/



























































































































































































































































Автор: Гавриленко В.Ф. Жигалова Т.В.
Теги: физиология растений ботаника биология
ISBN: 5-7695-1110-9
Год: 2003




Текст
ВЫСШЕЕ ОБРАЗОВАНИЕ
В.Ф.ГАВРИЛЕНКО, Т.В.ЖИГАЛОВА
БОЛЬШОЙ ПРАКТИКУМ
ПО ФОТОСИНТЕЗУ
Под редакцией профессора И. П. ЕРМАКОВА
Допущено
Министерством образования Российской Федерации
в качестве учебного пособия для студентов высших учебных заведений,
обучающихся по направлению 510600 «Биология» и специальностям
011600 «Биология», 012000 «Физиология»
Москва
ACADEMA
2003
УДК 581.132(075.8)
ББК 28.57я73
Г12
Рецензенты:
д-р биол. наук, проф. Ф. Ф. Литвин (зав. кафедрой физико-химической
биологии МГУ им. М. В. Ломоносова);
д-р биол. наук, проф. И. Ю. Усманов (зав. кафедрой физиологии растений
Башкирского государственного университета);
д-р биол. наук, проф. Т. Е. Кренделева (главн. научн. сотрудник кафедры
биофизики МГУ им. М. В. Ломоносова);
д-р биол. наук, проф. И. А. Шульгин (географический факультет МГУ
им. М. В. Ломоносова)
Гавриленко В.Ф.
Г12 Большой практикум по фотосинтезу: Учеб, пособие для студ.
вузов / В. Ф. Гавриленко, Т. В. Жигалова; Под ред. И. П. Ерма-
кова. — М.: Издательский центр «Академия», 2003. — 256 с.
ISBN 5-7695-1110-9
В книге рассмотрены современные методы комплексного исследова-
ния функциональной активности фотосинтетического аппарата, которые
позволяют получить детальную характеристику пигментных систем, оце-
нить фотохимическую активность работы ЭТЦ хлоропластов, определить
интенсивность и энергетическую эффективность фотосинтеза, получить
общую характеристику процесса фотосинтеза на уровне целого растения.
Изложены теоретические основы используемых методов исследования,
сведения о технике проведения лабораторных работ, об устройстве типо-
вых приборов и специальных установок, разработанных на кафедре фи-
зиологии растений МГУ им. М. В. Ломоносова. Приведены примеры рас-
четов и способы статистической обработки результатов исследований.
Для студентов высших учебных заведений, обучающихся по направле-
нию «Биология», специальностям «Биология» и «Физиология».
УДК 581.132(075.8)
ББК 28.57я73
© Гавриленко В. Ф., Жигалова Т. В., 2003
© Издательский центр «Академия», 2003
ISBN 5-7695-1110-9
Посвящается 250-летию
Московского государственного
университета им. М. В. Ломоносова
и 140-летию кафедры физиологии
растений МГУ
ПРЕДИСЛОВИЕ
Исследование фотосинтеза представляет собой одну из наи-
более важных и сложных проблем биологии. Достижения по-
следних лет позволили не только значительно расширить пред-
ставления о механизме фотосинтеза, но и понять сущность и
значение фотосинтеза в общем метаболизме как раститель-
ной клетки, так и целого растения.
Фотосинтез занимает центральное место в энергетике клет-
ки, поскольку именно этот процесс служит первичным ис-
точником всей энергии, используемой живыми организмами
в процессах жизнедеятельности. При фотосинтезе энергия
света, поглощаемая пигментными системами, преобразуется
в химический потенциал богатых энергией метаболитов. На-
копление энергии сопряжено с включением углерода в мета-
болизм клетки. Таким образом, фотосинтез, неразрывно свя-
занный с реакциями энергетического и пластического обме-
на, составляет основу метаболизма зеленой растительной
клетки.
На уровне целого растения фотосинтез выступает основ-
ным донором метаболитов, удовлетворяющим потребности в
них таких процессов, как дыхание, поглощение и ассимиля-
ция минеральных веществ, рост и развитие растений. Нахо-
дясь в центре донорно-акцепторных связей, фотосинтез иг-
рает ключевую роль в формировании общей и хозяйственной
продуктивности растения. Поэтому исследования фотосинте-
за лежат в центре комплексных работ, направленных на изу-
чение физиологии растений и моделирование растения как
целостной саморегулирующейся и самонастраивающейся си-
стемы.
Фотосинтез составляет основу первичной биологической
продуктивности природных экосистем и определяет форми-
рование урожаев в посевах сельскохозяйственных растений.
Исследования фотосинтетической деятельности сложных ра-
стительных сообществ приобретают практическое значение.
При этом возникает необходимость комплексного изучения
3
фотосинтеза, определения его количественных показателей,
а также изучения других, сопряженных с ним процессов —
роста, дыхания, ассимиляции азота и др.
Глубокий и постоянный интерес к исследованию меха-
низма фотосинтеза определяется не только его огромным
значением в жизнедеятельности живых организмов. Деталь-
ное изучение процесса фотосинтеза необходимо и для реше-
ния ряда вопросов общей биологии, например, касающихся
механизмов регуляции процессов в живой клетке, природы
возбужденных состояний молекул, механизмов миграции энер-
гии в живой системе, а также проблем биоэнергетики и др.
Большие успехи в изучении структурно-функциональной
организации фотосинтетических процессов и механизмов их
регуляции в последние годы связаны с новыми методически-
ми подходами к изучению фотосинтеза и развитием экспери-
ментальной техники.
Фотосинтез — сложный комплекс последовательно про-
текающих, различных по природе и скорости реакций. Его
специфика состоит в тесном взаимодействии фотофизичес-
ких, фотохимических и энзиматических реакций. В связи с
этим в современных исследованиях фотосинтеза наряду с
физическими и фотохимическими направлениями существен-
ное место занимает изучение функционирующих в хлоропла-
стах ферментных систем, а также регуляторных механизмов,
определяющих эффективность работы всего фотосинтетичес-
кого аппарата.
В зависимости от целей исследований и характера изучае-
мых процессов применяют ту или иную совокупность мето-
дов, весьма разнообразных, в том числе довольно сложных.
Они включают, как правило, различные виды спектрофото-
метрии и хроматографии, методы определения газообмена,
полярографию, электронную микроскопию, дифференциаль-
ное центрифугирование и др. При изучении первичных ста-
дий фотосинтеза для регистрации нестойких, короткоживу-
щих интермедиатов используют высокочувствительные мето-
ды импульсной спектрофотометрии, электронного парамаг-
нитного резонанса, лазерной техники и др. В последнее время
для исследования генетических основ организации, функци-
онирования и регуляции фотосинтетического аппарата ак-
тивно применяют методы биотехнологии и генной инжене-
рии.
При выполнении заданий практикума по фотосинтезу сту-
денты знакомятся с главными принципами современных ме-
тодов исследования фотосинтеза и осваивают методические
приемы, которые применяются в научных лабораториях. Ме-
тоды, включенные в практикум, дают возможность изучить
4
свойства пигментных систем, оценить интенсивность работы
фотосинтетического аппарата, получить общую характерис-
тику и выяснить особенности процесса фотосинтеза, а также
активности составляющих его звеньев. При проведении работ
создаются определенные экспериментальные условия, исполь-
зуются различные физиологические схемы с включением в
опыт специфических ингибиторов и стимуляторов отдельных
реакций в целях изучения механизмов эндогенной регуляции
фотосинтеза.
Зав. кафедрой физиологии растений МГУ
проф. И. П. Ермаков
ОТ АВТОРОВ
Успехи в развитии представлений о механизме фотосин-
теза, достигнутые в последние десятилетия, в значительной
мере обусловлены разработкой и внедрением новых принци-
пов и методов научных исследований, применением доволь-
но сложной аппаратуры для анализа структуры и функции
фотосинтетического аппарата. Все это требует существенной
модернизации и совершенствования специального практи-
кума по фотосинтезу, отражающего широкий спектр совре-
менных методов подготовки студентов физиолого-биохими-
ческого профиля. Поэтому создание настоящего учебного
пособия «Большой практикум по фотосинтезу» авторы счи-
тают своевременным и необходимым.
Пособие написано на основе материалов спецпрактику-
ма, проводимого на кафедре физиологии растений биологи-
ческого факультета МГУ.
Практикум отражает комплексный подход к исследованию
организации и функциональной активности фотосинтетичес-
кого аппарата, пигментных систем, активности работы элек-
трон-транспортной цепи и ее отдельных участков, энергети-
ческой эффективности фотосинтеза, процессов фотофосфо-
рилирования и сопрягающих систем хлоропластов, а также
темновых реакций фотосинтеза и фотосинтетического про-
цесса на уровне целого растения.
Непосредственному изложению практической части работ
предшествует краткое описание применяемых методов спек-
трофотометрии, хроматографии, полярографии, потенцио-
метрии, электрофореза. В ряде заданий дано описание типо-
вых приборов и специальных установок, разработанных на
кафедре физиологии растений МГУ.
В практикум включены подготовленные сотрудниками Ин-
ститута физиологии растений РАН задачи: «Определение све-
товой кривой скорости фотосинтеза с помощью газометри-
ческой установки открытого типа, включающей инфракрас-
ный газоанализатор» (Е.А. Егорова, И. С. Дроздова, Н.Г. Бу-
6
хов) и «Определение фотохимического и нефотохимическо-
го тушения флуоресценции хлорофилла при освещении лис-
тьев» (Е. А. Егорова, Н. Г. Бухов). Эти задачи студенты выпол-
няют в лаборатории фотосинтеза ИФР.
Авторы благодарят профессора Ф.Ф.Литвина за ценные
замечания по материалам рукописи, профессора М. Н. Мерз-
ляка за консультации и помощь в разработке ряда задач по
анализу пигментов в растении, кандидата биологических наук
Г. Г. Лысенко за помощь в разработке задач по определению
фотохимической активности хлоропластов на отдельных уча-
стках ЭТЦ хлоропластов, доцента О. Б. Беляеву за ценные
советы при подготовке рукописи.
Авторы выражают благодарность ведущим инженерам ка-
федры физиологии растений — Б. А. Соловьеву за разработ-
ку, конструирование и отладку установок, используемых при
проведении практикума по фотосинтезу, и Л.В.Кирпичевой
за техническую помощь в оформлении рукописи.
ОБЩИЕ УКАЗАНИЯ О ПРОВЕДЕНИИ РАБОТ
«БОЛЬШОГО ПРАКТИКУМА ПО ФОТОСИНТЕЗУ»
«Большой практикум по фотосинтезу» включает работы,
направленные на освоение основных методов изучения фи-
зиолого-биохимических аспектов фотосинтеза.
Объектами физиологических исследований при проведе-
нии практикума могут быть представители разных групп рас-
тений с достаточно быстрым начальным ростом — горох,
кукуруза, пшеница, овес и др. Для работы берут, как прави-
ло, растения в возрасте 10—20 дней. Их выращивают в кли-
матических камерах при контролируемых параметрах темпе-
ратуры, освещения, влажности и фотопериода в почвенной
или водной культуре, применяя питательные смеси, напри-
мер смеси Прянишникова, Хогланда или Кнопа. Для получе-
ния средней пробы растительного материала одного вида рас-
тений высаживают 100 — 200 семян, сходных по виду и раз-
мерам.
В некоторых работах, связанных с исследованием пигмен-
тного аппарата растений и общих показателей интенсивнос-
ти фотосинтеза, могут быть использованы травянистые рас-
тения, растущие в природных условиях, листья древесных и
кустарниковых растений, а также листья комнатных расте-
ний.
Возможно также проведение физиологических исследова-
ний зависимости формирования и активности фотосинтети-
ческого аппарата от возраста растений, условий освещения
(использование света разной интенсивности и качества при
выращивании растений и проведении функциональных оп-
ределений), элементов минерального питания, температуры,
действия дефицита воды и других стрессовых для растений
факторов.
Все практические задания студенты выполняют самостоя-
тельно, при этом неукоснительно соблюдая инструкции по
технике безопасности.
Перед началом работы необходимо освоить теоретические
основы выполняемого практического задания.
8
Определение физиологических и биохимических показа-
телей осуществляют в нескольких биологических и аналити-
ческих повторностях; экспериментальные данные подверга-
ют статистической обработке. Для получения достоверных ре-
зультатов при подготовке растительных проб необходимо сле-
дить за однородностью растительного материала. При этом
надо использовать растения одного возраста, выращенные в
одинаковых условиях, а также учитывать ярусность располо-
жения и возраст листьев.
Для выполнения заданий используют реактивы максималь-
ной степени чистоты (ХЧ, ОСЧ). В случае их отсутствия необ-
ходимо проводить самостоятельно очистку или перекристал-
лизацию реактивов. При работе с изолированными хлоро-
пластами и субчастицами, а также для определения активно-
сти ферментов следует готовить растворы на бидистиллиро-
ванной воде.
Ход работы, необходимые для ее выполнения расчеты,
полученные экспериментальные данные и конечные резуль-
таты записывают в рабочем журнале. При оформлении зада-
ний результаты представляют в виде таблиц, графиков, ди-
аграмм, на основании которых делают выводы из проведен-
ного исследования.
Глава 1
ОСНОВНЫЕ ПРИНЦИПЫ
ИСПОЛЬЗУЕМЫХ МЕТОДОВ
1.1. АБСОРБЦИОННАЯ СПЕКТРОФОТОМЕТРИЯ
Абсорбционная спектрофотометрия является одним из наибо-
лее широко применяемых в настоящее время методов количествен-
ного и качественного анализа в химии, биологии и медицине [2,
4, И, 14, 15, 18, 19] благодаря сравнительно высокой чувстви-
тельности, быстроте определений и высокой точности. В этом со-
стоят ее преимущества перед другими методами химического ана-
лиза. Помимо чисто аналитических целей спектрофотометричес-
кий метод в последнее время находит все более широкое приме-
нение в экспериментальной биологии и химии для исследования
механизма различных биологических процессов, кинетики отдель-
ных реакций, тонких структурных изменений биополимеров, для
регистрации окислительно-восстановительных изменений отдель-
ных компонентов, связанных с переносом электронов в элект-
рон-транспортной цепи фотосинтеза, дыхания и т.д.
Спектрофотометрическим анализом определяют вещества с
концентрацией 10-6—10'7 моль/л и ниже. Наименьшее количе-
ство вещества, которое может быть обнаружено при фотометри-
ческих определениях, находится в пределах 0,01 — 0,001 мкг/см3.
В руководстве даны краткое описание общих принципов спек-
трофотометрии и конкретные примеры ее использования при
физиологических исследованиях растительного организма.
Химический анализ методом абсорбционной спектрофотомет-
рии основан на измерении количества света, поглощенного ис-
следуемым веществом или раствором какого-либо вещества.
Если через однородный слой вещества (например, раствор) про-
пустить луч монохроматического света с начальной интенсивнос-
тью /0, то вследствие частичного поглощения лучей веществом ин-
тенсивность прошедшего через него света уменьшится до / (рис. 1).
Уменьшение интенсивности светового потока при прохожде-
нии через раствор зависит от количества молекул на его пути и их
способности поглощать лучи данной длины волны. Вероятность
акта поглощения в тонком слое раствора пропорциональна коли-
честву квантов, падающих на единицу поверхности в единицу вре-
мени (интенсивность света /0 [1/(см2-с)]), количеству молекул в
10
единице объема (концентрация п [1/см3]),
толщине слоя dl [см]: -dIQ = IQ<3ndl. Коэф-
фициент о [см2] характеризует поглоща-
тельную способность молекулы. Он зави-
сит от длины волны излучения, имеет раз-
мерность площади и называется сечением
поглощения молекулы. Для хлорофилла в
максимуме поглощения о = 0,037 нм2, что
значительно меньше площади хромофора
(~ 2,3 нм2). Произведение 10-а [с-1] равно
числу актов поглощения квантов за 1 с
одной молекулой.
Фотометрические методы количествен-
ного определения веществ основаны на
законах Бугера—Ламберта и Бера.
Закон Бугера—Ламберта устанавлива-
ет зависимость количества монохромати-
; ческого света, поглощенного веществом,
Рис. 1. Изменение интен-
сивности светового по-
тока при прохождении
через раствор вещества:
/0 — интенсивность пада-
ющего света; I — интен-
сивность прошедшего че-
рез раствор света; d — тол-
щина слоя
! от толщины поглощающего слоя, т. е. дли-
i ны оптического пути.
Закон Бера устанавливает прямую пропорциональность между
поглощением света и концентрацией вещества в растворе.
В соответствии с законами Бугера—Ламберта и Бера между по-
глощением света раствором, длиной оптического пути и концент-
рацией в растворе поглощающего вещества существует опреде-
ленная зависимость, которая выражается уравнением
1g у = ecd,
(1)
где Zo — интенсивность падающего света; I — интенсивность про-
шедшего света; е — коэффициент поглощения (или коэффициент
экстинкции) вещества; с — концентрация вещества; d — толщи-
на слоя. Детальный вывод уравнения (1) дан в работе [13].
Величину 1g у обозначают символом А (или D) и называют
поглощением (или оптической плотностью)'.
А = ted. (2)
Оптическая плотность раствора пропорциональна суммарному
сечению поглощения 1 моля вещества No, поскольку молярный
коэффициент поглощения е = oNA
(Na — число Авогадро,
равное 6,02- 1023 моль-1). Коэффициент 1/2300 является результа-
том перехода от натурального логарифма к десятичному и от мил-
лилитров к литру.
11
Если с — молярная концентрация раствора [моль/л], то £ оз-
начает молярный коэффициент поглощения {молярный коэффициент
экстинкции) и выражает количество света, поглощаемое раство-
ром вещества с концентрацией 1 моль/л при толщине d слоя в 1 см.
Из уравнения (2) следует, что при концентрации раствора 1 моль/л
и толщине слоя 1 см молярный коэффициент поглощения е чис-
ленно равен поглощению А. Молярный коэффициент характери-
зует внутренние свойства вещества и не зависит от интенсивности
падающего света, объема исследуемого раствора или толщины слоя.
Размерность молярного коэффициента может быть определена
^4
из уравнения е = — и выражена в [л • моль-1 • см-1], [М-1-см-1]
или [моль-1 • см2].
Если с выражено в [г/л], то вместо е используют а — удельный
коэффициент поглощения [л • г-1 • см-1]. Тогда уравнение (2) может
быть преобразовано следующим образом:
А = acd. (3)
Между £ и а имеется следующее соотношение:
а = ТГ’ £ = аМг,
Мг
где Мг — относительная молекулярная масса.
Определив на приборе величину поглощения раствора (Л) и
зная соответствующие значения е и а, можно легко рассчитать
концентрацию с этого вещества в растворе, используя уравнения
(2) или (3), откуда
А А
с = —-• или с = —-.
cd ad
Если в растворе есть несколько компонентов, то при данной
длине волны поглощение смеси равно сумме оптических плотно-
стей отдельных компонентов смеси:
А = Ai + Л2 + ... = axcxd+ a2c2d + ... .
На этом основано определение концентраций веществ в смеси
без их разделения. Обычно измеряют поглощение в характерных
для каждого вещества максимумах — X' и X".
Для двух компонентов с разными максимумами поглощения:
при Xх
А' = (ад + a2c2)d\
при X"
А^ = (a"ci + а2 c2)d,
12
где а\ и «2 — удельные коэффициенты поглощения компонентов
1 и 2 при V; а" и а" — удельные коэффициенты поглощения
компонентов 1 и 2 при X". Подставив соответствующие значения а
и А, получаем систему уравнений. Решая эту систему уравнений,
можно найти концентрацию двух веществ с1 и с2 даже при силь-
ном перекрывании спектров поглощения.
Количество поглощенной раствором энергии света может быть
определено величиной пропускания. Пропускание раствора (Г) —
это отношение интенсивности излучения, прошедшего через ра-
створ, к интенсивности падающего света:
T = Y = Ю™1. (4)
Таким образом, Т характеризует долю прошедшего через сис-
тему света, величина (1 - Т) — долю поглощенного света. Погло-
щение и пропускание связаны следующим уравнением:
Л = Igy-= Igy =-IgT. (5)
При измерении пропускания интенсивность поглощенного
объектом света может быть рассчитана из уравнения
г _ г _ г
1 погл 10 7
ИЛИ
/погл = /о(1-П- (6)
Некоторые указания о проведении спектрофотометрических
определений даны ниже.
Выбор «нулевого» раствора. При количественных определени-
ях в качестве «нулевого» раствора, или раствора сравнения, по
отношению к которому определяется поглощение исследуемых
растворов, обычно берут чистый растворитель (воду, ацетон и т. д.).
В этом случае поглощение опытного раствора определяется толь-
ко концентрацией растворенных в нем веществ.
При определении скорости ферментативной реакции, особен-
но если опытный раствор имеет некоторую мутность (например,
при работе с суспензией хлоропластов, гомогенатами или белко-
выми растворами), в качестве «нулевого» раствора удобнее взять
такой же раствор, но без добавления необходимых для реакции
реагентов. Вместо них вводят воду или буферный раствор. В этом
случае регистрируют поглощение веществ, изменяющихся в ходе
реакции (снижение или увеличение поглощения реагента в ре-
зультате окисления или восстановления, образование окрашен-
ных комплексов и др.).
Высокой степенью точности отличается метод дифференци-
альной спектрофотометрии, где в качестве «нулевого» раствора
13
используют не растворитель, а раствор, близкий по поглощению
к исследуемому.
Выбор длины волны при спектрофотометрическом анализе ме-
тодом абсорбционной спектрофотометрии необходим для работы в
области наибольшей спектральной чувствительности. Как правило,
измерения проводят при длине волны максимального поглощения
света исследуемым веществом. Этим достигаются высокая чувстви-
тельность и точность количественных определений. Иногда спектр
поглощения образующегося в реакции окрашенного комплекса мало
отличается от исходного реагента по положению основного макси-
мума, но имеет несколько иную форму спектра. В этом случае опре-
деления проводят при длине волны, где разность поглощения ис-
ходного и конечного продукта реакции максимальна.
Случаи отклонения от основных законов светопоглощения. При-
менение спектрофотометрических методов возможно только в том
случае, если исследуемый раствор или вещество подчиняется за-,
конам Бугера—Ламберта и Бера, т.е. существует пропорциональ-
ность между значениями оптической плотности, концентрации и
толщины слоя исследуемого вещества. В этом случае график, где
по оси абсцисс отложена концентрация вещества в растворе с, а
по оси ординат — поглощение А, имеет вид прямой линии, выхо-
дящей из начала координат (рис. 2). При нарушении пропорцио-
нальности, причинами которой могут быть высокая концентра-
ция молекул и их агрегация, высокая интенсивность света, недо-
статочная монохроматичность светового потока, изменение со-
стояния вещества, использование данного метода невозможно.
Оптимальным для точного определения концентрации веществ
является интервал поглощения в пределах 0,2 —0,8. При очень
низких и высоких значениях поглощения намного возрастает про-
цент ошибки. Относительная ошибка определения может быть
снижена при использовании кюветы большей длины, т.е. при уве-
Рис. 2. График зависимости по-
глощения от концентрации ве-
ществ в растворе (объяснение
в тексте)
личении толщины слоя исследуемого
вещества.
Применение и возможности аб-
сорбционной спектрофотометрии. Раз-
личают два основных вида спектро-
фотометрического анализа: фотоко-
лориметрию и спектрофотометрию.
В фотоколориметрии анализ ве-
ществ осуществляют на основе из-
мерения поглощения в довольно
широком диапазоне длин волн (по-
лихроматическое излучение), выде-
ляемом фильтрами. Метод отличает-
ся простотой и дешевизной аппара-
туры, легкостью измерений.
14
В спектрофотометрии используют монохроматическое излу-
чение (ширина спектрального интервала обычно не превышает
2 — 3 нм) монохроматоров, спектрофотометров, что значительно
повышает точность определений и позволяет производить анализ
смеси веществ даже с близкими и сильно перекрывающимися
спектрами поглощения. В связи с этим возможности применения
спектрофотометрических методов значительно шире и они отли-
чаются более высокой точностью.
Впервые метод спектрофотометрического анализа в исследова-
ниях физиологических проблем фотосинтеза был применен про-
фессором кафедры физиологии растений МГУ К. А. Тимирязевым,
что позволило ему установить прямую связь между энергетичес-
кой характеристикой отдельных лучей электромагнитного спект-
ра, активностью поглощения их пигментами и интенсивностью
фотосинтеза.
В настоящее время спектрофотометрические методы широко
применяют в экспериментальной биологии, например:
• при изучении спектров поглощения веществ;
• при определении концентрации веществ, поглощающих в ви-
димой и ближней ультрафиолетовой области (пигментов, актив-
ных групп ферментов, нуклеиновых кислот, различных кофакто-
ров и т.д.);
• при качественном анализе веществ по спектрам поглощения;
• при исследовании кинетики ферментативных процессов и оп-
ределении активности ферментов, где реакция протекает с уве-
личением или уменьшением оптической плотности реакционной
смеси.
Большое распространение в научных исследованиях получили
различные модификации методов абсорбционной спектрофото-
метрии, такие, как дифференциальная и импульсная спектрофо-
тометрия, производная спектрофотометрия и др.
Дифференциальная спектрофотометрия отличается высокой
степенью точности и чувствительности определений и использу-
ется для качественного и количественного анализа веществ, об-
ладающих характерным спектром поглощения. Высокая точность
метода дает возможность регистрировать незначительные и быст-
рые изменения в спектрах поглощения (до 0,001 %) на фоне вы-
сокой общей оптической плотности и сильного светорассеяния,
проводить определение веществ в общей смеси в присутствии дру-
гих поглощающих свет компонентов, например определение ци-
тохромов или пигментов непосредственно в суспензии хлоропла-
стов.
Принцип метода состоит в том, что в качестве «нулевого» ра-
створа (контрольного) берут не чистый растворитель, как при
обычных спектрофотометрических определениях, а раствор, со-
держащий все те же компоненты, что и опытный. Опытная проба
15
подвергается действию света или каких-либо химических реаген-
тов, окисляющих или восстанавливающих определяемое соедине-
ние, в то время как контрольная проба не изменяется. Измеряя
оптическую плотность опытного образца относительно контрольно-
го, регистрируют разность оптической плотности между контро-
лем и опытом, обусловленную только применяемыми воздействи-
ями.
Измерение дифференциальных спектров осуществляется на
двухлучевом спектрофотометре, который включает монохроматор,
источник света, камеру для образца, регистрирующее устройство.
Луч монохроматического света с помощью специальных призм
делится на два пучка света равной интенсивности. Один пучок
проходит через опытную, другой через контрольную кювету. На
приборе регистрируется разность оптической плотности для двух
световых потоков (Д1)= Д ~ А)-
Если оптическая плотность опытной пробы вследствие каких-
либо (например, окислительно-восстановительных) превращений
исследуемого вещества изменилась в определенной области спект-
ра, то при данной длине волны будет зарегистрирована разность
оптических плотностей опытной и контрольной пробы. Эта раз-
ность может быть положительной (если в опытной пробе про-
изошло увеличение оптической плотности по сравнению с конт-
рольной) или отрицательной (если произошло уменьшение оп-
тической плотности). Отсюда в дифференциальном спектре ис-
следуемого вещества (в отличие от обычного спектра) регистри-
руются максимумы в области как положительных, так и отрица-
тельных значений. Длина волны максимумов может служить для
идентификации изменяющегося вещества, положение максиму-
ма в положительной или отрицательной области — показателем
характера изменений (увеличение или уменьшение концентрации),
а по величине максимумов можно судить о величине изменений
концентрации вещества.
Дифференциальная спектрофотометрия в различных модифи-
кациях широко используется при исследовании индуцированных
светом или химическими агентами изменений спектров поглоще-
ния пигментов, цитохромов, хинонов и других компонентов элек-
трон-транспортной цепи фотосинтеза и дыхания [11, 19].
На рис. 3 представлены спектры поглощения пигментов этио-
лированного и освещенного вспышкой света листа и соответству-
ющий дифференциальный спектр. На дифференциальныххпект-
рах отчетливо видно фотоиндуцируемое превращение предшествен-
ников хлорофилла в хлорофилл (исчезновение максимума 655 нм
сопровождается появлением максимумов 635 и 688 нм).
При использовании дифференциальной спектрофотометрии
для количественного определения цитохромов опытную пробу
восстанавливают, а контрольную окисляют соответствующими
16
реагентами. В результате измере-
ния разности спектров при раз-
личных длинах волн получают
дифференциальный спектр «вос-
становленная форма — окислен-
ная форма» цитохромов, позво-
ляющий оценить содержание
того или иного цитохрома в хло-
ропластах.
Методом дифференциальной
спектрофотометрии исследуют
также кинетику индуцированных
светом процессов фотоокисления
или фотовосстановления цитох-
ромов, пластоцианина и других
компонентов электрон-транс-
портной цепи (ЭТЦ) [2, 18, 19].
В данном случае контрольная
проба находится постоянно в
темноте, а опытную освещают
кратковременной импульсной
вспышкой. Регистрируют измене-
ние разности оптической плот-
ности опытной и контрольной
проб во времени. Измерения про-
водят при одной и той же длине
волны, соответствующей макси-
муму поглощения исследуемого
соединения. При включении све-
та оптическая плотность резко
Рис. 3. Сравнение спектров погло-
щения (/) с соответствующим
дифференциальным спектром (//)
пигментов этиолированного (7) и
освещенного в течение 2 мин (2)
листьев кукурузы (по [10], с из-
менениями)
увеличивается (или снижается) вследствие быстрого нарастания
восстановленного (или окисленного) цитохрома. При выключе-
нии света соотношение форм восстановленного или окисленного
цитохрома в пробе возвращается к исходному уровню. Использо-
вание метода дифференциальной спектрофотометрии позволило
выяснить многие вопросы механизма фотосинтеза, в частности
локализацию отдельных компонентов в ЭТЦ фотосинтеза, неко-
торые кинетические закономерности, связанные с транспортом
электронов в ней, и др.
1.2. ХРОМАТОГРАФИЧЕСКОЕ РАЗДЕЛЕНИЕ ВЕЩЕСТВ
Хроматография — физико-химический метод разделения жид-
ких и газообразных смесей, при котором компоненты смеси вы-
деляются в виде отдельных полос или зон.
17
Основоположником хроматографического анализа является
профессор Петербургского университета М. С. Цвет. В 1904 г. он
впервые применил адсорбционную хроматографию для разделе-
ния пигментов листа. В настоящее время хроматографический ме-
тод разделения сложных смесей органических веществ является
одним из основных методов биохимического анализа [1, 5, 10,
21]. Преимущество этого метода заключается в возможности раз-
деления небольших количеств исследуемых веществ, при этом они
не подвергаются химическим изменениям.
Хроматография основана на распределении веществ между дву-
мя фазами — неподвижной, которая бывает твердой или жид-
кой, и подвижной (жидкой), содержащей исследуемую смесь ве-
ществ, — она пропускается через неподвижную фазу. Распределе-
ние веществ между двумя фазами (твердой и жидкой или двумя
несмешивающимися жидкими фазами) характеризуется «коэф-
фициентом распределения», т.е. отношением концентрации ве-
щества в подвижной фазе и его концентрации в неподвижной
фазе. Вещества с более высоким коэффициентом распределения
продвигаются по сорбенту с большей скоростью, отделяясь от
веществ с более низким коэффициентом.
Методы хроматографического анализа в зависимости от сил,
действующих между разделяемыми веществами и поверхностью
твердой или жидкой фазы, с которой они соприкасаются, можно
разделить на три группы: адсорбционная хроматография; распре-
делительная хроматография; осадочная хроматография.
1. Адсорбционная хроматография основана на различной ад-
сорбируемости компонентов смеси на адсорбенте. Адсорбционная
хроматография подразделяется на молекулярную и ионообмен-
ную хроматографию.
В молекулярной хроматографии (классический метод М. С. Цве-
та) вещества адсорбируются на материале колонки под действием
межмолекулярных сил. Для каждого компонента смеси эти силы раз-
личны. Поэтому если раствор, содержащий смесь пигментов, про-
пускать через слой (колонку) сорбента, то пигменты располага-
ются в колонке в виде отдельных различно окрашенных зон. В вер-
хней части колонки задерживаются наиболее прочно связывае-
мые сорбентом компоненты, а ниже будут находиться менее прочно
связываемые. Все анализируемые компоненты в зависимости от
относительного адсорбционного сродства к сорбенту образуют ад-
сорбционный ряд — А, В, С,.... При промывании колонки чистым
растворителем вещества будут вымываться из верхних слоев и вновь
адсорбироваться в нижних слоях по закону адсорбционного замеще-
ния М. С. Цвета: компонент смеси, обладающий большим адсорб-
ционным сродством к сорбенту, вытесняет другие компоненты.
При использовании полярных сорбентов (целлюлоза, сахаро-
за, силикагель) прочность связи пигмент — сорбент зависит от
18
полярности пигментов. Для разделения пигментов используют
систему неполярных растворителей с примесью определенного
количества полярного растворителя. В этой системе положение зоны
пигмента на хроматограмме определяется силой его взаимодей-
ствия с полярным сорбентом и неполярным (или малополярным)
растворителем и скоростью передвижения зоны при промывании.
Пигменты с наибольшей полярностью (хлорофиллы, неоксантин),
образующие прочные связи с полярным сорбентом, задерживают-
ся у линии старта, а неполярные пигменты (каротин), для которых
характерны более прочные связи с неполярным растворителем,
передвигаются с током растворителя и занимают наиболее отда-
ленное положение от места нанесения пигмента. Таким образом,
зональное распределение компонентов смеси в колонке адсорбен-
та отражает их относительное положение в адсорбционном ряду.
При разделении смеси пигментов методом молекулярной ад-
сорбционной хроматографии могут быть использованы: а) стек-
лянные колонки, заполненные сорбентом; б) бумага или в) пла-
стинки с тонким слоем сорбента.
Метод хроматографии на пластинках в тонком слое сорбента
получил название «тонкослойная хроматография» [1], которая
имеет ряд преимуществ по сравнению с обычными методами:
• быстрота разделения пигментов;
• возможность разделения пигментов в высоких концентрациях;
• получение более четко ограниченных зон пигментов;
• лучшая элюция пигментов с сорбента.
Для разделения растительных пигментов в тонком слое исполь-
зуют различные сорбенты: глюкозу, сахарозу, маннит, целлюло-
зу, силикагель, окись алюминия, окись магния и др. Сорбенты
различаются размерами частиц и степенью активности. Одним из
наиболее активных сорбентов является окись алюминия; сахароза
относится к группе менее активных сорбентов. Некоторые сорбен-
ты стабилизируют пигменты (маннит, сахароза, глюкоза) и сни-
жают опасность их разрушения при хроматографировании. При
использовании в качестве сорбента силикагеля к нему обычно
добавляют СаСО3 и аскорбиновую кислоту, чтобы предохранить
пигменты от разрушения. Особенно быстро окисляются ксанто-
филлы (лютеин, виолаксантин).
Толщина слоя сорбента в тонкослойной хроматографии дости-
гает 0,1 —0,5 мм; стандартная толщина слоя — 0,25 мм.
Для получения четкого разделения пигментов при использова-
нии метода тонкослойной хроматографии необходимо учитывать
влияние ряда факторов на процесс адсорбции и соблюдать при
хроматографировании определенные условия.
Основными факторами, влияющими на процесс адсорбции,
являются влажность сорбента, концентрация пигментов и темпе-
ратура.
19
Влияние влажности. При добавлении в систему воды активность
сорбента снижается, так как часть его связей насыщается водой.
При этом ослабляется энергия связей сорбента с пигментами,
последние образуют плохо отделенные, размытые зоны. Соответ-
ственно, приготовленная для хроматографирования вытяжка пиг-
ментов не должна содержать воды. На пластинку наносят пигмен-
ты в ацетоне, серном или петролейном эфире. Пигменты из аце-
тона рекомендуется переводить в безводный этиловый или петро-
лейный эфир. При необходимости раствор пигментов высушива-
ют безводным сернокислым натрием. Пластинку с тонким слоем
сорбента перед нанесением пигментов активируют в термостате
15 — 20 мин при температуре 40 °C (для сахарозы) или 100 — 110 °C
(для силикагеля), затем охлаждают в эксикаторе над прокален-
ным СаС12. После нанесения пигментов пятно на пластинке хоро-
шо высушивают струей воздуха.
Зависимость от концентрации пигментов. Концентрация пиг-
ментов, нанесенных на единицу площади сорбента, должна быть
достаточной, чтобы при элюции было получено необходимое ко-
личество пигмента, но не слишком высокой, так как это затруд-
няет разделение и снижает качество хроматограммы. Из уравне-
с
ния —п—- = k = /(с) следует, что адсорбционный коэффициент
р-ре
Оствальда к является функцией концентрации. При низких кон-
Рис. 4. Влияние концентрации пиг-
ментов, нанесенных на единицу
площади сорбента, на качество хро-
матограммы:
А — 0,2 мл вытяжки пигментов; Б —
0,4 мл; В — 0,6 мл; 1 — старт; 2 — хло-
рофилл Ь; 3 — хлорофилл а; 4 — вио-
лаксантин; 5 — лютеин; 6 — Р-каротин
центрациях вещества в раство-
ре значение к высокое и пигмен-
ты хорошо разделяются. При
увеличении концентрации пиг-
ментов значения к уменьшает-
ся, что ухудшает разделение
пигментов. Если концентрация
пигментов на единицу площа-
ди сорбента достигает предель-
ных значений, прочность связи
пигментов с сорбентом снижа-
ется и все пигменты, в том чис-
ле хлорофиллы, «ползут» с то-
ком растворителя вверх (рис. 4).
Пигменты на пластинку луч-
ше наносить не пятном, а по-
лосой, причем количество на-
несенного пигмента на едини-
цу площади сорбента должно
быть равномерным.
Влияние температуры. Ад-
сорбция является процессом эк-
зотермическим, обратный про-
20
цесс — десорбция — идет с поглощением тепла. Отсюда следует,
что при понижении температуры адсорбционное равновесие сдви-
гается в направлении адсорбции и количество адсорбированного
вещества увеличивается. Хроматографирование пигментов ведется
при пониженной температуре; оптимальной является температу-
ра 10—15 °C. Хроматографическую камеру с пластинкой помеща-
ют в кювету со льдом. В этих условиях зоны пигментов получаются
более компактными, а границы зон более четкими.
В ионообменной хроматографии для разделения веществ исполь-
зуют процесс обмена ионов между раствором и высокомолеку-
лярным ионообменным адсорбентом (например, разделение ве-
ществ на ионообменных смолах). Содержащиеся в растворе ионы
обмениваются на эквивалентное количество подвижных ионов,
входящих в состав ионообменника.
В зависимости от химической структуры адсорбента различают
катиониты и аниониты. Катиониты содержат активные кислые груп-
пы —SO3H (сильнокислые) или —СО2Н (слабокислые), Н+-ион
которых способен к обмену. При пропускании смеси веществ че-
рез катионит задерживаются положительно заряженные компо-
ненты. Аниониты содержат основные группы —CH2NR2 (слабо-
основные) или —CH2N+R3 (сильноосновные), благодаря чему
на анионите задерживаются отрицательно заряженные компо-
ненты.
Чем выше заряд молекулы, тем прочнее она связывается с ад-
сорбентом. На этом основано разделение смеси аминокислот. При-
меняя буфер с кислым значением pH (в кислой среде аминокис-
лоты несут положительный заряд), адсорбируют аминокислоты
на катионите. Затем, постепенно изменяя (увеличивая) pH буфе-
ра, последовательно вымывают из колонки отдельные группы ад-
сорбированных аминокислот.
Механизм ионообменной адсорбции отличается от механизма
молекулярной адсорбции. Он основан на электростатическом вза-
имодействии компонентов смеси и адсорбента; действующими
силами в данном случае являются ионные силы. Энергия связи
ионов с адсорбентом значительно выше энергии связи при моле-
кулярной адсорбции. ?
2. Распределительная хроматография основана на различном
распределении компонентов смеси между двумя жидкими несме-
шивающимися фазами — неподвижной и подвижной.
Твердый носитель (бумага, порошкообразное вещество колон-
ки) удерживает на своей поверхности неподвижную фазу раство-
рителя (чаще всего это вода или полярный растворитель). Какой-
либо органический растворитель (фенол, «-бутиловый спирт, кол-
лидин), частично или совсем не смешивающийся с водой, служит
подвижным растворителем. Исследуемую смесь вещества (напри-
мер, смесь аминокислот) наносят на бумагу или на верхнюю часть
21
колонки и пропускают чистый подвижный растворитель. В про-
цессе хроматографии в силу того, что разные компоненты имеют
различные коэффициенты распределения, отдельные аминокис-
лоты, увлекаемые подвижным растворителем, перемещаются с
различными скоростями. Наибольшая скорость будет у компонен-
тов смеси, которые имеют наибольшее сродство к подвижной фазе.
При этом компоненты отделяются друг от друга и распределяют-
ся на различных участках в виде зон или пятен. Для характеристи-
ки положения пятен вводится величина Rf:
где / — расстояние между стартом и положением центра пятна на
хроматограмме; L — расстояние между стартом и фронтом движе-
ния растворителя.
Обычно более полярные вещества, для которых характерно
большее сродство к полярной фазе, имеют меньшее значение Rf.
На принципе распределительной хроматографии основан хро-
матографический метод разделения на бумаге смеси аминокис-
лот, пептидов, органических кислот и др.
Распределение соединений между жидкой и газовой фазами
лежит в основе газожидкостной хроматографии. В этом методе жид-
кая фаза — органическое вещество с высокой температурой кипе-
ния — наносится на инертный носитель. Носитель с закреплен-
ной на нем жидкой фазой помещают в стеклянную или стальную
колонку и через нее пропускают инертный газ (например, аргон
или азот). Колонку ставят в термостат, позволяющий создавать
температуру, при которой анализируемые вещества испаряются.
Разделение исследуемых соединений происходит по мере их про-
движения по колонке вместе с газом-носителем на основе разли-
чий в их коэффициентах распределения между жидкой и газовой
фазами. Газожидкостная хроматография благодаря высокой чув-
ствительности и быстроте разделения находит широкое примене-
ние для количественного и качественного анализа многих веществ
в биохимических исследованиях, например для анализа жирно-
кислотного состава липидов мембран.
3. Осадочная хроматография основана на различной раствори-
мости образующихся осадков. В этом случае разделяемую смесь солей
пропускают через колонку с носителем, пропитанным раствором
осадителя. Носитель — обычно хорошо фильтруемое высокодис-
персное вещество. В качестве осадителя применяется вещество, с
которым осаждаемые ионы образуют трудно растворимые осадки.
Разделение происходит в результате различной растворимости
осадков, образующихся при взаимодействии ионов осадителя с
ионами разделяемых солей. Скорость движения веществ сверху вниз
по колонке выше у более растворимых соединений.
22
Кроме отмеченных выше методов в настоящее время широкое
применение в биохимии находит метод проникающей хроматогра-
фии, или метод гель-хроматографии (гель-фильтрации). В основе
разделения веществ в данном случае лежит принцип молекуляр-
ного сита. Применяемые для разделения веществ материалы —
органические полимеры с трехмерной сетчатой структурой, при-
дающей им свойства геля, при набухании образуют поры опреде-
ленного размера. Набухший гель помещают в стеклянную колон-
ку, уравновешивают с помощью соответствующего растворителя
и используют для разделения веществ. Разделение молекул проис-
ходит в соответствии с их размером и формой. Молекулы, по раз-
мерам соответствующие размерам пор, задерживаются гелем, а
более крупные молекулы, не проникающие в поры молекулярно-
го сита, проходят между частицами геля и могут быть получены в
чистом виде. Большой набор материалов для гель-хроматографии
(декстраны, агарозные гели, полиакриламидные гели и др.) по-
зволяет получать молекулярные сита с заданным размером пор и
разделять разные группы веществ.
1.3. ЭЛЕКТРОФОРЕЗ
Электрофорез — метод разделения органических молекул и
надмолекулярных комплексов в электрическом поле на основе
различий их заряда, размера (молекулярной массы), конфигура-
ции [4, 7, 10, 12, 13, 17]. Он применяется в различных областях
биологии, в том числе при изучении структуры и функций фото-
синтетического аппарата (разделение пигмент-белковых комплек-
сов хлоропластов, получение очищенных препаратов фотосистем
и светособирающих комплексов, исследование АТФ-синтазного
комплекса, состава белков хлоропластов и т.д.).
Возможность применения электрофореза в биологии основана
на том, что многие органические молекулы содержат кислые и
основные ионизирующиеся группы, количественное соотноше-
ние которых определяет суммарный электрический заряд. Вели-
чина и знак заряда зависят от химической природы вещества,
соотношения кислых и основных групп в молекуле, а также от pH
среды. Кроме того, молекулы с близкими по величине зарядами,
но разной молекулярной массой отличаются друг от друга отноше-
нием заряда к массе. Указанные различия определяют возможность
разделения заряженных частиц под действием электрического поля.
В электрическом поле происходит миграция заряженных час-
тиц, причем направление миграции к катоду или аноду будет зави-
сеть от знака суммарного заряда молекулы или надмолекулярного
комплекса, а скорость миграции — от величины заряда, а также от
размеров структуры. Взаимодействие частицы с окружающей сре-
23
дой (трение, электростатические взаимодействия) также являет-
ся важным фактором, влияющим на подвижность биологических
структур в электрическом поле. В результате компоненты смеси
будут разделяться в соответствии с их зарядом и (или) размером,
пространственной организацией, причем одинаковые структуры
будут сосредоточиваться в определенных зонах.
Таким образом, электрофорез позволяет провести фракциони-
рование заряженных органических соединений и препаративно
получить отдельные компоненты из смесей. Кроме того, он может
быть использован для исследования химической природы, заряда
и конфигурации биологических структур. Поэтому электрофоре-
тические методы широко используют для разделения и анализа
смесей низкомолекулярных и высокомолекулярных органических
соединений. В сочетании с иммуноферментным анализом его ис-
пользуют для идентификации органических соединений. Электро-
форез — важнейший метод получения чистых препаратов белков
и нуклеиновых кислот, надежный метод оценки чистоты биоло-
гического материала. Его применяют также для разделения и ис-
следования надмолекулярных комплексов.
В основе метода электрофореза лежит способность заряженных
частиц к движению в электрическом поле. Сила, определяющая
движение заряженной частицы, пропорциональна ее заряду (Q)
и напряженности электрического поля (Е), определяемой как раз-
ность потенциалов по концам рабочего канала (или его участка),
отнесенная к его длине [В/см].
Скорость движения катионов к катоду и анионов к аноду зави-
сит от соотношения между движущей силой электрического поля,
действующей на заряженные ионы, и замедляющими движение
силами взаимодействия между молекулами и окружающей средой.
Если силы, вызывающие движение частицы, уравновешены си-
лой трения частицы и среды, то
QE = 6 л пр,
где Q — электрический заряд частицы; Е — напряженность элект-
рического поля; г — радиус частицы; г] — вязкость среды; v —
скорость движения частицы.
Отсюда подвижность сферической частицы (и) можно опреде-
лить как отношение скорости движения частицы (v) к напряжен-
ности электрического поля [см2/(В-с)]:
и - — - ®
Е 6лгг| ’
Таким образом, подвижность заряженной частицы в однород-
ном электрическом поле прямо пропорциональна заряду и обрат-
но пропорциональна радиусу самой частицы и вязкости среды.
24
При электрофорезе в жидкости, где силы трения минималь-
ны, определяющими величинами для движения частицы будут
являться ее заряд и размер.
Движение частиц при электрофорезе определяют следующие
группы факторов:
• свойства самих частиц исследуемого образца — молекулярная
масса, размер и форма молекулы, электрический заряд, степень
гидратации и диссоциации;
• свойства среды — вязкость, pH, температура, ионная сила, а
в случае применения гель-электрофореза — размер пор.
Кроме того, для проведения электрофоретических исследова-
ний важны такие параметры, как напряженность электрического
поля, зависящая от силы тока, напряжения, длины пути, прой-
денного частицей, продолжительность электрофореза, сопротив-
ление.
Ввиду этого варьирование условий проведения электрофореза
позволяет проводить исследования с различными группами ве-
ществ, осуществлять их фракционирование и исследовать их свой-
ства.
Электрофоретические методы исследования. В настоящее время
в практике биохимических и физиологических исследований ис-
пользуют следующие разновидности электрофореза: высоковольт-
ный электрофорез, непрерывный (проточный) электрофорез, диск-
электрофорез, иммуноэлектрофорез, изоэлектрическое фокусирова-
ние, изотахофорез. В основе этих методов лежит либо свободный (в
свободном растворе) электрофорез, либо электрофорез с исполь-
зованием какого-либо носителя, насыщенного буфером. Подроб-
но об этих методах можно прочитать в специальных методических
руководствах [7, 10, 12, 13, 17].
При свободном электрофорезе сопротивление движению ионов
за счет трения между ними и раствором минимально. Продвиже-
ние ионов происходит быстро. Близкие по структуре и заряду мо-
лекулы передвигаются совместно в виде полос с границами раз-
дела, образованными веществами с несколько различающимися
электрофоретическими подвижностями. В соответствии с этим дан-
ный метод электрофореза назван методом подвижной границы. При-
мерами указанного метода являются изоэлектрофокусирование и
изотахофорез, используемые для определения изоэлектрической
точки органических молекул и комплексов, а также для проверки
чистоты и гомогенности полученных препаратов. Возможно при-
менение этих методов и для препаративной работы.
При электрофорезе с использованием неподвижного носителя
разделяемые вещества движутся в виде отчетливых зон, которые
затем легко обнаруживаются соответствующими аналитическими
методами. Ввиду этого данный метод называют зональным элект-
рофорезом. В качестве носителя применяют хроматографическую
25
бумагу, ацетат целлюлозы, тонкие слои окиси кремния или алю-
миния, крахмальные, агаровые и полиакриламидные гели. Носи-
тель насыщают соответствующим буфером. Выбор носителя и бу-
фера определяется целями эксперимента. Как правило, носители
инертны, хотя и возможно использование носителей, специфи-
чески взаимодействующих с ионами, что создает дополнитель-
ные силы, замедляющие движение тех или иных молекул.
На принципах зонального электрофореза основываются ши-
роко применяемые для анализа белков, нуклеиновых кислот и
надмолекулярных комплексов методы гель-электрофореза (им-
муноэлектрофорез, диск-электрофорез), где в качестве носителя
применяют гели крахмала, агара или агарозы, полиакриламида.
Использование гелей повышает эффективность электрофореза,
поскольку, обладая большей вязкостью, они увеличивают со-
противление трения и делают существенными для разделения
такие параметры фракционируемых веществ, как размер и кон-
фигурация.
Наибольшую разрешающую способность электрофореза уда-
ется получить, используя гели крахмала и полиакриламида. Они
обладают сетчатой структурой с порами определенного размера.
Электрофорез на этих носителях сочетает два принципа разделе-
ния — по электрофоретической подвижности и на основе мо-
лекулярного сита. Движущиеся в гелях частицы испытывают в
зависимости от своего размера различное сопротивление тре-
ния и, следовательно, не только разделяются по своим заря-
дам, но и «просеиваются» в зависимости от размера и форм
молекул.
Широкое распространение получил электрофорез в полиакри-
ламидном геле (ПААГ). Это связано с рядом преимуществ данного
синтетического полимера полиакриламида по сравнению с при-
родным полимером — крахмалом. При использовании ПААГ:
• исключается влияние адсорбции и электроосмоса;
• размер пор геля можно варьировать в широких пределах, при-
чем стандартизировать их величину путем использования опреде-
ленных концентраций полимера (концентрации акриламида от 3
до 30 % соответствуют размерам пор от 0,5 до 0,2 мм);
• сохраняется прозрачность геля даже при высоких концентра-
циях акриламида;
• можно применять калиброванные трубки, что обеспечивает
воспроизводимость результатов;
• разделение веществ проходит очень быстро.
Полиакриламидный гель инертен, устойчив к изменениям тем-
пературы, не поглощает ультрафиолетовый свет при 270 нм, что
позволяет определять белки в этом участке спектра.
Преимущества электрофореза в полиакриламидном геле наи-
более ярко проявляются при разделении белков, несущих одина-
26
ковый заряд, но различающихся по размерам, т.е. белков, не раз-
деляющихся с помощью обычного свободного электрофореза.
Полиакриламидный гель представляет собой продукт сополи-
меризации акриламида СН2=СН—СО—NH2 и какого-либо сши-
вающего агента, например, ^^-метилен-бмс-акриламида
CH2=CH-CO-NH-CH2-NH-CO-CH=CH2. ПАА Г имеет
структуру трехмерной сетки:
I
сн2
NH
СО
I
—сн2—сн—[сн2—СН—]„сн2—сн—[СН2—сн—]ИСН2—
СО со со
I I I
NH NH2 NH2
сн2
NH
I
со
-CH2-CH-[CH2-CH-]„CH2-CH-[CH2-CH-]„CH2-
со со со
I I I
NH2 NH NH2
Реакция сополимеризации акриламида и ^^-метилен-бмс-
акриламида проходит в присутствии катализатора, в качестве
которого используют обычно окислительно-восстановительные
системы, служащие источником свободных радикалов. Чаще всего
катализатором служит смесь 0,1 —0,3 % (весовые проценты) пер-
сульфата аммония либо с 0,1 —0,3% Н^^^'-тетраметилэти-
лендиамином (ТЕМЕД), либо 3-диметиламинопропиолнитрилом
(ДМАП). Возможно также применение системы рибофлавин —
N,N,N',N'-TeTpaMeTJUi3TmreHnnaMHH, которая действует на свету.
Эти катализаторы не изменяют характеристик буферных раство-
ров и гелей, обеспечивают быструю полимеризацию, что важно
для воспроизводимости результатов. Персульфат аммония в этих
системах наиболее предподчтителен, поскольку его можно полу-
чить в высокоочищенном виде. Он относительно устойчив при
температурах, близких к 0 °C, и практически не выделяет молеку-
лярный кислород. Молекулярный кислород, мелкодисперсные
примеси (например, металлы и др.), охлаждение могут замедлить
или подавить полимеризацию.
27
Рекомендуется подобрать условия сополимеризации и стандар-
тизировать их, чтобы гели полимеризовались в течение одного
часа.
Свойства ПААГ (размер пор, вязкость, пластичность) зависят
от степени полимеризации (т. е. от длины цепей) полиакриламида
и от степени сшивки (т. е. от количества включенного N,N'-MeTH-
лен-бис-акриламида). Установлено определенное соотношение меж-
ду размером пор и концентрацией акриламида и метилен-бис-ак-
риламида в геле. Подобрав подходящую концентрацию акрилами-
да, можно получить гели с порами нужного размера. Так, средний
диаметр пор в 7,5 %-х полиакриламидных гелях равен 0,5 нм, а в
30 %-х гелях — около 0,2 нм. Мелкопористые гели (от 10 %) ис-
пользуют при разделении макромолекул, различающихся по раз-
меру при более или менее одинаковом у них отношении заряда к
массе. Крупнопористые гели (5 %) применяют при исследовании
компонентов смеси, имеющих близкие значения молекулярной
массы, но различающихся отношением заряда к массе [13]. Вяз-
кость, пластичность геля зависят также от весового отношения
акриламид: метилен-^мс-акриламид. Эластичные и совершенно
прозрачные гели получаются при отношении 30:1, при этом кон-
центрация акриламида должна быть выше 3 %. Если отношение
ниже 10:1, то гели будут ломкими, твердыми и мутными, если
же оно превышает 100:1, то гели будут вязкими и очень нестой-
кими.
Наиболее широкое применение в биохимических и физиоло-
гических исследованиях нашел диск-электрофорез в полиакрил-
амидном геле (от англ, discontinuous — прерывистый). В этом методе
используется неоднородная (прерывистая) разделяющая система
с полиакриламидным гелем в качестве носителя [7]. Носитель со-
стоит из разных слоев геля, отличающихся друг от друга по раз-
мерам пор, и буферная система включает пару буферов разного
состава и с разными значениями pH.
Как правило, носитель состоит из двух слоев: верхний слой
(примерно 1/3 часть общей величины слоя) — крупнопористый
гель (концентрирующий гель, или гель-прокладка), а нижний —
мелкопористый гель (рабочий, или разделяющий, гель). Роль кон-
центрирующего геля заключается в концентрировании образца по
мере прохождения его через этот гель. В результате образец подхо-
дит к разделяющему гелю в виде очень узкой полосы. Рабочий гель
действует по принципу молекулярного сита и обеспечивает разде-
ление частиц не только в соответствии с их зарядом, но и по
величине и форме.
Таким образом, в ходе диск-электрофореза смесь сначала кон-
центрируется в узкой полосе крупнопористого полиакриламид-
ного геля, а затем в мелкопористом геле ее компоненты распре-
деляются по величине, форме и заряду молекул. Сочетание эф-
28
фекта концентрирования и эффекта молекулярного сита обуслов-
ливает высокую разрешающую способность этого метода.
Подбор концентрирующего и разделяющего гелей, а также со-
става буферных систем играет ключевую роль при диск-электро-
форезе. При подборе буферных систем важен pH и ионный состав
буфера. Как правило, при разделении белков pH буфера в верхнем
сосуде меньше pH буфера, насыщающего гель, и он обладает бо-
лее низкой ионной силой. Благодаря этим различиям образец по
мере прохождения через гель-прокладку концентрируется. Под-
робно механизм концентрирования образца при диск-электрофо-
резе изложен в [7].
В современных исследованиях все большее предпочтение отда-
ют сложным системам диск-электрофореза, где создается гради-
ент пористости ПЛАТ. Электрофорез в градиенте пористости име-
ет ряд преимуществ — самоограничение миграции белков в геле,
непрерывное сужение белковых зон. Это обеспечивает высокую
фракционирующую способность метода. Выбор буфера и электри-
ческого режима электрофореза играет в нем второстепенную роль.
Диск-электрофорез с применением детергентов. При проведе-
нии диск-электрофореза белков в ряде случаев используют детер-
генты. Это могут быть детергенты, несущие заряд, например до-
децилсульфат натрия (ДДС-Na), или нейтральные детергенты, на-
пример тритон Х-100. Цель внесения того или другого детергента
различна.
Внесение заряженного детергента (ДДС-Na) в образец и бу-
фер, насыщающий гель, играет двоякую роль. С одной стороны,
оно препятствует денатурации белков, а с другой — позволяет
разделять белки главным образом по размеру молекул и на основе
их молекулярной массы. Нейтрализуя собственный заряд белков,
додецилсульфат натрия заряжает их примерно одинаково за счет
собственного заряда. В этих условиях белки не денатурируют и раз-
деление их идет в соответствии с молекулярной массой и разме-
рами. Данный способ электрофореза применяют в аналитических
исследованиях для определения молекулярной массы белков.
Использование нейтральных детергентов типа тритон Х-100 или
цетавлона рекомендуется в случае плохой растворимости иссле-
дуемых белков в водных буферах. Это, как правило, имеет место
при анализе мембранных белков. Тритон Х-100 не вызывает дена-
турацию белков (в отличие от мочевины, также иногда использу-
емой при электрофорезе, избыточные концентрации которой мо-
гут вызвать денатурацию белков) и значительно улучшает их ра-
створимость. Образование комплексов с тритоном Х-100 можно
использовать не только для повышения растворимости белков,
но и для их фракционирования.
Возможности и применение диск-электрофореза в ПААГ. Диск-
электрофорез в ПААГ находит широкое применение для исследо-
29
вания физико-химических свойств белков, нуклеиновых кислот и
надмолекулярных комплексов, а также для получения высокоочи-
щенных препаратов этих соединений. Электрофорез в ПААГ обла-
дает рядом достоинств:
1. Определенные преимущества ПААГ как носителя при элект-
рофорезе (см. выше);
2. Отсутствие потребности в дорогостоящей аппаратуре и доро-
гостоящих реактивах. Установка для диск-электрофореза включа-
ет электроды, сосуды для электрофореза, заполненные буферны-
ми растворами, источник питания и маленькие колонки (трубоч-
ки) с полиакриламидным гелем. В последнее время применяется
пластинчатый электрофорез, когда гель вносят в пространство
между двумя стеклами и получают пластинку геля, однородную
по своим свойствам. Это позволяет вести одновременный анализ
ряда проб в стандартизованных условиях;
3. Быстрое выполнение (30 — 60 мин);
4. Использование малых количеств материала (1 — 100 мг);
5. Высокая разрешающая способность для молекул различного
размера (молекулярная масса от 104 Да и менее до 106 Да и более);
6. Высокая чувствительность (до нано- и пикограмм; сравнима
с иммуноэлектрофорезом);
7. Возможность работы с высокими концентрациями веществ;
8. Хорошая воспроизводимость разделения.
Благодаря этим достоинствам диск-электрофорез в ПААГ мо-
жет применяться для самых разнообразных целей. Его используют
как микроаналитический или ультрамикроаналитический метод
для испытания чистоты препаратов, определения относительных
концентраций белков в смеси, исследования изоферментного со-
става смеси белков, определения молекулярной массы, заряда
компонентов смеси и др., а также для препаративного получения
высокоочищенных белков, нуклеиновых кислот и надмолекуляр-
ных комплексов.
В исследованиях фотосинтеза диск-электрофорез в ПААГ ши-
роко используют для анализа белковых компонентов мембран и
водорастворимой фракции хлоропластов. Применяют этот метод
как для характеристики качественного состава белков фотосинте-
тического аппарата, так и для препаративного получения очи-
щенных ферментов хлоропластов, олигомерных комплексов и их
отдельных субъединиц.
1.4. ЦЕНТРИФУГИРОВАНИЕ
Центрифугирование — метод, широко используемый в биоло-
гии для разделения смесей на отдельные компоненты [4, 6, 9, 10].
Его применяют как для разделения и очистки органелл и частиц,
30
получаемых при разрушении клеток, так и для препаративного
выделения отдельных внутриклеточных структур {препаративное
центрифугирование), а также для проведения некоторых анали-
зов, например определения молекулярной массы биологических
макромолекул {аналитическое центрифугирование).
Метод центрифугирования основан на различном поведении в
центробежном поле структур, обладающих неодинаковой плот-
ностью, размерами или формой. Центробежное поле формирует-
ся при вращении ротора на валу привода центрифуги. В зависимо-
сти от радиуса ротора и угловой скорости его вращения создается
центробежная сила, которая и определяет скорость седиментации
частиц.
Центробежную силу {G) рассчитывают по формуле:
G= тго)2,
где г — радиус вращения, см; со — угловая скорость, рад/с; т —
масса частицы.
Угловая скорость может быть выражена через число оборотов в
минуту п [мин-1]. При этом исходят из того, что радиан — это
угол, дуга которого равна радиусу, и один оборот ротора состав-
ляет 2л радиан. В результате
тогда при т = 1
„ (Inn'?
Таким образом, центробежная сила при постоянном г может
быть охарактеризована числом оборотов в минуту.
Другое выражение центробежной силы — единицы g (ускорение
свободного падения, равное 981 см/с2). В этом случае ее называют
относительной центробежной силой (ОЦС), или фактором разделе-
ния {/), и рассчитывают по формуле:
оцс =
4л2«2г
3600-981
или
ОЦС = (1,11 • 10-5)«2г.
В практических целях для пересчета числа оборотов в минуту в
единицы g используют номограмму, построенную в логарифми-
ческом масштабе (рис. 5). Радиус, указанный в номограмме, пред-
ставляет собой средний радиус вращения столбика жидкости в
центрифужной пробирке (т. е. расстояние от оси вращения до се-
31
Радиус, см Относительная центробежная сила, g Частота вращения, мин-
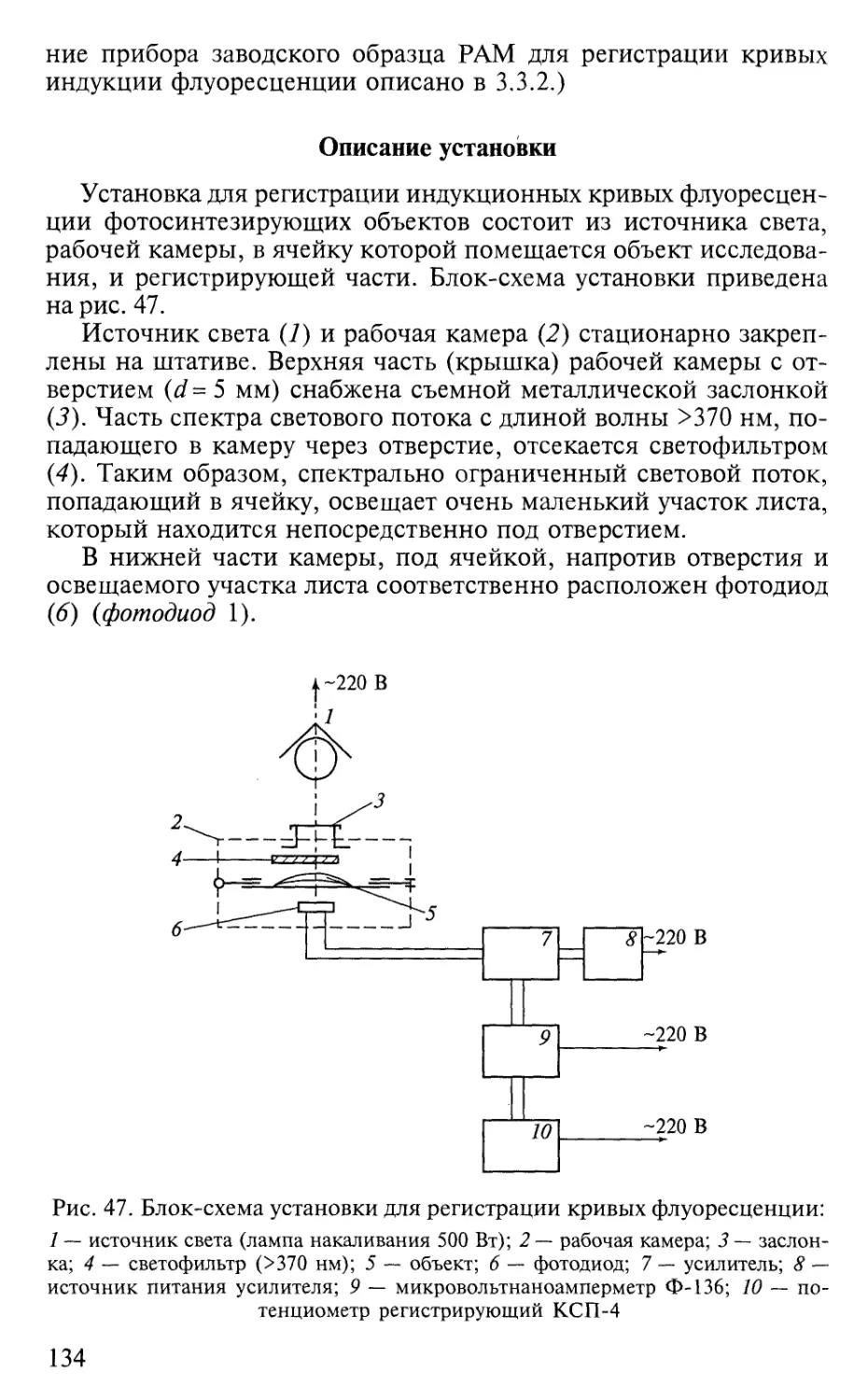
30 5000- г- 50 000
7000-1 1-700 000
6000- -600 000 4500 4 г 45 000
25 5000- -500 000
4000- -400 000 4000 7 40 000
зооо- -300 000 3500 7 35 000
20
19 2000- -200 000
18 3000- 30 000
17 1500- -150 000
16
15 1000- -100 000 2500 - - 25 000
14 800 - 2 80 000
11 700- -70 000
13 600- -60 000
12 500 - -50 000 2000- - 20 000
И — 400- -40 000
10 зоо - -30 000
о 1500 - - 15 000
У 200 - -20 000
- 1400 - - 14 000
8 — 150 - -15 000 1300 - - 13 000
- 1200 - - 12 000
7 100 - -10 000
80 - -8000 1100 - - 11 000
Г 70 - -7000 1000 - - 10 000
О 60 - -6000
50 ~ -5000 900 - - 9000
40 - -4000
5 800 - - 8000
30 - -3000
700 - - 7000
4 20 - -2000
15 - - 1500 600 - - 6000
10 - -1000
3 500 - - 5000
Рис. 5. Номограмма перевода частоты вращения в величину относитель-
ной центробежной силы
редины столбика жидкости). Для определения ОЦС в единицах g
соединяют прямой линией значения радиуса вращения г и часто-
ты вращения ротора центрифуги. Значение центробежной силы
устанавливают по месту пересечения линии со средней шкалой.
Левая сторона шкалы g соответствует левой стороне шкалы часто-
ты вращения, правая — правой.
Номограмму, приведенную на рис. 5, широко используют в
эксперименте для центрифуг разного типа.
32
Центробежная сила — один из факторов, определяющих ско-
рость седиментации частицы. При постоянной центробежной силе
скорость седиментации частицы зависит от размера, плотности
(массы), формы самой частицы, а также от вязкости и плотности
среды суспендирования.
Для частицы, по форме близкой к сферической, время осаж-
дения в жидкой среде рассчитывают на основе закона Стокса, с
изменениями Сведберга и Никольса.
Время седиментации от мениска жидкости до дна центрифуж-
ной пробирки определяется уравнением
2 2о)2гч2 (рч - р) гм’
где t — время седиментации; ц — вязкость среды; гч — радиус
частицЁц рч — плотность частицы; р — плотность среды; гм —
расстояние от оси вращения до мениска жидкости; гд — расстоя-
ние от оси вращения до дна пробирки.
Таким образом, время осаждения частицы пропорционально
вязкости среды и обратно пропорционально квадрату ее радиуса,
разности плотности частицы и среды.
При одной и той же силе разделение частиц по плотности и
(или) по размеру достигается либо путем варьирования времени
центрифугирования, либо за счет распределения седиментирую-
щихся частиц вдоль пробирки, устанавливающегося через опре-
деленный промежуток времени. Для разделения частиц, различа-
ющихся по плотности и (или) размеру, можно также изменять
центробежную силу. Центрифугируя суспензию при ступенчатом
увеличении центробежной силы, выбранной в соответствии со
свойствами частиц, можно добиться поэтапного осаждения час-
тиц разной плотности и (или) размера.
Плотность и вязкость среды являются определяющими факто-
рами при разделении компонентов гетерогенных систем. Варьируя
эти параметры (например, создавая градиенты плотности в цент-
рифужной пробирке), можно добиться разделения и очистки близ-
ких по размерам и (или) массе частиц.
Осаждение частиц несферической формы не подчиняется при-
веденному выше уравнению, поэтому частицы с одинаковой мас-
сой, но разной формы осаждаются при разных скоростях. Эту осо-
бенность используют для исследования конформации биооргани-
ческих молекул методом ультрацентрифугирования.
Способы центрифугирования. Центрифугирование в зависимос-
ти от целей его применения принято подразделять на препаратив-
ное и аналитическое.
Препаративное центрифугирование — метод разделения биоло-
гического материала с целью получения достаточно больших ко-
личеств гомогенных фракций внутриклеточных структур. Он мо-
2 Гавриленко
33
жет быть использован также для получения чистых препаратов
органических молекул, например белка, ДНК из предварительно
очищенных препаратов.
Препаративное центрифугирование подразделяют на диффе-
ренциальное, зонально-скоростное, изопикническое и равновес-
ное в градиенте плотности.
Дифференциальное центрифугирование основано на зависимос-
ти скорости седиментации частиц от их массы (плотности) и
размера от центробежной силы. Разделение гетерогенной систе-
мы на отдельные компоненты производят путем ступенчатого
увеличения центробежной силы. Режим центрифугирования под-
бирают таким образом, чтобы на каждом этапе происходило осаж-
дение на дно центрифужной пробирки лишь одного компонента
смеси. Однако получение абсолютно чистого компонента прак-
тически невозможно. Это связано с тем, что до начала центри-
фугирования все частицы независимо от их массы или размера
распределены равномерно по толще жидкости в центрифужной
пробирке и уже при малой частоте вращения мелкие частицы,
изначально близко находящиеся к дну пробирки, осаждаются.
Поэтому для большей очистки исследуемой фракции ее промы-
вают: осадок после удаления супернатанта гомогенизируют со
свежей порцией среды суспендирования и повторно центрифу-
гируют при соответствующей центробежной силе. Иногда в це-
лях повышения степени очистки фракции частиц промывание
производят 2 — 3 раза.
Метод дифференциального центрифугирования чрезвычайно
широко используют для разделения внутриклеточных структур. Он
позволяет получить фракции ядер, хлоропластов, митохондрий,
рибосом и т.д.
Центрифугирование в градиенте плотности дает возможность
провести дальнейшую очистку исследуемых структур. В основе ме-
тода лежит зависимость скорости седиментации частиц от плот-
ности (вязкости) среды, в которой происходит центрифугирова-
ние.
При использовании данного метода в центрифужной пробирке
готовят градиент плотности, на который наслаивают исследуе-
мый образец. В процессе центрифугирования частицы распределя-
ются в градиенте плотности в соответствии с их размером и раз-
ницей в плотности частицы и среды.
Выбор вещества, используемого для приготовления градиен-
та, зависит от целей исследования. При этом учитываются прони-
цаемость данного вещества внутрь разделяемых структур, его вли-
яние на структуру и функции исследуемых частиц, возможность
получения необходимой плотности. На практике часто применя-
ют растворы сахарозы, фиколла, декстрана или других высокомо-
лекулярных соединений.
34
Градиент плотности может быть ступенчатым или непрерыв-
ным (линейным). При подготовке ступенчатого градиента плот-
ности в центрифужную пробирку с помощью пипетки или шпри-
ца осторожно (по стенке) вносят несколько растворов с последо-
вательно уменьшающейся плотностью. На верхний слой, облада-
ющий наименьшей плотностью, наслаивают образец в виде узкой
зоны. В результате получают дискретные зоны с различной плот-
ностью. В ходе центрифугирования частицы распределяются меж-
ду зонами в соответствии с их размерами и плотностью.
Для приготовления непрерывного (линейного) градиента плот-
ности, как правило, используют специальные установки, кото-
рые автоматически смешивают растворы разных концентраций в
определенных количествах и подают их в центрифужные пробир-
ки. Плавный линейный градиент удается также получить из сту-
пенчатого градиента при длительном его стоянии, когда границы
между зонами сглаживаются. Этот процесс можно ускорить, ак-
куратно перемешивая содержимое пробирки тонкой проволокой
или слегка покачивая пробирку.
В настоящее время разработано несколько способов центрифу-
гирования в градиенте плотности. Подробное описание данных
методов можно найти в практических пособиях по биохимии и
молекулярной биологии [6, 9, 10].
Аналитическое центрифугирование (или седиментационный
анализ) применяют при исследовании свойств предварительно
очищенных внутриклеточных структур или органических молекул.
При использовании небольших количеств препарата метод по-
зволяет определить гомогенность материала, размеры и (или)
массу частиц, конформацию и молекулярную массу молекул. Се-
диментационный анализ проводят с использованием ультрацент-
рифуг, имеющих роторы и регистрирующие системы особой кон-
струкции, позволяющие наблюдать за седиментацией структур в
центробежном поле. Аналитические ультрацентрифуги позволяют
развивать высокие скорости и создавать центробежную силу до
500 тыс. g.
Возможности и применение методов центрифугирования. Цент-
рифугирование как метод фракционирования частиц, различаю-
щихся по массе (плотности) и (или) по размеру, находит широ-
кое применение в биологии. Дифференциальное центрифугирова-
ние и центрифугирование в градиенте плотности позволяют по-
лучить фракции отдельных органелл клетки из гомогенатов раз-
рушенных тканей и произвести очистку их от примесей. Изопик-
ническое центрифугирование в градиенте плотности делает воз-
можным разделение смеси близких по молекулярной массе и раз-
меру частиц на основе различий в их плавучей плотности (напри-
мер, фракционирование микросомальной фракции). Зонально-
скоростное центрифугирование и равновесное центрифугирова-
35
ние в градиенте плотности позволяют разделять высокомолеку-
лярные соединения (например, РНК, ДНК) на основе их моле-
кулярной массы (плотности). Таким образом, центрифугирование
является достаточно эффективным препаративным методом раз-
деления и очистки компонентов в жидких смесях. Кроме того,
ультрацентрифугирование лежит в основе седиментационного
анализа, используемого для определения молекулярной массы
частиц. Это делает центрифугирование важным аналитическим
методом.
1.5. ПОЛЯРОГРАФИЯ
Полярография — способ аналитического определения нейтраль-
ных веществ или ионов на основе измерения вольт-амперных кри-
вых (или кривых напряжение-сила тока), которые можно полу-
чить при электролизе электровосстанавливающегося или элект-
роокисляющегося вещества.
Метод основан на анализе зависимости между силой тока, ха-
рактеризующей скорость электрохимического процесса, и напря-
жением, приложенным к электродам [3, 4, 10, 16]; может быть
использован при условии, что определяемое вещество (или ион)
способно электрохимически восстанавливаться или окисляться на
поверхности электрода.
От внешнего источника на электроды подают возрастающее
напряжение. Если по оси абсцисс отложить значения приложен-
ной ЭД С, а по оси ординат — соответствующие значения силы
тока, то получится кривая, которую называют вольт-амперной
характеристикой, или полярографической волной. По характеру по-
лярографической волны можно определить как природу, так и
концентрацию веществ в растворе.
Физико-химические явления, составляющие электрохимичес-
кий процесс, довольно сложны. Основная особенность электрохи-
мических процессов — зависимость их скорости от потенциала
Г —г^п Е ^==- П Г1 я^АД I Рис. 6. Принципиальная электри- ческая схема подключения по- лярографической ячейки (объяс- нение в тексте) электрода. Это свойство лежит в ос- нове полярографического анализа. На рис. 6 изображена принципиаль- ная электрическая схема, исполь- зуемая в полярографии. Гальвано- метром (Г) измеряется сила тока, проходящего через ячейку (Я) в зависимости от напряжения (Е). Напряжение (1,5 В) создается с помощью источника постоянного тока и делителя напряжения (Hj) в гальванической цепи, состоящей из
36
рабочего (К) и вспомогательного (А) электродов. В качестве рабо-
чего электрода (катода) используют ртутный капельный или пла-
тиновый электрод; в качестве вспомогательного электрода — ка-
ломельный, хлорсеребряный или другой неполяризуемый элект-
род, который служит анодом.
Для получения вольт-амперной характеристики (зависимости
силы тока от напряжения) необходимо изменять напряжение, ре-
гистрируя одновременно силу тока по показаниям гальванометра.
Напряжение, поданное на электролитическую ячейку, вызы-
вает изменения потенциалов обоих электродов, а также расходу-
ется на прохождение тока в растворе:
£=(Фа-Фк) + /Д
где I — сила тока; R — сопротивление раствора; фА и фк — потен-
циалы анода и катода соответственно. Если уменьшить сопротив-
ление раствора добавлением постороннего электролита, то значе-
ние IR будет настолько малым, что им можно пренебречь. При
использовании вспомогательного (неполяризуемого) электрода его
потенциал во время электролиза остается практически постоян-
ным. Это связано с тем, что в данном случае поверхность анода во
много раз больше поверхности катода. Следовательно, все прило-
женное напряжение затрачивается на изменение потенциала ра-
бочего электрода (катода):
£=-Дфк.
Электродные реакции носят многостадийный характер. В каж-
дой из них можно выделить три последовательные стадии:
1) подвод реагирующих частиц вещества (А) из раствора к
поверхности электрода;
2) собственно электрохимическое превращение вещества А в
вещество В (п — число электронов);
3) отвод продуктов реакции в раствор:
Ар.р —> Аэл.д ± пе —> Вэл.д —> Вр.р,
1 2 3
Первая и третья стадии подчиняются законам диффузии, вто-
рая связана с химическим превращением и подчиняется законо-
мерностям электрохимической кинетики.
В условиях, когда электрохимическая (вторая) стадия протека-
ет быстро и скорость суммарного процесса определяется целиком
скоростью подтока вещества А к поверхности электрода, зависи-
мость силы тока от потенциала будет иметь вид кривой, изобра-
женной на рис. 7, называемой полярографической волной. В нача-
ле процесса, при небольших значениях приложенной ЭДС (Е),
когда потенциал электрода недостаточен для осуществления элек-
37
Рис. 7. Полярографиче-
ская волна (объяснение в
тексте)
трохимической реакции с заметной ско-
ростью, сила тока остается почти посто-
янной и лишь очень медленно возрастает
(участок а) — это так называемый кон-
денсаторный ток. Он обусловлен тем, что
при наложении потенциала, недостаточ-
ного для разрядки ионов, ионы подходят
к катоду, но не разряжаются, а образуют
здесь двойной электрический слой — кон-
денсатор. Концентрация вещества А у по-
верхности электрода (сА) при этом не от-
личается от его концентрации в глубине
раствора (сА).
При увеличении отрицательного потен-
циала на катоде до значения, достаточного для разряда ионов на
поверхности электрода, начинается электролитическая реакция вос-
становления. Скорость реакции быстро растет по сравнению со
скоростью увеличения потенциала (участок б). В этой области на-
чинается разряд частиц вещества А, находящихся вблизи поверх-
ности электрода. Поверхностная концентрация вещества А стано-
вится меньше его концентрации в растворе, и возникает диффу-
зионный процесс, направленный на выравнивание концентрации
вещества А и обеспечивающий его подачу к электроду.
Для вычисления скорости диффузионного процесса (/), выра-
женной в электрических единицах (плотности тока), применяют
первый закон Фика'.
J= nFDA И - d)/5A,
где F— число Фарадея; DA — коэффициент диффузии вещества А;
5а — толщина диффузного слоя.
По мере увеличения отрицательного потенциала поверхност-
ная концентрация сА падает, что приводит к увеличению плотности
тока J. Наконец, после того как поверхностная концентрация ве-
щества сА достигает нуля, скорость процесса становится максималь-
ной и перестает зависеть от потенциала электрода (участок в).
Величина Jd, называемая плотностью предельного тока диффузии,
пропорциональна коэффициенту диффузии DA и обратно пропор-
циональна величине диффузного слоя 5А:
Jd = nFD^cl/^.
Величина Z)A постоянна для данных частиц в растворе и обыч-
но имеет порядок IO-5 —10"6. Величина 5А зависит от коэффициен-
та диффузии, кинематической вязкости раствора и скорости дви-
жения жидкости. Для снижения 5А раствор перемешивают, что
резко уменьшает диффузный слой и увеличивает предельный ток
диффузии. При этом важен строго определенный режим переме-
38
шивания растворов, для поддержания которого используют маг-
нитные мешалки или вращающиеся электроды. Скорость диффу-
зионного процесса не зависит от природы электрода или состоя-
ния его поверхности.
Таким образом, при потенциале восстановления иона сила тока
резко возрастает, но после достижения определенной величины,
несмотря на увеличение потенциала, остается почти постоянной.
Плотность предельного тока диффузии, или предельного тока, как
правило, пропорциональна концентрации восстанавливающего-
ся вещества. Пропорциональная зависимость между предельным
током, концентрацией восстанавливаемого вещества в растворе и
является основой количественного полярографического анализа.
При определении природы вещества пользуются потенциалом
полуволны (Д/2), т.е. потенциалом середины полярографической
волны. Он определяется следующим образом: из середины поля-
рографической волны опускается перпендикуляр на ось абсцисс,
тогда расстояние от нуля потенциалов до точки пересечения пер-
пендикуляра с осью абсцисс и будет потенциалом полуволны.
Величина потенциала полуволны не зависит от концентрации
восстанавливающегося иона и определяется только природой са-
мого иона, являясь, таким образом, основой качественного поля-
рографического анализа.
При полном отсутствии в растворе анализируемого вещества
значение силы тока не равно нулю. Это так называемый остаточ-
ный ток. Поправку на остаточный ток необходимо вносить при
проведении анализа. Остаточный ток уменьшает точность опреде-
ления анализируемого вещества при низком содержании его в ра-
створе.
В отличие от диффузионных процессов скорость электрохими-
ческих стадий в большей мере зависит от материала электрода. На
скорость электродной реакции сильно влияет строение двойного
электрического слоя на границе между электродом и раствором.
К поверхности электрода, несущей заряд, притягиваются нахо-
дящиеся в растворе ионы с противоположным знаком заряда. Об-
разуется двойной электрический слой, который может быть рас-
смотрен как плоский конденсатор с расстоянием между обклад-
ками, равным радиусу ионов. Из-за малого расстояния между об-
кладками напряженность электрического поля может достигать
очень больших размеров (порядка 107 В/см), что в значительной
степени сказывается на скорости присоединения (отдачи) элект-
ронов. Строение двойного слоя зависит от состава раствора, нали-
чия в нем постороннего электролита, поверхностно-активных ве-
ществ. Все эти факторы оказывают влияние на скорость электро-
химического процесса.
При использовании твердых электродов скорость процесса оп-
ределяется физико-химическими свойствами поверхности элект-
39
рода. На поверхности твердого электрода обычно присутствуют
адсорбированные газы (О2 или Н2), что может привести к резко-
му изменению скорости реакции.
Недостатком твердых электродов (по сравнению с ртутно-ка-
пельными) является необновляемость поверхности, приводящая
к меньшей стабильности результатов. Для получения более вос-
производимых результатов применяется предварительная обработка
электродов в течение нескольких минут попеременно анодно-ка-
тодной поляризацией при потенциале начала выделения кисло-
рода и водорода. Этой же цели служит покрытие гладкого плати-
нового электрода платиновой чернью, что в сотни раз увеличива-
ет истинную поверхность электрода. На платинированном элект-
роде получается хорошо воспроизводимая кривая с площадкой
предельного тока. Воспроизводимости результатов способствует
также максимальная чистота растворов и поверхности электрода.
Для предохранения последней от механического загрязнения при-
меняют полупроницаемые защитные пленки из полимерных ма-
териалов (например, тефлон).
Количественный полярографический анализ можно осуществ-
лять на основе измерения предельных токов диффузии. При се-
рийных анализах нет необходимости каждый раз снимать полные
полярограммы. Достаточно ограничиться измерением силы тока
при потенциале, примерно соответствующем середине площадки
предельного тока. Однако необходимо убедиться в том, что изме-
ряемая величина действительно соответствует предельному току в
растворе данного состава и на данном электроде. Анализ необхо-
димо проводить при одинаковых условиях перемешивания раствора.
Метод полярографии для измерения концентрации кислорода в
среде в настоящее время широко используют для определения
скорости реакций, связанных с поглощением или выделением
кислорода, например при определении фотохимической актив-
ности хлоропластов.
Метод основан на реакции восстановления кислорода на като-
де. В качестве катода применяют платиновый электрод, в качестве
анода — хлорсеребряный. В этих условиях при электрохимическом
(катодном) восстановлении кислорода силу предельного тока
определяет присоединение четырех электронов в реакции
О2 + 4ё + 4Н+ -> 2Н2О
Начальная реакция на платиновом электроде (катоде) сопро-
вождается образованием ионов ОН“:
О2 + 4ё + 2Н2О -> 4ОН"
Затем ионы ОН" диффундируют к электроду сравнения и обра-
зуют с серебром форму Ag2O:
4ОН“ + 4Ag -> 2Ag2O + 2Н2О + 4е
40
12 3 4
5
6 7 8
Рис. 8. Электрод Кларка:
1 — платина (катод); 2 — сплав платины с медью; 3 — место прикрепления
пленки; 4 — чехол для прижима полупроницаемой пленки; 5 — серебро (анод);
6 — кожух с раствором КС1 в боратном буфере; 7 — пробка с запрессованными
выводами от платины и серебра; 8 — резиновый уплотнитель; 9 — прижимное
кольцо
Таким образом, реакция восстановления кислорода на плати-
не дает гидроксид, который затем вступает в реакцию с электро-
дом сравнения.
На основании полярографической волны, получаемой при
восстановлении кислорода, определяют его содержание в ра-
створе. Напряжение обычно поддерживают постоянным в ин-
тервале 0,6 —0,8 В. Сила тока, возникающего при этом, прямо
пропорциональна концентрации кислорода в среде.
Для анализа концентрации кислорода в среде наиболее часто
применяют платиновый закрытый электрод Кларка (рис. 8). Он со-
стоит из платинового катода и серебряного анода, погруженных в
раствор концентрированного КС1 и отделенных от исследуемого
раствора мембраной. Мембрана (обычно изготовленная из тефло-
на) защищает электроды от загрязнения химическими реагента-
ми, содержащимися в исследуемом растворе.
В течение опыта концентрация О2 в растворе постоянно меня-
ется. При этом прибор регистрирует изменение силы предельного
тока диффузии, что дает представление о динамике окислитель-
но-восстановительных процессов в препарате.
Глава 2
ХАРАКТЕРИСТИКА ПИГМЕНТНЫХ СИСТЕМ
ФОТОСИНТЕТИЧЕСКОГО АППАРАТА
К основным группам пигментов растительного происхожде-
ния, принимающих непосредственное участие в фотосинтезе, от-
носятся:
• хлорофиллы — соединения тетрапиррольной природы, маг-
ний-порфирины,
• каротиноиды — пигменты, относящиеся к группе полиизо-
преноидов,
• фикобилины — дополнительные пигменты водорослей, имею-
щие тетрапиррольную незамкнутую структуру.
Каждая группа пигментов включает большое число различных
пигментных форм, из которых наиболее важными у высших рас-
тений являются: протохлорофилл, хлорофиллы anb, каротины, ксан-
тофиллы (лютеин, виолаксантин, неоксантин и др.). Структурные
формулы некоторых пигментов даны на рис. 9.
В настоящее время любая работа в области фотосинтеза требует
более или менее детальной характеристики пигментных систем.
Это необходимо не только при проведении специальных исследо-
ваний метаболизма пигментов и изучении влияний условий рос-
та, питания и интенсивности света на развитие и фотосинтети-
ческую продуктивность растений. Оценка содержания фотосинте-
тических пигментов важна также в работах, связанных с изучени-
ем механизма фотосинтеза, где все расчеты активности исследуе-
мых реакций (выделение кислорода, скорость электронного транс-
порта, синтез АТФ, фиксация углекислого газа и др.) ведут на
единицу хлорофилла.
Характеристика пигментных систем включает количественную
оценку содержания в ткани хлорофиллов а и Ь, их суммы (а + Ь),
отношения (а/b), содержания каротина (кр), ксантофиллов (кс),
суммы всех каротиноидов (кр + кс), отношение зеленых пигмен-
тов к желтым (а + Ь)/(кр + кс). Во многих случаях необходимо полу-
чение данных об уровне содержания в ткани пигментов — пред-
шественников хлорофилла (протохлорофиллида, протохлорофил-
ла), о динамике превращения протохлорофиллида (протохлоро-
филла) в хлорофиллид (хлорофилл).
42
СН
Н2С
Н2С
Н2С
но^,
W
сн2 (СН=О Ь)
Хлорофилл а
СН
СН
СН
с
сн3
сн3
СН
С СН
С СН
СН СН
С
з
сн3
сн3
16 с
5
сн2
СН ' снпсн13сн15сн14
с
^сн3
12'
с сн
Р-Каротин
сн3
с
2>сн
2
СН
сн3
с
сн3
с сн сн
СН3 СН2
2
сн сн
сн3 сн
сн
сн сн
сн
сн3
сн сн
сн2
сн сн сн
с сн
с НС
СН3 сн3 сн н
СН2 СНз
Лютеин
сн3
Виолаксантин
Неоксантин
Рис. 9. Структурные формулы основных пигментов растений
43
Важнейшей характеристикой пигментных систем являются
спектры поглощения и флуоресценции. Они содержат информа-
цию об индивидуальных формах пигментов, присутствующих в
биологической системе, их состоянии, взаимодействии и взаимо-
превращении в процессах биосинтеза и т.д. Особенно перспектив-
ны в этом отношении низкотемпературные спектры поглощения
и флуоресценции. Регистрацию низкотемпературных спектров
широко применяют в последнее время для характеристики натив-
ных форм пигментов в растительной ткани, исследования пиг-
ментного состава отдельных фотосистем в структуре хлороплас-
тов и различных фракциях, полученных при обработке хлоропла-
стов детергентами.
Высокочувствительным методом исследования пигментных
систем является дифференциальная спектрофотометрия. Этот ме-
тод эффективно используется в настоящее время для количествен-
ной и качественной характеристики пигментов, цитохромов и
других компонентов электрон-транспортной цепи хлоропластов,
при исследовании фотоиндуцируемых изменений спектров погло-
щения пигментов и цитохромов в связи с изучением многих воп-
росов механизма фотосинтеза.
При исследовании процессов биосинтеза или распада пигмен-
тов важным показателем является активность хлорофиллазы —
фермента, участвующего в процессах деградации хлорофилла. Су-
щественное значение имеет характеристика прочности хлорофилл-
белковых комплексов.
Для количественной оценки содержания пигментов в настоя-
щее время используют спектрофотометрический метод, который
основан на регистрации характерных спектров поглощения от-
дельных групп пигментов. Он позволяет получить данные о содер-
жании хлорофиллов а и b без предварительного выделения их из
суммарной вытяжки пигментов. Возможно также определение ка-
ротиноидов в суммарной вытяжке пигментов. Однако для точного
количественного определения каротиноидов необходимо предва-
рительное отделение их от хлорофиллов, так как максимумы по-
глощения этих двух групп пигментов значительно перекрываются
в сине-фиолетовой области спектра. Для разделения пигментов
используют ряд методов хроматографии. Сочетание спектрофото-
метрического метода определения пигментов с хроматографией
позволяет получить полную качественную и количественную ха-
рактеристику пигментных систем хлоропластов.
Важнейшим условием количественного определения пигмен-
тов является полное экстрагирование их из растительной ткани.
Экстракция пигментов. Пигменты могут быть экстрагированы
из свежего или фиксированного материала. При выборе экстраги-
рующих веществ необходимо учитывать растворимость пигментов
и возможность их выделения данным растворителем из пигмент-
44
Таблица 1
Дипольный момент некоторых растворителей
Растворитель Химическая структура Дипольный момент, ед. Дебая (Д)
Ацетон н3сх с=о Н3СХ 2,71
Метанол СН3ОН 1,64
Этанол СН3СН2ОН 1,63
Этиловый эфир Н5С2\ О Н5С2 1,14
Хлороформ СНС13 0,95
Сероуглерод s=o=s 0
Гексан СН3(СН2)4СН3 0
Петролейный эфир — 0
но-липопротеидного комплекса, в виде которого пигменты нахо-
дятся в пластидах.
В зависимости от химического строения различают растворите-
ли полярные (спирты, ацетон) и неполярные (петролейный эфир,
гексан, бензин и др.). Степень полярности растворителя опреде-
ляется дипольным моментом его молекулы (табл. 1).
Хлорофиллы и каротиноиды — в основном липофильные со-
единения, хорошо растворяются во всех растворяющих липиды
веществах: спирте, ацетоне, серном эфире, бензине, петролей-
ном эфире и т.д. Однако полное извлечение пигментов из расти-
тельного материала достигается только при использовании по-
лярных органических растворителей или смесью полярных и не-
полярных растворителей. Полярные органические растворители,
вызывая денатурацию белка и нарушая связи пигментов с липо-
протеидным комплексом, обеспечивают быструю экстракцию всех
пигментов*. Чистые неполярные растворители не нарушают связи
пигментов с белком. Поэтому бензин, петролейный эфир и дру-
* Из сухого материала абсолютный спирт пигменты не экстрагирует, так как
для гидролиза связей с белком необходимо присутствие воды. При работе с сухим
материалом используют 80 %-й ацетон или 90 %-й спирт.
45
гие неполярные растворители экстрагируют в нормальных усло-
виях только каротин, часть ксантофиллов и очень небольшое ко-
личество (1 — 5 %) хлорофиллов. Для полной экстракции пигмен-
тов неполярными растворителями необходима примесь полярно-
го растворителя (например, 2 — 3% этанола в петролейном эфи-
ре). Для выделения пигментов используют 80 — 85 %-й ацетон или
90 %-й спирт.
Экстракцию проводят как можно быстрее. Пигменты экстраги-
руют последовательно несколькими порциями чистого раствори-
теля, пропуская каждый раз раствор пигментов через стеклянный
фильтр. При растирании растительного материала с растворите-
лем необходимо добавлять небольшое количество СаСО3, MgCO3
или 1 М раствора NH4OH для предотвращения феофитинизации
пигментов.
Всю подготовительную работу с пигментами ведут в затемнен-
ном помещении, на холоде.
2.1. КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ ПИГМЕНТОВ
МЕТОДОМ АБСОРБЦИОННОЙ СПЕКТРОФОТОМЕТРИИ
Характерным свойством растительных пигментов является их
способность поглощать лучи света определенной длины волны.
Поглощение (или экстинкция) в ультрафиолетовой и видимой
области связано с присутствием в молекулах хромофорных групп,
определяющих окраску вещества, форму спектра, интенсивность
поглощения. Как правило, это группы с системой сопряженных
связей, которые могут быть циклическими (ароматические ами-
нокислоты, порфирины) или линейными (полиены, каротинои-
ды и др.). У хлорофиллов таким хромофором является макроцикл,
образованный тетрапиррольной структурой.
Поглощение энергии света молекулой пигмента сопровожда-
ется изменением ее энергетического состояния (рис. 10). При по-
глощении кванта света молекула переходит из основного состоя-
ния (So) в одно из возбужденных состояний (S*), что обусловле-
но переходом электрона на более высокий энергетический уро-
вень. Электронно-колебательные переходы при поглощении кван-
тов сопровождаются увеличением энергии системы. Такие перехо-
ды берут начало на самых нижних колебательных уровнях основ-
ного невозбужденного состояния (обычно синглетного So) и за-
вершаются на одном из колебательных подуровней возбужденных
электронных уровней (S*). При этом возникают одна или несколько
довольно широких полос поглощения, характерных для молеку-
лярных спектров.
Для поглощения пигментом из общего потока электромагнит-
ного излучения определенных квантов необходимо строгое соот-
46
Рис. 10. Схема электронных переходов для магний-порфиринов в поляр-
ном растворителе:
50 — основное состояние; — первое синглетное возбужденное состояние; S2 —
второе синглетное возбужденное состояние; 7\ — первое триплетное возбужден-
ное состояние; Т2 — второе триплетное возбужденное состояние
ветствие энергии кванта и энергии перехода между основным и
возбужденным уровнем молекулы:
^^кванта = Д-^мол> ИЛИ /2VKBaHTa = Еп — Eq,
где Eq — энергия основного уровня; Е„ — энергия возбужденного
уровня; Д£мол — энергия перехода, разность энергий между уров-
нями.
Положение максимумов в спектре поглощения соответствует
условию равенства энергии кванта и разности энергии между ос-
новным и электронным или электронно-колебательным уровнем.
Так, хлорофилл а имеет два основных максимума поглощения,
что соответствует двум уровням возбуждения этой молекулы: в
красной области спектра (переход 50 ^*) и в синей области —
так называемая полоса Соре, характерная для циклических тетра-
пирролов (переход 50 -> Таким образом, положение максиму-
мов в спектре отражает структуру электронно-колебательных уров-
ней, которая специфична для молекулы и тесно связана с ее строе-
нием, химическими свойствами, а также с характером влияния
на нее окружения. Это и лежит в основе спектрального анализа
молекул.
Форма спектра поглощения служит для идентификации моле-
кул (качественный анализ) и исследования их состояния в клетке.
Поглощение (обозначается А или D) в максимуме используется
на основании законов Бугера—Ламберта и Бера для количествен-
ного анализа. Обычно спектр поглощения представляется в виде
47
зависимости поглощения от длины волны (X). Единицами измере-
ния длин волн служат микрометры (1 мкм = 10-4 см), нанометры
(1 нм=10-7 см=10-9 м).
В соответствии с формулой Планка E=h\ = hc/X энергия кван-
та прямо пропорциональна частоте электромагнитных колеба-
ний (v) и обратно пропорциональна длине волны (X). Длина вол-
ны и частота связаны соотношением: X = c/v, где с — скорость
света, равная 3 • 1О10 см-с-1, или 3 • 1017 нм-с-1. Постоянная Планка
h = 6,625 • 10-34 Дж-с. Частота измеряется в обратных секундах [с-1]
и выражается отношением скорости распространения излучения
с к длине волны X [нм]:
v = с/Х = 3 • 1017/Х.
В спектрофотометрии часто используют показатель «волновое
число» (v), характеризующий число волн в 1 см. Волновое число —
величина, обратная длине волны, измеряется в [см-1]:
v [см-1] = 1/Х [см] = 107/Х [нм].
Так, при Х = 250 нм v =40 000 см-1.
Волновое число (v), пропорциональное частоте электромаг-
нитных колебаний v = v/c и, следовательно, пропорциональное
энергии перехода Е (по формуле hv = &ЕМОЛ), часто используется
для энергетической характеристики уровней электронных пере-
ходов в молекуле. Соотношение между длиной волны (X), волно-
вым числом (v) и энергией квантов приведено в табл. 2.
Для определения концентрации вещества в растворе необхо-
димо знать молярный коэффициент поглощения (е) или удельный
коэффициент поглощения (а) данного вещества. В настоящее время
значения а (е) определены для многих пигментов. Значения ко-
эффициентов поглощения необходимо брать с учетом природы
используемого растворителя (табл. 3,4).
Таблица 2
Энергия видимой области спектра
Длина волны, нм Волновое ЧИСЛО, СМ’1 Энергия в расчете на 1 моль квантов Энергия 1 кванта, эВ
Дж ккал
700 14 300 17,10-104 40,87 1,77
600 16 600 19,95-104 47,68 2,07
500 20000 23,95- 104 57,24 2,48
400 25 000 29,93-104 71,53 3,10
48
Таблица 3
Максимумы поглощения и молярные коэффициенты поглощения (е)
для хлорофилла а в различных растворителях
(по Г. И.Гуриновичу и др., 1963)
Растворитель X Соре, нм X], нм 10*4, л • молы1- см-1
Метанол 432,0 665,7 6,66
Этанол 431,5 664,7 6,94
Этиловый эфир 428,8 660,6 8,51
Ацетон 430,1 662,0 7,66
Изопропанол 431,6 664,2 7,44
Бензол 432,8 665,6 7,82
Таблица 4
Удельные коэффициенты поглощения (л) хлорофиллов а и b
в различных растворителях
(по данным ряда авторов)
Растворитель Длина волны, нм Удельные коэффициенты поглощения, л-г’1-см-1 Автор
хлорофилла а хлорофилла b
Этиловый эфир 660 642,5 102,0 16,3 4,5 57,6 С. L. Comar, F. R. Zschheile, 1942
Ацетон (вод- ный 80 %-й) 663 645 82,04 16,75 9,27 45,6 G. Mac-Kinney, 1941
Метанол 665 650 74,5 27,6 18,3 36,4 G. Mac-Kinney, 1941
Ацетон 662 644 440,5\ 104,5 22,68 57,0 4,83 47,72 57,0 D.Wettstein, 1957
Ацетон 661,6 644,8 82,6 46,86 H. K. Lichtenthaler, 1987
Метанол 665,2 652,4 70,54 35,27 H. K. Lichtenthaler, 1987
49
На измерении количества света, поглощенного пигментами,
основаны методы их количественного определения [7, 9, 17, 18,
24, 25, 29, 32].
Основные принципы спектрофотометрии приведены в гл. 1.
^4
Общая формула спектрофотометрии с = — может быть исполь-
ad
зована лишь для определения концентрации чистых пигментов,
выделенных из общей смеси. В этом случае определяют поглоще-
ние раствора (Л) в максимуме поглощения пигмента и, подста-
вив в формулу соответствующее значение а (см. табл. 4), рассчиты-
вают концентрацию.
Для расчета концентраций хлорофиллов а и b при определе-
нии их в смеси пигментов без предварительного разделения ис-
пользуют более сложные формулы.
Вследствие перекрывания спектров поглощения хлорофиллов
а и b оптические плотности в красной области являются суммар-
ной величиной:
А ={а\сх + d2c2}d,
A'=(a\cx + a2c2}d,
где А и А' — поглощение в максимумах поглощения хлорофиллов
а и Ь; ах и а2 — удельные коэффициенты поглощения (экстинк-
ции) для хлорофиллов а и b при X'; а"х и 02 —удельные коэффици-
енты поглощения для хлорофиллов а и b при А.
Подставив в эти уравнения соответствующие значения удель-
ных коэффициентов поглощения, получим для хлорофиллов а и b
в ацетоне следующую систему уравнений:
А ббз= 82,04 • са + 9,27 • Q,
А 645 = 16,75 • са + 45,6 • С/,.
Решая эту систему уравнений относительно са и сь, имеем фор-
мулы для определения са и сь в общей смеси (формулы Дж. Мак-
Кинни, 1941).
На этом принципе основан двухволновой метод определения
концентрации хлорофиллов а и b в общей смеси пигментов. Двух-
волновой метод сводится, таким образом, к следующему: с помо-
щью спектрофотометра устанавливают величину поглощения (А)
суммарной вытяжки пигментов при двух длинах волн, соответ-
ствующих максимумам поглощения пигментов в данном раство-
рителе (-46бо,б42 — в этиловом эфире и А 663>645 — в ацетоне), и затем
рассчитывают концентрацию пигментов [мг/л] по одной из при-
веденных ниже формул:
(1) в 80 %-м ацетоне (G. Mac-Kinney, 194Гу.
са- 12,7-Лббз _ 2,69 • Лб45;
50
cb - 22,9 • Лб45 - 4,68 • Лббз;
са+— 8,02 • Л6бз + 20,2 • -^4б45>
(2) в 80%-м ацетоне (L.P. Vernon, 1960\.
са = 11,63 Лбб5 ~ 2,39 • -4б495
сь = 20,11 • Л649 - 5,18 •
са + ь — 6,45 • ^4бб5 + 12,72 • -4б49,
(3) в 85 %-м ацетоне (G. Robbelen, 1957Y
Са = 10,3 • ЛббЗ “ 0,918 • ^644;
сь= 19,7 • Лб44 - 3,87 • Л663;
Ca + b ~ ^;4 • Л663 + 18,8 • ^644;
(4) в 100 %-м ацетоне (D. Wettstein, 1957\.
са = 9,784 • Лбб2 - 0,990 • А^;
сь = ^ ,426 • Лб44 - 4,650 Лбб2j
са + ь= 5,134 • Лбв2 + 20,436 • А$44;
(5) в 96 %-м этаноле (I.F. Wintermans, A. De Mots, 1965Y
са~ 13,70-^665 ~ 5,76-449;
сь - 25,80 • Лб49 - 7,60 Лбб5?
са + ь= 6,10 • Л665 + 20,04 • Лб49 = 25,1 • Л654;
(6) в этиловом эфире (С. L. Comar, F. P.Zscheile, 1942\.
са = 9,93 • ЛббО “ 0,78 • ^642,55
СЬ= 12,6 -^642,5 “ 2,81-Лбб(Ь
са +— 2,12 • у4ббо + 16,8 • ^4б42,5 •
Формулы получены на основе экспериментальных данных мно-
гих лабораторий при использовании различной спектральной тех-
ники, поэтому имеют несколько отличающиеся коэффициенты.
Выбор формулы зависит от используемого растворителя и приме-
няемой в эксперименте спектральной аппаратуры.
Совершенствование техники спектрофотометрических измере-
ний позволило уточнить коэффициенты полученных ранее фор-
мул, а также сделало возможным определение в суммарном экст-
ракте пигментов не только хлорофиллов а и Ь, но и каротиноидов
[мг/л]:
(2) в 80%о-м ацетоне (Н. К. Lichtenthaler, 1987Y
са = 12,21 -Лббз _ 2,81 - А 64б;
51
сь - 20,13 • 446 - 5,03 -Л663;
_ 1000-470 -3,27-са -100-Q
сИр - 229
(8) в 100 %-м ацетоне (Н. К. Lichtenthaler, 1987):
са~ 11,24-4б1,б ~ 2,04-Лб44 8;
сь= 20,13 • Л644 8 - 4,19 • ^661,6?
_ 1000 • 4?о - 1,90 • са - 63,14 • сь
е“₽ ’ 1000
В работах Р.Порра с сотрудниками (R. J.Porra et al., 1989) полу-
чены более точные значения молярных и удельных коэффициен-
тов поглощения для хлорофиллов яи6в80%-м ацетоне (табл. 5).
На основании полученных коэффициентов поглощения рас-
считаны уравнения для определения молярных и весовых концент-
раций хлорофиллов а и Ь:
(9) расчет молярных концентраций хлорофиллов [мкмоль/л] в 80 %-м
ацетоне (R. J. Porra et al., 1989):
~ 13,71 • ^4ббз,б ~ 2,85 • -446,65
сь ~ 22,39 • 4>4б,б — 5,42 • 4>бз,б,
са + ь~ 19,54 Л646 6 + 8,29 -4бз,б;
(10) расчет весовых концентраций хлорофиллов [мг/л} в 80 %-м
ацетоне (R. J. Porra et al., 1989):
12,25 • 4бз,б 2,55 • -'446,бэ
с6 = 20,31 -446,6 - 4,91 -463,6;
са + ь~ 17,76 • 446,6 + 7,34 • 463,6-
Таблица 5
Коэффициенты поглощения хлорофиллов в 80 %-м ацетоне
(R.J. Porra et al., 1989)
Длина волны, нм Коэффициент поглощения
хлорофилла а хлорофилла b
миллимолярный удельный миллимолярный удельный
663,6 76,79 85,95 9,79 10,78
646,6 18,58 20,79 47,04 51,84
52
Для быстрого определения суммы хлорофиллов (а + Ь), напри-
мер в суспензии хлоропластов, могут быть использованы следую-
щие формулы [мг/л]:
(11) в 80 %-м ацетоне (D. I.Arnon, 1949)'.
_ ^652 Ю00 - 29 0 • А •
Са + Ь - -24^ “ Лб52’
(12) в 80 %-м ацетоне (I. Bruinsma, 1961)'.
_ 452-1000 _
с»+4’ 36,о”” ~27’8'^2-
Каротиноиды определяют после разделения пигментов мето-
дом хроматографии на бумаге или колонке (см. 2.2).
Удельные коэффициенты поглощения каротиноидов, получен-
ные в ряде работ, приведены в табл. 6 и 7.
Таблица 6
Основные максимумы поглощения и удельные коэффициенты
поглощения каротиноидов в различных растворителях [8]
Пигмент Растворитель Основной максимум поглощения, нм Удельный коэффициент поглощения
[3-Каротин Петролейный эфир 452 260
Лютеин Этанол 445 258
Виолаксантин Этанол 442 225
Неоксантин Этанол 438 227
Таблица 7
Удельные коэффициенты поглощения каротина и ксантофиллов
(по данным ряда авторов)
Растворитель Длина волны, нм Удельный коэффициент поглощения Автор
каротина ксанто- филлов суммы кароти- ноидов
80 %-й спирт + 20%-й этиловый эфир 450 445 235(236) 215 — К. Miller, 1934
Ацетон 440,5 — — 213 D.Wett- stein, 1957
53
Концентрацию пигментов [г/л] группы каротиноидов (кароти-
на — кр и ксантофиллов — кс) рассчитывают по формулам:
_ ^450 . _ ^445
кр 236 ’ кс 215 ‘
Для определения каротиноидов (кар) в суммарной вытяжке
пигментов [мг/л] может быть использована формула Д. Веттштей-
на (D. Wettstein, 1957):
^кар ~~ 4,695 • Д440 5 — 0,268 (са + /,).
2.1.1. Анализ пигментов с использованием экстрактов
пигментов в ацетоне и этиловом эфире
Спектрофотометрический анализ ацетоновой вытяжки пигмен-
тов — это наиболее распространенный метод определения их со-
держания в листе. Экстракцию пигментов из листьев производят
100 %-м или 80 %-м ацетоном. Экстракты содержат всю сумму
пигментов листа (хлорофиллы а и Ь, каротиноиды) и могут быть
использованы для количественной оценки содержания пигмен-
тов в листе. Эти экстракты можно также использовать для приго-
товления экстрактов пигментов в неполярных растворителях (на-
пример, в этиловом эфире).
Ход работы
Работа включает следующие этапы: 1) приготовление ацетоно-
вого экстракта пигментов; 2) спектрофотометрический анализ аце-
тонового экстракта пигментов; 3) приготовление эфирного экст-
Стеклянный
фильтр
Рис. 11. Колба Бунзена со
стеклянным фильтром для
получения экстракта пиг-
ментов
ракта пигментов.
1. Приготовление ацетонового экстрак-
та пигментов. Навеску свежего раститель-
ного материала (0,2 —0,3 г) тщательно
растирают в фарфоровой ступке с не-
большим количеством 80 %-го ацетона
(2—3 мл), чистого кварцевого песка и
мела или углекислого магния. После на-
стаивания (2 — 3 мин) экстракт перено-
сят на стеклянный фильтр и фильтруют
в стеклянную пробирку, вставленную в
колбу Бунзена, соединенную с водо-
струйным насосом (рис. 11). Экстракцию
пигментов небольшими порциями чис-
того растворителя повторяют на фильт-
ре 3 — 4 раза до полного извлечения пиг-
54
ментов. Последние порции фильтрата, собранные в пробирку, дол-
жны быть бесцветными.
Экстракты количественно переносят в мерную колбу на 50 мл
и объем вытяжки доводят чистым растворителем до метки. Полу-
ченная ацетоновая вытяжка содержит сумму зеленых и желтых
пигментов.
2. Спектрофотометрический анализ ацетонового экстракта пиг-
ментов. Концентрация пигментов в вытяжке может быть опреде-
лена на фотоэлектроколориметре по калибровочной кривой или
непосредственно на спектрофотометре с использованием для рас-
чета концентраций пигментов соответствующих формул (в зави-
симости от используемого растворителя).
Для количественного определения часть полученного экстрак-
та наливают в круглую кювету спектрофотометра диаметром 1 см.
Вторая кювета заполняется чистым растворителем (80 %-м ацето-
ном). Кюветы помещают в кюветную камеру спектрофотометра и
определяют поглощение (А) при длинах волн, соответствующих
максимумам определяемых пигментов.
Установив концентрацию пигмента в вытяжке, определяют его
содержание в исследуемом материале с учетом объема вытяжки и
массы пробы:
F = cV
где F — содержание пигмента в растительном материале, мг/г
сырой массы; с — концентрация пигментов, мг/л; V — объем вы-
тяжки пигментов, мл; Р — навеска растительного материала, г.
Количество пигментов выражают в миллиграммах на единицу
сырой или сухой массы, на единицу площади листа и в % от
сухой (сырой) массы.
Обычно в нормальных зеленых листьях содержание хлоро-
фйлла колеблется от 0,5 — 3 мг на 1 г сырой массы при отноше-
нии а/b = 2,5 — 3,0. Содержание каротиноидов — 0,1 — 0,5 мг/г сы-
рой массы. Отношение ксантофиллов (кс) к каротину (кр) равно
3 — 5, отношение (а + 6)/(кс + кр) = 3 — 8.
3. Приготовление эфирного экстракта пигментов. При работе с
пигментами часто используют не ацетоновый, а эфирный экст-
ракт пигментов. Для этого пигменты из ацетона переводят в эти-
ловый эфир, предварительно очищенный от перекисей и пере-
гнанный.
Внимание! Серный эфир является легковоспламеняющейся,
взрывоопасной жидкостью. При работе с ним следует соблю-
дать инструкции по технике безопасности работы с органичес-
кими растворителями и другими ЛВЖ. При нагревании и пере-
гонке использовать только закрытые нагревательные приборы,
55
перегонку осуществлять в вытяжном шкафу в круглодонных кол-
бах из тугоплавкого стекла с использованием водяной бани.
Предварительно необходимо очистить этиловый эфир от пере-
кисей.
Очистка серного эфира от перекисей. К этиловому эфиру в де-
лительной воронке приливают раствор FeSO4 (30 мл FeSO4 • 7 Н2О +
+ 55 мл кислой воды, содержащей 3 г концентрированной H2SO4 на
100 мл воды) из расчета 10 — 20 мл на 1 л эфира. Содержимое
воронки осторожно встряхивают и после отстаивания нижний слой
FeSO4 сливают. К эфиру добавляют 5 —10 мл 0,5 %-го водного ра-
створа КМпО4 для превращения примеси ацетальдегида в уксус-
ную кислоту и осторожно встряхивают. Нижний слой сливают.
Процедуру повторяют, если раствор КМпО4 заметно изменяет
окраску. Эфир промывают 5 %-м раствором NaOH (по 10 — 15 мл
до тех пор, пока промывные воды не станут совершенно бесцвет-
ными). Промытый эфир помещают в сухую темную бутыль, до-
бавляют 150 — 200 г безводного СаС12 и оставляют на 24 ч для
просушки.
Перегонка эфира осуществляется непосредственно перед опы-
том. Очищенный от перекисей эфир отфильтровывают и перего-
няют на водяной бане при температуре 35 °C (рис. 12). Перегнан-
ный эфир хранят в темноте.
Приготовление эфирного экстракта пигментов. Из ацетоново-
го экстракта берут 25 мл и переносят в делительную воронку. До-
бавляют 25 мл этилового (серного) эфира и около 1 мл воды (по
каплям). Смесь осторожно встряхивают. После расслоения ниж-
ний водно-ацетоновый слой сливают. Эфирный раствор пигмен-
тов 5 раз промывают дистиллированной водой. После промывания
в воронку добавляют немного безводного сернокислого натрия,
Рис. 12. Установка для перегонки этилового эфира
56
встряхивают. После расслоения водный слой сливают, а верхний
количественно переносят в мерную колбу, доводят до метки и
используют для анализа фотометрическим методом. Производят
измерения при длинах волн максимального поглощения хлоро-
филла аи b (660 и 642,5 нм) и по формулам, представленным на
с. 51, рассчитывают концентрацию пигментов в вытяжке.
Оформление результатов. Результаты представляют в виде таб-
лицы, где приводят первичные данные фотометрического анали-
за, расчет концентрации пигментов в вытяжке, содержание пиг-
ментов в листе [мг/г сырой массы], а также отношение хлоро-
филла а к хлорофиллу b (а/Ь).
2.1.2. Анализ пигментов с использованием смеси
хлороформа с метанолом
Метод количественного анализа пигментов с применением
смеси хлороформа с метанолом разработан и рекомендован в пер-
вую очередь для анализа пигментов в растительном материале,
содержащем высокие концентрации фенолов, антоцианов, фла-
воноидов и других соединений, поглощающих в области погло-
щения фотосинтетических пигментов и вносящих значительные
погрешности при количественном определении фотосинтетичес-
ких пигментов в спиртовых и ацетоновых экстрактах (A. R. Wellbum,
1994; А.Е.Соловченко и др., 2001).
Ход работы
Работа включает следующие этапы: 1) получение вытяжки
пигментов из листьев с использованием смеси хлороформа с ме-
танолом (J. Folch et al., 1957); 2) спектрофотометрический ана-
лиз фотосинтетических пигментов в хлороформе; 3) определение
содержания антоцианов.
I Внимание! Работу выполняют только после усвоения инст-
рукции по технике безопасности при работе с метиловым спир-
том, под наблюдением руководителя лабораторных работ.
1. Получение вытяжки пигментов из листьев. Для анализа пиг-
ментов берут 5 — 7 высечек из различных областей листа, сделан-
ных сверлом диаметром 10 — 11 мм (для микроанализа достаточно
1 высечки). Растительный материал растирают в фарфоровой ступке
с 10 мл смеси хлороформа с метанолом (2:1 по объему) в при-
сутствии 100 — 200 мг СаСО2 или MgO (для предотвращения фео-
фитинизации хлорофиллов). Хлороформ предварительно промыва-
ют в делительной воронке водой для удаления небольших количеств
HClu других примесей, образующихся при хранении. Гомогенат филь-
57
труют в мерную пробирку через бумажный фильтр, предварительно
смоченный смесью хлороформа с метанолом. Затем к фильтрату
добавляют дистиллированную воду (1 /5 часть от объема фильтра-
та) и смесь центрифугируют в стеклянных пробирках при 3000 g в
течение 10 мин до полного разделения хлороформной (ХФ) и
водно-метанольной (ВМ) фракций.
После центрифугирования нижнюю фракцию ХФ, содержа-
щую хлорофиллы и каротиноиды, аккуратно отбирают шприцем
в мерную пробирку, измеряют ее объем и используют для спект-
рофотометрирования.
Верхнюю ВМ-фракцию (ВМФ) можно использовать для опре-
деления фенолов, флавоноидов, антоцианов и других водно-ра-
створимых пигментов. В этом случае необходимо также измерить
ее объем. Для анализа антоцианов к ВМФ добавляют НС1 до ко-
нечной концентрации 0,1 %.
2. Спектрофотометрический анализ и расчет содержания хлоро-
филла а, хлорофилла b и суммы ксантофиллов и каротина в листе.
Для расчета концентрации пигментов в хлороформе [мг/л] ис-
пользуют формулы А.Вельбурна (A. Wellburn, 1994):
са = 10,91 -Л666 - 1,2-Дб48;
сь= 16,38 • Аб48 - 4,57 -Аббв',
скс+кр = (Ю00 • Л480 - 1,42 са - 46,09 • q)/202.
Экстракт пигментов в хлороформе промеряют на спектрофо-
тометре при длинах волн 666, 648, 480 и 750 нм (значение после-
днего измерения вычитают из предыдущих для учета возможного
рассеивания образца).
3. Определение содержания антоцианов. При анализе антоциа-
нов в ВМФ добавляют несколько капель концентрированной НС1,
после чего пробы спектрофотометрируют при 530 нм. Для расчета
концентрации пигментов используют миллимолярный коэффи-
циент поглощения, равный 30 мМч • см-1 (D. Strack, V. Wray, 1989).
Оформление результатов. На основании данных спектрофото-
метрического анализа рассчитывают содержание пигментов в еди-
нице площади листа [мг/см2], учитывая объем экстракта и пло-
щадь высечек листа, взятых для исследования. Результаты изме-
рений и расчетные значения вносят в таблицу.
2.2. ИССЛЕДОВАНИЕ ФОТОСИНТЕТИЧЕСКИХ
ПИГМЕНТОВ МЕТОДАМИ ХРОМАТОГРАФИИ
Хроматография широко используется для качественного и ко-
личественного анализа фотосинтетических пигментов. Ниже опи-
саны методы хроматографического разделения основных пигмен-
58
тов зеленого листа на бумаге и методы тонкослойной хроматогра-
фии на сахарозе и силикагеле.
2.2.1. Разделение и количественное определение основных
пигментов листьев методом хроматографии на бумаге
(по Д. И. Сапожникову)
Хроматография пигментов на бумаге позволяет получить в до-
статочном количестве и без значительной деградации все основ-
ные пигменты зеленого листа. Методом одномерной хроматогра-
фии удается из общей вытяжки пигментов выделить каротин,
лютеин, виолаксантин или неоксантин. Метод двумерной хрома-
тографии позволяет разделить хлорофиллы а и b и получить хро-
матографически чистые пигменты группы каротиноидов (каро-
тин, виолаксантин, лютеин и неоксантин).
Ход работы
Работа включает следующие этапы: 1) получение вытяжки пиг-
ментов; 2) нанесение пигментов на бумагу; 3) разделение пиг-
ментов на одномерной или двумерной хроматограмме; 4) элюция
пигментов с бумажного носителя; 5) спектрофотометрическое оп-
ределение содержания пигментов.
1. Получение вытяжки пигментов. Все работы по хроматографи-
ческому разделению пигментов проводят в затемненном помеще-
нии.
500 мг листьев (свободных от крупных жилок) тщательно рас-
тирают в охлажденной на льду фарфоровой ступке с безводным
Na2SO4 (около 1 г на 0,5 г сырой массы листьев) и небольшим
количеством мела. Измельченный в порошок растительный мате-
риал переносят на стеклянный фильтр и настаивают в течение 5
мин с 3 — 5 мл охлажденной смеси этилового спирта и ацетона в
отношении 3:1. Для окончательной экстракции пигментов поро-
шок промывают порцией этилового эфира. Общий объем экстрак-
та доводят до 10 мл. Экстракт содержит сумму зеленых и желтых
пигментов.
2. Нанесение пигментов на бумагу. Для разделения пигментов
используют листы хроматографической бумаги размером 16x16 см.
При одномерной хроматографии 1 мл полученной вытяжки на-
носят полосой длиной 12 см на расстоянии 2 см от нижнего края
листа.
При проведении двумерной хроматографии для разделения всех
пигментов 0,3 мл вытяжки наносят на лист хроматографической
бумаги короткой полосой длиной 2 см, расположенной в правом
углу на расстоянии 2 см от краев бумаги (рис. 13).
59
Рис. 13. Последова-
тельность разгон-
ки пигмента в двух
направлениях (/и
II) на двумерной
хроматограмме
Рис. 14. Хроматогра-
фическая камера для
разделения пигмен-
тов на бумаге
Рис. 15. Распределение от-
дельных зон пигментов на
одномерной хроматограмме:
1 — 0-каротин; 2 — лютеин;
3 — виолаксантин; 4 — хлоро-
филл о; 5 — хлорофилл Ь; 6 —
линия нанесения образца
Для удобства нанесения вытяжки бумагу можно закрепить на
специальном штативе. Участок бумаги, на который наносят ра-
створ пигментов, необходимо поместить над прорезью штатива.
Снизу бумагу подсушивают током воздуха.
Объем наносимой вытяжки зависит от плотности бумаги и кон-
центрации пигментов в вытяжке. Количество пигментов после
разделения должно быть достаточным для их последующего коли-
чественного определения. При этом надо учитывать, что с увели-
чением концентрации вещества на единицу площади бумаги за-
трудняется разделение пигментов на хроматограмме. Ввиду этого
необходимо предварительно подобрать объем вытяжки, оптималь-
ный для проведения хроматографии (см. рис. 4).
После нанесения вытяжки бумагу хорошо подсушивают, свер-
тывают в цилиндр (соединяя края бумаги скрепкой так, чтобы
нижние края не соединялись вместе) и помещают в хроматогра-
фическую камеру для разгонки (рис. 14).
За 15 — 20 мин до разгонки в хроматографическую камеру за-
ливают смесь растворителей, приготовленную в отдельной колбе.
Уровень растворителя в камере должен быть ниже нанесенной
полосы пигментов на 1 см. Камеру плотно закрывают и затемняют
черной бумагой.
3. Разделение пигментов на одномерной хроматограмме. На од-
номерной хроматограмме в зависимости от применяемой для раз-
гонки пигментов смеси растворителей удается отделить каротин,
лютеин и виолаксантин от зеленых пигментов или выделить хро-
матографически чистый неоксантин. Поэтому работу проводят с
двумя хроматограммами.
60
Отделение каротина, лютеина и виолаксантина. Хромато-
грамму помещают в камеру, заполненную 30 мл смеси, состоя-
щей из бензола и петролейного эфира (в объемном отношении
2:1)*. Через 30 — 40 мин, когда фронт растворителя поднимется на
14 см (2 см от верхнего края) и будет видно четкое разделение
пигментов, хроматограмму вынимают и подсушивают. Располо-
жение отдельных зон пигментов показано на рис. 15.
Окрашенные полосы, соответствующие каротину и двум ксан-
тофиллам, вырезают, измельчают и переносят в отдельные бюк-
сы на 25 мл. Для лучшей элюции бумагу обрабатывают этиловым
эфиром (порцией около 1 мл). Затем пигменты элюируют смесью
этилового спирта и ацетона (1:3). Растворитель добавляют неболь-
шими порциями, каждый раз настаивая 2 — 3 мин. Экстракт акку-
ратно сливают в мерные пробирки. Элюцию проводят до полного
обесцвечивания бумаги. Объем полученных элюатов не должен пре-
вышать 3 — 4 мл. Полученные экстракты используют для спектро-
фотометрического анализа.
Отделение неоксантина. Вышеописанным образом 1 мл вытяж-
ки наносят на хроматографическую бумагу. После подсушивания
бумагу помещают в камеру, содержащую 40 мл смеси: бензол —
петролейный эфир — этанол в отношении 18:6:1 (или в отноше-
нии — 29:10:1). Через 30 — 45 мин хроматограмму вынимают и
подсушивают. Все пигменты, кроме неоксантина, идут с фронтом.
Неоксантин располагается ниже фронта на 2—3 см. Участок бума-
ги, содержащий неоксантин, вырезают из хроматограммы, из-
мельчают и переносят в бюкс на 10 мл. После обработки этило-
вым эфиром (около 1 мл) пигмент элюируют смесью этанол—
ацетон (1:3). Объем вытяжки не должен превышать 3 — 4 мл. По-
лученный элюат используют для спектрофотометрического ана-
лиза.
4. Разделение пигментов на двумерной хроматограмме. Для пол-
ного разделения всех пигментов используют двумерную хромато-
графию. Последовательность разгонки пигментов в двух направле-
ниях на двумерной хроматограмме показана на рис. 13. Для разгон-
ки пигментов в первом направлении используют смесь 1 (40 мл):
бензол — петролейный эфир (3:1). После того как фронт подни-
мется на 14 см, хроматограмму вынимают, подсушивают и поме-
щают в другую камеру, содержащую смесь 2 (30 мл): петролейный
эфир — этанол (14:1). Хроматограмму помещают таким образом,
чтобы направление разгона было перпендикулярно первоначаль-
ному. Когда фронт растворителя поднимется на 14 см, хромато-
грамму вынимают и просушивают. Положение отдельных пигмен-
тов на хроматограмме показано на рис. 16. В стартовом пятне после
хроматографирования остаются хлорофиллиды.
* Иногда лучше использовать смесь 3:1.
61
Рис. 16. Распределение
пигментов в направле-
ниях I и II на двумер-
ной хроматограмме:
1 — исходное пятно; 2 —
неоксантин; 3 — хлоро-
филл Ь; 4 — хлорофилл а;
5 — лютеин; б — виола-
ксантин; 7 — 0-каротин
Для получения достаточного для спект-
рофотометрирования количества пигментов
необходимо иметь 3—4 хроматограммы.
Окрашенные зоны с 3 — 4 хромато-
грамм, соответствующие отдельным пят-
нам, вырезают, бумагу измельчают и по-
мещают в бюксы на 50 мл. Бумагу предва-
рительно обрабатывают этиловым эфиром
(1 мл), а затем пигменты элюируют сме-
сью этанол — ацетон (1:3). Вытяжку ко-
личественно переносят в мерные пробир-
ки. Конечный объем экстракта не должен
превышать 3—4 мл.
5. Спектрофотометрический анализ пиг-
ментов. Поглощение вытяжек, содержа-
щих каротин и ксантофиллы, измеряют
на спектрофотометре при следующих дли-
нах волн: 450 нм — каротин (в ацетоне);
445 нм — ксантофиллы (в ацетоне).
Концентрацию пигментов в вытяжке
рассчитывают по формулам, приведенным на с. 54.
Поглощение хлорофиллов определяют при длинах волн, ука-
занных в табл. 3; для расчета используют указанные там же удель-
ные коэффициенты поглощения.
Расчеты. Содержание пигментов F [мг/г сырой массы] рассчи-
тывают по формуле
1000Р ’ v '
где с — концентрация пигментов, мг/1000 мл; V3 — конечный
объем вытяжки, полученной после элюирования пигмента из хро-
матограммы; Р — навеска, соответствующая количеству материа-
ла, нанесенного на хроматограмму, г.
Пример. Требуется рассчитать содержание каротина в миллиграммах
на 1 г сырой массы (F).
Общая навеска растительного материала Р= 500 мг; объем получен-
ного экстракта И = 10 мл; объем экстракта, нанесенного на хромато-
грамму, К = 1 мл; объем элюата И3 = 3 мл.
Поглощение промеренного на спектрофотометре раствора Л450 = 0,29.
Определяем концентрацию каротина в элюате:
с = = о, 001229 г/л = 1,229 мг/л.
236 236
В 1 л раствора содержится 1,229 мг каротина, а в 3 мл:
(2)
сУз
1000
1 229 • 3
= 0,00369 мг.
62
Это количество каротина получено из нанесенной на бумагу вытяжки,
что соответствует 50 мг растительного материала (500 мг : 10 мл = 50 мг =
= 0,05 г). Следовательно, в 1 г сырого материала содержится каротина [мг]:
F = °?..9.9..^9 = 0,0738-
0,05
Таким образом, подставив в формулу (1) соответствующие значе-
ния, получим [мг/г сырой массы]
ск 1 729 • 3
F = = 0,0738.
1000Р 1000-0,05
Оформление результатов. Результаты оформляют в виде табли-
цы, куда внесены первичные данные по спектрофотометрическо-
му анализу пигментов и конечные данные о содержании отдель-
ных групп пигментов [мг/г сырой массы].
2.2.2. Разделение пигментов методом тонкослойной
хроматографии на сахарозе
Тонкослойная хроматография с использованием сахарозы по-
зволяет провести разделение пигментов листа с наименьшей их
деградацией и препаративно получить достаточные количества
хроматографически чистых пигментов.
Ход работы
Работа включает следующие этапы: 1) получение экстракта пиг-
ментов из листьев; 2) разделение (разгонка) пигментов на дву-
мерной хроматограмме; 3) элюция и количественное определе-
ние пигментов.
В работе используют листья 10 —14-дневного гороха.
1. Получение экстракта пигментов из листьев. Навеску листьев
в 0,5 г растирают в фарфоровой ступке с мелом и двойным (по
весу) количеством безводного Na2SO4. Пигменты экстрагируют аце-
тоном, фильтруют через стеклянный фильтр и объем экстракта
доводят до 10 мл.
2. Разгонка хроматограммы. Для тонкослойной хроматографии
на сахарозе можно использовать коммерческие пластинки разме-
ром 20 х 20 см или пластинки собственного изготовления.
Приготовление пластинки с тонким слоем, сахарозы. В качестве
сорбента используют сахарозу, просеянную через капроновую ткань
или сито с диаметром пор, равным 0,25 мм.
Жидкий сорбент готовят путем смешивания примерно рав-
ных количеств (масса/объем) сахарозы и метанола (13 г сахаро-
63
зы+ 12 мл метанола) в колбе Эрленмейера с притертой пробкой.
Смесь тщательно перемешивают при энергичном встряхивании в
течение 1 — 2 мин. Затем добавляют крахмал (3 % от сахарозы) и
встряхивают еще 1 мин.
Внимание! Работа с метанолом требует особой осторож-
ности и выполняется только после изучения инструкции по тех-
нике безопасности, в присутствии руководителя лабораторных
работ.
Стеклянную пластинку размером 13x18 см, предварительно
хорошо вымытую хромовой смесью и высушенную, помещают на
выровненную горизонтальную поверхность. На пластинку нано-
сят смесь сахарозы с метанолом и осторожным покачиванием рас-
пределяют по всей поверхности пластинки слоем толщиной 0,3 —
0,5 мм. Пластинку с нанесенным сорбентом высушивают 10 мин
на воздухе и затем в термостате при 40 °C в течение 2 ч. Пластинки
лучше приготовить за день до хроматографирования.
Нанесение пигментов на пластинку и разгонка хроматограм-
мы. Определенный объем экстракта (около 0,3 мл) наносят поло-
сой 2 см на расстоянии 2 см от края пластинки. Для этого пользу-
ются микропипеткой с оттянутым концом. Раствор наносят в виде
отдельных капель, не касаясь поверхности сорбента и не повреж-
дая его. В процессе нанесения экстракта пятно непрерывно подсу-
шивают током воздуха. Пятна располагают таким образом, чтобы
достичь равномерного распределения пигментов по всей длине
полосы нанесения экстракта.
Пластинку с нанесенным на сорбент раствором пигментов окон-
чательно высушивают в течение 10 мин и помещают в хромато-
графическую камеру. Предварительно камеру в течение 15 мин на-
сыщают соответствующей смесью растворителей:
I направление, петролейный эфир (точка кипения 70—100 °C) —
этиловый спирт (30:1);
II направление: бензол — петролейный эфир (2:1 или 3:1).
Общий объем каждой смеси должен быть около 60 мл.
Хроматограмму разгоняют в каждой смеси по 20 — 30 мин и
после четкого разделения пигментов вынимают из камеры и про-
сушивают на воздухе в темноте. Расположение пятен отдельных
пигментов на хроматограмме с тонким слоем сахарозы аналогич-
но расположению пигментов на двумерной хроматограмме на бу-
маге (см. рис. 16).
3. Элюция и количественное определение пигментов. Отдельные
окрашенные зоны сорбента снимают со стеклянной пластинки
скальпелем и собирают в стеклянные бюксы. Пигменты элюируют
ацетоном или этиловым эфиром (в зависимости от полярности
пигмента). Элюаты аккуратно собирают в мерные пробирки, ста-
раясь не взмучивать раствор в бюксах, доводят растворителем до
64
определенного объема (около 3 мл) и используют для спектрофо-
тометрического анализа.
При определении концентрации пигментов в растворе и рас-
чета их содержания в растительном материале промеряют полу-
ченные элюаты на спектрофотометре против соответствующих
растворителей. Длины волн, используемые для спектрофотомет-
рических измерений, и удельные коэффициенты поглощения,
необходимые для расчета концентрации пигментов, приведены в
табл. 4, 6, 7. Расчет концентрации пигментов в растворе и содер-
жания их в растительном материале проводят, как описано в ра-
боте 2.2.1.
Регистрируют спектры поглощения отдельных пигментов на
двухлучевом спектрофотометре с целью подтверждения чистоты
полученных препаратов пигментов. Полученные спектры погло-
щения могут быть также использованы для количественных оце-
нок концентрации пигментов в растворе и содержания их в рас-
тительном материале.
Оформление результатов. Первичные данные спектрофотомет-
рических измерений, а также результаты расчетов концентрации
отдельных пигментов в элюатах [мг/л] и их содержания в расти-
тельном материале [мг/г сырой массы листа] вносят в таблицу.
Проводят анализ спектров поглощения полученных препара-
тов пигментов, указывая положения основных максимумов по-
глощения и оценивая степень чистоты полученных препаратов.
2.2.3. Анализ пигментов методом тонкослойной
хроматографии на силикагеле
Тонкослойная хроматография на силикагеле находит широкое
применение при исследовании пигментов фотосинтетического ап-
парата, причем часто используют коммерческие пластинки с тон-
ким слоем силикагеля, например пластинки silufol (так называе-
мые силуфоловые пластинки). Особенно удобно использовать дан-
ные пластинки для анализа каротиноидов. При работе с зелеными
пигментами в метод тонкослойной хроматографии на силикагеле
вносят ряд модификаций (предварительная обработка пластинок
антиоксидантами, например аскорбиновой кислотой, примене-
ние антиоксидантов в смесях для разгонки пигментов и др.). В по-
следнее время появились коммерческие колонки (картриджи) с
обращенной фазой, изготовленные на основе силикагеля. Эти ко-
лонки содержат гидрофобную фазу и могут использоваться для
хроматографии и препаративного получения пигментов, в том
числе и хлорофиллов, без их существенной деградации.
В данной работе тонкослойную хроматографию на силуфоло-
вых пластинках используют для анализа каротиноидов.
3 Гавриленко
65
Ход работы
Работа включает следующие этапы: 1) экстракция пигментов
из листьев растений; 2) хроматография пигментов в тонком слое
на силуфоловых пластинках; 3) спектрофотометрический анализ
пигментов.
Для работы могут быть использованы листья разных видов ра-
стений.
1. Экстракция пигментов из листьев растений. Для выделения
пигментов из листьев может быть применен реактив Фолча (смесь
хлороформа с метанолом в соотношении 2:1). Навеску листьев
(2 — 3 г) растирают с 10 мл охлажденной смеси. Экстракт перено-
сят в мерный цилиндр или в мерную пробирку и после определе-
ния конечного объема экстракта добавляют к нему дистиллиро-
ванную воду из расчета 1/5 от общего объема экстракта. Смесь
перемешивают, переносят в стеклянную центрифужную пробир-
ку и центрифугируют 15 мин при 4000 мин'1 для разделения фаз.
После центрифугирования нижний слой хлороформа, содержа-
щий пигменты, аккуратно отбирают шприцем с длинной иглой и
подсушивают над безводным Na2SO4. Подсушенный экстракт филь-
труют через бумажный фильтр в колбочку со шлифом и упарива-
ют на роторном испарителе. После упаривания пигменты раство-
ряют в небольшом количестве хлороформа (200 — 250 мкл) и ис-
пользуют для хроматографии на силуфоловых пластинках.
В ряде случаев для хроматографии каротиноидов производят
предварительное омыление суммарного экстракта. При этом про-
исходит омыление сложных эфиров, в том числе и хлорофиллов,
что позволяет анализировать каротиноиды в отсутствие хлорофил-
лов, затрудняющих хроматографию желтых пигментов. Кроме того,
процедура омыления позволяет разделить каротиноиды на омы-
ляемую и неомыляемую фракции.
I Внимание! Работа выполняется только после усвоения ин-
струкции по технике безопасности при работе с метиловым
спиртом, под наблюдением руководителя лабораторных работ.
Омыление сложных эфиров производят следующим образом.
Часть суммарного экстракта, отобранного из слоя хлороформа (см.
выше), переносят в пенициллиновый пузырек, упаривают досу-
ха, после чего добавляют 3 мл щелочного метанола (3 н. NaOH в
90 %-м метаноле: 0,3 г Н2О + 22,5 мл метанола).
Пузырьки плотно закрывают тефлоновыми пробками с фикса-
торами и помещают на 1 — 2 ч в термостат (70 °C). После термоста-
тирования пузырьки с пробами охлаждают, открывают и произ-
водят экстракцию неомыляемой фракции гексаном (пробу про-
мывают несколько раз порциями гексана по 2 мл до тех пор, пока
гексан не станет бесцветным); слой гексана после встряхивания
66
пробы и расслаивания отбирают шприцем. Пробы упаривают на
роторном испарителе, растворяют в хлороформе и используют для
хроматографии на силуфоловых пластинках. Промытую гексаном
фракцию подкисляют, добавив к содержимому пенициллиновых
пузырьков 0,2 мл 6 н. НС1, и вновь промывают гексаном несколь-
ко раз. При этом в гексан переходят жирные кислоты, получен-
ные после омыления сложных эфиров. Данный препарат может
быть использован для анализа жирных кислот методом газожид-
костной хроматографии.
2. Хроматография пигментов в тонком слое на силуфоловых пла-
стинках. Для хроматографии пигментов на силуфоловых пластин-
ках могут быть использованы различные смеси. Выбор смеси зави-
сит от целей работы.
Для полного анализа пигментов в зеленых листьях можно ис-
пользовать одну из следующих смесей:
смесь Г. бензин — ацетон — петролейный эфир (точка кипе-
ния 40 —70°C) — гексан в объемном соотношении 10:10:3:10;
смесь 2: ацетон — гексан — бензол в соотношении 1:1:1;
смесь 3: петролейный эфир (точка кипения 40—70 °C) — эта-
нол в соотношении 16:1 (распределение отдельных зон пигмен-
тов при разделении в смеси 3 представлено на рис. 17).
При анализе пигментов в стареющих листьях, где появляются
дополнительные желтые пигменты, например эфиры каротинои-
дов, используют смесь 4\ гексан — хло-
роформ — бензол в соотношении 10:20:1.
После разделения пигментов пластин-
ки вынимают из хроматографической
камеры, отдельные зоны каротиноидов
вырезают и помещают в отдельные про-
бирки, заполненные элюирующей сме-
сью: хлороформ — метанол в соотноше-
нии 1:1. Соответствующие зоны с не-
скольких хроматограмм помещают в
одни и те же пробирки. Элюцию прово-
дят в темноте при достаточном количе-
стве элюирующего раствора (должен по-
крывать полностью вырезанные зоны).
После того как собрано достаточное ко-
личество материала, элюаты фильтруют
через бумажный фильтр, упаривают на
роторном испарителе, доводят до опре-
деленного объема (3 — 4 мл) необходи-
мым растворителем и анализируют спек-
трофотометрически. Применение различ-
ных растворителей (80 %-й ацетон, гек-
сан, этиловый эфир и др.) и анализ
Рис. 17. Распределение от-
дельных зон пигментов при
разделении на силикагеле:
1 — старт; 2 — неоксантин;
3 — виолаксантин; 4 — люте-
ин; 5 — хлорофилл Ь; 6 —
хлорофилл а; 7 — р-каротин
67
получаемых спектров поглощения проб позволяют идентифици-
ровать полученные каротиноиды.
3. Спектрофотометрический анализ пигментов. Производят за-
пись спектров поглощения хроматографически чистых пигментов
на регистрирующем двухлучевом спектрофотометре и идентифи-
кацию пигментов.
Полученные спектры используют также для количественного
определения полученных пигментов и расчета их содержания в
листьях.
Оформление результатов. При оформлении результатов необ-
ходимо отметить положение максимумов на спектрах поглощения
пигментов, определить величину поглощения в максимуме, про-
извести расчет концентрации пигментов в экстрактах и в листьях
[мг/г сырой массы]. Полученные данные вносят в таблицу.
2.2.4. Хроматографическое разделение и препаративное
получение пигментов на колонках силикагеля
с обращенной фазой
Хроматографические исследования хлорофиллов затруднены в
связи с чрезвычайной лабильностью этих пигментов. Широко ис-
пользуемые в настоящее время для анализа пигментов методы тон-
кослойной хроматографии на силикагеле требуют специальных мо-
дификаций в случае применения их при анализе хлорофиллов (пред-
варительная обработка пластинок с силикагелем антиоксиданта-
ми, например аскорбатом, добавление в жидкие разгоночные смеси
стабилизаторов, например ионола или гексадекана). Эти трудно-
сти смогла обойти хроматография на силикагеле с обращенной
фазой (силикагель, окруженный путем специальной обработки гид-
рофобной пленкой). В этом случае гидрофобное окружение сили-
кагеля стабилизирует пигменты во время разделения. Однако при-
менение тонкослойной хроматографии с обращенной фазой огра-
ничено из-за малой адсорбционной емкости пластинок. Ввиду этого
применение коммерческих колонок (картриджей) с обращенной
фазой представляется весьма перспективным для хроматографии
хлорофиллов. Данные колонки (Super clean solid phase extraction
tubes) в качестве основы имеют силикагель, модифицированный
путем присоединения к молекуле SiO2 остатков С18. Это, очевид-
но, приводит к изменению гидрофобного окружения пигментов и
к их стабилизации на силикагеле. Кроме того, применение данных
колонок позволяет работать с достаточно высокими концентраци-
ями пигментов и получать хроматографически чистые пигменты
препаративно. Немаловажным является также и быстрота получе-
ния достаточно больших количеств хлорофиллов (выделение чис-
тых пигментов проходит в одну стадию).
68
Ход работы
Работа включает следующие этапы: 1) подготовка хроматогра-
фической колонки к работе; 2) получение экстрактов пигментов
из листьев; 3) хроматография на колонке; 4) спектрофотометри-
ческий анализ хлорофиллов.
1. Подготовка хроматографической колонки к работе. Колонку
(картридж) закрепляют на штативе вертикально и промывают в
следующем порядке: 2 мл дистиллированной воды, 2 мл 100 %-го
метилового спирта, 2 мл 90 %-го метилового спирта.
2. Получение экстракта пигментов из листьев. Навеску листьев
5 г растирают в охлажденной на льду фарфоровой ступке с без-
водным Na2SO4 и небольшим количеством мела. Измельченный в
порошок растительный материал переносят на стеклянный фильтр
и настаивают в течение 3 — 5 мин с 5 мл охлажденной смеси аце-
тона и этилового спирта в отношении 1:3. Экстракт фильтруют
через стеклянный фильтр в пробирку, вставленную в колбу Бун-
зена. Для полной экстракции пигментов пробу промывают неболь-
шими порциями той же смеси. Полученный экстракт переносят в
колбу со шлифом и упаривают досуха на роторном испарителе.
Затем пигменты растворяют в 150 — 200 мл 90 %-го метилового
спирта. Данную пробу наносят за один раз на подготовленную
хроматографическую колонку.
3. Хроматография пигментов на колонке. После нанесения эк-
стракта пигментов на колонку дожидаются полного вхождения в
нее пигментов, после чего колонку начинают промывать 90 %-м
метанолом до полного отделения и выхода желтых пигментов (ксан-
тофиллов). Далее, промывая колонку тем же раствором, добива-
ются выхода отдельной полосой хлорофилла b (всего около 25 мл
90 %-го метилового спирта). После этого из суммы пигментов, ос-
тавшихся на старте, 95 %-м метиловым спиртом экстрагируют хло-
рофилл а (около 10 мл 95 %-го метанола). После отбора в отдель-
ные пробирки хлорофиллов полное удаление пигментов с колон-
ки производят 100 %-м метанолом. При этом удается получить также
чистый препарат каротина.
4. Спектрофотометрический анализ хлорофиллов. После полу-
чения хроматографически чистых препаратов пигментов произво-
дят их спектрофотометрический анализ.
Пробы упаривают на роторном испарителе и переводят в 80 %-й
ацетон (3 мл). Затем на двулучевом спектрофотометре осуществ-
ляют регистрацию спектров поглощения и отмечают положение
максимумов поглощения пигментов.
Производят оценку поглощения проб хлорофиллов при дли-
нах волн, соответствующих хлорофиллам а и b (663 и 646 нм со-
ответственно) и каротиноидам (470 нм). Используя формулы Лих-
тенталера (с. 51), рассчитывают концентрацию соответствующих
69
пигментов в пробах и оценивают чистоту полученных препара-
тов.
Оформление результатов. Представляют спектры поглощения
препаратов хлорофиллов с указанием положения максимумов
поглощения пигментов и значений поглощения при длинах волн,
соответствующих хлорофиллам а и b (663 и 646 нм соответствен-
но) и каротиноидам (470 нм). В таблицу вносят значения погло-
щения и расчетные данные по содержанию пигментов в препа-
ратах [мг/л] и [ммоль/л], используя соответствующие молеку-
лярные массы (для хлорофилла а — 893,48, хлорофилла b —
907,46).
2.3. КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ ПИГМЕНТОВ
ГРУППЫ ПРОТОХЛОРОФИЛЛА
При исследовании формирования пигментных систем фото-
синтетического аппарата важнейшим показателем является уро-
вень содержания в тканях листьев пигментов группы протохлоро-
филла.
Протохлорофиллид, а в ряде случаев его этерифицированная
форма — протохлорофилл являются предшественниками хлорофил-
ла в биосинтетической цепи (рис. 18).
Подавляющая часть пула предшественников хлорофилла пред-
ставлена бесфитольной формой пигмента — протохлорофилли-
дом. Протохлорофиллид образуется в результате серии фермента-
тивных реакций в темноте и при освещении восстанавливается в
хлорофиллид, что связано с гидрированием двойной связи между
атомами углерода 7 и 8 в IV пиррольном кольце (см. рис. 9).
Дальнейшее превращение хлорофиллида в хлорофилл проис-
ходит путем его этерификации спиртом фитолом (С20Н39ОН) или
присоединения к макроциклу геранилгеранилпирофосфата — по-
лиизопреноидной структуры (С20) с четырьмя двойными связя-
ми; при последующем гидрировании трех из них образуется фи-
тольная структура.
Пул предшественников хлорофилла представляет довольно
сложную гетерогенную систему. Он включает моновинил- и диви-
нилпроизводные протохлорофиллида и протохлорофилла, кото-
рые при освещении восстанавливаются до соответствующих моно-
и дивинилформ хлорофиллида и хлорофилла. В этиолированных
листьях обнаружено также несколько разных форм протохлоро-
филлида. Основными являются три формы пигмента с максиму-
мами поглощения 628, 638 и 650 (648) нм (Е.Dujardin, C.Sironval,
1970), из которых фотоактивны две наиболее длинноволновые фор-
мы — 638, 650 нм, прочно связанные с белком. Связь молекулы
предшественника с белком необходима для фотовосстановления
70
Протопорфирин IX
Mg-хелатаза Mg2+- АТФ
Mg-протопорфирин IX
Mg-протопорфирин
IX- метилтрансфераза
S-аденозилметионин
(метилирование пропионовой
кислоты III кольца)
S-аденозилгомоцистеин
Mg-протопорфиринмонометиловый эфир
t
। (0-окисление 6-пропионовой кислоты,
’ образование С=О, замыкание V кольца)
t
Дивинилпротохлорофиллид а
Винилредукгаза
НАДФН
(восстановление С-4 винильной
группы до этильной)
НАДФ+
Хлорофиллид а
Хлорофиллсинтаза
Хлорофилл а
фитил- (р)(р)
(присоединение фитола)
------------Хлорофилл b
Хлорофилл а-оксигеназа
Рис. 18. Схема заключительных этапов биосинтеза хлорофилла
71
протохлорофиллида в хлорофиллид. Как показали работы Гриф-
фитса (1974), реакция фотовосстановления двойной связи 7 = 8 в
IV кольце макроцикла осуществляется при участии специального
фермента НАДФН-протохлорофиллид оксидоредуктазы (ПОР), ак-
тивируемого у высших растений светом. В настоящее время выде-
ляют два типа фермента ПОР — А и В, кодируемых различными
генами. Фотоактивные формы протохлорофиллида образуют трой-
ной комплекс, содержащий протохлорофиллид 650 или прото-
хлорофиллид 638, фермент ПОР и донор водорода НАДФН (про-
тохлорофиллид 650 — ПОР — НАДФН). Предполагают, что ак-
тивный комплекс протохлорофиллида 638 с ферментом и доно-
ром водорода является димером, в то время как протохлорофил-
лид 650 представляет собой более сложный агрегат тройных ком-
плексов [1, 5, 13, 14, 17, 20 — 22].
Кодируемый ядром фермент НАДФН-протохлорофиллид ок-
сидоредуктаза составляет значительную часть проламеллярного тела
этиопластов. С общим содержанием белка этого фермента корре-
лируют и процессы накопления фотоактивных форм протохлоро-
филлида и формирование мембранных структур пластид. Устой-
чивый в темноте фотоактивный комплекс организован так, что
при поглощении фотона протохлорофиллид восстанавливается до
хлорофиллида в течение нескольких секунд, что сопровождается
перестройкой мембран проламеллярного тела.
По данным ряда исследований (Ф.Ф.Литвин с сотр., 1968,
2000; M.Gassman et al., 1968), цепь биосинтеза хлорофилла вклю-
чает две последовательные фотореакции с образованием различ-
ных спектральных форм пигментов, которые могут быть зарегис-
трированы с помощью высокочувствительных методов абсорбци-
онной и люминесцентной спектроскопии.
В последнее время значительно расширен круг вопросов, свя-
занных с изучением ближайших предшественников хлорофилла.
Количественные определения пигментов группы протохлорофил-
ла проводятся во многих лабораториях в связи с изучением за-
ключительных стадий биосинтеза хлорофилла и кинетики про-
цессов фотопревращения различных форм протохлорофиллида в
хлорофиллид, механизмов регуляции биосинтеза пигментов [4,
6, 16, 26, 30], а также общих вопросов метаболизма пигментов в
растении. Количественные определения различных форм прото-
хлорофиллида при одновременной регистрации их спектров флуо-
ресценции могут дать представление об эффективности процес-
сов миграции энергии между ними. Новым направлением работ
являются исследования реакций биосинтеза пигментов в связи с
формированием реакционных центров фотосистем I и II (ФС I и
ФС II) в хлоропластах (Ф.Ф.Литвин с сотр., 1993, 2000; F.Franck
et al., 1997), показавшие участие в них разных форм предшествен-
ников и разных типов протохлорофиллидоксидоредуктаз. Прове-
72
дение всех этих исследований требует разработки и применения
методов выделения и количественного определения отдельных
форм пигментов группы протохлорофиллида.
Для характеристики пигментного фонда предшественников
могут быть использованы методы хроматографии, разделение
фитольных и бесфитольных форм пигментов с использованием
полярных и неполярных растворителей, низкотемпературный
флуоресцентный анализ.
Возможность применения этих методов обусловлена тем, что
отдельные формы пигментов различаются по молекулярной струк-
туре, полярности и спектральным характеристикам. Большинство
пигментов группы протохлорофилла и хлорофилла имеют харак-
терные спектры поглощения и флуоресценции. Но фитольные и
бесфитольные формы пигментов нельзя разделить спектрально,
так как их спектры поглощения и флуоресценции идентичны,
однако они различаются по полярности и могут быть разделены
хроматографически и путем применения различных растворите-
лей.
«Кислые» бесфитольные формы (протохлорофиллид и хлоро-
филлид) более полярны, они прочнее удерживаются адсорбен-
том и в соответствующих системах растворителей менее подвиж-
ны, чем фитольные, менее полярные формы пигментов (прото-
хлорофилл и хлорофилл). Фитольные формы более липофильны и
растворимы в петролейном эфире, в то время как бесфитольные
формы нерастворимы в петролейном эфире, но растворимы в
разбавленных растворах щелочи.
На этих свойствах пигментов основаны методы выделения и
количественного определения отдельных форм пигментов группы
протохлорофилла и хлорофилла.
2. 3.1. Выделение протохлорофилла методом хроматографии
и его количественное определение
Ход работы
Работа включает следующие этапы: 1) получение ацетоновой
вытяжки пигментов из этиолированных листьев; 2) перевод пиг-
ментов в этиловый эфир; 3) хроматографическое разделение пиг-
ментов; 4) спектрофотометрический анализ пигментов.
1. Получение ацетоновой вытяжки пигментов. Этиолированные
проростки растений (овса, ячменя, кукурузы, фасоли) выращи-
вают в камере в полной темноте в течение 7—10 дней. В темноте
или при слабом зеленом свете (чтобы предотвратить превращение
предшественников хлорофилла в хлорофилл) берут навеску лис-
тьев в 1 г. Пробу помещают в мешочек из марли и фиксируют в
73
темноте в течение 2 мин в кипящей воде. После фиксации листья
подсушивают фильтровальной бумагой, растирают в ступке с не-
большим количеством СаСО3 и ацетона (3 — 4 мл), переносят в
стеклянный фильтр и пигменты экстрагируют небольшими пор-
циями ацетона до общего объема 10 —15 мл.
2. Перевод пигментов в этиловый эфир. Полученный экстракт
переносят в делительную воронку и пигменты переводят в рав-
ный объем этилового эфира, предварительно очищенного от пе-
рекисей и перегнанного (см. с. 55). Экстракцию эфиром повторяют
два раза. Эфирный раствор пигментов промывают в делительной
воронке 5 %-м раствором NaCl, высушивают над прокаленным
Na2SO4 и количественно переносят на хроматографическую бума-
гу (16 х 32 см) в виде полосы длиной 12 см. Бумагу высушивают в
токе воздуха, сворачивают в цилиндр и помещают в камеру для
разгонки.
3. Хроматографическое разделение пигментов. Для разделе-
ния пигментов используется смесь Годнева: бензин «Калоша»
(фракция 80—115 °C) — петролейный эфир — ацетон — изопро-
панол (17:5:: 7:0,8). Смесь в объеме 40 — 50 мл наливают в камеру
за 10 мин до начала разгонки.
Хроматограмму разгоняют в темноте в течение 1 —1,5 ч и после
высушивания просматривают в ультрафиолетовом свете. Флуорес-
цирующие красным светом зоны обводят карандашом. Обычно
пигменты располагаются в порядке, указанном на рис. 19.
Для количественного определения протохлорофилл и прото-
хлорофиллид элюируют этиловым эфиром или ацетоном. Объем
элюата не должен превышать 3 мл.
4. Спектрофотометрический анализ пигментов. Поглощение элю-
атов определяют на спектрофотометре при 623 — 625 нм. Удельный
коэффициент экстинкции для протохлорофилла в эфире я625 = 36,9
(V. Koski, I.H. Smith, 1948).
Для разделения пигментов эфирный раствор обрабатывают
0,02 н. NaOH или КОН. Бесфитольная форма пигмента — прото-
Рис. 19. Распределение пигментов на хро-
матограмме при выделении протохлоро-
филла:
1 — линия нанесения образца; 2 — зона крас-
ной флуоресценции протохлорофилла; 3, 5 —
нефлуоресцирующие желтые пигменты; 4 —
следы красной флуоресценции хлорофиллов;
6 — фронт растворителя
74
хлорофиллид — переходит в щелочной раствор, а протохлоро-
филл остается в эфире. После кратковременного освещения этио-
лированных растений (1 — 2 мин) количество протохлорофилла
резко уменьшается и появляется хлорофилл. Зона хлорофилла на
бумаге расположена над зоной протохлорофилла.
Оформление результатов. Результаты оформляют в виде табли-
цы, куда заносят данные спектрофотометрического анализа, рас-
чет концентрации пигментов в элюате и расчет содержания пред-
шественников хлорофилла в листе [мг/г сырой массы].
2. 3.2. Выделение и количественное определение
протохлорофиллида с использованием полярных
и неполярных растворителей
Ход работы
Работа включает следующие этапы: 1) экстракция пигментов
из листьев; 2) получение эфирного экстракта; 3) спектрофото-
метрический анализ пигментов; 4) исследование динамики на-
копления и активности новообразования протохлорофиллида.
1. Экстракция пигментов из листьев. Экстракцию протохлоро-
филлида из листьев 10— 15-дневной кукурузы, выдержанных в те-
чение 1 — 2 суток в темноте, проводят по несколько модифициро-
ванному методу А. А. Шлыка (1965).
Навеску зеленых листьев 1 — 5 г фиксируют в затемненном по-
мещении горячим паром или в воде при температуре 95 °C в тече-
ние 2 мин. Листья подсушивают фильтровальной бумагой, смачи-
вают в ступке небольшим количеством 0,1 н. NH4OH (для нейтра-
лизации органических кислот и дезактивации хлорофиллазы) и
растирают с песком и 80 %-м ацетоном (4—5 мл). Вся последующая
работа проводится при обычном освещении.
Пигменты экстрагируют 3 — 4 раза, фильтруя ацетоновые экст-
ракты через стеклянный фильтр. Фильтраты собирают, доводят до
объема 25 мл и добавляют 2 — 3 мл 0,02 н. NH4OH (из расчета 1 мл
NH4OH на 10 мл водно-ацетонового экстракта) для перевода про-
тохлорофиллида в водорастворимую форму.
2. Разделение фитольных и бесфитольных форм пигментов.
Для разделения фитольных и бесфитольных пигментов смесь пе-
реносят в делительную воронку, добавляют 0,2 мл насыщенно-
го раствора NaCl и 5 — 7 мл легкого петролейного эфира (точ-
ка кипения 40 — 60 °C). Содержимое делительной воронки встря-
хивают несколько раз. При этом в петролейный эфир переходят
содержащие фитол липофильные формы пигментов — протохло-
рофилл, хлорофиллы, а также каротиноиды. В щелочной водно-
ацетоновой фазе остаются гидрофильные, бесфитольные пигмен-
75
ты — протохлорофиллид, хлорофиллиды а и Ь. После разделения
слоев нижний слой, водно-ацетоновый, и верхний, петролейно-
эфирный, собирают в отдельные колбы. Экстракцию петролей-
ным эфиром повторяют 4 — 5 раз до полного извлечения липо-
фильных пигментов. При этом частично извлекаются протохлоро-
филлид и хлорофиллиды. Они вымываются из петролейно-эфир-
ных экстрактов 2 — 3 мл 0,02 н. NH4OH. Полученный экстракт объе-
диняют с первичным водно-ацетоновым раствором.
Оставшиеся в водно-ацетоновом экстракте бесфитольные пиг-
менты — протохлорофиллид и хлорофиллид извлекаются этило-
вым эфиром (20 мл водного ацетона и 30 —40 мл эфира). Предва-
рительно экстракт подкисляют 0,5 —1,0 мл насыщенного раство-
ра однозамещенного фосфата натрия NaH2PO4 (или добавлением
нескольких кристаллов сухой соли) до pH 4,5 — 5. При этом про-
тохлорофиллид и хлорофиллид переходят из водорастворимой
солевой формы в кислую форму (свободные СООН-группы), ко-
торая лучше растворима в серном эфире.
Трех-четырехкратная обработка раствора серным эфиром по-
зволяет полностью выделить протохлорофиллид в смеси с хлоро-
филл ид ом.
Общий эфирный экстракт доводят до определенного объема и
используют для спектрофотометрического анализа.
3. Спектрофотометрический анализ пигментов. Полученные
эфирные экстракты промеряют на спектрофотометре при мак-
симуме поглощения протохлорофиллида (623 — 625 нм), хлоро-
филлида а (663 нм) и хлорофиллида b (644 нм). Количество про-
тохлорофиллида в присутствии хлорофиллидов [мг/г сырой мас-
сы] рассчитывают по формулам Коски (V.M. Koski, 1950), ис-
пользуя для расчета концентрации бесфитольных форм коэффи-
циент 0,69:
. К(-0,1206463 -0,1619444 + 1,0057^24)
протохлорофилл =—---------.......
0,0399-1000 -Wd
хлорофилл а =
К(1,0152463 -0,0896444 -0,0037424).
0,095 • 1000 • Wd
хлорофилл b = ^-°-161^ + 1,0195Ли-0,03014.)
0,0575-1000 Wd
где V — объем раствора, мл; А — поглощение при соответствую-
щей длине волны; W — масса листьев, г; d — длина кюветы, см.
Протохлорофилл и протохлорофиллид имеют одни и те же
молярные коэффициенты поглощения (е), их спектры поглоще-
76
Таблица 8
Миллимолярные коэффициенты поглощения (емМ)
для хлорофиллов а и b и протохлорофилла в 80 %-м ацетоне
(M.Brouers, М. R. Michel-Wolwertz, 1983)
X, нм емМ, л • ммоль'1 • см-1
хлорофилла (ида) а хлорофилла b протохлорофилла (ида)
663 79,5 9,3 0,1
647 18,9 47,5 3,1
626 14,5 11,7 30,4
ния идентичны и в 80 %-м водном ацетоне оба пигмента погло-
щают при 626 нм. Однако, поскольку молекулярная масса прото-
хлорофилла (891,5) больше молекулярной массы протохлорофил-
лида (613,0), его удельный коэффициент поглощения ниже, чем
/ е \
у протохлорофиллида {а = ^-). Поэтому для расчета концентра-
ции бесфитольных форм пигментов используют коэффициент 0,69.
В ряде случаев концентрации пигментов удобнее сравнивать в
молярных, а не в весовых отношениях. В табл. 8 приведены значе-
ния миллимолярных коэффициентов поглощения (емМ) для хло-
рофиллов а и b и протохлорофилла.
Эти коэффициенты были использованы для расчета молярной
концентрации протохлорофилла (ида) в присутствии хлорофил-
лов а и b в 80 %-м ацетоне [нмоль/мл]:
хлорофилл (ид) а= 13,16Л664 - 2,63Л647 + 0,23Л626;
хлорофилл b = -4,95Л664 + 22,59Л647 - 2,29Л626;
протохлорофилл (ид) = -4,37Л664 - 7,44Л647 + 33,67Л626.
4. Исследование динамики накопления протохлорофиллида в ус-
ловиях темноты и активность новообразования протохлорофиллида
в листьях кукурузы разного возраста (10 — 15 дней).
Варианты опыта:
1) зеленые растения — 0 ч темноты;
2) зеленые растения — 24 ч темноты;
3) зеленые растения — 48 ч темноты.
Относительное содержание протохлорофиллида в пробе мож-
но также определить флуоресцентным методом (см. с. 94).
Оформление результатов. Результаты представляют в виде таб-
лицы, где приводят первичные данные спектрофотометрического
анализа и результаты расчета содержания различных пигментов в
листьях.
77
2.4. АНАЛИЗ СПЕКТРОВ ПОГЛОЩЕНИЯ
И ФЛУОРЕСЦЕНЦИИ ПИГМЕНТОВ
Спектром поглощения называют зависимость поглощения (Л)
или коэффициента поглощения (е или а) от длины волны (X)
или частоты электромагнитных колебаний (v), выражаемую кри-
вой поглощения света данным веществом. Различные по химичес-
кой природе вещества имеют характерные для них спектры по-
глощения.
Спектры поглощения характеризуются числом и положением
максимумов поглощения, интенсивностью отдельных полос по-
глощения, их полушириной. Положение главного максимума в
электромагнитном спектре определяется энергией поглощаемых
фотонов (Б) и зависит от разности энергии возбужденного (Е2) и
основного (Д) состояний молекулы вещества (А — постоянная
Планка):
£ 2 — 1
V = —----Ь
h
Положение максимума поглощения выражается обычно соот-
ветствующей длиной волны в нанометрах или волновым числом
в обратных сантиметрах. Интенсивность полосы поглощения оп-
ределяется вероятностью данного электронного перехода, т.е. ве-
роятностью поглощения кванта молекулой. Интенсивность по-
лосы поглощения обычно измеряется в единицах поглощения
H = lgy- или молярного е (удельного а) коэффициента погло-
щения. Положение максимума полосы поглощения лежит в ос-
нове качественного анализа вещества, а интенсивность полосы
поглощения — в основе количественного анализа.
Рис. 20. Схема расчета полуширины
полосы поглощения (объяснение
в тексте)
Для характеристики отдельных
полос поглощения используют
также значение полуширины
спектральной линии, т. е. рассто-
яния [нм], соответствующего по-
ловине максимальной оптической
плотности (рис. 20).
Спектр поглощения является
важнейшей физической характе-
ристикой фотохимически актив-
ных соединений, так как позво-
ляет судить о количестве энергии,
необходимой для активации ис-
следуемой фотореакции. Исследо-
вание спектров поглощения не-
78
Рис. 21. Спектр поглощения про-
тохлорофилла в этиловом эфире
Рис. 23. Спектр поглощения хло-
рофилла b (7) и феофитина b (2)
в этиловом эфире
а, л • г-1 • см-1
140>L
Рис. 22. Спектр поглощения хло-
рофилла а (7) и феофитина а (2)
в этиловом эфире
Рис. 24. Спектр поглощения вио-
лаксантина в этаноле
Рис. 25. Спектр поглощения лю-
теина в этаноле
79
обходимо во многих случаях работы с пигментами. Спектры по-
глощения отдельных пигментов в различных растворителях даны
на рис. 21 — 25.
В настоящее время анализ спектров поглощения широко при-
меняется при исследовании химической природы пигментов, про-
цесса биосинтеза пигментов, при анализе пигментного состава
ФС I и ФС II, а также для других целей.
Для исследования пигментов в нативном состоянии очень пер-
спективным оказался метод регистрации низкотемпературных спек-
тров поглощения, который в последние годы находит все более
широкое применение. Низкотемпературные спектры дают более
высокую степень разрешения в структуре спектральной кривой и
позволяют получить дополнительную информацию о состоянии
пигментов в живом листе или какой-либо модельной системе.
Использование этого метода в ряде лабораторий позволило обна-
ружить сложную структуру красной полосы поглощения, обус-
ловленную наличием большого разнообразия нативных форм пиг-
ментов с различным характером пигмент-пигментных и пигмент-
белковых взаимодействий (табл. 9, 10).
Спектры поглощения пигментов могут быть сняты с помощью
любого спектрофотометра. Спектры поглощения пигментов в ра-
Таблица 9
Основные максимумы поглощения пигментов группы хлорофилла
в различных растворителях, нм
Пигмент Этиловый эфир Этиловый спирт Ацетон Живой лист
Протохлорофилл 623 625 625 630, 650
Хлорофилл а 661 665 663 675-680
Хлорофилл b 642,5 645 645 650
Таблица 10
Максимумы поглощения каротиноидов в различных растворителях, нм
Пигмент Сероуглерод Бензин Хлороформ
Р-Каротин 450; 485; 520 426; 452; 483,5 - 466; 497
Лютеин 445; 475; 508 420; 448; 477,5 428; 456; 487
Виолаксантин 440; 470; 501 418; 443; 462 424; 452; 482
80
627
Рис. 26. Спектр флуоресцен-
ции раствора протохлорофил-
ла в этиловом эфире
668
Рис. 27. Спектр флуоресценции
хлорофилла а в ацетоне
створах, гомогенатах и живых тканях листа снимают с помощью
регистрирующего спектрофотометра с интегрирующей сферой.
Спектры флуоресценции, как и спектры поглощения, являются
важным физическим параметром, характеризующим химическую
природу и состояние пигментов. Для всех пигментов класса пор-
фиринов характерна интенсивная флуоресценция в красной об-
ласти спектра (рис. 26, 27).
Флуоресценция — излучение световой энергии при переходе
фотовозбужденной молекулы пигмента из первого возбужденного
синглетного состояния в основное, невозбужденное состояние.
Вероятность излучения поглощенной молекулой энергии в виде
света флуоресценции зависит от ряда факторов: химической струк-
туры вещества, его физического состояния, молекулярного окру-
жения, возможности миграции энергии в системе, эффективнос-
ти преобразования энергии в химическую форму и т.д.
Исследования спектров флуоресценции и кинетики флуорес-
ценции в настоящее время широко используют для характеристи-
ки пигментных систем, анализа динамики взаимопревращения
пигментов при фотобиологических процессах, характера взаимо-
действия молекул пигментов в системе, в частности при исследо-
вании первичных реакций фотосинтеза и процессов миграции
энергии, для характеристики пигментного состава и функциональ-
ной активности первой и второй фотосистем и решения многих
других проблем фотосинтеза.
Большим преимуществом использования метода флуоресцен-
ции является возможность проведения измерений in vivo; при этом
исследования можно проводить на интактных органах растения,
клетках, суспензии хлоропластов, предварительно обработанных
по определенной схеме.
Исследования флуоресценции в различных экспериментальных
условиях могут дать информацию о процессах миграции энергии в
81
Рис. 28. Спектр флуоресцен-
ции хлоропластов при 20 °C
(7) и -196°C (2) (по [8], с
изменениями)
фотосинтезирующей системе, о ки-
нетике процессов переноса элект-
ронов в электрон-транспортной
цепи и влиянии на этот процесс
различных агентов, изменяющих со-
стояние переносчиков ЭТЦ.
Анализ спектров флуоресцен-
ции при температуре жидкого азо-
та (-196 °C) дает возможность по-
лучить многостороннюю информа-
цию о характере протекающих при
фотосинтезе процессов. При темпе-
ратуре жидкого азота, вследствие
ингибирования биохимических про-
цессов и сопряженных с ними про-
цессов преобразования энергии в фотохимических реакциях, рез-
ко возрастает квантовый выход флуоресценции. В этих условиях
можно наблюдать интенсивную флуоресценцию не только вто-
рой, но и первой пигментной системы, которая в обычных усло-
виях очень слабо флуоресцирует. Как видно на рис. 28, в спектре
флуоресценции хлоропластов высших растений при 20 °C главный
максимум расположен при 685 нм и более слабая широкая полоса
появляется при 735 нм. При охлаждении хлоропластов до темпе-
ратуры -196 °C в спектре появляются три основные полосы: при
685, 695 и 735 нм (очень интенсивная полоса). Максимум 685 обус-
ловлен флуоресценцией коротковолновых форм пигментов, со-
ставляющих основную часть второй пигментной системы, в то
время как полоса 735 принадлежит длинноволновым формам хло-
рофилла первой фотосистемы. Отличаясь по составу и соотноше-
нию длинноволновых и коротковолновых форм пигментов, пер-
вая и вторая пигментные системы при возбуждении дают различ-
ные спектры, которые различаются по интенсивности флуорес-
ценции при 685 и 735 нм. На основании этого во многих работах
метод низкотемпературной флуоресценции используется для ха-
рактеристики фотохимической активности фотосистем в различ-
ных экспериментальных условиях.
Исследования низкотемпературных спектров флуоресценции с
успехом применяют в настоящее время для изучения структуры
ЭТЦ и природы акцепторов первой и второй фотосистем. Иссле-
дования показали, что ряд окислителей является эффективным
тушителем флуоресценции. Например, добавление феррицианида
калия в суспензию хлоропластов значительно снижает интенсив-
ность излучения, ускоряя регенерацию (окисление) акцептора.
Определение степени тушения флуоресценции при введении раз-
личных агентов может дать сведения о взаимодействии последних
с фотовозбужденной молекулой пигмента реакционного центра.
82
Метод флуоресцентной спектроскопии очень удобен для ис-
следования быстропротекающих процессов фотопревращения про-
тохлорофилловых пигментов в хлорофилл в процессе зеленения
этиолированных проростков. При изучении процессов биосинтеза
пигментов широко используют метод ингибирования отдельных
ферментных систем, приводящий к накоплению промежуточных
продуктов биосинтеза и снижению содержания конечного про-
дукта — хлорофилла.
Исследование стимулирующего и ингибирующего действия
отдельных групп веществ на процессы синтеза хлорофилла позво-
ляет выяснить участие ферментных систем в процессах биосинте-
за пигментов и зависимость последних от характера различных
метаболических процессов, протекающих в растении.
2.4.1. Характеристика спектров поглощения пигментов
в растворах, гомогенатах и живых тканях листа с помощью
спектрофотометрического метода
Спектры поглощения отражают химическую природу фотосин-
тетических пигментов, а также характер их взаимодействия с ок-
ружающими соединениями (молекулами растворителя в раство-
рах, белками и другими пигментами в системах in vivo). Сравни-
тельный анализ спектров поглощения позволяет получить инфор-
мацию о состоянии пигментов фотосинтетического аппарата, оце-
нить их структурные и функциональные свойства.
Ход работы
Работа включает следующие этапы: 1) приготовление экстрак-
тов пигментов и снятие спектров поглощения пигментов в ра-
створах; 2) приготовление гомогената листьев и снятие спектров
поглощения пигментов в гомогенате; 3) запись спектров погло-
щения листьев; 4) запись низкотемпературных спектров погло-
щения листьев.
Работу проводят на регистрирующем двухлучевом спектрофо-
тометре с интегрирующей сферой.
1. Приготовление экстрактов пигментов и снятие спектров по-
глощения пигментов в растворах. Растворы пигментов в ацетоне и
этиловом эфире готовят по методу, описанному ранее на с. 54.
Экстракт пигментов наливают в кювету, укрепляют в кюве-
тодержателе кюветного отделения. В контрольную кювету нали-
вают растворитель. Производят запись спектра. Спектры погло-
щения пигментов ацетонового и эфирного экстрактов пигмен-
тов, а также гомогената и ткани живого листа записывают на
одном бланке.
83
2. Приготовление гомогената листьев и снятие спектров погло-
щения пигментов в гомогенате. Навеску листьев 0,3 —0,5 г расти-
рают в охлажденной ступке с 10 мл (для лучшего растирания сна-
чала добавить 3 — 4 мл) 1/15 М Na2HPO4. Суспензию отжимают
через полотно и центрифугируют 15 мин при 100g. В работе ис-
пользуют надосадочную жидкость. Для просветления к раствору
гомогената добавляют глицерин (40 % по объему в конечной кон-
центрации). В качестве контрольного раствора используют раствор
глицерина той же концентрации.
3. Запись спектров поглощения листьев. Живой лист опытного
растения помещают в специальные круглые кюветы. В качестве
контроля для уравнивания светорассеяния непрозрачного листа
используют пергаментную или фильтровальную бумагу. Кюветы
ставят в специальные (круглые) прорези кюветного отделения,
соединяющие кюветное отделение с интегрирующей сферой, и
записывают спектр поглощения.
Различные положения максимумов поглощения пигментов в
растворе и в комплексе с белком (гомогенат, живой лист) обус-
ловлены изменением положения электронных уровней в резуль-
тате взаимодействия молекул пигмента с молекулами раствори-
теля, молекулами белка, пигмент-пигментных взаимодействий.
In vivo наблюдается заметный сдвиг максимума поглощения в длин-
новолновую область.
4. Запись низкотемпературных спектров поглощения листьев. Для
регистрации низкотемпературных спектров поглощения исследу-
емый объект (живые ткани, гомогенаты, суспензия изолирован-
ных хлоропластов) помещают в специальные маленькие стеклян-
ные сосуды с двойными стенками (сосуды Дьюара), в которые
добавляют жидкий азот. После достаточного охлаждения объекта
и прекращения кипения азота контрольный и опытный сосуды
Дьюара помещают в верхние отверстия интегрирующей сферы
спектрофотометра на пути световых пучков и производят запись
спектров.
Оформление результатов. Производят описание полученных
спектров поглощения, отмечают изменения в их характере и по-
ложении максимумов поглощения в гомогенатах и целом листе по
сравнению с раствором пигментов.
2.4.2. Регистрация спектров флуоресценции
пигментов в растворах, гомогенатах и живых
тканях листа
Анализ спектров флуоресценции является в настоящее время
эффективным методом исследования пигментных систем и со-
пряженных с ними реакций фотосинтеза.
84
Для регистрации спектров флуоресценции в данной работе
используют установку, собранную на кафедре физиологии расте-
ний МГУ на основе монохроматора МДР-3*.
Описание установки
Блок-схема установки для регистрации спектров флуоресцен-
ции представлена на рис. 29. Установка включает следующие ос-
новные части:
• ртутно-кварцевую лампу ДРС-50 с фокусной линзой и свето-
фильтрами в защитном кожухе (7) и с блоком питания (2);
• фокусирующую линзу (5);
• монохроматор МДР-3 (4);
• приставку с фотоэлектронным умножителем ФЭУ-38 (5);
• стабилизированный источник питания ФЭУ ВСВ-2 (6);
Рис. 29. Блок-схема установки для регистрации спектров флуоресценции
(объяснение в тексте)
Для регистрации спектров флуоресценции можно воспользоваться флуори-
метром заводского образца, чувствительным в области 600—1000 нм. Снятие низ-
котемпературных спектров флуоресценции требует специальной кюветы, позво-
ляющей работать с жидким азотом.
85
• универсальный лабораторный потенциометр ЛПУ-01 (7);
• контрольный самопишущий потенциометр КСП-4 (<?).
Система освещения состоит из источника возбуждающего све-
та — ртутно-кварцевой лампы ДРС-50 и системы линз, фокуси-
рующих свет на объект (Р) и щель монохроматора. Возбуждающий
свет от лампы проходит через светофильтры СЗС-8 и СЗС-9, про-
пускающих свет от 400 до 550 нм и отсекающих свет длинновол-
новой части спектра, где находятся максимумы флуоресценции
зеленых пигментов. Благодаря этому в систему монохроматора
попадает только свет флуоресценции объекта, и линии ртутно-
кварцевой лампы в красной области света не мешают измерениям.
Монохроматор МДР-3 построен по асимметричной схеме Фа-
сти со сферическими зеркалами в качестве объективов.
Оптическая схема монохроматора показана на рис. 30. Свет от
источника 1 через осветительную систему 2 попадает на свето-
фильтр 11 и далее на входную щель 3. Поворотное зеркало 4 и
зеркальный сферический объектив 5, в фокальной плоскости ко-
торого расположена входная щель, направляют параллельный
пучок на дифракционную решетку 6. После дифракции параллель-
ный пучок лучей направляется зеркальным сферическим объек-
тивом 7на одну или на другую выходную щель, расположенную в
его фокальной плоскости.
Рис. 30. Оптическая схема монохроматора МДР-3 (объяснение в тексте)
86
В зависимости от положения поворотного зеркала 8 пучок све-
та попадает либо на щель 9, либо на щель 10.
Рабочий диапазон монохроматора обеспечивается тремя смен-
ными дифракционными решетками с числом штрихов 1200, 600
и 300 на 1 мм, работающими соответственно в трех областях спек-
тра: 200-600, 400-1200 и 800-2200 нм.
Для проектирования источника света на щель монохроматора
служит конденсор; так как конденсор не ахроматичен, его фоку-
сировка его в различных областях спектра осуществляется изме-
нением расстояний между входной щелью и конденсором и меж-
ду конденсором и источником света; при этом увеличение изоб-
ражения источника излучения, обеспечиваемое конденсором, бу-
дет изменяться от 4,7 до 3,3х.
Положение конденсора в зависимости от длины волны указа-
но ниже:
Длина волны линии, приведенной на выходную щель, нм Расстояние от источника света до конденсора, мм Расстояние от конденсора до входной щели, мм
220 80,2 381,6
273 90,3 371,5
700 100,2 361,6
1000 101,9 359,9
1500 104,0 357,8
2000 106,5 355,3
Щелевая линза проектирует плоскость конденсора в зрачок
монохроматора с увеличением 1,6х.
Для срезания налагающихся на рабочий диапазон спектров
высшего порядка служат светофильтры 11. Светофильтр БС5 при-
меняется для работы в области спектра 360 — 500 нм, светофильтр
ОС11 — в области спектра 600— 1000 нм, светофильтр ИКС1 — в
области спектра 1000—1500 нм; в области спектра 1500 — 2200 нм
используется интерференционный светофильтр с коротковолно-
вой границей пропускания около 1250 нм.
Кинематическая система монохроматора обеспечивает поворот
дифракционной решетки при развертке спектра и отсчет длины
волны излучения, падающего на выходную щель. Развертка спек-
тра может осуществляться от руки или автоматически от электро-
двигателя.
Общий вид монохроматора представлен на рис. 31, Л. Монохро-
матор собран на жестком литом основании 1. Все оптические узлы
размещены на верхней плоскости основания, а внутри него смон-
тированы элементы кинематической схемы.
Входная и две выходные щели монохроматора симметричные,
с радиусом кривизны 118 и 122 мм соответственно и переменной
87
оо
оо
22
21
20
19
18
13 12
Рис. 31. Монохроматор:
А — общий вид; Б — расположение дифракционной решетки;
В — схематическое изображение передней панели (пульта уп-
равления монохроматора). Объяснение в тексте
шириной раскрытия от 0 до 4 мм. Отсчет ширины раскрытия осу-
ществляется по шкале барабанчика 2 входной щели и барабанчи-
ков 3 выходных щелей с ценой деления 0,01 мм.
Для дополнительной фокусировки каждую щель можно пере-
мещать вдоль оптической оси в пределах 6 мм. Перемещение ще-
лей осуществляется вращением колец 4, при этом отсчеты снима-
ются по шкалам 5 соответственно для каждой щели. Фиксация
выбранного положения щелей осуществляется винтами 6, распо-
ложенными на тубусе каждой щели.
В насадке 7, надеваемой на входную щель, могут быть установ-
лены диафрагма с фигурными вырезами, ограничивающая высо-
ту щели, и сменные светофильтры. За входной щелью находится
затвор, который включается рукояткой 8.
Направление света в одну из выходных щелей (9 или 10) осу-
ществляется с помощью поворотного зеркала, включаемого в све-
товой пучок рукояткой, расположенной под щелью 10. При рабо-
те с одной выходной щелью вторая щель закрывается крышкой.
Дифракционные решетки в оправах помещаются на кронштейн
И (рис. 31, Б) держателя. При этом шаровые опоры устанавли-
ваются в лунки 12 держателя, а упор прижимается к плоской
площадке держателя. Шаровые опоры закреплены контргайка-
ми 13.
Откидные крышки предохраняют решетки, находящиеся в не-
рабочем состоянии, от повреждения. Установка дифракционных
решеток в монохроматор осуществляется через окно в корпусе,
закрываемое крышкой 14 (рис. 31, Л).
Определение длины волны участка спектра, выходящего из
выходной щели, производится по шкале счетчика 15, градуиро-
ванного для решетки 600 штр/мм. При установке других решеток в
показании счетчика следует вводить коэффициент 0,5 для решет-
ки 1200 штр/мм или коэффициент 2 для решетки 300 штр/мм.
Внутри основания монохроматора расположены синусный ме-
ханизм, коробка скоростей, электромагнитные муфты и элемен-
ты схемы автоматики.
Все узлы монохроматора закрыты кожухом 16, закреплены вин-
тами 17(рис. 31, Л).
На основании монохроматора расположен пульт управления,
на котором имеются (рис. 31, Л): тумблер 7<? включения сети, тум-
блер 19 включения механизма развертки спектра, переключатель
20 скоростей развертки и тумблер 21 прямого или обратного хода
развертки, рукоятка 22 ручной установки длины волны.
Схематично передняя панель монохроматора (пульт управления)
показана на рис. 31, В.
Переключатель скоростей развертки имеет пять положений.
Положения 1—4 переключателя соответствуют различным скоро-
стям развертки спектра, значения которых приведены далее:
89
Положение переключателя скоростей развертки спектра Скорость развертки спектра, нм/мин
для решетки 1200 штр/мм для решетки 600 штр/мм для решетки 300 штр/мм
1 0,8 1,6 3,2
2 4 8 16
3 20 40 80
4 96 192 384
При положении 5 переключателя редуктор отключается и обес-
печивается поворот дифракционной решетки от руки с помощью
рукоятки 22.
Скорости, соответствующие положениям 1, 2 и 3 переключа-
теля, могут использоваться для регистрации спектров. Скорость,
соответствующая положению 4, предназначена для быстрого по-
ворота дифракционной решетки при выборе рабочего диапазона
или при возвращении решетки в исходное положение.
На концах рабочего диапазона для ограничения поворота ди-
фракционной решетки предусмотрены концевые выключатели.
С монохроматором соединена приставка с фотоумножителем
(см. рис. 29). Приставка снабжена микроскопом, что позволяет осу-
ществить визуальный контроль за разверткой спектра. Специаль-
ная рукоятка-переключатель осуществляет переключение направ-
ления потока света, вышедшего из монохроматора. Линии спект-
ра, попадающего в выходную щель монохроматора, направляют-
ся в микроскоп поворотом зеркала, которое включается при вытя-
гивании рукоятки вверх до упора. При выключении зеркала (руко-
ятка вдвинута) открывается окно фотоголовки, и свет флуорес-
ценции попадает на катод фотоумножителя, преобразующий све-
товой сигнал в электрический ток. Для работы в красной и ин-
фракрасной областях спектра (600 — 1100 нм) используют фото-
умножитель ФЭУ-38. Ток, возникающий в фотоумножителе, уси-
ливается потенциометром ЛПУ-01 и поступает на вход потенцио-
метра КСП-4, отклонение пера которого пропорционально ин-
тенсивности линий. Для питания ФЭУ-38 служит высоковольт-
ный стабилизированный выпрямитель ВСВ-2.
Стабилизированный источник питания (выпрямитель) ВСВ-2 яв-
ляется стабилизатором тока высокого напряжения от 600 до 4000 В.
Записывающее устройство — контрольный самопишущий по-
тенциометр КСП-4. Самопишущие потенциометры позволяют
измерять величину ЭД С. Показания приборов отсчитываются при
помощи указателя на шкале. В одноточечном приборе запись про-
изводится непрерывно на диаграммной ленте той же градуиров-
90
ки, что и шкала. Для работы удобны приборы со следующей ха-
рактеристикой: пределы измерения 0—10 мВ; время пробега ка-
реткой шкалы — 1,0 с; скорость движения диаграммной ленты —
360—- 720 мм/ч.
Порядок работы на установке
|| Внимание! Рабочее напряжение питания ФЭУ выше 1000 В.
1. Перед началом работы необходимо проверить, чтобы все
выключатели приборов были установлены в положении «Выкл.».
2. Включить в сеть усилитель ЛПУ-01, самописец КСП-4, вы-
соковольтный выпрямитель ВСВ-2, монохроматор МДР-3, блок
питания лампы ДРС-50.
3. Открыть кран подачи воздуха ддя охлаждения лампы ДРС-50.
4. Включением правого тумблера блока питания зажечь лампу
ДРС-50.
5. Включить сетевые тумблеры на приборах: ВСВ-2 (левый тум-
блер), ЛПУ-01, КСП-4, МДР-3.
6. Прогреть приборы 20 — 30 мин.
7. Убедившись в открытии дифракционной решетки, закрыть
крышку монохроматора и поднятием рукоятки-переключателя на
приставке закрыть шторку ФЭУ-38, включить на ВСВ-2 тумблер
высокого напряжения (правый тумблер).
8. Установить исследуемый объект, произвести, если необхо-
димо, юстировку так, чтобы проекция светового потока люми-
несценции совпадала с центром крышки коллиматора.
9. Установить на МДР-3 требуемую длину волны (в области
максимума флуоресценции образца).
10. Установить требуемые значения входной и выходной щелей
монохроматора.
11. Снять крышку входного коллиматора, открыть шторку ФЭУ-
38 опусканием рукоятки-переключателя приставки вниз, поста-
вить маховики справа МДР-3 в положение «Откр.», слева — в
положение «Вкл.».
12. На ВСВ-2 рукоятками «Грубо» и «Плавно» установить необ-
ходимое ддя работы напряжение (в пределах 1400—1600 В).
I Внимание! Работа с высоким напряжением требует большого
внимания и осторожности!
13. Произвести, если это нужно, установку пера самописца в
требуемое положение с помощью рукояток «Настройка по буфер-
ному раствору» или «температурная компенсация» на ЛПУ-01. В не-
обходимых случаях переключить диапазон измерений.
Сигнал на самописец следует регулировать одновременным изме-
нением размера входной и выходной щелей, а также изменением на-
пряжения на ФЭУ-38.
91
14. При закрытой шторке ФЭУ-38 произвести возврат в корот-
коволновую (или длинноволновую) часть спектра вручную, вра-
щая маховик 22 (рис. 31, В) на передней панели МДР-3 (при этом
переключатель развертки спектра должен быть в положении 5)
или быстрым ходом развертки спектра (положение 4).
15. Включить лентопротяжный механизм КСП-4 и развертку на
МДР-3 (рис. 31, А, тумблер 19), произвести запись в требуемых
пределах спектра. На диаграммной ленте самописца автоматичес-
ки регистрируются значения интенсивности флуоресценции ис-
следуемого объекта (в относительных единицах) в установленных
пределах длин волн.
Определенный режим работы всей системы (ширину входной
и выходной щелей монохроматора, напряжение для питания ФЭУ,
усиление на ЛПУ-01) устанавливают в зависимости от объекта
(раствор пигментов, живой лист).
Смену исследуемого объекта, юстировку и другие вспомога-
тельные операции следует проводить при закрытой шторке ФЭУ
(рукоятка-переключатель приставки поднята вверх).
Внимание! При появлении неисправностей во время работы
установки (ненормальный шум, искрение, запах горелой изоля-
ции и др.) немедленно отключить приборы установки от сети
(в первую очередь ВСВ-2).
16. По окончании работы:
• отключить тумблером на ВСВ-2 высокое напряжение;
• выключить сетевые тумблеры на ЛПУ-01, КСП-4, ВСВ-2,
МДР-3, блоке питания;
• отключить все приборы от сети;
• через 5 — 10 мин после выключения лампы ДРС-50 прекра-
тить подачу охлаждающего воздуха.
Запрещается
1. Включать установку в работу или работать на ней при от-
ключенном или неисправном заземлении.
2. Устранять самостоятельно неисправности приборов уста-
новки.
3. Производить какие-либо изменения в схеме, соединениях
приборов установки, а также вскрывать приборы.
4. Снимать кожух с лампы ДРС-50.
Ход работы
Работа включает следующие этапы: 1) регистрация спектров
флуоресценции суммарной вытяжки пигментов и растворов хро-
матографически чистых пигментов; 2) регистрация низкотемпера-
турных спектров флуоресценции гомогенатов и листьев растений.
92
Таблица И
Основные спектральные линии ртутно-кварцевой лампы ДРШ-500
и неоновой лампы
Тип лампы Длина волны линии, нм
ртутно-кварцевая ДРШ-500 589 (оранжевая) 607 (красная) 612 (то же) 623 (- » -) 671,6 (красная) 690 (то же) 708 (- » -)
Неоновая 540 (зеленая) 585 (оранжевая) 594 (то же) 609 (красная) 614 (то же) 626 (— » —) 640 (красная) 650 (то же) 659 (- » -) 667-670 (- » -) 693 (- » -) 703 (- » -)
Перед началом измерений для проверки работы установки рекомен-
дуется произвести запись спектра источника света с отдельными
линиями определенной длины волны. Обычно для этой цели берут нео-
новую лампу или ртутные лампы с известными спектрами (табл. 11).
Неоновая лампа устанавливается вместо объекта. Снимается
спектр неоновой лампы с одновременной точной регистрацией
соответствующих значений шкалы барабана. Пользуясь спектрами
неоновой лампы (рис. 32) и учитывая относительную интенсив-
ность полученных полос, идентифицируют отдельные основные
полосы спектра. По этим данным строят калибровочную кривую,
где на оси абсцисс откладывают деления шкалы барабана, а на
оси ординат — длину волны [нм].
1. Регистрация спектров флуоресценции растворов пигментов.
В работе используют раствор пигментов в ацетоне, приготовлен-
ный, как описано в 2.1.1 (суммарная вытяжка пигментов), и хро-
матографически чистые препараты хлорофиллов а и Ь, получен-
ные одним из методов хроматографии (см. 2.2). Образец помещают
в держатель и тщательно фокусируют его флуоресценцию на вход-
ную щель. Затем производят запись спектра флуоресценции. Поря-
док работы на установке см. выше.
2. Регистрация низкотемпературных спектров флуоресценции
зеленого листа и гомогената. Гомогенат листьев готовят методом,
описанным в 2.4.1. Замораживание гомогената производят жид-
ким азотом в небольшом сосуде Дьюара.
Внимание! При работе с жидким азотом необходимо соблю-
дать правила техники безопасности: не допускать разбрызги-
вания азота, удерживать сосуд только с помощью сухого поло-
тенца или рукавиц.
93
690
Рис. 32. Спектр неоновой лампы (7) и ртутной лампы ПРК-4 (2).
Цифрами указаны длины волн основных пиков (нм)
После замораживания образец закрепляют в держателе и тща-
тельно фокусируют на входную щель. Производят запись спектра
флуоресценции гомогената.
При работе с листом используют специальный зажим, в кото-
рый крепят лист, после чего зажим опускают в сосуд Дьюара с
жидким азотом. Образец тщательно фокусируют на входную щель,
подбирают необходимое напряжение на фотоумножителе (доби-
ваются максимального сигнала на самописце) и производят за-
пись спектра флуоресценции.
Оформление результатов. Производят описание спектров флуо-
ресценции с указанием положения максимумов флуоресценции
исследованных объектов, отмечают изменения в спектрах, свя-
занные с нарушениями связи пигментов с белком при экстрак-
ции пигментов.
2.4.3. Исследование превращений пигментов при освещении
этиолированных проростков методом регистрации
низкотемпературных спектров флуоресценции
Высокочувствительный метод флуоресцентной спектроскопии
очень удобен для исследования быстро протекающих процессов
фотопревращения протохлорофилловых пигментов в хлорофилл
в процессе зеленения этиолированных проростков. Благодаря это-
94
jjy методу удалось установить последовательность реакций пре-
вращения предшественников хлорофилла в хлорофилл и динами-
ку формирования пигмент-белковых комплексов хлоропластов [5,
16, 26]. Данный метод также широко используют для исследова-
ния механизмов регуляции синтеза хлорофилла и формирования
активной фотосинтезирующей системы под влиянием фитохром-
ной системы, в ответ на действие различных веществ (промежу-
точных метаболитов синтеза хлорофилла, например аминолевули-
новой кислоты, регуляторов ферментов синтеза порфиринов и т. д.).
Ход работы
Работа состоит из двух частей: 1) регистрация низкотемпера-
турных спектров флуоресценции этиолированного листа и иссле-
дование динамики превращения пигментов в ходе его зеленения;
2) исследование влияния ряда веществ на процессы синтеза зеле-
ных пигментов в растении.
Используют листья разных видов растений (кукурузы, конских
бобов, пшеницы), выращенные в темноте в течение 7—10 дней.
1. Регистрация низкотемпературных спектров флуоресценции
этиолированного листа и исследование динамики превращения пиг-
ментов в ходе его зеленения. При выполнении данной работы не-
обходимо снять низкотемпературные спектры этиолированного
листа и листьев, освещаемых в течение определенного интервала
времени (от 15 с до 1 ч). Фиксацию материала проводят жидким
азотом, как описано выше. Спектры флуоресценции снимают в
области от 600 до 800 нм.
Отмечают скорость превращения протохлорофиллида (прото-
хлорофилла) в хлорофиллид (хлорофилл) у разных видов расте-
ний, используя для расчетов относительные значения флуорес-
ценции в области максимума кривой протохлорофилла и хлоро-
филла.
В опытах с более длительным освещением (4 — 6 ч) процесс
накопления пигментов можно исследовать путем регистрации спек-
тров поглощения на спектрофотометре.
2. Исследование влияния ряда веществ на процессы синтеза зе-
леных пигментов в растении. При выполнении данной работы про-
ростки двудольных или однодольных растений выращивают в пол-
ной темноте в течение 10 дней. Затем часть этиолированных про-
ростков в темноте помещают в стаканы, содержащие 10—15 мл
опытного раствора (опытные растения), а другую часть растений —
в стаканы с дистиллированной водой (контрольные растения).
Растения оставляют в полной темноте на ночь.
На следующий день растения переносят на свет и через каж-
дые 2 ч снимают низкотемпературные спектры флуоресценции
листьев.
95
По выявленным различиям между контрольными и опытными
растениями судят о влиянии того или иного вещества на биосин-
тез зеленых пигментов.
Возможные варианты опыта представлены ниже:
Вариант опыта Соединение Концентрация, М
1. Влияние промежуточных метаболитов синтеза порфиринов Аминолевулиновая кислота 0,01
2. Влияние активаторов ферментов биосинтетиче- ской цепи Пиридоксальфосфат 6-ю-7
Глутатион, цистеин (активаторы SH-фер- ментов) 0,005-0,01
3. Влияние ферментных ядов Азид натрия (инги- битор Me-содержа- щих ферментов) 0,0075
Иодацетамид (ингибитор SH-фер- ментов) 0,001-0,02
Оформление результатов. При оформлении результатов опреде-
ляют положения максимумов в спектрах флуоресценции, исполь-
зуя калибровочную для барабана по длинам волн. Данные, полу-
ченные при исследовании динамики превращения пигментов и
по влиянию ряда веществ на синтез пигментов, представляют в
виде таблицы и графика.
2.5. ОПРЕДЕЛЕНИЕ АКТИВНОСТИ ХЛОРОФИЛЛАЗЫ
МЕТОДОМ РАЗДЕЛЕНИЯ ФИТОЛЬНЫХ И
БЕСФИТОЛЬНЫХ ФОРМ ЗЕЛЕНЫХ ПИГМЕНТОВ
Активность хлорофиллазы определяют во многих исследовани-
ях, посвященных вопросам метаболизма пигментов в высших ра-
стениях и водорослях. Особый интерес к хлорофиллазе в послед-
нее время связан с ее участием в процессах деградации хлорофил-
ла в листьях. Фермент катализирует реакцию отщепления фитола
от хлорофилла с образованием хлорофиллида:
, хлорофиллаза , ~ тт
хлорофилл ---------------> хлорофиллид + С20Н39ОН.
Именно эта реакция, очевидно, реализуется в процессах де-
градации хлорофиллов in vivo.
96
Гидролитическая функция фермента также проявляется в ус-
ловиях in vitro: хлорофиллаза расщепляет хлорофиллы с образо-
ванием соответствующих хлорофиллидов и свободного фитола.
Хлорофиллаза выделена из листьев разных видов растений и
изучены некоторые свойства фермента [10, 11]. Показано, что
для проявления активности фермент требует высокой концент-
рации органических растворителей. Максимальная активность in
vitro наблюдалась в гомогенате, содержащем 45 % ацетона. При
определении активности хлорофиллазы в тканях большинства
видов высших растений обычно используют гомогенат, содер-
жащий 50 — 60% ацетона. Более высокая концентрация ацетона
(80 %) останавливает действие фермента. Определение активно-
сти хлорофиллазы проводится при pH 7,0, температуре 21 — 23 °C;
время инкубации фермента с субстратом — 30—60 мин. Субстра-
тами для действия хлорофиллазы являются хлорофиллы а и Ь,
феофитины а и b и другие пигменты, содержащие эфирную связь
с фитолом.
Изучение свойств белковых препаратов хлорофиллазы показа-
ло, что фермент может существовать в нескольких формах. Моле-
кула фермента в условиях разведения диссоциирует на отдельные
субъединицы, обладающие более высокой удельной активностью,
чем агрегированная форма фермента. В опытах с гель-фильтраци-
ей хлорофиллазы через сефадекс G-100 обнаружены две фракции
фермента (низко- и высокомолекулярная), способные к взаимо-
обратимым превращениям. Предполагают, что состояние фермента
и степень ассоциации отдельных субъединиц определяют актив-
ность хлорофиллазы в нативных структурах и при выделении фер-
ментов из ткани.
Методы определения активности хлорофиллазы основаны на
учете количества хлорофиллида в пробе до и после действия фер-
мента. Об активности фермента судят по увеличению количества
хлорофиллида в пробе за определенный период инкубации пиг-
мента с препаратом фермента. Образующийся в ходе реакции
хлорофиллид отделяют от неизмененного хлорофилла путем рас-
пределения фитольных и бесфитольных форм пигмента между
полярными и неполярными растворителями. При этом исполь-
зуют способность фитольных форм пигмента извлекаться полно-
стью из смеси неполярными растворителями. В качестве непо-
лярного растворителя берут обычно петролейный эфир (точка
кипения 40—60 °C). Хлорофиллиды остаются в щелочном вод-
но-ацетоновом растворе. Активность хлорофиллазы можно оп-
ределить также по убыли хлорофилла в петролейно-эфирной
фракции.
Отдельные виды растений резко отличаются по активности
хлорофиллазы. Например, в листьях сахарной свеклы активность
хлорофиллазы обычно очень высокая (до 40 % хлорофилла может
4 Гавриленко
97
превращаться в хлорофиллид), тогда как листья примулы отлича-
ются чрезвычайно низким уровнем активности фермента (1 — 4 %).
Активность хлорофиллазы изменяется с возрастом листьев.
Ход работы
Работа состоит из следующих этапов: 1) активация хлорофил-
лазы в гомогенате листьев и получение ацетонового экстракта
пигментов; 2) определение хлорофиллида путем отделения фи-
тольных пигментов от бесфитольных с использованием органи-
ческих растворителей; 3) спектрофотометрический анализ пиг-
ментов в полученных пробах.
В работе могут быть использованы листья гороха разных ярусов.
1. Активация хлорофиллазы в гомогенатах листьев. Определе-
ние активности хлорофиллазы у высших растений и водорослей
обычно проводят путем инкубации гомогената растительной тка-
ни, содержащего 40 — 60% органического растворителя, с после-
дующим учетом количества хлорофиллида, образовавшегося за
время инкубации.
Берут две навески листьев гороха одного яруса по 0,3 г (сред-
няя проба). В первой навеске (контрольной) определяют сумму зе-
леных пигментов и исходное содержание хлорофиллида в пробе.
Для этого навеску листьев растирают с 80 %-м ацетоном в при-
сутствии СаСО3 или MgCO3, экстракт пигментов фильтруют че-
рез стеклянный фильтр и объем доводят в мерной колбе до 50 мл.
Во второй навеске (опытной) определяют содержание хлоро-
филлида после действия хлорофиллазы. Навеску листьев растира-
ют с 60 %-м ацетоном и СаСО3 (MgCO3), доводят в цилиндре до
объема 25 мл и инкубируют при комнатной температуре (23 °C) в
течение 1 ч в темноте. Реакцию останавливают, добавляя ацетон
до его конечной концентрации 80 %. Для этого к гомогенату до-
бавляют равный объем (25 мл) 100 %-го ацетона. Гомогенат филь-
труют через стеклянный фильтр и экстракт доводят в мерной кол-
бе до объема 50 мл.
2. Определение хлорофиллида. Количество хлорофиллида в кон-
трольном и опытном экстрактах определяют по методу Веста и
Мак-Кинни [32]. Экстракты контрольного и опытного вариантов
делят на две части. Первую половину каждого из растворов остав-
ляют в темноте для определения суммарного содержания зеленых
пигментов. Во второй половине экстрактов определяют количе-
ство хлорофиллида, удаляя пигменты, содержащие фитол, пет-
ролейным эфиром. Для этого к 25 мл водно-ацетонового экстрак-
та добавляют 2,5 мл 0,02 н. NH4OH (из расчета 1 мл на 10 мл
водно-ацетонового экстракта) и смесь переносят в делительную
воронку. Туда же приливают 0,2 мл насыщенного раствора NaCl и
5 мл легкого петролейного эфира (точка кипения 40 —60 °C) и
98
смесь энергично встряхивают несколько раз. При этом в петро-
лейный эфир переходят липофильные формы пигментов, содер-
жащие фитол: хлорофиллы и каротиноиды. В щелочном водно-
ацетоновом слое остаются гидрофильные, бесфитольные пигмен-
ты — хлорофиллиды. После разделения слоев нижний водно-аце-
тоновый слой собирают в отдельную колбу. Его переносят обрат-
но в делительную воронку и экстракцию повторяют 3 — 4 раза до
полного извлечения хлорофиллов. К экстракту каждый раз добав-
ляют ацетон до первоначального объема. После конечной экст-
ракции водно-ацетоновый слой сливают в мерную колбу на 25 мл
и доводят чистым ацетоном объем до метки.
3. Спектрофотометрический анализ проб. Поглощение обоих
ацетоновых экстрактов (необработанного и обработанного петро-
лейным эфиром) контрольной и опытной проб измеряют на спек-
трофотометре. Концентрацию суммы хлорофиллов а и b рассчиты-
вают по формуле Арнона (см. с. 53). Для расчета концентрации
хлорофиллидов используют коэффициент 0,69 (см. 2.3.2).
Об активности хлорофиллазы судят по количеству хлорофил-
лида, образовавшегося в пробе дополнительно после одного часа
инкубации, выраженного в процентах к общему содержанию зе-
леных пигментов в контрольной пробе.
Оформление результатов. Полученные первичные данные по
спектрофотометрическому определению пигментов и расчетные
результаты вносят в таблицу (табл. 12). Отмечают изменения ак-
тивности хлорофиллазы в зависимости от возраста растений.
Схема записи результатов опыта
Таблица 12
Вариант Оптическая плотность Содержание пигментов в пробе Активность хлорофил- лазы
мг %
1. Контроль (сумма пигмен- тов хлорофиллид + хлоро- филл)
2. Контроль (хлорофиллид)
3. Опыт (сумма пигментов хлорофиллид + хлорофилл)
4. Опыт (хлорофиллид)
Глава 3
ХАРАКТЕРИСТИКА СТРУКТУРНОЙ
ОРГАНИЗАЦИИ И ФУНКЦИОНАЛЬНОЙ
АКТИВНОСТИ ФОТОСИНТЕЗИРУЮЩИХ
ТКАНЕЙ И ОРГАНОВ РАСТЕНИЙ
Фотосинтетическая функция растений имеет определенную
структурную организацию. Основным фотосинтезирующим орга-
ном большинства растений является лист, где локализованы спе-
циализированные органоиды — хлоропласты, содержащие пиг-
менты и другие компоненты, необходимые для поглощения и пре-
образования энергии света в энергию химических связей. Лист был
сформирован в ходе эволюции как специальный орган фотосин-
теза, поэтому анатомия листа, расположение хлорофиллсодержа-
щих клеток и тканей, их соотношение с другими элементами мор-
фоструктуры подчинены наиболее эффективному течению про-
цесса фотосинтеза, и они в наибольшей степени подвергаются
адаптивным изменениям. Таким образом, структура листа и его
фотосинтетическая активность оказываются тесно связанными.
Организация фотосинтетического аппарата на уровне листа
может быть рассмотрена на основе анализа его мезоструктуры.
Понятие «мезоструктура» введено А. Т. Мокроносовым [10]. Оно
включает систему морфологических характеристик фотосинтети-
ческого аппарата листа, хлоренхимы и клеток мезофилла. Основ-
ными показателями мезоструктуры фотосинтетического аппарата
(по А. Т. Мокроносову) являются: площадь листа; число клеток
на 1 см2; объем клеток; число хлоропластов в клетке; объем хло-
ропласта, содержание хлорофилла. Функциональная активность ли-
ста характеризуется общей интенсивностью фотосинтеза, а также
содержанием и активностью фотосинтетических ферментов. На
основании этих параметров можно получить расчетным путем боль-
шое число важных для понимания организации и функциониро-
вания фотосинтетического аппарата характеристик: активность
фотосинтеза однсго хлоропласта, количество хлорофилла в од-
ном хлоропласте, индекс пластидной поверхности, соотношение
между объемом клетки и объемом хлоропластов и т.д. Оценки
фотосинтеза в отдельном хлоропласте и числа хлоропластов в еди-
нице площади листа очень важны, так как произведение этих двух
показателей определяет фотосинтетическую активность листа
(А.Т. Мокроносов, Р.А. Борзенкова, 1978).
100
Комплексные исследования структурной организации и функ-
циональной активности листа представляют особый интерес в тех
случаях, когда требуется сопоставить особенности организации
фотосинтетического аппарата, например, при сравнительных ис-
следованиях растений, принадлежащих к различным системати-
ческим и экологическим группам, при изучении разнообразия сор-
тов культурных растений, различных фенотипических вариантов
одного растения, листьев на разных этапах онтогенеза и т.д.
3.1. ИССЛЕДОВАНИЕ МЕЗОСТРУКТУРЫ
ФОТОСИНТЕТИЧЕСКОГО АППАРАТА РАСТЕНИЙ
(по А.Т.Мокроносову)
Оценка мезоструктуры листа включает ряд количественных
показателей, одни из которых получают экспериментально, дру-
гие же рассчитывают на основании полученных эксперименталь-
ных данных. Принципиальная схема изучения мезоструктуры лис-
та показана на рис. 33.
Показатели, определяемые экспериментально.
1. Площадь листа [см2 или дм2].
2. Количество клеток фотосинтезирующих тканей в единице пло-
щади листа [тыс./см2].
3. Объем клеток [тыс./мкм3].
4. Число хлоропластов в клетке.
5. Объем хлоропластов [мкм3].
6. Содержание хлорофилла в единице
площади листа [мг/дм2].
Показатели, определяемые расчет-
ным путем.
1. Количество хлоропластов в единице
площади листа [млн/см2]: произведение
числа хлоропластов в клетке и числа
клеток в 1 см2 листа.
Рис. 33. Основные показатели мезострукту-
ры фотосинтетического аппарата (А.Т. Мо-
кроносов, 1981):
I — площадь листа; // — число клеток на 1 см2
листа, фотосинтез на 1 дм2 листа, хлорофилл
на 1 дм2, ключевые ферменты на 1 дм2, белок
фракции 1 на 1 дм2; III — объем клеток, тыс.
мкм3, число хлоропластов в клетке; IV — объем
хлоропласта, мкм3, площадь проекции хлоро-
пласта, мкм2, поверхность хлоропласта, мкм2
101
2. Объем клетки, соответствующий одному хлоропласту [мкм3]:
отношение объема клетки к числу хлоропластов в ней.
3. Относительный объем хлоропластов в клетке: суммарный объем
всех хлоропластов в процентах к объему клетки.
4. Индекс проективной поверхности хлоропластов (ИПХ): отно-
шение площади проекции всех хлоропластов к единице площади
листа. Определяют по номограмме.
5. Индекс поверхности наружных мембран хлоропластов (ИМХ):
произведение поверхности мембран хлоропластов на число плас-
тид в 1 см2 листа. Этот показатель имеет значение для расчета ско-
ростей трансмембранного переноса углекислого газа при фото-
синтезе и оттока ассимилятов из хлоропластов. Определяют по
номограмме.
6. Содержание хлорофилла в одном хлоропласте [мг/109 хлоро-
пластов или 109молекул хлорофилла на хлоропласт]: отношение
количества хлорофилла к числу хлоропластов на единице площа-
ди листа. Определяют по номограмме.
7. Молярная концентрация хлорофилла в хлоропласте [моль хло-
рофилла на 1 л объема хлоропластов]: определяется из соотноше-
ния объема хлоропластов и содержания в них хлорофилла (по но-
мограмме). Показатель достаточно условный, поскольку хлорофилл
распределен в объеме хлоропластов гетерогенно и не образует
молекулярного раствора.
Имея количественные показатели структурной организации
фотосинтетического аппарата, при наличии экспериментальных
данных по интенсивности фотосинтеза листа [мг СО2/(дм2- ч)] мож-
но рассчитать:
• фотосинтез одного хлоропласта [мг СО2/( 109 хлоропластов • ч)
или млн молекул СО2/(хлоропласт - с)] — отношение интенсивности
фотосинтеза и количества хлоропластов в единице площади листа;
• фотосинтез единицы площади наружной мембраны хлороплас-
тов [мг СО2/(дм2 мембран • ч)] — отношение интенсивности фото-
синтеза листа к ИМХ;
• ассимиляционное число [мг СО2/(мг хлорофилла • ч) или моль
СО2/(моль хлорофилла • ч)] — отношение интенсивности фотосин-
теза к содержанию хлорофилла в листе.
Ход работы
Работа включает следующие этапы: 1) подготовка суспензии
клеток мезофилла листа; 2) экспериментальное определение и
расчет ряда характеристик мезоструктуры листа.
В работе в качестве физиологических вариантов можно исполь-
зовать листья светолюбивых (например, одуванчика, ежи сбор-
ной, фикуса, герани) и теневыносливых (например, копытня,
калужницы, бегонии) растений, листья одного вида растений,
102
расположенных на свету и в тени (например, листья сирени, рас-
положенные снаружи и внутри куста), листья различного возрас-
та одного вида растения.
1. Подготовка суспензии клеток листа. Для приготовления сус-
пензии клеток используют высечки листа, сделанные трубчатым
сверлом диаметром 1 см из центральной части листа по обе сто-
роны от центральной жилки. Число высечек, сделанных справа и
слева от центральной жилки, должно быть одинаковым (по 2 — 3
высечки). Диски помещают в мерную пробирку со шлифом и за-
ливают 1 мл НС1 (концентрация 0,5 — 1 н., подбирают с учетом
специфики объекта).
Мацерацию тканей листа проводят на водяной бане при тем-
пературе 60 — 100 °C в течение 10 — 30 мин под тягой.
Затем пробирки охлаждают, объем жидкости доводят дистилли-
рованной водой до 5 мл, закрывают стеклянными пробками и тща-
тельно взбалтывают до получения однородной суспензии клеток.
2. Определение ряда характеристик мезоструктуры листа. До
начала определений необходимо сделать поперечные срезы листа и
под световым микроскопом тщательно изучить степень дифферен-
цировки листа на палисадную и губчатую ткань. Это обозначит
дальнейшую методику получения количественных характеристик
структурной организации листа.
В работе проводят определение и расчет представленных ниже
показателей мезоструктуры листа.
Количество клеток фотосинтезирующих тканей
в единице площади листа
В работе используют камеры Фукса—Розенталя (в случае рабо-
ты с крупноклеточными объектами, например с картофелем, свек-
лой, табаком) или камеры Горяева (при работе с мелкоклеточ-
ными объектами, например с древесными растениями, злаками).
Учитывают клетки по всему полю камеры (только хлорофиллонос-
ные). При взятии проб суспензию клеток тщательно перемешива-
ют, следят за правильностью притирания стекла в камере.
• При отсутствии дифференцированных клеток палисадной и губ-
чатой паренхим подсчитывают все клетки, находящиеся в камере.
• Если клетки палисадной и губчатой паренхим отличаются по
форме и имеется четкая грань между губчатой и палисадной тканя-
ми, то палисадные (П) и губчатые (Г) клетки подсчитывают от-
дельно. Для контроля подсчитывают общее число клеток. Оно дол-
жно быть равно или очень близко к сумме подсчитанных отдельно
клеток обеих тканей.
• В том случае, когда клетки палисадной и губчатой паренхим не
отличаются по форме, но имеется четкая грань между этими тканя-
ми, подсчитывают общее число клеток. Соотношение между клетка-
103
ми палисадной и губчатой паренхим в суспензии определяют ис-
ходя из соотношения палисадных и губчатых клеток (П/Г), опре-
деленного заранее на поперечных срезах листа.
• При отсутствии четкой грани между палисадной и губчатой
паренхимами на поперечном срезе листа необходимо ввести услов-
ную границу между тканями или выделить третий промежуточ-
ный слой и определить отношение П/Г. Далее поступить, как в
предыдущем случае.
Анатомический анализ многих видов растений показал, что при
подсчете клеток в камере наиболее часто коэффициент вариаций
V= 20 %. При таком значении для определения среднего числа
клеток необходимо провести их подсчет не менее чем в 16 каме-
рах при заданной ошибке Sx, равной 10 % при р = 0,95.
Для пересчета количества клеток [тыс./см2] нужно умножить
среднее значение числа клеток в полном объеме камеры Фукса
на коэффициент К = 411, а для камеры Горяева — на коэффици-
ент К= 1462. Эти коэффициенты справедливы для условий, когда
проба листьев имеет площадь 3,8 см2, а объем суспензии — 5 мл.
Объем клеток мезофилла листа
Наиболее точно удается определить объем клеток палисадной
ткани, так как по форме они близки к цилиндру или эллипсоиду.
Клетки губчатой ткани имеют чаще всего неправильную форму, и
ошибка определения их размеров значительна. Объем клеток вы-
ражают в тыс. мкм3. Для измерения объема клеток каплю суспен-
зии помещают на предметное стекло и осторожно закрывают по-
кровным, не прижимая его.
Палисадные клетки фотографируют при увеличении 15 х 10 или
15 х20 (в зависимости от размера). При этом же увеличении фото-
графируют шкалу объект-микрометра. Полученные негативы про-
ектируют на экран фотоувеличителя и с помощью линейки объект-
микрометра измеряют длинную (Z) и короткую (D) оси клеток,
включая клеточную оболочку. Объем клеток (V) рассчитывают по
формуле цилиндра
V=Ttr2LK,
К — поправочный коэффициент, значение которого меняется
в зависимости от соотношения L/D.
Таблица 13
Поправочные коэффициенты для расчета объема палисадных клеток
L/D 2,6 2,8 3,0 3,2 3,4 3,6 3,8 4,0 4,2 4,4 4,6 4,8 5,0
К 0,69 0,70 0,73 0,76 0,78 0,82 0,83 0,86 0,89 0,92 0,94 0,97 1,00
104
Поправочные коэффициенты найдены эмпирически Ю. Л. Цель-
цикер и представлены в табл. 13. При отношении L/D больше 5
поправочный коэффициент можно не вносить, так как клетка
условно принимается за цилиндр. При отношении L/D меньше 2,5
объем клетки целесообразно определить по формуле эллипсоида
вращения
Средний объем клеток можно рассчитать двумя путями:
1) определить объем каждой отдельной клетки и затем вычис-
лить средний объем;
2) вычислить объем по средним значениям Ln D. Этот способ
расчета проще, но ошибка найденного среднего объема увеличи-
вается, так как он определяется по формуле произведения сред-
них.
При средних значениях Ln D, измеряя 50—100 клеток при
И= 15—20 %, имеем Sx не более 5 % при р = 0,95.
Определить объем клеток губчатой ткани можно двумя спосо-
бами, которые, однако, дают лишь приближенные значения.
1. Делают и фотографируют тангентальный срез с нижней по-
верхности листа. Снимки проектируют на экран и определяют
площадь контуров 50—100 клеток. Затем, пользуясь объект-мик-
рометрбм, измеряют высоту клеток на поперечном срезе. Объем
определяют по произведению площади проекции клетки на вы-
соту.
2. Получают серию снимков тангентальных и поперечных сре-
зов листа, на основании чего строят объемную модель клетки
из пластилина в удобном масштабе и определяют объем клетки
по объему модели, используя при этом удельный вес пласти-
лина.
При наличии компьютера и программ для фотоморфометри-
ческих измерений можно произвести обработку полученных нега-
тивов и фотографий с помощью сканера и компьютера.
Число хлоропластов
1. Число хлоропластов в клетке определяют на давленых препа-
ратах. Для приготовления препарата каплю суспензии клеток по-
мещают на предметное стекло и закрывают покровным. Неболь-
шого нажатия на стекло достаточно, чтобы хлоропласты в клетке
распределились по плоскости.
Число хлоропластов определяют отдельно для клеток палисад-
ной и губчатой ткани. При выборе клеток одного типа необходи-
мо стремиться, чтобы в поле зрения попадали клетки разной ве-
105
личины. Для облегчения измерений можно использовать окуляр-
ную сетку.
Средний коэффициент вариаций числа хлоропластов в клетке
равен 25 — 30%. Для определения среднего значения числа хло-
ропластов в клетке при р = 0,95 и Sx = 10% нужно провести под-
счет не менее 25 — 35 клеток, а при Sx = 5% — в 100 — 140 клетках.
2. Число хлоропластов в единице площади листа [млн/см2] рас-
считывают путем умножения числа хлоропластов в клетке на чис-
ло клеток в 1 см2.
Размеры хлоропластов
Для определения размеров хлоропластов используют препа-
раты свежевыделенных хлоропластов. Для получения суспензии
хлоропластов кусочки свежих предварительно освещенных лис-
Рис. 34. Номограмма для определения объема хлоропласта
106
тьев помещают в небольшой объем (около 0,5—1 мл) среды,
содержащей 0,35 М сорбитол, 20 мМ трис-HCl буфер (pH 7,4) и
0,2 мМ MgCl2. Смесь осторожно растирают так, чтобы клетки раз-
рушались, а хлоропласты оставались неповрежденными (изото-
ническую концентрацию раствора сорбитола подбирают эмпиричес-
ки с учетом специфики объекта). Каплю суспензии быстро поме-
щают на предметное стекло, закрывают покровным и фотографи-
руют при увеличении 15 х40. При этом же увеличении фотографи-
руют шкалу объект-микрометра. Все операции проводят очень быс-
тро, не допуская разрушения хлоропластов!
Полученные негативы проектируют на экран фотоувеличителя
и измеряют длинные (Z) и поперечные (D) оси хлоропластов,
пользуясь линейкой объект-микрометра.
При наличии компьютера и соответствующих программ для
фотоморфометрических измерений можно осуществлять обработ-
Рис. 35. Номограмма для определения площади проекции хлоропласта
107
о
оо
Площадь поверхности наружной мембраны хлоропласта, мкм2
3,6 3,8 4,0 4,2 4,4 4,6 4,8 5,0 5,2 5,4 5,6 5,8 6,0 6,2 6,4 6,6 6,8 7,0 7,2 7,4
2,4 2,6 24,4 26,9 25,4 28,0 26,5 29,2 27,6 30,3 28,7 31,5 29,8 32,7 30,9 33,9 32,0 35,1 33,2 36,3 34,3 37,5 38,7 48,3 49,0 55,0 58,9 60,5 66,3 70,5 76,7 81,3 83,1 89,8 94,7
2,8 3,0 30,7 31,9 34,7 33,2 36,1 34,4 37,4 35,7 38,7 37,0 40,1 38,2 41,4 39,5 42,8 40,8 44,2 42,1 45,6 43,4 46,9
3,2 3,4 37,6 39,0 42,1 40,1 43,6 41,9 45,1 43,3 46,6 44,7 48,1 46,2 49,7 47,6 51,2 49,1 52,7 50,6 54,3 52,0 55,8 53,5 57,4
3,6 3,8 46,9 50,2 48,4 51,9 50,0 53,5 51,6 55,2 53,2 56,8 54,8 58,5 56,4 60,2 58,1 61,9 59,7 63,6 61,3 65,4 63,0 67,0 64,6 68,8
4,0 4,2 56,2 59,0 57,1 60,8 58,8 62,6 60,6 64,4 62,3 67,2 64,1 68,1 65,9 69,9 67,7 71,8 69,5 73,7 71,3 75,5 73,1 77,4 74,9 79,3
4,4 4,6 64,5 66,4 70,4 68,3 72,3 70,2 74,3 72,1 76,3 74,1 78,3 76,0 80,3 78,0 82,3 79,9 84,4 81,9 86,4 83,9 88,5 85,8 90,5 88,8 92,6
4,8 5,0 76,4 78,5 82,7 80,5 84,9 82,6 87,0 84,7 89,2 86,8 91,3 88,9 93,5 91,0 95,7 93,1 97,9 95,3 100 97,4 102 99,6 105
5,2 5,4 91,6 96,2 93,8 98,5 96,0 101 98,3 103 100 105 103 108 105 110 107 112 ПО 115
5,6 5,8 103 109 106 111 108 ИЗ 110 115 113 118 115 121 118 123 120 125
Окончание
Площадь поверхности наружной мембраны хлоропласта, мкм2
D 7,4 7,6 7,8 8,0 8,2 8,4 8,6 8,8 9,0 9,2 9,4 9,6 9,8 10,0 10,2 10,4 10,6
5,4 115 117 120 122 124 127 129 131 133 133
5,6 120 123 125 128 130 133 135 138 141 143
5,8 128 131 133 136 139 141 144 147 149 151
6,0 136 139 142 144 147 149 152 155 158 161
6,2 142 145 148 150 152 153 155 157 160 163 165
6,4 151 153 156 158 161 163 166 169 171 174 176
6,6 157 159 162 165 167 170 172 175 178 180 182 185
6,8 166 168 171 173 175 178 180 182 185 187 190 193
7,0 175 177 179 182 185 187 189 191 193 195 198 201
7,2 181 183 185 188 190 193 195 197 199 201 203 205
7,4 190 192 195 197 199 202 205 207 209 211 213
7,6
7,8
Рис. 36. Номограмма-таблица для определения площади наружной мембраны хлоропласта
Рис. 37. Номограмма для определения индекса пластидной поверхности
ку полученных негативов и фотографий с помощью сканера и
компьютера.
Средний коэффициент вариаций при измерении хлоропластов
равен 20 —25 %. Измеряя 100 хлоропластов, получаем ошибку не
более 5 % при р = 0,95, поэтому необходимо измерить длинную и
поперечную оси у 100 — 200 хлоропластов.
На основании проведенных измерений рассчитывают средние
значения осей. Полученные средние значения используют для оп-
ределения трех величин — объема, площади проекции, поверх-
ности наружной мембраны хлоропласта.
1. Рассчитывают объем хлоропласта [мкм3], принимая его за
объем эллипсоида вращения. Это максимально возможный объем
хлоропласта (рис. 34). Если фактически хлоропласт имеет форму
уплощенного эллипсоида, то найденный объем превышает реаль-
ный на 20 —30 %.
ПО
Рис. 38. Номограмма для определения площади поверхности наружных
мембран хлоропластов в единице площади листа
2. Площадь проекции хлоропласта [мкм2] рассчитывают по фор-
муле площади эллипсоида (рис. 35):
3. Поверхность наружной мембраны хлоропласта [мкм2] опреде-
ляют как поверхность эллипсоида вращения (рис. 36).
Используя объем хлоропласта, определяют индекс проективной
поверхности хлоропластов (ИПХ). Он представляет собой отноше-
ние площади проекции всех хлоропластов к единице площади листа
(рис. 37) и имеет тот же смысл, что и индекс листовой поверхно-
сти в фитоценозе.
На основании площади поверхности наружной мембраны и чис-
ла хлоропластов в единице площади листа определяют общую по-
верхность наружных мембран пластид в единице площади листа (но-
мограмма на рис. 38).
Используя данные по определению суммарного объема хло-
ропластов и объема клетки, рассчитывают относительный объем
хлоропластов в клетке [%].
111
Содержание хлорофилла
1. Содержание хлорофилла в листе [мг/дм2] определяют описан-
ными ранее методами (см. гл. 2).
2. Содержание хлорофилла в одном хлоропласте рассчитывают как
отношение количества хлорофилла к числу хлоропластов в еди-
нице площади листа (номограмма на рис. 39) и выражают либо в
мг/109 хлоропластов, либо через число Авогадро, пересчитанное
в 109 молекул на хлоропласт. Данные последнего варианта расчета
могут быть использованы для приближенной оценки числа фото-
синтетических единиц в хлоропласте, если известны размеры фо-
тосинтетической единицы.
Рис. 39. Номограмма для определения содержания хлорофилла в одном
хлоропласте
112
Пример. Содержание хлорофиллов а+ b равно 3,7 мг/дм2. При числе
хлоропластов 14,7 млн/см2 оно составляет 2,5 мг/109 хлоропластов, или
1,695 млрд молекул на 1 хлоропласт, что соответствует 5,63 млн фото-
синтетических единиц при размере единицы 300 молекул хлорофилла.
Оформление результатов. Экспериментально полученные и рас-
четные показатели мезоструктуры листа заносят в таблицу (табл. 14).
Таблица 14
Показатели структурной организации фотосинтезирующих тканей листа
Показатель Тип ткани Численное выражение показателя Средняя арифме- тическая Отклонение от средней арифметиче- ской
1. Количество клеток фотосинтезирующих тканей, тыс./см2 Палисад- ная
Губчатая
2. Объем клеток, тыс.мкм3 Палисад- ная
Губчатая
3. Число хлоропластов в клетке Палисад- ная
Губчатая
4. Объем хлоропластов, мкм3
5. Площадь проекции хлоропласта, мкм2
6. Поверхность наружной мембраны хлороплас- тов, мкм2
7. Количество хлороплас- тов в единице площади листа, млн/см2
8. Относительный объем хлоропластов в клетке, %
9. Индекс проективной поверхности хлоро- пластов (ИПХ)
10. Индекс поверхности наружных мембран хлоропластов (ИМХ)
11. Содержание хлоро- филла в листе, мг/дм2
12. Содержание хлорофил- ла в одном хлороплас- те, мг/109 хлоропластов
113
3.2. ОПРЕДЕЛЕНИЕ ИНТЕНСИВНОСТИ ФОТОСИНТЕЗА
И ДЫХАНИЯ РАСТЕНИЙ
Определение интенсивности фотосинтеза и дыхания на целых
растениях, отдельных листьях и высечках может быть проведено
путем регистрации скорости газообмена образцов. Как известно,
в ходе фотосинтеза на свету растения выделяют кислород и по-
глощают углекислый газ, в темноте происходит обратный про-
цесс — поглощение кислорода и выделение углекислого газа за
счет дыхания. Поэтому, определяя скорость выделения и погло-
щения того или другого газа, можно судить о скорости фотосин-
теза и дыхания у растений [13].
Наиболее распространенные в настоящее время газометри-
ческие методы оценки суммарной интенсивности фотосинтеза
основаны на учете количества поглощенной углекислоты или вы-
деленного кислорода. Эти методы с достаточной точностью по-
зволяют исследовать фотосинтетическую активность целых рас-
тений или отдельных листьев, хотя некоторые трудности в ин-
терпретации данных могут возникнуть вследствие того, что на
свету возможны неполное ингибирование «темнового дыхания»,
а также активация у многих растений фотодыхания. Поэтому
наблюдаемые на свету изменения в концентрации исследуемых
газов являются интегральными показателями указанных про-
цессов.
Для определения интенсивности газообмена растений могут
быть использованы различные методы: определение интенсивно-
сти фотосинтеза с помощью инфракрасных газоанализаторов,
манометрический метод Варбурга, радиоуглеродный, катаромет-
рический, электрохимический и др. Выбор метода зависит от цели
работы и наличия той или иной лабораторной техники. Наиболее
широко для определения интенсивности фотосинтеза и дыхания
по углекислому газу используется инфракрасный газоанализатор,
а по скорости выделения или поглощения кислорода — поляро-
графический метод.
3.2.1. Определение интенсивности фотосинтеза и дыхания
с использованием потенциометрического метода
регистрации углекислого газа и полярографического
определения кислорода
В данной работе интенсивность фотосинтеза и дыхания опре-
деляют по кислороду и углекислому газу, причем для определе-
ния по кислороду используют полярографический метод (датчи-
ком является закрытый электрод Кларка), а по углекислому газу —
114
г
потенциометрический метод (СО2-чувствительным датчиком яв-
ляется стеклянный рН-электрод).
Полярографический метод определения концентрации кисло-
рода в среде основан на реакции восстановления кислорода на
Катоде и может быть использован для характеристики скорости
реакций фотосинтеза, связанных с выделением кислорода. Тео-
рия метода описана в 1.5.
В опытах измеряют силу тока, возникающего при потенциале,
примерно соответствующем середине площадки предельного тока
вольт-амперной характеристикой (650 мВ). Сила тока пропорцио-
нальна содержанию кислорода в среде.
Потенциометрический метод определения СО2 основан на при-
менении СО2-чувствительного датчика, представляющего собой
модификацию оборудования для измерения pH. Он состоит из
стеклянного pH-электрода, погруженного в смесь, содержащую
5 • 10"3 М NaHCO3 и 5 • 10'2 М КС1. Изменения в содержании СО2
приводят к изменению pH смеси: при его увеличении происходит
подкисление раствора, при уменьшении — подщелачивание. Из-
менения pH регистрируются pH-метром и передаются на много-
канальный самописец.
Описание установки
Регистрация количественных изменений кислорода и углекис-
лого газа производится на специальной установке, разработан-
ной и собранной на кафедре физиологии растений биологическо-
го факультета МГУ.
Установка для определения интенсивности фотосинтеза и ды-
хания целых растений и отдельных листьев — комплексная, т. е.
позволяет исследовать любой из этих процессов двумя методами:
полярографическим и потенциометрическим одновременно. Воз-
можность одновременного использования двух методических при-
емов обусловлена конструкцией комбинированной электродной
ячейки.
На рис. 40 представлена схема общего замкнутого воздушного
канала с комбинированной ячейкой, включающей датчик О2 и
датчик СО2. Как видно из представленной схемы, воздух из каме-
ры с проростками (5) по одному из отводящих шлангов поступа-
ет в комбинированную ячейку (7) сначала в отсек с электродом
Кларка (датчик О2) (5), затем — в отсек со стеклянным электро-
дом, погруженным в буферный раствор (датчик СО2) (2). Далее
из ячейки воздух поступает в микрокомпрессор (4) и снова на-
гнетается в камеру с проростками.
Блок-схема данной установки представлена на рис. 41.
Полярографическая часть установки включает: закрытый элект-
род Кларка (6) в ячейке (4), через которую продувается воздух,
115
Рис. 40. Схема воздушного канала с ком-
бинированной ячейкой датчиков О2 и СО2:
1 — комбинированная ячейка; 2 — стеклян-
ный электрод (датчик СО2); 3 — закрытый
электрод Кларка (датчик О2); 4 — микроком-
прессор; 5 — камера для растений
Рис. 41. Блок-схема уста-
новки с комбинирован-
ной ячейкой датчиков О2
и СО2 для определения
фотосинтеза и дыхания
растений:
1 — источник света; 2 —
тепловой фильтр; 3 — каме-
ра для растений; 4 — ком-
бинированная ячейка; 5 —
датчик СО2; 6 — датчик О2
(электрод Кларка); 7—мик-
рокомпрессор; 8 — микро-
вольтнаноамперметр Ф-136;
9- ЛПУ-01; 10- ЛПУ-01
(рН-метр); 11 — самописец
КСП-4 (многоканальный,
точечный)
116
рыходящий из герметически закрытой камеры с проростками (5);
микровольтнаноамперметр Ф-136 (<?), регистрирующий показа-
ния датчика О2; ЛПУ-01 (9), усиливающий мощность сигнала. Ре-
гистрацию показаний микровольтнаноамперметра (силу тока, со-
ответствующую концентрации О2 в воздухе) производят на мно-
гоканальном точечном самописце КСП-4 (77).
Потенциометрическая часть установки включает стеклянный
электрод (5), помещенный в ячейку с буфером (4), и ЛПУ-01
(рН-метр) (79). Изменения pH, вызванные изменениями концен-
трации СО2 в воздухе, регистрируются на том же самописце.
Порядок работы на установке
1. Полярографическая часть установки.
1.1. Проверить заземление.
1.2. Включить приборы в сеть.
1.3. Включить усилитель ЛПУ-01, микровольтнаноамперметр
Ф-136, самописец; подключить электрод Кларка к соответствую-
щим клеммам установки (панель регулировки: клеммы «+» и «-»),
включить батарею.
1.4. С помощью рукоятки регулировки напряжения подать на-
пряжение 650 мВ на электрод (контролировать по шкале ЛПУ-01),
при этом положение тумблера на ЛПУ-01 и тумблеров на панели
регулировки установлены в положения «Проверка» и «Имита-
ция».
1.5. Переключить тумблеры: на ЛПУ-01 — в положение «Рабо-
та», на панели регулировки — в положения «Работа» и «Элект-
род», установить необходимое усиление.
1.6. Прогреть установку в течение 30—40 мин, убедиться в ста-
билизации тока электрода.
1.7. Тумблером «Настройка по буферному раствору» на ЛПУ-
01 отрегулировать положение писчика на диаграммной ленте са-
мописца.
2. Потенциометрическая часть установки.
2.1. Проверить заземление, ввести штекер электрода (датчика
СО2) во входное отверстие ЛПУ-01 (рН-метр).
2.2. Установить тумблер на панели регулировки усиления в
правое крайнее положение и подключить приборы (ЛПУ-01 и са-
мописец) к сети.
2.3. Включить приборы.
2.4. Тумблером «Настройка по буферному раствору» отрегули-
ровать положение писчика на диаграммной ленте самописца.
После окончания работы на панели регулировки полярогра-
фической части установки переключить тумблеры в положения
«Проверка», «Имитация», выключить тумблер «Батарея», выклю-
чить тумблеры на приборах. Отключить приборы от сети.
117
Ход работы
Работа включает следующие этапы: 1) измерение фотосинтеза
и дыхания на целых растениях или отделенных листьях по скорос-
ти поглощения или выделения кислорода и углекислого газа;
2) калибровка кислородного и углекислотного датчиков; 3) рас-
чет интенсивности фотосинтеза и дыхания растений.
В работе используют проростки растений (пшеницы, кукурузы и
др.), выращенные в различных условиях водоснабжения, освеще-
ния, минерального питания, и листья растений разного возраста.
1. Измерение фотосинтеза и дыхания на исследуемых объектах.
Растения помещают в камеру так, чтобы в ней оказались лишь
листья проростков. Смазывают резиновые прокладки камеры ва-
зелиновым маслом и закрывают ее, следя за тем, чтобы винты
плотно прижимали крышку и обеспечили герметичность камеры.
Устанавливают камеру в штатив. Между камерой и лампой поме-
щают тепловой фильтр (стеклянный сосуд с водой). Подключают
камеру нижним шлангом к микрокомпрессору, а верхним шлан-
гом — к ячейке с электродом и включают компрессор.
На микровольтнаноамперметре Ф-136 отмечают начальную силу
тока, проходящего через электрод. Включают протяжку диаграм-
мной ленты самописца и записывают кинетику изменений силы
тока, которая соответствует изменениям концентраций кислоро-
да и углекислого газа в канале, связанным с темновым дыханием
(10—15 мин).
После записи темнового дыхания включают осветитель и реги-
стрируют на самописце изменения силы тока, вызванные изме-
нениями концентраций кислорода и углекислого газа в результате
фотосинтеза растений (10—15 мин). Запись фотосинтеза и дыха-
ния одних и тех же растений производят два-три раза. По оконча-
нии записи растения извлекают из камеры и определяют размер
фотосинтетической поверхности и ее биомассу.
Расчет площади листьев проводят весовым методом. Для этого
на листе бумаги обводят контур опытного листа, обрисованный
участок бумаги вырезают и взвешивают (Р). Взвешивают 1 дм2
этой же бумаги (р) и по отношению Р/р определяют площадь
листа S.
Площадь листьев злаковых можно рассчитать по формуле
s=jlb,
где / — длина листа; b — ширина листа в наиболее широкой его
части.
Затем определяют сырую массу листьев. Для этого их помещают
в предварительно взвешенный бумажный пакетик и взвешивают
на аналитических весах.
118
Для определения сухой массы листьев их вместе с пакетиками
помещают в сушильный шкаф с температурой 105 °C и высушива-
ют до постоянной массы.
2. Калибровка кислородного и углекислотного датчиков. Для
расчета интенсивности фотосинтеза и дыхания в миллилитрах О2
или СО2 необходимо осуществить переход от миллиметров диаг-
раммной ленты самописца к миллилитрам О2 или СО2, соответ-
ственно выделившихся или поглощенных за определенное время.
Для этого проводят калибровку датчиков.
Калибровку кислородного датчика осуществляют в режиме про-
верки (переключатели на панели регулировки «Работа —Провер-
ка» и «Электрод —Имитация» устанавливают в положения «Про-
верка» и «Имитация»). Тумблером делителя напряжения «650 мВ»
на ЛПУ-01 устанавливают потенциал, равный 650 мВ при грубой
чувствительности установки, отмеченной на микровольтнаноам-
перметре Ф-136. После фиксации потенциала установку перево-
дят в режим работы, фиксируют оптимальную чувствительность
на Ф-136 и проводят калибровку цены деления диаграммной лен-
ты в единицах силы тока. Калибровку осуществляют следующим
образом.
1. На диаграммной ленте самописца фиксируют начальное по-
ложение писчика, а на микровольтнаноамперметре Ф-136 отме-
чают силу тока, проходящего через электрод Кларка, которая со-
ответствует этому положению.
2. Регулятором напряжения на панели регулировки устанавли-
вают силу тока, отличающуюся от первоначального значения не
более чем на 0,2 мкА, и на диаграммной ленте фиксируют поло-
жение писчика при данном значении силы тока.
3. Установив разницу между начальным и конечным положе-
ниями писчика в миллиметрах, рассчитывают цену деления диаг-
раммной ленты в единицах силы тока.
Пример. Если разнице силы тока в 0,2 мкА соответствует смещение
писчика на 60 мм, то в 0,1 мкА — на 30 мм диаграммной ленты.
Важно учесть, что при калибровке величина потенциала может
измениться, поэтому перед опытом следует снова провести коррек-
цию потенциала.
Калибровку СО 2-чувствительного электрода проводят с исполь-
зованием либо смеси газов с известной концентрацией в них СО2,
либо инертного газа аргона. Для калибровки с помощью аргона
газ пропускают через систему и регистрируют значение pH, соот-
ветствующее исходному содержанию СО2 в воздухе (0,03%), и
значение pH, соответствующее его полному вытеснению. Зная
объем воздуха в замкнутой системе и процент содержания в ней
СО2, рассчитывают исходное содержание СО2, соответствующее
начальному уровню сигнала. По разнице между начальным поло-
119
жением точек на самописце и конечным, соответствующим прак-
тически нулевому содержанию СО2 в смеси, рассчитывают цену
деления диаграммной ленты в единицах СО2.
3. Расчет интенсивности фотосинтеза и дыхания. Интенсивность
фотосинтеза и дыхания измеряют в следующих единицах:
1) мл О2 (мл СО2) • см-2-ч-1;
2) мл О2 (мл СО2)г сырой массы-1-ч-1;
3) мл О2 (мл СО2)г сухой массы-1-ч-1.
Для расчета интенсивности дыхания и фотосинтеза в милли-
литрах СО2 и О2 необходимо знать объем воздуха, циркулирую-
щего в замкнутом воздушном канале. В установке, собранной на
кафедре физиологии растений биологического факультета МГУ,
он складывается из:
• объема камеры для проростков — 216 мл;
• объема воздушного пространства (незанятого буфером и элек-
тродами) комбинированной электродной ячейки — 9,41 мл;
• объема воздуха в 4 отводящих шлангах — 14,62 мл.
В итоге объем воздуха в канале составляет 252,03 мл.
Определение цены деления диаграммной ленты в миллилитрах О2.
При расчете исходят из следующего:
1. Начальная сила тока, проходящего через электрод в отсут-
ствие растений, отражает содержание кислорода, находящегося в
замкнутом объеме камеры, компрессора и отводящих шлангов
(канал). Если объем воздуха в канале составляет 252,03 мл, то,
зная содержание кислорода в воздухе — 21 %, можно рассчитать
объем кислорода в канале — 52,92 мл.
Начальную силу тока для канала необходимо отмечать перед
проведением измерений.
2. Известна калибровка цены деления диаграммной ленты в еди-
ницах тока (см. выше). Зная начальную силу тока, количество кис-
лорода, обусловливающее этот ток, и цену деления для 0,2 мкА
диаграммной ленты [мм], производят расчет.
Пример. Начальная сила тока равна 6,5 мкА. В канале содержится
52,92 мл О2. Сила тока 0,2 мкА соответствует 60 мм (0,1 мкА= 30 мм) ди-
аграммной ленты (см. выше). Тогда 6,5 мкА соответствует
= 1950 мм
V, 1
диаграммной ленты, или 52,92 мл О2. Отсюда 1 мм диаграммной ленты
равен 0,0271 мл О2 (52,92 : 1950).
Определение цены деления диаграммной ленты в миллилитрах СО2.
Предварительная калибровка (через систему незамкнутого воз-
душного канала пропускали аргон) показала, что разница между
начальным (СО2 в воздухе — 0,03%) и конечным положениями
(СО2 — 0 %) точек на диаграммной ленте составляет 35 мм. По-
скольку система воздушного канала разомкнута, при расчете учи-
120
a
рис. 42. Диаграммная запись изменения концен-
трации кислорода (углекислого газа) в воздухе
При определении интенсивности фотосинтеза X б
и дыхания с использованием комбинированной *-Вкл.
ячейки (объяснение в тексте)
тывается объем воздуха, вытесненного только из отсеков, не заня-
тых буфером и электродами, электродной ячейки. Их общий объем
составил 9,41 мл, объем СО2 в них — (2,823-10-3) мл. Отсюда 1 мм
(2 823 • 10-3)
диаграммной ленты равен [мл СО2]: —-——----.
Расчет интенсивности фотосинтеза и дыхания по диаграмм-
ной записи (рис. 42).
1. Соединяют на диаграммной ленте прямой линией точки от
одного канала.
2. Опускают перпендикуляр а из любой точки этой линии на
продолжение «нулевой линии» и измеряют [мм]. Его значение со-
ответствует изменению концентрации О2 (или СО2) в канале за
время, прошедшее с момента начала регистрации (отрезок б «ну-
левой линии»). Согласно калибровке диаграммной ленты [мл] О2
(или СО2) определяют количество выделившегося (или поглощен-
ного) газа [мл].
3. Измеряют отрезок б [мм] и, исходя из скорости движения
диаграммной ленты, выражают его в единицах времени [ч].
4. Зная количество газа, выделившегося или поглощенного за
время б, рассчитывают его количество за 1 ч [мл].
Далее пересчитывают количество определяемого газа, выделив-
шегося или поглощенного за 1 ч на единицу площади, а также на
единицу сырой и сухой массы растений.
Оформление результатов. При оформлении результатов необ-
ходимо представить данные по калибровке кислородного и угле-
кислотного датчиков, данные по определению площади листьев,
сырой и сухой массы растений, расчеты интенсивности фотосин-
теза и дыхания. Сравнить интенсивность фотосинтеза и дыхания у
растений разных физиологических вариантов.
3.2.2. Определение световой кривой скорости фотосинтеза
с помощью газометрической установки открытого типа,
включающей инфракрасный газоанализатор
Принцип газометрического определения скорости фотосинте-
за заключается в регистрации изменения концентрации СО2 в
потоке газа, проходящего через камеру, герметически отделен-
ную от атмосферы, в которую помещен лист растения [6]. При
121
освещении листа концентрация СО2 в потоке газа, контактирую-
щего с ним, уменьшается из-за фотосинтетического поглощения
углекислоты.
С технической точки зрения, наиболее удобно регистрировать
изменения концентрации СО2 в потоке газа с помощью инфра-
красного газоанализатора (ИКГ). Общий принцип работы ИКГ
основан на том, что большинство газообразных веществ обладают
специфическими спектрами поглощения в инфракрасной (ИК)
области. Будучи помещенным в замкнутый объем и освещенным
ИК-светом, газ нагревается и давление внутри замкнутого про-
странства увеличивается. Это изменение давления может быть за-
регистрировано и служит показателем количества ИК-радиации,
попавшей на светочувствительный элемент ИКГ, так называе-
мый лучеприемник. Очевидно, что при изменении потока ИК-
радиации, поглощенной газом в лучеприемнике, температура, а
следовательно, и давление внутри лучеприемника будут также
изменяться. Если интенсивность источника ИК-света постоянна,
изменение потока ИК-радиации, падающей на лучеприемник,
будет связано с изменением в той газовой среде, через которую
проходят ИК-лучи. В газоанализаторе, предназначенном для изме-
рения концентрации СО2, лучеприемник заполнен чистой угле-
кислотой, селективно поглощающей ИК-лучи с длиной волны
4,2 —4,3 мкм. Соответственно изменения интенсивности ИК-ра-
диации, достигающей лучеприемника, будут связаны с измене-
нием содержания СО2 в газовой среде, через которую проходят
ИК-лучи перед попаданием на лучеприемник.
Наиболее распространенными газометрическими установками,
различающимися по принципу поддержания концентрации угле-
кислоты в листовой камере, служат установки закрытого и от-
крытого типов. В установках закрытого типа газ, содержащий СО2,
постоянно циркулирует по замкнутому контуру, включающему
листовую камеру. Соответственно в процессе освещения листа в
газовом контуре концентрация СО2 постоянно снижается до дос-
тижения светового компенсационного пункта. Хорошо известно
между тем, что динамическая адаптация фотосинтетического ап-
парата в интактных листьях к изменению интенсивности света
или концентрации углекислоты развивается в течение нескольких
минут. Таким образом, при измерениях фотосинтеза в установках
закрытого типа фотосинтетический аппарат всегда находится в
нестационарных условиях. Этого недостатка лишены установки
открытого типа, в которых выбранная концентрация углекислоты
поддерживается постоянной на входе листовой камеры в течение
всего процесса измерения фотосинтетической активности. Это по-
зволяет исследовать как кинетику изменения фотосинтетической
активности при смене освещенности, так и зависимость активно-
сти фотосинтеза от интенсивности света (световая кривая скоро-
122
сти фотосинтеза) в стационарных условиях протекания фотосин-
тетических процессов.
Достоинствами метода являются его относительная простота,
высокая чувствительность, а также возможность измерения фо-
тосинтетической активности на листе, не отделенном от расте-
ния.
Описание установки
Функционально газометрическая установка открытого типа,
разработанная в Институте физиологии растений им. К.А.Тими-
рязева РАН, состоит из трех частей: газоанализатора, системы
подачи газа к листовой камере и газоанализатору, осветителя.
Основным узлом установки является ИКГ «Infralyt-З» (Junkalor,
Германия), в котором, как и в большинстве ИКГ, используется
дифференциальная схема регистрации (рис. 43). От двух излучате-
лей (7), которыми служат нихромовые спирали, два пучка ИК-
излучения, выровненные по интенсивности специальными атте-
нюаторами, проходят через цилиндрические золоченые кюветы
(3), прежде чем попасть на окошки двух лучеприемников (4). Прой-
дя через прозрачное окошко, сделанное из литий-фтористого стек-
ла, каждый из двух пучков ИК-
излучения поглощается углекис-
лым газом, герметически замк-
нутым в лучеприемнике, и на-
гревает его. В результате повыше-
ния давления в лучеприемнике
отклоняется мембрана конден-
сатора (5) и изменяется его ем-
кость. Емкость конденсатора
преобразуется в электрический
ток, который усиливается (6)
и регистрируется токовым само-
писцем (7). Для улучшения ка-
чества усиления потоки ИК-из-
лучения модулируются с помо-
щью вращающегося секторного
диска (2), что позволяет при-
менить в приборе усилитель не
постоянного, а переменного
тока.
Рис. 43. Схема детектирующего эле-
мента инфракрасного газоанали-
затора:
Существенным моментом,
увеличивающим стабильность и
чувствительность установки,
служит реализованная в ней схе-
ма дифференциального вклю-
1 — излучатели; 2 — вращающийся
секторный диск; 3 — золоченые кю-
веты для пропускания газовой смеси;
4 — окошко лучеприемника; 5 — мемб-
рана конденсатора; 6 — усилитель;
7 — самописец
123
чения кювет с лучеприемниками в газовую систему. В заводском
варианте одна из кювет ИКГ «Infralyt-З» заполнена азотом и запа-
яна, а через другую пропускается газовая смесь, в которой опре
деляется концентрация СО2. При дифференциальной схеме вклю-
чения газовая смесь постоянно пропускается с одинаковой ско-
ростью через обе кюветы. При этом через одну из кювет пропуска
ется газ, предварительно прошедший через листовую камеру, а
через другую — газ, не контактировавший с листом (рис. 44). Диф
ференциальная схема позволяет избежать шумов, связанных с воз-
можными колебаниями в скорости подачи газовой смеси во вре
Рис. 44. Схема газовой установки от-
крытого типа:
1 — осветительный блок; 2 — ватный
фильтр; 3 — компрессорный насос; 4 —
осушитель с СаС12; 5 — поглотительная
колонка с КОН; 6 — увлажнитель с на-
сыщенным раствором NaCl; 7 — гидро-
статические маностаты; 8— баллон с уг-
лекислотой; 9 — листовая камера; 10 —
водяной термостат; 11 — реометры; 12 —
инфракрасный газоанализатор; 13 — са-
мописец
мя измерения.
Газовая система установки
представляет собой совокуп-
ность последовательно и парал-
лельно соединенных хлорвини-
ловыми трубками элементов,
герметически отделенную от
окружающей среды (за исклю-
чением конечного сброса газа
в атмосферу). Воздух в систему
подается компрессорным насо-
сом (5) через ватный фильтр
(2), предотвращающий попада-
ние пылевых частиц. Проходя
через осушитель (4), представ-
ляющий собой стеклянную
трубку с гранулированным
СаС12, воздух освобождается от
паров воды и поступает в ко-
лонку, наполненную гранули-
рованным КОН (5), в которой
полностью поглощается содер-
жавшаяся в воздухе углекисло-
та. Проходя через колбу с на-
сыщенным раствором NaCl (6),
воздух увлажняется до относи-
тельной влажности 60 %. Осво-
божденный от углекислоты воз-
дух смешивается с СО2, пода-
ваемым из баллона (5). В зави-
симости от цели эксперимента
концентрацию СО2 в газовой
смеси можно варьировать в пре-
делах 0,01 —0,1 %. После этого
газовая смесь разделяется на два
потока. Один из них направля-
124
ется сначала в листовую камеру (9), температура в которой под-
держивается водяным термостатом (10). После листовой камеры
этот поток проходит через одну из кювет инфракрасного газо-
анализатора (72). Другой поток проходит через вторую кювету
ИКГ, минуя листовую камеру. Перед поступлением в ИКГ оба
воздушных потока осушаются пропусканием через колонки с гра-
нулированным СаС12 (4). Поскольку оба воздушных потока дол-
жны иметь одинаковые скорости на входе в ИКГ, последние ре-
гистрируются и уравниваются реометрами (77). Для изменения
давления в газовой системе, а также поддержания его постоян-
ным в процессе измерения используются гидростатические мано-
статы (7).
Осветительная установка (7) служит для освещения объекта,
помещенного в листовую камеру. Источником света служит проек-
ционный фонарь с лампой накаливания. Для поглощения инфра-
красного излучения от лампы пучок света от нее пропускается че-
рез сосуд с плоскопараллельными стенками, заполненный 3 %-м
раствором CuSO4. Конденсор формирует параллельный пучок све-
та для равномерного освещения всей площади листовой камеры.
Интенсивность света, падающего на листовую камеру, можно
регулировать как изменением раскрытия диафрагмы, стоящей на
пути пучка света, так и размещением на пути последнего нейт-
ральных светофильтров. Кроме того, применение различных цвет-
ных стеклянных или интерферированных фильтров позволяет ва-
рьировать спектральный состав света.
Ход работы
1. Проведение измерений. Перед началом работы ИКГ должен
быть прогрет в течение не менее 3 ч для обеспечения стабильной
работы лучеприемников. Кроме того, необходимо убедиться, что
в колонках 4 и 5 (см. рис. 44) находятся свежие гранулированные
СаС12и КОН. Начальным этапом приведения установки в рабочее
состояние является включение компрессорного насоса 3, подаю-
щего воздух в газовую систему. Перед этим следует убедиться, что
листовая камера 9 открыта. После включения насоса исследуемый
лист, не отделяя от растения, помещают в листовую камеру и
закрывают ее герметично. Устанавливают одинаковую скорость
потока газовой смеси через обе кюветы ИКГ, добиваясь осторож-
ным поворачиванием ручек реометров 77 одинаковых значений
на их шкалах. Открывают баллон с углекислотой и устанавливают
нужную концентрацию СО2 в газовой смеси подъемом или опус-
канием трубки в маностате 7.
Перед началом измерений необходимо прокалибровать уста-
новку. Для этого включают самописец, подъемом или опусканием
трубки в маностате изменяют концентрацию СО2 в газовом пото-
125
1450
2240
Рис. 45. Изменения концентрации СО2 при освещении листа огурца све-
том различной интенсивности, зарегистрированные с помощью уста-
новки открытого типа.
Стрелки с цифрами обозначают изменения интенсивности света [мкмоль/(м2-с)]
ке, проходящем через листовую камеру, на 0,01 % и регистриру-
ют на самописце соответствующие изменения потока. Процедуру
повторяют три раза.
После калибровки установки регистрируют темновой уровень
СО2, для чего лист затеняют черной тканью и включают самопи-
сец. После 10 мин регистрации темнового уровня включают свет
максимальной интенсивности. При этом концентрация СО2 в по-
токе газа, проходящего через листовую камеру, постепенно умень-
шается, указывая на развивающееся фотосинтетическое поглоще-
ние углекислоты (рис. 45). Приблизительно через 30 мин освеще-
ния сигнал достигает стационарного значения. Это означает, что
достигнута максимальная скорость фотосинтеза листа при данной
интенсивности света. Последовательно снижают интенсивность
света, падающего на лист, помещая на пути светового пучка раз-
личные нейтральные светофильтры или их комбинации. При каж-
дой интенсивности света необходимо дождаться, пока сигнал до-
стигнет стационарного значения, что происходит значительно
быстрее, чем после начала освещения затененного листа, — обычно
в течение 8 — 10 мин. После измерения при самой низкой из выб-
126
ранных интенсивностей выключают свет и затеняют лист черной
тканью. Вновь регистрируют темновой уровень сигнала. Заключи-
тельным этапом измерений является регистрация сигнала при за-
крытой камере, лишенной листа. Разница между последним уров-
нем и уровнем сигнала, зарегистрированным в темноте с листом,
является показателем скорости темнового дыхания листа.
Оформление результатов. Скорость фотосинтеза при определен-
ной интенсивности света в стационарных условиях, характерных
для установок открытого типа, вычисляется по формуле
F=Q(Cq-C), (1)
где F — стационарная скорость поглощения СО2 при данной ин-
тенсивности света; Q — скорость тока газовой смеси через листо-
вую камеру; Со — концентрация СО2 в контрольной смеси; С —
концентрация СО2 на выходе из листовой камеры. Величину С для
каждой интенсивности света вычисляют по расстоянию х [мм] от
темнового уровня СО2, зарегистрированного на ленте самописца,
до концентрации СО2 при данной интенсивности светопотока. При
этом расстояние а [мм], соответствующее изменению концентра-
ции СО2 на 0,01 %, уже известно из калибровки прибора. Значе-
ние С вычисляют по формуле
С=0,01х/я. (2)
Тогда формула определения скорости фотосинтеза (1) прини-
мает следующий вид:
F=0(Co-O,Olx/a). (3)
После вычисления значения Гдля каждой интенсивности све-
та строится график зависимости скорости фотосинтеза от количе-
ства света, падающего на лист.
3.3. АНАЛИЗ ФУНКЦИОНАЛЬНОГО СОСТОЯНИЯ
ФОТОСИНТЕТИЧЕСКОГО АППАРАТА РАСТЕНИЙ
МЕТОДОМ РЕГИСТРАЦИИ ИНДУКЦИИ
ФЛУОРЕСЦЕНЦИИ ХЛОРОФИЛЛА
Явление флуоресценции хлорофилла было обнаружено Д. Брюс-
тером в 1834 г. Затем появились первые доказательства тесной свя-
зи между флуоресценцией и процессами фотосинтеза, была вы-
явлена сложная кинетика флуоресценции при освещении адапти-
рованных к темноте листьев (Г. Каутский). Согласно современным
преставлениям, флуоресценция хлорофилла, и в первую очередь
Кинетические характеристики этого процесса, могут служить по-
казателями структурных и функциональных свойств фотосинте-
127
тических мембран у интактных растений и использоваться при
разработке методов экспресс-анализа влияния различных факто-
ров на фотосинтетическую активность хлоропластов [2, 3, 5, 18
21].
Основным источником люминесценции (флуоресценции и за-
медленной люминесценции, или послесвечения) в фотосинтези-
рующих организмах является хлорофилл, причем основным флу-
оресцирующим пигментом является хлорофилл а фотосистемы II
(ФС II). Это, по-видимому, хлорофилл реакционных центров, а
также хлорофилл антенного комплекса. Остальные пигменты флу-
оресцируют только тогда, когда не могут передать энергию хло-
рофиллу а.
Флуоресценция (ФЛ) — один из путей трансформации энер-
гии, поглощенной хлорофиллом во время облучения. В виде флу-
оресценции in vivo испускается лишь небольшая доля поглощен-
ной энергии (3—5 %). Частичная потеря энергии происходит так-
же в виде тепла. Однако большая часть поглощенной энергии све-
та в листе используется в первичных реакциях фотосинтеза (миг-
рация энергии в светособирающих комплексах к пигменту реак-
ционного центра, передача возбужденного электрона на акцептор
в реакционных центрах). Передача электрона от возбужденного хло-
рофилла (Хл*) к первичному акцептору (А) приводит к образова-
нию комплекса с разделенными зарядами (Хл* + А —> Хл+ + А~), что
является первым этапом консервирования энергии возбужденной
молекулой хлорофилла в электрон-транспортной цепи (ЭТЦ) хло-
ропластов.
Соотношение различных путей использования энергии погло-
щенных квантов света может изменяться в зависимости от функ-
циональной активности фотосинтетического аппарата, а также от
условий освещения, что находит отражение в интенсивности флу-
оресценции хлорофилла.
При освещении интактных фотосинтезирующих организмов,
находящихся предварительно в темноте, флуоресценция устанав-
ливается не сразу, а наблюдаются колебания ее выхода по слож-
ной кинетической кривой (эффект Каутского), получившей на-
звание индукционной кривой ФЛ (рис. 46).
Кинетическая кривая флуоресценции отражает сложный меха-
низм быстрых перестроек в листе — переход хлоропластов из тем-
нового состояния в световое. В ходе этого процесса происходит
разделение зарядов в реакционном центре (РЦ) фотосистем, ак-
тивация транспорта электронов в ЭТЦ фотосинтеза, формирова-
ние градиента электрохимического потенциала протонов на мем-
бранах, активация реакций цикла Кальвина и др. В условиях высо-
ких интенсивностей света возможно перераспределение энергии
света между ФС II и фотосистемой I (ФС I) благодаря перемеще-
нию светособирающего комплекса II (ССК II) (переход хлоро-
128
Б
Рис. 46. Индукционные кривые флуоресценции интактного листа:
А — при большей скорости движения ленты самописца; Б — при меньшей ско-
рости (объяснение в тексте)
пластов из состояния 1 в состояние 2), что также находит отраже-
ние в параметрах индукционной кривой ФЛ.
На кинетической кривой выхода ФЛ хлорофилла адаптирован-
ных к темноте листьев наблюдаются быстрые и медленные изме-
нения, при этом кривую изменений флуоресценции хлорофилла
можно разделить на ряд участков, для которых приняты стандарт-
ные обозначения (см. рис. 46):
О, Fo — постоянная ФЛ;
Fm — максимальная флуоресценция;
Fp (или /’пер, или Fv) — переменная ФЛ;
I — промежуточный пик (intermediary);
D — спад (dip);
Р — пик (peak);
S — промежуточный стационарный уровень (quasi steady state);
М — максимум (maximum);
Т — конечный (terminal) уровень;
R{ — скорость IP-подъема;
R2 — скорость РУ-тушения ФЛ;
В индукционной кривой ФЛ хлорофилла различают два компо-
нента: постоянную ФЛ и переменную.
Постоянная {фоновая) ФЛ {уровень FQ) излучается хлорофил-
лом антенного комплекса ФС II еще до передачи энергии в РЦ. Fo
изменяется пропорционально освещенности объекта. Увеличение
уровня Fq означает увеличение доли хлорофилла, энергия от ко-
торого не поступает в РЦ.
Переменная ФЛ (Рпер, Fp, Fv) — флуоресценция, амплитуда
которой возрастает в ответ на освещение сильным светом. Fp не-
5 Гавриленко
129
сет информацию о состоянии ЭТЦ фотосинтеза. Практически вся
Fp излучается ФС II.
Согласно гипотезе Дюйзенса—Свирса (L. Duysens, B.Sweers,
1963), выход ФЛ зеленых растений определяется окислительно
восстановительным состоянием акцептора электронов ФС II QA.
Восстановление QA вызывает возрастание ФЛ, а окисление — ее
уменьшение. В начале освещения у адаптированных к темноте объек-
тов не происходит оттока восстановленных эквивалентов от QA
ФС II по ЭТЦ из-за неактивного состояния ферредоксин-НАДФ
оксидоредуктазы и ферментов цикла Кальвина. Следствием этого
является быстрое заполнение пула электронных емкостей на ак-
цепторной стороне ФС I и восстановление всех первичных хинон-
ных акцепторов ФС II. При этом ФЛ хлорофилла возрастает до
максимального уровня Р (Fm).
Наблюдаемый на кинетической кривой ФЛ сложный характер
быстрых изменений в ходе достижения максимального уровня флу-
оресценции, по-видимому, может отражать гетерогенность ФС II:
быструю сигмовидную часть кривой {01D) относят к восстанов-
лению ос-центров ФС II, а более медленную DP — к восстановле-
нию p-центров ФС II. Соотношение ос- и p-центров в хлоропластах
клеток, относящихся к разному возрасту или функционирующих
в разных световых условиях, варьирует, что отражается на харак-
тере быстрых изменений ФЛ хлорофилла.
Возможно также, что переменная ФЛ включает люминесцен-
цию, возникающую при рекомбинации заряженной пары РЦ ФС
II, хлорофилла Рб8о и феофитина (Фео-), при фотохимическом и
темновом восстановлении QA, что отражается в сложной кинети-
ке возрастания ФЛ.
Таким образом, фоновая флуоресценция (Го) происходит в
условиях, когда все первичные хинонные акцепторы ФС II QA
находятся в окисленном состоянии, тогда как при полном их
восстановлении достигается максимальный уровень флуоресцен-
ции (7%). Затем флуоресценция постепенно снижается (затуха-
ет).
Природа медленного затухания ФЛ (участок PSMT) пока окон-
чательно не установлена. По времени этот период соответствует
активации цикла Кальвина в хлоропластах — реакций фиксации
СО2 и восстановления углерода.
Различают два механизма тушения ФЛ — фотохимическое и
нефотохимическое.
Их соотношение зависит от длительности и интенсивности ос-
вещения, а также от ряда других факторов.
Фотохимическое тушение ФЛобусловлено использованием энер-
гии возбуждения хлорофилла для фотохимических реакций и свя-
зано с окислением QA при активации работы ФС I. Полагают, что
скорость PS-тушения отражает светоиндуцированную активацию
130
ЭТЦ вследствие активации фермента ферредоксин-НАДФ+-окси-
доредуктазы.
Нефотохимческое PS-тушение ФЛ наблюдается при высоких
интенсивностях света. Согласно современным представлениям, по
Крайней мере три процесса, происходящих в хлоропластах в этих
условиях, могут обусловливать тушение флуоресценции.
• Переход хлоропластов из состояния 1 в состояние 2, когда
ССК II, передающий энергию света к антеннам ФС II в состоя-
нии 1, фосфорилируется и меняет свое расположение в мембране
тилакоидов, приближаясь к ФС I. При этом уменьшается началь-
ное преимущественное поступление энергии возбуждения в ФС II
за счет роста ее потока в ФС I, флуоресцирующей с более низким
квантовым выходом, что и воспринимается как тушение ФЛ ФС II.
Обратимое фосфорилирование ССК II происходит за счет акти-
вации соответствующей протеинкиназы в ответ на увеличение
уровня восстановленности пула пластохинонов в условиях высо-
ких интенсивностей света, когда поток электронов в ЭТЦ фото-
синтеза превышает использование НАДФН в цикле Кальвина.
Сенсором редокс-состояния ЭТЦ является цитохромный комп-
лекс.
• Энергозависимое тушение ФЛ, вызываемое светоиндуциро-
ванным градиентом концентрации протонов (ДрН) на тилакоид-
ной мембране, вероятно, обусловлено ростом тепловой дезакти-
вации возбужденных состояний хлорофилла а. Последнее может
быть следствием структурных изменений внутренней поверхности
тилакоидной мембраны в результате обмена Mg2+ на Н+. В настоя-
щее время роль протонного градиента в нефотохимическом туше-
нии флуоресценции связывают с активацией при определенных
pH виолаксантинового цикла и образованием зеаксантина — ту-
шителя возбужденного состояния хлорофилла.
• Тушение ФЛ, связанное с фотоингибированием фотосинте-
за — снижением квантового выхода ассимиляции СО2 в условиях
высоких интенсивностей света, может быть обусловлено рядом
деструктивных процессов в ФС II. Процессы, вызывающие фото-
ингибирование фотосинтеза, могут возникнуть также сразу после
включения света при индукционной фазе фотосинтеза после дли-
тельной темновой адаптации растений, когда в клетке нет такого
акцептора, как СО2, и вносить свой вклад в Р5-тушение ФЛ.
После перехода PS на индукционной кривой наблюдаются еще
колебания с одним SMT или несколькими максимумами. По-ви-
димому, эти колебания отражают изменения в скорости фикса-
ции СО2 при возникновении определенных условий. Установле-
но, что подъем ФЛ SM происходит в момент начала выделения О2
и поглощения СО2. Ингибиторы цикла фиксации СО2 устраняют
57И7-переход. SMTзависит от освещенности листа, времени тем-
новой предадаптации и возраста листа.
131
Медленные изменения ФЛ PSMT реагируют на соотношения
АТФ/АДФ, НАДФН/НАДФ+, циклического/нециклического
транспорта электронов, зависят от содержания О2 и СО2 в листе,
размеров пулов интермедиатов и активности ферментов цикла
темновой фиксации углерода.
Информативность параметров индукции флуоресценции
Обычно анализируют следующие параметры индукции ФЛ.
амплитуды в точках О (Fo), I (Т7), Р (Fm), М (FM), Т (FT) и их от-
ношения: Fj/F0, Fm/F0, (Fm-F0)/Fm, (Fj-Fo)/Fo, Fm/FT, (Fm-FT)/Fm,
скорость нарастания (Rx) или спада (Л2) ФЛ, площади над и под
кривой индукции ФЛ, время достижения Ми Т.
Уровень Fo характеризует долю хлорофилла фотосинтетическо -
го аппарата, энергия возбуждения которого не используется в
прямых процессах фотосинтеза. В условиях, когда воздействия внеш-
него фактора обратимы, величина Fo практически не изменяется.
В условиях слабой освещенности Fo увеличивается пропорциональ-
но интенсивности освещения.
Уровень Fm — максимальный уровень ФЛ, когда акцептор QA
ФС II максимально восстановлен. Это достигается, когда блоки-
рован транспорт электронов на акцепторной стороне ФС II (на-
пример, диуроном или облучением сильным светом). В этом слу-
чае уровень Fm пропорционален содержанию хлорофилла а объек-
та. Используют Fm/F(} или (Fm - FQ)/Fm как показатели фотохими-
ческой активности ФС II. Снижение Fm/F$ при Fo = const — показа-
тель уменьшения активности донорной стороны ФС II. Однако
снижение Fm/FQ может быть вызвано и другими причинами: а)
усилением обратных реакций в ФС II; б) снижением квантового
выхода фотохимической активности РЦ ФС II; в) уменьшением
количества световой энергии, достигающей РЦ.
Отношение (Fm - F^/Fm характеризует квантовый выход раз-
деления зарядов в ФС II (V.Genty et al., 1989). Уровни Fo и Fm,
регистрируемые у нативных объектов, длительное время адапти-
рованных к темноте, используют для расчета максимального кван-
тового выхода первичного разделения зарядов в ФС II. Длитель-
ное освещение листа светом умеренной или высокой интенсив-
ности приводит к активации работы ФС I и цикла Кальвина, в
результате чего квантовый выход флуоресценции хлорофилла
снижается.
Для наблюдения за функционированием ЭТЦ можно исполь-
зовать характеристики затухающей части кривой. При облучении
интактных объектов светом с интенсивностью ниже уровня све-
та, насыщающего фотосинтез, Р5-спад вызывается в основном
фотохимическим тушением, т.е. окислением акцептора QA ФС II.
Скорость Р5-затухания (Л2) используется как показатель актив-
132
ности ЭТЦ при солевом стрессе и водном дефиците. У интактных
объектов наблюдалась пропорциональная зависимость скорости
спада и устанавливающейся позже максимальной скорости выде-
ления О2. Таким образом, скорость спада PS может характеризо-
вать величину межсистемного электронного транспорта. Прямая
пропорциональность между этими показателями отмечена только
в норме.
3.3.1. Использование метода регистрации
индукционных кривых флуоресценции хлорофилла
для оценки функционального состояния
фотосинтетического аппарата
Анализ индукционных кривых флуоресценции хлорофилла в
настоящее время широко используют в исследованиях фотосин-
теза как показатель структурного и функционального состояния
фотосинтетического аппарата в различных физиологических ус-
ловиях. Показано изменение ряда параметров индукционных кри-
вых флуоресценции хлорофилла (Fo, Fm, Fp, Fp/F0, Rb R2h др.) в
ответ на действие засухи, засоления, гербицидов, тяжелых метал-
лов, ультрафиолета и прочих стрессовых факторов [3, 5, 14, 16,
18, 20, 21]. Это позволяет, с одной стороны, понять механизмы
повреждающего действия исследуемых факторов, а с другой сто-
роны, использовать параметры кинетической кривой флуоресцен-
ции для оценки устойчивости растений к неблагоприятным воз-
действиям. При этом важно выявить те параметры индукционной
кривой, которые коррелируют с действующим фактором, и по-
нять биофизические, биохимические и физиологические основы
наблюдаемых изменений флуоресценции. Возможность проведе-
ния исследования фотосинтетической активности без поврежде-
ния объектов исследования, относительная простота измерений
и достаточно высокая информативность получаемых данных со-
ставляют преимущества данного метода. Вместе с тем значитель-
ная вариабельность кривых индукции флуоресценции у разных
клеток одного листа, у листьев разного возраста делает необходи-
мым регистрацию индукционных кривых в многократных биоло-
гических повторностях и проведение статистической обработки
результатов.
Особое внимание следует уделять условиям проведения опыта
(время темновой адаптации растений, интенсивность освещения,
температура и т.д.), оказывающих значительное влияние на ха-
рактер кривых индукции флуоресценции.
Для регистрации кривых индукции флуоресценции хлорофил-
ла в листьях используют установку, собранную на кафедре физи-
ологии растений биологического факультета МГУ. (Использова-
133
ние прибора заводского образца РАМ для регистрации кривых
индукции флуоресценции описано в 3.3.2.)
Описание установки
Установка для регистрации индукционных кривых флуоресцен-
ции фотосинтезирующих объектов состоит из источника света,
рабочей камеры, в ячейку которой помещается объект исследова-
ния, и регистрирующей части. Блок-схема установки приведена
на рис. 47.
Источник света (7) и рабочая камера (2) стационарно закреп-
лены на штативе. Верхняя часть (крышка) рабочей камеры с от-
верстием (d = 5 мм) снабжена съемной металлической заслонкой
(5). Часть спектра светового потока с длиной волны >370 нм, по-
падающего в камеру через отверстие, отсекается светофильтром
(4). Таким образом, спектрально ограниченный световой поток,
попадающий в ячейку, освещает очень маленький участок листа,
который находится непосредственно под отверстием.
В нижней части камеры, под ячейкой, напротив отверстия и
освещаемого участка листа соответственно расположен фотодиод
(6) (фотодиод 1).
Рис. 47. Блок-схема установки для регистрации кривых флуоресценции:
7 — источник света (лампа накаливания 500 Вт); 2 — рабочая камера; 3 — заслон-
ка; 4 — светофильтр (>370 нм); 5 — объект; 6 — фотодиод; 7 — усилитель; 8 —
источник питания усилителя; 9 — микровольтнаноамперметр Ф-136; 10 — по-
тенциометр регистрирующий КСП-4
134
Ф-136
Рис. 48. Передняя панель усилителя фотодиода:
1 — тумблер включения усилителя; 2, 3 — тумблеры регулировки усиления; 4 —
тумблер включения контрольного фотодиода
Камера снабжена блокировочным микровыключателем, отклю-
чающим фотодиод 1 при открытой камере, а также дополнитель-
ным фотодиодом {фотодиод 2) в крышке камеры, позволяющим
контролировать световой поток, который проходит через обра-
зец.
К регистрирующей части установки относятся фотодиод 1 (б),
микровольтнаноамперметр Ф-136 (9), усилитель фотодиода 1 (7)
и самописец КСП-4 {10). Запись индукционных кривых ФЛ про-
изводится на бумажной ленте при скорости ее движения 1200 или
7200 мм/ч.
Чувствительность установки регулируется переключателем на
микровольтнаноамперметре Ф-136 и переключателями на усили-
теле фотодиода. Передняя панель усилителя фотодиода представ-
лена на рис. 48. Тумблер 1 предназначен для общего включения
усилителя, тумблеры 2 и 3 — для регуляции чувствительности,
тумблер 4 — для подключения дополнительного фотодиода (фо-
тодиод 2), контролирующего световой поток (см. выше).
Порядок работы на установке
1. Проверить заземление приборов. Включить лабораторный
щиток. Подключить приборы (источник света, блок питания уси-
лителя, микровольтнаноамперметр Ф-136, самописец КСП-4) к
сети.
2. Включить приборы (при грубой чувствительности Ф-136 во
избежание зашкаливания и поломки приборов).
3. Установить переключатель 1 усилителя фотодиода (рис. 48) в
положение «Вкл.», переключатели 2 и 3 — в необходимой для
усиления комбинации, переключатель 4 — в положение «Вкл.»
135
при контроле светопотока контрольным фотодиодом 2 и в поло-
жение «Выкл.» — при снятии кривых ФЛ (работа с фотодиодом 1).
4. Зафиксировать оптимальную чувствительность установки на
микровольтнаноамперметре Ф-136.
5. Поместить исследуемый объект в ячейку рабочей камеры.
6. Выполнить работу по описанию (лентопротяжный механизм
самописца удобнее включать за 10 — 20 с до регистрации кривой
ФЛ).
7. По окончании работы установить более грубую чувствитель-
ность установки на микровольтнаноамперметре Ф-136. Перевести пе-
реключатели усилителя сигнала фотодиода в исходное положение.
8. Выключить приборы. Отключить приборы от сети. Выключить
лабораторный щиток.
Ход работы
Работа включает следующие этапы: 1) регистрация индукци-
онных кривых ФЛ интактных листьев в различных условиях экспе-
римента — при различных условиях темновой адаптации и в усло-
виях одинаковой освещенности; в разных условиях освещенности
и одинаковой длительности темновой адаптации; в условиях по-
следовательного изменения интенсивности освещения; 2) опре-
деление параметров кривых индукции флуоресценции хлорофил-
ла и анализ механизмов фотоиндуцированных процессов в фото-
синтетическом аппарате исследованных растений (по параметрам
одной из кривых ФЛ).
В работе используют: 1) листья различных видов растений;
2) листья различного возраста одного вида растений; 3) листья
растений, выращенных в различных условиях минерального пи-
тания, освещения, водного режима.
1. Регистрация индукционных кривых ФЛ интактных листьев.
Приготовить исследуемые варианты к опыту. Для этого проростки
или листья растений (срезать лист непосредственно перед опы-
том) помещают в темноту. Отмечают время темновой адаптации
образцов.
Регистрация индукционных кривых ФЛ в условиях одинаковой
освещенности и разной длительности темновой адаптации. Лист
опытного растения помещают в ячейку рабочей камеры, отвер-
стие закрывают заслонкой и лист выдерживают в темноте в тече-
ние 15 мин. По истечении этого времени дают световую вспышку
длительностью 30 с и записывают индукционную кривую ФЛ. По
достижении стационарного уровня запись прекращают.
Не производя замены листа, последовательно записывают ин-
дукционные кривые после темновой инкубации в течение 2 и 5 мин.
По индукционным кривым ФЛ, полученным в данном опыте,
можно дать относительные характеристики изменений фотоинду-
136
цированных процессов фотосинтетического аппарата исследуемых
растений, связанных с длительностью темнового прединдукци-
онного периода.
Регистрация индукционных кривых ФЛ в условиях разной ин-
тенсивности освещения и одинаковой длительности темновой
адаптации. Свежий лист одного из опытных растений помеща-
ют во влажной фильтровальной бумаге в темноту не менее чем на
15 мин. Затем помещают лист в камеру, выдерживают около 5 мин,
по истечении этого времени включают запись и дают вспышку
света малой интенсивности. При установлении стационарного уров-
ня запись прекращают. Не производя замены листа, через 5 мин
снова включают запись и дают вспышку света той же длительнос-
ти, но большей интенсивности. После записи второй индукцион-
ной кривой через 5 мин записывают третью, освещая лист светом
наибольшей интенсивности. Изменения интенсивности действу-
ющего света производят, помещая на пути светового луча различ-
ные нейтральные фильтры.
При сравнении индукционных кривых ФЛ, полученных в дан-
ном опыте, следует обратить внимание на изменения стационар-
ного уровня (Ро) как показателя доли хлорофилла, энергия кото-
рого не задействована в прямых процессах фотосинтеза, а также
на отношение Fp/F0, отражающее активность ФС II.
2. Определение параметров кривых индукции флуоресценции
хлорофилла. Произведя запись кривых ФЛ всех опытных вариан-
тов, анализируют полученные кривые. Отмечают следующие па-
раметры: Ро, Fp, Fmn уровни S, М, Т. Результаты вносят в таблицу.
Для анализа чаще всего используются следующие показатели:
1) высота уровня Fo, отражающая долю хлорофилла, энергия
возбуждения которого не используется в реакциях фотосинтети-
ческого аппарата;
2) высота Fp, которая коррелирует с общим содержанием хло-
рофилла в исследуемом объекте, величина от Fo до Р указывает
на долю Qa, участвующего в фотохимических реакциях ФС II;
3) Fm (точка Р), в которой первичный стационарный акцептор
ФС II Qa максимально восстановлен;
4) показатель Fp /Fo, отражающий функциональную активность
ФС II;
5) отношение (Fm - FQ)/Fm, характеризующее квантовый выход
первичного разделения зарядов в ФС II;
6) измерив — скорость достижения максимума (от Ро до Р)
на кривой ФЛ — и определив скорость восстановления QA, мож-
но охарактеризовать донорные свойства ФС II, так как R{ корре-
лирует со скоростью электронного транспорта через ФС II;
7) R2, или скорость PS-тушения, — показатель величины элек-
тронного транспорта между двумя системами, пропорциональный
максимальной скорости выделения О2;
137
8) временной период PSMT, соответствующий периоду акти-
вации цикла фиксации СО2;
9) FM/FT — величина спада от М к Т, отражающая одновре-
менное увеличение скорости выделения О2 и фиксации СО2 ин-
тактными хлоропластами.
Оформление результатов. Параметры, определенные по кри-
вым индукции флуоресценции хлорофилла, вносят в таблицу,
проводят статистическую обработку данных и сравнительный ана-
лиз параметров кривых, полученных в различных условиях экспе-
римента.
3.3.2. Определение фотохимического и нефотохимического
тушения флуоресценции хлорофилла при освещении листьев
Подобно другим пигментам молекулы хлорофилла, входящие
в состав фотосистем I и II (ФС I и ФС II) хлоропласта, испускают
короткоживущую флуоресценцию. При комнатной температуре
практически вся флуоресценция излучается хлорофиллом ФС II.
При освещении в течение 1 — 2 с предварительно адаптированно-
го к темноте листа мощным светом выход флуоресценции хлоро-
филла возрастает примерно в 5 раз (7%) вследствие накопления
акцептора ФС II — QA в восстановленном состоянии (рис. 49). Бо-
лее длительное освещение листа постоянным, так называемым
действующим светом средней или высокой интенсивности приво-
дит к постепенному снижению выхода флуоресценции хлорофил-
ла до стационарного уровня флуоресценции {Fs) вследствие раз-
вития на свету двух процессов:
1) увеличения использования энергии электронного возбуж-
дения хлорофилла в фотохимических реакциях хлоропластов в связи
с усилением оттока электронов от ФС II при световой активации
ферментов цикла Кальвина {фотохимическое тушение флуоресцен-
ции)’,
2) потери энергии поглощенных хлорофиллом квантов света
или в виде теплового излучения, или в реакциях тушения возбуж-
денного состояния хлорофилла {нефотохимическое тушение флуо-
ресценции).
Фотохимическое тушение флуоресценции, развившееся на дей-
ствующем свету, может быть обращено при помощи дополнитель-
ного кратковременного освещения листа очень мощной вспышкой све-
та, которая переводит на непродолжительное время все реакци-
онные центры фотосистемы II в фотохимически неактивное со-
стояние вследствие восстановления их первичных хинонных ак-
цепторов Qa. При этом уровень флуоресценции возрастает {F^, не
достигая уровня, зарегистрированного у адаптированного к тем-
ноте листа. Разница между максимальным уровнем флуоресцен-
138
ции хлорофилла при освещении листа, адаптированного к тем-
ноте, (Fw) и уровнем флуоресценции листа на свету при дей-
ствии очень мощной вспышкой света длительностью около 1 с
(/%) является показателем нефотохимического тушения флуо-
ресценции, развившегося в процессе предварительного освеще-
ния листа.
Измерения флуоресценции хлорофилла в различных условиях
позволяют специалистам оценивать в дополнение к интегральной
фотосинтетической активности целый ряд фотосинтетических ре-
акций, протекающих в хлоропластах интактных листьев, а также
анализировать взаимосвязь световой и темновой стадий фотосин-
теза. При этом можно проводить измерения на листьях, не отде-
ленных от растения, что в свою очередь позволяет изучать изме-
нения фотосинтетической активности в онтогенезе индивидуаль-
ного листа или следить за изменениями фотосинтетической ак-
тивности одного и того же листа после воздействия на растение
А Б
Рис. 49. Кинетические кривые фотоиндуцируемых изменений выхода флу-
оресценции хлорофилла при освещении адаптированного к темноте ли-
ста при одиночном освещении односекундным импульсом мощного бе-
лого света (Л) и чередующимися односекундными импульсами белого
света на фоне продолжительного постоянного освещения действующим
светом (Б):
Fo — фоновый уровень флуоресценции хлорофилла у адаптированного к темно-
те листа, Fo' — фоновый уровень флуоресценции хлорофилла после продолжи-
тельного освещения листа; Fm — максимальный уровень флуоресценции хлоро-
филла у адаптированного к темноте листа; F„ — максимальный уровень флуо-
ресценции у освещенного длительное время листа; Fs — квазистационарный
уровень флуоресценции у адаптированного к свету листа. Светлыми стрелками
показано включение слабого модулированного света, темными стрелками, на-
правленными вниз, — подача односекундных импульсов мощного света
139
различных биотических или абиотических факторов. Более под-
робно с многочисленными возможностями флуоресцентных ме-
тодик для физиологических исследований в области фотосинтеза
можно ознакомиться в рекомендованных в конце книги публика-
циях по данному вопросу [1, 5, 15, 17, 18, 21].
Наиболее широкое распространение в физиологических иссле-
дованиях получили измерения фото- и нефотохимического туше-
ния флуоресценции [4, 19, 21, 22].
Фотохимическое тушение флуоресценции может быть охаракте-
ризовано коэффициентом фотохимического тушения qP (van Kooten
and Snel, 1990):
qP = (F'm - Fs)/(F'm - Fo'\
С помощью qP можно количественно охарактеризовать рео-
кисление Qa в процессе световой адаптации. У адаптированных к
темноте листьев, в которых QA полностью окислен, qP= 1. Значе-
ния параметра 1 - qP пропорциональны количеству восстанов-
ленного Qa.
Нефотохимическое тушение флуоресценции количественно ха-
рактеризуется коэффициентом нефотохимического тушения флуо-
ресценции qN, рассчитываемым по следующей формуле:
qN = (Fm - F^/(Fm - Fo').
Альтернативно для характеристики нефотохимического туше-
ния флуоресценции хлорофилла может быть использован пара-
метр NPQ, рассчитываемый по уравнению Штерна—Фольмера:
NPQ =Fm/F'm- 1.
В этом случае NPQ пропорционален количеству тушащих цен-
тров (A. Bilger, B.Bjorkman, 1990).
Описанный выше метод измерения фотохимического и нефо-
тохимического тушения флуоресценции хлорофилла реализован
технически в специализированном приборе РАМ 100 (Walz, Гер-
мания), краткое описание которого приведено ниже. Прибор по-
зволяет регистрировать флуоресценцию хлорофилла, возбуждае-
мую очень слабым, фотосинтетически неактивным светом, и из-
менения ее выхода в процессе освещения листьев с периодичес-
ким переводом всех реакционных центров ФС II в фотохимически
неактивное состояние посредством освещения объекта короткой
мощной вспышкой света.
Описание флуорометра РАМ 100
Общая схема устройства прибора представлена на рис. 50. Пара
«эмиттер—детектор» (на схеме обведена пунктирной чертой) пред-
назначена для генерации слабого возбуждающего света (эмиттер)
140
Рис. 50. Схема устройства прибора РАМ100
и регистрации излучения флуоресценции (детектор). Эмиттером
служит светодиод, испускающий свет с длиной волны 650 нм,
модулированный с частотой 1,6 или 100 кГц. Этот свет дополни-
тельно пропускается через светофильтр, отрезающий излучение све-
тодиода с длиной волны, больше 680 нм. Детектором испускаемой
объектом флуоресценции служит фотодиод. Светофильтр, который
помещен перед детектором, пропускает лучи с длиной волны только
больше 700 нм, предотвращая тем самым попадание на фотодиод
рассеянного возбуждающего света. Таким образом, комбинация двух
141
светофильтров обеспечивает спектральное разделение возбуждаю-
щего света и излучения флуоресценции хлорофилла. Модуляция
возбуждающего света осуществляется электрически посредством
подачи на световод тока от генератора импульсов, модулированно-
го с частотой 1,6 или 100 кГц. Возбуждающий свет представляет
собой последовательность 1-микросекундных импульсов, разделен-
ных в зависимости от выбранной частоты модуляции темновым
промежутком в 625 мкс (1,6 кГц) или 10 мкс (100 кГц).
Электрический сигнал от генератора последовательно усили-
вается импульсным и селективным усилителями. Последний ра-
ботает синхронно с подачей на объект возбуждающих флуорес-
ценцию хлорофилла микросекундных световых импульсов, по-
скольку электрический генератор импульсов одновременно фор-
мирует последовательность импульсов возбуждающего света и
поддерживает несущую частоту для селективного усилителя. Та-
кой принцип усиления позволяет регистрировать сигнал флуо-
ресценции, только модулированный с одной из двух вышеназ-
ванных частот, несмотря на присутствие в исходном сигнале от
детектора значительной составляющей, связанной с возбуждени-
ем флуоресценции хлорофилла постоянным действующим светом.
После усиления модулированный сигнал либо преобразуется
прибором в вид постоянного напряжения и может быть зарегист-
рирован любым самописцем, либо с помощью специально разра-
ботанной для прибора РАМ 100 приставки «Data Acquisition System»
PDA-100 может быть преобразован в форму компьютерных asc-
кодов. Если на самописце возможно регистрировать изменения
флуоресценции, протекающие быстрее 1 с (временной ход пера
самописца), то применение PDA-100 позволяет увеличить вре-
менное разрешение прибора до 20 мкс.
Характерной чертой флуорометра РАМ 100 является легкость
освещения объекта постоянным светом различной интенсивнос-
ти и спектрального состава, что позволяет широко варьировать
фотосинтетическую активность во время измерения флуоресцен-
ции хлорофилла. Такая легкость достигается при использовании
разветвленного световода (рис. 51). Световод, сплетенный из мно-
гочисленных тонких световодных волокон, имеет с одной сторо-
ны четыре отростка, которые с другой его стороны соединены в
общий торец, контактирующий с объектом. Два отростка проти-
воположной стороны световода используются для обеспечения
функционирования пары «эмиттер—детектор» и предназначены
для проведения возбуждающего света от эмиттера к объекту и пе-
редачи излучения флуоресценции хлорофилла от объекта к детек-
тору. Два остальных отростка используются для проведения к объек-
ту световых потоков, изменяющих его фотосинтетическую актив-
ность. При измерении коэффициентов тушения флуоресценции
хлорофилла один из этих отростков вставляется в специальный
142
Световод
к объекту
Электрическое соединение с РАМ 100
Пара «эмиттер—детектор»
Световод к источнику
насыщающих вспышек
Световод к детектору
Световод к эмиттеру
О
Световод к источнику
действующего света
Рис. 51. Светопроводящая система прибора РАМ 100
осветитель KL1500 (Schott, Германия). Этот осветитель использу-
ется для генерации очень мощных импульсов света длительнос-
тью не более 1 с, которые предназначены для кратковременного
перевода всех реакционных центров ФС II в фотохимически неак-
тивное состояние. В приборе РАМ 100 предусмотрена возможность
автоматического освещения объекта последовательностью мощ-
ных вспышек, интервал между которыми может в широких пре-
делах варьировать. Наконец, четвертый отросток предназначен для
подведения к объекту постоянного действующего света, изменя-
ющего состояние фотосинтетического аппарата. В конкретной ус-
тановке, представленной в Институте физиологии им. К. А. Тими-
рязева РАН, действующий свет обеспечивается вторым осветите-
лем KL1500, однако в принципе этот светопоток может быть по-
лучен с помощью любого источника света.
Ход работы
Работа состоит из следующих этапов: 1) измерение парамет-
ров индукции флуоресценции (Fo, Fm) у адаптированного к тем-
ноте листа; 2) регистрация стационарного уровня флуоресцен-
ции хлорофилла (Fs) при различных интенсивностях света и уровня
флуоресценции F^ при дополнительном освещении объекта мощ-
ной насыщающей вспышкой.
1. Измерения параметров индукции флуоресценции листьев. По-
скольку целью работы является исследование фотоиндуцирован-
ных изменений нефотохимического и фотохимического тушения
143
флуоресценции хлорофилла, в начале работы измеряют парамет-
ры флуоресценции у листьев, долгое время {не менее 30 мин) адапти-
рованных к темноте. Темновая адаптация приводит к релаксации
изменений в выходе флуоресценции, индуцированных предыду-
щим освещением листа. Неотчлененный лист растения, выдер-
жанного в темноте, зажимается в специальной листовой камере,
к окошку которой прикрепляется общий торец световода. Убедив-
шись, что все остальные отростки световода находятся в рабочем
положении (два соединены с парой «эмиттер—детектор», а два
других — с осветителями KL1500), можно включить прибор и
приступить к измерениям.
Прибор РАМ100 (рис. 52) включается тумблером «Power on»,
расположенным на задней стенке. Тумблер «Pulse on» на передней
стенке прибора (7) должен быть в нижнем положении, что соот-
ветствует отсутствию возбуждающего света. После этого необхо-
димо однократным нажатием в течение нескольких секунд крас-
ной кнопки «Zero offset» (5) на передней панели прибора приве-
сти к нулевому значению напряжение выходного сигнала, регис-
трируемое на цифровом индикаторе (5). Вслед за этим включают
лентопротяжный механизм самописца и регистрируют в течение
примерно 1 мин «нулевой» уровень сигнала, предварительно убе-
дившись в том, что размах шкалы самописца составляет 1 В.
На следующем этапе измерений регистрируют «фоновый» и
максимальный уровни флуоресценции (7^ и Fm) у адаптирован-
ного к темноте листа. Уровень Fq соответствует состоянию листа с
полностью окисленным QA, а уровень Fm — с полностью восста-
новленным. Для регистрации уровня Fq включают слабый возбуж-
дающий свет переводом тумблера «Pulse on» (7) в верхнее поло-
Рис. 52. Общий вид прибора РАМ100 в конфигурации РАМ101-103. Стрел-
ками обозначены различные элементы управления (объяснение в тексте)
144
жение. Предварительно следует убедиться, что частота модулиро-
ванного света составляет 1,6 кГц (тумблер 4 должен быть в поло-
жении «1,6 kHz»). Одновременно нужно также убедиться в том,
что ручки «Timer» на правой секции прибора «Flash Trigger Control»
находятся в положениях «1s» (ручка 7) и «1» (ручка <?), что отве-
чает длительности насыщающей вспышки света 1 с, а верхняя
ручка «Clock» (6) этой секции прибора находится в положении
«Stop», что соответствует выключенной автоматической програм-
ме подачи последовательных насыщающих вспышек света. Следу-
ет включить осветитель KL1500, соединенный электрическим ка-
белем с прибором РАМ 100. Это производится нажатием кнопки,
расположенной на нижней стороне осветителя. Поскольку увели-
чение выхода флуоресценции при освещении адаптированного к
темноте листа мощной вспышкой света превышает уровень Fo при-
мерно в 5 раз, следует выбрать такую интенсивность возбуждаю-
щего света, чтобы амплитуда фонового уровня не превышала 15 %
шкалы самописца. Последнее производится путем варьирования
интенсивности модулированного возбуждающего света поворотом
ручки «Light Int.» (2). Ручки «Gain» (усиление — 12) и «Damping»
(постоянная времени прибора — 11) должны находиться в поло-
жениях «2» и «9» соответственно.
После регистрации в течение примерно 1 мин фонового уров-
ня флуоресценции хлорофилла (Fo) измеряют максимальный уро-
вень флуоресценции (Fm) при освещении листа мощной насыщаю-
щей односекундной вспышкой. Однократное включение этой
вспышки достигается нажатием кнопки «Start» (9). После регист-
рации уровней Fo и Fm ручки «Clock» продвигаются в положения
«100» (6) и «1» (10), что соответствует набору автоматической
программы освещения объекта последовательными мощными на-
сыщающими вспышками с частотой 1 вспышка в 100 секунд. Про-
грамма активируется нажатием кнопки «Start» (9).
2. Регистрация амплитуд стационарного уровня флуоресценции
хлорофилла Fs при различных интенсивностях действующего света
и уровня флуоресценции F^ при дополнительном освещении объек-
та мощной насыщающей вспышкой. Эта процедура является фак-
тически заключительным этапом измерений. Нажатием кнопки на
нижней панели включают второй осветитель KL1500, предвари-
тельно убедившись, что правая ручка на верхней панели освети-
теля находится в положении «1», что соответствует самому низ-
кому значению интенсивности действующего света. Измерения про-
должают, пока уровни Fs и F'm не достигнут стационарных значе-
ний (обычно это наступает через 8—10 мин после включения дей-
ствующего света), вслед за чем увеличивают интенсивность дей-
ствующего света поворотом правой ручки на панели осветителя в
положение «2». После окончания измерений при данной интен-
сивности действующего света ее ступенчато увеличивают до до-
145
стижения максимального значения, соответствующего положению
«6» ручки осветителя, генерирующего действующий свет.
Анализ результатов измерений. После завершения измерений для
каждой использованной интенсивности действующего света оп-
ределяют амплитуды следующих уровней:
• Fq — как разницу между уровнем флуоресценции у адаптиро-
ванного к темноте листа на возбуждающем свету и уровнем сиг-
нала в отсутствие возбуждающего света («нулевой сигнал»);
• Fm — как разницу между уровнем флуоресценции при осве-
щении адаптированного к темноте листа насыщающей вспышкой
света и «нулевым сигналом»;
• F'm— как разницу между уровнем флуоресценции при осве-
щении адаптированного к действующему свету листа насыщаю-
щей вспышкой света и «нулевым сигналом»;
• Fs — как разницу между амплитудой стационарного уровня
флуоресценции на действующем свету и «нулевым» уровнем.
После нахождения амплитуд вышеуказанных уровней флуорес-
ценции рассчитывают:
• максимальный квантовый выход разделения зарядов в ФС II
как отношение
(Fm - F,)/Fm- (1)
• нефотохимическое тушение:
= (2)
• фотохимическое тушение:
= (3)
• квантовый выход фотохимического превращения поглощен-
ной световой энергии в ФС II при длительном освещении как
отношение
Л - Fs)/F'm. (4)
Значения нефотохимического тушения, рассчитанные по фор-
муле (2), представляющей собой формулу Штерна — Фолъмера, при-
мененную для оценки тушения флуоресценции хлорофилла, ва-
рьируют от 0 (в отсутствие нефотохимического тушения) до при-
мерно 10 в реальности. Значения фотохимического тушения, рас-
считанные по формуле (3), варьируют от 0 (полное восстановле-
ние первичного акцептора QA фотосистемы II) до 1 (полное окисле-
ние первичного акцептора QA). Значения квантового выхода варь-
ируют от 0,83—0,85 (адаптированные к темноте листья) до значе-
ний, близких к 0 (листья, адаптированные к очень сильному дей-
ствующему свету).
Оформление результатов. После проведения расчетов строят
графики зависимости NPQ, qP и квантового выхода от интенсив-
146
ности действующего света, показывающие, что при увеличении
освещенности листа активность темновой стадии фотосинтеза
уменьшается в сравнении со световой стадией (возрастает NPQ),
восстановленность первичного акцептора ФС II увеличивается (сни-
жается qP) из-за относительного замедления переноса электрона
между двумя фотосистемами, а квантовый выход фотохимическо-
го превращения световой энергии в ФС II снижается из-за общего
торможения электронного транспорта вследствие лимитирования
последнего скоростью функционирования темновых процессов
фотосинтеза.
3.4. ОПРЕДЕЛЕНИЕ АКТИВНОСТИ
РИБУЛОЗОБИСФОСФАТКАРБОКСИЛАЗЫ
СПЕКТРОФОТОМЕТРИЧЕСКИМ МЕТОДОМ
(по И. М. Магомедову)
Одним из подходов к оценке активности темновых реакций
фотосинтеза, осуществляющих фиксацию углекислого газа и син-
тез метаболитов, является определение активности ферментов
цикла Кальвина. Особый интерес при этом представляют фермен-
ты, регулирующие активность темновых реакций фотосинтеза в
целом.
Ключевым ферментом цикла Кальвина, регулирующим актив-
ность фотосинтетической ассимиляции углекислого газа, являет-
ся рибулозобисфосфаткарбоксилаза (РБФ-карбоксилаза, Рубис-
ко). При участии фермента РБФ-карбоксилазы в цикле Кальвина
осуществляется фиксация углекислоты на рибулезобисфосфате
(РБФ). Реакция карбоксилирования приводит к образованию двух
молекул трифосфоглицериновой кислоты (3-ФГК), которые в
последующих реакциях восстанавливаются до трифосфоглицери-
нового альдегида (3-ФГА) с использованием АТФ и НАДФН. Ввиду
этого для определения активности данного фермента обычно при-
меняют два способа: радиометрический, основанный на использо-
вании 14СО2, и спектрофотометрический, с использованием
НАДФН или НАДН в качестве кофактора реакции:
3-ФГК ........> 3-ФГА.
Первый метод предпочтителен, поскольку позволяет напря-
мую определить активность реакции карбоксилирования. Однако
радиометрический метод требует специального оборудования и
помещения для работы с изотопами. Второй метод косвенный, но
также может быть использован в серийных определениях. Ниже
описан спектрофотометрический метод определения активности
РБФ-карбоксилазы [7 — 9].
147
Ход работы
Работа включает следующие этапы: 1) подготовка препарата
фермента; 2) спектрофотометрическое определение активности
РБФ-карбоксилазы; 3) определение содержания белка в пробе;
4) расчет активности фермента.
Для работы используют листья 14 — 21-дневных проростков ку-
курузы.
1. Подготовка препарата фермента. Навеску листьев (5 г) рас-
тирают в ступке или измельчают в гомогенизаторе с 5 мл среды,
содержащей 0,05 М трис-HCl буфер (pH 8,0), 1 мМ MgCl2, 0,1 мМ
ЭДТА, 5 мМ дитиотрейтол (ДТТ) или 5 мМ меркаптоэтанол. Же-
лательно гомогенизировать листья быстро (1 — 3 мин). После филь-
трации через капрон фильтрат центрифугируют при 18000 g в те-
чение 20 мин. К осадку добавляют 2 мл исходной среды и снова
центрифугируют при тех же условиях; объединяют оба суперна-
танта, доводят объем до 8 мл и этот супернатант используют для
определения активности ферментов.
2. Определение активности РБФ-карбоксилазы спектрофотомет-
рическим методом. Для проведения опыта в две пробирки (одна —
контрольная, другая — опытная) вносят инкубационную среду,
содержащую 0,05 М трис-HCl буфер (pH 8,2), 10 мМ MgCl2, 10 мМ
ДТТ, 50 мМ NaHCO3; НАДФН (или НАДН) 0,15 мМ, 0,3 мл
экстракта.
Пробирки с инкубационной средой выдерживают в термостате
при 30 °C в течение 2 мин. Затем запускают реакцию путем внесе-
ния в пробирки 0,2 мл смеси АТФ и рибозо-5-фосфата (Р-5-Ф)*
(конечная концентрация компонентов в реакционной среде 1 мМ).
В контрольную пробу сразу вносят равный объем 30 %-й уксусной
кислоты. Опытную пробу инкубируют 4 мин, после чего реакцию
останавливают добавлением равного объема 30 %-й уксусной кис-
лоты.
Измеряют на спектрофотометре оптическую плотность опыт-
ного образца при 340 нм (в области поглощения НАДФН и НАДН).
Контролем служат пробы, зафиксированные уксусной кислотой
одновременно с внесением в них Р-5-Ф и АТФ.
Опыт проводят в 3-кратной повторности.
3. Определение содержания белка в пробе. Для определения
содержания белка в пробе используют метод Лоури (см. описание
в разделе 6.5.2).
4. Расчет активности фермента. За единицу активности фер-
мента А принимается изменение оптической плотности D на 0,01
* Для определения активности очищенного фермента в качестве субстрата
необходимо применять РБФ, но при использовании гомогената можно восполь-
зоваться Р-5-Ф и АТФ, так как в экстрактах содержатся рибозоизомераза (5.3.1.6)
и рибонуклеаза (2.7.1.19), которые синтезируют РБФ.
148
в 1 мин. Для пересчета активности с единиц оптической плотнос-
ти в микромоли НАДФН (НАДН) применяют коэффициент по-
глощения кофакторов, равный 6,22 мМ-1- см-1 при 340 нм.
Пример расчета. Пусть за 5 мин изменение оптической плотности
д/) = 0,25. Тогда за 1 мин AZ) = 0,05. Приняв за единицу активности
фермента изменение плотности 0,01/мин, получаем активность фер-
мента А = 5. При содержании белка в пробе 0,1 мг активность фермента
Л =50.
Оформление результатов. Результаты измерений оптической
плотности и расчеты активности фермента вносят в таблицу.
Глава 4
МЕТОДЫ ВЫДЕЛЕНИЯ И ХРАНЕНИЯ
ХЛОРОПЛАСТОВ, ТИЛАКОИДОВ
И ПИГМЕНТ-БЕЛКОВЫХ КОМПЛЕКСОВ
Прогресс в изучении фотосинтеза как в целом, так и отдель-
ных его реакций связан с возможностью получения изолирован-
ных фотосинтезирующих органелл клетки — хлоропластов, и от-
дельных структурно-функциональных компонентов фотосинте-
тического аппарата — тилакоидов и мультипротеиновых комп-
лексов мембран хлоропластов. В настоящее время разработаны
различные методы выделения названных компонентов. Они по-
зволяют получить препараты с различной степенью очистки, ин-
тактности и функциональной активности. Выбор методики вы-
деления фотосинтезирующих частиц определяется целями иссле-
дования.
Интактные, достаточно хорошо очищенные от других субкле-
точных структур и биохимически активные хлоропласты необхо-
димы для изучения метаболизма углерода, транспорта метаболи-
тов в хлоропластах и компартментации ферментов в клетке. При
установлении локализации ферментов, присутствующих в клетке
в очень небольших количествах, необходимы препараты хлоро-
пластов с очень высокой степенью очистки, функциональная ак-
тивность при этом оказывается не столь значимой. Напротив, для
других целей необходимо получение изолированных хлороплас-
тов с максимальной активностью фотосинтеза, наличие же не-
больших примесей цитоплазматических ферментов при этом до-
пустимо.
Изучение реакций, связанных с переносом электронов в элек-
трон-транспортной цепи (ЭТЦ) фотосинтеза и синтезом АТФ,
не требует сохранения высокой интактности внешней оболочки
хлоропластов, но предполагает интактность их внутренних мемб-
ран. Изолированные тилакоиды позволяют исследовать структур-
но-функциональную организацию фотосинтетических мембран,
исследовать первичные реакции фотосинтеза. Изолированные фун-
кциональные комплексы тилакоидных мембран используют для
установления их пептидного состава, локализации кофакторов,
исследования отдельных реакций, происходящих в этих комплек-
сах [1, 3, 4, 6, 13, 14].
150
В настоящее время существует определенная номенклатура пре-
паратов изолированных хлоропластов, в основу которой положе-
ны критерии морфологической целостности и функциональной
активности хлоропластов (D. О. Hall, 1972). Согласно современ-
ной классификации, можно выделить 6 типов хлоропластов [2, 9].
Хлоропласты типа А (иначе называемые интактные целые хло-
ропласты) имеют неповрежденную внешнюю оболочку, непрони-
цаемую для НАДФ и искусственных акцепторов электронов типа
феррицианида калия. Они содержат все ферменты цикла Кальвина
и проявляют высокую скорость фиксации СО2 в отсутствие экзо-
генных кофакторов (50 — 200 мкмоль СО2/мг хлорофилла в час).
Хлоропласты типа В (неразрушенные хлоропласты) имеют наруж-
ную оболочку (обнаруживается при фазово-контрастной микро-
скопии), но она незначительно нарушена, и это ведет к частич-
ной потере водорастворимых компонентов стромы хлоропластов.
Вследствие этого скорость фиксации углекислого газа у хлоропла-
стов данного типа значительно снижена (менее 5 мкмоль СО2/мг
хлорофилла в час). Нарушенная внешняя оболочка хлоропластов
становится проницаемой для экзогенных водорастворимых кофак-
торов (НАДФ, АТФ, феррицианид калия).
Хлоропласты типа С (разрушенные хлоропласты) — это структуры
с сильно нарушенной наружной оболочкой, но с интактной внут-
риламеллярной системой. Они не способны к фиксации углекисло-
го газа, но проявляют фотохимическую и фотофосфорилирующую
активность в присутствии экзогенных кофакторов. Хлоропласты
этого типа, как правило, выделяют механическим способом.
Хлоропласты типа D (препараты свободных ламелл) получают
из хлоропластов типа А путем осмотического шока с последую-
щим переносом в изотоническую среду. Хлоропласты теряют на-
ружную оболочку, но сохраняют интактную тилакоидную систе-
му и проявляют фотохимическую и фосфорилирующую активность
в присутствии экзогенных кофакторов.
Хлоропласты типа Е (фрагменты хлоропластов) представляют
собой свободные тилакоиды, отмытые от компонентов стромы
гипотоническим раствором.
Хлоропласты типа F (или субхлоропластные частицы) — это ча-
стицы, получаемые при обработке хлоропластов детергентами,
ультразвуком или с помощью Френч-пресса. Состав и функцио-
нальная активность частиц зависят от условий их выделения и
очистки. Возможно получение различных препаратов ФС I, ФС II,
Цитохромного ^//-комплекса, светособирающих комплексов.
При проведении большинства исследований, связанных с вы-
яснением механизма фотосинтеза (изучение углеродного метабо-
лизма, определение фосфорилирующей и восстановительной ак-
тивности хлоропластов, анализ их химического состава, структу-
ры и т.д.), необходимо получение функционально активных пре-
151
паратов изолированных хлоропластов. В настоящее время для вы-
деления хлоропластов используют методы, основанные либо на
механическом разрушении ткани листа, либо на разрушении про
топластов, предварительно полученных из нее. Любой из двух спо-
собов выделения хлоропластов имеет свои достоинства и недо
статки. Выбор метода зависит от цели эксперимента, а также во
многом определяется видом растения, используемого в экспери-
менте. Так, из листьев гороха и шпината удается получить доста -
точно высокий выход в значительной степени интактных хлоро-
пластов при механическом разрушении тканей, тогда как исполь
зование протопластов, согласно литературным данным, не дает
больших преимуществ. В то же время при работе с такими расте-
ниями, как пшеница, ячмень, табак, мягкое выделение хлоро-
пластов из протопластов позволяет получить более интактные и ак-
тивные хлоропласты, чем механическое разрушение листьев [1, 3].
Выделение хлоропластов из протопластов предпочтительно при
исследовании локализации ферментов в клетке, так как при этом
удается получить больший процент интактных хлоропластов, ос-
вобожденных от других клеточных органелл (митохондрий, мик-
ротелец), эпидермальных и проводящих тканей. Кроме того, при
выделении хлоропластов из протопластов снижается вероятность
агрегации органелл, неспецифического связывания ферментов с
органеллами и формирования комплексов белков с таннинами по
сравнению с механическим методом выделения хлоропластов.
К явным достоинствам ферментативного метода разрушения тка-
ней следует также отнести возможность получения гомогенного
препарата интактных, высокоактивных хлоропластов. Недостат-
ками ферментативного метода выделения хлоропластов, по-ви-
димому, являются: невозможность использования этого метода для
некоторых видов растений, большая его продолжительность из-за
предварительной мацерации ткани, необходимость подбора ми-
нимального времени мацерации и способов очистки протоплас-
тов с целью избежания побочного действия гидролизующих фер-
ментов, ограниченный выход органелл.
Методы выделения хлоропластов при механическом разруше-
нии тканей отличаются быстротой, достаточной простотой и бо-
лее высоким выходом хлоропластов, но при этом существует боль-
шая вероятность повреждения хлоропластов, снижения их актив-
ности и, как отмечалось выше, агрегации органелл.
Выделение хлоропластов любым из двух способов, как прави-
ло, сопряжено с использованием дифференциального центрифу-
гирования, позволяющего отделить их от остального содержимо-
го клетки. При необходимости получения высокоочищенных пре-
паратов хлоропластов в ряде случаев требуется дополнительная
очистка хлоропластов, которая достигается путем центрифугиро-
вания в градиенте плотности.
152
4.1. ВЫДЕЛЕНИЕ ХЛОРОПЛАСТОВ
ПРИ МЕХАНИЧЕСКОМ РАЗРУШЕНИИ
ТКАНЕЙ ЛИСТА
4.1.1. Выделение хлоропластов
методом дифференциального
центрифугирования
Данный метод включает растирание навески листьев со средой
выделения, приготовление гомогената и его последующее цент-
рифугирование со скоростями, позволяющими отделить фракцию
хлоропластов от других более тяжелых и более легких компонен-
тов клетки. В качестве среды выделения могут быть взяты водные
растворы хлористого натрия, сахарозы, сорбитола с определен-
ной величиной осмотического давления или безводные органи-
ческие среды (гексан, четыреххлористый углерод).
При выделении в водной среде хлоропласты сохраняют фото-
химическую активность, так как при этом не нарушаются ламел-
лярные структуры, но хлоропласты теряют часть водораствори-
мых комплексов, главным образом ферменты углеродного цикла.
Поэтому хлоропласты, изолированные в водные среды, мало спо-
собны к реакциям восстановления СО2.
При выделении в органической, безводной среде хлороплас-
ты сохраняют целостность стромы и содержащиеся в ней водо-
растворимые вещества, но теряют способность к ряду фотохи-
мических реакций вследствие экстракции жирорастворимых ком-
понентов ламелл (хиноны, каротиноиды) и нарушения систем
транспорта электронов. Применение органических сред выделе-
ния оказалось эффективным при исследовании локализации в
хлоропластах минеральных элементов, белков, ферментов, при
изучении продуктов фотосинтеза и ряда реакций, требующих
сохранения стромы.
При определении фосфорилирующей и восстановительной ак-
тивности хлоропластов обычно используют водные среды выде-
ления. Они должны быть изотоничны и химически инертны по
отношению к компонентам хлоропластов и иметь определенное
значение pH, что достигается добавлением фосфатного буфера,
трис-, трицин-буфера или других буферных систем.
Обычно для выделения хлоропластов используют водные среды
следующего состава: 1) 0,35 М NaCl в 0,05 М трис-HCl буфере,
pH 8,0 (D.I.Arnon et al., 1956); 2) 0,30 М NaCl в 0,06 М фосфат-
ном буфере, pH 6,9 (J. Miller, 1960); 3) 0,40 М сахароза и 0,01 М
КС1 в 0,05 М фосфатном буфере, pH 6,9 (A. Jagendorf, I960).
Для сохранения нативных свойств хлоропластов в среду выде-
ления иногда добавляют восстановители (аскорбат), сульфгидриль-
153
ные соединения (цистеин, глютатион), а также стабилизирую-
щие факторы (альбумин, фикол, полиэтиленгликоль).
Ход работы
Работа включает следующие этапы: 1) приготовление гомоге-
ната листьев с использованием различных сред выделения и вы-
деление хлоропластов из гомогената методом дифференциально-
го центрифугирования; 2) определение степени интактности по-
лученных хлоропластов.
В работе можно использовать листья С3- и С4-растений (горох,
пшеница, кукуруза и др.).
1. Выделение хлоропластов. Все процедуры выделения хлоро-
пластов ведут на холоду при 0 —4 °C. Используемые растворы и
посуду также предварительно охлаждают в холодильнике.
Ниже приведены два варианта выделения хлоропластов при ме-
ханическом разрушении тканей листа, различающиеся средами и
процедурами выделения.
Вариант 1
1. Навеску листьев в 2 г, предварительно охлажденную в холо-
дильнике в течение 20 мин в полиэтиленовом пакете или влажной
фильтровальной бумаге, измельчают в фарфоровой ступке 1 — 2 мин
с 5-кратным количеством (от массы) среды выделения. Возмож-
ные варианты среды выделения приведены ниже.
2. Гомогенат пропускают через двойной слой полотна и цент-
рифугируют 5 мин при 100 g.
3. Супернатант осторожно сливают в чистую центрифужную
пробирку, а осадок, содержащий обрывки тканей, ядра, разру-
шенные клеточные оболочки и другие фрагменты, отбрасывают.
4. Супернатант для осаждения хлоропластов центрифугируют
15 мин при 1000 g. (
5. Осадок, содержащий хлоропласты, промывают средой выде-
ления (10 — 15 мл) и центрифугируют 15 мин при 1000 g.
6. Хлоропласты аккуратно гомогенезируют со средой выделения
с использованием кисточки и тефлонового гомогенизатора. Неболь-
шими порциями гомогенат переносят в мерную пробирку или ци-
линдр. Объем гомогената доводят средой выделения до 30—40 мл
(при определении фотофосфорилирования — до 10 мл).
Полученный препарат хлоропластов следует держать при тем-
пературе 0 — 3 °C (в бане со льдом): в этих условиях активность
хлоропластов сохраняется в течение нескольких часов.
Дальнейшая очистка препаратов хлоропластов от компонентов
клетки может быть осуществлена путем центрифугирования их в
ступенчатом градиенте плотности сахарозы (см. ниже).
154
Состав сред выделения и используемый pH могут варьировать
В зависимости от объекта и целей исследования. При выделении
^поропластов с целью исследования их фотохимической активно-
сти используют обычно pH, близкий к нейтральному, увеличе-
ние pH при выделении хлоропластов создает оптимальные усло-
вия для функционирования системы сопряжения потока электро-
нов с синтезом АТФ.
Варианты среды выделения хлоропластов'.
1. 0,30 М NaCl в 0,06 М фосфатном буфере, pH 6,9;
2. 0,30 М NaCl в 0,06 М фосфатном буфере, pH 8,04;
3. 0,40 М сахароза и 0,01 М NaCl в 0,06 М фосфатном буфере,
pH 6,9;
4. 0,40 М сахароза и 0,01 М NaCl в 0,06 М фосфатном буфере,
pH 8,04.
Вариант 2
Для выделения хлоропластов используют следующие среды:
• Среда 7 (25 мл):
0,35 М сорбитол (1,5925 г);
20 мМ трицин-NaOH, pH 7,8 (0,0896 г)*;
0,2 мМ MgCl2 (50 мкл 0,1 М раствора).
• Среда 2 (10 мл):
0,35 М сорбитол (0,637 г);
1 мМ трицин-NaOH, pH 7,8 (0,5 мл 20 мМ трицин-NaOH,
pH 7,8).
• Среда 3 (25 мл):
0,35 М сорбитол (1,5925 г);
20 мМ трицин-NaOH, pH 7,8 (0,0896 г);
5 мМ MgCl2 (125 мкл 1 М раствора);
10 мМ КС1 (125 мкл 2 М раствора).
1. Охлажденные листья гороха (2 г) растирают в ступке с 20 мл
среды 1 в течение 15 —20 с.
2. Фильтруют суспензию через 2 слоя капрона и центрифугиру-
ют 15 — 20 с при 100 g.
3. Супернатант аккуратно сливают в новую центрифужную про-
бирку и центрифугируют 3 мин при 1000 g.
4. Промывают осадок средой 2 и центрифугируют еще раз при
1000 g.
5. Добавляют к осадку небольшие порции среды 3 и гомогени-
зируют с использованием тефлонового гомогенизатора. Конечный
объем суспензии — 10 мл.
* Можно приготовить 100 мл в расчете на все три среды (0,358 г трицина).
155
2. Определение степени интактности изолированных хлороплас
тов. Одним из показателей интактности мембран изолированных
хлоропластов может служить степень стимуляции потока элект-
ронов в присутствии фосфатакцепторной системы (АДФ+ФН) [1].
Отношение скорости выделения кислорода хлоропластами в ус -
ловиях сопряженного с синтезом АТФ транспорта электронов к
базальному транспорту электронов (т. е. в отсутствие фосфатак-
цепторной системы) позволяет охарактеризовать состояние АТФ-
синтетазной системы хлоропластов и оценить степень сопряжен-
ности потока электронов с синтезом АТФ в препаратах изолиро-
ванных хлоропластов. Оценку активности транспорта электронов
в отсутствие и в присутствии фосфатакцепторной системы можно
осуществить полярографическим методом (см. 5.2.1).
Оформление результатов. При оформлении результатов приво-
дят данные по фотохимической активности хлоропластов, опре-
деленной в различных условиях эксперимента, и рассчитывают
степень сопряженности потока электронов с синтезом АТФ в изо
лированных хлоропластах.
4.1.2. Выделение хлоропластов методом дифференциального
центрифугирования в градиенте плотности сахарозы
Дифференциальное центрифугирование с использованием гра-
диентов плотности позволяет провести дальнейшую очистку хло-
ропластов и получить более гомогенные препараты. Градиенты
плотности готовят с использованием сахарозы, перкола и других
соединений.
Ход работы
Проводят дальнейшую очистку суспензии изолированных хло-
ропластов.
В стеклянные центрифужные пробирки объемом около 46 мл с
помощью пипетки осторожно по стенке наслаивают столбик сту
пенчатого градиента сахарозы (растворы сахарозы готовят на ос-
нове буфера, используемого при выделении хлоропластов) сле-
дующей структуры:
Слой Концентрация, М Объем, мл
Нижний 1,5 5
Средний 1,0 10
Верхний 0,8
Сверху наслаивают около 6 мл хлоропластов, суспензирован-
ных в среде с 0,5 М сахарозой. Пробирки уравновешивают и цент-
156
рис. 53. Разделение суспензии хлороплас-
тов в градиенте плотности сахарозы на от-
дельные фракции
Фракция I
Фракция II
Фракция III
рифугируют 20 мин при 1000 g. Различные по величине фрагменты
хлоропластов и целые хлоропласты обычно распределяются по
фракциям, как показано на рис. 53. Полученные при фракциониро-
вании отдельные зоны (слои) хлоропластов осторожно отсасывают
шприцем (объемом около 3 мл) с длинной иглой, собирают их в
отдельные пробирки, разбавляют до конечной концентрации саха-
розы 0,5 М и просматривают под микроскопом.
Используют световой и фазово-контрастный микроскоп.
Оформление результатов. Зарисовывают распределение хлоро-
пластов в градиенте плотности и результаты микроскопических
наблюдений.
4.2. ВЫДЕЛЕНИЕ ИНТАКТНЫХ ХЛОРОПЛАСТОВ
ИЗ ПРОТОПЛАСТОВ С3-РАСТЕНИЙ
Выделение хлоропластов из протопластов в ряде случаев являет-
ся более предпочтительным, чем метод механического разрушения
тканей, несмотря на его большую трудоемкость, малый выход хло-
ропластов и высокую стоимость применяемых реактивов [1,3]. Ис-
пользование протопластов позволяет значительно расширить спи-
сок растений, из тканей которых удается выделять интактные хло-
ропласты. Данная методика успешно применяется при работе с не-
которыми С3-злаками, например с пшеницей и ячменем, некото-
рыми двудольными С3-растениями — подсолнечником, табаком,
горохом, шпинатом, а также со многими видами С4-растений. Это
связано с тем, что, во-первых, механическое усилие, необходи-
мое для разрушения плазмалеммы, намного меньше усилия, необ-
ходимого для разрушения клеточной стенки в тканях листа, в ре-
зультате степень повреждения органелл значительно снижается, и
хлоропласты, выделенные из протопластов, сохраняются в высо-
кой степени интактными. Во-вторых, в процессе выделения прото-
пластов удаляются соединения, находящиеся в клеточной стенке и
сосудистых тканях листа, неблагоприятно влияющие на хлоропла-
сты. При механическом разрушении тканей эти соединения высво-
бождаются, и их воздействие можно свести к минимуму только
путем быстрого отделения органелл центрифугированием. Напри-
мер, при работе с листьями подсолнечника, содержащими фено-
157
лы, механически выделенные хлоропласты ассимилируют СО2 до-
вольно медленно, несмотря на внесение в среду выделения очень
большого количества различных защитных соединений. И наобо-
рот, хлоропласты, выделенные из протопластов подсолнечника,
обладают высокой активностью. Точно так же активные хлоропла-
сты нельзя получить механическим способом из флаговых листьев
пшеницы, но они успешно выделяются из протопластов [1].
Выделение органелл с высокой степенью интактности позво-
ляет исследовать темновые реакции фотосинтеза, механизмы ре-
гуляции отдельных стадий фотосинтеза. Кроме того, сами прото-
пласты являются важным инструментом в исследовании таких
вопросов, как внутриклеточное распределение метаболитов в про-
цессе фотосинтеза, внутриклеточная локализация фотосинтети-
ческих ферментов, межклеточная компартментация фотосинтеза
у С3-, С4- и CAM-растений. Интактные протопласты и клетки
позволяют также проследить процесс фотосинтеза при высокой
концентрации СО2 в системе, т. е. избежать ограничений, возни-
кающих при работе с интактными листьями (например, ограни-
чения устьицами скорости газообмена в листе). При этом, одна-
ко, следует учитывать снижение транспорта ассимилятов из изо-
лированных клеток, а также довольно активное передвижение
углеводов к местам регенерации клеточных стенок вследствие ра-
невого эффекта, что может привести к некоторым нарушениям
метаболизма в клетке.
Оценка степени интактности изолированных хлоропластов мо-
жет быть осуществлена различными способами. Тестом на интакт-
ность внешней мембраны хлоропластов может служить их способ-
ность осуществлять ассимиляцию углекислого газа или СО2-зави-
симого выделения кислорода. Так, для высокоинтактных хлоро-
пластов пшеницы или шпината скорость фиксации углекислого
газа или скорость СО2-зависимого выделения кислорода состав-
ляет 80—150 мкмоль/мг хлорофилла в 1 ч и выше. Целостность
внешней мембраны определяет также чувствительность хлоропла-
стов к экзогенным добавкам. При неповрежденной внешней мем-
бране хлоропласты не чувствительны к экзогенным акцепторам
электронов, таким, как НАДФ, феррицианид калия и др. Поэто-
му наиболее простым методом оценки интактности хлоропластов
считают определение их фотохимической активности в присут-
ствии феррицианида калия.
Критерием интактности изолированных мембран может слу-
жить степень сопряженности потока электронов с синтезом АТФ.
Оценку сопряженности проводят путем регистрации изменения
скорости выделения хлоропластами кислорода в присутствии раз-
общителей, например NH4C1. Увеличение скорости транспорта
электронов в присутствии разобщителя в 10 и более раз указывает
на высокую интактность изолированных хлоропластов.
158
Ход работы
Работа включает следующие этапы: 1) получение протопластов
из мезофилла листьев гороха; 2) получение хлоропластов из про-
топластов гороха; 3) определение содержания хлорофилла в суспен-
зии хлоропластов; 4) определение степени интактности изолиро-
ванных хлоропластов по фотохимической активности в присутствии
феррицианида калия и разобщителя полярографическим методом.
В работе используют листья гороха.
1. Получение протопластов из мезофилла листьев гороха. Для
выделения протопластов готовят следующие среды.
• Среда 1 (200 мл):
0,5 М сорбит;
1 мМ СаС12;
5 мМ трицин-NaOH буфер, pH 6,0.
• Среда 2 (10 мл):
0,4 М сахароза;
0,1 М сорбит;
1мМ СаС12;
5 мМ трицин-NaOH буфер, pH 6,0.
• Среда 3 (10 мл):
0,5 М сахароза;
1 мМ СаС12;
5 мМ трицин-NaOH буфер, pH 6,0.
Среды, используемые для выделения и отмывки протоплас-
тов, охлаждают.
Листья гороха (0,5 г) разрезают по центральной жилке и на
каждой половине листа лезвием бритвы делают надрезы в виде
гребенки. Подготовленные таким образом листья помещают для
мацерации в 5 мл среды 7, содержащей 2 % (масса/объем) целлю-
лазы и 0,3 % (масса/объем) пектиназы (Macerozime R-10). Обра-
ботку ферментами ведут в чашках Петри, помещенных под лампу
накаливания. Время мацерации 1 —1,5 ч.
После обработки ферментами ткани отмывают в 20 мл среды 7,
фильтруют через нейлоновое сито с диаметром ячей 500 мкм (на-
пример, через чайное ситечко), чтобы отделить более крупные
куски, и через нейлоновое сито с диаметром ячей 200 мкм, чтобы
отделить сосудистые тяжи. Фильтрат центрифугируют 5 мин при
100 g. Осадок промывают еще два раза средой 7, причем перед вто-
рым промыванием смесь переносят в стеклянные пробирки на
15—20 мл (можно использовать стеклянные мерные пробирки на
5 мл). После последнего центрифугирования в течение 5 мин при
100 g аккуратно удаляют надосадочную жидкость, быстро пере-
вернув пробирки, и осторожно ресуспендируют осадок в несколь-
ких каплях среды 3. Добавляют еще 5 мл (2,5 мл) среды 3 и сверху
осторожно наслаивают 2 мл (1 мл) среды 2 (для отмывания про-
159
топластов), а затем 1 мл (0,5 мл) среды 1. После центрифугирова -
ния в течение 5 мин при 250g из слоя раздела фаз пастеровской
пипеткой (или шприцем с длинной иглой) отбирают чистую фрак -
цию протопластов. Осадок обычно состоит из хлоропластов и разру-
шенных клеток, но перед выбрасыванием его стоит проверить под
световым микроскопом на наличие протопластов. Если в осадке
обнаружено много протопластов, к сахарозной среде можно доба-
вить 5 — 10% декстрана Т20 (масса/объем) и повторить выделение.
Протопласты суспендируют в 5 мл среды 1 и хранят на льду.
В суспензии определяют содержание хлорофилла и активность вы-
деления кислорода на свету.
Для протопластов пшеницы можно использовать среду, содер-
жащую 0,4 М сорбита, 10 мМ NaHCO3, 5 мМ трицин-КОН буфер
(pH 7,6). В этой же среде определяют фиксацию СО2.
2. Получение хлоропластов из протопластов гороха. Для полу
чения хлоропластов протопласты в среде с сорбитом центрифу-
гируют в течение 5 мин при 100g. Затем для разрушения прото-
пластов их ресуспендируют в среде, состоящей из 0,4 М сорбита.
10 мМ ЭДТА и 25 мМ трицин-КОН буфер (pH 8,0); концентра-
ция протопластов составляет приблизительно 200 мкг хлорофил-
ла на 1 мл среды. После разрушения протопластов смесь центри-
фугируют в течение 60 с при 250 g, полученный осадок хлоропла-
стов осторожно ресуспендируют в этой же среде и хранят на льду.
Внесение NaHCO3 (5 — 10 мМ) в среду для разрушения протопла-
стов и ресуспендирования хлоропластов может дать благоприят-
ные результаты.
3. Определение степени интактности изолированных хлороплас-
тов. Работа включает подготовку исходной суспензии хлороплас-
тов для тестирования и определение скорости выделения кисло-
рода полярографическим методом в присутствии феррицианида
калия и NH4C1 у исходных хлоропластов и хлоропластов, подвер-
гнутых осмотическому шоку.
Подготовка хлоропластов для тестирования. Для проведения
анализа на интактность используют тест-среду, содержащую 0,33
М сорбитол, 1 мМ MgCl2, 1 мМ МпС12, 2 мМ ЭДТА и 50 мМ
HEPES-KOH буфер (pH 7,6). Эту среду используют для ресуспен-
дирования хлоропластов, а также в качестве реакционной среды
при определении степени интактности хлоропластов.
При подготовке хлоропластов к тестированию исходную сус-
пензию помещают в стеклянную центрифужную пробирку и цен-
трифугируют 5 мин при 1000g. Супернатант аккуратно удаляют, а
хлоропласты суспендируют в указанной выше тест-среде. Кон-
центрация хлоропластов должна соответствовать 0,5—1 мг хлоро-
филла в 1 мл.
Определение скорости выделения кислорода хлоропластами по-
лярографическим методом в присутствии экзогенных акцепторов
160
электронов и разобщителей. Для проведения теста производят срав-
нительный анализ активности хлоропластов исходной суспензии
и хлоропластов, подвергнутых осмотическому шоку. Анализ ведут
в присутствии 3 мМ феррицианида калия и при добавлении NH4C1
(конечная концентрация 2,5 мМ).
Осмотический шок хлоропластов проводят непосредственно в
полярографической ячейке. Для осуществления осмотического шока
к 0,1 мл суспензии хлоропластов добавляют 0,9 мл воды и пере-
мешивают в течение 1 мин. Затем добавляют 1 мл тест-среды с
удвоенным содержанием компонентов (0,66 М сорбитол, 2 мМ
MgCl2, 2 мМ МпС12, 4 мМ ЭДТА и 100 мМ HEPES-KOH буфер,
pH 7,6). После проведения осмотического шока к хлоропластам
добавляют 0,1 мл раствора феррицианида калия и необходимое
для полного заполнения полярографической ячейки количество
тест-среды. Производят запись выделения кислорода хлороплас-
тами на свету в течение 2 — 3 мин, затем добавляют в ячейку
шприцем NH4C1.
При определении активности исходных хлоропластов в поля-
рографическую ячейку вносят 0,1 мл исходной суспензии и 1,9 мл
тест-среды, перемешивают 1 мин, добавляют 0,1 мл феррициа-
нида калия и необходимое для заполнения ячейки количество
тест-среды. Производят запись выделения кислорода на свету в
отсутствие и присутствии разобщителя.
Расчет интактности [%] исходного препарата осуществляют
следующим образом:
(А - Б)/А-100,
где А — скорость выделения кислорода хлоропластов, подвергну-
тых осмотическому шоку; Б — скорость выделения кислорода ис-
ходных хлоропластов.
Оформление результатов. Данные полярографического иссле-
дования фотохимической активности хлоропластов и результаты
расчета степени интактности хлоропластов вносят в таблицу.
4.3. ВЫДЕЛЕНИЕ ТИЛАКОИДОВ ХЛОРОПЛАСТОВ
ИЗ ЛИСТЬЕВ РАСТЕНИЙ
Применение метода механического разрушения тканей листа
для получения хлоропластов позволяет получить хлоропласты типа
С. При воздействии на эти хлоропласты гипотонического раство-
ра наружная оболочка хлоропластов полностью разрушается, в
результате чего получают препараты тилакоидов хлоропластов (хло-
ропласты типа D и Е), способные осуществлять фотохимическую
и фотофосфорилирующую активности.
6 Гавриленко
161
В данной работе для получения фракции тилакоидных мембран
используют следующие среды.
• Среда 7 (25 мл):
0,4 М сорбитол (1,82 г);
10 мМ трицин-NaOH буфер, pH 7,6 (0,045 г);
10 мМ MgCl2 (вносят в виде концентрированного раствора
известной концентрации).
• Среда 2 (25 мл):
10 мМ трицин-NaOH буфер, pH 7,6 (0,045 г);
2 мМ №ЭДТА (0,0185 г).
Все процедуры выполняют при температуре 0 — 4 °C.
Ход работы
Охлажденные листья гороха (1г) растирают в ступке в тече-
ние 15 — 20 с с 10 мл среды 1. Суспензию фильтруют через 4 слоя
капрона и центрифугируют 2 мин при 1500 g. Супернатант акку-
ратно сливают. Осадок суспендируют в среде 2 кисточкой и го-
могенизируют с использованием стеклянного гомогенизатора.
Гомогенат центрифугируют 5 мин при 15 000g. Осадок гомоге-
низируют в среде 2 с использованием стеклянного гомогениза-
тора.
Конечный объем тилакоидов — 10 мл.
Препарат хранят на льду, в темноте.
Полученную суспензию хлоропластов используют для иссле-
дования фотохимической активности хлоропластов полярографи-
ческим методом, как описано в 5.2.
4.4. ВЫДЕЛЕНИЕ ПРЕПАРАТОВ ФОТОСИСТЕМЫ II
В исследованиях структурно-функциональной организации фо-
тосинтетического аппарата в настоящее время широко применя-
ют субхлоропластные частицы, получаемые при разрушении мем-
бран тилакоидов с использованием детергентов, ультразвука,
Френч-пресса и др. [8]. Последующее их разделение и очистка пу-
тем дифференциального и скоростного центрифугирования, элек-
трофореза и других процедур позволяет получить отдельные функ-
циональные комплексы фотосинтетического аппарата и исследо-
вать их организацию и активность. Состав и функциональная ак-
тивность фотосинтетических комплексов могут значительно раз-
личаться в зависимости от условий выделения субхлоропластных
частиц.
Ниже описана одна из методик выделения препаратов ФС II
(D.A.Bertold et al., 1981, с модификациями).
162
Ход работы
Работа включает следующие этапы: 1) выделение тилакоидов
хлоропластов; 2) определение концентрации хлорофилла в сус-
пензии хлоропластов; 3) солюбилизация мембран детергентом
тритон Х-100; 4) получение субхлоропластных частиц и их замо-
раживание в жидком азоте.
В работе используют листья 15 — 30-дневных растений гороха
или шпината.
Все процедуры ведут при температуре 4 °C.
1. Выделение тилакоидов хлоропластов. Листья гороха или шпи-
ната (10—15 г) гомогенизируют в гомогенизаторе с 10-кратным
количеством (по объему) среды 1, содержащей в 50 мМ трис-НС1
буфере (pH 7,8) 0,33 М сахарозу, 35 мМ NaCl, 2 мМ ЭДТА, 5 мМ
аскорбат натрия. Гомогенат фильтруют через 8 слоев марли и цен-
трифугируют в течение 10 мин при 4000 g.
Осадок ресуспендируют в среде 2, содержащей в 50 мМ трис-
НС1 буфере (pH 7,8) 15 мМ NaCl, 5 мМ MgCl2, и подвергают
двухстадийному центрифугированию: 1 мин при 500g и 10 мин
при 4000 g.
Тяжелый осадок с клеточными обрывками отбрасывают, гра-
ны тилакоидов, полученные при втором центрифугировании, ре-
суспендируют в среде 3, содержащей в 50 мМ MES или HEPES
буфере (pH 6,5) 0,33 М сахарозу и 35 мМ NaCl.
Для получения мембран можно использовать хлоропласты, замо-
роженные в жидком азоте. В этом случае суспензию хлоропластов,
эквивалентную 2 — 3 г хлорофилла, размораживают на льду, промы-
вают средой 2 и осаждают в течение 10 мин при 4000 g. Полученный
осадок хлоропластов ресуспендируют в среде 3 (3,5—4 мл) и ис-
пользуют для получения частиц ФС II.
2. Определение содержания хлорофилла в суспензии хлороплас-
тов. К 0,1 мл суспензии добавляют 0,9 мл Н2О и 4 мл этилового
спирта. Смесь перемешивают и затем фильтруют через стеклян-
ный фильтр с использованием колбы Бунзена (см. 2.1.1). Получен-
ный фильтрат измеряют на спектрофотометре при длинах волн
663 и 645 нм.
Данные измерения поглощения используют для расчета кон-
центрации хлорофилла в 80 %-м этаноле по формуле
са+ь - 8,02 • Л663 + 20,2 • Л645,
где са+ь — суммарная концентрация хлорофиллов, мг/л экстракта;
Л663 и Л645 — поглощение при длинах волн 663 и 645 нм соответст-
венно.
Рассчитывают содержание хлорофилла в полученной суспен-
зии хлоропластов [мг/мл суспензии].
3. Солюбилизация мембран детергентом и получение субхлоро-
пластных частиц. Обработку детергентом тритон Х-100 проводят на
163
льду в течение 20 мин при соотношении детергента к хлорофиллу
20:1 (объем детергента рассчитывают, исходя из полученных дан-
ных по содержанию хлорофилла в препарате хлоропластов).
После солюбилизации препарат центрифугируют в течение 1 ч
при 15 000g. Осадок дважды промывают средой, содержащей в
50 мМ MES или HEPES буфере (pH 6,5) 0,33 М сахарозу и 35 мМ
NaCl.
4. Замораживание препаратов ФС II. Полученные фрагменты
мембран (ФС II) ресуспендируют в небольшом объеме среды для
замораживания, содержащей в 50 мМ MES или HEPES буфере
(pH 6,5) 0,33 М сахарозу, 35 мМ NaCl и 20 %-й глицерин, и замо-
раживают в жидком азоте. Процедура замораживания препаратов
в жидком азоте описана в 4.5.1.
Возможно использование свежевыделенных препаратов ФС II
для определения их фотохимической активности полярографичес-
ким методом (см. 5.2.3).
4.5. ЗАМОРАЖИВАНИЕ И ХРАНЕНИЕ ИЗОЛИРОВАННЫХ
ХЛОРОПЛАСТОВ И ТИЛАКОИДОВ В ЖИДКОМ АЗОТЕ
4.5.1. Замораживание и хранение изолированных
хлоропластов в жидком азоте
Метод замораживания хлоропластов в жидком азоте позволяет
приготовить большое количество суспензии хлоропластов и ис-
пользовать однородный материал в серии опытов. Хранение изо-
лированных хлоропластов в жидком азоте практически не изме-
няет их активность.
Ход работы
Работа включает два этапа: 1) выделение хлоропластов; 2) за-
мораживание хлоропластов.
В работе можно использовать листья гороха, пшеницы и других
растений 10—14-дневного возраста.
1. Выделение хлоропластов. Выделение хлоропластов из листьев
производят ранее описанным механическим способом с использо-
ванием метода дифференциального центрифугирования (см. 4.1.1).
Среда для выделения и среда для хранения хлоропластов могут
варьировать в зависимости от объекта исследования. Варианты ис-
пользуемых сред приведены ниже.
На 15 г листьев необходимо 150 мл среды выделения.
• Среда для выделения хлоропластов пшеницы'.
400 мМ сахароза;
1 мМ NaCl;
164
1 мМ MgCl2;
2 мМ ЭДТА;
1 мМ МпС12;
0,05 М фосфатный буфер (pH 7,4 —7,5).
• Среда для выделения хлоропластов гороха.
Вариант Г.
0,35 М NaCl;
0,05 М трис-HCl буфер (pH 7,4).
Вариант 2\
400 мМ сахароза;
10 мМ NaCl;
0,05 М фосфатный буфер (pH 7,4).
В оба варианта среды выделения можно добавить 1 мМ MgCl2.
Навеска листьев — 10—15 г.
Для гомогенизации листьев используют 5-кратное (по объему)
количество среды выделения. Применяют электрический гомоге-
низатор.
Листья, охлажденные 15—20 мин в холодильнике в целлофа-
новом пакете и влажной фильтровальной бумаге, делят на 2 — 3
порции. Каждую порцию гомогенизируют в электрическом гомо-
генизаторе в течение 10—15 с при максимальных оборотах с соот-
ветствующим количеством среды выделения (40 — 50 мл).
Гомогенаты листьев фильтруют через капроновую ткань или
4 слоя марли и объединяют. Фильтрат центрифугируют 5 мин при
100g. Супернатант аккуратно сливают в новые центрифужные про-
бирки и центрифугируют 10 мин при 1000 g. Полученные осадки
гомогенизируют со средой выделения и объединяют в одной цен-
трифужной пробирке. Центрифугируют гомогенат 10 мин при 1000 g.
Супернатант отбрасывают, а осажденные хлоропласты использу-
ют для замораживания.
2. Замораживание хлоропластов. Осадок хлоропластов, получен-
ный после последнего центрифугирования, оставляют в центри-
фужной пробирке и с помощью кисточки осторожно разбивают,
постепенно приливая небольшими порциями среду замораживания.
Среда для замораживания хлоропластов гороха1, среда выделения +
глицерин (10 или 20 %).
Полученный гомогенат сливают в охлажденную пробирку (по-
мещена в лед).
В небольшой термос с широким горлышком наливают жидкий
азот (температура -196 °C) из сосуда Дьюара. Суспензию хлоро-
пластов набирают в пипетку на 2 мл и осторожно по каплям ска-
пывают в жидкий азот. Пипетку необходимо держать строго вер-
тикально, стараясь при этом, чтобы очередная капля не попадала
на предыдущую, в противном случае может произойти слипание
замороженной суспензии хлоропластов. Суспензия хлоропластов
165
замерзает в виде шариков. Зная концентрацию хлорофилла в сус-
пензии хлоропластов и ее массу, для дальнейших опытов можно
взвешивать на весах необходимое количество замороженных хло-
ропластов.
После замораживания всей суспензии хлоропластов образовав-
шиеся шарики переносят, не допуская размораживания, в ма-
ленькую центрифужную пробирку, которую плотно закрывают
ватным тампоном, или в мешочек из марли (пакетик из фольги),
помещают в сосуд Дьюара с азотом и замораживают. К пробе,
помещенной в большой сосуд Дьюара, прикрепляют этикетку с
необходимыми пометками.
Внимание! При всех операциях с жидким азотом нельзя до-
пускать размораживания суспензии хлоропластов. Так как тем-
пература жидкого азота равна -196 °C, необходимо строго со-
блюдать правила техники безопасности при работе с низкими
температурами:
нельзя дотрагиваться голыми руками до охлажденных пред-
метов или материалов; их следует брать длинным пинцетом
или использовать для этого несколько слоев ткани (полотенце).
4.5.2. Замораживание и хранение изолированных
тилакоидов в жидком азоте
Осадок тилакоидов, полученный ранее описанным методом (см.
4.3), ресуспендируют в среде, содержащей 6,2 мМ трис-HCl бу-
фер (pH 8,3), 48 мМ глицин, 10 %-й глицерин. Конечная концен-
трация суспензии: 1—1,1 мг хлорофилла в 1 мл. Суспензию замо-
раживают и хранят в жидком азоте, как описано в 4.5.1.
Глава 5
ОПРЕДЕЛЕНИЕ ФОТОХИМИЧЕСКОЙ
АКТИВНОСТИ ХЛОРОПЛАСТОВ
Способность хлоропластов осуществлять на свету перенос элек-
тронов от воды к НАДФ+ с образованием сильного восстановите-
ля НАДФН определяет их фотохимическую активность.
Впервые способность изолированных из клетки хлоропластов
при освещении восстанавливать ряд соединений с одновременным
выделением кислорода обнаружил Р. Хилл в 1937 г. [16]. Эта реак-
ция выделения кислорода хлоропластами на свету при наличии
акцепторов электронов (А) получила название «реакция Хилла»:
Н2О + А—^2—»АН2 + 1/2О2.
Реакция Хилла представляет собой комплекс световых реакций
фотосинтеза, связанных с фотоокислением воды, в которых мо-
билизованные из воды электроны по цепи переносчиков направ-
ляются на восстановление введенных в реакционную смесь ак-
цепторов электронов. Фотовосстановление акцепторов сопровож-
дается выделением кислорода. Реакция Хилла является показате-
лем фотохимической активности хлоропластов.
Общая система транспорта электронов в хлоропластах — это
последовательная цепь компонентов с различной величиной ре-
докс-потенциала, способных к окислительно-восстановительным
реакциям (см. приложение 2). Схема электрон-транспортной цепи
(ЭТЦ) хлоропластов высших растений (так называемая Z-схема)
показана на рис. 54. Редокс-агенты ЭТЦ входят в состав трех мульти-
пептидных, встроенных в мембрану комплексов (рис. 55) — фото-
система I (ФС I), фотосистема II (ФС II) и цитохром ^//-комп-
лекс, а также представлены рядом подвижных переносчиков элек-
тронов — это пластохиноны (PQ), пластоцианин (PC), ферре-
доксин (ФД).
ФС I при участии возбужденного пигмента реакционного цен-
тра Р700 обеспечивает образование соединений (Ло, с высоким
восстановительным потенциалом (Eq = —0,9 0,8 В). ФС II при
возбуждении пигмента реакционного центра Р680 генерирует об-
разование сильного окислителя P6to (Eq = 1,12 В), который ини-
циирует процессы, ведущие к фотоокислению Н2О. Цитохром
167
^//-комплекс обеспечивает перенос электронов и координиро-
ванное взаимодействие ФС I и ФС II, а также сопряженный с этим
энергозависимый процесс трансмембранного переноса протонов
из стромы во внутритилакоидное пространство [4 — 8, 13, 15, 19].
В результате поглощения энергии фотосистемами I и II в хло-
ропластах формируется нециклический поток электронов от Н2О
(Eq =+0,81 В) к НАДФ+ (Eq = —0,32 В). Кроме того, в хлоропла-
стах возможен циклический поток электронов, сопряженный с
работой комплекса ФС I, и псевдоциклический транспорт элект-
ронов от воды на кислород. Циклический поток электронов не
приводит к фотоокислению воды и выделению кислорода. Энер-
гия, освобождаемая в ходе циклического транспорта электро-
нов, используется исключительно на синтез АТФ. Псевдоцикли-
ческий транспорт электронов, хотя и сопряжен с фотолизом воды
и выделением кислорода, сопровождается его поглощением за
счет использования молекулярного кислорода в качестве конеч-
ного акцептора электронов [1, 2, 11, 25, 28]. Впервые перенос
А)'>в
(Аоо)*
-1,о
-0,8
-0,6
-0,4
0
+0,4
+0,6
+0,8
+ 1,0
+1,2
Рис. 54. Z-схема электрон-транспортной цепи хлоропластов:
5 — водоокисляющий комплекс; TyrZ — донор реакционного центра ФС II (ти-
розин 161); Фео — феофитин; QA, QB — пластохиноны — вторичные акцепторы
электронов ФС II; PQ — пул пластохинонов; Цит. Ь6 — цитохром b6, Fe2S2 —
центр Риске;/— цитохром/; PC — пластоцианин; Ао — хлорофилл а — первичный
акцептор электронов реакционного центра ФС I; А{ — витамин К\, Fx, FA, FB —
железосерные белки акцепторного комплекса ФС I; ФД — ферредоксин; ФАД —
ферредоксин-НАДФ+-оксидоредуктаза
168
Фотосистема II
АТФ-синтаза
Цитохром Фотосистема I
^/-комплекс н+
Рис. 55. Схема организации электрон-транспортной цепи в мембране ти-
лакоидов:
ССК I — светособирающий комплекс I; ССК II — светособирающий комплекс II;
CF0 — сопрягающий фактор 0; СЦ — сопрягающий фактор 1; Цит. ЬИ — цито-
хром Z>6 высокопотенциальный; Цит. bL — цитохром Z>6 низкопотенциальный;
FQR — ферредоксинхиноноксидоредуктаза. Остальные обозначения, как
на рис. 54
электронов на кислород в хлоропластах обнаружил А. Мелер в
начале 50-х гг. XX в. [24, 25], поэтому реакция восстановления
кислорода на свету получила была названа реакцией Мелера. В на-
стоящее время установлено, что фотовосстановление молекуляр-
ного кислорода может происходить на различных участках ЭТЦ
(на уровне ФС I — с участием железосерных белков, на уровне
ФС II — до восстановления QA и, вероятно, на уровне цитохром
^//-комплекса) (Р.М.Бекина, В. Е. Гусейнова, 1986; J.M. Robin-
son, 1988; К. Asada, 1993). На любом из этих участков электрон-
транспортной цепи вначале идет одноэлектронное восстановле-
ние кислорода и образование супероксидного анион-радикала.
Затем может происходить образование пероксида водорода либо
за счет реакции дисмутации с участием фермента супероксид-
дисмутаза (О2 + О2 + 2Н+ -э Н2О2 + О2), либо за счет последую-
щего одноэлектронного восстановления супероксидного анион-ра-
дикала (О2 + е" + 2Н+ -> Н2О2 + О2). Таким образом, реакция Меле-
ра приводит к образованию активных форм кислорода, которые
могут повреждать фотосинтетический аппарат растений.
В последние годы были получены данные о возможности хло-
ропластов осуществлять окисление восстановленных соединений,
находящихся в строме (например, НАДФН), и передавать элект-
роны кислороду через пул пластохинонов [27]. Этот процесс по-
169
лучил название хлоропластного дыхания (хлордыхания). Условия
реализации хлоропластного дыхания и его физиологическая роль
требуют дальнейшего изучения.
Основной поток электронов в хлоропластах осуществляется по
нециклическому пути, доля циклического и особенно псевдоцик-
лического переноса электронов, как правило, невелика, хотя при
некоторых условиях, например при избыточной интенсивности
света, активность альтернативных путей транспорта электронов
может возрастать [1].
Фотохимическая активность хлоропластов, соотношение аль-
тернативных путей транспорта электронов в ЭТЦ фотосинтеза
контролируются внешними и внутренними факторами и являют-
ся важнейшими показателями функционального состояния фото-
синтетического аппарата [5]. Скорость нециклического потока элек-
тронов определяет активность образования высоковосстановлен-
ных соединений (НАДФН, ФДВОсст)> участвующих в дальнейших
реакциях восстановления СО2, а также в других восстановитель-
ных процессах хлоропластов, и играющих важную регуляторную
роль при фотосинтезе. Транспорт электронов в ЭТЦ определяет
возможность формирования трансмембранного градиента прото-
нов (Дцн+), за счет энергии которого осуществляется синтез АТФ
в процессах фотосинтетического фосфорилирования [5, 7, 22].
Методы определения фотохимической активности изолирован-
ных хлоропластов основаны на регистрации количества выделен-
ного или поглощенного в реакции кислорода (полярографичес-
ким или манометрическим методом) или количества восстанов-
ленных акцепторов (спектрофотометрический метод) [4, 15, 17,
18]. В качестве акцепторов электронов (окислителей) в реакции
Хилла могут быть использованы соли трехвалентного железа (фер-
рицианид калия K3Fe(CN)6, оксалат железа K3Fe(C2O4)3), окисли-
тельно-восстановительные индикаторы (2,6-дихлорфенолиндофе-
нол — ДХФИФ, 2,3,6-трихлорфенолиндофенол — ТХФИФ и др.),
физиологические акцепторы электронов (хиноны, цитохромы, пла-
стоцианин, НАДФ+) и другие соединения [15, 17, 22, 30, 32].
В зависимости от величины окислительно-восстановительного
потенциала включение акцепторов электронов в цепь может про-
исходить в ее различных участках. На этом основано использова-
ние искусственных акцепторов электронов при исследовании со-
става электрон-транспортной цепи, последовательности включе-
ния в нее отдельных компонентов и других вопросов, связанных с
изучением механизма первичных реакций фотосинтеза.
Исследования активности псевдоциклического пути транспор-
та электронов в хлоропластах (реакции Мелера) могут включать
прямое определение образования пероксида водорода с помощью
этанол-каталазной ловушки, либо регистрацию активных форм
кислорода с использованием реагентов реакции Мелера — хими-
170
ческих соединений, способных активировать поглощение кисло-
рода хлоропластами. К реагентам реакции Мелера относятся как
физиологические соединения (ферредоксин, флавины, аскорби-
новая кислота, некоторые моно- и дикарбоновые кислоты), так и
нефизиологические соединения (виологеновые красители, адре-
налин и др.). Место включения в ЭТЦ хлоропластов и механизмы
действия разных групп реагентов реакции Мелера могут быть раз-
личны [1, 3]. Так, бензил- и метилвиологен, флавины взаимодей-
ствуют с кислородом на уровне ФС I и восстанавливают его до
супероксидного анион-радикала, тогда как органические кисло-
ты (глиоксилат, малонат, щавелевоуксусная кислота и др.) акти-
вируют поглощение кислорода, реагируя с образующимися на свету
его активными формами.
При исследовании световых реакций фотосинтеза наряду с
использованием искусственных акцепторов электронов широко
применяют введение в реакционную смесь искусственных элект-
рон-донорных систем (аскорбат + 2,6-дихлорфенолиндофенол,
аскорбат + диаминодурол и др.), которые в определенных экспе-
риментальных условиях могут служить источниками электронов
при нарушении процессов фотоокисления воды. Схема включе-
ния ингибиторов, искусственных акцепторов и доноров электро-
нов в ЭТЦ фотосинтеза дана на рис. 56.
В настоящее время определение фотохимической активности
изолированных хлоропластов при использовании различных элек-
трон-донорных и электрон-акцепторных систем, а также ингиби-
торов транспорта электронов в отдельных участках ЭТЦ широко
применяют в исследованиях механизма фотосинтеза. Определение
фотохимической активности целых хлоропластов или фрагмен-
тов, полученных при обработке хлоропластов детергентами или
ультразвуком, позволяет получить информацию о составе компо-
ФСП ФС1
NH,OH DCMU DBMIB KCN
Iz III
Рис. 56. Основные центры включения ингибиторов транспорта электро-
нов, искусственных акцепторов (А) и доноров (D) электронов:
— феррицианид калия, метилвиологен; Аг — фенилендиамин, диметилхинон,
дихлорфенолиндофенол; Dx — дихлорфенолиндофенол + аскорбат; Z)2 — дифе-
нилкарбозид; NH2OH — гидроксиламин; DCMU — диурон, 3-(3,4-дихлорфенил)-
1,1-диметилмочевина; DBMIB— 2,5-дибром-3-метил-6-изопропил-и-бензохинон;
KCN — цианистый калий; FeS — железосерные белки акцепторного комплекса
ФС I. Остальные обозначения, как на рис. 54
171
нентов и скорости потока электронов на исследуемом участке ЭТЦ,
расчленить участие ФС I и ФС II в фотохимических реакциях фо-
тосинтеза и т.д. Сравнение скорости потока электронов в отсут-
ствие АДФ и Фн (базальный транспорт электронов) и в присут-
ствии фосфатакцепторной системы (сопряженный с синтезом АТФ
транспорт электронов) позволяет оценить степень сопряжения
потока электронов с процессами фотофосфорилирования. Регист-
рация транспорта электронов в присутствии разобщителей пото-
ка электронов и синтеза АТФ (например, в присутствии NH4C1)
позволяет оценить максимально возможную скорость переноса элек-
тронов в ЭТЦ хлоропластов (разобщенный транспорт электронов).
Ниже рассмотрены некоторые методы определения фотохими-
ческой активности изолированных хлоропластов.
5.1. ОПРЕДЕЛЕНИЕ ФОТОХИМИЧЕСКОЙ АКТИВНОСТИ
ХЛОРОПЛАСТОВ СПЕКТРОФОТОМЕТРИЧЕСКИМ
МЕТОДОМ
Измерение восстановительной активности изолированных хло-
ропластов в присутствии акцепторов электронов может быть про-
ведено путем прямых спектрофотометрических определений ко-
личества образующихся восстановленных соединений. В качестве
акцепторов электронов в этих исследованиях могут быть исполь-
зованы 2,6-дихлорфенолиндофенол (ДХФИФ), феррицианид ка-
лия K3Fe(CN)6, НАДФ+, поскольку окисленная и восстановлен-
ная формы этих соединений различаются по спектральным ха-
рактеристикам. Так, ДХФИФ в окисленном состоянии при pH 6,5
имеет синюю окраску и поглощает при 620 нм, при восстановле-
нии он обесцвечивается. K3Fe(CN)6 в окисленном состоянии име-
ет желтую окраску, поглощает при 420 нм, а в восстановленном —
бесцветный. Восстановление НАДФ+ сопровождается изменения-
ми поглощения при 340 нм.
5.1.1. Определение фотохимической активности
хлоропластов по скорости восстановления феррицианида
калия в различных условиях эксперимента
Ход работы
Работа включает следующие этапы: 1) выделение хлороплас-
тов из листьев; 2) определение скорости базального (в отсутствие
АДФ и Фн) и 3) сопряженного (в присутствии АДФ и Фн) пото-
ков электронов; 4) определение содержания хлорофилла в сус-
пензии хлоропластов; 5) расчет активности реакции Хилла.
172
Рис. 57. Установка для определения
фотохимической и фотофосфорили-
рующей активности хлоропластов
Для работы могут быть ис-
пользованы проростки гороха,
пшеницы, кукурузы, выращен-
ные в различных условиях водо-
снабжения, минерального пита-
ния, освещения; растения раз-
ного возраста; семядоли тыквы,
обработанные регуляторами ро-
ста и развития растений.
1. Выделение хлоропластов.
Процедуры выделения хлоро-
пластов приведены в гл. 4. Навес-
ка листьев — 2 г.
Среда выделения хлоропластов'.
0,30 М NaCl в 0,06 М фосфат-
ном буфере (pH 6,9).
Конечный объем суспензии в
цилиндре — 12—15 мл.
Суспензию хлоропластов держат в темноте и на льду.
2. Определение скорости базального потока электронов в ЭТЦ
хлоропластов. Опыт проводят в конических колбах небольшого
объема на специальной установке (рис. 57). Для проведения опыта
берут 4 одинаковые колбы. В каждую колбу вносят реакционную
смесь.
Реакционная смесь в общем объеме 5 мл содержит 3 мл суспен-
зии хлоропластов, эквивалентные 50 — 100 мкг хлорофилла, 1 мл
водного раствора K3Fe(CN)6, содержащего 15 мкмоль акцептора
электронов, 1 мл среды выделения (объем среды выделения умень-
шают в случае внесения в реакционную смесь дополнительных
добавок).
Можно приготовить сразу на все пробы (из расчета 5 проб)
смесь феррицианида калия и среды выделения и внести в колбы
по 2 мл смеси. После этого в каждую колбу внести по 3 мл суспен-
зии хлоропластов. Схема проведения опыта приведена ниже:
Номер колбы Вариант
1 Свет
2 Свет
3 Темнота
4 Темнота
173
Темновые варианты помещают в темноту, а световые освеща-
ют в течение 5 мин на специальной установке.
После выключения света в световые пробы сразу же добавляют
1 мл 20 %-й ТХУ (трихлоруксусная кислота) и 2 мл 1 М CH3COONa.
После этого добавляют ТХУ и CH3COONa в темновые варианты.
Пробы фильтруют через двойные бумажные фильтры или цент-
рифугируют 10 мин при 2000 g. Полученные прозрачные растворы
промеряют на спектрофотометре при длине волны 420 нм. Конт-
роль — вода.
3. Определение скорости потока электронов в ЭТЦ хлороплас-
тов, сопряженного с синтезом АТФ, проводят в присутствии АДФ
и MgCl2. Добавление этих реагентов производят за счет уменьше-
ния в реакционной среде объема среды выделения.
Сравнивают скорости базального и сопряженного потоков элек-
тронов. Опыт проводят по следующей схеме:
Номер колбы
Вариант
1
2
3
4
5
6
7
8
Свет - АДФ
Свет - АДФ
Темнота - АДФ
Темнота - АДФ
Свет + АДФ
Свет + АДФ
Темнота + АДФ
Темнота + АДФ
Реакционную смесь для базального и сопряженного потоков
электронов можно приготовить сразу на 5 колб. Состав реакцион-
ной смеси представлен в табл. 15.
Ход проведения реакции Хилла описан выше.
4. Определение хлорофилла в суспензии хлоропластов. В мерную
пробирку вносят 1 мл суспензии, 1 мл дистиллированной воды и
объем доводят до 10 мл 100 %-м ацетоном. Раствор хорошо пере-
мешивают и фильтруют через стеклянный фильтр.
Полученный фильтрат исследуют на спектрофотометре при
длине волны 652 нм (контроль — 80 %-й ацетон). Измеренное по-
глощение раствора (й652) используют для расчета содержания хло-
рофилла в суспензии хлоропластов по формуле Арнона.
1 мл суспензии хлоропластов содержит (0,29 • А^) мг хлорофилла.
5. Расчет активности реакции Хилла. Активность переноса элек-
тронов в ЭТЦ фотосинтеза выражают в мкмолях K3Fe(CN)6/(Mr
хлорофилла • ч).
174
Таблица 15
Состав реакционной смеси
Компонент Вариант Конечная концентра- ция в реак- ционной смеси, мМ
-АДФ + АДФ
мл на 1 колбу мл на 5 колб мл на 1 колбу мл на 5 колб
Среда выделения 0,85 4,25 0,7 3,5 —
MgCl2 0,15 0,75 0,15 0,75 4
K3Fe(CN)6 1,0 5 1,0 5 3
АДФ — — 0,15 0,75 1
Хлоропласты 3,0 — 3,0 — —
Формула для расчета активности реакции Хилла:
(Лцо> темнота - Д420, свет) -60-8
1,04 • 5 • мг хлорофилла в пробе ’
где 1,04 — миллимолярный коэффициент поглощения K3Fe(CN)6.
Оформление результатов. Полученные данные вносят в табли-
цу. Сравнивают скорость базального и сопряженного потоков элек-
тронов в ЭТЦ хлоропластов, оценивают влияние АДФ на ско-
рость потока электронов в ЭТЦ. Схема записи результатов опыта
приведена ниже:
Номер колбы Вариант опыта Поглощение фильтратов (Азо) (Азо, темнота — Азо, свет) Активность реакции Хилла, мкмоль K3Fe(CN)6/MT хлорофилла-ч
5.1.2. Определение скорости нециклического потока
электронов в ЭТЦ хлоропластов по восстановлению НАДФ+
Фотохимическая активность хлоропластов in vivo связана с фо-
товосстановлением НАДФ+ — естественного конечного акцепто-
ра электронов в ЭТЦ хлоропластов. Его восстановление осуществ-
ляется при совместной работе двух фотосистем (ФС I и ФС II)
хлоропластов за счет нециклического потока электронов от воды.
175
Восстановленный НАДФН является донором электронов для вос-
становительных реакций цикла Кальвина.
Оценка скорости фотовосстановления НАДФ+ в электрон-
транспортной цепи хлоропластов на изолированных хлоропластах
может быть произведена путем определения количества образую-
щегося НАДФН спектрофотометрическим методом. Однако про-
ведение реакции фотовосстановления НАДФ+ на изолированных
в водной среде хлоропластах требует добавления в реакционную
смесь препарата растворимого ферредоксина, поскольку при вы-
делении хлоропластов слабо связанный с мембранами ферредок-
син легко вымывается в среду выделения. Ввиду этого определе-
ние фотохимической активности ЭТЦ хлоропластов по скорости
восстановления НАДФ+ предполагает предварительное получение
препарата растворенного ферредоксина (ФД) из листьев гороха или
использование коммерческих препаратов ФД [6, 32].
Ход работы
Работа состоит из следующих этапов: 1) выделение и очистка
на ДЭАЭ-целлюлозе ферредоксина из листьев гороха; 2) выделе-
ние хлоропластов из листьев гороха; 3) определение содержания
хлорофилла в суспензии хлоропластов; 4) определение скорости
восстановления НАДФ+ хлоропластами спектрофотометрическим
методом; 5) расчет фотохимической активности хлоропластов.
1. Выделение и очистка на ДЭАЭ-целлюлозе ферредоксина из
листьев гороха. Процедура выделения ФД из листьев гороха вклю-
чает:
1) подготовку колонки с ДЭАЭ-целлюлозой к работе;
2) выделение ФД из листьев гороха;
3) получение очищенного препарата ФД.
Подготовка колонки с ДЭАЭ-целлюлозой к работе
Для получения хроматографически чистых препаратов ферре-
доксина используют колонки размером 2x10 см, заполненные
ДЭАЭ-целлюлозой, подготовленной специальным образом.
Подготовка ДЭАЭ-целлюлозы. Около 30 см3 порошка ДЭАЭ-цел-
люлозы насыпают в цилиндр емкостью 1 л, заливают дистилли-
рованной водой и оставляют на ночь для набухания. После набу-
хания ДЭАЭ-целлюлозы избыток воды отсасывают пипеткой,
подсоединенной к водоструйному насосу.
ДЭАЭ-целлюлозу заливают 0,5 н. NaOH и оставляют в этом
растворе на 1 ч, периодически осторожно помешивая стеклянной
палочкой. После расслоения отсасывают жидкость водоструйным
насосом. Осадок отмывают дистиллированной водой до pH ис-
пользуемой дистиллированной воды.
176
Далее осадок заливают 0,5 н. НО и оставляют на 1 ч, периоди-
чески осторожно помешивая палочкой. После расслоения отсасы-
вают жидкость водоструйным насосом. Осадок отмывают дистил-
лированной водой до pH используемой дистиллированной воды.
Осадок заливают 0,5 н. NaOH и оставляют на 1 ч, периодичес-
ки осторожно помешивая палочкой. После расслоения отсасыва-
ют жидкость водоструйным насосом. Осадок отмывают дистилли-
рованной водой до pH используемой дистиллированной воды.
Отмучивание ДЭАЭ-целлюлозы от мелких и крупных частиц
производят следующим образом:
• от мелких частиц — путем отстаивания с дистиллированной
водой в течение 30 — 35 мин с последующей деконтацией раствора;
• от крупных частиц — путем отстаивания вновь залитой дис-
тиллированной воды в течение 30 с и последующего слива верх-
него слоя взвеси.
Заполнение колонки ДЭАЭ-целлюлозой. Колонку укрепляют в шта-
тиве строго вертикально и заливают наполовину дистиллирован-
ной водой. Энергичными движениями стеклянного поршня при
закрытом кране колонки освобождают пространство под перфо-
рированным диском колонки от пузырьков воздуха. На перфори-
рованный диск с помощью поршня помещают кружок фильтро-
вальной бумаги, затем вращательными движениями поршень из-
влекают из колонки.
Скорость тока жидкости из колонки устанавливают равной
1 мл/мин. Объем жидкости в колонке перед внесением ДЭАЭ-
целлюлозы должен составлять 1/3 объема колонки. Кран колонки
закрывают и в колонку наливают из стаканчика по палочке тща-
тельно взмученную взвесь адсорбента так, чтобы жидкость стека-
ла по стенке колонки и осаждающийся адсорбент не увлекал в
свою толщу пузырьки воздуха. Суспензии дают осесть, затем при-
мерно через 10 мин открывают кран колонки и продолжают на-
полнять колонку адсорбентом в токе жидкости, постепенно под-
ливая следующие порции адсорбента. Необходимо, чтобы самый
нижний слой адсорбента был ровным, а наполнение колонки рав-
номерным, без воздушных пузырей.
Колонку заполняют на 8 — 9 см при ее диаметре 2 см.
Верхний слой адсорбента должен быть ровным. Если он неров-
ный, около 1 см адсорбента размешивают стеклянной палочкой и
дают ДЭАЭ-целлюлозе отстояться.
Жидкости из колонки дают стечь настолько, чтобы над адсор-
бентом остался слой высотой 4—6 см. После этого колонку за-
крывают резиновой пробкой, в середине которой находится стек-
лянная трубка, соединенная с резервуаром, содержащим исход-
ный буферный раствор (0,005 М трис-HCl; pH 7,8). Нижний ко-
нец трубки, проходящей через пробку, должен быть ниже жидко-
сти, находящейся над адсорбентом.
177
Подготовка колонки к хроматографированию. Перед нанесени-
ем на колонку препарата ферредоксина колонку с ДЭАЭ-целлю-
лозой подготавливают следующим образом:
1) промывают 250 мл 0,5 М NaCl;
2) отмывают от ионов хлора дистиллированной водой (конт-
роль — по реакции с AgNO3);
3) заряжают колонку 0,005 М трис-HCl буфером (pH 7,8), про-
пуская его до тех пор, пока на выходе не получат pH 7,8 (около
250 мл).
Выделение ферредоксина из листьев гороха
Листья 7—10-дневных проростков гороха (около 80 г) измель-
чают в гомогенизаторе с охлажденной дистиллированной водой в
соотношении 1:1. К гомогенату добавляют трис-HCl буфер (pH 7,8)
до конечной концентрации 0,005 М и смесь оставляют на ночь в
холодильнике.
Гомогенат фильтруют через 4 слоя капроновой ткани. К зеле-
ному экстракту медленно, при постоянном перемешивании на
магнитной мешалке добавляют охлажденный в жидком азоте аце-
тон до конечной концентрации ацетона 35 %. Выпавший осадок
отделяют центрифугированием (5 — 7 мин при 1500g).
К полученному супернатанту так же медленно, при постоян-
ном перемешивании на магнитной мешалке добавляют охлажден-
ный ацетон до конечной концентрации 75 %. Осадок отделяют цен-
трифугированием (5 — 7 мин при 1500g).
Полученный осадок белков экстрагируют дважды 0,005М трис-
HCl буфером (pH 7,8). Экстракцию проводят при постоянном пе-
ремешивании на магнитной мешалке. Время каждой экстракции
1,5-2 ч.
Объединенные экстракты оставляют на ночь на диализ против
трис-HCl буфера 0,0005 М концентрации (pH 7,8).
Выпавший после диализа осадок удаляют центрифугировани-
ем при 5000g в течение 10 мин. Полученный супернатант аккурат-
но сливают в пробирку и используют для получения препарата
ферредоксина.
Очистка ферредоксина на колонке с ДЭАЭ-целлюлозой
Раствор, содержащий ферредоксин, наносят на колонку с
ДЭАЭ-целлюлозой, предварительно заряженной трисом и про-
мытой 10-кратным объемом воды. Ферредоксин осаждается на вер-
хней части колонки в виде плотного темно-коричневого кольца.
Для удаления примесей колонку последовательно промывают:
20 мл Н2О;
20 мл 0,02 М трис-HCl буфера (pH 7,3);
178
20 мл 0,2 М NaCl в 0,02 М трис-HCl буфере (pH 7,3);
20 мл 0,25 М NaCl в 0,02 М трис-HCl буфере (pH 7,3).
После этого ферредоксин элюируют 0,35 М NaCl в 0,02 М трис-
HCl буфере (pH 7,3).
Концентрацию ферредоксина рассчитывают, используя удель-
ный коэффициент поглощения 0,732 л/(г- см) при 420 нм.
2. Выделение хлоропластов из листьев гороха проводят, как опи-
сано ранее. Навеска листьев — 2 г. Среда выделения хлоропластов'.
0,35 М NaCl в 0,05 М трис-HCl буфере (pH 7,8). Конечный объем
суспензии — 10 мл.
Таблица 16
Состав реакционных смесей
Вариант Содержание компонента Объем, вносимый в реакционную смесь
Реакционная смесь 1 НАДФ+— 0,5 мМ 0,1 мл
MgCl2- 5мМ 0,1 мл
Ферредоксин — 100 мкг Исходя из концентрации ферредоксина в препарате
Хлоропласты — 50 мкг хлорофилла Исходя из концентрации хлорофилла в суспензии хлоропластов
Трис-HCl буфер (pH 7,8) - 0,05 М До конечного объема реакционной смеси 3 мл
Реакционная смесь 2 НАДФ+— 0,5 мМ 0,1 мл
MgCl2- 5мМ 0,1 мл
Ферредоксин — 100 мкг Исходя из концентрации ферредоксина в препарате
КН2РО4- 5мМ 0,1 мл
АДФ (конечная концентра- ция) — 0,5 мМ 0,1 мл
Хлоропласты — 50 мкг хлорофилла Исходя из концентрации хлорофилла в суспензии хлоропластов
Трис-HCl буфер (pH 7,8) - 0,05 М До конечного объема реакционной смеси 3 мл
179
Т абл ица 17
Определение скорости транспорта электронов в ЭТЦ хлоропластов
по восстановлению НАДФ+
Транспорт электронов Повторность Время, мин Ч' < НАДФН, мкмоль НАДФН, мкмоль/ мг хло- рофилла-ч Среднее значение из 3 повторностей, НАДФН, мкмоль / мг хлорофилла-ч Активность, %
Базальный 100
Сопряженный
3. Определение содержания хлорофилла в суспензии хлороплас-
тов описано на с. 174.
4. Определение скорости восстановления НАДФ+ хлоропласта-
ми спектрофотометрическим методом. Скорость восстановления
НАДФ+ определяют по изменению поглощения реакционной смеси
при 340 нм (максимум поглощения НАДФН). Исследуют два ва-
рианта нециклического транспорта электронов в хлоропластах: ба-
зальный поток электронов (реакционная смесь 7) и поток элект-
ронов, сопряженный с синтезом АТФ (реакционная смесь 2). Со-
став реакционных смесей приведен в табл. 16.
Конечный объем реакционной смеси — 3 мл.
Готовят реакционную смесь непосредственно в кварцевых кю-
ветах, используемых для спектрофотометрических измерений.
Для проведения опыта берут две кварцевые кюветы. В одну кю-
вету (опытную) вносят все компоненты реакционной смеси, во
вторую (контрольную) — трис-HCl буфер и изолированные хло-
ропласты.
Определяют исходное поглощение опытной кюветы (Ло), ис-
пользуя в качестве контроля реакционную смесь, содержащую
трис-HCl буфер и изолированные хлоропласты.
Пробу освещают лампой диапроектора в течение 5 мин непо-
средственно в кварцевой кювете, используемой впоследствии для
определения НАДФН на спектрофотометре. Кювету при этом по-
мещают в водяную термостатируемую баню (температура 12 —15 °C).
Затем определяют поглощение при 340 нм опытной кюветы (At).
Начальную скорость реакции определяют по разнице между
двумя измерениями поглощения (At- Ло). Для расчета используют
молярный коэффициент поглощения НАДФН, равный 6,22-103
л/(моль • см).
5. Расчет активности восстановления НАДФ+ проводят по сле-
дующей формуле:
количество восстановленного НАДФН [мкмоль] = 0,048 (At- Д,)/0,1,
180
где At — поглощение образца при 340 нм после освещения; Ао —
поглощение образца при 340 нм до освещения.
Для расчета скорости транспорта электронов НАДФН, мкмоль/
/(мг хлорофилла-ч), учитывают количество присутствующего в
реакционной смеси хлорофилла и время освещения реакционной
смеси.
Оформление результатов. Результаты проведенной работы офор-
мляют в виде таблицы (табл. 17).
5.2. ОПРЕДЕЛЕНИЕ АКТИВНОСТИ ТРАНСПОРТА
ЭЛЕКТРОНОВ В ЭТЦ ХЛОРОПЛАСТОВ ПО СКОРОСТИ
ВЫДЕЛЕНИЯ КИСЛОРОДА ПОЛЯРОГРАФИЧЕСКИМ
МЕТОДОМ
Нециклический транспорт электронов в ЭТЦ хлоропластов
связан с окислением воды и выделением кислорода. Восстанови-
тельная активность работы ЭТЦ прямо коррелирует с активнос-
тью фотоокисления воды и выделением кислорода в хлоропластах.
Поэтому регистрация выделения кислорода на свету полярогра-
фическим методом является удобным методом оценки активнос-
ти транспорта электронов в ЭТЦ хлоропластов. При определен-
ных условиях в хлоропластах происходит перенос электронов на
кислород (реакция Мелера), и тогда полярографическим методом
также можно наблюдать за поглощением кислорода.
Принцип метода полярографического определения кислорода
изложен в 1.5.
5.2.1. Определение базального, сопряженного
и разобщенного потоков электронов в ЭТЦ фотосинтеза
по активности выделения кислорода полярографическим
методом
Определение фотохимической активности хлоропластов по ско-
рости выделения (или поглощения) кислорода может быть про-
ведено полярографическим методом с помощью платинового за-
крытого электрода Кларка. Конструкция электрода Кларка пока-
зана на рис. 8 (см. с. 41).
Описание установки
На рис. 58 представлена блок-схема полярографической уста-
новки с термостатируемой ячейкой для регистрации кислорода в
181
Рис. 58. Блок-схема полярографической установки с термостатируемой
фотоячейкой:
1 — батарея; 2 — регулятор поляризационного напряжения; 3 — резистор, ими-
тирующий электрод; 4— переключатель («Электрод —Имитация»); 5— электрод
Кларка; 6 — ячейка термостатируемая; 7 — магнитная мешалка; 8 — тепловой
водяной фильтр; 9 — осветитель (диапроектор «Лектор 600»); 10 — микровольт-
наноамперметр Ф-136, регистрирующий показания датчика О2; 11 — регулятор
чувствительности установки 1:10; 12 — переключатель («Работа—Проверка»);
13 — ультратермостат, 14 — усилитель ЛПУ-01; 75 — самописец КСП-4
жидкой среде, собранной на кафедре физиологии растений био-
логического факультета МГУ*.
Порядок работы на установке
1. Проверить заземление.
2. Включить приборы в сеть.
3. Включить усилитель ЛПУ-01, микровольтнаноамперметр
Ф-136, самописец КСП-4.
4. Установить ячейку на мешалку, подключить шланги к ульт-
ратермостату.
5. Присоединить шланги ультратермостата к водопроводной
системе, включить термостат (положение «НО» без нагрева), от-
регулировать температуру (20 °C).
6. Включить мешалку.
7. Подключить электрод Кларка к соответствующим клеммам
установки: Ag, красный провод, «+»; Pt, синий провод, «-». Вклю-
чить блок питания электрода.
8. С помощью рукоятки регулировки напряжения подать напря-
жение 650 мВ на электрод (контролировать по шкале ЛПУ-01), при
этом положение тумблера на ЛПУ-01 и тумблеров на панели регу-
лировки установлены в положения «Проверка» и «Имитация».
* Для полярографических исследований можно воспользоваться полярогра-
фом заводского образца, снабженным самописцем и ячейкой с закрытым плати-
новым электродом Кларка.
182
9. Переключить тумблеры: на ЛПУ-01 в положение «Работа», а
на панели регулировки — в положения «Работа» и «Электрод»,
установить необходимое усиление.
10. Прогреть установку в течение 30 — 40 мин, убедиться в ста-
билизации тока электрода.
11. Заполнить ячейку реакционной смесью.
12. Тумблером «Настройка по буферному раствору» на ЛПУ-01
отрегулировать положение писчика на диаграммной ленте само-
писца.
13. Включить осветитель.
14. Произвести необходимые записи.
15. Выключить тумблеры. Отключить приборы от сети.
Чистка электрода Кларка
Разобранный электрод (т. е. освобожденный от пластмассовых ча-
стей корпуса и защитной пленки) промывают несколько раз дис-
тиллированной водой. После этого платиновый электрод осторож-
но протирают ватой, смоченной азотной кислотой (концентриро-
ванную HNO3 разбавляют водой в отношении 1:1), и многократно
промывают водой. Отмытый от кислоты электрод протирают петро-
лейным эфиром и вновь промывают дистиллированной водой. Пос-
ле этого электрод оставляют на ночь в дистиллированной воде.
При сборке электрода используют новую тефлоновую пленку.
Электрод заполняют электродным буфером, содержащим 50 мМ
Na2B4O2, 50 мМ Na2CO3, 50 мМ КС1.
Ход работы
Работа включает следующие этапы: 1) выделение хлороплас-
тов; 2) определение содержания хлорофилла в суспензии хлоро-
пластов; 3) определение скорости реакции Хилла на полярогра-
фической установке в различных условиях эксперимента; 4) ка-
либровка электрода; 5) расчет активности реакции Хилла.
В работе могут быть использованы следующие физиологичес-
кие варианты: проростки гороха, пшеницы, кукурузы, выращен-
ные в различных условиях водоснабжения, минерального пита-
ния и освещения; растения разного возраста; семядоли тыквы,
обработанные регуляторами роста и развития; свежевыделенные
хлоропласты и хлоропласты, хранящиеся в жидком азоте.
1. Выделение хлоропластов. Все процедуры выделения хлоро-
пластов описаны ранее (с. 154). Навеска листьев — 2 г. Среда выде-
ления: 0,30 М NaCl в 0,06 М фосфатном буфере (pH 6,9). Конеч-
ный объем суспензии — 5 — 10 мл.
2. Определение хлорофилла в суспензии хлоропластов описано
нас. 174.
183
3. Определение скорости реакции Хилла на полярографической
установке. Порядок работы на полярографической установке см.
выше.
Определение активности транспорта электронов в хлороплас-
тах проводят в различных экспериментальных условиях.
Реакционную смесь готовят непосредственно в ячейке. Объем
ячейки на установке, собранной на кафедре физиологии расте-
ний биологического факультета МГУ, — 3,4 мл.
В ячейку вносят:
0,1 мл K3Fe(CN)6 (конечная концентрация в реакционной сме-
си — 3 мМ),
0, 1 мл MgCl2 (конечная концентрация в реакционной смеси —-
1 мМ);
х мл суспензии хлоропластов;
у мл среды выделения.
Объем вносимой суспензии хлоропластов может варьировать
от 0,1 до 2 мл в зависимости от содержания в ней хлорофилла
(Содержание хлорофилла в ячейке должно быть в пределах от 0,05
до 0,1 мг.) Соответственно увеличивается или уменьшается коли-
чество вносимой среды выделения.
После заполнения ячейки плавно, ввинчивающим движением
опускают крышку ячейки, следя за тем, чтобы под крышкой не
осталось пузырька воздуха.
Включают движение диаграммной ленты самописца и в тече-
ние нескольких минут в темноте ведут запись исходного содержа
ния кислорода в реакционной смеси.
После установления прямой линии включают свет (отметить
момент включения света на диаграммной ленте) и регистрируют
изменения содержания кислорода в реакционной смеси в течение
2 — 3 мин. Запись отражает скорость базального потока электронов
в электрон-транспортной цепи фотосинтеза.
Для регистрации сопряженного потока электронов (потока элек-
тронов, связанного с синтезом АТФ в хлоропластах) в ячейку через
отверстие в крышке шприцем вносят 0,1 мл раствора АДФ (конеч-
ная концентрация в ячейке — 1 мМ). На диаграммной ленте отмена
ют момент внесения АДФ и проводят запись в течение 2—3 мин.
Для исследования разобщенного транспорта электронов в ячей-
ку вносят 0,1 мл раствора NH4C1 (конечная концентрация в ячей-
ке 10-3—10~4 М).
При изучении действия ингибиторов в реакционную смесь вводят
0,1 мл диурона (конечная концентрация в ячейке — 10~6—10-4 М)
или гидроксиламина (конечная концентрация в ячейке — 10~3 М).
Все добавки в ячейку делают, не выключая свет и движение
ленты самописца.
4. Калибровка электрода по кислороду. Цель калибровки — ус
тановление цены деления 1 мм (1 см) диаграммной ленты в еди-
184
ницах содержания кислорода и выяснение прямолинейной зави-
симости показаний электрода от концентрации кислорода в среде.
Калибровку можно проводить с использованием либо суспензии
дрожжей (биологическая калибровка по Чансу), либо Na2SO3. В обо-
их случаях обеспечивается обеднение среды кислородом в ячейке.
В первом варианте калибровки 1 г свежих дрожжей суспенди-
руют в водопроводной воде (10—15 мл) и суспензию центрифу-
гируют при 1000g в течение 5 мин. Полученный осадок суспенди-
руют в 1 мл 1 %-й глюкозы, приготовленной на 0,15 М КС1, и
преинкубируют в течение 15 мин. Для калибровки электрода 0,2 мл
суспензии впрыскивают шприцем в ячейку, предварительно за-
полненную 1 %-й глюкозой в 0,15 М КС1, и дожидаются полного
обеднения среды кислородом.
Для калибровки электрода сульфитом натрия используют либо
сухую соль, либо ее раствор (0,1 г Na2SO3 на 100 мл воды). При
использовании сухой соли в ячейку, заполненную дистиллиро-
ванной водой, Na2SO3 вводят небольшими порциями на кончике
скальпеля, дожидаясь всякий раз его полного растворения. В слу-
чае применения раствора сульфита реагент вводят в ячейку с во-
дой шприцем. Удаление кислорода сульфитом натрия происходит
быстро, в течение 5 мин.
Разница между начальным содержанием О2 в среде и конеч-
ным (нулевым) на диаграммной ленте будет соответствовать тому
содержанию кислорода, которое находится в среде при данных
условиях температуры, давления, ионного состава среды. Зная
растворимость О2 при этих условиях, по табл. 18 находят количе-
ство О2 на 1 л дистиллированной воды и рассчитывают его в объеме
ячейки. Основываясь на этих данных и зная отрезок диаграммной
ленты [мм или см], соответствующего данному содержанию О2,
можно рассчитать цену деления диаграммной ленты в мкмолях О2.
Пример расчета. При 20 °C в 1 л дистиллированной Н2О растворяется
9,39 мг О2, тогда в 3,4 мл — х мг О2:
Q 3Q.3 4
шоо 31’926 10‘’мг °2’
что равно 9,977 • 10"4 ммоль или 9,977 • 10"1 мкмоль О2.
Таким образом, в ячейке содержалось 9,977 • 10“’ мкмоль О2.
Если при калибровке отрезок на диаграммной ленте от началь-
ного до конечного (нулевого) уровня кислорода в ячейке равен
300 мм, то 300 мм диаграммной ленты соответствуют 9,977 • 10"1
мкмоль О2. Отсюда 1 мм диаграммной ленты соответствует
(9,977 • 10-7300) мкмоль О2.
5. Расчет активности реакции Хилла. Активность переноса элек-
тронов в ЭТЦ фотосинтеза выражают в мкмолях О2 • мг хлорофил-
ла-1-ч-1.
185
Таблица 18
Зависимость содержания растворенного в дистиллированной воде
кислорода от температуры при атмосферном давлении
1013 гПа и парциальном давлении О2 20,90 %
Температура, °C Содержание растворенного в дистиллированной воде О2, мг/л
18 9,74 9,70* 9,67** 9,63*** 9,60****
19 9,56 9,53 9,49 9,46 9,42
20 9,39 9,36 9,33 9,29 9,26
21 9,23 9,19 9,16 9,13 9,09
22 9,06 9,03 9,00 8,97 8,94
23 8,91 8,88 8,85 8,82 8,79
24 8,76 8,73 8,70 8,68 8,65
25 8,62 8,59 8,56 8,54 8,51
* Значения даны с интервалом в 0,2 °C.
** То же, в 0,4 °C.
*** То же, в 0,6 °C.
**** То же, в 0,8 °C.
Расчет по диаграммной записи (см. рис. 42).
1. Из любой точки экспериментальной линии опускают пер-
пендикуляр а к «нулевой линии» и измеряют [мм]. Его величина
соответствует изменению концентрации О2 в ячейке за время, со-
ответствующее отрезку б. Исходя из калибровки цены деления диа-
граммной ленты в мкмолях О2 (см. выше), определяют эту вели-
чину в мкмолях О2.
2. Измеряют отрезок б [мм] и, исходя из скорости движения
ленты, выражают его в единицах времени [ч].
3. Зная количество кислорода [мкмоль], выделившегося за вре-
мя б, рассчитывают количество О2 за 1 ч.
4. Пересчитывают количество кислорода, выделившегося за 1 ч,
на 1 мг хлорофилла (учитывая количество суспензии хлороплас-
тов, внесенное в ячейку, и рассчитанную концентрацию хлоро-
филла в суспензии).
Оформление результатов. Экспериментальные и расчетные дан-
ные вносят в таблицу, оценивают влияние фосфатакцепторной
системы, разобщителей и ингибиторов транспорта электронов на
фотохимическую активность хлоропластов (скорость базального
потока электронов принимают за 100 %).
186
5.2.2. Анализ транспорта электронов на отдельных участках
ЭТЦ хлоропластов с использованием искусственных
донорно-акцепторных систем и ингибиторов транспорта
электронов
При исследовании регуляторных механизмов и общих кинети-
ческих закономерностей функционирования ЭТЦ хлоропластов
широко используют метод моделирования работы всей цепи или
отдельных ее участков. При этом применяют ряд ингибиторов, ис-
кусственных электрон-донорных и электрон-акцепторных систем.
На рис. 56 дано линейное изображение ЭТЦ хлоропластов с указа-
нием центров действия наиболее часто используемых в экспери-
ментальной работе ингибиторов, а также места включения искусст-
венных электрон-донорных и электрон-акцепторных систем [29, 30].
Ингибиторы транспорта электронов на уровне ФС П:
• NH2OH (гидроксиламин) — отключает выделяющую кис-
лород систему S, прерывает транспорт электронов между S
(хлормарганцевый водоокисляющий кластер) и Z (остаток
тирозина Z, TyrZ);
• DCMU (диурон, 3-(3,4-дихлорфенил)-1,1-диметилмочеви-
на) — нарушает транспорт электронов на уровне первичных
хинонов, блокирует перенос электронов от QA к QB.
Ингибиторы транспорта электронов между двумя фотосисте-
мами'.
• DBMIB (2,5-дибром-3-метил-6-изопропил-п-бензохинон) —
ингибитор фермента пластохинолпластоцианиноксидоредук-
тазы (цитохром ^//-комплекса);
• KCN (цианистый калий) — специфический ингибитор
пластоцианина (PC).
Акцепторы и доноры электронов ЭТЦхлоропластов. Место вклю-
чения того или иного соединения в качестве акцептора или доно-
ра электронов определяется, с одной стороны, его окислитель-
но-восстановительным потенциалом, а с другой стороны, воз-
можностью его контакта с теми или иными переносчиками элек-
тронов ЭТЦ хлоропластов. С. Саха с сотр. (1970, 1971) выделяют
три основных класса акцепторов электронов. Акцепторы класса 1 —
гидрофильные акцепторы, принимающие электроны на внешней
поверхности мембраны. К ним относятся НАДФ+, метилвиологен
(MV), феррицианид калия K3Fe(CN)6. Акцепторы класса 2 прини-
мают электроны на уровне первичных пластохинонов Qa/Qb- К
этому классу относят 2,6-дихлорфенолиндофенол (ДХФИФ). Ак-
цепторы класса 3 — липофильные соединения, способные прони-
кать через мембрану тилакоидов и принимать электроны от пере-
носчиков ФС II, расположенных внутри мембраны или ближе к
поверхности люмена (например, пул пластохинонов). К акцепто-
187
рам этого класса относят фенилендиамин (PD), диметилхинон
(DMQ).
Для стимуляции переноса электронов на кислород {реакция
Мелера) используют метилвиологен, адреналин (сброс на кисло-
род на донорной стороне ФС I), органические кислоты глиокси-
лат, оксалат (стимуляция сброса электронов на кислород в обла-
сти донорного участка ФС II) [1, 2, 11, 28].
Донором электронов для ФС I в условиях ингибирования транс-
порта электронов от ФС II (например, в присутствии диурона)
может служить система ДХФИФ + аскорбат.
Ход работы
Работа включает следующие этапы: 1) выделение хлороплас-
тов; 2) определение содержания хлорофилла в суспензии хлоро-
пластов; 3) определение скорости транспорта электронов (актив-
ности работы ЭТЦ) полярографическим методом по приведен-
ной ниже схеме.
Схема проведения опыта
Нециклический транспорт электронов (ФС I + ФС II)
Акцептор электронов'. K3Fe(CN)g
ЭТЦ\ Н2О -э ФС II -э PQ -э (Цит. b6/f) -» PC -э ФС I ->
-э K3Fe(CN)6.
Транспорт электронов в ФС I
Акцептор электронов', метилвиологен (MV).
Диурон — ингибитор транспорта электронов в ФС II.
ДХФИФ+аскорбат — доноры ё для ФС I.
ЭТЦ Н2О -» ФС II -э PQ -э (Цит. bjf) -> PC -э ФС I ->
? Т
диурон ДХФИФ + аскорбат
-» MV^> О2 -> О;
Z.
Транспорт электронов в ФС II
А. Опыт с ДХФИФ.
Акцептор электронов'. ДХФИФ (1,5 мМ).
Б. Опыт с «-фенилендиамином (PD).
Акцептор электронов', «-фенилендиамин (PD).
ЭТЦ: Н2О -> ФС II -> ДХФИФ (или фенилендиамин).
Реакция Мелера
А. Опыт с адреналином.
ЭТЦ Н2О -> ФС II -> PQ -э (Цит. bjf) -» PC-э ФС I -> О2 ->
—> О2 -э адреналин.
Б. Опыт с глиоксилатом.
ЭТЦ: Н2О -> ФС II -» О2 -> Н2О2 -> глиоксилат.
188
1. Выделение хлоропластов. Хлоропласты из листьев гороха
(2 г) выделяют ранее описанным методом (см с. 154). Среда выде-
ления'. 0,30 М NaCl в 0,06 М фосфатном буфере (pH 6,9). Конеч-
ный объем суспензии 5 — 10 мл.
Для работы можно также использовать хлоропласты, заморо-
женные в жидком азоте. Перед работой из замороженной суспен-
зии отбирают несколько шариков, помещают их в пробирку и
держат при комнатной температуре в темноте до размораживания.
Размороженный хлоропласты хранят в темноте, на льду.
2. Определение концентрации хлорофилла в суспензии хлоро-
пластов. Определение хлорофилла в суспензии свежевыделенных
хлоропластов описано на с. 174.
Для определения концентрации хлорофилла в суспензии замо-
роженных хлоропластов из размороженной суспензии берут опре-
деленный объем (0,05 — 0,1 мл) и доводят его дистиллированной
водой до 1 мл. Затем добавляют 4 мл этилового спирта, хорошо
перемешивают и фильтруют через бумажный фильтр.
Концентрацию хлорофилла в полученном спиртовом экстрак-
те пигментов определяют на спектрофотометре в 1-сантиметро-
вых кюветах, длина волны 652 нм.
Для расчета концентрации хлорофилла [мг/л] используют фор-
мулу Арнона:
са+ь= 1000^-.
аЬ 34,5
Тогда содержание хлорофилла [мг] в пробе (5 мл экстракта)
равно 5 Са+Ь .
1000
Это количество хлорофилла содержится в объеме суспензии
хлоропластов, взятом для определения хлорофилла, например в
0,1 мл суспензии.
3. Определение активности работы ЭТЦ на отдельных ее участ-
ках. Анализ скорости потока электронов в хлоропластах проводят
полярографическим методом с использованием закрытого элект-
рода Кларка. Краткую теорию полярографии см. в 1.5, описание
используемой в работе установки — на с. 181. Калибровку электро-
да Кларка проводят, как описано в 5.2.1.
При определении активности работы ЭТЦ и отдельных ее уча-
стков руководствуются приведенной выше схемой опыта. Для каж-
дого варианта опыта используют соответствующую реакционную
смесь. Реакционная смесь должна содержать необходимую донор-
но-акцепторную систему и другие необходимые добавки, вноси-
мые в расчетных объемах, а также хлоропласты (около 0,05 мг
хлорофилла на 1 ячейку).
189
Таблица 19
Состав реакционных смесей в различных вариантах опыта
Вариант Реактив Мг Концентрация, мМ
ФС I+II K3Fe(CN)6 329 3
ФС I MV 257 0,2
ДХФИФ 266 0,15
Аскорбиновая кислота* 176,13 1
Диурон 233 0,01
ФС II, опытА ДХФИФ 266 1,5
ФС II, опыт Б PD** 108 0,007
Реакция Мелера, опыт А Адреналин — 0,003****
Реакция Мелера, опыт Б Глиоксилат*** 76,05 0,6
* Раствор аскорбиновой кислоты перед опытом нейтрализуют сухой солью
Na2CO3.
** 0,5 мМ PD в 10 мМ НО вносят в 0,05 мл после обязательного внесения в
ячейку 0,05 мл 3,2 мМ K3Fe(CN)6.
*** Свежеприготовленный раствор глиоксилата перед опытом нейтрализуют
сухой солью Na2CO3.
**** Концентрация дана в %.
При расчетах необходимо учитывать объем полярографической
ячейки. Для установки, собранной на кафедре физиологии расте-
ний МГУ, объем ячейки равен 3,4 мл.
Состав реакционной смеси и концентрация используемых в ней
компонентов приведены в табл. 19. Реагенты вносят в среду в объе-
ме 0,1 мл.
Реакционную смесь можно готовить в пробирках, причем для
каждого варианта используется отдельная пробирка, или непос-
редственно в полярографической ячейке.
После внесения донорно-акцепторной системы и необходимых
добавок непосредственно перед определением скорости транспорта
электронов в реакционную смесь добавляют хлоропласты. Конеч-
ный объем реакционной среды доводят до 3,4 мл инкубационной
средой (инкубационная среда равнозначна среде выделения, за
исключением сывороточного альбумина, если он добавлялся в сре-
ду выделения).
190
Реакционную смесь вносят в полярографическую ячейку, ак-
куратно закрывают ее крышкой, чтобы избежать образования пу-
зырька воздуха, и после стабилизации сигнала включают свет.
Проводят запись фотоиндуцируемого выделения (или поглоще-
ния — в опыте с метилвиологеном, адреналином и глиоксила-
том) кислорода в течение 3 — 5 мин.
Оформление результатов. На основании записей, полученных
на полярографической установке, и с учетом результатов калиб-
ровки электрода Кларка производят расчет фотохимической ак-
тивности хлоропластов в различных вариантах опыта. Пример рас-
чета приведен в 5.2.1. Полученные данные вносят в таблицу.
5.2.3. Определение фотохимической активности
субхлоропластных частиц ФС II полярографическим методом
Пигмент-белковые комплексы ФС II, изолируемые из хлоро-
пластов с помощью тритона Х-100 (D. Berthold et al., 1981, с мо-
дификациями), способны проявлять фотохимическую активность,
сопряженную с выделением кислорода (см. 4.4). Кислородвыделя-
ющую активность частиц ФС II определяют полярографическим
методом, используя в качестве акцепторов электронов 2 мМ
K3(FeCN)6* [12, 14], 2 мМ диметилбензохинон (DMBQ) или
0,2 мМ фенил-и-бензохинон с 1 мМ феррицианидом калия [9].
С другой стороны, ФС II способна передавать электроны на
кислород в присутствии реагентов реакции Мел ера [1, 2]. При
этом происходит поглощение О2, что также можно наблюдать
полярографическим методом.
Ввиду этого исследование фотохимической активности изоли-
рованных субхлоропластных частиц ФС II с использованием по-
лярографического метода в различных условиях эксперимента
позволяет получить информацию о функциональной активности
ФС II и исследовать регулирующие ее факторы.
Ход работы
Работа включает этапы: 1) определение скорости фотовыделе-
ния и 2) фотопоглощения кислорода (реакция Мелера) в субхло-
ропластных частицах ФС II полярографическим методом.
Измерения проводят на полярографической установке с исполь-
зованием закрытого электрода Кларка (описание установки и по-
рядок работы на ней см. в 5.2.1). Объем полярографической ячейки,
используемой на кафедре физиологии растений МГУ, — 3,4 мл.
* K3Fe(CN)6 может принимать электроны от ФС II только при значительном
нарушении мембран тилакоидов или при выделении комплекса ФС II из мембраны.
191
В работе используют препараты ФС II, полученные методом,
описанным в 4.4. Перед определением фотохимической активнос-
ти проводят определение содержания хлорофилла в препаратах,
как описано там же.
1. Определение скорости выделения кислорода частицами ФС II.
Для измерения активности выделения кислорода в препаратах
ФС II используют в качестве акцептора электронов K3Fe(CN)6 в
концентрации 2 мМ.
Реакционную смесь готовят в полярографической ячейке. Ко-
нечный объем среды — 3,4 мл. Реакционная смесь включает 2 мМ
K3Fe(CN)6, среду, используемую для суспендирования частиц ФС II
(50 мМ MES или HEPES буфер (pH 6,5), 0,33 М сахароза и 35 мМ
NaCl), суспензию частиц ФС II, эквивалентную 10 — 50 мкг хлоро-
филла. После заполнения ячейки реакционной средой ее аккуратно
закрывают крышкой и производят запись фотоиндуцируемых из-
менений содержания кислорода в ячейке, как описано в 5.2.1.
2. Определение скорости поглощения кислорода частицами ФС II.
Эндогенное поглощение кислорода частицами ФС II регистриру-
ют в присутствии реагентов реакции Мелера. Образование перок-
сида водорода Н2О2 можно фиксировать в присутствии 20 мМ гли-
оксилата, а супероксида анион-радикала — в присутствии 20 мМ
малоната (Р. М.Бекина с сотр., 1975).
В полярографической ячейке готовят реакционную смесь, содер-
жащую 20 мМ глиоксилат и суспензию частиц ФС II, эквивален-
тную 30 — 50 мкг хлорофилла. Конечный объем реакционной сме-
си доводят до 3,4 мл средой, используемой для суспендирования
частиц ФС II (50 мМ MES или HEPES буфер (pH 6,5), 0,33 М
сахароза и 35 мМ NaCl). Ячейку аккуратно закрывают крышкой и
производят запись фотохимической активности частиц ФС II, как
описано в разделе 5.2.1.
В случае малой активности реакции Мелера можно увеличить
содержание хлорофилла в реакционной среде до 100 мкг.
Оформление результатов. На основании произведенных запи-
сей изменений содержания кислорода в реакционной среде на
свету и данных по содержанию хлорофилла в ячейке делают рас-
чет скорости выделения (поглощения) кислорода [мкмоль О2/(мг
хлорофилла • ч)].
Глава 6
ЭНЕРГЕТИКА ФОТОСИНТЕЗА
Энергетическая характеристика процесса фотосинтеза (актив-
ность различных типов фотофосфорилирования и сопряженных с
ним процессов) является важнейшим показателем работы фото-
синтетического аппарата и широко используется при физиологи-
ческих исследованиях.
В настоящее время экспериментально исследуются два основ-
ных типа фотофосфорилирования — нециклическое и цикличес-
кое, сопряженное соответственно с нециклическим или цикли-
ческим потоком электронов в ЭТЦ хлоропластов. Псевдоцикли-
ческое фотофосфорилирование, сопряженное с нециклическим
транспортом электронов на кислород, по-видимому, имеет не-
большой энергетический выход в хлоропластах (около 50 мкмоль
поглощенного фосфора на 1 мг хлорофилла в 1 ч).
Фотофосфорилирование нециклического типа требует возбужде-
ния двух фотосистем и сопряжено с транспортом электронов от
воды к НАДФ+. Конечными акцепторами электронов могут слу-
жить НАДФ+ (физиологический вариант) или феррицианид ка-
лия K3Fe(CN)6 (нефизиологический вариант) [1, 2, 10].
Фотофосфорилирование циклического типа связано с циклическим
потоком электронов, осуществляемым фотосистемой I (ФС I). Цик-
лический поток электронов in vivo происходит благодаря переносу
электронов от железосерных белков акцепторной стороны ФС I на
пул пластохинонов, а с него через цитохромный комплекс и плас-
тоцианин — на окисленный первичный донор электронов ФС I Р^о-
При работе с изолированными хлоропластами в качестве кофакто-
ров циклического фотофосфорилирования могут быть использова-
ны феназинметосульфат (ФМС), пиоцианин, витамины К! и К3
(менадион), флавинмононуклеотид (ФМН), ферредоксин (ФД), 2,6-
дихлорфенолиндофенол (ДХФИФ), цитохромы [1, 2, 9, 13].
Разделение двух типов фотофосфорилирования в опытах с изоли-
рованными хлоропластами основано на использовании специфи-
ческих ингибиторов (моно- и диуроны, дезаспидин, антимицин А
и др.), избирательном возбуждении ФС I дальним красным светом,
введении соответствующих кофакторов, а также применении раз-
личных условий при проведении опыта (аэробные, анаэробные).
7 Гавр иленко
193
Фотосинтетическое фосфорилирование включает ряд сопряжен-
ных процессов, в ходе которых энергия поглощенного кванта света
преобразуется в химическую энергию фосфатных связей АТФ. В со-
ответствии с хемиосмотической теорией П. Митчела [32, 33] энер-
гия поглощенного кванта, первично преобразованная в редокс-
энергию в реакционных центрах, при переносе электронов на
отдельных участках ЭТЦ к более высокопотенциальным компо-
нентам ЭТЦ освобождается и может быть использована для энер-
гозависимого трансмембранного переноса протонов во внутрити-
лакоидное пространство хлоропластов. В результате на мембране
тилакоидов создается градиент электрохимического потенциала,
представляющего сумму осмотической и электрической энергии:
ДЦН* = -2,3 ЛТДрН + FA\g.
При обратном переносе протонов из внутритилакоидного про-
странства в строму через АТФ-синтазный комплекс осуществля-
ется синтез АТФ из АДФ и Фн [5, 6, 21, 36].
Градиент электрохимического потенциала, согласно развивае-
мой в настоящее время ротационной теории П.Бойера [15—17],
служит источником энергии для конформационных изменений бел-
ков АТФ-синтазного (сопрягающего) комплекса CF0— CF{, ито-
гом которых является образование молекулы АТФ из АДФ и Фн в
каталитическом центре сопрягающего фактора 1 (CFj) [11, 20,
25-27, 34].
Таким образом, синтез АТФ является результатом сложной
последовательности реакций, в связи с чем для оценки активно-
сти фотосинтетических процессов в растении могут быть исполь-
зованы различные показатели, сопряженные с фотофосфорили-
рованием и характеризующие отдельные стадии преобразования
энергии света в энергию макроэргических соединений:
1) непосредственное определение количества АТФ, образую-
щегося в реакциях фотофосфорилирования;
2) определение активности фотоиндуцированных изменений
pH, связанных с трансмембранным переносом Н+ при работе элек-
трон-транспортной цепи;
3) выделение сопрягающих белков, изучение их каталитичес-
ких и физико-химических свойств;
4) определение активности Са2+-АТФ-азы в изолированном CF{.
Для характеристики скорости фотофосфорилирования наибо-
лее широко используются методы определения синтеза АТФ по
убыли неорганического фосфора в реакционной смеси и по ско-
рости включения Р32 в АТФ.
Метод определения фотофосфорилирования по убыли неорга-
нического фосфора основан на учете количества неорганического
фосфора в реакционной смеси до и после освещения. Количество
поглощенного на свету Фн служит показателем активности про-
194
цесса фотофосфорилирования в соответствии со следующими урав-
нениями:
циклическое фотофосфорилирование
АДФ + Н3РО4 -----> АТФ+Н2О;
нециклическое фотофосфорилирование
АДФ+ Н3РО4+ НАДФ++ Н2О--------> АТФ + НАДФН + Н++ 1/2 О2.
В экспериментальных условиях активность фотофосфорилиро-
вания определяется в системе, включающей суспензию хлоро-
пластов, кофакторы циклического или нециклического транспорта
электронов, ионы магния, фосфатакцепторную систему (АДФ) и
неорганический фосфат (К2НРО4). Реакционная смесь освещается
в течение определенного периода времени в термостатных усло-
виях и фиксируется добавлением трихлоруксусной кислоты, в
фильтрате контрольной и опытной проб определяется содержа-
ние неорганического фосфора.
Специальные методические работы, проведенные в разные годы
[2], показали, что уровень фосфорилирующей активности препа-
ратов изолированных хлоропластов, выделенных из разных видов
растений, зависит от многих условий проведения опыта. Из них
наибольшее значение имеют перечисленные ниже факторы.
Состав среды выделения. При выделении хлоропластов из не-
которых растений необходимо добавлять стабилизирующие струк-
туру вещества — альбумин, полиэтиленгликоль, а также восста-
навливающие агенты — цистеин, восстановленный глютатион,
аскорбат. Отмеченные выше соединения сохраняют целостность
структур хлоропластов при выделении и их функциональную ак-
тивность.
Осмотическая концентрация растворов, в которых происходят
выделение хлоропластов и реакция фосфорилирования. В условиях
низкой или очень высокой осмотической концентрации хлоро-
пласты не фосфорилируют. Обычно используют следующие кон-
центрации осмотически активных веществ:
в среде выделения — 0,4—0,5 М сахароза, 0,3 М NaCl;
в реакционной смеси — 0,03 М сахароза, 0,015 М NaCl.
Значения pH среды играют решающую роль в определении ак-
тивности фотофосфорилирования. Оптимальным для большинства
растений является pH 7,8 —8,0. Однако при исследовании некото-
рых видов растений использование трицин-буфера с pH 8,8 обес-
печивало в ряде случаев более высокую скорость фотофосфори-
лирования.
Интенсивность и время освещения. Исследования показали, что
интенсивность освещения в 20 000 лк является насыщающей для
реакций фотофосфорилирования со многими кофакторами цик-
лического и нециклического процессов. Фотофосфорилирование,
195
катализируемое ФМС, отличается очень высоким уровнем свето-
вого насыщения (около 200 000 лк), которое в опытах обычно не
достигается. Оптимальное время экспозиции — 5—10 мин. С уве-
личением периода освещения активность фотофосфорилирования
при расчете на единицу времени снижается.
Содержание хлорофилла в пробе. Во многих исследованиях бе-
рется количество суспензии, эквивалентное 0,1 —0,2 мг хлоро-
филла. Дальнейшее увеличение количества хлорофилла нежела-
тельно, так как при расчете на единицу хлорофилла получаются
заниженные .данные.
Температура опыта. При определении скорости фотофосфори-
лирования температура реакционной смеси обычно поддержива-
ется около 18 —20 °C. Это достигается погружением сосудиков с
реакционной смесью в термостатную ванну.
Все процедуры выделения хлоропластов проводят при температу-
ре 0— 4 °C в возможно короткий срок.
6.1. ОПРЕДЕЛЕНИЕ АКТИВНОСТИ ЦИКЛИЧЕСКОГО ФО-
ТОФОСФОРИЛИРОВАНИЯ С ФЕНАЗИНМЕТОСУЛЬФАТОМ
Феназинметосульфат (ФМС) является искусственным кофак-
тором циклического фотофосфорилирования, широко применяе-
мым в современных исследованиях. В данном случае образование
ДрН обеспечивается передвижением через мембрану растворимо-
го в липидах переносчика электронов и протонов. Этот вариант
циклического фотофосфорилирования включает небольшое чис-
ло компонентов ЭТЦ, сопряженных с ФС I [1, 2, 9, 13].
ФМС, восстановленный на наружной стороне мембраны ком-
понентами акцепторного комплекса ФС I и протонированный,
передвигается во внутритилакоидное пространство, где отдает элек-
троны на пластоцианин или непосредственно на Р^, при этом
протоны освобождаются, участвуя в генерации протонного гра-
диента.
Ход работы
Определение фотосинтетического фосфорилирования в препа-
ратах изолированных хлоропластов включает следующие этапы:
1) выделение хлоропластов; 2) определение фосфорилирующей
активности хлоропластов; 3) определение неорганического фос-
фата в реакционной смеси; 4) определение хлорофилла в суспен-
зии хлоропластов; 5) расчет активности фотофосфорилирования.
Работу проводят на 10—14-дневных растениях гороха.
1. Выделение хлоропластов. Навеска листьев гороха 2 г охлажда-
ется в ступке в холодной комнате при температуре 3 — 5 °C в тече-
196
ние 15 — 20 мин. Одновременно охлаждают колбу со средой выделе-
ния, пробирки для центрифугирования, стакан и гомогенизатор.
Навеску листьев тщательно растирают с десятикратным коли-
чеством среды выделения (20 мл), и гомогенат фильтруют в ста-
кан через два слоя марли.
Среда выделения'.
NaCl 0,35 М, трис-HCl буфер 0,05 М (pH 7,8) или
сахароза 0,4 М, трис-HCl буфер 0,05 М (pH 7,8), NaCl 0,01 М.
Для приготовления трис-HCl буфера с определенным значе-
нием pH смешивают 25 мл 0,2 М раствора триса (2,42 г на 100 мл;
Мг 121,14) с х мл 0,1 н. НС1 (см. табл. 8 в приложении 2) и доводят
бидистиллированной водой в мерной колбе до 100 мл. Навеску
соли NaCl растворяют в буферном растворе и доводят до конеч-
ной концентрации 0,35 М.
Из фильтрата хлоропласты выделяют методом дифференциаль-
ного центрифугирования:
1) центрифугирование 3 мин при 100 g — осаждаются крупные
фрагменты клеток, клеточные стенки, ядра; осадок отбрасывают,
из надосад очной жидкости осаждают хлоропласты;
2) центрифугирование 10 мин при 1000 g; надосад очную жид-
кость сливают; осадок хлоропластов промывают один раз средой
выделения;
3) центрифугирование 10 мин при 1000 g; надосадочную жид-
кость сливают; осадок хлоропластов небольшими порциями сре-
ды выделения хлоропластов переносят в гомогенизатор, гомоге-
низируют, переносят в мерную пробирку или цилиндр и объем
суспензии доводят средой выделения до 10 мл.
Суспензию хлоропластов держат в темноте на холоде.
2. Определение фосфорилирующей активности хлоропластов.
Опыт проводят в конических колбах небольшого объема. В каждый
реакционный сосудик наливают по 3 мл реакционной смеси, со-
став которой приведен в табл. 20. Реакционную смесь готовят в
расчете на определенное количество сосудиков. При расчете кон-
центраций исходных растворов учитывают необходимое количе-
ство компонентов в пробе и объем раствора, вносимого в сосу-
дик.
Пример расчета для NaCl: 58,5 г — 1 моль
58,5 мг — 1 ммоль
58,5 мкг — 1 мкмоль
х_______— 60 мкмолей
х= 3,51 мг
В 0,1 мл раствора, вносимого в сосудик,
должно содержаться 3,51 мг NaCl,
в 50 мл исходного раствора — х.
х = 50 -3,51/0,1 = 1,755 г.
197
Таблица 20
Реакционная смесь для определения активности
циклического фотофосфорилирования с ФМС
Компонент Число микромолей в сосудике Количество раствора в сосудике, мл
Трис-HCl буфер (pH 7,8) 45 0,9
NaCl 60 од
MgCl2 10 од
АДФ 10 од
КН2РО4 10 од
ФМС (феназинметосульфат) од ОД
Н2О бидистиллированная — 0,6
Хлоропласты, эквивалентные 80—100 мкг хлорофилла — 1,0
Растворы АДФ, ФМС готовят непосредственно перед опреде-
лением. Опыт проводят в термостатных условиях при температуре
16—18 °C. Для этого пользуются установкой, показанной на рис. 57.
Лампа (500 Вт, 220 В) дает освещенность на уровне сосудиков
20 000 лк (800 Вт/м2). Схема проведения опыта приведена ниже:
Номер сосудика Вариант
1 Свет
2 То же
3 Контроль (темнота)
4 То же
В каждый сосудик приливают по 2 мл реакционной среды и по
1 мл суспензии хлоропластов. Контрольные сосудики ставят в тем-
ноту, а опытные сосудики освещают 5 мин в термостатных усло-
виях (16—18 °C), после чего в каждую колбу быстро добавляют по
1 мл 10 %-й ТХУ и по 1 мл 1 М уксусно-кислого натрия для дове-
дения смеси до pH 4. После этого в контрольные сосудики также
добавляют по 1 мл 10 %-й ТХУ и 1 мл 1 М уксусно-кислого натрия.
Содержимое сосудиков фильтруют в пробирки через беззоль-
ные фильтры или центрифугируют 10 мин при 2000 g. В фильтрате
198
(супернатанте) определяют содержание неорганического фосфо-
ра одним из методов, представленных ниже.
Для определения фосфора из каждого фильтрата берут по две
аналитические пробы.
3. Определение неорганического фосфора в реакционной смеси.
Для определения неорганического фосфора в растворах можно
воспользоваться одним из двух вариантов метода Лоури [30] или
методом Чена [19].
Определение неорганического фосфора методом Лоури по «сине-
му» фосфорномолибденовому комплексу.
В изложенных ниже вариантах метода Лоури о содержании не-
органического фосфора судят по интенсивности синей окраски
восстановленного фосфорномолибденового комплекса. Последний
образуется при строго фиксированном значении pH. Представлен-
ные варианты метода Лоури различаются условиями восстановле-
ния фосфорномолибденового комплекса.
Вариант 1
Реактивы'.
молибденовый раствор — 1 %-й (NH4)2MoO4, приготовленный
на 0,05 н. H2SO4;
восстанавливающая смесь — 1 %-я аскорбиновая кислота, при-
готовленная на 0,001 М CuSO4- 5Н2О;
ацетатный буфер (pH 4) — смешивают равные объемы 1 н. ук-
сусной кислоты и 0,25 М CH3COONa;
уксусно-кислый натрий — 1 М (82 г CH3COONa или 135 г
CH3COONa • ЗН2О растворить и довести объем до 1 л водой).
Ход определения. 0,5 мл фильтрата доводят в пробирке до объема
2 мл бидистиллированной водой (0,5 мл + 1,5 мл воды). Затем в
пробирку опытного раствора прибавляют:
2 мл 1 М CH3COONa;
4 мл ацетатного буфера (pH 4,0);
1 мл 1 %-й аскорбиновой кислоты, приготовленной на 0,001 М
CuSO4-5H2O.
Через 10 мин добавляют 1 мл 1 %-го (NH4)2MoO4, приготовлен-
ного на 0,05 н. H2SO4. Через 10 мин плотность раствора фосфорно-
молибденовой сини измеряют на ФЭКе с красным фильтром (кю-
веты с 1= 0,5 см) или на спектрофотометре при 660 нм (кюветы с
I- 1 см). В качестве контроля применяют воду.
Концентрацию фосфора в растворе определяют по калибро-
вочной кривой.
Вариант 2
Реактивы:
молибденовый раствор — 50 г молибденовокислого аммония —
растворить в серной кислоте (136 мл 98 % H2SO4 и 200 мл Н2О
бидистиллят) и довести водой до метки в литровой колбе;
199
восстанавливающая смесь — 1 г метола, 5 г безводного Na2SO3,
150 г метабисульфита натрия (Na2S2O5) или 175 г метабисульфита
калия — растворить и довести объем до 1 л бидистиллированной
водой;
уксусно-кислый натрий — 164 г CH3COONa или 270 г CH3COONa-
ЗН2О — растворить и довести объем до 1 л водой.
Ход определения. 0,5 мл фильтрата переносят в пробирку, добав-
ляют 1 мл восстанавливающей смеси, затем 1 мл молибденового
раствора. Смесь перемешивают и выдерживают 10 мин, после чего
вносят 2 мл ацетата натрия и бидистиллированную воду до ко-
нечного объема смеси 10 мл (все компоненты вносят только пи-
петками). Раствор в пробирке перемешивают и через 20 — 30 мин
фотометрируют на ФЭКе с красным фильтром (кюветы с 1=0,5 см)
или на спектрофотометре при 660 нм (кюветы с /= 1 см). В каче-
стве контроля применяют воду.
Концентрацию фосфора в растворе определяют по калибро-
вочной кривой.
Определение неорганического фосфора методом Чена.
Данный метод также основан на колориметрическом опреде-
лении восстановленного фосформолибденового комплекса, но
отличается от метода Лоури большей простотой и возможностью
работать с небольшими количествами растворов.
Для определения берут 0,1 мл фильтрата, к которому добавля-
ют 0,2 мл бидистиллированной воды и 0,7 мл реакционной сме-
си, состоящей из 1 части 10 %-й аскорбиновой кислоты и 6 час-
тей 0,42 %-го раствора (NH4)2MoO4, приготовленного на 1 н. H2SO4.
Смесь хорошо перемешивают и инкубируют 20 мин при 45 °C.
Затем пробы охлаждают до комнатной температуры в водяной бане
и спектрофотометрируют при 820 нм. Кюветы с /=0,1 см, конт-
роль — вода.
10 %-й раствор аскорбиновой кислоты и реакционную смесь
готовят непосредственно перед анализом; 0,42 %-й раствор
(NH4)2MoO4 на 1 н. H2SO4 можно приготовить заранее и хранить в
холодильнике.
Концентрацию фосфора в растворе определяют по калибро-
вочной кривой.
Построение калибровочной кривой. Калибровочную кривую стро-
ят, используя серию растворов, содержащих 5—100 мкг фосфора
в пробе. Для калибровки используют раствор, содержащий 50 мкг
фосфора в 1 мл, приготовленный из стандартного раствора со-
ответствующим разбавлением. Стандартный раствор фосфора —
0,25 мг фосфора в 1 мл, приготовленный из перекристаллизован-
ной соли КН2РО4.
При построении калибровочной кривой делают 3 — 4 аналити-
ческие повторности для каждой концентрации фосфора и конт-
роль на реактивы.
200
Измерения оптической плотности растворов проводят на ФЭКе
с красным фильтром или на спектрофотометре. Контролем слу-
жит вода.
4. Определение содержания хлорофилла в суспензии проводят
описанным ранее способом (с. 174).
5. Расчет активности фотофосфорилирования. Показателем
фосфорилирующей активности хлоропластов служит убыль неор-
ганического фосфора в реакционной среде за время инкубации
проб на свету. Поэтому на основании данных фотометрического
анализа и определения фосфора в пробах по калибровочной кри-
вой рассчитывают содержание фосфора в реакционных смесях тем-
новых и световых вариантов. Затем определяют разницу в содер-
жании неорганического фосфора между темновыми и световыми
вариантами. Фосфорилирующую активность изолированных хло-
ропластов выражают в микромолях Фн за 1ч в расчете на 1 мг
хлорофилла.
Оформление работы. Данные фотометрического определения
фосфора в фильтратах, результаты определения концентрации
фосфора по калибровочной кривой, а также расчетные данные по
активности фотофосфорилирования вносят в таблицу.
6.2. ОПРЕДЕЛЕНИЕ АКТИВНОСТИ НЕЦИКЛИЧЕСКОГО
ФОТОФОСФОРИЛИРОВАНИЯ, СОПРЯЖЕННОГО
С ВОССТАНОВЛЕНИЕМ ФЕРРИЦИАНИДА КАЛИЯ
При определении активности фотофосфорилирования нецик-
лического типа в качестве кофактора может быть взят феррициа-
нид калия K3Fe(CN)6. Фотовосстановление феррицианида калия
сопряжено с образованием АТФ. В этом случае обычно учитывают
не только количество поглощенного в опыте неорганического фос-
фата, но также количество восстановленного феррицианида ка-
лия. Отношение этих величин позволяет рассчитать коэффициент
Р/2ё, который дает представление о степени сопряженности тока
электронов в цепи с реакциями фотофосфорилирования [10, 18].
Ход работы
Работа включает следующие этапы: 1) выделение хлоропластов
и проведение опыта; 2) определение феррицианида калия и фос-
фора в реакционной смеси; 3) определение хлорофилла; 4) расчет
фотофосфорилирующей активности хлоропластов.
1. Выделение хлоропластов производят методом дифференци-
ального центрифугирования (см. 4.1.1).
Подробное описание проведения опыта см. в 6.1.
201
Реакционная смесь включает следующие компоненты (в микро-
молях):
трис-HCl буфер (pH 7,8) — 45;
NaCl - 60;
MgCl2 - 12;
КН2РО4 - 12;
АДФ-4;
K3Fe(CN)6 - 3,0.
Используемая суспензия хлоропластов эквивалентна 0,1 мг хло-
рофилла.
Общий объем реакционной смеси — 3 мл.
Реакционную смесь освещают 5 — 10 мин на специальной ос-
ветительной установке (см. рис. 57) при температуре 17—18 °C. Ре-
акцию прекращают выключением света и добавлением в реакци-
онную смесь 1 мл 10 %-й ТХУ и 1 мл 1 М CH3COONa.
Содержимое контрольных и опытных сосудиков центрифуги-
руют 10 мин при 2000 g или фильтруют через бумажные фильтры.
В супернатантах (фильтратах) определяют содержание феррициа-
нида калия и неорганического фосфора.
2. Определение содержания феррицианида калия в реакционной
среде. Количество восстановленного феррицианида измеряют по
разности оптических плотностей при 420 нм в контроле и опыте.
Для расчета используют миллимолярный коэффициент поглоще-
ния е для феррицианида при 420 нм, равный 1,04.
3. Определение неорганического фосфора в реакционной среде.
При высоком содержании феррицианида в реакционной смеси
(3 мкмоля и выше) феррицианид калия должен быть осажден пе-
ред определением фосфора. Осаждение проводится следующим об-
разом: к 2 мл исследуемого раствора добавляют 1 мл аскорбино-
вой кислоты (1,5 мкмоля) и 1 мл хлористого цинка (1,5 Мкмоля).
При этом феррицианид калия, восстановленный аскорбиновой кис-
лотой, полностью осаждается. Осадок отделяют фильтрованием че-
рез беззольный бумажный фильтр или центрифугируют 10 мин при
2000 g. В фильтрате (супернатанте) определяют неорганический фос-
фор одним из описанных выше методов Лоури или Чена (см. 6.1).
4. Определение содержания хлорофилла в суспензии проводят,
как описано на с. 174.
5. Расчет активности нециклического фотофосфорилирования
хлоропластов. Активность нециклического фотофосфорилирова-
ния хлоропластов оценивают по убыли содержания неорганичес-
кого фосфора в реакционной смеси за время инкубации хлороп-
ластов на свету. Количество поглощенного фосфора рассчитывают
в мкмолях/(мг хлорофилла • ч).
На основании данных по активности фотофосфорилирования
и восстановления феррицианида калия на свету определяют от-
ношение Р/2ё.
202
Оформление результатов. Полученные экспериментальные и
расчетные данные вносят в таблицу. Делают выводы о сопряжен-
ности потока электронов и синтеза АТФ в хлоропластах.
6.3. ОПРЕДЕЛЕНИЕ ФОТОИНДУЦИРУЕМЫХ
ИЗМЕНЕНИЙ pH В СУСПЕНЗИИ ХЛОРОПЛАСТОВ
ПОТЕНЦИОМЕТРИЧЕСКИМ МЕТОДОМ
Изолированные хлоропласты при освещении способны осуще-
ствлять процессы поглощения протонов, сопряженные с фотоин -
дуцированным потоком электронов в электрон-транспортной цепи.
Этот процесс сопровождается изменениями pH в наружном ра-
створе. В настоящее время индуцируемое светом поглощение про-
тонов хлоропластами рассматривается в связи с их функциями
запасания энергии при синтезе АТФ. В соответствии с теорией
П. Митчела транспорт электронов в хлоропластах индуцирует пере-
нос протонов через сопрягающую мембрану. В результате возникает
градиент протонов, который является движущей силой для реак-
ций синтеза АТФ, осуществляемых терминальной АТФ-синтези-
рующей (сопрягающей) системой, локализованной в мембране.
Амплитуда фотоиндуцируемого изменения pH в суспензии хло-
ропластов может быть показателем активности ряда сопряжен-
ных с потоком электронов реакций — образования трансмемб-
ранного протонного градиента, реакций синтеза или гидролиза
АТФ и др. В последние годы потенциометрический метод регист-
рации изменений pH в суспензии хлоропластов был разработан
для определения активности циклического фотофосфорилирова-
ния, нециклического фотофосфорилирования, сопряженного с
восстановлением феррицианида калия, активности АТФ-азы, ско-
рости реакции Хилла [4, 35].
Изменения pH реакционной смеси при освещении хлороплас-
тов описываются кривыми, представленными на рис. 59.
В смеси, не содержащей АДФ, фотоиндуцируемые изменения
pH полностью обратимы при выключении света. Добавление АДФ
в реакционную смесь приводит к увеличению амплитуды измене-
ния pH; при выключении света pH раствора снижается, не дости-
гая, однако, исходного уровня.
При освещении изолированных хлоропластов в среде, содер-
жащей фосфатакцепторную систему и кофакторы циклического
фотофосфорилирования, концентрация ионов Н+ в среде изме-
няется в соответствии с уравнением
АДФ3" + НРО42" + иН+ -» АТФ4" + Н2О. (1)
При фосфорилировании АДФ число кислотных групп умень-
шается, в результате чего среда подщелачивается, при этом имеет
203
Рис. 59. Кинетика фотоиндуцируе-
мого изменения pH в суспензии
хлоропластов (объяснение в тексте)
место необратимое поглощение
протонов из среды, сопровож-
дающееся синтезом АТФ.
Общее изменение pH (ДрН)
обусловлено двумя процессами:
1) обратимым в темноте процес-
сом поглощения Н+, который
происходит также в отсутствие
фотофосфорилирования (-АДФ)
и связан с образованием транс-
мембранного протонного гради-
ента и 2) необратимым погло-
щением Н+, сопровождающим
синтез АТФ в присутствии фосфа-
такцепторной системы (+АДФ).
Исследования показали, что
существует определенное стехио-
метрическое соотношение меж-
ду поглощением Н+, потоком
электронов и синтезом АТФ. При
добавлении в реакционную смесь
разобщающих агентов или инги-
биторов фотофосфорилирования
амплитуда фотоиндуцируемых
изменений pH резко уменьшается или изменения pH вообще от-
сутствуют. Следовательно, скорость фосфорилирования можно оце-
нить потенциометрически по изменениям pH в суспензии хлоро-
пластов, которые сопровождают процесс фосфорилирования АДФ.
Потенциометрический метод регистрации изменений pH в среде
может быть использован и для определения активности включае-
мой светом АТФ-азы. При добавлении АТФ к суспензии хлоро-
пластов, предварительно освещенной в течение нескольких ми-
нут, снижается pH вследствие выделения Н+ согласно уравнению
АТФ4" + Н2О -» АДФ3" + НРО42" + иН+. (2)
Активность АТФ-азы определяется по скорости освобождения
Н+, сопровождающего процесс гидролиза АТФ.
Краткая теория потенциометрического метода. Потенциомет-
рический метод определения активности водородных ионов осно-
ван на изменении электродвижущей силы (ЭДС) гальваническо-
го элемента, состоящего из двух электродов — индикаторного элек-
трода, потенциал которого зависит от активности водородных
ионов исследуемого раствора, и электрода сравнения с устойчи-
вым и известным потенциалом.
Электродами сравнения при измерении pH могут служить ка-
ломельный или хлорсеребряный электроды. В качестве индикатор-
204
ного электрода обычно используют стеклянный электрод. Стек-
лянный электрод представляет собой трубку, которая заканчива-
ется тонкостенным полым шариком из специального легкоплав-
кого стекла с высоким содержанием окиси лития или окиси на-
трия. Внутренняя часть стеклянного электрода заполнена буфер-
ным раствором с определенной концентрацией ионов Н+ или ра-
створом НС1 (0,1 н.), в который погружен хлорсеребряный элек-
трод. Мембрана стеклянного электрода проницаема для ионов во-
дорода и способна к обмену ионов натрия или лития на ионы Н+
из раствора. Вследствие этого на внутренней и наружной поверх-
ностях стеклянного шарика устанавливается ионное равновесие,
которое определяет потенциал обеих поверхностей шарика. По-
скольку состав раствора внутри шарика остается постоянным,
потенциал стеклянного электрода является функцией величины
pH исследуемого раствора. При погружении стеклянного электро-
да в раствор между поверхностью стекла и исследуемым раство-
ром возникает разность потенциалов Ех, величина которой опре-
деляется активностью ионов водорода в растворе:
Ех = -1пан = -2,3 —pH, (3)
F F
где R — универсальная газовая постоянная, равная 8,315 107 эрг/
/(°C-моль); Т — температура растворов, К; F= 96500 кулон/г-экв —
число Фарадея; схн — активность ионов водорода в растворе; pH
раствора = -lgcxH.
Система, состоящая из двух электродов, растворы которых со-
общаются между собой, образуют гальванический элемент (рис. 60).
В том случае, когда потенциалы электродов различны, в гальва-
Рис. 60. Гальванический элемент со стеклянным электродом:
А — стеклянный электрод; Б — испытуемый раствор; В — насыщенный раствор
КС1; Г — каломельный электрод; 1 — медная проволока; 2 — стандартный ра-
створ НС1; 3 — ртуть; 4 — серебряный — хлорсеребряный электрод; 5 — стеклян-
ная мембрана; 6 — раствор КС1; 7 — кристаллы КО; 8 — паста Hg2Cl2; 9 — ртуть;
10 — платиновая проволока
205
нической цепи возникает ток ЭДС, равный разности электро-
дных потенциалов. Суммарная ЭДС электродной системы линей-
но зависит от pH раствора. Измеряя ЭДС электронным милли-
вольтметром, определяют pH исследуемого раствора.
Описание установки
Опыт проводят на установке, блок-схема которой приведена
на рис. 61. Установка включает осветительную систему (7), реак-
ционную ячейку со стеклянным электродом и агаровым мости-
ком (2), соединенным с камерой с хлорсеребряным электродом
сравнения (4), ультратермостат (5), рН-метр-316 (5), самописец
КСП-4 (6) и магнитную мешалку.
Порядок работы на установке
1. Проверить заземление.
2. Включить приборы в сеть.
3. Включить тумблеры на приборах (рН-метр, КСП-4).
4. Заполнить ячейку реакционной смесью.
5. Подключить ячейку к ультратермостату, включить магнит-
ную мешалку.
6. Включить самописец и в темноте дождаться стационарного
уровня pH.
7. Включить свет и записать фотоиндуцируемые изменения pH.
8. После окончания работы хорошо промыть электрод водой,
выключить приборы.
Ход работы
Работа включает следующие этапы: 1) выделение хлороплас-
тов; 2) определение фотоиндуцируемых изменений pH в суспен-
Рис. 61. Схема установки для регистрации изменений pH в суспензии хло-
ропластов (объяснение в тексте)
206
зии хлоропластов потенциометрическим методом в отсутствие и
присутствии фосфатакцепторной системы; 3) определение кон-
центрации хлорофилла в суспензии хлоропластов; 4) расчет ак-
тивности фотоиндуцируемых изменений pH в отсутствие и при-
сутствии фосфатакцепторной системы и активности циклическо-
го фотофосфорилирования хлоропластов по фотоиндуцируемым
изменениям pH среды в полной фосфорилирующей среде.
В работе используют 10—14-дневные проростки гороха, пше-
ницы, кукурузы, выращенные в различных условиях водоснабже-
ния, минерального питания, освещения; растения разного возра-
ста.
1. Выделение хлоропластов. Все процедуры выделения хлоро-
пластов ведут при температуре 0 —4 °C.
Используют следующие среды.
Среда выделения 1 (25 мл):
сахароза — 0,4 М;
NaCl - 0,01 М;
трис-HCl буфер (pH 7,8-8,0) - 0,025 М.
Среда выделения 2 (100 мл):
сахароза — 0,4 М;
NaCl - 0,01 М.
pH среды 2 устанавливают около 7,8 —8,0, используя концент-
рированную щелочь NaOH.
Получение гомогената. Навеску листьев 2 г (предварительно
охлажденную в течение 10—15 мин в полиэтиленовом пакете и
влажной фильтровальной бумаге в холодильнике или на льду)
размельчают в фарфоровой ступке в течение 1 — 2 мин с 15 мл
среды выделения 1.
Получение суспензии хлоропластов методом дифференциально-
го центрифугирования. Гомогенат фильтруют через капроновую
ткань и переносят в центрифужную пробирку. Фильтрат в центри-
фужной пробирке уравновешивают с другой центрифужной про-
биркой водой. Проводят первое центрифугирование при 100 g в те-
чение 5 мин. Осадок, содержащий обрывки тканей, ядра, разру-
шенные клеточные стенки и другие фрагменты, отбрасывают. Су-
пернатант центрифугируют 10 мин при 1000g (второе центрифу-
гирование). Полученный осадок (содержит хлоропласты) с целью
промывания хлоропластов гомогенизируют кисточкой в центри-
фужной пробирке с 15 мл незабуференной среды выделения 2. Про-
водят третье центрифугирование в течение 10 мин при 1000 g. Су-
пернатант отбрасывают, а осадок перемешивают с 2 — 3 мл среды
выделения 2. После перемешивания со средой выделения 2 его пере-
носят в тефлоновый гомогенизатор, гомогенизируют до получе-
ния однородной смеси, а затем переносят в мерный цилиндр. Цен-
трифужную пробирку и гомогенизатор споласкивают небольши-
ми порциями среды выделения 2 для полного перенесения хлоро-
207
пластов в цилиндр. Конечный объем суспензии в цилиндре — 12 —
15 мл.
2. Определение фотоиндуцируемых изменений pH в суспензии
хлоропластов. Определение проводят потенциометрическим ме-
тодом в отсутствие и присутствии АДФ.
Схема опыта'.
-АДФ'. регистрация фотоиндуцируемых изменений pH хлороп-
ластами в среде, не содержащей АДФ. Опыт проводят при варьи-
ровании pH реакционной смеси в интервале pH 6,0 —7,0.
+АДФ'. регистрация фотоиндуцируемых изменений pH хлороп-
ластами в среде, содержащей АДФ, при активации циклического
фотофосфорилирования с ФМС. Опыт проводят при pH реакци-
онной смеси 8,0 —8,2.
Состав реакционных смесей приведен в табл. 21.
Таблица 21
Состав реакционных смесей
Вариант Компонент Конечная концентра- ция в среде, мМ Объем, вносимый в ячейку, мл
-АДФ MgCl2 1 0,1
NaCl 5 0,1
КН2РО4 1 0,1
ФМС 0,02 0,18
Н2О (pH 8,0)* — 7,52
Хлоропласты — 1
+АДФ MgCl2 1 0,1
NaCl 5 о,1
КН2РО4 1 0,1
АДФ (pH 7) 0,5 0,1
ФМС 0,02 0,18
Н2О (pH 8,0)* — 7,42
Хлоропласты —— 1
* pH воды доводят до нужного значения, используя концентрированный
раствор щелочи NaOH.
208
Реакционную смесь готовят непосредственно в ячейке. Конеч-
ный объем реакционной смеси в ячейке — 9 мл. Необходимое
значение pH реакционной смеси достигается путем добавления
небольших объемов 1 мМ НС1 или NaOH.
Хлоропласты и компоненты реакционной смеси обладают оп-
ределенной буферной емкостью. Поэтому для расчета фотоинду-
цируемых изменений концентрации протонов в опыте необходи-
мо предварительно определить, на сколько единиц изменяется
pH среды при добавлении в реакционную смесь известного коли-
чества протонов (ДН+/ДрН), а также цену деления диаграммной
ленты в единицах содержания протонов. Для определения буфер-
ной емкости в реакционную смесь после проведения каждого оп-
ределения микропипеткой добавляют определенное количество
(0,1 —0,2 мл) НС1 известной концентрации (10-4—10-3 М). Амп-
литуду изменений pH, соответствующую добавленному количе-
ству протонов, отмечают на pH-метре и диаграммной ленте.
3. Определение концентрации хлорофилла в суспензии хлоро-
пластов проводят, как описано ранее (с. 174).
4. Расчет активности реакций. Для определения величины об-
ратимого фотоиндуцированного процесса поглощения протонов
хлоропластами достаточно знать амплитуду изменений pH, цену
деления диаграммной ленты в значениях содержания протонов и
количество хлорофилла в опытной смеси. На основании этих ве-
личин рассчитывают число микромолей Н+, поглощенных в опы-
те, в расчете на 1 мг хлорофилла.
Активность циклического фотофосфорилирования определяют
по изменению pH при освещении суспензии изолированных хло-
ропластов в присутствии АДФ. Для расчета АТФ необходимо знать
амплитуду необратимого изменения pH, цену деления диаграмм-
ной ленты в единицах содержания протонов, время установления
стационарного уровня pH, содержание хлорофилла в пробе и зна-
чения коэффициента п в уравнении (1), где п — стехиометричес-
кое отношение числа молей поглощенных протонов к числу мо-
лей поглощенного неорганического фосфата (Фн) при фосфори-
лировании АДФ в условиях данного pH:
Значения п рассчитаны теоретически и определены эксперимен-
тально в работе М. Нишимура и др. (М. Nishimura et al., 1962): п=0,89
при pH 7,4 и п = 0,95 при pH 8,0. Фосфорилирующую активность
хлоропластов выражают в мкмолях Фн- мг хлорофилла-1 ч-1.
Оформление результатов. Полученные результаты вносят в таб-
лицу. Отмечают влияние фосфатакцепторной системы на уровень
и характер фотоиндуцируемых изменений pH.
209
6.4. РАЗОБЩЕНИЕ И РЕКОНСТРУКЦИЯ
ФОСФОРИЛИРУЮЩЕЙ СИСТЕМЫ ХЛОРОПЛАСТОВ
Синтез АТФ в хлоропластах осуществляет АТФ-синтазный ком-
плекс, включающий сопрягающий фактор О (CF0) и сопрягаю-
щий фактор 1 (CFJ. Оба белка имеют сложную четвертичную струк-
туру (рис. 62) [11, 14, 18, 23, 29, 38, 39]. CF0 — интегральная часть
АТФ-синтазы; состоит из четырех типов субъединиц — 1Ъ
IVi (индексы — количество субъединиц). При участии этих
субъединиц происходит транслокация протонов через мембрану,
сопряженная с вращением олигомера, состоящего из 12 субъеди-
ниц III, и изменением конформации белков сопрягающего комп-
лекса. CF} — водорастворимый белковый комплекс; расположен
на поверхности мембраны и обращен в стромальное простран-
Рис. 62. Модель структурно-функциональной организации сопрягающего
комплекса CF0 CFi (по [18], с изменениями)
CFq — интегральная мембранная часть комплекса (осуществляет перенос протонов
из люмена в строму); I, II, III, IV — субъединицы CF0; CFX — внешняя часть комп-
лекса (осуществляет синтез АТФ из АДФ и Фн); а, 0, у, 8, е — субъединицы CF\
210
ство, состоит из пяти типов субъединиц — а3, 03, Yb Ei и
непосредственно участвует в образовании АТФ.
Каталитические центры расположены на трех парах а-р-субъе-
диниц; у- и Е-субъединицы образуют «ствол» CF{ и необходимы
для энергетического сопряжения процессов транслокации прото-
нов и работы каталитических центров.
При вращении у- и Е-субъединиц в ходе катализа происходит
передача конформационных изменений на пары ос-(3-субъединиц,
в результате которых в каталитических центрах синтезируется АТФ.
В соответствии с ротационной теорией П. Бойера для нормально-
го функционирования АТФ-синтазы необходимо прочное сопря-
жение процессов транслокации Н+, вращательного движения у- и
Е-субъединиц и взаимодействия их с (ос(3)3-гексамером.
Связь CF{ с CFq зависит от гидрофильных и гидрофобных вза-
имодействий и требует участия ионов двухвалентных металлов —
Са2+ или Mg2+. Обработка хлоропластов растворами пирофосфата,
NaBr или ЭДТА нарушает связь между двумя компонентами со-
прягающего комплекса, в результате чего можно получить изоли-
рованный CF{. Выделение CFq происходит лишь при нарушении
структуры мембран тилакоидов детергентами или обработкой уль-
тразвуком. Обработка хлоропластов растворами ЭДТА приводит к
разобщению потока электронов в ЭТЦ и синтеза АТФ. Разобщен-
ные хлоропласты характеризуются меньшей фосфорилирующей
активностью по сравнению с исходными хлоропластами.
В определенных условиях, при добавлении изолированных пре-
паратов CFX к истощенным по CF\ мембранам, возможна рекон-
струкция системы фосфорилирования с частичным восстановле-
нием процессов образования АТФ.
Метод разобщения и реконструкции хлоропластов используют
при изучении механизмов взаимодействия отдельных компонен-
тов сопрягающего комплекса и общих механизмов регуляции фо-
тофосфорилирования, при специальных исследованиях структур-
ных и физико-химических свойств изолированного CFX в связи с
различным уровнем его каталитической активности и ряда других
вопросов механизма фотофосфорилирования [11, 14, 24, 28, 29,
37-39].
Ход работы
Работа включает следующие этапы: 1) выделение хлороплас-
тов из листьев гороха (или пшеницы) и определение их исходной
фосфорилирующей активности; 2) экстракция CFX из хлороплас-
тов путем обработки их раствором ЭДТА и определение остаточ-
ной фосфорилирующей активности мембран хлоропластов; 3) ре-
конструкция фосфорилирующей системы. Схема проведения опыта
приведена на рис. 63. На ней представлены последовательные эта-
211
Рис. 63. Схема опыта по разобщению и реконструкции фосфорилирующей системы хлоропластов
ю
пы разобщения и реконструкции сопрягающего комплекса и оп-
ределение активности фосфорилирования исходных препаратов
хлоропластов, а также после экстракции CFX и его обратного при-
соединения к мембранам. Схема демонстрирует возможность пред-
ставления хода выполнения задачи в виде отдельных блоков, что
облегчает проведение эксперимента.
Фосфорилирующую активность хлоропластов оценивают по
скорости циклического фотофосфорилирования с ФМС. Опре-
деление уровня фотофосфорилирования исходных, разобщенных
и реконструированных систем осуществляют по убыли неорга-
нического фосфора в реакционной смеси на свету. Определение
фосфора проводят методом Лоури (30) или Чена (19). Описа-
ние определения фосфорилирующей активности хлоропластов
см. в 6.1.
Определение содержания хлорофилла в суспензии хлоропластов
описано на с. 174.
Для проведения опыта с горохом используют следующие сре-
ды (в скобках указаны модификации для работы с хлоропластами
пшеницы).
• Среда Г.
0,05 М трис-HCl буфер (pH 7,8);
0,35 М NaCl.
• Среда 2\
0,05 М трис-HCl буфер (pH 7,8) (0,025 М трис-HCl буфер,
pH 8,0);
0,01 М NaCl;
0,75 мМ ЭДТА (0,15 мМ ЭДТА).
1. Выделение хлоропластов и определение их исходной фосфо-
рилирующей активности. Выделение хлоропластов производят опи-
санным ранее методом (см. 6.1) при температуре +4 °C. Охлажден-
ную навеску листьев гороха (5 г) измельчают в гомогенизаторе с
10-кратным (по массе) количеством среды 1. Гомогенат фильтру-
ют через капроновую ткань и фильтрат центрифугируют 5 мин
при 100g. Полученный супернатант центрифугируют при 1000g в
течение 10 мин.
Осажденные хлоропласты промывают 15 мл среды 1, центри-
фугируют при 1000 g в течение 10 мин. Полученный осадок хло-
ропластов суспендируют в 20 мл среды 1 (концентрация хлоро-
филла 0,05 — 0,1 мг/мл среды) с использованием тефлонового го-
могенизатора. Суспензию хлоропластов помещают на ледяную баню
и часть ее (5 мл) используют для определения исходной фосфо-
рилирующей активности хлоропластов и содержания хлорофилла
в суспензии. Оставшуюся суспензию хлоропластов (15 мл) исполь-
зуют для экстракции CFX.
2. Экстракция CFA из хлоропластов и определение остаточной
фосфорилирующей активности мембран. Суспензию хлороплас-
214
тов переносят в центрифужную пробирку и центрифугируют при
1000 g. Супернатант отбрасывают, а осадок хлоропластов обраба-
тывают 15 мл среды 2 в целях экстракции CF{.
Экстракцию CFt проводят в течение 15 мин на холоде, перио-
дически помешивая кисточкой. Затем проводят центрифугирова-
ние при 16 000g в течение 20 мин. Полученный супернатант, со-
держащий СД, сливают в пробирку, хранят на холоде и исполь-
зуют при последующей реконструкции сопрягающей системы.
Осадок мембран хлоропластов суспендируют в среде 1 (15 мл),
помещают на лед и используют для определения остаточной фос-
форилирующей активности хлоропластов и содержания хлорофил-
ла в суспензии.
Оставшуюся суспензию (10 мл) переносят в центрифужную
пробирку, центрифугируют при 2000 g и используют в опыте по
реконструкции сопрягающей системы.
3. Реконструкция фосфорилирующей системы. Осажденные хло-
ропласты суспендируют с содержащим CF{ ЭДТА-экстрактом хло-
ропластов. В ЭДТА-экстракт предварительно добавляют MgCl2flO
конечной концентрации 0,1 М (MgCl2 способствует процессу ре-
конструкции). Реконструкцию проводят при температуре 0 — 4 °C
в течение 20 мин. После этого проводят центрифугирование при
2000g в течение 20 мин. Полученный осадок хлоропластов суспен-
дируют в 10 мл среды 1 и используют для определения фосфори-
лирующей активности реконструированных хлоропластов и со-
держания хлорофилла в суспензии.
Оформление результатов. Полученные в ходе работы результа-
ты заносят в таблицу, где указывают фосфорилирующую актив-
ность исходных, обработанных ЭДТА и реконструированных хло-
ропластов [мкмоль Фн/(мг хлорофилла • ч)]. Степень разобщения и
реконструкции фосфорилирующей системы хлоропластов оцени-
вают в процентном выражении (за 100 % принимают исходную
фосфорилирующую активность хлоропластов).
6.5. ОПРЕДЕЛЕНИЕ АТФ-азной АКТИВНОСТИ
СОПРЯГАЮЩИХ БЕЛКОВ ХЛОРОПЛАСТОВ
Уровень синтеза АТФ в хлоропластах определяется каталити-
ческой активностью связанного с мембраной CF{. Вместе с тем
мембраносвязанный CFX проявляет свет- и SH-активируемую Mg2+-
АТФ-азную активность. Благодаря этому осуществляется гидро-
лиз АТФ с образованием АДФ и неорганического фосфора. АТФ-
азная активность комплекса CFq— CFx (гидролиз АТФ) связана с
использованием энергии АТФ для переноса протонов из стромы
хлоропластов во внутритилакоидное пространство с образовани-
ем протонного градиента. Физиологический смысл этой реакции
215
состоит в необходимости поддерживать концентрацию Н+ во внут-
ритилакоидном пространстве на определенном уровне, ниже ко-
торого система синтеза АТФ выключается.
Изолированный CF{ теряет способность синтезировать АТФ,
но сохраняет АТФ-азную активность, которая может быть выяв-
лена при нагревании препарата до 65 °C в присутствии ионов Са2+
и сульфгидрильных соединений или при обработке трипсином.
АТФ-азная активность сопрягающего белка хлоропластов, как
правило, находится в латентном состоянии и активируется SH-
соединениями, ионами Mg2+ или Са2+, нагреванием или обра-
боткой трипсином. Сопрягающий фактор 1, связанный с мемб-
раной, проявляет \^2+-АТФ-азную активность, в то время как
АТФ-азная активность изолированного CF\ проявляется в при-
сутствии ионов Са2+. Нагревание изолированного сопрягающего
фактора 1 до 65 °C в присутствии ионов Са2+ значительно повы-
шает его АТФ-азную активность, причем в присутствии сульф-
гидрильных соединений активация идет более энергично [8, 22,
24, 31, 37, 40].
Активность Са2+-АТФ-азы обычно прямо коррелирует с актив-
ностью АТФ-синтазной реакции и может служить одним из пока-
зателей каталитической активности сопрягающего фактора 1.
6.5.1. Определение активности светактивируемой
Mg2+-зависимой АТФ-азы хлоропластов
Для проявления АТФ-азной активности связанного с мембра-
ной сопрягающего комплекса необходимы определенные условия.
АТФ-аза активируется светом, требует присутствия сульфгидриль-
ных соединений и ионов Mg . Светактивируемую Mg -зависи-
мую АТФ-азную активность сопрягающего комплекса хлороплас-
тов определяют по увеличению неорганического фосфора в среде
инкубации.
Ход работы
Работа включает следующие этапы: 1) выделение хлоропластов;
2) определение хлорофилла в суспензии; 3) активация АТФ-азы
светом и определение АТФ-азной активности сопрягающего ком-
плекса.
1. Выделение хлоропластов из листьев гороха производят опи-
санным ранее методом (с. 154). Навеска листьев — 2 г, конечный
объем суспензии хлоропластов — 10 мл.
Среда выделения'. 0,35 М NaCl в 0,05 М трис-HCl буфере (pH 7,8).
2. Определение хлорофилла в суспензии проводят, как описано
ранее (с. 174).
216
3. Активация светактивируемой М£2+-зависимой АТФ-азы хло-
ропластов и определение ее активности. В работе используют сле-
дующие среды.
Инкубационная среда (15 мл):
трис-HCl (pH 8,0) - 25 мМ;
NaCl - 50 мМ;
MgCl2 - 50 мМ;
пиоцианин — 0,05 мМ;
дитиотрейтол (ДТТ) — 5,0 мМ.
Реакционная среда (15 мл):
трис-HCl (pH 8,0) - 25 мМ;
MgCl2 — 50 мМ;
АТФ — 5 мМ.
Для световой активации АТФ-азы 1 мл суспензии хлороплас-
тов (0,1 —0,2 мг хлорофилла) добавляют в сосудик с 1 мл инкуба-
ционной среды. Пробу (опытную) освещают 2 мин на осветитель-
ной установке, показанной на рис. 57, при температуре 19 — 20 °C
и освещенности 20 000 лк. Параллельно готовят аналогичную кон-
трольную (темновую) пробу, которую, в отличие от опытной (све-
товой), выдерживают 2 мин в темноте при той же температуре.
Сразу же после выключения света в опытную, а затем и в кон-
трольную пробы вносят по 1,5 мл реакционной среды. Смесь пере-
мешивают и помещают на 15 мин в термостат с температурой
37 °C. Реакцию останавливают 1 мл 6 %-й ТХУ.
Контрольную и опытную пробы центрифугируют 5 мин при
2000 g. После отделения осадка в супернатанте определяют неорга-
нический фосфор методом Лоури или Чена (см. 6.1).
Опыт проводят в 3-кратной повторности.
Активность свет- и SH-активируемой А^2+-АТФ-азы хлоро-
пластов рассчитывают по разнице в содержании неорганического
фосфора между световым и темновым вариантами и выражают в
микромолях Фн, освобожденного из АТФ, за 1 ч 1 мг хлорофилла.
Оформление результатов. Результаты фотометрического изме-
рения неорганического фосфора в реакционных смесях, а также
данные, полученные по калибровочной кривой и расчетным пу-
тем, вносят в таблицу.
6.5.2. Выделение сопрягающего фактора 1 из хлоропластов
и определение его Са2+-АТФ-азной активности
Изолированный CFX обладает латентной АТФ-азной активнос-
тью, для выявления которой требуется обработка белка трипси-
ном или нагреванием при температуре 60 — 65 °C. АТФ-азная ак-
тивность чувствительна к ионам Са2+ и сульфгидрильным соеди-
нениям [8, 40].
217
Ход работы
Работа включает следующие этапы: 1) выделение хлороплас-
тов из листьев гороха (или пшеницы); 2) экстракция CF\ из хло-
ропластов раствором ЭДТА и частичная его очистка путем пере-
осаждения с сульфатом аммония; 3) определение Са2+-АТФ-аз-
ной активности изолированного CF\ при активации ее нагрева-
нием; 4) электрофоретический анализ CF\ хлоропластов.
1. Выделение хлоропластов из листьев гороха проводят описан-
ным ранее методом (с. 154) с использованием электрического го-
могенизатора. Навеску листьев (20 — 25 г) измельчают в гомогени-
заторе со средой выделения, содержащей 0,05 М трис-HCl буфер
(pH 7,8) и 0,35 М NaCl (весовое соотношение навески листьев и
среды выделения Г. 5). Гомогенат фильтруют через капроновую
ткань, фильтрат центрифугируют 5 мин при 100 g. Полученный
супернатант центрифугируют в течение 10 мин при 1000 g. Осадок
хлоропластов суспендируют в 0,01 М NaCl (необходимый для горо-
ха pH 7,8 устанавливают непосредственно перед опытом, используя
концентрированный раствор щелочи NaOH). Суспензию центрифу-
гируют при 16000 g в течение 20 мин.
2. Экстракция сопрягающего белка из хлоропластов раствором
ЭДТА и частичная его очистка путем переосаждения с сульфатом
аммония.
Для экстракции CFj осадок хлоропластов обрабатывают 15 мл
0,75 мМ ЭДТА в 0,01 М NaCl, pH 7,8 (в случае хлоропластов
пшеницы 0,15 мМ ЭДТА в 0,01 М NaCl, pH 8,0). Необходимое
значение pH устанавливают, используя концентрированный ра-
створ NaOH. Экстракцию проводят в течение 15 мин при ком-
натной температуре, периодически помешивая кисточкой. Затем
проводят центрифугирование при 16 000g в течение 20 мин. По-
лученный супернатант сливают в пробирку, а осадок повторно
обрабатывают раствором ЭДТА. Полученный после второй об-
работки ЭДТА супернатант объединяют с первым и использу-
ют для выделения CF\ путем переосаждения с сульфатом аммо-
ния.
Переосаждение (высаливание) сопрягающего белка проводят на
магнитной мешалке. К экстракту ЭДТА добавляют сернокислый
аммоний до конечной концентрации 65 % насыщения. После это-
го осажденный сопрягающий белок отделяют путем центрифуги-
рования при 16000g в течение 20 мин. Полученный осадок ра-
створяют в 0,05 М трис-HCl буфере (pH 7,8) и ставят на диализ
против того же трис-HCl буфера, разведенного в 10 раз.
Препарат CFb полученный после диализа, если необходимо, упа-
ривают при температуре 0—4 °C в потоке воздуха (применяют ФЭН
или вентилятор) и используют для определения Са2+-АТФ-азной
активности и белка методом Лоури.
218
3. Определение Са2+-АТФ-азной активности изолированного со-
прягающего фактора 1 при активации ее нагреванием. Для иссле-
дования Са2+-АТФ-азной активности CF{ вначале необходимо ак-
тивировать его латентную АТФ-азную активность путем нагрева-
ния препарата при 60 — 65 °C, а затем определить активность фер-
мента при 37 °C.
Для проведения опыта берут 2 пробирки {контроль и опыт).
Активация Са2+ -ЛТФ-азной активности CFj. Для активации
Са2+-АТФ-азной активности изолированного CF} препараты тер-
мостатируют при 60 °C в течение 3 мин в среде активации, содер-
жащей:
10 мМ АТФ;
10 мМ СаС12;
25 мМ трис-HCl буфер (pH 7,8);
5 мМ дитиотрейтол (ДТТ).
Раствор сопрягающего белка в 0,05 М трис-HCl буфере (pH 7,8)
добавляют в максимально возможном объеме.
Общий объем среды активации — 1 мл.
Определение Са -ЛТФ-азной активности CF}. Для исследова-
ния Са2+-АТФ-азной активности образцы после нагревания пере-
носят в водяную баню с температурой 37 °C и быстро охлаждают
путем добавления равного объема (1 мл) ледяного 25 мМ трис-
HCl буфера (pH 7,8). В контрольный образец сразу после этого до-
бавляют 1 мл 8 %-й ТХУ, а опытный образец инкубируют при 37 °C
10 мин. После инкубации реакцию в опытном образце останавли-
вают добавлением 1 мл 8 %-й ТХУ. После добавления ТХУ образ-
цы фильтруют через двойной бумажный фильтр или центрифуги-
руют в течение 10 мин при 2000 g. В полученном фильтрате (супер-
натанте) определяют содержание неорганического фосфора, об-
разовавшегося в результате расщепления АТФ.
Определение неорганического фосфора. Определение произво-
дят одним из описанных ранее способов (см. 6.1) в двукратной
повторности. Ферментативную активность сопрягающего белка
определяют по разнице в содержании фосфора между опытными
и контрольными образцами. Расчет ферментативной активности
производят с учетом содержания белка в пробе и выражают в
микромолях Фн/(мг белка • ч).
Определение содержания белка в пробе методом Лоури. Для
определения белка используют:
реактив А — 10 г Na2CO3 на 100 мл дистиллированной воды;
реактив В — 0,5 % CuSO4- 5Н2О на 100 мл 1 %-го K-Na-вин-
но-кислого;
реактив С — 10 частей реактива Л на 1 часть реактива В;
реактив Д — 1 часть реактива Фолина на 10 частей воды;
3 М ТХУ;
0,5 н. NaOH.
219
К 1 мл раствора белка добавляют 0,15 мл 3 М ТХУ, хорошо
перемешивают. Через 2 мин пробы центрифугируют 10 мин при
9000g. К полученному осадку добавляют 1 мл 0,5 н. NaOH и при
встряхивании растворяют осадок в течение 10 мин. Затем добавля-
ют 1 мл реактива Си выдерживают 10 мин. После этого добавля-
ют 3 мл реактива Д и выдерживают 30 мин. Пробы измеряют на
спектрофотометре при 720 нм, используя в качестве контроля
дистиллированную воду.
Для определения содержания белка в пробе используют калиб-
ровочную кривую, построенную в интервале 0 — 250 мкг белка/мл
раствора для сывороточного альбумина.
4. Электрофоретический анализ сопрягающего фактора 1 хло-
ропластов. Электрофоретический анализ сопрягающего белка про-
водят с использованием полиакриламидного геля (ПААГ). Метод
описан в 1.3.
Реактивы для приготовления ПААГ:
реактив Г.
1 н. НС1 - 48 мл;
wpuc-оксиметиламинометан — 36,6 г;
Н2О — до 100 мл;
реактив 2\
акриламид — 30 г;
М,М'-метилен-6ис-акриламид — 0,368 г;
Н2О — до 50 мл;
реактив 3:
(NH4)2S2O8 - 21 мг;
Н2О — 15 мл.
Приготовление ПААГ и электродного буфера показано в табл. 22.
Анализ одной пробы белка проводят в 4 повторностях (исполь-
зуют 4 трубочки ПААГ): две повторности — для окраски на белок
и две повторности — для окраски на Са2+-АТФ-азу. В трубочку
заливают по 2,0 мл смеси геля*.
Количество наносимого на гель белка в пробе — около 100 мкг,
объем, наносимый на гель пробы, — не более 0,2 мл. Электрофо-
рез ведут при силе тока 4 мА на трубочку, напряжении 400 — 600 В.
Для окрашивания гелей на белок используют 0,05 %-й раствор
кумаси в 12,5 %-й ТХУ. Окраску проводят 4 ч, после чего гели
отмывают смесью этиловый спирт: уксусная кислота: вода в соот-
ношении 5:1:1 (по объему).
Выявление сопрягающего белка на гелях осуществляют по его
АТФ-азной активности [28]. Гели инкубируют 2—4 ч при 37 °C в
среде, содержащей 10 мМ СаС12 и 10 мМ АТФ, 0,25 мМ Pb(NO3)2;
0,1 М wpuc-малатный буфер (pH 7,9). После инкубации гели про-
мывают несколько раз дистиллированной водой в течение 20 мин,
* Возможно также проведение пластинчатого электрофореза.
220
Таблица 22
Приготовление ПААТ и электродного буфера
Смесь для геля, pH 8,9 Электродный буфер, pH 8,3
Реактив 7—1 часть Трис — 1,8г
Реактив 2—2 части Гликокол — 8,7 г
Реактив 3 — 4 части Н2О — до 3 л
Н2О — 1 часть
затем заливают раствором, содержащим сульфид аммиака (0,05 мл
желтого (NH4)2S растворяют в 100 мл воды). Через несколько ми-
нут гели промывают дистиллированной водой. Сопрягающий бе-
лок выявляется в виде темно-окрашенной полосы.
Окрашенные гели денситометрируют на денситометре, опре-
деляют Rf сопрягающего белка, фотографируют или зарисовыва-
ют положение окрашенных полос геля на миллиметровой бумаге.
Оформление результатов. Данные по определению Са2+-АТФ-аз-
ной активности в двух повторностях заносят в таблицу и рассчиты-
вают среднее значение. Результаты электрофоретического анализа
представляют в виде фотографий или рисунков с указанием Rf со-
прягающего белка.
ПРИЛОЖЕНИЕ 1
СТАТИСТИЧЕСКАЯ ОБРАБОТКА РЕЗУЛЬТАТОВ ОПЫТОВ
При проведении исследований по фотосинтезу в связи с определен-
ными физиологическими задачами первоочередное значение имеет по-
становка опыта в контролируемых условиях выращивания растений. Это
предполагает, с одной стороны, выращивание растений при неизменных
условиях температуры, освещенности, минерального питания, водоснабже-
ния и других внешних условий, а с другой стороны — возможность варьи-
рования условий выращивания и исследования процессов в измененных
условиях. Нестабильность условий выращивания приводит к изменению
того или иного показателя структурного и функционального состояния
фотосинтетического аппарата. Поэтому необходимо при проведении се-
рийных исследований выращивать растения в специальных климатичес-
ких камерах, позволяющих поддерживать достаточно стабильные условия
или производить контролируемое изменение условий выращивания рас-
тений. Но даже при общей выравненности условий выращивания индиви-
дуальные различия растений обусловливают определенную вариабельность
исследуемых признаков. Ввиду этого возникает необходимость проведе-
ния статистической обработки полученных экспериментальных данных.
В биологических исследованиях принято проводить статистическую
обработку получаемых экспериментальных данных в целях установления
степени достоверности (значимости) полученных различий между сери-
ями измерений (вариантами опыта), определения степени вариабельно-
сти показателя, характера зависимости измеряемого признака от опре-
деленных экспериментальных воздействий, внешних факторов и т.д. При
этом обычно используют стандартные статистические показатели — сред-
нюю арифметическую, среднее квадратическое отклонение, ошибку сред-
ней арифметической, критерий достоверности Стьюдента и т.д.; в ряде
случаев проводят более сложный анализ результатов [2, 3, 8].
Особое значение статистическая обработка данных и статистический
анализ имеют при моделировании физиологических процессов в расте-
нии, установлении взаимосвязи различных физиологических парамет-
ров и процессов в растительном организме. Для этого используют более
сложные приемы обработки и анализа экспериментальных результатов,
различные приемы оценки моделей и др.
Ниже дано краткое описание статистических методов, необходимых для
обработки результатов практических работ. Более детальное изложение ме-
тодов статистической обработки см. в специальной литературе [3—6].
222
Приемы первичной статистической обработки
результатов опытов
Для получения статистически достоверных результатов при определе-
нии любого физиологического показателя необходимо проводить несколько
(3 — 5 и более) измерений. Показатели повторных измерений составляют
выборку. Выборка, таким образом, включает ряд количественных показа^
телей, которые называются вариантами (вариансами): варианта х,- — ре-
зультат первичного z-ro измерения. Выборка представляет собой часть ге-
неральной совокупности — всего массива данных определенной категории.
Задачами первичной статистической обработки данных являются:
• определение основных параметров выборки, в первую очередь сред-
него значения варьирующего признака и степени его варьирования;
• обработка выборки с нормальным распределением или распределе-
нием Стьюдента путем соотнесения параметров выборки с параметрами
генеральной совокупности;
• сравнение двух выборок (в случае проведения сравнительных опы-
тов), определение критерия достоверности разности средних показате-
лей двух вариантов опыта либо контроля и опыта.
Определение основных параметров выборки
Средние значения
1. Средняя арифметическая (х) из ряда величин повторных определе-
ний (вариант) является наиболее часто употребляемой статистической
характеристикой:
(1)
п
где У х,- — сумма вариант выборки; п — число вариант.
2. Средняя геометрическая (хгеом) вычисляется в случае изучения из-
менения исследуемого признака во времени (например, при определе-
нии прироста содержания хлорофилла в ходе зеленения листа):
-^геом = \/^1 ' ^2 ‘ ^3 ‘ ‘ = ^/11 X > (2)
где хь х2, х3,..., хп — приросты за отдельные периоды; X — произведе-
ние всех частных приростов; п — число периодов.
Возможен расчет средней геометрической на основании измерений в
первый и последний периоды по формуле
= Д, (3)
где хп, X! — значения последнего и первого наблюдений соответственно.
3. Средняя гармоническая (хгарм) применяется для вычисления средней
характеристики признаков, которые представляют собой отношение двух
других варьирующих величин (например, при расчете отношения Fm/F0
по индукционным кривым флуоресценции хлорофилла):
223
•^•гарм — , (4)
где — — «обращенные» значения вариант; п — число вариант.
Xi
4. Средняя квадратическая (кубическая) вычисляется при исследова-
нии признаков, характеризуемых мерами площади (например, площади
листа) или объема (например, объема хлоропластов).
Общая формула для вычисления таких средних
где п — показатель степени, равный 2 для квадратической средней, 3 для
кубической; N — число наблюдений.
Показатели меры варьирования признака
1. Размах изменчивости (Н):
H—Xmax~Xmin, (6)
где хтах — максимальное значение варианты; xmin — минимальное значе-
ние варианты.
2. Среднее квадратическое отклонение (S) характеризует степень из-
менчивости (разнообразия) признака и величину ошибки у объектов
данной группы:
,_________ L JW
5 = + X.fo Z Х.Н = ±\ —----«—, (7)
V п-1 \ п-1
где У (х/ - х)2 — сумма квадратов отклонений отдельных вариант от
средней арифметической; п — число вариант; У х2 — сумма квадратов
вариант; (У х; ) — квадрат суммы вариант.
3. Коэффициент вариации (С) — характеристика относительной сте-
пени варьирования показателя:
С = ^100%, (8)
х
где S — среднее квадратическое отклонение; х — средняя арифметичес-
кая.
4. Среднее арифметическое отклонение (т|):
У(Х/-Х) ZQX
(9)
где У (х,- - х) — сумма отклонений отдельных вариант от средней ариф-
метической; п — число вариант.
5. Ошибка средней арифметической, или стандартная ошибка, (Sx) —
показатель степени варьирования средних, разности между средними:
224
S,=±-^, (10)
где 5 — среднее квадратическое отклонение; п — число вариант.
Данные первичной статистической обработки используют для после-
дующего выбора метода статистической обработки с целью установле-
ния, насколько достоверны выявленные различия между измерениями.
Предварительно получают доказательство того, что изучаемая выбор-
ка представляет совокупность с нормальным распределением и подчиняет-
ся закону нормального распределения. Для этого можно использовать один
из двух способов, приведенных ниже [6]:
1) соотнесение среднего арифметического отклонения ц со средним
квадратическим отклонением 5;
2) сравнение размаха изменчивости Н со средним квадратическим
отклонением 5.
Л
В первом случае находят отношение При п < 30 для приближенно
нормального закона распределения должно соблюдаться неравенство
— -0,7979 <^i.
Если это неравенство имеет место, изучаемая выборка может обраба-
тываться в соответствии с распределением Стьюдента.
Во втором случае находят отношение H/S и затем применяют специ-
альную таблицу (табл. 1), в которой приведены критические границы
этого отношения для двух уровней значимости: Р - 0,1 (10%) и Р = 0,05
(5 %). В случае нормального распределения значения отношения должны
находиться в пределах указанных границ. Особенно важен 10 %-й уровень
значимости.
Если установлена принадлежность данной выборки к нормальному
распределению, применяют критерии нормального распределения или
распределения Стьюдента.
Обработка выборки с нормальным распределением или распределением
Стьюдента путем соотнесения параметров выборки с параметрами
генеральной совокупности
Параметры генеральной совокупности
1. Генеральная средняя (ц):
ц = х±5^, (11)
где х — средняя арифметическая; S* — ошибка средней арифметичес-
кой [формула (10)]; t — коэффициент Стьюдента.
Коэффициент Стьюдента (0 определяют по табл. 2 с учетом необхо-
димого уровня значимости Р и числа степеней свободы v - п - 1.
Произведение S*t называется доверительным интервалом для гене-
ральной средней. Фактически это показатель точности определения ге-
неральной средней, получаемой для п вариант и уровня значимости Р.
8 Гавриленко
225
Таблица 1
Границы отношения Н/S для нормально распределенных выборок
Объем выборки Р=0,10 Р= 0,05 Объем выборки Р=0,10 0,05
3 1,782-1,997 1,758-1,999 30 3,59-4,70 3,47-4,89
4 2,040 - 2,409 1,98-2,429 35 3,70-4,84 3,58-5,04
5 2,22-2,712 2,15-2,753 40 3,79-4,96 3,67-5,16
6 2,37-2,949 2,28-3,012 45 3,88-5,06 3,75-5,26
7 2,49-3,143 2,40-3,222 50 3,95-5,14 3,83-5,35
8 2,59-3,308 2,50-3,399 55 4,02-5,22 3,90-5,43
9 2,68-3,449 2,59-3,552 60 4,08-5,29 3,96-5,51
10 2,76-3,57 2,67-3,685 65 4,14-5,35 4,01-5,57
11 2,84-3,68 2,74-3,80 70 4,19-5,41 4,06-5,63
12 2,90-3,78 2,80-3,91 75 4,24-5,46 4,11-5,68
13 2,96-3,87 2,86-4,00 80 4,28-5,51 4,16-5,73
14 3,02-3,95 2,92-4,09 85 4,33-5,56 4,20-5,78
15 3,07-4,02 2,97-4,17 90 4,36—5,60 4,24-5,82
16 3,12-4,09 3,01-4,24 95 4,40-5,64 4,27-5,86
17 3,17-4,15 3,06-4,31 100 4,44-5,68 4,31-5,90
18 3,21-4,21 3,10-4,37 150 4,72-5,96 4,59-6,18
19 3,25-4,27 3,14-4,43 200 4,90-6,15 4,78-6,39
20 3,29-4,32 3,18-4,49 500 5,49-6,72 5,37-6,94
25 3,45-4,53 3,34-4,71 1000 5,92-7,11 5,79-7,33
226
Таблица 2
Коэффициент Стьюдента t для уровней значимости Р
Число сте- пеней сво- боды (п - 1) Уровень значимости Число сте- пеней сво- боды (п - 1) Уровень значимости
0,05 0,01 0,001 0,05 0,01 0,001
1 12,71 63,66 — 21 2,08 2,83 3,82
2 4,30 9,93 31,60 22 2,07 2,82 3,79
3 3,18 5,84 12,92 23 2,07 2,81 3,77
4 2,78 4,60 8,61 24 2,06 2,80 3,75
5 2,57 4,03 6,87 25 2,06 2,79 3,73
6 2,45 3,71 5,96 26 2,06 2,78 3,71
7 2,37 3,50 5,41 27 2,05 2,77 3,69
8 2,31 3,36 5,04 28 2,05 2,76 3,67
9 2,26 3,25 4,78 29 2,04 2,76 3,66
10 2,23 3,17 4,59 30 2,04 2,75 3,65
11 2,20 3,11 4,44 40 2,02 2,70 3,55
12 2,18 3,06 4,32 50 2,01 2,68 3,50
13 2,16 3,01 4,22 60 2,00 2,66 3,46
14 2,15 2,98 4,14 80 1,99 2,64 3,42
15 2,13 2,95 4,07 100 1,98 2,63 3,39
16 2,12 2,92 4,02 120 1,98 2,62 3,37
17 2,11 2,90 3,97 200 1,97 2,60 3,34
18 2,10 2,88 3,92 500 1,96 2,59 3,31
19 2,09 2,86 3,88 оо 1,96 2,58 3,29
20 2,09 2,85 3,85
227
Доверительный интервал средней генеральной совокупности опреде-
ляется выражением
х - S*t <ц < х + Sxt.
2. Дисперсия генеральной совокупности (о2) — мера, характеризующая
рассеяние случайной величины. Один из показателей неоднородности
экспериментального материала
о2 = М (х2) - ц2, (12)
где М(х2) — среднее значение квадрата случайных величин; ^ — квад-
рат генеральной средней.
3. Стандартное (среднеквадратическое) отклонение (о) — квадратный
корень из дисперсии генеральной совокупности.
Среднее значение (ц) и степень варьирования опытных данных (о) —
наиболее точные характеристики генеральной совокупности.
Сравнение двух выборок
При проведении сравнительных исследований, когда изучается влия-
ние некоторого фактора на исследуемый показатель, необходимо произ-
вести оценку достоверности выявленных различий между вариантами
опыта или между опытным вариантом и контролем. В этом случае пользу-
ются определенными статистическими критериями. Для оценки сово-
купностей, имеющих нормальное распределение или умеренные откло-
нения от него, используют параметрические статистические критерии,
основанные на сопоставлении параметров (оценок) распределения (сред-
него арифметического, дисперсии и т.п.).
Достоверность различия между средними двух выборок оценивают с
помощью критерия Стьюдента (/):
(13)
Sd
где Х[-х2 ~ разность между средними двух выборок; Sd — ошибка раз-
ности средних.
При одинаковом числе наблюдений (п[ = п2) ошибку разности средних
(Sd) находят по следующим формулам:
Sd = + S* или Sd = (14)
где и 5^ — ошибки сравниваемых средних.
Тогда
t_ А- х2
7^+5?'
Т.е. при П[ = п2 t — число, показывающее, во сколько раз разность
между средними арифметическими величинами больше корня квадрат-
ного из суммы квадратов средних ошибок двух выборок.
Число степеней свободы (v) в этом случае равно 2(я - 1).
228
Значение t находят по табл. 2 для соответствующей степени свободы
вариационного ряда и уровня значимости Р. В биологических исследова-
ниях достоверность вывода является доказанной при уровне значимости
Р, равном 0,05, т.е. вероятность определений должна быть не менее 95 %.
Если вычисленное по экспериментальным данным значение t будет боль-
ше, чем найденное по табл. 2, то разность - х2 признается существен-
ной (достоверной). Если фактическое значение критерия Стьюдента рав-
но или меньше табличного, то разность между средними не выходит за
пределы ее случайных колебаний.
Более подробно методы статистической обработки изложены в спе-
циальной литературе, например [2 — 6, 8].
Первичная статистическая обработка данных,
полученных в эксперименте
Для первичной статистической обработки полученных в эксперимен-
те данных достаточно произвести расчеты, представленные в табл. 3.
Таблица 3
Операция Формула Конечное выражение расчета Примечания
1. Опреде- лить сред- нюю ариф- метическую (х) п где У Х[ — сумма вариант выборки; п — число ва- риант X При необходимос- ти рассчитывают другие средние (среднюю гармо- ническую, сред- нюю геометричес- кую и др.)
2. Рассчи- тать среднее квадрати- 5 = + |£(х;-х)2 = V п -1 х ± S
ческое отклонение (S} ч где квадра! отделы средне] п — чи U - вариан квадрап £x2_(X^L п
п -1 {Xj - х)2 — сумма гов отклонений шх вариант от л арифметической; ело вариант; - сумма квадратов г; (W - г суммы вариант
229
Окончание табл. 3
Операция Формула Конечное выражение расчетов Примечания
3. Рассчитать ошибку сред- ней арифме- тической (или стандартную ошибку) (5*) +1 II х ±
4. Определить значение критерия Стьюдента t t Находят по таблице при известном чис- ле степеней свобо- ды и заданном уровне значимости Р (для биологичес- ких исследований Р= 0,05)
5. Определить генеральную среднюю (ц), рассчитав доверитель- ный интервал для средней генеральной совокупности Ц = X +Sxt, где х — средняя арифметическая; 5* — ошибка среднего; t — коэффициент Стью- дента
6. Оценить значимость различия меж- ду средними двух выборок с помощью кри- терия Стью- дента t (при сравнитель- ных исследо- ваниях) , _ *1 - х2 _ sd ’ где (Х| - х2) — разность между средними двух выборок; Sd — ошибка разности средних. При одинаковом числе наблюдений (п{ = п2) Sd^S^+Sl, или о _ /^12 + $2 > N п где и 5^ — ошиб- ки сравниваемых сред- них. Если вычисленные по эксперименталь- ным данным значе- ния t будут больше, чем найденные по табл. 2, то разность (х! - х2) признается существенной; если фактическое значе- ние t равно или меньше табличного, то разность между средними не выхо- дит за пределы ее случайных колеба- ний
230
С применением компьютеров процедура статистической обработки
данных технически значительно упрощается. Статистический анализ про-
водят с использованием стандартных статистических программ, напри-
мер Stadia 6.0, Statgraph и др. [1, 7], позволяющих осуществлять не толь-
ко первичную статистическую обработку данных, но и более сложный,
специальный статистический анализ.
ПРИЛОЖЕНИЕ 2
ПРИГОТОВЛЕНИЕ РАСТВОРОВ.
СПРАВОЧНЫЕ ДАННЫЕ
I. ПРИГОТОВЛЕНИЕ БУФЕРНЫХ РАСТВОРОВ
Таблица 1
Ацетатный буфер 0,2 М, pH 3,6 —5,8
(ацетат натрия CH3COONa- ЗН2О; Мг 136,09)
pH при 18 °C Ацетат натрия 0,2 М, мл Уксусная кислота 0,2 М, мл pH при 18 °C Ацетат натрия 0,2 М, мл Уксусная кислота 0,2 М, мл
3,6 0,75 9,25 4,8 5,90 4,10
3,8 1,20 8,80 5,0 7,00 3,00
4,0 1,80 8,20 5,2 7,90 2,10
4,2 2,65 7,35 5,4 8,60 1,40
4,4 3,70 6,30 5,6 9,10 0,90
4,6 4,90 5,10 5,8 9,40 0,60
Таблица 2
Боратный буфер 0,2 М, pH 7,4 —9,0
(бура Na2B4O7- 0Н2О; Мг 381,43; борная кислота Н3ВО3; Мг 61,84)
pH Бура 0,05 М, мл Борная кислота 0,2 М, мл pH Бура 0,05 М, мл Борная кислота 0,2 М, мл
7,4 1,0 9,0 8,2 3,5 6,5
7,6 1,5 8,5 8,4 4,5 5,5
7,8 2,0 8,0 8,7 6,0 4,0
8,0 3,0 7,0 9,0 8,0 2,0
Таблица 3
Боратный буфер 0,05 М, pH 9,3 — 10,1
(бура Na2B4O7 - ЮН2О; Мг 381,43)
pH Бура 0,05 М, мл NaOH 0,2 М, мл Вода, мл pH Бура 0,05 М, мл NaOH 0,2 М, мл Вода, мл
9,3 50 0,0 До 200 9,8 50 34,0 До 200
9,4 50 11,0 » 200 10,0 50 43,0 » 200
9,6 50 23,0 » 200 10,1 50 46,0 » 200
232
Таблица 4
Глицин-HCI буфер 0,05 М, pH 2,2-3,6
(глицин H2NCH2COOH; Мг 75,07)
pH Глицин 0,2 М, мл НС1 0,2 М, мл Вода, мл pH Глицин 0,2 М, мл НС1 0,2 М, мл Вода, мл
2,2 50 44,0 До 200 3,0 50 11,4 До 200
2,4 50 32,4 » 200 3,2 50 8,2 » 200
2,6 50 24,2 » 200 3,4 50 6,4 » 200
2,8 50 16,8 » 200 3,6 50 » 200
Таблица 5
Глицин-NaOH буфер 0,05 М, pH 8,6 — 10,6
(глицин H2NCH2COOH; Мг 75,07)
pH Глицин 0,2 М, мл NaOH 0,2 М, мл Вода, мл pH Глицин 0,2 М, мл NaOH 0,2 М, мл Вода, мл
8,6 50 4,0 До 200 9,6 50 22,4 До 200
8,8 50 6,0 » 200 9,8 50 27,2 » 200
9,0 50 8,8 » 200 10,0 50 32,0 » 200
9,2 50 12,0 » 200 10,4 50 38,6 » 200
9,4 50 16,8 » 200 10,6 50 45,5 » 200
Таблица 6
Диэтилбарбитуратный (вероналовый) буфер pH 6,8 —9,6
(натрийдиэтилбарбитурат C(O)NHC(O)NHC(O)C(C2H5)2Na; Мг 206,2)
pH при 18 °C Натрий- диэтилбар- битурат 0,04 М, мл НС1 0,2 М, мл pH при 18 °C Натрий- диэтилбар- битурат 0,04 М, мл НС1 0,2 М, мл
6,8 100 18,4 8,4 100 5,21
7,0 100 17,8 8,6 100 3,82
7,2 100 16,7 8,8 100 2,52
7,4 100 15,3 9,0 100 1,65
7,6 100 13,4 9,2 100 1,13
7,8 100 11,47 9,4 100 0,70
8,0 100 9,39 9,6 100 0,35
8,2 100 7,21
233
Таблица 7
Сукцинатный буфер 0,05 М, pH 3,8 —6,0
(янтарная кислота С4Н6О4; Мг 118,09)
pH Янтарная кислота 0,2 М, мл Натриевая щелочь 0,2 М, мл Вода, мл pH Янтарная кислота 0,2 М, мл Натриевая щелочь 0,2 М, мл Вода, мл
3,8 25 7,5 До 100 5,0 25 26,7 До 100
4,0 25 10,0 » 100 5,2 25 30,3 » 100
4,2 25 13,3 » 100 5,4 25 34,2 » 100
4,4 25 16,7 » 100 5,6 25 37,5 » 100
4,6 25 20,0 » 100 5,8 25 40,7 » 100
4,8 25 23,5 » 100 6,0 25 43,5 » 100
Таблица 8
Трис-HCl буфер 0,05 М, pH 7,2-9,1
(трис; Мг 121,14)
pH при температуре Трис 0,2 М, мл Соляная кислота 0,1 М, мл Вода, мл pH при температуре Трис 0,2 М, мл Соляная кислота 0,1 М, мл Вода, мл
23 °C 37 °C 23 °C 37 °C
9,10 8,95 25 5,0 До 100 8,05 7,90 25 27,5 До 100
8,92 8,78 25 7,5 » 100 7,96 7,82 25 30,0 » 100
8,74 8,60 25 10,0 » 100 7,87 7,73 25 32,5 » 100
8,62 8,48 25 12,5 » 100 7,77 7,63 25 35,0 » 100
8,50 8,37 25 15,0 » 100 7,66 7,52 25 37,5 » 100
8,40 8,27 25 17,5 » 100 7,54 7,40 25 40,0 » 100
8,32 8,18 25 20,0 » 100 7,36 7,22 25 42,5 » 100
8,23 8,10 25 22,5 » 100 7,20 7,05 25 45,0 » 100
8,14 8,00 25 25,0 » 100
Таблица 9
Фосфатный буфер 0,1 М, pH 5,8 —8,0
(Na2HPO4 • 2Н2РО4; Мг 178,05; Na2HPO4 • 12Н2О; Мг 358,22;
NaH2PO4 H2O ; Мг 138,0; NaH2PO4 -2Н2О ; Мг 156,03)
pH Na2HPO4 0,2М, мл NaH2PO4 0,2 М, мл Вода, мл pH Na2HPO4 0,2М, мл NaH2PO4 0,2 М, мл Вода, мл
5,8 8,0 92,0 До 200 7,0 61,0 39,0 До 200
6,0 12,3 87,7 » 200 7,2 72,0 28,0 » 200
6,2 18,5 81,5 » 200 7,4 81,0 19,0 » 200
6,4 26,5 73,5 » 200 7,6 87,0 13,0 » 200
6,6 37,5 62,5 » 200 7,8 91,5 8,5 » 200
6,8 49,0 51,0 » 200 8,0 94,7 5,3 » 200
234
Таблица 10
Фосфатно-цитратный буфер (Мак Ильвейна), pH 2,2 —8,0
(Na2HPO4 • 2Н2О; Мг 178,05; лимонная кислота С6Н8О7 -2Н2О; Мг 210,14;
буфер не может быть использован, если в растворе присутствуют ионы
кальция или магния)
pH Na2HPO4 0,2 М, мл Лимонная кислота 0,1 М, мл pH Na2HPO4 0,2 М, мл Лимонная кислота 0,1 М, мл
2,2 0,40 19,60 5,2 10,72 9,28
2,4 1,24 18,76 5,4 11,15 8,85
2,6 2,18 17,82 5,6 11,60 8,40
2,8 3,17 16,83 5,8 12,09 7,91
3,0 4,11 15,89 6,0 12,63 7,37
3,2 4,96 15,06 6,2 13,22 6,78
3,4 5,70 14,30 6,4 13,85 6,15
3,6 6,44 13,56 6,6 14,55 5,45
3,8 7,10 12,90 6,8 15,45 4,55
4,0 7,71 12,29 7,0 16,47 3,53
4,2 8,28 11,72 7,2 17,39 2,61
4,4 8,82 11,18 7,4 18,17 1,83
4,6 9,35 10,65 7,6 18,73 1,27
4,8 9,86 10,14 7,8 19,15 0,85
5,0 10,30 9,70 8,0 19,45 0,55
Таблица 11
Фосфатная смесь Серенсена III 0,06 М, pH 4,87 — 9,18
(приготовление буферной смеси: в 1 л дистиллированной воды растворяют
11,876 г Na2HPO4 • 2Н2О и в 1 л — 9,078 г КН2РО4. Растворы смешивают
перед работой в различных соотношениях)
pH Na2HPO4, мл КН2РО4, мл pH Na2HPO4, мл КН2РО4, мл
4,87 0,00 10,00 6,98 6,00 4,00
4,94 0,10 9,90 7,17 7,00 3,00
5,29 0,25 9,75 7,38 8,00 2,00
5,59 0,50 9,50 7,73 9,00 1,00
5,91 1,00 9,00 8,04 9,50 0,50
6,24 2,00 8,00 8,34 9,75 0,25
6,47 3,00 7,00 8,67 9,90 0,10
6,64 4,00 6,00 9,18 10,00 0,00
6,81 5,00 5,00
235
Т абл ица 12
Фосфатный буфер 0,05 М, pH 5,8 —8,0
(однозамещенный фосфат калия КН2РО4; Мг 136,09)
pH при 25 °C КН2РО4 0,2 М, мл NaOH или КОН 0,2 М, мл Вода, мл pH при 25 °C КН2РО4 0,2 М, мл NaOH или КОН 0,2 М, мл Вода, мл
5,8 5 0,36 До 20 7,0 5 2,91 До 20
6,0 5 0,56 » 20 7,2 5 3,47 » 20
6,2 5 0,81 » 20 7,4 5 3,91 » 20
6,4 5 1,16 » 20 7,6 5 4,24 » 20
6,6 5 1,64 » 20 7,8 5 4,45 » 20
6,8 5 2,24 » 20 8,0 5 4,61 » 20
Таблица 13
Цитратный буфер 0,1 М, pH 3,0 —6,2
(лимонная кислота С6Н8О7 • Н2О; Мг 210,14;
трехзамещенный цитрат натрия Na3C6H5O7 • 2Н2О; Мг 294,12)
pH Лимонная кислота 0,1 М, мл Трехзамещенный цитрат натрия 0,1 М, мл pH Лимонная кислота 0,1 М, мл Трехзамещенный цитрат натрия 0,1 М, мл
3,0 16,4 3,6 4,8 8,0 12,0
3,2 15,5 4,5 5,0 7,0 13,0
3,4 14,6 5,4 5,2 6,1 13,9
3,6 13,7 6,3 5,4 5,1 14,9
3,8 12,7 7,3 5,6 4,2 15,8
4,0 11,8 8,2 5,8 3,2 16,8
4,2 10,8 9,2 6,0 2,3 17,7
4,4 9,9 10,1 6,2 1,6 18,4
4,6 8,9 И,1
236
II. ПИТАТЕЛЬНЫЕ СМЕСИ ДЛЯ ВЫРАЩИВАНИЯ РАСТЕНИЙ
Таблица 14
Состав питательных смесей Кнопа, Гельригеля и Хоглэнда
Соль Концентрация соли в смесях
Кнопа Гельригеля Хоглэнда
г/л ммоль/л г/л ммоль/л г/л ммоль/л
Ca(NO3)2 1,00 6,1 0,492 3,0 0,821 5
КН2РО4 0,25 1,8 0,136 1,0 0,136 1
MgSO4 0,125 1,0 0,060 0,5 0,120 1
КС1 — — 0,075 0,5 — —
KNO3 0,25 2,5 — — 0,506 5
FeCl3 Следы Следы 0,025 0,15 — —
Железо винно-кислое — — — — 0,005* 0,03
Общая концентрация 1,63 12,5 0,788 5,15 1,588 12,3
* Винно-кислое железо прибавляют периодически в течение роста растений.
Таблица 15
Состав питательной смеси Прянишникова и смесей Цинцадзе
со стабильной реакцией среды
Соль Концентрация соли в смесях
Прянишникова Цинцадзе, г/л, при значениях pH
г/л ммоль/л 4,0 5,0 5,5-6,6 7,3
NH4NO3 0,240 3 0,33 0,33 0,20 —
СаНРО4-2Н2О 0,172 1 — — — —
MgSO4 0,06 0,5 0,50 0,50 0,50 0,50
CaSO4-2H2O 0,344 2 1,46 0,50 — 0,28
КС1 0,150 2 0,61 0,61 0,36 —
FeCl3 0,025 0,15 — — — —
KNO3 — — 0,17 0,17 0,51 0,80
Fe2(SO4)3 — — 0,04 0,25 0,32 0,25
FePO4 — — 0,68 — — —
Ca3(POfl)2 — — — 0,70 5,00 0,70
Ca(NO3)2 — — — — — 0,21
KOH — — — — — 0,17
237
Таблица 16
Оптимальные значения pH среды для роста растений
Растение pH Растение pH
Пшеница 6,5—7,8 Капуста 5,0-6,0
Ячмень 6,5 —7,8 Картофель 5,0-7,2
Рожь 5,0—6,0 Конопля 6,0-7,0
Овес 5,0-6,0 Лен 5,0-6,0
Кукуруза 5,5-7,5 Огурцы 5,5-7,4
Бобы конские 6,0-7,0 Салат 6,0-8,0
Горох 6,0-7,0 Подсолнечник 5,7-6,5
Люпин 4,5-6,0 Сахарная свекла 7,0-8,0
Люцерна 6,2-7,8 Табак 5,2-6,5
Фасоль 6,7-7,4 Томат 5,6-6,5
Гречиха 5,0-6,5 Тыква 6,0-7,0
Хлопчатник 5,0-6,0
III. СПРАВОЧНЫЕ ДАННЫЕ
Таблица 17
Некоторые константы растворителей
Растворитель Температура кипения, °C „20 nD Относительная летучесть
Эфир диэтиловый 34,5 1,3527 1,0
Ацетон 56,0 1,3589 2,1
Спирт этиловый 78,3 1,3614 8,3
» метиловый 64,5 1,3286 —
» изопропиловый 2,4 1,3776 —
Хлороформ 61,2 1,4455 2,5
Бензол 80,1 1,5011 3,0
Петролейный эфир (легкая фракция) 40-60 — —
Петролейный эфир (тяжелая фракция) 60-80 — —
Бензин «Калоша» 85-115 — —
238
Таблица 18
Относительная плотность (rf) растворов трихлоруксусной кислоты
разных концентраций при 25 и 35 °C
Концентрация dis rf35 Концентрация rf>5 rf35
моль/л % моль/л %
0,1956 3,196 1,01616 1,01518 2,4580 40,16 1,2217 1,2170
0,2333 3,813 1,0196 1,0188 4,3550 71,15 1,4309 1,4229
0,5237 8,557 1,0435 1,0429 5,3150 86,84 1,5534 1,5440
0,9526 15,56 1,0807 — 5,8260 95,19 1,625 1,6159
Таблица 19
Производные группы хлорофилла
Пигмент Формула Молеку- лярная масса Максимум поглощения в красной области спектра в этиловом эфире, нм
Протохлорофилл а C55H70O5N4Mg 891,46 623
Хлорофилл а C55H72O5N4Mg 893,48 660
Хлорофилл b C55H70O6N4Mg 907,46 644
Феофитин а C55H74O5N4 871,18 667
Феофитин b C55H72O6N4 885,16 655
Феофорбид а Сз5Н36О5М4 592,67 667
Феофорбид b C35H34O6N4 606,67 656,2
Таблица 20
Стандартные окислительно-восстановительные потенциалы (Ец) компо-
нентов электрон-транспортной цепи хлоропластов
Компонент Ро, В
Р68о — реакционный центр ФС II + 1,12
5-система (водоокисляющий комплекс) +0,90
Н2О/О2 +0,82
Р700 — реакционный центр ФС I +0,52 (+0,49)
Цитохром f +0,37
Пластоцианин +0,37
Центр Риске +0,29 (+0,3)
Цитохром Ь559Н (высокопотенциальный) Z>559Z (низкопотенциальный) +0,33 +0,08
PQ/PQH2 (пул пластохинонов) 0,0
239
Окончание табл. 20
Компонент Е’о, В
Цитохром />6Я(высокопотенциальный) beL (низкопотенциальный) -0,05 -0,11
Qa — первичный пластохинон -0,13
QB — вторичный пластохинон -0,01
НАДФН -0,32
ФАД (ФД-НАДФ+-оксидоредукгаза) -0,36
н+/н2 -0,42
Ферредоксин -0,43
Fa (центр А в ФС I) -0,55
FB (центр В в ФС II) -0,59
Fx (акцептор электронов в ФС I) -0,70
Феофитин (первичный акцептор электронов в ФС II) -0,61
Д (филлохинон, витамин К,) -0,90
Д (первичный акцептор электронов в ФС I) -1,1
СПИСОК ЛИТЕРАТУРЫ
Глава 1
1. Ахрем А. А., Кузнецова А. И. Тонкослойная хроматография. — М.: На-
ука, 1964.
2. Бабко А. К., Пилипенко А. Т. Фотометрический анализ. Общие сведе-
ния и аппаратура. — М.: Химия, 1968.
3. Виноградова Е.Н., Галлай З.А., Финогенова З.М. Методы полярогра-
фического и амперометрического анализа. — М.: Изд-во МГУ, 1960.
4. Гавриленко В. Ф., Ладыгина М.Е., Хандобина Л.М. Большой практи-
кум по физиологии растений. — М.: Высшая школа, 1975.
5. Детерман Т. Гель-хроматография. — М.: Мир, 1970.
6. Кейл Б. Центрифугирование // Лабораторная техника органической
химии. - М., 1966. - С. 179-193.
7. Маурер Г. Диск-электрофорез. — М.: Мир, 1971.
8. Методы биохимического анализа растений / Под ред. В. В. Полево-
го, Г. Б. Максимова. — Л.: Изд-во ЛГУ, 1978.
9. Методы изучения мембран растительных клеток / Под ред. В. В. По-
левого, Г. Б. Максимова, Н. Ф. Синютиной. — Л.: Изд-во ЛГУ, 1986.
10. Методы практической биохимии / Под ред. Б.Уильямса, К.Уилсо-
на. - М.: Мир, 1978.
11. Николаева Л. Ф., Поршнева Е. Б. Изучение процессов, индуцируемых
кратковременным освещением в этиолированных листьях кукурузы //
Физиологии и биохимия здорового и больного растения / Отв. ред. С. С. Ан-
дреенко. - М.: Изд-во МГУ, 1970. - С. 241-256.
12. Пейве Я. В., Ягодин Б. А., Бакеева Н. М. Препаративный электрофо-
рез белков на полиакриламидном геле // Биофизические методы в фи-
зиологии растений. — М., 1971. — С. 5—13.
13. Остерман Л. А. Методы исследования белков и нуклеиновых кис-
лот: Электрофорез и ультрацентрифугирование (практическое пособие). —
М.: Наука, 1981.
14. Пешкова В.М., Громова М.И. Практическое руководство по спект-
рофотометрии и колориметрии. — М.: Изд-во МГУ, 1965.
15. Практикум по физико-химическим методам в биологии. Практи-
ческое пособие / Под ред. Ф.Ф. Литвина. — М.: Изд-во МГУ, 1981.
16. Руководство по изучению биологического окисления полярогра-
фическим методом. — М.: Наука, 1973.
17. Сафонов В. И., Сафонова М. И. Микроэлектрофорез белков в поли-
акриламидном геле. — М.: Наука, 1968.
241
18. Современные методы биофизических исследований: Практикум
по биофизике / Под ред. А. Б. Рубина. — М.: Высшая школа, 1988.
19. Спектроскопические методы исследования в физиологии и биохи-
мии. — Л.; М.: Наука, 1987.
20. Фотосинтез и биопродуктивность: методы определения. — М.: Аг-
ропромиздат, 1989.
21. Чмутов К. В. Хроматография. — М.: Химия, 1962.
Глава 2
1. Беляева О. Б., Литвин Ф. Ф. Фотобиосинтез хлорофилла. — М.: Изд-
во МГУ, 1989.
2. Гуринович Г. И., Севченко А. И., Соловьев К. Н. Спектроскопия хлоро-
филла и родственных соединений. — Минск: Наука и техника, 1968.
3. Игнатов Н. В., Литвин Ф. Ф. Фотоиндуцируемый биосинтез феофи-
тина а в листьях растений // Биохимия. 1993. — Т. 58. — С. 1469—1480.
4. Литвин Ф. Ф. Беляева О. Б., Гуляева Б. А., Синещеков В.А. Организа-
ция пигментной системы фотосинтезирующих организмов и ее связь с
первичными фотопроцессами // Проблемы биофотохимии. — М.: На-
ука, 1973.-С. 132-147.
5. Литвин Ф. Ф., Беляева О. Б. Исследование биохимических реакций био-
синтеза хлорофилла // Биохимия. — 1968. — Т. 33. — Вып. 5. — С. 928—936.
6. Литвин Ф. Ф., Беляева О. Б., Игнатов И. В. Биосинтез хлорофилла и
формирование реакционных центров фотохимических систем фотосин-
теза // Успехи биологической химии. ОНТИ АНЦ РАН: Пущино-на-Оке,
2000.-Т. 40.-С. 3-42.
7. Пигменты пластид зеленых растений и методика их исследования /
Под ред. Д. И. Сапожникова. — М.; Л., 1964.
8. Рубин А. Б. Биофизика: В 2 т. — М.: Изд-во МГУ, 1999. — Т. 2.
9. Соловченко А. Е., Чивкунова О. Б., Мерзляк М.Н., Решетникова И. В.
Спектрофотометрический анализ пигментов в плодах яблони // Физио-
логия растений. — 2001. — Т. 48. — № 5. — С. 801 — 808.
10. Судъина Е.Г. Образование и накопление хлорофилла в зависимос-
ти от активности хлорофиллазы // Проблемы фотосинтеза. — М.: АН
СССР, 1959.-С. 204-210.
11. Судъина Е. Г., Лозовая Г. И., Довбыш Е. Ф. и др. К вопросу о локали-
зации хлорофиллазы в хлоропласте // Физиология и биохимия культур-
ных растений. — 1973. — Т. 5. — Вып. 2. — С. 154—158.
12. Шлык А. А. Метаболизм хлорофилла в зеленом растении. — Минск,
1965.
13. Apel К., Santel И. Y., Redlinger Т. et al. The protochlorophyllide holo-
chrome of barley: isolation and characterization of the NADPH-protochloro-
phyllid oxidoreductase // Eur. J. Biochem. — 1980. — V. 111. — P. 251 — 258.
14. Armstrong G., Runge S., Frick G. et al. Identification of NADPH: pro-
tochlorophyllide oxidereductases A and B: a branched pathway for light-depen-
dent chlorophyll biosynthesis in Arabidopsis thaliana // Plant Physiol. — 1995. —
V. 108.-P. 1505-1517.
15. Amon D. I. Cooper enzymes in isolated chloroplasts. Polyphenoloxidase
in Beta vulgaris // Plant Physiology. — 1949. — V. 24. — N 1. — P. 1 — 15.
242
16. Belyaeva 0., Timofeev К., Litvin F. The primary reactions in the
photochlorophill(ide) photoreduction as investigated by optical and ESR spec-
troscopy// Photosynth. Res. — 1988. — V. 15. — P. 247 — 256.
17. Brouers M., Michel- Wolwertz M.R. Estimation of protochlorophyll
(ide) contents in plant extracts: re-evaluation of the molar absorption
coefficient of protochlorophyll (ide) // Photosynth. Res. — 1983. — V. 4. —
P. 265.
18. Bruinsma I. A comment on the spectrophotometric determination of
chlorophyll // Biochim. Biophys. Acta. — 1961. — V. 52. — P. 576 — 578.
19. Folch J., Lees M., Stoane Stenley G.H. A simple method for isolation
and purification of total lipids from animal tissues // J. Biol. Chem. — 1957. —
V. 226.-P. 497-509.
20. Fujita Y. Protochlorophyllide reduction: a key step in the greening of
plants // Plant Cell Phisiol. - 1996. - V. 37. - P. 411 -421.
21. Granick S., Gassman M. Rapid regeneration of protochlorophyllide //
Plant Cell Physiol. - 1970. - V. 45. - P. 201-205.
22. Griffits W. T. Protochlorophyllide photoreduction: Chlorophylls / Ed.
Scheer H., Boca Raton, Ann Arbor. — Boston; London: CRC Press, 1991. —
P. 433-449.
23. Koski V. M., Smith I. H. C. // Amer. Chem. Soc. - 1948. - V. 70. - N 11. -
P. 3558.
24. Lichtenthaler H.K. Chlorophyll and Carotinoids: Pigments of Photosyn-
thetic Biomembranes // Methods. Enzimol. — 1987. — V. 148. — P. 331 — 382.
25. Lichtenthaler H.K., Wellburn A.R. Determinations of total carotenoids
and chlorophylls a and b of leaf extracts in different solvents // Biochem. Soc.
Trans. - 1983. - V. 603. - P. 591.
26. Litvin F., Ignatov N., Belyaeva O. Photoreversibility of transformations of
protochlorophyllide into chlorophyllide // Photobiochem. Photobiophys. —
1981.-V. 2.-P. 233-237.
27. Mac-Kinney G. Absorption of light by chlorophyll solutions 11 J. Biol.
Chem. 1941.-V. 140, N 2. - P. 315-322.
28. Porra R.J. Recent advances and re-assessment in chlorophyll extraction
and assay procedures for terrestrial, aquatic and morine organisms, including
re-calcitrant algae // Chlorophylls / Ed. Scheer H., Boca Raton, Ann Arbor. —
Boston; London: CRC Press, 1991. — P. 31 —57.
29. Porra R.J., Tompson W.A., Kriedemann P. E. Determination of accurate
extinction coefficients and simultaneous equations for assaying chlorophylls a
and b extracted with four different solvent: verinfication of the concentration
of chlorophyll standards by atomic absorption spectroscopy 11 Biochem. Bio-
phys. Acta. - 1989. - V. 975. - P. 384.
30. Schoefs B., Bertrand V., Frank F. Spectroscopic properties of proto-
chlorophyll(ide) analysed in situ in the course of etiolation and in illuminated
leaves // Photochem. Photobiol. — 2000. — V. 72. — P. 85 — 93.
31. Strack D., Wray V. Antocyanins // Methods in Plant Biochemistry / Eds
Harbom J. B., Dey P. M. — London: Acad. Press, 1989. — V. 1. — P. 326 — 352.
32. Weast C.A., Mac-Kinney G. // J. Biol. Chem. — 1940. —V. 133. — P. 551.
33. Wellburn A.R. The spectral determination of chlorophylls a and b, as
well as total carotenoids, using various solvents with spectrophotometers of
different resolution // J. Plant Physiol. — 1994. — V. 144. — P. 307 — 313.
243
Глава 3
1. Бухов Н.Г. Старение листа. Выявление участков, лимитирующих
фотосинтез, с помощью коэффициентов тушения флуоресценции хло-
рофилла и редокс-изменений Р700 в листьях // Физиология растений. —
1997. - Т. 44. - № 3. - С. 352-360.
2. Веселовский В. А., Веселова Т.В. Люминесценция растений. Теорети-
ческие и практические аспекты. — М.: Наука. 1990.
3. Веселовский В.А., Веселова Т.В. Индукция флуоресценции. Теория и
практика использования для диагностики // Физиолого-биохимические
и биофизические методы диагностики степени устойчивости растений к
патогенам и другим факторам / Под ред. М.Е. Ладыгиной. — М.: Изд-во
МГУ, 1992.-С. 80-95.
4. Егорова Е.А., Бухов Н.Г., Кренделева Т.Е., Рубин А. Б. Гетерогенность
восстановления хинонных акцепторов в интактных листьях ячменя //
ДАН. - 2001. - Т. 337. - № 5. - С. 1 -4.
5. Карапетян Н. В., Бухов Н. Г. Переменная флуоресценция хлорофил-
ла как показатель физиологического состояния растений // Физиология
растений. — 1986. — Т. 33. — Вып. 5. — С. 1013—1026.
6. Карпушкин Л. Т. Применение инфракрасного газоанализатора для
изучения СО2-газообмена растений // Биофизические методы в физио-
логии растений. — М.: Наука, 1971. — С. 44.
7. Магомедов И.М. Определение активности рибулозодифосфаткар-
боксилазы / Под ред. В. В. Полевого, Г. Б. Максимова // Методы биохи-
мического анализа растений — Л.: Изд-во ЛГУ, 1978. — С. 68 — 69.
8. Методы выделения и исследования белков-компонентов фотосинте-
тического аппарата: Материалы Всесоюзного семинара (21 — 23 июня
1972 г.). — Пущино-на-Оке, 1973.
9. Методы исследования структуры фотосинтетического аппарата: Ма-
териалы Всесоюзного семинара (16—17 июня 1971 г.). — Пущино-на-
Оке, 1972.
10. Мокроносов А. Т. Онтогенетический аспект фотосинтеза. — М.: На-
ука, 1981.
11. Мокроносов А. Е., Борзенкова Р.А. Методика количественной оцен-
ки структуры и функциональной активности фотосинтезирующих тка-
ней и органов // Труды по прикладной ботанике, генетике и селекции. —
1978.-Т. 61.-Вып. З.-С. 119-133.
12. Чмора С. Н., Егоров В. П., Алехин В. И. Исследование газообмена листь-
ев растений на свету и дыхания при переходе свет-темнота по двум газам —
СО2 и О2 // Физиология растений. — 1992. — Т. 39. — Вып. 4. — С. 775—785.
13. Conroy J.P. et al. Clorophyll a fluorescence and photosynthetic and
growth responses of Pinus radiata to phosphorus deficiency, drought stress and
high CO2 // Plant Physiol. - 1986. - V. 81. - P. 423-429.
14. Genty B., Briantais J.-M., Baker N.R. The relationship between the quan-
tum yield of photosynthetic electron transport and quenching of chlorophyll
fluorescence // Biochim. Biophys. Acta. — 1989. — V. 990. — P. 87 — 92.
15. Havaux M., Lannoye R. In vivo clorophyll fluorescence and delayed light
emission as rapid screening techniques for stress tolerance in crop plants 11
Ztschr. Pflanzenzucht. 1985. — Bd. 95. — S. 1—13.
244
16. Horton P., Ruban A. V., Walters R.G. Regulation of light harvesting in
green plants// Ann. Rev. Plant Physiol., Plant Molecular. Biol. — 1996. — V. 47. —
P. 655-684.
17. Krause G.H., Weis E. Chlorophyll fluorescence and photosynthesis: The
Basics //Ann. Rev. Plant Physiol., Plant Biol. — 1991. — V. 42. — P. 313—349.
18. Schreiber U, Schliwa U, Bilger W. Continuous recording of photochemical
and non-photochemical chlorophyll fluorescence quenching with a new type of
modulation fluorescence // Photosynthesis Research. — 1986. — V. 10. — P. 51 — 62.
19. Smillie R. M., Hetherington S. E. Stress tolerance and stress-induced inju-
ry in crop plants measured by chlorophyll fluorescence in vivo: chilling, freezing,
ice cover, heat and high light // Plant Physiol. — 1983. — V. 72. — P. 1043—1050.
20. Snel J.F.H., Kooten O. van. The use of chlorophyll fluorescence and
other non-invasive spectroscopic techniques in plant stress physiology // Pho-
tosynthesis Res. — 1990. —V. 25. — P. 146 — 332.
21. Weis E., Berry J. A. Quantum efficiency of photosystem II in relation to
«energy»-dependent quenching of chlorophyll fluorescence // Biochim Bio-
phys. Acta. - 1987. - V. 894. - P. 198-200.
Глава 4
1. Лиигуд P. К., Уокер Д.А. Хлоропласты и протопласты // Фотосинтез
и биопродуктивность: методы определения. — М.: Агропромиздат, 1989. —
С. 202-225.
2. Лысенко Г. Г. О номенклатуре изолированных хлоропластов и опре-
делении степени их целостности // Физиолого-биохимические и биофи-
зические методы диагностики степени устойчивости растений к патоге-
нам и другим факторам / Под ред. М.Е. Ладыгиной. — М.: Изд-во МГУ,
1992.-С. 37-42.
3. Методы выделения и исследования белков-компонентов фотосинте-
тического аппарата: Материалы Всесоюзного семинара (21—23 июня
1972 г.). — Пущино-на-Оке, 1973.
4. Методы выделения хлоропластов: Материалы Всесоюзного семинара
16—18 июля 1969 г. / Под ред. Е.Б.Кириченко. — Пущино-на-Оке, 1970.
5. Albertson Р. A quantitative model of the domain structure of the photo-
synthetic membrane // Trends in Plant Sci. — 2001. — V. 6. — N 8. — P. 349 — 354.
6. Albertson P. et al. Fractionation of thylakoid membrane // Methods En-
zymol. - 1994. - V. 228. - P. 469-482.
7. Amon D. I., Allen M. B., Whatley F. K. // Bioch. Biophys. Acta. — 1956. —
V. 20. - N 3. - P. 449.
8. Bertold D.A., Babcock G. T., Yocum C. F. A high resolved oxygen-evolving
photosystem 2 preparation from spinach thylakoid membranes // FEBS Lett. —
1981.-V. 134.-P. 231-234.
9. Hall D. O. Nomenclature for isolated chloroplasts // Nature New Biol. —
1972. - V. 235. - N 56. - P. 125 -126.
10. Mustardy L. Development of thylakoid membrane stacking // Oxygenic
Photosynthesis: The Light Reaction I Eds. Ort D.R., Yocum C.F. — Kluwer
Acad. Pub., 1996. - P. 59-68.
11. Peter G., Thornber J.P. // J. Biol. Chem. — 1991. — V. 266. — N 25. —
P. 16745-16754.
245
12. Staehelin L., Staay W. Structure, composition, functional organization
and dynamic properties of thylakoid membranes // Oxygenic Photosynthesis:
The Light Reaction / Eds. Ort D.R., Yocum C.F. — Kluwer Acad. Pub., 1996. —
P. 11-30.
13. Weibull C., Albertson P. Ultrastructure of spinach thylakoid as seen in low-
temperature and conventional embeddings // J. Ultrastruct. Mol. Struct. Res. —
1998.-V. 100.-P. 55-59.
Глава 5
1. Бекина P. M., Гусейнова В. E. Фотосинтез и фотоокислительные про-
цессы // Физиология растений. — 1986. — Т. 33. — Вып. 1. — С. 171 — 184.
2. Бекина Р.М., Шувалов В. А., Лысенко Г. Г. и др. Исследование меха-
низма фотоокислительных превращений органических кислот в хлоро-
пластах // Физиология растений. — 1975. — Т. 22. — Вып. 4. — С. 680.
3. Любимов В.Ю., Застрижная О.М. Роль перекиси водорода в фотоды-
хании С4-растений // Физиология растений. — 1992. — Т. 39. — Вып. 4. —
С. 701-710.
4. Методы исследования фотосинтетического транспорта электрона:
Материалы Всесоюзного семинара (19 июня 1973 г.). — Пущино-на-Оке,
1974.
5. Мокроносов А. Т., Гавриленко В. Ф. Фотосинтез. Физиолого-экологи-
ческие и биохимические аспекты. — М.: Изд-во МГУ, 1992.
6. Мухин Е.И., Акулова Е.А. Об участии ферредоксина из проростков го-
роха в фотовосстановлении НАДФ //ДАН. — 1966. — Т. 167. — № 5. — С. 1177.
7. Рубин А. Б., Кренделева Т.Е. Фотосинтетический перенос электронов
и сопряженные с ним процессы фосфорилирования у растений // Успе-
хи современной биологии. — 1972. — Т. 73. — Вып. 3.
8. Шувалов И. Ф. Первичное преобразование световой энергии при
фотосинтезе. — М.: Наука, 1990.
9. Шутилова Н.И., Кадашникова И.Г., Смолова Т.Н., Климов В.В. Реак-
тивация функций выделения кислорода у субхлоропластных фрагментов
фотосистемы II, отмытых от белков с молекулярной массой 17, 23 и 33
kDa // Биохимия. - 1987. - Т. 52. - Вып. 12. - С. 1958-1964.
10. Albertson Р. A quantitative model of the domain structure of the photosyn-
thetic membrane // Trends in Plant Science. — 2001. — V. 6. — N 8. — P. 349—354.
11. Asada K., Kisok, Yoshikawa K. Univalent Reduction of Molecular Oxy-
gen by Spinach Chloroplasts on Illumination // J. Biol. Chem. — 1974. — V. 249. —
N7.-P. 2175-2181.
12. Bumman D., Oesterhelt D. Destruction of a single chlorophyll is correlat-
ed with the photoinhibition of photosystem II with a transiently inactive donor
side // Proc. Nat. Acad. Sci. USA. - 1995. - V. 22. - P. 12195-12199.
13. Duysens L., Amesz J. Function and identification of two photochemical
systems in photosynthesis // Biochim. Biophys. Acta. — 1962. — V. 64. — N 2. —
P. 243.
14. Ghanotakis D.F., Demetriou D.M., Yocum C.F. Isolation and character-
ization of an oxygen evolving photosystem II reaction center core prepara-
tion and a 28 kDa Chi а-binding protein // Biochim. Biophys. Acta. — 1987. —
V. 891. —P. 15-21.
246
15. Govindjee R. Emerson enhancement effect and! two light reactions in
photosynthesis // Photosynthetic mechanisms of green plants. — Washington,
1963.
16. Hill R. Oxygen evolution by isolated chloroplasts // Nature. — 1937. —
V. 139.— P. 881 — 882.
17. Hill R. Oxydoreduction in chloroplasts // Adv. In Enizymol. — 1951. — V. 12.
18. Hill R., Bendall F. Function of the two cytochrome compounds in chlo-
roplasts: a working hypothesis 11 Nature. — 1960. — V. 186. — N 4719. — P. 136.
19. Hind G., Olson J. Electron transport pathways in photosynthesis // Ann.
Rev. Plant Physiol. — 1968. — V. 19.
20. Horton P., Crofts J., Oxborongh K. et al. Regulation of photosystem II by
metabolic and environmental factors // Phil. Trans. R. Soc. bond. — 1989. —
В 323. —P. 43-52.
21. Izawa S., Norman E., Good N.E. // Biochim. Biophys. Acta. — 1968. —
V. 162. - P. 380.
22. Krogmann D., Jagendorf A., Avron M. Uncouplers of spinach chloroplast
photosynthetic phosphorylation // Plant Physiol. — 1959. — V. 34. — N 3. — P. 272.
23. McCarty, Racker E. Partial resolution of the enzymes catalyzing photo-
phosphorylation. III. Activation of adenosine; triposphatase and 32P labeled ortho-
phosphate adenosine triphosphate exchange in chloroplasts // J. Biol. Chem. —
1968. - V. 243. — N 1. — P. 129.
24. MehlerA.H. Studies on reactions of illuminated chloroplasts. 1. Mecha-
nism of reduction of oxygen and other Hill reagents // Archives of Biochem.,
Biophys. - 1951,-V. 33.-N l.-P. 65 — 77.
25. Mehler A.H., Brown A.H. Studies on reactions of illuminated chloroplasts.
III. Simultaneous photoproduction and consumption of oxygen studied with
O2 isotops //Arch. Biochem., Biophys. — 1952. — V. 38!. — P. 365.
26. Park R., Pon N. Correlation of structure with function in Spinacea olera-
cia chloroplasts // J. Mol. Biol. — 1961. — V. 3. — N 1. — P. 1.
27. Peltier G., Comae L. Chlororespiratin //Ann. Rev. Plant Biol. — 2002. —
V. 53.-P. 523-550.
28. Robinson J.M. Does O2 photoreduction occur within chloroplasts in
vivo // Plant Physiol. — 1988. — V. 72. — P. 666 — 680.
29. Saha S., Izawa S., Good N. Photophosphorylation as a function of light
intensity // Biochim. Biophys. Acta. — 1970. — V. 223. — N 158.
30. Saha S., Quitrakul R., Izawa S., Good N. Electron transport and photo-
phosphorylation in chloroplasts as function of the electron acceptor // J. Biol.
Chem. - 1971. - V. 246. - N 10. - P. 3204-3209.
31. San Pietro A., Lomg H.M. Photosynthetic pyridine nucleotide reductase. I.
Partial purification and properties of the enzyme from spinach // J. Biol. Chem.
1958.-V. 231.-N l.-P. 211.
32. Vishniak W., Ochoa S. Photochemical reduction of pyridine nucleotides
by spinach grana and coupled carbon dioxide fixation // Nature. — 1951. —
V. 167.-N 4254.-P. 768.
Глава 6
1. Гавриленко В. Ф., Гусев М. В., Никитина К. А., Хоффман П. Избранные
главы физиологии растений. — М.: Изд-во МГУ, 1986. — С. 168 — 297.
247
2. Методы исследования фотофосфорилирования. Материалы Всесоюз-
ного семинара 24 — 26 июня 1970 г. / Под ред. Е. Б. Кириченко. — Пущи-
но-на-Оке, 1970.
3. Марченко 3. Фотометрическое определение элементов. — М.: Мир,
1971.
4. Молотковский Ю. Г., Дзюбенко В. С. Индуцируемое светом поглоще-
ние Н+ изолированными хлоропластами // Молекулярная биология. —
1970. - Т. 4. - Вып. 3.
5. Николас Д. Биоэнергетика. Введение в хемиосмотическую теорию. —
М.: Мир, 1985.
6. Рэкер Э. Биоэнергетические механизмы: новые взгляды. — М.: Мир,
1979.
7. Скулачев В. П. Энергетика биологических мембран. — М.: Наука, 1989.
8. Степанова А.М., Никифорова Л. Ф. Методика выделения латентной
Са2+-АТФ-азы из хлоропластов гороха // Методы биохимического анализа
растений / Под ред. В.В.Полевого, Г.Б.Максимова. — Л.: Изд-во ЛГУ,
1978. - С. 62—66.
9. Arnon D., Allen М., Whatley F. Photosynthesis by isolated chloroplast 11
Nature. - 1954. - V. 174. - N 24.
10. Arnon D., Whatley F, Allen M. Photosynthetic phosphorylation by isolat-
ed chloroplast in coupled with TPN reduction // Science. — 1958. — V. 127. —
N 3305.
11. Avron M. A coupling factor in photophosphorylation // Biochim. Bio-
phys. Acta. - 1963. - V. 77. - N 77.
12. Avron M. Energy transduction in photophosphorylation // FEBS Lett. —
1978. - V. 96. - N 2. - P. 225-232.
13. Bendall D., Manasse R. Cyclic photophosphorylation and electron trans-
port // Biochim. Biophys. Acta. — 1995. — V. 1229. — P. 23 — 38.
14. Berzbom R., SchroerP. Photophosphorylation: mechanism of reconstitution
by coupling factor 1 (CFj) // FEBS Lett. - 1976. - V. 70. - N 1. - P. 271-275.
15. Boyer P. Coupling mechanism in capture, transmission and use energy //
Ann. Rev. Biochem. — 1977. — V. 46. — P. 957—966.
16. Boyer P. The binding change mechanism for ATP synthase — some
probabilities and possibilities // Biochim. Biophys. Acta. — 1993. — V. 1140. —
P. 215-250.
17. Boyer P., Kohlbrender W. The present status of the binding-change mecha-
nism and the relation to ATP formation by chloroplasts // Energy coupling in
photosynthesis. — Amsterdam. — 1981. — P. 231 — 240.
18. Buchanan B.B., Gruissem W., Jones R.L. / Biochemistry and Biology of
Plants. Amer. Soc. Plant Physiologists. — Rockville, Maryland, 2000.
19. Chen P., Toribara T., Warner U. Microdetermination of phosphorus //
Anal. Chem. - 1956. - V. 28. - P. 1756-1763.
20. Cross R. The rotary binding change mechanism of ATP synthases //
Biochim. Biophys. Acta. - 2000. - V. 1458. - P. 270-275.
21. Davenport J., McCarty R. An analysis of proton fluxes coupled to elect-
ron transport and ATP synthesis in chloroplast thylakoids // Biochim. Biophys.
Acta. - 1984. - V. 766. - N 2. - P. 363-374.
22. Dunhum K., Selman B. Regulation of spinach coupling factor 1 ATPase
activity//J. Biol. Chem. - 1981. -V. 256. - N 1. - P. 212-218.
248
23. Groth G. Molecular models of the structural arrangement of subunits
and the mechanism of proton translocation in the membrane domain of FjFo
ATP synthase // Biochim. Biophys. Acta. — 2000. — V. 1458. — P. 417 — 427.
24. Farron F. Izolation and properties of a chloroplast coupling factor and
heat-activated adenosine triphosphotase // Biochemistry. — 1970. — V. 9. —
P. 3823.
25. Futai M., Omote H., Sambongi Y., Wada Y. Synthase (H+-ATPase): coup-
ling between catalysis, mechanical work and proton translocation // Biochim.
Biophys. Acta. - 2000. -V. 1458. - P. 276-288.
26. Jung W. ATP-synthase: etectro-chemical transducer with stepping rota-
tory mechanics // Proc. 11-th Intern. Congr. Photosynthethis. — 1998. — P. 34.
27. Jung W. ATP-synthase and other motor proteins // Proc. Natl. Acad. Sci.
USA. - 1999. -V. 96. - P. 4735-4737.
28. Karu A., Mondrianakis E. Fractionation and comparative studies of en-
zymes in aqueous extracts of spinach chloroplasts // Arch. Biochem., Biophys. —
1969. - V. 129. - P. 655.
29. Lien S., Racker F. Preparation and assay of chloroplast coupling factor
CF[ // Meth. Enzym. - 1971. - V. 23. - Part A. - P. 547-555.
30. Lowry O., Rosenbrought N., Farr A., Randall R. Protein measurement with
Folin phenol reagent//J. Biol. Chem. - 1951. -V. 193. - N 1. - P. 265-275.
31. McCarty R., Racker E. Partial resolution of the enzymes catalyzing photo-
phosphorylation.III.Activation of adenosine triposphatase and P32 labeled ortho-
phosphate adenosine triphosphate exchange in chloroplasts // J. Biol. Chem.—
1968.-V. 243.-N1.-P. 129.
32. Mitchell P. Chemiosmotic molecular mechanism for proton-translocating
adenosine triphosphotases // Biol. Rev. Cambrige Phill. Soc. — 1966. — V. 41.—
N3. -P. 445-502.
33. Mitchell P. A chemiosmotic coupling in oxidative and photosynthetic
phosphorylation // FEBS Lett. - 1974. - V. 43. - N 1. - P. 189-194.
34. Nakamoto R., Ketchum Ch., Kuo Ph. et al. Molecular mechanisms of rota-
tion catalysis in the FjFq ATP synthase // Biochim. Biophys. Acta. — 2000. —
V. 1458.-P. 289-299.
35. Nishimura M., Jto J., Chance B. Studies on bacterial photophosphoryla-
tion. III. A sensitive and rapid method of determination of photophosphoryla-
tion I/ Biochim. Biophys. Acta. — 1962. — V. 59. — N 1. — P. 177.
36. Oster G., Wang H. Reverse engineering a protein: the mechanochemistry
of ATP synthase // Biochim. Biophys. Acta. — 2000. — V. 1458. — P. 482 — 510.
37. Pick U., Racker E. Purification and reconstitution of the N,N'-dicyclo-
hexil-carbodiimide — sensitive ATPase complex from spinach chloroplasts // J.
Biol. Chem. - 1979. -V. 254. - N 8. - P. 2793-2799.
38. Shoshan V., Shavit N. On the reconstitution of photophosphorylation in
chloroplast membranes 11 Eur. J. Biochem. — 1973. — V. 37. — N 2. — P. 355—360.
39. Strotmann FL, Hesse H., Edelman K. Quantitative determination of cou-
pling factor CF[ of chloroplasts // Biochim. Biophys. Acta. — 1973. — V. 314. —
N l.-P. 202-210.
40. Vambutas V., Racker E. Partial resolution of the enzymes catalyzing
photophosphorylation. I. Stimulation of photophosphorylation by a prepara-
tion of latent Ca2+-dependent adenosine triphosphotase from chloroplasts // J.
Biol. Chem. - 1965. - V. 240. - N 6. - P. 2660.
249
Приложение 1
1. Кулачев А.П. Методы и средства анализа данных в среде Windows.
Stadia 6.0. — М.: НПО «Информатика и компьютеры», 1996.
2. Лакин Г. Ф. Биометрия. — М.: Высшая школа, 1990.
3. Максимов В.Н. Многофакторный эксперимент в биологии. — М.:
Изд-во МГУ, 1980.
4. Маслов Ю. И. Статистическая обработка данных биохимических ис-
следований // Методы биохимического анализа растений / Под ред.
В.В.Полевого, Г.Б.Максимова. — Л.: Изд-во ЛГУ, 1978. — С. 163—187.
5. Мятлев В.Д., Панченко Л. А., Терехин А. Т. Основы математической
статистики. — М.: МАКС Пресс, 2002.
6. Мятлев В.Д., Панченко Л. А., Терехин А. Т. Основы теории вероятно-
сти. — М.: МАКС Пресс, 2002.
7. Пакет электронных таблиц Quatro Pro. — Обнинск: Фирма «Элис»,
1992.
8. Плохинский Н.А. Алгоритмы биометрии. — М.: Изд-во МГУ, 1980.
Приложение 2
1. Баславская С.С., Трубецкова О.М. Практикум по физиологии расте-
ний. — М.: Изд-во МГУ, 1964.
2. Справочник химика: В 6 т. — Л.: Ленингр. отд. АН СССР, 1965. —
Т. III. - 1967. - Т. IV.
СОДЕРЖАНИЕ
Предисловие..................................................3
От авторов...................................................6
Общие указания о проведении работ «Большого практикума
по фотосинтезу»......................................... 8
Глава 1. Основные принципы используемых методов.............10
1.1. Абсорбционная спектрофотометрия....................10
1.2. Хроматографическое разделение веществ..............17
1.3. Электрофорез.......................................23
1.4. Центрифугирование..................................30
1.5. Полярография.......................................36
Глава 2. Характеристика пигментных систем фотосинтетического
аппарата....................................................42
2.1. Количественное определение пигментов методом
абсорбционной спектрофотометрии.........................46
2.1.1. Анализ пигментов с использованием экстрактов
пигментов в ацетоне и этиловом эфире............54
2.1.2. Анализ пигментов с использованием смеси
хлороформа с метанолом...............................57
2.2. Исследование фотосинтетических пигментов методами
хроматографии...........................................58
2.2.1. Разделение и количественное определение основных
пигментов листьев методом хроматографии на бумаге
(по Д. И. Сапожникову)..........................59
2.2.2. Разделение пигментов методом тонкослойной
хроматографии на сахарозе.............................63
2.2.3. Анализ пигментов методом тонкослойной
хроматографии на силикагеле...........................65
2.2.4. Хроматографическое разделение и препаративное
получение пигментов на колонках силикагеля
с обращенной фазой...................................68
2.3. Количественное определение пигментов группы
протохлорофилла..........................................70
2.3.1. Выделение протохлорофилла методом хроматографии
и его количественное определение..................73
2.3.2. Выделение и количественное определение
протохлорофиллида с использованием полярных
и неполярных растворителей...........................75
2.4. Анализ спектров поглощения и флуоресценции пигментов.78
2.4.1. Характеристика спектров поглощения пигментов
в растворах, гомогенатах и живых тканях листа
с помощью спектрофотометрического метода..............83
251
2.4.2. Регистрация спектров флуоресценции пигментов
в растворах, гомогенатах и живых тканях листа........84
2.4.3. Исследование превращений пигментов при освещении
этиолированных проростков методом регистрации
низкотемпературных спектров флуоресценции............94
2.5. Определение активности хлорофиллазы методом разделения
фитольных и бесфитольных форм зеленых пигментов..........96
Глава 3. Характеристика структурной организации и функциональной
активности фотосинтезирующих тканей и органов растений.... 100
3.1. Исследование мезоструктуры фотосинтетического аппарата
растений (по А. Т. Мокроносову)........................101
3.2. Определение интенсивности фотосинтеза и дыхания
растений...............................................114
3.2.1. Определение интенсивности фотосинтеза и дыхания
с использованием потенциометрического метода
регистрации углекислого газа и полярографического
определения кислорода...............................114
3.2.2. Определение световой кривой скорости фотосинтеза
с помощью газометрической установки открытого
типа, включающей инфракрасный газоанализатор.........121
3.3. Анализ функционального состояния фотосинтетического
аппарата растений методом регистрации индукции
флуоресценции хлорофилла...............................127
3.3.1. Использование метода регистрации индукционных
кривых флуоресценции хлорофилла для оценки
функционального состояния фотосинтетического
аппарата............................................133
3.3.2. Определение фотохимического и нефотохимического
тушения флуоресценции хлорофилла при освещении
листьев.............................................138
3.4. Определение активности рибулозобисфосфаткарбоксилазы
спектрофотометрическим методом (по И. М. Магомедову) ... 147
Глава 4. Методы выделения и хранения хлоропластов, тилакоидов
и пигмент-белковых комплексов..............................150
4.1. Выделение хлоропластов при механическом разрушении
тканей листа.............................................153
4.1.1. Выделение хлоропластов методом
дифференциального центрифугирования..................153
4.1.2. Выделение хлоропластов методом дифференциального
центрифугирования в градиенте плотности сахарозы.... 156
4.2. Выделение интактных хлоропластов из протопластов
С3-растений............................................157
4.3. Выделение тилакоидов хлоропластов из листьев растений ... 161
4.4. Выделение препаратов фотосистемы II...............162
4.5. Замораживание и хранение изолированных хлоропластов
и тилакоидов в жидком азоте............................164
252
4.5.1. Замораживание и хранение изолированных
хлоропластов в жидком азоте........................164
4.5.2. Замораживание и хранение изолированных
тилакоидов в жидком азоте..........................166
Глава 5. Определение фотохимической активности хлоропластов.167
5.1. Определение фотохимической активности хлоропластов
спектрофотометрическим методом........................172
5.1.1. Определение фотохимической активности хлоропластов
по скорости восстановления феррицианида калия
в различных условиях эксперимента.............172
5.1.2. Определение скорости нециклического потока
электронов в ЭТЦ хлоропластов по восстановлению
НАДФ+..............................................175
5.2. Определение активности транспорта электронов в ЭТЦ
хлоропластов по скорости выделения кислорода
полярографическим методом.............................181
5.2.1. Определение базального, сопряженного и разобщенного
потоков электронов в ЭТЦ фотосинтеза по активности
выделения кислорода полярографическим методом.181
5.2.2. Анализ транспорта электронов на отдельных участках
ЭТЦ хлоропластов с использованием искусственных
донорно-акцепторных систем и ингибиторов
транспорта электронов............................ 187
5.2.3. Определение фотохимической активности
субхлоропластных частиц ФС II полярографическим
методом............................................191
Глава 6. Энергетика фотосинтеза...........................193
6.1. Определение активности циклического
фотофосфорилирования с феназинметосульфатом.......196
6.2. Определение активности нециклического
фотофосфорилирования, сопряженного
с восстановлением феррицианида калия..................201
6.3. Определение фотоиндуцируемых изменений pH
в суспензии хлоропластов потенциометрическим методом... 203
6.4. Разобщение и реконструкция фосфорилирующей системы
хлоропластов...........................................210
6.5. Определение АТФ-азной активности сопрягающих белков
хлоропластов...........................................215
6.5.1. Определение активности светактивируемой Mg2+-
зависимой АТФ-азы хлоропластов................216
6.5.2. Выделение сопрягающего фактора 1 из хлоропластов
и определение его Са2+-АТФ-азной активности........217
Приложение 1. Статистическая обработка результатов опытов.222
Приложение 2. Приготовление растворов и справочные данные.232
Список литературы.........................................241
Учебное издание
Гавриленко Вероника Феодосиевна,
Жигалова Татьяна Викторовна
Большой практикум по фотосинтезу
Учебное пособие
Редактор Г. Г. Есакова
Технический редактор Н. И. Горбачева
Компьютерная верстка: Д. В. Федотов
Корректоры Г. Н. Петрова, Н. В. Савельева
Диапозитивы предоставлены издательством
Изд. № А-445. Подписано в печать 31.03.2003. Формат 60x90/16.
Гарнитура «Таймс». Бумага тип. № 2. Печать офсетная. Усл. печ. л. 16,0.
Тираж 20000 экз. (1-й завод 1 - 5 100 экз.). Заказ №2895.
Лицензия ИД № 02025 от 13.06.2000. Издательский центр «Академия».
Санитарно-эпидемиологическое заключение № 77.99.02.953.Д.002682.05.01 от 18.05.2001.
117342, Москва, ул. Бутлерова, 17-Б, к. 223. Тел./факс: (095)330-1092, 334-8337.
Отпечатано на Саратовском полиграфическом комбинате.
410004, г. Саратов, ул. Чернышевского, 59.