков (в к-рых сжигается газ). На торцах ка-
меры имеются съемные двери для выдачи кокса.
Продолжительность К. составляет 14—16 час. и за-
висит от темп-ры обогревательных простенков (в
среднем .1350—1380°) и ширины камеры К. Для обо-
грева коксовых печей используются доменный, ге-
нераторный или коксовый газы, их смеси либо к.-л.
другие газы. Экономичность работы коксовых печей
повышается путем применения теплообменников-ре-
геператбрОв, в к-рых физич. тепло продуктов го-
рения, выходящих из отопительных простенков,
используется для подогрева воздуха, а в случае
отопления низкокалорийным газом й для подогрева
газа.
Передача тепла в коксовой печи осуществляется в
направлении от стенок камеры к ее середине, поэтому
в каждый данный момент на развых расстояниях от
стенок проходят разные стадии К. Находящиеся
на разных стадиях процесса слои коксующегося угля
передвигаются навстречу друг другу, причем при
слиянии в конце К. слоев кокса в осевой плоскости
камеры образуется шов, к-рый делит массив кокса на
две части. Готовый кокс выдается из печей коксовы-
талкивателем и поступает в тушильный вагон, где
раскаленный кокс тушится водой или инертными
газами. Потушенный кокс сортируется на крупный
кокс (больше 25 или 40 мм), коксовый орешек (25—
10 мм) и коксовую мелочь (10 мм и меньше).
В результате процесса К. получают (в расчете на
тонну угля): 650—750 кг кокса, 310—340 л<3 высоко-
калорийного коксового газа, 3—4% смолы, 1,0—
1,1% сырого бензола, 0,25—0,34% аммиака. Выходы
продуктов К. зависят от состава, свойств исходного
угля и режима процесса К.
Лит.: М е й к с о ц Л. В., Шварц С. А., Производ-
ство кокса, Харьков, 1055; Агроскин А. А., Химия и
технология угля, М., 1961; Т айц Е. М., Свойства камен-
ных углей и процесс образования кокса, М., 1961; Агроскин
А. А., Шелков А. К., Расширение угольной базы коксова-
ния, М., 1962; Дмитриев М. М., Обуховский Я. М.,
Краткий справочник коксохимика, М., i960; Онусайтис
Б. А., Образование и структура каменноугольного кокса,
М„ 1960; Мот Р. А., Уилер Р. В., Качество кокса,
пер. с англ., М., 1947. А. А. Агроскин.
КОКСОХИМИЧЕСКОЕ ПРОИЗВОДСТВО — пере-
работка твердых топлив с целью получения из кок-
сующихся углей металлургия.кокса и химич.продуктов,
образующихся при коксовании. Главными из этих
продуктов являются: газ коксовый — высококалорий-
ное газообразное топливо, а также сырье для химич.
пром-сти; каменноугольная смола, состоящая в основ-
ном из ароматич. соединений, из к-рой выделяют:
нафталин, фенантрен, антрацен, карбазол, метилнаф-
талин, аценафтен, фенолы, пиридиновые- основания,
Хинолин и его гомологи, а также технич. масла (по-
глотительное, шпалопропиточное, газгольдерное и др.)
и пек; сырой бензол — жидкая смесь легкокипящих
ароматич. углеводородов, являющаяся источником
получения бензола, толуола, ксилолов, ароматич.
растворителей (сольвенты) и ряда других соединений;
аммиак, частично растворяющийся в надсмольной
(аммиачной) воде, служащий в основном для полу-
чения азотных удобрений; сероводород, при удале-
нии к-рого из газа (см. Газов очистка) получается
элементарная сера или серная к-та. Кроме этих основ-
ных, могут быть получены такие продукты, как эти-
лен пли простейшие продукты его переработки (ди-
хлорэтан и др.), кумароновая смола и др.
Выход основных химич. продуктов коксования из 1 т
коксового угля: ок. 140 кг (~ 300 н'м3/т) коксового
газа, 30 кг кам.-уг. смолы, 10 кг сырого бензола, 3 кг
аммиака и сероводород (ок. 20% от серы, содержав-
шейся в угле). Приведенные выходы могут колебаться
в определенных пределах в зависимости от характера
угля, подвергавшегося коксованию, и режима про-
цесса. Эти выходы тем выше, чем больше содержание
летучих веществ в исходном угле, а .также чем ближе
темп-ра коксования к нижнему пределу темп-p, ха-
рактерных для процесса.
Летучие продукты коксования отводятся из печей
(см. схему) в виде смеси паров и газов, нагретых
до 700°, в общий газосборник, где они подвергаются
предварительному охлаждению до 80° за счет распы-
ления в нем воды. Газ обогащается водяными парами,
смола (60—70%) конденсируется, из газа удаляется
увлеченная пыль; паро-газовый поток охлаждается
до 25—35° в трубчатых холодильниках, где конден-
637
КОКСОХИМИЧЕСКОЕ ПРОИЗВОДСТВО — КОЛАМИНФОСФАТИДЫ
638
сируются остатки смолы и аммиачная вода. Конден-
сат отстаиванием делится на смоляной и водный слои.
Коксовый газ, освобожденный от смолы и воды, но
содержащий еще пары бензола, аммиак, сероводород
и другие продукты, отсасывается газодувкой и про-
талкивается затем через аппаратуру для улавливания
перечисленных продуктов. После освобождения от
Схема улавливания продуктов коксования: 1 — коксо-
вая печь; 2 — газосборник; 3 — газопровод; 4 — отде-
литель фусов (густых осадков из угольной и коксовой
пыли и тяжелой смолы); 5 — холодильник; 6 — труб-
чатые холодильники; 7 — отстойник конденсата; 8 —
сборник смолы; 9 — сборник аммиачной воды; 10 — га-
зодувка; 11 — электрофильтр; 12 и 13 — аммиачные
колонны; 14 — фенольная установка; 15 — центрифуга
для сульфата; 16 — сатуратор; 17 — каплеотбойник;
18 — конечный холодильник; 19 и 20 — скрубберы;
31 — бензольная колонна; 22 — подогреватель масла;
23 — дефлегматор; 24 — колонна для разделения лег-
кого и тяжелого бензолов; 25 — конденсатор; 26 — се-
паратор; 27 — холодильник; 28 — холодильник масла;
29 — отстойник; 30 — регенератор масла.
них чистый коксовый газ направляется для исполь-
зования в качестве топлива или химического сырья
гл. обр. на азотно-туковые заводы (газ содержит
до 55% водорода).
Методы улавливания из газа несконденсировав-
шихся летучих продуктов коксования зависят от
свойств последних. Аммиак и сероводород улавли-
ваются методами хемосорбции: первый — кислотами,
второй — слабыми щелочами; бензольные продукты
улавливаются методами абсорбции или адсорбции.
Для улавливания этилена может быть использована
его высокая реакционная способность. На нек-рых
новейших предприятиях для интенсификации сорб-
ционных процессов применяются повышенные дав-
ления.
Выработка аммиачных продуктов в К. п. связана
с улавливанием NH3, содержащегося в газе, и отгоном
NH3 из аммиачной воды. Аммиак обычно связывается
серной к-той в барботажных аппаратах, наз. сатура-
торами, и дает удобрение — сульфат аммония, содер-
жащее до 25% NH3. Для получения из него смешанных
удобрений начато использование и фосфорной к-ты;
производятся также жидкие удобрения. Параллельно
с улавливанием NH3 улавливаются легкие фенолы
и пиридиновые основания. Удаление сероводорода,
мешающего использованию газа, производится ме-
тодами мокрой или сухой очистки газов (см. Газов
очистка); особый интерес представляет улавливание
H2S аммиачной водой — продуктом К. п.
Сырой бензол улавливают промывкой газа в аб-
сорберах (скрубберах) маслами (кам.-уг. или нефтя-
ным) с последующей отдувкой бензола острым паром
в отгонных (бензольных) колоннах.
Сырой бензол и кам.-уг. смола поступают для полу-
чения из них товарных продуктов в цехи переработки,
первый — в цех ректификации, вторая — в цех смо-
лоразгонки. Переработка бензольных продуктов со-
стоит в комбинировании процессов их ректификации
и химич. очистки от примесей. Смола нагревается в
трубчатых печах и разгоняется на кам.-уг. масла и
пек. Из масел методами кристаллизации, повторной
перегонки, химич. обработки и очистки кристаллич.
продуктов выделяют различные ароматич. соединения
(гл. обр. нафталин), фенолы и основания. По весу
химич. продукты коксования составляют примерно
четвертую часть от веса угля, пошедшего на коксо-
вание. В ценностном выражении они равны примерно
половине стоимости продуктов К. п.
Лит.: Коляндр Л. Я., Улавливание и переработка
химических продуктов коксования, 2 изд., Харьков, 1962;
Улицкий Л. И., Вопросы экономики коксохимической
промышленности, М., 1960; Караваев Н. М. [и др.], Ма-
шины и аппараты коксохимического производства, М., 1955.
Д. Д. Зыков.
КОЛАМИН —• см. Этаноламины.
КОЛАМИНФОСФАТИДЫ. (этаноламинфосфатиды,
фосфатидилэтаноламины) — сложные эфиры этанол-
амина и диглицеридфосфорных к-т (фосфатидных к-т),
важнейшие представители фосфолипидов. В зависи-
мости от расположения остатка фосфоэтаноламина у
а- или Р-углеродного атома глицерина различают
а- » Р-коламинфосфатиды:
а'
Н2С-О-СОК'
ROC—О^СН
н2с-ох он
“ Уч
о och2ch2nh2
а'
h2c-ocor
HC-Os ОМ
У
О OCH2CM2nh2:
h2c-o-cor
а
а —фосф^идилзтаколаиин ft - фосфзтмдипэййодаиии
R и R*-остатки жирных кислот
Поскольку p-углеродный атом в а-коламинфосфа-
тидах является асимметрическим, возможно сущест-
вование двух стереоизомерных форм а-К. Все природ-
ные К. являются L-а-коламинфосфатидами. К. раз-
личают по входящим в их состав остаткам жирных
к-т. Чаще всего в состав К. входят: из насыщенных
жирных к-т стеариновая и пальмитиновая, из не-
насыщенных — олеиновая, а также полиеновые
С20—С22— жирные к-ты. Обычно природные К. содер-
жат в молекуле по одному остатку насыщенной и нена-
сыщенной к-ты. В большинстве К. ненасыщенная к-та
располагается у а-углеродного атома, а насыщенная —
у Р-углеродного атома остатка глицерина.
К., выделенные из природных источников, — твер-
дые, очень гигроскопичные, бесцветные или слабо-
желтые вещества, быстро темнеющие на воздухе. К.
растворимы в хлороформе, петролейном эфире, серо-
углероде, бензоле, горячей уксусной к-те и спирте
и нерастворимы в ацетоне. С водой К. образуют эмуль-
сию, из к-рой они осаждаются минеральными к-тами,
Ва(ОН)2, Са(ОН)2, CdCl2, PtCl4 и ацетатом свинца.
Свойства и мол. веса нек-рых К., полученных син-
тетически, приведены в таблице (см. стр..,6^Х
К. легко окисляются кислородом и присоединяют
по двойным связям бром. При нагревании в воде
639 КОЛАМИНФОСФАТИДЫ —КОЛИЧЕСТВЕННЫЙ АНАЛИЗ 640
и особенно в слабокислых или слабощелочных рас-
творах К. легко гидролизуются до жирных к-т, гли-
церофосфорной к-ты и этаноламина.
Наименование Мол. вес Т. пл., °C
D, L-1,2-Днстеарил-К. . . . Ь, L-1,2-Дипалыиитолеил-К. D, Ь-1,2-Димиристил-К. . . L>, L-1,2-Дилаурил-К. . . . L-1,2-Дистеарил-К Н~1,2-Дипальмитил-К. . . . Ь-1,2-Димиристил-К 748,01 687,87 635,79 579,67 748,01 691,91 637,79 198 203 207 208 173—174 172,5—175 175-177 -Ь6,0’ -Ь6,4° 4-6,7°
К. могут быть выделены из природных источников,
в частности из мозговой ткани, экстракцией петро-
лепным эфиром с последующим фракционированием
растворителями. Более совершенными способами вы-
деления К. и разделения их на индивидуальные со-
единения являются методы хроматографии в колонке
(на А12О3 или силикагеле) и хроматографии на бумаге.
Синтетически К. могут быть получены, напр., ацили-
рованием а-иодгидрина глицерина, обработкой полу-
ченного продукта серебряной солью фенилфосфорил-
N-карбобензооксиэтаноламина и последующим ката-
литич. гидрированием.
К. широко распространены во всех растительных
и животных тканях, биологич. жидкостях (крови,
молоке, желчи), а также в микроорганизмах. Осо-
бенно богата К. нервная ткань, печень, надпочечники
и семена растений (соевые бобы). Подобно другим фос-
фатидам, К. входят в состав структурных элементов
клеточных мембран и участвуют в регулировании их
проницаемости. Большое значение имеют К. для пе-
реноса в организме жирных к-т и регулирования их
содержания в крови и тканях.
К., подобно другим фосфолипидам, находятся в
организме в динамич. состоянии, подвергаясь постоян-
ным процессам обмена и обновления. Как и в случае
лецитинов, скорость обмена различных частей мо-
лекулы К. различна: в частности, в то время как ос-
таток насыщенной жирной к-ты в p-положении мета-
болически довольно устойчив, остаток ненасыщенной
к-ты в a-положении подвергается обмену со значи-
тельно большей скоростью. Скорость обмена остатка
фосфорной к-ты в молекуле К., так же как у лецити-
нов, является довольно низкой по сравнению со скоро-
стью обмена ее в случае инозитфосфатидов и фосфа-
тидных к-т. К., подобно лецитинам, синтезируются
в организме из соответствующих диглицеридов и фос-
фоэтаиоламина при участии кофермента цитидинтри-
фосфорной к-ты (см. Лецитины). Расщепление К.
происходит при действии специфич. ферментов ле-
цитиназ (фосфолипаз). Образующиеся при этом лизо-
коламипфосфатиды обладают, подобно лизолецитинам,
сильным гемолитич. действием (вызывают разрушение
эритроцитов).
К. иногда называют также фракцию фосфатидов,
впервые выделенных из головного мозга и отличаю-
щихся от лецитинов плохой растворимостью в спирте.
Впоследствии из этой фракции, названной кефа-
линами, наряду с К. были выделены серинфос-
фатиды и инозитфосфатиды. Разделение кефалинов
па отдельные группы фосфатидов может быть достиг-
нуто методами, указанными выше (фракционирование
растворителями и хроматография). Так, при добав-
лении спирта к раствору кефалинов в хлороформе
вначале осаждаются инозитфосфатиды, при повыше-
нии концентрации спирта в осадок выпадают серин-
фосфатиДЫ; К. осаждается из этого р-ра после добав-
ления ацетона.
Лит.: Трусов В. И., Усп. соврем, биол., 1958, 45,
вып. 1, 28; Крепе Е. М., Усп. соврем, биол., 1956,
41, вып. 3, 261; Deuel Н. J., The lipids. Their chemistry
and biochemistry, v. 1—3, N. Y.—L., 1951—57; Malkin
T., Bevan T. H., The synthesis of phospholipids, в кн.:
Progress in the chemistry of fats and other lipids. Ed. by R. T.
Holman, W. O. Lundberg, T. Malkin, v. 4, L.—N. Y., 1957;
Hanahan D. J., Lipide chemistry, N. Y.—L., 1960.
В. Б. Спиричев.
КОЛИМИЦИН — см. Неомицины.
КОЛИЧЕСТВЕННЫЙ АНАЛИЗ — раздел ана-
литической химии, в задачу к-рого входит определе-
ние количества (содержания) элементов (ионов), ра-
дикалов, функциональных групп, соединений или фаз
в анализируемом объекте. В отличие от качественного
анализа, позволяющего установить, из каких химич.
элементов (ионов) или соединений состоит анализи-
руемый материал, К. а. имеет целью установить эле-
ментарный и молекулярный состав исследуемого
объекта или содержание отдельных его компонентов.
В тех случаях, когда неизвестно происхождение п
качественный состав анализируемого материала, коли-
чественному анализу всегда предшествует Качест-
венный. Вообще, уже качественное испытание в ка-
кой-то мере ориентировочно позволяет оценить содер-
жание компонента, напр. по количеству осадка, интен-
сивности окраски.
Различают макро-, полумикро-, микро- и ультра-
микрометоды по количеству вещества (в граммах),
взятого для анализа; подробнее см. Аналитическая
химия. Помимо веса образца, необходимого для вы-
полнения анализа, существенное значение имеет от-
носительное содержание, компонентов в анализиру-
емом материале. Различают главные (основные) ком-
поненты в анализируемом образце (содержание в пре-
делах от 100% до 1%) и неглавные компоненты (при-
меси), содержащиеся в пределах от 1 до 0,01%. Эти
две группы составных частей принято считать макро-
компонентами. Если содержание компонента меньше
0,01%, то его рассматривают как следы вещества
(микросоставную часть). Выбор метода К. а. зависит
от содержания определяемого и сопутствующих ком-
понентов. Микро- и ультрамикрометоды требуют осо-
бенно тщательной работы и специальной техники
эксперимента (см. Микрохимический анализ и Ультра-
микрохимический анализ).
К. а. можно свести к следующим четырем прин-
ципиальным стадиям: 1) отбор и подготовка образца
к анализу; 2) переведение определяемой составной
части в состояние, удобное для измерения; 3) изме-
рение количества компонента; 4) расчет и интерпре-
тация количественных данных. Обычно наиболее
трудная часть аналитиу. процесса — изолирование
определяемой составной части объекта, количество
к-рой затем должно быть измерено. Сам процесс из-
мерения относительно прост. Количественные методы
анализа развиваются в сторону создания, по возмож-
ности, специфич. методов определения с тем, чтобы
избежать илп свести до минимума сложные операции
по отделению. Одно из важнейших требований при
отборе образца для К. а. состоит в том, чтобы проба,
взятая от большого количества исследуемого мате-
риала, точно отражала его средний состав. Определе-
ние количественного содержания компонентов, как
правило, связано с его превращением в форму, при-
годную для последующего измерения. При этом про-
изводят выделение мешающих примесей или же изо-
лирование выделяемого компонента в удобной форме.
В последнем случае используемые методы должны быть
как можно более селективными и чувствительными
относительно определяемого элемента (иона) или
радикала, соединения или класса соединений. Боль-
шое значение имеют методы групповых разделений
и методы выделения определяемых компонентов путем
осаждения (электроосаждения), комплексообразова-
ния, экстракции, хроматографии, перегонки, электро-
фореза и др.; очень полезным является регулирование
641
КОЛЛАГЕН— КОЛЛОДИОННЫЙ ПРОЦЕСС
642
pH среды, изменение валентного состояния элемента
и т. д. В количественных методах аналитич. химии
широко используют разнообразные органич. реагенты
и маскирующие вещества.
В зависимости от объекта исследования различают
неорганич. и органич.. анализ. В свою очередь, неор-
ганич. и органич. анализ разделяют на элементарный
анализ, ставящий своей целью установить, в каком
количестве содержатся элементы (ионы) в анализи-
руемом объекте, и на молекулярный и функциональный
анализы, дающие ответы о колич. содержании ради-
калов, соединений, а также определенных (функцио-
нальных) групп атомов в анализируемом объекте. Для
количественного определения форм нахождения эле-
ментов (простых и сложных веществ) в анализируемых
материалах для определения вещественного состава
исследуемого объекта пользуются методами вещест-
венного (фазового) К. а. При элементарном анализе
органич. соединений анализируемое в-во разлагают
тем или иным способом с получением чаще всего неор-
ганич. соединений (Н2О, СО2, N2 и др.), удобных для
измерений. Для определения функциональных групп
в органич. соединениях используют как характерные
реакции, так и различные специальные методы К. а.
Способ выражения результатов К. а. зависит от
цели, для какой он был выполнен.
Классич. методами К. а. (т. наз. химич. методами
К. а.) являются весовой анализ, основанный на изме-
рении веса продукта реакции, в к-рой участвует оп-
ределяемое вещество, и объемный анализ, заключаю-
щийся в измерении количества реактива, израсходо-
ванного на реакцию с определяемым в-вом (титримет-
рич. анализ), или изменении объема анализируемого
газа после поглощения к.-л. составной части (газо-
вый анализ). Известны и мн. др. методы определения,
основанные на измерении физич. величин, зависящих
от количества вещества. К этим методам относятся
т. наз. физико-химич. и чисто физич. методы анализа.
В их числе электрометрии, методы анализа, напр.
полярография, кондуктометрия, потенциометрия, куло-
нометрия и др.; оптич. методы анализа, напр. колори-
метрия, спектрофотометрия, спектральные методы и
др. (перечень основных методов приведен в ст. Ана-
литическая химия, т. 1, стр. 218). За нек-рыми исклю-
чениями все эти методы являются инструментальными,
т. к. они требуют использования иных измерительных
инструментов, чем весы и бюретки. Подробнее см.
статьи об отдельных методах анализа, а также ст.
Инструментальные методы анализа.
Лит.: Бабко А. К., Пятницкий И. В., Коли-
чественный анализ, М., 1956; Крешков А. П. Основы
аналитической химии, кя. 2, М., 1961; Belcher R., Nut-
ten A. J., Quantitative inorganic analysis, 2 ed., L., 1960;
Treatise of analytical chemistry, pt 1, N. Y., 1959; Szabad-
vSry F., Az analltlkai kemla mddszeretnek klalakuldsa,
Bdspt, 1960; Бобранский Б., Количественный анализ
органических соединений, пер. с польск., М., 1961; Хим.
наука и пром-сть, 1959; 4, № 2; Chariot G., В a d о г -
LamblingJ., Tr emi 1 1 on В., Electrochemical reacti-
ons. The elektrochemical methods of analysis, Amst.—N. Y., 1962.
См. также литературу при ст. Аналитическая химия, Инстру-
ментальные методы анализа, а также при других статьях
о методах анализа. А. 11. Бусев.
КОЛЛАГЕН — фибриллярный белок группы скле-
ропротеинов, составляет основную массу коллагено-
вых волокон соединительной ткани животных. Ха-
рактерными для коллагеновых волокон являются
резкое сокращение при гидротермич. обработке (т. н.
сваривание К.), рентгеноструктурный рефлекс, соот-
ветствующий периоду 2,9 А вдоль оси волокна, и
период поперечной исчерченности ~ 640 А, наблю-
даемый в электронном микроскопе.
Большая часть К. в составе естественных колла-
геновых волокон не растворяется в воде и органич.
растворителях. При длительном нагревании К. в воде
он превращается в желатину. Неуплотненная в зрелые
11 к. X. Э. т. 2
волокна часть К. (т. н. п р о к о л л а г е н) раство-
ряется в кислых р-рах, а также в конц. нейтральных и
щелочных р-рах. Мол. вес проколлагена ок. 0,5 млн.,
длина молекул 3000 А, толщина ок. 15 А, характе-
ристик. вязкость [ц] = 15 г 1 • 100 см3; [a]jj = —350°.
При умеренном нагревании в растворах молекулы
проколлагена распадаются на а- и в-компоненты.
К. выделяют из измельченной ткани (обычно из кожи
животных) путем экстракции кислым буферным р-ром.
Аминокислотный состав К. бычьей кожи
(в г/100 г белка)
Аланин.......10,32
Глицин ........26,57
Валин........ 2,46
Лейцин....... 3,73
Изолейцин .... 1,88
Пролин ........14,42
Фенилаланин . . . 2,35
Тирозин...... 0,99
Триптофан .... 0
Серин........ 4,27
Треонин......... 2,26
Оксипролин .... 12,83
Оксилизин....... 1,00
Метионин........ 0,97
Цистин ......... 0
Аргинин......... 8.22
Гистидин........ 0,70
Лизин........... 3,96
Аспарагиновая к-та 6,95
Глутаминовая к-та 11,16
Иногда в научной литературе неправильно отож-
дествляют коллагеновые волокна и К., что приводит
к ошибочному описанию и пониманию свойств этого
белка.
Лит.: Кендрью Дж., Структурные белки, в кн.;
Белки. Под ред. Г. Нейрата и К. Бэйли, пер. с англ., т. 3,
ч. 2, М., 1959, с. 400—435; Орехович В. Н., Прокол-
лагены, их химический состав, свойства и биологическая роль,
М., 1952; Орехович В. Н., Шпикитер В. О.,
Биохимия, 1958, 23, вып. 2, с. 285—90; Gustavson К. Н.,
The chemistry and reactivity of collagen, N. Y., 1956.
В. О. Шпикитер.
КОЛЛАГЕНАЗА — протеолитич. фермент бакте-
риального происхождения, гидролизующий белки
группы коллагена, к-рые не расщепляются в природ-
ном состоянии другими протеиназами. К. — белок,
мол. в. ок. 100 000; оптимум действия при pH 6,5—
7,8, изоэлектрич. точка при pH 9. К. гидролизует
пептидные связи между теми двумя аминокислотами,
рядом с к-рыми в полипептидной цепи стоят остатки
аминокислот пролина или оксипролина:
прол.-Х—Y—прол.
Такая последовательность аминокислот в поли-
пепти^ных цепях встречается только в белках группы
коллагена. К. продуцируется нек-рыми видами ана-
эробных бактерий рода Clostridium. В животных и
растительных тканях К. не обнаружена. К. приме-
няют для лечения ожогов и удаления рубцов.
Лит.: Кунина О. В., Шпикитер В. О., Усп.
совр. биол., 1960, 50, 3. О. В. Казакова.
КОЛЛИДИНЫ — см. Метилпиридины.
КОЛЛОДИЙ — четырехпроцентный р-р нитроцел-
люлозы в смеси этилового спирта и эфира (1:7). Бес-
цветная или слегка желтоватая, иногда несколько
опалесцирующая, сиропообразная жидкость. При-
меняется в малой хирургии для фиксирования повя-
зок. Для эластичности пленки к К. добавляют 3%
касторового масла. К. огнеопасен.
КОЛЛОДИОННЫЙ ПРОЦЕСС — получение фото-
графии. изображений на коллодионных галогеносереб-
ряных слоях (мокром коллодионе и коллодионных
эмульсиях). В мокром колодиокном процессе свето-
чувствительным веществом служит гл. обр. иодид
серебра, а защитным коллоидом и механич. средой
(носителем) — нитроклетчатка (тетранитроцеллюлоза).
При изготовлении мокрых коллодионных слоев в
2—4%-ный р-р нитроклетчатки в смеси равных объ-
емов спирта и эфира вводят растворимые галогениды
(NH4J, CdJ2, NH4Br); слой такого иодированного
коллодия наносят на стекло. Застудневшцй (но не
сухой) слой сенсибилизируют (погружают его в р-р
AgNO3) для образования в слое иодида серебра. Сырой
светочувствительный слой экспонируют, а затем
643
КОЛЛОИДНАЯ ХИМИЯ
644
проявляют кислым р-ром FeSO4 за счет избыточного
нитрата серебра в слое (физич. проявление):
3AgNOs+3FeSO4—>3Ag+Fe3(SO4)3+Fe(NO3)3
Выделяющееся металлич. серебро осаждается на цен-
трах конденсации, к-рыми служат частицы серебра
скрытого изображения. Фиксирование проявленного
изображения проводят растворением остаточного га-
логенида серебра в р-ре тиосульфата натрия или
цианида калия.
Кроме мокроколлодионных слоев, применяют кол-
лодионные эмульсии, несколько более чувствитель-
ные вследствие того, что светочувствительным ве-
ществом служит в основном бромид серебра. Колло-
дионные эмульсии получают по схеме изготовления
эмульсий для сухих желатиновых бром-иодосеребря-
ных эмульсий. Коллодионные эмульсии лучше воспри-
нимают оптич. сенсибилизацию (эозин, эритрозин),
чем мокрые коллодионные слои. Для коллодионных
эмульсий применяют химич. проявление. Негативы,
полученные на коллодионных слоях, в случае необхо-
димости подвергают обычным процессам усиления и
ослабления.
Коллодионные слои в тысячи раз менее чувстви-
тельны, чем современные желатиновые галогеносереб-
ряные эмульсии, но в связи с высокой разрешающей
способностью и большой кроющей способностью эти
слои длительное время применяли в репродукционной
фотографии. В последнее время в полиграфия, тех-
нике для фотомеханич. процессов коллодионные слои
применяют сравнительно редко.
Лит.: Катушев Я. М., Шеберстов В. И.,
Основы теории фотографических процессов, 2 изд., М., 1954.
В. С. Чельцов.
КОЛЛОИДНАЯ ХИМИЯ — раздел физической хи-
мии, в к-ром рассматриваются процессы образования
и разрушения дисперсных систем, а также их харак-
терные свойства, связанные в основном с поверхност-
ными явлениями на границах раздела фаз в этих сис-
темах. Термин «К. х.» связан с тем, что по традиции
коллоидами называют наиболее высокодисперсные
системы с предельно развитой поверхностью раздела
фаз (коллоидные системы). В современном ее зна-
чении К. х. является физико-химией дисперсных
систем и поверхностных явлений. Особое значение
К. х. определяется тем, что: 1) Природные тела —
горные породы, организмы растений и животных, а
также строительные, конструкционные и др. мате-
риалы техники — являются обычно высокодисперс-
ными, что и определяет многие их особенности, напр.
высокую прочность. 2) Основой многих технологич.
процессов и важнейших процессов в природе служат
образование и разрушение дисперсных систем (сус-
пензий, эмульсий, пен, туманов, дымов и пр.) и свя-
занные с ними процессы диспергирования и конденса-
ционного образования новой фазы, процессы адсорб-
ции, коалесценции, коагуляции и образования про-
странственных структур, определяющиеся взаимодей-
ствием дисперсных частиц — поверхностными яв-
лениями на границе фаз в дисперсных системах.
3) Величиной поверхности раздела определяются ско-
рости всех гетерогенных процессов межфазного массо-
и теплообмена (испарение, растворение, конденсация,
кристаллизация, химич. взаимодействие и разру-
шение, напр. коррозия). Скорости всех этих процессов
являются наибольшими для предельно высокодисперс-
ных (коллоидных) систем.
Исторически развитие физико-химии полимеров и
в особенности их растворов началось в качестве
раздела К. х. В дальнейшем эта область выдели-
лась как самостоятельная глава физико-химической
науки. Типичные высокополимеры и их растворы
Являются однофазными, термодинамически устой-
чивыми системами. Однако в плохих растворителях
(или при их добавлении) растворы полимеров стано-
вятся дисперсными двухфазными системами, как и
сами эти вещества в частично 'закристаллизованном
состоянии. С другой стороны, сами процессы поли-
меризации часто целесообразно проводить в дис-
персных системах, напр. в эмульсиях мономера с
обязательным участием поверхностно-активного мы-
лообразного стабилизатора (т. наз. эмульсионная по-
лимеризация). При этом полимер получается в виде
латекса — коллоидной водной дисперсии (см. Латек-
сы синтетические). Полимеры часто применяются в
изделиях и лакокрасочных покрытиях не в чистом
виде, а Для повышения прочности, улучшения ме-
ханич. свойств смешиваются с активными дисперс-
ными наполнителями (сажей, минеральными напол-
нителями и Др.). При этом полимер используется в виде
связующего в дисперсной системе. Тесное смешение
(объединение) полимеров происходит не только путем
их истинного взаимного растворения, но и путем их
взаимного диспергирования с образованием коллоид-
ной смеси. Таким образом, К. х. как учение о дисперс-
ных, т. е. микрогетерогенных двух- или многофазных
системах тесно соприкасается с физико-химией высо-
комолекулярных соединений.
К. х. — научная основа ряда геологич. процессов —
генезиса горных пород, выветривания, образования
глинистых пород, иловых отложений, седиментаци-
онных процессов, процессов миграции. Выводы К. х.
используются в учении о почвенных структурах и
управлении этими структурами с целью повышения
урожайности. На основе представлений К. х. осу-
ществляют процессы закрепления грунтов, придания
им прочности и водостойкости (в строительном деле,
в строительстве дорог и аэродромов). Теория аэродис-
персных систем (аэрозолей) — один из разделов К. х.,
играет важную роль в современной физике атмосферы
и управлении процессами выпадения осадков, рассе-
яния тумана и др. Живые организмы состоят из
сложных дисперсных систем, образованных высоко-
молекулярными соединениями. Т. обр., К. х. вместе
с физико-химией полимеров составляет основу изу-
чения механизма процессов в живых организмах.
Основные разделы К. х. в ряде случаев можно рас-
сматривать и как самостоятельные области физико-
химической науки. К ним относятся: 1) Молекулярно-
кинетические явления (броуновское движение, диффу-
зия) в дисперсных системах, содержащих частицы
больших размеров, аналогичные молекулам по своему
участию в тепловом движении. Сюда же относится
дисперсионный анализ, гидродинамика дисперсных
систем. 2) Поверхностные явления, включая теорию
адсорбции и адсорбентов, изучение строения и свойств
поверхностных (адсорбционных) слоев, кипетики об-
разования адсорбционных слоев, смачивания, поверх-
ностно-химических процессов в дисперсных системах.
3) Теория образования новой (дисперсной) фазы, воз-
никновения и роста зародышей в метастабильной
среде. Конденсационные методы образования дисперс-
ных систем. 4) Теория устойчивости, коагуляции и
стабилизации различных дисперсных систем, вклю-
чающая строение частиц дисперсной фазы (см. Ми-
целлы). 5) Физико-химическая механика дисперс-
ных систем, включающая теорию механического
диспергирования, образования новых поверхностей
в процессах Деформации и разрушения твердых
тел, влияние понижения поверхностной энергии (в
результате адсорбции) на механические свойства и
дисперсную структуру Деформируемого твердого
тела, явления дисперсного упрочнения. Образование
пространственных структур в дисперсных систе-
мах и механич. свойства таких структур (тиксотроп-
ные коагуляционные структуры, конденсационные и
кристаллизационные структуры), течения структури-
645
КОЛЛОИДНАЯ ХИМИЯ-КОЛЛОИДНЫЕ СИСТЕМЫ
646
рованных систем с различной степенью разрушения
структуры в потоке (реология дисперсных систем).
Этот раздел науки становится основой управления
процессами получения материалов с заданными свой-
ствами и процессами обработки твердых тел. 6) Элек-
трич. свойства поверхностных слоев в дисперсных
системах. Электрокинетич. явления. Строение диф-
фузных двойных слоев ионов на поверхностях раздела
фаз (область, пограничная между К. х. и электрохи-
мией). 7) Оптика дисперсных систем (коллоидная
оптика). Явления рассеяния света в дисперсных систе-
мах. К. х. фотографических процессов — область на
границе между К. х. и фотохимией (процессы образо-
вания и проявления скрытого изображения).
К. х. подразделяется также на ряд областей по
наиболее важным группам дисперсных систем: учение
об эмульсиях и пенах, суспензиях и коллоидных р-рах,
пористых дисперсных телах (адсорбентах, катализа-
торах и их носителях), учение об аэрозолях, К. х.
структурированных систем (гелей), К. х. лиофильных
коллоидов — полуколлоидов типа мыл и их растворов.
Очень велико значение современной К. х. в ряде наи-
более актуальных отраслей техники, где К. х. служит
научной основой важнейших технологич. процессов.
Таковы: технология строительных материалов и си-
ликатов (керамич. производств), особенно огнеупоров
и тонкой керамики для новой техники; технология
переработки полимеров и особенно произ-ва пластмасс
и резин с активными, всегда высокодисперсными
наполнителями; лаков и красок, а также лакокрасоч-
ных (полимерных) защитных покрытий с использовани-
ем пигментов, служащих активными наполнителями в
качестве дисперсной фазы; технология различных
процессов разрушения твердых тел и в особенности
их тонкого измельчения, а также процессов бурения
горных пород, включая и реологию тиксотропно-
структурированных промывочных жидкостей (диспер-
сий), процессов шлифовки и полировки; технология
процессов обогащения полезных ископаемых, их от-
деления в дисперсном состоянии от пустой породы,
особенно методами флотации; технология обработки
волокон и тканей, процессы моющего действия, кра-
шения и полиграфия, процессов печатания; произ-во
бумаги; почти все области пищевой пром-сти.
Такие разделы К. х., как учение об образовании и
разрушении устойчивых пен и эмульсий, о процессах
смачивания и моющего действия, приобретают все
большее значение почти во всех областях народного
хозяйства. Разрушение высокодисперсных эмульсий
воды в сырых нефтях — основной прием их обезво-
живания и обессоливания, столь важного для разви-
тия нефтяной пром-сти. Образование высокоустой-
чивых пен является самым эффективным средством
огнетушения, особенно при борьбе с нефтяными
пожарами; пеногашение, т. е. разрушение устойчивых
пен, необходимо в котлах высокого давления и в ряде
произ-в, где возникновение стойких пен может вы-
звать аварии. С.-х. яды для борьбы с вредителями ра-
стений обычно применяются в виде дисперсий с до-
бавками поверхностно-активных веществ — смачива-
телей. Направленная кристаллизация, напр. метал-
лов и сплавов из жидкой фазы, а также при спекании
высокодисперсных масс или при термообработке
стекол, приводит к образованию высокопрочных тонко-
дисперсных конструкционных материалов.
Особенности поверхностных явлений и высокодис-
персных систем служат причиной своеобразных яв-
лений и процессов, изучаемых К. х. Таковы проявле-
ния термодинамической (агрегативной) неустойчивости
дисперсных систем (коагуляция и коалесценция),
самопроизвольного разрушения жидких пленок и
пен, явления тиксотропного коагуляционного струк-
турообразования, влияние моаомолекулярных ад-
сорбционных слоев на свойства дисперсных систем
и взаимодействия соприкасающихся тел (адгезию,
трение). Эти специфич. особенности вызвали появ-
ление ряда новых методов исследования, характери-
зовавших развитие К. х. как самостоятельной области
науки. Таковы: методы разделения и фракциониро-
вания дисперсных систем — ультрацентрифугирова-
ние, ультрафильтрация; диализ и электродиализ, элек-
троосмос и электрофорез, оптич. методы исследова-
ния — нефелометрия и ультрамикроскопия, электрон-
ная микроскопия, применение рентгенография, и элек-
тронография. анализа для определения дисперсности.
Дисперсное и в пределе коллоидно-дисперсное
состояние является вполне универсальным состоя-
нием, в к-рое в подходящих условиях может быть
переведено любое вещество в двух- или многофазной
системе. История, значение К. х. для развития ес-
тествознания и современных представлений о молеку-
лярном строении вещества весьма велико. Благодаря
развитию К. х. и ее методов исследования оказалась
возможной экспериментальная проверка выводов
молекулярной статистики — теории флуктуаций и
броуновского движения, что привело к определению
числа Авогадро и к доказательствам реальности
существования молекул.
Лит..- Песков Н. П., Физико-химические основы
коллоидной науки, 2 изд., М.—Л., 1934; Жуков И. И.,
Коллоидная химия, ч. 1, Л., 1949; Думанский А. В.,
Учение о коллоидах, 3 изд., М.—Л., 1948; Наумов В. А.,
Химия коллоидов, 3 изд., Л., 1932; Ребиядер П. А.,
Конспект общего курса коллоидной химии, [сост. К. А. Пос-
пелова], 2 изд., М., 1950; Песков Н. П., Алексан-
дров а-П рейс Е. М., Курс коллоидной химии, 2 изд.,
М.—Л., 1948; Сведберг Т., Коллоидная химия, пер.
с англ., 2 иад., М., [1930]; Freundlich Н., КарШаг-
chemle, Bd 1—2, 4 Aufl., Lpz., 1930—32; Colloid chemistry.
Theoretical and applied, collected and ed. by J. Alexander,
v. 1—6, N. Y., 1926—46; Alexander J., Colloid chemistry.
Principles and applications, 4 ed., N. Y., [1945]; Colloid
science, ed. by H. R. Kruyt, v. 1—2, N. Y. — [a. o.]. 1949—1952;
Наука о коллоидах, пер. с англ., т. 1, М., 1955; Шелудко
А., Коллоидная химия, пер. с болг., М., 1960; Руцкой
А. П., Краткий курс коллоидной химии, Л., 1958; Пасын-
ский А. Г., Коллоидная химия, М., i959.
И. А. Ребиндср.
КОЛЛОИДНЫЕ СИСТЕМЫ (коллоиды, коллоид-
ные растворы) — дисперсные системы с предельно
высокой дисперсностью при условии сохранения ге-
терогенности, т. е. поверхности раздела между дис-
персной фазой и дисперсионной средой. Термин
«коллоиды» (клееподобные тела), введенный Грэмом
в 1861, давно устарел и лишь для краткости и по
история, традиции применяется вместо правильного
современного научного термина «предельно высоко-
дисперсные микрогетерогенные системы» (ультрамик-
рогетерогенные системы). Основные своеобразные
свойства К. с. определяются не только дисперсно-
стью, но и характером физико-химич. взаимодействия
обеих фаз (1) и (2) системы на их общей границе раз-
дела, что определяет по величине уд. свободной по-
верхностной энергии <т12 на границе раздела фаз боль-
шую или меньшую лиофильность К. с. Вследствие
наличия избытка свободной энергии в виде поверхност-
ной энергии К. с. при не очень малых'<т12 обычно яв-
ляются термодинамически неустойчивыми системами,
самопроизвольно разрушающимися в процессах агре-
гирования частиц (коагуляции и коалесценции), со-
провождающихся уменьшением свободной энергии.
Препятствием для протекания таких процессов служат
защитные стабилизующие оболочки на поверхности
частиц или отверждение К. с. в целом, напр., когда
частицы дисперсной фазы связаны молекулярными
силами в пространственную сетку геля. Только при
очень низких значениях <т12 (а;2-<0,1 эрг/см2) при
обычной темп-ре К. с. становятся лиофильными или
термодинамически устойчивыми (самопроизвольно об-
разующиеся эмульсии, суспензии, см. также Семи-
коллоиды).
647
КОЛЛОКСИЛИН — КОЛОРИМЕТРИЧЕСКИЙ АНАЛИЗ
648
К. с. получают двумя способами. Первый, наиболее 1
универсальный, — конденсация (начальная стадия об-
разования новой фазы из пересыщенного р-ра, пара
или переохлажденной жидкости). Пересыщение в
растворе может быть достигнуто в результате химич.
реакции с образованием малорастворимого вещества,
напр. при образовании нерастворимых в воде солей,
используемых в количественном анализе (сульфат
бария, галогениды серебра). К. с. образуются при
этом, если рост возникающих зародышей новой фазы
тормозится уже на начальной стадии, например при
возникновении большого числа зародышей в едини-
це объема с последующим резким падением пере-
сыщения или при введении добавок адсорбирующихся
веществ, замедляющих рост зародышей. Второй —
диспергирование, обычно механич. разрушение — тон-
кое измельчение твердых тел или распыление жидкости.
Диспергирование обычно ведет к образованию гру-
бодисперсных систем и может привести к получению
высокой дисперсности только в достаточно лиофиль-
ных К. с., переходных к самопроизвольно образую-
щимся (наир., диспергирование бентонитовых глин
в воде).
К. с. имеют ряд характерных свойств: механи-
ческих, выражающихся в возникновении про-
странственных структур (см. Структурообразование в
дисперсных системах) и в явлении тиксотропии; элек-
трических, связанных с возникновением заряда
на поверхности раздела — образованием двойного
электрич. слоя ионов (см. Электрокинетические яв-
ления); оптических — рассеяние света (см. Не-
фелометрия и турбидиметрия, Оптические свойства
коллоидных систем), двойное лучепреломление в по-
токе вследствие ориентации анизометричных частиц.
Лит. см. при ст. Коллоидная химия. П. А. Ребиндер.
КОЛЛОКСИЛИН — один из видов промышленных
нитратов целлюлозы (см. Нитроцеллюлоза) с содер-
жанием нитратного азота до 12%. К. применяют в
произ-ве порохов и взрывчатых веществ, а также
нек-рых лаков и пленок.
КОЛОРИМЕТРИЧЕСКИЙ АНАЛИЗ — визуаль-
ный метод фотометрия, анализа, основанный на пе-
реведении определяемого компонента в окрашенное
соединение и установлении концентрации окрашенно-
го соединения по интенсивности или оттенку окраски.
В первой ступени имеют значение те же факторы, что
и в фотометрич. анализе вообще: выбор реактива, ус-
ловий реакции для возможно более полного переве-
дения определяемого компонента в окрашенное со-
единение с определенными спектрофотометрии, ха-
рактеристиками, а также устранение влияния мешаю-
щих компонентов. Во второй стадии применяется ряд
приемов, основанных на сравнении цвета и интенсив-
ности окраски испытуемого и стандартных р-ров с из-
вестной концентрацией. При всех приемах К. а.
необходимо получить одинаковую окраску обоих рас-
творов; при этом достигается равное количество по-
глощающих свет центров в поле зрения наблюдателя
в Данных условиях.
Во всех методах К. а. используют различные окра-
шенные соединения. Наиболее важны следующие
типы этих соединений: 1) Окрашенные галогенидные
и роданидные комплексы — для определения Fe, Со,
V, Мо, W, Bi, UO2 и др. Вследствие ступенчатого ком-
плексообразования необходимо строгое соблюдение
одинаковой концентрации реактивов в испытуемом
и стандартном растворах. 2) Комплексы металлов
с перекисью водорода — для определения Ti,UO2, V
и др. 3) Аммиачные комплексы — для определения Си
и Ni. 4) Гетерополикислоты — Для определения Р,
Si и As, а также W, Ti, Nb и др. 5) Комплексы основ-
ных красителей с галогенокислотами металлов —
для определения Sb в виде соли метилового фиолето-
вого (основание) с ЗЬС1ё; Для определения Та в виде
соли метилового фиолетового с H2TaF7, трехвалент-
ных TI, In, Ga и т. п. Комплексы оснований с ацидо-
кислотами металлов применяют для определения Си
в виде пиридин-роданидного комплекса, Ti в виде
диантипирилметан-роданидного комплекса и др. Обе
последние группы обычно применяют в К. а.
после экстрагирования окрашенных комплексов
органич. растворителями. 6) Комплексы металлов с
органич. реагентами — как, напр., ализарин, различ-
ные металлохромные индикаторы, нитрозонафтол,
дитизон и др. 7) Окислительно-восстановительные
реакции, напр. определение Мп в виде перманга-
ната, и др. 8) Для определения анионов применя-
ют часто реакции обесцвечивания ими окрашенных
комплексов металлов; так, для определения фто-
рид-ионов измеряют ослабление окраски растворов
перекисного комплекса титана вследствие связывания
титана во фторидный комплекс. 9) Применяют также
реакции синтеза; напр., нитриты определяют путем
измерения окраски азокрасителя, синтезированного
в определенных условиях с помощью измеряемого
нитрита. Кроме обычных типов реакций комплексо-
образования, окисления-восстановления и синтеза,
используются также каталитич. реакции, в к-рых
определяемый компонент является катализатором
реакции с образованием или исчезновением окрашен-
ного соединения.
Наиболее часто применяют следующие приемы сравнения
окраски. Стандартная шкала (стандартная серия).
Заранее или одновременно с испытуемым р-ром готовят серию
растворов с различным содержанием определяемого компо-
нента. Все растворы получают при одинаковых условиях,
добавляя необходимые реактивы и разбавляя до одинакового
объема, после зтого испытуемый р-р сравнивают с серией
стандартных р-ров. Совпадение окраски испытуемого р-ра
с одним из р-ров стандартной серии означает равенство их
концентраций относительно определяемого компонента. При-
нимая во внимание аликвотную часть исходного испытуемого
р-ра, вычисляют содержание определяемого компонента.
Этим путем определяют концентрацию различных катионов
металлов, анионов и т. п., а также pH раствора; в последнем
случае в качестве стандартного р-ра используют буферные
р-ры с определенным значением pH. Метод стандартной шкалы
применяется широко: он неприменим, когда окрашенное ве-
щество неустойчиво во времени. В таких случаях готовят ис-
кусственные или имитирующие стандартные р-ры. Так, при
определении кремнекислоты в виде желтого кремнемолибде-
нового комплекса берут в качестве стандартного р-ра легко
приготовляемый р-р хромата калия определенной концентра-
ции. Аналогичный прием используется при определении pH
без буферных р-ров с применением одноцветных индикаторов
типа нитрофенолов, к-рые в молекулярной форме бесцветны,
а в ионной — окрашены (см. Индикаторы).
В нек-рых случаях необходимо определять концентрацию
нек-рого компонента (или pH раствора) в таких испытуемых
р-рах, к-рые имеют собственную окраску. Для зтой цели приме-
няют компаратор (рис. 1) — коробка с шестью отверстия-
ми — гнездами для пробирок. В гнез-
до 2 ставят испытуемый р-р, в гнездо
5 — воду (растворитель). При рас-
смотрении в направлении стрелок
наблюдают суммарный оптич. эффект
от окраски определяемого компонента
и окраски примеси в испытуемом р-ре.
В гнезда 4 и 6 устанавливают про-
бирки с исходным испытуемым р-ром,
в к-ром определяемый компонент не
переведен в окрашенное соединение.
В гнезда 1 и 3 попеременно устанав-
ливают различные пробирки из шка-
лы стандартных р-ров. Рассматривая
в направлении стрелок, наблюдают
суммарный эффект от окраски опре-
деляемого компонента и окраски примеси, т. к. в пробирках
(44-1) и (64-3) находятся соответствующие компоненты. Сов-
падение окрасок одной из пар пробирок с окраской проби-
рок (54-2) означает равенство концентрации определяемого
вещества.
Этот прием используется, напр., при определении мар-
ганца в сталях, т. к. раствор последней обычно имеет соб-
ственную окраску от коллоидных частиц углеродистых соеди-
нений, основных солей железа и др. Аналогичный прием при-
меняют при определении pH почвенных вытяжек или др. слабо
окрашенных р-ров. Все основные приемы метода стандартной
шкалы широко используют такжевдругих методах фотометрии,
напр. для приготовления калибровочной кривой в спектро-
фотометрии и др.
/ 2 3
Рис. 1-
649
КОЛОРИМЕТРИЯ - КОЛХИЦИН
650
Разбавление. Необходимо иметь 2 пробирки или
2 цилиндра, один из них с испытуемым р-ром, а другой — со
стандартным. Пользуясь тем или другим приспособлением, рас-
сматривают пробирки только в отраженном свете через окна
коробки или соответствующего оптич. устройства. Раствор,
окрашенный более сильно, постепенно разбавляют до тех пор,
пока окраски станут одинаковыми. Учитывая степень разбав-
ления, можно вычислить концентрацию определяемого компо-
нента в испытуемом р-ре. Метод разбавления менее точен, чем
предыдущий, т. к. при разбавлении нередко изменяется равно-
весие между определяемым компонентом, реактивом и окра-
шенным соединением, что нарушает пропорциональность между
общей концентрацией определяемого компонента и окраской.
Дублирование (колориметрическое
титрование). Пользуются устройством, в к-ром находится
два цилиндра — с испытуемым и стандартным р-рами. В один
цилиндр вносят испытуемый р-р после переведения определя-
емого компонента в окрашенное соединение и разбавления
до определенного объема. В другой цилиндр вносят все необ-
ходимые реактивы, а затем постепенно прибавляют из бюретки
неокрашенный стандартный р-р, представляющий раствор
определяемого компонента без реактивов. Достижение одина-
ковой окраски в обоих цилиндрах (при одинаковых объемах)
означает, что в цилиндре с испытуемым р-ром содержится
столько же определяемого компонента, сколько его прибавлено
во второй цилиндр. Метод колориметрич. титрования не тре-
бует приготовления серии стандартных р-ров и в этом отно-
шении он проще, чем первый метод; этот метод применим также
в случае образования окрашенных соединений, для к-рых
шкала недостаточно устойчива во времени. С другой стороны,
дублирование в указанной простой форме неприменимо, если
окрашенное соединение образуется медленно или необходимо
нагревание и т. п. В этих случаях иногда применяют другой
вариант, при к-ром в качестве стандартного р-ра пользуются
приготовленным заранее р-ром окрашенного соединения или
имитирующим р-ром. Так, при определении марганца исход-
ный испытуемый р-р обрабатывают персульфатом в присут-
ствии катализатора, нагревают до полного превращения мар-
ганца в перманганат, разбавляют до определенного объема и
переносят в цилиндр для испытуемого р-ра. В цилиндр для стан-
дартного р-ра постепенно прибавляют из бюретки р-р перманга-
ната известной концентрации до получения одинаковой окрас-
ки. В случае окрашенных р-ров пользуются компаратором.
Колориметры погружения. Эти приборы
позволяют уравнивать окраски двух растворов изменением
толщины слоя одного из них. Сравниваемые-растворы нали-
вают в цилиндры I и 2 с прозрачным дном (рис. 2). В эти ци-
линдры погружают два других стеклянных цилиндра 3 и 4.
Цилиндры 3 и 4 закреплены, цилиндры 1
и 2 могут передвигаться в вертикальном
направлении (в нек-рых приборах, наобо-
рот, передвигаются цилиндры з и 4). По-
ток света и^Ьт снизу и поступает в глаз
наблюдателя; над цилиндрами устанавли-
вают оптич. устройство, которое сближает
два световых потока и позволяет наблю-
дать их в виде двух половин круга, разде-
ленных линией. Если растворы окрашены
неодинаково, поднимают с помощью зуб-
чатки, снабженной нониусом, цилиндр с
раствором, окрашенным более интенсив-
но. Т. обр., толщина слоя этого раствора
уменьшается и можно достичь одинаковой
окраски обоих половин в поле зрения
объектива. Толщины слоев /г, и hz при этом обратно пропор-
циональны концентрациям л С2 (закон Бэра). Отсюда, если
известна концентрация одного из растворов, можно вычис-
лить концентрацию второго.
Диафрагмы (приборы типа фотометра
П у л ь ф р и х а). Метод занимает промежуточное положение
между обычной колориметрией и спектрофотометрией, т. к.
дает возможность не только сравнивать два раствора с различ-
ной окраской, но и устанавливать степень поглощения света
окрашенным раствором по отношению к растворителю. При-
бор снабжен светофильтрами с узкими полосами пропускания
отдельных участков спектра. В результате наблюдатель видит
в поле зрения не цвет испытуемого р-ра, а цвет светофильтра.
Уравнивание световых потоков достигается изменением отвер-
стия диафрагмы. Поэтому в один из цилиндров можно поместить
растворитель, в другой — испытуемый р-р; тогда ноле зрения,
соответствующее растворителю, будет более светлым. Умень-
шают отверстие измерительной диафрагмы над цилиндром
с растворителем до уравнивания освещенности полей. Отсчет
по диафрагме позволяет рассчитать степень поглощения света
в данном участке спектра, обусловленную испытуемым р-ром.
Нет необходимости каждый раз готовить стандартные р-ры;
последние готовят только один раз, как в методе стандартных
шкал, составляют калибровочную кривую при данном свето-
фильтре и толщине слоя, после чего определения выполняются
без стандартного р-ра.
Лит.: Бабко А. К., Пилипенко А. Т., Колори-
метрический анализ, М.—Л., 1951; Сен дел Е. Б., Коло-
риметрическое определение следов металлов, М.—Л., 1949;
Юинг Г. В., Инструментальные методы химического ана-
лиза, пер. с англ., М., 1960. А. К. Бабко.
КОЛОРИМЕТРИЯ — метод анализа, основанный
на определении концентрации вещества по интен-
сивности окраски р-ров. Интенсивность светового
потока уменьшается по сравнению с первоначальной
в зависимости от толщины слоя раствора, степени его
окраски и концентрации (см. Поглощение света).
Определение интенсивности окраски производят либо
визуально (см. Колориметрический анализ), напр.
методом стандартных шкал и др., либо инструмен-
тальными методами (см. Спектрофотометрия).
Лит. см. при ст. Колоргшетпрический анализ.
КОЛХАМИН С21И2БО5Х, мол. вес 371,34 — алкало-
ид, выделенный из луковиц безвременника велико-
лепного (Colhicum Speciosum .. —
stev.) и безвременника осей- CHgO-ri^^yy-NHCH3
него (Colhicum Autumnale L.)
семейства лилейных (Liliace- 1 ]
ae); кристаллы; т. пл. 181— 0СН3.|1^ J=o •
182° (из этилацетата); легко
растворим в хлороформе, мета- осн3
ноле и спирте, труднее—в ацетоне, нерастворим в эфире.
Дает хлоргидрат, т. пл. 216—217° (с разл.), и перхло-
рат, т. пл. 264° (с разл.). Вторичное основание, близко
по строению к колхицину (является дезацетилметилкол-
хицином). Физиология, действие К. то же, что у кол-
хицина, но К. в 7—8 раз менее токсичен. К. — силь-
ное антимитотич. средство. К. задерживает развитие
злокачественной ткани; при нанесении на пораженную
раком кожу вызывает распад злокачественных клеток.
К. также угнетает лейко- и лимфопоэз. В медицине
применяется в виде 0,5%-ной мази для лечения рака
КОЖИ. А. Е. Васильев.
КОЛХИЦИН C22H26OeN, мол. в. 399,45 — алкалоид
безвременника (Colhicum) семейства лилиевых (Li-
liaceae), найден также в других родах лилиевых;
светло-желтое аморфное вещество; т. пл. 143—147°;
дает хорошо кристаллизующиеся сольваты: с водой
(1,5 Н2О), бензолом (СвН6), т. пл. 140°, этилацета-
том (СН3СООС2Н6), т. пл. 155—157°; [а]у5 = —120,8°
(СНС13) и —429° (Н2О). Легко растворим в водном
спирте и хлороформе, трудно — в воде и бензоле.
Чистый К. получают хроматографированием. При
УФ-облучении К. изомеризуется в люмиколхицин,
т. пл. 220°.
К. (I) — слабое основание (азот в амидвой форме),
обычно солей с кислотами не дает и экстрагируется
из кислых р-ров. Один из немногих алкалоидов, со-
держащих нециклич. азот и трополоновое кольцо
(С). При перегонке К. с Zn-пылью происходит изоме-
ризация углеродного скелета и образуется 9-метил-
фенантрен. Действие на К. иода в присутствии щелочи
приводит к N-ацетилиодколхинолу, к-рый при вос-
становительном удалении иода и обработке спирто-
вым р-ром НС1 дает колхинол (П). Строение К. под-
тверждено синтезом.
К. весьма токсичен, действует даже через кожу.
Вызывает возбуждение, а затем паралич центральной
нервной системы. Ранее применялся при лечении по-
дагры. К. обладает значительной антимитотич. актив-
ностью, являясь кариокластич. ядом. Одновременно
угнетает лейко- и лимфопоэз. Однако избирательность
К. относительно злокачественных клеток невелика.
Как средство для лечения новообразований в меди-
цине используется близкий к К. алкалоид колхамин.
К. вызывает полиплодию у растений и применяет-
ся для этой цели.
Лит. см. при ст. Алкалоиды, а также: Schreiber J.,
[а. о.], Angew. Chem., 1959, 71, № 20, 637. А. Е. Васильев.
651
КОЛЬБЕ ЭЛЕКТРОХИМИЧЕСКИЙ СИНТЕЗ-КОЛЬРАУША ЗАКОН
652
КОЛЬБЕ ЭЛЕКТРОХИМИЧЕСКИЙ СИНТЕЗ —
реакция образования углеводородов и нек-рых их
производных, оспованная на рекомбинации углево-
дородных радикалов, образующихся при электролизе
растворов солей карбоновых к-т. Предполагается три
последовательных стадии процесса: происходящая
на аноде разрядка аниона к-ты с образованием кар-
боксил-радикала (1); элиминирование СО2 из кар-
боксил-радика ла с образованием углеводородного
радикала (2); рекомбинация углеводородных радика-
лов (3):
rcoo-—— RCOO- (1)
rcoo--~c°2.r- (2)
2R----R-R (3)
К. э. с. обычно осуществляют пропусканием посто-
янного тока через насыщенный водный или метаноль-
ный р-р соли щелочных металлов соответствующей
к-ты. Электролиз ведут в широком интервале плот-
ностей тока при 20—50° с применением платинового
анода. Материал катода на характер процесса су-
щественно не влияет (обычно платина или ртуть).
Применение К. а. с. к смесям карбоновых к-т позволило
наряду с симметрии, получать также и несимметрич. углево-
дороды:
RC О О Na+R'C О О Na элект.Р°л?3R-R4-R'-R'4-R-R'
Наличие в углеводородной цепи заместителей, удаленных
от карбоксильной группы, не препятствует К. э. с. Так, элек-
тролиз солей кислых эфиров алифатич. дикарбоновых к-т
приводит к полным эфирам высших дикарбоновых к-тз
2ROOC(CHa)nCOONa^S2-°^ROOC(CHa)2nCOOR
Смеси солей монокарбоновых к-т с солями кислых эфиров
дикарбоновых к-т в условиях К. э. с. образуют сложные
эфиры высших монокарбоновых к-тз
RCOONa+R'COO(CHa)nCOONa?5^2255S?R(CHa)nCOOR'
Наличие алкильного заместителя в a-положении к карбоксиль-
ной группе снижает выход в К. э. с. с 50—90% до 10%. К а-га-
логено-,а- окси-,а-алкокси- и а-цианкарбоновым к-там К. э. с.
неприменим. Неудачными оказались попытки применить К. э. с.
к кислотам, содержащим карбоксильную группу в ароматич.
ядре, а также к а, 0- и 0,у-ненасыщенным н-там этиленового
и ацетиленового рядов.
При электролизе алифатич. к-т с кратной связью, удаленной
от карбоксильной группы, образуются ненасыщенные соеди-
нения с изолированными кратными связями:
C2H5OOCCH=C(CH3)CH2CH2COONa^^^~
—[С2Н5ООССН=С(СН8)СН2СНа—]а
Применение К. э. с. к оптически активным карбоновым к-там
с асимметрии, атомом углерода в a-положении к карбоксильной
группе приводит к получению неактивных углеводородов. Кар-
боновые к-ты, содержащие асимметрия, центр, удаленный от
карбоксильной группы, в условиях К. э. с. образуют оптически
активные углеводороды. К. э. с. открыт А. Кольбе в 1849.
Лит.: Quart. Revs, 1952, в, М 4, 380; Advances in organic
chemistry, v. 1, N. Y., 1960. Л. С. Герман.
КОЛЬБЕ —ШМИДТА РЕАКЦИЯ — метод синтеза
ароматич. или гетероциклич. оксикислот, заключаю-
щийся в действии СОа на щелочную соль соответствую-
щего оксисоединения:
Ar-ONa—^Ar<f°H
'COONa
К.— Ш.р: осуществляется посредством электрофильной
атаки СО2 на активированные орто- ti пара-положения
фенолят-иона, причем возможность образования про-
межуточного 6-членного реакционного комплекса спо-
собствует вступлению карбоксильной группы преим.
в орто-положение к оксигруппе:
Заместители 1-го рода в ароматич. ядре (особенно в
.мета-положении к оксигруппе) облегчают, а замести-
тели 2-го рода затрудняют либо вообще ингибируют
К. —Ш. р.
Природа щелочного катиона заметно влияет на
характер образующихся продуктов. Так, если фено-
лят натрия в К.— Ш. р. образует салициловую кислоту,
то карбонизация фенолята калия в тех же условиях
дает смесь салициловой и n-оксибензойной к-т. Ярко
выражено влияние катиона на направление реакции
в ряду производных нафталина и хинолина:
COONa
Предполагается, что в нек-рых случаях образования
n-оксикарбоновых к-т из калиевых фенолятов происхо-
дит изомеризация промежуточно образующейся о-окси-
кислоты (особенно при высокой темп-ре). К.—Ш. р.
осуществляют либо действием СО2 на сухие щелоч-
ные соли соответствующих фенолов при 120—130°
и 80—90° (вариант Шмидта), либо нагре-
ванием под давлением смеси фенола с избытком без-
водного К2СО3 (метод М а р ас се). Присутствие
воды в реакционной среде в общем случае нежелатель-
но, однако карбонизация оксисоединений, содержа-
щих заместители первого рода в лета-положении к
оксигруппе, может быть произведена действием вод-
ного р-ра бикарбоната щелочного металла:
К.— Ш. р. применяют в пром-сти как основной метод
получения салициловой к-ты из фенола; п-аминоса-
лициловой к-ты (ПАСК) из л-аминофенола, а также
0-оксинафтойной к-ты из 0-нафтола. К.— Ш. р. была
открыта А. Кольбе в 1860 и усовершенствована
Р. Шмидтом в 1884.
Лит.: Lindsey A., Jeskey Н., Chem. Revs.,
1957, 57, JsS 4, 583; Шилов Е. А. [и др.], Укр. хим. ж.,
1055, 21, вып. 4, с. 484. Л. С. Герман.
КОЛЬРАУША ЗАКОН — закон независимости дви-
жения ионов, или закон аддитивности электропро-
водности при бесконечном разведении р-ров электро-
литов. Был установлен экспериментально Ф. Коль-
раушем в 1879, т. е. еще до возникновения теории
электролитической диссоциации, но объяснение полу-
чил позднее, на ее основе. По этой теории причиной
электропроводности р-ров электролитов служит дви-
жение ионов растворенного вещества в электрич. поле.
Согласно К. з., эквивалентная электропроводность
электролита при бесконечном разведении, Асо, склады-
вается из электропроводности, обусловленной дви-
жением катионов, Х+, и электропроводности, обуслов-
ленной движением анионов, Х_, т. е. Асо = Х+4~ Х_,
причем величины Х+ и являются характерными
постоянными (при данной темп-ре) соответствующих
ионов. Их наз. ионными электропроводностями, или
подвижностями ионов в бесконечно разбавленных р-рах,
и значения их приводятся обычно в справочных таб-
лицах. Подвижности ионов определяют путем изме-
рений электропроводности р-ров различных концен-
траций и параллельных измерений чисел переноса
ионов; экстраполяцией величин, измеренных при ко-
нечных концентрациях, находят подвижности ионов
при бесконечном разведении. Числа переноса опре-
деляют долю участия катионов и анионов в общем
653
КОМАРОВСКОГО РЕАКЦИЯ - КОМБИНАЦИОННОЕ РАССЕЯНИЕ СВЕТА
654
переносе электричества через р-р. К. з. применим лишь
для разбавленных р-ров. С увеличением концентрации
увеличивается межионное притяжение и начинает
сказываться изменение степени сольватации ионов
и степени ионизации электролита. В результате
перемещение ионов затрудняется, что приводит к
уменьшению эквивалентной электропроводности.
Лит.: Бродский А. И., Физическая химия, т. 2,
6 изд., М.—Л., 1948; Киреев В. А., Курс физической хи-
мии, 2 изд., М., 1956; Скорчеллетти В. В., Теорети-
ческая электрохимия, Л., 1959. А. И. Молодо».
КОМАРОВСКОГО РЕАКЦИЯ — образование ок-
рашенных в желтый, оранжевый или красный цвета
высокомолекулярных соединений при взаимодействии
высших алифатич. или гидроароматич. спиртов с аро-
матич. альдегидами в среде конц. H2SO4. При этой
реакции, вероятно, происходят окисление алифатич.
спирта до соответствующего альдегида или кетона
и его конденсация с ароматич. альдегидом:
АгСНО+R'R''CHOH—>ArCH2OH+R'R"CO
R'R"CO-|-ArCHO—-высокомолекулярный продукт
конденсации
(Аг—ароматич. радикал, R'—алифатич. радикал,
R"—алифатич. радикал или атом водорода)
Легче других реагируют вторичные спирты, в част-
ности изобутиловый, и изомерные амиловые спирты.
В качестве реагентов пользуются бензальдегидом,
n-диметиламинобензальдегидом, салициловым альде-
гидом, фурфуролом и др. Реакцию применяют для
колориметрия, определения небольших количеств
высших спиртов (сивушных масел) в этиловом спирте.
Этот метод включен в ГОСТ на ректификованный этиловый
спирт, в соответствии с к-рым определение выполняют следую-
щим образом: 10 мл спирта смешивают с 25 каплями 1%-ного
спиртового р-ра салицилового альдегида и 20 мл конц. х. ч.
серной к-ты и через 20 мин. колориметрируют появляющуюся
окраску, сравнивая ее с окрасками стандартной серии рас-
творов, содержащих известные количества изоамилового спирта.
Полученный окрашенный р-р можно также фотометрировать
с помощью фотоэлектроколориметра или спектрофотометра.
А. А. Черкасский.
КОМБИНАЦИОННОЕ РАССЕЯНИЕ СВЕТА —
рассеяние света исследуемым веществом, связанное
со структурой его молекулы; К. р. с. сопровождается
заметным изменением длины волны рассеиваемого
света.
К. р. с. было открыто в 1928 Л. И. Мандельштамом и Г. С.
Ландсбергом при исследовании света в кристаллах и одновре-
менно В. Раманом и С. Кришнаном при изучении рассеяния
света в жидкостях. В зарубежной литературе К. р. с. обычно
называют «эффектом Рамана».
При значительной разнице частоты колебаний све-
товой волны и частоты электронных полос поглоще-
ния вещества ее энергия, не поглощаясь, может
перейти на короткое время в энергию деформации
электронной оболочки молекулы. В большинстве
случаев эта энергия отдается молекулой в виде вто-
ричного излучения с фазой, изменяющейся одинако-
вым образом у разных молекул. Световая волна, об-
разовавшаяся в результате интерференции всех вто-
ричных, распространяется далее в прежнем направ-
лении. Слабое рассеяние в стороны с неизмененной
частотой вызывается флуктуациями плотности ве-
щества.
В очень редких случаях энергия световой волны
hv е, заключенная в деформированной электронной
оболочке, может перераспределиться на другие виды
внутримолекулярного движения, напр. на возбужде-
ние колебательного движения. Для этого нужна энер-
гия AvB. Остаток энергии hv = hve — hvv отдается в
пространство в виде вторичного излучения с частотой
v = ve — vv (стоксово рассеяние). Воз-
можен и обратный процесс — объединение колеба-
тельной энергии с электронной и отдачи ее в виде
излучения с суммарной частотой v = ve + vv (а н-
тистоксово рассеяние). Такое перерас-
пределение может происходить также и с вращатель-
ным движением. В этом случае v = ve ± 2vrot. Из
сказанного следует, что спектр К. р. с. отличается
от спектра обычного рассеяния появлением по обе
стороны от линии возбуждающего монохроматич.
света двух симметрич- i
но расположенных ли-
ний - спутников. На
рис. 1 для иллюстра-
ции приведен спектр . ЛММ1Им
К. р. с. четыреххло- рис j спектр комбинационного
ристого углерода. Та- рассеяния четыреххлористого угле-
кой акт перераспреде- рода.
ления энергии имеет
случайный характер. Случайны поэтому и началь-
ные фазы вторичного испускания у разных молекул.
В этих условиях единая световая волна образоваться
не может и свет с измененной частотой рассеивается
во все стороны.
Акт К. р. с. является маловероятным; этим объяс-
няется его очень малая интенсивность и необходимость
устранения даже очень слабой флуоресценции, на
фоне к-рой спектр К. р. с. может оказаться неразли-
чимым. По той же причине интенсивность сложных
колебаний, например с частотой v = v0 ± vv ±
определяемых вероятностью одновременного наступ-
ления двух маловероятных событий, еще гораздо
меньше.
Сказанным выше объясняются и другие свойства
К. р. с.: 1) Стоксовы спутники более интенсивны, чем
антистоксовы. С повышением темп-ры возрастет интен-
сивность последних. 2) Спектры К. р. с. построены
проще, чем инфракрасные спектры. Вращательно-
колебательная полоса поглощения свободной моле-
кулы обычно состоит из двух ветвей (Р и R), значи-
тельно реже из трех (Р, QnR). При К. р. с. интенсив-
ность сильно сжатой Q-ветви значительно превышает
интенсивность остальных двух и обычно наблюдается
только эта ветвь. Только у самой возбуждающей
линии сравнительно легко наблюдается вращатель-
ный спектр К. р. с. В жидкостях и твердых телах
основные полосы инфракрасного спектра и спектра
К. р. с., будучи изображены в одинаковой шкале ча-
стот, очень похожи и по расположению и по своему
контуру. 3) При условии, когда vej>vv, интенсивность
полос К. р. с. растет в коротковолновую область
спектра примерно пропорционально v*. Однако,
несмотря на значительно большую интенсивность
спектров К. р. с. в УФ-области, их наблюдение
и использование представляет дополнительные труд-
ности из-за легко возбуждающейся флуоресцен-
ции излучаемого вещества при коротковолновом об-
лучении, а также отсутствия удобного источника
для монохроматич. облучения. По мере приближения
частоты возбуждающего излучения к полосе погло-
щения вещества явление К. р. с. усложняется. В
пределе должно возникнуть электронное возбуждение,
связанное с поглощением падающего излучения, а
при соответствующих условиях — и флуоресценция.
При этом наблюдается значительное увеличение ин-
тенсивности полос К. р. с., не пропорциональное v*.
Для возбуждения спектров К. р. с. обычно применяют
различные ртутные лампы, из спектра которых с помошц,10
светофильтра чаще всего выделяют синюю линию А. = 4358
к-рой сопутствуют две гораздо более слабые при 4348 и 4334 А,
практически не вызывающие наблюдаемых полос К. р. с.
На рис. 2. приведена схема установки кюветы 1 с жидким
или газообразным образцом и ртутной лампы 2. Между лампой
и кюветой находится светофильтр з, выделяющий возбуждаю-
щую линию. Наблюдение производится со стороны горизон-
тальной стрелки справа, т. е. в направлении, более или менее
строго перпендикулярном к освещающему пучку. Вследствие
перпендикулярности световых колебаний к направлению рас-
пространения света поляризованная составляющая световых
колебаний в плоскости чертежа, по направлению двойной
655 КОМБИНАЦИОННОЕ РАССЕЯНИЕ СВЕТА — КОМПЛЕКСНЫЕ СОЕДИНЕНИЯ 656
стрелки, должна отсутствовать в освещающем пучке света.
Колебания, параллельные горизонтальной стрелке, не могут
дойти до наблюдателя. Значит, при идеальном соблюдении опи-
санных условий и отсутствии деполяризации при К. р. с.
наблюдатель воспринял бы ли-
нейно поляризованный свет с
колебаниями, перпендикуляр-
ными плоскости чертежа. В дей-
ствительности свет частично де-
поляризуется. Степень этой депо-
ляризации дает дополнительную
информацию о строении молекул
изучаемого вещества.
Рис. 2. Схема наблюдения
спектра комбинационного
рассеяния света: 1 — кювета,
2 — лампа, з — светофильтр.
Для наблюдения спектров
К. р. с. обычно используют-
ся светосильные спектрогра-
фы с несколькими стеклян-
ными призмами и регист-
рацией на фотопластинке.
Лишь в последнее время ста-
ли широко применять фото-
электрические приемники (фотоумножители), с по-
мощью к-рых измерение интенсивности выполняется
легче, скорее и точнее, а самый диапазон непосредствен-
ного сравнения интенсивности двух линий становится
гораздо более широким. Еще более эффективным ока-
залось использование дифракционных решеток с двой-
ной монохроматизацией и фотоэлектрической реги-
страцией (как это делается в приборе ДФС-12). С
помощью такого прибора становятся легче доступ-
ными изучению даже такие трудные объекты, как
порошкообразные образцы, дающие К. р. с. в поверх-
ностном слое рассеивающего вещества.
Трудности использования методов К. р. с. заклю-
чаются: 1) в том, что измерение интенсивности не носит
абсолютного характера и требует эталонов, что прак-
тически не всегда возможно, а также применения
гетерохромной фотометрии; 2) в малой интенсивности
спектров и значительно меньшей чувствительности
методов спектроскопии К. р. с., чем, напр., методов
ИК-спектроскопии. Кроме того, успешная работа
возможна только с веществами, не флуоресцирующими
и не изменяющимися при интенсивном облучении.
Как сказано выше, в области собственного поглощения
(окраска) может наступить значительное увеличение
интенсивности К. р. с., к-рое повышает чувствитель-
ность метода, напр. при обнаружении малых при-
месей.
Несмотря на указанные выше трудности, методы
К. р. с. сохраняют свое большое значение при иссле-
довании строения и свойств вещества, т. к. они ха-
рактеризуют его по другим свойствам, чем, напр., ме-
тоды ИК-спектроскопии. Для симметрично построен-
ных свободных молекул, таких, как молекулы водо-
рода, азота, кислорода и др. или нек-рых симметрич-
ных колебаний более сложных молекул, в и-рых
валентные колебания не вызывают изменения вели-
чины дипольного момента, спектр вообще не может
быть наблюден в инфракрасном поглощении. В К. р. с.
появление спектра и его интенсивность зависят от
поляризуемости электронной оболочки и ее связи с
колебанием молекулы, и в указанных выше случаях
спектр К. р. с. наблюдается. В этом и в нек-рых других
отношениях спектры К. р. с. и инфракрасные хоро-
шо дополняют друг друга.
Лит.: Ландсберг Г. С.. Оптика, 4 изд.. М., 1057.
(Общ. курс физики, т. 3); Ч у л а н о в с к и й В. М., Вве-
дение в молекулярный спектральный анализ, 2 изд., М.—Л.,
1951; Кельрауш К., Спектры комбинационного рассе-
яния, пер. с нем., М., 1952; Волькенштейн М. В.,
Ельяшевич М. А. и Степанов Б, И., Колебания
молекул, т. 2, М.—Л., 1949; Шорыгин П. П. и И в а н о-
в а Т. М., ДАН СССР, 1958, 121, № 1, 70; Ландсберг
Г. С., Б а ж у л и н П. А., С у ш и н с к и й М. М., Ос-
новные параметры спектров комбинационного рассеяния
углеводородов, М., 1956; Л а н д с б е р г Г. С. [и др.], Оп-
ределение индивидуального углеводородного состава бензинов
прямой гонки комбинированным методом, М., 1959; U 1 1-
m а п п, 3 Aufl., Bd 2/1, MUncb. — В., 1961, S. 236,
В. Id. Чила'новский,
КОМПЛЕКСНЫЕ ИОНЫ — см. Комплексные со-
ил
КОМПЛЕКСНЫЕ СОЕДИНЕНИЯ. Содержа-
ние:
Введение .............................. 656
Классификация ......................... 656
Координационная теория ................ 657
Номенклатура .......................... 659
Изомерия .............................. 662
Исследование комплексных соединений в растворе . . . 665
Практическое применение ............... 668
Введение. В определении понятия «К. с.» («коор-
динационное соединение») у химиков нет полного
единства. Основатель координационной теории строе-
ния комплексных соединений А. Вернер делил все
химич. соединения на соединения первого порядка
(простые, или атомные, типа СоС13, NH3, Н2О и т. п.)
и соединения высшего порядка (иначе молекулярные),
представляющие собой продукты сочетания соеди-
нений первого порядка (напр., СоС13 • 6NH3). Вернер
называл К. с. наиболее устойчивые соединения выс-
шего порядка, к-рые в водном р-ре либо вообще не
распадаются на свои составные части, либо распа-
даются в самой незначительной степени. Впоследствии
разница между терминами «К. с.» и «соединение
высшего порядка» стерлась и они стали идентичными.
Трудности, возникающие при попытке четкого
разграничения К. с. и соединений первого порядка,
в значительной степени обусловлены тем, что часто,
в зависимости от условий, одно и то же соединение
можно рассматривать и как простое и как комплекс-
нре. Так, многие вещества (в частности, галогениды
щелочных и щелочноземельных металлов), будучи
ассоциированными в твердом состоянии, в газообраз-
ном существуют в виде отдельных молекул, а в рас-
творе диссоциируют на составляющие их ионы. Т. к.
в лабораторной практике имеют дело с К. с. в твердом
или растворенном состояниях, важно охарактеризо-
вать их способность к существованию именно в этих
условиях, что и отражено в приведенном ниже опре-
делении.
Комплексным соединением наз. соеди-
нение, в узлах кристаллич. решетки к-рого находятся
комплексные ионы, способные к существованию
в р-ре. Комплексным ионом наз. сложный
ион, состоящий из атома металла (иногда неметалла)
в определенном валентном состоянии, связанного
с одним или несколькими способными к самостоя-
тельному существованию молекулами или ионами.
К. с. могут содержать комплексный анион, комплекс-
ный катион, либо вообще не диссоциировать на ионы
(соединения типа неэлектролитов). В иностранной
литературе (особенно в английской или американской)
комплексный ион, безотносительно к его заряду, назы-
вают «комплекс». Этот термин получил достаточно
широкое распространение и в отечественной лите-
ратуре.
Классификация. 1) Комплексные соеди-
нения, в состав комплексного иона к-рых входят
молекулы, содержащие атомы с донор-
ной функцией (S, N, О, As, Рит. п.). Сюда
относятся многочисленные аммиакаты, соединения
с оргаиич. производными аммиака, арсина, фосфина
и т. п. Нек-фые из этих К. с. являются циклическими.
Среди них встречаются и неэлектролиты, напри-
мер [NH2CH2CH2NH2PtCl2], и комплексы анионного
или катионного типов, например K[Co(NH3)2(NO.,)4]
и [{(NH2)2CS }4Pt]Cl2 соответственно.
2) Двойные соли — соединения, в состав
комплексного иона к-рых входят, кроме иона металла-
комплексообразователя, только анионы кислот. На-
ряду со сравнительно устойчивыми двойными солями,
напр. Kj[PtCL], существуют и малоустойчивые, напр.
квасцы К.[А1(8О4)а]. Устойчивость последних в рас-
G57
КОМПЛЕКСНЫЕ СОЕДИНЕНИЯ
658
творе существенно зависит от темп-ры и природы
растворителя. Поэтому можно ожидать, что при пони-
женных темп-рах в специально подобранных раство-
рителях квасцы могут оказаться столь же устой-
чивыми, как и наиболее стабильные из двойных
солей.
3) Внутрикомплексные соединения — циклич. комп-
лексы типа неэлектролитов, содержащие во внутрен-
ней сфере органич. группы, связанные с атомом-
комплексообразователем, во-первых, посредством ато-
мов с донорной функцией, а, во-вторых, путем заме-
щения водорода координируемой органич. молекулы,
металлом, напр.:
хКН2х nh2x
Н2Сч Си сн2
4c-oz чо-с'
II II
о о
4) Многоядерные комплексные соединения, содержа-
щие несколько центральных атомов с окружающими
их координационными сферами, соединенными посред-
ством отдельных атомов или групп атомов, называе-
мой.
мыхмостиками, напр. [(NH3)4Co< ;Co(NH3)41X4
хон/
5) Пол и соединен и я, содержащие в составе
своего аниона более чем одну молекулу ангидрида.
Различают изополисоединения, содержащие ангид-
риды одного типа, напр. К2Сг2О7 и гетерополисоедине-
ния, в состав аниона к-рых входят ангидриды различ-
ных кислот, напр. K4SiW12O40.
6) Комплексы типа полигалогенидов,
полисульфидов ит. п. Такие К. с. содержат
в качестве центрального иона J , S2 и т. п., а в каче-
стве координируемых групп — молекулы галогенов
(С12 и др.) или атомы S, Se и т. п. Напр., KJCL, CsJ6,
NaHS8.
7) Сверхкомплексные соединения, содержащие коор-
динированные группы в количестве, большем, чем это
характерно для комплексообразующего иона. Таковы,
напр., многие кристаллогидраты комплексных соеди-
нений, молекулярные соединения типа enCl2Pt-
• enCl4Pt (где еп — этплендиамин NH2CH2CH2NH2) ,
Координационная теория. Свойства К. с. опреде-
ляются их геометрич. конфигурацией и характером
связи в молекуле (прочность, степень ионогенности
или ковалентности связи). Координационная теория,
выдвинутая Вернером в 1891 (см. также Координа-
ционная связь), лишь в самых общих чертах указывала
на различие в характере связи отдельных атомов или
групп атомов в молекулах К. с. Согласно этой теории,
молекула К. с. имеет центрич. строение (имеется
в виду размещение отдельных заместителей около иона
металла — комплексообразователя), в большинстве
случаев октаэдрическое, тетраэдрическое нли плоское.
Атом комплексообразователя наз. централь-
ным атомом или центральным ионом.
Чтобы подчеркнуть разницу между центральным
ионом в К. с. и ионом того же металла в свободном
состоянии, центральный ион обозначают символом
элемента, рядом с к-рым в скобках римскими цифрами
ставится его валентность., Молекулы и ионы, непо-
средственно связанные с центральным ионом, наз.
координированными группами (С1 —
хлорогруппа, NH3 — амминогруппа, Н2О — акво-
группа), или внутрисферными замести-
телями (иначе аддендами, или лиган-
дами). Центральный ион в совокупности с коорди-
нированными группами образует внутреннюю
сферу К. с. При написании формул К. с. централь-
ный ион вместе с внутрисферными заместителями за-
ключают в квадратные скобки, напр. [Co(NH3)6]Cl3.
Если заряд центрального иона не равен по величине
сумме зарядов всех входящих во внутреннюю сферу
ионов, то К. с. содержит ионы, образующие его внеш-
нюю сферу и компенсирующие его заряд. Заряд
комплекса может нейтрализоваться либо простыми,
либо комплексными ионами:
[Pt(NH8)4]Cls; K2[PtCl4]; (Pt (NH8)4 [PtCl4]
В растворе К. с., содержащие внешнесферные ионы,
диссоциируют след, образом:
[Pt(NH8)4]Cla^(Pt(NH8)4]2+ + 2Cl-
Ka(PtCl4]^[PtCl4]3- + 2K+
(Pt (NH3)4] [PtCl4]j±[Pt(NH8)4]3 ++ [PtChJs-
B К. с. центральный ион проявляет т. наз. глав-
ную и побочную валентности. Глав-
ной валентностью Вернер назвал силы, аналогичные
тем, к-рые проявляются при образовании простых
соединений первого порядка. За счет сил побочной
валентности происходит сочетание молекул соедине-
ний первого порядка в соединения высшего порядка.
Часто при написании структурных формул К. с.
главные валентности обозначают сплошными линиями,
FNH3 .СГ
^nh/ хС1_
две хлорогруппы присоединены к Pt (II) за счет глав-
ной валентности, а две молекулы NH3 — за счет по-
бочной. Уже Вернер, основываясь на таких фактах,
как отсутствие изомерии у K2PtCl4 и т. п. соединений,
а побочные — пунктиром. Так, в
не проводил принципиального различия между
заместителями, присоединенными за счет главной
и побочной валентностей. В дальнейшем это предпо-
ложение было подтверждено рентгеноструктурным
анализом, изучением изотопного обмена и т. п.
Проявление побочной валентности часто приводит
к упрочнению связей, образованных за счет главной
валентности. Чаще встречаются случаи стабилизадии
высщего валентного состояния центрального атома,
хотя иногда имеет место упрочнение и его низшего
валентного состояния. Напр., в [Co(NH3)e]Cl3 состоя-
ние Со (III) более устойчиво, чем трехвалентное со-
стояние кобальта в СоС13, СоВг3. Двухвалентное со-
стояние для серебра нехарактерно, но комплексное
соединение Ag (II) с а, а'-дипиридилом (dp) вполне
устойчиво. Соли закисного железа весьма легко под-
вержены окислению, но [Fedp3]Cl3 окисляется с боль-
шим трудом. С другой стороны, характер насыщения
главной валентности существенно влияет на проч-
ность связи за счет побочной валентности. Напр.,
в ряде случаев термич. устойчивость К. с. (темп-ра
удаления внутрисферных групп) существенно зави-
сит от характера внешнесферного аниона. Так,
[Ni(NH3)e]Cl2 отщепляет аммиак при 164°, тогда как
[Ni(.NH3)e] 12 устойчив до 22Г.
Количество атомов или групп атомов, связанных
с центральным атомом, наз. его координаци-
онным числом (сокращенно к. ч.). Координа-
ционное число является характеристикой элемента
в определенном валентном состоянии. Двухвалентная
Pt проявляет к. ч. 4, четырехвалентная — к. ч. 6.
Координационное число может быть больше валент-
ности (наиболее частый случай, см. вышеприведенные
примеры), равно валентности (напр., у ч углерода
в многочисленных органич. соединениях) и, наконец,
иногда бывает меньше валентности. Последнее чаще
всего проявляется в соединениях, содержащих коор-
динированные многовалентные ионы типа О2-, напр.
в соединениях СО|~, ClOj и т. п. (к. ч. углерода в ионе
СО8" равно трем, а его валентность — четырем;
к. ч. хлора и его валентность в ионе CIO7 равны четы-
рем и семи соответственно). Нек-рые ионы металлов
характеризуются постоянным, другие — переменным
координационным числом. К первым относятся: Pt(II)
659
КОМПЛЕКСНЫЕ СОЕДИНЕНИЯ
660
(к. ч. 4), Pt (IV) (к. ч. 6), Со (III) (к. ч. 6), Rh (III)
(к. ч. 6), Ir (III) (к. ч. 6), Ir (IV) (к. ч. 6), Pd (II)
(к. ч. 4), Au (III) (к. ч. 4). Ко вторым: Ni (II)
(к. ч. 4; 6), Си (II) (к. ч. 6; 4; 3) и большинство других
элементов.
Проявляемое данным ионом координационное число
зависит от природы внутрисферных и внешнесферных
групп и от темп-ры, а в растворе К. с., кроме того, —
от концентрации адденда и центрального иона, а также
от природы растворителя. Понижение темп-ры спо-
собствует проявлению более высокого координацион-
ного числа. Так, при нагревании [Co(NH3)e]Cl2
в равновесии находятся след, соединения:
150’ 200°
[Co(NH3)„]Cl2^[Co (NH3)2Cl2] + 4NH3»-CoCla + 6NH3
к. ч. Со (П) = 6 к. ч. Со (П) = 4
Наиболее часто встречаются комплексы, содержащие
центральный ион с к. ч. 4 и 6, реже с к. ч. 3 и 2.
Предполагают существование соединений с к. ч. 5 и 7.
Наконец, к соединениям с к. ч. более высокими, чем 6,
относятся Me4[Mo(CN)s] и Me4[W(CNs)J, где Me —
однозарядный катион.
Одновалентные кислотные остатки, NH3, RNH2,
C5H5N и многие другие группы, входящие в состав
К. с., связаны с центральным ионом посредством одной
связи и занимают одно координационное место. Но
возможен случай, когда один заместитель связан
с центральным ионом посредством двух или больше-
го числа входящих в него атомов. Такие К. с. наз.
циклическими. Количество координационных
мест около центрального атома, к-рое может занимать
тот или иной заместитель, наз. координацион-
ной емкостью, а сами адденды — одно-,
двух-, трех- и т. д. поликоординационными замести-
телями. В качестве поликоординационных замести-
телей могут выступать нейтральные молекулы, содер-
жащие два или несколько атомов со свободной элект-
ронной парой (NH2—СН2—СН2—NH2 и т. п.), кислот-
ные остатки многоосновных кислот (SO|~, СО|~ и т. п.).
Возможен случай, когда один и тот же заместитель
содержит и атом с донорной функцией и по способу
своего образования является кислотным остатком.
Таковы кислотные остатки аминокислот, например
NH2CH2COO-b
zNH24 zNHjч
НгСч 'с/ сн2
с-о' чо-с'
II II
В табл. 1 приводятся при-
меры циклич. соединений.
Как видно из последних
двух примеров, координа-
ционная емкость замести-
теля может меняться.
Номенклатура. Основы
современной номенклату-
ры К. с. были даны Вер-
нером. Построение назва-
ния по этой номенклатуре
можно проследить, поль-
зуясь примером
[Co(NH3)4NO2Cl]NO3.
Прежде всего в алфавит-
ном порядке перечисляют
находящиеся во внутрен-
ней сфере кислотные остат-
ки, причем к их названи-
ям прибавляют окончания
«о» (сначала нитро-, а затем
хлорогруппы в данном слу-
чае). Затем, также в алфа-
Таблица I. Циклические комплексные соединения.
Адденд Услов- ное обозна- чение адден- да Koop- дина- цион- ная ем- кость К. ч. цен- траль- ного иона Комплексные соединения
сн2-ына 1 ch2-nh2 en 2 4 [en2Pt]Cl2
сна—nh2 1 СН—NHa 1 ch2-nh2 tn 3 6 [tn2Co] Cl3
/CH2-CH2-NH2 N^-CHa- CH2- NH2 \ch2-ch2—nh2 tren 4 4 [Pt tren] Cl2
zch2-coo- CH»-N< I XCH2-COO- 1 ZCH2-COO- ch2-n< ч>н2-соо- ЭДТА 5 6 6 6 [Со ЭДТАВг]К2 [Со ЭДТА] К
витном порядке указывают внутрисферные нейтраль-
ные молекулы (NH3 в данном примере). Внутрисфер-
ные NH3 называются аммино-, а Н2О — аквогруппами;
остальные нейтральные молекулы имеют свое обычное
наименование. Если во внутренней сфере содержится
несколько однотипных молекул или кислотных остат-
ков, то перед названием однородных заместителей
ставят греческие числительные: ди, три, тетра, пента,
гекса и т. д. (в данном случае «тетра» перед названием
групп NH3). После этого указывают центральный
ион, причем его валентность обозначают суффиксами,
приведенными в таблице 2. Если соединение содер-
жит комплексный катион (рассматриваемый случай),
то затем следует название анионов внешней сфе-
ры (нитрат-ион в данном примере). Таким образом,
Таблица 2. Названия комплексных соединений.
Заряд цент- раль- ного иона Суффикс, употребля- емый для обозначе- ния ва- лентности Формула соединения Название соединения
по номенклатуре, предложенной Вернером по номенклатуре Между- народного союза по чистой и прикладной химии
1 а [Ag(NH3)a]Cl Ка [СиС1„] диамминоаргента- хлорнд трихлорокупрат калия диамминоаргенто (I)- хлорид трихлорокупрат (I) калия
2 о [Со (NH3)0] Clj K2[Pt(NO2)a Cl2] [Pt (NH3)2C12] гексамминокобаль- тохлорид динитродихлоропла- тоат калия дихлородиаммино- платина гексамминокобальто (III)- хлорид динитродихлороплатинат ЦТ) калия дихлородиамминоплатина
3 и [Co [NH3)e] Cla [Co [NH,)5H2O]C13 K3 [Co (N02)o] гексамминокобаль- тихлорид аквопентамминоко- бальтихлорид гексанитрокобаль- тиат калия гексамминокобальто (III)- хлорид аквопентамминокобальто- (III) хлорид гексанитрокобальтат (III) калия
4 е [Pt(CbHBN)4NO2Cl](NO3)a нитрохлоротетра пи- ридиноплатине хлорид нитрохлоротетр апиридино- платина(1У) хлорид
5 ан Ka[VOFs] оксипентафторова- наданат калия оксипентафторованадат (V) калия
6 он Ka[UO2Cl4] диокситетрахлоро- уранонат калия диокситетра хлороуранат (VI) калия
661
КОМПЛЕКСНЫЕ СОЕДИНЕНИЯ
662
[Co(NH3)4NO2C1]NO3 согласно принципам номенкла-
туры, предложенной Вернером, наз. «нитрохлоротетр-
амминокобальтинитрат».
Если соединение содержит комплексный анион, то
после названия суффикса, указывающего на валент-
ность центрального атома, ставится суффикс «ат»,
а затем следует название внешнесферного катиона.
Напр., K[Co(NH3)2(NO2)4] наз. «тетранитродиаммино-
кобальтиат калия». Наконец, если соединение яв-
ляется неэлектролитом, то в его названии обозначение
валентности отсутствует. Так, [Pt(NH3)2Cl4] наз.
«тетрахлородиамминоплатина».
Кроме этой номенклатуры, к настоящему времени
стали довольно часто пользоваться номенклатурой,
принятой Международным союзом по чистой и при-
кладной химии. Она отличается от номенклатуры,
предложенной Вернером, только тем, что валентность
центрального иона обозначается не суффиксом, а рим-
ской цифрой в скобках. В названиях комплексных
катионов и неэлектролитов римская цифра следует
за названием металла. Напр., [Co(NH3)4NO2C1]NO3
наз. «нитрохлоротетрамминокобальто (Ш)нитрат. При
обозначении комплексных анионов римская цифра
пишется после суффикса «ат». Если центральный ион
имеет нулевой заряд, то его валентность обозначается
арабской цифрой (0). Напр. K4[Ni(CN)4] наз. «тетра-
цианоникелат (0) калия». В дополнение к принципам
номенклатуры Вернера предписывается положитель-
но заряженные группы называть после того, как пере-
числены нейтральные молекулы. В многоядерных
соединениях названия мостиковых групп следуют
после названий всех других координированных групп,
причем перед названием мостиковой группы ставится
символ «р,». Все мостиковые группы, кроме ОН, наз.
обычным образом; мостиковая ОН-группа, в отличие
от гидроксильной, наз. оловой группой, напр.:
.NH2 ч
[(NH,)» Со< >Со (NH3)3] С13
\OH)Z
наз. гексаммино- р-аммино-диол кобал ьтихл орид.
Для обозначения строения геометрич. изомеров
употребляются символы цис и транс, например
NHS Cl
изомер Pt наз. цис-дихлородиамминоплатина, а
NHS Cl
NH3 Cl
C1₽S?H — Иране-дихл ородиамминоплатина. Более
сложные случаи геометрич. изомерии номенклату-
рой не предусматриваются. В этом последнем случае
полезно пользоваться способом, предложенным
И. И. Черняевым, согласно к-рому адденды указы-
вают не в алфавитном порядке, а называют те адденды,
к-рые находятся в транс-положении друг к другу.
Этот же принцип можно применять и при написании
формул. Так, два геометрич. изомера согласно выше-
сказанному называют и записывают следующим об-
разом: (NH3)2Cl4Pt — диамминотетрахлороплатина,
a (NH3Cl)2Cl2Pt — амминохлороамминохлородихлоро-
платина.
Кроме указанных способов обозначения К. с.,
следует отметить, что в литературе до сих пор встре-
чаются старые названия солей, напр. (NH3)2Cl4Pt—
соль Жерара, (NH3Cl)2Pt—соль Пейроне, (NH^C^Pt—
хлорид второго основания Рейзе. Особенно употре-
бительны эти названия для К. с., содержащих
многовалентные центральные ионы. (Довольно полная
сводка старых названий К. с. приводится в книге
Д. Бейлара, см. лит. в конце статьи).
Изомерия. Для К. с. большинства элементов
VIII группы, а также хрома и нек-рых других, обра-
зующих прочные связи, по природе близкие к кова-
лентным, характерна изомерия. Известны различные
виды изомерии К. с.
Геометрическая изомерия (цис-,
транс-изомерия) состоит в различном пространствен-
ном расположении аддендов около центрального иона
и проявляется только у К. с., обладающих октаэдрич.
или'плоским строением, напр. [Pt(NH3)2Cl4] сущест-
вует в виде след, изомерных форм:
NH3
Cl
NH3
Pi
Ci
Тс'
Cl
II
Формуле [Pt(NH3)2Cl2] [две аммино- и две хлоро-
группы находятся в вершинах квадрата, в центре
к-рого расположен атом Pt (IV)] отвечают геометрич.
изомеры III и IV:
NH, Cl
Pt
Cl NH,
III
NH, Cl
Pt
NH, Cl
IV
Соединения II и IV наз. цис-изомерами, а I и III —
транс-изомерами. Чем сложнее состав молекулы, тем
больше возможностей для проявления изомерии. Так,
для К. с., содержащего во внутренней сфере 6 различ-
ных аддендов, могут существовать 15 геометрически
изомерных форм. У многоядерных соединений воз-
можности проявления геометрич. изомерии особенно
широки. В большинстве случаев геометрич. изомеры
различаются: химич. свойствами (см. Трансвлияние),
растворимостью, магнитной восприимчивостью, ди-
польными моментами.
В случае наиболее прочных К. с., напр. производ-
ных Pt (IV) и Pt (II) в той или иной реакции образует-
ся, как правило, только один изомер, напр. реакция
ГС1 СП fNH3 С11
К2 Pt |-|-2NH,-»| Pt +2КС1
Lei cij Lnh3 cij
приводит только к цис-изомеру; транс-форма обра-
зуется след, образом:
NH, NH3] INHj
Pt C12 + 2HC1
NH, NH,J
°1 1
Pt H-2NH4C1
Cl NH,J
или при нагревании
[NH, NH,1
Pt Cl2
[NH, NH3J
В случае менее прочных соединений, напр. производ-
ных Со (III), Сг (III) и др., в реакции образуется смесь
двух или нескольких изомеров, к-рые можно разде-
лить перекристаллизацией.
Оптическая (зеркальная) изоме-
рия наблюдается у К. с., молекулы к-рых не обла-
дают ни центром симметрии, ни плоскостью симмет-
рии. Причиной появления оптич. активности может
служить: 1) асимметрия центрального иона, напр.
иона Pt (IV) в
Вг
С)
663
КОМПЛЕКСНЫЕ СОЕДИНЕНИЯ
664
где еп соответствует NH2—СН2—СН2—NH2; 2) асим-
метрия молекулы в целом вследствие координации
однородных поликоординационных аддендов.
3) асимметрия одного из атомов адденда, обусловлен-
ная двумя причинами: а) координацией адденда,
уже содержащего асимметрия, атом, напр.
где рп соответствует nh2—сн2—сн—сн3. б) Асимметрия
адденда, возникающая вследствие координации, на-
пример в приведенном ниже соединении, в котором
атом серы асимметричен, тогда как в исходной моле-
куле адденда S(CH2CH2NH2)2 атом серы не обладает
асимметрией:
С|
-------7NH2-CH2
Pt / /
—сн2
Cl CH8-CH8-NH3Ci
При синтезе оптически активных К. с. в большинстве
случаев получаются их рацематы, к-рые можно раз-
делить на отдельные оптич. изомеры следующими спо-
собами: 1) кристаллизацией; 2) добавлением к р-ру
рацемата затравки вещества той же конфигурации,
что и выделяемое соединение; 3) получением диасте-
реоизомерных форм, к-рые можно далее разделить
перекристаллизацией; для разделения оптич. изоме-
ров, содержащих оптически активный комплексный
анион, применяют оптически активные основания
(стрихнин, бруцин, цинхонин), а для разделения
К. с. катионного типа — оптически активные кислоты
(винная к-та, камфорсульфоновая к-та и т. п.); 4) на
основе разной адсорбируемости оптич. изомеров на
поверхности оптически активного кварца; 5) поль-
зуясь различной реакционной способностью изоме-
ров и др. свойствами (см. также Активность оптиче-
ская, Антиподы оптические, Асимметрический син-
тез, Диастереоизомеры, Изомерия').
Сольватная изомерия состоит в раз-
личном распределении молекул воды или какого-
нибудь другого вещества между внутренней и внеш-
ней координационными сферами, напр. СгС13 • 6Н»О
существует в 3 изомерных формах:
[Сг(Н2О)а]С13 — серо-синего цвета,
[Сг(Н2О)5С1]С12 • Н2О — зеленого цвета,
[Сг(Н2О)4С12]С1 • 2Н»О — зеленого цвета.
В растворах этих соединений соблюдается рав-
новесие:
[Cr(H2O)3J Cl3z±[Cr(H2O)cJ3 + + ЗС1 -
[Сг(Н2О)3С1] Cl2s±[Ст (Н»О)5 Cl Р + + 2С1-
[Сг(Н2О)4С12] Cis [Cr(H2O)4 С12] + + С1-
[Cr(HaO)1Cl2]+ + H2Oi±[Cr(H2O)5Cl]3 + +Cl-
[Cr(H2o)5ci]2++H2o^[cr(H2oj0]3+-|-ci-
Различный характер диссоциации этих соединений
сказывается, напр., на их взаимодействии с AgNO3
и на электропроводности р-ра.
Координационная изомерия ха-
рактерна для веществ, содержащих несколько комп-
лексных ионов, и заключается в различном распреде-
лении аддендов во внутренних координационных
сферах этих ионов. Напр., известны два координа-
ционных изомера состава CoCr(NH3)e(CN)e, к-рые обра-
зуются по реакциям:
[Co(NH3)3] Cl3-[-K3[Cr(CN)3] =
= [Co(NH3)e] [Cr(CN)e] + 3KCl
K3[Co(CN)e] + [Cr(NH3)0]Cl3 =
= [Co(CN)0][Cr(NH3)()]-|-3KCl
При взаимодействии с AgNO3 первое из этих сое-
динений дает труднорастворимый осадок Ag3[Cr(CN)e]
и в растворе [Co(N H3)e|(NO3)3; второе — осадок
Ag3[Co(CN,)e] и в растворе [Cr(NH,)e](NO3)3.
Оба центральных иона могут быть ионами одного
и того же элемента, напр.
[Со (NH3)S (NO2)2] [Со [NR,),(NO3)J
И [Co(NH3)0][Co(NO»)0]
Валентности элемента могут быть различными в том
и другом комплексном ионе, напр.
[Pt(NH3)4Cl2][PtClj] и [Pt(NH3)4][PtCle]
Иногда координационная изомерия может быть ослож-
нена геометрич. или оптич. изомерией. Наряду с коор-
динационными изомерами существуют и координа-
ционные полимеры, отличающиеся числом комплекс-
ных ионов (мономер, димер, тример). В табл. 3 при-
ведены примеры координационных полимеров (у каж-
дого из них, в свою очередь, проявляется координа-
ционная изомерия).
Таблица 3. Координационные полимеры
Соединение Степень по- лимеризации
(NH3)»CbPt и (NII3Cl»)Pt Мономеры
[(NH3)iPt][PtCl4] [(NH.,)3ClPt][PtNJI3Cl3] Димеры
[(NHs):<ClPt].,[PtCl4] [(NHs)1Pt][PtNHiCl3] Тримеры
Ионизационная метамерия харак-
теризуется различным распределением ионов между
внешней и внутренней сферами К. с. и как следствие
этого — различным характером диссоциации на ионы.
Строение изомеров выясняется на основании изучения
их реакций и методов получения. Примеры таких реак-
ций приведены в табл. 4:
Таблица 4. Реакции ионизационных метамеров
Ионизационные
метамеры
[Pt(NII.,)4 Вг2]С12
[Pt(NH3)4Cl2[Br2
[Со (NH3)-,Br]SO4
[Со (NH3)5SO4] Вг
Продукты реакции с AgNO3
[Pt(NH,),Br»](NO3)» + AgCl
[Pt(NH,)4ClJ](NO3)2 + AgBr
[Co(NH1)5Br](NO3)»+Ag»SO4
[Co(NH1)-,SO4]NO3 + AgBr
Изомерия положения характерна для
многоядерных соединений, напр.:
(NFI3)3 ZOH, (NH3)3T
Со'' )Со Cl»
Cl XOIIZ Cl 1
;(nii3)2 oh,
Coz >Co(NH3)4
Cl2 О1Г
Cl2
665
КОМПЛЕКСНЫЕ СОЕДИНЕНИЯ
666
Солевая изомерия проявляется у соеди-
нений, содержащих кислотные остатки из нескольких
атомов; к центральному иону такие кислотные остатки
могут присоединяться посредством различных вхо-
дящих в их состав атомов. Напр. [Co(NH3)5NO2]X2 —
ксантосоль и [Co(NH3)6ONO]X2 — изоксантосоль.
Группа ONO связана с центральным ионом Со(Ш)
через кислород, а группа NO2 — через азот (это пока
единственный достоверный случай солевой изомерии).
Некоторые особые типы изоме-
рии. Вещества могут обладать одинаковым соста-
вом, но содержать во внутренней и внешней сферах
различные группы, напр.:
[Co(NH3)4C108N03B И [Co(NH3)4C104N02]+
Этот тип изомерии иногда называют суммар-
ной изомерией. Известны два соединения состава
[Co(NH3)6NO]C12. Одно из них парамагнитно и окра-
шено в черный цвет, тогда как другое диамагнитно
и обладает красной окраской. Предполагают, что
в одном из этих соединений присутствует двухзаряд-
ный ион кобальта, а в другом — трехзарядный. Такой
тип изомерии наз. нек-рыми исследователями элект-
ронной изомерией.
Наконец, нек-рые виды изомерии связаны с изоме-
рией аддендов. Примерами изомеров такого типа
являются [Pt(NH2C6H4COO)2Cl2], в состав внутренней
сферы к-рых входят о-, м- или п-аминобензойные
кислоты.
Исследование комплексных соединений в растворе.
В растворе К. с. подвергаются превращениям след,
типов: 1) диссоциации на комплексный и внешнесфер-
ные ионы; 2) замещению внутрисферных групп моле-
кулами растворителя; 3) диссоциации внутрисферного
заместителя.
Диссоциация К. с. на комплекс-
ный и внешиесферные ионы (равно-
весие ионного типа). Такого типа равновесие устанав-
ливается в растворах практически моментально,
напр.:
K3[Co(N02)e]rt3K+ + [Co(N02)e]8-
[Сг (NH8)BJ С18 й [Сг (NHB)B]»+-J-3CI-
К. с. типа неэлектролитов, напр. [Pt(NH3)2ClJ
или [Co(NH3)3(NO2)5], по указанной схеме не диссо-
циируют. Количество ионов, на к-рые диссоциирует
данное К. с., можно установить путем измерения
молекулярной электропроводности р-ров, определяе-
мой суммой подвижностей составляющих ионов; т. к.
подвижности комплексных ионов с одинаковой вели-
чиной зарядов приблизительно равны, а подвижности
таких однозарядных ионов, как К+, NO3_, С1“, также
практически одинаковы, то величина молекулярной
электропроводности р-ров в целом определяется
количеством и зарядами ионов, на к-рые диссоции-
рует данное К. с. Вернером и Миолати была установ-
лена след, зависимость между молекулярной электро-
проводностью 0,001 М растворов К. с. и количеством
ионов, на к-рые оно диссоциирует (простые ионы обя-
зательно однозарядны): соединения, дающие в
0,001 М водном р-ре один однозарядный комплексный
и один простой ион, характеризуются величиной элект-
роводности р, р-ра ок. 100 олг1 • см2; один двухзаряд-
ный комплексный и два простых иона — ок. 250
оаг1- см2; один трехзарядный комплексный и три
простых иона — ок. 400 ом~2 см2; один четырехза-
рядный комплексный и четыре простых иона — ок.
500 омг1 см2 и т. д. (примеры даны в табл. 5).
Ряд К. с. какого-либо металла в определенном
валентном состоянии (координационное число металла
на протяжении всего ряда остается постоянным),
каждый предыдущий член -к-рого отличается от по-
следующего содержанием во внутренней сфере лишней
Таблица 5. Молекулярная электропроводность К. с.
Количество
ионов, иа
которые
диссоции-
рует К. с.
Пример К. с.
д ом-1 СМ3
в 0,001 М р-ре
0
»
2
»
»
3
»
4
»
5
»
[Co(NH3)3(N02)8]
[Pt(NHB)»Cl4]
K[Co(NH3)2 (NO2)4]
[Co(NH,)4(NO2)2]C1
K[PtNH3Cl5]
[Pt(NH3)BClB]Cl
[Co(NH3)5NO 2] Cl2
[Pt(NH3)4Cl»J Cl2
K3[Co<N02)„]
[Co(NH8)bJC1,
[Pt(NHB)5Cl]Cls
K4[Fe(CN)B]
[Pt(NH8)0]Cl4
0
0
99,3
98,4
108,5
96,8
246,2
229
430
431,6
404
558
522,9
нейтральной молекулы адденда, наз. рядом Вернера—
Миолати, напр. ряд:
[Pt(NH8)0]Cl4, [Pt(NH8)5Cl]Cl8, [Pt(NH8)4С12]С12,
[Pt(NH3)3Cl3]Cl, [Pt(NH8)2Cl4], K[PtNH3Clo], K*[PtCle]
Молекулярная электропроводность соединений, обра-
зующих ряд Вернера—Миолати, меняется закономер-
ным образом, как указано выше. Путем измерения
чисел переноса можно, таким образом, определить
заряд комплексного иона. Количество и химическую
природу внешнесферных ионов можно установить
химическим путем. Так, при взаимодействии р-ров
[Pt(N Н3)6]С14 и AgNO3 сразу количественно осаж-
дается 4 моля AgCl. Из растворов [Pt(NH3)BCl]Cl3
или [Pt(NН3)4С12]С12 при действии AgNO3 выделяется
три или два моля AgCl соответственно. Из холодного
р-ра [Pt(NH3)2Cl4] хлорид серебра в первый момент
не осаждается. Только в результате длительного стоя-
ния раствора или при нагревании выпадает нек-рое
количество AgCl (о причинах этого явления см.
Т рансвлияние).
Замещение внутрисферных групп
К. с. молекулами растворителя.
Устойчивость комплексного иона (комплекса), в смыс-
ле замещения аддендов молекулами растворителя,
определяется рядом факторов и, в первую очередь,
характером и прочностью связи центральный ион—
адденд. В зависимости от природы этой связи комплек-
сы делятся на ионные (нормальные) и ковалентные
(комплексы проникновения). Разница в свойствах
комплексов того и другого типа видна при сопоставле-
нии двух соединений: [Co(NH3)6]C12 и [Co(NH3)6]Cl3.
Первое из них — комплекс ионного типа — при наг-
ревании обратимо отщепляет NH3:
159° 200°
[CO(NH3)b]C12^[Co(NH8)2C12] + 4NH3^CoC12 + 6NH3.
В растворе комплексный ион обратимо распадается
по схеме:
[Co(NHB)8]=++6H2O;i[Co(H2O)0P++6NH8
Реакции, протекающие при нагревании комплекса
ковалентного типа [Co(NH3)6]Cl3, необратимы:
175-180’
[Co(NHs)B] С1в------ [Co(NH3)5 Cl] С12 + NHa
180—220’
6[Co(NH3)5C1]C12-----•6CoC12 + 6NH4C1 + 22NH3+N2.
Это соединение практически не разрушается водой
даже в сильнокислых растворах.
Устойчивость комплексного иона в растворе зави-
сит от свойств центрального иона — от его заряда,
размера и электронной структуры. Как правило,
чем выше заряд центрального иона, тем устойчивее
ионные К. с. Известны, однако, случаи обратного
соотношения, что, по-видимому, связано с изменением
природы связи центрального иона и адденда в сторону
687
КОМПЛЕКСНЫЕ СОЕДИНЕНИЯ
668
увеличения ее ковалентности. Между размером цент-
рального иона и адденда существуют нек-рые опти-
мальные соотношения. Устойчивость комплексов с
аддендами небольшого размера (радиус аддендов
меньше 1,6А) уменьшается с увеличением радиуса
катионов, тогда как в случае комплексов с крупными
заместителями наблюдаются обратные соотношения.
Говоря о влиянии электронной оболочки центрального
иона на устойчивость К. с., необходимо иметь в виду
изменение характера связи центральный ион—адденд
от ионного к ковалентному типу при переходе от эле-
ментов с законченными электронными оболочками
типа инертных газов к элементам с недостроенной
18-электронной оболочкой и, наконец, к элементам
с 18-электронной оболочкой. Метал лы-комплексообра-
зователи, склонные к образованию ковалентных свя-
зей, дают более устойчивые в растворе К. с.
На устойчивость К. с. в растворе влияют и нек-рые
свойства адденда. Чем больше заряд адденда или его
дипольный момент, чем меньше его радиус, тем ста-
бильнее ионные К. с., образуемые этим аддендом. Чем
выше тенденция адденда к образованию ковалентных
связей, тем устойчивее соответствующие К. с. И нако-
нец, К. с. тем устойчивее, чем выше координационная
емкость заместителя. В случае наиболее прочно по-
строенных ковалентных комплексных ионов (напр.,
К. с. Pt (IV) или Pt (II) подвижность внутрисферных
заместителей существенно определяется закономер-
ностью трансвлияния, согласно к-рой заместители,
находящиеся в транс-положении, оказывают друг на
друга влияние, проявляющееся в лабилизации или
упрочнении их связей с центральным атомом (об
устойчивости К. с. в растворах см. также Комплекс-
ных соединений константы нестойкости).
Диссоциации внутрисферных за-
местителей. Кроме превращений указанного
типа, К. с. в растворе могут претерпевать изменения,
связанные с изменением химич. природы аддендов.
В частности, в результате координации изменяются
кислотно-основные свойства водородсодержащих адден-
дов (NH3, НаО, NHaCH3 и т. п.). Так, амминосоеди-
нения [Pt(NH3)e]Cl, [Pt(NH3)6Cl]Cl3 и др. в растворе
подвергаются диссоциации, напр.
[Pt(NH8)5Cl]»+s£[Pt(NH3)4NH2Cip++H+
В результате этого р-ры амминосоединений приобре-
тают кислую реакцию. Кислотно-основные свойства
К. с. определяются след, факторами: размером и заря-
дом центрального иона и, следовательно, его поляри-
зационными свойствами; размером и зарядом образую-
щегося комплексного катиона или аниона; устойчи-
востью комплекса в растворе; строением К. с. и харак-
тером трансвлияння координированных групп; степе-
нью диссоциации комплексно связанных аддендов.
Превращения двух последних типов (замещение
внутрисферных групп молекулами растворителя и дис-
социация аддендов) приводят к след, процессам:
1) изомеризация К. с., напр.:
TNHa Brl TNHa Вг 1+
Н2О-Н Pd 1^1 Pd +Br-s±
|_NH3 BrJ 1_NH3 H2Oj
TNHa Br "I INH3 Br 1
Pd Pd +H2O
L Br H20j L Br NH3J
2) расщепление К. с., напр.:
[PtCI4(OH)2]3~-|-2H2O J±[ptci2 (H2O)2 (OH)2]+2C1-
[PtCl2 (H2O)2 (OH)2] [Ptcis (OH),]!- _|_ 2H+
[Ptcij (OH)2]2- + 2H+s± [PtClj (II2O)2]
[PtCl4 (H2O)2] + 2Cl-5±[PtCl0]2- +2H2O
3) обратная реакция, образование нз 2 веществ
одного К. с.:
Г Cl NH3-| TNO2 ХН.Л ГС1 NH3-|
Pt H- Pt =2 Pt
Lnh3 ci J Lnh3 no2 J Lnh3 no2 J
Практическое применение. К. с. применяются:
1) в аналитической химии для определения многих
химич. элементов весовым, колориметрическим или
др. методами; 2) для разделения ряда металлов и
получения их в состоянии высокой степени чистоты
(разделение металлов VIII группы, лантанидов, полу-
чение золота, серебра, меди и т. д.; см. соответствую-
щие статьи об отдельных химич. элементах); 3) для
получения плотных и прочных покрытий в гальвано-
стегии и гальванопластике; 4) в качестве красителей;
5) в процессах умягчения воды (полифосфаты, поли-
аминокислоты, фитаты). К числу К. с. относятся
многие вещества, несущие в организме животных
и растенйй самые ответственные функции (гемоглобин,
хлорофилл, энзимы и др.).
О современных взглядах на строение К. с, см. Кван-
товая химия, Магнетохимия, Химическая связь и др.
общие статьи. О К. с. важнейших металлов-комплек-
сообразователей см. Железо, Золото, Иридий, Кобальт
и т. д.
Лит.; Г р и н б е р г А. А., Введение в химию комплекс-
ных соединений, 2 изд., Л.—М., 1951; Некрасов Б. В.,
Курс общей химии, 13 изд., М.—Л., 1961; Вернер А., Но-
вые воззрения в области неорганической химии, пер.’ с нем.,
5 изд., Л., 1936; Химия координационных соединений, под ред.
Дж. Бейлар, Д. Буш, пер. с англ., М., 1960; Чугаев Л. А.,
Избранные труды, М., 1954; Вознесенский С. А.,
Внутрикомплексные соединения и их значение для аналитиче-
ской химии, М.—Л., 1938; Яцимирский К. Б., Термо-
химия комплексных соединений, М., 1951; Яцимирский
К. Б., Васильев В. П., Константы нестойкости комплекс-
ных соединений, М., 1959. Н. Н. Желиговская.
КОМПЛЕКСНЫХ СОЕДИНЕНИЙ КОНСТАНТЫ
НЕСТОЙКОСТИ — величины, характеризующие ус-
тойчивость комплексных соединений в растворе.
Комплексы ионного типа в растворе равновесным
образом распадаются по схеме:
[MAn ]m+ + nH2O [М (Н2О)П Г"+ + nA
где М — центральный ион, а А — координируемая
молекула; или, не учитывая сольватационные про-
цессы:
ГМАпГп+ ^Mm+ +nA
Положение равновесия этой реакции, иначе устойчи-
вость комплекса [МАп]т+в растворе можно охаракте-
ризовать константами нестойкости А:
г. см™+ -са
С1МАП]«+
где С — концентрации соответствующих молекул или
ионов. Напр., в реакции [Ag(NH3)2]+ Ag+ -ф- 2NH3
константа равновесия к ти есть константа нестойкости
комплекса [Ag(NH3)2]+:
!_ CAg+'cNH3
c[Ag (NH8)2]+ ’
где CAg. CNHa И C[Ag(NHa)2]+ концентрации централь-
ного иона, адденда и недиссоциированного комплекса
соответственно.
Строго говоря, в приведенных выражениях, следо-
вало бы писать не концентрации, а активности соот-
ветствующих ионов. Однако при определении кон-
стант нестойкости применяют растворы с постоянной
ионной силой, что позволяет пользоваться выраже-
ниями в приведенной выше форме. Иногда для харак-
теристики устойчивости комплекса служат кон-
станты устойчивости К (иначе константа
образования комплексного соединения) — величины,
обратные константам нестойкости: к = 1/А. Встре-
чающиеся иногда термины «константы диссоциации
комплекса», «константы комплексности», «константа
закомплексованности» являются синонимами понятия
«константа нестойкости» и пока еще не получили
достаточно широкого распространения.
669
КОМПЛЕКСОМЕТРИЯ
670
Строго говоря, процесс диссоциации комплексного
соединения является ступенчатым. Напр., диссоциа-
ция иона [Fe(SCN)e]3~ протекает по ур-ниям:
[Fe(SCN)e]«-s±[T'e(SCN)5]3-+SCN-
[Fe(SCN)5]2-^[Fe(SCN)4]-+SCN-
[Fe (SCNhJ-^[Fe (SCNM1+SCN-
[Fe (SCN)aJ ss [Fe (SCN)2]+ + SC N"
[Fe (SCN)j]+ s± [Fe (SCN)]2 + + SCN~
[Fe (SCN)P+t± Few- 4- SCN“
Для каждого из этих равновесий можно найти соот-
ветствующие константы равновесия, к-рые называются
последовательными, промежуточными или ступенча-
тыми константами нестойкости (klt к2, ..., кп и т. д.),
связанными с общей константой нестойкости к след,
соотношением:
fe=ki-k2 • ••• • kn .
Константы нестойкости можно определить с по-
мощью различных физико-химич. методов.
Метод растворимости: о равновесной концентра-
ции центрального иона или аддендов судят на основании измере-
ния растворимости малорастворимой соли в растворе, содержа-
щем комплексообразующее вещество, или растворимости комп-
лексообразующего вещества в растворе соли металла-комплек-
сообразователя.
Метод экстракции (метод распределения): основан
на изучении распределения центрального иона, адденда или всего
комплекса между двумя несмешивающимися растворителями.
Ионнообменный метод: о равновесных концент-
рациях компонентов судят в результате изучения гетерогенного
равновесия между раствором и катионитом или анионитом.
Потенциометрический м е т о д: равновесные
концентрации центрального иона и адденда вычисляют, измеряя
pH растворов веществ определенных концентраций.
Полярографический метод: константа не-
стойкости вычисляется на основании полярограмм, полученных
в отсутствии и в присутствии комплексообразующих веществ.
Кинетический метод: данные относительно рав-
новесных концентраций компонентов получаются в результате
измерения скорости- реакции, протекающей с участием цент-
рального иона, адденда или комплекса.
Колориметрический метод: о равновесной
концентрации одного из компонентов комплексообразования
судят на основании измерения оптич. плотности раствора окра-
шенного вещества, находящегося в равновесии с аддендом, цент-
ральным ионом или комплексом.
Метод радиоактивных индикаторов: о
равновесных концентрациях судят по скорости изотопного об-
мена между комплексом и аддендом, содержащим меченый атом,
а также между комплексом и гидратированным центральным
ионом.
Метод замораживания: в случае медленно про-
текающей реакции комплексообразования о равновесных кон-
центрациях компонентов судят по количеству одного из компо-
нентов, к-рое может быть быстро и количественно выведено ив
сферы реакции.
Спектрофотометрический метод: констан-
та нестойкости и состав образующегося комплекса определяются
на основании изучения спектров поглощения растворов.
Метод электропроводности: о константе
нестойкости судят на основании отклонения электропроводности
от аддитивного значения.
Калориметрический метод: по количеству
тепла, выделившегося в результате смешения растворов компо-
нентов комплексообразования, судят относительно количества
частиц, образующихся в растворе.
Криоскопические и эбулиоскопические
методы: относительно числа частиц, образующихся в рас-
творе в результате реакции комплексообразования, судят по
изменению темп-ры кипения или замерзания растворов.
Лит.: Яцимирский К. Б., Васильев В. П.,
Константы нестойкости комплексных соединений, М., 1959;
Stability constants of metal-lon complexes -with solubility products
of inorganic substances, pt 1—2, L., 1957—58; см. также при ст.
Комплексные соединения. Н. Н. Желиговская.
КОМПЛЕКСОМЕТРИЯ (хелатометрия, тригоно-
метрия) — титримет^ич. метод, основанный на обра-
зовании комплексных соединений с этилендиаминтет-
рауксусной к-той и другими аминополикарбоновымп
к-тами, называемыми комплексонами. Ионы металлов
взаимодействуют с комплексонами в большинстве
случаев практически мгновенно с образованием рас-
творимых, малодиссоциированных соединений посто-
янного состава. Этим методом можно определять
практически все катионы и многие анионы. Подавляю-
щее большинство определений основано на использо-
вании двунатриевой соли этилендиаминтетрауксусной
к-ты — ЭДТА (комплексон Ill)', другие комплексоны
применяются значительно реже.
К. введена в аналитич. химию Г. Шварценбахом,
к-рый предложил метод титрования ионов кальция
и магния (1945), а впоследствии и ионов ряда других
металлов. Им же разработана теория К. При титро-
вании протекают следующие реакции;
Mes++H2Y2-=MeY2-4-2H+
Me’++H2Y2-=MeY-+2H+
Me4++H2Y2-=MeY+2H+
где H2Y2~ — анион двунатриевой соли ЭДТА. Из этих
уравнений видно, что грамм-эквивалент металла,
независимо от его валентности, связывает один грамм-
эквивалент комплексона. Обычно конечную точку
титрования устанавливают по исчезновению определя-
емого катиона в растворе с помощью т. н. металл-ин-
дикаторов или комплексометрич. индикаторов. В ка-
честве последних применяют вещества, образующие
с определяемым катионом окрашенное соединение,
разрушающееся под действием комплексона. Конеч-
ную точку титрования определяют в этом случае
либо по обесцвечиванию раствора, либо, если инди-
катор окрашен, по появлению его собственной ок-
раски.
Если для определяемого иона металла неизвестен
индикатор с четким переходом окраски в конечной
точке, то прямое титрование комплексоном может быть
заменено обратным титрованием избытка комплек-
сона раствором соли магния, цинка или другого ме-
талла. Этим способом широко пользуются, напр.,
при определении стронция и галлия; этот прием по-
лезен и в тех случаях, когда вследствие гидролиза
определяемый катион не может быть удержан в рас-
творе при pH, необходимом для проведения титрова-
ния. При титровании с индикатором точку эквива-
лентности обычно устанавливают визуально или
спектрофотометрии., пользуются также амперомет-
рии. или потенциометрии, методами.
Хотя комплексон и не является избирательным реа-
гентом, однако во многих случаях, когда анализи-
руемый материал содержит лишь один взаимодействую-
щий с комплексоном элемент, применение К. является
весьма эффективным, т. к. дает возможность получать
результаты простыми средствами за короткий срок.
При анализе продуктов более сложного состава влия-
ние мешающих титрованию элементов часто удается
легко и просто устранить маскировкой цианидом,
фторидом, триэтаноламином и т. п. или выведением
в осадок в виде оксалата, сульфата и т. п. Избиратель-
ность К. может быть в отдельных случаях повышена
увеличением кислотности раствора, т. к. при этом
уменьшается число элементов, взаимодействующих
с комплексоном. Можно,, напр., титровать Fe3+ при
pH 2—3 в присутствии Са и Mg, к-рые в этих условиях
комплексонатов не образуют; титрованию циркония
в 2 н. солянокислой среде практически не мешают все
другие элементы, не считая тех, к-рые гидролизуются
в указанных условиях. Зависимость устойчивости
комплексов от кислотности используется и для после-
довательного титрования двух, а иногда и более эле-
ментов в одном и том же растворе. Так, напр., тит-
руют торий при pH 1.6, а затем повышают pH до 4,5
и титруют в том же растворе редкоземельные элементы.
В тех случаях, когда пет возможности устранить влия-
ние сопутствующих элементов непосредственно в тит-
руемом растворе, прибегают к их предварительному
отделению с помощью обычных методов. Применение
К. в таких случаях упрощает определение.
Комплексометрич. определение жесткости воды широко
применяют на практике. Также часто применяют К. для оп-
ределения металлов в различных материалах: минеральном
сырье, различных продуктах технологич. и металлургич.
671
КОМПЛЕКСОН III — КОМПЛЕКСООБРАЗОВАНИЯ МЕТОДЫ
672
производств, фармацевтич. препаратах и мн. др. Особенно
ценным является комплексометрия, определение металлов,
для к-рых ранее не было известно удовлетворительных титри-
метрич. методов (Са, Mg, Al, Zn, Bi, Zr, Th, Pb и др.). К. в
ряде случаев целесообразно, применять и для косвенного
определения анионов (SO|-, РО3 -, F-), после осаждения мало-
растворимой соли кальция, магния или свинца.
Лит.: Комплексометрия. Теоретические основы и прак-
тическое применение, сб. переводов, М., 1958; Горюши-
н а В. Г., Бюллетень цветной металлургии, 1959, № 13 (138),
29; см. также при ст. Комплексоны. В. Г. Горюшина.
КОМПЛЕКСОН III [этилендиаминтетрауксусной
кислоты (ЭДТА) двунатриевая соль, трилон Б, хе-
латон III; титриплекс III, иргалон, версен]
C10H18O10N2Na2,i сокращенно Na2H2Y • 2Н3О, мол. в.
372,25 — белый’ кристаллич. порошок, хорошо рас-
творимый в воде и щелочах; pH водного раствора
ок. 6. -В табл. 1 приведена растворимость ЭДТА
[(HOOCCH2)2N—СН2—CH2N(CH2COOH)2] и ее натрие-
вых солей (в г на 100 мл р-ра) при разных темп-рах.
Таблица 1
Вещество
80°
ЭДТА (H,Y)..................
Мононатриевая соль (NaH3Y)
Динатриевая соль (Na2H2Y) .
Тринатриевая соль (Na,HY) .
Тетранатриввая соль (Na4Y) .
0.2 0,2 0,5
1,4 1,4 2,1
10,8 13,7 23,6
46,5 46,5 46,5
60,0 59,0 61,0
ЭДТА имеет след, значение констант диссоцпации:
рК1 Р-Ка Рйз Р-Г<4
(H4Y) (H3Y-) (H2Y3-) (HY8-)
2,00 2,76 6,16 10,26
Комплексон III образует очень устойчивые внутри-
комплексные соединения с большинством катионов,
прочность к-рых можно характеризовать константами
устойчивости. В табл. 2 приведены соответствующие
данные, полученные при 20° и ионной силе р-ра р, =
= 0,1 в среде KNO3.
Таблица 2
Катион Комплекс log K, Катион Комплекс log Ki
•Na+ NaY3-- 1,66 Al3+ . A1Y- 16,13
♦L1+ L1Y3~ 2,79 Y3+ YY- 18,09
Ag+ AgY’- 7,32 La3+ LaY- 15,50
*Ма2+ MgY2“ 8,69 Ce3+ CeY- 15,98
•Са2+ CaY2 - 10.70 РГЗ + PrY- 16,40
•Sr2+ SrY2- 8.63 Nd3+ NdY~ 16,61
*Ва2+ BaY2~ 7,76 Sm3+ SmY- 17,14
МП2Ь MnV" 14.04 Eu3+ EuY- 17.35
•Fe2+- FeY2- 14,33 Gd3+ GdY- 17,37
*Fe3+ FeY- 25,1 Tb3+ TbY~ 17,93
Со2- CoY2- 16,31 Dy3+ DyY- 18,30
Ni2*- NiY2 18,62 Ho3+ HoY~ 18,74
Cu2+ CuY2- 18.80 Er3+ ErY- 18,85
Zn2+- ZnY2- 16,50 Tu3+ TuY- 19,32
Cd2+ CdY2~ 16,46 Yb3+ YbY- 19,51
Hg2+ HgY2- 21,80 Lu3+ LuY- 19,83
Pb2+ PbY2- 18,04 Sc’+ ScY- 23,1
V2I- VY2- 12,7 Ga3+ GaY- 20.27
узд- VY- 25.9 In3± InY- 24.9
vo!+ VOY2- 18,77 Thi+- ThY- 23,2
• 0,1 М р-р КС1.
Комплексон III широко применяют в аналитич.
химии в комплексометрич. титрованиях для опреде-
ления многих катионов и анионов, напр. Са, Mg, Со,
Си, Ni, Zn, Fe, Al, Mn, Ga, редкоземельных элементов,
Tb, UIV и др.; SO|“, CN~, PO|~ и др., при потенцио-
метрия., полярография, и амперометрия, определе-
ниях, для маскировки при фотометрия, и титриметрич.
определениях для разделения близких по свойствам
элементов с использованием хроматографии. К. ис-
пользуют также для умягчения воды, для маскировки
следов металлов в фармацевтич. и химич. произ-вах;
известны и другие области применения.
Лит. см. при ст. Комплексоны. И. П. Ефимов.
КОМПЛЕКСОНЫ — аминополикарбоновые к-ты,
у к-рых с атомом азота связано неск. алкилкарбо-
ксильных групп. К. можно рассматривать как произ-
водные иминодиуксусной к-ты HN(CH2COOH)2, у
к-рых атом водорода иминогруппы замещен органич.
радикалом. В молекуле К. имеется неск. функцио-
нальных групп, способных одновременно связывать
центральный атом комплекса несколькими коорди-
национными связями (клешневидные сое-
динения). К. образуют весьма прочные, хорошо
растворимые в воде, комплексные соединения с боль-
шинством катионов, в том числе с катионами щелоч-
ноземельных металлов.
К настоящему времени (1962) известно св. 50 К.
Лучшей по свойствам и получившей наибольшее рас-
пространение остается пока эти лен диаминтетр ауксус-
ная к-та
(HOOCCH»),N-CH2CH2-N(CH2COOH)2
(ЭДТА, комплексон II)
обладающая среди других К. лучшей из возможных
структур — число атомов-аддендов равно 6, что соот-
ветствует наиболее часто встречающемуся координа-
ционному числу металлов. Эта кислота используется
чаще в виде двунатриевой соли, обладающей большей
растворимостью (см. Комплексон III).
Впервые и наиболее детально изучением свойств и
условий синтеза К. занимался Г. Шварценбах и сотр.
Ими выяснены особенности образования комплексных
соединений с К., определены константы диссоциации
отдельных К. и их комплексов с различными катио-
нами и др. Теоретич. работы Шварценбаха привели
к возникновению комплексометрии. В дальнейшем
была установлена эффективность применения К.
в анализе для маскировки.
С образованием комплексов щелочноземельных металлов
связано первоначальное (1936) практич. применение Н. для
смягчения воды. К. широко используются благодаря легкости
образования легкорастворимых прочных комплексов, когда
требуется связать (замаскировать) ионы металлов в растворе,
как, напр., в крашенин, приготовлении и проявлении цветной
кинопленки и т. п. Различие в константах нестойкости ком-
плексонатов отдельных редкоземельных элементов позволяет
в сочетании с ионным обменом добиться аффективного разде-
ления элементов этой группы. В с. х-ве К. пользуются для
лечения железного хлороза растений и др. В медицине К. при-
меняют для лечения лучевой болезни, свинцового и ртутного
отравлений, а также других болезней, связанных с отложением
в организме человека малорастворимых солей. К. широко при-
меняют в фотометрии, полярографии, анализе и др. Примене-
ние К. позволило существенно упростить большое число ранее
известных методов анализа и разработать ряд принципиально
новых. См. также КомплексоЛетрия, Комплексон III,
Внутрикомплексные соединения.
Лит..: П р ш и б и л Р., Комплексоны в химическом ана-
лизе, пер. с чеш., 2 изд., М., 1960; Горюшина В. Г.,
Завод, лабор., 1954, 20, „V 6, 647; Я ними рений К. Б.,
там же, 1955, 21, № 10, 1149; W е 1 с h е г F. J., The analy-
tical uses of elhylenediamine-tetraacetic acid, N. Y., 1958;
В j er rum J., Schwarzenbach G., Sillen L.,
Stability constants of metal-ion complexes..., pt 1. Organic
ligands, L., 1957; Яцимирский К. Б., Васильев
В. П., Константы нестойкости комплексных соединений,
М., 1959. В. Г. Горюшина.
КОМПЛЕКСООБРАЗОВАНИЯ МЕТОДЫ (в хи-
мическом анализе) — методы количественного тит-
рпметрич. анализа, основанные на различных реакциях
образования комплексных соединений. К ним отно-
сятся, напр., меркуриметрия, основанная на образо-
вании комплексов двухвалентной ртути, фторометрия
(образование фторидных комплексов). К К. м. от-
носятся такжЬ нек-рые аргентометрия, методы. Осо-
бенно большое значение имеет комплексометрия, ос-
нованная на образовании многими катионами ком-
плексных соединений с Na-солью этилендиаминтет-
рауксусной к-ты и др. комплексонами. Пользуясь
К. м., можно определять многие катионы (Ag+, Fe3,
Cu2+, Ni2+ и др.) и анионы (Ch, F , SCN и др.), образую-
щие комплексы. Равновесие в р-ре, содержащем ком-
плексные соединения, определяется общими уравне-
ниями, основанными на действующих масс законе.
673
КОМПЛЕКСООБРАЗОВАНИЯ МЕТОДЫ — КОМПОНЕНТЫ
674
Можно выделить две группы реакций комплексооб-
разования: 1) реакции, в результате к-рых образуются
моноядерные соединения MLn и 2) реакции образо-
вания многоядерных соединений MmLn (М — цен-
тральный ион, например ион металла, L — лиганд,
комплексообразующий анион). В наиболее простом
случае для мономолекулярных комплексов (п = 1)
константа устойчивости комплексного
соединения, получающегося в результате реакции
М + L = ML (заряд ионов для простоты опущен),
определяется ур-нием:
Kml = Дм] • [L]
(квадратные скобки обозначают молярные концентра-
ции). Если с центральным ионом металла связано
несколько лигандов, то комплексообразование идет
ступенчато н равновесие определяется несколькими
последовательными константами устойчивости соот-
ветствующих соединений. Общая константа устойчи-
вости определяется уравнением:
[MLn]
1<п = 1мГКгг
Принято также использовать величины, обратные на-
званным, — константы диссоциации комплексных со-
единений (константы нестойкости).
Подобно комплексам MLn, полиядерные комплексы
образуются ступенчато, но этот процесс гораздо слож-
нее н обычно связан с образованием нескольких видов
комплексных частиц. Реакции комплексообразования
обычно иллюстрируются кривыми титрования, выра-
жающими зависимость рМ (—lg [М]) как функции
добавленного количества комплексообразующего аген-
та. Основная трудность в построении таких кривых —
Отклонение от точми~эквива/>ентности
оценка влияния раз-
личных факторов на
реакцию комплексо-
образования. На рис.
представлены кривые
титрования при обра-
зовании комплексных
соединений металл-
лиганд 1:1 в зависи-
мости от различной
устойчивости. С по-
нижением значения
константы устойчиво-
сти К скачок на кривой титрования выражен менее
четко. Положение точек на кривой титрования может
быть рассчитано по уравнению:
[L]
[ML]
pM=lg кml + 1g
В точке эквивалентности [М]экв = [Б]экв и [ML]№B =»
См — общая концентрация металла в р-ре. Отсюда
ГШ / [ML]3KB. \ !/э / См \*/2
[М]ЭКВ. = I J
Если константа устойчивости комплексного соеди-
нения мала (К < 107), то для вычисления кривой
титрования принимают след, значения:
[ML] = CM-[M] [ML] = CL-[L] [ML] = [М] [L] KML
Для определения конечно!! точки при такого типа
титрованиях используют обычно физико-химич. ме-
тоды.
Если центральный ион металла присоединяет не-
сколько лигандов, то равновесное состояние опреде-
ляется ступенчатыми константами устойчивости. На
кривых титрования подъем (скачок) в между раз-
личными ступенями обычно невелик и только послед-
няя ступень может быть использована для титрования.
В таких случаях необходимо знать значение послед-
ней константы устойчивости, прежде чем решить,
возможно ли определение или нет. Теоретич. обра-
ботка результатов таких титрований имеет много
общего с обработкой кривых нейтрализации много-
основных к-т. Последний комплекс MLn преобладает,
остальными можно пренебречь. Величииу[М]энв
тогда можно рассчитать по уравнению
/ [MLn] \i/n + l
[М]ЭКВ. ~
\ tl KMLrt )
В визуальных методах определения конечной точки
титрования применяют индикаторы, изменяющие ок-
раску с изменением концентрации ионов металла
в р-ре. В качестве индикаторов могут быть исполь-
зованы многие металлохромные индикаторы, образую-
щие с металлами окрашенные комплексные соедине-
ния, менее устойчивые, чем соединения иона металла
с комплексообразующим веществом, или бесцветные
вещества, реагирующие с металлом с образованием
окрашенных соединений, напр. SCN~ или сульфосали-
циловая к-та при титровании Fe (III). Конечная точка
титрования может быть также установлена по появ-
лению мути, напр. при определении цианидов по
Либиху—Дениже:
2С N+Ag+T= Ag(C N)i
Ag(CN)7=rrAg[Ag(CN)2]
Для определения конечной точки титрования в
К. м. часто пользуются инструментальными методами.
Лит.: К о л ь т г о ф И. М., Стенгер В. А., Объем-
ный анализ, пер. с англ., т. 1—2, М., 1050—52; Крешков
А. П., Основы аналитической химии, кн. 2, М., 1961; К о 1 t-
h о f f I. M., E 1 v 1 n g P. J., Treatise on analytical che-
mistry, v. 1, pt 1, N. Y., 1959; Stability constants of metal—
ion complexes with solubility products of inorganic substances,
pt 1—2, L., 1957—58; Яцимирский К. Б., Василь-
ев В. IL, Константы нестойкости комплексных соединений,
М., 1959. См. также при ст. Аналитическая химия, Объемный
анализ, Внутрикомплексные соединения. В. П. Ефимов.
КОМПОНЕНТЫ (независимые компоненты) — хи-
мически индивидуальные вещества, наименьшее чис-
ло к-рых достаточно для образования всех фаз, т. е.
гомогенных частей данной системы (см. Фаз правило).
Понятие К. введено в 1873—76 У. Гиббсом. Характер-
ной особенностью К. является то, что масса каждого
из них в системе не зависит от массы других. Если
составные части системы не вступают друг с другом
в химич. реакции, то она наз. системой 1-го класса,
или физической; число К. такой системы равно
числу ее составных частей. Если же составные части
системы реагируют друг с другом, то она наз. систе-
мой 2-го класса, или химической; число К.
такой системы равно числу ее составных частей, умень-
шенному на число независимых химич. реакций, мо-
гущих идти в этой системе.
Примеры физических систем: 1) Система, обра-
зованная льдом, жидкой водой и водяным паром; число К. —
один (вода). 2) Система, образованная солью, ее насыщенным
водным р-ром и водяным паром; если соль не взаимодействует
с водой, то число К. — два (соль и вода).
Примеры химических систем: 1) Система лед —
жидкий раствор — водяной пар, когда соль дает гидрат, т. е.
химич. соединение с водой. Число составных частей — три
(соль, ее гидрат, вода); число реакций — одна (образование
гидрата из соли и воды); число К. 3—1 = 2 (вода и соль).
2) Система из окиси кальция и двуокиси углерода, образую-
щих соединение — углекислый кальций по реакции: СаО +
+ С02ггСаС0я. Число составных частей — три, число реак-
ций — одна; число К. 3 — 1 = 2. 3) Система хлорид калия —
нитрат натрия, в к-рой может идти реакция: KCl+NaNO^
^tNaCl-l-KNOs. Число составных частей— четыре (КС1, NaNO%
NaCl, KNO3); число реакций — одна; число К. 4 — 1 = 3. 4) Та
же система в присутствии воды; число составных частей—
пять (КС1, NaNOs, NaCl, KNO3, Н2О); число реакций—одна,
число К. 5 — 1 = 4.
Какие составные части принимать за К., принципи-
ально безразлично. Напр., если в системе СаО — С02
за К. принять СаО и С02, то СаС03 следует рассмат-
ривать как продукт их соединения. Если же в той же
системе принять за К. СаСО3 и СаО, то С03 надо счи-
675
КОНВЕРСИЯ ГАЗОВ
676
тать продуктом термин. диссоциации СаСО8. В зави-
симости от числа К. различают: однокомпонентные
системы, двухкомпонентные, или двойные системы,
трехкомпонентные, или тройные системы, и т. д.
Системы с числом К. более трех наз. многокомпонент-
ными системами.
Лит.: Каблуков И. А., Правило фаз в применении
к насыщенным растворам солей, Л., 1933; Аносов В. Я.
иПогодин С. А., Основные начала физико-химического
анализа, М.—Л., 1947; Терминология физико-химического
анализа, ч. 1, М., 1951.
КОНВЕРСИЯ ГАЗОВ — процесс переработки га-
зов с целью изменения состава исходной газовой смеси.
Конвертируют обычно газообразные углеводороды
(метан и его гомологи) и окись углерода, с целью полу-
чения водорода или его смесей с окисью углерода.
Эти смеси используются для синтеза органич. про-
дуктов и в качестве газов-восстановителей в метал-
лургии или подвергаются дальнейшей переработке
для получения чистого водорода.
Конверсия газообразных угле-
водородов. Наиболее экономичным сырьем для
конверсии является метан (природный газ). Конвер-
сию проводят с применением различных окислителей.
Выбор последних и их возможные сочетания опреде-
ляются назначением процесса и технико-экономич.
соображениями. В качестве окислителей используют
кислород, водяной пар, двуокись углерода и их смеси.
Возможно также использование для этой цели окис-
лов металлов.
Конверсия метана различными окислителями может
быть описана следующими уравнениями:
СН4+Н2О-=ьСО+ЗН2-49,3 ккал (1)
СН4%СО2=±2СО+2Н2—59,1 ккал (2)
СН4+0,5О2=СО+2Н2+8,5 ккал (3)
СО+Н2От=±СО2+Н2+9,8 ккал (4)
Реакции окисления гомологов метана идут анало-
гичным образом. Изучением термодинамич. равнове-
сия реакций (1) и (2) установлено, что, при прочих
равных условиях, степень конверсии метана увеличи-
вается с ростом темп-ры и отношения Н2О/СН4 или
СО2/СН4 в исходной газовой смеси. Изменение дав-
ления практически не влияет на состав получаемого
газа при темп-рах выше 900° и соотношении Н2О/СН4
или СО2/СН4 больше 5. Скорость реакций (1) и (2)
очень мала, поэтому для достижения
полной конверсии метана требуются вы-
сокие темп-ры или применение катализа-
торов. Наилучшим катализатором являет-
ся никель, активированный добавками
окислов алюминия, магния, хрома и др.
Каталитич. конверсия метана с водя-
ным паром производится в трубчатых
аппаратах с внешним обогревом трубок
с катализатором. Примерный состав по-
лучаемого конвертированного газа: 8%
СО2, 15% СО, 75% Н2, 1,5% СН4, 0,5%
Na. Темп-ра реакционного слоя 700—
800°, молярное отношение Н2О/СН4 в
дутьевой смеси 2:1, объемная скорость
по метану 300—500 объемов в час на
1 объем катализатора.
В СССР разработан и получил про-
мышленное развитие автотермич. про-
цесс каталитической конверсии метана
смесью водяного пара и кислорода в аппаратах шахт-
ного типа (рис. 1). Смесь метана с водяным паром
подогревают в теплообменнике до 500—600° и направ-
ляют в смеситель, куда добавляют чистый кислород
(получение безазотистого газа) или воздух, обогащен-
ный кислородом (получение газа для синтеза аммиака).
Бессажевый режим процесса достигается перемеши-
ванием реагентов без воспламенения смеси, для чего
IV
Рис. 1. Схема шахтной
печи для каталитич. кон-
версии метана с кисло-
родом: I — ввод кисло-
рода; II — ввод парога-
зовой смеси; III — выход
продуктов реакции; IV—
ввод газов для разогрева
печи; V — ввод инерт-
ного газа (азота); 1 —
печь; 2 — смеситель.
создается скорость потока, значительно превышаю-
щая скорость распространения пламени (линейная
скорость смешанных газов на
выходе из смесителям! 00 м/сек}.
Примерный состав сухого га-
за, получаемого после конвер-
сии метана парокислородной
смесью, следующий: 9% СО»,
22% СО, 67% Н2,1% СН4,1%Ns“.
Расход реагентов на 1000 нм3
конвертированного газа: 322 и.ч3
природного газа; 163 нм3 кис-
лорода (95%-ного); 217 кг во-
дяного пара. Получает все
большее признание метод вы-
сокотемпературной некаталити-
ческой кислородной конверсии
углеводородов. Термодинамич.
анализом установлено, что при
1450° и давлениях до 30—35 ат
происходит практически пол-
ное окисление метана и других
углеводородов с получением СО
и Н2 при отсутствии свободного
углерода в продуктах реакции.
Преимущество метода — отсут-
ствие катализатора и неслож-
ное аппаратурное оформление
процесса при высоком давлении. Недостаток метода—
повышенный уд. расход кислорода (250—300 -и3 на
1000 нм3 конвертированного сухого газа). Примерный
состав сухого газа, получаемого при высокотемпера-
турной некаталитич. конверсии метана: 3—4,5% СО2,
36—38% СО, 57—59% Н2, 0,2—0,4% СН4, 2,0% N2.
Конверсия окиси углерода основана
на реакции (4), к-рая широко применяется при
произ-ве водорода. С понижением темп-ры реакция идет
в сторону образования Н2и СО2, т. е. степень превра-
щения СО возрастает. Т. к. при снижении темп-ры
скорость реакции уменьшается, процесс конверсии
обычно проводят с участием катализаторов. Опти-
мальная темп-ра процесса выбирается в зависимо-
сти от типа и активности катализатора, необходимой
степени конверсии СО, давления процесса и экономия,
соображений. Наиболее эффективными катализато-
Рис. 2. Схема одноступенчатой конверсии окиси углерода: 1 — сатуратор;
2 — газодувка; 3 — водогазосмеситель; 4 — теплообменники; 5 — отводной
трубопровод; 6 — конвертор; 7 — пусковой подогреватель; 8 — водонагрева-
тельная башня; 9 — холодильник; 10 — циркуляционные насосы.
рами оказались железо-окисные с различными до-
бавками Сг, Pb, Mg, Си и др. металлов. В промыш-
ленности процесс ведут при 425—450° и соотношении
пар: газ — 3:1.
Схема одноступенчатой конверсии окиси углерода
дана на рис. 2.
Лит.: Л е й б у ш А. Г., Химическая наука и пром-сть,
1956, 1, N 6; Альтшулер В. С., Шафир Г. А.,
677
КОНГО КРАСНЫЙ — КОНДЕНСАЦИИ РЕАКЦИИ
678
Тр. Ин-та горючих ископаемых АН СССР, 1959, 11; Л е ft-
бу ш А. Г. [и др.], Тр. Гос. и.-и. и проектного ин-та азотной
пром-сти, 1954—56, вып. 3—5; Лобачев А. Г., Достиже-
ния современной промышленности синтетического аммиака,
М., 1958; Иоффе В. Б., Основы производства водорода,
Л., 1960. В. С. Альтшулер.
КОНГО КРАСНЫЙ с32н24OeN2S2Na2, мол. вес
642,70 — азокраситель, получаемый сочетанием ди-
азотированного бензидина с двумя молекулами наф-
тионовой кислоты. Раствор К. к. в нейтральной или
слабокислой среде имеет красный цвет, в сильнокис-
лой среде (pH 5,2—3,0) — синий.
К. к. используют в химич. анализе как индикатор.
Лит.: Коган И. М., Химия красителей, 3 изд., М.,
1956, с. 136.
КОНДАКОВА РЕАКЦИЯ — присоединение гало-
генангидридов карбоновых к-т к олефинам и аце-
тиленам с образованием соответственно 0-галогено-
кетонов и 0-галогеновинилкетонов:
R-c/°+')C =С<"---- R-C-C-C-X
/ х II I I
о
R—С^°+ - СНС------R-C-C=C-X
Хх II
Катализаторами обычно служат апротонные к-ты,
например А1С13, ZnCl2, SnCl4, BF8. К. р. — типичное
электрофильное присоединение по кратной связи.
Первоначально образующийся комплекс (I) гало-
генангидрида с катализатором МеХп диссоциирует,
давая ацилкатион (II), к-рый атакует олефин (или
ацетилен) с образованием карбониевого иона (III)
или соответственно (IV). Последний, взаимодействуя
с комплексным анионом [MeX^J-, стабилизуется
присоединением аниона галогена, причем регенери-
руется катализатор:
//°
R—С? +МеХп—>RCOMeXn + 15=±R-C=O+[MeXn + ц-
(I) (И)
R—
(И)
—CZC-
I I [MeXn+1]~ I |
R-C-C-C+-----—Х-1—-R—С—С—С—Х-]-МеХ
О 6
(III)
I ! [MeXn + 1I~ ( I
R-С—С=С+--------It---- R—С—С=С—Х-]-МеХ
о
(IV)
о
При наличии атома водорода в a-положении к кар-
бонильной группе в молекуле 0-галогенокетона может
происходить отщепление галогеноводорода с обра-
зованней а, 0-ненасыщенного кетона:
I 1 —НХ I I
R—С-СН-С—X —S±»R-C-C=C-
А 1 А
Иногда элиминирование галогеноводорода приводит
к 0, у-ненасыщенным кетонам, В ряде случаев, в
зависимости от строения олефина и условий реакции,
непредельные кетоны оказываются главными продук-
тами К. р. а-Галогеновинилкетоны в условиях К. р.
галогеноводорода не отщепляют.
Из побочных продуктов К. р. в нек-рых случаях
могут быть выделены насыщенные кетоны, образова-
ние к-рых, по-видимому, связано с присоединением
карбониевым ионом (III) гидрид-иона, отрываемого
от молекулы растворителя:
I I II
С-С-С++Н-—-R—С—С—С-
V 1 А 1 1
(Ш)
Обычно для К. р. готовят комплекс галогенангидрида
с катализатором и затем вводят олефин или ацетилен;
при получении ₽-галогенвинилкетонов катализатор
часто прибавляют постепенно, небольшими порци-
ями. Средой служит или избыток галогенангидрида,
или инертный растворитель, хорошо растворяющий
вышеупомянутый комплекс, напр. хлористый метилен,
дихлорэтан, хлороформ, четыреххлористый углерод,
нитробензол. Применение петролейного эфира или
сероуглерода, в к-рых комплекс нерастворим, дает
худшие результаты. Темп-ру реакции варьируют от
—10 до -+-20°; в отдельных случаях нагревают до
70—80°. Повышение темп-ры способствует образова-
нию ненасыщенных кетонов.
В К. р. вступают: ациклич. олефины — этилен,
пропилен, бутилены, триметилэтилен, тетраметил-
этилен; алициклические — циклопентен, циклогек-
сен и их гомологи; галогенозамещенные этилены —
хлористый винил, 1, 2-дихлорэтилен, трихлорэтилен,
фтористый винилиден, трифторэтилен; терпеновые
и сесквитерпеновые углеводороды. Из алкинов были
использованы ацетилен, фенилацетилен, моно- и
диалКилацетилены. В качестве ацилирующих агентов
обычно применяют хлорангидриды, реже — броман-
гидриды (в последнем случае катализатор — А1Вг3).
Хорошие результаты получены с хлорангидридами
уксусной, пропионовой, масляной, изомасляной,
кротоновой, хлоруксусной, бромуксусной, а- и 0-
хлорпропионовой к-т. Вступают в К. р. хлоран-
гидриды фенилуксусной, трихлоруксусной и дру-
гих к-т.
Описаны варианты К. р. с применением уксусного
ангидрида вместо хлористого ацетила, однако при
этом образуются те же продукты реакции, что и в
случае хлористого ацетила, т. к. ион карбония III
или IV, взаимодействуя с комплексным анионом
[ZnCl2(CH3COO)] ~, стабилизуется присоединением ани-
она галогена, а не ацетата. Для реакции с уксус-
ным ангидридом в качестве катализатора мо-
жет быть применена серная к-та. При этом
п образуются непредельные кетоны. К. р. откры-
та в 1892 И. Л. Кондаковым. К ацетиленам
ее впервые применили Корнилло и Алкейр в
1934.
п Лит.: Белов В. Н., Рудольфи Т. А.,
в сб.: Реакции и методы исследования органических
соединений, кн. 7, М., 1958, с. 255; Кочет-
ков Н. К., Усп. хим., 1955, 24, вып. 1, 32; К н у-
н н н ц И. Л. 1и др.], Изв. АН СССР. Отд. хим. наук,
1958, № 3, 296. В. Л. Дяткин.
КОНДЕНСАЦИИ РЕАКЦИИ — исторически за-
крепившееся в органич. химии название большой
группы реакций самого различного характера как
в отношении природы реагентов, так и в отношении
существа имеющих место превращений. В более огра-
ниченном значении К. р. представляют собой внутри-
молекулярный (а) или межмолекулярный (6) про-
цесс образования новой С—С-связи, сопровождаю-
щийся элиминированием к.-л. простой неерганич.
679
КОНДЕНСАЦИИ РЕАКЦИИ
680
или органич. молекулы:
ь
К. р. обычно протекают при участии конденсирующих
средств, роль к-рых может быть самой различной:
они могут оказывать чисто каталитич. действие, или
давать реакционноспособные промежуточные продукты,
или просто связывать отщепляющуюся частицу X—Y,
смещая равновесие в системе. Чаще всего их действие
является комбинированным. Строгой специфичности
конденсирующие средства не проявляют, однако их
можно приблизительно сгруппировать по типам К. р.,
соответственно характеру элиминируемой частицы.
К. р. с отщеплением воды могут проходить по одной
из следующих схем:
\ । I I
^С-ОН+Н-С-------С-С- +Н2О (I)
^>с=о+н2с<^—Ас =с<<рн2о • (И)
\ н \ /с^
\с = О+ —Ч-Н2о (III)
Н-С^ с\
Схеме (I) соответствуют реакции алкилирования аро-
матич. и непредельных соединений спиртами, напр.:
CeH3CH2OH+CeHe-^-^CeH3CH2CeH3
н+
(СНз)зСО Н-рН2С=С(СНз)2—*(СНз)аС—СН=С(СНз)2
реакции хлорметилирования:
CGH0-f-CH2=O-|-HCl-------.СвН5СН2С1+Н2О
самоконденсации жирных спиртов:
С Н8
2СНзСН2СНаОН-5^-^-»СН3СН2СН2СНСН2ОН
И Т. П.
По схеме (II) протекают кротоновая конденсация
и многочисленные родственные процессы, напр. Пер-
кина реакция, Кнёвенагеля реакция и т. д., а по схеме
(III) — многие синтезы соединений ряда трифенил-
метана:
ZnCb .C6H4N(CH3)2
CeH3CHO+2CeH3N(CH3)s—-^CeH3CH<
\свНаН(СНз)з
Отщепление воды катализируется разнообразными
реагентами — сильными протонными и апротонными
к-тами, напр. H2SO4, НС1, HF, А1С13, ZnC^, сильными
щелочами, напр. гидроокисями, алкоголятами, ами-
дами, гидридами щелочных металлов, ZnO, РЬО,
аммиаком, первичными и вторичными аминами.
К К. р. с отщеплением галогеноводородов относят,
напр., алкилирование и ацилирование по Фриделю—
Крафтсу (см. Фриделя — Крафтса реакция}, синтезы
Гаттермана — Коха и Гёша (см. Гаттермана —
Коха реакция, Гёша синтез}, катализируемые гл. обр.
апротонными к-тами, а также процессы, происходя-
щие под действием щелочей, напр. Реймера—Тиманна
реакция и Дарзана реакция, алкилирование кетонов
иодистыми алкилами, напр.:
C2H5J+CHsCOCoH5-^i^CHsCH2CH2COCeH5 ИТ. п.
К. р. с отщеплением неорганич. солей — это син-
тезы с патрмалоновым и натрацетоуксусным эфирами,
взаимодействие галогенопроизводных с солями си-
нильной к-ты и Na-производными ацетиленов:
RX+NaCN—-RCN+NaX
RX+NaC=C—R’—>RC=CR’+NaX
С-алкилирование фенолятов:
и т. д. Сюда же можно отнести Реформатского реак-
цию:
RCHO+BrCH2COOC2H5+Zn(+H2O)—-
--»RCHCH2COOC2H3+Zn(OH)Br
I
он
и значительную часть других синтезов с металлоор-
ганич. соединениями:
\c=O+RMgX (+Н2О)---.\C-R+Mg(OH)X
ОН
К. р. с отщеплением галогенов происходит в основном
при действии различных металлов — Ag, Na, К, Си,
Mg, Zn. К этому типу К. р. относятся реакции Вюрца,
Фиттига, Ульмана. Соответственно, К. р. с отщепле-
нием металла происходят либо в отсутствие конден-
сирующих средств, либо при действии галогенов:
2СН3-С=С-Си----. СНз-С^С--С = С-СНз+2Си
2NaCH(COOC2H3)2i(C2H3OOC)2CH-CH(COOC2H3)2+2NaJ
К. р. с отщеплением азота включают многочислен-
ные реакции жирных и ароматич. диазосоединенип,
напр.:
/О
R—СУ 4-CH2N2—»R-C-CH3-FNs
ЧН II
о
^C=C<^+CH2N2-.\с----C^+Ns
\сн2^
ArN=NCl+\c=C<^—>Аг—С—С— Cl+N2
К. р. с отщеплением водорода представляют по су-
1 зству процессы окисления, независимо от того,
происходят ли они пирогенно или с участием окисли-
телей:
^с-н+н-с^—Ус-с^+н2
-хс-н + о + н-с^—> ^с-с^+ н2о
Типичным примером пирогенной реакции является
образование дифенила из бензола:
Такие реакции катализируются активным углем,
Ni, Pt, Pd, SiO2, A12O3, в ряде случаев — А1С13,
SbCl3, SnCl4, ZnCl2. Примером К. р. с участием окис-
лителя, О2, может служить превращение индоксила
в индиго:
Окислителями являются: FeCl3, NaOCl, H2SO4, HNO3,
KMnO4, Na2Cr2O7, PbO, PbO2, S, галогены, нитробен-
зол, нитрозобензол. К К. p. с отщеплением кислорода
или серы относятся нек-рые восстановительные про-
631
КОНДЕНСАЦИИ РЕАКЦИИ — КОНДЕНСАЦИЯ
682
цессы, напр.:
(CH3)2NC6H4 (СН3)2\С6Нч xCf,H,,N(CH3)2
хС=О ----------- с=с
(CH3)2NC6h/ (CH3)2NC6h/ 4C6H4N(CH3)2
Ряд процессов К. р. сопровождается отщеплением
органич. молекул, напр. спиртов. К этому типу
принадлежат сложноэфирная конденсация, Клайзена
конденсация и Дикмана реакция. Конденсирующими
средствами.нвляются щелочные металлы, их гидрооки-
си, алкоголнты, амиды, гидриды. Разделение К. р. по
роду элиминируемой частицы во многом условно,
т. к., с одной стороны, часто происходит отщепление
двух или более частиц, а с другой — такое разделение
не учитывает механизма реакции.
Самым существенным распространением термина
К. р. по сравнению с рассмотренным выше является
включение в это понятие всех процессов образовании
новой С—С-связи, независимо от того, происходит
элиминирование к.-л. простой частицы или нет. Сюда
относятся, напр., многочисленные случаи альдольной
конденсации, зачастую представляющей собой пред-
варительную стадию кротоновой конденсации:
СН3СОСН3+СНС13—>(СН3)2С(ОН)СС13
СН3СНО—.CH3CH(OH)CII»CHO^-!i^LcH3CH-.CIICHO
бензоиновая конденсация:
ArCHO-f-Ar’CHO—-Аг—С--СН--АГ'
II I
О он
различные случаи бимолекулярного восстановлении:
СНзСОСНз
сн3. ,сн3
\q___QX
CH3ZI ЛсНз
он он
2CH2=CHCN—►ncch2ch2ch2ch2cn
диеновый синтез:
реакции заместительного присоединения:
хСН2 о
СН "сн - с.
II I
сн2 сн2-с'
'о
различные реакции раскрытия кратной связи и окис-
ного кольца соединениями с подвижным атомом водо-
рода:
-Ас-Н + СН2—CH—CN—.AC-CH2CH2CN
Ас—Н-|-СН2-СН3---«Ас—СН2СН2ОН
о
В органической химии К. р. наз., кроме того, все
реакции, в результате к-рых происходит образование
гетероциклов. В этих процессах могут возникать но-
вые связи: углерод—углерод, углерод—гетероатом,
гетероатом—гетероатом. Обычно они сопровождаются
элиминированием одной или нескольких частиц и осу-
ществляются в присутствии конденсирующих средств
рассмотренных выше типов. По аналогии с кротоновой
конденсацией применяют название К. р., напр. по
отношению к таким реакциям:
CcH^NHa-pOHCCoIIs-.CeII5N=CHCeH3+H2O
CeH5NHs+ONCeHj—.CcII5N=NC6H5+H2O
Обычно к К. р. не относят этерификацию, переэтери-
фикацию, алкилирование и ацилирование по кислороду
или по азоту и т. п., однако происходящие по этим
схемам процессы образования полимеров объединяют
под термином поликонденсация.
Лит.: Houben J., Die Methoden der organischen Che-
mie, Bd 2, 3 Aufl., Lpz., 1925, S. 716. Б. Л. Дяткин.
КОНДЕНСАЦИЯ — переход вещества из газооб-
разного (парообразного) состоянии в жидкое или
твердое. К. происходйт при изотермич. сжатии газа
(пара), охлаждении его при постоянном давлении или
таком одновременном изменении его давлении и
темп-ры, что они достигают значений, при к-рых кон-
денсированная фаза более устойчива. Если при этом
давление и темп-ра выше их значений в тройной точке
данного вещества, оно переходит из газообразного
состонния в жидкое (сжижение), если же давление и
температура ниже, чем в тройной точке, то вещество
переходит из газообразного состонния в твердое,
минуя жидкое (кристаллизация из паров). В зависи-
мости от условий К. (естественные, в промышленных
аппаратах и т. п.) конденсированная фаза может об-
разовываться в объеме пара или на поверхности более
холодного тела, с к-рым соприкасается пар.
Конденсация в объеме пара (инди-
видуального или содержащегося в многокомпонент-
ной смеси) наблюдается при его расширении, охлаж-
дении парогазовой смеси вследствие лучеиспускания,
смешении влажных газов, имеющих разную темп-ру,
и химич. взаимодействий газообразных веществ.
Конденсированная фаза образуется в этих случаях
в виде мелких капель жидкости (тумана) или мелких
кристаллов. Для образования конденсированной фазы
в объеме необходимо наличие центров (ядер)
К. При их отсутствии изменение параметров пара
может приводить к длительно сохраняющемуся мета-
стабильному пересыщенному его состоянию. При этом
изменения фазового состонния не происходит, несмотря
на то, что при достигнутых параметрах часть пара
должна была бы сконденсироваться. Степень пересы-
щении характеризуется отношением: П = р!рн, где
р — действительное давление пара, />и—давление на-
сыщения пара при данной темп-ре.
Центрами К. могут служить очень мелкие капельки
жидкости (зародыши), самопроизвольно образующиеся
в результате флуктуаций плотности газовой фазы,
взвешенные в ней частички твердых примесей (пыли)
или газовые частицы, имеющие электрич. заряд (ионы).
Так как равновесное давление пара над поверхностью
частиц с очень малым радиусом кривизны выше дав-
ления пара того же вещества над плоской поверх-
ностью (см. Капиллярные явления), К. в объеме и при
наличии центров К. может начаться лишь при опре-
деленном пересыщении пара П > Пкр = рк/рн, где
рк — равновесное давление пара над зародышами.
Величину Пкр наз. критич. пересыщением пара. При
П с Пкр образующиеся в объеме зародыши конден-
сированной фазы вновь испаряются, а при П > Пкр
возникает устойчивая К. После того как К. началась,
в результате роста капель жидкости быстро достигает-
ся равенство рк я» рн .Критич. пересыщение состав-
ляет, напр., для водяного пара в воздухе, освобож-
денном от взвешенных твердых частиц "и ионов, от
5 до 8. С увеличением темп-ры, а особенно при нали-
683
КОНДЕНСАЦИЯ
684
чии твердых частиц и ионов, величина критич. пере-
сыщения уменьшается.
Когда К. в объеме нежелательна, она может быть
предупреждена путем создания условий, при к-рых
максимальное пересыщение пара ниже критич. Необ-
ходимость в этом может возникать, напр., при улав-
ливании летучих растворителей (этилового спирта,
эфира, бензола, толуола или др.) путем К. на поверх-
ности, получении серной к-ты нитрозным методом,
осушении газа серной к-той и т. п.
Конденсация на поверхности
твердого тела или жидкости происходит
при любом состоянии пара — насыщенном или пере-
гретом, — когда темп-ра поверхности ниже темп-ры
насыщения при данном давлении. В присутствии по-
ристых тел К. может происходить и при давлении
пара, меньшем, чем давление насыщения при данной
темп-ре (см. Капиллярная конденсация). К. на поверх-
ности имеет место во многих технологических и теп-
лообменных аппаратах (конденсаторы выпарных, рек-
тификационных, холодильных и энергетич.установок,
скрубберы для охлаждения газов, кондиционеры
и т. д.). Эти аппараты разделяются на две основные
группы: поверхностные, в к-рых подлежащий К. пар
и более холодная среда (вода или другая жидкость,
пар или газ) разделены между собой твердой стенкой,
и контактные (смешивающие), в к-рых пар непосред-
ственно соприкасается с более холодной жидкостью.
Скорость К. в поверхностных аппаратах (количе-
ство пара, конденсирующегося в единицу времени на
единице поверхности) при данной разности темп-р
пара и твердой стенки Дг зависит в основном от ско-
рости отвода теплоты фазового перехода, выделяю-
щейся при К., и характеризуется величиной коэфф,
теплоотдачи
GK Л i ...
а = -=-— (кал/сек • см2 • граЭ) (1)
Г “I
где Ск — количество сконденсировавшегося пара
(г/сек); Д£— разность энтальпии пара и конденсата
(кал/г); К — поверхность охлаждения (смг). Скорость
отвода теплоты, выделяющейся при К., зависит от
ряда факторов, среди к-рых весьма существенным
является характер К.
При образовании на твердой стенке жидкой фазы
возможны: пленочная К., при к-рой стекающий
конденсат образует сплошную пленку, покрывающую
всю поверхность, и капельная К., при к-рой
на поверхности образуются мелкие капли конденсата,
а между ними пленка, разрывающаяся после дости-
жения очень небольшой толщины (для воды — по-
рядка микрона). Первый вид К. наблюдается на по-
верхности, хорошо смачивающейся конденсатом, вто-
рой — на несмачивающейся (гидрофобной) поверх-
ности. Образующиеся на гидрофобной поверхности
очень мелкие капли затем растут в результате слияния
их между собой, подтягивания конденсата из разо-
рвавшейся пленки и образования нового конденсата.
Наиболее крупные капли стекают вниз, объединяясь
(коалесцируя) с нижележащими мелкими каплями,
после чего на освободившейся поверхности опять
образуются мелкие капли, и цикл повторяется вновь.
Несмачиваемость металлич. поверхности достигается
при помощи различных гидрофобизаторов (напр.,
при К. водяного пара — жирных к-т, меркаптанов,
пленкообразующих аминов или др., см. также Гид-
рофобные покрытия), непосредственно наносимых
на поверхность или вводимых в пар. В случае ка-
пельной К. коэфф, теплоотдачи а намного выше,
чем при пленочной К. Однако поддержание в усло-
виях эксплуатации промышленных аппаратов устой-
чивой капельной К., особенно когда поверхность
стальная, связана с рядом трудностей, вследствие
чего в этих аппаратах наблюдается, как правило,
пленочная К. При пленочной К. коэфф, теплоотдачи
существенно зависит от режима течения пленки кон-
денсата: при ламинарном течении отвод тепла фазо-
вого перехода через пленку происходит в результате
молекулярной теплопроводности, в случае же тур-
булентного течения преобладающее влияние приоб-
ретает турбулентное перемешивание жидкости, резко
повышающее интенсивность переноса тепла через
пленку.
Режим течения конденсата зависит от величины критерия
Рейнольдса для пленки Нек = ^ =>—Г_ , где w— средняя
по сечению скорость пленки конденсата; 6 — толщина пленки;
Г — весовой расход конденсата на единицу периметра (или
ширины) поверхности; vK и цк — кинематич. и динамич. вяз-
кость конденсата; g — ускорение силы тяжести. При относи-
тельно малых ReK течение ламинарное, при неподвижном паре
иЯек= 10—30 оно переходит в т.н. волновое(псевдоламинарное)
течение, а приНек =а 400 — в турбулентное. В случае движу-
щегося пара в результате внешнего возмущения, вызываемого
механич. взаимодействием между паровым потоком и жид-
костью, переход от одного режима течения пленки конденсата
к другому происходит при более низких значениях ReK. При
пленочной К. на вертикальных поверхностях (трубах, стенках)
величина ReK растет по мере движения пленки конденсата свер-
ху вниз в результате увеличения расхода конденсата Г и в
зависимости от высоты поверхности могут наблюдаться раз-
личные режимы ее течения. В области ламинарного и волнового
течения увеличение толщины пленки вызывает уменьшение
местных значений коэфф, теплоотдачи а, а переход к турбулент-
ному течению — их рост. При К. неподвижного пара на наруж-
ной поверхности горизонтальной трубы течение пленки кон-
денсата обычно ламинарное, но движущийся пар может и в
этом случае вызывать возмущение ее течения. Когда имеется
большое число расположенных друг под другом рядов гори-
зонтальных труб, конденсат, стекающий с верхних труб,
увеличивает толщину пленки конденсата на нижних трубах,
что уменьшает коэфф, теплоотдачи. Но этому противостоит
действие парового потока, увеличивающее местные и среднее
значения коэфф, теплоотдачи.
Коэфф, теплоотдачи при пленочной К. неподвижных чистых
(одиокомпонентных) насыщенных паров, и ламинарном течении
пленки конденсата составляет по теоретической ф-ле Нус-
сельта
.1 К ' п- I
а = А I --туг-
\ /
(2)
где — коэфф, теплопроводности (кал7сеК‘СМ’град.)9
плотность (г/слг8), динамич. вязкость (г/сек-слг2) конденсата;
г — теплота К. (кал/г); — температурный напор (разность
темп-р пара и стенки) (°C); I — характерный размер поверх-
ности (высота вертикальной трубы или стенки, диаметр гори-
зонтальной трубы, см); А — постоянный множитель, равный
в случае вертикальной поверхности 0,943 и в случае горизон-
тальной трубы 0,725. При волновом течении пленки конденсата
на вертикальной поверхности А — 1,14.
Эта ф-ла справедлива для паров жидкостей с числом Пран-
дтля PrK = vK/aK > 1 и при параметре К = r/cKAt > 5 (где
аки ск — козфф. температуропроводности и уд. теплоемкость
конденсата). При более низких значениях Ргк и К, наблюдаю-
щихся, как правило, только при К. паров жидких металлов,
козфф. теплоотдачи может быть значительно меньшим, чем это
следует из приведенной ф-лы. При турбулентном течении плен-
ки конденсата эта ф-ла также непригодна; для этого случая
предложен ряд полуэмпирич. и чисто экспериментальных за-
висимостей.
При нисходящем потоке пара средняя скорость течения
ламинарной пленки конденсата увеличивается, а толщина ее
уменьшается, при направлении же потока пара снизу вверх
пленка конденсата подтормаживается и утолщается, но повы-
шение скорости пара сверх нек-рого предела может привести
к изменению направления (обращению) движения пленки и
уменьшению ее толщины. Большее влияние на величину коэфф,
теплоотдачи оказывает, однако, режим течения пленки и,
следовательно, скорость пара. При движении пара внутри
труб пли в межтрубном пространстве трубных пучков скорость
его падает от значительной величины до очень малой, вплоть
да нуля. В этих случаях начальная скорость пара и пределы
ее изменения оказывают большое влияние на величину сред-
него коэфф, теплоотдачи а.
В случае К. перегретого пара возникает
конвективный теплообмен между паром и конденсатом,
имеющим на наружной поверхности темп-ру насы-
щения при данном давлении. Однако суммарный теп-
ловой поток при этом лишь незначительно возрастает
685
КОНДЕНСАЦИЯ
686
по сравнению с К. насыщенного пара при том же дав-
лении и той же темп-ре стенки. Это позволяет поль-
зоваться для определения коэфф, теплоотдачи при
К. перегретого пара зависимостями, полученными для
К. насыщенного пара, в частности формулой (2),
если принимать в ней Al = tB — tCT , где tB — темп-ра
насыщенного пара при данном давлении и гст —
температура стенки.
При К. смеси паров условия могут быть
существенно различными в зависимости от того,
является ли конденсат гомогенной или гетерогенной
смесью. В первом случае наблюдается обычно пленоч-
ная К. При этом коэфф, теплоотдачи приближенно
определяют по ф-ле для чистых однокомпонентных
паров, предполагая, что на поверхности раздела фаз
существует равновесие между жидкой и паровой фа-
зами каждого компонента смеси. Во втором случае
наблюдаются одновременно оба типа К., напр. при
К. смеси, состоящей из паров органич. вещества (бен-
зола, толуола или др.) и водяного пара, на поверх-
ности образуется пленка жидкого органич. вещества,
покрывающаяся каплями воды. Коэфф, теплоотдачи
при этом приблизительно равен:
a = aifl + —----Г2У/4 (3)
\ ‘ 1 — Г1 J ' '
где 04 — коэфф, теплоотдачи для случая К. чистых
паров органич. вещества; у2 — весовое содержание
воды в конденсате; rt и г2 — теплота К. соответственно
органич. и водяных паров. Множитель при аг в
ф-ле (3) учитывает увеличение количества теплоты,
передаваемой через пленку жидкости.
К. пара на поверхности жидкости
того же вещества характеризуется обычно значительно
большей интенсивностью по сравнению с пленочной К.
на поверхности твердого тела. Скорость К. на поверх-
ности медленно движущейся жидкости примерно того
же порядка, что при капельной К. на поверхности
твердого тела; значительно выше скорость К. на по-
верхности быстродвижущейся турбулентной струи
жидкости, когда теплота фазового перехода очень ин-
тенсивно отводится от поверхности раздела. При не-
посредственном контакте пара и охлаждающей жид-
кости поверхность К. может быть увеличена путем
раздробления последней на капли или пленки.
Присутствие неконденсирующих-
ся газов в паре уменьшает скорость К., так
как затрудняет доступ пара к поверхности раздела
фаз. Концентрация инертных примесей у последней
возрастает по сравнению с основной массой парога-
зовой смеси, и перенос пара к поверхности К. проис-
ходит путем диффузии и конвекции. В этом случае на
поверхности раздела фаз как темп-ра, так и парци-
альное давление пара соответственно ниже темп-ры
и парциального давления пара в основной массе паро-
газовой смеси. Поэтому при пленочной К. на твердой
стенке между парогазовой смесью и пленкой конден-
сата возникают одновременно тепло- и массообмен.
Теплота, отданная смесью в результате конвектив-
ного теплообмена и конденсации пара, передается
через пленку конденсата к стенке.
Весовое количество пара, достигающего поверх-
ности К., определяется для элемента поверхности
dF ф-лой:
= (Рп Рп. гр.)^ (4)
где Рр — коэфф, массоотдачи;рв ирв гр— парциальные
давления пара в основной массе парогазовой смеси
и у поверхности раздела фаз. Общее количество
теплоты, отдаваемой парогазовой смесью на элементе
поверхности, составляет:
= аж(ггр. — гст.) dF=rdGu-\-4t (tu — ггр)dF, (5)
где аж — коэфф, теплоотдачи от наружной поверх-
ности пленки конденсата к стенке; 1П, 1гр и 1СТ —
темп-ры пара (парогазовой смеси) на границе раздела
фаз и стенки; аг — коэфф, теплоотдачи от парогазовой
смеси к пленке конденсата. Коэфф. аж зависит от
толщины пленки конденсата, характера ее течения
‘и физич. параметров жидкости, коэфф. Рр и аг — от
характера течения, состава и физич. параметров ком-
понентов парогазовой смеси. Кроме того, все назван-
ные коэфф, зависят от формы и размеров поверхности К.
Если, как и в случае К. чистого пара, ввести общий
коэфф, теплоотдачи, отнесенный к разности темп-р
пара и стенки, т. е. a = dQ/(tB— lCT)dF, то с учетом
(4) и (5):
я=м_+
I “ж
(6)
Рп — Рп. гр.
р
п ~ !гр.
Отсюда видно, что в рассматриваемом случае К.
коэфф, теплоотдачи а определяется интенсивностью
взаимосвязанных процессов различной физич. при-
роды (теплообмена и массообмена) и находится в слож-
ной зависимости от параметров и условий течения
парогазовой смеси и жидкости. При прочих одинако-
вых условиях он всегда ниже коэфф, теплоотдачи при
К. чистого пара и умень-
шается тем значительнее,
чем больше содержание
инертного газа в паро-
газовой смеси и чем
меньше скорость (число
Рейнольдса) последней
(см. рисунок). Для рас-
чета процесса К. в при-
сутствии неконденсирую-
щихся газов пользуются
ур-ниями (4) и (5) и опыт-
ными зависимостями для
ВХОДЯЩИХ в эти ур-ния
коэфф, тепло- и массоот-
дачи.
Условия К. в твер-
дое состояние су-
щественно отличаются от
рассмотренных выше. В
этом случае конденсиро-
ванная фаза не может сте-
Объемное содержание воздуха
Влияние примеси воздуха на
коэфф, теплоотдачи при К. во-
дяного пара на горизонтальной
трубе. ReCJlt — Число Рейнольд-
са парогазовой смеси.
кать по поверхности и толщина ее слоя непрерывно
возрастает, вследствие чего процесс К. является неста-
ционарным и его скорость все время понижается.
Кроме того, при очень глубоком вакууме (когда сред-
няя длина свободного пробега молекул соизмерима
с характерным размером аппарата) изменяется меха-
низм переноса пара и тепла в газовой фазе, что меняет
условия К. как чистого пара, так и пара, содержащего
примесь неконденсирующихся газов. Зависимости для
скорости К. меняются при этом не только количест-
венно, но и качественно.
К. широко используется в различных технологич.
процессах. В природе с К. связано образование росы,
инея, облаков, тумана и др. На явлении К. основано
устройство одного из важнейших физич. приборов —
камеры Вильсона. См. также Аэрозоли.
Лит.: Амелин А. Г., Теоретические основы образо-
вания тумана в химических производствах, М-.—Л., 1951;
Берман Л. Д., Изв. Всес. теплотехнического ин-та,
1953, М 3; е г о же, Теплоэнергетика, 1957, №6; Берман
Л. Д. и Ф у к с С. Н., Теплоэнергетика, 1958, N 8;Гр е-
б е р Г., Эрк С. и Григулль У., Основы учения о
теплообмене, пер. с нем., М., 1958; Кутателадзе С. С.,
Теплопередача при конденсации и кипении, 2 изд., М.—Л.,
1952; Михеев М. А., Основы теплопередачи, 3 изд.,
М.—Л., 1956; Ш у м с к и Й К. П., Вакуумные конденсаторы
химического машиностроения, М., 1961. Л. Д. Берман.
687
КОНДЕНСАЦИЯ ФРАКЦИОНИРОВАННАЯ
688
КОНДЕНСАЦИЯ ФРАКЦИОНИРОВАННАЯ -
метод разделения паровых и газовых смесей на фрак-
ции или практически чистые компоненты путем их
последовательной парциальной конденсации. Так как
по мере охлаждения газовой смеси конденсируются
преимущественно высококипящие компоненты, то
несконденсировавшийся остаток обогащается низко-
кипящими компонентами. Различают К. ф. прямо-
точную, сопровождающуюся длительным соприкосно-
вением образовавшегося конденсата с остаточным
паром, и противоточную, характеризующуюся немед-
ленным удалением конденсата из системы.
При прямоточной К. ф., Dt кг-молей исход-
ной бинарной газовой смеси с концентрацией низко-
кипящего компонента у1 моль/моль охлаждаются
в конденсаторе 1 до t2, к-рая ниже начала их конден-
сации (рис. 1 и 2). Da кг-молей несконденсировав-
шегося газа с концентрацией у2 моль/моль и (Dj— D2)
Рис. 1. Схема прямоточной фракционированной
конденсации: 1,3 — конденсаторы; 2, 4 — сепа-
раторы.
Многократно повторяя процессы парциальной кон-
денсации и механического разделения фаз, можно
получить то или иное количество практически чи-
стого низкокипящего компонента или ряд фракций
с нарастающей концентрацией указанного компо-
нента.
Противоточная К. ф. производится чаще
всего в вертикальных трубчатых конденсаторах при
движении газа (пара) снизу вверх (рис. 3). Образую-
щийся при этом конден-
сат стекает вниз, не со-
прикасаясь с той газовой
смесью, из которой он
выделился. Если прене-
бречь межфазовым обме-
ном между стекающим
конденсатом и восходя-
щим потоком газа, то
материальный баланс по
низкокипящему компо-
ненту процесса конден-
сации на элементарной
поверхности охлаждения
dF выражается в том,
что происшедшее изме-
нение количества и кон-
центрации компонента в газовой фазе d (Dy)
няется количеству низкокипящего компонента,
решедшему в образовавшийся конденсат:
♦'г
Хладоагент
Пар
Хладоагент
О,
Рис. 3. Схема противоточной
фракционированной конденса-
ции.
рав-
пе-
d (Dy) — xdD (5)
откуда получается уравнение Рэлея
кг-молей конденсата с концентрацией х2 моль/моль
ботанически разделяются в сепараторе 2. Ур-ние
материального баланса рассматриваемого процесса
DiJ/i = D2y2 (Dt— D2) xs (1)
откуда
4?S___V1 ^2 /Q\
£>1 1/2 — «2
«г УЛг Уг /2' ".У
Рис. 2. Диаграмма (I — х, у) прямо-
точной фракционированной конден-
сации.
При необходимости D2 кг-молей несконденсировав-
шегося газа дополнительно охлаждают в конден-
саторе 3 до t3, и вновь образовавшийся конденсат
в количестве (D2—D/) кг-молей отделяют от остатка
газа в сепараторе 4.
Материальный баланс
этого процесса пов-
торной парциальной
конденсации выра-
жается уравнением,
аналогичным (1). Если
начальному состоя-
нию газовой смеси
на диаграмме I— х,у
(рис. 2) соответствует
точка А и во всех
сечениях конденсато-
ра достигается фазо-
вое равновесие, то
после понижения темп-ры смеси до t2 ее состояние
изобразится точкой С, а состояние конденсата и
несконденсировавшегося газа — соответственно точ-
ками А и В. Согласно правилу рычага, между коли-
чествами конденсата и остаточного газа существует
отношение:
Р1-Р8 _ вс
Р2 _ АС ( '
а после повторной конденсации при темп-ре t3
d2 - Pj____ df
D'a ED
D±
D
где у и х — равновесные концентрации низкокипя-
щего компонента в газовой и жидкой фазах, ух и
у2 — начальная и конечная концентрации низко-
кипящего компонента в газовой фазе.
Уравнение (6) может быть использовано для опре-
деления количества газового остатка Da, если за-
дано у2, и наоборот. Если зависимость между у и х
выражается простым ур-нием, то интеграл решается
аналитически, в противном случае он решается гра-
фически при помощи диаграммы равновесия у — х.
В случае многокомпонентных паровых (газовых)
смесей прямоточная и противоточная конденсации
рассчитываются способом постепенных приближений.
Дефлегмация, применяемая для частичного
обогащения воздуха кислородом, представляет разно-
видность противоточной К. ф.
К. ф., не обеспечивая в большинстве случаев раз-
деления газовых смесей на чистые компоненты, состав-
ляет физич. основу ряда промышленных методов раз-
деления газовых смесей на фракции, обогащенные
отдельными компонентами. Самостоятельное прак-
тическое значение К. ф. имеет при разделении нек-рых
многокомпонентных газовых (напр., газы коксовых
печей, пиролиза и т. п.) и бинарных смесей, компо-
ненты к-рых имеют значительно отличающиеся темп-ры
конденсации (напр., СН4—Не, N2—Не, N2—Н2ит. д.).
См. также Газов разделение.
Л.ит:: Гельперин Н. И., Дестилляция и ректифика-
ция, М.—Л., 1947; Г е р ш С. Я., Глубокое охлаждение,
ч. 1—2, 3 изд., М.—Л., 1957—60; Павлов К. Ф. и М а л-
к о в М. П., Холод в химической промышленности, М
1937 (ИПН-ВСНИТО. Хим. ф-т, вып. 18); Фастовский
В. Г., Петровский Ю. В., Автогенное дело, 1945,
11—12; Фастовский В. Г., Разделение газовых
смесей, М.—Л., 1947; Klrscli Ьгиит Е., Destillier- imd
Rektlflzlertechnik, В., 1950; Robinson С. S., Gil-
liland E. R., Elements of fractional distillation, N. Y. —
la. oJ, 1950 (Chem. Engng. ser.).
H. 11. Гелыгерин, В. Л. Дебалк.
689
КОНДЕНСИРОВАННАЯ СИСТЕМА
690
КОНДЕНСИРОВАННАЯ СИСТЕМА — система,
состоящая только из твердых или жидких (т. е. кон-
денсированных) фаз, а также их смесей. К. с. не со-
держит газообразных частей. Характерная особен-
ность К. с. — малая зависимость происходящих в них
процессов от давления (напр., повышение давления
на 1 атм понижает точку плавления льда лишь
на 0,0075° и повышает точку плавления олова лишь
на 0,0033°). Это резко отличает К. с. от систем с газо-
образными частями. Благодаря указанной особен-
ности исследования К. с. при атмосферном давлении
с большой точностью могут считаться произведен-
ными при постоянном давлении. К числу К. с. при-
надлежат сплавы металлов, сплавы солей, растворы
нелетучих веществ (напр., обычных солей) в нелету-
чих растворителях, сплавы силикатов и т. д.
КОНДЕНСИРОВАННЫЕ АРОМАТИЧЕСКИЕ СО-
ЕДИНЕНИЯ — соединения, содержащие в моле-
куле два и более бензольных ядра, соединенных между
собой общими атомами углерода. Простейшим пред-
ставителем этого типа соединений является нафталин.
В отличие от бензола, в К. а. с. реакционная способ-
ность атомов углерода различна в зависимости от их
положения. В нафталине, напр., реакции электро-
фильного замещения протекают гораздо легче с а-ато-
мами углерода. Антрацен имеет три типа атомов угле-
рода, наиболее реакционноспособными являются ато-
мы в мезо- или 9- и 10-положениях.
Если бензольные ядра в молекуле сочленяются
(аннелируются) так, что их центры лежат на одной
прямой, то образуются т. н. линеарные си-
стемы, напр. антрацен (соединения этого типа
называют также по-
лиаценами). Если же
центры бензольных
ядер не лежат на
одной прямой, а мо-
гут быть соединены
'только ломаной ли-
(а)
пинеарно; аинелирование ангулярное аниелированив нией, то такое анне-
лирование наз. а н-
гулярным (угловым), напр. у фенантрена', к та-
кого рода соединениям относятся также системы с
большим числом колец, как пирен, коронен, овален.
Соединения линеарного типа известны вплоть до
гептацена. Для октацена и нонацена описаны нек-рые
производные. С ростом числа колец окраска этих со-
единений углубляется, т. е. максимум поглощения
света сдвигается из УФ-области в видимую.
лением. При облучении р-ра антрацена в сероуглероде
в присутствии кислорода образуется т. наз. фото-
окись антрацена. Нафтацен и пентацен еще легче
подвергаются фотоокислению, растворы гексацена и
гептацена на свету окисляются мгновенно. Анало-
гично растет реакционная способность и по отношению
к малеиновому ангидриду (в реакции Дильса —
Альдера). Антрацен дает аддукт при кипячении в кси-
лоле; нафтацен реагирует настолько легче, что это
обстоятельство используется для очистки антрацена
от примеси нафтацена. Пентацен, гексацен и гепта-
цен реагируют с малеиновым ангидридом почти
мгновенно.
Многие производные антрацена, относящиеся к со-
единениям ангулярного типа, также реагируют с мале-
иновым ангидридом, однако медленнее, чем соеди-
нения линеарного типа с тем же числом колец. Такие
высококонденсированные системы, как
пирен, 3,4-бензпирен и т. д., также яв-
ляются чрезвычайно реакционноспособ-
ными соединениями, однако они не реа-
гируют с малеиновым ангидридом, т. к.
реакционные центры молекул располо-
жены неблагоприятно для этой реак-
ции. Напр., в пирене (I) реакционными центрами мо-
лекулы являются положения 3, 5, 8 и 10.
В то время как в бензоле длина всех связей С—С
одинакова и равна 1,40 А, в конденсированных си-
стемах длины связей различны, напр. в антрацене
длина связей С—С меняется от 1,37 до 1,44 А; в пери-
лене — от 1,38 до 1,50 А. Аналогичная картина на-
блюдается и для других конденсированных циклич.
систем, таких, напр., как пирен, перилен, коронен
и т. д. Различию в длинах связей между атомами
С—С соответствует и различие их реакционной спо-
собности, напр. 9, 10-связь в фенантрене по своему
характеру напоминает олефиновую связь; фенантрен
в относительно мягких условиях присоединяет бром,
образуя 9,10-дибромфенантрен. Продукт присо-
единения — вещество нестойкое, отщепляет НВг и
дает 9-бромфенантрен. По другим связям фенантрена
бром не присоединяется. Подобные результаты по-
лучены и со многими другими полициклич. арома-
тич. углеводородами при использовании таких ре-
агентов на двойную связь, как диазоуксусный эфир,
озон, тетраокись осмия. Соединения такого типа не
обладают полностью ароматич. системами связей, напр.
фенантрен можно представить в виде следующих
изомерных форм:
Соединение Число колец Цвет
Нафталин 2 Бесцветный
Антрацен 3 Бесцветный
Нафтацен 4 Оранжевый
Пентацен 5 Сине-фиолетовый
Г ексацен в Темно-зеленый
Гептацен 7 Почти черный, в тонком
слое травянисто-зеле- ный
Одновременно с ростом числа колец повышается ре-
акционная способность соединений, особенно склон-
ность к реакциям присоединения. Уже антрацен
способен присоединять в -иезо-положепие водород,
галогены, азотную к-ту и двуокись азота, давая дигид-
роантрацен и его производные. Высшие члены этого
ряда еще легче присоединяют указанные реагенты.
Стабильность образующихся при этом дигидропроиз-
водных растет с увеличением числа колец, так же как
растет и стабильность соответствующих хинонов.
Большая реакционная способность высших членов
соединений этого ряда иллюстрируется их фотоокис-
Связь 1—2 представлена как двойная связь в трех
случаях из пяти и, следовательно, она на 60% является
олефиновой; аналогично связь 2—3 на 40%, связь
3—4 на 60% и связь 9—10 на 80% имеют олефиновый
характер. В свете этих данных понятна способность
связи 9—10 к реакциям присоединения. Подобный
полуколичественный анализ (метод изомерных форм)
может быть применен и к более сложным конденси-
рованным системам. Пользуясь методами молеку-
лярных орбит (см. Квантовая химия) и изомерных
форм, можно предвидеть значительное различие
в характере двойных С—С-связей, содержащихся
691
КОНДЕНСИРУЮЩИЙ ФЕРМЕНТ-КОНЕССИН
692
в К. а. с. Было найдено также, что между характером
двойной связи и ее длиной существует определенная
зависимость. Эта зависимость была использована
для расчета длин связей в различных К. а. с. Полу-
ченные расчетным путем данные по длинам связей
очень хорошо согласуются с данными рентгенострук-
турного кристаллография, анализа.
Нек-рые представители К. а. с. встречаются в кам.-
уг. смоле и в продуктах пиролиза нефти, большин-
ство же было получено синтетически; многие из них,
а также их производные обладают канцерогенной
активностью; ряд этих соединений и их производных
обладают свойствами красителей. Частично гидриро-
ванная структура фенантрена входит в молекулы
таких биологически важных соединений, как сте-
роиды. Частично гидрированная система тетрацена
лежит в основе строения важнейших антибиотиков —
террамицина, биомицина, тетрациклина и др.
Лит.: Chemistry of carbon compounds, ed. E. H. Rodd,
v. 3, pt B, Amst. — L. — N. Y., 1956, p. 1476; Чичибабин
A. E., Основные начала органической химии, т. 2, 5 изд.,
1957, с. 484; С 1 а г Е„ Вег., 1942, 75, 1330.Л. С. Поваров.
КОНДЕНСИРУЮЩИЙ ФЕРМЕНТ (оксальацетат-
транс-ацетаза, цитрогеназа, цитратсинтезирующий
фермент) — фермент, относящийся к группе транс-
ацилаз, катализирующий образование лимонной к-ты
путем конденсации щавелевоуксусной к-ты и ацетил-
кофермента А.:
СН2СО-КоА сн2соон
+ I
СО-СООН —-НО-С—СООН-4-КоА
I I
СН2СООН СНаСООН
Реакция, катализируемая К. ф., является ключевой
реакцией цикла трикарбоновых кислот (цикл
Кребса), с помощью к-рой ацетат, образующийся
в качестве промежуточного продукта в процессе
обмена углеводов, жиров и белков (аминокислот),
включается в вышеуказанный цикл, где и окис-
ляется до СО2 и Н2О. При этом химич. энергия, осво-
бождающаяся в процессе окисления, резервируется
благодаря сопряженным реакциям окислительного
фосфорилирования в макроэргич. связях аденозин-
трифосфорной к-ты (АТФ).
Помимо конденсации ацетил-КоА со щавелевоуксус-
ной к-той, К. ф. со значительно меньшей скоростью
может катализировать конденсацию щавелевоуксус-
ной к-ты с фторацетил-КоА или с пропионил-КоА
с образованием соответственно фторлимонной или
гомолимонной к-ты.
К. ф. присутствует во всех животных, раститель-
ных и бактериальных клетках с аэробным обменом
веществ. Кристаллич. К. ф., выделенный из сердеч-
ной мышцы свиньи, — гомогенный белок с высокой
ферментативной активностью: число оборотов —
5 - 103 молей лимонной к-ты в минуту при 25°. Ми-
хаэлиса константа (Км) для ацетил-КоА составляет
2,2 • 10’3 М. Кристаллич. К. ф. устойчив и при хра-
нении в виде суспензии в водном р-ре (NH4)2SO4
при 16° не утрачивает активности годами.
Лит.: Stern J. R., в кн.: The enzymes. Ed. Р. D.
Boyer, H. Lardy, К. Myrback, v. 5, 2 ed., N. Y., 1960, p. 367.
В. Б. Спиричев.
КОНДУКТОМЕТРИЯ — один из электрохимии,
методов анализа, основанный на измерении электро-
проводности растворов. Различают собственно кон-
дуктометрию и кондуктометрии, титро-
вание. Под кондуктометрией понимают измерение
электропроводности непосредственно для определе-
ния концентрации солей, кислот, оснований в раство-
рах известного состава, для определения плотного
остатка в природных водах, а также для контроля
промышленных р-ров постоянного состава (в нек-рых
случаях кондуктометрии, метод позволяет автомати-
зировать этот процесс). Конечная точка при кондук-
тометрии. титровании устанавливается по изменению
электропроводности, к-рое может наступить: 1) При
замене одного иона другим, обладающим иной подвиж-
ностью, напр., при титровании сильной к-ты раство-
ром сильной щелочи (HCI + NaOH=NaCl + Н2О),
при к-ром ионы водорода, обладающие большой под-
вижностью, заменяются менее подвижными ионами
натрия. В результате электропроводность раствора
уменьшается и достигает минимума в конечной
точке титрования, после чего снова возрастает вслед-
ствие поступления в раствор избыточных ионов Na+
и ОН~ (кривая 1). 2) При
изменении концентрации
ионов вследствие изме-
нения степени диссоциа-
ции соответствующих со-
единений, например, при
титровании слабой к-ты
раствором сильной щело-
чи (СН3СООН + NaOH=
= CH3COONa. + Н2О);
вместо слабо диссоции-
рованной уксусной к-ты
при титровании обра-
зуется ее натриевая соль, посылающая в раствор
ионы натрия (кривая 2), вследствие чего электропро-
водность возрастает. 3) В случае выпадения осадка,
понижающего концентрацию ионов, обусловливаю-
щих электропроводность раствора, напр., при титро-
вании сульфата магния раствором гидроокиси бария
[MgSO4 + Ba (ОН)2= jMg(OH)2 + BaSOJ; ход кри-
вой при этом такой же, как в первом случае (кривая 1).
Если в растворе присутствует несколько ионов, обла-
дающих разной подвижностью, но способных осаж-
даться одним и тем же титрующим р-ром, то возможно
последовательное их определение: на кривой титро-
вания наблюдается несколько переломов, отвечаю-
щих оттитровыванию каждого вида ионов.
Кондуктометрии, определения проводят на обычных
установках для измерения электропроводности. Так
как последняя обусловливается всеми присутству-
ющими в растворе ионами, то определение одного
какого-либо иона в присутствии всех других может
оказаться недостаточно точным. Современным видо-
изменением кондуктометрия, титрования является
высокочастотное титрование.
Лит.: Кольтгоф И. М., Лайтинен Г. А.,
Определение концентрации водородных ионов и электротит-
рование, пер. с англ., М., 1947; Л я л и к о в Ю. С., Физико-
химические методы анализа, 3 изд., М., 1960; Б е р л ь-Л у н-
г е, Химико-технические методы исследования, пер. с нем.,
т. 1, вып. 1, Л., 1937; Sa nd era К., Konduktometrie,
Urah 9 4 Q Б 7 А ОстготI'uci
КОНЕССИН c24h40n2, мол. в. 356,58 — бесцветные
кристаллы; т. пл. 125—126°;[а]0= —1,9° (СНС13),
рат(2НС1 2Н2О), т. пл. ок.
+ 21,6 (спирт); подоб-
но пирролу К. окраши-
вает сосновую лучинку,
смоченную соляной к-той,
в красный цвет.
К. имеет стероидную
конфигурацию. Как дву-
третичное основание К.
образует соли: хлоргид-
10°, [a]D = + 9,3’ (вода);
бромгидрат, [а]с = -|- 7,4°; хлороплатинат; пикрат,
т. пл. 224° (с разл.); дииодметилат 2CH3J • ЗН2О,
т. пл. 303—304° (с разл.), [aJD = -|- 11,5’ (вода),
К. — важнейший и наиболее хорошо изученный
среди 19 родственных алкалоидов, выделенных из
семян и коры растения Holarrhena antidysentherica
Wall (Индия), т. наз. «курчи». Из 100 г высушенной
коры обычно получают 0,5—1,7 г алкалоидов. К. най-
ден также в растениях Н. congolensis S., Н. africana
693
КОНИИН —КОНОВАЛОВА РЕАКЦИЯ
694
D, С. и др. Экстракт коры курчи с древних времен
применяется в Индии для лечения больных амебной
дизентерией. Недавно появился препарат «курчи-
иодистый висмут» (смесь алкалоидов курчи с BiJ3).
Конессин подавляет рост туберкулезных палочек in
vitro.
Лит.: Орехов А. П., Химия алкалоидов, 2 изд., М ,
1955, с. 713; Haworth R. D. [а. о.]. Chem. Ind., 1952,
№ 10, 215. В. Н. Фросин.
КОНИИН CsH17N, мол. в. 127,14 —главный алкало-
ид и ядовитое начало болиголова Conium maculatum
(сем. Umbelliferae). К. содержится во всех частях
этого растения; в незрелых плодах содержание дохо-
дит до 1%. К. — бесцветная жидкость с резким запа-
хом; т. кип. 165,7—165,9°/759 мм; т. пл. —2°; п~^
1,4505; <7‘80,8438, [aJ}J = + 15,7° (d-форма); хорошо
растворим в органических растворителях.К. немного
растворим в воде (1 : 90), с повышением темп-ры рас-
творимость К. уменьшается; К. растворяет до 25%
воды. При стоянии на воздухе медленно окисляет-
ся. Соли d-К.: хлоргидрат, т. пл. 220° (из воды);
бромгидрат, т. пл. 211°; кислый <7-тартрат (дигидрат),
т. пл. 54°; хлороплатинат, т. пл. 175°. К. — а-н-
пропилпиперидин (II) имеет сильно основной ха-
рактер; синтезирован конденсацией a-пиколина (I)
с паральдегидом и энергичным восстановлением про-
дукта реакции натрием в спирте (реакция Ладен-
бурга):
0Хсн3 +(снзсн0)з " С1сн=сн—
I
” СХсн2сн2сн3
1Чп
II
Разделение К. на оптич. изомеры достигнуто через
кислый <7-тартрат. В болиголове содержатся также
другие алкалоиды, имеющие тот же углеродный ске-
лет, что и К.: N-метил-К., коницеины и др.
К. сильно ядовит, вызывает паралич окончаний
двигательных нервов, сначала возбуждает, а затем
парализует нервную систему, дыхание сначала уча-
щается, а затем ослабевает. В медицине не приме-
няется.
Лит. см. при ст. Алкалоиды. А. Е. Васильев.
КОНИФЕРИЛОВЫЙ СПИРТ [3- (4-окси-З-мет-
оксифенил)-2-пропен-1-ол], мол. в. 180,22 — кристал-
лы (призмы), т. пл. 73,4°; труд-
Г’г|>-СН=СНСН2ОЯ но растворим в холодной воде
НО-%.J* * и спирте, легче—в горячей воде
дС и эфире. При окислении К. с.
хромовой смесью образуются
ванилин и ацетальдегид; при восстановлении—изоэв-
генол. Сплавление со щелочами приводит к образова-
нию протокатеховой (3,4-диоксибензойной) к-ты. Со
щелочами К. с. образует хорошо кристаллизующиеся
соли; под действием минеральных к-т превращается
в аморфный смолистый полимер. К. с. образуется
при гидролизе глюкозида, содержащегося в коре
хвойных деревьев; при воздействии фермента эмуль-
сина на глюкозид кониферин наряду с К. с. полу-
чается глюкоза. Доказана тесная генетическая связь
К. с. и лигнина. Дегидрированием К. с. в водной
среде в присутствии окислительно-восстановитель-
ных ферментов или солей меди было получено веще-
ство, по физич. и химич. свойствам идентичное хвой-
ному лигнину. См. также Лигнин.
Лит.: Каррер П., Курс органической химии, пер.
с нем., Л., 1960, с. 547; Роговин 3. А., Шорыгина
И. Н., Химия целлюлозы и ее спутников, М.—Л., 1953, с. 633.
А. В. Арбатский.
КОНОВАЛОВА ЗАКОНЫ — два закона, выра-
жающие связь между составом однородной жидкой
смеси двух летучих компонентов, составом насыщен-
ного (равновесного с ней) пара и его давлением. Вы-
ведены теоретически и подтверждены эксперимен-
тально в 1881—84 Д. П. Коноваловым. Несколько
более общее положение было высказано в 1876—78 Дж.
Гиббсом. В современной формулировке К. з. выра-
жаются так: в двойной жидкой системе пар относи-
тельно богаче тем компонентом, прибавление к-рого
повышает общее давление пара (первый закон);
экстремуму (максимуму или минимуму) давления
пара двойной жидкой системы соответствует жидкая
смесь, насыщенный пар к-рой обладает одинаковым
с ней составом (второй закон). Под словами «отно-
сительно богаче» следует понимать только то, что
пар более богат указанным компонентом, чем
жидкость, а не то, что содержание этого компонента
в паре больше, чем содержание другого. Упомянутая
во втором К. з. жидкая смесь, имеющая пар того же
состава, наз. постоянно кипящей, или азеотропной
смесью. Из К. з. вытекают 2 следствия, также назы-
ваемые К. з.: в двойной жидкой системе пар относи-
тельно богаче тем компонентом, прибавление к-рого
понижает температуру кипения (первый закон); экс-
тремуму температуры кипения двойной жидкой си-
стемы соответствуют жидкость и пар с одинаковым
качественным и количественным составом (второй
закон).
Для двойной жидкой системы, давление пара к-рой
изменяется монотонно с изменением состава жидкости
(т. е. не проходит через экстремум), первый К. з.
может быть формулирован проще: в двойной жидкой
системе пар относительно богаче тем компонентом,
к-рый в чистом виде обладает большим давлением
пара, т. е. более низкой темп-рой кипения. К. з. дают
научную основу для чрезвычайно важных в химич.
пром-сти процессов перегонки и ректификации жидких
смесей.
Лит.: Коновалов Д., Об упругости пара растворов,
3 изд., Л., 1928; Аносов В. Я. и Погодин С. А.,
Основные начала физико-химического анализа, М.—Л.,
1947, с. 122—44; Киреев В. А., Курс физической химии,
2 изд., М., 1956.
КОНОВАЛОВА РЕАКЦИЯ — замещение водорода
нитрогруппой в алифатич. и циклоалифатич. соеди-
нениях, а также в боковой цепи жирноароматич.
соединений, при прямом нитровании азотной к-той:
r-h+hno8=r—no2+h2o
R — алкил, циклоалкил, арилалкил
Собственно К. р. представляет собой жидкофазное
нитрование азотной к-той различных концентраций
(6,5—70%-ной, преим. 12—20%-ной) при 100—150°
под давлением или, реже, в открытых сосудах. Со-
вершенно чистая, свободная от окислов азота, азот-
ная к-та не обладает нитрующим действием, по-
скольку активным агентом процесса является радика-
лоподобная двуокись азота -NO2. Азотная к-та слу-
жит лишь источником ее возникновения:
RH-i-NOa—-R-f-HNO2; R-f-NO2—-RNOa
HNOa + HNO2—>H2O-f-N2O4; N2O4—>2-NOa
Рекомбинация R- и мономерной двуокиси азота с не-
спаренным электроном на атоме кислорода -О—N=O
приводит к алкилнитритам:
R-+-O-N=o R-O-N=O
Алкилнитриты в условиях К. р. могут окисляться
до алкилнитратов и подвергаться гидролизу с образо-
ванием спиртов, к-рые далее окисляются азотной
к-той, давая гл. обр. карбоновые к-ты. Рекомбина-
ция R- и обычно присутствующей в смеси радикало-
подобной окиси азота - NO приводит к нитрозосоеди-
695
КОНОПЛЯНОЕ МАСЛО — КОНРАДА—ЛИМПАХА РЕАКЦИЯ
еэ8
нениям:
R.-L.N=O—-R—N=O
Третичные нитрозосоединения окисляются далее в
третичные нитросоединения, а вторичные и первичные
изомеризуются в соответствующие оксимы, к-рые либо
гидролизуются до кетонов или альдегидов с после-
дующим окислением, либо взаимодействуют с дву-
окисью азота, превращаясь в ге.и-динитро- (1) и гем-
тринитросоединения (II):
)>C(NO2)2
I
-C(NO2)3
II
Образующиеся при жидкофазном нитровании нитро-,
производные обычно имеют, то же число атомов угле-
рода, что и исходный углеводород. Число образую-
щихся побочно продуктов окисления возрастает
с увеличением относительной массы азотной к-ты
и продолжительности нагревания. Нитрование угле-
водородов с нормальной углеродной цепью направ-
ляется ко второму атому углерода. От других ра-
дикальных реакций К. р. отличается Гем, что замеще-
ние при асимметрии, атоме углерода не приводит
к исчезновению оптич. активности. Напр., нитрова-
ние 1-3-метилоктана приводит к оптически деятель-
ному З-нитро-З-метилоктану.
Ориентация при К. р. определяется стабильностью
промежуточно образующегося радикала, к-рая, в свою
очередь, зависит от сопряжения неспаренного элек-
трона со связями С—Н. Легче всего происходит заме-
щение при третичном атоме углерода, труднее — при
вторичном, наиболее трудно — при первичном:
КОНРАДА - ЛИМПАХА РЕАКЦИЯ — метод
синтеза 4-оксихинолинов конденсацией эфиров (3-кето-
кислот с ароматич. аминами через промежуточное
образование анила (5-кетоэфира.
Ариламин конденсируют с Р-кетоэфиром при комнат-
ной темп-ре, а превращение в 4-оксихинолин осущест-
вляют либо кипячением образовавшегося промежу-
точного продукта — анила Р-кетоэфира (с выделением
или без выделения из реакционной массы) с неорганич.
к-той, либо нагреванием при 240—250 . Применение
повышенной темп-ры на первой стадии К. — Л. р.
приводит к образованию анилидов (5-кетокислот, при
последующей циклизации к-рых образуются 2-окси-
хинолины (см. Кнорра реакция). Конденсация этило-
вого эфира формилуксусной к-ты с ариламинами при-
водят к 4-оксихинолинам, не содержащим замести-
телей в положении 2:
Жирноароматич. углеводороды легко нитруются
в a-положение боковой цепи. Галогенопроизводные
нитруются несколько легче, чем соответствующие
углеводороды. Карбоновые к-ты образуют нитрокар-
боновыё к-ты и продукты их превращений, а также
продукты окисления. Как и в случае углеводородов,
легче всего происходит замещение при третичном
атоме углерода. Легко нитруются производные мало-
новой и ацетоуксусной к-т:
Анилы (J-альдегидокарбоновых эфиров циклизуют
при большом разбавлении, в противном случае обра-
зуются значительные количества диарилмочевин;
СН2(СООС»Н5).>—.О,ХСН(СООС2Н5)2
СН3-С-СН2СООС2Н5—► O2NCH2COOC»H5
о
Жидкофазное нитрование парафинов, циклопара-
финов и боковой цепи жирноароматич. углеводородов
открыто М. И. Коноваловым в 1888. Дальнейшим
развитием К. р. является парофазное нитрование
углеводородов при атмосферном давлении и 400—
450°. Реакция промотируется следами О2 или гало-
генов. При этом образуются смеси первичных, вторич-
ных или третичных нитросоединений, содержащих
то же или меньшее число атомов углерода, чем исход-
ный углеводород. Процесс парофазного нитрования
парафинов и циклопарафинов осуществлен в про-
мышленном масштабе. Подробнее см. Нитросоедине-
ния алифатические.
Лит.: Забродина А. С., Забродина К. С.,
в сб.: Реакции и методы исследования органических соединений,
кн. 7, М., 1958, с. 133; Топчиев А. В., Нитрование угле-
водородов и других органических соединений, М.—Л., 1949;
Химия углеводородов нефти, пер. с англ., т. 3, М., 1959;
Гольдштейн Р., Химическая переработка нефти, пер.
с англ., М., 1952. Б. ,Л. Дятхин.
КОНОПЛЯНОЕ МАСЛО— см. Жиры растительные.
Взаимодействие щавелевоуксусного эфира с арилами-
нами легко приводит по К.—Л. р. к 4-окси-2-карбэто-
ксихинолинам, к-рые при последующем гидролизе и
декарбоксилировании могут быть переведены в цен-
ные 4-оксихинолины:
СООС2Н5
+• сн2
О=С-СООС2Н5
СН2С(2илн
СН3СООН
СООС2Н5
СН2 250°
,-г—СООСЕН инертный
2 ° растворится
1)КаОН-Н2О
СООС2Н5 нагревание *"
При конденсации ацетоуксусного эфира с нафтиламть
нами образуются эфиры а-нафтиламинокротоновых
к-т, к-рые при последующем нагревании превращаются
697 КОНСЕКУТИВНЫЕ РЕАКЦИИ— КОНФОРМАЦИОННЫЙ АНАЛИЗ
698
в оксипроизводное метилбензхинолина:
^СН3-С-СН2СООС2Н5
Аналогично в реакцию с нафтиламинами можно ввести
этоксиметиленмалоновый, ацетилмалоновый либо ща-
велевоуксусный эфиры. Описано применение гетеро-
циклич. аминов в К. — Л. р. (синтез изофенантро-
линов из 5-аминоизохинолина). К. — Л. р. открыта
М. Конрадом и Л. Лимпахом в 1887.
Лит.: Surrey A. R., Name reactions in organic che-
mistry, N. Y., 1954; Э лдерфил ьд P., в кн.: Гетероцик-
лические соединения. Сб. ст., пер. с англ., т. 4, М., 1955, с. 25,
317, 478. Л. С. Герман.
КОНСЕКУТИВНЫЕ РЕАКЦИИ — см. Последо-
вательные реакции.
КОНСИСТЕНТНЫЕ СМАЗКИ — см. Пластичные
смазки.
КОНСТАНТА РАВНОВЕСИЯ — см. Равновесие
'Г 1J At UUPCKfiP
КОНСТАНТА СКОРОСТИ РЕАКЦИИ — см. Ки-
нетика химическая.
КОНТАКТ ПЕТРОВА — см. Сульфокислоты неф-
тяные.
КОНТАКТНЫЕ АППАРАТЫ — устройства для
взаимодействия (контактирования) гетерогенных сред:
газов или паров с жидкостями; газов (паров) с твер-
дыми веществами; жидкостей с твердыми веществами;
несмешивающихся жидкостей; твердых веществ с
твердыми; газов с жидкостью и твердым веществом
И Т. д.
В К. а. могут протекать чисто физич. процессы
массообмена (ректификации, абсорбции, адсорбции,
экстракции и др.), теплообмена с гетерогенными
теплоагентами смешения, сочетание химич. реак-
ций с физич. процессами массо- и теплообмена. Во
всех случаях непременным условием успешности
контактирования является наличие высокоразвитой
поверхности фазового контакта и достаточной ско-
рости транспортировки вещества (тепла) к этой
поверхности и от нее. Названия К. а. обычно отобра-
жают природу проходящих в них процессов, цель
взаимодействия контактируемых сред, напр.: ректи-
фикационные колонны, абсорберы, адсорберы, эк-
стракторы, мешалки, контакторы, генераторы, кон-
тактные нагреватели, обжиговые печи, конверторы,
химич. реакторы для гетерогенных реакций и т. д.
В химии под К. а. обычно понимают химические
реакторы для каталитич. превращений. О методах
управления К. а., их принципиальных схемах и кон-
струкциях см. Реакторы химические. Д- И. Орочко.
КОНТРОЛЬНО-ИЗМЕРИТЕЛЬНЫЕ ПРИБОРЫ —
см. Приборы контрольно-измерительные.
КОНФОРМАЦИОННЫЙ АНАЛИЗ — область сте-
реохимии, изучающая химич. и физич. свойства
изомеров (структур), возникающих в результате вра-
щения в молекуле атомов или групп атомов вокруг
простых (ординарных) связей. Такие структуры наз.
конформациями (констелляциями). Одни
структуры переходят в другие без разрыва связей
путем поворота, поэтому их иногда наз. поворот-
ными изомерами (этот термин принят гл.
обр. в спектроскопия, исследованиях). Причиной
существования конформаций является взаимное от-
талкивание атомов, в том числе и атомов водорода,
и атомных группировок. Это отталкивание прояв-
ляется только при сближении атомов на расстояние,
близкое к сумме их вандерваальсовых радиусов.
Поэтому даже в такой простой молекуле, как этан,
возникают энергетически неравноценные различные
положения метильных групп друг относительно
друга. Атомы водорода вследствие взаимного оттал-
кивания стремятся занять положение, при к-ром
один тетраэдр повернут на 60° по отношению к дру-
гому. Такая конформация энергетически выгоднее,
чем конформация, в к-рой атомы водорода располо-
жены друг против друга. При средних темп-рах запас
энергии достаточен, чтобы переходы из одного поло-
жения в другое могли совершаться до одного раза
в 1О~10 сек.
Для обозначения конформаций применяют два вида
формул: перспективные, в к-рых два атома
углерода, связанные друг с другом, рассматривают
сбоку (рис. 1, а), и проекционные, в к-рых
при наблюдении в направлении связи С—С второй
атом углерода находится точно позади первого
(рис. 1, б). Устойчивость конформационных изомеров
б
Рис. 1. Способы изображения конформаций
этана: а — перспективная формула; б — проек-
ционная формула.
к о н ф о р-
различных конформа-
Заслоненная форма
Заторможенная форма
Рис. 2. Потенциальная
энергия внутреннего вра-
щения и конформаций
этана.
устанавливают в основном физич. методами исследо-
вания (рентгеноструктурный анализ, термодинамич.
расчеты, термохимич. исследования, измерения ди-
польных моментов, изучение спектров).
Теоретически молекула может существовать в виде
множества конформаций; однако поскольку разные
конформации обычно энергетически неравноценны,
большинство молекул существует преим. в одной
или немногих устойчивых конформациях. Остальные
конформации не устойчивы и являются переходными
формами при превращении одной устойчивой конфор-
мации в другую. В условиях химич. реакции соеди-
нение может временно принять одну из неустойчивых
конформаций (т. н. р е а к ц и о н н у ю
м а ц и ю). При образовании
ций требуется энергия актива-
ции порядка 3 ккал/моль} этим
конформации отличаются от
цис-, транс-изомеров, для пре-
вращения к-рых друг в друга
требуется энергия активации
порядка 40 ккал/моль.
При повороте двух атомов
углерода вокруг С—С-связи
потенциальная энергия моле-
кулы меняется приблизительно
по синусоидальной кривой.
Если проектировать молекулу
на плоскость, перпендикуляр-
ную оси С—С, то максимумам
кривой потенциальной энергии
соответствует попарно парал-
лельное положение связей, со-
единяющих углеродные атомы
с заместителями. В такой проек-
ции заместители попарно пол-
ностью заслоняют друг друга,
поэтому соответствующее поло-
жение заместителей называют
заслоненным (рис. 2).
энергии соответствует такое положение заместителей,
когда молекула попадает в потенциальную яму и
Минимумам значений
699
КОНФОРМАЦИОННЫЙ АНАЛИЗ
700
вращение вокруг С—С-связи как бы затормажи-
вается. Поэтому конформации, соответствующие ми-
нимумам кривой потенциальной энергии, наз. з а-
торможенными. Переход от максимума к ми-
нимуму энергии осуществляется поворотом оси С—С
на 60°, поэтому для молекулытипа R1R3R3C—CR4R5R6
мыслимы три различные конформации, отвечающие
минимумам потенциальной кривой. Если нек-рые
из заместителей одинаковы, то и нек-рые из конфор-
маций эквивалентны и число их сокращается.
Несмотря на то, что представления об отдельных конформа-
циях возникли уже давно (предположение о двух конформа-
циях циклогексанового кольца высказано Заксом еще в 1890),
К. а. как область стереохимии начал развиваться лишь в 40 гг.
20 в., после того как рентгеноструктурными, электронографич.
и термодинамич. исследованиями была экспериментально до-
казана и теоретически обоснована предпочтительность формы
кресла для циклогексана (Хассель, Полинг, Питцер). Особое
развитие К. а. получил в работах Бартона (полициклич. сое-
динения, стероиды, сесквитерпены), Прелога (макроциклич.
соединения), Ривса (углеводы) и др.
Е, ккал/молъ
0° 120° 240° 360°
ШШ1
а б в г в б а
Рис. 3. Потенциальная энер-
гия и конформации соедине-
ний типа н-бутана и дихлор-
этана: айв — заслоненные
конформации, б — скошен-
ная конформация, г — тран-
соидная конформация.
Устойчивые конформации алифатических и цикли-
ческих соединений. Легко видеть, что в случае этапа
возможна лишь одна устойчивая конформация (рис.2);
разность энергий между нею и заслоненной формой
составляет 2,8 ккал/моль. Если молекула не имеет
оси симметрии, совпадающей с осью вращения, как,
например, в случае и-бута-
на или дихлорэтана, то кри-
вая потенциальной энергии
становится несимметричной
(рис. 3). На этой кривой
значениям минимума также
соответствуют заторможен-
ные конформации;однако не
все они между собой равно-
ценны. Абсолютному мини-
муму энергии отвечает кон-
формация, в к-рой метиль-
ные группы или атомы хлора
занимают наиболее удален-
ное траис-положение, по-
этому такую конформацию
можно назвать трансо-
идной. Относительным же
минимумам соответствуют
конформации, в к-рых метильные группы или атомы
хлора занимают скошенное положение (скошен-
ные, гош конформации). Разность энергии тран-
соидной конформации и наиболее неустойчивой,
заслоненной формы молекулы превышает 3,5 ккал/молъ
для бутана и 5 ккал/молъ для дихлорэтана. Разность
энергии трансоидной и скошенной конформаций зна-
чительно меньше и составляет (в газообразном со-
стоянии) всего лишь ок. 0,8 ккал/молъ для бутана
и 1,2 ккал/молъ для дихлорэтана.
Высшие нормальные алифатич. углеводороды можно
рассматривать как ряд и-бутановых систем, к-рые
должны существовать преим.
в заторможенной конфор-
мации. Поэтому наиболее
устойчивая конформация та-
ких углеводородов пред-
ставляет собой копланар-
ную зигзагообразную цепь
(рис. 4).
Кольцо циклогексана не
может быть плоским, т. к.
для построения плоского
кольца требуется значи-
нтных углов. При соблюде-
Рис. 4. Устойчивая конфор-
мация высшего нормального
алифатического углеводо-
рода.
тельная деформация вал<
нии тетраэдрич. углов для циклогексана возможны
две конформации: форма кресла и форма ванны
(рис. 5, а). Поскольку «кресло» состоит из шести
вааимоскошенных метиленовых групп, а «ванна» —
из четырех скошенных и двух взаимозаслоненных,
форма кресла энергетически выгоднее формы ванны
(разность энергий обеих форм составляет 5,6 ккал/молъ)
и последняя при обычных условиях почти не реали-
зуется. Если представить себе кресло жестким, то
молекула хлорциклогексана может быть изображена
„кресло" „вакка"
б
Рис. 5. Конформации циклогексана (а), ак-
сиальная и экваториальная конформации
хлорциклогексана (б).
в виде двух моделей: в одной из них связь С—С парал-
лельна оси симметрии 6-членногр кольца (аксиаль-
ная связь), в другой связь С—С направлена к пери-
ферии молекулы (экваториальная связь)
(рис. 5, б). Из электронографич. измерений расстоя-
ний между атомом хлора и всеми атомами углерода
в кольце следует, что хлор при обычных условиях
занимает экваториальное положение; как видно,
аксиальная конформация более богата энергией, чем
экваториальная (см. ниже).
В полициклич. соединениях устойчивость отдель-
ных конформаций может быть значительно выше,
чем в ряду циклогексана. Так, ' " н
транс-декалин практически су-
ществует только в одной кон- /
формации, в к-рой оба кольца '—-/
имеют форму кресла (рис. 6), Д
поэтому аксиальные замести-
тели монозамещеннЫХ транс- Рис. 6. Устойчивая кон-
декалинов не могут переходить формация^транс-дека-
без разрыва ковалентной свя-
зи в экваториальное положение и наоборот. г^ис-Де-
калин способен существовать в двух конформациях
с кресловидными кольцами, поэтому любой моно-
замещенный цис-декалин представляет собой равно-
н
Рис. 7. Конформации чис-декалина.
весную смесь аксиальной и экваториальной конфор-
мации, причем равновесие сдвинуто в сторону послед-
ней (рис. 7).
Для циклогексена, исходя из принципа наимень-
шего отклонения валентных углов, могут быть по-
строены две конформации «полукресло» и «полу-
ванна» (рис. 8), соответствующие кресловидной и
ваннообразной конформациям циклогексана. Согласно
термодинамич. расчету, конформация полукресла
устойчивее полуванны. Однако разность энергии этих
конформаций примерно в два раза меньше, чем в слу-
чае циклогексана, и составляет всего 2,7 ккал/молъ.
Поэтому уже при обычных условиях доля молекул,
имеющих конформацию полуванны, может быть
значительной, а у замещенных циклогексенов даже
преобладающей. В отличие от циклогексенового
кресла, в циклогексеновом полукресле нет параллель-
701
КОНФОРМАЦИОННЫЙ АНАЛИЗ
702
пых связей С—Н и понятие «аксиальная связь» здесь
неприменимо. Однако поскольку отклонение четырех
(связанных с насыщенными атомами углерода) атомов
водорода от аксиального направления незначительно
(см. рис. 8), эти атомы называют ,
«к в а з и а к с и а л ь н ы м и». ,[
Остальные четыре атома водорода н
могут быть названы «к в а з и- н—74. н
экваториальными». ч- ._/ -'I 1______________:
Эти же названия применяют У —
для обозначения двух типов свя- и \/.н
зей С—Н в циклич. соединениях -
с числом звеньев более 6. Не-
устойчивость средних циклов (с
7—12 С-атомами) по сравнению
С циклогексаном обусловлена тем, Рис. 8. Устойчивая
что внутреннее пространство та- конгфе°к₽с^И(«по5?™°'
ких циклов недостаточно для кресло»),
того, чтобы вместить все квазиак-
сиальные Н-атомы при сохранении выгодной кон-
формации цепи (рис. 9). Результатом этого может
быть либо сжатие вандерваальсовых радиусов атомов
водорода при сохранении выгодной формы цепи,
либо изменение формы цепи с появлением заслонен-
ных конформаций. Это
явление, наз. некласси-
ческим, или конфор-
мационным нап-
ряжением, является
причиной повышенного
содержания энергии в
средних циклах. В мак-
роциклич. соединениях
(13 звеньев и более)
внутреннее пространство
достаточно для того, что-
бы в нем свободно могли
разместиться водород-
ные атомы и полимети-
Рис. 0. Устойчивая конфориа-
ция циклодекана.
леновая цепь не содержала невыгодных заслоненных
конформаций. Поэтому соединения с большими цик-
лами по своей устойчивости почти не отличаются
от соответствующих алифатич. соединений.
Поскольку замещение одного или нескольких атомов
углерода гетероатомами мало влияет на форму кольца,
а р-орбиты азота и кислорода расположены примерно
тетраэдрически, выводы К. а., сделанные при изу-
чении алициклич. соединений, могут быть перене-
сены и на азотистые или кислородные гетероциклич.
соединения. На основе такой аналогии были объяс-
нены стереохимия, свойства сахаров и многих алка-
лоидов.
Причины предпочтительности отдельных конфор-
маций. Потенциальная энергия Е каждой из конфор-
маций зависит от угла поворота <р и может быть
выражена ур-нием:
Е = — l,3cos3<p — 0,5cos<p
Если принять потенциальную энергию заторможен-
ной конформации, напр., и-бутана равной нулю,
то энергии остальных конформаций будут состав-
лять:
Конформация Заслоненная Скошен- ная Частично заслоненная
Избыток энергии, ккал/моль 3,6 0,8 2,9
Вероятность разных конформаций любой молекулы
определяется коэфф. Больцмана е~~где Е —
энергия, Т — абс. темп-ра, к — постоянная Больц-
мана. Скорость превращения конформаций друг
в друга (скорость внутреннего вращения) зависит от
высоты потенциального барьера внутреннего вра-
щения, т. е. от разности энергий наиболее устойчивой
трансоидной конформации и наименее устойчивой
заслоненной конформации молекулы. Если высота
барьера равна Е, то, согласно уравнению для кине-
тики мономолекулярного процесса, возможное число
переходов через барьер в единицу времени равно;
п е
ki — константа скорости реакции. Для большинства
обычных молекул барьеры внутреннего вращения
не особенно велики (чаще всего они находятся в об-
ласти 600—6000 кал/молъ) и скорость вращения имеет
порядок величины Ю10 сек-1. Поэтому отдельные
конформации таких молекул при обычных условиях
не могут быть выделены.
Энергетич. неравноценность конформаций вызвана
тем, что из-за изменения расстояния силы отталки-
вания между непосредственно не связанными между
собой заместителями в различных конформациях
неодинаковы. В общем случае эти силы складываются
из стерич. и электростатич. отталкиваний. Сравни-
тельно большая разность энергий между заслонен-
ными и заторможенными конформациями (т. е. вы-
сота потенциального барьера свободного вращения)
определяется в основном не электростатич., а стерич.
отталкиванием. О преобладающей роли стерич. фак-
тора при создании потенциальных барьеров свиде-
тельствует, напр., то обстоятельство, что гексахлор-
этан имеет высокий барьер свободного вращения
(ок. 10 ккал/моль), тогда как в аналогично построен-
ном гексахлордисилане барьер практически отсут-
ствует. В заслоненной конформации расстояния
Cl—С1 равны: в С13С—СС13 2,72 Айв Cl3Si—SiCl3
3,20 А. Увеличение электростатич. отталкивания,
вызванное таким небольшим уменьшением расстоя-
ния, недостаточно для создания столь большого
барьера; очевидно, последний обусловлен в основном
стерич. силами отталкивания. В отношении произ-
водных этана важно отметить, что для любых засло-
ненных заместителей (кроме водорода) межатомные
расстояния меньше суммы их вандерваальсовых радиу-
сов. Поэтому при обычных условиях заслоненные
конформации практически отсутствуют.
В заторможенных конформациях силы стерич.
отталкивания, как правило, уже сравнительно неве-
лики и соизмеримы с электростатич. отталкиванием
и силами водородных связей. Роль стерич. отталки-
вания в создании различных энергетич. уровней
трансоидной и скошенной конформаций приближенно
может быть оценена сравнением разностей энергии
скошенной и трансоидной форм (Д7Г) дибромэтана
(1,4 ккал/молъ) и н-бутана (0,8 ккал/моль). Поскольку
н-бутан неполярен, а вандерваальсовы радиусы
СН3-группы и атома Вг одинаковы, значение ЕЕ
бутана примерно соответствует «стерической» доле
ЕЕ в дибромэтане.
Причину различной устойчивости конформаций
хлорциклогексана следует искать в различной сте-
пени стерич. взаимодействия экваториального или
аксиального атома хлора с водородными атомами
кольца. Рассмотрение моделей показывает, что ак-
сиальный атом хлора расположен близко к двум
экваториальным атомам водорода при С(2) и С(6)
и очень близко к двум аксиальным атомам водорода
при С(3) и С(5), тогда как вблизи экваториального
атома хлора находятся лишь экваториальные атомы
водорода при С(2) и С(6). Особенно велико стерич.
взаимодействие аксиального атома хлора с аксиаль-
ными атомами водорода при С13) и т. к. расстоя-
703
КОНФОРМАЦИОННЫЙ АНАЛИЗ
704
ние между ними меньше суммы вандерваальсовых
радиусов атомов хлора и водорода. То же самое отно-
сится к любым другим аксиальным заместителям,
поэтому в общем случае аксиальная конформация
менее устойчива, чем экваториальная, а разность
энергии обеих конформаций возрастает с объемом
заместителя.
Влияние конформации на физико-химические свой-
ства органических соединений. Зависимость физико-
химич. свойств от конформации обычно изучают на
примере соединений с фиксированной ориентацией
заместителя. К таким соединениям относятся заме-
щенные циклогексаны с большим по объему замести-
телем (напр., mjoem-бутильной группы), всегда зани-
мающем экваториальное положение, или соединения
dl
мезо
Рис. 10. Устойчивые конформации
типа транс-декалина и высшие полициклич. системы.
Выявленные в результате этих исследований законо-
мерности используются, в свою очередь, в К. а. для
определения предпочтительной конформации молекул.
Важнейшие закономерности этого типа рассмотрены ниже.
1) Среди ациклич. диастереоизомеров мезо- (или эритро-)-
форма всегда устойчивее рацемич. (или трео-)-формы. В устой-
чивой конформации мезо-
изомера большие и сред-
ние заместители зани-
маютнаиболееудаленное,
трансоидное, положение,
тогда как в рацемич.
изомере при трансоидном
положении больших за-
местителей средние заме-
стители скошены (рис. 10)
и сильнее отталкивают
ДРУГ друга.
2) В ряду 1,2- и 1,4-
ациклических мезо- и рацемич. изо- дизамещенных циклогек-
меров: Б — большие, С — средние, санов транс-изомер ус-
М — малые заместители. тойчивее цис-изомера, а в
ряду 1,3-замещепных бо-
лее стабильна цис-форма. Эти энергетич. соотношения объясня-
ются тем, что в устойчивой конформации транс-1,2- и 1,4-ди-
замещенных циклогексанов оба заместителя экваториальны,
тогда как в цис-изомерах один из заместителей всегда аксиа-
лен. В ряду 1,3-дирамещенных циклогексанов имеет место
обратное явление (рис. 11).
в г
Рис. 11. Устойчивые конформации дизаме-
щенных циклогексанов: а — 1, 2- и 1,4-
транс-изомеры, б — 1,2- и 1,4-цис-изомеры,
в — 1,3-цис-изомер, г — 1,3-транс-изомер.
3) Соединения с экваториальными карбоксильными или
аминогруппами обладают большей кислотностью или соот-
ветственно основностью, чем аксиальные изомеры. Кислот-
ность и основность тех или иных групп увеличиваются с их
сольватацией. Аксиальные заместители сольватированы меньше
экваториальных, т. к. доступ к ним молекул растворителя
пространственно затруднен.
4) Как правило, характерная ИК-частота данной связи
повышается при переходе от аксиального к экваториальному
заместителю. Так, частота колебаний С—О экваториальных
гидроксильных, ацетоксильных или метоксильных групп при-
мерно на 40 еле-1 выше, чем у аксиальных заместителей. Это
явление объясняется тем, что колебания экваториального
заместителя вызывают значительное расширение и сжатие
всего циклогексанового кольца, тогда как колебания акси-
ального заместителя происходят преим. в плоскости циклогек-
санового кольца и оказывают меньшее влияние.
5) Поскольку экваториальные гидроксильные и карбо-
ксильные группы менее экранированы, чем аксиальные, они
этерифицируются быстрее. Точно также экваториальные слож-
ноэфирные группы омыляются быстрее аксиальных.
6) Аксиальные вторичные гидроксильные группы окисля-
ются в кетонные быстрее, чем экваториальные. Стадией, опре-
деляющей скорость таких реакций, является атака окисли-
теля, направленная на связь С—Н. В случаях аксиального
гидроксила эта связь экваториальна и поэтому более доступна.
7) При реакциях бимолекулярного отщепления <Е2) ацик-
лич. Л1езо-соединения реагируют быстрее рацемич. изомеров,
а в ряду циклогексана аксиальные заместители отщепляются
быстрее экваториальных. Нак известно, реакции этого типа
представляют собой процессы транс-отщепления, причем
минимуму потенциальной энергии соответствует такое переход-
ное состояние, в к-ром 4 центра, участвующие в реакции, кои-
ланарны, а отщепляющиеся группы антипараллельны. В соот-
ветствующей такому переходному состсянию реакционной
Рис. 12. Реакционные конформации диастереоизомеров
.при Е2-реакциях отщепления заместителей X: а-мезо-
форма; б-рацемическая форма; в — 1,2-дизамещенные
циклогексаны; Б — большие, М — малые заместители.
Рис. 13. Реакционная конформация
ациклич. диастереоизомеров при ре-
акциях 8^2: а — мезоформа; б —
рацемическая форма. Б—большие,
М — малые заместители.
конформации л1сзо-изомера ббльшие по объему заместители
трансоидны, в рацемич. же изомере они занимают скошенное
положение (рис. 12, а и б) и сильнее отталкивают друг друга,
что связано с большей затратой энергии на достижение пере-
ходного состояния. В ряду же циклогексана копланарная
реакционная конформация соответствует 1,2-диаксиальному
положению отщепляющихся групп (рис. 12, в) и протекает
лишь медленно, если переход экваториального заместителя
в аксиальное положение затруднен.
8) При электрофильных реакциях присоединения к этиле-
новым связям, протекающих через л-комплекс, и при реакциях
раскрытия эпоксидного кольца образуются преим. транс-
диаксиально замещенные продукты. Эти закономерности также
определяются планарностью переходного состояния.
9) При реакциях бимолекулярного замещения (S^2) ацик-
лич. .и^зо-изомеры реагируют медленнее рацемич. изомеров,
а в циклогексановом ряду экваториальные группы замещаются
медленнее аксиальных.
Реакции этого типа про-
ходят через переходное
состояние, в к-ром заме-
щающая и замещаемая
группы расположены на
одной прямой, а три, не
участвующие в реакции
заместителя, размещают-
ся в плоскости, перпен-
дикулярной этой прямой.
В случае ациклич. соеди-
нений реакционной кон-
формацией, соответствую-
щей этому переходному
состоянию, будет та, в
которой трансоидное по-
ложение к замещаемой
группе занимает наи-
меньший из заместителей
соседнего С-атома (рис. 13). Такая конформация менее выгодна
для «мезо-изомера, т. к. в этом случае оба больших заместителя
скошены. В случае циклогексановых производных подход к
экваториальному заместителю с «тыльной» стороны аатруд-
Рис. 14. Переходные состояния при реак-
циях S^q2 в ряду циклогексана: а — ак-
сиальная форма, б — экваториальная
форма.
няется двумя аксиальными Н-атомами в положении 3 и 5
(рис. 14). Подход к аксиальному заместителю менее затруднен
и последний реагирует быстрее.
10) При реакциях, протекающих через стадию карбония
иона (8^-1), аксиальные электроотрицательные заместители
отщепляются быстрее, чем экваториальные. Скорость таких
реакций определяется склонностью к ионизации, при к-рой
атом углерода переходит из тетрагонального состояния в
705 КОНЦЕНТРАТЫ СУЛЬФИТНО-СПИРТОВОЙ БАРДЫ — КОНЦЕНТРАЦИОННЫЕ ЦЕПИ 706
тригональное. При ионизации аксиальной группы выигрыш
энергии больше, т. к. больше уменьшается конформационное
напряжение, поэтому такие заместители реагируют быстрее.
Значительные успехи, достигнутые в К. а. за по-
следнее десятилетие, позволили количественно оце-
нить взаимодействие непосредственно не связанных
атомов и групп в органич. молекулах и объяснить
стереохимия, свойства не только простых органич.
соединений, но и таких сравнительно сложных ве-
ществ, как стероиды, белки, углеводы, алкалоиды.
Во многих случаях данные К. а. позволяют предска-
зать пространственную направленность реакций и
зарапее оценить относительную реакционную спо-
собность конформаций. Л. Д. Бергельсон.
Конформации высокомолекулярных соединений.
Конформации макромолекул реализуются при враще-
нии вокруг каждой ординарной связи в цепи. Напр.,
в полиэтилене при вращении вокруг каждой орди-
нарной связи возникают поворотные изомеры того же
типа, что и в случае 1,2-дихлорэтана или н-бутана.
Ниже приведены эти конформации, СН2 — означает
продолжение полимерной цепи. Разность энергий
НИ Н СН2~ Н В
~H2C-^--CH2~ -Н2С-^—Н ~Н2С—4^-Н
НН НН н СН2~
Рис. 15. Конформации полиэтилена.
поворотных изомеров этилена, определенная спектро-
скопически и из термоэластических свойств поли-
мера, составляет 600—800 кал на 1 моль звеньев.
Важнейшие свойства полимерных цепей, связанные
с их гибкостью, определяются существованием кон-
формаций. Механизм растяжения каучукоподобных
полимеров сводится к поворотной изомеризации
макромолекул. Свободная макромолекула в растворе
или макромолекула в блочном полимере непрерывно
изменяет свои конформации, приобретая в среднем
форму клубка. При растяжении такого клубка одни
конформации переходят в другие т. обр., чтобы гиб-
кая цепь приобрела большую длину. Если в моле-
куле имеется несколько ординарных связей, по отно-
шению к к-рым реализуются различные конформа-
ции (в макромолекуле таких связей очень много),
то повороты вокруг соседних связей не являются
независимыми и при соответствующих расчетах мо-
лекулярных констант необходимо учитывать корреля-
цию внутренних вращений. Поэтому поворотная изо-
мерия в полимерах имеет кооперативный характер —
конформация каждого мономерного звена зависит
от конформаций соседних звеньев и изменение кон-
формаций происходит согласованно. Такие коопера-
тивные или согласованные изменения происходят
в биополимерах — белках и нуклеиновых кислотах
(см. Молекулярная биология). В- Волькепгитейн.
Лит.: Каррер П., Курс органической химии, пер. с
нем., Л., 1960; Мидзусима С., Строение молекул и
внутреннее вращение, пер. с англ., М., 1957; Вольней-
штейн М. В., Конфигурационная статистика полимерных
цепей, М., 1959, с. 44—131; Стереохимия производных цикло-
гексана, сб. статей, под ред. В. Ф. Кучерова, пер. с англ, и
франц., М., 1958; Д обен У., Питцер К., в кн.: Про-
странственные эффекты в органической химии, пер. с англ.,
М., 1960, с. 9; Бартон Д., в кн.: Перспективы развития
органической химии, пер. с англ, и нем., М., 1959, с. 63;
П р е л о г В., там же, с. 77; Б е р г е л ь с о п Л. Д., Усп.
химии, 1958, 27, вып. 7, 817; Гольдфарб Я. Л. и
Беленький Л. И., там же, 1960, 29, вып. 4, 470.
КОНЦЕНТРАТЫ СУЛЬФИТНО - СПИРТОВОЙ
БАРДЫ (концентраты сульфитно-бардяные) — тех-
ническое название кальциевых солей лигносулъфоно-
еых кислот, образующихся при сульфитной варке
целлюлозы и переходящих совместно с нецеллюлоз-
ными углеводными компонентами древесины в р-р
сульфитного щелока. В СССР выпускаются три марки:
КБЖ жидкие (50% сухих веществ), КБТ твердые
12 К, х. э, т. 2
(76% сухих веществ), КБП порошкообразные (87%
сухих веществ). Концентраты — малогидратированные
лиофильные коллоиды, несущие отрицательный элек-
трический заряд и состоящие из полидисперсных
фенилпропановых цепочек с сульфогруппами в про-
пановом остатке, с мол. вес. от 250 до 25 000 (и более);
растворимы в воде в любых соотношениях; повышают
точку кипения р-ра и понижают точку его замерза-
ния. При повышении содержания сухих веществ
непропорционально возрастает вязкость р-ра (при 20°:
Ю%. сухих веществ 1,4 спуаз, 35% сухих веществ
12,5 спуаз, 50% сухих веществ 515 спуаз); вязкость
резко снижается при повышении темп-ры (50% сухих
веществ: 40°—135 спуаз, 80°—20 спуаз).
Концентраты — сильные поверхностно-активные ве-
щества, легко вступают в реакции замещения катио-
нов, конденсации с карбонильными соединениями,
полимеризации; с соединениями 6-валентного хрома
образуют водонерастворимые гели.
Качественные реакции: при сжигании — запах
меркаптана; под кварцевой лампой — голубое или
зелено-голубое свечение; в водном р-ре солянокислого
анилина или 0-нафтиламина — желтые хлопья; в
р-ре 1 % желатины и 10% поваренной соли — желтый
осадок.
К. с.-с. б. применяют в качестве разжижителя це-
ментно-сырьевого шлама и пластификатора цементных
и бетонных р-ров, в произ-ве силикатных и абразив-
ных изделий, для интенсификации процесса произ-ва
и повышения прочности фарфоро-фаянсовых изделий;
для стабилизации суспензий и эмульсий, в качестве
вяжущего, клеящего и дубящего средства, для полу-
чения ванилина, протокатеховой к-ты и др.
Лит.: Hen ей ин Н. Н., Технология целлюлозы, т.1,
М.—Л., 1956; Сапотницкий С. А., Использование
сульфитных щелоков, М.—Л., 1960. С. А. Сапотницкий.
КОНЦЕНТРАЦИОННЫЕ ЦЕПИ (концентра-
ционные элементы) — один из видов гальванич.
элементов; различают два типа: с переносом ионов
и без переноса ионов. В К. ц. первого типа оба элек-
трода изготовлены из одного и того же металла и
погружены в раствор соли данного металла, но с раз-
ными концентрациями ионов. Напр., один серебря-
ный электрод помещен в раствор AgNO3 концентра-
ции mlt а второй, тоже серебряный электрод, — в ра-
створ концентрации т2. Условно К. ц. изображается
следующим образом:
Ag I AgNOaCmO ([ AgNO3(ms) | Ag
Одной чертой принято обозначать границу между
металлом и раствором, двумя чертами (или пунктир-
ной линией) — границу между растворами разной
концентрации. Электрич. ток в такой цепи возникает
за счет выравнивания концентрации ионов серебра
в обоих растворах: на электроде, погруженном в ра-
створ с меньшей концентрацией ионов серебра (mJ,
идет растворение металлич. серебра, вследствие чего
электрод заряжается отрицательно, а раствор обога-
щается ионами Ag+; на электроде, погруженном
в раствор с концентрацией т2, наоборот, выделяется
металлич. серебро, вследствие чего электрод заря-
жается положительно, а концентрация ионов Ag~
в р-ре уменьшается. Потенциалы обоих электродов
зависят от концентрации ионов серебра в растворе:
, RT , , ВТ,
Ч>1 = tPo1 + In mt <р2 = ?o2 + -p In ms
T. к. оба электрода состоят из одного и того же ме-
талла, то их нормальные потенциалы <р01 и <р02 равны,
вследствие чего электродвижущая сила Е равна
RT т2
Е = ®2 — = -- 1П —
Т F mi
т. е. зависит от соотношения концентрации ионов
в обоих растворах. Для точных расчетов следует
707
КОНЦЕНТРАЦИЯ — КООРДИНАЦИОННАЯ СВЯЗЬ
708
исходить не из концентраций, а из активностей
ионов. Равным образом при точных расчетах необхо-
димо учитывать диффузионный потенциал. Однако
в большинстве случаев, преследующих практич.
цели, эта поправка может быть опущена.
Ко второму типу относятся К. ц., состоящие из
двух электродов, изготовленных из разных металлов,
погруженных в один и тот же электролит, ионы к-рого
могут участвовать в процессах, происходящих на
каждом электроде. Напр., если погрузить в р-р соля-
ной к-ты один серебряный, а другой платиновый
электроды, то серебро будет окисляться и, переходя
в раствор, соединяться с ионами хлора; на платино-
вом электроде, наоборот, будет идти восстановление
ионов водорода. Иначе говоря, один электрод будет
хлоро-серебряным, другой — водородным. Потен-
циалы обоих электродов определяются концентрацией
соответствующих ионов, Н+ и С1~, т. е. концентра-
цией соляной к-ты. Если соединить два таких эле-
мента, в одном из к-рых концентрация соляной к-ты
равна вц, а в другом — т2, то создастся К. ц.:
Ag I AgCl, HCl(mi) I Pt—Pt | HCl(m2), AgCl | Ag
Эдс такой цепи выражается ур-нием:
£=2RT-nm1
nF mt
Вместо платиновых электродов применяют также
ртутные, погружая их в р-р хлорида щелочного ме-
талла. Последний восстанавливается на ртутном
электроде, образуя амальгаму, вследствие чего подоб-
ные элементы называют амальгамными, концентра-
ционными элементами или К. ц., если они соеди-
нены между собой. В качестве вторых электродов и
в этом случае используются хлоро-серебряные:
Ag I AgCl, КСЦтО | Hg—Hg | КС1(т2), AgCl 1 Ag
К. ц. без переноса ионов не имеют жидкостного
контакта, поэтому при работе с ними не приходится
считаться с ошибкой за счет диффузионного потен-
циала. Из приведенных уравнений видно, что эдс
элемента без переноса вдвое больше, чем эдс цепи
с переносом вследствие того, что в первом случае
в электродной реакции участвуют оба иона, тогда как
во втором — только один.
Лит.: Глесстон С., Введение в электрохимию, пер.
с англ., М., 1951; Скорчеллетти В. В., Теоретическая
электрохимия, Л., 1959. О. А. Сангина.
КОНЦЕНТРАЦИЯ величина, выражающая от-
носительное содержание данного компонента (состав-
ной части) в смеси или растворе. Применяются раз-
личные способы выражения К. Количество данной
составной части и количество всей смеси могут быть
выражены или в весоных единицах (граммы, кило-
граммы и пр.), или в атомных, молекулярных, экви-
валентных и прочих единицах (грамм-атом, моль,
грамм-эквивалент, грамм-ион), или в объемных едини-
цах (литры, миллилитры и др.). При любом из этих
способов относительное содержание может быть
выражено в долях, в процентах или в отношениях
(напр., 1 : 10). При этом наряду со способами, в к-рых
количества данного компонента и количества всей
смеси выражаются в одинаковых величинах, при-
меняются и способы, в к-рых они выражаются в раз-
ных величинах (напр., в граммах на литр). Наиболее
употребительными являются следующие способы.
Весовая доля — отношение весового коли-
чества данной составной части к сумме весовых
количеств всех составных частей, т. е. к общему весо-
вому количеству смеси. Мольная доля — отно-
шение числа молей данной составной части к сумме
числа молей всех составных частей, т. е. к общему
числу молей смеси. Аналогично определяется и атом-
ная доля. Объемная доля — отношение
объема данной составной части к сумме сбьемов всех
составных частей, причем каждый из них относится
к индивидуальному состоянию этой составной части
при тех же темп-ре и давлении.
Определение К. в весовых, мольных или атомных,
или же объемных процентах вполне аналогично опре-
делению ее в соответствующих долях, будучи численно
больше их в 100 раз, т. к. в этом случае рассматри-
ваемое количество смеси принимается не за единицу,
а за сто.
Молярность — число молей данной состав-
ной части в 1 л раствора. Модальность —
число молей данной составной части в 1000 г раство-
рителя. Нормальность — число грамм-экви-
валентов данной составной части в 1 л раствора.
Титр-— большей частью число граммов растворен-
ного вещества в 1 мл раствора или число граммов
определяемого элемента, отвечающих ,1 мл данного
раствора (см. Объемный анализ). Кроме того, нередко
применяются К., выраженные числом граммов раство-
ренного вещества в 100 г или в 1000 г или в 1 л раство-
рителя, а также — числом молей растворенного
вещества в 1000 молей растворителя. В обычной
практике для выражения К. чаще применяются
весовые единицы. При рассмотрении закономерностей
зависимости свойств смеси или раствора от их К.
чаще пользуются мольными или атомными единицами.
Впрочем, при рассмотрении зависимости уд. свойств
смеси (плотность, уд. объем и др.) естественно вы-
ражать К., используя весовые единицы. В аналитич.
химии широко применяют выражения К. нормаль-
ностью или титром.
При определении К. растворов используются раз-
личные методы аналитической химии: весовые, объем-
ные, а также методы, основанные на измерении
плотности, показателя преломления и других физико-
химич. свойств.
Лит.: Киреев "В. А., Курс физической химии, 2 изд.,
М.—Л., 1956; Виноградов Г. В., Номограммы пере-
счета концентраций, М.—Л., 1948. В. А. Киреев.
КОНЪЮГАЦИЯ — см. Сопряжение.
КООРДИНАЦИОННАЯ СВЯЗЬ — термин, обозна-
чающий один из способов образования химич. связи
между атомными и (или) молекулярными части-
цами (заряженными или нейтральными), обычно
не имеющими неспаренных электронов. Одна из
частиц представляет собой донор, а другая — акцеп-
тор пары электронов, вследствие чего К. с. часто
наз. донорно-акцепторной связью.
Понятие о К. с. впервые возникло в связи с необходимостью
объяснить «необычные» валентные состояния комплексообра-
зующих ионов. К началу 20 в. в неорганич. химии был накоп-
лен большой экспериментальный материал, показывающий,
что наряду с обычными соединениями, существует обширная
группа т. наз. комплексных соединений, состав к-рых не может
быть понят в рамках учения о постоянной валентности. Так,
напр., существование устойчивых аммиакатов общей ф-лы
[Pt(NH3)nCl6_П]С1П_2 ПРИ п=2, 3, 4,5, 6, казалось бы, проти-
воречит хорошо установленному факту 4-валентности платины.
Изучение молекулярной электропроводности водных р-ров
таких соединений показало, что электропроводность обуслов-
лена не всеми ионами хлора, а лишь частью их. Подобная
неравноценность ионов хлора казалась весьма удивительной.
Многие исследователи безуспешно пытались объяснить строе-
ние и свойства этих «необычных» неорганич. соединений. А.
Вернер был первым, кто понял необходимость отказа от учения
о постоянной валентности. Им была создана (1893) коорди-
национная теория, основной постулат к-рой Вер-
нер формулировал след, образом: «Даже тогда, когда согласно
учению о валентности связующая способность определенных
атомов исчерпана, атомы эти все же обладают ещё способно-
стью участвовать в дальнейшем построении комплексных мо-
лекул, с образованием совершенно определенных атомных
сочетаний. Причина этой способности атомов к дальнейшему
присоединению заключается в том, что в них, кроме главных
валентных сил, имеются ещё силы — силы побочной валент-
ности». Первоначально побочные валентности считались резко
отличающимися от главных валентностей, но уже Вернер пока-
зал, что между ними нет существенной разницы. В дальнейшем
это было многократно подтверждено экспериментально и объяс-
нено теоретически.
В теории Вернера вопрос о природе главных
и побочных валентностей не ставился. Это ока-
709
КООРДИНАЦИОННАЯ СВЯЗЬ — КООРДИНАЦИОННЫЕ ПОЛИМЕРЫ
710
залось. возможным лишь после возникновения электронной
теории валентности. Теория электронных пар Льюиса (см.
Валентность) сделала гораздо более приемлемыми постулаты
Вернера о главных и побочных валентностях. Напр., в моле-
куле воды HSO атом кислорода предоставляет по одному элек-
трону каждому из двух атомов водорода, образуя ковалентные
связи и проявляя, по Вернеру, свою главную валентность.
При образовании иона оксония Н3О+ связь между атомом кис-
лорода и третьим протоном образуется за счет пары электронов,
предоставляемой атомом кислорода, к-рый, т. обр., проявляет
побочную валентность. Хотя способ образования этих двух
типов связей различен, связи всех трех атомов водорода стано-
вятся одинаковыми, как только они образуются.
Основные положения теории К. с. были разрабо-
таны Сиджвиком (1927). Согласно теории Сиджвика,
К. с. может образоваться между любым атомом или
ионом, способным принять пару электронов (акцеп-
тор), и любым другим атомом или ионом, к-рый может
предоставить для совместного обладания пару элек-
тронов (донор). Эти представления были распростра-
нены и на молекулярные системы. В настоящее время
под акцептором понимают молекулярную (атом-
ную) систему, обладающую свободными (вакантными)
уровнями (орбитами) и положительным сродством
к электрону. К акцепторам относятся любые положи-
тельно заряженные, а в нек-рых случаях и нейтраль-
ные системы. Донором называют такую систему, к-рая
имеет свободные (неподеленные) пары электронов.
При образовании К. с. неподеленная пара электронов
становится общей для донора и акцептора. К. с. ча-
сто обозначают стрелкой, указывающей направление
смещения неподеленной электронной пары.
Когда К. с. уже образована, она практически ничем
не отличается от обычной ковалентной связи. Классич.
примером К. с. может служить взаимодействие трех-
фтористого бора BF3 и аммиака NH3, связь между
к-рыми возникает благодаря вакантной орбите атома
бора и неподеленным электронам атома азота. Акцеп-
торы часто именуют (особенно в органич. химии)
электрофильными (электроноакцепторными, реже ка-
тионоидными) реагентами, а доноры — нуклеофиль-
ными (электронодонорными, реже анионоидными)
реагентами. Наиболее часто донорами являются
молекулярные системы, содержащие атомы N, О, F
и С1 (а также, но реже, С, Р, S, As, Вг, J). Донорно-
акцепторные связи играют большую роль в химии
комплексных соединений, в к-рых центральный атом
(как правило, ион металла с незаполненной оболочкой)
обладает свободными орбитами, а лиганды — непо-
деленными парами электронов. Особенность К. с.
состоит в том, что они повышают число связей между
атомами путем вовлечения большего числа электро-
нов и свободных орбит.
Способность образовывать координационные соеди-
нения не ограничивается переходными металлами,
вакантные орбиты ионов к-рых обладают большим
сродством к электрону. В настоящее время известно
большое число координационных соединений ионов
с замкнутыми внешними sp-оболочками (ионы щелоч-
ных и щелочноземельных металлов и галогенов).
Представления о К. с. позволяют объяснить струк-
туру и свойства сольватов (если растворителем яв-
ляется вода — гидратов) этих ионов. Атомы галогенов
могут исполвзовать свои неподеленные пары и обра-
зовывать не одну связь (как это предполагается
в элементарной теории валентности), а по крайней
мере, две, а то и три. Это и определяет строение т. наз.
мостиковых соединений, в к-рых, как, напр., в поли-
мерных цепях PdCl2, атом Pd связан с четырьмя ато-
мами G1, а каждый атом С1 является мостиком между
двумя атомами Pd.
Центральное место занимает проблема прочности
К. с.; она обусловливается многими факторами и
ее нельзя непосредственно привести в соответствие
с типом связи. Как правило, прочность К. с. тем
12*
выше,чем меньше разница между энергиями вакант-
ных и донорных орбит. Так, в сольватах прочность
К. с. уменьшается в ряду Н+ > Li+ > Na+ > К+ >
> Rb+ > Cs+. Среди многих других факторов на
прочность К. с. оказывают сильное влияние заряд
атома-акцептора, природа атома-донора, циклообразо-
вание и др. факторы. Квантово-механич. теория К. с.
дает возможность исследовать механизм ее образова-
ния и зависимость энергии связи от различных фак-
торов.
Лит.: Химия координационных соединений, под ред.
Дж. Бейлар, Д. Буш, пер. с англ., М., 1960; С ы р к и н Я. К.,
Усп. химии, 1959, 28, вып. 8, 903; Измайлов Н. А.,
Кругляк Ю. А., ДАН СССР, 1960, 134, № 6, 1390;
С аг tm ell Е., Fowl es G. W. A., Valency and mole-
cular structure, 2 ed., L., 1961. Ю. А. Кругляк.
КООРДИНАЦИОННАЯ ТЕОРИЯ — см. Коор-
динационная связь, Комплексные соединения.
КООРДИНАЦИОННОЕ ЧИСЛО — число атомов
или атомных групп, связанных с данным атомом,
называемым центральным. См. Комплексные соеди-
нения.
КООРДИНАЦИОННЫЕ ПОЛИМЕРЫ (клешне-
видные или хелатные полимеры) — гетероцепные вы-
сокомолекулярные соединения, содержащие коорди-
национно связанные ионы металла в основной цепи
макромолекулы. Подобно другим высокомолекуляр-
ным соединениям, макромолекулы К. п. состоят из
повторяющихся звеньев, число которых выражает
степень полимеризации.
Получают К. п. конденсацией бифункционального
лигапда (низкомолекулярное вещество, способное
к образованию координационных, т. е. комплексных
соединений с металлами) и металлич. соединения. При
этом координационно-связанные ионы металла обус-
ловливают образование самого полимера, входя в его
основную цепь (поликоординация), напр.
образование К. п. на основе бис-(Р-днкетона), взятого
в качестве лиганда'.
О=С-В’—С=О
I I
НС сн
II 1L
НО—С С-ОН +Ме2+—-
I I
В в
О=С-В'—0=0- „О---С—R’ —
I I \ / I
---- НС СН >Ме< СН
II II z \ II
О=с C-Oz ХО-с
I I I
R R R
Этот метод, позволяет получать К. п., макромоле-
кулы к-рых построены как за счет ковалентных свя-
зей (характерных для обычных органич. полимеров),
так и за цчет координационных связей.
Для осуществления поликоординации и получения
линейного К. и. необходима молекулярная структура
где X и Y являются атомами-донорами, причем два
из них должны быть связаны с водородом, обладающим
протонной подвижностью. Тогда при взаимодействии
с двухвалентным ионом металла (Ме2+), имеющим
координационное число 4, каждая молекула лиганда
(I) теряет два протона и образуется линейный поли-
мер типа II.
При помощи поликоординации, в зависимости от
строения исходного лиганда и природы иона металла,
можно получить К. п. линейной, плоскостной или
трехмерной структуры, а также сополимеры, блок- и
привитые полимеры с широким комплексом физич.
и физико-химич. свойств. Существует также большая
группа карбоцепных полимеров, содержащих в боко-
711
КООРДИНАЦИОННЫЕ ПОЛИМЕРЫ — КОПА ПЕРЕГРУППИРОВКА
712
вых цепях группировки атомов, способные к комп-
лексообразованию с металлами. Эти полимеры из-
вестны как ионообменные смолы (см. Иониты).
При потере ионов металла у этих полимеров сохра-
няется неизменной основная полимерная цепь, в то
время как К. п. при потере ионов металла разру-
шаются. Разнообразие К. п. обусловливается харак-
тером координационных групп лиганда. Синтезиро-
ваны К. п., содержащие как органич., так и неорганич.
лиганды. Так, напр., хлористый палладий существует
в виде полимера линейного типа:
К числу органич. соединений, образующих К. п., от-
носятся следующие: бис-0-дикетоны RCOCH2CO—R'—
—COCH2COR, бис-а-аминокислоты HOOCH(NH2)—
—R—СН (NH2) СООН, бис-о-окспальдегпды (I и II)
диоксихиноны (III, IV, V), бис-салицилальдегиддп-
амины (VI), бис-о-оксиазосоединения (VII), бис-гидр-
оксамовые кислоты (HON)CO—R—CO(NOII), бис-
нитрозофенолы (VIII) и мн. др.
Синтезировано большое число однородных и смешан-
ных К. п. на основе бис-(Р-дикетонов) различного
строения, хинизарина, бис-8-оксихинолилметана, бис-
(а-тиоппколиламидов), фталоцианинов, производных
ферроцена, различных диокси- и диметоксидикарбо-
новых к-т и др. лигандов, а также ионов Си, Zn,
Be, Mn, Ti, TI, Co, Ni, Cd и др. металлов.
К. п. представляют собой твердые окрашенные про-
дукты, термин, стойкость к-рых, в зависимости от
строения и состава полимера, лежит в пределах
200 — 400°. К. п. плохо растворяются в органич.
растворителях и большинство из них разлагается
не плавясь. Наиболее растворимыми являются К. п.
бериллия, к-рые также плавятся без разложения.
Можно предполагать, что К. п., являясь как бы пере-
ходным мостом между органич. и неорганич. поли-
мерами, будут сочетать термостойкость и электро-
проводность последних с пластичностью и эластич-
ностью органич. полимеров. К. и., полученные на
основе хинизарина, бис-8-оксихинолилметана- и
4,4'-бис-(ацетоацетил) дифенилоксида, и пек-рые дру-
гие представляют собой парамагнитные вещества.
Электрич. свойства нек-рых из них (величина электро-
проводности 10 ~7—10~13о.м- см-1 и энергия актива-
ции проводимости) свидетельствуют об их близости
к полупроводниковым материалам. К. п. могут быть
получены, как в расплаве, взаимодействием лиганда
с ацетилацетонатом соответствующего металла, так
и в растворе, реакцией между лигандом и солями
металлов; возможна также реакция в растворе на гра-
нице раздела фаз.
Изучение закономерностей поликоординации, про-
веденное на примере взаимодействия 4,4'-бис-(ацето-
ацетил) дифенилоксида с ацетилацетонатами берил-
лия и цинка, с уксуснокислым цинком и аммиакатом
цинка в расплаве и р-ре, показало большую аналогию
полпкоординации с обычной равновесной поликонден-
сацией. Поликоординация тоже является обратимым
процессор. К. п. 4,4'-бис~(ацетоацетил) дифенилоксида
и бериллия при нагревании с избытком ацетилацето-
пата разрушается с образованием исходного лиганда;
К. п. вступает в обменные реакции не только с низко-
молекулярными продуктами реакции, но и с низко-
молекулярными веществами, близкими к полимеру
по химич. природе. Так, при взаимодействии К. п.
4,4'-бис-(ацетоацетил)дифенилоксида и бериллия с
ацетилацетонатом меди имеет место обменная реакция,
приводящая к образованию К. п., содержащего медь
и ацетилацетопат бериллия. Для получения К. п.
высокого мол. веса необходимо проводить поликоор-
динацию в условиях, обеспечивающих возможно
более полное удаление из реакционной смеси низко-
молекулярного продукта реакции. При соблюдении
этого условия удалось получить из 4,4'-бис-(ацето-
ацетил)дифенилоксида и ацетилацетоната Be в рас-
плаве полимер с мол. весом более 125 000. Алифатич.
бис-(Р-дпкетоны) наряду с линейными полимерами
образуют циклич. структуры. Преобладание циклич.
или линейного продукта зависит как от условий реак-
ции (разбавление реакционной среды), так и от при-
роды исходных веществ (длина метиленовой цепочки,
ионный радиус металла).
Лит.: Химия и технология синтетических высокомоле-
кулярных соединений, вып. 1, кн. 2, М., 1959 (Итоги науки.
Хим. науки, т. 3), с. 831; тоже, вып. 7, М., 1961, с. ЗЗГ,
Берлин А. А., в кн.: Успехи химии и технологии поли-
меров, сб. 3, М„ I960; К о р ш а к В. В., С о с и н С. Л.,
Чистякова В. М., Реакция полирекомбинации как
метод получения полимеров, там же; X а й д У к И., Усп.
химии, 1961, 30, вып. 9, 1124; Берлин А. А., Мат-
веева Н. Г., там же, 1960, 29, вып. 3, 277; Marvel
С. S, [а. о.], t. Amer. Chem. Soc., 1957, 79, К» 22, 6000; 1958,
80, Ki 5, 1197, Ki 24, 6600; Martin К. V., там же, 1958, 80,
М 1; Коршак В. В. [и др.], Высокомолекулярные соеди-
нения, 1959, 1, Ki 12, 1764; 1960, 2, К« 4, 492 — 98, М 5, 662;
1961, 3, К 3; К 1 u 1 b е г R. W., Lewis J. W., J. Amer.
Chem. Soc., 1960, 82, 5777. E. С. Кронгауэ.
КОПА ПЕРЕГРУППИРОВКА (Коупа перегруп-
пировка) — миграция аллильной или замещенной
аллильной группы в 1,5-диенах (I)
от а- к у-атому углерода с одно-
временным сдвигом двойной свя-
зи из Р, у- в а,р-положение (II).
К. п. происходит при нагревании
до 150—300° в течение несколь-
ких часов, выход продукта состав-
ляет обычно 68—100%- К. п. об-
X и Y=CN, COOR,
СвН5 и т. д.
легчается, если X и Y — заместители 2-го рода, по-
вышающие электроотрицательность а-атома углерода;
при этом различные заместители по силе влияния
могут быть расположены в ряд:
CN > CN + COOR > 2 COOR
Отмечалась структурная аналогия К. п. Клайзена
перегруппировке аллиловых эфиров енолов и фенолов,
имеющих электроотрицательный атом кислорода:
Однако К. п. отличается от перегруппировки Клай-
зена тем, что требует наличия в 1,2-положении соеди-
713
КОПА ПЕРЕГРУППИРОВКА — КОРДИАМИН
714
пения I лишь ациклич. или алициклич. двойной связи;
перегруппировка не происходит в случае систем,
у к-рых 1,2-двойиая связь является частью ароматич.
системы (II). Так, напр., в К. п. невсту-
хч xy пает система II, где заместителями X и
а^хСН Y могут быть: а) X = Y = COOR;
I 2 б) X = COOR, Y=CN; в) X=Y=CN;
qi г) X=Y=C6H5. В случае же производ-
н ных фенантрена и нафталина, у к-рых
связь в положении а,|3 имеет, по По-
лингу, повышенный характер двоесвязности, происхо-
дит перегруппировка, отличная от К. п., напр.:
COOR
----Ch2COOR
К. п. является некаталитической, впутримолекуляр
ной реакцией 1-го порядка, протекающей через циклич.
переходное состояние с инверсией аллильной группы,
напр.:
(COOR)2
Н3Сч ,с' Н3(% CICOOR).,
“7 185\ СН2
СН2 СН СН, ^СН
СН сн
сн3 сн3
Механизм К. п., включающий диссоциацию 1,5-диспа на 2 ал-
лильных радикала с последующей их рекомбинацией, не согла-
суется с наблюдениями:
1) К. п. происходит только в системе диаллила (1,5-диена)
и, напр., соединения Ш в К. н. не вступают:
CN хСООСоН5
НзС\Х^
J R R = CH3, С3Н7, С4Н9
Н3С
111
2) К. п. однозначна и, как правило, приводит лишь к одному
из возможных продуктов рекомбинации аллильных радикалов.
Исключение представляет К. п. соединения IV:
IV
При одновременной К. п. двух различных 1,5-диепов обмена
аллильными группами не наблюдается.
3) Энергия активации К. п. значительно ниже энергии
диссоциации С—С-связи в системе диаллила (ок. 41 ккал/мглД
Для К. п. соединения V
найдены следующие зна-
чения энергии и энтро-
пии активации: Еакт =
= 25,8ккал/л1оль, Д8^ =
= —11,1 кал/град - моль.
v
Взаимная ориентация двух аллильных групп в пере-
ходном состоянии К. п. может соответствовать двум
конформациям — «ванны» и «кресла», что отвечает
или 6-центровому (А), или 4-центровому (В) переход-
ному состоянию.
Было показано, что при К. п. мезо- и рацемических
изомеров 3,4-диметплгексадпепа-1,5 реализуется толь-
ко 4-центровое переходное состояние (В), к-рое на
~5,7 ккал/молъ выгоднее 6-цептрового переходного
состояния (А).
Лит..- Соре А, С., Hardy Е. М., J. Amer. Chem.
Soc., 1940, 62, № 2, 441; Соре А. С., Hoyle К. Е„
Н е у 1 D., там же, 1941, 63, № 7, 1843; Соре А. С.,
Field L., там же, 1949, 71, № 5, 1589; Соре А. С.,
Meili J. Е., McDowell D. W. Н., там же,
1956, 78, АГ» И, 2551; Соре А. С., F i е 1 d L., М с D о w е 1 1
D. W. Н., Wright М. Е., там же, 1956, 78, И, 2547;
Деринг В. Э„ Ж. Всес. хим. об-ва им. Д. И. Менделеева,
1962, 7, № 3. Ю. Д. Корешков.
КОПАЛЫ — ископаемые природные смолы. К. от-
личаются большой твердостью, высокой темп-рой
плавления (от 100° до 360° и выше), химич. стойкостью.
К. в основном состоят (до 90%) из кислот смоляных;
кроме того, в К. содержится 6—12% неомыляемых
(резенов), эфирные масла, небольшие количества
минеральных примесей и др. К. классифицируют по
твердости и темп-ре плавления, по цвету, степени про-
зрачности, величине кусков. Наиболее употребитель-
ны следующие виды К. (названия их даются чаще
всего по месту добычи): твердые — Занзибар, Мозамбик
и Мадагаскар; средней твердости — линдп, сиерра-
леопе, ангола, конго каури и др.; мягкие — манила,
борнео, Сингапур, бразильский. Твердые и средние
сорта применяются гл. обр. для варки масляных ла-
ков; мягкие, растворимые — для произ-ва спирто-
вых лаков.
Цвет различных К. — от бесцветного до коричне-
вого; нек-рые К. обладают своеобразным запахом;
плотн. 1,03—1,07; растворимость: в спирте от 7,4%
до 98%, в бензине от 11,7% до 49,5%, в скипидаре
от 0 до 52%. Кислотное число К. — от 50 до 160;
число омыления — от 75 до 215; иодное число от
50 до 180.
В маслах К. не растворяются; для растворения их
предварительно подвергают частичному термин, раз-
ложению с достижением т. наз. «градуса раствори-
мости» и потерей веса от 3 до 30%. При термин, раз-
ложении К. отгоняется т. наз. копаловое масло. Чем
глубже деструкция К., тем легче он растворяется
в масле и тем стабильнее, но темнее получается его
раствор. В связи с появлением разнообразных син-
тетпч. смол, К. утратил свое прежнее значение.
И. И. Головистиков.
КОПРОСТЕРИН, см. Стерины.
КОРАЗОЛ (кардиазол, а,|3-пентаметилентетразол)
C6lI10N4, мол. в. 138,17 — бесцветные кристаллы;
т. пл. 59—61°; т. кип. 194°/12 мм; легко
растворимы в большинстве органпч.
растворителей и воде; водные р-ры К.
имеют нейтральную реакцию на лакмус.
При нагревании К. с водным НС1 при
высокой темп-ре образуется пентаметилендиамин; с
водным р-ром сулемы К. образует двойную соль,
т. пл. 175—180°.
К. может быть получен из капролактама следующим
образом:
Схн (СИ;,)а5О,_
\___An-NH3
К. — антагонист наркотиков, применяется для сти-
мулирования деятельности центральной нервной си-
стемы, возбуждения дыхания и кровообращения.
Лит.: Глушков Р. Г., Г о л о в ч и н с к а я Е. С.,
Мед. пром-сть СССР, 1960, № 1, 12. Р. Г. Глушков.
КОРДИАМИН (корампп, никетамид) — 25%-ный
раствор диэтиламида никотиновой к-ты в воде; бес-
цветная или яселтоватая жидкость с своеобразным за-
пахом, смешивается с водой и спиртом во всех отно-
шениях; d 1,023 — 1,024.
,CH2s.
(СН2)3 N—N
\ ,С N
715
КОРДИТЫ — КОРИЧНАЯ КИСЛОТА
716
Диэтиламид никотиновой кислоты
C10H14ON2, мол. в. 178,20 — светло-желтая маслянистая жид-
кость; хорошо растворима в воде и ор-
ганич. растворителях; т. кип. 280°.
При его нагревании с NaOH образует-
ся Na-соль никотиновой к-ты и диэтила-
мин. С сульфатом меди диэтиламид ни-
котиновой к-ты дает интенсивное синее
окрашивание. Количественное определение диэтиламида ни-
котиновой к-ты основано на омылении щелочью и отгонке
выделяющегося диэтиламина с паром в избыток титрованного
р-ра соляной к-ты. Диэтиламид никотиновой к-ты получают
из никотиновой к-ты и диэтиламина:
QCOOH+NH(C2H5)2^ <YCON(C2iy2
ТУ
К. стимулирует центральную нервную систему, воз-
буждает дыхание, тонизирует сердечно-сосудистую
систему; по характеру действия близок к камфаре,
кофеину и коразолу; применяется при острых и хро-
нич. расстройствах сердечной деятельности,сердечной
слабости и ослаблении дыхания при инфекционных
заболеваниях, коллапсе и асфиксии, а также при
отравлении наркотиками, снотворными, окисью угле-
рода и синильной к-той.
Лит.: Машковский М. Д., Лекарственные сред-
ства, 4 изд., М., 1960. Л. Н. Яхонтов.
КОРДИТЫ — винтовочные и орудийные пороха на
основе нитроцеллюлозы, пластифицированной нитро-
глицерином и летучим растворителем. Зерна К. имеют
форму бесканальных шнуров. В Англии приняты сле-
дующие К.:
Название пороха Содержание компонентов, %
высоко азотной нитроцел- люлозы нитрогли- церина вазелина аце- тона ВОДЫ
Кордит № 1 36 57 5 1,5 0,5
Кордит МД 63 30 5 1,5 0,5
Темп-ра горения кордита № 1 при плотности заря-
жания 0,5 достигает 3100°; применение такого К.
в артиллерии и для автоматич. оружия приводит
к быстрому износу (разгару) каналов стволов. При 7°
и ниже на поверхности зерен К. выделяется нитрогли-
церин, что делает его опасным в обращении; при 38°
на поверхности зерен выделяется вазелин, к-рый вво-
дится в состав пороха в качестве стабилизатора. Эти
недостатки в значительно меньшей степени выражены
в кордите МД.
Для различных видов ствольного огнестрельного
оружия готовятся пороха кордитного типа различных
марок с зернами разных форм и размеров (трубки,
пластинки, ленты и т. п.) (см. табл.).
Содержание компонентов, %
Название пороха низкоазот- ная нитро- целлюлоза высоно- азотная нитроцел- люлоза нитро- глице- рин стабили- затор остаточ- ный лету- чий рас- твор итель вода KNO3
Эксайт, охотничий (Анг- 0,7
ЛИЯ) . . Maxim, орудийный — 63 29 5 (вазе- лин) 0,3 2
(США) Соленит, винтовочный 8 78 9 2 (моче- вина) 1,5 1,5
(Италия) 30 35 33 1 (цен- тралит) 0,5 0,5
Лит.: Брунсвиг Г., Бездымный порох, пер. с нем.,
М.—Л., 1933. А. С. Бакаев.
КОРИАНДРОВОЕ МАСЛО — см. Жиры расти-
тельные и Эфирные масла.
КОРИНАНТЕИН C22H2eO3N2, мол. в. 366,45 —
алкалоид, содержащийся в африканском ложнохин-
ном дереве (Pseudocinhona afri-
cana Chew.) и в тех же расте-
ниях, что и иохимбин; выделя-
ется из маточников после отде-
ления иохимбина. Существует
в двух переходящих друг н
ДРУга . )
т. пл. 170—171°
чивая) и 117°
изоморфных формах,
(более устой-
(из абс. спирта),
[а]п = —28,1° (СН3ОН). К. дает дигидрат, т. пл. 71°
(из разб. спирта), и хлоргидрат, т. пл. 205°, раствори-
мый в хлороформе.
По строению К. схож с иохимбином. Циклич. си-
стема К. отличается от системы иохимбина отсутствием
одного цикла. При омылении К. щелочью расщеп-
ляется сложноэфирная группа. Действие разб. к-т
на К. вызывает гидролиз обоих метоксилов, обра-
зующаяся 0-альдегидокислота декарбоксилируется с
образованием коринантеаля (I), к-рый может быть
далее превращен в коринантеан (II). Дегидрирование
К. с Se приводит к коринантирину, идентичному аль-
стерину. При каталитич. гидрировании К. восста-
СНдООС
ill
Н3'
ОСН3
образуется
IR=CHO: Il R=CH3
навливается только виипльная группа и <
дигидрокоринантеин (III), строение к-рого подтвер-
ждено синтезом. По физлологич. активности К. подо-
бен иохимбину; спазмолитич. средство.
Лит.: Т a m е 1 е n Е. van, Hester I. В., J. Amer.
Chem. Soc., 1959, 81, Jxft 14, 3805; См. также при ст. Алкалоиды.
_________ _ ____________ п А. Е. Васильев.
кис-
(I) и
КОРИЧНАЯ КИСЛОТА ([5-фенилакриловая
лота), мол. в. 148,15 — существует в транс-
ifuc-формах (II):
н-с-соон
II
с„н5-сн
I
т.
кип.
н-с-соон
II
н-с-сен5
II
транс-изомер более устойчив; т. пл. 133°;
300°; теплота сгорания 1040 ккал/моль. Цис-К. к. об-
разует три полиморфные формы: аллокоричная к-та
Либермана, т. пл. 68°, изокоричная к-та Либермана,
т. пл. 58° и изокоричиая к-та Эрленмейера, т. пл. 42°.
Аллокоричная к-та более устойчива; все три формы
могут быть легко переведены друг в друга (йапр.,
внесением соответствующей зат-
равки в расплав). Константа дис-
социации г{ис-изомера, К =
= 1,4- 10 4 (25°), значительно
выше, чем транс-изомера, К ==
= 3,5.10-5 (25°).
Транс-К. к. можно получить
несколькими способами: 1) С по-
мощью Перкина реакции из бен-
зальдегида и уксусного ангид-
рида в присутствии ацетата нат-
рия(190°):
СсН5СНО+(СН3СО)8О СНз—00-g-
-->С0Н5СН=СНСООН-|-СНзСООН
2) Нагреванием хлористого бензилидена с ацетатом
натрия (180—200°):
С8ЩСНС12+2СН3СООХа—-
—>C8H3CH=CHCOOH-j-CH3COOH-f-2NaCL
717
КОРИЧНЫЙ АЛЬДЕГИД —КОРРЕЛЯЦИЯ ГЕОХИМИЧЕСКАЯ
718
3) Окислением бензилиденацетона хлорноватистой
к-той; этот кетон, в свою очередь, получают конден-
сапией бензальдегида с ацетоном:
с8н5сно+сн3сосн3—>с8н5сн=снсосн3
С8113С11==С11СОСП3-1Н^С1-!-С8113С11==С11СОО11
4) Конденсацией бензальдегида с этилацетатом по
Клайзену:
С8Н5СНО+СН3СООС2Н5 СгН5° — »С8Н5СН=СНСООС;Н5
В пром-сти К. к. получают одним из трех первых
способов. В последнее время отдают предпочтение
синтезу через бензилиденацетон. Для получения
цис-К. к. целесообразнее исходить из смеси кислот,
являющейся отходом в произ-ве кокаина (эти кислоты
образуются при омылении сопутствующих кокаину
алкалоидов). К. к. содержится (гл. обр. в виде эфи-
ров) в ряде эфирных масел и смол, в перуанском и
толуанском бальзамах и др. При декарбоксилирова-
нии (ок. 146°) К. к. образуется стирол; окисление
приводит к смеси бензойной и уксусной к-т, а восста-
новление — к гидрокоричной к-те СвН6СН2СН2СООН.
К. к. легко присоединяет галогены, напр.:
С8Н5СН=СНСООН-|-Вг3—>С8Н5СНВгСНВгСООН
Дибромкоричная к-та при нагревании в растворе
соды или поташа дает [5-бромстирол. При нитровании
К. к. образуется смесь нитрокоричных к-т; сульфиро-
вание идет гл. обр. в пара-положение. В табл, приве-
дены свойства нек-рых эфиров, имеющих практич.
значение или применяющихся для идентификации.
Эфир Молеку- лярный вес Темп-ра пл., °C Темп-ра кип., °С/мм . d4
Метиловый3 . . . 162,18 33,4 263/760 1,0700 (35°)
Этиловый®.... 176,21 12 271/760 1,0490
Пропиловый . . 190,23 285/760 1,0435 (0°)
Изобутиловый 204,26 — 165/17 —
н-Амиловый . . . 218,28 — 192/29 0,999 (20°)
Фениловый . . . 224,25 72 73 205—207/15 —
Циннамиловый . 264,31 44 • — —
Бензиловый . . . 238,27 39 244/25 —
О п 1,5736. с 1,5598.
К. к. используется преим. для синтеза бромстирола,
фенилацетальдегида и эфиров К. к. (метилового, этило-
вого и бензилового и циннамилового); эти соединения
применяются как душистые вещества. В небольших
количествах К. к. используется в произ-ве нек-рых
фармакологич. препаратов. Н. А. Несмеянов.
КОРИЧНЫЙ АЛЬДЕГИД (Р-фенилакролеин)
CeH6CH = СНСНО, мол. в. 132,16 — бесцветная или
бледно-желтая подвижная жидкость с сильным за-
пахом корицы и жгучим вкусом; входит в состав ко-
ричного и многих других эфирных масел; т. пл.
—7,5°; т. кип. 252°/760 мм (с частичным разл.), 128—
130°/20 мм; d|» 1,1102; 1,61949; 1 часть К. а. раст-
воряется в 25 частях 50%-ного спирта или в 2—3 ча-
стях 70%-ного спирта; плохо растворим в воде и пет-
ролейном эфире, с серным эфиром смешивается во
всех отношениях. К. а. перегоняется с водяным паром.
К. а. проявляет обычные свойства а,р-ненасыщен-
ных альдегидов, хотя двойная связь в нем гораздо
слабее активирована, чем в акролеине. При стоянии
на воздухе К. а. медленно окисляется в коричную к-ту.
Восстановление К. а. может быть осуществлено либо
только по альдегидной группе, либо по двойной
связи, либо по обоим направлениям сразу. В реак-
циях К. а. с бисульфитом и с гидразином участвует
как альдегидная группа, так и двойная связь. По-
скольку К. а. является винилогом бензальдегида (см.
из бензола;
1,430А, длины
соответственно
Винилогия), он обладает многими свойствами арома-
тич. альдегидов, напр. вступает в Перкина реакцию;
для него могут быть осуществлены все реакции, ха-
рактерные для альдегидной группы. Бензольное ядро
К. а. нитруется в орто- и пара-положения. К. а. со-
держится в коре, листьях и масле корицы, пачули,
гиацинта и др. В пром-сти К. а. получают конденса-
цией бензальдегида с ацетальдегидом в присутствии
оснований; подчиненное значение имеют окисление
коричного спирта, восстановление хлорангидрида
коричной к-ты по Розенмунду, формилировапие сти-
рола по Вильсмайеру.
Применяют К. а. в парфюмерной пром-сти самостоя-
тельно и для произ-ва коричного спирта, в органич.
синтезе И др. Н. И. Гамбарян.
КОРИЧНЫЙ СПИРТ С6Н6СН=СНСН2ОН, мол.
в. 114,08 — кристаллы в виде игл; т. пл. 33°; т. кип.
257,5°/760 мм; 143,5°/14 мм; d*> 1,0440; 1,5819;
растворим в спирте, эфире и других органич. раство-
рителях; в воде растворим мало. К. с. содержится
в виде эфиров в гиацинтовом масле, перуанском баль-
заме, стираксе (смоле амбрового дерева Liquid ambra,
произрастающего в Малой Азии, Сирии, Китае).
В пром-сти К. с. получают восстановлением коричного
альдегида алкоголятами аммония. К. с. обладает
запахом гиацинтов; применяют в произ-ве душистых
веществ. Производные К. с. обладают сильными инсек-
тицидными свойствами, их используют в качестве репе-
Л6НТ0В при борьбе С МОСКИТаМИ. А. В. Арбатский.
КОРОНЕН (гексабензобензол) С24Н12, мол. вес
300,34 — длинные светло-желтые иг,
т. пл. 438—440°; 1 вес. ч. раство-
ряется в 1290 вес. ч. холодного бен-
зола и в 315 вес. ч. кипящего; не-
растворим в конц. H2SO4; растворы
в обычных органич. растворителях
обладают сине-фиолетовой флуорес-
ценцией; длина связей центрального
кольца и связей, соединяющих это
кольцо с внешним —с—, равна
связей внешних колец а и Ь равны
1,385 и 1,415 А. Пикрат—темно-красные кри-
сталлы из бензола, разлагается ок. 250°; комп-
лекс с 1, 3, 5-тринитробензолом — красно-оранжевые
иглы из бензола, разлагается выше 250°. К. термически
устойчив. При восстановлении К. натрием в амиловом
спирте образуется тетрадекагидрокоронен; окисление
хромовой к-той дает короненхинон; при действии
HN03 (а? 1,2) образует мононитрокоронен; кипячение
с конц. HNO3 дает динитрокоронен; в уксусной к-те
присоединяет бром с образованием ди- и тетрабромко-
ронена; при действии бензоилхлорида в присутствии
А1С13 в среде CS2 был получен бензоилкоронен. К. со-
держится в кам.-уг. смоле; в продуктах гидрогенеза-
ции каменного или бурого угля, или гудрона. Известны
синтетич. способы получения К.
Лит.: Ф и з е р Л., Ф и в е р М., Органическая химия,
пер. с англ., М., 1949, с. 717. Л. С. Поваров.
КОРРЕЛЯЦИЯ ГЕОХИМИЧЕСКАЯ — сопостав-
ление слоев или массивов горных пород различных
территорий по особенностям их химич. состава. К. г.
наибольшее применение нашла при изучении т. н.
«немых толщ» осадочных пород, т. е. толщ, не содер-
жащих остатков фауны или флоры, что не позволяет
точно определить их геологич. возраст и увязать слои
разных районов. Для отдельных горизонтов, слоев,
свит осадочных пород характерны т. н. «ведущие
элементы». Так, в Куйбышевской области (в Завол-
жье) в разрезе пермских отложений установлено три
горизонта, обогащенных стронцием, содержащих ми-
нерал целестин SrSO4. Средний целестиновый гори-
зонт был обнаружен в керне буровых скважин, рас-
положенных друг от друга на 200 км. Это позволило
719
КОРРОЗИОНИОСТОЙКИЕ МАТЕРИАЛЫ— КОРРОЗИОННЫЕ ИСПЫТАНИЯ
720
«увязать» разрезы, т. е. установить, что данный гори-
зонт развит в обоих районах. В палеозойских отложе-
ниях Заволжья выделяется ряд горизонтов с харак-
терным содержанием V, Мп, Mg, что позволяет выде-
лить породы этого возраста в разных районах, даже
если они не содержат ископаемой фауны. На Север-
ном Кавказе по составу редких элементов намечается
четкое различие между средне- и верхнеюрскимп от-
ложениями, между юрскими и меловыми. Караган-
ский горизонт и сарматский ярус третичных отложе-
ний Северного Кавказа постоянно обогащены литием,
что позволяет местами идентифицировать породы
этого возраста.
Для корреляции песчаных горизонтов наибольшее
значение имеют Zr, Ti, Сг, Ga, Fe, Al, для корреляции
глинистых — Zr, Ga, Ст, V, Ni, Со, Си, Мп, Sr, Ва,
Mg, Са, Li, Rb, К, Na, Ge, Be, для карбонатных по-
род — Sr, Ва, Мп, Си, Ni, Na, Mg, Al, Si. Важное
значение при К. г. имеет качественный и количест-
венный спектральный анализ, позволяющий быстро
определить в образцах горных пород многие элементы.
В ряде случаев для К. г. используются отношения
между двумя элементами: Na : К; К : Li; Си : Sr;
Са : Sr; Sr : Ва; V : Сг; Си : Ni; Fe : Мп и т. д. Так,
~ равно 31,
напр., в морской воде отношение Са ; Sr
в породах кунгурского яруса Заволжья —
32, в породах артипского яруса — 611, ка-
занского яруса — 248. Это позволяет де-
лать вывод об условиях образования пород
в нормальных морских бассейнах или в за-
соленных лагунах. К. г. используется так-
же при изучении изверженных пород и руд-
ных месторождений. Так, в породах, со-
держащих месторождения свинца, цинка и
серебра на Алтае, повышено содержание
этих элементов, что позволяет на основе
анализа пород искать и сами рудные место-
рождения. Аналогично, к гранитам, обога-
щенным оловом, бериллием и редкими зем-
лями, приурочены оловянные месторож-
дения.
Методика К. г. состоите отборе проб гор-
ных пород (10—15 г) иэ естественных обна-
жений или чаще из керна и шлама буровых
скважин. Отбор обычно производят из всех
слоев, с интервалом не более, чем 5—20 м
(по вертикали). Порошки пород подвергают
спектральному анализу, данные к-рого сум-
мируются в таблице или в особой геохимия,
диаграмме, аналогичной каротажной ди-
аграмме буровых скважин. Анализ этой
диаграммы позволяет выделить ведущие
элементы отдельных слоев и горизонтов.
При этом важно не абсолютное содержание
того или иного элемента, а относительное
изменение концентрации между соседними
слоями- Иногда для анализа берут не пробы
пород, а отдельные механич. фракции (напр.,
глинистая ^фракция) или водные, солевые,
кислотные вытяжки из пород.
Лит.: Методы изучения осадочных пород, т. 2,
М., 1957; Катченков С., Новый метод кор-
реляции осадочных толщ по данным спектраль-
ного анализа, Новости нефтяной техники. Нефте-
промысловое дело, 1955, вып. 3.
А. И. Перельман.
КОРРОЗИОННОСТОЙКИЕ МАТЕРИАЛЫ — ус-
тойчивые в агрессивных средах материалы, применяе-
мые для изготовления аппаратов, сооружений, трубо-
проводов, арматуры и т. п. изделий, предназначенных
для эксплуатации в условиях воздействия кислот,
щелочей, солей, газов и др. В зависимости от их при-
роды К. м. делятся на металлические и неметалличе-
ские. Металлич. К. м. применяют в качестве самостоя-
тельных конструкционных материалов или в виде
специальных покрытий (тонким слоем высоколегиро-
ванной стали, цветного металла или сплава). Неметал-
лпч. К. м. применяют в виде конструкционных, футе-
ровочных, обкладочных и прослоенных материалов,
лакокрасочных покрытий и композиций. Неметаллич.
К. м. подразделяют на материалы неорганич. проис-
хождения и материалы органич. происхождения.
Подробно о К. м. см. Химически стойкие материалы.
И. Я. Клинов.
КОРРОЗИОННЫЕ ИСПЫТАНИЯ — определение
опытным путем скорости и характера коррозии метал-
лов и сплавов; во многих случаях при К. и. изучают
также состав и свойства продуктов коррозии. Часто
к К. и. относят определение существенных для корро-
зионной стойкости свойств защитных покрытий: пори-
стости, адгезии и др. К. и. проводят с целью: выбора
материалов и методов их защиты от коррозии для к.-л.
заданной среды; нахождения агрессивных сред, в
к-рых могут быть использованы новые материалы;
выявления причин коррозионных повреждений; вы-
яснения механизма коррозии (в исследовательских
работах) и др. случаях. Различают эксплуатационные!!
лабораторные К. и.; испытания машин, механизмов, ап-
паратов и их моделей иногда называют натурными К. и.
^5 20
.18
о ’гД
способов коррозионных испытаний:
И
Схематическое изображение нек-рых ... - - __ ___________
а — при полном погружении образца и естественной аэрации жидкости;
б — при частичном погружении образца; в — при периодическом смачивании
образца; г — при принудительной аэрации жидкости; д — при перемеши-
вании жидкости; е — при термостатировании; ж — на контактную корро-
зию; з — на щелевую коррозию; и — на коррозионное растрескивание при
постоянной деформации образца; к — на коррозионное растрескивание при
постоянной нагрузке на образец; л — на газовую коррозию методом пе-
риодического взвешивания; м — на коррозионную усталость: н — на кор-
розионную кавитацию; о —• установка для изучения фреттинг-коррозип.
1 — образец; 2 —• коррозионная среда; 3 — трубка для подвода воздуха;
4 — мешалка; 5 — термостатирующая среда; в — нагреватель; 7 — 1целе-
вой зазор; 8 — болт; 9 — скоба; 10 — рычаг; 11 — груз; 12 — печь; 13 —
весы; 14 — электродвигатель; 15 — магнитострикционный вибратор с сер-
дечником 1в; 17 — поверхности трения; 18 — прозрачный колпачок; 19 и
20 — трубки для ввода и вывода газа; 21 — электромагнит для вертикаль-
ных колебаний образца; 22 —• катушка переменного тока для продольных
колебаний образца.
Эксплуатационные К. и. представляют
собой испытания образцов или небольших элементов
конструкций в действующих установках (произ-
водственные испытания) или в природных
условиях (полевые испытания). Последние
обычно проводятся на специальных станциях, раз-
мещаемых в различных условиях (в промышленной,
сельской, тропич. атмосфере, у различных морей и
721
КОРРОЗИОННЫЕ ИСПЫТАНИЯ — КОРРОЗИЯ МЕТАЛЛОВ
722
т. д.) для изучения атмосферной, морской и почвенной
коррозии.
Натурные К. п. большей частью сводятся
к тщательному обследованию машин и конструкций
после определенных периодов их работы и допол-
няются лабораторным изучением деталей или образ-
цов металла, вырезанных из стенок трубопроводов,
реакционных камер и т. д.
При лабораторных К. и. стремятся искус-
ственно воспроизвести реальные условия для воз-
можно более точного определения скорости коррозии
в процессе эксплуатации, или выяснить роль отдель-
ных факторов, влияющих на коррозионную стойкость.
Особое значение среди лабораторных К. и. имеют
т.н. ускоренные испытания, в к-рых необ-
ходимые данные по коррозионной стойкости полу-
чают за короткие сроки с помощью предвари-
тельно разработанных способов увеличения скорости
коррозии (повышения концентрации агрессивного
вещества во внешней среде, поляризации металла
и др.), не оказывающих существенного влияния па
механизм коррозии. Нек-рые способы выполнения
лабораторных К. и. схематически показаны на ри-
сунке (см. стр. 720).
Помимо указанной классификации, К. и. принято
также разделять по типу агрессивной среды: испыта-
ния на коррозию в газах при высоких темп-рах, в
нефтепродуктах, расплавленных солях, жидких ме-
таллах и пр.
Для определения характера и измерения скорости корро-
зии при К. и. наиболее часто употребляют следующие методы:
1) Контроль внешнего вида металла (наблюдение за возникно-
вением и развитием очагов коррозии, потускнением и др.).
2) Измерение глубины коррозионных язв с помощью иглы,
укрепленной на индикаторной головке. 3) Микроскопии, ис-
следование металла (выявление межкристаллитной коррозии,
селективного окисления, определение размеров питтинга и
др.). 4) Определение потери веса на единицу поверхности (при
удалении продуктов коррозии с поверхности). 5) Измерение
увеличения веса на единицу поверхности (при сохранении
всех образовавшихся продуктов коррозии; используется гл.
обр. при изучении газовой коррозии). 6) Количественное оп-
ределение содержания продуктов коррозии в жидкой среде
(при полной их растворимости). 7) Определение изменений
механич. свойств металла в результате коррозии (уменьшение
предела прочности на разрыв, числа возможных перегибов
образца до разрушения и др.). 8) Измерение количества выде-
ляющегося водорода при коррозии с водородной деполяриза-
цией. 9) Измерение количества кислорода, расходуехмого при
коррозии с кислородной деполяризацией, при окислении в
воздухе или в кислороде. 10) Измерение увеличения электрич.
сопротивления образца (в результате уменьшения сечения
металла при коррозии). И) Определение времени до разруше-
ния образца (при испытаниях на коррозионное растрескива-
ние).^) Определение числа циклов изменения напряжений
до разрушения образца (при испытаниях на коррозионную
усталость).
При изучении электрохимии, коррозии наибольшее
значение для К. и. имеют измерение электродных по-
тенциалов металлов и сплавов и снятие поляриза-
ционных кривых. Измерения потенциалов проводят
в лабораторных исследованиях, а также в реальных
условиях, если заранее известна зависимость скорости
коррозии от потенциала, напр. для выбора и контроля
катодной и анодной защиты металлич. сооружений.
Поляризационные кривые используются для уста-
новления механизма коррозии, для ускоренного опре-
деления устойчивости нек-рых сплавов против пит-
тинговой коррозии и в нек-рых случаях для опре-
деления скорости коррозии. Широкое применение
нашел потепциостатич. метод поляризационных изме-
рении, при к-ром определяют зависимость скорости и
характера коррозии от потенциала, заданная величина
к-рого поддерживается с помощью потенциостата.
Потенциостатич. кривые в определенных условиях
являются наиболее полной характеристикой корро-
зионной стойкости металла. При К. и. часто поль-
зуются также измерением силы тока коррозионных
макропар, составленных, напр., из разных металлов
(при изучении контактной коррозии) или из металла
с покрытием и металла без покрытия (при ускорен-
ных определениях защитных свойств некоторых по-
крытий).
Основными критериями химич. стойкости неметал-
лич. материалов являются изменения внешнего вида,
веса стандартных образцов и механич. свойств в ре-
зультате испытаний.
Лит.: Коррозия металлов, пер. с апгл., под ред. В. В. Скор-
челлетти, кн. 2, М., 1952, ч. 6; Т о м а ш о в Н. Д.
[и д р.1, Лабораторные работы по коррозии и защите метал-
лов, М., 1961; Жук Н. П., Коррозия и защита металлов.
Расчеты, М., 1957; Новые методы и приборы для коррози-
онных испытаний, М., 1959; (Тр. Ин-та физ. химии АН СССР,
вып. 7); Якубович С. В., Испытания лакокрасочных
материалов и покрытий, М.—Л., 1952; Методы исследования
ингибиторов коррозии металлов, [под ред. С. А. Балезипа и
В. Б. Ратинова, М., 1958] (Всес. совет научно-технич. обществ,
сб. 7); Колотыркин Я. М. [и д р.], Потенциостати-
ческий метод исследования в электрохимии, в сб.; Проблемы
физической химии, вып. 3, М., 1962; см. также лит. при
статьях Коррозия металлов и Защитные покрытия.
Л. И. Фрейман.
КОРРОЗИЯ МЕТАЛЛОВ. Содержание:
Введение .................................. 722
Классификация и механизм коррозионных процессов . 722
Типы коррозионных разрушений............... 726
Количественная характеристика коррозионных про-
. цессов................................... 727
Электрохимическая коррозия в различных средах . . 727
Методы защиты ............................ 729
Введение. К. м. — разрушение металлов вслед-
ствие физико-химич. воздействия внешней среды; при
этом металл переходит в окисленное (ионное) состоя-
ние и теряет присущие ему свойства. По имеющимся
данным, примерно ок. 10% ежегодной добычи металла
расходуется на покрытие безвозвратных потерь вслед-
ствие коррозии и последующего распыления. Основ-
ной ущерб от К. м. связан не только с потерей боль-
ших количеств металла, но и с порчей или выходом из
строя самих металлич. конструкций, т. к. вследствие
коррозии они теряют необходимую прочность, пла-
стичность, герметичность, тепло- и электропровод-
ность, отражательную способность и другие необхо-
димые качества. К потерям, которые терпит народ-
ное хозяйство от коррозии, должны быть отнесены
также громадные затраты на всякого рода защит-
ные антикоррозионные мероприятия, ущерб от ухуд-
шения качества выпускаемой продукции, выход
пз строя оборудования, аварий в производстве и др.
Для большинства металлов металлич. состояние
является термодинамически неустойчивым. Стремле-
ние металлов переходить из металлич.состояния в ион-
ное весьма различно для разных металлов и может
быть, в общем случае, охарактеризовано по уменьше-
нию свободной энергии при протекании соответствую-
щей коррозионной реакции в данных условиях. Такую
характеристику в отношении коррозии металлов в
р-рах можно приближенно получить па основании
величины стандартного электрохимии, потенциала
металлов (табл. 1).
На рисунке 1 (стр. 723) дается характеристика кор-
розионной стойкости металлов в связи с их место-
нахождением в периодической системе элементов Мен-
делеева.
Классификация и механизм коррозионных процес-
сов. По механизму коррозионного процесса различа-
ют два основных типа коррозии: химическую и элек-
трохимическую .
Под химической коррозией подразу-
мевают взаимодействие металлич. поверхности с ок-
ружающей средой, не сопровождающееся возникнове-
нием электрохимия, (электродных) процессов на гра-
нице фаз. Механизм химич. коррозии сводится к реак-
тивной диффузии атомов или ионов металла сквозь
постепенно утолщающуюся пленку продуктов корро-
723
КОРРОЗИЯ МЕТАЛЛОВ
724
2
&11
6,940:
п
ГРУППЫ ЭЛ E M E H T О В
vni
ш
IV
3
3
22,991
5
® в
е 10,82
Я
и
И
4
И
39,100 i
oW
21
400$
мам»
30
Zn
65,38
ИГ
26.98
v.'.v.v
21
Sc
44,96
• 31
Ga
69.72
~39
® C
g 12,011
з Л
§ Si
„ 28.09
N
14,008
15
P
30,98
50,95;
VI
8
О
16,000
° 2
* S
„32,066
VII
5
85.48:
я
® о»
2 107.88
87,63.
е
48
Cd
112,41
а;
6
Ж
g||
7
87
Fr
(223)
88,92
g In
g 114,76
~57
La
138,92
81“
TI
®204,39
~89
Ac
(227)
47.90 -
. SWws а
о 32
® Ge
g 72,60
W
О 50
I Sn
> a 08,70
’^7Z§®
о 82
Pb
„207.21
90 •
(Th)
232,05g
o 33
g As
a 74,91
Ж 8
.ж
о 51
I Sb
) a 121,76
180,95 „
. .ww a
о 83
I Bi
I a 209,0
~»i
(Pal
231
g&s
о 34
8 Se
a 78.96
Ж
55,'^5 в
2
3
Со;в
$Ni$®
5’8’, 69.®
4
Mog
95,95. „
O 52
® Те
э127.61
о
®
....®
183.92-
84
Po
(209)
~92~
(U)
238,07®
H
1.008
9“
F
19,00
Й“
Cl
35.457
“25
Mn
54.94
зГ"
Вг
79,916
~43
Tc
(98)
53~
J
126,91
~75
Re *
186.31 o
85
At
(210)
'О?
Rhjg
’Э~
PdJ
\Иъ'т. ®
//z/zzzz О
5
6
7
ч
6s;®
vzzzzzZ х
190.2; g
. -z/zzyzcO
WI
193,2' ®
. ZZZ^Zz J
8
=Rt|g
19(1.23®
9
10
УСЛОВНЫЕ ОБОЗНАЧЕНИЯ:
устойчивые
ш Благородные и полублагородные • Металлы
Металлы легко пассивирующиеся „ ”
в окислителях ”
Очень неустойчивые металлы
Мало устойчивые металлы
е
о
створе NaCl
в азотиой кислоте
в серной ».
в соляной „
в органич. кислотах
в щелочах
в аэрированном ра-
Рис. 1. Приближенная характеристика коррозионной
в связи с их местонахождением в периодич. системе . .
(Приведенные данные относятся к обычным темп-рам и средним или повышен-
ным концентрациям кислот нли щелочей). Металлы, не имеющие в таблице
значков устойчивости, недостаточно устойчивы в соответствующих средах,
либо — гл. обр. для более редких металлов — недостаточно обследованы.
устойчивости металлов
элементов Менделеева.
ва различных добавок (хрома, алюми'
ния, кремния и др.). Например, сталь,
содержащая 25% Сг, 20% Ni, 1% Мп,
0,15% С и до 1% Si, является жаро-
стойкой (до 900—950°), а при наличии в
стали 30% Сг и 4—5% А1 она выдер-
живает температуру 1100—1150°. Добав-
ки алюминия, бериллия и магния к меди
повышают ее сопротивление газовой
коррозии в окислительных средах. Для
защиты железных и стальных изделий
от газовой коррозии поверхность изде-
лия покрывают алюминием (алити-
рование).
Под электрохимической
коррозией подразумевают процессы
взаимодействия металлов с электроли-
тами (в виде водных растворов, реже
с неводными электролитами, напр. с
нек-рыми органич. электропроводными
соединениями или безводными распла-
вами солей при повышенных темп-рах).
Процессы электрохимии, коррозии про-
текают по законам электрохимии, кине-
тики, когда общая реакция взаимодей-
ствия может быть разделена на след.,
в значительной степени самостоятель-
ные, электродные процессы: а) Анодный
процесс — переход металла в раствор
в виде ионов (в водных р-рах, обычно
гидратированных) с оставлением экви-
валентного количества электронов в ме-
талле. б) Катодный процесс — ассими-
ляция появившихся в металле избыточ-
ных электронов к.-л. деполяризаторами.
На рис. 2 приведена схема химич. (А)
и злектрохимич. (Б) коррозионного про-
цесса.
В зависимости от того, происходит ли
катодная деполяризация в основном за
счет разрядов ионов водорода, иониза-
цией растворенного в р-ре кислорода
или восстановлением к.-л. окислителей,
различают коррозию с водород-
ной, кислородной или окис-
лительной деполяризацией.
Изучение механизма и разработка тео-
рии процессов злектрохимич. коррозии
металлов в значительной мере основы-
ваются на общих закона^ злектрохимич.
кинетики и, в частности, на изучении электродных по-
тенциалов, кинетики электродных реакций и общих
законов работы коррозионных гальванич. элементов.
зии (напр., окалины) и встречной диффузии атомов или
ионов кислорода. По современным воззрениям этот
процесс имеет ионно-электронный механизм, аналогич-
ный процессам электропроводности в ионных кристал-
лах. Примером химич. коррозии является взаимодей-
ствие металла с жидкими неэлектролитами или сухими
газами в условиях, когда влага на поверхности ме-
талла не конденсируется, а также воздействие на
металл жидких металлич. расплавов. Практически
наиболее важным видом химич. коррозии является
взаимодействие металла при высоких темп-рах с ки-
слородом и др. газообразными активными средами
(H2S, SO2, галогены, водяные пары, СО2 и др.). По-
добные процессы химич. коррозии металлов при повы-
шенных темп-рах носят также название газовой
коррозии. Многие ответственные детали инже-
нерных конструкций сильно разрушаются от газовой
коррозии (лопатки газовых турбин, сопла ракетных
двигателей, элементы электронагревателей, колос-
ники, арматура печей и т. д.). Большие потери от
газовой коррозии (угар металла) несет металлурги-
ческая промышленность. Стойкость против газовой
коррозии повышается при введении в состав спла-
Рис. 2. Схема химического (А) и электрохими-
ческого (Б) окисления металлов.
Для процессов злектрохимич. коррозии величина
коррозионного тока (прямо пропорционального ско-
рости коррозии) определяется выражением:
Vk - Va
Pk + pa + R
725
КОРРОЗИЯ МЕТАЛЛОВ
726
где Vfa — — разность начальных потенциалов (ка-
тодного деполяризующего процесса VnK и анодной
реакции растворения металла в условиях корро-
зии. Знаменатель данного выражения, представляю-
щий собой общее кинетич. торможение системы, выра-
жен след, величинами, имеющими омическую раз-
мерность: средней катодной (Pg) и анодной (РА) по-
ляризуемостью, характеризующими кинетику катод-
ного и анодного процессов, и общим омическим сопро-
тивлением системы (Я).
Таблица 1. Стандартные электродные потенциалы металлов
(F®) ПРИ 25О (в вольтах) для электродных реакций
ЛГе Jfen+ 4- пе.
Металл и число электронов (пе), участвующее в элек- тродной реакции V°,e Металл и число электронов (пе), участвующее в элек- тродной реакции V°c’ *
Первая гру и п а а Вторая группа^
Li —е —3,045 Cd — 2е -0,402
Rb — е —2,925 In — Зе -0,342
К — е —2,925 Т1 — е -0,336
Cs — е -2,923 Мп — Зе -0,283
Ra — 2е —2,92 Со — 2е -0,277
Ва — 2е —2,90 Ni - 2е -0,250
Sr - 2е —2,89 Мо — Зе -0,2
Са — 2е —2.87 Ge - 4е -0.15
Na — е -2,714 Sn — 2е -0,136
La — Зе -2,52 РЬ — 2е -0,126
Mg — 2е Ат — Зе -2,37 -2,32 Fe — Зе -0,036
Рп — Зе Th — 4е -2,07 —1,90 Н — е -0,000
Np - Зе —1,86 т.р етья группа"
Ве— 2е —1,85 Bi — Зе -0,226
U - Зе -1,80 Sb — Зе -0,24
Ш-- 4е —1,70 As — Зе -0.30
AI - Зе —1,66 Си — 2е -0,337
Ti — 2е —1,63 Си — е -0,521
Zr — 4е —1,53 Hg — е -0,789
U — 4е — 1,50 Ag — е +0,799
Np - 4е Ри — 4е —1,354 —1,28 Rh — Зе +0,80
Ti — Зе V- 2е -1,21 -1,18 Четвертая группа1.
Мп — 2е —1,18 Hg - 2е -0,854
Nb - Зе -1.1 Pd — 2е -0,987
Сг — 2е -0,913 1г - Зе -1,000
V- Зе —0.876 Pt — 2е -1,19
Zn — 2е —0,762 Аи — Зе И,50
Сг — Зе Ga — Зе Fe - 2е —0,74 —0.53 —0,440 Аи — е -1,68
а Металлы повышенной термодинамич. нестабильности;
могут корродировать даже в нейтральных водных средах,
не содержащих кислорода и окислителей.
б Металлы термодинамически нестабильные; устойчивы
в нейтральных средах при отсутствии кислорода; в кислых
средах могут корродировать и в отсутствии кислорода.
в Металлы промежуточной термодинамич. стабильности
(полублагородные); в отсутствии О8 и окислителей устойчивы
в кислых и нейтральных средах.
г Металлы высокой термодинамич. стабильности (благо-
родные); в нейтральных средах не корродируют и при наличии
кислорода; в кислых средах или средах, содержащих комплексо-
образователи, могут корродировать при наличии кислорода
или окислителей.
Процесс электрохимической коррозии представляет
собой замкнутый цикл из отдельных более простых
последовательно соединенных (сопряженных) про-
цессов. Поэтому установление реальной скорости кор-
розионного процесса для данного металла и среды
(Vjj — = const) зависит только от суммарного
торможения процесса на каждой из более простых
сопряженных звеньев. Доля торможения процесса
коррозии каждой элементарной ступенью, равная
при электрохимия, коррозии падению потенциала на
данной, ступени, называется степенью конт-
роля коррозионного процесса дан-
ной ступенью. Таким образом можно отличать корро-
зионные процессы, имеющие в основном катодный,
анодный или омический контроль. К первому виду
коррозии относится большинство практич. процессов
коррозии металлов в активном состоянии в морской
и речной воде, в почве, а также коррозия во многих
средах химической промышленности и др. Коррозия
металла из пассивного состояния (см. Пассивность
металлов') имеет, наоборот, преимущественный анодный
контроль.
Типы коррозионных разрушений. При равномерном
распределении коррозионных разрушений по всей
поверхности металла коррозию называют равно-
мерной; если же
значительная часть по-
верхности металла сво-
бодна от коррозии и
последняя сосредоточе-
на на отдельных участ-
ках, то ее называют
местной. Наиболее
часто встречающиеся в
практике типы мест-
ной коррозии описаны
ниже.
Рис. 3. Язвенная коррозия сталь-
ной трубы с промыслов морской
нефти: а — продольный разрез
трубы (продукты коррозии не
удалены); б —поперечный разрез
трубы (продукты коррозии уда-
лены).
Язвенная (рис. 3),
точечная, или пит-
тинговая, корро-
зия (рис. 4). Основной
причиной подобного кор-
розионного разрушения яв-
ляется работа коррозион-
ных пар, у к-рых катодами
являются участки поверх-
ности, находящиеся в пас-
сивном состоянии, а анодами — участки поверхности в ак-
тивном состоянии. Этот вид коррозии особенно опасен для кон-
струкций, где важно сохранение герметичности или непрони-
цаемости, напр. различного рода
емкостей, аппаратов, трубопрово-
дов и т. д. Развитию точечной и яз-
венной коррозии благоприятствует,
1
Рис. 4. Примеры питтинговой (точечной) коррозии; а —
коррозия внутренней поверхности трубы из алюминие-
вого сплава в морской воде; б — коррозия нержавеющей
стали при анодной поляризации в растворе 0,5н. NaCI-J-
+0,001и. НС1.
напр., одновременное присутствие в р-ре пассиваторов (кис-
лород, окислители) и сильных активаторов (ионы хлора, брома
или иода).
Щелевая коррозия. Причиной развития такой
коррозии является преим. анодная работа частично экрани-
рованной поверхности металла по отношению к открытой
поверхности металла, играющей роль катода; коррозионному
разрушению в основном подвергается участок конструкции,
находящейся в зазоре (щели), или участок каким-то образом
частично экранирован-
ный от внешней среды.
Контактная кор-
розия — разновидность
электрохимической мест-
ной коррозии, когда пре-
имущ. разрушается уча-
сток конструкции, изго-
товленной из материала,
имеющего более отрица-
тельный стационарный
электрохимии, потенпиал.
Межкристал-
литная коррозия
(рис. 5). В этом случае
коррозионное разруше-
ние локализуется по гра-
ницам зерен структуры
сплава.
Рис. 5. Пример межкристаллитной
коррозии металлов; а — латунь;
б — нержавеющая сталь.
727
КОРРОЗИЯ МЕТАЛЛОВ
728
Коррозионное р а с’т рос кивание (рис. 6)
возникает при одновременном воздействии на металл агрессив-
ной среды и механич. напряжений; в металле появляются тре-
щины транскристаллитного характера, к-рые часто приводят
к полному разрушению изделий. Последние 2 вида коррозион-
ного разрушения наибо-
лее опасны длн конструк-
ций, несущих механич.
нагрузки (мосты, тросы,
рессоры, оси, автоклавы,
паровые котлы и т. д.).
Рис. 6. Пример коррози-
онного растрескивания
(трапскристаллитная кор-
розия) при наличии рас-
тягивающих напряжений:
а — латунь; б — нержа-
веющая сталь.
Количественная характеристика коррозионных про-
цессов. При относительно равномерном типе корро-
зионного разрушения скорость коррозии может быть
выражена весовым показателем коррозии (7Г), равным
весу металла (в граммах;, превращенного в продукты
коррозии за единицу времени (час или сутки) с еди-
ницы его поверхности (м2 или дм2). Иногда скорость
коррозии выражают также глубинным показателем
коррозии (77), т. е. средним проникновением коррози-
онного разрушения в металл (в миллиметрах) за еди-
ницу времени (1 год). Пересчет скорости коррозии
от весового показателя (К) в глубинный (77) или об-
О 7Q
ратно производится по формуле: П = • К, где у —
плотность металла, К — весовой показатель коррозии,
г/м2 • час, П — глубинный показатель коррозии,
мм/год.
При резко выраженной местной коррозии скорость
коррозионного разрушения, как правило, уже не мо-
жет быть достаточно точно характеризована этими
показателями. При точечной коррозии необходимо
определять максимальный глубинный показатель.
Скорости межкристаллитной коррозии, а также кор-
розионного растрескивания могут быть количественно
характеризованы механич. показателем коррозии,
напр. по потере прочности металла (в %) за определен-
ное время. Прочностной показатель коррозии {Ка)
определяется по формуле:
где Од — предел прочности металла до коррозии;
Од — кажущийся предел прочности после коррозии
(т. е. рассчитанный па начальное сечение образца).
Применяют также и другие приемы для количествен-
ной характеристики общей или местной коррозии
(напр., измерение возрастания электросопротивления
образцов и нек-рые др.).
Электрохимическая коррозия в различных средах.
Различают след, тпиы электрохимич. коррозии, имею-
щие наиболее важное практич. значение.
Коррозия в электролитах. К этому
типу относятся коррозия в природных водах (морской
и пресной), а также различные виды коррозии в жид-
ких средах. В зависимости от характера среды раз-
личают кислотную, щелочную, соле-
ную и морскую коррозию. По условиям
воздействия жидкой среды па металл этот тип корро-
зии также характеризуется как коррозия при
полном погружении, при неполном
погружении (или коррозия по ватерлинии),
при переменном и о г р уж он п и, имею-
щие свои характерные особенности.
II о ч в е и п а я (г р у п т о в а я, п о д з е м и а я)
коррозия — воздействие па металл грунта, к-рый
в коррозионном отношении должен рассматриваться
как своеобразный дисперсно-пористый, иногда кол-
лоидный электролит. Характерной особенностью под-
земной электрохимич. коррозии является большое
различие в скорости доставки кислорода (основной
деполяризатор) к поверхности подземных конструк-
ций в разных почвах (в десятки тысяч раз). Значитель-
ную роль при коррозии в почве играет образование и
функционирование макрокоррозиоиных пар вследст-
вие неравномерной аэрации отдельных участков кон-
струкции, а также наличие в земле блуждающих то-
ков. В ряде случаев на скорость электрохимич. корро-
зии в подземных условиях оказывает существенное
влияние также развитие биологич. процессов в почве.
Атмосферная коррозия — коррозия ме-
таллов в условиях атмосферы, а также любого влаж-
ного газа; наблюдается под конденсационными види-
мыми слоями влаги на поверхности металла (м о к-
рая атмосферная коррозия) пли под
тончайшими невидимыми адсорбционными слоями
влаги (влажная атмосферная корро-
зия). Особенностью атмосферной коррозии является
сильная зависимость ее скорости и механизма от тол-
щины слоя влаги на поверхности металла или сте-
пени увлажнения образовавшихся продуктов коррозии.
Коррозия в условиях механич.
воздействия. Этому типу разрушения подвер-
гаются многочисленные инженерные сооружения, ра-
ботающие как в жидких электролитах, так и в атмо-
сферных п подземных условиях. Наиболее типичными
видами подобного разрушения являются: а) К о р р о-
з и о п н о е растрескивание; при этом ха-
рактерно образование трещин, к-рые могут распростра-
няться не только межкристаллитно, по также и транс-
кристаллитно (см. рис. 6). Примером подобного разру-
шения является щелочная хрупкость котлов, сезон-
ное растрескивание латуней, а также растрескивание
нек-рых конструкционных высокопрочных сплавов.
В табл. 2 приведены нек-рые условия, в к-рых наблю-
дается коррозионное растрескивание различных ме-
Таблица 2. Коррозионные среды, в к-рых установлено
наличие коррозионного растрескивания металлич. сплавов
Сплавы Коррозионные среды
Малоуглеродистые стали Р-ры NaOH, Na2SO3, азотнокислых солей кальция, аммония или натрия
Углеродистые п низко- Кипящие концентрированные р-ры
легированные стали MgCl2, водные р-ры HCN
Нержавеющие высоко- Р-р NaCl + Н2О2, морская вода,
хромистые стали р-р H2S
Нержавеющие Cr-Ni- Хлориды (MgC]2, ZnC]2, LiCl), дис- тиллированная вода при высоких темп-рах и давлениях
аустенитные стали
Никель Р-ры NaOH
Си-сплавы Пары аммиака, р-ры ртутных со- лей, атмосфера, загрязненная SO2 пли NH3
М он ел ь-м ета л л Р-р HF И H2SiFe
Свинец Р-р РЬ (СН3СОО)2
Al-сплавы Расплавы NaCl, р-р NaCl, морская вода, водяной пар, атмосфера, загрязненная SO2
Mg-сплавы Р-ры Nad 4- К2Сг2О7» морская ат- мосфера, дистиллированная вода
Титан и его сплавы Концентрированная IINO3, р-р иода в метаноле
таллпч. сплавов, б) Коррозионная уста-
лость, вызываемая воздействием коррозионной
среды и знакопеременных или пульсирующих меха-
нических напряжений. Этот вид разрушения также
характерен образованием меж- и транскристаллитных
трещпп. Разрушения металлов от коррозионной уста-
лости встречаются при эксплуатации различных ин-
женерных конструкций (валов гребных винтов, рес-
729
КОРРОЗИЯ МЕТАЛЛОВ — КОРТИЗОН
730
сор автомобилей, канатов, штанг глубинных насосов,
охлаждаемых валков прокатных станов и др.). В табл. 3
приведены данные по коррозионно-усталостной про-
чности некоторых металлов и сплавов в различных
агрессивных средах, в) Коррозионная ка-
витация, являющаяся обычно следствием энер-
гичного механич. воздействия коррозионной среды
на поверхность металла. Подобное коррозионно-
механич. воздействие может приводить к весьма силь-
ным местным разрушениям металлич. конструкций
и образованию глубоких каверн, как это, напр.,
наблюдается для гребных винтов морских судов.
Механизм разрушения от коррозионной кавитации
близок к разрушению от поверхностной коррозион-
ной усталости, г) Коррозионная эрозия,
вызываемая механич. истирающим воздействием дру-
гого твердого тела при наличии коррозионной среды
или непосредственным истирающим действием самой
коррозионной среды. Это явление иногда называют
также коррозионным истиранием, или ф р е т т и и г-
коррозией.
Таблица 3. Коррозионно-усталостная прочность
(выносливость) различных металлов и сплавов в различных
агрессивных средах -
Металл или сплав, его состав и обработка Предел проч- ности исход- ного ме- талла, кг/мм2 Предел выносливости, кг /мм2
на воз- дух е в пре- сной воде в соле- ной воде (0,5% хлоридов)
Низкоуглеродистая сталь (0,14%С), отожженная. . И 14 6
Хромо-никелевая сталь (1,5% Ni, 0,73% Сг, 0,28% С), закаленная и отпущенная 97 47 12 10
Хромистая нержавеющая сталь (13% Сг, 0,11% С), закаленная и отпущенная 62 38 26 21
Хромо-никелевая нержа- веющая сталь (17% Сг, 8% Ni, 0,16% С), горяче- катаная 88 35 35 16
Хромо-никелевая нержа- веющая сталь (18% Сг, 25% Ni, 0,39% С), горя- чекатаная 81 38 31
Чугун (3,3% С, 1,5% Si, 0,9% Мп), литье 30 13 9 8
Никель (98,9%), отожжен- ный 54 23 17 15
Монель-металл (71% Ni, 26% Си), отожженный . . 57 2i 16 19
Чистая медь (99,9%), отож- женная 22 7 7 7
Купро-никель (48% Си, 48% Ni), отожженный . . 55 22 15 17
Латунь (62,3 % Си, 37% Zn), отожженная 37 15 11 11
Бронза (94,5 % Си, 5,4 % Sn), отожженная 56 16 15 15
Сплав АМЦ (98% А1, 1,2% Мп), холоднокатаный . . 17 7 4 3
Дюралюмин (4% Си, 0,5% Mg, 0,3% Si, 0,5% Мп, 0,3% Ее), отожженный . . 24 10 5 4
* База испытаний: для алюминия и его сплавов 1 • 10;
циклов, для остальных металлов и сплавов 5 • 107 циклов.
Методы защиты. Широко применяются след, основ-
ные методы защиты металлич. конструкций от корро-
зии: 1) Защитные покрытия. 2) Обработка коррозион-
ной среды с целью снижения коррозионной актив-
ности (особенно при постоянных объемах коррозион-
ных сред). Примерами такой обработки могут служить:
нейтрализация или обескислороживание коррозион-
ных сред, а также применение различного рода инги-
биторов коррозии. 3) Защита металлов электрохими-
ческая. 4) Разработка и произ-во новых металлич.
конструкционных материалов повышенной коррозион-
ной устойчивости путем устранения из металла или
сплава примесей, ускоряющих коррозионный процесс
(устранение железа из магниевых или алюминиевых
сплавов, серы из железных сплавов и т. д.), или вве-
дения в сплав новых компонентов, сильно повышаю-
щих коррозионную устойчивость (напр., хрома —
более 12% — в железо, марганца в магниевые сплавы,
никеля в железные сплавы, меди в никелевые сплавы
и т. д.). Для жаростойких сплавов на основе железа
основными легирующими компонентами являются
хром, кремний, алюминий, сильно тормозящие диффу-
зионные процессы в защитных окисных слоях. 5) Пе-
реход в ряде конструкций от металлич. к химически
стойким материалам (пластич. высокополимерные
материалы, стекло, керамика и др.). 6) Рациональное
конструирование и эксплуатация металлич. соору-
жений и деталей (исключение неблагоприятных ме-
таллич. контактов или их изоляция, устранение ще-
лей и зазоров в конструкции, устранение зон застоя
влаги, ударного действия струй и резких изменений
скоростей потока в конструкции и др.).
Классификацию защитных мероприятий можно
также осуществить, исходя из механизма их защитного
действия (теории электрохимия, коррозии). При такой
классификации все защитные мероприятия по борьбе
с коррозией можно разделить след, образом: а) умень-
шающие степень термодинамич. нестабильности си-
стемы (легирование металла более благородным ком-
понентом, изоляция его от коррозионной среды и др.Д
б) повышающие катодный контроль коррозионной
системы (уменьшение катодных компонентов в сплаве,
введение катодных ингибиторов в р-р, снижение кон-
центрации катодных деполяризаторов в р-ре, приме-
нение катодной электрохимия, защиты и др.); в) по-
вышающие анодный контроль (легирование сплава
пассивирующими компонентами, введение в сплав
эффективных катодов, добавление анодных ингибито-
ров в раствор, анодная электрохимия, защита и Др );
г) повышающие омич, сопротивление системы (повы-
шение омич, сопротивления коррозионной среды, сло-
ев продуктов коррозии или защитных покрытий).
Лит.: Акимов Г. В., Теория и методы исследования
коррозии металлов, М.—Л., 1945; его же, Основы учения
о коррозии и защите металлов, М., 1946; Фрумкин А. Н.
[ и др.], Кинетика электродных процессов, [M.J, 1952; Тома-
шов Н. Д., Теория коррозии и защиты металлов, М., 1959;
его же, Коррозия металлов в химической промышленности,
в сб.: Методы и процессы химической технологии, сб. 1, М.—•
Л., 1955; Розенфельд И. Л., Атмосферная коррозия
металлов, М., 1960; Клинов И. Я., Коррозия химической
аппаратуры и коррозионностойкие материалы, 3 изд., М.,
1960; Коррозия реакторных материалов. Сб. статей, под ред.
В. В. Герасимова, М., 1960; Батраков В. П., Коррозия
конструкционных материалов в агрессивных средах, М., 1952;
Дятлова В. Н., Золотницкий И. М., Коррозионная
и химическая стойкость материалов. Справочник, М., 1954;
Данков П. Д., И г н а т о в Д. В., Ш и ш а к о в Н. А.,
Электронографнческие исследования окисных н гидроокисных
пленок на металлах, М., 1953; Рыбасенко И. Д.,
Якубовский Л. А., Каган И. 3., Технология
изготовления химической аппаратуры из нержавеющей стали,
Киев — М., 1951; Кубашевский О., Гопкинс Б..
Окисление металлов и сплавов, М., 1955; Шварц Г. Л.,
Кристаль М. М., Коррозия химической аппаратуры, М.,
1958; Коррозия металлов, пер. с англ., под ред. В. В. Скор-
челлетти, кн. 1—2, Л. — М., 1952; Evans U. R., The
corrosion and oxidation of metals, L., [I960]; Todt F.
(Hrsg.), Korrosion und Korrosionschutz, B., 1955; Lee J. A.,
Materials of construction for chemical process industries, N. Y.,
1950; R a b a 1 d E., Corrosion guide, N. Y., 1951; U h 1 i g H.
(ed.), Corrosion handbook, N. Y. 1948. H. Д. Ромашов.
КОРТИЗОН (Д4-прегнендиол-17а, 21-трион-З, 11, 20;
вещество Е Кендалла) C2iH28O5, мол. в. 360,46 —
один из активных кортикостероидов. К. (I) принято
рассматривать как гормон коры надпочечников, хотя
истинным гормоном является гидрокортизон
(II — кортизол, вещество Е Кендалла).
К. — бесцветные кристаллы, т. пл. 215°, [а]о =
= +209° (С2Н6ОН), практически нерастворим в воде
и с трудом растворяется в большинстве органич. рас-
творителей. В медицине К. применяют исключительно
731
КОРТИЗОН — КОРТИКОСТЕРОИДЫ
732
в форме кортизон-ацетата (R = СОСН3, мол. в.
402,5) — бесцветные кристаллы, без запаха, горькие
на вкус; его темп-ра плавления сильно зависит от
скорости нагревания. Согласно Государственной Фар-
макопее СССР (IX изд.), т. пл. К.-ацетата должна
быть 235—245° (в пределе 3°,с разл.), [a]D = + 178—
----(-194° (0,5%-ный раствор в. ацетоне) и уд. показа-
тель поглощения в УФ-свете при длине волны
238 ммк 390 ± 10(0,001 %-ный раствор в 95%-ном
спирте). К.-ацетат нерастворим в воде и петролейном
эфире, слегка растворим в серном эфире, относи-
тельно хорошо в хлороформе и диоксане, трудно раст-
ворим в холодном спирте (1 г в 350 мл), значительно
легче — в кипящем спирте, что используется обычно
при перекристаллизации.
Химич, свойства К. определяются наличием двух
реакционноспособных кетогрупп (кетогруппа в поло-
жении 11 химически инертна), диоксиацетонной це-
почки в положении 17 (с ее присутствием связана
неустойчивость вещества к щелочам и окислителям)
и Д4-двойпой связи, находящейся в сопряжении с
3-кетогруппой. К.-ацетат значительно устойчивее
к действию окислителей, чем свободный К. Иденти-
фикацию К.-ацетата производят хлористым трифенил-
тетразолием (красное окрашивание) и сернокислым
фенилгидразином (желтое окрашивание). Количест-
венно К. и К.-ацетат определяют обычно методом
УФ-спектрофотометрии в сочетании, если необходимо,
с хроматографией на бумаге.
Биосинтез К. в организме осуществляется из уксусной к-ты
через мевалоновую к-ту, сквален, холестерин н прогестерон.
Последний подвергается ферментативному гидроксилированию
последовательно в 17, 21 и lip-положения с образованием
гидрокортизона (II), при окислении к-рого получается К.
К. был выделен в 1936 из экстрактов надпочечников,
однако его ничтожное содержание в надпочечниках
и трудности выделения делают этот источник практи-
чески бесполезным. Промышленное получение К. ба-
зируется на природном стероидном сырье раститель-
ного или животного происхождения (диосгенйн,
желчные к-ты, стигмастерин, холестерин и др.).
В СССР наиболее перспективным сырьем для полу-
чения К. и его аналогов является, по-видимому, с о-
л а с о д и н, выделяемый из растения «паслен птичий»
(Solanum aviculare Forst). Синтез К.-ацетата из сола-
содина ведется через прогестерон (18 стадий) или
через вещество S Рейхштейна, Д4-прегнендиол-17а,
21-дион-З,20 (12 стадий) с применением микробиоло-
гии. гидроксилирования; выход К.-ацетата из исход-
ного продукта 6—10%.
К. оказывает сильное и многостороннее действие
на организм. Он усиливает образование углеводов
из белков, выделение азотсодержащих продуктов с мо-
чой и отложение гликогена; вызывает задержку в ор-
ганизме ионов натрия и хлора и усиленное выделение
ионов калия; угнетает гиалуронидазу, сильно влияет
на состояние соединительной ткани, угнетает функции
лимфоидных органов. Для практич. целей особое зна-
чение имеют противовоспалительные и противоаллер-
гич. свойства К. (Ф. Хенч, 1949). К.-ацетат применяют
для лечения острого и хронич. ревматизма, полиар-
тритов разнообразной этиологии, диссеминированной
красной волчанки, разнообразных воспалительных за-
болеваниях кожи и глаз, аллергия, болезней, злока-
чественных новообразований лимфатич. и кроветвор-
ной систем и т. д. К.-ацетат вытесняется более эф-
фективными и дающими меньшее число побочных
явлений стероидными аналогами (см. Кортикосте-
роиды) — преднизоном (Д1-дегидрокортизон), предни-
золоном (Д1-дегидрокортизол) и фторпроизводными
(дексаметазон, триамцинолон).
Лит.: Назаров И. Н., Бергельсон Л. Д.,
Хнмня стероидных гормонов, М., 1955; Fieser L. F.,
F i е s е г М., Steroids, N. Y. — L., 1959; Heftin ann Е.,
Mosettlg Е., Biochemistry of steroids, N. Y., 1960; Госу-
дарственная фармакопея СССР, 9 нзд., М., 1961, с. 138; Су-
воров Н. Н., Хнм. паука н пром-сть, 1956, 1, № 4, 406;
его яг е, Мед. пром-сть СССР, 1956, At 3, 22; Глин Дж. X.,
Кортнзонотерапия, пер. с англ., М., 19 60. Н. Н. Суворов.
КОРТИКОСТЕРОИДЫ — производные циклопен-
танопергидрофенантрена, выделяемые из коры над-
почечников и содержащие в положении 17 стероид-
ного ядра одну из группировок;
а-кетольную
^сн—СО—СН2ОН,
диокенацетонную
^С(ОН)—СО—СН2ОН,
а-гликольную
\СН—СН(ОН)—СН2ОН,
нли глицериновую
^>С(ОН)—СН(ОН)—СН2ОН.
Из экстрактов надпочечников, лишенных адреналина
(т. наз. «к о р т и н а»), выделено ок. 46 кристаллич.
стероидов, из к-рых способными в той или иной сте-
пени продлевать жизнь адреналэктомированных жи-
вотных оказались след. К.; дезоксикортик о-
с т е р о н (I) — кортексон, Д4-прегненол-21-дион-3,20,
т. пл. 142°, [a]D = +178° (С2Н5ОН); кортико-
стерон (II) — Д4-прегнендиол-11р, 21-дион-3,20,
т. пл. 182°, [a]D = +222° (С2Н5ОН); 11-д е г и д р о-
кортикостерон (III) — Д4-прегненол-21-три-
он-3,11,20, т. пл. 180°, [a]D = +258° (С2Н5ОН);
вещество S Рейхштейна (IV) — кортек-
солон, Д4-прегнендиол-17а, 21-дион-З, 20, т. пл. 213°,
[a]D = +116° (СН3СОСН3); гидрокортизон
(V)—кортизол, Д4-прегнентриол-11|3,17а,21-дион-З,20,
т. пл. 220°, [a]D = +167° (С2Н5ОН); кортизон
(VI)—вещество/? Кендалла, Д4-прегнендиол-17а,21-три-
он-3, 11,20, т. пл. 215°, [a]D = + 209° (С2Н5ОН);
альдостерон (VII) — электрокортин, гемиаце-
таль Д4-прегнендиол-11Р, 21-аль-18-диона-3,20, т. пл.
199°, [a]D= + 122° (СНС13); 19-о кейкортикосте-
р о н (VIII) — Д4-прегнентриол-11р, 19, 21-дион-З, 20,
т. пл. 187° — 192°.
Выделение индивидуальных К. из «кортина» может
быть осуществлено применением фракционной кри-
сталлизации, хроматографии и противоточного рас-
пределения.
Химич, свойства К. определяются наличием актив-
ных групп в кольце А (а, fj-кетонепредельная группи-
ровка или вторичный гидроксил) и a-кетольной, дио-
ксиацетонной, a-гликольной или глицериновой цепо-
чек в кольце D. Кетогруппа и вторичный гидроксил
в положении 11 из-за стерич. препятствий химически
довольно инертны: по 11-кетогруппе не образуются
гидразоны и семикарбазоны, lip-гидроксильная груп-
па в обычных условиях не ацетилируется. Наличие
Д4-3-кетонепредельной группировки сообщает соот-
ветствующим К. способность селективно поглощать
УФ-излученио в обл.асти 240 ммк; a-кетольная и дио-
ксиацетонная цепочки придают им восстанавливаю-
щие свойства; 21-ацетаты К. значительно более устой-
чивы к окислителям.
733
КОРТИКОСТЕРОИДЫ — КОРУНД
734
Для идентификации К.„широко применяют хрома-
тографию на бумаге (алифатич. или ароматич. угле-
водород — низшие спирты — вода или углеводо-
роды — формамид). Пятна на хроматограммах выяв-
ляют путем просмотра бумаги в УФ-свете (напр.,
посредством ультрахемископа) с помощью аммиачного
р-ра окиси серебра или тетразолового синего. Нали-
чие диоксиацетонной цепочки устанавливают при
помощи фенилгидразина. Количественное определе-
ние К. производят измерением оптич. плотности их
растворов при 240 ммк или количественных мо-
дификаций вышеуказанных качественных цветных
реакций.
их синтезы из других стероидных соединений. Напр.,
вещество S Рейхштейна (IV) может быть получено
из ацетата дегидропрегненола — продукта перера-
ботки соласодина или диосгенина, выделяемых из
растительного сырья. Микробиология, гидроксили-
рованием с помощью нек-рых грибков вещество S
Рейхштейна с удовлетворительным выходом может
быть превращено в гидрокортизон (V). Для получе-
ния кортизона (VI) проводят микробиология, гидро-
ксилирование в положение Нас образованием эпигид-
рокортизона (Д4-прегнентриол-11а, 17а, 21-дион-3,20),
21-ацетат к-рого окисляют хромовой к-той. Дезок-
сикортикостерон (I) и 11-дегидрокортикостерон (III)
могут быть сравнительно легко получены соответст-
венно из прегненолона (X) и 11-кетопрогестерона кон-
денсацией с диэтплоксалатом, иодированием и кетон-
ным расщеплением до 21-иодопроизводного с после-
дующей заменой иода на ацетоксигруппу. Кортико-
стерон (II) и альдостерон (VII) до сих пор синтети-
чески труднодоступны.
Биосинтез. Ближайшим предшественником
К. является холестерин (IX), к-рый в надпочечниках
превращается последовательно в прегненолон (X) и
прогестерон (XI). Последний подвергается фермента-
тивному гидроксилированию в двух направлениях:
а) в положение 17 с образованием 17а-оксипрогесте-
рона (XII), превращаемого далее ферментативным
путем в вещество S Рейхштейна (IV), гидрокортизон
(V) и кортизон (VI); б) в положение 21 с образованием
дезоксикортикостерона (I) и далее кортикостерона
(II) и альдостерона (VII).
Фармакология и медицинское при-
менение К. Из перечисленных веществ истинными
гормонами считают; гидрокортизон (V), кортикосте-
рон (II) и альдостерон (VII); остальные, в том числе
и Кортизон (VI), являются продуктами их метабо-
лизма. К числу веществ, сильно влияющих на мине-
ральный обмен, относятся альдостерон и дезоксикор-
тикостерон. Последний вызывает в организме задержку
воды, ионов натрия и хлора и усиленное выделение
ионов калия, повышает артериальное давление и
усиливает воспалительные процессы; применяется
в виде ацетата (ДОКСА) или триметилацетата при
хронич. недостаточности надпочечников (болезнь Ад-
дисона) .
Альдостерон обладает сильно выраженным минера-
локортикоидным действием, значительно превосходя-
щим действие дезоксикортикостерона (по разным пока-
зателям в 5—100 раз). В отличие от последнего, ока-
зывает заметное влияние на углеводный обмен (в
3 раза слабее, чем кортизон), не вызывает повышения
кровяного давления у здоровых животных. Альдосте-
рон — эффективное средство для лечения Аддисоновой
болезни. Кортикостерон действует подобно альдосте-
рону, но значительно менее активен. Он не оказывает
противовоспалительного действия. Действие 11-дегид-
рокортикостерона (III), а также 19-оксикортикосте-
рона (VIII) на организм в качественном и количест-
венном отношениях напоминает действие кортикосте-
рона. Для вещества S Рейхштейна (IV) характерна
слабо выраженная минералокортикоидная актив-
ность. Практич. интереса для медицинского примене-
ния оно не представляет. Гидрокортизон и кортизон
обладают, сильным противовоспалительным дейст-
вием.
Аллопрегнандиол-Зр, 1(?а-он-20 отличается замет-
ным влиянием на солевой обмен — вызывает выведе-
ние из организма ионов натрия.
Практич. применение нашли синтетич. аналоги К.:
дегидро-, метил- и фтораналоги кортизона и гидро-
кортизона, обладающие повышенной противовоспа-
лительной активностью (преднизон, преднизолон, три-
амцинолон, дексаметазон). Стероидные 17-спиролак-
тоны — антагонисты альдостерона — применяются
как диуретики.
Лит.: Назаров И. Н., Бергельсон Л. Д.,
Химия стероидных гормонов, М., 1955; Fieser L.,
Fleser М., Steroids, N.Y. — L., [1959]; Юдаев H. А.,
Биохимия стероидных гормонов коры надпочечников, М., 1956;
его же, Химические методы определения стероидных гор-
монов в биологических жидкостях, М., 1961; Thorn G. W.
[а. о.], Pharmacologic aspects of adrenocortical hormones in
man, and their effects in adrenal insufficiency, в кн.: Medical
uses of cortisone, N. Y. — Toronto, [1954], p. 46—176; H a n 6
O., Hormone, Jena, 1959. H. H. Суворов.
КОРУНД (а-А12О3) — единственная встречающаяся
в природе наиболее устойчивая кристаллич. модифи-
кация глинозема (см. Алюминия окись); применяется
в виде природного и искусственного йрбдукта. При-
735
КОРУНД — космохимия
736
родный К. встречается в виде цветных драгоценных
камней (рубин, сапфир и др.), обыкновенного ко-
рунда и наждака. В пром-сти применяют преимущест-
венно искусственный К. (электрокорунд, синтетич. ру-
бин), получаемый из окиси алюминия или глинозем-
содержащих материалов. Основные свойства искус-
ственного К. приведены в таблице.
Свойство Электро- корунд Синтетический рубин
Плотность 3,93-4.01 3,99-4,01
Т. пл., °C Прочность, кг/лш2: 1900-1950 2010-2030
на сжатие 75.7 1 304,2
на изгиб 8,7 1 58,0 || 33,0
Модуль упругости, кг/мм* . — J 32 400 || 32 800
Микротвердость, кг/.и.и2 . . 1900-2700 2400-2600
Коэфф, трения — 0,14
Теплоемкость, кал'г • град . 0.280 0,1827-0,200
Термин, расширение, град-1 Электропроводность при — 1 54 • 10-7 || 62 • 10“?
565°, ом~1 с.и-1 Диэлектрическая проницае- — 2,74 • 10-12
мость Коэфф, преломления: — 1 13,38 || 11,03
«е — 1,750
”о 1,758
Существует 3 вида искусственного К.: монокристал-
лический, абразивный и спеченный. Монокри-
сталлический К. (рубин, сапфир, лейкосап-
фир) получают из порошка окиси алюминия, непре-
рывно подаваемого в струю пламени гремучего газа;
расплавленные частицы падают на кристаллич. за-
травку и обеспечивают рост монокристалла. Моно-
кристаллич? К. применяют для опор и подшипников
в точном приборостроении, для изготовления фильер
в производстве искусственного волокна, калибров,
игл для звукозаписи, контактов индикаторов и микро-
метров, наконечников твердомеров и др.
Абразивный К. получают нагреванием смеси
боксита с углем в дуговой электропечи. Полученный
расплав, остывая, образует блок, к-рый дробят, сор-
тируют, а затем измельчают в порошок. Изготовлен-
ный т. обр. абразивный К. в СССР носит название
электрокорунда (за границей он имеет мно-
жество названий: алунд, корракс, а л о к-
сид, абразит, диаментин, дура л, ко-
ру иди и, электрорубин и др.). Различают
след, электрокорунды: белый (98—99,5% А13О3), ро-
зовый (96—97% А12О3), нормальный (91—95% А12О3)
и черный, или искусственный наждак (75—85% А12О3).
Белый электрокорунд получают из глинозема, а осталь-
ные сорта — из бокситов. Электрокорунд приме-
няют для изготовления шлифовальных кругов, лент,
брусков, оселков, шлифовальной шкурки на бумаге
и полотне, пасты для полировки, абразивных порош-
ков ит. п. Спеченный К. обычно получают из
тонкозернистых порошков технич. глинозема или
электрокорунда. Порошок окиси алюминия смеши-
вают с пластификатором, формуют в изделия и, в за-
висимости от дисперсности порошка и примесей в нем,
спекают при 1700—1900°. Свойства спеченного К.
не только не уступают свойствам монокристалличе-
ского, но в ряде случаев значительно превосходят их,
напр. сопротивление износу выше у специальных ви-
дов спеченного К. (микролит, синтокс и др.). Спечен-
ный К. применяют в качестве огнеупорных изделий,
электрич. изоляторов, тиглей, конструкционного и
инструментального материала (втулки, сопла, резцы
по металлу, нитеводптели, подшипники и пр.). О при-
родном К. и его основных свойствах см. Алюминия
окись.
Лит.: Ryshkewitsh Ё., Oxide ceramics. Physical
chemistry and technology, N. Y. — L., 1960; Тресвят-
c к и й С. Г., Черепанов А. М., Высокоогнеупорные
материалы и изделия из окислов, М., 1957; Павлушкин
Н. М., Спеченный корунд, М., 1961; Полубояринов
Д. Н., Балкевич В. Л., Попильский Р. Я.,
Высокоглиноземистые керамические и огнеупорные материалы,
М., 1960. Н. М. Павлушкин.
КОСМОХИМИЯ (астрохимия) — раздел астрофи-
зики, включающий изучение химич. и изотопного
состава космич. тел, а также межпланетной и меж-
звездной среды; изучение распространенности химич.
элементов в космосе и их генезиса, процессов радио-
активного распада и ядерных реакций и др.
Космич. тела распадаются на две группы: излучаю-
щие собственный свет (Солнце, звезды, внегалактич.
туманности и др.) и излучающие отраженный, солнеч-
ный или звездный, свет (планеты и их спутники, асте-
роиды, диффузные туманности и др.). В соответствии
с этим к изучению химич. состава каждой группы тел
применяются принципиально разные методы, но в том
и другом случае в основе метода лежит спектральный
анализ. Для косвенного суждения о природе, а следо-
вательно, отчасти и о минеральном и химич. составе
планет, их поверхностей, спутников, астероидов, ко-
мет имеют значение измерения поляризации отра-
женного света. О важном значении К. можно судить
по тому факту, что неизвестный ранее химич. элемент
гелий первоначально был открыт на Солнце и уже
после этого был обнаружен на Земле.
Первые исследования химич. состава космич. тел
относятся к середине 19 в., когда Г. Кирхгоф совместно
с Р. Бунзеном ввели в 1860 спектральный анализ.
Однако систематич. исследования получили развитие
в последней четверти 19 в., т. е. со времени усовер-
шенствования фотографии и применения ее в астро-
физике. Развитию К. способствовали разработка уже
в 20 в. новых методов исследования и усовершенство-
вание астрономия, инструментов, особенно введение
мощных телескопов и светосильных астроспектрогра-
фов, и, наконец, значительное повышение чувстви-
тельности фотоматериалов, используемых для фото-
графирования спектров. Огромное значение имеет
зарождение в начале 40-х гг. 20 в. радиоастрономии,
посвященной изучению космич. тел путем исследова-
ния либо излучаемых ими радиоволн, либо отра-
женных радиосигналов, посылаемых с Земли.
К. тесно связана с геохимией, результаты к-рой
используются в К. для интерпретации получаемых
сведений о космич. телах. В свою очередь и геохимия
использует данные космохимии. К. находится в со-
стоянии выделения из астрофизики в самостоятельную
научную дисциплину. Этому способствует быстро и
успешно развивающееся освоение космич. простран-
ства и возникшая реальная возможность получения
в недалеком будущем непосредственных образцов
космич. тел. Пока единственными подобными образ-
цами являются метеориты.
Величайшим достижением К. является открытие
единства химич. элементов в космосе. Так, устано-
влено, что в космосе распространены (составляют кос-
мич. тела и рассеяны в пространстве) те же самые
химич. элементы, к-рые известны и на Земле. Ника-
ких новых химич. элементов не обнаружено. Важное
значение К. (наряду с астрофизикой) состоит в том,
что исследование химич. состава и физич. состояния
звезд (звездных атмосфер) представляет пока единст-
венную возможность теоретически изучать синтез и
ядерные реакции химич. элементов при таких давле-
ниях и темп-рах, к-рые недостижимы в настоящее
время в земных лабораторных условиях.
Ниже приводятся нек-рые черты химич. состава
основных космич. тел. В спектре Солнца обнаружено
ок. 60 химич. элементов. Из них наиболее обильным
является водород; его содержание в 4—5 раз превы-
шает содержание гелия и в 1000 раз — всех остальных
737
КОТАРНИН —КОФЕРМЕНТ А
738
химич. элементов, вместе взятых. Далее идут гелий,
кислород и азот. Затем выделяются магний, кремний,
железо, натрий, калий, кальций, алюминий и др.
Имеются также химич. соединения СН, CN, ON, NH
и др.
Химический состав звездных атмосфер практически
одинаков и подобен составу солнечной атмосферы.
Однако спектры звезд изменяются с изменением
температуры. Так, в спектрах самых «горячих» звезд
(классы «О» и «В», темп-ра 25000° и значительно выше)
наблюдаются линии ионизованных гелия и кислорода,
а также слабые линии водорода; появляются линии
ионизованного азота. В спектрах менее горячих звезд
(класс «А», темп-ра 11000°) наблюдаются исключи-
тельно интенсивные и широкие линии водорода, появ-
ляются слабые линии ионизованного кальция и др.
металлов. В спектрах звезд подобных или близких
Солнцу (классы «F» и «G», темп-ра 6000—7500°) видны
полноразвитые многочисленные линии металлов. На-
конец, в спектрах «холодных» звезд (классы «К» и
«М», темп-ра 3600—4500°) очень сильны линии каль-
ция, появляются молекулярные полосы, в том числе
окиси титана, а также CN, СН и ОН. В недрах звезд,
так же как и Солнца, происходят реакции преобра-
зования водорода в гелий с освобождением термоядер-
ной энергии.
Оболочки т. наз. планетарных туманностей состоят
гл. обр. из водорода. Присутствует также гелий,
кислород, азот, углерод и другие легкие элементы;
имеется и железо. Галактич. газовые (эмиссионные)
туманности в основном также состоят из водорода.
Вообще водород обильно распространен в космосе.
Он является главной составной частью межзвездной
среды (газа), где наблюдается также гелий и незначи-
тельные количества кальция, натрия, кислорода,
калия, титана, углерода и молекулярных соединений
СН и CN. Планеты делятся на две группы: внутрен-
ние (планеты земной группы) и внешние (планеты-
гиганты). Из первых, Венера и Марс, а также Луна
состоят из плотного каменистого вещества и металлов;
Меркурий — из еще более плотного вещества. Пла-
неты второй группы состоят в основном из легких ве-
ществ: водорода и его соединений с углеродом и азо-
том; меньшую часть составляют каменистые вещества.
В спектрах ядер комет наблюдаются полосы угле-
рода и циана и нек-рых других химич. соединений
(СН4, ОН, NH, СН и NH2). В спектрах голов комет
(оболочек вокруг ядер) обнаружены полосы углерода,
линии натрия, железа и никеля. Наконец, в спектрах
хвостов комет видны полосы молекул окиси углерода
и азота.
Лит.: Ферсман А. Е., Геохимия, т. 1, М., 1933;
Фесенков В. Г., Современные представления о вселен-
ной, М.—Л., 1949; Унзольд А., Физика звездных ат-
мосфер, пер. с нем., М., 1949; Гольдберг Л. иАллер
Л., Атомы, звезды и туманности, пер. с англ., М.—Л., 1948;
Пэйн-Гапошкина Ц., Рождение и развитие звезд,
пер. с англ., М., 1956; Северный А. Б., Физика Солнца,
М., 1956; Всехсвятский С. К., Физические характе-
ристики комет, М., 1958; журнал «Geochlmlca et cosmoclilmlca
ecta», Лондон (e 1950). E. Л. Кринов.
КОТАРНИН CI2H15O4N, мол. в. 237,26 — продукт
распада алкалоида наркотина, получается из послед-
него при действии разб. азотной к-ты. Кристаллы,
т. пл. 132—133° (с разл.), 125° (из бензола, с разл.),
100° (при высушивании на водяной бане). Легко раст-
ворим в спирте и эфире, почти нерастворим в воде и
щелочах.
К. — слабое вторичное основание, содержит аль-
дегидную группу (образует оксим), что соответствует
формуле IA. Однако в твердом состоянии К., вероятно,
отвечает формуле IB, а в растворах К. частично при-
сутствует в аммонийной форме IB. При образовании
солей с кислотами К. выделяет молекулу воды, так
что соли соответствуют формуле C12H13O3N • НХ.
Хлоргидрат К. (II), стиптицин, т. пл. 192°,
кислый фталат, стиптол, т. пл. 115°, применяются
N N
I
СН,
в медицине. Результаты окисления К. подтверждают
наличие изохинолиновой системы.
По своему строению К. является 8-метоксигидрасти-
нином и может быть превращен в гидрастинин (см.
Гидрастин). Стиптицин при-
меняется для тонизации мус- / j Т |+ С1
кулатуры внутренних орга- Н2О
нов, в особенности матки, qCH
а также местно в виде 1— 3 II
2%-ного раствора для остановки кровотечений. Ме-
нее активен, чем гидрастинин. X. Е. Васильев.
КОФЕИН CsHioO2N4, мол. в. 180,20 — алкалоид,
содержащийся в кофе (Coffea arabica L. семейства
мареновых — Rubiaceae), чае
(Thea sinensis семейства Thea- 7
сеае) и др. Зерна кофейного де- CH3-Nfe^_n-CH3
рева и листья чая содержат 1—
3% К. Бесцветные кристаллы,
т. пл. 235—237°; т. возг. 180°;
с водой образует моногидрат,
к-рый'при 100° теряет кристаллизационную воду.
К. растворим в воде (1 : 80 при 15° и 1 : 2 при 100°),
хлороформе (1 : 9), спирте (1 : 50), плохо растворим
в эфире (1 : 1300). Растворимость К. в воде сильно
повышается в присутствии нек-рых солей, напр.
бензоата натрия, с к-рым К. образует двойное соеди-
нение. К. — слабое основание, да^т неустойчивые
соли с кислотами, является 1,3, 7-триметильным про-
изводным алкалоида ксантина (2,6-диоксипурина).
Получают К. как из природного сырья (отходов
чайного произ-ва — чайной пыли, формовочного ма-
териала и т. д.), так и полным или частичным синтезом.
При выделении К. из отходов чайного произ-ва по-
следние экстрагируют водой, а из водной вытяжки К.
извлекают дихлорэтаном (коэфф, распределения К.
между дихлорэтаном и водой — 1,88). Синтезируют К.
из мочевой к-ты. Полный синтез К. произведен из мо-
чевины и циануксусного эфира через теофиллин (вы-
ход К. до 50%). К. широко применяется в медицине
как стимулятор центральной нервной системы, вызы-
вает повышение жизнедеятельности всех тканей орга-
низма, усиливает общий обмен, дыхание и кровооб-
ращение путем возбуждения корковых процессов;
обладает также диуретич. действием. Помимо самого
К., применяются (в тех же случаях) его двойное со-
единение с бензоатом натрия (лучше растворимо в воде
и быстрее выводится из организма) и 8-метилкофеин,
к-рый аналогичен по действию, но технически более
доступен, чем К.
Лит. см. при ет. Алкалоиды. А. Е. Васильев.
КОФЕРМЕНТ А (КоА, КоА—SH, кофермент аци-
лирования) C2iH36Oi6N,P3S, мол. в. 767,56 — производ-
ное Р-меркаптоэтиламида пантотеновой к-ты (панте-
теина) и нуклеотида — аденозин 3,5-дифосфата; осу-
ществляет перенос и активирование кислотных остат-
ков при ряде важнейших ферментативных реакций
в живой клетке. Чистый (95%) КоА — белый, аморф-
739
КОФЕРМЕНТ А —КОФЕРМЕНТЫ
740
иый порошок, хорошо растворим в воде, нерастворим
в спирте, ацетоне и эфире. УФ-спектр КоА характери-
зуется пиком поглощения при 260 ммк, Е = 16 400.
пантетеин
i---------------
аденол.а —3 х, 5Z-» дифосфат
КоА — довольно сильная к-та, в химич. отношении
совмещает свойства нуклеотидов и тиоловых соедине-
ний. КоА дает характерную окраску с нитропрусси-
дом, с тяжелыми металлами образует нерастворимые
в воде Мёркаптиды, легко окисляется с образованием
дисульфидов с помощью J2, Н2О2, КМпО4, а также
кислородом воздуха, особенно в присутствии следов
тяжелых металлов. С реагентами, алкилирующими
тиолы, йодацетатом и малеинимидами КоА образует
соответствующие тиоэфиры. Каталитич. активность
КоА обратймо подавляется п-хлормеркурбензоатом.
КоА активирует карбоновые к-ты, к-рые сами по себе
мало реакционноспособны, образуя с ними в процессе
ферментативных реакций промежуточные соединения,
являющиеся ацилпроизводными КоА; они построены
по типу Тиоэфиров RCO—SKoA. Процесс образования
ацилпройзйодных КоА сопряжен с затратой энергии.
Если аЦйлпроизвоДные КоА образуются из свободных
к-т, источником этой энергии служит сопряженное рас-
щепление богатых энергией связей аденозинтрифос-
форнрй к-ты (АТФ). llpii образований ацйлйроизвод-
ных КоА в процессе окислительного декарбоксилиро-
вания й-кётойислот источником этой ЭнерТйи служит
сам процесс окисления. ТйОэфирпая свйзь ацил-КоА
обладает большим запасом свободной энергий, ее рас-
щепление дает приблизительно такой же эффект, что
и расщепление макроэргич. связи АТФ (от 7,65 до
8,25 ккал/моль). Эта энергия используется в реакциях,
протекающих с участием ацилпроизводных КоА,
напр. при ацилировании окси- и аминосоединений.
Кроме того, образование тиоэфира способствует повы-
шению положительного заряда на углеродном атоме
карбонильной группы ацила и возрастанию подвиж-
ности атома водорода в a-положении к карбонилу.
Первое обстоятельство облегчает
ло н4 превращения, осуществляемые по
КдА 5 У’ ГС Г механизму нуклеофильного заме-
<1 щения у карбонильного атома
Н* углерода (реакций ацилирования),
второе — облегчает реакции с отщеплением водорода
в a-положении, напр. конденсацию ацетил-КоА со
щавелевоуксусной к-той.
Роль КоА и его ацилпроизводных в обмене веществ
исключительно велика: с их участием осуществляется
более 60 различных ферментативных реакций, важней-
шими из к-рых являются: 1) окислительное декарбо-
ксилирование пировиноградной к-ты; 2) {З-окислсние
жирных к-т; 3) синтез жирных к-т; 4) синтез ацетил-
холина из холина и ацетил-КоА; эта реакция очень
важна для нормального функционирования нервной
системы; 5) синтез лимонной к-ты конденсацией аце-
тил-КоА со щавелевоуксусной к-той:
соон
соон
I
сн,
I
со
I
соон
I
сн2
I
4-CH3CO-SKoA-|-H2O = HO-C-COOH-t-HSKoA
СН2
I
СООН
Эта реакция занимает одну из ключевых позиций
в общей системе окислительных процессов, поскольку
с ее помощью ацетат, образующийся в результате
частичного окисления жиров и углеводов, включается
в цикл трикарбоновых к-т, где происходит его оконча-
тельное окисление до СО2 и Н2О.
Ацетил-КоА, как показано на схеме, занимает
положение центрального промежуточного звена, через
к-рое проходят процессы окислительного распада
различных веществ и со стадии к-рого начинаются
многочисленные синтетич. реакции, осуществляемые
в клетке:
|со2+Нго|
КоА принимает участие также в процессах расщеп-
ления и синтеза остатков карбоновых к-т, углеродный
скелет к-рых лежит в основе ряда аминокислот.
С участием КоА идет ацилирование ароматич. аминов,
синтез гиппуровой к-ты, порфиринов, Терпенов, жи-
ров, фосфолипидов, стероидов й ряда других соеди-
нений.
КоА широко распространен в животных и расти-
тельных тканях, а также в микроорганизмах. Он мо-
жет быть получен синтетически или выделен из при-
родных источников, в частности из дрожжей — путем
экстракции теплой водой с последующей адсорбцией
на активированном угле и очисткой на ионообменных
смолах.
Лит.: Березовский В. М., Химия витаминов, М.,
1959, с. 91; Т р У ф а й о в А. В., Биохимия и физиология
витаминов и антивитаминов, М., 1959; Methods in enzymology,
ed. S. P. Colowick, N. O. Kaplan, v. 3, N. Y., 1957, p. 907;
The enzymes, ed. P. D. Boyer, H. Lardy, K. Myrbfick, v. 3,
2 ed., N. Y., 1960, p. 3; W 1 e 1 a n d O., Klin. Wochenschr.,
1954, 32, H. 17/18, 385—92. В. Б. Спиричев.
КОФЕРМЕНТЫ — низкомолекулярные органич.
соединения, необходимые наряду с апоферментом (бел-
ковой частью фермента) для осуществления биоката-
литич. процессов. В отличие от высокомолекулярной
белковой части фермента, денатурируемой при нагре-
вании и не способной к диализу в силу больших раз-
меров молекулы, К. характеризуются термостабиль-
ностью и обычно могут быть отделены от апофермента
посредством диализа. Сами по себе К. не обладают
каталитич. активностью, так же как не обладают ею
отделенные от К. апоферменты. Ферментативная ак-
тивность присуща лишь системе в целом, содержащей
741
КОФЕРМЕНТЫ
742
как апофермент, так и К. В ряде случаев удается
обратимо отделить К. от апофермента, не нарушая
структуру последнего. Если к полученному т. о. фер-
ментативно неактивному апоферменту добавить соот-
ветствующий К., отделенный от него ранее или полу-
ченный из других источников, в том числе путем син-
теза, то активность фермента восстанавливается.
Не все ферменты требуют для своего действия присут-
ствия К. Многие ферменты, относящиеся к классу гидро-
лаз (пептидазы, эстеразы, гликозидазы, фосфатазы
и др.), являются простыми белками, не нуждающимися
для проявления своих ферментативных функций в к.-л.
дополнительных кофакторах органич. природы. Од-
нако большинство ферментов и среди них прежде
всего ферменты, относящиеся к классу трансфераз
(дегидрогеназы, оксидазы, цитохромы, аминоферазы,
фосфокиназы, ацилферазы и др.), активны лишь при
наличии соответствующих К.
Механизм действия К. очень сложен и разносторо-
нен. Как правило, К. непосредственно взаимодейст-
вуют с субстратом. При этом они могут играть роль
промежуточных переносчиков (акцепторов и доноров)
определенных химич. группировок (ацильных, фос-
фатных, аминных и т. п.), а также атомов водорода и
электронов (аденозинтрифосфорная к-та, кодегидроге-
назы и др.). Наряду с этим К. участвуют в процессе
активирования субстрата, образуя с его молекулами
реакционноспособные промежуточные соединения, в
составе к-рых молекула субстрата претерпевает опре-
деленные химич. превращения. Такова, в частности,
роль тиаминдифосфата при декарбоксилировании
пировиноградной к-ты и роль пиридоксалъфосфата при
многочисленных и разнообразных превращениях ами-
нокислот. Активирование молекулы субстрата К.
обычно имеет двоякий характер: во-первых, промежу-
точное соединение субстрата с К. может обладать
избыточным запасом свободной энергии, т. е. нахо-
диться на более высоком энергетич. уровне, что со-
здает выгодные термодинамич. предпосылки для нуж-
ных реакций; во-вторых, электронная конфигурация
молекулы субстрата или определенной ее части при
образовании промежуточного соединения с К. изме-
няется т. о., что это благоприятствует в кинетич. от-
ношении нужным реакциям вследствие снижения энер-
гии активации. Обычно К. совмещают роль активатора
и переносчика молекулы субстрата или ее части (см.,
напр., Кофермент А).
Характерная для ферментов исключительно высо-
кая специфичность в отношении субстратов и катали-
зируемых реакций определяется белковой частью
фермента. К., число к-рых по сравнению с ферментами
очень йёвелико, значительно менее специфичны.
В пределах данного класса суббтратов й данного типа
превращений К. мОГут каГализировёть разные реак-
ции, в зависимости от Toto, в совокупности с какими
апоферментами они функционируют. Так, пирИдоксаль-
фосфат, функционируя в качестве К.различных ами-
нофераз (трансаминаз), принимает участие в переа-
минировании различных амино- и кетокислот; высту-
пая в качестве К. декарбоксилаз а-аминокислот, пи-
ридоксальфосфат катализирует процесс декарбокси-
лирования а-аминокйслот, но не может служить К.
при декарбоксилировании а-кетокислот, к-рые декар-
боксилируются различными ферментными системами
при участии другого К. — тиаминдифосфата.
К. различаются по типу и прочности их связи с
апоферментом. Ряд К. образует с белковой частью
ферментов очень прочные, недиссоциирующие со-
единения. К числу их относятся железопорфирины,
биотин, пиридоксальфосфат и флавиновые нуклео-
тиды. Эти К., называемые Обычно простетич. группами
соответствующих ферментов, не могут отщепляться от
оДйого фермента и переходить на другой; они взаимо-
действуют с субстратом, оставаясь постоянно в составе
одной определенной молекулы фермента —• протеида.
С другой стороны, типичные К., примером к-рых могут
служить кодегидрогеназы I и II, образуют с белковой
частью ферментов весьма непрочные и легко диссо-
циирующие комплексы. Это позволяет К. осуществ-
лять их функцию переноса групп от донора к акцеп-
тору, от одного фермента к другому, связывая послед-
ние в единую ферментную систему. Такая система
может быть схематически изображена след, образом:
D—R—11— К ------1 г—*-A —R
D-*—’ ’—*-К—R— —А
Ф, Ф2
В первой реакции, катализируемой ферментом Фх,
происходит перенос остатка R с донора D на К.
Кофермент, акцептировавший группу R, отделяется
от фермента Фх, и в реакции, катализируемой фермен-
том Ф2, передает группу R акцептору А. В этом слу-
чае в известном смысле стирается грань между К. и
субстратом: в каждой отдельной реакции К. выступает
как субстрат, подвергающийся тому или иному прев-
ращению. Только по отношению к системе в целом он
оказывается истинным катализатором, поскольку
после каждого оборота цикла он принимает исходную
форму и в процессе суммарной реакции не расхо-
дуется.
Весьма своеобразная форма взаимоотношений между
субстратом и К. наблюдается у фосфомутаз. Одна из
них — фосфоглицеромутаза, катализирует обратимое
превращение 3-фосфоглицериновой к-ты в 2-фосфогли-
цериновую. Коферментом этого процесса является
2,3-дифосфоглицериновая к-та. В ходе ферментатив-
ной реакции происходит перенос фосфатного остатка
из положения 3 кофермента (2,3-дифосфоглицериновой
к-ты) в Положение 2 субстрата (3-фосфоглицериновой
к-ты); при этом К. превращается в продукт реакции —
2-дифосфоглицериновую к-ту, а из субстрата обра-
зуется новая молекула К.
СН2ОРО(ОН)2 СН2ОРО(ОН)2
СНОН +СНОРО(ОН)2
соон соон
СН2ОРО(ОН), СН2ОН
I I
= СНОРО(ОН)2 + СНОРО(ОН)2
соон соон
Аналогичную коферментную роль выполняет 1,6-ди-
фосфоглюкоза при катализируемом фосфоглюкомута-
зой превращении 1-фосфоглюкозы в 6-фосфоглюкозу.
Химич, строение К. весьма разнообразно. Многие
К. являются нуклеотидами. В состав большинства
К. входят витамины.
Ниже приводится краткая классификация важнейших
К. с указанием типа ферментативных реакций, в к-рых они
участвуют.
1. Железопорфириновые коферменты
(см. HopgJupuHw) — простетич. группы каталазы, пероксидазы,
цитохромов и цитохромоксидазы; осуществляют перенос элек-
тронов к кислороду на завершающих этапах процессов био-
химич. окисления.
2. Кофермент ы-н уклеогиды: а) Адениловые
рибонуклеотиды — аденозин-5-моно-, ди- и трифосфорные
к-ты (см. Аденозинфосфорные кислоты) — участвуют в реак-
циях фосфорилирования в качестве доноров орто- и пирофос-
фатных остатков, активируют аминокислоты в процессе био-
синтеза белков и пептидов путем образования аминоациладе-
нилатов, осуществляют перенос и активирование остатков
угольной и серной к-т. б) Гуаниловые рибонуклеотиды — гуа-
нозин-5-фосфорные к-ты (см. Гуаниловая кислота) — осущест-
вляют перенос остатка янтарной к-ты, играют роль К. при
биосинтезе адениловой к-ты из инозиновой, в) Цитидиловые
рибонуклеотиды — цитидин-5-фосфорные к-ты — осуществля-
ют активирование й перенос остатков фосфохолина и фосфоэта-
ноламина при биосинтезе фосфатидов, г) Уридиловые нуклео-
тиды — уридин-5-фосфорные к-ты — осуществляют перенос
743
КРАСИТЕЛИ СИНТЕТИЧЕСКИЕ
744
остатков моноз при биосинтезе ди- и полисахаридов и прини-
мают участие в других ферментативных реакциях, в частности
при взаимопревращении глюкозы в галактозу, д) Кодегидро-
геназы — участвуют в важнейших для клеточного обмена про-
цессах переноса водорода в качестве К. многочисленных
дегидрогеназ, е) Флавиновые коферменты (флавинмононуклеотид
и флавинаденипдинуклеотид) — участвуют в процессах пере-
носа водорода в качестве простетич. группы различных оксидаз.
ж) Кофермент А — осуществляет перенос и активирование ос-
татков карбоновых к-т в ряде ферментативных реакций (аци-
лирование холина и гиппуровой к-ты, синтез лимонной к-ты,
окисление и синтез жирных к-т).
3. Другие коферменты, содержащие фосфатные остатки:
а) Тиаминдифосфат — осуществляет коферментные функции
при простом и окислительном декарбоксилировании а-кето-
кислот, а также при реакциях расщепления и синтеза угле-
родной цепи ряда кетоз, б) Пиридоксальфосфат — простетич.
группа ферментов, катализирующих разнообразные превра-
щения аминокислот (переаминирование, декарбоксилирование,
расщепление, реакции замещения и конденсации).
4. Коферменты пептидной природы: а) Ко-
фермент формилирования — КоФ, тетрагпдрофолевая к-та,
у - (тетрагидроптеридил-парааминобензоил)-Ь-г л у т аминовая
к-та — играет роль К. в промежуточном обмене т. н. одно-
углеродиых остатков (формила и оксиметила), участвует как
в переносе этих остатков, так и в их взаимопревращении;
осуществляет функции К., образуя с этими остатками соответ-
ствующие формильные и оксиметильные производные КоФ,
являющиеся «активными формами» муравьиной к-ты и формаль-
дегида. б) Глютатион — К. глиоксалазы, катализирующей
превращение метилглиоксаля в молочную к-ту; входит также
в состав активного центра дегидрогеназы 3-фосфоглицерино-
вого альдегида.
5. Кофермент ы-г о р м о н ы. Нек-рые стероидные
гормоны, в частности эстрон и андростерон, играют роль К.
в трансгидрогеназной реакции, осуществляют перенос водорода
между кодегидрогеназой I и кодегидрогеназой II по схеме:
6. Прочие коферменты: а) Липоевая кислота — выполняет
наряду с тиаминдифосфатом функции К. при окислительном
декарбоксилировании а-кетокислот, осуществляя промежуточ-
ный перенос водорода и ацильных остатков, б) Биотин — вхо-
дит в состав ферментов, осуществляющих обратимые реакции
декарбоксилирования и карбоксилирования ряда карбоновых
к-т. в) Витамин В12; производные витамина В12, в к-рых циано-
группа заменена на нуклеозид аденина, а кобальт, по-види-
мому, находится в 2-валентном состоянии, функционируют
в качестве К. в реакциях изомеризации глутаминовой к-ты
в fj-метиласпарагиновую и метилмалонил-кофермента А в сук-
ципил-кофермент А.
Лит.: Нейландс Дж., Штумпф П., Очерки по
химии ферментов, пер. с англ., М., 1958; Диксон М.,
Уэбб Э., Ферменты, пер. с англ., М., 1961; Fermente, Hor-
mone, Vitamine und die Beziehungen dieser Wirkstoffe zueinan-
der, hrsg. v. R. Ammon und W. Dlrscherl, Bd 1, Stuttg., 1959;
The enzymes, ed. by P. D. Boyer [a. o.J, v. 1—3, N. Y. — L.,
1959—60. В. Б. Спиричев.
КРАСИТЕЛИ СИНТЕТИЧЕСКИЕ — вещества для
окраски различных материалов, получаемые методами
оргапич. синтеза. В широком смысле сюда относятся
также бесцветные соединения, из к-рых окрашенные
вещества образуются уже после нанесения на окраши-
ваемый материал, напр. индигозоли (см. Кубовые
красители растворимые), диазоаминовые красители
(см. Диазоаминолы), фталогены, нек-рые красители
для меха.
Синтетич. красители, производство к-рых было начато
в 1857 выпуском мовеина (Перкин), практически полностью
вытеснили из промышленной практики природные красители,
при этом наиболее ценные природные красители — ализарин,
индиго — стали объектами промышленного произ-ва. Первые
К. с. — мовеин, фуксин — были получены окислением н раз-
ных условиях технич. анилина, содержащего много толуиди-
нов, откуда пошло название «анилиновые красители» как си-
ноним К. с.
Термин «краситель» был введен в 1908 А. Е. Порай-
Кошицем взамен ранее применявшихся терминов
«краска» и «пигмент», к-рым теперь придают иное,
более ограниченное значение. Краской называют
смесь красящих и связующих веществ, применяемую
для поверхностного окрашивания разных изделий или
для печатания на тканях, бумаге, пластических плен-
ках и др. материалах. Название «пигменты» сохраняет-
ся для нерастворимых красителей, а также нек-рых
неорганич. веществ.
На мировой рынок выпускается более 5000 различ-
ных красителей, основная часть к-рых предназначена
для расцветки волокнистых материалов. Необходи-
мость большого числа красителей определяется не
только разнообразием цветов и оттенков, а и различи-
ем в свойствах окрашиваемых материалов, методах
нанесения окраски/ условиях службы окрашенных
изделий, значительной разницей в стоимости красите-
лей, что стимулирует в ряде случаев практику при-
менения менее прочных, но дешевых красителей. О
методах применения красителей см. Крашение тек-
стильных материалов.
Красители выпускаются в продажу обычно под
условными названиями, к-рые как фирменная торго-
вая марка в капиталистич. странах защищены патен-
тами. При этом одинаковые по строению красители
разные фирмы выпускают под различными названиями.
Перечень выпущенных на рынок красителей с указа-
нием фирмы изготовителя, свойств и, когда это из-
вестно, строения, приведены в справочнике Colour
Index.
В Советском Союзе красители выпускаются также
под условными названиями, к-рые образуются в со-
ответствии с определенными правилами. По техниче-
ским свойствам и областям применения красители де-
лят на: прямые; сернистые; водорастворимые производ-
ные сернистых красителей; кубовые; водораствори-
мые производные кубовых красителей; протравные
красители для хлопка; компоненты, образующие кра-
сители на волокне; красители для полушерсти; кис-
лотные; протравные красители для шерсти; специ-
альные красители для химич. волокон; красители,
образующие ковалентную связь с волокном; красители
для кожи и шубной овчины; красители для меха; ос-
новные красители; нигрозины и индулины; красители,
растворимые в органич. веществах; красители для ано-
дированного алюминия; красители для дерева; опти-
чески отбеливающие препараты; пигменты и лаки.
В пределах каждой группы красители различаются
по цветам получаемых окрасок на желтые, золоти-
сто-желтые, оранжевые, красные, розовые, рубино-
вые, бордо, красно-фиолетовые, фиолетовые, синие,
голубые, бирюзовые, зеленые, оливковые, желто-
коричиевые, красно-коричневые, коричневые, серые,
сине-черные, черные.
Название красителей состоит из двух и более слов.
Первое слово указывает на способ применения кра-
сителя или его свойства (прямой, кислотный, кубовый,
сернистый, пигмент, лаковый, легкосмываемый и
т. п.), второе — характеризует цвет, в отдельных слу-
чаях вводится третье слово, характеризующее спе-
цифические свойства красителя (напр., светопрочный)
или его строения (напр., антрахиноновый). Следую-
щие за словами цифра и буква указывают на откло-
нение оттенка красителя (напр., желтый К или жел-
тый 3 обладают красноватым или зеленоватым оттен-
ком желтого цвета, у желтого 2К и желтого 23 —
соответствующие отклонения оттенка выражены силь-
нее и т. д.). Вторая и последующие буквы в названии
служат для обозначения специфич. свойств красителя
и их зпачение неодинаково в разных классах и даже
группах, они могут указывать на особенности при-
менения, наличие в молекуле красителя металла и
т. п. Для части красителей отечественного ассорти-
мента сохранены традиционные названия: индиго,
тиоиндиго, родамин и т. д.
По физич. свойствам К. с. можно разбить на две
основные группы: растворимые и нерастворимые
в воде.
Красители, растворимые в воде. Почти все раст-
воримые красители имеют ионогенный характер;
известно лишь ограниченное число окрашенных непо-
лярных соединений, растворимых в воде, рекомендо-
745
КРАСИТЕЛИ СИНТЕТИЧЕСКИЕ
746
ванных для крашения ацетатного шелка и синтетич.
волокон. Соли аммониёвых, реже сульфониевых и ок-
сониевых оснований, составляющие класс основных
или катионоидных красителей, находят применение
для окраски натурального шелка, кожи, полиакрил-
нитрильного волокна, приготовления чернил, ка-
рандашей, в полиграфии, косметике, а также в ка-
честве сенсибилизирующих красителей, для галогепо-
серебряных фотоэмульсий.
Раньше основные красители применялись для окрас-
ки хлопка, предварительно обработанного веществами,
образующими с красителями на волокне нераствори-
мые соединения (крашение по протраве). Нераство-
римые соли основных красителей с фосфорно-вольфра-
мовой и фосфорно-молибденовой к-тами, известные
под названием фаналевых лаков, находят применение
как яркие пигменты, гл. обр. в полиграфии. По химич.
строению катионоидные красители пред-
ставляют собой диарилметановые красители, три-
арилметановые красители, ксантеновые красители,
акридиновые красители, хинониминовые красители,
оксазиновые красители, азиновые красители, цианино-
вые красители, моноазокрасители, триазиновые кра-
сители, реже дисазокрасители, содержащие солеоб-
разующие группы основного характера. Для свето-
прочной окраски полиакрилпитрильного волокна
выпускают специальные цианиновые красители, ге-
тероциклич. азокрасители, а также замещенные
фталоцианина, антрахинона и др. полициклич. со-
единений, имеющие характер солей четвертичных ам-
мониевых соединений.
Анионоидные красители, за неболь-
шим исключением, представляют собой соли сульфо-
кислот. В зависимости от размера, копланарности и
других особенностей молекулы красители обладают
способностью преим. окрашивать целлюлозные (хло-
пок, лен, вискоза, медно-аммиачное волокно) или бел-
ковые (шерсть, шелк) и полиамидные волокна. Рас-
творимые красители, пригодные для непосредственного
окрашивания целлюлозного волокна, наз. прямыми,
или субстантивными. Первым прямым красителем
для хлопка, открытым в 1884, явился конго красный,
получаемый сочетанием диазотированного бензидина
с 1,4-нафтиламинсульфокислотой. Сродством к цел-
люлозному волокну обладают азокрасители, получен-
ные сочетанием диазотированного бензидина с дру-
гими амино- и оксисоединениями ароматич. ряда, а
также производных бензидина, не замещенных в по-
ложениях 2 и 6 дифенила. Наличие заместителей в
указанных положениях препятствует расположению
молекулы в одной плоскости и такие красители обла-
дают большим сродством к белковым волокнам, чем
к целлюлозе. В качестве прямых красителей исполь-
зуются и другие типы дисазокрасителей и полиазо-
красителей, в том числе содержащие атомы меди или
других тяжелых металлов, нек-рые производные ан-
трахинона. С целью получения прямых красителей
с высокой светопрочпостью сульфируют яркоокра-
шенные полициклич. соединения, например фталоциа-
нин меди.
Сорбция прямых красителей волокном — процесс
равновесный. Для повышения устойчивости окрасок
прямыми красителями к стирке окрашенные изделия
обрабатывают растворимыми искусственными смолами
основного характера, образующими с красителями
нерастворимые или труднорастворимые соединения.
1 Повышение устойчивости окрасок как к мокрым об-
работкам, так и к действию света при одновременном
изменении цвета достигается, в случае наличия в мо-
лекуле прямого красителя групп, способных к ком-
плексообразованию, обработкой солями меди или
других комплексообразующих металлов, а при нали-
чии свободной первичной аминогруппы — диазотирова-
нием и последующим сочетанием на волокне (диазо-
тируемые красители). Кислотные красители для шер-
сти представляют собой сульфокислоты моноазо-,
реже дисазокрасителей, аминоокси-, ариламиноантра-
хинона, ариламино-антрапиридона и -антрапирими-
дина (см. Антрахиноновые красители, Аминоантра-
хиноновые красители). Яркие сульфокислоты триарил-
метановых и азиновых красителей ввиду малой свето-
прочности потеряли значение для крашения шерсти.
С целью упрочнения окрасок здесь шире, чем в краше-
нии целлюлозы, используют образование комплексных
соединений с металлами, особенно с хромом: приме-
няется последующая обработка окрасок солями хро-
ма — хромирующиеся красители, хро-
мирование в процессе крашения (однохромовые кра-
сители) и использование для крашения готового ком-
плекса красителя с металлом. Особенное значение для
прочного крашения шерсти и капронового штапеля из
слабокислых и нейтральных ванн приобрели комплек-
сы двух молекул диоксиазокрасителей с отрицательно
заряженным 4-валентным атомом
хрома или кобальта (I). При этом
молекулы азокрасителей, содержа-
щих обычно сульфамидные груп-
пы, расположены в взаимно пер-
пендикулярных плоскостях. Для
прочного крашения шерсти из
нейтральных и слабокислых ванн
в более яркие цвета наряду с ме-
таллокомплексами используются
антрахиноновые и азокрасители, содержащие алифа-
тич. цепи, включающие до 12 углеродных атомов.
С целью прочной фиксации растворимых красите-
лей на волокне в их молекулу вводят группы, способ-
ные образовывать прочную ковалентную связь с ок-
сигруппами целлюлозы или аминогруппами белковых
волокон. Красители такого типа, выпущенные в по-
следние годы впервые фирмой Ай-Си-Ай (Англия)
под названием проциопов, фирмой Циба (Швей-
цария) под названием цибакронов для хлопка
и брил лиантци бал ан для шерсти, фирмой
Хехст (ФРГ) под названием р ем а зо л о в, теперь
в разпых вариантах и под различными названиями вы-
пускаются многими другими иностранными фирмами.
Согласно принятой в СССР номенклатуре, эти краси-
тели носят название активных. А ктивные кр а-
с и т е л и представляют собой азо-, антрахиноновые
или фталоцианиновые красители, обычно близкие по
строению к обычным кислотным красителям, но наряду
с сульфогруппой они содержат лабильно связанные
атомы хлора, сульфоэфирные, винилсульфамидпые
или другие аналогичные группировки, способные при
увеличении щелочности среды в процессе крашения
или при последующей обработке взаимодействовать
с активными функциональными Группами окрашивае-
мого волокна. Преимуществом активных красителей
является простота применения, недостатком — па-
раллельный взаимодействию с волокном гидролиз
активных группировок, что, помимо потери значитель-
ной части красителя, для достижения высокого пока-
зателя прочности к мокрым обработкам требует тща-
тельной отмывки окрашенных продуктов гидролиза.
Красители, нерастворимые в воде. К числу нерас-
творимых в воде относятся кубовые красите-
ли и сернистые красители, использова-
ние к-рых связано с временным превращением в рас-
747
КРАСИТЕЛИ СИНТЕТИЧЕСКИЕ
748
творимые соединения. Для кубовых красителей ха-
рактерно наличие парных карбонильных групп, со-
единенных системой сопряженных связей, способных
восстанавливаться в щелочном р-ре гидросульфита
натрия с образованием растворимого в воде продукта,
легко и нацело окисляющегося кислородом воздуха
в исходный краситель. Щелочной раствор продукта
восстановления красителя называется кубом, продукт
восстановления с незамещенными гидроксильными
группами — лейкосоединением или, как - принято в
текстильной практике, кубовой кислотой. Природные
кубовые красители — индиго и тирийский пурпур
(6,6'-диброминдиго) — были известны в древности.
Установление строения индиго, синтез и освоение
произ-ва синтетич. индиго оказали существенное
влияние на развитие органич. химии и химич. пром-сти.
Само индиго в настоящее время утеряло прежнее значе-
ние, однако индигоидные красители и тиоиндигоид-
ные красители широко применяют для печати по хлоп-
чатобумажным тканям, крашения меха и шерсти из
слабощелочного аммиачного куба. Особенно высокая
прочность окрасок целлюлозных волокон достигается
применением антрахиноновых красителей и полици-
клических или, как их не вполне точно называют,
полициклокетоновых кубовых красителей. Для кра-
шения и печати по текстильным материалам кубовые
красители используют также в виде растворимых в
воде и достаточно устойчивых солей кислых сернокис-
лых эфиров лейкосоединений, выпускаемых в СССР
под названием кубозолей и индигозолей.
Превращение сернистых красителей в растворимую
форму связано с восстановлением сернистым натрием
дисульфидных групп в меркаптидные. Сернистые
красители, несмотря на тусклый цвет, малую интенсив-
ность и только удовлетворительную прочность окрасок,
вследствие низкой стоимости и относительной про-
стоты крашения все еще применяют в крашении хлоп-
ка, особенно в черный и темные цвета. Для окраски
вискозного штапеля в массе нашли использование
стабильные растворимые формы сернистых красителей.
Нерастворимые органические пигменты, как и
пигменты минеральные, отличаясь от последних боль-
шей яркостью и особенно интенсивностью окрасок,
используются для лакокрасочных покрытий, в поли-
графии, произ-ве карандашей, художественных кра-
сок, окраски пластич. масс, резины, крашения ис-
кусственных и синтетич. волокон в массе в процессе
их формования. Развитие химии полимерных ма-
териалов открыло возможность использования пиг-
ментов вместе со связующими для печати и даже ок-
раски текстильных материалов. Нанесенная окраска
после полимеризации связующего прочно удерживается
на ткани, заметно не изменяя свойств последней.
В качестве органич. пигментов используют нерас-
творимые азокрасители, производные антрахинона и
полициклич. хинонов, в том числе кубовые красители.
Особенное значение в качестве ярких голубых и зе-
леных пигментов приобре-
ли фталоцианиновые краси-
тели. Как красный пигмент
в последнее время с успе-
хом используется линейный
хинакридон (II).
Яркость, интенсивность, иногда и цвет окраски
пигментов зависят от размеров частиц, а часто, в
связи с полиморфизмом, и от кристаллич. структуры.
В связи с этим технология выпускных форм наряду
со стадиями синтеза является важнейшим элементом
произ-ва органич. пигментов. Для приготовления не-
растворимых форм часто пользуются приемом осаж-
дения иопогенных красителей в виде нерастворимых
в воде солей. Выше упоминалось об изготовлении
фаналевых лаков катионоидных красителей. Сульфог
кислоты красителей осаждают обычно в форме не-
растворимых бариевых, кальциевых и марганцевых
солей, также носящих название лаков. Иной характер
комплексного соединения имеет красный краплак,
получаемый взаимодействием ализарина С солями
алюминия и кальция, и зеленый пигмент из нитро-
зобетанафтола и солей железа. Для крашения аце-
татного шелка и синтетич. волокон применяют нерас-
творимые в воде дисперсные красители, способные
выбираться из водной суспензии и дифундировать
в массу окрашиваемого волбкпа. Они представляют
собой азо- и антрахиноновые красители небольшого
мол. веса, часто содержащие оксиэтильные группы.
Для крашения полиэфирных волокон используют
переносчики, способствующие набуханию волокна,
или процесс ведут при темп-ре выше 100° под давле-
нием. С целью повышения прочности окрасок поли-
амидного волокна в молекулу дисперсных красителей
вводят активные группировки, обычно содержащие
лабильные атомы хлора, способные взаимодействовать
с аминогруппами полиамидного волокна (п р -о ц и-
н и л ы — Ай-Си-Ай). Крашение ацетатного волокна
при прядении осуществляют также введением краси-
телей, растворимых в ацетоне. Известны опыты кра-
шения капрона и лавсана красителями, раствори-
мыми в расплавленных полимерах, поступающих на
формование волокон.
Растворимые в органич. растворителях К. с. при-
меняют для маркировки специальных сортов бензина,
окраски пластич. масс, лаковых покрытий и в других
отраслях техники. Термически стойкие и легко
сублимирующиеся К. с. используют для создания
цветных сигнальных дымов.
• Большое распространение имеет образование обычно
красящего вещества из бесцветных или слабоокрашен-
ных соединений непосредственно на окрашиваемом
материале. Получение на ткани ярко-красного алюми-
ниево-кальциевого лака (кумач) с использованием
природного ализарина было известно задолго до от-
крытия К. с. В 1863 был найден способ получения
глубоко-черной и чрезвычайно прочной окраски хлопка
окислением анилина на волокне; только через 50 лет
Р. Вилыптеттером и А. Грином был раскрыт механизм
процесса образования «черного анилина» и строение
образующегося азинового красителя. Окисление ами-
нов, фенолов и аминофенолов является основным при-
емом крашения меха. Для печати по хлопчатобу-
мажным тканям широко применяется прием образо-
вания непосредственно на волокне нерастворимых
азокрасителей. Чрезвычайно прочную и яркую ок-
раску тканей достигают синтезом на волокне фтало-
цианина меди из дииминоизоиндолина и солей меди
с применением ряда вспомогательных веществ. Для
этой цели используют также растворимые, уже со-
держащие металл комплексы. Примером образования
красителей непосредственно на материале служит
цветная фотография. В процессе цветного проявления
в многослойной фотоэмульсии совместным окисле-
нием при действии активированного светом галоген-
ного серебра из ранее введенной цветообразующей
компоненты и проявляющего вещества образуются
хинониминовые красители. Светочувствительность
диазосоединений используют для светокопирования
чертежей. Диазосоединение, сохранившееся после эк-
спозиции в затемненных изображением местах, после
проявления образует на бумаге или пленке азокраси-
тель (см. Диазотипия}.
Многие К. с. нашли применение, не связанное
с окраской. Напр., метиленовый синий, бриллианто-
вый зеленый, фенолфталеин в качестве лекарственных
веществ, индиго, фталоцианин и нек-рые полициклич.
кубовые красители для консистентных высоко-
температурных смазок и т. п.
749
КРАСКИ
„750
Как правило, глубокая и интенсивная окраска ор-
ганич. веществ определяется системой сопряженных
двойных связей, наиболее устойчивой в изо- и гетеро-
циклич. соединениях (см. Цветности теория). По-
этому К. с., за исключением полиметиновых, приме-
няемых не для крашения, а для сенсибилизации фото-
графия. эмульсий, представляют собой циклич., часто
полициклич. соединения.
Основным исходным сырьем в произ-ве К. с. служат аро-
матич. углеводороды: бензол, толуол, ксилол, нафталин,
антрацен, в меньшей степени мезитилен, псевдокумол, аце-
нафтен, пирен и гетероциклич. соединения — пиридин и
карбазол. Большое разнообразие красителей и многостадий-
ность их синтеза из продуктов коксохимич. пром-сти опреде-
лили необходимость организации произ-ва товарных промежу-
точных продуктов: сульфокислот, нитро-, галогено-, амино-,
оксипроизводных названных углеводородов, карбоновых кис-
лот, более сложных соединений, содержащих различные
группы в ароматич. ядре антрахинона и его замещенных.
Нек-рые промежуточные продукты — анилин, фенол, бета-
нафтол, фталевый ангидрид и т. д. — широко применяют
в других отраслях химич. произ-ва: пластич. масс, синтетич.
смол, химикатов для произ-ва резины, гербицидов, взрывча-
тых, лекарственных, душистых и текстильно-вспомогательных
веществ.
Русские ученые внесли крупный вклад в развитие
химии ароматич. соединений, в частности первый
синтез анилина восстановлением нитробензола был
осуществлен Н. Н. Зининым, а название «анилин»
введено Ю. Фрицше, установившим тождественность
соединения, полученного Зининым, с веществом, по-
лученным им ранее при Нагревании со щелочью при-
родного индиго. М. А. Ильинский открыл каталитич.
влияние ртути при сульфировании антрахинона, что
явилось началом развития большой группы прочных
кубовых и кислотных красителей—альфапроизводных
антрахинона.
Советская анилинокрасочная промышленность,
созданная после Октябрьской революции, в настоя-
щее время занимает второе место в мире по объему
произ-ва красителей, но пока в недостаточном ассор-
тименте выпускает нек-рые группы прочных краси-
телей и красителей для новых видов синтетич. воло-
кон. В последнее время наблюдается тенденция к за-
мене мапоинтенсивных сернистых красителей более
яркими прочными и интенсивными красителями.
Лит.: Colour index, [v. 1], 2 ed., Bradford, [1956]; Во-
рож ц о в Н. Н., Основы синтеза промежуточных продуктов и
красителей, 3 изд., М., 1955; Коган И. М., Химия кра-
сителей, 3 изд., М., 1956; Венкатараман К., Химия
синтетических красителей, пер. с англ., т. 1—2, Л., 1956—57;
Хим. наука и пром-сть, 1958, 3, № 2. Н. С. Докунихин.
КРАСКИ — лакокрасочные материалы, состоящие
из пленкообразующего вещества (связующего) и тонко-
дисперсных пигментов. В состав К. могут входить
также минеральные наполнители, матирующие ве-
щества, пластификаторы, растворители и др. до-
бавки. К. масляные содержат в качестве плен-
кообразующего олифы, К. эмалевые — синте-
тические смолы или эфиры целлюлозы в виде рас-
творов в органич. растворителях (лаков). Эмалевые
К. также содержат различные добавки, ускоряющие
процесс пленкообразования, повышающие устойчи-
вость жидкой К. и пленки покрытия и т. д. При нане-
сении на поверхность тонким слоем К. образуют не-
прозрачные, окрашенные прочные пленки, имеющие
хорошее сцепление с поверхностью изделия. Приме-
нение К. преследует две основные цели: придание
поверхности изделия красивого внешнего вида и
предохранение материалов от вредного воздействия
внешней среды — солнечной радиации, влаги, химич.
реагентов, высокой температуры, плесени и т. д. По-
крытия К. выполняют иногда и другие функции, па-
пример увеличивают отражение света поверхностью,
повышают видимость изделий и знаков (светящие-
ся К.) и т. д.
Свойства К. зависят от вида пленкообразующего,
а также от природы и содержания пигментов и напол-
нителей. Роль пленкообразующего (связующего) в-ва
заключается в обволакивании частиц пигмента и на-
полнителя, в создании монолитной, твердой и эластич-
ной, газо- и влагонепроницаемой пленки, в обеспе-
чении хорошего сцепления пленки с окрашиваемой
поверхностью, предохранении ее от внешних воз-
действий.
Пигменты обеспечивают получение непрозрачного
(укрывистого) покрытия, придают необходимый цвет,
повышают светостойкость и стойкость к атмосферным
воздействиям, способствуя отражению ультрафиоле-
товых лучей, снижению влагонабухаемости путем об-
разования водостойких химических соединений со
связующим (напр., свинцовый сурик или свинцовые
белила с олифой) и т. д. Пигменты уменьшают и влаго-
проницаемость, уплотняя пленку К. Особенно заметно
это проявляется в случае пигментов и наполните-
лей, имеющих чешуйчатую структуру (алюминиевая
пудра, слюда и др.). Некоторые пигменты повышают
термостойкость покрытий благодаря образованию
с пленкообразующим металлоорганпч. соединений
(напр., алюминиевая пудра с кремнийорганич. смо-
лами) либо путем упрочнения структуры пленки,
изменяют коэффициент отражения света от 2% до 90%,
а также способность покрытия излучать тепловую
энергию. Нек-рые пигменты имеют избирательные
отражательные или поглотительные свойства в раз-
личных частях спектра, что позволяет создавать мас-
кировочные К., оптически недещифрируемые и визу-
ально трудно различимые на фоне местности. Приме-
няя в качестве пигментов вещества, способные к фос-
форесценции или флуоресценции и излучающие свет
в видимой части спектра, можно получать светящиеся
К. Нек-рые пигменты способны замедлять электро-
химич. коррозию путем пассивирования поверхности
металла (хроматы цинка, стронция и др.) или катодной
защиты (цинковая пыль по отношению к железу).
Пигмент должен находиться в К. в тонкодисперсном
состоянии (размер частиц от 0,2 до 5 мк). Такие К.
обладают лучшей укрывистостью, яркостью, атмосфе-
ростойкостью и гладкостью покрытия; при хранении
они менее склонны к седиментации. Однако чрезмерно
большая дисперсность (диаметр частиц меньше раз-
мера половины длины световой волны) снижает ук-
рывистость К. Пигменты диспергируют вместе со
связующим (в оптимальных соотношениях) на краско-
терках, в шаровых мельницах или на валках методом
сухого вальцевания. Последний способ особенно
эффективен для диспергирования ассоциированных
частиц мелкодисперсных пигментов (сажи, милори
и нек-рых органич. пигментов). В качестве пигментов
применяют металлы, окислы и соли металлов, нераст-
воримые в связующем. Пигменты металлические:
порошки алюминия, бронзы, нержавеющей стали,
цинка, свинца; белые: цинковые белила, титаповые
белила (анатазной и рутильной формы), литопон;
красные: железпый сурик, мумия, марс, кадмопоны,
свинцовый сурик, молибдатный крон, киноварь;
синие: милорь, синий кобальт, ультрамарин; желтые:
охра, свинцовый, цинковый и стронциевый крона,
окислы железа, желтый кадмий; зеленые: окись
хрома, смеси желтых кронов с милорыо, соединения
кобальта; черные: сажа, окислы марганца, окислы
железа и др. Применяются также органич. пигменты
(фталоцианиновые, крапплакп, азокрасители индантре-
иовые, тиоиндиговые и др ), которые, в отличие от
растворимых органич. красителей, находятся в К.
в дисперсном состоянии. Сульфиды цинка, кальция,
кадмия, а также их смеси применяются в качестве
пигментов для светящихся фосфоресцирующих К. вре-
менного действия. Смолы, окрашенные нек-рыми ор-
ганич. красителями, переведенные в нерастворимое
состояние и измельченные до размера частиц 3—10 мк-
751
КРАСКИ
752
применяют в качестве пигментов для флуоресцирую-
щих К.
Наполнители, в отличие от пигментов, не обладают
укрывистостыо и добавляются в К. для удешевления
(барит, каолин, тальк) или повышения термостойкости
и стойкости к атмосферным воздействиям (тальк,
асбестит, слюда). Матирующие добавки позволяют
уменьшить или устранить блеск пленки. С этой целью
применяют стеараты цинка, кальция или алюминия.
Увеличение содержания наполнителей (талька) и
пигментов также уменьшает блеск пленки К. Стеараты,
тальк, бентонитовые глины и тонкодисперсный сили-
кагель препятствуют, кроме того, осаждению и рас-
слаиванию пигментов в К. при хранении. Пластифи-
каторы вводят для повышения эластичности пленок.
Чаще других применяют касторовое масло, дибутил-
фталат, трикрезилфосфат, полихлордифенил (совол).
Растворителями в К. служат летучие органич. ве-
щества (уайт-спирит, ацетон, толуол, ксилол, спирты,
ацетаты и др.) или вода.
Качество К. существенно зависит от весовых со-
отношений пигментов, наполнителей и пленкообра-
зующего. В зависимости от свойств пигмента (плот-
ность, маслоемкость, укрывистость и др.) содержание
его колеблется от 0,2 до 1,2 вес. ч. на 1 вес. ч.
пленкообразующего; содержание наполнителей —до
1,2 вес. ч. на 1 вес. ч. пленкообразующего.
В зависимости от процессов, происходящих при
образовании пленки, различают (см. табл.) два вида
К.: 1) Непревращаемые К., у к-рых обра-
зование пленок происходит в результате улетучивания
растворителей, повышения вязкости и отвердевания.
Тип пленки Пленкообразующие вещества
Непревра- щаемые Низкомолекулярные Канифоль, эфир гар- пиуса, шеллак, би- тум, асфальт
Низкомолекулярные, степень конденсации не более 10—12 Новолачные смолы, гли- кольфталат
Высокомолекулярные, степень полимериза- ции 50000 — 150000 Полистирол, перхлор- винил, эфиры целлю- лозы, полиакрилаты, хлоркаучук и т. п.
Превра- щаемые Низкомолекулярные — мономерные Высыхающие масла
Низкомолекулярные — полимерные Алкидные смолы, фе- нольно-альдегидные резолы, эпоксидные, полиуретановые, мо- чевино- и меламино- формальдегидные и ДР.
Высокомолекулярные (цепные полимеры) Вулканизуемые каучу- ки, поливинилбути- раль в сочетании с ре- зольпыми смолами и т. п.
Такая пленка остается растворимой. 2) Превра-
щаемые К., к-рые при образовании пленки пре-
терпевают глубокие структурные и хнмич. изменения
и становятся нерастворимыми. При этом происходит
превращение мономеров или линейных полимеров н
полимеры сетчатого строения в результате поликон-
денсации (феноло-альдегидныс резолы, мочевипо- и
меламино-формальдегидные смолы при горячей сушке)
и полимеризации или сополимеризации по месту не-
насыщенных связей как непосредственно, так и через
кислород (высыхающие масла, алкидные, фенольные
и др. смолы, модифицированные маслами, каучуки).
Сетчатые полимеры получаются также в результате
присоединения полигидроксильных алкидов к диизо-
цианатам (уретаны) и полиэфиров, содержащих
эпоксидное кольцо, к веществам, имеющим в моле-
куле подвижный атом водорода (напр., гексам тилен-
диамин, меламино-формальдегидная смола в другие
соединения).
Образование пленки К. сопровождается постепен-
ным увеличением вязкости. Особенно быстро возра-
стает вязкость эмалевых К., что обусловлено лету-
честью растворителей. У масляных К., не содержащих
летучих растворителей, вязкость пленки растет посте-
пенно, по мере протекания окислительных и полиме-
ризационных процессов. Продолжительность перехода
слоя К. из жидкого в гелеобразное состояние зависит
от природы пленкообразующего и темп-ры. Для не-
превращаемого пленкообразующего, где образовщше
пленки связано с химич. процессами, решающую роль
играет темп-ра. Алкидные, полиуретановые, феноло-
алкидные и другие эмалевые К. образуют высокока-
чественные пленки только с применением горячей
сушки (80—150°).
В процессе эксплуатации пленка К. подвергается
воздействию солнечных лучей, тепла, кислорода
воздуха и др. факторов, вызывающих в ней'химич.
и структурные изменения: деструкцию и структу-
рирование («сшивание» макромолекул пленкообразую-
щего). Интенсивная термоокислительная деструкция
нитроцеллюлозных пленок происходит при 80—90°,
пленок из виниловых полимеров — при 100°, акри-
ловых — при 180—200°, кремнийорганических — при
250—300°. Термин, деструкцию резко замедляет до-
бавление термостабилизаторов. Глубина фотохимии,
деструкции пропорциональна интенсивности ультра-
фиолетового облучения. Светостойкость К. увеличи-
вают добавлением пигментов, отражающих свет (алю-
миниевая пудра) или поглощающих коротковолновую
часть спектра (титановые белила рутпльной модифи-
кации, сажа). Фотохимии, деструкцию предотвращают
введением в состав К. специальных стабилизаторов
(напр., для хлорвиниловых К. это — 2,4-тидрокси-
бензофенон, тетраоксисебацефеноп, для нитроцел-
люлозы — дифениламин). В результате деструкции
пленка «стареет» — делается хрупкой и склонной к
растрескиванию, особенно при глубоком охлаждении.
В жидком состоянии (при хранении) в К. также про-
исходят процессы деструкции и структурирования
полимеров. В первом случае К. разжижается, во
втором — загустевает. Механизм этого явления ана-
логичен старению пленки К. и резко ускоряется под
влиянием света, тепла и при наличии электролитов,
попавших в К. вместе с пигментами, плеикообразую-
щим веществом, а также под влиянием продуктов раз-
ложения пленкообразующих веществ (NO2 для ни-
троцеллюлозы, НС1 для хлорсодержащих полимеров
и т. д.). Структурирование ускоряется при наличии
в связующем активных групп, напр. СООН, ОН,
и солей или окислов поливалентных металлов. К.,
перешедшая в гелеобразное состояние, к применению
не пригодна. Храпение К. рекомендуется при темп-ре
0—+5°.
Для обеспечения защиты от коррозии пленка К.
должна обладать рядом специфических свойств, к-рые
необходимо сочетать со свойствами грунтовочного
покрытия. Спстема антикоррозионного покрытия со-
стоит из слоя грунта и внешних слоев эмалевой К.
Грунтовочный слой обеспечивает покрытию хорошую
адгезию, пассивирует поверхность металла в случае
проникновения влаги, или анодно защищает металл.
Слой эмалевой К. препятствует проникновению влаги,
газов и электролитов к слою грунта. Влагопроница-
емость пленки эмалевой К. зависит прежде всего от
природы цлепкообразующего; она минимальна у ли-
нейных полимеров с малыми боковыми группами
(напр., сополимер хлорвинила с винплиденхлоридом,
753
КРАСКИ
перхлорвинил, эпоксидные, полиуретановые, алкидные
и фенольные смолы). Высокой влагопроницасмостью
обладают акриловые и эфироцеллюлозные пленки.
Пигменты, содержащиеся в эмалевой К., тоже сущест-
венно влияют на проницаемость влаги и ионов элек-
тролитов сквозь пленку. Лакокрасочная пленка пред-
ставляет собой избирательно ионопроницаемые мем-
браны, где стенки капилляров имеют-электрич. заряд
определенного знака вследствие содержания в плеп-
кообразующем веществе ионогенных групп. Наличие
тех или иных пигментов в эмалевых К. сказывается
на зарядности стенок капилляра и тем самым замедля-
ет или ускоряет проницаемость тех или иных ионов
через пленку эмали.
Масляные К. (МА) готовят на олифе из высыхаю-
щих или полувысыхающих масел (см. Масла высы-
хающие). Масляные К. выпускают в виде густотертых
паст. Для доведения К. до рабочей консистенции
применяют синтетич., комбинированные или нату-
ральные олифы. Масляные К. наносят кистью или
распылением; они медленно высыхают, пленки их
мягкие и приобретают твердость через 10—15 суток.
Обладают невысокой водостойкостью и разрушаются
от действия щелочей. С применением горячей сушки
(80—120°) качество покрытия резко улучшается. Ос-
новная область применения — строительство и же-
лезнодорожный транспорт.
Эмалевые К. (эмали, лаковые К.) подразделяются
в зависимости от вида лака, на основе к-рого они
изготовлены. (Описание состава и свойств соответ-
ствующих лаков см. в ст. Лаки).
Масляные эмали (КФ) готовят на основе масля-
ных лаков. Растворители: скипидар, уайт-спирит, сольвент,
ксилол. Эмали на жирных лаках (с содержанием до 75 % масла)
образуют эластичную и устойчивую к атмосферным воздей-
ствиям пленку. Со снижением содержания масла до 50—70%
эластичность и атмосферостойкость пленок резко уменьшаются.
Поэтому эмали на тощих лаках применяют для окраски пред-
метов, находящихся внутри помещений. Масляные эмали при-
годны для холодной и горячей сушки при 80—120°. Пленки
получаются глянцевые, твердые, с умеренной водостойкостью.
Эмали алкидные (ГФ, или ПФ) готовят на алкид-
ных лаках. В зависимости от вида алкидной смолы, входящей
в состав лака, различают'глифталевые и пентафталевые эмали.
Растворители — ксилол, сольвент, уайт-спирит и их смеси.
Эмали, приготовленные на смолах с малым содержанием масел
(50—59%), образуют твердые, хрупкие, с высоким глянцем
пленки со сравнительно малой стойкостью к атмосферным
воздействиям — около года; эмали на жирных смолах обра-
зуют эластичные, твердые и атмосферостойкие покрытия.
Высыхают при 20° за 24 часа, при 100—120° в течение
1,5—2 часов. После горячей сушки приобретают высокую
водостойкость, выдерживают периодич. воздействие минераль-
ных масел, керосина, бензина. Применяют для окраски изде-
лий, подвергающихся атмосферным воздействиям: автомоби-
лей, с.-х. машин, а также различных приборов и электрома-
шин. Специальные марки эмалей, пигментированных алюми-
ниевой пудрой, обладают термостойкостью до 350°. Тиксо-
тропные алкидные эмали, помимо глифталевых или пентафта-
левых смол, содержат совмещенную с ними низкомолекуляр-
ную полиамидную смолу, вызывающую образование гелеоб-
разной структуры вследствие возникновения водородной связи
полиамидной цепи с карбоксильными группами алкида. Рас-
творитель — уайт-спирит. При встряхивании или перемеши-
вании К. переходит в жидкое состояние, а затем обратно
в гелеобразное. Тиксотропные эмали имеют следующие пре-
имущества перед обычными; при хранении пигменты не осе-
дают, не требуется разжижение перед употреблением, К. не
капают и не стекают с кисти, допускается нанесение эмали
слоем примерно в два раза толще, чем для обычных К. Пред-
назначаются в основном для окраски пористых поверхностей
(строительных материалов) и антикоррозийной защиты метал-
лич. изделий. Эмульсионные алкидные эмали представляют
собой суспензию пигментов в эмульсии, состоящей из глифта-
левого лака, воды и эмульгатора, с добавлением сиккативов
и растворителя. Перед нанесением эмали разбавляют уайт-
спиритом или же сольвентом, скипидаром, ксилолом в коли-
честве до ,15%. В первой стадии высыхания К. улетучиваются
растворители и вода. Частички эмульсии сливаются, образуя
сплошную, монолитную красочную пленку. Пленка перестает
быть липкой уже через два часа, полное высыхание происхо-
дит в течение 24 часов. Применяются для окраски внутренних
поверхностей помещений по штукатурке и древесине. Эмуль-
сионные эмали более экономичны и менее вредны, чем обыч-
ные алкидные эмали.
Эмали фенольно-масляные и фенольно-
ал к ид н ы е (ФЛ, ФА) готовят на масляных или алкидных
754
I лаках; пленкообразующее — феноло-альдегидные смолы, сов-
мещенные с растительными маслами (ФЛ), или модифициро-
ванные высыхающими алкидными смолами (ФА), Темп-ра
сушки 140—160° (в течение 1—2 часов). Пленки покрытия
обладают высокой твердостью, отличной адгезией, водо-.
масло- и бензостойкостью, удовлетворительной стойкостью
к действию слабых кислот и малой щелочестойкостью. Вслед-
ствие темной окраски связующего эмали приготовляют только
темных цветов. Применяются для окраски приборов и машин,
эксплуатирующихся в любых климатич. условиях.
Эмали эпоксидные (ЭП) готовят на основе эпо-
ксидных лаков. В качестве отвердителей применяют гексаме-
тилендиамин, полиэтиленполиамин, низкомолекулярные по-
лиамиды или аддукты (модифицированные амины), ортофос-
форную к-ту, а также феноло-, мочевино- или меламино-фор-
мальдегидные смолы. Все отвердители вводят в эмалевую
К. в строго определенных количествах. Феноло-, мочевино- и
меламино-формальдегидные смолы вводят в процессе при-
готовления лака, получаемые затем эмали имеют длительную
жизнеспособность (однокомпонентные эмали). Темп-ра отвер-
ждения их — не ниже 150°. Амины и другие отвердители вво-
дят в К. перед употреблением. Такие двухкомпонентные
эмали после смешения с отвердителем пригодны к употребле-
нию в течение 8—20 часов, после чего они загустевают.Пл<нка
образуется при 15° и выше. Сушка при нагревании до 80—
дает более качественные покрытия. Растворители — кетоны,
эфиры и ароматич. углеводороды. При отверждении эпоксид-
ных эмалей образуется пленка сетчатого строения, обладаю-
щая высокой адгезией, водостойкостью, твердостью, стой-
костью к концентрированным горячим щелочам и слабым к-там.
Термостойкость пленок эпоксидных эмалей: длительная при
180—200° и кратковременная до 250°. Эпоксидные эмали при-
меняют для окраски различных приборов, машин и химич. аппа-
ратуры, эксплуатируемых в любых климатич. условиях. Эмали,
содержащие амины, обладают значительной токсичностью.
Эмали эпоксиэфирные (ЭФ) приготовляют на
эпоксидных смолах, этерифицированных жирными к-тами
высыхающих масел или кислыми алкидами, модифицирован-
ными жирными к-тами. Растворители — кетоны, эфиры и
ароматич. углеводороды. В отличие от эмалей ЭП эмали ЭФ
не требуют отвердителей и высыхают с добавлением сиккативов
при-(-15° и выше. Пленки эмалей ЭФ обладают высокой атмо-
сферостойкостью, эластичностью, хорошим внешним видом,
но несколько худшей, чем эмали ЭП, химич. стойкостью и
водостойкостью. Применяются для окраски приборов и машин,
эксплуатируемых в любых климатич. условиях. Токсичность
их значительно меньше, чем эмалей ЭП.
Эмали полиуретановые (УР) готовят на
полиэфирах, содержащих гидроксильные группы, способные
взаимодействовать с диизоцианатами, образуя полиуретаны
(уретанизация). Сразу же после смешения пигментированного
р-ра полиэфира в органич. растворителях (смесь кетонов, эфи-
ров и ароматич. углеводородов) со вторым компонентом —
диизоцианатом — начинается уретанизация. Такая смесь
пригодна к употреблению только в течение 6—8 часов. Поэтому
эмаль выпускают в виде двух компонентов, смешиваемых перед
употреблением или при нанесении двухсопловыми распылите-
лями (т. наз. двухкомпонентная эмаль). В пленке эмали обра-
зование полиуретанов особенно энергично происходит при
80—120°. Если применять блокированные малоактивные изо-
цианаты, то значительно уменьшается токсичность материала.
Такая полиуретановая эмаль может сохраняться длительное
время. В этом случае изоцианат становится химически актив-
ным только после нагрева до 140—160° (т. наз. однокомпонент-
ные эмали). Пленки полиуретановых эмалей, особенно после
горячей сушки, обладают хорошей адгезией к различным ме-
таллам, высокой эластичностью, водостойкостью, твердостью,
абразивостойкостью, исключительной щелочестойкостью и
бензостойкостью, высокими диэлектрич. свойствами; их тер-
мостойкость — в пределах 140—180°. Пригодны для защиты
химич. аппаратуры различных приборов и машин, эксплуати-
руемых в нормальных и тропич. условиях.
Эмали мочевино-формальдегидные (МЧ)
готовят на лаках, содержащих мочевино-формальдегидные
смолы, совмещенные с алкидными смолами. В качестве рас-
творителей применяют смесь бутанола с ксилолом. Прозрач-
ность и светостойкость мочевино-формальдегидных смол по-
зволяет готовить на их оенпве эмали любых расцветок. Сушка
эмалей горячая, с постепенным подъемом темп-ры до 120°;
при этом происходит поликонденсация с образованием нерас-
творимых пленок. Эмали склонны к коагуляции, устраняе-
мой нагреванием. Благодаря высокой светостойкости, твер-
дости, эластичности их широко применяют для окраски раз-
личных приборов, машин и оборудования; наносят обычными
методами.
Эмали меламино-формальдегидные (МЛ)
готовят на лаках, содержащих меламино-формальдегидные
смолы, совмещенные с алкидными смолами. Пленки обладают
длительной цветостойкостью при нагреве до 100° и кратковре-
менной — при 150°. Сушка при +120° с постепенным подъемом
темп-ры. Превосходят эмали МЧ по стойкости к атмосферным
воздействиям, водостойкости и щелочестойкости, по скорости
сушки. Широко применяются для окрарки холодильников,
стиральных машин, осветительной аппаратуры, автомобилей,
медицинского оборудования и т. д.
Эмали перхлорвиниловые (ХВ) готовят на
основе перхлорвиниловых лаков. В качестве растворителей
755
КРАСКИ
756
применяют кетоны, ацетаты и ароматич. углеводороды. Пленки
эмалей обратимы и термопластичны, если смола не образовала
трехмерной структуры (что бывает при нагревании до
130—140°). Время высыхания эмалей при 12—20° 2—3 часа.
Нанесение К. и сушка возможны при темп-ре не ниже +5°.
Покрытия обладают большой стойкостью к атмосферным воз-
действиям (до 6 лет) в системе с акриловыми или алкидными
грунтами. К. светлых расцветок под действием солнечного света
желтеют. Устойчивы к действию р-ров щелоцей (с концентра-
цией до 40—50%) и кислот (с концентрацией 30—40%), р-ров
солей, промышленных газов, бензина, керосина и минеральных
масел. В эмалях, стойких к химич. средам, применяют инертные
пигменты — железный сурик, окись хрома, титановые белила.
Термостойкость ограниченная (до 100°) вследствие отщепления
хлористого водорода при дальнейшем нагревании. Это может
активизировать коррозию металла под слоем лакокрасочного
покрытия. Сильно повышает термостойкость введение стабили-
заторов, особенно эпоксидных смол. Эмали ХВ применяют для
антикоррозионных покрытий железнодорожных вагонов, само-
летов, с.-х. машин, металлоконструкций, работающих в любых
климатич. условиях, а также для защиты химич. аппаратуры.
Эмали на основе растворов сополиме-
ров винилхлорида (ХС). В качестве пленкообра-
зующих веществ наиболее часто применяют: а) Сополимер
винилхлорида с винилиденхлоридом. Эти эмали близки по
свойствам к перхлорвиниловым, но обладают лучшей адге-
зией, водостойкостью и несколько большей химич. стойкостью,
особенно в кислых средах. Применяют в основном для окраски
химич. аппаратуры, б) Сополимер винилхлорида с винилаце-
татом, иногда с добавкой алкидных или меламино-формальде-
гидных смол. Пленки обладают хорошей адгезией и повышен-
ной светостойкостью, ограниченно бензостойки, пригодны для
окраски изделий, эксплуатируемых на открытом воздухе.
Высыхают в течение 1—2 часов.
Эмали масляно-стирольныеи алкидно-
стирольные (МС) на основе р-ров сополимеров масел
или алкидов со стиролом; в качестве растворителей применяют
ароматич. углеводороды. В зависимости от жирности лака
эмали бывают быстросохнущими при комнатной темп-ре или
же горячей сушки. Пленки обладают высокой светостойкостью
и термостойкостью (примерно до 150°), высокими диэлектрич.
свойствами (е = 5—6), водостойкостью, стойкостью к действию
слабых к-т и щелочей. Применяют в качестве электроизоля-
ционных покрытий, для печати по бумаге, для окраски дре-
весины, стиральных машин, станков и т. д.
Эмали кр емн ийо р ган ическ ие (КО, поли-
силоксановые, силиконовые) готовят на основе кремнийорга-
нич. лаков, в к-рые иногда вводят еще эпоксидные, алкидные,
акриловые смолы или этилцеллюлозу. Применяют термостой-
кие пигменты: титановые белила, зеленую окись хрома, строн-
циевый крон, окислы железа, кадмопоны, окись кобальта,
алюминиевую пудру, цинковую пыль и др. Растворители —
смесь толуола, ксилола с ацетоном, этил- и бутилацетатом.
При сушке в течение 2—3 часов при 200—250° пленкообра-
зующее вещество приобретает сетчатое строение, и эмаль ста-
новится устойчивой и к воздействию минеральных масел и
растворителей. Эмали, пигментированные алюминиевой пуд-
рой, обладают длительной термостойкостью при 550° и кратко-
временной — при 700°, цветные эмали — длительной термо-
стойкостью при 400°, кратковременной при 500° без суще-
ственного изменения цвета и защитных свойств. Обладают
хорошими электроизоляционными свойствами, сохраняющи-
мися в условиях высокой темп-ры и большой влажности.
Стойкость к атмосферным воздействиям — ок. двух лет.
Покрытия склонны к растрескиванию при толщине пленки
выше 40—50 мк. Применяют для антикоррозионной защиты и
электроизоляции приборов и машин, подвергающихся нагреву.
Эмали на основе акриловых смол (АК)
и акриловых сополимеров (АС) готовят, исполь-
зуя акриловые лаки, содержащие небольшие количества пла-
стификатора. Растворители — кетоны, эфиры, ароматич. угле-
водороды. Пленки обладают исключительно большой свето-
стойкостью, водостойкостью, хорошей адгезией и твердостью.
Высыхают при комнатной темп-ре. Пленки термопластичны и
неустойчивы к действию органич. растворителей. Термостой-
кость — до 180°, стойкость к атмосферным воздействиям —
до 3 лет. Применяют для окраски приборов, машин и самоле-
тов. Эмульсионные акриловые эмали представляют собой
эмульсии типа «масло в воде», содержат эмульгатор, стабили-
затор, загуститель. Содержание смолы в эмульсии ок. 45—48%,
водной фазы 9,0—9,5%. Эмульсионные К. применяют для
окраски пористых материалов, древесины, штукатурки, бе-
тона, бумаги. Пленки устойчивы к действию воды и атмосфер-
ным воздействиям. Благодаря ничтожному содержанию орга-
нич. растворителей экономичны, безопасны и безвредны.
Эмали эфироцеллюлозные: нитроцеллюлоз-
ные (НЦ), этилцеллюлозные (ЭЦ), бензилцеллюлозные (ВЦ),
ацетилцеллюлозные (АЦ), ацетобутиратцеллюлозные (АБ).
Готовят на соответствующих лаках. Эфироцеллюлозные эмали
высыхают при 12—20° в течение 30—60 мин. Покрытия эма-
лями НЦ обладают хорошей стойкостью к атмосферным воз-
действиям, водо-, бензо- и маслостойкостью, хорошо поли-
руются, имеют хорошую адгезию к древесине и металлу. Недо-
статок — сильная горючесть, низкая термостабильность (до
80°) и склонность к старению при действии солнечных лучей.
Применяют для окраски автомобилей, различных приборов и
машин, кожи, тканей, деревянных изделий. Покрытия эма-
лями ЭЦ и АБ термостойки до 100—120°, их горючесть в де-
сятки раз меньше, чем нитроцеллюлозных. Применяют для
окраски тканевых обшивок на самолетах. Эмали НЦ, ЭЦ
и БЦ применяют также в качестве электроизоляционных по-
крытий. С увеличением содержания алкидной высыхающей
смолы (до 50—100% по отношению к нитроцеллюлозе) НЦ
по свойствам приближаются к алкидным эмалям, сохраняя
способность быстро высыхать. Такие эмали называют нитро-
алкидными. Благодаря повышенному содержанию пленко-
образующего, лучшей адгезии, блеску и др. свойствам нитро-
алкидные эмали широко применяют.
Эмали див ин и л а цетил ено в ые (ВН) готовят
на дивинилацетиленовом лаке. Эти смолы иногда содержат
добавку битумов, эпоксидных или перхлорвиниловых смол,
а также пластификаторов — хлорпарафина, дибутилфталата,
хлорированного дифенила и др. Добавка пластификатора
несколько снижает водостойкость пленки эмали. Раствори-
тели: ксилол, сольвептнафт. Для предотвращения окисления
полимера при хранении вводят антиоксидант. Высыхание
эмали происходитв течение 1—2 часов при темп-ре от —10э до
+ 20° и сопровождается образованием сетчатого полимера.
Пленки обладают длительной стойкостью к воздействию воды,
кислот: серной, соляной, азотной и щелочных растворов;
устойчивы к действию бензина, масла и к микологическим
воздействиям. Широко применяются для окраски металлич.
конструкций, эксплуатируемых в воде и во влажных условиях
без воздействия солнечных лучей (подводная часть кораблей,
трюмы и т. п.).
Эмали поливинилацетальные (ВЛ) готовят
па основе растворов поливинилбутираля в смеси с резольными
или меламино-формальдегидными смолами. Растворители —
этиловый спирт, эфиры, ароматич. углеводороды. После сушки
при 120—150° образуется очень твердая пленка, стойкая
к воде, бензину, керосину, маслам, и с высокой адгезией к ме-
таллу. Термостойкость 200—250°. Эмали ВЛ применяют для
окраски металлич. изделий, подвергающихся длительному
воздействию бензина, керосина, масла, горячей воды и тропич.
атмосферы.
Эмали поливинилацетатные (ВА) готовят
на основе растворов поливинилацетатной смолы в органич.
растворителях с добавками фенольных смол или эфиров цел-
люлозы или эмульсий их в воде. Эмали первого типа приме-
няют сравнительно редко. Эмульсионные эмали состоят из
тонкодисперсной, пластифицированной эмульсии смолы (диа-
метр частиц 0,3—4 мк), пигмента, эмульгатора, стабилизатора
и дистиллированной воды. Различают т. наз. однокомпоненг-
ные и двухкомпонентные эмали ВА. Однокомпонентная содер-
жит все компоненты К. и после разведения водой пригодна
к употреблению; двухкомпонентная состоит из двух отдельных
частей: красочной пасты и эмульсии с пластификатором, к-рые
перед употреблением смешивают. Эмульсионные эмали высы-
хают в течение 1—2 часов при -f-15°. Пленка матовая, эла-
стичная, с хорошей адгезией, светостойкая, сравнительно
неводостойкая. Применяют для окраски кистью, распылением
или валиком пористых поверхностей, тканей, кожи, строитель-
ных материалов и металлич. поверхностей, предварительно
загрунтованных. Благодаря высокому содержанию пленко-
образующего, отсутствию органич. растворителей и дешевизне
эмульсионные К. ВА весьма перспективны.
Эмали битумные (БТ). Пленкообразующими яв-
ляются асфальты, битумы или их смеси, растворенные в расти-
тельных маслах с добавкой сиккативов. В качестве пигментов
применяют сажу, милорь, алюминиевую пудру. Раствори-
тели: ксилол, толуол, сольвент или уайт-спирит. Благодаря
темной окраске пленкообразующего эмали изготовляют только
черного цвета. Пленки, полученные при 100—160°, обладают
высокой прочностью, хорошей адгезией, водостойкостью и
термостойкостью (до 150—180°), устойчивы к воздействию
слабых растворов соляной, серной и уксусной к-т, мало устой-
чивы к воздействию солнечного света. Эмали, пигментирован-
ные алюминиевой пудрой, благодаря большой отражательной
способности частичек алюминия, более атмосфероустойчивы.
Применяются для окраски металлич. изделий, подвергаю-
щихся периодич. увлажнению и загрязнениям, без воздействия
прямого солнечного света.
Художественные К. и применяемые в живописи К.
подразделяют на акварельные, гуашевые, темперные,
масляные. Эти К. содержат светостойкие пигменты.
Акварельные К. являются разновидностью клее-
вых. Пленкообразующим в них служат водорастворимые
камеди (гуммиарабик, вишневый клей). В качестве пластифика-
торов вводят глицерин и т. д. Акварельные К. наносят на
бумагу.
Гуашевые К. готовят на водорастворимом связую-
щем — декстрине. К. укрывисты, их наносят на бумагу, кар-
тон, ткань.
Темперные К. готовят на водной эмульсии яичного
желтка, гуммиарабика (или вишневого клея) или казеина и
масла. Темперные К. после высыхания нерастворимы. Их при-
меняют в основном для живописи по дереву, т. к. для холста
они недостаточно эластичнее
Масляные К. готовят растиранием пигментов на ра-
финированном отбеленном льняном масле, иногда с добавкой
орехового или подсолнечного. При высыхании они претерпе-
вают необратимые превращения. Применяют по грунтован-
ному холсту и древесине.
757
КРАСКИ — КРАСЯЩИЕ ВЕЩЕСТВА ПРИРОДНЫЕ
758
Декоративные К. служат для окраски деталей
приборов и аппаратуры и отличаются своеобразным
красивым рисунком. К декоративным К. относятся:
а) эмали «Муар», образующие после горячей сушки
морщинистый рисунок. Они содержат и качестве
пленкообразующих веществ алкидные смолы и тун-
говое масло, б) Молотковые эмали, образующие ри-
сунок, напоминающий вмятины от чеканки молотком.
Их готовят на основе меламино-формальдегидной смо-
лы, совмещенной с алкидной, с добавлением алюминие-
вой пудры и рисункообразователя — кремнийорга-
нич. жидкости, в) Трескающиеся эмали, образующие
рисунок наподобие крокодиловой кожи. При нане-
сении на поверхность грунта эмаль в процессе высы-
хания растрескивается, и в местах трещин становятся
видными прожилки грунта. Грунт и трескающиеся
эмали готовят на основе нитроцеллюлозы.
Светящиеся К. обладают способностью к люмине-
сценции (холодному свечению). С этой целью в лаки
вводят люминофоры — вещества, способные к свече-
нию. Различают флуоресцирующие и фосфоресцирую-
щие К. Флуоресцирующие акриловые К. содержат спе-
циально приготовленные органич. красители. Пленка
такой эмали обладает большой яркостью свечения
при освещении ее УФ-лучами. Применяются для
повышения видимости наземного и воздушного транс-
порта в условиях тумана и слабой освещенности, а
также на рекламных вывесках.
Фосфоресцирующие К. существуют двух видов:
а) постоянного действия, содержащие радиоактивные
вещества и не требующие источника световой энер-
гии; б) временного действия, содержащие сульфиды
цинка, кадмия, кальция с добавками солей меди
и серебра (активаторы). Такие К. для свечения тре-
буют предварительного возбуждения дневным светом.
Первые применяют для обозначения цифр и знаков на
часах и приборах. Вторые—в качестве декоративных К.
Термочувствительные К. служат для контроля
темп-ры на поверхности окрашенного изделия. При-
меняются в виде пастообразных карандашей или К.,
где в качестве связующего применены р-ры смол
(шеллак или синтетич. смолы). Существуют два вида
термочувствительных К.: обратимые и необратимые.
У обратимых (рабочая темп-ра до 100°) цвет после
охлаждения превращается в первоначальный. Содер-
жат в качестве пигмента двойные соли иодистоводо-
родной к-ты, к-рые при нагревании переходят из одной
модификации в другую, а после охлаждения сразу
возвращаются в первоначальную модификацию с со-
хранением начального цвета. Применяются также
соединения солей кобальта и никеля с гексаметиленте-
трамином. При нагревании такие соединения теряют
кристаллизационную влагу, а после охлаждения
снова поглощают влагу из воздуха и через 2—4 часа
восстанавливают свой цвет. Существуют К. для темп-р
в пределах от 35 до 100° с интервалом 5—10°.
Применяют как сигнальные К. в пром-сти для кон-
троля темп-p на поверхностях котлов, теплоизоля-
ции, корпусов подшипников и т. д. Необратимые К.
(рабочая темп-ра до 950°) после охлаждения не при-
обретают первоначальный цвет. Изменение цвета
происходит в результате химич. процессов, приводя-
щих к образованию новых соединений, окрашенных
в другие цвета, напр. NiNH4PO4 . 6Н2О при 120°
меняет цвет из светло-зеленого в серо-зеленый,
CONH4PO4 • Н2О при 140° — из пурпурно-красного
в темно-голубой, Cd(OH)2 при 200° из белого перехо-
дит в желтый, а СиСО3 при 400° из светло-зеленого
в темно-коричневый, и т. д. Существует набор К.,
работающих до 950° с интервалом темп-р 30—50 —
100°. Применяют для контроля степени нагрева там,
где установка термопар невозможна или сложна,
напр. внутри тепловых аппаратов, двигателей и т. д.
Необрастающие К. наносят на подводную часть
морских судов по слою антикоррозионного грунта
для защиты обшивки от обрастания раковинами мор-
ских организмов, сильно замедляющими скорость хода
судов. Такие К. содержат закись меди, окись ртути
и др. вещества, способные реагировать с хлористым
натрием, содержащимся и морской воде, с образова-
нием водорастворимых соединений, токсичных для
морских организмов. Связующим служат лаки КФ,
ХВ, ХС и др.
О К., применяемых в полиграфия, произ-ве, см.
Печатные краски.
Лит.: Дринберг А. Я., Технология пленкообра;
зующих веществ, Л.—М., 1948; Д р и н б е р г А. Я., Гуре-
вич Е. С., Тихомиров А. В., Технология неметалли-
ческих покрытий, Л., 1957; Беленький Е. Ф., Рис-
кин И. В., Химия и технология пигментов, 3 изд., М.,
1960; Якубович С. В., Испытания лакокрасочных мате-
риалов и покрытий, М.—Л., 1952; Любимов Б. В., Спе-
циальные лакокрасочные покрытия в машиностроении, М,—Л.,
1959; Органические защитные покрытия. Сб. статей, пер. с
англ., М,—Л., 1959; Лакокрасочные материалы. Справочник,
М., 1961; Пэ йн Г. Ф., Технология органических покрытий,
пер. с англ., т. 1, М., 1959; Ш ампетье Г., Рабат э Г.,
Химия лаков, красок и пигментов, пер. с франц., т. 1—2,
М., 1960—62; Чеботаревский В. В., Лаки и краски
в народном хозяйстве, .М., 1960. В. В. Чеботаревский.
КРАСЯЩИЕ ВЕЩЕСТВА ПРИРОДНЫЕ — слож"
ные органич. соединения, вырабатываемые живыми
организмами и окрашивающие различные животные
и растительные клетки и ткани. До 2-й половины
19 в. многие К. в. п. применялись для окраски тек-
стильных изделий, кожи, бумаги, парфюмерных изде-
лий, пищевых продуктов и др. С развитием пром-сти
органич. синтеза, и особенно анилино-красочной про-
мышленности, К. в. п. не выдержали конкуренции
с красителями синтетическими и в подавляющем
большинстве случаев утратили былое практич. зна-
чение. Однако значение К. в. п. не исчерпывается их
практич. использованием для целей крашения. Боль-
шинство из них играет существенную роль в жизни
организмов, к-рыми они продуцируются. Многие
К. в. п. обладают значительной физиология, и анти-
биотич. активностью и часто применяются как ле-
чебные средства.
К. в. п. широко распространены и в химич. отно-
шении крайне многообразны. Классификация их
возможна по различным признакам, напр. по проис-
хождению и нахождению в природе (животные, расти-
тельные, крыльев бабочек, цветов и т. д.). Во мно-
гих случаях в различных природных источниках
встречаются одни и те же или близкие по строению
К. в. п., и поэтому наиболее целесообразной классифи-
кацией (принятой и в данной статье) является обычная
классификация органич. соединений по функциональ-
ным признакам. В приведенных ниже структурных
формулах заместители расшифрованы только для
индивидуальных соединений.
К. в. п. алифатического и алициклического рядов.
Сюда в основном относятся т. наз. каротиноиды,
содержащие цепи углеродных атомов с системой конъ-
югированных двойных связей, наличие к-рой и обу-
словливает окраску. С увеличением длины конъюги-
рованной системы окраска углубляется. Среди краси-
телей этой группы встречаются углеводороды (в том
числе с алициклич. ядрами на концах углеродной
цепи), спирты (иногда встречающиеся в виде сложных
эфиров), кетоны, кетоспирты, альдегиды, кислоты,
сложные эфиры окиси и др. Встречаются каротино-
иды как в растительном, так и в животном мире. Мно-
гие из них, как, напр., каротин, обладают высокой
физиология, активностью (провитамин А). К поли-
енам неустановленного строения относится ряд ан-
тибиотиков (канацидин, пентамицин, филипин, ме-
лиоцидин, кандимицин, трихомицин и др.), проду-
цируемых различными видами Streptomyces.
759
КРАСЯЩИЕ ВЕЩЕСТВА ПРИРОДНЫЕ
760
К. в. п. ароматического ряда. В основном эти кра-
сители представляют собой окси- и алкоксизамещецные
непредельные кетоны, производные диароилметана
[4,4'-диоксидициннамоилметан, т. пл. 224°, желтого
цвета; 4-оксицпннамоилферулоилметан, т. пл. 168°,
оранжевого цвета и диферулоилметан (куркумин),
т. пл. 181—183°, красного цвета, встречающиеся в
корнях Curcuma longa, С. tinctoria и др.], халковы
(I), хиноны: бензохиноны (II), нафтохинопы (III),
антрахиноны (IV), антроны: хризоробин (1,8-диокси-
З-метилантрон-9), т. пл. 203,5—204°, желтый краси-
тель древесины Andi га araroha и эмодипантрон
(1, 6, 8-триокси-3-метилаптрон-9), т. пл. 250—258°, жел-
тый краситель древесины Rhamnus dahurica. Многие
из этих красителей в природе встречаются в виде
।
О
О
гликозидов. Антрахинонам близки по строению гипе-
рицин [V, R=CH3] и псевдогиперицин [V, R =
= СН(ОН)СН3], темно-фиолетовый и . темно-красный
красители цветов Hypericum perforatum, обладающие
фотодинамич. свойствами. Среди К. в. п. ароматич.
ряда имеются также соединения, обладающие физио-
логии. и антибиотич. активностью (папр., витамин К,
куркумин, многие хиноны и др.). К частично гидри-
рованным ароматич. К. в. п. относятся антибиотики
группы тетрациклина, окрашенные в желтый цвет.
Многие К. в. и. ароматич. ряда раньше широко
применялись для целей крашения, напр. ализарин
(основное красящее вещество краппа, содержащего
также пурпурин, ксаитопурпурин, мунджистип и
рубиадин), картамин, куркумин (применяемый в на-
стоящее время в качестве индикатора в ацидометрии),
кошениль и др.
К. в. п. гетероциклического ряда, а) Кислород-
содержащие гетероциклы. Известны
К. в. п., содержащие фурановое ядро, напр. краси-
тель понгамон, т. пл. 128—129° — 4-метокси-5-бен-
зоилацетилбензофуран, выделенный из корня Те-
phrosia lanceolate и др. К производным дигидрофу-
рапа относятся лактон пульвицовой к-ты (VI, R = II),
т, пл. 221—222°, светло-желтый краситель лишайника
Sticta coronata и др., салицин (VI, R=OH), т. пл.
244—245°, оранжево-красный краситель лишайника
Calycium chrysocephalum и др. красители (напр.,
VIII), а также кумарононы (VII) — 5-членные цик-
лнч. изомеры халкопов.
Наиболее широко представлены К. в. п., производ-
ные хромана. К ним относятся флаваноиды: флавоны
(IX, Х=Н), флаванолы (IX, Х=ОН, ОСН3 или OSO2K),
флаванолы (X, Х=Н) и флаванонол (X, X =ОН),
антоцианы (XI) и изофлавоны (XII). Среди К. в. п.
этой группы имеются производные кумарина, напр.
еллагоная к-та (XIII, R=OH), широко распростра-
ненная в древесине многих пород, напр. Quebracho
Colorado, ее дпметиловый эфир (XIII, R —ОСН3),
т. пл. 337—338°, встречающийся в корнях Euphorbia
formosana Hag, и др. К этой же группе относятся
ксантоны (XIV).
Кислородсодержащим гетероциклом оказалась так-
же телефоровая к-та (XV), черно-фиолетовый краси-
тель многих видов Thelephora, к-рому ранее приписы-
валось строение производного фенантрена. Несколько
обособленно стоят бразилип (XVI, R=H) и гематок-
силин (XVI, R = OH), выделенные из древесины крас-
ного дерева (см. Leguminosae) и кампешевого дерева
(Haematoxylon campechianum). Эти вещества, будучи
сами бесцветными, широко применялись для краше-
ния хлопка по протраве (синевато-красные и фиоле-
товые выкраски).
Многие К. в. п. рассмотренных групп встречаются
в виде гликозидов (нек-рые исключительно). Чаще
всего углеводом является глюкоза, рамноза, галактоза,
арабиноза и др.
б) Азотсодержащие гетероциклы.
Производные индола. К числу самых
старых К. в. п., применявшихся для крашения, от-
носятся индиго и пурпур древних (античный пурпур).
Индиго добывалось из листьев тропич. растения Indi-
gofera tinctoria, а также Isatis tinctoria и Polygonum
tinctorium, произрастающих в Западной Европе и
па юге СССР. В растениях содержится не само ин-
761
КРАСЯЩИЕ ВЕЩЕСТВА ПРИРОДНЫЕ — КРАХМАЛ
762
диго, а глюкозид индоксила — индикан, распадаю-
щийся при гидролизе на глюкозу и индоксил, окисляю-
щийся кислородом воздуха в индиго. Ему сопутствует
изомер индирубин (также образующийся из индо-
ксила) и другие К.в.п. (в том числе кемферол). Пур-
пур древних, содержащийся в моллюсках Мпгех Ьап-
daris, является 6,6'-диброминдиго.
Производные пиррола. Сюда относятся
три важнейшие группы природных пигментов: пиг-
менты крови (гемоглобин), хлорофиллы и желчные
пигменты. Все они содержат скелет, состоящий из
4 пиррольных ядер, связанных метиновыми мости-
ками, только в первых двух система замкнута в т.
наз. порфириновый цикл, а в последних—пиррольные
ядра расположены линейно.
С ogHogOOC
XVIII
ноос—сн, сн2~соон
I “ I
В природе встречаются различные порфирины
(XVII). Близок по строению к протопорфирину (одна
винильная группа заменена на формильную) хло-
рокруоропорфирин — простетич. группа пигмента хло-
рокруорина из Spirographis. В хлоропластах зеленых
частей растений наряду с каротином и ксантофиллом
содержатся в соотношении 3:1 два пигмента, близкие
по строению к пигментам крови:сине-зеленый хлоро-
филл а (XVIII, R=CH3) и желто-зеленый хлорофилл
б (XVIII, R=CHO), играющие важнейшую роль в
фотосинтезе. Кроме того, в растениях содержится про-
тохлорофилл а (XVIII, Н=С2Н5), у к-рого кольцо А
дегидрировано. К этой же группе К. в. п. относится
широко распространенный в природе витамин BX2,
содержащий комплексно связанный кобальт (см.
Цианкобаламин). К желчным пигментам относятся
билирубин (XIX, R = СН=СН2), мезобилирубин (XIX,
Н=С2Н5) и уробилин, отличающийся от последнего
тем, что ядра А, Б и В гидрированы. К производным
пиррола относится также красный пигмент бактерий
Serratia marinorubra (XX), т. пл. 151—152°.
Производные феназина — желтые кра-
сители гемипиоцианин (1-оксифеназин), т. пл. 158°,
а-феназинкарбоновая к-та, т. пл. 242° и пиоцианип
(1-оксо-9-метилфеназин), т. пл. 242°, темно-синего
цвета, содержащиеся в Pseudomonas pyocianea и Р.
aureofaciens. К этим К. в. п. примыкает также широко
распространенный в природе витамин В2 (лактофла-
вин, рибофлавин) — 9-1Э-рибитил-6,7-диметилизоалло-
ксазин.
Производные птеридина (XXI), впер-
вые обнаруженные в пыльце крыльев бабочек, также
оказались довольно широко распространенной груп-
пой К. в. п.
Лит.: Майер Ф., Естественные органические красящие
вещества, пер. с нем., М., 1940; Вильштедт Г., Кароти-
ноиды и красящие вещества бактерий и грибов, пер. с нем.,
М., 1936; Венкатараман К., Химия синтетических
красителей, пер. с англ., т. 2, Л., 1957, с. 892—93, 914—18,
950—54; Ш и м а н Г., Химия естественных и искусственных
красителей, пер. с. нем., М., 1939; Вульфсон Н. С.,
Усп. химии, 1948, 17, вып. 2, 249;. Березовский В. М.,
там же, 1953, 22, вып. 2, 191; К а г г е г Р., в сб.: Fortschritte
der Chemie organischer Naturstoffe, Red. L. Zechmeister, Bd 5,
Wien, 1948, S. 1; Inhoffen H., S 1 e m e r H., там яге,
Bd 9, 1952, S. 1; Zechmeister L., там же, Bd 18, 1960.
S. 223; Hoffnian-Ostenhof О., там же, Bd 6, 1950,
S. 154; Brockmann H., там же, Bd 14, 1957, S. 141,
Venkataraman К., там же, Bd 17, 1959, S. 1;
Schmid H., там же, Bd 11, 1954, S. 124; F r e u d e n-
li erg K., Welnges К., там же Bd 16, 1958, S. 1; L e m-
b e г g R., там же, Bd 11, 1954, S. 299; Schroeder W. A.,
там же,Вй 17, 1959, S. 322; Stoll A., Wiedemann E.,
там же, Bd 1, 1938, S. 159; S i e d e 1 W., там же, Bd 3, 1939,
S. 81; R u d у H., там же, Bd 2, 1939, S. 61; Albert A.,
там же, Bd 11, 1954, S. 350; VO 1 k er О., там же, Bd 18,
1960, S. 177; Geissman T., The chemistry of flavonoid
compounds, L., 1962; Bentley K. W., The natural pigments,
N. Y., 1960. H. С. Вульфсон.
КРАТНЫХ ОТНОШЕНИЙ ЗАКОН — Один пз
основных законов химии, заключающийся в том, что
если 2 элемента образуют 2 или несколько химич.
соединений, то в них на одно и то же количество одной
составной части приходятся количества другой, отно-
сящиеся между собой как простые целые числа. При-
мером могут служить метан СН4 и этилен С2Н4,
количества углерода в к-рых, приходящиеся на 1 ве-
совую часть водорода, относятся как 1 к 2. К. о. з.
выведен в 1803 Дж. Дальтоном.
КРАХМАЛ — смесь полисахаридов, встречающих-
ся в растениях в виде зерен; с иодом дает харак-
терное синее окрашивание; белый порошок. К. —
самый распространенный запасной углевод расте-
ний; образуется в листьях в результате фотосинтеза
и откладывается в корнях, клубнях и семенах в виде
зерен, имеющих величину, форму и внутреннее
строение, характерные для каждого вида растения.
Крахмальные зерна неоднородны, помимо полисаха-
ридов они содержат воду (10—20%) и в очень неболь-
ших количествах фосфаты, кремнезем, жирные к-ты,
липиды и др. Полисахариды К. состоят из остатков
D-глюкозы в ее a-D-глюкопиранозной форме и отли-
чаются степенью полимеризации и характером свя-
зей a-D-глюкопиранозных единиц. Полисахариды К.
можно разделить на 2 главные фракции: амилозу
и амилопектин. (Схему строения их макромо-
лекул см. на рис. 1).
Макромолекулы амилозы представляют собой линей-
ные или очень слабо разветвленные цепи, состоящие
из 200—1000 остатков D-глюкозы (см. рис. 1,а), к-рые
связаны между собой а-1,4-глюкозидными связями
(рис. 2), в местах ветвления глюкозные остатки
в амилозе образуют а-1,6-связи.
В амилозе содержится также небольшое количество
Р-1,2-, Р-1,3- и р-1,4-глюкозидных связей. По типу
строения амилоза похожа на клетчатку, отличаясь
от последней наличием a-связей (в клетчатке глюко-
пиранозные остатки образуют p-связи) и пространст-
венной конфигурацией макромолекул; макромолекулы
амилозы образуют спирали. Мол. вес полисахаридов
фракции амилозы — от 32 000 до 160 000.
Макромолекулы амилопектина сильно разветвлены
(рис. 1,6), они содержат от 600 до 6000 остатков D-глю-
козы, связанных между собой а-1,4-глюкозидными
связями, а в местах ветвления а-1,6 связями (рис. 3).
В амилопектине обнаружено также небольшое коли-
763
КРАХМАЛ
764
чество 1,3-связей. Мол. вес амилопектина 100 000—
1 000 000 и выше. По типу строения амилопектин по-
хож на животный крахмал — гликоген.
Рис. 1. Схема строения макромолекул амилозы (а)
и амилопектина (б).
Амилозу и амилопектин разделяют, используя их
различную растворимость в воде и способность ами-
лозы давать нерастворимые комплексы с иодом, бу-
тиловым спиртом и др. веществами. Амилоза и амило-
Н НО Н НО н но
Рис. 2. Участок цепи макромолекулы амилозы.
пектин неоднородны, они содержат полисахариды,
отличающиеся мол. весом. Соотношение амилозы и
амилопектина в К. зависит от вида растения. В сред-
нем К. содержит 25% амилозы и 75% амилопектина.
Путем селекционирования получают сорта кукурузы,
К. к-рых содержит до 75%
амилозы. К. восковидной
кукурузы содержит свыше
95% амилопектина. Макро-
молекулы полисахаридов
расположены в крахмаль-
снюн
Рис. 3. Участок цепи амилопектина с точкой ветвления.
пых зернах радиально и слоями. Крахмальные зерна
имеют микрокристаллич. структуру и дают два ос-
новных тииа рентгенограмм: тип «А», характерный
для К. злаков, и тип «В», характерный для клубней.
Промежуточные типы относят к типу «С». Крахмальные
зерна обнаруживают двойное лучепреломление.
К., амилоза и амилопектин нерастворимы в холод-
ной воде, спирте и эфире. При нагревании в воде
зерна К. разрушаются с образованием клейстера.
Клейстеризация К. — сложный процесс, идущий в
три основные стадии. Сначала крахмальные зерна
обратимо набухают, присоединяя небольшие коли-
чества воды. При повышении темп-ры присоединяется
большое количество воды, сопровождающееся силь-
ным набуханием зерен с увеличением их объема в
сотни раз и повышением вязкости раствора; эта стадия
необратима. Набухание К. происходит вследствие
разрыва водородных связей и гидратации макромо-
лекул полисахаридов. На последней стадии раствори-
мые полисахариды извлекаются водой, зерна теряют
форму, превращаясь в мешочки, суспендированные
в растворе. Клейстеризация картофельного К. проис-
ходит при 55—65°, пшеничного при 60—80°, кукуруз-
ного при 64—71°, рисового при 70—80°. Из крахмаль-
ного клейстера и из растворов амилозы при длительном
хранении выпадает осадок амилозы (ретрогра-
дация). К. и амилоза, выделенные из растворов
при выпаривании, ретроградации или осаждении спир-
том, дают рентгенограммы типа «А», «В», «С» или «V»
(последний типичен только для клейстеризованного
К.) в зависимости от условий выделения осадка. Рас-
творы К. в воде оптически активны,[а]р =180—210°.
Восстанавливающие способности К. ввиду того,
что в макромолекуле К. содержится лишь одна свобод-
ная альдегпдпая группа, очень малы. К. гидролизуется
при действии кислот вначале до декстринов — поли-
сахаридов с небольшой степенью полимеризации, а
при полпом гидролизе — до D-глюкозы. При действии
различных ферментов гидролиз К. идет разными пу-
тями и с образованием различных продуктов (дек-
стринов, мальтозы, глюкозы). Ферменты, гидроли-
зующие К., наз. амилазами.
К. и его компоненты образуют ряд сложных и про-
стых эфиров. Амилоза легко образует нерастворимые
кристаллические комплексы со спиртами (бутило-
вым, амиловым и др.), жирными кислотами, фено-
лами, нитропарафинами, пиридином. Амилопектин об-
разует нерастворимый комплекс с гидроокисью алю-
миния.
К. окрашивается иодом в синий цвет; амилоза с
иодом дает интенсивпое синее окрашивание с макси-
мумом поглощения при 620—650 ммк, а амилопектин—
красно-фиолетовое с максимумом поглощения при
520—580 ммк. Для качественного обнаружения К.
используют иодную реакцию, микроскопия, исследо-
вание. Количественное определение К. производят
пегидролитич. и гидролитич. методами. Первые ос-
нованы на определении К., извлеченного раствори-
телями. В качестве растворителей используют холод-
ную соляную к-ту, надхлорпую, трихлоруксусную,
сульфосалициловую к-ты, растворы СаС)2, ZnCl2,
AlgClg, щелочи, глицерин, формамид и др. К. осаж-
дают спиртом и определяют весовым путем, поляри-
метрически или иодометрически. К. осаждается также
в виде производных, напр. в виде комплекса с иодом.
Гидролитич. методы основаны на определении восста-
навливающих веществ (глюкозы), образующихся прп
гидролизе К. кислотами или ферментами.
Получают К. в промышленном масштабе из карто-
феля и кукурузы. Меньшее значение имеет К. пшеницы,
риса, батата, сорго, саго и др. растений. Для полу-
чения картофельного К. (картофельной муки) массу
перетертого картофеля—кашку — обрабатывают во-
дой, к-рая извлекает крахмальные зерна, образуя
т. наз. крахмальное молоко. К. отделяют
отстаиванием этой взвеси или центрифугированием.
На нек-рых заводах от картофельной массы вначале
отделяют клеточный сок, а затем извлекают крахмал.
Сырой крахмал повторно промывают, высушивают и
измельчают. Для выделения К. из кукурузы зерна
замачивают в воде (40—50°), содержащей 0,15—0,3%
сернистого газа. Замоченное зерно дробят для удале-
ния ростков, используемых для получения кукуруз-
ного масла (ростки всплывают в воде и отделяются).
Кукурузную массу повторно измельчают п получен-
ную кашку обрабатывают водой для вымывания К.,
к-рый затем отделяют отстаиванием или центрифугиро-
ванием. Картофель используется также Для производ-
ства амилозы (суперлоза) и амилопектина (рамалин).
Амилозу и амилопектин выделяют, действуя на К.
765
КРАШЕНИЕ ИСКУССТВЕННЫХ ВОЛОКОН — КРАШЕНИЕ КОЖИ
766
р-рами солей [MgSO4, (NH4)2SO4, Na2SO4], содержа-
щих н-бутиловый спирт, при нагревании до 120°
с последующим осаждением амилозы при 70° и ами-
лопектина при 20°. Для нроиз-ва амилозы исполь-
зуются также специальные сорта кукурузы, К. к-рых
содержит 55—75% амилозы. Амилопектин (амиока)
производят также из восковидной кукурузы.
К. имеет чрезвычайно широкое применение в раз-
личных отраслях пром-сти. Его перерабатывают в
патоку и глюкозу, используют для приготовления
кулинарных и кондитерских изделий, колбас. К.
пищевых продуктов (хлеба, круп, картофеля) по-
крывает в основном потребность человека в углеводах.
К. является сырьем для произ-ва этилового спирта,
н-бутилового спирта, ацетона, молочной, лимонной и
глюконовой к-т, глицерина, 2,3-бутиленгликоля и
др. продуктов (см. Брожение и Глюкоза). К. приме-
няется также в составе питательных сред в произ-ве
антибиотиков, витаминов и др. К. используется для
шлихтования текстильных тканей, загустки красок,
для проклеивания бумаги и картона, произ-ва дек-
стринов и клеев. Из амилозы получают прочные
пленки типа целлофановых, нерастворимые в воде
и органич. растворителях. Амилопектин применяют
в качестве клеев и в пищевой пром-сти. Применяют
также модифицированные К., к-рым соответствующей
обработкой, напр. термической или окислением ги-
похлоритами, придают специальные промышленные
свойства. Ацилированный К. применяют для приго-
товления покрытий и загустителей, а ацетилирован-
ная амилоза используется для произ-ва пленок и во-
локна. Алкилпроизводные К. применяют в качестве
пластификаторов и клеев, а растворимые в холодной
воде оксиэтилированные производные К. применяют
в текстильной и бумажной пром-сти вместо К.
Лит.: Степаненко Б. Н. [и др.], «Усп. современной
биол.», 1951, 32, вып. 2 (5), 193; Френкель С. Я., Усп.
химии, 1950, 19, вып. 4, 488; Химия и технология крахмала,
под ред. Р. В. Керра, пер. с англ., 2 изд., М., 1956;
Успехи химии целлюлозы и крахмала, под ред. Дж. Хониме-
на, пер. с англ., М., 1962; Whistler R. L., Smart С. L.,
Polysaccharide chemistry, N. Y., 1953, p. 229—74; Radley
J. A., Starch and its derivatives, [3 ed.], v. 1—2, L., 1953;
Industrial gums. Polysaccharides and their derivatives, ed. R. L.
Whistler, N. Y.—L., 1959, p. 675—740; Handbuch der Pflan-
zenphyslologle, Bd 6, B.—Gottingen—Hdlb., 1958, S. 137,
154; French D., в кн.; Blochemle des glucides, P., 1961,
p. 349, 151. См. также лит. при статьях Амилазы, Декстрин,
Гликоген. Л. И. Линевич.
КРАШЕНИЕ ИСКУССТВЕННЫХ ВОЛОКОН —
крашение ацетатного волокна, полиамидных, поли-
эфирных, полиакрилонитрильных, поливинилхлорид-
ных волокон. См. Крашение текстильных материалов.
КРАШЕНИЕ КОЖИ — придание коже определен-
ной окраски. В зависимости от назначения кожи для
ее окрашивания применяют либо две последователь-
ные операции — крашение в барабапе р-ром краси-
телей и покрытие затем поверхности кожи пленкой из
красящих веществ, либо ограничиваются одной из
операций.
Для К. к. применяют красители: прямые, кислотные,
протравные для шерсти, основные, нигрозины, актив-
ные красители, образующие химич. связь с молекула-
ми белка, и специальные для кожи и шубной овчины.
Прямые красители (обычные и диазотирующиеся)
используют гл. обр. для крашения кожи хромового
дубления. Эти красители неустойчивы к действию
кислот и не проникают внутрь кожи, а связываются
лишь ее поверхностным слоем. Они непосредственно
окрашивают кожу в красильном р-ре при pH 6—7.
Исключение составляет прямой диазочерный С, к-рый
после крашения в барабане диазотируют и сочетают
с .«-толуилсндиамином. Кислотные красители при-
меняют для крашения всех видов кожи; они хорошо
растворимы, обладают удовлетворительной прочностью
окраски к различным обработкам; вследствие высокой
дисперсности эти красители проникают внутрь кожи.
Кислотные протравные красители для шерсти при
крашении кожи используют без обработки солями
металлов. Наиболее равномерную окраску дают ки-
слотные металлосодержащие красители (комплексы
1:1 и 1 : 2, т. е. на 1 молекулу'красителя 1 или2 атома
металла), к-рые обладают особенно высокой проч-
ностью к трению и даже в светлых тонах к свету. Спо-
собность их к проникновению в глубину кожи меньше,
чем у обычных кислотных красителей. Специальные
красители для кожи и для шубной овчины являются
также кислотными и прямыми красителями или их
смесями, синтезированными для крашения этих ма-
териалов. Крашение кислотными красителями ведут
при pH, равном 4—4,5. Через 30—60 мин. после на-
чала крашения в ванну добавляют муравьиную или
уксусную к-ту (50—70% от веса взятых красителей).
Все указанные классы красителей окрашивают кожу
при температуре 60—70°; расход красителя состав-
ляет 1—2%; отношение веса жидкости к весу кожи
(жидкостный коэфф.) — 200—300% от веса строга-
ной кожи.
Основные красители применяют гл. обр. для углуб-
ления окраски кожи хромового дубления после кра-
шения кислотными или прямыми красителями ио
таннидной протраве. В крашении велюровых кож и
кож растительного дубления их применяют без про-
травы. Основные красители дают яркую окраску,
непрочную к действию света; их нельзя применять
в смеси с прямыми и кислотными красителями, т. к.
при этом основные красители выпадают в осадок.
Для получения равномерной окраски кожу обрабаты-
вают 15—20 мин. перед началом крашения в светлые
тона диспергатором НФ и в темные тона — препа-
ратом ОП-Ю, к-рых берут 1,5% и 0,5% от веса стро-
ганой кожи соответственно. Нигрозин применяют
в обоих видах крашения. Активные красители пока
не нашли широкого применения для К. к.; по-види-
мому, они найдут применение в будущем, т. к. дают
яркую окраску, прочную к различным обработкам, и
глубоко проникают в ткань кожи. Активные краси-
тели взаимодействуют с кожей в слабощелочной сре-
де. По химич. строению все перечисленные красители
относятся к азокрасителям, арилметановым, хинони-
миновым и активным красителям.
Азокрасители. Для К. к. наиболее широко исполь-
зуют коричневые и черные красители, являющиеся гл. обр.
дисазо- и полиазокрасителями, в к-рые в качестве концевой
компоненты вводится -м-фепилендиамшг, резорцин, Аш-кис-
лота, а-нафтол. Широко применяют также желто-коричневые
хромовые комплексы и синие медьсодержащие красители.
Нек-рое применение находят и моноазокрасители, производ-
ные бензола, нафталина, бензолазонафталина, пиразолона.
Старейшим представителем этого класса является кислотный
желтый метаниловый — производный азобензола. К моноазо-
красителям принадлежит новый краситель — кислотный ко-
ричневый Ж для кожи, обладающий глубокой прокрашиваю-
щей способностью; его получают азосочетанием сульфаниловой
к-ты и фенил-гамма к-ты. К моноазокрасителям относится и
хризоидин (I) — основной краситель,
получаемый сочетанием хлористого бен-
зол-диазония с .и-фенилендиамином. Так НО N=N% y-NO,
как у хризоидина группы NH2 непо- I___1 \ / 2
Qr~\ НО N=N-Z \-ОН NH2
N=N-/ V-NHs
HOaS-LXJ-SOaH
7 .
средственно связаны с ароматич. ядром, он чувствителен
к кислотам. Из дисазокрасителей находит применение, напр.,
кислотный коричневый ЗК для кожи (II), устойчивый к из-
менению pH среды, проникающий в глубь волокна.
Новый краситель — кислотный коричневый 2К для кожи
получают последовательным азосочетанием сульфаниловой
к-ты с резорцином и .и-ксилидином. При применении в каче-
стве конечной компоненты фенилперикислоты получают ряд
прочных красителей, дающих ровные окраски в слабокйслой
среде. Полиазокрасители образуются при сочетании ^-ди-
аминов, оксиаминов или оксибензолов с двумя диазотирован-
ными аминами; полученный диазокраситель сочетается далее
767
КРАШЕНИЕ КОЖИ—КРАШЕНИЕ МЕХА
768
с образованием трисазокрасителя, напр. кислотный коричне-
вый К для кожи (III) получают при последовательном соче-
зочерного С и последующего сочетания
получают черный краситель (ГУ).
тании резорцина, пик-
раминовой к-ты и п-
нитроанилина. К по-
лиазокрасителям от-
носятся также неко-
торые прямые кра-
сители, например пря-
мой диазобордо све-
топрочный С окраши-
вает перчаточную ко-
жу и велюр в цвет
бордо; при диазоти-
ровании прямого диа-
с л-толуилепдпампном
IV
Из азокрасителей, хромирующихся на волокне, наиболее
часто применяют хромовый желтый Н (V), хромовый оранже-
вый и хромовый коричневый К. Для крашения кожи при-
меняют комплексы азокрасителей с хромом или медью, хром
вводится р-ром формиата хрома и образует группировку,
напр. для комплекса 1 : 2СУа).
СООН
N з О з S
ОН
V
Уа о
Красителями этой группы являются, напр., кислотный темно-
коричневый МШ (хромовый комплекс 1 : 1 красителя из
пикраминовой к-ты и .и-фепилдиаминсульфокислоты), кислот-
ный коричневый МШ (хромовый комплекс 1 : 2 из красите-
лей п-аминобензоил-З-амино-5-сульфосалициловой к-ты и
фенил-гамма-кислоты), прямой синий КМШ (медный комп-
лекс красителя из дисдиа-
зобензидина, Р-соли и Аш-
кислоты).
Арилметановые
красители. Для К. к.
применяют гл. обр. ос-
Оновные красители — рода-
СН3 хшны. Функцию хромофо-
ра в этих красителях вы-
„ полняет хиноидное ядро,
“2‘ содержащее ауксохромы,
Vi например основной бирюзо-
вый (VI).
Хинониминовые красители. Наибольшее
применение находит метиленовый голубой ц (VII), к-рый слу-
жит для подцветки при получении черного велюра. Это тиази-
новый краситель из нитрозодиметиланилина, выделенный
в виде двойной соли с хлористым цинком
К этой группе относятся и нигрозины, к-рые получают спла-
влением анилина, солянокислого анилина и нитробензола
в присутствии чугунной стружки с последующим сульфирова-
нием моногидратом. Нигрозин растворим в воде и приме-
няется для К. к. растительного и хромового д; блсния
Активные красители. Среди красителей, обра-
зующих связь с белком, следует назвать активные красители,
содержащие в молекуле остатки дихлортриазина, например
ярко-голубой (VIII) или красно-фиолетовый медный ком-
плекс f-оксиэтилсульфата (IX).
Первичный акт крашения активными красителями состоит
в равномерной адсорбции иона красителя межмицеллярной
поверхностью кожи. Реагирующий атом углерода красителя,
приходя в непосредственное соседство с аминогруппой белка,
отщепляет в первом случае хлорид-ион, во втором — сульфат-
ион, образуя ковалентную связь: краситель — белок. Низкая
темп-ра крашения имеет значение не только из-за снижения
скорости гидролиза, но и потому, что при низкой темп-ре рав-
новесие между растворенным и абсорбированным красителем
смещено в сторону волокна.
Покрывное крашение. Этот виц краше-
ния производят щеткой или под давлением (при помощи
распылителей) для получения наиболее равномерной
окраски кожи и для создания на коже с пораженной
поверхностью искусственного лица. В состав покрыв-
ных красок входят: а) красящие вещества — неорга-
пич. и органич. пигменты; б) связующие или клеющие
вещества (нитроцеллюлоза, акриловые смолы, казеин
и др.); в) растворители для связующих компонентов
(амил- и бутилацетаты, толуол) и разбавители (эти-
ловый спирт, бензол и др.); г) пластификаторы, при-
дающие гибкость и эластичность пленкам (касторо-
вое масло, дибутилфталат, трифенилфосфат и др.);
д) вспомогательные вещества — для смягчения, кон-
сервации и блеска плепок (фенол, формалин, аммиак,
мыло, парафин, воск и др.). Покрывными красками
окрашивают по преимуществу верхние хромовые
кожи. В качестве пигментов применяют гл. обр.
минеральные: сажа, свинцовый крон, двуокись ти-
тана, железоокисные пигменты и др. Для получения
красных и синих окрасок применяют оргапич. пиг-
менты и лаки, напр. нерастворимые азокрасители.
Лит.: Коган А. Б., Окраска кожаных и резиновых
изделий, М., 1953; Коган И. М., Химия красителей, 3 изд.,
М., 1956; Венкатараман К., Химия синтетических
красителей, пер. с англ., т. 2, М., 1957; Цоллингер Г.,
Химия азокрасителей, пер. с нем., Л., 1960.
И. Н. Алатырцева.
КРАШЕНИЕ МЕХА—физико-химический процесс,
главная цель которого—крашение волосяного по-
крова и имитирование цепных видов пушпины, на-
пример шкурок кролика под норку, соболя и котика,
меховой овчины — под выдру, бобра, хоря и др.
Целью К. м. является также подцветка или выравни-
вание природной окраски волосяного покрова. Окрас-
ка во всех случаях должна быть устойчивой к свету,
светопогоде, сухому и мокрому трению. Для правиль-
ного крашения пмеют значение pH среды, темп-ра
ванны, продолжительность крашения и отношение
объема раствора к весу окрашиваемого полуфабрика-
та, так. наз. жидкостной коэффициент; в большинстве
случаев он равен 10—15. Различают крашение:
а) красителями, образующими на волосе соединения,
содержащие хиноидные связи, характерные для окра-
шенных продуктов (оксидациопное крашение); б) ку-
бовыми красителями (кубовое крашение); в) кислот-
ными хромирующимися или др. азокрасителями (ки-
слотное крашение). Наиболее распространенным яв-
ляется оксидационное крашение. Во всех случаях
крашение состоит из подготовки полуфабриката (ме-
ховой шкурки), собственно крашения, промывок и
солки (обработки солями). Все процессы проводят
последовательно, часто в одном оборудовании, без
промежуточных выгрузок, загрузок и отжимов.
Оксидациопное крашение. Подготовка
полуфабриката состоит пз след, операций: а) обработ-
ки разбавл. р-рами аммиака, соды или смеси аммиака
и перекиси водорода при 20—25° (т. н. нейтрализа-
ция); целью этой операции является создание соот-
ветствующего pH среды, удаление механич. загряз-
нений и частично жира, увеличение смачиваемости
волосяного покрова; б) разрушения натурального
пигмента волосяного покрова для его последующей
769
КРАШЕНИЕ МЕХА
770
окраски в различные цвета (отбеливание). Процесс
ведут при 32—35° и pH 5,5. Отбеливанию предшест-
вует формалиновое дубление; в) обработки слабыми
р-рами хромпика и серной к-ты при 30° и pH 3,5—4.
Эта операция способствует получению разной степени
насыщенности светопрочных окрасок, вплоть до черно-
го цвета. Иногда для светлых тонов используют желез-
ную протраву (FeSO4 • 7Н2О и уксусная к-та).
К. м. производят окуночным или «намазным» спосо-
бом. В первом случае шкурки погружают в водный р-р
красителя, содержащий перекись водорода и аммиак.
Крашение ведут при pH среды 7,5—8,5 и темп-ре 35—
40°. При намазном способе более конц. р-ры краси-
телей наносят жесткой волосяной щеткой, при этом
остается незакрашенной сама кожа; или же, пользуясь
распылителем, наносят красящий р-р на поверхность
остевого волоса после окуночного крашения (верховое
крашение). Этот способ применяют для имитации
ценной пушнины. Известен способ нанесения красиль-
ного р-ра с помощью спец, оборудованной аэрографной
машины, дающей плоскую или круглую струю (аэро-
графное крашение); его применяют для имитации шкур
бобра, хоря или норки па меховой овчине. Для ими-
тации шкур леопарда, барса, тигра и т. п. используют
трафаретное крашение; в случае необходимости оно
может быть многоцветным; распространен метод фото-
фильмпечати. Для защиты кончиков волос от закраши-
вания на них наносят р-р хлористого олова в спец,
замеске (резерв). Так получают, напр., имитацию
серого каракуля на белой мерлушке.
Для оксидационного крашения применяют бесцвет-
ные или слабоокрашенные соединения под общим наз-
ванием «красители для меха» (СССР), урзолы (ГДР,
ФРГ), фурамины (Франция) и т. д. Эти красители
являются аминами, фенолами,, нафтолами и, соответ-
ственно, аминофеполами и аминонафтолами. Все эти
соединения легко окисляются в продукты хиноидной
структуры, к-рые при низкой темп-ре окисляются
далее в глубокоокрашенные в-ва более сложной
структуры, нерастворимые в воде.
Для крашения меха используют смесь двух или более
оксидационных красителей, из к-рых основным почти
всегда является черный для меха Д (га-фенилендиамин
H2N—СвН4—NH2). В процессе окисления полупродукт
легко реагирует с ароматич. аминами и фенолами,
образуя индофенолы и индоамины. При окислении
n-фенилдиамина получают тетраминодифенил-п-азо-
фенилен, сильно окрашенное соединение, т. наз.
основание Бандровского (I).
При дальнейшем окислении основания Бандровского
возможно образование азинового производного. Строе-
ние продуктов окисления n-фенилендиамина нельзя
считать вполне установленным.
При крашении смесью черного для меха Д с корич-
невым для меха Т (.и-толуилендиамин) или с серым
для меха ДА (л-диаминоанизол) в волосяном покрове
образуется индамин (сине-черный цвет), постепенно,
в результате дальнейшего окисления, превращающий-
ся в феназин (красный пвет).
Поэтому такие смеси для полу-
1| I Т 1 чения черного цвета нецелесо-
н Nобразны. При окислении смеси
2 черного для меха Д с резорци-
u ном образуется феноксазин (II),
волосяной покров окрашивается в насыщенный корич-
невый цвет. Большой практич. интерес представляет
13 К. X. Э. т. 2
окисление смеси черного для меха Д и пирокатехина
для получения прочного черного цвета на волосяном
покрове. К числу красителей для меха относятся также
коричневый для меха А (n-аминофенол), серый для
меха Д (диметил-л-фенилендиамин), желтый для меха Н
(4-нитро-о-фенилендпамин), желтый для меха А
(о-аминофенол), серый для меха А(аминодифениламин)и
др. полупродукты. Недостатки окспдационного краше-
ния: относительно слабая прочность окрасок, особенно
светлых; черный цвет устойчив; возможны профес-
сиональные заболевания дерматитом или урзольной
астмой.
К оксидационному крашению относится также про-
цесс, в к-ром сначала мех обрабатывают смесью соляно-
кислого анилина CeH5NH2 • НС1 и окислителя (берто-
летовой соли). В анилиновый р-р входят также наша-
тырь и препарат ОП-Ю. Крашение — трехкратное
нанесение с одновременным втиранием р-ра в волося-
ной покров с последующими после каждого раза цик-
лами сушки, обработки паром (запарки), сушки. При
этом происходит накопление эмеральдина в волосе
и он окрашивается в зеленый цвет. После цикла ани-
линового крашения следуют протравление в р-ре хром-
пика (происходит образование нпгранилияа) и окси-
дациояное крашение смесью черного для меха Д с
коричневым для меха Т или с другими красителями,
в присутствии аммиака (pH 7,5—8; 40°) образуется
черный незеленеющий анилин.
Кубовое крашение. Этот процесс обес-
печивает получение на волосяном покрове коричне-
вого, серого и черного цветов высокой прочности;
позволяет обрабатывать полуфабрикат непрерывным
методом. Процесс состоит из след, стадий: а) нейтрали-
зации р-ром соды; б) пропитки волосяного покрова
в предварительно подготовленном р-ре лейкосоеди-
нения; в) окисления лейкосоединения в волосяном
покрове. В конце крашения вводят перекись водорода.
Затем следуют промывка, солка и др. операции. Кубовые
красители восстанавливают и растворяют в р-ре гидро-
сульфита и едкой щелочи. Последнюю в нек-рых слу-
чаях заменяют содой. Крашение ведут при темп-ре
45—50° и pH ок. -10, а в конце крашения 8,6—9.
Для меха применимы красители несложной структуры,
лейкосоединения к-рых находятся в основном в мо-
лекулярно-дисперсном состоянии при невысоких кон-
центрациях щелочи. Сюда относятся индигоидные,
тиоиндигоидные, а также кубовые красители, произ-
водные бензохинона, нафтохинона и нек-рые производ-
ные антрахинона с несложной структурой. Основные
красители для крашения меха: тиоиндиго черный,
тиоиндиго красно-коричневый Ж, индиго, кубовый
красно-коричневый 4 ЖМ, кубовый красно-коричне-
вый 2 ЖМ, кубовый коричневый 2 ЖШ, кубовый се-
рый М (III).
Крашение кислотными красителями.
Для меха принято крашение кислотными красите-
лями с последующей обработкой хромпиком в кислой
среде. Образующийся комплекс сообщает прочную
окраску меху. Обработку мехового сырья можно вести
непрерывным способом. Цикл крашения включает
процессы: нейтрализацию, крашение, обработку
смесью красителей (pH 6,8—7,2), хромирование —
обработка р-ром хромпика в среде уксусной к-ты
(52—55°), промывка и солка. Крашение и хромиро-
вание можно проводить одновременно.
771
КРАШЕНИЕ ТЕКСТИЛЬНЫХ МАТЕРИАЛОВ
772
Для крашения используют смесь (1 : 1) красителей:
кислотного однохром коричневого 3 (IV) и однохром
оливкового Т (V):
Лит.: Родионов А. М., Крашение меха, М., 1954;
Якобсон А. М., Красители для меха, М., 1955; Ко-
ган И. М., Химия красителей, 3 изд., М., 1956; Бого-
словский Б. М., Лаптев Н. Г., Химия красителей,
М., 1960; Беленький Л. Н., Теория крашения и опыт
ее практического применения, М., 1958; [Г о л а н д Н. И.],
Крашение меха кубовыми красителями, М., 1959; Кож.-обувн.
пром-сть, 1959, № 4; Меховая пром-сть, 1961, вып. 1—2;
Чацкий П. И., Мягкова 3. В., там же; Чацкий
П. И., Мягкова 3. В., Фетискина Л. И., там же,
1962, вып. 2; Ч а ц к и й П. И., Легкая пром-сть, 1956, № 12;
Тр. н.-и. ин-та меховой пром-сти, 1960—62, № 1—12;
Справочник по меховой и овчинно-шубной промышленности,
т. 1—2, М., 1954—59. II. И. Чацкий.
КРАШЕНИЕ ТЕКСТИЛЬНЫХ МАТЕРИАЛОВ
Содержание:
I. Введение............................... 771
II. Крашение целлюлозных волокон........... 773
III. Крашение белковых волокон ............. 777
IV. Крашение химических волокон............. 779
V. Крашение смешанных волокон............. 784
I. Введение. Крашение текстильных материалов —
физико-химич. процесс взаимодействия красителей
с текстильными волокнами, в результате к-рого волок-
на окрашиваются более или менее прочно к действию
света, водным и мыльным обработкам, к трению и др.
Крашение волокнистых материалов осуществляют, как
правило, в водной среде. Крашение состоит из диф-
фузии красителя к поверхности волокна, адсорбции
его на поверхности, диффузии красителя внутрь суб-
страта и фиксации или закрепления его1 в волокне.
В отдельных случаях крашение осуществляют, синте-
зируя краситель непосредствепно в волокне. Способы
крашения определяются химпч. природой красите-
лей и волокнистых материалов (см. Красители синте-
тические).
Крашение заключается в самопроизвольном пере-
ходе красителя из красильной ванны в волокно до
установления равновесия. В соответствии с этим,
двумя главными сторонами почти всех процессов
крашения являются: а) распределение красителя
между волокном и раствором в момент равновесия и
б) скорость достижения равновесного распределения.
Тенденция перехода красителя из одной фазы в дру-
гую (химич. сродство красителя к волокну) изме-
ряется разностью химич. потенциалов (Др.°) красителя
в волокпе и в растворе в стандартном состоянии. Для
различных случаев крашения значения химич. срод-
ства будут иметь выражения, зависящие от механизма
взаимодействия красителя с волокном: 1) краситель
растворяется в волокпе с образованием твердого р-ра:
[П]ф
2) Краситель притягивается к поверхности раздела
волокно—растворитель без адсорбции на активных
центрах с возможностью образования многомолеку-
лярного слоя:
[Na]* • [D]®
-ДрД = КТ In у___________
V* + 4-[Na]*. [D]3
3) Краситель адсорбируется па активных центрах по-
верхности раздела волокно—раствор в виде мономо-
лекулярного слоя:
“ “ КТ !„ [ f | V [In [HJHDI.
Здесь^а]ф, [П]ф,'[Н]о, [Na]o, [D]o—соответственно
концентрации ионов натрия, водорода и красителя
в волокне и в растворе при установившемся рав-
новесии, z — валентности иона красителя, V — эф-
фективный объем волокна; вн и gD—доля активных
центров волокна, занятых соответственно ионами
водорода и красителя. Две другие термодинамич.
характеристики процессов крашения — теплота и
энтропия — рассчитывают по известным в термоди-
намике соотношениям. Сродство красителя к волокну
не зависит пи от начальной концентрации красителя,
ни от добавок в красильную ванну посторонних элект-
ролитов, опо определяется строением молекул краси-
теля и темп-рой крашения.
Наряду с равповесным распределением красителей
между волокном и раствором, к-рое регулируется
химич. сродством, в нек-рых случаях может происхо-
дить практически необратимое замещение функцио-
нальной группы волокнистого материала реакционно-
способной частицей красителя. Это характерно для
«активных» красителей в стадии их фиксации на волок-
не. При крашении химич. волокон и бумаги в массе,
а также резины и пластич. масс краситель или пиг-
мент механически распределяется в субмикроскопич.
пространстве окрашиваемого материала. Иногда при
крашении и печатании используют т. н. пигментное
крашение—приклеивание частиц красителя к окраши-
ваемому материалу с помощью синтетич. сеткообразую-
щих связующих веществ. Достижение равновесия в
распределении красителя между волокном и раствором
за висит от скорости процесса; эта скорость в значитель-
ной степени определяет глубину проникновения краси-
теля с периферии волокна к его центру. Особенно
важное значение эта величина имеет в том случае,
когда крашение осуществляется по непрерывно-по-
точному методу при малом времени пребывания окра-
шиваемого материала в красилыюй ванне. Наиболее
существенным показателем скорости перемещения
красителей в волокнистых материалах является
коэфф, диффузии. В общем виде эту зависимость мож-
но представить как
Д = 1 - Ае - ^ - Се- - Ge-Hfe - ...
^оо
где А, В, С и т. д. — постоянные величины, k = Dt/r2,
t — время, г — радиус волокна. Конкретные частные
решения можно пайти в монографии Виккерстаффа
(см. лит.).
Коэфф, диффузии красителей в волокне в сотни и
тысячи раз меньше, чем в водной среде. Объясняется
это прежде всего тем, что частицы красителя на опре-
деленные промежутки времени задерживаются на
активных центрах молекул волокна. Скорость диффу-
зии красителей в волокне снижают также стерич. за-
труднения и во многих случаях силы электростатич.
отталкивания, обусловленные наличием одноимен-
ного заряда волокна и красителя. Коэфф, диффузии,
а следовательно, и скорость крашения в целом, можно
увеличить двумя путями: 1) повышением темп-ры
крашения (на каждые 10° скорость крашения повы-
шается примерно в два раза); 2) введением в красиль-
ный р-р нек-рых поверхностно-активных веществ и
гидрофильных органич. растворителей. Таким спо-
собом можно увеличить коэфф, диффузии в 10—20 раз,
что открывает широкие возможности интенсификации
процесса, не прибегая к значительному повышению
темп-ры. Весьма благоприятно сказывается на равно-
мерности окраски наличие в растворе гидрофильных
органич. растворителей и текстильных вспомогатель-
ных веществ, напр. сульфопроизводных гомологов
нафталина (некаль) и др. По современным представ-
лениям, красители удерживаются на волокне силами
773
КРАШЕНИЕ ТЕКСТИЛЬНЫХ МАТЕРИАЛОВ
774
химич. связи, ван-дер-ваальсовыми связями и водород-
ными связями. В зависимости от сил, удерживающих
краситель на волокне, а также от строения красителя
и субстрата прочность окрасок к различным видам
воздействия может меняться в широких пределах.
В соответствии с целевым назначением к окраши-
ваемым текстильным материалам предъявляют раз-
личные требования. Наибольшей прочностью должны
обладать окраски материалов, к-рые при использова-
нии подвергаются воздействию света, погоды, много-
кратных стирок и т. д. Прочность окрасок регламенти-
руется ГОСТом; по действующим в СССР нормам наи-
высшая прочность определяется баллом 5, низшая — 1.
Крашение волокон пряжи и тканей производят в аппара-
тах периодич. и непрерывного действия. Предпочтительно
используют непрерывно-поточные методы крашения, обеспе-
чивающие более высокую производительность труда и обору-
дования, однако при крашении небольших партий, особенно
волокна и пряжи, в один цвет применяются также и аппараты
периодич. действия. Качество окраски в основном зависит от
свойств красителей, условий их применения и подготовки
текстильных материалов перед крашением. Крашение по
периодич. способу осуществляют гл. обр. в различных барках
и красильных роликовых машинах. Способ характеризуется
экономным расходованием красителей и вспомогательных
материалов, хорошим проникновением красителя в толщу
отдельных волокон, но вместе с тем отличается большой дли-
тельностью и трудоемкостью. Низкая производительность
труда и оборудования послужила стимулом к переходу на
непрерывно-поточные способы крашения. В последнем случае
с целью интенсификации проникновения красителя в волокно,
при малом времени пребывания его в красильной ванне, окра-
шиваемый материал прогревают при высоких темп-рах (термо-
золь-процесс), запаривают (плюсовочно-запарные способы),
обрабатывают в расплавленном металле (степдфаст-процесс)
или в горячем масле. С этой же целью в красильную ванну
вводят различные гидрофильные органич. растворители и
текстильные вспомогательные вещества. Значительного повы-
шения скорости крашения можно добиться применением ва-
куума для удаления воздуха из субмикроскопич. пор во-
локна, а также проводя процесс под давлением или при по-
вышенных темп-рах.
Для унификации непрерывно-поточных методов крашения
созданы универсальные линии для крашения всех видов тка-
ней из целлюлозных волокон красителями различных клас-
сов. В этом агрегате ткань пропитывается раствором или
суспензией красителя, подвергается запариванию, последую-
щим обработкам, промывке и сушке. При крашении непрерыв-
ным способом большое значение имеют контроль и регулиро-
вание технологич. параметров, а именно: концентрации рас-
творов, pH среды, темп-ры, уровня жидкости в ваннах и др.
И. Крашение целлюлозных волокон. Хлопок, лен и
гидратцеллюлозные волокна (вискозное и медпо-амми-
ачпое) можно окрашивать из водных р-ров описан-
ными ниже группами красителей. Кроме того, непо-
средственно на волокне синтезируют нерастворимые
оксиазокрасители и «черный анилин».
Крашение прямыми красителя-
м и. В этом виде крашения принимают участие моле-
кулы или ионы красителя; по мере их перехода в во-
локно происходит дезагрегация более крупных
частиц красителя. Поглощение красителя хлопковым
волокном определяется его сродством it целлюлозе и
условиями крашения: концентрацией красителя и
электролита в ванне, темп-рой, продолжительностью
крашения. Равновесное поглощение красителя падает
с повышением темн-ры, а скорость выбирания краси-
теля из ванны возрастает. Поэтому в аппаратах пери-
одич. действия с длительным временем пребывания
окрашиваемого материала в ванне наблюдается тем-
пературный оптимум (обычно 70—90°). Оптимальное
содержание электролита также будет различным.
В условиях непрерывно-поточного крашения интен-
сивность окраски возрастает при повышении темп-ры
и до определенного предела содержания электролита
в ванне. Лучшие результаты получаются при после-
дующей обработке окрашенной ткани паром (запари-
вании^. Расход красителя составляет 1—4% и пова-
ренной соли до 20% от веса волокна, в зависимости от
интенсивности получаемой окраски. При наличии
жесткой воды добавляют соду. Окраски прямыми
красителями не обладают достаточно высокой проч-
ностью. Значение этого класса красителей возросло
благодаря появлению светопрочных металлосодержа-
щих красителей, а также разработке различных
методов упрочения окраски к действию водных и
мыльных обработок. Для этой цели широко применя-
ют препараты ДЦУ и ДЦМ (см. Закрепляющие веще-
ства). Прямые красители, помимо получения гладко-
окрашенных тканей, широко применяют для получе-
ния вытравных расцветок.
Крашение активными красите-
лями. Рассматриваемые красители растворимы в
воде, они содержат активные группы, способные взаи-
модействовать с гидроксильными группами целлюлозы
в слабощелочной среде; к этой группе красителей от-
носят т. н. «проционовые» красители.
При крашении протекают одновременно две ре-
акции
S—R-Х+НО-цел.—-S-R-O-цел.+НХ (1)
S—R—Х-рНОН -S- R—0Н-|-НХ (2)
Здесь S—R—X—молекула красителя, в к-рой S—одна
или песк. сульфогрупп, R — окрашенная органич.
часть молекулы, X — реакционноспособный атом
галогена; НО-цел. — целлюлоза. Реакция (1) при-
водит к фиксированию красителя волокном; реакция
(2) является побочной, ведущей к образованию нереак-
ционноспособных гидроксильных производных исход-
ного красителя. В практич. условиях крашения ско-
рость первой реакции значительно больше скорости
второй, и фиксация красителя волокном составляет
до 70%. Окраски, полученные с помощью активных
красителей, отличаются яркостью, разнообразием
цветов и оттенков и повышенной прочностью. Красить
этими красителями можно как непрерывным, так и
периодич. способом. В свою очередь, крашение на
аппаратах непрерывного действия может быть про-
ведено по двухстадийному' процессу (обработка ткани
нейтральным р-ром красителя и затем р-ром щелочи)
и по одностадийному; последний — более приемлем.
Для создания стабильного щелочного р-ра красителя
пользуются слабой щелочью — бикарбонатом нат-
рия. Для улучшения выбираемости красителя полезно
добавлять нейтральный электролит в красильную
ванну. В целях предотвращения миграции красителя
на ткани в процессе сушки в красильный р-р рекомен-
дуется вводить альгинат натрия.
Крашение кубовыми красите-
лями. Кубовые красители дают весьма прочные
окраски с широкой гаммой цветов и оттенков. Кубовые
красители нерастворимы в воде, их предварительно
восстанавливают в щелочной среде гидросульфитом
натрия, в результате чего получаются растворимые
в воде натриевые соли лейкосоединений напр.:
Na2S2O4 + 2H2O 2NaHSO3+2H
Лейкосоединения кубовых красителей избирательно
поглощаются целлюлозными волокнами из водных
р-ров. Окислением кислородом воздуха на волокне
лейкосоединения переводят в исходные кубовые кра-
сители. Крашение ведут при темп-рах от 30 до 80°
в зависимости от применяемого красителя.
13’
775
КРАШЕНИЕ ТЕКСТИЛЬНЫХ МАТЕРИАЛОВ
776
Широко распространен также суспензионный метод
крашения, разработанный М. А. Ильинским в 1911.
Сущность способа состоит в обработке окрашиваемого
материала высокодисперсной суспензией невосстанов-
ленного кубового красителя; последний восстанавли-
вают непосредственно на волокне обработкой щелочно-
гидросульфитным р-ром. Окончательная окраска об-
разуется при последующем окислении лейкокраси-
теля на волокне. Суспензионный метод крашения
имеет ряд преимуществ по сравнению с описанным
выше; в частности, он позволяет получить равномерные
интенсивные окраски с хорошим прокрашиванием
волокна на аппаратах непрерывного действия. Успех
суспензионного способа крашения обеспечивается
применением кубовых красителей в виде высокодис-
персных паст или порошков с размером частиц ок.
1 мк.
Особую группу кубовых красителей составляют ку-
бозоли — растворимые в воде натриевые соли серно-
кислых эфиров лейкосоединений кубовых красителей.
Целлюлозные материалы обрабатывают водными
р-рами кубозолей с последующим проявлением окраски
гидролизом и окислением эфиров. В результате па
волокне фиксируется исходный кубовый краситель,
из к-рого был получен соответствующий кубозоль.
В зависимости от способа проявления окраски разли-
чают нитритный, бихроматный, запарной и др. спо-
собы крашения. Кубозоли выгодно применять при
крашении в светлые оттенки. При крашении кубовыми
красителями применяют различные вспомогательные
вещества, в частности выравниватели и диспергаторы,
для улучшения качества окраски (см. Выравнивающие
вещества).
Крашение сернистыми красите-
л я м и. Широкое применение сернистых красителей
обусловлено их низкой стоимостью и сравнительно
прочными окрасками; недостатками являются непол-
нота гаммы цветов и неяркость окрасок. Нек-рые
красители дают низкую прочность окрасок к дейст-
вию света. Сернистые красители нерастворимы в воде
и применяются для крашения в виде лейкосоединепий
с последующим окислением их на волокне. В каче-
стве восстановителя и растворителя обычно приме-
няют сернистый натрий. При восстановлении серни-
стых красителей дисульфидные группы переходят в
сульфгидрильные и в них водород замещается натрием;
RS-SR,—►RSH—HSRi—►RSNa—NaSR[
В состав красильных ванн входят также поваренная
соль, сода, иногда едкий натр и смачиватели. Приме-
нение смачивателей особенно ваяшо при крашении
хлопка, пряжи и недостаточно подготовленных хлоп-
чатобумажных и льняных тканей. Красящая способ-
ность сернистых красителей невелика, поэтому при
крашении в темные оттенки берут до 20% красителя
от веса волокна. Крашение ведут при темп-ре, близкой
к 100°. Существуют и растворимые в воде препараты
сернистых красителей, т. н. водорастворимые. При
применении их текстильный материал пропитывают
водным р-ром красителя, высушивают горячим возду-
хом и обрабатывают в р-ре, содержащем сернистый
натрий и едкий натр. В этом р-ре при 80—90° в тече-
ние 30 сек происходит проявление, красителя, т. е.
переход его в исходную нерастворимую в воде форму.
Описаны также однованные способы применения сер-
нистых водорастворимых красителей. Для повышения
прочности окраски к действию света обрабатывают
окрашенную ткань препаратом ДЦМ; иногда обработ-
ку совмещают с аппретированием ткани.
Крашение основными красите-
лями. Основные красители не обладают свойством
непосредственно окрашивать целлюлозные волокна.
Они фиксируются целлюлозными волокнами с по-
мощью кислых протрав с образованием солей и об-
ладают высокой красящей способностью. Для краше-
ния в светлые тона берут десятые доли процента кра-
сителя от веса волокна и не более 3% для получения
интенсивных окрасок. Основные красители дают
красивые и яркие окраски, однако прочность окрасок
к водным и мыльным обработкам, а также к действию
света весьма незначительна. Поэтому применение их
для гладкого крашения ограничено. Нек-рое повыше-
ние прочности окрасок к действию света достигается
последующей обработкой окрашенной ткани фосфор-
номолибденовой к-той. Тем не менее и после такой
обработки окраски под действием света быстро меняют
свой оттенок и становятся тусклыми.
«Холодное крашение». При этом способе
крашения нерастворимые оксиазокрасители обра-
зуются непосредственно на волокне. Этот вид краше-
ния широко распространен, т. к., пользуясь им, можно
получать яркие и прочные окраски в широкой гамме
оттенков. Сущность процесса состоит в том, что окра-
шиваемый материал обрабатывают при темп-ре ок. 50°
щелочным р-ром азотола (азосоставляющая) и затем
па холоду — р-ром диазотированного' первичного
ароматич. амина (диазосоставляющая). При этом на
волокне протекает азосочетание и образуется нерас-
творимый оксиазокраситель:
I
R-OH+R^Cl----*R—N=N—Ri
Цвет окраски определяется исходными азо- и диазо-
компонентами. Известно более 20 азотолов и ок. 40
различных диазокомпонеятов, как в форме азоаминов,
так и в форме диазолей, пользуясь к-рыми можно син-
тезировать на волокне до 800 красителей различных
цветов и оттенков. Выход красителя на волокне, т. е.
степень использования азо- и диазосоставляющих,
определяется в основном pH среды реакции азосочета-
ния. Оптимальное значение pH среды составляет 7—9;
поддерживается буферными р-рами, чаще всего ацетат-
ным буфером. В кислой среде азосочетание замедляется
вследствие того, что азосоставляющая переходит
в нерастворимое состояние; в щелочной среде реакция
не идет вследствие перехода диазосоставляющей
в неактивную форму — антидиазотат или яитрозамип.
Хлопковое волокно красят на аппаратах перподич.
действия без сушки волокна после обработки его азо-
толом. Для этой цели предложен также аппарат не-
прерывного действия. Ткань обрабатывают р-ром азо-
тОла на плюсовке (С -- Ю г/л, темп-ра 5С°), агреги-
рованной с сушилкой. После этого ткань обрабатывают
па холоду диазораствором, промывают и высушивают.
Мыльная обработка при темп-ре, близкой к 100°, повы-
шает яркость и прочность окраски к действию света.
Вместо синтеза нерастворимого красителя на волокне
можно применять готовый азопигмент в растворимой
форме, папр. неокотоны, к-рые проявляют на волокне
пропусканием ткани, пропитанной р-ром неокотона,
через р-р щелочи.
«Черноаяилиновое» крашение. Ма-
териал, предварительно пропитанный солянокислым
анилином, окрашивается в результате окисления его
непосредственно на волокне. В результате получается
пигмент, известный под названием черного анилина.
Это — хинониминовый краситель, отличающийся ис-
ключительной прочностью ко всем факторам воздей-
ствия и глубиной черного цвета
КРАШЕНИЕ ТЕКСТИЛЬНЫХ МАТЕРИАЛОВ
777
В качестве окислителя применяют хлорат натрия и
бихромат натрия или калия; катализаторами окисли-
тельного процесса являются медный купорос, желези-
стосинеродистый калий и др.
На практике используют два способа крашения:
окислительный и запарной. Первый применяют гл. обр.
для крашения одежных тканей, второй — для краше-
ния легких тканей, в частности идущих для последую-
щей расцветки. Разница состоит в том, что черный
окислительный получается при обработке ткани в те-
чение не более 30 мин в специальных зрельниках
в воздушной среде при 50—60° и относительной влаж-
ности 40%. После этого следует дополнительное окис-
ление раствором бихромата. Черный запарной обра-
зуется в течение не более 1 мин в окислительных зрель-
пиках в паровой среде при 97—98°. Имеется разница
и в рецептуре при изготовлении растворов, к-рыми
ткань обрабатывают перед пропуском на зрельные
аппараты. Недостаток этого вида крашения — некото-
рое ослабление ткани и неблагоприятные санитарно-
гигиенич. условия при произ-ве. Степень поврежде-
ния ткани можно снизить, применяя вместо солянокис-
лого анилина производные сульфаминовой кислоты
(сульфаматы).
Крашение гидратцеллюлозных во-
локон. Особенности крашения гидратцеллюлоз-
ных волокон (вискозное и медноаммиачное) опреде-
ляются тем, что они по своей физич. структуре менее
однородны по сравнению с природными и отличаются
более рыхлым строением; в результате получаются
неравномерные окраски. В связи с этим перед краше-
нием выравнивают структуру этих волокон обработкой
в растворах, вызывающих нек-рое набухание (напр.,
едкий натр, сода). Крашение, как правило, ведут при
более высоких темп-рах и более низком содержании
электролита в ванне, чем при крашении хлопка. Все
это способствует выравниванию окраски. Еще лучшие
результаты в этом отношении получаются при формо-
вании волокна из окрашенного прядильного раствора,
т. н. «крашение в массе». Метод получил довольно
широкое распространение при произ-ве вискозного
волокна. Чаще всего для этой цели применяют водо-
растворимые сернистые красители.
О крашении волокон из ацетата целлюлозы см.
ниже «крашение химич. волокон».
III. Крашевие белковых волокон. Для крашения
протеиновых (белковых) волокон наибольшее приме-
нение получили кислотные, кислотно-протравные,
металлосодержащие и прямые красители. Взаимодей-
ствие протеиновых волокон с кислотными и кислотно-
протравными красителями основано на реакции:
NH3 NH3 NH3C1- NH3-O3SR'
I । +СЩ1 R'SOU +CI-
Illi
coo- соон соон COOH
Крашение проводят в кислой среде, при этом подав-
ляется кислотная диссоциация протеина и анионы
могут сорбироваться на положительно заряженных
аминогруппах. Вначале волокно адсорбирует более
подвижные и быстродиффундирующие в субстрат
неорганич. анионы, к-рые затем вытесняются с актив-
ных мест анионами красителей, обладающими большим
сродством к волокну. С увеличением кислотности
среды растет положительный заряд аминогрупп про-
теина, а следовательно, повышается и количество
сорбированного волокном красителя. Максимум пог-
лощения красителя волокном для шерсти составляет
0,90, для шелка—0,24—0,26 г-акв/кг волокна. При
уменьшении pH среды ниже 0,8 положительный заряд
приобретают амидные группы волокна; в результате
наблюдается дополнительное (сверх максимального
в обычных условиях) поглощение красителя. Однако
'778
в этих условиях возможна деструкция волокнистого
материала. Поэтому крашение при столь высокой ки-
слотности ванны, как правило, не ведут. Для посте-
пенного уменьшения величины pH красильной ванны
и равномерного поглощения красителя волокном
в нек-рых случаях раствор подкисляют не кислотой,
а легко гидролизующимися кислыми солями. При
нагревании р-ра в процессе крашения медленно по-
вышается концентрация водородных ионов, а следова-
тельно, и растет поглощение красителя волокном.
Постепенное повышение содержания нейтрального
электролита в красильном р-ре приводит к накопле-
нию неорганич. анионов в ванне и к созданию благо-
приятных условий для конкуренции между двумя
типами анионов. Это уменьшает поглощение краси-
теля волокном и способствует выравниванию окрасок.
Иногда для этой цели используют специальные вспо-
могательные вещества, напр. сульфокислоты нафта-
лина. Применяют и такие выравниватели, влияние
к-рых основано на взаимодействии с самим красите-
лем (см. Выравнивающие вещества). Уменьшение пог-
лощения кислотных красителей волокном под влия-
нием нейтральных электролитов характерно только
для крашения в кислой среде. В нейтральной и тем
более слабощелочной средах повышение концентра-
ции соли в растворе, наоборот, способствует сорбции
красителя волокном. Такое влияние электролита наи-
более резко проявляется при крашении шелка пря-
мыми красителями.
В зависимости от кислотности среды и содержания
нейтрального электролита в красильной ванне кислот-
ные красители подразделяются на хорошо-, плохо- и
средневыравнивающиеся. Для повышения равномер-
ности окраски и сокращения длительности крашения
применяют суспензии бариевых солей кислотных кра-
сителей. В этом случае процесс состоит из двух ста-
дий — обработка материала в суспензии красителя и
кипячение в воде в течение 3—7 мин.
Нек-рые кислотные красители взаимодействуют на
волокне с солями хрома, образуя при этом комплекс-
ные соединения — лаки, очень прочные ко всем видам
воздействия. Окрашиваемый материал можно обраба-
тывать солями хрома до крашения, после него или
одновременно с крашением. В соответствии с этим
различают способы крашения с предварительным,
последующим и одновременным хромированием. В ка-
честве хромирующего вещества используют соли
6-валентного хрома К2Сг2О, или Na2Cr2O7. Для пере-
вода хрома в 3-валентное состояние в ванну добав-
ляют восстановители — муравьиную, щавелевую к-ты
или калиевую соль виннокаменной к-ты. Иногда вос-
становителем служит сам краситель.
Крашение белковых волокон кислотно-протравными
красителями значительно сложнее, чем обычными
кислотными красителями; при крашении шелка поло-
жение осложняется еще и тем, что хромирование при-
дает жесткость волокну. Для преодоления недостат-
ков, присущих кислотно-протравным красителям,
прибегают к использованию металлосодержащих кра-
сителей, которые делятся на две группы в зависимо-
сти от числа молекул красителя, приходящихся
на один атом хрома. По этому признаку различают
1:1 и 2 : 1-металлосодержащие красители. К пер-
вой группе относятся, в частности, палатиновые, или
неолановые, красители, отличающиеся тем, что рав-
номерно окрашивают материал лишь при увеличен-
ном расходе серной к-ты (до 8—10% к весу волокна).
Т. обр., они проявляют свойства, противоположные
обычным кислотным красителям, к-рые при повыше-
нии кислотности ванны быстрее выбираются и ухуд-
шают равномерность окраски. При повышении кон-
центрации водородных ионов меньшее число амино-
групп кератина остается в незаряженном, доступном
779
КРАШЕНИЕ ТЕКСТИЛЬНЫХ МАТЕРИАЛОВ
780
для взаимодействия с атомом хрома, состоянии. Это
ухудшает условия связывания красителя волокном и
способствует получению более равномерных окрасок
с комплексом 1:1. Металлосодержащие красители
красят шерсть в слабокислой или нейтральной среде;
при этом волокно не теряет прочности. В кислой среде
между положительно заряженными центрами кератина
и отрицательными ионами красителя происходит соле-
образование. В нейтральной и слабощелочной среде
краситель фиксируется за счет межмолекулярных сил
и водородных связей. Для крашения по периодич. спо-
собу берут 0,2—1,0% красителя и 2—3% сульфата
аммония от веса волокна.
Белковые волокна окрашиваются основными кра-
сителями непосредственно; этими красителями чаще
окрашивают шелк. Прямые красители при крашении
шелка дают более прочную окраску, чем кислотные.
В светлые тона шелк красят в слабощелочной ванне,
содержащей 2—3% (от веса материала) олеинового
мыла и иногда 5—7% соли, а в средние и темные цве-
та — в нейтральной ванне в присутствии электролита
(10% NaCl или 20% Na2SO4). Крашение начинают при
40°, постепенно нагревают раствор до 90—95° и красят
при этой темп-ре 1 час. Применение кубовых краси-
телей ограничено ввиду разрушающего действия
щелочи на белковые волокна. Чтобы снизить щелоч-
ность красильного раствора до pH 9,5—10 и этим рас-
ширить возможность применения кубовых красителей
для крашения белковых волокон, можно брать орга-
нич. основание—триэтаноламин—N(C2H4OH)3, являю-
щееся в нек-рой степени растворителем и дисперга-
тором кубовых красителей. При крашении с триэтано-
ламином окраски белковых волокон кубовыми краси-
телями отличаются высокой яркостью и прочностью;
степень же деструкции волокна примерно такая же,
как и при крашении кислотно-протравными краси-
телями. Этот способ крашения пока еще широкого
распространения в практике не получил.
Шелк и шерсть можно окрашивать кубозолями.
Первую стадию процесса — обработку материала
в р-ре кубозоля — ведут в присутствии 4—5% ук-
сусной к-ты и 5—10% сульфата натрия. Вторую ста-
дию—проявление окраски—осуществляют в растворе
нитрита натрия и серной к-ты или хромпика и серной
к-ты. Для крашения белковых волокон можно исполь-
зовать образование нерастворимых азопигментов не-
посредственно на волокне. С этой целью синтезиро-
ваны растворимые в воде азосоставляющие и раство-
римые формы готовых красителей, получивших наз-
вание солазолевых. Разработаны способы крашения
этими красителями. Белковые волокна обычно окра-
шивают в аппаратуре периодич. действия (барки и
красильные роликовые машины); запарной метод кра-
шения начинают применять на аппаратах непрерыв-
ного действия.
IV. Крашение химических волокон. При разработке
способов крашения волокон, полученных из различных
синтетич. полимеров, а также изделий из них, встрети-
лись значительные затруднения, вызванные малым на-
буханием волокон, их высокой кристалличностью и
низким содержанием активных групп, способных
к взаимодействию с активными группами красителей.
В зависимости от химич. природы волокна крашение
его производят различными красителями.
Крашение полиамидных волокон.
Получаемые из капролактама (капрон, перлон, ней-
лон-6, силон, трилон и др.) или из гексаметиленди-
амина и адипиновой к-ты (анид, нейлон-66 и др.)
волокна красятся в наименьшими трудностями; эти
волокна не слишком кристалличны и обладают нек-рой
способностью к набуханию в воде; их кондиционная
влажность ок. 4%. Благодаря наличию в структуре
полиамидных волокон гидрофильных групп (NH2,
СООН и др.), а также гидрофобных парафиновых це-
пей они обладают способностью накрашиваться кра-
сителями различной природы: дисперсными, кислот-
ными, основными, прямыми, металлосодержащими,
кислотно-протравными, кубовыми и нек-рыми дру-
гими видами красителей. Кислотные и основные кра-
сители закрепляются на волокне путем образования
ионной связи; остальные виды красителей фикси-
руются гл. обр. за счет образования водородных свя-
зей с амидными группами. Наиболее легко эти
волокна накрашиваются дисперсными красителями,
обычно используемыми в крашении ацетатного шелка.
Краситель диспергируют в холодной или теплой воде
и вводят в красильную ванну, содержащую мыло
или, лучше, неионогенный диспергатор, напр. ОП-Ю
(1 г/л). Крашение начинают при 40—50° и закапчивают
в течение 1 час при 80°.,Широкое использование диспер-
сных красителей при крашении капронового волокна
объясняется гл. обр. способностью их обеспечивать
равномерную окраску тканей. Для получения окрасок
с более высокой прочностью к мокрым обработкам
применяют дисперсные красители, подвергающиеся
последующему диазотированию и сочетанию непо-
средственно на волокне с бетаоксинафтойной к-той
или к.-л. другой азосоставляющей. С этой же целью
синтезируют нерастворимые оксиазокрасители непо-
средственно на волокне: сначала изделия обрабаты-
вают в высокодисперсной суспензии азотолов и азо-
аминов, а затем проявляют окраску пропусканием про-
питанного материала через подкисленный р-р нитрита
натрия. При этом диазотирование азоамина и сочета-
ние полученного диазония с азотолом идут в волокне
одновременно при темп-ре ок. 50°.
Применение новой технологии крашения в этом слу-
чае вызвано тем, что при обычных способах холодного
крашения нельзя обеспечить удовлетворительную
сорбцию азотолов волокном, а также достаточное
проникновение в него холодных диазорастворов при
проведении сочетания. Взамен азоаминов могут быть
использованы специальные виды стойких к повышен-
ной темп-ре диазоаминосоединений (офнаперсоли).
Сочетание их с азотолами достигается при обработке
пропитанного волокна в горячем р-ре серной к-ты,
т. е. без диазотирования.
Полиамидные волокна можно окрашивать и кислот-
ными красителями. Поглощение последних волокном
очень сильно зависит от pH среды: в области pH от 8
до 5 наблюдается медленный рост сорбции кислотного
красителя волокном. В интервале pH 5—2 поглоще-
ние красителя практически не изменяется, а при
pH 2 — резко возрастает. Это обусловлено тем, что
при pH 5—2 происходит сорбция анионов красителя
на концевых аминогруппах, а при pH 2 сорбция воз-
можна и за счет заряжающихся положительно амидных
групп волокна. Обычно крашение кислотными краси-
телями проводят в слабокислой среде (pH 5—2).
При этом получаются малоинтенсивные окраски, т. к.
число концевых аминогрупп у полиамидных волокон
невелико. Крашение в сильнокислых средах с целью
получения интенсивных окрасок невозможно вслед-
ствие потери механич. прочности волокна.
Кислотные красители дают неравномерную окраску,
обусловленную различной степенью вытягивания и
неодинаковой структурой разных партий полимера.
Для устранения этого недостатка иногда применяют
способ крашения (способ кемнайл), основанный на
осаждении этих красителей катионоактивными про-
дуктами типа четвертичных аммониевых оснований
с одновременным переводом образовавшихся осадков
в состояние устойчивой суспензии. Полученными
суспензиями красят изделия из полиамидных волокон
в муравьинокислом р-ре (pH 3,5) при темп-ре ок. 80°.
В таких условиях анионные красители приобретают
781
КРАШЕНИЕ ТЕКСТИЛЬНЫХ МАТЕРИАЛОВ
782
свойства, близкие к дисперсным. Вариантом данного
способа является крашение суспензией бариевых
солей кислотных красителей.
Прямые красители хотя и применяются для краше-
ния изделий из полиамидных волокон, но не обладают
заметными преимуществами по сравнению с кислот-
ными красителями. Большой интерес для крашения
полиамидных волокон представляют металлосодер-
жащие анионные комплексы, свободные от сульфо-
групп (1 : 2). Эти красители позволяют получать
окраски выдающейся прочности к действию света и
мокрых обработок. Крашение ими производят из
нейтральной или содержащей аммонийные соли ванны
с добавкой выравнивающих веществ. Для крашения
полиамидного волокна и штапельной пряжи иногда
применяют кислотно-протравные красители. Красят
ими так же, как и кислотными красителями. После-
дующее же хромирование проводят в р-ре муравьиной
к-ты с добавкой тиосульфата натрия для перевода
хрома в 3-валентное состояние. Основные красители,
в связи с их низкой светопрочностью, для крашения
полиамидных волокон не рекомендуются. Также почти
не применяют для крашения и кубовые красители,
к-рые на этом волокне не дают светопрочных окрасок.
Однако водорастворимые препараты кубовых краси-
телей — кубозоли — используют несколько шире. В
этом случае сначала материал пропитывают р-ром
кубозоля в присутствии уксусной к-ты и сульфата
аммония, а затем окраску проявляют в растворе сер-
ной к-ты и хромпика.
Для прочного крашения полиамидных волокон могут
быть использованы и активные красители, вступаю-
щие в химич. связь с волокном (проционовые, проци-
ниловые, цибакроновые и ремазолевые). Однако только
проциниловые красители образуют на тканях равно-
мерную окраску. Они применяются как дисперсные
красители, но отличаются от последних тем, что в на-
чальной стадии окрашивают полиамид не из нейтраль-
ной, а из слабокислой ванны. Крашение заканчивают
в щелочной ванне, где и происходит фиксация краси-
теля волокном с образованием ковалентной связи.
Крашение полиэфирных волокон.
Крашение волокон этого типа (лансан, терилен, дак-
рон, ланон) затруднено тем, что они еще менее гигро-
скопичны (кондиционная влажность 0,4%), чем поли-
амидные волокна; они не набухают в воде и неспособны
к поглощению водорастворимых красителей. Эти
волокна красят обычно дисперсными красителями.
Но, даже пользуясь этими красителями в обычных
условиях при 95—100°, получают только светлые
оттенки, что связано с большой трудностью проник-
новения красителей внутрь волокна. Для ускорения
крашения и получения темных оттенков прибегают
к т. наз. «переносчикам», или интенсификаторам кра-
шения, и процесс ведут при темп-рах выше 100°.
Механизм действия переносчиков заключается в том,
что они, адсорбируясь волокном, вызывают его набу-
хание и этим обеспечивают расширение пор и капилля-
ров волокна. Сродство переносчиков как к синтетич.
волокну, так и к дисперсным красителям создает
условия к быстрому извлечению красителя из ванны
пленкой переносчика, адсорбированной на поверх-
ности полимера. Возникающий высокий концентра-
ционный градиент обеспечивает повышенную скорость
диффузии красителя внутрь волокна. В качестве
интенсификаторов крашения практич. применение по-
лучили о-фенилфенол, дифенил, моно- и дихлорбен-
золы, бензойная и салициловая к-ты, бетанафтол.
Для быстрого и интенсивного окрашивания поли-
эфирных волокон без использования переносчиков
применяют также повышенные темп-ры. Крашение
при темп-рах выше 100° сопровождается сравнительно
большим отложением красителя на поверхности во-
локна. В связи с этим для повышения прочности ок-
раски к трению и мокрым обработкам поверхностно-
сорбированный краситель разрушают обработкой
изделия при 70° в щелочном р-ре гидросульфита.
С целью интенсификации проникновения дисперсных
красителей в полиэфирное волокно применяют краше-
ние в парах трихлорэтилена. Для этого волокнистый
материал пропитывают водной дисперсией красителя,
высушивают и пропускают через закрытую камеру,
заполненную парами трихлорэтилена. Последний
растворяет краситель и одновременно вызывает зна-
чительное набухание волокна; в результате краситель
быстро диффундирует в набухшее волокно. Для окра-
шивания полиэфирных волокон применяют также обра-
зование в них нерастворимых оксиазокрасителей.
Делается это также, как и для полиамидных волокон.
Разница состоит в том, что пропитку волокна в высо-
кодисперсной суспензии азо- и диазосоставляющих
ведут при темп-ре не ниже 100°, а последующее прояв-
ление окраски в кислом р-ре нитрита натрия при 85°.
Полиакрилонитрильные волок-
н а. Такие волокна, как нитрон, орлон, пан, волькри-
лон и др., окрашиваются дисперсными красителями
в светлые тона при 95—100°. Более интенсивные ок-
раски дисперсными красителями могут быть получены
при применении переносчиков (о-фенилфенола) или
путем крашения при 100—115°. Благодаря слабокис-
лотному характеру полиакрилонитрильные волокна
способны поглощать красители катионного типа (основ-
ные). На этих волокнах светопрочность основных кра-
сителей гораздо выше, чем на других типах текстиль-
ных материалов. Для крашения полиакрилонитрило-
вых волокон этого типа были созданы особые
катионные красители (астразоны, севроны, деорлены,
максилоны и др.); пользуясь этими красителями, можно
получать на акрилонитрильных волокнах яркие и
насыщенные оттенки довольно высокой прочности.
Крашение производят без использования переносчи-
ков, в уксуснокислой среде, в присутствии глауберо-
вой соли и неионогенных выравнивающих веществ;
темп-ра ванны 95—100°, время крашения 60—90 мин.
В присутствии солей закиси меди полиакрилонит-
рильные волокна способны интенсивно окрашиваться
красителями анионного типа — кислотными и пря-
мыми. Вначале ионы одновалентной меди присое-
диняются к азоту нитрильной группы, придавая во-
локну положительный заряд, а затем на этих поло-
жительно заряженных активных центрах сорбируются
красители анионного типа. Крашение полиакрилонит-
рильных волокон кислотными красителями произ-
водят в кислой красильной ванне в присутствии суль-
фата меди и гидрокси ламин а. Последний можно заме-
нить металлич. медью (проволочной сеткой). Для
крашения по медно-ионному методу были предложены
специальные кислотные антрахиноновые красители
(сандокриловые). Полиакрилонитрильное волокно с
примесью винилпиридина — акрилан — хорошо ок-
рашивается не содержащими сульфогрупп азокраси-
телями типа металлокомплексов 1 : 2 в минерально-
кислой среде при 100° с применением специального
переносчика. Перед крашением материал отваривают
в р-ре серной к-ты (2 г/л).
Для значительного повышения восприимчивости
нитрона по отношению к красителям всех классов
предложено проводить крашение свежесформованного,
но не высушенного волокна, особенно содержащего
нек-рое количество неудаленного диметилформамида.
Предложено также модифицировать волокно посред-
ством переводанек-рой части нитрильных групп в амид-
оксимные. Для этого волокно или пряжу обрабаты-
вают при 90° в растворе сульфата гидроксиламина
(10—20 г/л) с добавкой небольших количеств едкого
натра (до pH 6,5). Модифицированное волокно приоб-
783
КРАШЕНИЕ ТЕКСТИЛЬНЫХ МАТЕРИАЛОВ
784
ретает способность непосредственно накрашиваться
кислотными и прямыми красителями.
Поливинилхлоридные волокна.
Волокна этого типа — хлорин, пеце, ровил и др.,
а также поливинилиденхлоридные волокна (совиден,
саран и др.) не содержат активных групп в своих
молекулах и поэтому неспособны к набуханию и
почти не поглощают красители. Низкая темп-ра их
размягчения (65—90°) не позволяет применять высо-
котемпературные методы крашения. Наиболее при-
годными красителями в данном случае являются
дисперсные. В качестве вспомогательных веществ,
ускоряющих крашение, применяют этилцеллозольв,
хлорбензол, ароматич. амины. Нек-рые поливинилхло-
ридные волокна можно окрашивать образованием на
них нерастворимых оксиазокрасителей, так как это
было описано ранее.
Волокна из ацетата целлюлозы.
По отношению к красителям эти волокна напоминают
синтетич. материалы и неспособны окрашиваться кра-
сителями, обычно применяемыми для крашения цел-
люлозных волокон. Наибольшее распространение для
крашения как обычного ацетатного волокна, так и
триацетатного волокна нашли дисперсные красители.
Для крашения ацетатного шелка применяют гл. обр.
дисперсные формы нерастворимых или малораствори-
мых в воде азокрасителей или антрахиноновых краси-
телей. Крашение ткани этими красителями начинают
при 40°, затем темп-ру медленно в течение 90 мин
доводят до 85° и красят при этой темп-ре еще 60 мин.
Дисперсные красители, содержащие диазотируемую
аминогруппу, можно диазотировать на волокне и
сочетать с резорцином, 0-пафтолом, 2,3-оксинафтойной
к-той или метафенилендиамином. При этом получают
более прочные и более глубокие по оттенку окраски.
Дальнейшим достижением следует считать крашение
натриевыми солями сернокислых эфиров — производ-
ных этаноламина, напр.:
С2Н5
NX
4CH2CH2OSO3Na
Эти соединения получили название солацетовых кра-
сителей. Они растворяются в воде, не теряя сродства
к ацетатному волокну. Солацетовые красители не
претерпевают никаких химич. превращений в процессе
крашения, поэтому ими красят аналогично крашению
хлопка прямыми красителямй. Аналогия с прямым
крашением хлопка увеличивается также вследствие
чувствительности солацетовых красителей к измене-
нию концентрации соли в красильной ванне. Процесс
можно регулировать прибавлением поваренной или
глауберовой соли, чем достигают желательной степени
выбираемости красителя из ванны. Остаток алкилсер-
ной к-ты сообщает красителю поверхностно-активные
свойства, повышая его способность проникать в во-
локно. Солацетовые красители являются особенно
ценными при крашении плотных тканых изделий.
Один из способов крашения ацетатного шелка со-
стоит в применении в одной ванне смеси азотола и
ароматич. амина, т. к. это было описано ранее. Для
крашения изделий из триацетатного волокна, и отчасти
из обычного ацетатного волокна, предложен след, спо-
соб: ткань пропитывают слегка загущенным р-ром
дисперсного красителя и пропускают после сушки че-
рез камеру, заполненную парами трихлорэтилена.
В этих условиях полная фиксация дисперсного краси-
теля волокном достигается за 30—60 сск. При краше-
нии триацетатного волокна дисперсными красителями
иногда с целью интенсификации процесса прибегают
к темп-ре выше 100° и к использованию в качестве
переносчика диэтилфталата.
Химич, волокна можно также окрашивать в процессе
их изготовления. В этом случае в расплав или раствор
высокополимера вводятся высокодисперсные пигмен-
ты и из этой окрашенной массы формуют волокно.
Такой способ получил название «крашение в массе».
V. Крашение смешанных волокон. Для крашения
пряжи и ткани, состоящих из смеси целлюлозных и
белковых волокон, напр. хлопка и шерсти или смеси
химич. волокон с целлюлозными или белковыми,
напр. полиакрилонитрильного с шерстяным, приме-
няют красители разных групп. Ткань сначала окраши-
вают красителем, выбирающимся одним из волокон
и не затрагивающим второе; затем ткань окрашивают
другими красителями, действующими на оставшееся
неокрашенным волокно и не затрагивающими уже
окрашенное (двухванный способ). Возможна окраска
смесью красителей из одной ванны, напр. смесь хлоп-
ка и шерсти можно окрашивать одновременно смесью
кислотных и прямых красителей. Такие виды смесей
как, напр., хлопок и гидратцеллюлозные волокна,
шелк и шерсть, целлюлозные волокна и шерсть, аце-
татное волокно с хлопком, вискозой, шелком или
шерстью применялись уже давно. В настоящее время
все более широкое распространение приобретают
смеси полиакрилонитриловых волокон с шерстью,
полиэфирных волокон с шерстью, хлопком, нейлоном
и др. волокнами, полиамидных волокон с шерстью
и т. д.
При крашении тканей из хлопка и гидратцеллюлоз-
ных волокон обычно используют прямые, сернистые,
кубовые красители, а также кубозоли и образующиеся
на волокне нерастворимые оксиазокрасители. Для
того чтобы на обоих волокнах получить окраску оди-
наковой интенсивности, уменьшают повышенную вос-
приимчивость гидратцеллюлозных волокон к краси-
телям. С этой целью уменьшают содержание электро-
лита в ванне и одновременно повышают ее темп-ру по
сравнению с условиями крашения изделия из одного
хлопка.
Изделия из хлопка и шерсти часто окрашивают в две
стадии: сначала кислотными красителями окрашивают
протеиновое волокно, а затем прямыми красителями—
целлюлозное волокно при 65—70°. В этих условиях
(нейтральная среда при темп-ре ниже 100°) шерсть
практически не закрашивается прямыми красителями
и краситель переходит только на целлюлозные волок-
на. Полушерстяные ткани можно также окрашивать
смесью кислотных и прямых красителей из одной
ванны, а также кубовыми красителями в присутствии
триэтаноламина вместо едкого натра. Смешанные
ткани из ацетатного волокна и хлопка или вискозы
можно окрашивать из одной ванны и получать окраску
в один или в два разных цвета, применяя смесь прямых
красителей для хлопка и дисперсных для ацетилцел-
люлозы. При этом, если необходимо, то прибавляют
диспергирующее вещество.
Изделия из полиакрилонитриловых волокон в смеси
с шерстью окрашивают по двухванному способу основ-
ными и кислотными красителями. Обычно вначале ве-
дут крашение синтетич. волокна основными краси-
телями при кипячении в присутствии уксусной к-ты
и препарата О П-10 и затем шерсти — кислотными
красителями, пользуясь обычными методами. Полиак-
рилонитриловые волокна в смеси с целлюлозным
волокном практически не окрашиваются прямыми,
кубовыми, сернистыми, активными и кубозолевыми
красителями. Более или менее однородные окраски
можно получить только при использовании смесей
прямых и дисперсных красителей.
При крашении смеси шерсти и полиэфирных воло-
кон (терилен, лавсан) синтетич. волокно окрашивают
дисперсными красителями в присутствии переносчи-
ков, напр. о-фенилфенола, а шерсть — кислотными,
785
КРЕАТИН — КРЕАТИНФОСФОРНАЯ КИСЛОТА
786
металлосодержащими, кислотно-протравными краси-
телями. Для крашения в светлые тона можно приме-
нять однованный метод, а в темные — двухванпый;
сначала полиэфирное волокно окрашивают дисперс-
ными красителями, потом шерсть — кислотными
или другими анионными красителями.
Изделия из смеси полиэфирных волокон с хлопком
можно окрашивать в одной ванне быстрокрасящими
дисперсными и прямыми красителями. С целью повы-
шения прочности окраски на хлопке лучше использо-
вать двухванный способ крашения. Сначала окраши-
вают полиэфирное волокно дисперсными красителями
при 130°, а затем хлопок — кубовыми красителями
в свежеприготовленной ванне. Кубовые красители
можно применять как по обычному способу в лейко-
форме, такипо суспензионному методу. Для закраши-
вания хлопка используют также активные и нераство-
римые оксиазокрасители. Крашение ими ведут обыч-
ными методами, но при этом обязательно вначале
окрашивают полиэфирное волокно дисперсными кра-
сителями.
Смеси полиэфирных волокон с хлопком можно окра-
шивать в светлые тона кубозолями с последующим
проявлением окраски нитритом. Очень хороших
результатов удается достигнуть при применении ку-
бозолей для крашения тканей из смеси капрона
с шерстью. Изделия из полиамидных и полиэфирных
волокон окрашивают дисперсными красителями по
однованному методу. Иногда с целью повышения ин-
тенсивности окраски полиамидного волокна его под-
крашивают кислотными или металлосодержащими
красителями.
Если волокнистые материалы в смеске резко раз-
личаются по своим свойствам и отношению к краси-
телям, целесообразно их красить раздельно по спосо-
бам, описанным в соответствующих разделах, или же
следует применять пигментные красители. Закрепле-
ние этих красителей на волокне не зависит от природы
субстрата и производится с помощью синтетич. свя-
зующих веществ. См. также Красители синтетиче-
ские.
Лит.: Садов Ф. И., Корчагин М. В., Матец-
к и й А. И., Химическая технология волокнистых материа-
лов, 2 изд., М., 1956; Виккерстафф Т., Физическая
химия крашения, пер. с англ., М., 1956; Мор ы гав ов П. В.,
Мельников Б. Н., Усп. химии, 1956, 25, вып. 9, 1149;
их же, Изв. Высших учебных заведений. Технология тек-
стильной пром-сти, 1958, М 5, 36; Беленький Л. И.,
Автоматический контроль и регулирование технологических
процессов отделочного производства, М., 1960; Гордон
Н. Б., Борисов Н. А., Отделка льняных тканей, М.,
1956; Соловьев Н. П., Крашение хлопка сернистыми
и кубовыми красителями, М., 1955; Применение кубовых
красителей, пер. с англ., М., 1957; Цоллингер Г.,
Химия азокрасителей, пер. с нем., Л., 1960; Шиканова
И. А., Матецкий А. И., Отделка шерстяных тканей, М.,
1958; Виккерстафф Т., в кн.: Волокна из синтетиче-
ских полимеров, под ред. Р. Хилла, пер. с англ., М., 1957,
с. 463; Венкатараман К., Химия синтетических кра-
сителей, пер. с англ., т. 1, М., 1956, с. 337, 330, 366, 338;
Коган И. М., Химия красителей, 3 изд., М., 1956, с. 235—36,
247—50; Крашение изделий из химических волокон. Зарубеж-
ная техническая информация. Бюро технической информации
легкой промышленности, [Л.], 1957; Handbook of biological
data, ed. W. S. Spector, Phil.—L., [1956]; KramrischB.,
J. Soc. Dyers Colourists, 1959, 75, № 5, 242; H i n d 1 e W. H.,
там же, 1958, 74, M 3, 151; Partridge H. W., там же,
1959, 75, М 7, 373. II. В. Морыганов, Б. Н. Мельников.
КРЕАТИН (N-метилгуанидинуксусная кислота,
, ,NH2
гуанидинметилглицин) HN==C < , мол.
\N(CHS)-CHS-COOH
вес 131,14—содержится главным образом в мышечной
ткани животных, найден также в нервной ткани,
крови и в моче. К. кристаллизуется с 1 молекулой Н.2О
из водных р-ров в виде моноклинич. призм, т. пл. 315°
(с разл.). К. растворяется в горячей воде, плохо —
в холодной, мало растворим в спирте, нерастворим
в эфире. При действии минеральных к-т на К. послед-
ний теряет молекулу воды и превращается в лактам —
креатинин, на чем основано качественное и коли-
чественное определение К. В щелочной среде К. разру-
шается с образованием мочевины и метилглицина
(саркозина); с диацетилом К. дает цветную окраску,
используемую для колориметрия, определения К.
К. получают синтетически при взаимодействии
цианамида и метилглицина:
N^C-NH24-HOOCCH2NHCH3—>к
Кроме того, К. можно выделить из водного мышечного
экстракта. В организме К. образуется в результате
обмена белков; ферментативная реакция переноса
аминокарбонимидной (амидинной) группы от аргинина
к глицину приводит к образованию гуанидинуксус-
ной к-ты, к-рая затем метилируется с образованием К.:
HN.X .СООН
>C-NH(CH2)3CH< 4-H2NCHoCOOH------►
H..NZ 'NHa
нх
—► >C—NHCH2COOH >K
H2Nz
Обычно К. превращается в организме в креатинин,
к-рый выводится с мочой.
Лит.: Мешкова Н. П., Северин С. Е., Практи-
кум по биохимии животных, М., 1950; Handbook of biological
data, ed. W. S. Spector, Phil.—L., 1956; Methods in enzy-
mology, v. 3, N. Y., 1957; Blochem. Preparation, 1957, 5,
9; White A. [a. o], Principles ot biochemistry, 2 ed.,
N. Y. — [a. o.], 1959; Archibald R. M., J. Biol. Chem.,
1962, 237, № 2, 612; F a w a z E. N. [a. o.], Proc. Soc. Exptl.
Biol, and Med., 1962, 109, 38. Л. H. Боброва.
,NH-----CO
КРЕАТИНИН HN=C< | , мол. в. 113,12—
'N(CHS)-CH2
лактам креатина, один из конечных продуктов азоти-
стого обмена позвоночных, нормальная составная
часть мышц и др. тканей, а также мочи. К. — бес-
цветные кристаллы, т. пл. 260° (с разл.); хорошо раст-
ворим в воде, растворим в спирте, нерастворим в эфире.
Двойная соль К. с хлористым цинком плохо раство-
рима в воде и нерастворима в спирте. При действии
на К. щелочи образуется креатин. С пикриновой
к-той в щелочной среде К. образует пикрат (т. пл.
220°) красно-оранжевого цвета (реакция Яффе);
с нитропруссидом натрия в щелочной среде раствор К.
окрашивается в красный цвет, переходящий при
стоянии в желтый. При кипячении или подкислении
желтая окраска переходит в зеленую, а затем в синюю
(реакция Вейля — Сальковского). Коли-
чественное определение К. основано на цветных реак-
циях с пикриновой к-той и 3,5-динитробензойной
к-той. К. получают действием минеральных к-т
на креатин. В организме К. образуется из креатина.
Лит- см. при ст. Креатин. Л. Н. Боброва.
КРЕАТИНФОСФОРНАЯ КИСЛОТА (фосфокреа-
, , ,КП~РО3Н2
тин, фосфаген) HN = С< ,мол. в. 211,12—
'N(CH3)CH2COOH
нормальный продукт обмена в мышечной, нервной
и др. тканях позвоночных. К. к. — очень нестойкое
соединение, легко гидролизуется с образованием
креатина и фосфорной к-ты. Na- и Ва- соли К. к.
растворимы в воде и нерастворимы в спирте. Качест-
венное и количественное определение К. к. проводят
по креатину и фосфорной к-те, образующихся при
кислотном гидролизе, а также энзиматически. К. к.
получают при действии РОС13 на креатин в щелочной
среде. К. к. — важный источник фосфатных остатков
при ферментативном синтезе аденозинтрифосфорной
к-ты (АТФ) из аденозиндифосфорной к-ты (АДФ).
В качестве макроэргич. соединения (изменение сво-
бодной энергии при отщеплении фосфата составляет
ок. 10 ккал/моль) К. к. участвует в биохимич. реак-
циях, необходимых в различных физиологич. про-
цессах (напр., при работе мышц).
Лит. см. при ст. Креатин. Л. И. Боброва»
787
КРЕЗИДИН — КРЕКИНГ
788
КРЕЗИДИН (З-амино-4-метокситолуол) мол. в.
137,29, почти бесцветные иглы; т. пл. 51,5°, т. кип.
235°; легко растворяется в большинстве
.1 органич. растворителей; трудно растворим
[ Д в воде; перегоняется с водяным паром.
К. — первичный ароматич. амин; с силь-
qth ними кислотами образует устойчивые соли;
® легко вступает в реакции диазотирования,
ацилирования, алкилирования. Из реакций электро-
фильного замещения практич. значение имеют азо-
сочетание и нитрование. С целью введения нитро-
группы К. предварительно ацетилируют; после нит-
рования в полученном 3-ацетиламипо-4-метокси-6-
нитротолуоле обычным путем удаляют ацетильный
остаток.
К. получают из га-толуидина или 4-хлор-З-нитро-
толуола. К. — важный полупродукт для произ-ва
фиолетовых, синих, черных и коричневых азокраси-
телей для шерсти, хлопка и ацетатного волокна. К.
вначале используют как азосоставляющую, после чего
в полученном азокрасителе диазотируют аминогруппу
крезидинового остатка и вводят в дальнейшее сочета-
ние с другими азокомпонентами. в. А. Пучков.
КРЕЗОЛ СН3—СвН4—ОН, мол. в. 108,14 — жид-
кость; известны все три изомера: орто (1-метил-2-
оксибензол), мета (1-метил-З-оксибензол) и пара
(1-метил-4-оксибензол).
Свойства орто мета пара
т. пл., °C 30,8 12,0 34,7
Т. кип., °C, 760 мм 190,8 202,8 201,8
200 мм 146,7 157,3 157,7
100 лиг 127,4 138,0 140,0
10 мм 76,7 87,8 88,6
1,0465 1,0344 1,0347
<1«° 0,994 0,986 0,986
20 nD 1,5453 1,5438 1.5359
Вязкость при 80°, ест 1,47 1,76 2,0
Растворимость в воде, г/100 мл Константа диссоциации в вод- 3,1 (40°) 2,35 (20°) 2,4 (40°)
ных р-рах (25°) Теплота сгорания при i?=const, 6,3 • 10-п 9,8Л0~Ц 6,7-10-и
ккал/моль 883 881 883
Т. пл. N-фенилкарбамата, °C . 142,5 124,5 113
Все изомеры хорошо растворимы в обычных органич.
растворителях. Подобно фенолу, К. обладает свойст-
вами слабых к-т и растворяется в щелочах с образо-
ванием солей — крезолятов. Химич, свойства К.
связаны с наличием бензольного кольца, метильной
группы и гидроксильной группы. К. легко вступает
в реакции электрофильного замещения, легко гало-
генируется, нитруется, сульфируется и т. п. с обра-
зованием соответствующих моно- и полипроизводных;
подобно фенолу, К. легко вступает в реакции конден-
сации.
Для идентификации К. используют как цветные
реакции, так и образование характерных производ-
ных. Кроме того, К. способен давать аддукты с раз-
личными соединениями, напр. с пиридиновыми осно-
ваниями, нафталином, уротропином, щавелевой к-той.
Основным источником промышленного получения
К. являются крезольные фракции смол, образующихся
при термич. обработке различных топлив: коксова-
нии каменного углп, полукоксовании бурого угля
или торфа, горючих сланцев, сухой переработки дре-
весины. К. образуется также при гидрогенизации
топлива. Для разделения К. на изомеры используют
различные методы фракционирования. Из смеси К.
можно выделить о-К.; для разделения м- и га-К. ис-
пользуют различные методы, основанные на разнице
в их химич. свойствах.
Синтетически К. получают либо из толуолсульфо-
кислот, либо из толуидинов. К. является сырьем для
произ-ва крезоло-альдегидных смол. Довольно боль-
шие количества К. расходуются на синтез различных
красителей, медицинских препаратов, дезинфекцион-
ных средств, взрывчатых веществ, для произ-ва ду-
бящих веществ и флотореагентов. о-К. является сырьем
для произ-ва салициловой и о-крезотиновой к-т;
.«-К. — исходный продукт для получения амбрового
мускуса, .«-толуидина и метилциклогексанола; га-К.—
для произ-ва га-оксибензальдегида, га-крезидина,
га-крезотиновой к-ты.
Лит.: Ворожцов Н. Н., Основы синтеза промежуточ-
ных продуктов и красителей, М., 1950. В. Н. Александров.
КРЕКИНГ. Содержание:
I. Крекинг термический.................... 788
II. Крекинг каталитический................. 793
III. Крекинг с водяным паром............... 798
IV. Крекинг под давлением водорода (гидрокрекинг) . . 798
Крекинг — процесс деструктивной переработки неф-
ти или ее фракций, проводимый для увеличения выхода
легких продуктов и повышения их качества, гл. обр.
для получения легких моторных топлив, иногда для
других целей. При К. преобладает распад тяжелых
молекул, но его нельзя отождествлять только с де-
струкцией, т. к. наряду с этим при К. происходят слож-
ные процессы синтеза и перестройки молекул углево-
дородов. Различают два основных вида: К., осуществ-
ляемый только под воздействием высокой темп-ры,—
термический — и К., происходящий при одновре-
менном воздействии высокой темп-ры и катализато-
ров — каталитический. Дальнейшая классификация
процессов производится в зависимости от условий
ведения процесса (темп-ра, давление), назначения,
вида сырья и технологии, оформления. Известны
нек-рые другие виды К., напр. с водяным паром или
под давлением водорода (гидрокрекинг), или же
окислительный К.
I. Крекинг термический. Первым по времени возник
термич. К.; он был известен еще в 19 в., однако в связи
с отсутствием спроса на бензин не получил промыш-
ленного применения. Строительство промышленных
установок началось в 20 гг. сначала в США, а затем
в СССР. Современная нефтеперерабатывающая
пром-сть применяет след, разновидности термич. К.:
1) под высоким давлением для получения бензина;
2) остаточного сырья под низким давлением (кок-
сование) с получением широкой фракции, облегчен-
ного состава для последующей переработки, и нефтя-
ного кокса; 3) высокотемпературный под низким дав-
лением (пиролиз) для получения газа с высоким со-
держанием олефинов и ароматич. углеводородов.
Крекинг под высоким давлени-
е м. Процесс ведут при 470—540° и давлении 40—•
60 ат, используют его для переработки различного
сырья — от лигроинов до гудронов включительно.
Этот вид К. получил наибольшее распространение.
Получаемый бензин обладает октановым числом
60—70 (моторный метод), однако вследствие высокого
содержания олефинов он не стабилен и мало восприим-
чив к тетраэтилсвинцу, а поэтому нуждается в до-
полнительной обработке — риформинге. В термич.
К. в основном используют низкосортные тяжелые
остатки для получения бензина и более широко вы-
кипающей фракции для каталитич. К. и газа для
нефтехимии, синтеза.
Превращение углеводородов различных классов при К.
проходит с различной трудностью. Скорость распада углево-
дородов одного и того же класса возрастает с увеличением мол.
веса. Поэтому более легкое сырье в промышленных установ-
ках перерабатывают обычно в более жестких условиях; напр.,
К. лигроина при 530—540°, керосино-газойлевых фракций
при 500—510°, а мазута ок. 470°. Наиболее легко распадаются
предельные углеводороды (молекулы углеводородов разры-
ваются преим. посередине), первоначальными продуктами
распада являются предельный углеводород меньшего мол.
веса и олефин. Продукты первичного распада реагируют с дру-
гими углеводородами и между собой, а также распадаются
дальше, поэтому даже при К. индивидуальных углеводородов
789
КРЕКИНГ
790
образуется сложная смесь продуктов. Изомеризации парафи-
нов при термин. К. практически не происходит. Нафтены устой-
чивее парафинов; при их К. происходит дегидрогенизация,
раскрытие циклов с образованием ароматич. и парафиновых
углеводородов и деалкилирование. Поэтому бензин из нафте-
нового сырья обладает большей детонационной стойкостью.
Непредельные углеводороды, содержащиеся во вторичном
сырье, распадаются труднее парафинов; они легко полимери-
зуются и конденсируются с ароматич. углеводородами с обра-
зованием высокомолекулярных продуктов уплотнения. В жест-
ких условиях олефины дают ароматич. углеводороды. Неалки-
лироваиные ароматич. углеводороды наиболее стойки. Аро-
матич. углеводороды склонны к уплотнению с образованием
конденсированных колец. В этих реакциях могут участвовать
и олефины. В результате образуются смолисто-асфальтовые
вещества и кокс. Поэтому сырье с высоким содержанием аро-
матич. углеводородов мало пригодно для переработки, т. к.
требует жестких условий крекирования; при этом происходит
большое коксообразоваиие. По мере углубления К., получае-
мые продукты обогащаются ароматич. углеводородами за
счет дегидрогенизации циклич. парафинов, циклизации оле-
финов и пр.; имеет значение также термич. устойчивость аро-
матич. углеводородов. Поскольку последними обогащаются и
остаточные фракции крекинг-продуктов, то их повторная пере-
работка (повторный К., крекинг е рециркуляцией) требует
более жестких условий, чем переработка исходного сырья.
Сернистые соединения нефти при К. разлагаются легче, чем
углеводороды. Наиболее стойки тиофены и тиофаны. В про-
дуктах К. сернистого сырья содержатся H2S, элементарная
сера, меркаптаны и др. Содержание серы в жидких крекинг-
продуктах ниже, чем в исходном сырье, т. к. часть серы пере-
ходит в виде H2S в газ, а также в виде высокомолекулярных
соединений концентрируется в крекинг-остатке.
Объяснение механизма термич. К., представляю-
щего совокупность реакций, протекающих со сложной
смесью углеводородов, состав к-рой известен непол-
ностью, затруднительно. В более простых случаях
при невысоких давлениях и небольших глубинах
превращения экспериментальные данные удовлетво-
рительно истолковываются с точки зрения радикально-
цепного механизма. Первичным актом распада али-
фатич. углеводорода должен быть разрыв одной из
С—С связей с образованием двух одновалентных ра-
дикалов:
Сп + т н (2п + т) + 2 * CnH2n + 1 +СтН2т + 1
Радикалы, содержащие более двух атомов углерода,
диспропорционируют на меньший радикал и соот-
ветствующий олефин:
Рц + т ^2(т 4- п) +1 * <-'nH2n+<-'m®2m | 1
Распад радикалов продолжается до образования
метильных и этильных радикалов или же олефинов
и атомарного водорода. Метильный и этильный ради-
калы, обладающие достаточной продолжительностью
жизни, реагируют с молекулами исходного углево-
дорода. Наиболее вероятным направлением их рас-
пада является образование метана или этана и нового
радикала. Образующийся радикал может распадаться
далее с образованием олефина и новых радикалов.
Радикал может взаимодействовать также с молеку-
лой исходного сырья:
^п^2п 2+^ '®'H-pCnHgn j
с образованием парафинового углеводорода и нового
радикала. Расчет показывает, что наиболее вероятным
представляется участие в этой реакции третичного
атома углерода, затем вторичного; наиболее трудно
в реакцию вовлекается первичный атом углерода.
Взаимодействие алифатич. радикалов с молеку-
лами исходных алкановых углеводородов (CnHm)
по приведенным выше схемам приводит к цепной
реакции. Состав получающихся продуктов мало за-
висит от реакций между первично образовавшимися
радикалами и молекулами, т. к. каждый первичный
радикал приводит к образованию большого числа
вторичных, третичных и т. д. радикалов, реакции
к-рых и определяют состав крекинг-продуктов. Состав
конечных продуктов определяется параметрами цеп-
ного процесса и не зависит от направления первичного
распада молекул исходного углеводорода. Расчет
состава продуктов сводится к нахождению коэфф.,
определяющих относительную вероятность образо-
вания радикалов при расщеплении углеводородов.
Для этого необходимо вычислить относительную
вероятность отрыва атома водорода от исходного угле-
водорода с образованием предполагаемого радикала
и статистич. фактор, означающий число равноценных
атомов водорода в углеводороде, отщеплением к-рых
может образоваться данный радикал. Для простей-
ших алифатич. углеводородов — пропана, бутанов,
пентанов получено удовлетворительное совпадение
расчетных и опытных данных.
Хотя К. представляет собой совокупность ряда
последовательных и параллельных реакций, тем не
менее скорость его с хорошим приближением описы-
вается ур-нием 1-го порядка:
к = -±- In—5—
т а — х
где к—константа скорости распада, а — количество мо-
лей исходного сырья в единице реакционного объема,
х — количество молей, распавшихся за время т.
Это ур-ние хорошо описывает реакции первичного
распада парафинов, высших олефинов и деалкили-
рования. Низшие олефины склонны к полимеризации
и поэтому порядок реакции для них обычно выше
первого. Иногда наблюдается торможение К. продук-
тами распада, а также замедление его по мере углуб-
ления процесса и возрастания термич. устойчивости
сырья. В этом случае скорость К. лучше описывается
ур-нием:
к = — In —------Вх
т а — х г
где 0 — постоянная, характеризующая степень тор-
можения. В зависимости от рода сырья и условий
величина 0 находится в пределах 0—1. Результаты,
получаемые при кинетич. обработке опытных данных,
сильно зависят от того, что принимается за превра-
щенный продукт. Часто за превращенный продукт
принимают все продукты реакции, кипящие выше
и ниже исходного сырья, а иногда, что более удобно
для практики, только выход целевого продукта —
бензина. На промышленных установках наблюдается
примерно линейная зависимость выхода бензина от
продолжительности процесса.
Важнейшими факторами, определяющими течение
и результаты К., являются характер сырья, темп-ра,
давление, время реакции и достигнутая глубина
превращения. Темп-рой определяется скорость и на-
правление реакций К. Компенсировать влияние тем-
пературы изменением продолжительности процесса
и наоборот можно только в известных пределах.
Температурный коэфф, скорости К. сильно зависит от
условий процесса и обычно ниже 2. Напр., в интер-
вале 400—450° скорость К. возрастает вдвое при
повышении темп-ры на каждые 11—14°, в интервале
500—520° на каждые 14—20°. Зависимость между к
и темп-рой К. описывается уравнением Аррениуса.
Величины энергий активации зависят от характера
сырья и условий процесса. Для высокомолекуляр-
ного сырья они ниже, напр. для керосино-газойлевых
фракций они равны 53—58 ккал/молъ, а для лигрои-
новых — 65—70 ккал/моль. Наиболее высокие энер-
гии активации у ароматич. углеводородов — ТО-
98 ккал/молъ и ниже у парафинов и нафтенов — 60—
65 ккал/молъ.
К. является псевдомономолекулярным процессом
и давление не влияет на скорость распада, однако
влияет на скорость вторичных реакций — полиме-
ризации и конденсации, протекающих по 2-му по-
рядку, благодаря чему уменьшается выход газа и лег-
ких олефинов и содержание непредельных в газе
и в бензине. В жидкофазном процессе, ввиду практич.
791
КРЕКИНГ
792
несжимаемости жидкости, давление не влияет на ре-
зультаты К. От давления зависит объем паров кре-
кируемого в-ва, а следовательно и продолжительность
реакции, а тем самым глубина процесса и произво-
дительность установки. Поэтому установки паро-
фазпого К. под низким давлением из-за недостаточной
компактности и мощности не получили широкого
применения.
Продолжительность К. наряду с темп-рой и видом
сырья определяет глубину распада, т. е. выход и ха-
рактер продуктов. В начале К. преобладает распад
наиболее нестабильных компонентов сырья, а затем
начинаются процессы уплотнения. Образовавшиеся
вначале бензин и легкий газойль разлагаются дальше
на газ, смолу и кокс. Поэтому при слишком большой
продолжительности процесса выходы бензина па-
дают, а газа и кокса чрезмерно возрастают. Для полу-
чения максимального выхода бензина практикуют
т. наз. К. с рециркуляцией или повторный К. При
однократном К. лигроин дает 70—75% бензина, 15—
20% керосино-газойлевой фракции и 7—15% ма-
зута. После отгонки бензина, напр. при К. керосино-
газойлевого дистиллята, получают остаток и 50—60%
фракции, выкипающей в тех же пределах, что и ис-
ходное сырье. Последнюю смешивают в определенном
соотношении со свежим сырьем и повторно крекируют.
Отношение количества рециркулируемой фракции
к свежему сырью называют коэффициентом
рециркуляции,а отношение общего количества
крекируемого сырья к количеству свежего сырья —
коэффициентом загрузки. К. с рецир-
куляцией повышает выход бензина из солярового
масла до 60% и из мазута до 34% при выходе кокса
до 0,2%. Однако при повторном К. вследствие боль-
шей стабильности крекинг-флегмы выход бензина за
один проход сырья несколько снижается.
Для термич. К. в СССР применяют двухпечные установки
с раздельным крекированием тяжелого и легкого сырья: ма-
зутов, широких фракций с атмосферно-вакуумных установок,
тяжелых и смолистых нефтей. Упрощенная схема двухпечной
установки для работы на мазуте или на широкой фракции
приведена на рис. 1. Мазут подают в колонну 4, где он кон-
Рис. 1. Принципиальная схема
двухпечной установки для кре-
кинга мазута: 1 и 2 — нагрева-
тельно-реакционные печи легко-
го (Г) и тяжелого (2) крекинга;
3 — испаритель; 4 — первая ко-
лонна; 5 — вторая колонна; I —
охлаждение; II — отвод крекинг-
остатка; III—отвод газов; IV —
крекинг-бензин; V—подача сырья.
тактирует с парами кре-
кинг-продуктов, поступаю-
щих из испарителя з при
420°, нагревается и из него
удаляются легкие соляро-
вые фракции (до 350°), а
тяжелые фракции конден-
сируются и переходят в
мазут. В колонне 4 обра-
зуется смесь остатка сырья
и тяжелых фракций кре-
кинг-продуктов — тяжелая
флегма, поступающая да-
лее в печь легкого К. 1,
назначение к-рой — сниже-
ние вязкости остатка, по-
лучение дополнительного
количества фракций для
глубокого К. и бензина.
Продукты крекинга из этой
печи вместе с соляровыми
фракциями, отогнавшими-
ся из мазута с верхней час-
ти колонны 4 в виде па-
ров, поступают во вторую
колонну 5, где происходит
разделение на газ и крекинг-бензин, легкую флегму — смесь
промежуточных фракций К. и соляровых фракций, отогнан-
ных от мазута. Легкая флегма нижней части колонны 5 по-
ступает в печь 2 для глубокого К. Продукты из обоих печей
поступают в испаритель 3, откуда пары направляются в ко-
лонну 4, а остаток жидкого крекинг-продукта выпускается
из нижней части испарителя. Эта установка работает с рецир-
куляцией промежуточных фракций К. В дальнейшем крекинг-
установки были модифицированы с учетом изменения харак-
тера сырья. Солярово-газойлевые фракции, сами ставшие
целевыми продуктами, стали заменять утяжеленными мазу-
тами, гудронами и полугудронами с вакуумных установок.
Поэтому было изменено соотношение мощностей между печами
легкого и глубокого К. Ранее процесс происходил непосред-
ственно в печах, новые установки строят с выносными реак-
ционными камерами, увеличивающими продолжительность
пребывания в зоне реакции без затраты дополнительного топ-
лива; изменена конструкция печей и увеличена мощность уста-
новок (до 1600—2200 т/сутки). В дальнейшем были созданы
установки, комбинирующие К. с прямой перегонкой нефти,
и многопечные установки, на к-рых отбирается большое число
узких фракций, подвергаемых К. в отдельных печах или
змеевиках в условиях, оптимальных для К. каждой фракции.
Крекинг термический низкого
давления (коксование). Целью процесса яв-
ляется получение кокса, содержащего 2—6% водо-
рода и одновременно получение 60—65% широкой
фракции (бензип-керосин-газойль) и газа. Сырьем
служат гудроны, крекинг-остатки, битумы и т. д.
Широкая фракция может служить сырьем для ката-
литич. или для термич. К., благодаря чему повышается
выход светлых продуктов в расчете на перерабатывае-
мую нефть. Коксование, напр. гудрона, дает более
20% кокса, до 15% бензина, более 50% широкой
фракции и до 10% газа. Коксование можно проводить
как периодически (в кубах), полунепрерывно (в ке-
рамич. печах и коксовых камерах) или же непрерывно
с твердым гранулированным или порошкообразным
теплоносителем. Коксование в кубах утратило свое
значение. Коксование в керамич. печах используется
гл. обр. для получения беззольного электродного
кокса. Коксование в коксовых камерах (т. наз. «за-
медленное коксование») применяется гл. обр. для
получения дистиллята для дальнейшей переработки,
а получаемый кокс используют как топливо.
Наиболее совершенно коксование осуществляется
в виде непрерывного процесса, причем кокс полу-
чается в диспергированном виде и легко выводится
из реактора. Коксование происходит на поверхности
нагретых частиц гранулированного кокса с размером
зерен 5—15 мм, или же порошкообразного с зернами
0,02—0,3 мм. В этом случае коксование происходит
в т. наз. «кипящем слое» теплоносителя — кокса.
Основными аппаратами установок коксования яв-
ляются: реактор, где происходит коксование при
темп-ре 510—550° и давлении до 3 ат, и коксонагре-
ватель, где кокс нагревается до 550—620° за счет
сожжения газообразного топлива или же части самого
кокса. При коксовании количество кокса и размер
гранул возрастают, поэтому принимают меры к со-
хранению постоянными отношение кокса к сырью
(10—14 : 1) и гранулометрия, состава путем размола
и сортировки зерен. Нагревание при помощи тепло-
носителя позволяет снизить до минимума темп-ру
предварительного подогрева сырья, что очень важно
при переработке тяжелых смолистых остатков. При
этом способе выход газойлевой фракции выше, а кокса
ниже. Напр., при непрерывном коксовании сырья,
содержащего 1,25% серый 9% кокса, получают выход
до. 72% газойля и 10% бензина и только 11% кокса,
а при коксовании в камерах — 53% газойля и 22%
кокса при несколько большем выходе бензина —
17%. Получаемый газойль выкипает в пределах
200—500°, содержит меньше серы, чем исходное
сырье, и дает более низкое коксообразование — до 2%,
а газ содержит до 30% непредельных. Кокс применяют
для изготовления электродов, как топливо и др.
Крекинг высокотемпературный
под низким давлением (пиролиз). Глав-
ная цель этого вида К. — получение газа для нужд
химич. пром-сти и ароматич. углеводородов. Процесс
проводят при 650—750°, давлении, близком к атмо-
сферному; при этом образуется до 50% газа (считая
на сырье) и 40—60% смолы, содержащей до 10%
бензола, до 6% толуола, а также ксилолы, нафталин
и антрацен. Сырьем служат низкосортные тяжелые
остатки, дающие, кроме кокса и ароматич. углеводо-
родов, также газ, содержащий до 30% этилена. Про-
цесс проводят в пирогенных установках, основный
элементы к-рых—трубчатая печь, в змеевиках к-рое
сырье нагревают и происходит его частичный пиролиз,
793
КРЕКИНГ
794
и реакционная камера, где процесс завершается.
Продукты пиролиза — газ и пары — отмыкаются от
выносимой ими сажи и кокса водой в т. наз. «гидрав-
лике», газ поступает на разделение, а жидкие про-
дукты на ректификацию, проводимую в колонне на
самой установке или же в специальном цехе. Из смолы
пиролиза отбирают легкое масло — наиболее ценный
продукт, из к-рого перегонкой отбирают «бензольную
головку» (до 75°) и фракции: бензольную (75—90°),
толуольную (95—125°), ксилольно-хвостовую (125—
180°) и остаток — сольвент. Из смолы получают также
зеленое масло (175—350°) и остаток — пек. Зеленое
масло используют для получения нафталина и сажи,
пек дает при коксований электродный кокс. Газ
содержит до 45 об. % непредельных (до 40% этилена,
выше 5% пропилена), остальное водород, метан и про-
пан. При проведении пиролиза на получение аро-
матич. углеводородов содержание непредельных в газе
ниже — до 30%. Как и коксование, пиролиз наиболее
целесообразно проводить с твердым теплоносителем,
напр. коксом.
П. Крекинг каталитический. Этот процесс проводят
с участием катализаторов, что дает возможность по-
лучать продукты лучшего качества и с большими вы-
ходами, Современное назначение процесса — произ-во
высококачественного базового бензина с октановым
числом до 85 (моторный метод) для автотранспорта.
Кроме того, каталитич. К. дает керосино-газойлевые
франции, которые можно использовать в качестве
топлива для дизелей или газовых турбин, и газ,
содержащий большое количество предельных и не-
предельных углеводородов С3—С4.
Катализаторы каталитич. К. готовят обработкой
природных глин или же из соответствующих солей,
синтетически. Природные глины, типа флоридина,
обладают достаточной активностью в естественном
состоянии и нуждаются только в формовании. Боль-
шинство же бентонитовых глин активируют кисло-
тами или же нек-рыми солями, напр. сульфатом алю-
миния или хлоридом аммония; в результате такой
обработки из катализатора удаляется избыточное
количество катионов металлов, и, кроме того, они
приобретают более пористую структуру. Затем произ-
водит их термин, активацию прокаливанием при
450—500°; при этом происходит удаление гигроскопич.
и частично структурной воды, обусловливающее
дальнейшую полимеризацию алюмосиликата. Полу-
чаемые катализаторы обладают значительно большей
активностью, чем лучшие природные неактивирован-
ные глины. Применяют также катализаторы, приго-
товляемые из каолинитовых глин, отличающиеся
высоким — 30% и выше — содержанием окиси алю-
миния, против 10—15% в обычных катализаторах
(остальное SiO2 и нек-рые другие окислы в незначи-
тельных количествах). Из синтетич. катализаторов
наибольшее применение получили алюмосиликатные
катализаторы.
В пром-сти катализаторы готовят обычно совместным оса-
ждением гелей и исходят из водных растворов сульфата алю-
мцрия и силиката натрия, смешением к-рых получают гидро-
гель алюмокремневой к-ты. Гель подвергают синерезису,
напр. в р-ре сульфата алюминия, отмывают от натрий-иона,
освобождают от избыточной воды и формуют, напр. в виде
цилиндриков диаметром 3—4 jiw, длиной 4—5 мм. Гранулы
сушат сначала в атмосфере перегретого водяного пара, во
избежание растрескивания их при слишком быстром удалении
воды, а затем топочными газами, и, наконец, прокаливают
при 500—700°. Для получения катализатора сферич. формы
смесь растворов сульфата алюминия и силиката натрия по
каплнм вводят в нагретое до 80—90“ масло или керосин, где
образуются гранулы, обрабатываемые далее как указано
выше. Микросферич. катализатор можно готовить инжекцией
смешанных растворов сульфата и силиката в горячие топочные
газы с последующей обработкой получающихся зерен катали-
затора. Катализаторы сферич. формы применяют в системах
К. с подвижным катализатором, т. к. они обладают большей
«текучестью» и прочностью и вызывают меньшую эрозию
аппаратуры. Концентрация исходных р-ров, условия их сме-
шения, синерезиса, отмывки и высушивания имеют важней-
шее влияние на свойства готового катализатора, основными
характеристиками к-рого являются активность, стабильность,
избирательность, регенерируемость и механич. прочность.
Активность алюмосиликатных катализаторов на
практике оценивают обычно по индексу ак-
тивности, т. е. по выходу бензина (а иногда
и нек-рых других продуктов) в стандартных условиях.
Наблюдаемая активность зависит не только от химич.
состава катализатора, но также и от величины по-
верхности, достигающей нескольких сотен м2'г,
от размера пор катализатора (50—ЮОА) и иногда
от размера гранул. Индекс активности промышленных
катализаторов лежит в пределах 30—50%; это озна-
чает, что на данном катализаторе из легкого газойля
при 450° за 10-минутный цикл можно получить 30—
50 об.% бензина с концом кипения 210°. Более трудно
распадающееся легкое сырье требует применения ка-
тализаторов с большим индексом активности, чем
легче распадающееся тяжелое.
Избирательность катализаторов К. определяет со-
отношение выходов различных продуктов (газ, бен-
зин, кокс) и их состав; она зависит от химич. состава
и структуры катализатора. Напр., магний-силикатные
катализаторы в равных условиях дают обычно не-
сколько большие выходы бензина, но с меньшим со-
держанием ароматич. и изопарафиновых углеводоро-
дов, чем алюмосиликаты. Мелкопористые катализа-
торы дают большие выходы газа, чем крупнопористые,
т. к. их поверхность меньше доступна для молекул
исходного сырья и больше для уже превратившихся
молекул. Поэтому применение крупнопористых ката-
лизаторов предпочтительнее, особенно в случае тя-
желого сырья.
Регенерируемость катализатора, определяемая лег-
костью удаления углистых отложений с его поверх-
ности при минимальной потере активности в значи-
тельной мере зависит от размера пор. Крупнопористые
катализаторы регенерируются легче, чем мелкопо-
ристые. Для ускорения выжига к катализатору до-
бавляют иногда немного окиси хрома. В промышлен-
ных условиях полного удаления кокса не достигают,
а лишь снижают его содержание до нек-рого допу-
стимого уровня, принятого в данном процессе
Стабильность катализатора определяет сохранение
его активности и избирательности в процессе эксплуа-
тации. Пар и высокая темп-ра разрушают структуру
катализатора — уд. поверхность его сокращается,
размер пор возрастает ва счет разрушения мелких пор,
активный алюмосиликатный комплекс разлагается,
образуя неактивные окиси алюминия и кремния.
Тяжелые металлы, особенно в присутствии серы
в сырье, резко изменяют селективность катализатора—
увеличивается выход газа и содержание в нем метана
и водорода, а также возрастает коксообразование.
Механич. прочность катализатора на раздавливание
и истирание, а также склонность его к растрескиванию
при регенерации имеют большое значение, особенно
для систем с подвижным катализатором, определяя
потери катализаторов в процессе.
Каталитич. К. проводят обычно при 450—520°,
давлении 1—2 ат и продолжительности контакта
в неск. секунд. Каталитич. реакции проходят в этих
условиях значительно быстрее термических и яв-
ляются более сложными. Основные из них (помимо
распада) — это реакции изомеризации и перераспре-
деления водорода; эти реакции определяют высокое
качество крекинг-продуктов. Меньшее значение имеют
реакции трансмутации алкильных групп, дезалкили-
рования, полимеризации и дегидрогенизации. Про-
исходят также процессы уплотнения, приводящие
к образованию кокса на катализаторе. Для различ-
ных классов углеводородов скорость указанных реак-
ций различна. Наиболее реакционноспособны в уело-
795
КРЕКИНГ
796
виях каталитич. К. олефиновые и нафтеновые угле-
водороды, далее следуют нафтеново-ароматич., аро-
матич. с длинными боковыми цепями и, наконец,
чисто ароматич. углеводороды. Распад олефинов
и парафинов обычно происходит сразу в нескольких
местах с образованием олефинов и парафинов, содер-
жащих три и более атомов углерода в молекуле.
Нафтеновые и нафтеново-ароматич. углеводороды
претерпевают разрыв нафтенового кольца или его
отщепление от ароматич. кольца, а также дезалкили-
руются. Алкилароматич. углеводороды отщепляют
алкильную группу и образуют дезалкилированный
ароматич. углеводород и олефин. Парафины и нафтены
в условиях каталитич. К. почти не изомеризуются,
олефины же изомеризуются очень легко и реакция
часто достигает равновесия. У ароматич. углеводо-
родов происходит сравнительно медленно трансмута-
ция алкильных групп.
Наиболее важной реакцией каталитич. К. является
перераспределение водорода, обусловливающее на-
сыщенный характер получаемых бензинов и высокое
содержание в них изопарафинов. Реакция заклю-
чается в насыщении олефинов водородом за счет
образования бедных водородом продуктов уплотне-
ния. Избирательность этой реакции в отношении
преимущественного насыщения олефинов с третич-
ным атомом углерода способствует образованию
в крекинг-продуктах высокоразветвлепных изопара-
финов. Реакции алкилирования и дегидрогенизации
не играют большой роли при каталитич. К. Полиме-
ризация олефинов в силу термодинамич. ограничений
не может протекать в значительной степени, тем не
менее она может быть первой ступенью в образовании
ароматич. углеводородов и кокса даже в условиях
невысокой равновесной концентрации полимера. Кон-
денсация ароматических и олефиновых углеводоро-
дов является, вероятно, основной реакцией, ведущей
к образованию кокса. Поэтому наиболее подходящим
для каталитич. К. будет нафтеновое или нафтеново-
парафиновое сырье. Большое содержание ароматич.
углеводородов затрудняет переработку и дает боль-
шое коксообразование. Поскольку скорость распада
возрастает с увеличением мол. веса, то легкое сырье
требует более жестких условий переработки, чем тя-
желое. При каталитич. К. протекает также интенсив-
ный обмен атомами водорода между катализатором
и углеводородами.
Для объяснения реакций, протекающих при ката-
литич. К., наиболее удовлетворительные результаты
дает теория карбонил ионов. Активным началом алю-
мосиликатного катализатора можно считать соедине-
ние кислотного типа — (HAlSiO4)x, в к-ром отноше-
ние кремния к алюминию равно 1. В промышленных
образцах катализаторов содержание окиси кремния
обычно значительно выше и поэтому они могут рас-
сматриваться как соединение (HAlSiO4)x, нанесенное
на неактивный силикагель. Это соединение является
источником протонов, образующих при присоедине-
нии к молекулам углеводородов ионы карбония.
Нестабильный ион карбония или реагирует с другими
молекулами, или же подвергается внутримолекуляр-
ной перестройке с отщеплением протона или иона
карбония меньшего размера. Из насыщенных молекул
ион карбония может возникать только после предва-
рительного распада молекулы с получением олефина
или же при отнятии от нее атома водорода протоном
и образования молекулы водорода. Из олефина же
возникновение иона карбония происходит при прос-
том присоединении протона по правилу Марковнико-
ва, т. е. к наиболее гидрированному атому углерода,
например
СН2=СН-СН34-Н+—> СНз -СН -СН2-СН3
Ион карбония реагирует с молекулой углеводо-
рода с образованием новой молекулы и нового иона
карбония:
СН3СН-СНз-СН3+В1Н—►СНзСН(Е)-СНгСНз4-в1’
В соответствии с обычными представлениями теории
ионов карбония могут быть представлены, кроме рас-
пада, и другие, протекающие при каталитич. К. реак-
ции — изомеризации, полимеризации, перераспре-
деления водорода и т. п.
Хотя каталитич. К. является очень сложным про-
цессом, кинетика его, так же как и в случае термич.
К., в ряде случаев удовлетворительно описывается
уравнением 1-го порядка, напр. вида
1/о1пТ4^=^о?/ + а
где Г’о — объем сырья, подаваемого в реактор в еди-
ницу времени на единицу объема катализатора, у —
степень превращения, а и Р — величины, независящие
от Го, из них а — величина, пропорциональная кон-
стапте скорости реакции, и р — коэфф., отражающий
замедление реакции продуктами превращения или же
в результате увеличения объема в процессе, часто
близкий к единице. Доказано, что К. может тормо-
зиться переносом массы в глубь пор катализатора;
хотя вид кинетич. ур-ния при этом не меняется, од-
нако коэфф, аир приобретают иной физич. смысл.
Наибольшее влияние на результаты К. имеет темп-ра
процесса, поскольку крекирующее действие катали-
заторов проявляется в определенном, сравнительно
узком интервале темп-p. Ниже нек-рой темп-ры, в за-
висимости от природы катализатора и сырья, К.
вообще не происходит, хотя могут протекать некото-
рые сопутствующие ему реакции, напр. полимериза-
ция или перераспределение водорода. С повышением
темп-ры наряду с увеличением глубины распада
начинают играть большую роль реакции термич. К.,
благодаря чему увеличивается выход газа и содер-
жание в нем метана, этана и олефина, непредельность
продуктов К. возрастает. Темп-рный коэфф, скорости
К. мал, для увеличения скорости процесса вдвое
необходимо повысить темп-ру на 50—100°. В соот-
ветствии с этим энергия активации низка и составляет
6 000—15 000 ккал/молъ, редко больше. Давление не
имеет значения при каталитич. К., т. к. процесс
протекает в адсорбционном слое на поверхности ката-
лизатора, а не в объеме и, кроме того, в промышлен-
ных системах давление вообще варьирует очень не-
значительно. Однако повышение давления способст-
вует полимеризации, перераспределению водорода
и коксообразованию. Существенное влияние на ре-
зультаты К. оказывает его продолжительность. Чем
больше продолжительность К. при прочих равных
условиях, тем большая глубина превращения может
быть достигнута за один проход. Глубина превраще-
ния при каталитич. К. может изменяться в широких
пределах и ее лимитирует гл. обр. слишком большое
газообразование при высокой конверсии. Поскольку
одна и та же глубина превращения может быть до-
стигнута при различном сочетании указанных выше
факторов, то оптимальное ее значение, а также выбор
надлежащих условий К. определяются соображениями
практич. целесообразности, напр. заданной произ-
водительностью установки. Для получения лучших
выходов целевых продуктов при приемлемом газо-
образовании каталитич. К., так же как и термич.,
проводят с рециркуляцией части крекинг-остатка.
Первые промышленные установки каталитич. К.
являлись системами с неподвижным слоем катализа-
тора с несколькими реакторами, в к-рых попеременно
производились циклы К. и регенерации катализатора.
Трудность поддержания заданной темп-ры во время
797
КРЕКИНГ
798
рабочего цикла и регенерации, необходимость нали-
чия нескольких реакторов для сохранения непре-
рывности процесса, сложность смены циклов и т. п.
привели к тому, что эти установки были вытеснены
более экономичными с подвижным катализатором,
в к-рых последний непрерывно циркулирует через
реактор и регенератор. В свою очередь, системы с по-
движным катализатором подразделяются на системы
с поступательно движущимся катализатором (система
«термофор»), в которых К. происходит в медленно
опускающемся слое гранулированного или таблети-
рованного катализатора, попутно или противоточно
к к-рому подаются пары сырья,а перемещение ката-
лизатора из реактора в регенератор производится
смесью воздуха и дымовых газов (т. наз. «пневмо-
транспорт» катализатора) и на системы, в к-рых К.
происходит в псевдоожиженном, т. наз. кипящем
слое пылевидного или мелкосферич. катализатора
с размером зерен 20—100 мк (система «флюид»).
В системах с подвижным катализатором последний,
помимо своей прямой роли, выполняет также функции
подвода тепла для эндотермич. процесса К. и вывода
кокса из реактора. Кроме того, в этих системах до-
стигается истинная непрерывность процесса, легкая
регулировка темп-ры К. и регенерации. Активность
катализатора благодаря непрерывному выводу части
дезактировапного катализатора и подводу свежего
поддерживается на нек-ром постоянном уровне (такая
активность наз. «равновесной активностью»). Боль-
шую роль для результатов процесса в системах с по-
движным катализатором играет т. наз. «кратность
циркуляции катализатора», т. е. от-
ношение количества катализатора, поступающего в ре-
актор в единицу времени, к количеству сырья, вво-
димому за то же время. В системах термофор крат-
ность равна 2—6, в системах флюид 8—14. Чем выше
кратность циркуляции при прочих равных условиях,
тем меньше содержание кокса на катализаторе, что
очень важно при переработке тяжелого сырья. Одно-
временно, однако, увеличиваются расход энергии на
транспорт катализатора и потери его от истирания.
Принципиальная схе-
ма крекинг-установки си-
стемы флюид приведена
на рис. 2. Регенерирован-
ный горячий катализатор
из регенератора 1 спус-
кается по стояку 2 в узел
смешения з, где, контак-
тируя с нагретым в печи
19 сырьем, к-рое при этом
испаряется, и смесь ка-
тализатора и паров по
трубопроводу 4 посту-
пает в реактор 5, где ско-
рость потока резко умень-
шается и частицы ката-
лизатора, осаждаясь, об-
разуют плотный кипя-
Рис. 2. Схема каталити-
ческой крекинг-установ-
ки системы «флюид»: 1 —
регенератор; 2 — стояк;
з — узел смешения; 4 —
трубопровод для смеси
сырья с катализатором;
•5 — реактор; в — плот-
ный кипящий слой; 7—
отстойная зона; 8 — циклонные сепараторы; 9 — спускные тру-
бы; 10 —отпарная колонна; 11 — отводящий стояк; 12 — узел
смешения; 13 — трубопровод; 14 — циклонные сепараторы;
15— барабан для разделения воды и пара; 16— котел-утилиза-
тор; 17 — задвижка, регулирующая количество выводимого
катализатора; 18— колодец для отвода катализатора; 19 — печь
для нагрева сырья; 20 — воздуходувка; 21 — распределитель-
ная решетка; I — пылеосадитель; II — вода; III —газы реге-
нерации; IV — водяной пар; V — продукты реакции; VI —
возврат уловленного катализатора; VII — сырье; VIII — во-
дяной пар; IX — воздух; X — рециркулирующий газойль;
XI — продукты крекинга на разд еление.
щий слой 6, в к-ром и протекает К. Высоту слоя подбирают
так, чтобы обеспечить нужное время реакции. Выходящий из
этого слоя газо-паровой поток продуктов К. проходит через
отстойную зону 7, где оседает значительная часть выносимых
частиц катализатора, а расположенные внутри его регенера-
тора циклонные сепараторы 8 улавливают выносимые частицы
и возвращают по трубам 9 в плотный слой катализатора. Отра-
ботанный катализатор из реактора поступает через отпарную
колонну 10 в отводящий стояк 11. Назначение колонны 10 —
отдувка перегретым водяным паром паров углеводородов из
катализатора. Нижний конец стояка 11 присоединен ко вто-
рому узлу смешения 12, где отработанный катализатор под-
хватывается током воздуха и транспортируется в регенератор.
В последнем, также в кипящем слое, выжигается кокс с ката-
лизатора и регенерированный катализатор через колодец 18
отводится в стояк г. Предварительно в колодце, расположен-
ном над распределительной решеткой 21, катализатор отду-
вается водяным паром от продуктов сгорания. Уносимые га-
зами регенерации частицы катализатора улавливаются в ци-
клонных сепараторах 14, газы проходят через котел-утилиза-
тор 16 и затем через пылеосадитель выбрасываются в атмос-
феру. Жидкие продукты К. и газ из верхней части реактора
поступают на разделение (на схеме не показано), а тяжелый
газойль из ректификационной колонны рециркулируется.
Выход продуктов при каталитич. К. зависит от
принятой схемы переработки, условий ведения про-
цесса и рода сырья. При однократном К. выход со-
ставляет: сухого газа 6—9 вес. %, бутан-бутиленовой
фракции 6—9 вес. %, бензина (конец кипения 205°)
30—40 вес. %, каталитич. газойля 45—55 вес. %
и кокса 3—6 вес. %. При К. с рециркуляцией выхода
всех продуктов повышаются. Бензин (30—205°) со-
держит много (до 50%) парафинов, 20% и более
ароматич. углеводородов и обычно не более 30%
олефинов. Бензин каталитич. К. значительно ста-
бильнее бензина термич. К. и обладает большей при-
емистостью к тетраэтилсвинцу. Октановое число его
76—82 (без ТЭС, моторный метод). Легкий каталитич.
газойль (200—350°) можно применять как дизельное
топливо, однако по цетановому числу он несколько
уступает соответствующим фракциям прямой пере-
гонки, содержи^ больше ароматич. и меньше нафте-
новых углеводородов и поэтому применяется обычно
в смеси с ними. Состав получаемого газойля сильно
зависит от состава исходного сырья; из парафинового
сырья получают лучшие результаты, чем из нафтеново-
ароматич. Тяжелый каталитич. газойль (выше 300°)
используют как сырье для термич. К. Кокс, обра-
зующийся при каталитич. К., представляет собой
смесь трудно летучих углеводородов с атомным от-
ношением Н : С в пределах 0,3—1 и иногда выше.
Теплоту выжига кокса используют в энергетич.
установках. Газы, образующиеся при крекинг-про-
цессах, являются сырьем для органич. синтеза (см.
Газы нефтепереработки).
III. Крекинг с водяным паром. Процесс, осущест-
вляемый в виде чисто термического или же термока-
талитич. процесса, развивается в последние годы для
произ-ва гл. обр. легких олефинов для нужд нефте-
химия. синтеза из самого различного углеводородного
сырья — от нефтезаводских газов до тяжелых фрак-
ций нефти включительно. Преимущество процесса
перед обычным пиролизом — значительно более низ-
кое коксообразование при более высоком выходе
олефинов, особенно при применении катализатора
(обычно окись никеля на огнеупоре, напр. на окиси
магния). Технология, оформление процесса может быть
весьма различным. Темп-ра К. 650—800° (выше —
для более легкого сырья). Основным продуктом яв-
ляется этилен, получаются также пропилен, бута-
диен, ацетилен, но в значительно меньших коли-
чествах. Высоконепредельные жидкие продукты при-
меняют для получения термопластич. смол. Крекинг-
установки работают совместно со сложными установ-
ками для разделения и очистки газообразных оле-
финов.
IV. Крекинг под давлением водорода (гидрокре-
кинг). Процесс переработки нефти и ее средних и тя-
желых дистиллятов с большим содержанием сернистых
799
КРЕМНЕВОДОРОДЫ
800
и смолистых соединений, непригодных для перера-
ботки путем обычного каталитич. К. По своему ха-
рактеру гидрокрекинг близок к процессу деструктив-
ной гидрогенизации, однако проводится при относи-
тельно невысоких давлениях водорода (30—140 ат)
и температурах (350—450°). Катализаторами служат
окислы или сульфиды молибдена и никеля, молибдат
кобальта, обычно на кислых носителях, напр. на
активированных фтористым водородом глинах. Ос-
новными реакциями являются гидрирование и после-
дующее расщепление полициклич. углеводородов с об-
разованием изопарафинов, изомеризация и распад
высокомолекулярных парафинов с образованием лег-
ких изопарафинов, деструкция сернистых соединений
с образованием сероводорода. В отличие от деструк-
тивной гидрогенизации, где применением высоких
давлений водорода стремятся максимально подавить
образование кокса на катализаторе, при гидрокре-
кинге допускается образование кокса на катализа-
торе, регенерируемом' затем выжигом кокса в токе
воздуха. Назначение водорода — обеспечить эффек-
тивное гидрирование высокомолекулярных и сер-
нистых соединений с их последующим распадом на
крекирующем компоненте (алюмосиликат) катали-
затора и снизить образование кокса до допустимого
предела. От каталитич. риформйнга гидрокрекинг
отличается типом применяемых катализаторов и ха-
рактером протекающих реакций. Основное назна-
чение гидрокрекинга состоит в повышении выхода
легких фракций, непосредственно пригодных в ка-
честве моторного топлива (бепзин, дизельное и реак-
тивное топлива), и дистиллятов, пригодных для даль-
нейшей переработки обычными методами (крекинг,
риформинг). Расход водорода составляет 170—350л«3М3
сырья, поэтому строительство установок гидрокре-
кинга лимитируется наличием источников дешевого
водорода, напр. установок каталитич. риформинга
или конверсии природного газа с водяным паром.
При гидрокрекинге выход светлых дистиллятов повы-
шается до 70% от нефти, при этом сильно снижается
содержание в них серы и непредельных углеводородов.
Помимо рассмотренных в статье, существуют и
практически используются другие виды пиролитич.
расщепления углеводородного сырья, напр. электро-
крекинг, К. в присутствии кислорода, К. в плазме
и др., применяемые гл. обр. для получения олефи-
нов, напр. ацетилена. См. также Пиролиз, Риформинг.
Лит.: Обрядчиков С. Н., Технология нефти, ч. 2,
3 изд., М.—Л., 1952; Пархоменко В. Е., Технология
переработки нефти и газа, 2 изд., М., 1959; Пичугин А. П.,
Переработка нефти, М., 1960; Бондаренко Б. И.,
Никулин Д. Д., Суханов В. П., Каталитический
крекинг, М., 1956; Б он д а р ен к о Б. И., Установки ка-
талитического крекинга, М., 1958; Агафонов А. В.,
Алюмосиликатные катализаторы, М.—Л., 1952; Катализ
в нефтехимической и нефтеперерабатывающей промышлен-
ности, пер. с англ., под ред. П. Эмметта, кн. 2, М., 1961; Н а-
г и е в М. Ф., Химия, технология и расчет процессов синтеза
моторных топлив, т. 1, 12 изд.], Баку, 1961; Г у р в и ч В. Л.
и Смидович Е. В., Каталитический крекинг-флюид за
рубежом, М., 1960. В. В. Щекин.
КРЕМНЕВОДОРОДЫ (силаны) — соединения крем-
ния с водородом. Известны предельные К. — силаны,
аналоги предельных углеводородов, общей ф-лы
SinH2n_|.2’ предполагают, что существуют и непредель-
ные К. — силены, аналоги этиленовых углеводородов,
SinH2n, и силины, аналоги ацетиленовых углеводо-
родов, SinH2n_2.
К. отличаются от углеводородов неустойчивостью
I. I.
силоксановых цепей —Si—Si—; высшим известным
I I
членом гомологии, ряда силанов является гексасилан
SieH14. В таблице приведены основные свойства
нек-рых низших силанов.
Силаны T. пл., °C Т. кип., °C d, г/см3
SiH4 Sl2He Sl3Hs Si4Hio -185,0 -132,5 -117 — 93,5 -112,0 — 15,0 53 109,0 0,68 (-185”) 0,686 (—25°) 0,743 (0°) 0,825 (0°)
Плотности силанов выше плотности соответствующих
углеводородов; темп-ры кипения и плавления повы-
шаются более резко, чем у углеводородов.
Моносилан SiH4 и дисилан Si2He при комнатной
темп-ре — газы с неприятным запахом; трисилан
Si3H8 и тетрасилан Si4H10 — легко подвижные, лету-
чие, ядовитые жидкости с еще более неприятным за-
пахом. Силаны растворимы в спирте, бензине, тетра-
замещенных силанах и сероуглероде; при попадании
воздуха в закрытые сосуды с р-ром силана в сероугле-
роде происходит взрыв. Характерным свойством си-
ланов является их чрезвычайно легкое окисление;
для соединений, содержащих три и более атомов Si
в молекуле, реакция протекает с сильным взрывом.
Моносилан в присутствии кислорода окисляется со
вспышкой даже при темп-ре жидкого воздуха. В за-
висимости от условий реакции, продуктом окисления
является либо SiO2, либо промежуточные вещества.
Силаны — хорошие восстановители: они переводят
КМнО4 в MnO2, Hg(II) в Hg(I), Fe(III) в Fe(II) и т. д.
Другим характерным свойством силанов является
легкость гидролиза, особенно в щелочной среде, наир.:
SiH4-(-2H2O->SiO2+4H2
SiH4-|-2Na0H+H2O—►Na2SiOs+4H2
Реакция протекает количественно и может служить
для определения силанов. Под действием щелочи воз-
можно также расщепление связи Si—Si, напр.
H3Si-S1H2-SiH3-|-6H2O—►3SlO2-f-10H2
С галогенами силаны реагируют co взрывом, при
низких темп-рах образуются галогеносиланы (см.
Кремния галогениды). С галогеноводородами, ацил-
или алкилгалогенидами силаны также образуют га-
логеносиланы (катализатор А1С13 или А1Вг3, темп-ра
100—200°), напр.:
SlH44-2HBr A,B--SiH5Br2-:-2H2
Sl3H8+5CHCl3---<^Si3H3Cl5-(-5CH2Cl2
R3SiH-|-R'Cl —^-RaSiCl+R'H
С кислородсодержащими соединениями, напр. аце-
тоном, эфиром и т. п., силаны реагируют в газовой
фазе при высокой темп-ре с образованием алкоксила-
нов ROSiH3. С алифатич. спиртами в присутствии
ОН-ионов (катализатор) силаны образуют сложные
тетраэфиры.
Для получения силанов применяют след, способы:
1) Разложение силицидов металлов кислотами или
щелочами. Для этой цели обычно применяют силицид
магния, к-рый разлагают соляной к-той в инертной
атмосфере. Вероятная схема реакции:
Mg.>Sl+2HaO—*'H2Si(MgOH)2
H2Si(MgOH)2-f-4HCl—►2MgCl2-|-2H2O-(-H2+(SiH2)2
(SiH2)2+H2O—-H2SiO+SlH4
Образующиеся силаны разделяют ступенчатой кон-
денсацией, а затем отдельные фракции разгоняют при
низкой темп-ре.
2) Восстановление галогеносилаиов гидридом лития
или алюмогидридом лития. В среде этилового спирта
при нормальной темп-ре идут, напр., след, реакции:
SiCh-f-LlAlHj—►SiII4+LiCl+AlCl3
2Si2Cle+3LIAlH4—-2Si2Hc4-3LlCl+3AlCl3
С алюмогидридом лития образуются чистые силаны
с высоким выходом.
801
КРЕМНЕВОЛЬФРАМОВАЯ КИСЛОТА — КРЕМНИЙ
802
3) Восстановление галогеносиланов водородом. Про-
цесс ведут в присутствии галогенидов алюминия (ка-
тализатор) или же добавляют алюминий или цинк
в качестве компонентов реакции.
Силены SinH2n и силины SlnH2n_2 в мономер-
ной форме не выделены. Получен субиодид SiJ2, производное
S1H2, что служит косвенным доказательством существования
последнего. Образование силикоэтилена Si2H4 не доказано;
вероятно, он существует в полимерной форме. Полимерные ве-
щества, отвечающие общей ф-ле Sin Н2П_2j выделены при раз-
ложении моносилана в калориметрии, бомбе. Предполагается,
что при обработке силицида кальция разб. к-тами выделяется
силикоацетилен — полимерное кристаллич. вещество желтого
цвета со взрывчатыми свойствами. Ненасыщенные
полимерные гидриды (SiH) х образуются при само-
произвольном разложении высших силанов и при разложе-
нии силицидов. Из образовавшейся мазеподобной массы сна-
чала выделяют S1H4, Sl2Ha и Н2. При разгонке выстоявшейся
массы образуется пористый, желтый аморфный остаток, из
к-рого выделены продукты состава от SiH, t2 до SIH, 22 Поли-
мерные гидриды разлагаются слабощелочной водой на Н2 и
SiH4, едкое кали разлагает их на Н2 и S1O2.
Лит.: Бажант В., Хваловски В., Р а т о-
У с к и И., Силиконы, пер. с чеш., М., 1960; П е т р о в А. Д.
[и др.], Синтез кремнийорганических мономеров, М., 1961.
См. также лит. при ст. Кремний.
КРЕМНЕВОЛЬФРАМОВАЯ КИСЛОТА — белые или
желтовато-белые кристаллы триклинич. системы,
H3[SiW12O42] хН2О. При нагревании до 100° частично
плавится в кристаллизационной воде, при 200° теряет
почти всю воду (кроме 2 молекул),образуя тонкий
порошок, легко растворимый в воде с выделением
тепла; при 600—650° разлагается. К. к. хорошо
растворяется в воде, растворима в эфире,что исполь-
зуется для получения свободной кислоты из ее натрие-
вой соли. К. к. можно получить по реакции:
Na2SiO34-12Na8WO14-26HCl=H8[SlWj2O42H-26NaC14-9HsO
Применяют К. к. в аналитич. химии для разделения
Cs и Rb; для микрохимия, обнаружения калия; для
осаждения алкалоидов; для определения атропина,
никотина, для идентификации стеринов.
И. П. Ефимов.
КРЕМНЕФТОРИДЫ (фторсиликаты) — соли крем-
нефтористоводородной к-ты H2SiF6. Кремнефто-
ристоводородная кислота не сущест-
вует как индивидуальное соединение ни в жидком,
ни в газообразном состоянии, т. к. распадается на HF
и SiF4. В водном р-ре при упаривании под вакуумом
при комнатной темп-ре может быть получена до кон-
центрации 61%. Продажная конц. кислота содер-
жит 30—35% H2SiF6. Получены кристаллогидраты:
H2SiFe • пН2О, где n = 1, 2, 4. Раствор H2SiF6
обладает высокой концентрацией ионов водорода,
по своей силе эта кислота не уступает серной. Как
II2SiF6, так и ее соли ядовиты.
К. разделяются на трудно- и легкорастворимые
в воде. К первым относятся соли Na, К, Rb, Cs и Ва.
Р-ры К. имеют кислую реакцию (pH 2—4). При на-
гревании К. подвергаются термич. диссоциации на
SiF4 и соответствующий фторид. Нерастворимые К.
термически более стойки. При действии щелочей К.
разлагаются: Na2SiFe +6 NaOH — 6NaF + Na2SiO3 +
+3H2O. Аммиак действует аналогично, но при этом
выделяется свободная кремневая к-та. Жидким HF
и конц. H2SO4 К. разлагаются с выделением газооб-
разного SiF4. К. получают действием плавиковой к-ты
на смесь кремнезема и фтористого металла, действием
кремнефтористоводородной к-ты на гидроокиси (или
карбонаты) соответствующих металлов, а также че-
тырехфтористого кремния на фтористые металлы.
Наибольшее практич. значение имеет кремне-
фторид натрия Na2SiFe, представляющий
белые кристаллы гексагональной системы; отношение
параметров элементарной ячейки а : с = 1 : 0,5635;
плотн. 2,755. Растворимость (г на 100 г воды): 0,65
(17°), 2,46 (100°). Теплота образования Na2SiF6 (из
NaF и Si F4) составляет 37,27 ккал/молъ. Термич.
диссоциация происходит при 600°. В пром-сти NaaSi Fe
получают из отходящих газов суперфосфатных за-
водов (на 1 т суперфосфата образуется обычно 7—8 кг
Na2SiF6). Находящийся в газах SiF4 гидролизуется,
превращаясь в туманообразпую H2SiFe. Последняя
абсорбируется распыляемой водой с образованием
8—10%-ного р-ра H2SiF6, который затем вливается
в р-р NaCl, причем образуется мелкокристаллич.
Na2SiFe. Применяют Na2SiF6 в качестве инсектицида.
Значительная часть его перерабатывается на NaF.
Он входит также в состав сырьевых смесей для
произ-ва кислотоупорных цементов, эмалей и «глухого»
стекла; служит источником получения «белой сажи» —
тонкодисперсного кремнезема, применяемого в ка-
честве наполнителя в резиновом произ-ве; применяют
как коагулянт латекса. Кремнефториды ка-
лия, рубидия и цезия кристаллизуются
в кубич. системе по типу хлороплатината калия с пе-
риодом элементарной ячейки соответственно 8,168;
8,446; 8,867 А, и плоти. 2,746; 3,338; 3,375. Раствори-
мость KaSiF6 (в г на 100 г воды): 0,12(17,5°), 0,462(78°);
то же для Rb2SiFe: 0,16 (20°), 1,35(100°); для Cs2SiF6
0,6(17°). Термич. устойчивость растет от калия к цезию.
Кремнефторид бария BaSiFe кристаллизуется
вбесцветныхромбич.иглах; плотн. 4,279, растворимость
(г на 100 г воды) 0,025 (25°), 0,044(78°). Кремнефто-
риды других металлов хорошо растворимы и кристал-
лизуются обычно с гидратной водой. Большая группа
К. 2-валентных металлов общей формулы MeSiFe •
• 6Н2О, где Me — Mg, Мп, Fe, Со, Ni, Zn, Си дает
изоморфные между собой кристаллы с ромбоэдрич.
элементарной ячейкой. Двухводные К. Li2SiFe • 2Н2О,
CaSiF6 • 2Н2О, SrSiFe • 2Н2О, PbSiFe • 2Н2О образуют
призматич. моноклинные кристаллы.
Легкорастворимые кремнефториды Mg, Zn, Al под
техническим названием ф л ю а т о в применяют в
строительном деле для придания водонепроницае-
мости и закрепления поверхности строительных кам-
ней, гл. обр. известняков и мраморов. Процесс осно-
ван на взаимодействии К. с СаСО3 с образованием
малорастворимых фторидов и кремнезема. Благодаря
характерным формам хорошо выраженных кристал-
лов, К. используются в микрохимия, анализе.
Лит.: Рысс И. Г., Химия фтора и его неорганических
соединений, М., 1956; Позин М. Е., Технология минераль-
ных солей, 2 изд., М., 1961; Евстропьев К. С., Торо-
пов Н. А., Химия кремния и физическая химия силикатов,
2 изд., М., 1956; Kausch О., Flussaure, KieselflussaUre
und deren Metallsalze. Elgenschaften, Herstellung und Ver-
wendung, Enke, 1936. В. П. Варзаковский.
КРЕМНЕФТОРИСТОВОДОРОДНАЯ КИСЛОТА —
см. Кремнефториды.
КРЕМНИЙ (Silicium) Si — химич. элемент IV гр.
периодич. системы Менделеева; п. н. 14, ат. в. 28,086.
Состоит из трех стабильных изотопов: Si28 (92,27%),
Si29 (4,68%) и Si30 (3,05%). Сечение захвата тепловых
нейтронов атомом К. 0,13 барн. Получены искус-
ственно радиоактивные изотопы, в т. ч. Si32 (Т1/3 =
= 710 лет). Внешняя электронная оболочка атома К.
имеет строение 3s23p2. Энергии ионизации (в эв)
Si° —► Si+ —> Si2+ —> Si3+ —► Si4+ соответственно равны
8,15; 16,34; 33,46; 45,13. Сродство к электрону
Si° + е — Si' 1,22 эв.
Наиболее давно известным соединением К. является его
двуокись S1O2, К-рую химики 18 в. называли кремнеземом и
причисляли к «землям», считавшимися простыми телами.
Сложность состава кремнезема показал Берцелиус. Им же
был получен и подробно изучен элементарный К. Новому
элементу было дано название «силиций» (от лат. silex — кре-
мень). Русское название «К.» введено в 1834.
Содержание К. в земной коре составляет 27,6
вес. % и уступает только кислороду. В свободном
виде К. в природе не встречается и находится преим.
в виде двуокиси SiO2 (см. Кремния окислы) и силикатов.
Соединения К. входят в состав растительных и жи-
вотных организмов, где они необходимы для обра-
803
КРЕМНИЙ
804
зования твердых скелетных частей и тканей. Осо-
бенно много К. могут накапливать нек-рые морские
организмы, как растительные — диатомовые водо-
росли, так и животные — солнечники, радиолярии,
кремневые губки, скелет к-рых состоит из SiO2.
Эти организмы способны не только поглощать К.
из р-ров, но и разлагать силикаты алюминия (глины),
используя освобождающийся при этом SiO2. Диато-
мовые водоросли и инфузории, погибая, образуют на
дне морей в громадных количествах тонкий пористый
кремнезем, наз. кизельгуром, трепелом и др. Нек-рые
наземные растения (злаки, осоки, пальмы, хвощи)
также могут концентрировать К. Растения получают
К. из почвы (при недостатке его происходит задержка
роста нек-рых растений — кукурузы, овса, ячменя,
бобов и др.). В присутствии солей кремневой к-ты
повышается усвояемость растениями фосфорной к-ты.
Животные получают К. с пищей. В наибольших коли-
чествах К. обнаружен в плотной соединительной ткани,
в почках, поджелудочной железе.
Физические и химические свойства. Сплошной кри-
сталлич. К. — темно-серое металловидное тело. К.
имеет кубич. гранецентрированную кристаллич. ре-
шетку типа алмаза с периодом а = 5,4297 А. Есть
указания на существование нестабильной высоко-
температурной гексагональной модификации. Корич-
невый т. наз. «аморфный» К. представляет мелко-
кристаллич. форму кубич. модификации. Плоти. К.
2,328, т. пл. 1423°, т. кип. прибл. 2600°. Давление
пара {мм рт. ст.) 7,6 • 10 4 (1207°); 7,6 • 10~3 (1327°);
7,6 • Ю~2 (1467°); 7,6 - 10“i (1647°); 7,6 (1867°); 760
(2477°). Теплота плавления 11,1 ккал/г-атом, теплота
парообразования 71 ккал/г-атом. Атомная теплоем-
кость (в кал/г-атом град) твердого К.: 2,44 (138°К);
4,10(234°К); 4,84(297°К): 5,142(350°К); 5,301 (400°К);
5,585(500°К); 5,827(600°К); 6,178 ' (800°К); 6,354
(1000°К); жидкого 6,62 (при т. пл.). Теплопроводность
0,20 кал/см . сек град (20°). Температурный коэфф,
линейного расширения К. отрицателен при низких
температурах: — 0,6 • 10‘6 (—189°) и возрастает до
+2,4-10’® при 35°, средняя величина для области
18—1000° составляет 3,72 • 10’®. Твердость К. по
шкале Мооса 7, по Бринеллю 240 кГ/мм2, модуль
упругости при комнатной темп-ре 10890 кГ/мм2,
предел прочности при сжатии 9,47 кГ/мм2.
К., не содержащий примесей, с идеально правиль-
ной кристаллич. структурой, при абс. нуле должен
быть совершенным изолятором. При темп-ре, отличной
от абс. нуля, возникает собственная проводимость,
причем носителями электрич. тока являются не только
свободные электроны, но и т. наз. «дырки», образую-
щиеся после ухода электронов. Дефекты структуры
также влияют па электропроводность, вызывая обычно
дырочную проводимость. Принимается, что для чи-
стого К. при комнатной темп-ре уд. сопротивление
должно составлять величину порядка 10® ом см.
Электрич. свойства К. очень сильно зависят от при-
месей. Добавляя элементы V гр. периодич. системы,
замещающие К. в решетке с освобождением электро-
нов, получают кристалл К., к-рый проводит ток почти
полностью с помощью электронов (т. наз. n-тип про-
водимости); введение элемента III группы приводит
к созданию дырочной проводимости (р-тип). Ди-
электрич. проницаемость К. равна 12. К. диамагнитен,
с атомной магнитной восприимчивостью —5,3 • 10~®.
Для К. характерна прозрачность для длинноволно-
вых ИК-лучей. Показатель преломления К. 3,87.
Ат. радиус (при четверной координации и ковалентной
связи) 1,175 А, ионный радиус Si4+ 0,39А. Ввиду
большого радиуса атома К. заряд его ядра экрани-
рован, поэтому К. имеет тенденцию отдавать свои
валентные электроны. В связи с этим электроотрица-
тельность К. значительно меньше, чем у его ближай-
шего аналога углерода, и близка к таковой для метал-
лов IV гр. периодич. системы (электроотрицатель-
ности элементов IV гр. составляют, по Полингу:
С 2,5, Si 1,8, Sn 1,7, РЬ 1,5). Различие химич. свойств
К. и углерода видно из сопоставления прочности
пек-рых связей.
Таблица 1. Энергия некоторых связей Si и С
Связь Энергия связи по Полингу, ккал/моль Связь Энергия связи по Полингу, ккал/моль
Si—О 88,2 с—о 84,0
Si—Вг 69,1 С—Вг 65,9
Si—Cl 85,7 С—С1 78,5
Si—F 129,3 С—F 105,4
Si—J 50,9 С—I 57,4
Si—C 69,3 С—S 62,0
Si—S 54,2 с—с 83,1
Si—Si 42,2 с—н 98,8
Si—H 70,4 С—N 69,7
Большая прочность связи Si—О обусловливает
главные черты химии К.: чрезвычайное многообразие
его кислородных соединений, в частности силикатов,
составляющих основу земной коры. Напротив, связь
Si—Si значительно менее прочна, чем связь С—С,
и цепочки из атомов К. разрываются гораздо легче,
чем углеродные, особенно, если имеется возможность
образования связи Si—О. Большой интерес представ-
ляет сравнение полярности нек-рых связей С и Si.
Таблица 2. Степень «ионности» некоторых связей
Связь Степень «ионно- сти», % Связь Степень «ионно- сти», %
Si —H 2,5 C—H 4
Si —0 50 с—о 22
Si —c 12
Si — Cl 30 с—C1 6
SI — F 70 С—F 43
В соединениях К. (аналогично углероду) 4-валентен.
Однако, в отличие от углерода, К. наряду с коорди-
национным числом (к. ч.) 4 проявляет к. ч. 6, что
объясняется большим объемом его атома (примером
таких соединений являются кремнефториды, содер-
жащие группу Si ’). Соединения с формально 2-ва-
лентным К. (например, SiO) содержат, как теперь
полагают, связь Si—Si и являются полимерными.
Между атомами К. (в отличие от углерода), а также
между атомом К. и атомом углерода устойчивые
двойные и тройные связи не образуются.
При низких темп-рах К. химически инертен, но
при повышенных темп-рах реагирует со многими ве-
ществами. Реакционная способность К. сильно зави-
сит от его физич. состояния. Т. наз. аморфный К. зна-
чительно легче вступает в реакции, чем сплошной
кристаллический. Особенно химически активен рас-
плавленный К. На воздухе К. устойчив, даже при
повышенных температурах. В кислороде окисляется,
начиная с 400°, с образованием SiO2 (см. Кремния
окислы). Гидратные формы SiO2 представляют собой
слабые, очень мало растворимые в воде к-ты. Наряду
с моно- и изополикремневыми кислотами К. образует
гетерополикислоты, например кремнемолибденовую
Н8 [Si (Мо2О,)в], кремневольфрамовую Hg [Si (W2O7)eI
и др. Пары воды действуют на К. при ярко-красном
калении, но образующаяся пленка SiO2 замедляет
процесс окисления. При нагревании К. восстанавли-
вает большое число окислов, что связано с значитель-
ным сродством его к кислороду и трудной летучестью
SiO2. Возможно даже получение Mg путем восстанов-
ления окиси магния кремнием. В р-рах отдельных
кислот К. нерастворим, но растворяется в р-рах,
805
КРЕМНИЙ
806
содержащих смеси плавиковой к-ты с азотной к-той,
с хлоратом или нитратом калия. К. энергично раство-
ряется в р-рах щелочей с выделением водорода (наи-
более подходящим считается 35%-ный р-р NaOH).
Фтор действует на К. при комнатной темп-ре и реакция
сопровождается раскаливанием. Также интенсивно
действует хлор, но реакция начинается при 300°.
Взаимодействие с бромом и иодом начинается выше
500° и протекает спокойнее. Сухой HF действует
на К. при комнатной темп-ре, газообразные НС1,
НВг и HJ при 400—500°. Во всех этих процессах
получаются кремния галогениды общей формулы SiX4.
Кроме того, К. образует с галогенами соединения
типа SinX2n_^2’ пРичем в этом случае могут быть полу-
чены более длинные цепи, чем в кремневодородах
(см. ниже). SiF4 образует при гидролизе кремнефто-
ристоводородную к-ту H2SiF6, производные к-рои—
фторсиликаты, или кремнефториды, имеют разнообраз-
ное практич. значение.
С парами серы К. реагирует при 600°. В качестве
первичного продукта образуется дисульфид К. SiS2 —
бесцветные шелковистые нити, устойчивые в сухом
воздухе и медленно гидролизующиеся во влажной
среде. По своей структуре они представляют цепочки,
состоящие из тетраэдров SiS4. Моносульфид К. SIS
получается из дисульфида при высокой темп-ре. Селен
и теллур образуют с К. соединения, аналогичные
сульфидам, по отличающиеся меныпей устойчивостью.
Водород непосредственно не реагирует с К., и
кремневодороды (силаны) получают разложением
силицидов (см. ниже). Кремневодороды, подобно угле-
водородам ряда метана, имеют общую формулу
SinH2n_|_2>нообразуют сравнительно малоустойчивые
и короткие цепи, что связано с меньшей прочностью
связи Si—Si по сравнению со связью С—С (см. табл. 1).
К. образует две группы кислородсодержащих сила-
нов — силоксаны и силоксены. К сило-
ксанам относятся т. наз. кремнещавелевая HOOSi—
SiOOH и кремнемезоксалевая HOOSi — SiO — SiOOH
к-ты. Силоксены, производные полимера (8!6О3Н6)Л,
имеют циклич. строение и по кристаллич. форме
сходны с цеолитами. С азотом К. реагирует выше
1000°. Нитрид К. Si3N4 — бесцветные кристаллы,
плотн. 3,44; плавится (одновременно сублимирует)
при 1900°. Его уд. электрич. сопротивление 1,43-10®
ом-см, теплопроводность 0,045 кал/см сек град,
термин, коэфф, линейного расширения 2,47 • 10 s.
Нитрид К. не окисляется на воздухе и исключительно
стоек к действию химич. реагентов, даже таких, как
плавиковая к-та, расплавы щелочей, расплавленные
металлы, что делает его ценным материалом для
химич., огнеупорной и др. отраслей пром-сти. С фос-
фором К. образует фосфид SiP, с мышьяком — арсе-
ниды SiAs2 и SiAs, отличающиеся химич. стойкостью.
Большое промышленное значение имеет единственное
бинарное соединение К. с углеродом — кремния
карбид (карборунд) SiC. С бором К. образует
два соединения, B3Si и BeSi, отличающиеся твердостью
и химич. стойкостью.
К. растворяется во многих расплавленных метал-
лах, часто образуя химич. соединения — силициды.
Металлы Ag, Au, Be, Al, Ga, Sn, Zn, In, Sb растворяют
К., но не образуют с ним химич. соединений. В наи-
более тяжелых металлах, расположенных внизу
периодич. системы, — Bi, Pb, Т1 и Hg, К. нераство-
рим. Силициды К. образует со щелочными и щелочно-
земельными металлами (за исключением Be), с Ti,
Zr, Сг, Мо, W, Мп, металлами VIII гр. и лантанидами.
Силициды переходных металлов — металлич. соеди-
нения с высокой электропроводностью. Для многих
силицидов характерны смешанные металлически-ко-
валентные связи. Силицид магния Mg2Si имеет соле-
образный характер с четырехзарядным отрицательным
ионом Si4’ (см. Силициды).
Аналитическое определение. При
химич. анализе веществ, содержащих К., последний
мешает определению других элементов и должен быть
предварительно удалей, напр. в виде летучего SiF4
путем обработки плавиковой к-той. Нерастворимые
в кислотах соедипения К. — силикаты — обычно
разлагаются сплавлением со щелочами. При обработке
сплавов водой (в присутствии NaOH) в р-р переходит
кремнекислота, где она обнаруживается по образова-
нию желтого кремнемолибденового комплекса с мо-
либдатом аммония. Обнаружению мешает присутствие
фосфора, дающего аналогичное комплексное соедине-
ние с молибдатом аммония. Количественно К. опре-
деляют выделением кремневой к-ты из р-ров путем
дегидратации или с помощью коагулянтов, а также
осаждением кремнемолибденового комплекса органич.
основаниями. Для определения малых количеств К.
в р-ре применяется колориметрии, метод, основанный
на образовании желтого кремнемолибденового ком-
плекса или голубого продукта его восстановления
(подробнее см. Кремния определение).
Получение. К. технич. чистоты (95—98°/о) полу-
чают в электрич. печах восстановлением находящегося
в избытке кремнезема коксом. В связи с развитием
полупроводниковой техники, большое значение при-
обрели методы получения чистого и особо чистого К.
Это требует предварительного синтеза чистейших ис-
ходных соединений К., из к-рых К. извлекают путем
восстановления или термин, разложения. В практике
нашли применение след, методы получения чистого К.:
1) Восстановление газообразного SiCl4 цинком. Реак-
ция протекает в трубах из чистого плавленого кварца
при 950° между парами Zn и SiCl4. Образующийся
К. оседает на внутренних стенках реактора в виде
игл-дендритов, к-рые затем измельчают и тщательно
промывают р-ром НС1. 2) Восстановление SiCl4 или
SiHCl3 водородом. Эта реакция инициируется с по-
мощью электрич. разрядов. К. в расплавленном виде
накапливается на нижнем электроде и затем кристал-
лизуется в форме стержня. 3) Термич. разложение
SiJ4. Пары SiJ4 разлагаются на танталовой ленте на-
гретой до 1000°. 4) Термич. разложение моносила-
на SiH4. Несмотря на значительную чистоту полу-
чаемого указанными методами К., его дополнительно
подвергают зонной плавке. Наконец, из полученного
поликристаллич. материала выращивают монокри-
сталлы К. Указанными методами возможно получить
К. с содержанием примесей 10’5—10“6!1/о. Для опре-
деления таких малых количеств примесей в К. при-
меняют специальные методы: радиоактивационный
анализ и масс-спектрометрию. Вдыхание мельчайшей
пыли SiO2 и других соединений К. (напр., асбеста)
вызывает опасную профессиональную болезнь — си-
ликоз. Полагают, что частицы кремнезема дей-
ствуют химически на ткани легкого.
Применение. К. имеет все расширяющиеся области
применения. В металлургии благодаря своему боль-
шому сродству к кислороду К. применяют для удале-
ния растворенного в расплавленных металлах кисло-
рода (раскисления). К. является составной частью
большого числа железных и цветных сплавов. Обычно
К. придает повышенную устойчивость к коррозии,
улучшает литейные свойства и повышает механич.
прочность, однако в больших количествах К. может
вызвать хрупкость металлич. сплавов. Наибольшее
значение имеют железные, медные и алюминиевые
сплавы, содержащие К. В сплавах К. может присут-
ствовать в виде твердых р-ров, силицидов или в виде
сплава эвтектич. типа. К. является одним из элемен-
тов для произ-ва легированных сталей и чугунов (см.
Железа сплавы). Медно-кремнистые сплавы—бронзы
807
КРЕМНИЙОРГАНИЧЕСКИЕ КАУЧУКИ
808
(2—5»/0 К.) являются заменителями оловянной бронзы.
Сплавы алюминия с К. — силумины (4,5—14% К.),
обладают хорошей прочностью при малой плотности.
Для придания эвтектич. алюминиевому сплаву более
тонкого строения производят т. н. модифицирование,
что достигается добавкой, напр., натрия (0,1%).
В последнее время К. используют для изготовления
фотоэлементов, выпрямителей и транзисторов. В ка-
честве полупроводника К. имеет преимущество перед
германием, т. к. максимальная рабочая темп-ра у К.
составляет 250° (у германия 75°). Все увеличиваю-
щееся количество К. используется для получения
кремнийорганических соединений и нек-рых силици-
дов. Кремнезем и многие силикаты в очень больших
количествах (гл. обр. глины, полевые шпаты, слюды,
тальки и т. д.) перерабатываются стекольной, цемент-
ной, электротехнич. и др. отраслями пром-сти. Прак-
тически безграничное количество К. в земной коре
требует нахождения новых областей его применения.
Лит.: Бережной А. С., Кремний и его бинарные
системы, Киев, 1958; Красюк Б. А. иГрибовА. И.,
Полупроводники — германий и кремний, М., 1961; В в-
стропьев К. С. и Торопов Н. А., Химия кремния
и физическая химия силикатов, 2 изд., М., 1956; Кремний.
Сб., под ред. Д. А. Петрова, пер. с англ., М., 1960; Электрофи-
зические свойства германия и кремния. Сб. переводов, под
ред. А. В. Ржанова, М., 1957; Бажант В., Хвалов-
ски В., Ратоуски И., Силиконы, [пер. с чеш.],
М., 1960; Вопросы металлургии и физики полупроводников.
Полупроводниковые соединения и твердые сплавы, М., 1957;
Шашков Ю. М., Металлургия полупроводников, М.,
1960; Pauling L., The nature of the chemical bond and the
structure of moleculs and crystals, 3 ed., L., 1960, p. 85;
S n e e d С. M., В г a s t e d R. C., Comprehensive inorga-
nic chemistry, v. 7, The elements and compounds of group 4 a,
1958; Gmelin, 8 Aufl., Syst.-Nummer 15, TI B., Sllicium,
B., 1959; Mellor, v. 6, L.—N. Y.—Toronto, 1947; то же,
v. 6, 1953; Rare metals handbook, ed. C. A. Hampel, 2 ed., L.,
1961; Conwell E. M., Properties of silicon and germanium,
II, Proc. I. R. E., 1958, 46, Кэ 6, 1281—300. В. П. Борзаковский.
КРЕМНИЙОРГАНИЧЕСКИЕ КАУЧУКИ (сили-
коновые, полиорганосилоксановые, силастомеры) —
продукты поликонденсации циклич. силоксанов или
линейных силоксандиолов, получаемых гидролизом
диалкилдихлорсиланов. Макромолекулы К. к. имеют
линейное строение и содержат в основной цепи чере-
дующиеся атомы кремния и кислорода:
R R R
I I I
. . . —Si—О —Si—О—Si-О-. . .
I I I
R R R
По внешнему виду К. к. представляют собой бес-
цветную эластичную массу, способную к кристаллиза-
ции. Мол. в. 500000—1000000, а отдельные фракции
до 2 800'000. К. к. отличаются повышенной термо-
стойкостью, обусловленной значительной прочностью
силоксановой связи Si—О; энергия этой связи ок.
89,3 ккал, тогда как связи С—С в органич. каучуках
58,6 ккал. К. к. обладают также высокой морозостой-
костью.
Промышленное значение имеют К. к.: метилсил-
оксановый, метилфенилсилоксановый, метилвинилсил-
оксановый и метилсилоксановый, содержащий нитриль-
ные группы.
Метилсилоксановый каучук получают из диметил-
дихлорсилана.
Сначала проводят гидролиз диметилдихлорсилана, сопро-
вождающийся образованием полимерных соединений — ци-
клич. силоксанов и линейных силоксандиолов;
''si-0-Sl
Si-О—Si
(CH8)2S1C12—^(СН8)з81(ОН)8/ +IIC1
^Н- [-О-Si—]ПОН
Процесс ведут в среде водного этанола. Продукты гидролиза
образуют два слоя: верхний — слой силоксанового масла и
нижний — вода, спирт и кислота. Из силоксанового масла
отделяют перегонкой фракции циклич. диметилсилоксанов —
тетрамера и пентамера, кипящие при 174—208°. Следующей
стадией процесса является раскрытие циклов диметилсил-
оксанов (90—100°) и поликонденсация (20—25°, катализаторы—
конц. H2SO4, А1С13, FeCl, и др ). Полученный полимер отмы-
вают от катализаторов и сушат.
Метилфенилсилоксановый сополимер получают сов-
местным гидролизом (CH3)2SiCl2 и (СН3) С6Н5—SiCl2
и дальнейшей поликонденсацией полученных продук-
тов в присутствии основных катализаторов [КОН,
NaOH]. Метилсилоксановый полимер, содержащий
винильные группы, получают при поликонденсации
продуктов совместного гидролиза (CH3)2SiCl2 и
(СН3)(СР2=СН) SiCl2 или поликонденсацией смеси
диметилсилоксанов и метилвинилснлоксанов в присут-
ствии H2SO4 или FeCl3.
К. к. усиливают активными наполнителями (кол-
лоидная кремнекислота, белая сажа, аэросил, дву-
окись титана и др.). К. к., содержащие предельные
боковые группы (СН3, С2Н5 и т. д.), вулканизуют пере-
кисными соединениями, чаще всего — перекисью бен-
зоила. К. к. вулканизуются также под влиянием
7-излучения (Со60). Полученные таким путем резины
отличаются повышенной температуростойкостью (ма-
лое падение прочности с повышением темп-ры) и
термостойкостью (сохранение свойств после теплового
старения), но несколько уступают по механич. свой-
ствам резинам, полученным при вулканизации пере-
кисями. Метилвипилсилоксановый каучук способен
вулканизоваться серой. Он совмещается и совулка-
низуется (при наличии не менее 1% винильных
групп) с органич. каучуками и, в частности, с бутил-
каучуком. Совулканизаты обладают свойствами, про-
межуточными между свойствами ревин на основе
соответствующих органич. и метилвинилсилоксано-
вых каучуков. Введение в резиновые смеси из бутил-
каучука 25% метилвинилсилоксанового К. к. сущест-
венно улучшает морозе- и термостойкость резин, вы-
зывая нёк-рое снижение их механич. прочности.
Введение в макромолекулы К. к. небольшого коли-
чества (ок. 10%) фенильных групп повышает морозо-
стойкость резин до минус 90—100° вследствие нек-рого
нарушения регулярности структуры полимера и
затруднения кристаллизации. При увеличении содер-
жания фенильных групп св. гО’/о каучуки приобре-
тают негорючесть. Введение в макромолекулы К. к.
нитрильных групп обеспечивает получение термо-
маслостойких резип, сохраняющих работоспособ-
ность от —83° до +260° и по маслостойкости
близких к бутадиен-нитрилъным каучукам. К. к.,
содержащие в органич. радикалах атомы фтора, сохра-
няя высокую термо- и морозостойкость, обеспечивают
получение резин, обладающих хорошей стойкостью
к топливам и маслам. Нек-рые свойства вулканизатов
различных К. к. приведены в табл. (см. стр. 809).
Метилсилоксановый каучук обладает высокими элек-
троизоляционными свойствами (1015 ом). Благодаря
исключительной термостойкости, наряду с высокими
диэлектрич. свойствами и хорошей морозостойкостью,
резины, изготовленные на основе К. к., применяются
для жароупорных формовых прокладок, уплотнений,
диафрагм, мембран, клапанов, деталей мощных про-
жекторных установок, электроизоляции и др. рези-
новых технич. изделий, предназначенных для работы
в условиях низких и высоких темп-p. Эластичность
резин на основе К. к. сохраняется длительное время
при темп-рах от —60 до +225°, а кратковременно
до +250—300°. Резины из К. к. имеют невысокую
прочность на разрыв (40—50 кг/см2) и применяются
в деталях, работающих в основном на сжатие. Иногда
К. к. выпускаются в виде готовых резиновых смесей,
содержащих наполнители и вулканизующие агенты,
809
КРЕМНИИОРГАНИЧЕСКИЕ ПОЛИМЕРЫ
810
Показатели Метил- силокса- новый Метил- этилси- локса- новый Этил- силок- сановый Метил- винил- силокса- иовый Метил- фенилси- локса- новый
Предел прочности при разрыве, кг /см2 * Относит, удлинение, % . . . . Эластичность по отскоку, % . Темп-ра стеклования, °C ... Коэфф, морозостойкости вулка- низатов (100% растяжения) . 45-49 225-250 50 40-42 220-240 45 38—40 200—230 43 44-47 210-230 0,7 (-60е) 45-48 225-250 115 0,81 (—75°)
Лит.: Андрианов К. А. иСоболевский М. В.,
Высокомолекулярные кремнийоргапические соединения, М.,
1949; Бородина И. В., Н и к и т и н А. К., Технические
свойства советских синтетических каучуков, Л.—М., 1952;
Андрианов К. А., Г л у х о в а А. И., Хим. пром-сть,
1957, М 6, 27; С т а в и ц к и й И. К., Хим. наука и пром-сть,
1957, 2, № 3, 331; Поддубный И. Я. [и др.], Каучук
и резина,1960, № 9, 5; К а р ц е в В. Н., Ф о м и ч е в а М. М.,
там же, 1959, М 4, 3; и х ж е, там же, 1959, Xi 8, 25; Синте-
тический каучук, под ред. Г. С. Уитби, пер. с англ., Л., 1957,
с. 888—96. А. К. Никитин.
КРЕМНИИОРГАНИЧЕСКИЕ ПОЛИМЕРЫ — вы-
сокомолекулярные элементоорганич. вещества, содер-
жащие атомы кремния. По химич. составу и строе-
нию основной цепи макромолекулы К. п. делятся на
3 класса:
1) К. п. с неорганич. цепями макромолекул, обрам-
ленными органич. группами. Главные цепи макро-
молекул состоят из атомов: кремния — кислоро-
да, кремния — азота, кремния — кислорода — азота,
кремния — кислорода — алюминия, кремния — кис-
лорода — титана, а углерод входит в состав групп,
обрамляющих главную цепь. К таким К. п. относятся:
полиорганосилоксаны (или силиконы) (I), полиоргано-
силазаны (II), полиорганосилазаносилоксаны (III),
полиалюмоорганосилоксаны (IV), полититанооргано-
силоксаны (V).
R R R R
1 1 -Si -О—Si— 1 1 —Si—NH—Si—
1 1 R R 1 1 R R
(I) (II)
R R R R
1 I 1 —Si—NH—Si—O—Si— —Al—O—Si—O-
1 1 1 R R R 1 1 0 R
(Ш) (IV)
R
I I
-Ti-О-Si - 0-
I I
R
(V)
Из них наибольшее практич. значение имеют полиор-
ганосилоксаны и полиалюмоорганосилоксаны.
2) К. и. с органонеорганич. цепями макромолекул.
Цепи макромолекул содержат наряду с углеродом
атомы кремния и кислорода, напр.:
I I I
-СН2-Si-СН2-, —СН2—Si—О -Si-CH2-
I I I
3) К. п. с оргапич. цепями, обрамленными груп-
пами, содержащими кремний (карбоорганосиланы),
например:
-сн»-сн-
I
Si(CH3)3 ,
сн3
I
-сн2-с-
I
OCOSiR3
По форме и строению главные цепи
макромолекул К. п. могут быть ли-
нейными, иногда более или менее раз-
ветвленными, либо циклолинейными,
напр. полифенилсилоксан (VI) и поли-
алюмоорганосилоксан (VII).
К. п. в природе не встречаются и яв-
ляются синтетич. веществами.
Важнейшие методы получения К. п.
с неорганич. цепями макромолекул:
1. Совместный гидролиз различных
кремнийорганич. соединений с после-
дующей конденсацией образующихся олигомеров в вы-
О
Si
I
О
I
Si
о
с6н5 Свн5
.1/°'
Si
I
о
I
Si
I Чо'
5-6^5 свн3
VI
о
о.
R
.1 .
Si
I
О
I
Si
'I
R
О R О
\1 / ч
SI
6
।
Ai
OZR О
VII
сокомолекулярные соединения, напр. (А):
2 C6HsSlCl3 + 2 (CH3)2SiCl2 + 4 Н2О ZTfiQ
О СвН5
/ XI 5
(СНз), s‘ Si-OH
- “о О
HO-Si Si(CH3)2
Н3Сс О
нагрез.
Sl(CH3)2
6 о
I /
Si
CeH&
А
лН2О
Этот метод применяется для синтеза К. п. с развет-
вленными макромолекулами. Подобные К. п. исполь-
зуют в качестве смол (силиконовые смолы). Развет-
вление полимерной цепи макромолекул К. п. происхо-
дит за счет частичного раскрытия циклов и окисления
метильных групп у атома кремния.
2. Гидролиз или совместный гидролиз кремнийорга-
нич. соединений (Б) и каталитич. полимеризация
получаемых при гидролизе циклич. соединений (В):
4 (CH3)2SiCl2 + 4 н2о —-
(СН3)2 о (СН3)2
Si Si СН3 СН3 СН3
—^8НС14- о 6 - Si-O-Sl-O-Si-O - Б
II III
^Si с^з сн3 сн3
(СН3)2 О (СН3)2
2 C6H5SiCl3 + 2(CH3)2SiCl2 + 5Н2О
О С6Н5
(CH3)s Si Si-OH
I I
О о
I I
НО—Si xSi (СН3)2
Н5С6 0х
С6Н5
О | О (СН3)2
sr Si
I I
о о
I I
(CH3)2 C6H5
Катализаторами полимеризации служат серная к-та
и ее соли, щелочи, органич. аммониевые основания
[R4N]+OH_ и др. Этот метод приводит к образованию
К. п. с линейными цепями макромолекул-, которые
811
КРЕМНИЙОРГАНИЧЕСКИЕ ПОЛИМЕРЫ
812
являются эластомерами (см. Кремнийорганические
каучуки).
Практич. значение приобрели методы синтеза поли-
органосилоксанов и полиметаллоорганосилоксанов,
основанные на реакции обменного разложения. При
этом вначале исходный кремнийорганич. мономер
гидролизуют, а затем получают натриевые соли.
Обработанные алкил(арил)хлорсиланами натриевые
соли при нагревании вступают в реакцию поликон-
денсации (Г):
RSiCl3 + Н2О
Si Si
+NaOH
-H2O
RSl(OH)2ONa -^^7
СП нагрев.
RSi(OH)2O SiR —*
-*3 “HoO
полимер
К. п. с органонеорганич. цепями макромолекул по-
лучаются методами, основанными на поликонденсации
кремнийорганич. полифуикциональных мономеров с
органич. мономерами (Д):
с0н5 с0н5
I I
nR'OSi—О- Si-OR'-f-nHOCHa-СН2ОН—.
I I
СНз СН3
R'
СвН6 СвН5 -]
I I
—О—Si—О—Si—О—СН2—СН2— 0Н+2П-1R0H
I I
СН3 СНз Jn
Полимеры с органич. главными цепями макромоле-
кул, содержащими кремний в обрамляющих цепи
группах, получают обычными методами радикальной
или ионной полимеризации непредельных кремний-
органич. соединений, содержащих кратные связи
в органич. группах. Так, винилтриметилсилан поли-
меризуется в присутствии перекисей или таких ката-
лизаторов, как триалкилалюминий и четыреххло-
ристый титан (Е):
-г—сн-сн,-1—
n(CH3)3SiCH=CH2—- | Е
L Si(CH3)3 Jn
Свойства К. п. определяются в значительной сте-
пени составом и структурой главной цепи макромоле-
кулы. Полимеры с неорганическими главными цепя-
ми макромолекул обладают более высокой теплостой-
костью, чем полимеры с органонеорганич. или орга-
нич. цепями молекул, обрамленных кремнийорганич.
группами. Плотпость К. п. от 1,10 до 1,30 г/см3
(зависит от состава обрамляющих оргавич. групп
и структуры главных цепей макромолекул); она уве-
личивается, если главную цепь обрамляют ароматич.
группы или группы, содержащие гетероатомы в угле-
водородном радикале, напр. хлор, кислород в виде
эфирных групп и т. д. Все К. и. с линейной и цикло-
линейной структурой макромолекул хорошо раство-
римы в различных органич. растворителях: ароматич.
и алифатич. углеводородах, галогенопроизводных
углеводородах, кетонах, эфирах. К. п. с разветвлен-
ной и пространственной структурой макромолекул
не растворяются в органич. растворителях, но набу-
хают в них при нагревании. Сильно действующими
растворителями являются ароматич. углеводороды и
их галогенопроизводные.
Химич, свойства К. и. зависят от структуры и химич.
состава главных цепей молекул и химич. состава ор-
ганич. обрамляющих групп. Полиорганосилоксаны
с углеводородными обрамляющими группами устой-
чивы к действию кислот, щелочей, и только конц.
серная к-та и конц. щелочи вызывают расщепление
—Si—О—Si— связей. Полиметаллоорганосилоксаны
по своим химич. свойствам близки к полиоргано-
силоксанам; они более чувствительны к гидролизу,
чем полиорганосилоксаны, слабыми кислотами гидро-
лизуются, но устойчивы к щелочам. Линейные поли-
меры, макромолекулярные цепи к-рых построены из
атомов кремния и кислорода (полиорганосилоксаны),
по эластич. свойствам не уступают органич. поли-
мерам и превосходят их по теплостойкости и ряду
других свойств. Полидиметилфенилсилоксаны (VIII)
сохраняют эластические свойства при 180° в течение
2000 час, при 220°—150 час, а полиамиды, соответ-
ственно, 28 и 1,5 час. Полифенилсилоксан (IX) при
350° за сутки терял 3% веса, а капрон 94,3%, феноло-
формальдегидная смола 68%, полистирол 98,5%, поли-
тетрафторэтил 2,9%.
Высокая термостабильность полиорганосилоксанов
сочетается с рядом других ценных технич. свойств.
Они обладают хорошей хладостойкостью. Жидкие
полидиметилсилоксаны и их сополимеры, несмотря
на то, что они замерзают при —95°, нелетучи и не пере-
гоняются в вакууме. Темп-ры стеклования (хруп-
кости) эластомеров и смол полиорганосилоксапового
ряда от —60° до —95°, при этом тепловая устойчи-
вость резин на их основе высокая; они могут длительно
работать при 200° и кратковременно до 350°. Нек-рые
полимеры, напр. полифенилсилоксаповый с наполни-
телем (алюминий), образуют лакокрасочные пленки,
устойчивые в диапазоне темп-р от —60° до +550°.
Большой атмосфероустойчивостью и светостойкостью
обладают все известные полиорганосилоксаны и
полиметаллоорганосилоксаны. Полиорганосилоксаны
обладают высокими диэлектрич. свойствами. Так,
жидкие чистые полидиметилсилоксаны имеют тангенс
угла диэлектрич. потерь 0,0002, уд. объемное электро-
сопротивление 1014 ом • см, электрическая прочность
до 20 кв/мм. Эластомеры и смолы также обладают
высокими диэлектрич. свойствами. Пленки, получен-
ные из кремнийоргапических смол, имеют уд. объем-
ное сопротивление 1 1016 — 1 • 1016 ом • см. Тангенс
угла диэлектрич. потерь от 0,01 до 0,0005, в зависи-
мости от чистоты полимера. Электрич. прочность
пленок 40—60 кв/мм. Электрич. и др. свойства поли-
органосилоксанов мало изменяются с темп-рой; при
темп-рах до 200° уд. объемное сопротивление сни-
жается только до 1 • 1012—1 • 1013 ом • см, в то время
как у органич. полимеров электрич. свойства при ука-
занных темп-рах меняются очень резко. Действие
влаги оказывает незначительное влияние па электрич.
свойства полиорганосилоксанов.
Полиорганосилоксаны выпускают в виде жидких
полимеров, эластомеров и смол. Жидкие полимеры
широко используются для приготовления теплостой-
ких смазок, работающих при 200° и выше, для полу-
чения гидрожидкостей, работающих в широком диа-
пазоне темп-р (от—60 до +250°)и хладостойких гидро-
жидкостей и смазок для работы при минус 95—100°.
К. п. широко применяют для гидрофобизации раз-
личных материалов, тканей, бумаги, стекла, кера-
мики, строительных материалов и т. д. (см. Гидро-
фобные покрытия). Смолы применяют в произ-ве
лаков, пластмасс (композиционные пластмассы и слои-
стые пластики, включая и стеклопластики), электрич.-
изоляции.
813
КРЕМНИЙОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
814
О К. п., содержащих Al, Ti, В и др. элементы, см.
Высокомолекулярные соединения элементоорганические.
Лит.: Андрианов К. А., Кремнийорганические со-
единения, М., 1955; Бажант В., Хваловски В.,
Ратоуски И., Силиконы, [пер. счет.], М., 1960.
КРЕМНИЙОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ8 —
соединения, в молекулах к-рых имеется непосред-
ственная связь кремний-—углерод. Органич. соеди-
нения, содержащие кремнии, впервые были получены
еще в 1845 из спирта и SiCl4:
SiCl< +-4С2Н5ОН —- Si(OC2H5)4 + 4НС1
В 1863 при действии диэтилцинка на SiCl4 было синте-
зировано первое К. с.:
S1CU + 2Zn(C2H5)3 —. Si(C2H5)4 + 2ZnCI2
К К. с. примыкают нек-рые неорганич. соединения
кремния, аналогичные по строению органич. соеди-
нениям,— силаны (гидридыкремния), SimHn и гало-
генсиланы — соединения от SiHX3 до SiH3X (см.
Кремневодороды и Кремния галогениды). Все К. с. раз-
деляют на след, типы: алкил(арил)силаны RnSiH4_n;
алкил(арил)галогенсиланы RnSiX4_n; алкил(арил)-
алкоксисиланы RnSi(OR)4_n; алкил(арил)замещенные
силаны R4Si; алкил(арил)гидроксиланы RnSi(OH)4_m;
алкил(арил)аминосиланы RnSi (NH2)4_n. Получены
К. с., в к-рых атом кремния связан с металлом, напр.
трифенилсилиллитий (C6H6)3SiLi. Известны органич.
соединения, в к-рых кремний связан с азотом Si—NH
(силазаны) или с серой Si—S (силтианы). Важнейшими
органич. соединениями, содержащими кремний, явля-
ются эфиры и галогепэфиры ортокремневой к-ты,
соответственно Si(OR)4 и XnSi(OR)4_n.
К. с. приобрели большое значение в связи с разра-
боткой методов синтеза высокомолекулярных кремний-
органич. соединений, нашедших широкое применение
в различных областях техники (см. Кремнийорганиче-
ские каучуки, Кремнийорганические полимеры). Для син-
теза высокомолекулярных К. с. широко используют
алкил(арил)галогенсиланы, эфиры ортокремневой
к-ты, алкил(арил)замещенные силаны.
К. с. могут быть синтезированы несколькими спо-
собами, напр. взаимодействием хлорсиланов с цинк-
или ртутьорганическими соединениями, взаимодей-
ствием литийорганич. соединений с алкилхлорсила-
нами, алкилированием галогенгидридсили-
нов углеводородами и др. Промышленный
интерес представляло получение К. с. с по-
мощью реактивов Гриньяра, напр.:
C0H5CI-|-Mg—► C0H5MgCl; Si+2CI2—.SiClj
C0H5MgCI+SiCl4—>C0H5SiCIs+MgCl2
особенно прямой синтез из галогеналкила
или галогенарила и кремния, напр.:
2CH3C1+Si —- (CH3)2S1C13
Последний способ дает смесь SiCl4, CH3SiCl3,
(CH3)2SiC]2, (CH3)3SiCl, CH3SiHCl2. Методы,
основанные на присоединении непредельных и аро-
матич. углеводородов к галогенгидридсиланам:
CHE=CH-|-HSiCI3 —► CH2=CHS1C13
2C0H0+2HSiCl3 —► 2CeH5SiCI3+H2
Алки л(а р и л)с иланы получают действием
цинкорганич. соединений на хлорсиланы:
H3S1CI+Zп(СН3)2 —* CH3SiH3+CH3ZnCl
2HSlCl3+4Zn(C3H7)2 —►
—* HSi(C3H7)34-(C3H7)4Si-t-3ZnCl2+Zn4-C3Hs
или гидрированием алкил(арил)хлорсиланов:
RSlCIa-F3Ha — RS1H3+3HC1
В табл. 1 приведены нек-рые свойства простейших алкил(арил)силанов. Таблица 1
Соединение Т. пл., °C Т. кип., °C
Метилсилан, а CH3S1H3 . . Диметилсилан, (CH3)2S1H2. Триметилсилан, (CH3)3S1H Этилсилан, C2H5S1H3.... Бутилсилан, C,H9SiH3 . . . Трифенилсилан, (C9Hs)3SiH -156,5 —150 -135,9 -179,7 -138,2 36—37 - 57 —20 6,7 - 13,7 56,4 152—162 6
а d0,64 (—54°). ® При 2 мм рт. ст.
Алкил(арил)силаны в присутствии щелочей легко
гидролизуются:
4(CHj)2SiHs4-4HaO^22-[(CHs)2SiO]4+8Hs
и легко окисляются на воздухе, вступают во взаимо-
действие с литийорганич. соединениями, с бромом и
непредельными углеводородами.
Алкил(арил)галогенсиланы можно
получить взаимодействием галогеналкилов или гало-
генарилов с элементарным кремнием или галоген-
силанов с металлоорганич. соединениями или при-
соединением непредельных и ароматич. углеводородов
к галогенгидридсиланам. Интересен в техническом
отношении синтез алкил(арил)галогенсиланов дей-
ствием метилхлорида, этилхлорида, хлорбензола и др.
на элементарный кремний в присутствии меди или
сплавов кремния с медью и другими металлами; при
этом образуется смесь продуктов:
2RX+S1—-RaSiXj; 3RX+S1—>RSiX3+R-R
3RX4-S1—>R3SiX4-X2
где R=CH3, С2Н6, С6Н6 и т. д.; Х=С1 или Вг. Эта
реакция в случае галогеналкилов протекает при 280—
300°, а с галогенарилами при 420—450°; катализато-
ром обычно служит медь, реже Ni, Sn, Sb, Мп и др.
Подавляющее большинство алкил- и арилгалоген-
силанов представляет собой бесцветные жидкости,
дымящие на воздухе. Лишь отдельные представители,
главным образом высшие триалкил- и триарилгало-
генсиланы, при обычных условиях являются твер-
дыми кристаллич. продуктами. В табл. 2 приведены
свойства важнейших алкил(арил)силанхлоридов.
Таблица 2
Соединение Т. пл., °C Т. кип., °C 4 20 nD
Метилтрихлорсилан, CH3S1CI3 — 77,8 65,7 1,273
Диметилдихлорсилан, (CH3)2S1CI2 - 76,1 70,1 1,0663 1,4002
Триметилхлорсилан, (CH3)3S1C1 - 57,7 57,5 0,846 —
Этилтрихлорсилан, C2H5S1CI3 —105,6 97-100 1,2388 —
Диэтилдихлорсилан, (CaH5),,SiCl2 - 96,5 129-130 1.1050 1.4809
Триэтилхлорсилан, (C-?H5)3SiCI — 143,5 0,8986 1,4314
Винилтрихлорсилан, СН2=СН—SiCl3 . . — 92 1,2650 —
Фенилтрихлорсилан, C0H5SiCI3 — 201 1,3256 —
Алкил- и арилгалогенсиланы хорошо растворимы
в органич. растворителях: ароматич. углеводородах,
галогенопроизводных углеводородах, эфирах и др.
Алкил(арил)галогенсиланы легко разлагаются водой:
R2S1C12-|-2H2O —- R2Si(OH)2-|-2HCl
Продукты гидролиза легко конденсируются с потерей
молекулы воды и образуют циклич. или линейные
полимерные соединения. Алкил(арил)галогенсилапы
легко реагируют с аммиаком и аминами, со спиртами
и ангидридами кислот с образованием азотсодержа-
щих соединений кремния, замещенных эфиров, ацил-
производных кремнийорганич. соединений.
Замещенные эфиры ортокремне-
вой кислоты, или алкил(арил)алкоксисиланы.
815
КРЕМНИЙОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ — КРЕМНИЯ ГАЛОГЕНИДЫ
816
Подобно алкил(арил)галогенсиланам, эти соединения
приобрели большое значение в пром-сти. В отличие
от алкил(арил)галогенсиланов, они гидролизуются
труднее и процессы образования из них полимеров
легче регулируются. На основе этого класса соеди-
нений получены жидкие полимеры, гидрофобизирую-
щие и клеящие вещества, смолы, лаки. Важнейшие
методы получения замещенных эфиров ортокремне-
вой к-ты основаны на реакциях алкил(арил)галоид-
силанов со спиртами:
RSiCI3+3R'OH---► RS1(OR')3+3HC1
на взаимодействии магнийорганич. соединений с тет-
р аэтоксис планами;
RMgX+Si(OR')4 RSi(OR')1---MgXOR
или замещении водорода в алкилсиланах на алкоксиль-
ные группы при действии спиртов в присутствии алко-
голятов:
RgSiH-pR'OH---- R'OSiR3+H2
Алкил(арил)алкоксисилацы — бесцветные жидкости;
они хорошо растворяются в спиртах, ароматич. и
хлорированных углеводородах, кетонах и эфирах;
с водой не смешиваются. Нек-рые из них образуют
азеотропные смеси со спиртами и ароматич. углеводо-
родами. Физич. свойства нек-рых алкил(арил)алко-
ксисиланов приведены в табл. 3.
Таблица 3
Соединение Т.кип., °C J20 4 ,20 п£)
Метилтриметоксисилан, CH3S1(OCH3)3 103—105 1,3679
Триметилметоксисилан, (СН3)Э S1OCH3 57,2 — 1.3649
Метилтриэтоксисилан, С H3Si (ОС 2Н5) 3 151 0,9383 1,3861
Диметилдиэтоксисилан, (CH3)2Si(OC2H3)2 114 0,830 —
Триметилэтоксисилан, (CH3)3SiOC2H3 75 а 0,7573 1.3741
Этилтриметоксисилан, C2H-,Si(OCH3)3 126 0,9747 —
Этилтриэтоксисилан, C2H3Si (ОС2Нг,)3 159 0,9207 1,3853
Диэтилдиэтоксисилан, (С2Н3)3 Si (ОС2Н3)2 155-156 0,8752 —
Триэтилэтоксисилан, (СгН5)381ОС2Нг, . 153 0,8414 —
Фенилтриэтоксисилан, CsH3Si (ОС2Н3)3 233 1,0133 —
а При 745 Л1Л1 рт. ст.
Алкил(арил)алкоксисилапы легко гидролизуются в
присутствии к-т и щелочей:
R2Si(OR')2-2Н2О ^5^ R»Si(OH)2+2R'OH
Продукты гидролиза — диалкилдигидроксисилапы,
конденсируются в полимеры. Алкпл(арил)алкокси-
силаны легко реагируют с металлоорганич. соедине-
ниями.
Замещенные силаны (табл. 4) получают
действием на SiCl, или алкил(арил)галогенсиланы илн
алкоксисиланы металлоорганич. соединениями:
SiCl4+2Zn(C2H5)2.—►Sl(C»Hr,)1-|-2ZnCls
.ОС2Н3
Si(OCsH.,)4-|-4RMgBr —- R4Si-p4Mg<
'Вг
Кроме того, они могут быть получены присоединением
непредельных углеводородов к различным гидридси-
ланам.
Таблица 4
Соединение Т. кип., °C dat) 4 20 П£)
Тетраметилсилан, (CH3)4Si 26,5 0,646 1,3478
Тетраэтилсилан, (СаНо)4 Si Тетрапропилсилан, (С3Н?)4 Si ... . 153 0,7662 1,424
213 0,785 —
Тетрабутилсилан, (С4Нэ)4 Si 157 а 0,8008 1,4465
Тетрафенилсилан, (C3Hj)4Si 530 1.078 —
а При 22 мм рт. ст.
А л кил (а рил) гидроксисиланы
(табл. 5) получают гидролизом алкил(арил)алкокси-
силанов водой в нейтральной среде:
R2Si(OC2H5)2+2H,O—>R2Si(OH)2+2C2H5OH
Они легко конденсируются при нагревании с образова-
нием полимеров; важнейшие из них: триметилгидро-
ксисилан (CH3)3SiOH, т. кип. 100°, <7|° 0,8112; триэтил-
гидроксилан (С2Н5)3 SiOH, т. кип. 153—154°, <7|° 0,8647;
трифенилгидроксисилан (C6H6)3SiOH, т. кип. 155°;
диметилдигидроксисилан (CH3)2Si(OH)3, т. кип. 100—
101°; диэтилдигидроксилан (C2H6)2Si(O-H)2, т. кип. 96°;
дифенилдигидроксисилан (C6H6’)2Si(OH)2, т. пл. 132°.
Таблица 5
Соединение Т. кип., °C 4 20 Пр
Тетраметоксисилан, (CH3O)4Si . . 121-122 а 1,0523 1.3681
Тетраэтоксисилан, (C2H;jO)4Si. . . 166,5 0,9676 1,3837
Тетрапропоксисилан, (CgHzO^Si . 225-227 0,918 1,4015
Тетрабутоксисилан, (C4HoO)4Si . . 163 6 0,899 1,4128
Тетрафёноксисилан, (CeHjO)4Si . 415-420 — —
Тетра-о-крезилоксисилан,
(CH3C0H4O)4Si 443-445 — —•
а При 759 мм рт. ст. 6 При 20 лип рт. ст.
Алкил (а рил) аминосиланы получают
взаимодействием аммиака с алкил(арил)хлорсилапамп;
из них изучены лишь нек-рые представители, напр.
триэтиламиносилан (C2H6)3SiNH2, т. кип. 136—137°,
nff 1,4259 или трифениламиносилан (C6H6)3SiNH2,
т. пл. 55—56°.
Эфиры орто кремневой кислоты
(тетраалкоксисиланы) получают действием на гало-
генсиланы абс. спиртом:
SiX4+4C2H5OH —► Si(OC2H5)4+4HX
где Х=С1, Вг, F.
Тетраалкоксисиланы (табл. 5) — бесцветные про-
зрачные жидкости; тетраароксисиланы — густые мас-
ла или кристаллич. продукты. Все они обладают
характерным эфирным запахом, хорошо растворимы
в различных органич. растворителях. С увеличением
молекулярного веса соединения уменьшается его плот-
ность и повышается темп-ра кипения примерно на 12°
на каждую СН2-группу. Наиболее важной реакцией,
как и в случае алкилгалогенсиланов, является гидро-
лиз, ведущий к отщеплению алкоксильных групп:
I I
- Si-OR+HOH—> —Si—OH-J-ROH
I I
Полученные силанолы склонны в дальнейшем к кон-
денсации с образованием полимерных соединений.
Алкоксисоединения легко реагируют с ангидрида-
ми, хлорангидридами, хлористым алюминием, борной
к-той, четыреххлористым кремнием и т. д.; в послед-
нем случае образуются алкоксихлорсиланы, напр.:
(C2Hr,O)4Si+SiCl4—>2C(2H5O)2S1CI2
Лит.. Андрианов К. А., Кремнийорганические соеди-
нения, М., 1955; Петров А. Д. [и др.], Синтез кремний-
органических мономеров, М., 1961; Б аж ант В., X ва-
лов с к и В., Р а т о у с к и И., Силиконы, [пер. с чеш.],
М., 1960; Крешков А. П., Борн В. А., Практическое
руководство по анализу мономерных и полимерных кремний-
органических соединений, 2 изд., М., 1962.
К. А. Андрианов.
КРЕМНИЯ ГАЛОГЕНИДЫ — соединения крем-
ния с галогенами общей ф-лы SinX2n_(_2’ гДе X — гало-
ген. Часто К. г. рассматривают как производные
кремневодородов (силанов); продукты полного (SiC.l,)
или частичного (SiHCl3) замещения водорода галоге-
ном в силанах объединяют под общим названием
галогеносиланы. Наиболее изучены простейшие К. г.
SiX4, к-рые могут быть синтезированы из элементов:
Si 2Х2 =. 31Х4.’Фтор реагирует уже при обычных
817
КРЕМНИЯ ГАЛОГЕНИДЫ—КРЕМНИЯ КАРБИД
818
условиях, остальные галогены — при нагревании.
(Условия взаимодействия, а также энергии связей
кремния с галогенами см. в ст. f/ремний). Из химич.
свойств галогенидов SiX4 наиболее характерно их
энергичное взаимодействие с водой по общей схеме:
SiX4 + 2Н2О Z: SiO2 + 4НХ (гидролиз SiF4 идет
по более сложной схеме, см. ниже). При X — С1, Вг и J
равновесие практически нацело смещено вправо,
тогда как в случае F реакция заметно обратима.
Вследствие образования при гидролизе твердых ча-
стиц SiO2 (точнее xSiO2 • г/Н2О) пары таких К. г. ды-
мят во влажном воздухе. При гидролизе галогено-
силанов, содержащих водород, в нейтральном или
кислом р-ре одновременно происходит конденсация и
образуются полимеры с силоксановой цепью, напр.:
2SiH3CI+H2O—>H3Si—О—SiII34-2HCl
nSiH2CI2-|-r!H2O — (SiH2O)n-|-2nHCI
(SiH2O)„ — т. наз.просилоксан. SiCl4n его аналоги обра-
зуют с молекулами нек-рых других веществ продукты
присоединения, напр. SiCl4 • 6NH3. Большинство подоб-
ных комплексных соединений мало устойчиво. Важ-
ным исключением являются производные Si F4 и
фторидов — кремнефториды.
Кроме простейших К. г., изучены производные Si2X6.
Общим способом их образования является взаимо-
действие при высокой темп-ре паров SiX4 и «аморф-
ного» кремпия: 3SiX4 + Si z7 2Si2X6. Галогениды
Si2X6 — бесцветные жидкие или твердые вещества.
Для хлора и брома известны соединения и с более
длинными цепями атомов Si. Наивысшим известным
представителем гомология, ряда является Si25Cl52
(пластичная масса). Все сложные К. г. легко разла-
гаются водой с образованием в конечном счете Si(OH)4
и соответствующей галогеноводородной к-ты. При
проведении реакции на холоду и без избытка воды
могут быть выделены неустойчивые промежуточные
продукты гидролиза, еще сохраняющие связи Si—Si
в своем составе.
Кроме простых К. г., известны смешанные, напр.
SiF3Cl, SiBr2J2 и т. д., свойства к-рых являются про-
межуточными между свойствами соответствующих про-
стых К. г. Фторохлориды кремния образуются, напр.,
при взаимодействии SiCl4 и SbF3 при нагревании.
Получены также окси галогениды кремния, напр.
Si2OBr6, Si4O4Cl8 и т. д. Так, оксихлориды кремния —
бесцветные летучие и малоустойчивые соединения,
образуются при пропускании смеси паров SiCl4 с воз-
духом через сильно накаленную трубку. Известны
также полимерные субгалогениды кремния, формально
не отвечающие его валентности 4. Таков субхлорид
кремния, состав которого примерно соответствует
ф-ле SiCl0,5_2;6.
Ниже описаны простейшие К. г. — тетрагалогениды
кремния, или тетрагалогеномоносиланы. Некоторые
свойства галогеномоносиланов сопоставлены в таблице.
Кремния тетрафторид SiF4 — бесцвет-
ный ядовитый газ с резким запахом. При атмосферном
давлении сублимируется приблизительно при —95°;
плавится под давлением 2 атм при —77°; кипит (дав-
ление 1810 мм рт. ст.) при —65°. Теплота образова-
ния ДН298 = —370 ккал/молъ; термически стоек.
Водой гидролизуется с образованием кремнефтористо-
водородной кислоты: 3SiF4 + 2H2O=SiO2+2H2SiF6.
В пром-сти получается как побочный продукт при
переработке природных фосфатов, содержащих фто-
риды (образуется при действии крепкой H2SO4 на
CaF2 и фторапатит). Используется для получения
H2SiF'6 и кремнефторидов.
Кремния тетрахлорид SiCl. — бесцвет-
ная ядовитая жидкость, т. пл. —70°, т. кип. 57,6°,
плотн, 1,48 (20°). Теплота образования ДН288 =
= —153 ккал/молъ; термически стоек. Водой Легко
гидролизуется. Реагирует с окислами многих метал-
лов, образуя соответствующие хлориды. В пром-сти
SiCl4 получают хлорированием кремния, ферросили-
ция или смеси кремнезема с углем при высокой
темп-ре. В смеси с аммиаком дает очень густой дым.
Как SiCl4, так и др., более сложные галогеносиланы,
применяют для синтеза кремнийорганических соеди-
нений.
Кремния тетрабромид SiBr4 — бес-
цветная жидкость, т. пл. 5°, т. кип. 153°. Теплота
образования ДН298 = —95,1 ккал/молъ. Кремния
тетраиодид SiJ4 — белые кристаллы, т. пл.
121°, т. кип. 290°. Теплота образования ДН298 =
= —31,6 ккал/молъ.
Соединение T. пл., °C T. кип., °C/760 мм pm. cm. d
SiF4 ok.— 95,0 (субл.) -05/1810
SiHF3 -131 -95 —
SiH2F2 -122 —77,8
SiH3F — —98,6 —
SiCI4 - 70 57,6 1.48 (20°)
S1HC13 —127 31.8 1.35 (0°)
SiH2CI2 -122 8,3 1,42 (-122°)
S1H3CI -118 -30,4 1,15 ( — 113е)
SiBr4 5,0 153 2,81 (20°)
SiHBr3 —73,5 111,8 —
SiH2Br2 -70,1 ~ 66 2,17 (0°)
SiH3Br -94 1,9 1,53 (0°)
SiJ4 121 290
S1HJ3 8 220 3,31 (20°)
SiH2J2 — 1,0 149,5 2,729 (20,5°)
S1H3J - 57 45,4 2,035 (14.8°)
Лит. CMV при ст. Кремний, Кремния гидриды.
В. П. Варзаковский.
КРЕМНИЯ КАРБИД (карборунд) SiC — соеди-
нение кремния с углеродом, один из важнейших кар-
бидов, применяемых в технике. В чистом виде К. к. —
бесцветные кристаллы, обладающие алмазным блес-
ком; технич. продукт окрашен в зеленый или сине-
черный цвет. К. к. существует в двух основных
кристаллич. модификациях: кубической, стабильной
до прибл. 2000° и гексагональной, стабильной при
более высоких темп-рах, однако существующей также
и при низких темп-рах. Кубич. модификация имеет
одну единственную кристаллич. форму, обладающую
алмазоподобной элементарной ячейкой с периодом
а = 4,359бА, гексагональная модификация имеет
большое число форм (ок. 20), различающихся перио-
дом с гексагональной или ромбоэдрцч. элементарной
ячейки: а = 3,080бА и с = п • 2,512А, где п — елей-
ность упаковки (количество слоев в элементарной
ячейке). Причиной образования различных форм
гексагонального карборунда являются примеси и
дефекты роста кристаллов — число и распределение
винтовых дислокаций. К. к. плавится при 2830°
(с разл.). Темп-рпый коэфф, линейного расширения
повышается с ростом темп-ры, составляя для кубич.
модификации 4,5 • 106 (300°) и 5,4 • 106 (1200°).
Теплопроводность 0,04—0,05 кал/сек-см-град. Тепло-
емкость 6,46 (27°) и 12,65 кал/молъ-град (1227°).
В чистом виде К. к. является изолятором. В зави-
симости от характера примесей приобретает полупро-
водниковые свойства с п- или р-проводимостью.
Уд. электросопротивление технич. образцов может
меняться на много порядков в зависимости от коли-
чества примесей металлич. кремния. Температурный
коэфф, электросопротивления — отрицательный. Твер-
дость К. к. по Моосу 9,5—9,7 (по твердости уступает
только алмазу и карбиду бора).
Химич, стойкость К. к. очень высока. Из кислот на
него действуют только смесь азотной и плавиковой,
а также фосфорная к-та при 230°. Хлор действует на
819
КРЕМНИЯ КИСЛОТЫ-КРЕМНИЯ ОКИСЛЫ
820
К. к. уже при 400—600° и полностью разлагает его
при 1200°. От окисления К. к. предохраняет поверх-
ностная пленка кремнезема. Медленное окисление
К. к. начинается при 800°. Скорость окисления в токе
кислорода связана с чистотой К. к. и в зависимости
от количества примесей при 950° колеблется в преде-
лах 0,27—1,7% в час. Выше 1700° К. к. энергично
окисляется. Окисление К. к. при недостатке кисло-
рода и в присутствии растворителей кремнезема (ще-
лочи, фосфорная к-та и др.) приводит к разложению
К. к. с выделением графита при относительно низкой
темп-ре (200—250°). Расплавленные едкие и угле-
кислые щелочи в присутствии кислорода воздуха
взаимодействуют с К. к. по схеме:
PiC+Na2CO34-2O2=Na2SlO3-|-2CO2
SiC-|-2KOH-|-2O2=K2SiO3-|-H2O4-CO2
При взаимодействии К. к. с перекисью натрия обра-
зуются силикат натрия и карбонат натрия. С кремне-
земом К. к. не реагирует даже при 1900°. Расплавы
стекол и кислых шлаков, растворяя пленку кремне-
зема, ускоряют окисление К. к. Основные окислы
разрушают К. к.: окислы щелочноземельных метал-
лов при 1000°, окислы тяжелых металлов (FeO, NiO,
MnO) — при 1300—1370°, Fe2O3 — при 1000—1200°.
Расплавы металлов, взаимодействуя с К. к., обра-
зуют карбиды и силициды металлов.
К. к. получают в электропечах при 2000—2200° из
смеси кварцевого песка (51—55%), кокса (35-.-40%),
с добавкой NaCl (1—5%) и древесных опилок (5—10%).
Реакция образования К. к. протекает в 2 фазы:
SiO2-|-2C—»Si-|-2CO; Sl-|-C—-SIC
. Взаимодействие кварца с углеродом начинается уже
ок. 1000°, при 1600° оно становится заметным и уско-
ряется с дальнейшим повышением темп-ры. После
охлаждения (5—6 суток) продукт тщательно сорти-
руют, примеси удаляют промыванием водой и разб.
серной к-той. Как абразив К. к. более успешно, чем
корунд, применяют для шлифовки материалов с низ-
ким сопротивлением разрыву: латуни, бронзы, алю-
миния, чугуна, твердых сплавов и др. Из порошков
К. к. изготовляют шлифовальные круги, бруски,
шлифовальную бумагу и полотно. К. к. отличается
высокой огнеупорностью (1980°), теплопроводностью
(в 8—10 раз выше обычных огнеупоров), термостой-
костью и сопротивлением истиранию, вследствие чего
его применяют для получения огнеупорных изделий
специального назначения. Из К. к. изготовляют
огнеупорные плиты, муфели для туннельных печей,
футеровку пода коксовых печей, защитные обмазки
и др. Из смеси порошкообразного К. к., металлич.
кремния и глицерина изготовляют нагревательные
(силитовые) стержни для электропечей. Смесь прес-
суют и обжигают в среде СО или СО2 при 1700°.
Из К. к. изготовляют также плиты и покрытия в соору-
жениях с интенсивным движением (метро, вокзалы
и пр.). Кристаллы К. к. применяют в радиотехнике
(детекторы).
Лит.: Миклашевский А., Карборунд. Химический
анализ и свойства, Л.—М., 1938; Каменц ев М. В.,
Искусственные абразивные материалы, М., 1950; Береж-
ной А. С., Кремний и его бинарные системы, Киев, 1958;
Самсонов Г. В., Силициды и их использование в тех-
нике, Киев, 1959; Харман Дж. К., Карбид кремния,
в кн.: Ядерные реакторы. III. Материалы для ядерных реак-
торов, пер. с англ., М., 1956; Будников П. П. [и др.],
Технология керамики иогнеупоров, 3 изд.,М.,1962,с. 359; Silicon
carbide. A high temperature semiconductor. Proceedings of
the conference on silicon carbide, Boston, April, 1959, ed. J. R.
O’Connor and J. Smiltens, N. Y.-—Oxf.—L.—P., 1960; G m e-
1 1 n, 8 Aufl., Syst.—Summer 15, Silicium, Ti B., 1959.
S. 761 — 864. H. M. Павлушкин, В. П. Борзаковский.
КРЕМНИЯ КИСЛОТЫ — производные кремне-
вого ангидрида SiO2, очень слабые, мало растворимые
в воде кислоты. Выделены след, твердые К. к. опре-
деленного стехиометрического состава: SiO2-2,5H2O;
SiO2 • 2HgO — ортокремневая к-та; SiO4 • 1,5Н2О —
пирокремневая кислота; SiO2 • Н2О — метакремневая
к-та и 2SiO2 • Н2О — метадикремневая к-та. Эти гид-
раты, получаемые, напр., осторожным гидролизом
эфиров кремневой к-ты, весьма неустойчивы, и их
существование можно установить по наличию пере-
гибов на кривой зависимости давления пара от состава
твердой фазы.
Растворением в воде аморфного кремнезема (напр.,
обезвоженного геля К. к.) можно получить насыщен-
ный р-р H4SiO4 или H2SiO3; ее равновесная кон-
центрация (считая на SiO2) составляет 125 мг/л (26°),
170 мг/л (38°) и 416 мг/л (98°). Гидролизом Sid4 (см.
Кремния галогениды) или эфиров кремневой к-ты
можно получить пересыщенный р-р H2SiO3. В таких
пересыщенных р-рах постепенно происходит полиме-
ризация мопокремневой к-ты с образованием поли-
кремневых к-т, из к-рых в истинно растворимом со-
стоянии может находиться дикремпевая к-та. Даль-
нейшая полимеризация приводит к образованию кол-
лоидных частиц кремневой к-ты, находящихся в раст-
воре в виде золя. Эти частицы следует рассматривать
как соединение водосодержащего кремнезема с иони-
зированной кремневой к-той x(SiO2 • nH2O) ?/SiO3H +
+ г/Н+, причем отношение х : у может изменяться
от 600 до 1500, а мол. вес частиц равен приблизи-
тельно 1500. В зависимости от pH среды золь кремне-
вой к-ты либо может быть устойчивым, либо посте-
пенно агрегироваться, переходя в гель. Область с pH
5—6 является наименее устойчивой, и здесь гелеобра-
зование происходит быстро. При pH выше 7 коллоид-
ные частицы приобретают сильный отрицательный
заряд, и образование геля замедляется, а затем и сов-
сем прекращается. Устойчивость золей повышается
также при изменении pH от 5 до 2 и снова понижается
при pH < 2. Подбирая значения pH и применяя спе-
циальные вещества — стабилизаторы, удается полу-
чить устойчивые высококонцентрированные золи (до
30% SiO2). Золи кремневой к-ты находят применение
в произ-ве бумаги, в текстильной пром-сти, для обра-
ботки воды, в качестве связующего материала при
изготовлении литейных форм и т. д.
Гель кремневой к-ты, или силикагель, представляет
собой структуру, образованную сочленением гид-
ратированных групп [SiO4]4 . Как показали рентге-
нографии. исследования, имеется нек-рая упорядо-
ченность структуры геля, причем обнаруживается
сходство со строением кристобалита. При высушива-
нии геля образуются пористые продукты, служащие
в качестве адсорбентов.
Лит.: Ай л ер Р. К., Коллоидная химия кремнезема
и силикатов, пер. с англ., М., 1959; Думанский А. В.,
Учение о коллоидах, 3 изд., М.—Л., 1948; см. также лит.
при ст. Кремний. В. П. Борзаковский.
КРЕМНИЯ ОКИСЛЫ — соединения кремния с
кислородом, из к-рых устойчивым является двуокись,
или кремнезем SiO2; получена также неустойчивая
моноокись Si О и есть указания на существование полу-
торной окиси Si2O3.
Кремния моноокись SiO получают воз-
гонкой смеси SiO2 и Si в вакууме при 1250—1300°
в виде темпого смолоподобного аморфного продукта.
Накаливанием этой смеси выше 1800° удалось полу-
чить кристаллич. моноокись — кубич. кристаллы,
а = 5,16 А, плотн. 2,13. Твердая SiO находится в мета-
стабильном («замороженном») состоянии; термодина-
мически SiO устойчива только в виде газа при высо-
ких темп-рах. При 400—700° диспропорционирует
на Si и SiO2. При обычных темп-рах SiO в сплошной
массе устойчива к действию кислорода, особенно благо-
даря образованию предохраняющей пленки кремне-
зема. В последнее время оспаривается существование
моноокиси в кристаллич. состоянии.
Кремния двуокись SiO2 имеет большое
число кристаллич. модификаций. Кроме давно из-
821
КРЕМНИЯ окислы
822
вестных кварца, тридимита и кристо-
балита, недавно получены волокнистый
кремнезем и две новые модификации, названные
коэзит и к и т и т. Кремнезем легко переходит
в стеклообразное состояние (см. Стекло кварцевое)',
с помощью высокого давления получены своеобраз-
ные модификации стеклообразного кремнезема, т. наз.
супрапьезостекло и конденсирован-
ное стекло (с плотн. 2,61, тогда как обычное
кварцевое стекло имеет плотн. 2,2). У кварца, три-
димита и кристобалита при нек-рых темп-рах наблю-
даются фазовые переходы, протекающие с большой
скоростью и не сопровождающиеся изменением ха-
рактера связей тетраэдров SiO4, из к-рых построены
все кристаллич. формы кремнезема. Принимается,
что при этих фазовых переходах происходят сдвиги,
а также возникает или прекращается вращение струк-
турных единиц. Обычно самую высокотемпературную
форму обозначают буквой а (а-кварц, а-кристобалит),
затем следуют 0-форма и у-форма (имеется только
у тридимита). Взаимные переходы кварца, тридимита
и кристобалита происходят медленно и требуют
обычно присутствия легкоплавких веществ—т. н. мине-
рализаторов. Обычно принимается, что в соответствии
с диаграммой состояния кремнезема, предложенной
Феннером в 1913, а-кварц при 870° переходит в а-три-
димит, а последний при 1470° — в а-кристобалит.
Однако в самое последнее время высказывается пред-
положение, что тридимит не является стабильной
фазой на диаграмме состояния кремнезема. Опыт по-
казывает, что в области температурной устойчивости
тридимита часто образуется кристобалит (напр.,
при кристаллизации стеклообразного или прокали-
вании аморфного кремнезема ниже 1470° и т. д.).
Устойчивая при обычных условиях низкотемпера-
турная 0-модификация кварца — бесцветные кри-
сталлы призматич. формы, тригональной системы
с периодами решетки а =.- 4,913А и с = 5,405А (25°) и
показателями преломления ng= 1,553 и пр = 1,544.
Плотность 2,65068 (0°); теплота образования ДЯ298 =
= —210,3 ккал/молъ (получено новое значение
ан:98 = — 217,6 ккал/молъ; однако расчет теплот
образования других соединений кремния основан
на прежней величине теплоты образования 0-кварца).
При 573° происходит переход 0-формы в а-фор-
му, сопровождающийся поглощением теплоты (250
кал/молъ) и увеличением объема. Кристаллы а-фор-
мы, гексагональной системы имеют периоды решетки:
а == 4,989А и с = 5,44бА (575°); показатели прелом-
ления: ng = 1,540 и пр = 1,532; плотн. 2,533. Кварц
обладает пьезоэлектрич. свойствами. Растворимость
кварца в воде при обычной темп-ре очень мала —
порядка 0,0005%, но, начиная с 150°, возрастает,
достигая 0,25% при 500°. В последнее время разра-
ботаны методы выращивания крупных кристаллов
кварца очень высокой чистоты в бомбах высокого
давления при 300—500° и давлении 15000 атм. Рост
кристалла происходит на зародыше определенной
формы, обычно из р-ра NaOH, питание р-ра происхо-
дит за счет находящихся в бомбе мелких кусков
кварца.
Низкотемпературная у-форма тридимита кристаллизуется
в ромбич. системе; периоды решетки: а = 9,88 А, Ь = 17,1 А
и с = 16,3 А; показатели преломления; ng = 1,473, пр =
= 1,469; плотн. 2,264 (25°); теплота образования ДН°98 =
= — 204,8 ккал/моль. Тридимит имеет фазовые превращения
при 117° (у-> 3) и 163° (Р-> а). Обнаруженное нек-рыми
исследователями превращение при 210° объясняется примесью
кристобалита. В лабораторных условиях тридимит может
быть получен нагреванием кварца при 1300—1400° в присут-
ствии большого избытка Ka2W0, (150% от веса кварца).
Низкотемпературная p-форма кристобалита кристаллизуется
в триклинной системе, периоды решетки: а = 4.971А, с =
= 6,929 А; показатели преломления: ng = 1,487, пр = 1,484;
плотн. 2,32; теплота образования ДН'°2о8 ~ — 209,3 ккал/моль.
Высокотемпературная а-форма — кристаллы кубич. системы,
а = 7,143 А (541°); показатель преломления п — 1,466;
плотн. 2,21.
Полиморфные превращения кристобалита сильно зависят
от структурных нарушений его кристаллов и предваритель-
ной термич. обработки; р —>- а переход обнаруживается обычно
в пределах 220—272° при нагревании и 238—198° при охла-
ждении. Кристобалит можно получить, напр., прокаливанием
при 1150° аморфного кремнезема в присутствии небольшого
количества соды или поташа. Полиморфные превращения
кварца, тридимита и кристобалита сопровождаются измене-
нием объема. Это обстоятельство имеет большое значение при
эксплуатации огнеупорных изделий, содержащих свободный
кремнезем. Темп-ра превращения одной модификации в дру-
гую и происходящие при этом изменения объема приведены
ниже, знак «+» соответствует увеличению (объема).
Модификация кремнезема и темп-ры превращения Объемный эффект превращения при данной темп-ре, %
573° + 0,82
р-кварц —♦ а-кварц
117° Y-тридимит —► р-тридимит + 0,20
250° 0-кристобалит —* а-кристобалпт .... + 3,70
о 163° + 0,20
р-тридимит —* а-тридимит 870° а-кварц —► а-тридимит
+ 16,0
а-кварц —*• а-кристобалит + 15,4
1723° а-кварц ► кремнеземистое стекло . . + 15,5
кремнеземистое стекло —» а-кристобалит — 0,90
Новая, с плотностью 3,01, нерастворимая в плавиковой
к-те модификация кремнезема — коэзит, получена в бомбе
высокого давления (750° и 35 000 атм). Т. наз. китит, полу-
чаемый в гидротермальных условиях, схож с кварцем, но
кристаллизуется в тетрагональной системе. Волокнистый
кремнезем, кристаллизующийся в орторомбич. системе, полу-
чается окислением газообразной моноокиси S1O при
1200—1400°. В 1961 С. М. Стишов и С. В. Попова получили
в условиях высоких давлений (160—180 тысяч атм) и тем-
ператур (1200—1400°) новую рутилоподобную модификацию
кремнезема с плотностью 4,35 и шестерной координацией
щэемния, причем октаэдр Si О. сильно вытянут по диагонали:
для четырех атомов кислорода расстояние S1—О состав-
ляет 1,716А, для двух— 1,872А. Новая модификация по твер-
дости близка к корунду и устойчива к кислотам, включая
плавиковую, растворима в щелочах, имеет показатели пре-
ломления rig = 1,826 и Пр = 1,799; найдена в породах
аризонского метеоритного кратера (США).
Кремнезем имеет т. пл. 1728°, т. кип. не ниже
2950°. Химич, активность модификаций SiO2 возра-
стает от кварца к кристобалиту, тридимиту и осо-
бенно т. наз. аморфному кремнезему, получаемому
обезвоживанием геля кремневой к-ты. Из галогенов
на кремнезем действует только фтор. Плавиковая
к-та и газообразный HF энергично взаимодействуют
как с чистым кремнеземом, так и с силикатами. Крем-
незем растворим в щелочных р-рах. Аморфный крем-
незем растворяется в нек-рых органич. основаниях
(напр., этилендиамине), а также в ацетоне. Кремнезем
реагирует с многими окислами с образованием сили-
катов, причем этот процесс может происходить до
начала плавления, т. е. между твердыми частицами.
В природе из модификаций Si О2 широко распро-
странен кварц; кристобалит и тридимит встречаются
редко; недавно найден образец коэзита. Кварц обра-
зует т. наз. кварцевые пески, жильный кварц, песча-
ники, кварциты и др., а также входит в состав многих
горных пород (граниты, пегматиты и др.). Наиболее
чистой природной разновидностью кварца является
горный хрусталь. Окрашенные разновидности кварца:
дымчатый — раухтопаз, черный — марион, фиолето-
вый — аметист, желтый — цитрин. Встречаются так-
же скрыто-кристаллич. формы кремнезема (халцедон,
агат, яшма, кремень) и аморфные (опал, гейзерит).
Кремнезем широко применяется в силикатной про-
мышленности: в произ-ве стекла (кварцевое стекло, си-
ликатное стекло и др.), керамики (фарфор, фаянс,
динас и т. д.), абразивов, бетонных изделий, сили-
823
КРЕМНИЯ ОПРЕДЕЛЕНИЕ
824
катного кирпича и др. Важная область применения
кварца, в к-рой используются его пьезоэлектрич.
свойства, — изготовление радиотехнич. приборов и
ультразвуковых установок. В качестве пьезо-кварца
применяют кристаллы горного хрусталя, дымчатого
кварца и мариона, не содержащие дефектов. Для этой
же цели применяют специально выращиваемые кри-
сталлы искусственного кварца.
Лит.: Кукол ев Г. В., Химия кремния и физическая
химия силикатов, М., 1951; Зальманг Г., Физико-хими-
ческие основы керамики, пер. с нем., М., 1959; Евстро-
п ь е в К. С,, Торопов Н. А., Химия кремния и физи-
ческая химия силикатов, 2 изд., М., 1956; Белянкин Д. С.,
Иванов Б. В., Л а п и и В. В., Петрография технического
камня, М., 1952; Е i t е 1 W., The physical chemistry of the
silicates, Chicago, 1954; Шубников А. В., Кварц и его
применение, М,—Л., 1940; Будников П. П. [и др.],
Технология керамики и огнеупоров, 3 изд., М., 1962;
С т и ш о в С. М., Белов Н. В., О кристаллической
структуре новой плотной модификации кремнезема SiCL,
ДАН СССР, 1962, 143, № 4, 951; см. также лит. при ст.
Кремний. В. П. Борзаковский.
КРЕМНИЯ ОПРЕДЕЛЕНИЕ. Обычно соединения
кремния нерастворимы в воде, напр. природные и ис-
кусственные силикаты, а также входящие в состав
металлич. сплавов силициды п др. Некоторые из этих
соединений разлагаются сильными кислотами. Боль-
шая часть силикатов требует сплавления ‘ со щелоч-
ными материалами, чаще всего с углекислым натрием;
сплавляют в платиновом тигле, после чего разлагают
кислотой. Наиболее важные для анализа свойства
кремния: а) Образование коллоидных р-ров кремневой
к-ты. Гель SiO2 • иН2О выделяется при подкислении
и выпаривании р-ра досуха. Из слабощелочных
р-ров гель кремневой к-ты выделяется только при
действии нек-рых электролитов, аммиаката цинка
и т. п., причем выделение идет медленно и непол-
ностью. б) Образование в кислых р-рах летучего SiF4,
а при обычных темп-рах и при избытке HF — ком-
плексной кислоты H2SiF6, к-рая образует трудно-
растворимые соли с калием и с барием, в) Образование
гетерополикомплексов с молибденовой к-той. Кремне-
молибденовая к-та Н4 [Si (МО3О10)4] интенсивно жел-
того цвета, устойчива в кислой среде, разлагается
щелочами на соли молибденовой ц кремневой к-т.
В кислой среде она образует труднорастворимые соли
с рядом органич. оснований: способна реагировать
с восстановителями, образуя окрашенные синие
соединения.
Для количественного определения Si пользуются
гравиметрия., титриметрич. и фотометрия, методами.
Гравиметрия, методы применяют при определении
относительно больших количеств Si, напр. в сплавах,
силикатах и др. После переведения материала в кис-
лый р-р значительная часть кремнекислоты остается
в коллоидном состоянии. Для количественного выде-
ления кремнекислоты выпаривают до удаления воды.
Нерастворимый остаток представляет гидрат крем-
невой кислоты в порошкообразном виде. Для уско-
рения и более полного выделения кремнекислоты
часто прибавляют желатину, которая в кислом р-ре
ускоряет коагуляцию кремнекислоты. Нерастворимый
остаток обрабатывают кислотой, отфильтровывают,
промывают и прокаливают. Ввиду значительной ад-
сорбционной способности кремнекислоты и др. при-
чин выделенная выпариванием кремнекислота со-
держит 0,2—2% примесей. Поэтому при точных
определениях прокаленный осадок двуокиси кремния
обрабатывают фтористоводородной и серной к-тами
и выпаривают до удаления образующегося Si F4,
а затем прокаливают сульфаты металлов для превра-
щения их в окислы. По разнице в весе находят содер-
жание SiO2. В нек-рых случаях последний прием
используют непосредственно для определения крем-
ния. Анализируемый материал выпаривают с HF
и H2SO4 и по потере в весе рассчитывают содержа-
ние SiO2. Однако этот метод часто приводит к ошибкам
из-за летучести фторидов нек-рых металлов, а также
из-за образования сульфатов металлов вместо окис-
лов. Если в анализируемом материале имеется фтор,
описанные методы выделения кремнекислоты непри-
годны из-за летучести SiF4. В этих случаях для при-
ближенных определений вводят борную к-ту, к-рая
образует летучий BF3, удаляя, т. обр., фтор из сферы
реакции. При точных определениях для выделения
кремнекислоты из растворов, содержащих фториды,
применяют длительное их стояние при pH ~ 9 в при-
сутствии аммониевых солей, солей цинка и др.
Титриметрич. методы основаны гл. обр. па переведе-
нии кремния в кремнефторид, к-рый затем титруют
щелочью: SiF% + 40 Н~ -> Si(OH)4 -ф- 6F~. Предвари-
тельно выделяют кремнефторид из кислых растворов,
в к-рых он образуется, в форме кремнефторида калия.
В других методах нейтрализуют свободную к-ту.
Ряд титриметрич. методов основан на осаждении мало-
растворимых солей кремнемолибденовой к-ты (напр.,
пиридиниевой соли) и определением количества мо-
либдена в осадке.
Большинство фотометрических методов основано на
переведении кремнекислоты в кремнемолибденовую
к-ту. Эти методы применяют гл. обр. для определения
малых количеств кремния в различных материалах.
Кислый р-р обрабатывают избытком молибдата, при-
чем в определенных условиях образуется желтая
кремнемолибденовая к-та, количество которой опре-
деляют фотометрически. В отличие от аналогичного
соединения фосфора, она является более прочным
комплексом, в частности не разрушается при действии
2—4 н. р-ра сильной к-ты, при введении фосфорной
к-ты и др. Фосфорномолибденовую к-ту отделяют
также экстракцией смесью бутанола с хлороформом.
Аналогичные приемы используют также для опреде-
ления кремния в присутствии нек-рых других ме-
шающих компонентов. При необходимости увеличе-
ния чувствительности, а также в присутствии некото-
рых посторонних окрашенных компонентов кремнемо-
либденовую к-ту восстанавливают в определенных
условиях, при этом образуются интенсивно окрашен-
ные в синий цвет кремнемолибденовые комплексы.
Эти соединения интенсивно поглощают свет в красной
и в инфракрасной областях спектра, где поглощение
ряда компонентов, напр. солей железа (III), а также
избытка молибдена значительно меньше, чем в фиоле-
товой и ультрафиолетовой частях спектра. При вы-
полнении таких методов строго соблюдают ряд усло-
вий, чтобы достичь полного восстановления кремнемо-
либденовой к-ты,не восстанавливая избыток молибдата.
А. К. Бабко.
Определение кремния в органич. соединениях осно-
вано на разрушении вещества и выделении кремния
в виде двуокиси или в виде солей кремневой к-ты.
Дальнейшее определение содержания кремния прово-
дится методами, принятыми в неорганич. анализе
(см. выше).
В зависимости от свойств кремнийорганич. соеди-
нений применяются след, способы разложения: 1) На-
гревание с копц. кислотами (серной, олеумом, азот-
ной, хлорной) с добавлением иногда окислителей
(перманганат калия, бром). 2) Сплавление с карбона-
тами щелочных металлов. 3) Сплавление с щелочами.
4) Сожжение в токе кислорода в трубках из прозрач-
ного кварца. 5) Нагревание с перекисью натрия в гер-
метичной аппаратуре (бомба). 6) Нагревание с щелоч-
ным металлом в герметичной аппаратуре (бомба).
7) Гидролиз водой, разб. к-тами и щелочами. Наибо-
лее употребительны три первых способа. Наиболее
распространены весовое определение SiO2 и опреде-
ление по интенсивности окраски р-ра (колориметрии,
или спектрофотометрии ). Предложены методы одно-
825
КРЁНКЕ РЕАКЦИЯ—КРЕОЛИН
826
временного определения кремния и галогенов, а также
весовое определение кремния, углерода, водорода,
галогенов или серы из одной навески сожжениехМ
в токе кислорода. Анализ легколетучих соединений
выполняют в герметически закрытой аппаратуре
(бомбе или кварцевой трубке). Для нек-рых соедине-
ний возможен предварительный перевод в нелетучие
производные, напр. выдерживанием навески с олеумом
и др. реагентами при охлаждении и последующим
кислым разложением в обычных условиях. Разрабо-
таны специфич. методы определения, пригодные для
нек-рых типов соединений или отдельных веществ.
Н. Э. Гельман.
Лит.: Егорова Е. Н., Методы выделения кремневой
кислоты и аналитического определения кремнезема, М.—Л.,
1959; Анализ минерального сырья, под ред. Ю. Н. Книпсвич
и Ю. В. Морачевского, 2 изд., Л., 1956; Гиллебранд В.Ф.
[и др.], Практическое руководство по неорганическому ана-
лизу, пер. с англ., М., 1960; Бабко А. К., Пили-
пенко А. Т., Колориметрический анализ, М.—Л., 1951;
Климова В. А., Коршун М. О. и Березнич-
ка я Е. Г., ДАН СССР, 1952, 84, № 6, 1175; 1954, 96, № 1,
81; Ж. аналит. химии. 1956, И, вып. 2, 223; Крешков
А. П. [и др.], Анализ кремнийорганических соединений,
М., 1954, с. 106—43; Крешков А. П., Борк В. А., Усп.
химии, 1959, 28, вып. 5, 576.
КРЁНКЕ РЕАКЦИЯ — превращение четвертичных
пиридиниевых солей в альдегиды через нитроны по
схеме:
RCH3-N
n-(CH3)3N—CeH4—NO
Н3О
И*
Исходные пиридиниевые соли (X—обычно галоген)
чаще всего получают из соответствующих галогено-
производных и пиридина; иногда удобно получать
пиридиниевые соли иодированием гомологов гетеро-
пиклич. соединений и метилкетонов в присутствии
пиридина:
(выход 100Х)
(выход 74^)
Для получения нитрона четвертичную пиридини-
евую соль обрабатывают/г-нитрозодиметиланилином( А)
в воднопиридиновом р-ре в присутствии эквивалент-
ного количества едкого натра. Нитрон обычно выпа-
дает в осадок. Иногда нитрон получают иными спо-
собами. Для омыления нитрона в альдегид его обычно
обрабатывают разб. соляной либо серной кислотой,
и полученный альдегид извлекают эфиром. Метод
применяется для получения ароматич., гетероциклич.
а, ^-ненасыщенных и а-кетоальдегидов:
ОНС
I. A; NaOH
2. Н8О: H2SO4
(выход 77 %)
СНО
1. A; NaOH
Г 2.H2O;H2SO4
(выход 91 Ж)
I. A; NaOH
2. H2O: H2SO4
(Выш 87 %>
Механизм получения нитрона из нитрозосоединепия
и четвертичной пиридиниевой соли предполагает про-
межуточное образование продукта присоединения (I)
и гидрата нитрона (II):
ArNO
О
RCHOH
Аг -NOH
RCH
ArN—О
II
Механизм омыления нитрона до альдегида, вероят-
но, аналогичен механизму омыления шиффовых осно-
ваний:
R-СН НЛ Н2<Д
Аг-|Ч-О
R-CH-OH
Ar-N+-OH
I
Н
Н
—- RCHO + Аг —N+—ОН
I
Н
Применяя К. р., можно довольно просто и в мягких
условиях получать альдегиды, малодоступные при
использовании других путей синтеза. К. р. открыта
в 1936 Ф. Крёике.
Лит.: К г ft h n k е F., Angew. Chem., 1953, 65, № 24,
612. Е. М. Рохлин.
КРЕОЗОТ — маслянистая желтоватая жидкость
с запахом древесного дегтя и жгучим вкусом; перего-
няется при 200—230°, плотн. 1,037—1,087; малорас-
творим в воде, хорошо — в спирте, эфире, маслах.
К. состоит из смеси различных эфиров фенолов, гл. обр.
гваякола и креозола. Получают К. сухой перегонкой
древесины бука (реже других лиственных пород).
К. обладает сильным антимикробным действием, при-
меняют для предохранения лесных материалов от
гниения и в качестве флотореагента при обогащении руд.
КРЕОЗОТОВОЕ МАСЛО — см. Фенольные масла.
КРЕОЛИН — концентрированная водная эмульсия
мыльного эмульгатора и фракции масел (т. кип. 190—
230°), получаемых при термич. разложении угля, древе-
сины, торфа или сланцев. Действующим началом К.
являются входящие в состав масла органич. соединения
преим. ароматич. ряда; бактерицидным действием
обладают только фенолы и крезолы, инсектицидным —
нейтральная часть масел, фенолы и крезолы. К. под-
разделяют на фенольный (содержание фенолов
при применении К. для дезинфекции 26—33%, для
дезинсекции 10 ± 1%), бесфенольный (содер-
жит нейтральную часть масел) иактивирован-
н ы й (состоит из креолиновых масел, обогащенных
Д ДТ, у-изомером гексахлорциклогексана, хлорофосом,
пиретрумом и др. ядохимикатами).
К. — коричневато-черная маслянистая жидкость,
прозрачная в тонком слое, легко растворима в ацетоне,
эфире и хлороформе, нерастворима в кислотах; с водой
образует стойкую эмульсию. К. приготовляют след.
827
КРЕПИТЕЛИ ЛИТЕЙНЫЕ — КРИОЛИТ
828
образом: к 40—45 вес. ч. подогретого до 60—70°
эмульгатора (омыленная канифоль, хозяйственное или
технич. мыло, поверхностно-активные вещества —
ОП-7, ОП-Ю, контакт Петрова, нейтрализованные
сульфонафтеновые к-ты и др.) добавляют 55—60 вес. ч.
креолинового масла, смесь перемешивают в течение
40—60 мин. до получения однородной жидкости.
К. в виде водных эмульсий различных концентра-
ций применяют для борьбы с эктопаразитами и вреди-
телями с.-х. животных и растений, дезинфекции, дез-
инсекции, а также в ветеринарной практике для
лечения различных заболеваний.
Лит.: Дмитриев М. М., Обуховский Я. М.,
Краткий справочник коксохимика, М., 1960; Ефимов А. Л.,
Краткий справочник по применению ядов для борьбы с вре-
дителями и болезнями растений, 2 изд., М., 1958; Ци-
цин Н. В., Ч е р к а с с к и й Е. С., ДАН СССР, 1952,
84, № 3; Ц и ц и и Н. В., Черкасский Е. С., Ков-
туненко В. Ф., там же, 1962, 145, № 1.
КРЕПИТЕЛИ ЛИТЕЙНЫЕ — связующие для фор-
мовочных материалов, применяемые в литейном
произ-ве, гл. обр. при изготовлении стержней и форм,
с целью придания им необходимой прочности. В ка-
честве К. л. применяют растительные и минеральные
масла и их смеси с другими веществами: канифолью,
жидким стеклом, декстрином, сульфитной бардой,
продуктами термич. переработки древесины, торфа,
сланцев, каменных углей, нефти и др.
К. л. классифицируются по природе материала
(органические и неорганические, неводные и водные)
и характеру затвердевания (необратимый, обратимый,
промежуточный). К неводным относятся К. л., нера-
створимые в воде и несмачиваемые ею (напр., масла);
к водным — растворимые в воде или смачиваемые ею
(напр., сульфитно-спиртовая барда). К необратимо
затвердевающим относятся К. л., претерпевающие
в результате однократного нагрева при сушке стерж-
ней необратимые химич. изменения, приводящие к об-
разованию прочной пленки; к обратимо затвердеваю-
щим — К. л., к-рые при многократном нагревании и
охлаждении не меняют своих первоначальных свойств
(папр., канифоль). Промежуточные К. л. обычно
являются смесями необратимо и обратимо затверде-
вающих материалов.
Связующее действие различных К. л. характери-
зуется удельной прочностью, т. е. прочностью сухого
образца из формовочной смеси, отнесенной к 1%
связующего материала, введенного в смесь, из к-рой
изготовлен образец.
Наиболее качественными К. л. являются льняное
масло, олифа, пульвербакелит. Из К. л. на основе про-
дуктов переработки твердых топлив и нефти наиболее
распространены: а) крепитель ГТФ (тяжелая
фракция генераторной сланцевой смолы — ГОСТ
5339—50); б) крепитель ЗИС (сплав ГТФ и
нефтебитума, растворенных в уайт-спирите); в) кре-
питель КТ (суспензия торфяного пека и глины
в водном р-ре сульфитно-спиртовой барды) — раство-
римая в воде однородная твердая масса темного цвета,
состоящая из 50—55% торфяного пека, 28—30% суль-
фитно-спиртовой барды и 15—22% формовочной гли-
ны; г) древесный пек (остаточный продукт
после отгонки масел из смол, полученных при гази-
фикации древесины) в порошкообразном виде; д) д р е-
весный трехкомпонентный крепи-
тель (ДП), состоящий из 50% древесного пека,
25% сухой сульфитной барды и 25% формовочной
глины; е) крепитель КВ, получаемый при упа-
ривании необессмоленной кислой воды газогенератор-
ных станций, работающих на древесном топливе.
Разработаны высококачественные крепители на
базе продуктов переработки кам.-уг. смолы.
Лит.: Справочник машиностроителя, т. 5, 2 изд., М.,
1956; Сумароков В. П., Гордон Л. В., Химико-тех-
ническии контроль лесохимических производств, М.—Л.,
1956. А. М. Кунин.
КРИЗАНОЛ (олеокризин) — взвесь ауртиопропа-
нолсульфоната кальция и глюконата кальция в масле.
Действующее начало — ауртиопропанолсульфонат
кальция (AuSCH2CHOHCH2SO3)2Ca, мол. в. 774,51,
бледно-желтый порошок, не плавится, легко раство-
рим в воде, нерастворим в органич. растворителях,
устойчив при стоянии. Количественное определение
К. основано на определении Ан сжиганием, а Са—
оксалатным методом. Ауртиопропанолсульфонат каль-
ция получают, исходя из глицерина:
НОСНа-СНОН—СН2ОП—I^CICH2CHOH-CH2CI NaaS°3 •
--►ClCH3CHOHCH2SO3Na —HSCH2CHOHCH2SO3Na—-
IL'VtlCI’,AuscH2CHOH -CIbSO3Na CaC4-
------*(AuSCH2CHOHCH2SOg)2Ca.
К. применяют преим. при красной волчанке, а
также при лечении инфекционного полиартрита.
Лит.: Машковский М. Д., Лекарственные средства,
4 изд., М., 1960. В. Г. Яшунский.
КРИОГИДРАТЫ — продукты затвердевания вод-
ных р-ров эвтектич. состава. Практически имеются
в виду системы, состоящие из воды и соли. В таких
системах эвтектич. точка наз. иначе криогидрат-
ной точкой. Выделение К. при охлаждении вод-
ного р-ра (другими словами, одновременная кристал-
лизация всех растворенных солей и льда) происходит,
как и кристаллизация любой жидкой эвтектики, при
постоянной темп-ре (см. Двойные системы). Следует
подчеркнуть принципиальное отличие К. от к р и-
сталлогидрата, заключающееся в том, что К.
представляет собой механич. смесь кристаллов воды
и кристаллов соли (или солей), в то время как в кри-
сталлогидратах они образуют общую решетку кри-
сталла, отличную от решеток безводной соли и льда.
Равновесное давление водяного пара над К. равно
давлению насыщенного пара льда при данной темп-ре,
а над кристаллогидратом опо ниже. Образованием К.
пользуются для получения охлаждающих смесей.
КРИОЛИТ Na3AlF6 — одна из наиболее важ-
ных в технике солей фтора. Природный криолит —
редкий минерал, встречающийся в пегматитах. Обра-
зуется из горячих водных р-ров, обогащенных фто-
ром. Бесцветные кристаллы со слабым стеклянным
блеском, моноклинная сингония. Плотность минерала
2,95—3,01; твердость 2—3; хрупок. Природный К.
часто окрашен в серовато-белый, желтоватый, красно-
ватый, изредка черный цвета. Единственное крупное
месторождение К. находится в Ивигтуте, на юго-зап.
побережье Гренландии. Встречается в одной из топа-
зовых копей Ильменских гор на Южном Урале.
В системе NaF— A1F3 установлено существование
двух химич. соединений — К., плавящегося конгруэнт-
но при 1009 ± Г, и хиолита Na5Al3F14, составу
к-рого отвечает скрытый максимум с точкой перехода
725°. Ранее принималось, что К. плавится при 1ОО0°,
и в литературе все свойства были даны для этой
темп-ры. Плотность жидкого К. 2,080; поверхностное
натяжение 145,4 дин/см', вязкость 4,72 спуаза; уд.
электропроводность 3,23 обр.ом/см3. Истинная моле-
кулярная теплоемкость в интервале 0—1000° выра-
жается уравнением: Ср = 54,2 + 0,02781. При 565°
моноклинная сингония переходит в кубическую; те-
плота превращения 1294 кал/молъ. Теплота плавления
16640 кал/молъ. Растворимость окислов тяжелых метал-
лов в К. весьма ограничена. В системах Na3AlF6 — МеО
для хорошо растворимых окислов наблюдаются эвтек-
тики. В системе Na3AlFe — А12О3 эвтектика лежит
при темп-ре 962° и 10 вес. % А12О3. При 1050° К.
растворяет 16% А12О3. В пром-сти сырьем для полу-
чения искусственного К. служит плавиковый шпат
CaF2, к-рый разлагают конц. H2SO4 во вращающейся
печи при 220—280°. Выделяющийся HF поглощают
829
КРИОСКОПИЯ — КРИПТОЦИАНИН
830
водой в башнях с коксовой насадкой. Полученную
плавиковую к-ту очищают от кремния добавкой соды.
Чистую плавиковую к-ту нейтрализуют Na2CO3 и
А1(ОН)3 для осаждения К.После отстаивания и декан-
тации р-ра пульпу фильтруют и криолитовую пасту
высушивают во вращающихся барабанах при 130 —
140°. Побочным продуктом в произ-ве К. является
кремпефтористый натрий Na, SiFe {см. Кремнефториды).
Разработан также щелочной способ произ-ва К.,
заключающийся в спекании плавикового шпата с со-
дой и трепелом, водном выщелачивании спека для
получения р-ра NaF и карбонизации смеси р-ра NaF
и алюмината натрия. Основная область применения
К. — электролитич. произ-во алюминия, при к-ром
он в смеси с A1F3 служит электролитом, растворяю-
щим А12О3. К. применяют как компонент флюсов при
произ-ве алюминиевых сплавов, а также в произ-ве
молочно-белого стекла, эмалей и для др. целей.
Известны также калиевый (K3AlFe) и литиевый
(Li3AlF6) К., но они не имеют практич. применения.
Лит.: Бетехтин А. Г., Курс минералогии, 2 изд.,
М„ 1956; Беляев А. И., Рапопорт М.Б.,Фир са-
нов а Л. А., Электрометаллургия алюминия, М., 1953;
Беляев А. И., Металлургия легких металлов, 3 изд., М.,
1949; Беляев А. И., Жемчужина Е. А., Фирса-
нов а Л. А., Физическая химия расплавленных солей, М.,
1957; Phillips N. W. F., Singleton R. Н., Hol-
lingshead Е. A., J. Electrochem. Soc., 1955, 102, № 11,
648—49. А. Я. Фишер.
КРИОСКОПИЯ — измерение понижения темп-ры
замерзания раствора по сравнению с чистым раство-
рителем (точнее — измерение понижения темп-ры
равновесия кристаллов растворителя с раствором по
сравнению с темп-рой равновесия их с чистым раство-
рителем при постоянном давлении). Метод К., предло-
женный в 1882—88 Ф. Раулем, применяется для опре-
деления молекулярного веса растворенного в-ва, а также
его активности в растворе, что дает возможность в со-
ответствующих случаях рассчитывать осмотич. давле-
ние, относительное понижение давления насыщенного
пара растворителя или степень электролитич. диссо-
циации растворенного слабого электролита.
На основании Рауля закона между концентрацией
молекул растворенного в-ва и понижением темп-ры
начала кристаллизации Агкрист бесконечно разб. р-ра
(при отсутствии электролитич. диссоциации) суще-
ствует зависимость
А^крист. = (1)
где с — концентрация раствора, выражаемая обычно
в молях растворенного в-ва на 1000 г (реже на 100 г)
растворителя. Коэфф. К — величина постоянная для
данного растворителя, наз. криоскопической
постоянной, или молекулярным понижением
темп-ры замерзания растворителя. При определении
мол. веса М растворенного в-ва пользуются тем, что
вес его (в г), приходящийся на 1000 г растворителя,
равен М и, следовательно, число граммов g вещества,
растворенного в G граммах растворителя, определяется
„ McG
соотношением g = —откуда
в юоо J
м= gK 1000
д^крист. ®
(2)
Значения К (см. таблицу) могут быть вычислены из
соотношения
дткрист.
(3)
где Ткрист — темп-ра кристаллизации (отвердева-
ния) чистого растворителя по абсолютной шкале,
I — уд. теплота плавления его в кал/г и R — универ-
сальная газовая постоянная. Постоянную К данного
растворителя можно также определить опытным путем
из соотношения (2), измеряя понижение темп-ры на-
чала кристаллизации раствора вещества с известным
мол. весом. Криоскопические постоянные нек-рых
растворителей приведены в табл.
Растворитель ^крист. К-теор. К ОПЫТ.
Вода 0,00 1,859 1,853
Бензол 5,53 5,069 5,7
Уксусная кислота . . . 16,55 3,57 3,9
Диоксан 11,80 4.71 4,63
Камфора 178,4 37,7 40,0
Так как для точности результата имеет значение
точность измерения разности темп-р, то для К.
пользуются термометром Бекмана, предназначенным
для измерения температурных разностей с точностью
до 0,002° (или соответствующими сочетаниями тер-
мопар).
Для определения мол. веса методом К. берут отве-
шенное количество чистого растворителя и измеряют
его темп-ру отвердевания. Затем вновь расплавляют
растворитель (в том же сосуде), вносят в него навеску
определяемого вещества и измеряют темп-ру начала
кристаллизации полученного раствора при помощи
того же термометра (или той же термопары). Метод К.
получил в настоящее время применение также для
количественного определения содержания примесей
при приготовлении веществ высокой степени чистоты.
Лит.: Киреев В. А., Курс физической химии, 2 изд.,
М„ 1956.
КРИПТОН (Krypton) Кг — химич. элемент нуле-
вой гр. периодич. системы Менделеева, инертный газ;
п. н. 36, ат. в. 83,80. Состоит из смеси 6 стабильных
изотопов: Кг78, Кг80, Кг82, Кг83, Кг84 (56,90%) и
Кг86 (17,37%); приведенные в скобках числа относятся
к К. атмосферного происхождения. Поперечное сече-
ние поглощения тепловых нейтронов атомом К. 28
барн. Искусственно получены радиоактивные изотопы
К. Нек-рые изотопы К. образуются при делении урана.
Конфигурация внешних электронов атома 4s24p6.
Энергия ионизации (в эв): Кг° —► Кг+ —► Кг2+ —► Кг31"
соответственно равна 14,00; 24,57; 36,94. К. состоит
из одноатомных молекул: ат. радиус 1,98 А. Плотн.
газа 3,745 г/л (при 0° и 760 ммрт. ст.). Т. пл. — 157,1°
и т. кип. — 153,2° (760 мм рт. ст.). Теплота плавле-
ния 391 кал/молъ. Теплота испарения (в точке кипе-
ния) 2158 кал/молъ. Плотность жидкого К. (в точке
кипения) 2,155 г/см3 * *; «;рит — 63,8°; Ркрпт
55,9 кг/см3 и <7крит.0,909 г/л. Тройная точка: 115,95°К;
549 мм рт. ст.; плотность в тройной точке <4ГВ
2,821 кг/см3, d.K 2,451 г/см3. Как и другие инертные
газы, К. образует только соединения, связь в к-рых
осуществлена межмолекулярными силами: Кг • 6Н2О,
Кг • ЗС6Н5ОН и др. (подробнее см. Инертные газы).
В пром-сти К. получают из воздуха (см. Воздуха раз-
деление). Применяют К. в электровакуумной технике
для заполнения ламп накаливания, тиратронов, рек-
ламных трубок (чисто белый свет).
Лига. см. При ст. Инертные газы.
КРИПТОЦИАНИН (1,1'-диэтил-4,4'-хинокарбоциа-
ниниодид) C23H25N2J, мол. в. 480,40 — синтетический
краситель группы триметинцианинов:
К. получают конденсацией двух молекул иодэтилата
лепидина с этиловым эфиром ортомуравьиной к-ты
в пиридиновом р-ре. К. кристаллизуется из хлоро-
форма в виде мелких коричневых кристаллов; спир-
товый р-р зеленовато-синего цвета с = 7050 А.
831
КРИСТАЛЛИЗАЦИЯ
832
К. обладает способностью сенсибилизировать гало-
геносеребряную эмульсию к красной и ближней
ИК-областям спектра (X сенсибилизации 7500 А),
вследствие чего раньше применялся в пром-сти кино-
фотоматериалов; заменен более эффективными сенси-
билизаторами. Н. С. Вульфсон.
КРИСТАЛЛИЗАЦИЯ — процесс образования и
роста кристаллов из расплава, раствора или из газо-
вой фазы. К. возникает в результате пересыщения
или переохлаждения исходной фазы по отношению
к возникающей в ней твердой фазе. Газовая фаза наз.
пересыщенной, если (при постоянной темп-ре) давле-
ние Р превышает давление Ps насыщенных паров твер-
дой фазы, объем к-рой достаточно велик. Количественно
величина пересыщения выражается как &Р=Р — Ps,
или как A7’/Ps=y — 1, гДе Y = Р/Р3 наз. степенью
пересыщения. Для пересыщенного р-ра Ps заменяется
равновесной концентрацией насыщения (раствори-
мостью) Cs, а Р — концентрацией С. Метастабильный
расплав характеризуется переохлаждением &T=TS—Т
или Д77Т8, где Т— темп-ра переохлажденного рас-
плава, a Ts — темп-ра плавления.
Общая термодинамич. теория образования зароды-
шей новой фазы в ме'гастабильной переохлажденной
или пересыщенной исходной среде была развита Гибб-
сом в 1878. Изменение Дф свободной энергии системы,
обусловленное появлением в ней зародыша новой фазы
радиуса г:
Дф=г ^(|XS-H) +’-? = /(/•) (1)
где V — объем зародыша, р — его плотность, М —
мол. в., р2 — химич. потенциал новой фазы, pt —
химич. потенциал исходной среды, а — поверхност-
ное натяжение на границе фаз и S — поверхность
зародыша. При темп-рах, превышающих темп-ру фа-
зового перехода, ц2 > щ и величина Дф > 0 при
любых размерах зародыша и в процессе его роста.
Следовательно, образование и рост зародыша но-
вой фазы в этих условиях исключаются. Если
исходная фаза оказывается переохлажденной или
пересыщенной, то — Ц; <0 и функция ДФ =
= /(г) утратит монотонный характер и при
некотором критич. размере зародыша rm будет иметь
максимум ДФт, после прохождения к-рого Дф
резко уменьшается с дальнейшим ростом г. Следо-
вательно, в этих условиях должно начинаться обра-
зование новой фазы, причем скорость этого процесса
будет ограничена необходимостью пройти через по-
тенциальный барьер ДФт, играющий роль, аналогич-
ную энергии активации в процессах химич. превра-
щения. Для зародышей простейшей сферич. или ку-
бич. формы г = ——— - . Разность химич. потен-
1 1 m Hi—из р
циалов ц, — ц2 может быть выражена через величину
переохлаждения ц2 — р1=£(7' — Ts)/Ts и, следо-
вательно:
2М0-Т-
Гт= flAT
где L — скрытая теплота фазового перехода. Зародыш
новой фазы, имеющий критич. размеры, находится
в состоянии неустойчивого равновесия: если размеры
его немного увеличатся, он будет самопроизвольно
расти дальше, т. к. его рост сопровождается убылью
свободной эвергии системы; если же он немного
уменьшится, то за этим последует самопроизвольное
его уменьшение вплоть до исчезновения, что также
приведет к убыли свободной энергии.
Образование равновесного зародыша новой фазы
из пересыщенной газовой фазы или раствора (а также
из расплава) связано с законом Кельвина, выражаю-
щим зависимость упругости пара от размеров частиц.
При К. из газовой фазы этот закон выражается
ур-нием:
(3)
Ps Rr?r ' '
где рг — давление насыщенных паров над частицей
радиуса г, a ps — давление насыщенных паров твер-
дой фазы, объем (и размеры) к-рой достаточно велик.
Для К. из раствора в этом ур-нии отношение pr/ps
заменяется на отношение концентраций Cr/Cs. Равно-
весие между исходной фазой и образующимися в ней
зародышами новой фазы имеет неустойчивый харак-
тер. Если степень пересыщения исходной фазы отве-
чает равновесию по отношению к зародышу радиуса гт
определяемому из ур-ния (3), то при незначительном
уменьшении размеров зародыша давление его пара
окажется выше окружающего, что вызовет дальней-
шее все более и более быстрое его испарение.
Наоборот, при незначительном увеличении радиуса
зародыша давление его пара окажется меньше давле-
ния пара в окружающей среде, и зародыш будет про-
должать расти за счет конденсации этого пара. Сле-
довательно, каждой данной степени пересыщения
(или переохлаждения) исходной фазы соответствует
свой размер равновесного зародыша новой фазы,
определяемый из закона Кельвина:
__ 2Ма ...
Го—’ RTln(pr/ps)
От величины пересыщения в этом выражении
можно легко перейти к величине переохлаждения,
воспользовавшись ур-нием Клапейрона — Клаузиуса:
RTln p/ps == 1ЛТ/Т$, и тогда вновь получаем уже
известное выражение для размера равновесного за-
родыша. Из ур-ния, определяющего критич. размеры
зародыша новой фазы (2), следует, что чем больше
переохлаждение ДУ, тем меньше величина равновес-
ного зародыша и, следовательно, тем больше вероят-
ность его спонтанного образования.
Возникновение зародыша новой фазы и началь-
ная стадия его роста связаны с возрастанием сво-
бодной энергии системы (преодолением потенциаль-
ного барьера) и в рамках термодинамики не могут
быть объяснены. Фольмером, Странскими др. было дано
молекулярно-статистич. решение этой проблемы на
основе теории флуктуаций. Вероятность образования
равновесного зародыша новой фазы по Фольмеру
определяется из след, выражений: 1) кристаллизация
переохлажденного расплава
ВьЗ
W = Ае~ Т(^тг-
где А и В — постоянные;
2) конденсация пересыщенного пара
В рЗ
W — Ае ~
3) кристаллизация пересыщенного р-ра
BiJ3
рр- _тз ln-'(c'Cs)
Из этих ур-ний следует, что определяющую роль
в процессе образования зародышей новой фазы играет
межфазное поверхностное натяжение а. В связи с этим
часто наблюдается «ступенчатое» образование новой
фазы. Если к.-л. вещество может конденсироваться
из пара или выделяться из раствора в двух различных
модификациях, одна из к-рых стабильна при данной
темп-ре, а другая стабильна при более высокой темп-ре,
то необязательно сразу же возникнет стабильная мо-
дификация. Поверхностное натяжение а модифика-
ции, устойчивой при более высокой темп-ре, как пра-
833
КРИСТАЛЛИЗАЦИЯ
834
вило, ниже поверхностного натяжения низкотемпера-
турной модификации, и, следовательно, вероятность
образования нестабильной в данных условиях моди-
фикации может оказаться выше, чем стабильной. Так,
напр., при К. пересыщенных паров воды при темп-ре
не намного ниже точки замерзания, часто вначале
пары конденсируются в воду и лишь затем происхо-
дит К. Этим объясняется образование града. Однако
при больших переохлаждениях, т. е. когда темп-ра
значительно ниже точки замерзания, имеет место
прямая К. пересыщенных паров, без промежуточного
перехода в жидкую фазу, чем объясняется образова-
ние снега.
Существенную роль в процессах К. играют готовые
поверхности раздела фаз — стенки сосуда, взвешен-
ные коллоидные частицы примесей и т. п. Наличие
таких готовых поверхностей раздела значительно об-
легчает процесс образования кристаллич. зародышей
в результате адсорбции молекул исходной фазы на
этих поверхностях и снижения тем самым энергетич.
барьера, связанного с возникновением равновесного
зародыша. Особенно отчетливо роль готовых поверх-
ностей раздела проявляется при К. расплава. Дли-
тельная выдержка расплава при темп-рах, превышаю-
щих темп-ру К., повышает величину переохлаждения
расплава, при к-рой происходит образование кри-
сталлич. зародышей и в тем большей мере, чем выше
была темп-ра перегрева. Объясняется это тем, что
перегрев способствует растворению или дезактивации
твердых частиц примеси, на к-рых прежде всего воз-
никают кристаллич. зародыши при небольшом пере-
охлаждении, и основную роль приобретает истинная
спонтанная кристаллизация, т. е. образование трех-
мерных зародышей в объеме жидкой фазы, что тре-
бует гораздо большей степени переохлаждения.
Кинетика процесса К. характеризуется двумя ос-
новными параметрами: 1) числом центров К., т. е.
числом равновесных кристаллич. зародышей, возни-
кающих в единице объема исходной фазы в единицу
времени; 2) линейной скоростью роста кристаллов, т.е.
скоростью перемещения граней кристалла в направле-
нии .нормали к этим граням, выражаемой в слгсек.
При изотермич. К. оба параметра являются функцией
только степени переохлаждения. Однако практически
число центров К. и линейная скорость роста кристал-
лов могут меняться в результате выделения тепла К.
и вызываемого этим местного повышения темп-ры.
Молекулярно-кинетич. рассмотрение К. приводит к вы-
воду, что число пентров К. пропорционально произведению
вероятности образования равновесного зародыша на вероят-
ность его роста. Т. к. трехмерный кристаллич. зародыш ра-
стет не путем присоединения единичных молекул (атомов)
из окружающей среды (что имеет место при росте зародышей
жидкой фазы), а через образование и рост на его гранях двух-
мерных равновесных зародышей, то вероятность роста опре-
деляется вероятностью возникновения двухмерных зародышей
на его гранях. Т. обр. число центров К.:
Ваз В' (о')‘2
Т(ДТ)-7 ТАГ
п = Ае е
где А, В и В' — постоянные, а а' - периферийная межфазная
поверхностная энергия двухмерного зародыша. Отсюда видно,
что число центров К. существенно зависит от величины меж-
фазного поверхностного натяжения на границе кристаллич.
зародыш — исходная фаза. При заданной степени переохла-
ждения ДТ, число пентров будет тем меньше, чем выше меж-
фазное поверхностное натяжение. Следовательно, чем выше о
и о', тем в большей мере исходная фаза склонна к переохла-
ждению перед К. Металлы, у к-рых о и о' достаточно малы,
кристаллизуются уже при малых степенях переохлаждения,
тогда как соли, силикаты, органич. вещества в связи со сравни-
тельно высокими значениями о и о' весьма склонны к значи-
тельным переохлаждениям и могут вообще не закристаллизо-
вываться, образуя аморфные твердые тела — переохлажден-
ные жидкости. Можно понизить о и о' введением в кристал-
лизующийся расплав малых количеств поверхностно-активных
веществ. Молекулы (атомы) поверхностно-активных веществ,
адсорбируясь на гранях кристаллич. зародышей, значительно
снижают межфазное поверхностное натяжение и тем самым
резко уменьшают склонность данного расплава к переохла.-
14 к. X. Э. т. 2
ждению и вместе с тем повышают число центров К. в резуль-
тате уменьшения размеров равновесных зародышей, что при-
водит к возникновению высокодиснерсной микроструктуры
в закристаллизовавшейся жидкости. Этот метод, получивший
название модифицирования, широко используется в метал-
лургия для измельчения зерна в слитках.
Линейная скорость роста кристаллов теснейшим образом
связана с равновесной формой кристалла. Форма является
равновесной, если свободная энергия кристалла минимальна.
Для идеального кристалла среди всех кристаллич. форм рав-
ного объема (следовательно, и равной объемной энергии)
равновесной является та, к-рая обладает наименьшей свобод-
ной поверхностной энергией (для реальных кристаллов этого
требования недостаточно, т. к. в равных по объему кристаллах
объемная свободная энергия может быть различной в зависи-
мости от числа, характера и распределения дефектов струк-
туры). Она определяется след, вариационным выражением
Гиббса: ScjjSj—► min (при V = const), где OjSj. — свободная
поверхностная энергия и площадь i-той грани, V — объем
кристалла. Решение этой вариационной задачи, известное
под названием теоремы Вульфа, может быть представлено
след, образом: == const, где — расстояние г-той
грани от центра кристалла. Из этой теоремы следует, что
линейные скорости роста различных граней кристалла
пропорциональны величинам их поверхностной энергии.
Следовательно, быстрее всего растут грани, обладающие наи-
большей поверхностной энергией, и медленнее — грани с мень-
шим значением о. В результате этого равновесная форма кри-
сталла содержит преим. грани с наименьшей поверхностной
энергией.
Большое значение в процессе роста кристаллов
имеют винтовые и краевые дислокации, выходящие на
поверхность растущего кристалла. Наличие винтовой
дислокации резко снижает работу образования двух-
мерного зародыша на гранях растущего кристалла,
а если длина края винтовой дислокации превышает
длину края равновесного двухмерного зародыша, то
рост кристалла может происходить без образования
двухмерных зародышей путем прямого присоединения
к образующемуся дислокационному выступу молекул
(атомов) исходной фазы. Вследствие особой формы
поверхности грани при наличии винтовой дислокации
на ней образуется характерная спираль роста.
В. И. Лихтман.
Технологические методы. Методы К. и их аппара-
турное оформление зависят от физико-химич. свойств
исходной фазы и от характера решаемых практич.
задач. Ниже эти вопросы рассматриваются примени-
тельно к К. из р-ров, наиболее распространенной
в химич. пром-сти.
Неорганич. вещества чаще всего кристаллизуют из
водных р-ров, реже из спиртовых и водно-спиртовых
смесей, а органические —
также из р-ров в разно-
образных органич. жид-
костях. В зависимости
от природы растворенно-
го вещества и темп-ры К.
из водных р-ров могут
выделяться безводные
кристаллы или кристал-
логидраты с разным чис-
лом молекул воды. К. и з
р-ров производят: 1) ох-
лаждением горячих кон-
центрированных раство-
ров (если растворимость
вещества значительно
уменьшается с пониже-
нием темп-ры); 2) удале-
нием части растворителя
выпариванием р-ра; 3) од-
новременным охлажде-
нием и выпариванием го-
рячих р-ров; 4) пониже-
нием растворимости вы-
г,соли
Температура
Рис. 1. Зависимость раствори-
мости в воде нек-рых солей от
темп-ры.
деляемого вещества добавлением к р-ру др. веществ
(спиртов, кислот, солей и др.). Выбор метода в значи-
тельной мере зависит от изменения растворимости
.кристаллизующегося вещества с темп-рой (рис. 1). В
835
КРИСТАЛЛИЗАЦИЯ
836
ряде случаев выбор метода К. зависит от свойств
выделяемого вещества и сопряжен с температурным
диапазоном. Так, К. Na2SO4 -10 Н2О возможна охлаж-
дением насыщенного р-ра ниже 32°, a Na2SO4 —вы-
париванием при темп-рах выше 32°.
Если К. имеет целью лишь выделение твердого
вещества из р-ра, то важнейшим его технич. показа-
телем является степень К., выражаемая отношением
количества выделившегося в твердую фазу вещества
к его содержанию в исходном р-ре. Если же в резуль-
тате К. должна быть еще достигнута очистка основ-
ного растворенного вещества от сопутствующих при-
месей, то важнейшим показателем процесса является
также степень очистки.
Примесь, находящаяся в р-ре в ионной или моле-
кулярно-дисперсной форме, может попадать в кри-
сталлы в виде: включений маточного р-ра; компонента
твердого р-ра с основным веществом (в частности,
в результате внутренней адсорбции твердой фазой).
Примесь распределяется определенным образом между
основным веществом, выделившимся в виде твердой
фазы и оставшимся в маточном р-ре. Отношение кон-
центраций примеси (в расчете на основное вещество)
в обеих этих фазахназ. коэфф, распределения^).
Величина последнего определяется экспериментально;
в зависимости от природы твердых веществ и раство-
рителя, а также от условий проведения испытаний,
в частности от конечной темп-ры, она колеблется от
значений, близких к нулю, до 100 и выше. Очевидно,
только при D < 1 К. будет сопровождаться очисткой
основного вещества от примеси и концентрированием
последней в маточном р-ре. При D = 1 концентрация
примеси в образовавшихся кристаллах не будет от-
личаться от ее концентрации в исходном р-ре, а при
D > 1 примесь будет концентрироваться в твердой
фазе, убывая в маточном р-ре. Изменение концентра-
ции примеси в твердой фазе и солевой части маточ-
ного р-ра в результате К. выражается количественно
коэфф, изоморфного захвата примеси кристаллами <рк
и маточным р-ром <рм. Первый из этих коэфф, равен
отношению концентраций примеси в выкристаллизо-
вавшейся твердой фазе и в солевой части исходного
р-ра, а второй — отношению концентраций примеси
в солевой части маточпого р-ра и солевой части исход-
ного р-ра. Иэ условий материального баланса сле-
дует:
D 1
'Рк aD + 1 — а Н aD + 1 — “
где а — степень К. основного вещества. Наконец,
коэфф, захвата примеси кристаллами с маточным
р-ром фк, равный отношению концентрации примеси
в продукте К. за счет остатка маточного р-ра к ее
концентрации в солевой части исходного р-ра при D
значительно меньше 1, выражается ур-ппем:
। __ 100— ^кр 1 _1_
Скр ’ 100 — см ’ “О + 1 - а ’ Iх
где Скр — конц. основного вещества в кристаллах
после их отделения от маточного р-ра; См — концен-
трация основного вещества в маточном р-ре; ц —
коэфф., учитывающий дополнительный эффект очистки
кристаллов при их промывке после центрифугирова-
ния (при центрифугировании без промывки р, = 1,
с промывкой р > 1), Величины концентраций Скр
и См берутся соответственно составу твердой фазы
(напр., % CuSO4 • 5Н2О, а не CuSO4).
Суммарный коэфф, захвата примеси кристаллами
'Psk = + Фв
где tpSK — отношение концентрации примеси в отжа-
тых кристаллах к их концентрации в солевой части
исходного р-ра. Если в процессе К. происходит очистка
твердой фазы (D с 1)., то применяется понятие коэфф,
очистки кристаллов: Ккр ~ l/(psk.
Важной областью применения процессов К. яв-
ляется выделение отдельных веществ из их смесей.
Возможности и оптимальные условия такого процесса
разделения в основном зависят от характера веществ
и определяются диаграммой равновесия. С этой точки
зрения следует различать: 1) системы с неизоморфными
веществами, не образующими твердых р-ров; 2)' си-
стемы с изоморфными или изодиморфными вещест-
вами, образующими в широких интервалах концен-
траций твердые р-ры.
Простейшим примером разделения веществ, не об-
разующих твердые р-ры, может служить получение
хлористого калия и хлористого натрия из их природ-
ной смеси — сильвинита. При охлаждении р-ра,
насыщенного относительно КС1 и NaCl выделяется
в твердую фазу только КС1, a NaCl остается в маточ-
ном р-ре. Примерами фракционирования изоморфных
или изодиморфных веществ могут служить: 1) выде-
ление солей радия из солей бария, образующихся при
обработке минералов, содержащих радий; 2) разде-
ление солей редкоземельных элементов.
При К. из водных р-ров смесей веществ, образую-
щих твердые р-ры, выделяется в твердую фазу не
один, а одновременно несколько растворенных ком-
понентов. Принцип фракционированной К. основан
на различии отношений концентраций компонентов
смеси й твердой фазе и в маточном р-ре. Для получе-
ния изоморфных веществ иЗ их смесей в достаточно
чистом виде на практике обычно прибегают к много-
кратной перекристаллизации. Технологии, схемы К.
относительно многочисленны и весьма разнообразны.
Основные схемы изображены на рис. 2.
В случаях, когда необходимо выделить лишь растворенное
вещество, а не очистить его от сопутствующих примесей, при-
менима схема однократной К. с полным возвратом маточного
р-ра (рис. 2,х). Эта схема применима также для очистки основ-
ного вещества от примесей до тех пор, пока концентрация по-
следних в маточном р-ре не превышает допустимого предела,
после чего он выводится из системы и процесс повторяется.
Если же полный возврат маточного р-ра не может быть осу-
ществлен из-за высокой концентрации примесей, то прибегают
к его частичному возврату (рис. 2,ц), сопряженному с мень-
шей степенью К., т. е. с меньшим выходом продукта. Для
повышения выхода продукта без ухудшения степени его очистки
от сопутствующих примесей применяют схему, изображенную
на рис. 2,щ. По этой схеме маточный р-р (MJ после первич-
ной К. (KJ выпаривается до насыщенного состояния, а полу-
ченный насыщенный р-р (М2) подвергается вторичной К. (К2),
после чего кристаллич. продукт возвращается в растворитель,
а грязный маточный р-р (М3) выводится из системы. Если
концентрация примеси и коэфф, ее распределения позволяют,
то маточный р-р М3 может быть подвергнут вторичному выпа-
риванию и последующей К.
На рис. 2,iv изображена простейшая схема двукратной К.,
состоящая Из 2 растворителей (Pt и Р2) и 2 кристаллизаторов
(Ki и К2). По этой схеме из-за отвода из системы двух маточ-
ных р-ров (Mj и М2) выход продукта может оказаться низким.
Для повышения выхода продукта при двукратной К. приме-
няется схема с возвратом маточных р-ров в каждой из двух
стадий К. (рис. 2,у). При наличии в р-ре, кроме основного
вещества, сопутствующих примесей, последние будут непре-
рывно накапливаться в маточных р-рах, и их придется перио-
дически выводить из системы. Применение этой схемы целе-
сообразно в тех случаях, когда однократная К. не обеспечи-
вает требуемой очистки основного вещества, а двукратная —
дает эффект очистки выше требуемого.
В большинстве случаев оказывается выгодной схема, изо-
браженная на рис. 2,ух, по к-рой весь маточный р-р после
вторичной К. (М2) возвращается в растворитель исходного
вещества, а весь первичный маточный р-р (MJ выводится из
системы. Более высокий выход продукта, часто без существен-
ного ущерба для степени очистки основного вещества, может
быть достигнут по схеме, изображенной на рис. 2,уц где на
растворение исходного вещества возвращается не только весь
вторичный маточный р-р, но и часть первичного.
В ряде случаев, с целью повышения выхода продукта,
степень возврата первичного маточного р-ра в цикле можно
увеличить, если направить вторичный маточный р-р не только
на растворение исходного вещества, но частично также во
837
КРИСТАЛЛИЗАЦИЯ
838
второй растворитель. Схема такого цикла двукратной К. изо-
бражена на рис. В данной схеме, по сравнению с пре-
дыдущей, будет происходить повышенное накопление при-
месей в маточных р-рах. Для процессов К., имеющих целью
тонкую очистку основного вещества от примесей, рассматри-
ваемая схема может иметь весьма ограниченное применение.
Повышение выхода продукта в процессах двукратной К.
часто может быть достигнуто путем выпаривания первичного
маточного р-ра с последующей К. и возвратом кристаллов
в цикл. «Здесь возможны две технологич. схемы, изображенные
на рис. 2,IX и 2>Х- По первой из этих схем вторичный ма-
точный р-р полностью возвращается в растворитель исходного
вещества (Pi), а первичный маточный р-р после выпаривания
подвергается К. (К3), причем кристаллы присоединяются
к исходному веществу, а маточный р-р М3 выводится из си-
стемы. Отличительная особенность второй схемы состоит в том,
что часть первичного маточного р-ра возвращается непосред-
ственно на растворение исходного вещества и только осталь-
ная часть подвергается выпариванию и последующей К.
Рис. 2. Технологические схемы кристаллизации из р-ров:
А — исходное вещество; Р — растворение; К — кристал"
лизация; В — выпаривание; П — продукт кристаллиза-
ции; М — маточный р-р.
Кратность К. на практике в нек-рых случаях бывает
больше двух.В принципе при этомвозможно столько же
основных вариантов, сколько и при двукратной К. Выбор
кратности К. и оптимальной технологич. схемы может
быть произведен на осноне материальных и энергетич.
расчетов.
В пром-сти К. проводят в аппаратах различных конструк-
ций. Для К. выпариванием (удаление части растворителя
в виде пара) применяют обычные выпарные аппараты, чаще
всего аппараты с принудительной циркуляцией (см. Выпари-
вание). К. методом охлаждения горячих конц. р-ров при
небольших масштабах производства осуществляется в аппара-
тах периодич. действия (рис. 3), представляющих собою ци-
линдрич. сосуды, снабженные охлаждающими рубашками,
змеевиками и механич. мешалками, заменяемыми часто вибри-
рующими змеевиками, посредством которых одновременно
охлаждают р-р. Частая вибрация предотвращает инкруста-
цию змеевика и внутренней поверхности кристаллизатора. Так
14»
как корпус кристаллизатора выполнен в виде цилиндрич.
колонки, то кристаллы по мере своего роста спускаются вниз.
Рис. 3.
При значительных масштабах произ-ва
применяют кристаллизаторы непре-
рывного действия, чаще всего —
трубчатые, с водяным или воздушным
охлаждением (рис. 4). К., основанную
на одновременном охлаждении и вы-
паривании р-рой, производят часто
в многоступенчатых вакуум-кристал-
лизаторах (рис. £>). В последних имеет
место адиабатное испарение раство-
рителя при переходе от начального
давления (^1 ат абс.) к значитель-
ному вакууму; одновременно проис-
ходи! значительное понижение темпе-
ратуры р-ра. Число последовательно
соединенных кристаллизаторов (сту-
пеней) в одном агрегате доходит до
15; давление и темп-ра кипения в
кристаллизаторах падают в направ-
лении движения р-ра. Вторичный пар,
Рис. 4.
Рис. 3. Кристаллизатор периодического действия: 1 — ви-
брационный механизм; 2 — центральная труба; 3 — ру-
башка; 4 — кристаллизатор; 5 — гуммированная цилинд-
рич. стенка; 6 — кольцевое пространство; 7 — змеевик;
8 — кольцевой канал; & — выгружающий механизм.
Рис. 4. Трубчатый кристаллизатор с водяным охлажде-
нием: 7 — вход насыщенного р-ра; 2 — выход охлаждаю-
щей воды; з — выход кристаллов и маточного р-ра; 4 —
вход охлаждающей воды; 5 — кристаллизатор; 6 — водя-
ная рубашка; 7 — опорные бандажи; 8 — опорные роли-
ки; о — зубчатый венец для вращения кристаллизатора.
образующийся в отдельных кристаллизаторах, обычно исполь-
зуется для нагревания возвращаемого маточного р-ра. С уве-
Рис. 5. Многоступенчатые вакуумкристаллизаторы: i —
вход насыщенного пара; 2 — переточные трубы; 3 — вы-
ход кристаллов и маточника; 4 — кристаллизаторы;
5 — отвод вторичного пара; 6 — отвод конденсата;
7 — поверхностные конденсаторы; 8 — вход охлаждаю-
щей жидкости; & — Выход охлаждающей жидкости.
личеиием числа последовательно соединенных кристаллиза-
торов возрастает степень использования теплосодержания
исходного р-ра и увеличиваются раз-
меры кристаллов за счет уменьшения
степени пересыщения в каждой сту-
пени. Рассматриваемые кристаллиза-
торы бывают вертикальные и горизон-
тальные, причем в последнем случае
их обычно снабжают мешалками.
В тех случаях, когда особое зна-
чение имеет получение продукта в виде
крупных однородных зерен, применяют
кристаллизаторы специальной конст-
рукции (рис. 6) с принудительной цир-
куляцией очень слабо пересыщенного
р-ра через слой взвешенных растущих
в жидкостй кристаллов. Этот аппарат
Рис. 6. Кристаллизаторы специальной
конструкции с принудительной цирку-
ляцией: 1 — испаритель; 2 — опускной
канал для насыщенного р-ра; 3 —
t
кристаллизатор; 4 — выгрузка кристаллов; 5 — насос с мо-
тором; б — кипятильник; 7 — вход греющего пара; 8 — вы-
ход греющего пара.
839
КРИСТАЛЛИЗАЦИЯ ВЫСОКОМОЛЕКУЛЯРНЫХ СОЕДИНЕНИЙ
840
представляет собой сочетание испарителя и кристаллизатора.
Кипятильник 6 испарителя соединен циркуляционной трубой
с пространством испарителя 1, откуда вторичный пар уходит
через штуцер 8, а р-р опускается по трубе 2 в пространство
под решетчатым дном кристаллизатора з. На решетке всегда
находится слой кристаллов, к-рые смываются потоком насы-
щенного р-ра (снизу вверх), продолжая расти за этот счет.
Р-р, пройдя через слой кристаллов, поступает в трубки кипя-
тильника, а оттуда забирается циркуляционным насосом и
подается снова в пространство испарителя. Кристаллы вы-
гружаются через штуцер 4.
Лит.: Кузнецов В. Д., Кристаллы и кристаллиза-
ция, М., 1953; Данилов В. И., Строение и кристаллиза-
ция жидкости, Киев, 1956; Френкель Я. И., Собрание
избранных трудов, т. 3, М.—Л., 1959; Ш е ф т а л ь Н. Н., в сб/.
Рост кристаллов, т. 1, М., 1957, т. 3, М., 1961; X о н и г-
м а н Б., Рост и форма кристаллов, пер. с нем., М., 1961;
Бакли Г., Рост кристаллов, пер. с англ., М., 1954;
Варма А., Рост кристаллов и дислокации, пер. с англ.,
М., 1958; Горштейн Г. И., в кн.: Сборник работ лабо-
раторий института, М,—Л., 1951 (Тр. Всес. ин-та хим. реакти-
вов 1ИРЕА), вып. 20); Рост кристаллов, т. 1—3, М., 1957 —
1961; Хлопни В. Г., Избранные труды, т. 1, М.—Л.,
1957; Старик И. Е., Основы радиохимии, М.—Л., 1959;
Горштейн Г. И., Силантьева Н. И., в кн.: Изо-
тоны и излучения в химии [Сб.], М., 1958; Matz G., Die
Kristallisation in der Verfahrenstechnlk, B.—Gott. — Heidel-
berg, 1954. Г. И. Горштейн, И. И. Гельперин.
КРИСТАЛЛИЗАЦИЯ ВЫСОКОМОЛЕКУЛЯРНЫХ
СОЕДИНЕНИИ — образование кристаллич. фазы
в высокомолекулярных веществах. О закономер-
ностях К. в. с. и кристаллич. структуре полимеров
см. Кристаллическое состояние полимеров. _
КРИСТАЛЛИЧЕСКИЙ ФИОЛЕТОВЫЙ (кри-
сталлвиолет) — краситель класса триарилметановых;
C,5H3ON3C1; 9Н2О, мол. в. 570,2; кристаллы с бронзовым
блеском (при кри-
сталлизации с 9 мол.
Н2О) или с зеленым
металлическим блес-
ком (безводный краси-
тель). Краситель хо-
рошо растворяется в
воде и спирте с об-
разованием интенсив-
но фиолетовой окраски. Максимумы поглощения
в воде X =- 591,0 и 540,5 ммк. От добавления НС1 к
фиолетовому водному р-ру красителя окраска через
нек-рое время исчезает. При действии NaOH обра-
зуется фиолетовый осадок. К. ф. может быть получен
из окиси углерода и диметиланилина под давлением
250 ат в присутствии А1С13 или FeCl3, а также по
следующей схеме; сначала конденсируют диметилани-
лин с фосгеном (а); получившийся тетраметилди-
аминобензофенон (кетон Михлера) снова конденси-
руют с диметиланилином, напр. прп помощи хлор-
окиси фосфора (б):
a) 2C„H.-,N(CHS)3 + СОС12—.OC[C0H4N(CH3)2]2
6) ОС[СВЩН(СН3).,]2+С0Н-Л(СН3)2—-HO-C[CeH1N(CH3)3]3
Образующееся основание К. ф. представляет собой
гексаметилтриаминотрифенилкарбинол; последний с
соляной к-той образует соль, являющуюся красителем.
К. ф. применяют как индикатор для определения pH
с переходом окраски от зеленой до синей в пределах
pH от 0,5 до 2,0; для осаждения, отделения и цветных
реакций на Zn, Cd, Sb, TI, W, Au, Hg и др. элементы,
способные образовывать комплексные галогенные или
кислородные анионы с большим мол. весом; для
фотометрии, определения Zn, Hg, W; в микроскопии —
для окрасок; К. ф. применяется также как краситель
в произ-ве химич. карандашей, чернил, лент для пишу-
щих машинок, штемпельной краски; ограниченно
применяется для крашения хлопчатобумажного во-
локна по танниновой протраве. И. п. Ефимов.
КРИСТАЛЛИЧЕСКОЕ СОСТОЯНИЕ — устойчи-
вое фазовое состояние твердого тела, у к-рого струк-
тура обладает правильной периодич. трехмерной по-
вторяемостью расположения частиц — атомов, ионов
или молекул. Строгая повторяемость в расположении
частиц, распространяющаяся подряд на бесконечное
число периодов, наз. дальним порядком;
структуру, обладающую дальним порядком в трех
пространственных измерениях наз. кристалли-
ческой решетке й. Все другие известные со-
стояния вещества — газообразное, жидкое, ротационно-
кристаллическое (или газокристаллическое) и жидко-
кристаллическое — существенно отличаются от К. с. от-
сутствием дальнего порядка и наличием элементов бес-
порядка в расположении частиц. Реальные кристаллы,
в том числе и имеющие совершенную огранку, почти
всегда содержат большое число дефектов строения,
отклонений от идеальной периодичности кристаллич.
решетки (см- Дефекты структуры, Дислокации).
На диаграмме состояния вещества (см. Фаза) К. с.
занимает совершенно определенную область, соответ-
ствующую, как правило, относительно более низким
темп-рам и более высоким давлениям. Образуя кри-
сталлич. решетку, частицы занимают положения,
соответствующие минимуму их потенциальной энер-
гии взаимодействия. При кристаллизации всегда вы-
деляется нек-рое количество тепла. Такое же коли-
чество тепла требуется затратить на плавление кри-
сталлич. решетки. Одпа из самых характерных осо-
бенностей К. с. — анизотропия, к-рая про-
является в том, чтомеханич., оптич., электрич. и др.
свойства кристаллов в общем случае зависят от на-
правления, т. е. различаются по непараллельным на-
правлениям. К. с. противопоставляется аморфному
состоянию жидкостей и твердых тел, обладающему
лишь ближним порядком в расположении частиц и
изотропией — одинаковыми свойствами по веем
направлениям.
По степени распространенности среди твердых тел
К. с. является основным: почти все твердые тела,
включая минералы и металлы, являются кристалличе-
скими. В природе иногда встречаются крупные оди-
ночные кристаллы (монокристаллы), к-рые легко отли-
чить по их внешней огранке. Монокристаллы могут
быть получены и в искусственных условиях. Однако
большинство кристаллич. тел является поликристал-
лами — сростками большого числа по-разному ориен-
тированных мелких кристалликов неправильной
формы.
Кристаллы могут возникать различными путями
(см. Кристаллизация). Они могут вырастать из газо-
вой фазы (см. Сублимация) и из жидкой фазы — при
охлаждении расплава, насыщенного р-ра или при
испарении насыщенного р-ра. Они могут образоваться
и из других твердых тел — кристаллич. или аморф-
ных — при изменении темп-ры или давления, либо
просто при их хранении. Наконец, кристаллы могут
появиться непосредственно в химич. процессе образо-
вания данного вещества. Если не приняты специаль-
ные меры, то кристаллизация обычно приводит к об-
разованию не монокристалла, а поликристаллич. тела.
Отдельные маленькие кристаллики («зерна») в таком
теле четко видны под микроскопом на отполирован-
ной и подвергнутой травлению кислотой поверхности
металла. Обычные размеры зерна в поликристаллич.
телах — металлах и сплавах — 19 3—ПУ5 см. Каждое
зерно-кристаллит — это кристалл, принявший не-
правильную форму, т. к. его дальнейшему росту пре-
пятствовали соседние кристаллы. Зерна отделены друг
от друга т. н. межкристаллитной прослойкой, в к-рой
частицы расположены в нарушенном порядке. Меж-
кристаллитные прослойки содержат значительное
число чужеродных частиц, вытесненных туда в про-
цессе роста зерен. Поскольку в поликристаллич. теле
отдельные маленькие кристаллы имеют совершенно
случайные ориентации, то такое тело как целое
(в объеме, содержащем достаточно много зерен) пред-
ставляется изотропным. Однако иногда (напр., в ре-
841
КРИСТАЛЛИЧЕСКОЕ СОСТОЯНИЕ ПОЛИМЕРОВ
842
зультате пластич. деформации) может возникать тек-
стура — преимущественная ориентация кристал-
литов в определенном направлении, а вместе с нею и
анизотропия свойств поликристаллич. тела. Реаль-
ные кристаллы, как правило, имеют блочное строение.
Отдельные блоки с идеальной кристаллич. решеткой
обычно имеют размеры в несколько сот или тысяч А
(несколько сот периодов элементарной ячейки кри-
сталлич. решетки). Эти блоки слегка дезориентированы
друг относительно друга на уголдо одного градуса и сме-
щены на величины порядка межатомных расстояний.
В зависимости от того, какой из типов связей между
частицами преобладает — ионный, металлический, ко-
валентный или вандерваальсовский (см. Химическая
связь, Межмолекулярное взаимодействие), твердые
кристаллич. тела делят на ионные кристаллы, металлы,
валентные кристаллы, молекулярные кристаллы. Ион-
ные кристаллы отличаются хорошей ионной проводи-
мостью при высоких темп-рах, сильным поглощением
в ИК-области спектра и часто хорошо выраженной
спайностью. Они образуются сочетанием ионов сильно
электроположительных и сильно электроотрицатель-
ных элементов. Типичным примером ионного кристалла
является NaCl. Отличительными чертами металлов
являются хорошая электро- и теплопроводность. Ме-
таллы состоят из атомов электроположительных
элементов. Валентные (атомные) кристаллы, наир,
алмаз и карбид кремния, обладают малой электрон-
ной и ионной проводимостью, весьма высокой твер-
достью и плохой спайностью. Молекулярные кри-
сталлы, к-рые образуются из органич. молехгул (напр.,
метана, бензола) или атомов благородных газов,
характеризуются низкими точками плавления и ки-
пения и, как правило, испаряются в виде устойчивых
молекул. Имеется большое число кристаллич. тел,
относящихся по своим свойствам одновременно к двум
или нескольким группам. Поэтому перечисленные
выше группы не могут быть резко разграничены. По
электрич. свойствам кристаллич. тела делятся на
проводники, полупроводники и изоляторы. Различие
в электрич. и оптич. свойствах кристаллов хорошо
объясняется т. н. зонной теорией твердых тел, осно-
ванной на квантово-механич. расчете движения сво-
бодных электронов в периодич. потенциальном поле
идеальной кристаллич. решетки.
Ю. В. Мнюх> А. И. Китайгородский.
КРИСТАЛЛИЧЕСКОЕ СОСТОЯНИЕ ПОЛИМЕ-
РОВ—фазовое состояние полимеров, характеризующее-
ся образованием высохсоупорядоченных кристаллич.
областей (кристаллитов) и специфическими
физич. свойствами. По сравнению с кристалли-
ческим состоянием низкомолекулярных веществ К. с. и.
отличается рядом особенностей, связанных с природой
полимера, состоящего из длинных макромолекул.
К. с. п. может быть достигнуто различными путями;
охлаждением расплава, осаждением из раствора,
непосредственно в результате химич. процесса их
образования и, наконец, при растяжении аморфного
материала. Кристаллизация последнего типа является
специфич. свойством ряда полимерных веществ.
Структура полимеров в кристал-
лическом состоянии. Низкомолекулярное
вещество может быть получено в виде монокристаллов,
в к-рых размер молекулы на много порядков меньше
размера кристалла. Обычный кристаллич. полимер
представляет собой поликристалл, размеры кристал-
литов в к-ром порядка нескольких сот ангстрем.
Т. обр., длина вытянутой макромолекулы больше раз-
мера кристаллита, в к-ром повторяющейся единицей,
образующей кристаллич. решетку, является, собствен-
но, не целая макромолекула, а ее звено.
Структуру полимеров в кристаллич. состоянии во
многих случаях удалось расшифровать методами рент-
геноструктурного анализа и электронной микроско-
пии (полиэтилен, полипропилен, полиэтилентерефта-
лат, ряд полиамидов и т. д.).Если при кристаллизации
охлаждением образуется тело, состоящее из беспоря-
дочно ориентированных кристаллич. областей, то
при одноосном растяжении кристаллизация идет одно-
временно с образованием текстуры. В этом случае
оси цепей в кристаллич. областях вытягиваются преим.
по направлению растяжения. Такого рода текстуру
имеют также естественные и синтетические волокна.
Кристаллич. области в полимерах, как правило,
обладают высокой дефектностью, что вызывается
искажениями на границах области, а также диспер-
сией по мол. весам (см. Молекулярный вес высокомоле-
кулярных соединений) и наличием случайных боковых
ответвлений в макромолекулярных цепях. В резуль-
тате этого атомы внутри кристаллич. области оказы-
ваются смещенными из положений, соответствующих
идеальному кристаллу. В зависимости от причин
дефектность может проявляться по-разному; в раз-
бросе азимутальных поворотов макромолекулярных
цепей, в нек-рой неупорядоченности их продольных
смещений, в разбросе межцепных расстояний, в умень-
шении размеров кристаллич. областей.
Существуют структуры, в к-рых имеются только
нек-рые элементы кристаллич. упорядоченности, в то
время как другие частично или полностью отсутствуют.
Напр., у полиакрилонитрила и у нек-рых полиамидов
обнаружены структуры, где сохраняется дальний по-
рядок в расстояниях между осями цепей, однако по-
вороты макромолекул вокруг своих осей и их сдвиги
вдоль оси полностью беспорядочны. При вытяжке
аморфной пленки полиэтилентерефталата при темп-рах
ниже 80° возникает текстура, где имеется дальний
порядок в направлениях осей макромолекул, однако
в межцепных расстояниях дальний порядок отсут-
ствует. Именно поэтому проведение четкой границы
между кристаллич. и аморфным состоянием полиме-
ров вызывает иногда затруднения.
За редким исключением, ни один полимер не
является полностью кристаллическим. Помимо кри-
сталлических, имеются аморфные области, места пере-
ходов между кристаллитами и другие неупорядоченные
участки. Признаком существования аморфных обла-
стей служит появление на рентгенограмме наряду
с кристаллич. рефлексами аморфного гало (полиэти-
лен, полиамиды). Сравнивая интенсивность аморф-
ного гало и кристаллич. рефлексов, определяют т. н.
процент кристалличности.
Вопросы сопряжения различных областей в струк-
туре и характер расположения макромолекул в не-
упорядоченных областях во многом пока еще не выяс-
нены. Структурными элементами всякого полимера,
помимо макромолекул, являются продолговатые об-
разования— пачки, состоящие из упорядоченно
расположенных макромолекул. Поперечное сечение
пачки содержит 100—200 макромолекул. Внутри
пачки цепи макромолекул не переплетаются друг с дру-
гом, а расположены приблизительно параллельно
ДРУГ ДРугу.
Тенденция к образованию пачек цепей приводит
к высокой упорядоченности даже в аморфных поли-
мерах. При кристаллизации в ряде случаев (напр.,
полиэтилен) пачки на расстояниях порядка 100 А
накладываются сами на себя (за счет изгиба на 180°,
сопровождающегося возникновением неупорядочен-
ной области), смыкая кристаллич. области в единое
целое и образуя ленту, в к-рой направления осей
макромолекул перпендикулярны ее продольному на-
правлению. Т. обр., лента является следующей сту-
пенью надмолекулярной структуры в кристаллич.
полимерах. Существование всех этих, а также других
форм надмолекулярной структуры в полимерах под-
843
КРИСТАЛЛИЧЕСКОЕ СОСТОЯНИЕ ПОЛИМЕРОВ
844
тверждается электрониомикроскопическими исследо-
ваниями.
Путем осаждения из разб. р-ров получены поли-
мерные монокристаллы правильной формы. Эти моно-
кристаллы в случае полиэтилена состоят из плоских
слоев, образованных примкнувшими друг к другу
лентами. При этом ленты соединяются в слои т. обр.,
что направления осей макромолекул оказываются
перпендикулярными плоскости слоя. Следовательно,
для монокристаллов полимеров характерны сложные
структуры, возникающие и разрушающиеся в не-
сколько стадий (пачка, лепты, слои, монокристаллы).
Кроме ограненных монокристаллов, у ряда полимеров
{гуттаперча, нек-рые полиамиды и полиэфиры и др.)
на электронномикроскопич. снимках обнаружены
различные другие своеобразные структурные образо-
вания, к-рые, очевидно, также построены из сложен-
ных в ленты пачек макромолекул. Доказана тесная
связь этих ленточных форм со строением сферолитов,
наблюдающихся в кристаллич. полимерах очень
часто, но пока еще мало изученных.
Существует также и другой крайний тип полимер-
ных кристаллов — глобулярные криста л-
л ы. Макромолекулы (напр., у белков) способны свер-
тываться в глобулы, к-рые могут укладываться в трех-
мерном порядке. Возникают хорошо ограненные
кристаллы с большими периодами повторяемости,
порядка сотен ангстрем. Своеобразие подобных гло-
булярных кристаллов, образуемых многими белками
и вирусами, заключается в том, что конформации
макромолекул внутри глобул не в точности повторяют
ДРУГ друга- Детали атомного расположения в двух
соседних макромолекулах в нек-рых случаях могут
отличаться незначительно, с разбросом порядка 1 А,
но иногда и очень сильно — с разбросом порядка 10 А.
Существуют и такие кристаллич. образования, в к-рых
атомная повторяемость почти полностью отсутствует.
Такие «кристаллы» могут иметь ограненные формы,
не обнаруживая при этом типичной для кристаллов
дифракционной картины рентгеновских лучей.
Кристаллизация полимеров, подоб-
но кристаллизации низкомолекулярвых соединений,
имеет две стадии: стадию образования центров (или
зародышей) кристаллизации и стадию роста этих
центров.
Кинетика кристаллизации характеризуется ур-нием:
, - МЛ
а = 1 — е
где t — время; а — доля закристаллизовавшегося вещества;
k — константа роста центров кристаллизации; п — характе-
ризует как тип возникновения центров кристаллизации (тер-
мич. или атермич.), так и форму растущего из них кристаллич.
образования (сферическая, дискообразная, фибриллярная,
сферолитная);
« = (у( — va)!{vl — vj
где va — уд. объем аморфного материала;
и, — УД- объем в конце кристаллизации;
— УД- объем в момент времени t.
Наряду с непосредственным электронномикроскопич.
наблюдением одним из основных методов иссле-
дования кинетики кристаллизации является получе-
ние зависимостей уд. объема от времени при различ-
ных темп-рах и определение из них констант к и п,
что позволяет судить о механизме обеих стадий кри-
сталлизации.
Кристаллизация полимеров осложняется след, фак-
торами: большой размер и сложное строение макро-
молекул, большие времена механич. релаксации и
большая вязкость среды. Однако быстрое развитие
кристаллизации полимеров по сравнению со скоро-
стями релаксационных процессов перегруппировок
макромолекул указывает на то, что уже в аморфном
фазовом состоянии полимера (стеклообразное или
высокоэластич. состояние, расплав, раствор) имеются
местные упорядочения длинных цепных макромоле-
кул — пачки, в к-рых созданы условия для возник-
новения зародышей кристаллизации. Т. обр., в поли-
мерах с хорошо выраженным пачечным строением
стадия зародышеобразования не лимитирует процесс
кристаллизации. Вторая стадия — рост центров кри-
сталлизации — состоит в установлении дальнего по-
рядка уже в какой-то степени упорядоченных цепей
внутри пачки и в построении более сложных надмо-
лекулярных структур (лент, слоев, монокристаллов
и др.). Т. обр., из-за сложности строения полимера
вторая стадия кристаллизации является многосту-
пенчатым процессом и по различным причинам (напр.,
вследствие возникновения механич. напряжений) мо-
жет останавливаться на любой промежуточной сту-
пени. Так, монокристаллы образуются только в осо-
бых условиях (пока обнаружены монокристаллы
микроскопия, размеров), а наиболее распространенной
в полимерах является сферолитная структура, обра-
зованная из кристаллич. плоских слоев и лент.
Как и у низкомолекулярных веществ, обе стадии
кристаллизации полимеров сильно зависят от темп-ры.
Понижение темп-ры благоприятствует образованию
зародышей кристаллизации, но в то же время умень-
шает молекулярную подвижность, что снижает ско-
рость роста кристаллов. Поэтому кривая температур-
ной зависимости скорости кристаллизации проходит
через максимум. Поскольку подвижность макромоле-
кул исчезает вблизи темп-ры стеклования {Тс), то
температурным интервалом кристаллизации является
область между Тс и Тпп (темп-рой плавления поли-
мера). Ниже Тс кристаллизация принципиально идти
не может. В полимерах, имеющих Тс значительно
ниже комнатной темп-ры, максимум скорости кристал-
лизации находится при отрицательных темп-рах,
и в нормальных условиях такие полимеры нельзя полу-
чить в аморфном состоянии (полиэтилен, изотактич.
полипропилен, полиамиды), если только скорость
кристаллизации не слишком мала. Полимеры, имею-
щие Тс выше комнатной темп-ры, могут существовать
при нормальных условиях как в аморфном, так и
в кристаллич. состоянии (полиэтилентерефталат, изо-
тактич. полистирол). В зависимости от химич. при-
роды полимера и связанной' с ней упорядоченностью
макромолекул в аморфном состоянии скорость кри-
сталлизации может меняться в значительной степени.
Так, в натуральном каучуке в оптимальных условиях
кристаллизация полностью проходит за 8 час., в но-
лиэтилентерефталате — за несколько минут, а в по-
лиэтилене—за доли секунды.
Обе стадии кристаллизации зависят от гибкости
макромолекул. Вез определенной гибкости длинных
макромолекул кристаллизация вообще невозможна,
т. к. для обеспечения перегруппировок макромолекул
необходима подвижность участков цепей. Поэтому
полимеры с жесткими макромолекулами или вообще
не кристаллизуются или кристаллизуются очень
медленно, причем лимитирующей является стадия
роста центров кристаллизации. Но и в случае очень
гибких макромолекул вследствие большой их подвиж-
ности пачки могут быть плохо упорядоченными. От-
сутствие в аморфной фазе хорошо упорядоченных па-
чек требует значительного времени для упорядочения
гибких скрученных макромолекул. Поэтому кристал-
лизация протекает медленно как из-за малой скорости
возникновения центров, так и из-за малой скорости
их роста. Т. обр., быстрая кристаллизация характерна
для полимеров со средними величинами гибкости
макромолекул. Другим фактором, необходимым для
кристаллизации, является регулярность строения
макромолекул (См. Стереорегулярные полимеры, Изо-
тактические полимеры). Полимеры с нерегулярными
845
КРИСТАЛЛИЧЕСКОЕ СОСТОЯНИЕ ПОЛИМЕРОВ — КРИСТАЛЛЫ
846
макромолекулами нс кристаллизуются вовсе; редкие
нарушения порядка расположения или типа звеньев
в цепи макромолекулы замедляют кристаллизацию.
Химич, строение макромолекул также влияет на ско-
рость кристаллизации. Напр., низкая симметрия и
большие размеры звеньев замедляют кристаллизацию.
При достаточно медленной кристаллизации может
быть достигнуто равновесное кристаллич. состояние,
нс зависящее от пути проведения кристаллизации.
При обычных условиях легко возникают неравновес-
ные состояния, являющиеся результатом много-
стадийности кристаллизации в полимерах. Следствием
неравновесного состояния являются расширение ин-
тервала плавления и его смещение к более низким
темп-рам. Поэтому более медленно закристаллизо-
ванные полимеры имеют более высокую темп-ру пла-
вления и более узкий интервал плавления (вследствие
уменьшения возможности возникновения внутренних
напряжений и облегчения их релаксации). Большое
влияние на неравновесные состояния оказывают также
внешние механич. воздействия.
Как следует из термодинамики, одноосное растяжение
аморфного полимера благоприятствует кристаллизации. Усло-
вие равновесия фаз в точке плавления состоит в равенстве их
свободных энергий, т. е. в выполнении равенства ДН—ТДЗ=0,
где ДИ — разность теплосодержаний, a AS — разность энтро-
пий сосуществующих фаз. При кристаллизации ДН < 0 и
AS < 0. При одноосном растяжении вследствие раскручива-
ния свернутых макромолекул AS уменьшается. Поэтому пере-
ход из аморфного в кристаллич. состояние смещается к более
высоким темп-рам, если аморфный полимер предварительно
растянут, т. е. при этом повышается темп-ра плавления. В тех
случаях, когда выполнение равенства ДИ—ТПл AS =0 для
значений 7'пл> >ТС возможно только при уменьшенных вслед-
ствие растяжения значениях AS, кристаллизация происходит
только при одноосном растяжении (полиизобутилен). При
растяжении возрастает также и скорость кристаллизации:
у полиэтилентерефталата она увеличивается с 10~3 г/см3/сек
для нерастянутого полимера до 10 г/см3/сек для полимера, рас-
тянутого в 2—3 раза. С увеличением растягивающего напря-
жения эффект влияния растяжения возрастает. Всестороннее
давление увеличивает скорость и глубину кристаллизации
до определенного предела давлений, выше к-рого скорость и
глубина кристаллизации заметно падают вследствие умень-
шения подвижности макромолекул.
Результатом большого разнообразия неравновесных
состояний является существенное влияние предысто-
рии кристаллич. полимера на все его физич. свойства
(это влияние заметно даже после повторной кристал-
лизации вследствие сохранения кристаллич. зароды-
шей намного выше темп-ры плавления). Термич. и
механич. предыстория, темп-ра кристаллизации, а
в случае кристаллизации из р-ров — начальная кон-
центрация р-ра и тип растворителя — определяют
предкристаллизационное состояние полимера, а сле-
довательно, скорость и тип зародышеобразования.
Все эти факторы являются причиной громадного раз-
нообразия структур и размеров образующихся при
кристаллизации полимеров надмолекулярных струк-
тур. Об особенностях механич. свойств полимеров
в кристаллич. состоянии см. Деформация полимеров
и Механические свойства полимеров.
Характерным для процесса одноосного растяжения
кристаллич. полимера является внезапное возникно-
вение «шейки», в ходе развития к-рой происходит
рекристаллизация полимера с образованием ориенти-
рованно расположенных кристаллов. Возникновение
шейки связано со скачкообразным уменьшением тол-
щины и ширины образца, к-рые сохраняются неиз-
менными в дальнейшем процессе развития шейки.
Развитие шейки продолжается до завершения рекри-
сталлизации во всем объеме исходного образца. Соот-
ношение скоростей процессов рекристаллизации и
растяжения определяет глубину кристаллизации по-
лимера, возникающей в процессе растяжения. При
растяжении кристаллич. полимера ниже его Т , когда
кристаллизация практически невозможна, происхо-
дит амортизация полимера.
Лит.: Манделькерн Л., Усп. химии, 1958, 27,
вып. 2, 193; Каргин В. А., Слонимский Г. Л.,
там же, 1955, 24, вып. 7, 785; Каргин В. А., Китай-
городский А. И., С л о н и м с к и й Г. Л., Коллоидн.
ж., 1957, 19, 131; Каргин В. А., Слонимский Г. Л.,
Краткие очерки по фиэико-хпмии полимеров, М., 1960; Кар-
гин В. А., Вести. АН СССР, 1961, вып. 4; А V г a m I М.,
J. Chem. Phys., 1939, 7, 1103; 1940, 8, 212; 1941, 9, 177;
Вуд Л. А., в сб.: Химия больших молекул, [пер. с англ.],
т. 2, М., 1948; Stuart Н. A., Die Physik der Hocnpolymeren,
Bd 3, В., 1955; Keller A., Philos. Mag., 1957, 2, № 21,
1171; Makromolek. Chem., 1960, 41, 86—173.
КРИСТАЛЛОГИДРАТЫ — кристаллы, включаю-
щие молекулы воды. В зависимости от термодинамич.
условий одна и та же соль может кристаллизоваться
с разным числом молекул воды. По прочности связи,
координации и положению в кристаллич. решетке
различают разные типы К. 1) Атомы кислорода моле-
кул воды координированы вокруг центрального ка-
тиона. Вода способствует увеличению координацион-
ионов, что
энергии и
Я1 о2 /
iCo
Со'
Со
Я С1 С1 £
х12
Структура СоС12 • 2Н2О.
Н—о. .0
Си--ОС >S<
Структура CuSO, • Н2О.
этих случаях кислород
.о, . .н
/ н \ Хо-С
\ /' н
ного числа, образованию комплексных
сопровождается снижением свободной
стабилизацией. Так, \ н »т
в СоС12 . 2Н2О име- \ о2 Я] о2
ются бесконечные
ленты, в которых
каждый атом Со ок-
ружен 4 атомами
CI, а каждый атом
С1 связан с 2 ато-
мами Со. Вода занимает 2 дополнительных коорди-
национных места вокруг Со, доводя координацию
до октаэдра, характерного для гибридных связей
d2spa. 2) Вода в К. является мостиком, соединяющим
катион с анионом (напр., CuSO4-H2O), или связую-
щим звеном между водой,
окружающей катион, и анио-
ном. В таких случаях часто К.
содержит нечетное число моле-
кул воды, причем последняя
нечетная молекула своим кислородом образует водо-
родные связи с водой первой координационной сферы, а
атомами водорода связывается с анионом. 3) В ряде К.
молекула воды своим кислородом связывает 2 катиона
[Li(OH)-H2O, ВаС12 Н2О]. В
воды в К. имеет тетра-
эдрич. координацию.
4) Если ионы упако-
ваны слоями, то кри-
сталлизационная во-
да способствует свя-
зыванию слоев меж-
ду собой (CaSO4 •
2Н2О). В нек-рых таких К. кислород воды имеет
координационное число, равное трем; кислород свя-
зывает катион, а водороды образуют две водородные
связи. 5) В К. со слабо связанной водой после удале-
ния воды зачастую молекулярный объем меняется
незначительно. Это значит, что вода размещена в пу-
стотах кристалла, причем после удаления воды
остаются дырки [(NH3)4 PtCl2-H3O]. Существуют К.,
наз. тектогидратами, в к-рых воды много, а ионов
мало (Na2SO4 • 10Н2О). Для ряда таких К. харак-
терна непрерывная структура льда, стабилизованного
присутствием стягивающих ионов [Al2(SO4)g-27HsO].
Лит.; Бернал Дж., Роль воды в кристаллических
веществах, Успехи химии, 1956, 25, вып. 5; У эл лс А. Ф.,
Строение неорганических веществ, пер. с англ., М., 1948,
гл. XI, Я. К. Сыркин.
н\
н/
>Li
Н
II
Xof \ н /
н/ \
Структура Ll(OH) Н2О.
КРИСТАЛЛЫ. Содержание:
Введение ........................... 846
Кристаллография....................... 847
Кристаллохимия ....................... 853
Физические свойства кристаллов ....... 859
Введение. Кристаллы — твердые тела, характери-
зующиеся закономерным периодич. расположением
частиц (молекул, атомов или ионов) в пространстве.
847
КРИСТАЛЛЫ
848
В этом смысле кристаллическое состояние — нормаль-
ное равновесное состояние твердого тела — можно
считать прямой противоположностью газообразного,
целиком неупорядоченного. Строгая периодичность
расположения частиц в К. определяет почти все основ-
ные законы и свойства К. Основной особенностью
индивидуального К., отличающей его от агрегатов и
аморфных тел, является анизотропия физич. и физико-
химич. свойств. В классич. определение К. наряду
с анизотропностью обычно входит и однородность —
одинаковость свойств К. во всех его точках. Одно-
родным в применении к К., как веществу дискрет-
ному, следует считать такое тело, в к-ром для любой
точки всегда найдется совершенно аналогичная ей
по свойствам и по окружению другая точка, отстоя-
щая от первой на конечное, экспериментально опре-
деляемое расстояние; для неорганич. веществ оно
обычно ~ 10А и не превышает 50 А.
Наука о К. — кристаллография — изу-
чает законы образования К., их морфологию и тонкую
(атомную) структуру, физич. свойства как моно-
кристаллов, так и кристаллич. агрегатов, специфику
разнообразных явлений в К., а также взаимодей-
ствие К. с внешней средой. Из кристаллографии при-
нято выделять кристаллохимию и кри-
сталлофизику. Первая, определяя различ-
ными, преим. дифракционными, методами простран-
ственное расположение атомов (ионов, молекул)
в К., стремится выяснить природу химич. связи
в К. и установить зависимость структуры К. от со-
става и условий образования, а также связь между
тонкой структурой К. и его физико-химич. свойствами.
Кристаллофизика изучает физич. свойства К., в осо-
бенности анизотропные, т. е. такие, к-рые резко из-
меняются в разных направлениях. Совместно с кри-
сталлохимией кристаллофизика выясняет зависимость
этих свойств от тонкой (атомной) структуры.
Кристаллография; Наиболее известным внешним
признаком К. является геометрия, форма, к-рую
К. принимает при образовании в соответствующих
условиях (см. Кристаллизация). В идеальных слу-
чаях К. ограничен плоскими гранями, сходящимися
в точечных вершинах и прямолинейных ребрах.
Способность самоограняться является отражением
решетчатого строения К., грани К. отвечают нек-рым
плоским сеткам кристаллич. структуры, ребра —
рядам. Грани в процессе роста К. передвигаются
параллельно самим себе, однако из-за неравномер-
ного притока отлагающегося на гранях вещества
скорости роста даже одинаковых граней обычно раз-
личны. Кроме того, грани растут тем быстрее, чем
выше их поверхностное натяжение па границе с окру-
жающей средой. Поэтому гра-
ни растущего К. могут изме-
нять свои размеры, очертания,
а быстро растущие грани —
вообще исчезнуть (рис. 1); не-
изменными остаются лишь
углы между гранями. Закон
о постоянстве углов
К., конкретно сформулирован-
ный Роме де Л’Илем (1783),
а на отдельных примерах —
Стенопом (1669), Ломоносовым
(1749) и др., строго справед-
лив лишь при одинаковой тем-
пературе. Этот закон положен
Е. С. Федоровым (1910) в основу кристаллохи-
Рис. 1. Рост граней кри-
сталла. Грани с большей
скоростью роста (а)
уменьшаются в процессе
роста п могут исчезнуть
с поверхности кри-
сталла.
мического анализа — определения вещест-
ва по комплексу гранных углов К.
Решетчатое строение К. находит свое отражение
в законе рациональности парамет-
ров (Гаюи, 1784), Если OAlt ОВ1, ОС1 — отрезки,
Рис. 2. Рациональность
двойных отношений от-
резков, отсекаемых дву-
мя гранями на трех реб-
рах.
отсекаемые какой-либо гранью К. на трех непарал-
лельных ребрах К. (параметры грани), а ОА2,ОВ2,
ОС2 — соответствующие па-
раметры другой грани того
же К., то
ОА; , ОВ] . OCj__
ОАз '• бв7 ' ОС2 ~Р:1:Г
где р, q, г — небольшие це-
лые числа (рис. 2). Это по-
ложение, выведенное на ос-
новании измерения большого
числа К., было первым в
истории химии законом це-
лых чисел, одним из ранних
доказательств дискретного
строения материи. Закон
рациональности параметров
позволяет при помощи т. наз.
символов однозначно фиксировать положение любой
грани и ребра К. относительно нек-рой исходной
грани и трех кристаллографии, направлений, приня-
тых за координатные оси.
Отрезки, отсекаемые нек-рой гранью на координатных
осяхл измеряют в масштабных единицах, представляющих
собой отрезки, к-рые отсекает на соответствующих осях
исходная грань. Полученное отношение параметров грани
(Р ; 9 : г), для характеристики ее положения, удобнее заме-
нить обратными числами: h : k : I = 1/р: 1/q: l/г. Эти три
индекса (h k I) и являются символами грани. Если грань
параллельна какой-либо координатной оси, то соответствую-
щий индекс равен 0. Так, грань куба, к-рая параллельна
двум осям, имеет символ (100). Символ ребра
[uvw] представляет совокупность трех наименьших целых
чисел, пропорциональных координатам любой точки этого
ребра. Координаты ее измеряются параметрами единичной
грани. Зависимость, существующая между символом грани
и ребра, в ней лежащего (hu Д- fiv -f- lw ~ 0), позволяет
найти символ ребра пересечения двух граней и символ
грани, параллельной двум ребрам К. (закон поясов
или зо н). Существуют графич. (напр., метод поя-
сов или метод косинусов Вульфа) и ана-
литич. способы определения символов граней, исходящие
из сферич. координат, к-рые получают при измерении К.
на двухкружном гониометре. Для упрощения тригонометрия,
расчетов используют различные типы проекций, в особен-
ности стереографическую.
Важной особенностью большинства К. является
их симметрия, к-рая описывается при помощи
элементов симметрии (оси симметрии, плоскости
симметрии, центра инверсии), каждый из к-рых
производит одну или несколько операций, совме-
щающих равные части фигуры друг с другом так, что
и вся фигура совмещается сама с собой.
Для полного описания симметрии любого кристал-
лич. многогранника достаточно пользоваться пово-
ротными (2V или L ) и инверсионными
осями (А или Lin). Порядком оси п наз. число
совмещений К. с самим собой при повороте на 360°
(L2 — ось 2-го порядка, L3 — 3-го, и т. д.). Инвер-
сионная ось предполагает одновременно с поворотом
на нек-рый угол, определяемый порядком оси, также
и отражение (инверсию) в точке, лежащей на этой
оси. Иногда вместо инверсионных осей используют
более наглядные зеркально-поворотные оси (А, Лп),
к-рые, поворачивая фигуру, одновременно отражают
ее в плоскости, нормальной к оси поворота. Решет-
чатое строение К. не допускает существования в К.
осей 5-го порядка и порядка выше 6-го (закон
симметрии).
В классич. кристаллографии вместо нек-рых ин-
версионных осей предпочитают более наглядные их
эквиваленты: зеркальную плоскость (т или Р = 2)
и центр инверсии (С = 1) и описывают симметрию,
используя лишь оси 2-го, 3-го, 4-го и 6-го порядка,
зеркальную плоскость и центр инверсии, а также
инверсионную, или что то же самое, зеркально-пово-
849
КРИСТАЛЛЫ
850
ротную ось 4-го порядка. Зеркальная п л о с-
к ость делит фигуру на две половины так, что одна
половина является зеркальным отражением другой.
Центр инверсии — точка внутри К., по обе’ сто-
роны от к-рой на одинаковом расстоянии лежат
кристаллографически тождественные точки. При на-
личии центра инверсии каждой грани К. соответ-
ствует параллельная и обратно расположенная грань.
Таким образом, различают симметрические преобра-
зования 1-го рода (простые повороты),к-рые правую
руку (грань) превращают в правую же, и 2-го рода
(отражение в точке или плоскости, или поворот
с отражением), превращающие правую руку в левую
и наоборот. К. может иметь несколько одинаковых
элементов симметрии, но центр инверсии всегда
только один.
Возможны только 32 различных сочетания элемен-
тов симметрии (32 точечные группы симметрии).
В таблице приведены их обозначения.
близостью некоторых физических свойств, например
оптических.
Если все грани К. кристаллографически тождественны
(переходят одна в другую с помощью элементов симметрии),
то такая фигура относится к простой форме, в общем случае
К. представляет собой комбинацию простых форм. 32 видам
симметрии соответствует 47 простых форм.
Поскольку симметрия реального К. строго проявляется
лишь в угловых величинах, для определения симметрии
К. следует измерить на гониометре. Симметрию легче выя-
вить, если, воспользовавшись сеткой Вульфа, нанести
результаты измерения на стереография, проекцию, так как
идеальный К. и тот же К., искаженный в процессе роста,
дадут одинаковую проекцию. Так как одни и те же формы
могут принадлежать разным видам симметрии, то оконча-
тельное заключение об истинной симметрии часто можно
сделать лишь на основании изучения оптич. и др. физич.
свойств К., а также морфология, особенностей его граней,
напр. фигур травления.
Вещества, кристаллизующиеся в классах, в кото-
рых отсутствуют симметрии, преобразования второго
рода, могут давать энантиоморфные К.; левые и
правые формы этих К. относятся друг к другу, как
Обозначения и названия 32 видов симметрии
Катего- рии Сингония Символ вида симметрии Название (по общей простой форме)
международный по Шён^лису развернутый (формула симметрии)
Низшая Триклинная 1 г Ci C\, So С Моноэдрический Пинакоидальный
Моноклинная m 2/m Cs C-i C-A p l.2 L-jPC Диэдрический безосный Диэдрический осевой Призматический
Ромбическая mm, mm2 222 2/m 2/m. 2/m, mmm 1 ° L%2P 3L3 3L2 3PC Ромбо-пирамидальный Ромбо-тетраэдрический Ромбо-дипирамидальный
Тетраго- нальная 4 4/m 4mm 42, 422 4/m 2/m 2/m, 4.'mmm I 42m Ct cth Cw Dih St V,, a1 '2a Ci LiPC LftP L^4L2 Li4L2bPC л$ Ь{^Ь22Р, Л42Р22Р Тетрагонально-пирамидальный Тетрагонально-дипирамидальный Дитетрагонально-пирамидальный Тетрагоналыю-трапецоэдричесний Дитетрагонально-дипирамидальный Тетрагонально-тетраэдрический Тетрагонально-сналеноэдричесвиЙ
Средняя Гексаго- нальная 3 3 3m 32 3 — , 3m m 6 6/m 6mm 62, 622 6/m 2/m 2/m, б/ттщ 3/m 6 3/ni2m, 62m » <□ ' s » "S О Л -C О Q u Cs. Liss, L3C, JIS L33P LS3L., Lis3L.23P, LS3L.,3PC, Л0ЗЬ3ЗР Lo LePC L,fiP T(j6T2 L66L27PC Ьц» Ь3Р, Л3 Li63L23P, L^LAP Л-лЗЬ,ЗР Тригонально-пирамидальный Ромбоэдрический Дитригонально-пирамидальный Тригонально-трапецоэдрнческпй Дптригонально-скаленоэдрический Гексагонально-пирамидальный Гексагон ально-дипирамидальный Дигексагонально-пирамидальный Гексагонально-трапецоэдрический Дигексагонально-дипирамидальный Тригонально-дипирамидальныи Дитригональ но-ди пирамидальный
Высшая Кубическая 2/m3, m3 43m 43, 432 4/m32/m, m3m T Th Td 0 Oh 3L>4L3 3L24L33PC SLi^Lgep 3Li4L36L2 3Lj 4L36L29PC Центагонтритетраэдрический Дидодекаэдрический Гексатетраэдрический Пентагонтриоктаэдрический Гексаоктаэдрический
По общности особых направлений 32 вида симмет-
рии принято делить на 6 сингоний (систем), каждой
из к-рых отвечает своя координатная система. Син-
гонии объединяют в три категории: низшая — три-
клинная, моноклинная и ромбическая, средняя —
тетрагональная и гексагональная, и высшая — куби-
ческая. К, ОДНОЙ и ТОЙ же категории характеризуются
предмет и его зеркальное изображение. Энантиомор-
физм характерен для кварца, киновари, винной ки-
слоты и др.
Для реальных К. характерно образование сростков
по различным законам (двойники). Двойники нек-рых
типов К. показаны на рис. 3. Они могут быть опоз-
наны н<? входящим углам, к-рые отсутствуют у моно-
851
КРИСТАЛЛЫ
852
кристаллов, но иногда двойник может быть принят
и за монокристалл более высокой или более низкой
симметрии. Нередки закономерные срастания К.
Рис. 3. Различные случаи двойникования: а, Ь,
с — двойники кварца; d —двойник кальцита; е —
двойник пирита; f, g — двойники алмаза.
различных веществ (эпитаксия). Примерами
могут служить слюда и иодистый калий или халько-
пирит и тетраэдрит (рис. 4). Весьма интересным
является нарастание игольчатых кристалликов ру-
Рис. 4. Закономерное сра-
стание тетраэдрита и халь-
тила на п. гематита, ко-
рунда или слюды с обра-
зованием так называемых
сагенитовых треугольни-
ков. Причиной эпитаксии
является геометрия, ана-
логия в строении некото-
рых плоских сеток кри-
сталлич. структур этих
соединений.
Обязательным элемен-
том симметрии, отличаю-
щим кристаллич. струк-
туру, как систему беско-
нечную, от конечного кри-
копирита. сталлич. многогранника,
являются оси трансляций
(переносов). Каждому рациональному направлению
(оси трансляции) в кристаллич. структуре
отвечает определенный отрезок — трансляция, при
переносе на который вся структура совмещается
сама с собой (рис. 5). Совокупность всех трансляций
образует трансляционную группу (группу переносов)
пли кристаллическую (пространственную) решетку.
Рис. 5. Различные по величине и направлению транс-
ляции в одной и той же кристаллич. структуре. При
построении кристаллич. решетки за узел можно при-
нять любую точку структуры.
(Термин «пространственная» и «кристаллическая»
решетки целесообразно употреблять как синонимы).
Под кристаллической структурой
следует понимать конкретное расположение мате-
риальных частиц в пространстве. Только в случае
нек-рых простых веществ, в основном металлов, сама
пространственная решетка дает представление о
кристаллич. структуре.
Существует 14 топологически различных трансля-
ционных групп — 14 решеток Браве. Для характе-
ристики любой решетки Браве на трех, но лежащих
в одной плоскости и кратчайших для данного направ-
ления трансляциях строят т. наз. параллелепипед
повторяемости. Поскольку любой решетке отвечает
бесчисленное множество таких параллелепипедов,
при выборе его пользуются определенными ограничи-
тельными правилами (сингония выбранного паралле-
лепипеда отвечает сингонии К., число прямых углов —
максимальное, объем — минимальный), позволяю-
щими иметь для каждой решетки единственный парал-
лелепипед повторяемости, к-рый обычно наз. эле-
ментарной ячейкой Браве (теми же пра-
вилами пользуются, выбирая элементарную ячейку
кристаллич. структуры).
14 решеток Браве отличаются друг от друга не
только по сингонии, но и по комплексу трансляций,
т. е. по типу центрировки: помимо примитивных
(т. е. пустых) ячеек (Р), существуют центрированные,,
по паре противоположных граней (базоцентрирован-
ные — С, оокоцентрированные —-А, В), по всем гра-
ням (гранецентрированные— Г) или по объему (объем-
ноцентрированные — I). Каждая решетка характе-
ризуется осевыми параметрами, т. е. отрезками а, Ъ, с
(выражаемыми в А), и тремя координатными углами
А „ А А
а, р, у (а = Ьс, р = са, у — ас). На рис. 6 представ-
лены 14 решеток Браве и даны их геометрич. харак-
теристики.
Для кристаллич. структур наряду с элементами
симметрии кристаллич. многогранников характерны
и специфические для бесконечных узоров элементы
симметрии: плоскости скользящего отражения и вин-
.товые оси. Эти элементы симметрии наряду с отраже-
нием и поворотом одновременно включают и сколь-
жение (сдвиг) на нек-рую долю трансляции, парал-
лельной плоскости отражения или соответствующей
винтовой оси.
Совокупность всех элементов симметрии данной
кристаллич. структуры наз. ее пространственной
группой. В 1890 Е. С. Федоров впервые доказал,
что 32 видам симметрии (точечным группам) соответ-
ствует 230 пространственных групп симметрии, к-рые
часто называют федоровскими. Результат раз-
множения одной точки всеми элементами симметрии
пространственной группы наз. правильной
системой точек.
32 вида симметрии предопределяют образование
47 простых форм, и каждый многогранник представ-
ляет собой либо одну простую форму, либо комбина-
цию нескольких, число таких многогранников бес-
конечно. Аналогично 230 пространственных групп
предопределяют образование конечного числа пра-
вильных систем точек, любая же кристаллич. струк-
тура представляет собой либо одну правильную
систему точек, либо совокупность нескольких. Струк-
турные исследования показали, что в химич. соеди-
нении атомы одного и того же элемента пе всегда
равноценны кристаллохимически, т. е. не обязательно
располагаются по положениям единственной пра-
вильной системы точек. Эго касается пе только соеди-
нений, но и многих простых веществ (В, S8, а - и
[5-Мп, графит). С другой стороны, в случае образова-
ния смешанных кристаллов (см. Изоморфизм') и не-
упорядоченных интерметаллич. фаз разные атомы
могут занимать одну правильную систему точек, по
к-рой они распределяются статистически. Правиль-
ную систему точек характеризуют координатами,
выраженными в долях ребер ячейки. Координаты
всех атомов при известных константах ячейки
853
КРИСТАЛЛЫ
854
(а, Ь, с и а, р, у) дают возможность вычислить любые
межатомные расстояния и угловые величины — ко-
нечную цель формального исследования кристаллич.
структуры вещества. По полученным данным можно
Рис. 8. Решетки Браве.
Низшая категория а Ф b Ф <:: 1— триклинная
примитивная (а ф 3 фу ф 90°); 2 — моноклинная прими-
тивная (а=у=90°, ₽ ф 90°); 3 — моноклинная базоцент-
рированная (а=у = 90°; р Ф 90°); 4 — ромбическая
примитивная (а=р=у = 90°); 5 —ромбическая базоцент-
рированная (a=p=v = 90°); 6 — ромбическая объемно-
цевтрированная (a=p=V = 90°); 7 — ромбическая гране-
центрированная (a=P=Y = 90°).
Средняя категория а — b Ф а в — гекса-
гональная примитивная (а=р = 90°, у = 120°); 9 — гек-
сагональная дважды центрированная (a=3 = 90°, у=120°);
10 — тетрагональная примитивная (а = р=у=90°); 11 —
тетрагональная' объемноцентрированная (а = р=т=90°).
Высшая категория а = Ь = с: 12 — куби-
ческая примитивная (а=р=у = 90°); 13 — кубическая
объемноцентрированная (а=р=у = 90°); 14—кубическая
гранецентрированная (ц=р=у = 90°).
делать заключения о силах и характере связи частиц,
о переменной валентности и др. Результаты рентгено-
структурных исследований подтвердили подчинение
всех кристаллич. структур 230 пространственным
группам. Хотя среди почти 10 000 известных структур
еще не найдены представители для всех 230 простран-
ственных групп, но нот и ни одного случая им противо-
речащего.
Кристаллохимия. В подавляющем большинстве
(> 95%) неорганич, соединений отсутствуют обособ-
ленные группы атомов, к-рые можно было бы назвать
молекулами. На рис. 7 изображены нек-рые основные
структурные типы неорганич. соединений и простых
веществ. Вместе с тем, структурный анализ подтвер-
дил существование радикалов в большинстве тех слу-
чаев, когда последние были предсказаны на основе
химич. исследований. С помощью рентгеноетруктур-
ного анализа определяются межатомные расстояния
и углы между связями в радикалах. На рис. 8 пока-
зано строение нек-рых химич. радикалов, а также
приведены структуры соединений, содержащих эти
радикалы.
Употребляемые для изображения соединений не-
молекулярного типа структурные формулы, в к-рых
связи между отдельными атомами изображают ва-
лентными черточками, например,
,ОЧ
Са< >Т1=О,
Oz
яв-
ляются лишь условной записью валентных отношений,
из к-рых не следует, что около Са располагается
2 атома О, вокруг Ti — 3 атома О и что атомы О двух
Рис. 7. Основные структурные типы неорганических
соединений и простых веществ: 1 — NaCl; 2 — CsCl;
3 — ZnS (тип сфалерита); 4 — ZnS (тип вюртцита); 5 —
NiAs; 6 — флюорита CaF2; 7 — пирита FeS2; 8 — рутила
Т1О2; 9 —СО2; 10 — меди; 11 — а-железа; 12— магния;
13— алмаза; 14—графита.
сортов (см. рис. 9). Кристаллохимии, структурные
ф-лы должны отражать такую важную геометрич.
характеристику структуры, как координационные
числа ее компонентов, определяемые числом ближай-
ших к данному атому соседей (в высокосимметрич-
855
КРИСТАЛЛЫ
•855
ных структурах все атомы данной координационной
сферы находятся на одинаковом расстоянии от коор-
динируемого, в менее симметричных структурах
к одной координационной сфере принято относить
и атомы, находящиеся на разных, но близких расстоя-
Рис. 8. Строение химических радикалов и примеры со-
единений, содержащих такие радикалы,
Г.а — структура СаСО3, б—строение радикала СО3;
П:а — структура CaWO4, б — строение радикала WO4;
1П:а — структура K2PtCl4, б — строение радикала PtCI4;
I V:a — структура K2PtCI„, б — строение радикала PtC le.
ОСа *Tl Qo
Рис. 9. Структурный
тип перовскита
CaTiO,.
пиях). С этой точки зрения хлористому натрию, напр.,
можно приписать «развернутую» ф-лу [NaCle/J3 =°,
указывающую на шестерную координацию обоих ком-
понентов; знак «Зоэ» говорит о бесконечном в трех из-
мерениях мотиве. Для хлористого
цезия — формула [CsCb/J300, для
фтористого кальция — [CaFs/4]3=o.
Дробь 8/4 указывает, что вокруг
атома Са расположено 8 атомов F,
а атом F окружен четырьмя ато-
мами Са. Приведенные типы струк-
турных формул отражают геомет-
рию структуры, но гораздо боль-
шее значение имеют те структур-
ные формулы, которые, в отличие
от валовых, позволяют расшифро-
вать сущность химич. соединения, напр. валентность
его компонентов. Структура селенида таллия указы-
вает, что таллию нельзя приписать не свойственную ему
валентность 2, как это может показаться из валовой
ф-лы TISe. Так как половина атомов Т1 находится
в крупных восьмивершинниках, а половина — в те-
траэдрах, то очевидно, что первому типу окружения
отвечает ТР (радиус Т1+ 1,49 А), а второму — Т13!’
(радиус Т13+ 1,05 А). Т. обр., с учетом структуры
ф-ла пишется: Tl+Tl3,Se2. Аналогичные по валовой
ф-ле пятибромистый и пятихлористый фосфор описы-
вают совершенно различными структурными ф-лами,
поскольку в структуре первого соединения чередуются
тетраэдрич. группы [PBrJ+ и одиночные попы Вг ,
а второго — комплексные тетраэдрические катионы
[РС14]+ и октаэдрические анионы [РС16] ~. Т. обр., для
РВг5 имеем [РВг4|+ Вг, а для РС15— [РС14]+ [РС1е]д
Сопоставление структуры кальцита СаСО3 и перов-
скита CaTiO3 (см. рис. 9 и рис. 8, 1, а) показывает,
что кальцит действительно является карбонатом
кальция с дискретными группами СО3 и, наоборот,
отсутствие радикалов TiO3 в структуре перовскита
заставляет считать его двойным окислом кальция и
титана. Структурные ф-лы, отражающие это, можно
записать: Са [С()3] и Са [ТЮв,„]3х?.
Структурными можно считать и формулы, приме-
няемые в случае т. п. дефектных соединений, харак-
теризующихся теми или иными отклонениями от
законов классич. химии и кристаллохимии, в част-
ности от закона рациональных отношений. Для
дефектных соединений ведущим признаком следует
считать не состав, а структуру. Дефекты могут быть
обусловлены, напр., наличием вакантных положений
для одного типа компонентов при переменной валент-
ности другого. Так, Na-вольфрамовые бронзы харак-
теризуются переменным содержанием Na при соот-
ветствующем изменении валентности W. Так как
любая Na-вольфрамовая бронза имеет структуру
перовскита (рис. 9), где положению Са соответствует
Na, а положению Ti — W, то нет смысла подбирать
ф-лы с разнообразными коэфф, и можно ограничиться
единой: Na,_T (Wp^, W®+) О3, каждому составу
отвечает свое значение х; запятая в ф-ле означает,
что оба типа W статистически замещают друг друга.
Крайними случаями этого ряда твердых р-ров (см.
Изоморфизм) являются химич. соединения WO3 (х = 1)
и NaW03 (х = 0). При описании природных соеди-
нений, к-рые в связи с изоморфными замещениями
обычно характеризуются переменным составом, по-
добный способ записи весьма плодотворен, он исклю-
чает, напр., открытие «новых» минералов. Так,
Fe6S7, Fe5Se, Fe8S9 и другие, встречающиеся в различ-
ных месторождениях, надо считать вариантами одного
и того же пирротина FeS, к-рып существует только
в дефектном состоянии за счет частичного окисления
Fe2' в Fe3 ' (структурная формула Fe^tx ре
Пе меньшее значение этот подход имеет и для интер-
металлпч. соединений, т. к. широкий интервал со-
ставов является одной из их основных особенностей,
и нек-рые из них вообще могут существовать лишь
в дефектном состоянии.
Кристаллич. структуры принято делить по приз-
наку «неравноценности» межатомных расстояний на
пять классов: координационные, островные, цепочные,
слоистые и каркасные. В к о о р д и н а ц и о н н ы х
структурах расстояния между всеми струк-
турными единицами одного порядка. Примерами
могут служить металлич. медь, алмаз, NaCl, ZnS
и др. Чисто координационные структуры гомодес-
мичны, т. е. характеризуются каким-то одним типом
связи. Такой (см. Химическая связь, Межмолекулярные
взаимодействия) связью может быть ионная (NaCl),
ковалентная (алмаз), металлическая (медь) и даже
ван-дер-ваальсовская (кристаллы инертных газов).
Координационные структуры чаще всего характери-
зуются симметричной и высокой координацией.
В островных, цепочечных и слои-
стых структурах можно выделить группы атомов,
к-рыс образуют соответственно изолированные остро-
ва, непрерывно простирающиеся в одном измерении
цепи или бесконечные в двух измерениях слои. Такие
обособленные группы атомов могут быть либо ней-
тральными, либо заряженными. К островным можно
отнести ромбическую серу (S8), иод (J2), двуокись
углерода (СО2), а также подавляющую массу органи-
ческих соединений, поскольку они обладают молеку-
лярной структурой. Примерами структур с несущи-
ми электрический заряд островами являются: бе-
рилл Be3Al2[Si6O18], кальцит Са[СО3], пирит FeS2.
Цепочечными можно считать Se (нейтральная цепочка),
Na [ПСО3], диопсид CaMg[Si2O6] (анионные цепочки).
Нейтральные слои характеризуют графит, мышьяк,
тальк [Mgj(Si4O10)(OH)2], анионные — мусковит
K[Al2(Si3AlOI0)(OH)2],
Каркасные структуры можно предста-
вить состоящими из трехмерного анионного остова,
построенного по координационному принципу, и
нейтрализующей «начинки» из отдельных катионов
или атомных групп. Расстояние между структурными
единицами в предедах остова меньше, чем между
857
КРИСТАЛЛЫ
858
структурными единицами остова и «начинки». В ка-
честве примера можно привести структуру перов-
скита Са [ТЮ3] или ортоклаза К [AlSi3Osl. Струк-
туры островные, цепочечные, слоистые и каркасные,
в отличие от координационных, гетеродесми-
ческие. Так, островные структуры элементарных
иода или серы характеризуются ковалентными свя-
зями между атомами в молекулах J2 и S8 и остаточ-
ными (ван-дер-ваальсовскими) связями между от-
дельными молекулами. В цепочечной структуре
Na [НСОа] существует ионное взаимодействие между
Na+ и цепочками [НСО3]~, в которых треугольные
группы СО3 сцементированы воедино водородными
связями. Структуры цепочечные и слоистые обычно
характеризуются довольно резко выраженной анизо-
тропией всех свойств, включая внешнюю форму (вытя-
нутость, слоистость). Для остальных типов структур,
в особенности координационных, анизотропия редко
бывает значительной.
Изложенная классификация в значительной сте-
пени условна; так, берилл, относимый обычно к остров-
ным структурам, можно считать и координационным,
поскольку расстояния в пределах 6-членного кольца
[Si6OI8] не очень резко отличаются от всех прочих.
Тот же берилл мы можем считать каркасным, если
Ве-тетраэдрам приписать ту же роль, что и Si-тетра-
эдрам. В структуре мышьяка слоистость выражена
особенно четко, поскольку ковалентным связям
внутри слоя с расстоянием между атомами в слое
2,51 А противостоят остаточные связи между слоями
с расстоянием между атомами соседних слоев 3,15 А.
При переходе от As к Bi, относящемуся к тому же
структурному типу, доля металлич. связи в межслое-
вом взаимодействии растет, и поэтому разница между
расстояниями обоих типов становится не столь резкой
(3,10 и 3,47 А); это дает основание считать структуру
висмута не слоистой, а координационной.
В К. встречаются все четыре типа химич. связи —
ионная, ковалентная, ван-дер-ваальсовская и метал-
лическая, но нужно всегда учитывать, что в большин-
стве веществ связи имеют промежуточный, переход-
ный характер. Структурный тип не дает еще возмож-
ности всегда однозначно определять тип химич. связи.
Так, напр., к одному структурному типу относятся
такие пйры, как CsCl и CuZn, медь и аргон. Но во
многих случаях структура дает совершенно определен-
ные указания на тип химич. связи. Так, ионная
связь, характеризующаяся ненаправленностью и не-
насыщаемостью, приводит к образованию структур
с высокой и симметричной координацией. То же
обычно характерно и для соединений с металлич.
связью, однако в интерметаллических соединениях в
ближайшую к данному атому координационную сфе-
ру могут попадать атомы как другого, так и того
же сорта. В соединениях ионного типа из-за анта-
гонизма между компонентами такие сочетания ис-
ключены.
Ненаправленный и ненасыщаемый характер ионной
и металлич. связи приводит к широкому распростра-
нению т. иаз. плотнейших упаковок среди этих
классов соединений. В ионных соединениях часть
атрмов и атомных групп — обычно такими являются
анионы — располагается по центрам тяжести плот-
нейшей упаковки, более мелкие частицы размещаются
в пустотах, между крупными частицами (NaCl, TiO2,
Li2O и др.). Для металлов такую плотнейшую упа-
ковку могут создавать либо атомы разного типа
(CuaAu), либо одного. Подавляющее большинство
металлов относится к двум структурным типам —
плотнейшим гексагональной и кубич. упаковкам.
Третьим основным структурным типом чистых метал-
лов является структура a-Fe, в к-роп атомы распо-
лагаются Цо узлам объемноцентрированного куба,.
Плотнейшие упаковки характеризуют и соединения
с остаточной связью, поскольку она также ненаправ-
ленна и ненасыщаема. Так, все К. благородных газов
образуют плотнейшие упаковки; по таким же или
близким законам располагаются друг относительно
друга молекулы в ряде молекулярных соединений
(Н2, СО и др.). С другой стороны, структуры соеди-
нений с ковалентной связью, связью насыщаемой и
направленной, характеризуются небольшой и асим-
метричной координацией. Так, в элементарном мышья-
ке As связи внутри слоя заведомо ковалентные,
о чем говорит как низкое координационное число 3,
определяемое стремлением каждого атома создать
устойчивый электронный октет, так и одностороннее
размещение этих трех соседей относительно любою
атома As. Однако направленность ковалентной связи
не исключает и возникновения симметричной коорди-
нации (структуры алмаза и элементарных Si, Ge
и a-Sn).
Вопрос о причинах возникновения того или иного
структурного типа составляет одну из главных до
конца еще не решенных задач кристаллохимии так же,
как и связанный с ним вопрос о причинах возникно-
вения полиморфных модификаций. Многими выска-
зывалось положение, что проявление полиморфизма
можно считать скорее правилом, чем исключением.
Нек-рые указания на «выбор» соединением того или
иного структурного типа дает правило, сформулиро-
ванное еще в 1927 Гольдшмидтом п гласящее, что
структура К. определяется числом его структурных
единиц, соотношением их размеров и их поляриза-
ционными свойствами; правило это иногда называют
основным законом кристаллохи-
м и и, хотя оно до сих пор носит чисто качественный
характер и охватывает лишь соединения ионные или
близкие к ним. Для многих простых веществ и неко-
торых соединений, между атомами к-рых существует
ковалентная связь, указание иа тип структуры часто
дает правило Юма-Розери, по к-рому
координационное число атомов, стремящихся создать
устойчивый электронный октет, определяется раз-
ностью между числом 8 и номером группы периодич.
системы. Так, структуры хлора, брома и иода харак-
теризуются двухатомными молекулами (координа-
ционное число равно 1), для серы и селена коорди-
национное число равно 2, для структурного типа
As оно равно 3, а для алмаза 4. В реальгаре (AsS),
координационное число As равно 3, а S — 2. Тип
электронов (s, р, d), участвующих в ковалентной
связи, часто дает возможность предугадать величины
валентных углов. Структуры ряда интерметаллич.
соединений определяются электронной концентрацией
(отношение числа валентных электронов к общему
числу атомов). Так, напр., AgZn, Сн3А1, Cii8Sn
(электронная концентрация 3/2) имеют одинаковую
структуру объемноцентрированного куба.
Правило Гольдшмидта показывает, какое значение
для кристаллохимии имеют радиусы частиц, в осо-
бенности ионные радиусы. Их следует считать столь же
важной числовой характеристикой химич. элемента,
как и атомный еес. Величина ионных радиусов позво-
ляет не только объяснить образование тех или иных
соединений, но и предсказать в какой-то степени воз-
можность или невозможность синтеза нек-рых типов
соединений. Так, из соотношения радиусов была пред-
сказана возможность образования ВаСеО3 и BaThO3
(структурный тип перовскита) и невозможность по-
лучения соответствующих стронциевых соединений.
Ф-лу теллуровой к-ты по аналогии с серной писали
Н2ТеО4 • 2Н2О. Соотношение радиусов Те и О пока-
зывает, что координация теллура должна быть пе
четверной, а шестерной. Рентгенометрически действи-
тельно было доказано, что ее ф-ла Н6ТеО6. Аналогично
839
КРИСТАЛЛЫ
86Q
формула иодной к-ты HJO4 • 2Н2О превратилась
в H5JOe. От ионных радиусов в значительной степени
зависят изоморфные отношения между элементами,
определяющие нахождение редких и рассеянных
элементов в природе. Наряду с ионными радиусами
для кристаллохимии представляют интерес ковалент-
ные радиусы, радиусы металлические и ван-дер-вааль-
совские. Здесь справедлив принцип аддитивности:
каждое межатомное расстояние является суммой
более или менее постоянных величин, отождествляе-
мых с радиусами. Радиусы ковалентные и металли-
ческие близки между собой, поэтому их иногда упро-
щенно наз. «атомными радиусами».
Важной кристаллохимии, характеристикой, исполь-
зуемой в основном для К. с ионной связью, является
энергия кристаллической решетки, позволяющая вы-
числить величины электронного сродства атомов, полу-
чить данные о растворимости К., теплотах гидрата-
ции ионов, об устойчивости химич. соединений.
Большого успеха достигла кристаллохимия в изу-
чении силикатов, заменив прежние гипотетич. и спе-
кулятивные ф-лы на конкретную стереохимию как
природных, так и искусственных соединений (цемен-
ты). Кристаллохимии, исследо-
вания органич. соединений
подтвердили существование в
большинстве из них дискрет-
ных молекул, причем распо-
ложение атомов в молекулах
в основном соответствует клас-
сич. структурным формулам
(см., напр., рис. 10). Кристал-
лохимия помогла распростра-
нить структурные представле-
ния Бутлерова на такие орга-
нич. вещества, ’ как каучук,
белки, целлюлоза и др. В этой
очень сложной области иссле-
дования достигнуты значитель-
Рис. 10. Кристалличе-
ская структура металь-
дегида. Расположение
молекул в ячейке (ато-
мы водорода не пока-
заны).
ные успехи, к числу их сле-
дует отнести, напр., расшифровку кристаллографич.
методами химич. строения гемоглобина, йодистого
холестерила, пенициллиновых солей, ряда витаминов,
вирусов и др.
Для интерметаллич. соединений, как правило, не
подчиняющихся валентным соотношениям и харак-
теризующихся широкой областью гомогенности, кри-
сталлохимия. методы позволили дать рациональные
химич. формулы. Выяснение связи между структурой
и физико-химич. особенностями вещества, причин
кристаллизации различных химич. соединений в том
или ином структурном типе, позволяет кристалло-
химии вносить большой вклад в решение проблемы
создания твердых тел с заданными свойствами. Серьез-
ными трудностями на пути к решению этой проблемы
являются различия между теоретически вычисляемы-
ми свойствами идеального К. и свойствами К. реаль-
ного, обусловленные различного рода искажениями
кристаллич. структуры (см. Дефекты структуры,
Дислокации). Ю- Г. Загальская.
Физические свойства К. Такие свойства К., как
спайность, механич., тепловые, электрич., оптич. и
магнитные — анизотропны, т. е. меняются в зависи-
мости от направления в К.
Спайность — способность многих К. раскалы-
ваться при ударе по определенным кристаллографич.
плоскостям спайности. Различают спай-
ность: весьма совершенную, совершенную, среднюю,
несовершенную и • весьма несовершенную. Браве,
исходя из развитой им теории кристаллич. решеток,
высказал гипотезу, что плоскости спайности проходят
параллельно сеткам с наибольшей атомной плот-
ностью, т. к. такие сетки отстоят друг от друга на
максимальных расстояниях. Обычно это действитель-
но так, но не всегда.
Механические свойства К. зависят
от характера связей между частицами. В К. с ковалент-
ной, ван-дер-ваальсовой или ионной связью, как
правило, слабо выражены пластич. свойства. Пластич.
деформация, предшествующая разрушению, в обыч-
ных условиях весьма мала или вовсе отсутствует.
Однако в условиях значительного всестороннего
сжатия такие К. приобретают способность к пластич.
деформированию. К. с металлич. связью обладают
значительной пластичностью, что обеспечивает воз-
можность их механич. обработки — ковку, прокатку,
волочение, и т. п. Наибольшая твердость и прочность
присущи К. с ковалентной связью (алмаз, нек-рые
бориды и карбиды металлов), К. с ван-дер-ваальсовой
связью наименее прочны. Механич. свойства зависят
от структуры К., что отчетливо выявляется при
полиморфных превращениях. Полиморфное превраще-
ние, сопровождающееся изменением структуры, всегда
влечет за собой и более или менее резкое изменение
механич. (и других) свойств К. (Более подробно см.
в ст. Механические свойства материалов).
Тепловые свойства. При равномерном
нагревании К. в общем случае расширяются неравно-
мерно по разным направлениям, однако симметрия
его сохраняется. Термич. расширение, а также и
теплопроводность одинаковы в двух противополож-
ных направлениях, т. е. обладают собственным цен-
тром симметрии. Если придать шарообразную форму
К. высшей (кубические), средней (гексагональные,
тетрагональные) и низшей симметрии и нагревать их,
то первые сохранят шарообразную форму, вторые
превратятся в эллипсоид вращения с осью, совпадаю-
щей с главной осью К., а третьи — в трехосный
эллипсоид. Однако при очень длительном нагревании
при высокой темп-ре кубич. К. так же изменяют свою
форму, приобретая свойственную им огранку, что
связано с диффузионной подвижностью атомов и стрем-
лением системы к минимуму свободной энергии.
Линейное тепловое расширение К. описывается урав-
нением I = (1 + 0/), где обе основные величины
I и 10 — тензоры, р — линейный коэфф, расширения,
равный по порядку величины 10 6—10 е. Значение р
минимально в направлении кратчайших расстояний
между атомами. Анизотропия расширения особенно
сильна в цепочечных и слоистых структурах. В струк-
туре кальцита СаСОа величина Р в слое из плотно
упакованных атомов кислорода равна 6 • 10"6, а в
перпендикулярном направлении 26 • 10 6. Если из
точечного источника тепла, помещенного в К., с по-
стоянной скоростью распространяется по радиусам
тепло, то поверхности равных темп-p (изотермы)
будут иметь ту же форму, что и при тепловом расшире-
нии. Коэфф, теплопроводности А является функцией
структуры и зависит от направления. В слоистых и
цепочечных структурах величина X больше в слое и
вдоль цепочки, чем в перпендикулярных к ним на-
правлениях.
Электрические свойства. Электро-
проводность К. меняется в чрезвычайно широком
диапазоне: разница в проводимости хороших диэлек-
триков и хороших проводников достш ает величины
порядка 1020. В К. имеют место оба крайних типа
электропроводности: ионная и электронная. Ионная
проводимость является характерной для NaCl, элек-
тронная — для металлов. Обычно в К. имеет место
промежуточная форма электропроводности. К. со
смешанной проводимостью, как, например, полупро-
водники, играют исключительную роль в технике.
В теории ионной и электронной проводимости важная
роль отводится дефектам структуры. В ионных соеди-
нениях со стехиометрия, составом различают 4 тица
861
КРИСТАЛЛЫ
862
дефектов: 1) катионы в междоузлиях решетки, при
вакантных положениях катионов; 2) то же для анионов;
3) катионы и анионы в междоузлиях; 4) вакансии
в положениях катионов и анионов. В ионных соеди-
нениях, не отвечающих стехиометрия, составу, избы-
ток катионов или анионов для электронейтральности
должен компенсироваться эквивалентным количеством
электронов или электронных вакансий (дырок).
В цинките ZnO избыток катиона в междоузлиях ком-
пенсируется эквивалентным количеством электронов
(электронная проводимость). В куприте Си2О вакансии
в положениях катиона создают избыток кислорода.
Выравнивание зарядов и, соответственно, проводи-
мость осуществляются в результате перехода электро-
на от Си+ к Си2+ (дырочная проводимость). Проводи-
мость за счет лишних электронов или электронных
вакансий принципиально возможна и в случае про-
стых веществ. Проводимость морокристалла германия
при комнатной темп-ре незначительна, но увеличи-
вается при вхождении (замещении) в структуру чуже-
родных атомов, напр. Р, Аз (появляются лишние
электроны) или В, А1 (появляются электронные ва-
кансии).
Если К. 0-кварца сжать не в направлении полярной
оси 2-го порядка, то на противоположных параллель-
ных поверхностях К., перпендикулярных к этой
оси, появятся разноименные заряды (продольный
пьезоэффект). При растяжении знаки зарядов меня-
ются. На тех же поверхностях К. появятся аналогич-
ные заряды и при растяжении (сжатии) К. в направ-
лении, перпендикулярном оси второго порядка (по-
перечный пьезоэффект). При атом соблюдается строгая
пропорциональность между напряжением и возни-
кающим зарядом. Явление пьезоэлектриче-
ства обратимо: если К. внести в переменное электрич.
поле, так что вектор поля лежит в направлении по-
лярной оси, то К. в этом направлении будет попере-
менно сжиматься и растягиваться. Здесь также разли-
чают пьезоэффект продольный и поперечный. Пьезо-
эффект наблюдается лишь у К. с полярным направле-
нием, у к-рых отсутствует центр симметрии. Таких
классов симметрии 20 (из 32). Кроме кварца, пьезо-
эффект наблюдается у К. турмалина, цинковой обман-
ки, NaClOj, винной к-ты, сахарозы, сегнетовой соли
и т. д. Пьезоэффект, особенно К. кварца и сегнетовой
соли, широко используется в технике (для стабилиза-
ции радиочастот, для получения ультразвука, изго-
товления основных деталей в микрофонах, телефонах,
адаптерах и т. п., для измерения механич. усилий,
ускорений, давлений и т. д.).
При равномерном нагревании на полярных концах
К. появляются электрич. заряды. При охлаждении
знаки зарядов меняются. Это явление наз. пиро-
электричеством. Пироэффект проявляется
в К. тех 10 классов симметрии, где есть одно поляр-
ное направление. Пироэффект наблюдается у турма-
лина, сахарозы, 0-борацита Mg6[Cl2B14O26], каламина
Zn4 [(OH)2Si2O7] Н2О. Диэлектрич. проницаемость е
К., не обладающих металлич. проводимостью, обычно
имеет значение в пределах от 1 до 100. Особыми
диэлектрич. свойствами обладают К. так наз. сег-
нетоэлектриков, получивших свое название
от сегнетовой соли NaKC4H4O6 • 2Н2О. У нее вне
интервала темп-р от —16° до 4- 24° диэлектрич. свойства
соли обычны, и е не зависит от напряженности внеш-
него поля. Внутри же указанного интервала вели-
чина е обнаруживает сильную зависимость от напря-
женности поля, достигая значений десятков тысяч.
Крайние точки интервала наз. точками Кюри.
В точках Кюри изменяется не только е, но и коэфф,
расширения, электропроводность, пьезоэффект и т. д.
Явление сегнетоэлектричества объясняется тем, что
в соответствующих К. имеются области с постоянными
диполями, к-рые ориентируются в электрич. поле
и тем самым по природе своей явление сегнетоэлектри-
чества очень близко к ферромагнетизму. Эффект
сегнетоэлектричества найден также у К. BaTiOa,
КН2РО4 и др.
Магнитные свойства. Диамагнитными
свойствами обладают К. NaCl, СаСОа, лед. Для них
магнитная восприимчивость х < 0, т. е. создаваемый
магнитный момент М направлен против внешнего
поля. Величина к для диамагнетиков не зависит от
напряженности поля Н. Атомы или ионы диамагнит-
ных К. не обладают начальным магнитным моментом.
Он создается индукцией в магнитном поле. К пара-
магнитным К. относятся, напр., Al, FeCO3, берилл
Al2Bea[Si6O18], диоптаз Cu6[Si6O18]6H2O. Для них
х > 0, т. е. создаваемый магнитный момент М на-
правлен вдоль поля Н (Н — вектор напряженности
магнитного поля). Величина х у этих К. также не за-
висит от Н. Атомы или ионы таких К. обладают
постоянным магнитным моментом^ Однако из-за теп-
лового движения он неупорядочен, так что в целом К.
не обнаруживает магнитного момента. Магнитная
восприимчивость х диа- и парамагнетиков .зависит от
направления, т. е. определяется структурой К. Так,
разница в значениях восприимчивости у СаСО3,
имеющего слоистую структуру, в слое и перпенди-
кулярно к нему, равна 4,6 • 10*’ (на моль), тогда
как у 0-SiO2, имеющего более изометричную струк-
туру, анизотропия восприимчивости незначительна
(0,12 10-6). К ферромагнитным К. относятся Fe,
Со, Ni, их сплавы, ферриты и др. Для них х 2> О,
как и у парамагнетиков, но, в отличие от них, дости-
гает весьма высоких значений, Кроме того, она
сильно зависит от И. Особенностью ферромагнетиков
является их способность сохранять состояние намаг-
ничения после снятия поля. (Подробнее о магнитных
свойствах К. см. в ст. Магнитные свойства).
Оптические свойства. Анизотропия К.
резко проявляется в онтич. свойствах. Луч све^а,
проходящий через К., не только преломляется, но и
распадается на два поляризованных луча (явление
двупреломления), плоскости колебаний к-рых взаим-
но перпендикулярны. Если откладывать в направле-
нии колебаний соответствующего поляризованного
луча показатели преломления п, то можно получить
геометрич. фигуру — оптич. индикатрису, точно ха-
рактеризующую оптич. свойства данного кристаллич.
вещества. Симметрия оптич. индикатрисы связана
с симметрией К. У К. кубич. сингонии она имеет форму
шара, т. е. кубич. К. характеризуются одним значе-
нием показателя преломления п. Оптич. индикатриса
гексагональных и тетрагональных К. имеет форму
эллипсоида вращения. Эллипсоид вращения характе-
ризуется двумя радиусами и, соответственно, К. имеют
два главных значения п : па — обыкновенный (соот-
ветствующий радиусу кругового сечения) и щ —
необыкновенный (перпендикулярный к круговому
сечению). В К. низшей симметрии индикатриса будет
трехосным эллипсоидом, т. е. описывается тремя
значениями п : ng, пт, пр, причем ng > пт > пр.
В таком эллипсоиде имеются 2 круговых сечения,
расположенных косо друг к другу и к главным сече-
ниям эллипсоида. Нормаль к круговому сечению
эллипсоида наз. оптической осью, вдоль
к-рой свет проходит без двупреломления. В К. сред-
ней симметрии одна оптич. ось, они оптически одно-
осны. К. низшей симметрии, обладающие двумя оптич.
осями, наз. двухосными. Угол между оптич. осями
у двухосных К. является важной индивидуальной кон-
стантой вещества. Поляризационный микроскоп позво-
ляет определять все главнейшие оптич. характеристики
К.: положение оптич. индикатрисы, ее знак, одпо-
или двухосностъ К., величину двупреломления и т. д.
863
КРИТИЧЕСКАЯ ТЕМПЕРАТУРА РАСТВОРЕНИЯ
864
В нек-рых К. при пропускании плоско поляризо-
ванного света вдоль оптич. оси наблюдается вращение
плоскости поляризации. Такие К. наз. оптически
активными. К. нек-рых веществ вращают плоскость
поляризации только в одну сторону — вправо или
влево, а К. кварца, напр., встречаются и в правой,
и в левой формах. Это явление находит применение
при изготовлении поляроидов и в специальных при-
борах (сахариметрах), служащих для определения
концентрации сахара в растворе. В оптически изо-
тропных К. поглощение света одинаково во всех
направлениях, в анизотропных — зависит от направ-
ления. Анизотропия поглощения вызывает различие
в окраске и интенсивности двух поляризованных
лучей, выходящих из К. При этом окраска меняется
при вращении К. Это явление наз. плеохроиз-
мом. Плеохроизм наблюдается только в одноосных
и двухосных К.
Практически важным свойством является люми-
несценция К., т. е. свечение К. под влиянием
излучения от внешнего источника после прекращения
освещения. Продолжительность послесвечения ме-
няется от ничтожных долей секунды (10 10 сек) до
многих часов. Продолжительное послесвечение назы-
вается фосфоресценцией, в отличие от
флуоресценции, когда послесвечение про-
должается доли секунды. К., способные фосфоресци-
ровать, наз. к р и с т а л л о ф о с ф о р а м и; к ним
относятся ZnS, ZnO, виллемит Zn2SiO4, шеелит CaVV(.)4,
сульфиды щелочноземельных элементов, галогениды.
Для возникновения люминесценции необходимо хо-
тя бы ничтожное количество постороннего активи-
рующего вещества (Си, Zn, Мп, Bi, редкие земли
и т. д.). Явление люминесценции наблюдается в К.,
у к-рых имеются дефекты во внутреннем строении,
напр. вакантные положения, атомы в междоузлиях.
Люминесценция К. может возникать под действием
света, катодных, рентгеновских, радиоактивных лу-
чей (фото-, катодо-, рентгено- и радиолюминесценция),
при создании в К. электрич. поля (электролюминесцен-
ция), при раздавливании нек-рых К.,напр. As2O3 (трибо-
люминесценция), при кристаллизации, напр., сахарина
(кристаллолюминесценция), при химич. процессах, в
особенности при окислении (хемолюминесценция).Све-
чение К. связано с испусканием квантов света при воз-
вращении возбужденного атома в нормальное состоя-
ние. При этом длина волны света люминесценции
равна или больше длины волны возбуждающего света.
На явлении люминесценции основан люминесцентный
анализ, позволяющий определять состав смеси. Лю-
минесценция широко используется в различных
электровакуумных приборах (катодные осциллографы,
люминесцентные лампы, телевизоры и т. д.).
Е. М. Po-uanoia.
Лит.: Федоров Е. С., Симметрия и структура
кристаллов, М., 1 949; Вульф Г. В., Кристаллы, их
образование, вид и строение, М., 1917; Ш у б н и к о в А. В.,
Флинт Е. Е., Б о к п й Г. Б., Основы кристаллографии,
М., 1940; Аншелес О. М., Начало кристаллографии,
Л., 1952; Попов Г. М., Шафрановский И. II.,
Кристаллография, М., 1955; Белов Н. В., Структура
ионных кристаллов и металлических фаз, М., 1947; Б е-
лов Н. В., Структурная кристаллография, М., 1951;
Б о к и й Г. Б., Кристаллохимия, М., I960; Эванс Р.,
Введение в кристаллохимию, М., 1948; Ормонт Б. Ф.,
Структуры неорганических веществ, М., 1 950; Китай-
городский А. И., Органическая кристаллохимия, М.,
1955; Вустер У., Практическое руководство по кристалло-
физике, пер. с англ., М., 1958; Пай Дж., Физические
свойства кристаллов..., пер. с англ., М., I960; Кузне-
цов В. Д., Физика твердого тела, т. 1 —4, 1 937—1947;
Татарский В. Б., Кристаллооптика и иммерсионный
метод определения вещества, Л., 1949; Kleber W.,
Einliihrung in die Kristallographie, В., 1961; Winkler
Ст. F., Struktnr und Eigenschatten der Kristallc, B., 1955.
КРИТИЧЕСКАЯ ТЕМПЕРАТУРА РАСТВОРЕ-
НИЯ — температура, при к-рой достигается неогра-
ниченная взаимная смешиваемость компонентов, огра-
ниченно растворимых друг в друге при более высоких
или при более низких темп-рах. К. т. р. впервые
была изучена В. Ф. Алексеевым (1876) в системах
вода — фенол, вода — анилин и других. Растворимость
анилина в воде и воды в анилине возрастает с тем-
пературой; при повышении темп-ры анилиновый слой
обогащается водой, а водный слой — анилином. При
достижении К. т. р. оба слоя (при любых относитель-
ных количествах компонентов) становятся тождест-
венными и жидкость — однородной. К. т. р. системы
вода — анилин 167°. Состав равновесных слоев при
подходе к этой темп-ре приближается к 51,4% воды
и 48,6% анилина. Прибавление любого из компонен-
тов смеси при этой (и более высоких) темп-ре не
вызывает расслоения смеси.
.На рис. 1 нанесена кривая ABCDE, наз. кривой
расслоения, разделяющая области гомогенных
и гетерогенных систем. Об- 18д
ласть, ограниченная этой кри-
вой и осью абсцисс, отвечает '60
двухслойной системе (гетеро- 140
генной), а область, лежащая
вне этой кривой, — одпослой- >->'20
ной (гомогенной) системе. Точ- ~ 1дд
ки на кривой характеризуют |
составы равновесных слоев, во
напр. при 100° составы опре-
деляются точками В и D. Пря-
мые, соединяющие точки рав- 4Р
повесных (сопряженных) меж- 1
ду собой слоев, например BD,
— —* никотин
называются коннодами, или связующими прямыми
(см. также Прямолинейного диаметра правило).
Различают верхнюю и нижнюю К. т. р. В первом
случае расслаивание происходит при темп-рах ниже
критич. темп-ры, во втором случае — выше критич.
темп-ры. Системы с нижней К. т. р. встречаются зна-
чительно реже, чем системы с верхней К. т. р. При-
мером систем с верхней К. т. р. может служить рас-
смотренная выше система вода — анилин, а с нижней
К. т. р. — система вода — диэтиламин. Известны и
такие системы, которые обладают верхней и нижней
К. т. р., например система вода — никотин. На
рис. 2, представляющем эту систему, внутренняя
область замкнутой кривой (Л)
является областью гетероген-
ной системы, а область вне этой
кривой (В) — областью гомо-
генной системы.
К. т. р. еильво зависят от
примесей. Если примесь раст-
воряется только в одной из
сосуществующих фаз, то верх-
няя К. т. р. повышается, а
нижняя понижается. Так, 0,19
весовых % нафталина повы-
шает К. т. р. в системе вода —
фенол на 3,2°. В этой системе
существует только верхняя
К. т. р. Если же примесь раст-
воряется в обеих фазах приблизительно одинаково,
то верхняя К. т. р. понижается, а нижняя повы-
шается. Няпр., прибавление к системе вода — фенол
0,98 вес. % олеиновокислого натрия снижает К. т. р.
на 21,6°. Это явление используется для проверки
чистоты веществ. Изменение критической температу-
ры растворения при изменении давления на 1 атм
составляет обычно сотые доли градуса, причем ниж-
няя с повышением давления всегда повышается, а
верхняя в Одних системах повышается, а в других
понижается.
И. С. Курпаков и С. Ф. Жемчужный (1905) впервые
обнаружили К. т. р. в твердых р-рах КС1 и NaCl.
865
КРИТИЧЕСКОЕ СОСТОЯНИЕ
866
И. Г. Кричевский и П. Е. Большаков (1941) впервые
изучили критич. явления в р-рах двух газов (аммиак—
азот).
Вблизи критич. точки наблюдается ряд характер-
ных явлений: флуктуации состава, появление оптич.
неоднородности и т. д. (см. Критическое состояние),
В таблице приведены К. т. р. и составы нек-рых
двухкомпонентиых растворов.
Система х2, мольные доли К. т. р., °C
Метиловый спирт — н-гексан . . . 0,46 42.8
Нитробензол — н-гексан 0,583 20,0
Нитробензол — н-гептан 0,528 20,0
Метиловый спирт — циклогексан . 0,489 46,14
Иодистый метилен — циклогексан . — 34.5
Бензол — гексафторгептан — 13,5
Гептан — дихлорэтиловый эфир . — 16,50
Октан — дихлорэтиловый эфир . . — 2'0,67
Декан — ацетон — 18,0
Камфен — этилоксалат Метаксилол — малеиновый ап гид- — 23,8
РИД — 15
Гексан — о-толуидин — 21,2
Циклопентан — анилин — 18
Циклогексан — анилин 0,4656 29,428
Бензол — анилин — 20
Терпентин — анилин — 18,04
Сероуглерод — уксусный ангидрид — 29,8
Бензол — триэтиленгликоль .... —
Триэтилбензол — метиловый спирт — 19
Гептан — фенол 0.56 23,5
Фенол — вода 0.91 66,4
Триэтиламин — вода 0.875 18,3
Гексаметиленимин — вода 0,942 68,1
Лит. см. при ст. Критическое состояние.
КРИТИЧЕСКОЕ СОСТОЯНИЕ — состояние, при
к-ром две фазы вещества, находящиеся между собой
в термодинамич. равновесии, становятся тождествен-
ными ио ( v -
р
своим физич. свойствам. Точка на кривой
фазового равновесия К (рис.), в к-рой
наступает К. с., наз. критиче-
ской точкой. Эта точка харак-
теризуется: критическим дав-
ен и е
м п е
ски
Гетерогенный и гомогенный пе-
реход от пара к жидкости.
м
м Ркрит.’ критической
р а т у р о Й <крит , к р ити-
м объемом Ёкрит .кри-
тической плот-
ностью йкрит Пере-
двигаясь по кривой фа-
зового равновесия из об-
ласти низких давлений
в область более высоких
давлений, легко можно
обнаружить, что разница
в свойствах сосущест-
вующих фаз уменьшает-
ся, а по достижении
критич. точки она вовсе исчезает. В этой точке
кривая равновесия жидкость — пар р — <р (г) обры-
вается и t = гкрит , = dT = </крит , = Уг =
=. Екрит, число степеней свободы / = 0.
Впервые К. с. наблюдал Каньяр де ла Тур (1822).Суще-
ствование критической температуры первоначально установил
Д. И. Менделеев (1880), к-рый назвал ее темп-рой абс. кипе-
ния. Диаграмму равновесия жидкость — пар для СО2, вклю-
чавшую критич. точку, впервые опубликовал Т. Эндрюс
(1869). Термодинамич. теорию К. с. разработал Дж. Гиббс
(1876).
На рис. показаны гетерогенный и гомогенный
переходы от пара к жидкости (по оси абсцисс отложен
уд. объем У, по оси ординат — давление р). Кривая
ABCD соответствует изотермич. переходу пара в жид-
кость при темп-ре ниже критической: часть АВ —
газообразному состоянию вещества (переход от раз-
реженного газа к насыщенному пару); горизонталь-
ная часть ВС — равновесию газовой и жидкой фаз
(давление пара при конденсации остается постоянным);
часть CD — жидкому состоянию. Если проводить
подобный же изотермич. процесс при более высокой
темп-ре, то его графич. изображением будет кривая
A^BiCiD^. Длияа горизонтального участка, на к-ром
обе фазы сосуществуют в равновесии, с повышением
темп-ры уменьшается, и при нек-рой темп-ре горизон-
тальный участок изотермы стягивается в критич.
точку К. Изотерма MKN, проходящая через критич.
точку, наз. критической изотермой. От
жидкости к пару (и обратно) можно перейти и непре-
рывным путем, минуя двухфазную область (огибая
сверху заштрихованную область) так, что на всем пути
перехода вещество остается однородным, и нельзя
установить момент перехода из одной фазы в другую.
Критич. давление />крит и критич. темп-ра «крит
могут быть определены опытным путем. Кроме того,
одна из этих величин может быть найдена (графически
или аналитически), если известна зависимость р =
= <р (г). Критич. объем Р'крит непосредственно не под-
дается точному определению, т. к. ничтожные изме-
нения давления при t = «крит и р = Р1!рит влекут
за собой огромные изменения объема; Икриг обычно
определяют на основании прямолинейного диаметра
правила.
Установлено, что вблизи критич. точки жидкость —
пар резко возрастают теплоемкость вещества С и
изотермич. сжимаемость Р(, что может быть объяс-
нено увеличением флуктуации плотности. Последние
легко возникают благодаря малой энергии их обра-
зования вследствие сглаживания различий между
двумя сосуществующими фазами в К. с. Наличие
флуктуаций плотности приводит к оптической неод-
нородности системы, к рассеянию света. Это явле-
ние носит название критической Опалес-
ценции. Рассеяние света служит источником све-
дений о величине и характере флуктуаций в крити-
ческой области.
Установление понятия о критич. состоянии сыграло
большую роль в технике сжижения газов. (Как ясно
из предыдущего, сжижение газов возможно лишь
в области темп-р, лежащих ниже критич. точки).
В таблице приведены критич. темп-ры, давления
и объемы нек-рых веществ.
Вещество *крит., °C Ркрит., атм ^крит., мл/моль
Простые вещества и неорганические соединения
Не .............. -267,9 2,26 57,8
и, -239,9 12,80 65,0
Ne -228.7 26,9 41,7
n2 -147,0 33,5 90,1
Аг -122 48.0 75,2
О2 -118,4 50.1 78
Кг - 63,8 54,3 92,2
Хе 16.59 5S.0 118,8
С12 . . . . 144,0 76,1 124
Вг, 311 102 144
СО -140 34,5 93,1
NO - 93 64 58
BF3 - 12.3 49,2
СО2 31.0 72,9 94.0
HCI 51,4 81,5 48
НВг 90 84.0
H2S 100.4 88,9 97,7
NH3 132,3 111,3 72,5
HJ 150 81
so2 157,5 77,8 122
no2 158 100 82
COC12 182 56 190
HCN 183,5 53,2 139
so3 218,2 83,8 126
cs2 279 78 170
SnCl. 318.7 37,0 351
H2O 374,2 218,3 56
867
КРОВЯНАЯ СОЛЬ-КРОНЫ
868
Продолж е и и е
Вещество *крит., Ркрит.» ^крпт.,
СС атм мл/моль
[Органическ ие соединения
Метан - 82 45,8 99,0
Этилен 9,00 50,50 124
Этан 32,3 48,2 148
Ацетилен 36 61,6 113
н-Пропан 96,8 42,0 200
м-Бутан 152,0. 37,5 255
к-Пентан 196,6 33,3 311
н-Гексан 231,7 29,9 368
н-Гептан 267,0 27,0 426
Циклогексан 280 40 308
Бензол . • 289 48,6 260
н-Октан 296,2 24,6 490
Толуол 320,8 40 320
н-Декан 346 20,8 602
и-Ундекан 367 19,2 660
и-Додекан 386 17,9 718
Метиловый спирт 240,0 78,5 118
Этиловый спирт 243 63,0 167
н-Пропиловый спирт .... 264 50,2 220
Диметиловый эфир 126,9 53 190
Диэтиловый эфир 194 35.6 281
Ацетон 235,5 46,6 213
Четыреххлористый углерод 283,2 45,0 276
Хлортрифторметан 28,8 39 180
Фтористый метил 44,6 58,0 113
Хлороформ 263,4 54 240
Хлорбензол 359,2 44,6 308
Анилин 425,6 52,3 274
Для нек-рых веществ эти данные могут отличаться в по-
следнем знаке от значений критич. постоянных, указанных
в статьях, относящихся к отдельным соединениям. Это связано
с естественным небольшим расхождением данных разных авто-
ров и отвечает возможной их погрешности.
Лит.: Киреев В. А., Курс физической химии, М.,
1955; Аносов В. Я., Погодин С. А., Основные на-
чала физико-химического анализа, М.—Л., 1947; Финд-
лей А., Правило фаз и его применение, пер. с англ., 2 изд., М.,
1935; Т а м м а н Г., Руководство по гетерогенным равнове-
сиям, пер. [с нем.], Л., 1935; ЛеонтовичМ. А., Введение
в термодинамику, 2 изд., М.—Л., 1952; К р и ч е в с к и й И. Р.,
Фазовые равновесия в растворах при высоких давлениях,
2 изд., М.—Л., 1952; Карапетьянц М. X., Химиче-
ская термодинамика, 2 изд., М.—Л., 1953; Ландау Л.
и Л и ф ш и ц Е., Статистическая физика (классическая и
квантовая), М.—Л., 1951; (Теоретическая физика, т. 4);
Кириллин В. А. и Ш е й н д л и н А. Е., Термодина-
мика растворов, М.—Л., 1956; Шахпаронов М. И.,
Введение в молекулярную теорию растворов, М., 1956; Сто-
летов А. Г., Заметки о критическом состоянии тел, в его
кн.: Избранные сочинения, М.—Л., 1950; Гиббс Дж. В.,
Термодинамические работы, пер. с англ., М.—Л., 1950; Кри-
тические явления и флюктуации в растворах. Труды совеща-
ния, М., 1960. М. И. Шахпаронов.
КРОВЯНАЯ СОЛЬ — 1) Желтая кровяная
соль, железистосинеродистый калий, ферроцианид
калия, калия гексацианоферроат К4[ 1’е(С.\%]. 2) К р а-
с и а я кровяная соль, железосинеродистый ка-
лий, калия феррицианид, калия гексацианоферриат
K3[Fe(CN)6],
КРОНЫ (крона) — минеральные пигменты, ок-
раска к-рых обусловлена присутствием иона СгО('.
К. различают по составу: свинцовые, свинцовомолиб-
датные, цинковые, стронциевые, бариевые, а также
по цвету (желтые, оранжевые, красные).
Желтые свинцовые К. представляют со-
бой изоморфную смесь хромата и сульфата свинца,
пнет к-рой изменяется от светло-лимопиого (45—50%
РЬСгО4) до темпо-желтого (90—100% PbCrO4). С уве-
личением содержания РЬСгО, повышается также и
укрывпстость пигмента. Желтые К. обладают высо-
кими пигментными свойствами, атмосфероустойчиво-
стью, термостойкостью, но недостаточно светопрочны
(па свету темнеют); растворяются в минеральных
к-тах и щелочах. Ппотн. ок. 5,9, укрывистость 45—
60 г/л»2, маслоемкость 12—16 (укрывистость,
или кроющая способность, пигмента —
способность закрывать групт при окраске изделия
так, чтобы грунт не просвечивал через слой краски;
маслоемкость — количество масла в г на
100 г пигмента, при к-ром образуется однородная
паста). К. желтые получают совместным осаждением
хромата и сульфата свинца из растворов свинцовых
(ацетат, нитрат, оксихлорид) солей смесью бихро-
мата натрия с серной к-той. Желтые К., несмотря
на нек-рую ядовитость, широко применяют для полу-
чения красок и эмалей всех типов, а также при про-
изводстве карандашей, клеенки и др.
Оранжевые и красные свинцовые
К. представляют собой оксихромат свинца РЬСгО4 • РЬО.
В высших сортах содержится 80% РЬО и 17%
СгОа. Чем крупнее частицы К., тем более глубокий
красный оттенок они имеют; укрывистость 45—50 г/л»2,
маслоемкость 7,5—8,5, плотн. 6,75—7,0. Оранже-
вые К. отличаются атмосферостойкостью, термостой-
костью (не разлагаются при нагревании до 600°) и
довольно хорошей светостойкостью. Практически не-
растворимы в воде, полностью растворяются в мине-
ральных к-тах и конц. щелочах. Оранжевые К. полу-
чают осаждением основных свинцовых солей бихрома-
том натрия в щелочной среде (pH > 9) при ~ 90°.
Применяют оранжевый К. гл. обр. для антикоррозион-
ных и грунтовочных красок, нек-рых эмалей; крас-
ный К. применяют редко.
Красный свинцовомолибдатный К.
является изоморфной смесью хромата, молибдата и
сульфата свинца, содержащей в среднем 70—80 мол. %
РЬСгО4, 10—15% РЬМоО4, 3—10% PbSO4. Наиболее
яркий и насыщенный цвет имеет пигмент состава
7РЬСгО4 • РЬМоО4 • PbSO4. Хромат свинца в этом
пигменте находится в виде кристаллов тетрагональ-
ной системы красного цвета, чем и обусловлен цвет К.
Это единственный яркий, красно-оранжевого цвета
минеральный пигмент, отличающийся высокой интен-
сивностью и укрывистостью (15—20 г/л»2) и удовлетво-
рительной светопрочностью. Его получают осажде-
нием нитрата свинца смесью хромата и сульфата
натрия с молибдатом аммония в строго контролируе-
мых условиях (pH 2,5—3,0; 20—25°) с последующей
стабилизацией осадка гидроокисью алюминия и др.
( агентами. Применяют самостоятельно и в смеси с ор-
ганич. красителями (придает непрозрачность и боль-
шую светопрочность) в произ-ве красок, пластмасс,
искусственного волокна, бумаги, кожи и др.
Цинковые К. представляют собой основные
хроматы цинка: тетраоксихромат 5ZnO • СгО3 • 4Н2О,
триоксихромат 4ZnO • СгО3 • ЗН2О и двойные хроматы
цинка и калия, гл. обр. 4ZnO • 4СгОа • К2О • ЗН„О.
Плотность соответственно: 3,84; 3,67 и 3,41. Чем
выше содержание СгО3, тем насыщеннее цвет и выше
пигментные свойства К. Тетра- и триоксихроматы
цинка хотя и имеют блеклый желтый цвет, высокую
маслоемкость, низкую укрывистость (180 г/л»2), но
отличаются очень высокими антикоррозионными свой-
ствами и поэтому используются в защитных грунтах
по стали, алюминию и легким сплавам. Пассивирую-
щее действие таких грунтов связано с медленным вы-
щелачиванием водой иона СгО|~. Двойные хроматы
цинка и калия (малярные цинковые К.) имеют кра-
сивый лимонный оттепок. Укрывистость 120 г/л»2,
они более светопрочны, чем свинцовые К., частично
растворимы в воде и полностью в к-тах и щелочах.
Применяют гл. обр. для художественных красок.
Цинковые К. получают обработкой диспергированных
в воде цинковых белил раствором бихромата калия
или хромового ангидрида.
Стронциевый К. SrCrO4 — лимонно-желтый
светостойкий пигмент, укрывистость 90 г/ле2, плотн.
3,75; растворимость в воде 0,8 г/л. Полностью рас-
творяется в минеральных к-тах и разлагается ще-
лочами. Применяется в художественных красках и
в грунтах специального назначения.
869
КРОТОНАЗА- КРОТОНОВАЯ КОНДЕНСАЦИЯ
870
Бариевый К. — двойная соль хромата бария
и калия ВаСгОд • К2СгО4, тусклого желтого цвета;
плотн. 3,65; в минеральных к-тах растворяется полно-
стью, вода медленно выщелачивает из него К2СгО4.
Барий-калиевый хромат обладает очень низкими
пигментными и исключительно высокими антикорро-
зионными свойствами. Пигмент получают прокалива-
нием смеси карбоната бария и бихромата калия при
650°. Применяют только для грунтов и антикорро-
зионных покрытий по железу, алюминию, магнию и
их сплавам. Используется также в качестве примеси
к другим пигментам для повышения их атмосферо-
устойчивости.
Лит.: Беленький Е. Ф. иРискин И. В., Химия
и технология пигментов, 3 изд., М., 1960. М. А. Штерн.
КРОТОНАЗА — фермент, катализирующий обра-
тимую реакцию гидратирования] а, [^ненасыщенных
к-т в форме тиоэфиров кофермента А:
RCH2CH=CHCO-SKoA-|-H2Os=iRCH2CH(OH)CH2CO-SKoA
Катализируемая К. реакция является важнейшим
звеном в процессе биология, окисления жирных к-т
(см. Жиры), а также разветвленных ненасыщенных к-т,
образующихся при окислительном дезаминировании
таких аминокислот, как валин, лейцин, изолейцин.
К. широко распространена в животных и раститель-
ных тканях; кристаллич. К. имеет мол. в. ок. 210 000,
обладает типичным для простого белка спектром с мак-
симумом поглощения при 280 ммк. Оптимум активно-
сти К. соответствует pH 9,4. Число оборотов (коли-
чество молей вещества, превращаемых 1 молем фер-
мента в единицу времени при 25° и pH 7,5) составляет
для К. 730 000 молей/мин; при оптимальном значении
pH это число возрастает до 1 400 000.
К. строго стереоспецифична, хотя и не обладает
специфичностью в отношении геометрич. изомеров:
она способна гидратировать как транс-, так и цис-
а,Р-ненасыщевные к-ты, но при этом всегда обра-
зуются только L-p-оксикислоты. При осуществлении
обратной реакции К. также катализирует дегидрата-
цию только L-0-оксикислот и не действует ва кислоты
D-ряда. Величина Михаэлиса константы в случае,
если субстратом К. является кротонил-КоА, соста-
вляет 2,0 - 10“6 М, а в случае т/?анс-2-гексеноил-
КоА — 2,5 • 10 6 М. Активность К. тормозится реа-
гентами, блокирующими SH-группы: п-хлормеркури-
бензоат в концентрации 10 6 М снижает активность К.
на 28%, а в концентрации 10-3Л/ — более, чем на
95%. Активность заторможенной К. восстанавли-
вается при добавлении тиоловых соединений, напр.
восстановленного глутатиона. Активность К. опре-
деляют различными спектрофотометрич. методами,
простейший из к-рых основан на том, что а, р-ненасы-
щенные к-ты обладают характерным пиком поглоще-
ния при 263 ммк, отсутствующим у р-оксикислот;
это позволяет следить за ходом реакции по уменьше-
нию поглощения при этой длине волвы.
Лит.: The enzymes, ed. Р. D. Boyer, H. Lardy, К. Myr-
back, v. 5, 2 ed., N. Y., 1961, p. 511. В. Б. Спиричев.
КРОТОНОВАЯ КИСЛОТА (Р-метилкарбоновая ки-
слота) СН3СН=СНСООП, мол. в. 86,09 — кристаллы
в виде мопоклинвых игл или призм; т. пл. 71,4—
71,7°; т. кип. 184,7°/760 мм, 81°/13 мм; <%' 0,964;
п8д 1,4228; константа диссоциации К = 2,0 -105
(25°); в 100 г воды растворяется 7,61 г (20°), 65,6 г
(40°). К. к. имеет г^аис-конфигурацию (I): ее цис-
изомер — изокротоновая к-та (II) [т. пл. 15,5°;
т. кип. 169°; 1,0265; 1,4456, константа диссо-
циации К = 3,5 • 10 ’6 (25°)] менее устойчива и при
нагревании переходит в К. к.
н-ссоон н-с соон
II II
CHS-CH Н-С-СН3
I и
Кротоновая и изокротоновая к-ты восстанавли-
ваются до масляной к-ты, а при окислении конц.
азотной к-той дают смесь щавелевой и уксусной к-т.
К. к. является винилогом уксусной (см. Винилогия);
ее эфиры (алкилкротонаты), подобно этилацетату,
вступают в сложноэфирную конденсацию:
с2н5ооссоос2н3+сн3сн=снсоос2н3—►
--Щ»Н5ООССОСН,СН=СНСООС2Н3
К. к. может быть приготовлена конденсацией малоно-
вой кислоты с ацетальдегидом в присутствии пири-
дина, а также окислением кротонового альдегида
СН3СН=СНСНО; последний метод имеет технич. зна-
чение.
К. к. присоединяет по двойной связи водород,
галогены, галогеноводородные к-ты с образованием
соответственно масляной к-ты или галогеномасляных
к-т (иод присоединяется труднее).
К. к. используется при получении нек-рых фарма-
цевтич. препаратов (напр., d, 1-треонина); аллиловый
и гераниевый эфиры применяют в парфюмерии.
Амид К. к., т. пл. 102°; анилид, т. пл. 115—118° (его N-ал-
кильные производные являются инсектицидами); хлоранги-
дрид, т. кип. 124—125°; метиловый эфир, т. кип. 119—120°,
d“° 0,9444, пд 1,4242; этиловый эфир, т. кип. 136—138°,
0,9175, пд 1,4245. И. А. Несмеянов.
КРОТОНОВАЯ КОНДЕНСАЦИЯ — конденсация
двух молекул альдегида с отщеплением воды и обра-
зованием ненасыщенного альдегида. В зависимости от
условий уплотнение может останавливаться на ста-
дии образования альдоля (см. Альдольная конденсация)
или же сопровождаться дополнительным отщеплением
молекулы воды с получением кротонового альдегида,
напр. для ацетальдегида:
СН3СНО4-СН3СНО—>сн8снонсн2сно—►
—->СН8СН=СНСНО-|-Н2О
Аналогичная конденсация может в более жестких
условиях наблюдаться и в случае взаимодействия
двух молекул кетонов, а также альдегида и кетона
с образованием ненасыщенного кетона, напр.:
СсН5СНО-|-СНзСОСН3—-
--►CeH5CHOHCH2COCH8^S^CeH5CH=CHCOCH3
Образовавшийся кротоновый альдегид может, естест-
венно, вступать в конденсацию еще с одним молем
уксусного альдегида по схеме К. к. с образованием
сорбинового альдегида:
СН3СН=СНСНО1-СНзСНО---->СН3СН=СНСН=СНСНО+Н2о
к-рый может реагировать далее с СН3СНО.
Действительно, из продуктов высших фракций, об-
разовавшихся при К. к. уксусного альдегида,
был выделен ряд ненасыщенных альдегидов до
СН3(СН=СН),СНО, к-рые были использованы для
получения полиенов. Из ацетона по схеме К. к. можно
получить мезитила окись (I) и форон (II):
2СН3СОСН8---(СН8)2С^СНСОСН3 <СН»)2С.<Ц
I
—-(СН8)2С=СНСОСН=С(СН8)2
II
Обычно в качестве катализаторов применяются
агенты основного характера (СН3СООК, К2СО3, KCN).
В кислой среде реакция протекает по-другому, с пред-
варительной енолизацией и участием в реакции атома
водорода при наименее гидрогенизированном атоме
углерода. Так, при конденсации бензальдегида с ме-
тилэтилкетоном могут образоваться след, изомерные
ненасыщенные кетовы
—i-CeH5CH=C(CH8)COCH8
С0Н5СНО )-СН3СОСН»СН8—
——>СаН6СН=СНСОСН2СН8
871
КРОТОНОВЫЙ АЛЬДЕГИД - КСАНТИН
S72
Гидрированием кротонового альдегида можно полу-
чить н-бутиловый спирт. Взаимодействие смеси двух
альдегидов с последующим гидрированием продуктов
К. к. может служить методом получения различных
спиртов.
Лит.: Grignard V., Dupont G„ L о с q u 1 n R.,
Traite de chimie organlque, v. 7, P., 1950, p. 127.
Я. Ф. Комиссаров.
КРОТОНОВЫЙ АЛЬДЕГИД (2-бутеналь, 0-метьл-
акролеин) СН3СН=СНСНО, мол. вес 70,09 — проз-
рачная жидкость с резким запахом, вызывает слезо-
течение; известен в виде цис- и транс-изомеров,
продажный К. а. — транс-изомер; т. пл. —69°;
т. кип. 102,2°; d-,'' 0,848; 1,436; в 100 г воды раство-
ряется 18,1 г (20°), 19,2 г (5°); в 100 г К. а. растворяется
9,5 г воды (20°); с обычными органич. растворителями
К. а. смешивается во всех отношениях; образует
азеотроп с водой (т. кип. 84°, Н2О 24,3%). Теплота
испарения 123,5 кал/г', теплота сгорания 775,3 кал/г
(р = const); т. воспл. безводного К. а. +8°; взрыво-
опасная концентрация паров в воздухе 2,95—15,5
об. %.
Благодаря наличию альдегидной группы и двойной
связи К. а., подобно акролеину, чрезвычайно реак-
ционноспособен. Он быстро димеризуется и полиме-
ризуется при хранении, на воздухе медленно окис-
ляется. В зависимости от условий опыта К. а.
можно окислить в кротоновую, малеиновую, уксус-
ную или щавелевую кислоты. К. а. может быть
также восстановлен либо по альдегидной группе, либо
по двойной связи, либо по обоим направлениям одно-
временно. К. а., подобно другим а,[5-непредельным
альдегидам, имеющим активированную двойную связь,
легко присоединяет галогены, галогеноводороды, воду,
сцирты, амины; при конденсации К. а. с аммиаком
образуются пиридиновые основания (Чичибабина ре-
акция)' с анилином — хинальдин (Дёбнера — Миллера
реакция)', вступает в Михаэля реакцию, в диеновый
синтез. Кроме того, К. а. вступает во все реакции,
характерные для альдегидов. В пром-сти К. а. полу-
чают кротоновой конденсацией ацетальдегида; огра-
ниченное значение имеют другие методы получения,
напр. пиролиз солей кротоновой и муравьиной к-т,
отщепление галогеноводородов от [5-галогеномасляных
альдегидов и изомеризация 1,4-бутендиола:
H2SO4
сн2-сн=сн-сн2---------»сн2-сн-сн=сн2 —-
I I ! I
ОН он он он
—- сн2=сн-сн2-сно —> сн3сн=сн-сно
К. а. используют гл. обр. для произ-ва бутилового
спирта; кроме того, из него получают масляный аль-
деги д, малеиновую к-ту, кротиловый спирт и хиналь-
дин. К. а. добавляют к топливному газу для предупреж-
дения о просачивании его- в помещение.
Н. Я. Гамбарян.
КРУГОВЫЕ ПРОЦЕССЫ (или циклы) — процессы,
при к-рых система, претерпев ряд изменений, в резуль-
тате вновь возвращается в исходное состояние. Наи-
более характерным отличием К. п. является то, что
любая термодинамич. функция, являющаяся пара-
метром состояния (внутренняя энергия, энтальпия,
изохорный и изобарный изотермич. потенциалы,
энтропия и др.), в результате К. п. вновь принимает
первоначальное значение и, следовательно, ее изме-
нение при К. п. равно нулю (АТ/ = 0, ..., AS = 0).
Первый закон термодинамики приводит при этом
к выводу, что алгебраич. сумма количеств теплоты,
полученных и отданных системой в ходе К. п., должна
быть равна работе, произведенной системой или над
системой: Q = .4. Рассмотрение К. п. широко приме-
няется в термодинамике при исследовании различных
вопросов. Наиболее известным из таких процессов
является Карно цикл, A. Kupege. ।
КСАНТЕНОВЫЕ КРАСИТЕЛИ — красители, со-
держащие группировку (I), сходную с группировкой
самого ксантена. К. к. относятся к клас-
СУ арилметановых красителей — про-
Е I Т д изводных трифенилметана (СвН6)3СН.
К. к. окрашены в желтые, красные,
R I розовые, красно-фиолетовые и синие
цвета, отличающиеся большой ярко-
стью; однако светопрочность их невелика. Среди
К. к. имеются основные, кислотные и протравные;
большинство из них обладает флуоресценцией. К. к.
делят на неск. групп. Наиболее важной из них
является группа фталеинов. Фталеины, содержащие
в положениях 3 и 6 аминогруппы, наз. родамипами,
а содержащие оксигруппы — флуоресцеинами.
Красители группы родамина. Ти-
пичным представителем красителей этой группы яв-
ляется родамин С (II). Этот краситель образуется спла-
влением л«-диэтиламинофенола с фталевым ангидри-
дом либо при нагревании 3,6-дихлорфлуорана с ди-
этиламином. Строение родаминов, так же как и строе-
ние других К. к., принято изображать с помощью
хиноидных формул. Среди родаминов имеются и не-
симметрично построенные красители. Нек-рые марки
родаминов содержат вместо карбоксильной группы
сложноэфирную группировку, напр. родамин Ж.
Этерификация карбоксильной группы в ряде случаев
способствует улучшению колористич. свойств. Рода-
мин С и родамин Ж являются основными красителями
и применяются для окраски текстильных волокон,
кожи, бумаги, мыла, а также для получения лаков.
Из числа кислотных красителей можно назвать суль-
фородамин С и прочный кислотный синий Р; послед-
ний относится к числу т. н. виоламивов. К группе ро-
даминов принадлежат также хромоксановые краси-
тели, напр. хромоксан ярко-красный БЛ. Эти краси-
тели применяются в качестве протравных и образуют
окраски, прочные к свету и стирке.
Красители группы флуоресцеина.
3,6-Диоксифлуоран—флуоресцеин (III) образуется при
сплавлении фталевого ангидрида с резорцином в при-
сутствии водоотпимающих средств. Флуоресцеин тру-
дно растворим в воде, но легко — в растворах щело-
чей. Хорошо растворима в воде двунатриевая соль
флуоресцеина, известная под названием уранина А.
Эозин, иодэозип, эритрозин и бенгальский розо-
вый являются д'алогепопроизводными флуоресцеина.
Шерсть и особенно шелк окрашиваются этими красите-
лями в очень яркие и красивые красные тона, однако
окраски недостаточно прочны к действию света. Эо-
зин и иодэозин используют в лакокрасочной пром-сти
и полиграфии. К этой же группе красителей отно-
сятся галлеин и церулеин, обладающие протравными
свойствами. Галлеин получают сплавлением фталевого
ангидрида с пирогаллолом, а церулеин образуется
из галлеина при нагревании последнего с серной к-той.
Лит.: Коган И. М., Химия красителей, 3 изд., М.,
1956, с. 297; Венкатарамап К., Химия синтетиче-
ских красителей, пер. с англ., т. 2, Л., 1957, с. 851.
О. Ф. Гинзбург.
КСАНТИН (2,6-ди оксо-1, 2, 3, 6-тетрагидропурип,
2,6-диоксипурин) C5H4N4O2, мол. в. 152,12 — алка-
лоид, бесцветные или желтоватые микроскопия, кри-
сталлы в форме чешуек или пластипок (из воды); при
темп-ре >-150° разлагается не плавясь, частично
сублимируется; растворимость в 100 мл воды: 0,008 г
873
КСЛНТИНОКСИДАЗА — КСАНТОГЕНАТ ЦЕЛЛЮЛОЗЫ
874
(17°), 0,018 г (40°); растворимость в 100 мл спирта
0,33 г (17°); растворяется в формамиде, горячем гли-
церине, легко растворяется вщелочах. УФ-спектр К.
в кислых р-рах (pH 2,0) характеризуется максимумом
поглощения при % = 267 ммк (молярный коэфф, по-
глощения е267 = 10,2 • 103 л/молъ см); в щелочных
р-рах (pH 10,0) К. обнаруживает два пика поглоще-
ния: при % = 240 ммк (е240 = 8,9 • 103) и % = 277 ммк
(г277 = 9,3-103). При УФ-облучении р-ры К. флуорес-
цируют.
К. обладает слабоосновными свойствами; рйГа1=0,8;
рА'а =7,44; рйГаз = 11,12; с холодной конц. HCI, HNO3,
НС1О4 образует кристаллич. легко гидролизующиеся
соли. При кипячении в разб. щелочах К. устойчив;
при кипячении с конц. НС1 и нагревании сухого про-
дукта выше 150° разлагается с выделением СО2 и
NH3. При окислении К. КМпО4 в кислой среде или
хлорной водой образуется аллоксан, на чем основано
качественное определение К. При бромировании К.
образуется 8-бром-К.; действием на К. диметилсуль-
фата при pH 4—7 получают теобромин (выход 70%),
а при pH 8—9 — кофеин (выход 90%). При действии
кони. HNO3 К. окрашивается в желтый цвет. Коли-
чественно К. определяют осаждением его серебряной
соли из водного р-ра добавлением аммиачного р-ра
AgNO3. Количественное определение К. в биологич.
материале основано на окислении К. в мочевую ки-
слоту с помощью ксантиноксидазы и манометрич.
определении количества поглощенного О2 или же
колориметрии, определении количества образовав-
шейся мочевой к-ты.
К. может быть получен из циануксусного эфира и
мочевины через 4,5-диаминоурацил, конденсируемый
далее с формамидом, или же восстановлением мочевой
к-ты формамидом. К. можно получить также дезами-
нированием гуанина или гидролизом нуклеиновых
к-т, выделенных из дрожжей.
К. применяют для синтетич. получения пуриновых
алкалоидов теобромина и кофеина. В природе К.
в свободном состоянии встречается в небольших коли-
чествах во многих животных и растительных тканях
и у микроорганизмов. К. — важное звено в процессе
обмена пуриновых соединений: образуясь в резуль-
тате окислительного дезаминирования гуанина под
действием гуаназы, а также в результате окисления
гипоксантина, К. окисляется далее с помощью ксан-
тиноксидазы в конечный продукт пуринового обмена —
мочевую к-ту. К. обладает сильным диуретич. дейст-
вием.
Лит.: Май оф ис Л. С., Технология химико-фарма-
цевтических препаратов, Л., 1958; Збарский Б. И.,
Иванов И. И., Мардашев С. Р., Биологическая хи-
мия, 3 изд., М., 1960, с. 360. В. В. Спиричев.
КСАНТИНОКСИДАЗА (альдегидоксидаза, фермент
Шардингера) — фермент, катализирующий окисление
гипоксантина в ксантин, и ксантина в мочевую ки-
слоту молекулярным кислородом; относится к группе
оксидаз. Наряду с гипоксантином и ксантином окис-
ляет нек-рые другие пуриновые основания, птерины,
а также многие алифатич. и ароматпч. альдегиды.
При окислении указанных субстратов К. может пере-
носить электроны и водород не только нд но и на
другие акцепторы, в частности на обратимо восста-
навливающиеся красители (напр., метиленовый си-
ний), нитрат, хиноны и цитохром С.
К. содержится гл. обр. в молоке и печени животных,
а также в нек-рых растительных тканях и играет
существенную роль в обмене пуриновых оснований,
завершая процесс их превращения в конечный про-
дукт обмена — мочевую к-ту. К. получена из молока
в кристаллич. состоянии, мол. в. 290000, изоэлектрич.
точка р/ 5,3—5,4; представляет собой металлофлаво-
протеид, т. е. белок, связанный с флавинадениндинук-
леотидом (ФАД) и ионом металла. С одной молеку-
лой К. связаны 2 молекулы ФАД и 2 иона молибдена,
к-рые образуют активный центр (простетическую
группу) фермента, осуществляющий перенос электро-
нов и водорода от субстрата на акцептор в катализи-
руемых К. реакциях. Кроме того, с молекулой К.
связано до 8 ионов железа, функция которых неясна.
1 моль К. катализирует окисление 660 молей ксан-
тина в минуту при 38°. Михаэлиса константа для
ксантина 3,0-10 5 М. Ингибиторами К. являются ксан-
топтерин и 2-амино-4-окси-6-птеридинальдегид. Циа-
ниды необратимо подавляют активность К. Опреде-
ление К. производят след, методами: восстановле-
нием нитрата с помощью К. в нитрит, определяемый
по Гриссу; колориметрически — по обесцвечиванию
красителя 'метиленового синего; манометрически —
измерением количества О2, поглощенного в процессе
реакции; спектрофотометрически — определением ко-
личества образующейся мочевой к-ты (фотометриро-
вание при 290 л<к).
Лит.: Михлин Д. М., Биохимия клеточного дыхания,
М., 1960; Диксон М., Уэбб Э.. Ферменты, пер. с англ.,
М., 1961; Biochemist’s handbook, ed. С. Long, L., 1961, p. 367.
В. Б. Спиричев.
КСАНТОГЕНАТ ЦЕЛЛЮЛОЗЫ — натриевая соль
сложного кислого эфира целлюлозы и тиоугольной
к-ты. К. ц. получают обработкой щелочной целлюлозы
жидким сероуглеродом или его парами:
Г /ОСвНоОГ
[СвН9О4ОН • NaOH] —-A. c=S -|-пНОН
_ ^SNa _п
Одновременно с основной реакцией (ксантогенирова-
нием) происходит также образование побочных при-
месей, в основном тритиокарбоната, соды и сульфида
натрия. В обычных условиях ксантогенирования (20—
30°, 32—40% сероуглерода от веса целлюлозы, про-
должительность 0,75—3,0 час) ок. 75% сероуглерода
расходуется на образование К. ц., а остальное коли-
чество — на побочные примеси. Ксантогенирование
щелочной целлюлозы имеет целью получение К. ц.,
растворимого в воде или в слабом (4%-ном) р-ре ед-
кого натра. Для получения водорастворимого К. ц.
в обычных условиях (степень полимеризации целлю-
лозы не выше 400—450) необходимо, чтобы К. ц. со-
держал в связанном виде не менее 25 тиоугольных
групп на 100 глюкозных остатков, т. е. гамма-число
К. ц. (число эфирных групп в целлюлозе на 100 глю-
козных остатков) не должно быть ниже 25. Обычно
получают К. ц. с гамма-числом 50—75. С повышением
темп-ры ксантогенирования ускоряется связывание
сероуглерода как в главной, так и в побочных реак-
циях; отношение полезно связанного сероуглерода
к общему его расходу, определяющее эффективность
ксантогенирования, зависит от степени отжима щелоч-
ной целлюлозы и от особенностей аппаратуры. К. ц.
получают в медленно вращающихся ксантат-бараба-
нах или в аппаратах месильного типа (аппараты ВА,
ксантат-мешалки и т. п.). В первом случае К. ц. полу-
чается в рыхлом, волокнистом виде и растворяется в
отдельном аппарате в 4%-ном р-ре едкого натра, обра-
зуя вискозу; во втором получается сильно набухший
тестообразный К. ц., к-рый в т°М же аппарате, смеши-
875
КСАНТОГЕНОВАЯ РЕАКЦИЯ — КСАНТОГЕНОВЫЕ КИСЛОТЫ
876
ваясь с водой и щелочью, переходит в вискозу. Цвет
К. ц. обычно оранжево-красный от присутствия сильно
окрашенных примесей три- и пертиокарбонатов Na2CS3
и Na2CS4. В чистом виде К- П- имеет светло-желтый
цвет. Ввиду взрывоопасности и ядовитости сероугле-
рода при получении К. ц. применяются специальные
меры предосторожности и усиленная вентиляция.
Как и другие соли кислых (неполных) эфиров тио-
угольной к-ты со спиртами (т. наз. ксантогеновых
кислот общей формулы R—О—-н + > гДе R —спир-
товый остаток), К. ц. неустойчив в водных р-рах и
подвергается гидролизу или омылению. При этом
К. ц. распадается на целлюлозу (спирт), сероуглерод
и едкий натр. Гидролизом К. ц. в воднощелочной среде
объясняется «созревание» вискозы.
В то время как натриевые и калиевые соли К. ц.
растворимы в воде (если гамма-число выше 25) и легко
подвергаются гидролизу, соли с двухвалентными ме-
таллами, напр. с Zn2+, мало растворимы и медленно
гидролизуются в воде и щелочи. Это объясняется обра-
зованием через Zn24' поперечных связей между макро-
молекулами целлюлозы и используется на практике,
особенно при формовании высокопрочных вискозных
волокон, когда в осадительпой ванне содержится
большое количество ZnSO4. Легкий гидролиз К. ц.
объясняется общим свойством всех кислых эфиров
спиртов с многоосиовными кислотами — гидролизо-
ваться в воде из-за присутствия в этих эфирах под-
вижного атома водорода. Средние эфиры тех же спир-
тов значительно более стойки. На этом основании
делаются попытки получения стойких К. ц. в виде
метильных, ацетильных и других производных. Стой-
кие К. ц. могут быть использованы для формования
волокон и пленок без разложения, т. е. без превраще-
ния К. ц. в гидратцеллюлозу. Однако практпч. затруд-
нения при получении стойких К. ц. до сих пор не
позволили использовать их для произ-ва волокон и др.
изделий из вискозы. К. ц. применяют при произ-ве
вискозного шелка, кордного и штапельного волокон
(см. Вискозные волокна), а также целлофана и других
иеволокнистых изделий (см. Вискозные неволокнистые
изделия) из вискозы, к-рая является 7—10%-ным
воднощелочным р-ром К. ц.
Лит.: Роговин 3. А., Основы химии и технологии
производства химических волокон, 2 изд., М., 1957,
с. 287—308. А. Б. Пакшвер.
КСАНТОГЕНОВАЯ РЕАКЦИЯ (Чугаева реак-
ция) — способ образования углерод-углеродной крат-
ной связи, заключающийся в термич. разложении эфи-
ров ксантогеновой к-ты:
II II
—С—С—О—С- SR---► -С=С - + COS + RSH
VI, 1
Наличие водородной связи между атомом водорода
в p-положении алкильной группы и одним из атомов
серы ксантогеновой группировки способствует обра-
зованию промежуточного 6-членного комплекса, к-рый
при нагревании распадается (гщс-элиминирование) с
образованием олефина, серонкиси углерода и алкил-
меркаптана:
ы. sz И Т S
_л+ ? _^ + RSH
с d~SR *
О f
Наиболее легко (уже в момент образования) происхо-
дит разложение ксантогенатов третичных спиртов и
несколько труднее — вторичных. Применение К. р.
к первичным спиртам ограничено.
Исходные спирты обычно превращают в ксантоге-
наты обработкой соответствующего алкоголята серо-
углеродом с последующим метилированием образовав-
шейся ксантогеновой соли иодистым метилом или
диметилсульфатом:
RJJ_ONa^R-tLo-C-SNa^M™^°ib
II I I II
Н Н S
I I
-->R—С-С-О-С-SCH,
i 1
Как правило, К. р. не сопровождается побочными про-
цессами, типичными для других способов дегидрата-
ции спиртов (изомеризация углеродного скелета, пере-
мещение кратной связи и др.). Эта особенность К. р.
позволила успешно применить метод разложения
ксантогенатов для дегидратации спиртов терпенового
ряда (превращение ментола в Д3-ментен; борнеола и
изоборнеола в борнилен и др.) и особенно для уста-
новления строения камфары.
Приложение К. р. к пинаколиновым спиртам, осо-
бенно склонным к изомеризациям с глубоким измене-
нием углеродного скелета (см. Пинаколиновая и ретро-
пинаколиновая перегруппировки), позволило перейти
(при нагревании) к mpem-алкилэтиленам:
r\C-Ch-ch3 нагреванис-цх>с-сн^сп8
R/ I R/
OCSSCHs
Значительный интерес представляет использование
К. р. для получения соединений с семициклич. крат-
ной связью:
>СНЭ
СН2ОС$ЗСНз • шгршш .
н
Аналогично разложение ксантогената фенилциклогек-
силкарбинола приводит к бензилиденциклогексану.
При термич. разложении ксантогенатов двухатомных
спиртов образуются ацетилен и его гомологи:
RCH СН2 R—СНСН
I I ----------к
HsCSSCO OCSSCHs R=H, CHS
Исходные бме-ксантогенаты были получены действием
ксантогената натрия на соответствующие дибромпды,
т. к. обычный метод получения ксантогеновых эфиров
в данном случае приводит к неудовлетворительным
результатам. К. р. была открыта Л. А. Чугаевым в
1899.
Лит.: Сысоева Н. Д., Усп. хим., 1951, 20, вып. 4,
498; Alexander Е. R., М u d г a k A., J. Amer. Chem.
Soc., 1951, 73, № 1, 59. Л. С. Герман.
КСАНТОГЕНОВЫЕ КИСЛОТЫ — кислые эфиры
RO—С—SH
дитиоугольной к-ты | , нестойкие масла,
в присутствии влаги разлагаются на сероуглерод и
соответствующий спирт. Соли К. к. вполне устойчивы;
ксантогенаты щелочных металлов хорошо раство-
ряются и кристаллизуются из воды, соли тяжелых
металлов плохо растворимы.
Соли К. к. легко образуются из алкоголятов и серо-
углерода:
CS2+C2H5ONa—► C»H:.OC(-S)SNa
В пром-сти их получают взаимодействием сероугле-
рода, безводной щелочи и соответствующего спирта.
Наибольшее значение из К. к. имеют ксантогенаты
целлюлозы, к-рые используются при получении вис-
козного волокна. Соли амил-, бепзил- и нек-рых
других К. к. употребляются в качестве коллекторов
при флотации сульфидных руд. К. к. используются
как ускорители вулканизации каучука, при произ-
водстве инсектицидов и для аналитич. определения
молибдена. Эфиры К. к. (полные эфиры дитиоуголь-
пой к-ты) могут быть получены из солей К. к. и гало-
геналкилов:
C2HdOC(=S)SK4 C2H5J—> CiHiOC^SlSCaHs+KJ
877
КСАНТОН —КСЕНОН
878
При пагревании эти эфиры разлагаются, напр.:
C5H11OC(=S)SC2H5----------- C2H5SH+COS+-C5H10
Аммиак и амины взаимодействуют с эфирами К. к.
с образованием амидов К. к.:
C2H5OC(=S)SC2H5+RNH2 —- C2H3OC(=S)NHR 4 C2H3SH
Ариламиды К. к. получают из тиоангидридов К. к. и
аминов:
(C2H5OCS)2S4-CeH5NH2—-
—>C2H5OC(=S)NHCeH5 +cs2 + С2Н5ОН
Тиоангидриды К. к. получают действием фосгена на
ксантогенаты натрия:
ROC(=S)SNa + СОС12 —- (ROCS)2S + COS + 2NaCl
Соли К. к. реагируют с ароматич. диазосоедине-
ниями, образуя эфиры К. к.:
ArN2Cl+KSC(^S)OC2H3---► ArS-C(=S)-OC2H5 + N2 + КС!
Эту реакцию применяют для синтеза ароматич. тио-
фенолов:
CeH5S-C(=S)OC2H5 —-®-2. CeHjSH
К. к.— сильные восстановители; они легко окисляются
до диксантогенатов:
ROC(=S)SK4-K2S2O8 —. [ROC(=S)]2S2 + 2K2SO4
ROC(—S)SH + CeH5SO2Cl —- (ROCS)2 + C6H5SO2K + HCl
Этилксантогеновая к-та C2H5OC(=S)SH,
наз. также ксантогеповой к-той, — нестабильная жид-
кость, разлагается при 25° на сероуглерод и спирт;
метиловый эфир, т. кип. 182—183°; этиловый эфир,
т. кип. 199—200°; ангидрид, т. пл. 55°.
Н. А. Несмеянов.
КСАНТОН (дибензо-у-пирон) С13Н8О2, мол. вес
196,19 — кристаллы; т. пл. 174°; т. кип. 351°; немно-
го растворим в горячей воде, в холодном спирте
(0,55 г/100 мл), эфире и бензоле, растворим в горячем
спирте (6,71 г/100 мл).
К. (I) обладает свойствами ароматич. соединений и
не вступает в большинство реакций, характерных для
л ли карбонильной группы.
V Jr С SOCL, или (СОС1)2
образует 9,9-дихлор-
K^sJL ксантен, а с P2S5 —
0 ксантион. Щелочные
1 " восстановители(напр.,
Zn _|_ NaOH) превра-
riljlj Г II 1 J щают К. в ксантигид-
рОЛ (jj), н0 Na +
ш + C2II5OH восстанав-
I JI JI J ливает К. в ксантен
ч/ч/ч (III), а с кислыми вос-
” становителями (напр.,
Zn + HCl) К. дает диксантилен (IV). При ката-
литич. восстановлении К. переходит в ксантен, а
в более жестких условиях происходят восстановле-
ние бензольных ядер и расщепление молекулы.
С магнийорганич. соединениями К. образует 9-заме-
щенные ксангидролы. К. — весьма слабое основание,
его продукты присоединения к А1Вг3, SnCl4 и НС1О4
легко гидролизуются. При нитровании К. дает смесь
2,4-, 2,7- и 2,8-динитроксантонов, при галогенирова-
нии образуются 2,7-производные. Аминирование
(с NH2OH) и амидометилирование (с HOCH2NHCOR)
идут в положение 2. К. термически вполне устойчив
и только при 860° разлагается с образованием дибен-
зофурана. Получают К. пиролизом салициловой к-ты
или ее производных, а также из о, о’-диоксибензофено-
нов. К. обладает инсектицидным действием.
Лит.: Вавзонек С., в кн.: Гетероциклические со-
единения, под ред. Р. Элдерфилда, пер. с англ., т. 2, М.,
1954, с. 320. А. Е. Васильев.
КСАНТОПРОТЕИНОВАЯ РЕАКЦИЯ (Мульдера
реакция) — цветная качественная реакция на белки;
заключается в появлении желтой окраски при воз-
действии на белок азотной к-ты. К. р. обусловлена
образованием нитросоединеиий ароматич. и нек-рыми
гетероциклич. (триптофан) группами белка. При про-
ведении К. р. к р-ру белка прибавляют конц. азотную
к-ту до тех пор, пока не прекратится образование
осадка, затем нагревают. При нагревании осадок
окрашивается в желтый цвет и растворяется, сообщая
жидкости желтую окраску. При добавлении к охла-
жденной жидкости избытка аммиака или едкой щелочи
появляется оранжевое окрашивание, обусловленное
образованием солей нитроновых к-т:
nh2
civ- сн - соон
nh2
сн,- сн-соон
+ нго
Не дают этой реакции клупеин и сальмин. Окрашива-
ние кожи в желтый цвет при попадании на нее HNO3
обусловлено К. р. С. И. Пехтерева.
КСАНТОПТЕРИН (уроптерин, 2-амино-4,6-диок-
сиптеридин) C6H5O2N5, мол. в. 179,14 — природный
желтый пигмент, относящийся к н
классу птеринов. 9
К. — кристаллич. продукт, т. пл. HNj'fZZs'yO
выше 410®; практически нерастворим JliJL J
в воде, растворяется в разб. NaOH нгг'|'т>Гч5г
и NH4OH с сильной флюоресценцией.
При действии на К. водорода образуется лейкоптерин.
К. выделен из крыльев бабочек Gonopterix rahmi
(лимонница), содержится в наружных покровах ос
и шершней, обнаружен также в моче млекопитающих
и человека. Синтетически К. получают конденсацией
2, 4,5-триамипо-6-оксипиримидина с глиоксиловой
к-той или с дихлоруксуспой к-той, с последующей
циклизацией промежуточно образующегося 2,4-ди-
амино- 5-дихлорацетиламино-6-оксипиримидина. К. яв-
ляется витамином для рыб; обладает витаминной ак-
тивностью, во многом напоминающей таковую у фо-
лиевой к-ты (стимулирует процесс кроветворения
у млекопитающих, улучшает лактацию у мышей и
крыс и др.).
Лит.: С о б ч у к В., Синтез ксантоптерина и его побоч-
ных продуктов и их влияние на экспериментальные опухоли
животных, Киев, 1959 (Диес.); Albert A., Reich F.,
J. Chem. Soc., 1960, [№ 3], р. 1370; Ciba foundation symposium
on the chemistry and biology of pteridines. Ed. G. E. W. Wol-
stenholme, M. P. Cameron, L., 1954.
E. В. Будницкая-Павлова.
КСАНТОФИЛЛЫ — см. Каротиноиды.
КСЕНОН (Xenon) Хе — химич. элемент нулевой
гр. периодич. системы Менделеева, инертный газ;
п. п. 54, ат. в. 131,30. Природный К. состоит из смеси
9 стабильных изотопов, из которых наиболее рас-
пространены Хе129 (26,44%), Хе1®1 (21,18%), Хе132
(26,89%). Приведенные в скобках числа относятся
к образцу К. атмосферного происхождения. Изотоп-
ный состав К., выделенного из урановых минералов,
иной, т. к. нек-рые изотопы К. образуются при деле-
нии U238 и U235. Поперечное сечение поглощения теп-
ловых нейтронов атомом К. 35 барн. В ядерном реак-
торе при делении U235 образуются изотопы К., из
к-рых радиоактивный Хе1®3 является сильнейшим по-
глотителем тепловых нейтронов. Конфигурация внеш-
них электронов атома 5s25p6. Энергии ионизации
(в эв): Хе° —> Хе+ —»• Хе2+ —» Хе3+ —► Хе1+ соответст-
венно равны 12,13; 21,21; 32,13; 46,62. К. состоит
из одноатомных молекул. Ат. радиус 2,18 А. Плот-
ность газа 5,85 1 г/л (при 0° и 760 мм рт. ст.); т. пл.
—111,8° и т. кип. —108,1 (760 мм рт. ст.). Тепло-
та плавления 548,5 кал/.иоль. Теплота испарения
879
КСЕРОГЕЛИ - КСИЛИТ
880
3020 кал/молъ (в точке кипения). Плотность жидкого
К. 2,987 г/см3 (в точке кипения), твердого 2,7
(-140°). «крит 16,59°; Ркрит 60,16 кг/см* и
^крит. 1>155 кг/л. Тройная точка: 161,36° К.,
611 мм рт. ст., плотность в тройной точке
с?тв 3,540 г/см3, а?ж 3,076 г/см3.
Как и другие инертные газы, К. не вступает
в обычные химич. реакции. Получены гидрат
Хе - 6Н2О и нек-рые др. соединения, в к-рых
связь осуществляется межмолекулярными си-
лами (подробнее ем. Инертные газы). В пром-сти
К. получают из .воздуха (см. Воздуха разделе-
ние). К. используют в электровакуумной тех-
нике и газосветных лампах.
Лит. см. при ст. Инертные газы.
КСЕРОГЕЛИ -- коллоидно-пористые твердые тела,
образующиеся при обезвоживании (высушивании)
гелей или студней. В обоих случаях К. являются
двухфазными дисперсными системами. К., получаю-
щиеся из студней эластомеров, могут обладать доста-
точно высокой эластичностью.
КСЕРОФОРМ (висмутовая соль 2, 4, 6-трибромфе-
нола) (C6H2Br3O)2 BiOH • Bi2O3, мол. в. 1353,67 —
желтый порошок; нерастворим в воде и спирте; в виде
присыпок и мазей применяется как антисептик, сред-
ство; входит, в частности, в состав мази Вишневского.
КСИЛЕНОЛ (СН3)2 С6Н3ОН, мол. в. 122,17 —
известны все 6 изомеров К. В зависимости от положе-
ния метилытых групп различают орто-, мета- и пара-
изомеры.
Изомер Т. пл., °C Т. кип., °C Теплота сгорания, ккал/моль
Орто- рядовой (виц), 2,3-диме- тилфенол 75 218
несимм. (асимм.), 3,4-ди- метилфенол а 62,5 225/757 1085,4
Мета- рядовой (виц), 2,6-диме- тилфенол 49 203
несимм. (асимм.), 2,4-ди- метилфенол 0 27-28 211,5/776 ММ
симм., 3,5-диметилфенол 68 97—98/14 219,5 мм 1037
Пара- 1,4-диметилфенол в . . . 74,5 211,5/762 мм 1035,6
а Константа диссоциации в воде 5,2 • 10 11 (25°).
6 dj4 1,0276. 1,5420.
в Константа диссоциации в воде 4,8 10-11 (25°).
К. растворимы в обычных органич. растворителях,
мало растворимы в воде, растворяются в водных р-рах
едких щелочей с образованием солей, на воздухе мед-
ленно окисляются.
Основным источником промышленного получения К.
являются крезольные (фенольные) фракции смол,
образующихся при термич. обработке топлив. Обычно
К. не разделяют на изомеры, а используют технич.
продукт, содержащий их смесь, а также значительные
количества крезолов. Синтетич. способы получения
(гидролиз ксилолсульфокислот, диазотирование кси-
лидинов) пе имеют практич. значения. Основным по-
требителем К. является произ-во феноло-альдегидпых
смол, где К. применяют в качестве заменителей фено-
лов, однако такие смолы обладают меньшей скоростью
отверждения и пониженной механич. прочностью.
Нек-рое количество К. расходуется на изготовление
дезинфицирующих и моющих средств.
В. Н. Александров.
КСИЛИДИНЫ (диметилапилины, аминоксилолы
(CH3)2C6H?NH?, мол. р. 121,11 — твердые продукты
или жидкости с запахом анилина, бесцветные, быстро
темнеют на воздухе; известны все 6 изомеров.
г/см3
Положе- ние групп Название Т. кип., °C
СНз nh2
1,2 3 Рядовой о-К. 221 0,0910 в (15°) 1,5706 (15,3°)
1.2 4 Несимм. о-К. а 226 1,0755 (17,5°) —
1.3 9 Рядовой м-К. 214/739 мм 0,9796 (20°) 1,5612 (20°)
1.3 4 Несимм. .и-К. 216/728 мм 0,9783 (19,6°) 1,5607 (20°)
1,3 5 Симм. м-К. 220 0,9935 (0°) 1,5618 (12,1°)
1,4 2 п-К. 6 213,4 0,9790 (21,3°) 1,5593 (20°)
а Т. пл. 51°. 6 Т. пл. 15,5°. Е d}%
К. хорошо растворимы в спирте, эфире, ацетоне, хло-
роформе, бензоле; ограниченно — в поде; горят и
образуют взрывоопасные паровоздушные смеси (т. всп.
несимм. о-К. 97°); ядовиты, вызывают поражение пе-
чени и являются ядами крови; предельно-допустимая
концентрация в воздухе производственных помещений
0,005 мг/л. К. — органич. основания (по степени ос-
новности близки к анилину); легко образуют соли
с кислотами (минеральными и карбоновыми) и алкпл-
галогенидами (кроме рядового ..u-К.). С ангидридами
и галогенапгидридами кислот образуют соответствую-
щие амиды кислот. При действии азотистой к-ты обра-
зуют диазогидраты и соли диазония.
Получают К. нитрованием ксилолов и последующим
восстановлением нитросоединений; технич. продукт
состоит в основном из 40—60% симм. ж-К. п 10—20%
n-К.; разделение изомеров производят фракционной
кристаллизацией их солей (хлоргидрата, ацетата).
Аналогично несимм. о-К. получают из о-ксилола,
а «-К. — из /г-ксилола. Симм. as-K. получают нитро-
ванием несимм. а«-К., дезаминированием и последую-
щим восстановлением.
К. используют как высокооктановые добавки к авиа-
ционным бензинам, как ускорители вулканизации
каучука, как антиоксиданты (в частности, для про-
питки деревянных изделий), как вспенивающие сред-
ства при флотации руд. Индивидуальные К. при-
меняют в качестве исходных веществ при произ-ве
азокрасителей. Г. Сокольский.
ксилил — см. Тринитроксилол.
КСИЛИТ, мол. в. 152,15 — пятиатомный спирт;
бесцветные гигроскопич. кристаллы, псевдооктаэдры,
СН.ОН известны в двух кристаллических фор-
I “ мах: метастабилыюй (моноклинной синго-
НСОН нии), т. пл. 61—61,5°, и стабильной (ром-
НОСН бич. сингонии), т. пл. 93—94,5°. К. опти-
I чески неактивен, растворим в воде, метило-
НСОН воми этиловом спиртах, гликолях, пириди-
сн.>он не> уксусной к-те; нерастворим в эфирах,
диоксане, хлороформе, углеводородах, бу-
тиловом и пропиловом спиртах; обладает пластифици-
рующими свойствами. Наличие пяти гидроксильных
групп обусловливает большую реакционную способ-
ность К. Окислением могут быть получены ксилоза и
ксилооксикарбоновые к-ты. При дегидратации К. при
темп-ре выше 100° в присутствии дегидратирующих
катализаторов образуются сто моно- или диангидриды.
В условиях каталитич. гидрирования К. выше 150° и
при избыточном давлении водорода происходит его
гидрогенолиз с разрывом связи С—С преим. между
2-м и 3-м и, в незначительной степени, 1-м и 2-м ато-
мами углерода с образованием соответственно этилен-
гликоля и глицерина (1) и метанола и эритрита (2):
СН.,ОН(СНОН)3СНгОН—-
---------------CH2OHCH»OH-|-CH„OHCHOUCII2OH (1)
СН2ОН(СНОН)3СН2ОН--->СН8ОН-|-СН2ОП(СПОН)2СН4ОН(2)
К- обладает молодящим с,падким вкусом.
881
КСИЛОЗА —КСИЛОЛ
882
Для качественного и количественного определения
пользуются хроматографией в ее различных модифи-
кациях, а также применяются методы, основываю-
щиеся на способности К. к образованию в определен-
ных условиях медного или ванадиево-оксихиноли-
вового комплекса.
К. образуется при восстановлении ксилозы (промышлен-
ный метод). Сырьем для произ-ва К. могут служить пенто-
зансодержащие растительные отходы с. х-ва (кукурузная ко-
черыжка, хлопковая шелуха, подсолнечная лузга и др-),
а также древесшга лиственных пород. Произ-во К. состоит из
в одно-кислотного облагораживания сырья, его пентозного
гидролиза (при 100—120° в присутствии разб. серной к-ты),
нейтрализации и очистки гидролизата при помощи ионо-
обменных смол и активного угля, упаривания, гидрирования
15—20%-ного водного р-ра ксилозы при 115—120° и давле-
нии водорода 50—150 ат в присутствии никелевых катали-
заторов, адсорбционной очистки раствора К., его упарива-
ния и выделения кристаллич. продукта.
К. и его ангидрид широко применяют как стабили-
заторы влажности и пластификаторы (бумажная
пром-сть, парфюмерия, произ-во целлофана и т. д.).
К. является одним из основных компонентов в полу-
чении ксифталевых олиф, поверхностно-активных
веществ, неионогенпых детергентов, лаков, клеев и др.
Лит..' Соболева Г. Д., в сб.: Химическая перера-
ботка древесины. Научно-техн, сборник, №3, М., 1962
(ЦИ11ТИ бумажн. и деревообр. пром-сти); Ч е и и г о С. В.,
Химическая переработка растительных материалов методом
гидролиза, М., 1960; Адаскин Е. М., Заманская
Р. И., Гидролизная и лесохим. пром-сть, 1960, № 1; Як о-
в е н к о Г. 3. [и др.], там же, 1962, № 2. С. В. Чепиго.
КСИЛОЗА С6Н!0О6, молекулярный вес 150,13 —
одна из четырех изомерных альдопентоз. К. существу-
ет в D- и L-формах,
каждая из к-рых об-
разует открытую аль-
дегидную и несколь-
ко циклических тауто-
мерных форм. В кри-
сталлич. виде получе-
ныа-D-K., т. пл. 145°,
[a]D = +93,6°; p-L-K ,
т. пл. 143°, [alD =
= —79,3° и D, L-K.,
гворах вследствие му-
таротации из a-D-K. образуется равновесная смесь
таутомеров с [a]D = +18,8°, а из 0-L-K. смесь с
[a]D = —18,8°. В природе встречается D-К. (древес-
ный сахар), она входит в состав полисахаридов к си-
лан о в, к-рые сопровождают в растениях целлюлозу
и содержатся в гемицеллюлозной фракции, различ-
ных гликозидов (примвероза) и олигосахаридов; в
свободном состоянии К. встречается в растениях в
небольших количествах.
К. растворима в воде, нерастворима в эфире и
большинстве органич. растворителей. К. дает все
общие реакции на альдозы: восстанавливает раствор
Фелинга, аммиачный раствор AgNO3, образует оза-
зон, п-бромфенилозазон, фенилозазон. Фенилозазон К.
идентичен фенилозазону ликсозы и ксилулозы. К.
дает цветные реакции, характерные для пентоз, ре-
акцию с орсином (реакция Биаля) и др. При действии
на К. разб. к-т из нее образуется фурфурол (фур-
фураль). При окислении альдегидной группы в D-K.
образуется D-к силоновая к-та; из L-ксилозы
получается L-к силоновая к-та. Плохо раст-
воримая в воде двойная соль ксилоната кадмия
и бромида кадмия (C5HeO6)2Cd • CdBr2 2Н2О слу-
жит для идентификации К. При более глубоком
окислении D-К. и L-К. образуется оптически неак-
тивная двухосновная триоксиглутаровая
к-та. При восстановлении D-К. и L-К. образуется
оптически неактивный спирт ксилит.. Обыкновенными
дрожжами К. не сбраживается, сбраживается некото-
рыми бактериями и специальными расами дрожжей.
Продуктами различных типов брожения К. являются
И ОН
II
СНС н
н-с-он
но-с-н
н-с-он
I
СН2ОН
I
I — альдегидо-Б-ксилоза;
II — a-D-ксилопироноза.
т. пл. 129—131°. В водных
молочная, уксусная, лимонная, янтарная и др. кис-
лоты, спирт и др. продукты. Человеком К. усваи-
вается плохо. Введение К. в пищу животным вызывает
у них катаракту.
D-К. производят из древесины и отходов с. х-ва
(лузга подсолнуха, кукурузные кочерыжки, солома)
гидролизом содержащихся в них ксиланов. Применяют
К. для получения фурфурола и триоксиглутаровой
к-ты, которую используют как заменитель лимонной
к-ты в пищевой пром-сти. Синтетически D- и L-K.
получены соответственно из D- и L-гулоновых к-т
и др. способами.
Лит.; The carbohydrates. Chemistry, biochemistry, phy-
siology, ed. W. Pigman, N. Y., 1957. Л. И. Линевич.
КСИЛОКАИН (ксикаин, хлоргидрат диэтиламипо-2,
6-диметилацетанилида) CI4H23ON2C1, мол. в. 270,8 —
местный анестетик; бе-
лый или слегка желто- ЬпСОСНгК(С2Нз)2- НС1
ватый порошок горького н3С -fS-CHa
вкуса, т. пл. 128—129°. II \
К. хорошо растворим
в воде, спирте и ацетоне, нерастворим в эфире;
гигроскопичен, при длительном стоянии на свету
желтеет. К. весьма устойчив вследствие экрани-
рующего эффекта метильных групп, выдерживает
многочасовое кипячение с 30%-ной соляной к-той
или нагревание со щелочью. Основание К. — твер-
дое вещество, т. пл. 65—67°, т. кип. 183°/3 мм, нерас-
творимо в воде, растворимо в спирте и хлороформе.
К. получают ацилированием 2,6-ксилидина хлор-
ацетилхлоридом и обработкой продукта реакции ди-
этиламином. Образующееся основание К. переводят
в К. действием сухого хлористого водорода в спирте
или эфире:
nh2 nhcoch2ci
Н3С-Т|Ч-СН3— HaC-f^VCHa —-
NHCOCH2N(C2H5)2
К. — наиболее устойчивый местный анестетик; он
в 1,5—2 раза эффективнее новокаина, несколько
более токсичен, действует быстрее и более продол-
жительно (в 2—3 раза), не уменьшает антибактериаль-
ного действия сульфаниламидов. Токсичность К. по-
нижается в разб. р-рах (0,5%), а также в присутствии
хлоргидрата адреналина, к-рый замедляет всасывание
и усиливает действие К. Применяется К. для поверх-
ностной и инфильтрационной анестезии, в последнем
случае — в виде 0,25—0,5%-ного р-ра.
Лит.: Машковский М. Д., Лекарственные сред-
ства, 4 изд., М., 1960; BudJSin sky Z., Protiva М.,
Synthetlsche Arzneimittel, В., 1961; Кудряшова Н. И. и
Хромо в-В о р и с о в Н. В., «Мед. пром-сть СССР», 1959,
М 7, с. 32; Жу рав л ев С. В. и Н и к о л а е в Е. В.,
Ж. общ. химии, 1960, 30, вып. 4, 1155. А. Е. Васильев.
КСИЛОЛ (диметилбензол) (СН3)2С6Н4, мол, вес
106,16 — бесцветная жидкость с ароматичным запа-
хом; существует в виде трех изомеров: орто-, мета-
ть пара-ксилола.
. Все изомеры К. смешиваются со спиртом, эфиром,
ацетоном, хлороформом, бензолом; плохо растворя-
ются в воде (менее 0,015%); горят и образуют взрыво-
опасные паро-воздушные смеси в интервале 3,0 —
7,6%; темп-ра самовоспламенения 553°; т. всп. 29°;
ядовиты: при контакте с кожей вызывают дерматиты,
при ингаляции — острые и хронич. поражения крове-
творных органов (костного мозга и селезенки) и почек;
предельно допустимая концентрация 0,1 мг/л.
К. — типичные ароматич. соединения: легко алки-
лируются, хлорируются, сульфируются и нитруются;
883
КУБОВЫЕ КРАСИТЕЛИ —КУБОВЫЕ КРАСИТЕЛИ РАСТВОРИМЫЕ
884
Свойства Изомеры
орто мета пара
Т. пл., °C -25,18 —47,87 +13,26
I. кип., °C 144.41 139,10 138,35
0,8802 0,8642 0,8611
nD 1.5054 1.4972 1,4958
С?1р(13 0.809 0.617 0,644
t Q(' ‘Крит, 359,0 346,0 345,0
Ркрит, ат 36 35 34
Диэлектрич. проницаемость (20°) 2,26 9 ‘V< 2,23
Теплота парообразования, кал/г . 82,9 82,6 81,2
Теплота образования (25°),
ккал/моль -5,841 - 6,075 -5,838
Энтропия (25°), ккал/град - моль . 58,91 60,27 59,12
Максимум поглощения света:
ИК, мк 13,48 13,00 12,59
УФ, ммк 270,7 272,5 274,3
относительная легкость реакций замещения характе-
ризуется рядом: м-К. > о-К. > га-К. Частичное окис-
ление К. (азотной к-той, хромовым ангидридом, пер-
манганатом калия) приводит к соответствующим
толуиловым кислотам; при более глубоком окислении
(катализ над V2O5) образуются соответственно фтале-
вый ангидрид, изофталевая и терефталевая к-ты;
озонирование и обработка хромовой смесью приводят
к разрушению бензольного ядра.
К. (совместно с этилбензолом) выделяют из «легкого
бензола», образующегося при коксовании угля или
ароматизации нефти; при термич. обработке смеси
К. содержание n-К. повышается до 20%. Разделение
изомеров производят фракционной кристаллизацией
(га-К. образует с м-К. эвтектич. смесь состава п : м =
= 12,9 : 87,1%, т. пл.—52,9°); п-К. кристаллизуется
совместно с СС14 (т. пл. —3,9°) и последующей фрак-
ционной ректификацией (выделение о-изомера). Изо-
меры можно разделить также селективным сульфи-
рованием и последующим гидролизом; с л:-К. про-
цессы протекают при более низких темп-рах.
К. — растворитель лаков, красок, мастик, фарма-
цевтич. препаратов, а также высокооктановая добавка
к авиационным бензинам. Индивидуальные К. и их
смеси используют для получения ксилидинов (нитро-
ванием и последующим восстановлением), фталевых
к-т (окислением) и толуола (обработкой хлористым
водородом и треххлористым алюминием при высокой
темп-ре). Наибольшее практич. значение имеет п-К. —
исходное в-во для произ-ва терефталевой к-ты.
Лит.: Ворожцов Н. Н., Основы синтеза промежу-
точных продуктов и красителей, 4 изд., М., 1956.
Г. А. Сокольский.
КУБОВЫЕ КРАСИТЕЛИ — группа красителей,
нерастворимых в воде, к-рые восстановлением в ще-
лочной среде гидросульфитом переводят в раствори-
мое состояние (т. наз. лейкосоединение). Образовав-
шуюся натриевую соль лейкосоединения после нане-
сения на ткань окисляют кислородом воздуха и пере-
водят в нерастворимое состояние; см. Антрахиноно-
вые кубовые красители, Индигоидные красители,
Кубовые красители растворимые, Полициклические
кубовые красители, Тиоиндигоидные красители.
КУБОВЫЕ КРАСИТЕЛИ РАСТВОРИМЫЕ — рас-
творимые в воде соли кислых сернокислых эфиров
лейкосоединений кубовых красителей. Для К. к. р.
характерно наличие парных карбонильных групп,
соединенных системой сопряженных связей, способ-
пых восстанавливаться в щелочном р-ре гидросуль-
фита натрия с образованием растворимого в воде
продукта, легко и нацело окисляющегося кислородом
воздуха в исходный краситель (см. Антрахиноновые
кубовые красители, Полициклические кубовые кра-
сители, Индигоидные красители, Тиоиндигоидные кра-
сители). Щелочной р-р продукта восстановления ку-
бового красителя, используемый для крашения тек-
стильных материалов, наз. кубом, а продукт восста-
новления с незамещенными гидроксильными группа-
ми — лейкосоединепием. С целью облегчения текстиль-
ной практики предпринимались многочисленные по-
пытки получения стабилизированных восстановлен-
ных форм кубовых красителей, непосредственно рас-
творимых в воде или водной щелочи. Иностранные
фирмы выпускают под названием «Индиго куб 60%
и 80% » упаренный в вакууме, содержащий глюкозу
раствор мопонатриевой соли лейкоиндпго в форме
плотных зереп, только очень медленно окисляющегося
на воздухе. Под названием «Индиго белое» поступает
в продажу 50%-пая водная паста лейкосоединения
индиго, также сравнительно устойчивая к окисле-
нию воздухом. К. к. р. выпускают в СССР под назва-
нием индигозолей (для производных индиго и тиоин-
диго) или кубозолей (для производных ПОЛИЦИКЛИЧ.
кубовых красителей). Эти растворимые в воде веще-
ства, устойчивые в щелочной среде, при действии
кислоты и окислителей (обычно нитрита натрия или
хромпика) количественно выделяют соответствующий
кубовый краситель. «Проявление» кубового красителя
на текстильных изделиях, окрашенных кубозолями
или индигозолями, при действии кислоты и окисли-
теля не является последовательным гидролизом сер-
нокислого эфира и окислением лейкосоединения, по-
скольку скорость «проявления» во много раз превы-
шает скорость кислотного гидролиза.
Первоначально для получения индигозолей и кубозолей
краситель восстанавливали гидросульфитом в среде водной
щелочи: из образовавшегося куба подкислением выделяли
свободные лейкосоединения, к-рые после азеотропной сушки
в хлорбензоле этерифицировали продуктом присоединения
серного ангидрида к диметиланилину, полученному взаимо-
действием последнего с хлорсульфоновой к-той. После доба-
вления соды или Поташа диметиланилин отгоняли с водяным
паром и из раствора с добавкой соответствующих солей оса-
ждали щелочную соль кислого сернокислого эфира лейко-
соединения:
OSO3C6HsN(CH3)2 OSO3Na
6sO3C6H5N(CH3)2 OSO3Na
Технология получения индигозолей и кубозолей может
быть упрощена, если краситель восстанавливать в безводной
среде, напр. водородом в присутствии катализатора, но и
при этом в ряде случаев не удается получить удовлетворитель-
ных результатов.
Лучшие результаты дает общепринятый в настоящее
время метод одновременного восстановления и этери-
фикации кубовых красителей действием порошков
металлов (железа, меди, цинка) и продукта присоеди-
нения серного ангидрида к пиридину. В этом случае
есть основание считать, что промежуточными про-
дуктами образования сернокислых эфиров служат не
лейкосоединения, а комплексы кубового красителя,
металла и этерифицирующего агента; более актив-
ными этерифнпирующими агентами, видимо, являются
продукты присоединения серного ангидрида не к тре-
885
КУБОВЫЕ КРАСИТЕЛИ РАСТВОРИМЫЕ — КУЛОНОМЕТРИЯ
886
тичным аминам, а к диалкиламидам, напр. к диметил-
формамиду.
В отличие от других «Кубозоль голубой К.», являю-
щийся одной из основных марок кубозолей, не гото-
вят из соответствующего красителя, а получают из
соли кислого сернокислого эфира 2-амино-З-хлорап-
трагидрохинопа окислением двуокисью свинца в ще-
лочной среде:
Кубозоли и индигозоли выпускают в виде натрие-
вых или калиевых солей, обычно содержащих кристал-
лизационную воду. Труднорастворимые сернокислые
эфиры лейкосоединений дибромантрона и дихлоризо-
виолантрона готовят в виде солей с органич. основа-
ниями, полученными оксиэтилированием триэтанол-
амина. Выделение кислого сульфата при разложении
кубозолей и индигозолей определяет автокаталитич.
характер этого процесса. Для повышения стабиль-
ности при хранении в технич. продукт всегда добав-
ляют вещества основного характера: соду, мочевину
и т. п. Продажные продукты содержат также вещества,
защищающие от переокисления в процессе проявления
кубового красителя на нолокне, чаще всего N, N-ди-
метилсульфаниловую к-ту. Все индигозоли и нек-рые
кубозоли при действии света распадаются с выделе-
нием кубового красителя, причем скорость фотохимия,
процесса мало зависит от pH среды. Светочувствитель-
ность индигозолей предлагалось использовать для
светокопирования изображений на ткани.
Названия кубовых красителей и соответствующих
им солей сернокислых эфиров лейкосоединений при-
ведены ниже.
Кубовый
краситель
Кубовый золотисто-желтый
ЖХ
Кубовый золотисто-желтый
КХ
Тиоиндиго оранжевый КХ
Кубовый ярко-оранжевый
КХ
Тиоиндиго ярко-розовый Ж
Тиоиндиго красно-коричне-
вый Ж
Кубовый коричневый СК
Тиоиндиго красно-фиолето-
вый С
Кубовый ярко-фиолетовый К
Броминдиго
Кубовый голубой К
Кубовый ярко-зеленый С
Кубовый ярко-зеленый Ж
Кубовый темно-зеленый Ж
Тиоиндиго черный
Сернокислый
эфир
Кубозоль золотисто-желтый
ЖХ
Кубозоль золотисто-желтый
КХ
Индигозоль ярко-оранже-
вый К
Кубозоль ярко-оранжевый
КХ
Индигозоль ярко-розовый Ж
Индигозоль красно-коричне-
вый Ж
Кубозоль коричневый СК
Индигозоль красно-фиолето-
вый С
Кубозоль ярко-фиолетовый
К
Броминдигозоль
Кубозоль голубой К
Кубозоль ярко-зеленый С
Кубозоль ярко-зеленый Ж
Кубозоль темно-зеленый Ж
Индигозоль серый С
Прочность окрасок кубозолями и индигозолями
определяется свойствами взятого для их изготовления
кубового красителя. Как правило, при высокой проч-
ности к мокрым обработкам индигозоли показывают
хорошую и удовлетворительную, а кубозоли •— отлич-
ную и хорошую прочность к свету, стирке и погоде.
Кубозоли и индигозоли пригодны для крашения и
печати по тканям из растительных и животных во-
локон, регенерированной целлюлозы и нек-рых видов
синтетич. волокон. Если раньше они применялись в
основном для печати, то в последнее время особенно
широко используются для гладкого крашения хлоп-
чатобумажной ткани в светлые тона. В этом случае
легче, чем при применении соответствующих кубовых
красителей, достигается равномерная окраска; при от-
носительно малом расходе красителя более высокая
стоимость кубозолей не имеет существенного значения.
Лит.: Коган И. М., Химия красителей, 3 изд., М.,
1956; Венкатараман К., Химия синтетических кра-
сителей, пер. с англ., т. 2, Л., 1957. II. С. Докунихин.
КУКУРУЗНОЕ МАСЛО —см. Жиры растительные.
КУЛОМЕТР (вольтаметр) — прибор для опреде-
ления количества электричества Q но количеству
вещества т, выделившегося в ходе электролиза на
одном из электродов (иногда и на обоих). Величину Q
находят из соотношения Q = т/е, являющегося вы-
ражением законов Фарадея, где е — электрохимия,
эквивалент выделившегося вещества (см. Фарадея
законы). К. представляет собой электролитич. ячейку,
включаемую последовательно в цепь постоянного
тока. В качестве К. используются такие ячейки, в
к-рых для измеряемого вещества выход по току ра-
вен 100%. Последнее достигается подбором подходя-
щих электродов и электролита, высокой чистотой
применяемого электролита, сохранением в ходе элек-
тролиза определенных значений плотности тока,
pH р-ра и темп-ры. Количество выделившегося ве-
щества определяется взвешиванием (напр., серебро,
выделившееся на катоде из р-ра AgNO3 в серебряном
К.) либо титрованием р-ра (иод, выделившийся иа
аноде из р-ра KJ иодного К.), или же измерением
объема выделившихся газов (водорода на никелевом
катоде и кислорода на платиновом аноде из р-ра КОН
В газовом К.). Н. Е. Хомутов.
КУЛОНОМЕТРИЯ — один из электрохимических
методов анализа, основанный на измерении количества
электричества, расходуемого на электролитич. вос-
становление или окисление. Необходимое условие
для применения К. — 100%-ный выход по току дан-
ного вещества. В частности, при катодном процессе
должны отсутствовать такие побочные процессы, как
восстановление ионов водорода или растворенного
кислорода, а также продуктов, образующихся на аноде.
Первый из этих процессов устраняется применением
ртутного катода, обладающего высоким перенапряже-
нием для выделения водорода, остальные — работой
в атмосфере инертного газа и применением серебря-
ного анода (при электролизе галогенидов) или соответ-
ствующих анодных деполяризаторов. Сила тока
во время электролиза не остается постоянной; поэ-
тому для измерения количества электричества обычно
пользуются кулонометрами различных типов (медным,
серебряным, газовым); предложены электронные схе-
мы приборов.
Различают собственно кулонометрический анализ
и кулонометрическое титрование. Кулономет-
рический анализ восходит к М. Фарадею,
но практически был впервые применен лишь в 1938
для определения тяжелых металлов в пищевых про-
дуктах. Ранее метод применяли i
деления толщины металлических
покрытий или оксидных пленок
на металлах. Кулономет-
рическое титрование
заключается в том, что электро-
литически генерируется реактив,
вступающий во взаимодействие
с определяемым веществом. Ге-
нерацию можно проводить как
непосредственно в титруемом ра-
створе, так и в отдельном прибо-
ре, подобном изображенному на рисунке. В первом слу-
чае необходимо принять меры для того, чтобы исклю-
чить влияние второй электродной реакции (анодной—
при катодной генерации и катодной — при генера-
гл. обр. для опре-
887
КУМАРИН-КУМАРОНОВАЯ СМОЛА
888
ции реактива на аноде). Для этой цели в раствор вво-
дят соответствующие деполяризаторы или проводят
электролиз с разделенными катодным и анодным про-
странствами, используя для этого два сосуда, соеди-
ненные солевым мостиком. В качестве электродов
генерирующей цепи применяют чаще всего платину.
Для генерации реактива из трудно восстанавливаю-
щихся веществ применяют ртутный катод. Ртутный
анод используют для генерации ионов ртути; с его
помощью можно определять минимальные количества
галогенидов. Для аналогичных целей применяют
и серебряный анод. Известны и другие случаи при-
менения растворимых анодов. Конечную точку можно
определять визуально, фотометрически или электро-
химически. Последний способ применяется чаще всего.
В таких случаях, кроме генерирующей цепи, в уста-
новку включают индикаторную цепь — амперо- или
потенциометрическую. Индикаторный электрод и сое-
динительный мостик электрода сравнения (иногда
пользуются двумя платиновыми индикаторными элек-
тродами) помещают непосредственно в ту часть элек-
тролизера, в к-рой происходит титрование генери-
рованным реактивом. При кулонометрич. титровании
электролиз можно проводить при постоянной силе
тока, т. к. концентрация вещества, из к-рого генери-
руется реактив, может быть взята достаточно высокой,
благодаря чему она остается практически постоянной
во время „титрования11. Количество электричества в
этом случае рассчитывают по силе, тока и продолжи-
тельности электролиза. Силу тока обычно определяют
потенциометрически, по падению напряжения на
стандартном сопротивлении, последовательно вклю-
ченном в генерирующую цепь, а не прямым измерением
ее амперметром. Кулонометрич. титрование обладает
высокой чувствительностью и легко может быть авто-
матизировано.
Для определения As, Se, Sb и нек-рых органич. соедине-
ний применяют методы, основанные на анодной генерации
галогенов. Трехвалентный Т1, генерированный на ртутном
катоде, используют для восстановления U, Fe, Се, V. Генери-
рование марганца высших валентностей или 2-валентного
железа применяют для различных окислительно-восстанови-
тельных титрований. Электрогенерированный ион водорода
используют для изучения кинетики гидролиза органич. со-
единений и для титрования органич. оснований. Известны
работы по генерированию на ртутном катоде комплексона III
из его ртутных комплексных соединений для определения
различных металлов, к-рое можно проводить последовательно,
изменяя pH среды.
Лит.: Козловский М. Т., Ртуть и амальгамы
в электрохимических методах анализа, Алма-Ата, 1956;
Делахей П., Новые приборы и методы в электрохимии,
пер. с англ., М., 1957; А г а с я н П. К., Завод, лабор., 1956,
22, М 1, 7; Мир кин В. А., там же, 1959, 25, № 3, 292;
1961, 27, № 9, 1063. О. А. Сонгина.
КУМАРИН (лактон о-оксикоричной кислоты)
С9Н6О2, мол. вес 146,14—бесцветные кристаллы с
запахом свежего сена, т. пл. 70°; т. кип.
| 2917760 мм, 153,9710 мм; малораство-
LJL. ^С=О рим в воде, лучше — в 50%-ном вод-
0 ном спирте, растворим в спирте и эфире.
К. найден во многих растениях, в бобах тонка его
содержится 1,5%. При действии щелочи на К. обра-
зуется цис-о-НОС6Н4СН=СНСООК, соль цис-о-окси-
коричной к-ты. При сильном нагревании со щелочью К.
расщепляется, образуя смесь солей салициловой
и уксусной кислот. К. при восстановлении Na в
спирте превращается в у-(2-оксифенил)-пропанол
о-НОС6Н4(СН2)2СН2ОН, а при восстановлении амаль-
гамой Na образует о-оксигидрокоричную кислоту
о-НОС6Н4СН2СН2СООН. Бисульфит натрия реагирует
с К., образуя продукт присоединения по двойной связи
гетероциклич. ядра. К. получают в пром-сти несколь-
кими способами, в частности по реакции Перкина из
салицилового альдегида и уксусного ангидрида в при-
сутствии СН3СООК или CHjCOONa. К. — одно из
широко известных душистых в-в, применяется в пар-
фюмерных композициях для духов и одеколонов,
отдушек Для мыла, пищевых эссенций, ароматизации
табака. в. М. Дашунин.
КУМАРИНОВАЯ КИСЛОТА — см. Кумаровая кис-
лота.
о-КУМАРОВАЯ КИСЛОТА (транс-о-оксикоричная
кислота) С9Н8О3, мол. в. 164,16 — стабильная форма
одного из двух геометрич. изомеров (I); второй изо-
мер — лабильная цис-
форма — наз. кума-
риновой кисло-
той (II).
Кумариновая к-та из-
вестна только в виде со-
лей и производных. Ее соли получают кратковремен-
ным нагреванием кумарина со щелочами; при их
подкислении снова образуется кумарин. Кумаровая
кислота более устойчива; т. пл. 207—208°, растворима
в спирте и воде, плохо—в эфире. Константа диссоциа-
ции К.=2,14-10-5 (25°); при нагревании выше темп-ры
плавления декарбоксилируется с
i при стоянии
ОН
он
сн
нс-соон
он
9Н
ноос-сн
II
образованием о-
в крепких к-тах
Вг
Вг
О
'О
IV
н=сн2
III
винил-фенола (III), а
переходит в кумарин;
на свету постепенно
димеризуется. Броми-
рование К. к. в серо-
углероде приводит к
дибромкумарину (1V).
К. к. находится во многих растениях, часто вместе
с кумарином. Получают ее в виде о-ацетильного про-
изводного из салицилового альдегида и уксусного
ангидрида (см. Перкина реакция). Другим способом
синтеза может служить конденсация салицилового
альдегида с малоновой к-той в пиридине в присутствии
пиперидина. Соли кумариновой кислоты изомеризу-
ются в соли о-К. к. при нагревании с алкоголятами.
Т. обр., К. к. может быть получена и из кумарина.
Известно большое число функциональных производных
К. к. и кумариновой к-ты как по окси-, так и по карбоксиль-
ной группе. Производные кумаровой к-ты: О-ацетил, т. пл. 146°;
метиловый эфир (сложный), т. пл. 184—185°; диметиловый
эфир, т. кип. 304—305°. Промышленное применение находит
кумарин. Производные кумариновой к-ты: о-метиловый эфир,
т. пл. 93,6°; диметиловый эфир, т. кип. 247°'25О мм\ диэтило-
вый эфир, т. кип. 290—291°; лактон, см. Кумарин,
Н. А. Несмеянов.
КУМАРОНОВАЯ СМОЛА (индеи-кумароновая смо-
ла) — смесь полимеров типа
где п меняется от 5 до 10; средний мол. в. от 500 до
1200. К. с. — смола от светло-желтого до черного
цвета, от жидкой до твердой консистенции (в зависи-
мости от исходного сырья и способа получения).
Чем выше мол. вес К. с., тем выше темп-ра размягче-
ния; для жидких смол она равна («по кольцу и шару»)
25—35°, для особо твердых 160—180°. Окраска смол,
определяемая по иодной шкале, меняется от 5—15 для
особо светлых смол до 100 и более для темных смол.
К. с. получают сополимеризацией смолообразую-
щих веществ — кумарона, индена, стирола и их го-
мологов, содержащихся во фракциях сырого бензола
и каменноугольной смолы, а также в соответствующих
фракциях, получаемых при ароматизации нефтепро-
дуктов. Для получения К. с. соответствующие фрак-
ции, содержащие смолообразующие в-ва, подвергают
полимеризации при 30—120° в присутствии катализа-
торов (серная к-та, хлористый алюминий, хлорное
железо, фтористый бор или его комплексы) или ини-
циирующих веществ (напр., перекись бензоила).
Полимеризат отделяют от катализатора, иногда ста-
билизируют гидрированием; под вакуумом от него от-
гоняют растворитель и низкомолекулярные компонен-
889
КУМИНОВЫЙ АЛЬДЕГИД - КУМУЛЯТИВНЫЙ ЭФФЕКТ
890
ты, остаток— К. с. —разливают в блоки или получают
в виде чешуек на охлаждаемом барабане. Путем сопо-
лимеризации смолообразующих веществ с фенолами
и другими активными в-вами получают модифициро-
ванные. К. с., обладающие повышенной растворимо-
стью.
К., с. применяют гл. обр. в качестве связующего
или клеящего вещества для изготовления линолеума,
лаков, эмалей, электроизоляционных материалов, кож-
заменителей, абразивных изделий и др.
Лит.: Белов К. А., Переработка химических продук-
тов коксования, Харьков — М., 1949; Ко лян др Л. Я.,
Переработка сырого бензола, Харьков, 1960; Литви-
ненко М. С., Ное алев ич И. М., Химические про-
дукты коксования для производства полимерных материалов,
Харьков, 1962. _ И. М. Носалевич.
КУМИНОВЫИ АЛЬДЕГИД (куминаль, п-изопро-
пилбензальдегид) п-(СН3)2СН—СвН4—СНО, мол. в.
148,21 — бесцветная жидкость; т. кип. 235°;
0,978; zip 1,5301; нерастворим в воде, хорошо рас-
творим в спирте и эфире. К. а. является составной
частью куминового цейлонского коричного масла.
К. а. проявляет все свойства альдегидов ароматич. ряда,
но активность карбонильной группы по отношению к
нуклеофильным реагентам несколько понижена вслед-
ствие влияния электронодонорной изопропильной
группы. В промышленности К. а. получают окислением
цимола. Кроме того, он может быть получен из
n-хлорметилкумола по Соммле реакции или окислени-
ем, введением альдегидной группы в кумол (1), одновре-
менным карбонилированием и изопропилированием
бензола (2), а также через п-изопропилфенилмагпий-
бромид (3):
BFq
(СН3)дСН-СсН5-|-СО---i п-(СН3)2СН-С„Н4-СНО (1)
СвН0+(СН3)2СНС1+СО —k, п-(СН3)2СН-СвН4-СНО ( )
n-(CH3)2CH-C3H4-MgBr-|-HC(OR)3--
—>;CH3bCH-CcH4-CH(OR)s —► n-(CH3)oCH—CeHj—СНО (3)
К. а. применяют в парфюмерии при составлении
композиций с запахом ландыша, гиацинта и др.
Н. П. Гамбарян.
КУМУЛЕНЫ — соединения, содержащие систему
более чем двух кумулированных углерод-углеродных
двойных связей. Простейший представитель — угле-
водород бутатриен СН2=С=С=СН2 — получен из
1,4-дибромбутина СН2Вг—С-.-.С—СН2Вг действием
цинка. Дегидробромированием 1,1,4,4-тетрафтор-!,
4-дибромбутена-2 с помощью КОН получен тетра-
фторбутатриен CF2=C=C=CF2. Бутатриен чрезвы-
чайно легко полимеризуется и окисляется. Бромиро-
вание его приводит к СН2Вг—СВг=СВг—СН2Вг,
гидратация — к метилвинилкетону. Тетраарилкуму-
лены — устойчивые соединения, окисляются КМпО4
и озоном, присоединяют галогены; не взаимодействуют
с хиноном и малеиновым ангидридом.
Теоретич. рассмотрение показывает, что К. с нечет-
вым числом атомов С в кумуленовой цепи, как и
аллены, могут существовать в виде d- и 1-изомеров,
тогда как для К. с четным числом атомов С в цепи
должна существовать 1{ис-транс-изомерия, как для
олефинов. Действительно, для приведенного ниже
соединения были получены две формы, являющиеся
цис-(1) и транс (П)-изомерами:
Общими методами получения замещенных К. являются
восстановление ацетиленовых 1,4-диолов P2J4, VC12,
CrCl2, PBr3, Na в жидком аммиаке, или SnCl2:
/С-(С=С)П-С\—у(Н<С С ],г(д
он он
и действие на ацетиленовые спирты уксусного ангид-
рида в присутствии КОН:
2 \c-(C=C)n-H^S»k.^>C=[=C=C=]jn=C\
ОН
Последним способом можно получать несимметрич. К.
Лит.: Kuhn R., Wallenfels К., Вег., 1938, 71,
Ki 4, 783; 71, № 7, 1510; Kuhn R„ Scholler К., Chem.
Вег., 1954, 87, № 4, 598; С a d 1 о t Р., Ann. Chlmie, 1956,
ser. 13, 1, 215; Schubert W. M. [a. о.], J. Amer. Chem.
Soc., 1954, 76, № 7, 1929; M a r t 1 n E. L., S h а г k e у W. H.,
там же, 1959, 81, № 19, 5256. Б. Л. Дяткин.
КУМУЛИРОВАННЫЕ СВЯЗИ — связи, соединя-
ющие в органич. соединениях каждый последующий
атом цепи с предыдущим двумя парами электронов
(двойной связью). Классы органич. соединения, со-
держащие К. с.: 1) аллены >С=С=С<^; 2) кумулены
/С=[=С=]п=с/; 3) кетены \с=С=О иихимины
X)C=C=N—; 4) карбодиимиды —N=C=N—; 5) изо-
цианаты — N=C=O; 6) изотиоцианаты —N=C=S.
Двуокись углерода О=С=О также можно рассмат-
ривать как соединение с К. с., однако это понятие
др ). Такие заряды
вряд ли приложимо к таким семиполярным системам,
как нитрогруппа, сульфогруппа и др. К. с. более
активны, чем соответствующие изолированные связи.
Б. Л. Дяткин.
КУМУЛЯТИВНЫЙ ЭФФЕКТ — существенное по-
вышение местного (пробивного, дробящего) действия
взрыва заряда взрывчатого в-ва (ВВ) в определенном
направлении. К. э. достигается при устройстве на од-
ном из концов заряда (противоположном месту уста-
новки детонатора) выемки той или иной формы (по-
лусфера, конус, параболоид и
носят название кумулятивных.
Если поверхность выемки в за-
ряде покрыть сравнительно тон-
кой металлич. облицовкой, то
К. э. во много раз увеличивается
(рис. 1).
Рис. 1. Действие на стальную под-
ставку сплошного заряда (а), заряда
с кумулятивной выемкой (б) и заряда
с кумулятивной выемкой при наличии облицовки (в): 1 — за-
ряд; 2 — отверстие в стальной подставке, образованное взры-
вом; з — стальная подставка; 4 — металлическая облицовка.
К. э. известен еще с 60-х гг. 19 в., однако широкое
применение он получил лишь в годы второй мировой
войны в различных боеприпасах (кумулятивные сна-
ряды, мины) и подрывных средствах, предназначен-
ных для борьбы с танками и фортификационными
сооружениями. К. э. широко используют также в
мирной технике, особенно для перфорации нефтяных
скважин. Сущность явления кумуляции заключается
в следующем.
1. Для необлицованной металлом
выемки. При истечении через поверхность выемки
продукты детонации кумулятивного заряда образуют
сходящуюся вдоль оси выемки кумулятивную струю,
обладающую, по сравнению с разлетающимися про-
дуктами взрыва обычного заряда, повышенной ско-
ростью и плотностью энергии, а следовательно, и
891 КУМУЛЯТИВНЫЙ ЭФФЕКТ - КУНА-ФРЕЙДЕНБЕРГА ПРАВИЛО 892
значительно более сильным местным разрушительным
действием. Процесс формирования кумулятивной
струи схематически показан на рис. 2. Расстояние
от торца заряда до места наибольшего уплотнения
струи называется фокусным (/<’). При установке за-
ряда на расстоянии от преграды, равном фокусному,
наблюдается .. .. -
2. Фор-
Рис,
мирование ку-
мулятивной
струи при от-
сутствии обли-
цовки над вы-
емкой.
максимальный эффект. В образовании
кумулятивной струи принимает участие
лишь некоторая доля заряда, прилегаю-
щая к поверхности кумулятивной вы-
емки, т. н. активная часть кумулятив-
ного заряда (заштрихованный участок
на рис. 2). Для кумулятивных зарядов
с выемками, радиус основания которых
равен полукалибру г0 заряда, объем
активной части Va пропорционален rj.
С увеличением калибра заряда масса ку-
мулятивной струи и ее бронепробивпое
действие могут быть существенно уве-
личены. В современных кумулятивных
зарядах вес активной части колеблет-
ся в пределах 10—25% от общего веса
заряда.
2. Для выемки, облицо-
ванной металлом. При нали-
чии на поверхности выемки металлич.
облицовки последняя под действием про-
дуктов детонации обжимается и отдель-
ные ее элементы последовательно захлопываются (смы-
каются) с образованием тонкой металлич. струи, в к-рой
сконцентрирована основная доля энергии активной
части кумулятивного заряда. Струя образуется исклю-
чительно за счет течения металла, прилегающего к
внутренней поверхности облицовки, что является след-
ствием сильного соударения ее элементов в момент
захлопывания. Основная часть облицовки при этом
превращается в т. н. «пест» (рис. 3). Диаметр куму-
лятивной струи, как правило,
г ’’ * * 1 не превышает 1—3 лги, а масса
ее составляет 6—11% от массы
всей облицовки. В кумулятнв-
Рис. 3. Формирование кумулятивной струи при обжатии
конической облицовки: а — до взрыва; б — через 6 мксек
после взрыва; в — через 17 .иксе/: после взрыва: 1 — пест;
2 — кумулятивная струя.
ной струе достигается очень высокая объемная плот-
ность энергии и она приобретает резко повышен-
ную пробивную способность. Последовательные ста-
дии процесса образования кумулятивной струи прп
металлич. облицовке выемки, зафиксированные с по-
мощью импульсной рентгеносъемки, показаны на
рис. 3. Как видно из рисунка, кумулятивная струя
в процессе своего развития отрывается от песта и
заметно растягивается, что объясняется наличием
градиентов скоростей вдоль нее. Для типичных куму-
лятивных зарядов с конич. выемкой и облицовкой из
стали скорость головной части струи 7,0—7,5 км/сек,
а скорость песта и хвостовой части струи всего
лишь ок. 0,5—1,0 км/сек. Пробивная способность
струи тем выше, чем больше она может растягиваться
без потери монолитности, что определяется свойствами
металла облицовки. Наилучшими материалами для
облицовки кумулятивных выемок являются сталь и
медь.
К. э. повышается с увеличением уд. энергии
взрыва, плотности ВВ и скорости детонации за-
ряда. Для изготовления кумулятивных зарядов'при-
меняют тротил, гексоген, тэн и др. высокобризант-
иые ВВ.
Согласно гидродипймич. теории кумуляции М. А.
Лаврентьева, глубина бронепробивания£=1аффлР1/Р2’
где pj и р2 — плотности металла струи и брони
(преграды), £аф — эффективная длина струи, т. е.
длина той ее части, в к-рой скорость металла превы-
шает нек-рое критич. значение 77кр, зависящее от
свойств пробиваемой преграды. Современные куму-
лятивные заряды способны пробивать стальную броню
толщиной 5—5,5 калибров заряда.
Лит.: Баум Ф. А., Станюкович К. П., III е х-
т е р Б. И., Физика взрыва, М., 1959; Лаврентьев М. А.,
Кумулятивный заряд и принципы его работы, Успехи матем.
наук, 1957, 12, вып. 4 (76), 41—56; Андреев К. К. и
Беляев А. Ф., Теория взрывчатых веществ, М., 1960.
Ф. А. Баум.
КУНА — ФРЕЙДЕНБЕРГА ПРАВИЛО (вициналь-
ное правило) — закономерность оптич. вращения
органич. соединений, вытекающая как из обобщения
опытного материала, так и из теоретич. представлений
Куна и Фрейденберга о механизме возникновения
оптич. вращения и его связи с полосами поглощения в
УФ-области спектра. Согласно этим представлениям
оптическое вращение появляется как результат
анизотропий определенных полос поглощения, свой-
ственных функциональным группам (хромофорам);
анизотропия возникает в хромофоре под влиянием
асимметрического окружения. С этой точки зрения
любой заместитель, вводимый в оптически активное
соединение, влияет па вращение двумя путями:
1) вносит собственный вклад за счет новых, свойст-
венных данному заместителю полос поглощения;
2) оказывает вицпнальное действие — изменяет анизо-
тропию уже имевшихся в исходном соединении полос
поглощения других заместителей. Это влияние осу-
ществляется в соответствии с вицинальным правилом,
к-рому Кун и Фройденберг придали след, наиболее
общую формулировку: изменения вращения, вызыва-
емые замещением, возникают главным образом вслед-
ствие изменения анизотропии полосы поглощения
группы, подвергшейся замещению, и лишь в малой
степени являются следствием изменения влияния
(вицинального действия) данной группы на полосы
поглощения других заместителей.
Часто вицинальное правило формулируют иначе:
изменения анизотропии полосы поглощения (т. е.
изменения ее вклада во вращение) тем значительнее,
чем в большей близости к соответствующему хромо-
фору производится химич. изменение. В этой форме
вицинальное правило более конкретно и легче при-
менимо к экспериментальному материалу, однако при
этом вицинальное правило превращается в правило
положения вводимого заместителя (правило удаления),
сформулированное Чугаевым задолго до работ Куна
и Фрейденберга. Последним принадлежит заслуга
теоретич. истолкования рассматриваемой закономер-
ности и широкого применения ее при сравнении от-
носительной конфигурации оптически активных в-в
в виде правила оптического сдвига. Согласно послед-
нему разница во вращении аналогично построенных
производных для конфигуративно сходных соединений
одинакова по знаку и близка по величине. Правило
удаления можно иллюстрировать след, рядом соеди-
нений, в к-рых видно ослабление влияния карбо-
нильной группы на вращение по мере роста расстоя-
ния между карбонильной группой и асимметрии.
893
КУНЖУТНОЕ МАСЛО — КУРЦИУСА РЕАКЦИЯ
894
центром; для сравнения приведено значение [А/] для
соответствующего углеводорода.
СН3
I
Н—С—R
I
С2н5
R = СОС2Н5 СН2СОСН3 (СН2)2СНО н-С3Н7
[М]и +36,3 +11,0 +12,0 +9,8
На основании общих закономерностей оптич. вра-
щения, открытых Чугаевым, Куном и Фрейдепбергом,
был в дальнейшем сформулирован ряд правил при-
менительно к отдельным типам оптически активных
веществ. Сущность связи между оптической активно-
стью и химич. строением была установлена в резуль-
тате успехов спектрополярометрии, когда вращение
стало характеризоваться не просто числом, величина
к-рого зависит от многих внешних факторов, а кривой
дисперсии вращения, более тесно связанной со строе-
нием вещества.
Лит.: Freudenberg К., Stereochemie, Lpz.— W.,
1933, S. 398, 693; Kuhn W., Ber., 1930, 63, № 1, 207; Angew.
Chemie, 1956, 68, № 3, 91; Brewster J. H., J. Amer.
Chem. Soc., 1959, 81, № 20, 5475 (и последующие работы);
Чугаев Л. А., Избранные труды, т. 2, М., 1955; Д же-
р а с с и К., Дисперсия оптического вращения, пер. с англ.,
М., 1962. В. М. Потапов.
КУНЖУТНОЕ МАСЛО — см. Жиры растительные.
КУПОРОСНОЕ МАСЛО — см. Серная кислота.
КУПОРОСЫ — техническое название кристалло-
гидратов сернокислых солей нек-рых тяжелых метал-
лов. Наиболее широкое применение нашли следую-
щие К.: железный—FeSO4 • 7Н2О (см. Железа сульфа-
ты), медный — CuS04 • 5НаО (см. Меди сульфат), ни-
келевый — NiSO4 7Н2О (см. Никеля сульфаты), цин-
ковый — ZnSO4 • 7Н2О (см. Цинка сульфат).
КУПФЕРОН (N-нитрозофенилгидроксиламин, ам-
монийная соль) C6H9O2N3, мол. вес 155,16 — белые
до или буроватые кристаллы (чешуйки),
V'-ONH т- пл- 163—164°; растворим в воде, бензо-
4 ле, спирте, эфире; при нагревании разла-
Г J гается с образованием нитробензола. При
длительном хранении К. медленно разла-
гается и приобретает более темную окраску; разложе-
ние реагента в растворе может быть обнаружено по
появлению мути. Для стабилизации растворов К. до-
бавляют по 50 мг ацетофенетидина на каждые 150 мл
р-ра. К. обычно применяют в виде водного р-ра (ок. 6%);
константа диссоциации К = 5,3 • 10 + К. может быть
получен из N-фенилгидроксиламина действием NaNO2
с последующей обработкой аммиаком. К. образует
малорастворимые внутрикомплексные соединения
с различными элементами (Си, Fe, Al, Bi, Ga, Hg,
Мп, Nb, Sn, Ta, Th, Ti, V, Zr и др-)- Это свойство К.
широко используется в аналитич. химии для обнару-
жения, весового определения и отделения одних эле-
ментов от других. Купферонаты металлов растворимы
в эфире, хлороформе и др. органич. растворителях,
что широко используется в настоящее время для раз-
деления элементов экстракцией. Ввиду того, что К.
и образуемые им соединения с катионами металлов
могут разлагаться при нагревании, экстракцию про-
водят на холоду. Для Th4+ методом распределения
между водой и органич. растворителем при р, = 0,1 М
и 25° найдены след, значения констант устойчивости:
1g К, = 7,35; 1g К2 = 6,95; 1g К3 = 6,55 и lg =
= 6,15. Практически важным гомологом К. является
неокупферон (N-нитрозонафтилгидроксиламин, аммо-
нийная соль), к-рый ведет себя подобно К.
Лит.: Welch er F. J., Organic analytical reagents,
v. 3, N. Y. — la. o.l, 1947; Моррисон Дж. и Фрей-
зер Г., Экстракция в аналитической химии, пер. с англ.,
Л., 1960. И. П. Ефимов.
КУРЦИУСА РЕАКЦИЯ — метод получения пер-
вичных аминов (III) превращением азидое карбоновых
кислот (I) в эфиры изоциановой к-ты (II) с последую-
щим гидролизом до аминов (III):
+ - — N-, И>0
R-C-N=N=N-----=.R N—С—О -t++R -NII2
II Нт
0 I II III
Механизм превращения азидов к-т в эфиры изоциано-
вой к-ты (перегруппировка Курциуса) во многом
сходен с аналогичными перегруппировками N-гало-
геиамидов (см. Гофмана реакция) и гидроксамовых
к-т. Первоначально ацилазид (I) выделяет молекулу
азота с образованием промежуточной частицы (IV),
содержащей на атоме азота секстет электронов:
R-C-N
II "
О
IV
заключительная стадия перегруппировки состоит в
миграции группы R со своей электронной парой атома
углерода к атому азота:
{КЙС-N/—‘►O'-C-N-R
”'i6
При этом R кинетически несвободен, что доказано
образованием оптически активных аминов при пере-
группировке азидов карбоновых к-т с асимметрич.
атомом углерода в a-положении к карбоксильной
группе;
CSH3CH-C-N3—-CeH5-CH-NH2
СН3 i CHS
Азид о-(2-метил-6-нитрофенил)-бензойной к-ты, оптич.
активность к-рого обусловлена невозможностью сво-
бодного вращения ароматич. ядра в орто-положении
к карбоксильной группе, в условиях К. р. также об-
разует оптически активный амин;
Миграция углеводородного радикала в виде свобод-
ного аниона неизбежно привела бы к рацемизации.
Азиды к-т превращают в эфиры изоциановой к-ты
обычно в инертном растворителе (бензол, хлороформ,
диоксан) при 20—150°. Показано, что эта реакция
катализируется сильными к-тами. Разложение азидов
в присутствии спиртов или рассчитанного количества
воды приводит соответственно к уретанам или N-за-
мещенным производным мочевины:
R-C-Na—R—N—С—О -
„NH . СООСЛ13
IT /V
—R-NH-C-NH-R
О
Далее эфиры изоциановой к-ты, а также уретаны и
мочевины превращают в соответствующие амины
гидролизом в кислой среде. Азиды а, ^-ненасыщенных
к-т (V) в условиях К. р. образуют легко полимеризую-
щиеся виниловые эфиры изоциановой К-ты (VI),
гидролиз к-рых приводит к альдегидам или кетонам
(VII или VIII):
R-CH=CR'-CN3 —>RCH=CR'-N=C=O
II
V 0 VI
RCH=CR'—N=C-=O—*RCH2—CO—R' R=H, алкил
VII VIII
Перегруппировка азидов а-галогенокислот (в кислой
895
КУЧЕРОВА РЕАКЦИЯ
896
среде) также приводит в конечном счете к карбониль-
ным соединениям:
Вг /Вг н+
R2CZ --------R.C< ----R..C=O l-HBr+HNCO
XC-N3 XNCO
И
о
В том случае, когда галоген находится в более уда-
ленном положении относительно карбоксильной груп-
пы, К. р. нормально приводит к соответствующим га-
логенозамещенным аминам. Азиды а-оксикислот при
нагревании перегруппировываются в а-оксиалкил-
изоцианаты, к-рые легко отщепляют молекулу циано-
вой к-ты и переходят в альдегиды или кетоны:
.он ,он
R,CZ —»R>C( ---► R.CO-f-HNCO
XCON3 ^NCO
Азиды карбоновых к-т, содержащие оксигруппу, уда-
ленную от карбоксильной, в условиях К. р. образуют
циклич. или полимерные уретаны:
х 0
НО —R -CONg — R0.C=O или (-O-R -NH-C-)X
NH
Термич. разложение бис-азидов дикарбоновых к-т
с последующим гидролизом было предложено в ка-
честве эффективного метода получения диаминов али-
фатич. и ароматич. ряда:
Ns-C-(CH2)4 -C-Ns—.H.,N(CH2)4-NH2
II II
о о
Исключение составили замещенные малоновые к ты,
бис-азиды к-рых в условиях'К. р. переходят в карбо-
нильные соединения:
С0НиСН2СН(СО\3)2/-т,‘..СсНзСНаСН(КНСООС2Н,).,
Н+ | Н2О
сещсн.,сн
II
О
К. р. была с успехом применена к синтезу различных
аминокислот:
усоок
сн3-сн —>сн3-сн-соон
Xc0N3 NH2
Реакция открыта в 1890 Т. Курциусом и в дальней-
шем усовершенствована П. Шмидтом (см. Шмидта ре-
акция).
Лит.: Смит П. А. С., в кн.: Органические реакции.
Сб. 3, пер. с англ., М., 1951, с. 322; X ю к к е л ь В., Тео-
ретические основы органической химии, пер. с нем., т. I,
М., 1955, с. 348. Л. С. Герман.
КУЧЕРОВА РЕАКЦИЯ — гидратация ацетилена,
его гомологов и производных, приводящая к синтезу
карбонилсодержащих соединений:
О О
Н2О II II
RCZCR'-—-- RCCH2R'4-RCH2CR'
Гидратацию обычно проводят в кислой среде в при-
сутствии солей двухвалентной ртути в водном или
водноспиртовом р-ре. Ацетальдегид, наир., получают,
пропуская ацетилен в горячий р-р сульфата ртути
в разб. серной к-те. Монозамегценные ацетилены, как
правило, превращаются исключительно в кетоны (1);
лишь при наличии сильно электронооттягивающих
заместителей образуются наряду с кетонами и альде-
гиды (2):
I—►СР3СН2СНО
RCHCH—►RCOCHs (1) CF3C+CH- (2)
I--*CF3COCH8
Направление гидратации несимметричных дквамвг.
щепных ацетиленов соответствует Марковникова пра-
вилу. Соединения с неясно выраженной полярностью
тройной связи дают смесь двух кетонов:.
СН3СНССН2СН3 — СН3СОСН2СН2СН8+СН3СН2СОСН2СН3
Аминогруппа в а-, 0- и у-аминоацетиленовых соеди-
нениях направляет атом кислорода к наиболее отда-
ленному атому углерода; влияние аминогруппы падает
с увеличением ее расстояния от тройной связи:
R\ R4 ,о
'NCI+CCR'—- )N(CH2)2Cf
Rz Rz \R
R\ R. /zO
)NCH2CH2C=CR'---. )N(CH2)3CZ
Rz Rz 'R'
R. R\ //0
,NCH2(CH2)2C=CR'—» }NCH2(CH.)aCf (77%)+
Rz Rz NR'
0
II
+ NNcHe(CH2)2CCH<.R' (23%)
Rz
На направление гидратации также влияют окси-
и алкоксигруппы:
R’ R’
I I
R-C-C CR'"---► R—С—CH»C0R"’
I I
OR" OR"
где fl' + H, алкил. Только сложные эфиры а-ацетиле-
новых спиртов превращаются в а-ацетоксикетоны:
R R
СН3СОО-С—G_.GR”---► СН3СОО—С—COCH2R"
R' R^
а-0ксипроизводные образуются и из хлорацетило-
повых спиртов:
R R
НО —С—С=СС1—>НО-С-СОСН2С1
R' XV
Особенно легко гидратируется тройная связь в алко-
ксиацетиленах, что дало возможность использовать
последние в качестве мягкого дегидратирующего сред-
ства при синтезе пептидов:
I I
RCOXH- С-СООН + H.,NCCOOR + HCrCOR'----•
I 'l
I I
--* RCONH—C—CONH—C—COOR + CH3C00R'
I I
Гидратация алкоксиацетиленовых спиртов приводит
к образованию 0-окси- и а,0-ненасыщенных к-т:
R\
R ZHOC-CH2COOR”
HO^C-C+COR"-----/
R,/ 'g —CH- COOR''
R'Z
Механизм К. p. до сих пор точно не установлен; предпо-
лагают, что продукт гидратации получается в результате
гидролиза промежуточно образующегося продукта присоеди-
нения соли ртути к тройной связи:
С С
Ш +HgX2——- нгх--х -
I I
fb,HgX'Q' ihHgX vH
XHg-'-X-’-C —- х-с —-HOC—- о=с
На К. р. основано промышленное произ-во ацеталь-
дегида. Реакция открыта М. Г. Кучеровым в 1881.
Лит.: Петров А. Д., Усп. хим., 1952, 21, вып. 2, 25Q;
Мацони С. Г., ЧухаджянГ. А., Вартанян С. А.,
Ж. общ. химии, 1959, 2», вып. 2, 451; Фишер Л. Б., Усп.
ХИМ., 1958, 27, вып. 5, 589. Н. П. Гамбарян. '
897
КЬЕЛЬДАЛЯ МЕТОД-КЮРИЙ
898
КЬЕЛЬДАЛЯ МЕТОД — метод количественного
определения содержания азота в органич. соединениях
и различных материалах растительного и животного
происхождения, основанный на разложении вещества
конц. серной к-той при нагревании до темп-ры кипения
в присутствии передатчиков кислорода (ртуть или ее
соли), окислителей (K2Cr2O7, Н2О2, CuS04 и др.) и суль-
фата калия, повышающего темп-ру кипения кислоты.
При разложении вещества в этих условиях содержа-
щийся в нем азот количественно образует аммиак, даю-
щий с серной к-той бисульфат аммония. Реакционную
смесь затем охлаждают, разбавляют водой, прибав-
ляют избыток щелочи, пемного тиосульфата или суль-
фида натрия и цинковой пыли (для разложения азо-
тистых соединений ртути) и отгоняют аммиак, погло-
щая его титрованным р-ром к-ты (HCl, H2SO4, Н3ВО3).
Избыток последней титруют р-ром щелочи в присут-
ствии индикатора — метилового красного или бромфе-
полового синего, или титруют образовавшийся, напр.,
борнокислый аммоний р-ром Н2ЗО4или HCI в присут-
ствии метилового оранжевого. К. м. применяют гл.
обр. для анализа аминов и их производных. Азот
выделяется количественно в виде аммиака только из
аминов и их производных. Другие азотсодержащие в-ва
(нитро-, нитрозо-, азосоединения и т. п.) образуют на-
ряду с аммиаком также молекулярный азот, что при-
водит к получению пониженных результатов. Для
определения азота в этих веществах пх предварительно
восстанавливают до аминов, нагревая с цинковой
пылью и кислотой, или проводят разложение в среде
смеси серной и фосфорной к-т (3:1), содержащей не-
большие количества селена и сульфата меди, или
в среде серной к-ты в присутствии глюкозы и суль-
фата калия. Предложено много вариантов К. м., де-
лающих его пригодным для разнообразных типов
соединений.
Лит.: Ластовский Р. П., Вайнштейн Ю. И.,
Технический анализ в производстве промежуточных продук-
тов и красителей, 3 изд., М., 1958; В а й б е л ь С., Иденти-
фикация органических соединений, пер. с англ., М., 1957,
с. 29. А. А. Черкасский.
КЮРИЙ (Curium) Cm — химич. элемент с п. н.
96 из семейства актинидов.
15 К, X, ЭА т, 2
.1
ЛАВАНДОВОЕ МАСЛО — см. Эфирные масла.
ЛАВСАН — см. Полиэфирные волокна.
ЛАДЕНБУРГА РЕАКЦИЯ — метод синтеза алкил-
и аралкилпиридинов термич. изомеризацией соответ-
ствующих четвертичных солей пиридиния:
В результате Л. р. образуется, как правило, смесь
а- и у-изомеров с преобладанием а-изомера. 0-Замещен-
ные пиридины образуются лишь в незначительных
количествах. Л. р. проводят нагреванием галогенал-
килата (аралкилата) пиридиния либо смеси галогенал-
кила (аралкила) с пиридином при 200—300°. В ряде
случаев Л. р. катализируется медью или ее солями.
Применение хлоргидрата пиридина в качестве разба-
вителя позволяет снизить темп-ру реакции и умень-
шить смолообразование.
Изомеризация йодистого Х-(и-пропил)-пиридиния
приводит к смеси а- и у-изопропилпиридинов:
СН3^СНЭ
1,2-Дииодэтан в условиях Л. р. образует смесь
этилпиридинов, т. к. промежуточно образующийся
Р-иодэтилпиридин легко восстанавливается присут-
ствующим в реакционной смеси иодистым водородом.
Аналогично пиридину реагируют хинолин, изохино-
лин и гомологи пиридина. Выходы в Л. р. достигают
70—75% от теоретического. Реакция открыта А. Ла-
денбургом в 1883.
Лит.: Ladenburg А., Вег., 1883, 16, 1410, 2059;
Чичибабин А. Е., О продуктах действия галоидных
соединений на пиридин и хинолин, М., 1902; Crook К. Е.,
J. Amer. Chem. Soc., 1948, 70, № 1, 416. Л. С. Герман.
ЛАДЕНБУРГА — ВЫШНЕГРАДСКОГО РЕАК-
ЦИЯ — восстановление пиридина и его производных
натрием в спирте с образованием соответствующих
соединений ряда пиперидина. Легко восстанавлива-
ются алкилпиридины, пиридинкарбоновые к-ты и т. д.;
3- и 4-аминопиридины образуют соответствующие ами-
нопиперидины, в то время как восстановление 2-ами-
нопиридина приводит к пентаметилендиамину:
. -^1- H2N(CH2)5NH,
nh2 '
Гидрирование пиридинового кольца сопровождается
восстановлением функциональных групп, чувстви-
тельных к действию натрия в спирте, напр.:
Аналогично можно восстановить пиридиновое кольцо
хинолина и его производных:
I
Н
Л. — В. р. обычно проводят, добавляя избыток
металлич. натрия к раствору вещества в кипящем эти-
ловом или бутиловом спирте. См. также Буво-Блана
восстановление.
Лит.: Мошер Г., в кн.; Гетероциклические соединения,
под ред. Р. Эльдерфилда, пер. с англ., т. 1, М., 1953, с. 315.
Б. Л. Деткин.
ЛАКИ — растворы пленкообразующих веществ в ор-
ганич. растворителях, применяемые для защитных и
декоративных покрытий или электроизоляционной
пропитки различных материалов, а также для приго-
товления эмалевых красок (см. Краски). Основная
составная часть Л. — пленкообразователь, вещество,
к-рое при нанесении на поверхность тонким слоем
способно образовывать достаточно прочную пленку
в результате химич. или физич. процессов. Помимо
пленкообразователя и растворителя, в состав Л.
в ряде случаев входят пластификаторы, а также ката-
лизаторы и инициаторы процесса образования пленки
(соли металлов, органич. перекиси). В зависимости от
типа пленкообразователя Л. разделяют па: масля-
ные, смоляные, эфиро целлюло зные.
В зависимости от применения различают Л.: для
наружных работ; для внутренних работ; для художе-
ственных работ; стойкие к агрессивным средам; тер-
мостойкие; электроизоляционные и Л. специального
назначения (напр., для отделки кожи, лакировки
жести для консервных банок, для изготовления свето-
составов маркировочных красок и др.). Л. выпуска-
ются б. ч. в виде одного раствора (однокомпонентные
или одноупаковочные) и в нек-рых случаях в виде
двух растворов, смешиваемых перед употреблением
(двухкомпонентные или двухупаковочные Л.). Вторым
компонентом может быть раствор катализатора или
инициатора. Процесс образования лаковой пленки
(высыхание) может проходить при разных темп-рах,
и, в зависимости от этого, Л. обычно подразделяют
на Л. холодной сушки и Л. горячей сушки. Однако
такое деление не является достаточно точным, т. к.
покрытия одним и тем же Л. иногда могут высыхать
как без применения подогрева, так и при повышенной
темп-ре. Процесс высыхания Л., если пленкообразо-
901
ЛАКИ
902
ватель нереакционноспособен, сводится к улетучи-
ванию растворителя; такие Л. наз. летучими, после
высыхания они образуют пленку из линейных полиме-
ров, плавкую и растворимую. Если же пленкообразо-
ватель реакционноспособен, помимо улетучивания
растворителя, в ном протекают химич. реакции, при-
водящие к образованию пленки сетчатого строения,
неплавкой и нерастворимой.
Качество Л. характеризуется его свойствами в виде
раствора и свойствами образующейся пленки. К числу
первых относятся: цвет, прозрачность, вязкость, кон-
центрация пленкообразователей (содержание нелету-
чих веществ), поверхностное натяжение, скорость
высыхания (пленкообразования). Показатели лаковых
пленок: блеск, твердость, эластичность, адгезия, проч-
ность на разрыв, сопротивление истиранию, стойкость
к действию удара, во до- и газопроницаемость, атмо-
сферостойкость, стойкость .к действию агрессивных
сред. Пленки электроизоляционных Л., кроме того,
характеризуются по диэлектрич. показателям — про-
бивное напряжение, уд. и объемное сопротивление,
уд. поверхностное сопротивление, величина диэлект-
рич. потерь. В ряде случаев лаковые покрытия испы-
тывают на стойкость в условиях низких или вы-
соких темп-р (морозостойкость и термостойкость
пленок).
Л. наносят на покрываемую поверхность кистью,
распылением, погружением в Л., обливом и с помощью
вальцов. Выборы способа нанесения зависят как от
физико-малярных характеристик Л., так и от рода
окрашиваемых изделий.
Лаки масляные — р-ры таких пленкообразующих
веществ, как растительные масла и природные или
синтетич. смолы. Пленки этих Л. относятся к реакцион-
носпособным самоокисляющимся. В состав масляных
Л., кроме масел и смол, входят растворители и сикка-
тивы (катализаторы окисления масел). В качестве
растворителей для масляных Л. применяются легкие
фракции нефтяных углеводородов, ароматич. углеводо-
роды, терпены и нитропарафины. Иногда использу-
ются хлорированные углеводороды и кетоны (цик-
логексанон). Различают Л. на маслах высыхающих,
полу высыхающих и на смеси масел. В зависимости
от жирности (процентное содержание масла в пленко-
образующей части без учета растворителя) все эти
Л. делят на жирные (80—64%), средние (60—55%)
и тощие (50—30%). Жирные Л. образуют высокоэла-
стичные пленки, их применяют для внешних покры-
тий, эксплуатируемых в условиях переменных темп-р,
подвергающихся атмосферным воздействиям. Л. то-
щие, образующие твердые, блестящие и малоэластич-
ные пленки, применяют для покрытий внутри помеще-
ний. Применение Л. средней жирности очень разно-
образно. Масляные Л. можно наносить любыми спо-
собами. Они могут подвергаться холодной и горячей
сушке. Холоднан сушка длится сутки и более, горя-
чая — несколько .часов (конвекционная) или 10—
30 минут (радиационная).
Основные стадии изготовления масляных Л.:
1) подготовка масла (рафинирование, полимеризация,
оксидация); 2) растворение смолы и получение т. наэ.
лаковой основы; 3) разбавление лаковой основы раст-
ворителем (или смесью растворителей); 4) отстаивание
и центрифугирование. В ряде случаев для приготов-
ления масляных Л. применяют неполимеризованные
(сырые) масла; их полимеризация происходит в про-
цессе варки со смолой.
Применение светлых природных смол сильно сокра-
тилось в связи с заменой их синтетич. смолами. Огра-
ниченно используют копалы, продукты облагоражива-
ния канифоли (эфиры канифоли) и янтарь (послед-
ний — преим. в Л. для консервной жести). Широко
применяют масляные Л. на основе черных смол —
15*
асфальтов и битумов, пленки к-рых после горячей
сушки обладают хорошими механич. и электроизоля-
ционными свойствами. Их используют для защитных
покрытий и электроизоляционных пропиток. Из син-
тетич. смол в масляных Л. применяют масло раствори-
мые фенольные смолы, получаемые конденсацией
алкилфенолов с формальдегидом или же модифициро-
ванием фенольной смолы канифолью с последующей
этерификацией полиатомным спиртом (глицерином,
пентаэритритом). Л. на основе модифицированных
смол используют для консервной жести, а на основе
алкилфенольных смол — в коррозионностойких грун-
тах и эмалях холодной и горячей сушки.
Лаки смоляные — р-ры природных или синтетич.
смол в летучих растворителях. Все смоляные Л. на
природных смолах наз. летучими. Значительную
подгруппу летучих Л. составляют спиртовые
лаки — р-ры смол в этиловом спирте. Для приго-
товления спиртовых Л. из природных смол применяют
шеллак, нек-рые сорта копалов, сандарак, акароид,
мастике, канифоль, а из синтетич. смол — феноло-
альдогидные, кетоно-альдегидные и др. Спиртовые
Л. раньше употреблялись для отделки мебели, лаки-
ровки кожи, бумаги, стекла и др. В связи с развитием
произ-ва синтетич. смол их промышленное значение
уменьшилось. Применявшийся ранее для покрытия
картин даммарный Л. (р-р природной смолы — дам-
мары в скипидаре) также заменен акрило-фисташковым
Л. Нек-рое применение, в частности в строительстве,
имеют летучие битумные Л., напр. кузбасс-лак.
Большую группу летучих смоляных Л. составляют Л.
на основе синтетич. полимеров (особенно виниловых) и сопо-
лимеров. В СССР наиболее распространены перхлорви-
ниловые Л. на основе смолы, получаемой при дополни-
тельном хлорировании поливинилхлорида. Преимуществом
перхлорвиниловой смолы перед поливинилхлоридной с точки
зрения ее применения в Л. является достаточно хорошая раст-
воримость при нормальной темп-ре в ряде органич. раствори-
телей (кетоны, ацетаты, ароматич. углеводороды), а также
более высокая адгезионная способность пленок. В состав
перхлорвиниловых Л. входят также пластификаторы (фталаты,
фосфаты и др. эфиры) в количестве до 40% от веса смолы и
различные другие синтетич. смолы, преим. алкидные. Кроме
того, добавляют стабилизаторы, обеспечивающие повышенную
светостойкость пленок, как, например, феноксипропеноксид,
эпоксидированные растительные масла, выполняющие одно-
временно роль пластификаторов, и органич. и неорганич. со-
единения нек-рых металлов (гл. обр. свинца). Перхлорвиннло-
вые Л. используют для приготовления красок для атмосферо-
стойких и химически стойких покрытий (окраска самолетов,
металлич. конструкций, химич. аппаратуры, машин, фасадов
зданий). Наибольшую химич. стойкость имеют Л., не содер-
жащие других смол, кроме перхлорвиниловой.
Применяют также Л. на основе сополимеров винилхлорида
с випилиденхлоридом и с винилацетатом, содержащие значи-
тельное количество первого мономера, а также Л. на основе
сополимеров винилхлорида с простыми виниловыми эфирами
и с метилакрилатом. Пленки этих Л. по своим защитным свой-
ствам близки к перхлорвиниловым, но обладают более высокой
эластичностью и адгезией. Для изготовления Л. начинают
применять привитые виниловые полимеры, напр. получаемые
при полимеризации винилхлорида в латексе сополимера бутил-
метакрилата и метакриловой к-ты.
Лаки поливинилацетальные характери-
зуются сильной адгезионной способностью к металлу. Их
применяют для изоляции медных проводов (Л. винифлеке на
основе поливинилформальэтилаля и феноло-формальдегидной
смолы) и изготовления фосфатирующих грунтов (на основе
поливин илбутираля).
Лаки полиакриловые готовят на основе поли-
меров и сополимеров эфиров акриловой или метакриловой
к-т, чаще всего сополимеров бутилметакрилата. Покрытия
отличаются бесцветностью, прозрачностью, хорошей адгезией
к металлу и атмосферостойкостью. Растворителями служат
кетоны, эфиры, ароматич. углеводороды.
Лаки хлоркаучуковые — растворы хлоркау-
чука в ароматич. углеводородах, хлорированных углеводоро-
дах и нек-рых сложных эфирах (напр., циклогенсанолацетат).
Эти Л. применяют для получения специальных кислотостой-
ких покрытий, эксплуатируемых в условиях повышенных
темп-р и действия агрессивных сред (р-ров солей, жирных к-т,
сероводорода, хлористого водорода, хлора, а также при дейст-
вии смазочных масел, спирта и бензина).
К числу смоляных Л. на основе реакционноспособных смол
относятся алкидные, полиэфирные, эпоксидные, полиурета-
новые, феноло-апьдегидные, мочевино- и меламино-формаль-
дегидные, дивинилацетиленовые и кремнийорганич. Л.
903
ЛАКИ
904
Лаки алкидные — р-ры алкидных смол (глифтале-
вых, пентафталевых), модифицированных жирными к-тами
растительных масел (см. Смолы алкидные) в растворителях
(уайтспирите, сольвенте, ксилоле). В состав Л. иногда вводят
сиккативы. Содержание и степень ненасыщенности жирных
к-т растительных масел в алкидных смолах определяют их
«жирность» и применение алкидных Л. для холодной или горя-
чей сушки. Л. с высоким содержанием этих кислот применяют
для наружных покрытий. Л. на «тощих» алкидных смолах
используют для покрытий внутри помещений. Нанесение
алкидных Л. можно производить любыми способами. Свойства
алкидных Л. и покрытий на их основе можно изменять в широ-
ких пределах, используя различные типы алкидных смол и
добавки других синтетич. смол (напр., фенольных, мочевино-
и меламино-формальдегидных). Комбинирование алкидных
и фенольных смол позволяет получать Л., образующие покры-
тия с повышенной химич. стойкостью и термостойкостью.
Алкидные Л., содержащие меламиновые и мочевинные
смолы, используют для приготовления эмалевых красок —
автомобильных, велосипедных, для холодильников и стираль-
ных машин и т. п.
Лаки алкидно-стирольные представляют
собой р-ры сополимеров алкидных смол со стиролом. Отли-
чаются быстрым высыханием при комнатной темп-ре; приме-
няют для покрытий внутри помещений. В качестве раствори-
телей применяются ароматич. углеводороды.
Лаки полиэфирные подразделяют на гликоль-
малеинатные и полиэфир-акрилатные. Гликоль-малеинатные
Л. — растворы ненасыщенных полиэфирных смол на основе
гликолей н малеиновой к-ты в мономерах (стирол, винилто-
луол, эфиры акриловой и метакриловой к-т, монометилмалеи-
нат, диаллилфталат, диаллилмалеинат, триаллилфосфат, три-
аллилцианурат, винилацетат и др.). Триаллилцианурат повы-
шает термостойкость покрытий. Непосредственно перед их
нанесением на окрашиваемую поверхность к этим Л. добав-
ляют инициаторы полимеризации (перекиси или гидропере-
киси). Благодаря этому сополимеризация линейных ненасы-
щенных полиэфиров с мономером сопровождается образова-
нием трехмерной структуры. В состав гликоль-малеинатных Л.,
кроме того, входят: ускорители, вызывающие быстрый распад
инициаторов в условиях образования пленки (третичные амины
или нафтенат кобальта), добавки, позволяющие изолировать
покрытие от ингибирующего влияния кислорода воздуха (пара-
фин,-пчелиный воск, спермацетовое масло, стеариновая к-та),
стабилизаторы, обеспечивающие приемлемый для практич.
применения срок годности лакового р-ра (гидрохинон, пирока-
техин и их алкилпроизводные). Иногда в гликоль-малеинат-
ные Л. вводят инертные растворители, пластификаторы и
тиксотропные добавки, предотвращающие стекание нанесен-
ного на вертикальные поверхности Л. (напр., аэросил). Из
растворителей применяют ацетон, метилэтилкетон, метанол,
этилацетат. Для получения Л., у к-рых процесс пленкообразо-
вания не замедляется под воздействием воздуха, используют
твердые ненасыщенные полиэфиры, синтезируемые из поли-
циклич. двухосновных к-т и полициклич. гликолей. Эти Л.
могут быть получены при введении в их состав соединений,
способных к окислительному высыханию и к сополимеризации
с мономерами (напр., диаллиловым эфиром диметилолмоче-
вины, пенталлиловым эфиром гексаметилолмеламина), а также
при использовании для Л. полиэфиров, получаемых с приме-
нением указанных метилольных производных и аллилового
спирта. Тот же эффект может быть достигнут при применении
для отверждения полиэфиров полиизоцианатов. Гликоль-мале-
инатные Л., отверждаемые изоцианатами, рекомендуются для
окраски металла. Благодаря отсутствию летучего раствори-
теля при работе с гликоль-малеинатными Л. можно получать
сразу покрытие значительной толщины (200—300 мк), приобре-
тающее после полирования зеркальный блеск.
Лаки полиэфир-акрилатные представляют
собой 70%-ные р-ры гликолевых полиэфиров метакриловой
к-ты в растворителях. Для достижения эластичности полиэфиры
комбинируют между собой или вводят в Л. алкидные смолы.
После введения инициатора (перекись бензоила, гидроперекись
кумола) и ускорителя (линолеат кобальта) в Л., нанесенных
на поверхность, начинается полимеризация. При комнатной
темп-ре она длится около суток, при 100°—2 часа. Полиэфир-
ные Л. применяют для отделки мебели, радиоаппаратуры,
а также для покрытий по бетону и по древесно-стружечным
плитам, их используют для окраски стеклопластиков, полу-
ченных на основе ненасыщенных полиэфирных смол. Покрытия
из акрилатных полиэфиров имеют сильный блеск и без поли-
рования.
Лаки полиуретановые представляют собой
композиции р-ров полифункциональных гидроксил-содержа-
щих соединений и диизоцианатов (см. Смолы полиуретановые).
При нанесении полиуретанового Л. на поверхность образуется
полимер трехмерной структуры за счет реакции изоцианатов
с гидроксилсодержащим компонентом. В качестве гидроксил-
содержащих соединений применяют неполные сложные поли-
эфиры, получаемые при реакции избытка полиатомных спир-
тов с двухосновными к-тами или со смесью двухосновных и
одноосновных жирных к-т. Используют также частично омы-
ленный поливинилацетат, эфиры целлюлозы, неполные полн-
ацетали, фенол- и ксилоло- формальдегидные смолы. Из изоциа-
натов применяют 2,4-толуилендиизоцианат, смеси 2,4-толуи-
лендиизоцианата с 2,6-толуилендиизоцианатом (80 : 20 или
65 : 35), 1,6-гексаметилендпизоцпанат и различные продукты
взаимодействия избытка 2,4-толуилендиизоцианата с много-
атомными спиртами или с гидроксилсодержащими полиэфи-
рами. Работа с изоцианатом требует мер предосторожности,
поэтому удобнее применять «блокированные», или «скрытые»,
изоцианаты, представляющие собой соединения, разлагаю-
щиеся при нагревании с выделением изоцианатов. К ним отно-
сятся фенилуретаны, продукты взаимодействия изоцианатов
с ацетоуксусным и с малоновым эфирами, а также с этилеп-
имином. Растворителями для полиуретановых Л. служат слож-
ные эфиры, кетоны, хлорированные углеводороды, нитро-
парафины. В качестве разбавителей применяют толуол, кси-
лол, алифатич. углеводороды. Полиуретановые Л. выпускают
в виде двухкомпонентных и однокомпонентных композиций.
В первом случае отдельно изготовленные р-ры гидроксилсодер-
жащих соединений (или смеси этих соединений) и раствор
изоцианата смешивают перед употреблением Л. После сме-
шивания Л. может храниться от 4 часов до полутора-двух
суток. Двухкомпонентные Л. применяют для покрытий как
холодного, так и горячего отверждения. Неограниченно ста-
бильны однокомпонентные Л. горячего отверждения на основе
«блокированных» изоцианатов.
Полиуретановые Л. образуют эластичные покрытия с высо-
кой твердостью, адгезией, химич. стойкостью, исключительной
стойкостью к истиранию, достаточно высокими диэлектрич.
показателями. Их применяют для защиты металлич. бетонных
и деревянных поверхностей. Полиуретановые Л. рекомен-
дуются для окраски аппаратуры на нефтеперерабатывающих
и химич. заводах, в литейных цехах, на заводах пищевой
пром-сти, а также для отделки паркета, мебели, бумаги, ре-
зины, кожи и пластич. масс и тканей из синтетич. волокна.
Однокомпонентные Л. горячей сушки применяют для покры-
тия медных проводов; такие провода легко паяются без пред-
варительной зачистки концов.
Лаки эпоксидные состоят из р-ров различных
типов эпоксидных смол и отвердителей. При нанесении их на
поверхность образуются полимеры с трехмерной структурой.
Из эпоксидных смол чаще всего применяют твердые диэпок-
сиды с мол. весом в пределах 900—4000, полиэпоксиды на
базе продуктов конденсации фенолов с формальдегидом, а так-
же различные эпоксиэфиры (продукты этерификации диэпокси-
дов и полпэпоксидов жирными к-тами растительных масел).
Роль отвердителей выполняют различные полиамины, низко-
молекулярные полиамиды (см. Смолы полиамидные), аддукты
аминов с эпоксисоединениями, полиизоцианаты, феноло-,
меламино- и мочевино-альдегидные смолы. Для получения
покрытий с повышенной термостойкостью отвердителями слу-
жат ангидриды к-т. Из растворителей применяют кетоны
(метилизобутилкетон, циклогексанон), простые эфиры (целло-
зольв), ароматич. углеводороды и их смеси, а также нитропара-
фины. Л., отверждаемые аминами и полиамидами, применяют
для покрытий холодной и горячей сушки; они относятся к числу
двухкомпонентных. Работа с аминами требует мер предосто-
рожности. Фенольные, мочевинные и меламиновые смолы от-
верждаются только при 150—180°; Л. этого типа однокомпо-
пентны. Стабильность двухкомпонентных Л. после смешения
р-ров в основном зависит от типа отвердителя; наиболее ста-
бильные системы (до 48 часов годности) получают при отвержде-
нии полиамидами и аддуктами. Покрытия на основе эпоксидных
Л. обладают высокой твердостью и адгезией, хорошей эластич-
ностью. Применяют для окраски деревянных, металлич.,
а также бетонных поверхностей. Высокая водоустойчивость,
химич. стойкость (конц. р-ры щелочей, слабые р-ры к-т) п
стойкость к действию растворителей дают основание применять
их для защиты химич. аппаратуры или аппаратуры, работаю-
щей в условиях высокой влажности. Л., содержащие эпокси-
эфиры, не требуют отвердителя и высыхают при комнатной
темп-ре, их применяют для электроизоляции медных прово-
дов и др.
Лаки феиоло-альдегидные — спиртовые р-ры
резольпых смол (см. Смолы феноло-альдегидные). После нанесе-
ния и горячего отверждения 160—170° они образуют пленки,
обладающие хорошими электроизоляционными свойствами,
водо- и кислотостойкостью, а также термостойкостью. Недо-
статками таких покрытий являются темный цвет и хрупкость.
Применяются для защиты химич. аппаратуры, а также в ка-
честве связующего для слоистых пластиков.
Лаки меламино- и мочевино-формаль-
дегидные представляют собой композиции, содержащие
алкоксилированные (гл. обр. бутоксилированные) продукты
конденсации меламина или мочевины с формальдегидом (см.
Смолы мочевино-формальдегидные и Смолы меламино-формаль-
дегидные) и растворители (спирты и ароматич. углеводороды).
Они образуют бесцветные, прозрачные, но очень хрупкие
покрытия. В связи с этим их пластифицируют либо насыщенной
полиэфирной смолой с добавлением перед нанееением кислот-
ного катализатора отверждения, либо алкидной или эпоксид-
ной смолой. Л. первого типа (холодной сушки) служат для
покрытия по дереву (лыжи, паркет), а лаки второго типа,
как таковые или в виде эмалевых красок горячей сушки
(темп-ра 120—150°), — для защитных покрытий по металлу.
Покрытия на основе этих Л. обладают высокой твердостью и
хорошей светостойкостью.
Лак дивинилацетиленовый (этиноль) — р-р
в ксилоле полимеров дивинилацетилена. Образует покрытия,
высыхающие на воздухе и обладающие большой водонепрони-
цаемостью, стойкостью к химич. реагентам, бензину, маслам.
Применяется в судостроении.
905
ЛАКМУС — ЛАКТАМЫ
906
Лаки кремнийорганические (полисилок-
сановые силиконовые) представляют собой р-ры различных
кремнийорганических полимеров в органич. растворителях
(чаще всего — в ароматич. углеводородах). Кремнийорганич.
Л. после горячей сушки образуют термостойкие покрытия
с высокими диэлектрическими показателями, сохраняющимися
в условиях большой влажности. Их недостаток — малая
механич. прочность и слабая адгезия пленок. С целью улучше-
ния механич. свойств в состав Л. вводят другие синтетич.
смолы, чаще всего алкидные. В последнем случае удается
значительно повысить адгезионные свойства покрытий, хотя
термостойкость несколько уменьшается. Силиконовые Л. при-
меняют для пропитки электроизоляционных обмоток, защиты
горячих металлич. поверхностей, наир, дымовых труб, кипя-
тильников, нагревательных котлов, выхлопных труб и г. п.
Используют также при окраске стеклянных и керамич. по-
верхностей.
Лаки эфироцеллюлозные — р-ры эфиров целлюлозы
в летучих растворителях, содержащие смолы и пла-
стификаторы. Все они быстро высыхают при комнат-
ной темп-ре; их наносят обычно распылением. Разли-
чают нитратцеллюлозные Л‘. (или нитролаки), ацетил-
целлюлозные, ацетобутиратцеллюлозные, этилцел-
люлозные.
Лаки нитратцеллюлозные (нитролаки)
обычно получают на основе лакового коллоксилина — нитро-
целлюлозы с содержанием от 10,7 до 12,5% азота. Л. и полу-
чаемая пленка очень горючи. Наиболее широко применяют
нитролаки, в состав к-рых, кроме коллоксилина и растворите-
лей, входят смолы и пластификаторы. Из смол используют:
модифицированные маслом алкидные смолы, мочевино-фор-
мальдегидные и Меламино-формальдегидные, модифицирован-
ные канифолью фенольные, простые поливиниловые эфиры,
полиакрилатные смолы, кетонные и нек-рые природные смолы.
Из пластификаторов применяют касторовое масло, фталаты,
фосфаты и др. Летучие растворители, входящие в состав
нитролаков, представляют собой смесь истинных растворите-
лей (сложные и простые эфиры, кетоны), скрытых раствори-
телей (спирт) и разбавителей (углеводороды). Разбавители
снижают стойкость Л. и часто являются растворителями смол
и пластификаторов, присутствующих в Л. Нитроцеллюлозные
Л. быстро высыхают при нормальной темп-ре и образуют
хорошо полирующиеся пленки. При комбинировании коллок-
силина с такими смолами, как алкидные, образуются покрытия,
устойчивые в атмосферных условиях. Механич. свойства
нитролаковых покрытий, их адгезия, а также цвет и свето-
стойкость в значительной степени зависят от входящих в состав
Л. смол и пластификаторов. Л. этого типа применяют для
покрытий по дереву (мебельные, карандашные Л.) и по ме-
таллу. На их основе готовят, например, эмалевые краски
для отделки автомобилей. Л., содержащие в нелетучей части
только коллоксилин и пластификаторы, применяют для отдел-
ки кожи.
Лаки ацетилцеллюлозные и ацет.о бу-
тиратцеллюлозные представляют собой р-ры со-
ответствующих эфиров целлюлозы в кетонах, сложных эфирах,
хлорированных углеводородах и нитропарафинах, а также в
смеси хлористого метилена с этиловым или метиловым спир-
том. Основное преимущество этих Л. — светостойкость и не-
горючесть их пленок. К недостаткам, значительно ограничи-
вающим их применение, особенно ацетилцеллюлозных, отно-
сятся трудность подбора хорошо совмещающихся смол и
пластификаторов. Основное назначение ацетилцеллюлозных
Л. — специальные светостойкие покрытия по металлу, бумаге
и по тканям. Ацетобутиратцеллюлозные Л. применяют как
покрытия по бумаге и текстилю.
Лаки этилцеллюлозные можно применять
для замены нитроцеллюлозных Л. Покрытия из этилцеллю-
лозных Л. обладают большей химич. стойкостью и меньшей
горючестью, чем нитроцеллюлозные Л. Для растворения этил-
целлюлозы применяют спирты, простые и сложные эфиры,
кетоны.
Лит.: Дринберг А. Я., Технология пленкообразую-
щих веществ, 2 изд., Л., 1955; Киселев В. С., Олифа и
лаки, 3 изд., М.—Л., 1940; Дринберг А. Я., Г у р е-
в'и ч Е. С., Тихомиров А. В., Технология неметалли-
ческих покрытий, Л., 1957; Пэйн Г. Ф., Технология орга-
нических покрытий, пер. с англ., т. 1, Л., 1959; Якубович
С. В., Испытания лакокрасочных материалов и покрытий,
И.—Л., 1952; Хим. наука и пром-сть, 1959, 4, К» 3 (статьи
Штанько Н. Г., Благонравовой А. А., Раскина Я. Л..Свер-
длина М. С., Лубмана А. М.); Благонравова А. А.,
П рон ина И. А., Лакокрасочные материалы и их применение,
1961, № 2,3; Благонравова А. А., там же, 1960, № 6,
68; Киттель Г., Целлюлозные лаки, пер. с нем., Л.,
1957; Ш ампетье Г., Рабата Г., Химия лаков, красок
и пигментов, пер. с франц., [т. 1] —2, М., 1960—62; Лакокрасоч-
ные материалы. Сырье и полупродукты. Справочник, под ред.
И. Н. Сапгира, М., 1961; Д о м б р о у В. А., Полиуретаны,
пер. с англ., М., 1961. А.’Л. Благонравова.
ЛАКМУС — красящее вещество, добываемое Л не-
которых видов лишайника. Состав Л. сложен и окон-
чательно не установлен. Красящее вещество Л. —
слабая кислота, азолитмин (индофенол) C7H7NO4; соли
ее имеют синий цвет. Л. применяют как индикатор для
определения реакции среды: в кислой фазе Л. окрашен
в красный цвет, в щелочной — в синий, в нейтральной
остается без изменений. Для получения Л. растения
измельчают и сбраживают в водных р-рах поташа или
аммиака, смесь постепенно приобретает темно-синий
цвет, затем в нее добавляют мел или гипс; получив-
шуюся массу формуют в виде небольших кубиков и
высушивают на воздухе. На практике применяют
водный настой Л., чаще в виде бумаги реактивной.
ЛАКТАМЫ — внутренние циклич. амиды амиио-
карбоновых кислот, содержащие группировку
—СО—NH — в кольце; таутомерная «енольная» форма
Л. наз. лактимной:
/”С=О <С-ОН
: । -у* • а
V, NM \.N
В зависимости от типа аминокислот, образующих
Л., различают р-, у-, 6-, е-лактамы и т. д.
Свойства нек-рых Л. приведены в таблице.
Название Формула Т. пл., °C Т. кип., сС/лш
₽-Пропиолактам СНо-СНв Ан—со 73-74 —
у-Бугиролактам (пирроли- дон) ^л-СН^со CH2-NH'' 24,6 245/760 133/12 •
-
б-Валеролактам (а-пипе- ридон) Н NH ° 39—40 256/760 137/14 64/0,4
е-Капролактам (2-кето- гексаметиленимин) Н2С-'СН8 н8с' сна Hed с-о 74 Н 68—69 139/12
Основными методами синтеза, общими для боль-
шинства Л., являются циклизация аминокарбоновых
к-т или их производных (1) и циклизация амидов
галогене- или оксикарбоновых к-т (2):
/В
,(СН2)Л
\*Н2
.хСО
— (СнХ j (П
^^NH
,Х ^N-R
(СН2)„ — (СШ„ | (2)
'CONHR ^-СО
В=СООН, COOR, CONHa, CN
X— галоген, ОН
Для циклизации у- и б-аминокарбоновых к-т доста-
точно уже нагревания их выше темп-ры плавления.
Тенденция к замыканию лактамного кольца часто
настолько велика, что не удается получить соответ-
ствующую аминокислоту, как, напр., в случае о-ами-
нофенилглицина, о-аминофеноксиуксусной к-ты, у-аМи-
нокротоновой к-ты и т. п. у-Л. иногда можно полу-
чать и из N.N-дизамещенных аминокарбоновых к-т:
СН3-СН-СНа-С(СвН5)2 SOCia
N(CH3)a СООН
СН3-СН-СН3
I > C(C3H5)a+SOa+ HCi-t-CH3Cl
CHj-N.—CO
907
ЛАКТАМЫ
908
P-Аминокислоты'циклизуются только при нагрева-
нии в присутствии PGla, SOC12 или дициклогексил-
карбодиимида. Особое значение приобрел последний
метод, с помощью к-рого впервые удалось получить
биологически активный пенициллин:
zS\
С6Н5ОСН2СОЫНСН-СН C(CHg)z ---
НООС .NH-ibiCOOH
ZS4
CeH5OCH2CONHCH-CH С(СН3)а
' СО-N—СНСООН
При дегидрогалогенировании анилида а-хдорфонил-
укс.усцой к-ты промежуточно образуется неустойчи-
вый а-Л.:
c6h5nhx
СО
СВН5СН-С1
CeHb-N4
I Xе0
С6н5-сн
CeH5NHCHCONH2
с6н5 .
е-Аминокарбоновые к-ты, а также такие аминокисло-
ты, в к-рых аминогруппа еще более удалена от кар-
боксильной, при нагревании в основном превра-
щаются в полиамиды и лишь при соблюдении особых
условий удается выделить соответствующие Л.
Аналогично свободным аминокислотам, циклизуют-
ся их эфиры, напр.:
’ СНа-СН2-COOR - NH3 СН2-СН2
НООС-СН HOOC-QH СО
\'Н2 'NH
Эфиры 0-аминокислот превращаются в 0-Л. под дей-
ствием металлоорганич. соединений:
CeH5CH2CONHCH-CHi ’ ,CH3MgiT,
0=6 NHC6HS'
OCgHB
СбН5СН2СОЫН-СН-СН2
0=6—n-c6h5 .
CeH5N=CH-C6H5 Гс6н5- N CH-C6H5
+ ZnBr 6h3
ROCOCHaBr ROCOZ
CgHg-N-CH-CeHg
—- о=с-снг
Амиды и нитрилы аминокислот превращаются в Л.
при сильном нагревании:
Амиды у-, б- и е-галогенокислот циклизуются под
действием таких оснований, как аммиак, водная
щелочь или алкоголяты металлов, напр.:.
zCH2Br NH ' ^СН2
(СН^ ----------^(CHaU /МН
4CONHj ^"CO
c6h5n
CeH5C
со
^сн-с6н5
Н ^N-CgHg
со
Удобным методом синтеза Л. является обработка
лактонов аммиаком или аминами при сильном нагре-
вании:
ОН _^NR
(СНз)„ | +h2nr—-<сн2)„ —- (СН2)„ I
^CO 'cONHR "'~''СО
К менее общим методам синтеза Л. относятся цикло-
димеризация кетенов с иминами и нитрозосоединения-
ми, циклизация амидов непредельных к-т, перегруп-
пировки Шмидта и Бекмана для циклич. кетонов,
перегруппировка изонитронов и т. д.
r2c=co „ r2C=C0
R N-О —------- R’N=CR2 — -----
CH2=CH сн2-сна
(СН2)„ *-1 Z(CH2)„
RNH-СО R-N CO
R2C-NR'
RzC-CO
(CH2)„ C-NR
— О
Превращения Л. протекают либо с раскрытием лак-
тамного кольца, либо с его сохранением. Так, цикл
раскрывается мри гидролизе, аммонолизе, гидроге-
нолизе и полимеризации:
нон ,-<оон
н+ или ОН' l''NH2
r2nh ,--conr2
<NH2
|5-Л. образуются только под действием едких щело-
чей либо амидов щелочных металлов; при исполь-
зовании аминов или алкоголятов получаются лишь
амиды непредельных к-т:
KNH2j R-CH-CHa
NH3 ’ r'-n—io
R-CH-CH2CONHR —
* R"OK
о,,,,,,*- R-CH=CH-CONHR'
CH2-(JH-C6H5 Н , сн2-сн2-свн5
co—n-c6h5 [n.j co-nh-c6h5
^co
(CHa)„ I —- н [NH(CH2)„COJx oh
Устойчивость лактамного кольца зависит как от его
величины, так и отколичества и расположения замести-
телей. Наиболее устойчивы 5- и 6-членные Л., наи-
менее устойчивы 4-членные (если не считать 3-членных,
909
ЛАКТАТДЕГИДРОГЕНАЗА - ЛАКТОЗА
910
к-рые вообще не выделены в свободном виде). Полиме-
ризуются легче всего Л. с семью и более атомами в цик-
ле; напр., при полимеризации е-капролактама легко
получается капрон. Лактамное кольцо сохраняется
в случае таких реакций, как алкилирование, ацили-
рование, галогенирование и нитрозироваиие, а также
при различных превращениях карбонильной группы:
,--со
• I
’•«-NR
с°
Г9°
NCOR
c=s
NH
C=NR
I
NH
,--со
N—Hal
—со
I
1-N-NO
z—CO
' I
'-NH
cs2 | (ch3i2so,
,--с-осн3
' II
'--N
I rnh2
-C-NHR
JI
-N
Л. широко используются в органич. синтезе; многие
из них являются биологически активными в-вами.
Очень важная область применения лактамов — произ-
водство полиамидных волокон.
Лит.: Но ube в—W еу 1, Bd 11/2, Stuttgart, 1958, S. 516;
К н у н я я ц И. Л., Д я т к и н Б. Л., Гамбарян Д. П.,
Усп. химии, 1956, 25, вып. 7, 785. Н. П. Гамбарян.
ЛАКТАТДЕГИДРОГЕНАЗА (ла ктико дегидрогена-
за) — фермент, катализирующий взаимопревращение
пировиноградной и молочной к-т.
СНзСОСООН + ДПН-Н + Н+ - СН3СН(ОН)СООН + дпн
(ДПН — дифосфопиридиннуклеотид).
В физиологических условиях при pH около 7,0 равно-
весие практически сдвинуто в сторону образования
молочной кислоты. В щелочной среде (pH 10,0), а
также при добавлении веществ, которые связывают
пировиноградную кислоту (KCN, гидразины), равно-
весие сдвигается в сторону образования пировино-
градной кислоты. Л. играет важную роль в обмене
веществ; она является одним из ферментов процесса
анаэробного гликолиза и широко распространена в
животных тканях, особенно в сердечной и скелетных
мышцах. Л., полученная из сердца крупного рогато-
го скота, — кристаллич. продукт, мол. в. 135000 ±
± 15000, Михаэлиса константа Ам для пировиноград-
ной к-ты 5,2 • 10 5 М, число оборотов 3,7 • 104 мин"1
при pH 7,0. Помимо пировиноградной к-ты, Л. ка-
тализирует восстановление гомологичных а-кето-
кислот (а-кетомасляной, а-кетовалериановой, а-кето-
капроновой), однако скорость каталитич. реакции
в этом ряду уменьшается с увеличением длины угле-
водородной цепи; в отношении щавелевоуксусной,
а-кетоглутаровой, а также р- и б-кетокислот, кетонов
и ацетальдегида Л. неактивна. Л. обладает строгой
стереоспецифичностью: она окисляет только L-молоч-
ную к-ту, а при катализируемом ею восстановлении
оптически неактивной пировиноградной к-ты также
образуется только L-молочная к-та.
Коферментом Л. является никотинамид-
адениндинуклеотид (дифосфопири-
диннуклеотид, ДПН — см. Кодегидрогеназы).
Активность Л. обычно определяют спектрофотомет-
рия. методом, основанным на измерении поглощения
ДПН-Н при 310л(«с. Кристаллич. Л., полученная из
дрожжей или бактерий (цитохром Ъ*), отличается от
Л., подученной из животных тканей, Дрожжевая Л.
(мол. в. 100000) катализирует окисление L-молочноц
к-ты в пировиноградную: СНаСО(ОН)СООН + окис-
ленный цитохром С -»• СНаСОСООН + восстановлен-
ный цитохром С. Дрожжевая Л. — белой с двумя
простетич. группами: флавинмононуклеотидом и ге-
мом. Первичным акцептором водорода окисляемой
молочной к-ты при ферментативной реакции является
флавинмононуклеотид, с к-рого электроны переходят
на гем фермента и затем на цитохром С.
Лит.: Михлин Д. М., Биохимия клеточного дыхания,
М., 1960; Диксон М., Уэбб Э., Ферменты, пер. с англ.,
М., 1961; Methods ip enzymology, ed. S. P. Colowick and N. O.
Kaplan, v. 1, N. Y., 1955. В. Б. Спиричев.
а- ЛАКТОАЛЬБУМИН — один из белков молока;
константа седиментации s = 1,75 единиц Сведберга;
мол. в. 17000; [а]д = —61°. Л. содержит 15,9% азота,
1,9% серы, 7,2% триптофана. В коровьем молоке
количество Л. составляет 2,4% от всех белков. Выде-
ляют Л. осаждением при 2/3 насыщения сульфата
аммония (при pH 6,6). Л. соответствует а-компонецту
на седиментационной диаграмме белков сыворотки
молока (отсюда и его название а-Л.).
Лит. см. при ст. $~Лактоглобулин. В, О. Шпикитер.
Р-ЛАКТОГЛОБУЛИН — один из белков сыворотки
молока; константа седиментации s = 3,1 единицы
Сведберга; мол. в. 35000; изоэлектрич. точка при
pH 5,2. Приблизительный химич. состав Л. из коровь-
его молока (в в на 100 а белка): 15,6 общего азота;
1,6 общей серы; 1,24 аминного азота; 1,07 амидного
азота; 1,4 глицина; 7,4 аланина; 5,8 валина; 15,6 лей-
цина; 6,1 изодейцина; 4,1 пролина; 3,5 фенилаланина;
1,1 цистеина; 2,3 цистина; 3,2 метионина; 1,9 трип-
тофана; 2,9 аргинина; 1,6 гистидина; 11,4 лизина;
11,4 аспарагиновой к-ты; 19,5 глутаминовой к-ты;
5,0 серина; 5,8 треонина; 3,8 тирозина. В коровьем
молоке содержание Л. составляет 11% от всех белков.
Л. выделяют из сыворотки молока солевым фракцио-
нированием; при диализе р-ра Л. при pH 5,2 белок
выпадает в виде кристаллов. Л. соответствует р-ком-
поненту на седиментационной диаграмме белков сыво-
ротки молока (отсюда и его название р-Л.).
Лит.: Белки, под ред. Г. Нейрата и К. Бэйли, пер. с англ.,
т. 3, ч. 1, М., 1958, с. 689. В. О. Шпикитер.
ЛАКТОЗА (4-0-р-П-галактопиранозил-П-глюкопи-
ракоза, молочный сахар) C12II22011,mo.’i. в. 342,3 — вос-
станавливающий дисахарид, построенный из остатков
D-галактозы и D-глюкозы. Вследствие наличия полу-
ацетального гидроксила Л. может существовать в а- и
p-формах. В кристаллич. виде Л. получена в трех мо-
дификациях: в виде a-формы, т. пл. 223°; [а]D — -j-90°;
p-формы, т. пл. 252°; [a]D= +34,9°; моногидрата а-фор-
мы, т. пл. 202°; [a]D = -|-83,50—85°. В водных р-рах
идет мутаротация и образуется равновесная смесь
с [a]D = +52,3°—55,3°. Л. растворима в воде значи-
тельно хуже сахарозы и других дисахаридов; у р-фор-
мы Л. растворимость больше, чем у a-формы. В эфире,
абсолютных этиловом и метиловом спиртах Л. нераст-
ворима; в разбавленном спирте и пиридине раство-
ряется умеренно.
Л. гидролизуется под влиянием кислот и фермен-
тов—р-галактозидаз (лактаз), содержащихся в некою-
911
ЛАКТОНОВ ПРАВИЛО — ЛАКТОНЫ
912
рых видах бактерий, грибов п в кишечнике млекопи-
тающих. Спиртовому брожению Л. не подвергается,
она сбраживается специальными молочнокислыми
дрожжами, под влиянием к-рых Л. вначале гидроли-
зуется, а затем превращается в молочную к-ту (глав-
ный продукт брожения). Аналогично Л. подвергается
лимоннокислому брожению.
Л. восстанавливает р-р Фелинга, аммиачный р-р
AgNO3. Ряд реакций обусловлен также наличием
потенциальной альдегидной группы: образование фе-
нилозазона, бензилфенилозазона и .др. производных.
При окислении концевой альдегидной группы Л.
образуется лактобионовая к-та. При окислении Л.
азотной к-той идет гидролиз ее до моносахаридов
с последующим окислением их через D-галактоновую и
D-глюконовую к-ты до двухосновных D-глюкаровой
(сахарной) и D-галактаровой (муциновой, или слизе-
вой) к-т. При гидролитич. восстановлении Л. обра-
зуются дульцит (галактитол) и сорбит (D-глюцитол).
Л. содержится в молоке млекопитающих в свободном
виде (1,8—7,6%) и в виде олигосахаридов, в частности
трисахаридов нейраминолактоз (см. Нейраминовая
кислота). Обнаружена Л.в пыльце нек-рых растений.
Синтетически Л. получена конденсацией D-глюкозы и
D-галактозы и др. способами. Производят Л. из
молочной сыворотки, к-рая является отходом при из-
готовлении масла и сыра. Применяют Л. в фармации
как наполнитель в порошках, таблетках и экстрактах.
Сладость Л. в 4—5 раз меньше, чем у сахарозы, и как
заменитель сахара ее не используют.
Лит.: Инихов Г. С., Биохимия молока, М., 1956; The
carbo-hydrates. Chemistry, biochemistry, physiology, ed. W.
Pigman, N. Y., 1957; Proceedings ot the 4-h International
congress of biochemistry. Vienna, 1—6 September 1958, v. 1,
L.—N. Y.—P., 1959; Advances in carbohydrate chemictry,
1961, 16, 159. Л. И. Линевич.
ЛАКТОНОВ ПРАВИЛО (лактонов вращения пра-
вило) — эмпирич. правило, согласно к-рому лактоны
альдоновых к-т вращают плоскость поляризованного
света: вправо, если в образовании лактонового кольца
принимает участие гидроксил, расположенный справа
от углеродной цепи; влево, если в образовании лакто-
нового кольца принимает участие гидроксил, распо-
ложенный слева от углеродной цепи. Так, у- и б-лак-
тоны D-глюконовой к-ты (I и II) и D-манноновой к-ты
вращают вправо, а у-лактон D-галактоновой к-ты
(III) вращает влево.
°? 1 ОС со
неон 1 неон 6 1 неон
1 0 1 0 1
НОСН 1 НОСН НОСН
НС неон 1 1 СН
неон НС HlflOH
н»сон Н2С0Н щеон
+ 67,5° + 63,4“ -73“
I II III
Исключением из этого правила являются: лактон
D-аллоновой к-ты (IV), D-дигитоксоновой к-ты (V)
и D-дигитоксонкарбоновой к-ты (VI).
0| 1 °?—i ОС f
неон | СН.> 1 НОСН
1 0 1 0 1 6
неон । неон I
1 1 нс ! 1 НС— ! нс 1
неон неон неон
н2сон СН, неон
сн3
— 12° -28,7“ - 13,7°
IV V VI
Л. п. пользуются для подтверждения конфигурации
у атомов углерода в соответствующих сахарах.
Лит.: М 1 с h е е 1 F., Chemie der Zucker und Polysaccha-
ride, 2 Aufl., Lpz., 1956; The carbohydrates. Chemistry, bioche-
mistry, physiology,ed. W. Pigman, N. Y., 1957. Л. II. Линевич.
ЛАКТОНЫ — внутренние циклич. эфиры оксики-
слот. В зависимости от типа оксикислот, образующих
Л., различают Р-, у-, б-, е-лактоны и т. д. В таблице
приведены свойства нек-рых Л.
Название и формула т. пл., °C Т. кип., °C/ai.u
(З-Пропиол актол СН2-СН2 1 1 0 СО -33,4 155/760 51/10 — 1.4131 (20“)
у-Бутиролактон (тетрагидро- 2-фуранон) СН2—СН2—СО 1 1 СН2 0 203- 204/760 89/12 1,1441 (0“)
6-Валеролактон (тетпагидро- 2-пирон) СН2—(СН2)2—СО 1 1 СН2 0 -12,5 218- 220/760 88/4 1.0794 (20°) 1.4503 (20“)
е-Капролактон СН2—(СН2)3—СО 1 1 СН2 0 98—99/2 1,0693 (20°) 1.4611 (22“)
Л. как по методам синтеза, так и по свойствам напо-
минают соответствующие лактамы. Основными мето-
дами синтеза Л. является циклизация галогено- и
оксикислот. Галогенокислоты в присутствии средств,
отщепляющих галогеноводород (карбонаты или би-
карбонаты щелочных металлов, едкие щелочи, тре-
тичные амины, влажная окись серебра ит. п.), цикли-
зуются, образуя соответствующие Л.:
zHal ,0
(сн,)< -.(CHs)n-; I
VcOOH Vco
Получаемые этим методом р-Л. сразу же удаляют из
сферы реакции, т. к. они могут взаимодействовать
с солями щелочных металлов. При получении дру-
гих Л., включая и высшие, таких предосторожностей
не требуется. у-Л. можно получить даже простой пере-
гонкой у-галогенокислот:
С1СН2СН2СН2СООН CH2CH2CH2io + HCI
у- и б-Л. образуются при нагревании серебряных
солей дикарбоновых к-т с галогенами:
/СОО Ag 4-Hal Г „ zHaI 1
<сн2) h\c00 Ag > [( H2)^\cOOAg ] - Aglfii
б- и у-Оксикислоты циклизуются чрезвычайно легко,
последние уже при растворении в воде:
ZOH zo
(СН2) -»(СН2)п< |
Vcooh со
поэтому многие у-оксикислоты в свободном виде
неизвестны. р-Оксикислоты, как правило, при дегид-
ратации образуют не Л., а непредельные к-ты, п
ко р, р-бис-(трифторметил)-Р-оксипропионовая
при нагревании с Р2О5 циклизуется в р-Л.:
CF3 CF3.
VC-OIL, СООН )C-CH,
CF3Z | ' CF/ I I
OH o-co
а-Л. неизвестны; при нагревании а-оксикислот
зуются лактиды:
толь-
к-та
обра-
/Ч /R
2 %-СООН __ °C 4R
к 6н С- /СО
913
ЛАКТОНЫ — ЛАМИНАРАН
914
е-Оксикислоты и высшие оксикислоты также не
циклизуются в Л. и при дегидратации превращаются
в непредельные к-ты, лактиды или полиэфиры. По-
добно свободным оксикислотам, но в более жестких
условиях, циклизуются эфиры у- и ё-оксикислот
(внутримолекулярная переэтерификация).
Менее общими методами синтеза Л. являются циклизация
непредельных к-т (1), циклодимеризация кетонов с карбониль-
ными соединениями (2), окисление циклич. кетонов или оки-
сей (3, 4), восстановление внутренних ангидридов дикарбоновых
к-т (5), дегидрирование к-т (6), внутримолекулярная реакция
Канниццаро (7), свободнорадикальное присоединение спир-
тов к акриловой к-те или ее эфиру (8) и т. п.
R СН=СН RCH—QH2
\.Н2 ---»- \н2 (1)
НО-СО О—со
R2CO + R’2C=CO —- r2c-o
R'2C-CQ (2)
«ислотз Каро
(CHg)n I —г— (СН2)„ О (3)
^СО ^СО
^-СН2
<СН2>„ \)
'-со
аСН(С6Н8)2
СООН
Na
<f2H5OH
С6Н5-СН-СН2СНО RQNa_ С6Н5-СН-СН2СООН
с6н6-сн-сно с6н5-(!:нсн2он
___свН5-С.Н-С.Н2-^__
” свн5-сн—с.н2—сг"
R'CH2OH + CH2=CHCOOR (СН3)3СООС(СН3)3
R'—СН—СН2
I /СН2
о—со
Л. обладают многими свойствами сложных эфиров.
Так, при нагревании с кислотами или щелочами,
а иногда и просто с водой они {гидролизуются в соот-
ветствующие оксикислоты, со спиртами переэтерифи-
цируются в эфиры оксикислот, с аминами дают амиды,
с гидразином и фенилпгдра'зином — гидразиды окси-
кислот:
V „СО
V>-COOH
X=OH, RO'R2N, rnhnh
Т. обр., при реакциях Л., связанных с размыканием
кольца, разрывается обычно связь ацил — кислород,
а не алкил — кислород.
В присутствии избытка галогеноводородной к-ты
образуются эфиры галогенокислот:
< О
R I
1 СО
НН.П
R'OH
,—Hal
-R
K-COOR'
В случае р-Л., в зависимости от строения исходного
вещества, характера реагента и условий, возможны
два направления реакции, причем получаются либо
производные оксикислот (9), либо замещенные кисло-
ты (10):
r,c-cr2
| : I +МХ —MOCR3CRSCOX (9)
О у СО
Rs С--С Rs
-I- | + MX - XCRsCRoCOOM (10)
О---СО
MX = ROH, RSH, MH, RCOC1, SOC12, HHal,
RSO2H, NaCN, RCOONa, неорганич. соли и пр.
Другой характерной особенностью р-Л. является
способность распадаться при умеренном нагревании
на олефины и двуокись углерода:
r2c—со
I I -» RsC=CRs + СО2
R,C- О
у- и ё-Л. не отщепляют СО2 даже при нагревании до
500°. Высшие Л. при повышенной темп-ре изомери-
зуются в непредельные к-ты. Общие свойства Л. —
способность восстанавливаться в жирные к-ты (11),
конденсироваться с ароматич. углеводородами (12),
полимеризоваться в линейные полиэфиры: (13)
^-СО
(СНДл | Л. Н(СН2)„СООН (11)
^0
AlClg
(СН2)„СООН
(12)
о ’
но (сн2)Ло н
J *
(13)
Л. широко используются в органич. синтезе, в
произ-ве душистых и лекарственных веществ. Многие
Л., напр. иодофиллотоксин или антибиотик клавацин,
являются биологически активными веществами.
Лит.: Chemistry of carbon compounds, ed. E. H. Rodd,
v. 1, pt B, Amst., 1952, p. 796; 3 а у г г Г. Э., в кн.:
Органические реакции, пер. с англ., сб. 8, М., 1956, с. 392;
Н и к и ш и н Г. И., Воробьев В. Д., Петров А. Д.,
ДАН СССР, 1961, 136, № 2, 360. Н. П. Гамбарян.
ЛАЛЛЕМАНЦИИ МАСЛО — см. Жиры расти-,
тельные.
ЛАМИНАРАН (ламинарии) — полисахарид бурых
водорослей (Laminaria, Fucaceae), построенный из
остатков D-глюкозы, связанных преим. р-1,3-связями
(нек-рые остатки D-глюкозы образуют р-1,6-связи).
В состав Л. входит также D-мапнит, занимающий кон-
цевое положение в полисахаридной цепи.
Участок цепи молекулы ламинарана.
Очищенные препараты Л. — белый аморфный поро-
шок. Мол. вес Л. 1900—5000. Получены 2 формы
Л.: 1) Л., осажденный при стоянии подкисленных
водных р-ров, очень плохо растворим в холодной, но
растворим в горячей воде; 2) Л., осажденный из
водных р-ров спиртом, хорошо растворим в холодной
воде и разб. спиртовых р-рах. Из водных р-ров Л.
осаждается также глицерином. В воде Л. образует
коллоидные р-ры; [a]Jj от —6 до —16° (повышается
при разведении р-ров Л.).
Л. легко гидролизуется при нагревании с разб.
неорганич. к-тами до D-глюкозы (99%) и маннита (1%).
При неполном гидролизе Л. щавелевой к-той или
915
ЛАНОЛИН — ЛАНТАНИДЫ
916
ферментами образуется преим. дисахарид ламин-
арибиоза — 3-(р-В-глюкопиранозил)-В-глюкоза.
Ферменты, гидролизующие Л., — л а м и н а р а и-
а з ы (ламинариназы) обнаружены в водорослях и
нек-рых плесенях, в экстрактах пшеницы, овса, яч-
меня, картофеля, луковицах гиацинтов и в др, расте-
ниях, а также улитках. Сульфатированный Л., со-
держащий по две сульфатные группы на глюкозную
единицу, подобно гепарину уменьшает свертываемость
крови. Сульфаты Л., содержащие 0,62 моля H2SO4
на глюкозную единицу, влияют на липидный обмен
человека и животных. Л. является запасным полиса-
харидом водорослей, его количество осенью достигает
42—49% от сухого веса растения и резко снижается
весной. Водоросли, содержащие Л., используются как
пищевой продукт (морская капуста) и для произ-ва
Л. и альгиновых к-т. Л. может быть использован для
произ-ва глюкозы, 2-дезокси-0-рибозы и сульфатиро-
ванных препаратов.
Лит.: Whistler К. L., S mart С. L., Polysaccharide
Chemistry, N. Y., 1953; Handbuch der Pflanzenphyslologie,
Bd 6, B.-[u. a.], 1958; Percival E. &. V., Structural car-
bohydrate chemistry, L., 1950; Industrial gums. Polysaccharides
and their derivatives, N. Y.—L., 1959. Л. И. Линевич.
ЛАНОЛИН — см. Воски.
ЛАНТАН (Lanthanum) La — химич. элемент III гр.
периодич. системы Менделеева с п. н. 57; ввиду близо-
сти свойств с лантанидами описан в ст. Лантаниды.
ЛАНТАНИДЫ (лантаноиды). Содержание:
Введение..........................................915
Физические свойства...............................916
Электронная структура, валентность, свойства ионов . . 918
Химические свойства........................... . . 920
Характеристика некоторых соединений лантанидов, вы-
деляемых из растворов...........................921
Источники сырья и извлечение редкоземельных эле-
ментов .........................................923
Разделение редкоземельных элементов ........... 925
Получение металлов..............................927
Методы анализа..................................927
Области применения..............................928
Введение. Лантаниды — семейство из 14 химич.
элементов с порядковыми номерами 58—71, располо-
женных в VI периоде системы Менделеева за лан-
таном и сходных с ним по свойствам а. Эту группу
элементов вместе с лантаном и иттрием (к-рый всегда
сопутствует лантанидам в минеральном сырье), а
также скандием часто объединяют общим названием
«редкоземельные элементы» (РЗЗ).
Лантаниды (РЗЗ) подразделяются на две подгруппы:
цериевую [(La), Се, Pr, Nd, Pm, Sm, Ей] и ит-
триевую [Gd, Tb, Dy, Ho, Er, Tu, Yb, Lu, (Y)].
По физико-химич. свойствам Л. весьма сходны между
собой, что объясняется строением их электронных
оболочек: по мере увеличения заряда ядра структура
двух внешних электронных уровней у атомов Л. не
изменяется, т. к. происходит заполнение электронами
глубоколежащего 4/-уровня. Максимально возможное
число электронов на /-уровне (14) и определяет число
элементов семейства Л.
Природный изотопный состав лантана и Л. приведен
в табл. 1 (искусственно полученные радиоактивные
изотопы см. на цветной вклейке в ст. Изотопы). Из
радиоактивных изотопов в качестве изотопных инди-
каторов для контроля разделения Л. и др. целей
используют La140 (Ti/2 = 40,22 час), Се141 (Т:/2 =
= 33,1 дня), Рг442 (Ti/2 = 19,2 час), Nd44’ (Ti/2 =
= 11,9 дня), Sm467 (Ti/2=47 час), Eu452 (Ti/2 = 9,2 час),
Ho499 (Ti/S = 27,3 час), Er499 дня), Yb179
(Ti/2 = 102 час) и др.
Радиоактивный элемент прометий получен искус-
ственно. Вопрос о нахождении его в земной коре
окончательно еще не решен. Эффективные сечения
а Для удобства изложения лантан также описан в данной
статье.
захвата тепловых нейтронов атомами лантана и Л.
также приведены в табл. 1. Особенно высокие сечения
захвата имеют Gd, Sm и Eu. По сечению захвата Gd
(44000 барн) превосходит все известные элементы.
Таблица 1. Порядковый номер, атомный вес и природные
изотопы лантанидов
I Порядно- ] выйномер Элемент Атомный вес (по данным на 1962) Массовые числа при- родных изотопов н их содержание (в скобках) Сечение захвата тепловых нейтронов барн/атом
57 Лантан La 138,91 138 ( 0,089); 139 (99,911) 8,9
58 Церий Се 140,12 136 (0,195); 138 (0,265); 140 (88,45); 142.(11,10) 0,70
59 Празеодим Рг 140,907 141 (100) 11,2
60 Неодим Nd 144,24 142(23,17); 143(12,2); 144 (23,87); 145 (8,29); 146 (17,18); 148 (5,72); 150 (5,60) 44
61 Прометий Рт (147) не стабилен, в при- роде не обнаружен —
62 Самарий Sm 150,35 144 (2,87); 147 (14,94); 148(11.24); 149 (13,85); 150 (7,36); 152 (26,90); 154 (22,84) 6500
63 Европий Ей 151,96 151 (47,77); 153 (52,23) 4500
64 Гадолиний Gd 157,25 152 (0,20); 154(2,15); 155 (14,78); 156 (20,59); 157 (15,71 ; 158 (24,78); 160 (21,79) 44000
65 Тербий ТЬ 158,924 159 (100) 44
66 Диспрозий Dy 162,50 156 (0,052); 158 (0,0902); 160 (2,29); 161 (18,88); 162 (25,52); 163 (24,97); 164 (28,18) 1100
67 Гольмий Но 164,930 165 (100) 64
68 Эрбий Ег 167,26 162 (0,136); 164 (1,56); 166 (33,41); 167 (22,94); 168 (27,07); 170 (14,88) 166
69 Тулий Tu 168,934 169 (100) 118
70 Иттербий Yb 173,04 168 (0,14); 170 (3,03); 171 (14,34); 172 (21,88); 173(16,18); 174 (31,77); 176 (12,65) 36
71 Лютеций Lu 174,97 175 (97,5); 176 (2,5) 108
«Иттриевые земли» открыты Гадолином в 1794 в минерале,
найденном в Швеции (близ Иттербю) и названном позже гадо-
линитом. Спустя несколько лет, в 1803, Клапрот и одновре-
менно Берцелиус выделили из «тяжелого камня бастенза»
новую «цериевую землю». В последующем, на протяжении
примерно ста лет были, открыты и выделены из иттриевых и
цериевых земель все Л. Так,Берцелиус в 1814 получил из це-
риевых земель церий, а спустя двадцать пять лет Мозандер
выделил из цериевых и иттриевых земель окись лантана,
«дидима» (смесь окислов празеодима и неодима), тербиевую
и эрбиевую земли. Дальнейшее «расщепление» смесей окислов
и открытие новых элементов было ускорено открытием спект-
рального анализа. Из эрбиевых земель в 1878 мариньяк выде-
лил иттербий, в 1879 Клеве открыл в них эрбий, туллий и голь-
мий. В том же году Лекок де Буабодран получил из цериевых
земель самарий, а в 1880 Мариньяк выделил гадолиний. Д. И.
Менделеев в 1873 впервые применил для разделения лантана
и «дидима» дробную кристаллизацию двойных нитратов. Ис-
пользуя этот метод, Ауэр фон Вельсбах в 1885 показал, что
«дидим» представляет собой смесь неодима и празеодима.
В последующие годы были открыты диспрозий, европий и
лютеций. К 1907 оставался неоткрытым лишь элемент с Z — 61.
Последний, оказавшийся радиоактивным, был получен лишь
в 1947 И. Маринским и Л. Глендениным из осколков деления
урана в ядерпом реакторе и назван ими прометием.
Хотя открытие Л. было завершено к началу 20 в.,
многие из них не были выделены в достаточно чистом
виде и были мало исследованы. В последние 15 лет
разработаны новые, более эффективные методы разде-
ления .Л. В настоящее время (1962) все Л. получены
не только в виде чистых соединений, но и в виде чистых
металлов.
физические свойства. РЗЭ — металлы серебристо-
белого цвета. Нек-рые из них имеют слегка желтова-
тый цвет (напр., Рг и Nd). Металлы кристаллизуются
917
ЛАНТАНИДЫ
918
в плотной гексагональиой или кубич. граиецеитриро-
ванной решетках. Исключение составляют Sm (ром-
боэдрич. структура) и Ей (кубич. объемноцентриро-
ванная структура) (см. табл. 2). La, Се, Рг, Nd, Sm,
Gd, Tb, Yb имеют аллотропия, модификации. В табл. 2
ны, нек-рые из них проявляют ферромагнитные свой-
ства (Gd, Dy, Но). а-Лантан переходит в состояние
сверхпроводимости при 4,9° К, p-La — при 5,85° К.
Для Л. сверхпроводимость не обнаружена даже при
темп-рах ниже десятых долей °К.
Таблица-2. Физические свойства лантанидов, лантана, иттрия и скандия3
а [еталл Кристаллич, структура Параметры решетки, А Атомный радиус (для координ. числа 12), А Плотность (рентге- новская), г/см3 Т. пл., °C Т. кип. (ориентиро- вочные данные), °C Теплота плавления, ккал/г-атом Теплота испарения (при 25°), ккал /г-атом Атомная теплоем- кость (при 0°), кал/г-атом • град Уд. электросопро- ' тнвление (при 25°), ом • см • 10° Работа выхода элек- трона, эв
57 a-La Гексагональная плотно упа- кованная а = 3,770 с = 12,159 1,877 6,162 920 ± 5 3470 1,6 99,5 6,27 56,8 3,33
58 у-Се Гранецентрированная куби- ческая (тип Си) а = 5,1612 1,825 6,768 804 + 5 3470 1,238 + 4 97,6 6,37 75,3 2,84
59 a-Рг Гексагональная плотно упа- кованная (тип La) а = 3,6725 с = 11,8354 1,828 6,769 935 + 5 3017 1,650 84,7 6,38 68,0 2,7
60 a-Nd То же а = 3,6579 с = 11,7992 1,821 7,007 1024 ±5 3210 1,705 + 19 75,6 6,52 64,3 3,3
62 a-Sm Р омбоэдричес кая а = 8,996 а=23°13' 1,802 7,536 1072 + 5 1670 2,061 + 15 50,6 11,80 88 3,2
63 Ell Объемноцентрированная ку- бическая (тип W) а = 4,5820 2,042 5,245 826 + 10 1430 2,0 42,2 6,20 81,3 2,54
64 a-Gd Гексагональная плотно упа- кованная (тип Mg) а = 3,6360 с = 5,7826 1,802 7,886 1312 ± 15 2830 2,1 80,9 9,633 140,5 3,07
65 a-Tb То же а =3,6010 с =5,6936 1,782 8,253 1368 + 10 2480 2,2 72,0 7,136 — 3,09
66 Dy а = 3,5903 с = 5,6475 1,773 8,559 1380 ± 20 2330 3,8 69,8 6,72 56 3,09
67 Ho » » а= 3,5773 с = 5,6158 1,776 8,779 1500 ± 25 2380 4,1 69,0 6,45 87 3,09
68 Er а= 3,5588 с = 5,5874 1,757 9,062 1525 ±25 2390 4,1 73,6 6,66 107 3,12
69 Tu » » а = 3,5375 с = 3,5546 1,746 9,318 1600 ± 50 1720 4,3 58,4 6,50 79 3,12
70 a-Yb Гранецентрированная куби- ческая (тип Си) а =5,4862 1,940 6,953 824 ± 5 1320 1,5 41,5 6,32 27 2,59
71 Lu Гексагональная плотноупа- коваиная (тип Mg) а = 3,5031 с = 5,5509 1,734 9,849 1675 ± 25 2680 4,5 77,0 6,23 79 3,14
21 a-Sc То же а = 3,3090 с = 5,2733 1,641 2,985 1538 ±20 2900 4,2 80,8 6,00 — 3,23
39 Y » » а = 3,6474 с = 5,7306 1,801 4,472 1525 + 25 3025 4,2 80,0 6,13 69 + 3 3,07
а Структура, плотность и ряд других свойств приведены для модификации, устойчивой при комнатной темп-ре, если нет
дополнительных указаний. Большая часть свойств приводится по последней сводке (см. Пробл. совр. металлургии,
№ 2, 1960).
приведены нек-рые физич. свойства РЗЭ. Точки плав-
ления элементов подгруппы церия значительно ниже,
чем элементов подгруппы иттрия. Примечательно,
что у элементов Sm, Ей и Yb, проявляющих валент-
ность + 2 (см. ниже), точки кипения значительно
ниже, чем у других Л. Все Л. и лантан парамагнит-
Таблица 3. Механические свойства лантанидов,
лантана и иттрия
Металл Твер- дость no Бри- неллю, кГ/мм2 Коэфф, сжи- маемости, смг/кГ 10е мо- дуль упру- гости, кГ/мм* Мо- дуль сдвига, кГ/мм2 Коэфф. Пуас- сона Предел проч- ности, кГ/мм2
a-La 36 3,24 3915 1518 0,288 И,2
у-Се 19 4,95 3058 1223 0,248 10,3
a-Pr 5э 3,28 3592 1378 0,305 8,9
a-Nd 33 3,02 3860 1476 0,306 13,9
a-Sm __ 2,56 3480 1286 0,352 —
a-Gd 2,52 5730 2278 0,259 15,9
a-Tb __ 2,45 5864 2327 0,261
Dy 2,39 6433 2587 0,243 24,4
Ho 2,14 6850 2720 0,255 —.
Er 2,11 7474 3016 0,238 19,5
a-Yb 7,12 1815 717 0,284 —
Y 45-50 2,09 6700 2672 0,265 14,0
Металлы высокой чистоты пластичны и легко под-
даются деформации (ковке, прокатке). Механич. свой-
ства сильно зависят от содержания примесей, в осо-
бенности таких элементов, как кислород, сера, азот
и углерод. Механические свойства Л. приведены в таб-
лице 3. Значения предела прочности и модуля упру-
гости элементов иттриевой подгруппы (за исклю-
чением Yb) выше, чем для элементов цериевой под-
группы.
Электронная структура, валентность, свойства ио-
нов. Электронная конфигурация атомов Л. может быть
выражена общей ф-лой 4/214 5s2 5pe 5d01 6s2
(см. табл. 4).
В нормальном состоянии у Л. (за исключением гадо-
линия, лютеция и, возможно, тербия) нет 5с?-электро-
нов. Однако для перехода одного (а в нек-рых случаях
двух) электронов с уровня 4/ на уровень 5d требуется
небольшая затрата энергии. Характерная для всей
группы Л. валентность 4-3 основана на возбужденных
состояниях 5с?1 6s2 или 5 с?2 6s1, возникающих при таком
переходе. Остальные /-электроны в химич. связи не
участвуют. Нек-рые Л. проявляют, кроме валентности
+3, также валентность +4 или +2 (табл. 4). Эти «ано-
мальные» валентности объясняются различиями в проч-
919
ЛАНТАНИДЫ
920
Таблица 4. Электронная структура (по Меггерсу), энергии
ионизации, валентность и ионный радиус R3+ лаитаиа
и лантанидов
Эле- мент if 5s 5p 5d 6s Энергия иониза- ции, R»_>R34-J Ва- лент- ность Ионный радиус Я3+ по Гольд- шмид- ту б, А Стан- дартный элек- тродный потен- циал в
67 La 2 6 1 2 36,2 3 1,22 -2,4
58 Се 2 2 6 — 2 37,2 3,4 1,18 — 2,335
59 Рг 3 2 6 — 2 37,5 3,4 1,16 - 2,2
60 Nd 4 2 6 2 37,8 3 1,15 - 2,246
61 Pm 5 2 6 — 2 38,2 3
62 Sm 6 2 6 2 38,2 2,3 1,13 — 2,2
63 Eu 7 • 2 6 2 38,8 2,3 1.13 - 2,2
64 Gd 7 2 6 1 9 38,6 3 1,11 — 2,2
65 Tb 9 2 6 — 9 39.4 3,4 1,09 - 2,2
66 Dy 10 2 6 — 9 39,5 3(4) 1,07 — 2.2
67 Ho 11 2 6 — 2 40,0 3 1,05 — 2.1
68 Er 12 2 6 — 9 40,2 3 1,04 — 2,1
69 Tu 13 2 6 2 40,3 3(2) 1.04 - 2,1
70 Yb 14 2 6 —— о 40,8 2,3 1.00 — 2,1
71 Lu 14 2 6 1 2 41,0 1 0,99 - 2,1
а Значения первой энергии ионизации Я° -» Я+ см. в табл,
ст. Атом.
б Значения ионных радиусов по другим системам см. в ст.
Ионные радиусы.
ности связи электронов на 4/-уровне в зависимости от
их числа. Прочность связи электронов возрастает по
мере заполнения /-уровня на половину (до семи элект-
ронов) или при полном его заполнении (до 14 электро-
нов). Поэтому наиболее устойчивой конфигурацией
/-уровня отличаются атомы Gd и Lu. Валентность
(+4) проявляется у Се и Рг (первые /-электроны легко
Таблица 5. Окислительно-
восстановительные потенциалы
Ео при образовании ионов
R2t или R1+
Реакция
Sm2 г = Sm3+ 4- е~
Yb2+ = Yb3+ + e~
Ец2+- = Еи3+ 4- е-
Рг3+ = Рг4+ 4- е~
Се3+ = Се4+ + е~
в
переходят на 5 d-уровень)
и у ТЬи Dy, следующих
за Gd. Валентность (+2)
наблюдается у Sm, Eu и
Yb, т. е. у элементов с
числом электронов на /-
уровне, равным или близ-
ким к 7 и 14.
Значения окислитель-
но-восстановительных по-
тенциалов Л., проявляю-
щих переменную валент-
ность, приведены в табл.5.
Среди свойств Л. и лантана, обусловленных осо-
бенностями их электронной структуры, следует отме-
тить: а) парамагнитную восприимчивость, к-рая объяс-
няется наличием неспаренных электронов;
б) характерные для этих элементов резкие
полосы в спектрах поглощения в инфра-
красной, видимой и ультрафиолетовой ча-
стях спектра. Полосы поглощения и маг-
нитные восприимчивости Л. и лантана очень
мало изменяются при изменении окружаю-
щей ионной среды (напр., при изменении
состава соединения или растворителя). Это
объясняется экранированием /-электронов
от внешних влияний электронами наруж-
ных оболочек.
Деление РЗЭ на две подгруппы — це-
риевую и иттриевую, к-рое первоначально
было основано на различии в растворимо-
сти двойных сульфатов, образуемых Л.
с сульфатами натрия или калия (см. ниже),
согласуется с периодичностью в изменении
нек-рых свойств внутри семейства Л. Так,
наблюдается примерно аналогичное изме-
нение устойчивости валентных состояний
в обеих подгруппах (рис. 1), а также в ок-
раске ионов: окраска растворов 3-валент-
ных ионов первых семи элементов близка к окраске
последующих семи ионов, расположенных в обрат-
ном порядке (см. табл. 6). Изменение магнитных
свойств ионов R3+ также носит периодич. характер.
Sm Eu Gd * _ Tu Yb Lu
La Ce Pr Nd Pm | | Tb Dy Ho Er *
Рис. 1. Валентность лантанидов.
В противоположность перечисленным выше, нек-рые
свойства при переходе от одного элемента к другому
изменяются непрерывно. Так, по мере увеличения по-
рядкового номера Л. непрерывно уменьшаются их
ионные радиусы (см. табл. 4). Это явление, называемое
«лантанидным сжатием», объясняет постепенное пони-
жение основности в ряду элементов, а также обуслов-
ливает различия в растворимости солей Л. и в устой-
чивости их комплексных соединений.
Химические свойства. Л. и лантан отличаются высо-
кой химич. активностью. Они образуют весьма проч-
ные окислы, галогениды, сульфиды, реагируют с во-
дородом, углеродом, углеводородами, окисью и дву-
окисью углерода, азотом, фосфором и рядом других
’элементов. Металлы разлагают воду (медленно на
холоду, быстрее при нагревании) и легко растворяются
в соляной, серной и азотной к-тах. В плавиковой и
фосфорной к-тах Л. устойчивы, что объясняется обра-
зованием защитных пленок малорастворимых солей.
При комнатной темп-ре в сухом и влажном воздухе
La, Се, Рг и Nd быстро корродируют, тогда как дру-
гие металлы окисляются мало и длительное время
сохраняют металлич. блеск. При темп-рах выше 180—
200° все Л. быстро окисляются па воздухе с образова-
нием окислов типа R.,O3 (за исключением Се, Рг и
ТЬ, образующих СеО2, Рг6Оц и ТЬ4О7). Се отличается
от других Л. тем, что образующаяся при его окисле-
нии окись Се2О3 легко окисляется до двуокиси СеО2,
что служит причиной пирофорности самого Се и бога-
тых Се сплавов.
ОкислыЛ. и лантана отличаются высокой проч-
ностью и плавятся при высоких темп-рах. Напр.,
СеО2 плавится ок. 2500°, La2O3 — выше 2000°. Се2О3,
Рг2О3 и Nd2O3 кристаллизуются в решетке гексаго-
нального типа, окислы всех других элементов имеют
кубич. решетку. Окислы типа Й2О3 элементов La, Се,
Таблица 6. Окраска ионов трехвалентных лантанидов
Электронная конфигурация иона R3+ Порядковый помер
57 La 58 Се 59 Рг 60 1 61 62 63 Eu 64 1 65 Gd Th 66 By 67 Ho 68 Er 69 I 70 Tu 1 Yb 71 Lu
Nd Pm Sm
41° и 4/ы Бц 1 1 1 i । i i 1 j Бц
4/1 и 4/13 | Бц 1 1 ! i i ' 1 Бц
4/- и 4/12 1 Жз 1 Бз I
4/3 ц 4/11 i p j ! ;
4/4 и 4/m 1 lp Кж \ i
4/з и 4/о i ж 1 Б?к i i
4/3 и 4/8 | I Бр i Бр i ! i
I N i i I I К I I ! I I i
Условные обозначения окрасок: Бц — бесцветная, Жз — желто-зеленая,
Кф — красно-фиолетовая, Р — розовая, Ж — желтая, Бр — бледно-розовая,
Бж — бледно-желтая, Бз — бледно-зеленая, Кж — коричневато-желтая.
921
ЛАНТАНИДЫ
922
Gd, Tb, Dy, Lu, Y — бесцветны; Tu2O3 — белый с зеле-
новатым оттенком, Ho2O3 — бледно-желтый, Рг2О3 —
зеленовато-желтый, Еи2О3 — бледно-розовый, Ег2О3—
розовый, Nd2O3 — лиловый, ТЬ4О7 — темно-коричне-
вый, РгвОы — коричнево-черный, СеО2 (без примесей
других Л.) — в нагретом состоянии желтый, после
охлаждения — белый.
Все галогены выше 200° активно реагируют с Л.
Образующиеся галогениды типа RX3 имеют
сравнительно высокие темп-ры плавления и кипения
(табл. 7). Все галогениды, за исключением фторидов,
весьма гигроскопичны и легко гидролизуются с обра-
зованием оксигалогенидов ROX. Низшие галогениды
RX2 известны для Sm, Ей и Yb.
Таблица 7. Температуры плавления и кипения галогенидов
редкоземельных элементов типа RX3 (в СС)
Эле- Фториды Хлориды Бромиды Йодиды
мент T. ПЛ. T. кип. г. пл. т. кип. т. пл. Г. кип. т. пл. т. кип.
La 1430 2330 855 1750 786 1580 784 1405
Се 1435 2330 805 1730 735 1560 755 1400
Рг 1373 2330 779 1710 696 1550 736 1380
Nd 1413 2330 773 1690 687 1540 778 1370
Pm 1410 2330 740 1670 680 1530 800 1370
Sm 1400 2330 681 разл. 667 разл. 823 разл.
Eu 1390 2280 626 705 880
Gd 1380 2280 612 1580 778 1490 929 1340
Tb 1370 2280 591 1550 830 1490 955 1330
Dy 1360 2230 657 1530 884 1480 958 1320
Ho 1360 2230 721 1510 917 1470 1013 1300
Er 1350 2230 777 1500 953 1460 1023 1289
Tu 1340 2230 824 1490 955 1440 1018 1260
Yb 1330 2230 857 разл. 943 разл. 1030 разл.
Lu 1320 2230 895 1480 960 1410 1048 1210
Y 1390 2230 703 1510 907 1470 1003 1310
Водород поглощается Л. уже при комнатной темп-ре;
быстрое взаимодействие наступает при 250—300° с об-
разованием гидридов типа RH2,8 (для Се, La,
Рг) или RH2. Гидриды разлагаются при нагревании
в вакууме (выше 1000°) и не устойчивы во влажном
воздухе. Азот реагирует с Л. при 750—1000° с образо-
ванием нитридов, преим. типа RN. Их свойства
мало изучены. С углеродом, СО, СО2, СН4 и др. Л.
взаимодействуют при нагревании с образованием
карбидов типа RC2. Они не устойчивы на воздухе
и разлагаются водой с выделением углеводородов —
преим. ацетилена, частично метана. При нагревании
Л. в парах серы образуются сульфиды состава
R2S3, R3S4 и RS. Сульфиды тугоплавки и жаростойки;
т. пл. La2S3 2100°, Ce3S4 2500°, Nd2S3 2200°.
Характеристика некоторых соединений лантанидов,
выделяемых из растворов. Гидроокиси Л.
R(OH)3 имеют основной характер и малорастворимы
в воде и щелочах. Соответственно понижению основ-
ности в ряду Л. от Се к Lu pH начала осаждения гидро-
окисей и произведения их растворимости понижаются
(табл. 8).
Таблица 8. Значение pH осаждения и произведения
растворимости гидроокисей R (ОН)3
Элемент pH осажде- ния из р-ра нитратов Произведение ра- створимости для R (ОН)3 (при 25°)
La 7,82 1,0 • 10“*»
Се 7,60 1,5 • 10-2°
Рг 7,35 2,7 10-а»
Nd 7,31 1.9 • 10-21
Sm 6,92 6,8 • 10"22
Eu 6,82 3,4 ♦ 10-22
Gd 6,83 2,1 • IO”22
Ег 6,75 1,3 • 10-23
Tu 6,40 3,3 . 10-24
Yb 6,30 2,9 10-21
Lu 6.30 2,5 • 10-24
Y 6,95 —
Гидроокись Се4+ осаждается из р-ров при pH 0,9—1,
что позволяет отделить Се (после окисления Се3+ до
Се4+) от других Л.
Хлориды, сульфаты и нитраты 3-ва-
лентных Л. растворимы в воде и кристаллизуются б. ч.
Рис. 2. Растворимость суль-
фатов нек-рых лантанидов в
зависимости от темп-ры.
в виде кристаллогидра-
тов различного состава.
Растворимость сульфатов
Бг(8О4)3- 8Н2О сильно по-
нижается при повышении
темп-ры от 0 до 100° (см.
рис. 2). Фториды и
оксалаты малора-
створимы в воде и разб.
минеральных к-тах. Фто-
риды осаждаются в виде
кристаллогидратов соста-
ва RF3- 0,5 Н2О (La, Се
и др.) или безводных солей
(напр., Рг и Nd). Для окса-
латов наиболее характерен
состав R2(C2O4)3 • ЮН20.
Растворимость оксалатов
элементов иттриевой груп-
пы выше, чем цериевой
(рис. 3). При нагревании
до 500—600° оксалаты разлагаются с образованием
окислов типа R2O3. К труднорастворимым в воде со-
лям Л. относятся также фосфаты, карбона-
Nd Sm СИ Dy Er Vb
Растворимость глдратиро-
оксалатов лантанидов в
воде при 25°.
Се
Рис. 3.
ванных
ты и феррици-
аниды.
Большинство про-
стых солей лантони-
дов склонно к обра-
зованию двойных или
комплексных солей с
солями щелочных ме-
таллов и аммония, а
также рядом солей
двухвалентных эле-
ментов. К важнейшим
из них относятся:
двойные нитра-
ты Л. с нитратом аммония R(NO3)3 • 2NH4NO3 4Н2О
или нитратом магния 2R(NO3)3- 3Mg(NO3)2 • 24Н2О,
растворимость к-рых уве-
личивается от La к Gd;
двойные сульфа-
ты с сульфатами натрия
или калия (напр., состава
R2(SO4)3- Me2SO4- nH2O,
п= 1 или 2). По раство-
римости в воде двойные
сульфаты Л. подразде-
ляются на три подгруп-
пы: малорастворимые (La,
Се, Рг, Nd, Sm), средне-
растворимые (Eu, Gd, Tb,
Dy) и растворимые (Но,
Er, Tu, Yb, Lu, Y). Это
широко используется для
предварительного разде-
ления Л. на цериевую и
иттриевую подгруппы.
Л. образуют комплекс-
ные соединения с мно-
гими органнч. вещества-
ми. Среди них важное
значение имеют комп-
лексы, образуемые с ли-
монной к-той (H3Cit) и
Рис. 4. Константы устойчивости
комплексных соединений ланта-
нидов с ЭДТА (белые кружки)
и НТА (черные кружки). Для
сравнения даны константы ус-
тойчивости комплексов с др. эле-
ментами.
рядом аминополиуксусных кислот: нитрилотриук-
сусной кислотой (НТА), этилендиаминтетрауксусной
923
ЛАНТАНИДЫ
924
к-той (ЭДТА) и другими «комплексонами». Среди
лимоннокислых комплексных ионов наиболее устой-
чивыми в водных р-рах являются трехзарядные ионы
RCitl-. С ЭДТА и НТА лантаниды образуют внутри-
комплексные (клешневидные) соединения, структур-
ные формулы к-рых приведены ниже.
Устойчивость комплексных соединений с органич.
к-тами возрастает в ряду Л. от La к Lu (рис. 4), что
широко используется в нек-рых методах разделения Л.
Источники сырья и извлечение редкоземельных
элементов. Ресурсы Л. в земной коре относительно
велики. Суммарное весовое содержание Л. (включая
лантан) в земной коре 0,01%, что равно .
содержанию меди. К наиболее распростра-
ненным из них относятся La, Се и Nd.
57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 39
La Се Рг Nd Pm Sm Eu Od Tb Dj Ho Bf Tv Yb Le V
Рис. .5. Относительная распространенность лантани-
дов в земной коре по А. Е. Ферсману (жирная линия),
В. М. Гольдшмидту (тонкая линия) и в литосфере по
А. П. Виноградову (пунктирная линия).
Элементы с нечетными атомными номерами содержатся
в земной коре в меньших количествах, чем их ближай-
шие соседи с четными номерами (рис. 5).
Известно более 250 минералов, содержащих РЗЭ.
К собственным минералам можно отнести 60—65 из
них, в к-рых содержание суммы редких земель выше
5—8%. По химич. природе они представляют собой
гл. обр. фосфаты, фториды или фторокарбонаты, сили-
каты и силикотитанаты, ниоботанталаты, титанонио-
баты. Минералы обычно содержат нек-рое количество
тория, иногда урана. В табл. 9 приведен состав важ-
нейших минералов.
Соотношение между отдельными элементами в ми-
нералах сильно колеблется. В одних преобладают эле-
менты цериевой группы и только до 5% иттриевых
земель (напр., монацит, бастнезит, лопарит), в дру-
гих — иттриевой группы (ксенотим, эвксенит, гадоли-
нит). Л. концентрируются в различных типах магмато-
генных, осадочных и метаморфогенных месторожде-
ний. До настоящего времени пром-сть базируетсй
в основном на разработке монацитовых россыпей и
нек-рых типов эндогенных месторождений (бастнезит-
кальцитовые жилы, пегматиты с метамиктными тита-
но-тантало-ниобатами). Однако в последние годы
выявлен ряд перспективных месторождений иного
типа (гидротермальные карбонатные или гематитовые
тела с бастнезитом, флюоритом и баритом).
Один ив основных источников элементов цериевой под-
группы — монацит — обычно встречается в пегматитах,
иногда гранитах и гнейсах. При разрушении коренных пород
он переходит в россыпи (речные и морские) вместе с ильме-
нитом, цирконом, магнетитом и др. Минимальное содержание
монацита в разрабатываемых россыпях — 1 %. Наиболее круп-
ные месторождения найдены в Индии, Бразилии, США, Австра-
лии, Мадагаскаре, Цейлоне. Применяя гравитационные и
магнитные способы обогащения, получают концентраты
с содержанием 58—65 % R2O3. Из них попутно с торием извле-
кают Л. В последнее время большое промышленное значение
приобрел бастнезит. Одним из минералов сложного
комплексного состава является лопарит, к-рый распро-
странен в нефелиновых сиенитах, а также во многих пегмати-
товых жилах. Лопаритовые руды легко обогащаются с получе-
нием концентратов, содержащих 80—90% минерала. При их
переработке Л. извлекают попутно с ниобием, танталом и тита-
ном. К минералам, к-рые служат богатым сырьем для извлече-
Таблица 9. Состав некоторых минералов редкоземельных элементов
Название минерала Формула Примерный состав Плотность, г/см3
Монацит ......... (Се, La ...) Ро< (Се, La ...)2О2 50—60%; Р2О5 22—31,5%; ТЬО2 4—12%; изредка ZrO2 (до 7%) и SIO2 (до 6%) 4,9—5,5
Ксенотим YPO4 Y2O, 52—62,6%; примеси Ег, Се и других Л.; иногда ThO2, ио2 (до 5%), ZrO2 (до 3%), SnO2, SiO2 (до 9 %) 4,45—4,59
Бастнезит (Се, La...) FCO3 (Се, La, Рг...)203 73—77%; F 6,2—8,5%; СО2 19,8—20,2% 4,8-5,2
Гадолинит Y2FeBe2SisOi0 FeO 10—13,7%; Y2O3 30,7—46.5 %; (Се, La ...)2О3 5—23%; SiO2 23-24,5 %; ThO30,3-0,4%; ВеО 9—10,2%; примеси Са, Mg 4,1—4,5
Ортит (алланит) (Са, Се)2 (Al, Fe)3-SlsOi3 (О, ОН) Се2О3 до 6%; (La, Се...)3 О3до 7%; иногда ВеО (до 3,8%), Y2O3 до 8% (пттроортит), примеси ТЬО3 4,1
Лопарит (Na, Са, Се...)2 • (Tl,Nb, Та)2Ов TiO2 39,2—40 %; (Се, La...)2O3 32—34 %; (Nb, Та)2О5 8—10%; СаО 4,2—5,2%; NasO 7,8—9%; примеси Sr, К, Si, ТЬО2 (0,5—0,6%) 4,75-4,89
Эвксенитполикраз а . . . (Y, Се, Са...) . (TI, Nb, Та)2Ов (Y, Er...)2O3 18,2—27,7%; (Се, La...)2O3 0,2—4,3%; TiO2 16—30%; Nb2Os 4,3—41,4%; Ta2O5 1,3—23%; ThOa 1 —5%; НО, 0,4—12% 4,78—5,37
Фергюсонит (Y, Er, Се, U) • (Nb, Та, Ti) О4 (Nb, Та)аО3 57,5—46%; Y2OS 31—42%; (Ce, La...)2O3 0,9—6%; Er2O3 до 14%; ThO2 1—3,4%; UO2 1,2—6%; TiO2 до 6%; примеси ZrO2, SnO2, WO3 5,58—6,23
Самарскит (Y, Ег ...)4 (Nb,Ta)eO31 Y2O3 6,4—14.5%; Er2O3 2,7—13,4%; (Ce, La.,.)2O3 до 8%; Nb2O5 27 7—46,4%; Ta2O-, 1,8—27,0%; ThO2 0—4,2%; UO2 4—16%; примеси Ti, Zr, Sn и др. 5,6-5,8
а При отношении (Nb, Та)2О3 : TiO2 = 1 : 4 — 1 ; 6 и ниже минерал наз. поликразом, при отношении выше 1:3 —
эвксенитом.
925
ЛАНТАНИДЫ
920
ния редких земель группы иттрия, относятся фергюсонит,
ксенотим и др. (см. табл. 9).
Технология переработки сырья зависит от его минераль-
ного состава. Наиболее распространенным способом вскрытия
монацитовых и бастнезитовых концент-
ратов является обработка их концентрированной серной
к-той при нагревании до 200° с последующим выщелачиванием
массы водой. В результате получают сернокислые растворы,
содержащие все РЗЭ вместе с торием и др. элементами. Для
вскрытия монацитовых концентратов, кроме того, используют
обработку р-рами щелочи при 140—160°. Образующуюся при
этом смесь гидроокисей РЗЭ и тория растворяют в соляной
к-те. Из полученных сернокислых или солянокислых р-ров
первоначально выделяют торий. Большей частью для этой
цели используют различия в pH выделения фосфатов или гид-
роокисей тория и Л. (фосфат тория выделяется из р-ров при
pH 1—1,2; фосфаты РЗЭ — при pH ок. 2,3; гидроокиси тория и
РЗЭ выделяются при pH 4,6—5 и pH 6,5—7,8 соответственно).
После отделения Th из р-ров осаждают РЗЭ в виде оксалатов
щавелевой к-той или в виде двойных сульфатов с помощью
сульфата натрия. Оксалаты или двойные сульфаты обработкой
щелочью превращают в гидроокиси, к-рые поступают на даль-
нейшие операции разделения Л.
Сложное комплексное сырье типа лопарита вскрывают хло-
рированием в присутствии угля при 700—850°. Летучие хло-
риды титана, ниобия, тантала и ряда других элементов уда-
ляются с газами. В печи остается расплав хлоридов РЗЭ.
Хлориды растворяют в воде и из раствора выделяют смесь Л.,
напр., как описано выше.
Разделение редкоземельных элементов. Получение
Л. в чистом виде относится к труднейшим задачам
химич. технологии. Сравнительно легко достигается
отделение Се от других РЗЭ.
Отделение Се обычно основано на окислении Се в смеси
гидроокисей Л. до 4-валентного состояния кислородом воздуха,
хлором или перекисью водорода. Гидроокиси 3-валентных РЗЭ
затем растворяют в разб. азотной к-те, в то время как гидро-
окись Се (IV) остается в осадке. Последний растворяют в конц.
HNO3 и из р-ра выделяют чистые соединения Се кристаллиза-
цией гексанитрата церия-аммония (NH4)2Ce(NO3)e или гидро-
литич. осаждением основного нитрата Се(ОН)Х • (NO3)4_X.
Для очистки Се от 3-валентных РЗЭ применяют также избира-
тельную экстракцию Се (IV) из 6—8 н. р-ра HNO8 диэтиловым
эфиром или трибутилфосфатом.
Разделение РЗЭ после отделения Се ранее преимущественно
осуществлялось сочетанием методов фракционного
осаждения и фракционной кристалли-
зации. В дополнение к ним использовали способность
нек-рых Л. (Sm, Eu, Yb) восстанавливаться до валентности
-4-2. Среди методов осаждения наиболее распространено дроб-
ное осаждение двойных сульфатов (для разделения РЗЭ на
цериевую и иттриевую подгруппы) и дробное осаждение гид-
роокисей, осуществляемое постепенной нейтрализацией р-ра
нитратов РЗЭ аммиаком, напр. по методу Тромба, состоящему
в пропускании через р-р аммиака в смеси с воздухом. Отделяя
фракции, получаемые в различных интервалах pH, получают
при более низких pH фракцию, обогащенную Sm, Eu и иттрие-
выми землями, затем промежуточные фракции (Nd 4- Рг и
примесь La) и при более высоких pH (-5= 8,6) — богатые ланта-
новые концентраты (98—99% La). В практике разделения при-
меняют также фракционное осаждение оксалатов из их р-ра
в .ЭДТА (для элементов цериевой группы) и осаждение фер-
рицианидов (для иттриевой группы). Из методов кристалли-
зации широко используются дробная кристаллизация двой-
ных нитратов Л. с нитратом аммония и нитратом магния (при
разделении элементов цериевой группы) и кристаллизация
броматов RBrO3 • 9Н2О (для разделения элементов иттриевой
группы).
С целью извлечения Sm, Eu и Yb из соответствующих
фракций восстанавливают 3-валентные ионы этих элементов
в солянокислом р-ре, содержащем уксусную к-ту, амальгамой
натрия или электролизом с ртутным катодом. Из амальгамы
обработкой ее соляной к-той извлекают Sm, Eu и Yb. Затем
Eu от Sm может быть отделен избирательным восстановлением
Еи8+ амальгамой цинка в присутствии серной к-ты. При этом
2-валентный Ей осаждается в составе труднорастворимого
сульфата.
Для получения большинства Л. в относительно чистом
виде по схемам, основанным на использовании дробного
осаждения или кристаллизации, требовалось проведение
многих сотен и даже тысяч фракционирований. Это особенно
относится к элементам иттриевой группы, разделение к-рых
сложнее, чем элементов цериевой группы. В последние годы
схемы разделения Л. значительно упрощены и усовершен-
ствованы применением методов ионообменной хро-
матографии и экстракции органич. растворите-
лями. Для разделения РЗЭ методом ионообменной хрома-
тографии применяют различные типы сульфостирольных
смол (в СССР катионит КУ-2, за границей — Дауэкс-50 и
др.), Хроматографич. разделение Л. состоит из двух стадий;
а) сорбции разделяемой смеси,катионов в верхней части слоя
смолы в колонке; б) вымывания смеси катионов р-рами комп-
лексообразующих соединений. Десорбция происходит в после-
довательности, соответствующей прочности анионных комп-
лексов Л. По мере движения вымывающего р-ра вдоль колон-
ки (или ряда последовательно соединенных колонок) смесь
катионов разделяется на отдельные сорбционные зоны (по-
лосы), перемещающиеся с определенной скоростью к ее выходу.
Смолу заряжают ионами Н^, NH+, Си-+ или Zn2+, что зависит
от используемого для вымывания комплексообразователя. Для
вымывания применяют лимонную к-ту, р-ры аммонийных или
натриевых солей нитрилтриуксусной к-ты (трилон А) или
этилендиаминтетрауксусной к-ты (трилон Б) и нек-рые другие
органич. соединения. Из них трилон-Б наиболее эффективный
элюент. При работе с ним применяют смолу в Си2+-форме, вы-
мывание ведут 2—5%-ным р-ром трилона-Б при pH 7,5—8.
Хроматография, методом удается
получить почти все РЗЭ чисто-
той 99,99%. Недостатком метода
является малая его производи-
тельность.
В последнее время важное
место в технологии разделения
РЗЭ заняли экстракционные ме-
тоды, основанные на избира-
тельном извлечении нитратов
органич. растворителями из вод-
ных р-ров в присутствии высали-
вателей. Последними в случае
экстракции нитратов РЗЭ слу-
жат азотная к-та и нек-рые нит-
раты [A1(NO3)3B др.1. Одним из
лучших экстрагентов для разде-
ления нитратов является трибу-
тилфосфат (ТБФ) — (С4Н8О)3РО,
образующий с нитратами РЗЭ
комплексы R(NO3)3 • ЗТБФ.
Козфф. распределения нитратов
между водной и органич. фазами
возрастают по мере увеличения
порядк. номера элемента (рис. 6),
причем для растворов, содержа-
щих 12н. HNO3, значения коэфф.
Рис. 6. Зависимость коэфф,
распределения лантанидов
между раствором 15,6 н.
HNO3 и трибутилфосфатом
от атомного номера элемента
(К = _S°Pr- ). Начало коор-
^водн.
динат Z = 57 (лантан).
разделения двух соседних эле-
ментов составляют 1,6. Благоприятным является возрастание
коэфф, распределения с увеличением концентрации Л. в рас-
творе, что делает процесс пригодным для переработки больших
количеств. Разделение осуществляется как в процессе много-
ступенчатой экстракции, так и на стадии реэкстракции нитра-
тов из растворителя. Экстракцию ведут в противоточных экс-
тракторах непрерывного действия (смесители — отстойники,
колонны). В современных схемах разделения РЗЭ методы
экстракции и ионного обмена в большой мере заменили ме-
тоды фракционного осаждения и кристаллизации. Использо-
вание последних ограничивается б. ч. начальными стадиями
грубого фракционирования. На рис. 7 и 8 в качестве примера
приведены принципиальные схемы разделения.
отделение; II — отделение La методами нейтрализации или
кристаллизацией двойных нитратов; III — дробная кри-
сталлизация нитратов (наибольшее число серий); IV —
экстракция, хроматография; V — восстановление амальга-
мой натрия; VI — восстановление, экстракция, хромато-
графия; VII —восстановление амальгамой цинка; Yttr—
сумма элементов иттриевой группы.
927
ЛАНТАНИДЫ
928
ть+ Сег. Dy Y Но Ег Tu Yb Lu
।
Рис. 8. Примерная схема
последовательности опе-
раций разделения ланта-
нидов иттриевой груп-
пы: I — дробное осажде-
ние комплекснообразую-
щимися реагентами; II,
III, IV — экстракция,
хроматография; V, VI —
восстановление амальга-
мой натрия или электро-
лизом; VII—экстракция,
хроматография; Сег —
сумма элементов церие-
вой группы.
Получение металлов. Ввиду высокой химич. проч-
ности соединений Л. (галогенидов, окислов) для по-
лучения металлов применяют металлотермия, мето-
ды восстановления или электролиз расплавленных
сред. Металлотермический метод состоит
в восстановлении безводных гелогенидов чистым
кальцием. Процесс проводят в стальных бомбах, фу-
терованных окисью кальция, или тиглях из тантала,
не взаимодействующего с расплавленными Л., в атмо-
сфере чистого аргона. Сравнительно легкоплавкие ме-
таллы группы церия получают восстановлением хло-
ридов или фторидов, в то время как более тугоплавкие
элементы иттриевой группы восстанавливают только из
фторидов, т. к. при темп-ре плавления этих металлов
(1400—1700°) вследствие высокой летучести хлоридов
РЗЭ получается низкий выход металла. После пере-
плавки в вакууме в тиглях из тантала металлы, полу-
ченные восстановлением галогенидов, содержат лишь
0,01—0,02% Са. Восстановлением галогенидов могут
быть получены в чистом виде все Л., кроме Sm, Eu и
Yb, восстанавливающихся только до низших галоге-
нидов. Поэтому для получения Sm, Eu и Yb разра-
ботан метод восстановления их окислов лантаном в ва-
кууме с одновременной дистилляцией образующихся
металлов, к-рые кипят при сравнительно низких
темп-рах (см. табл. 2).
Электролизом из расплавов могут быть по-
лучены все Л. Металлы цериевой группы, а также их
сплав (мишметалл) получают электролизом безводных
хлоридов в расплаве, состоящем из К.С1 + СаС1.2 или
КС1 NaCl. Электролиз б. ч. ведут в графитовых
тиглях, служащих одновременно анодом; катоды —
стальные или молибденовые стержни. Для исключения
прямого соприкосновения расплавленного металла
с графитом на дне тигля под катодом устанавливают
тигель-сборник из окисей бериллия или циркония.
Для тугоплавких металлов иттриевой группы прове-
дение электролиза хлоридов, как описано выше,
невозможно из-за испарения хлоридов при высоких
темп-рах. Чтобы обеспечить выделение этих Л. в жид-
ком виде при темп-рах ванны не выше 1100° ведут
электролиз с жидким катодом из кадмия или цинка,
образующих легкоплавкие сплавы с выделяемыми
металлами. Zn и Cd затем удаляют отгонкой в вакууме.
По чистоте металлы, полученные электролизом, усту-
пают металлотермическим.
Методы анализа. Определение общего содержания
Л. в рудном сырье или других материалах не пред-
ставляет трудностей. Исходный материал разлагают
конц. серной к-той пли соляной к-той. Из слабокис-
лых р-ров щавелевой к-той осаждают оксалаты Л.
вместе с'/ГЬ. Для отделения ТЬ оксалаты прокаливают,
растворяют смесь окислов в азотной к-те и осаждают
Th в виде йодата из сильнокислого р-ра или в виде
перекиси-гидроокиси из слабокислого р-ра. В филь-
трате после отделения Th может быть определен Се.
Методы его определения основаны на окислении Се3,_
до Се4+,что осуществляется с помощью бромата калия,
персульфата аммония или висмутата натрия. Затем Се
определяют весовым (в виде йодата или основного
бромата), объемным (титрование перманганатом избыт-
ка добавленной соли Мора) или колориметрич. ме-
тодом .
Для определения всех других Л. применяют преим.
спектральные методы. Большинство Л. при высоком
их содержании может быть определено методом адсорб-
ционной спектроскопии (спектры поглощения) с точ-
ностью ±1,5%. Однако для определения малых ко-
личеств интенсивность полос поглощения оказывается
недостаточной. Большей чувствительностью обладают
методы дуговой спектроскопии и особенно рентгено-
спектрального анализа. Широко распространенными
способами контроля процессов разделения Л. служат
измерения магнитной восприимчивости, а также радио-
метрии. метод, использующий введение радиоактивных
изотопов. Последний особенно удобен при хромато-
графии. разделении Л. Простым способом оценки со-
става бинарных смесей Л. является определение
среднего «атомного веса» смеси.
Области применения. Потребителями Л. (в виде
металлов, сплавов и химич. соединений) являются
различные отрасли техники. Ниже рассмотрены наи-
более важные области их применения. В черной и цвет-
ной металлургии находят все возрастающее приме-
нение присадки Л. в стали, чугуны и сплавы цветных
металлов (магниевые, алюминиевые и др.). Они улуч-
шают механич. свойства, коррозионную устойчивость
и жаропрочность сплавов. В качестве присадок ис-
пользуют гл. обр. ферроцерий и сплав Л. с преим. со-
держанием Се или Се и La. Стекольная пром-сть —
один из крупных потребителей Л. Добавки окислов
Се, Рг и Nd сообщают стеклу способность поглощать
УФ- и ПК-лучи. Стекло, содержащее Се, не тускнеет
под действием излучений и применяется в атомной
технике. Окислы нек-рых Л. вводятся в состав оптич.
стекла, применяются для окраски стекла и его обес-
цвечивания. Двуокись церия (под названием «поли-
рит») широко применяется в оптической промышлен-
ности для полировки стекла. В керамике окислы Л.
применяют для окраски фарфора, а также глазурей и
эмалей.
В химич. и легкой пром-сти используют соединения
Л. для изготовления лаков и красок, светящихся со-
ставов (люминофоров), катализаторов, фотореагентов
в произ-ве кожи, текстильной пром-сти. В осветитель-
ной технике для изготовления дуговых прожекторов
и кинопроекционных углей применяют фториды Л.
(в основном церия), усиливающие интенсивность све-
чения.
В радиоэлектронике Се или сплав Л. (мишметалл)
применяют в составе нераспыляющихся поглотителей
газов (геттеров). Окись неодима используют в электрон-
ных приборах как диэлектрик с малым коэфф, расши-
рения. Перспективно применение боридов нек-рых Л.
для изготовления катодов в нек-рых электронных при-
борах. Изотоп прометия Рт147 применяют для изго-
товления микробатарей, в к-рых p-излучение превра-
щается в электроэнергию. В атомной технике исполь-
зуют Л. с высокими сечениями захвата тепловых нейт-
ронов (Gd, Sm, Eu). Их окислы входят в состав защит-
ных керамич. покрытий ядерных реакторов. Они
могут также применяться для управления работой
атомного реактора. Важное применение имеет радио-
активный изотоп тулия Tu170 (излучает у-лучи) для
929
ЛАНТАНОИДЫ - ЛАТЕКСЫ СИНТЕТИЧЕСКИЕ
930
изготовления портативных рентгеновских установок
медицинского назначения и для металлодефектоскопии.
В с. х-ве в последние годы соединения Л. стали при-
менять в качестве инсектицидов и микроудобрений,
ускоряющих рост растений. Несмотря на разно-
образие областей использования Л., современный уро-
вень их потребления значительно ниже возможных
масштабов произ-ва этих металлов. По нек-рым дан-
ным, в 1956 в капиталистич. странах добыча редкозе-
мельно-ториевого сырья составляла 32’000 т.
Лит.: Серебренников В. В., Химин редкозе-
мельных елементов, т. 1—2, Томск, 1959—61; Vickery
К. С., Chemistry of the lanthanons, L., 1953; Зеликман
A. H., Металлургия редкоземельных металлов, тория и урана,
М., 1961; Редкоземельные металлы. Сб. переводов, под ред.
Л. Н. Комиссаровой и В. Е. Плющева, М., 1957; Методы разде-
ления редкоземельных металлов. Сб. статей, под ред. Л. Н.
Комиссаровой и В. Е. Плющева, М., 1961; Редкоземельные
влементы. Сб., М., 1958; Свойства и применение редкоземель-
ных металлов. Материалы конференции по редкоземельным
металлаз, ноябрь 1959, Чикаго, пер. с англ, под ред. Е. М. Са-
вицкого, М., I960; Трифонов Д. Н., Редиоземельпые
элементы, М., 1960; Ельяшевич М. А., Спектры редких
земель, М., 1053; Коган Б. И., Экономические очерки по
редким землям, М-, 1961; Редкоземельные элементы в сталях
и сплавах, М., 1959; Рябчиков Д. И., С е и я в и и М. М.,
Скляренко Ю. С., Выделение индивидуальных редко-
земельных элементов, в кн.: Труды 2 Международной конф,
по мирному использованию атомн. энергии, Женева, 1958,
т. 4, М., 1959, с. 75; Gmelin, 8 Anti., System-Nummer 39 —
Seltene Erden, Lfg 1, B., 1938, то же, 1955; Mellor, v. 5,
L.—N. Y.—Toronto, 1946; то же, v. 5, L., 1956; Pascal,
t. 7,P., 1959; К I r k, v. 11, N. Y., 1953; Rare metals handbook,
ed. C. A. Hampel, 2 ed., L., 1961. A. H. Зеликман.
ЛАНТАНОИДЫ — см. Лантаниды.
ЛАТЕКС НАТУРАЛЬНЫЙ — млечный сок кау-
чуконосных растений. Практич. значение имеет толь-
ко Л. н. бразильской гевеи. Находится в млечниках,
расположенных в коре растения, и добывается подсоч-
кой. Молочно-белая жидкость с желтым, розовым или
сероватым оттенком. Средний состав: 52—60% воды,
34—37% каучука, 2—2,7% белков, 1,65—3,4% смолы,
1,5—4,2%, сахара, 0,7—0,2% минеральных веществ.
Состав Л. н. зависит от возраста дерева, климатич.
условий, времени года и т. д. Свежий Л. н. имеет ще-
лочную реакцию (pH 7,2). Каучук находится в Л. н.
в виде отрицательно заряженных глобул — взвешен-
ных частиц шарообразной или грушеобразной формы,
со средним размером 0,17—0,2о мк. Основная масса
каучука содержится в частицах со средним размером
ок. 1 мк. На поверхности глобул находится защитный
адсорбционный слой поверхностно-активных веществ
(белков, мыл жирных к-т и др.), обусловливающий
устойчивость Л. н. и препятствующий его коагуляции.
Дисперсная фаза свежего Л. н., кроме глобул, содер-
жит «желтую фракцию» в виде коллоидных частиц
неправильной формы, по-видимому, белковой при-
роды.
Во избежание коагуляции при хранении, вызывае-
мой действием кислот, образующихся в результате
жизнедеятельности микроорганизмов, Л. н. консерви-
руют прибавлением аммиака (0,5—0,7%). Л. н. коагу-
лирует при прибавлении кислот и солей 2- и 3- ва-
лентных металлов.
Основная масса Л. н. применяется для получения
каучука натурального и ок. 7%—для непосредственного
произ-ва резиновых изделий. В последнем случае для
облегчения перевозки Л. н. концентрируют центрифу-
гированием (до 60—62%) или выпариванием (до 70—
75%), реже — отстаиванием. Из Л. н. производят
изделия, получение которых обычными методами из
твердого каучука невозможно: пенистую резину, нити
круглого сечения, толстостенные изделия без шва,
нек-рые виды искусственной кожи, прорезиненные
ткани и др.
При вулканизации изделий из Л. н. применяют ак-
тивные ускорители вулканизации и вулканизуют при
более низких темп-рах. В большинстве случаев при-
меняют ненаполненные смеси, т. к. обычные активные
наполнители (сажа и др.) не усиливают резину из
Л. н. Ингредиенты вводят в Л. н. в виде стабилизо-
ванных водных суспензий или эмульсий. Водораство-
римые ингредиенты (нек-рые ускорители вулканиза-
ции) вводят непосредственно в латекс. Тонкостенные
изделия без шва производят многократным маканием
форм в латексную смесь, высушиванием слоя латекса
с последующей вулканизацией. Для получения таких
изделий часто применяют предварительно подвулкани-
зованный Л. н. (вультекс). В этом случае вулканиза-
ция заканчивается в процессе сушки при 60—70°.
При получении изделий без шва толщиной более
0,2 мм применяют т. н. ионное отложение.
Форму предварительно макают в р-р коагулирующего
вещества, напр. хлористого кальцин, а затем ее опускают
в латекс и держат в нем определенное время. При этом коагу-
лирующие ионы диффундируют с поверхности формы в латекс
и вызывают вокруг нее отложение слон каучука. Толщина слон
увеличивается с продолжительностью пребывания фопмы
в латексе. Изделия либо промывают и высушивают на форме
(перчатки и др.), либо снимают с формы, промывают и разду-
вают до требуемых размеров (метеорологии, шары). После
высушивания их вулканизуют.
Изделия со стенками значительной толщины полу-
чают т. н. желатинированием. В Л. н. вводят вещества,
постепенно выделяющие в нем астабилизующие ионы,
напр. окись цинка и соли аммония, кремнефтористый
натрий и др. При повышении темп-ры Л. н. превра-
щается в гель. При дальнейшей обработке (промывка,
высушивание, вулканизация) гель сохраняет свою
форму. Желатинированием получают также пенистую
резину — основной вид изделий, получаемых непо-
средственно из Л. н. Для этого латексную смесь вспе-
нивают, механически взбивая ее. Пену заливают
в форму и, нагревая, желатинируют и вулканизуют.
Латексная губка обладает рядом преимуществ перед
резиновой губкой из твердого каучука (большая пори-
стость, бблыпая гигиеничность и др.). Нити круглого
сечения получают, выдавливая Л. н. в раствор коагу-
лирующего вещества. Л. н. широко применяют в
произ-ве ковров, нетканых материалов и т. п.
Лит.: Догадкин Б. А., Химия и физика каучука,
М.—Л., 1947; Черная В. В., Хим. наука и пром-сть,
1959, 4, М 1, 50; Нобль Р. Д., Латекс в технике, пер. с
англ., Л., 1962. Д. М. Сандомирский.
ЛАТЕКСЫ СИНТЕТИЧЕСКИЕ — водные диспер-
сии каучукоподобных полимеров, получаемые эмуль-
сионной полимеризацией или сополимеризацией; к
Л. с. иногда относят, кроме того, дисперсии каучуков,
получаемые поликонденсацией (напр., дисперсии тио-
колов), диспергированием в воде готовых полимеров
(бутилкаучука и др.), а также дисперсии пластич.
масс, образующиеся при эмульсионной и суспензион-
ной полимеризации (напр., дисперсии поливинил-
ацетата). Эмульсионную полимеризацию проводят в
смеси, содержащей воду, мономеры, эмульгаторы,ини-
циатор, а также, как правило, регулятор, стабилиза-
тор и др. После полимеризации обычно производится
отгонка из латекса непрореагировавщих мономеров.
Синтез товарных Л.' с. имеет ряд отличий от синтеза
эмульсионных каучуков: в составе исходной смеси
мономеров, природе и количестве эмульгаторов,
степени конверсии, ограниченном применении регуля-
торов, прерывателей и противостарителей и др.
Обычно получают латексы, содержащие 20—35%
каучука. Однако можно получать непосредственно
в реакторе нек-рые латексы с концентрацией 50%
,и даже 60%. Л. с. с невысоким содержанием полимера
могут быть сконцентрированы упариванием, «сливко-
отделением» и др. методами. Верхний предел концент-
рации ограничивается снижением в ряде случаев агре-
гативной устойчивости латексов, а также быстрым
ростом вязкости. Вязкость Л. с. при концентрациях
полимера до 20—40% мало отличается от вязкости
воды; это одно из основных их преимуществ перед
высоковязкими р-рами соответствующих каучуков.
931
ЛАТЕКСЫ СИНТЕТИЧЕСКИЕ—ЛАУРИНОВАЯ КИСЛОТА
932
Вязкость латекса может быть повышена введением
загустителей (казеинатов, полиакрилатов, альгинатов,
производных целлюлозы и др.). Вязкость конц. Л. с.
сильно зависит от размеров частиц полимера; для этих
латексов, кроме того, характерно явление тиксо-
тропии.
Л. с. — коллоидные системы, содержащие сферич.
частицы полимера диаметром порядка 10 е—10-6 см.
Своей агрегативной устойчивостью Л. с. обязаны
молекулам или ионам эмульгаторов, адсорбированным
на поверхности частиц. Устойчивость, как правило,
возрастает с увеличением степени покрытия поверхно-
сти частиц эмульгаторами, которая, в свою очередь,
связана с поверхностным натяжением латекса. В за-
висимости от насыщенности защитных слоев и их
природы поверхностное натяжение латексов колеб-
лется обычно в пределах от 30 до 70 дин/см. Т. к.
подавляющее большинство Л. с. производят с при-
менением анионных эмульгаторов, частицы каучука
в них обычно заряжены отрицательно. В зависимости
от типа эмульгатора устойчивость Л. с., кроме того,
зависит от pH водной фазы. Л. с., стабилизованные
мылами на основе жирных к-т, канифоли и др., устой-
чивы лишь при pH выше 7—8. Латексы, стабилизо-
ванные сульфонатами (напр., некалем), устойчивы и
при более низких значениях pH. Наиболее распро-
странены дивинил-стирольные Л. с., полу-
чаемые сополимеризацией дивинила и стирола («го-
рячие» — при 50°, «холодные» — при 5—20°). Уве-
личение относительного количества стирола приводит
к повышению темп-ры стеклования содержащегося
в латексе полимера и прочности получаемых из ла-
текса вулканизованных ненаполненных пленок. Для
достижения прочности 150—180 кг/см2 полимер «горя-
чего» латекса должен содержать не менее 40% сти-
рола; повышение содержания стирола в полимере до
60—65% позволяет получать пленки с хорошими фи-
зико-механич. показателями в невулканизованном
состоянии и повышенной устойчивостью к окислитель-
ному старению. В случае необходимости сочетания
удовлетворительной прочности и морозостойкости
изделий целесообразно применение «холодных» Л. с.,
обеспечивающее хорошие физико-механич. показатели
пленок при более низком содержании связанного сти-
рола.
Дивинил-нитрильные Л. с. получают
обычно сополимеризацией дивинила с акрилонитри-
лом при 30° или при 5°. Содержание акрилонитрила
в мономерной фазе от 18 до 50%. Увеличение относи-
тельного количества акрилонитрила также приводит
к повышению физико-механич..показателей латексных
пленок с одновременным падением их морозостойко-
сти. Изготовляемые на основе дивинил-нитрильных
латексов изделия способны противостоять действию
масел. Пленки из этих латексов обладают высокой
адгезией к полярным поверхностям и хорошо совме-
щаются с полярными термопластичными (напр.,
поливинилхлоридными) и термо реактивными (напр.,
феноло-формальдегидными) смолами.
Дивинил-акрилатные и д и ви-
ни л-м-ста крила тные Л. с., получаемые
сополимеризацией дивинила с метилакрилатом или
метилметакрилатом, дают при высушивании пленки
с хорошим блеском, высокими физико-механич. пока-
зателями и устойчивостью к окислительному и ультра-
фиолетовому старению.
Дивинил-винилпиридиновые и
дивинил-метилвинилпиридиновые
латексы получают сополимеризацией дивинила с не-
большими количествами (5—15%) винилпиридинов
(напр., 2-метил-5-винилпиридина). Благодаря хоро-
шим адгезионным свойствам содержащегося в них
полимера применение этих Л. с. для пропитки тканей
обеспечивает очень высокую прочность связи между
элементами многослойных резино-тканевых изделий,
в том числе шинных покрышек.
Хлоропреновый латекс, как и нату-
ральный, при желатинировании образует прочный
гель; разрывное усилие ненаполненных пленок дости-
гает 200—300 кг/см2. Тонкостенные изделия из хлоро-
пренового Л. с. отличаются от соответствующих изде-
лий из натурального латекса повышенной масло-, огне-
и озоностойкостью и более низкой газопроницае-
мостью, но значительно уступают им в отношении мо-
розостойкости (температура стеклования полихлоро-
прена — минус 40°).
Большие перспективы открывает модифицирование
свойств Л. с. путем добавления в полимеризуемую
систему небольшого количества (1—5%) карбоксил-
содержащих мономеров, напр. метакриловой к-ты,
что приводит к получению латексов, содержащих кар-
боксильные группы в полимере. Введение в эти латек-
сы окисей или гидроокисей поливалентных металлов:
ZnO, MgO, Са(ОН)2 и др. — приводит к своеобразной
вулканизации полимера с образованием солевых
связей между его макромолекулами. Такие полимеры
отличаются исключительно высокой прочностью в не-
наполненном состоянии; добавление к полимеризуе-
мому дивинилу метакриловой к-ты в количестве 2%
повышает прочность плёнок из такого латекса от
15 кг/см2 до 150—300 кг/см2. Дивинил-нитрильные
карбоксилсодержащие латексы могут давать пленки
с прочностью 400—600 кг/см2 в сочетании с высокой
бензомаслостойкостью.
Среди полимерных материалов Л. с. занимают одно
из первых мест по возможностям применения. Они
используются для пропитки шинного корда, для из-
готовления широкого ассортимента губчатых, формо-
вых, тонкостенных и др. резиновых изделии, для
произ-ва водоразбавляемых красок, для отделки кож,
пропитки и покрытия бумаги, изоляции проводов;
применяются в виде цементно-латексных смесей
в строительстве, в обувной, полиграфической, хими-
ческой и других отраслях промышленности в каче-
стве клеев. Л. с. используются в производстве за-
менителей кожи, нетканых текстильных материа-
лов и т. д.
Лит.: Литвин О. Б., Синтетические латексы, Л.—М.,
1953; Синтетический каучук, под ред. Г. Уитби, пер. с англ.,
М„ 1957; Воюцкий С. С., Ш т а р х Б. В., Физико-химия
процессов образования пленок из дисперсий высокополимеров,
М., 1954; Лебедев А. В., Ф е р м о р Н. А., Хим. наука и
пром-сть, 1957, 2, 3, 339; Производство и применение син-
тетических латексов. Сб., М.—Л., 1953; Синтез латексов и их
применение. Сб. статей, Л., 1961; Нобль Р. Д., Латекс
в технике, пер. с англ., Л., 1962; Ullmann, 3 Autl., Bd 9,
Munch.—В., 1957, S. 351 — 58. А. В. Лебедев, А. И. Езриелев.
ЛАТУНЬ — см. Меди сплавы.
ЛАУРИЛМЕРКАПТАН — см. Додецилмеркаптан.
ЛАУРИНОВАЯ КИСЛОТА (додекановая кислота)
СН3(СН2)10СООН, мол. в. 200,31 —кристаллы; т. пл.
44,2; т. кип. 225°/100 мм, 141°/0,6 мм; 0,875;
п*& 1,4191; кислотное число 280,23. Л. к. обладает
общими свойствами карбоновых к-т. В виде глице-
Эфир Мол. вес Т. кип., °C/мм d (°C) пЬ
Метиловый 214,34 214/760а — 1,4220г
Этиловый 228,36 163/25 0,867 (19) 1,4326 Д
и-Бутиловый 256,42 194/30 0,855 (25) 1,4354
н-Амиловый 270,44 0,858 1,4380
н-Гептиловый 298,49 184/2 0,8635 (15) 1,441/20
Фениловый 276,40 210/15 ° — —
n-Тол иловый 290,43 219,5/15в — —
а Т. пл.5°. 6 Т. пл. 24,5°. в Т. пл. 28°. г При 15°. «При
12,9°.
933
ЛАУРИНОВЫЙ АЛЬДЕГИД - ЛЕВОПИМАРОВАЯ КИСЛОТА
934
ридов содержится в кокосовом и лавровом маслах, из
к-рых ее и получают. В технич. Л. к. содержатся
примеси миристиновой и олеиновой к-т. Цинковую
соль Л. к. применяют в резиновой промышленности;
натриевая соль — основная составная часть многих
мыл. Эфир этиленгликоля- и лауриновой кислоты
СН3(СН2)10СООСН2СН2ООС(СН2)10СН3 используют в
качестве пластификатора. Свойства эфиров Л. к. при-
ведены в табл, на стр. 932.
Метиловый и этиловый эфиры Л. к. применяют как
душистые вещества с фруктовым запахом.
См. также Жиры растительные, Мыла, Высшие
жирные кислоты. Н- А. Несмеянов.
ЛАУРИНОВЫЙ АЛЬДЕГИД (н-додеканаль), моле-
кулярный вес 184,31, СН3(СН2)10СНО — бесцветные
кристаллы с неприятным запахом; т. пл. 44,5°; т. кип.
185°/100 мм; df 0,8532; пр 1,433; семикарбазон,
т. пл. 105°; 2,4-динитрофенилгидразон, т. пл. 106°.
Л. а. находится в ничтожном количестве в сосновых
иглах, маслах лимона и руты. На воздухе Л. а. по-
степенно окисляется в лауриновую к-ту; под влиянием
кислот образует димер с т. пл. 57,5°. Л. а. получают
в пром-сти пропусканием смеси лауриновой и мура-
вьиной к-т над медью при 300—330° либо окислением
лауринового спирта. Раствор Л. а. при значительном
разбавлении приобретает цветочный запах; приме-
няют его как душистое в-во, вводится в очень неболь-
ших количествах (0,05—0,1%) в парфюмерные ком-
позиции. в. М. Дашунин.
ЛАУРИНОВЫЙ СПИРТ (додеканол-1, и-додеци-
ловый спирт) СПН23СН2ОН, мол. в. 186, 33— бесцвет-
ные кристаллы; т. пл. 26°; т. кип. 255—259°/760 мм,
150°/20 мм; d)'1 0,8201; нерастворим в воде, раство-
рим в спирте и эфире. Л. с. получают восстановлением
этилового эфира лауриновой к-ты натрием в спирто-
во-толуольном р-ре. При кипячении Л. с. с 48%-ной
бромистоводородной к-той в присутствии конц. H2SO4
образуется м-додецилбромид (бромистый лаурил
С12Н25Вг, т. кип. 145°/15 мм), применяемый в синтезе
додецилмеркаптана; сложные эфиры Л. с. и серной
к-ты используются в качестве моющих средств и де-
тергентов. ' В. Н. Фросин.
ЛЕВОМИЦЕТИН (хлоромицетии, хлорамфеникол),
молекулярный вес 327,14 — кристаллы, бесцветные
Н СН2ОН
N°2 \==/ <Н~Н
он NHCOCHCij
иглы, очень горького вкуса;
т. пл. 150,5—151,5°; кри-
сталлизуется из воды или
дихлорэтана, легко раство-
рим в низших спиртах,
ацетоне, уксусной кислоте, П,П-диметилацетамиде,
труднее — в воде (2,5 мг/мл при 25°), практически
нерастворим в углеводородах. В молекуле Л. со-
держатся два асимметрич. атома углерода: =
= —25,5° (этилацетат), [a]|J = -rl8,6° (С = 4,9,
спирт). Устойчив при хранении как в порошке, так и
в водных р-рах, к-рые не теряют активности после
часового кипячения. Л. устойчив при pH р-ра от 2,0
до 9,5; в более щелочной среде быстро инактивируется.
Л. является первым Аследованным природным соеди-
нением, содержащим в своей структуре п-нитрофениль-
ную и дихлорацетильную группы, и первым антибио-
тиком, крупное промышленное произ-во к-рого осу-
ществлено синтетическим путем. Промышленный син-
тез Л. в СССР основан на использовании стирола:
сн=сн3
снз°н'с|г.
осн3
СН-СН2С1
осн3
СН-СН2С1
no2
COCHgNHg'HCI
(сн3со)2о
no2
coch2nhcoch3
OH',CH2O
NOa
NHCOCH3
CO-CH-CH2OH
OH NHCOCH3
< CH-CH-CH3OH
AI(OC3H,-M0)s
деление
стереоизоиерое
no2
OH NH3
CH-CH-CH
no2
2OH
Полученный рацемат mpeo-амииодиола делят иа
оптические антиподы, пользуясь различной раство-
римостью их кислых виннокислых солей или методом,
основанным на чередующемся пересыщении р-ра ра-
цемата одним из антиподов. D-изомер действием ме-
тилового эфира дихлоруксусной к-ты переводят в Л.;
биологически неактивный L-изомер рядом реакций
превращают обратно в рацемат треоамииодиола.
Стереохимически Л. аналогичен псевдоэфедрину
и относится к соединениям mpeo-ряда; конфигурация
атома углерода, с к-рым связана дихлорацетиламино-
группа, такая же, как и в природных аминокислотах.
Антибактериальное действие Л. весьма специфично
и связано, вероятно, с подавлением образования бак-
териальных протеинов, содержащих аминокислоты
L-конфигурации. Три возможных стереоизомера Л.
(L-mpeo-, D-apumpo- и L-apumpo-) практически неак-
тивны. Препарат синтомицин — рацемат Л.
(D, Lmpeo-форма) — содержит 50% действующего
начала и, следовательно, вдвое менее активен, чем Л.
Синтомицин имеет т. пл. 149—153° и несколько менее
растворим, чем Л. Даже небольшие изменения в
структуре молекулы Л. ведут к уменьшению или
полной потере активности, и среди многих сотен
синтезированных аналогов и производных Л. не най-
дено веществ, равных по ценности исходному анти-
биотику. Л. — высокоактивный антибиотик с широ-
ким спектром антибактериального действия, выделен
в 1947 из актинбмицета Streptomyces venezuelae.
Л. весьма активен против многих грамположительных и
грамотрицательных бактерий, риккетсий, спирохет и нек-рых
крупных вирусов; действует на штаммы микроорганизмов,
устойчивых к сульфаниламиду, пенициллину и др. антибиоти-
кам. Л. задерживает развитие микроорганизмов. Применяют
его длн лечения брюшного тифа и паратифов, дизентерии,
бруцеллеза, коклюша, пневмонии, гонореи, гнойных инфекций,
туляремии, сыпного тифа, трахомы, пситтакоза и др. Хотя
Л. относительно мало токсичен, применение его допускается
только по назначению и под наблюдением врача из-за возмож-
ного влияния его на кроветворные органы и развивающихся
иногда (как и при приеме других антибиотиков) грибковых
поражений кожи и слизистых оболочек.
Количественно Л. в биологич. объектах определяют коло-
риметрически, сравнением интенсивности окраски красителя,
полученного в результате восстановления, диазотирования
и азосочетания исследуемого образца с соответствующим стан-
дартом.
В педиатрии иногда применяют эулевомицетин, т. пл. 87—
89°, или эусиитомицин, т. пл. 84—87°, сложные эфиры (по
первичной оксигруппе) соответствующих препаратов и пальми-
тиновой к-ты. Эти соединения практически безвкусны; в орга-
низме происходит их гидролиз с освобождением действующего
начала — Л.
Лит.: Шемякин М. М. [и др.], Химия антибиоти-
ков, т. 1, 3 изд., М., 1961, с. 337. А. П. Сколдинов.
ЛЕВОПИМАРОВАЯ КИСЛОТА С80Н30О2, мол. в.
302,46 — одна из основных кислот смоляных; белый
935
ЛЕВУЛИНОВАЯ КИСЛОТА —ЛЕЙКАРТА РЕАКЦИИ
936
кристаллический порошок; т. пл. 150°; [а]|$ = —280,4°
(спирт), коэффициент удельного поглощения а: 19,6
Н С СООН (272—273 мк) и 0,8 (218 л/.ик);
3 хорошо растворима в бензоле,
р ®1 диэтиловом эфире, ацетоне,
скипидаре, хуже — в этаноле
Н3сТ ХСН3 и метаноле, нерастворима в
%в,АсНч воде. Л. к. медленно окис-
СН3 ляется на воздухе в темноте,
быстрее — на свету, соли щелочных металлов и ам-
мония растворимы в спирте и в воде, лучше — при
нагревании; соли двух (и более)-валентных металлов
нерастворимы в воде.
Л. к. чрезвычайно легко изомеризуется при дейст-
вии сильных минеральных к-т, органич. к-т, особен-
но при нагревании. Основными продуктами изомери-
зации Л. к. являются абиетиновая, яеоабиетииовая
и палюстровая к-ты. При более длительном нагрева-
нии, до 180° и выше, происходит диспропорциони-
рование водорода и получаются дегидро-, дигидро-
и тетрагидроабиетиновые кислоты. Под действием
УФ-лучей Л. к. превращается в правовращающий
изомер.
Л. к. содержится в смоле (живице), выделяющейся
при ранении деревьев большинства хвойных. Проще
ее можно получить из свежей живицы сосны обыкно-
венной кристаллизацией ее натриевых солей; еще луч-
ше — кристаллизацией борниламиновых, бутанолами-
новых или бруциновых солей. В процессе производст-
венного получения канифоли Л. к. большей частью
изомеризуется, а поэтому в канифоли ее содержание
составляет, как правило, менее 10%. Аддукты Л. к.
и малеиновой или фумаровой к-т и их эфиры — хоро-
шие пленкообразователи лакокрасочных материалов.
ЛЕВУЛИНОВАЯ КИСЛОТА — простейшая кето-
карбоновая к-та, СН3—СО—(СН2)2—СООН, мол. в.
116,11, бесцветные кристаллы; т. пл. 37°; плотн. 1,140;
т. кип. ок. 250°; 1,442; хорошо растворима в воде,
спирте и эфире. Л. к. обладает всеми реакциями кето-
нов и типичными свойствами карбоновых к-т; с си-
нильной к-той дает соответствующий циангидрид,
с фенилгидразином — гидразон, с гидроксиламином —
оксим и т. д. При продолжительном нагревании теряет
воду, образуя внутренний ангидрид (еноллактон):
СНа-СО-СНз СН = С-СН3
I -Ч >°
СН2-СООН СН3-СО
Л. к. очень легко восстанавливается (с образованием
у-валеролактона). Л. к. образуется в значительном
количестве при нагревании сахаров — гексоз—с
соляной к-той. Первым продуктом реакции является
оксиметилфурфурол, к-рый может быть количественно
превращен в левулиновую и муравьиную к-ты:
CgHi 3Ов->СоН6Оз-»СН3—СО—(С На) а—COOH-J-HCOOH
При гексозном гидролизе в жестких условиях расти-
тельных материалов, содержащих полисахариды в
присутствии минеральных к-т, выход Л. к. может
составлять 60—70% от веса гексоз. Из солей Л. к.
наибольшее распространение получила натриевая,
применяемая в качестве антифриза. Добавка солей
кальция и лития в смазочные масла повышает их
качество. Кальциевая, магниевая и аммониевая соли
этой к-ты стимулируют рост растений. Нек-рые слож-
ные эфиры Л. к. применяют в качестве растворителей
смол, циклогексиловый и алкильные эфиры являются
пластификаторами. При взаимодействии Л. к. и ор-
ганич. аминов образуются термостойкие смолы. При
конденсации Л. к. с альдегидами получаются полу-
продукты, из к-рых можно синтезировать себаципо-
вую и др. к-ты. При полимеризации продуктов конден-
сации ненасыщенных эфиров Л. к. с формальдегидом
получаются твердые, неплавкие, химически стойкие
СМОЛЫ. С. В. Чепиго.
ЛЕВУЛИНОВЫЙ АЛЬДЕГИД (пентанон-4-ал-1,
у-кето-н-валериановый альдегид) СН3СОСН2СН2СНО,
мол. в. 100,12—кетоальдегид жирного ряда; бесцвет-
ная жидкость; т. кип. 186—188° (разл.); т. пл. — 21°;
1,0184; 1,42567; растворяется в воде,
спирте, эфире и др. органич. растворителях, летуч с
водяным паром. Л. а. дает реакции на альдегидную и
кето-группы, напр. образует диоксим, дисемикар-
базон, ди-2,4-динитрофенилгидразон, диацетали; обла-
дает восстанавливающими свойствами: восстанавли-
вает аммиачный р-р AgNO3, фелингов р-р. При окис-
лении альдегидной группы Л. а. образуется левули-
новая кислота. Л. а. дает йодоформную реакцию,
характерную для метилкетонов. Эту реакцию и реак-
цию образования 2,4-динитрофенилгидразона исполь-
зуют для колориметрич. определения Л. а. Реакция
может служить методом количественного определения.
Л. а. может быть получен синтетически и озонолизом
метилгептанона, изопренового каучука, гуттаперчи
II Нек-рых др. Природных продуктов. Л. И. Липевич.
ЛЕГИРОВАННЫЕ СТАЛИ — см. Железа сплаеы.
ЛЕГКОЕ МАСЛО — см. Каменноугольная смола.
ЛЕЙКАРТА РЕАКЦИИ — 1) замена карбонильной
группы аминогруппой прп действии на альдегиды и
кетоны формиата аммония, формамида или их про-
изводных:
/О-» -н
Ч Г
,)С--N-COH
Н
rT
,C-N-COH
R А
Rx ^O -
—^^C=N-C; .C-N-CH-0
R XH R>
H,0 . - Л.0
NH2COH » NH, + o-cf
XH
Rv - -
c—n=ch—о + о-с; —-
Rz XH
R4?~C0H
r,zc-n=cho
R\ - NH, Rx rs
CH—N“CHO-----— ,CHNHCOH----- CHNH„
R Rz R" 2
Л. p. применима к алифатич., алициклич., жирно-
ароматич., ароматич. и гетероциклич. альдегидам и
кетонам. N-алкил- и N, N-диалкилформамиды реа-
гируют труднее формамида, а N-арилформамиды —
значительно легче. Вместо замещенных формамидов
или формиата аммония можно исходить из равномо-
лекулярной смеси аммиака или амина и муравьиной
к-ты. В качестве растворителей используют муравьи-
ную или уксусную к-ты, их эфиры, этиленгликоль
и др. Иногда применяют катализаторы: хлориды маг-
ния, цинка и железа, сульфат аммония и катализаторы
гидрирования.
Механизм Л. р. до сих пор точно Же установлен; наиболь-
шим признанием пользуется след, схема реакции при образо-
вании первичных и вторичных аминов:
ГО—-Н он
RM, I R4I +НСОО
C-—'-N-COH---- C-N-COH -и О~*~
Rz l„ R % 2'
R К
R\ *
—► , CH-N-COH—,ZCHNHR
R- l„
Реакция открыта P. Лейкартом в 1885.
Лит.: Богословский Б. М., в сб.: Реакции и
методы исследования органических соединений, кн. 3, М.,
1954, с. 255; Кост А. Н., Г р а н д б с р г И. И., Ж. общ.
химии, 1955, 25, вып. 7, 1436. И. П. Гамбарян.
937
ЛЕЙКОГЕН — ЛЕЙКОСОЕДИНЕНИЙ
938
2) Получение арилмеркаптанов из солей диазония и
этилксантогената калия гидролизом промежуточно об-
разующегося арилового эфира этилксантогеновой к-ты:
ArN.x+^E^j ArSCSOC2H5
-N2;-KX -KSCOOC2H5;
— НоО
- ArSK ±S ArSH
Обычно солянокислый р-р соли диазония нейтрали-
зуют содой до слабокислой реакции по конго и затем
приливают к нагретому до 70° водному р-ру этилксан-
тогената калия (при более низкой темп-ре создается
опасность накопления промежуточно образующегося
взрывчатого S-азосоединения). Ариловый эфир ксан-
тогеновой к-ты, выпадающий обычно в виде масла,
кипятят с 15%-ным р-ром NaOH или КОН. После
подкисления и добавления сернистого натрия, предот-
вращающего окисление, арилмеркаптан выделяют
обычным образом, часто перегонкой с паром. Л. р.
применима для синтеза тпофенола и замещенных тио-
фенолов, а также тионафтолов:
Гопкинсом в 1895 из пыльцы крыльев бабочки ка-
пустницы (Pieris brassicae), ошибочно принявшем его
за мочевую кислоту. Л. встречается в пыльце многих
других видов бабочек; строение его было в основном
установлено работами Виланда и Пуррмана и под-
тверждено синтезом, осуществленным в 1940 конден-
сацией 2,4, 5-триамино-6-оксипиримидина с щавеле-
вой к-той. Л. может быть получен также окислением
ксашпоптерина перекисью водорода, каталитически
над платиной или ферментативно в присутствии ксан-
тиндегидразы. Л. токсичен для мышей при внутривен-
ном введении в дозе 50 мг/кг веса.
Лит.: Вульфсон Н. С., Усп. химии, 1948, 17, вып. 2,
с. 249; Березовский В. М., там же, 1953, 22, вып. 2,
с. 191; Purrmann R., Fortschritte der Chemie organlscher
Naturstolte, 1945, Bd 4, S. 64; A 1 b e г t А., там же, 1954,
Bd 11, S. 350. H. С. Вульфсон.
ЛЕЙКОСОЕДИНЕНИЯ (лейкооснования) — вос-
становленные формы нек-рых красителей. Примерами
могут служить Л. триарилметановых, хинонимпповых,
индигоидных, антрахиноновых, полициклокетоновых
и сернистых красителей. Ниже приведены нек-рые Л.
I. KSCSOC2H5
2.КОН; 3, Н*
Cl
I. KSCSOC2H,
2. КОН; 3. Н*
l>KSCSOC2H5
2, КОН; 3. Н*
дарафуксин (парарозанmm
«ейиопарафуисин
Ррз -п - ахттрифекил^ц]
По Л. р. первоначально образуется S-азосоеди-
пение, к-рое в условиях реакции распадается с отщеп-
лением азота и образованием арилового эфира этил-
ксантогеновоп к-ты. Последний под действием основа-
ния расщепляется, образуя соль арилмеркаптана и
этилтнокарбонат (см. выше).
Л. р. используют для препаративного получения
замещенных тиофенолов; в пром-сти не применяется.
Реакция открыта Р. Лейкартом в 1890.
Лит.: Saunders К. Н., The aromatic diazo-compounds
and their technical applications, 2 ed., L., 1949, p. 325; S c h o-
berl A., Wagner А., в пн.: Houben—Weyl, 4 Anti.,
Bd 9,. Stuttgart, 1955, S. 12. E. M. Рохлин.
ЛЕЙКОГЕН [Ь-2-(а-фенил-а-карбэтоксиметил)-тиа-
золидин-4-карбоновая кислота]. Cj4Hj7O4NS, мол. в.
295,36 — бесцветные кристаллы,
CH2-Ss т. пл. 186—169°; трудно раство-
I ZCH-CH-C6H5 рим в воде и спирте, легко раст-
CH-NH СООС2Н6 ворим в щелочах. Водные р-ры Л.
СООН нестойки; с кислотами Л. дает
соли, легко гидролизующиеся во-
дой. Получают Л. конденсацией L-цистеина с этило-
вым эфиром а-формилфенилуксусной к-ты. Способ-
ность Л. увеличивать в крови содержание лейкоцитов
используется для лечения лейкопении.
Лит.: Струко» И. Т., Ж. общ. хим., 1952, 22, Ns 6,
1025. Р. Г. Глушков.
ЛЕЙКОМИЦИН — см. Магнамицин.
ЛЕЙКОПТЕРИН (2-амино-4,6,7-триоксиптеридин)
CeH6O3N5, молекулярный вес 195,14 — природный бе-
Он 5 ” ~
лыи пигмент, относящийся к клас-
су птеринов. Л. трудно растворим
в воде (1 вес. ч. в 750000 вес. ч.
воды), т. пл. выше 350°, растворы
его сильно флуоресцируют; УФ-
полосы поглощения (в воде при
pH 13): 240 ммк (Е 4,20),285 ммк (Е 3,84), 340 им»
(Е 4,02). Л. устойчив при двухчасовом нагревании
с конц. H2SO4, но разрушается при нагревании с 10 и.
НС1 при 165° с образованием глицина, СО, СО2 и
NH3 в соотношении 1:1:3:4. Л. впервые выделен
ОН
он
HaN
спектр имеет три
939
ЛЕЙКОТРОПЫ — ЛЕКАРСТВЕННЫЕ ВЕЩЕСТВА
940
Л. легко, многие даже под действием кислорода
воздуха, окисляются в исходные красители. Л. не-
растворимых в воде красителей (сернистых и содер-
жащих карбонильные группы — индигоидных, поли-
циклокетоновых и др.) хорошо растворяются в водных
щелочах и потому широко используются в кубовом
крашении. Нек-рые красители (индигоидные, серни-
стые и др.) поступают на рынок в виде Л. Так как
последние неустойчивы при хранении и транспорти-
ровке, то на практике часто применяют более стойкие
к окислению кислые эфиры Л., т. наз. кубозоли
(напр., индигозоль — натриевая соль кислого сер-
нокислого эфира лейкоиндиго). Кислые эфиры Л.
получают либо ацилированием Л. в присутствии пи-
ридина, либо непосредственно из красителя: обработ-
кой красителя железом в среде пиридина и ацили-
рующего агента (хлорсульфоновой к-ты и т. п.).
К Л. часто относят также гидр азосоединения, полу-
чающиеся восстановлением азокрасителей, например:
HOSO3-С0Н4-N = N-С0Н4—N (CH3)2JU
гелиантин (метилоранж)
—* HOSOg—С2Н4—NH—NH—С3Н4—N (СН3)2
лейкогелиантин
НООС-С„Н4-N = N-С0Н4-N(CHa)3 Si
метиловый красный
— HOOC-C0H4-NH-NH-Ci1H4-N (СН3)2
лейкооснование метилового красного
См. также Индиго.
Лит.: Коган И. М., Химия красителей, 3 изд., М.,
1956; Ворожцов Н. Н., Основы синтеза промежуточных
продуктов и красителей, 4 изд., М., 1955. В. Н. Фросин.
ЛЕЙКОТРОПЫ — вещества, применяющиеся для
получения вытравных рисунков по тканям, окрашен-
ным кубовыми или холодными красителями. Известны:
лейкотроп O,N — метилбензилфениламмоний хлорид
[C0H5N(CHa)2CH2C6Hs]Cl_, лейкотроп В.кальц. соль его
дисульфоната [(C6H4SO3^)N(CH*3)2CH2CeH4SO3^]Cl-.
При запаривании напечатанной ткани Л. реаги-
руют с восстановленным кубовым красителем,
образуя нерастворимые слабоокрашенные продукты
(лейкотроп О) или растворимые соединения (лейко-
троп В). Марка В образует водорастворимые соеди-
нения, легко смываемые с поверхности ткани при
промывании ее горячим р-ром силиката натрия (5—
15 г/л) и водой; в результате на окрашенной ткани
остается рисунок белого цвета. Марка О образует
нерастворимые соединения, ее применяют для полу-
чения окрашенных вытравок, в частности желто-
оранжевого цвета по тканям, окрашенным индиго.
Л. образуют с лейкосоединением кубовых красителей
простые бензиловые эфиры или же бензильное производ-
ное при атоме углерода гидроксильной группы лейкосоедине-
• I I
ния красителя НО—С—СН2С2Н2 или НО—С—CH2CeH4SOaH.
I I
При вытравке фонов, окрашенных холодными кра-
сителями, лейкотроп В взаимодействует с реакцион-
ными группами красителя (в частности, с гидроксиль-
ной группой), образуя растворимые и легко вымы-
ваемые соединения. Готовят Л. из N-диалкиланилина
(диметил- или диэтиланилина) и бензилхлорида. Л.
гигроскопичны и требуют хранения в хорошо закры-
ваемой таре.
Лит.: Порай-Кошиц Е. А., Сидоров И. И.,
ЖРФХО, 1910, 42, 1079; Чиликин М., Исследования
в области индиго [ч. 1], М., 1914; С а д о в Ф. И., Корча-
гин М. В. |и Матецкий А. И., Химическая техноло-
гия волокнистых материалов, 2 изд., М., 1956, с. 728;
Фротшер Г., Химия и физическая химия текстильных
вспомогательных материалов, пер. с нем., т. 1, М., 1958,
с. 165. Н. А. Штелинг.
ЛЕЙЦИН (а-аминоизокапроновая кислота), мол.
вес 131,18, (CH3)2CHCH2CH(NH2)COOH — в приро-
де встречается преим. в L-форме, входя в состав
почти всех белков животного и растительного проис-
хождения. Л. — бесцветные кристаллы, т. пл. 293—
295° (с разл.); растворим в кислотах и щелочах,
умеренно растворим в воде, плохо — в холодном
спирте и нерастворим в эфире. Для L-Л. [a]D = —11°
(в воде) и -ф-16о (в 5 н. HCI); pAaj 2,36; pKas 9,60;
р/ 6,04. Л. дает качественные цветные реакции на
аминокислоты, в частности с нингидрином. Для коли-
чественного определения Л. используют хроматогра-
фия. и микробиологич. методы.
D, L-Л. получают из изовалерианового альдегида,
из a-бромизокапроновой к-ты и др. общими способами
синтеза аминокислот; L-Л. получают из гидролизатов
белков. L-Л. — незаменимая аминокислота, приме-
няется в качестве лечебного препарата в смеси с др.
аминокислотами при лечении нек-рых нарушений
обмена веществ и психич. заболеваний.
Лит.: Белки, под ред. Г. Нейрата и К. Вейли, пер. с
англ., т. 1—2, М., 1956; Майстер А., Биохимия амино-
кислот, пер. с англ., М., 1961; Biochimie comparee des acl-
des aminds basiques, Р.‘, 1 960; GreenstetnJ. Ph. and
Wl n 1 t z M., Chemistry of the amino acids, v. 1—3, N. Y.—L.,
[1961]; Greenberg D. M. led.], Metabolic pathways, v.
2 — Amino acids, nucleic acids, porphyrins, vitamins and
coenzymes, N.Y. — L., 1961. См. также литературу при статье
Аминокислоты. Л. И. Линевич.
ЛЕЙЦИНАМИНОПЕПТИДАЗА — фермент, гидро-
литически расщепляющий пептиды со свободной
аминогруппой у N-копцевого аминокислотного ос-
татка:
R—СН—СО—NH—СН —СО— +Н2О -
I I
NH2 R
— R— СНСООН + R—СНСО-
nh2 nh2
где R — боковая цепь аминокислотного остатка.
Л. с наибольшей скоростью расщепляет лейцил-
пептиды, однако она действует и на пептиды с др.
алифатич. N-концевыми аминокислотами, причем ско-
рость гидролиза уменьшается с уменьшением длины
алифатич. цепи в ряду: лейцин —- норвалин — ва-
лин —- аланин — глицин. Со значительно меньшей
скоростью Л. расщепляет пептиды с N-концевыми
остатками фенилаланина, тирозина, гистидина или
триптофана. Помимо пептидов, Л. также гидролизует
амиды аминокислот, в частности лейцинамид. Дейст-
вие Л. строго стереоспецифично и направлено только
на остатки L-аминокислот. Л. широко распростра-
нена в животных и растительных тканях, а также
у нек-рых микроорганизмов. Л., выделенная из почек
свиньи и очищенная путем фракционирования ацето-
ном и (NH4)2SO4, а также с помощью электрофореза
в крахмальном геле, имеет оптимум активности при
pH 7,8—9,3, обладает электрофоретич. подвижностью
6,0-10~6 смг/в-сек (в 0,1Л/ вероналовом буфере, pH
8,5) и характеризуется изоэлектрич. точкой р/ 4,0—
5,0. Мол. в. фермента составляет ок. 300 000. Л.
является Mg-протеидом и при удалении Mg2+ теряет
свою активность. Константа диссоциации комплекса
Mg-фермент 1,2-10“4 М• Mg моя?ет быть заменен Мп
без существенного изменения активности Л. Актив-
ность Л. подавляют такие связывающие Mg соедине-
ния, как цитрат, этилендиаминтетраацетат и др.,
а также ионы тяжелых металлов: Zn2+, Pb2+, Hg2 r и
Fe2+. Л. применяют при изучении расположения ами-
нокислотных остатков в молекулах различных белков
путем последовательного отщепления их с помощью
Л., атакже для определения абс. оптич. конфигурации
этих остатков.
Лит.: Smith Е. L., Н1 11 R. L., Leucine aminopepti-
dase, в кн.: The Enzymes. Ed. Р. D. Boyer, H. Lardy, К. Myr-
bdck, v. 4, 2 ed., N. Y., 1960, p. 37. В. Б. Спиричев.
ЛЕКАРСТВЕННЫЕ ВЕЩЕСТВА синтетиче-
ские — соединения, применяемые для лечения или
предупреждения заболеваний.
941
ЛЕКАРСТВЕННЫЕ ВЕЩЕСТВА
942
В древности люди применяли для лечения бо-
лезней растения в разных видах (настойки, отвары
и др.), высушенных насекомых, пресмыкающихся,
органы животных и т. п. С развитием научных зна-
ний из природных источников были получены ин-
дивидуальные, достаточно чистые вещества, отли-
чающиеся постоянством действия, поддающиеся опре-
деленной дозировке и удобные* для употребления.
Так были получены широко применяемые алкалоиды,
антибиотики, гормоны, витамины и др. Наряду
с этими веществами в медицине еще широко поль-
зуются природными препаратами в виде экстрактов,
настоек, отваров из растений (например, экстракты
красавки, чилибухи, крушины, горечавки и др.) или
органов животных (например, тиреоидин, питуитрин,
липокаин и др.). В современном ассортименте меди-
каментов подобных препаратов 25—30%, но с каждым
годом число их уменьшается за счет бурного р азвития
синтетич. Л. в.
Первые ценные синтетич. лекарственные вещества
появились в последней четверти 19 в. Так, в 1887
было открыто жаропонижающее действие ацетанил-
ида (антифебрин); вслед за ним появляется фенаце-
тин, в 1896 — пирамидон, в начале 20 в. — веро-
нал и т. д. Целые группы новых синтетич. препаратов
не имеют себе подобных среди природных веществ
(напр., жаропонижающие, наркотические, снотвор-
ные, противогистаминные и др.). Исключительные
успехи в области синтеза Л. в., давшие возможность
вести эффективную борьбу с большинством заболева-
ний и значительно удлинить среднюю продолжитель-
ность человеческой жизни, обусловлены были разви-
тием химии и медико-биологич. наук (физиологии,
фармакологии, микробиологии, биохимии), позволяю-
щих исследовать действие химич. соединений на фи-
зиологии. процессы животного организма и на возбу-
дителей инфекций. Большое значение в создании Л. в.
имеет разработанный И. П. Павловым метод экспери-
ментальной терапии, позволяющий изучать действие
препарата на животных, у к-рых искусственно вызы-
вается болезненный процесс, сходный с соответствую-
щим заболеванием человека. Не менее важное значе-
ние имело развитие экспериментальной химиотера-
пии, изучающей воздействие Л. в. на инфекционные
процессы у животных. Практически развитие экспе-
риментальной химиотерапии началось после того, как
П. Эрлих показал плодотворность метода биологиче-
ского испытания определенных рядов соединений,
отличающихся друг от друга по своему строению
(характеру функциональных групп, их положению в
молекуле и т. д.). В результате длительных иссле-
дований мышьякорганич. соединений Эрлиху уда-
лось синтезировать эффективный препарат против
сифилиса — сальварсан. Метод экспериментальной
химиотерапии дал возможность внедрить в медицин-
скую практику ряд сульфамидных, противомалярий-
ных, противотуберкулезных и др. препаратов, а также
антибиотики.
Совместной работой химиков и биологов выявлен
ряд закономерностей, связывающих биологич. эф-
фект с химич. строением, но эти закономерности не
носят общего характера и действуют только в пределах
определенных рядов соединений.
Классификации Л. в., построенной по единому прин-
ципу, не существует. Л. в. делят на группы по фарма-
кология. принципу, т. е. по характеру их избиратель-
ного действия на организм (наркотические,снотворные,
анальгезирующие, мочегонные, спазмолитические и
т. д.), или по механизму действия (ганглиоблокирую-
щие, антихолинэстеразные, антигистаминные и т. д.).
Нек-рые группы выделены по признаку их физиоло-
гия. значения (гормональные препараты, витаминные
препараты), по Лечебному применению (противосьфи-
литич. средства, противоглистные средства и т. д.),
нек-рые — по химич. принципу (сульфаниламидные
препараты, фенотиазиновые производные и т. д.).
Краткая характеристика наиболее важных групп
Л. в. приводится ниже (см. также специальные ста-
тьи, посвященные описанию отдельных Л. в.).
Наркотичес.кие и снотворные ве-
ще с т в а. К наркотич. веществам, применяемым при
хирургия, операциях, относят гл. обр. летучие ве-
щества, к-рые вводят в организм путем вдыхания.
Чаще всего для этих целей пользуются этиловым эфи-
ром,применяют также виниловый эфир, а в последнее
время и галотан (фторотан) CF3CHBrJ, предложенный
в 1956. Из газообразных Л. в. для наркоза приме-
няют также циклопропан и закись азота. Для кратко-
временного наркоза пользуются хлористым этилом
и трихлорэтиленом (триленом).Для кратковременного
наркоза путем внутривенного введения используют
натриевые соли некоторых барбитуратов (гексенал)
и тиобарбитуратов (тиопентал). Хлороформ, ранее
широко применявшийся в качестве наркотич.средства,
употребляется редко в связи с его токсичностью.
В качестве снотворных веществ чаще всего поль-
зуются производными барбитуровой кислоты, боль-
шинство к-рых применяется в виде растворимых нат-
риевых солей. Изучены закономерности,связывающие
силу наркотич. действия барбитуратов, продолжи-
тельность и быстроту наступления этого действия с
их химич. строением. Снотворное действие у барби-
туратов (I) проявляется, если сумма атомов углеро-
да в алкильных заместителях В' и R" у
С(5) не меньше 4. С увеличением этого
числа растут сила и скорость их дейст-
вия, а продолжительность укорачивает-
ся. Тот же эффект достигается при вве-
дении метильного заместителя у N(J,
или у N(3„ а также при замене атома
кислорода у С(2, атомом серы. Барбитураты, со-
держащие у С(5) алкильные группы с разветвленной
цепью, действуют сильнее, чем соединения с нераз-
ветвленными цепями, при этом для повышения силы
действия выгодно, чтобы разветвление цепи проис-
ходило у углеродного атома, ближайшего к С(5).
Барбитураты можно классифицировать по продол-
жительности их действия.По этому принципу их делят
на барбитураты длительного действия (8—12 час),
напр. веронал, мединал, люминал; барбитураты сред-
ней продолжительности действия (6—8 час), напр.
барбамил, этаминал; барбитураты кратковременного
действия (2—3 час), напр. квиэтал [R' = —СН(СН3)2;
R" = —СН2СВг=СН2]; барбитураты сверхкратко-
временного действия, напр.
В качестве снотворных средств
нек-рые производные пиридина,
и пиперидина, напр. димер и
диэтил-5-метилпиперидин).
Противосудорожные
меняют для лечения эпилепсии. К таким Л.
относятся люминал (II), недостатком к-рого является
!Г в
NH-CO
3 4
* IV ‘
гексенал, тиопентал.
применяют также
напр. тетридин,
н (2,4-диоксо-3,3-
NH-eo„ nh-co
I Г I I /СоН.
H III
вещества при-
” ”. в.
NH
(fO4C(CeHs),|
NH-CO
IV
Н§Св~^Н2 СНаСНгС|
' NH-CO
v
о
(^о (р(Сн3)2
H3c-N — СО
VI
вызываемая им сонливость, гексамидин (III), д и ф е-
н и н (IV) и хлоракон (V). Эти препараты эф-
943
ЛЕКАРСТВЕННЫЕ ВЕЩЕСТВА
944
фективны при такой форме заболевания эпилепсией,
при к-рой у больного периодически наступают судо-
рожные припадки. При эпилепсии, протекающей без
судорожных припадков, эффективен т р и м е т и н
(VI). В структуре перечисленных противосудорож-
ных Л. в. имеется группировка —С—N—СО—С—.
В последнее время в качестве противосудорож-
ного средства предложена р-окси-у-аминомасляная
кислота (NH2—СН2СН(ОН)СН2СООН). К противосу-
дорожным веществам иногда относят также соеди-
нения, оказывающие лечебное действие при повышен-
ном тонусе скелетной мускулатуры и мышечных дро-
жаниях, зависящих от поражения подкорковых узлов
головного мозга (напр., при болезни Паркинсона).
К этой группе принадлежат т. наз. х о л и н о л и ти-
пе с к и е вещества (т. е. вещества, блокирующие
чувствительность тканей к передатчику нервных им-
пульсов ацетилхолину), к-рые хорошо проникают в
центральную нервную систему (центральные холпно-
литики). Такими Л. в. являются тропацин, динезин,
циклодол (артан).
Седативные и нейроплегические
вещества действуют успокаивающе на центральную
нервную систему. Такое действие оказывают малые
дозы барбитуратов и других снотворных (напр., ада-
лин). К средствам, к-рые не оказывают снотворного
действия (их обозначают терминами: нейроплегикп,
транквилизаторы и др.), относится аминазин, дейст-
вие к-рого связывают с его способностью блокировать
чувствительность тканей к передатчику нервных им-
пульсов — норадреналину. По действию близки к ами-
назину нек-рые другие фенотиазиновые производные
(напр., пропазин и мепазин). В неврологич. и психи-
атрической практике в качестве средств, устраняю-
щих внутреннюю напряженность и состояние трево-
ги, используют также мепротан и амизил —
(С0Н6)2С(ОН)СОО(еН2)2А'(С2Н5)2 • НС1.
Анальгезирующие (болеутоляющие) в е-
щ е с т в а напоминают по своему действию п строе-
нию морфин (VII). Типичными Л. в. подобного рода
IX х
являются дроморан (VIII), фенадон (IX) и
промедол (X). Анальгезирующнми свойствами обла-
дают также производные анилина (например, ан-
тифебрин, фенацетин), пиразолона (антипирин,
анальгин) и салициловой кислоты, в частности аспи-
рин. Применение последних не связано с опасно-
стью развития болезненного пристрастий, характер-
ного при употреблении препаратов VII—X. Все они
оказывают также жаропонижающее действие, а про-
изводные салициловой к-ты и пиразолона, в особен-
ности бутадиен, проявляют выраженное противорев-
матич. действие.
Стимуляторы центральной нерв-
ной системы, устраняющие состояние психич.
депрессии (т. наз. психолептики, или т и мо-
ле п т и к и), и вещества, возбуждающие дыхатель-
ный и сосудодвигательный центры (а н а л е п т и к и).
Соединения первого типа встречаются среди фенил-
изопропиламинов (см. Фенамин, Первитин, Фенатин)
и гидразидов (напр., Ипразид). Выраженное антиде-
прессивное действие оказывает также м е р и д и л
(XI) и пиридрол (XII). Из синтетич. аналепти-
ОСОСН3
Х>
Н5С6 ОН
XII
ков к числу наиболее активных относятся коразол
и кордиамин. В качестве аналептика, устраняющего
токсич. эффекты наркотиков, в особенности барби-
туратов, большую ценность представляет б е-
м е г р и д (Р, р-м етилэтилглутаримид)
НдС, .С Н 2—СО,
>NH
H5cz хон2-со/
,СН3
HCOCH2N(C2H5)2 • HCI
сн3
• xill
СНЯ
Н3С
NHCOCH2N(C2H5)^HCI
сн’ XIV
ствуюшие на медиа-
Местноанестезирующие вещее т-
в а. Среди них наиболее популярны многочисленные
диалкиламиноалкиловые эфиры п-аминобензойной
к-ты n-RHNC6HiCOO(CH2)nN(R')2. К ним относится
широко применяемый для инфильтрационной и про-
водниковой анестезии новокаин, а также дикаин,
к-рый значительно более токсичен, но в отличие от
новокаина пригоден для поверхностной анестезии.
Анестетики этого класса вызывают относительно не-
продолжительную анесте'зию; более длительную ане-
стезию вызывают ди-
алкиламиноалкилами-
ды ароматич. кислот.
К их числу относит-
ся применяемый для
спинномозговой ане-
стезии совкаин. Отно-
сительно длительную
анестезию вызывают
также производные
ацетанилида: ксило-
каин (XIII) и три-
мекаин (XIV).
Вещества, дей
торные процессы, имитируют действие пе-
редатчиков нервных импульсов или, наоборот, изо-
лируют органы от соответствующих нервных влияний.
Передатчиком нервного возбуждения в нек-рых си-
напсах центральной нервной системы, в вегетативных
узлах и в окончаниях парасимпатич. нервных волокон
и двигательных нервов является ацетилхолин (см.
Ацетилхолинхлорид). Вещества, действующие по-
добно ацетилхолину, называют холиномимети-
ческими, а вещества, препятствующие действию
ацетилхолина, — холинолитическими. Пе-
редача импульсов с окончаний симпатич. волокон,
осуществляется посредством норадреналина. Веще-
ства, действующие подобно норадреналину, наз.
адреномиметическими, а вещества, пре-
пятствующие действию этого медиатора, — а д ре-
но л и т и ч е с к им и.
К холиномиметич. веществам относится карбахо-
лин; применение его усиливает сокращения кишеч-
ника и мочевого пузыря и вызывает сужение зрачка
и понижение внутриглазного давления, такое же
действие оказывает бензамон-, эти средства применяют-
ся при лечении глаукомы. При «утяжелении» струк-
945
ЛЕКАРСТВЕННЫЕ ВЕЩЕСТВА
946
туры холиномиметиков могут быть получены холино-
литич. вещества; напр., при утяжелении структуры
карбахолина образуется холинолитич. соединение —
дибутолин (CH3)2C2H5N(CH2)2OCON(C4H9)2.
Холиномиметич. эффекты могут быть также полу-
чены под влиянием веществ, блокирующих актив-
ность холинэстеразы — фермента,гидролизующего аце-
тилхолин. Наибольшее значение имеют а н т и х о-
линэстеразные вещества, в структуре
которых (подобно алкалоиду физостигмину) содер-
жится N-алкилкарбаминовая к-та, находящаяся в
эфирной связи с фенольным гидроксилом (см., напр.,
Прозерин). Мощным антихолинэстеразным действием
обладают нек-рые фосфорорганич. соединения, напр.
диизопропилфторфосфат (ДФФ), армии, пирофос,
фосфакол. Антихолинэстеразные вещества применяют
для лечения глаукомы, миастении, острой атонии ки-
шечника и мочевого пузыря, для стимулирования
родовой деятельности. Фосфорорганич. соединения,
ввиду их токсичности, применяют только местно для
лечения глаукомы.
К адреномиметическим вещест-
вам, применяемым для сужения сосудов и расши-
рения бронхов, относятся фенилалкиламины, причем
для сосудосуживающего действия важным является
наличие в структуре первичной аминогруппы (см.
Норадреналин) или вторичной аминогруппы с корот-
ким алкильным заместителем (напр., адреналин).
С увеличением числа атомов углерода в алкильном
заместителе при азоте сосудосуживающее действие
ослабевает. В отношении бронхорасширяющей ак-
тивности наблюдается обратная закономерность; ад-
реналин является сильным бронхорасширяющйм сред-
ством, изадрин действует еще сильнее, а норадреналин
не расширяет бронхов. Стойкость соединений подоб-
ного рода в организме обусловлена отсутствием гид-
роксилов'в ароматич. ядре и разветвленным характе-
ром алкильной цепи, соединяющей ароматич. ядро
с аминогруппой (см. Адреналин, Мезатон, Эфедрин).
К адренолитич. средствам относятся хлорэтиламины,
например дибензамин (C„H5CH2)2N(CH2)2C1. Адреноли-
тич. свойства проявляют также апрессин и бензолин;
их применяют при сосудистых заболеваниях.
Курареподобные вещества препят-
ствуют передаче импульсов с двигательных нервов
на скелетные мышцы. Простейшим соединением с
высокой курареподобной активностью является д е-
каметоний (CH3)3N(CH2)10N(CH3)2 • 2Х, где X —
атом галогена. Примерно таким же расстоянием (ок.
15 А) разделены атомы азота в других курарепо-
добных веществах, например в парамионе.
(CH3)3NC9H4CH(C2H5)CH(C2H5)C„H4N(CH3)3- 2J-, ди-
тилине и др. Эти вещества применяют в основном
в хирургии для расслабления скелетных мышц при
операциях.
Ганглиоблокирующие вещества за-
щищают внутренние органы и кровеносные сосуды
от нервных влияний, вызывая расслабление спазмов
гладких мышц внутренних органов. Такими свойст-
вами обладают четвертичные аммониевые основания,
напр. тетамон (C2H5)4NJ\ Более активны бис-
аммониевые основа-
ния, у к-рых расстоя-
ние между четвертич-
ными атомами азота
равно 7,5—9 А (гексо-
ний, пантамин). Ганг-
лиоблокирующими
свойствами обладают
также нек-рые третичные и вторичные амины, напр.
цемпидин (XV) и мекамиламин (XVI), преимущест-
вом к-рых является их лучшая всасываемость из
кишечника. Ганглиоблокирующие вещества исполь-
зуют при лечении гипертопич. болезни.
Спазмолитические вещества рас-
слабляют спазмы гладких мышц (мускулатуру кро-
веносных сосудов, бронхов и органов брюшной по-
лости). Наиболее активными веществами этого класса
являются диалкиламиноалкиловые эфиры дифенил-
уксусной и бензиловой кислот или соответствующих
тиокислот (см. Апрофен, Дипрофен, Тифен, Спазмо-
литин).
К мышечным спазмолитич. веществам относят такие,
к-рые, подобно папаверину, оказывают прямое дейст-
вие на гладкие мышцы и ослабляют их сократитель-
ные свойства. Подобное действие оказывают гете-
роциклич. соединения, содержащие бензильный за-
меститель; простейшим йз них является бензил-
бензимидазол (см. Дибазол). Большое прак-
тическое значение имеют спазмолитические веще-
ства, оказывающие избирательное сосудорасширяю-
щее действие на коронарные сосуды сердца, напри-
мер нитроглицерин, нитропентон (тетранит-
рат пентаэритрола) C(CH2ONO2)4 и нитранол
N(CH2CH2ONO2)3.
Противогистаминные средства
применяются для лечения аллергич. заболеваний
(крапивница, сенной насморк, сывороточная болезнь
и др.), в возникновении к-рых, вероятно, участвует
освобождающийся из неактивных тканевых комплек-
сов гистамин. Большинство противогистаминных
I I
средств имеет общее строение А—X—С—С—N<(
л 1 1
где А — полициклич. структура или система из одной
или двух ароматич. или гетероциклич. групп; X —
азот, кислород или углерод; концевой атом азота
содержит 2 метильных заместителя или включен в
гетероциклич. систему. В качестве противогистамин-
ных средств применяют димедрол, нек-рые производ-
ные фенотиазина (дипразин, ятизин) и производные
тетрагидрокарболина {диазолин). Большинство анти-
гистаминных средств обладают седативным (успокаи-
вающим) действием, а нек-рые из них (димедрол,
дипразин) являются эффективными средствами при
морской и воздушной болезнях.
Гормональные препараты — синтетич.
продукты, близкие по физиология, действйю к при-
родным гормонам. Из большого числа синтетич. гор-
мональных препаратов значительный интерес пред-
ставляют: синэстрол, диэтилстилъбэстрол и др.,
проявляющие активность, свойственную фоликуляр-
ному гормону; соединения, к-рые при приеме внутрь
понижают содержание сахара в крови, напр. бута-
мид n-CH3C„H4SO2NHCONHC4H9; полипептиды — ва-
зопрессин, окситоцин и др. Для лечения гиперфунк-
ции щитовидной железы пользуются т. н. антитире-
оидными средствами, как правило, имеющими в своей
структуре рядом расположенные атомы С, Sh N;
их типичными представителями являются метилтио-
урацил и мерказолил.
Большое практич. значение имеют получаемые син-
тетич. путем гормональные препараты коры надпо-
чечников. Из них дезоксикортикостерон, влияющий
на минеральный обмен (минералокортико-
и д), применяют при лечении Адисоновой болезни.
Еще более активный минералокортикоид — альдосте-
рон, вследствие трудности синтеза пока еще недо-
ступен, как лечебный препарат. За последнее время
широкое лечебное применение получили гормональ-
ные препараты коры надпочечников, влияющие на
обмен углеводов, белков и жиров (глюкокортикоиды),
к-рые повышают содержание сахара в крови и обога-
щают печень гликогеном за счет неуглеводных про-
947
ЛЕКАРСТВЕННЫЕ ВЕЩЕСТВА
948
дуктов (гликонеогенез). Важной особенно-
стью глюкокортикоидов, напр. кортизона, является их
способность ослаблять воспалительные и аллергия, ре-
акции организма. Поэтому соединения с глюкокорти-
коидной активностью с успехом применяются при лече-
нии ревматизма, бронхиальной астмы, нек-рых кожных
заболеваний и т. д. Глюкокортикоиды проявляют
также более или менее выраженные минералокорти-
коидные свойства, с чем связаны нек-рые осложнения
при их лечебном применении. Синтетически получают
как природный глюкокортикоид коры надпочечни-
ков— г идрокортизон (XVII), так и его про-
изводные. Многие синтетич. вещества с глюкокортико-
идной активностью более активны и менее токсичны,
чем природные глюкокортикоиды коры надпочечни-
ков. Так, преднизолон (XVIII) в 4—5 раз,
а дексаметазон (XIX) в 30 раз более активен,
чем гидрокортизон; при этом дексаметазон почти
лишен минералокортикоидной активности.
Химиотерапевтическими средст-
вами обычно называют вещества, действующие
губительно на возбудителей болезней, проникших в
организм. Первыми синтетич. химиотерапевтич. сред-
ствами были органич. соединения мышьяка. Позднее
были синтезированы противомалярийные средства
(акрихин, плазмоцид), в структуре к-рых имеется
сходство с хинином. В годы второй мировой войны
были созданы противомалярийные средства другого
типа (см. Бигумалъ, Хлоридин). В середине 30-х гг.
20 в. было открыто химиотерапевтич. действие суль-
фаниламидов, чем было положено начало химиотера-
пии бактериальных инфекций. Эти соединения яв;
ляются производными стрептоцида (XX). Противо-
г микробное действие оказывают
/==\ только такие соединения, в струк-
H2N-/ y-SOaNH2 туре к-рых оба заместителя в аро-
'—\ матич. ядре находятся в пара-по-
хх' ложении друг к другу. Амино-
группа должна быть обязательно
свободной или легко освобождаемой. Соединения с за-
мещенной аминогруппой, напр. фталазол, становятся
активными в организме после отщепления от азота ос-
татка фталевой к-ты. Наиболее активными оказались
соединения, в к-рых в амидную группу введен гетеро-
цнклич. заместитель. Наиболее ценными являются
пиримидинзамещенные сульфаниламиды (см. Сульфа-
зин, Сульфадимезин, Сульфаниламидные препараты).
Сульфаниламидные препараты останавливают рост
чувствительных к ним микроорганизмов в силу струк-
турного сходства этих веществ с парааминобензойной
к-той, к-рая необходима микробам для синтеза фоли-
евой к-ты, входящей в ферментную систему биосинтеза
нуклеиновых к-т.
Синтетич. химиотерапевтич. средства имеют боль-
шое значение в лечении туберкулеза. Наиболее актив-
Фтивазид).
CONHNH2
N
ххр
ными синтетич. противотуберкулезными средствами
являются т у б а з и д, или изониазид (XXI), и его
производные (см- Метазид, Салюзид,
Широко применяется также для химио-
терапии туберкулеза Na-соль параами-
носалициловой к-ты (ПАСК).
Лечение злокачественных новообразо-
ваний и близких к ним заболеваний кро-
ветворных органов при помощи средств,
тормозящих опухолевый рост, также
называют химиотерапией. Идеальные средства
для химиотерапии рака должны избирательно по-
вреждать опухолевые клетки и не оказывать вредного
влияния на клетки нормальных тканей; подобные
вещества до сих пор не найдены. В качестве противо-
опухолевых средств иногда применяют нек-рые анти-
метаболиты, препятствующие биосинтезу нуклеино-
вых к-т, необходимых для клеточного размножения,
напр. аминоптерин, являющийся антиметаболитом
фолиевой к-ты, и меркаптопурин (б-тиопурин) —
антиметаболит аденина и гипоксантина. Особое вни-
мание как противоопухолевые средства привлекли
к себе т. н. алкилирующие агенты, эффект к-рых схо-
ден с эффектом рентгеновых лучей. Эти агенты, бла-
годаря их электрофильным свойствам, алкилируют
нуклеофильные центры клеток. К алкилирующим ве-
>СН2СН2С1
ществам относятся соединения типа В—N/
• ХСН2СН2С1
например э м б и х и н (R=CH3), н о в э м б и-
х и н (В=СН2СНС1СН2), д о п а н (R—остаток 4-ме-
тилурацила), сарколизин (R — остаток фенил-
аланина). К алкилирующим веществам относят так-
же соединения, содержащие этилениминные группы,
например тиофосфамид (Т и о Т Э Ф — XXII). Зна-
чительным успехом в изыскании противоопухолевых
средств явилось обнаружение противоопухолевого
действия циклофосфана (эндоксан — XXIII), от-
личающегося меньшей токсичностью и лучшей пере-
носимостью по сравнению с другими противоопухо-
левыми средствами.
С
H2CXn_»n<9H2
Hac/N I хсна
zNx
XXII сн^сн2'
nh-ch2
ClCHaCHa^N_p^ ЧСН2
ClCH2CHa Xq—CHZ
XXIII
течение длительного времени но-
Создание Л. в. в
сило эмпирич. характер и основывалось гл. обр. на
варьировании строения либо природного вещества,
обладающего лечебным действием, либо' синтетич.
вещества, лечебный эффект к-рого был случайно об-
наружен. Успехи физиологии и биологич. химии в
расшифровке механизмов и химич. основ жизненных
функций и их нарушений при заболеваниях, а также
достижения в изучении биохимии микробов и вирусов,
открыли для фармакологии новые возможности в
поисках Л. в. Огромное значение приобрело изучение
закономерностей, связывающих химич. строение Л. в.
с их биологич. действием. Руководствуясь этими зако-
номерностями, удается в пределах данного ряда соеди-
нений вести направленный синтез. Исследование био-
химического механизма действия Л. в., установле-
ние природы их взаимодействия с биологическими
структурами позволит в еще большей степени при-
близиться к разработке рациональных основ синтеза
Л. в. с заданными свойствами. См. также ст. Алкало-
иды, Антибиотики, Витамины, Гликозиды сердечные,
Гормоны.
Лит.: Государственная фармакопея СССР, 9 изд., М.,
1961; Барлоу Р., Введение в химическую фармакологию,
пер. с англ., М., 1959; Машковский М. Д., Лекарствен-
949
ЛЕКЛАНШЕ ЭЛЕМЕНТ — ЛЕСОХИМИЧЕСКИЕ ПРОИЗВОДСТВА
950
ные средства, 4 изд., М., I960; Преображенский
Н. А., Генкин Э. И., Химия органических лекарственных
веществ, М.—Л., 1953; Сонов П. Л., Курс фармацевтиче-
ской химии, №., 1952; Dyson G. М., May's chemistry of
synthetic drugs, 5 ed., L., 1959; Fleet H. R., Synthetic
drugs, L., 1955; Negwer M., Organise!}—chemjsche Arz-
neimittel und Hire Synonyma, B., 1959. M. Л. Беленький.
ЛЕКЛАНШЕ ЭЛЕМЕНТ — см. Химические ис-
точники тока.
ЛЕМОНГРАССОВОЕ МАСЛО — см. Эфирные масла.
ЛЕМОРДН (дроморан, битартрат Е-З-окси-N-Me-
тилморфинаиа), С21Н2вО7 • 2НаО, молекулярный вес
443,50 '— бесцветные кристаллы умеренно
| 1 растворимы в воде, трудно в спирте, не-
. растворимы в эфире;
I 1 А СйНвОв-2НаО Т.пл.ИЗ—115°; [a]D=
хТ'''Х| снз =—13,8°. Рацемич. ос-
I Ч---- нование Л. получают
след, образом: мети-
ловый эфир n-метоксифенилглицидной кислоты, полу-
чаемый конденсацией анисового альдегида с мети-
ловым эфиром хлоруксусной к-ты, при взаимодей-
ствии с р-циклогексен-1-ил-этиламином дает 1-п-мето-
ксибензил-10-оксидекагидроизохинолин. При его об-
работке формальдегидом и муравьиной к-той обра-
зуется N-метильное производное, которое действием
НВг превращают в П,С-3-окси-Х-метилморфинан.
4 - СН3ОСвН4СНО + С|СН2СООСН3 —-
о
4-СН3ОС6Н4СН-СНСООСНз
Разделение рацемата на оптич. антиподы при помощи
винной к-ты приводит к Л. Леморан — сильное обез-
боливающее средство, в несколько раз превосходя-
щее морфин. D-Изомер Л. лишен анальгетич. свойств,
но применяется в качестве лечебного средства против
кашля.
Лит.: Преображенский Н. А., Генкин
Э. И-, Химия органических лекарственных веществ, М.—Л.,
1953; Hen ее k а Н., Liebigs Ann. Chem., 1953,583, Н. 2,
S. 110. А. И. Травин.
СН3
Л JZ
ЛЕПИДИН (у-или 4-метилхинолин) C10HeN, мол. в.
143,18; т. пл. 9—10°; т. кип. 260°; 1,0852; 1,6206,
константа диссоциации К = 2,9 - 10 ®;
практически нерастворим в воде, раст-
ворим в спирте и эфире. Л. способен
к реакциям электрофильного и нуклео-
фильного замещения: нитрования (4-
метил-8-нитрохинолин), сульфирования
(Л.-6-сульфокислота) и др. Нагревание Л. с КОН
приводит к 2-окси-4-метилхинолину. Окислением Л.
получают, в зависимости от условий, либо 4-метил-
гн соон хинолиновую кислоту (I),
с 3 ги либо цинхониновую к-ту
ЧООС /К Ц (см. Цинхонин).
L J . Метильная группа Л.
N • легко окисляется дву-
'll окисью селена до альде-
гидной. Электроноакцепторные свойства связи C=N
обусловливают активирование атомов водорода ме-
ноос
тильной группы:
нсно
(C2H5)aNH-HCl
С гадогеналкилами Л. образует галогеналкилаты,
к-рые конденсируются легче, чем сам Л., напр.:
Л. и его производные синтезируют конденсацией ани-
лина с различными карбонильными соединениями.
Л. содержится в кам.-уг. дегте. Применяют Л. для
синтеза цианиновых красителей и нек-рых фармацев-
тич. препаратов.
Лит.: Эльдерфилд Р., в кн.: Гетероциклические
соединения, подред. Р. Эльдерфилда, пер. с англ., т. 4, М., 1955.
Н. В. Гнучев.
ЛЕСОХИМИЧЕСКИЕ ПРОИЗВОДСТВА — хими-
ческие произ-ва, исходным сырьем для к-рых является
древесина. К числу важнейших Л. п. относятся:
процз-во целлюлозы и бумаги, гидролиз древесины (см.
Гидролиз растительных материалов), произ-во кани-
фоли И скипидара, дубящих веществ, термич. разло-
жение древесины (сухая перегонка, газификация),
энергохимич. ее переработка, произ-во древесных пла-
стиков.
По отраолевому делению к Л. п. относят лишь канифольно-
скипидарные произ-ва и отрасли, основанные на термич. раз-
ложении древесины.
Сырьем для Л. п. служат: 1) специально заготовля-
емые сортименты древесины (напр., балансы для цел-
люлозного произ-ва, технологич. дрова для сухой
перегонки); 2) неликвидная дровяная древесина лист-
венных пород, лесосечные отходы (вершины, сучья,
пни) и отходы лесопиления и деревообработки (рейки,
горбыли, опилки, стружки).
951
ЛЕСОХИМИЧЕСКИЕ ПРОИЗВОДСТВА
952
При механизированных лесозаготовках и первичной
механич. обработке древесины на складах леспром-
хозов общее количество древесных отходов достигает
25% от объема товарной древесины, причем около
половины отходов приходится на сучья. При лесопи-
лении и деревообработке общее количество отходов
достигает 45% от объема переработанного круглого
леса. Пока эти отходы используются в небольшом
объеме, но в ближайшие годы их доля достигнет 60—
65% сырьевого баланса Л. п. Наибольшее количество
отходов предполагается использовать в произ-ве
целлюлозы, бумаги и картона. Далее (по масштабу
потребления отходов) следуют: гидролиз древесины,
произ-во древесных плит, энергохимич. переработка
и сухая перегонка древесины. По предварительным
подсчетам потребление сырья в лесохимия, произ-вах
всех видов в СССР в 1970 значительно возрастет
гл. обр. за счет древесных отходов. Выработка
основных продуктов химич. переработки древесины
и другого растительного сырья в тыс. тонн (этиловый
спирт в млн. декалитров) показана в таблице.
Продукты 1958 г. 1960 г. 1965 г. (план)
Целлюлоза 2093 2282 5104
Бумага 2236 2421 3903
Картон 720 806 2676
Кормовые дрожжи 9 17 263
Этиловый спирта 21 24 31
фУРфУрол Канифоль и канифольные про- 3 4 23
дукты (включая талловое масло и клей-пасту) 130 139 219
Древесно-волокнистые плиты б 35 68 220
Древесно-стружечные плиты в Уголь древесный (по крупным 34 157 1902
заводам) 117 121 142
Уксусная кислота 21 23 32
Этилацетат и бутилацетат . . . 36 44 101
а Существенный рост произ-ва не планируется в связи с
развитием произ-ва синтетич. спирта. ® В млн. м‘. в В тыс. .н3.
Целлюлозно-бумажное производ-
ство занимает главное место среди Л. п. как по
объему перерабатываемого сырья, так и готовой про-
дукции. Основными полуфабрикатами для произ-ва
бумаги и картона являются целлюлоза (сульфитная,
сульфатная, моносульфитная) и древесная масса.
В последнее время усиленно развивается произ-во
таких полуфабрикатов, как полуцеллюлоза и хими-
ческая древесная масса; при этом стремятся в качестве
полуфабриката получить продукт, близкий к х о л о-
целлюлозе (полиозный комплекс, состоящий из
22—25% пентозанов, 70—75% целлюлозы и др. гек-
созанов), т. е. использовать по возможности весь
комплекс содержащихся в древесине полимерных
углеводов. Это позволяет значительно повысить выход
полуфабрикатов, сократить уд. расход древесины
в целлюлозно-бумажном произ-ве и снизить себесто-
имость бумажной продукции. Кроме древесины,
в качестве сырья в целлюлозно-бумажном произ-ве
применяют также солому, тростник и др. Важное
значение приобретает также произ-во специальных
видов целлюлозы для химич. переработки на искус-
ственное волокно (вискозной и кордной целлюлозы).
Из щелоков — отходов от варки целлюлозы — полу-
чают этиловый спирт, кормовые дрожжи, канифоль-
ные продукты, крепители литейные, активный лиг-
нин для пластич. масс и др. продукты.
Гидролиз древесины и др. раститель-
ных материалов лежит в основе гидролизно-спир-
тового, гидролизно-дрожжевого, гидролизно-фурфу-
рольного и др. произ-в. В качестве сырья в гидролиз-
ном произ-ве применяют опилки и смесь измельчен-
ной в щепу древесины и отходов лесопиления (рейки,
горбыли), а также нек-рые растительные отходы (ку-
курузная кочерыжка, подсолнечная лузга и др.).
Путем химич. переработки водорастворимых сахаров,
образующихся при гидролизе растительных материа-
лов, можно получить: глюкозу, техническую ксилозу,
многоатомные спирты (сорбит, ксилит, глицерин,
этиленгликоль, пропиленгликоль), фурфурол, леву-
линовую, триоксиглутаровую и глюконовую кислоты.
Биохимич. переработкой сахаров можно получить
этиловый и бутиловый спирты, белково-витаминные
кормовые дрожжи, антибиотики и другие продукты.
Основные направления дальнейшего развития гидро-
лизной промышленности—получение кормовых белков
из отходов древесины методом гидролиза, а также
произ-во фурфурола. При гидролизе древесины полу-
чается негидролизуемый остаток, представляющий
собой лигнин, к-рый частично используется в основном
как топливо; намечается также использование лиг-
нина для произ-ва активного угля и лигноволокни-
стых строительных плит.
Сырьем для канифольно-скипидарно-
го производства служат: живица, получаемая
при подсочке сосны и других хвойных пород древесины;
осмол — просмолившаяся после рубки леса ядровая
древесина сосновых пней; мыло сульфатное — побоч-
ный продукт сульфатцеллюлозного произ-ва. В СССР
вырабатывается в основном живичная канифоль,
в дальнейшем будет возрастать уд. вес более дешевых
экстракционной и талловой канифоли. Сопутствую-
щими канифоли продуктами являются скипидар и
масло сосновое флотационное. Основное направление
развития канифольно-скипидарного произ-ва — со-
здание широкого ассортимента модифицированных и
синтетических продуктов на основе канифоли, и ски-
пидара.
Дубильно-экстрактовое производ-
ство — источник получения дубящих веществ ра-
стительного происхождения (основной группы дуби-
телей для кожевенного произ-ва). В СССР в дубильно-
экстрактовом произ-ве для выработки дубителей при-
меняют кору ивы, дуба, ели, лиственницы, древесину
дуба, каштана и др. Подробнее см. Дубящие вещества.
Сухая перегонка, или пиролиз древе-
сины, — одна из старейших отраслей Л. п. Основные
продукты, получаемые при этом, — древесный уголь
и уксусная к-та. В качестве сырья применяют дрова
лиственных пород, преим. березы, не поврежденные
(или мало поврежденные) гнилью. Новые возможности
открываются перед сухой перегонкой древесины при
использовании катализаторов, в результате чего,
кроме угля, уксусной к-ты и других продуктов этого
процесса, может быть получен в значительном ко-
личестве фурфурол.
Энергохимическая переработка
древесины — газификация древесины в газо-
генераторах или в топках-генераторах с выделением
лесохимия, продуктов в процессе очистки газа. Очи-
щенный газ используется для сжигания в топках
котельных или в двигателях внутреннего сгорания.
Энергохимич. переработка древесины в газогенера-
торах позволяет использовать отходы любых пород
и любой формы, вплоть до несортированных лесо-
сечных отходов. Энергохимич. установки могут стро-
иться на нижних складах лесозаготовительных пред-
приятий, при котельных лесопильно-деревообрабаты-
вающих предприятий и на лесохимических заводах
(где газ нужен для технологических целей). Основным
продуктом энергохимической переработки древе-
сины являются древесные смолы. Древесина как источ-
ник фенолосодержащих смол занимает особое место,
являясь неограниченным и постоянно возобновля-
емым сырьем; выход фенолов до 2% от веса древесины,
953
ЛЕСОХИМИЯ — ЛЕЦИТИНЫ
954
т. е. в 20 раз больше, чем выход фенолов от веса угля
в коксохимия, произ-ве. Древесные смолы примерно
наполовину состоят из фенолов (преим. 2- и 3-атом-
ных), в значительной мере в виде неполных метило-
вых эфиров; сюда же включаются фенолокислоты и
естественно образовавшиеся новолачные феноло-аль-
дегидные смолы; на основе фенолов можно синтези-
ровать дубители, понизители вязкости глинистых
р-ров, используемых при бурении нефтяных и газо-
вых скважин, и связующие для древесных плит.
Древесные пластики — материалы, в
к-рые древесина входит как основной компонент.
Особенно широкое распространение приобретают дре-
весные плиты — древесноволокнистые и древесно-
стружечные, применение к-рых (особенно изготов-
ленных с применением синтетич. связующих) в ме-
бельной пром-сти и в строительстве обеспечивает
большую экономию деловой древесины и повышение
производительности труда в этих отраслях народного
хозяйства. Подробнее см. ст. Древесные пластики
и Древесные плиты.
Существуют еще нек-рые направления химич.
переработки древесины — произ-во на основе дре-
весных опилок азотистых удобрений, лимонной кис-
лоты и др.
Лит.: Кейси Д. П., Целлюлоза и бумага. Химия и
химическая технология, пер. с англ., т. 1, кн. 2, М., 1958;
Шарков В. И., Гидролизное производство, ч. 1—3, М.,
1945—50; Козлов В. Н., НиМвицкий А. А., Техно-
логия пирогенетической переработки древесины, М.—Л.,
1954; Васечкин В. С., Технология экстрактивных веществ
дерева, М,—Л., 1953: Гордон Л. В. [и др.], Технология
лесохимических производств, 2 изд., М,—Л., 1960.
Л. Б. Гордон.
ЛЕСОХИМИЯ — область знаний, включающая на-
уку о химии древесины, а также произ-вах, в к-рых
исходным сырьем для получения разнообразных хи-
мич. продуктов служит древесина — один из важней-
ших и непрерывно возобновляющихся источников при-
родного сырья. Химич, произ-ва, основанные на дре-
весном сырье, являются самостоятельными отраслями
пром-сти. К их числу относятся: целлюлозно-бумаж-
ная, гидролизная, канифольно-скипидарная, энерго-
химич. переработка древесины и др. Подробнее см.
Лесохимические производства.
ЛЕТТСА СИНТЕЗ — способ получения нитрилов
взаимодействием соответствующих карбоновых к-т
или солей с роданидами металлов:
R—COOH + KSCN - R-CHN4-CO2 + KSH
(RCOO)2Pb4-Pb(SCN)2 -► 2R-C=N + 2PbS +2СО2
Л. с. с применением щелочных роданидов дает удов-
летворительные выходы (до 80% от теоретического)
лишь в случае ароматич. к-т. Алифатич. к-ты с рода-
нидом калия образуют, как правило, смесь нитрила
и соответствующего амида. Применение тиоцианатов
тяжелых металлов, напр. Pb(SCN)2, позволяет рас-
пространить Л. с. и на алифатич. ряд. Наилучшие
результаты были достигнуты при взаимодействии
Zn-солей карбоновых кислот с 20%-ным избытком
Pb(SCN)2 (бензонитрил из Zn-бензоата — 91%-ный
выход; пропионитрил из Zn-пропионата — 71%-ный
выход).
При проведении Л. с. сухие компоненты нагревают,
а образующийся нитрил отгоняют. Метод применим
для синтеза ароматич. динитрилов и нитрилов «^-не-
насыщенных к-т. В случае окси-, амино- и нитрокар-
боновых к-т применение Л. с. к успеху не привело.
Метод открыт Е. Леттсом в 1872.
Лит.: Letts Е., Вег., 1872, 5, 669; Epps. G. van,
Reid Е. Е., J. Amer. Chem. Soc., 1916, 38, 2120; Mowry
D. T., Chem. Revs, 1948, 42, № 2, 264. Л. С. Герман.
ЛЕТУЧЕСТЬ — см. Фугитивность.
ЛЕЦИТИНАЗЫ (фосфолипазы) — ферменты, ката-
лизирующие гидролитическое расщепление эфирных
связей в лецитинах, коламин- и серинфосфатидах.
В зависимости от того, на какую из четырех эфирных
связей, имеющихся в молекуле указанных фосфати-
дов, направлено действие фермента, различают Л.:
А, В, С и D.
лецитиназа А ~
лецигиназаВ « CH2-O~CO~R
R-CO-6-^CH О п
I и г ОН
аСН2-О-P-O-CH2CH2N(CH3)3
лецитиназа С | ОН | леимтинэзэ р
Лецитиназа А (фосфолипаза А) — гидроли-
зует эфирную связь в «'-положении молекулы леци-
тина или коламинфосфатида, в результате чего от-
щепляется 1 молекула жирной к-ты и образуется
а-л изо лецитин или а-л изокефалин. По-
следние обладают сильным гемолитич. действием
(вызывают распад эритроцитов). Лецитиназа А со-
держится в соке поджелудочной железы животных
и человека. Очень активная лецитиназа А содержится
в ядах нек-рых насекомых и змей (скорпионов, пчел,
ос, кобр), с чем связано их токсич. действие. Лизоле-
цитины и лизокефалины, образующиеся в желудочном
тракте при действии лецитиназы А, сразу же инак-
тивируются в результате действия на них лецити-
назы В.
Лецитиназа В (лизолоцитиназа В, фосфо-
липаза В, лизофосфолипаза) — катализирует гидро-
литическое расщепление эфирной связи в Р-положе-
нии молекулы а-лизолецитина или а-лизокефалина
е образованием молекулы жирной кислоты и гли-
церилфосфорилхолина или глицерилфосфорилэтанола-
мина. Лецитиназа В содержится в соке поджелу-
дочной железы, а также в тканях животных и рас-
тений.
Лецитиназа С (фосфолипаза С, а-токсин) —
катализирует гидролитич. отщепление фосфорилхо-
лина от молекулы лецитина с образованием а, |3-ди-
глицерида. Лецитиназа С входит в состав бактериаль-
ных токсинов. Продукты, образующиеся из лецити-
на при гидролизе его этой Л., сами по себе неток-
сичны. Токсич. действие фермента при инфекции ран
соответствующими микроорганизмами, по-видимому,
связано с катализируемым им быстрым разрушением
лецитинов, входящих в состав клеточных оболочек.
Лецитиназа С активируется ионами Са2+ и в меньшей
степени ионами Mg2+. Ионы Си, Sr, Fe и Ва сильно
тормозят этот фермент. Оптимум действия лецитина-
зы С лежит при pH 6,7—7,0. В небольших количест-
вах лецитиназа С обнаружена в связанном состоя-
нии в тканях животных, в частности в тканях
мозга.
Лецитиназа D (фосфолипаза) — гидроли-
тически расщепляет эфирную связь между азоти-
стым основанием и фосфорной к-той в молекуле ле-
цитина, серин- и коламинфосфатида с образованием
свободного основания и фосфатидной к-ты. Лецитиназа
С содержится в кишечном соке, а также во многих
растениях. Физиологич. роль лецитиназ А, В и D,
присутствующих в секретах желудочно-кишечного
тракта, состоит в расщеплении фосфолипидов пищи,
что обеспечивает возможность их всасывания и
усвоения организмом.
Лит.: Диксон М. и Уэбб Э., Ферменты, пер. с
англ., М., 1961; Progressin the chemistry of tats and other lipids,
V. 4, L.—N. Y.—P., 1957, p. 142. В. В. Спиричев.
ЛЕЦИТИНЫ (холинфосфатиды, фосфатидилхоли-
ны) — сложные эфиры аминоспирта холина и дигли-
церидфосфорных (фосфатидных) к-т; являются важ-
нейшими представителями фосфолипидов. В зависи-
955
ЛЕЦИТИНЫ —ЛЕ ШАТЕЛЬЕ ПРИНЦИП
956
мости от расположения остатка фосфохолина у а-
или в-углеродного атома глицерина различают а-
или р-лецитины:
N(CH3)3
снг
r'oc-o-ch2 снг
ROC-O-CH О
I I
н„с-о-р~он
£ II
о
он
а — децитиа
N(CH3)3
СН2
СН2
R’OC-O~CH2 о
нс-о-р-он
roc-o-ch2 о
р - лецитин
RCO и R СО-остатки мирны» кислот
Все природные Л. являются L-a-лецитинами. Из-
вестно большое число Л., различающихся природой
входящих в их состав остатков жирных к-т. Наиболее
часто в построении молекулы Л. участвуют пальмитино-
вая и стеариновая, а из ненасыщенных — линолевая и
олеиновая к-ты. Преобладающее число природных Л.
содержит в молекуле один остаток насыщенной и один
остаток ненасыщенной жирной к-ты, причем, как
правило, насыщенная к-та располагается у а-углерод-
ного атома, а ненасыщенная у Р-углеродного атома
остатка глицерина. Мол. вес Л. колеблется в зависи-
мости от входящих в их состав жирных к-т от 750 (ди-
пальмитоиллецитин) до 870 (арахидоноил-клупано-
доноиллецитин).
Л. широко распространены во всех животных и
растительных тканях, а также в микроорганизмах.
Особенно велико их содержание в органах животных
с высокой интенсивностью обмена, в частности в
печени и сердечной мышце. Значительные количества
Л. содержатся в яичном желтке и эритроцитах. Из
растительных тканей наиболее богаты Л. бобы сои.
Вопрос о физиологич. роли Л. еще не решен. Наряду
с другими фосфолипидами Л., по-видимому, прини-
мают участие в обмене жирных к-т. Кроме того, Л.,
совместно с прочими фосфолипидами, входят в состав
белково-липидных комплексов — липопротеидов, об-
разующих клеточные мембраны и регулирующих
перенос через эти мембраны ионов и молекул различ-
ных веществ.
Л., выделенные из природных источников, представ-
ляют собой белые или светло-желтые, воскообразные
продукты, хорошо растворимые в метиловом и эти-
ловом спиртах, хлороформе, петролейном эфире, се-
роуглероде, четыреххлористом углероде, растворимые
в эфире и 90%-ном водном ацетоне и нерастворимые
в ацетоне и метилацетате. Л. очень гигроскопичны,
с водой образуют коллоидные р-ры, коагулирующие
при добавлении ионов 2-валентных металлов. Точка
плавления большинства природных Л. лежит в пре-
делах 230—250° и обычно сильно растянута, поскольку
указанные вещества являются смесями индивидуаль-
ных Л. с различным жирнокислотным составом. При-
родные Л. оптически активны: [a]D для а-дипальми-
толеиллецитина, выделенного из дрожжей, составляет
+ 6,6° (8,2%-ный р-р в смеси СНС13 — СН3ОН 1:1).
Л. гидролизуются кислотами и щелочами. При щелоч-
ном гидролизе Л. быстро отщепляются жирные к-ты,
отщепление же холина происходит значительно мед-
леннее. Гидролиз Л. сопровождается частичной ми-
грацией остатка фосфорной к-ты из а- в [1-положение
в результате промежуточного образования циклич.
а, Р-диэфира глицерина и фосфохолина. Л., содержа-
щие ненасыщенные жирные к-ты, способны присоеди-
нять по двойной связи галогены и водород.
Ряд Л. получен синтетически, они представляют
собой белые мелкокристаллич. продукты.
Расщепление Л. в организме, а также в раститель-
ных тканях происходит под действием ферментов
лецитиназ (фосфолипаз), последовательно отщепляю-
щих от молекулы Л. остатки жирных к-т, холин или
фосфохолин. Образующиеся при действии лецитиназы
А лизолицетины имеют след, строение:
1 <^Н2ОН
RCO-O-CH ОН
H2C-O-P-OCH2CH2N(CH3)3
о
ОН"
Эти соединения весьма ядовиты в связи с их гемоли-
тич. (разрушающим эритроциты) действием. Био-
синтез Л. из соответствующих диглицеридов и хо-
лина протекает при участии особого кофермента —
цитидинтрифосфорной к-ты (ЦТФ). Первой стадией
в процессе биосинтеза Л. является фосфорилирование
холина, осуществляемое соответствующей киназой
путем перенесения на него фосфатного остатка аде-
нозинтрифосфорной к-ты (АТФ):
[НОСН2СН2Й(СН3)3]ОН4-АТФ->
холин
"НО
-> O=P-OCHaCH2N(CH3)3
_нох
фосфохолин
он-
Образующийся фосфохолин реагирует далее с ЦТФ
с образованием цитидинфосфохолина (ЦДФ-холина)
фосфохолин + ЦТФ -> ЦДФ-холинпирофосфат.
ЦДФ-холин осуществляет перенос холина на дигли-
церид, в результате чего синтезируется молекула Л.:
диглицерид-(-ЦДФ-холин -* лецитин -|- цитидинмонофосфат.
Л. могут образовываться также в результате фер-
ментативного метилирования коламинфосфатидов или
ацилирования соответствующих лизолицетинов.
Л. широко применяют в лечебных целях (при мало-
кровии, ряде заболеваний нервной системы), в пи-
щевой пром-сти (при произ-ве маргарина), а также
в текстильной, кожевенной, резиновой и косметич.
пром-сти (в качестве эмульгатора и смягчающего сред-
ства). Используемый для промышленных нужд т. наз.
«коммерческий» Л., получаемый экстракцией соевых
бобов органич. растворителями, представляет собой
смесь различных липидов след, состава: 21% леци-
тинов (холинфосфатидов), 8% коламинфосфатидов,
20% серинфосфатидов, 11% инозитфосфатидов, 2%
стеринов, токоферола и др., 33% соевого масла, 5%
свободных углеводов.
Лит. см. при ст. Коламинфосфатиды. В. В. Спиричев.
ЛЕ ШАТЕЛЬЕ ПРИНЦИП (принцип подвижного
равновесия) — правило, характеризующее влияние
изменения условий существования термодинамич.
системы на положение равновесия. Этот принцип
в частной форме высказал в 1884 Я. Вант-Гофф, в об-
щем виде сформулировал в том же году А. Ле Шателье
и теоретически обосновал в 1887 К. Браун, к-рый
показал, что Ле Ш. п. есть следствие второго закона
термодинамики. Ле Ш. п. может быть формулирован
след, образом: если на систему, находящуюся в устой-
чивом равновесии, возаействовать извне, изменяя
957
ЛИБЕРМАНА РЕАКЦИЯ —ЛИГНИН
958
какое-нибудь из условий, определяющих положение
равновесия, то равновесие смещается в том направ-
лении, при к-ром эффект произведенного воздей-
ствия уменьшается.
Важнейшими изменениями такого рода при химич. и фазо-
вых превращениях являются изменения темп-ры и давления,
а также концентрации веществ, участвующих в реакции. Теп-
ловые эффекты прямого и обратного направлений процесса
всегда противоположны по знаку. Повышение темп-ры вызы-
вает смещение равновесия в направлении того из процессов,
течение к-рого сопровождается поглощением теплоты, а пони-
жение темп-ры действует в противоположном направлении.
Напр., в равновесной системе из воды и ее насыщенного пара
повышение темп-ры усиливает испарение и повышает давление
насыщенного пара. Подобно этому, повышение давления сме-
щает равновесие в направлении процесса, сопровождающегося
уменьшением объема, а понижение давления действует в про-
тивоположном направлении. Напр., в равновесной системе
из воды и льда повышение давления смещает равновесие в сто-
рону образования воды и понижает темп-ру плавления льда,
т. к. плавление льда сопровождается уменьшением объема.
Введение в систему, находящуюся в устойчивом химич. равно-
весии, дополнительных количеств к.-л. из веществ, участвую-
щих в реакции, смещает равновесие в том направлении, при
к-ром концентрация этого вещества уменьшается.
Все это объясняется тем, что устойчивое равновесие всегда
отвечает равенству скоростей прямого и обратного процессов;
смещение равновесия происходит тогда, когда произведенное
воздействие неодинаково изменяет скорости прямого и обрат-
ного процессов. Так, повышение темп-ры, вызывая увеличение
скоростей и прямого и обратного процессов, в большей степени
увеличивает скорость того из них, к-рый происходит с погло-
щением теплоты.
Количественным выражением Ле Ш. п. для химич.
равновесий служат действующих масс закон, изобары
реакции уравнение, изохоры реакции уравнение, а
для фазовых превращений —Клапейрона — Клаузиуса
уравнение.
Лит.: Киреев В. А., Курс физической химии, 2 изд.,
М., 1956; Карапетьянц М. X., Химическая термоди-
намика, 2 изд., М.—Л., 1953. В. А. Киреев.
ЛИБЕРМАНА РЕАКЦИЯ •— реакция, применяе-
мая для открытия нитрозогруппы в алифатич. и аро-
матич. С-нитрозосоединениях и ароматич. N-нитроз-
аминах; основана на взаимодействии нитрозосоеди-
нений с фенолом и конц. серной к-той при нагревании.
Образующиеся продукты конденсации типа индофе-
нолов растворяются в присутствии избытка едкой
щелочи с образованием р-ров, интенсивно окрашенных
в фиолетовый, синий или зеленый цвет. Пример Л. р.:
Ill
I—нитрозофенол; II—таутомерная его форма—хинонмо-
нооксим; III—индофенол.
Несколько сантиграммов нитрозосоединения смешивают
в пробирке с небольшим количеством фенола и несколькими
миллилитрами конц. H2SO4, смесь нагревают, затем охлаждают,
выливают в воду, прибавляют конц. р-р NaOH до сильнощелоч-
ной реакции и наблюдают появление окраски.
Л. р. применяют также для открытия одноатомных
и многоатомных фенолов с незамещенным пара-
положением в ароматич. ядре; сначала фенол нитро-
зируют и полученное нитрозосоединение открывают,
как описано выше.
0,1—0,2 е кристаллич. NaNO2 растворяют в 1—2 мл конц.
H2SO4, прибавляют 0,1—0,2 г вещества, содержащего фенол,
и наблюдают появляющуюся синюю или зеленую окраску
сначала на холоду, затем при кратковременном нагревании
до темп-ры несколько ниже 100°; потом выливают смесь
в 20 мл воды (переход окраски в красную) и прибавляют избы-
ток NaOH (переход окраски снова в синюю или зеленую).
Лит.: Мейер Г., Анализ и определение строения
органических веществ, пер. с нем., Харьков—Киев, 1935;
Вайбель С., Идентификация органических соединений,
пер. с англ., М., 1957. А. А. Черкасский.
ЛИГАНДЫ (адденды) — молекулы и ионы,
связанные с центральным ионом в комплексном со-
единении. Л. наз. также координированными груп-
пами, или внутрисферными заместителями. См.
Комплексные соединения.
ЛИГНИН — природный полимер, инкрустирующее
вещество одревесневших растительных тканей, со-
держится в древесине (ок. 30%). Л. может быть вы-
делен из древесины двумя способами: растворением
ее углеводных компонентов (напр., при гидролизе
полисахаридов древесины) или же растворением
самого Л.
Природный Л., не изолированный из растительной
ткани, нерастворим в органич. растворителях. В вод-
ных р-рах щелочей Л. растворяется только при дли-
тельном воздействии и при нагревании. Природный
Л. приобретает способность частичной растворимости
в органич. растворителях после интенсивного размола
древесной муки на шаровой вибрационной мельнице
в индифферентной жидкости, напр. в толуоле. После
достаточно длительного размола еловой древесины
можно извлечь диоксаном при комнатной темп-ре до
50% содержащегося в ней Л. (т. наз. лигнин Бьёрк-
мана). Этот Л., по-видимому, является наименее хи-
мически измененным видом изолированного Л. В спир-
те этот «природный» Л. растворяется в очень незначи-
тельных количествах. Для выделения Л. из расти-
тельных тканей с помощью спиртов (алкоголиз) или
фенола необходимо присутствие минеральных к-т
и более или менее длительное кипячение. При этом
Л. претерпевает значительные изменения и вступает
со спиртами во взаимодействие. Аналогично полу-
чают фенол-и диоксан-Л. Природный Л. растворяется
в т. наз. гидротропных р-рах (напр., водные р-ры
Na-соли бензол- или толуолсульфокислот) при нагре-
вании и под давлением. Л. из таких р-ров может быть
выделен разбавлением их водой (гидротропный Л.).
В виде кальциевых солей лигносульфоновых к-т Л.
получается в качестве отхода при произ-ве целлюлозы
по сульфитному способу. Этот способ заключается
в варке древесины при темп-ре ок. 140° с бисульфитом
кальция или других оснований в присутствии свобод-
ного SO2. При сульфатной варке древесины Л. рас-
творяется в щелочной варочной жидкости и можетоыть
высажен из нее подкислением раствора (щелочной Л.,
сульфатный Л.).
Сложность структуры Л. и его химич. неустойчи-
вость являются причинами того, что строение Л.
далеко еще не выяснено. Строение и свойства Л. резко
отличают его от основных компонентов древесины —
целлюлозы и гемицеллюлоз. Л. — это нерегулярно
построенный полимер с разветвленными макромоле-
кулами. Структурная единица «природного» Л. Бьёрк-
мана (еловая древесина) — СвН8>8О2)4(ОСН3)0 88. Его
молекулярный вес равен примерно’ 11000 (ультра-
центрифугальный метод). Мономерами Л. являются
жирноароматические соединения — производные фе-
нилпропана. Т. обр., Л. имеет в основном ароматич.
природу. Л. не является индивидуальным химич.
соединением со строго определенными свойствами,
составом и строением. Л. различного происхожде-
ния заметно отличаются Друг от друга. Для Л.
хвойных пород характерны структурные элементы,
представляющие собой производные гваяцилпропана.
Л. лиственных наряду с этими элементами содер-
жат большое количество производных сирингилпро-
пана. В состав Л. однолетних, кроме того, входят
производные п-оксифенилпропана. Предполагают,
что Л. хвойных образован в основном из остат-
ков кониферилового спирта (I), Л. лиственных —
из кониферилового и синапового спирта (II), а Л.
однолетних, кроме того,и из п-оксикоричного спир-
та (III), к-рый в небольших количествах входит в
959
ЛИГНИН
960
состав также и первых двух видов Л.:
r=ch=ch-ch2oh
Лигнин содержит разнообразные функциональные
группы. Содержание метоксильных групп в Л. раз-
личных типов неодинаково. Л. лиственных содержит
больше метоксильных групп. Содержание гидроксиль-
ных групп в Л. разного происхождения тоже неоди-
наково и зависит также от способа выделения Л. из
растительных тканей. Результаты определения тех
или иных функциональных групп в Л. часто весьма
отличаются в зависимости от используемого метода,
что обусловлено сложностью и неустойчивостью струк-
туры Л., а также наличием в нем функциональ-
ных групп, занимающих разное положение в моле-
кулах. Лигнин Бьёркмана содержит ок. 1,15 ОН на
одну ОСН3-группу, из них примерно 0,3 — феноль-
ные. Содержание алифатических ОН-групп, следо-
вательно, составляет ок. 0,85. Часть этих групп —
первичные.
Значительные количества спиртовых групп — вто-
ричные, из них часть находится в боковой цепи у
С-атома, находящегося в a-положении к ароматич.
ядру (бензилспиртовые ОН-группы). Эти гидроксиль-
ные группы являются особенно реакционноспособ-
ными и их наличием обусловлены многие характерные
для Л. реакции. К таким реакциям относится сульфи-
рование Л. бисульфитом (сульфитная варка), где
сульфогруппа, по-видимому, гл. обр. становится в бо-
ковую цепь к С-атому, находящемуся в а-положении
к ядру, вместо соответствующих ОН-групп. Эти
группы легко, уже при 20°, реагируют с метиловым
спиртом в присутствии 0,5% НС1, образуя эфир.
Количественное определение ОН-групп различной природы
в Л. связано с большими трудностями и до сих пор для этой
цели нет вполне надежных методов. Количественное опреде-
ление фенольных ОН-групп проводят спектрофотометрия.
Де-методом, основанным на смещении УФ-спектров в щелочной
среде, методом потенциометрич. титрования в неводных раст-
ворителях, окислительным деметоксилированием с помощью
NaJ04 и др. методами. Для лигнина Бьёркмана все эти методы
дают сравнительно близкие значения.
Л. содержит значительное число ОН-групп, свя-
занных в форме простых эфиров. Эфирные мостики
соединяют в молекулах Л. отдельные мономеры друг
с другом (наряду с эфирными мостиками в Л. имеются
также С—С-связи между мономерами). В виде эфи-
ров связана большая часть фенольных ОН-групп Л.
В Л. хвойных почти полностью этерифицированы (ме-
тиловым спиртом) фенольные ОН-группы в .и-поло-
жении к боковой цепи и ок. 0,7 всех фенольных ОН-
групп, находящихся в га-положении к боковой цепи.
Предполагают, что значительная часть этих феноль-
ных гидроксильных групп связана в виде алкил-
арильных эфиров типа fi-кониферилового эфира
а-гваяцилглицерина (IV)
ОСНЭ
НО-АА-СН(ОН)-СН-СН2ОН
о_________________
И ¥-сн=сн-сн2-он
оснэ
IV
В Л., кроме того, имеют место и другие типы эфирных
связей: фенилкумароновый, связи алкиларильного
типа, образованные за счет гидроксильной группы
остатка гваяцилглицерина, находящейся в а-положе-
нии к ядру, а также а-алкилалкильные связи (типа
бензиловых эфиров). Эти эфиры легко разлагаются,
напр. при сульфировании Л. в условиях сульфитной
варки, и легко подвергаются переэтерификации..
Л. содержит карбонильные (кетонные и альдегидные)
группы. Он обладает характерными для СО-групп
полосами поглощения в ИК-спектрах, указывающими
на наличие очень небольшого количества конъюги-
рованных альдегидных групп типа кониферилового
альдегида (1 — на 35 структурных элементов) и конъю-
гированных с двойной связью ароматич. кольца ке-
тонных групп, а также неконъюгированных кетонных
групп, занимающих в боковых цепях [J-положение
по отношению к ароматич. кольцу. Число СО-групп,
определяемых в лигнине Бьёркмана по гидроксил-
аминному способу, соответствует ~ 0,2 на ОСН3-
группу. Двойных связей в лигнине Бьёркмана мало.
Их присутствие может быть отнесено за счет неболь-
ших количеств кониферилового альдегида, входяще-
го в структуру Л. Наличие большого числа актив-
ных функциональных групп различного типа делает
Л. способным к многочисленным химич. превраще-
ниям. Являясь фенолом, Л. легко нитруется. Даже
очень разб. (3—8%-ная) HNO3 способна нитровать Л.
При этом на каждые 3 элементарных звена нитролиг-
нина приходится ок. 2 NO2-rpynn. Одновременно Л.
окисляется, приобретая карбоксильные группы, число
к-рых определяется условиями нитрования. Л. легко
хлорируется. Хлор вступает как в ароматич. ядро, так
и в боковые цепи. В ароматич. ядрах (с закрытыми
фенольными группами) Л. хвойных хлор занимает
положение 6 относительно боковой цепи. Положение
хлора в боковых цепях не установлено. В присутст-
вии влаги одновременно с хлорированием происходит
окисление Л. Получаемый хлорлигнин содержит
карбоксильные группы. При сплавлении Л. со ще-
лочью образуются пирокатехин и протокатеховая
к-та. Если метилированный Л. сплавить со щелочью
и снова метилировать, а затем окислить КМпО4,
получается ряд к-т, среди к-рых преобладают вера-
тровая, изогемипиновая и дегидродивератровая. Ка-
талитич. гидрирование Л. приводит к получению
мономерных продуктов гидрогенолиза. Л. В зависи-
мости от условий и катализатора получаются различ-
ные смеси соединений ароматич. или гидроароматич.
рядов. Так, при гидрировании водородом (Л. осины) в
присутствии медно-хромового катализатора при 250 —
260° получается смесь производных пропилциклогек-
сана: 4-н-пропилциклогексанол-1, 4-м-пропилцик-
логександиол-1,2, 3-(4-оксициклогексил)-пропанол-1.
В присутствии Ni-Ренея при 225—250° получаются
также производные пропилциклогексана, производ-
ные этил- и пропилбензола, а также гидрированные
полимеры. При более низкой темп-ре в присутствии
скелетного никеля образуются фенолы. Л. легко
окисляется. При окислении Л. нитробензолом в ще-
лочной среде (при 160°) образуются ароматич. альде-
гиды. Из Л. хвойных получается ванилин (до 25%
от веса Л.). Из Л. лиственных получается смесь си-
реневого альдегида и ванилина, соотношения выходов
к-рых несколько различаются в зависимости от по-
роды древесины. В небольшом количестве получается
также га-оксибензальдегид. Из изолированных Л.
выходы ванилина значительно ниже. Ванилин из Л.
может быть получен и окислением кислородом в ще-
лочной среде в присутствии нек-рых окислов металлов
(СнО, Ag2O, HgO). Из сульфитных щелоков ванилин
с выходом до 4—6% получается также при щелочном
гидролизе (нагревание с водными р-рами щелочей).
При действии на Л. других окислителей (перекиси
водорода, перманганата, хромовой к-ты и др.) полу-
961
ЛИГНИН — ЛИГНОСУЛЬФОНОВЫЕ кислоты
962
чаются ароматич. к-ты, в том числе небольшое коли-
чество бензолполикарбоновых к-т и щавелевая к-та.
Предпринятые в последнее время попытки предста-
вить строение «природного» Л. в виде химич. формулы
(Фрейденберг, Адлер), однако, отражают лишь воз-
можные принципы строения Л. и далеко еще не обосно-
ваны существующими экспериментальными данными.
Не ясно еще, состоит ли Л. из вполне идентичных
макромолекул или, что вероятнее, составляющие
его макромолекулы более или менее значительно от-
личаются друг от друга порядком расположения от-
дельных звеньев и связей между ними.
Вопросы образования Л. в растительных клетках,
его роль в жизнедеятельности растительных орга-
низмов далеко еще не ясны. Большинство исследова-
телей склонны считать конифериловый спирт источ-
ником образования Л. в растениях. Согласно теории
Фрейденберга, находящийся в камбиальном соке
древесины глюкозид кониферин гидролизуется под
влиянием энзима — fj-глюкозидазы. Образующийся
конифериловый спирт при действии дегидрогеназ
теряет водород и далее конденсируется, образуя
полимер — Л. Конденсация происходит без участия
энзим и поэтому приводит к образованию оптически
неактивного неоднородно построенного полимера с раз-
ветвленной формой молекул. В качестве предшествен-
ника кониферилового спирта в растениях можно рас-
сматривать шикимовую к-ту (V), к-рая образуется из
углеводов — первичных продуктов при-
СООН родного фотосинтеза. Введение в ра-
стения меченой С14 шикимовой кислоты
11о Г 1 q приводит к образованию в растении ра-
диоактивного Л. Процесс одревеснения
но Н v и появление Л. в растениях связаны с их
филогенетич. развитием, т. к. Л. от-
сутствует в водорослях, мхах и явно обнаруживается
в папоротниках.
В одревесневшем растении Л. играет роль инкру-
стирующего вещества, скрепляющего целлюлозные во-
локна. Л. вместе с гемицеллюлозами определяет проч-
ность стволов и стеблей растений. Пока остается от-
крытым вопрос о характере связи между Л. и угле-
водными компонентами древесины.
Л. пока не нашел еще широкого применения.
В силу особенностей строения Л. непригоден для полу-
чения нитей и пленок. Без существенных химич.
изменений его нельзя применять в качестве пласти-
ков и клеев. Отходы гидролизной пром-сти (гидро-
лизный Л.) и бумажной пром-сти (лигносульфоновые
к-ты) являются сильно измененными, трудно исполь-
зуемыми формами Л. Более интересным с точки зре-
ния использования является Л. сульфатных щелоков,
однако этот Л. нельзя считать отходом, т. к. он участ-
вует в цикле регенерации щелочи в сульфат-целлюлоз-
ном произ-ве. Попытки найти рациональные способы
применения громадных отходов Л. пока еще не до-
стигли существенных успехов. Использование гидро-
лизного Л. является большой народнохозяйственной
задачей. Гидролизный Л. может быть использован
в строительном деле (получение прессованных досок
и плит, термоизоляционных плит, где он служит
наполнителем вместе с другими дешевыми отходами).
Л., особенно полученный осаждением к-той из черных
сульфатных щелоков, может применяться в качестве
активного усилителя каучуков взамен газовой сажи
в резиновой пром-сти. Гидролизный Л. для этой цели
следует предварительно активировать, напр. нагре-
ванием со щелочью в автоклаве. Являясь полимером
с трехмерной структурой макромолекул и обладая
фенольными функциями, Л. может быть использован
в произ-ве пластмасс как наполнитель при получении
прессизделий, а также в качестве компонента термо-
реактивных смол, в к-рых он частично может заменить
16 к. X. Э. Т. 2
фенол.Смолы бакелитового типа удовлетворительного
качества могут быть получены при замене 30% фенола
Л. Из Л., в том числе гидролизного, путем его моди-
фицирования (напр., окислением) могут быть полу-
чены заменители природных и синтетич. дубителей
(регуляторы реологич. свойств глинистых растворов,
комплексообразователи и т. д.) и др. Гидролизный
Л. — хорошее сырье для получения активных углей.
Л. применяют в произ-ве пористого кирпича. Лигно-
сульфоновые к-ты, остающиеся в барде после сбра-
живания содержащихся в сульфитных щелочах са-
харов на спирт, находят применение в нек-рых от-
раслях пром-сти (как дешевые крепители и связующие
в литейном произ-ве, при формовании керамич. и аб-
разивных изделий, добавки к цементу, пластифици-
рующие добавки в бетон, для брикетирования уголь-
ной пыли, рудной мелочи, для улучшения реологич.
свойств глинистых р-ров при бурении, в дорожном
строительстве). Лигносульфоновые к-ты обладают
слабыми дубящими свойствами, и поэтому сульфит-
спиртовая барда используется при дублении кожи.
Из Л. могут быть получены ценные продукты:
ванилин, ванилиновая к-та, протокатеховый альде-
гид, протокатеховая к-та, пирокатехин. Выходы
этих веществ из гидролизного Л., однако, малы. Из
природного Л. (древесные опилки) те же продукты
могут быть получены с более высокими выходами.
Лит.: Никитин Н. И., Химия древесины и целлюлозы,
М,—Л., 1962; Роговин 3. А., ШорыгинаН. Н., Хи-
мия целлюлозы и ее спутников, М.—Л., 1953, гл. XI; Химия
древесины, под ред. Л. Э. Уайза, Э. С. Джана, пер. с англ., т. 1,
М.—Л., 1959, гл. XI; Brauns F. Е., Brauns D. А.,
The chemistry ot lignin, suppl. volume, N. Y., 1960; Чуда-
ков M. И., Усп. химии, 1961, 30, вып. 2, 184; III о p ы-
гина Н. Н., Изумрудова Т. В., Хим. наука и
пром-сть, 1959, 4, Ка 6, с. 747; Сапотницкий С. А.,
Использование сульфитных щелоков, М.—Л., 1960; Ники-
тин В. М., Лигнин, М., 1961. Н. Н. Шорыги-на.
ЛИГНОСТОН — см. Древесные пластики.
ЛИГНОСУЛЬФОНОВЫЕ КИСЛОТЫ — кислоты,
получающиеся при произ-ве сульфитной целлюлозы
в виде солей — лигносульфонатов. Состав Л. к. за-
висит от глубины сульфирования и методов выде-
ления; средний состав: 53,46% С, 5,37% Н, 5,02% S,
12% ОСН3. Строение к-т, как и лигнина, являющегося
их основой, недостаточно изучено. Наиболее вероятно,
что при сульфировании лигнина происходит заме-
щение сульфогруппой спиртового гидроксила, на-
ходящегося у а-атома углерода боковой цепи фенил-
пропановых структурных звеньев, образующих слож-
ную и неоднородно построенную молекулу лигнина
(см. формулу). В процессе суль-
фирования лигнина протекают
как реакции расщепления, так
и конденсации. Поэтому Л. к.
представляют полидисперсную ОСН3
коллоидную систему с широ-
ким диапазоном мол. весов (2 000—100 000). Л. к. рас-
творимы в воде, титруются как сильные минеральные
к-ты (диссоциированы на 60%), обладают лиофиль-
ным характером и являются полиэлектролитами.
Свободные к-ты получают из солей диализом с по-
следующим выделением путем ионообмена. После
выпаривания в вакууме образуется серо-коричневый
порошок сухих кислот. Из растворов они осаждаются
солями тяжелых металлов или ароматич. аминами.
Соли Л. к. (сконцентрированные выпариванием
в вакууме лигносульфонаты Na, NH4, Са, Mg) широко
применяют как диспергаторы, клеи, комплексообра-
зователи, дубители, в произ-ве ванилина и его про-
изводных. Значительное количество лигносульфонатов
сжигают или сбрасывают со сточными водами.
Лит.: Brauns F. Е., The chemistry ot lignin, N. Y.—L.,
1952—60; Никитин H. И., Химия древесины, M.—Л.,
1951; F е 1 i с е 11 а V. F. [а. о.], J: Amer. Chem. Soc., • 1956,
78, № 9, 1899; Goring D. A. I., Pulp and Paper Mag. Ca-
nada, 1957, Б8, № 5, 165. M. И. Чудаков.
963
ЛИГРОИН —ЛИЗЕРГИНОВАЯ КИСЛОТА
964
ЛИГРОИН — смесь жидких углеводородов, полу-
чаемая при прямой перегонке нефти или крекинге
нефтепродуктов и выкипающая от 120° до 240°. Л. —
прозрачная, бесцветная или желтоватая жидкость,
плотн. 0,760—0,795, поверхностное натяжение 23,5
дин/см (20°), давление пара ок. 150 мм рт. ст. (40°),
скрытая теплота испарения ок. 54 ккал/кг.
В табл. 1 приведены фракционный состав и нек-рые
др. константы Л., полученных из нефтей различного
месторождения.
Таблица 1
Лигроин из нефти Й20 4 Фракционный состав (перегоняется при темп-ре не выше), °C Ко- нец кипе- ния Окта- новое число (мотор- ный метод)
10% 50% 90%
Карачухарской. . 0,796 151 164 189 220 56
Бинагадинской. . 0,758 101 121 165 206 61
Еиби-Эйбатской . 0,803 148 181 212 228 52
Грозненской(пара- фин) 0,781 172 184 205 219 17
Туймазинской . . 0,765 147 164 187 198 —
Краснокамской . 0,778 148 168 188 200 —
Химич, состав Л. зависит от состава нефти и тех-
нологии получения. В табл. 2 приведены нек-рые
констайты и групповой химич. состав Л., полученных
из различных нефтей.
Таблица 2
Лигроин из нефти <120 , Начало кипения, j °C Конец кипе- ния, •с Групповой химич. состав, %
арома- тич. нафте- новых пара- фино- вых
Сураханской отборной 0,7920 147 175 11,8 51,5 36,7
Биби-Эйбатской легкой 0,7890 139 227 14,1 41,5 44,4
Сураханской тяжелой 0,7846 141 228 21,7 35,3 43,0
Бинагадинской тяжелой 0,8060 0,7720 140 226 12,6 57,7 29,7
Ставропольской .... 150 200 3,3 33,8 61,9
Краснокамской .... 0,7780 14В 200 15 38 49
Л. применяют: 1) в качестве тракторного горючего.
Для этой цели используют Л. с октановым числом
не ниже 54 и т. кип. 120—230°; 2) как растворитель
для лакокрасочной пром-сти (уайт-спирит).
В этом случае берут фракцию с пределами кипения
165—200°, плотностью не более 0,795 при 20°, содер-
жанием ароматич. углеводородов не выше 16%, серы
не выше 0,025% и т. всп. не ниже 33°; 3) в качестве
гидравлич. жидкости для нек-рых приборов (Л. при-
борный). Для этого применяют фракцию с пре-
делами кипения 120—240°, плотн. 0,785—0,795 (20°),
вязкостью не менее 1,2 сст (20°) и не более 6 сст
(—50°), иодным числом не более 0,4 и содержанием
серы не более 0,02%. В первом случае разрешается
применять продукты крекинга; в остальных исполь-
зуют только фракции прямой перегонки нефти или
гидрированные.
Лит.: Брусянцев И. В., Автотракторные топлива и
смазочныематериалы, М., 1958; К а р п о в П. II., Переработка
нефти, 2 изд., М.—Л., 1953; Технические нормы на нефтепро-
дукты, М., 1957. А. Д. Фатьянов.
ЛИЗЕРГИНОВАЯ КИСЛОТА CI6H16O2N2, мол. в.
268,32 — кристаллы; т. пл. 238° (с разл.), [a]g =
= + 40° (пиридин); входит в состав алкалоидов
спорыньи (наряду с диастереоизомерной изолизерги-
новой к-той, отличающейся от Л. к. конфигурацией
у С(8)) и получается из них при энергичном щелочном
гидролизе. Л. к. образует хлоргидрат, т. пл. 208—
210°, кислый сульфат, т. пл. 220°. И з олизер г и -
новая к-та, т. пл. 218° (с разл., дигидрат),
= -ф281о (безводная, пиридин), образует нит-
рат, т. пл. 185°. Л. к. при кипячении в водном р-ре
переходит в изолизергиновую; обратное превращение
происходит при обработке щелочью. При каталитич.
гидрировании Л. к. дает дигидролизергиновую к-ту.
Амид Л. к. (эргин) образуется наряду с изоэргином
при мягком гидролизе алкалоидов спорыньи. Строение
Л. к. подтверждено синтезом:
СОС6Н5
СОС6Н5 н
I I
Н СОСНз
ОН CN
Получены многочисленные синтетич. производные
и аналоги Л. к. Физиологич. действие эргина и изо-
эргина то же, что и у алкалоидов спорыньи, но бо-
лее слабое, однако все же более сильное, чем са-
мой Л. к.
Важное значение имеет не сама Л. к., а ее диэтил-
амид (препарат «LSD-25»), кристаллич. вещество
с т. пл. 80—85°; [= ^30° (С = 0,44; пиридин) —
965
ЛИЗИН —ЛИКВИДУС И СОЛИДУС
966
сильное галлюциногенное средство. Доза для человека
50—100 мг/кг. Галлюцинации сопровождаются веге-
тативными расстройствами. На гладкую мускулатуру
диэтиламид Л. к. оказывает эрготаминоподобное
действие, но более слабое, чем в случае эргоалкалои-
дов. Обладает нек-рой противогистаминной активно-
стью. Из организма быстро выводится с желчью. Яв-
ляется конкурентным антагонистом серотонина', гал-
люциногенное действие связано с недостатком серо-
тонина. Применяется в клинике для лечения шизо-
френии (выведения из заторможенного состояния).
Лит.: Орехов А. П., Химия алкалоидов, 2 изд., М.,
1955, с. 630; Генри Т. А., Химия растительных алкалоидов,
пер. с англ., М., 1956, с. 555; Kornleld Е. С. [а. о.],
J. Amer. Chem. Soc., 1954, 76, Ki 20, 5256; 1956, 78, № 13,
3087. A. E. Васильев.
ЛИЗИН (а, е-диаминокапроновая кислота), мол. в.
146,19, CH2(NH2)—СН2—СН2—СН2—CH(NH2)COOH
— бесцветные кристаллы. Л. известен в виде двух
оптически активных D-и L-формирацемич. D, L-фор-
мы. Т. пл. L-Л. 224—225° (с разл.). Л. очень хорошо
растворяется в воде, в кислотах и основаниях. Для
L-Л. [a]D = +25,9° (2%-ный р-р в 6 н. HCl), p/fai =
= 2,18; p^ao = 8,95; рГаз = 10,53; р/= 9,74. С кис-
лотами Л. дает два ряда солей, напр. с НС1 L-Л.
образует монохлоргидрат, т. пл. 263—264°, и дихлор-
гидрат, т. пл. 193°. Л. образует; нерастворимые соли
с пикриновой и фосфорномолибденовой к-тами, дает
характерные реакции аминокислот, напр. с нингид-
рином и др. L-Л. входит в состав почти всех белков
животного и растительного происхождения, в боль-
ших количествах содержится в гистонах и протами-
нах. Получают L-Л. из гидролизатов белков, осаждая
эту аминокислоту в виде пикрата. Синтетически Л.
получают из циклогексанона через е-капролактам,
через a-аминопимелиновую к-ту по способу Арштейна
и др. способами. В пром-сти Л. производят микро-
биология. путем. Л. является незаменимой амино-
кислотой', применяется в животноводстве для под-
кормки скота.
Лит.: Красильников И. А., Усп. совр. биологии,
1961, 52, вып. 2 (5), 149; Biochemical preparation, v. 1, N.Y.—
L., [19501, p. 63; Blochlmie сотрагёе des acldes amlnSs basl-
ques, P., 1960; см. также лит. при ст. Лейцин и Незаменимые
аминокислоты. Л. И. Линевич.
ЛИЗОЛ — раствор крезола (50%) в калийном мыле
(50%); красно-бурая, сильно пенящаяся при встря-
хивании жидкость с запахом крезола, легко раство-
ряется в воде, спирте, бензине. Водные р-ры Л.
прозрачны, бесцветны, слегка опалесцируют. Р-ры Л.
обладают бактерицидными свойствами; применяют
в виде 3—10%-ных водных р-ров для дезинфекции
различных предметов.
ЛИЗОЦИМЫ — белки, ферменты, широко рас-
пространенные в животном мире; находятся почти во
всех тканях и жидкостях живого организма, особенно
много в печени, селезенке, слюне, слезах; найдены
также в ряде микроорганизмов. По-видимому, часто
встречаются и в растениях. Основное свойство, от
к-рого и происходит название Л., — способность
растворять, «лизировать», оболочки ряда бактерий.
Л. выделены в 1922 А. Флемингом, впервые обстоя-
тельно исследованы в 30-х гг. 3. В. Ермольевой и
И. С. Буяновской.
Л. разного происхождения, обладая биологич.
активностью одного и того же характера, отличаются
несколько по интенсивности действия, а также имеют
небольшие различия в аминокислотном составе.
Л. куриных яиц относится к числу наиболее изучен-
ных белков, и все приводимые здесь данные относятся
к Л. этого происхождения. Мол. в. ок. 14 800, содер-
жание азота 18,7%. Молекула Л. состоит из одной
полипептидной цепи, включающей 127—130 амино-
кислотных остатков, из них: 11 глицина, 10 аланина,
9 серина, 10 цистеина, 2 метионина, 7 треонина,
16’
2 пролина, 6 валина, 8 лейцина, 6 изолейцина, 3 фе-
нилаланина, 3 тирозина, 6—8 триптофана, 20 аспара-
гиновой к-ты, 5 глутаминовой к-ты, 6 лизина, И ар-
гинина, 1 гистидина; 18 свободных карбоксильных
групп амидировано. На аминном конце цепи нахо-
дится лизин, на карбоксильном — лейцин. Установ-
лена последовательность связи 60 аминокислот,
считая с аминного конца. Преобладание свободных
аминных групп над свободными карбоксильными при-
дает белку сильно основной характер: изоэлектрич.
точка его находится между 10,5—11,0. Л. как белок
очень устойчив; хорошо растворяется в воде и в бу-
ферных р-рах при всех значениях pH, кроме изо-
электрич. области; [а]р = —49,6° ± 0,5° (вода).
В нейтральных и кислых р-рах он выдерживает крат-
ковременное кипячение без денатурации, за 50 мин
активность падает лишь на 40%; в щелочной среде
менее Стоек, однако при pH 12 и комнатной темп-ре
активность в течение нескольких часов остается без
изменений.
Л. проще всего получается из цельного яичного
белка непосредственной кристаллизацией, легко про-
текающей при pH 9,5 с добавкой 5% NaCl и неболь-
шого количества кристаллов изоэлектрич. лизоцима
(для затравки); через 4—7 дней выпавшие кристал-
лы отделяют, их перекристаллизовывают в тех же
условиях и лиофилизуют, выход — ок. 3 г Л. из 1 л
исходного белка (35—40 яиц). При перекристаллиза-
ции при pH 4,5 (НС1) получается Л.-хлорид, а при
pH 8,5 (NaHCO3) — Л.-карбонат. Менее удобны для
получения — адсорбция на бентоните или фракцион-
ное осаждение ацетоном. Чистейший Л. можно вы-
делять также прямым хроматографированием яичного
белка на ионообменной целлюлозе.
Характерная реакция на Л. — лизис им суспен-
зии микробов Microccocus lysodeikticus, к-рый ин-
тенсивно протекает при pH 5—8 и концентрации Л.
1 мкг/мл и выше (используется для количественного
определении Л.). Разрушая клеточные оболочки мик-
робов, Л. разрывает в мукополисахаридах оболочек
р-глюкозидную 1—4 связь N-ацетилмураминовой
к-ты и N-ацетилглюкозамина:
СН2ОН СН2ОН
о Н NHCOCHg Н NHCOCH3.
CHg-CH-COOH
Менее интенсивно Л. гидролизует хитин и его произ-
водное гликольхитин. Л. действует на ряд грамполо-
жительных бактерий (Вас. megatherium, Вас. anthra-
cis, Вас. Iluorescens, Sarcina lutea, Sarcina flava,
Staphylococcus aureus); на нек-рые грамотрицательные
(Azotobacter venelandii, E. coli, Ps. aeruginosa,
Vibrio cholerae и др.) действует лишь в присутствии
динатриевой соли этилендиаминтетрауксусной к-ты
(трилон Б, комплексон III) или таурохолевокислого
натрия. Л. находит применение при лечении воспали-
тельных заболеваний глаз, носоглотки, ожогов, ран
и в акушерской практике. В микробиологии приме-
няется для разрушения клеточных оболочек бакте-
рий. Показано консервирующее действие Л. на икру
рыб; начинают использовать как добавку к молоку
с целью консервации и лучшей усвояемости.
Лит.: Ермольева 3. В., Усп. соврем, биол., 1938,
9, вып. 1, 68; S а 1 t о n М. R. J., Bacterlol. Revs, 1957, 21,
№ 2, 82; В 1 I а п о U., Clin, terap., 1957, 13, 159; Biochemi-
cal preparations, v. 1, N. Y.—L., [1950],^). 67.^
ЛИКВИДУС И СОЛИДУС— графическое изобра-
жение темп-р начала и конца равновесной кристалли-
зации растворов или сплавов в зависимости от их
967
ЛИКОРИН — ЛИНАЛООЛ
968
состава. Ликвидус — геометрич. место точек,
отвечающее темп-рам начала равновесной кристалли-
зации.. Солидус — геометрич. место точек, от-
вечающее темп-рам конца равновесной кристаллиза-
ции. В зависимости от того, представляют ли Л. и с.
кривую, поверхность и т. д. применяются термины:
«линия ликвидуса», «линия солидуса» — для двойных
систем, «поверхность солидуса» — для тройных си-
стем и т. д. На диаграммах состояния двойных систем
Л. и с. изображаются комплексом линий, число к-рых
равно числу твердых фаз, кристаллизующихся в си-
стеме. Л. и с. диаграмм состояния тройных систем —
комплекс поверхностей, число к-рых также равно
числу твердых фаз, кристаллизующихся из жидкости.
ЛИКОРИН CleH17O4N, мол. в. 286,30 — алкалоид,
выделенный из многих растений, принадлежащих
к семейству амариллисовых (Amarylli-
НО-г^Х- । daceae). Основной скелет Л. содержит-
HO-Lу ся во многих алкалоидах Amaryllida-
• |Н сеае. Кристаллы, т. пл. 275° (с разл.),
[«]р = —129° (из спирта). Образует
J J соли: хлоргидрат, т. пл. 217° (с разл.);
lY* пикрат, т. пл. 196°; перхлорат, т. пл.
Сн2~О 230° (с разл.) и др., а также два иод-
метилата: а, т. пл. 247° (с разл.),
[а]5р = —46,1° (в воде), и р, т. пл. 198° (с разл. моно-
гидрат), [a]|J = +128° (в воде). Л. сравнительно мало
токсичен. В небольших дозах при введении per os
вызывает слюнотечение, в больших — рвоту и понос.
В медицине не применяется.
Лит.: Орехов А. П., Химия алкалоидов, 2 изд., М.,
1955, с. 421; С о о k J. W., London J. D., McCloskey
Р., Chem. Ind., 1954, № 39, 1199 ; Taylor W. I., T h о m a s
B. R., Uyeo S., там же, 1954, № 30, 929; Nakagawa J.,
Uyeo S., J. Chem. Soc., 1959, 3736. A. E. Васильев.
ЛИМАЦИДЫ — химические средства борьбы со
слизнями; для этих целей чаще всего применяют от-
носительно безопасные для растений вещества, ожи-
гающие кожу слизней. К Л. относятся: суперфосфат,
железный купорос, гашеная известь и нек-рые другие;
нормы расхода 50—250 кг/га. Эффективность этих
препаратов относительно мала; более эффективен
для этих целей метальдегид (кристаллич.
полимер ацетальдегида), к-рый используется в виде
2—5%-ной смеси (дуста) с отрубями. Такой состав
легко поедается слизнями, после чего они гибнут.
Н. Н. Мельников.
ЛИМОНЕН (1-метил-4-изопропенилциклогексен-1)
C10Hle,f мол. вес 136,24 — бесцветная, летучая, лег-
сн ко окисляющаяся на воздухе жидкость
I 3 с приятным, напоминающим лимонный
или апельсиновый запахом; хорошо рас-
J творяется во всех неполярных органи-
Тг ческих растворителях, не растворяется
СН в воде. Л. существует в виде трех форм:
1'зС/^СН2 d, 1 и d,l.
Соединение Т. КИП., °C/AtAt t nD [а]Ь“
d-Лимонен .... 1-Лимонен .... d.l-Лимонен . . 175,5-176/763 68,2/20 175,5-176,5/763 68,2/20 175,5-176,5/763 68,2/20 0,8411 0,8422 0,8402 а 1,4743 (21") 1,4713 (20°) 1,4719 (20") 4-126,84° -123,7° 0,00°
а При 20,84°.
Л. образует азеотропные смеси (в скобках темп-ра
кипения и содержание d-Л.): с фурфуролом (155,95°,
65%); ментолом (64,63°, 0,8%), н-гексиловым спиртом
(155,5°, ~21 %), циклогексанолом (159,25°, 26,5%,) и др.
Л. весьма реакционноспособное соединение. На
воздухе окисляется в карвон и карвеол (хранят в за-
паянных стеклянных ампулах); окисление гидропере-
кисью ацетила приводит к образованию моноокиси
и диокиси, при гидратации к-рых разб. серной к-той
образуются соответственно гликоли и эритриты.
Окисление Л. перманганатом калия приводит к об-
разованию эритрита, а дальнейшее окисление—к ди-
кетокислоте и трикарбоновой к-те. Все минеральные
к-ты на холоду гидратируют Л. с образованием тер-
пинеола и терпингидрата. Гидрогенизация на Pt-
или Ni- катализаторе при 180—200° приводит к пара-
ментану; при дегидрогенизации Л. на тех же катали-
заторах (350—400°) образуется цимол. При пропуска-
нии Л. над нагретыми до красного каления платиновой
или нихромовой проволоками и последующем быстром
охлаждении получается изопрен.
Л. весьма распространен в природе; входит в состав
почти всех без исключения скипидаров хвойных
и эфирных масел, получаемых из хвои различных
хвойных деревьев, содержится в эфирных маслах:
тминном, поручейника (Sium latilolium), цитронел-
ловом и мн. др. Фракционной перегонкой масел
на эффективных колонках можно выделить сравни-
тельно чистый Л. Весьма чистый Л. получают разло-
жением тетрабромидов цинком и уксусной к-той.
Л. применяют как душистое вещество, в качестве
сырья для синтеза а-терпинеола, терпингидрата,
карвона, флотационного масла и др.
Лит.: Simonsen J. L., The terpenes, v. 1, Camb.—
L., 1953; Пигулевский Г. В., Химия терпенов, Л.,
1949; Braun J. V., Lemke Вег., 1923, 53, № 7, 1562.
И. Й. Бардышев.
ЛИМОННАЯ КИСЛОТА (СН2СООН)2 С(ОН)СООН,
мол. в. 192,12 — одноводный кристаллогидрат; т. пл.
безводной 153°; в воде хорошо растворима; константы
диссоциации (18°): К\ = 8,4- 10“4, = 1,7 • 10~5
и К3 = 4,0- 10~в. При нагревании до 175° Л. к. пе-
реходит в аконитовую (I) и ацетондикарбоновую (II)
кислоты:
сн-соон II с-соон сн2соон 1 с=о
СН2СООН СН2СООН
I II
выше 175 образует итаконовую кислоту. При прокали-
вании Л. к. со щелочью образуются соли щавелевей
и уксусной к-т. Л. к. обнаруживает обычные свойства
многоосновных к-т, давая три ряда солей и эфиров —
кислых и полных; плохо растворимы соли щелочно-
земельных металлов. Л. к. может быть проацили-
рована по ОН-группе.
Л. к. широко распространена в природе, относи-
тельно много ее содержится в нек-рых ягодах, фрук-
тах, особенно в цитрусовых (в лимоне 6—8%,). Зна-
чительные количества Л. к. содержатся также в ли-
стьях хлопчатника и стеблях махорки, откуда ее и вы-
деляют (один из промышленных способов). Л. к.
выделяют в. виде лимоннокислого кальция из продук-
тов переработки хвои ели или плодов лимона, а также
получают из р-ров сахара с помощью нек-рых гриб-
ков. Л. к. находит значительное применение в фар-
макологии и пищевой промышленности (в приготовле-
нии напитков, кремов, желе и т. д.). В небольшом
количестве Л. к. употребляют в произ-ве нек-рых
алкидных смол. Триэтиловый и трибутиловый эфи-
ры Л. к. служат пластификаторами при произ-ве
лаков и пр. Триметиловый эфир, т. пл. 78—79°,
т. кип. 283—287°; триэтиловый эфир, т. кип. 294°,
dl° 1,1369, «5$ 1,44554. И. А. Несмеянов.
ЛИМОННОЕ МАСЛО — см. Эфирные масла.
ЛИНАЛООЛ С10Н18О, мол. в. 154,23 — бесцветная
жидкость с запахом ландыша. В природе встреча-
ются правовращающая (Coriandrol, I), левовращаю-
969
ЛИНДАН —ЛИОТРОПНЫЕ РЯДЫ
970
щая (Licareol, II) и неактивная (Myrcenol) d,1-формы:
I. снзх
>С= СН- СН2СН2 С (ОН) (СН3)-СН=СН2
СНз
2,6-диметил-2,7-октадиен-6-ол
II. СН2.
+С-СН2-СН2СН2С(ОН) (СН3)-СН = СН2
СН3
2,6-диметил-1,7-октадиен-6-ол
Свойства Изомер
d 1 dl
Т. пл., СС . . . . Т. кип., 0С/лш . 86-87 198 - 200/760 85-90/20 197-200/756 86—87/14 197—199/760 89-91/15
df 0,8679 (20°) 0,8622 (20°) 0,870(15")
SO nD 1,4652 1,4604 (22") 1,4627
[alb° +19,18 -19,37 —
Превращение d-изомера в 1-изомер происходит при
последовательном действии РС15 и CH3ONa. Л. рас-
творим в спирте, эфире и других органич. растворите-
лях. При окислении Л. окисью хрома образуется
смесь цитраля и метилгептенона; окисление перман-
ганатом приводит к образованию левулиновой кис-
лоты и ацетона. Для Л. характерна способность к
изомеризации. Под действием кислых реагентов Л.
изомеризуется в гераниол; одновременно происходит
частичная циклизация с образованием терпинеола.
Уксусный ангидрид превращает Л. в смесь гераниола,
нерола, а-терпинеола и терпингидрата.
Л. широко распространен в эфирных маслах, осо-
бенно в форме сложных эфиров (ацетатов и др.).
Является главной составной частью мексиканского,
бергамотного, кориандрового, лавандового масел,
где находится в смеси оптич. активных Л. и изомерных
2,6-диметилоктадиен-1,7-олов-6. Кайенское масло «ли-
налое» содержит 60—80% левовращающей формы.
В кориандровом масле, получающемся из культиви-
руемого в СССР растения, содержится до 70% право-
вращающей формы.
Л. получают фракционированной разгонкой эфир-
ных масел, в СССР чаще всего кориандрового (фрак-
ция 190—199°). Если Л. присутствует в виде эфиров,
то эту фракцию предварительно омыляют; выделяют
Л. через натриевую соль кислого эфира фталевой к-ты.
Известны и другие способы выделения Л. из эфирных
масел. Л. можно синтезировать нагреванием гера-
ниола до 200° (изомеризация) и др. методами.
Л., содержащийся'в кориандровом масле, служит
сырьем для получения цитраля. Л. применяют в пар-
фюмерной промышленности при составлении многих
цветочных композиций. Л. обладает бактерицидными
свойствами. Ацетилированием Л. получают его эфир —
линалилацетат — жидкость с приятным за-
пахом цветов; т. кип. 220°/760 мм; 98—100710 мм;
dr 0,8998; ng 1,450; найден в эфирных маслах
бергамота, лаванды и др. растений. Линалилацетат
широко применяют как составную часть духов и оде-
колонов высших сортов, для приготовления искус-
ственных масел — бергамотного и лавандового.
Лит.: Белов В. Н. [и др.1, Химия и технология души-
стых веществ, М., 1953. А. В. Арбатский.
ЛИНДАН — см. Гексахлоран.
ЛИНЕЙНАЯ ПЕРЕДАЧА ЭНЕРГИИ (ЛПЭ) — см.
Передача анергии линейная.
ЛИНОЛ — смесь метиловых эфиров жирных к-т
льняного масла; бесцветная или слабоокрашенная
маслообразная жидкость; т. кип. 180—185°/8 мм,
206—216720 мм; 0,8860—0,8891; «у 1,4633 —
1,4641; иодное число 170—186; легко растворяется
в большинстве органич. растворителей, трудно —
в воде. В состав Л. входят метиловые эфиры линоле-
новой, линолевой, олеиновой, пальмитиновой и стеа-
риновой к-т. Получают Л. путем нагревания льня-
ного масла с метанолом в присутствии 10,5%-ной
серной к-ты. Применяют Л. для лечения повреждений
кожи.
Лит.: Зиновьев А. А., Химия жиров, М., 1952.
Р. Г. Глушков.
ЛИНОЛЕВАЯ КИСЛОТА, мол. в. 280,43 — кар-
боновая к-та с двумя изолированными двойными свя-
зями, СН3(СН2)4СН=СН—СН2—СН=;СН(СН2)7СООН;
т. пл. —5,2°; т. кип. 230—233715 мм, 21075 мм;
0,9038; ng 1,4699; кислотное число 200,06; иодное
число 181,03. Л. к. присоединяет галогены, обра-
зуя тетраметилгалогеностеариновую к-ту; на воз-
духе постепенно окисляется (аутоокисление), образуя
смесь капроновой, азелаиновой и щавелевой к-т;
иодистым водородом восстанавливается до стеари-
новой к-ты. Для Л. к. возможно 4 стереоизомера;
встречающаяся в природе имеет ifuc-ifuc-конфигура-
цию. Л. к. относится к незаменимым жирным кисло-
там. Л. к. распространена в природе в виде тригли-
церидов и в смеси с триглицеридами других кислот
входит в состав важнейших высыхающих и полу-
высыхающих масел, которые применяют в качестве
основы в произ-ве лаков, красок, эмалей и олиф. В
соевом масле содержится ок. 55% Л. к., в перилловом
и подсолнечном — до 52—53%, в льняном — до 30%,
в конопляном — до 50—65%, в маковом — до 60%.
Метиловый эфир, т. кип. 211—212716 мм,
168—17071 мм; этиловый эфир, т. кип.
270—275° /180 мм. См. также Высшие жирные ки-
слоты, Жиры, Масла высыхающие.
И. А. Несмеянов.
ЛИНОЛЕНОВАЯ КИСЛОТА С18НзоО2 мол. вес
278,42 — бесцветная жидкость, т. пл. -(11—11,3$
0,9046; ng° 1,4800; хорошо растворима в эти-
ловом спирте, эфире, ацетоне и др. органич. раствори-
телях. Л. к. содержится в виде триглицеридов во
многих растительных маслах: льняном (до 50%),
перилловом (до 55%), конопляном, соевом и др.
Л. к. содержит три изолированные двойные связи
СН3(СН2СН=СН)3(СН2)7СООН, обладает общими свой-
ствами кислот с изолированными двойными связями;
на воздухе окисляется еще легче, чем линолевая
кислота; бром присоединяется по двойным связям,
при этом образуется гексабромид, т. пл. 180°, желтые
кристаллы, нерастворимые в эфире. Л. к. необходима
для нормальной жизнедеятельности животного и че-
ловеческого организма и относится к незаменимым
жирным кислотам. Л. к. — главная составная часть
масел высыхающих, служащих основой олиф, масля-
ных красок И др. Н. А. Несмеянов.
ЛИОТРОПНЫЕ РЯДЫ — ряды, в к-рых ионы
последовательно располагаются по величине их влия-
ния на свойства растворителя в растворе или диспер-
сионной среды в дисперсной системе (в случае воды —
гидротропные ряды, термин сравнительно редко при-
меняемый). Таковы Л. р. ионов по их возрастающему
или убывающему влиянию на вязкость и поверхност-
ное натяжение водных р-ров, и в том числе растворов
поверхностно-активных веществ, на растворимость
в воде, набухание высокомолекулярных веществ
(белков, пектинов, агар-агара, крахмала и др.),
на застудневание водных р-ров таких веществ, а
также на их высаливание из растворов, на устойчи-
вость коллоидных растворов (гидрозолей) при их
коагуляции и на др. свойства растворов, студней
и дисперсных систем. Известны Л. р. по гемолизи-
рующему действию по отношению к взвесям эритро-
цитов, подавлению возбудимости мышц и нервов
и др. Наиболее изучен ряд неорганических анионов:
SO*-, F-, JO3, BrO3, Cl-, СЮу, Вг-, NO,, С1О4,
971
ЛИОТРОПНЫЕ РЯДЫ - ЛИПАЗЫ
972
J~, CNS~. Менее ясно выражены различия в Л. р.
однозарядных (Li+, Na+, КА, Rb+, Cs+) и двухзарядных
(Mg2+, Са2+, Sr2+, Ва2+) катионов.
Л. р. по высаливанию яичного альбумина натрие-
выми солями различных кислот был установлен
Г. Гофмейстером (1888). Он определял наименьшие
концентрации натриевой соли, вызывающие помут-
нение р-ра альбумина; полученные им концентрации
(в скобках) представлены в ряду анионов: цитрат
(0,56); тартрат (0,78); сульфат (0,80); ацетат (1,69);
хлорид (3,62); нитрат (5,42);хлорат (5,52); иодид;
роданид.
Как видно, высаливающее действие наиболее сильно
в случае введения лимоннокислого натрия, а иоди-
стый и роданистый натрий ни при каких возможных
концентрациях не вызывают помутнение р-ра альбу-
мина.
Расположение ионов в Л. р. определяется их гид-
ратацией — способностью связывать воду, отнимая
ее от гидратированных молекул растворенного ве-
щества или частиц дисперсной фазы. Изучение меха-
низма влияния ионов неорганич. солей на свойства
водных р-ров и дисперсных систем показало наличие
тесной связи между энергией гидратации ионов и спо-
собностью их солеи повышать поверхностное натяже-
ние воды. Интенсивное взаимодействие ионов с водой
означает, что энергия связи между ионом и молекулой
воды больше энергии взаимного притяжения молекул
воды (т. е. ион сильнее втягивает молекулы Н2О
с поверхности вглубь, чем это имеет место в чистой
воде, что и повышает поверхностное натяжение).
Энергия гидратации ионов возрастает при переходе
от ионов низшей валентности (зарядности) к ионам
высшей валентности, а при одинаковой валентности —
с уменьшением радиуса ионов (см. Ионный радиус).
В Л. р. катионы расположены в порядке возрастаю-
щей величины их радиуса, что совпадает с располо-
жением их в периодич. системе элементов Д. И. Мен-
делеева (в данном случае существен закономерно
нарастающий объем этих ионов). Анионы обычно
слабее гидратируются, чем катионы, т. е. их стремле-
ние уйти в глубь раствора с его поверхности выражено
слабее. В результате этого поверхностный слой вод-
ных р-ров солей обычно заряжен отрицательно.
В Л. р. закономерно нарастает способность аниона
отрицательно заряжать поверхность водного р-ра
по отношению к воздуху. Л. р. ионов определяют их
способность вызывать коагуляцию коллоидных р-ров,
причем различия в пороге коагуляции, особенно для
золей с отрицательно заряженными частицами, могут
быть очень значительными. Чем слабее гидратация
ионов, тем больше их способность адсорбироваться
на гидрофобных поверхностях. Способность ионов
к адсорбции растет в Л. р. в направлении от SO(
к CNS-, поэтому ионы CNS- оказывают обычно ста-
билизирующее действие на дисперсные системы.
У катионов различия в адсорбируемости выражены
слабее. Места членов Л. р. ионов не являются строго
постоянными и могут изменяться в зависимости от
условий (pH р-ра, концентрации соли, темп-ры).
Действие Л. р. ионов на высаливание или набухание
белков зависит прежде всего от pH раствора,
напр. анионы в кислой среде, когда ионы белков за-
ряжены положительно, по высаливающему действию
располагаются в ряд; CNS“>J~>... и т. д., т. е.
имеет место обращение Л. р. Подобное обращение
наблюдается у Л. р. катионов на щелочной стороне
от изоэлектрич. точки, где высаливающее действие
ионов падает от Cs+ к Li+. Количественная характе-
ристика закономерности Л. р. выражается ур-нием:
N — к (Н — Н^), в к-ром Н и — соответственно
энергии гидратации иона и высаливаемого вещества
(напр., желатина), к — константа, а А — величина,
оценивающая лиотропное действие этого иона (лио-
тропное число). Лиотропные числа сохраняют свое
значение и для количественного описания изменений
любого свойства раствора. Числовые значения лио-
тропных чисел нек-рых анионов приведены в таблице.
Анион N Ав ион N
Сульфат 2,0 Бромид 11,3
Фторид 4.8 Нитрат 11.6
Иодат 6,2 Перхлорат 11.8
Бромат 9,5 Иодид 12,5
Хлорид Хлорат 10 10,6 Роданид 13,2
Лит.: Рабинерсон А. И., Проблемы коллоидной
химии, Л., 1937; Freandllch Н., Kapillarchemie,
Bd 1—2. 4 Aufl., Lpz., 1930—32; Пасынок ий А. Г.,
Коллоидная химия, М., 1959; Руцкой А. П., Краткий
курс коллоидной химии, Л., 1958. И. Н. Путилова.
ЛИОФИЛЬНОСТЬ И ЛИОФОБНОСТЬ коллои-
дов — характеристика интенсивности молекуляр-
ного взаимодействия вещества дисперсной фазы (в кол-
лоидно-дисперсных системах) с дисперсионной средой
(в частности, с водой — гидрофильные и гидрофобные
коллоиды). Чем более лиофильна поверхность дис-
персной фазы, тем больше сольватация (см. также
Гидрофильность и гидрофобность, Смачивание).
ЛИОФОБНЫЕ КОЛЛОИДЫ — коллоидно - дис-
персные системы, в которых диспергированное веще-
ство инертно по отношению к дисперсионной среде.
См. также Лиофильность и лиофобность коллоидов,
Мицеллы, Коллоидные системы, Золи.
ЛИПАЗЫ — ферменты, катализирующие гидро-
литич. расщепление сложных эфиров глицерина и выс-
ших жирных кислот. Подобно большинству эстераз,
Л. не отличаются строгой специфичностью и, помимо
жиров, расщепляют триглицериды низших алифатич.
к-т (триацетин, трибутирин), а также нек-рые слож-
ные эфиры одноатомных спиртов. Л. расщепляют
триглицериды последовательным отщеплением остат-
ков жирных к-т, при этом с максимальной скоростью
происходит гидролиз триглицеридов до соответствую-
щих диглицеридов; диглицериды расщепляются зна-
чительно медленнее; еще медленнее гидролизуются
моноглицериды. В определенных условиях Л. спо-
CH.OCOR сн,.он СН2ОН СН2ОН
сносок ^сносок —а5снон —2--снон
1111
CH2OCOR CHoOCOR CH2OCOR СН2ОН
собны синтезировать жиры из глицерина и жирных
к-т, однако в клетках организма роль Л. состоит
только в расщеплении жиров, синтез же последних
осуществляется иными ферментными системами.
Л. играют важную роль в переваривании жиров
и содержатся в желудочном и кишечном соках, соке
поджелудочной железы и в грудном молоке. Л. най-
дены в тканях животных и растений, а также у микро-
организмов. Богаты Л. семена масличных растений,
в частности клещевины и сои; эти Л. играют важную
роль в период прорастания растения.
Л., выделенные из различных источников, отличаются друг
от друга нек-рыми свойствами, в частности оптимумом pH
и субстратной специфичностью. Панкреатическая
Л., полученная в высокоочищенном состоянии из свиного пан-
креатина, представляет собой гомогенный белок, р! 5,2, число
оборотов 1,2 • 10е мин~1 при 37° и pH 9,0. Панкреатич. Л.,
как, по-видимому, и другие Л., катализирует гидролиз жиров
и др. сложных эфиров лишь на межфазной поверхности раздела
системы вода—субстрат и слабо действует на молекулярно-
дисперсные системы. Активность Л. значительно повышается
в присутствии солей желчных и жирных к-т, СаС12 и альбуми-
нов, способствующих эмульгированию жиров. Альдегиды,
надуксусная к-та, n-хлормеркурибензоат тормозят фермента-
тивную активность. В покоящихся семенах клещевины при-
сутствует т. н. сперматолипаза, нерастворимый
в-воде фермент с оптимумом pH 4,7—5,0. В период прорастания
сперматолипаза превращается в бластолипазу, также
нерастворимую в воде (оптимум pH 7,0). Бластолипаэа может
973
ЛИПИДЫ — ЛИПОЕВАЯ КИСЛОТА
быть получена из сперматолипазы искусственным путем при
воздействии на последнюю пепсином.
Своеобразной Л. является липопротеидлипаза
(просветляющий фактор), обнаруживаемая в сыворотке крови
и тканях животных. Действие липопротеидлипазы направлено
на расщепление нек-рых липопротеидов и хиломикро-
нов (микроскопия, частицы жировой эмульсии, появляющие-
ся в лимфе и крови после переваривания и всасывания жиров).
Высокоочищенная липопротеидлипаза — белок, содержащий
мукополисахарид типа гепарина; оптимум активности фермен-
та при pH 6,4—7,5.
Активность Л. определяют: титрованием щелочью
жирных к-т, образующихся при ферментативном гид-
ролизе; манометрич. измерением количества СО2,
вытесняемой из бикарбонатного буфера кислотой, об-
разующейся в результате гидролиза; денситометри-
ческими методами, основанными на измерении умень-
шения оптич. плотности жировой эмульсии под
действием Л.
Растительные Л., гл. обр. из семян клещевины,
используют для мягкого расщепления жиров при по-
лучении концентрированных препаратов нек-рых ви-
таминов (в частности, при выделении витамина А из
жира печени трески), а также для получения из соот-
ветствующих масел нек-рых ненасыщенных жирных
к-т, напр. рицинолевой. Л. применяют также в коже-
венной пром-сти (для очистки шкур от остатков жира),
при произ-ве сыров (для ускорения их созревания) и
в лечебных целях (при плохом переваривании жиров).
Лит.: Fermente, Hormone, Vitamlne, hrsg. R. Ammon,
W. Dirscherl, Bd 1, 3 ed., Stuttgart, 1959; The enzymes, ed.
P. D. Boyer, H. Lardy, K. Myrback, v. 4, 2 ed., N. Y.—L.,
1960. См. также лит. при ст. Мальтаза. В. В. Спиричев.
ЛИПИДЫ — жиры и жироподобные вещества, ор-
ганич. соединения растительного или животного
происхождения, различные по химич. составу, строе-
нию и выполняемой физиологии, или биохимич.
функции и объединяемые на основе общих физико-
химич. свойств. Л. нерастворимы или плохо раство-
римы в воде, растворяются в типичных жирораство-
рителях — бензине, бензоле, хлороформе, четырех-
хлористом углероде, ацетоне, эфире, петролейном
эфире, горячем спирте и т. п. Не все Л. одинаково
хорошо растворимы в указанных растворителях.
В частности, фосфолипиды, в отличие от нейтральных
жиров, очень плохо растворимы в ацетоне; ацеталь-
фосфатиды и сфингофосфатиды нерастворимы в эфире.
Наиболее распространенная классификация Л. при-
водится ниже.
Простые липиды. В эту группу включают эфиры
высших жирных к-т и нек-рых спиртов. К ним относятся:
а) нейтральные жиры, представляющие собой эфиры жирных
к-т и глицерина; б) воски, являющиеся эфирами жирных к-т
и высших одноатомных спиртов (сюда же относятся выделяе-
мые иногда в особую группу стериды — эфиры стеринов и
высших жирных к-т, в частности эфиры холестерина).
Сложные липиды. Эта группа соединений отли-
чается наличием в их молекулах, помимо спирта и жирной
к-ты, других составных частей (остатков фосфорной или
серной к-т, азотистых оснований, нек-рых сахаров и др.).
В нее входят: а) фосфолипиды, характеризующиеся присутст-
вием в их молекуле остатка фосфорной к-ты. К ним относятся
лецитины, коламинфосфатиды, серинфосфатиды, инозитфос-
фатиды, ацетальфосфатиды, сфингофосфатиды и фосфатидные
к-ты; б) цереброзиды, характеризующиеся наличием в их моле-
куле остатка аминоспирта—сфингозина, сахара — обычно
галактозы — и высшей жирной к-ты, а также отсутствием фос-
форной к-ты и глицерина; в) ганглиозиды, родственные цереб-
розидам и отличающиеся от них большей сложностью строения
и наличием в молекуле остатка нейраминовой кислоты; цере-
брозиды и ганглиозиды обычно объединяют под общим назва-
нием гликолипидов; г) сульфолипиды, характери-
зующиеся наличием в их молекуле остатка серной к-ты.
Прочие липиды. В эту группу входят соединения,
являющиеся структурными элементами простых и сложных
Л. и обычно представляющие собой промежуточные продукты
в биосинтезе или расщеплении последних. Соединениями ука-
занной группы являются; а) моно- и диглицериды — произ-
водные глицерина, в к-рых одна или две спиртовые группы
этерифицированы высшими жирными к-тами; б) высшие жирные
кислоты; в) высшие спирты, в частности высшие алифатич.
спирты, входящие в состав восков, стерины, а также спирты,
содержащие кольцо g-ионона (витамин А, зеаксантин); г) жи-
рорастворимые витамины D, Е и К; д) высшие углеводороды,
в том числе каротины и каротиноиды, а также высшие алифа-
тич. углеводороды (напр., изооктадекан CigHjs, присутствую-
974
I щий в жирах печени) и нек-рые твердые углеводороды, встре-
чающиеся в природных восках, например н-пентакозан
СНз(СН2)2зСН3 и др.; е) простые эфиры глицерина, напр. бати-
ловый СН3(СН2)1оСНгОСН2СН(ОН)СН2ОН, входящий в состав
неомыляемой фракции печени.
Л. относятся к числу важных в биологич. отношении
веществ, входящих в состав всех живых клеток.
Нек-рые Л. в той или иной степени специфичны для
определенных тканей или органов (напр., цереброзиды
для мозговой ткани), другие (напр., нейтральные
жиры) встречаются во всех тканях. Особенно богата
Л. нервная ткань: содержание фосфолипидов и гли-
колипидов в белом веществе мозга достигает 7,5—9,0%
от веса ткани. Л. в живых организмах находятся в сво-
бодном или в связанном состоянии — в виде комп-
лексов с белками: липопротеидов и протеолипидов.
Биохимич. и физиологич. функции отдельных групп
Л. довольно разнообразны и далеко еще не изучены.
Важнейшее физико-химич. свойство Л. — нераство-
римость в воде — определяет их роль основного
структурного элемента протоплазмы: из Л. и липо-
протеиновых комплексов построены поверхностные
мембраны клеток и клеточных органоидов — ядер,
митохондрий, рибосом. Л., входящие в состав мем-
бран, принимают непосредственное участие в процес-
сах активного переноса через эти мембраны ионов
и молекул различных веществ. Нейтральным жирам
принадлежит важная роль источника энергии и эко-
номичной формы, в к-рой организм запасает эту
энергию.
Для выделения Л. из биологцч. источников поль-
зуются методами экстракции органич. растворите-
лями. Индивидуальные Л. выделяют с помощью
хроматографии, методов.
Различные Л. имеют широкое практич. применение
в качестве продуктов питания, в медицине и в раз-
личных отраслях пром-сти. Более подробно вопросы
о биологич. роли различных Л., о методах разделения
Л. на отдельные группы и индивидуальные соединения,
а также об их практич. применении изложены в спе-
циальных статьях, посвященных этим Л. (см. ст.
Воски, Жиры, Инозитфосфатиды, Коламинфосфа-
тиды, Лецитины, Серинфосфатиды, Стерины, Сте-
роиды, Сульфолипиды, Сфингофосфатиды, Фосфоли-
пиды, Цереброзиды, Витамины).
Лит.: Зиновьев А. А., Химия жиров, М., 1952;
Deuel Н. J., The lipids. Their chemistry and biochemistry,
V. 1, N.Y.—L., 1951. В. Б. Спиричев,
ЛИПОЕВАЯ КИСЛОТА (I) (тиоктовая кислота)
C8H14O2S2, мол. в. 206,33 — кристаллы, т. пл. 59—60°
(рацемич. Л. к.); выделенный из природных источни-
ков (печень крупного рогатого скота) изомер кристал-
лизуется в виде желтых пластинок, т. пл. 47,5° (из
гексана), [а]|§ = +96,7° (бензол); т. пл. (+)-Л. К.,
полученной синтетически, 49—50°, [а]р = +113°.
Л. к. обладает характерным пиком поглощения при
335 ммк, умеренно растворима в воде и хорошо рас-
творима в большинстве органич. растворителей.
Л. к. легко восстанавливается с образованием ди-
гидролипоевой к-ты (II):
Q(CH2)*COOH i^S-ICHaliCOOH
-2Н HS SH
1 II
Этот процесс в биологич. системах осуществляется
ферментативно. При восстановлении (+)-Л. к.
образуется биологически активная (—)-дигидроли-
поевая к-та, [a]^J = —14,5° (бензол). Кислород
и в особенности перекиси окисляют Л. к. до сульф-
оксида (т. наз. Р-Л. к.). (+)-Л.к. широко распростра-
нена в растениях, животных и микроорганизмах.
В клетках (-J-) -Л. к. прочно связана с белками ацил-
амидной связью с е-аминогруппой лизина на участке
975
ЛИПОИДЫ — ЛИПОПРОТЕИДЫ
976
пептидной цепи с такой последовательностью амино-
кислотных остатков: аланин-лизин-аспарагиноваяк-та.
(+)-Л. к. может быть выделена из печени крупного
рогатого скота путем гидролиза разб. H2SO4 с после-
дующей экстракцией органич. растворителями и очист-
кой на А12О3 (из 10 т исходного сырья получают ок.
30 мг кристаллич. продукта). Синтетически рацеми-
ческая и оптически активная Л. к. могут быть полу-
чены, напр., из СН2=СН2 и С1СО(СН2)4СООС2Н6
(выход до 36%).
(+)-Л. к. играет важную роль в биохим. превра-
щениях, протекающих в живой клетке, функциони-
руя в качестве кофермента систем, осуществляющих
окислительное декарбоксилирование а-кетокислот. Роль
Л. к. в этих системах состоит в промежуточном пере-
носе водорода и ацильных остатков. Л. к., по-ви-
димому, играет также важную роль в процессе фото-
синтеза.
Лит.: Нейландс Дж., Штумпф П., Очерки по
химии ферментов, пер. с англ., М., 1958, с. 211; The enzymes,
ed. by P. D. Boyer [a. o], v. 3, N.Y.—L., 1960, p. 195; Methods
In enzymology, ed. by S. P. Colowlck, N. O. Kaplan, v. 3,
N. Y., 1957, p. 941. В. Б. Спиричев.
ЛИПОИДЫ — см. Липиды.
ЛИПОКСИДАЗА (каротиноксидаза) —' фермент,
катализирующий окисление нек-рых ненасыщенных
жирных к-т (линолевой, линоленовой, арахидоновой)
молекулярным кислородом с образованием гидропере-
кисей этих к-т, к-рые затем распадаются на эпоксиды,
альдегиды, кетоны и др. продукты окисления. Дейст-
вие Л. строго специфично и направлено лишь на не-
насыщенные жирные к-ты с двумя или более двойными
связями в молекуле, причем эти двойные связи должны
быть разделены одной СН2-группой и иметь цис-
конфигурацию. Перекиси, образующиеся в резуль-
тате действия Л., могут вызывать вторичное окисление
нек-рых соединений, не являющихся истинными
субстратами Л., в частности полифенолов, 0-каротина
и др. пигментов.
Л. широко распространена в растительном мире,
особенно высоко ее содержание в семенах бобовых
растений, в частности сои. Л. соевых бобов, получен-
ная в кристаллич. состоянии, представляет собой
белок с мол. в. 102400; изоэлектрич. точка р/ 5,4;
практически нерастворима в воде, хорошо раство-
ряется в разб. солевых р-рах, при диализе к-рых
против воды вновь выпадает в осадок. 1 моль Л. ка-
тализирует окисление 360 молей линолевой к-ты
в секунду. Михаэлиса константа для линолеата Na
2,0 • 10-5 Л7; оптимум действия Л. при pH 9,0 (30°,
парциальное давление О2 — 160 мм). Окисле-
ние ненасыщенных жирных кислот Л. подавляется
такими антиоксидантами, как пирогаллол, гидрохи-
нон, а-токоферол (в частности, пирогаллол в концент-
рации 1 • 10-4 М подавляет активность Л. на 100%,
а а-токоферол в концентрации 2 • 10-4 М — на 40%).
Ингибиторами Л. являются такие жирные к-ты, как
олеиновая, элаидолинолевая и др. Активность Л.
определяют манометрически — по количеству погло-
щаемого в час на 1 мг Л., а также иодометрически —
по количеству J2, выделяющегося из K.J под действием
перекисей, образующихся в результате реакции.
Один из методов определения Л. основан на вторич-
ной реакции окисления 0-каротина. На способности
Л. окислять некоторые пигменты основано примене-
ние соевой муки, содержащей активную Л., для
отбеливания нек-рых пищевых продуктов, в част-
ности теста.
Лит. см. при ст. Ксактинохсидаза. В. В. Спиричев.
ЛИПОПРОТЕИДЫ (липопротеины) — высокомо-
лекулярные соединения, липидно-белковые комп-
лексы, по растворимости обладающие свойствами
белков. Л. широко распространены как в животных,
так и в растительных организмах. Отдельные пред-
ставители этой большой группы веществ различаются
по физич. свойствам (мол. вес, плотность, раствори-
мость и т. д.) и химич. строению липидных и белковых
компонентов. Липидная часть Л. может быть пред-
ставлена нейтральными жирами, жирными к-тами,
фосфолипидами, холестерином и его эфирами, каро-
тиноидами и др. или, чаще всего, комбинацией раз-
личных липидных веществ. Относительное содержание
обоих компонентов у разных Л. весьма различно;
нек-рые Л. содержат до 75% липидов.
Характер связи между липидным и белковым ком-
понентами Л. может быть различен. В одних случаях
липидные молекулы (обычно наиболее полярные ли-
пиды — жирные к-ты, лизофосфатиды, нек-рые сте-
роиды) связаны с определенными функциональными
группами белковой молекулы. Наиболее распростра-
ненной, по-видимому, является такая форма связи,
когда белок соединяется с целым комплексом липид-
ных молекул, образующих мицеллярные агрегаты
или пленочные структуры (на границе раздела сред)
с определенной ориентацией своих полярных и непо-
лярных групп. Наименее полярные липиды (например,
триглицериды) образуют сферич. капли, покрытые
белковой оболочкой.
Л. очень чувствительны к действию различных
факторов, денатурирующих как белковый, так и ли-
пидный компоненты Л. (нагревание, замораживание,
высушивание, резко кислая или резко щелочная
реакция среды, протеолитич. и липолитич. ферменты,
органич. растворители и т. д.); сравнительно легко под-
вергаются аутоксидации.
В животном организме Л. обнаруживаются во всех
тканях. Наиболее полно изучены Л. плазмы крови
млекопитающих. Были выделены две фракции Л.
плазмы, названные на основании их электрофоретич.
свойств а4-Л. и 04-Л. и отличающиеся помол, весу,
содержанию и составу липидного компонента, рас-
творимости в воде и в водноспиртовых смесях. а4-Л.
плазмы растворимы в воде в отсутствии солей, имеют
мол. вес порядка 200 000, молекула эллипсоидной
формы, размер 300 X 50 А. 04-Л. плазмы в изоэлект-
рич. точке нерастворимы в воде в отсутствии солей,
мол. вес безводного белка 1 300000 (по другим данным,
порядка 3 Q00 000), молекула сферич. формы диа-
метром 185 А. Каждая из этих фракций путем электро-
фореза на бумаге может быть подразделена на ряд
еще более мелких групп.
Наиболее распространенным способом выделения
и разделения Л. плазмы крови является метод ультра-
центрифугирования в средах с определенной (одно-
родной или градиентной) плотностью. При этом
получаются фракции Л., характеризующиеся раз-
личной плотностью и значительно отличающиеся
друг от друга как по составу своих липидных компо-
нентов, так и по характеру белковой части. Л. с вы-
сокой плотностью (1,149) обладают электрофоретич.
подвижностью ai-глобулинов, содержат 21% фосфо-
липидов, 57% белка, 5% триглицеридов, 17% хо-
лестерина. Л. с низкой плотностью (1,035) обладают
электрофоретич. подвижностью 04-глобулинов, со-
держат 22% фосфолипидов, 21% белка, 10% тригли-
церидов и 46% холестерина. В основном выделенная
из плазмы крови ультрацентрифугированием фракция
Л. с высокой плотностью соответствует по своим свой-
ствам а4-Л., с низкой плотностью — 01-Л.
Л. входят гл. обр. в состав различных структур
клеток тканей (ядер, митохондрий, микросом) и ча-
стично находятся в растворенном виде в цитоплазме.
Принимая участие в образовании мембран, Л. играют
важную роль в явлениях проницаемости и в переносе
ионов и различных органич. соединений. Примерами
пластинчатых структур, построенных с участием Л.,
служат миелиновая оболочка нервов, хлоропласты
977
ЛИТИЙ
978
растений и фоторецепторные элементы сетчатки глаза.
Л. играют важную роль в транспортировке с кровью
нерастворимых в воде веществ.
Лит.: Троицкий Г. В., Липопротеиды плазмы крови
и некоторых тканей, в сб.: Успехи биологической химии, т. 3,
М., 1958; Lipoproteins, Discuss. Faraday Soc., 1949, К» 6; The
lipoproteins. Methods and clinical significance. Ed. F. Hom-
burger, P. Bernfeld, N. Y., 1958; Deuel H. J., The lipids,
their chemistry and biochemistry, v. 2, N. Y., 1955; Hana-
han D. J., Llpide chemistry, N. Y.—L., [i960].
Д. А. Четвериков.
JJIITIlfl (Lithium) Li — химич. элемент I гр. пе-
риодич. системы Менделеева, п. н. 3, ат. в. 6,939.
Природный Л. состоит из двух стабильных изотопов
с м. ч. 6 (7,52%) и 7 (92,48%). Искусственные радиоак-
тивные изотопы Л.: Li8 (Ti/2 = 0,841 сек.) и Li9
(Ti/2 = 0,168 сек.). Важной для технич. использова-
ния особенностью Л. является резкое различие в зна-
чениях поперечного сечения поглощения тепловых
нейтронов (о) его изотопами. Значение а (в барнах):
Li® 910, Li7 0,033 (при 67 для естественной смеси изо-
топов). Электронная конфигурация атома Л.: ls22sx.
Энергии ионизации (в ае): Li® -* Li+ —> Li2+ —>• Li3+
соответственно равны 5,390; 75,619; 122,419.
Л. был открыт в 1817 А. Арфведсоном в минерале петалите,
металлич. Л. впервые получен в 1818 Г. Дзви разложением
окиси Л. электрич. током. В значительных количествах Л. был
получен в 1854 Р. Бунзеном и О. Матиосенои электролизом
расплавленного хлорида Л.
Л. достаточно широко распространен в природе.
Его содержание в земной коре составляет 6,5 • 10*3 вес.
% — более, чем Hg, Ag, Au, Sn, Pb, As, Sb, Bi.
JI. — типично литофильный элемент.
Л. входит в состав многих горных пород, содержится
в минеральных источниках, морской, озерной и подземных
водах, в каменных углях, почве, в животных и растительных
организмах. Наибольшая концентрация Л. наблюдается в руд-
ных месторождениях, генетически связанных с поздними ста-
диями дифференциации магмы. Важнейшие Промышленные
месторождения Л. относятся к гранитным пегматитам, в к-рых
Л. тесно аесоциирует с натриеи. Меньшее значение имеют
образования, для к-рых характерна ассоциация Л. с фтором.
Месторождения руд Л. часто комплексные и содержат, помимо
минералов Л., минералы Cs, Be, Sn, Nb, Ta, W и др., а также
драгоценные камни. Ионный радиус L1+ (см. ниже) близок
не к Na+, а к Mg2+, Fe2+ и А13+ и в минералах Л. играет роль
щелочноземельного элемента. Изоморфизм Л. с Mg, Fe, реже
с А1, объясняет вхождение Л. в состав многих магнезиально-
железистых минералов. Кроме того, Л. часто является спут-
ником калия и присутствует во многих полевых шпатах, лей-
ците и др. минералах.
Л. обнаружен более чем в 150 минералах, из них
собственных минералов Л. ок. 30. Большая часть
их — силикаты и фосфаты (преобладают первые).
Промышленную ценность имеют (в порядке убываю-
щего значения): сподумен, лепидолит, петалит, амб-
лигонит и циннвальдит.
Сподумен ЫА1[81гО0] — силикат Л. и алюминия.
Теоретич. содержание окиси Л. 8,1%, фактич. 6,0—7,5%,
т. к. в результате гипергенных процессов часть L1 замещается
Na. Нек-рые ив замещающих L1 и А1 элементов придаютсподу-
мену красивую окраску, и его отдельные разновидности счи-
таются драгоценными камнями. Ценной примесью в сподумене
является галлий (0,03—0,1%). Твердость сподумена 6—7,
плотн. 3,13—3,20. Характерен необратимый переход споду-
мена ив его природной моноклинной (а) модификации в тетра-
гональную (Р) при 950—1100°. Монотропный а—»₽ переход
сопровождается резким уменьшением плотности сподумена до
2,4 и, соответственно, увеличением объема (на 24%), что при-
водит к сильному измельчению минерала, позволяющему легко
отделить его от вмещающей породы. Это ценное свойство
используется при обогащении сподуменовых руд (см. ниже).
Лепидолит KLli.sAlbsISlaAlOiohF, ОН)2 — алюмо-
силикат Л. и калия. Химич, состав непостоянный; содержание
окиси Л. 1,2—5,9%. Очень ценными примесями являются руби-
дий (иногда до 3,7% Rb,O) и цезий (до 1,5% Cs2O).
Петалит Ll[AlSl40ie] или (LI, Na) [AlSi4O)0]— алю-
мосиликат Л. Теоретич. Содержание окиси Л. 4,9%, фактич.
не более 3—4%. При 680° разлагается на Р-сподумен и S1O2,
что может быть использовано в технологии его переработки
на соединения Л.
Амблигонит L1A1IPO4](F,OH) — фосфат Л. и алю-
миния. Теоретич. содержание окиси Л. 10,1 %, фактич. 7—9,5%.
Циннвальдит KLl(Fe, Mg)Al[Si3A10iol(F,ОН)2 —
промежуточный член непрерывного изоморфного ряда биотит—
лепидолит. Химич, состав весьма непостоянный. Бедные желе-
зом разновидности наз. криофилитом, богатые — железный
лепидолитом. Содержание окиси Л. 1—5%. ценными при-
месями могут быть рубидий и цезий.
Важнейшие промышленные месторождения мине-
ралов Л. находятся в Канаде, США, Ю.-Зап. Африке,
Ю. Родезии, Испании, Швеции, Бразилии, Австра-
лии, СССР.
Вынос Л. из минералов водами приводит к образо-
ванию осадочных месторождений Л. Эти месторожде-
ния перспективные, хотя промышленное значение
имеет пока рапа высохшего озера Сирлс в Калифорнии
(США). Рапа содержит 0,02% хлорида Л., большие
количества солей Na, а также соли К и В. Из озерной
рапы добывается примерно столько же Л., сколько из
амблигонита и циннвальдита вместе взятых.
Физические и химические свойства. Компактный
Л. — серебристо-белый металл, быстро тускнеющий
на воздухе вследствие образования темно-серого на-
лета, состоящего из нитрида Л. и его окиси. Л. —
самый легкий металл; плотп. Л. в твердом состоянии
0,534 (20°), в расплавленном 0,507 (200°) и 0,441
(1000°). При обычной темп-ре устойчива кубич.
объемноцентрированная решетка, а — 3,5089 А (20°),
ат. радиус 1,57 А, ионный радиус Li+ 0,68 А, ниже
—195° — гексагональная плотноупакованная, а =
= 3,111 А, с = 5,093 А. Т. пл. 179°, т. кип. 1370°.
Вязкость Л. (спуаз): 0,5918 (183,4°) и 0,4548 (285,5°).
Летучесть Л. на воздухе относительно невелика;
давление пара (мм рт. ст.): 1(745°), 100 (1084°),
200 (1156°), 400 (1252°) и 760 (1370°), в вакууме
0,04 мм рт. ст. Л. перегоняется выше 600°. Теплота
плавления 103,2 кал/г, теплота испарения 4680—
5275 кал/г-, уд. теплоемкость 0,784—0,905 кал/г • град
(0—100°); среднее значение 0,790. Термич. коэфф,
линейного расширения 5,6 • 10~5. Теплоемкость в жид-
ком состоянии 0,975 кал/г • град; теплоемкость паров
0,714 кал/г - град. Теплопроводность 0,17 кал/см-
• сек • град (0—100°), уд. электрич. сопротивление
(мком- см): 1,149 (—192°); 8,55 (0°); 9,29 (20°);
12,269 (100°); температурный коэфф, электросопротив-
ления между 4,58- 10*® и 4,35- 10“3 (0-—100°); сред-
нее значение 4,50 • 10*®. Л. парамагнитен, уд. маг-
нитная восприимчивость (18—20°) +0,5- 10“8. Л. —
весьма пластичный и вязкий металл, легко протяги-
вается в проволоку. Твердость по Моосу 0,6 (тверже
др. щелочных металлов), легко режется ножом;
давление истечения 1,7 кГ/мм2 (15—20°). Модуль
упругости 500 кГ/мм2; предел прочности при растя-
жении 11,8 кГ/мм2, относительное удлинение 50—70%.
Л. стоит первым в ряду напряжений, его нормаль-
ный потенциал — 3,02 в; электродный потенциал Л.
в расплаве —2,1 в. Пары Л. окрашивают пламя
в карминово-красный цвет. Во всех известных со-
единениях Л. одновалентен. Ион Л. характеризуется
наибольшим коэфф, поляризации (1,64) и наименьшим
коэфф, поляризуемости (0,075) из всех щелочных
элементов, и это определяет особое положение Л.
среди них и его сходство со щелочноземельными
элементами (особенно кальцием) и магнием, ионный
радиус к-рого (0,74 А) лишь незначительно отли-
чается от ионного радиуса Л. При непосредственвом
взаимодействии с кислородом или при нагревании
(горит голубым пламенем) Л. образует окись (см.
Лития окись и гидроокись). Образование перекисных
соединений при окислении не характерно. Пере-
кись Л. Li2O2 получается косвенно в виде перокси-
гидрата Li2O2 • Н2О2- ЗН2О при действии Н2О2 на
насыщенный спиртовой р-р гидроокиси Л. При вы-
сушивании пероксигидрата в вакууме над Р2О5
получается безводная Li2O2. С водой Л. реагирует
с образованием LiOH и Н2; реакция менее энергична,
чем у др. щелочных металлов, но при недостаточном
охлаждении может произойти воспламенение. Мине-
ральные кислоты энергично взаимодействуют с Л.
с образованием соответствующих солей. В водном
р-ре ион Л. гидратирован сильнее иовов др. щелочных
079
ЛИТИЯ
980
влементов и имеет наибольший радиус и наименьшую
подвижность.
Л. непосредственно соединяется с галогенами с об-
разованием солей галогеноводородных к-т (см. Лития
галогениды}. Известны также соли Л., образованные
кислородсодержащими к-тами галогенов, — LiClO,
LiBrO, LiC102, LiBrO2, LiC103, LiBrO3, LiJO3,
LiC104, LiJO4. При взаимодействии нагретого Л.
с парами серы или при нагревании Л. и серы выше
темп-ры плавления получается растворимый в воде
сульфид Л. Li2S. Кислород и др. окислители при
300° легко окисляют Li2S до лития сульфата. При
нагревании Л. реагирует с Н2, образуя лития гидрид.
Для Л. характерно также образование двойных гид-
ридов (см. Лития алюмогидрид). Л. медленно реаги-
рует с азотом уже при комнатной темп-ре с образо-
ванием нитрида Li3N; реакция резко усиливается
при 250°. В токе сухого азота реакция протекает
быстро с полным переходом Л. в нитрид, а при на-
гревании — с воспламенением. Нитрид Л. плавится
при 845° и является реакционноспособным соедине-
нием: при 800° разъедает Fe, Си, Ni, Pt, кварц и фар-
фор. Растворяясь в жидком аммиаке, Л. образует
лития амид (р-р при этом окрашивается в синий
цвет), к-рый в процессе термич. разложения дает
(с хорошим выходом) имид Li2NH — малостойкое
соединение, изоморфное Li2O.
Кислородсодержащими соединениями Л. с азотом
являются лития нитрат и н и т р и т LiNO2, к-рый
может быть получен восстановлением нитрата и пред-
ставляет собой легкорастворимое в воде и абс. спирте
соединение, разлагающееся при 185° (до плавления)
и кристаллизующееся из р-ров в виде LiNO2- Н2О.
Известен и LiNO2- !/2Н2О. С фосфором Л. непо-
средственно не реагирует; фосфид переменного состава
(LinPm) образуется при взаимодействии карбида Л.
с парами фосфора. Л. дает многочисленные средние
и кислые соли с различными фосфорными, к-тами;
важнейшим соединением является ортофосфат
Л. Li3PO4 — одна из наиболее труднорастворимых
солей Л., имеющая значение для извлечения Л. из
р-ров небольших концентраций (маточников литие-
вого произ-ва); при 0° в 100 г воды растворяется 0,022 г
Li3PO4. При нормальной темп-ре из водных р-ров
выделяется Ы3РО4 • 2Н2О; длительной сушкой при
60° его переводят в полугидрат; безводный фосфат Л.
может быть получен прокаливанием водной соли
при 120°.
При нагревании Л. легко реагирует с углеродом
с образованием карбида Li2C2 — производного
ацетилена; в больших масштабах Li2C2 может быт!
получен восстановлением карбоната Л. углем в элект-
ропечи или непосредственно из элементов в вакууме
при высокой темп-ре. Карбид Л. — сильный восста-
новитель, диссоциирующий при нагревании на Л.
и графит. При нагревании избытка Л. с кремнием
образуется силицид Л., к-рому обычно припи-
сывают ф-лу LieSi2, хотя вопрос о составе силицида
Л. и о числе и природе фаз в системе Li—Si остается
неясным. При 600° в вакууме силицид Л. разлагается
на исходные компоненты; при темп-ре красного кале-
ния силицид Л. легко восстанавливает Fe, Мп и А1
из их окислов. Л. образует ряд кремнекислых со-
единений — моносиликат (Li2O • SiO2) и полисили-
каты (nLi2O • mSiO2); алюминийсодержащие си-
ликаты Л. (двойные силикаты и алюмосиликаты)
встречаются в природе в виде минералов.
Характерной для Л. является его способность
образовывать металлоорганич. соединения, что опре-
деляет большую роль Л. в современном органич.
синтезе (см. Литийорганические соединения).
Л. является компонентом многих сплавов.
С нек-рыми металлами (Mg, Zn, Al) он образует твер-
дые р-ры значительной концентрации, ео многими
металлами (Al, Zn, Mg, Cd, Hg, TI, Pb, Bi, Ag, Sn)—
интерметаллиды (напр., AgLi, LiHg, LiMg2, LiAl
и мн. др.). Последние часто обладают большой твер-
достью и тугоплавкостью, незначительно изменяются
на воздухе; нек-рые из них обладают константами,
характерными для полупроводников. В настоящее
время изучены бинарные системы, образованные Л.
со щелочнымиметаллами,Си, Ag, Au, Zn, Cd, Hg, Mg,
Ca, Ba, Ga, In, TI, Al, Si, Ge, Sn, Pb, As, Sb, Bi, Se,
Те и др. — более чем с 30 элементами. Технич. зна-
чение получили сплавы Л. с Al, Mg, Pb, Zn, Си и Са.
Содержание Л. в большинстве сплавов очень невелико,
но обычно добавка ок. 1% Li улучшает свойства ос-
новного металла, сообщая ему вязкость или твер-
дость; предел прочности и упругие свойства сплавов,
содержащих Л., имеют высокие значения. Сплавы
с алюминием при содержании до 2,94% Li обладают
высокой пластичностью и повышенной устойчивостью
против коррозии. Наибольший интерес представляет
сплав склерон (12% Zn, 2% Си, 0,5—1% Мп, 0,5% Fe,
0,5% Si, 0,1% Li, остальное — Al); его предел проч-
ности, упругость и твердость выше, чем у сплавов
типа дюралюминия, и по своим физич. свойствам он
подобен мягкой стали илп латуни. Такими же свойст-
вами обладает аэрон (4% Си, 0,1% Li). Эти сплавы
применяют для изготовления деталей автомашин
и основных рам трамвайных и ж.-д. вагонов. Нек-рые
сплавы Л. с алюминием сохраняют свои важнейшие
свойства при относительно высоких темп-рах (250°)
и считаются перспективными в авиатехнике.
Присадки небольших количеств Л. к магнию уве-
личивают его прочность, а при содержании 1% Li
магний приобретает хорошие литейные свойства
и повышенную коррозионную стойкость на воздухе.
Сплавы магния, содержащие ок. 10% Li, имеют ку-
бич. объемноцентрированную решетку и характе-
ризуются высокими физическими свойствами; к тому
же присутствие Л. облегчает процессы прессования
и прокатки при обработке деталей, изготовляемых
из этих сплавов, и понижает их плотность. Свинцу Л.
придает более мелкозернистую структуру, повышает
твердость, замедляет его рекристаллизацию. При-
садка 0,05 % Li заметно улучшает механич. и физич.
свойства свинцовых баббитов (увеличиваются вяз-
кость и твердость, сопротивление разрыву и модуль
упругости). Сплавы Л. и свинца (с добавками Cd
и Sb) применяют для изготовления оболочек электрич.
кабелей. В сплавах с цинком Л. (0,005%) улучшает
структуру; добавка 0,6% Li в сплавы цинка для литья
под давлением снижает пористость. Л. улучшает
электропроводность и механич. свойства меди. Вы-
сокопроводящим является сплав 2%Li и 98% Си.
Сплавы Л. с кальцием представляют ценность в ме-
таллургии Си, Pb, Ni и Fe, т. к. применяются в про-
цессах получения этих металлов и их сплавов. Ис-
пользуют два сплава: 50% Li -г 50%Са и 30%Li
+ 70% Са. Добавки сплавов Li — Са к кадмиевым,
кремнистым и оловянным бронзам применяют для
улучшения их механич. свойств п повышения электро-
проводности.
Из тройных систем изучались А1—Mg—Li, Al—Zn—
Li, Pb—Na—Li, Mg—Ag—Li и Pb—Ca—Li. Нек-рые
соответствующие им сплавы уже нашли применение
в технике. Напр., сплавы Al, Mg и Li под общим
названием «магналий» ценятся как коррозионноустой-
чивые материалы с хорошими физич. свойствами;
сплав Pb, Na и Li обладает высокими антифрикцион-
ными качествами и давно получил известность в
произ-ве т. наз. банметалла (безоловянистого сплава
для подшипников). Несмотря на небольшое содер-
жание Л. в этом сплаве (0,04% Li, 0,73% Са, 0,66%
Na, 0,03% К, 0,2% А1, остальное — РЬ), он снижает
981
ЛИТИИ
982
коэфф, трения и устраняет «задирание» подшипников.
Сплавы системы РЬ—Са—Li приобрели значение в
связи с проблемой замены сплавов Sb—Pb в аккуму-
ляторах и полиграфия, пром-сти. Представляют ин-
терес и сплавы Ag, Си и Li (0,002—3% Li), применяе-
мые в ювелирном деле и для изготовления электро-
контактов и серебряных припоев. Последние (до
0,25% Li) лучше обычных в смысле жидкотекучести,
смачивающей способности, предела прочности и удар-
ной вязкости. Они представляют большую ценность
для пайки металлич. изделий, содержащих окисляю-
щиеся компоненты (Сг, Мо, W).
Аналитическое определение. Ка-
чественно Л. обнаруживается по окрашиванию пла-
мени горелки его летучими солями и по наиболее
четко выраженным спектральным линиям Л.: 6707,84
А и 6103,64 А. Чувствительность определения Л.
спектральным методом составляет 1,25- 10 6 мг Li.
Для определения Л. в присутствии др. щелочных
элементов может быть использована высокочувстви-
тельная микрокристаллоскопич. реакция образования
крупных желтых октаэдрич. кристаллов при действии
на каплю р-ра соли Л. капли 15%-ного р-ра уротро-
пина и капли 15%-ного р-ра красной кровяной соли.
Для количественного определения Л. большое зна-
чение имеют весовые методы, предполагающие пред-
варительное отделение Л. от др. щелочных элементов.
Чаще всего Л. отделяют, используя хорошую раство-
римость его хлорида во многих органич. раствори-
телях и в конц. соляной к-те, в к-рых хлориды Na
и К. весьма плохо растворимы. Процесс отделения Л.
сводится к экстрагированию LiCl из смеси с др.
хлоридами с помощью амилового спирта, пиридина,
ацетона и др. растворителей. Хорошие результаты
дает при экстракционном разделении н-пропиловый
спирт, к-рый после насыщения его сухим газообразным
НС1 оказывается селективным экстрагентом для хло-
рида Л. После отделения хлорид Л. переводится
в сульфат, который прокаливается и взвешивается.
Другими, но менее пригодными, весовыми формами
для определения Л. являются фторид (см. Лития га-
логениды), фосфат Li3PO4 и др., к-рые, в отличие от
соответствующих соединений Na и К., трудно раство-
римы в воде.
Из объемных методов определения Л. лучшим
является перйодатный, основанный на осаждении Л.
перйодатом калия в щелочной среде, разложении
осадка смесью солярой и серной к-т и титровании
выделившегося иода Na2S2O3 в присутствии KJ.
Другие щелочные элементы определению не мешают.
Меньшее значение имеют колориметрич. методы опре-
деления Л., т. к. он почти не дает окрашенных соеди-
нений. Прямое колориметрич. определение Л. воз-
можно на основе реакции с тороном, косвенное ос-
новано на колориметрич. определении железа после
осаждения труднорастворимого феррипериодата со-
става LiK.FeJO6. В последнее время большое рас-
пространение приобрел пламеннофотометрич. метод
определения Л. Однако определение Л. продолжает
оставаться одной из' труднейших задач аналитич.
химии, т. к. вполне селективного реагента для Л.
не найдено.
Получение. Руды Л. содержат 0,25—3,0% окиси
Л. и перед переработкой должны быть обогащены
с целью получения концентратов промышленных ми-
нералов. Для обогащения применяются: ручная
рудоразборка, магнитная сепарация, разделение в тя-
желых суспензиях, декрипитация (термич. обогаще-
ние, использующее а —. 0 переход сподумена при
950—1100°, сопровождаемый увеличением объема
на 24% и след, сильным измельчением минерала)
и флотация, получившая наибольшее значение. Со-
временные процессы обогащения, особенно комплекс-
ных руд Л., почти всегда комбинированные. После
обогащения получаются концентраты с устойчивым
содержанием окиси Л. (%): 4—6 (сподуменовые),
6—8 (амблигонитовые), 3—4 (лепидолитовые и пета-
литовые), 3—3,5 (циннвальдитовые). Последующая
гидрометаллургии, переработка концентратов сили-
катных минералов может быть осуществлена самыми
различными методами. Для сподумена важнейшими
являются: известковый, сульфатный и сернокислот-
ный. В основе первого лежит разложение известняком
при 1150—1200°: Li2O • А12О3 • 4SiO2 % 8СаСО3 =
= Li2O • А12О3 % 4 (2СаО • SiO2) + 8 СО2. При вы-
щелачивании спека водой в присутствии избытка
извести происходит разложение алюмината Л. с об-
разованием гидроокиси Л.: Li2O • А12О3 + Са(ОН)2 =
= 2LiOH СаО • А12О3. Метод успешно применяется
за рубежом и при промышленной переработке лепи-
долита.
По сульфатному методу концентрат сподумена
вскрывается при 1050—1100° с получением в опеке
сульфата Л. (см. Лития сульфат) и алюмосиликата
калия (лейцита); Li2O • А12О3 • 4SiO2 + K2SO4 =
= Li2SO4 + К2О • А12О3 • 4SiO2. После выщелачива-
ния сульфата Л. из его раствора содой при 80—90°
осаждается карбонат Л.: Li2SO4 + Na2CO3 = Li2CO3H>
+ Na2SO4. Метод приложим и к переработке лепидо-
лита, циннвальдита и может дать хорошие результаты
при извлечении Л. из петалита. Сернокислотный метод
также рассчитан на получение раствора сульфата Л.
и последующее получение в качестве первичного про-
дукта карбоната Л., но вскрытие в данном случае
проводится H2SO4 при 250—300°. Реакция применима
только для р-сподумена: P-Li2O • А12О3 • 4SiO2
+ H2SO4 = Li2SO4 Н2О • А12О3 • 4SiO2. Метод, од-
нако, используется не только для переработки кон-
центратов сподумена, но и его руд, если содержание
в них окиси Л. не менее 1%. При условии успешного
решения аппаратурных вопросов следует считать пер-
спективным метод возгонки хлорида Л. из споду-
мена после разложения его смесью СаСО3 и СаС12
при 1200°. В отличие от силикатных минералов, фос-
фатные минералы Л. легко разлагаются кислотами.
Однако современными способами предусматривается
разложение их смесью гипса и извести при 950—1050°
с последующим выщелачиванием спеков и. осаждением
из р-ров карбоната Л.
Основой промышленного произ-ва металлич. Л.
является электролиз расплавленной смеси хлоридов
Л. и калия. Электролиз проводят при 400—450°,
обычно при весовом соотношении компонентов смеси
1:1, что близко к эвтектич. составу. Электролизные
железные ванны футеруются изнутри графитом, магне-
зитом, алундом или обожженным тальком; анодом
служат графитовые, а катодом — железные стержни.
Электролиз позволяет получать Л. с выходом по току
95%, по металлу — 98%; при высоком качестве LiCl,
а также футеровки Л. загрязняется гл. обр. только
натрием (0,5—0,6%). Обычно черновой металлич.
Л. содержит механич. включения и примеси (помимо
натрия) К, Mg, Са, Al, Si, N, Fe и др., из к-рых
наиболее трудно удаляется магний. Включения уда-
ляются переплавкой под слоем вазелинового или
парафинового масла (или в атмосфере инертного газа)
в железных тиглях нли (для получения весьма чи-
стого Л.) тиглях из ZrO2, футерованных изнутри фто-
ридом Л. Очистку от примесей осуществляют в ваку-
уме (ок. 10~5 мм рт. ст.) при 600—800° и темп-ре
конденсации 340—420°. В этих условиях можно полу-
чать дистиллят, содержащий 0,001—0,003% натрия
с выходом 85—90%. Рафинирование при пониженном
давлении имеет в настоящее время большое значение,
т. к. значительное различие в упругости паров Л.
и большинства примесей благоприятствует почти пол-
983
ЛИТИЙ — ЛИТИЙОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
984
ному отделению от них Л. Дистилляцию возможно
совмещать с ректификацией или использовать для
очистки Л. зонную плавку.
Получение сплавов Л. осуществляется также элек-
тролизом солевых расплавов, компонентами к-рых
являются хлориды Л. и калия, если легируются тяже-
лые металлы (в этом случае катодом служит распла-
вленный легируемый металл, если он легкоплавкий,
или применяются дополнительные растворимые аноды
из легируемого металла, если он тугоплавкий), или
хлориды Л. и легируемых металлов, если легируются
легкоплавкие легкие металлы (таким путем получают,
напр., сплавы Л. с кальцием).
В настоящее время большое внимание уделяется
металлотермич. методам получения Л. Хорошие ре-
зультаты достигаются на смеси окисей Л. и кальция
при использовании в качестве восстановителей фер-
росилиция или алюминия; в этом случае вакуумный
процесс позволяет получить Л., содержащий сотые
доли % Si, Al и Са, с выходом 80—85%; с выходом
чистого Л. 90% протекает восстановление Li2O • А12О3
алюминием в вакууме при 1150—1200°. Металлотер-
мия. метод пригоден и для получения сплавов Л.'
(напр., с магнием), но возможности в данном случае
невелики, т. к. число подходящих восстановителей
ограничено. Получение металлич. Л. восстановлением
продуктов -химич. переработки его минералов пока не
может конкурировать с электролизом расплавленных
солей. Однако металлотермия, вакуумные процессы
восстановления соединений Л. представляют особый
интерес для решения проблемы прямого получения Л.
из его руд.
Металлич. Л. выпускается в виде слитков, стерж-
ней, гранул или проволоки и сохраняется в герметич-
ной таре под слоем вазелинового или парафинового
масла. Для длительного хранения герметично запрес-
совывается в тонкостенные оболочки из меди или алю-
миния.
Применение. Важнейшей областью применения Л.
является ядерная энергетика. Изотоп Li6 — единст-
венный промышленный источник для произ-ва трития
по реакции: 3LiG + on1 = jH3 + 2Не4; он применяется
также при изготовлении регулирующих стержней
в системе защиты реакторов благодаря большому
сечению захвата тепловых нейтронов. Высокая тепло-
емкость, большой диапазон жидкого состояния и вы-
сокая теплопроводность при малой плотности и вяз-
кости жидкого Л. являются благоприятным сочета-
нием свойств для использования Л. (в виде изотопа Li’
с малым сечением захвата тепловых нейтронов) в ка-
честве теплоносителя в урановых реакторах. Расплав-
ленный Li’F применяется как растворитель соедине-
ний U и Th в гомогенных реакторах.
Крупнейшим потребителем соединений Л. является
силикатная пром-сть, в к-рой используются фосфат-
ные и силикатные минералы Л., специально полу-
чаемые соединения Л. (алюминат, кобальтит, манга-
нит, молибдат, силикат, титанат, цирконат, метабо-
рат и др.), а также фторид и имеющий наибольшее
применение в произ-ве стекла и керамики лития
карбонат. В черной металлургии Л., его соединения
и сплавы (особенно сплав Li — Са) находят широкое
применение для раскисления, легирования и модифи-
цирования многих марок черных сплавов. Небольшие
добавки Л. улучшают физич. свойства чугуна и по-
вышают его литейные качества — благодаря сродству
кН, N, О Л. удаляет эти газы. В цветной металлур-
гии Л. обрабатывают сплавы для получения хорошей
структуры, пластичности и высокого предела проч-
ности. Л., реагируя с водородом, окислами и сульфи-
дами. образует нерастворимые в металлах легко флю-
сующие соединения с низкой плотностью и низкой
темп-рой плавления и, в отличие от многих добавок,
не оставляет в металлах вредных примесей. Поэтому
он оказывается незаменимым для раскисления, дега-
зации и десульфурации расплавленных металлов и
сплавов на основе Си, Zn, Pb, Mg, Al, Ni, бронз, мо-
нель-металла и др. ценных сплавов. С этой целью Л.
применяют в виде лигатур с Си, Ag, Zn, Al и Са.
В химич. пром-сти наибольшее значение Л. при-
обрел в реакции Гриньяра (вместо Mg) и реакциях
конденсации и ацетилирования (напр., при синтезе
витамина А). В диспергированном состоянии Л. при-
меняют в качестве катализатора при полимеризации
изопрена. Прочие области применения соединении Л.:
произ-вофармацевтич.и косметич. препаратов, произ-во
катализаторов и стабилизаторов пластмасс, техника
очистки газов, текстильная пром-сть (отбеливание
тканей, пропитка тканей для придания водонепрони-
цаемости), пищевая пром-сть (консервирование мяса,
изготовление напитков), с. х-во (инсектициды и фунги-
циды, стимуляторы роста растений) и др. По значи-
мости в современной технике Л. является одним из
важнейших редких элементов.
Лит.: III а м р а й Ф. И., Литий и его сплавы, М., 1952;
Требования промышленности к качеству минерального сырья.
Справочник для геологов, вып. 41 — К о г а н Б. И., Литий,
2 изд., М., 1959; Коган Б. И., Литий. Области освоенного
и возможного применения, М., 1960; Литий. Сборник перево-
дов, М., 1954, 1959 (Редкие металлы); Остроушко Ю. И.,
[и др.], Литий, его химия и технология, М., 1960; Gmelin,
8 Aufl., Syst.—Num. 20. Lithium, В., 1960; Mellor, v. 2,
L.—N. Y.—Toronto, 1952; Ullmann, 3 Aufl., Bd 11,
Munch. —B., 1960; Rare metals handbook, ed. C. A. Hampel,
L., 1961; Hansen M., Constitution of binary alloys, 2 ed.,
N. Y., 1958. В. E. Плющее.
ЛИТИЙОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ — со-
единения, содержащие связь углерод — литий. Аро-
матич. Л. с. — твердые кристаллич. продукты; али-
фатич. Л. с. (за исключением CH3Li и C2H5Li) — ассо-
циированные неперегоняющиеся жидкости, разлагаю-
щиеся при нагревании. Л. с. растворимы в углеводо-
родах (кроме CH3Li) и в эфире; с бромистым литием
и эфиром образуют комплексы типа 2C2H5Li •
• LiBr • (С2Н5)2О. Л. с. отличаются высокой реакцион-
ной способностью; в одних и тех же реакциях Л. с.
по химич. активности превосходят магнийорганические
соединения. Л. с. крайне чувствительны к кислороду,
влаге и СО2; все операции с ними проводятся в атмо-
сфере азота или аргона. Водой, спиртами, кислотами
и т. п. разлагаются: RLi + R'OH— R'OLi + RH;
с CO2 дают соли соответствующих карбоновых к-т;
RLi 4- СО2 — RCOOLi (побочно — кетоны и третич-
ные спирты).
Алифатич. Л. с. медленно расщепляют простые
эфиры, напр.:
(CgH^l^O 4- С2НзЫ —> С2Н5ОЫ С 2II, Ц- С2Н0
(CH3Li и ArLi в этой реакции неактивны); легко
расщепляются галогенами: RLi + Х2—>RX + LiX;
с галогеналкилами (особенно с иодидами) вступают
в реакцию Вюрца: RLi + R'J —> R—R' -ц LiJ. Для
Л. с. эта реакция менее характерна, чем для более
реакционноспособных натрийорганических соедине-
ний, однако она часто является побочным процессом
при получении Л. с. и во многих реакциях этих со-
единений. Взаимодействие Л. с. с карбонильными со-
единениями (альдегиды, кетоны, сложныеэфиры, хлор-
ангидриды) протекает аналогично соответствующим
реакциям магнийорганич. соединений, напр.:
RLi + R'R''CoS49>RR’R"COH
Эти реакции имеют преимущества в тех случаях,
когда RMgX реагируют с трудом или не реагируют
вовсе. Так, в отличие от uao-C3H7MgBr, w?o-C3H7Li
с изо- (С3Н7)2СО дает (мао-С3Н7)3СОН. Реакция нитри-
лов с Л. с. служит методом получения кетонов:
RCN + R'Li -»RR'C=NH RCOR'
985
ЛИТИЙОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ - ЛИТИЯ ГАЛОГЕНИДЫ
986
(бензонитрил с Л. с. дает триазины). С О-алкилгидрок-
силаминами Л. с. образуют первичные амины:
RLI + R'ONHa -► RH + R'ONHLl -У
- RNHLl + R’OLi —5 RNHs
Л. с. присоединяются по азометиновой связи азотсо-
держащих гетероциклич. соединений с последующим
отщеплением гидрида лития; в результате образуются
а-алкильные производные гетероциклич. соединений,
например
R Н R
^~^N + RLi —•- /NLi —- + L1H
Если a-положение занято, металлирование направ-
ляется в боковую цепь. Л. с. присоединяются к двой-
ной связи, особенно, если последняя активирована
соседством ароматич. групп или непредельных систем,
напр.
(С9Н9)2С =СН2-|-C4H9Li —* (С9Н8)2CL1CH2C4H9
К сопряженным системам типа дивинила Л. с. легко
присоединяются в 1,2- или в 1,4-положение; в резуль-
тате многократно повторяющихся актов присоедине-
ния реакция сопровождается полимеризацией диена.
Л. с. широко применяют для синтеза других эле-
ментоорганич. соединений, преим. в реакциях обмена.
В большинстве случаев выходы элементоорганич. со-
единений выше, чем при магнийорганич. синтезе. Ал-
килирующая способность Л. с. значительно выше, чем
алкилмагнийгалогенидов, что сказывается, например,
при стерически затрудненном алкилировании галоге-
нидов кремния — (uso-C3H7)4Si получается только
с помощью иао- C3H7Li — и при получении таллий-
триалкилов (RMgX с Т1С13 образует лишь R2T1C1).
Со многими элементоорганич. соединениями Л. с.
' образуют комплексные соединения, напр.. RLi -f-
+ R3A1 —► LiAIR4. Известны также комплексы типа
LiAIR3H, LiAIR2Ha и т. п.; аналогичные комплексные
соединения получены с органич. соединениями бора,
цинка и сурьмы. Реакции, аналогичные металлирова-
нию, протекают при взаимодействии Л. с. с гидридами
бора и их производными, напр. В2Нв 4- RLi —> RH 4~
4- LiBH4.
Следующие характерные типы реакций позволяют
осуществить замену алкильной (арильной) группы
в Л. с.: а) Металлирование (реакция Шорыгина):
RLi -4 R'H -» R'Li + RH. Обмен идет в том случае,
если углеводород R'H обладает более подвижным
атомом водорода, чем RH. Чаще всего R' — ароматич.
или гетероциклич. радикал, a R — С4Н9. б) Замена
галогена атомом лития: RX4- R'Li --RLi + R'X; реак-
цию проводят обычно с ароматич. или гетероциклич.
соединениями, содержащими активный атом галогена,
в) Обмен радикалами с другими металлоорганич. со-
единениями, напр.
R2Hg + 2R'Li — 2RL1 + R'2Hg
(удобный способ получения нерастворимого в углево-
дородах CH3Li), или
(CH2 = CH)4Sn + 4C4H9Ll-»(C4H9)4Sn + 4СН2 = СНЫ
Важнейшими способами получения Л. с. являются:
а) Взаимодействие металлич. лития с хлористыми и
бромистыми алкилами (арилами): RX 2Li —> RLi +
+ LiX. Хлориды дают лучшие результаты, чем бро-
миды; иодиды (кроме CH3J) для реакции непригодны.
Обычно Л. с. получают таким путем в среде углеводо-
родов (или в эфире) и используют для дальнейших ре-
акций в растворах, б) Нагревание лития со ртутьор-
ганич. соединениями (также обычно в среде углево-
дородов): R2Hg 4- 2Li —< 2RLi + Hg. Метод удобен для
получения индивидуальных Л. с.
Л. с. (в частности, бутиллитий) применяют при по-
лимеризации дивинила. Л. с. широко пользуются
в синтезе вместо магнийорганич. соединений, особенно
в тех случаях, когда нужно применить более энергич-
ный реагент, чем соответствующий реактив Гриньяра.
Лит.: Синтетические методы в области металлоорганиче-
ских соединений, под ред. А. Н. Несмеянова и К. А. Кочеш-
кова, вып. 1—4, М., 1945—50; Rochow Е. G., Hurd
D. Т., Lewis R. N., The chemistry of organometallic com-
pounds, N. Y.—L., [1957]; Coates G. E., Organometallic
compounds, 2 ed., L.—N.Y., 1961. О. Ю. Охлобыстин.
ЛИТИЯ АЛЮМОГИДРИД (лития аланат) LiAlH4—
бесцветные кристаллы; плотн. 0,917; теплота образо-
вания АТ/298 = —24,2 ккал/молъ. Во влажном воз-
духе гидролизуется, водой бурно разлагается с выде-
лением Н2 и воспламенением; при 120° на воздухе
начинает разлагаться на LiH, Н2 и А1. При быстром
нагревании плавится с разложением при 150°. Раство-
рим во многих органич. растворителях. Л. а. получают
взаимодействием лития гидрида с А1С13 (или А1Вг3)
в эфирном р-ре при комнатной темп-ре: 4LiH + А1С13=
= LiAlH4 + 3LiCL Л. а. широко используют в
органич. синтезе как быстрый и сильный (даже при низ-
кой темп-ре) селективный восстановитель. Л. а. вос-
станавливает кислородные органические соединения
(альдегиды, кетоны, кислоты и их ангидриды, слож-
ные эфиры) в спирты; нитрилы — в первичные ами-
ны; галогенопроизводные углеводородов — в соответст-
вующие углеводороды. При этом двойные или трой-
ные связи в исходных соединениях не нарушаются.
Л. а. ценен и для восстановления тех соединений,
к-рые вследствие стерич. препятствий восстанавли-
ваются с,трудом. В неорганич. синтезе реакцией Л. а.
с хлоридами пользуются для получения летучих гид-
ридов бора, алюминия, кремния, германия, олова,
мышьяка, сурьмы.
Лит.: Херд Д., Введение в химию гидридов, пер.
с англ., М., 1955; Минович В., Михайлович М.,
Алюмогидрид лития и его применение в органической химии,
пер. с англ., М., 1957. В. Е. Плющев
ЛИТИЯ АМИД LiNH2 — бесцветные кристаллы,
тетрагональная решетка, а — 5,016 А, с = 10,22 А;
плотность 1,178 (17,5°); т. пл. 373—375° (в запаян-
ном капилляре); т. кип. 430°. Теплота образования
ДТ/288 = — 43,50 ккал/молъ. Растворим в холодной
воде, раствор вследствие гидролиза имеет щелочную
реакцию; при растворении в горячей воде выделяет
NH3. Плохо растворяется в жидком аммиаке, слабо —
в спирте; при нагревании вспирте, растворяясь, отщеп-
ляет аммиак. На воздухе Л. а. разлагается медленно,
при нагревании очень энергично, однако без воспла-
менения; при 450° разлагается на имид лития Li2NH
и аммиак. Л. а. может быть получен электролизом
р-ров галогенидов лития в жидком аммиаке, а также
реакцией между последним и металлич. литием в при-
сутствии железа (катализатор). В пром-сти Л. а.
получают взаимодействием лития гидрида с газооб-
разным аммиаком при 440—460°: LiH -f- NH3 =
= LiNH2 + H2. Л. а. применяют в органич. синтезе
для введения аминогруппы в соединения и в качестве
катализатора в реакциях конденсации.
Лит. см. при ст. Литий. В. Е. Плющев.
ЛИТИЯ ГАЛОГЕНИДЫ — соединения Li с галоге-
нами, соли галогеноводородных к-т.
Фторид лития LiF — бесцветные кристаллы,
, гранецентрированная кубич. решетка, а = 4,0279 А;
плотн. 2,635 (20°); т. пл. 870° (в токе HF); т. кип. 1681°.
Теплота образования ДЯ2°98 = —146,3 ккал/молъ.
При 1000° начинает испаряться, пары имеют щелочную
реакцию. Кристаллогидратов не образует, трудно
растворим в воде: 0,13 г в 100 г Н2О (25°). Раствори-
мость в воде понижается в присутствии NH3 и особенно
NH4F. В отличие от др. Л. г., LiF не растворяется
в органич. растворителях. Легко растворим в HNO3
и H2SO4. Плавиковая к-та переводит LiF в бифторид
987
ЛИТИЯ ГАЛОГЕНИДЫ —ЛИТИЯ ГИДРИД
988
LiHF2, выделяющийся при выпаривании р-ра; при
нагревании до 200° бифторид снова переходит в LiF.
Для перевода LiF в растворимые соединения лития
применяют нагревание с известковым молоком или
спекание с СаО (с последующим выщелачиванием
водой); в обоих случаях образуется LiOH.
Получают LiF добавлением NaF или NH4F к р-рам
солей Li. Применяют как компонент многих флюсов,
используемых при плавке металлов и сварке Mg, Al
и легких сплавов; в произ-ве специальных стекол,
кислотоупорных и прозрачных для УФ-лучей. Моно-
кристаллы LiF нашли применение в произ-ве оптич.
приборов, т. к. они прозрачны для лучей с % до 1000 А
и имеют практически постоянную дисперсию в пре-
делах всего видимого спектра.
Хлорид лития LiCl — бесцветные кристаллы,
гранецентрированная кубич. решетка, а = 5,1398 А;
плотн. 2,07 (25°); т. пл. 614°; т. кип. 1382°. Теплота
образования ДЯ298 = — 97,70 ккал/молъ. При 1000°
LiCl начинает заметно испаряться. LiCl весьма гигро-
скопичен, расплывается на воздухе, легко растворим
в воде (г/100 г Н2О): 67,0 (0°), 78,5 (20°), 127,5 (100°).
Растворим во многих органич. растворителях (спир-
тах, сложных эфирах, ацетоне, пиридине и др..),
с нек-рыми из них образует сольваты. На этом свой-
стве основано отделение Li от остальных щелочных
металлов в аналитич. химии.
Из водных р-ров безводная соль может быть выде-
лена лишь выше 94°; ниже этой темп-ры кристалли-
зуются гидраты; точки перехода: LiCl - Н2О (94°),
LiCl - 2Н2О (19°), LiCl - ЗН2О (—20,5°), LiCl • 5Н2О
(—65,6°). В отличие от NaCl и КС1, хлорид лития не
выделяется из р-ра при пропускании НС1 или прибав-
лении соляной к-ты, так как ионы Li+ в водном р-ре
сильно гидратированы. По той же причине LiCl
является энергичным высаливающим и дегидратирую-
щим агентом. В аммиачном р-ре LiCl образуются ком-
плексные ионы типа [Li (NH3)4]+. При действии NH3
на сухой LiCl образуются соединения LiCl - nNH3,
в к-рых п = 1—4. Хлорид Li легко поглощает газо-
образные амины, реагирует со многими органич. со-
единениями, образует с хлоридами многих элементов
двойные соли и типично комплексные соединения.
LiCl может быть получен при непосредственном
соединении лития с хлором и др. способами. При дей-
ствии хлора на LiOH наряду е LiCl могут быть полу-
чены также гипохлорит LiClO, хлорит LiC102 и хло-
рат LiClO3. Наиболее распространенный промышлен-
ный способ получения LiCl основан на растворении
Li2CO3 или LiOH в соляной К-те. Хлорид Li имеет
важное промышленное значение для получения метал-
лич. Li электролизом из расплава. Благодаря способ-
ности поглощать аммиак, амины и др. примеси и
обратимо поглощать пары воды при изменении темп-ры
LiCl (обычно в виде 40%-ного р-ра) применяют для
кондиционирования воздуха. Другие области примене-
ния: произ-во фотореагентов, сухих батареи, флю-
сов для плавок металлов и сварки Mg, Al и легких
сплавов.
Бромид лития LiBr — бесцветные кристал-
лы, гранецентрированная кубич, решетка, а = 5,501 А;
плотн. 3,46 (25°); т. пл. 549°; т. кип. 1310°. Теплота
образования ДЯ288 = — 83,72 ккал/молъ. Весьма
гигроскопичен и хорошо растворяется в воде (г/100 г
Н2О): 143 (0°), 177 (20°), 266 (100°). Растворим также
во многих органич. растворителях. Из водных р-ров
LiBr выделяется в виде кристаллогидратов, содержа-
щих 1,2 или 3 молекулы воды.Полное обезвоживание
достигается с большим трудом. При действии аммиака
на сухой LiBr образуются соединения LiBr - reNH3,
в к-рых п — 1—4. С галогенидами многих элементов
LiBr дает ряд двойных солей. В жидком броме LiBr
не растворяется и полибромидов не образует. В рас-
плавленном состоянии сильно разъедает стекло, фар-
фор и платину. LiBr образуется при непосредственном
соединении лития с бромом. Промышленное получе-
ние основано на реакции: Li2CO3 + 2НВг — 2LiBr -р-
+ СО2 + Н2О. Потребляется бромид Li обычно в виде
55%-ного р-ра в установках по кондиционированию
воздуха. Другие области применения — произ-во
фотореагентов и медицина (антиподагрич. средство).
Иодид лития LiJ — бесцветные кристаллы,
гранецентрированная кубич. решетка, а = 6,012 А;
плотн. 4,06 (25°); т. пл. 450°; т. кип. 1171°. Теплота
образования Afl[og — —64,79 ккал/молъ. Весьма гиг-
роскопичен и хорошо растворяется в воде (г/100 г Н2О):
151 (0°), 164 (19°), 263 (75°), 476 (100°). Хорошо раст-
ворим в органич. растворителях. Из водных р-ров
LiJ выделяется в виде кристаллогидратов с 0,5, с 1, с
2 или 3 молекулами воды. Р-р иодида, подкисленный
соляной, серной или азотной к-той, выделяет иод,
особенно под действием солнечного света. С жидким
аммиаком LiJ образует комплексные соединения
LiJ • nNH3 (n = 1—5; 5,5; 7). Ниже 0° LiJ образует
продукты присоединения с SO2 типа LiJ • nSO2 (n =
= 1; 2; 4). При действии LiJ на расплавленный иод
образуются полииодиды LiJ„ (re = 3—9). В расплав-
ленном состоянии LiJ разъедает стекло, фарфор и
платину. LiJ образуется при взаимодействии NH4J
с Li в жидком аммиаке или при растворении Li2CO3
в иодистоводородной к-те. LiJ в виде р-ра с HgJ2
образует т. н. тяжелые жидкости, используемые для
разделения минералов. Аналогично бромиду, LiJ при-
меняется в медицине и произ-ве фотореактивов.
Лит. см. прист. Литий. В. Е. Плющев.
ЛИТИЯ ГИДРИД LiH — бесцветные кристаллы
с гранецентрированной кубич. решеткой, а = 4,093 А;
плотн. 0,776 (25°). Теплота образования ДЯ208 =
— —21,61 ккал/молъ. Л. г. устойчивее всех гидридов
щелочных и щелочноземельных металлов. В отсутст-
вии воздуха плавится при 680—697° почти без разло-
жения, при более высокой темп-ре разлагается; давле-
ние диссоциации достигает атмосферного при 850°.
При хранении на свету или под действием УФ-лучей
Л. г. приобретает голубую окраску вследствие частич-
ного разложения с выделением тонкодиспергирован-
ного лития. С водой Л. г. бурно реагирует: LiH 4-
-|- Н2О = LiOH + Н2. При нагревании Л. г. с азотом
образуются нитридлитияи аммиак. С кислородом Л. г.
реагирует только при темп-ре красного каления.
В этих же условиях взаимодействует со взрывом с су-
хим хлором, образуя LiCl. Длительное нагревание
Л. г. до темп-ры красного каления с С, Si, Р или S
приводит к получению соответственно карбида, сили-
цида, фосфида или сульфида лития. При взаимодей-
ствии Л. г. с аммиаком образуется лития амид LiNH2
(с жидким NH3 уже при обычной темп-ре). При высо-
кой темп-ре Л. г. бурно реагирует с SiO2 и силика-
тами, поэтому Л. г. разрушает аппаратуру из кварца,
стекла и фарфора. Расплавленный JI. г. проводит
электрич. ток с выделением водорода на аноде. Л. г.
взаимодействует с галогенидами многих металлов,
образуя лития галогениды и новые гидриды. Л. г. дает
двойные гидриды с алюминием (см. Лития алюмогид-
рид), с таллием (LiTlH4), бором (LiBH4) и др., имею-
щие восстановительные свойства. Боргидрид лития
представляет интерес как вещество с большим запа-
сом водорода (содержит 19 вес. % Н) и как источник
получения диборана (В2Н6).
В пром-сти Л. г. обычно получают непосредствен-
ным гидрированием расплавленного лития чистым
водородом. Присутствие примесей в исходных про-
дуктах приводит к воспламенению или взрывам. Гид-
рирование начинается при 500°, оптимальная темп-ра
680—700°, выход 98,5% при чистоте продукта 99,65%.
В США промышленное произ-во Л. г. базируется также
989
ЛИТИЯ ГИДРООКИСЬ - ЛИТИЯ ОКИСЬ И ГИДРООКИСЬ
990
на восстановлении окиси или гидроокиси лития алю-
минием или магнием под небольшим давлением водо-
рода. Л. г. нашел широкое применение как легкий и
портативный источник простого и быстрого получе-
ния водорода (1 кг Л. г. дает 2,8 м3 водорода) для за-
полнения аэростатов и автоматич. заполнения мор-
ского и воздушного спасательного снаряжения при
авариях самолетов в открытом море. В органич. син-
тезе Л. г. используется как сильный восстановитель,
а также для получения высококалорийного специаль-
ного топлива (бороводороды).
Лит.: Херд Д., Введение в химию гидридов, пер.
с англ., М., 1955; Литий, сб. переводов, М., 1954, 1959. См.
также лит. при ст. Литий. В. Е. Плющев.
ЛИТИЯ ГИДРООКИСЬ — см. Лития окись и гид-
роокись.
ЛИТИЯ КАРБОНАТ (литий углекислый) Li2CO3 —
бесцветные кристаллы моноклинной сингонии; плотн.
2,11 (0°). Теплота образования М//.., = —290,54
ккал/моль; т. пл. 732°; выше этой темп-ры начинается
диссоциация. Давление СО2 при диссоциации состав-
ляет (мм рт. ст.): 1 (610°) н 760 (1270°). Образую-
щаяся при диссоциации окись лития разрушает даже
платину. Л. к. нс образует кристаллогидратов. В воде
трудно растворим. Растворимость (г/100 г Н2О):
1,54 (0°); 1,33 (20°); 0,73 (100°). В присутствии солей
щелочных металлов и аммония (кроме карбонатов)
растворимость Л. к. повышается вследствие образова-
ния комплексных соединений. Пропусканием СО2
через водную суспензию Л. к. можно перевести послед-
ний в бикарбонат LiHCO3, к-рый растворим в воде
значительно лучше средней соли; при нагревании
р-ра LiHCO3 вновь образуется Li2CO3. Такой прием
может быть использован для очистки Li2CO3. При
нагревании Л. к. с Mg или А1 можно восстановить
Л. к. до металла; реакция бурная, сопровождается
воспламенением или даже взрывом (с магнием).
Чистый Л. к. получают пропусканием СО2 в р-р
LiOH; в пром-сти — действием поташа или преим.
соды (в виде сухих солей или р-ров) на р-ры солей
лития вблизи их точки кипения. Обычно осаждение
проводится из технич. р-ров сульфата лития, т. к.
они непосредственно получаются после разложения
рудных концентратов (см. Литий). Л. к. — важней-
шая промышленная соль лития, источник для полу-
чения большинства других его соединений. Самостоя-
тельное значение Л. к. имеет в пиротехнике, произ-ве
пластмасс (катализатор), в черной металлургии (де-
сульфурация стали). Однако наибольшее применение
Л. к. находит в произ-ве керамики и стекла. При этом
используется способность образующейся из Л. к.
окиси лития давать с многими окислами легкоплавкие
эвтектики без потери кислотоупорности основного
материала. Окись лития придает материалу специ-
фич. особенности, повышает его качество или сообщает
новые свойства.
Применение Л. к. для изготовления керамич. масс, эмалей,
глазурей и различных кислотоупорных покрытий сокращает
продолжительность обжига, понижает коэфф, термич. расши-
рения, повышает термин, и химич. устойчивость, твердость и
динамич. прочность материала. Литийсодержащая керамика
оказалась ценной для произ-ва высоковольтного фарфора и
керамич. материала («ступалит»), применяемого для аэродина-
мич. покрытий и защиты реактивных двигателей. Термо- и
кислотоупорные эмали с большим содержанием лития служат
для покрытия алюминия и для изготовления легкоплавких
эмалей для фарфора, а также для грунтовки и покрытия листо-
вой стали и чугуна. В произ-ве стекла соединения лития повы-
шают вязкость силикатных масс (что упрощает технологию
изготовления стекла), увеличивают прочность стекла и сопро-
тивляемость действию атмосферной коррозии, уменьшают
расстекловывание и коэфф, термич. расширения, повышают
проницаемость для УФ-лучей. Поэтому многие соединения LI
(фторид, минералы и особенно Л. к.) нашли применение в
произ-ве специальных стекол для телевизоров, водомеров кот-
лов высокого давления и рентгеновских установок (стекла
Линдемана).
Лит.: Литий, сб. переводов, М., 1954, 1959, см. также лит.
при ст. Литий. В. Е. Плющев.
ЛИТИЯ НИТРАТ (литий азотнокислый) LiNO3 —
бесцветные кристаллы, гексагональная решетка, а =
= 4,674 А, с = 15,199 А; плотн. 2,36 (20°); т. пл. 261°,
теплота образования ДЯ298 = —115,279 ккал/молъ.
При 600° начинает разлагаться с выделением кисло-
рода и окислов азота. Весьма гигроскопичен, хорошо
растворяется в воде (г/100 е Н2О): 53 (0°) и 227 (100°).
Легко растворим в жидком аммиаке и многих органич.
растворителях. Из водных р-ров безводная соль вы-
деляется выше 60°; в интервале 30—60° кристалли-
зуется LiNO3- 1/2Н2О, ниже 30°— LiNO3- ЗН2О.
Л. н. получают взаимодействием Ы2СО3 или LiOH
с разб. HNO3, последующим глубоким упариванием
р-ра и нагреванием остатка в вакууме до 200°. Основ-
ное применение Л. н. находит в пиротехнике (сигналь-
ные ракеты, трассирующие составы); используется
и для целей стабилизации жидкого аммиака, потреб-
ляемого в сельском хозяйстве и в холодильных ус-
тановках.
Лит. см. при ст. Литий. В. Е. Плющев.
ЛИТИЯ ОКИСЬ И ГИДРООКИСЬ — важнейшие
кислородные соединения лития.
Окись лития 1л2О — бесцветные кристаллы
с гранецентрированиой кубич. решеткой, а = 4,628 А;
плотн. 2,013 (25°); т. пл. 1570°; т. кип. 2600°. Теплота
образования А77298 = —142,4 ккал/молъ. Термически
устойчива. Li2O легко, но менее энергично, чем окислы
др. щелочных элементов, соединяется с водой, обра-
зуя LiOH. Легко поглощает СО2, образуя Li2CO3.
Выше 1000° Li2O может быть восстановлена до металла
с помощью Al, Mg или Si. Окись лития корродирует
большинство материалов, выше 1000° — даже пла-
тину. Li2O можно получить нагреванием нитрата,
гидроокиси или карбоната лития в токе сухого Н2
выше 800° или окислением металлич. Li выше 200°;
очень чистую окись — прокаливанием Li2O2 ок. 200°.
Самостоятельное применение Li2O незначительно.
Однако ценными свойствами ее определяется приме-
нение многих др. соединений Li, к-рые при высоких
темп-рах разлагаются с выделением Li2O (см. Лития
карбонат).
Гидроокись лития LiOH — бесцветные
кристаллы тетрагональной сингонии; а = 3,549 А,
е = 4,334 А; плотн. 1,46 (25°); т. пл. 462°, теплота
образования АН298 = —116,45 ккал/молъ. При 1000°
полностью диссоциирует на Li3O и Н2О. В воде LiOH
растворима менее гидроокисей др. щелочных элемен-
тов. Растворимость (г/100 г Н2О): 12,7 (0°), 17,5 (100°).
Плотность водных р-ров:
L1OH, вес. % . . 1 2 4 6 8 10
Плотность .... 1,010 1,021 1,044 1,065 1,086 1,107
Из водных р-ров кристаллизуется LiOH • Н2О, к-рый
полностью обезвоживается лишь выше 600°. LiOH
и ее конц. р-ры разъедают стекло и фарфор даже при
обычной темп-ре, а при нагревании — многие металлы
(кроме Ni и Au), окислы и силикаты. LiOH может
быть получена растворением в воде Li и его окиси,
электролизом LiCl на ртутном катоде, а также обмен-
ной реакцией в р-ре между Li2SO4 и гидроокисью К,
Са или Ва; в пром-сти ее получают янустификацией
карбоната Li известью в растворе: LisCO3 +
+ Са(ОН)2 2LiOH + СаСО8 и последующим упа-
риванием р-ров LiOH в вакууме на моногидрат
LiOH • Н2О. Гидроокись лития потребляется в боль-
ших количествах как добавка к электролиту щелоч-
ных аккумуляторов, повышающая их емкость на 20—
22%, расширяющая температурный диапазон дейст-
вия и удлиняющая в 2—3 раза срок службы. LiOH
является основой для получения стеарата, оксистеа-
рата, олеата и пальмитата Li, используемых в произ-ве
морозо- и термостойких смазок, получивших широкое
991
ЛИТИЯ СУЛЬФАТ — ЛОБРИ ДЕ БРУИНА И ВАН ЭКЕНШТЕЙНА РЕАКЦИИ
992
распространение в авиации и военной технике (см,
Литийорганические соединения). На подводных лодках
LiOH применяют для поглощения СО- из воздуха.
Лит. см. при ст. Литий. В. Е. Плющев.
ЛИТИЯ СУЛЬФАТ (литий сернокислый) Li2SO4 —
бесцветные кристаллы; при обычной темп-ре устойчива
моноклинная a-модификация, а — 8,44А, Ь = 4,95А,
с = 8,24 А, Р = 107°54'; ниже 500° она переходит
в гексагональную P-модификацию, к-рая при 575—
578° превращается в кубическую у-модификацию,
существующую до температуры плавления. Плотн.
2,22 (20°); т. пл. 860°. Теплота образования ДЯ298 =
= —342,83 ккал/молъ. В термич. отношении Л. с.
более устойчив, чем другие растворимые соли лития.
Л. с. хорошо растворим в воде (г/100 г Н2О): 35,3 (0°),
30,0 (100°). Из водных р-ров при обычных условиях
выделяется (Li2SO4 • Н2О), в интервале 100—130° —
безводная соль. Л. с. образует двойные соли типа
MeLiSO4 (Me = К+, Na+, NH+); Me4Li2(SO4)3 • 9Н2О
(Me = K+, Na+) и др. Л. с. получают по реакции:
Li2CO3 + H2SO4 = Li2SO4 + СО2 + Н2О; выделив-
шийся Li2SO4 • Н2О прокаливают при 500° до полу-
чения мелких призматич. кристаллов безводной соли.
Л. с. находит применение в ультразвуковой дефекто-
скопии для изготовления головок щупов.
Лит. см. при ст. Литий. В. Е. Плющев.
ЛИТОПОН — см. Пигменты белые.
ЛИТОСФЕРА (земная кора) — твердая внешняя
оболочка Земли, средней условной мощности 16 км.
Толщина Л. на равнинах составляет 30—40 км, в об-
ласти горных хребтов 50—75 км, в пределах впадин
морей и океанов 5—6 км. Л. состоит на 95% из извер-
женных пород (гл. обр. базальтов и гранитов), на 5%—
из осадочных пород, причем 2,9% приходится на долю
глинистых сланцев, 1,10% — на долю песчаников,
1,0% — на долю карбонатных пород. Средний химич.
состав Л. см. в табл. 2 ст. Геохимия. Наиболее распро-
страненными элементами Л. являются О, Si, Al, Fe,
Са, Na, К, Mg, к-рые образуют многочисленные окислы
и кислородные соли (гл. обр. силикаты, алюмосили-
каты, карбонаты, сульфаты и др.), входящие в состав
различных пород. Наиболее важной формой нахожде-
ния химич. элементов в Л. являются минералы.
В связи с геологич. процессами в Л. протекают раз-
личные геохимические процессы, вызывающие мигра-
цию элементов, концентрацию одних и рассеяние дру-
гих. Накопление к.-л. элемента выше его среднего
содержания в Л. ведет к образованию месторождения
полезного ископаемого.
Все различные химич. процессы в Л. сводятся
к трем основным геохимич. циклам: магматический;
цикл земной поверхности; метаморфический. Между
тремя циклами существует определенная связь, вы-
ражающаяся в закономерном и последовательном раз-
витии земной коры. Наиболее ранний магматич.
цикл, связанный с остыванием расплавленной магмы
(см. Геохимические процессы), заканчивается образо-
ванием изверженных горных пород, новых форм рель-
ефа, месторождений полезных ископаемых. Далее
наступает второй цикл, связанный с действием внеш-
них сил. В результате процессов этого цикла (вывет-
ривание, перенос, осадконакопление и др.) горные
породы разрушаются, рельеф земной коры сглажи-
вается, образуются осадочные породы и новые место-
рождения полезных ископаемых. Процессы второго
цикла далее сменяются различными процессами тре-
тьего цикла. Осадочные породы подвергаются мета-
морфизму, в результате к-рого возникают новые гор-
ные породы и иногда новые месторождения полезных
ископаемых.
Лит. см. при ст. Геохимия.
ЛИТОФИЛЬНЫЕ ЭЛЕМЕНТЫ — см. Геохимиче-
ская классификация элементов.
ЛОБЕЛИИ (I) [1 -метил-2-фенацил-6-(2'-фенил-
2'-оксиэтил)-пиперидин] C22H27O2N, мол. в. 337,41 —
главный алкалоид ра-
стения лобелии (Lo- R. /X. r" r™
belia inflate и др. ви- \ z I _u Vх r h-
ды), сем. колоколь- C6H6-C-CHa-4VNxJ~C 2 e s
чиковых (Campanula- '
ceae), где встречаются
как левовращающий Л., так и рацемический. 1-Л. —
бесцветные кристаллы, т. пл. 130—131° (из спирта);
[alp = —42,85°; трудно растворим в воде и петро-
лейном эфире, легко — в спирте, хлороформе и бен-
золе; хлоргидрат, т. пл. 182°, растворим в воде
(1 : 10), спирте (1 : 15) и в хлороформе. dl-JI., т. пл.
110°; хлоргидрат, т. пл. 170°. Прочие алкалоиды
лобелии, принадлежащие к группе Л.,— лобеланин
(II), лобеланидин (III), норлобеланин (IV) и норлобе-
ланидин (V)—отличаются от Л. и друг от друга сте-
пенью окисления в боковых цепях и (или) метильной
группой у азота.
I. R = CH3; R’ + R" = O; R'" = OH; R'"' = H
II. R = CH3; R’ + R" = R"'-|-R'"' = O
III. R = CH3; R’ = R'” = OH; R" = R’”' = H
IV. R = H; R’ + R” = R”’ + R'"'=0
V. R = R" = R"” = H; R' = R"'=OH
Получают Л. как из растительного сырья, так и синте-
тически (dl-Л.) из а, а'-лутидина,
Л. является сильным стимулятором дыхания. В ме-
дицине применяют хлоргидрат Л. Показанием для
его применения служит остановка дыхания или ослаб-
ление дыхательной деятельности. Особенно показан
при рефлекторных остановках дыхания (при вдыха-
нии раздражающих веществ, при наркозе). Приме-
няется при асфиксии новорожденных. Возбуждение
дыхания не сопровождается общим возбуждением
(в отличие от кофеина) и является кратковременным.
Недостатки лобелина — сильная токсичность и малая
терапевтич. широта.
Лит.: Орехов А. П., Химия алкалоидов, 2 изд.,
М., 1955, с. 95. А. Е. Васильев.
ЛОБРИ ДЕ БРУИНА И ВАН ЭКЕНШТЕЙНА РЕ-
АКЦИИ — процессы эпимеризации альдоз (изме-
нение пространственного расположения групп Н и
ОН у углерода, соседнего с карбонилом), эпимериза-
ции кетоз и взаимного
превращения альдоз и
кетоз,
к-рые идут под влия-
нием оснований.
Реакции 1—3 идут
одновременно, но с
различной скоростью;
обычно самой быст-
рой является реак-
ция 2, наименее быст-
нс=о
неон
неон
R
-
НС-0
НОСН
д СН2ОН
М=О
н<рон
//
, СН2ОН
2_ I
—
НОСН
8
рой реакция 3. Han- НСОН
равление реакций и I
выход продуктов за- R
висят от природы сахара и основания. Процесс про-
ходит под влиянием неорганич. оснований [NaOH,
КОН, NH4OH, Ва(ОН)2], слабых органических осно-
ваний (пиридин, хинолин), ионообменных смол ос-
новного характера, а также некоторых ферментов;
в некоторых случаях процесс удается провести под
влиянием кислот. При хроматография, разделении
сахаров и их производных следует учитывать воз-
можность трансформации под влиянием анионооб-
менных смол. Реакции используются как препаратив-
ный метод получения сахаров, напр. кетоз из соответ-
ствующих альдоз (с выходами 10—50%).
Ферментативные превращения имеют большое зна-
чение в обмене углеводов; к реакциям этого типа отно-
сят изомеризацию 3-фосфоглицеринового альдегида
993 ЛОССЕНЯ ПЕРЕГРУППИРОВКА - ЛЭНГМЮРА УРАВНЕНИЕ ИЗОТЕРМЫ АДСОРБЦИИ 994
в фосфат диоксиацетоиа под влиянием фермента
мышц — изомеразы триозофосфата, превращение глю-
козо-6-фосфата во фруктозо-6-фосфат (см. Гликолиз)
и ряд других промежуточных реакций обмена углево-
дов. Реакции указанного типа идут также и с окси-
альдегидами и с оксикетонами неуглеводного харак-
тера (превращение З-окси-4-гептанона в 4-окси-З-геп-
танон по Фаворскому, превращения альдегидов и
кетонов в ряду стероидов).
Лит.: The carbohydrates, chemistry, biochemistry physio-
logy, ed. W. Pigman, N. Y., 1957; Advances In carbohydrate
chemistry, v. 13, N. Y., 1958, p. 63. Л. И. Линевич.
ЛОССЕНЯ ПЕРЕГРУППИРОВКА — превращение
гидроксамовых к-т и их производных в изоцианаты
или в продукты взаимодействия образующихся изо-
цианатов с компонентами среды, напр. в присутст-
вии воды в амины:
о
RC^NHOR' —*RN=C=O —5 RNH2-J CO2
Л. п. проводят в присутствии дегидратирующих аген-
тов, напр. Р2О6, SOC12 и др., или же простым нагре-
ванием. Ацильные производные гидроксамовых к-т
разлагают обычно с применением в качестве ката-
лизаторов оснований. Производные а-оксигидроксамо-
вых кислот превращаются в альдегиды, а, 0-нена-
сыщенных кислот (в присутствии спирта) — в нена-
сыщенные уретаны, о-оксиарилгидроксамовых к-т —
в бензоксазолоны; алифатич. дигидроксамовые к-ты
при нагревании в инертных растворителях образуют
диизоцианаты, а карбоксигидроксамовые к-ты — по-
лиамиды:
1 н„о I
RC-№C“O * - RC~NHS
ОН ОН
II
—— RCH
О
RCH“CH-C-NHOR'— [RCH=CH-
Механизм Л. п. аналогичен механизму родственных
перегруппировок Гофмана и Курциуса:
II ОН“ г ч- II
RC^NHOR'-*-[RC-NOR'] —*-R“C-N + RO— RN=C=O
I II III
Определяющей скорость стадией Л. п. является пере-
ход сильноосновного аниона I в менее основной, ста-
бильный аниоя III и промежуточное соединение II
с электронным секстетом у атома азота. В согласии с
этим скорость Л. п. солей диб.ензгидроксамовых к-т ти-
па IV увеличивается при на-
линии электроноакцептор-
ных и уменьшается при на-
личии электроиодонорных
заместителей в отщепляю-
IV
щейся бензоилоксигруппе.
Напротив, введение электроноакцепторных заместите-
лей в мета- и пара-положения мигрирующей фениль-
ной группы замедляет Л. п., а электронодонорных —
ускоряет ее. При переходе от углерода к азоту карба-
нион не отщепляется в виде кинетически независимой
I. r-ch2oh, r -h
IL R*H; R’-CHgOH
частицы, что доказывается сохранением конфигура-
ции производных оптически активных гидроксамо-
вых к-т при Л. п.
Лит.: Franzen V., Chemlker—Ztg., 1956, 80, Н. 1, 8;
Eckstein Z., Roczn. chem., 1954, 28, z. 4, 549.
H. П. Гамбарян.
ЛОССЕНЯ ПРОБА — см. Aaoma определение.
ЛОУРЕНСИЙ (Lw) — искусственно полученный
радиоактивный химич. элемент, замыкающий семейство
актинидов; п. н. 103. Назван в честь изобретателя
циклотрона Э. Лоуренса (Е. Lawrence). Синтезиро-
ван в 1961 А. Гиорсо, Т. Сиккеландом, А. Ларшем и
Р. Латтимером в виде изотопа Lw267 (a-активен, энер-
гия а-частиц 8,6 Mae, Ti/„t=s 8 сек) при бомбарди-
ровке смеси изотопов калифорния многозарядными
ионами бора, в реакции типа Cf (В, неск. n) Lw.
Идентифицирован чисто физич. методами, без иссле-
дования ХНМИЧ. СВОЙСТВ. В. И. Гольданский.
ЛУПИНИН Ci0Hi9ON, мол. в. 169,27 — алкалоид,
выделенный из Anabasis aphylla (сем. Chenopodiaceae)
и различных видов Lupinus (сем. Le-
guminosae), причем в первом расте-
нии сопутствует анабазину, от к-рого
его отделяют химич. методами (нитро-
зированием, бензоилированием). Л.
представляет собой 1-оксиметилхино-
лизидин с аксиальной оксиметильной
группой (I), его более стабильный
изомер с экваториальной оксиметиль-
ной группой наз. эпилупинин (II). Л. — кри-
сталлы, т. пл. 68—69° (из петролейного эфира), т. кип.
269—270°, [аЗр = —19°; хорошо растворим в воде и
органич. растворителях. Л. — сильное основание,
рА03,84, вытесняет аммиак из его солей; образует хлор-
гидрат, т. пл. 212—213°, хлораурат, т. пл. 196—197°,
пикрат, т. пл. 137°.
Лит.: Садыков А. С., Химвя алкалоидов Anabasis
aphylla, Ташкент, 1956; Bolt Н. G., Ergebnlsse der Alkalold-
Chemle bls 1960 unter besonderer Bertlckslchtlgung der Fort-
schrltte selt 1950, B., 1961. A. E. Васильев.
ЛУТИДИН — см. Метилпиридины.
ЛЬНЯНОЕ МАСЛО — см. Жиры растительные.
ЛЭНГМЮРА УРАВНЕНИЕ ИЗОТЕРМЫ АДСОРБ-
ЦИИ — простейшее уравнение изотермы локализо-
ванной адсорбции, при к-рой молекулы фиксируются
у определенных мест поверхности (адсорбционных
центров). Частным случаем локализованной адсорб-
ции является хемосорбция, когда молекулы фикси-
руются на поверхности благодаря образованию химич.
связей. По отношению к молекулам, способным к об-
разованию водородных связей, адсорбционными цент-
рами являются гидроксильные группы. При физич.
адсорбции местами локализации являются места
с повышенной потенциальной энергией молекул ад-
сорбата, напр. в случае адсорбции на базисной грани
графита — места над центром шестиугольника из
атомов углерода, а в случае неоднородных поверхно-
стей — тонкие поры, трещины. Помимо условия лока-
лизации, ур-ние основано на допущении о равноцен-
ности всех адсорбционных центров (т. е. об однород-
ности активной поверхности, о постоянстве сил взаи-
модействий адсорбат — адсорбент) и об отсутствии
взаимодействия между адсорбированными молекулами
(адсорбат — адсорбат).
Простейший вывод ур-ния Лэнгмюра основан на
рассмотрении адсорбционного равновесия как равно-
весия при обратимой квазихимич. реакции:
молекула адсорбата в газе + адсорбционный центр
на поверхности — адсорбционный комплекс
Константа равновесия этой реакции при допущениях
теории Лэнгмюра:
к=р^ или р== W
995
ЛЭНГМЮРА УРАВНЕНИЕ ИЗОТЕРМЫ АДСОРБЦИИ — ЛЮИЗИТ
996
где 9 = а/ат — доля заполнения поверхности, а —
адсорбция при давлении газа р, ат — предельная
адсорбция, соответствующая заполнению всех адсорб-
ционных центров, активных по отношению к данному
адсорбату. Из (1) следует Л. у. и. а. — зависимость 6
от р при постоянной темп-ре в форме:
8==i+KF или (2)
Ур-ние (2) выражается кривой, приведенной на
рис. 1. С уменьшением р ур-ние Лэнгмюра прибли-
жается к ур-нию Генри, 6 — Кр, при больших р
6 —>• 1, а —► ат. Зависимость константы равновесия
от темп-ры: К = e^s°/R eQ“/R37 где Аб10 и Q° -
стандартные (напр., при
р = 1, 6 — 0,5) величины
энтропии и теплоты ад-
сорбции, позволяет найти
6 при разных темп-рах.
Более подробный вывод
ур-ния Л. у. и. а. на основе
статистич. термодинамики свя-
зывает константу равновесия К
е функциями распределения
(суммами состояний) молекулы
адсорбата в газе и на поверхно-
сти. Ур-ние Лэнгмюра выводит-
ся также кйнетически прирав-
ниванием скорости адсорбции и
десорбции. Скорость адсорбции иа определяется числом столк-
новений с поверхностью z = pV 2ftmkT, пропорциональным
давлению газа р (т — масса молекулы газа, к — константа
Больцмана) и долей незанятой поверхности 1 — 5
«а=
V 2nmkT
Рис. 1.
(3)
где <т — вероятность того, что столкновение с поверхностью
приведет к адсорбции (неупругий удар; при физич. адсорбции
и невысоких темп-рах обычно а = 1).
Скорость десорбции ид с однородной поверхности пропор-
циональна доле занятой поверхности $ и уменьшается с рос-
том потенциального барьера, вызванного притяжением к ад-
сорбенту, к-рый молекулам необходимо преодолеть, чтобы
покинуть поверхность (т. е. с ростом потенциальной энергии
адсорбции, приблизительно равной теплоте адсорбции Q):
иД = Ме-0/ЛТ (4)
где — константа скорости десорбции (приближенно соответ-
ствующая произведению частоты колебаний молекулы перпен-
дикулярно поверхности на число центров на единице поверх-
ности).
При равновесии иа равно ид, так что
Q/RT
2 . • р
к- V 2xmkT
’”77--------(5)
кд у 2лткТ
Это и есть Л. у. и. а. (2), в к-ром
К=—(6)
кд V2nmkT
В случае хемосорбции вероятность а связана также с необ-
ходимостью благоприятной ориентации молекулы по отноше-
нию к реагирующему центру поверхности адсорбента. Так как
иа зависит от энергии активации адсорбции Еа, а ид — от энер-
гии активации десорбции Ед, то Q — Ед — Еа. Хемосорбция
часто сопровождается диссоциацией молекул адсорбата, что
усложняет зависимость и от 0. Так, при диссоциации молекулы
адсорбата на два атома, занимающие на поверхности два
центра, Л. у. и. а. принимает вид:
S + J£2^
1+кГр
(7)
где К — соответствующая константа.
Соответствие экспериментальной изотермы ур-нию
Лэнгмюра проверяют построением эксперименталь-
ных точек в диаграмме линейной формы ур-ния Лэнг-
мюра (2):
(8)
а атК ат
откладывая p/а как функцию (рис. 2). Отсекаемый
на оси ординат отрезок, равный 1/ят К, и наклон
прямой (8) позволяют определить емкость мономоле-
кулярного слоя (число адсорбционных центров) ат
и константу равновесия ад-
сорбции К. Однако само по р/а
себе спрямление экспери- s
ментальной изотермы в коор-
динатах p/а и р еще не до- у'
статочно для суждения о у'
применимости теории Лэнг- S'
мюра, т. к. влияние неодно- s
родности поверхности (не-
равноценность адсорбцион-
ных центров, непостоянство р
взаимодействий адсорбат — рис. 2.
адсорбент) и влияние взаи-
модействий адсорбат — адсорбат, компенсируя друг
друга, могут привести к чисто формальному описанию
опыта ур-иием Лэнгмюра. Поэтому наряду с изотер-
мой адсорбции следует определить также Дифферен-
циальную теплоту адсорбции Q, которая "по теории
Лэнгмюра не должна зависеть от заполнения поверх-
ности fl или я.
Лит.: Брунауер С., Адсорбция газов и наров, пер.
с англ., т. 1, М., 1948; Трепне л Б., Хемосорбция, пер.
с англ., М., 1958. А. В. Киселев.
ЛЮИЗИТ — смесь Р-хлорвинилдихлорарсина
(C1CH=CHAsC12—а-люизит); ди-(0-хлорвинил)-хлор-
арсина [(GICII=CH)2 AsCI—Р-люизит] и три-(р-хлор-
винил)-арсина [(C1GH = СН)3 As — у-люизит]. Все
три соединения представляют собой бесцветные тя-
желые жидкости, нерастворимые в воде и хорошо
растворимые в органич. растворителях. а-Л. — смесь
Ck /AsC12 Ск ,н
цис- >С=С< а транс- >с=с<
н/ \н н/ 'AsC12
изомеров. Основные физич. свойства отдельных ком-
понентов Л. приведены в таблице.
Соединение Т. пл., °C Т. кип., °C .20 d4 nD
а-Л. цис -44 170 1,86 1,6092 (23,7°)
транс -1,2 196 1,88
₽-Л — 230 (с разл.) 1,692 (24,1°) 1,6080 (23,4°)
у-Л +18 260 (с разл.) 1,5664(23,7°) 1,6005 (23,7°)
а-Л. легко гидролизуется водой с образованием
токсичного р-хлорвиниларсиноксида:
С1СН = CHAsCla + Н3О -CICH = CHAsO + 2НС1
При действии окислителей (НаО2, HNOS и др.) а-Л. об-
разует нетоксичную Р-хлорвинилмышьяковую к-ту.
Галогены (С12, Вг2) разлагают а-Л. с образованием
1,2-дихлорэтилена и AsCl3. С сероводородом а-Л. об-
разует нерастворимый в воде Р-хлорвиниларсинсуль-
фид (эта реакция часто используется для качествен-
ного определения Л.). Характерно отношение а-Л.
к щелочам — транс-а-Л. легко разлагается щелочью
на холоду с выделением ацетилена:
ClCH = CHAsCl24-6NaOH — СН_-СН4-As(ONa)3 + 3NaCl
цис-а-Л. в этих условиях превращается в натриевую
соль Р-хлорвинилмышьяковистой к-ты, к-рая при
дальнейшем действии более конц. щелочи разлагается
с выделением винилхлорида:
CICH = CHAsCl2+4NaOH - CICH =СН As (ONa)2 +2NaCl+2H2O
CICH = CHAs (ONa)s + NaOH - CH2 = CHC1 + As (ONa)3
Реакция а-Л. co щелочами используется для качест-
венного и количественного определения Л.; при про-
997
ЛЮМИНАЛ - ЛЮМИНЕСЦЕНТНЫЙ АНАЛИЗ
пускании ацетилена через аммиачный р-р закиси меди
или окиси серебра образуются окрашенные осадки;
по объему выделившегося ацетилена определяют со-
держание а-Л.
Л. получают из ацетилена и AsCl3 в присутствии
катализаторов (А1С13, HgCl2, CuCl2 и др.):
CH^CH + HgCl2 -С1СН= CHHgCl
С1СН = СН HgCl + AsCls - С1СН = CHAsCls + HgCI2
Технич. Л. представляет собой темно-коричневую,
почти черную жидкость с чрезвычайно резким раздра-
жающим запахом, в малых концентрациях напоми-
нающим запах герани.
Наиболее сильным физиология, действием обладает
а-Л. и особенно его транс-изомер. В отличие от ип-
рита (см. Дихлордиэтилсулъфид), Л. практически
не обладает скрытым периодом действия. Концентра-
ция 3 • 10 4 мг/л вызывает непереносимое раздражение
верхних дыхательных путей; доза в 0,2—0,3 мг/см2,
поверхности кожи вызывает покраснение (наряду
с сильным болевым эффектом), а затем появление
пузырьков, сливающихся в волдыри. Аналогичное
поражение вызывают и пары Л. в концентрации
0,3.3 мг!л и выше. Смертельная доза при нанесении
капельножидкого Л. на кожу 35 жг/кг; концентрация
0,12 мг!л вызывает смертельное поражение при дейст-
вии через органы дыхания. Эффективным средством
лечения поражений Л. является димеркаптопропанол
и его производные (см. Антидоты). Л. был предложен
в качестве отравляющего в-ва в конце первой мировой
войны А. Льюисом, откуда и получил свое название.
Л. производился в США в заводских масштабах, но
в боевых операциях использован не был.
Лит.: Соборовский Л. 3., Эпштейн Г. Ю.,
Химия и технология боевых химических веществ, М.—Л.,
1938; Немец В. Г., С о ч и л и н Е. Г., Химия отравляю-
щих веществ, Л.—М., 1941. Р. Н. Стерлин.
ЛЮМИНАЛ (фенобарбитал, 5-этил-5-фенилбарби-
туровая кислота) C12H12O3N2, мол. в. 232,24 — бес-
цветные кристаллы горьковатого вку-
свКъч ZCO-NH са, т. пл. 174—177°. Л. очень трудно
ZCO растворим в холодной воде, раство-
С2Н5 СО-NH рим в 40 частях кипящей воды; легко
растворим в большинстве органич.
растворителей и в р-рах щелочей; pH водного р-ра
ок. 5. При добавлении к р-ру Л. в спирте спиртовых
р-ров нитрата кобальта и аммиака появляется лило-
вая окраска.
Получают Л. конденсацией а-этнл-а-фенилмалоно-
вого эфира с мочевиной в спиртовом р-ре этилата
натрия, а также при взаимодействии а-этил-а-фенил-
а-циануксусного эфира с дициандиамидом в метаноль-
ном р-ре метилата натрия с последующим кислот-
ным гидролизом промежуточного карбимидного про-
изводного.
Л. — снотворное средство; его применяют также
для лечения эпилепсии и ряда др. заболеваний.
Р. Глушков.
ЛЮМИНЕСЦЕНТНЫЙ АНАЛИЗ — совокупность
методов анализа, основанных на наблюдении люминес-
ценции — излучения тел, представляющего избыток
над температурным излучением тела и обладающего
длительностью ок. 10 10 сек и больше. Для возбужде-
ния свечения вещество должно предварительно погло-
тить нек-рое количество энергии и перейти в возбу-
жденное состояние, более богатое энергией. При воз-
вращении вещества в нормальное состояние часть
избыточной энергии выделяется в виде излучения
с большей длиной волны. Длительность возбужден-
ного состояния отличает явление люминесценции от’
рассеяния света, отражения и др. кратковременных
видов свечения.
Для Л. а. имеют значение след, законы люминесценции:
1) интенсивность люминесценции возрастает с увеличением
концентрации вещества только до нек-рого предела, после
098
чего она начинает уменьшаться (концентрационное тушение);
2) на интенсивность люминесценции сильно влияет присутствие
примесей, к-рые могут вызывать ее гашение; 3) спектры погло-
щения и люминесценции являются зеркальными, т. е., как
правило, они не перекрывают друг друга; т. обр., поглощение
света веществом не мешает наблюдению его люминесценции;
4) спектры люминесценции сужаются, иногда они даже могут
трансформироваться в квазилинейные, что облегчает иденти-
фикацию вещества и определение его концентрации по интен-
сивности линий.
Для возбуждения люминесценции применяют обычно
фильтрованный УФ-свет (ртутно-кварцевая лампа ПРК и све-
тофильтр марки УФС-3 и УФС-4, почти полностью погло-
щающий видимый,свет, но пропускающий ближний ультрафио-
летовый, К 3660 А). Люминесценцию лучше всего наблюдать
в темном помещении. Наблюдения люминесценции ведут визу-
ально или пользуясь спец, приборами типа колориметров или
фотоколориметров, в которых источник УФ-свста и приемник
люминесценции расположены под прямым углом (подробнее см.
Хемилюминесценция, Фотолюминесценция). Для исследования
спектров люминесценции пользуются спектрографами или
специальными фотоэдектрич. установками.
По характеру решаемых задач Л. а. разделяют на
сортовой и химический — качественный и
количественный. Основная задача сортового анализа—
обнаружение различия между предметами, к-рые
в видимом свете кажутся одинаковыми. Сортовой Л. а.
основан иа разной люминесценции веществ под дей-
ствием возбуждения. Сюда относятся, напр., способы
сортировки стекол, семян, обнаружение битумов в по-
родах, мнкродефектов в металлич. изделиях, выявле-
ние подделок документов и др. На наблюдении люми-
несценции под микроскопом основана т. наз. люминес-
центная микроскопия, имеющая особое значение при
исследовании биологич. объектов, в медицине, фарма-
кологии ит. п.; люминесцентная микроскопия в при-
менении к биологии и медицине выделилась в само-
стоятельную отрасль науки. Для хнмич. анализа
имеют значение люминесцентные индикаторы, приме-
няемые для титрования в окрашенных средах, и лю-
минесцентная хроматография — наблюдение люминес-
ценции различных зон, разделенных методами хро-
матографии.
Для количественного Л. а. необходимо знать меха-
низм реакций, учитывать возможность образования
нефлуоресцирующих веществ, напр. озон можно коли-
чественно определять по образованию ярко флуорес-
цирующего акридина в результате взаимодействия
озона с дигидроакридином. Многие неорганич. вещест-
ва флуоресцируют в твердом состоянии, в растворах
же флуоресцируют лишь соли уранила и соли редко-
земельных элементов. Наиболее интенсивно в р-рах
флуоресцируют тербий, гадолиний и церий. Спектры
редкоземельных элементов состоят из характерных для
каждого элемента линий и полос; с большой точностью
можно определить семь из них (Се, Sm, Eu, Gd,
Tb, Dy, Pr). Чувствительность Л. а. очень большая,
напр. соли тербия можно определить в концентрации
10 е—10-8 г!мл. В твердых р-рах редкоземельные
элементы сохраняют типичные линейчатые и полоса-
тые спектры, что было использовано для их опреде-
ления. Люминесценцию твердых р-ров можно исполь-
зовать для открытия сурьмы, висмута и свинца;
уран определяют в виде перлов, чувствительность
метода 10 5—1010 г урана в 0,3 г фторида натрия.
Катионы, не обладающие собственной люминесцен-
цией, определяют с помощью флуоресцентных реак-
ций, основанных на наблюдении люминесценции ком-
плексов, образуемых катионами с различными орга-
нич. реагентами. Большинство предложенных органич.
реагентов недостаточно селективно и образует люми-
несцирующие комплексы с различными катионами,
напр. 8-оксихинолин с Li, Al, In, Zn, Cd, Ga; морин
c Be, Ga, In, Pb, Zn, Mo и редкоземельными элемен-
тами. Подбирая условия реакции, можно повысить
селективность и использовать эти реактивы для опре-
деления нек-рых катионов, напр. 8-оксихинолин для
определения лития, и морин — для определения бора.
999
ЛЮТЕОСТЕРОН - ЛЮЦИФЕРАЗЫ
1000
Спектры люминесценции р-ров большинства орга-
нич. веществ представляют собой широкие размытые
полосы, только нек-рые соединения имеют спектры,
состоящие из узких характерных полос (хлорофилл,
порфирины). Преобладающие цвета люминесценции —
фиолетовый и синий, реже — зеленый; красным цве-
том люминесцируют лишь немногие соединения (хло-
рофилл, порфярины). В большинстве случаев для
идентификации органич. соединений приходится со-
четать непосредственное наблюдение люминесценции
с частичным разделением смесей и проведением про-
верочных реакций на отдельные компоненты. Для
идентификации канцерогенных веществ из числа по-
лициклич. углеводородов (напр., 3, 4-бензпирен, 3, 4,
6, 7-дибензпирен) наблюдают спектры их свечения в
р-рах нейтральных, легко кристаллизующихся пара-
финов (пентан, гексан, гептан) при низких темп-рах
(77,3° К и 20° К). В указанных условиях полосы
значительно сужаются и по типичным квазилиней-
ным спектрам можно идентифицировать и количест-
венно определять канцерогенные углеводороды. Напр.,
бензпирен можно обнаружить уже при концентрации
10-® г на 1 г вещества. При наблюдении флуоресцен-
ции органич. веществ необходимо учитывать след,
факторы: 1) если молекула обладает кислыми или
основными свойствами, ее люминесценция меняется
с изменением величины pH, т. к. люминесценция не-
диссоциированной молекулы и иона различны, напр.
ион акридина люминесцирует зеленым цветом, а не-
диссоциированное основание — лиловым; 2) спектры
флуоресценции углеводородов почти не изменяются
при перемене растворителя; спектры веществ, способ-
ных ассоциировать с растворителем, могут меняться
с его переменой. Известны люминесцентные групповые
реакции на фенолы, спирты, эфиры фталевой к-ты,
перекиси, монокарбоновые к-ты и др.
Л. а. широко используют для определения порфи-
ринов, витаминов (В2, Bj, никотиновой к-ты и ее
производных), антибиотиков (террамицина, ауреоми-
цина и др.), эстрогенных веществ (эстрон, эстрадиол,
фолликулин и др.), гормона адреналина (чувстви-
тельность до 0,005 мкг в 1 мл водно-щелочного р-ра)
и др. Пользуясь Л. а., можно определять некото-
рые лекарственные средства (хинин, риванол, акри-
хин, стильбамидин и др.) в биологич. средах (крови,
моче и др.).
Лит.:- Константинова-Шлезингер М. А.,
Люминесцентный анализ, М.—Л., 1948,- Материалы 4-го совеща-
ния по люминесценции (Минск, 20—25 июня 1955 г.), Минск,
1956; 6-е совещание по люминесценции (молекулярная люми-
несценция и люминесцентный анализ) 17—22 февр. 1958 г.
Тезисы докладов, Л., 1958; Материалы 7-го совещания по люми-
несценции (кристаллофоры). Москва, 26 июня — 3 июля
1959, Тарту, 1959; Константинова-Шлезин-
гер М. А., Реферативный сборник по люминесцентному ана-
лизу, М., 1951, вып. 2, М., 1954; Прингсхейм П.,
Фогель М., Люминесценция жидких и твердых тел и ее
практические применения, пер. с англ., М., 1948; Люми-
несцентный анализ, под ред. М. А. Константиновой-Шлезин-
гер, М., 1961; Danckwortt Р. W., El senbrand J.,
Lumlneszenz-Analyse fm filtrie'rten nltravloletten Licht, 6 Aufl.,
Lpz., 1956; Radley J. A., Grant J., Fluorescence ana-
lysis in ultra-violet light, 4 ed., L., 1954; White C., Analyt.
Chem. (Обзоры по Л. a. c 1951). H. А. Горбачева.
ЛЮТЕОСТЕРОН — см. Прогестерон.
ЛЮТЕЦИЙ (Lutetium) Lu — химич. элемент с п. н.
71, относится к лантанидам.
ЛЮЦИФЕРАЗЫ — ферменты, катализирующие
процессы биолюминесценции, являющиеся частным
случаем хемилюминесценции. Основное отличие био-
люмипесцентных процессов от прочих хемилюминес-
центных заключается в том, что биолюминесцентные
реакции протекают не самопроизвольно, а катализи-
руются особыми ферментами — Л. Всякая биолюми-
несцентная система, кроме Л., обязательно включает
в себя люциферин — вещество, подвергающееся
окислению и излучающее энергию в виде света. В
большинстве случаев окисленный люциферин затем
вновь восстанавливается с помощью вспомогательных
ферментных систем. Т. обр., люциферин в этих слу-
чаях подвергается циклич. превращениям, выступая
в роли кофермента Л. Схематически указанный про-
цесс может быть представлен след, образом:
У”
sr (L*)L —x а н 2
----X
4---LU 2*-•У ^A
где LH2 — восстановленный люциферин, L* — оки-
сленный люциферин в возбужденном состоянии, L —
окисленный люциферин в нормальном состоянии,
hv — квант света, АН2 — донор водорода.
Явление люминесценции чаще всего встречается
у морских животных (ракообразных, медуз, моллю-
сков, жгутиковых, глубоководных рыб и др.), а также
у бактерий, нек-рых червей и насекомых (светляки,
жуки, мухи). Спектр излучения света у разных видов
люминесцирующих организмов различен: излучение
светляка имеет максимум при 565 ммк, различных
бактерий — в пределах 470—550 ммк, ракообразного
Cypridina hilgendorfii при 480 ммк. Уругвайский «же-
лезнодорожный червь» замечателен своей способностью
испускать свет двух цветов: зеленый — вдоль каждой
стороны тела и красный — в головной части.
Наиболее изучены биолюминесцентные системы: 1) Рако-
образного животного Cypridina, к-рая включает в себя Л. и
люциферин. Л. Cypridina, выделенная в кристаллич. состоя-
нии, представляет собой простой белок типа альбуминов, мол. в.
37 000—45 000, pf 3,28. Люциферин Cypridina также выделен
в кристаллич. состоянии в виде оранжево-желтых игл; змпи-
рич. ф-ла CjtHn-nOjN, • 2НС1, т. пл. 182—195°, представляет
собой циклич. хромополипептид. Люциферин обладает свой-
ствами слабого основания и легко окисляется кислородом
воздуха с утратой своей способности к люминесценции в при-
сутствии Л. 2) Л. светляка Photinus получена в кристаллич.
виде (длинные иглы); представляет собой эвглобулин (мол. в.
100 000, р/ 6,2—6,3); оптимум действия фермента при pH 7,8.
Фермент не содержит к.-л. простетич. группы и обладает спек-
тром простого белка. Функция Л. состоит в каталитич. дейст-
вии при образовании адениллюциферина, а также при окис-
лении последнего молекулярным кислородом. Люциферин
светляка Photinus pyralis CnHj^OaS^,!. пл. 196°, [а] д =
— —29° (в диметилформамиде), полученный в кристаллич, со-
стоянии, имеет след, строение:1
В состав люминесцентной системы бактерий входит Л. —
флавинмононуклеотид (ФМН), высокомолекулярный жирный
альдегид с числом атомов углерода от С, до С„, напр. пальми-
тиновый, и восстановленный дифосфопиридиннуклеотид
(ДПН-Н). Кристаллич. Л., выделенная из бактерии А с h-
romobacter tlscherl, представляет собой флаво-
протеид с ФМН в качестве простетич. группы, ее мол. в. 85 000,
оптимум действия при pH 6,7. Активность фермента подав-
ляется n-хлормеркурибензоатом, что свидетельствует о суще-
ственной роли SH-группы в осуществлении ферментативных
функций.
Лит.: Рид С., Возбужденные электронные состояния
в химии и биологии, пер. с англ., М., 1960; Harvey Е. N., в
кн.: Comparative biochemistry, v. 2, N. Y.—L., 1960, p. 545—87;
White E. H. [a. o.J, J. Amer. Chem. Soc., 1961, 83,
№ 10, 2402. В. Б. Спиричев.
МАГНАМИЦИН (карбомицпн) C42He7NOle, мол. в.
841,5 — антибиотик нз группы антибиотикое-лшкро-
«зримбозз
лидов; кристаллы (основание); т. пл, 212—214° (с разл.);
[а1В = —58,6° (1°/0, СНС13); рАа 7,2. Основание М. дает
устойчивые соли с минеральными к-тами; хлоргидрат,
т. пл. 149—150°. Основание М. хорошо растворимо
в большинстве органич. растворителей, нерастворимо
в воде и гексане; хлоргидрат хорошо растворим в воде.
Получен ряд биологически активных производных М.:
диацетильное производное, оксим, тиосемнкарбазон.
Восстановление М. приводит к получению тетра-
гидр опроизв одного.
М. образуется при глубинной ферментации штамма
Str. halstedii. М. выделяют нз культуральной жидкости
после отделения мицелия и подкисления до pH 3,0
экстракцией органич. растворителем, не смешиваю-
щимся с водой, напр. метилизобутнлкетоном. Оста-
ток после удаления растворителя экстрагируют серной
к-той при pH 2,0, промывают бензолом п экстрагируют
эфиром при pH 6,5, высушивают и удаляют раствори-
тель и кристаллизуют технич. магнамицин из этанола.
При изучении противоточного распределения образцов
технич. М. и анализе по растворимости был обнаружен анти-
биотик более растворимый в бензоле и спирте — магнами-
цин В, к-рый может быть выделен из маточных р-ров после
кристаллизации технич. М. Разбавлением водой и осаждением
аморфного М. с последующим растворением в абс. этаноле
удается выделить из спиртового р-ра магнамицин В. После
переосаждения его из ацетона водой выделяется технический
магнамицин В, его кристаллическое основание выделяется пос-
ле трехкратной кристаллизации из водного ацетона. Чистый
препарат основания выделен в результате противоточного рас-
пределении в системе: бензол—ацетатный буфер (pH 4,5);
магнамицин В C42H«7NOis, т. пл. 141—142°; в молекуле имеется
а-, ₽-, V", б-ненасыщенная система. При обработке магнами-
пина В иодистым калием в уксусной к-те выделяется иод и
появляется новая полоса в УФ-спектре поглощения при
278 Л1л<к, характерная для системы сопряженных двойных
связей в a-положении к карбонильной группе. Продукт реак-
ции C42H,7NO1S оказался идентичен природному магнамицину
В, но отличался от исходного М. отсутствием одного
атома кислорода. В магнамицине В отсутствует этилеиоксид-
ная группировка и имеется вторая двойная связь — С=С—,
обусловливающая наличие в его молекуле а-, Р-, у-, б-нена-
сыщенной системы.
Магнамицин В весьма близок к М. по антибактериальному
спектру и низкой токсичности. Получены его производные,
обладающие биологич. активностью. Оба антибиотика легко
образуют комплексы с ароматич. углеводородами при pH
7,5—9,5, плохо растворимы в воде и хорошо — в органич.
растворителях, они легко разлагаются в кислой среде и могут
быть использованы для получения очищенных, кислых солей
антибиотиков и выделения М. в виде основания из щелочных
растворов.
М. подавляет рост грамполоЖительных бактерий
при концентрации 0,19—1,2 мкг/мл, грамотрицатель-
ных — при 100 мкг!м.1. Он действует также на рлккет-
сни и нек-рые вирусы. М. обладает низкой токсич-
ностью. При внутривенном введении мышам хлоргид-
рата М. ЛД50 = 550 мг!кг. Клинич. применение М.
показало его эффективность при лечении заболева-
ний, вызванных штаммами стафилококка, устойчи-
выми к др. антибиотикам. М. эффективен при лечении
амебиозов.
Лит.: Т a n n е г F. W. [а. о.], Antibiot, and Chemotherapy,
1952, 2, 8, 441; Wagner R. L. [a. o.J, J. Amer. Chem.
Soc., 1953, 75, M 19, 4684; R e g n a P. P. [а. о.], там же,
1953, 75, Ай 18, 4625; H о c h s t e 1 n F. A., Regna P. P.,
там же, 1955, 77, Ай 12, 3353; HochstelnF., Mural К.,
там же, 1954, 76, № 20, 5080; Woodward R. В., Angew.
Chemie, 1957, 69, K2 1/2, 50; Perlman D., Science, 1953,
118, № 3073, 628; Трахтенберг Д. M., в кн.: Антибио-
тики, сб. переводов, 1956, Alt 5(55), 22; Т рахтенбергД. М.,
Родионовская 9. И., там же, 1959, Ай 6(74), 42.
Д. М. Трахтенберг.
ОН
МАГНЕЗОНЫ — группа органич. реактивов, при-
меняемых в основном для обнаружения и определения
магния. Все М. являются азосоединениями. Известны
магнезоны I и II и магнезон ИРЕА.
Магнезон I (n-нитробензолазорезорцин) Ci2H2O4N2,
мол. в. 259,23 — темно-красный кристаллич. порошок, т. пл.
199—220°; нерастворим в воде,
мало растворим в кипящем НО
спирте, ацетоне, уксусной к-те
и толуоле, раствор окраши- _
вается в желтый цвет; хоро- ° 2
шо растворим в щелочах с ма-
линовой окраской. Получают
М. диазотированием n-нитроанилина с последующим взаимо-
действием с резорцином. В присутствии магния красная окрас-
ка раствора реактива переходит в синюю.
Магнезон II (n-нитробензолазо-а-нафтол) Ci«HuO3Ns,
мол. в. 293,29 — темно-коричневые или красные иглы со сталь-
ным блеском; т. пл. 234—235°, —.
при нагревании до 255—260° ы-ы_/ Х-ОН
разлагается; плохо растворим в O2N N-N-C г~
обычных органических раствори-
телях, лучше—в кипящем нитро-
бензоле; хорошо — в щелочах с
образованием красно-фиолетово-
го р-ра. Получают диазотированием n-нитроанилина с после-
дующим взаимодействием с а-нафтолом. Применяют для цвет-
ной реакции на магний^ подобно магнееону I.
Магнезон ИРЕА [2-нафтол-(1-азо-2')-4'-хлор-
фенол-6-сульфокислота, натриевая соль], мол. в. 418,8,
CleH10O6N2SClNa • Н2О — мелкокристаллич. корич-
нево-красный порошок; мало
растворим в воде, этиловом
и пзоамнловом спиртах; не-
растворим в хлороформе, бен-
золе, толуоле и эфире. Синте-
зируют магнезон ИРЕА ана-
логично описанным выше ре-
активам. Водный 0,01%-ный р-р
шен в ярко-красный цвет, а 0,01 %-ный ацетоновый р-р—
в оранжевый цвет; в щелочной среде раствор реактива
окрашен в синий цвет. Раствор магнезона ИРЕА с ио-
нами Mg, Са, Al, Fes+, Со, Ni, Zn, Pb, Cd, Си, Мп,
ОН НО SO3Na
Н,О
магнезона ИРЕА окра-
1003
МАГНЕТОХИМИЯ
1004
V, Се, La и Pd образует при различных значениях pH
окрашенные соединения. Применяют его для обнару-
жения и фотометрия, определения малых количеств
магния. В присутствии магния окраска раствора
изменяется от сине-фиолетовой до ярко-красной
(pH 9,8—11,2). Благодаря быстрому образованию
окрашенного комплекса реактив может быть исполь-
зован для фотометрия, микротитроваиия магния.
Чувствительность реакции 5 10"7 г магния в 5 мл
раствора. Реактив можно использовать также в ка-
яестве индикатора при комплексонометрпч. титро-
ваниях. Применяют реактив в виде ацетонового р-ра;
устойчив не менее двух месяцев.
Лит.: Welcher F. J., Organic analytical reagent, v. 4,
N. Y., 1948, p. 379, 381; Тр. Всес. н.-и. ин-та химических реак-
тивов, вып. 23, М., 1959. И. П. Ефимов.
МАГНЕТОХИМИЯ — раздел физич. химии, пред-
метом к-рого является изучение: а) зависимости между
магнитными свойств’ами и химич. строением веществ п
б) влияния магнитных явлений на кинетику химия,
реакций. При не очень низких темп-рах энергии ато-
мов и небольших молекул вещества непрерывно под-
вергаются столь сильным случайным флюктуациям
вследствие хаотич. теплового движения, что измене-
ния энергии, вызываемые обычными (до 104 эрстед)
магнитными полями, оказываются незаметными. Поэ-
тому влияние такого рода магнитных полей на ско-
рость химич. реакций является незначительным по
сравнению с влиянием темп-ры. Заметного эффекта
можно было бы, по-видимому, ожидать для неболь-
ших молекул лишь при магнитных полях в согни и
тысячи раз более сильных, чем обычные поля,
создаваемые в лабораторных условиях. Одиако
подобного рода сверхсильные поля в настоящее время
удается поддерживать лишь в течение очень коротких
промежутков времени (10~3—10~4 сек), так что опытная
проверка влияния этих магнитных полей на кинетику
химич. реакций до сих пор не производилась. Иначе
обстоит дело у макромолекул с молекулярным весом
106 и выше. Изменения энергии, вызываемые в них
даже полями около 104 эрстед, оказываются величи-
нами того же порядка, что и средняя кинетич. энергия
теплового движения. Поэтому можно ожидать влия-
ния обычных магнитных полей на кинетику химич.
реакций в растворах макромолекул, но эксперимен-
тально этот вопрос еще не изучен. Поскольку вторая
из указанных выше задач (б) до сих пор не привлека-
ла внимания, магнетохимич. исследования сосредото-
чены пока в основном на первой задаче (а).
Магнитные свойства вещества характеризуются
для слабомагнитных тел магнитной вос-
приимчивостью хи собственным магнит-
ным моментом атомов и молекул р. Характе-
ристиками сильно магнитных тел служат намаг-
ниченность насыщения о'00, а также
величины, определяющие гистерезис.
Результаты экспериментальных исследований маг-
нитных свойств анализируются теоретически с помощью
современной квантовой теории магнетизма и квантовой
химии. При образовании двухэлектронной химич. свя-
зи происходит, как известно, взаимная компенсация
магнитных моментов (спинов) входящей в связь пары
электронов. Ввиду этого через всю М. красной нитью
проходит правило, согласно к-рому каждый акт обра-
зования прочной двухэлектронной связи соответ-
ственно снижает результирующий собственный маг-
нитный момент молекулы. Напр., у каждого атома
водорода имеется магнитный момент, равный спино-
вому моменту одного электрона (магнетон БораЛ/в,
равный 0,9273- 10~20CGSM). У молекулы Н2 магнитный
момент отсутствует из-за образования двухэлектрон-
ной связи. У каждого атома кислорода имеется спино-
вый магнитный момент, равный двум Л/в. При образо-
вании молекулы Н2О оба валентных электрона атома О
вступают в двухэлектрониую связь с атомами Н, и
результирующий момент оказывается равным нулю.
Исследование магнитных моментов до и после образо-
вания химич. соединения позволяет т. обр. судить
о степени насыщения валентностей.
Важным обстоятельством является то, что свободные
радикалы, т. е. химически активные состояния атомов
или молекул, всегда характеризуются одним или
несколькими (бирадикалы) нескомпенсированными
(неспаренными) спиновыми электронными моментами.
Молекулы химически насыщенных веществ, как пра-
вило, лишены магнитного момента, наличие же маг-
нитного момента свидетельствует о неполной насыщен-
ности данного соединения. Неспаренные спины обус-
ловливают появление свойства, именуемого парамаг-
нетизмом (см. Магнитные свойства). Диамагнетизм
(см. там же) наблюдается при отсутствии момента.
Основным в изучении химически ненасыщенных ве-
ществ является вопрос о количестве нескомпенсиро-
ванных электронных спинов, о природе их носителей
и о взаимодействии этих электронов с остальной
частью вещества.
Наиболее чувствительным методом обнаружения
нескомпепсированных электронных спинов является
метод электронного парамагнитного резонанса, по-
зволяющий обнаружить уже 1014 спинов на грамм
(см. Радиоспектроскопия). Определение числа неском-
пенсированных электронов, приходящегося на моле-
кулу химич. соединения, наиболее удобно осущест-
влять путем измерения магнитной восприимчивости у.
Однако это возмояшо лишь тогда, когда концентрация
молекул—носителей неспаренных спинов—составляет
не менее 1020 на грамм вещества. Следует также заме-
тить, что сама магнитная восприимчивость у веще-
ства слагается из двух величин % = %р~|~ Xd, где /р—
доля магнетизма, зависящего от самих неспаренных
спинов (парамагнетизм), и '/ri — доля магнетизма,
зависящего от всей остальной части вещества (диамаг-
нетизм). Поскольку '/р падает с ростом темп-ры, а
/(] не зависит от темп-ры, то при очень низких темп-рах
Хр )> yd. В этих условиях можно приближенно считать
X — %р и не учитывать ’/d вовсе.
Исследования, произведенные на координационных
(комплексных) соединениях, значительно продвинули
наши познания о химич. связи в этих веществах. Так,
напр., атом железа обнаруживает в растворах FeCl3
магнитный момент, свидетельствующий о наличии у
Fe3+ 5 неспарепных электронов. В комплексном же
соединении K3[Fe(CN)6] железо обнаруживает момент,
свойственный лишь 1 неспаренному спину. Соедине-
ние K4[Fe(CN)6] вовсе лишено магнитного момента, что
объясняется отсутствием неспаренных'электрояов. Или,
напр., у иона Ni2+ оказывается момент, свойственный
двум неспаренпым электронам, а в квадратных плос-
ких комплексах типа K„[Ni(CN)4] момент атома ни-
келя равен нулю. Карбонил железа Fe(CO)6 лишен
магнитного момента, что показывает, что он имеет
структуру, при к-рой все 6 связей, имеющихся у атома
Fe, насыщены. Магнетохимич. исследования позво-
лили этим путем выяснить [электронное строение
таких сложных веществ, как гемопротеины. [Магнит-
ный момент молекулы гемоглобина, как оказалось,
соответствует 5 неспаренным спинам, между тем как
оксигемоглобин во всех случаях лишен магнитного
момента. Это означает, что при окислении гемоглобина
насыщаются все свободные валентности.
Между тем как в парамагнитных ненасыщенных со-
единениях отдельные носители магнитного момента
практически независимы друг от друга, в ферромаг-
нитных и антиферромагнитных веществах они очень
1005
МАГНЕТОХИМИЯ
сильно взаимодействуют друг с другом. К числу таких
соединений относятся окислы, сульфиды, теллуриды,
особенно со структурой ферритов, манганитов, грана-
тов, и т. п. соединения, содержащие атомы (или ионы)
переходных элементов.
В этих антиферромагнетиках при низких темп-рах
силы взаимодействия между носителями момента столь
велики, что в отсутствии внешнего поля магнитные мо-
менты оказываются ориентированными по отношению
друг к другу. В одних антиферромагнетиках эта ориен-
тация приводит к полной взаимной компенсации мо-
ментов (напр., MnF2, NiF2), в других антиферромагпе-
тиках получается неполная компенсация (ферриты,
манганаты, гранаты). В ферромагнетиках наблюдается
полный взаимный параллелизм всех магнитных мо-
ментов. Измерение максимальной намагниченности
этих веществ в очень сильных полях (так наз. намаг-
ниченность насыщения ак) позволяет определить маг-
нитный момент, к-рый характеризует валентность
носителя момента. Т. обр., изучение магнитных свойств
позволяет делать важные заключения о химич. связях
переходных атомов в этого рода соединениях. То об-
стоятельство, что многие из этих веществ играют роль
катализаторов, дало возможность применить М. к ис-
следованию процессов гетерогенного катализа. Вся-
кий раз, когда на поверхности катализатора происхо-
дит хемосорбция посторонних атомов или молекул,
возникает изменение магнитных моментов катализа-
тора за счет образования двухэлектронных связей.
Т. обр., исследование магнитных свойств катализато-
ров как в процессах их изготовления, так и в самих
каталитич. реакциях позволяет вскрыть весьма ин-
тересные стороны механизма подобных процессов.
Разумеется, обнаружение этих изменений в магнитных
свойствах возможно только в том случае, если катали-
затор изучается в мелкодисперсной форме, при к-рой
роль поверхностных слоев доминирует над ролью
объема вещества. Однако нек-рые вещества в очень
мелкодисперсном виде обнаруживают крайне не-
ожиданные свойства, резко отличные от свойств тех
же сплошных веществ, что сильно затрудняет интер-
претацию опытов. Среди исследователей нет еще уста-
новившегося мнения относительно всех этих опытных
данных. Возможно, что некоторые из этих результа-
тов обусловлены ферромагнитным загрязнением (об
этих загрязнениях см. ниже), внесенным в образец
в процессе их изготовления. Впрочем, теория маг-
нетизма показывает, что процессы намагничивания
ферро- и антиферромагнитных веществ в мелко-
дисперсном виде имеют свою специфику, которую
также необходимо учитывать в такого рода иссле-
дованиях.
Среди химически ненасыщенных веществ особую
группу образуют как слабомагнитные, так и сильно-
магнитные металлы в твердом и жидком состоянии.
Хотя изучение именно магнитных свойств впервые по-
зволило вскрыть химич. природу связи в интерметал-
лич. соединениях, магнетохимия сплавов, в силу
своей крайней сложности, до сих пор еще не
разработана. Наиболее многочисленную группу не-
металлич. химических соединений образуют соеди-
нения с полностью насыщенными валентностями.
Будучи лишены собственных магнитных моментов,
они обнаруживают, как правило, диамагнитные свой-
ства. Таковы, напр., галогениды щелочных и щелочно-
земельных металлов, где как катион, так и анион
приняли конфигурации атомов инертных газов, пол-
ностью лишенные магнитных моментов. Полностью
лишено магнитного момента громадное большинство
органич. молекул с ковалентной связью, в к-рых
отсутствуют неспаренные электроны.
Паскаль разработал эмпирич. схему, позволяющую
заранее предвычислять величину диамагнитной вос-
1006
приимчивости (на I моль) %м органич. соединения,
если известны его химич. состав и его структурная
ф-ла. Эта схема может быть использована для проверки
структурной формулы путем измерения восприимчи-
вости '"/.у на опыте, если химич. состав заранее изве-
стен. Согласно этой схеме
%M==SXa+S^ (1)
где %А — «атомный инкремент», т. е. вклад восприим-
чивости, приходящийся на один атом, а ). — структур-
ная поправка, зависящая от вида связи между ато-
мами. В табл. 1 приведён новейший вариант значений
уАи X в этой схеме (вариант Паскаля — Оарб).
Таблица 1. Схема Паскаля (вариант Паскаля—Оарб)
Атомы и группы атомов -ХА • ‘0е Вид связи к 10»
н 2 Двойная связь 4- 5,5
с 7,4 Связь в метиле — 0,85
0 (алкоголи) 5,3 Кольцо бензольное —15,0
С=»О (альдегиды и кетоны) /0 с (кислоты) 6,4 » антраценовое -49,1
15,15 » тиофеновое -14,22
^0 S 16,9 C=N + 4.-0
N 9
CI 18,5
Вг 27,8
J 42,8
Аддитивная схема Паскаля нередко применяется хи-
миками и дает удовлетворительное согласие с опытом,
однако необходимо иметь в виду, что применяемые в ней
численные значения как инкрементов %А (табл. 1), при-
писываемые атомам, так и X имеют чисто условный
характер и в различных вариантах схемы они сильно
отличаются друг от друга. Поэтому никаких дру-
гих выводов о строении органич. молекул, кроме
проверки структурных формул, из этой схемы делать
нельзя.
В последнее время начало, однако, развиваться
новое направление в этой области М.; стремятся
выяснить реальные значения восприимчивостей от-
дельных атомов и отдельных связей. Это направление
исходит из того факта, что восприимчивость диамаг-
нитных веществ слагается из двух членов — одного
чисто диамагнитного (т. е отрицательного) и
другого (положительного) %рМ. Первый член зависит
от габаритов электронных орбит молекулы, но слабо
зависит от структуры. Второй член, наоборот,
является структурно-чувствительным; он зависит от
характера связи, от полярности и других особенностей
строения. Оба члена не зависят от темп-ры. Оказы-
вается, что %dM может быть приближенно рассчита-
на, если иэ опыта известны состав и молекулярная
рефракция данного вещества
•х<ш=—t07'10 0 VRk (2)
где 7? — молекулярная рефракция соединения, а к —
полное число электронов в молекуле соединения. По-
скольку полное число, электронов в атоме равно
его атомному номеру, то, зная состав молекулы, не-
трудно найти к. Если измерена на опыте, то из
соотношения уру = %м—xdy можно получить важ-
ные сведения о строении. При наличии в молекуле
нескольких связей различного типа с различными
Х рМ суммарная %рМв большинстве случаев аддитивно
складывается из них, т. е. уру = S%'pM. Следовательно,
магнитная восприимчивость (на 1 моль) соединения
выражается формулой
Хм = Хам+2 Х'рм (3)
1007
МАГНЕТОХИМИЯ - МАГНИЙ
1008
имеющей внешнее сходство с формулой Паскаля, но
отличающейся от нее тем, что все входящие в нее
величины имеют реальный физич. смысл. В табл. 2
приведены нек-рые приближенные значения %рМ1
наблюденные у различных видов ковалентных Связей
(новая схема по Я. Г. Дорфману).
Величина %рМ характеризует распределение элект-
ронов на связи. При сферич. распределении электро-
нов Хрм = 0- Чем больше Хрм> тем сильнее отличается
распределение электронов от сферического. Из табл. 2
можно видеть, что наиболее близким к сферич. яв-
ляется, как правило, распределение электронов на оди-
нарной связи, сильнее всего отличаются от сферич.
симметрии двойные связи С = С, С= N, С= О ит. п.
Таблица 2. Новая схема (Я. Г. Дорфман)
Вид связи 4м •1ов Вид СВЯЗИ 4м •1ов Вид СВЯЗИ 4м •106
с—с 0 С —С1 2,0Д C-N 0
с—н оа с=с 4,0е р+О 8,4
с—н с—н 0,25 0,30 0,8в Q Q II III о 2,5 8 N/ О ‘No 16,5
о—н 1,8 с— -N 3 s=o 9,5
N —Н О—С 0 2,5Г с = о 9,5 C-.-U.C 1,6Ж
а В метиле. 6 В метилене. в В метине. г В эфире. Д В од-
нохлорзамещенных углеводородах. е В этилене. т Ароматич.
связь в бензольных и нафталиновых кольцах.
Ниже рассмотрено применение обеих схем к кон-
кретным примерам.
Новая схема
Схема Паскаля
Предварительный расчет
на основании имеющейся
структурной формулы:
ХМ=10Хс + 20хм 4-
+ ^снз'Ь^с— с~ Н2,75*
♦ 10-е.
Опыт дает — 112,6 • 10*6
Структурная формула вер-
на. Однако, поскольку
5Хсн3=—4,25 • 10-’, а
1ЛС^О = -5,5 • 10-’,
сумма ЕЛ составляет лишь
+ 1,25 -10-’, т. е. 1% от
Хщ, а это есть точность
экспериментального зна-
чения хм-
Расчет на основании имею-
щейся структурной фор-
мулы:
X = 6хс4-5Хн+хс1+3*с=с+
4“ ^бензол = — *^’9 * Ю-6.
Опыт дает 70 * 10-в. Струк-
турная формула верна.
Однако 2 А. = ЗАС=С 4-
4- ^бензол =4-1 * Ю-e, что
составляет лишь 1,4% от
величины
1. Три метил - 2,4,6 - гептен - ЗСщНго
Расчет на основании имею-
щейся структурной форму-
лы с учетом опытных зна-
чений молекулярной реф-
ракции и магнитной вос-
приимчивости:
R = 47,9
fe = 80
ХМ =-112,6 -10-’
XdM =—122 • 10-’
ХрМ “ХМ - XdM = 9’4 •
• 10-’
ХрМ=Хр мс =с+ЗХэдсс_н+
+ *рМсн2= %М =(R'R +
+ 2,4 + 0,6) • 10'’=9,6 • 10-’
Структурная формула вер-
на, причем 2 ХрМ состав-
ляет 9% от величины
2. Хлорбензол СвН&С1
Расчет на основании имею-
щейся структурной фор-
мулы с учетом опытных
данных молекулярной ре-
фракции и опытн. значения
восприимчивости:
R ==31,6
fe = 58
Ход 70 -10-е
XdM =- 84 • 10 ’
Хрм = 1\ • ю-’
ХрМ = еХрМс::-.с+
+хрМс -CI = S*pM = (9’S +
+ 2). 10-’ = llj6 . 10-в
Структурная формула вер-
на, причем 1>/ составляет
14% от величины хм.
Структурная формула проявляется в схеме Паскаля
на величине SX, а в новой схеме на величине 2%’рМ, но
надежность проверки структуры тем больше,чем больше
численные значения этих структурных поправок по срав-
нению с погрешностью в измерении %м. Во многих
случаях, проверка структурных формул по новой
схеме более надежна, чем по схеме Паскаля, ибо
^Хрм значительно превосходит погрешность измере-
ния %м. По мере накопления опыта и уточнения дан-
ных табл. 2 преимущества новой схемы будут, по-
видимому, возрастать. Схема Паскаля не может быть
применена к ионным соединениям без особых огово-
рок, в то время как новая схема применима и к ним.
Напр., известно, что в органич. (ковалентных) со-
единениях нитрогруппа обнаруживает Хрmno2 = 16,5 •
•10-6. Новая схема позволяет проверить, правильно
ли предположение, что в ионе (NO3)~ каждая связь
между N и О на 1/3 соответствует нитросвязи. Из
аддитивности ионных восприимчивостей в нитратах
XmnOs^ — 2®’ Ю-6- Из аддитивности ионных рефрак-
ций определяется рефракция иона NO3“, позволяю-
щая рассчитать X^mno^ ~ ^5 • 10-8. Оказывается
XmnoT — XdMNOJ = XpMNOj^ 15 • 10~e , что действи-
тельно соответствует предполагаемой структуре.
Аналогичным методом могут изучаться структурные
особенности и более сложных молекулярных ионов.
Поскольку молекулы с диамагнитными свойствами
зачастую анизотропны, анизотропией обладают и диа-
магнитные молекулярные кристаллы. Изучение диа-
магнитной анизотропии ароматич. соединений откры-
вает путь к выяснению делокализации л-электронов
в ароматических кольцах. Делокализация л-электро-
нов играет, возможно, активную роль в некоторых
биохимич. процессах, связанных с образованием зло-
качественных опухолей, поэтому магнетохимич. иссле-
дование ароматич. соединений представляет большой
интерес для биохимии и хемотерапии. Вопрос этот до
сих пор недостаточно изучен.
При магнетохимич. изучении диамагнитных и пара-
магнитных веществ необходимо обращать самое
серьезное внимание на удаление нерастворимых ферро-
магнитных примесей. Если, напр., исследуемое твер-
дое диамагнитное вещество содержит всего лишь 10'6%
металлич. железа или 10 4 % ферромагнитных окислов
железа (Fe3O4, Fe2O3 и т. п.),то намагниченность,
приобретаемая крупинками примеси, сравнима с на-
магниченностью самого образца и вносит серьезные
искажения. Если частички примеси имеют удлинен-
ную форму или форму пластинок, то при протяжке или
прокатке исследуемого вещества они могут ориенти-
роваться определенным образом (напр., вдоль направ-
ления протяжки или прокатки) и создавать анизот-
ропию магнитных свойств. В заключение следует под-
черкнуть, что в М. имеется еще большое число нере-
шенных вопросов. Основной причиной этого положе-
ния является то, что магнетохимич. эксперименталь-
ные исследования зачастую ведутся без надлежащего
учета всех достижений современной физики магнитных
явлений.
Лит.: Клемм В., Магнетохимия, пер. с нем., М., 1939;
Д о р ф м а н Я. Г., Магнитные свойства и строение вещества,
М., 1 955; Селвуд П., Магнетохимия, пер. с англ., М., 1958;
Дорфман Я. Г., Диамагнетизм и химическая связь, М.,
1961. Я. Г. Дорфман.
МАГНИЙ (Magnesium) Mg — химич. элемент II гр.
периодич. системы Менделеева; п. п. 12, ат. в. 24,312.
Природный М. состоит из трех устойчивых изотопов;
Mg24 (78,6%), Mg25 (10,11 %) и Mg26 (11,29%). Сечение
захвата тепловых нейтронов атомом М. 0,059 барн.
Известны три искусственных радиоактивных изотопа
Mg23 (7i/2 = 12,3 сек), Mg27 (71/2 = 9,45 сек), Mg28
(Ti/2 = 21,2 часа). Последний может быть использо-
ван как индикатор. Конфигурация внешних электро-
нов атома М. 3s2. Энергии ионизации (в ав)'. Mg° —<•
1009
МАГНИЙ
1010
— Mg+—► Mg2+ —► Mg3+ —► Mg1+—»Mgs+ соответственно
равны 7,64; 15,03; 80,12; 109,29; 141,23.
В 1808 Дэви, исходя из известной давно магнезии, получил
с помощью вольтова столба амальгаму нового металла, к-рый
он назвал по исходному веществу magnesium. Это наимено-
вание сохранилось за М. на всех языках. Металлич. М. был
впервые получен в 1828 А. Бюсси действием паров калия на
расплавленный хлорид М.
Содержание М. в земной коре 2,10 вес. %. По рас-
пространенности М. занимает6-е место среди элементов.
Самородного М. в природе нет. В первичных горных
породах М. находится, за редкими исключениями, в со-
ставе силикатных минералов, среди к-рых преобладает
форстерит, или оливин, Mg2SiO4 (34,6% Mg;
плотн. 3,2), обычно содержащий в виде изоморфной
примеси 8—25% фаялита Fe2SiO3. Как главный поро-
дообразующий минерал, оливин в составе дунитов и
перидотов образует горные массивы, к которым при-
урочены важнейшие месторождения хрома, никеля,
платины. От выветривания оливиновых пород проис-
ходят столь же широко распространенные змее-
вик и с главным минералом серпентином
[SiO4]2Mg2[Mg(OH)]H3 (26,3% Mg; плотн. 2,6), в к-ром
Mg2+отчасти замещен Fe21'iiNi2''. Змеевики вместе с
первичными породами слагают горные массивы на Ура-
ле, на главном Кавказском хребте, в Поднепровье, в
Монче-Тундре, на Саянах и т. д. Они широко распро-
странены и в других странах. К змеевикам приурочена
их волокнистая разновидность—асбест. Мощные отва-
лы змеевика и оливинита в местах добычи асбеста слу-
жат (особенно в странах, бедных магнезитом) сырьем
для произ-ва MgO. При частичном разложении змееви-
ков горячими глубинными углекислыми водами образу-
ются тальковые породы, а при полном разложении —
кристаллич. магнезит MgCO3 (28,8% Mg; плотн.
2,9—3,1) с изоморфными примесями других карбона-
тов (см. Магния карбонаты). От действия поверхност-
ных вод на Оливины и змеевики образуются залежи
аморфного магнезита, далеко уступающие по запасам
месторождениям первого рода. Магнезитами богат
СССР (крупнейшее в мире Саткинское месторождение
около Челябинска), а также Китайская Народная
Республика, Корейская Народно-Демократическая
Республика, Индия, Чехословакия, Австрия, Югосла-
вия, Греция (о-в Эвбея) и др. Известняки под действием
рассолов, содержащих MgCl2, переходят в доло-
мит MgCa(CO3)2 (13,2% Mg; плотн. 2,8—2,9). На
долю доломита приходится до 40% всех карбонатных
пород. Хотя главное назначение магнезита и доломита
в пром-сти — произ-во огнеупоров, они пригодны и
как магниевые руды либо непосредственно, либо после
переработки (на MgCl2 и MgO). Источником MgCl2
и MgO служат горько-соленые воды — от морской,
содержащей до 0,38% MgCl2, до рапы в замкнутых
водоемах в засушливых странах, содержащей до 30%
MgCl2 (в СССР — озера Баскунчак, Эльтон и Индер
в Юж. Заволжье, Сакское и Красное — в Крыму,
Кучук — на Алтае и мн. др., Мертвое море — в Иор-
дании, Б. Соленое оз. — в Юта, США).
К магниевым рудам относятся также ископаемые
соли М., залегающие поверх толщ каменной соли во
многих месторождениях (напр., в СССР в обширнейшем
Верхне-Камском).
Физические и химические свойства. М. — серебри-
сто-белый блестящий металл, тускнеющий на воздухе
вследствие окисления. Кристаллическая решетка М.
гексагональная плотноупакованная, а — 3,2028 А,
с — 5,1998 А; полиморфных превращений нет. Атомный
радиус 1,60 А, ионный радиус Mg2+ 0,74 А. Плотн.
99,9%-ного М. 1,739. Т. пл. 651°, т. кип. 1107°. Теп-
лоты плавления и испарения (при т. кип.)в кал/г-атом
соответственно равны 2100 и 30500. Уд. теплоемкость
в кал/г -град-. 0,241 (0°); 0,248 (20°); 0,254(100°); 0,312
(650е). Теплопроводность 0,37 кал/см • сек • град (20°).
Термич. коэфф, линейного расширения 25,0 • 10~6
0,0188 / (в интервалеО—550°), термич. коэфф, объем-
ного расширения в жидком состоянии 380 • 10-6
(651—800°). Уд. электрич. сопротивление 4,5 10~6
ом- см (20°). Давление насыщенного пара М.
в мм рт. ст.-. 1,66 (627°); 8,71 (727°); 407,4 (1027°);
760 (1107°). М. парамагнитен, его уд. магнитная
восприимчивость -)-0,5 • 10~6. М. — сравнительно мяг-
кий и пластичный металл. Его механич. свойства
сильно зависят от обработки. Ниже даны механич.
свойства М. при 20° соответственно для литого и
деформированного образца. Предел текучести (в
кГ/мм2): 2,5 и 9,0; предел прочности (в кГ/мм2): 11,5
и 20,0; относительное удлинение (в %): 8,0 и 11,5;
относительное сужение (в %): 9,0 и 12,5; твердость по
Бринеллю (в кГ/мм2): 30 и 36.
Во всех своих стойких соединениях М. 2-валентен.
Химически металл весьма активен; на воздухе от окис-
ления его защищает окисная пленка даже при нагрева-
нии примерно до 350°. Едва заметное окисление уско-
ряется с дальнейшим нагреванием, что отвечает усиле-
нию испарения М. и нарушению цельности пленки. При
600—650° М. воспламеняется и, испаряясь, сгорает на
воздухе с образованием густого дыма окиси и отчасти
нитрида, с ослепительным белым светом (см. Магния
окись). Нитрид Mg3N2 — зеленоватый порошок,
устойчивый без плавления до 600°, образуется при
нагревании М. в атмосфере азота до 500°. От влаги он
легко разлагается с образованием Mg(OH)2 и NH3,
поэтому окалина, образующаяся при плавлении маг-
ниевых сплавов и на зеркале жидкого М., пахнет ам-
миаком. Жидкий М. во влажной атмосфере разлагает
Н2О, насыщаясь водородом, к-рый почти нацело вы-
деляется при застывании. Однако в среде водорода
М., нагретый до 400—500°, образует гидрид
MgH2. При малых давлениях в среде водорода пар М.
дает спектр с полосами, к-рые указывают на образо-
вание субгидрида MgH. При нагревании М. соеди-
няется с галогенами (с влажным хлором на холоду),
образуя галогениды MgX2, из к-рых важнейшим яв-
ляется магния хлорид. При нагревании М. до 500—
600° с серой или с SO2 и H2S образуется магния суль-
фид MgS. В тех же условиях М. разлагает углеводоро-
ды с образованием карбидов MgC2 и Mg2C3.
Водой MgC2 разлагается с выделением ацетилена, а
Mg2C3 — с выделением преим. аллилена СН3—С =
= СН. При нагревании выше 500° MgC2 распадается
на сажу и Mg2C3, к-рый, начиная с 800°, распадается
на элементы. Известны соединения Mg2Si, MgsSi2,
Mg3P2, Mg3As2 и Mg3Se2. Все они разлагаются разб.
к-тами с выделением соответственно ядовитых сила-
нов, фосфина, арсина и стибина. За малыми исключе-
ниями, М. сплавляется с металлами и образует интер-
металлиды (см. Магния сплавы). Будучи сильным вос-
становителем, М. при нагревании вытесняет из окислов
и галогенидов другие металлы, включая щелочные,
бериллий, алюминий, а также неметаллы — бор, крем-
ний и углерод.
Стандартный электродный потенциал М. относи-
тельно водорода равен — 2,4 в, поэтому М. вытесняет
большинство металлов из растворов их солей в воде.
Холодная вода, свободная от воздуха, на М. почти не
действует; из кипящей воды он медленно выделяет
водород. Водяной пар сильно реагирует с М., начиная
с 400°. В разб. к-тах М. легко растворяется уже на
холоду. В плавиковой к-те, однако, он нерастворим
вследствие образования нерастворимой защитной
пленки MgF2. В конц. серной к-те и в смеси ее с азотной
М. почти нерастворим. Водные р-ры щелочей на холоду
на М. не действуют. В р-рах щелочных бикарбонатов и
солей аммония М. растворяется; с последними он обра-
зует в растворе комплексные катионы [Mg(NH4)2]2+.
Почти все соли М. бесцветны, хорошо растворимы
1011
МАГНИЙ - МАГНИЙОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
1012
в воде и имеют горький вкус. Щелочи осаждают из
р-ров солей М. гидроокись Mg(OH)2 (см. Магния
окись), растворимость к-рой в воде ничтожна. Из солей
М. в воде труднорастворимы MgF.2, MgCO3, Mg,(PO4)2,
MgNH4PO4, MgNH4AsO4 (о важнейших солях М. см.
Магния сульфат, Магния карбонаты, Магния хлорид).
Аналитическое определение. М.
относится к аналитич. группе элементов, у к-рых
трудно растворимы карбонаты и фосфаты. Нов присут-
ствии NH3 карбонат М. не выпадает, что позволяет
отделить М. от прочих катионов этой группы. Как
правило, М. осаждают в виде фосфата магния и аммо-
ния MgNH4PO4 • 6Н2О и после прокаливания взвеши-
вают в виде пирофосфата магния Mg2P2O7. Перед
определением М. из раствора осаждают сульфиды
тяжелых металлов, гидроокиси железа и марганца
(аммиаком с добавкой окислителя), оксалат кальция
(оксалатом аммония). Качественно М. открывают по
его окрашенным соединениям с хинализарином, тита-
новым желтым, магнезоном, а также по выпадению
в осадок MgNH4PO4 и MgNH4A«O4. Количественно
М. может быть определен весовым методом — осажде-
нием MgNH4PO4 с последующим прокаливанием его
до весовой формы пирофосфата, а также в виде окси-
хинолята. Объемные определения М. основаны на
осаждении Mg(OH)2 щелочью и обратном титровании
ее избытка кислотой или на титровании оксихинолята
М. р-ром бромата. Удобнее новый метод титрования
комплексонами.
Получение. В пром-сти М. получают тремя спосо-
бами: электролитическим, металлотермическим и уг-
летермическим. По электролитическому
методу производят наибольшее количество М.
В этом случае исходным сырьем служит безводный
хлористый магний или обезвоженный карналлит.
В состав электролита входят также хлориды натрия,
калия и кальция и небольшое количество фторидов
натрия или кальция. Содержание MgCl2 в электро-
лите должно быть не ниже 5—7%. По мере расходо-
вания MgCl2 и накопления других солей в электролите
часть его выводят из электролизера и добавляют рас-
плавленный хлористый магнии или карналлит.
Темп-ра электролиза 720—750°. Аноды изготовляют
из графита, катоды — из стали. Катодное простран-
ство отделяют от анодного перегородкой, не доходящей
до дна электролизера. Расплавленный М. легче элект-
ролита. Он всплывает на поверхность и периодически
извлекается из катодного пространства вакуум-ков-
шом. Выход по току 83—85%; расход электроэнергии
18—21 кет-ч/кг металла. В магнии-сырце содер-
жится ок. 2% примесей. Его рафинируют в тигельных
электрич. печах под слоем флюсов и затем разливают
в чушки. В лучшем сорте первичного металла содер-
жится только 99,8% Mg. Для дальнейшей очистки
металл подвергают сублимации в вакууме. После 2- и
3-кратной сублимации можно получить металл чисто-
той 99,999%. Хлор, получающийся на аноде, по тру-
бам поступает на очистку конц. серной к-той, после
чего идет в шахтные электропечи для получения без-
водного MgCl2 из магнезита или из хлорокиси М.
(2MgCO3 -j- 2С12 -j- С = 2 MgCl2 -j- ЗСО2) или на по-
лучение четыреххлористого титана (TiO2 2С13 -|-
4~ С = TiCl4 -f- СО2), а также на произ-во хлорной
извести и других хлоропроизводных.
При металлотермическом спо-
собе произ-ва М. сырьем служит доломит, а восста-
новителем — ферросилиций или силикоалюминий.
Доломит прокаливают до полного разложения карбо-
натов, затем размалывают, смешивают с размолотым
восстановителем и брикетируют. Брикеты загружают
в ретортные или во вращающиеся печи с графитовыми
или угольными нагревателями и выдерживают их там
в вакууме (остаточное давление 1—2 мм рт. ст.)
при темп-ре 1280—1300°. Полученный в парообразном
состоянии М. поступает в конденсаторы, нагретые до
400—500°, где собирается в виде кристаллов. Такой
металл для дальнейшей очистки подвергают пере-
плавке под флюсом или в вакууме и затем разливают
в чушки. В последнем случае получают М. достаточно
высокой чистоты, мало склонный к коррозии, а потому
служащий хорошим восстановителем или материалом
для произ-ва сплавов.
По углетермическому способу М.
получают в герметичных 3-фазных электрич. печах
при темп-ре выше 2100°. Окись магния смешивают
с углем и брикетируют. В результате восстановления
(MgO -j- С Mg %- СО) металл получается в паро-
образном виде. При выходе из печи газообразную
смесь быстро охлаждают инертным газом для предот-
вращения обратного взаимодействия СО с парами М.
Дальнейшее охлаждение смеси проводят в холодиль-
никах. Полное отделение твердых частиц М., MgO
и углерода от смеси газов производят с помощью ме-
шочных фильтров. Полученную смесь — т. н. пусьеру,
состоящую примерно из 80% Mg, 15% MgO и 5% С,
направляют в ретортные печи для очистки М. субли-
мацией. М. переплавляют и отливают в чушки, а ос-
таток, состоящий из MgO и С, используют как напол-
нитель резины.
Техника безопасности. При получении М. возможны пора-
жения электрич. током, ожоги расплавленным электролитом
и воздействие вредных газов: Cl2, НС1 и СО. Мерами предупре-
ждения против электрич. тока являются хорошая изоляция
электролизеров и печей (застилка полов асфальтом, диабазом,
установка на фарфоровых изоляторах); против ожогов — при-
менение соответствующей шерстяной спецодежды (костюмы,
валенки, фартуки) и защитных очков. Усиленная вентиляция
и подача кондиционированного воздуха почти полностью
устраняют воздействие вредных газов на организм человека.
Применение. Основная область применения метал-
лич. М. — произ-во сверхлегких сплавов (см. Магния
сплаеы). В металлургии М. используют для раскисле-
ния и обессеривания нек-рых металлов и сплавов, для
получения трудновосстанавливаемых металлов — ва-
надия, титана, урана, циркония и др., а также в про-
изводстве высокопрочного т. наз. «магниевого» чугуна
с включенным графитом. Смеси порошка М. с окисли-
телями применяют для изготовления осветительных
и зажигательных ракет, снарядов и авиабомб, в кино-,
фото- и осветительной технике. Широкое применение
находят и соединения М. (см. Магния окись, Магния
карбонаты, Магния сульфат, Ангидрон, Магнийорга-
нические соединения).
Лит.: Гуськов В. М., Производство магния, Л.—М.,
1938; Стрелец X. Л., Тайц А. Ю., Гуляниц-
к и й Б. С., Металлургия магния, 2 изд., М., 1960; П о р т-
н о й К. И., Л е б е д е в А. А., Магниевые сплавы, М., 1952;
Roberts С. Sb., Magnesium and Its alloys, N. У.—L.,
1960; Б e л я e в А. И., Металлургия легких металлов, 4 изд.,
М., 1954; Пазухин В. А., Ф и ш е р А. Я., Вакуум в ме-
таллургии, М., 1956; Гиллебранд В. Ф. [и др.1, Прак-
тическое руководство по неорганическому анализу, пер.
о англ., М„ 1957; Gmelin, 8 Aufl., Syst.-Num. 27, Mag-
nesium, TI А—В, В., 1937—53; M e 1 1 о r, v. 4, L.—N. Y. —
Toronto, 1946; то же, v. 4, 1952; Pascal, t. 4, P., 1958;
Ullmann, 3 Aufl., Bd 12, Milnch. — B., 1960, S. 75—133;
Kirk, v. 8, N. Y., 1952, p. 554—617. В. M. Гуськов.
МАГНИЙОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ — сое-
динения общей ф-лы R2Mg или RMgX. Первые из
них известны со 2-й половины 19 в. Следы диэтилмаг-
ния были получены нагреванием йодистого этила
с магнием в запаянной трубке. Магнийдиалкилы полу-
чались также обменной реакцией диалкилртути с маг-
нием (CH3)2Hg 4- Mg — (CH3)2Mg-f- Hg. Этим путем
получены диметил-, диэтил- и дифенилмагний — твер-
дые продукты, возгоняющиеся в высоком вакууме,
растворимые в эфире с образованием эфиратов. Со-
единения эти весьма чувствительны к воздействию
кислорода, влаги и СО2; способны к самовоспламене-
нию не только на воздухе, но и в атмосфере СО2.
По указанным причинам магнийдиалкилы не получили
практич. применения.
1013
МАГНИЙОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ - МАГНИТНЫЕ СВОЙСТВА
1014
Громадное значение в синтетич. органич. химии
получили смешанные М. с. RMgX (X — галоген),
впервые введенные в практику П. Барбье и В. Гринья-
ром. В 1900 Гриньяр установил, что мелкораздроблен-
ный магний в среде абс. этилового эфира без доступа
влаги реагирует с галогенопроизводными органич.
соединений с образованием соединений RMgX, перехо-
дящихв эфирный р-р, к-рые получили название реакти-
вов Гриньяра. Стойкость этих реактивов, при условии
исключения влаги и кислорода атмосферы, не ограни-
чена. Применяемый в реакции эфир образует с реакти-
вом Гриньяра эфираты состава RMgX(C2H5)2O или,
еще чаще, RMgX • 2(С2Н5)2О, к-рые прочно удержи-
вают молекулы эфира. Наиболее вероятно след, строе-
ние эфиратов, напр.:
гН»С., .1-12 —
ю>(С2Н5)аО • Mgs+ jMg-'.' .’Mg;'’
L j’’ ‘'-jJ
где m, вероятно, равно 2,
Исследованиями электропроводности RMgX в эфир-
ных р-рах было установлено, что магний находится
в р-ре в виде катиона и что содержащийся всегда
в р-ре иодистый магний не является, однако, составной
частью магнийорганич. комплекса. Степень ассоциа-
ции М. с. зависит от концентрации р-ра. При образо-
вании RMgX возможен радикальный механизм реак-
ции:
2RX-f-Mg-»MgX2-p R2
или 2RX-|-Mg-»MgXs + R(+H)+R (- Н)
MgX2-|-Mg-2MgX- RX4-MgX.MgX24-R-
R- -f-MgX • — RMgX
Установлено, что в эфирном р-ре реактива Гриньяра
существует равновесие: 2RMgX R2Mg -|- MgX2, при-
чем RMgX и MgX2 могут быть количественно осаждены
из эфирного р-ра с помощью диоксана; в фильтрате
остается R2Mg. Этим путем получены чистые R2Mg.
Положение равновесия зависит от характера ради-
кала, его строения, природы галогена, растворителя,
но не зависит от концентрации р-ра. Пользуясь радио-
активным Mg28, можно показать, что более правильной
ф-лой является не RMgX, a R2Mg • Mg, находящийся
в равновесии с R2Mg и MgX2.
М. с. получаются также в присутствии третичных
оснований (диметиланилин, хинолин), причем обра-
зуются вещества типа аммониевых соединений. Алкил-
тиоэфиры, диэтилселенид и диметилтеллурид при обра-
зовании RMgX могут заменить эфир. Однако в их
присутствии реакция не идет так гладко н они не
имеют практич. значения. Замена эфира на бензол,
толуол, ксилол или петролейный эфир с добавкой
небольшого количества этилового эфира обычно при-
водит к снижению выхода реактива Гриньяра. Этило-
вый эфир можно заменить высшими эфирами, а также
эфирами этиленгликоля и тетрагидропираном. Широ-
кое применение получил тетрагидрофуран, в среде
к-рого с магнием реагируют винилгалогениды, не
образующие реактива Гриньяра в эфирной среде.
По аналогии со строением эфиров и здесь предпола-
гается образование оксониевой структуры (Норман):
| -Mg(X)-C = c/
В нек-рых случаях реакция идет в отсутствии раство-
рителя. Так, нагреванием хлорбензола с магнием
в автоклаве получен хлористый фенилмагний.
Медленно протекающие реакции образования реак-
тивов Гриньяра обычно ускоряют добавлением к смеси
небольшого количества иода. Образование RMgX
катализируется эфиром ортокремневой к-ты. Наиболее
часто для получения реактивов Гриньяра применяют
бромистые алкилы и арилы. Йодистые алкилы реаги-
руют наиболее энергично, однако часто дают худшие
выходы из-за побочных реакций, главной из к-рых
является Вюрца реакция-. 2RX Mg —- MgX2 RR.
Если в иодалкиле имеются вторичные и третичные
радикалы, то, кроме того, происходит отщепление Н J
с образованием непредельных соединений. В случае
третичных радикалов рекомендуется использование
хлоридов. Трудно реагирующие галогенопроизводные
(напр., пентаметилбромбензол) часто удается ввести
в реакцию с магнием методом «сопровождения», путем
добавки к смеси легко реагирующего с магнием бро-
мистого этила. Получены смешанные М. с., содержа-
щие перфторалкильные радикалы: CF3MgJ, C3F,MgJ
и CF2=CFMgJ.
Ацетилен и его однозамёщенные производные реа-
гируют с бромистым этилмагнием, заменяя водо-
род на остаток MgBr с образованием реактивов
Гриньяра ацетиленового ряда, типа BrMgC =CMgBr и
RC.~CMgBr, к-рые широко используются в органич.
синтезе. Подобное замещение подвижного водорода на
остаток MgX с образованием соответствующих реакти-
вов Гриньяра наблюдается при реакции C2H5MgBr
и циклопентадиеном, инденом, флуореном, 2-метил-
бензотиазолом, тиофеном, а также с азотистыми гете-
роциклами, содержащими «активный водород»: пир-
ролом, индолом, карбазолом, имидазолом и пиразо-
лом. Так, при реакции пиррола с CH3MgJ в эфирном
р-ре образуется реактив Гриньяра, к-рый при дей-
ствии СО2 образует а-пирролкарбоновую к-ту. Не
реагируют с магнием СНС13, СНВг3 и СС14. При попыт-
ке получения XMgCH3CH3MgX происходит элимини-
рование этилена.
Качественной реакцией на присутствие в р-ре реак-
тива Гриньяра служит реакция Гильмана: образова-
ние зеленовато-голубой окраски с 1%-ным бензольным
р-ром кетона Михлера. Количественно RMgX опреде-
ляют волюмометрически, если выделяющийся углево-
дород газообразный, напр. CHsMgBr -|- Н2О —► СН4
MgBrOH, либо титрованием к-той, причем выделив-
шийся MgXОН переходит в р-р в виде MgX2, а избы-
ток кислоты оттетровывают щелочью. О реакциях и
свойствах RMgX см. Гриньяра реакция.
Лит. см. при ст. Гриньяра реакция. С. Т. ИоаЗдОе.
МАГНИТНАЯ ВОСПРИИМЧИВОСТЬ — см. Маг-
нитные свойства, Магнетохимия.
МАГНИТНЫЕ СВОЙСТВА вещества — способ-
ность вещества взаимодействовать с внешним магнит-
ным полем. В более узком смысле магнитными свой-
ствами наз. способность вещества создавать собствен-
ное магнитное поле либо самостоятельно, либо под
действием внешнего магнитного поля. Мерой этой спо-
собности вещества является его магнитный мо-
мент или намагниченность а, мерой маг-
нитного поля служит его напряженность Н. Намагни-
ченность а тела слагается11 из намагниченности,
бывшей у него до появления внешнего магнитного
поля, и из намагниченности, вызванной полем. Од-
нако у большинства веществ в отсутствии поля намаг-
ниченность отсутствует.
Различают соответственно: 1) намагниченность 1 г
вещества стт, 2) намагниченность 1 моля ам и
3) намагниченность 1 см3 I. У большинства веществ,
наз. слабомагнитными, при обычных темпе-
ратурах и не слишком сильных полях намагничен-
ность, вызванная внешним магнитным полем, пропор-
циональна напряженности поля Н (рис. 1):
О = ХН (1)
Коэфф, пропорциональности
Х = а/Н (2)
*1 Необходимо иметь в виду, что поскольку и магнитный
момент, и магнитное поле характеризуются как числевной
величиной, так и направлением, и намагниченность, и напря-
женность магнитного поля являются величинами векторными.
1015
МАГНИТНЫЕ СВОЙСТВА
1016
носит название магнитной восприимчи-
во с т и и представляет собою, как видно из ф-лы (2),
намагниченность при Н = 1. Различают магнитную
0 восприимчивость 1 г вещества /т, магнитную
восприимчивость 1 моля %м и магнитную вос-
приимчивость 1 см3 х.
Намагниченность, подсб-
но магнитной воспрпим-
чивости, может иметь
как положительный, так
-------------------н и отрицательный знак, в
рис j--------------зависимости от того, на-
магничивается ли данное
тело параллельно направлению внешнего ноля (поло-
жительный знак) или противоположно (антипарал-
лельно) направлению внешнего поля (отрицательный
а знак).У сильномагнитных веществ
намагниченность а при обычных темп-рах имеет
сложную зависимость от Н и при достаточ-
но сильном поле до-
1 ... стигает оиределенно-
/ го значения, называе-
/ мого магнитным
/ насыщением цот
/ данного вещества (ри-
J сунок 2).
-------------(! Основные разиовид-
Рис 2.-------ности магнетизма. Из-
вестны 4 основных
вида магнитных процессов, протекающих в веще-
стве под действием внешнего магнитного поля,
или 4 разновидности магнетизма: 1) диамагне-
тизм; 2) парамагнетизм; 3) ферро-
магнетизм; 4) антиферромагнетизм.
Характерное отличие диамагнетизма состоит в том,
что диамагнитная намагниченность всегда направле-
на противоположно (навстречу) внешнему полю и,
следовательно, как и диамагнитная восприимчивость,
имеет отрицательный знак.
Зачастую делят все тела на 4 класса — диамагнит-
ные, парамагнитные, ферромагнитные и антиферро-
магнитные. Однако в большинстве веществ одновре-
менно сосуществуют и налагаются друг на друга
несколько из указанных процессов, а диамагнетизм,
напр., неизбежно присутствует во всех телах. Ука-
занная классификация тел говорит лишь о том, какая
из четырех разновидностей магнетизма преобладает
в данном теле. Т. обр., в диамагнитных телах преоб-
ладает диамагнетизм, в парамагнитных доминирует
парамагнетизм; ферромагнетизм перекрывает прочие
виды магнетизма в ферромагнитных телах, а в анти-
ферромагнитных телах сильнее всего проявляется
антиферромагнетизм.
Все 4 разновидности магнетизма связаны с тем об-
стоятельством, что элементарные частицы — электрон,
протон, нейтрон, входящие в состав атомов и молекул
вещества, обладают постоянными собственными маг-
нитными моментами (спиновый момент),
а кроме того, заряженные частицы (напр., электроны)
создают добавочный магнитный момент, двигаясь
в атоме или в молекуле по замкнутой орбите (орби-
тальный момент). Поскольку собственные
(спиновые) магнитные моменты протонов или нейтро-
нов примерно в 1000 раз меньше спинового магнит-
ного момента электронов, то магнитные моменты
атомов, молекул и макроскопия, тел определяются
в основном спиновыми и орбитальными моментами
электронов. Все атомы и молекулы можно разбить
на два вида по их магнитному моменту. Если
в атоме (или в молекуле) все моменты, создаваемые
отдельными электронами, взаимно компенсируются,
то результирующий момент данного атома (или мо-
лекулы) рсген нулю, в противном случае он отличен
от нуля.
Диамагнетизм и парамагнетизм.
Если атомы (или молекулы) данного вещества лишены
собственного магнитного момента, то в присутствии
внешнего магнитного поля они обнаруживают исклю-
чительно диамагнетизм. Возникающая при диа-
магнетизме намагниченность, противоположная (анти-
параллельная) направлению внешнего поля, обуслов-
лена тем обстоятельством, что все орбиты электронов в
атомах или молекулах приходят во вращение (процес-
сию) вокруг направления поля. Исследование этого
явления показывает, что диамагнитная восприимчи-
вость 1 моля
А’в- V-2
/-dM‘ emca (°)
где N — число Авогадро, т — масса покоящегося
электрона, с — скорость света в вакууме, г2 — сред-
ний квадрат расстояния данного электрона от ядра
атома. Суммирование произведено по всем электронам
данного атома. Как видно из ф-лы (3), диамагнитная
восприимчивость пропорциональна числу электронов
в атомах и квадратам их расстояний от ядра. Действи-
тельно, напр., магнитная восприимчивость гелия,
в атоме к-рого лишь два электрона, примерно равна
—2 • 10 в, между тем как магнитная восприимчивость
ксенона, в атоме к-рого 54 электрона, составляет
—43- 10 е. Диамагнитная восприимчивость мо-
лекул, лишенных магнитного момента
XdM = -®S?s-^₽) (*)
где р зависит от строения молекулы. Т. обр., диамагне-
тизм молекулы меньше диамагнетизма входящих
в ее состав атомов на величину р. Как видно из (3) и
(4), диамагнетизм атомов и молекул не зависит от
темп-ры.
До сих пор нами рассматривались атомы и молекулы,
у к-рых, в отсутствии внешнего поля, собственный маг-
нитный момент равен нулю. Однако существуют атомы
и молекулы, обладающие собственным магнитным мо-
ментом. Напр., молекула О2 обладает постоянным соб-
ственным магнитным моментом. Газ, состоящий из
молекул, обладающих магнитными моментами, в от-
сутствии поля не намагничен, т. к. вследствие хаотич-
ности теплового движения частиц их магнитные мо-
менты направлены беспорядочно и результирующий
момент равен нулю. Однако если этот газ поместить
в магнитное поле, то появляется преимущественная
ориентация моментов параллельно полю.
Наличие этой ориентации моментов означает, что у
газа оказывается результирующий магнитный момент,
т. е. намагниченность, направленная параллельно
внешнему полю. Эта намагниченность возрастает как
с усилением поля, так и с понижением темп-ры, т. е.
с уменьшением теплового движения молекул; она убы-
вает с ростом темп-ры или с ослаблением поля. Это
явление паз. парамагнетизмом. Выяснено,
что намагниченность в этом случае при не слишком
низких темп-рах и в слабых полях
„ — I N2V°H
арм — т знт (°;
где р — магнитный момент одной молекулы, /? —
газовая постоянная, а Т — абс. темп-ра. Парамагнит-
ная восприимчивость в этом случае
Ее так же можно выразить (закон Кюри)
ХРм = +т. где С = ^1Г V
В парамагнитном веществе, помещенном в магнитное
поле, происходит вращение электронных орбит вокруг
1017
МАГНИТНЫЕ СВОЙСТВА
1018
направления поля. Это вращение вызывает, в свою
очередь, диамагнетизм, к-рый накладывается на пара-
магнетизм, так что в результате мы наблюдаем сум-
марную восприимчивость
Хм — Хрм + Хам (8)
Диамагнетизм от темп-ры не зависит, поэтому при
не очень высоких темп-рах парамагнитный член (8)
всегда преобладает над диамагнитным. При очень
низких темп-рах, близких к абс. нулю, или при сверх-
сильных магнитных полях восприимчивость %рМ
перестает следовать закону Кюри(7)и намагниченность
приближается к насыщению (рис.2). При этихисключи-
тельных условиях магнитные моменты всех молекул
оказываются ориентированными параллельно направ-
лению магнитного поля, т. е. тогда
о = *Р’+ Хам# (9)
Величина момента р' (9) несколько отличается от
момента р (5), (6) или (7).
В жидких и особенно твердых телах, атомы или мо-
лекулы к-рых обладают собственным магнитным мо-
ментом, парамагнетизм несколько иначе зависит от
темп-ры, чем в газах, благодаря более или менее
сильному взаимодействию частиц друг с другом. В
этом случае вместо закона Кюри (7) восприимчивость
Хрщ следует формуле
ХрМ ‘ зд^т-рд) Г^+Д ' '
именуемой законом Кюри — Вейсса, где
А — величина, характерная для данного вещества
(константа Вейсса), может быть и положительной и
отрицательной. Примерами твердых веществ, следую-
щих закону Кюри — Вейсса, могут служить кристаллы
FeSO4 • 7Н2О с А = +13,7° или FeSO4 с А = —31°
или CuS04 с А = -(-60°, где носителями магнитного
момента являются атомные ионы Fe2+ и Сп2+ соответ-
ственно.
Зная температурный ход восприимчивости, можно
из формул (6) или (9) определить магнитный момент р
атома или молекулы. Обычно принято выражать р
через величину собственного спинового момента элект-
рона, равного 0,9273 • 10-20. Этот момент наз. элек-
тронным магнетоном Бора и обоз-
начается через Л/в. Т. обр., напр., момент р иона Fe2+
равен 5,8 Л/в, р иона Сп2+ 1,99Л/В, а р молеку-
лы Оа 2,86МВ, Для вычисления момента р из
формул (6) или (10) полезно нанести графически 1/^ в
зависимости от темп-ры Т. В таком случае у веществ,
следующих закону Кюри, получается прямая, про-
ходящая через начало координат (рис. 3, а). У ве-
ществ, следующих закону Кюри — Вейсса, получается
прямая, пересекающая ось темп-р либо вправо от нуля
(отрицательное А), либо влево от нуля (положительное
А) (рис. 3, б). Из наклона прямой определяется С
(7), откуда вычисляется р.
Особый вид пара- и диамагнетизма наблюдается
у т. н. свободных электронов в металлах и полупроеод-
никах. Как известно, свободные электроны в металлах
и полупроводниках образуют своеобразный газ. На-
личие собственного момента (спинового момента) у
электрона приводит к тому, что в газе свободных
электронов появляется парамагнетизм, вызванный
преимущественной ориентацией спинов во внешнем
магнитном поле. С другой стороны, тогда как в от-
сутствии внешнего магнитного поля электроны дви-
жутся хаотически по прямолинейным траекториям,
в присутствии поля они начинают описывать замкну-
тые орбиты. Это орбитальное движение свободных
электронов вызывает диамагнетизм. Т. обр., магнит-
ная восприимчивость свободных электронов в металле
слагается из пара- и диамагнетизма. В одних металлах,
как Li, К, Na, преобладает парамагнетизм; в других,
как Sb, Bi, доминирует диамагнетизм. Если концентра-
ция свободных электронов мала (что наблюдается у по-
лупроводников), то восприимчивость сильно зависит от
темп-ры. Если, напротив, концентрация свободных
электронов велика (металлы), то восприимчивость
почти независима от темп-ры.
Фактически движение электронов в кристаллич.
решетке металла или полупроводника значительно
сложнее, чем это описывается данной теорией. Чем
сильнее отличается кристаллич. решетка металла от
кубической, тем своеобразнее ее влияние на магнитные
свойства свободных электронов. Вот почему именно
такие металлы, как Sb и Bi, полупроводники, графит
и др., кристаллизующиеся в гексагональной, ромби-
ческой и тригональной си-
стеме, обнаруживают ано- х
мально большой диамаг-
нетизм и притом очень зна-
чительную анизотропию
магнитных свойств. Так,
у кристаллов графита диа-
магнетизм в направлении,
параллельном оси, превос-
ходит примерно в 6> раз
диамагнетизм, перпенди-
кулярный оси кристалла.
н
Почти все пара- и диамаг- рис. 4.
нитные металлы обнаружи-
вают при очень низких темп-рах своеобразную ано-
малию — периодич. зависимость восприимчивости от
напряженности поля (рис. 4).
Ферромагнетизм и антиферро-
магнетизм. Парамагнетизм, рассмотренный вы-
ше, наблюдается в твердых телах тогда, когда атомы
или ионы обладают магнитным моментом и когда
между этими носителями момента либо нет взаи-
модействия вовсе, либо существует слабое взаимодей-
ствие. Но в кристаллич. решетках твердых тел наи-
более часто имеет место весьма сильное взаимодей-
ствие между атомами (или ионами). Если эти частицы
обладают собственным магнитным моментом, то в ре-
зультате их взаимодействия даже при полном отсут-
ствии внешнего магнитного поля, но при достаточно
низких темп-рах, магнитные моменты частиц оказы-
ваются определенным образом ориентированными
по отношению друг к друту.
Возможны два вида взаимной самопроизвольной
ориентации магнитных моментов частиц: а) параллель-
а б
Рис. 5.
ная ориентация и б) антипараллельная (т. е. встреч-
ная) (рис. 5). Физич. явление (в) наз. ферромагнетиз-
мом, а явление (б) — антиферромагнетизмом. Взаимо-
1019
МАГНИТНЫЕ СВОЙСТВА
1020
действие между атомами, приводящее как к ферромаг-
нетизму, так и к антиферромагнетизму, имеет в основ-
ном не магнитное, а электрич. происхождение. Силы
этого взаимодействия получили в квантовой механике
название обменных сил. Выбор между ферро-
магнитным и антиферромагнитным характером взаи-
модействия определяется спецификой строения дан-
ного вещества.
Как ферромагнетизм, так и антиферромагнетизм мо-
гут существовать лишь при темп-рах ниже нек-рой
критич. точки 6, характерной для данного вещества
(точка Кюри для ферромагнетизма, точка
Н е э л я для антиферромагнетизма). Выше этой темп-ры
тепловое движение оказывается достаточно интен-
сивным, чтобы разбить самопроизвольную взаимную
ориентацию моментов частиц. Тогда оба эти вида маг-
нетизма переходят в парамагнетизм. Обменное взаи-
модействие между магнетиками можно для нагляд-
ности представить себе в виде нек-рого «магнитного
внутреннего» поля. Т. обр., в отсутствии внешнего
поля в ферромагнитном теле нак бы действует очень
сильное «внутреннее поле» Hi (в железе или никеле
оно эквивалентно магнитному полю в 10е—107 эрстед),
пропорциональное числу ориентированных электрон-
ных спинов. Оно и вызывает самопроизвольную на-
магниченность, или, как ее обычно называют, спон-
танную Намагниченность. Опыт по-
казывает, что маленькие ферромагнитные кристаллики
(до 105 атомов) действительно всегда самопроизвольно
намагничены при темп-рах ниже точки Кюри. Однако
этого не наблюдается в больших кристаллах. Внешне
они кажутся ненамагниченными, между тем при
детальном изучении выясняется, что фактически от-
дельные небольшие области кристалла, содержащие
103—10* атомов (домен ы), намагничены в различ-
ных направлениях. Поэтому результирующая сум-
марная намагниченность всего кристалла оказывается
равной нулю.
При темп-рах, близких к абс. нулю, в каждом домене
практически имеется полное магнитное насыщение,
в этих условиях при достаточно сильном магнитном
поле ферромагнитный кристалл намагничивается до
максимального возможного значения, при к-ром все
имеющиеся магнитики параллельны. Измерив в этих
условиях молярную намагниченность стоо = Nр, можно
определить магнитный момент атома ферромагнетика.
Опыты такого рода дали для Fe р = 2,2 Л/в, а для
Ni р = 0,6 M-q. С возрастанием теМп-ры намагничен-
ность в каждом отдельном домене уменьшается.
а ' 6 в .г
Рис. 6.
Разработаны различные экспериментальные методы,
позволяющие непосредственно обнаруживать границы
доменов на поверхности ферромагнетика и определять
направление намагниченности в отдельном домеве.
Если поместить ферромагнетик, состоящий из несколь-
ких доменов, в слабое внешнее магнитное поле Н, то в
нем происходит рост объема тех доменов, в к-рых на-
правление намагниченности наиболее близко к направ-
лению Н, за счет менее «благоприятно» ориентирован-
ных доменов. Этот процесс осуществляется путем
смещения границмежду доменами (рис. 6,6). По мере
усиления внешнего поля этот процесс быстро воз-
растает, что приводит к крутому подъему намагничен-
ности (рис. 7). Одновременно происходит постепенный
поворот направлений намагниченности в доменах в при-
ближении к направлению поля Н (рис. 6, в). В силь-
ных полях «процессы вращения» доминируют над «про-
цессами смещения
границ». И нако-
нец, при достаток- -
но сильном поле все а __________
домены сливаются, ~ г
причем намагничен-
ность во всем кри- /
сталле оказывает- L
ся ориентирован- /
ной параллельно /
полю (намагничен- ау
ность насыщения) '----------------Н
(рис. 6, г). Если рис. 7.
теперь уменьшить
интенсивность внешнего поля, то в идеальном нена-
пряженном кубич. кристалле все описанные выше
процессы протекают строго в обратном порядке и при
исчезновении поля кристалл оказывается снова нена-
магничеНным.
На практике обратимое размагничивание встре-
чается сравнительно редко, и в большинстве случаев
наблюдаются гистерезисные явления,
так что при выключении поля образец оказывается
остаточно намагниченным (рис. 8).
Если приложить
поле встречного на-
правления, то ос-
таточная намагни-
ченность убывает.
При нек-ром зна-
чении поля, зави-
сящем от природы
образца и от его
предыстории (ме-
ханич. или термич. г//
обработки и т. д.)
и вариирующем в
очень широких пре-
делах, остаточная намагниченность исчезает. Это
встречное поле наз. коэрцитивной силой Ни. Течение
процессов намагничивания и размагничивания зависит
в сильной степени от быстроты изменения внешнего
магнитного поля. При быстрых изменениях поля раз-
личные ферромагнетики в большей или меньшей сте-
пени поспевают за полем, обнаруживая так наз.
магнитную вязкость, т. е. запаздывание.
Таковы основные свойства ферромагнетизма.
Антиферромагнетизм бывает двоякого вида: ском-
пенсированный и нескомпенсиро-
ванный. В веществах со скомпенсированным анти-
ферромагнетизмом при темп-рах, близких к абс. нулю,
в отсутствии внешнего поля намагниченность равна
нулю даже в самых малых объемах. Т. обр., хотя в от-
сутствии поля ферромагнитный кристалл внешне как
будто не отличается от антиферромагнитного, их внут-
реннее состояние существенно различно. Путем эк-
спериментального изучения дифракции нейтронов
в кристаллах удалось определить детальную картину
ориентации элементарных магнитиков в различных
телах. Силы между элементарными магнитиками
в скомпенсированных антиферромагнетиках препят-
ствуют намагничиванию во внешнем поле. Поэтому
при очень низких темп-рах намагниченность, дости-
гаемая в этих телах во внешнем поле, как правило,
крайне мала. По мере роста темп-ры восприимчивость
скомпенсированного антиферромагнетика возрастает,
т. к. тепловое движение уменьшает препятствующее
намагничиванию взаимодействие между частицами.
В критич. точке Неэля (0) взаимодействие между ча-
1021
МАГНИТНЫЕ СВОЙСТВА —МАГНИЯ КАРБОНАТЫ
1022
стицами ослабляется тепловым движением до такой
степени, что взаимная ориентация магнитиков пол-
ностью разрушается. Выше точки Неэля скомпенсиро-
ванный антиферромагнетик становится парамагнети-
ком и подчиняется закону Кюри—Вейсса: Х =
(см. выше). Т. обр., температурный ход магнитной
восприимчивости скомпен-
* С сированного антиферрома-
/л гнетика обнаруживает ха-
I \ рактерный максимум в точке
। \ Неэля (рис. 9).
] \ Нередко силы антиферро-
। \ магнитного взаимодействия
। направлены вдоль какого-
। нибудь одного или несколь-
___________J______н ких немногих направлений
®_________________в кристалле. В таком случае
Рис. 9. во всех прочих направ-
лениях скомпенсированный
антиферромагнетик намагничивается внешним полем
подобно парамагнетику. Примерами скомпенсирован-
ных антиферромагнетиков могут служить FeO, МпО,
СоО, FeS, FeF2, CoF2, NiFe.
Нескомпенсированный антиферромагнетизм (иногда
наз. ферримагнетизмом, в отличие от
ферромагнетизма) характеризуется тем, что в этом
случае магнитные моменты атомов, ориентирован-
ных навстречу друг другу, неполностью компенси-
руют друг друга. Такие тела разбиваются на домеиы
и ведут себя во многих отношениях подобно феррома-
гнетикам. В зависимости от степени декомпенсации по-
лучаются вещества со свойствами от очень сильно-
го до очень слабого ферромагнетизма. К числу сильно
магнитных веществ этого рода относятся, напр.,
т. наз. ферриты (соединения типа Fe2O8- NiO,
Fe2Oa • МпО и т. д.), получившие широкое примене-
ние в современной радиотехнике.
Таблица 1. Некоторые диамагнитные вещества
(при комнатной темп-ре). Восприимчивость /.,•
И Хщ в ед. CGSM м
— хм 108 xm 10»
Газы
Водород 3,96 1.98
Азот 11,8 0,42
Гелий 1,91 0,04
Аргои 18,7 0,494
Углекислый газ Жидкости 20,88 0,46
Вода 13 0.721
Бензол 55 0,698
Ацетон 33,8 0,585
Этиловый алкоголь Твердые тела 33,6 0,744
NaCl 30,8 0,0
KCI 39,0 0,516
Ag 21.56 0,20
Au 29,59 0,15
Bl 288.4 1,38
Sb 80,0 0.66
С (графит) 36 3,0
С (алмаз) 5,9 0,49
Нафталин 93,5 0,717
Как уже отмечалось, рассмотренные явления диа-
магнетизма, ферромагнетизма и антиферромагнетизма
очень редко встречаются в чистом виде и налагаются
друг на друга самым причудливым образом, что, разу-
меется, очень осложняет картину. Особенно важно
иметь в виду, что в обычных условиях жидкие и твер-
дые тела зачастую содержат в себе мельчайшую
пыль, в состав которой входят частички окали-
ны, т. е. преимущественно ферромагнитных и фер-
римагнитных окислов железа. Так как эти вещества
намагничиваются в сотни тысяч раз сильнее диама-
гнитных и парамагнитных тел, то содержание в этих
телах Fe в количестве от 10 5% (если примесь при-
сутствует в виде нерастворимых твердых частиц)
полностью искажает, магнитные свойства изучаемых
диамагнитных и парамагнитных веществ. При изу-
чении магнитных свойств сильномагнитных веществ
необходимо иметь в виду, что наряду с влиянием
внешнего магнитного поля на образец на него дейст-
вует также создаваемое им самим магнитное поле.
Оно зависит от формы образца и, как правило, ос-
лабляет действие внешнего поля, т. е. затрудняет
намагничивание. При намагничивании, напр., ци-
линдра вдоль оси саморазмагничивающее его действие
тем больше, чем меньше отношение длины цилиндра
к его диаметру.
В отношении твердых тел необходимо также иметь
в виду, что магнитные свойства в поверхностных слоях
и в мелкодисперсной фазе могут существенным обра-
зом отличаться от свойств сплошных тел. Далее, они
зависят от кристаллич. модификации и обладают зна-
чительной анизотропией.
Таблица 2. Некоторые парамагнитные вещества (при комнат-
ной темп-ре). Восприимчивость %т в ед. CGSM
Xm ’ 106 Xm-10’
Гааы Твердые тела
Кислород 100,1 NISO, 27,1
NO 49,07 FeSO, • 7H2O .... Al 42,4 0,58
Жидкости Pt 0,982
MnBr2 MnPO4 FeJ2 71,0 69,0 40,0 К Uyb2
Таблица 3. Некоторые ферромагнитные вещества
(а и I в ед. CGSM; в эрстедах; 8 В “К)
°макс экстр ап ол. к 0°K ^макс акстрапол. к 0°К 1 оста- точн. «к б
Fe 221,89 1752 1043
Co 162,5 1446 — 1400
Ni Gd Сплав «Альнико» Ферриты 57,50 510 1980 800 480 975 631 289
BaFeiaOie - . . MnO Fe,O3. . . FeO FeaO3 . . . NiO Fe2O, . . . 300 360 500 250 125 2000 723 783 848 863
Лит.: Вонсовский С. В., Современное учение о маг-
нетизме, М.—Л., 1952; Дорфман Я. Г., Магнитные свой-
ства и строение вещества, М., 1955; Б о з о р т 3. Р., Ферро-
магнетизм, пер. с англ., М., 1956; Антиферромагнетизм и фер-
риты, М., 1962 (Итоги науки. Физ.-мат. науки. 4).
Я. Г. Дорфман.
МАГНИТНЫЙ МОМЕНТ — см. Магнитные свой-
ства.
МАГНИТНЫЙ РЕЗОНАНС — см. Радиоспектро-
скопия.
МАГНИЯ КАРБОНАТЫ — углекислые соли маг-
ния, из к-рых наибольшее практич. значение имеют:
нормальный карбонат MgCO3 (магнезит), основ-
ной карбонат 3MgCO3 • Mg(OH)2- ЗН2О (белая
магнезия), а также двойной карбонат магния и
кальция MgCO3 • СаСО3 (доломит).
Нормальный карбонат MgCOs широко
распространен в природе в виде минерала маг-
незита. MgCO3 — бесцветные ромбоэдрические кри-
сталлы, а = 5,61 А, а = 48°10'; плотн. 3,0—3,1;
твердость по Моосу 4,0—4,5. Теплота образования
— — 266,0 ккал/молъ. Разложение на MgO и СО,
1023
МАГНИЯ ОКИСЬ —МАГНИЯ СПЛАВЫ
1024
становится заметным ок. 500°. Давление диссоциации
достигает атмосферного при 650°. В воде MgCO3 трудно
растворим: при 25° для различных давлений СО2
растворимость (в мг/л) составляет: 0,5 (0,0001 атм),
22 (0,968 атм), 55 (15 атм). С повышением темп-ры
растворимость уменьшается. Известен гидрат MgCO3-
• ЗН2О, получаемый при взаимодействии р-ров
MgCl2 и КНСО3 ниже 80°. При насыщении двуокисью
углерода водной суспензии MgCO3 последний раство-
ряется благодаря образованию растворимого бикар-
боната: MgCO3 СО2 Н2О = Mg (НСО3)2. Из вод-
ных р-ров нормальный М. к. выделяется лишь при
одновременном присутствии в растворе большого
избытка СО2; в противном случае выпадают основные
соли. Последние всегда и образуются при действии
на соли Mg растворами щелочных карбонатов. При
нагревании с крепким р-ром КНСО3 основные кар-
бонаты переходят в нормальный. С карбонатами ще-
лочных и щелочноземельных металлов MgCO3 обра-
зует двойные соли, к числу к-рых относится и природ-
ный доломит.
Магнезит имеет широкое практич. применение.
В пром-сти его обжигают в шахтных, трубчатых или
механич. полочных печах, причем, в зависимости от
условий обжига, получают технич. окись магния раз-
личного качества, идущую гл. обр. на изготовление
огнеупоров, вяжущих веществ, металлич. магния
(подробнее об условиях обжига, качестве и приме-
нении MgO см. Магния окись).
Основной карбонат 3MgCO3 • Mg(OH)2 •
• ЗН2О — бесцветные кристаллы, плотн. 2,16. Плохо
растворим в воде (0,04 г в 100 г Н2О), но растворим
в водных р-рах аммониевых солей. При нагревании
до 900—1000° разлагается с образованием MgO.
Основной карбонат может быть получен из магне-
зита, доломита и хлорида магния морской воды. Для
этого магнезит, напр., обжигают и переводят в
Mg(OH)2, к-рую (в виде суспензии) подвергают карбо-
низации (насыщению СО2) при 20—25°. Из получен-
ного р-ра бикарбоната магния нагреванием при 45—
50° выделяют основной карбонат. Основной карбонат
магния (т. наз. «белая магнезия», или «магнезия
альба») применяют как наполнитель и усилитель в ре-
зиновых смесях, для изготовления высококачествен-
ных теплоизоляционных материалов и в медицине
(принимается внутрь при повышенной кислотности,
входит в состав зубного порошка).
Двойной карбонат магния и каль-
ция MgCO3 • СаСО3 образует широко распростра-
ненный в природе минерал доломит. MgCO3-CaCO3 —
бесцветные ромбоэдрические кристаллы, а = 6,05 А,
а = 46°54'; плотн. 2,86; твердость 3,5—4,0; нераство-
рим в воде. При нагревании разлагается ступенчато:
при 600—750° распадается MgCO3, при 900° — СаСО3.
Благодаря дешевизне доломит находит широкое при-
менение. Доломит, прокаленный «намертво», при
1500—1600°, применяется в качестве «металлурги-
ческого порошка» для наварки подов томасовских
конверторов и мартеновских печей при основном
процессе получения стали. Доломит, обожженный
при 850—950°, идет на изготовление «магнезии альба»
методом карбонизации, а также металлич. Mg. Доло-
мит используется также в произ-ве магнезиальных
цементов, в стекольной и керамич. пром-сти, для из-
готовления щебня, облицовочного камня и пр.
Лит. см. при ст. Магния окись.
МАГНИЯ ОКИСЬ (жженая магнезия) MgO — про-
стейшее соединение магния с кислородом. В при-
роде встречается в виде минерала периклаза —
бесцветных кристаллов с гранецентрированной кубич.
решеткой, а = 4,213А; плотн. 3,56—3,65; т. пл.
2800°, т. кип. 3600°, твердость по Моосу 5—6. Уд.
теплоемкость 8,96 кал/г-град. Теплота образования
А7/%ч = — 143,84 ккал/молъ. Такую же структуру
имеет и т. н. «аморфная» (мелкокристаллическая)
М. о. — легкий белый порошок, поглощающий на
воздухе Н2О и СО2 с образованием гидроокиси и кар-
боната магния. «Аморфная» М. о. легко растворяется
в кислотах, даже в уксусной, и в р-рах солей аммония.
Растворимость в воде 0,00062 г MgO на 100 г Н2О (20°).
Рекристаллизация М. о. начинается при нагревании
выше 500°, и при 1200—1600° образуются крупные
кристаллы. Сильно прокаленная М. о. становится
очень твердой и теряет способность соединяться с во-
дой и растворяться в кислотах. Летучесть М. о. за-
метна в восстановительной среде при 1800—2000°,
а в окислительной — при 2000—2100°. О получении
и применении MgO см. ниже.
Магния гидроокись встречается в при-
роде в виде минерала брусита. Кристаллич.
решетка гексагональная, а = 3,12 А, с = 4,73 А;
плотп. 2,35—2,46; твердость по Моосу 2,5. Теплота
образования ДТ7298 = — 221,0 ккал/молъ. Mg(OH)2—
слабое основание, выпадает в виде объемистого сту-
денистого белого осадка при действии щелочей на
р-ры солей магния; pH осаждения 10,5. Насыщенный
водный р-р содержит Mg(OH)2 0,019 г/л (20°) и
0,04 г/л (100°). Mg(OH)2 легко растворима в кисло-
тах. Давление диссоциации при 300° достигает 10 мм
рт. ст. При 500° Mg(OH)2 переходит в MgO.
В пром-сти MgO получают обжигом магнезита и
доломита, термич. разложением MgSO4, гидролизом
MgCL,, а также осаждением Mg(OH)2 и основного
карбоната магния с их последующей термич. обра-
боткой.
Свойства технич. продукта зависят от условий получения.
Различные сорта отличаются по своей легкости (объемному
весу), химич. активности, адсорбционной способности и др.
свойствам. Чем более дисперсна и пориста магневия, тем
меньший объемный вес она имеет, поэтому о качестве М. о.
судят по ее легкости. Обжигом магнезита при 700—900°
получают сравнительно тяжелую форму магнезии, содержащую
все примеси, имевшиеся в магнезите, т. н. «каустический маг-
незит». При высокотемпературном обжиге магнезита (1500—•
1800°) получают еще более тяжелую крупнозернистую неак-
тивную форму MgO — т. н. «металлургический порошок».
Для получения легких магнезий обожженный магнезит гасят
водой и полученную суспензию подвергают карбонизации под
давлением: Mg(OH)2+ 2СО2 = Mg (НСО3)2; при дальнейшем
кипячении р-ра бикарбоната последний разлагается и в осадок
выпадает основной карбонат магния, прокаливанием к-рого
можно получить легкие сорта магнезии. Для получения М. о.
широко используются также природные и искусственные
рассолы, содержащие соли Mg (морская вода, рапа, щелока
калийного произ-ва и т. д.). При обработке этих рассолов
известью или известковым молоком выпадает Mg(OH)2, при
обжиге к-рой получаются, в зависимости от ее дисперсности
и условий обжига, различные сорта магнезии.
Легкие сорта магнезий применяют в качестве на-
полнителя и усилителя в резиновых смесях, а также
для очистки нефтепродуктов. Менее легкие сорта
(напр., каустический магнезит) — для изготовления
магнезиальных цементов, строительных материалов,
искусственных камней (мельничные жернова) и т. д.
Тяжелые сорта магнезий используют в произ-ве
огнеупоров. М. о. служит также для получения
магния. Чистую М. о. применяют в медицине при
повышенной кислотности желудочного сока, как сла-
бительное и пр.
Лит.: По зин М. Е., Технология минеральных солей,
2 изд., Л., 1961, гл. IX; Стрелец X. Л., Т а й ц А. Ю.,
Г у лян иц кий Б. С., Металлургия магния, 2 изд., М.,
1960; Сборник работ по комплексному использованию природ-
ных растворов и получению перманганата калия из пиролю-
зита, Л., 1960 (Тр. Гос. ин-та прикл. химии, вып. 47).
МАГНИЯ ПЕРХЛОРАТ — см. Ангидрон.
МАГНИЯ СПЛАВЫ — сплавы на основе Mg с до-
бавками (в сумме до 12%) Al, Zn, Мп, Zr, Th, Са, Се
и др. элементов (см. таблицу). Mg в чистом виде как
конструкционный материал не применяют из-за низ-
кой механич. прочности. Легирующие добавки вводят
в Mg гл. обр. для повышения прочности. Легирующие
элементы входят в твердый раствор замещения на
1025
МАГНИЯ СПЛАВЫ— МАГНИЯ СУЛЬФАТ
1026
Составы, термическая обработка и механические свойства нек-рых магниевых сплавов
Марка сплава Содержание легирующих элементов, % Термическая обработка Типич. механич. свойства
А1 Zn Мп прочие ав , кГ/мм2 с0»2» кГ/мм2 «,: % Нв, кг/мм2
Магний литой — — — 11,5 2,5 8 30
Литейные сплавы
МЛ 4 5,0-7,0 2,0—3,0 0,15-0,5 — Закалка с 380° (12 час.) с ох-
7,5-9,0 лаждением на воздухе 2э 8,5 9 55
МЛ5 0,2-0,8 0,15-0,5 — Закалка с 415° (12 час.) с ох-
МЛ 12 лаждением на воздухе 25 8,5 9 60
— 4,0—5,0 — Zr 0,6-1,0 Закалка с 500° (3 час.) с ох-
лаждением на воздухе; ста- рение при 150°, 24 час. 27 10 6 80
Деформируемые сплавы *
МА1 1.3 -2,5 21 12 8 45
МА2 3,0—4,0 0,2—0,8 0,15-0,5 28 18 10 55
МА5 7,8-9,2 0,2-0,8 0,15—0.5 — 32 2 ' 14 65
МА 8 —— 1,5 -2,5 Се 0.15-0,35 26 15 7 55
ВМ65-1 — 4,0-5,5 — Zr 0,3-0,9 Старение при 170°, 10 час. 33 28 10 60
ав — предел прочности при растяжении, *д о — предел текучести, 6 — относительное удлинение, Hq — твердость
по Бринеллю.
• Свойства указаны для горячепрессованных прутков.
оснсве Mg, а также образуют металлические соедине-
ния (Mg4Al3, MgZn, MgxAl!/Znz, Mg9Ce). Основу сплава
составляет твердый р-р, Интерметаллич. соединения,
если и имеются, то содержатся в очень небольшом ко-
личестве, т. к. они хрупки и сильно снижают пластич-
ность М. с. Промышленные М. с., называемые элек-
тронами, относятся к трем системам: Mg—Al—Zn,
Mg—Мп и Mg—Zn—Zr.
Наиболее широко применяют M, с, системы Mg—
Al—Zn, содержащие от 3 до 10% А1 и от 0,2 до 3%
Zn. Для повышения стойкости против коррозии в эти
сплавы добавляют Мп 0,15—0,5%. М. с., содержащие
более 8% А1 или более 5% А1 и 2% Zn, можно упроч-
нять термич. обработкой. При нагреве под закалку
до 380—420° происходит переход включений Mg4Al3
в твердый р-р на основе Mg. С понижением темп-ры
растворимость А1 в Mg в твердом состоянии сильно
уменьшается, но даже при медленном охлаждении
сплава на воздухе интерметаллид Mg4Al3 не успевает
выделиться из магниевого р-ра и происходит само-
закалка (без охлаждения в воде). Структура закален-
ного М. с. состоит из кристаллитов пересыщенного
твердого р-ра А1 и Zn в Mg. Такой раствор термо-
динамически неустойчив, но при комнатной темп-ре
он не распадается, М, с. не склонны к естественному
старению. При нагреве закаленного сплава до 170—
180° происходит частичный распад пересыщенного
твердого р-ра с выделением высокодисперсных ча-
стиц Mg4Al3 и прочностные характеристики М. с,
несколько возрастают (искусственное старение). На-
грев до более высоких темп-р приводит к интенсивному
распаду твердого р-ра и укрупнению частиц интер-
металлида, в результате чего прочностные свойства
снижаются. Чаще всего ограничиваются одной закал-
кой без старения, т. к, повышение прочности при
старении невелико, а пластичность снижается. М. с.
системы Mg — Мп содержат от 1 до 2,5% Мп; в нек-рые
из них добавляют Се (до 0,35%). Эти сплавы, по срав-
нению со всеми М. с., имеют наиболее высокую стой-
кость против коррозии. М. с. системы Mg — Zn — Zr,
содержащие до 5,5% Zn и до 1,0% Zr, отличаются
сочетанием повышенного предела текучести и хоро-
шей пластичности (Zr сильно измельчает зерно).
В последнее годы для повышения прочности, в особен-
ности при высоких темп-рах, М. с. легируют небольши-
ми добавками Th, Nd, La, Y и др. редких элементов.
17 к. х. э, т. 2
Все М. с. делят на литейные и деформируемые. Из
литейных М. с. (типа МЛ2, МЛ5, МЛ12 и др.) отли-
вают фасонные детали. Из деформируемых М. с.
(типа MAI, МА5, МА8, ВМ65-1 и др.) обработкой дав-
лением (прокаткой, прессованием, ковкой и т. д.)
изготавливают листы, плиты, прутки, профили, по-
ковки и штамповки. Так как М. с. при комнатной
темп-ре сравнительно малопластичны, то их обраба-
тывают давлением при темп-рах выше 250°. М. с.
в расплавленном состоянии сильно окисляются и
склонны к загоранию. Для защиты от окисления и
загорания расплав в тигле покрывают флюсом (напр.,
смесью 75% плавленого карналлита, 17,5% CaF3
и 7,5% MgO), а струю металла прилитье припыли-
вают порошком серы. Для защиты изделий от окис-
ления при термич. обработке создают в печи атмосферу
смеси воздуха с 1% сернистого газа.
Большим недостатком М, с. является низкая стой-
кость против коррозии во влажной атмосфере, в прес-
ной и морской воде. М. с. сильно корродируют
в органич. и минеральных к-тах (за исключением хро-
мовой и плавиковой) и в солях. Ионы хлора ускоряют
коррозию М. с, в водной среде, М. с. устойчивы
в р-рах фторидов, хроматов и бихроматов, в мине-
ральных маслах, нефти, бензине и керосине, слабо
корродируют в разб, щелочах. Детали из М, с. защи-
щают от коррозии оксидированием и нанесением лако-
красочных покрытий.
Основное достоинство М. с. — малая плотность
(1,8). По прочности, в расчете на 1 м.и2 сечения детали,
М. с. уступают алюминиевым, но при равном весе
деталь из М. с,, имея большее сечение, может оказать-
ся прочнее. М. с. хорошо воспринимают ударные
нагрузки, прекрасно обрабатываются резанием, шли-
фуются и полируются. М. с. недефицитны и в большом
количестве потребляются в ракетной технике и авиа-
строении, где вопросы веса деталей имеют первосте-
пенное значение. Применение М. с. расширяется в
авто-, мото- и приборостроении, на железнодорожном
транспорте, в текстильной пром-сти и других отраслях
техники.
Лит.: Б о ч в а р А. А., Металловедение, 5 изд., М., 1956;
Справочник по машиностроительным материалам, т. 2, М.,
1959. И. И. Новиков.
МАГНИЯ СУЛЬФАТ (сернокислый магний)
MgSO4 — бесцветные кристаллы, плотн. 2,66. Теп-
лота образования ДН388 = — 305,5 ккал/молъ. Рас-
1027
МАГНИЯ СУЛЬФИД —МАГНИЯ ХЛОРИД
1028
творимость в воде (в %): 25,2 (20°); 37,1 (69°);
33,5 (100°); 13,0 (160°) и 0,5 (240°). Плотн. водных
р-ров:
% MgSO4 . . 10 20 26
d3°....... 1,1034 1,2998 1,2961
4
Насыщенный р-р кипит при 108° и содержит 75 г
MgSO4 на 100 г Н2О. Криогидратная точка — 3,9°
(при 17,0% MgSO4).
М. с. образует кристаллогидраты с 1, 2, 3, 4, 5,
6, 7 и 12 молекулами воды. При комнатной темп-ре
из водных р-ров кристаллизуется MgSO4 • 7Н2О,
а выше 48° — MgS04 • 6Н2О, к-рый в интервале
87—92° плавится с образованием метастабильных
MgSO4 • 5Н2О и MgSO4 • 4Н2О. Твердый MgSO4 •
• 4Н2О при 106° переходит в MgSO4 • ЗН2О, а послед-
ний при 122—124° — в MgSO4 • 2Н3О, к-рый при
161—169° превращается в MgSO4 • Н2О. Из водных
р-ров стабильный моногидрат кристаллизуется выше
67,5°. Обезвоживание моногидрата наблюдается при
320—330°. Безводный MgSO4 при 1100—1200° раз-
лагается с заметной скоростью на MgO, SO2 и О2.
В присутствии восстановителей (С, S, Н2, СО, СН4)
темп-ра термич. восстановления снижается. Известны
основные соли М. с.: MgSO4 • 3MgO • 11Н2О; MgSO4 •
•5MgO• 8Н2О.
В природе М. с. встречается в виде минералов ки-
зерита и эпсомита. Кизерит MgSO4 • Н2О — бес-
цветные кристаллы, моноклинная решетка, а = 7,53А,
b = 7,69 А, с = 6,89 А, Р = 116°5'; плотн. 2,6; твер-
дость по Моосу 3,5. В воде, даже при нагревании,
растворяется очень медленно (по-видимому, лишь
после перехода в более богатый водой кристаллогид-
рат). Эпсомит (горькая соль) MgSO4 •
• 7Н2О — бесцветные прозрачные кристаллы4 орто-
ромбич. решетка, а = 11,94 А, Ь = 12,03а, с =
= 6,865 А; плотн. 1,68; твердость по Моосу 2—2,5.
В воде хорошо растворим. С солями щелочных метал-
лов MgSO4 образует двойные соли; нек-рые из них
также встречаются в природе: каинит К.С1 •
• MgSO4 • ЗН2О; астраханит Na2SO4 • MgSO4-
• 4Н2О; полигалит K2SO4 • MgS04 • 2CaSO4;
лангбейнит 2 MgSO4 • K2SO4. Сульфат магния
добывают из природных рассолов морского типа и
твердых солевых отложений. Его получают также
нагреванием магнезита и доломита в р-ре (NH4)2SO4
или как побочный продукт при получении борной
к-ты обработкой ашарита 2MgO • В2О3 • Н2О и гидро-
борацита CaMgBeOu • 6Н2О серной к-той. MgSO4,
подобно CaSO4, служит сырьем для сернокислотного
произ-ва. Восстановление обычно ведут углем в ин-
тервале 700—900°: 2MgSO4 С = 2MgO 2SO3 -{-
СО2. Получаемый при обжиге сернистый газ пере-
рабатывают на серную к-ту, а огарок представляет
собой высококачественную магния окись, пригодную
для изготовления огнеупоров и ксилолита. М. с.
применяют также в произ-ве магнезиальных цемен-
тов, в качестве утяжелителя хлопка и шелка, напол-
нителя для бумаги, как протраву при крашении и т. д.
В медицине М. с. используют как наркотич. средство
для борьбы с судорожным состоянием и пр. (введе-
ние под кожу, в мышцу, в вену) и как слабительное
(«горькая соль»).
Лит.: П о з и н М. Е., Технология минеральных солей,
2 изд., Л., 1961.
МАГНИЯ СУЛЬФИД (сернистый магний) MgS —
бесцветные кристаллы кубич. системы, а = 5,20 А;
плотн. 2,82; т. пл. выше 2000°. Теплота образования
&Н 29S = — 83,0 ккал/молъ. В воде М. с. нерастворим,
но постепенно разлагается ею на Mg (ОН)2 и Mg (SH)2.
При высоких темп-рах разлагается парами воды с об-
разованием MgO и H2S. Легко растворяется в кисло-
тах. При нагревании MgS с серой получаются поли-
сульфиды MgS4 и MgS6. М. с. образуется из элемен-
тов при температуре выше 600°. При быстром нагре-
вании смеси порошкообразного Mg с S развивает-
ся очень бурная реакция; удобнее пропускать пары
S над нагретым Mg; можно исходить также из
MgO, пропуская над ней при 700—900° пары S
или CS2. М. с., содержащий некоторые примеси (Мп,
Sb, Bi, Cd), образует желтоватые или розовые
кристаллы, обладающие люминесцентными свойства-
ми. Применяется для изготовления светящихся со-
ставов.
Магния гидросульфид Mg (SH)2 из-
вестен только в водных р-рах. Постепенно разла-
гается с выделением H2S. Образуется при пропуска-
нии H2S в водную суспензию Mg (ОН)2.
Лит.: Руководство по препаративной неорганической
химии, под ред. Г. Брауера, пер. с нем., М., 1956.
Д. С. Стасиневич.
МАГНИЯ ХЛОРИД (хлористый магний) MgCl2 —
бесцветные кристаллы гексагональной системы, а =
= 6,22 А, а = 33°30'; плотн. 2,325; т. пл. 713°;
т. кип. 1412°. Теплота образования ДЯ298 =
= — 153,40 ккал/моль. Растворимость в воде (в %):
34,5 (0°); 35,3 (20°); 42,2 (100°); очень гигроскопичен.
Плотность водных р-ров:
% MgCl2. . . 14 20 32
d2°....... 1,1198 1,1757 1,2979
4
Насыщенный р-р, содержащий 62,9 a MgCl2 на
100 г Н2О, кипит при 130,0°. Криогидратная точка
—33,6° (20,6% MgCl2). М. х. образует кристаллогидраты
с 1, 2, 4, 6, 8 и 12 молекулами воды. В интервале
темп-р от —3,4° до 116,7° М. х. устойчив в равно-
весии с р-ром MgCl2 • 6Н2О — бесцветными моноклин-
ными кристаллами, а = 9,90 А, b = 7,15 А, с = 6,10 А,
р = 94°; плотность 1,560. В интервале 116,7—181,5°
устойчив MgCl2 • 4Н2О, при 181,5—240° — MgCl2 •
• 2Н2О, при 240—285° — MgCl2 • Н2О; при нагре-
вании моногидрата выше 285° образуется основной
хлорид Mg (ОН) С1. Выше 500° последний разлагается
на MgO и НС1. Давление паров воды над насыщенным
р-ром MgCl2 • 6Н2О (в мм рт. ст.) 4,11 (15°); 9,95
(30°); 44,33 (60°); 165,06 (100°). При действии конц.
водных р-ров MgCl2 на MgO образуются основные
хлориды магния. При темп-ре ниже 100° из таких р-ров
кристаллизуются MgCl2 • 3Mg (ОН)2 • 8Н2О и MgCl2 •
• 5Mg(OH)3‘- 2Н3О, а выше 100° — MgCl2 • 9Mg(OH)2-
• 5Н3О и MgCl3 • 2Mg (ОН)2 • 4Н2О. Эти соединения
входят в состав т. наз. магнезиальных цементов.
М. х. образует двойные соли, напр. природный карнал-
лит MgCl2 • КС1 • 6Н2О.
Шестиводный М. х. (бишофит) полу-
чают упариванием Морских рассолов, предварительно
сконцентрированных в испарительных бассейнах до
содержания в них 18—20% MgCl2. Растворы с высо-
ким содержанием MgCl2 (28—31%) образуются также
в качестве побочного продукта при получении КС1
(см. Калия хлорид) из карналлита или же при раство-
рении магнезита или Mg(OH)2 в соляной к-те. Mg (ОН)2
может быть также превращена в MgCl2 при взаимо-
действии с СаС12 и СО2. Полученный любым способом
хлормагниевый щелок упаривают в открытых аппара-
тах с огневым обогревом (чренах) или котлах с погру-
женными горелками, а затем разливают в тару, где
при охлаждении он закристаллизовывается в твердую
массу. Для получения чешуйчатого М. х. выпаренный
щелок кристаллизуют на вращающемся стальном
барабане, охлаждаемом изнутри водой. Безвод-
ный М. х. получают путем постепенного обезвожи-
вания бишофита до дигидрата. Обезвоживание ведут
при 100—200° в токе горячего воздуха, постепенно
повышая темп-ру, чтобы избежать плавления. Полу-
ченный MgCl2 • 2НаО подвергают дегидратации в токе
1029
МАДЕЛУНГА РЕАКЦИЯ—МАЙЕРА РЕАКЦИЯ
1030
НС1 при 100—200°. Другой способ получения безвод-
ного М. х. заключается в хлорировании MgO в при-
сутствии угля при 800—1000°. Его получают также
как побочный продукт в произ-ве титана по реакции:
TiCl4 2Mg = 2MgCl2 -J- Ti.
Безводный M. х. является
сырьем в произ-ве металлич.
магния. Шестиводный М. х. при-
меняют для получения магнези-
альных цементов, служащих ос-
новой для изготовления многих
строительных материалов, напр.
ксилолита, фибролита и т. п.
М. х. используют также в тек-
стильной пром-сти, для прида-
ния огнестойкости дереву и т.д.
Лит.: Позин М. Е., Техноло-
гия минеральных солей, 2 изд., Л.,
1961.
МАДЕЛУНГА РЕАКЦИЯ —
внутримолекулярная конденса-
ция о-толуидидов с образованием
производных индола:
- СН8 _Нг0
чм-с^°
I
Н
R
Таблица 3
Мазут Смолы, % Асфаль- тены, % Карбены и кар- боиды, % Акциз- ные смо- ЛЫ, % Кокс, %
ФС-5 (прямой гонки, сернистый) 13,60 0,94 0,03 28 7,97
Ф-12 (прямой гонки, малосернистый). . 14,03 0,11 0,03 28 5.79
Ф-12 (крекинг, сернистый) 10,59 4,30 0,19 40 10.22
М-40 (крекинг, сернистый) 8,12 6,64 1,32 72 15,20
М-200 (крекинг-остаток, малосернистый). 16,60 14,5 1,19 — 17,60
н
Катализаторами служат алкого-
ляты, амиды, анилиды, о-толуи-
диды щелочных металлов и р-ры
щелочных металлов в жидком
аммиаке. Механизм М. р., по-ви-
димому, аналогичен механизму
других конденсаций кротоново-
го типа в присутствии сильных
оснований. В М. р. могут быть
успешно применены о-толуиди-
ды как алифатических, так и
ароматич. карбоновых к-т. Сам
индол получают из N-формил-
о-толуидина в присутствии трет-
бутилата калия. N-Замещенные
о-толуидиды циклизуются с пло-
хими выходами. Наличие нитро-
группы в бензольном кольце пре-
пятствует образованию индола.
Лит.: Джулиен П., Мейер Э.,
Пр инти Э., в кн.: Гетероцикли-
ческие соединения, под ред. Р. Эль-
дерфилда, пер. с англ., т. 3, М.,
1954,с. 13. М. Н. Преображенская.
МАЗУТ — жидкий продукт, остаток после отгона из
нефти топливных фракций (бензина, лигроина, кероси-
на и дизельного топлива); используют для произ-ва
масел (см. Масла минеральные), топочных мазутов и
битума. Значительная часть М. перерабатывается
на легкое моторное топливо путем крекинга и коксо-
вания.
Физич. свойства и химич. состав М. зависят от
глубины отбора легких фракций и состава исходной
нефти (парафинистая, сернистая, смолистая и др.).
М. характеризуется след, данными: rfj" 0,89—0,995;
поверхностное натяжение: 30—40 эрг/см2 (40°), 27—
36 эрг/см2 (70°), 24—32 эрг/см2 (100°); теплота сгорания
9100—ЮОООккол/ке; теплота испарения 40—50 ккал/кг',
теплопроводность 0,35—0,40 кал)см сек • град-, эле-
ментарный состав: 83,5—88,5% С и 10,5—12,5% Н.
Нек-рые физико-химич. свойства М., полученных из
нефтей различных месторождений после отгона фрак-
ций, выкипающих до 300°, приведены в табл. 1.
Для произ-ва топлив используют М. прямой пере-
гонки и крекинга нефти, а также продукты коксо-
вания сланцев и углей. Товарные сорта топочных М.
и нек-рые их свойства приведены в табл. 2.
В табл. 3 приведено содержание асфальтовых соеди-
нений в нек-рых сортах М.
Таблица 1
Нефть Выход на нефть, % Плот- ность при 20° Вязкость услов- ная, °ВУ при Т. заст., °C Т. всп. в закры- том тиг- ле, °C Содержа- ние смол (акциз- ных), %
50° 100°
Сураханская отборная 52,44 0.8915 5,24 20—24 12,4
Сураханская тяжелая 64,68 0,9269 15,92 2,42 -(8-10) 202 36,8
Бинагадинская тяжелая 65,51 0,9530 — 3,41 -(9-12) 200 45,0
Биби-эйбатская легкая Биби-эйбатская парафи- 52,00 0,9213 — 2,1 ниже —20 174 36,0
нистая 61.92 0,9340 — 2,53 13—16 196 40,0
Ишимбайская Туймазинская (девон- 60,0 0,9550 — 3,5 3 167 60,0
ского горизонта) . . . 59,0 0,950 —— 3,3 6 178 68,0
Ставропольская 54,0 0,9380 11,3 2,5 —7 128 60,0
Таблица 2
Марка мазута Свойства
Плот- ность при 20° Вязкость условная, ВУ (°C) Золь- ность, О/ /о Сера, % Т. заст., °C Т. всп., °C Теплота сгорания ккал/кг
Флотский ФС-12 0,900 5 (50") 0,10 2,0 -5 80 9870
Ф-12 0,930 12 (50°) 0,15 0,8 —8 90 9870
Ф-20 0,930 6 (75°) 0,15 0,8 —5 90 9870
Топливо нефтяное М-20 5 (80°) 0,30 ДО 3,5 4-5 80 9680
М-40 8 (80°) 0,30 3,5 --10 100 9610
М-60 И (80°) 0,30 3,5 --15 110 9560
М-80 0,990 13 (80°) 0,30 3,5 -20 120 9510
М-100 0,990 15,5 (80°) 0,30 3,5 --25 125 9500
М-200 9,5 (100°) 0,30 3,5 --36 140 9450
Ухтинский — 13(100°) 0,50 1,4 --25 110 —
Парафинистый — 25 (80°) 0,20 1,0 --42 95 9870
Сланцевый — 3,5 (75°) 0,30 2,0 —5 65 —
Угольный I — 5 (75°) 0,30 0,5 ti5 100 —
Угольный II — 3 (75°) 0,30 0,5 70 —-
М. применяют в качестве топлива для паровых кот-
лов, промышленных печей различного назначения и
газовых турбин. В последнем случае важное значение
имеет величина и состав зольного остатка топлива.
В золе топлива должно быть минимальное количество
ванадия (0,001%) и натрия (0,0005%), являющихся
основными коррозионными агентами. Этим требова-
ниям отвечают М. из несернистых нефтей. Борьба
с отложениями и ванадиевой коррозией поверх-
ностей нагрева котлов и лопаток газовых турбин
ведется с помощью различных присадок к топливам.
Лит.: ГуревичИ. Л., Технология нефти, ч. 1, 2 изд.,
М.—Л., 1952; Лосиков В. В., Пучков Н. Г., Эн-
глин В. А., Основы применения нефтепродуктов, 2 изд.,
М., 1959; Технические нормы на нефтепродукты, 16 изд.,
М., 1957; Моторные, реактивные и ракетные топлива, под ред.
К. К. Папок и Е. Г. Семенидо, 4 изд., М., 1962.
А. Д. Фатьянов.
МАИСОВОЕ МАСЛО — см. Жиры растительные.
МАЙЕРА РЕАКЦИЯ (Мейера реакция) — метод
синтеза алкилмышьяковых к-т, состоящий во взаимо-
действии арсенитов щелочных металлов с алкилирую-
17*
1031
МАЙЕРА РЕАКЦИЯ — МАКРОМОЛЕКУЛА
1032
щими агентами в спирто-водной щелочной среде.
Алкилирующий агент атакует арсенит по месту сво-
бодной электронной пары атома мышьяка:
NaOJ' ^*’n„oh NaO\
NaO-^As'-+R-X ——- NaO -^As-R + NaX+H2O
HO"
Эта реакция является общей для галогенопроизвод-
ных предельных углеводородов алифатического ряда,
галогенопроизводпых жирных карбоновых к-т, их
производных, галогенопроизводных спиртов, кетонов,
сульфидов, эфиров и др. Т. обр., с помощью М. р.
могут быть синтезированы алкилмышьяковые к-ты,
содержащие в радикале различные функциональные
группы. Применяя дигалогенпроизводные жирного
ряда, удалось синтезировать соединения, содержащие
два остатка мышьяковой к-ты:
о
NaO, 2NaOH 'NaO, // \
2NaO xAs4-XCII.,CIbX-----I \AsCH.>- +2NaX+2H2O
СИ/ ' \NaOz /2
Наиболее энергично реагируют с арсенитами иодо-
производные, далее следуют бром- и, наконец, хлоро-
производные, одпако при взаимодействии хлоро-
производных с арсенитами в автоклаве при давлении
10—15 ат получают соответствующую алкилмышьяко-
вую к-ту с отличным выходом.
Взаимодействие ароматич. галогенопроизводных с
арсенитами щелочных металлов лишь в редких слу-
чаях и в достаточно жестких условиях ведет к син-
тезу солей ароматич. мышьяковых к-т с очень малыми
выходами. Наличие в орто- или параположепии арил-
галогенида ориентантов II рода облегчает реакцию
с арсенитами. Так, при взаимодействии о-бромбен-
зойной к-ты и арсенита натрия получается о-карбо-
ксифениларсеновая к-та с выходом более 40%.
В качестве алкилирующих агентов в М. р. могут
быть использованы, помимо галогеналкилов, диал-
килсульфаты, эфиры арилсульфокислот и др. Обра-
зование основного продукта М. р. — соли алкил-
мышьяковой к-ты — сопровождается побочными про-
цессами и в первую очередь отщеплением элементов
галогеноводородпой к-ты от галогенопроизводного:
О
/-
(KO)»AsCH2CH2Br
I ' О
КОН| //
(KO)-.As(HO)4-BrCH..CH.>Br-O[(K0)»AsCH2-]2
I, СИ2 = СНВг
омылением галогенопроизводпого до спирта и обра-
зованием простого эфира.
Распространением М. р. явилась открытая Оже
(Auger, 1903) реакция солей первичных органич.
мышьяковистых к-т с жирными галогенопроизвод-
ными, приводящая к образованию солей диалкил-
мышьяковой к-ты:
RAs(ONa)»+R'X — RR'AsO (ONa) + NaX
Соли алкилмышьяковых к-т, полученные М. р.,
им ользовались для синтеза алкилмышьяковистых
к-т и арилхлорарсипов. Последние получают восста-
новлением RAsO(ONa)2 сернистым газом в р-ре конц.
соляной к-ты в присутствии небольших количеств
иода, как катализатора:
RAsO (ONa)a + SO2 + HCl RAsCl2 + Na2SO4 + H2O
M. p. открыта В. Майером в 1883 при взаимодей-
ствии йодистого метила с тринатрийарсенитом.
Лит.: Синтетические методы в области металлоорганиче-
ских соединевий, вып. 7, М.—Л., 1945; Реутов О. А.,
Теоретические проблемы органической химии, М., 1956;
Немец В. Г., С очи л ив Е. Г., Химия отравляющих
веществ, Л.—М., 1941. Р. Н. Стерлин.
МАЙОНЕЗ — вязкая концентрированная эмульсия,
состоящая в основном из растительного масла, яич-
ного желтка, уксуса и воды. Примерный состав
(один из рецептов) М.: 67% растительного масла,
10,1% желтка, 0,7% казеина, 3,3% р-ра однопроцент-
ной соды, 2% сахара, 2,3% горчицы, 1,2% поварен-
ной соли, 6,7% столового уксуса, 6,7% воды. Стаби-
лизаторами в эмульсиях М. являются яичный желток
(иногда в сочетании с казеином) или сухое обезжи-
ренное молоко, которые образуют прочные адсорб-
ционные оболочки вокруг диспергированных капель
масла, удерживая их во взвешенном состоянии в вод-
ной части эмульсии. Прочность эмульсии майоне-
за возрастает с увеличением степени дисперсности
масла.
ОН
Процесс изготовления М. состоит из подготовки
материала, приготовления эмульсии, расфасовки и
упаковки. Готовый М. во избежание окисления
жировой фракции должен быть быстро расфасован
в герметичную тару. В микробиологии, отношении
М. очень устойчив благодаря наличию в нем уксус-
ной к-ты. Расфасованный в герметичную тару М. ре-
комендуется хранить в темноте от минус 5° до -|-7°,
В этих условиях он может сохраняться до 8 месяцев.
Н. И. Козин, А. А. Смотрин.
МАКЛУРИН (2,3', 4, 4', 6-пентаоксибензофенон)
С1зН10О6, мол. вес 262,13 — кристаллогидрат (с одной
молекулой Н2О), кристалли-
зуется из воды в виде он он
светло-желтых призм, крн-
сталлизационную воду те- н0
ряет при 130—140°; т. пл. _
безводного 200°; 1г М. рас- °
творяется при 14° в 190 г воды, хорошо растворим в
спирте и эфире; обладает сладким вяжущим вкусом.
М. является одним из красящих начал желтого де-
рева, откуда он может быть извлечен кипящей водой.
М. растворяется в щелочах и концентрированной сер-
ной к-те; с хлорным железом окрашивается в зеленый
цвет, переходящий под действием соды в красный.
Все пять гидроксильных групп М. можно прометилиро-
вать диметилсульфатом в присутствии щелочей или
диазометаном. Наличие пяти гидроксильных групп
ослабляет связь карбонильной группы с бензольным
кольцом. М. распадается на флороглюцин и прото-
катеховую к-ту при нагревании с водными щелочами
с разб. серной к-той или при сплавлении с содой.
Синтетически М. может быть получен конденсацией
флороглюцина с нитрилом протокатеховой к-ты в
условиях Гёша синтеза:
маклура#
В экстракте желтого дерева М. содержится вместе
С морином. Я- Я- Гамбарян.
МАКОВОЕ МАСЛО — см. Жиры растительные.
МАКРОМОЛЕКУЛА — совокупность большого
числа атомов, соединенных химическими связями.
Вещества, построенные из М., наз. высокомолеку-
лярными (см. Высокомолекулярные соединения). В
отличие от молекул низкомолекулярных веществ,
к-рые характеризуются постоянством мол. веса, М.
одного и того же высокомолекулярного вещества мо-
гут иметь различный мол. вес. Это свойство наз.
полидисперсностью или полимолекулярностью (см.
Молекулярный вес высокомолекулярных соединений).
Строго монодисперсные высокомолекулярные веще-
ства пока получаются только в условиях биосинтеза.
Число атомов, входящих в состав М., может быть
очень большим (сотни тысяч и миллионы). Однако
1033
МАКРОМОЛЕКУЛА
1034
в пределе при неограниченном возрастании мол. веса
понятие М. теряет смысл, т. к. по существу та-
кая частица перестает отличаться от тела макро-
скопия. размеров. Мол. веса обычных М. колеблются
в широких пределах — от 103 до 10е.
Линейные М. построены из мономерных звеньев
одного или- разных типов, соединенных регулярно
или нерегулярно химич. связями в длинные цепи.
Длина таких цепей обычно составляет 103—105 А
при поперечнике 3—7,5 А. Примером линейных М.
могут служить М. каучука натурального, регуляр-
ного полиэтилена, полиэфиров и полиамидов, полу-
ченных полимеризацией или поликонденсацией би-
функциональных мономеров, М. нуклеиновых кислот,
ряда белков, целлюлозы и др. В разветвлен-
ных М. к основной цепи присоединены боковые
цепи большей или меньшей длины. Примером развет-
вленных М. могут служить М. привитых сополиме-
ров, крахмала, нек-рых белков, полиэфиров и поли-
амидов, полученных из три- и более функциональных
мономеров и др.
Специфические свойства высокомолекулярных сое-
динений — проявление в определенных условиях
высокоэластич. деформаций (см. Эластичность поли-
меров), способность к образованию пленок и анизо-
тропных волокнистых структур, к набуханию и обра-
зованию высоковязких р-ров, аномальные термоди-
намич. свойства растворов высокомолекулярных сое-
динений — обнаруживаются только у высокомолеку-
лярных соединений, построенных из линейных или
умеренно разветвленных М. Эти свойства целиком
обусловлены большой длиной и гибкостью цепей М.,
т. е. способностью М. в широких пределах изменять
свою форму (конформацию) за счет вращения обра-
зующих цепь звеньев относительно ординарных хи-
мич. связей с сохранением валентных углов (см.
Гибкость цепных молекул).
Граница между низкомолекулярным и высокомоле-
кулярным веществами определяется минимальным мол.
весом, при к-ром начинают проявляться перечислен-
ные выше специфич. свойства полимеров. Эта граница
связана с химич. природой мономерных звеньев,
образующих М. (в частности, с величинами энергетич.
барьеров для внутримолекулярного вращения, от
к-рых зависит гибкость М.), и поэтому для различных
веществ может изменяться в весьма широких пределах
(от мол. весов порядка тысяч — для гибких, до сотен
тысяч — для жестких М.).
М. в пределе может иметь форму вытянутой нити
или компактного клубка (глобулы). Однако
наиболее характерным являются промежуточные кон-
формации случайного или упорядоченного характера
(статистич. клубок, спираль, разнообразные складчатые
формы и т. д.).
Изменение конформации М. представляет собой один из
видов теплового движения в высокомолекулярном веществе.
Большая длина и гибкость М. позволяют отдельным частям
М. в определенных пределах двигаться независимо от других.
Поскольку число таких частей — сегментов — достаточно
велико, их поведение подчиняется статистич. закономерностям.
Это дает возможность при теоретич. рассмотрении свойств и
поведения отдельной М. использовать статистич. метод, пол-
ностью аналогичный методу рассмотрения броуновского дви-
жения. Поскольку М. могут иметь различные конформации
и каждой конформации отвечает определенный размер М.,
то рассматривается распределение М. по их длинам, характе-
ризуемым расстоянием h между концами М. Функция распре-
деления абсолютных значений длины М., описывающая вероят-
ность трго, что величина h лежит в интервале (Л, h + dh), по
форме аналогична функции распределения молекул газа по
скоростям:
з/ 3/12
/ 3 \ 22 2Zb-
W)^= h-dh.,
где W(h)dh — искомая вероятность, Z — число сегментов
в цепи и Ъ — длина сегмента (длина предельно растянутой м.
^мако. =
Вследствие гибкости М. J' /i2 -7iAlaKC , где—среднее ква-
дратичное расстояние меязду концами М., вычисленное с по-
мощью приведенной функции распределения. Пользуясь
методами статистич. физики, на основе распределения М.
по длинам, удается вывести основные закономерности термо-
динамически равновесной высокоэластич. деформации тел,
построенных из М., а также ряд свойств растворов полиме-
ров. Современные данные о размерах М. и их строении по-
зволяют связать Н* с энергетич. и структурными характери-
стиками М.
Форма М. в конденсированной фазе, конц. р-рах
и студнях тесно связана с характером надмолекуляр-
ных структур, от к-рого существенно зависят свойства
высокомолекулярного вещества, в первую очередь
механические. В кристаллич. полимерах (см. Кри-
сталлическое состояние полимеров) конформации М.
упорядочены. Для аморфных полимеров, в отличие
от кристаллич., характерным является набор различ-
ных конформаций М. Изменение конформаций М. иг-
рает очень большую роль в биологич. системах. Так,
напр., процессы денатурации белков и нуклеиновых
к-т связаны с изменением формы их М. (в частности, с
переходами спираль — клубок). Изменение формы М.
мышечного миозина обусловливает сокращение мышц
и т. ц.
Согласованного изменения конформаций М. в из-
делиях из полимерного материала достигают путем
приложения к образцу механич. поля нужной сим-
метрии (ориентация). Придание М. заданной формы
является одной из важнейших сторон процесса пере-
работки полимерных материалов. Иногда ориенти-
рованные структуры из М. возникают непосредственно
при синтезе М. Это особенно характерно для биологич.
структур. Конформации М. могут меняться при из-
менении свойств среды, в к-рой находится М. Так,
напр., в разб. р-рах высокомолекулярных соединений
в случае растворителей, хорошо смачивающих поли-
меры, преобладают вытянутые конформации, а в плохо
смачивающих растворителях (так называемых «пло-
хих» растворителях) преобладают свернутые (гло-
булярные) конформации. Эти различия обусловлены
взаимодействием между молекулами растворителя и
М., к-рое приводит к появлению разностей энергии
различных конформаций, обладающих одной и той же
энергией в чистом полимере. В случае хорошего сма-
чивания полимера растворителем для перевода М.
из вытянутой конформации в свернутую требуется
затратить энергию на преодоление взаимодействия
звеньев М. с растворителем. При плохом смачивании,
наоборот, необходима работа для распрямления
М. В идеальных растворителях, т. е. в растворителях,
для к-рых энергия межмолекулярного взаимодействия
равна или близка к энергии взаимодействия их мо-
лекул со звеньями М., все конформации остаются
изоэнергетическимп. Глобулярная М. в плохом рас-
творителе уподобляется коллоидной частице. При
замене хорошего растворителя на плохой раствор
превращается в эмульсию. Добавление нераствори-
теля к относительно конц. р-рам может приводить
к образованию фибрилярных агрегатов, построенных
из распрямленных М. (особенно в том случае, когда
при агрегировании происходит кристаллизация), или
смеси фибрилярных и глобулярных форм.
Самопроизвольный переход от неупорядоченных
к упорядоченным конформациям М. в конденсиро-
ванной фазе имеет место при кристаллизации поли-
меров. При плавлении происходит обратный процесс.
Форма М. полиэлектролитов в растворах и студнях
обратимо меняется при изменении pH среды. На этом
свойстве М. полиэлектролитов основано действие
систем, способных непосредственно превращать химич.
энергию в механич. и представляющих собой простей-
шие модели мышц. Сокращение реальных мышц
связано с изменением конформаций белковых М. при
1035
МАКРОМОЛЕКУЛА
1036
изменении окислительно-восстановительного потен-
циала среды. Прямое превращение химич. энергии
в механич. может быть осуществлено только в си-
стеме, построенной из М. Способность М. обратимо
изменять свою форму обусловливает явления «меха-
нической» памяти.
М. не могут быть переведены в газовую фазу путем
испарения, т. к. работа отрыва М. от соседних М.
или молекул растворителя, равная практически сумме
энергий межмолекулярного взаимодействия с сосе-
дями ее многочисленных звеньев, всегда значительно
больше энергии химич. связи. Поэтому процесс хи-
мич. деструкции всегда происходит значительно ниже
гипотетич. темп-ры кипения высокомолекулярного
вещества.
Для определения формы М. в конденсированной
фазе и в растворах используют различные физич.
и физико-химич. методы исследования (электронная
микроскопия, двойное лучепреломление, светорас-
сеяние, вискозиметрия, рентгеноструктурный ана-
лиз и др.).
При переходе от линейных и малоразветвленных
М. к сильно разветвленным их специфич. свойства
постепенно утрачиваются. Сильно разветвленная М.
может иметь форму глобулы, однако, в отличие от
глобулярных форм линейных М., такие глобулы обра-
зуются в результате возникновения достаточно боль-
шого числа внутримолекулярных химич. связей
(напр., глобулы крахмала). При увеличении числа
и частоты разветвлений М. утрачивает гибкость и
в пределе превращается в коллоидную частицу.
Если увеличение числа разветвлений сопровождается
неограниченным возрастанием мол. веса, то в пре-
деле М. превращается в пространственно-структури-
рованное твердое тело. Примерами таких тел могут
служить отвержденные феноло-альдегидные смолы,
вулканизаты каучуков, алмаз и т. п. Для оценки
разветвленности М. пользуются различными физич.
и физико-химич. методами исследования (светорас-
сеяние, вискозиметрия, осмометрия и др.).
М. может иметь регулярное и нерегулярное строе-
ние. Регулярной наз. М., в к-рой химич. звенья и их
пространственные конфигурации периодически чере-
дуются (см. Стереорегулярные полимеры). Частным
случаем регулярных М. явл. М., построенная из
одинаковых химич. звеньев, находящихся в одина-
ковых пространственных конфигурациях (см. Изо-
тактические полимеры). При соединении молекул
мономеров в М. даже в простых случаях возникает
ряд возможностей для построения М. различной
структуры.
Простейший мономер винилового ряда — этилен, при
полимеризации в положении 1,2 образует линейные регуляр-
ные М. полиэтилена:
пСН2 = СН2-»-[СН2-СН2 -]п -
Наряду с этим часть молекул этилена может соединиться
в положении 1,1 с образованием цепи
I 2 1 1 S I 1
—СН2—СН2—СН—СН2—СН2—СН2—СН—
I I I
2СН3 2СН3 2СН3
Возможно также образование более длинных боковых раз-
ветвлений в результате реакций передачи цепи через полимер.
Монозамещенные этилены при образовании линейной цепи
присоединяются по типу «голова к хвосту»...
* * *
... —СН2—СНХ—СН2—СНХ—СН2—СНХ—...
или «голова к голове»
* * # *
... —сн2—снх—снх—сн2—сн2—снх—снх—сн2 -...
Необходимым условием построения регулярной цепи
является отбор в процессе полимеризации конфигураций
одного типа, либо нек-рой строго периодич. последователь-
ности чередования конфигураций обоих типов. Однако это
необходимое условие не является достаточным. При соедине-
нии молекул монозамещенного этилена в М. в каждом звене
М. возникает асимметрический атом углерода (помечен звез-
дочкой), вследствие чего звено может иметь либо <1, либо 1
конфигурацию.
М. будет регулярной лишь в том случае, если при
соблюдении первого условия в процессе полимериза-
ции осуществлен отбор конфигураций одного типа
(только d или только 1) или нек-рой правильной
периодич. последовательности чередования d- и
1-коифигураций. Случайное чередование конфигура-
ций «голова — хвост», «голова — голова», d, 1-кон-
фигураций, а также случайное возникновение раз-
ветвлений в результате реакций передачи цепи через
полимер приводит к нарушению регулярности М.
При переходе от виниловых мономеров к диеновым
возрастает число возможных изомерных структур
М. и, соответственно, возможностей нарушения регу-
лярности строения М.
Регулярность М. является одним из необходимых
условий кристаллизации полимеров. Высокоупоря-
доченные надмолекулярные структуры (кристаллич.
ленты, сферолиты, монокристаллы) могут образо-
вываться только из регулярных М.
Физич. и химич. свойства М. зависят от ее химич.
состава, мол. веса (а также количества и длины раз-
ветвлений в случае разветвленной М.), последователь-
ности чередования химич. звеньев и их взаимного рас-
положения в пространстве. Все эти характеристики
М. задаются непосредственно при синтезе М. и в даль-
нейшем не могут быть изменены без дополнительных
воздействий, приводящих к разрыву и образованию
новых химич. связей. Наиболее строгое и тон-
кое регулирование состава, строения и длины М.
достигается при синтезе М. в клетках живых орга-
низмов. В лабораторных условиях и в промышлен-
ности такое регулирование пока возможно лишь
в относительно простых случаях. Однако использо-
вание даже этих возможностей в процессах стерео-
специфич. полимеризации позволяет коренным обра-
зом воздействовать на свойства полимерных веществ
и синтезировать материалы с разнообразными жела-
тельными свойствами.
Химич, свойства М. связаны с природой функцио-
нальных групп, входящих в состав М. Специфиче-
скими химич. реакциями М. являются: 1) деструкция
полимеров, приводящая к разрыву цепей и снижению
мол. веса; 2) структурирование (см. Вулканизация),
т. е. возникновение химич. связей между различ-
ными М., приводящее к возрастанию мол. веса и
в пределе к образованию сплошной сетчатой струк-
туры (см. Структурирование полимеров пространст-
венное); 3) реакции присоединения и отщепления
низкомолекулярных веществ без изменения степени
полимеризации, приводящие к образованию поли-
мераналогов (напр., этерификация целлюлозы с полу-
чением простых и сложных эфиров целлюлозы, омы-
ление поливинилацетата с получением поливинило-
вого спирта, внутримолекулярное отщепление воды
от полиакриловой кислоты с получением полиан-
гидрида и т. п.).
Важной особенностью М. является высокая чув-
ствительность к очень незначительным изменениям
химич. состава, напр. к введению в М. одного или не-
скольких звеньев другого типа. Поскольку при та-
ких изменениях подавляющее число химич. звеньев
не затрагивается, М. в основном сохраняет комплекс
присущих ей свойств. Однако химич. превращение
даже в одном из многочисленных звеньев или введе-
ние даже одного постороннего звена может привести
к появлению какого-либо нового свойства или изме-
нению одного из существующих (напр., появлению
повышенной склонности к структурообразованию или
деструкции, изменению растворимости и т. п.). Близ-
ким к этому свойством, присущим только М., яв-
1037
МАКРОРАДИКАЛЫ СВОБОДНЫЕ
1038
ляется т. н. «химическая память», т. е. способность
запасать и хранить информацию о тех или иных воз-
действиях, послуживших причиной структурио-химич.
изменения одного или нбскольких звеньев М. Явле-
ние «химической памяти» лежит в основе мута-
ций, приводящих к изменению наследственности.
Незначительные изменения химич. структуры одного
из звеньев М. дезоксирибонуклеиновой кислоты, вы-
полняющей основные генетические функции, закреп-
ляются при ее редупликации в процессе деления кле-
ток и приводят к изменению и закреплению определен-
ных наследственных признаков живого организма.
Лит..-В олькенштейн М. В., Конфигурационная
статистика полимерных цепей, М.—Л., 1959; Гейлорд Н.,
Марк Г., Линейные и стереорегулярные полимеры, пер.
с англ., М., 1962; Современные проблемы биофизики. Сб. ст.,
перевод, т. 1, М., 1961; F 1 о г у Р. J., Principles of polymer
chemistry, N. Y., 1953; Каргин В. А., Слоним-
ский Г. Л., Краткие очерки по физико-химии полимеров,
М., 1960. В. А. Кабанов, Г. Л. Слонимский.
МАКРОРАДИКАЛЫ СВОБОДНЫЕ (полимерные
радикалы) — полимерные цепи, имеющие один или
несколько неспаренных электронов; последние могут
быть в середине или конце основной цепи, если поли-
мер не разветвлен, или в боковой цепи. М. с. могут
образовываться двумя способами: 1) из низкомоле-
кулярных соединений (мономеров) путем радикальной
полимеризации в результате присоединения исход-
ного инициирующего радикала свободного к двойной
связи молекулы мономера с образованием свободного
радикала большего размера, к-рый, в свою очередь,
по аналогичной схеме реагирует с другой молекулой
мономера; 2) из полимеров — при действии различ-
ных деструктирующих факторов (см. Деструкция
полимеров), в результате действия к-рых происходит
разрыв основной полимерной цепи, отрыв боковых
групп, атомов водорода или мономерных звеньев
(см. Деполимеризация) с образованием М. с. Эти
процессы могут обусловливаться как механохимич.
реакциями, имеющими место, напр., при вальцевании
полимеров, действии на них ультразвука (см. Механо-
химия полимеров), так и действием на полимеры иони-
зирующих излучений (радиационная деструкция),
УФ-лучей, тепла, кислорода (термич. и термоокис-
лительная деструкция) и нек-рых других факторов.
М. с., как и низкомолекулярные свободные ради-
калы, имеют нечетное число электронов, обладающих
спиновым моментом, вследствие чего М. с. характери-
зуются парамагнитными свойствами. Наличие послед-
них дает возможность использовать электронные
парамагнитные спектры для идентификации М. с.
и изучения их реакций. Реакционноспособность М. с.
определяется, - в первую очередь, свойствами звена,
несущего неспаренный электрон, и не зависит от
длины цепи; реакционноспособность М. с. не отли-
чается от реакционноспособности подобных низко-
молекулярных свободных радикалов.
Характерной особенностью М. с. является их спо-
собность образовывать полирадикалы, т. е. каждая
полимерная цепь может обладать несколькими сво-
бодными валентностями (радикальными центрами).
Это свойство М. с. является одной из главных причин
большого разнообразия и сложности вторичных про-
цессов в полимерах при различных воздействиях.
Число (концентрация) таких свободно-радикальных
центров может меняться в широких пределах и опре-
деляется соотношением скоростей образования и
«гибели» М. с. Большой мол. вес М. с. обусловливает
малую скорость их диффузии. Скорость различных
химич. процессов с участием М. с. нередко опреде-
ляется скоростью их диффузии и зависит от вязкости
среды. В таких реакциях решающим фактором яв-
ляется скорость диффузии отдельных полимерных
цепей друг относительно друга или скорость переме-
щения сегментов полимерной цепи. Возрастание вяз-
кости приводит к резкому изменению скорости реак-
ций с участием М. с., а при очень высоких значениях
вязкости — к образованию «долгоживущих», или «за-
стрявших», М. с. Если обычно при установлении в про-
цессе полимеризации стационарного состояния кон-
центрация М. с. порядка Ю 8—10~в молъ/л-сек, то
концентрация таких «застрявших» М. с. может дости-
гать величины 10~4 молъ/л сек, а время жизни от
долей секунды увеличивается до десятков и сотен
секунд. В отдельных случаях время жизни таких
долгоживущих М. с. может быть очень велико. Так,
парамагнетизм многих облученных пластиков сохра-
няется в вакууме многие месяцы; в случае со-поли-
меризации (процессе, приводящем к образованию
нерастворимых полимеров с пространственной струк-
турой) активность М. с., находящихся в инертной
атмосфере, также сохраняется сколь угодно долго.
Введение в такую систему мономера через много дней
после прекращения полимеризации приводит к возоб-
новлению этого процесса. Во многих случаях происхо-
дит увеличение скорости процесса или дополнитель-
ное превращение в полимерах, прогретых после
облучения. Это обусловливается тем, что в результате
увеличения подвижности цепей в реакциях начинают
принимать участие «застрявшие» М. с.
М. с., также как и низкомолекулярные свободные
радикалы, могут реагировать между собой двумя спо-
собами — вступать в реакцию рекомбинации или дис-
пропорционирования. При рекомбинации М. с. проис-
ходит соединение двух полимерных радикалов с обра-
зованием полимерной молекулы с большим мол.
весом (I). В случае диспропорционирования мол. вес
образующегося неактивного полимера практически
очень мало отличается от мол. веса М. с., ио полови-
на полимерных молекул имеет на концах двойную
связь (II):
'—СН2-СН21 А^СНа-СНа-СНа-СНа (I)
~~ СНа-СНа I \ СНа-СН8+СН2=СН ~~ (II)
Обе эти реакции имеют очень низкую энергию акти-
вации и часто идут одновременно (напр., при полиме-
ризации). Однако особенности строения М. с. в каж-
дом отдельном случае могут благоприятствовать
течению одной из этих реакций. Так, радикалы типа
СН-СН3 аналогичные полистирольным радика-
лам, вступают преимущественно в реакцию (I). Одной
из наиболее характерных реакций М. с. является взаи-
модействие их с соединениями, обладающими двой-
ной связью, что и определяет цепной характер
радикальной полимеризации. Эта реакция может
быть инициирована различными способами, но по-
сле первых немногих актов присоединения моле-
кул мономера радикал можно рассматривать как
полимерный, а его реакционную способность — не за-
висящей от длины цепи. В случае совместной полиме-
ризации (см. Сополимеризация) двух мономеров в си-
стеме находятся два вида М. с., к-рые отличаются
конечными группами, несущими свободную валент-
ность, что и определяет их реакционную способность.
Эти М. с. могут реагировать с обоими присутствую-
щими в системе мономерами, но, в зависимости от
соотношения между реакционными способностями
М. с. и мономера, обычно происходит или обогаще-
ние сополимера к.-л. одним мономером, или образо-
вание сополимера с правильным чередованием разных
мономерных звеньев.
Если М. с., образующиеся из полимеров, полиме-
ризуются с мономерами иной природы, то получаются
привитые полимеры, к-рые характеризуются тем,
что к основной цепи полимера присоединяются боко-
вые цепи с другим химич. строением. Это легко осу-
1039
МАКРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
1040
ществляется облучением полимера в среде другого
мономера; возникающие при облучении полимера
М. с. служат активными центрами для инициирова-
ния полимеризации мономера. Если же М. с., обра-
зовавшиеся из полимера, содержат активные центры
на концах цепи (к-рые могут быть получены, напр.,
при отщеплении, или распаде нек-рых группировок —
атомов Вг, перекисных групп), то при полимеризации
получаются блок-сополимеры. Последние получаются
также рекомбинацией М. с., образующихся в поли-
мерах (чаще всего в результате механохимич. про-
цессов или при обработке смесей полимеров ультра-
звуком). Взаимодействие М. с. с полимерной цепью,
т. е. реакция передачи цепи через полимер при поли-
меризации, приводит к образованию разветвленных
полимеров, а в нек-рых частных случаях — поли-
меров с трехмерной структурой.
С образованием М. с. связаны вулканизация, а также
вулканизация радиационная, приводящие к образова-
нию в полимере сплошной пространственной сетки.
Противоположными реакциями М. с. являются
деполимеризация и деструкция. Инициирование де-
полимеризации происходит обычно по концевым
группам или «слабым» местам (осколкам инициатора,
перекисным группам и т. д.), причем М. с. для каж-
дого полимера в этом процессе аналогичны М. с.,
образующимся из цопомера. При деструкции М. с.
участвуют обычно в реакциях передачи цепи, а при
окислительной деструкции — быстро окисляются, да-
вая промежуточные перекисные и гидроперекисные
соединения. Распад последних сопровождается чаще
всего разрывом цепи с образованием кислородсодер-
жащих М. с. или неактивных продуктов.
Лит.: БагДасарьян 5^. С., Теория радикальной
полимеризации, М., 1959; Г р а с с и Н., Химия процессов
деструкции полимеров, пер. с англ., М., 1959; Б е м ф о р д К.,
Барб Л., Дженкинс А., О н ь о н П., Кинетика ради-
кальной полимеризации виниловых соединений, пер. с англ.,
М., 1961. Т. С. Никитина.
МАКРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ — сое-
динения, молекулы к-рых имеют замкнутое циклич.
строение, причем циклы состоят из 8 и более атомов.
Среди М. с. различают средние (8—12 атомов) и
большие циклы (13 и более атомов в цикле). Известны
карбоциклич., гетероцпклпч., а также неорганич.
М. с., не содержащие углерода в цикле. Наряду с мо-
ноциклич. существуют би- и полициклич. М. с., в мо-
лекулах к-рых два или песк. атомов являются общими
для макроцикла и другого цикла (или других циклов).
__________________ В случае полициклического
Ю/ \ М. с. эти другие циклы могут
р \ / > быть, в частности, многочпс-
I /_______________\ J ленными. Рассматриваемые
\ / системы могут быть конден-
f ' сированнымп (напр., I) или
11 мостиковыми (напр., II).
Названия М. с. строятся по тем же принципам,
что и названия соединений с малыми (3-, 4-членными)
и обычными (5-, 6-, 7-члепными) циклами, напр.
циклодекан (III), S-энаптолактам (IV), пентадекано-
лид (V), азациклононан (VI). Кроме того, многие М. с.
имеют и тривиальные названия, напр. экзальтолид
или тибетолид (V).
важнейших природных веществ, как гемин (см. Гемоглобин),
хлорофилл, витамин В12 (см. Цианкобаламин)].
М. с. и прежде всего соединения со средними циклами
обладают своеобразными физич. и химич. свойствами,
связанными с особенностями их геометрии (простран-
ственной близостью заместителей, находящихся на
противоположных сторонах цикла, и наличием в цепи
энергетически невыгодных затененных конформаций).
Указанные особенности наиболее ярко проявляются
в свойствах, связанных с т. наз. трансаннуляр-
ным эффектом и «неклассическим» напряже-
нием (см. Напряжения теория). Большой интерес
для теории органич. химии представляют ненасы-
щенные М. с., особенно те из них, к-рые имеют арома-
тич. характер (см. Ароматические системы).
Образование М. с., как правило, затруднено, по-
этому для их синтеза используются специфич. приемы.
Методы синтеза М. с. весьма разнообразны и могут
быть разбиты на 4 основных типа.
1. Циклизация бифункциональных соединений,
которая является наиболее общим методом и исполь-
зуется для синтеза любых М. с. Наряду с цикли-
зацией всегда идет нежелательная реакция поликон-
денсации, причем последняя более вероятна, чем
циклизация.
Причина этого явления заключается в том, что из мно-
жества конформаций, в виде к-рых может существовать би-
функциональное соединение с длинной цепью, лишь очень
немногие (т. н. свернутые) удобны для циклизации. В случае
средних циклов неклассич. напряжение еще более затрудняет
циклизацию, что проявляется в характерном снижении вы-
ходов при синтезе средних циклов почти по всем способам
циклизации. Поэтому циклизация бифункциональных соеди-
нений проводится, как правило, в разб. р-рах, что затрудняет
межмолекулярную и делает более вероятной внутримолеку-
лярную реакцию (принцип разбавления Руггли—Циглера).
Циклизация облегчается при наличии в цепи бифункциональ-
ного соединения, т. наз. «жестких» группировок, напр. аро-
матич. колец или кратных связей. Это объясняется пониже-
нием подвижности молекулы, т. е. уменьшением общего числа
возможных конформаций, и, след., увеличением вероятности
осуществления выгодных для циклизации «свернутых» кон-
формаций. Последние более вероятны и при наличии в цепи
циклического соединения заместителей, особенно объемистых.
Циклизация гетероцепных бифункциональных соединений,
содержащих двухвалентные атомы (кислород или серу), про-
ходит легче, чем у их аналогов, содержащих в цепи только
метиленовые группы, что связано с уменьшением конформа-
ционного (неклассич.) напряжения при замене 4-валентного
атома на 2-валентный («кислородный эффект»).
К числу методов циклизации относятся, напр., конден-
сация динитрилов в присутствии натриевых производных
моноалкиланилина по Циглеру (1), внутримолекулярная
сложноэфпрная конденсация по Дикману (2), внутримолеку-
лярное алкилирование эфиров со-галоген-р-кетокислот (3) и
ряд др. методов.
/CN C«"r>'<'CH;! НгО t
(СН2)„ ---------- (CHg)„_, <Lh_cn -- (СНаЕп Q=O (!)
ZCOOR ^-^9°
(CH2)„ ----- (Cflgh-t I
XCOOR ^'~~~~CH-COOR
J(CH2)n.COCH2COOR ---►
(3)
in
М. с. встречаются в природе и играют важную биологич.
роль. Среди природных М. с. — алкалоиды (тубокирарин,
протопин, бруцин, ряд сенецио-алкалоидов и др.), гормоны
(окситоцин, инсулин), антибиотики (грамицидин С, макро-
лиды — группа антибиотиков — лактонов, в к-рую входят
такие вещества, как эритромицин, магнамицин и др.), ду-
шистые вещества (см. Мускус), макроциклич. группировка,
включающая четыре пиррольных ядра [входит в состав таких
Наиболее удобным в препаративном отношении
является, по-видимому, синтез М. с. ацилоиновой
конденсацией диэфпров (4):
COOR
(СН2)„
COOR
^-С=О
(СИЛ, j
'ХЛ1ОН
(4)
Разновидностью циклизации бифункциональных соеди-
нений является образование М. с. из двух или боль-
шего числа молекул. Такие процессы часто протекают
1041 МАКРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ-МАКрОЭРГИЧЕСКИЕ СВЯЗИ Ю42
одновременно с внутримолекулярной циклизацией и
подавляются при увеличении разбавления.
Примерами препаративных реакций такого типа
являются синтез циклооктатетраена из ацетилена
по Реппе (5), а также тримеризация бутадиена в цикло-
додекатриен-1,5,9 в присутствии смешанного металло-
органич. катализатора, подобного использованному
Циглером для полимеризации этилена (6):
сн_- сн -сн
I II
4СН^СН - СН СН (5)
II I
сн-сн=сн
сн2-сн=сн-сн»
I I
ЗСН >=СН- С1НСН< -► сн2 сн2
I I
сн сн
II II
СН-СН.,-СН2-СН
По своему механизму эти реакции близки к рас-
сматриваемым ниже синтезам М. с. из би- или поли-
пиклич. соединений.
2. Деполимеризация различных гетероцепных поли-
меров, напр. полиэфиров, полиангидридов, к-рая
позволяет получать гл. обр. гетероциклич. М. с.
Деполимеризацию по Карозерсу проводят в вакууме, что
позволяет выводить М. с. из сферы реакции и смещать тем
самым равновесие между полимером и мономером в сторону
последнего. Примером таких реакций может служить синтез
высших лактонов из полиэфиров (7):
[-СМСНД-СО-]*—~Х (CiyJ (7)
^0
3. Синтез М. с. из би- или полициклич. соединений, напр.
CHj-CH---СН2
I I I
СН2 N(CH3)CO
I I I
сн2-сн—сн2
VII
осуществленное Вильштеттером превра-
щение псевдопельтьерина (VII) в углево-
дороды ряда циклооктана — циклоокта-
диен, циклооктатриен и циклооктатетра-
ен. Другой пример — окисление декалина
кислородом воздуха в циклодеканол-
1-оп-б (8).
4. Расширение цикла. Для осуществления этого процесса
часто применяют реакцию диазометана с циклическими кетона-
МИ (0): !-----------, ,--------,
(СН2)ПСО i (CHs)n+iCO (9)
I_____I I________1
Другой важный способ — Демьянова перегруппировка. Для
получения нек-рых М. с. может быть использовано и сужение
цикла, если соединение с большим циклом более доступно.
Наконец, синтез М. с. может быть основан на соче-
тании разных методов, что открывает особенно широ-
кие синтетич. возможности. Так, одним из способов
циклизации получают бициклич. М. с., включающие
тиофеновое кольцо, которые при восстановительной
десульфуризации действием скелетного никеля превра-
щаются в многочленные алициклич. соединения (10).
Использование тиофена для синтеза алициклич.
М. с. упрощает построение скелета и введение заме-
стителей, облегчает циклизацию из-за наличия жест-
кой группы, а также позволяет использовать для
циклизации специфич. реакции ароматич. соединений.
Многие М. с. производятся в пром-сти; нек-рые из
ццх получают синтетически. К числу последних отно-
сятся лактогенный гормон — окситоцин, душистые
вещества — циклопентадеканон (экзальтон), цикло-
гексадеканон, циклогептадеканон (дигидроцибетон)
и лактоны высших w-оксикислот (С15—С17); промыш-
ленным или полупромышленным способом получают
пиклооктатетраен и циклододекатриен-1,5,9, исполь-
зуемые в качестве полупродуктов для синтеза раз-
личных соединений с 8- и 12-членными циклами.
Лит.: Ziegler К., в кн.: Methoden der organisclien
Chemie, brsg. E. Miiller, Bd 4, TI 2, 4 Aufl., Stuttgart, 1955:
II P e л о г В., в кн.: Перспективы развития органической
химии, пер. с англ, и нем., М., 1959; Hulsgen R.,Angew.
Chemie, 1957, 69, № 11, 341; Гольдфарб Я. Л., Бе-
ленький Л. И., Усп. химии, 1960, 29, вып. 4, 470;
Белов В. Н. [и др.1, Успехи химии душистых веществ,
М„ 1 956; Г о л ь д ф а р б Я. Л., Таиц С. 3., Белень-
кий Л. И., Ж. общ. химии, 1959, 29, 3564; Колино-
кий Р., Усп. химии, 1961, 30, вып. 6, 701.
Л. И. Беленький.
МАКРОЭРГИЧЕСКИЕ СВЯЗИ (высокоэргическпе
связи) — богатые энергией химич. связи в соедине-
ниях, входящих в состав живых организмов; расщеп-
ление М. с. сопровождается освобождением значи-
тельного количества свободной энергии — от 7 до
12 ккал/моль в стандартных условиях (при расщеп-
лении обычных связей освобождается от 2 до
4 ккал/моль). М. с. обозначают значком ~. Известны
следующие типы М. с.: полифосфаты (I), ацил-
фосфаты (II)., енолфосфаты (III), фосфосульфат (IV),
амииофосфаты (V), S-ацилмеркаптаны (VI) и К-карбо-
кспсоединения (VII).
О О
II II
-О-Р-О-Р-ОП
I I
он он
I
о о
II |[
-О- Р -O-S-OH
‘ II
он о
IV
В живых организмах М. с. используются для осу-
ществления самых разнообразных функций, тре-
бующих затраты энергии. Освобождение энергии
М. с. происходит не путем гидролиза, при к-ром
свободная энергия обесценивается, переходя в тепло-
вую, а путем переноса групп, участвующих в образо-
вании М. с.
Наиболее распространены соединения с М. с. по-
лифосфатного типа (I). К ним относятся свободные
рибо- и дезоксирибонуклеозид-ди- и трифосфаты
аденина, гипоксантина, гуанина, урацила, цитозина,
тимина, имеющие строение:
ООО ОО
II II II II II
R—О—Р—О—Р—О-P-ОН и R-O-P-O-P-OII
III II
ОН он он он он
нук лео зидтрифосфат нуклео зиддифосфат
где R — остаток соответствующего рибо- или дез-
оксирибонуклеозида. Особенно важную роль в живых
организмах играют адениновые нуклеозидполифос-
фаты — аденозинтрифосфориая к-та (АТФ) и адено-
зиндифосфорная к-та (АДФ) (см. Аденозинфосфорные
кислоты). АТФ представляет собой универсальный
аккумулятор химич. энергии в живых организмах,
поскольку вся энергия окислительного распада пита-
тельных веществ в процессе фосфорилирования, со-
пряженного с дыханием и брожением (гликолизэм),
переходит в энергию М. с. Аналогичен механизм
аккумуляции энергии солнечных лучей у фотосинте-
зирующих организмов — зеленых растений и неко-
торых бактерий. Вместе с тем АТФ является универ-
сальным донатором энергии в организме для осущест-
вления механич. работы, переноса веществ против
градиента концентрации (осмотическая работа), био-
1043
МАКРОЭРГИЧЕСКИЕ СВЯЗИ
1044
электрических токов, свечения (биолюминесценция)
и всех эндергонич. синтезов.
Значение нуклеозидполифосфатов с различными
пуриновыми и пиримидиновыми основаниями состоит,
прежде всего, в том, что из них в живых клетках
строятся важнейшие компоненты протоплазмы —
нуклеиновые кислоты. Кроме этой общей роли, боль-
шинство нуклеозидполифосфатов неаденинового типа
играет специальную роль в реакциях обмена веществ.
Так, уридиновые, гуаниновые и тиминовые нуклео-
тиды принимают участие в механизме активирования
простых углеводов и их производных, к-рые в виде
соединений с нуклеотидами становятся способными
участвовать в реакциях синтеза более сложных моле-
кул и соответствующих полимеров. Гуаниновые ну-
клеотиды, кроме того, играют существенную роль
в процессах окислительного фосфорилирования, а
также в реакциях синтеза белка. Цитидиновые
нуклеотиды принимают непосредственное участие
в биосинтезе фосфолипидов. Различные нуклеозид-
полифосфаты выходят из подобных реакций энерге-
тически обесцененными, дефосфорилируясь до нукле-
озид-ди- или монофосфатов. «Перезарядка» их, т. е.
превращение снова в нуклеозидтрифосфаты, осущест-
вляется путем рефосфорилирования их за счет кон-
цевой фосфатной группы АТФ. Образующийся при
этом АДФ, в свою очередь, «перезаряжается» до АТФ
в процессе сопряженного с дыханием или гликолизом
фосфорилирования.
Важное биологич. значение имеют вещества, отно-
сящиеся к ацилфосфатам, примерами к-рых являются
1,3-дифосфоглицериновая к-та и ациладенилаты:
О
сн2-о-р-он о п_
I \ II /ОН
СНОН Он C-0-P--0
I //° 0 I \он
С< ||-он nh2
\о~р—он
1,3-дифосфоглицериновая карбамилфосфат
кислота
R О
I II .ОН
N=C—NHg О=С-О-р/
НС С—N I----О-----1 О
J || ^сн |нохН0 ОН | |
N-C-N-----С—С-С-С— С-СН2
I I I I I
н н н н н
ациладенилаты
1,3-Дифосфоглицериновая к-та возникает в процессе
гликолиза и спиртового брожения, и ее фосфорильный
остаток переносится далее на АДФ с образованием
АТФ. Ациладенилаты являются активной формой
органич. к-т, промежуточными продуктами при син-
тезе соответствующих ацилпроизводных кофермента А.
Образование аналогичных производных аминокис-
лот — аминоациладенилатов является первым эта-
пом в цепи реакций синтеза белка. Карбамилфосфат
имеет важное значение при синтезе мочевины и пи-
римидиновых оснований.
Представителем соединений с М. с. енолфосфат-
ного типа (III) является фосфоенолпировиноградная
к-та (ФЕП), образующаяся, как и 1,3-дифосфогли-
цериновая к-та, при гликолизе и спиртовом бро-
жении. В ходе этих процессов М. с. ФЕП превра-
щается в М. с. АТФ.
СН2 ОН
II /
С—О~р—О (ФЕП)
I \
СООН он
Фосфосульфатная М. с. (IV) содержится в «ак-
тивном» сульфате, представляющем собой аденозин
3'-фосфат-5'-фосфосульфат. Активный сульфат служит
донатором сульфатных групп в организме.
К соединениям с М. с. аминофосфатного типа (V)
относятся фосфагены — креатинфосфат и аргининфос-
фат:
HN-P
HN=C
/ОН
\он
II
IIN-P
I
HN=C
/ОН
Чн
H3C-N-CH2-COOH
I
HN-CHs-CHj-CH
NHj
i-CH-COOH
креатинфосфат
аргининфосфат
Они образуются в результате переэтерификации
между соответствующим азотистым основанием и
АТФ. Благодаря обратимости этих реакций может
происходить перенос фосфата с фосфагенов на АДФ
и поэтому фосфагены способны играть роль резер-
вуара макроэргич. фосфатных групп, поддерживая
уровень АТФ при большом ее расходовании на функ-
циональную деятельность (мышцы, мозг).
Наряду с соединениями с М. с. полифосфатного
типа огромное биологич. значение имеют макроэргич.
соединения, содержащие М. с. тиоэфирного типа (VI).
Особенно широко распространены в живой природе
S-ацильные производные кофермента А, представ-
ляющие собой ту форму, в к-рой органич. к-ты не-
посредственно подвергаются разнообразным превра-
щениям в организме. Активной группой КоА является
SH-группа меркаптоэтаноламина, по к-рой и происхо-
дят присоединения ацилов.
Ацетил-КоА является общим, как бы обезличенным
промежуточным продуктом окислительного распада
жиров, углеводов и белков. Взаимодействуя с щаве-
левоуксусной к-той, он вводит остаток уксусной к-ты
в цикл трикарбоновых кислот, в к-ром этот остаток
сгорает до СО2 и Н2О. В то же время ацетил-КоА
является исходным продуктом при биосинтезе выс-
ших жирных к-т и функционирует в качестве ацетили-
рующего агента, при участии к-рого образуются
такие важные для организма соединения, как ацетил-
холин.
Из других S-ацилмеркаптанов существенное зна-
чение имеют S-ацильные производные дигидролипое-
вой к-ты (6, 8-димеркаптоктановой к-ты) и глутатиона.
Первые непосредственно образуются при окислитель-
ном декарбоксилировании а-кетокислот, а вторые
возникают при окислении фосфоглицеринового аль-
дегида (гликолиз и спиртовое брожение), катализи-
руемого триозофосфатдегидрогеназой, одним из ко-
ферментов к-рой является глутатион.
Недавно открыт новый тип макроэргич. соедине-
ния, образованный витамином N-карбокси-биотин,
связанный с ферментным белком. Вся эта система
функционирует как карбоксилирующий агент в соот-
ветствующих ферментативных реакциях (см. Карбо-
ксилирование).
Необходимо указать, что в структуре макроэргич.
соединений положение М. с. строго локализовано,
причем у одного и того же соединения это положение
может быть различным в зависимости от реакции,
в к-рой оно участвует в том пли другом случае, напр.
в карбамилфосфате связь С—О разрывается при
переносе карбамила на орнитин (синтез цитруллина)
пли на аспарагиновую к-ту (образование уреидоян-
тарной к-ты — предшественника пиримидиновых ос-
нований), а при переносе фосфорила на АДФ рвется
связь —О—Р. В то же время в молекуле АТФ в реак-
ции ферментативного фосфорилирования глюкозы
рвется связь —О—Р между (3- и у-фосфатными груп-
пами, а в реакции образования ациладенилата из
АТФ и уксусной к-ты, идущей с вытеснением пиро-
1045
МАКСВЕЛЛА — БОЛЬЦМАНА ЗАКОН РАСПРЕДЕЛЕНИЯ
1046
фосфатной группировки из АТФ, разрывается связь
Р—О— между а- и 0-фосфатными группами АТФ.
Лит.: Брей Д ж., Уайт К., Кинетика и термодина-
мика биохимических процессов, пер. с англ., М., 1959; Дик-
сон М., Уэбб Э., Ферменты, пер. с англ., М., 1961, с. 582;
Котельникова А. В., Успехи соврем, биол., 1957,
43, вып. 2, 133; К р е б с Г., К о р н б е р г Г., Превращения
энергии в живых системах, пер. с англ., М., 1959; Энгель-
гардтВ. А., Изв. АН СССР. Сер. биол., 1945, № 2, 182;
Ш а п о т В. С., в кп.: Фосфорилирование и функция. Симпо-
зиум 1958 г., Л., 1960.
А. В. Котельникова, В. С. Шапот.
МАКСВЕЛЛА—БОЛЬЦМАНА ЗАКОН РАСПРЕДЕ-
ЛЕНИЯ — один из основных законов классич. ста-
тистик. механики, определяющий наиболее вероятное
распределение молекул по различным возможным
энергетич. уровням при статистик, равновесии в сис-
теме с неизменной общей энергией:
бп = Спё~z/hT ..bq/$P16pi ... брг
где б/г — наиболее вероятное число молекул, находя-
щихся в заданном состоянии с энергией е; п — общее
число молекул в системе; Т — абс. темп-ра; к —
Больцмана постоянная^ 6q1&q2... bqr&p1bp2... &pf —
элемент объема т. н. ^пространства, т. е. области
всех возможных значений координат q и импульсов р*
для г различных степеней свободы движения отдельной
молекулы; С — константа, определяемая при интег-
рировании по условию:
^бп = $•••$ Спё~e/kTbqi ...bqrbpt ...f>pr — n
исходя из конкретной зависимости е от всех q и р.
М.— Б. з. р. выводится методами классич. статисти-
ки при след, допущениях: 1) координаты и импульсы
молекулы непрерывны и могут быть определены
с любой степенью точности; 2) числа молекул, рас-
пределяющихся по различным энергетич. уровням,
достаточно велики, чтобы считать их непрерывными
величинами; 3) кванты энергии достаточно малы,
чтобы считать е также непрерывной величиной; 4) силы
межмолекулярного взаимодействия достаточно малы
и могут быть игнорированы.
С помощью теории флуктуаций можно показать,
что при наличии большого числа молекул в системе
наиболее вероятное распределение, определяемое
М.—Б. з. р., является неизмеримо более вероятным, чем
любое другое распределение, отличающееся от него
в сколько-нибудь заметной степени; это служит гаран-
тией отражения действительного поведения системы
при условии выполнения перечисленных выше допу-
щений. М. — Б. з. р. не налагает ограничений на форму
энергии молекул и приложим как к общей энергии,
так и к отдельным ее формам (колебательной, враща-
тельной, поступательной), лишь бы энергия (или
рассматриваемая ее форма) всей системы в целом
оставалась постоянной. В частности, при рассмотрении
лишь поступательной энергии из М.—Б. з. р. можно
получить закон распределения скоростей Максвелла:
dn = 4лп (т/2л/сТ)3/-0 е— vzdv
где dn — число молекул, обладающих скоростью v
(независимо от ее направления).
М.—Б.з.р. наз. иногда просто распределением Больц-
мана, к-рыи вывел этот закон в 1877. Однако его част-
ный случай, относящийся к поступательному движе-
нию молекул, был получен Максвеллом еще в 1860
при помощи кинетич. теории газов. М.—Б. з. р. поз-
воляет рассчитывать средние, среднеквадратичные
и наивероятнейшие значения энергии и скорости
молекул. Он служит основой статистич. толкования
многих основных термодинамических соотношений и
в частности уравнения состояния идеального газа.
Из М.—Б. з. р. следует т. н. принцип равного распре-
деления энергии по степеням свободы: = J/a кТ,
где Bj — энергия, приходящаяся на любую г-тую
степень свободы движения молекулы.
М.—Б. з. р. является частным случаем более общего
Гиббса распределения, относящегося не только к от-
дельным молекулам, но к любой относительно малой
части замкнутой системы.
Лит.: Г л е с т о н С., Теоретическая химия, пер. с англ.,
М., 1950;Ландау Л.,Лифшиц Е., Статистическая физика
(Классическая и квантовая), М.—Л., 1951 (Теоретич. физ.,
т. 4); Больцман Л., Лекции по теории газов, пер. с нем.,
М., 1953. С. 9. Вайсберг.
МАКСИМАЛЬНАЯ РАБОТА РЕАКЦИИ — работа,
к-рая производится при термодинамически обратимом
течении химич. реакции. М. р. р. Лмаке слагается из
двух частей — собственно работы химич. процесса —
т. наз. максимальной полезной (или внутренней) рабо-
ты А' макс и работы против внешнего давления Лмех =
2
= Величина Л'макс при изобарно-изотермич.
1
протекании процесса равна убыли изобарного потен-
циала системы:
^'макс.= (1)
В качестве примера можно указать на осуществле-
ние при данных темп-ре Т и давлении Р реакции в галь-
ванич. элементе, в к-ром химич. энергия превращается
в электрическую. Если представить себе, что этот
элемент работает т. обр., что его электродвижущая
сила компенсируется электродвижущей силой другого
элемента, включенного против него, тогда в данном
элементе в каждый момент будет существовать равно-
весие между электродами и растворами и
Лакс. - Лакс. + Лех. = nFE + PEV &
где п — валентность иона, переносящего заряд, F —
число Фарадея, Е — электродвижущая сила элемен-
та, ДУ — изменение объема при реакции. В таком
гальваническом элементе (его следует назвать обра-
тимым) при пропускании тока в противоположном
направлении будет протекать процесс, обратный то-
кообразующему.
Для вычисления М. р. р. служит уравнение изотер-
мы реакции (см. Изотермы реакции уравнение)-, для
процесса &В Д- сС ••• — гВ Д- sS Д- ... оно имеет
вид:
Г 9
Яр •
Лакс. = RT Ь Ка - RT In (3)
“в “с...
здесь Ка — константа химич. равновесия, ов, ас, ...,
°r> 0S’"’—активности реагентов в реакционной смеси,
R — универсальная газовая постоянная.
С помощью уравнения (3) определяют: а) направле-
ние процесса (если Л'макс. > 0, то процесс может про-
текать только слева направо; при /Гмакс <0 — только
в противоположном направлении; если же -4^акс = 0,
то реагенты находятся в равновесии); б) его «глубину»
(чем больше Ка, тем больше «химическое сродство»
исходных веществ); в) влияние на равновесие давле-
ния, концентраций исходных веществ и продуктов
реакции (оно отражено на втором члене правой
части уравнения); в частности, можно выявить те
условия, при к-рых при данной темп-ре процесс
из неосуществимого становится осуществимым, и
наоборот.
Уравнение (3) не передает влияния темп-ры на
М. р. р.; для этого служит уравнение изобары реакции
(см. Изобары реакции уравнение):
dT ~ RT*
1047 МАК-ФАДИЕНА—СТИВЕНСА РЕАКЦИЯ — МАЛЕИНОВАЯ И ФУМАРОВАЯ КИСЛОТЫ Ю48
здесь АН — изобарный тепловой эффект процесса.
В соответствии с (4), М. р. р. возрастает с темп-рой
для эндотермич. и уменьшается для экзотермич. ре-
акций.
Для сравнения различных процессов по величине
М. р. р. введена величина т. наз. нормального (стан-
дартного) сродства. Под ней подразумевается величина
Лмакс. ПРИ условии, что активности всех веществ в
реакционной смеси равны единице. Тогда в соответ-
ствии с (3)
Л.аке. = RT 1п Ка (5)
При переходе от условия Р, Т = const к условию
У, Т = const А^акс = Лмакс , а в уравнении (4) еле-
дует пользоваться изохорным тепловым эффектом At/.
Для вычисления М. р. р. может служить также
Гиббса—Гельмгольца уравнение.
Лит.: Киреев В. А., Курс физической химии, М.—Л.,
2 изд., 1956; Карапетьянц М. X., Химическая термо-
динамика, 2 изд., М.—Л. 1953; Partington J. R., Ап
advanced treatise on physical chemistry, v. 1, section 2, L. — N.Y.—
Toronto 1949. M. X. Карапетьянц.
МАЙ-ФАДИЕНА - СТИВЕНСА РЕАКЦИЯ — пре-
вращение карбоновых к-т в альдегиды разложением
арилсульфонилгидразидов:
RCOOH -> RCOOC2H3 (или RCOC1) -» RCONHNH* -
RCONHNHSOiAr —RCHO+N»-|-ArSO2Na
Обычно бензолсульфонилгидразид карбоновой к-ты,
полученный из ее гидразида и бензолсульфохлорида
в пиридиновом р-ре, нагревают несколько минут
в этиленгликоле до 150—165° в присутствии поташа
или соды. При этом выделяется азот и образуются
альдегид и соль бензолсульфиновой к-ты. После добав-
ления горячей воды альдегид извлекают из реакцион-
ной смеси подходящим органич. растворителем или
отгоняют с водяным паром.
Реакция применима гл. обр. для получения арома-
тич. альдегидов, причем выходы обычно хорошие, за
исключением случаев, когда ароматич. ядро содержит
электронооттягивающие заместители в о- пли и-ноло-
жении. С худшими выходами получаются гетероцик-
лич. альдегиды; описано также получение формпл-
циклоиропана:
CeHsCONHNHSO2C6H5
выход 77Я
CONHNHSO2C6H5
с6н5сно
СН,Ч
I CH-CONHNHSO2C6H5
выход 16Х
СН2х
I сн-сно
Механизм реакции не изучен. Установлено, что
реакция осуществляется лишь в том случае, когда
сода или поташ находится в твердой фазе; при взаи-
модействии в гомогенной среде (нагревание р-ра нат-
риевой соли бензолсульфопплгидразида карбоновой
к-ты в этиленгликоле) требуется присутствие толченого
стекла. Реакция не получила широкого распростра-
нения; открыта в 1936 Дж. С. Мак-Фадиеном и Т. С.
Стивенсом.
Лит.: Мозеттиг Э., в кн.: Органические реакции,
Сб. 8, пер. с англ., М., 1956, с. 288; Newm an М. S., С a f-
I i s с h Е. G., J. Amer. Chem. Soc., 1958, 80, № 4, 862; Surrey
A. R., Name reactions in organic chemistry, N. Y., 1954, p.
115. E. M. Рохлин.
МАЛАТДЕГИДРОГЕНАЗА (маликодегидрогеназа,
дегидрогеназа L-яблочной кислоты) — фермент, ка-
тализирующий взаимопревращение яблочной и щаве-
левоуксусной к-т;
НООС-СН (ОН)-СНз-СООН-РДПН-
-» НООС-С(ОН)=СН~СООН + ДПН-Н-РН+
Положение равновесия реакции зависит в значитель-
ной мере от pH. В физиология, условиях при pH 7,2
кажущаяся константа равновесия имеет величину
2,33 10 5; практически равновесие полностью сдви-
нуто в сторону образования яблочнойк-ты. В щелочной
среде (pH 9,0), а также при удалении щавелевоуксус-
ной к-ты из сферы реакции равновесие сдвигается
в сторону образования последней. М. — фермент цикла
трикарбоновых кислот и играет исключительно важ-
ную роль в обмене дикарбоновых к-т, имеющих
большое значение при превращении углеводов, жиров
и белков. Образующаяся в результате катализируе-
мой М. реакции щавелевоуксусная к-та служит ис-
точником образования лимонной к-ты и дает, т. обр.,
начало циклу трикарбоновых кислот. Кроме того,
щавелевоуксусная к-та может превращаться в аспа-
рагиновую к-ту, участвующую в многочисленных
реакциях переаминирования.
М. широко распространена в животных и расти-
тельных тканях, а также у микроорганизмов. Особенно
высоко содержание М. в сердечной и скелетных мыш-
цах животных. Кристаллич. М., полученная из
сердца свиньи, имеет мол. в. 40 000; очищенные пре-
параты М. из сердца свиньи катализируют окисление
ц,5 • 104 молей/мин ДПН-Н на 100 000 е белка. Помимо
L-яблочной к-ты, М. способна катализировать окисле-
ние ряда других а-окепдикарбоновых к-т, в частности
щавелево-гликолевой и а-оксиглутаровой. М. активна
лишь в отношении L-пзомеров соответствующих к-т.
Коферментом М. служит никотинамидадениндинук-
леотид (дифосфопиридиннуклеотид, ДПН, см. Коде-
гидрогеназы). Активность М. подавляется фумаровой
к-той, а также реагентами на SH-группу. Активность
М. определяют спектрофотометрически, измеряя ин-
тенсивность поглощения света ДПН-Н при 340 ммк.
Лит. СУ1. при ст. Лактатдегидрогеназа. В. Б. Спиричев.
МАЛЕИНОВАЯ И ФУМАРОВАЯ КИСЛОТЫ (цис-
и траие-бутен-2-диовая-2,3-кислота), мол. в. 116,07,
НООССН=СНСООН — геометрич. изомеры этилен-
дикарбоновой к-ты; бесцветные моноклинные призмы.
Малеиновая кислота (I, цис-изомер) легко переходит
в фумаровую (II, транс-изомер) на свету или при
нагревании выше 200°. Фумаровая к-та превращается
в малеиновую при УФ-облучении:
нссоон нссоон
II II
нссоон HOOCH
I II
Кислоты Т. пл., °C Т. кип., °C ,20 а 4
Малеиновая 131 160 1.590
Фумаровая 287а 290 1,635
а В запаянном капилляре.
Малеиновая к-та легко растворяется в воде (г/100 г):
78,8 (25°), 392,6 (97,5°); легко растворима в эфире,
трудно — в бензоле. Фумаровая к-та трудно раство-
рима в воде (е/100 г): 0,69 (17°), 9,8 (100°) и почти во
всех растворителях, несколько лучше — в эфире.
Вследствие удаленности карбоксильных групп в фу-
маровой к-те первая и вторая константы ее диссоциа-
ции различаются меньше (Кг = 1 • 10'-3, К2 = 3 • 10”5),
чем первая и вторая константы диссоциаций малеи-
новой к-ты (при 25°): А, = 1,5- 10~2, К2 = 1,3 -10 6.
При нагревании фумаровой к-ты с Р2О5 образуется
малеиновый ангидрид (ангидрид фумаровой к-ты
неизвестен). Двойная связь в обеих к-тах легко
восстанавливается, напр. амальгамой натрия, с обра-
зованием янтарной к-ты НООС(СН2)2СООН; гидрата-
ция приводит к рацемической яблочной кислоте
НООССН(ОН)СН.2СООН.
1049
МАЛЕИНОВЫЙ АНГИДРИД-МАЛОНОВЫЙ ЭФИР
1050
Малеиновую к-ту получают окислением бензола или
фурфурола (промышленные методы), напр.: СвНв -|-
4- О2-гС4Н2О3 -|- СО2 -|- Н2; реакция протекает с вы-
делением теплоты (ДАТ = —428 ккал/молъ). Фумаровая
к-та может быть получена кипячением 30—40%-ного
водного р-ра малеиновой к-ты с НС1. Фумаровая
к-та содержится во многих растениях, особенно в гри-
бах; она образуется при брожении сахаристых в-в
посредством Aspergillus fumaricus.. Основное приме-
нение находит малеиновый ангидрид. Эфиры М. к.
с многоатомными спиртами образуют вязкие жидкости,
отверждаемые при нагревании.
Производные малеиновой кислоты:
диметиловый эфир, т. кип. 205°; диэтиловый эфир, т. кип. 223°;
имид, т. пл. 93°. Производные фумаровой кис-
лоты: диметиловый эфир, т. пл. 102°, т. кип. 192°; диэтиловый
эфир, т. пл. 70°, т. кип. 147°/16 лш.
Лит.: Ullman п, 3 Aufl., Bd 12, Munchen—В., 1960,
S. 185. Н. А. Несмеянов.
МАЛЕИНОВЫЙ АНГИДРИД (ангидрид этилен-1,
2-цис-дикарбоновой кислоты), мол. в. 98,06 — бес-
цветные кристаллы (ромбовидные иглы); т. пл. 52,8°;
СН2—сох т. кип. 199,9°; с?тв 1,48; вязкость (в
1 ~_с /° пуазах): 0,0153 (70°),0,0099(100°); теп-
СН2 Со лота образования 112,2 кал/молъ; теплота
сгорания 333,9 кал/молъ; теплота кристаллизации
2,75 кал/г; теплота парообразования 10,5 кал/г; смеши-
вается с диоксаном, водой (с образованием малеиновой
к-ты, теплота гидратации 8,33 кал/молъ); спиртами (с об-
разованием диалкилмалеинатов); хорошо растворяется
в ацетоне(70%), этилацетате (53%), хлороформе (34%),
бензоле (33%); ограниченно растворяется в четырех-
хлористом углероде (0,6%), керосине (0,25%); горит
на воздухе; т. всп. 103°; т. самовоспламенения 477°.
М. а. — чрезвычайно реакционноспособное соедине-
ние. При взаимодействии с сопряженными диеновыми
соединениями образуются циклич. аддукты типа
ангидридов циклогександикарбоновых к-т (см. Дие-
новый синтез). Сополимеризация М. а. с олефинами
(гл. обр. с соединениями, содержащими виниль-
нЫе группировки) протекает по свободно-радикаль-
ному механизму с образованием насыщенных поли-
меров линейного строения; взаимодействует при
высокой темп-ре с несопряженными ненасыщенными
соединениями, содержащими метильные или метиле-
новые группы при кратной связи, напр.:
сн2=сн-сн3 + сн-со. сн5=сн-сн3-сн—со.
II >0 - I >О
СН-СО/ сн2-сох
Аналогично -взаимодействует с толуолом и другими
замещенными в ядро алкилбензолами. При восстанов-
лении М. а. образуется янтарный ангидрид. При дей-
ствии аммиака и аминов на М. а. образуются соответ-
ственно аспарагиновая кислота и ее N-алкильные заме-
щенные.
М. а. получают парофазным каталитич. окислением
бензола, а также фурфурола над V2O5; является основ-
ным продуктом перегонки малеиновой, фумаровой
(над Р2О5) и яблочной к-т; образуется как побочный
продукт при каталитич. окислении олефиновых и
ароматич. углеводородов и их производных, напр.:
кротонового альдегида, нафталина; перспективный
способ получения М. а. — окисление бутана (буте-
на-2, бутадиена-1,3) при 400° над V2O5.
М. а. используют в пром-сти при произ-ве пластич.
масс (сополимеров со стиролом и акрилатами), синте-
тич. волокон, фармацевтич. препаратов, детергентов
(на основе производных сульфоянтарной к-ты), раз-
личных химич. препаратов (малеатов, фумаратов,
сукцинатов, N-хлорсукцинилимида и др.); при очистке
ароматич. углеводородов от серосодержащих и ааото-
содержащих веществ; при идентификации диеновых
соединений (напр.: т. пл. аддукта с бутадиеном 104°,
с изопреном 64°, с 2,3-диметилбутадиеном 79°).
Лит.: К л етцел ьМ. С., в кн.: Органические реакции.
Сб. 4, пер. с англ., М., 1951, 7—85; Norton J. A., Chem.
Revs, 1942, 31, ATj 2, 319. Г. А. Сокольский.
МАЛОНОВАЯ КИСЛОТА (метандикарбоновая кис-
лота) НООССН2СООН, мол. в. 104,6 — бесцветные
кристаллы; т. пл. 135,6° (с разл.); 1,6305; константа
диссоциации (25°); А’4 = 1,77- Ю-3,^ = 4,72- 10-в.
Вследствие близкого расположения карбоксильных
групп М. к. сравнительно легко декарбоксилируется,
переходя в уксусную. Декарбоксилирование проис-
ходит при нагревании несколько выше темп-ры плав-
ления или при кипячении в р-ре разб. соляной к-ты
(темп-ра несколько выше 100°). Подобно этому декар-
боксилируются и С-аамещенные М. к., напр.:
КСН(СООН)2 -> RCHaCOOH+CO2
При нагревании М. к. с Р2О6 образуется простейший
кетен— недоокись углерода С3О2, очень реакционно-
способное соединение. Как двухосновная к-та М. к.
дает два ряда производных (кислых и полных) —
эфиров, нитрилов, амидов, хлорангидридов. Дейст-
вием SOC12 на М. к. в зависимости от условий реакции
можно получить полный хлорангидрид(малонилхло-
рид) и полухлорангидрид, напр.:
HOOCCH2COOH+SOC12 - ClOCCH2COOH-|-SOs+HCl
М. к. бромируется до бром- и диброммалоновой к-ты;
окисляется азотистой к-той до мезоксалевой к-ты:
сн2(соон)2 —о=с <соон)2
конденсируется с мочевиной с образованием барбиту-
ровой к-ты:
/.о
-NH-С/
NH2CONH2 + С1-С-СН»-СС1 - ос< >сн2
II II 'NH—С<
0 0 о
М. к. и ее производные имеют два подвижных атома
водорода в a-положении и поэтому могут вступать
в ряд конденсаций, напр. в Манниха реакцию и в кон-
денсации с альдегидами:
HOOCCH2COOC2H3+CH2O + HN(CHs)2 —
— C2H5OOC(CH2)2N(CH3)2
CH2(COOH)2+RCHO^RCH(OH)CH(COOH)2-
-RCH = C(COOH)2
Полные эфиры и нитрил М. к. образуют с натрием
металлич. производные, к-рые широко используются
в ортанич. синтезе. Омылением малонового эфира или
циануксусной к-ты получают саму М. к. В пром-сти
М. к. и ее производные используют в синтезе витами-
нов В4 и Вв, для синтеза аминокислот, при получении
барбитуровой и мочевой к-т, для получения ряда дру-
гих кислот и в синтезе нек-рых гетероциклов.
Производные М. к.: малонилхЛорид, т. кип.
58°/26 мм, пд 1,45915; нитрил, т. пл. 29—30°, т. кип. 218—
219°; диметиловый эфир, т. кип. 181°. См. также Диэтилма-
лонат. Н. А. Несмеянов.
МАЛОНОВЫЙ ЭФИР — см. Диэтилмалонат.
АЛФАВИТНО-ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ*
Абиетиновая кислота 587
Абразит 735
Абрикосовое масло 73, 75
Автобим 629
Автокоагуляция 607
Агар 385
Агглютинация 224
Аглопорит 81
Агрегатное состояние вещества 59
Адалин 943
Адденды — см. Лиганды
Адекс^метазон 947
Аденин, производные 405
Адреналин 945
Адренокортикотропный гормон 405
Адренолитические вещества 944
Адреномиметические вещества 944, 945
Адсорбированный слой идеальный 566
Адсорбционное равновесие 994
Адсорбционные центры 994
Адсорбция, Лэнгмюра уравнение изо-
термы А. 994
6-Азаурацил 404
Азолитмин 906
Азот, магнитная восприимчивость 1021
Азот-илиды 212
Азотирование 97
Азотные удобрения 58
Азотциклопентадиенилиды 213
Азофиолетовый 250
Азурит 439
Аквопентахлориридиты 327
Аконитовая кислота 968
Акридин 250
Акринит 628
Акролеин 63, 65
Аксиальная связь 700
Активность оптическая 155, 175
Актиноксантин 405
Актиномицины 405
Актор 265
Акцептор 265, 709
Р-Аланил-Ь-гистидин — см. Карнозин
Алитирование 97, 724
Алифатические соединения
— устойчивые конформации 699
Алициклические соединения 209
Алкалакс 303
Алкалоз 579
Алкилалкоксисиланы 814, 815
Алкиламиносиланы 816
Алкилгалогенсиланы 814
Алкплгидроксисиланы 816
Алкилсиланхлориды 814
Алкилсиланы 813
Алнилфенолы 235
Алкоксисоединения 816
Алконсихлорсиланы 816
Алланит — см. Ортит
Аллены 890
Аллокоричная кислота Либермана 716
Алмаз, магнитная восприимчивость 1021
Алнико 619
---магнитные свойства 1022
Алоксид 735
Алудрин — см. Изадрин
Алунд 735
Альбертоли 395
Альдегидоксидаза — см. Ксантинокси-
даза
Альдокетены 547
Альдольная конденсация 681
Альдостерон 732, 946
Альфа-распад 203
Альфа-частицы 90
Алюминий 28
— карбид 425
— магнитная восприимчивость 1022
Амальгамация 116
Амальгамные концентрационные элемен»
ты 707
Амберлайты 303
Амбиполярная диффузия ионов 321
Амблигонит 977
Америпол Эс — Эн — см. Изопреновый
каучук
Аметоптерин 404
Амизил 943
Амилазы 429, 764
* О том, как пользоваться Алфавитно-
предметным указателем — см. т. I,
стр. 1229.
Амилоза 762
Амилопектин 762
Аминазин 943
Аминоанфоль 404
n-Аминобензойная кислота, диалкил-
аминоалкиловые эфиры 944
2-Амино-4,6-диоксиптеридин — см. Ксан-
топтерин
а-Аминоизокапроновая кислота — см.
Лейцин
е-Аминокапроновая кислота, лактам —
см. е-Капролактам
Аминокислоты заменимые 80
o-Аминокоричная кислота, лактам — см.
Карбостирил
Аминоксилолы — см. Ксилидины
6-Амино-2-меркаптопурин 405
а-Амино-р-метил-р-этилпропионовая кис-
лота — см. Изолейцин
З-Амино-4-метонситолуол — см. Крези-
дии
6-Аминоникотинамид 403
Аминоптерин 404, 948
4-Аминоптероилглутаминовая кислота —
см. Аминоптерин
Аминотероптерин 404
2-Амино-4, 6, 7-триоксиптеридин — см.
Лейкоптерин
Аминофенолы 235
Аминофосфаты 1042
З-Аминофталевая кислота, гидразид —
см. Люминал
Аммиак, критич. постоянные 866
Аммиак жидкий 59
Аммиакаты 59
Аммиачная вода 59
Аммониевые основания четвертичные,
закрепители окраски 79
Аналептини 944
Анальгеаирующие вещества 943
Анальгин 943
Ангидрит 371, 375, 381
Анестезирующие вещества 944
Анизотропия 840
Анилин, критич. постоянные 867
Анионитовая колонна 305
Анионитовые диафрагмы 298
Аниониты 300, 303
Анионная полимеризация 422
Анионный контакт 310
Анионоидный реагент — см. Донор
Анионы 314
— аналитич. классификация 502
Анкерит 439
Аннелирование ангулярное 689
Анодные ингибиторы 228, 229
Антибиотики 405
Антигены 220 —224
Антикоагулянты 122
Антиметаболиты 403, 948
Антиокислители 234
--- древесно-смольные 235
Антипирены, защитная обработка дре-
весины 93
Антипирин 943
Антиподы оптические 155
Антисептики, защитная обработка дре-
весины 93
Антитела 220
Антитиреоидные средства 946
Антиферромагнетизм 1005, 1018
--- нескомпенсированный 1020
--- скомпенсированный 1020
Антихолинэствразные вещества 945
Антоцианы 759
Антраден 386, 387, 388
— фотоокись 690
Антраценовое масло 386
Антрацит 331
Антроподезоксихолевая кислота — см.
Хвиодезоксихолевая кислота
Апоэимаза 107
Апоферменты 429, 740
Арагонит 371, 379, 439
Арахиновая кислота 75
Арахисовое масло 73, 75
Аргининфосфат 1044
Аргон 266
— критич. постоянные 866
— магнитная восприимчивость 1021
Арзамит 580, 581
Арилалкоксисиланы 814, 815
Ариламиносиланы 816
Арилгалогенсиланы 814
Арилгидроксисиланы 816
Арилсиланхлориды 814
Арилсиланы 813
«Армко железо» 46
Ароматические соединения 209 — см. так-
же Ароматические соединения (т. 1)
--- конденсированные 689
Аррениуса уравнение 564
Артан — см. Циклодол
Артериодон — см. Кардиотраст
Асболан 612
Асболит — см. Асболан
Астразоны 259, 782
«Астрафлоксин ФФ» 259
Астраханит 1029
Астрохимия — см. Космохимия
Атмосфера, ионы в А. 322
Атом, магнитный момент 104, 519, 1003.
10.15
— состояния валентные 519
— состояния стационарные 518
— спонтанное деление ядра 203
— энергия А. в магяитаом поле 104
— уровни энергии 104, 519
Атом водорода, вероятности локализации
электрона 518; волновая функция 511
---центральный 657
Атомная доля 707
Атомные кристаллы — см. Валентные
кристаллы
Атомные орбиты 527
Атомный радиус 859
--- для инертных газов 312
«Атомов в молекулах» метод 532
Атомы квазиаксиальные 701
--- квазиэкваториальные 701
Атропоизомерия 157
Аурантин 405
Аураты 114
Ауриды 112
Ауриты 114
Аустенит 19, 44
Аценафтен 386
Ацетальфосфатиды 973
Ацетилацетон 421
Ацетилен, Илосвая реактивы для откры-
тия А. 214
— критич. постоянные 867
Ацетилиды 424
Ацетилхолин 944
Ацетилцеллюлоза 130, 131, 132
Ацетильное число 65, 69
Ацетоин 431
Ацетон, критич. постоянные 867
— магнитная восприимчивость 1021
Ацетондикарбоновая кислота 968
Ацидоз 579
Ациладенилаты 1043
2-Ацилиндандионы 239
S-Ацилмеркаптан 1042
Ацилфосфаты 1042
Аэрозоли 109
— коагуляция 607
Аэрон 980
Б
Бандровского основание 769
Банметалл 980
Барбан — см. Карбин
Барбитураты 942
Барий, карбид 425
— кремнефторид 802
Барий углекислый 122
Баротал — см. Калиппон
Бастнезит 924, 925
Бегеновая кислота 75
Бемегрид 944
Беиэальнитрозокарвакрол 448
1,2-Бенэантрацен 398, 399, 400
6-Бензилтиопурин 404
Бензилцеллюлоэа 130, 131, 132
Бензоилноронен 718
Бензол, криоскопия, постоянные 830
— критич. постоянные 867
— магнитная восприимчивость 1021
Р; у-Бензопиридин — см. Изохинолин
2,3-Бензопиррол — см. Индол
Бензпиразол — см. Индазол
3,4-Бензпирен 398, 399, 400
3,4-Бензфенантрен 400
Бентониты 106
Бериллий, карбид 425
Берлинская лазурь — см. Железная ла-
зурь
Бертолетова соль — см. Калий, хлорат
Бета-излучение, источники 335
Бета-распад 203
Бета-частицы 90, 91
Бетон жаростойкий 15
Биберит 620
Бикарбонаты 438, 439
Биксин 455
Биливердин 50
Билирубин 50, 761
Бимолекулярные реакции 563, 564
Биндон 238
Биокоррозия 87
Биолюминесценция 1000
Биотин 743
Бишофит — см. Магний, хлорид
Бластолипаза 972
Бозе распределение 525
-- частицы 520
Бозе — Эйнштейна конденсация 526
--- статистика 524
Больцмана коэффициент 701
--- распределение 525, 1045
Бор, карбид 425, 428
— фторид, критич. постоянные 866
Бора магнетон 519, 1017
Боронений радиус 511
Браве решетки 852
Бразилии 760
Браннерит 337
Бренстеда теория кислотно-основного
равновесия 583
Бренстедовские кислоты и основания
582, 585
Бриллиантцибалан 746
Бройля де соотношения 506
Бром, критич. постоянные 866
Броминдиго 243
о-Бромфенолиндофенол 261
Брусит 1026
Буковое масло 73, 75
Бурое масло 105
Бурый железняк 41
Бурый уголь 331
Бутадиен 496
Бутадиен-нитрильный каучук 131, 132
Бутадиеновый научук 130, 132
Бутадиен-стирольный каучук 130, 131,
132
Бутамид 946
н-Бутан, критич. постоянные 867
1,4-Бутандиизоцианат 207
Бутатриен 889
2-Бутеналь — см. Кротоновый альдегид
н-Бутилизоцианат 207
Бутилкаучук 132, 498
Бутиллитий 986
Бутилсилан 814
у-Бутиролактам 906
у-Бутиролактон 912
Буфодезоксихолевая кислота 49
Бьеркмана лигнин 958
В
Вагнера — Меервейна перегруппировка
392
Валентная структура 529
Валентность главная 658, 708
--- лантанидов 917, 920
--- побочная 658, 708
Валентные кристаллы 841
Валентных связей метод 527
б-Валеролактам 906
б-Валеролактон 912
Ванадий, карбид 425
— пятиокись 34
Ван-ден-Берга реакция 51
Вариационный принцип в квантовой
химии 527
Вапфарин 122
Вебера проба 131
Вейля — Сальковского реакция 786
Вернера номенклатура 659
Вероятность локализации влектрона 511
Версен — см. Комплексон Ш
Весовая доля 707
Весовой анализ 641
Взрывчатые вещества инициирующие 269
Виллемит 863
Винилтриметилсилан 811
Винилтрихлорсилан 814
Винные кислоты 156
Виологены 251
Вискоза 875
Висмут, магнитная восприимчивость 1021
Виталлиум 15, 619
Витерит 439
Виттига реакция 213
Вицинальное правило — см. Куна —
Фрейденберга правило
Внутрикомплексные соединения 657
Внутрисферные заместители — см. Ком-
плексные соединения
Вода 60
— жесткость 51
— криоскопия, постоянные 830
— критич. постоянные 866
— магнитная восприимчивость 1021
Вода жесткая 52
--- кристаллизационная 846
--- Морская, жесткость 53; кальций,
содержание 371
мягкая 52
природная 51
Водоподготовка 309
Водород, изотопный обмен 193
— критич. постоянные 866
— магнитная восприимчивость 1021
— поглощение В. железом 43
Водород бромистый, критич. постоянные
866
--- иодистый 289; критич. постоянные
866
--- хлористый, критич. постоянные 866
Водородная хрупкость 43
Волновая функция 509, 527
--- антисимметричная 520
--- симметричная 520
--- синглетная 528
---триплетная 528
Волновой пакет 510
Волокна текстильные, крашение 771
Вольтаметр — см. Кулометр
Вольфрам 28
— карбиды 425, 428
Ворвань 70
Воронение 43
Вофатиты 303
«Временная жесткость» воды 51
Время коагуляции 608
---контакта (в реакционной зоне) 562
Вуда сплав 346
Вулканизация 494, 496
Вультекс 930
Вырождения критерий 525
Высокомолекулярные соединения, кон-
формации 705
— окраска по реакции Либермана—Штор-
ха 131, 132
— свойства 1033
Высоновргические связи — см. Макро-
зргические связи
Вязкости коэффициент 61
Вязкость, индекс 240
--- двойных жидких систем 57
--- жидкостей 59, 61
--- латексов синтетических 930
Г
Гадолиний 916
— магнитная восприимчивость 1021
Гадолинит 337, 925
Газ коксовый 636
Газовый анализ 641
Газы 60, 62
— ионы в Г. 314
— конверсия 675
— очистка 462
Галактоманнаны 384
Галлеин 872
Галлодезоксихолевая кислота «— см. Хе-
нодезоксихолевая кислота
Галогеномоносиланы 817
Галогены, изотопный обмен 193
— определение в органич. соединениях
451
Галотан 942
Гамильтониан 515
Гамма-излучение, источники 335
Гамма-лучи, облучение, доза 90
— защита от облучения 91
Гаммета уравнение 263
--- функция кислотности 587
Ганглиоблокирующие вещества 945
Ганглиозиды 973
Гарпиуса эфир 396
Гафний, карбид 425
Гексабензобензол — см. Коронен
Гексабромное число 66
Гексагональная сингония симметрии (в
кристаллах) 849
Гексагональный вид симметрии (дипи-
рамидальный, пирамидальный, трапе-
цоэдрический) 850
Гексамидин 942
н-Гексан, критич. постоянные 867
1,6-Гександиизоцианат 207
н-Гексановая кислота — см. Капроновая
кислота
1053 - 1054
Гексаоксициклогексан — см. Инозит
Гексаоктаэдрический вид симметрии 850
Гексатетраэдрический вид симметрии 850
Гексенал 942
Гексоний 945
Гелий 266
— критич. постоянные 866
— магнитная восприимчивость 1021
Гематит 18, 41
Гематитовые руды — см. Железняки
красные
Генера число 65, 69, 73
Геометрическая изомерия 153
--- комплексных соединений 662
Геохимическая корреляция 718
Геохимические процессы 991
«'Гептан, критич. постоянные 867
Германий, очистка 122
Гетеровалентный изоморфизм 161
Гетерополисоединения 657
Гетит 41
Гибкость цепных молекул 1033
6-Гидразинопурин 404
Гидратцеллюлоза 131
Гидринден 241
Гидроборацит 371
Гидрогетит 41
Гидрозоли 109
— коагуляция 607
Гидрокоричная кислота 717
Гидрокортизон 732, 947
Гидрокрекинг — см. Крекинг под дав-
лением водорода
Гидроксиапатит 300
Гидролиз древесины 951
--- жиров 64, 67
--- эфиров сложных 184
а-Гиодезоксихолевая кислота 49
₽-Гиодезоксихолевая кислота 49
Гиперицин 759
Гипертонические растворы 179
Гипоиодиты 285
Гппоксантозин — см. Инозин
Гипоксантозинфосфорные кислоты — см.
Инозинфосфорные кислоты
Гипотонические растворы 179
Гипс 371, 375, 381
Гистерезис капиллярно-конденсацион-
ный 410, 411
--- магнитный 1020
Глазерит 358
Глазурь 538
Гликозидазы 429
Гликолипиды 973
Гликонеогенез 946
Глинистый фаянс 538
Глины отбеливающие 106
Г.чиоисалин — см. Имидазол
Глицериновый альдегид 155
Глицероген 227
Глобулины 223
Глобулы макромолекул 1033
Глобулярные кристаллы 843
а-Глюкозидазы 226, 227
Глюкокортикоиды 946
Глюкоманнаны 385
3-(Р-0-Глюкопиранозил)-С-глюкоза —
см. Ламинарибиоза
Глютатион 743
Гмелина реакция 51
Гольдшмидта правило — см. Кристалло-
химии основной закон
Гольмий 916
Гормон адренокортикотропный 405
Гормональные препараты 946
Гормоны 405
— иофврменты-Г. 743
Горчичное масло 73, 75
Горчичные масла — см. Изотиоцианаты
Горькая соль — см. Эпсомит
Гош-конформации — см. Конформации
скошенные
Гралекс 629
Графит, магнитная восприимчивость
1021
Гремучее золото 114
Гринокит 342
Гриньяра реакция 1013
Гуанидинметилглицин — ем. Креатин
Гуанозин-5-фосфорные киелоты 742
Гуаран 385
Гульдберга — Гюи правило 671
Гуминовые кислоты 301
Гумиты 330
Гумми — см. Камеди
Гуммиарабик 385
Гумолиты 330
1055-1056
д
Давление капиллярное 412
--- пара жидкой системы 56
Дайайон 303
Дацэкс 303
Двойники кристаллов 848
Двойные соли 656
Дегидроабиетиновая кислота 587
Дегидрогеназы 623
11-Дегидрокортикостерон 732
Деготь каменноугольный — см. Каменно-
угольная смола
Дегранол 402
Дезинтегратор 140
Дезоксикортикостерон 732, 946
Дезоксипиридоксин 403
Деэоксихолевая кислота 49
Действующих масс закон 563
Дейтерий 199
Декагидронафталин — см. Декалин
Декалин 700
Декаметоний 945
n-Декан, критич. постоянные 867
Декановая кислота — см. Каприновая
кислота
Декстропимаровая кислота 53, 146
Деметон 277
А-Деноптерин 404
Деполимеризация 1037, 1039, 1041
Дерматин 629
Десорбция 410
Деструкция полимеров 1037, 1039
Детерминанты (в иммунохимии) 220
Дефектные соединения 855
Дефекты структуры 861
Дефлегмация 688
Джепурит 621
1,3-Диазол — см. Имидазол
Диаэолин 946
Диазотипия 748
Диакарб 429
Диамагнетизм 1004, 1015, 1016
Диамагнитная восприимчивость 1016
Диаментин 735
а, е-Диаминокапроновая кислота — см.
Лизин
1,5-Диаминопентан — см. Кадаверин
2^6-Диаминопурин 404
Диатомовые земли 106
2,5-Диацетамидо-3,6-диэтиленимино-1,4-
бензохинои 403
Диацетилдифенилизатин — см. Изафе-
нин
Дибенамин 945
1, 2, 5, 6-Дибензакридин 401
1, 2, 5, 6-Дибенэантрацен 398, 399, 400
1, 2, 5, 6-Дибенэ-карбазол 401
Дибенэо-у-пирон — см. Ксантон
Дибензопиррол см. Карбазол
3, 4, 6, 7-Дибензпирен 999
6,6'-Диброминдиго 242, 761
2,6-Дибромфенолиндофенол 261
Дибромфлуоресцин 254
2, 6-Ди-трет-бутил-4-метилфеиол — см.
Ионол
N, Г'Г-Ди-втор-бутил-п-фенилен-диамин
235
Дибутолин 945
Дигалогенокарбены 423
Дигексагона льно-дипирамидальный вид
симметрии 850
Дигексагонально-пирамидальный вид
симметрии 850
Дигидроабиетиновая кислота 587
№, №-Дигидро-1, 2, 1', 2'-антрахинон-
аэии — см. Индантрон
Дигидрокоринантеин 716
Дигидрокортизон 405
3, 4-Дигидропиранокумарины 123
Дидодекаэдрический вид симметрии 850
Дииэопропиловый эфир — см. Изопро-
пиловый эфир
Дииэопропилфторфосфат 945
Дииодбенэол 289
Дикаин 944
Дикакодил 347
Дикальцийфосфат 382
Дикмана реакция 589
Дикона процесс 461
Диксантилен 877
§икумарол 122
-L-1,2-Дилаурилколаминфосфатид 639
Дильса — Альдера реакция 565
Димедрол 946
Димерин 942
1,4-Ди-(метансульфонилокси)-бутан —
см. Милеран
п-Диметиламиноазобензол 401
ЪГ-(3-Диметиламинопропил)-иминодибен-
зил, хлоргидрат — см. Имизин
Диметиланилины — см. Ксилидины
Диметиларсин — см. Какодиловый во-
дород
Диметиларсиновая кислота — см. Ка-
кодиловая кислота
Диметиларсиноксид — см. Какодил,
окись
5,9-Диметил-3,4-бенэакридин 401
9, 10-Диметил-1, 2-бензантрацен 400
Диметилбенэол — см. Ксилол
4,9-Диметил-5,6-бенэтиофантрен 401
2,4-Ди-метил-б-трет-бутилфенол 235
Диметилдигидроксисилан 816
Диметилдихлорсилан 814
Диметилдиэтоксисилан 815
Диметилкальций 375
Диметилмагний 1012
0,0-Диметнл-З-метил амидометилдитио-
фосфат 277
3,3-Диметил-2-метилен-бицикло-[1, 2, 2]-
гептан — см. Камфен
ДимегиловыЙ эфир, критич. постоянные
867
2,2-Диметилпропан 165
Диметилсилан 814
Диметилхлорарспн — см. Какодил хло-
рид
Диметилцианарсин — см. Какодил циа-
нид
Диметилэтилметан 165
Диметоат 277
Димефокс 277
D, L-1,2-Димиристилколаминфосфатид
639
L-1,2-Димиристилколаминфосфатид 639
Динамитрон 335
Динезин 943
Диодон — см. Кардиотраст
Диодраст — см. Кардиотраст
Диоксан, криоскопии, постоянные 830
1,8-Диокси-3-метилантрон-9 — см. Хри-
зоробин
2,6-Диоксипурин — см. Ксантин
1-(3', 4'-Диоксифенил) -2-иэопропилами-
ноэтанол, хлоргидрат — см. Иэадрин
2,4-Диоксо-3,3-диэтил-5-метилпиперидин
— см. Димерин
2,6-Диоксо-1, 2, 3, 6-тетрагидропурин —
см. Ксантин
Дипаксин 239
L-1, 2-Дипальмитилколаминфосфатид 639
D, L-1, 2-Дипальмитолеилколаминфос-
фатид 639
Дипин 403
Дипразин 946
2,5-Дипропокси-3,6-диэтилениминобензо-
хинон-1,4 403
Дисистон 277
Дислокации в кристаллах 834
Диспергирование 138
Дисперсность 139
Дисперсные системы 643, 646
Диспрозий 916
D, L-1, 2-Дистеарилколаминфосфатид 639
L-1, 2-Дистеарилколаминфосфатид 639
Дитетрагоиальный вид симметрии (ди-
пирамидальный, пирамидальный) 850
Дитилии 945
Дитригональный вид симметрии (дипира-
мидальный, пирамидальный, скалено-
эдрический) 850
Дифенацин 239
Дифенилацетилиндандион 239
Ди фенилдигидроксисилан 816
Дифенилмагний 1012
4, 4'-Дифенилметандиизоцианат 207
Дифенин 942
Диферулоилметан — см. Куркумин
1, З-Дифосфоглицериновая кислота 1043
Дифракция электронов 506
---рентгеновских лучей 506
Диффузионная область (протекания реак-
ции) 566
Диффузия ионов 321
Ди-(р-хлорвинил)хлорарсин — см. Лю-
изит
Дихлоркарбен 423
п-Ди-(2-хлорэтил)аминобенэилфосфино-
вая кислота, диэтиловый эфир 402
п-Ди-[2-хлорэтиламино]фенилаланин
402
Y"I п-Ди-(2-хлор этил) амино] фенилмаслн-
ная кислота — см. Хлорамбуцил
Диэдрический вид симметрии (безосный
и осевой) 850
Диэтиламино-2,6-диметил ацетанилид,
хлоргидрат — см. Ксилокаин
Диэтилдигидроксисилан 816
Диэтилдихлорсилан 814
Диэтилдиэтоксисилан 815
Диэтиленимино-ЪГ,ЪГ-диэтилфосфорамид
403
Диэтилмагний 1012
Диэтиловый эфир, критич. постоянные 867
Диэтилстильбэстрол 405
— дифосфат — см. Хонван
1,1 '-Диэтил-4,4'-хинокарбоцианинио-
дид — см. Криптоцпанин
w-Додекан, критич. постоянные 867
н-Додеканаль — см. Лауриновый аль-
дегид
Додекановая кислота — см. Лауриновая
кислота
Додеканол-1 — см. Лауриновый спирт
Додецилиэоцианат 207
н-Додециловый спирт — см. Лаурино-
вый спирт
Доза ионизирующего излучения 89
Доломит 137, 439, 1009, 1022
Доменный процесс 47
Домены 1019
Донор 709
Донорно-акцепторная связь — см. Коор-
динационная связь
Допан 402, 948
Дорил — см. Карбохолин
Древесина, гидролиз 951
— защитная обработка 92
— сухая перегонка (пиролиз) 952
— энергохимич. переработка 952
Древесные пластики 953
Древесный сахар 881
Дробилки 140, 141
Дробление 138, 140
Дроморан — см. Леморан
Дубильно-экстрактовое производство 952
Дубление 627
Дублирование (колориметрии, титрова-
ние) 649
Дурал 735
ДЦМ (закрепитель окрасок) 79
ДЦУ (закрепитель окрасок) 79
Европий 916
Едкое кали — см. Калий, гидроокись
Еллаговая кислота 759
Енол фосфаты 1042
Ж
Жаропрочность 13, 427
Жаростойкость 15
Жасмон 16
Желатина 16, 17, 211
Желатинирование — см. Застудневание
Железистая кислота 39, 19
Железистосинеродистая кислота 39
Железистосинеродистый калий — см. Ка-
лий, гексацианоферроат
Желеэная кислота 40, 18
Железная лаэурь 40
Железные квасцы — см. Железо, суль-
фаты
Железные руды 41
Железный блеск — см. Гематит
Железный купорос — см. Железо, супы
фаты
Железняки красные 41
---магнитные 41
Железо 40
— аналитич. определение 46
— в природе 40
— водорода поглощение 43
— галогениды 43
— гексацианоферраты 45
— гексацианоферриаты 45
— гексацианоферроаты 45
— гидриды 43
— гидроокиси 18
— дисульфид—см. Железо двусернистое
— закись 18, 42
— закись-окись 18, 42
— карбиды 425, 427 — см. также Же-
лезо, сплавы
— карбонил 44, 441
— карбонилгидрид 441
— магнитные свойства 1022
— моносульфид — см. Железо сернистое
— нитраты 17, 44
— нитриды 43
— окислы 18, 42
— окись 18, 42
— — сульфат 38
— пентакарбонил 44
— получение 46
— применение 48
— сплавы 19, 48
— сульфаты 37
— сульфиды 38
— тр-ехокись 18
— трисульфид — см. Железо серное
— физич. и химич. свойства 41
— фосфаты 45
— фосфиды 44
— хлориды 39
Железо губчатое 48
--- двусернистое 38
--- карбонильное 46
--- метеорное 40
--- самородное 40
--- сварочное 48
•—» сернистое 38
--- серное 39
---теллурическое 40
--- техническое 46
--- хлористое 39
--- хлорное 39
--- чистое 46, 48
а-Железо 19, 41, 44
Э-Железо 41
6-Железо 41
у-Железо 19, 41, 44, 613
Железо — углерод (система) 19
Железо-аммониевые квасцы 38, 534
Железоокисные пигменты — см. Пиг-
менты желеаоокисные
Железопорфириновые коферменты 742
Железосинеродистая кислота 49
Железосинеродистый калий — см. Ка-
лий, гексацианоферриат
Железо-титанс-ванадиевые руды 41
Желтая кровяная соль — см. Калий,
гексацианоферроат
Желтый фермент Варбурга — см. Фла-
виновые кислоты
Желчные кислоты 49
Желчные пигменты 50
Женевская номенклатура 445
Жесткость воды 51
--- временная 51
--- кальциевая 51, 53
--- карбонатная 51, 52, 53
--- магниевая 51, 53
--- морской 53
--- некарбонатная 51, 52, 53
--- общая 51, 52, 53
--- постоянная 51
Живица 53
Жидкие кристаллы 53, 59
Жидкие металлы 60
Жидкие системы 55
— вязкость 57
— давление пара 56
— Коновалова законы 694
— растворимость взаимная 55
— темп-ра кипения 57
Жидкие удобрения 58
Жидкости 59
— вязкость 59, 61
— кипение 569
— механич. свойства 61
— растворимость взаимная 55
— сжимаемость 61
— структура 60
— текучесть 59, 61
— физич. свойства 60
— электропроводность 61
«Жидкость Кадэ» 347
Жирные кислоты — см. Карбоновые кис-
лоты и Высшие жирные кислоты (т. I)
Жировой обмен — см. Обмен веществ
Жиротопление 68
Жиры 62
— аналитич. показатели 65
— биологич. роль 66
— высыхание 62
— гидрирование 64
— гидролиз 64, 67
— окисление 66, 67
— омыление 64
— переэтерификация 64
— прогоркание 65
— • расщепление 64, 65, 66 — см. также
Расщепление жиров
— синтез в организме 67
— титр Ж. 63
— физич. свойства 63
— химич. свойства 64
— э«чгльгирование 63, 66
Жиры животные 68, 62
--- костные 70
--- морского зверя 69
--- протоплазматические 66
--- растительные 71, 62
---расщепленные — см. Расщепление
жиров
--- рыб 69
--- технические 70
3
Зажигательные составы 77
Зайцева правило 77
Закрепляющие окраски вещества 78
Замазки кислотоупорные 580
--- андезитовые 580
--- диабазовые 580
--- на органич. основе (арзамиты) 581
--- силикатные 580
Заменимые аминокислоты 80
Замша 629
Зандмейера реакция 80
Заполнения числа 520
Заполнители пористые 81
Зарин 82
Застудневание (растворов высскомоле-
куляр. соединений) 82
Заурановые элементы — см. Трансура-
новые элементы
Защита глаз 84
--- индивидуальная (промышленная) 83
--- органов дыхания 83
---открытых участков кожи 85
Защита «жертвенная» (в радиац. химии
95
--- типа «губки» (в радиац. химии) 95
Защита металлов (электрохимическая) 86
--- анодная 89
--- катодная 86
--- протекторная 86, 88
Защита от излучений радиоактивных
веществ и других излучений высоких
энергий 89
Защитная обработка древесины 92
Защитная одежда 93, 85
Защитное действие (в радиац. химии) 94
Защитные добавки (в радиац. химии) 94
Защитные коллоиды 95, 608
Защитные покрытия 96
--- гальванические 96
--- горячим способом получаемые 96
--- гуммированные 101
--- диффузионным способом получае-
мые 97
--- лакокрасочные 98
--- металлические 96
--- неметаллические 98
---пластмассовые 99
---эбонитовые 101
Защитные рукавицы и перчатки 86
Зеаксантин — см. Каротиноиды
Зеемана явление 103, 519
Зеин 104
Зеленое масло 105
Зеленый купорос — см. Железо, суль-
фаты
Зелинского—Стадникова реакция 105
Земли отбеливающие 106
Земляной воск — см. Озокерит
Земная кора — см. Литосфера
Зеркальная плоскость симметрии (в кри-
сталлсгр.) 848
Зеркально-поворотные оси симметрии
(в кристаллогр.) 848
Зимаза 107
Зимогены 108
Зимозан 108
Зинина реакция 109
Змеевики (минералы) 1009
Зола 352
Золи 109
— коагуляция 607—610
Золи гидрофобные 110
--- лиофильные 110
---лиофобные 110
---твердые ПО
Золотая соль 118
Золото 112
— амальгамация 116
— аналитич. определение 115
— галогениды 110
— магнитная восприимчивость 1021
— монсгалогениды 110
— нитраты 114
— скислы 113
— окись 113
— получение 115
— применение 117
— роданид 114
— селенат 115
1057 — 1058
— соли 114
— сплавы 111
— сульфаты 114
— сульфиды 114
— тиокарбамид 115
— тригалогениды 110
— физич. и химич. свойства ИЗ
— цианиды 114
Золото бромное 111
--- гремучее 114
--- иодное 111
--- самородное 112
--- фторное 110
--- хлористое ИЗ
--- хлорное 110, ИЗ
Золотое число 96
Золотоорганические соединения 117
Золотосинеродистая кислота 114
Золотохлориотоводородная кислота 118,
ИЗ
Зоман 118
Зонгорин 118
Зонная очистка — см. Зонная плавка
Зонная перекристаллизация — см. Зон-
ная плавка
Зонная плавка 119
—- дуговая 120
--- методом «в клетке» 121
--- методом «плавающей» воны 121
--- с магнитным подвешиванием 121
--- с «подхватом» 120
--- электронной бомбардировкой 120
Зонная теория гетерогенного ката-
лиза 466
Зонное выравнивание (в зонной плавке)
121
«Зонно-пустотная» плавка 121
«Зонно-транспортная» плавка 121
Зооциды 122
Зрительный пурпур — см. Родопсин
И
Иванова реакция 125
Идеальные растворы — см. Растворы
Идентификация 127, 999
--- высокомолекулярных соединений
128
Идоза 133
Идозаиы 133
Иервин 134
Изадрин 134, 945
Изатин 134, 242
а-Изатинанилид 136
Изатиновая кислота, лактам — см. Иза-
тин
Изатин-р-оксим 136
Изафенин 137
Изацен — см. Изафенин
Известковая вода 137, 381
Известковое молоко 381
Известняки 137, 371
Известь 137, 371
--- воздушная 137
--- гашеная 381
--- гидравлическая 138
Излучение электромагнитное 506
Излучения радиоактивных веществ, за
щита 89
--- ядерные источники 335
Иэмольчаемости коэффициент 142
Измельчение 138
Измельчители 139
Изо (приставка) 144
Изоамилацетат — см. Амилацетат и изо-
амилацетат (т. I)
Изоамплеиы, дегидрирование 168
N-Изсамилкадаверин, дихлоргндрат —
см. Изоверин
Изоамиловый спирт — см. Амиловые
спирты (т. I)
Изобарный потенциал — см. Термодина-
мические потенциалы
Изобары (атомы) 201, 204
Изобары реакции уравнение 145, 1046
Изобары ядерные — см. Изотопы
Изобутилацетат — см. Бутилового спир
та эфиры (т. I)
Изобутилен 145
Изоверин 145
Изовиолантрон — см. Изодибензантрон
Изогуанин 405
Изодекстропимаровая кислота 146
Изодибензантрон 146
Изодиморфизм — см. Изоморфизм
Изодиморфные вещества 162
1059 —1060
Изоиндолы 147
Изоионная точка 147, 211
Изокарбостирил 206
Изокоричная кислота Либермана 716
--- Эрленмейера 716
Изоксазол 148
Изолейцин 148
Изолизергиновая кислота 963
Изолимонная кислота, дегидрогеназа —
см. Цикл трйкарбоновых кислот
Изолированные системы 149
Изологические ряды 149
Изомеразы 149
Изомеризация 150
Изомерия 152
--- геометрическая 153, 662
--- комплексных соединений 662
--- координационная 664
--- оптическая 153, 155, 662
--- поворотная 158, 157
-— положения 152, 664
~— пространственная 153
--- скелета 152
--- солевая 665
--- сольватная 663
--- структурная 152
--- цис-транс см. Изомерия геомет-
рическая
--- ядерная 158
Изомеры поворотные 697
Иэомолярных серий метод 159
Изоморфизм 161
Изомочевина — см. Мочевина
Изониазид — см. Тубазид
1-Изоникотиноил-2-изопропилгидразин—
см. Ипразид
Изонитрилы К—N —С 162
Изонитрильная проба по Гофмаиу 164
Изонитрозоацетон 164
Изооктан 164
Изоосмотические растворы — см. Изо-
тонические растворы Ьм
Изопентан 165
Изополикислоты 166
Изополисоединения 166, 657
Изопрен 166
— полимеризация 169, 485
— получение 168
Изопреналин — см. Изадрин
Изопреновый каучук 169
Изопропанол — см. Изопропиловый спирт
п-Изопропилбензальдегид — см. Куми-
новый альдегид
Изопропилбензол 171
Изопропиловый спирт 172
Изопропиловый эфир 173
Изопротонная точка — см. Изоионная
точка
Изорибофлавин 403
Изосафрол 173
Изостильбен — см. Стильбен
Изоструктурность 161
Изотактические полимеры 173
Изотермы адсорбции Лэнгмюра уравне-
ние 994
--- вязкости двойных жидких систем
57
----- реакции уравнение 177, 1046
Изотиоцианаты 177, 890
Изотонические растворы 179
Изотоны 201
Изотопного разбавления метод 180, 184
Изотопные индикаторы 181
Изотопные источники излучений 335
Изотопные эффекты 186
--- кинетические 188
--- термодинамические 187
Изотопный обмен 189, 185
— кинетика 190
— механика 191
— термодинамика 189
Изотопный обмен азота 194
--- ассоциативный 192
--- водорода 193
--- галогенов 193
--- диссоциативный 192
--- кислорода 193
--- серы 194
--- углерода 194
Изотопов разделение 195, 188
--- диффузией 195, 197
--- каскадное 195
---кинетическое — см. Изотопов разде-
ление электролизом
--- ректификацией 195, 197, 198
--- термодиффузией 195, 197, 198
--- химическим обменом 195, 197, 199
--- центрифугированием 195, 197, 199
--- электролизом 195, 197, 199
--- электромагнитное 195, 200
Изотопов стабильных анализ 200
Изотопы 201
--- радиоактивные 182, 202
--- ^-радиоактивные 203
--- стабильные 182, 202
--- р-устойчивые 203
Изотропия 840
Изофлавоны 759
Изофталевая кислота — см. Фталевые
кислоты
Изохинолин 205
Изохорный потенциал — см. Термодина-
мические потенциалы
Изохоры реакции уравнение 206
Изоцианаты 206, 179, 890
Изоцианиновые красители — см. Циа-
ниновые красители
Изоциануровая кислота — см. Циануро-
вая кислота
Изоциклические соединения 209
Изоцитратдегидрогеназа — см. Цикл три-
карбоновых кислот
Изоэвгенол 210
Изоэлектрическая точка 211, 147
Изупрел — см. Изадрин
Илиды 212, 421
Илосвая реактивы 214
Йльновича уравнение 215
Имидазол 215
Имидгалогениды 216
Имидоэфиры — см. Иминоэфиры
Имиды кислот 217
Имизин 218
Имииазол — см. Имидазол
Иминоэфиры 218
Имипрамин — см. Имизин
Иммерсионный анализ 219
Иммунополисахариды 220
Иммунохимия 221
Иммуноэлектрофоретический метод Гра-
баря 224
Имония соли 225
Импульс микрочастицы 506, 507, 508
Инвар 27
Инверсии центр (в кристаллогр.) 848
Инверсия сахаров 226
Инвертазы 227, 226
Инвертин — см. Инвертазы
Инвертный сахар — см. Инверсия сахаров
Ингибиторы 228, 458
--- анодные 228
--- катодные 228, 229
--- коррозии металлов 228, 230
--- кроющие 229
--- летучие 228, 230
--- окисления нефтепродуктов 234
--- опасные 229
--- ржавления 230, 234
--- ферментов — см. Ферменты
Индазол 236
Индамины 237
Индан — см. Гидринден
Индандион-1,3 237
Индантрен — см. Индантрон
Индантреновые красители — см. Кубо-
вые красители
«Индантреновый синий Р» — см. Индан-
трон
Индантрон 239
Индекс вязкости 240
Инден 240
Инден-кумароновая кислота — см. Кума-
роновая кислота
Индивид химический 241
Индиго 241, 243, 747, 760
«Индиго белое» 884
Индигозоли 242, 884
Индигокармин 243
Индиготин — см. Индиго
Индий 244
— аналитич. определение 245
— гидроокись 245
— окислы 244
— получение 245
— применение 246
— сульфат 245
— сульфид 245
— физич. и химич. свойства 244
— хлорид 245
Индикан 246, 242, 761
Индикаторная реакция 567
Индикаторные электроды — см. Электро- !
ды сравнения
Индикаторы 247
---• адсорбционные 253
--- внешние 248
--- внутренние 248
--- для титрования в неводных средах
250
--- кислотно-щелочные 248
--- комплексометрические 252, 670
---необратимые 247
--- обратимые 247
--- окислительно-восстановительные 251
--- смешанные 249
---универсальные 254, 251
--- флуоресцентные 250
---хемилюминесцентные 254
Индирубин 243, 255, 761
Индоаиилины 260
Индокарбоцианины — см. Красители ин-
долениновые
Индоксил 254
Индоксилсульфат — см. Индикан
Индол 255, 136
Индолениновые красители 259
Индоленины 259
Индолилмасляная кислота — см. Гете-
роауксин (т. I)
Индолин 259
N-Индолкалий 255
N-Индолмагнийгалогениды 256
N-Индолнатрий 255
Индолхиноксалин 135
Индофенин 135, 260
Индофениновая реакция 260
Индофенолы 260
Индуктомерный эффект 261
Индуктор 265
Индукции период 262
Индукционный эффект 262
--- динамический — см. Индуктомер-
ный эффект
Индукция химическая 265
Инертные газы 266
— атомный радиус 312
Инжекторы 268
Инжекция 268
Инициаторы 474
---полимеризации — см. Полимериза-
ции инициаторы
Инициирующие взрывчатые вещества 269
Инконель 15
Инозин 271
Инозиновые кислоты — см. Инозинфос-
форные кислоты
Инозинфосфорные кислоты 271
Инозит 273
Инозитфосфатиды 273, 639
Инсектициды 275
---кишечные 275
--- контактные 276
--- системные 277
--- фосфорорганические 277
- - фумигационные 276
Инструментальные методы анализа 277
Инсулин 278
Интегральная доза — см. Доза ионизи-
рующего излучения (т. I)
Интерметаллические соединения 280, 856,
858, 859
Интерферометрия 280
Интерферометры 280, 281
Интратион 277
Инулазы 282
Инулин 282
Инулиназы — см. Инулазы
Инулобиоза 282
Инулиды 282
Инулосахараза 282
Инфракрасная спектроскопия 283
Иод 285
— аналитич. определение 286
— получение 287
— применение 287
— пятиокись 286
— техника безопасности 287
— физич. и химич. свойства 285
Иодаллилуротропин 287
Иодатометрия 287
Иодаты 285, 286
Иодацетамид 288, 294
Иодбензол 288
Иодиды 285, 286
Иодистоводородная кислота 289
Иодистый водород 289
— критич. постоянные 866
Иодистый метил 289
Иодистый метилен 290
Иодистый этил 290
Иодная кислота 286
Йодноватая кислота 286
Иодноватистая кислота 286
Иодное число 290, 65, 69, 73
Иодобензол 288, 291
Иодогност — см. Сульфопиридазин
.и-Иодозобензойная кислота 291
1061 -1062
о-Иодозобензойная кислота 290
n-Иодозобензойная кислота 291
Иодозобензойные кислоты 290
Иодозобензол 291, 288
7-Иод-8-оксихинолин-5-сульфокислота
291
Иодолипол — см. Рентгеноконтрастные
препараты
Иодометрия 292
Йодоформ 292
Йодоформная реакция 293
Иодуксусная кислота 294
— амид — см. Иодацетамнд
.м-Иодфенилацетат 294
о-Иодфенилбензоат 294
Иодфенол 294
Иодциан — см. Галогеноцианы (т. I)
Иодэтан — см. Йодистый этил
Ион карбония 442
Ионизации потенциал 295
-- сечение 315
--- энергия 295
Ионизационная метамерия 664
Ионизация в электрическом поле 317
--- на поверхности 317
--- при соударениях нейтральных ча-
стиц 316
Ионизирующее излучение, дозы 89
Ионикс-200 303
Ионитовая колонна 305
Ионитовые диафрагмы 298
Ионитовые сита 299
Иониты 299
— коэфф, внутренней диффузии ионов
в И. 309
— свойства 303
Иониты полимерные 301
--- селективные 308
Ионная атмосфера 306
Ионная пара 444
Ионная связь 857 — см. также Хими-
ческая связь
Ионная сила раствора 307
Ионное облако — см. Ионная атмосфера
Ионное отложение 930
Ионно-молекулярные реакции 317, 321
Ионные реакции 475
Ионный выход 307
Ионный обмен 307, 305
Ионный радиус 310, 858
Ионол 235, 236
Иононы 312
Ионообменная сорбция 305, 308, 309
Ионообменное сродство — см. Ионный
обмен
Ионообменные сорбенты 308, 309
Ионы 314
— аналитич. классификация 502
— в фотохимии 321
— в электрич. разрядах 321
Ионы (в газах) 314
— в атмосфере Земли 322
— в радиационной химии 321
— • образование 315
— при высоких темп-рах 322
— реакции И. с молекулами 317
— рекомбинация 319
Ионы-радикалы 323
Йохимбин 324
Иоцича реакция 324
Ипраэид 325
Иприт 401, 402 — см. также р, р' Ди-
хлордиэтилсульфид (т. I)
Ипрониазид — см. Ипразид
Иралия — см. Иононы
Иргалои — ом. Комплексон Ш
Ирган НТ 276
Иридий 326
— аналитич. определение 328
— гидроокись 827
— гидросульфид 327
— комплексные соединения 327
— окислы 327
— получение 328
— применение 329
— сульфат 328
— сульфиды 327
— физич. и химич. свойства 326
— хлориды 327
— шести окись 327
Иридий осмистый 326, 329
Иридий-З-гексахлориды — см. Хлорири-
диты
Иридий-4-гексахлориды — см. Хлорири-
даты
Ирон 313
Ископаемое твердое топливо 330
Исландский шпат 371, 374
Испарение 331
Испарители 334
Источники ядерных излучений 335
Итаконовая кислота 336
Иттербий 337, 915, 916
Иттриалит 337
Иттриевая группа элементов 915, 919,
923
Иттрий 337, 915, 917, 918
— аналитич. определение 338
— гидроокись 338
— карбонат 338
— нитрат 338
— нитрид 338
— окись 338, 339
— оксалат 338
— получение 339
— применение 340
— соли 338
— сульфат 338
— феррицианид 338
— физич. и химич. свойства 337
— хлорид 338
Иттропаризит 337
Иттрофлюорит 337
Ихтиол 340
К
Кадаверин 341
Кадионы 341
Кадмиевые пигменты —- см. Пигменты
кадмиевые
Кадмий 342
— аналитич. определение 343
— бромиц 345
— галогениды 345, 343
— гидроокись 343, 345
— иодид 345
— карбонат 343
— нитрат 343
— окись 345
— получение 343
— применение 344
— сплавы 345
— сульфат 343
— сульфид 346, 343
— физич. и химич. свойства 342
— фторид 345
— хлорид 345
— цианид 343
Кадмийорганические соединения 344
Казеин 346, 130, 211
Казеиноген 347
Каинит 348, 351, 1029
Кайенское масло «линалое» 969
Какао масло — см. Жнры растительные
Какодил 347
— окнсь 347
— соединения 347
Какодил хлорид 347
--- цианид 347
Какодиловая кислота 347
Какодиловый водород 347
Кали едкое — см. Калий, гидроокись
Калиевый хромпик — см. Калий, би-
хромат
Калий 348
— азид 349
— аналитич. определение 350
— бисиликат 357
— бисульфат 358
— бисульфит 359
— бифторид 360
— бихромат 361
— борогидрид 354
— бромид 354, 284
— гексацианоферриат 354
— гексацианоферроат 354, 408
— гидрид 349
— гидроокись 355, 349
— гидросульфид 358
— иодид 355, 408
— карбонат 356
— кремнефторид 802
— магнитная восприимчивость 1022
— метабисульфит — см. Калий, пиро-
сульфит
— метасиликат 357
— метафосфат 359
— нитрат 356
— нитрид 349
— окись 349
— перекись 349
— перманганат 356
— персульфат 357
— перхлорат 357
— пиросульфит 359
— пирофосфат 359
— полисульфиды 358
— получение 350
— применение 351
— роданид 357, 408
— роданохромиат — см. Калий, хромо-
роданид
— силикаты 357
— сульфат 358, 352
— сульфид 358, 349
— сульфит 359
— тетрасиликат 357
— техника безопасности 351
— триполифосфат 359
— феррицианид — см. Калий, гексациа-
ноферриат
— ферроцианид — см. Калий, гексациа-
ноферроат
— физич. и химич. свойства 348
— фосфаты 359
— фторид 359
— хлорат 360, 155
— хлорид 360, 351
— — магнитная восприимчивость 1021
— хромат 361
— хромороданид 361
— цианид 361
— этилксантогенат 362, 408
Калий азотнокислый — см. Калий нитрат
--- бромистый — см. Калий, бромид
--- двухромовокислый — см. Калий,
бихромат
--- железистосинеродистый — см. Ка-
лий, гексацианоферроат
--- железосинеродистый — см. Калий,
гексацианоферриат
--- иодистый — см. Калий, иодид
--- иодноватокислый 286
--- марганцовокислый — см. Калий,
перманганат
--- надсернокислый — см. Калий, пер-
сульфат
--- роданистый — см. Калий, роданид
--- сернистокислый — см. Калий, суль-
фит
---сернистый — см. Калий, сульфид
--- сернокислый — см. Калий, сульфат
--- углекислый — см. Калий, карбонат
— фтористый — см. Калий, фторид
---хлористый — см. Калий, хлорид
--- хлорноватокислый — см. Калий,
хлорат
--- хлорнокислый — см. Калий, пер-
хлорат
--- хромовокислый — см. Калий, хро-
мат
-- цианистый — см. Калий, цианид
--- этилксантогеиовокиолый — см. Ка-
лий, этилксантогенат
Калий — магний, сульфат 352
Калий — натрий 349
Калийная селитра — см. Калий, нитрат
Калийные соли 348, 352
Калийные удобрения 351
Калийорганические соединения , 352
Калипнеттен — см. Калийное
Калипнон 353
Калифорний 354
Каломельный влектрод 362
Калориметр 363
--- адиабатный 364, 368
--- двойной 368
--- жидкостной 365
--- Кальве 370
--- массивный 366
--- с изотермической оболочкой 364
— с переменной температурой звз
— с плавящимся твердым телом 369
— с постоянной температурой 369
Калориметрическая бомба 365
Калориметрическая система 363
Калориметрия 362
Калориметр-контейнер 367
Кальциевая селитра — см. Кальций, нит-
рат
Кальций — 370
— амид 373
— аналитич. определение 373
— арсенат 375
— арсенит 375
— ауртиопропанолсульфонат 828
— бикарбонат 380
— бисульфит 382
— борид 373
— бромид 377
— галогениды 376, 372
— гидрид 377, 372
— гидроокись 381
— гидросульфид 382
— иодид 377
1063 -1064
— карбид 378, 373, 425, 428
— карбонат 379, 439
— комплексный аммиакат 373
— нитрат 380
— нитрид 372
— окись 380, 372
— перекиси 372
— пернитрид 373
— пирофосфат 383
— полисульфиды 372
— получение 373
— применение 374
— силициды 373
— субгалогениды 376
— сульфат 381
— сульфид 382, 372
— сульфит 382
— физич. и химич. свойства 372
— фосфаты 382
— фосфид 373
— фторид 376 — см. также Флюорит
— хлорид 376
— цианамид 383
Кальций азотнокислый — см. Кальций,
нитрат
Кальций двууглекислый — см. Кальций,
бикарбонат
---сернистый — см. Кальций, сульфид
•-- сернокислый — см. Кальций, суль-
фат
— углекислый — см. Кальций, кар-
бонат
Кальцийорганические соединения 375
Кальцинированная сода — см. Натрий,
карбонат
Кальцит 137, 139, 371, 379, 439
Камеди 384
Каменная соль 139, 284
Каменноугольная смола 385, 636
Каменноугольные масла — см. Масла
каменноугольные технические
Каменноугольный деготь — см. Смола
к аменноугольная
Каменный уголь 331
Кампса реакция 388
Камфан 389
Камфаноновая кислота 389
Камфара 389
— криоскопия, постоянные 830
Камфарная кислота 389
Камфарное масло — см. Эфирные масла
Камфароновая кислота 389
Камфен 390
Камфенгликоль 391
Камфенилановый альдегид 391
Камфениловая кислота 391
Камфенилон 391
Камфеновая кислота 391
Камфеновые перегруппировки 392
Канамицин 394
Канифоль 395, 587, 827
— эфиры 396
Канифоль гидрированная 395
--- диспропорционированная 395
--- живичная 395
--- конденсированная 396
--- модифицированная 395
--- окисленная 396
--- полимеризованная 396
--- сульфированная 396
--- талловая 395
--- экстракционная 395
Канифольное мыло 395, 396
Канифольно-скипидарное произ-во 952
Канниццаро реакция 397
Каноническое распределение — см. Гиб-
бса распределение (т. I)
Кантаридин 398
Канцерогенные аещества 398
— идентификация 999
Канцеролитические вещества 401
Каолин 406
Каолинит 406
Капельная колориметрия 407
Капельный анализ 406
Капиллярная конденсация 409, 413
Капиллярная постоянная 413
Капиллярное давление 412
Капиллярно-конденсационный гистере-
зис 410, 411
Капиллярные явления 412
Каприловая кислота 414
Каприловый альдегид 414
Каприновая кислота 414
е-Капролактам 414, 906
е-Капролактон 912
Капрон — см. Полиамидные волокна
Капроновая кислота 417
Капсаицин 417
КйпСЮль-детонатор 270 — см. также Сред-
ства инициирования
Каптакс 418
Карагеенан 385
Каран 418
Карбазол 419, 386
Карбазол-9-карбоновая кислота 419
Карбаматы 420
Карбамидные пластики — см. Аминопла-
сты (т. I)
Карбамилхлорид 420
Карбаминовая кислота 420
— аммониевая соль 420
— калиевая соль 420
— кальциевая соль 420
— хлорангидрид — см. Карбамилхло-
, РНД
Карбаминоилхолин — см. Карбохолин
Карбаминоилхолинхлорид — см. Кар-
бохолин
Карбанион 420
Карбены 423
Карбидные сплавы 428
Карбиды 424, 25
— получение 427
— применение 428
Карбиды с ковалентной связью 425
--- солеобразные 424
--- типа Fe.iC 427
Карбиды — фазы внедрения 426
Карбиламины — см. Изонитрилы
Карбин 428
Карбоангидраза 428
Карбогидразы 429
Карбодиимиды 430, 890
.Карбоиды 431
Карбоксигемоглобин — см. Гемоглобины
(т. I)
Карбоксилаза 431
Карбоксилатные каучуки 432
Карбоксилирование 434
Карбоксильная группа — см. Карбоно-
вые кислоты
Карбоксиметилцеллюлоза 436
Карбоксипептидаза А 437
Карбоксипептидаза В 438
N-Карбокси-соединения 1042
Карболинеум 438
Карболовая кислота — см. Фенол
Карболовое масло — см. Фенольные масла
Карбометилен — см. Кетены
2р-Карбометокси-зр-бензоилокситропан
— см. Кокаин
Карбомицин — см. Магнамицин
Карбонаты 438
Карбонилгалогениды металлов 439, 441
Карбонилгидриды металлов 441
Карбонилы металлов 439
Карбонильная группа — см- Альдегиды
(т. I) и Кетоны
Карбония ион 442, 795
— образование 442
— свойства 443
Карбоновые кислоты 445
Карбоорганосиланы 809
Карборунд — см. Кремний, карбид
Карбостирил 447
Карбофос — см. Фосфорорганические ин-
сектициды
Карбохолин 448
Карбоцепные высокомолекулярные со-
единения — см. Высокомолекулярные
соединения (т. I)
Карбоцианины — см. Цианиновые краси-
тели
Карбоциклические соединения — см. Изо-
циклические соединения
Карвакрол 448
Карвоментолы 448
Карвон 449
Кардамоновое масло — см. Эфирные мас-
ла
Кардиазол — см. Коразол
Кардиотраст 449
Карен 449
Кариофиллен 450
Кариуса метод 451
Каркасные структуры 856
Кармин 451
Карминовая кислота 451
Карналлит 348, 360
Карнаубский воск — см. Воски (т. I)
Карнитин 452
Карно цикл 452
Карнозин 453
Карнозиназы 453
Каробан 385
Каротиноиды 453, 758
] Каротиноксидааа — см. Липооксидааа
Каротины — см. Каротиноиды
Касторовое масло — см. Жиры раститель-
ные
Кастрикс 122
Каталаза 455, 742
Катализ 456
— в живой природе 463
— в пром-сти 461
— классификация процессов 458
— мультиплетная теория К. 464
— открытие явлений К. 460
— промоторы в К. 460, 470
— яды в К. 460, 470
Катализ гетерогенно-гомогенный 464
— гетерогенный 463, 459, 561; кинети-
ка 471; применение промышл. 472
--- гомогенный 473, 459
--- и кристаллическая структура 470
--- ионообменный 477
--- кислотно-основной 478, 458
--- -— гетерогенный 465
--- координационно-комплексный 475
--- микрогетерогенный 459
--- на металлах 468
--- на полупроводниках 466
--- окислительно-восстановительный
458
----- --. гетерогенный 466
--- отрицательный 456
--- положительный 456
Катализаторы 483, 456, 457, 560
— активность - 460, 465, 794, 797
— избирательность действия 459, 794
— модифицирование 460, 487
— отравление 460, 487
— подбор 483
— приготовление 484
— промотирование 460
Катализаторы крекинга 793; активность
794, 797
--- полимеризации — см. Полимериза-
ции катализаторы
----- Циглера — Натта 485, 462, 483
Каталитические яды 486, 460, 470
Каталитический крекинг 793
Катепсины 488
Катехины 489
Катионактивные закрепляющие окраски
вещества 78
Катионитовые диафрагмы 298
Катиониты — см. Иониты
Катионоидный реагент — см. Акцептор
Катионотропия 490
Катионотропные превращения — см. Ка-
тионотропия
Катионы — см. Ионы
Катодолюминесценция кристаллов 863
Каустическая сода — см. Натрий, гидро-
окись
Каучук, гидрохлорид 499, 492
— определение типа 131
— старение 493
— цветные качественные реакции на К.
132
Каучук (и) бутадиен-нитрильный 131,
132
--------карбоксилатный 433
--- бутадиеновый 130, 132
--- бутадиен-стирольный 130, 131, 132
--------- карбоксилатный 432
----- изопреновый 169; вулканизаты 171
---карбоксилатные 432
--- кремнийорганические 807
--- маслонаполненные 500
--- метилвинилсилоксановый 808, 809
--- метилсилоксановый 807, 809
—— метилфенилсилоксановый 808, 809
---метилэтилсилоксановый 809
--- натуральный 491, 130, 131, 132
---полиорганосилоксановые — см. Кау-
чуки кремнийорганические
--- полисульфидные 131
--- саженаполненные 500
--- силиконовые — см. Каучуки крем-
нийорганические
--- синтетический 495
--- хлорированный 131
--- хлоропреновый 131
--- этилсилоксановый 809
Каучукогены 495
Качественный анализ 500
--- анионов 502
--- катионов 502 '’
--- «мокрым путем» (в растворах)
501
--- неорганический 501
--- органический 503 ‘
— «сухим» путем 501
Квазиаксиальная связь 701
Квазиаксиальные атомы 701
Квазикомплексные соединения 503
Квазиэкваториальная связь 701
Квазиэкваториальные атомы 701
Квантование 513, 517
Квантовая механика 505
Квантовая статистика 523
Квантовая химия 526
Квантовые переходы 522
Квантовые системы 519
Квантовые числа — см. Атом (т. I)
Кварц 533 — см. также Кремний, окислы
Кварцевое стекло — см. Стекло кварце-
вое
Квасцы 533
Квебрах 273
Квебрахин — см. Йохимбин
Квенстетит 38
Кверцетин 534
Квиэтал 942
Кельвина закон 831
Кемферол 761
Кендалла вещество F — см. Кортизон
Керамзит — см. Заполнители пористые
Керамика 535
--- бытовая 538
высокоглиноземистая 539, 541
-----для токов высокой частоты 539
------для токов промышленной частоты
(твердый фарфор) 539, 541
---клиноэнстатитовая (стеатитовая) 539,
540, 541
--- кордиеритовая 539, 540, 541
--- корундовая 539, 541
•--магнезиальная 539, 541
•--муллитовая 539, 541
--- муллито-корундовая 539, 541
--- строительная 536
--- техническая 538
---титаносодержащая 540, 541
—•— форстеритовая 539, 540, 541
•--цельзиановая 539, 541
---шпинелевая 539, 540, 541
Керамикометаллические материалы —
см. Керметы
Керамические краски 542
Кератеины 543
Кератины 543
Кератосульфат 543
Кермесовая кислота 452
Керметы 543
Керобит — см. Ионол
Керосин 545
--- авиационный 546
--- осветительный 546
---тракторный 546
Кетены 547, 890
Кетимины 549
Кетоальдегиды 550
у-Кето-н-Валериановый альдегид —см.
Левулиновый альдегид
2-Кетогексаметиленимин — см. «-Капро-
лактам
а-Кетоглутаратдегидрогеназа 552
а-Кетоглутаровая кислота 552
З-Кетодигидроиндол — см. Индоксил
Кетозы — см. Моносахариды
З-Кетоиндолин — см. Индоксил
Кетокетены 547
Кетокислсты — см. Альдегидокислоты и
кетокислоты (т. I)
Кетоны 552
Кетостероиды — см. Стероиды
р-Кетотиолазы — см. Трансацилазы
Кефалинфосфолипид 273
Кефалины — см. Коламинфосфатиды
К-захват 203
Кижнера реакция 555
Кижнера — Вольфа реакция 556
Кизерит 1027
Киназы 558
Кинга реакция 559
Кинекс — см. Сульфопиридазин
Кинетика гетерогенных реакций 565
--- гомогенных реакций 562
--- изотопного обмена 190
---катализа гетерогенного 471
--- крекинга каталитического 796
--- кристаллизации 833
--- кристаллизации полимеров 843
---химическая 559
Кинетическая область (протекания реак-
ции) 566
Кинетические изотопные эффекты 188
Кинетические методы анализа 567
Кинетический метод разделения изотопов
195, 197, 199
Кинуренин 569
Кинурениназа 569
Кинуреновая кислота 569
Кипение 569
— темп-ры К. жидких систем 57 ,
Кипящий слой — см. Псевдоожижение
Кирза 628
Кислород 572
— атом 572
— изотопный обмен 193
— изотопы 572
— критич. постоянные 866
— магнитная восприимчивость 1022
— молекула 572
— определение 575
— получение 575
— применение 575
— физич. и химич. свойства 573
Кислородный электрод 577
Кислот и оснований теории — см. Кис-
лоты и основания
Кислотное число 578
Кислотно-основное равновесие 579
Кислотности константа 583
--- функция Гаммета 587
Кислотно-щелочные индикаторы 248
Кислотостойкие материалы — см. Хими-
чески стойкие материалы
Кислотоупорные замазки 580
Кислоты жирные синтетические — см.
Высшие жирные кислоты (т. I)
Кислоты и основания 581
— электронная теория 585
Кислоты смоляные 587
Китит 821
Клавацин 914
Клайэена конденсация 588
--- перегруппировка 589
Клайзена — Шмидта реакция 591
Клапейрона — Клаузиуса уравнение 592
Клапейрона уравнение 591
Кларки элементов 593
Клатратные соединения — см. Соедине-
ния включения
Клеи 594
— вязкость 595, 599
— жизнеспособность 595
— клеящее действие 602, 594, 599
— концентрация К. 595, 599
Клеи альбуминовые 596
---глютиновые 596
--- животные 596
---изоцианатные 598
--- казеиновые 596
--- карбамидные 597
--- комбинированные 596
--- конфекционные 599
— костные 596
--- кремнийорганические 598
— мездровые 596
---меламиновые 597
---минеральные 5 96
---полиуретановые 598
--- полиэфиракрилатные 598
--- природные 595
---растительные 596
-— резиновые 598
--- рыбьи 596
--- самовулканизующиеся 599
---синтетические 597
--- смоляные — см. Клем синтетические
--- термопластичные 598
--- термореактивные 597
--- феноло-каучуковые 597
--- феноло-поливинилацетальные 597
--- феноло-формальдегидные 597
--- эластомерные 598
---эпоксидные 598
Клейстер 763
Клемменсена реакция 599
«Клетки» эффект 601
Клетчатка — см. Целлюлоза
Клешневидные полимеры — см. Коорди-
национные полимеры
Клешневидные соединения — см. Внутри-
комплексные соединения (т. 1)
Клеящее действие 602, 594
Клиноэнстатит 539, 540, 541
Клоны 223
Клостеры — см. Сольваты
Кневеяагеля реакция 604
Кнорра реакция 606
Коагулянты 607
Коагулят 607
Коагуляция 607
— время К. 608
— порог К. 609
— неправильные ряды К. 609
— скорость К. 608
Коагуляция ортокинетическая 608
•-- скрытая 607
--- явная 607
Коалесценция 610
I 1085 — 1066
Коацерват 611
Коацервация 610
Кобальт 611
— аналитич. определение 614
— бромиды 617
— галогениды 616
— гексаммины 613, 614
— гидрид 613
— гидроокиси 618
— иодиды 617
— карбонаты 618
— карбонил 441, 613
— карбонилгидрид 441
— комплексные соединения 613
— критич. постоянные 866
— магнитные свойства 1022
— нитрат 618
— окислы 618
— получение 615
— применение 616
— сплавы 619
— сульфаты 620
— сульфиды 621
— трифторид 617
— физич. и химич. свойства 612
— фториды 616
— хлориды 617
Кобальтиаки — см. Кобальтиаммины
Кобальтиаммины 614
Кобальтин 612
Кобальтовые цветы — см. Эритрин
Кобальтовый блеск — см. Кобальтин
Кобальто-платиновые сплавы 619
Кобальт-пирит 621
Ковалентная связь — см. Химическая
связь
Ковалентный радиус 621, 859
Когезия 622, 602
Кодегидрогеназы 622, 742, 743
Кодеин 624
Кожа 625
— крашение 765
Кожа искусственная 628
Козимаза 107 — см. также Кодегидроге-
назы
Кокаин 629
Коквимбит 38
Кокосовое масло — см. Жиры раститель-
ные
Кокс 630
— горючесть 632
— прочность 631
— реакционная способность 632
— ситовый состав 631
— химич. свойства 631
Кокс доменный 631
--- каменноугольный 630
---литейный 631
--- нефтяной 633
---электродный пековый 632
Коксование 634, 792
Коксовая смола — см. Каменноугольная
смола
Коксовые печи 635
Коксохимическое производство 636
Коламин — см. Этаноламины
Коламинфосфатиды 638
Колеманит 371
Колимицин — см. Неомицины
Количественный анализ 640
Коллаген 641, 16
Коллагеназа 642
Коллидины — см. Метилпиридины
Коллодий 642
Коллодионный процесс 642
Коллоидная химия 643
Коллоидные растворы — см. Коллоидные
системы
Коллоидные системы 646
— защитное действие 95
— коагуляция 607
— лиофильность и лиофобность 972
— старение 607
Коллоидные системы гидрофильные 972
--- гидрофобные 972
---лиофобные 972; изоэлектрич. точка
211
Коллоиды — см. Коллоидные системы
--- защитные — см. Защитные колло-
иды
Коллоксилин 647 — см. также Нитро-
целлюлоза
Колориметрический анализ 647
Колориметрическое титрование 649
Колориметрия 649
--- капельная 407
Колориметры погружения 649
1067 -1068
Колхамин 650
Колхицин 650
Кольбе электрохимический синтез 650
Кольбе — Шмидта реакция 651
Кольрауша закон 652
Комаровского реакция 653
Комбинационное рассеяние света 653
Компаратор 648
Комплексные ионы — см. Комплексные
соединения
Комплексные соединения 656, 708
— валентности 658, 708
— диссоциация К. с. на комплексный и
внешнесферный иоиы 665
— замещение внутрисферных групп К. с.
молекулами растворителя 666
— изомерия 662
— исследование К. с. в растворе 665
— кислотно-основные свойства 667
— классификация 656
— константы нестойкости 668, 673
— константы устойчивости 668, 679
— координационная теория 657, 708
— молекулярная электропроводность
666
— номенклатура 659
— применение 668
— теория поля лигандов 532
Комплексные соединения иридия 327
--- кобальта 613
--- лантанидов 922
--- многоядерные 657
--- содержащие атомы с донорной
функцией 656
--- циклические 659
Комплексометрические индикаторы 252,
670
Комплексометрия 669, 672
Комплексон 11 672
Комплексон III 671, 670
Комплексоны 672
Комплексообразования методы (в химич.
анализе) 672
Компоненты (системы) 674
Конверсия газов 675
---газообразных углеводородов 675
--- окиси углерода 676
Конго красный 677, 745
Кондакова реакция 677
Конденсации реакции 678
Конденсация 682
--- в объеме пара 682
--- в присутствии неконденсирующихся
газов в паре 685
---в твердое состояние 686
--- капельная 683
-—• капиллярная 409, 413
---на поверхности жидкости 685
---на поверхности твердого тела 683
--- перегретого пара 684
-— пересыщенного пара 832
--- пленочная 683
--- смеси паров 685
•—- фракционированная 687, 688
Конденсация Клайзена 588
---Клайзена — Шмидта 591
--- кротоновая 870, 679, 681
Конденсированная система 689
Конденсированные ароматические соеди-
нения 689
Конденсирующий фермент 691
Кондуктометрическое титрование 691
Кондуктометрия 691
Конессин 692
Кониин 693
Конифериловый спирт 693, 958, 961
Коновалова законы 693, 56, 57
Коновалова реакция 694
Конопляное масло — см. Жиры расти-
тельные
Конрада — Лимпаха реакция 695
Консекутивные реакции — см. Последо-
вательные реакции
Консистентные смазки — см. Пластич-
ные смазки
Константа кислотности 583
--- нестойкости комплексных соедине-
ний 668, 673
--- основности 583
--- протолиза 583
--- равновесия — см. Равновесие хими-
ческое
--- равновесия адсорбционного 994,
995
--- равновесия изотопного обмена 189
--- скорости реакции — см. Кинетика
химическая
--- устойчивости комплексного соеди-
нения 668, 673
Констелляции — см. Конформации
Контакт Петрова — см. Сульфокислоты
нефтяные
Контактные аппараты 697
Контактные вещества 459
Контактные процессы 459
Контактные яды — см. Каталитические
яды
Контрольно-измерительные приборы —
см. Приборы контрольно-измеритель-
ные
Конфигурация абсолютная 156
Конформации 174, 697
— влияние на физико-химич. свойства
органич. соединений 703
— потенциальная энергия 701
— причины предпочтительности отдель-
ных К. 701
Конформации аксиальные 700
--- высокомолекулярных соединений
705
--- заслоненные 698, 701
--- заторможенные 699
--- макромолекул 1035
---• реакционные 698
--- скошенные 699, 701
--- трансоидные 699
--- устойчивые алифатических и цикли-
ческих соединений 699
--- экваториальные 700
Конформационное напряжение 701
Конформационный анализ 697
Концентраты сульфитно-спиртовой бар-
ды 705
Концентрационные цепи 706
Концентрационные элементы — см. Кон-
центрационные цепи
Концентрация 707
Конъюгация — см. Сопряжение
Координационная емкость 659
Координационная изомерия 664
Координационная связь 708, 657
Координационная теория — см. Коорди-
национная связь, Комплексные соеди-
нения
Координационное число 710, 658
Координационные полимеры 710
Координационные структуры 856
Координированные группы — см. Ком-
плексные соединения
Копа перегруппировка 712
Копаловое масло 714
Копалы 714
Копростерин — см. Стерины
Коразол 714, 944
Корал — см. Изопреновый каучук
Корамин — см. Кордиамин
Кордиамин 714, 944
Кордиерит 539, 540, 541
Кордиты 715
Кориандровое масло — см. Жиры расти-
тельные и Эфирные масла
Коринантеаль 716
Коринантеан 716
Коринантеин 716
Коричная кислота 716, 154
Коричный альдегид 717
Коричный спирт 718
Коронен 718
Короненхинон 718
Корпускулярно-волновая природа микро-
частиц 506
Корракс 735
Корреляция геохимическая 718
Коррозионная кавитация 729
Коррозионная усталость 728
Коррозионная устойчивость металлов 723
Коррозионная эрозия 729
Коррозионное истирание 729
Коррозионное растрескивание 726, 728
Коррозионностойкие материалы 719
Коррозионно-усталостная прочность 729
Коррозионные испытания 720
--- лабораторные 721
--- натурные 721
--- полевые 720
--- производственные 720
--- ускоренные 721
--- эксплуатационные 720
Коррозионные процессы, классифика-
ция 722
— контроль 725
— механизм 722
— характеристика 727
Коррозионные разрушения 726
Коррозионные среды 728
Коррозия металлов 722
— весовой показатель К. 727
— глубинный показатель К. 727
— защита 729
— ингибиторы 228—234
— прочностной показатель 727
— скорость К. 725, 727
Коррозия металлов атмосферная 728
--- в условиях механич. воздействия
728
--- в электролитах 727
--- газовая 723
--- кислотная 727
--- контактная 726
--- межкристаллитная 726
--- местная 726
--- морская 727
--- питтинговая — см. Коррозия ме-
таллов точечная
--- почвенная 727
--- равномерная 726
--- с водородной деполяризацией 724
--- с кислородной деполяризацией 724
— с окислительной деполяризацией
724
--- солевая 727
--- точечная 726
--- химическая 722
--- щелевая 726
--- щелочная 727
--- электрохимическая 724, 727
--- язвенная 726
Кортексолон — см. Рейхштейна веще-
ство S
Кортексон — см. Дезоксикортикостерон
(т. I)
Кортизол — см. Гидрокортизон
Кортизон 730, 405, 732
Кортизон-ацетат 731
Кортикостероиды 732, 405
— биосинтез 734
— фармакология и медицин, применение
К. 734
Кортикостерон 732
Кортин 732
Корунд 734, 139
--- абразивный 735
--- монокристаллический 735
---спеченный 735
Корундин 735
Космохимия 736
Котарнин 737
Коупа перегруппировка — см. Копа пе-
регруппировка
Кофеин 738
Кофермент А 738, 743, 1044
Кофермент Ф 743
Коферменты 740
--- железопорфириновые 742
--- пентидной природы 743
--- флавиновые 743
Коферменты-гормоны 743
Коферменты-нуклеотиды 742
Кошениль 451
Коэзит 821
Коэрцитивная сила 1020
Коэффициент активности электролита
307
•-- Больцмана 701
--- вязкости 61
--- диффузии красителей в волокне 772
--- захвата примеси кристаллами 835
--- измельчаемости (разламываемости)
142
---инжекции 269
--- массоотдачи 333, 334
--- ослабления 91
--- очистки кристаллов 836
--- разделения изотопов 196, 197
--- распределения изотопов 189, 190
--- распределения примеси (при кри-
сталлизации) 835
--- рекомбинации ионов 319, 320
--- рециркуляции (при крекинге) 791
--- теплоотдачи 333, 334, 683
Красители, коэфф, диффузии красителей
в волокне 772
--- азо- 745, 766
--- активные 746, 766,, 768, 774
-— анилиновые — см. Красители син-
тетнч.
--- анионоидные 745
--- арилметановые 767
--- группы родамина 872
--- группы флуоресцеина 872
--- диазотируемые 746
--- для меха 769
--- индигоидные 243
---индолениновые 259
--- катионоидные 745
--- кислотные 765, 770, 780
--- ксантеновые 872
-- кубовые 888, 748, 770, 774
— кубовые растворимые 883
-- нерастворимые в воде 748
-- оксидационные 769
--- основные 766, 775
--- проциниловые 781
---прямые 745, 773
--- растворимые в воде 744
--- сернистые 746, 775
--- синтетические 743
-- солацетовые 783
--- субстантивные — см. Красители
прямые
--- хинониминовые 748, 767
-- хромирующиеся 746
--- хромоксановые 872
Краски 749, 743
--- акварельные 756
--- гуашевые 756
--- декоративные 757
--- керамические 542
--- лаковые — см. Краски эмалевые
--- масляные 749, 753, 756
--- необрастающие 758
--- непревращаемые 751
--- превращаемые 751
--- светящиеся 757
--- темперные 756
--- термочувствительные 757
--- флуоресцирующие 757
--- фосфоресцирующие 757
--- художественные 756
--- эмалевые 749, 753, 754, 755, 756
Красная кровяная соль — см. Калий,
гексацианоферриат
Красные железняки 41
Красящие вещества природные 758
--- алифатич. и алициклич. рядов 758
--- ароматич. ряда 759
--- гетероциклич. ряда 759
Кратных отношений закон 762
Крахмал 762
Крахмальное молоко 764
Крашение, переносчики К. 781
--- ацетатного волокна 783
.— белковых волокон 777
--- волокон пряжи и тканей 773
--- гидратцеллюлозных волокон 777
-- искусственных волокон 765
--- кожи 765, 768
--- меха 768, 770
--- пигментное 772
--- покровное 768
-- полиакрилонитрильных волокон 782
--- полиамидных волокон 779
--- поливинил хлоридных волокон
783
--- полиэфирных волокон 781
--- смешанных волокон 784
--- суспензионное 775
--- текстильных материалов 771, 772
--- химич. волокон 779
--- целлюловных волокон 773, 774,
775, 776
---черноанилиновое 776
Креатин 785
Креатинин 786
Креатинфосфат 1044
Креатинфосфорная кислота 786
Кребса цикл 691
Кревидин 787
Кревол 787
о-Крезолиндофенол 261
Крекинг 788
--- высокотемпературный под низким
давлением (пиролиз) 792
- — каталитический 793
--- под высоким давлением 788
под давлением водорода (гидро-
крекинг) 798
• с водяным паром 798
-- термический 788, 792
Кремень 822
Кремневая кислота 820, 823
Кремневодороды 799, 805, 813
--- вамещенные 815
Кремневольфрамовая кислота 801,
804
Кремнезем — см. Кремний, двуокись
Кремнемезоксалевая кислота 805
Кремнемолибденовая кислота 804, 823
Кремнефториды 801
Кремнефтористоводородная кислота — см.
Кремнефториды
Кремнещавелевая кислота 805
Кремний 802, 29
— аналитич. определение 806
— арсениды 805
— галогениды 816, 805, 813
— двуокись 802, 804, 807, 820
— дисульфид 805
— карбид 818, 425, 428
— кислоты 819
— моноокись 820
— моносульфид 805
— нитрид 805
— окислы 820
— оксигалогениды 817
— оксихлориды 817
— определение 823
— очистка 122
— получение 806
— применение 806
— субгалогениды 817
— субхлорид 817
— тетрабромид 818
— тетрагалогениды 817
— тетраиодид 818
— тетрафторид 817
— тетрахлорид 817
— физич. и химич. свойства 803
— фосфид 805
— фторохлориды 817
Кремний аморфный 804
Кремнийорганические каучуки 807
Кремнийорганические полимеры 809
Кремнийорганические соединения 813,
130
Крёнке реакция 825
Креозот 826
Креозотовое масло — см. Фенольные
/масла
Креолин 826
Крепители литейные 827
Кризанол 828
Криогидратная точка 828
Криогидраты 828
Криолит 828
Криоскопическая постоянная 829
Криоскопия 829
Криофилит 977
Криптоксантин 453, 455
Криптон 830, 266
— критич. постоянные 866
Криптоцианин 830
Кристаллвиолет — см. Кристаллический
фиолетовый
Кристаллизаторы 838
Кристаллизационная вода 846
Кристаллизация 831, 840
--- высокомолекулярных соединений—
см. Кристаллическое состояние поли-
меров
--- двукратная 836
--- из газовой фазы 832
--- однократная 836
--- полимеров 843
---расплава переохлажденного 832
--- раствора переохлажденного 832
Кристаллиты 841
Кристаллическая решетка 840, 851
Кристаллическая структура 852
--- лантанидов 917
Кристаллический фиолетовый 839, 250
Кристаллического поля теория 468
Кристаллическое состояние 839
---полимеров 841
Кристаллогидраты 846, 828
Кристаллография 846
Кристаллолюминесценция 863
Кристаллофивика 846
Кристаллофосфоры 863
Кристаллохимический анализ 847
Кристаллохимия 846, 853
— основной закон К. 858
Кристаллы 846, 840
— дислокации в К. 834
— люминесценция 863
— магнитные свойства 862
— механич. свойства 860
— образование и рост 833, 834
— оптич. свойства 862
— постоянство углов К. 847
рациональность параметров 847
— симметрия 847—853
— спайность К. 859
— тепловые свойства 860
— физич. свойства 859
— электрич. свойства 860
— энантиоморфизм К. 848
Кристаллы валентные (атомные) 841
--- глобулярные 843
--- двухосные 862
--- диамагнитные 862
---жидкие — см. Жидкие кристаллы
--- ионные 841
--- молекулярные 841
--- оптически активные 863
--- парамагнитные 862
--- полимерные 843
1069 —1070
--- энантиоморфные 848
Кристобалит 821
Критическая изотерма 866
Критическая опалесценция 866
Критическая плотность 865
Критическая температура 865
--- растворения 863, 55
Критическая точка 865
--- растворения 55
Критический объем 865
Критическое давление 865
Критическое пересыщение пара 682
Критическое состояние 865
Кровельные материалы 536
Кровяная соль 867
Кроны 867
--- бариевые 869
--- свинцово-молибдатные 868
--- свинцовые 867, 868
--- стронциевые 868
--- цинковые 868
Кросс-реакция (перекрестная) 220
Кротоназа 869
Кротоновая кислота 869
Кротоновая конденсация 870, 679, 681
Кротоновый альдегид 871
Кроцетин 455
Кроющая способность пигмента 867
Круговые процессы (или циилы) 871
Ксантин 872
Ксантиноксидаза 873
Ксантогенат целлюлозы 874
Ксантогеновая реакция 875
Ксантогеновые кислоты 876, 875
Ксантон 877, 760
Ксантопротеиновая реакция 877
Ксантоптерин 878
Ксантофиллы — см. Каротиноиды
Ксенон 878, 266
— критич. постоянные 866
Ксенотим 337, 925
Ксерогели 879
Ксероформ 879
Ксикаин — см. Ксилокаин
Ксипаны 881
Ксиленол 879
Ксиленоловый оранжевый 253
Ксилидины 879
Ксилил — см. Тринитроксилол
Ксилит 880, 881
Ксилоза 881
Ксилокаин 882, 944
Ксилол 882
Ксилоновые кислоты 881
Куб (в крашении) 884
Кубическая сингония симметрии (в крп-
сталлогр.) 849
Кубовые красители 883, 746, 770, 774
Кубовые красители растворимые 883
«Кубовый синий 0» — см. Индантрон
Кубозоли 775, 779, 884, 939
Кузбасс-лак 902
Кукурузное масло — см. Жиры расти-
тельные
Кулометр 886
Кулоновский интеграл 528, 531
Кулонометрический анализ 886
Кулонометрическое титрование 886
Кулонометрия 886
Кумарин 887
Кумариновая кислота — см. о-Кумаро-
вая кислота
о-Кумаровая кислота 888
Кумароновая смола 888
Кумарононы 759
Кумахлор 122
Куминаль — см. Куминовый альдегид
Куминовый альдегид 889
Кумол — см. Изопропилбензол
Кумулены 889
Кумулированные связи 890
Кумулятивный варяд 890
Кумулятивный эффект 890
Куна — Фрейденберга правило 892
Кунжутное масло — см. Жиры расти-
тельные
Купоросное масло — см. Серная кисло-
та
Купоросы 893
Купферон 893
Курареподобные вещества 945
Куркумии 759
Курциуса реакция 893
«Курчииодистый висмут» 693
Кучерова реакция 895
Кьельдаля метод 897
Кюри—Вейсса закон 1017
1071 -1072
Кюри закон 1016
Кюри точки 861, 1019
Кюрий 898
Л
Лавандовое масло — см. Эфирные масла
Лавсан — см. Полиэфирные волокна
Лагодезоксихолевая кислота 49
Ладенбурга реакция 899
Ладенбурга — Вышнеградского реакция
899
Лаки 900
--- алкидно-стирольные 903
---алкидные 903
--- ацетилцеллюлозные 905
--- ацетобутиратцеллюлозные 905
--- гликоль-малеинатные 903
--- дивинилацетиленовый 904
--- жирные 901
-—- кремнийорганические 905
-- летучие 901, 902
---масляные 901
--- меламино-формальдегидные 904
--- мочевино-формальдегидные 904
— нитроцеллюлозные 905
--- перхлорвиниловые 902
--- полиакриловые 902
---поливинилацетальные 902
--- полисилоксановые — см. Лаки крем-
нийорганические
--- полиуретановые 903
--- полиэфир-акрилатные 903
--- полиэфирные 903
--- силиконовые — см. Лаки кремний-
органические
--- смоляные 902
--- спиртовые 902
--- тощие 901
--- феноло-альдегпдные 904
--- хлоркаучуковые 902
--- эпоксидные 904
--- этилцеллюлозные ‘905
— - эфироцеллюлозные 905
Лакмус 905
Лактамы 906
Лактатдегидрогеназа 909
Лактикодегидрогеназа — см. Лактатде-
гидрогеназа
о-Лактоальбумин 910
р-Лактоглобулин 910, 211
Лактоза 910
Лактонов вращения правило — см. Лак-
тонов правило
Лактонов правило 911
Лактоны 912
Лаллеманции масло — см. Жиры расти-
тельные
Ламинаран 914
Ламинараназы 915
Ламинарибиоза 915
Ламинарии — см. Ламинаран
Ламинариназы — см. Ламинараназы
Лангбейнит 348, 358, 1027
Ланолин — см. Воски (т. I)
Лантан 915, 916, 917, 918
— карбид 425
— окислы 920
Лантаниды 915
— анализ, методы 927
— бромиды 921
— валентность 917
— галогениды 921
— гидриды 921
— гидроокиси 921
— иодиды 921
— ионы, свойства 917
— источники сырья и извлечение Л. 923
— карбиды 921
— карбонаты 922
— комплексные соединения Л. 922
— нитраты 922
— нитриды 921
— окислы 920
— оксалаты 922
— оксигалогениды 921
— получение 924, 926
— применение 928
— разделение 924
— соединения Л., выделяемых из ра-
створов 921
— структура кристаллич. 917
— структура электронная 917
— сульфаты 922
— сульфиды 921
— феррицианиды 922
— физич. и химич. свойства 916, 920
— фосфаты 922
фториды 921, 922
— хлориды 921, 922
Лантаноиды — см. Лантаниды
Латекс натуральный 929, 491
Латексы дивинил-акрилатные 931
---дивинил-винилпиридиновые 931
--- дивинил-метакрилатные 931
---дивинил-метилвинилпиридиновые
931
--- дивинил-нитрильные 931
--- дивинил-стирольные 931
--- синтетические 930, 498
--- хлоропреновые 932
Латунь — см. Медь, сплавы
Лаурилмеркаптан — см. Додсцилмеркап-
тан (т. I)
Лауриновая кислота 932
Лауриновый альдегид 933
Лауриновый спирт 933
Леватиты 303
Левомицетин 933
Левопимаровая кислота 934, 53, 587
Левулиновая кислота 935
Левулиновый альдегид 936
Легированные стали — см. Железо,
сплавы
Легирующие элементы 25, 27
Легкое масло — см. Каменноугольная
смола
Ледебурит 21
Лейкарта реакции 936
Лейкеран — см. Хлорамбуцил
Лейкоген 937
Лейкоиндиго 241
Лейкомицин — см. Магнамицин
Лейконит 598
Лейкооснования — см. Лейкосоединения
Лейкоитерин 937
Лейкосапфир 735
Лейкосоединения 938, 883, 884
Лейкотропы 939
Лейции 939
Лейцинаминопептидаза 940
Лекарственные вещества 940
--- анальгезирующие (болеутоляющие)
943
--- адренолитические 944
--- адреномиметические 944, 945
--- алкилирующие 948
--- антихолинэстеразные 945
—- возбуждающие 944
--- ганглиоблокирующие 945
--- действующие на медиаторные про-
цессы 944
---курареподобиые 945
--- местноанестезирующие 944
---наркотические 942
--- нейроплегпческие 943
--- противосудорожные 942
--- седативные 943
--- синтетические 940
--- снотворные 942
--- спазмолитические 946
---стимулирующие центральную нерв-
ную систему 944
--- холинолитические 943, 944
--- холиномиметические 944
Лекланше элемент — см. Химические
источники тока
Лемонграссовое масло — см. Эфирные
м.асла
Леморан 949, 943
Леонит 358
Лепидин 949
Лепидолит 977
Лесохимические производства 950
Лесохимия 953
Леттса синтез 953
Летучесть — см. Фугитивность
Лецитиназы 953, 956
Лецитины 954, 639
Ле Шателье принцип 956
Либермана аллокоричная кислота 716
--- изокоричная кислота 716
--- реакция 957
Либермана — Шторха реакция 131, 132
Лиганды 958, 532 — см. также Комплекс-
ные соединения
Лигнин 958, 131, 693
Лигностон — см. Древесные пластики
(т. I)
Лигносульфонаты 962
Лигносульфоновые кислоты 962, 961
Лигроин 963
Лизергиновая кислота 963
Лизин 965
Лизис (в иммунохимии) 224
а-Лизокефалин 954
Лизоколаминфосфатиды 639
Лизол 965
Лизолецитиназа В — см. Лецитиназы
Лизолицетины 954, 956
Лизофосфолиназа — см. Лецитиназы
Лизоцимы 965
Ликвидус и солидус 966
Ликопин 455
Ликорин 967
Лимациды 967
Лимонен 967
Лимонит — см. Гидрогетит
Лимонная кислота 968
Лимонное масло — см. Эфирные масла
Линалилацетат 969
Линалоол 968
Линдан — см. Гексахлоран (т. I)
Линеарные системы 689
Линейная передача энергии (ЛПЭ) —
см. Передача энергии линейная
Линейной комбинации атомных орбит
метод 527, 531
Линол 969
Линолевая кислота 970, 75
Линоленовая кислота 970, 75
Лиотропные ряды 970
Лиотропные числа 972
Лиофильность и лиофобность коллоидов
972
Лиофобные коллоиды 972, 211
Липазы 972
Липиды 973
Липовица сплав 346
Липоевая кислота 974, 743
Лппозитол 274
Липоиды — см. Липиды
Линооксидаза 975
Липопротеидлипаза 973
Липопротеиды 975, 955
Липопротеины — см. Липопротеиды
Липохромы — см. Каротиноиды
Липтобиолиты 330, 331
Литейные крепители 827
Литий 977
— аланат — см. Литий, алюмогидрид
— алюмогидрид 986
— амид 986
— аналитич. определение 981
— боргидрид 988
— бромид 987
— галогениды 986
— гидрид 988
— гидроокись 990
— иодид 988
— карбид 979
— карбонат 989
— моносиликат 979
— нитрат 990
— нитрид 979
— нитрит 979
— окись 990
— ортофосфат 979
— перекись 978
— полисиликаты 979
— получение 981
— применение 983
— силицид 979
— сплавы 979
— сульфат 991
— сульфид 979
— физич. и химич. свойства 978
— фосфат 979
— фосфид 979
— фторид 986, 284
— хлорид 987
Литий азотнокислый — см. Литий, нит-
рат
--- сернокислый — см. Литий, сульфат
---углекислый — см. Литий, карбонат
Литийорганические соединения 984
Литопон — см. Пигменты белые
Литосфера 991
Литофильные элементы — см. Геохими-
ческая классификация элементов (т. 1)
Литохолевая кислота 49
Лобеланидин 992
Лобеланин 992
Лобелии 992
Лобри де Бруина и Ван Экенштейна реак-
ции 992
Лопарит 924, 925
Лоретин — см. 7-Иод-8-оксихинолин-5-
сульфокислота
Лоссеня перегруппировка 993
Лоссеня проба — см. Азот, определение
(т. I)
Лоуренсий 994
Лужение 97
Лупинин 994
Лутидин — см. Метилпиридины
1073 -1074
Льняное масло — см. Жиры раститель-
ные
Льюисовские кислоты и основания 582,
586
Лэнгмюра уравнение изотермы адсорбции
994
Люиэит 996
Люминал 997, 942
Люминесцентный анализ 997
-- сортовой 998
--- химический 998
Люминесценция 997
--- кристаллов 863
Люминол 254
Лютеостерон — см. Прогестерон
Лютеций — см. Лантаниды
Люцигенин 254
Люциферазы 999
Люциферин 1000
м
Магналий 980
Магнамицин 1001
Магнезит 439, 1009, 1022
--- каустический 1024
Магнезия белая 1022
--- жженая — см. Магний, окись
Магнезоны 1002
Магнетит 18, 41
Магнетон Бора 519, 1017
Магнетохимия 1003
Магний 1008
— аналитич. определение 1011
— гидрид 1010
— гидроокись 1024
— гидросульфид 1028
— карбид 425, 1010
— карбонаты 1022, 439
— нитрид 1010
— окись 1023
— перхлорат — см. Ангидрон (т. I)
— получение 1011
— применение 1012
— силицид 805
— сплавы 1024
— сульфат 1026
— сульфид 1027
— техника безопасности 1012
— физич. и химич. свойства 1009
— хлорид 1028
Магний сернистый — см. Магний, сульфид
--- сернокислый — см. Магний, сульфат
--- хлористый — см. Магний, хлорид
Магнийорганические соединения 1012
Магнитная восприимчивость — см. Маг-
нитные свойства, Магнетохимия
Магнитная вязкость 1020
Магнитное насыщение — см. Намагничен-
ность насыщения
Магнитное поле 1014
Магнитные железняки 41
Магнитные свойства 1014, 1003
--- кристаллов 862
Магнитный момент — см. Магнитные
свойства
Магнитный резонанс — см. Радиоспектро-
скопия
Маделунга реакция 1029
Мазут 1029
Маисовое масло — см. Жиры раститель-
ные
Майера реакции ЮЗО
Майонез 1032
Маклурин 1032
Маковое масло — см. Жиры раститель-
ные
Макромолекула 1032
— конформации М. 1033
— физич. и химич. свойства 1036
Макромолекулы линейные 1033
—- разветвленные 1033
--- регулярные 1035
Макрорадикалы свободные 1037
Макроциклические соединения 1039
Макроэргические связи 1042
Максвелла — Больцмана закон распре-
деления 1045
Максимальная работа реакции 1046
Мак-фадиена — Стивенса реакция 1047
Малатдегидрогенаэа 1047
Малахит 439
Малеиновая и фумаровая к-ты 1048, 154
Малеиновый ангидрид 1049
Маликодегидрогеназа — см. Малатдегид-
рогенаэа
Малоновая кислота 1050
Малоновый эфир 421 —см. также Диэтил-
малонат (т. I)
Марассе метод 652
Марганец 29 .
— бромид, магнитная восприимчивость
1022
— карбид 425
— фосфат, магнитная восприимчивость
1022
Марказит 38
Марсилид — см. Ипраэид
Мартенсит 21
Мартит 41
Масла растительные жирные — см. Жи-
ры растительные
Маслоемкость пигмента 868
Масса атома изотопа 187, 202
Массовое число 202
Масс-спектрометры для анализа изотопов
стабильных 200
Мастики кислотоупорные — см. Кислото-
упорные замазки
Матирующие вещества 751
Матричные теории 223
Медина л 942
Межгщисталлитная прослойка 840
Мезобилирубин 761
Мезоформы 156
Мейера реакция — см. Майера реакция
Мекамиламин 945
Мел 371, 374, 380
Мелфалан 402
Мельницы 140, 141
Мембранное равновесие 308
Менделеева — Клапейрона уравнение —
см. Клапейрона уравнение
Мениск 409, 410, 411
Меншуткина реакция 185
Мепаэин 943
Мепротан 943
Мергели 137
Меридил 944
2-Меркаптобензтиаэол — см. Каптакс
6-Меркаптопурин 40 4
6-Меркапто-9-₽-Б-рибофураноэилпурин
404
Меркаптотетраэолл 179
Меркаптофос 277
Метадикремневая кислота 820
Метакремневая кислота 820
Металлизация 98
Металлиндикаторы 670
Метальдегид 967
Метамерия 153
--- ионизационная 664
Метан, конверсия 675
— критич. постоянные 867
Метандикарбоновая кислота — см. Мало-
новая кислота
Метапериодаты 286
Р-Метилакролеин — см. Кротоновый аль-
дегид
N-Метил-п-аминоазобенэол 401
2-Метилбутадиен-1,3 — см. Изопрен
2-Метилбутан 165
Метилглиоксаль, монооксим — см. Иэо-
нитроэоацетон
N-Метилгуанидинуксусная кислота — см.
Креатин
4-Метил-4'-диметиламиноаэобенэол 401
Мтилен 423
3,3"-Метилеибис-4-оксикумарин — см.
Дикумарол .
3,4-Метилендиоксипропенилбенэол — см.
Изосафлор
Метиленовый синий 252
1-Метилиэатин 135
1-Метил-4-иэопропенилциклогексен-1 —
см. Лимонен
1-Метил-4-изопропенил-Д’-циклогексен-
2-он — см. Карвон
2-Метил-5-иэопропилфенол — см. Кар-
вакрол
З-Метилиндол 257
Метилиононы 313
Р-Метилкарбоновая кислота — см. Кро-
тоновая кислота
1-Метилнафталии 386
2-Метилнафталин 386
8-Метилнонен-6-овая кислота, ванилил-
амид — см. Капсаицин
Метиловый красный 248
Метиловый оранжевый 248
Метиловый спирт, критич. постоянные
867
Метиловый фиолетовый 250
Метил-оисибенэолы — см. Крезол
6-Метилпурин 404
Метилсилан 814
6-Метилсульфонилурацил 404
Метилтестостерон 405
2-(Метилтио)-аденин 405
6-(Метилтио)-пурин 404
Метилтриметоксисилан 815
Метилтрихлорсилан 814
Метилтриэтоксисилан 815
Р-Метилумбеллиферон 250
1 -М етил-2-фенацил-6-(2'-фени л-2 '-окси-
этип)-пиперидин — см. Лобелии
Метилфосфиновая кислота, изопропило-
вый эфир фторангидрида — см. Зарин
— пинаколиновый эфир фторангидри-
да —• см. Зоман
у-Метилхинолин — см. Лепидин
4-Метилхинолин — см. Лепидин
20-Метилхолантрен 399, 400
Метилцеллюлоза 132
Р, Р-Метипэтилглутаримид — см. Бемег-
рид
Метимеркаптофос 277
4-Метокси-5-бензоилацетилбензофуран —
см. Понгамон
Метокси дифторметилизоцианат 207
2-Метокси-4 (пропенилфенол) — см. Изо-
эвгенол
Метсистокс 277
Меченых соединений синтез 183
Миелосан — см. Милеран
Микролит 735
Микрочастицы, корпускулярно-волновая
природа 506
— состояние 509
Милеран 403
Милори — см. Железная лазурь
Миндальное масло 73, 75
Минералокортикоиды 946
Миристиновая кислота 75
Митин ФФ 276
Митомицин С 405
Михаэлиса метод 249
Михаэлиса уравнение 147
Мицеллы 109
Модификаторы катализаторов 471
Модифицирование катализаторов 460,
487
--- кристаллизации 834
--- химическое поверхности 412
Модальность 708
Молекулы, потенциал ионизации 297;
степень изоморфизма 162
--- «энергетиэированные» 563
Молекулярный вес, определение 829
Молекулярных орбит метод 527, 530
Молибден 28
— карбид 425
Молоко 63
Молочный сахар — см. Лактоза
Мольная доля 707
Молярная теплота испарения 331
Молярность 708
Монацит 923, 925
Моноаэокрасители 766
Моногалогенокарбены 424
Монокальцийфосфат 383
Моноклинная сингония симметрии (в
кристаллогр.) 849
Монокристаллы 840
--- полимеров 843
Мономолекулярные реакции 563, 565
Мононитрокоронен 718
Монофосфоиноэитиды — см. Фосфатидп-
линоэпты
Моноэдрический вид симметрии 850
Монтепонит 342
Мора соль 37, 38
Морфин 943
Мостиковые соединения 709
Моющее действие 54
Мрамор 137, 371
Муллит 536
Мульдера реакция — см. Ксантопротеи-
новая реакция
Мультиплетная теория катализа 464
Мыло канифольное 395, 396
н
Наждак 735
Намагниченность 1014
--- насыщения 1003, 1005, 1015
--- спонтанная 1019
Напалм 77
Наполнители (в составе красок) 749, 751
Наркотические вещества 942
Натрий, кремнефторид 801
— фторацетат 122
Натрий хлористый, магнитная восприим-
чивость 1021
1075 — 1076
Натрийкамфара 390
Натсин — см. Изопреновый каучук
Нафталин 210, 386, 387, 689
— магнитная восприимчивость 1021
Нафтацен 399, 689
а-Нафтиламин 250
р-Нафтиламин 399
1,5-Нафтилендиизоцианат 207
1-Нафтилизоцианат 207
а-Нафтилтиомочевина 122
2-Нафтол-6,8-дисульфокислота 250
Нафтохиноны 759
Невьянскит 326
Нейлон 176
Нейроплегические вещества 943
Нейтрино 203
Нейтроны 201, 203
• — дозы облучения 90
— защита от облучения 92
Неоабиетиновая кислота 53, 587
Неодим 916
Неон 266
— критич. постоянные 866
Неопентан — см. 2,2-Диметилпропан
Неопределенностей соотношения 508
Неорганические вещества, идентифика-
ция 127
Неоцид 405
Непрерывных изменений метод — см.
Изомолярных серий метод
Неэпя точка 1019
Нигрозины 767
Никель, карбонил 439, 441
— карбонилгидрид 441
— магнитные свойства 1022
Никетамид — см. Кордиамин
Никотинамидадениндинуклеотид 622
Никотинамидаденинди нуклеотидфосфат
622
Никотиновая кислота, диэтиламид 715
Нимоник 15
Ниобий, карбид 425
Нитранол 946
о--Нитроанилин 250
п-Нитробензолазо-а-нафтол — см. Магне-
зоны
п-Нитробензолазорезорцин — см. Магне-
зоны
Нитроглицерин 946
п-Нитродиазоаминоазобензол — см. Ка-
дионы
n-Нитро диазо амино азо бен зо лсуль фокис-
лота, динатриеван соль — см. Кадионы
Нитрозокарвакрол 448
N-Нитрозофенилгидроксиламин, аммо-
нийная соль — см. Купферон
5-НитроиЗохинолин 205
Нитрокамфаны 389
З-Нитрокарбазол 419
Нитролаки — см. Лаки нитроцеллюлоз-
ные
Нитромин 402
Нитрон 825
Нитропентон 946
Нитроцеллюлоза 130
Нихром 15
Новаин — см. Карнитин
Новокаин 944
Новоэмбихин 402, 948
Норадреналин 944, 945
Норлобеланидин 992
Норлобеланин 992
Нутриахолеван кислота 49
Нормальность 708
Носители 470, 484
Нуклеозидполлфосфаты 1043
Нуклеотиды, коферменты-Н 742
Нуклеофильный реагент — см. Донор
Нуклиды 202, 204
Нуклоны 201, 203
Нулевая энергия 516
Нулевого заряда точка 211
О
Обермейера реакция 247
Облицовочные материалы 536
Обменная энергия 522
Обменное взаимодействие 522
Обменные силы в квантовой механике
1019
«Объединенного атома» метод 532
Объемная доля 707
Объемный анализ 641
Одежда защитная 93, 85
Оже реакция 1031
Окраски, закрепление 78
Оксаль-ацетат-транс-ацетаза — см. Кон-
денсирующий фермент
п-Оксиабенэол 250
З-Оксииндол — см. Индоксил
З-Оксииндол-Р-О-глюкозид — см. Инди-
кан
транс-о-Оксикоричная кислота — см.
о-Кумаровая кислота
о-Оксикоричная кислота, лактон — см.
Кумарин
19-Оксикортикостерон 732
Ь-3-Окси-№-метилморфинан, битартрат—•
см. Леморан
1-Окси-2-метокси-4-пропенилбензол —
см. Изоэвгенол
3-(4-Окси-3-метоксифенил)-2-пропен-1-ол—
см. Конифериловый спирт
Оксипираконовая кислота 336
17 а-Оксипрогестерон 734
2-Оксихинолин — см. Карбостирил
у-Окси-а-хинолинкарбоновая кислота —
см. Кинуренован кислота
4-Оксициннамоилферулоилметан 759
а-Оксоглутаровая кислота — см. а-Ке-
тоглутаровая кислота
1-Оксо-9-метилфеназин — см. Пиоцианин
Октадецилизоцианат 207
Октаметил 277
и-Октан, критич. постоянные 867
Октаналь — см. Каприловый альдегид
Октановая кислота — см. Каприловая
кислота
Олеиновая кислота 75
Олеокризии — см. Кризанол
Оливин — см. Форстерит
Оливковое масло 73, 75
Олово хлорное, критич. постоянные 866
Омаии 403
Омыления число 65, 69, 73
Ониевые соединения 443
Операторы (в квантовой механике) 513
Оптическая активность 155, 175
Оптическая изомерия 153, 155
--- комплексных соединений 662
Оптическая ось кристаллов 862
Оптическая плотность 283
Оптически-активные соединения 155, 156
Оптические антиподы 155
Оптические свойства кристаллов 862
Оптического сдвига правило 892
Орбитальный момент 1015
Органические реактивы 408
Органические соединения, влияние кон-
формации на физико-химич. свойства
О. с. 703
— идентификация 127, 999
— критич. постоянные 867
Органозоли 109
Ореховое масло 73, 75
Ортит 925
Ортокремневая кислота 819
— эфиры 814, 816
Ортопериодаты 286
Оси симметрии (в кристаллогр.) 848
--- трансляций (в кристаллогр.) 851
Осмий иридистый 326
Основания — см. Кислоты и основания
Основности константа 583
Островные структуры 856
Остромысленского — Жоба метод — см.
Изомолярных серий метод
Осциллятор гармонический 516
--- квантовый 516
--- классический 516
Отавит 342
Отбеливающие земли 106
Относительная биологическая эффектив-
ность (ОБЭ) 89, 90
Отравление катализатора 460, 487
Открываемый минимум 408
Павинол 629
Пальмитиновая кислота 75
Пальмовое масло 73, 75
Пальмоядровое масло 73, 75
Палюстровая кислота 587
ПАН (индикатор) 253
ПАР (индикатор) 253
Пар, давление в жидких системах 56
— конденсация 682
Параказеин 347
Параллелепипед повторяемости (в кри-
сталлогр.) 852
Парамагнетизм 1004, 1015, 1016
Парамагнитная восприимчивость 1016
Парамион 945
Параоксидифениламин 235
Парапериодаты 286
Парижская синяя — см. Железная ла-
зурь
Парообразование — см. Испарение
Паскаля схема (в магнетохимии) 1006
Паули принцип 521, 528
Пек древесный 827
--- каменноугольный 387, 388, 632
Пемза шлаковая 81
Пемпидин 945
Пентагонтриоктаэдрический вид симмет-
рии 850
Пентагонтритетраэдрический вид симмет-
рии 850
н-Пентакозан 974
Пентаметилендиамин — см. Кадаверин
а, р-Пентаметилентетразол — см. Кора-
зол
и-Пентан, критич. постоянные 867
Пентанон-4-ал-1 — см. Левулиновый аль-
дегид
2, 3’, 4, 4’, 6-Пентаоксибензофенон —
см. Маклурин
3, 5, 7, 3’, 4’-Певтаоксифлавон — см.
Кверцетин
Пентацен 689
Пентаэритрол, тетранитрат — см. Нитро-
пентон
Перегруппировка
---Вагнера — Меервейна 392
--- камфеновые 392
--- Клайзена 589
--- Копа 712
--- Лоссеня 993
--- пинаколиновая 392
--- ретропинаколиновая 392
Перезарядка 317
--- резонансная 318
--- эндотермическая 319
Перекрестная реакция 220
Переноса процессы (в кинетике химич.)
566
«Переносчики» крашения 781
Переход тяжелых частиц 317
Переходное состояние 565
Периклаз 102 3
Перилловое масло 73, 75
Период полураспада 203
Перйодаты 285, 286
Перлит 21
---вспученный 81
Пермутиты 300
Перовскит 371
Пероксидаза 742
Персиковое масло 73, 75
Перспективные формулы 698
Перхлорвинил 131
Пестокс 277
Петалит 977
2-Пивалилиндандион-1,3 — см. Пиваль
Пиваль 122, 123
Пигменты 743, 747, 749, 750
— кроющая способность 867
— маслоемкость 867
Пигменты желчные 50
--- керамические 542
Пимантрен 588
а-Пимаровая кислота 587
Пинакоидальный вид симметрии 850
Пинаколиновая и ретропинаколиновая
перегруппировка 392
а-Пииен 390
Пинит 273
Пиоцианин 761
а-Пиперидон — см. б-Валеролактам
Пирен 386, 399, 690
2-Пиридилуксусная кислота 434
Пиридин 386
Пиридинийфлуоренилид 421
Пиридинийциклопентадиенилид 213, 421
Пиридоксальфосфат 741, 743
Пиридрол 944
Пирит 38
Пировиноградная кислота 431
Пирозоли 110
Пирокатехиновый фиолетовый 253
Пирокремневая кислота 820
Пирофосфокиназы 558
Пирофосфорная кислота, октаметилтетра-
амид — см. Октаметил
— эфиры 431
Пирохлор 371
Пироэлектричество 861
Пиррол, производные 761
Пирролидон — см. у-Бутиролактам
Плавиковый шпат — см. Флюорит
Плазмин 108
Пластификаторы 751
Пласткожа 628
1077 —1078
Платина, магнитная восприимчивость
1022
Платинит 27
Пленкообразующее вещество 749, 751
Плеохроизм 863
Плотнейшие упаковки 857
Плотность вероятности локализации
электрона 511
— тока 560
Повеллит 371
Подсолнечное масло 73, 75
Полевой шпат 139
Поленске число 65, 69, 73
Полиазокрасители 766
Полиазы 429
Полиакрилаты 130
Полиакриловые эфиры 131
Полиакрилонитрил 131
Полиалюмоорганосилоксаны 809, 810
Полиамиды 130, 131, 132
Полиацены 689
Полибутадиен атактический 131
Полибутен-1 175
Поливинилацетали 131, 132
Поливинилацетат 130, 131, 132
Поливинилбутираль 101, 130, 131, 132
Поливинилиденхлорид 130, 131, 132
Поливиниловый спирт 130, 131, 132
Поливинилформаль 130, 131, 132
Поливинилхлорид 130, 131, 132
Поливинилхлоридацетаты 131
Полигалит 358, 1027
Полигексен-1 175
Полидиметилсилоксан 812
Полидиметилфенилсилоксан 812
Полидисперсность 1032
Полиизобутилен 130, 131, 132
Полиизопрен — см. Изопреновый кау-
чук
Полииодбензолы 289
Полииодиды 286
Полииодфенолы 295
Поликапролактам 101
Поликарбонаты 130, 131, 132
Поликонденсация 682
Поликоординация 710
Поликраз 925
Полимеризация анионная 422
--- эмульсионная 644
Полимерные иониты 301
Полимерные радикалы — см. Макрора-
дикалы свободные
Полимеры, деструкция 1039
— идентификация 128
— кристаллизация 843
— кристаллич. состояние 841
— оптическая активность 175
— плотность 128, 131
— цветные реакции на П. 131
Полимеры диизотактические 175
--- изотактические 173
-- клешневидные — см. Полимеры
координационные
---координационные 710
--- кремнийорганические 809
--- привитые 1038
--- трео-диизотактические 176
---хелатные — см. Полимеры коорди-
национные
--- эритро-диизотактнческие 176
Полиметакрилаты 130
Полиметаллоорганосилоксаны 811, 812
Полиметилакрилат 132
Полиметилметакрилат 131, 132
Поли-З-метилбутен-1 175
Поли-4-метилгексен-1 175
Поли-4-метилпентен-1 175
Полимолекулярность — см. Полидисперс-
ность •
Полиорганосилазаносилоксаны 809
Полиорганосилазаны 809
Полиорганосилоксановые каучуки — см.
Кремнийорганические каучуки
Полиорганосилоксаны 809, 811
Полипентен-1 17$
Полипропилен 130, 131, 132, 175, 176
Полипропиленоксид 175
Полирадикалы 1037
Полисоединения 657
Полистирол 130, 131, 132, 175
Политетрафторэтилен 130, 131, 132
Полигитаноорганосилоксаны 809
Политрифторхлорэтилен 131
Полиуретаны 130
Полифенилсилоксаи 810, 812
Полифенолы 235
Полиформальдегид 130, 132
Полифосфаты 1042
Полихлоркамфен 391
Полиэтилен 130, 131, 132
— конформации 705
— кристаллизация 842
Полупроводники, каталитическая актив-,
ность 466, 467, 483
Полуфарфоровые изделия 537, 538, 539
Понгамон 759
Поролон 628
Порфирины 761
Порядок ближний 60
--- дальний 840
--- оси симметрии (в кристаллогр.) 848
--- реакции 563
Поташ — см. Калий, карбонат
Потенциал ионизации элементов 295
--- молекул 297
--- радикалов 297
Потенциальная яма 516
Потенциальный барьер 516
--- свободного вращения 702
Поясов закон (в кристаллогр.) 847
Правильная система точек (в кристал-
логр.) 852
Празеодим 916
Д’-Прегнендиол-l 1 р,21-аль-18-дион-3,20—
см. Альдостерон
Д4-Прегнендиол-11Р,21-дион-3,20 — см.
Кортикостерон
Д4-Прегнендиол-17а,21-дион-3,20 — см.
Рейхштейна вещество S 732
Д4-Прегнендиол-17а, 21-трион 3,11,20 —
см. Кортизон
Д4-Прегненол-21-дион-3,20 — см. Дезокси-
кортикостерон
Прегненолон 734
Д4-Прегненол-21-трион-3,11,20 — см.
11-Дегидрокортикостерон
Д4-Прегнентриол-11 а, 17а, 21-дион-3,20—
см. -Эпигидрокортизон
Д4-Прегнентриол-11 ₽, 17 а,21-дион-3,20 —
см. Гидрокортизон
Д4-Прегнентриол-11р, 19, 21-дион-3,20 —
см. 19-Оксикортикостерон
Предел ползучести 13
Предельная концентрация 502
Предельно допустимые дозы облучения 90
Предельное разбавление 408
Предельный заряд инициирующих ВВ 270
Преднизолон 405, 732, 947
Преднизон 405, 732
Предэкспоненцнальный фактор 564
Препарат (закрепитель окрасок), 79
Преципитация 220, 224
Преципитины 220
Призматический вид симметрии 850
Призмы для инфракрасной области 284
Примвероза 881
Приставка изо- 144
Проекционные формулы 698
Проколлаген 642
Промедол 943
Прометий 916
Промотирование катализаторов 460
Промоторы 460, 470
Промурит 122
Пропазин 943
м-Пропан, критич. постоянные 867
Пропанол-2 — см. Изопропиловый спирт
Пропердин 109
н-Пропиловый спирт, критич. постоян-
ные 867
р-Пропиолактам 906
Р-Пропиолактон 912
Просилоксан 817
Пространственная решетка — см. Кри-
сталлическая решетка
Пространственные (федоровские) группы
симметрии (в кристаллогр.) 852
Протеиназы 108
Противогазы промышленные 83, 84
Противогистаминные средства 946
Противостарители резины 493, 497
Противосудорожные вещества 942
Протокатеховая кислота 693
Протолиза константа 583
Протолитическая реакция 479, 581
Протолитическое кислотно-основное рав-
новесие 583
Протоны 201, 203
Протромбин 108
Прототропия 490
Проферменты 108
Процинилы 748
Проционы 746
Прямолинейного диаметра правило 55
Псевдогиперицин 759
Псевдоиндолы — см. Индоленины
Пси-функция — см. Волновая функция
Психолептики — см. Стимуляторы цен-
тральной нервной системы
Птеридин 762
Пульвиновая кислота, лактон 759
Пурин 404
Пурпур античный (Тирийский) — см.
6,6'-Д иброминдиго
Пьезоэлектричество 861
Р
Равновесие адсорбционное 994
--- кислотно-основное (в организме) 579
--- кислотно-основное протолитическое
583
---химическое 560
Равновесия подвижного принцип — см.
Ле Шателье принцип
Радиационный захват электрона 319
Радикалы, потенциал ионизации 297
---полимерные — см. Макрорадикалы
свободные
--- свободные 323, 1037
Радикальные реакции 474
Радикальные центры 1037
Радиоавтография 183
Радиоактивность 203
Радиоактивные вещества, защита от из-
лучений 89
Радиолюминесценция кристаллов 863
Радон 266
Разбавления метод (в колориметрич. ана-
лизе) 649
Рамана эффект — см. Комбинационное
рассеяние света
Рамнетин 534
Рапсовое масло 73, 75
Рассеяние света антистоксово 653
--- комбинационное 653
--- стоксово 653
Раствор Рингера 180
--- Рингера — Локка 180
--- Рингера — Тироде 180
Растворение, критич. темп-ра 863, 55
— критич. точка 55
Растворимость жидкостей взаимная 55
Растворы, ионная сила 307
--- гипертонические 179
--- гипотонические 179
--- изоосмотические — см. Растворы
изотонические
--- изотонические 179
--- физиологические 180
Ратиндан 239
Рауля закон 56
Рацематы 155
Рациональности параметров закон (в
кристаллогр.) 847
Реактор безграднентный 562
--- проточный перемешиваемый 562
Реакции бимолекулярные 563, 564
---гетерогенные 560, 565
--- гомогенные 560, 562
--- индикаторные 567
--- индофениновые 260
--- ионные 475
--- капельные, открываемый минимум
408
--- конденсации 678
--- коисекутивные 431
--- кросс- 220
--- ксантогеновые 875
--- ксантопротеиновые 877
—- молекулярные гомогенно-каталити-
ческие 477
--- мономолекулярные 563, 565
--- обратимые 560
--- перекрестные 220
--- протолитические 479, 581
--- радикальные 474
—- сложные 560
--- тепловые 561
--- тримолекулярные 563
---цепные 561
--- электродные (электрохимия.) 560
--- элементарные 560
Реакционная способность 194
Реакция, кинетика 560
— максимальная работа 1046
— порядок 563
— скорость 457, 560, 561, 568
Реакция Ван-ден-Берга 51
—— Вейля — Сальковского 786
--- Виттига 213
---Гмелина 51
--- Гриньяра 1013
--- Дикмана 589
— Дильса — Альдера 565
--- Зандмейера 80
1079 — 1080
--- Зелинского — Стадникова 105
--- Зинина 109
--- Иванова 125
---Иоцича 324
--- Кампса 388
--- Канниццаро 397
--- Кижнера 555
--- Кижнера — Вольфа 556
---Кинга 559
--- Клайзена — Шмидта 591
--- Клемменсена 599
--- Кневенагеля 604
--- Кнорра 606
--- Кольбе — Шмидта 651
----- Комаровского 653
--- Кондакова 677
--- Коновалова 694
--- Конрада — Лимпаха 695
•-- Крёнке 825
--- Курциуса 893
--- Кучерова 895
--- Ладенбурга 899
---Ладенбурга — Вышнеградского
899
--- Лейкарта 936
--- Либермана 957
--- Либермана—Шторха 131, 132
--- Лобридебруина и Ванэкенштейна
992
--- Маделунга 1029
---Майера (Мейера) 1030
--- Мак-Фадиена — Стивенса 1047
--- Меншуткина 185
--- Мульдера — см. Реакции ксанто-
протеиновые
--- Обермейера 247
--- Оже 1031
--- Соммеле 422
--- Стивенса 422
--- Чугаева — см. Реакции ксантоге-
новые
--- Шорыгина 422
--- Яффе 786
Редкоземельные элементы — см. Ланта-
ниды
Резина, противостарители 493, 497
— старение 493
Резины из изопренового каучука 170,
171
--- из карбоксилатных каучуков
432
--- из каучука синтетического 497
--- из кремнийорганич. каучуков 808
Резинаты 543
Резонанс магнитный 103
Резонансный интеграл 531
Рейхерта — Мейссля число 65, 59, 73
Рейхштейна вещество S 732
Рекомбинация ионов 319
Рекристаллизация 13
Ремазолы 746
Рентгенолюминесценция кристаллов
863
Респираторы 83, 84
Ретен 588
Рециркуляции коэффициент (при крекин-
ге) 791
Решетка кристаллическая 840, 851
--- Браве 852, 853
Рибонуклеаза 211
9-р-В-Рибофуранозил-гипоксантин — см.
Инозин
9-р-В-Рибофуранозил-гипоксантин-5'-фос-
форные кислоты — см. Инозинфосфог -
ные кислоты
Риверсия сахаров 227
Рингера раствор 180
Рингера—Локка раствор 180
Рингера — Тироде раствор 180
Рогор 277
Родамины 872
Роданистый калий — см. Калий, рода-
нид
Родановое число 65, 69, 73
Родохрозит 439
Ромбическая сингония симметрии (в кри-
сталлогр.) 849
Ромбический вид симметрии (дипирами-
дальный, пирамидальный, тетраэдри-
ческий) 850
Ромбоэдрический вид симметрии 850
Ротационнокристаллическое состояние
54
Рубидий, кремнефторид 802
Рубин 735
Румера теорема 529
С
Сагенитовые треугольники 851
Салициловая кислота 434
Салициловый альдегид, семикарбазон 250
Салицин 759
Самарий 916
Самарскит 925
Самозащита (в радиац. химии) 95
Санитарно-технические изделия 537
Сапропели 331
Сапропелиты 330, 331
Сапфир 735
Сарколизин 402, 948
Саркомицин 405
Сафлоровое масло 73, 75
Сафранин Т 252
Сахар солодовый — см. Мальтоза
Сахара, инверсия 226
— риверсия 227
Сахаразы — см. Инвертазы
Сверхкомплексные соединения 657
Светлый креп (каучук) 491
Свинец, применение в защитном покры-
тии 97
Связь аксиальная 700
--- донорно-акцепторная — см. Связь
координационная
--- ионная 857
--- квазиаксиальная 701
---квазиэкваториальная 701
--- ковалентная 521
--- координационная 708
--- кумулированная 890
--- макроэргическая 1042
--- химическая 529, 857
— - экваториальная 700
Севроны 259, 782
Сегнетоэлектрики 861
Седативные вещества 943
Сезамовое масло 73, 75
Секвойтол 273
Сенсибилизация 608
Сера, двуокись, критич. постоянные 866
— изотопный обмен 194
— окислы 574
— определение 451
— трехокись, критич. постоянные 866
Серебро, магнитная восприимчивость
1021
Серинфосфатиды 639
Сероводород, критич. постоянные 866
Сероуглерод, критич. постоянные 866
Серпентин 1009
Сечение ионизации 315
Сжимаемость жидкостей 61
Сидерит 41, 439
Сидеритовые руды 41
Сиджвика теория 709
Силазаны 813
Силаны — см. Кремневодороды
Силастомеры — см. Кремнийорганиче-
ские каучуки
Силены 801
Силикагель 409, 412, 820
Силикатные железные руды 41
Силикаты 806, 807
Силиконы — см. Полиорганосилоксаны
Силикоэтилен 801
Силины 801
Силициды 805
Силицирование 98
Силоксаны 805
Силоксен 254, 805
Силтианы 813
Силумины 807
Сильвин 348
Сильвинит 348, 351, 360
Сильхромы 14
Симметрия (в кристаллогр.) 847—853
— пространственные (Федоровские) груп-
пы С. 852
— сингонии С. 848
— точечные группы С. 852
Синглетное состояние 528
Сингонии симметрии (в кристаллогр.)
848
Синерезис студня 83
Синильная кислота, критич. постоянные
866
Синтокс 735
Синтомицин 934
Синькали — см. Калий, гексациаяофер-
роат
Синэстрол 405
Систокс 277
Скандий 917
Скатол 257
СКИ — см. Изопреновый каучук
1 Скиллиразид 122
Склеивание — см. Клеящее действие
• Склерон 980
Сланцы 331
Сливовое масло 73, 75
Смачивание 413
Смеси постояннокипящие 57
Смитсонит 439
Смокедшит 491
Смола каменноугольная 385, 636
---коксовая — см. Смола каменно-
угольная
----- кумароновая 888
--- меламино-формальдегидная 130
--- мочевино-формальдегидная 130
--- феноло-формальдегидная 130
Смолуховского уравнение 608
Смолы анилино-формальдегидные 130,
131
--- глифталевые 131
— ионообменные 301
--- канифольно-малеиновые 396
--- канифольно-фумаровые 396
--- кумароно-инденовые 131, ,132
-— меламино-формальдегидные 131, 132
--- мочевино-формальдегидные 131, 132
---полиэфирные 130, 131, 132
—-^езорцино-формальдегидные 130,
--- силиконовые 810
--- синтетические, растворимые в воде
79
--- сульфонамидные 131
--- тиомочевино-формальдегидные 131
--- феноло-кумароно-инденовые 132
---феноло-формальдегидные 131, 132
---феноло-фурфурольные 130, 132
---фуриловые 130, 132
--- эпоксидные 130, 132
Смоляные кислоты 587
Снотворные вещества 942
Сода 439
Соевое масло 73, 75
Сокатализаторы 485
Соласодин 731
Солевая изомерия 665
«Солевая ошибка» 249
Соленит (порох) 715
Соли двойные 656
Солидогены (закрепители окрасок) 79
Солидус — см. Ликвидус и Солидус
Сольватная изомерия 663
Сольваты 318
Сольвосистем соединений теория 583
Соммеле реакция 422
Сополимеризация 1038
Сорбиерит 133
Сорбция 410
--- ионообменная 305, 308
Состояние переходное 565
Состояния стационарные связанные 515
Спазмолитические вещества 946
Спайность кристаллов 859
Спекание 535
Спектроскопия инфракрасная 283
Спектры звезд 737
--- интерференционные 281
--- инфракрасного поглощения 283
--- комбинационного рассеяния света
654
---комет 737
--- люминесценции 998
Сперматолипаза 972
Спиновой момент 1015
Спираны 209
Сплав Вуда 346
---Липовица 346
Сплавы жаропрочные 13
--- железа 19, 48
—- золота 111
--- карбидные 428
--- кобальта 619
---магнитные 32
Сподумен 977, 981
Спонтанное деление 203
Сродство нормальное (стандартное) 1047
Стадия (элементарная реакция) 5би
Стайонит ФК 303
Сталь 19, 21, 47
— жаропрочность 13, 14, 31
— жаростойкость 31
— коррозионно-усталостная прочность
729
— коррозионные среды 728
— механич. свойства 1 — 176
— прокаливаемость 27
Сталь аустенитная 14, 26
--- быстрорежущая 30
— динамная 33
доэвтектоидная 26
11)6'1 --- 1UO2.
- жаропрочная 13, 14, 31
- жаростойкая 31
- заэвтектоидная 26
- износоустойчивая 32
- инструментальная 26, 29, 48, 428
- карбидная 26
- конверторная 47
- конструкционная 13, 26, 48
- легированная 14, 428 — см. также
Железо, сплавы
— ледебуритная 26
— мартеновская 47
- - нержавеющая 30
— никелевая 27
— полуастенитная 26
— полуферритная 26
— с особыми свойствами 26, 30, 48
— трансформаторная 33
— углеродистая 13, 14, 21, 47, 48, 428
— ферритная 26
- хромистая 27
— хромомарганцовокремнистая 29
— хромомолибденоалюминиевая 28
— хромоникелевая 28
гарение,каучука и резины 493
— коллоидных систем 607
гациоцарные связанные состояния 515
гериламидометилперидинит 79
геариновая кислота 75
геатит — см. Клиноэнетатит
текло 284
-- кварцевое 821
— конденсированное 821
теллиты 620
теновые материалы 536
тереоизомерия — см. Изомерия про-
странственная
тереохимическая индикация 443
териды 973
терический фактор 564
тивенса реакция 422
тимуляторы центральной нервной систе-
мы 944
;тинтицин 738
!типтол 738
Itokcobo рассеяние света 653
Столкновений теория 564
Стронцианит 439
Стронциевый крон 868
Структура, дефекты 861
— валентная 529
— кристаллическая 851, 917
— тонкая 519
Структурная изомерия 152
Структурные формулы кристаллохимич.
854, 855
Структуры гетеродесмические 857
— каркасные 856
— координационные 856
---- островные 856
---- слоистые 856
— цепочечные 856
ССтудень 82, 83
— синерезис 83
Зтупалит 989
Сульфамиды 429
Сульфаниламидные препараты 947
Сульфитно-спиртовая барда 827
Сульфокиназы 558
Сульфокислоты, эфиры 401, 403
Сульфолипиды 973
Сульфоугли 301, 303
Суперпозиции принцип 512
Суперфосфат 383
Супрапьезостекло 821
Сурепное масло 73, 75
Сурьма, магнитная восприимчивость 1021
Суспензионный метод крашения 775
Сферофизин 146
Сысертскит 326
т
Таллий, сульфат 122
Талловое масло 587
Тальк 139
Тантал, карбид 425
Таутомерия 153
Тафта формулы 264
Текстовинит 628, 629
Текстура 841
Текучесть жидкостей 59, 61
Телефоровая кислота 760
Температурная депрессия 571
Теплообмен при испарении 331, 333
Теплота испарения 331
Тербий 916
Термит железо-алюминиевый 77
Термодинамика изотопного обмена 189
Термодинамики первый закон 871
Термодинамические изотопные аффекты
187
Термодиффузионный метод разделения
изотопов 195, 197, 198 •
«Термофор» система (в крекинге) 797
Тероптерин 404
Терпентин — см. Живица
Тестостерон 405
Тетамон 945
Тетраалкоксисиланы 816
Тетраарилкумулены 889
Тетрабромкоронен 718
Тетрабутилсилан 815
Тетрабутокснсилан 816
Тетрагалогеномоносипаны — см. Крем-
ний, тетрагалогениды
Тетрагидроабиетиновая кислота 587
Тетрагидронафтапин — см. Тетралин
Тетрагидро-2-пирон — см. 6-Валеролак-
тон
7-(Тетрагидроптеридип - парааминобензо-
и л )-1.-глутаминовая кислота — см. Ко-
фермент Ф
Тетрагидрофолевая кислота — см. Ко-
фермент Ф
Тетрагидро-2-фуранон — см. у-Бутиро-
лактон
Тетрагональная сингония симметрии (в
кристаллогр.) 849
Тетрагонапьно-трапецоэдрический вид
симметрии 850
Тетрагональный вид симметрии (дипира-
мидальный, пирамидальный, скалено-
эдрический, тетраэдрический, трапецо-
эдрический) 850
Тетрадекагидрокоронен 718
Тетра-о-крезилоксисилан 816
Тетралин 210
Тетраметилдиамидофторфосфат — см. Ди-
мефокс
Тетраметилдиарсин — см. Дикакодил
Тетраметипендисульфотетрамин 122
Тетраметилпентан — см. 2,2-Диметил-
пропан
Тетраметилсилан 815
Тетраметоксисилан 816
3,7,8,12-Тетраоксихолевая кислота 49
Тетрапропилсилан 815
Таленит 337
Тетрапропоксисилан 816
Тетрафенилсилан 815
Тетрафеноксисилан 816
Тетрафторбутатриен 889
Тетраэтипсилан 815
Тетраэтоксисилан 816
Тиаминдифосфат 431, 741, 743
Тиамин-пирофосфокиназа 558
Тимет 277
Тимкенаплой 14
Тимолептики — см. Стимуляторы цен-
тральной нервной системы
Тимоловый синий 250
Тимолфталеин 248
Тинидур 14
Тно ТЭФ — см. Тиофосфорная кислота,
триэтиленимид
6-Тиогуанин 405
6-Тиогуанозин 405
Тиоиндиго 243
Тиоктовая кислота — см. Липоевая к-та
Тионафтен 386
2-Тиоурацил 404
Тиофосфорная кислота, триэтиленимид
403, 948
Титан,' карбид 425
Титано-магнетитовые руды 41
Титр 708
Титриметрический анализ 641
Титриплекс III — см. Комплексон III
Титрование колориметрическое 649
Тождественности принцип 520
Токсафен 391
а-Токснн — см. Лецитиназы
п-Толилизоцианат 207
2,4-Толуилендиизоцианат 207
Толуол, критич. постоянные 867
Томсона уравнение 409
Тонкая структура 519
Топаз 139
Топанол-О — см. Ионол
Топливо твердое ископаемое 330
Торий, карбид 425
Тормозное излучение 91
Торон 408
Торф 331
Тофранил — см. Имизин
Транквилизаторы 943
Трансаннулярный эффект 1040
Трансляции (в кристаллогр.) 851
Трансляций оси (в кристаллогр.) 851
Трансферазы 741 1
Трегеры — см. Носители
Третамин 403
Триамепин — см. Третамин
Триамцинолон 732
Триариламминий, сопи 323
Три-(Р-хлорвинил) арсин — см. Люизит
Триболюминесценция кристаллов 863
2,4,6-Трибромфенол, висмутовая соль —
см. Ксероформ
Тригональный вид симметрии (дипнрами-
дальный, пирамидальный, трапецоэдрп-
ческий) 850
Тридимит 821
Трикальцийфосфат 382
Трикапроин 417
Триклинная сингония симметрии (в кри-
сталлогр.) 849
Трилаурин 63
Трилон Б — см. Комплексон Ш
Трипонометрпл — см. Комплексометрия
Тримекаин 944
Триметиламмонийметилилид 213, 421
Триметиламмонийфлуеренилид 213
1,7,7-ТриметилбициклО-[1,2,2]-гептан —
см. Камфан
1,7,7-Триметилбицикло-[ 1,2,4 .гептанон-2
— см. Камфара
4,7,7-Триметллбицикло-[0,1,4]-г?птан —
см. Каран
Триметилгидроксисилан 816
Триметилзолото 117
Триметилметоксисилан 815
Триметилолмеламин, триглнколевый
эфир — см. Препарат 105
2,2,4-Триметилпентан — см. Изооктан
Триметилсипан 814
Триметилхлорсилан 814
Триметилэктосисилан 815
Триметин 943
Триоксиглутаровая кислота 881
1,6,8-Триокси-3-метилантрон-9—см. Эмо-
динантрон
3,3', 4-Триоксифуксон-2 ’ '-сульфоксило-
та — см. Пирокатехиновый фиолетовый
Трипальмитин 63
Триплетное состояние 528
Трипсин 108
Трипсиноген 108
Триптамин 135
Тристеарин 63
Трифениламиносилан 816
Трифенилгидроксисилан 816
Трифенилен 399
4,4',4’'-Трифенилметантринзоцианат 207
Трифенилсилан 814
Трифенилсипиллитий 813
Трифосфоинозитиды 274
Трифосфопиридиннуклеотид 622
Трихлортриэтиламин 402
Три этиламиносилан 816
Триэтилгидроксисилан 816
2,3,5-Триэтиленимино-1,4-бензохинон 403
2,4,6-Триэтиленимино-8-триазин — см.
Третамин
Триэтилхлорсилан 814
Триэтилэтоксисилан 815
Тромбин 108
Тромбокиназа 108, 559
Тропеолин ОО 248
Трутона — Пикте правило 571
Тубазид 948
Тулий 916
Тунговое масло 73, 75
Туннельный эффект 518
«Турнбуллева синь» 46
ТЭТ — см. Третамин
ТЭФ — см. Фосфорная кислота, тризти-
ленимид
У
Уайт-спирит 963
Углеводороды газообразные, конверсия
675
Углерод, изотопный обмен 194
— окислы 574
— окись, конверсия 676
— химич. свойства 804
Углерод четыреххлористый, критич. по-
стоянные 867
Уголь 331
Угольная ангидраза — см. Карбоанги-
драза
Удаления правило 892
1083 — 1084
Удобрения азотные 58
--- жидкие 58
--- сложные 58
Укрывистость — см. Кроющая способ-
ность пигмента
Уксусная кислота, криоскопия, постоян-
ные 830
Ультрамикрогетерогенные системы — см.
Коллоидные системы
м-Ундекан, критич. постоянные 867
Уравнения состояния 61
Уран, карбид 425
Уранин А 872
Урацил 404
Урзолы 769
Уридин-5-фосфорные кислоты 742
Уробилин 51, 761
Уробилиноген 51
Уровни энергии, вырождение 519
— расщепление 103, 519
— тонкая структура 519
Уровни энергии дискретные 515
Уроптерин — см. Ксантоптерин
Урсодезоксихолевая кислота 49
Ускорители частиц 335
Ф
Фаворского метод получения изопрена
168
Фактор накопления 91
Фяолит, применение в защитном покры-
тии 100
Фарфоровые изделия 537, 538, 539
— окраска 542, 543
Фаялит 1009
Фаянсовые изделия 537, 538, 539
— окраска 542, 543
Федоровские группы симметрии (в кри-
сталлогр.) 852
Фенадон 943
Феназин, производные 761
а-Феназинкарбоновая кислота 761
Фенантрен 386, 690
о-Фенантролин 252, 408
Фенилалкиламины 945
p-Фенилакриловая кислота — см. Корич-
ная кислота
р-Фенилакролеин — см. Коричный аль-
дегид
Фенил-п-аминофенол — см. Параоксиди-
фениламин
Фенилантраниловая кислота 252
3-(1-Фенил-2-ацетилэтил)-4-оксикума-
рин — см. Варфарин
п-Фенилендиизоцианат 207
Фенилизотиоцианат 179
Фенилизоцианат 207
Фенилин — см. 2-Фенилиндандион
2-Фенилиндандион 239
Фенилиодиддихлорид 288, 291
Фенилкальцийиодид 375
Ь-2-(а-Фенил-а-кароэтоксиметил)-тиазо-
лидин-4-карбоновая кислота — см.
Лейк о ген
Фенилтрихлорсилан 814
Фенилтриэтоксисилан 815
Фенобарбитал — см. Люмииал
Феноксазин 769
Фенолиндофенол 261
Фенол-крезолы 826
Феноловый красный 248
Фенолсульфофталеин — см. Феноловый
красный
Фенолфталеин 248
Фенолы 386, 387, 388
Фергюсонит 337, 925
Фермент конденсирующий (цитратсинте-
зирующий) 691
•--Шардингера — см. Ксантиноксидаза
Ферменты 108, 463, 741
Ферми уровень 466
--- частицы 520
Ферми — Дирака статистика 524
Ферраты 18, 46
Ферри-гипс — см. Феррон
Ферримагнетизм 1021
Ферринитрат 17
Феррисульфат — см. Железо, сульфаты
Феррит — см. а-Железо
Ферриты 19, 1021, 1022
Ферробор 33
Феррованадий 34
Ферровольфрам 34
Ферроии — см. о-Фенантролии
Ферромагнетизм 1005, 1018
Ферромарганец 34
Ферромолибден 35
Феррон — см. 7-ИОД-8-ОКСИХННОЛИН-5-
сульфокислота
Феррон (ферри-гипс) 38
Феррониобий 35
Ферронитрат 17
Ферросилиций 35
Ферросплавы 33
Ферросульфат — см. Железо, сульфа-
ты
Ферротитан 36
Феррофосфор 37
Феррохром 36
Ферроцирконий 37
Физалиен 455
Физиологические растворы 180
Физические системы 674
Фитии 273
Фитогликолипид 275
Флаваноиды 759
Флаванолы 759
Флаванонол 759
Флаваноны 759
Флавинадениндинуклеотид 743
Флавинмононуклеотид 743
Флавоны 759
Флоридины 106
Флотация 413
Флуорен 386
Флуоресцеин 254, 872
Флуоресценция кристаллов 863
Флюаты 802
«Флюид» система (в крекинге) 797
Флюорит 139, 284, 371, 375
р-Фокохолевая кислота 49
Форконтакт 485, 487
Формилциклопропан 1047
Формулы перспективные 698
--- проекционные 698
Форстерит 539, 540, 541, 1009
Фосфаген — см. Креатинфосфорная к-та
Фосфамид 277
Фосфатидилинозиты 274
Фосфатидилхолины — см. Лецитины
Фосфатидилэтаноламины — см. Коламин-
фосфатиды
Фосфоглицеромутаза 742
Фосфоенолпировиноградная кислота
(ФЕП) 1043
Фосфокиназы 558
Фосфокреатин — см. Креатинфосфорная
кислота
Фосфолипазы — см. Лецитиназы
Фосфолипиды 638, 973
Фосфомутазы 742
Фосфор, окислы 574
Фосфор белый 77
Фосфоресценция кристаллов 863
Фосфорная кислота, триэтиленимид 403
Фосфосульфат 1042
Фосфоферазы — см. Фосфокиназы
Фотоионизация 298, 316
Фотолюминесценция кристаллов 863
Фотосинтез 434
Фотохимия, ионы 321
Франка и Рабиновича эффект — см.
«Клетки» эффект
Франка — Кондона принцип 296
Фреттинг-коррозия 729
Р-Фруктофуранозидаза 226, 227, 429
Фталеинкомплексон 253
Фталеины 872
Фтористый метил, критич. постоянные
867
Фторкаучуки 498
Фторопласты для защитного покрытия
101
5-Фторпиримидины 404
Фторсиликаты — см. Кремнефториды
5-Фторурацил 404
Фукоидин 385
Фуллеровы земли — см. Флоридины
Фумарин 123
Фумаровая кислота — см. Малеиновая и
фумаровая кислоты
Функциональный анализ 641
3-( 1 -Фурил-2-ацетил этил)-4-оксикума-
рин — см. Фумарин
Фурамины 769
Фурфурол 881, 951
Футор 628
X
Халконы 759
Халцедон 822
Хелатометрия — см. Комплексометрия
Хелатои III — см. Комплексон III
Хемолюминесценция кристаллов 863
Хемосинтез 434
Хемосорбция 467, 994, 995
— кинетика 566
Хенодезоксихолевая кислота 49
Хиломикроны 973
Химиотерапевтические средства 947
Химиотерапия 948
Химическая память 1037
Химические системы 674
Химотрипсин 211
Хинализарин 408
Хинолин 386
— производные 135
4-Хинолинкарбоновая кислота 135
Хиноны 759
Хиолит 828
Хлопковое масло 73, 75
Хлор, критич. постоянные 866
Хлоракон 942
Хлорамбуцил 402
Хлорамфеникол — см. Левомицетин
Хлораураты 111, 118
Хлорбензол, критич. постоянные 867
4-Хлорбутинил-Х-3-хлорфенилкарбамат—
см. Карбин
₽-Хлорвинилдихлорарсин — см. Люизит
2-Хлор-4-диметиламино-6-метил пирими-
дин — см. Кастрикс
Хлоридиты 327
Хлоринат — см. Карбии
Хлоринация 114
Хлориридаты 327
а-Хлоркамфара 389
Хлоркаучук 492
Хлорокруорин 761
Хлорокруоропорфирин 761
ХлоромиЦетин — см. Левомицетин
Хлорофилл 761
Хлороформ 942
— критич. постоянные 867
Хлорпромурит 122
6-Хлорпурин 405
Хлортрифторметан, критич. постоянные
867
Хлорфен 391
3-(2-п-Хлорфенил-2-ацетилэтил)-4-оксн-
кумарин — см. Кумахлор
4-Хлорфенилдиазотиомочевина — см.
Промурит
и-Хлорфенилкарбаминовая кислота,
4-хлорбутиниловый эфир — см. Карбин
о-Хлорфенолиндофенол 261
Хлорциклогексан 700, 702
Хлорэтиламины 402, 945
Холановая кислота 49
Холантрен 400
Холевая кислота 49
Холеиновые кислоты 50, 66
Хопестерилбензоат 54
Холинолитические вещества 943, 944
Холиномиметические вещества 944
Холинфосфатиды — см. Лецитины
Холинэстераза 945
Холоцеллюлоза 951
Хонван 405
Хризен 386, 400
Хризоидин 766
Хризоробин 759
Хром, карбиды 425, 426
--- хлористый, гексагидраты 157
Хроман 759
Хромансиль — см. Сталь хромомарган-
цовокремнистая
X роматография ионообменная 305, 309,
310
Хромирование 97
Хромотроповая кислота 408
ц
Цветная фотография 748
Цветные реакции на полимеры 131
--- на каучуки качественные 132
Цезий, иодид 284
— кремнефторид 802
Целлюлоза 130, 131
— ацетобутират 130, 131, 132
— ацетопропионат 131, 132
— ксантогенат 874
— нитрат 131
— эфиры простые 131
Целлюлозно-бумажное производство 951
Цемент андезитовый 580
--- базальтовый 580
--- бештаунитовый 580
--- диабазовый 580
--- кислотоупорный — см. Кислото-
упорные замазки
1U6O — 1U6D
Цементит — см. Железо, сплавы
Центральный ион — см. Атомцентральный
Цеокерб 303
Цепочечные структуры 856
Цереброзиды 973, 974
Цериевая группа элементов 915, 919, 923
Церий 916
— карбид 425
Церолит 303
Церулеин 872
Церуссит 439
Цианирование 116
Т4-(Р-Цианзтил)-индол 256
Цибакроны 746
Циклизация бифункциональных соедине-
ний 1040
Циклические соединения, устойчивые
конформации 699
Циклобутен 209
Циклогексан 209, 415, 416, 699, 703
— дегидрирование 464
—• критич. постоянные 867
Циклогексаноноксим 414, 415
Циклогексен 700
Циклогексилизоцианат 207
Циклодол 943
Циклокаучук 492, 499
Циклопентадиен 209, 422
Циклопентадиениланион 421
Циклопентадиенилиды 213, 421
Циклопропан 209
Циклотроны 335
Циклофосфан 948
Цинк, фосфид 122
Цинкование 96, 97
Цинковые кроны 868
Циннвальдит 977
Цирконий, карбид 425
— фосфат 300, 303
Цис-транс-изомерия — см. Изомерия
геометрическая
Цитидин-5-фосфорные кислоты 742
Цитохром-с 211
Цитохромоксидаза 742
Цитохромы 742
Цитраконовый ангидрид 336
Цитратсинтезирующий фермент — см.
Конденсирующий фермент
Цитрогеназа — см. Конденсирующий фер-
мент
ч
Частицы, ускорители 335
---тяжелые, переход 317
Частотный фактор — см. Предэкспонен-
циальный фактор
Числа заполнения 520
Чугаева реакция — см. Ксантогеновая
реакция
Чугун 19, 21, 23, 47, 428
--- алюминиевый 24
--- белый 23
•-- высококремнистый 24
-- высок омарганцовый 24
ковкий 24
легированный 24
литейный 47
медистый 24
— ‘ передельный 47
природнолегированный 47
-- серый 24
---титановый 24
--- хромистый 24
--- хромоникелевый 24
ш
Шарголин 628
Шардингера фермент — см. Ксантино-
ксидаза
Шеелит 371, 863
Шеллак 131
Шёнит 352, 358
Шёнфлиса символ симметрии (в криётал-
логр.) 849
Шикимовая кислота 961
Шорыгина реакция 422
Шпатовые железняки — см. Сидеритовые
РУДЫ
Шпинель 539, 540, 541
Шрадан 277
Шредингера уравнение 513
Штарка эффект 519
щ
Щавелевоуксусная кислота 435
э
Эбонит 495
Эбулиоскопия 571
Эвксенит 337, 925
Эжекторы 268
Эжекция 268
Эйлан НК 276
Эйлан НЦ 276
Экатин 277
Экваториальная связь 700
Экгонин 630
Экзопептидазы 488
Эксайт (порох) 715
Экстракристаллический обмен 308
Электрический дренаж 88
Электрический разряд, ионы 321
Электрод 560
--- кислородный 577
Электродные потенциалы металлов
725
Электроионитовое обессоливание 299
Электроионитовые установки 299
Электрокортин — см. Альдостерон
Электрокорунд 735
Электролиты 307
— ионный обмен 307
— коагулирующее действие 608
— козфф. активности 307
Электролюминесценция кристаллов 863
Электронная теория кислот и оснований
585
Электронное облако 512
Электронный магнетон Бора 519, 1017
Электронный удар 297, 315
Электронных пар метод — см. Валентных
связей метод
Электроиоакцепторный реагент — см.
Акцептор
Электронодонорный реагент — см. До-
нор
Электроны (сплавы) 77, 1025
Электроны, вероятность локализации
511
— дифракция 506
Электроположительность элемента 296
Электропроводность жидкостей 61
--- жиров 63
Электрорубин 735
Электросталь 47
Электрофильный реагент — см. Акцеп-
тор
Элементарный анализ 641
Эмали — см. Краски эмалевые
Эмбихин 401, 402, 948
— N-окись — см. Нитромин
Эм^динантрон 759
Эмульсин 429
Энантиоморфизм кристаллов 848
Эндоксан 402, 948.
Эндопептидазы 488
Энергетизированные молекулы 563
Энергии равного распределения по сте-
пеням свободы принцип 1045
Энергия активации 457, 458, 564
--- ионизации 295
--- нулевая 516
--- обменная 522
Эозин 254, 872
Эпигидрокортизон 733
Эпикатехин 489
Эпилупинин 994
Эпитаксия 851
3, б-Эпокси-1,2-диметилциклогександи-
карбоновая-1,2 кислота, ангидрид —
см. Кантаридин
Эпоксиды 401
Эпсомит 1027
Эрбий 916
Эргин 964
Эриохромчерный Т 253
Эритрин 612
Эритрозин 872
Эрленмейера изокоричная кислота 716
Эруковая кислота 75
Эстрон 743
Этаминал 942
Этан, критич.'-ярстояниые 867
Этаноламинфосфйящды — см. Коламин-
фосфатиды
2-Этиламино-1,3,4-тйвдиазол 403
Этилен, критич. постоянные 867
— полимеризация 485, .’86
Этилендиаминтетрауксусна кислота
(ЭДТА) 672
— двунатриевая соль — см. Комплек-
сон III
Этиленимин 401, 403
Этилениминохиноны 403
Этилен-1,2-цис-дикарбоновая кислота, ан-
гидрид — см. Малеиновый ангидрид
Этилизоцианат 207
5-Этил-5-кротилбарбитуровая нислота —
см. Калипнон
Этилксантогеновая кислота 877
Этиловый спирт, критич. постоянные
867
Этилсилан 814
Этилтриметоксисилан 815
Этилтрихлорсилан 814
Этилтриэтоксисилан 815
5-Этил-5-фенилбарбитуровая кислота —
см. Люминал
Этилцеллюлоза 130, 131, 132
Этиноль — см. Лак дивинилацетилено-
вый
4-Этоксиакридон 250
Эулевомицетин 934
Эусинтомицин 934
Эухризин 3R 250
Эфир гарпиуса 396
Эфирное число 65
Эфиры сложные, гидролиз 184
--- сульфокислот 401, 403
Эффект Франка и Рабиновича 601
ГО
Юма —Розери правило 858
L-Яблочная кислота, дегидрогеназа —
см. Малатдегидрогеназа
Ягоды метод 407
Ядерная изомерия 158
Ядерные излучения, источники 335
Ядерные изомеры 158
Ядерные превращения изотопов 202
Ядерные реакторы 336
Ядро атома, уровни возбужденные 158
— уровни метастабильные 158
Ятрен 292
Яффе проба 247
— реакция 786
ОПЕЧАТКИ И ИСПРАВЛЕНИЯ В 1-м и 2-м ТОМАХ КРАТКОЙ ХИМИЧЕСКОЙ ЭНЦИКЛОПЕДИИ
Ко- лонка Статья Строка статьи Напечатано Должно быть
В первом томе
499 Буферные растворы 21 и 22 снизу кн2О ^кисл.
1018— Дальтониды и бертолиды — Рис. 1 и 2 Рисунки следует поменять местами
1019
п 2 / п
1136 Дипольный момент 16 снизу в = 1/ mo + 2 (т1) •₽! II 3 © м м 3 Н“
г Ln—1 J г 1
Во втором томе
248 Индикаторы 8 снизу Тимолфталеин, 9,8—10,5 Тимолфталеин, 9,3—10,5
342 Кадмий 19 сверху (гакмей) (галмей)
357 Калия персульфат 9 сверху металлы, способные окисляться до металлы, способные окисляться до окисей
843 Кристаллическое состоя- 27 снизу а = 1 -е-«а а =1 -е-«"
ние полимеров
970 Линоленовая кислота 2 сверху т. пл. 11—11,3 т. пл. — (11 — 11,3)
1 О .
1 ононон
1043 Макроэргические связи 22—26 снизу ...-С-С—с-с-с—... '1111 С-С-С-С-С-. ..
н i н н н А А А А А
Краткая химическая эипиклопедия. Ред. кол. И. Л. Кнунянц
(отв. рёд.) и др. Т. 2. М., «Советская Энциклопедия», 1963
т. (Энциклопедии. Словари. Справочники).
т. 2. Ж—Малоновый эфир. 1963. 1088 стб. с илл. 1 л. табл.
Сдано в набор 14 мая 1962 г. Том подписан к печати 27 октября 1962 г.
Государственное научное издательство «Советская Энциклопедия». Москва, Ж-28. Покровский бульвар, д. 8.
Т-09691. 27/Х 1962 г. Тираж 85 тыс. экз. Заказ Ка 1497. Формат 82X108>/ie- Объем 34 физич. п. л. 55,76 усЛ. п. л.
+ вклейка 0,41 усл. п. л. Всего 56,17 усл. п. л. уч.-изд. л. 96,18. Цена 1 экз. книги 3 р. 30 к.
Ленинградский Совет народного хозяйства. Управление целлюлозно-бумажной и полиграфической промышленности.
Типографии 1 «Печатный Двор» имени А. М, Горького. Ленинград, гатчинская, 26.