/












































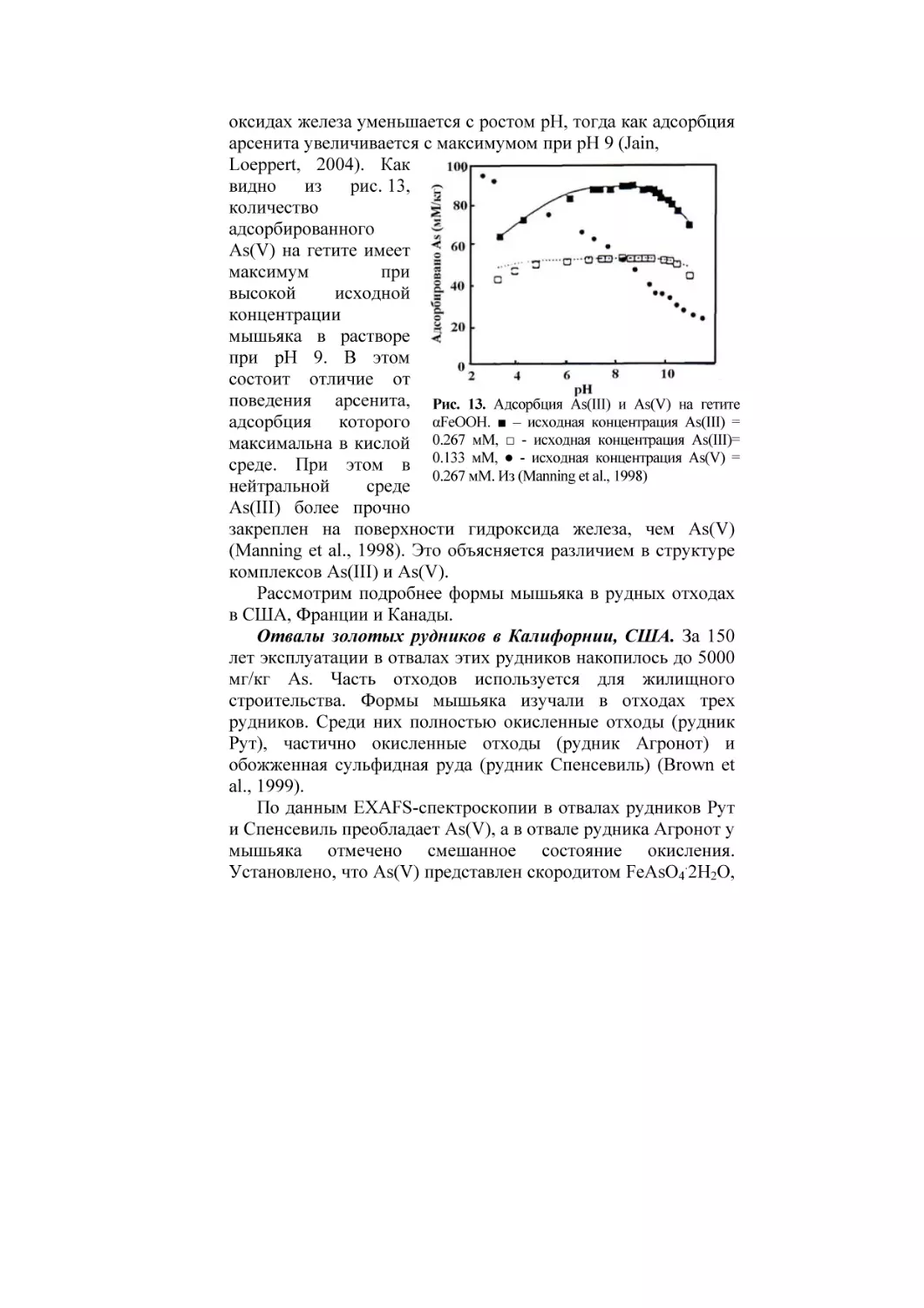


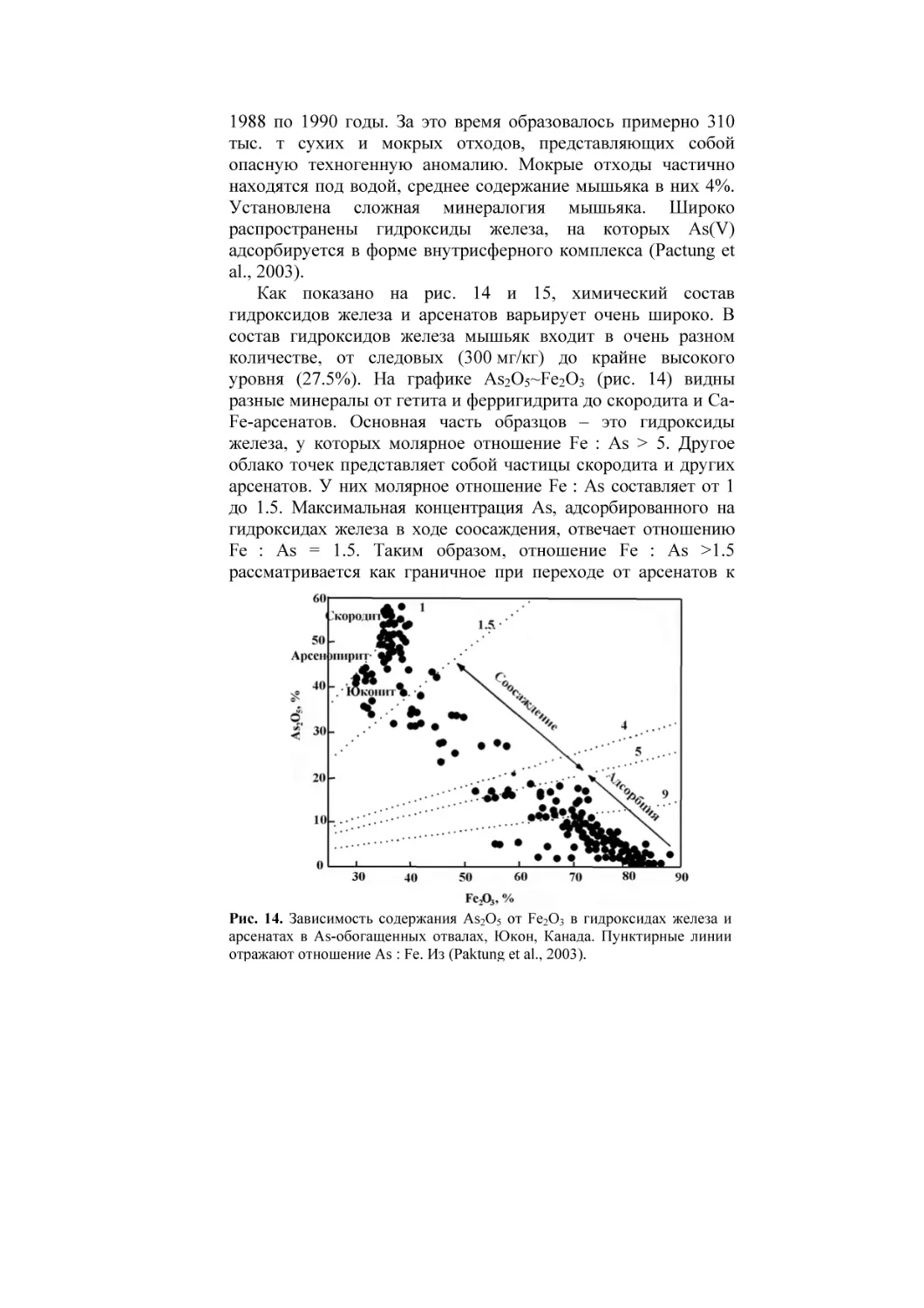





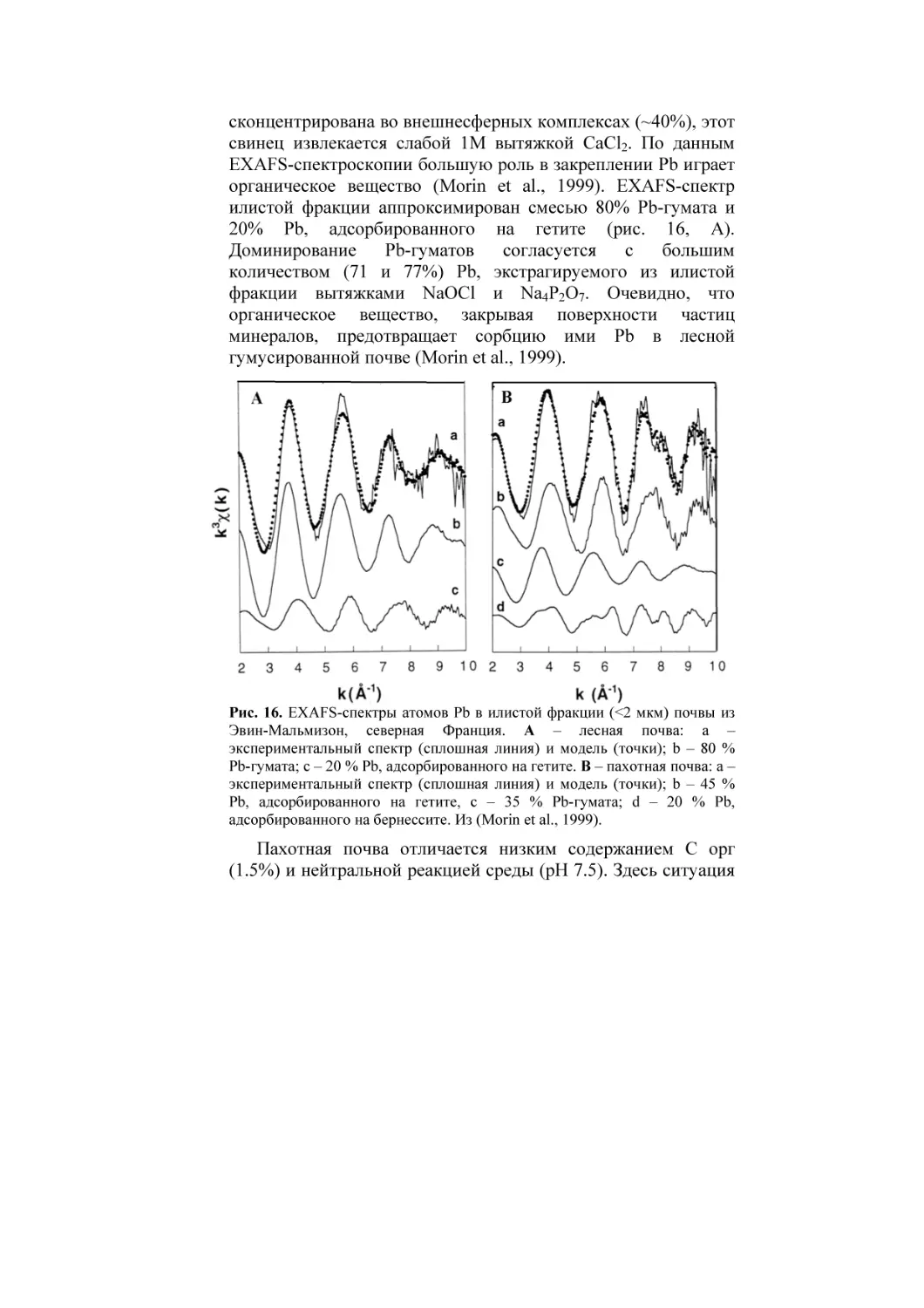

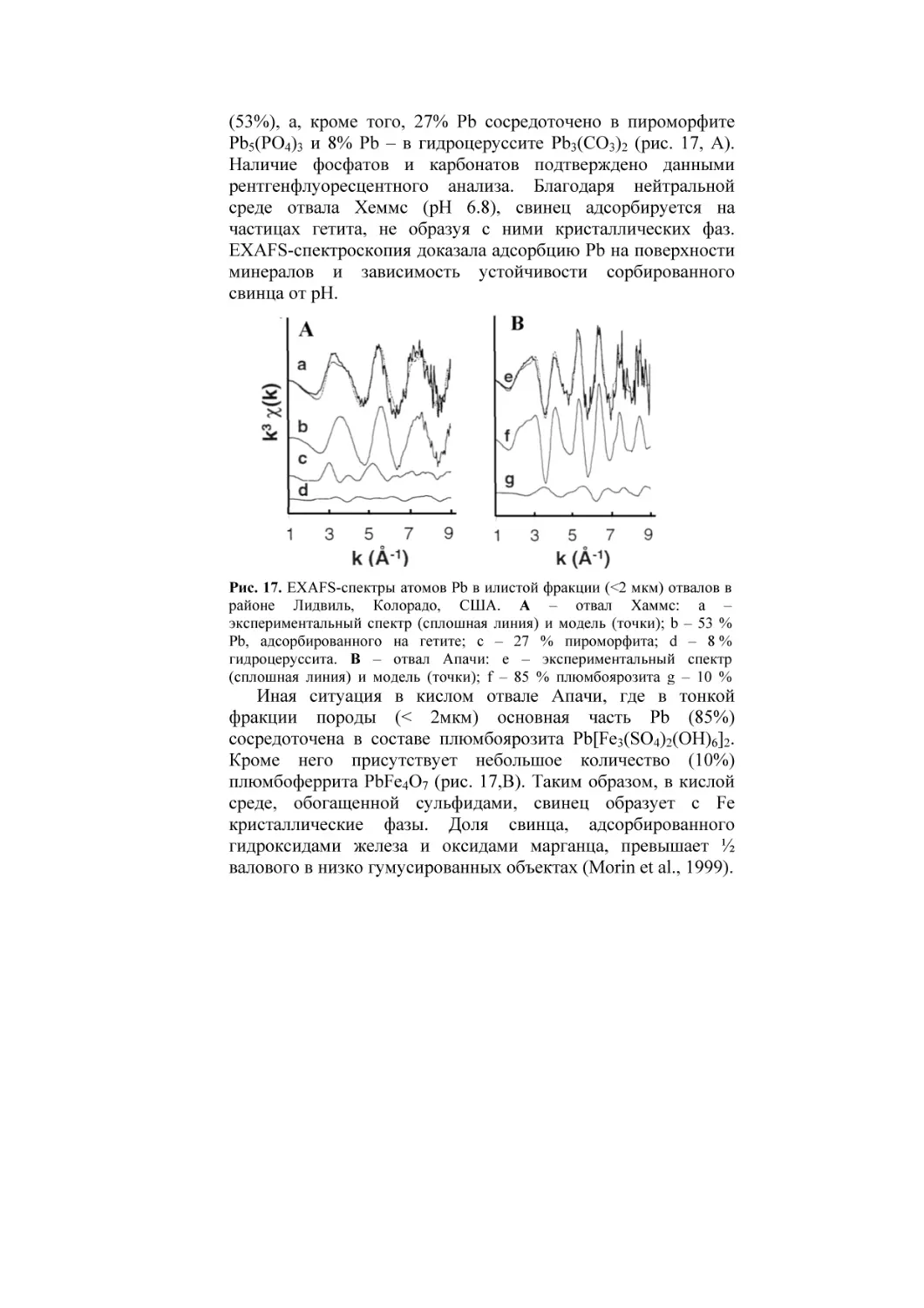

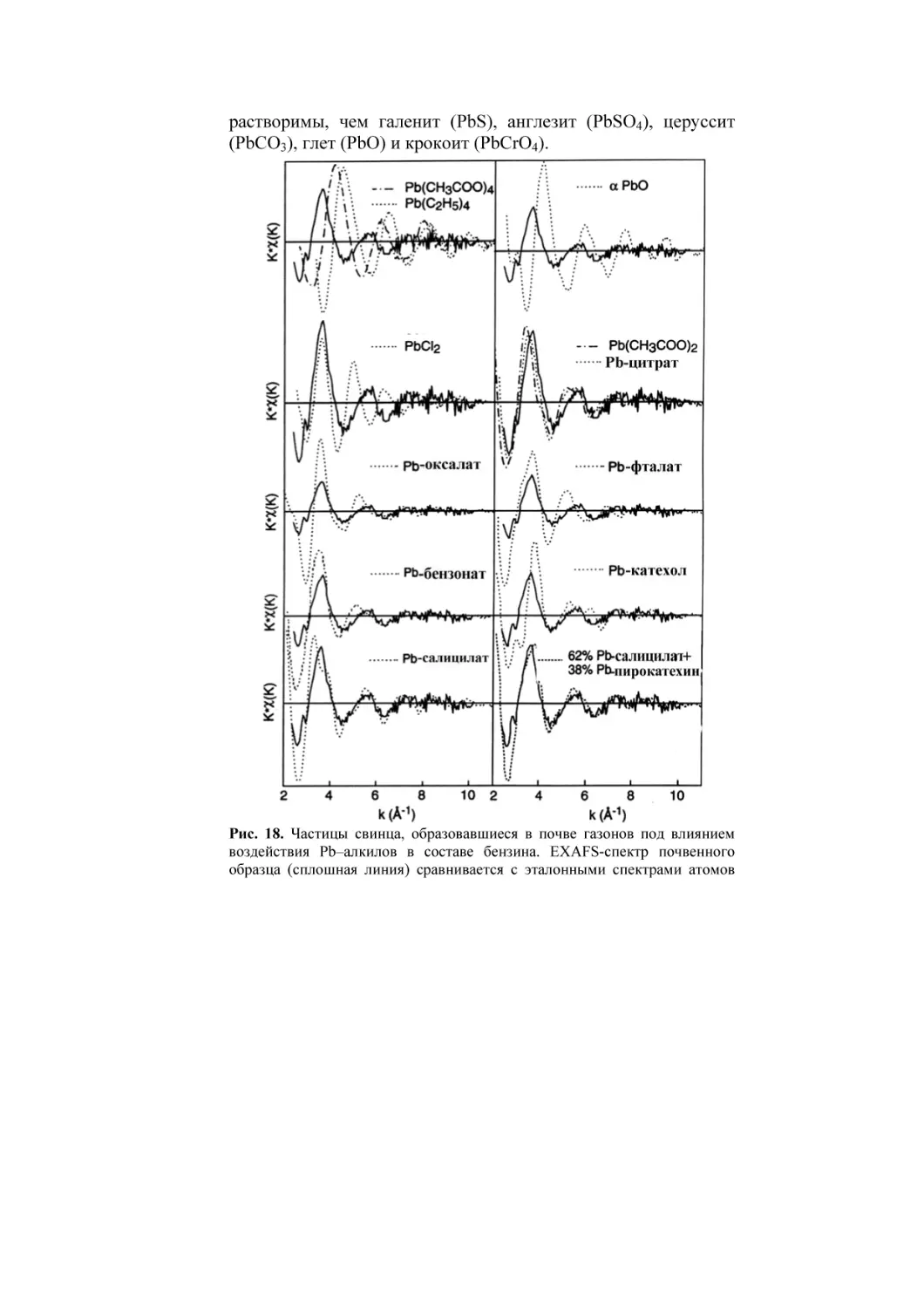





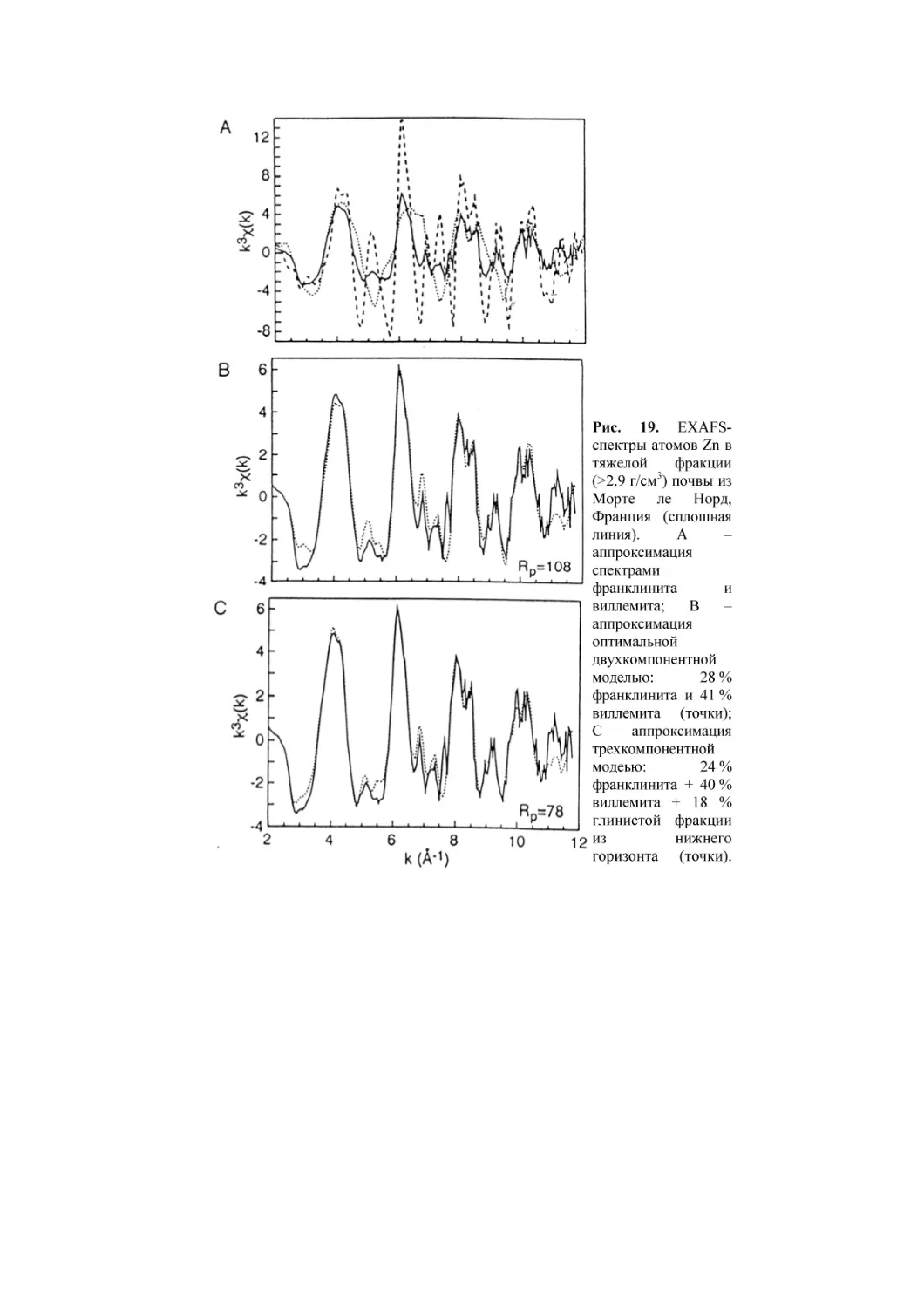
















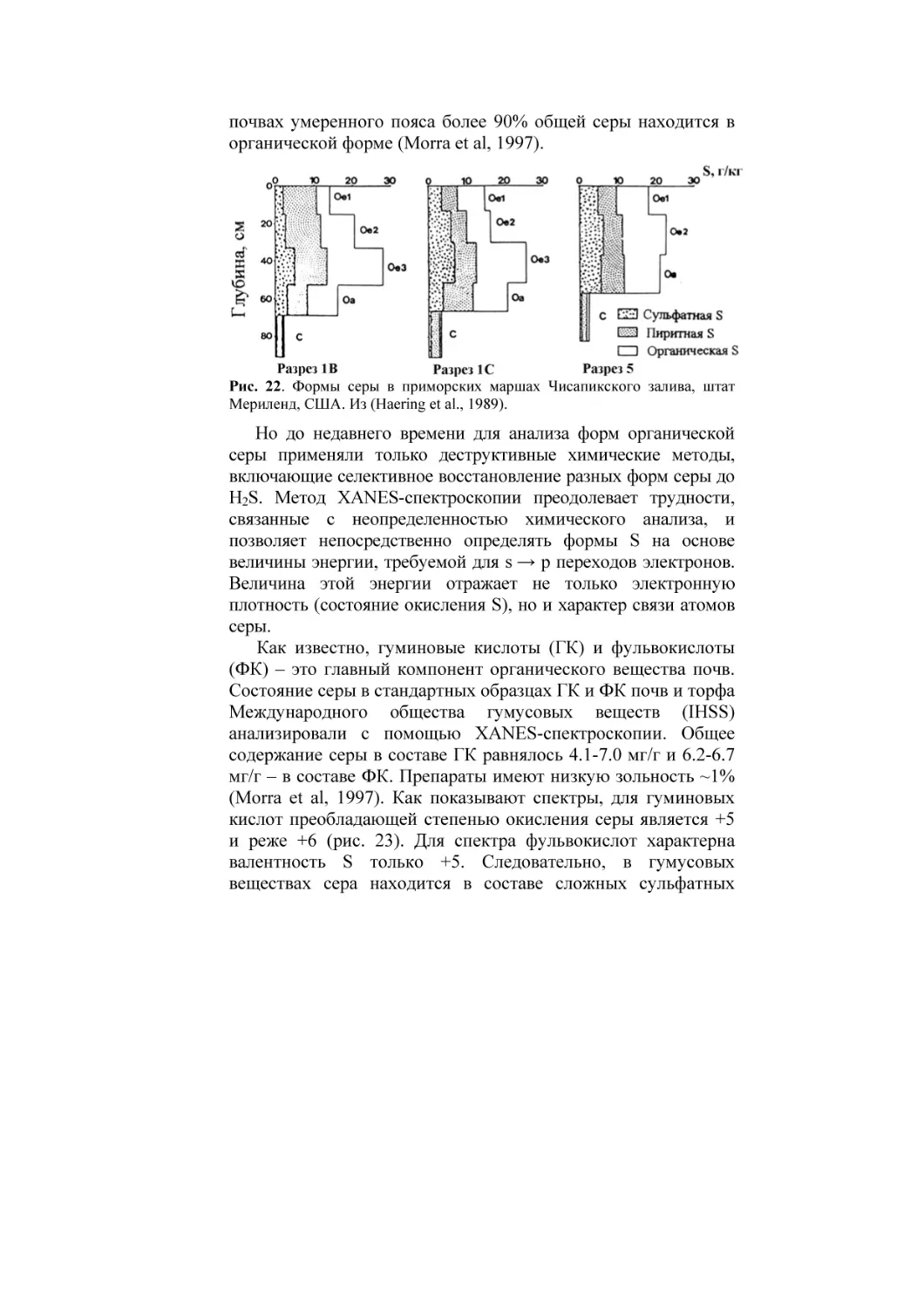




















Текст
РОССИЙСКАЯ АКАДЕМИЯ СЕЛЬСКОХОЗЯЙСТВЕННЫХ НАУК
ПОЧВЕННЫЙ ИНСТИТУТ имени В.В. ДОКУЧАЕВА
Ю.Н. ВОДЯНИЦКИЙ
ИЗУЧЕНИЕ
ТЯЖЕЛЫХ МЕТАЛЛОВ
В ПОЧВАХ
Москва
2005
ББК П03
В62
УДК 631.41
Рецензенты: доктор сельскохозяйственных наук, профессор
В.А. Большаков, доктор биологических наук Д.Л. Пинский,
кандидат биологических наук Л.К. Садовникова.
Ю.Н. Водяницкий
В62 Изучение тяжелых металлов в почвах. – М.: ГНУ
Почвенный институт им. В.В. Докучаева РАСХН, 2005.
ISBN 5
Отражены достижения современных методов исследования
форм тяжелых металлов и металлоидов. Обобщены
современные данные о химии и минералогии тяжелых
металлов для ряда техногенных геохимических аномалий.
Приводятся многочисленные сведения о формах тяжелых
металлов и их носителях в почвах Западной Европы и
Северной
Америки.
Основное
внимание
уделено
синхротронной рентгеновской технике третьего поколения. С
ее помощью получены принципиально новые данные о формах
металлов,
часто
расходящиеся
с
представлениями,
полученными методами химического фракционирования. Это
требует
пересмотра
основных
схем
химического
фракционирования тяжелых металлов. Анализ форм металлов
в
молекулярном
масштабе
позволяет
эффективно
мелиорировать загрязненные почвы и контролировать
состояние опасных техногенных геохимических аномалий.
ББК П03
3702040000 − 002
В
без объявл.
1Р8(03) − 05
©Почвенный институт
им. В.В. Докучаева
ISBN 5
©Водяницкий Ю.Н., 2005
ВВЕДЕНИЕ
Условно все почвоведение можно разделить на
«фундаментальное», изучающее в первую очередь генезис
почв, и «прикладное», призванное отвечать на следующие
вопросы: обеспечить население доброкачественной пищей и
дать ему здоровую среду обитания. Первую прикладную
проблему решают специалисты агропочвоведы. Второй
проблемой занимаются почвоведы-экологи. В сферу
экологического почвоведения попадают самые различные
проблемы, в том числе загрязнение почв тяжелыми металлами
и металлоидами, различными органическими поллютантами и
т.п. Оставив в стороне влияние выбросов органических
поллютантов, в этой книге сосредоточимся на проблеме
загрязнения почв тяжелыми металлами и металлоидами.
В Западной Европе и США проблема тяжелых металлов
возникла с началом технической революции, хотя ее масштабы
долго не осознавались общественностью. В 50-60-х гг. ХХ в.
парламенты этих стран принимают строгие законы,
направленные на повышение качества жизни, включая меры
по очистке воздуха, воды и почвы. В США Федеральный закон
о государственной политике в области окружающей среды был
принят в 1970 г. На базе этого комплексного закона в
последующие годы появились и другие: о чистой воде,
воздухе, почве и т.д. С их принятием экологическая ситуация в
стране резко изменилась к лучшему, выброс новых
поллютантов резко сократился.
Меры, принимаемые в промышленно развитых странах,
благоприятно отразились на экологии планеты. В глобальном
масштабе началось уменьшение загрязненности биосферы
тяжелыми
металлами,
что
обусловлено
закрытием
предприятий с устаревшей технологией и строительством
экологически чистых заводов. Но остались места,
сильнозагрязненные в результате предыдущей неразумной
деятельности человека.
Таким образом, надо различать глобальное, региональное и
локальное загрязнения тяжелыми металлами. На локальном
уровне проблема загрязнения почв тяжелыми металлами
остается. Достаточно вспомнить критическую экологическую
ситуацию на Кольском полуострове и в таких городах, как
Череповец, Норильск и Магнитогорск. В северной Франции и
Бельгии сильно загрязнены почвы, попавшие в зону
воздействия ныне снесенных металлургических заводов
(Manceau et al., 2000). В юго-восточной части Франции
образовались техногенные геохимические аномалии в виде
рудных отходов, обогащенных мышьяком (Morin et al., 2003).
Изучению тяжелых металлов посвящено множество работ.
Подробно изучены техногенные источники тяжелых металлов.
В почвах анализируется валовое содержание разных металлов
(Большаков и др., 1993; Орлов и др., 2002). Но оценить
опасность загрязнения на основе определения только валового
содержания не возможно. Токсическое действие поллютантов
зависит от их форм, от степени окисления элемента с
переменной валентностью, от характера закрепления металлов
минеральными и органическими носителями и др. Среди
носителей тяжелых металлов основную роль играют
гумусовые вещества и глинистые минералы, а также оксиды
марганца и железа. Последние выполняют важную роль в
фиксации тяжелых металлов такими новообразованиями как
Fe-Mn-ортштейны (Водяницкий, 2005).
Традиционно формы тяжелых металлов в почвах
анализируют косвенно, путем химического анализа и
последующего привлечения методов термодинамического
расчета (Горбатов, 1988; Пинский, 1997). Но в последние годы
активно развиваются прямые методы определения форм тяжелых
металлов и металлоидов в почвах. Среди них ведущее место
занимает синхротронная рентгеновская техника. Именно с ее
помощью получена важнейшая информация о формах тяжелых
металлах в почвах.
Автор благодарит И.В. Перминову за консультации.
Особая признательность рецензентам В.А. Большакову, Д.Л.
Пинскому и Л.К. Садовниковой за полезные советы.
Глава 1. ИЗУЧЕНИЕ ТЯЖЕЛЫХ МЕТАЛЛОВ И
МЕТАЛЛОИДОВ МЕТОДАМИ СИНХРОТРОННОЙ
РЕНТГЕНОВСКОЙ РАДИАЦИИ
Идентификация форм тяжелых металлов в почвах
сопряжена
с
рядом
трудностей.
Традиционная
рентгендифрактометрия часто оказывается бесполезной, так
как не способна выявлять малое количество минералов этих
металлов и устанавливать их связь с фазами-носителями.
Просвечивающая электронная микроскопия, сопровождаемая
микродифракцией электронов, позволяет выявить замещение
тяжелыми металлами железа и марганца в составе их оксидов
(Чухров и др., 1989; Водяницкий, 2005). Но просвечивающая
электронная
микроскопия
дает
лишь
качественную
информацию. Кроме того, она предполагает диспергацию
почвенного образца, что в ряде случаев искажает реальные
отношения между почвенными фазами. Наиболее эффективно
применение синхротронного рентгеновского излучения.
Методы синхротронной радиации, основанные на
использовании ускорителей, сообщающих заряженным
частицам огромную энергию, сейчас используются в
различных отраслях науки, в том числе в почвоведении. В
синхротронах, огромных сооружениях диаметром в сотни
метров, элементарные частицы ускоряются в магнитном поле,
образуя мощное рентгеновское излучение очень высокой
яркости и чистоты.
Третье поколение источников синхротронной радиации
используется в Европейском центре синхротронной радиации
(ESRF) в Гренобле, Франция, а также в США: в центре
Advanced Light Source (ALS) в университете Беркли, в
Стенфордской синхротронной радиационной лаборатории
(SSRL) и в центре National Synchrotron Light Source (NSLS) в
Брукхевенской Национальной Лаборатории, шт. Нью-Йорк.
Исследования тяжелых металлов в почвах, выполненные в
этих центрах, начались с 1994 г. (Cotter-Howells et al., 1994;
Manceau et al., 1996; Manceau et al., 2000; Manceau et al., 2002;
Jain, Loeppert, 2004).
В настоящее время методы синхротронной радиации
позволяют изучать состав твердой фазы в микрообъеме,
состояние окисления элементов с переменной валентностью,
распределение тяжелых металлов и металлоидов в
ненарушенных почвенных образцах и характер их связи с
фазами-носителями. Для этого используется рентгеновская
микрофлуоресценция (µXRF), рентгеновская микродифракция
(µXRD), анализ рентгеновских спектров вблизи полосы
поглощения (XANES) и расширенный анализ тонкой
структуры спектров поглощения (EXAFS). Эта структурная
техника
имеет
необходимую
специализацию:
чувствительность к слабоупорядоченным частицам и
достаточный предел идентификации форм тяжелых металлов
при их содержании свыше ~0.01 %. При обычном режиме
съемки на облучение одной точки уходит от нескольких
десятков секунд до нескольких минут, в результате на анализ
одного почвенного образца требуется несколько часов.
Тонкая структурная информация позволяет обнаружить
большинство доминирующих форм тяжелых металлов. При
благоприятных условиях идентифицируется и определяется
содержание рассеянных фаз (Scheinost et al., 2002). Обычно
разложение экспериментального спектра позволяет выявить до
3-4 основных фаз.
Рентгеновская микрофлуоресценция
Метод
рентгеновской
микрофлуоресценции
давно
используется
для
определения
соотношения
между
различными металлами в микромасштабе. Но применение
техники синхротронного излучения и улучшение качества
фокусирующей аппаратуры резко увеличило эффективность
метода. Исключительная чувствительность микроэлементов к
синхротронному рентгеновскому излучению и высокое
пространственное разрешение (несколько мкм2) объясняют
растущий интерес почвоведов к этой технике.
Первый шаг анализа частиц минералов состоит в
составлении картин распределения химических элементов в
пределах гетерогенного почвенного образца. Массив данных
представляется в виде картин распределения элементов. Они
обычно даются в обратном контрасте, т.е. высокая
концентрация элемента представлена темной областью
(Manceau et al., 2002). При этом для каждого элемента
выбирается своя шкала светлоты, поэтому сравнивать
содержание различных элементов по степени светлоты нельзя.
Прямые
и
обратные
корреляции
концентраций
микроэлементов с Fe, Mn, P и S позволяют отнести тот или
иной микроэлемент к (гидр)оксидам железа или марганца,
фосфатам и/или сульфидам (Manceau et al., 2002).
Важно затем адекватно обработать большой массив
данных. Наиболее часто при картировании используют шкалу
серых тонов для каждого элемента, после чего картины
сравнивают (Manceau et al., 2002). Следующий уровень
обработки данных – составление трехцветных карт. Обычно
картина включает красный, зеленый и голубой цвета. Яркость
исследуемой области зависит от суммарного вклада
компонентов, а цветовой тон – от их соотношения (Manceau et
al., 2002).
Более
точную
информацию
дает
использование
статистических
методов,
в
частности,
применение
кросскорреляционной
функции.
Строятся
диаграммы
рассеяния элементов. На них по одной оси откладывается
концентрация одного элемента, а по другой – другого
элемента. Для установления характера статистической связи
между
элементами
применяют
модифицированный
коэффициент корреляции Пирсона для непараметрических
показателей ρ (Manceau et al., 2002). Метод позволяет
установить статистические связи между содержанием тяжелых
металлов и макроэлементами (Fe, Mn), образующими
минералы-носители. Высокая корреляция между элементами в
пространстве указывает на вероятность закрепления тяжелого
металла конкретным минералом-носителем.
В качестве примера рассмотрим распределение химических
элементов в бурой лесной почве из района Монте де Норд,
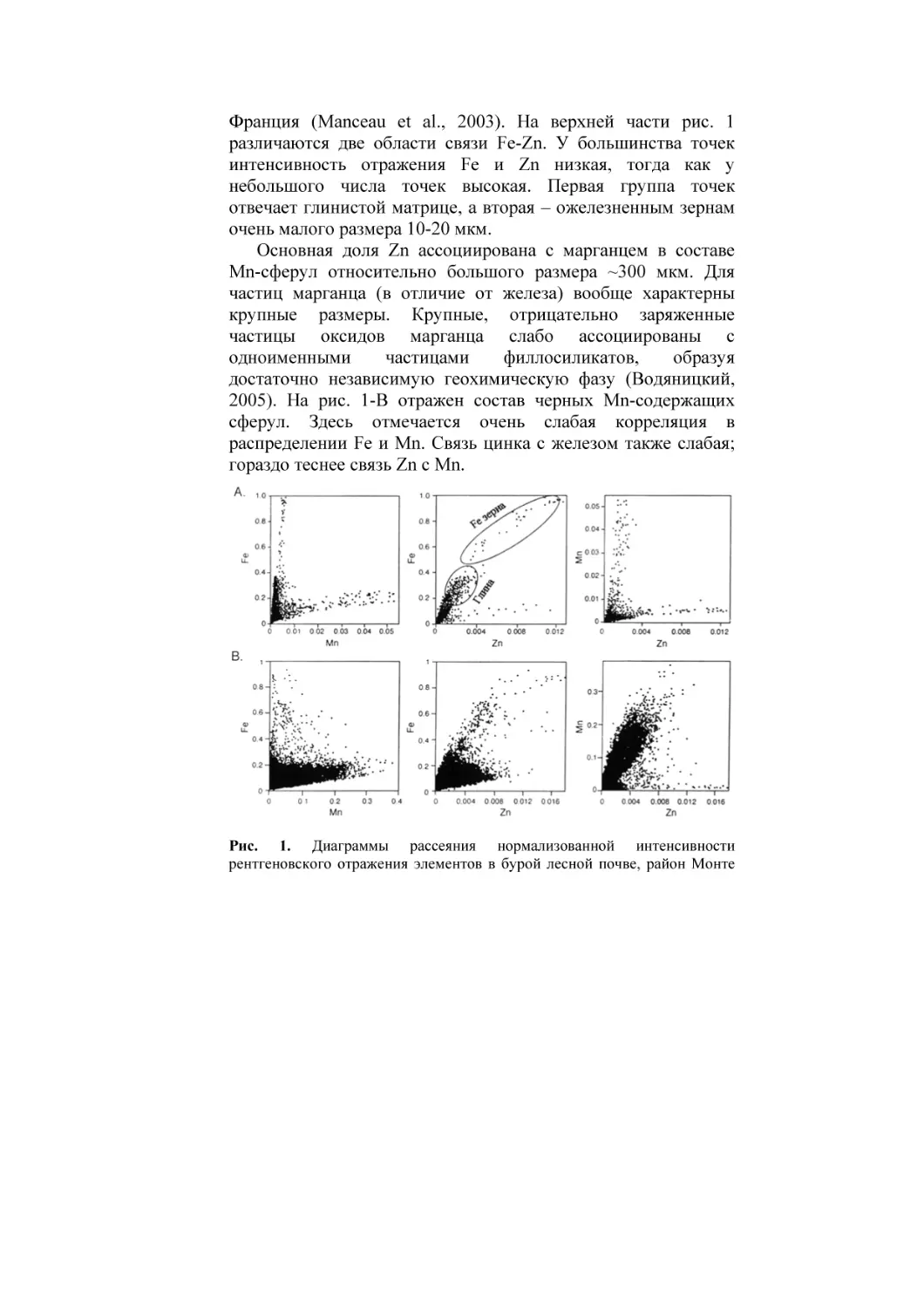
Франция (Manceau et al., 2003). На верхней части рис. 1
различаются две области связи Fe-Zn. У большинства точек
интенсивность отражения Fe и Zn низкая, тогда как у
небольшого числа точек высокая. Первая группа точек
отвечает глинистой матрице, а вторая – ожелезненным зернам
очень малого размера 10-20 мкм.
Основная доля Zn ассоциирована с марганцем в составе
Mn-сферул относительно большого размера ~300 мкм. Для
частиц марганца (в отличие от железа) вообще характерны
крупные размеры. Крупные, отрицательно заряженные
частицы оксидов марганца слабо ассоциированы с
одноименными
частицами
филлосиликатов,
образуя
достаточно независимую геохимическую фазу (Водяницкий,
2005). На рис. 1-В отражен состав черных Mn-содержащих
сферул. Здесь отмечается очень слабая корреляция в
распределении Fe и Mn. Связь цинка с железом также слабая;
гораздо теснее связь Zn с Mn.
Рис. 1. Диаграммы рассеяния нормализованной интенсивности
рентгеновского отражения элементов в бурой лесной почве, район Монте
де Норд, Франция. А – глинистая матрица, В – Fe-Mn-ортштейн. Из
(Manceau et al., 2003).
Таким образом, устанавливают зависимости между
содержанием тяжелых металлов и макроэлементами (Fe и Mn),
образующими минералы-носители. Высокая корреляция
между элементами в пространстве указывает на вероятность
закрепления тяжелого металла конкретным минераломносителем.
Рентгеновская микродифракция
Рентгеновская микродифракция позволяет не только
идентифицировать отдельные почвенные частицы, но и, что
более важно, фиксировать положение минералов в пределах
почвенного образца, что не в состоянии дать ни традиционная
рентгендифрактометрия, ни просвечивающая электронная
микроскопия, анализирующие нарушенные образцы. Основная
ценность рентгеновской микродифракции состоит в
возможности сравнивать картины распределения рассеянных
элементов и минералов-носителей. Если химический элемент и
минерал приурочены к одной и той же области, как, например,
в случае Zn и бернессита, то можно заключить, что Zn
ассоциирован с бернесситом.
Рентгеновское синхротронное излучение на несколько
порядков интенсивнее излучения, которое генерируется
трубкой с вращающимся анодом в лабораторном
оборудовании. Высокая коллимация и яркость синхротронного
излучения обеспечиваются использованием совершенной
микрофокусирующей оптики. Это позволяет исследовать
структуру кристалла микронного размера. Но почвы – это
гетерогенные многофазные системы с нонометровыми
размерами частиц, в результате часто их дифракционные
картины содержат только один широкий рефлекс. Достоинство
синхротронного излучения выражается в возможности
получения исключительно тонкого высокоинтенсивного и
параллельного пучка рентгеновских лучей, что уменьшает
гетерогенность материала в данной области почвенного
образца и позволяет собрать большое количество
дифракционных картин.
Активные исследования почв этим методом проводятся в
центре Advanced Light Source в университете Беркли, США.
Рентгеновский луч фокусируется до субмикронного размера с
использованием пары эллиптических зеркал особой
конфигурации. Энергия луча меняется в широком интервале
без смещения его фокуса. Диапазон изменения энергии луча
(Е) 5.5 – 14 кэВ, что соответствует длине волны λ = 0.885 –
2.25Å. На экспериментальной установке анализируются
образцы размером 9 х 9 см с размером пикселя 88 мкм.
Итоговый угловой диапазон 2θ составляет около 30º-40º при
разрешении Δθ = 0.03º, что достаточно для получения hkl
отражения разных минералов (Manceau et al., 2002).
На освещаемой площади 14 х 11мкм объем дифракции
составляет ~7 х 10-3 мкм3. В почвах фиксируются два
основных типа дифракционных картин: острые точечные
отражения от микронных и субмикронных кристаллов и
дебаевские кольца от нанометровых частиц. Грубые зерна
дают острые и прерывистые кольца. В почвах кристаллы
кварца, полевых шпатов, карбонатов и оксидов титана
формируют точечную дифракционную картину. Слюды и
каолинит обычно дают умеренно структурированную картину.
Структурные эффекты используют для распознания минералов
в случае наложения рефлексов, например, каолинита и
бернессита (Na,Ca)Mn7O14.2.8H2O. Эти два минерала трудно
различаются в почвах, так как оба дают интенсивный рефлекс
00l при 7.1Å. Когда анализируют образец, обогащенный Mn,
то с этим рефлексом связывают присутствие бернессита. Так
как кристаллы бернессита разупорядочены, то они образуют
сплошные дифракционные кольца, тогда как достаточно
крупные и упорядоченные частицы каолинита и могут давать
пятнистые кольца. Так, рентгеновская микродифракция
различает эти два типа минералов без разрушения почвенного
образца.
Распределение высокодисперсных минералов получают
при растровом плоскостном анализе образца, суммируя всю
дифракционную информацию. Подчеркнем, что рентгеновский
микродифракционный анализ по чувствительности уступает
электронному. Но рентгеновская синхротронная техника имеет
важные преимущества: анализировать неразрушенные образцы
почв и определять в них количество минералов. При этом
выполняется
параллельно
несколько
анализов:
микрофлуоресцентный,
микродифракционный,
анализ
спектров вблизи края полосы поглощения и расширенный
анализ тонкой структуры спектров поглощения. Это позволяет
на одной экспериментальной установке изучить распределение
элементов,
определить
степень
их
окисления,
идентифицировать минералы-носители и их количество в
почве, а также определить механизм закрепления тяжелого
металла в молекулярном масштабе.
Анализ рентгеновских спектров вблизи
края полосы поглощения
Соотношение двух видов синхротронного исследования:
анализа спектров вблизи края полосы поглощения
рентгеновских лучей (XANES-спектроскопии) и расширенного
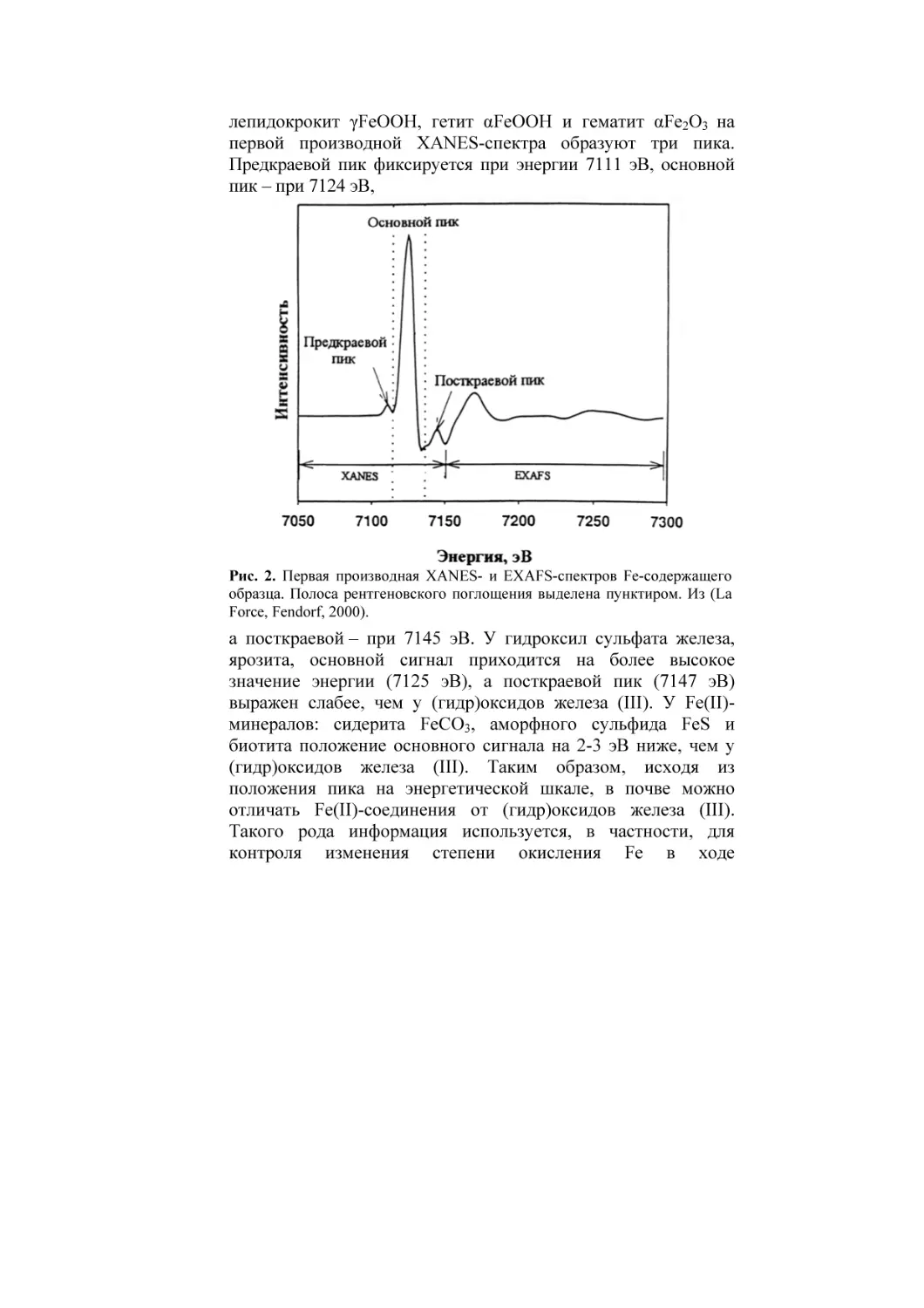
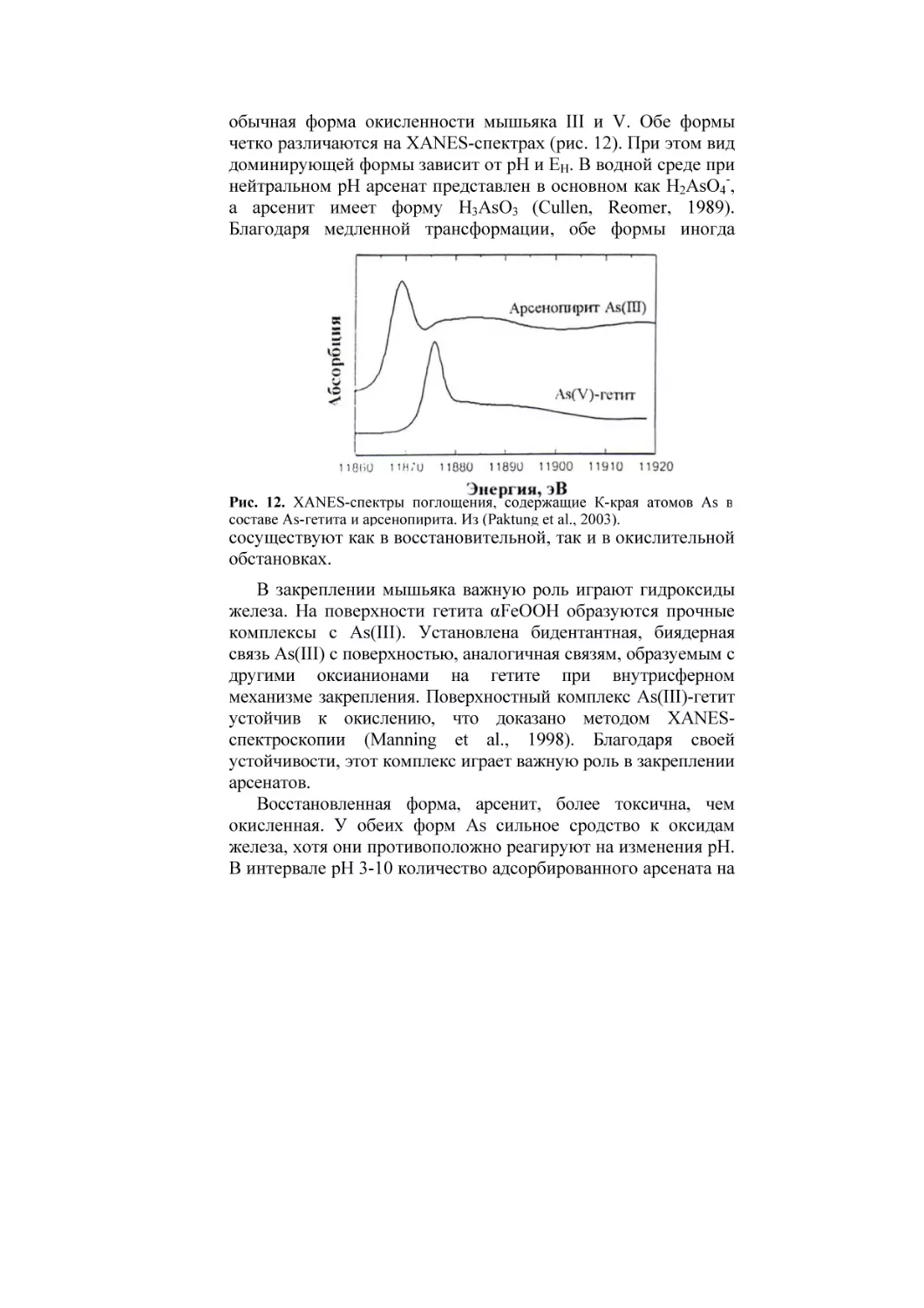
анализа тонкой структуры спектров поглощения (EXAFSспектроскопии) для железа показано на рис. 2 (La Force,
Fendorf, 2000). На первой производной полного спектра
выделена пунктиром полоса поглощения рентгеновского
излучения для Fe. Основной пик XANES-спектра лежит в
пределах, а два дополнительных – до и после этой полосы.
Предкраевой сигнал отражает электронные переходы 1s → 3d
и характеризует окружение атомов Fe. Так, у тетраэдрически
координированного Fe в составе магнетита предкраевой
сигнал больше, чем у октаэдрически координированного Fe.
Основной сигнал отражает электронное состояние элемента, а
посткраевой используется для определения форм элемента.
Обсудим особенности XANES-спектров гидроксиды
железа (III). Ферригидрит FeOOH . 2Fe2O3 . 4H2O,
лепидокрокит γFeOOH, гетит αFeOOH и гематит αFe2O3 на
первой производной XANES-спектра образуют три пика.
Предкраевой пик фиксируется при энергии 7111 эВ, основной
пик – при 7124 эВ,
Рис. 2. Первая производная XANES- и EXAFS-спектров Fe-содержащего
образца. Полоса рентгеновского поглощения выделена пунктиром. Из (La
Force, Fendorf, 2000).
а посткраевой – при 7145 эВ. У гидроксил сульфата железа,
ярозита, основной сигнал приходится на более высокое
значение энергии (7125 эВ), а посткраевой пик (7147 эВ)
выражен слабее, чем у (гидр)оксидов железа (III). У Fe(II)минералов: сидерита FeCO3, аморфного сульфида FeS и
биотита положение основного сигнала на 2-3 эВ ниже, чем у
(гидр)оксидов железа (III). Таким образом, исходя из
положения пика на энергетической шкале, в почве можно
отличать Fe(II)-соединения от (гидр)оксидов железа (III).
Такого рода информация используется, в частности, для
контроля изменения степени окисления Fe в ходе
последовательного химического экстрагирования железа и
марганца из почв и осадков.
XANES-спектроскопия очень эффективна при определении
степени окисления элементов с переменной валентностью,
таких как железо, марганец или сера (Schulze et al., 1995; Xia et
al., 1998). Состояние окисления элементов предварительно
устанавливается на моделях. Затем экспериментальные
спектры, обычно включающие элемент в различных стадиях
окисления, разлагают на составляющие согласно эталонам,
находящимся в базе данных.
В органогенных почвах судьба тяжелых металлов тесно
связана с серой. Важные исследования проведены по
изучению состояния окисления серы в составе гумусовых
субстанций, извлеченных из почв и водных осадков (Xia et al.,
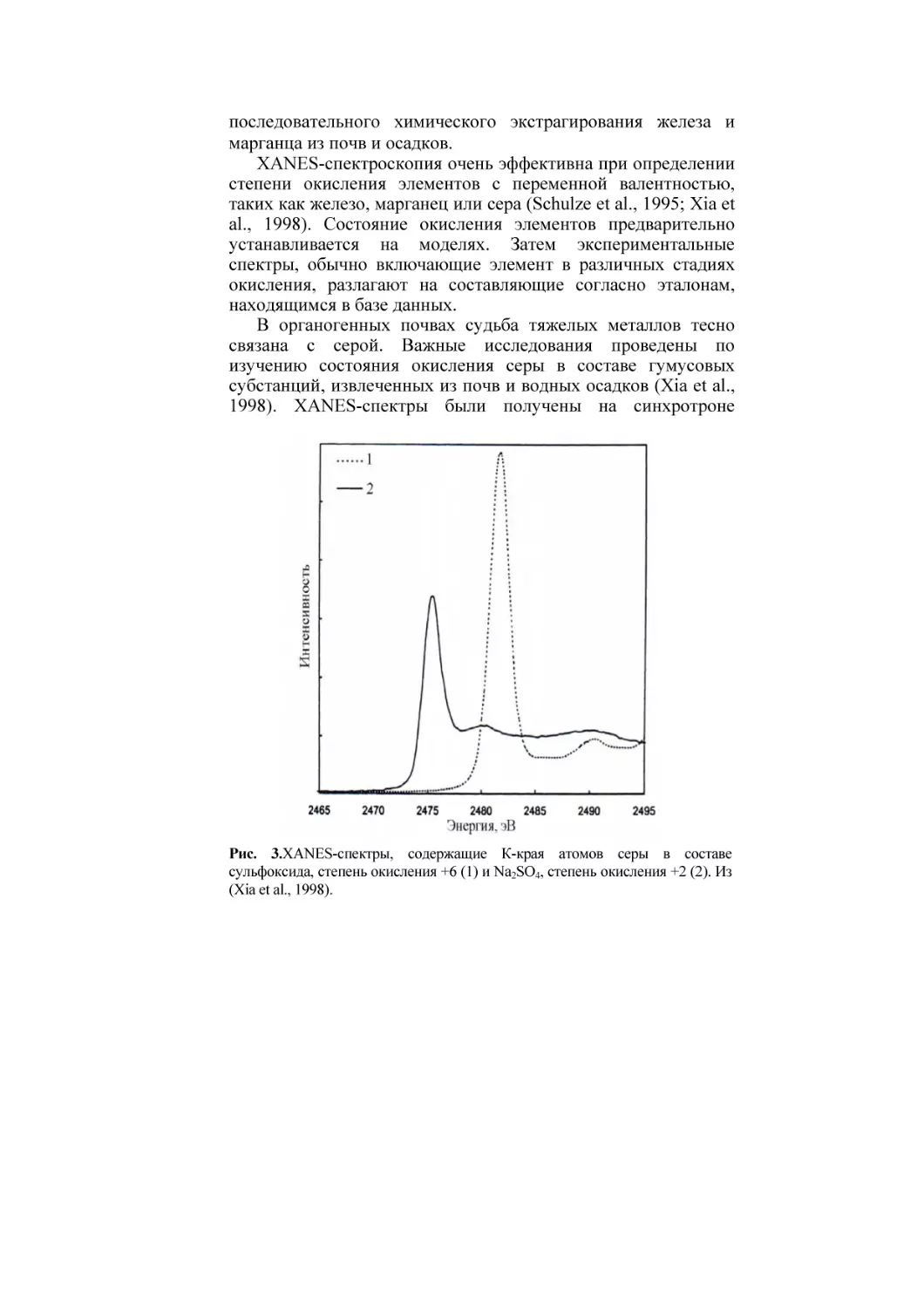
1998). XANES-спектры были получены на синхротроне
Рис. 3.XANES-спектры, содержащие К-края атомов серы в составе
сульфоксида, степень окисления +6 (1) и Na2SO4, степень окисления +2 (2). Из
(Xia et al., 1998).
Брукхевенской национальной лаборатории, штат Нью-Йорк,
США. По положению пика s → p перехода электронов
устанавливают степень окисления серы (рис. 3). Полученные
значения энергии для пика отражающего s → p переход
электронов, сравнивают с положением основного пика
элементной серы (2472 эВ). Сканирование образца начинается
на 20 эВ ниже абсорбционного пика и ведется до 50 эВ выше
его значения с шагом 0.2 эВ (Xia et al., 1998).
В силу того, что в почве сера обычно находится в разном
состоянии окисления, на экспериментальном спектре
наблюдается несколько пиков или перегибов. Поэтому
XANES-спектр аппроксимируют серией гауссовых пиков,
отражающих индивидуальные s → p переходы, а также –
функцией арктангенса, отражающей подъем нулевой линии
экспериментального спектра (Xia et al., 1998). Само
разложение спектра на гауссовы компоненты проводят из
условия минимума среднего квадратичного отклонения
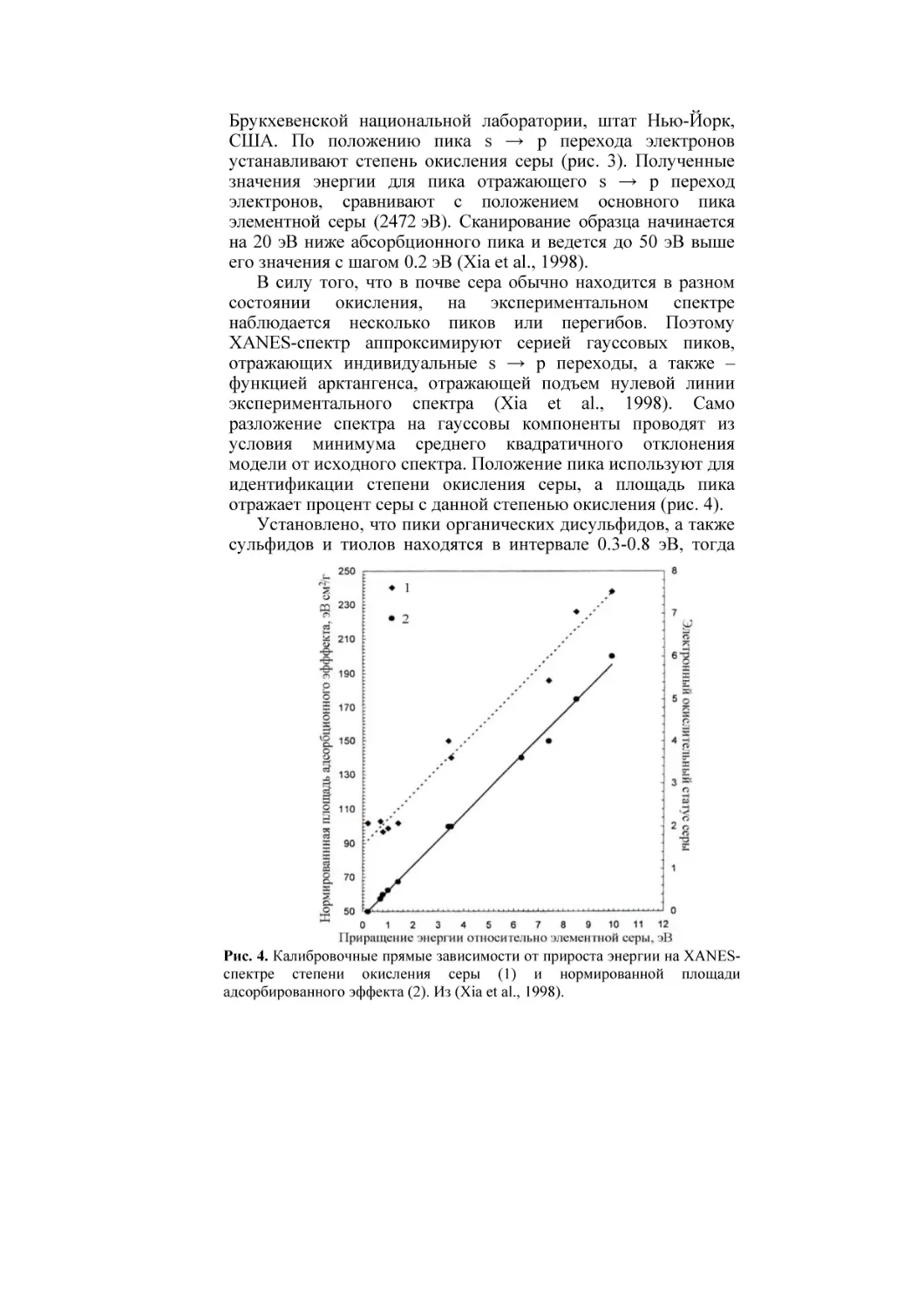
модели от исходного спектра. Положение пика используют для
идентификации степени окисления серы, а площадь пика
отражает процент серы с данной степенью окисления (рис. 4).
Установлено, что пики органических дисульфидов, а также
сульфидов и тиолов находятся в интервале 0.3-0.8 эВ, тогда
Рис. 4. Калибровочные прямые зависимости от прироста энергии на XANESспектре степени окисления серы (1) и нормированной площади
адсорбированного эффекта (2). Из (Xia et al., 1998).
как пики органических тиофенов в интервале 1.3 - 1.8 эВ. Хотя
идентифицировать специфические химические формы каждого
типа восстановленных частиц серы не удается, но можно
отличать формы органосульфидов и тиолов от других форм
органических тиофенов. В результате XANES-спектроскопия
успешно выявляет степень окисления серы. Точность
установления доли серы в каждом окислительном состоянии
составляет от ±5 до ±10% (Xia et al., 1998).
Фактическое состояние окисления, в отличие от
формального, отражает реальную электронную плотность в
валентной оболочке элемента. Например, формальное
состояние окисления серы равно 0 в составе органосульфидов
(R-S-R) и –1 в тиолах (R-SН). Но фактическая степень
окисления серы в органосульфидах и тиолах колеблется от 0
до 0.5 в зависимости от того, связана сера с S, H или с C или с
комбинацией этих элементов (табл. 1).
Таблица 1. Структура компонентов серы с различной степенью окисления.
Компонент
Структура
Степень окисления
формальная
фактическая
Сульфид
R–S–R
0
≈ 0.5
Тиол
R–SH
-1
≈ 0.5
Тиофен
Сульфоксид
Сульфит
Сульфон
Сульфонат
S
O
R–S–R
O
R–O–S–OH
O
R–S–R
≈1
2
≈2
4
≈ 3.6
4
≈4
5
5
6
6
O
O
H–O–S–R
Сульфат
0
O
O
H–O–S–O–R
O
Разница между фактическим и формальным окислением не
существенна для органической серы с высокой валентностью
(≥ +4). Поэтому значение окисления высоковалентной серы
округляют до целого числа.
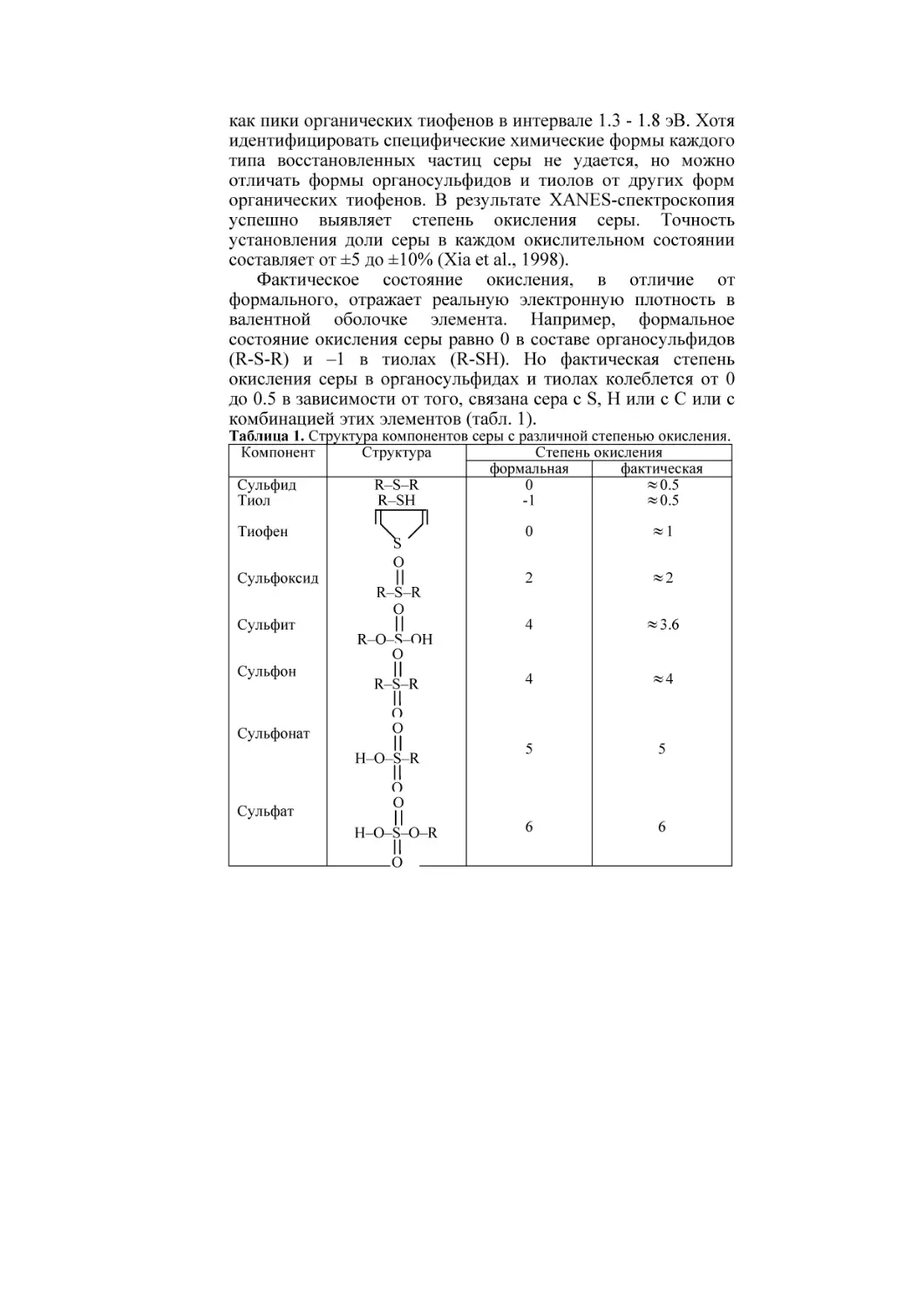
В качестве примера спектры после корректировки нулевой
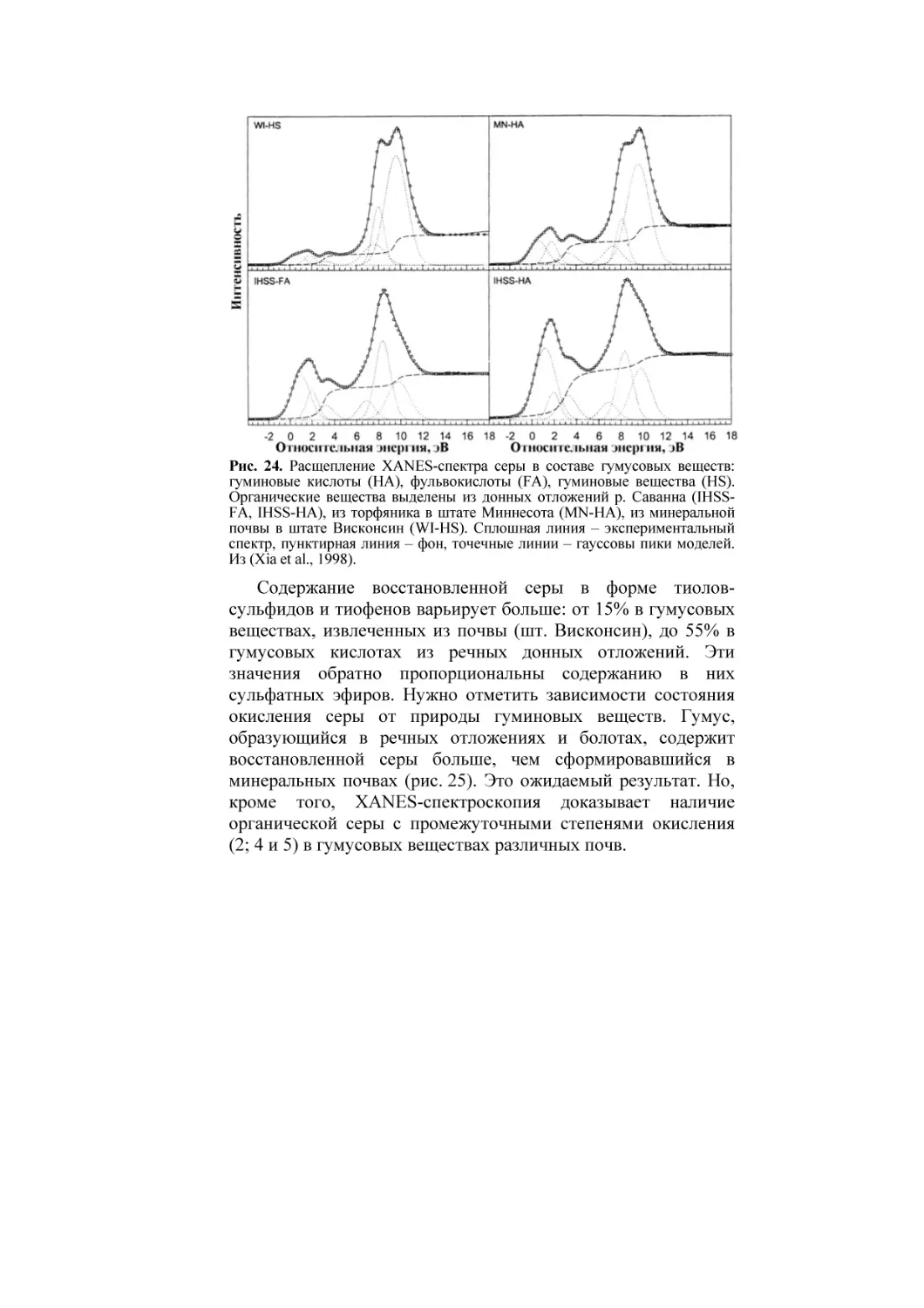
линии для 4-х гумусовых образцов даны на рис. 5 (Xia et al.,
1998). На каждом спектре отмечено четыре очевидных
максимума (b, c, e и f) превышения энергии поглощения Ккраем атомов серы в интервале от 0 до 10 эВ. Два других
максимума (a и d) не очевидны, но вторая производная спектра
на рис. 5 фиксирует эти скрытые пики. Наличие максимумов
Рис. 5. XANES-спектры, содержащие К-края атомов серы в составе гуминовых
кислот, выделенных из речной воды (1), из торфяной почвы шт. Миннесота (2), из
минеральной почвы, шт. Висконсин (3). На второй производной спектра
гуминовых кислот (4), выделенных из торфяной почвы, шт. Миннесота, скрытые
пики а и d показаны стрелками. Энергия выражена относительно края атомов
элементарной серы. Из (Xia et al., 1998).
при разных значениях энергии показывает, что сера
существует в разных состояниях окисления во всех четырех
гумуссодержащих образцах.
Расширенный анализ тонкой структуры
спектров поглощения
Метод
очень
эффективен
при
изучении
плохоокристаллизованных минералов. EXAFS-спектроскопия
дает информацию о металлах в твердой фазе почв в
молекулярном масштабе, в частности, позволяет определить
формы сорбционных комплексов и характер закрепления
тяжелых металлов минералами-носителями.
Физически этот метод основан на следующих
предпосылках (Тео, 1986). Известно, что при прохождении
пучка рентгеновских лучей через образец интенсивность I
пучка ослабевает за счет влияния двух различных процессов:
изменения первоначального направления фотонов (рассеяния)
и их поглощения (абсорбции). Именно последний процесс и
изучается в ходе анализа EXAFS.
Исчезновение фотонов в процессе поглощения происходит
вследствие ионизации атома, когда энергия кванта тратится на
удаление электрона с внутренних орбит атома. Возбужденный
атом возвращается в нормальное состояние путем целой серии
разных переходов. Ослабление интенсивности рентгеновских
лучей, прошедших через образец толщиной t, при условии, что
на поверхности частицы при t = 0 интенсивность I = I0,
выразится так:
It = I0 exp (-λμ),
где μ - коэффициент ослабления первичного потока
рентгеновских лучей. EXAFS-спектроскопия основана на
изучении изменения коэффициента поглощения μ для
химического элемента в зависимости от длины волны λ.
Коротко, в ходе EXAFS-спектроскопии измеряется
абсорбции веществом рентгеновских лучей в зависимости от
прилагаемой энергии вплоть до 800 эВ за границами края
полосы поглощения данного элемента. За этим краем
наблюдаются осцилляции. Если бы атомы изучаемого элемента
были изолированы, кривая μ (λ) была бы без осцилляций. При
анализе тонкой структуры рентгеновских спектров поглощения
исследуется только осциллирующая часть абсорбционного
коэффициента
Δμ,
нормированного
на
коэффициент
поглощения μ0 атома, т.е. χ(k) = (μ - μ0) : μ0, где k - вектор
фотоэлектронной функции.
Фурье-преобразование функции χ(k) приводит к функции
радиального распределения F(R), пики которой содержат
информацию о расстояниях от центрального до соседних
атомов,
составляющих
первую,
вторую
и
третью
координационные сферы.
Первый максимум на кривой F(R) соответствует
расстояниям металл–анион, а второй _ расстояниям между
ближайшими катионами (Ме–Ме), находящимися в соседних
полиэдрах. Полиэдры могут контактировать друг с другом
гранью, ребром или вершиной. Расстояния между атомами
металлов при каждом из таких контактов разные, они
устанавливаются методом EXAFS-спектроскопии. Для того,
чтобы выделить расстояния между атомами металлов и число
атомов в каждой координационной сфере, проводят операцию
обратного Фурье-преобразования отдельно для каждого из
пиков на кривой F(R). У новой функции амплитуда (А) и
частота осцилляций электронной волны зависят от числа
атомов N данной координационной сферы и расстояния d от
центрального
атома
до
соответствующего
атома
координационной сферы, т.е. А = f (N, d). Определение
искомых величин N и d ведется методом подбора таких
расстояний от центрального атома до атомов анализируемой
координационной сферы, при которых рассчитанные функции
χ`(k) в максимально возможной степени совпадают с
экспериментальными.
В отличие от дифракционной техники, эффективность
EXAFS-спектроскопии не зависит от дальнего порядка
вещества и используется для анализа тонкой (локальной)
структуры как кристаллических, так и аморфных тел. Другое
достоинство – химическая специализация метода. Если
изучаемый
элемент
сконцентрирован,
то
EXAFSспектроскопия способна идентифицировать минерал-носитель
или
определить
локальную
структуру
слабоокристаллизованных частиц. Если же элемент рассеян, то
EXAFS-спектроскопия различает механизм его распределения
в матрице носителя в молекулярном масштабе (Brown et al.,
1999).
Идентификация минералов-носителей. Рентгеновская
дифракция, безусловно, наиболее адекватный метод для
идентификации минералов. Но если у фазы-носителя имеются
обширные кристаллические дефекты (замещения, вакансии,
смещения пакетов), то размер когерентно рассеивающих
доменов может уменьшиться до десятков ангстрем, что
затрудняет использование дифракции.
Несмотря на слабую окристаллизованность частиц,
например ферригидрита, в них металлсодержащие полиэдры
(обычно октаэдры) все же контактируют вполне идентичным
для данного типа структуры образом – гранями, ребрами или
вершинами, что позволяет определить расстояние металл–
металл (Ме–Ме). Это резко противоречит стеклам, в которых
углы между полиэдрами варьируют вплоть до нескольких
десятков градусов, что приводит к неопределенности
расстояний Ме–Ме. Например, в кварце тетраэдры SiO4
формируют трехмерную сетку со средним углом связи Si–O–
Si в 144º. Но в противоположность кристаллу, в стеклофазе
угол связи варьирует в широком интервале от 120 до 180º.
Данное различие между истинно аморфным состоянием и
дальним порядком структуры очень важно. Даже в
слабоупорядоченных
природных
частицах
EXAFSспектроскопия выявляет характер сферы у данного металла, из
чего можно вывести локальную структуру атома.
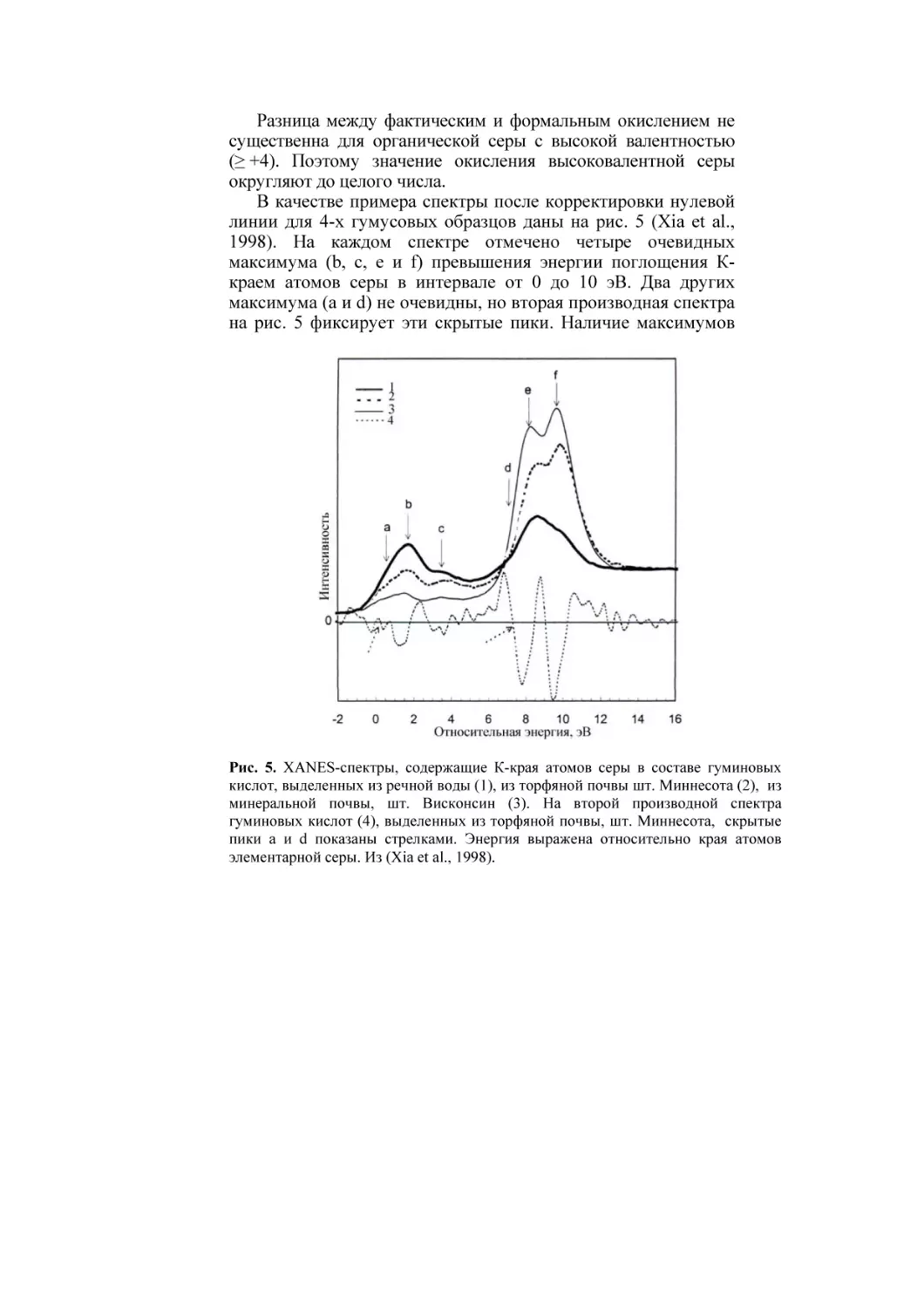
На рис. 6 сравниваются картины порошковой
рентгендифрактометрии
и
EXAFS-спектры
окристаллизованного оксида германия αGeO2 (со структурой
кварца), аморфного оксида германия αGeO2, фероксигита и
ферригидрита. Аморфное сили
Рис. 6. Рентгендифрактограммы ферригидрита и Si-стекла (А). Функция
радиальной структуры ЕXAFS-анализа упорялоченного фероксигита
(δFeOOH) и разупорядоченного ферригидрита, а также аморфного и
окристаллизованного оксида германия (В). Ферригидрит и аморфное стекло
ренгеноаморфны. На ЕXAFS-спектрах видны различия в упорядоченности
ферригидрита и аморфного стекла. Из (Mаnceau et al., 2002).
.
.
катное стекло, как и ферригидрит FeOOH 2Fe2O3 4H2O, дают
широкие полосы рассеяния на рентгендифрактограмме.
Но на EXAFS-спектрах видны сильные различия между
аморфным и слабоупорядоченным веществами. У аморфного
оксида αGeO2 небольшой второй пик говорит о слабом
взаимодействии Ge-Ge, тогда как у слабоупорядоченного
ферригидрита имеется два четких пика, аналогичных по
интенсивности с пиками у более упорядоченного фероксигита
δFeООН (Manceau et al., 2002).
Из этого обсуждения ясно, что EXAFS-спектроскопия,
благодаря способности регистрировать связи металл-металл,
обладает необходимой чувствительностью для идентификации
фаз-носителей, а также любых осадков сорбата. Этот метод
позволяет уловить даже малые вариации в геометрии,
межатомных расстояниях и состоянии окисления химического
элемента.
Способность EXAFS-спектра различать структурное
окружение атома хорошо иллюстрируется на примере
семейства манганатов, которое включает огромное количество
минералов. Диагностика основывается на том, что манганаты
различаются по ряду характеристик: по числу октаэдров,
соединенных ребрами и вершинами, по отношению Mn3+ : Mn4+,
по особенностям структуры. Все эти особенности EXAFSспектроскопия успешно выявляет (Чухров и др., 1989).
Но различать филлосиликаты по EXAFS-спектрам трудно,
так как эти минералы в химическом и структурном отношении
слишком сложные: включают большое количество катионов
(Al, Si, Fe, Mg и др.) и имеют различные структуры – три- и
диоктаэдрические.
Идентификация форм металлов. При обсуждении этого
вопроса рассмотрим
модельную систему «Zn +
филлосиликаты», которая выбрана благодаря значительной
распространенности в почвах и высокой химической
активности поверхности филлосиликатов. На примере этой
модели покажем, как EXAFS-спектроскопия способна
различать механизмы сорбции металлов.
Как известно, филлосиликаты сложены из одного
центрального слоя октаэдров и двух слоев тетраэдров.
Существует два типа мест на поверхности: постоянный
отрицательный заряд структуры базальных плоскостей,
образующийся в результате гетеровалентного изоморфного
замещения катионов в решетке, и рН-зависимый заряд на
ребрах слоев, образующийся за счет свободных атомов
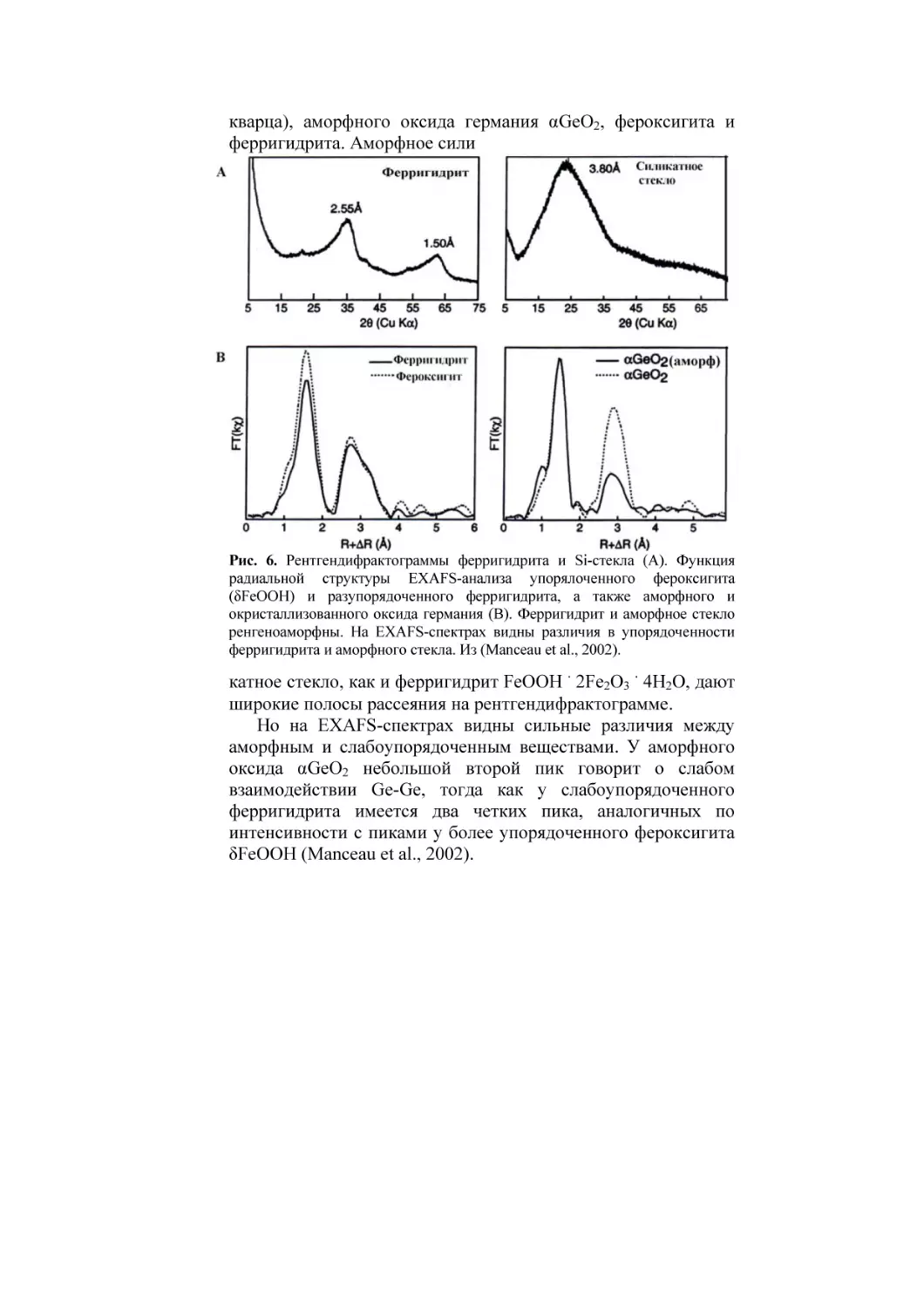
кислорода. Металл, сорбируемый на базальной плоскости,
формирует внешнесферный поверхностный комплекс и поэтому
легко обменивается с ионами раствора при изменении его
состава (ионной силы) или же металл может диффундировать
на другие места (т.е. на ребра слоев), где закрепляется прочнее
(рис. 7).
Закреплению металла на ребрах слоев способствуют
нейтральное значение рН и высокая ионная сила раствора.
Таким путем образуется внутрисферный комплекс. Внутри- и
внешнесферные комплексы вполне четко дифференцируются
EXAFS-спектроскопией, так как металл-сорбат соседствует с
Рис. 7. Механизм сорбции Zn на смектите при низкой и высокой ионной
силе (справа) и ЕXAFS-спектры моделей (слева). При низкой ионной силе
Zn вначале сорбируется в виде внешнесферного поверхностного комплекса
в межслоевом пространстве филлосиликата, а затем латерально
диффундирует и образует внутрисферный комплекс на кромке слоя. При
высокой ионной силе межслоевое пространство занято щелочными и
щелочно-земельными катионами и Zn прямо сорбируется на кромке слоя. В
присутствие Si цинк эпитаксиально растет на кромке филлосиликата.
Трансформация
внешнесферного
поверхностного
комплекса
во
внутрисферный отражается на ЕXAFS-спектрах в виде высокочастотных
перегибов, интенсивность которых растет по мере роста ионной силы
(показано стрелками). Из (Mаnceau et al., 2002).
оболочкой металла-сорбента при внутрисферном механизме и
не соседствует с ней при внешнесферном механизме
закрепления (Manceau et al., 2002). При высоком значении рН
или высокой концентрации катионов металл-сорбат
полимеризуется и образует различные виды осадков
(новообразованные филлосиликаты, гидроксиды металлов),
закрепленные или не закрепленные на поверхности минераланосителя. Все эти осадки различаются по форме EXAFSспектров, что позволяет их идентифицировать в почве.
В заключение отметим, что в последние годы удалось
преодолеть недостатки EXAFS-спектроскопии, обусловленные
малой чувствительностью к рассеянным частицам и к
металлсодержащим филлосиликатам (Manceau et al., 2000).
Первая проблема в настоящее время решается путем
совмещения
EXAFS-спектроскопии
с
микрорентгенфлуоресценцией, а вторая – путем применения
поляризованной P-EXAFS-спектроскопии. Рассмотрим обе
проблемы.
1. Сигнал EXAFS представляет собой средневзвешенный
вклад всех форм данного металла в почвенном образце.
Основные формы выявляют, исходя из линейной средней
квадратичной
аппроксимации
известных
соединений
микроэлементов.
Но
здесь
имеются
определенные
ограничения. Во-первых, легко выявляются только главные
фазы почвы. Во-вторых, нельзя включать слишком много
компонентов для аппроксимации почвенного спектра:
информационное содержание EXAFS-спектра не позволяет его
линейно расщеплять более чем на четыре индивидуальных
компонента. В-третьих, у частиц металла с сильным
рентгеновским рассеянием (с высоким атомным номером Z)
сигнал мощнее, чем у частиц низкой плотности. Такая
ситуация встречается в почвах, когда данный металл не только
входит в состав минерала, но и связывается органическими
лигандами, Например, 6% ошибка при определении ZnO
маскирует присутствие 20% слаборассеивающих органических
соединений Zn. Для решения этих проблем используется
пространственно-разрешающая
структурная
техника,
пригодная для «точечного» анализа, в первую очередь, микроEXAFS-спектроскопия.
2. Филлосиликаты, такие как смектиты, – главный
компонент почвы. Они чрезвычайно сильно сорбируют
тяжелые металлы. Но идентификация металлсодержащих
филлосиликатов затрудняется тем, что обычно изучаемая
илистая фракция (<2мкм) содержит также неглинистые
минералы: в частности, (гидр)оксиды железа и марганца.
EXAFS-сигнал от легких элементов алюмосиликатов (Si, Al,
Mg) маскируется интенсивным рассеянием Fe и Mn в составе
оксидов-носителей тяжелых металлов.
Чтобы повысить чувствительность EXAFS к определению
металлсодержащих
филлосиликатов
используется
поляризованные
Р-EXAFS-спектры
ориентированных
глинистых
пленок.
Р-EXAFS-спектры
обеспечивают
ориентационно-зависимую структурную информацию и
позволяют анализировать локальную структуру слоев
силикатов параллельно и перпендикулярно базальной
плоскости (001) путем варьирования угла между вектором
электрического поля и поверхностью глинистой пленки
(Manceau et al., 2000).
Глава 2. МЕТОДЫ ПОСЛЕДОВАТЕЛЬНОГО
ЭКСТРАГИРОВАНИЯ ТЯЖЕЛЫХ МЕТАЛЛОВ
При химическом мониторинге загрязнения почв тяжелыми
металлами наиболее широко используются системы
последовательного фракционирования (Пампура и др., 1993;
Пампура, 1996; Мотузова, 1999; Переломов и др., 2003;
Ладонин, 2002). В России наряду с другими известна система
Тессиера с соавт. (Tessier et al., 1979). В Европе используют
систему Тессиера и более новую, разработанную в 1994 г.,
европейскую систему (BCR) (Whalley, Grant, 1994; Raksasataya
et al., 1996). Система BCR часто используется для
идентификации форм тяжелых металлов в почвах и осадках на
территории Европейского Союза.
Из обширного обзора, составленного Филгуэрасом с
соавторами (Filgueiras et al., 2002), можно выявить наиболее
популярные вытяжки, применяемые для анализа форм
тяжелых металлов. В обзоре анализу тяжелых металлов в
почвах посвящено всего 136 публикаций. Абсолютное
большинство работ включает оригинальные, «авторские»
методы экстрагирования тяжелых металлов, не находящие
последователей. Исключение составляют две схемы: Тессиера
и BCR, причем схема Тессиера наиболее популярна.
Эффективность этих двух популярных схем и контролируется
чаще всего методами минералогического анализа. Мы также
уделим основное внимание этим схемам.
В настоящее время принцип селективности экстрагентов по
отношению к конкретным фазам-носителям тяжелых металлов
заменяется общим «принципом действия», например,
«редуцирующее» действие гидроксиламина; «окисляющее»
действие пероксида водорода; «ионный обмен» и «кислотное
растворение» при обработке уксусной кислотой (Raksasataya et
al., 1996).
Таким
образом, современное
направление
работ
выражается не в поиске «наилучших» химических
экстрагентов, а в установлении особенностей действия
«стандартных» экстрагентов на почвы резко различающихся
по химии и минералогии. Различие почв основывается на
преобладании тех или иных индивидуальных, контрастных по
природе «геохимических» фаз, носителей тяжелых металлов.
Таких фаз выделено немного: карбонаты, (гидр)оксиды железа
и марганца, глинистые минералы и гумусовые вещества (Shen,
Chen, 1993). На них и проверяют селективность основных
реактивов. Отметим отсутствие в этом списке оксидов
алюминия, которым ранее придавалось преувеличенное
значение в закреплении тяжелых металлов и металлоидов
(Ковда, 1985).
Химическое
действие
реактивов
контролируют
различными
способами.
Наиболее
эффективный
минералогический и физико-химический контроль действия
реактивов обеспечивают методы синхротронной радиации.
Процедура
последовательной
экстракции
давно
используется для изучения форм металлов в почвах.
Считается, что она позволяет идентифицировать и определять
содержание стольких форм металла, сколько используется
этапов экстракции (Scheinost et al., 2002), хотя как будет
показано ниже, применение кинетического анализа процесса
экстракции позволяет расширить число искомых форм
металла. Но у экстракционного метода анализа есть ряд
недостатков. Среди них – неполное растворение фаз-носителей
(La Force, Fendorf, 2000), растворение второстепенных
геохимических фаз (Ostregren et al., 1999), неполное удаление
растворенного металла за счет его реадсорбции (Ostregren et
al., 1999), переосаждение и изменение степени окисления
элемента. Поэтому химическое фракционирование способно
только
оперативно
разделять
металл
по
формам,
соответствующим процедурам экстракции, но не всегда
отражает их природные химические формы.
Система Тессиера
В Европе она широко используется, начиная с 1979 г. В
табл. 2 отражены условия последовательной экстракции
тяжелых металлов по схемам Тессиера и по новой европейской
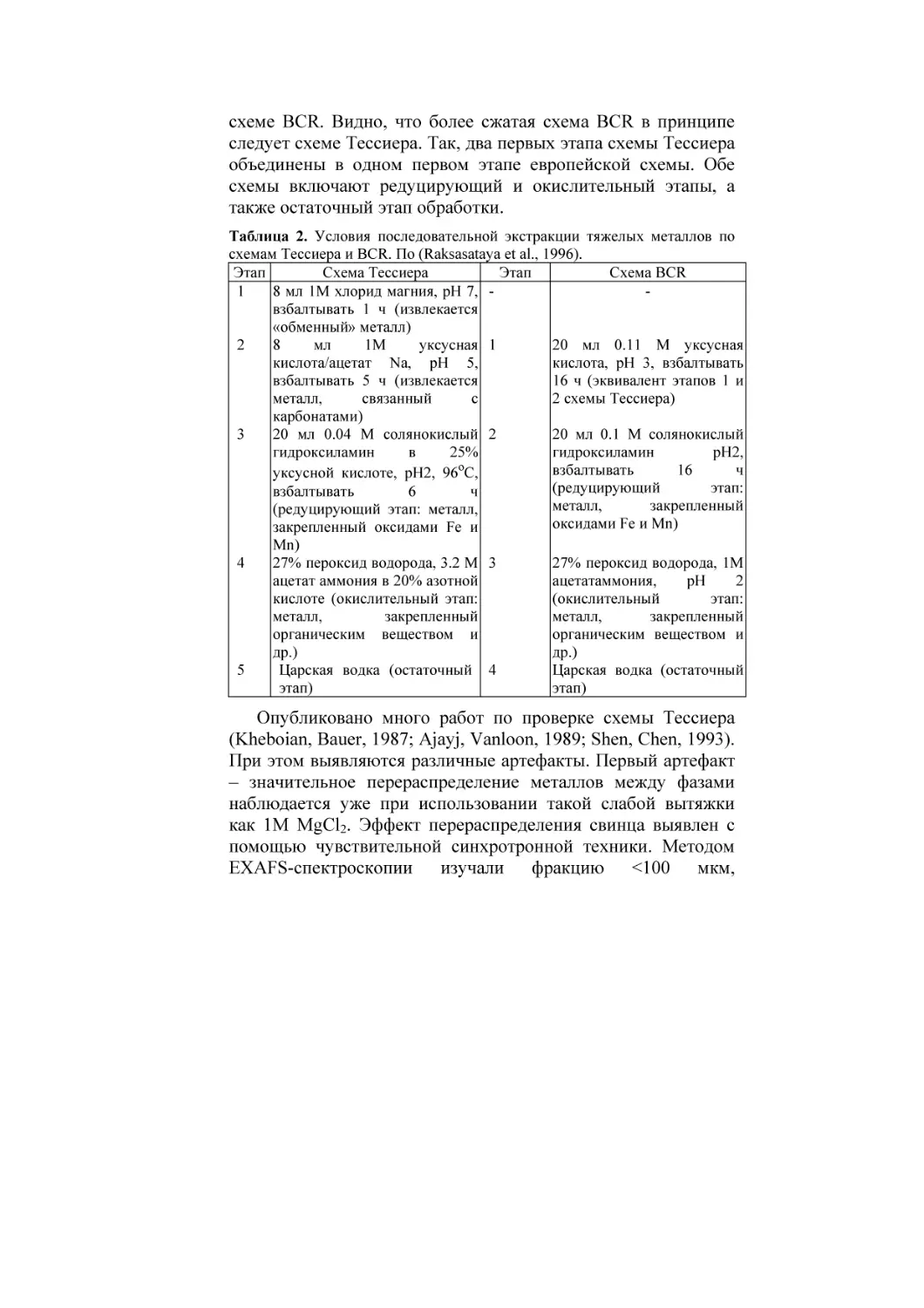
схеме BCR. Видно, что более сжатая схема BCR в принципе
следует схеме Тессиера. Так, два первых этапа схемы Тессиера
объединены в одном первом этапе европейской схемы. Обе
схемы включают редуцирующий и окислительный этапы, а
также остаточный этап обработки.
Таблица 2. Условия последовательной экстракции тяжелых металлов по
схемам Тессиера и BCR. По (Raksasataya et al., 1996).
Этап
Схема Тессиера
Этап
Схема BCR
1
8 мл 1М хлорид магния, рН 7, взбалтывать 1 ч (извлекается
«обменный» металл)
2
8
мл
1М
уксусная 1
20 мл 0.11 М уксусная
кислота/ацетат Na, рН 5,
кислота, рН 3, взбалтывать
взбалтывать 5 ч (извлекается
16 ч (эквивалент этапов 1 и
металл,
связанный
с
2 схемы Тессиера)
карбонатами)
20 мл 0.1 М солянокислый
3
20 мл 0.04 М солянокислый 2
гидроксиламин
рН2,
гидроксиламин
в
25%
взбалтывать
16
ч
уксусной кислоте, рН2, 96ºС,
(редуцирующий
этап:
взбалтывать
6
ч
металл,
закрепленный
(редуцирующий этап: металл,
оксидами Fe и Mn)
закрепленный оксидами Fe и
Mn)
4
27% пероксид водорода, 3.2 М 3
27% пероксид водорода, 1М
ацетат аммония в 20% азотной
ацетатаммония,
рН
2
кислоте (окислительный этап:
(окислительный
этап:
металл,
закрепленный
металл,
закрепленный
органическим веществом и
органическим веществом и
др.)
др.)
5
Царская водка (остаточный 4
Царская водка (остаточный
этап)
этап)
Опубликовано много работ по проверке схемы Тессиера
(Kheboian, Bauer, 1987; Ajayj, Vanloon, 1989; Shen, Chen, 1993).
При этом выявляются различные артефакты. Первый артефакт
– значительное перераспределение металлов между фазами
наблюдается уже при использовании такой слабой вытяжки
как 1М MgCl2. Эффект перераспределения свинца выявлен с
помощью чувствительной синхротронной техники. Методом
EXAFS-спектроскопии изучали фракцию <100 мкм,
выделенную из отвала породы Апачи, штат Колорадо, США
(Morin et al., 1999; Ostregren et al., 1999). У материала отвала
реакция среды кислая. Спектры свинца снимали до и после
обработки фракции раствором MgCl2 (табл. 3).
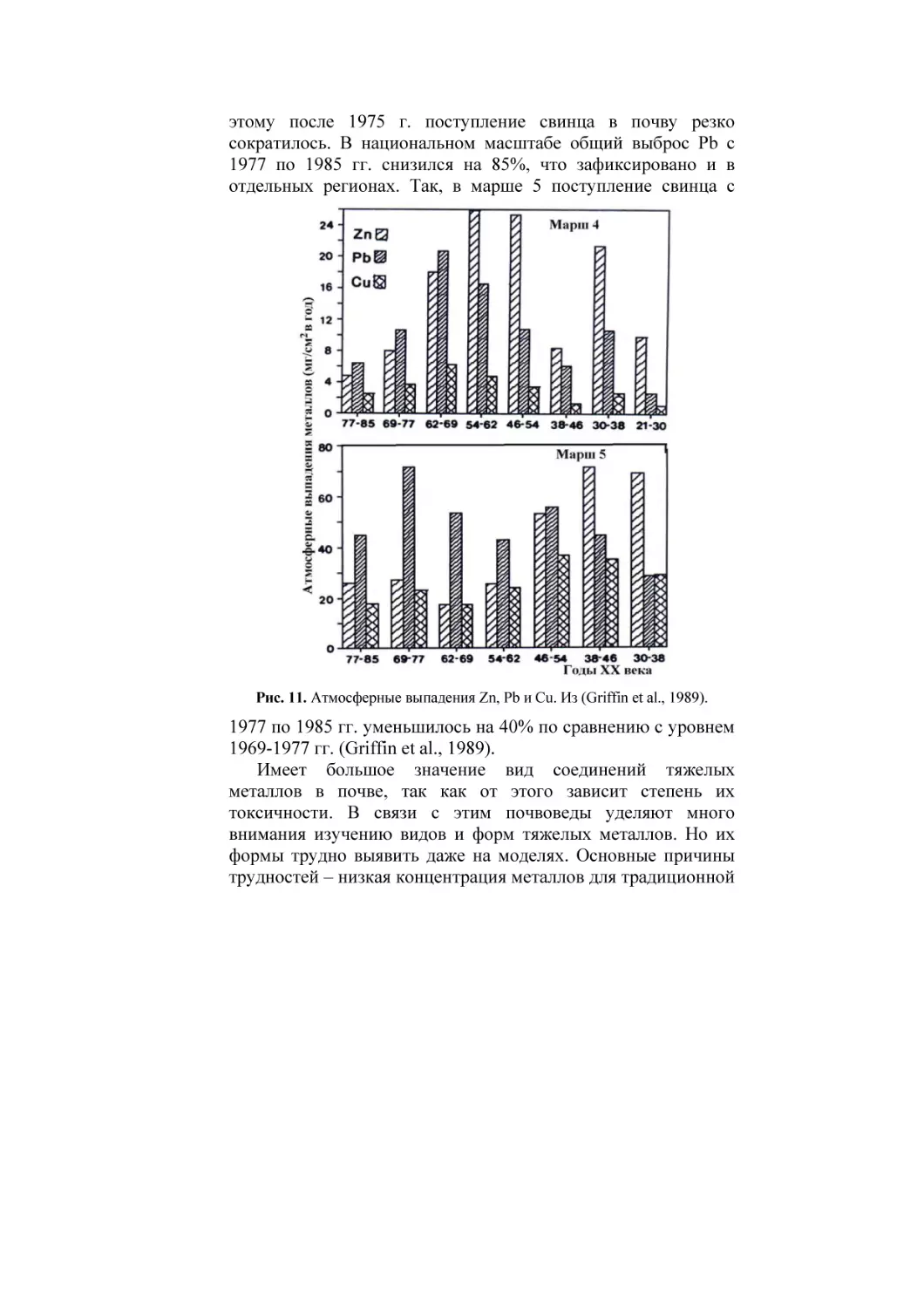
Как видно из табл. 3, состав Pb-содержащих минералов в
почве значительно изменился после обработки раствором
MgCl2. Самое главное изменение – это появление частиц Pbгетита (45%), которых не было в исходной почве. Поскольку в
раствор MgCl2 свинец практически не перешел, то можно
считать, что произошло перераспределение частиц свинца за
счет растворения Pb-ярозита и последующей адсорбции Pb на
частицах гидроксида железа – гетита αFeOOH. Растворение
ярозита KFe3(SO4)2OH6 в ходе обработки раствором MgCl2
легко объяснимо неустойчивостью минерала в нейтральной
среде и согласуется с падением рН раствора с 7 до 6 после
взаимодействия с образцом (Ostregren et al., 1999). Таким
образом, на некоторые образцах даже слабая обработка
провоцирует артефакт – перераспределение металла между
фазами-носителями.
Таблица 3. Формы Pb во фракции <100мкм отвала Апачи, Колорадо, США,
до и после обработки 1М MgCl2 по данным EXAFS-спектроскопии
Исходный образец
Не обнаружен
45% Pb-ярозит
28% Pb-феррит
10% ванадинит
Не обнаружен
После обработки MgCl2
45% Pb-гетит
20% Pb-ярозит
19% Pb-феррит
10% ванадинит
5% галенит
Важные исследования по проверке схемы Тессиера
провели китайские ученые (Shen, Chen, 1993). Вначале они
определяли концентрацию металлов, переходящих в раствор
при растворении отдельных геохимических фаз-носителей:
гематита, кальцита, пиролюзита, иллита, монтмориллонита и
гуминовых кислот. Выяснилось, что реактивы в схеме
Тессиера достаточно селективно извлекают тяжелые металлы
из этих фаз, обработанных в отдельности.
Затем приготовили модель почвы, смешав фазы в
определенном соотношении и обработав их теми же
реактивами. Полученные результаты представим для
наглядности в виде отношения количества фактически
экстрагируемого металла к расчетному его содержанию,
полученному на основании суммарной экстракции из
отдельных фаз: СМ-факт : СМ-расч. Оказалось, что в смеси фаз ни
один из экстрагентов не проявил своей специализации.
Металл, растворимый в ходе первых этапов обработки,
реадсорбируется на более устойчивых фазах (табл. 4).
Таблица 4. Отношение количества фактически экстрагированного металла
к расчетному его содержанию СМ-факт : СМ-расч. Смеси твердых фаз
включают: гематит, кальцит, пиролюзит, иллит, монтмориллонит и
гумусовые кислоты. Экстракция выполнена по схеме Тессиера. По данным
(Shen, Chen, 1993).
Элемент
Этап 1
Этап 2
Этап 3
Этап 4
Этап 5
Са
0.8
1.0
2.1
0.8
4.4
Fe
Не опр.
0.1
0.2
1.0
1.0
Cu
»
Не опр.
0.5
1.0
2.0
Mn
1.0
0.5
0.5
104
7.6
Ni
Не опр.
0.5
0.5
38
1.5
Zn
»
Не опр.
0.4
1.2
1.1
Co
»
»
0.5
47
1.8
Pb
»
0.2
2.0
7
1.2
Cr
»
0.3
0.6
3.7
1.1
Конкретные результаты таковы. Для кальция высокое
значение ССа-факт : ССа-расч = 2.1 на третьем этапе экстракции
указывает на то, что часть обменного кальция на первых двух
этапах не переходит в раствор, а реадсорбируется на частицах
(гидр)оксидов железа, марганца и глинистых минералов. Для
железа низкое значение СFe-факт : СFe-расч = 0.1-0.2 на
начальных этапах экстракции указывает на то, что часть
железа реадсорбируется на остающихся твердых фазах.
Марганец на третьем этапе переходит в раствор из состава
пиролюзита не полностью (СMn-факт : СMn-расч) = 0.5.
Значительная часть растворенного Mn реадсорбируется на
других фазах, в основном на гумусовых кислотах и в меньшей
степени на глинах, переходя в раствор позже: на 4 и 5 этапах.
Почти для всех тяжелых металлов кроме Pb, на третьем
этапе обработки отношение <1, а на последующих этапах
значительно возрастает, становясь >1. Следовательно, тяжелые
металлы, освободившиеся вначале из неустойчивых фаз
(карбонатов и слабоупорядоченных (гидр)оксидов железа и
марганца) не экстрагируются, а вновь сорбируются уже на
других, более устойчивых фазах, в первую очередь на
гумусовых кислотах. Действительно, на четвертом этапе, когда
гумус окисляют, указанное отношение достигает высоких
значений: от 1.2 для Zn до 104 для Mn.
Несмотря на появление новой европейской схемы, интерес
к классической схеме Тессиера сохраняется: появляются
свежие публикации, посвященные контролю действия
вытяжек. Недавно кроме перераспределения металлов между
фазами-носителями, обнаружен новый артефакт – неосинтез
минерала свинца в ходе экстракции (Scheckel et al., 2003). В
опытах исходный свинец был представлен соединениями с
различной растворимостью: ацетатом свинца, англезитом
PbSO4, церусситом PbCO3, галенитом PbS и пироморфитом
Pb5(PO4)3. Их растворимость в целом отвечала интерпретации
Тессиера. Но добавка к соединениям свинца фосфата кальция
резко изменила характер распределения Pb по фракциям. В
присутствии фосфата резко снизилась доля обменного свинца
и возросла доля нерастворимых соединений. Это объясняется
извлечением Pb из обменного состояния и последующей
реакцией с фосфатом, и образованием труднорастворимого
пироморфита,
что
было
подтверждено
данными
рентгендифрактометрии и EXAFS-спектроскопии.
Таким образом, постоянно выявляются новые особенности
действия схемы Тессиера на фазы-носители тяжелых металлов.
Оказалось, что в одних почвах нарушение селективности
реагентов очень существенно, в других – незначительно.
Вероятно, со временем можно будет выявить типы почв
абсолютно не пригодные для такого фракционирования,
пригодные условно и, наконец, пригодные вполне. Уже сейчас
ясно, что абсолютно не пригодны для фракционирования
органогенные почвы, где закрепление тяжелых металлов
определяется во многом химией и минералогией серы (Xia et al.,
1997; Martinez et al., 2002).
Европейская система BCR
В 1994 г. Европейским бюро по стандартам была
утверждена трехстадийная последовательная экстракция
металлов (BCR). Эту систему уже используют для
мониторинга окружающей среды (Raksasataya et al., 1996).
Но и новая система имеет недостатки. Отмечают слабую
селективность солянокислого гидроксиламина в отношении
синтетического гетита αFeOOH, соосажденного с Ni, а также –
пероксида водорода в отношении гумусовых кислот,
адсорбировавших Zn (Raksasataya et al., 1996). На модельных
почвах
наблюдали
уже
известный
артефакт
–
перераспределение Pb и Cu между фазами-носителями.
Например, доказано, что для смеси каолина, гумусовых кислот
и кальцита, насыщенных свинцом, концентрация Cu в
уксусной вытяжке снижается при увеличении доли гумусовых
кислот. Очевидно, что медь реадсорбируется на гумусовых
кислотах в ходе химической обработки.
Рассмотрим подробнее опыт по оценке селективности
системы BCR при последовательном экстрагирования
металлов (Cu, Ni, Zn) из различных фаз-носителей. Среди них
были кальцит СаСО3, ферригидрит FeOOH . Fe2O3 . 4Н2О,
гумусовые
кислоты,
каолинит,
Na-полевые
шпаты,
монтмориллонит и диоксид марганца (Whalley, Grant, 1994).
Тяжелые металлы, выделенные из морской воды, закреплялись
на модельных фазах. Предполагается, что впоследствии
металл должен полностью высвобождаться из фазы-носителя
под воздействием конкретного абсолютно селективного
реагента.
Результаты экспериментов приведены в табл. 5. В ней
сравниваются предполагаемое действие разных реагентов с
фактическим. Ожидаемым было вытеснение уксусной
кислотой основной массы металла, ассоциированного с
кальцитом. Также естественно, что металл, ассоциированный с
диоксидом марганца, успешно извлекался за счет деструкции
фазы-носителя солянокислым гидроксиламином. Но видно
также, что действие реагентов не всегда согласуется с
ожидаемым. Характер высвобождения тяжелых металлов,
связанных с ферригидритом, оказался неожиданным, большая
доля металла переходила в уксуснокислую вытяжку, тогда как
предполагаемое
влияние
гидроксиламина
оказалось
минимальным.
Таблица 5. Сравнение ожидаемого и экспериментального действия
реагентов по схеме BCR на отдельные фазы-носители тяжелых металлов
(медь, никель, цинк). По (Whalley, Grant, 1994).
Фаза-носитель
Реагент,
Реагент,
фактически Фактический
предположите экстрагирующий
механизм
большую часть металла действия
льно
экстрагирую
реагента
щий большую
часть металла
Кальцит
НОАс
НОАс
Кислотное
растворение
Ферригидрит
NH2OH . HCl НОАс
Редукция
Гумусовые
Н2О2 / NH4Ac Варьирует:
Cu – Н2О2 / NH4Ac
Окисление
кислоты
Ni – NH2OH . HCl
?
Zn – НОАс и NH2OH . HCl ?
Каолинит
? НОАс
НОАс
Ионный обмен
Полевые шпаты ? НОАс
НОАс
Ионный обмен
Монтмориллонит ? НОАс
NH2OH . HCl
Ионный обмен
Диоксид марганца NH2OH . HCl NH2OH . HCl
Редукция
Примечание. Знак вопроса обозначает отсутствие достоверной информации
о селективности и механизме действия реактива.
Второй неожиданный эффект – это экстракция одной меди
из всей группы металлов, связанных с гумусовыми кислотами,
в ходе третьей обработки Н2О2 /NH4Ac. Но Ni и Zn,
предположительно связанные с гумусовыми кислотами, ведут
себя иначе и в основном удаляются в результате первых двух
(оказавшихся неспецифическими) экстракций. Столь легкое
разрушение органо-минеральных связей означает, что Ni и Zn
были адсорбированы непрочно, и их высвобождение
произошло до начала деградации органического вещества.
Очевидно, что органические комплексы с Ni и Zn непрочные и
их доли в почвах вряд ли существенны.
Из изученных минералов только каолинит и Na-полевой
шпат высвобождают большую долю адсорбированных
металлов в уксусную кислоту. В тоже время значительная доля
Cu
и
Zn,
предварительно
адсорбированного
монтмориллонитом, извлекалась гидроксиламином во второй
экстракции.
Особенно
прочно
закрепляется
монтмориллонитом никель, всего было освобождено только от
1/3 до 2/3 металла, причем преимущественно в ходе
экстракции солянокислым гидроксиламином (Whalley, Grant,
1994).
Таким образом, реагенты действуют неселективно. При
обсуждении причин обращается внимание на следующее.
Переход металлов (Cu, Ni и Zn) в раствор из силикатов может
быть скорее результатом ионного обмена, чем химически
обусловленного изменения поверхности. Не ясно, какой
механизм растворения соединений металлов доминирует.
Среди механизмов могут быть реакции переноса электронов,
протонов и комплексообразования. Полученные данные все же
позволяют прояснить специфику новой европейской системы
фракционирования форм тяжелых металлов.
Cравнение действия двух систем на примере свинца
провели Раксасатая с соавт. (Raksasataya et al., 1996). Артефакт
в виде перераспределение Pb в обеих схемах был
значительным, что исключает любую попытку установить
исходные формы свинца чисто химическим путем. Главные
сорбенты, на которые переосаждается Pb в ходе анализа – это
оксиды марганца и гумусовые кислоты (табл. 6).
Но выявлены и отличия в результатах фракционирования
свинца в двух схемах. Процедура Тессиера приводит к
большему выходу Pb на редуцирующем этапе, а BCR – на
окислительном этапе, независимо от природы частиц свинца.
Это различие обусловлено высокой концентрацией уксусной
кислоты (25%) и высокой температурой обработки (96°С) на
редуцирующем этапе схемы Тессиера.
Таблица 6. Количество меди, высвобождаемой из моделей в вытяжки при
последовательной экстракции по схемам Тессиера и BCR, %. Содержание
этапов экстракции дано в табл. 2. По данным (Raksasataya et al., 1996).
Схема Тессиера
Модель
Этап 1 Этап 2 Этап 3
Этап 4
Этап 5
Кальцит+кварц
4
0
58
40
17
Галлуазит+кварц
3
1
48
31
13
Гетит+кварц
3
1
44
37
24
Гаусманит+кварц
0
1
53
36
12
Гумусовые кислоты+кварц
2
4
50
41
10
Схема BCR
Этап 1
Этап 2
Этап 3
Этап 4
Кальцит+кварц
0
1
87
10
Галлуазит+кварц
0
0
73
8
Гетит+кварц
0
0
73
17
Гаусманит+кварц
0
0
83
10
Гумусовые кислоты+кварц
0
1
100
9
Примечание. Превышение суммы экстрагируемой меди 100% обусловлено
неточным определением суммарного содержания адсорбированного металла.
Жесткие условия обработки влияют на гумусовые кислоты,
снижая их сорбционную способность более чем на половину.
У системы BCR выявилась другая особенность. Свинец
реадсорбируется на органическом веществе, (гидр)оксидах
железа и марганца (Raksasataya et al., 1996). Значит, пока не
будет решена проблема экстракций без сопутствующего
перераспределения металлов между фазами, идентифицировать
характер природного распределения свинца в загрязненной
почве
нельзя.
Например,
фактически
удерживаемый
карбонатами свинец может быть представлен как закрепленный
оксидами железа и марганца при использовании схемы
Тессиера, и – органическим веществом при экстракции по схеме
BCR.
Наконец, надо отметить недостаток данных по
химическому фракционированию металлоидов. В частности,
нет надежных данных о химическом фракционировании форм
мышьяка.
Расчет вклада фаз-носителей в закрепление
тяжелых металлов
Обратимся теперь к другой проблеме. Даже без учета
описанных выше артефактов, имеются неопределенности в
участии разных фаз-носителей на первом и втором этапах
экстракции по схеме BCR. На первом этапе при обработке
почвы уксусной кислотой, возможно извлечение тяжелых
металлов, ассоциированных как с кальцитом, так и
ферригидритом. На втором этапе при обработке почвы
солянокислым гидроксиламином, возможно извлечение
тяжелых металлов, ассоциированных или с оксидами железа,
или марганца (Whalley, Grant, 1994). Вклад каждой из этих фаз
в закрепление металла при любой из двух экстракций остается
неясным, и роль «дополнительной» фазы игнорируется, что
приводит к завышению роли основной фазы. Так, на
основании данных химического фракционирования, считалось,
что примерно ½ всего количества тяжелых металлов в почвах
связано с гидроксидами железа (Добровольский, 2003). При
этом роль оксидов марганца, растворимых одновременно с
гидроксидами железа, в закреплении тяжелых металлов
вообще не учитывалась. Но исключая из рассмотрения оксиды
марганца, мы получаем искаженный перечень основных фазносителей тяжелых металлов. Игнорирование вклада оксидов
марганца особенно неоправданно при изучении Fe-Mn
ортштейнов.
Попытаемся решить эту проблему. Очевидно, что для
вычленения участия каждой фазы в данной вытяжке необходимо
знать каков переход в раствор основных элементов из состава
фаз-носителей, если высвобождение металла сопровождается их
растворением, хотя бы частичным. Конкретно, чтобы разделить
фазы-носители тяжелого металла, на которые воздействует
уксусная кислота, нужно определить в вытяжке кроме этого
металла, еще и содержание Са и Fe. Соответственно, при
обработке почвы раствором солянокислого гидроксиламина
нужно дополнительно определить содержание Fe и Mn.
В дальнейшем, чтобы выявить долевое участие фазносителей,
необходимо
подсчитать
коэффициенты,
характеризующие количество растворенного тяжелого металла
(ТМ), приходящегося на 1 моль растворенного Са и Fe в
уксусной кислоте и на 1 моль Fe и Mn в солянокислом
гидроксиламине. Обозначим эти коэффициенты как αТМ-Са,
αТМ-Fe и αТМ-Mn. Чтобы установить значения коэффициентов,
нужно иметь более широкий набор аналитических данных в
вытяжке, кроме принятого сейчас единственного измерения в
конце обработки почвы. Для этого проще всего использовать
данные кинетики экстракции ТМ (и сопутствующих элементов
из состава фаз-носителей) в раствор. Методики Тессиера и
BCR дают для этого возможности, поскольку экстракция
металлов на первых этапах ведется длительно в течение 4-6
или даже 16 часов (табл. 2).
Дадим новую методику расчета доли металла,
ассоциированного отдельно с каждой из фаз-носителей. Ее
удобно изложить на конкретном примере. В качестве такого
примера используем опыт Пампуры (1996) по обработке
чернозема раствором солянокислого гидроксиламина NH4OH .
HCl при Т = 96ºС, согласно третьему этапу схемы Тессиера.
Предварительно чернозем насыщали солями меди и цинка. В
опытах использовали образцы почвы, сорбировавшие
минимальное (0.5 мМ Cu/кг и 1.0 мМ Zn/кг) и максимальное
(11.0 мМ Cu/кг и 17.9 мМ Zn/кг) количество металлов. Автор
изучала кинетику экстракции до 12 ч. Мы в расчетах
анализировали кинетику экстракции на трех этапах: на
начальном через 30 мин и 4 ч, на среднем через 4 и 8 ч и на
конечном через 8 и 12 ч взаимодействия почвы с реактивом.
Для каждого из этапов допускали, что кинетика развивается по
линейному закону. Тогда, например, для начального этапа
можно составить два таких уравнения:
ТМ0.5 = αТМ-Fe . Fe0.5 + αТМ-Mn . Mn0.5,
ТМ4 = αТМ-Fe . Fe4 + αТМ-Mn . Mn4,
где индексы 0.5 и 4 характеризуют содержание элементов,
полученное через 0.5 и 4 ч обработки. Решая эти уравнения,
получали искомые коэффициенты αТМ-Fe и αТМ-Mn. Затем,
подставив эти коэффициенты в любое из уравнений, определяли
величину FeТМ = (100 . αТМ-Fe Fe) / ТМ, характеризующую в %
долю участия (гидр)оксидов железа в закреплении тяжелого
металла, и величину MnТМ = (100 . αТМ- Mn Mn) / ТМ,
характеризующую в % долю участия оксидов марганца в
закреплении тяжелого металла. Аналогичным образом
составляются уравнения для среднего и конечного этапов
кинетики экстракции.
Данный расчет необходимо контролировать, так как
возможна ошибка. В качестве контроля можно использовать
значения коэффициентов, характеризующих долю тяжелого
металла (Zn), приходящегося на 1 моль элемента,
составляющего минерал-носитель, в нашем обозначении это
αZn-Fe и αZn-Mn. Эти коэффициенты не должны превышать
некоторых предельных величин. Связано это с ограниченной
сорбционной емкостью минералов-носителей. В самом деле,
(гидр)оксиды железа и оксиды марганца не могут
адсорбировать чрезмерно большие массы тяжелых металлов.
Так, известно, что максимальная сорбция цинка оксидом
марганца – бернесситом составляет 0.123 моль Zn на 1 моль
Mn (Manceau et al., 2000). Близкое значение можно получить,
анализируя данные по сорбции Zn гетитом: 0.11 моль Zn на 1
моль Fe (Kuo, 1986). Заметное превышение этих критических
величин указывает на ошибку, допущенную в расчете.
Вначале рассчитаем долю участия (гидр)оксидов Fe и Mn в
закреплении цинка при его низком содержании в черноземе
(табл. 7). На начальном этапе экстракции коэффициент,
характеризующий связь Fe с Zn, αZn-Fe = 0.045, значительно ниже
коэффициента для связи Fe с Mn: αZn-Mn = 0.140. Следовательно,
при растворении 1 моля Fe в вытяжку переходит только 0.045
моля Zn, тогда как при растворении 1 моля Mn в вытяжку
переходит уже 0.14 моля Zn. Такое резкое различие в
коэффициентах отражается в различном участии в закреплении
цинка фазами-носителями. (Гидр)оксиды железа закрепляют
всего FeZn = 16%, тогда как оксиды марганца – весь остальной
цинк (MnZn = 84%).
Таблица 7. Количество молей Zn и Cu, приходящихся на 1 моль Fe (αТМ-Fe)
и Mn (αТМ-Mn), и процентное содержание отдельно (гидр)оксидов железа
(FeТМ) и оксидов марганца (MnТМ), участвующих в закреплении Zn и Cu в
черноземе за разное время экстракции солянокислым гидроксиламином
при 96°С. Опыт с черноземом, сорбировавшим 1мМ Zn/кг и 0.5 мМ Cu/кг.
По данным (Пампура, 1996).
Fe
Mn
Часы
αТМ- Fe
FeТМ, %
αТМ- Mn
MnТМ, %
Zn
0.5-4
0.045
16
0.140
84
4-8
0.000
0
0.0165
100
8-12
0.000
0
0.000
0
Cu
0.5-4
0.0016
79
0.0003
21
4-8
0.0039
100
-0.0011
0
8-12
0.0730
100
-0.0500
0
Контроль расчета показывает, что коэффициент αZn-Fe =
0.045 сомнений не вызывает. Второй коэффициент (связи Fe с
Mn αZn-Mn = 0.140) немного выше предельного и его можно
признать реальным с небольшой натяжкой.
На среднем этапе экстракции доля марганца в закреплении
цинка возросла до 100%. При этом доля марганца (0.165 М),
приходящегося на 1 моль цинка
чрезмерно высока.
Нереальное значение указывает на растворение оксидов
марганца и перераспределение Zn между другими более
устойчивыми фазами. На конечном этапе экстракции выход Zn
в раствор прекратился.
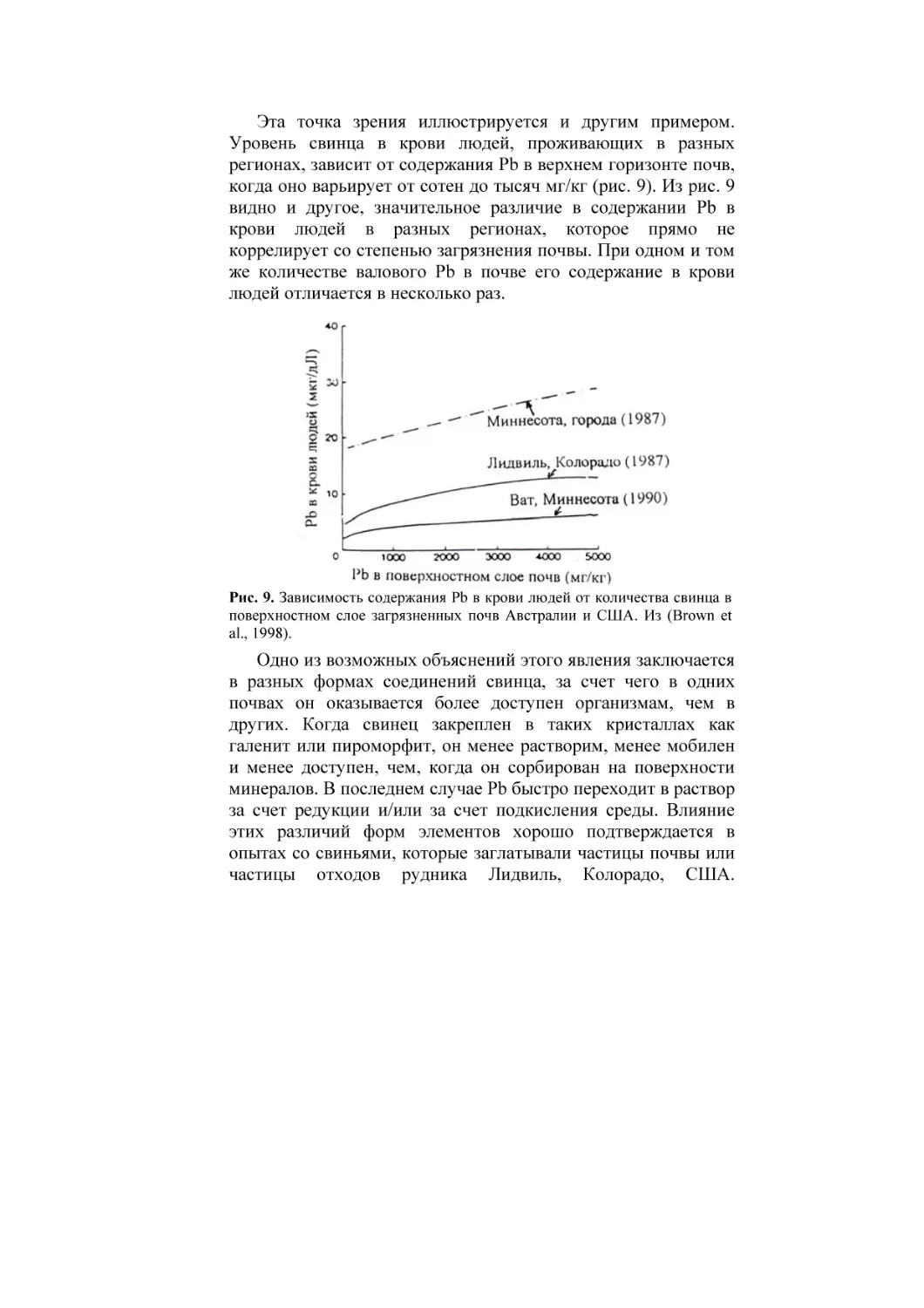
Обсудим результаты расчета. Интересно, что хотя кларк
Mn в 38 раз ниже кларка Fe, доля участия Mn в закреплении
цинка оказывается значительно выше (данные начального
этапа экстракции). Это легко объяснить. Доля реакционноспособных соединений Mn в почвах гораздо выше доли
соответствующих соединений Fe. Об этом говорит более
высокая доля легкорастворимого Mn, чем Fe (Водяницкий,
2003, 2005). Во-вторых, имеет значение более значительная
емкость поглощения катионов оксидами марганца, чем
гидроксидами железа, обусловленная высоким отрицательным
зарядом оксидов марганца в широком интервале значений рН.
Наш вывод о более высоком сродстве Zn к оксидам марганца,
чем к гидроксидам железа вполне согласуется с данными
рентгеновского флуоресцентного анализа лесной почвы из
района Эви-Мальвес, северная Франция (Manceau et al., 2000).
В микромасштабе распределение Mn и Zn сильно коррелирует
друг с другом, а распределение Fe и Zn коррелирует слабо.
Рассчитаем теперь долю участия (гидр)оксидов Fe и Mn в
закреплении меди черноземом (табл. 7). На начальном этапе
растворения значение коэффициента, отражающего связь Fe с
Cu, составляет αCu-Fe = 0.0016, а коэффициента,
характеризующего связь Mn с Cu, снижается до αCu-Mn =
0.0003. Характер закрепления меди в черноземе отличается от
закрепления цинка. В основном (на 79 %) медь фиксируется
гидроксидами железа и только 21% – оксидами марганца. На
среднем этапе экстракции доля железосодержащих носителей
меди увеличилась до 100%. Она осталась максимальной и на
позднем этапе экстракции. Очевидно, мы столкнулись с
эффектом перераспределения меди, когда оксиды марганца
полностью растворились и закрепленные ими частицы Cu
реадсорбировались на оставшихся частицах (гидр)оксидов
железа. Этот эффект наблюдали ранее со свинцом,
адсорбировавшимся после растворения частиц оксидов
марганца на частицах гидроксидов железа (Raksasataya et al.,
1996).
Известно, что в кислой среде частицы гидроксида марганца
растворяются с высокой скоростью; в зависимости от
концентрации редуктантов константа скорости составляет k =
10-5 – 10-7 М/м2.с, что соответствует скорости растворения
карбонатов. При том же значении рН частицы оксидов железа
растворяются гораздо медленнее: k ≅ 10-11 М/м2.с, что
соответствует
скорости
растворения
слоистых
алюмосиликатов (Zinder et al., 1986; Jun, Martin, 2003).
Поэтому при обработке почвы таким восстановителем, как
гидроксиламин, оксиды марганца растворяются быстро, а
(гидр)оксиды железа медленно. За время обработки тяжелые
металлы, исходно закрепленные оксидами марганца,
переходят в раствор, а затем переосаждаются на
нерастворенных твердых фазах, включая наиболее устойчивые
частицы (гидр)оксидов железа.
Таким образом, предлагаемый метод расчета позволяет
разделить тяжелые металлы на две группы, различающиеся по
прочности связи с минералами-носителями, на менее
устойчивые к редукции, связанные с оксидами марганца, и на
более устойчивые, связанные с (гидр)оксидами железа.
Но есть граничные условия, за которыми возможны
серьезные ошибки в расчетах. Во-первых, следует
использовать данные, полученные только в первые часы
экстракции металла. Более поздние результаты могут отражать
эффект перераспределения металла между геохимическими
фазами. Вероятно, длительный срок экстракций, принятый для
схем Тессиера и BCR способствует
перераспределению
металла между геохимическими фазами.
Во-вторых, концентрация тяжелого металла в почве
должна быть низкой. Так, при высоком насыщении чернозема
(17.9 мМ Zn / кг) расчет, основанный на начальном этапе
экстракции, дает абсурдный результат: при растворении 1
моля Mn растворяется 0.92 моля Zn. Как отмечалось выше,
закрепить такое большое количество Zn оксиды марганца не в
состоянии.
Возможная
причина
ошибки
лежит
в
перераспределении Zn между фазами (Ostergren et al., 1999;
Dupont et al., 2002; Manceau et al., 2002). В этом случае расчет
фиксирует только масштабы артефакта перераспределения
металла между фазами. В рамках данных схем
фракционирования интерпретация результатов более надежна
для слабозагрязненных почв, чем для сильнозагрязненных.
Глава 3. ХИМИЯ И МИНЕРАЛОГИЯ ТЯЖЕЛЫХ
МЕТАЛЛОВ И МЕТАЛЛОИДОВ
Почвы – это многокомпонентные, открытые и динамичные
системы, где полное равновесие между компонентами никогда
не достигается. В почвах всегда можно обнаружить самые
разные формы элементов. Твердая фракция почв (а только она
и рассматривается в монографии) представляет собой
гетерогенный ансамбль минералов, малых органических
молекул
и
высокополимеризованных
органических
компонентов, образующихся в результате деятельности живых
организмов: бактерий, грибов, корней растений и т.п.
Минералы обычно делят на первичные и вторичные.
Первичные минералы: кварц, полевые шпаты, оксиды титана и
т.п. – наследуются в почвах от материнской породы. В
последние годы в почвы вблизи заводов и рудников попадает
значительное количество первичных техногенных минералов и
среди них: оксиды цинка и никеля, виллемит, франклинит и
т.п.
Вторичные минералы представлены филлосиликатами,
оксидами железа, алюминия и марганца, а также карбонатами,
которые, впрочем, могут быть и литогенными.
Органическое вещество включает живые организмы
(мезофауна и микроорганизмы), мертвый растительный
материал (в лесу это подстилка, а в степи – дернина) и
коллоидальный гумус. Твердые компоненты объединяются
вместе в виде агрегатов, создающих в почве систему пор
разного размера, наполненных газом и водным раствором. Как
у неорганической, так и органической твердой фазы
химический состав и дисперсность сильно варьируют. С
химической точки зрения коллоиды представляют собой
наиболее реакционноспособную часть почвы, и она наиболее
важна при оценке влияния загрязнения металлами в
экосистеме.
Минеральные коллоиды включают преимущественно
филлосиликаты и разное количество оксидов железа и
марганца. Органические коллоиды представлены гумусовыми
субстанциями. Коллоидальные наночастицы с высокой
удельной поверхностью обладают зарядом, образованным за
счет действия поверхностных функциональных групп, которые
закрепляют лабильные ионы. Физические, химические и
биологические условия в почве непрерывно меняются, что
приводит к постепенной трансформации педогенных,
литогенных и техногенных частиц тяжелых металлов.
Последние термодинамически неустойчивы в почвенных
условиях и относительно быстро переходят в более
стабильные формы. Основной процесс их первичной
трансформации в почвах – переход в жидкую фазу за счет
растворения. Со временем лабильные формы тяжелых
металлов превращаются в более стабильные, лучше
отвечающие новым условиям (Пинский, 1997; Han et al., 2001).
Главные механизмы закрепления тяжелых
металлов в почвах
Выделяют пять главных механизмов закрепления тяжелых
металлов, которые четко идентифицируются методом EXAFSспектроскопии (Brown et al., 1999; Ford et al., 2001; Manceau et
al., 2002).
1.
Образование
внешнесферных
поверхностных
комплексов. При этом механизме ион-сорбат со своей водной
оболочкой закрепляется на заряженной поверхности среди
массы диффузных ионов с помощью электростатического
взаимодействия. Частицы сорбата изолированы от металла
сорбента двумя слоями кислорода, так что расстояние металл–
металл (Ме–Ме) оказывается большим, не менее 4.5 Å.
Образование внешнесферного поверхностного комплекса
типично для минералов с постоянным отрицательным зарядом
(филлосиликатов). Напомним, что постоянные заряды
образуются в результате изоморфного замещения металлов в
решетке минералов.
Для обмена катионов наибольшее значение имеют
глинистые минералы типа 2:1. Поскольку межпакетные связи в
трехслойных минералах ослаблены из-за симметричной
неполярной структуры пакетов, то пространство между
пакетами может варьировать в широких пределах и
становиться доступным для большинства катионов (Пинский,
1997). В результате емкость катионного обмена у
монтмориллонита достигает ~100 смоль(+)/кг, у иллита ~30
смоль(+)/кг, а у каолинита только ~8 смоль(+)/кг. У оксидов
железа и алюминия этой емкости почти нет. У органического
вещества емкость катионного обмена достигает ~200
смоль(+)/кг. Филлосиликаты совместно с оксидами железа –
два главных компонента большинства почв. Емкость
катионного обмена в основном зависит от содержания
смектитов (монтмориллонита, бейделлита, нонтронита) в
почве.
Неудивительно,
что
обменносвязанных
с
филлосиликатами неустойчивых частиц тяжелых металлов
сравнительно много в глинистых почвах.
Так как внешнесферный поверхностный комплекс
образован непрочными связями, то он может легко замещаться
за счет ионного обмена. Если говорить о количестве, то
фракция тяжелых металлов, закрепленных этим механизмом,
может превышать 10% в загрязненных кислых почвах (Roberts
et al., 2002). Но в большинстве почв эта фракция не превышает
нескольких процентов. Этот мобильный обменный пул
тяжелых металлов может легко опустошаться или переходить
в иные формы при изменении почвенных условий, например,
при росте рН или искусственном обогащении почвы
минералами-сорбентами.
2.
Образование
внутрисферных
изолированных
комплексов. При этом механизме катионы-сорбаты или
оксианионы связываются изолированно с поверхностью
сорбента через один или несколько лигандов, обычно это
кислород. Затем в ходе роста кристалла сорбированный ион
может постепенно проникать в структуру сорбента (Watson,
1996). Этот сорбционный механизм развивается вследствие
дефектов структуры твердого тела на поверхности. Свободные
химические связи на ребрах пакетов образуют рН-зависимый
переменный заряд, величина которого зависит от удельной
поверхности частицы: чем тоньше частица, тем больше у нее
реакционных поверхностных мест на единицу массы. За счет
этого механизма сорбция металла происходит в гораздо
большем объеме, чем можно ожидать из величины емкости
катионного обмена почвы. Сорбируемые внутрисферно ионы, в
гораздо меньшей степени способны вновь возвращаться в
окружающую среду, чем ионы, закрепляемые за счет ионного
обмена. Такая форма закрепления металла минераламиносителями характерна для начальной стадии загрязнения почв.
Полиэдры металлов и металлоиодов (обычно это октаэдры
или тетраэдры) часто контактируют ребрами и вершинами и
редко гранями с полиэдрами сорбата. Для системы «металл +
сорбат», кристаллографическая природа сорбционных мест
может быть установлена на основе величин расстояния Ме–
Ме, что доступно методу EXAFS-спектроскопии.
Хотя в загрязненных почвах тяжелых металлов много, они
обычно плохо идентифицируются с помощью традиционной
рентгеноструктурной техники. Но, благодаря высокой энергии
источников синхротронного излучения третьего поколения,
сейчас удается их выявлять и анализировать. Первый
поверхностный комплекс, идентифицированный в почвах, –
это тетраэдрически координированный Zn, сорбированный в
межслоевом пространстве гексагонального бернессита на
кристаллографических позициях слоев, свободных от Mn
(Manceau et al., 2002).
3.
Образование
многоядерных
поверхностных
комплексов.
Этот
механизм
реализуется
в
ходе
гетероэпитаксиального роста сорбата. (Термин «эпитаксия»
обозначает совместную ориентировку кристаллов разных
видов, когда один кристалл ориентирован в зависимости от
другого, на который он нарастает). Сорбент действует как
структурная подложка для нового осадка (Schlegel et al., 2001).
Минеральная поверхность выполняет важную функцию, ее
присутствие снижает уровень перенасыщения, необходимого
для осаждения за счет сходства размеров двух решеток
(McBride, 1989). Затем катионы сорбата полимеризуются на
субстрате.
Гетерогенный синтез ядер – это наиболее важный
механизм кристаллообразования в почвах. Поверхность
минерала-носителя снижает высокий энергетический барьер
для образования зародышей новых кристаллов из раствора.
Подложка и новые кристаллы координируют анионы, если две
твердые частицы контактируют за счет электростатического
взаимодействия (рис. 8). В результате анионная структура
подложки и поверхностного осадка ориентирована одинаково.
Таким образом, кристаллизуются малые кристаллы, которые
из-за высокой межфазной энергии между кристаллом и
раствором быстрее растворимы, чем крупные частицы. Без
данной матрицы тонкие частицы более склонны к
растворению, чем к росту.
Рис. 8. Схема основных сорбционных процессов на примере СоООН.
Многоядерный поверхностный комплекс (MSC) показан в виде эпитаксии
частиц гетита αFeOOH (слева). Одноядерный (ISC) монодентантный
комплекс (справа в центре), одноядерный бидентантный (справа вверху) и
двуядерный бидентантный (справа внизу) представляют внутрисферные
комплексы. Внешнесферный комплекс (OSC) показан вверху. Из (Manceau
et al., 2002).
Многоядерный поверхностный комплекс может образовать
эпитаксиальный осадок, если фазы сорбента и сорбата растут
аналогично, или если они растут в форме разных слоев, образуя
смешаннослойные образования (Chiarello et al., 1997). Среди
последних преобладают глинистые минералы (гекторит и
корренсит) и оксиды марганца (литиофорит и асболан). Так, у
литиофорита переслаиваются слои MnO2 и Al(OH)3, а у
обогащенного никелем асболана слои MnO2 и Ni(OH)3.
Гетерогенное
образование
ядер
–
это
широко
распространенный
механизм
покрытия
поверхности
минералов подложки. Считается, что в процессе участвуют
микроорганизмы (Manceau et al., 2002). Образующиеся
поверхностные пленки обычно мало растворимы и могут
рассматриваться как формы закрепления тяжелых металлов.
4. Гомогенное осаждение происходит, когда растворенные
катионы полимеризуются и осаждаются из раствора без
образования структурной связи с сорбентом. Дело в том, что
концентрация сорбата может опуститься ниже критической,
при которой растворяются смешанные фазы, но сохраняются
«чистые» (Scheckel et al., 2000; Dahn et al., 2002). Этот
механизм называют «гомогенное осаждение индуцируемое
растворением». Гомогенные осадки в дальнейшем покрывают
субстрат оболочкой. Так как твердые растворы и смеси менее
устойчивы, чем чистые металлсодержащие фазы, то этот
механизм существенно закрепляет тяжелые металлы в почвах.
В качестве примера можно указать на эволюцию часто
встречаемого в почвах Fe-вернадита (Fe-δMnO2). Его
рассматривают как самый ранний и неустойчивый продукт
оксидогенеза железа и марганца. В ходе раскристаллизации
образуются более устойчивые отдельные фазы: безжелезистый
вернадит и безмарганцевый фероксигит (Чухров и др., 1989).
5. Диффузия в решетке. С течением времени
сорбированный единичный ион диффундирует в решетку
сорбента, заполняя вакансии или замещая атомы сорбента.
Совместно с механизмом гомогенного осаждения, это явление
постепенно уменьшает подвижность тяжелых металлов в
почвах. Оба механизма приводят к постепенной стабилизации
тяжелых металлов в почвах. Прямое доказательство диффузии
металлов в решетку почвенных минералов установлено для
гетита и кальцита.
Формы тяжелых металлов в почвах
Токсичность и биологическая доступность тяжелых
металлов в почвах зависит от реакционной способности и
химической формы элемента (Вrown et al., 1999). Понятие
«химическая форма» важнейшее, оно охватывает широкий
спектр свойств элемента, то, что ранее называлось «химией и
минералогией элемента». Среди этих свойств: 1) степень
окисления элемента; 2) физический статус, который включает
множество параметров: фазовый состав; аморфное или
кристаллическое строение твердофазных частиц; разделение
на коллоиды, животные и растительные клетки; нахождение на
поверхности или в виде тонкой пленки на твердой частице;
идентификация вида сорбционного комплекса (моно- или
полимерного) на минеральной частице или органической
субстанции; 3) эмпирическая химическая формула; 4)
детальная молекулярная структура.
Важность
подробной
характеристики
элемента
иллюстрируется на примере с хромом. Валовое содержание
хрома мало, что говорит об опасности загрязнения почвы. Но
имеет значение степень окисления хрома. Когда Cr(VI)
выступает как оксианион в составе тетраэдров в водном
растворе, он полностью растворим, легко подвижен в грунтовой
и почвенной водах и токсичен для организмов. Напротив,
восстановленный до степени окисления (III), хром в составе
октаэдра окружен 6 атомами кислорода, относительно мало
растворим, стабилен и мало опасен для организмов (Вrown et al.,
1999). Как видно из этого примера, фундаментальное понимание
особенностей металла должно обязательно включать описание
его химических форм.
Эта точка зрения иллюстрируется и другим примером.
Уровень свинца в крови людей, проживающих в разных
регионах, зависит от содержания Pb в верхнем горизонте почв,
когда оно варьирует от сотен до тысяч мг/кг (рис. 9). Из рис. 9
видно и другое, значительное различие в содержании Pb в
крови людей в разных регионах, которое прямо не
коррелирует со степенью загрязнения почвы. При одном и том
же количестве валового Pb в почве его содержание в крови
людей отличается в несколько раз.
Рис. 9. Зависимость содержания Pb в крови людей от количества свинца в
поверхностном слое загрязненных почв Австралии и США. Из (Brown et
al., 1998).
Одно из возможных объяснений этого явления заключается
в разных формах соединений свинца, за счет чего в одних
почвах он оказывается более доступен организмам, чем в
других. Когда свинец закреплен в таких кристаллах как
галенит или пироморфит, он менее растворим, менее мобилен
и менее доступен, чем, когда он сорбирован на поверхности
минералов. В последнем случае Pb быстро переходит в раствор
за счет редукции и/или за счет подкисления среды. Влияние
этих различий форм элементов хорошо подтверждается в
опытах со свиньями, которые заглатывали частицы почвы или
частицы отходов рудника Лидвиль, Колорадо, США.
Оказалось, что биологическая доступность свинца колебалась
от 5% для рудных отходов, где доминирует галенит (PbS), до
45% для частиц почвы, где большинство Pb входит в состав
оксидов железа, марганца и свинца (Вrown et al., 1999).
Хотя корреляция между характером форм поллютантов и
их биодоступностью показана во многих работах (Davis et al.,
1992; Davis et al., 1997; Gasser et al., 1996), пока собрано мало
сведений о составе этих форм при высоком уровне загрязнения
почв (Davis et al., 1996). Например, современный стандарт в
США ограничивает содержание As в питьевой воде в 50 мг/л
независимо от состава частиц. Но мышьяк может
присутствовать в разных формах: как неорганический
оксианион As(III)O33- и As(V)O43-, как органическое
соединение и т.д. Трехвалентная форма мышьяка, независимо
от характера лигандов, более токсична, чем пятивалентная
(Brown et al., 1999). В прошлом, мероприятия по очистке
загрязненных почв или рудных отвалов планировались только
на основе значений валового содержания элемента, без учета
его форм, токсичности и биодоступности (Davis et al., 1992,).
Такой примитивный подход был обусловлен трудностью
получения информации о молекулярном уровне концентрации
рассеянных элементов и в переводе этой информации в
обычные биогеохимические понятия. В результате недавних
исследований было установлено, что биологическая опасность
часто преувеличивается, так как многие формы тяжелых
металлов и металлоидов биологически не доступны.
Глава 4. ФОРМЫ ТЯЖЕЛЫХ МЕТАЛЛОВ
И МЕТАЛЛОИДОВ В ЗАГРЯЗНЕННЫХ ПОЧВАХ
Глобальное, региональное и локальное загрязнение почв
Необходимо различать разные виды загрязнения почв
тяжелыми металлами: глобальное, региональное и локальное.
Глобальное загрязнение не является постоянным, оно
заметно меняется со временем. Динамику глобального
загрязнения изучают по-разному. Для этого часто используют
датированные с помощью изотопов образцы снега или торфа.
В первой программе глобального мониторинга в 1973 г.
среди наиболее опасных элементов фигурировало три металла:
ртуть, свинец и кадмий. Но уже в 1980 г. среди металловполлютантов помимо этих трех рассматривались семь новых:
марганец, олово, медь, молибден, хром, никель, кобальт
(Добровольский, 2003). После активного обсуждения
научными кругами проблемы роста загрязнения окружающей
среды тяжелыми металлами в Стокгольме в 1972 г. сессия
ООН приняла итоговые материалы, что способствовало
общему снижению загрязненности биосферы. В 1991 г. в
журнале Nature опубликована статья под характерным
названием «Уменьшение антропогенных Pb, Cd и Zn в снеге
Гренландии после 1960-годов» (Bouton et al., 1991). Эти
результаты согласуются с данными НПО Тайфун о сохранении
уровня содержания тяжелых металлов в почвах 20 км зон
вокруг городов России за последние 5 лет (Бабкина и др.,
2004). Глобальное снижение загрязненности обусловлено
закрытием предприятий с устаревшей технологией и
строительством экологически чистых заводов.
Региональное загрязнение анализируют по изменению
состава речных, озерных или морских отложений, которые
формируются за счет смыва дождевыми и талыми водами
тонких почвенных частиц. В донных отложениях рек, озер и
прибрежных частей морей определяют содержание тяжелых
металлов. Согласно анализу металлов в илах Рейна,
протекающего через промышленные районы Западной
Европы, с конца ХVII в. по 1975 г., концентрация хрома
возросла в 9, меди и свинца – в 13, цинка – в 19, ртути – 50 и
кадмия – в 100 раз (Добровольский, 2003). Результаты
ужесточения национальных экологических законов не
замедлили сказаться. Уже в начале 80-годов высокие уровни
содержания металлов в донных отложениях рек и озер
существенно снизились (рис. 10).
Рис. 10. Изменение содержания тяжелых металлов в пойменных
отложениях низовьев Рейна на протяжении ХХ в. (Добровольский, 2003).
Сейчас наибольшую опасность представляет локальное
загрязнение почв, в ряде случаев формирующее техногенную
геохимическую аномалию. С точки зрения влияния на
здоровье человека и вообще на культуру ландшафта
Перельман (1975) различает три основных вида техногенных
аномалий.
1.
Полезные
техногенные
аномалии,
играющие
положительную роль в культурном ландшафте. Примером
такой аномалии может служить повышение содержания
кальция в кислых лесных почвах.
2. Нейтральные техногенные аномалии. К ним условно
можно отнести повышение содержания железа и алюминия в
почвах.
3. Вредные техногенные аномалии, обусловливающие
нежелательные явления в культурном ландшафте. Их
примером могут служить повышенное содержание свинца
вдоль автомобильных дорог, накопление ряда тяжелых
металлов вблизи старых промышленных предприятий и в
составе рудных отвалов. Такие аномалии требуют
наибольшего внимания.
Наиболее опасные источники локального загрязнения
тяжелых металлов – мощные промышленные объекты, не
прошедшие реконструкцию. В Европе, где техническая
революция началась раньше, чем в России, раньше и
произошло загрязнение почв тяжелыми металлами. Начиная
со второй половины ХIХ столетия в почвы Западной Европе
попало огромное количество металлических поллютантов. К
1996 г. во Франции было выявлено несколько сотен отдельных
мест сильнозагрязненных металлами (Manceau et al., 2000). В
Европе в последние десятилетия многие заводы с устаревшей
технологией модернизируются или закрываются. Так, в Морте
де Норд (северная Франция) в 1963 г. был ликвидирован завод
по выплавке цинка и свинца, работавший с 1906 г. На другом
металлургическом заводе в том же районе в 1970 г. установили
фильтры, сократившие выброс пыли с нескольких тонн до 300
кг в сутки. Но почвы, за многие годы, вобравшие в себя
большое количество поллютантов, требуют изучения и
мелиорации.
В России к числу наиболее загрязненных городов
относятся Мончегорск, Ревда, Белово, Дальнегорск и др.
(Большаков и др., 1993). К регионам с неблагоприятным
санитарным состоянием за счет загрязнения тяжелыми
металлами принадлежит Кольский полуостров (Орлов и др.,
2002). Здесь свой вклад в загрязнение вносят предприятия
черной и цветной металлургии, энергетики и производства
минеральных удобрений.
Площадь локальных геохимических аномалий составляет
единицы и десятки километров. Характерна техногенная
геохимическая аномалия, образовавшаяся в результате
длительных
аэральных
выбросов
Череповецкого
металлургического комбината (Водяницкий и др., 1995).
Особую группу аномалий формируют почвы, образовавшиеся
в поймах промышленно загрязненных рек (Трухина, 1988;
Водяницкий и др., 2004).
Важные исследования динамики локального загрязнения
тяжелыми металлами проведены на маршевых почвах,
окружающих Чесапикский залив на Атлантическом побережье
США, штат Мериленд (Griffin et al., 1989). Здесь были
отобраны образцы торфа с глубины 1 м. Вначале по изотопу
210
Pb определили геохронологию нескольких участков маршей.
На разных участках скорость роста торфа составляла от 0.35
до 0.75 см/год. Эти данные были использованы для
хронологии загрязнения территории тяжелыми металлами
почти за весь ХХ в. Наиболее интересные результаты
получены на маршах 4 и 5 (рис. 11).
На марше 4 поступление Zn и Pb постоянно росло с начала
20 годов, достигнув, пика в конце 50-х и начале 60-х годов.
Этот марш расположен всего в 5 км от Бетлехемского
сталелитейного
завода,
загрязнявшего
близлежащую
территорию тяжелыми металлами. С 1930 по 1969 были годы
максимального выпуска стали, при этом росли и выбросы. За
время с 1970 по 1980 годы выбросы на этом заводе
значительно сократились: Zn на 91%, Cu на 86% и Pb на 93%.
Уменьшение заводских выбросов за 15 лет привело к
заметному снижению содержания металлов в торфе. В
отложениях марша это отразилось в снижении накопления Zn
и Pb на 70 %, а Cu на 66% (Griffin et al., 1989).
На марше 5 вблизи автодороги повышенное содержание Pb
связано с выхлопными газами автомобилей. Оно росло,
начиная с 1930-х годов, достигнув пика в 1969-1977 гг. В начале
70-х годов в бензине начали сокращать содержание свинца, а с
1975 г. в США начали выпускать бензин без Pb. Благодаря
этому после 1975 г. поступление свинца в почву резко
сократилось. В национальном масштабе общий выброс Pb с
1977 по 1985 гг. снизился на 85%, что зафиксировано и в
отдельных регионах. Так, в марше 5 поступление свинца с
Рис. 11. Атмосферные выпадения Zn, Pb и Cu. Из (Griffin et al., 1989).
1977 по 1985 гг. уменьшилось на 40% по сравнению с уровнем
1969-1977 гг. (Griffin et al., 1989).
Имеет большое значение вид соединений тяжелых
металлов в почве, так как от этого зависит степень их
токсичности. В связи с этим почвоведы уделяют много
внимания изучению видов и форм тяжелых металлов. Но их
формы трудно выявить даже на моделях. Основные причины
трудностей – низкая концентрация металлов для традиционной
кристаллографической аппаратуры, влияние сосуществующих
минералов, трудности идентификации минералов-носителей,
многообразие механизмов сорбции. Эти проблемы решаются
при использовании синхротронной техники третьего
поколения. Ниже мы рассмотрим формы соединений мышьяка,
свинца, цинка и ртути, в основном полученные при
использовании синхротронной техники.
Соединения мышьяка
В настоящее время идентифицировано более 300 арсенатов
и минералов, закрепляющих As (Manceau et al., 2002). Часть
мышьяка включается в промышленные циклы, и затем,
благодаря растворению минералов, поступает в почву и
почвенные воды. В Национальном приоритетном списке США
опубликована информация о 1000 мест потенциально опасных
для здоровья людей. В этом списке As по частоте цитирования
занимает второе место после Pb среди неорганических
поллютантов (Manceau et al., 2002). Основным источником
техногенного мышьяка являются As-содержащие отвалы руд.
Этот параграф составлен на основе материалов,
посвященных анализу форм мышьяка в пустой породе,
извлеченной из разных рудников, а также – в рудных отходах,
которые представляют собой вредные техногеохимические
аномалии.
Присутствие мышьяка в отходах руды, хранящихся
длительное время, – одна из главных современных проблем
окружающей среды. Недавние эпидемии в Бангладеш и Индии
в результате токсического влияния мышьяка привели к
повсеместному пересмотру стандартов содержания As в
питьевой воде. Увеличилось внимание к состоянию отвалов,
обогащенных мышьяком (Pactung et al., 2003).
Пустая порода, извлеченная из урановых и золотых
рудников, где As заключен в форме (арсено)пирита,
представляет собой очень опасную техногенную аномалию.
Частицы мышьяка водных коллоидов и вторичной фазы из
кислых дренажных вод и осадков многократно анализировали
методами EXAFS- и XANES-спектроскопии. Наиболее
обычная форма окисленности мышьяка III и V. Обе формы
четко различаются на XANES-спектрах (рис. 12). При этом вид
доминирующей формы зависит от рН и ЕН. В водной среде при
нейтральном рН арсенат представлен в основном как Н2AsО4-,
а арсенит имеет форму Н3AsО3 (Cullen, Reomer, 1989).
Благодаря медленной трансформации, обе формы иногда
Рис. 12. XANES-спектры поглощения, содержащие К-края атомов As в
составе As-гетита и арсенопирита. Из (Paktung et al., 2003).
сосуществуют как в восстановительной, так и в окислительной
обстановках.
В закреплении мышьяка важную роль играют гидроксиды
железа. На поверхности гетита αFeOOH образуются прочные
комплексы с As(III). Установлена бидентантная, биядерная
связь As(III) с поверхностью, аналогичная связям, образуемым с
другими оксианионами на гетите при внутрисферном
механизме закрепления. Поверхностный комплекс As(III)-гетит
устойчив к окислению, что доказано методом XANESспектроскопии (Manning et al., 1998). Благодаря своей
устойчивости, этот комплекс играет важную роль в закреплении
арсенатов.
Восстановленная форма, арсенит, более токсична, чем
окисленная. У обеих форм As сильное сродство к оксидам
железа, хотя они противоположно реагируют на изменения рН.
В интервале рН 3-10 количество адсорбированного арсената на
оксидах железа уменьшается с ростом рН, тогда как адсорбция
арсенита увеличивается с максимумом при рН 9 (Jain,
Loeppert, 2004). Как
видно
из
рис. 13,
количество
адсорбированного
As(V) на гетите имеет
максимум
при
высокой
исходной
концентрации
мышьяка в растворе
при рН 9. В этом
состоит отличие от
поведения арсенита, Рис. 13. Адсорбция As(III) и As(V) на гетите
адсорбция которого αFeOOH. ■ – исходная концентрация As(III) =
максимальна в кислой 0.267 мМ, □ - исходная концентрация As(III)=
среде. При этом в 0.133 мМ, ● - исходная концентрация As(V) =
0.267 мМ. Из (Manning et al., 1998)
нейтральной
среде
As(III) более прочно
закреплен на поверхности гидроксида железа, чем As(V)
(Manning et al., 1998). Это объясняется различием в структуре
комплексов As(III) и As(V).
Рассмотрим подробнее формы мышьяка в рудных отходах
в США, Франции и Канады.
Отвалы золотых рудников в Калифорнии, США. За 150
лет эксплуатации в отвалах этих рудников накопилось до 5000
мг/кг As. Часть отходов используется для жилищного
строительства. Формы мышьяка изучали в отходах трех
рудников. Среди них полностью окисленные отходы (рудник
Рут), частично окисленные отходы (рудник Агронот) и
обожженная сульфидная руда (рудник Спенсевиль) (Brown et
al., 1999).
По данным EXAFS-спектроскопии в отвалах рудников Рут
и Спенсевиль преобладает As(V), а в отвале рудника Агронот у
мышьяка отмечено смешанное состояние окисления.
Установлено, что As(V) представлен скородитом FeAsO4.2H2O,
а также мышьяком (V), адсорбированным на частицах гетита
αFeOOH и гиббсита γAl(OH)3. Содержание каждой из форм
мышьяка в разных отходах варьирует. В отходах рудника Рут
гидроксиды железа и глинистые минералы связывают равные
доли As(V). В отходах рудника Аргонот 20% мышьяка в
восстановленной форме входит в состав арсенопирита FeAsS и
As-пирита (FeS2-xAsx), а 80% находится в окисленной форме и
входит в состав ферриарсенатов, таких как скородит (Brown et
al., 1999).
В обожженной сульфидной руде (рудник Спенсевиль)
содержится окисленный As(V), замещающий сульфат в
кристаллах
ярозита
[KFe3(SO4)2(OH)6],
и
мышьяк,
сорбированный на поверхности гематита αFe2O3. Определение
методом EXAFS-спектроскопии форм мышьяка – это важный
шаг на пути установления его биодоступности, подвижности и
токсичности, так как эти свойства зависят от степени
окисления As. В целом можно сказать, что мышьяк,
находящийся в составе кристаллических и аморфных осадков,
менее доступен для организмов, чем, сорбированный на
поверхности минералов (Brown et al., 1999).
Мышьяк, адсорбированный на гидроксидах железа,
скородит и Аs5+-содержащий ярозит, все эти формы стабильны
в кислых условиях, типичных при окислении сульфидов, но их
нельзя рассматривать как надежные формы закрепления As.
Ярозит как обычный минерал кислых ландшафтов не устойчив
при рН > 3 и тогда быстро растворяется, высвобождая As.
Аналогично, растворимость скородита сильно зависит от
кислотности, скорость растворения минимальна при рН 4 и
резко возрастает при увеличении рН (Savage et al., 2000). Хотя
внутрисферные комплексы арсената с гидроксидами железа
отличаются прочностью (Waychunas et al., 1993; Manning et al.,
1998), всегда есть опасность десорбции поверхностного
комплекса при изменении условий: при появлении в растворе
сильных лигандов, при усилении бактериальной активности и
в результате сезонных изменений рН и ЕН в гидроморфных
почвах и в рудных отвалах.
Отвалы свинцово-цинкового рудника Карно, юго-восток
Франции. Рудник был закрыт в 1962 г. (Morin et al., 2003). За
все годы работы скопилось 1.5 млн. т сернистых отходов,
содержащих в среднем 0.7% Pb; 10% Fe и 0.2% As. Отходы
складировались в дамбу высотой 6 м. В кислом ручье,
вытекающим из-под дамбы, концентрация As(III) достигает
80-280 мг/л. На расстоянии первых десятков метров ручья
быстрое окисление Fe(II) ведет к соосаждению большого
количества As и Fe(III), образующих «бактериальные маты» на
дне. Анализ минеральных и органических осадков показал
образование редкого обогащенного мышьяком сульфатгидроксида железа – туелита (tooeleite) с химической
формулой Fe6(AsO3)4(SO4)(OH)4.4H2O. Туелит образуется
совместно с аморфным гидроксидом железа, обогащенным
мышьяком в разной степени окисления As(III) и As(V). Во
влажный сезон взвесь в истоке ручья содержит туелит
совместно с аморфным гидроксидом As(III)–Fe(III). В сухой
сезон ситуация меняется, доминируют гидроксиды As(V)–
Fe(III), в которых молярное отношение этих элементов
достигает 0.6-0.8 (Morin et al., 2003).
Как уже отмечалось, в закреплении мышьяка большую
роль играют гидроксиды железа. Окисление Fe(II) до Fe(III)
катализируется при активном метаболизме микроорганизмов,
таких как Acidithiobacillus ferrooxidans. Образуются биогенные
гидроксиды железа, которые включают в свою структуру или
адсорбируют на своей поверхности токсические элементы
(Brown et al., 1999; Morin et al., 2003). В кислых дренажных
водах микробное окисление железа и нейтрализация
кислотности приводит к осаждению сначала сульфатов, а
затем
гидроксидов
в
последовательности:
ярозит
KFe3(SO4)2(OH)6, швертманит Fe8O8(ОН)6SO4, ферригидрит
2Fe2O3.FeOОН.4H2O, гетит αFeOОH или лепидорокит γFeOОН.
Такие химические реакции сильно ограничивают подвижность
мышьяка в почвах.
Отвалы золотого рудника Кетца Ривер, центральный
Юкон, Канада. Рудник эксплуатировался короткое время с
1988 по 1990 годы. За это время образовалось примерно 310
тыс. т сухих и мокрых отходов, представляющих собой
опасную техногенную аномалию. Мокрые отходы частично
находятся под водой, среднее содержание мышьяка в них 4%.
Установлена сложная минералогия мышьяка. Широко
распространены гидроксиды железа, на которых As(V)
адсорбируется в форме внутрисферного комплекса (Pactung et
al., 2003).
Как показано на рис. 14 и 15, химический состав
гидроксидов железа и арсенатов варьирует очень широко. В
состав гидроксидов железа мышьяк входит в очень разном
количестве, от следовых (300 мг/кг) до крайне высокого
уровня (27.5%). На графике As2O5~Fe2O3 (рис. 14) видны
разные минералы от гетита и ферригидрита до скородита и СаFe-арсенатов. Основная часть образцов – это гидроксиды
железа, у которых молярное отношение Fe : As > 5. Другое
облако точек представляет собой частицы скородита и других
арсенатов. У них молярное отношение Fe : As составляет от 1
до 1.5. Максимальная концентрация As, адсорбированного на
гидроксидах железа в ходе соосаждения, отвечает отношению
Fe : As = 1.5. Таким образом, отношение Fe : As >1.5
рассматривается как граничное при переходе от арсенатов к
Рис. 14. Зависимость содержания As2O5 от Fe2O3 в гидроксидах железа и
арсенатах в As-обогащенных отвалах, Юкон, Канада. Пунктирные линии
отражают отношение As : Fe. Из (Paktung et al., 2003).
B
B
B
B
B
B
B
B
гидроксидам железа.
Содержание СаО в составе гидроксидов железа зависит от
содержания Fe2O3 и As2O5. Количество СаО прямо зависит от
As2O5 (рис. 15). Содержание железа высокое при низком
количестве СаО, когда Fe входит в состав гидроксидов, и
низкое – при высоком количестве СаО для частиц арсенопирита
и юконита (Pactung et al., 2003).
Мышьяк гораздо сильнее выщелачивается из сухих
отходов (его содержание в фильтрующей воде 18-35 мг/л), чем
из мокрых отходов (содержание в воде 1-4 мг/л).
Растворимость мышьяка сильно зависит от состава минералов
в рудных отходах. Арсенопирит и пирит находятся в малых
концентрациях и эти частицы обычно окружены вторичными
арсенатами и гидроксидами железа, что ограничивает
окислительное растворение сульфидов. Поэтому участие
сульфидов в высвобождении As незначительное. Скородит
занимает ⅓ от массы всех As-минералов в сухих отходах и ⅛ –
в мокрых отходах. При этом мокрые отходы богаче
гидроксидами железа, а сухие – рентгеноаморфными
Рис. 15. Зависимость содержания СаO от As2O5 в гидроксидах железа и
арсенатах в As-обогащенных отвалах, Юкон, Канада. Из (Paktung et al.,
2003).
арсенатами Fe(III) (Pactung et al., 2003).
Исследования
показали
устойчивость
As(V)
в
восстановительных условиях, если мышьяк закреплен на
частицах гидроксидов железа, хотя в свободных условиях (в
отсутствие гидроксидов) редукция до As(III) происходит
быстро. Интересно, что если на поверхности гидроксидов
осаждается восстановленный As(III), он быстро окисляется до
As(V) при высыхании отходов (Manning et al., 1998).
Сильное выщелачивание мышьяка из сухих отходов
обязано высокой доле As-минералов и низкой доле
гидроксидов железа. Величина рН воды, фильтрующейся из
отходов, в среднем 8.3. В слабощелочных условиях
ферриарсенат термодинамически не устойчив. В особенности
это справедливо для соединений с Fe : As < 4. Растворимость
арсенатов возрастает с ростом доли кальция: Са-Fe-арсенаты
гораздо более растворимы, чем бескальциевые Fe-арсенаты.
Поскольку Са-Fe-арсенаты менее устойчивы, чем гидроксиды
железа,
интенсивное
высвобождение
мышьяка
из
ферриарсенатов в сухих отходах не удивительно.
Другой фактор, определяющий вынос As из отходов – это
форма мышьяка на поверхности частиц гидроксидов железа.
По данным EXAFS-спектроскопии арсенат-ионы образуют, в
основном, прочные бидентантные комплексы на частицах
гидроксидов железа (Pactung et al., 2003). После быстрого
высвобождения арсенатов с поверхности гидроксидов железа,
дальнейшая десорбция As с течением времени замедляется.
В опытах по совместному соосаждению Fe и As начальное
закрепление мышьяка было значительно выше, чем при
адсорбции на уже синтезированных частицах гидроксидов
железа. Скорость поглощения As не имела диффузного
характера, так как As(V) координировался на поверхности до
начала роста кристаллов. После начальной адсорбции As(V)
медленно, в течение месяца, высвобождался из осадка
гидроксида железа, так как рост кристаллов ферригидрита
приводит к десорбции As(V). Адсорбционная плотность
достигала 0.7 моля As(V) на моль Fe(III) в совместно
осажденных осадках по сравнению с 0.25 моля As(V) на моль
Fe(III) на предварительно синтезированном ферригидрите
(Fuller et al., 1993).
Свежеосажденные частицы гидроксида представляют
собой дискретные цепи полимера диоктраэдрического железа с
минимумом поперечных связей (Waychunas et al., 1993).
Совместное осаждение с арсенатом тормозит рост частиц
ферригидрита. Поперечные связи в структуре ферригидрита
образуются только в отсутствие арсената. Другими словами,
при старении ферригидрит полимеризуется лишь при
высвобождении мышьяка в раствор. Пока отношение Fe : As
ниже 5, гидроксиды железа не устойчивы и продолжается
постепенное высвобождение мышьяка, сопровождаемое
полимеризацией и старением частиц гидроксидов железа
(Pactung et al., 2003).
Основные эксперименты с адсорбцией мышьяка проводят
на
таком
гидроксиде
железа
как
ферригидрит
.
.
2Fe2O3 FeOOН 4H2O. Но, как показали наши исследования, в
почвах умеренного пояса ферригидрит распространен гораздо
меньше, чем фероксигит δFeOOН (Водяницкий, 2003). В
будущем необходимо изучить адсорбцию As именно на
фероксигите, который образуется в результате окисления
Fe(II), в отличие от ферригидрита, который в лаборатории
синтезируют путем подщелачивания раствора соли Fe(III).
Прочному закреплению мышьяка частицами (гидр)оксидов
железа препятствует конкуренция со стороны других
оксианионов: PO43-, SO42-, MoO42-. Так, при высокой
концентрации в растворе фосфатов адсорбция As(V) почвой
резко снижается (Manning, Goldberg, 1996). Поэтому фосфаты
применяют для экстрагирования мышьяка из почв (Jаckson,
Miller, 2000).
Органические молекулы подобно фосфатам повышают
биологическую
доступность
мышьяка.
Исследования,
проведенные на гетите αFeOOH, показали, что адсорбция
As(III) уменьшается в присутствии гуминовых кислот и
фульвокислот и, особенно, лимонной кислоты (Grafe et al.,
2001). Что касается арсената, то его адсорбция снижалась при
участии гуминовой кислоты и фульвокислот, но не лимонной
кислоты. На адсорбцию влияет тип, плотность и кислотные
функции органических полимеров. Кроме того, органические
молекулы затрудняют электростатическое взаимодействие
As(V) c поверхностью гетита. Влияние конкуренции
органических лигандов зависит от рН среды.
Многие участки почвы, загрязненные мышьяком, слишком
токсичны,
чтобы
можно
было
рассчитывать
на
самопроизвольное заселение и рост самых устойчивых
растений. Современные – западноевропейская и американская
мелиорации ориентируются, скорее, на инактивацию почв, чем
на удаление загрязненной почвы (Van der Lebie et al., 2001).
Последнее мероприятие дорого, требует мест для захоронения
грязной почвы, а также запасов чистой почвы. Все
минералогические и геохимические исследования доказывают,
что нет простого средства, которое продолжительно закрепит
мышьяк в окисленных и умеренно восстановленных почвах.
Современный подход ориентируется на кооперацию и поиск
активных мелиорантов для максимального снижения
биологической доступности экзогенных токсичных металлов и
металлоидов.
Соединения свинца
Свинец попадает в почву при добыче свинцовых руд, как
отход металлургии, из автомобилей, потребляющих бензин с
присадкой свинца, а также из свалок, куда попадают
использованные электрические аккумуляторы, краски, сплавы
металлов. Изучение истории аэрального выпадения Pb за
последние 12370 лет, выполненное на образцах торфа из
болота в Юрских горах Швейцарии (Shotyk et al., 1998),
показало, что наибольшая эмиссия Pb в размере 15.7 мг / м2 в
год приходится на 1979 г. Этот уровень в 1570 раз выше
природного фона, который составляет 0.01 мг Pb / м2 в год.
После строгих ограничений на добавку в бензин тетраэтил
свинца, введенных в 70-х гг. в США и других развитых
странах, глобальный уровень загрязнения почв свинцом
значительно упал (Brown et al., 1999). Но он превышает на
порядки природный фон в локальных аномалиях в зоне
воздействия рудников и металлургических заводов, а также в
городских почвах за счет попадания свинцовых красок и
продуктов сгорания этилированного бензина.
Состав форм Pb широко варьирует, что объясняет различие
его биологической доступности в геохимических ландшафтах.
Понимание зависимости между химической формой и
биодоступностью элемента возможно только после полной,
точной и прямой идентификации форм Pb в первую очередь в
почвах и в рудных отвалах. Для достижения этой цели начали
применять синхротронную рентгеновскую технику (Ostergren
et al., 1999; Morin et al., 1999; Cotter-Howells et al., 1994;
Manceau et al., 1996).
Свинец – первый тяжелый металл, частицы которого в
загрязненных почвах были изучены методом EXAFSспектроскопии (1994 г.). В дальнейшем идентифицировано
пять вторичных соединений свинца: Pb2+-органические
комплексы, Pb2+-сорбированный на Fe(III)-оксидах, Pb2+сорбированный на оксидах марганца, плюмбоярозит и
фосфаты свинца (Manceau et al., 1996; Manceau et al., 2002;
Morin et al., 1999; Ostergren et al., 1999). Исследования свинца
методом синхротронной рентгеновской радиации уже нашли
практическое применение, снижая стоимость работ и повышая
эффективность мелиорации почв. Рассмотрим подробнее
данные о формах свинца в техногенных геохимических
аномалиях во Франции и США.
Загрязненные почвы в районе Эвин-Мальмизон, север
Франции. Методом EXAFS-спектроскопии изучали илистую
фракцию (< 2мкм) лесной и пахотной почв. Лесная почва
отличается высоким содержанием С орг (6.4%) и имеет
слабокислую реакцию среды (рН 5.5). Теоретически в илистой
фракции Pb либо входит в состав минералов, либо сорбируется
как прочный внутрисферный комплекс на поверхности
минералов. Но в иле этой почвы значительная доля Pb
сконцентрирована во внешнесферных комплексах (~40%), этот
свинец извлекается слабой 1М вытяжкой СаСl2. По данным
EXAFS-спектроскопии большую роль в закреплении Pb играет
органическое вещество (Morin et al., 1999). EXAFS-спектр
илистой фракции аппроксимирован смесью 80% Pb-гумата и
20% Pb, адсорбированного на гетите (рис. 16, А).
Доминирование
Pb-гуматов
согласуется
с
большим
количеством (71 и 77%) Pb, экстрагируемого из илистой
фракции вытяжками NaOCl и Na4P2O7. Очевидно, что
органическое вещество, закрывая поверхности частиц
минералов, предотвращает сорбцию ими Pb в лесной
гумусированной почве (Morin et al., 1999).
Рис. 16. EXAFS-спектры атомов Pb в илистой фракции (<2 мкм) почвы из
Эвин-Мальмизон, северная Франция. А – лесная почва: а –
экспериментальный спектр (сплошная линия) и модель (точки); b – 80 %
Pb-гумата; с – 20 % Pb, адсорбированного на гетите. В – пахотная почва: а –
экспериментальный спектр (сплошная линия) и модель (точки); b – 45 %
Pb, адсорбированного на гетите, с – 35 % Pb-гумата; d – 20 % Pb,
адсорбированного на бернессите. Из (Morin et al., 1999).
Пахотная почва отличается низким содержанием С орг
(1.5%) и нейтральной реакцией среды (рН 7.5). Здесь ситуация
иная. Об отсутствии Pb во внешнесферных комплексах
говорит неэффективность 1 М вытяжки СаСl2. Наилучшая
аппроксимация EXAFS-спектра достигнута для смеси,
включающей 45% Pb, адсорбированного на гетите, 35% Pbгумата и 20% Pb-адсорбированного на диоксиде марганца –
бернессите (рис. 16, В). Незначительное участие бернессита
объясняется его крупными размерами (2-50 мкм), вследствие
чего минерал в основном попадает в пылеватую
фракцию.Фосфаты и карбонаты свинца в этой нейтральной
почве не обнаружены. Содержание Pb-гуматов (35%)
согласуется с масштабами экстракции вытяжками NaOCl (35%
Pb). Другая, пирофосфатная вытяжка оказалась мало
селективной, помимо Pb-гуматов она также извлекала свинец,
адсорбированный оксидами марганца, что установлено по
изменению характера EXAFS-спектра илистой фракции (Morin
et al., 1999).
Таким образом, в почвах образуются самые разнообразные
соединения свинца; их состав и количество сильно варьируют
в лесных и пахотных почвах, различающихся по степени
гумусированности.
Отвалы свинцовых рудников в районе Лидвиль,
Колорадо, США. В этом районе рудная и металлургическая
промышленности более 130 лет загрязняли ландшафт
отходами, включающими пустую породу и шлаки.
Образовавшаяся геохимическая аномалия в 1983 г. внесена в
Национальный приоритетный список США как опасная.
Методом EXAFS-спектроскопии детально изучали состав
породы в двух отвалах: Хеммс и Апачи (Morin et al., 1999).
Валовое содержание Pb в отвалах варьирует от 6000 до 10000
мг/кг (Ostergren et al., 1999), что явно опасно для здоровья
человека и окружающей среды. Но биологическая доступность
и химическая подвижность Pb на территории геохимической
аномалии различны и слабо коррелирует с валовым
содержанием свинца.
В тонкой фракции породы (< 2мкм) отвала Хеммс
доминирует свинец, адсорбированный на частицах гетита
(53%), а, кроме того, 27% Pb сосредоточено в пироморфите
Pb5(РO4)3 и 8% Pb – в гидроцеруссите Pb3(СО3)2 (рис. 17, А).
Наличие фосфатов и карбонатов подтверждено данными
рентгенфлуоресцентного анализа. Благодаря нейтральной
среде отвала Хеммс (рН 6.8), свинец адсорбируется на
частицах гетита, не образуя с ними кристаллических фаз.
EXAFS-спектроскопия доказала адсорбцию Pb на поверхности
минералов и зависимость устойчивости сорбированного
свинца от рН.
Рис. 17. EXAFS-спектры атомов Pb в илистой фракции (<2 мкм) отвалов в
районе Лидвиль, Колорадо, США. А – отвал Хаммс: а –
экспериментальный спектр (сплошная линия) и модель (точки); b – 53 %
Pb, адсорбированного на гетите; с – 27 % пироморфита; d – 8 %
гидроцеруссита. В – отвал Апачи: е – экспериментальный спектр
(сплошная линия) и модель (точки); f – 85 % плюмбоярозита g – 10 %
Иная ситуация в кислом отвале Апачи, где в тонкой
фракции породы (< 2мкм) основная часть Pb (85%)
сосредоточена в составе плюмбоярозита Pb[Fe3(SO4)2(OH)6]2.
Кроме него присутствует небольшое количество (10%)
плюмбоферрита PbFe4O7 (рис. 17,В). Таким образом, в кислой
среде, обогащенной сульфидами, свинец образует с Fe
кристаллические фазы. Доля свинца, адсорбированного
гидроксидами железа и оксидами марганца, превышает ½
валового в низко гумусированных объектах (Morin et al., 1999).
Городские почвы, загрязненные свинцом. В загрязненных
высокогумусированных почвах низкая подвижность свинца
объясняется образованием стабильных Pb2+-органических
комплексов, но тип этих комплексов не был ясен до
использования EXAFS-спектроскопии. Мансо с соавт.
(Manceau et al., 1996) изучали структурную химию свинца в
почвах городских газонов,
загрязненных частицами
тетраалкила, используемого как добавка к бензину. Так как у
алкиловых частиц период полураспада всего несколько часов,
то Pb быстро связывается почвенными частицами,
преимущественно органическими молекулами.
Подтверждение деградации исходных частиц тетраалкила
Pb было получено путем сравнения EXAFS-спектров почв
газонов с эталонными спектрами органических соединений
(рис. 18). Но самое главное, что почвенный спектр резко
отличен от спектров Pb с карбоксилатами. В тоже время, для
смеси 60% Pb-салицилата и 40% Pb-пирокатехина получено
хорошее согласие с почвенным спектром. Несмотря на
высокую долю карбоксильных групп в органическом веществе
почв, Pb предпочтительнее хелатируется функциональными
группами ароматических колец, формируя бидентантные
комплексы (Manceau et al., 1996, Manceau et al., 2002). Свинец
стабилен в органических почвах, где его средний срок
сохранения исчисляется от сотен до тысяч лет (Heinrichs,
Mayer, 1977).
Мелиорация почв, загрязненных свинцом. Очень
перспективна химическая мелиорация с применением
ортофосфатов. Ион ортофосфата образует труднорастворимые
фазы с такими токсичными металлами как Cd, Zn и Pb. Многие
исследователи
используют
фосфаты
для
снижения
подвижности
и
биодоступности
тяжелых
металлов,
находящихся в различных промышленных отходах, или
попавших в почву. Обсудим образование и биодоступность
Pb-фосфатов.
Плюмбофосфаты – это наименее растворимые фазы Pb(II),
образующиеся в зоне гипергенеза (табл. 8). В стандартных
условиях Pb-фосфаты, по крайней мере, в 44 раза менее
растворимы, чем галенит (PbS), англезит (PbSO4), церуссит
(PbCO3), глет (PbO) и крокоит (PbСrO4).
Рис. 18. Частицы свинца, образовавшиеся в почве газонов под влиянием
воздействия Pb–алкилов в составе бензина. EXAFS-спектр почвенного
образца (сплошная линия) сравнивается с эталонными спектрами атомов
Pb. Сравнение показывает, что в почве доминируют комплексные хелаты
Pb2+ с салицилатом и пирокатехином. Из (Manceau et al., 2002).
Доказано, что Pb-фосфаты способны контролировать
растворимость Pb в некарбонатных почвах (Santillan-Medrano,
Jurinak, 1975). В карбонатных почвах формируются более
растворимые карбонаты свинца.
Таблица 8. Константы растворимости некоторых минералов свинца (Traina,
Laperche, 1999).
lоg Kp
Минерал
Химическая формула
Глет
PbO
12.9
Англезит
PbSO4
-7.7
Церуссит
PbCO3
-12.8
Пироморфит
Pb5(PO4)3Cl
-84.4
Гидроксилпироморфит Pb5(PO4)3ОН
-76.8
Флюоропироморфит
Pb5(PO4)3F
-71.6
Бромопироморфит
Pb5(PO4)3Br
-78.1
Коркит
PbFe3(PO4)3(SO4)(OH)6
-112.6
Гинсдалит
PbAl3(PO4)3(SO4)(OH)6
-99.1
Плюмогуммит
PbAl3(PO4)3(OH)5.H2O
-99.3
Плюмбофосфаты образуются в почвах на свинцовых
месторождениях, вблизи автодорог и в городских условиях
(Cotter-Hewellls, 1996). Исходя из этого, для снижения
растворимости усилия были направлены на образование Pbфосфатов через внесение в почву фосфора. Обработка
загрязненных свинцом почв путем внесения таких
высокорастворимых форм ортофосфатов как Na2HPO4 или
К2HPO4 снижает биодоступность Pb за счет образования
фосфатов. К сожалению, обработка высокорастворимым
фосфором увеличивает риск его выноса из почв, что приводит
к зарастанию водорослями окружающих водоемов. Чтобы
этого избежать, используют менее растворимый фосфат –
апатит. Гексагональные кристаллы группы апатитов
Са5(РО4)3Х содержат три основных конечных члена: в
гидроксилапатите Х = ОН, во флюорите Х = F, в хлорапатите
X = Cl. Константы растворимости наиболее распространенных
апатитов равны у гидроксилапатита 10-3.1 и у флюороапатита
10-25.
Апатиты широко используются для удаления ионов
токсичных металлов из сточных вод. Механизм удаления
металла зависит от типов сорбата и сорбента и состава
раствора. Реакцию растворенного Pb2+ с гидроксилапатитом с
образованием гидроксилпироморфита записывают так (Ma et
al., 1993)
Сa5(PO4)OH + 5 Pb2+ → Pb5(PO4)3OH + 5Ca2+.
Эта экзотермическая реакция, так как приращение величины
потенциала Гиббса ΔG0 = -137.1 кДж/моль (Lower et al., 1998).
Группа пироморфитов включает минерал пироморфит
Pb5(PO4)3Сl,
флюоропироморфит
Pb5(PO4)3F,
бромопироморфит Pb5(PO4)3Br и др. Их растворимость
уменьшается в таком порядке: флюоропироморфит >
гидроксилпироморфит > бромопироморфит > пироморфит.
Пироморфиты изоструктурны с апатитами. Пироморфиты
обнаружены в отходах добычи руды, почвах газонов, почвах
вблизи заводов по выработке фосфорной кислоты, а также в
почвах, образовавшихся на природных геохимических
аномалиях. Пироморфит надежно идентифицируют с
помощью
электронной
микроскопии,
методами
микродифракции электронов и EXAFS-спектроскопии.
Превращение Pb в почвах из химически доступных форм в
менее доступные фосфаты приводит к снижению его
биодоступности. В одном из опытов добавляли природный
апатит из штата Северная Каролина (США) к почве,
загрязненной Pb-содержащим порошком (Traina, Laperche,
1999). Эти смеси использовали в вегетационных опытах с
последовательным выращиванием кукурузы и ячменя. При
внесении апатита содержание экстрагируемого Pb в почве и в
растениях уменьшилось.
В другом опыте сильнозагрязненные почвы (37 г Pb / кг
почвы) обработали разными дозами гидроксилапатита или
природного флюорапатита, из расчета получения 33, 66 и 100 %
пироморфита (Laperche et al., 1997) и выращивали на них разные
сорта трав. Во всех случаях внесение апатитов снижает
концентрацию Pb в наземных частях травы до 87-98 % от уровня
немелиорированной почвы.
Влияние добавок апатита на содержание Pb в корнях было
более сложным. В целом, увеличение дозы апатита
сказывается на увеличении содержания Pb, усвоенного
корнями. С помощью электронной микроскопии установлено
присутствие Са-замещенного пироморфита на поверхности
корней растений, выращенных на загрязненной почве,
мелиорированной апатитом. Но таких частиц не было
выявлено в контрольном варианте. Оказалось, что внесение
апатита привело к осаждению пироморфита неравномерно во
всем объеме почвы, а только в ризосфере растений. Локальная
кислотность внутри ризосферы спровоцировала локальное
растворение зерен апатита, вызывая осаждение пироморфита.
Тот же эффект осаждения Са-замещенного пироморфита на
корнях растений отмечали в загрязненных свинцом почвах.
Пироморфит осаждается в результате выделения корнями
фосфатазы, которая увеличивает местную концентрацию
фосфора в ризосфере. Во всех этих опытах образование
пироморфита снизило биологическую доступность свинца
(Traina, Laperche, 1999). Закрепление Pb в виде фосфатов
наиболее надежный путь стабилизации свинца в загрязненных
почвах и в местах складирования промышленных отходов.
Соединения цинка
Цинковые удобрения, осадки сточных вод и воздушная
пыль промышленного происхождения – это основные
источники поступления Zn в почву (Robson, 1993). Как
важный компонент клеток цинк участвует в биохимических
процессах и может стать токсичным при избыточном
содержании. В свое время много почв было загрязнено Zn в
результате деятельности плавильных заводов с устаревшей
пирометаллургической технологией, когда выбрасывалась
масса пыли и дыма, обогащенных Zn и Pb. Рассмотрим
подробнее загрязненные цинком почвы во Франции и США.
Загрязненные почвы в районе Морте де Норд, север
Франции. В этих нейтральных почвах (рН > 5.5) легкого
гранулометрического состава цинк накапливался более 100 лет
в
результате
аэрального
загрязнения
несколькими
металлургическими заводами. Образцы загрязненной почвы
фракционировали
по
гранулометрическому
и
денсиметрическому принципам (Manceau et al., 2000).
На поверхности почва настолько загрязнена, что образовался
слой шлака. EXAFS-спектр атомов Zn в тяжелой фракции (2.9
г/см3) верхнего шлакового горизонта и спектры модельных Znминералов: франклинита ZnFe2O4 и виллемита Zn2SiO4
представлены на рис. 19, А. Эффективность расщепления
исходного спектра оценивается по двум основным критериям.
Суммарное содержание металла в модели должно приближаться
к 100 %, а «остаток», обусловленный ошибкой модели, должен
стремиться к 0. Как видно из рис. 19, А, почвенный спектр
занимает промежуточное положение между спектрами двух
минералов. Амплитуда и частота волны экспериментального
спектра воспроизведены в виде смеси двух минералов,
содержащих 41% виллемита и 28 % франклинита (рис. 19, В).
Согласие модельного и экспериментального спектров высокое.
Но суммарное содержание Zn равное 70% в модели
предполагает присутствие третьей неучтенной фазы Zn.
Улучшение модели было достигнуто путем добавления
спектра глинистой фракции из более глубокого горизонта. На
рис. 19, С показан суммарный спектр, содержащий 40 %
виллемита, 25 % франклинита и 18 % глинистой фракции.
После добавления третьего компонента остаток снизился со
108 до 78, а сумма компонентов Zn увеличилась 83 %. Таким
образом, выявляется присутствие Zn, ассоциированного с
глинистой фракцией.
Затем методом микро-EXAFS-спектроскопии изучали
частицы Zn в нижних (незагрязненных) и в пахотных
горизонтах почв. Анализировали состав Zn-фаз в глинистой
фракции. EXAFS-спектры образцов глинистой фракции
песчаных горизонтов с глубины 8-70 см очень близки, что
говорит о схожести Zn-фаз.
Рис. 19. EXAFSспектры атомов Zn в
тяжелой
фракции
(>2.9 г/см3) почвы из
Морте ле Норд,
Франция (сплошная
–
линия).
А
аппроксимация
спектрами
франклинита
и
–
виллемита;
В
аппроксимация
оптимальной
двухкомпонентной
моделью:
28 %
франклинита и 41 %
виллемита (точки);
С – аппроксимация
трехкомпонентной
модеью:
24 %
франклинита + 40 %
виллемита + 18 %
глинистой фракции
из
нижнего
горизонта (точки).
Органоцинковые
соединения
в
гумусированных
горизонтах отсутствуют, что доказано сходством спектров
исходных почв и обработанных Н2О2. Вначале спектры
глинистой фракции аппроксимировали однокомпонентной
моделью (рис. 20, А-С). Наилучшее совпадение получено для
Zn-содержащего керолита Si4(Mg2.25Zn0.75)O10 . (OH)2 . nH2O
(остаток 126, сумма 91%) или для Zn, адсорбированного на
гекторите (остаток 208, сумма 98%).
Спектры этих цинксодержащих филлосиликатов хорошо
согласуются с фазой волны экспериментального спектра, но
различаются амплитудой (Manceau et al., 2000).
Затем сопоставили спектр почв со спектрами других Zn-фаз
(рис. 20, D-F). Сравнивали спектры Zn-замещенного гетита
(остаток 292, сумма 87%), цинка, адсорбированного на
бернессите (остаток 356, сумма 68%), Zn-гидроталькита
Zn3Al(OH)8(CO3)0.5 (остаток 260, сумма 63%) и гидроцинкита
Zn5(OH)6(CO3)2 (остаток 342, сумма 83%). Как видно, все они
плохо соответствуют почвенному спектру.
Кроме значительной разницы в частоте экспериментальной
волны, у эталонных спектров выше амплитуда, в результате
чего в однокомпонентной модели суммарное содержание
каждого компонента низкое. Иногда оно опускается до
нереального уровня, как, например, для бернессита (68%) или
для
(Zn,Al)-гидроталькита
(63%).
Однокомпонентное
моделирование показывает, что гетит, бернессит, Znгидроталькит и гидроцинкит как цинксодержащие минералы,
если и присутствуют в почве, то не доминируют.
Минеральную природу Fe- и Mn-носителей цинка изучали
также методом микро-EXAFS-спектроскопии. Было записано
три спектра железа: два в области ожелезненных зерен, а
третий – в Fe-содержащей глинистой матрице. Все спектры
выглядят схоже и соответствуют спектру фероксигита
δFeООН. Спектр марганца в составе Mn-сферул отвечает
гексагональному бернесситу (Manceau et al., 2002; Morin et al.,
1999).
Таким образом, франклинит, виллемит, гемиморфит и Znсодержащий магнетит, обнаруженные в тяжелой фракции
загрязненного
главными
поверхностного
горизонта
почвы,
являются
Рис. 20. EXAFS-спектры атомов Zn в илистой фракции (<2мкм) почвы из
Морте ле Норд, Франция (сплошная линия) и эталонных образцов (точки):
А – Zn-содержащего керолита; В – Zn-содержащего гекторита; С –
гидроталькита; D – гидроцинкита; Е – Zn-содержащего гетита; F – Zn,
адсорбированного бернесситом. Из (Manceau et al., 2000).
техногенными
Zn-содержащими
поллютантами
металлургических заводов. Вторичные соединения Zn
сосредоточены в илистой фракции почв. Среди них
доминируют Zn-содержащие филлосиликаты, и в меньшем
количестве присутствует цинк, закрепленный оксидами
марганца
(бернесситом)
и
гидроксидами
железа
(фероксигитом). Незначительное участие бернессита в составе
илистой фракции обусловлено его крупными размерами, в
результате чего значительная доля частиц бернессита при
гранулометрическом фракционировании попадает в пылеватые
фракции почвы.
Загрязненные почвы в Пальмертоне, шт. Пенсильвания,
США. Почвенный разрез заложен в 1 км на юго-восток от
бывшего завода, переплавлявшего 80 лет сфалерит ZnS. Почва
аэрально загрязнена тяжелыми металлами, в особенности
цинком, а также серной кислотой. Верхний слой почвы (0-10
см) представляет собой черные, сухие органические куски (в
среднем 32% С орг). Куски, обогащенные тяжелыми металлами
(Zn и Pb), отличаются большей прочностью, чем обедненные
ими. Слой 10-30 см имеет суглинистый состав и содержит ~15%
илистых частиц (Scheinost et al., 2002).
Для анализа форм цинка использовали последовательную
химическую экстракцию и синхротронную рентгеновскую
технику. Экстракцию проводили по Цейну и Брюммеру, этапы
экстракции даны в табл. 9 (Scheinost et al., 2002).
Синхротронный анализ выполняли в Национальной
Лаборатории NSLS в Артоне, шт. Нью-Йорк.
Таблица 9. Последовательная химическая экстракция тяжелых металлов по
Цейну–Брюммеру (Scheinost et al., 2002).
Этап
Экстрагент
Условия
Экстрагируемые формы
обработки
1 1М NH4NO3
24 ч, 20ºC Обменные
ионы,
водорастворимые соли
2 1М NH4OАс (рН 6)
24 ч, 20ºC Металлы
слабоскомплексированные
и
связанные с карбонатами
3 0.1М NH3OHCl +1 M 0.5 ч, 20ºC Металлы, связанные с оксидами
Mn
NH4OАс (рН 6)
4 0.25 M NH4-ЭДТА 1.5 ч, 20ºC Металлы,
связанные
с
(рН 6)
органическим веществом
5 0.2 М NH4-оксалат 4 ч, 20ºC
Металлы,
связанные
со
(рН 3.2)
слабоокристаллизованными
оксидами Fe
6 0.1 М аскорбиновая 0.5 ч, 97ºC Металлы,
связанные
с
кислота + 0.2 М
сильноокристаллизованными
7
NH4-оксалат (рН 3.2)
Остаток
оксидами Fe
Металлы,
связанные
остаточной фракцией
Такое сочетание методов анализа было обусловлено
следующими соображениями. В ходе химического удаления
форм металла (Zn) число фаз снижается, что облегчает
спектроскопическую идентификацию остающихся фаз.
Последовательная
химическая
экстракция
наиболее
эффективна для выявления самых подвижных форм, которые и
наиболее токсичны.
Верхний слой почвы. В этом слое с кислой реакцией среды
(рН 3.2) содержатся 6200 мг/кг Zn и 7000 мг/кг Pb. Результаты
последовательного химического экстрагирования показали,
что в основном цинк экстрагируется на последнем (6-м) этапе
обработки, а также остается в остатке (рис. 21). Из этого
следует, что Zn наиболее прочно связан с (гидр)оксидами
железа и другими стабильными минералами. Обменный Zn
составляет 7%, а остальные 7% высвобождаются на
промежуточных этапах, со второго по пятый. Значительное
количество Mn высвобождается на всех этапах, что не
согласуется с представлениями об однородной минералогии
марганца в почвах. Удаление 23% Fe на четвертом этапе
Рис. 21. Процент Zn и Fe, удаленных при последовательном
экстрагировании. Этапы химической обработки даны в табл. 9. Из (Scheinost
t l 2002)
с
экстракции, возможно, обязано разрушению комплексов
железа с органическим веществом. Но, главным образом,
железо высвобождается на 6-м этапе, т.е. за счет растворения
окристаллизованных оксидов железа, хотя большое его
количество остается в остатке после полного окончания
экстрагирования. Контроль растворимости цинка методами
синхротронной техники показал следующее (табл. 10).
После 4-х первых экстракций спектры почв изменялись
слабо, что согласуется с низким выходом Zn в раствор на этих
этапах. Данные XANES- и EXAFS-спектроскопии указывают,
что основной Zn-содержащий минерал – это франклинит (Zn, Fe,
Mn)II(Fe, Mn)III2O4).
Таблица 10. Исходные и новообразованные формы цинка в разных слоях
почвы по данным синхротронной техники до и после 6-и этапов
последовательного химического фракционирования (% от выявленного
цинка). По данным (Scheinost et al., 2002).
ZnZn-литиоЭтап
Франклинит Сфалерит Оксалат Zn2+
Zn
водный монтмофорит
обработки
риллонит
Слой почвы 0-10 см
0
59
28
14
1
69
34
2
68
34
3
71
33
4
72
32
5
35
21
47
6
23
50
25
Слой почвы 10-30 см
0
56
44
1
77
31
6
104
Примечание. Образование оксалата цинка представляет собой артефакт в
ходе пятого этапа экстракции. Несовпадение суммы со 100 % связано с
недостаточной точностью аппроксимации спектров модельными фазами.
Так как пятая экстракция извлекла мало цинка, то можно
было ожидать сохранения вида спектра. Но фактически он
изменился сильно, что указывает на дифференциацию форм Zn
после обработки оксалатом аммония. Обратный эффект
отмечен после шестой обработки, когда удаляется более 1/3
цинка, но спектральные изменения Zn в почве оказались не
значительными. Спектры образцов после 5-й и 6-й обработок
показывают новые сигналы, относящиеся к сфалериту, тогда
как сигналы франклинита снижаются. Четко изменяется
отношение сфалерит/франклинит в остатках почвы после 5-й и
6-й экстракций: за счет частичного растворения франклинита
доля сфалерита возрастает. На исходном спектре присутствие
сфалерита маскируется сильным сигналом франклинита.
Следует отметить как артефакт эффект образования новой
фазы в ходе 5-й обработки. Образуется новая октаэдрически
координированная форма при участии Zn, высвобождаемого при
частичном растворении франклинита, – оксалат цинка. Хотя
идентификация форм с учетом констант растворимости
предполагает, что экстрагент слегка не насыщен по отношению к
оксалату цинка, присутствие Pb2+ и других ионов металлов
может изменить ситуацию и вызвать синтез смешанных
металлоксалатных фаз менее растворимых, чем оксалат цинка.
Инструментальный контроль выявил следующие процессы,
протекающие в ходе химической экстракции. Как ожидалось,
первая обработка удаляет ионы цинка (гидратированные или
сорбированные в виде внешнесферных комплексов).
Франклинит и сфалерит устойчивы к первым четырем
обработкам, но пятая растворяет примерно половину обоих
минералов, а именно те частицы, которые слабо
окристаллизованы. При этом растворенный Zn немедленно
осаждается в виде оксалата цинка, в результате на пятом этапе
экстрагируется всего 5% цинка. Наконец, шестая вытяжка
удаляет 38% общего цинка, растворив часть оксалата и больше
франклинита, чем сфалерита. В значительной степени оксалат
цинка удаляется в ходе шестой обработки, когда к оксалату
аммония добавляют аскорбиновую кислоту и используют
более высокую температуру (Scheihost et al., 2002). После
последнего этапа обработки в качестве остаточной фазы
доминирует сфалерит (50%).
В нижнем слое почвы значение рН увеличилось до 3.9,
содержание Zn сократилось до 900 мг/кг, а Pb – до 62 мг/кг.
Результаты последовательного химического экстрагирования
показали, что 56% общего Zn находится в обменной форме
(рис. 21). Другие формы Zn частично экстрагируются в ходе
шестой (аскорбино-оксалатной) вытяжки, а частично
сохраняются после нее.
Спектроскопическое содержание гидратированного Zn2+
прекрасно согласуется с экстракцией цинка первой, азотноаммонийной вытяжкой. После этой обработки спектры
гидратированного Zn2+ больше не проявляются, а выявляется
форма Zn, связанная в виде внутрисферного комплекса,
преимущественно оксидом марганца - литиофоритом. Такой
комплекс образуется при закреплении цинка в кислой среде,
характерной для данной почвы. Известно, что литиофорит
полностью растворяется в ходе третьей, гидроксиламинной
обработки.
Обратимся к формам цинка после последней, шестой
Химическое
фракционирование
не
обработки.
дифференцирует формы остатка после всех экстракций.
Поэтому
из
химического
анализа
можно
данных
предположить, что состав остатка одинаковый как для
верхнего, так и для нижнего горизонтов почвы. Но это не так и
спектры остатков почв, отобранных с разной глубины, сильно
различаются (Scheinost et al., 2002). Как отмечалось, в остатке
верхнего слоя почвы сохраняется франклинит, сфалерит и
новообразованный оксалат цинка. В горизонте 10-30 см
ситуация иная. Цинк входит в состав слоя гидроокиси Al,
расположенного между отрицательно заряженными слоями
монтмориллонита.
Таким образом, Zn способен закрепляться в слабо кислых и
нейтральных почвах за счет образования Zn-содержащих
филлосиликатов
в
составе
Al-гидроокисного
слоя
неупорядоченных филлосиликатов (Manceau et al., 2000; Dahn
et al., 2002). В сильно кислых почвах образование таких форм
затруднено. При рН <5 цинк остается полностью в обменной
форме в течение длительного периода времени. Поэтому
можно допустить, что Zn, который переходит в подпочву за
счет растворения техногенных минералов (франклинита и
сфалерита),
остается
преимущественно
в
виде
внешнесферного комплекса. Но очень неожиданным оказалось
то, что почти половина Zn закрепляется в составе весьма
малорастворимой фазы – в межслоевой гидроокиси алюминия
и сохраняется в ходе обработки 0.2 М СаСl2, что снижает
токсическое влияние подвижного Zn2+. Ранее в лаборатории
было доказано, что таким путем закрепляется никель и,
вероятно, другие 3d металлы (Scheinost et al., 2002).
Соединения ртути
Основные источники загрязнения окружающей среды
ртутью – это отходы золоторудных и ртутных рудников и
промышленных предприятий. Для прогноза растворимости,
условий транспорта и потенциальной биодоступности
необходимо знать формы ртути. Методы изучения форм ртути
со временем становятся все более сложными. Начинали с
визуального анализа ртутьсодержащих фаз, затем применяли
методы
последовательной
химической
экстракции,
последовательной температурной десорбции, электронного
микроанализа и, наконец, использовали неразрушающую
технику
EXAFS-спектроскопии.
Как
самый
легкий,
эффективный и воспроизводимый прием наиболее часто
применяют последовательную химическую экстракцию.
Рассмотрим последовательную химическую экстракцию
ртутьсодержащих частиц, выполненную по Блюму (Bloom et al.,
2003). Схема включает пять этапов и основана на постепенном
увеличении силы экстрагентов, растворяющих все более
устойчивые соединения ртути (табл. 11). Масса образца
составляет 0.4 г, отношение раствор: образец = 100 : 1,
суспензия взбалтывается в течение 18 ч при комнатной
температуре. Затем суспензию центрифугируют, а супернатант
фильтруют через фильтр 0.2 мкм с последующим определением
ртути в растворе.
Химическое экстрагирование дает информацию о характере
распределения форм ртути в почвах и имеет достаточно
высокую чувствительность 0.5 мг/кг, что позволяет выявить
степень загрязнения почв.
Таблица 11. Схема последовательной экстракции частиц ртути по (Bloom et
al., 2003).
Этап
Экстрагент
Характеристика
Типичные растворимые
соединений ртути
соединения
1 DI-вода
Водорастворимые
HgCl2
2 HCl/HOAc, pH 2 Кислоторастворимые HgO, HgSO4
3 1 н. KOH
Органокомплексы
Гуматы Hg, Hg2Сl2, CH3Hg
4 12 н. HNO3
Прочносвязанные
В
решетке
минераловносителей, Hg2Сl2, Hg0
5
Царская водка
Сульфиды
HgS, HgSe
Но как в случае любой простой техники метод
последовательной химической экстракции имеет свои
ограничения. Среди них:1) возможная трансформация частиц
ртути в ходе экстракции, 2) неспецифическое удаление Hg-фаз в
ходе последовательных этапов экстракции (Barnet et al., 1997).
Как и для других элементов, химическая экстракция,
оперативно разделяющая частицы ртути, не способна точно их
идентифицировать.
Из-за этих ограничений метод последовательной
экстракции полезно объединить с другими видами анализа, в
особенности с методом EXAFS-спектроскопии. EXAFS-анализ
показал свою эффективность при общей концентрации ртути
>100 мг/кг; точность анализа порядка 10% и поэтому Hg-фазы
в количестве менее 10% не идентифицируются (Kim et аl.,
2000).
Такое совмещение двух техник применили к образцам с
содержанием Hg от 132 до 7539 мг/кг. Анализировали
следующие образцы: летучую золу медеплавильного
производства, смесь нескольких стандартных Hg-фаз с
каолинитом, 3 вида отходов золотых рудников и морские
отложения (Kim et al., 2003). EXAFS-спектры исходных
образцов расщепляли на сумму спектров Hg-фаз, взвешенных
согласно их атомной доли от всей ртути в образце (табл. 12).
В целом доля нерастворимых сульфидов HgS (киноварь и
матациннабарит) и HgSe, определенная с помощью EXAFSспектроскопии, коррелирует с суммой соединений HgS +
HgSe, экстрагированной царской водкой.
Таблица 12. Состав моделей ртутьсодержащих фаз (Kim et al., 2003).
Состав модели
Сумма, %
Остаток
43 % тимоннита HgSe
93
0.076
33% метациннаборита HgS(cub)
17 % киновари HgS(hex)
48 % хлорида ртути HgCl2
107
0.075
32 % оксида ртути HgO
27 % киновари HgS(hex)
60 % метациннаборита HgS(cub)
31 % киновари HgS(hex)
49 % киновари HgS(hex)
30 % метациннаборита HgS(cub)
14% шутеита Hg3O2SO4
64 % метациннаборита HgS(cub)
28 % оксида ртити HgO
62 %метациннаборита HgS(cub)
38 % киновари HgS(hex)
91
0.063
93
0.060
92
0.590
100
0.196
Но для легкорастворимых Hg-фаз (HgCl2, HgO, Hg3S2O4)
такого согласия методик нет. Отклонение в содержании
химических фракций может быть обусловлено влиянием
матрицы,
размером
частиц
и
степенью
их
окристаллизованности.
Контроль селективности вытяжек
Один из видов контроля селективности экстрагентов –
сравнение состава выявленных при химическом анализе форм
тяжелых металлов с реально существующими в почвах.
Синхротронная техника третьего поколения позволяет это
сделать. Больше всего информации получено о формах свинца
и цинка.
С помощью синхротронной техники идентифицировано
пять вторичных форм соединений свинца: Pb2+-органические
комплексы, Pb2+-сорбированный на оксидах железа, Pb2+сорбированный на оксидах марганца, плюмбоярозит и
фосфаты свинца (Ostergren et al., 1999; Hansel et al., 2001;
Dupont et al., 2002; Manceau et al., 2002).
В отношении цинка установлено следующее. Главными
техногенными Zn-содержащими минералами являются
франклинит, виллемит, гемиморфит и Zn-содержащий
магнетит. Вторичные соединения Zn представлены: Znсодержащими филлосиликатами и в меньшей степени –
цинком, закрепленным оксидом марганца – бернесситом
(Na,Ca)Mn7O14 . 2.8H2O и гидроксидом железа – фероксигитом
δFeOOH.
Схемы Тессиера и BCR. Данные о формах Pb и Zn в почвах
сведем в табл. 13. Видно, что часть реальных форм тяжелых
металлов выявляетcя при последовательной экстракции
методами Тессиера и BCR.
Таблица 13. Разделение форм Рb и Zn, выявленных в почвах с помощью
синхротронной рентгеновской техники (Manceau et al., 2000, 2002; Scheinost
et al., 2002), на интерпретируемые в рамках химического фракционирования
схем Тессиера и ВСR и не интерпретируемые этими схемами.
Формы Pb и Zn
Минералы
Интерпретируемые
Гуматы тяжелых металлов
Pb-гуматы
Оксиды Fe и Mn
Pb-содержащие
Pb на гетите
Pb на бернессите
Плюмбоярозит
Pb[Fe3(SO4)2(OH)6]2
Плюмбоферрит
PbFe4O7
Zn-содержащие
Франклинит
ZnFe2O4
Zn-магнетит
Zn-гетит
Zn-фероксигит
Zn-литиофорит
Карбонаты
Гидроцеруссит
Pb-содержащие
Pb3(CO3)2(OH)2
Zn-содержащие
Zn-гидроталькит (?)
Zn3Al(OH)8(CO3)6
Гидроцинкит (?)
Zn5(OH)6(CO3)2
Неинтерпретируемые
Фосфаты
Пироморфит
Pb5Cl(PO4)3
Силикаты
Виллемит Zn2SiO4
Керолит
Si4(Mg2.25Zn0.75)O10
Zn-гекторит
Zn-монтмориллонит
Гемоморфит
Zn4(Si2O7)(OH)2
Среди них гуматы свинца, карбонаты Pb и Zn, а также
металлы, закрепленные оксидами железа и марганца. Но
другая
часть
реальных
форм
тяжелых
металлов
экстракционными системами не выявляется. Это относится к
найденным в почвах, но отсутствующим в схемах фосфатам Pb
и силикатам Zn. Особенно ощутимо отсутствие в схемах
химического фракционирования фосфатов, которые относятся
к
общим
осадителям,
способным
образовывать
кристаллические или аморфные осадки с большим числом
двух- и более заряженных ионов. Силикаты относятся к
групповым осадителям, осаждающим небольшую подгруппу
ионов.
Свинец. Важно разобраться, как себя ведут при
последовательном химическом фракционировании фосфаты и
силикаты свинца. Известно, что фосфаты свинца – наиболее
устойчивая форма Pb в почвах самого разного генезиса. Согласно
термодинамическим расчетам в почвах с нейтральной реакцией
образуются такие фосфаты как Pb3(РО4)2, Pb5(РО4)3ОН,
Pb4О(РО4)2
(Santillan-Medrano,
Jurinak,
1975).
Термодинамические расчеты, выполненные для Pb в дерновоподзолистой, черноземной и сероземной почвах показали
преобразование оксидов свинца в гидроксид или в
гидроксокарбонат свинца (Горбатов, 1988).
Но данные синхротронной техники показывают отсутствие
в почвах упомянутых соединений и присутствие не
учитываемых соединений Pb-салицилата и Рb-пирокатехина, а
также фосфата свинца – пироморфита Pb5(РО4)3Cl –
высокостабильного минерала с константой растворимости
Краств = 10-84.4. Пироморфит найден в отходах добычи руды, в
почвах газонов, вблизи заводов по выработке фосфорной
кислоты, а также в почвах, образовавшихся на геохимических
аномалиях. Пироморфит надежно идентифицируется методом
микродифракции электронов и EXAFS-спектроскопии на
внешней поверхности корней различных растений (Manceau et
al., 2002). Но о растворимости пироморфита вытяжками по
схемам Тессиера и BCR ничего не известно. Также остается
неясным характер растворимости этими вытяжками силикатов
цинка.
Цинк. Существенное противоречие имеет место и для форм
Zn. Согласно термодинамическим расчетам в почвах
образуется гидрокарбонат цинка Zn5(ОН)6(СО3)2 (Горбатов,
1988). Но синхротронная техника показывают вместо него
новообразованные
минералы:
Zn-содержащий
монтмориллонит или Zn-содержащий силикат – керолит.
Очевидно,
в
расчетах
необходимо
учитывать
термодинамические характеристики реально найденных
соединений тяжелых металлов.
Приведем
термодинамические
характеристики
Zn.
содержащего филлосиликата – керолита Si4(Zn3)O10 (OH)2.
Для реакции растворения керолита
Si4(Zn3)O10 (OH)2 + 6Н+ + 4Н2О → 3Zn2+ + 4Si (OH)4
подсчитана стандартная свободная энергия образования. Она
оказалась равной ΔGo = -4682 ± 30 кДж/моль при 95 % уровне
вероятности (Manceau et al., 2000).
Факт образования в почвах силиката цинка – керолита
требует пересмотра систем последовательных химических
экстракций, так как в них нет места для фосфатов тяжелых
металлов (Ладонин, 2002).
Рассмотрим вопрос о Zn, закрепленном органическим
веществом. Эту форму всегда обнаруживают при
последовательном химическом фракционировании. Так, при
экстрагировании Zn из серых лесных почв Среднерусской
равнины по схеме Тессиера доля Zn-органической формы
колеблется от 4 до 35 % (Переломов, 2001). Считается, что
аккумуляция цинка органическим веществом начинается
только после исчерпания фиксирующих возможностей
(гидр)оксидов железа и марганца. Но странно, что в почве,
содержащей 35 % Zn-органических форм, количество С орг
(1.8%) почти не отличается от той почвы, где доля Znорганической форм всего 7 %. Отметим, что EXAFS-спектры
почв Zn-органических соединений не обнаруживают. Таким
образом, имеются противоречия между результатами
последовательного
химического
фракционирования
и
данными физического анализа, что оставляет вопрос об
образовании Zn-органических соединений в почвах открытым.
Следовательно, методы последовательной химической
экстракции по Тессиеру и BCR устанавливают точное
содержание легкоподвижных форм цинка, но они не способны
идентифицировать и отразить содержание Zn в минеральных
фазах из-за не селективности растворителей. Более того,
обнаруженный артефакт – осаждение оксалата цинка – не
позволяет определить цинк, адсорбированный (гидр)оксидами
железа и марганца. Но в сочетании с синхротронной техникой,
метод последовательной химической экстракции помогает
определять состав и количество форм цинка.
Схема Цейна–Брюммера. Эффективность схемы обсудим
на примере форм цинка (табл. 14). Контроль экстрагированных
форм также велся методами синхротронной рентгеновской
техники (Scheinost et al., 2002). Как видно из табл. 14, хорошо
выявляются гидратированные ионы Zn2+, которые адекватно
растворимы
1М
раствором
NH4NO3.
Солянокислый
гидроксиламин в смеси с уксусной кислотой эффективен при
растворении оксида марганца (литиофорита). Кислый оксалат
аммония растворяет слабоокристаллизованные частицы
франклинита, а с добавкой аскорбиновой кислоты и с нагревом
до 97°С – растворяет остальные, сильноокристаллизованные
частицы франклинита. Но на этом положительные стороны
химического фракционирования исчерпываются.
Выделены
следующие
недостатки
химического
фракционирования (табл. 14). Во-первых, значительное
образование оксалата цинка. Это очень серьезный недостаток
кислого оксалата аммония. Неудивительно, что популярные
методики Тессиера и BCR обработку кислым оксалатом
аммония не включают. Во-вторых, химическая экстракция не
выявляет присутствие Zn-содержащего слабоупорядоченного
монтмориллонита. В-третьих, схема фракционирования не
выявляет присутствие сульфида цинка сфалерита.
Таблица 14. Разделение форм Zn, выявленных с помощью синхротронной
рентгеновской техники (Scheinost et al., 2002), на интерпретируемые в рамках
химического
фракционирования
систем
Цейна–Брюммера
и
интерпретируемые этой системой.
Формы Zn
Минералы
Интерпретируемые
Обменный
Гидратированный Zn2+
Сорбированный оксидами Мn
Zn-литиофорит
В составе слабоокристаллизованных
Франклинит
оксидов Fe
В составе сильноокристаллизованных
Франклинит
оксидов Fe
Неинтерпретируемые
Новообразованный в ходе обработки
Оксалат Zn
оксалатом
Филлосиликаты
Zn-монтмориллонит
Сульфиды Zn
Сфалерит
не
Его частичное растворение в ходе пятого этапа экстракции
(кислым оксалатом аммония) только вносит ошибку,
содержание Zn, связанного со слабоупорядоченными
соединениями, оказывается завышенным. При выявлении
форм тяжелых металлов обработка кислым оксалатом аммония
не эффективна. Этот реактив слабо селективен к формам
тяжелых
металлов
и
может
образовывать
новую
искусственную фазу – оксалат некоторых тяжелых металлов.
Таким образом, факт обнаружения в минеральных почвах
не учитываемых форм тяжелых металлов ставит перед
почвоведами новые задачи. Необходимы модельные
эксперименты по химической обработке фосфатов и силикатов
тяжелых металлов. В зависимости от результатов этих опытов,
следует уточнить интерпретацию действия реагентов в составе
основных систем фракционирования.
Глава 5. ФОРМЫ СЕРЫ И ТЯЖЕЛЫХ МЕТАЛЛОВ
В ОРГАНОГЕННЫХ ПОЧВАХ
Формы и подвижность состояние тяжелых металлов в
органогенных почвах и торфах во многом зависят от степени
окисления серы. Поэтому формам серы в составе органических
веществ уделяется большое внимание.
Степени окисления серы в органическом веществе почв
Долю органической серы в торфяниках определяют обычно
химическими методами. Общую серу определяют, применяя
смесь азотной, хлорной и фтористоводородной кислот в
присутствии катализаторов (Cu и V), что позволяет окислять
все формы S (Haering et al., 1989). Растворимые сульфаты
извлекаются слабым раствором ацетата Mg. Сульфатное
железо окисляется 0.5 М НСl и затем Н2О2. Содержание серы в
составе сульфатов подсчитывают, исходя из количества
окисленного Fe, используя стехиометрическую формулу
пирита FeS2, как наиболее распространенного сульфида.
Органическую серу определяют как разницу между общей
серы и суммой S в составе пирита и сульфатов.
Распределение форм серы в трех разрезах в приморских
маршах на побережье Чисапикского залива Атлантического
океана, шт. Мериленд показано на рис. 22. Органическая сера
доминирует, при этом ее содержание зависит от количества
общей серы в маршах. Но прямая зависимость наблюдалась
только при высоком содержании органического вещества (С орг
>10%) (Haering et al., 1989).
Ранее найдена высокая корреляция между содержанием
органического углерода и общей серы в приморских маршах (r
= 0.73-0.64). Считается, что связь С орг и S общ проявляется
благодаря органической сере. При этом нет существенной
корреляции между содержанием пирита и S общ. Именно с
количеством органической серы связано варьирование
содержания общей серы в таких маршах. Биохимический цикл
серы до сих пор не достаточно ясен в силу трудно
определяемого состояния окисления серы в разных почвах. В
почвах умеренного пояса более 90% общей серы находится в
органической форме (Morra et al, 1997).
Рис. 22. Формы серы в приморских маршах Чисапикского залива, штат
Мериленд, США. Из (Haering et al., 1989).
Но до недавнего времени для анализа форм органической
серы применяли только деструктивные химические методы,
включающие селективное восстановление разных форм серы до
Н2S. Метод XANES-спектроскопии преодолевает трудности,
связанные с неопределенностью химического анализа, и
позволяет непосредственно определять формы S на основе
величины энергии, требуемой для s → р переходов электронов.
Величина этой энергии отражает не только электронную
плотность (состояние окисления S), но и характер связи атомов
серы.
Как известно, гуминовые кислоты (ГК) и фульвокислоты
(ФК) – это главный компонент органического вещества почв.
Состояние серы в стандартных образцах ГК и ФК почв и торфа
Международного общества гумусовых веществ (IHSS)
анализировали с помощью XANES-спектроскопии. Общее
содержание серы в составе ГК равнялось 4.1-7.0 мг/г и 6.2-6.7
мг/г – в составе ФК. Препараты имеют низкую зольность ~1%
(Morra et al, 1997). Как показывают спектры, для гуминовых
кислот преобладающей степенью окисления серы является +5
и реже +6 (рис. 23). Для спектра фульвокислот характерна
валентность S только +5. Следовательно, в гумусовых
веществах сера находится в составе сложных сульфатных
эфиров (+6) и сульфоновых кислот (+5). Ранние исследования
с помощью деструктивных методов показали, что в почвенных
гумусовых кислотах доля сложных сульфатных эфиров
варьирует от 25 до 93% от общей серы (Morra et al, 1997), но
эти данные были уточнены с использованием метода XANESспектроскопии. Подчеркнем, что в отличие от деструктивных
методов, которые определяет сульфонаты (со степенью
окисления серы +5) по разнице масс, XANES-спектроскопия
определяет эту форму прямым методом.
Рис. 23. XANES-спектры серы в составе стандартных гуминовых кислот
(ГК) и фульвокислот (ФК), извлеченных из почвы и торфа. Из (Morra et al.,
1997).
Низковалентная сера также присутствует в ГК и ФК
(табл. 15). Степень ее окисления варьирует от –0.3 до +2. В этот
интервал попадают органические сульфиды и дисульфиды,
цистин, метионин, тиофены и сульфоксиды (Waldo et al., 1991).
Для формального восстановленного состояния серы
характерна степень окисления между –1 и +2.
В работе (Xia et al., 1998) изучали степень окисления серы
в образцах гумусовых кислот, извлеченных из почв и речных
осадков.
Методом
XANES-спектроскопии
установлено
преобладание четырех главных групп: сера сульфидов и тиолов со
степенью окисления (0.2-0.6), сера сульфонидов (1.8), сера
сульфонатов (5) и сера сульфатов (6). При этом соотношение
двух высокоокисленных форм серы (5) и (6) у разных
гумусовых кислот сильно варьирует.
Таблица 15. Значения энергии, полученные путем разложения XANESспектров стандартных гуминовых и фульвокислот почвы и торфа (IHSS), и
степень окисления серы в препаратах (Morra et al, 1997).
Энергия, эВ Степень
Источник Энергия, эВ Формальная
степень
окисления S
ГК и ФК
окисления S
гуминовые кислоты
фульвокислоты
Почва
2472.1
0.0
2471.8
-0.3
2473.6
+0.8
2475.0
+1.7
2475.9
+2.3
2479.8
+5
2480.3
+5
2481.5
+6
Торф
2471.1
-0.3
2471.7
-0.3
2473.5
+0.8
2475.0
+1.7
2480.1
+5
2479.8
+5
2481.5
+6
В то же время доли тиофена (степень окисления 1.0) и
сульфона (степень окисления 4.0) низкие. Химические формулы
для каждой группы серы даны ранее в табл. 1.
По преобладающей валентности все группы серы можно
подразделить на: а) восстановленную серу, включающую
сульфиды, тиолы и тиофенилы, b) серу промежуточной
степени окисления, включающую сульфониды, сульфонилы и
сульфонаты и с) окисленную сульфатную серу. Такое
упрощенное деление позволяет сопоставлять результаты
синхротронного анализа с химическим фракционированием
соединений серы.
По содержанию серы разных степеней окисления можно
сравнивать образцы различных органических веществ. Такие
исследования описаны в работе (Xia et al., 1998). XANESспектры серы в четырех препаратах гумусовых веществ
показаны на рис. 24. Степень окисления серы в составе
гумусовых веществ, извлеченных как из водной среды, так и
из почв, лежит в диапазоне от +2 до +5. Массовая доля серы в
этих степенях окисления колеблется от 26 до 38%.
Рис. 24. Расщепление XANES-спектра серы в составе гумусовых веществ:
гуминовые кислоты (НА), фульвокислоты (FA), гуминовые вещества (HS).
Органические вещества выделены из донных отложений р. Саванна (IHSSFA, IHSS-HA), из торфяника в штате Миннесота (MN-HA), из минеральной
почвы в штате Висконсин (WI-HS). Сплошная линия – экспериментальный
спектр, пунктирная линия – фон, точечные линии – гауссовы пики моделей.
Из (Xia et al., 1998).
Содержание восстановленной серы в форме тиоловсульфидов и тиофенов варьирует больше: от 15% в гумусовых
веществах, извлеченных из почвы (шт. Висконсин), до 55% в
гумусовых кислотах из речных донных отложений. Эти
значения обратно пропорциональны содержанию в них
сульфатных эфиров. Нужно отметить зависимости состояния
окисления серы от природы гуминовых веществ. Гумус,
образующийся в речных отложениях и болотах, содержит
восстановленной серы больше, чем сформировавшийся в
минеральных почвах (рис. 25). Это ожидаемый результат. Но,
кроме того, XANES-спектроскопия доказывает наличие
органической серы с промежуточными степенями окисления
(2; 4 и 5) в гумусовых веществах различных почв.
Рис. 25. Относительная доля (%) органической серы в различных степенях
окисления в составе гуминовых кислот (НА), фульвокислот (FA) и
гумусовых веществ (HS). Органические вещества выделены из донных
отложений р. Саванна (IHSS-HA, IHSS-FA), из торфяника в штате
Миннесота (MN-HA), из минеральной почвы в штате Висконсин (WI-HS). У
восстановленной серы (1) степень окисления от 0.2 до 1.1, у
«промежуточной» (2) – от 1.8 до 5, у окисленной (3) – 6. Из (Xia et al., 1998).
Детально исследовали фульвокислоты, выделенные из
сточных городских вод (Hundel et al., 2000). Чтобы лучше
понять химию S-содержащих лигандов использовали
селективное
растворение
и
XANES-спектроскопию.
Предварительно фульвокислоты фракционировали на два
класса: общие гидрофильные и гидрофобные кислотные
фракции.
Затем
фракции
разделяли
диализом
по
молекулярным
массам.
Обработка
йодоводородной кислотой дает содержание восстановленной
серы, а разница между валовой и восстановленной определяет
количество окисленной серы.
Селективное растворение показало, что в гидрофильной
фракции доминирует окисленная форма серы, а в кислой
гидрофобной – восстановленная; XANES-спектроскопия это
подтверждает (рис. 26). Между относительной долей
восстановленной и окисленной серы, определенной двумя
методами, нет разницы при 95% уровне достоверности.
Соотношение между восстановленной и окисленной формами
серы согласуется с распределением функциональных групп,
доминирующих в гидрофильных и гидрофобных веществах.
XANES-спектроскопия показывает, что у большей части
восстановленной серы степень окисления ≤2, что говорит об
органических полисульфидах как важном компоненте в
структуре фульвокислот.
Рис. 26. Сопоставление степени окисления серы в гидрофильных и
гидрофобных
фракциях
фульвокислот,
определенное
методами
селективного растворения и XANES-спектроскопии. 1 – восстановленная S;
2 – окисленная S. Из (Hundel et al., 1998).
Обнаружение органической восстановленной серы очень
важно в методическом отношении. Именно ее наличие делает
заметным низкую эффективность использования дитионитцитрат-бикарбоната (ДЦБ) при растворении (гидр)оксидов
железа по Мера–Джексону. В результате искажается роль
(гидр)оксидов железа как носителей тяжелых металлов.
При обработке почв с определенным содержанием
органической восстановленной серы низкая эффективность
ДЦБ становится очевидной, когда сравнивают его действие с
другими реактивами, обычно извлекающими меньше Fe, чем
ДЦБ: кислым оксалатом аммония по Тамму или
уксуснокислым дитионитом по Баскомбу. В качестве примера
можно
указать
на
необычно
высокое
отношение Fe окс /Fe дит
>1 в отложениях с
высоким
содержанием
органического вещества
(рис. 27) (Karathanasis et
al.,
1995).
Резкое
возрастание содержания Рис. 27. Влияние органического углерода
отложениях
на
отношение
оксалаторастворимого Fe в
оксалаторастворимого железа Fe(окс) к
окс
отмечается
в дитионитрастворимому Fe(дит). Из
присутствии в почве (Karathanasis et al., 1995).
восстановленного Fe(II),
например, сидерита или магнетита (Водяницкий, 2003).
Вероятно, эффективность реактива Тамма возрастает и за
счет другого восстановителя – серы со степень окисления <2.
Дитионит Na2S2O4 нельзя рассматривать как источник
предельно восстановленной серы, в нем степень окисления
атомов серы составляет +2 и +4. Если органическая сера в
почве находится в более восстановленном состоянии (<2), то
эффективность дитионита как восстановителя снижается.
Другими словами, в присутствии более сильного восстановителя,
чем дитионит, роль последнего уходит на второй план, а
возрастет роль кислотности экстрагента. Так как у реактива
Тамма рН 3.4, то из почв с высоким содержанием
восстановленных элементов он извлекает больше железа, чем
ДЦБ с его нейтральной реакцией. Возможно, наличие
восстановленной серы увеличивает выход тяжелых металлов в
раствор экстрагента с низким рН. Следовательно, методики,
разработанные для минеральных почв, не годятся для
фракционирования тяжелых металлов в органогенных почвах.
Но, кроме того, стандартная методика не пригодна и для
минеральных почв, если они загрязнены техногенной серой.
Так, в загрязненных пойменных почвах Ивановской области
установлен другой дисбаланс железа, хотя и менее грубый
(Трухина, 1988). Оказалось, что в ряде оглеенных горизонтов
аллювиальных почв содержание слабоокристаллизованных
соединений железа по Баскомбу (дитионит Nа в буферном
растворе уксуснокислого натрия) выше, чем их суммарное
содержание по Мера–Джексону. Указанные образцы почв
отличаются повышенным общим содержанием SO3 = 0.4-1.4%.
Из этого следует, что для минеральных почв с высоким
содержанием органической восстановленной серы степень
извлечения
металлов
определяется
кислотностью
дитионитсодержащих
реактивов.
Необычно
высокая
эффективность реактивов Тамма и Баскомба на образцах почв,
содержащих восстановленную серу, связана с кислой реакцией
реактивов с рН 3.4 и 3.8 по сравнению со слабым действием
реактива Мера–Джексона с нейтральной реакцией. Это
обстоятельство следует иметь в виду при использовании ДЦБ,
практикуемого в России для определения тяжелых металлов,
ассоциированных с (гидр)оксидами железа.
характеризует
Конечно,
XANES-спектроскопия
органические соединения серы более детально, чем
химическое фракционирование. Она позволяет тонко
дифференцировать группы в рамках широких категорий
восстановленных и окисленных групп серы.
Для органогенных почв разработаны собственные
методики химического фракционирования, отличные от
методик, предназначенных для минеральных почв. Эти
методики
дифференцируют
формы
металлов,
ассоциированных с серой, находящейся в разной степени
окисления (Xia, 1998).
Ассоциации тяжелых металлов с серой в почвах
В торфе наиболее хорошо изучены формы нахождения
металлов-халькофилов:
Zn и Cd. Их подвижность
увеличивается после введения дренажа и освоения торфяных
почв, одновременно с окислением органического вещества и
сульфидов в аэробных условиях. Состояние подвижных Zn и Cd
в торфах во многом зависят от изменения окислительного
состояния серы.
Методом XANES-спектроскопии изучали торфяники,
образовавшихся на богатых металлами доломитах, в штате
Нью-Йорк, США и провинции Онтарио, Канада (Martinez, et
al., 2002). Установлено, что 35-45% серы находятся в наиболее
восстановленном состоянии (0.02-0.9) в составе сульфидов (R–
S–R) и тиолов (R–S–H). Доля серы в промежуточных степенях
окисления (1.2-1.9 и 5) еще значительнее: от 50 до 70%; в
основном это сульфоксиды (R–SО–R) и сульфонаты (R–SО3–
Н). Таким образом, в торфянике сера доминирует в
восстановленной и промежуточной формах.
Молярное отношение валового Zn к восстановленной S (со
степенью окисления 0.02-0.3) колеблется от 0.02 до 2.82, а
отношение Cd к восстановленной S составляет 0.0001-0.008.
Это допускает теоретическую возможность связывания всего
цинка и кадмия с восстановленной формой серы в составе
сульфидов и тиолов (Martinez, et al., 2002).
Рентгенфлуоресцентные картины подтверждают тесное
пространственное подобие в распределении Zn и S в разных
почвенных агрегатах, что предполагает их химическую
ассоциацию. В целом, в почвенном агрегате доминируют Si, Al
и Ca, тогда как Zn и S находятся в меньшем количестве, а Fe
отсутствует вовсе. Но в агрегате имеются отдельные зоны, где
Zn и S превалируют. Следовательно, определенные участки
почвенной матрицы обогащены Zn и S, представляя собой
геохимическую локализацию в пределах почвенного агрегата.
Цинк может быть связан с серой, образуя неорганическую Zn–
S твердую фазу или же, образуя Zn–S органический комплекс
(Martinez, et al., 2002).
Окисление восстановленной серы в осушаемых торфах
высвобождает значительную долю рассеянных металловхалькофилов за счет перехода в мобильные сульфаты. Большая
часть серы остается в восстановленной форме в органогенных
почвах даже через 60 лет после их дренирования.
Цинк и сера одинаково распределены внутри почвенных
частиц. Опыты показали высокую растворимость Zn и его
доступность растениям, но низкую растворимость и
доступность кадмия, несмотря на высокое валовое содержание
Cd в почве. Вероятно, кадмий прочно закреплен органической
серой или же находится в составе сульфидов. Таким образом,
сера играет важную роль в биогеохимическом цикле Zn и Cd в
органогенных почвах.
Возможна также ассоциация ртути с серой. Это
подтверждается выявлением мелкокристаллических частиц
HgS в почвах штата Теннеси (Barnett et al., 1997). Интересно,
что положительная корреляция на макроскопическом уровне
между валовыми содержаниями ртути и серы отмечается
только в наиболее загрязненном слое почвы (13-81 см), тогда
как в поверхностном слое (0-8см) такой корреляции нет.
Очевидно, при низкой концентрации данных элементов в
почве, снижается вероятность образования подобных
ассоциаций.
ЗАКЛЮЧЕНИЕ
До появления синхротронной техники, т.е. до 1994 г.,
почти всю информацию о формах тяжелых металлов
почвоведы
получали
из
данных
химического
фракционирования. Длительное использование таких методик,
как схема Тессиера, позволило выявить их слабые места:
неселективное растворение индивидуальных соединений
металлов, а также перераспределение тяжелых металлов среди
фаз-носителей и образование новых форм металлов в ходе
экстракции. Над устранением, или смягчением, или просто
осознанием этих фактов нам надо задуматься.
С использованием синхротронной техники третьего
поколения открылись совершенно новые перспективы. За
прошедшие 10 лет удалось установить в почвах состав и
количество доминирующих фаз-носителей тяжелых металлов
и металлоидов, в первую очередь, свинца, цинка, ртути,
мышьяка.
Обнаружение
некоторых
форм
оказалось
неожиданным, оно никак не следовало из результатов
химического анализа почв. В то же время некоторые
ожидаемые формы тяжелых металлов обнаружены не были.
Пока с помощью новой техники проанализировано
небольшое количество сильнозагрязненных почвенных
образцов и промышленных отходов. Вероятно, с расширением
географии исследований можно ожидать и новых результатов
в области минералогии и химии тяжелых металлов и
металлоидов. Но уже сейчас полученные данные можно
использовать
для
корректировки
схем
химического
фракционирования тяжелых металлов, что позволит лучше
использовать этот простой и доступный метод. В частности,
анализ кинетики экстрагирования химических элементов
позволит расширить идентификацию фаз-носителей тяжелых
металлов.
СПИСОК ЛИТЕРАТУРЫ
Бабкина Э.И., Сатаева Л.В., Сурнин В.А. Загрязнение почв Российской
Федерации пестицидами и тяжелыми металлами за 10 лет // Современные
проблемы загрязнения почв. Межд. научн. конф. 24-28 мая 2004. М.: Издво Моск. ун-та, 2004. С. 28-30.
Большаков В.А., Краснова Н.М., Борисочкина Т.И., Сорокин С.Е.,
Граковский В.Г. Аэрогенное загрязнение почвенного покрова тяжелыми
металлами: источники, масштабы, рекультивация. М.: Почв. ин-т им. В.В.
Докучаева, 1993. 90 с.
Водяницкий Ю.Н. Химия и минералогия почвенного железа. М.: Почв.
ин-т им. В.В. Докучаева, 2003. 238 с.
Водяницкий Ю.Н. Оксиды марганца в почвах. М.: Почв. ин-т им. В.В.
Докучаева, 2005. 95 с.
Водяницкий Ю.Н., Большаков В. А., Сорокин С.Е., Фатеева Н.М.
Техногеохимическая
аномалия
в
зоне
влияния
Череповецкого
металлургического комбината // Почвоведение. 1995. № 4. С. 498-507.
Водяницкий Ю.Н., Васильев А.А., Кожева А.В. Тяжелые металлы в
аллювиальных почвах Среднего Предуралья // Докл. РАСХН. 2004. № 5. С.
23-25.
Горбатов В.С. Устойчивость и трансформация тяжелых металлов (Zn,
Pb, Cd) в почвах // Почвоведение. 1988. № 1. С. 35-43.
Добровольский В. В. Основы биогеохимии. М.: ACADEMIA, 2003. 397 С.
Ковда В.А. Биогеохимия почвенного покрова. М.: Наука, 1985. 263 с.
Ладонин Д.В. Соединения тяжелых металлов в почвах – проблемы и
методы изучения // Почвоведение. 2002. № 6. С. 682-692.
Мотузова Г.В. Соединения микроэлементов в почвах: системная
организация, экологическое значение, мониторинг. Эдиториал УРСС. М., 1999.
166 с.
Орлов Д.С., Садовникова Л.К., Лозановская И.Н. Экология и охрана
биосферы при химическом загрязнении. М.: Высшая школа, 2002. 334с.
Пампура Т.В. Поглощение меди и цинка черноземом типичным в условиях
модельных экспериментов: Дис. … канд. биол. наук. Пущино, 1996. 170 с.
Пампура Т.В., Пинский Д.Л., Остроумов В.Г., Гершевич В.Д., Башкин В.Н.
Экспериментальное изучение буферности чернозема при загрязнении медью и
цинком // Почвоведение. 1993. № 2. C. 104-110.
Переломов Л.В. Факторы иммобилизации тяжелых металлов в серых
лесных и аллювиальных почвах Среднерусской возвышенности: Автореф.
дис. … канд. биол. наук. М., 2001. 20 с.
Перельман А.И. Геохимия ландшафта. М.: Высшая школа, 1975. 341 с.
Пинский Д.Л. Ионообменные процессы в почвах. Пущино, 1997. 164 с.
Трухина Л.Ф. Почвы пойм малых рек и пути повышения их плодородия
и производительности (на примере Ивановской области): Автореф. дис. ...
канд. c.-х. наук. М., 1988. 23 с.
Чухров Ф.В., Горшков А.И., Дриц В.А. Гипергенные окислы марганца.
М.: Наука, 1989. 208 с.
Ajayi S.O., Vanloon G. W. Studies of redistribution during the analytical
fractionation of metals in sediments // Sci. Total Environ. 1989. V. 87/88. P.
171-187.
Barnett M.J., Harris L.A., Tufner R.R., Stevenson R.J., Henson T.J., Melton
R.C., Hoffman D.P. Formation of mercuric sulfide in soil // Environ. Sci.
Technol. 1997. V. 31. P. 3037-3043.
Bloom N.S., Preus E., Katon J., Hiltner M. Selective extractions to
biogeochemically relevant fractionation of inorganic mercury in sediments and
soils // Anal. Chim. Acta. 2003. V. 479. N 2. P. 233-248.
Boutron C.F., Gorlasch U., Candelone J.P., Bolshov M.A., Delmas R.J.
Decrease in anthropogenic lead, cadmium and zinc in Greenland snows since late
1960s // Nature. 1991. V. 353. P. 153-156.
Brown G.E., Foster A.L., Ostergren J.D. Mineral surface and bioavailability
of heavy metals: A molecular-scale perspective // Proc. Natl. Acad. Sci. USA.
1999. V. 96. P. 3388-3395.
Chiarello R.P., Sturchio N.C., Grace J.d., Geissbuhler P., Sorensen L.B.,
Cheng L., Xu S. Otavite-calcite solid-solution formation at the calcite-water
interface in situ by synchrotron X-ray scattering // Geochim. Cosmochim Acta.
1997. V. 61. P. 1467-1474.
Cotter-Howells J.D., Cheampness P.E., Charnock J.M., Pattrick R.A.D.
Identification of pyromorphite in mine-waste contaminated soils by ATEM and
EXAFS // Eur. J. Soil Sci. 1994. V. 45. P. 393-402.
Cullen W.R., Reomer K.J. Arsenic speciation in the environment // Chem.
Rev. 1989. V. 89. P. 713-764.
Dahn R., Scheidegger A.M., Mаnceau A., Schlegel M., Baeyens B., Bradbary
H., Morales M. Neoformation of Ni phyllosilicate upon Ni uptake on
montmorillonite. A kinetic study by powder and polarized EXAFS // Geochim.
Cosmochim Acta. 2002. V. 66. P. 2335-2347.
Davis A., Ruby M.V., Bergstrom P.D. Bioavailability of arsenic and lead in
soils from the Butte, Montana, Mining District // Environ. Sci. Technol. 1992. V.
26. P. 461-468.
Dupont L., Guillon E., Bouanda J., Dumonceau J., Aplincourt M. EXAFS and
EXANES studies of retention of copper and lead by lignocelluloses biomaterial //
Environ. Sci. Technol. 2002. V. 36. P. 5062-5066.
Filgueiras A.D., Lavilla I., Bendicho C. Chemical sequential extraction for metal
partitioning environmental soil samples // J. Eviron. Monit. 2002. V. 4. P. 823-857.
Ford R.G., Scheinost A.C. Sparks D.L. Frontiers in metal/precipitation
mechanisms on soil mineral surfaces // Adv. Agron. 2001. V. 74. P. 41-62.
Fuller C.C., Davis J.A., Waychunas G.A. Surface chemistry of ferrigydrite:
Part 2. Kinetics of arsenate adsorption and coprecitation //Geochim. Cosmohim.
Acta. 1993. V. 57. P. 2271.
Grafe M., Eick M.J., Grossel P.R. Adsorption of arsenate (V) and arsenite
(III) on goethite in the presence and absence of dissolved organic carbon // Soil
Sci. Soc. Am. J. 2001. V. 65. P. 1680-1687.
Griffin T.M., Rabehorst M.C., Fanning D.S. Iron and trace metals in some tidal
marsh soils of the Chesapeake Bay // Soil Sci. Soc. Am. J. 1989. V. 53. P. 1010-1019.
Haering K.C., Rabenhorst M.C., Fanning D.S. Sulfur speciation in Chesapeake Bay
tidal marsh soils // Soil Sci. Soc. Am. J. 1989. V. 53. P. 500-505.
Han F.X., Banin A, Triplett G.B. Redistribution of heavy metals in arid-zone soils
under a wetting-drying cycle soil moisture regime // Soil Sci. 2001. V. 166. P. 18-28.
Hansel C. M., Fendorf S., Sutton S., Newville M. Characterization of Fe
plaque and associated metals on the roots of mine-waste impacted aquatic plants //
Environ. Sci. Technol. 2001. V. 35. P. 3863-3868.
Heinrichs H., Mayer R. Distribution and cycling of major and trace elements
in two Central European forest ecosystems // J. Env. Qual. 1977. V. 6. P. 402-407.
Hesterberg D., Savers D.E., Zhou W., Plummer G.M. Robarg W.P. X-ray
adsorption spectroscopy of lead and zinc speciation in a contaminated groundwater
aquifer // Environ. Sci. Technol. 1997. V. 31. P. 2840-2846.
Hundall L.S., Carmo A.M., Bleam W.L., Thompson M.L. Sulfur in biosolisderived fulvic acid: characterization by XANES spectroscopy and selective
dissolution approaches // Environ. Sci. Technol. 2000. V. 34. P. 5184-5188.
Jackson B.P., Miller W.P. Effectiveness of phosphate and hydroxide for
desorbtion of arsenic and selenium species from iron oxides // Soil Sci. Soc. Am.
J. 2000. V. 64. P. 1616-1622.
Jain A., Loeppert R.H. Effect of competing anions on the adsorption of
arsenate and arsenite by ferrigydrite // J. Env. Qual. 2004. V. 29. P. 1422-1430.
Jun Y.S., Martin S.T. Microscopic observations of reductive manganite
dissolution under oxic conditions // Environ. Sci. Technol. 2003. V. 37. P. 23632370.
Karathanasis A.D., Thompson Y.L. Mineralogy of iron precipitates in a
constructed acid mine drainage wetland // Soil Sci. Soc. Am. J. 1995. V. 59. P. 17731781.
Kheboian C., Bauer C.F. Accuracy of selective extraction procedures for metal
speciation in model aquatic sediments // Anal. Chem. 1987. V. 59. P. 1417-1423.
Kim C.S., Bloom N.S., Rytuba J.J., Brown G.E. Mercury speciation by X-ray
absorption fine structure spectroscopy and sequential chemical extractions: A
comparison of speciation methods // Environ. Sci. Technol. 2003. V. 37. P.
5102-5108.
Kneebone P.E., O/Day P.A., Jones N., Hering J.G. Deposition and fate of
arsenic in iron- and arsenic-enriched reservoir sediments // Envizon. Sci. Technol.
2002. V. 36. P. 381-386.
Kuo S. Concurrent sorption of phosphate and zinc, cadmium or calcium by
hydrous ferric oxide // Soil Sci. Soc. Am. J. 1986. V. 50. N 6. P. 1412-1419.
Labrenz M., Druschel G.K., Thomsen-Ebert T., Gilbert B., Weich S.A., Kemner
K.M., Logan G.A.,Summons R.E., De Stasio G., Bond P.L., Lai B., Kelly S.D.,
Banfield J.F. Formation of sphalerite (ZnS) deposits in natural biofilms of sulfateredusing bacteria // Science. 2000. V. 290. P. 1744-1747.
La Force M.J., Fendorf S. Solid-phase iron characterization during common
selective sequential extraction // Soil Sci. Soc. Am. J. 2000. V. 64. P. 1608-1615.
Laperche V., Logan T.L., Gaddam P., Ptaina S.J. Effect of apatite
amendments on plant uptake of lead from contaminated soil // Environ. Sci.
Technol. 1997. V. 31. P. 2745-2753.
Lower S.K., Maurice P.A., Traina S.J. Simultaneous dissolutoin of
hydroxylapatite and precipitation of hydroxypyromorphite: Direct evidence of
homogeneous nucleation // Geochim. Cosmohim. Acta. 1998. V. 62. P. 17731780.
Mа Q.Y., Traina S.J., Logan T.J., Ryan J.A. In situ lead immobilization by
apatite // Environ. Sci. Technol. 1993. V. 27. P. 1803-1810.
Mаnceau A., Boisset M.C., Sarret G., Hazemann J.L., Mench M., Cambier
P., Prost R. Direct determination of lead speciation in contaminated soils by
EXAFS spectroscopy // Environ. Sci. Technol. 1996. V. 30. P. 1540-1552.
Mаnceau A., Lanson B., Schlegel M.L., Harge J.C., Musso M., EybertBerard L., Hazemann J.L., Chateigner D., Lamble G.M. Quantitative Zn
speciation in smelter-contaminated soils by EXAFS spectroscopy // Am. J. Sci.
2000. V. 300. P. 289-343.
Mаnceau A., Marcus M.A., Tamura N. Quantative speciation of heavy metals in
soils and sediments by synchrotron X-ray techniques // Applications of Synchrotron
Radiation in Low-Temperature Geochemistry and Environmental Science. Reviews
in Mineralogy and Geochemistry. Washington, DC. 2002. V. 49. P. 341-428.
Mаnceau A., Tamura N., Celestre R.S., Macdowell A.A. Geoffroy N., Sposito
G., Padmore H.A. Molecular-scale speciation of Zn and Ni soil ferromanganese
nodules from loess soils of the Mississippi Basin // Environ. Sci. Technol. 2003.
V. 37. P. 75-80.
Mаnning B.A., Fendorf S.E., Goldberg S. Surface structures and stability of
arsenic(III) on goethite: spectroscopic evidence for innersphere complexes //
Environ. Sci. Technol. 1998. V. 32. P. 2383.
Mаnning B.A., Goldberg S. Modeling competitive adsorption of arsenate
with phosphate and molybdate on oxide minerals // Soil Sci. Soc. Am. J. 1996.
V. 60. P. 121-131.
Martinez C.E., McBride M.B., Kandianis M.T., Duxbury J.M., Yoon S.J., Bleam
W.F. Zinc-sulfur and cadmium-sulfur association in metalliferrous peats:
evidence from spectroscopy, distribution coefficients and phytoavailability //
Environ. Sci. Technol. 2002. V. 36. P. 3683-3689.
McBride M.B. Reactions controlling heave metal solubility in soils // Adv.
Soil Sci. 1989. V. 10. P. 1-47.
Morin G., Juillot F., Casiot C., Bruneel O., Persone J.C., Elbaz-poulichet
F., Leblanc M., Ildefonse P., Calas G. Bacterial formation of tooeleite and mixed
arsenic (III) or arsenic (V) – iron (III) gels in the Carnoules acid mine drainage,
France. A XANES, XRD, and SEM study // Environ. Sci. Technol. 2003. V. 37.
P. 1705-1712.
Morin G., Ostergren J.D., Juillot F., Ildefonse P., Calas G., Brown J.E.
XAFS determination of the chemical form of lead in smelter-contaminated soils
and mine tailings: Importance of adsorption process // Am. Mineral. 1999. V. 84.
P. 420-434.
Morra M.G., Fendorf S.E., Brown P.D. Speciation of sulfur in humic and
fulvic acids using X-ray absorption near-edge structure (XANES) spectroscopy //
Geochim. Cosmohim. Acta. 1997. V. 61/ 683-688.
Ostergren J. D., Brown G.E., Parks G.A., Tingle T.N. Quantitative
speciation of lead in selected mine tailing from Leadvill, Co. // Environ. Sci.
Technol. 1999. V. 33. № 10. P. 1627-1636.
Paktung D., FosterА., Laflamme G. Speciation and characterization of
arsenic in Ketza River mine tailings using X-ray adsorption spectroscopy //
Environ. Sci. Technol. 2003. V. 37. P. 2067-2074.
Raksasataya M., Langon A.G., Kim N.D. Assessment of extent of lead
redistribution during sequential extraction by two different methods // Analyt.
Chem. Acta. 1996. V. 332. P. 1-14.
Roberts D.R., Scheinost A.C., Sparks D.L. Zink speciation in a smeltercontaminated soil profile using bulk and microscopic techniques // Environ. Sci.
Technol. 2002. V. 36. P. 1742-1750.
Robson A.D. Zinc in soil and plants. Klumer: Acad. Publ. Australia, 1993.
Sаntillan-Medrano J., Jurinak J.J The chemistry of lead and cadmium in
soil: solid phase formation // Soil Sci. Soc. Am. Proc. 1975. V. 39. P. 851-858.
Savage K.S., Tingle T.N., O/Day P.A., Waychunas G.A., Bird D.K. Arsenic
speciation in pyrite and secondary weathering phases, Mother Lode Gold District,
Tuolumne County, California // Appl. Geochem. 2000. V. 15. P. 1219-1244.
Scheckel K.G., Impellitterri C.A., Ryan J.A., Mcevoy T. Assessment of
extraction procedure for perturbed lead-contaminated samples with and without
phosphorus amendments // Environ. Sci. Technol. 2003. V. 37. P. 1892-1898.
Scheckel K.G., Scheinost A.C., Ford R.G., Sparks D.L. Stability of layered
Ni hydroxide surfase precipitates – A dissolution kinetics study // Geochim.
Cosmochim. Acta. 2000. V. 64. P. 2727-2735.
Scheinost A.C., Kretzchmar R.S., Pfister S. Combining selective sequential
extractions, X-ray absorptoin spectroscopy, and principal component analysis for
quantitative zinc speciation in soil // Environ. Sci. Technol. 2002. V. 36. P. 50215028.
Schlegel M.L., Mаnceau A., Charlet L., Chateigner D., Hazemann J.l. Sorption of
metal ions on clay minerals. III. Nucleation and epitaxial growth of Zn phyllosilicate on
the edges of hectorite // Geochim. Cosmochim. Acta. 2001. V. 65. P. 4155-4170.
Schulze D.G., Sutton S.R., Bajt S. Determing manganese oxidation state in
soils using X-ray absorption near-edge structure (XANES) spectroscopy // Soil
Sci. Soc. Am. J. 1995. V. 59. P. 1540-1548.
Shen Xiao-Quan, Chen Bin. Evaluation of sequential extraction for
speciation of trace metals in model soil containing natural minerals and humic
acid // Analyt. Chem. 1993. V. 65. P. 802.
Shotyk W., Weiss D., Appleby P.G., Cheburkin A.K., Frei R., Gloor M.,
Kramers J.D., Reese S., Van Der Knapp W.O. History of atmospheric lead
deposition since 12.370 14C yr BP from a peat bog, Jura mountains, Switzerland
// Science. 1998. V. 281. P. 1635-1640.
Sutton S.R., Rivers M.L. Hard X-ray synchrotron microprobe techniques and
applications / Synchrotron X-ray methods in clay science // Clay Min. Soc. Am.
1999. P. 146-163.
Тeo B.K. EXAFS: Basic Principles and Data Analysis. Inorganic Chemistry
Concepts 9. Berlin: Springer-Verlag, 1986.
Tessier A., Campbell P.G.O., Bisson M. Sequential extraction procedure for the
speciation of the particulate trace metals // Analyt. Chem. 1979. V. 51. P. 844-851.
Trаina S.J., Laperche V. Contaminant bioavailability in soils, sediments, and
aquatic environments // Proc. Natl. Acad. Sci. USA. 1999. V. 96. P. 3365-3371.
Van der Lebie D., Schwitzguebel J.P., Glass D.J. Vangronsveld J., Baker A.
Assessing phytoremediation/s progress in the United States and Europe //
Envizon. Sci. Technol. 2001. V. 35. P. 446A-452A.
Waychunas G.A., Rea B.A., Fuller C.C., Davis J.A. Surface chemistry of
ferrigydrite: Part 1. EXAFS studies of the geometry of coprecipitated and adsorbed
arsenate // Geochim. Cosmochim. Acta. 1993. V. 57. P. 2251-2269.
Watson E.B. Surfase enrichment and trace-element uptake during crystal
growth precipitation of Co(II)(aq) on Al2O3 // Geochim. Cosmochim. Acta.
1996. V. 60. P. 5013-5020.
Whalley C., Grant. A. Assessment of the phase selectivity of the European
Community Bureau of Reference (BCR) sequential extraction procedure for metals in
sediment // Analyt. Chem. Acta. 1994. V. 291. P. 287-295.
Xia K., Blem W., Helmke P.A. Studies of nature of Cu2+ and Pb2+ binding
sites in soil humic substances using x-ray adsorption spectroscopy // Geochim.
Cosmochim. Acta. 1997. V. 61. P. 2211-2221.
Xia K., Weesner F., Bleam W.F., Bloom P.R., Skyllberg U.L., Helmke P.A.
XANES studies of oxidation states of sulfur in aquatic and soil humic substances
// Soil Sci. Soc. Am. J. 1998. V. 62. P. 1240.
Zinder B., Furrer G., Stumm W. The coordination chemistry of weathering. 2.
Dissolution of Fe(III) oxides // Geochim. Cosmochim. Acta. 1986. V. 50. P. 18611869.
СОДЕРЖАНИЕ
Введение ...................................................................//.............................................3
Глава 1. Изучение тяжелых металлов и металлоидов методами
синхротронной рентгеновской радиации .......Error! Bookmark not defined.
Рентгеновская микрофлуоресценция .............Error! Bookmark not defined.
Рентгеновская микродифракция .....................Error! Bookmark not defined.
Анализ рентгеновских спектров вблизи края полосы поглощенияError! Bookmark not defined.
Расширенный анализ тонкой структуры спектров поглощенияError! Bookmark not defined.
Глава 2. Методы последовательного экстрагирования тяжелых
металлов ............................................................Error! Bookmark not defined.
Система Тессиера..............................................Error! Bookmark not defined.
Европейская система BCR ...............................Error! Bookmark not defined.
Расчет вклада фаз-носителей в закрепление тяжелых металловError! Bookmark not defined.
Глава 3. Химия и минералогия тяжелых металлов и металлоидовError! Bookmark not defined.
Главные механизмы закрепления тяжелых металлов в почвахError! Bookmark not defined.
Формы тяжелых металлов в почвах ...............Error! Bookmark not defined.
Глава 4. Формы тяжелых металлов и металлоидов в загрязненных
почвах ................................................................Error! Bookmark not defined.
Глобальное, региональное и локальное загрязнение почвError! Bookmark not defined.
Соединения мышьяка .......................................Error! Bookmark not defined.
Соединения свинца ...........................................Error! Bookmark not defined.
Соединения цинка .............................................Error! Bookmark not defined.
Соединения ртути .............................................Error! Bookmark not defined.
Контроль селективности вытяжек ..................Error! Bookmark not defined.
Глава 5. Формы серы и тяжелых металлов в органогенных почвахError! Bookmark not defined
Степени окисления серы в органическом веществе почвError! Bookmark not defined.
Ассоциации тяжелых металлов с серой в почвах ........................................... 1
Заключение......................................................................................................... 3
Список литературы............................................................................................ 4